/






























































































































































































































































































































































Author: Уэйлес С.
Tags: химия основные процессы и аппараты химической технологии
ISBN: 5—03—001105—6
Year: 1989
Similar




Text
С.Уэйлес ФАЗОВЫЕ
РАВНОВЕСИЯ
В ХИМИЧЕСКОЙ
ТЕХНОЛОГИИ
ИЗДАТЕЛЬСТВО «МИР»
PHASE EQUILIBRIA IN
CHEMICAL ENGINEERING
Stanley M. Walas
Department of Chemical and Petroleum
Engineering
University of Kansas
and
The C.W. Nofsinger Company
BUTTERWORTH PUBLISHERS
Boston • London
Sydney • Wellington • Durban • Toronto
С. Уэйлес
ФАЗОВЫЕ
РАВНОВЕСИЯ
В ХИМИЧЕСКОЙ
ТЕХНОЛОГИИ
В 2-х частях
2
Перевод с английского
канд. техн, наук А.В. Беспалова,
канд. техн, наук А.П. Жукова и
В. В. Паукова
под редакцией
д-ра техн, наук, проф. В.С. Бескова
МОСКВА «МИР»
1989
2*
УДК 541
Уэйлес С.
У97 Фазовые равновесия в химической технологии: В 2-х
ч. Ч. 2. Пер. с англ. — М.: Мир, 1989.— 360 с., ил.
ISBN 5—03—001105—6
Издание (автор проф. Канзасского университета, США, С. Уэйлес), в кото-
ром собраны и систематизированы все практические методы расчета равновесий
для широкого круга систем (пар (газ) — жидкость; жидкость — жидкость; жид-
кость —твердое тело). Отличительная особенность книги — энциклопедическая
широта охвата обсуждаемых проблем. Формульный, графический и цифровой
справочный материалы, включая алгоритмы для расчета на ЭВМ, удачно допо-
лняют друг друга.
В русском издании выходит в 2-х частях.
В ч. 2 рассматриваются различные типы равновесия (пар — жидкость, жид-
кость — жидкость и др.), методы оценки изменений энтропии и энтальпии и
основные экспериментальные методы исследования фазового равновесия.
Для исследователей, инженеров-практиков, использующих существующие
разработки и методы при создании новых технологий и обеспечивающего их ин-
женерного оборудования, а также для студентов химико-технологических вузов.
2801000000-335 147 „„
041(01)-89
ББК 35.114
Редакция литературы по химии
ISBN 5—03—001105—6 (русск.)
ISBN 5—03—001107—2
ISBN 0—409—95162—5 (англ.)
© 1985 by Butterworth
Publishers. All rights reserved.
© перевод на русский язык,
«Мир», 1989
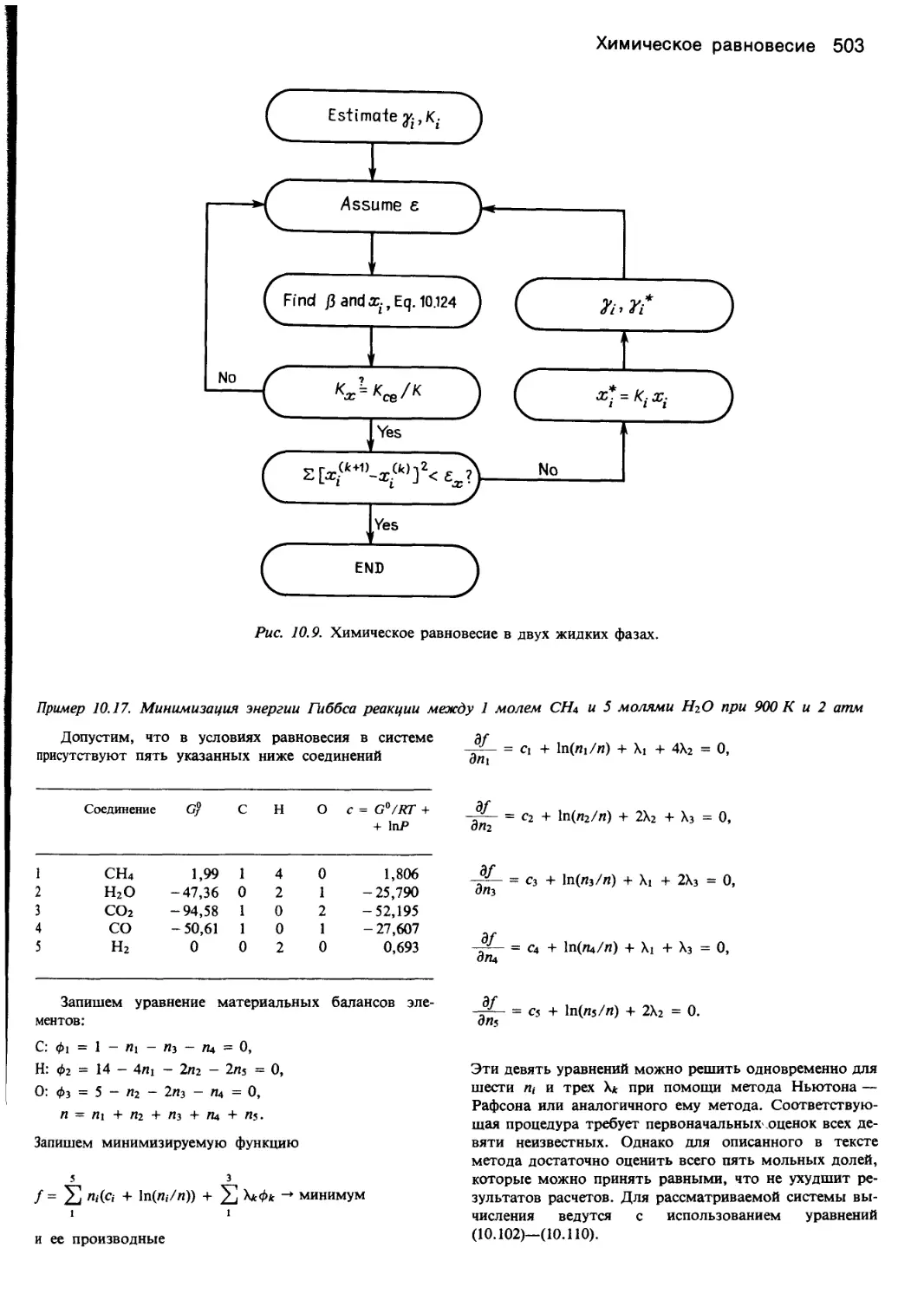
6. РАВНОВЕСИЕ В СИСТЕМАХ ПАР—ЖИДКОСТЬ
Системы пар — жидкость широко распространены
и имеют важное практическое значение, равновесия та-
кого вида интенсивно изучаются и, вероятно, исследова-
ны лучше, чем любые типы равновесий.
где коэффициент Пойнтинга
(PF), = exp yiL /R Т) dP,
(6.8)
6.1. Паровая и жидкая фазы
Эта глава посвящена определению состояния рав-
новесия между паровой и жидкой фазами при заранее
заданных составе и относительном количестве фаз, тем-
пературе, давлении, энтальпии, энтропии или других
термодинамических свойствах. Наиболее часто равнове-
сие оценивают посредством коэффициентов фугитивнос-
ти (летучести) и активности.
Межфазовое равновесие требует равенства Т, Р и
парциальных фугитивностей отдельных компонентов в
любой точке системы, т. е.
Ли = 7(2) = ...
Р(1) = Р(2) = ...
Л(1) = /1(2) = •••
для каждой фазы (/)
для каждой фазы
для каждой фазы (номер
компонента i = 1,2, .... п)
(6.1)
(6.2)
(6.3)
(см., однако, в разд. 2.10.2 об исключении равенства
давления в этих условиях). Для удобства мы рассмот-
рим отдельно системы, в которых паровая фаза нахо-
дится в контакте с одной и несколькими жидкими
фазами.
6.1.1. Равновесие в системе с одной жидкой фазой.
Выразим условия равновесия пар — жидкость
(6.4)
и преобразуем это равенство к выражению, содержаще-
му коэффициенты фугитивности,
У&уР = х,фцР,
(6.5)
где парциальные коэффициенты фугитивности могут
быть функцией уравнения состояния. В том случае, если
для жидкой фазы это условие невыполнимо, необходи-
мо использовать коэффициенты активности:
У,фгК7’ = xi1ifiL
= xi7i<t>SiPsi (PF),,
(6.6)
(6.7)
представляет собой поправку величины фугитивности
жидкой фазы в интервале от давления компонента до
давления пара в системе.
Уравнение (6.7) можно преобразовать и предста-
вить в виде пропорциональной зависимости между со-
ставом фаз в системе:
г, 0jP’(PF),
" = х,. (6.9)
Однако, так как коэффициенты парциальной фугитив-
ности и активности зависят от состава каждой из фаз,
а также от Т и Р, равновесный состав в конкретных ус-
ловиях можно успешно определить аппроксимацией, как
будет показано позднее. В ряде случаев уравнение (6.9)
можно несколько упростить:
1) при относительно низком давлении (PF), стре-
мится к единице;
2) отношение коэффициентов фугитивности может
приближаться к единице. Некоторые варианты уравне-
ний для определения коэффициентов фугитивности и ак-
тивности будут рассмотрены позднее.
6.1.2. Равновесие в системе с несколькими жидкими
фазами. Если в системе присутствует более одной жид-
кой фазы, уравнение (6.4) применяется к каждой из них.
Для системы с двумя жидкими фазами (параметры вто-
рой из них обозначим звездочками) уравнения (6.5)—
(6.7) примут следующий вид:
y^ivP = х&цР = xfofLP
= xiTifiL = x*y*fiL
= xzyz0jPf(PP)z = x*y*0fP«(PF)z.
(6.10)
C6.ll)
(6.12)
Системы с более чем двумя или тремя жидкими фазами
редко представляют практический интерес. Многофаз-
ность жидкости наиболее характерна для органических
систем, содержащих воду. Чаще всего необходимо опре-
делить количество и состав каждой из фаз, находящихся
в равновесии. Проблемы, обусловленные несмешивае-
мостыо (расслаиванием) жидкостей, описываются в
этой главе, а поведение более сложных систем жид-
кость — жидкость — пар — в гл. 7.
6.1.3. Равновесия в системах, содержащих пары со
сверхкритической температурой. Уравнение парциаль-
ной фугитивности жидкой фазы
fiL=xilifiL (6.13)
описывает жидкое состояние, для которого фугитив-
ность чистого компонента оценивается по гипотетиче-
скому состоянию, если вещество представляет собой
пар со сверхкритической температурой. Оценить пове-
дение жидкости в этой ситуации можно несколькими
способами.
1. Вопрос о сверхкритической температуре исклю-
чается путем использования уравнения (6.5) с одним и
тем же уравнением состояния для обеих фаз. Такой под-
ход применяется для легких углеводородов и других не-
полярных веществ.
2. С помощью подходящих параметров устанавли-
вают корреляцию между значениями /(£, полученными
из выражения
(6.14)
Ji xi
и экспериментальными данными, полученными в до-
ступной для измерений области, и допускают возмож-
ность экстраполяции. Пример такой корреляции описан
в разд. 6.5.
3. Корреляцию fiL проводят, используя ацентриче-
ский коэффициент и приведенные температуру и давле-
ние, как, например, в методе Чао — Сидера (разд.
6.2.3).
4. Путем экстраполяции точного уравнения давле-
ния пара (не уравнения Антуана) оценивают фугитив-
ность жидкости из выражения
fiL = P^Si(PF)iQ, (6.15)
и коррелируют как
ln/}L = Си + C2i/T + C3ZT + С4г- In Т+ C5iT2 (6.16)
согласно Праузницу и др. [563] (см. приложение Б, где
приведены значения этих коэффициентов уравнения для
92 веществ). Коэффициент Пойнтинга (PF)® — это по-
правка на давление, величина которой меняется от дав-
ления насыщения Pf до 0.
5. Парциальный коэффициент фугитивности жид-
кости можно выразить при помощи константы типа
константы Генри
fiL = kHixi- (6.17)
Зависимость кш от состава смеси и других характери-
стик описана в разд. 3.5, она детально изучена ван Нес-
сом и Эбботом [703] и Праузницем и др. [563].
6. Кинг и др. [397] успешно применяли уравнение
(6.7) в сочетании с уравнением Скэтчарда — Гиль-
дебранда — Флори — Хаггинса и Антуана для получе-
ния необходимых данных о веществах, чьи критические
температуры превышают температуру системы не бо-
лее чем на 50°С. Эта работа обсуждается в разд. 6.5.
Первый и третий из перечисленных методов рассмотре-
ны в данной главе довольно подробно.
6.2. Константа фазового равновесия
Коэффициент распределения
Ki = yi/xi, (6.18)
называемый также константой фазового равновесия
(КФР), — это ключевое понятие в количественном ана-
лизе парожидкостного равновесия. Многие равновесные
соотношения компактно выражаются через К, но, по-
скольку Kt зависит от Т, Р и состава обеих фаз, реше-
ния типичных задач парожидкостного равновесия,
выраженные через К, должны включать эти зависимос-
ти. Следует заметить, что идеальные значения К,, не
зависящие от состава, рассчитать легко и что они могут
служить отправными величинами для более точных
расчетов.
Исходя из уравнений (6.5) — (6.7), КФР можно
представить в виде следующих зависимостей:
Ki — yi/xi - <t>iL I<t>iV (6.19)
: (б.:»,
- ЪФиУФгУ
y^P^PF); (6'21)
02 vp (6.22)
В ряде случаев выражение ф!(PF)i/<t>iv мало отличается
от единицы (см пример 6.1), отсюда
(6.23)
Это выражение часто справедливо при низких, поряд-
ка 5—6 атм, давлениях. Идеальное значение коэффици-
ента распределения определяют как
(Ki) ideal = Р* /Р- (6.24)
Некоторые способы оценки К по уравнениям
(6.19) —(6.22) будут рассмотрены отдельно. Исчерпыва-
ющая оценка многих методов дана в работе Доберта и
др. [31]; некоторые выводы этих авторов суммированы
в разд. 6.2.6. Константа фазового равновесия Ki будет
применена для расчета точки росы, температуры нача-
ла кипения и температуры испарения.
6.2.1. Уравнения состояния. К числу уравнений со-
стояния пригодных при определенных условиях для рас-
чета коэффициентов фугитивности обеих фаз, относятся
уравнения Соава, Пенга — Робинсона, Бенедикта —
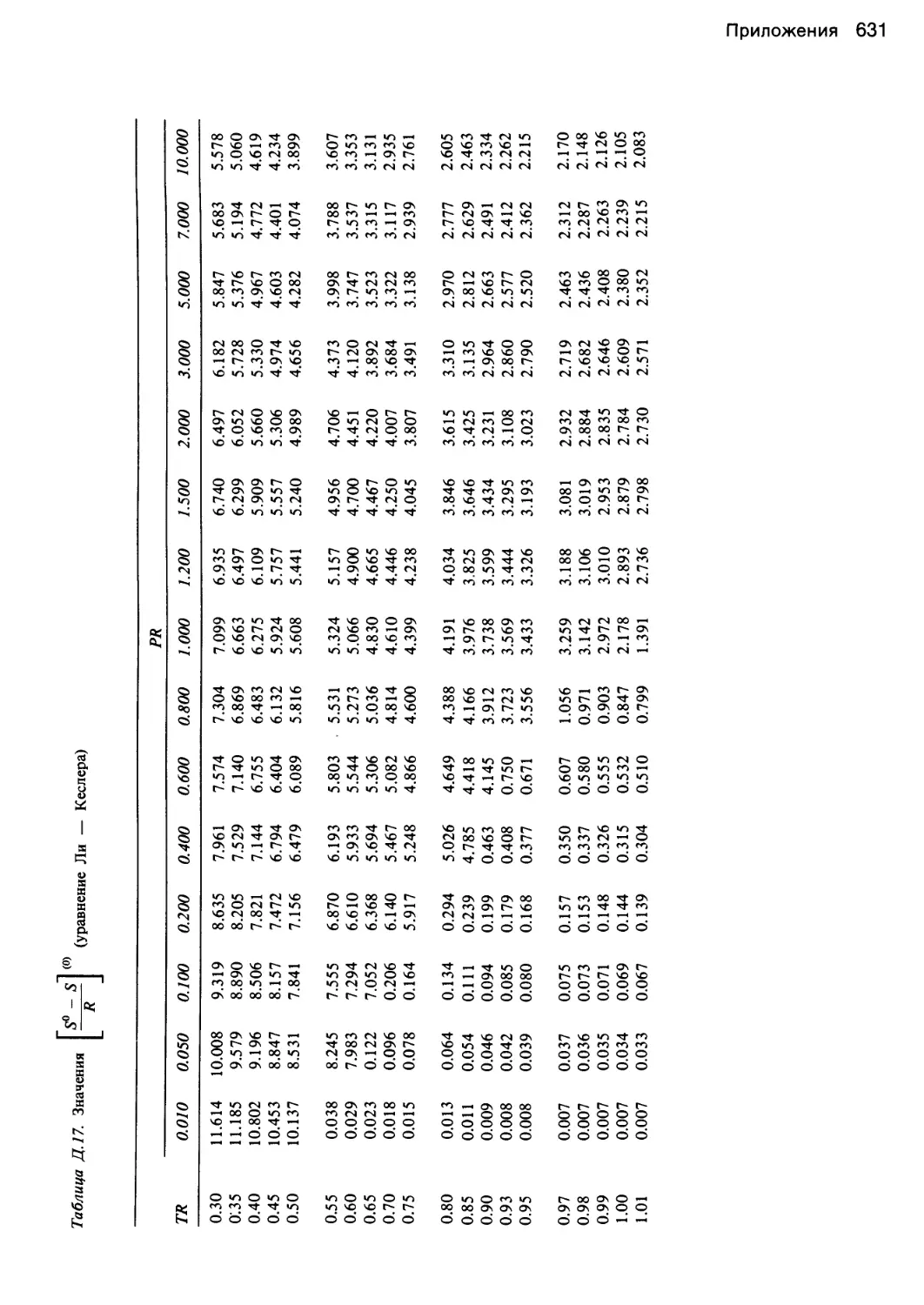
Уэбба — Рубина — Старлинга и Плекера — Ли — Кес-
лера. Первым из них мы воспользуемся в иллюстратив-
ных целях. Соответствующие формулы приведены в
табл. 1.11, 3.3 и 3.4. Полиномная форма уравнения
z3 -z2 + (А - В ~B2)z - АВ = 0 (6.25)
Равновесие пар —жидкость 311
разрешима для самых больших корней при фиксирован-
ном составе фазы и для самых малых корней при за-
данном составе жидкости. Два индивидуальных
парциальных коэффициента фугитивности определяют
далее по уравнению Соава
— In z
In ф/
(6.26)
аа Гб/ 2 V7aa), "I / b \
+ 1 о <7^ , /-- In I 1 + I ,
bRT \_b \аа \ V J
после подстановки соответствующих серий данных о
составах и давлениях (сжимаемостях) смесей.
Уравнение Бенедикта — Уэбба — Рубина долгое
время использовалось как стандартное для определения
К, обеих фаз, однако, как считают некоторые исследова-
тели, оно слишком сложно, чтобы его имело бы смысл
применять при повторяющихся расчетах, например при
решении задач, связанных с дистилляцией. В настоящее
время для решения такого рода задач разработаны бо-
лее простые методы расчета, примером может служить
программа Кристиансена и др. [222] для многокомпо-
нентной дистилляции, включая уравнение Соава. Ре-
зультаты, полученные по основному алгоритму с
акцентом на критические области и зоны высокого дав-
ления, рассмотрены на основе уравнения Соава —
Асселина и др. [165]. Схема дистилляции с применением
уравнения Соава или Пенга —Робинсона для оценки К,
в задачах криогенной техники превосходит метод
Чао — Сидера [632]. Сим и Доберт [637] пришли к вы-
воду, что метод Соава наиболее пригоден для расчетов
процессов испарения нефтяных смесей. Они разделяли
смесь на фракции с интервалом по температуре кипения
в 25 ° С и соотносили среднюю точку кипения Ть и плот-
ность 5 с молекулярной массой М и критическими ха-
рактеристиками, необходимыми для решения уравнения
Соава. Ниже приведены эти эмпирические зависимости:
М = 5,8О5(1О“5) Th2’3776/5°>9371, (6.27)
Рс = 6,1483(1012)£2>4853/Т2’3177, (6.28)
Тс = 0,5776 ехр (4,2009 T^08615S0’04614). (6.29)
(единицы измерения Па, К).
6.2.2. Методика расчета через давление пара. В
уравнении (6.9) оба коэффициента фугитивности отно-
сятся к паровой фазе, поэтому для определения коэффи-
циентов фугитивности газовой фазы достаточно одного
любого приемлемого уравнения состояния. Например,
если используется уравнение (6.26), фугитивность насы-
щенного пара находят путем подстановки а, = а и
bi = b. Для определения коэффициентов активности
можно воспользоваться любым приемлемым уравнени-
ем, но уравнение Скэтчарда — Гильдебранда наиболее
удобно, поскольку в него входят свойства только чис-
тых компонентов. Если уравнение достаточно точно, то
допустима некоторая экстраполяция значений давления
пара ряда компонентов до температур, превышающих
критические. Другие методы расчета при сверхкритиче-
ских температурах приведены в разд. 6.1.3 и 6.5.
6.2.3. Соотношение коэффициентов фугитивности
чистых жидкостей. Первая методика такого типа была
разработана Чао и Сидером [218] (см. также [318, 598,
428, 455]. Здесь в качестве иллюстрации мы воспользу-
емся методикой, предложенной Робинсоном и Чао, а
также Маффиоло и сотр. которая предусматривает ис-
пользование основных соотношений Чао — Сидера с
числовыми значениями коэффициентов, найденными
Грейсоном и Стридом, и расширенного уравнения Скэт-
чарда — Гильдебранда. Эта методика применяется, в
частности, в примере 6.7. В табл. 6.1 приведены необ-
ходимые для расчета формулы и числовые коэффициен-
ты и указаны ограничения, которые первоначально
были выдвинуты в оригинальных статьях, а позднее
обобщены Ленором и Коппани [435]. В уравнении (6.21)
парциальные коэффициенты фугитивности получены из
уравнения Редлиха — Квонга, а коэффициенты фугитив-
ности чистых веществ скоррелированы с ацентрическим
коэффициентом посредством соотношения типа предло-
женного Питцером — Керлом
,П 0j£ = 1П 0£O) + ф 1П ф(£. (6.30)
Логарифмические члены, стоящие в уравнении справа,
выражены через значения ТГ и Рг. Вначале коэффициен-
ты активности определяли из уравнения Скэтчарда —
Гильдебранда
In у,- = Г,(5(- - 8)2/R Т, (6.31)
но более полная форма уравнения, приведенная в табл.
6.1, гораздо удобнее; в данном выражении объемные
доли обозначены 0(.
В табл. 6.2 представлены величины растворимости,
ацентрических коэффициентов и молярных объемов, ко-
торые входят в указанные выше уравнения. Предпола-
гается, что эти величины не зависят от температуры.
Для некоторых легких веществ величины не совпада-
ют с истинными их значениями, но они позволяют свя-
зать между собой величины давления пара и
экспериментальные значения Ki; для разработки этих
корреляций было использовано более 2000 эксперимен-
тальных данных. Модификация этой системы, прове-
денная Робинсоном и Чао, усложнила методику, но эти
авторы включили метан в число основных веществ, и
при температурах вплоть до —45 ° С данная методика
дает в общем лучшие результаты.
Для веществ с критической температурой, превы-
шающей используемый диапазон, значительно более
точна усовершенствованная методика Маффиоло и др.
[445]. Эти авторы рассчитывают коэффициент актив-
ности по уравнению Скэтчарда — Гильдебранда (урав-
нение (2) табл. 6.1), а фугитивность жидкости
определяют как
fiL = ф*Р*(РГ)(, (6-32)
где фугитивность насыщенных паров получена по урав-
нению Редлиха — Квонга. Если давление паров не пре-
вышает двух атмосфер, применяют уравнение Антуана,
при более высоком давлении, вплоть до критического,
используют пятипараметрическое уравнение. Для описа-
Таблица 6.1. Метод Чао — Сидера
1. К, = 7(0ii/0i. _
2. у, = ехр[К,(<5 - tf/RT + ln#i
+ 1-0,-], 6i = Vi/'LxiVi.
3. 4>i рассчитывают по уравнению Редлиха—Квонга
(табл. 3.4).
4. Iog0,£ = log</>i£) + wlogjp.
5. Iog</><2? — >4 о +• /41/7У,- + АгТг, 4- AjT"^ 4-
4- (As 4- А£ГГ, 4- AiT^)Pri
4- A8 4- A*Tri)P* - logPr,.
6. Iog0$? = >4io 4- ЛцГГ( 4- Лц/Т, 4- А/зТ^
4- Ai4(Pr, 4- 0,6).
7. Значения коэффициентов:
Стандартная жидкость, w = 0 Метан Водород
Ао 2,05135 1,36822 1,50709
А1 - 2,10899 - 1,54831 2,74283
А2 0 0 - 0,02110
А3 -0,19396 0,02889 0,00011
А4 0,02282 - 0,01076 0
А5 0,08852 0,10486 0,008585
А6 0 - 0,02529 0
А7 - 0,00872 0 0
А8 - 0,00353 0 0
А9 0,00203 0 0
А10 А11 = - 4,28393 = 8,65808 Л12 = - 1,22060 Л13 = - 3,15224 14 - 0,025
8. Ограничения:
a. T < 260°C, T> - 18°C.
б. P < 6,89 МПа.
в. Для углеводородов (кроме метана) 0,5 < 77, <1,3
и приведенное критическое давление смеси <0,8.
г. Для систем, содержащих метан и(или) водород, ус-
редненное молярное значение 77 < 0,93 и мольная доля
метана <0,3. Мольная доля других растворенных га-
зов < 0,2.
д. Если определяется К для парафинов или олефи-
нов, мольная доля ароматической жидкой фазы должна
быть <0,5. И наоборот, если определяется К арома-
тических соединений, мольная доля ароматической жид-
кой фазы должна быть > 0,5.
9. Внимание: В этих уравнениях используются деся-
тичные логарифмы.
ния систем, содержащих пары со сверхкритическими па-
раметрами, указанные авторы рекомендуют исходные
корреляции Чао — Сидера.
Введение уравнения Редлиха — Квонга в методику
Чао — Сидера, как показали авторы работы [236], поз-
воляет получить несколько значений точки росы и точ-
ки начала кипения. Авторы данной работы предлагают
несколько возможных подходов при оценке физического
смысла частных решений.
Таблица 6.2. Свойства чистых компонентов, используе-
мых в методе Чао — Сидера (дополнительные данные
Хенли и Сидера [52], заимствованные в основном из
FLOWTRAN Data Bank)
Модифицирован- 6, V,
ный w-фактор (кал/мл)1/2 мл/моль
Водород 3,25 31
Парафины
Метан — 5,68 52
Этан 0,1064 6,05 68
Пропан 0,1538 6,40 84
Изобутан 0,1825 6,73 105,5
Бутан 0,1953 6,73 101,4
Изопентан 0,2104 7,02 117,4
Пентан 0,2387 7,02 116,1
Неопентан (0,195) 7,02 123,3
Гексан 0,2927 7,27 131,6
Гептан 0,3403 7,43 147,5
Октан 0,3992 7,551 163,5
Нонан 0,4439 7,65 179,6
Декан 0,4869 7,72 196,0
Ундекан 0,5210 7,79 212,2
Додекан 0,5610 7,84 228,6
Тридекан 0,6002 7,89 244,9
Тетрадекан 0,6399 7,92 261,3
Пентадекан 0,6743 7,96 277,8
Гексадекан 0,7078 7,99 294,1
Гептадекан 0,7327 8,03 310,4
Олефины
Этилен 0,0949 6,08 61
Пропилен 0,1451 6,43 79
1-Бутен 0,2085 6,76 95,3
1/ис-2-Бутен 0,2575 6,76 91,2
транс-2-Бутен 0,2230 6,76 93,8
Изобутен 0,1975 6,76 95,4
1,3-Бутадиен 0,2028 6,94 88,0
1-Пентен 0,2198 7,05 110,4
г/ыс-2-Пентен (0,206) 7,05 107,8
транс- 2-Пентен (0,209) 7,05 109,0
2-Метил-1 -бутен (0,200) 7,05 108,7
З-Метил-1-бутен (0,149) 7,05 112,8
2-Метил-2-бутен (0,212) 7,05 106,7
1-Гексан 0,2463 (7,40) 125,8
Нафтены
Циклопентан 0,2051 8,11 94,7
Метилциклопентан 0,2346 7,85 113,1
Циклогексан 0,2032 8,20 108,7
Метилциклогексан 0,2421 7,83 128,3
Аром ати ческие
соединения
Бензол 0,2130 9,16 89,4
Толуол 0,2591 8,92 106,8
о-Ксилол 0,2904 8,99 121,2
м-Ксилол 0,3045 8,82 123,5
л-Ксилол 0,2969 8,77 124,0
Этилбензол 0,2936 8,79 123,1
Равновесие пар —жидкость 313
6.2.4. Методики, предусматривающие применение
вириальных уравнений. Для описания паровой фазы
при давлении
Рг < 0,5 TcrLyiTci
(6.33)
или соответственно при
Р/Рс 0,5 (6.34)
вполне приемлемо В-усеченное вириальное уравнение.
Эта методика наиболее удобна при проведении чис-
ловых расчетов, так как уравнение имеет только один
корень и коэффициент фугитивности определяется отно-
сительно простым соотношением
р
1п 0Z = ррС! £ уiB;j — В), (6.35)
J
где
В = WyiyjBij, (6-36)
и для чистых веществ
In 0, = BiP/R Т. (6.37)
Во многих опубликованных работах приводятся
установленные расчетным путем величины второго ви-
риального коэффициента и необходимые корреляции,
полученные исходя из свойств чистых компонентов. Так
что применение вириального уравнения для решения
большинства задач, связанных, в частности, с дистил-
ляцией, вполне оправданно. Объединение вириального
уравнения и уравнения Скэтчарда — Гильдебранда для
расчета коэффициента распределения дает следующие
выражения:
yz0fPf(PF)z
0/ уР
Pf(PF)z ехр ”-(5z-3)2
IX 1
(6.22)
В&
exp
RT
(6.38)
Р ехР R т (2 ? yjBij ~ в)
Р^РР)'
п ехр
1
RT
(KZ(5Z- З)2
—I *
+ BzPj + BP - 2P S yjBij
j J .
(6.39)
В эти уравнения включены свойства лишь чистых ком-
понентов. При необходимости исходное уравнение
Скэтчарда — Гильдебранда можно заменить расширен-
ным уравнением Скэтчарда — Гильдебранда — Фло-
ри — Хаггинса.
Если информация доступна, для определения коэф-
фициентов активности более удобными могут оказаться
и другие уравнения, например уравнение Вильсона.
Праузниц и др. [567] объединяют вириальное уравнение
и уравнение UNIQUAC, которое приводится также в
книге Фреденслунда и др. [286]. В статье этих авторов
[287] описана программа расчета дистилляции, в кото-
рой используются коэффициенты активности, рассчиты-
ваемые по уравнению UNIFAC, и вторые вириальные
коэффициенты Хайдена и О’Коннела [336]. Драго и др.
[258] объединили последнее общее уравнение состояния
и модель «поверхность регулярных решений», применя-
емую для описания коэффициентов активности, такая
методика требует учета одного из двух параметров би-
нарной системы и рекомендуется для использования
при температурах 127—500 К и давлениях вплоть до 0,9
конвергентного* давления смеси. Уравнение NRTL на-
иболее предпочтительно применять к жидким фазам,
для которых можно получить необходимые данные.
6.2.5. Графические методы. В ряде работ расчет
параметров систем проводится при помощи номограмм
и сетевых графиков, демонстрирующих зависимость
равновесных соотношений испарения от температуры,
давления и некоторых параметров, характеризующих
состав смеси. В настоящее время к ним чаще всего при-
бегают в целях быстрой проверки результатов и прове-
дения отдельных ручных расчетов, поскольку имеются
эквивалентные им аналитические методы, позволяющие
использовать компьютеры. Тем не менее графические
методы до сих пор сохраняют некоторое значение при
рассмотрении высококипящих нефтяных смесей и для
изучения состояния или модернизации устаревших за-
водских установок. Ниже кратко описаны два из рас-
пространенных сейчас графических методов.
6.2.5.1. Конвергентное давление В критических
точках составы паровой и жидкой фаз смеси становятся
одинаковыми, или, другими словами, коэффициент рас-
пределения каждого из компонентов становится равным
единице, т. е. Ki = 1. Например, для приведенных на
рис. 1.30 и 1.31 (гл. 1) кривых зависимостей критическо-
го давления бинарных смесей имеется широкая область
температур и составов, в которой К\ = Кг = 1. Анало-
гичным образом ведут себя и многокомпонентные сис-
темы. Давление, при котором все константы фазового
равновесия становятся равными единице, называется
конвергентным. Вполне понятно, что эта характеристи-
ка состава смеси относится к определенной температуре
и что ее можно использовать в корреляциях коэффици-
ентов распределения в сложных смесях.
Так, на рис. 6.1 данные о составе фаз смеси пропа-
на и бензола представлены в виде зависимости К от
давления при двух различных температурах; показа-
тельным является резкое изменение значения Ki для тя-
желого компонента вблизи критической точки. На рис.
1.32 представлены аналогичные данные для тройной
системы; из этого графика можно найти температуру
и состав, при которых К\ = Кг = К$ = 1.
Истинное критическое давление многокомпонент-
ной смеси можно, используя некоторые уравнения со-
стояния, рассчитать при помощи компьютера, однако
* О конвергентном давлении см. разд. 6.2.5.1.—
Прим, перев.
применение Л'-графиков для массовых определений кри-
тического давления непригодно из-за больших затрат
времени. На практике вместо этого оценивают конвер-
гентное давление. При некоторой определенной темпе-
ратуре конвергентное давление берется равным
конвергентному давлению эквивалентной бинарной сме-
си, выбираемой по некоторым определенным прави-
лам. Приведем правила, в соответсвии с которыми
проводят расчет по методикам API и NGPSA.
1. Оцениваем состав жидкой фазы.
2. В качестве легкого компонента эквивалентной
бинарной смеси выбираем самое легкое вещество (но не
водород), содержание которого должно достигать по
крайней мере 0,1 мол. %.
3. Принимаем, что критическая температура и дав-
ление более тяжелого компонента — это средневзве-
шенные критические давления и температура всех
остальных компонентов жидкой фазы, исключая самые
тяжелые (2 мол.%).
4. Определяем конвергентное давление по критиче-
ским точкам диаграммы бинарной смеси, подобной по-
казанной на рис. 6.2; другие диаграммы приведены в
литературе (см. подпись к рис. 6.2).
5. Далее используем найденное конвергентное дав-
ление, Pcv и соответствующие ^/-графики для всех ве-
ществ смеси.
6. Вычисляем фазовый состав по температурам ки-
пения. Если полученные результаты не соответствуют
полученным на этапе 1, повторяем расчет.
На практике используют и другие схемы разделе-
ния смесей на два экивалентных компонента. Согласно
одной из них, компоненты смеси просто делят на две
группы и рассчитывают их средние температуры кипе-
ния. В статье [264] дана история рэ™»тия хыр^црв *
конвергентного давления.
6.2.5.2. Методика NGPSA {Natural Gasoline Proces-
sors Suppliens Association). NGPSA получены графики
типа приведенного на рис. 6.4 для ряда органических
соединений (от метана до декана и далее), некоторых
неорганических газов, исключая водород, и нефтяных
фракций со средними температурами кипения до 800°F
(426, 67 ° С) при дискретных величинах конвергентного
давления от 800 до 10 000 фунт/кв. дюйм. Опубликова-
ны также специальные графики для нескольких бинар-
ных систем, содержащих метан, и один график для
системы этан — азот. В указанных пределах может
быть проведена интерполяция по температуре или дав-
лению при помощи типичных уравнений
1п/Г = А - В/{Т + С), t6-40)
{К/Т)х/3 = а + ЬТ+ сТ2, (6.41)
\пК = а + b/Т + с\пР. <6-41а>
6.2.5.3. Методика API {American Petroleum Instit-
ute). Эта методика предусматривает применение не-
скольких номограмм для смесей углеводородов с
неуглеводородами и для водорода в различных темпе-
ратурных диапазонах. Идентичность веществ, темпера-
туры и давления «сетки» * однозначно определяет Ki.
Давление «сетки» зависит от давления системы и кон-
вергентного давления, как на рис. 6.3. При высоких
конвергентных давлениях (или низких давлениях систе-
мы) давление системы и давление «сетки» становятся
одинаковыми.
Считается, что методики API и NGPSA неудобны
для решения задач равновесия на компьютере. Обычно
принято поступать так: вручную вводят несколько взя-
тых из графика величин, соответствующих поставлен-
ной задаче, и осуществляют обычную интерполяцию,
пользуясь уравнениями (6.40) или (6.41) или более слож-
ным уравнением следующего вида:
\пКР= а + Ь/Т+ с 1пТ+ dT+ е Т2. (6.42)
Несомненно, существует несколько вариантов ком-
пьютеризации этих схем, но они являются собственнос-
тью разработчиков. Об одном из таких вариантов
упоминают, в частности, Доулинг и Тодд [257], но
лишь в общих чертах. С целью распространения мето-
дики API на высококипящие нефтяные фракции Жва-
нецкий и Платонов [257] построили подгоночные
кривые, которые, по-видимому, можно применить и к
определенным легким углеводородам. Как справедливо
указывают эти авторы, при тех давлениях, при которых
обрабатывается сырая нефть, необходимость в коррек-
тировке конвергентного давления отсутствует. Как пра-
вило, температуру кипения смеси делят на интервалы
в 25 ° С, каждый из которых характеризуют средней тем-
пературой кипения. Значение К в каждом интервале свя-
зано с К этана {Ке) и гептана {Kh) посредством
* Вспомогательная величина.— Прим. ред.
Равновесие пар — жидкость 315
уравнения
К=к1+Х/К%.
(6.43)
Величины К эталонных веществ зависят от температу-
ры (°C) и давления (бар)
lnX = <z0 + ах/Т + а2/Т2 + д3/Т3 + а4/Р
+ а5/Р2 + д6/ТР + а7/Т2Р + а8/Т3Р,
(6.44)
Две следующие формулы рекомендованы Лесли и
Хейном [429] как особенно удобные для проведения ин-
терполяции в ограниченных пределах Т и Р. Первая из
них
1пХ'Р = kx + к2 lnPsat
(6.45а)
дает хорошие результаты при Т = 50—100°С и Р 3
атм, а вторая
lntf = кх + к2 lnPsat + к3 1пР
(6.456)
где 32 < Т < 473°С, 1 < Р < 10 атм. Коэффициенты
для этого уранения приведены ниже. Коэффициент ле-
тучести /3 — это функция средней температуры кипения
(Г°С) погона нефти:
/3 = -0,723 + О,692(1О"2)Г - 0,291(10“5)Т2 +
+ 0,263(10 "ЪГ’. (6.45)
Разброс значений К,, рассчитанных по этим уравнениям
относительно найденных по графику, достигает 7%
Этан Гептан
«0 О] + 1,750 + 3,608 • 102 + 2,708 - 6,377-102
«2 - 6,260-104 + 4,032-103
«3 + 2,702-106 + 8,807-105
«4 + 4,750 + 4,782
а5 -2,156 + 2,198
«6 - 1,386-102 - 1,723-102
а7 + 2,192-104 + 2,328-104
«8 + 1,060-106 - 8,993-105
удобна при повышенных давлениях. Влияние темпера-
туры учитывает слагаемое, содержащее давление пара.
6.2.6. Сравнение методик. В гл. 1 (разд. 1.9) даны
сравнительные оценки различных уравнений состояния.
Указаны те из них, которые применимы в ряде случаев
для описания состояния как жидкой, так и паровой фаз,
в первую очередь это уравнения Соава, Пенга — Робин-
сона, Бенедикта — Уэбба — Рубина — Старлинга,
Плюкера — Ли — Кеслера. Уравнения для определения
коэффициентов активности сопоставлены в гл. 4 (разд.
4.13). Преимущество уравнений Скэтчарда — Гйльде-
бранда, UNIFAC и ASOG состоит в том, что для описа-
ния поведения многокомпонентных систем достаточно
иметь данные только для чистых компонентов, хотя на-
до отметить, что уравнение Скэтчарда — Гильдебранда
применимо к меньшему количеству веществ, чем два
других. Если имеется исходная информация, характери-
зующая жидкую фазу, то лучше воспользоваться урав-
нением Вильсона или его модификациями.
Доберт и др. [31] совместно с Американским нефтя-
ным институтом провели тщательное сравнение девяти
Пример 6.1. Относительная фугитивность 4>i/4>i и коэффициент Пойнтинга {PF} при нескольких величинах
давления, вычисленных по В-вириальному уравнению
В данном примере рассмотрена эквимолярная
смесь метан (1) + пентан (2) при 37,78°C(1OO°F). Коэф-
фициенты вириального уравнения заимствованы из ра-
боты Когана (1967):
Вп = -38 мл/моль
Ви = -196,
В22 = -1220.
Молярные объемы жидкости:
И = 52 мл/моль, у2 = 116,1.
Уравнение для расчета коэффициентов фугитивнос-
ти дано в табл. 3.4. Коэффициент Пойнтинга рассчиты-
вается для избыточного давления в 1 атм.
1п(ф,/ф,) = _g_ <7 _ 7,-)2(2Bi2 - Вц - В22) =
= 0,25( —392 + 38 + 1220)Р = Q ^5р
82,05(310,9)
(РП f ехр (Р - 1) .
Р = 5 Р = 10 Р = 25
ф/ф PF ф/ф PF ф/ф PF
СН4 1,0433 1,0082 1,0886 1,0185 1,2364 1,0501
С5Н12 1,0433 1,0184 1,0886 1,0412 1,2364 1,1154
В этом случае отношение (РР)(/(ф(7ф1) заметно отлича-
ется от единицы даже при давлении 5 атм. Однако
обычно коэффициент Пойнтинга рассчитывают для
давлений, превышающих давление пара, и полученные
значения могут быть ближе к единице, чем при Р = 1,
выбранном в качестве нижнего предела интегри-
рования.
аналитических методов. Ими были рассмотрены ме-
тодики:
1. Чао — Сидера и ее модификации, предложенные
Грейсоном — Стридом, Каветтом, Ли — Эдмисте-
— Эрбаром, а также Маффиоло с corp.; 2) Ли —
Кеслера; 3) Хана — Старлинга — Бенедикта — Уэб-
ба — Рубина; 4) Соава; 5) Пенга — Робинсона. Для
сравнения указанные авторы использовали информаци-
онный банк, содержащий более 4000 эксперименталь-
ных данных. В целом коэффициенты распределения
лучше описывались уравнениями Соава и Пенга —
Робинсона; последнее из этих уравнений несколько луч-
ше описывало плотность жидкости, а уравнение Ли —
Кеслера — изменение энтальпий. Уравнение Соава бы-
ло упрощено для «Справочника Американского нефтя-
ного института». Параметры бинарного взаимодейс-
твия были получены для смесей H2S, СО2, N2 и СО с
углеводородами. Были систематизированы данные о
фракциях водорода и нефтяных погонов, характеризуе-
мых определенными Тс, Рс и ш. Уравнение Пенга —
Робинсона было проанализировано не столь основа-
тельно, как другие перечисленные уравнения, поскольку,
когда оно было разработано, работа уже велась пол-
ным ходом. Тем не менее Gas Processors Association,
TUlsa представляет за плату компьютерные программы
для расчета коэффициентов распределения и других со-
отношений при помощи уравнения Пенга — Робинсона.
Для расчета конвергентного давления в методиках
NGPSA и API использованы одинаковые подходы. Дан-
ные для смесей углеводородов с Н2, Н2О, NH3, SO2,
НС1 и некоторыми другими веществами имеются толь-
ко в методике API. Никаких сопоставлений графических
и аналитических методик в обзоре API не проводится.
6.3. Равновесия с К, не зависимыми
от состава
Такие задачи, как определение температуры начала
кипения, точки росы и испарения, формулируются исхо-
дя из данных о коэффициенте распределения. Если по-
следний может быть аппроксимирован как независимый
от состава, решения становятся относительно просты-
ми. В любом случае такие решения могут служить хо-
рошими начальными условиями для получения точных
решений последовательной аппроксимацией, когда необ-
ходимо учитывать влияние состава, неизвестного к на-
чалу аппроксимации.
Согласно определению,
Фц
К( = —— —--------------- , (6.46)
0/К <t>ivP
парциальная фугитивность и коэффициенты активности
зависят от состава. В ряде случаев влияние состава пара
относительно мало (см. пример 6.1), но обычно это не-
обходимо проверить.
В дальнейшем мы будем исходить из допущения,
что коэффициент распределения (константа фазового
равновесия) получен из диаграмм API или NGPSA.
Для проведения расчетов на калькуляторе или ком-
пьютере необходимы уравнения, характеризующие вли-
яние температуры и давления. Для узкого диапазона
частных случаев можно применить следующие уравне-
ния с двумя либо тремя коэффициентами:
1пК,= А,--Bj/(T+ С,), (6.47)
к1К( = А{-В,/(Т+ 18 — 0.197*), (6.48)
К(=а(Рь1, (6.49)
где Ть — нормальная температура кипения, К. Коэффи-
циенты оцениваются по 2—3 отсчетам из Х'-графиков.
В приложениях к этому разделу нам потребуются
производные перечисленных выше уравнений, поэтому
приведем их.
= №.— (6.50)
дТ (T+Ci)2 ’
d2Kj = Д,[В/-2(Т+С,)]Х/
ВТ2 (Т+С^ ’ (6-51)
ВК,
--~=biKi/P, (6.52)
д2К-
-^i=bi(bi - \)Ki/P2. (6.53)
Решать уравнения, помещенные в этой главе, обыч-
но удобно методом Ньютона — Рафсона. Например, ес-
ли известна оценка корня х(г) уравнения
Л*) = 0,
(6.54)
то улучшенное значение корня определяется выра-
жением
Х(г+1) = x(r) _ /(x(r))//'(x(r)).
(6.54а)
Если необходимо выполнить множество расчетов тако-
го вида, как, например, при решении задач, связанных
с дистилляцией, становится важной эффективность про-
цесса поиска корней. Елинек и Главачек [371] применили
для расчета температур начала кипения метод, автором
которого можно считать Ричмонда (1944). Этот метод
часто дает самые лучшие результаты. Алгоритм его
таков:
х(г+1) = х(г)_ 2/(2/7/-/'7Г).
(6.55)
Необходимые производные легко получить, пользуясь
уравнениями, подобными уравнению (6.47). Способ ди-
фференцирования такого уравнения, основанный на тео-
реме Тейлора, показан в книге Лапидуса [74].
6.3.1. Точка начала кипения. Температура, при ко-
торой жидкость известного состава начинает кипеть,
определяется по уравнению
/(Г) = ЕХ,х, -1 = 0,
(6.56)
где К — известная функция температуры. Если исхо-
Равновесие пар —жидкость 317
2
«о
10000
9000
8000
7000
6000
5000
4000
3000
S
2000
Е
1000
900
800
700
600
500
400
300
200
0
100 200 300 ЧОО 500 600 700 800 900
100
-300 -200 -100
а
Т емпература, °F
Стабилизационные
колонны
4000 I—г—[—j—г f - | -|—1—|—।—]—।—г
Колонны легких
углеводородов
Г1 I I I I I I I I I I
-100 0 100 200 300400 500 600
-100 0 100 200 300 400
Температура, °F
Рис. 6.2. Конвергентное давление
углеводородных смесей, а — бинар-
ные смеси [756]; б — типичные смеси
нефтепродуктов [318]
-100 0 100 200 300400500 6007ПОЯЛО 300400500600700800900
Температура,°F
6
дить из уравнения (6.47), алгоритм Ньютона —Рафсона
приводит к выражению
f(P) = Zy./Ki -1 = 0.
(6.61)
- 1 + SA'/X,
г = т —---------------
BjKjXj
S (T+Ci)2
(6.57)
Запишем, исходя из уравнений (6.47) и (6.49), алгорит-
мы Ньютона — Рафсона для определения Т и Р:
Т=Т +
- 1 + ^y./Ki
Аналогичным образом алгоритм Ньютона — Рафсона
позволяет определить давление в точке начала кипения
из уравнения (6.49), описывающего влияние давления
на Кг.
dKi
дТ
(6.62)
f(P) = XKjXj -1 = 0,
(6.58)
Р = Р +
- 1 + Zyi/Ki
Р = Р
- 1 + T.aiPbi х(
ZaibiP^i~l Х{
(6.59)
~У, dKi
к2 др
= р +
(- 1 + Zyi/Kj)P
^biy./Ki) •
(6.63)
6.3.2. Точка росы. Температуру или давление, при
которых пар определенного состава начинает конденси-
роваться, находят решением соответствующих уравне-
ний:
/(Г) = zyi/Ki -1 = 0,
6.3.3. Испарение при фиксированных значениях Т. и
Р. В интервале температур от точки начала кипения до
точки росы (и соответствующем этим точкам интервале
давлений) в системе сосуществуют две фазы. Составы
и количества этих фаз зависят от условий, в которых
находится эта система. Как правило, эти условия опре-
деляются фиксированными значениями Т и Р, или Н и
(6.60)
100
10 20 4 0 6 0 80 100
Системное давление Р, фунт/кв.дюйм
Рис. 6.3. Давление «сетки» как функция давления системы и конвергентного давления (применяется вместе с номо-
граммой, показанной на рис. 6.5). (© Gulf Publishing Со.)
Рис. 6.4. Кривые NGPSA этана при конвергентном дав-
лении 1 000 (а) и 5 000 (б) фунт/кв. дюйм.
Равновесие пар —жидкость 319
Р, или 5 и Р. На рис. 6.6 представлены такие возмож- ные варианты, и мы их все по очереди рассмотрим, на- чиная с варианта фиксированных Т и Р. Материальный баланс по любому компоненту и равновесие системы определяются уравнениями - 1 + Е ' 1 + № - 1) ?-? + " (K-nz ’ <6’67) v* i ')zi 2 [1 + P(Ki- I)]2
Fz^LXj + Vyt, (6-64) После нахождения последовательной аппроксимацией значения /3 определяют состав фаз:
y, = X,xz. (6.65) z i Х;= „ , (6.68)
Объединяя эти уравнения и вводя коэффициент испаре- ния /3 = V/F, получаем уравнение, определяющее уело- 1 + /3(^-1)
вия испарения: ^(=А'(х/. (6.65)
f(li) = ~ 1 + ъК,х,- = “ 1 + 2 1 + _ 1) - °’ (6.66) Начальное значение /3 = 1 всегда приводит к сходимос- ти решения по этой методике. Рэчмонд и Райс [574] предложили целевую функцию, которая часто имеет лучшую сходимость при итерациях:
Запишем далее соответствующий алгоритм Ньюто- на — Рафсона: /(/?) = Ел-2*/ = о. (6.69)
а Д авление, фунт /кв. дюйм
Рис. 6.4. Кривые NGPSA этана при конвергентном дав-
лении 1 000 (а) и 5 000 (б) фунт/кв. дюйм.
laaaaiiiiniiiiiH
(iiiiiiiiiiuiHiiiiiaiaiiaiaiiiiiniHiiiiiiiiiiii i
aaaai inn ih 11 a ва а и t 11 к it и i г <
aaiiiiiin шин in inn ши 11П1ННИИ
Ma~MiLi 1111 iiiiiiiiiiHiiniiMttttttlt
«i inn inn......
UKWBHBaaaitmiiiiiiiiiiiiiiiiiiimiiiiiiiiiiiiiiiiiiiiiiiiMHaBaaaiiiiiiiiii i ; .IT. in 1111111111 niiiuuiimtiiii
'Чкч.чяааааа11111111111111111111111111111111111111111111иш1яваваа1111И11п n । 111111 ihiiiiuhimiiiii
K4k^4MHiiiiiiiiiiiii|iiiiiiiiiiiiiiiiiiiiiiiiiiiniiiiiiiiHBaaiMaiiinnni iiiiiii 1 u 1111111111 ......................11111111111
^^^«!!!!Н!Н*ШШ!Н!!!!!!!Н!!ШШ!!!!!!!’|,||||"8881!!!!!Шин|||Н11111Ч11111111111|||||||||| bi iiiiiiniiши1111111111111111111
.ж .................... .'la lllk. ИП1. ЧШ1. I
lгa88ьчlnчllh''lll:aaгu'lиl»'llh:'llll> 'iniii
lьa8al.:ulh'lllЬ''llьЧhЧИl.;'l»''lll.<lllll< ‘ill
4Wb4Ba№aiiiHiih?iiiii;4iii^n?ii;'ii!h:ii>:4iii.'luir!
£vi№ii№uiiii:4iiii;4ih!m>4ii:<ii;Mii!'ii:|iili
iiiiiiiiiiiiiiiiiiil
inn hhiiiiii 1
hiu iiiii iiiii I
jnillllHIIIIII
...Inn iiiii iiih 1
iiiiiii iiiiiniQi
ii^B^aBiiiiiiiiiiiiinimuiiiuiiiiiiiiiiiimiiiuiiiiii
mcihMiiiiiiiiiimiiiiiimimiiitiiiiii................
.-□aaiiiimiiiiiiiiiiiuiiiiiiiiii
—laaaaiuniiiiii hhiiiiii iiiii nil
laaaaalllllllllHlllnlllllllil
lвaallllllllllllllllHllllllllllll
aaaiiiiiiiiiiiiiuiiiiiiiiiiiiii
laHliiiiiiiiiiuiiiiiiniiiiiimiii
K=
10
1,0
MiiuHiiiiimiininiiHiiiiiliiiiiiiiiiiiii
iiiiiii
iiiiiii
m.'iiii:iiii
BmiiiiiniiiiiaaaaamiiiuiiiiiiiiiiiiiiHii
immiiiiiiiiii
.Minin
laaa aaiiiinii iiHniniiaiiiiiiil
inn nmaaaaa inil
^BbMiiik'iiin;'iiiii
min
iiiiiii
IllinillUIIIIIIIIII.'Ulllllllt
iiiiiiiiiiiiaiiiiiiiiu;!iiiiiiiiHi
iiiiiiiniiiii»'iiiiiiiiiih:'iinii
""iiiiinuiiii:4iiniiiiiiir~
I mu uni inn inn mn 1
(I Mill lilt I inn inn inn I
.....laiaaaiiaiiiiiiiiitiiiiiiiiiiiii
1111111111111111111111111111111111111111
iiiiiiiiihiiiiiiii iiiii uni inn iiili i_.
__________________mi mu 111 ai uni inn inn 11 ni mu hi
aaai iiiaiaiin hhiiiiii iiiaiuiii mu itni pin him n
I I I I 111111
f f I I 111
0,1
10 6
iaaalllllllllllllllllмll
......................................................_uaaaaaiiiiiniiiiii iiiiiiiiiiiiiiiiiii
lallllllllllllllllllll iiiiiuiii iiiiiiiiiiiiiiiiniiaiaiflaBiaaaiiiiiiiiijifiniiHu HJi j fin
laiiiiiiiiiimiiiiiiimiiiiiiiiiiiiiiiiiiiiiiiiiiih ь luianiiiiiiiiiiiiiiiiiiiiiiiiiiii
.□Baaaaaaiiiiiiiiiiiiiiiiiaaaiiiiiiiiin iiiiiiiiiinnii
l88aalllllllllшllllllllllllшllllпllilllllllllllиlll
100
Давление, фунт/кв.дюйм
1000
5000
Рис. 6.4 (продолжение)
В этом случае уравнение испарения имеет следующий писывает^я там:
вид:
{Ki-
f(Q}=T.------------= О,
J 1+0(XZ-1)
а соответствующий алгоритм Ньютона —
. (g4- l)rf
Х1+ДЖ/-1)
(6.71)
2-864
Н2 в лигроине ф
Азот®
Ф Н2 в бензоле
-40 (мол.масса195)
(6.76)
6.3.4. Испарение при фиксированной энтальпии. За-
дача формулируется для определенных конечных значе-
ний величин давления и энтальпии и при допущении,
что энтальпия — величина аддитивная (т. е. энтальпия
смешения равна нулю) и температурные зависимости
при заданном давлении известны. Запишем энтальпий-
ный баланс:
HF = (1 - 0)S XiHiL + /З^у^и
ziHiL. _ KfZjHjy
= а - ю s . . + _ n • <6-72)
1 т pi-Kj 1) It p(Jii 1)
Это уравнение и уравнение испарения (6.66) составляют
систему:
Z(<i.r)--.+Zi + ^_i) = 0, (6.73)
1 + p(Ki 1)
KpiH^
-^—/1(^-1)-°- <6'?4>
из которой, если известны температурные зависимости
энтальпии и коэффициентов распределения (констант
фазового равновесия), можно определить фазовое раз-
деление /3 и температуру. Применяя методику Ньюто-
на — Рафсона к уравнениям (6.73) и (6.74), находят
поправки для начальных оценок 3 и Т решением систе-
мы линейных уравнений
df df
h + k~~+f=O, (6.75)
dg dg
h + k -~ + g = Q,
dp d 1
где все члены рассчитаны при предполагаемых значени-
ях двух неизвестных /Зо, То. Корректировка значений
для следующего этапа расчета, если таковой необходим,
осуществляется следующим образом:
/3 = /3О+Л,
(6.77)
Т = То + к.
(6.78)
Другой вариант расчета /3 и Т (см. пример 6.3) заключа-
ется в следующем. Сначала задаются значением Т, да-
лее, используя алгоритм Ньютона — Рафсона, находят
из уравнения (6.73) значение /3 как единственной пере-
менной в этом уравнении. Правильность найденных Т
и /3 проверяют подстановкой их значений в уравнение
(6.74). При таком способе расчета может потребоваться
задание нескольких значений Т.
6.3.5. Изоэнтропное испарение Расчет расширения
или сжатия потока начинают с выяснения того, что
происходит в условиях обратимости адиабаты, т. е. в
изоэнтропных условиях. Последующий анализ возмож-
ных изменений относительной изоэнтропной эффектив-
ности в нашу задачу не входит.
Исследуемый процесс представлен на рис. 6.6,в. Ис-
ходные данные: при заданных значениях 7} и Р/ данной
жидкой фазы ее скрытая теплота парообразования рав-
на X,. При Tf и Pf происходит изоэнтропное расширение
пара до конечного давления Р, при котором относитель-
ная доля пара равна /3 = V/F. Запишем уравнение ба-
в
Рис. 6.6. Равновесное испарение при фиксированных
значениях Т и Р, или Н и Р, или S и Р.
а — испарительная камера с фиксированными значе-
ниями температуры и давления; б — дросселирование
при фиксированных значениях энтальпии и давления;
в — сжатие или расширение при фиксированных зна-
чениях энтропии и давления.
Равновесие пар —жидкость 323
ланса энтропий:
(1 - Д) Ех,
-
(6.80)
Р
— R In — R Inx,
Pf
+ Mi
Р
— R In — R In^
Pf
и проведем его преобразование:
р ( \
f{T, р, Xi, = R In— + Zzj I — - R lnz(-
Pf V/
-(l-/3)Sx(.
R Inx,-
(6.79)
После исключения х,- и у, путем использования уравне-
ний (6.68) и (6.65) полученное уравнение можно решить
совместно с уравнением (6.73), чтобы определить Р и
Г и в итоге фазовый состав, если, конечно, известны
зависимости величин энтальпии и К, от Т. Для решения
этой задачи пригодна любая из методик, описанных в
разд. 6.3.4.
Определение условий изоэнтропного испарения
можно выполнить при помощи алгоритма, показанного
на рис. 6.12, только в блоке 10 вместо указанного усло-
вия ставится вопрос:
«Is Eq. 6.80 Satisfied?»
6.3.6. Пар и несмешивающиеся жидкости. Несмот-
ря на то что присутствие следов углеводородов в воде
и следов воды в углеводородах в некоторых ситуациях
играет важную роль, работа конденсаторов и испарите-
лей таких смесей исчерпывающе проанализирована на
основе допущения, что эти жидкости не смешиваются.
Пример 6.2. Применение методики API для
коневергентного давления и без его учета
расчета температур начала кипения и точек росы с учетом
Давление равно 300 фунт/кв. дюйм. Состав и свойства
смеси приведены ниже.
Компонент х Тс, К Рс, атм
с2н6 0,05
С3Н8 0,20 370,0 42,0
Н-С4 0,30 425,3 37,4
н-С? 0,20 470,1 32,6
н-С6 0,25 508,1 29,4
Средневзвешен-
ные значения 453 К 32,8 атм
355,8°F 482 фунт/кв.дюйм
530 фунт/кв. дюйм 300°F 400° F
К Кх х/К К Кх х/К
с2н6 3,3 4,0
С3 1,8 2,5
с4 1,05 1,65
с5 0,54 1,02
с6 0,30 0,62
Суммарное значение 1,020 1,616 1,554 0,874
В состав легких компонентов входит более 0,1 мол.%
этана, поэтому он может служить эквивалентом легких
компонентов; эквивалент тяжелых компонентов имеет
Тс = 355,8°F (453 К) и Рс = 482 фунт/кв. дюйм (32,8
атм). Как следует из рис. 6.2, конвергентное давление
равно 600 фунт/кв. дюйм и в первом приближении не
зависит от температуры. Температуру начала кипения
и точку росы находят с помощью графика, приведенно-
го на рис. 6.3, при давлении «сетки» 530 фунт/кв. дюйм
и также для сравнения при 300 фунт/кв. дюйм, при ко-
тором не учитывается поправка на конвергентное
давление.
В результате интерполяции получают:
при 530 фунт/кв. дюйм Твр - 296°F, Tdp = 383°F;
при 300 фунт/кв. дюйм Твр = 226°F, Tdp = 312°Е
300 фунт/кв. дюйм 200°F 300°F
К Кх х/К К Кх х/К
с2 3,8 5,2
С3 1,65 2,7
с4 0,70 1,43
с5 0,30 0,75
с6 0,14 0,40
Суммарное значение 0,83 2,76 1,48 1,19
Очевидно, что в данном случае поправка на конвергент-
ное давление очень важна.
Пример 6.3. Испарение и конденсация тройной смеси
Давление 100 фунт/кв. дюйм. Общий состав смеси, нор-
мальные температуры кипения, К, при двух температу-
рах и энтальпии чистых жидкостей и паров при двух
температурах заданы. Температура исходной-смеси, на-
ходящейся в жидком состоянии, равна 100°Е Необходи-
мо определить температуру начала кипения и точку
росы, составы и количества паровой и жидкой фаз по-
сле изотермического испарения при 100°F и 100 фунт/
кв. дюйм и после адиабатического испарения при 100
фунт/кв. дюйм.
К Коэффициенты
Z Тх.
°F 100°F 40°F А В С
с2 0,3 -128 5,4 3,2 5,7799 2167,12 -30,6
н-С, 0,3 31 0,53 0,21 6,1418 3382,90 -60,8
h-Cs 0,4 97 0,18 0,057 6,4610 3978,36 -73,4
В /(-уравнение входят следующие коэффициенты:
1пА7, = Ai - Bi/(T + Ci), (!)
С = 32,4 - 0,197H°R), (2)
В = In (K2 /Ki) 1 1 (3)
Ti + C T2 + C
А = InKi + B/(Ti + C). (4)
Zi
* ~ 1 + 0(К, - 1) ’
У. = KiXi.
V (^ - 1)2.
R - В 4 — 1 4- 6(Ki - 1)
Ki - 1
1 4- 0(Ki - 1) Zi 1
Состав рассчитывается по уравнениям (13) и (14):
(12)
(13)
(14)
Числовые значения коэффициентов представлены в за- дании. Запишем алгоритмы для определения темпера- тур начала кипения. Алгоритм Ньютона — Рафсона:
- 1 + S к‘х‘
т ~ т (5)
У BjKjXi ’ (Г + Ci)2
Алгоритм Ричмонда F = -1 + S к‘х‘> (6)
F' = £ В‘К,Х‘ 2, (7)
(Т + Ci)2
F" = у ~ 2(Т 4- Ci)]KjXi (8)
(Т + Ci)2
Т — Т (9)
2F' F"
F F'
Алгоритм Ньютона — Рафсона для определения точки
росы
- 1 + S
By,
К,(Т + С)2
Результаты расчетов температур начала кипения и точ-
ки росы представлены ниже. Методика Ричмонда требу-
ет несколько меньше итераций для сходимости.
Температура начала кипения
методика Ньютона — Рафсона методика Ричмонда Точка росы
1000,0000 1000,0000 700,0000
695,1614 620,3217 597,8363
560,1387 504,4799 625,9790
506,5023 495,8020 635,3072
496,1742 495,7963 636,0697
495,7968 636,0743
495,7963 636,0743
Типичный алгоритм для равновесия испарения
-1 + У Ъ
0 = V/F = 0 +------------1 + ~ 0
у Bjy,
К,(Т + Ci)2
и алгоритм Рэчфорда — Райса
(П)
Последовательность итераций, проведенных по обеим
методикам, показана ниже.
Обычная Рэчфорда — Райса
1,0000 1,0000
0,8257 0,7890
0,5964 0,4610
0,3986 0,2720
0,3038 0,2818
0,2830 0,2819
0,2819
Состав паровой и жидкой фаз:
Zi Xi У>
С2 0,3 0,1339 0,7231
Н-С4 0,3 0,3458 0,1833
h-Cs 0,4 0,5203 0,0936
Адиабатическое испарение: значение энтальпий для
жидкости и пара взяты из графиков, приведенных в
справочнике API, и уточнены по температуре согласно
Равновесие пар — жидкость 325
линейным уравнениям
h = а + ЬТ, (15)
Н = с + dT, (16)
где Т измеряется в градусах Фаренгейта. Значения эн-
тальпий и соответствующих коэффициентов приведены
ниже. В испарительную колонну поступает жидкость с
Т - 100°F и Но = 8575,8 брит. тепл. ед./фунт-моль. На
этой стадии расчеты проводят по уравнению испарения
(11). Запишем уравнение энтальпийного баланса:
Но = 8575,8 (17)
= (1 - /3)S MiXthi + /32 W*
Процедура расчета включает следующие этапы:
1) задают некоторую температуру Т,
2) определяют К,, hi, Н,
3) находят из уравнения (11) /3,
4) оценивают энтальпию смеси и сравнивают с Но, рас-
считанной по уравнению (18).
Результаты нескольких таких расчетов показаны
ниже.
T°,R в H
530,00 0,1601 8475,70
532,00 0,1681 8585,46
531,82 0,1674 8575,58 (-8575,8 кон-
трольная величина)
Конечные значения коэффициентов распределения и
составов жидкой и паровой фаз таковы:
К X У
c2 4,2897 0,1935 0,8299
Н-Сд 0,3534 0,3364 0,1189
н-Cs 0,1089 0,4701 0,0512
Коэффициенты корреляции энтальпии:
Л
брит. тепл,
ед./фунт Н
М 0°F 100°F 0°F 100°F a b c d
С2 30 122 195 290 335 122 0,73 290 0,45
Н-Сд 58 96 152 267 301 96 0,56 267 0,34
h-Cs 72 90 145 260 300 90 0,55 260 0,40
Точка начала кипения
10 Т=600
20 READ А1,А2,АЗ,В1,В2,ВЗ,С1,С2,С2,Х1Л2,ХЗ
30 DATA 5.7799,6.1418,6.461,216,7.12,3382.9,3978.36,
30.6,60.8,73,4 , ,3,.3,. 4
40 GOSUB 150
50 H = F/F1
60 H1 = 2/(2*F1/F-F2/F1)
70 PRINT USING 75; T
75 IMAGE DDDD.DDDD
80 T = T—H
90 IF ABS(H/T) < =.0000000001 THEN 110
100 GOTO 40
110 END
150 K1=EXP(A1—B1/(T— Cl))
160 K2 = EXP(A2—B2/(T—C2)
170 КЗ = EXP(A3—B3/(T— C3))
180 F= —1+X1 *K1+X2*K2 + X3*K3
190 Fl =В1 *Х1 *K1/((T— С1)Л2+В2*Х2*К2/(Т—C2)
л2+B3 * X3 * K3/(T—С3)л2
195 G = (T—С3)л4
200 F2 = Bl * (Bl—2 * (T—Cl)) * KI * XI/(T— С1)л4+B2 *
(B2—2 * (T— C2)) * K2 * X2/(T—С2)л4 + B3 * (B3—2 *
(T—C3))*K3*X3/G
210 RETURN
220 END
Испарение
(включая методику Рэчфорда — Райса)
10 В=1
20 READ А1,А2,АЗ,В1,В2,ВЗ,С1,С2,СЗ,Х1,Х2,ХЗ,Т
30 DATA 5.7799,6.1418,6.461,2167.12,3382.9,
3978.36,30.6,60.8,73.4,.3,.3,.4,560
40 GOSUB 150
50 H = F3/F4
70 PRINT USING 75;В
75 IMAGE D.DDDD
80 B = B+H
90 IF ABS(H/B)< =.0001 THEN 110
100 GOTO 40
110 END
150 KI =EXP(A1—B1/(T—Cl))
160 K2=EXP(A2—B2/(T— C2))
170 K3=EXP(A3—B3/(T—C3))
180 D1 = 1 + B*(K1—1)
190 D2=l+ B*(K2—1)
200 D3 = 1 + B*(K3—1)
210 F= — 1+ X1/D1+ X2/D2+X3/D3
215 F1 = (K1—1)*X1/D1A2+(K2—1)»X2/D2A2+
+ (K3—1)*X3/D3A2
220 F3 = (KI—1) * XI /DI + (K2—1) * X2/D2+
+ (K3—1)*X3/D3
230 F4=((K1—1)/D1)A2*X1 +((K2—V)/D2)*2*X2 +
+ ((K3—1)/D3)A2*X3
240 RETURN
5 ! ADIABATIC FLASH
10 INPUT T
20 B=1
30 READ A1A2A3,B1,B2,B3,C1,C2,C3,X1,X2,X3
40 DATA 5.7799,6.1418,6.461,2167.12,3382.9,3978.36,
30.6,60.8,73.4,,3,.3,.4
50 GOSUB 200
60 H = F/F1
70 B=B + H
80 IF ABS(H/B) < = .0001 THEN 100
90 GOTO 50
100 H1 = 122+.73*(T—459.6)
НО Н2 = 96+.56* (Т—459.6)
120 НЗ = 90+.55*(Т—459.6)
130 Н4 = 290+.45*(Т—459.6)
140 Н5=267 + .34* (Т—459.6)
150 Н6=260+.4*(Т—459.6)
160 Н = (1—В)♦ (9* H1/D1 +17.4* H2/D2+28.8♦ H3/D3)+
+B*(9*H4*K1/D1 + 17.4*H5*K2/D2+28.8*H6*K3/D3)
170 PRINT USING 180; Т,В,Н
180 IMAGE DDD.DD,3X,.DDDD,3X,DDDDD.DD
190 RND
200 KI=EXP(A1—B1/(T—C1))
210 K2=EXP(A2—B2/(T—C2))
220 K3=EXP(A3—B3/(T—C3))
230 D1 = 1+B*(K1—1)
240 D2=1 + B*(K2—1)
250 D3=1 + B*(K3—1)
260 F= -1+X1/D1+X2/D2+X3/D3
270 Fl=(K1—1)*X1/D1A2+(K2—1)*X2/D2A2+
+ (K3—1)*X3/D3A2
280 RETURN
Рассматривая конденсацию, следует иметь в виду, что
такая смесь имеет две различные точки росы — одна из
них соответствует началу конденсации углеводорода, а
другая —началу конденсации воды. Расчет кривых кон-
денсации, влияния температуры на количество жидкос-
ти и пара, зависит от того, чья точка росы ниже.
Приведем одну из возможных методик расчета.
1. Постепенно понижают температуру пара и фик-
сируют значения 2 нс7//Х,- углеводородов и парциаль-
ное давление F»/P воды.
2. Отмечают, какие из указанных ниже условий ре-
ализуются первыми:
a. = 1, (6.81)
б. РЪ/Р = Ужо (начальные мольные доли воды в
парах). (6.82)
За. Если точка росы углеводородов выше, продол-
жают расчеты при более низких температурах, отмечая
температуру, при которой начинается конденсация во-
ды. Расчет продолжают, понижая температуру до тако-
го значения, при котором конденсируется вся смесь.
36. Если точка росы воды выше, заменяют углево-
дородную мольную долю на
Zi = Zi( 1 - Psw/P) (6.83)
и определяют точку росы углеводородов, характеризу-
ющуюся температурой, при которой
(l-n/P)S^= 1. (6.84)
НС
После достижения точки росы углеводорода продолжа-
ют расчеты, понижая температуру до тех пор, пока кон-
денсация не закончится.
Эта методика использована в примере 6.4. Равно-
весные кривые конденсации и парообразования выбран-
ной смеси идентичны.
Коэффициент распределения воды можно, сохраняя
достаточную точность, рассчитать по упрощенной фор-
муле
, (6.85)
~ Д р ~ Р^Р-
Точка начала кипения — это температура, при ко-
торой начинается парообразование или же заканчивает-
ся конденсация. Для смешивающихся жидкостей
условия, отвечающие точке начала кипения, определя-
ются как
Syz=l (6.86)
а для несмешивающихся жидкостей
Х„ + 2^=1, (6.87)
НС
или
Р£>/Р+ Sxzxz= 1. (6.88)
НС
Точка росы. Определение понятия и способ опреде-
ления точки росы воды или углеводородов приведены
в начале раздела.
Испарение при постоянном давлении. Запишем
уравнение материального баланса:
F = V + L + W, (6.89)
W = Fywo - Vyw, (6.90)
W/F = Ко - ltyw = доля водной (жидкой) фазы, (6.91)
L/F = 1 - ywo - /3(1 - yw) =
= доля жидких углеводородов в смеси. (6.92)
Для каждого углеводородного компонента
Fzi = Lxi + Vyi = (L/Ki + K)yz.
(6.93)
Поскольку
S у, = 1 - yw = S KiZiltLIF + pKi), (6.94)
нс HC
уравнение парообразования принимает следующий вид:
ЛР) l + yw+ S i-ywo+/?(X,.- l + yw) °-
(6.95)
Это уравнение применимо для температур ниже точки
росы углеводородов, которая может быть выше или
ниже точки росы воды. В примере 6.4 рассматриваются
кривые конденсации для обоих случаев.
Равновесие пар — жидкость 327
6.4. Равновесия с К, зависящими от состава
В формулах для определения коэффициента распре-
деления
X; = yz0fPf(PF)(/0zP, (6.96)
К,= $iL/<t>iV (6.97)
коэффициенты фугитивности и активности существен-
ным образом зависят от состава, особенно при экстре-
мальных Т и Р или в критических областях, или если
смесь содержит сильнополярные или ассоциирующиеся
вещества. Поскольку коэффициент Пойнтинга вносит
лишь незначительный вклад, колебаниями парциально-
го молярного объема, обусловленными изменением со-
става, можно пренебречь. Однако влияние давления на
парциальный молярный объем может быть очень зна-
чительным (см. также гл. 10).
Если изменение коэффициентов распределения, вы-
зываемое изменением состава, принимается во внима-
ние, для решения задачи используется последовательная
аппроксимация. Поскольку состав одной или обеих фаз
неизвестен, первый пробный расчет проводят при допу-
щении идеальности, т. е. принимают, что фугитивности
и коэффициенты активности равны единице, после чего
определяют состав смеси и далее уточняют величины
коэффициентов для следующей итерации.
На рис.6.7—6.12 приведены блок-схемы, используе-
мые для расчетов процессов известных типов. В основу
этих программ положено уравнение (6.96), которое,
впрочем, можно заменить уравнением (6.97) после неко-
торой модификации метода. В этих программах не да-
ется специальных уравнений для расчета у, и фг, в
указанных целях можно воспользоваться любым из
уравнений из числа упомянутых в разд. 6.2 и других
разделах книги. В примере 6.5 применяют вириальное
уравнение и уравнение Вильсона, тогда как в примере
6.6 для расчета обеих фаз используют уравнение Соава.
Пример 6.4. Кривые конденсации смесей воды и углеводородов
Углеводородные смеси (пример 6.3) с 1) 5 мол.% и
2) 10 мол.% водяных паров конденсируются при 100
фунт/кв. дюйм (7,8 атм). При температуре ниже точки
росы мольную долю оставшихся водяных паров рассчи-
тывают, исходя из величины давления паров, по урав-
нению Антуана
= «/₽ = ехр (18.3036 - ^13 ) •
Уравнение для определения величины К, углеводородов
заимствуют из предыдущего примера. При температуре
ниже точки росы углеводородов (уравнение (6.95) и шаг
300 программы) решение выполняется по методу Нью-
тона — Рафсона. Доля смеси, оставшейся в парообраз-
ном состоянии, и доли сконденсировавшихся воды и
углеводородов, а также величина отношения Kw =
= yw = Р^/Р показаны в приведенной ниже таблице и
в виде кривых зависимости от температуры.
1. В первом случае ^у,/К, = 1 при 631,77°R, тогда как
нс
Р^/Р = 0,0629, что значительно превышает у^ = 0,05.
При 626,23°R начинается конденсация воды, а конденса-
ция всей смеси заканчивается при 495,71 °R.
2. Во втором случае Р„/Р = 1 при 652,89°R. При ука-
занной температуре но^/Х, < 1. Углеводороды на-
чинают конденсироваться при 630,76°R, в то время как
конденсация всей смеси заканчивается при 495,71 °R.
L — доля жидких углеводородов в исходной смеси.
V — доля исходной смеси, оставшейся в парообраз-
ном состоянии.
W — доля воды (жидкость) в исходной смеси.
Результаты вычислений и программа приведены
для первого случая — смесь содержит 5 мол.% воды.
Чтобы найти точку росы для углеводородов, следует
заменить К4 = Р^/Р на ?4 = 0,05 в шагах 220, 230 и
300. S в шаге 100 при этом равно 1.
T H L W K„
631,77 1,0000 0,0629
631,76 0,9998 0,0002 0,0000 0,0629
630,00 0,9924 0,0176 — 0,0604
626,23 0,9022 0,0978 0,0000 0,0554
626,22 0,9020 0,0980 0,0001 0,0554
625,00 0,8756 0,1215 0,0029 0,0538
620,00 0,7798 0,2075 0,0127 0,0478
610,00 0,6319 0,3418 0,0263 0,0375
600,00 0,5236 0,4417 0,0347 0,0292
590,00 0,4408 0,5191 0,0401 0,0224
580,00 0,3750 0,5814 0,0436 0,0171
570,00 0,3207 0,6334 0,0459 0,0129
560,00 0,2740 0,6787 0,0474 0,0096
550,00 0,2320 0,7196 0,0484 0,0070
540,00 0,1926 0,7584 0,0490 0,0051
530,00 0,1539 0,7966 0,0494 0,0036
520,00 0,1141 0,8362 0,0497 0,0026
510,00 0,0713 0,8789 0,0499 0,0018
500,00 0,0231 0,9269 0,0500 0,0012
496,00 0,0017 0,9483 0,0500 0,0010
495,71 0,0000 0,9500 0,0500 0,0010
20 READ
A1,A2,A3,A4,B1,B2,B3,B4,C1,C2,C3,C4,Z1,Z2,Z3,Z4
30 DATA 5.7799,6.1418,6.461,18.3036,2167.12,3382.9,
3978.36,6869.6,30.6,60.8,73.4,83.03
40 DATA .285,.285,.38,.05
50 INPUT T
60 K1=EXP(A1—B1/(T—Cl))
70 K2=EXP(A2—B2/B2/(T—C2))
80 K3 = EXP(A3—B3/(T—C3))
90 K4 = EXP(A4—B4/(T—C4))/5170
100 S = .3 « (1—Z4)/K1 + .3 * (1—Z4)/K2 + .4 «(1—Z4)/K3
110 B=.l
120 GOSUB 300
130 F1 = F
140 B=1.0001»B
150 GOSUB 300
160 F2 = F
170 H = .0001*B*F1/(F2—Fl)
180 B=B/1.0001
190 IF ABS(H/B) < = .0001 THEN 220
200 B=B—H
210 GOTO 120
220 L = .95—B*(l—K4)
230 W=.O5—B*K4
240 PRINT USING 245; T,B,L,W,K4
245 IMAGE
ddd.dd,x,d.dddd,x,.dddd,x,.dddd,x,d.dddd
250 GOTO 50
260 END
300 F= — l + K4+Kl*Zl/(.95 + B*(Kl.l + K4))+K2*Z2/
(.95 + В* (K2—1 + K4))+КЗ *Z3/(.95 + В«(КЗ—1 + K4))
310 RETURN
Результаты вычислений и программа для второго
случая — смесь содержит 10 мол.% воды. Точка росы
воды выше чем точка росы углеводородов.
80 КЗ =ЕХР(АЗ—В3/(Т— СЗ))
90 К4=ЕХР(А4—В4/(Т— С4))/5170
100 S = .3 ♦ (1—К4)/К1 + .3 * (1—К4)/К2 + .4* (1—К4)/К3
110 В=.1
120 GOSUB 300
130 F1 = F
140 В= 1.0001* В
150 GOSUB 300
160 F2=F
170 H=.0001*B*F1/(F2—Fl)
180 B=B/1.OOO1
190 IF ABS(H/B)< =.0001 THEN 220
200 B=B—H
210 GOTO 120
220 L = .9—B*(l—K4)
230 W=.l—B*K4
240 PRINT USING 245; T,B,L,W,K4
245 IMAGE
DDD.DD,X,D.DDDD,X,.DDDD,X.DDDD,X>.DDDD,
X.D.DDDD
250 GOTO 50
260 END
300 F= -l+K4+Kl*Zl/(.9+B*(Kl—l + K4)) + K2*Z2/(.9+
+В ♦ (K2—1 + K4))+КЗ ♦ Z3/(.9+В ♦ (КЗ—1 + K4))
310 RETURN
т /3 L W /Си.
652,85 0,9999 0,0001 0,0000 0,1000
630,80 0,9590 0,0000 0,0410 0,0615
630,76 0,9589 0,0001 0,0410 0,0615
630,00 0,9402 0,0166 0,0432 0,0604
620,00 0,7388 0,1966 0,0647 0,0478
610,00 0,5987 0,3238 0,0775 0,0375
600,00 0,4960 0,4185 0,0855 0,0292
590,00 0,4175 0,4918 0,0906 0,0224
580,00 0,3553 0,5508 0,0939 0,0171
570,00 0,3038 0,6001 0,0961 0,0129
560,00 0,2595 0,6430 0,0975 0,0096
550,00 0,2198 0,6817 0,0985 0,0070
540,00 0,1825 0,7184 0,0991 0,0051
530,00 0,1458 0,7547 0,0995 0,0036
520,00 0,1081 0,7922 0,0997 0,0026
510,00 0,0675 0,8326 0,0999 0,0018
500,00 0,0219 0,8782 0,1000 0,0012
495,71 0,0000 0,9000 0,1000 0,0010
20 READ
A1,A2,A3,A4,B1,B2,B3,B4,C1,C2,C3,C4,Z1,Z2,Z3,Z4
30 DATA 5.7799,6.1418,6.461,18.3036,2167.12,3382.
9,3978.36,6869.6,30.6,60.8,73.4,83.03
40 DATA .27,.27,.36,.1
50 INPUT T
60 KI = EXP(A1—B1/(T—Cl))
70 K2 = EXP(A2—B2/(T— C2))
При y„o = 0,05 точка росы углеводорода выше, чем,
у воды.
При уию = 0,10 точка росы воды выше, чем у угле-
водорода.
Равновесие пар — жидкость 329
= Xi =
Рис. 6.7. Температура начала кипения.
Ус
6.4.1. Температура начала кипения (рис. 6.7 и 6.8).
В рассматриваемой ситуации состав жидкой фазы изве-
стен, а состав паровой фазы не известен. В п. 2 блок-
схемы, приведенной на рис. 6.7, указаны исходные до-
пущения, с принятия которых начинают определение
температуры начала кипения, хотя в ряде случаев удоб-
нее начать с допущения, что ф1/фг = 1, которое больше
соответствует реальной картине. Используя методику
итераций, расчеты при определенной температуре про-
водят до тех пор, пока S = 2 Ус не будет иметь посто-
янного значения. Если значение У1, у; больше единицы,
температуру следует снизить и провести следующую се-
рию итераций. После того как в результате двух серий
итераций получены два значения температуры, линей-
ной интерполяцией можно найти улучшенное значение
температуры для следующей итерации:
Т= Т2 + (1 - 52)
Т2 - Тг
S2 - Si
(6.98)
Когда наберется достаточно информации, может ока-
заться целесообразным воспользоваться более сложной
интерполяционной методикой. Чтобы все же ограни-
чить общее число итераций, следует сократить их число
по константе 5 для каждой пробной температуры, заме-
няя эту процедуру прямым обращением к другой темпе-
ратуре после единичной пробы, до тех пор пока не
будет достигнута точная температура.
Отбор переменных для следующих подстановок
при расчете давления начала кипения, точки росы и со-
ответствующего давления однотипен. Для расчета дав-
ления начала кипения может быть сделано лишь одно
начальное допущение, а именно: ф> = 1, так как давле-
ние практически не влияет на значения коэффициентов
активности.
6.4.2. Точка росы (рис. 6.9, 6.10). При определении
точки росы или соответствующего ей давления возмож-
но лишь одно исходное допущение: у, = 1. Блок-схемы
и способы расчета для всех четырех вариантов опреде-
ления температуры начала кипения и точки росы во
многом одинаковы.
6.4.3. Испарение при фиксированных Т и Р (рис.
6.11). Составы обеих фаз неизвестны, поэтому исход-
ным допущением является следующее: Ф, = у, = 1.
1. Read T, Pf, y0
I-
2. $j =1_________
I
3. Assume P
I
YES
NO
NO
I yES~
Э. Syz=1?
| YES
1П END
1U. Print Pfyi
Puc. 6.8. Давление, соответствующее температуре начала
кипения.
Рис. 6.9. Точка росы.
Рис. 6.10. Давление, соответствующее точке росы.
Рис. 6.11. Испарение при фиксированных температуре и
давлении.
Равновесие пар — жидкость 33
1. Read Hq, P, zz-
= Vi= 1
3. Assume T
4. Ф1 Pi (PF)i
12. $t, Jj
7. xt, y.
YES
8. First trial?
NO
NO
6. 0, Eq. 6.73
9. Е|у(*+1) -у(*)| < £?
4 YES
NO ,
- ( 10. H(Eq. 6.72) = Но-
YES
И END
Print /3, Т, Xj, у.
Рис. 6.12. Испарение при
давлении.
фиксированных энтальпии и
Критерий сходимости — постоянство фазовых соста-
вов (см. п. 7 блок-схемы (рис. 6.11), установленное пу-
тем последовательных расчетов мольных долей пара.
Сумма мольных долей автоматически равна единице из
расчета коэффициента фазового разделения /3, который
определяется по методике Ньютона — Рафсона или ей
подобной.
6.4.4. Испарение при. фиксированных Н и Р (рис.
6.12). Начальное допущение, указанное в п. 2, то же,
что и в предыдущем случае. Принимают некоторое зна-
чение температуры и проверяют сходимость по посто-
янству состава. Если энтальпийный баланс неудовлетво-
рителен, задается другая температура и цикл повторя-
ется. После проведения двух таких циклов расчетов для
двух заданных величин температуры следующее значе-
ние температуры определяют, используя два вычислен-
ных значения энтальпии, линейной интерполяцией.
Т=Т2+ (Hmix - Н2)(Г2 - т,)/(!{2 -Нх). (6.99)
6.4.5. Методика Чао — Сидера [21SJ. В этой мето-
дике равновесие парообразования представлено тради-
ционной комбинацией трех членов:
YiL <t>iL
$iV
(6.100)
но оценка отдельных членов осуществляется иначе.
Польностью набор уравнений приведен в табл. 6.1 и
6.2, а отдельные уточнения даны в разд. 6.2.3.
Согласно современному варианту данного метода,
коэффициенты активности получают по уравнению
Скэтчарда — Гильдебранда — Флори — Хаггинса:
УМ ~ 8)2
RT
+ ln(ViL/VL) + l-ViL/VL
УгТ = ехр
(6.101)
Равновесие пар — жидкость 333
Пример 6.5. Температура начала кипения тройной смеси,
Вильсона
Смесь ацетон(1) + 2-бутанон(2) + этилацетат(З)
при 20 атм имеет следующий состав: X/ = Х2 = 0,3 и
хз = 0,4. По формулам Эббота (рис. 1.16) рассчитыва-
ют вириальные коэффициенты, перекрестные критиче-
ские характеристики оценивают по правилу
Лоренца — Бертло (табл. 1.5). Формулы для определе-
ния коэффициентов фугитивности и активности приве-
дены в табл. 3.3 и 3.4 и 4.7 соответственно.
Экспоненциальные коэффициенты уравнения Вильсона,
указанные в справочнике DECHEMA (Vol.l, parts 3 and
4, р. 389), вносят в программу на шаге 160. Поскольку
давление системы близко к величине давления паров
всех компонентов, то коэффициент Пойнтинга принима-
ют равным единице. Соответственно коэффициент рас-
пределения рассчитывают по формуле Kt = у,ф!Pf/c^P.
Последующий расчет ведется в соответствии с
блок-схемой, приведенной на рис. 6.7.
Распечатка результатов показывает, что температу-
ра начала кипения должна быть равна 468,7 К, а паро-
вая фаза должна иметь следующий состав: 0,3779,
0,2951, 0,3270. Как показал анализ нескольких темпера-
тур, в этом случае, по-видимому, отсутствует необходи-
мость в шаге 8 блок-схемы, приведенной на рис. 6.7.
Поскольку переход сразу к температуре 468,7 К дает те
же результаты за меньшее число итераций, нежели при
проведении расчетов, начиная с температуры 500 К с
определенным ее уменьшением с шагом в 10 К. Эконо-
мия в этом случае достигается на стационировании в
итерационной вычислительной процедуре. Однако
= 4,1 при 500 К, и сравнение отношений коэффи-
циентов фугитивности при двух температурах также по-
казывает значительное различие:
i ф1/ф, при 500 К при 468,7 К
1 1,0003 1,0001
2 1,1058 1,0018
3 1,2411 1,0078
Конечные значения давления пара,
идеальные коэффициенты распре-
деления, парциальные коэффициенты
фугитивности, коэффициенты фуги-
тивности чистых компонентов, отно-
шение коэффициентов фугитивности
и коэффициентов активности при
468,7 К
25,1151633414 (1) Pi
14,4904039759 (2)
15,4532211081 (3)
1,25575816707
0,724520198795
0,772661055405
Pf/P = (KJideal
Сходимость
значений У‘
при каждой
температуре.
При 468,7 К
значение сум-
мы сходится к
единице
500 К
1,8044 5 = ^у,
0,3317 я
0,2955 у2
0,3728 уз
500 К
1,7844
0,3351
0.2993
0,3657
рассчитанная по вириальному уравнению и уравнению
500 К
1,25991956422 X, 1,7815
0,984045189215 0,3356
0,817621023225 0,2993
0,3651
470 К
0,843532547419 0,- 1,0362
0,790714279772 0,3724
0,785362161074 0,2973
0,3304
470 К
0,843632255947 ф! 1,026
0,792192034686 0,3760
0,791524258278 0,2957
0,3284
470 К
1,00011820353 ф!/ф. 1,0253
1,00186888608 0,3762
1,0078461855 0,2954
0,3283
468,7 К
1,00319527112 у, 1,0007
1,35566896974 0,3777
1,04995036875 0,2952
0,3270
468,7 К
1,0000^
0,3779 Конец
0,2951
0,3270
Программа расчета температуры начала кипения на
компьютере НР-85
10 !«FLASH» BUBBLEPOINT Т
20 SHORT S
30 OPTION BASE 1
40 READ P,R
50 DATA 20,.08205
60 DIM B(3,3),D(3),F(3),F1(3),G(3),K(3),1L(3,3),P(3),Y(3)
70 ! MATRICES ENTERED IN ORDER:
T11,T12,T13,T21,T22,T23,T31,T32,T33
80 ! T1(I,J) CRIT TEMP;W(I,J) A CENT FACTOR;
P1(I,J) CRIT PRESS
90 DIM T1(3,3),W(3,3),P1(3,3)
100 DATA
508.1,521.7,515.6,521.7,535.6,529.4,515.6,529.4,523.2
110 DATA.309,.319,.336,.319,.329,.346,.336,.346,.363
120 DATA 46.4,43.5,40,43.5,41,37.8,40,37.8,37.8
130 MAT READ T1,W,P1
140 ! A(I3) WILSON EXPONENTIAL
PARAMETERS
150 DIM A(3,3)
160 DATA 0,1371.31,—292.975,—650.152,0,—405.21,
644.481,2704.427,0
170 MAT READ A
180 ! V(I) MOLAL VOLUMES
190 DIM V(3)
200 DATA 73.52,89.57,97.79
210 MAT READ V
220 ! Al,Bl,Cl, ANTOINE COEFFS
230 DIM A1(3),B1(3),C1(3)
240 DATA 16.6513,16.5986,16.1516,2940.46,3150.42, 520 G(I)=EXP(1—LOG(D(I)—X(l) ♦ L(1,I)/D(1)—X(2)
2790.5,—35.93,—36.65,—57.15 ♦ L(2,I)/D(2)—X(3)*L(3,I)/D(3))
250 MAT READ Al,Bl,Cl 530 NEXT I
260 INPUT T 540 ! В MIXTURE VIRIAL COEFF
270 ! B(I,J) VIRIAL COEFFS 550 B2=Y(1)A2*B(1,1)+Y(2)A2*B(2,2)+Y(3)A2*B(3,3)
280 ! L(I,J) WILSON PARAMS 560 В=B2+2 * (Y(l) * Y(2) * B( 1,2) +Y(1) ♦ Y(3) * B(1,3) + Y(2)
290 J = 1 *Y(3)*B(2,3))
300 FOR 1 = 1 TO 3 570 !F1(I) PURE COMP FUG COEFF
310 B(I, J)=R « T1(I, J)/P1(I. J)* (.083—.422 «(T1(I, J)/T)* 580 ! F(I) PARTIAL FUG COEFF
1.6+W(I,J)*(.139—.172*(T1(I,J)/T)M.2)) 590 ! P(I) VAPOR PRESS
320 L(I,J)=V(J)/V(I)*EXP(—(A(I,J)/1.987/T)) 600 ! K(I) VAP EQUILIB RATIO
330 NEXT I 610 FOR 1 = 1 TO 3
340 J = J + l 620 F1(I)=EXP(B(I,I)*P/R/T)
350 IF J<=3 THEN 300 630 F(I)=EXP(P/R/T* (2 « (Y(l)« B(I,1 > +Y(2)« B(I,2) +Y
360 DIM X(3) (3)*B(I,3)-B))
370 DATA ,3,.3,.4 640 F2(I) = F1(I)/F(I)
380 MAT READ X 650 P(I) = 1/76O*EXP(A1(I)—B1(I)/(T+ C1(I)))
390 FOR J = 1 TO 3 660 K(I) = G(I)*F1(I)*P(I)/F(I)/P
400 D( J)=X(l) ♦ L(J,1) + X(2) * L(J,2) + X(3) * L(J,3) 670 Y(I)=K(I)*X(I)
410 P(J) = l/760*EXP(Al(J)—B1(J)/T—C1(J)) 680 NEXT I
420 Y(J) = P(J)*X(J)/P 690 S=Y(1)+Y(2)+Y(3)
430 NEXT J 700 PRINT T
440 S=Y(1)+Y(2)+Y(3) 710 PRINT S
450 DISP S 720 FOR 1 = 1 TO 3
460 FOR J = 1 TO 3 730 Y(I)=Y(I)/S
470 Y(J)=Y(J)/S 740 NEXT I
480 NEXT J 750 MAT PRINT USING «2X,.DDDD»;Y
490 MAT DISP Y 760 INPUT T
500 ! G(I) ACTIVITY COEFFS 770 GOTO 510
510 FOR 1 = 1 TO 3 780 END
Пример 6.6. Расчет равновесия в системе пар — жидкость с применением уравнения Соава к обеим фазам
Для системы пропилен(1) + изобутан(2) зависи-
мость х — у при 20 атм рассчитана по всему диапазону
концентраций с использованием формул, приведенных в
табл. 1.11 и 3.4. Компоненты системы имеют следую-
щие основные характеристики:
U Вещество Tc Pc Vc Zc a b
11 C3H6 365 45,6 181 0,275 0,148 8,4078 0,0569
22 цзо-СдНю 408,1 36,0 263 0,283 0,176 13,3125 0,0806
ai = [1 + 0,7113(1 - V 77365) ]2 ,
а2 = [1 + 0,7533(1 - V 77408.1) ]2 ,
аа = (yi V aiai + Уг агаг )2,
b = yibi - у2Ьг,
1пф> = -ln[z(l - b/V)] + -у- (z - 1) +
+ (bi/ь - (g°9i_\in(i + b/vi
b RT у < aa /
Методика. Уравнение (9) из табл. 1.11 решают относи-
тельно наибольших и наименьших корней, которые ха-
рактеризуют сжимаемость жидкой и паровой фаз
соответственно. В начале необходимо провести проб-
ную оценку состава паровой фазы при заданном составе
жидкой фазы. После нахождения z определяют парци-
альные коэффициенты фугитивности, с этой целью
уравнение (27) решают совместно с уравнением (2) (оба
уравнения даны в табл. 3.4). Коэффициенты распреде-
ления находят по выражению К, = ф./ф!'. Температура
начала кипения — контрольная точка расчета. На по-
следующих стадиях рассчитанное значение у, = К,х,
нормализуется и используется для пересчета коэффици-
ентов фугитивности паровой фазы. Полученные резуль-
таты осуществленного таким образом расчета даны
ниже в виде таблицы, далее приведена программа
расчета.
Равновесие пар — жидкость 335
Xi Т У1 Zv ZL фГ 02 Фг 02
0 373,42 0 0,6636 0,1026 0,9005 0,7538 1,5060 0,7538
0,1 367,00 0,1606 0,6793 0,0993 0,8823 0,7403 1,4165 0,6904
0,2 360,84 0,3067 0,6924 0,0963 0,8664 0,7281 1,3286 0,6311
0,3 354,93 0,4376 0,7024 0,0935 0,8525 0,7168 1,2434 0,5759
0,4 349,34 0,5535 0,7100 0,0909 0,8401 0,7063 1,1626 0,5256
0,5 344,05 0,6553 0,7154 0,0885 0,8290 0,6962 1,0866 0,4799
0,6 339,06 0,7445 0,7190 0,0862 0,8187 0,6865 1,0157 0,4385
0,7 334,39 0,8221 0,7211 0,0839 0,8093 0,6772 0,9504 0,4016
0,8 330,00 0,8898 0,7246 0,0817 0,8003 0,6687 0,8901 0,3683
0,9 325,89 0,9486 0,7215 0,0796 0,7920 0,6590 0,8348 0,3387
1,0 322,02 1 0,7202 0,0775 0,7840 0,6502 0,7839 0,3121
10 ! X—Y DATA WITH «SOAVE» EQN 350 DISP «S = »;S
20 OPTION BASE 1 360 FOR 1 = 1 TO 2
30 DIM X(2),Y(2),Z(2),F(2),G(2),H(2) 370 Y(I)=Y(I)/S
40 DATA ,8,.2,.9,.l 380 NEXT I
50 MAT READ X,Y 390 DISP USING 400; X(1),Y(1),T,F(1),F(2),G(1),G(2)
60 READ P,R 400 IMAGE
70 DATA 20..08205 D.D,X,D.DDD,X,DDD.D,X,D.DDD,X,D.DDD,
72 DIM B(2),A(2) X,D.DDD,X,D.DDD
74 DATA .0569,.0806 405 IF ABS (S-l)< =0.0001 THEN 420
80 MAT READ В 410 GOTO 90
90 INPUT T 420 END
100 A(l) = 8.4078 * (1 + .7113 * (I—(T/365)A.5))A2 460 !SR FOR SOAVE COEFFS
НО A(2) = 13.3125 *(1 + .7533 *(1—(Т/408.4Г.5))л2 470 A = (Z(1)*A(1)a.5 + Z(2)*A(2)a.5)a2
120 FOR 1 = 1 TO 2 480 B=Z(1)*B(1) + Z(2)*B(2)
130 Z(I) = X(I) 490 RETURN
140 NEXT I 500 ! SR FOR COMPRESSIBILITY Z
150 GOSUB 460 510 C=A*P/Ra2/Ta2
160 Z = .05 520 D=B*P/R/T
170 GOSUB 500 530 Fl = ZA3—ZA2 + (C—D—DA2) * Z—C * D
180 DISP «ZL = »;Z 540 F2 = 3 * ZA—2 ♦ Z + C—D—DA2
190 GOSUB 600 550 H1 = F1/F2
200 FOR 1 = 1 TO 2 560 IF ABS(H1/Z)< =.0001 THEN 590
210 G(I)=H(I)!0f 570 Z = Z^-H1
220 Z(I)=Y(I) 580 GOTO 530
230 NEXT I 590 RETURN
240 GOSUB 460 600 !SR FOR FUGACITY COEFFS
250 Z = 1 610 FOR 1 = 1 TO 2
260 GOSUB 500 620 V=Z*R*T/P
270 DISP «ZV=»;Z 630 H(I) = —LOG(Z « (1—B/V)) + B(I)/B * (Z—1)
280 GOSUB 600 640 H(I)=H(I) + LOG(1 + В/V)/B/R/T * (A « B(I)/B—
290 FOR 1 = 1 TO 2 2*(Z)(1)*(A(I)*A(1))a.5+Z(2)*(A(I)*A(2))a.5))
300 F(I)=H(I)!# 650 H(I) = EXP(H(I))
310 K(I) = G(I)/F(I) 660 NEXT I
320 Y(I)=K(I)*X(I) 670 RETURN
330 NEXT I 680 END
340 S=Y(1)+Y(2)
Парциальные коэффициенты фугитивности ф1(/ находят
по уравнению Редлиха — Квонга. Коэффициент фуги-
тивности чистой жидкости — это эмпирическая
функция ацентрического коэффициента и приведенных
температуры и давления. Принятые в настоящее время
коэффициенты разработаны Г]рейсоном и Стридом
[318]. Ленуар и Коппани [435] подробно исследовали
этот способ; представленные в табл. 6.1 ограничения
выявлены главным образом ими. Другие примеры ее
использования можно найти в книге Хенли и Сидера
[52]. Методика Чао — Сидера входит в число проанали-
зированных Американским нефтяным институтом (API)
[31]. Этот анализ показал общие недостатки методики
расчета, в которой уравнение Соава применяется к обе-
им фазам, тем не менее указанное уравнение до сих пор
еще широко применяют для расчетов равновесий смесей
легких углеводородов как в депропанизаторах, так и в
дебутанизаторах, а в сочетании с коэффициентами Грей-
сона — Стрида оно удобно -для моделирования систем
синтетического топлива.
Пример 6.7. Расчет точки росы по методике Чао — Сидера
Определяем точку росы смеси пропилен + изобутан
с yi = 0,607 при 20 атм. Сжимаемость находят мето-
дом проб по уравнению Редлиха — Квонга (уравнение
(7) табл. 1.9):
Z3 - z2 + (А - В - B2)z - АВ = 0, (7)
А = aP/R^T2'5,
В = bP/RT,
R = 0,08205.
Парциальные коэффициенты фугитивности определяют
по уравнению (18), приведенному в табл. 3.4:
1п0; = -ln(z - В) + Bi(z - 1)/В +
+ (А/В)(В,/В - 2\'A,/A)ln(l + B/z).
Фугитивность жидкостей и активность жидких компоне-
нтов рассчитывают по формулам, указанным в табл.
6.1. Точку росы устанавливают методом проб по блок-
схеме, показанной на рис. 6.9. Эта блок-схема преду-
сматривает определение температуры, оценку состава
при допущении, что коэффициенты активности равны
единице, и последующее повторение расчета для улуч-
шения значений коэффициентов активности до их схо-
димости.
В этом частном случае коэффициенты активности
действительно близки к единице, так что сходимость
достигается быстро.
В результате расчета получают Г = 346,4,
Xi = 0,541. Результаты расчета, показанного в примере
6.6: Т = 346,6, Xi = 0,451. Используя приведенную на
рис. 6.5 номограмму API, находим значения у,/К
при
350 К 0,670/1,50 + 0,393/0,68 = 0,9826
340 К 0,607/1,30 + 0,393/0,56 = 1,1687.
Интерполяция дает Т = 349 К, Xi = 0,41.
Существенные различия в результатах расчетов,
основанных на трех указанных методиках, вызывают
беспокойство. Как указывалось в разд. 1.9, исследова-
ния, проведенные Американским нефтяным институтом
на основе статистических данных, показали, что метод
Соава в том варианте, который рассмотрен в примере
6.6, нельзя считать удачным. В каждом конкретном
случае необходимо решать, что предпочтительнее —
завышенные или заниженные результаты.
Например, исходя из того, что коэффициенты активнос-
ти равны единице, путем проведения итераций было по-
лучено при 340 К Xi = 0,5383. Коэффициенты распреде-
ления рассчитывают по уравнению
Ki = 7i0ji/0i.
Vi 72 К кг Xi
T = 346
0,993 0,992 1,111 0,850 1,0085 0,542
0,993 0,992 1,111 0,850 1,0085 0,542
0,993 0,992 1,111 0,850 1,0085 0,542
T = 346,5
0,993 0,992 1,120 0,855 1,0015 0,541
0,993 0,992 1,120 0,855 1,0015 0,541
0,993 0,992 1,120 0,855 1,0015 0,541
T = 347
0,993 0,992 1,124 0,863 0,9954 0,542
0,993 0,992 1,124 0,863 0,9954 0,542
0,993 0,992 1,124 0,863 0,9954 0,542
10 ! DEWPOINT BY FIG. 6.9
20 READ T,L1,L2
30 DATA 347,1.132,.87
40 INPUT X
50 Fl = 79*X + 105.5*(l—X)
60 F=79*X/F1
70 T1 = 79/F1
80 T2=105.5/Fl
90 D3 = 6.43*F+6.73*(1—F)
100 G1 = EXP(79 ♦ (6.43—D3)A2/1.987/T+ LOG(T1)+1—T2)
110 G2=EXP(105.5#(6.73—D3)A2/1.987/T+LOG(T2)+1
—T2)
Tc Pc Ы 5 V а b
СзНб 365 45,6 0,1451 6,43 79 160,64 0,0569
U3O-C1 Смесь 408,1 36 0,1825 6,73 105,5 268,96 199,90 0,0806 0,0662
T z 0i 02 01£ 02£ 011/01 021/02
320 0,5827 0,7976 0,6277 0,6145 0,3565 0,7704 0,5679
330 0,6535 0,8121 0,6656 0,7310 0,4441 0,9001 0,6672
340 0,7000 0,8266 0,6957 0,8564 0,5441 1,0361 0,7821
350 0,7352 0,8401 0,7212 0,9886 0,6565 1,1768 0,9103
Равновесие пар — жидкость 337
120 K1 = G1*L1
130 K2=G2*L2
140 Х1 = .607/К1
150 Х2=393/К2
160 S=X1 + X2
170 X = X1/S
180 PRINT USING 190; G1,G2,K1,K2,S,X
190 IMAGE
.ddd,x,.ddd,x,d.ddd,x,.ddd,x,d.dddd,x,.ddd
200 GOTO 50
210 END
6.4.6. Энтальпия как функция T, Р и состава. Для
приемлемой оценки энтальпийных балансов необходимо
располагать значениями парциальных молярных эн-
тальпий в виде функций состава, а также величинами
температуры и давления. Такие характеристики систе-
мы можно выразить через коэффициенты фугитивности
и активности, которые получают из уравнений состоя-
ния или из обобщенных корреляций. Детально такие за-
висимости обсуждаются в гл. 11, а здесь лишь
перечисляются те методики, которыми можно воспо-
льзоваться, и приводятся соответствующие формулы.
Ниже указано основное выражение, связывающее
энтальпию смеси с нулевым давлением и темпера-
турой То:
корреляциях коэффициентов активности, если таковые
известны. Основным состоянием является следующее:
fi = 7iXiPsi <rf(PF)i. (6.106)
Коэффициент ф! чистого насыщенного пара получен из
соответствующего уравнения состояния типа
у/Х/Р/ехр
RT
(6.107)
nr пР /дН \
Н = Н%+ / C*dT + / — ] dP
JTo Р Jo \дР/Т
(6.102)
(6.103)
где звездочками обозначены величины, отвечающие
идеальному состоянию газов. Второй интеграл можно
рассчитывать из уравнения состояния смеси, например,
^-усеченного вириального уравнения
Я=
+ Гс'-odT
JjQ
(6.104)
где вириальный коэффициент смеси В зависит от ее со-
става (см. табл. 1.8).
Парциальную молярную энтальпию можно рассчи-
тать из следующего уравнения:
Н, = Hi- RT2
(6.105)
которые справедливы как для жидкой, так и для паро-
вой фазы. Для некоторых веществ уравнения Соава и
Бенедикта — Уэбба — Рубина, например, применимы к
обеим фазам. Уравнения для расчета парциальных ко-
эффициентов фугитивности приведены в табл. 3.4. Рас-
чет широкого спектра смесей может быть основан на
Энтальпийные зависимости, выведенные из этих урав-
нений, даны в гл. 11. Из-за сложности этих уравнений
требования, которым должны отвечать программы их
обработки на ЭВМ, могут быть чрезвычайно строгими-.
Как всегда, здравый смысл должен подсказать, на чем
следует остановить выбор — на лучшей из доступных
методик или более простой, но позволяющей получить
вполне удовлетворительные результаты. Наиболее об-
щее допущение состоит в том, что парциальные моляр-
ные значения энтальпий эквивалентны аналогичным
значениям энтальпий чистых компонентов и при прове-
дении расчетов необходимо учитывать лишь влияние
температуры и давления.
6.5. Растворимость газов в жидкостях
Растворы — это гомогенные смеси. Компоненты,
присутствующие в этих смесях в значительно большей
концентрации, обычно называют растворителями, а
все прочие компоненты — растворенными вещества-
ми. К последним относятся, в частности, газы и твер-
дые вещества, растворенные в жидкостях. В особых
рассматриваемых здесь растворах газов в жидкостях
давление паров растворителя может быть пренебрежи-
мо малым, так что весь растворитель может находить-
ся в жидком состоянии. Или, другими словами, у всех
компонентов растворителя, если он представляет собой
смесь, коэффициенты распределения равны нулю. Если
температура газа ниже критической, его растворимость
может быть установлена либо оценена рассмотренными
в этой главе методами. Если же температура газа пре-
вышает критическую, необходимы специальные ме-
тоды.
Ряд важных зависимостей получен на основе закона
Генри. Недавний пример — несколько уточненная схема
Линии и др. (1976), которые используют уравнение в
форме
%2 = G\S j + G2S2,
где G — характеристики растворенного вещества и
S —характеристики растворителя.
Многие из широкоиспользуемых единиц раствори-
3-864
Таблица 6.3. Растворимость газов (IO4* мольные доли растворенного вещества) при 25°С и парциальном
давлении 1 атм (данные Battino, Clever Solutions and Solubilities, Dack (ed.), pt. 1, 1976). Приведены парамет-
ры растворимости и молярные объемы
Растворитель Не Ne Аг н2 n2 о2 СО2 СНд 5 V
1. Вода 0,068 0,082 0,254 0,142 0,119 0,231 — 0,248 23,53 18,1
2. н-Гексан 2,604 3,699 25,12 6,315 14,02 19,3 — 50,37 7,27 131,6
3. н-Октан 2,397 3,626 24.26 6,845 13,04 20,83 — 29,27 7,54 163,4
4. н-Тетрадекан 2,249 3,340 26,50 — — — — — — —
5. Изооктан 3,083 4,593 29,21 7,832 15,39 28,14 138,7 29,66 — —
6. Циклогексан 1,217 1,792 14,80 4,142 7,61 12,48 76,0 32,75 8,19 108,7
7. Бензол 0,771 1,118 8,815 2,580 4,461 8,165 97,30 20,77 9,16 89,4
8. Толуол 0,974 1,402 10,86 3,171 5,74 9,09 101,3 24,14 8,93 106,8
9. л<-Ксилол 1,121 1,619 — 4,153 — — — — 8,88 123,4
10. н-Перфторгептан 8,862 — 53,22 14,03 38,80 55,08 208,2 82,56 — —
11. Гексофторбензол 2,137 3,455 23,98 — ' 17,95 24,18 220 38,42 — —
12. Тетрахлорид углерода — — 13,51 3,349 6,480 12,01 105,3 28,70 8,55 97,0
13. Хлорбензол 0,691 0,979 8,609 2,609 4,377 7,910 98,06 20,47 9,67 102,2
14. Нитробензол 0,350 0,436 4,448 — — 4,95 99,80 — 10,8 102,7
15. Метанол 0,595 0,814 4,491 — 2,747 4,147 55,78 8,695 14,5 40,7
16. Этанол 0,769 1,081 6,231 2,067 3,593 5,841 63,66 12,80 12,78 58,7
17. Ацетон 1,081 1,577 9,067 2,996 5,395 8,383 185,3 18,35 9,62 74,0
18. Диметилсуль- фоксид 0,284 0,368 1,54 0,761 0,833 1,57 90,8 3,86 12,0 71,3
19. 6 — — 5,33 0 4,44 4,0 7,12 5,68
20. V — — — 31,0 53,0 28,4’ 44,0 52,0
мости газов систематизированы Круисом [410], им так-
же выполнен теоретический обзор и приведено много
справочных данных. Расширенные теоретические разра-
ботки этого вопроса можно найти в книгах Шиноды
[121] и Декка [27].
Вне границ идеальных состояний понятие «пара-
метр растворимости» может быть полезным для коли-
чественного выражения растворимости газов и других
фаз. В балансе фугитивности, где индексом 2 обозначен
газ,
flG =flL = 12x2f2L > (6.108)
jiL — фугитивность чистого растворенного в жидкой
фазе вещества. Это величина гипотетическая, характе-
ризующая такую систему, в которой температура газа
превышает критическую. Однако, как будет показано
далее, ее можно оценить. Разрешая уравнение (6.108)
относительно растворимости хг и вводя уравнение
Скэтчарда — Гильдебранда, получаем
Л(7 Л(7
*2 7 А Al ехр
V2L
1\ 1
(6.109)
Если поведение газовой фазы по существу идеально, фу-
гитивность можно заменить на давление
*2-т£г«ч>
1\ 1
(6.110)
Однако это уравнение неудовлетворительно в тех случа-
ях, когда температура растворенного вещества выше
критической. Фугитивность чистого газа /го можно
найти из подходящего уравнения состояния, которое,
кроме того, можно использовать для оценки фугитив-
ности насыщенной жидкости при температуре системы
и, следовательно, для оценки /н. при температуре и дав-
лении системы, если ввести коэффициент Пойнтинга,
Равновесие пар — жидкость 339
f°2L=fS2L exp(V2L(P-Ps2)/RT).
(6.111) f°2i= 7.224 - 7.534(ГС,/Г) - 2.598 ln(T/Tci), (6.115)
Описанный подход был принят Кингом и др. [440] для
описания растворимости таких газов, как СО2, H2S и
СзНв в нескольких алканах. Поскольку критические
температуры этих газов превышают 25°C, экстраполя-
ция величин давления паров возможна лишь в умерен-
ных пределах —максимум до 70°С. Тем не менее даже
в таком случае лучше использовать расширенный вари-
ант уравнения Скэтчарда — Гильдебранда, т. е. уравне-
ние Флори — Хаггинса — Скэтчарда — Гильдебранда,
включающее параметры перекрестного взаимодействия:
У21
1пу2 = 0?К^1 - 5г)2+ 2/12^1 S2
J\ 1
+ 1 — 0]/х] + 1п(ф]/Х])]. (6.112)
Обычно считается, что коэффициент перекрестного вза-
имодействия /и равен нулю, но в работе Кинга его при-
нимают равным примерно 0,02 для легких
углеводородов и 0,1 для СОг и H2S. Дальнейшее улуч-
шение в пределах 1% данных эксперимента было полу-
чено подгонкой значения параметра растворимости
далекого от его теоретических значений. В предельном
случае — система H2S — подгоночное значение <5 соста-
вило 5,6, тогда как действительное значение равно 8,36.
Если экспериментальные данные доступны, а молярные
объемы и параметры растворимости известны, эффек-
тивную фугитивность перегретых жидкостей можно пе-
ресчитать из уравнения (6.109), что и было проделано
Праузницем и Шеером [571] и Праузницем [564]. Полу-
ченную ими графическую корреляцию, в которую вхо-
дят приведенная температура и критическое давление,
можно представить уравнением
In (Д/РС2) = 7.81 - 8.06/ Тг - 2.94 In Тг,
0.7 <Гг <2.5. (6.113)
Праузниц и др. [98] распространили аналогичную зави-
симость на многокомпонентные смеси. Согласно авто-
рам [98], растворимость может быть представлена
уравнением
Xi = fiv/jliyi (6.114)
Вклады отдельных компонентов в фугитивность даются
уравнением
вклады парного взаимодействия рассчитываются как
= ехр(а,у + д(7/Г), (6.116)
где коэффициенты ау и by имеют характерные для каж-
дой системы значения. В работе [98] приведены эмпири-
ческие значения этих коэффициентов для четырех газов
и двух жидкостей, но каких-либо обобщений не сделано.
В этой же работе сформулированы правила определе-
ния суммарных свойств /h и yt.
Если газ растворен в смеси, растворимость каждо-
го компонента можно выразить посредством многоком-
понентного уранения Скэтчарда — Гильдебранда, урав-
нение (4.195), так что в итоге уравнение (6.110), напри-
мер, принимает для каждого газа такой вид:
Р
*i= 7iZexp(- Vtfi-S^/RT) (6.117)
Одна из принципиальных схем расчета смесей, ком-
поненты которых находятся в сверхкритическом состоя-
нии, разработана ван Нессом и Эбботом [708]. В
данной ситуации необходимо знать парциальные фуги-
тивности всех пар компонентов в виде функции опреде-
ленного диапазона составов смеси и принять допущение
о специфической форме зависимости коэффициентов от
состава, например в виде уравнения Вильсона. Эти дан-
ные были восстановлены, поэтому параметры уравне-
ния Вильсона и получаемые экстраполяцией стан-
дартные значения фугитивности чистых компонентов
находят одновременно, проводя эту процедуру последо-
вательно для каждой пары компонентов. Достоинство
такой методики состоит в том, что эталонное состояние
всех компонентов однотипно: чистая жидкость при на-
личии компонентов в сверхкритическом состоянии и ги-
потетическая чистая жидкость во всех других случаях.
В примере 6.12 изложены основы метода и показано его
применение для расчета температуры начала кипения.
Как отмечалось в примере 6.8, результаты, полу-
ченные по методикам, рассмотренным в данном разде-
ле, имеют смысл лишь при условии, что критические
температуры незначительно разнятся от температуры
системы, в противном случае они менее приемлемы.
Как учитывать компоненты, характеристики которых
превышают критические, окончательно еще не решено.
Исследования по этому поводу ведутся весьма активно.
В связи с этим полезно познакомиться с работами
О’Коннели [528], Брандани и Праузница [197].
Пример 6.8. Растворимость СОг и H2S в н-декане при
Экспериментальные значения взяты из работы
Кинга и др. [400]. Они будут проверены расчетами по
уравнению параметра растворимости с использованием
фугитивности перегретых жидкостей путем 1) экстрапо-
ляции давления пара по уравнению Антуана и 2) прове-
дения расчетов по соответствующему уравнению
состояния Праузница и Шэра [571].
Исходные и расчетные данные приводятся в табл.
2, в табл. 1 сравниваются величины растворимости.
1 атм
Методика расчета включает следующие этапы.
1. Рассчитывают давление пара по уравнению
Антуана.
2. Оценивают коэффициенты фугитивности перегре-
той жидкости по таблицам Ли — Кеслера.
3. Оценивают значение коэффициента Пойнтинга по
уравнению PF = ехр(Кг(1 - Р“,)//?Г).
4. Рассчитывают фугитивность перегретой жидкос-
3*
ти fu. = Ргт <i>2M(PF)2, которую называют также экс-
траполяционным значением фугитивности.
5. Рассчитывают также эту величину по уравнению
(6.113)
Jil = Рсехр(7,81 - 8,06/77 - 1п77).
6. Определяют коэффициенты активности раство-
ренных веществ по упрощенному уравнению
72 = ехр(Иг(7,72 - 82)2/РТ,
пренебрегая влиянием небольших величин х2.
7. В заключение рассчитывают растворимость по
уравнению
х2 = /гг/ул/ь = l/yzfiL,
в котором фугитивность принимают равной давлению
в 1 атм.
Сравнение. Данные экстраполяционного метода
лучше согласуются с экспериментальными данными для
Таблица 1. Величины растворимости
СО2 H2S
20°С 50°С 20°С 50°С
Эксперимент 0,0133 0,0099 0,0502 0,0325
Уравнение Прауз- ница и Шэра Экстраполяция 0,021 0,013 0,060 0,031
давления пара 0,011 0,0082 0,052 0,031
СОг, но результаты расчетов, проведенных для H2S по
различным методикам, близки. Используя расширенное
уравнение Скэтчарда — Гильдебранда, Кинг и др. [400]
достигли точности расчета с разбросом в 1% от экспе-
риментальных значений.
Таблица 2. Исходные и расчетные данные
С10Н22 СО2 H2S
20°С 50°С 20°С 50°С
Ре 72,8 88,2
Тс 304,2 373,2
0) 0,225 0,1
5 7,72 7,12 8,8
V 196 44 34,3
psu 214 572 17,3 33,7
ф™' 0,6 0,53 С,91 0,88
Коэффициент
Пойнтинга 0,68 0,39 0,98 0,96
Jl (экстраполяция) 87 118 15,5 28,5
У? (корреляция) 46,7 76,1 15,5 30,1
7 1,0276 1,0250 1,0711 1,0643
Пример 6.9. Диаграммы х — у, рассчитанные при помощи однопараметрического уравнения Маргулеса для по-
стоянной температуры и постоянного давления
1. Диаграммы х — у первого типа (Т = const)
строят для давления паров Р% = 1,5 и Р% = 1. Коэффи-
циенты активности задают уравнениями
Inyi = Ах2, 1п72 = Ах2,
где А = 3? 2, 1 или 0. Состав пара определяют так же,
как в примере 6.10. Отделение жидкой фазы присходит
при А = 3. Штриховая часть кривой физически не ре-
ализуется.
2. При постоянстве давления расчет коэффициентов
активности проводят по уравнениям, учитывающим
влияние на эти коэффициенты температур:
1 М 1 Af 7
1П71 = — хг, 1пу2 = т х{.
Проверяют несколько значений М: 1000, 500 и 0. Давле-
ние пара рассчитывают по уравнениям
1пР? = 12,5 - 4300/Г, 1пР£ = 12 - 4500/Г.
При установленном значении Р = 0,5 температуру нача-
ла кипения каждой жидкой композиции определяют из
условий
Р — У1Х1Р^ + у2х2Р%
по методике Ньютона — Рафсоца. Состав пара находят
из уравнения:
У1 = 1/(1 + у2Х2Р%/у!Х1Р^).
На рис. б штриховой кривой показана область сосу-
ществования двух жидких фаз при М = 1000.
3. Графики зависимости температура — состав (в
частности, рис. б) построены при помощи части про-
граммы. Для М - 500 при Xi = 0,86 имеется темпера-
турный минимум (326°С). Смесь двух жидких фаз с
М - 1000 кипит при 319°С.
Равновесие пар — жидкость 341
Диаграмма х — у при постоянной температуре. Для области,
ограниченной штриховой линией, действительная ситуация,
имеющая физический смысл, соответствует горизонтальной
линии.
Диаграмма х — у при постоянном давлении. Для области,
ограниченной штриховой линией, действительная ситуация,
имеющая физический смысл, соответствует горизонтальной
линии.
Зависимость состав — температура. Для области, ограничен-
ной штриховой линией, действительная ситуация, имеющая
физический смысл, соответствует горизонтальной линии.
Пример 6.10. Расчет изотермического парожидкостного равновесия по уравнению Маргулеса
Иследуется влияние на форму кривых х — у, Р — х Маргулеса. Давление пара Pi = 3 и = 1. Параметр
и 7 — х нескольких комбинаций параметров уравнения д уравнения Маргулеса во всех случаях равен единице,
Диаграмма х — у. Штриховая линия получена расчетным пу-
тем, а действительный физический смысл ситуации в этой
области передает горизонтальная линия.
Равновесие пар — жидкость 343
Диаграмма Р — х. Зависимость общего давления от состава.
Для области, ограниченной штриховой линией, действитель-
ная ситуация, имеющая физический смысл, соответствует
горизонтальной линии ab.
Коэффициенты активности.
параметр В принимают равным 4, 2, - 2 или - 6. Коэф-
фициенты активности определяют по уравнениям
ln7i = [А + 2(В - ln72[B + 2(А - В)х2]х?.
а. Состав пара рассчитывают по формуле yi =
= 1/(1 + 72X2^/7iXiP?). При А и В, равным 1 и 4 или
1 и — 6, образуются две жидкие фазы. Кроме того, во
втором случае, так же как и при А = 1 и В — 2, образу-
ется азеотропная смесь.
6.6. Бинарные диаграммы равновесия
пар — жидкость
Хотя бинарные смеси встречаются на практике не-
часто, количественная оценка их поведения важна, по-
скольку методики, описывающие поведение многоком-
понентных систем, обычно базируются на данных, по-
лученных для отдельных бинарных систем. Такие пред-
ставления не отличаются точностью, а иногда и не
совсем адекватны, однако пока это единственно воз-
можный подход. Данных даже по трех- или четырех-
компонентным системам очень мало; исключение
составляют лишь тройные системы жидкость — жид-
кость, поскольку они играют важную роль в процессах
экстракции.
В последние годы опубликовано несколько обшир-
ных подборок данных о равновесии пар — жидкость
преимущественно для бинарных систем. К числу наибо-
лее интересных публикаций такого рода относятся сле-
дующие:
1. Landolt-Bornstein, II 2а: 661 серия данных о двухком-
понентных системах, 49 наборов данных о трехкомпо-
нентных системах, описание около 3000 бинарных
азеотропных систем [73]; IV4L (1972): 138 серий данных
о двух- и 29 — о трехкомпонентных системах; New
Series Group IV, vol. 3(1975): 557 серий данных о двух-
компонентных, 86 — о трехкомпонентных и 4 — о че-
тырехкомпонентных системах.
2. Коган, Фридман, Кафаров. Парожидкостные равно-
весия. — М.: «Наука», 1966: 1765 серий данных о двух-
компонентных, 358 — о трехкомпонентных и 44 — о
четырехкомпонентных системах [69].
3. Hirata, Ohe, Nagahama Computer-Aided Data Book of
Vapor-Liquid Equilibria, 1976: 800 серий данных о двух-
компонентных системах и 133 серии данных о двухком-
понентных системах при повышенных давлениях [57].
4. Maczynski et al. Verified Vapor-Liquid Equilibria Data,
4 volumes, 1977—1979 [83].
5. Gmehling, Onken, Arlt DECHEMA Vapor-Liquid Eq-
uilibrium Data Collection, 1977: около 12 томов, содержа-
щих 8000—9000 серий данных главным образом о
двухкомпонентных системах и нескольких трех- и четы-
рехкомпонентных системах. Отдельный том посвящен
равновесиям при высоких давлениях.
Корреляция данных по равновесию в системах
пар —жидкость с использованием фугитивности и ак-
тивности рассматривалась в предыдущих главах. В
большинстве случаев отклонение жидкой фазы от иде-
альности значительнее, чем аналогичное отклонение, на-
блюдаемое для паровой фазы; последним при
б. Общее давление рассчитывают по формуле
Р — 71Х1Р? + 72Х2^Э2.
Области сосуществования двух жидких фаз ограничены
точками а и Ь, а также d и е (рис. б). Максимальное
давление азеотропа соответствует точке с, а минималь-
ное — точке f.
в. Коэффициенты активности рассчитывают непо-
средственно по уравнениям Маргулеса. Характерные
особенности кривых, приведенных на рис. в, — наличие
максимумов и минимумов.
умеренных давлениях часто пренебрегают (см., напри-
мер, справочник DECHEMA или книгу Хираты [57]).
Эти корреляции, которые неточны из-за действитель-
ной неидеальности паровой фазы, требуют введения
группового параметра 7/ф/ (PF)i/ф,, называемого эффек-
тивным коэффициентом активности.
Формы диаграмм х — у и Р — х, Т — х довольно
разнообразны. Вероятно, наиболее важной представля-
ется классификация систем на 1) не содержащие азео-
тропную смесь, 2) содержащие азеотропную смесь, 3)
содержащие две жидкие фазы. Многочисленные приме-
ры систем всех указанных видов можно найти в пере-
численных публикациях, однако данных о равновесия
пар — жидкость для несмешивающихся жидкостей
опубликовано меньше всего. Так, в справочнике Лан-
дольда-Бернштейна [73] приведено лишь 9 (№60, 62, 65,
68, 90, 152, 378, 634 и 635) диаграмм, на которых име-
ются такие области, где входящие в исследуемую систе-
му жидкости не смешиваются.
На форму диаграмм равновесия наибольшее влия-
ние оказывают, безусловно, коэффициенты активности.
Показанные в этих примерах изотермы и изобары рас-
считаны по симметричному уравнению Маргулеса.
Одна из таких систем имеет двойной азеотроп, редко
наблюдаемый на практике (см. рис. 4.24, а и 5.22, ж).
Расчетные кривые х — у систем, для которых характер-
ны области несмешиваемости, всегда имеют максимум
и минимум, а участки кривой в окрестностях экстрему-
ма соответствуют областям нестабильности. Для на-
хождения истинных кривых равновесия, которые
представляют собой горизонтальные линии в двухфаз-
ной области, необходимо выполнить расчеты по равно-
весию в системах жидкость — жидкость, что и
проделано в примере 7.12, где также показаны области
нестабильности на кривых Р — х и Р — у.
6.7. Минимизация энергии Гйббса
Метод нахождения условий равновесия минимиза-
цией энергии Гиббса смеси не всегда является самым
простым, тем не менее область его применения доста-
точно широка (см., в частности, гл. 7 и 10). В данном
разделе, рассматривая использование этого метода, мы
ограничимся системами, содержащими паровую и всего
одну жидкую фазу.
Для каждого компонента паровой фазы парциаль-
ная энергия Гиббса определяется выражением
G/K= GiV + RT\n(fiVlfiV), (6.118)
Равновесие пар — жидкость 345
Для каждого компонента жидкой фазы справедливо вы-
ражение
GiL= GiL + RT In (fiL/fiL)
= GiV + RT\n(JiL/fiv).
(6.119)
(6.120)
Если за стандартное состояние выбран газ с единичной
фугитивностью, энергия Гиббса чистых компонентов
становится такой стандартной характеристикой, Gw.
Более того, если энергия Гиббса элементов произвольно
принимается равной нулю, то энергия Гиббса компоне-
нтов смеси становится равной энергии Гиббса образова-
ния. Соответственно уравнения (6.118) и (6.120) можно
использовать в форме
Giv= G^v + RT\nfiV,
GiL = GQiV + RT\nfiL.
(6.121)
(6.122)
Запишем уравнения для парциальных фугитивностей ис-
ходя из реально рассчитанных значений:
= Ф{РУь
fiL = YZ0fPj(PF)z.
(6.123)
(6.124)
Общая энергия Гиббса смеси, содержащей в целом и,
молей компонента i, v, молей в паровой фазе и h молей
в жидкой фазе, с долей паровой фазы
Д = S Vj/Z tii - V/F
(6.125)
определяется выражением
nG I Gjy
+ (1-/?)£//
Pr+ln(y/-0fPJ(PF),x/)
(6.126)
которое можно записать как
G-
RT
(6.127)
= Д by, In 0fPyz + (1 - Д) SXi In (yi$SiPSi(PF)iXi),
(6.128)
где zt = п> — общая мольная доля компонента i
смеси. Для данного общего содержания величина
S ZiGw — константа.
Состав пара можно исключить из этого уравнения
путем замены
Yi<tfPj(PF)i
(6.129)
хотя для этого может потребоваться последующая ап-
проксимация, если отношение ф°/ф1 заметно отличается
от единицы, поскольку в этом случае ф, зависит от со-
става пара. Коэффициент фазового разделения /3 оцени-
вается исходя из материального баланса:
j3 = (z(-x()/O,-x/). (6.130)
Какой из компонентов использовать для подобной под-
становки — безразлично, однако лучше, чтобы в резуль-
тате подстановок знаменатель значительно отличался
от нуля. Имеется еще одно условие:
Sx,-l = o. (6.131)
При введении множителя Лагранжа X с учетом этого ус-
ловия минимизация уравнения (6.128), включающего
уравнение (6.131), эквивалентна минимизации ф:
ф ~g + X(Sx(- — 1) — минимум. (6.132)
Производные ф относительно мольных долей жид-
кой фазы совместно с уравнением (6.131) составляют
систему уравнений, из которых X и состав могут быть
найдены, например, по методике Ньютона — Рафсона
либо подобной ей, или могут быть установлены пря-
мым поиском при минимальном значении ф.
Определение условий равновесия минимизацией
значений энергии Гиббса — процедура во многих случа-
ях трудоемкая по сравнению с другими методиками
расчетов испарения, приведенными в этой главе. Для
бинарных смесей нет необходимости применять множи-
тели Лагранжа, так как энергия Гиббса смеси обычно
выражается исходя из единственного молярного соста-
ва. Это обстоятельство учитывается в примере 6.11.
Примеры методик с использованием множителей Ла-
гранжа подробно описаны в гл. 10.
Пример 6.11. Равновесие как условие минимума энергии
Равновесие смесей циклогексан + л<-ксилол при
25 ° С хорошо описывается уравнением G^/RT =
= 0,48 XiXz(DECHEMA I/6a, р. 316). Система характе-
ризуется следующим давлением паров: Pf = 97,6 мм рт.
ст. Равновесный состав при общем давлении 50 мм рт.
ст. можно определить минимизацией энергии Гиббса.
При таком низком давлении, как указанное, можно
допустить, что 0; = 0f = (PF), = 1, так что состав пара
Гиббса
можно исключить из уравнения (6.128) путем подста-
новки у, - (yiPi/P)Xi. Однако до осуществления указан-
ной подстановки необходимо нормализовать значения
состава паров. Такая процедура гарантирует правиль-
ность нахождения минимума энергии Гиббса, однако
это не означает, что его величины справедливы при
других составах. Полученные в результате кривые (см.
рисунок) показывают, что состав при минимальном
значении энергии Гйббса не зависит от общего состава
смеси ъ, как это должно быть. Значение Xi = 0,379,
найденное графически, вполне сравнимо с результатами
вычислений: Xi = 0,3786, yi = 0,8895, /3 = 0,2376 при
Zi = 0,5.
5 EXAMPLE 6.11
10 SCALE 0,1,-4.5,-3.5
20 XAXIS -4.5..1
22 XAXIS -3.5..1 •
24 YAXIS l,.l
30 YAXIS 0,.l
40 READ Z,P,P1,P2
50 DATA .2,.0658,.1284,.0109
60 FOR X=.001 TO .999 STEP .01
70 G1 = EXP(.48*(1—Х)Л2)
80 G2=EXP(.48*XA2)
90 Yl = G1 * Pl * X/(G1 * Pl * X+G2 * P2 * (1—X))
100 Y2=l—Yl
110 B=(Z—X)/(Y1—X)
120 Y= В * (Yl * LOG(P * Yl) + Y2* LOG(P * Y2))+(1—B) *
(X * LOG(G1 * Pl * X)+(1—X) * LOG(G2 * P2 * (1—X)))
130 DRAW X,Y
140 NEXT X
150 MOVE 0,0
160 BEEP
170 END
Кривая g = f(xi) (см. уравнение (6.128)). Минимум соответ-
ствует Xi = 0,379.
Пример 6.12. Коэффициенты активности тройной смеси, компоненты которой находятся в сверхкритическом
состоянии, и использование этих коэффициентов для расчета температуры начала кипения
Компонент, находящийся в сверхкритическом со-
стоянии, обозначают индексом 1, а прочие компоненты
обозначают индексами 2 и 3 соответственно. Экстрапо-
ляционную фугитивность чистых компонентов обозна-
чают У12. В условиях равновесия
Л = (1)
Расчет избытка энергии Гиббса проводят по уравнению
Вильсона.
Соответственно
= g^/RT
= 2 Х/1П7, = 2 /Xifi} =
= S х,1п(фГР/у?) (2)
= Xiln^f + Xzln^ + InP - Xiln/12 - Х21Ц/2 (3)
= -Xiln(Xi + Л12х2) - X2ln(A2lx2 + x2) (4)
Неизвестные величины в выражениях (3) и (4) — это па-
раметры уравнения Вильсона и экстраполированные
значения фугитивности чистого компонента J12 для ком-
понента 1 в сверхкритических условиях в присутствии
компонента 2, находящегося в докритических условиях.
Их можно найти экстраполяцией, если по крайней мере
три серии значений фугитивности паровой фазы оцене-
ны из соответствующего уравнения состояния при со-
ставе жидкой фазы xi, Хг. Гипотетическую фугитив-
ность жидкой фазы У1з определяют аналогичным
образом.
Для рассматриваемой смеси эталонную фугитив-
Равновесие пар — жидкость 347
ность компонента 1 можно принять равной
7? = ((У?2)Х2(У?3)Хз]^ (5)
и использовать для определения коэффициентов сле-
дующие выражения:
1п71 = 1п71(Вильсои), (6)
1П72 = 1п72(В“> +. - Х1Хз 1п($/Л), (7)
(х2 + Хз)
1п7з = 1п7з(Внльсон) - - Х1Х2 2 1п(/?2/Лз), (8)
(Х2 + Хз)
где -у,(Вильсон> — коэффициент активности, рассчитанный
по уравнению Вильсона (табл. 4.6) с применением най-
денных здесь параметров.
В качестве приложения здесь приводятся уравнения
для расчета температуры начала кипения. Исходные
условия:
= S^x, = 1. (9)
Для компонента 1
Ki = yrf/tfP, (Ю)
7i рассчитывают по уравнению (6), а 7? — по уравне-
нию (5). Для других компонентов
Ki = 7,^a,P?a,(PF)i/^P, (11)
7i определяют по уравнениям (7) и (8), а другие члены
уравнения — обычными способами.
Смеси, содержащие более одного компонента с
сверхкритическими характеристиками, рассмотрены ван
Нессом и Эбботом [708].
6.8. Задачи
6.1. Давление, соответствующее точке начала кипе-
ния смесей сероуглерода (CS2) и циклогексана при
19,8°С, равно 150,8 мм рт. ст., если xi = 0,25, или 249,5
мм рт. ст., если xi = 0,727. Уравнение Редлиха —
Квонга применяют для расчета равновесия паровой фа-
зы, а уравнение ван Лаара — для расчета равновесия
жидкой фазы. Определите состав паровой фазы и кон-
станты уравнения ван Лаара.
6.2. Эквимолярная смесь этанола и бензола при
200°С представляет собой жидкость. Если давление
смеси адиабатически понизить до 12,6 атм, образуется
некоторое количество паровой фазы. Какова будет в
этом случае температура смеси? При 179°С коэффици-
енты активности 7Г и 7Г имеют следующие значения:
1,03 и 3,30. Вириальное уравнение с корреляциями Пит-
цера и Керла для В применимо для расчета равновесия
паровой фазы. Можно предположить, что энтальпия не
зависит от давления. Теплоемкости жидких бензола и
этанола равны 0,7 и 1,4 кал/г • °C соответственно. Дру-
гие необходимые физические величины приведены в
табл. Г.2.
6.3. Эквимолярная смесь при 600° R представляет
собой наполовину жидкость, наполовину пар. Коэффи-
циенты активности можно определить из уравнения
7i = 1,5(1 - Xi)2. Вириальные коэффициенты: Вп =
= -1,8, Bn = -6,0, В22 = -3,4 куб.фут/фунт-моль.
Давление пара: Р? = 30, = 20 атм. Определите дав-
ление системы.
6.4. Имеется эквимолярная смесь ацетона (7), эта-
нола (2) и этилового эфира (3). Температура смеси
55°С. Плотность жидкостей 0,790, 0,789 и 0,713 г/мл
соответственно. Поведение паровой фазы можно при-
нять за идеальное. Коэффициенты Вильсона при нижеу-
казанных температурах имеют следующие значения:
Смесь Т, °C Л<7 Ау.
12 55 0,30771 1,20101
13 40 0,49880 0,86494
23 30 0,29207 0,77045
Определите величины давления, соответствующие тем-
пературе начала кипения и точке росы, и состав паро-
вой фазы.
6.5. Имеется жидкая смесь, состоящая на 1/3 из ме-
танола (7) и на 2/3 из этил ацетата (2). Температура сме-
си 150°С. Каково давление паров при температуре
начала кипения и каков состав пара?
Коэффициент Пойнтинга можно принять равным
единице. Давление пара: Р? = 13,85 атм, Р? = 6,65 атм.
Параметры уравнения ван Лаара: Л12 = 0,4227,
Д21 = 0,4470. Вириальные коэффициенты: Вц =
= -0,3306, В22 = -0,7569, Bn = -0,4739 л/моль.
6.6. Ниже приведены истинные точки кипения при
дистилляции смеси керосин + нефть.
Дистиллят, Т, °C
мол. %
0 5
10 38
20 69
30 100
40 130
50 156
60 182
70 212
80 224
90 282
100 320
Используя метод Жванецкого и Платонова (разд. 6.2.5),
определите 1) температуру начала кипения при 5 атм,
2) температуру, при которой 50 мол.% смеси испаряет-
ся при атмосферном давлении.
6.7. Для смеси, описанной в примере 6.3, постройте
кривую конденсации при давлении 100 фунт/кв. дюйм,
т. е. кривую зависимости /3 = V/F от температуры.
6.8. Определите растворимость сероводорода в бен-
золе при 400 К и 12 атм, используя формулу Эббота
(рис. 1.16) для определения второго вириального коэф-
фициента и уравнение (6.110) для нахождения фугитив-
ности гипотетического жидкого сероводорода и
приведенные ниже данные
5 VL Тс Рс и)
H2S 8,80 43,1 373,2 88,2 0,100
С6Н6 9,16 89,4
Давление пара, мм рт. ст.: ацетон: 230,9(25 °C),
349,2(35°С) и 1020,3(65°С); циклогексан: 97,6(25°С), то-
луол: 46,8(35°С); гептан: 253,8(65°С). Молярные объ-
емы и константы уравнения UNIQUAC R и Q при-
ведены ниже.
Размерности применяемых единиц: кал, мл, моль,
К, атм.
6.9. Постройте диаграммы х - у при помощи каж-
дого из пяти уравнений для каждой из трех указанных
ниже систем.
Параметры уравнений Вильсона, NRTL и UNIQUAC
имеют следующие значения (кал/моль):
Уравнение А 12 А 21
Вильсона (Ап - Хц) (Х12 — Х22)
NRTL (#12 ~ S11) t?21 “Sil)
UNIQUAC (“12 ~ “22) (“21 ~ “11)
Ацетон + циклогексан, 25 ° С (DECHEMA, pt. 3 and 4,
216)
Параметры уравнения А 12 А 21 ALPHA12
Маргулеса 2,0522 1,7201
Ван Лаара 2,0684 1,7174
Вильсона 1114,6929 448,6899
NRTL 693,7917 881,9712 0,4841
UNIQUAC -55,5703 587,6497
Ацетон + толуол, 35°C (DECHEMA, pt. 3 and 4, 232)
Параметры уравнения А 12 А 21 ALPHA 12
Маргулеса 0,6277 0,5020
Ван Лаара 0,6480 0,5017
Вильсона 604,4247 -199,5906
NRTL -50,4587 446,0895 0,3017
UNIQUAC -248,2777 454,3634
Ацетон + гептан, 65°С (DECHEMA, pt. 3 and 4, 239)
Параметры уравнения А 12 А 21 ALPHA12
Маргулеса 1,4370 1,4455
Ван Лаара 1,4370 1,4455
Вильсона 1092,9664 157,4597
NRTL 553,7388 545,2523 0,2911
UNIQUAC -146,9628 628,9213
Компонент V R Q
Ацетон 74,05 2,5735 2,3360
Циклогексан 108,75 4,0464 3,2400
Толуол 106,85 3,9228 2,9680
Гептан 147,47 5,1742 4,3960
6.10. Температура эквимолярной смеси пропана и
гексана соответствует ее точке росы при 20 атм. Давле-
ние смеси изоэнтропно снижается до 5 атм. Определите
конечную температуру смеси, коэффициент фазового
разделения и фазовый состав. Примите следующие до-
пущения: теплоемкость смеси не зависит от давления,
уравнение Соава применимо к обеим фазам.
6.11. Используя метод Чао — Сидера, постройте
диаграммы х—у и Т—х для смесей циклогексана и толу-
ола при 5 атм.
6.12. Используя следующие значения параметров
уравнения Вильсона (кал/моль):
этанол + бензол 1 297,90 131,47
этанол + метанол -511,39 598,44
метанол + бензол 1 620,36 153,86
определите фазовые составы и температуру смесей, об-
щие составы которых эквимолярны, давление равно 2
атм, а температура лежит посредине интервала от тем-
пературы начала кипения до точки росы. Неидеальнос-
тью паровой фазы можно пренебречь.
6.13. Смесь следующего состава: Сг - 0,10,
С3 = 0,14, изо-С4 = 0,19, н-С4 = 0,54, изо-С5 = 0,03
при 100°F (37,78°С) на 1/4 представляет собой жид-
кость. Определите ее давление и фазовый состав. Для
решения задачи можно воспользоваться номограммой,
приведенной на рис. 6.5, на которой координатное дав-
ление равно давлению системы.
6.14. Рассматривается разделение в дистилляцион-
ной колонне. На выходе из конденсатора температура
составляет 120°F, а давление на 5 фунт/кв.дюйм мень-
ше, чем давление в верху колонны. Давление в подогре-
вателе на 10 фунт/кв.дюйм больше, чем давление в
верху колонны. Определите: 1) давление в верху колон-
ны, 2) температуру на верхней тарелке, 3) температуру
в кубе. Используйте методику Чао — Сидера. Примите,
что для паров ф, = ф,.
F D В
с2 10 10 0
С3 14 13 1
изо-С4 19 10 9
н-С4 54 4 50
изо-Сз 3 0 3
Равновесие пар — жидкость 349
6.15. Термокомпрессионная дистилляционная уста-
новка позволяет получить верхний погон состава 90%
этилена + 10% пропилена; в кубовом остатке содер-
жится 10% этилена + 90% пропилена. В качестве сырья
в колонну поступает эквимолярная смесь. Давление в
верхней части колонны повышают настолько, чтобы
температура начала кипения дистиллята была на 30° F
выше, чем точка росы кубового остатка. Давление Р в
изознтропном компрессоре 20 атм, а в кубе — на 1 атм
выше. Давление на выходе из компрессора необходимо
рассчитать. Энтальпию смеси можно считать величи-
ной аддитивной; необходимые для расчета данные мож-
но взять, например, из книги Старлинга [127].
Определите 1) давление на выходе из компрессора; 2)
температуру в кубе и на выходе из него; 3) соотношение
между давлением на выходе из компрессора и степенью
конденсации в кубе, если флегмовое число в верху ко-
лонны равно 2:1. Предпочтительна полная кон-
денсация.
6.16. Коэффициент распределения компонентов сме-
си представлен уравнением следующего вида:
InXjP = А, - Bi/T (где Т дана в кельвинах). Ниже ука-
заны состав смеси и величины коэффициентов.
Z А В
СбН14 0,1 11,15 3,811
СбНб 0,3 11,00 3,885
с7н16 0,4 10,85 4,030
С8Н18 0,2 11,22 4,471
Определите температуру начала кипения и точку росы
при давлении 3 атм, а также фазовый состав смеси при
температуре, равноотстоящей от этих двух температур.
б. 7 7. Коэффициенты распределения и значения эн-
тальпий в уравнении, полученном для давлений 120 и
264,7 фунт/кв. дюйм, даны в работе [353]. Значения ко-
эффициентов распределения даны относительно изо-
пентана.
а' ~ Ki/Ku3o-Cs'
Основная формула справедлива для изопентана. Рас-
сматривается эквимолярная смесь четырех веществ.
1. Определите температуру начала кипения и точку ро-
сы при давлении 264,7 фунт/кв.дюйм.
2. Определите температуру, распределение фаз и фазо-
вый состав после снижения давления насыщенной жид-
кости, осуществляемого адиабатически от 264,7
фунт/кв.дюйм (18,63 кг/см2) до 120 фунт/кв. дюйм
(8,437 кг/см2).
3. Определите все параметры на выходе, получаемые
после изоэнтропного снижения давления насыщенного
пара от 264,7 до 120 фунт/кв.дюйм.
кизо-с5 = 0,37088 - 0,55786(77100) + 0,44841(77100)2 -
- 0,03704(77100)3 и
а, = ап + </2.(77100) + а3/(77100)2,
где Т выражена в градусах Фаренгейта.
Компонент «2 аз
120 фунт/кв.дюйм Сз Н8 11,06095 «зо-С4 4,69290 н-С4 3,07033 H-Cs 0,73827 -5,20067 -1,82431 -0,83565 0,05246 0,92489 0,31755 0,12144 0,00189
264,7 фунт/кв.дюйм С3Н8 3,64797 изо-Сл 1,46077 н-С4 1,00000 н-С5 0,17185 -1,06605 -0,18097 0,00000 0,22019 0,16197 0,02721 0,00000 -0,02826
hi = C2i + 01/(77100) + С3/(77100)2, Hi = еи + 62/(77100) + е3/(77100)2, где Т выражена в градусах Фаренгейта.
Компонент су с2 с3
120 фунт/кв.дюйм С3Н8 0,00000 изо-С4 0,00000 н-С4 0,00000 н-С5 0,00000 2 521,92 175,417 3 345,00 150,000 2 960,00 400,000 3 845,00 250,000
264,7 фунт/кв.дюйм
С3Н8 4 425,16 2 968,64 48,868
изо-С4 5 320,76 3 525,03 90,076
н-С4 5 553,24 3 641,27 90,076
h-Cs 6 663,21 4 161,81 160,848
Компонент е1 е2 е3
120 фунт/кв.дюйм
С3Н8 7 488,65 1 750,51 79,273
изо-С4 9 592,01 1 843,56 221,433
н-С4 8 002,89 4 382,70 -401,587
h-Cs 12 004,88 3 168,66 67,456
264,7 фунт/кв.дюйм
С3Н8 11 806,71 1 797,38 112,6239
U3O-C4 13 726,02 2 261,20 167,3186
Н-С4 14 851,06 2 312,00 155,9965
н-Сз 18 757,52 2 903,45 233,0578
6.23. Даны значения коэффициентов распределения
и давление паров пропана и изобутана для некоторых
условий.
Р, фунт/кв .дюйм Т°, F Пропан Изобутан
100 150 100 150
150 1,2* 2,05 0,52* 0,94
200 1,0 1,55* 0,42 0,72*
psat 186,7 333,5 71,8 139,7
Коэффициенты Антуана (мм рт.ст., К) имеют следую-
щие значения:
6.18. Смеси циклогексана и .м-ксилола при 25 ° С хо-
рошо описываются уравнением Gn/BT = 0,48X1X2. Дав-
ление паров Pi — 97,6 мм рт.ст., Рг = 8,3 мм рт.ст.
Постройте диаграммы х — у и Р — х смеси (DECHE-
MA, 1/6а, 314).
6.19. Смеси ацетона и гексана при 760 мм рт.ст.
можно описать уравнением Вильсона с Хп = 1156,5822
кал/моль и Х21 = 262,7197 кал/моль. Давление паров
(мм рт.ст.) компонентов можно рассчитать по урав-
нениям:
log Pi = 7,11714 - 1210,595/(229,664 + Т),
log Я = 6,91058 - 1189,640/(226,280 + Т),
где Т выражено в градусах Цельсия.
Молярные объемы составляют соответственно 73,52 и
130,77 мл/моль. Постройте кривые зависимости х — у
и Т — х (DECHEMA, I, 3 and 4, 226).
6.20. Коэффициенты активности при заданной тем-
пературе определяются уравнениями 1п71 = Axl и
1пуг = Вх\. Давление паров компонентов составляет со-
ответственно Р1 = 1,2и/>2=1. Постройте диаграммы
х —у, Р — х и Р — у для: 1) А = В = 1,5 и 2) А = 2,
В = 1. Объясните, почему в последнем случае, которому
соответствует изоэнтропный состав смеси, давление в
системе максимальное. Подчиняются ли условия, ука-
занные в п.2, уравнению Гиббса — Дюгема при посто-
янных Т и Р?
6.21. Проверьте соответствие данных по раствори-
мости (табл. 6.3) правилу Скэтчарда — Гильдебранда,
уравнения (6.109) и (6.113).
6.22. Бинарное парожидкостное равновесие может
быть описано любым из двух однопараметрических от-
ношений: либо через относительную летучесть
у - <хх/[1 + (а - 1)х], либо с помощью симметричных
уравнений Маргулеса коэффициентов активности со сле-
дующим результирующим уравнением:
P?XiexpA(l - xi)2 + Рг(1 - хЦехр х? = Р.
Постройте графики функции аР%/Р% = /(xi) при значе-
ниях А от 0 до 5. Определите, в каком диапазоне кон-
центраций значение постоянной величины относитель-
ной летучести не соответствует постоянным значениям
А.
А В С
Пропан 15,726 1 872,46 -23,16
Изобутан 15,5381 2 032,73 -33,15
1. Используя два набора помеченных звездочками зна-
чений Ki, определите константы уравнения
InKiP = ки + кц\пР?х.
2. Оцените значение К, для не отмеченных звездочками
позиций по результирующей формуле и сравните их с
заданными значениями.
3. Определите температуру начала кипения эквимоляр-
ной смеси при 160 фунт/кв.дюйм.
6.24. Для смеси, описанной в задаче 6.7, определите
температуру кипения при 20 атм и значении Xi =0,5.
6.25. Эквимолярная бинарная смесь испаряется до
получения 50 мол. % паров. Каков состав фаз?
Данные:
Gn/RT = 1,8x1x2,
1п0, = 0,5 - 0,15yi,
02 = 0,8,
<t>iPi/P = 0,4,
^Pi/P = 1,2.
Обратите внимание, что необходим следующий шаг: не-
обходимо по уравнению Гиббса — Дюгема найти урав-
нение ДЛЯ 02.
6.26. Вследствие поверхностного натяжения давле-
ние внутри капли жидкости выше, чем вне ее. Точное
соотношение такое: Рвнут - Рвн = 4o/d, где а — по-
верхностное натяжение и d — диаметр капли. Как
следствие, относительная летучесть двух веществ зави-
сит от диаметра капли. Определите это влияние для
смеси метанол(1) + вода(2) на основе следующих исход-
ных данных: Т = 25°С, Xi = 0,3973. Исследуйте влия-
Равновесие пар —жидкость 351
ние изменения d вплоть до 1 А.
Vi = 1,186, 72 = 1,100 [68];
PiaI = 127,2 мм рт.ст., Иа‘ = 22,9 мм рт.ст.,
а = 34,9(Е - 3)Н/м (CRC Handbook, F44,60th ed.);
= Х1Хл[-4041 - 428(xi - хг) + 644(xi - X2)2 +
+ 990(xi - хг)3((мм3/моль)
(Int. Data Series, Series B, 4, 1978].
6.27. Исходя из уравнения состояния, применимого
к обеим фазам, коэффициент распределения задается
уравнением К, = ф^/ф^у. В таких случаях невозможно
начинать расчет условий испарения, принимая фугитив-
ности, равными единице, как в блоке 2 блок-схемы,
представленной на рис. 6.11. Тем не менее действитель-
ные значения К, можно взять из рис. 6.5 или из других
номограмм Американского нефтяного института, или
можно использовать идеальные значения К, = X?at /Р.
Попытайтесь модифицировать рис. 6.11 для таких
случаев.
7.1. Жидкие смеси
Жидкости, не относящиеся к одному гомологическо-
му ряду, как правило, имеют ограниченную взаимную
растворимость. Подобное явление — показатель неиде-
альности, поэтому, рассматривая вопросы равновесия
между жидкими фазами, необходимо руководствовать-
ся теми же термодинамическими приемами, что и при
изучении равновесия в системах жидкость — пар, по-
скольку оба указанных случая соответствуют условиям
минимума энергии Гиббса. Термодинамического разли-
чия между этими двумя типами равновесия не суще-
ствует, но некоторые фактические различия, несомнен-
но, имеются.
1. Равновесия жидкость — жидкость определяются
экспериментально обычно намного легче, особенно при
температурах близких к комнатной, т. е. при условиях,
представляющих наибольший интерес. Однако данные
экспериментов зачастую получены при решении кон-
кретных задач, и их количество и диапазон исследова-
ний недостаточны для получения необходимых корре-
ляций.
2. Уравнения состояния жидкостей, как правило, не-
пригодны для расчета фугитивности и активности.
3. Влияние температуры на равновесие системы жид-
кость — жидкость выражено наиболее заметно. Равно-
весия пар — жидкость определяются при температурах
кипения, которые обычно отличаются от комнатных
температур, т. е. от тех условий, при которых обычно
принято описывать равновесия жидкость — жидкость.
Интерес представляют три основных аспекта равно-
весий жидкость — жидкость:
1. Как получить необходимые данные исходя из
свойств отдельных компонентов или пар компонентов?
2. Как скоррелировать ограниченные данные, чтобы
их можно было интерполировать или экстраполировать
или комбинировать для описания поведения многоком-
понентных систем?
3. Как получить экспериментальные данные?
Первым двум проблемам и посвящена в основном
данная глава. Они также подробно рассмотрены в
серии обзорных статей Соренсена и др. [125]. В пер-
вых статьях этих авторов обсуждены публикуемые
DECHEMA данные о равновесии в системах жидкость
— жидкость (DECHEMA Liquid-Liquid Equillibrium Da-
ta Collection, 1979, 3 тома), в которые включены дан-
ные о 800—900 двух-, трех- и нескольких четырехком-
понентных системах.
Экспериментальные методы, используемые для реше-
ния данной проблемы, относятся к числу наиболее
простых. В первую очередь используют методы двух
следующих типов.
1. Пределы растворимости определяют при постепен-
ном добавлении известных количеств вещества к рас-
твору с фиксированным составом до тех пор, пока не
появятся первые следы помутнения или какие-либо дру-
гие признаки насыщения.
2. Равновесные составы находят следующим обра-
зом: вначале тщательно перемешивают смесь известно-
го общего состава, далее разделяют фазы, отбирают
пробы и анализируют их подходящими методами. Для
более точных исследований подбирают сложные мето-
дики; детальные описания таких методик приводятся в
статьях, публикуемых в The Journal of Chemical
Termodynamics либо в подобных научных изданиях.
7.2. Фазовые диаграммы
Примеры фазовых диаграмм двух- и трехкомпонент-
ных смесей приведены в гл. 5 (рис. 5.20 и 5.30). Обычно
на таких диаграммах переменными являются только
температура и состав, так как влияние давления на кон-
денсирующиеся фазы невелико. На рис. 5.28 показано
влияние давления на изменение растворимости на при-
мере системы твердая фаза — жидкость, однако подоб-
ное можно ожидать и для систем жидкость —
жидкость.
Анализ порядка 900 данных о бинарных системах,
опубликованных в DECHEMA Collection, показывает,
что свыше 41% систем имеют верхнюю критическую
температуру растворения, в то время как 53% систем
не имеют ни верхней, ни нижней критической темпера-
туры растворения. В некоторых случаях, наличие ниж-
ней критической температуры растворения обусловлено
ассоциацией молекул и дипольным взаимодействием,
которое усиливается с понижением температуры. Семь-
десят пять процентов тройных систем из числа рас-
смотренных имеют по одной одно- и двухфазной
области, как на рис. 5.36,а а 20% систем имеют диа-
граммы с двумя однофазными и одной двухфазной об-
ластями, как на рис. 5.36,6. Важное прикладное
значение имеют диаграммы других типов, хотя они не
распространены; к числу таких смесей относятся:
вода + 2-бутанол + 2-бутанон (рис. 5.36,в). Несколько
тройных систем были изучены в достаточно широком
Равновесие жидкость — жидкость 353
интервале температур для выявления верхней критиче-
ской температуры растворения (см. рис. 5.35). Почти
в 60% описанных двухкомпонентных систем и в 80%
трехкомпонентных систем присутствует вода как один
из компонентов, это характеризует важность воды как
растворителя, ее доступность в этом качестве и просто-
ту экспериментальных работ с водными системами.
Некоторые системы классификаций диаграмм трой-
ных систем показаны на рис. 5.5,г. Кривые распределе-
ния по составу в системах жидкость — жидкость могут
быть нескольких определенных видов подобно приве-
денным на рис. 5.29 и 5.32 для систем жидкость — пар.
В ряде случаев более удобны зависимости другого вида,
ряд таких зависимостей описан, например, Трейболом
[135]. Разработанная Мепстоном [463] схема должна
привлечь внимание тех, кого интересуют как вопросы
интерполяции соединительных линий, так и номограм-
мы. Бинодальные кривые тройных систем, имеющих
одну частично смешивающуюся пару, были описаны в
виде особых эмпирических уравнений с тремя и более
константами [351]. Разработанные еще в 30—40-х гг. но
все еще популярные корреляционные методы Хенда
(1930) и Отмера и Тобиаша (1943) были проверены на
ПО системах [214]. Проверка показала, что эти методы
не соответствуют принятым стандартам. Метод Хенда
использован в задаче 7.13. Классические и современней-
шие методы расчета или прогноза равновесия в систе-
мах жидкость — жидкость с учетом коэффициентов
активности будут рассмотрены в этой главе.
7.3. Смешиваемость и термодинамическая
стабильность
Для бинарных смесей присущи разнообразные виды
зависимости давления паров от состава; некоторые из
них показаны на рис. 7.1. Изотерма Т\ на этом рисунке
проходит маркированную звездочками область, в кото-
рой в равновесии сосуществуют две жидкие фазы. Со-
стояние равновесия характеризуется наличием мини-
мума энергии Гиббса при данных Т, Р и составе. По-
этому на рис. 7.2 точке d соответствует менее стабиль-
ная смесь, чем смеси, отвечающие точкам е или /, кото-
рые ближе к условиям равновесия. На штриховой
кривой от а до h имеются точки, отвечающие значени-
ям энергии Гиббса, меньшим чем G/, но не для каждого
из этих случаев состав соответствует заданному. Пере-
секающая рис. 7.2 пунктирная кривая GikGi характери-
зует нестабильные состояния двух не смешивающихся
жидкостей, равновесное состояние которых соответ-
ствует прямой GimG\. Кривая GinGi относится к пол-
ностью смешивающейся смеси, тогда как линия GzmGi
представляет энергию Гиббса этой смеси до того, как
произошло полное смешение компонентов.
Как будет объяснено далее, отрезок adh промежуточ-
ной кривой показывает нестабильное состояние, а соот-
ветствующее равновесное состояние демонстрирует
линия я/7г. Такие кривые всегда имеют по крайней мере
один локальный максимум либо два и более локальных
минимума, их принято называть кривыми с выпуклыми
участками. Математически это условие описывается
так: dG/dx = 0,
d2G/dx2 < 0.
Если композиции смеси, удовлетворяющие этим ус-
ловиям, возможны, может наблюдаться полная несме-
шиваемость компонентов.
Составы смесей, для которых вторая производная
равна нулю, отвечают точкам перегиба — с и g на
рис. 7.2. Участки а — с и h — g, крайние точки кото-
рых и есть точки перегиба, соответствуют метаста-
бильному состоянию. Анализ подобных кривых
проведен в работах [78, 86]. Участок с — d — g соот-
ветствует действительно нестабильному состоянию.
Локальные экстремумы, обнаруженные при приведении
первой производной к нулю, — это точки b, d и j. Со-
став фаз, находящихся в равновесии, определяется сле-
дующим образом: координаты пересечения касательной
общей для двух точек кривой с осями координат — это
парциальные молярные характеристики Gi и G2 (объяс-
нения см. в разд. 3.3).
Из следующих соотношений:
G= Ex,G, = Gex + Gideal (7.1)
= RT E Xj ln(y(xz) = RT E Xj In a,
= RT Zx^nifi/ff), (7.2)
отвечающих данной системе, становится ясно, что две
точки такой касательной имеют ту же самую величину
парциальной молярной энергии Гиббса и ту же самую
парциальную фугитивность, и, следовательно, они на-
ходятся в равновесии друг с другом. В действительнос-
ти вся гамма смесей с составом от ха до х* имеет
одинаковую парциальную фугитивность и отличается
друг от друга только соотношением двух фаз, имею-
щих такие составы. Точки перегиба, локальные мини-
мумы и точки касательной, общей для двух точек
кривой, исследованы в примере 7.1. В задаче 7.26 гово-
рится о спинодальной кривой, которая является локу-
сом предельных метастабильных составов или точек
перегиба; см. также пример 7.14.
Иногда возможны такие ситуации, когда к кривой
энергии Гиббса бинарной смеси можно провести более
чем одну касательную. Именно такой случай разобран
в примере 7.2. Касательная с тремя точками касания
отвечает равновесию трех фаз; специально подобранная
такого рода система показана в примере 7.25.
Кривые изменения энергии Гиббса смешения, полу-
ченные с использованием ряда уравнений коэффициен-
тов активности и несколько значений их параметров,
представлены в примере 7.3.
Возможен и другой подход к рассмотрению разделе-
ния фаз, основанный на изучении характера изменения
химических потенциалов или парциальных молярных
значений энергии Гиббса, наглядно представленный на
рис. 7.3 и 7.4. Внутри области равновесных составов —
Xi и х* на рис. 7.3 — величина химического потенциала
постоянна и равна значению щ, так как график зависи-
мости химического потенциала от Xi представляет со-
бой горизонталь во всем интервале значений хь Если
область несмешиваемости постепенно уменьшается
из-за изменения температуры или другого фактора, в
конце концов она вырождается в точку, как это показа-
но на кривой 2 рис. 7.4. Эта точка зарождения несме-
шиваемости или критического раствора, если ее
рассматривать в свете математических представлений,
4-864
Состав, мол. доли
Рис. 7.1. Давление паров бинарной смеси. При температуре Т\ прове-
денной штриховой линией участок кривой ABCDE отвечает нестабиль-
ному состоянию; в этом интервале составов присутствуют две жидкие
фазы состава ха и хе, давление паров постоянно вдоль горизонтальной
линии АСЕ. Две жидкие фазы существуют везде в пределах куполоо-
бразной зоны (пунктирная линия). Точка F на изотерме Тг — это точка
появления несмешиваемости; температура Тг называется критической
температурой растворения. Штриховые прямые линии соответствуют
значениям, рассчитанным по закону Рауля.
является точкой перегиба, которая характеризуется д2в d3G
уравнением. = 7 = 0. дх{ dxf Поскольку, согласно уравнению (2.54), (7.3) дх2 или d2g dxj = УТ = 0’ (7-5) ОХ] d3g (7-6)
dG Pl— G + (l - *1)~, (7.4) эквивалентные условия для зарождения растворимости определяются как Такие условия применяются в примере 7.4 для нахожде- ния параметрического диапазона, в котором бинарные смеси полностью растворимы. Эти рассуждения наводят на мысль о том, что вза- имную растворимость двух жидкостей можно устано-
Равновесие жидкость — жидкость 355
Рис. 7.2. Энергия Гиббса бинарной смеси как функция состава. Участок
кривой adh представляет область нестабильности, стабильные значения
энергии Гиббса показаны прямой afh; а и h — точки касательной, об-
щей для двух точек кривой, определяющие состав фаз в равновесных
условиях, b и j — локальные минимумы энергии Гиббса, с и g — точки
перегиба. Области ас и gh метастабильные.
вить путем построения графиков зависимости энергии
Гиббса от х (Gi — х) или энергии смешения Гиббса
от х (Sx/G, — х) с последующим определением точек
касательной, общей для двух точек кривой. Однако по-
следнее осуществимо только для бинарных смесей, в то
время как применение принципа равенства парциальных
фугитивностей совершенно неограниченно. Для бинар-
ной смеси
fi=f* (7.7)
где звездочкой помечена вторая фаза. Применив урав-
нение (4.57) и сократив общую переменную J?, по-
лучаем
(7.8)
У2(1 -*1)= У1(1 -**)•
(7.9)
Если зависимость между составом и коэффициентами
активности известна, эти уравнения можно разрешить
относительно равновесных составов. Возьмем в каче-
стве примера симметричное уравнение:
Сех//?Т = Лх1(1 -xj,
из которого получаем
In У! = А(1 -Х02,
In у* = Л(1 -xf)2,
In у 2 = Axj,
In у* = Ах*2.
Вначале исследуем условия сходимости.
g = (G- ExtG,)/RT= Gex/RT+ Ex,
Берем производные:
/ х< \
= А(1 - 2xi) + In (------- I,
dx 1 у 1 - х j у
d2g 1 ?
(7.Ю)
(7.11)
(7.12)
(7.13)
(7.14)
Допустим, что
flnX;. (7.15)
(7.16)
(7.17)
4:
Ясно, что неравенство наблюдается при всех значениях
Xi, если А 2.
На рис. 7.5 показана идентификация двух- и однофаз-
ных областей, проведенная в соответствии с этими па-
раметрами. Приведенные на рис. 7.6 кривые изменения
энергии Гиббса указывают на то, что касательная, об-
щая для двух точек кривой, существует при А > 2. По-
скольку эти кривые симметричны, точки касания такой
касательной совпадают с локальными минимумами, и,
следовательно, эти точки находят, решая уравнение
Л(1 - 2*0 +In [^/(l= 0. (7.19)
К такому же результату приводит подстановка уравне-
ний (7.8) и (7.9) в уравнения (7.11)—(7.14):
Х]ехр[Л(1 — Xi)2] = х*ехр[Л(1 — х*)2], (7.20)
(1 — Xj)exp(^xf) = (1 — х*)ехр(Лх*2). (7-21)
Симметрия этих уравнений свидетельствует о том,
что
x^l-xf, (7.22)
так что уравнение (7.20) можно записать следующим
образом:
Х1вхр[Л(1 ~*1)2] = (1 — xt)exp(Axj), (7.23)
прологарифмировав это выражение, можно получить
уравнение (7.19).
Специфика касательной, общей для двух точек кри-
вой, также может быть использована для расчета рав-
новесного состава. Если уравнение симметрично, точки
касания представляют собой локальные минимумы, но
в других случаях необходимо решить систему из двух
уравнений
g ~ g* dg
------» = — (7.24)
Xj — х’ dXj
где g* — функция g, записанная через xj в качестве пере-
менной. Если для расчетов необходимо лишь аппрокси-
мированное значение равновесного состава, касатель-
ную, общую для двух точек кривой, удобнее всего опре-
делить графически.
В большинстве случаев числовое решение легче ис-
кать исходя из равенства фугитивностей (см. уравнение
(7.7)], т. е. по методике, на которой акцентируется вни-
мание в этой главе. Уравнения (7.24) и (7.25) применя-
ются в примере 7.9, они необходимы и для решения
задачи 7.24.
Рис. 7.3. Кривые энергии Гиббса смешения и соответствующих значе-
ний химического потенциала. Внутри области составов, соответствую-
щих равновесию жидкость — жидкость, последний имеет постоянное
значение. Для расчетов использовано уравнение Маргулеса с А= 2 и
В=3.
Равновесие жидкость — жидкость 357
Рис. 7.4. Кривые химических потенциалов, полученные по уравнению
Маргулеса (А =2, значения В указаны на рисунке). По мере уменьшения
В диапазон составов, для которых /о является константой, сужается
и при В=2 это просто точка перегиба, в которой
dm/dxi = d2m/dxl = 0.
Рис. 7.5. Пределы смешиваемости бинарных смесей, установленные по
симметричному уравнению избыточной энергии Гиббса. Указаны пре-
делы смешиваемости и значения величины А.
Рис. 7.6. Значения энергии Гиббса смешения при нескольких значениях
параметра А (указаны на рисунке) симметричного уравнения для избы-
точной энергии Гиббса G*X/RT = ЛХ1Х2. Раздел фаз возможен только
при А > 2.
Пример 7.1. Изучение кривой зависимости энергии Гиббса бинарной смеси
Бинарная смесь описывается уравнением ван Лаара
G'X/RT = AxiX2/(Axi/B + х2), (1)
где А = 3 и В = 2 для данного случая. Далее будут по-
строены следующие графики:
1) зависимость энергии Гиббса G/RT = Gex/RT +
+ xilnxi + х21пх2 от xr, 2) зависимость производной
d(G/RT)/dxi от х\.
Кроме того, количественно будут определены 3) рав-
новесные составы двух жидких фаз; 4) локальные мини-
мумы энергии Гиббса двух жидких фаз; 5) точки
перегиба на кривой зависимости энергии Гиббса.
Энергию Гиббса можно определить при помощи
уравнения
G/RT = A/D + xilnxi + х21пх2, (2)
где
D = А/Вх2 + 1/х2. (3)
Допуская, что
получают первую и вторую производные:
3(G//?T)=_^+ln(Xi/X2)> (5)
dxi D
и
d2{G/RT) л Г 1 f 2А 2лИ 11 ...
dxi |_£>2 \Вх% D3 J Xi х2
Локальные минимумы определяют, приравнивая пер-
вую производную нулю, а точки перегиба находят, при-
равнивая вторую производную нулю. Состав фаз в
условиях равновесия находят либо решением уравнений
(7.24), (7.25), либо анализом графика (рис. 7.7), либо по
точкам касательной, общей для двух точек кривой, на
графике зависимости энергии Гиббса. Точки перегиба
кривой зависимости энергии Гиббса — это экстремумы
на кривой функции производной. Приведем краткое
описание различных композиций:
пределы растворимости (хх, х*) = (0,080, 0,755);
локальные минимумы при Xi = 0,11185 и 0,8071;
точки перегиба при Xi = 0,17178 и 0,5913.
Подобную информацию можно получить, используя
симметричное уравнение ван Лаара при А = В = 2,5.
Энергию Гиббса в этом случае можно определить по
уравнению
G/RT = 2, 5xix2 + xilnxi + х21пх2 (7)
и его производным
= 2,5(1 - 2x0 + ln(xi/x2), (8)
0X1
^G/PT) = _2A+± + ±
dxi *1 х2
Пределы растворимости и локальные минимумы оди-
наковы, а именно (xi, Xi) = (0,1448, 0,8552). Точки пере-
гиба определяются из уравнения
xj = 0,5 [1 ± <(1 - 2/Л) ] = 0,2764; 0,7236.
Эти числовые варианты могут быть определены на
графиках.
Равновесие жидкость — жидкость 359
или
Кривые энергии Гиббса смеси, полученные по уравне-
нию ван Лаара при А = 3 и В = 2.
Кривые энергии Гиббса смеси, полученные по симмет-
ричному уравнению ван Лаара при А - В = 2,5.
Пример 7.2. Множество касательных, общих для двух точек кривой
Для определения состава равновесных фаз необходи-
мо знать условия существования такой касательной, но
этого не всегда достаточно, так как кривые зависимос-
ти энергии Гиббса могут иметь несколько подобных ка-
сательных. Например, на рисунке данного примера
имеется шесть касательных, общих для двух точек кри-
вой, но при определенном составе лишь одна из них
может соответствовать минимуму энергии Гиббса для
смеси.
При использовании обычных уравнений избыточной
энергии Гиббса графики с более чем двумя минимума-
ми встречаются нечасто, однако подобные варианты
наблюдаются, согласно данным Соренсона [124], для
модели NRTL при экстремальной нерастворимости.
Правда, Соренсон не приводит конкретных примеров.
Такого рода ситуация смоделирована и здесь. Пред-
положим, что
g = (Gex + G,d)/RT = xxxiftxi) + xjnxi + x2lnx2, (1)
запишем соответствующую производную:
A. = X1(1 _ X1) + (1 _ ZxO/CxO + In . (2)
uX[ 1
На рисунке кривая б — это график зависимости
= In , X1- + 5000(xi - 0,1) (xi - 0,3) (xi - 0,5) • (3)
axi 1 — Xi
• (xi - 0,7) (xi - 0,9) = 0.
Это уравнение имеет пять корней, отвечающих пяти
позициям минимакса на кривой g = /(xi). В данном
случае функцию /(xi) находят решением линейного диф-
ференциального уравнения первого порядка
*1(1 - xi) + (i - 2X1)/= 5000(xi - 0,1) (xi - 0,3) • (4)
• (Xi - 0,5) (xi - 0,7) (xi - 0,9).
Аналитическое решение этого уравнения приводится в
задаче 7.22. Такое решение используется для построе-
ния графика зависимости уравнения (1) (см. рисунок а).
Очевидно, что нулевые значения зависимости производ-
ной уравнения (3) на рисунке б соответствуют экстре-
мумам на графике зависимости, которую описывает
уравнение (1).
Касательная, общая для трех точек кривой, соответ-
ствующая равновесию трех фаз, определяется в задаче
7.25 путем замены числа 5000 в уравнении (4) на число
4150.
Кривая энергии Гиббса с шестью касательными, об-
щими для двух точек кривой. Линия б соответствует
минимальным значениям g в данной области xi.
Равновесие жидкость — жидкость 361
Кривые производных энергии Гиббса, показывающие
положение нулевых значений и, следовательно, экстре-
мумов g = G/RT.
Пример 7.3. Энергия смешения Гиббса, представленная различными уравнениями для определения избыточной
энергии Гиббса
Смесь определена коэффициентами активности ком-
понентов при бесконечном разбавлении
1п7Г = 2, G - Sx,G,p
1п7Г = 3, 8 RT
G"
RT
+ Sxjnxi .
Формулы для определения избыточной энергии Гибб-
са представлены в табл. 4.2, а формулы для определе-
ния параметров уравнений на основе коэффициентов
активности при бесконечном разбавлении — в табл.
4.5. Ниже приведены величины параметров уравнений
Маргулеса и ван Лаара: Л12 = 2, A2i = 3,
Вильсона: А\2 = 0,3346, Л21 = 0,0968,
Цубоки — Катаямы — Вильсона: К2/Г1 = 3, А\2 = 0,459,
A2i = 0,2106,
NRTL: ап = 0,2, т12 = 2,5805, т21 = 0,4598,
NRTL: ai2 = -1, Т12 = 0,5881, т21 = 0,9411,
Равновесные составы, определенные по точкам двой-
ного касания с использованием уравнений Маргулеса,
ван Лаара, NRTL, Цубоки — Катаямы — Вильсона и
Вильсона, имеют следующие приблизительные со-
ставы:
уравнение Маргулеса: (0,20, 0,93),
уравнение ван Лаара: (0,23 , 0,92),
уравнение NRTL 0,2: (0,26, 0,91),
NRTL -1: (0,55, 0,87),
уравнение Цубоки — Катаямы — Вильсона: (0,38, 0,87),
уравнение Вильсона: (компоненты смешиваются пол-
ностью). Эти данные хорошо согласуются с установ-
ленными по рис. 7.7—7.12, но между собой
согласуются плохо.
Уравнение NRTL с ац2 = — 1 имеет три набора пара-
метров, соответствующих коэффициентам активности
при бесконечном разбавлении. В добавление к получен-
ным ранее наборам параметров известны еще такие,
как (3,0000, —58,26) и (—11,78, 2,0000). Однако только
из значений (0,5881, 0,9411) можно прийти к выводу о
разделении фаз. Графики энергии смешения Гиббса по-
казаны для всех трех случаев.
Кривые энергии Гиббса смешения, рассчитанные
по различным уравнениям для избытка энергии
Гиббса. Использованные уравнения: 7 — Маргулеса,
2 — ван Лаара, 3 — NRTL с ап = 0,2, 4 — Цубоки —
Катаямы — Вильсона с И2/И1 = 3, 5 — Вильсона.
Кривые энергии Гиббса смешения, полученные по урав-
нению NRTL при двух различных величинах параметра
«12-
Кривые энергии Гиббса смешения, полученные по уравнению NRTL при
ап = - 1. Этому уравнению удовлетворяют три следующие серии величин пара-
метров: 7 — (3000, - 58,26), 2 — ( - 11,78; 2,000), 3 — (0,5881, 0,9411). Только
кривая 3 показывает разделение фаз.
(G-E x-G:)^ExL bn(y-X:)
G It v / G G
Кривые энергии Гиббса смешения бинарных смесей, коэффициенты активности
для которых получены по уравнению ван Лаара с различными величинами пара-
метров. Величины параметров и пределы смешиваемости показаны в таблице;
кривые для двух последних в списке композиций обладают минимумами, по-
скольку в этом случае разделения фаз не происходит.
Кривые энергии Гиббса смешения, полученные по урав-
нению Цубоки — Катаямы — Вильсона. Графики по-
строены для К1/Г2 = 3. Величины параметров
Ап = Л21 указаны на рисунке.
ттттттт
Кривые энергии Гиббса смешения, полученные по урав-
нению Цубоки — Катаямы — Вильсона Л12 = Л21 = 0,05.
Значения V\/Vz указаны на рисунке.
Равновесие жидкость — жидкость 365
Кривые энергии Гиббса смешения, полученные по урав-
нению Цубоки — Катаямы — Вильсона, которое мож-
но преобразовать в уравнение Вильсона, поскольку в
данном случае И1/И2 = 1. Величины параметров
Ли = Л21 указаны на рисунке.
Влияние объемного отношения V\/ V2 на форму кривых
энергии Гиббса смешения, полученных по уравнению
Цубоки — Катаямы — Вильсона 1П7“ = 3 и ln7“ = 4.
Влияние величины параметра сш уравнения NRTL на
форму кривых энергии Гиббса смешения, 1пу” = 3 и
1пу” = 4.
Пример 7.4. Границы взаимной растворимости, определяемые по уравнениям ван Лаара и Маргулеса
Пределы взаимной растворимости соответствуют точкам перегиба на кривой щ = f(xt). Математически эти точки определяются так: В = - 9xt-t-10xi. j . (6) 6X1(1 - Xi)2
_ д _ л dxi дх$ 1 ’ Производные уравнения ван Лаара (табл. 7.2) записы- ваются так:
или Й = й = 0 • (2) dxi dxi Э2^ _ -2А2В2 l1/ _п гп Их, + вхг)1 °’ , М^(Л - В) _ 1/;rf + ,/xj = 0
Для уравнения Маргулеса (табл. 7.2) эти производные Эх? (ЛХ1 + Вх2)4
таковы: д2е = 2(3xi - 2) А + 2(1 - 3xi) В + 1/Х1Х2 = 0, (3) ан Коган [68] решает эти уравнения следующим образом: А = 13,5(1 ~ Х1>— , (9) (2 - xi)2 (1 + Xi)
^4 = 6А - 6В - 1/х? + 1/х1 = 0 . (4) ЙХ1 В = 13,5x1 . (Ю) (2 - xi) (1 + xi)2
Данная система двух уравнений легко решается мето- дом детерминантов: Полученные значения А и В больше, чем соответствую-
А = ~9х* 1 , (5) 6X1(1 - Xi)2 щие областям полной взаимной растворимости. Эти результаты показаны на рис. 7.7 и 7.8.
Равновесие жидкость — жидкость 367
7.4. Бинарные смеси
Выпуклость. Кривые большинства основных зависи-
мостей коэффициентов активности могут иметь выпук-
лые отрезки при некоторых значениях составов и
параметров и, следовательно, могут представлять рав-
новесия систем жидкость — жидкость. Математически
условие выпуклости
d2g/dX2! < 0 (7.26)
требует того, чтобы по крайней мере некоторые из чле-
нов второй производной g = Ga/RT + Sx,lnxi имели
отрицательные значения. Уравнения таких вторых
производных ряда общих зависимостей собраны в
табл. 7.1. Ясно, что все эти уравнения, исключая урав-
нение Вильсона, содержат положительные и отрица-
тельные члены и, следовательно, могут удовлетворить
уравнению (7.26). Уравнение UNIQUAC не включено в
эту таблицу, но оно также может представлять равно-
весие системы жидкость — жидкость. Для уравнения
Маргулеса, например, при xi = 0,5 условия таковы:
-(А + В) + 4 < 0, (7.27)
что, бесспорно, справедливо, если сумма коэффициен-
тов больше четырех. Для симметричного уравнения
этот вывод не противоречит условию (7.18).
На практике результаты определения равновесия
жидкость — жидкость, проведенного с использованием
нескольких зависимостей коэффициентов активности,
могут до некоторой степени различаться, как это пока-
зано в примере 7.7.
7.4.1. Определение параметров. Поскольку все основ-
ные корреляции для коэффициентов активности дают
непосредственно ln?j, очень удобно пользоваться урав-
нениями (7.8) и (7.9), выраженными следующим
образом:
Р = Iny! - InIn(xj7xj) - 0, (7.28)
<7 = lny2-lny?-ln[(l - х?)/(1 - Xi)] — 0. (7.29
Если коэффициенты активности известны как функции
состава, эти уравнения могут быть разрешены относи-
тельно состава равновесных фаз; обычно из-за слож-
ности уравнений их решают методом подбора. Если же
равновесные составы установлены экспериментально,
эти уравнения можно решить относительно двух пара-
метров корреляционного уравнения.
Таблица 7.1. Вторые производные g = (G - XxiG^/RT = G^/RT + Ixjnx,- не-
скольких уравнений, используемых для определения банарных смесей
Уравнение (G^/RT)
d2g/dx2
Симметричное — 2А + l/XjX2
Маргулеса
Ван Лаара
2(—2 + Зх,)Л + 2(1 - Зх,)В + 1/х,х2
-2А2В2
Мх,+Вх2)3 + 1/ХЛ
Вильсона
Цубоки—
Катаямы—
Вильсона
T2lG2l т12^12
xi + G21x2 G12Xi + х2
T21G21(i ~ ^21) г12°12(* + ^12)
_(x1 + G21x2)2 + (Gi2x\+x2>2 _
т21^21(^ ~ ^21)2 г12^12(* + ^12)2 \ 1
(х । + G21X2)3 + (<712X1 +*2)3 / *1*2
Для решения последней задачи можно воспользовать-
ся уравнениями Маргулеса и ван Лаара. Уравнения
Маргулеса преобразуются следующим образом:
1п(У1/у*) = 1п(х’|’/х1)= [А + 2(В ~А)Х1]Х2
— [А + 2(В — А)х*]х*2 (7.30)
После того как отношение А /В определено, его под-
ставляют в уравнение (7.31), чтобы найти значение А,
далее по уравнению (7.33) находят В, либо просто опре-
деляют В из соотношения В = А/(А/В).
Решение уравнения ван Лаара проводят подобным
методом. Так, например,
1п(У1/у*) = 1п(х1’/х1) =
АВ2
х2
Axi + Вх2
1п(у2/у?) = 1п(х2*/х2)
ПУг/У*) = 111 - (В + 2(А - B)x2)xj
1 Х1
- (В + 2(А - В)х2)х*2 (7.32)
(7.38)
(7.33)
Отношение А /В равно
/*1 х* \ /1п(х?7х}) \
Л \х2 + х? / \1п(х2/х2) / 2
В xi х* 2XjX* ln(x*/xj)
х2 х2 х2х$1п(х2/х$)
(7.39)
и, следовательно,
Когда эти уравнения упорядочены, единственной пере-
менной в них остается отношение А /В, значение кото-
рой можно определить по методу Карлсона и
Кольберна [211] как
ln(xf/X!)
А 2а2 ln(x*/x2) +/Jj ln(x*/xi)
В 2а! ln(xf/x!) + р2 1п(х^/х2) ’
Л \2 / а \2
1+^(х1/х2) 1+-(х?/х?)
(7.39а)
где
Располагая более чем одним набором равновесных
составов, можно для расчета параметров применить не-
которые методы нелинейной регрессии. Соответствую-
щая целевая функция
a.=x2_Xi*2_x3+Xf*3j (7.35)
д. = х2_Х1*2 -2х^+ 2х*3-
(7.36)
F(A, В) = Е (уххх - y?xf)2 + S (у2х2 -
— minimum. (7.40)
Пример 7.5. Определение параметров уравнения Маргулеса при известных равновесных составах системы жид-
кость — жидкость
Примем, что х и х* — два возможных состава фаз.
Исходя из условий равновесия, вытекающих из уравне-
ния Маргулеса,
1п(у1/у1) = 1п(х7х),
1П(72/Т2) = 1П[(1 - х*)/(1 - X)],
получаем
[Л. + 2(В - Л)х](1 - х)2 - [А + 2(В - Л)х*](1 - х*)2
-1п(х*/х) = 0,
[В + 2(Л - В)(1 - х)]х* - [В + 2(Л - В)(1 - х‘)]х*2
- 1п[(1 - х*)/(1 - х)] = 0.
Сгруппировав коэффициенты при Л и В, имеем
[(1 - 2х)(1 - х)2 - (1 - 2х*)(1 - х*)2]Л
+ [2х(1 - х)2 - 2х*(1 - х*)2]В - 1п(х7х) = 0,
[2x2(1 - х) - 2х*2(1 - х*)]Л
+ [х2(2х - 1) - х*2(2х* - 1)]В
- 1п[(1 - х*)/(1 - х)] = 0.
Математическая форма этих уравнений такова:
aiA + 61В = ci,
агА + ЬхВ = с2,
и они имеют следующие решения:
Л = (biCi - b\c2)/D,
В = (oiC2 - a2c{)/D,
D = а^Ьг - а2Ь\.
Равновесие жидкость — жидкость 369
Ниже приводится программа решения этих уравнений,
разработанная на языке Бейсик, и результаты, получен-
ные при xi -- 0,95 и нескольких значениях х*.
10 ! LIQUID-LIQUID EQUILIBRIA WITH
THE MARGULES EQNS
20 X = .95
30 Y = .05
40 Al = (1 - 2*X)»(1 - X)A2 - (I - 2»Y)*(1 - Y)A2
50 Bl = 2.X.(1 - X)A2 - 2.Y*(1 - Y^2
60 A2 = 2»XA2»(1 - X) - 2.YA2.(1 - Y)
70 B2 = XA2.(2»X - 1) - YA2»(2»Y - 1)
80 Cl = LOG(Y/X)
90 C2 = LOG((1 - Y)/(l - X))
100 D = A1.B2 - A2.B1
110 A = (C1.B2 - C2»B1)/D
120 В = (A1.C2 - A2»CI)/D
130 PRINT USING 140; Y,A,B
140 IMAGE.DD,2X,DDD.DDDD,2X,DDD.DDDD
150 Y = Y+.05
160 GOTO 40
170 END
x‘ A в
0,05 3,2716 3,2716
0,10 2,6668 3,2889
0,15 2,2649 3,2795
0,20 1,9165 3,2553
0,25 1,5714 3,2170
0,30 1,2012 3,1618
0,35 0,7820 3,0847
0,40 0,2888 2,9781
0,45 -0,3101 2,8310
0,50 -1,0571 2,6268
0,55 -2,0155 2,3407
0,60 -3,2791 1,9331
0,65 -5,004 1,3397
0,70 -7,4392 0,4479
0,75 -11,0735 -0,9522
0,80 -16,8752 -3,2943
0,85 -27,1285 -7,6122
0,90 -48,5854 -17,0037
0,95 — —
Пример 7.6. Применение методики Ньютона — Рафсона для определения равновесных составов систем жид-
кость— жидкость на базе уравнений Маргулеса и ван Лаара
Для этого примера выбраны значения параметров
А = 3 и В = 2. Оценка значений коэффициентов актив-
ности проводится с использованием следующих уравне-
ний Маргулеса:
ln7i = [А + 2(В - A)xi]x22 = (3 - 2X1X1 - х02, (1)
1п-у2 = [В + 2(А - В)х2]х1 = 2(2 - xi)x?. (2)
Определяем
f = ln(71/-уТ) - ln(x*/xi) (3)
= (3 - 2x0(1 - xi)2 - (3 - 2xi‘)(l - х*)2
- 1п(Х17х0 = 0, (4)
g = 111(72/72) - 1П[(1 - Х1*)/(1 - Xi)l (5)
= 2(2 - xOxi - 2(2 - хГ)х*2 - 1п[(1 - хГ)/(1 - Xi)] = 0.
(6)
Частные производные этих величин таковы:
Л = - (1 - х0(2 - Х1), (7)
= - (1 - xi*)(2 - хГ), (8)
gx = 2xi(4 - ЗхО, (9)
gx. = 2х*(4 - 3xi‘). (10)
Для определения поправок й и к следует решить указан-
ные уравнения для установочных величин xi и х*.
hfx + kfx.+f=0, . (11)
hgx + kgx- + g = 0. (12)
В результате получаем
Л - - (fgx> - gfx-)/D, (13)
к = - (fxg - gxf)/D, (14)
D = fxgx> - fx-gx. (15)
Критерием сходимости служит шаг 200 программы. Ес-
ли начинают с установочных оценок для xi и х* (0,1,
0,8), для решения необходимо 15 итерационных шагов.
Результаты, xi = 0,07195, хГ = 0,79206, соответствуют
величинам, определяемым их графика рис. 7.7, с точ-
ностью, допускаемой масштабом.
Программы для решения уравнений Маргулеса и
ван Лаара приведены ниже. По ван Лаару, предельной
взаимной растворимости соответствуют значения
xi = 0,0786, xj* = 0,7574. Уравнения Маргулеса и
ван Лаара дают несколько отличающиеся результаты,
хотя их параметры оцениваются исходя из одного и то-
го же значения 7".
10 ! NEWTON-RAPHSON SOLUTION OF
MARGULES EQUATION
20 SHORT F,G,X,X1,H,K,D
30 READ X,X1
40 DATA.1,.9
50 F=(3-2.X).(l-XT2-(3-2.Xl).(l-Xl)A2-
LOG(XIZX)
60 G = 2.(2-X).XA2-2.(2-X1).X1A2-LOG((1-XI)/
(1 - X))
70 Fl = - ((I — X)»(2 —X))+l/X
80 F2 = - ((1 - Xl).(2 - XI)) - 1/XI
90 G1 = 2.X.(4-3.X)-1/(1 -X)
100 G2 = 2.X1 .(4-3.Xl) +1/(1 -XI)
110 D = F1.G2-F2.G1
120 H = - ((F*G2- G*F2)/D) •
130 К = - ((F1.G-G1.F)/D)
140 DISP F;G
150 DISP X;X1
160 DISP H;K;D
5-S64
170 DISP
180 XI = XI+K
190 X = X+H
200 IF ABS(H/X)+ABS(K/X1)< =.0001/2 THEN 220
210 GOTO 50 .
220 END
10 INEWTON-RAPHSON FOR VAN LAAR, WITH
A = 3, B’= 2
20 READ A,В
30 DATA 3.2
40 INPUT X.X1
50 Hl = А»ВЛ2»((1-X)/(A»X + B»(1-Х)))л2
60 H2 = АЛ2»В*(Х/(А»Х + В*(1 — X))^2
70 H3 = А»ВЛ2»((1 - X1)/(A»X1 + B»(l - Х1)))л2
80 H4 = АЛ2*В*(Х1/(А*Х1+В*(1—Xl)))*2
90 F = Hl — H3 —LOG(X1/X)
100 G = H2 - H4 - LOG((1 - Xl)/(1 - X))
110 F3 = - (2*АЛ2*ВЛ2»(1 -X)/(A*X + B»(1 -Х))л3)
120 F1 = F3 + 1/X
130 F4 = - (2*АЛ2»ВЛ2»(1 -X1)/(A»X1 +B»(1 -Х1)ГЗ)
140 F2 = F4 —1/XI
150 G1 = - (F3 »X/(1 - X)) -1/(1 - X)
160 G2 = - (F4*X1/(1 - XI)) +1/(1 - XI)
170 D = F1»G2 —F2»G1
180 H = (G*F2-F»G2)/D
190 К = (G1»F-F1»G)/D
200 PRINT USING 210; X.X1
210 IMAGE.DDDD.3X..DDDD
220 X = X + H
230 XI = XI+K
240 IF ABS(H/X) + ABS(K/X1)< = .00005 THEN 260
250 GOTO 50
260 END
После дифференцирования
dF dF
dA~ dB = °
(7.41)
параметры можно определить по методу Ньютона —
Рафсона или симплексному методу. Первый метод был
применен, в частности, в сборнике данных по равнове-
сиям жидкость — жидкость, выпущенном DECHEMA.
В примере 7.5 параметры уравнения Маргулеса опре-
делены исходя из данных о равновесии в системе жид-
кость — жидкость.
7.4.2. Определение состава. В тех случаях, когда
уравнения коэффициентов активности известны, для
установления равновесных составов бинарных смесей
можно использовать несколько методик. Одна из на-
иболее общих схем предусматривает решение числовым
способом систем таких уравнений, как (7.28) и (7.29).
Однако иногда вполне приемлемые результаты можно
получить при помощи менее точных, но более удобных
методик. Графические методики суммированы в приме-
ре 7.7.
а. Метод решения системы уравнений. Уравнения
(7.28) и (7.29) — это трансцедентные и довольно слож-
ные уравнения и прямое их решение для определения
состава (xi и хГ) обычно невозможно. Рассмотрим чис-
ловые приемы, основанные на методике Ньютона —
Рафсона. Начинают с пробных значений составов, да-
лее для этих значений составов оценивают функции р
иди четыре частные производные, после чего находят
поправки h и к относительно xi и х* путем решения ли-
нейных уравнений
hpx + кРх* + Р ~ 0,
hqx + kqx* + q = 0.
На следующем этапе расчета принимают, что система
имеет состав (xi + h, х* + к). Уравнения частных про-
изводных имеют следующий вид:
др д In У1 _1_
Рх 5xj dxi Xj
(7.44)
Рх* = др 5х* 5 In у* 5x* 1 ~ Y* ’ X1 (7.45)
5? 5 In y2 1 (7.46)
Чх ~ 5xi 5xi 1 -Xi
dq 5 In y* 1 (7.47)
Чх* ~ 5х? 5xf + 1 - ? • 1 *1
В итоге получают соответствующие частные производ-
ные уравнения Маргулеса
5 in /!
—----= 2х2[(В-А)(1 - Зх^-Л], (7.48)
дх ।
д In у2
—----= 2x^(5-Л)(1 - Зх2) + Л], (7.49)
дх ।
производные уравнения ван Лаара
д In yi — 2Л2Л2х2
-----1 =-----------, (7.50)
5X1 (ЛХ1 4-Лх2)3
5 1пу, 2Л2Л2Х1 xt ainyi
-----=-------------7=----------;---. (7.51)
5x1 (Ах\ + Вх2)3 х2 °х\
Такая методика использована в примере 7.6, там же
приведена простая программа для решения на ЭВМ
уравнений Маргулеса и ван Лаара. В приложении В.9
и В. 10 дана программа для определения состава двух-,
трех- и четырехкомпонентных смесей, основанная на
уравнении Цубоки — Катаямы — Вильсона.
б. Графический метод. Равновесный состав или зна-
чения параметров уравнения могут быть найдены при
помощи диаграмм, приведенных на рис. 7.7—7.13.
Однако уравнение UNIQUAC нельзя представить таким
простым способом. Поскольку параметры всех уравне-
ний известны, если известны коэффициенты активности
при бесконечном разбавлении, в целях единообразия на
осях координат во всех случаях откладываются значе-
ния последних. Пара таких величин (Iny? и 1пу”) опреде-
Равновесие жидкость — жидкость 371
Рис. 7.7. Пределы смешиваемости, установленные по уравнению Маргулеса (данные Когана,
1968 [68]). Точки пересечения в пределах сетки показывают равновесный состав в мольных до-
лях (xi, хГ), соответствующий каждой паре логарифмов коэффициентов активности при беско-
нечном разбавлении или параметров уравнения А и В. Например, показанная на рисунке точка
(xi, х*) = (0,10, 0,98) соответствует (А,В) - (2,5; 4,03).
Рис. 7.8. Пределы смешиваемости, установленные по уравнениям ван Лаара и Скэтчарда —
Гильдебранда (см. подпись к рис. 7.7).
In у?
Рис. 7.9. Пределы смешиваемости, установленные по уравнению NRTL
с ай = 0,2 (данные С. Негахбана). In?” = Т21 + ri2exp( - 0,2ti2),
Iny” = Ti2 + T2iexp( - 0,2т21) (см. подпись к рис. 7.7).
Рис. 7.10. Пределы смешиваемости, установленные по уравнению
NRTL при ап = - 1 (данные С. Негахбана). Iny” = t2i + mexpfru),
lny“ = Т12 + T2iexp(T2i) (см. подпись к рис. 7.7).
Рис. 7.11. Пределы смешиваемости, установленные по уравнению Цу-
боки—Катаямы—Вильсона для К1/К2 = 2 (данные С. Негахбана). При
построении использованы те же значения Xi и xf, что и на рис. 7.7
(см.
подпись к рис. 7.7).
8 гч
Рис. 7.12. Пределы смешиваемости, установленные по уравнению Цу-
боки—Катаямы—Вильсона для V1/V2 = 3 (данные С. Негахбана).
При построении использованы те же значения xi их*, что и на рис. 7.7
(см. подпись к рис. 7.7).
Рис. 7.13. Пределы смешиваемости по уравнению Цубоки — Катая-
мы — Вильсона для К1/Р2 = 4 (данные С. Негахбана). При построе-
нии использованы те же значения Xi и х*, что и на рис. 7.7 (см. под-
пись к рис. 7.7).
Таблица 7.2. Пределы взаимной растворимости, расчитанные по уравнениям, параметры
которых установлены исходя из величин коэффициентов активности при бесконечном раз-
бавлении
**) Уравнение44^ (0,05, 0,90) (0,05, 0,70) (0,05, 0,50) (0,5, 0,5)
In 77 In 72 In 77 In 72 In 77 In 7 2 In 77 In 7 2
Маргулеса 3,29 2,67 3,16 1,20 2,63 -0,11 2 2
Ван Лаара Цубоки— 3,30 2,69 3,33 1,75 3,31 1,26 2 2
Катаямы— 4,39 3,42 4,81 2,60 5,30 2,31 2,71 2,63
Вильсона, 2 4,10 3,09 4,78 2,55 5,43 2,42 2,72 2,48
3 4,25 3,20 5,52 2,93 6,69 2,91 3,41 2,87
4 3,35 2,75 3,56 1,76 3,23 1,09 2,05 2,05
NRTL, 0,2 - 1 3,61 3,17 3,33 2,39 3,39 1,45 2,84 2,85
“Числовая индексация ния NRTL — а 12- для уравнения Цубоки — Катаямы — Вильсона означает Ь/И, для уравне-
ляет состав смеси (xi и хГ). В табл. 4.5 показано
соотношение между параметрами уравнений и 1пу”.
Результаты определения равновесных составов, про-
веденного с использованием нескольких уравнений, в
некоторых областях значительно расходятся. Эти раз-
личия отчетливо видны в табл. 7.2, в которой показаны
величины 1пу”, соответствующие нескольким комбина-
циям (xi, хГ). Приведем в качестве другого примера
равновесные составы, определенные графическим мето-
дом при Iny” = 2 и 1пу” = 3:
Равновесие жидкость— жидкость 375
Уравнение Х1 Х[
Маргулеса 0,21 0,92
ван Лаара 0,24 0,91
NRTL, ай = 0,2 0,27 0,91
NRTL, an = - 1 0,57 0,84
Цубоки—Катаямы—Вильсона, Взаимораст-
И2/К1 = 2 Цубоки—Катаямы—Вильсона, воримы
и2/и = 3 Цубоки—Катаямы—Вильсона 0,39 0,87
И2/И1 = 4 0,40 0,86
Итак, прогнозы, основанные на значениях 1пу”, име-
ют лишь приближенный качественный характер, хотя
известно, что в определенных случаях можно довольно
точно подобрать любое из этих уравнений для заданно-
го диапазона концентраций. В отсутствие дополнитель-
ной информации следует руководствоваться обычным
инженерным правилом — отдавать предпочтение на-
иболее устоявшимся результатам. Корреляции данных,
полученных для бинарных систем, приведенные в спра-
вочнике DECHEMA (DECHEMA LLE Data Collection),
основаны лишь на уравнении UNIQUAC и уравнении
NRTL с ап = 0,2, поскольку такие данные применимы
для тройных и более сложных смесей. Значения Q12,
равные 0,1, 0,3 и -1, считаются менее приемлемыми,
а уравнение Цубоки — Катаямы — Вильсона исследо-
вано недостаточно полно.
Диаграммы, приведенные на рис. 7.11—7.13, показы-
вают высокую степень зависимости результатов расче-
тов, основанных на уравнении Цубоки — Катаямы —
Вильсона, от объемных соотношений. Действительно,
при Ki/K2 = 1 указанное уравнение преобразуется в
уравнение Вильсона, посредством которого нельзя опи-
сывать системы с взаимонерастворимыми компонента-
ми; однако существуют взаимонерастворимые бинар-
ные смеси, в которых молярные объемы компонентов
различаются в пределах нескольких долей процента.
Уравнение UNIQUAC, как следует из работы [470], так-
же преобразуется в уравнение Вильсона при Ri = R2 и
Qi = Qi> однако при разработке корреляций взаимной
нерастворимости это уравнение не обмануло ожиданий,
по крайней мере применительно к серии более чем 2000
данных о равновесии в системах жидкость — жидкость,
заимствованных из справочника DECHEMA.
Не вполне решен вопрос и о правильности выбора ве-
личины ай в уравнении NRTL. Проверка 10 тройных
систем, полученных из 28 различных двойных систем,
показала [640], что уравнение NRTL с ап = -1 дает в
среднем несколько лучшие результаты, чем уравнения
NRTL, в которых для каждой исследуемой системы
применяются свои специфические положительные пара-
метры, или уравнение UNIQUAC.
в. Метод построения касательной, общей для двух
точек кривой. Как было показано в разд. 7.3, значения
равновесных составов (xi и хГ) соответствуют точкам
касания касательной, общей для двух точек кривой, за-
висимости величины энергии смешения Гиббса Sx/lny/x,
от xi. При наличии надежного графопостроителя по-
строить такую кривую достаточно легко, а точность
получаемых результатов приемлема для решения мно-
гих задач; кроме того, их можно использовать как на-
чальные данные при проведении точных числовых
расчетов. Эта методика используется в примере 7.7а
для нахождения равновесных составов. В примере 7.3
показано влияние зависимости результатов расчетов по
уравнениям NRTL и Цубоки — Катаямы — Вильсона
соответственно от значений величин ап и К1/К2.
г. Метод построения прямоугольника. Эта методика
расчета применена в примере 7.76. Построены отдель-
ные кривые зависимостей 71X1 и 72X2 от хь Построение
прямоугольника ведут методом проб так, чтобы одно-
временно удовлетворялись следующие условия:
71X1 = 7*х* и 72(1 - xi) = 72(1 - хГ). Такое построение
возможно, если каждая кривая имеет один минимум и
один максимум.
д. Метод петли. Если в исследуемой системе проис-
ходит разделение фаз, на кривой зависимости 71X1 от
72(1 - *1), где xi — переменная, при равновесных соста-
вах (xi и хГ) образуется петля, как это показано
в примере 7.7в и рис. 7.14. Точка пересечения дает
два значения xi, при которых 71X1 = 71Х* и
72(1 - xi) = 72(1 - хГ). Необходимость градуировки
петлеобразных кривых с величинами xi, соответствую-
щими каждому значению ординаты и абсциссы, можно
избежать, построив на той же диаграмме кривую
71X1 =/(xi) [525].
7.4.3. Влияние температуры. На взаимную раствори-
мость компонентов жидкофазной системы существен-
ное влияние оказывает только температура, хотя
иногда некоторую, впрочем незначительную, роль игра-
ют и примеси. Типичные диаграммы температура —
состав для бинарных смесей приведены на рис. 5.20.
Максимальную температуру, при которой возможно су-
ществование двух фаз, называют верхней критической
температурой растворения, а соответствующую мини-
мальную температуру — нижней критической темпера-
турой растворения.
Форма диаграмм тройных систем часто чрезвычайно
сильно зависит от температуры, как это видно, в част-
ности, из рис. 5.34 и 5.39. Очевидно, что математичес-
ки эту их особенность можно выразить лишь в том
случае, если зависимость коэффициентов активности от
температуры выражена достаточно точно, что, к сожа-
лению, не имеет места в используемых в настоящее
время уравнениях коэффициентов активности.
То обстоятельство, что коэффициенты активности,
полученные на основе данных о равновесии в системах
пар — жидкость, во многих случаях недостаточно точ-
но описывают равновесие систем жидкость — жид-
кость, отчасти обусловлено тем, что равновесия
первого типа исследуются при относительно высоких
температурах, тогда как равновесия жидкость — жид-
кость предпочтительнее исследовать при температурах,
близких к комнатным. Один из способов преодоления
этих трудностей, используемых при рассмотрении
тройных систем, упоминается в разд. 7.5.
Учесть при построении фазовых диаграмм влияние
температуры в том случае, если оно известно, доста-
точно просто. Несколько теоретических примеров влия-
ния температуры на бинарные системы с верхней и
нижней критическими температурами растворения ис-
следованы в примере 7.10. Простейший способ описа-
ния влияния температуры на параметры уравнений —
это линейная зависимость от Г-1.
Тгхг
Рис. 7.14.а — петлеобразные зависимости 71X1 = /(72X2) уравнения
Маргулеса о различными величинами параметров (А и В):(1 — 3,5 и
2,0, 2—2 и 2, 3—1 и 2). По обеим осям отложены в качестве парамет-
ров величины xi, начиная с xi = 0 внизу справа и кончая xi = 1 вверху
слева. Два значения xi, соответствующее точке пересечения, — это со-
ставы равновесных фаз; б — петлеобразные зависимости 71X1 = /(72X2)
уравнения ван Лаара с различными величинами параметров (Л и В):
1—3,5 и 2, 2—2 и 2, 3—1 и 2. Принцип построения кривых и возмож-
ности их применения см. выше, а в примере 7.4 (рисунок в) объясняет-
ся, как избежать необходимость градуировки таких кривых.
Равновесие жидкость — жидкость 377
Пример 7.7. Графические методы определения равновесных составов бинарных систем жидкость — жидкость
В этом частном случае мы используем уравнение
ван Лаара с Л12 = 3, А21 = 2, но в принципе можно
применить любое уравнение.
а. Точки касательной общей для двух точек кривой
на графике зависимости энергии смешения Гиббса дают
такой равновесный состав: (0,08 и 0,75).
б. Исходя из зависимостей 71X1 = / (xi) и 72X2 = f (xi),
строят прямоугольник и определяют состав, при кото-
ром 71Х, = у*х*.
в. Горизонтальная проекция отрезка кривой зависи-
мости 71X1 от 72X2 пересекает кривую 71X1= /(xi) в точ-
ке равновесного состава.
г. Сетевая диаграмма уравнения ван Лаара (рис. 7.8)
дает такой же равновесный состав (0,08, 0,75).
д. Числовое решение по методике Ньютона — Рафсо-
на (пример 7.6) дает следующие величины: (0,07855,
0,75628).
Определение равновесных со-
ставов систем жидкость —
жидкость как точек касатель-
ной, обшей для двух точек
кривой энергии Гиббса.
Определение равновесных со-
ставов систем жидкость —
жидкость построением прямо-
угольника на графике зависи-
мости У Xi и 7*Х* от Xi (см.
также задачу 7.27).
0
О
1
0,5
5
5
Jill 16 - Xi =1 1111 Illi 1111 11II 1111 ini 1111 1111 1111 1 1 1 L
тттт Г2Х2 *1 = 0.75^ 1111
тттт — — — — — Г IT 1
1111 08 Ух И X, х,=0,0 / 1 8 1 1111
1111 1111
тттт 1 1 ГТ
1111 1111
J 1II1
2
Z -
Э_1_1_1_ -L1..1 1 1111 1111 1 1 1-1 мм 1111 1.111 .1 1 1.1 1111 *1=0 1 1 1 Г
1
0,5
у2хг или х}
Петлеобразная кривая, построенная с использованием
xi в качестве параметра, начиная с xi = О (внизу) и за-
канчивая xi = 1 (слева). Непосредственно при входе в
петлю Xi = 0,08 и непосредственно на выходе из петли
Xi = 0,75. Построение кривой зависимости 71 от xi по-
могает избежать градуировки петлеобразной кривой
(см. также задачу 7.27).
Пример 7.8. Алгебраический метод определения равновесных составов
В данном случае задача из примера 7.7 будет решена
прямым применением уравнений (7.24) и (7.25). Соглас-
но определениям,
D = 1,5/хг + 1/xi,
2V = 1,5/хг - I/X1.
Функция g энергии Гиббса и ее производная определя-
ются следующими выражениями:
g = 3/D + xilnxi + X2I11X2,
dg/dx\ = -3N/D1 + \п(Х1/хг).
После подстановки
g ~ g* _ dg _ dg**
xi - x* dxi dxi
получаем
(3/D + xilnxi + Х21пхг) - (3/D* + x’lnx* + xzinx*)
Xi - xi
= -3N/D2 + ln(xi/x2)
= -3N7D*2 + ln(xi7x2‘).
Поскольку графический метод, использованный в пред-
шествующем примере, позволяет оценить xi и х* доста-
точно надежно, эти довольно сложные уравнения легко
решаются методом подбора. Результаты расчета
xi = 0,0785; х? = 0,7560; (g - g‘)/(xi - х?) = -0,1296;
dg/dxi = —0,1293; dg /dxi = —0,1295 вполне согла-
суются.
Пример 7.9. Определение условий равновесия в системе жидкость — жидкость методом минимизации энергии
Гиббса
В примере 7.7 показано, что если параметры уравне-
ния ван Лаара имеют следующие значения: Л = 3 и
В = 2, взаимная растворимость компонентов такой
смеси равна (xi, хГ) = (0,08, 0,755). Эти значения будут
подтверждены минимизацией энергии Гиббса для сме-
си. Примем, что
D = 1,5/хг + 1/Х1, (1)
ДГ = 1,5/xj - 1/х?, (2)
и запишем избыточную энергию Гиббса и ее произ-
Равновесие жидкость — жидкость 379
водные:
Gn/RT = 3/D, (3)
= -3N/D2.
dxi
Исходя из уравнения материального баланса
Z1 = 0X1 + (1 - а)х*, (4)
определим фракционный состав смеси в фазе 1:
а = (zi - %Г)/(Х1 - хГ). (5)
Функция Гиббса g = G/RT для смеси имеет следующий
вид:
gt = ag + (1 - a)g* (6)
= a(3/D + Sxilnx,) + (1 - a)(3/D* +
Zx‘lnx‘). (7)
Поскольку уравнение материального баланса вынуж-
денной минимизации имеет такую простую форму
[уравнение (5)], использовать множители Лагранжа нет
необходимости. Вместо этого о просто исключается из
уравнения (7). Запишем условия равновесия
dgi/dxi = dg*/dx* = 0. (8)
Далее нам будут необходимы следующие промежуточ-
ные значения:
да/dxi = -(Zi - xi)/(xi - хГ)2, (9)
да/дх! = (zi - xi)/(xi - хГ)2, (10)
dg/dxi = -3N/D2 + ln[xi/(l - xi)], (11)
dg’/dxi = 3N*/Dn + ln[xi/(l - xj)]. (12)
Запишем производные функции общей энергии Гиббса:
=о «8. + (g-g-)|SL.0, (13)
= (1 - а) + U - Z) = о. (14)
0X1 0X1 0X1
Коэффициент фазового разделения а и несколько произ-
водных в правой части можно исключить, выразив их
через xi и х* при помощи соответствующих уравнений.
Эти неизвестные можно определить совместным реше-
нием уравнений (13) и (14). Правильность определения
равновесного состава, Xi = 0,07855 и хГ = 0,75595, мож-
но проверить, проведя подстановку в эти уравнения и
оценив, насколько близка к нулю правая часть уравне-
ния. Результаты расчета должны быть независимыми
от суммарного состава zi. Сведенные в таблицу данные
10 000 значений правой части уравнения показывают,
что нулевые условия достаточно удовлетворяются.
Z1 а Уравнение (13) Уравнение (14)
0,07855 1,000 -0,2 0,0
0,10000 0,968 -0,2 -0,0
0,20000 0,821 -0,2 -0,1
0,30000 0,673 -0,2 -0,1
0,40000 0,525 -0,1 -0,1
0,50000 0,378 -0,1 -0,2
0,60000 0,230 -0,1 -0,2
0,75595 0,000 0,0 -0,3
7.5. Трех- и многокомпонентные смеси
При фиксированных температуре и давлении макси-
мальное число фаз, способных находиться в равнове-
сии, равно числу компонентов системы; на практике
это утверждение справедливо для конденсирующихся
систем даже при небольших изменениях давления.
Так, в трехкомпонентных смесях могут сосущество-
вать одна, две или три фазы; трехфазные системы
распространены менее всего, тем не менее несколько
таких примеров показано на рис. 5.38 и 5.39.
Боуден [24] упоминает в своей книге такую редкую
систему, как устойчивая шестикомпонентная система с
шестью жидкими фазами — ртутью, галлием, фосфо-
ром, анилином, водой и бензолом. Формы границ и
возможность появления и исчезновения фаз, несомнен-
но, связаны с влиянием температуры, как это показано,
например, на рис. 5.38. Твердая и паровая фазы могут
контактировать с множеством жидких фаз, см., напри-
мер, рис. 5.39; с такого рода сложными системами ча-
сто имеют дело при изучении криогенных процессов.
7.5.1. Определение параметров. При описании равно-
весий жидкость — жидкость в многокомпонентных сис-
темах в качестве уравнений общего характера можно
рассматривать только уравнения Цубоки — Катая-
мы — Вильсона, поскольку лишь в них непосредственно
используют данные о равновесии двухкомпонентных
систем. Хотя в уравнение NRTL для трехкомпонентной
смеси входит девять параметров, как выяснилось, для
расчетов достаточно универсального значения ац = 0,2,
поэтому все три уравнения можно рассматривать как
имеющие всего шесть параметров при заданной темпе-
ратуре трехкомпонентной смеси. В настоящее время
влияние температуры не принимается в расчет в систе-
матических исследованиях, исключение составляют
лишь метод ASOG и работа Ренона и др. [109], кото-
рые представляют параметры уравнения NRTL для ря-
да систем как линейные функции температуры.
В отсутствие экспериментальных данных о трехком-
понентных смесях для их приближенного описания
можно воспользоваться тремя сериямй параметров для
бинарных систем. Однако дополнительное использова-
ние некоторых экспериментальных данных, полученных
для трехкомпонентных смесей, несомненно может за-
метно улучшить полное представление. На значитель-
ное различие между параметрами, полученными для
бинарной смеси, и теми же параметрами, определенны-
ми исходя из . данных о трехкомпонентной смеси (для
одной и той же системы) указывают типичные резуль-
таты примера 7.12.
Для расчета шести параметров необходимо распола-
гать либо двумя сериями данных о соединительных ли-
ниях тройной системы, либо шестью равновесными
составами. Метод регрессии позволяет обработать
большее количество данных, что естественно более
предпочтительно. Можно также использовать данные о
равновесии тройных систем жидкость — жидкость и
бинарных систем пар — жидкость. Целевая функция в
этих случаях включает данные о равновесии пар —
жидкость и жидкость — жидкость.
OF = S[(Axi)? + (Дх2)Л (бинарная система 1),
+ S[(Axi)? + (Дхг)?] (бинарная система 2),
+ S[(Axi)f + (Дхг)? + (Дхз)?] (тройная система),
(7.52)
где каждая из трех разностей определяется как
Дх = х„о„ - х . (7.53)
Если параметры бинарной системы, основанные на их
взаимной растворимости, известны, можно предполо-
жить, что эти данные действительны и для тройной
системы. В этом случае одна соединительная линия и
данные о равновесии пар — жидкость для остальных
пар компонентов позволяют определить все парамет-
ры. Множество тонкостей регрессионных методов об-
работки данных приводится в литературе. В справоч-
нике DECHEMA (DECHEMA LLE Data Collection) в
большинстве случаев шесть параметров оцениваются
непосредственно из данных о тройных системах. Для
одинаковых пар компонентов, встречающихся в не-
скольких смесях, авторы справочника смогли разрабо-
тать универсальные параметры, которые они назвали
общими. Для детального ознакомления с методиками
оценки параметров можно рекомендовать следующие
статьи [159, 286, 125, 657 , 711].
7.5.2. Определение состава. Для решения указанной
задачи необходимо провести расчет соединительных ли-
ний и бинодальных кривых, а это возможно в том слу-
чае, если известны уравнения коэффициентов активнос-
ти. Теоретически все виды равновесий можно опреде-
лить нахождением минимума энергии Гиббса, соответ-
ствующего данному полному составу смеси, однако для
таких систем, как состоящие из двух жидких фаз, трех
жидких фаз, двух жидких и одной паровой фазы, разра-
ботаны специальные методики. Эти методики будут
представлены поочередно.
а. Система, состоящая из двух фаз. Равенства фуги-
тивностей, уравнения (7.4) и (7.5), распространяются на
все остальные компоненты. Следовательно, для каждо-
го компонента
7iXi = y*x*. (7.54)
Преобразуем это выражение:
х?= (Ji/y^Xi = KiXi, (7.55)
где Xi(= yt/yi) — коэффициент распределения, или
константа фазового равновесия. Исходя из величины
полного состава zt и относительной доли /3 вещества,
присутствующего в первой фазе, запишем уравнение
материального баланса:
zi = Дх,- + (1 - /3)х*= [(3 + Kt( 1 - (3)]х,-. (7.57)
Суммируя эти доли, получим уравнение, решением ко-
торого можно определить условия равновесия:
f(B) = Z------------- 1 - 0.
J(P’ p + Kj(l-p)
(7.58)
Этот метод в основном совпадает с используемым для
расчета испарения в системе пар — жидкость, где Ki за-
висит от состава. Указанный расчет удобно, в частнос-
ти, проводить по следующей схеме.
1. Оцениваем состав xi, Хг, ... первичной фазы.
2. Оцениваем фазовое разделение, /3.
3. Исходя из материального баланса, рассчитываем со-
став другой фазы:
х*= (zz-— Дх,)/( 1 — Д). (7.59)
4. Определяем все коэффициенты активности у, и у* и
коэффициенты распределения К,.
5. Методом подбора из уравнения (7.58) находим фазо-
вое разделение /3.
6. После того как /3 определено, отдельные члены сум-
мы обозначим Xi. С этим новым значением состава воз-
вращаемся в п. 3 и продолжаем расчеты до достижения
необходимой сходимости.
Даже для двухфазных трехкомпонентных систем та-
кой расчет провести нелегко, так как вероятность схо-
димости значительно зависит от начальных оценок
переменных. В табл. В.8 и В.9 приложения дана про-
грамма для расчетов четырехкомпонентных систем,
основанных на уравнении Цубоки — Катаямы — Виль-
сона. В книге Ренона и др. [109] проведена программа,
базирующаяся на уравнении NRTL.
Пользуясь программой, разработанной Фреденслун-
дом [286], в основу которой положено уравнение
UNIQUAC, можно осуществлять расчеты двухфазных
систем, содержащих до пяти компонентов, а програм-
ма Праузница и др. [98], базирующаяся на этом пред-
ложении, позволяет рассчитывать системы,' содержа-
щие 20 компонентов.
Программы В.8 и В.9 позволяют определить состав
фаз, находящихся в равновесии. Наиболее важное раз-
личие между этими расчетами и расчетами испарения
систем пар — жидкость состоит в том^ что в первом
случае нельзя начинать с допущения идеальности систе-
мы, так как разделения жидкостей в этой ситуации не
происходит. На стадии определения параметров соеди-
нительной линии суммарный состав также условен, хо-
тя эксперименты показывают, что сходимость дости-
гается быстрее, если полный состав приблизительно ра-
вен количеству двух фаз, т. е. /3 = 0,5. Полезными мо-
гут также оказаться следующие дополнительные
правила.
1. Идентифицируйте пару с наименьшей взаимной
растворимостью как состоящую из компонентов 1 и 3
и назовите их растворителями, компонент 2 в этой сис-
теме соответственно будет растворенным веществом.
2. Примите в качестве исходных следующие величи-
ны; xi = 0,90, хг = хз = 0,05 и xi = 0,1; однако эти ве-
личины до некоторой степени можно варьировать с
целью улучшения сходимости.
3. Задайтесь значением полного состава: zi = Z3 = 0,5,
z2 = 0, или примите некий промежуточный состав в
диапазоне взаимной растворимости двух раствори-
телей.
4. По методике Ньютона — Рафсона сходимость
Равновесие жидкость — жидкость 381
можно достигнуть при помощи дробных расчетных
приращений для каждой следующей попытки, т. е. при-
нимая Л = ah, где 0 < а 1.
В примере 7.11 сравниваются несколько результатов
расчетов для тройной смеси.
б. Система, содержащая более двух жидких фаз. В
качестве примера рассмотрим трехфазную систему с
тремя компонентами. Наллом [88] разработан специфи-
ческий вариант расчета состава такой системы с про-
граммой на языке Фортран. Предположим, что /31 и
02 — доли двух фаз. Запишем уравнение полного мате-
риального баланса по каждому компоненту:
Zi = + р2х*+ (I - Д1 - Д2А**-
(7.60)
4. Оцениваем сначала хГ, используя уравнение (7.63), а
затем исходя из равновесных соотношений (уравнения
(7.61) и (7.62)) X/ и х*.
5. Находим из уравнений (7.64) и (7.65) величины i/q и
^2, если они отличаются от нуля.
6. Определяем по методике Ньютона — Рафсона, поль-
зуясь линейными соотношениями, поправки Д01 и Д02
для фазового разделения
0^1
дц, (7-66’
дф2
*’ +д/’’+7₽, А/!’ "0' <7-67’
Перегруппируем соотношения, описывающие условия
равновесия, аналогично уравнениям (7.8) и (7.9)
у**
~--- V-** —
ЛI X j j X j
У*
(7.61)
(7.62)
Состав третьей фазы можно определить из уравнения
1 + (/с, - 1)0! +(К?~ 1)02 •
(7.63)
Требование равенства единице суммы мольных долей в
каждой фазе можно записать в виде однотипных
уравнений
^ = ЦхГ-^) = 2^**(1 -7С,)=0, (7.64)
h = S(xJ* - xf) = Ех**(1 - К?) = 0. (7.65)
Собственно расчет может включать следующие этапы.
1. Задаемся определенными значениями фазового разде-
ления /31 и 02 и состава х, и х*.
2. Из уравнения материального баланса, уравнение
(7.60), определяем состав третьей фазы.
3. Используя соответствующие зависимости, находим
все коэффициенты активности.
7. Принимаем приблизительные значения производных
с учетом допущения, что состав, согласно уравнениям
(7.64) и (7.65), в первом приближении влияет лишь на
значения 01 и 02. Эти производные определяются таки-
ми выражениями:
= х—Б?— <768>
а#, дф2 (к, -lx*:;-i)z,
ni------' (7М>
а#2 i>* 1 2 3z,
---Б?----• <7™>
где
Dj= 1 + (Ki- 1)0! + (КТ- 1)02. (7.71)
В этом методе расчета имеются два слабых места:
а) на первом этапе приходится принимать 6 допущений,
б) сходимость часто трудно достигается. Системы с
тремя жидкими фазами встречаются нечасто, тем не
менее в примере 7.12 мы рассмотрим одну йз них.
Четырехкомпонентная смесь с двумя фазами проана-
лизирована по методике UNIQUAC в примере 7.13.
Пример 7.10. Взаимная растворимость бинарных смесей с симметричным уравнением для коэффициентов актив-
ности как функция температуры
Коэффициенты активности представлены следующи-
ми выражениями:
In-yi = Ахг, (1)
ln?2 = Axi. (2)
Кривые зависимости температуры от состава имеют
нужную симметричность и критические температуры
растворения при Xi = 0,5. Температурную зависимость
А можно установить для параболической и эллиптиче-
ской зависимости температура — состав. Такие кривые
с верхней и нижней критическими температурами рас-
творения показаны на рисунках.
Зависимость между величиной параметра А и соста-
вом дана в уравнении (7.19).
А = —Ц- ln(l/xi - 1). (3)
1 “ 2X1
Запишем уравнение параболы с нижней критической
температурой растворения
Т = TL + a(xi - 0,5)2 (4)
и верхней критической температурой растворения
Т = Tv - a(xi - 0,5)2. (5)
Уравнение эллипса с верхней и нижней критическими
температурами растворения имеет следующий вид:
( Т- Тм\2
\Гм- TL)
( Xi - 0,5 \ 2
\хм - 0,5)
= 1,
(6)
где Тм = 0,5(71 + Ти).
На рисунке а показаны три вида зависимости темпе-
ратура — состав, соответствующие уравнениям (4) —
(6). Значения констант Ти = 400, TL = 300, а = 400 и
хм = 0,35. Соответствующие кривые зависимости па-
раметра А от температуры показаны на рисунках б и
в. Параметры уравнений коэффициентов активности ча-
сто меняются пропорционально обратной температуре.
Модельные графики взаимной растворимости бинарных
смесей, показывающие системы с верхней и нижней
критической температурой растворения и систему с
обоими вариантами критической температуры рас-
Температурная зависимость величины А при параболи-
ческой форме графика температура — состав.
Равновесие жидкость — жидкость 383
Температурная зависимость величины А при эллиптиче-
ской форме графика температура — состав.
Пример 7.11. Равновесие жидкость — жидкость в системе вода — ацетонитрил — этилацетат согласно уравне-
ниям Цубоки — Катаямы — Вильсона (ЦКВ), NRTL, LEMF и UNIQUAC
Характеристики этой системы при 60 °C приведены
в работе [668]. Одна из соединительных линий будет
проверена оценкой целевой функции
Ц I yiXi - у*Х* I /(yiXi + 7iW)]
для каждого из четырех уравнений. Соединительная ли-
ния имеет следующие характеристики.
Компонент системы X; X* V, мл/моль
1. Вода 0,9049 0,4733 18,07
2. Ацетонитрил 0,0800 0,2749 173,0
3. Этилацетат 0,0151 0,2518 286,0
Параметры уравнений:
Уравнение
Система ЦКВ NRTL LEMF UNIOUAC
Ni TV Hi
Kji TJi a TJi TJi R Q
Ацетонитрил н К вода 0,4920 0,6750 0,30 0,5361 0,6146 0,92 1,40
0,3532 1,8559 0,8222 0,6490 (вода)
Этилацетат + 0,6326 0,1791 0,30 0,1755 0,5461 1,87 1,72
ацетонитрил 1,2060 0,2391 0,1928 1,2834 (ацето- нитрил)
Вода + этилацетат 0,2802 4,3581 0,20 0,0621 0,6174 3,48 3,12
0,6039 0,1464 1,2593 0,4052 (этил- ацетат)
Значения коэффициентов активности и целевой функции
приведены ниже в виде таблицы. Для этого случая луч-
шим признано уравнение NRTL с заданным а и а = - 1
(уравнение LEMF)
Уравнение Компоненты системы Ti V* Целевая функция
Цубоки — Вода 1,20034 4,1603 0,2890
Катаямы — Ацетонитрил 33,5505 1,5457 0,7253
Вильсона Этил ацетат 488,196 2,2260 0,8587
1,8730
Вода 1,0225 2,0321 0,0193
Ацетонитрил 4,0178 1,3967 0,0887
NRTL Этилацетат 31,421 1,9615 0,0201
0,1281
Вода 1,0424 1,9783 0,0037
Ацетонитрил 5,0790 1,3642 0,0401
LEMF Этилацетат 20,931 1,4792 0,0819
0,1257
Вода 7,2666 3,9606 0,5563
Ацетонитрил 66,080 3,8030 0,6698
UNIQUAC Этилацетат 2393,9 7,4611 0,90119
2,1273
7.5.3. Метастпабильные и стабильные равновесия. В
ряде случаев в системе устанавливается состояние пере-
насыщения, и в отсутствие зародышей, на которых
способны расти пузырьки, капли или кристаллы, это
состояние может сохраняться неопределенно долго.
Как указывают, например, авторы работ [86, 2], для
небольших ядер (зародышей) характерно более высокое
давление паров или более высокие величины химиче-
ских потенциалов, а не большие массы, и поэтому та-
кие зародыши сохраняют стабильность, только нахо-
дясь в контакте с пересыщенными фазами.
Состояние метастабильного равновесия можно опи-
сать термодинамически. Как уже указывалось, истин-
ная, или абсолютная стабильность характеризуется
равенством Химических потенциалов отдельных компо-
нентов смеси в каждой фазе. Для бинарной смеси усло-
вия метастабильного равновесия характеризуются
термодинамически следующим образом: (d2G/dxi)rp>0.
Области метастабильного равновесия для бинарной
смеси выделены на рис. 7.2. С этой же темой связана
и задача 7.26.
Математическое описание метастабильности много-
компонентной смеси с использованием производной G
относительно мольной доли — задача более сложная,
подробно она рассмотрена в ряде публикаций, см., на-
пример, [78, 86]. В примере 7.14 с учетом проведенных
здесь соотношений обсуждаются условия существова-
ния в трехкомпонентной системе метастабильной и ста-
бильной (для сравнения) областей.
7.5.4. Литература, посвященная расчетам равнове-
сия в системах жидкость — жидкость. В литературе
опубликовано много примеров расчетов равновесия
трехкомпонентных и ряда четырехкомпонентных сис-
тем типа жидкость — жидкость. Справочник DECHE-
MA (DECHEMA LLE Data Collection), изданный в 1979
и 1980 гг. — наиболее полное издание такого рода, в
нем приведены параметры уравнений NRTL и
UNIQUAC для более чем 1000 серий данных. В табл.
7.3 показана в качестве примера страница из этого
справочника. О том, что легло в основу этой работы,
говорится в статьях Соренсона [124] и Соренсона и со-
авторов [657]. Несколько примеров приводят Праузниц
и др. в работах 1980 г. [567]. Авторы [668, 700] разра-
ботали несколько методов прогнозирования условий
равновесия систем исходя из данных о бинарных систе-
мах. Ряд примеров приводится в книгах Ренона и др.
[98] и Фреденслунда [109].
Параметры бинарного взаимодействия уравнений
NRTL и UNIQUAC можно использовать для прогнози-
рования условий равновесия жидкость — жидкость в
многокомпонентных системах, но полученные результа-
ты редко имеют высокую точность. Если нет надежной
информации даже о бинарных системах, параметры
уравнения UNIQUAC можно получить при помощи ме-
тода групповых вкладов UNIFAC. Специальная таблица
параметров, основанная на данных о равновесии жид-
кость — жидкость разработана Магнуссеном и др.
[454]; она дана в приложении Д (табл. Д.13). Эти же
авторы изучали точность расчетов по уравнениям
UNIQUAC hUNIFAC на примере 17 равновесных систем
жидкость — жидкость и оценили среднее абсолютное
отклонение (d, мол. %). При подстановке шести пара-
метров тройных систем d = 0,5%, а при использовании
параметров бинарный систем, полученных из данных о
Равновесие жидкость — жидкость 385
равновесии пар — жидкость, d = 3,7%; применение па-
раметров UNIFAC, полученных исходя из данных о рав-
новесии пар — жидкость, дает d = 9,2%, но при
использовании новых параметров UNIFAC, найденных
исходя из данных о равновесии жидкость — жидкость,
d = 1,7%. Во многих случаях результаты расчетов на
базе параметров бинарного взаимодействия или корре-
ляций UNIFAC могут рассматриваться лишь как оце-
ночные. Эти исследования точности результатов
расчетов можно сравнить с аналогичными исследовани-
ями, выполненными для приведенных в сборнике
DECHEMA данных о равновесии пар — жидкость в
водных трехкомпонентных системах. Необходимо,
однако, отметить, что для водных систем обычно
трудно подобрать корреляции, а большинство данных
о равновесии жидкость — жидкость получены для вод-
ных смесей.
Как способ разделения смесей экстракция занимает
второе место после дистилляции. В последнее время
объем литературы о равновесии жидкость — жидкость
существенно вырос, тем не менее одной из проблем, а
именно влиянию температуры, не уделено достаточно-
го внимания. Температурные уровни процессов дистил-
ляции и экстракции, осуществляемых как в промышлен-
ных масштабах, так и в масштабах лабораторных ис-
следований, значительно различаются. Температуры,
при которых изучается равновесие жидкость — жид-
кость, слишком низки, а температуры, обычно приме-
няемые для изучения равновесия пар — жидкость,
слишком высоки. Но в последнее время много удалось
выяснить, и расчеты, основанные на данных о равнове-
сии жидкость — жидкость, дают значительно лучшие
результаты, чем прогнозы, базирующиеся на ранней не-
четкой классификации, в основу которой положены та-
кие представления, как природа химических связей и
тенденции к образованию водородной связи. В настоя-
щее время расчет позволяет решить, имеет ли смысл
проводить экспериментальное исследование каких-то
конкретных систем.
Пример 7.12. Параметры уравнения UNIQUAC для трехкомпонентной системы с тремя жидкими фазами
Данные о системе 1-гексанол(1) + нитрометан(2) + во-
да(3) взяты из справочника DECHEMA (V/2, р. 69). Из-
вестны коэффициенты уравнения UNIQUAC для трех
пар. Опубликованы характеристики пятнадцати экспе-
риментально установленных соединительных линий, из
которых непосредственно определены шесть парамет-
ров тройного взаимодействия системы. Расчет прово-
дился по двухстадийному методу, рекомендованному
Соренсоном [657], и начинается с оценки параметров
бинарного взаимодействия. На первой стадии целевая
функция определяется выражением
(0.1)! = Х1[(хГп^ - х’^ “)2
+ (x'k'p.k ~ X*iky'*k )2
4- (x'ty'k - хТ?***)2] Minimum.
При соблюдении этого условия обеспечивается улуч-
шенная опенка параметров. На второй стадии целевая
функция минимизирует разность между измеренными и
рассчитанными значениями составов, отвечающими
всем соединительным линиям:
(OJ); = SSE(xyft)exp - (хце)са1с]г Minimum,
где i — индекс компонента, j — индекс фазы, к — ин-
декс соединительной линии.
Негахбан рекомендует пользоваться при рассмотре-
нии трехфазных систем симплексным методом. Ниже
приведены три набора параметров уравнения
Л,7 = (Uy - Ujj)/R (К).
Бинарная смесь (OJ), (OJ);
и А» Ал Atj Aji Aij A/i
12 302,15 59,312 359,80 26,843 331,76 468,898
13 109,38 276,54 81,979 321,77 64,177 360,85
23 421,70 229,72 403,03 198,85 499,65 159,11
Диаграмма трехкомпонентной системы построена с
использованием последней серии параметров; они более
всего соответствуют измеренным составам фаз. Точка-
ми показаны экспериментальные данные, на основе ко-
торых построены соединительные линии. Римские
цифры — это число фаз в отдельных областях систе-
мы, заштрихованный участок соответствует трехфаз-
ной области.
Вода
Трехкомпонентная диаграмма с трехфазной областью,
построенная в соответствии с найденными величинами
параметров уравнения UNIQUAC (данные С. Негахбана).
6-864
Пример 7.13. Двухфазная четырехкомпонентная система
Для четырехкомпонентной системы известны харак-
теристики двух экспериментально полученных соедини-
тельных линий и все коэффициенты бинарного взаимо-
действия (см. таблицу). Данные получены путем регрес-
сии в соответствии с целевыми начальными функциями,
использованными в примере 7.12. Для расчета соедини-
тельных линий в качестве исходных данных используют
коэффициенты бинарного взаимодействия и серию из
двенадцати параметров, полученных методом регрес-
сии^ На основе параметров* полученных методом ре-
грессии, построены четыре диаграммы трехкомпонент-
ных систем. Программа расчетов, в основу которой по-
ложено уравнение UNIQUAC, разработана Негахбаном.
Значения A(I, J) определены в примере 7.12.
Характеристики компонентов:
Начальные и конечные значения регрессионных пара-
метров взаимодействия
Начальные значения Конечные значения
I J A(I, J), К A(J, I), К A(I, J),K A(J, I), К
1 2 -108,50 -120,28 -105,46 -115,85
1 3 344,41 132,92 527,51 179,76
1 4 234,59 17,402 298,39 -41,402
2 3 447,87 29,002 554,16 2,7937
2 4 -191,05 -98,426 -135,53 -371,20
3 4 15,163 -145,01 -18,649 -178,21
1. Гептан R = 5,1742 Q = 4,396
2. Бензол 3,1828 2,400
3. 2-Этиламин 2,5736 2,360
4. 1-Метил-2-пирролидон 3,9810 3,200
Экспериментальные и расчетные соединительные
линии:
Экспериментальные данные
Левая фаза Правг 1я фаза
(1) (2) (3) (4) (1) (2) (3) (4)
70,870 24,240 0,290 4,600 2,190 11,560 54,420 31,830
49,590 40,200 1,650 8,560 2,930 19,700 50,520 26,850
Данные, рассчитанные с использованием исходных параметров взаимодействия
Левая фаза
Правая фаза
(1) (2) (3) (4) (1) (2) (3) (4)
70,870 22,113 2,510 4,507 2,421 4,509 61,239 31,830
49,590 40,121 3,247 7,042 2,021 10,257 60,872 26,850
Данные, рассчитанные с использованием серии из 12 регрессионных коэффи-
циентов
Левая фаза Правая фаза
(1) (2) (3) (4) (1) (2) (3) (4)
70,870 23,956 0,702 4,473 2,455 11,491 54,224 31,830
49,590 40,288 1,628 8,493 2,547 19,876 50,727 26,850
Равновесие жидкость — жидкость 387
Четыре трехкомпонентные диаграммы четырехкомпонентной системы. Что-
бы построить тетраэдр, нужно сложить кромки вдоль ребер внутреннего тре-
угольника и соединить три вершины, помеченные словом «гептан». Экспери-
ментальные данные четырех трехкомпонентных систем можно взять из спра-
вочника DECHEMA (DECHEMA LLE Data Collection, V/2, pp. 438, 439, 440,
V/3, p. 241).
7.6. Прогнозирование условий равновесия в
системах жидкость — жидкость с примене-
нием уравнений UNIFAC или ASOG
Большинство полученных к настоящему времени ре-
зультатов говорят о том, что равновесие систем жид-
кость — жидкость прогнозируется хуже, чем равнове-
сие систем пар — жидкость, для предсказания которого
используются коэффициенты активности, рассчитывае-
мые по одному из двух имеющихся методов групповых
вкладов, но работы в этой области активно ведутся, и
методика будет усовершенствована. В книге Фреденс-
лунда и др. [40] при помощи методики UNIFAC иссле-
довано пятьдесят трехкомпонентных систем. Хотя
раздел фаз был нё зафиксирован только в одной из этих
систем, точность определения характеристик соедини-
тельных линий и бинодальных кривых была различной.
В предыдущем разделе говорилось о полученной Маг-
нуссеном и др. [436] новой серии параметров уравнения
UNIFAC, базирующейся на результатах измерения рав-
новесия жидкость — жидкость. О дальнейшем развитии
этих исследований сообщается в еще одной статье Маг-
нуссена и др. [456]. Эти исследователи связаны с соста-
вителями сборника «DECHEMA LLE Data Collection».
Несколько меньшее внимание уделяется методике
ASOG, предназначенной для расчета равновесия жид-
кость — жидкость. Анализ 31 тройной системы [689]
показал, что прогнозирование можно считать точным
лишь в шести случаях, рассчитанные бинодальные кри-
вые оказались точными в 24 случаях, а прогноз воз-
можного раздела фаз оказался верным во всех случаях.
Достоинство методики ASOG состоит в том, что в ней
учитывается значительное влияние температуры на па-
раметры уравнения — фактор, который разработчики
и исследователи уравнения UNIFAC предполагают
включить в свою методику. Тем не менее, исходя из
опубликованных исследований, результаты расчетов,
проводимых по методике ASOG, можно рассматривать
только как качественные, в то время как методика
UNIFAC дает по крайней мере полуколичественные ре-
зультаты. Безусловно, что включение корректной тем-
пературной зависимости параметров уравнений
рассматривается как важный шаг в разработке таких
параметров, которые были бы в равной степени приме-
6*
Пример 7.14. Пределы фазовой нестабильности трехкомпонентных смесей
Термодинамические условия существования трехком-
понентных систем в метастабильном состоянии опреде-
лены Луписом [78] следующим образом:
(1 тгй 6+х>
\ ОЛ2 1 \ дЛз ) ОЛ3 ОЛ2
При ф - 0 достигается предел существования метаста-
бильного состояния, который показан на рисунке бино-
дальной кривой, ограничивающей затемненную
область. В пределах этой области разделение на две фа-
зы происходит мгновенно. Внешняя бинодальная кри-
вая ограничивает область существования истинной
стабильности. В области, ограниченной двумя бино-
дальными кривыми, разделение может произойти очень
быстро, если в системе присутствуют зародыши, одна-
ко в отсутствие зародышей немедленного разделения не
происходит.
Бинодальные кривые системы метанол + морфо-
лин + гептан построены с целью получения параметров
уравнения UNIQUAC из данных, взятых из справочни-
ка DECHEMA (LLE Data, Collection, volume V/2, 96,
1980).
Морфолин
Метастабильное и стабильное состояния системы
метанол + морфолин + гептан (данные С. Негахбана).
нимы как к системам жидкость — жидкость, так и к
системам пар — жидкость. Из материалов исследова-
ний, цитируемых здесь, однозначно следует, что рабо-
ты в этой области ведутся и что большинство их
базируется на данных DECHEMA по равновесиям жид-
кость — жидкость и пар — жидкость. Число систем,
проверенных по методикам UNIFAC или ASOG, — это
всего лишь крошечная доля более чем 2000 серий дан-
ных, приведенных в этом справочном издании.
7.7. Растворимость жидкостей
Различие между растворенным веществом и раство-
рителем совершенно однозначно, если в качестве перво-
го выступает твердое или газообразное вещество, а
растворитель в интересуемых условиях практически не-
летуч. Однако, если рассматриваются жидкие смеси,
различие это, возможно, до некоторой степени услов-
но. Так, термином растворенное вещество принято на-
зывать тот компонент, концентрация которого отно-
сительно мала, даже если эти вещества в некоторых
ограниченных пределах взаимно растворимы. Поэтому
принято говорить о растворимости углеводородов в во-
де и воды в углеводородах, как будто бы взаимной рас-
творимости не существует.
Говоря о взаимной растворимости жидкостей — а
это, безусловно, главная тема данной главы, — нельзя
не касаться коэффициентов активности и их зависимо-
стей. Так, в примере 7.10 рассматривается температур-
ная зависимость растворимости и в этих целях
Таблица 7.3
(1) C7H8 TOLUENE
(2) C3H6O 2-PROPANONE
(3) H2O WATER
HACKL A., SOLAR W. , ZIEBLAND G.
EUR.FED.CHEM.ENG.,RECOMM.SYST.LIQ.EXTR.STUD., EDITOR:T.MISEK
(1978)
TEMPERATURE = 10.0 DEG C TYPE OF SYSTEM = 1
EXPERIMENTAL TIE LINES IN MOLE PCT (GRAPH.INTER POL.)
LEFT PHASE RIGHT PHASE
(1 ) (2) (3) (1 ) (2) (3)
99.587 0. 158 0.255 0. 010 0,056 99.934
98. 110 1.637 0.254 0. 010 0.537 99.453
96.558 2.938 0.504 0. 010 0.91 3 99.072
95.497 4.001 0.502 0. 010 1.227 93.763
94.612 4.887 0.500 0. 010 1 . 434 98.506
91.710 7.796 0.495 0. 010 2.324 97.666
88.300 10.969 0.732 0. 021 3.27 0 96.709
33.486 15.558 0.956 0. 022 4.599 95.330
80.002 18.820 1. 178 0. 033 5.515 94.451
76.752 21.854 1.393 0. 034 6.432 93.534
72.635 25.768 1.597 0. 046 7.470 92.434
66.126 31.880 1.994 0. 059 9.158 90.783
SPECIFIC MODEL PARAMETERS IN KE LVIN
UNIQUAC NRTL(ALPHA:.2)
I J AIJ AJI AIJ AJI
1 2 115.14 29.585 21 1 .69 14.071
1 3 814.64 334.88 1057.6 1643.2
2 3 317.21 -22.287 366 .44 250.69
R1 = 3 .9228 R2 = 2.5735 Rj 1 = 0 .9200
Q1 = 2 .968 Q2 = 2.336 Q3 1 = 1 .400
MEAN DEV. BETWEEN CALC. AND EXP CONC. IN MOLE PCT
UNIQUAC (SPECIFIC PARAMETERS) 0.05
NRTL (SPECIFIC PARAMETERS) 0.09
100
MOLE PER CENT OF (3)
MOLE PER CENT OF (2) IN RIGHT PHASE
EXP. DISTR. RATIO (>
CALC.DISTR. RATIO UNIQ(SP)-------------NRTL (SP)--------------
Равновесие жидкость — жидкость 389
используется симметричное уравнение коэффициентов
активности.
В этом разделе, однако, некоторое внимание будет
уделено таким системам, в которых взаимная раство-
римость компонентов низка и растворенное вещество
можно рассматривать как вещество, остающееся совер-
шенно чистым. Такая «односторонняя» растворимость
математически описывается упрощенно, поскольку фу-
гитивность растворенного вещества остается постоян-
ной, а его мольная доля и коэффициент активности в
твердой фазе остаются, равными единице. Следователь-
но, условия равенства фугитивностей можно записать
следующим образом:
У2*2 = У**2 = 1, '
где подстрочным индексом помечено растворенное ве-
щество, а звездочкой — его чистая фаза. Исходя из
уравнения Скэтчарда—Гильдебранда растворимость
можно определить так:
/ Г2 \
х2 = 1/у2 = ехр I- “(^1 ~ 52)2Ф? I (7.72)
\ /С 1 /
/ V2 \ I0?
= ехр I- (Sj — S2)2 I . (7.73)
\ пт /
Влияние объемной доли значительно, если объемное
отношение Pi/Pi велико (рис. 7.15).
Важную группу соединений, обладающих «односто-
ронней» растворимостью, представляют жидкие поли-
меры: они растворяют обычные жидкости, но сами в
них нерастворимы. Взаимодействие воды и углеводоро-
дов также можно рассматривать как пример «односто-
ронней» растворимости. Данной проблеме посвящено
много работ, обширный библиографический список
приведен в API Tfechnical Data Book (1970). Раствори-
мость воды в углеводородах обычно можно рассчитать
с точностью в пределах порядка величины, но сходи-
мость оценок может выходить за пределы нескольких
порядков величины, хотя числовые значения в обоих
случаях невелики. Обычно для оценки растворимости
используют довольно специфические уравнения. Напри-
мер, растворимость воды в углеводородах и углеводо-
родов в воде оценивается по уравнениям API:
logxw = -(4200Я/С + 1050)( 1,8/7’ - 0,0016) (7.74)
-logxAc = а + ЬС2
(7.75)
где Н/С — массовое соотношение водорода и углерода,
С — число атомов углерода в углеводороде; константы
а и b зависят от класса углеводорода; температура Т
измеряется в градусах Ренона.
Хотя на установление связей между относительной
растворимостью и параметрами растворимости было
затрачено много усилий, к особому успеху это не приве-
ло. Две такие корреляции показаны на рис. 7.16а и
7.166. В ряде случаев такие корреляции можно улуч-
Рис. 7.15. Растворимость хг (мол. доли) в системе, описываемой урав-
нением Скэтчарда — Гильдебранда. Предполагается, что растворенное
вещество остается чистым (см. уравнение (7.72)).
(я)
Рис. 7.16а. Растворимость иода в растворителях с различными параметрами раство-
римости (14,1 — параметр растворимости иода) (Shinoda, 1978).
Параметр растворимости растворителя
Рис. 7.166. Растворимость газов как функция параметра
растворимости растворителя, <51 при 25°С и 1 атм.
[Jolley, Hildebrand JACS, 80, 1051 (1958)]. (©Am. Chem.
Soc.).
Равновесие жидкость — жидкость 39
шить путем подгонки параметров растворимости к дан-
ным о растворимости, как в примере 6.8, но каких-либо
общих рекомендаций разработать при этом не удалось.
Ввиду обычно ограниченной растворимости органиче-
ских веществ в воде эмпирические корреляции для таких
систем можно выразить исходя из их относительной
растворимости в октаноле и в воде — так называемого
коэффициента распределения октанол/вода. Этот коэф-
фициент получают из молекулярных групповых вкла-
дов. Соответствующие уравнения и данные представ-
лены в книге Лимана [80].
Источники информации. В этом разделе мы попыта-
лись кратко представить растворимость исходя из
простой теории. Результаты, которые дает эта теория,
соответствуют диапазону оценок — от посредственных
до плохих. Универсальной обоснованной теории, по-ви-
димому, пока не разработано, так что представить вли-
яние температуры, давления и состава на раствори-
мость можно только посредством эмпирических уравне-
ний. Причины отклонения растворимости от моделей
простой теории — неадекватное воздействие РКГ-фак-
торов, а именно разная степень межмолекулярного при-
тяжения растворенного вещества и растворителя.
Однако попытки учесть взаимодействие полимер —
растворитель и поведение поверхностно-активных ве-
ществ привели к созданию даже менее адекватных тео-
рий. Данных о растворимости опубликовано очень
много.
Укажем несколько очень обстоятельных обзоров, по-
явившихся в 80-х годах.
1. Landolt-Bornstein, II/2b, с (1975).
2. Freier, Aqueous Solutions: Data for Inorganic and Or-
ganic Compounds (1975 ff).
3. Stephen, Stephen, Silcock, Solubilities of Inorganic and
Organic Compounds (7 parts, 1979).
4. IUPAC Solubility Data Series (1979 ff).
Цель издания — собрать всю опубликованную инфор-
мацию; к 1980 г. выпущено около 20 томов серии.
7.8. Равновесие пар — жидкость —
жидкость
При постоянном значении либо Т, либо Р максималь-
но возможное число фаз в системе равно числу компо-
нентов, но часто число фаз меньше возможного.
Проблема заключается в том, чтобы найти составы
всех фаз, которые могут находиться в равновесии при
заданном общем составе и при определенной величине
Т, или Р, или Н, или S. Возможны следующие частные
случаи.
1. Точка начала кипения при заданном значении Т
или Р.
2. Точка росы при заданном значении Т или Р.
3. Испарение при заданных значениях Т и Р.
4. Испарение при заданных значениях Н и либо Т,
либо Р.
5. Испарение при заданных значениях S и либо Т,
либо Р.
Как и при определении равновесия в системе жид-
кость — жидкость или пар — жидкость, при рассмот-
рении равновесия пар — жидкость — жидкость воз-
можны два различных подхода.
1. Непосредственное решение системы уравнений, опи-
сывающих материальные балансы и равновесие между
фазами.
2. Определение минимума энергии Гиббса для всей сме-
си. В данном случае переменными являются количество
и состав всех фаз, и чтобы получить один минимум,
сумму мольных долей в каждой фазе принимают рав-
ной единице.
Пример 7.15. Взаимная растворимость воды и некото рых углеводородов
Растворимость рассчитывают по уравнению (7.42) и
сравнивают с округленными данными, приведенными в
справочнике API Technical Data Book (1970, Ch. 9). Под-
гоночные значения параметра растворимости воды, со-
поставляемые с данными API, маркируются звездоч-
кой; они сравниваются с истинным значением 23,53.
Компонент V 5
Гексан 131,6 7,27
Бензол 89,4 9,16
Вода 18,1 23,53
Растворимость при 300 К
Система Расчет API 6*
Вода в гексане 0,00032 0,00070 13,0
Вода в бензоле 0,0019 0,0033 15,3
Гексан в воде 4(Е-26) 1(Е-6) 15,2
Бензол в воде 4(Е-14) 4(Е-4) 16,4
Универсального подгоночного значения параметра
растворимости воды, соответствующего этим данным,
вероятно, не существует. Последующее изучение может
иметь смысл.
7.8.1. Двухкомпонентные смеси. Если смесь содер-
жит всего два вещества, для расчета равновесия легко
применить комбинацию уравнений материальных ба-
лансов и равновесий. Для иллюстрации рассмотрим
изотермическую систему, как в примере 7.16. Принятая
система обозначений показана на рис. 7.17. Первый
этап — это расчет взаимной растворимости, осущест-
вляемый как в разд. 7.4.2. Далее рассчитывают равно-
весие паров смеси:
у,ф^Р]Л
Ф{Р
X/ = KjXj
(7.76)
Равновесие жидкость — жидкость 393
Рис.7.17. Номенклатура трехфазных равновесий:
F = V + L + £*, а = V/F, 0 = L/(L + £*),
L = (1 - a)PF, L* = (1 - а)(1 - 0)F,
V = аЕ
y*0$at P$at
-М------— х?= К?х?.
Ф/Р 1 1 1
(7.77)
Диаграмму Р — xi устанавливают серией расчетов то-
чек начала кипения, используя выражение
P = yjX]P]at+ y2x2P^at. (7.78)
После этого при помощи формулы
У1 = У1Х1Р1а‘/Р. (7.79)
устанавливают соотношения Р — ji.
Если давление постоянно, необходимо знать, как
влияет изменение температуры на коэффициенты ак-
тивности, или же можно принять, что включенная в
уравнения NRTL или UNIQUAC, или Цубоки — Катая-
мы — Вильсона температурная поправка корректна. За-
висимость давления паров от температуры также
должна быть известна, например, должна отвечать
уравнению Антуана. Соответствующая методика расче-
та равновесия систем при постоянном давлении вклю-
чает следующие этапы.
1. Определяют взаимную растворимость во всем диапа-
зоне температур.
2. Определяют давление, отвечающее точке начала ки-
пения при каждой из этих температур, из выражения
Р = SyzxzP«at
(7.80)
и устанавливают температуру, при которой достигает-
ся определенное давление. Эта наименьшая температу-
ра, при которой паровая фаза может существовать при
данном давлении.
3. При температурах выше минимальной и при составе
смеси в диапазоне смешения температура, соответ-
ствующая данному составу, — это точка начала кипе-
ния, и ее находят при помощи уравнения (7.78).
Поскольку коэффициенты активности и давления паров
зависят от температуры, решение методом подстано-
вок необходимо для каждого варианта состава жидкой
смеси. Соответствующий состав паров находят из урав-
нения (7.79), Две ветви кривых Р — сходятся вместе
при минимальной температуре.
Другой путь поиска условий равновесия основан на
следующем: G, имеет одинаковое значение для конкрет-
ных компонентов во всех фазах. В соответствии с мето-
дом отсечения отрезков касательной, равновесные
составы определяются точками касаний кривых измене-
ния энергии Гиббса паровой и жидкой фаз. В том слу-
чае, если две жидкие фазы находятся в равновесии с
одной паровой фазой, на диаграмме изменения энергии
Гиббса при определенных Т и Р (см. пример 7.25), име-
ется касательная общая для трех точек кривой.
7.8.2. Трехкомпонентные системы. Рассмотренные
выше методы можно легко распространить и на много-
компонентные смеси. Оба расчетных метода, упомяну-
тые в начале этого раздела, применимы к
многокомпонентным системам; они будут рассмотрены
далее преимущественно применительно к изобариче-
ским условиям и системам, содержащим две жидкие
фазы.
Материальный баланс и равновесие. Равновесия жид-
кость — жидкость описываются уравнением
7ixi = У*х*
(7.81)
а равновесия пар — жидкость — уравнениями (7.76) —
(7.77). Материальный баланс испарения определяется
выражением
г, = ay, + (1 - а)[0 xz + (1 - Р)х*],
(7.82)
где а — доля общего количества вещества в паровой
фазе, Р — доля первой жидкой фазы в общем количе-
стве жидкости. Примем, что А, = у;/х„ К* = у,/х* и
Ki/K* = yi/y*.
Для каждого состава фаз
Zj
аК, + (1 - а)[0 + (1 - p)Ki/Kf] ’
(7.83)
Zj
аК*+ (1 - a)[pKf/Ki + 1 - p] ‘
После подстановки этих выражений в мольные фракци-
онные балансы получаем
Е yi- Е xz = 0, (7.85)
Еу(-Ех?=0. (7.86)
Уравнения материальных балансов принимают следую-
щий вид:
l)Zj
= Е-----:---------------------z- = 0, (7.87)
аК, + (1 - а)[Д + (1 - P)Ki/Kf] k ’
(К?~ l)zz
аК* + (1 - a)[pKf/Ki + 1 - р]
(7.88)
Для точки начала кипения
S у{ = S KjXi = S К*х*= 1, (7.89)
а для точки росы
S x,-=S xz*=S yi/Ki-b yi/Kf= 1. (7.90)
Уравнение (7.89) или (7.90) можно использовать для на-
хождения условий соответственно начала конденсации и
начала кипения. Практические приемы проведения та-
ких расчетов описаны в гл. 6.
При заданной температуре соединительные линии и
бинодальные кривые жидкой фазы определяют, как
описано в разд. 7.5. Для каждой соединительной линии
давление, отвечающее точке начала кипения, находят
по уравнению
Р = SyzxzP’at = Sy*x*P?at,
(7.91)
в котором истинная температура — это температура,
соответствующая определенному давлению системы.
Поскольку все смеси вдоль соединительной линии име-
ют одинаковый состав и температуру, двухфазная
(жидкость — жидкость) область на поверхности темпе-
ратура начала кипения — состав (Т — х,) соответствует
единственной линии на поверхности точка росы — со-
став (Т — yi).
Прежде чем приступить к расчету испарения для
определенных смесей, рекомендуется определить темпе-
ратуру начала кипения и точку росы. В уравнениях
(7.87) и (7.88) основные неизвестные — это коэффициен-
ты фазового разделения а и /3, но поскольку коэффици-
енты распределения Ki и К* являются функциями
состава, их следует оценить вначале. Далее находят
значения а и /3 и подставляют в уравнения (7.83), (7.84),
(7.86) и (7.77), чтобы определить состав и, следователь-
но, уточнить значения Kt и АГ* и т. д. Такая методика
расчета использована в примере 7.17. Пример, показан-
ный на рис. 7.18, заимствован из статьи Маури [471].
Пример 7.16. Равновесие пар — жидкость — жидкость в двухкомпонентной системе
Зависимости, определяющие условия равновесия жид-
кость — жидкость при 30 °C в системе мета-
нол(1) + гептан(2) + морфолин(3) приведены в справочни-
ке DECHEMA (V/2, р. 97). Согласно этим данным, вза-
имная растворимость бинарной смеси метанол + гептан
соответствует составу (xi, х*) = (0,1681, 0,8835). Пара-
метры уравнения NRTL имеют следующие значения:
bgn/R = 565,80; bsn/R = 345,19; «и = 0,2.
Для этой бинарной системы будут построены диа-
граммы
1) зависимости энергии смешения Гиббса от состава xi;
2) зависимости коэффициентов активности от соста-
ва xi;
3) зависимости давление — состав при 30 °C и
4) диаграмма х — у.
Давление пара, мм. рт. ст.: Р° = 164,3, Рг = 58,7.
Используя указанные значения параметров уравнения
NRTL, получают: ти = 1,8661, m = 1,1385,
Gu = 0,6885, G2i = 0,7964. Уравнения для коэффициен-
тов активности после соответствующих подстановок
имеют такой вид:
, , ( 0,7221
IlTyi = Х2 I ----------------?
\ (X! + 0,7964х2)2
1,2848_____
(0,6885xi + хг)2
, -г ( 0,8846
ln?2 = Xi I ---------------Т
\(Х1 + 0,6885х2)2
0,9067
(0,7964x1 + хг)2
1. Точки касания касательной общей для двух точек
кривой на графике зависимости энергии Гиббса соот-
ветствуют (xi, X*) = (0,14 и 0,88), т. е. они приблизи-
тельно подтверждаются приведенными эксперимен-
тальными данными.
2. Полученные из приведенных выше уравнений коэф-
фициенты активности в диапазоне 0,14 < xi < 0,88 яв-
ляются величинами гипотетическими, так как гомоген-
ных композиций в указанном диапазоне не существует.
3. Давление находят из выражения Р = 71X1Р? +
+ V2X2P2 и представляют в виде зависимости от хь
График Р — yi строят исходя из зависимости
У1 = У1Х1Р1/Р.
Условия равновесия пар — жидкость — жидкость,
найденные из этих графиков, таковы: Р = 200 мм рт.
ст., yi = 0,73. Участки кривых Р — xi и Р — yi при
Р = 200 мм рт. ст. дают неравновесные значения.
4. Кривую зависимости х — у получают исходя из
выражения
Ji = 7iXiP?/(yiXiP? + 72Х2Р2).
Горизонтальный участок кривой при yi = 0,73 соответ-
ствует области сосуществования двух жидких фаз.
Штриховые участки кривой, построенной по указанно-
му уравнению, — неравновесные области.
Равновесие жидкость — жидкость 395
Проверка экспериментальных значений растворимости
метанола и гептана.
Зависимость значений коэффициентов активности от состава. Штрихо-
вые линии ограничивают двухфазную область, поэтому указанные зна-
чения не имеют в данном случае физического смысла.
Р, мм. рт. ст.
Диаграмма состав — давление при 30° С. Штриховые линии соответ-
ствуют двухфазной области, поэтому указанные значения давлений не-
реальны.
Диаграмма составов паровой и жидкостной фаз при 30°С. Горизон-
тальная линия, пересекающая штриховую кривую, соответствует ис-
тинному составу пара в двухфазной жидкостной области.
Равновесие жидкость — жидкость 397
Пример 7.17. Один из этапов расчета равновесий в системе пар — жидкость — жидкость для трехкомпо-
нентной смеси с двумя жидкими фазами
Фазовое разделение и состав будут определены из
пробных значений коэффициентов распределения (конс-
тант фазового равновесия) двух фаз.
Исходные данные таковы:
Компонент К, к‘
1 10 20
2 0,2 2
3 5 0,2
Графическое решение уравнений (7.87) и (7.88) показа-
но на рисунке. Исходя из коэффициентов фазового раз-
деления а = 0,475 и 0 = 0,470, определяем относитель-
ные количества трех фаз системы: V = 0,475, L - Q,2A7
и L* = 0,278. Следовательно, фазы должны иметь сле-
дующий состав:
Ком- понент X X* у у"
1 0,065 0,032 0,649 0,649
2 0,903 0,080 0,181 0,159
3 0,035 0,871 0,174 0,174
1,003 0,983 1,004 0,982
Прежде чем приступать к следующим попыткам, со-
став фаз следует нормализовать и использовать для
оценки Ki и К* по уравнению
= ъфГ'РГ'/ф.Р
и аналогичному уравнению для К*. Очевидно, что для
решения такого типа задач необходима компьютерная
программа.
Графический метод нахождения соотношений пар —
жидкость — жидкость для тройной смеси с известными
коэффициентами распределения.
7.8.3. Минимизация энергии Гиббса. В разд. 2.10 гл.
2 были рассмотрены условия равновесия при постоян-
стве величин некоторых взятых в виде различных ком-
бинаций независимых характеристик системы. При
независимых значениях Т и Р условия равновесия харак-
теризуются наличием минимальной величины энергии
Гиббса. Нахождение этого минимума — один из воз-
можных методов определения условий равновесия.
Простой вариант решения такой задачи приведен в при-
мере 7.9.
Для компонента паровой фазы
Gi = Gi + ЛТ1п(Л/Л) = Gi + ЛГ1п(ф^,/ф,), (7.92)
а для одного компонента жидких фаз
Gi = Gi 4- RTXn^/fi) = Gi 4- RT х{/ф{Р),
(7.93)
где Gi — энергия Гиббса чистого компонента, находя-
щегося в газообразном состоянии, которое принято за
эталонное состояние; величина эта одинакова как для
паровой, так и для жидкой фаз. Следовательно, фуги-
тивность чистых компонентов также одинакова для
обеих фаз, т. е. ft = ф,Р. Если одно и то же уравнение
состояния можно применить к обеим фазам, уравнение
(7.92) используют применительно к жидкой фазе с х,
вместо у,.
Если смесь содержит всего п, молей, в том числе р,
молей в паровой фазе и 4 и I* молей в двух жидких фа-
зах, общее значение энергии Гиббса смеси описывается
уравнением
ntG niGi
= 2 —— 4- Е Vi 1п(ф(у(/ф{)
К1 1
4- Е Ц \п(у{ф^Р^Xi/ф{Р)
4-Е Iflntyfy^P^x^iP). (7.94)
Если химических превращений в системе не происходит,
л,- = Vi 4- Ц 4- Z* (7.95)
Исходя из выражений, определяющих долю паровой
фазы,
а = Е г/(/Е Е Vilnt, (7.96)
соотношение жидких фаз
/? = Е Z,/(E Z,-4-Е ZJ), (7.97)
Энергию Гиббса можно представить в виде следую-
щей зависимости:
<7 ~ Е ZjGj
g~ RT
= аЕ У{ \п(ф{У{/ф{)
4- (1 - а)[/ЗЕ х{ 1п(у,ф?а1Р?а1х,/Ф,Р)
4- (1 - Д)Е xflntyfa^P^xfaiP)].
Это выражение можно упростить, если принять значе-
(7.98)
(7.99)
ния энергии Гиббса чистых компонентов за соответ-
ствующие характеристики газов в стандартных
условиях, когда фугитивность равна единице. В этом
случае уравнение (7.99) принимает вид
g =
G - Е ziG?
RT
(7.100)
Рис. 7.18. Поверхности зависимостей точки росы и тем-
пературы начала кипения от состава частично раствори-
мой трехкомпонентной смеси акрилонитрил — ацето-
нитрил — вода (Mauri, 1980). (© Am. Chem. Soc.).
Две поверхности в действительности соприкасающи-
еся вершинами треугольников, показаны раздельно,
чтобы упростить картину. Все составы вдоль конкрет-
ной соединительной линии имеют одну и ту же темпе-
ратуру начала кипения и равновесный состав пара, что
соответствует только одной точке на поверхности тем-
ператур точки росы. Семейство соединительных линий
соответствует единственной линии на поверхности тем-
ператур точки росы.
Равновесие жидкость — жидкость 399
= а£ у, 1п(0,Ру,)
+ (1 - «)[/ЗХ х,- ln(y,-0|atP-atx,)
+ (1 - Д)Е x*ln(y/0l?atPl?atx*)] (7.101)
где G? — это обычное значение энергии Гиббса образо-
вания из элементов, которое приводится в таких спра-
вочниках, как [130].
Для данного полного состава смеси, любой из коэф-
фициентов фазового разделения а или <3 можно исклю-
чить, если использовать уравнение материального
баланса
=ау, + (1- а)[рх{ + (1 ~ р)х?]
(7.102)
а мольную долю паровой фазы можно исключить, ис-
ходя из равновесных соотношений
У, = К{Х{ =
y,-0?atP?at
0,Р
(7.103)
Если отношение фТ^/ф, практически равно единице, это
можно сделать непосредственно, в противном случае
можно прибегнуть к методу итераций, поскольку ф, —
функция состава паровой фазы. И наконец, сумма
мольных долей жидких фаз, входящих в систему, долж-
на быть равна единице, т. е.
2 х,- — S х*= 0.
(7.104)
Задачи подобные нахождению минимума величины g
по уравнению (7.101) в условиях, ограниченных уравне-
нием (7.104), рассмотрены в гл. 10. В одной из методик
решения таких задач используются множители Лагран-
жа. Теория утверждает, что минимум функции
ф = g + X(S х, — S х*) — minimum, (7.105)
определяет также минимум системы, описываемой
уравнениями (7.101) и (7.104). Множитель Лагранжа X
в начале расчета неизвестен, его находят путем мини-
мизации энергии Гиббса одновременно с х„ х* и одним
из коэффициентов фазового разделения а или /3.
Производные ф относительно каждого из неизвест-
ных системы, приравненные к нулю, вместе с уравнени-
ем (7.104) составляют систему уравнений, из которой
неизвестные и множитель можно определить либо ме-
тодом Ньютона — Рафсона, либо аналогичным ме-
тодом!
Методов поиска экстремумов и решения нелинейных
уравнений в настоящее время разработано много.
Однако единого метода, в котором была бы исключена
возможность появления таких осложнений, как медлен-
ная или недостаточная сходимость, появление отрица-
тельных величин мольных долей на промежуточных
стадиях, или исчезновение на промежуточных же стади-
ях фаз, которые в начале расчета рассматривались как
наличиствующие, пока, по-видимому, не существует.
Для решения подобных задач необходима практика.
Простой пример минимизации энергии Гиббса рас-
смотрен в примере 7.9, а более сложные примеры разо-
браны в гл. 10.
7.8.4. Литература, посвященная расчетам равнове-
сия в многофазных системах. Методы расчета равнове-
сий многофазных систем тесно связаны с методами
расчета равновесий таких систем, в которых происхо-
дят многочисленные химические реакции.
Среди работ, посвященных второй из указанных про-
блем, внимания заслуживают в первую очередь книги
Голуба и Вонки [58], Розенброка и Сторея [111], ван Зег-
герна и Сторея [136], а также Смита и Миссена [123].
Много статей и обзоров по указанной проблематике
опубликовано сравнительно недавно, и мы остановимся
на некоторых из них. Список публикаций, в которых
обсуждаются чисто химические равновесия, приведен в
гл. 10.
1. Кастилло и Гроссман [216]. Нелинейный програм-
мированный метод выявления идентичности фаз в
условиях одновременного фазового и химического
равновесия.
2. Дим и Мэддокс [243]. Рассчитаны условия равнове-
сия в трехфазной системе с независимым от соста-
ва коэффициентом распределения.
3. Джордж и др. [298]. Минимизацией энергии Гиббса
по методике Пауэлла найдены фазовые составы.
Разработанная программа позволяет оперировать с
четырьмя фазами и пятнадцатью компонентами.
4. Хейдеман [341]. Рассчитано трехфазное равновесие
в трехфазных водных и нефтяных смесях при помо-
щи модифицированного уравнения Редлиха —
Квонга, применимого к жидким и паровым фазам.
5. Хенли и Розен [54] (гл. 8). Рассмотрены как фазо-
вые, так и химические равновесия. Приведены алго-
ритмы решения в виде блок-схемы для
многожидкостного испарения при фиксированных Т
и Р или Н и Р.
6. Лахири [419]. Методика Ньютона — Рафсона при-
менена для нахождения минимума энергии Гиббса,
а следовательно, и фазовых составов при высоко-
температурных металлургических процессах.
7. Маури [471]. Решена система уравнений, включаю-
щая уравнения материальных балансов и равнове-
сия трех сосуществующих фаз. Описаны расчеты
точки росы, точки начала кипения и испарения.
8. Пенг и Робинсон [546]. Для расчета равновесия в
водных и углеводородных системах применено раз-
работанное этими авторами уравнение состояния.
Непосредственно использован критерий равенства
парциальных фугитивностей индивидуальных ком-
понентов во всех фазах.
9. Сандерсон и Чиен [607]. Приведены отношения,
описывающие фазовые и химические равновесия,
результирующие нелинейные уравнения решены ме-
тодом Марквардта.
10. Готэм и Сейдер [297]. Проведено сравнение не-
скольких методов расчета на ЭВМ фазового и хи-
мического равновесия смесей, включая смеси
электролитов. Сделаны некоторые выводы относи-
тельно пригодных методов расчета.
11. Сейдер, Готэм и Уайт [627]. Обзор последних пуб-
ликаций.
12. Росс и Сейдер [600]. Разработана схема моделиро-
вания трехфазной дистилляционной колонны.
13. Тиноко-Гарсиа и Кано-Домингуез [687]. Для реше-
ния систем уравнений, описывающих равновесия
жидкость — жидкость и пар — жидкость примене-
на итерационная подгонка значений фугитивностей
индивидуальных компонентов во всех фазах.
14. Нидзвески и др. [519]. Описана трехфазная дистил-
ляция углеводородов в присутствии воды.
7.9. Равновесия в системе пар—жидкость—
твердое вещество
Смеси веществ с широким диапазоном летучести мо-
гут сосуществовать в равновесии как три фазы пар —
жидкость — твердое вещество. Например, в природных
газах при низких температурах твердой фазой может
быть СОг, НгБ, тяжедые углеводороды или гидраты.
В таких случаях обычно принято считать твердую фазу
чистой, что до некоторой степени упрощает расчеты.
Кроме того, фугитивности можно определить из таких
уравнений состояния, как уравнения Бенедикта — Уэб-
ба — Рубина, Соава или Пенга — Робинсона. В услови-
ях равновесия парциальные фугитивности отдельных
компонентов равны друг другу
7Г=Л=Л, (7106)
где парциальная фугитивность твердой фазы равна фу-
гитивности чистого компонента и определяется урав-
нением
f. =0?atp?at(PF),- (7.107)
в которое входят давление пара и коэффициент Пойн-
тинга. Фугитивности жидких фаз получают непосред-
ственно из уравнения состояния, например из уравнения
Соава [табл. 3.4, уравнение (22)]
bi ( b \
In ф,= — (z - 1) - Inz I 1 - — I
О \ V j
Из этого уравнения коэффициенты для паровой фазы
получают после подстановки наибольшего значения
сжимаемости z, а для жидкой фазы — после подстанов-
ки трех наименьших корней уравнения Соава.
Условия, при которых твердая фаза начинает выде-
ляться из данной по составу жидкой фазы, находят,
сравнивая фугитивность твердой фазы, рассчитанную
по уравнению (7.107), с полученной для того же компо-
нента в паровой фазе по уравнению (7.108). Давление
или температура согласовываются до тех пор, пока эти
условия не будут удовлетворены. На рис. 7.19 дана об-
щая схема расчета. Если фугитивность твердой фазы
меньше, чем парциальная фугитивность паровой фазы,
возможно выпадение осадка. Для того чтобы опреде-
лить количество образовавшейся твердой фазы, умень-
шают в жидких фазах количество компонента,
образующего твердую фазу и повторяют расчет. Схема
проведения расчета показана на рис. 7.20. Методика
осуществления таких операций описана Пентом и Ро-
бинсоном [546].
Рис. 7.19. Температура в момент начала отвердевания
в равновесных системах пар — жидкость — твердая фа-
за (г, — суммарный состав).
Равновесие жидкость — жидкость 401
Рис. 7.20. Состав сжиженной фазы при заданных темпе-
ратуре и давлении в равновесных системах пар — жид-
кость — твердая фаза (л, — число молей г, ns — число
молей, способных к отвердеванию в жидкой фазе).
7.10.3адачи
7.1. Приращение энергии Гиббса смеси как функция
температуры (К) описывается выражением
Gn/RT = [2 + 0,009(Т - 31О)]Х1Х2.
Постройте график зависимости взаимной растворимос-
ти от температуры.
7.2. Зависимость параметра А симметричного уравне-
ния коэффициентов активности от температуры (К)
определяется следующими выражениями:
. = <2 + 0,0016 (Т - 325)2, Т < 325,
|^2,4 - 0,0016 (Т - 325)2, Т > 325.
Постройте график зависимости взаимной растворимос-
ти компонентов от температуры.
7.3. Зависимости параметров уравнения ван Лаара от
температуры описываются следующими выражениями:
Л12 = 2 + 0,009 (Г - 310),
Л21 = 3 — 0,008 (Т - 310).
Постройте график зависимости взаимной растворимос-
ти от температуры.
7.4. Применяя алгебраический метод касательной об-
щей для двух точек кривой, уравнения (7.24) и (7.25),
определите равновесные составы по уравнению Маргу-
леса с Л = 3 и В = 2,5, если
g = Х1Х2(ЛХ2 + Bxi) + Х11ПХ1 + х21пх2,
dg/dxi = Лх2(1 - 3xi) + Bxi(3x2 - 1) + ln(xi/x2)
Чтобы упростить определение исходных оценок рав-
новесных составов, на рисунке показаны кривые, опи-
сывающие зависимость этих уравнений.
7-864
7.5. Необходимые для расчета равновесия системы
гексан(1) + этанол(2) + ацетонитрил(3) при 40 °C данные
и зависимости получены авторами работы [668]. Ис-
пользуя установленные ими величины параметров урав-
нения Цубоки — Катаямы — Вильсона, определите при
помощи программы В-3 число соединительных линий и
сравните их с экспериментальными [668] соединитель-
ными линиями и бинодальной кривой.
Л12 = 0,0818; Л21 = 0,4308; Лп = 0,1002;
Аз1 = 0,1641; Л23 = 0,4159; Л32 = 0,5677.
Координаты бинодальной кривой
XJ х2 Х1 х2 XJ х2
0,0857 0,0000 0,1966 0,2682 0,5118 0,2142
0,0943 0,0410 0,2525 0,2884 0,5595 0,2005
0,1077 0,0971 0,3040 0,2832 0,6092 0,1723
0,1213 0,1614 0,3525 0,2680 0,6931 0,1255
0,1181 0,2060 0,3845 0,2578 0,7587 0,0848
0,1657 0,2414 0,4315 0,2422 0,9015 0,0000
Координаты соединительной линии
Правая ветвь Левая ветвь
Х1 х2 Х1 х2
0,8831 0,0166 0,0968 0,0879
0,8674 0,0251 0,1003 0,1299
0,8546 0,0332 0,1054 0,1622
0,8372 0,0433 0,1250 0,1942
0,8101 0,0567 0,1367 0,2169
0,7821 0,0772 0,1522 0,2331
0,7272 0,1086 0,2053 0,2695
0,6912 0,1269 0,2249 0,2748
7.6. Следуя методике, принятой в примере 7.6, опре-
делите равновесные составы по уравнению Цубоки —
Катаямы — Вильсона при 1п7~ = 4, 1п72 =3 и
Ki/K2 = 2. Рассчитанные по этим данным параметры
имеют следующие величины: Л12 = 0,0579 и
Л21 = 0,1566. Для пробной оценки составов можно ис-
пользовать зависимость Gn/RT + Sx/lnxi, приведенную
ниже на рисунке.
7.7. Взаимная растворимость н-бутанола и воды оце-
нивается значениями xi = 0,9765 и х* = 0,6770. Опреде-
лите параметры уравнений Маргулеса, ван Лаара и
Цубоки — Катаямы — Вильсона (Ответ. Параметры
уравнения ван Лаара: Ап = 0,334, Л21 = 1,60.)
7.8. Для смеси с объемным соотношением Ki/K2 = 2
параметры уравнения Вильсона имеют следующие зна-
чения: Ап = 0,2 и Д21 = 1,5. Оцените величины пара-
метров уравнения Цубоки — Катаямы — Вильсона.
7.9. Для системы анилин(1) + метилциклогексан(2):
7“ = 16,1; у? = 11,14, И1/И2 = 0,7137. Определите вза-
имную растворимость этих соединений по уравнениям
ван Лаара и Цубоки — Катаямы — Вильсона, исполь-
зуя методики, описанные в примерах 7.3 и 7.5.
7.10. Имеется равновесная двухкомпонентная система
жидкость — жидкость. Покажите, что состав одной
фазы можно определить исходя из коэффициентов ак-
тивности обоих компонентов в обеих фазах:
Х1 71(72 - 72)
7172 - 717г
7.11. Смесь СНС13 и C7F16 при 60 °C имеет следую-
щие характеристики: взаимная растворимость xi = 0,1
и х* = 0,8. Параметры растворимости, (кал/мл):
61 = 9,26 и <52 = 5,9. Молярные объемы, мл/моль:
Ki = 80,7, V2 = 227,3.
1. Определите коэффициенты активности при беско-
нечном разбавлении.
2. Определите взаимную растворимость по уравнению
Скэтчарда — Гильдебранда.
3. Определить взаимную растворимость по уравнени-
ям ван Лаара, Маргулеса, NRTL (для ап = 0,2 и
c*i2 = -1) и Цубоки — Катаямы — Вильсона, ис-
пользуя одно и то же значение 1п7“.
7.12. Постройте графически соединительную линию
исходя из приведенных ниже данных и определите со-
став фаз, находящихся в равновесии с фазой, содержа-
щей 10, 20 или 30 мол.% ацетальдегида. Состав в точке
полного смешения частично смешивающихся жидко-
стей: х = 0,33 их = 0,49.
вода ’ ацетальдегид ’
(1) С4Н10О диэтиловый эфир,
(2) СгЩО ацетальдегид,
(3) Н2О вода.
Температура = 15,1 °C, тип системы = 1.
Экспериментальные значения координат (мол. %) со-
единительных линий
Левая фаза Правая фаза
(D (2) (3) (1) (2) (3)
76,700 15,500 7,800 2,500 8,300 89,200
63,700 27,400 8,900 3,300 17,500 79,200
56,600 32,700 10,700 4,500 23,800 71,700
46,100 40,600 13,300 5,700 29,500 64,800
37,300 46,100 16,600 7,000 34,600 58,400
Задача составлена по данным работы Suska J. Collect.
Czech. Chem. Commun., 44, 1999 (1979).
7.13. В работе Хэнда [Hend J. Phys. Chem., 34, 1961
(1930)] даны графики зависимости
1о®(Храств. в-во/Храстворитель 1> фйЗЫ’ обогащенной раСТВО-
рителем 1, от log(x /х ,) фазы, обога-
r раств. в-во растворитель 2' * ’
щенной растворителем 2. Подобная зависимость часто
имеет линейный характер и может применяться для на-
хождения экспериментальных ошибок. Проведите по-
добный анализ данных предыдущей задачи (7.12) и с
этой целью постройте зависимость log(x2/xi) для левой
фазы от log(x2/x3) для правой фазы.
Равновесие жидкость — жидкость 403
К задаче 7.6
7.14. В справочнике DECHEMA (DECHEMA LLE Da-
ta Collection, V/2, p. 326) дана информация о системе
октан(1) + 2,4,4-триэтилпентан(2) + нитроэтан(З). Взаим-
ная растворимость пар компонентов оценивается так:
Xi = 0,81, х3 = 0,19, Х2 = 0,20, Хз = 0,80.
Известна одна тройная соединительная линия. Фаза 1:
xi = 0,48157, х2 = 0,30806, х3 = 0,21037, фаза 2:
xi = 0,08476, х2 = 0,05863, х3 = 0,85661.
Используя уравнение NRTL (cm = 0,2), определите 1)
713 и тз1 из данных о бинарных смесях; 2) пз и 732 из
данных о бинарных смесях; 3) тп и 721 из данных о
тройной системе.
7*
7./5. В оозоре [109] приведены следующие величины параметров уравнения NRTL для смеси циклогек- сан(1) + фурфурол(2) при 30 °C: тц = 2,2531, Т, °C 10’4 Н, атм/мол. доля
ти = 1,3772, ан = 0,1973. Определите взаимную рас- п 2 24
творимость компонентов при этой температуре.
(Экспериментальные значения: (0,15, 0,94).) 1U 2,97
20 3,76
7.16. В приведенной ниже таблице указана взаимная 30 4,49
растворимость компонентов смеси — равновесные 40 5,20
мольные доли в обеих фазах первого из компонентов 50 5,77
смеси. (Там же даны номера страниц справочника 60 6,26
DECHEMA (DECHEMA LLE Data Collection, V/2), от- 70 6,66
куда эта информация заимствована. 80 6 82
Определите параметры уравнения Маргулеса и пара- 90 6 92
метры ти и 721 уравнения NRTL при ац = 0,2. 100 7*01
Система Х1 xj Т, °C Страница справочника
4,4-Диметил-1,3-диоксан + вода 0,80369 0,03590 20 59
Нитрометан + вода 0,93873 0,03039 26,7 66
1-Нонанол + нитрометан 0,82952 0,01821 20 72
Метанол + гексан 0,88637 0,10361 5 86
Метанол + гексан 0,80164 0,23226 25 88
Циклогексан + метанол 0,87090 0,17520 25 115
2-Бутанон + вода 0,63617 0,07311 26,7 218
Циклогексан + 1,2-диаминоэтан 0,90960 0,07050 40 443
Циклогексан + 1,2-диаминоэтан 0,80680 0,133310 60 444
3-Метил-1-бутанол + вода 0,65418 0,00517 25 535
7.17. Коэффициенты уравнения Антуана для метано-
ла и гептана имеют следующие значения:
А В С
Метанол 18,5875 3626,55 —34,29
Гептан 15,8737 2911,32 -56,51
Постройте диаграммы Т — xi и Т — yi для этой смеси
при Р = 400 мм рт. ст. Все необходимые данные приве-
дены в примере 7.16.
7.18. Покажите, что включение множителя С в урав-
нение Вильсона для определения избыточной энергии
Г иббса
Ge*/RT = -C[xiln(xi + Ахг) + x?ln(Bxi + хг)]
позволяет согласовать требование выпуклости кривой
d2G/dxj < 0 с, некоторыми значениями А, В и xi, если
С # 1:
G = Gex + /?T(xilnxi + хДпхг).
7.19. Постоянная Генри Н = Р/х для системы
НгБ + вода зависит от температуры (данные Int. Crit.
Tilbies, 3, 259).
а. Определите константы уравнения Валентинера
1пЯ = А + В1пГ + С/Т,
при помощи которых можно наиболее точно предста-
вить эти данные как для всего диапазона температур,
так и в отдельных областях.
б. Определите также константы уравнения Гильде-
бранда
1пЯ = А + В/(Т + Q.
7.20. Ниже представлена взаимная растворимость и
параметры уравнений NRTL и UNIQUAC при несколь-
ких температурах (DECHEMA, V/1, р. 205) для системы
3-бутен-2-он(1) + вода(2). Покажите, что эти данные со-
вместимы с условиями yiXi = 7*х*. Параметры уравне-
ний: ти = 7*х* (уравнение UNIQUAC) и ту = kgy/R
(уравнение NRTL с а = 0,2).
°C *1 X? UNIQUAC NRTL
*12 *21 *12 *21
/?1 = 3,0178 61 = 2,664
*2 = 0,9200 62 = 1,400
30,0 18,7 66,8 194,17 26,799 -158,67 967,16
40,0 13,2 59,7 177,67 50,291 -166,13 1038,2
50,0 12,3 58,6 175,50 59,128 -174,13 1084,2
60,0 11,8 60,2 148,64 81,307 -207,84 1157,4
70,0 12,0 64,8 97,844 119,48 -268,92 1256,6
80,0 13,1 74,4 -7,8069 214,27 -393,29 1435,0
7.21. Смесь содержит 10% СОг и равные количества
метана и этана при 27,2 атм. Используя уравнения Со-
ава, определите температуру начала образования твер-
дой фазы. Согласно данным Пенга и Робинсона [Adv.
Chem. Series, 182, p.p. 185—196, (1979)], это произойдет
при температуре около 177 К.
7.22. Найдите аналитическое решение дифференциаль-
ного уравнения, приведенного в примере 7.2, для
/(xi) = 1 при xi = 0,05. График этой зависимости пока-
Равновесие жидкость — жидкость 405
К задаче 7.22
зан на рисунке.
(Ответ. f(x) = 1--- [1,5876 + 5000(х6/6 - 2,5x75
+ 2,Зх4/4 - 0,95x73 + 0,1689x72 - 0,00945х)].)
7.23. Постройте изотермическую диаграмму х — у
смеси при Pi = 3 и Рг = 1 и следующих величинах па-
раметров уравнения Маргулеса
а) А = 2,5, В = 0,5; б) А = 4,0, В = 0,5.
Определите предельную растворимость по построен-
ным кривым. Обратите внимание, что известны приме-
ры систем с частичной растворимостью компонентов,
которые а) не образуют азеотропов, б) образуют гете-
рогенные и гомогенные азеотропы.
Смеси вода + метилэтилкетон и вода + фенол — реаль-
ные примеры смесей типа а.
7.24. Покажите, что величина энергии Гиббса чистых
компонентов бинарной смеси не оказывает влияние на
состав, найденный по методике касательной, общей для
двух точек кривой, т. е. покажите, что
ga = %Xig° + SXilnX; + g“
И
gb = Sxjnxj + gex
имеют одни и те же абсциссы касательной, общей для
двух точек кривой.
7.25. Для зависимости f (xi) (см. задачу 7.22) найдите
методом подбора замену для числа 5000, которая при-
ведет к нахождению касательной, общей для трех точек
кривой, g = /(Xi).
7.26. Состав бинарной смеси при ограниченной мета-
стабильности определяется уравнением d2g/dxl = 0, где
g = (G - SxiG?)/KT = Ga/RT + Sx/lnxi.
Графическая зависимость таких составов от темпера-
туры называется спинодальной кривой. Постройте та-
кую кривую, приняв, что приращение энергии Гиббса
определяется уравнением Маргулеса, параметры кото-
рого зависят от температуры:
Ge*/RT = XiX2<Bxi + Лхг),
где А = 700/Г и В = 1000/Т (Т измеряется в кельви-
нах). Покажите также стабильные границы раствори-
мости. .Перенесите эти результаты на температуру
начала фазового раздела.
7.27. Исходя из графиков, приведенных в примере 7.7
(рис. а и б), можно сделать следующие выводы:
a) g имеет минимакс на верхней кривой при
71X1 = 72X2 на нижних кривых;
б) минимаксы 71X1 и 72X2 наблюдаются при одинако-
вых значениях xi;
в) минимаксы 71X1 и 72X2 встречаются при одинако-
вых значениях xi как точки перегиба g-кривой.
Докажите правильность этих выводов аналитически
при помощи уравнения Гиббса — Дюгема.
8.1. Введение
Из числа возможных видов равновесия между жид-
костями и твердыми веществами наиболее важны
два — равновесие растворения и равновесие плавления.
1. Равновесие растворения имеет место между раз-
личными жидкими и твердыми химическими компо-
нентами.
2. Равновесие плавления имеет место между рас-
плавленными и твердыми формами одних и тех же хи-
мических компонентов.
Количественно характер изменения равновесия вы-
ражается через коэффициенты активности. В принципе
коэффициенты активности жидкой фазы, выведенные
исходя из результатов измерения равновесия пар —
жидкость или жидкость — жидкость, применимы и к
жидким фазам при рассмотрении равновесия жид-
кость — твердое вещество, если они скорректированы
по температуре. На практике они достаточно точны для
описания растворимостей, однако менее пригодны для
равновесия плавления, поскольку в последнем случае
может наблюдаться воздействие усложняющих факто-
ров. Укажем некоторые из них.
1. В зависимости от температуры и давления твер-
дые вещства могут находиться в нескольких кристалли-
ческих формах, как, например, кристаллическая вода
(рис. 5.11) или система, показанная на рис. 5.25,ж.
2. Изучение равновесия плавления может ослож-
няться присутствием межмолекулярных соединений,
что, в частности, характерно для смесей ССЦ со многи-
ми углеводородами.
3. Твердые фазы могут быть или практически чис-
тыми, или ограниченно смешанными.
4. Температуры равновесия плавления могут выйти
за пределы разумной экстраполяции результатов изме-
рений равновесия пар — жидкость или жидкость —
жидкость.
Из-за указанных выше и, возможно, других причин
поведение систем расплав — твердая фаза может быть
более сложным, чем даже поведение систем, состоящих
из нескольких жидких и паровой фазы. Некоторые из
перечисленных выше особенностей систем твердая фа-
за — расплав положены в основу классификации равно-
весия плавления; так, в табл. 5.1 и 5.2 основным
критерием идентификации являются образование соеди-
нения и растворимость (смешиваемость) твердой фазы.
Данные о равновесии плавления представляют наиболь-
ший интерес для тех областей технологии, которые свя-
заны с применением металлов и их сплавов, кера-
мических материалов и водных систем неорганических
солей; однако довольно много такого рода данных со-
брано и для органических систем. Самая обширная
сводка этих данных содержится в обзорах Ландольта-
Бернштейна (П/2Ь, 1962; П/2с, 1964; П/З, 1956) [73].
Различные типы и несколько важных примеров фа-
зовых диаграмм двух- и трехкомпонентных систем
представлены в гл. 5 (рис. 5.21—5.26 и рис. 5.40—5.41
соответственно) и в данной главе. Прекрасные стерео-
скопические рисунки многих тройных систем выполне-
ны Тамасом и Палем [132], а в книге Принца [103] даны
очень четкие цветные рисунки.
До сих пор попытки выразить равновесие плавле-
ния через термодинамические принципы и данные пред-
принимались в большинстве случаев лишь применитель-
но к простейшим типам этого равновесия; широкое рас-
пространение получила лишь классификация, основан-
ная на правиле фаз. Для описания сложных ситуаций,
связанных с образованием твердых растворов, исследо-
ватели металлургических процессов привлекают теорию
регулярных растворов; ссылки на работы такого типа
приведены Луписом [78]. Ценные результаты может
дать теоретическое рассмотрение ряда промышленно
важных органических систем.
8.2. Растворимость твердых веществ
в жидкости
Если растворитель не входит в твердую фазу, фуги-
тивность твердого растворенного вещества остается
равной фугитивности чистого твердого вещства, так
что условие равенства парциальных фугитивностей при
равновесии можно записать следующим образом:
Z?(solid) — -A) solid) — У2хif2(subcooled liquid)- (8-1)
(s — solid — твердое вещество, scl — subcooled liquid —
переохлажденная жидкость).
Упростим выражение (8.1)
f2s
х2 = ~ ,
Y2/2(scl)
где Х2 — мольная доля растворенного твердого веще-
ства в растворе и /2(SCi) — фугитивность чистого раство-
ренного твердого вещества в состоянии переохлажден-
ной или гипотетической жидкости, температура кото-
рой ниже его точки плавления. На диаграмме, приве-
(8.2)
Равновесие жидкость — твердое вещество 407
денной на рис. 4.1, показано местонахождение такой
переохлажденной жидкости (точка Ь).
Отношение фугитивностей твердого веще-
ства и переохлажденной жидкости можно выразить че-
рез условия, соответствующие тройной точке. Если
фундаментальное уравнение
АН АГ
d 1п/= - —7 dT + л“ dP (8.3)
RT2 RT
1
— ехр
72
&S'p
~^-Т,р/Т)
(8.10)
применить к каждой из фаз и вычесть полученные ре-
где = ДН(р/Т(р — энтропия плавления в тройной
точке.
3. Поскольку температура тройной точки обычно
очень близка к температуре плавления при атмосфер-
ном давлении, а последняя известна гораздо чаще (см.,
например, табл. 3.5), уравнение растворимости после
зультаты, то получим
подстановки принимает вид
f2S
d In
•'Sfscl)
HL - Hs yL - vs
dT — _ dP.
RT2 RT
(8-4)
Практически можно принять, что разность удельных
объемов конденсированных фаз не зависит от давления,
однако энтальпия плавления Hl - Hs может заметно
изменяться с температурой. Такое поведение описыва-
1
х2 = ехр
72
1
= ехр
72
— (l-Tm/T)
л
(8.11)
ется при помощи уравнения
HL-Hs = (HL-Hs)'p+ ГТ (CpL-Cps)dT (8.5)
гт J ,р
= АН1р + / \CpdT (8.6)
JTtP
— АН[р + &Ср(Т — Ttp), (8.7)
где индекс tp означает тройную точку. Выражение (8.7)
применимо в случае, когда разность теплоемкостей не
где индекс m означает условия, отвечающие точке плав-
ления при атмосферном давлении.
4. Компактное уравнение растворимости, требую-
щее знания только свойств чистых компонентов, полу-
чено исходя из уравнения Скэтчарда — Гильдебранда
для коэффициентов активности растворенного ве-
щества:
зависит от температуры.
После подстановки уравнения (8.7) в уравнение (8.4)
полученное выражение интегрируют в пределах от (Ttp,
Ptp) до температуры и давления системы (Г, Р). Резуль-
тирующее отношение фугитивностей принимает сле-
дующий вид:
^201(^1 - g2)2
RT
где 0i — объемная доля растворителя,
01 = ^1Х 1/(^1* 1 + ^2*2)-
(8-12)
(8.13)
|П-^-
/2(scl)
/_J_
R \Т‘Р
&CP / Ttp \ AK
R (iny-^/r+1
(8.8)
Подстановка этого соотношения в уравнение (8.2) при-
водит к общему уравнению растворимости:
Иногда поправка Флори — Хаггинса может дать луч-
ший результат, однако для неуглеводородных смесей,
приведенных в примере 8.4, ни она, ни основное уравне-
ние Скэтчарда — Гйльдебранда, не вполне приемлемы.
Смешанные растворители можно представить при по-
мощи многокомпонентной формы уравнения Скэт-
чарда — Гильдебранда.
5. Вариант уравнения растворимости для идеаль-
ных растворов с коэффициентами активности, равными
единице,
" &Нт / 1 1 \ '
х 2 — ехр I — I
2 L R \Тm Т /
ЛСр
R
Ttp
\n~^-Ttp/T+\
АГ
(8.9)
Иногда правомерны одно или несколько из следующих
упрощений этого уравнения.
1. Поправкой на давление, как правило, можно пре-
небречь.
2. Вклад разности теплоемкостей, хотя и более су-
ществен, чем поправка на давление, тем не менее часто
он также невелик и им можно пренебречь, при этом
уравнение растворимости можно записать так:
1 Г АН,р /1 1 \
ехр I — ~ I
Г2 [ R \Т‘Р Т)
связан с именами Шредера (1893) и ван Лаара (1908)
[48], а в основе его лежат уравнения Клаузиуса — Кла-
пейрона <1850, 1834) или Вант-Гоффа.
Уравнение (8.13) и другие варианты уравнения рас-
творимости применимы также к равновесию плавления.
Если коэффициенты активности не учитываются, точ-
ность его недостаточна. Например, как показано на рис.
8.16, идеальная растворимость нафталина равна 44,10b,
однако действительное значение растворимости зависит
от природы растворителя и в этом примере изменяется
от 4,4 до 44,10о. Для других систем, приведенных на
рис. 8.1, наблюдается сходная картина.
Природа растворителя, энтальпия и температура
плавления растворенного вещества являются основны-
Параметр растворимости растворителя
Рис. 8.1а. Растворимость антрацена в растворителях с различными
параметрами растворимости.
Соединение 5 Соединение 6
Антрацен 9,9 Циклогексан 8,19
Нафталин 9,38 Циклогексанол 11,4
Фенантрен 10,52 Диэтиловый эфир 7,54
Уксусная кислота 10,05 Этанол 12,92
Ацетон 9,51 н-Гексан 7,27
Анилин 11,46 Метанол 14,51
1 -Бутанол 11,44 Фенол 12,11
Сероуглерод 9,86 1-Пропанол 12,05
Тетрахлорид углерода 9,34 2-Пропанол 11,57
Хлороформ 9,24
50
Параметр растворимости растворителя
Рис. 8.16. Растворимость нафталина (о) и фенантрена
(д) в растворителях с различными параметрами рас-
творимости. Показаны идеальная растворимость этих
соединений и кривая, лучше всего соответствующая экс-
периментальным данным.
Равновесие жидкость — твердое вещество 409
ми факторами, влияющими на растворимость. Даже
химически похожие вещества могут существенно разли-
чаться по своим характеристикам при плавлении и со-
ответственно по растворимости. Приведем в качестве
примера величины разности энтальпии плавления, тем-
пературы плавления и идеальные растворимости трех
изомеров хлорнитробензола при 300 К.
Изомер &Нт, кал/моль Тт, К X
орто 4,546 307,5 0,8303
мета 4,629 317,6 0,6503
пара 4,965 356,7 0,2661
Поскольку коэффициенты активности изомеров в
одном и том же растворителе часто имеют примерно
одинаковые величины, расчеты идеальных растворимо-
стей могут указать тенденции в изменении раствори-
мости, хотя возможно, что полученные при этом
значения не будут абсолютно точными.
Влияние разности теплоемкостей ДСР иллюстриру-
ют результаты определения растворимости пара-
ксилола в фиксированном интервале температур, полу-
ченные в задаче 8.1.
Ясно, что вклад теплоемкости в вычисленную рас-
творимость мал при умеренных отклонениях от темпе-
ратуры плавления, равной 286,39 К.
Т, ° К X
с учетом ДСр без учета ДСР
200 0,0558 0,0448
250 0,3616 0,3513
286,39 1 1
В тех случаях, когда давление паров невелико, его
в уравнении (8.2) можно заменить на фугитивность.
Давление паров твердого вещества должно быть из-
вестно, а давление переохлажденной жидкости можно
найти экстраполяцией кривой давления паров или по-
средством уравнения (8.2). В результате уравнение (8.2)
принимает такой вид:
^2 = ^at/y2^lat. (8.15)
Если экстраполяция уравнения давления паров прово-
дится в умеренных пределах, точность упрощенного ме-
тода может быть достаточно высокой. Это показано в
примерах 8.2 и 8.3, где найдено, что растворимость на-
фталина в бензоле составляет примерно 20%.
8. 2.1. Влияние температуры и давления. Уравнение
(8.14) можно записать в такой форме:
\пх2=А~В/Т, (8.16)
где и Л и В имеют в интервале средних температур
практически постоянные значения, характерные для
определенных смесей. Это широко известная форма
уравнения, характеризующая температурную зависи-
мость других свойств, например давления паров (выво-
дится из уравнения Клаузиуса — Клапейрона) и
вязкости, а также растворимостей газов и жидкостей.
Емкое представление растворимостей в виде номограм-
мы, основанной на уравнении (8.16), дано авторами
статьи [300]. Хотя уравнение (8.14) для идеальных рас-
творов предсказывает, что растворимость увеличивает-
ся с повышением температуры, влияние температуры
на коэффициент активности может, согласно уравнению
(8.11), привести к противоположному эффекту. Влияние
температуры, подобное показанному на рис. 5.20,а — ж
на примере смесей жидкость — жидкость, возможно и
для смесей жидкость — твердое вещество, однако труд-
но найти конкретные примеры.
Воздействие давления на растворимость в первую
очередь выражается через член ( Vl - К )/RT уравнения
(8.9), хотя изменение давления может также влиять и
на коэффициент активности. Объединяя уравнения (8.2)
и (8.4) и принимая температуру постоянной, получаем
VL ~ Vs
t/lny2x2 = ------dP. (8.17)
ix. 1
Если ввести
VL ~ VL
d\ny2 = ’ dP, (8.18)
i\ 1
указанное в табл. 4.1, выражение, определяющее зави-
симость растворимости от давления, принимает сле-
дующий вид:
д lnx2 _ _ VL-VS _VL-VL __ Vl-Vs 19)
дР RT RT RT
Последнее выражение из этой серии уравнений применя-
ется в тех случаях, когда парциальный молярный объем
растворенного вещества является по существу тем же
самым, что и для переохлажденной жидкости; подобная
аппроксимация может быть допустима и для разбавлен-
ных растворов. Слабое влияние давления показано в за-
даче 8.3, в которой dx/dP = -2,7(Е - 4) мол.
доли/атм и на примере гидрохлорида гидроксиламина
(рис. 5.28), у которого коэффициент давления равен
-0,4(Е - 4) при температурах выше 40° С и
+ 1,34(Е - 4) при температурах ниже указанной. Эффек-
ты влияния действительно высокого давления проиллю-
стрированы рис. 8.7.
8. 2.2. Смешанные растворители. В идеальных слу-
чаях состав растворителя не влияет на растворимость,
однако обычно она зависит от коэффициентов актив-
ности, которые в свою очередь зависят от природы рас-
творителя. Использование многокомпонентной формы
уравнения Скэтчарда — Гильдебранда применительно к
смешанным растворителям демонстрируется в примере
8.2. При наличии экспериментальных данных предпо-
чтительнее пользоваться уравнением NRTL или
Вильсона.
8. 2.3. Несмешивающиеся растворители. При испо-
льзовании уравнения Скэтчарда — Гильдебранда отно-
шение концентраций растворенного вещества в двух
жидких фазах (одна из которых помечена звездочкой)
записывается так:
V2
= «Ф
IX. 1
(8.21)
Концентрация растворенного вещества
в водной фазе, масс, доли
Рис. 8.2а. Коэффициент распределения муравьиной кис-
лоты между водой и бензолом при нескольких темпера-
турах [73].
моль растворенного в-ва/л в водной фазе
/Г — ----------------------------------------.
моль растворенного в-ва/л в бензольной фазе
Концентрация растворенного вещества в водной фазе,масс?/.
Рис. 8.26. Коэффициенты распределения спиртов между
водой и циклогексаном при 25°C [73].
моль спирта/л в водной фазе
моль спирта/л в циклогексановой фазе
где
01 = Г1Х1ДК1Х! + К2х2), (8.22)
0f= ГУх?/(Г?х?+К?х?). (8.23)
Поскольку коэффициенты активности зависят от соста-
ва, то и коэффициент распределения К также обычно
зависит от состава, хотя для разбавленных растворов
или для растворов с узким диапазоном изменения кон-
центраций часто допустимо принимать К постоянным
(К = хг/хг). На рис. 8.2, например, К метанола являет-
ся постоянным в широком диапазоне концентраций,
однако К этанола и муравьиной кислоты не постоянны.
При использовании симметричного уравнения Мар-
гулеса коэффициент распределения определяется следу-
Равновесие жидкость — твердое вещество 411
ющим образом:
х = х2/** = у!/у2
= (уГ/У2)ехр[(1 - х£)2 - (1 -х2)2! (8.24)
— К" ехр[(1 ~х2)2 - (1 -Хх^)2], (8.25)
где
к -у 2/Y2- (8.26)
Несколько возможных вариантов графического изобра-
жения дано в примере 8.1.
8. 2.4. Коэффициенты активности. В гл. 4 показа-
но, что коэффициенты активности связаны с нескольки-
ми различными типами уравнений, одно из них — урав-
нение Скэтчарда — Гильдебранда — использовалось в
качестве примера в настоящей главе. Применяя симмет-
ричное уравнение Маргулеса, можно опять-таки путем
аппроксимации провести дальнейшее исследование вли-
яния коэффициентов активности на растворимость:
1пу2 = (1 - х2)2 In уз - (8.27)
Соотношение реальных и идеальных растворимостей
выражается так:
ln(x2/x2 ideal) = “ ln?2 = ~ (1 ~ Х2)2 1пу2. (8.28)
Особенно полезными характеристиками равновесной
системы являются коэффициенты активности при беско-
нечном разбавлении уГ, поскольку параметры многих
распространенных корреляций выражаются именно че-
рез них. Эта форма уравнения растворимости графичес-
ки изображена в примере 8.1, из которого следует, что
относительная растворимость стремится к идеальному
значению при у°° < 1 и снижается в противоположном
случае; отношение растворимостей также зависит от ве-
личины идеальной растворимости.
Помимо уравнения Скэтчарда — Гильдебранда для
корреляции и предсказания растворимости используют-
ся более или менее успешно и другие уравнения. Неко-
торые из публикаций, посвященных этой теме, мы
кратко прокомментируем.
1. Коэффициенты активности, полученные по мето-
ду UNIFAC, использовались для определения раствори-
мости и условий образования эвтектики Гмелингом и
др. [46]; полученные результаты в основном хорошо со-
ответствуют эксперименту. Некоторые из этих данных
приводятся в примере 8.4, на рис. 8.1 и рис. 8.5,а.
2. Отношение фугитивностей входящее в
уравнение (8.2), было найдено непосредственно из урав-
нения Ли — Кеслера авторами статьи [469] и применено
для расчета растворимостей и эвтектических диаграмм.
Для каждой пары веществ необходимо было ввести два
параметра взаимодействия; эти параметры были даны
для 32 пар веществ и грубо скоррелированы через соот-
ношения критических объемов. Рис. 8.5,в заимствован
из статьи Музуоки и др. [469].
3. Используя уравнение Вильсона, Морими и Нака-
ниши [494] установили условия фазового равновесия
твердое вещество — жидкость. Они обнаружили, что
уравнение Вильсона дает лучшее соответствие с экспе-
риментом, чем уравнение Скэтчарда — Гильдебранда.
Результаты, полученные при исследовании бинарного
равновесия жидкость — твердое вещество, указанные
авторы применили для предсказания тройного равнове-
сия расплавленных солей. Рис. 8.9,6 — это результат
такой работы.
4. Уравнение Цубоки — Катаямы — Вильсона с па-
раметрами, рассчитанными по данным о равновесии
пар —жидкость, было использовано авторами статьи
[497] для расчета бинодальной кривой, характеризую-
щей равновесие трехфазной системы жидкость — твер-
дое вещество (рис. 8.9,а).
5. Несколько систем расплавленных солей были
проанализированы Наллом [88] с использованием урав-
нения Вильсона; при этом было учтено влияние темпе-
ратуры на параметры. Коэффициенты активности были
оценены по результатам измерения линии ликвидуса
при низких концентрациях и затем применены для пред-
сказания равновесия обеих фаз, жидких и твердых, во
всем диапазоне составов. Рис. 8.5,а заимствован из ра-
боты Налла [88].
6. Равновесие трехфазных смесей (пар — жид-
кость — твердое вещество) диоксида углерода с углево-
дородами было точно предсказано Соавом [652] с
использованием варианта уравнения состояния Ред-
лиха — Квонга. Параметры бинарного взаимодействия
были выведены из результатов измерения равновесия
смесей диоксида углерода с индивидуальными углеводо-
родами.
7. Метод групповых вкладов ASOG применен Унно
и др. [701] для предсказания равновесия жидкость —
твердое вещество для нескольких органических систем.
Рис. 8.5,г иллюстрирует один из таких примеров.
8.3. Равновесие плавления
В свете термодинамических представлений непо-
средственное растворение твердого вещества и раство-
рение расплава этого же вещества (плавление
предшествует растворению) идентичны, если темпера-
тура, давление и состав в обоих случаях одни и те же.
Соответственно уравнения растворимости, приведенные
в разд. 8.2, также применяются к равновесию плавле-
ния. Влияние температуры имеет, возможно,большее
значение для равновесия плавления, в частности, из-за
высоких значений и широкого диапазона температур, с
которыми часто приходится иметь дело. Минимальные
и максимальные температуры могут быть самыми раз-
ными. Ситуация может также осложняться вследствие
образования межмолекулярных соединений. Необходи-
мо также учитывать равновесие между несколькими
твердыми фазами; из-за малой скорости диффузии в
твердых фазах здесь особенно необходимы дополни-
тельные экспериментальные исследования. На рис.
12.14 и ряде других рисунков этой главы показаны экс-
периментальные кривые нагревания — охлаждения; на
диаграмме для установления точки замерзания бензола
появления «впадины» можно, например, избежать при
низкой скорости охлаждения.
При разработке соотношений, описывающих рас-
творимость, начиная с уравнения (8.1), и мольная доля,
и коэффициент активности твердой фазы принимались
равными единице, однако в условиях плавления могут
иметь место различные степени смешиваемости твер-
дых фаз: от полной смешиваемости до полной несме-
шиваемости. В таких случаях условие равенства
Пример 8.1. Растворимость и коэффициенты распределения в неидеальных условиях
Поскольку коэффициент активности зависит от со-
става раствора, в уравнение вводится понятие относи-
тельная растворимость — отношение действительной
растворимости к растворимости в идеальных условиях
(при коэффициенте активности, равном 1).
X2/Xid = I/72 = /(х2).
Рассмотрим особый случай, когда
1П72 = Л(1 - х2)2 = 1П72°(1 - х2)2.
В этой ситуации относительная растворимость опреде-
ляется следующим выражением:
X2/Xid = (1/72>(1-Х2)2.
Отношение реальной и идеальной растворимости.
Коэффициент активности растворенного вещества вы-
ражается симметричным уравнением Маргулеса.
На рисунке а показаны кривые, полученные решени-
ем этого уравнения относительно хг по методу Ньюто-
на — Рафсона для нескольких значений идеальной
растворимости х«/. Величины коэффициента активности
растворенных веществ часто имеют значения меньше
единицы, так что определенный интерес представляет
вид графика.
Если растворенное твердое вещество контактирует
с двумя растворителями, в которых его коэффициенты
активности при бесконечном разбавлении равны 72° и
72*”, коэффициент распределения выражается уравнени-
ем (8.25). Эта зависимость графически изображена для
нескольких значений растворимости х2 в той из фаз, в
которой при более низких концентрациях изменение
7“/72” сказывается в меньшей степени.
Распределение твердого вещества между двумя несме-
шивающимимя растворителями, для которых известны
коэффициенты активности твердого вещества при бе-
сконечном разбавлении.
Пример 8.2. Растворимость нафталина в бензоле, н-гексане и смеси, содержащей 10% этанола и 90% бензола
при 16°С
Свойства веществ:
Нафталин: 8 = 9,9, V = 111,5 мл, = 4558
кал/моль,
Тт = 353,4 К.
Бензол: 6 - 9,2, V = 89,4 мл.
н-Гексан: 8 = 7,3, V - 131,6 мл.
Этанол: 8 = 12,8, V = 58,7 мл.
Известна одна экспериментально найденная величина: в
бензоле хнафТ = 0,217, R = 1,987 кал/моль • К. Испо-
льзуем уравнения (8.13) и (8.14):
После подстановки соответствующих данных получаем
следующие уравнения:
для бензола
х2 = ехр
— 1,4409 +
0,0762
1,2472X2 V
1 - Хг I
= 0,2273,
(2)
для н-гексана
хг = ехр
хг = ехр
^1 +
1,5481
0,8473x2
1 - хг
= 0,0585. (3)
Уг Si — 8г
КГ < , УгХг
Т7(Г - х2)
Применительно к смеси уравнение записывается так:
x2 = ехр
У2(6г - 6)2'
RT
(4)
Равновесие жидкость — твердое вещество 413
Выразим мольные доли растворителей через мольную
долю нафталина х2:
Xi = 0,9(1 - х2), бензол,
хз = 0,1(1 - Хг), этанол.
Параметр средней растворимости определяется следую-
щим выражением:
(89,4)(0,9)(1 - х2)9,2 + 111,5х2(9,9) + 58,7(0,1X1 - х2)12,8
89,4(0,9X1 - х2) + 111,5x2 + 58,7(0,1X1 - х2)
815,37 + 288,48х2
86,33 + 25,7х2 ’ ( ’
Таким образом, растворимость в смеси определяется
выражением
« = ехр Г- 1,4409 - + (9,9 - «’1 • («>
Решаем уравнения (5) и (6) методом подбора.
В итоге получаем следующие значения х2:
Растворитель
Эксперимен-
тальные
Вычисленные
коррек- аппроксимиро-
тные ванные
Бензол
н-Гексан
Смесь
0,217 0,2273 0,2190
0,0585 0,0503
0,2315 0,2273
Аппроксимированные значения получены решением
уравнения (1) по методу Рафсона — Гильдебранда —
Скэтчарда при пренебрежении х2.
Пример 8.3. Давление паров нафталина и его растворимость в бензоле при 16 °C
Давление паров (мм рт. ст.) твердого и жидкого на-
фталина выражается уравнениями
1пЛ = 26,708 - 8712/7,
1пРь = 16,1426 - 3992,01/(7 - 71,29).
При таких низких давлениях отношение фугитивностей
равно отношению давлений паров. Соответственно
fls
угх2 = —--------
J2(scl)
0,0328
0,1134
= 0,2892.
Используя метод подбора, получаем
х2 = 0,2892ехр -=-------0,0762 , = 0,2793.
Г 1,2472X2 12
1 + —--------
I 1 ~ х2 I
Если в методе Рафсона — Гильдебранда — Скэтчарда
пренебречь х2, прямое решение дает х2 = 0,2680, что
вполне приемлемо. Пример 8.2 дает х2 = 0,2273.
Из примера 8.2 следует, что
72 = ехр
0,0762
1,2472х2 Р
1 - х2
Пример 8.4. Растворимость нафталина, установленная по методу UNIFAC, и методы определения параметра
растворимости
В 1978 г. Гмелинг, Андерсон и Праузниц [307] со-
брали экспериментальные данные о растворимости на-
фталина и обработали их по методу UNIFAC (см.
таблицу). При проведении расчетов использовались сле-
дующие исходные данные: Нт = 4494 кал/моль,
Тт = 80,2°С, V = 111,5 мл/моль, 8 — 9,738 (кал/мл)1/2.
Температура 40°С. Растворимость определяется уравне-
нием (8.12).
хг = ехр " у") ~ 012(51 " 9’738>2]
I \ л т * j -KI I
= ехр 1 -0,8214 - 0,1792
Si - 9,738
111,5х2
Эти же авторы провели аналогичные расчеты по урав-
нению Скэтчарда — Гильдебранда с учетом поправки
Флори — Хаггинса 1п02 + 1 - в2, где 02 = (1 - ф1)/х2.
, Как видно из сводной таблицы, погрешность вы-
числений по методу UNIFAC находится в пределах 10%
(или около того) по отношению к экспериментальным
данным, тогда как расчет по уравнениям Скэтчарда —
Гильдебранда и Скэтчарда — Гильдебранда — Фло-
ри — Хаггинса во многих случаях не дает такой точнос-
ти. Объяснить это различием в полярности, по-видимо-
му, нельзя, о чем, в частности, говорят результаты
измерения дипольных моментов, показанные в послед-
ней колонке таблицы.
И(1 - х2)
Растворимость (мол. %) нафталина
Расчет по уравнению, мол. %
Растворитель Экспери- мент UNIFAC Скэтчар- да — Гиль- дебранда Скэтчар- да — Гиль- дебранда— Флори — Хаггинса И 6 Диполь- ный мо- мент
Метанол 4,4 4,8 0,64 0,64 40,5 14,51 1,7
Этанол 7,3 5,4 4,9 4,9 58,4 12,92 1,7
1-Пропанол 9,4 9,3 11,3 11,3 74,7 12,05 1,7
2-Пропанол 7,6 9,3 16,3 16,3 76,5 11,57 1,7
1-Бутанол 11,6 Н,1 18,8 18,7 91,5 11,44 1,8
н-Гексан 22,2 25,9 11,5 11,5 131,6 7,27 0
Циклогексанол 22,5 20,5 20,0 20,0 160,0 11,40 1,7
Уксусная кислота 11,7 12,5 40,1 38,3 57,2 10,05 1,3
Ацетон 37,8 35,8 42,2 41,4 73,5 9,51 2,9
Хлороформ 47,3 47,0 37,8 37,5 80,2 9,24 1,1
Идеальный рас-
творитель 44,1
Пример 8.5. Определение коэффициентов активности
Как показывают приведенные ниже диаграммы, в
системах уксусная кислота + мочевина и фенол +
+ ацетамид наблюдается образование межмолекуляр-
ных соединений. Энтальпии плавления соединений оце-
ниваются с использованием средневзвешенных величин
энтропии плавления исходных соединений.
Компонент ДЯт Тт Шт/Тт
Уксусная кислота (У) 2,755 289,8 9,51
Мочевина (М) 3,472 406,0 8,55
У2М 2,888 314,2 9,19
Фенол (Ф) 2,695 314,1 8,58
Ацетамид (Ац) 3,396 355,1 9,56
Ф2Ац 2,810 315,5 8,91
ФАц 2,786 307,2 9,07
Коэффициенты активности при эвтектических со-
ставах, найденные из выражения
ln7, = ln(l/Xi) - (1 - T/Tm)bHm/RT,
имеют следующие значения:
Шры Т Xi Х2 T1 T2
У + У2М 284,2 0,85 0,15 1,0706 4,0911
У2М + м 310,2 0,87 0,13 1,0829 2,0361
Ф + Ф2Ац 300,7 0,6,1 0,39 1,3524 2,0565
ФАц + Ац 305,7 0,82 0,18 1,1925 2,5524
В системе уксусная кислота + мочевина образуется
межмолекулярное соединение состава 2:1 с температу-
рой плавления 41 °C.
В системе фенол + ацетамид образуются межмолеку-
лярные соединения состава 2:1 (Тт = 42,3°С) и 1:1
(Тт = ~ 34°С). Данные, использованные для построе-
ния диаграмм, взяты из справочника Ландольта—
Берншейна [73].
Равновесие жидкость — твердое вещество 415
Активность
Рис. 8.3. Термодинамические характеристики ряда рас-
плавленных металлов при повышенных температурах.
а — активности железа и никеля при 1600°С [747];
б — активность меди и железа при 1550°С [495];
в — коэффициенты активности смесей расплавленного
таллия и свинца. Необычное поведение отчасти обус-
ловлено присутствием соединения PbTh [344];
г — парциальные и полные энтальпии смешения в сис-
теме железо + никель [179];
д — энтропии смешения некоторых смесей металлов
[71].
0.2
парциальных фугитивностей растворенного вещества
определяется выражением
12sx2sf2s = Y2Lx2Z./2(scl), (8.29)
и эквивалент уравнения (8.9), полученного из уравнения
(8.4) замещением без члена, учитывающего давление,
имеет следующий вид:
12LX2L
In
12sx2s
Л \тт т)
- —£ [\n(Tm/T)-Tm/T+ 1], (8.30)
J\
Это уравнение можно использовать для нахождения ко-
эффициентов активности в твердой фазе, но при этом
все другие данные должны быть известны. В некото-
рых случаях коэффициенты активности жидкой фазы,
полученные из данных по равновесию пар — жидкость
(как на рис. 8.3 для некоторых расплавленных метал-
лов), могут быть экстраполированы до температурного
диапазона, допускающего отвердевание. Хотя данных
по коэффициентам активности твердой фазы опублико-
вано сравнительно немного, можно предположить, что
их поведение сходно с поведением жидкой фазы, как это
подтверждают зависимости, показанные на рис. 8.4
(CsCl + CsBr). Примеры полной смешиваемости твер-
Равновесие жидкость — твердое вещество 417
Рис. 8.4. Коэффициенты активности твердых
растворов хлорид цезия (1) + бромид цезия (2)
при 25°С [405].
На рис. 8.6 система уксусная кислота (Л) — мочевина
(В) состоит из двух бинарных систем А + АгВ и
АгВ + В. Часто информация о термических и других
свойствах межмолекулярных соединений отсутствует.
Кроме того, попытки оценить энтропии плавления и эв-
тектические точки по уравнению Шредера зачастую не
дают разумных величин, как показывает, в частности,
задача 8.17.
Полностью смешивающиеся твердые и жидкие фа-
зы. Диаграммы Т - х полностью смешивающихся сис-
тем, например показанная на рис. 8.5,6, похожи на
аналогичные диаграммы паровых и гомогенных жидких
смесей. Коэффициенты активности при бесконечном
разбавлении обеих фаз можно найти методом, анало-
гичным описанному в разд. 4.15, что и сделал, напри-
мер, Налл [525]. Далее он вывел параметры уравнения
Вильсона и использовал их, чтобы вычислить полную
смешиваемость жидкостей и твердых веществ.
Несмешивающиеся твердые фазы. Большинство
распространенных органических систем относится имен-
но к этой категории фаз, поскольку даже изомеры, в
том числе и оптические (см. задачу 8.14), редко образу-
ют смешанные кристаллы. Вычисления для несмешива-
ющихся систем можно легко выполнить, и часто они
дают ценные результаты.
8.4.1. Эвтектические составы. Определить эвтекти-
ческую температуру и состав идеальных смесей можно в
том случае, если известны их энтальпии и температуры
плавления. Если уравнения Шредера для каждого ком-
понента
X! = ехр
/ 1
_ R \Тт 1
(8.31)
дых органических веществ показаны на рис. 5.22,а при-
меры частичной их смешиваемости — на рис. 5.22,е и
ж. Для органических веществ наиболее характерна пол-
ная несмешиваемость твердых фаз; именно такой тип
поведения этих соединений был выявлен и изучен специ-
алистами, и несколько соответствующих примеров по-
казано в гл. 5 и в данной главе.
Идеальное поведение. Поскольку равновесие плав-
ления часто выводится по существу из идеального пове-
дения, для точной работы необходимы данные по
коэффициентам активности. Если такая информация от-
сутствует, в целях ориентации или ради интереса можно
выполнить вычисления, основанные на уравнении Шре-
дера и связанных с ним уравнениях. Несколько приме-
ров такого типа расчета даны и в тексте главы, и в
разделе «Задачи».
8.4. Бинарные смеси
Классификация типов равновесия плавления основа-
на в первую очередь на числе присутствующих жидких
и твердых фаз, степени их смешиваемости, природе и
температурах плавления межмолекулярных соединений,
которые могут образовываться.
Образование соединения. Диаграммы нескольких
систем, для которых характерно образование межмоле-
кулярных соединений, показаны в задаче 8.17. Такие фа-
зовые диаграммы можно рассматривать как составлен-
ные из нескольких более простых бинарных диаграмм.
1 — *1 = ехр
АНт2 / 1
R V m2
(8.32)
графически изображаются как Г = /(xi), кривые пересека-
ются в эвтектической точке(Ге, jq), и, крометого, они явля-
ются кривыми плавления. Если необходимо определить
только эвтектические условия, эти два уравнения объ-
единяют
(8.33)
Из полученного выражения Те можно найти методом под-
бора, после чего провести соответствующую подстановку
в одно из индивидуальных уравнений Шредера, с тем чтобы
найти состав хе. Аналогичное вычисление может быть вы-
полнено для нахождения эвтектических условий для любо-
го числа компонентов.
Если известны коэффициенты активности как функции
температуры и состава, для нахождения эвтектических ус-
ловий следует одновременно решить нижеследующие
уравнения:
/ 1 1
У1Х1 = ехр I -
к \7ml 1е
(8.34)
8-864
Концентрация бензола хг мол. доли
Рис. 8.5.а — расчетная кривая равновесия жидкость — твердая фаза для системы этанол + бензол, полученная
с использованием коэффициентов активности [307].
7 — интерполяция данных Пикеринга [95] и Виало (1914); 2 — расчет по методу UNIFAC; 3 — идеальная зависи-
мость. б — фазовая диаграмма системы карбонат натрия + сульфат натрия, рассчитанная с использованием урав-
нения Вильсона [525]. в — расчетная кривая равновесия жидкость — твердая фаза для систем
бензол + циклогексан, полученная с использованием уравнения Ли — Кеслера [469]. г — расчетная кривая равнове-
сия жидкость — твердая фаза для систем нафталин + фенол и нафталин + 1-нафтол, полученная с использованием
метода ASOG и данных о равновесии пар — жидкость в системе бензол + фенол [701].
Т2< 1 ~ *1) = ехР
АЯт2
R
(8.35)
In у2 = Ах]/Т,
(8.37)
В одном из самых простейших случаев коэффициенты ак-
тивности представляют уравнениями
1пУ1 = А(1 ~хг)2/Т, (8.36)
и эти уравнения для эвтектических условий можно решить
методом подбора
Л(1-Х!)2
+ lnXj-
АЯОТ1
R
(8.38)
Равновесие жидкость — твердое вещество 419
Рис. 8.6. Система уксусная кислота (Л) + мочевина (В),
в которой образуется межмолекулярное соединение АгВ.
Общая диаграмма разделена на две, на одной из кото-
рых показано вещество А.
Ах?
V~ + In(
* е
(8.39)
Поскольку 1 / Те меняется в этих уравнениях линейно, эту ве-
личину легко исключить, и задача сводится к решению
уравнения с одним неизвестным xi.
Хотя для идеальных смесей расчеты легко выполни-
мы, полученными результатами следует пользоваться
осторожно, поскольку поведение многих смесей сущест-
венно отличается от идеального. Например, приведенные
выше расчеты не обнаруживают наличие некоторой сме-
шиваемости твердых фаз, хотя в действительности в неко-
торых случаях органические системы могут ее проявлять.
На рис. 8.5 сравниваются экспериментальные и расчетные
диаграммы плавления. Пока мы не располагаем таким ме-
тодом, который позволял бы исходя из молекулярной
структуры решать, насколько близка какая-либо конкрет-
ная смесь к идеальной, так как даже смеси изомеров могут
быть неидеальными; примером сказанному служат систе-
мы 2 и 3, приведенные в задаче 8.16. Лучшими методами
предсказания являются методы ASOG и UNIFAC, которые
были применимы к смесям, показанным на рис. 8.5,а и г,
однако ни один из них не подходит для рассмотрения смеси
изомеров, и проблема экстраполяции в широком диапазо-
не температур еще не решена.
Экстремальные давления могут в значительной сте-
пени влиять на свойства и температуры эвтектики; не-
сколько таких примеров дано на рис. 5.28 и 8.7.
Понижение температуры при предполагаемом фиксиро-
ванном составе можно оценить по уравнению Клаузиу-
са — Клапейрона, однако в обычном химическом
процессе это влияние мало, и им, как правило, прене-
брегают.
8.4.2. Коэффициенты активности. Коэффициенты
активности можно оценить, если известны эвтектиче-
ские условия, посредством уравнения
I ГаяШ( /I I \
г ехр R I Т ~ Т I
L \* mt 1 е /
(8.40)
8'
Р, кбар
Р, кбар
Рис. 8.7. Влияние давления на температуры плавления
бензола и уретана и на температуру и состав их эв-
тектик.
Далее их можно использовать для нахождения парамет-
ров корреляционного уравнения, например уравнения
NRTL, или Вильсона, с тем чтобы определить полные
кривые плавления. Практические затруднения с испо-
льзованием уравнения Вильсона могут возникнуть в тех
случаях, когда параметры устанавливаются исходя из
ограниченного числа данных, например из эвтектиче-
ских условий. В таких ситуациях можно получить отри-
цательные величины, которые неприемлемы, если
уравнение используется для представления коэффициен-
тов активности в широком диапазоне состава. Напри-
мер, коэффициент активности и параметры уравнений
Вильсона и NRTL, полученные исходя из условий обра-
зования равновесной эвтектической смеси изомеров нит-
роанилина (задача 8.16), имеют следующие значения:
11 12 13 Уравнение Вильсона NRTL, а = 0.2
лц Tij Та
0,9539 0,7039 — 1,6472 1,0582 3,0059 -2,2628
0,9394 — 1,6371 -1,2463 10,2530 2,1591 -1,0630
— 0,6123 1,6523 -0,9108 17,3190 3,4799 -1,2842
На рис. 8.8 показаны кривые изменения коэффици-
ентов активности, построенные исходя из этих уравне-
ний. И как следует из приведенных графиков, уравнение
Вильсона не может представлять коэффициенты актив-
ности в полном диапазоне изменения состава. Анало-
гичные графики, полученные исходя из данных о
равновёсии пар — жидкость, показаны в гл. 4.
Далее в этой главе описывается применение данных
о бинарных эвтектиках для предсказания характера
плавления тройной смеси.
8.4.3. Энтальпия плавления межмолекулярных со-
единений. Предположив, что поведение межмолекуляр-
ного соединения является идеальным, что в принципе
маловероятно, энтальпию плавления можно найти по Хе
и Те из уравнения Шредера:
R 1п( 1/хе)
\/Те- \/Тт ‘
(8.41)
Однако такой расчет не даст сколько-нибудь полезных
результатов. В исследуемых в задаче 8.17 ситуациях эн-
тальпии плавления, рассчитанные из данных, получен-
ных для двух эвтектик, одним из компонентов которых
является данное соединение, различаются вдвое или
около того, а энтропии плавления находятся в интерва-
ле от 20 до 140, тогда как энтропии плавления исход-
ных соединений лежат в интервале 10—15 Дж/моль • К
или около того. В то же время известно, что, согласно
правилу Вальдена, все вещества имеют одну и ту же
энтропию плавления, равную приблизительно 13
Дж/моль • К.
8.4.4. Частичная смешиваемость фаз и другие воз-
можные осложнения. Примеры систем, в которых две
жидкие фазы находятся в равновесии с твердыми фаза-
ми, показаны на рис. 5.23, г и д. Частично смешиваю-
щиеся твердые органические фазы менее распростране-
ны, тем не менее мы сочли необходимым представить
два таких примера на рис. 5.25, е и ж. Для получения
необходимых корреляций и расширения области испо-
льзования данных о равновесии двух твердых фаз осо-
бенно необходима надежная теория, поскольку равно-
Равновесие жидкость — твердое вещество 421
Пример 8.6. Фазовая диаграмма этанол — вода, полученная из эвтектических условий
При температуре 150,2 К в эвтектическую смесь
указанных соединений входит 79,61 мол. % этанола.
Соединение
кал/моль ТтК,
Этанол 1461,9 158,7
Вода 1436,0 273,2
Для проведения расчетов используется уравнение Виль-
сона, поскольку оно имеет важное преимущество: в нем
учитывается влияние температуры. Коэффициенты ак-
тивности определяются по уравнениям (8.34) и (8.35)
In 0,7961 yi = - 14^£’9 fl - { \
' RT \ 158,7 /
30 SHORT Fl, F2
40 INPUT Т
50 А=18,1/58,4*ЕХР(219,904/Т)
60 В = 58,4/18,1 *ЕХР(—(95,33/Т))
70 С = А/(Х1+А*Х2) + В/(В*Х1 + Х2)
80 Gl = —LOG(X1+A*X2) + C*X2
90 G2=—LOG(B*X1—Х2)—С*Х1
100 F2=—G2—LOG(X2)—1436/1,987/Т * (1—Т/273,2)
ПО F1 = —G1—LOG(X1)—1461,9/1,987/Т* (1—Т/158,7)
120 DISP Fl; F2
130 GOTO 40
140 END
1 П1ПОП 1436 ( . Т
In 0,2039-уг -----I 1 -
' RT \ 273,2
При Т = 150,2 К коэффициенты активности имеют сле-
дующие значения: yi = 0,9663, 72 = 0,5621. Используя
метод, изложенный в примере 4.7 и табл. 8.5, находим
параметры уравнения Вильсона: Л12 = 1,3400, Л21 =
= 1,7104. Параметры экспоненциального уравнения
Вильсона:
Хп/R = - 150,2 In = -219,90,
1 о, 1
. /„ 18,1(1,7104)
Х21/Л = - 150,2 In —-~-у.—-
58,4
Уравнения для определения параметров Вильсона как
функции температуры принимают следующий вид:
Ац = 0,3099ехр(219,90/Т), (1)
Л21 = 3,2265ехр(- 95,33/Т). (2)
= 95,33.
Х1 Т, К
слева справа
0 273,2
0,1 261,4
0,2 247,8
0,3 233,4
0,4 218,4
0,5 203,0
0,6 187,0
0,7 169,5
0,75 159,9 147,8
0,7961 150,2 150,2
0,85 137,1 152,75
0,90 121,8 154,9
0,95 156,9
1 158,7
Уравнения для определения коэффициентов активности
записываются так:
In-yi = - 1п(Х1 + Л12Х2) + /3X2, (3)
1П-У2 = -1п(Л21Х1 + Хг) - /3X1, (4)
/3 = Л1г/(Х1 + Л12Х2) — Л21/(Л21Х1 + хг). (5)
Если концентрация превышает эвтектическую, темпера-
туру при заданной концентрации xi находят методом
подбора из выражения
_ 1461,9 Л Т
71 1 RT ( 1 158,7
(6)
используя уравнения Вильсона.
Если концентрация ниже эвтектической, то применяют
следующее выражение:
1П72Х2
1436 Л
RT у
Температуры подбирают при помощи приведенной ни-
же программы до тех пор, пока не будут удовлетворе-
ны следующие условия: для левой ветви Fi = 0 или для
правой ветви Fz = 0. Результаты табулируются и нано-
сятся на график вместе с данными, взятыми из справоч-
ника Ланге (р. 10, 69, 11-е издание). Введение поправки
на различие теплоемкостей может несколько улучшить
результаты.
10 INPUT XI
20 Х2=1 — XI
Концентрация этанола, мол. доли
Кривая отвердевания смеси этанол + вода. Кружком
помечены точки, нанесенные по данным, взятым из
справочника Ланге; остальные точки — расчетные
данные.
Пример 8.7. Нахождение эвтектического условия по идеальному уравнению и уравнениям Маргулеса и Вильсона
Рассмотрим смесь двух веществ со следующими ха-
рактеристиками: энтропия плавления 13,0, температуры
плавления 71 = 350 К и Т2 = 450 К, отношение объ-
емов К1/К2 = 0,8, коэффициенты активности при беско-
нечном разбавлении 1П7Г = 2 и In 7“ = 0,5. Определим
посредством уравнения для идеальной смеси (о), уравне-
ния Маргулеса (б) и уравнения Вильсона (в) температу-
ру образования эвтектики и ее состав.
а) Уравнение идеальной смеси
Используя метод подбора, получаем Т = 339, Г, Xi =
= 0,8103.
б) Уравнение Маргулеса
Принимаем, что параметры обратно пропорциональны
температуре и что данные 7“ соответствуют температу-
ре плавления.
Л 425 , оо 850
А — —=— 1П71 = - -,
Т 1 Т
D 350 . 00 175
В = 1п72 = —
Запишем уравнения коэффициентов активности:
1П71 = [А + 2(В - Л)Х1]Л2,
1П72 = [В + 2(А — B)x2]xi
и уравнения равновесия плавления:
/1 ч 1
(1 - Xi) =-------ехр
72
(2)
(3)
Эту систему уравнений решают графически, как показа-
но на рисунке а, и получают: Т = 344,0, Xi = 0,897.
в) Уравнения Вильсона
Значения параметров находят из коэффициентов ак-
тивности при бесконечном разбавлении
Л и = 0,0804 при 425 К,
Л21 = 1,5204 при 350 К.
Соответственно экспоненциальные параметры уравне-
ния Вильсона имеют такие значения:
Хи = -425 In 0,8(0,0804) = 1166,15
\21 = -350 In 1,25(1,5204) = -224,74,
а параметры уравнений Вильсона в виде функций тем-
пературы записываются следующим образом:
Л12 = 1,25 ехр (—1166,15/7'),
Л21 = 0,8 ехр (224,74/7).
Уравнения (2) и (3) — это уравнения равновесия плавле-
ния. Решение (Т = 343,1, Xi = 0,871) и в этом случае на-
ходят графически. Уравнения Маргулеса и Вильсона
прогнозируют сходные эвтектические условия, однако
результаты, полученные по уравнению идеальной сме-
си, существенно отличаются.
Графический метод определения эвтектической точки по
уравнениям Маргулеса (7 = 344 К, Xi - 0,897).
Графический метод определения эвтектической точки по
уравнению Вильсона (Т = 343,1 К, Xi = 0,871).
Рис. 8.8. Графики коэффициентов активности, полученных по уравнениям Вильсо-
на и NRTL, параметры уравнений выведены исходя из эвтектического состава
смесей о- и л-нитроанилина. Эвтектический состав: Xi = 0,83; параметры уравне-
ния Вильсона: Ли = -1,2463 и Л21 = 10,2530; параметры уравнения NRTL:
«12 = 0,2, Т12 = 2,1591 и 721 = -1,063. Данные по составу приведены в задаче
8.16.
Кривые 7 (In-yi) и 2 ОП72) рассчитаны по уравнению NRTL, кривые 3 (In-yi)
и 4 (1пуг) — по уравнению Вильсона. Как следует из графика, корреляция Вильсо-
на недейственна, по крайней мере в большей части диапазона концентраций; этот
результат подчеркивает, насколько опасно обосновывать параметры уравнения
Вильсона на результатах единственного измерения равновесия.
Рис. 8.9. Прогнозирование равновесия тройной смеси жидкость — твердое вещество.
а — построение бинодальной кривой бензол + капролактам + вода с использовани-
ем уравнения Цубоки — Катаямы — Вильсона [497].
б — фазовая диаграмма NaF + NaCl + Nal, рассчитанная на ЭВМ с использованием
уравнения Вильсона [494].
весне в таких системах устанавливается очень медленно,
эксперимент провести сложно, данных такого рода ма-
ло и получены они лишь для считанных специфических
систем.
Некоторые фазовые диаграммы металлов рассчита-
ны исходя из результатов немногих осуществленных из-
мерений на основе теории регулярных растворов
(уравнение Скэтчарда — Гильдебранда); примеры даны
Кауфманом и Бернштейном [62], Радменом [604] и Лу-
писом [78]. Кауфман и Бернштейн [62] рассмотрели так-
же влияние образования межмолекулярного соединения
на бинарные фазовые диаграммы.
Если в системе образуются твердые растворы,
уравнение (8.1), равенство фугитивностей необходимо
модифицировать, чтобы включить коэффициенты ак-
тивности в твердой фазе, и применить к обоим компо-
нентам. Обозначим твердую фазу звездочкой. Запишем
упрощенные уравнения, которые получают, пренебрегая
влиянием ДСР и давления:
Ylxl = (n^l)(/l(scl)//l)
= У1х1ехр[(Д5т1/Л)(1-Ги1/Г)], (8.42)
У*х2 = (Y2x2)(/2(scl)//2)
= У2х2ехр[(Д$и2/Я)(1 - Тт2/Т)]. (8.43)
Суммирование мольных долей приводит к двум урав-
нениям:
*1 + х2 = (У1/У1)^*ехР[(Д5т1//?)(1 - ТтХ/Т)}
+ (у?/у2)^*ехр[(Д5т2/Л)(1 - Tm2/T)] = 1,
(8.44)
х* +х*= (у1/у*)х1ехр[(Д5т1//?)(1 - Гт1/Г)]
+ (У2/У2)^ехр[(Д5т2/Л)(1 - Tm2/T)\ = 1.
(8.45)
В тех случаях, когда коэффициенты активности входят
как функции состава в двухпараметрическое уравнение,
эти параметры можно найти решением двух последних
уравнений, как это объясняется, например, в гл. 4. В ис-
следованиях, посвященных решению задач, связанных с
проблемами металлургии, использовались и уравнение
Скэтчарда — Гильдебранда, и симметричное уравнение
Маргулеса.
8.5. Многокомпонентные системы
Большинство поставленных в данном разделе во-
просов будет рассмотрено на примере тройных смесей,
а затем полученные выводы будут, как правило, непо-
средственно распространены на многокомпонентные
смеси. На рис. 5.40 и 5.41 представлены типичные диа-
граммы Т - х тройных смесей. В отстутствие межмо-
лекулярных соединений может образоваться только
одна тройная эвтектика. Поскольку парциальная сме-
шиваемость органических твердых фаз в отличие от ме-
таллических систем неизвестна, далее будут рассмотре-
ны только простейшие случаи несмешиваемости твер-
дых фаз и единственная тройная эвтектика. Диаграммы
состояния таких систем изображены на рис. 8.10, на ко-
тором имеются бинарные и тройные эвтектики и эвтек-
тические «впадины». Каждый из компонентов много-
компонентной системы представляется уравнением сле-
дующего вида:
1
х, = — ехр[(Д5И1/Л)(1 - ТИ1/Г)], (8.46)
У<
где Д5т1- = Mimi/Tmi и каждый коэффициент активнос-
ти зависит от температуры и полного состава жидкой
фазы.
8.5.1. Тройные эвтектики. Тройная эвтектика —
это возможный вариант системы, в которой жидкая фа-
Равновесие жидкость — твердое вещество 425
Рис. 8.10. Схематическая диаграмма равновесия тройной смеси и связанных с ней бинарных диаграмм равновесия
жидкость — твердое вещество. Е\, Ег и Ез — бинарные эвтектики; Е* — тройная эвтектика. Вдоль эвтектической
«впадины» Е\Ел находятся в равновесии твердые компоненты Л и В и жидкая фаза, вдоль «впадины» Е2Е4 —
твердые компоненты Л и С и жидкость, вдоль «впадины» Е3Е4 — твердые компоненты В и С и жидкость.
за находится в равновесии с тремя твердыми фазами.
Температура эвтектики представляет собой минимум на
поверхности ликвидуса диаграммы Т - хь
Условия, соответствующие образованию эвтектики,
находятся путем одновременного решения уравнения
(8.46), записанного для каждого из компонентов при со-
ответствующей температуре и числе мольных долей xi
и Х2. Можно применить следующие уравнения:
1
*! = — exp[(ASml/B)(l - ТтХ/Т)}, (8.47)
Y1
1
х2 = —ехр[(Д«и2/Л)(1-Ги2/Г)], (8.48)
1
х3 = 1 - xi -х2 = ехр|(Д5т3//?)(! - Tmi/T)\,
Тз
(8.49)
Ясно, что этот метод в принципе распространим на сис-
темы с любым числом компонентов. Если смесь иде-
альная, температуру можно найти из единственного
уравнения:
Е х, = Е ехр[(Д5т1/Л)(1 - Tmi/T)\ = 1. (8.50)
Потом мольные доли определяют из индивидуальных
уравнений Шредера. В примере 8.10 для решения такого
вида задач применяются уравнения Маргулеса и
Вильсона.
п-Д ибромбензал
(87,5 °C)
( 83,5°C) (32,5°С)
Рис. 8.11. Равновесие в системе л-нитрохлорбензол + о-нитрохлорбензол + л-ди-
бромбензол. Первое и последнее из этих соединений образуют молекулярное соеди-
нение с эвтектиками в точках Ез и Е* [240].
8.5.2. Эвтектические «впадины». Эвтектические
«впадины», или «долины», связывают индивидуальные
бинарные эвтектики с тройной эвтектической точкой.
Расположенные вдоль какой-либо одной из этих «впа-
дин» две твердые фазы находятся в равновесии с жид-
кой фазой. Например, на диаграмме, показанной на рис.
8.10, вдоль линии Е\Ец вещества А и В являются твер-
дыми. Связывающие линии часто представляют собой
почти прямые, так что их можно непосредственно ап-
проксимировать, если известны эвтектические точки.
Однако условия, определяющие, например, область
вдоль кривой Е1Е4, могут быть исследованы при опре-
деленных концентрациях Хс в диапазоне от нуля до кон-
центрации при тройной эвтектике. Для расчета можно
воспользоваться следующими уравнениями:
1
ха = ~ exp[(ASwa/2J)(l - Тта/Т)], (8.51)
Та
1
xb ~ ехР[(~ ТтЬ/Т)\ ~ 1 — ха ~ хс-
Уь (8.52)
После определения Хс оставшиеся переменные Ха и Т на-
ходят одновременным решением этих уравнений. Для
идеальных смесей задача сводится к решению един-
ственного уравнения для температуры:
1 хс ха + хь ехр[( ASma//J)( 1 ~ Тта/Т)]
+ ехр[(ASmb/R)( 1 - Tmb/R)] (8.53)
при каждом определенном значении Хс.
8.5.3. Изотермы. Аппроксимацию изотерм можно
выполнить путем соединения прямыми близких темпе-
ратур на двух сторонах треугольника, как, например,
это показано на рисунке б к примеру 8.8. Однако реаль-
ные системы, приведенные на рис. 5.40,6 и в, рис. 5.41
и рис. 8.11, имеют значительно искривленные изотер-
мы. Температура, при которой смесь данного состава
начинает кристаллизоваться, — это максимальная тем-
пература из числа определяемых для отдельных компо-
нентов по уравнению
100
l-(/?/ASmZ)ln(yfxf) '
Равновесие жидкость — твердое вещество 427
Рис. 8.12. Путь кристаллизации при понижении температуры. Осаждение чистого
компонента С начинается в точке D при температуре Td и продолжается до тех пор,
пока не будет достигнута точка F на эвтектической «впадине» Е3Е4. Осаждение обо-
их компонентов С и В продолжается до тех пор («впадина» Е3Е4), пока не будет
достигнута тройная эвтектическая точка Ец, которой соответствует полное отверде-
вание смеси. Только осаждение компонента С описывается линией CF, которая пред-
ставляет постоянные соотношения других компонентов, тех же самых, что в исход-
ной смеси, представленной точкой G.
Компонент с максимальной температурой — это тот
компонент, который начинает плавиться первым. Пол-
ные изотермы могут быть построены путем графиче-
ского изображения в виде пересекающихся линий
результатов серии таких расчетов. На рисунках к приме-
ру 8.8 «впадины» невелики, а на рисунках к примеру 8.9
отчетливо выражены.
8.5.4. Процесс охлаждения. Отвердевание смеси в
ходе понижения температуры можно проследить на
рис. 8.12, где исходный состав представлен точкой D.
В этом случае первым начинает осаждаться компонент
С. Температура Td начала его осаждения определяется,
как объяснено в предыдущем разделе. Только осаждение
компонента С по-прежнему отвечает линии CG, которая
соответствует фиксированному отношению других ком-
понентов 8 - Хао/хьо. Эти последние остаются в жид-
кой фазе до тех пор, пока после прохождения ЕъЕь не
достигается точка F, после этого компонент В также на-
чинает осаждаться совместно с С.
Температуру и состав в точке F находят решением
уравнения материального баланса:
(Хао/хъо + l)xb + хс - 1 = 0, (8.55)
После введения уравнения Шредера это выражение при-
нимает следующий вид:
(xao/xbo+ l)exp[(SSmb/R)(l ~ Tmb/TF)]
+ exp[(ASmc/R)(l — Tmc/TF)] — 1 = 0. (8.55)
Решение этого уравнения автоматически дает Tf, хь и
Хс и в итоге приводит к выражению
ха (xao/xbo)xb. (8.55а)
Заметим, что компоненты, входящие в уравнение
(8.53), относятся к числу тех, осаждение которых отве-
чает линии Е3Е4. Если бы кривая кристаллизации пере-
секла ЕгЕ^, то эти уравнения выглядели бы так:
(xbo/xaQ + 1)ха + хс - 1 = 0, (8.56)
и
(xbo/xao + l)exp[(AS та /Я)(1 Тта /TF)]
+ ехр[(Д5тс/Л)( 1 - Tmc/TF)\ -1 = 0. (8.56а)
Условия, соответствующие линии FEt, находят путем
решения уравнения (8.53).
Если коэффициенты активности известны как функ-
ции температуры и составов, их можно включить в эти
рассуждения.
Пример 8.8. Построение диаграмм двух- и трехкомпонентных смесей орто-, мета- и пара-изомеров хлорнитро-
бензола
Температуры плавления и энтальпии изомеров
Изомер Тт, К ЬНт, кал/моль bSm/R
Орто 307,5 4546 7,4383
Мета 317,6 4629 7,3327
Пара 356,7 4965 7,0055
Расчеты выполняют для значений х, слева в диапазоне
от нуля до тройных эвтектических составов; пары
мольных долей определяются автоматически, если из-
вестны численные значения температур. Результаты
расчетов представлены ниже в виде таблицы. Графики,
построенные по этим результатам, показывают, что эв-
тектические «впадины» смесей изомеров орто — мета
и орто — пара несколько несимметричны.
Равновесный состав двухкомпонентных жидких смесей
находят, как в примере 8.6. Состав бинарной эвтектики,
в данном случае смеси орто- и л/е/ла-изомеров, опреде-
ляют следующим образом:
= Хо
ехр 7,4383 I 1 -
307,5
Т
+ ехр 7,3327 ( 1 -
Аналогичным образом устанавливают и состав эвтек-
тик, образованных другими парами изомеров.
Пара Т, к Хо хм хп
Орто + мета 285,41 0,5628 0,4372 0
Мета + пара 303,34 0 0,7084 0,2916
Орто + пара 296,51 0,7590 0 0,2410
Температуру тройной эвтектики находят из выражения
307,5 \ 1
1 = ехр 7,4383 ( 1 -
Т
+ ехр
+ ехр
7,33271 1
7,0055 I 1
317,6
Т
356,7
подбора находим: Т = 279,97, Хо = 0,4811,
Методом
хм = 0,3731 и хП = 0,1458.
Уравнения трех эвтектических «впадин» имеют сле-
дующий вид:
= ехр 7,4383 / 1 - -Зу’5
+ехр 7,3327 I 1 -
317,6
Т
+ хп = ехр 7,3327 I 1 -
317,6
Т
+ехр 7,0055 I 1 -
356,7
Т
= Хо + х„ = ехр 7,4383 I 1 -
307,5
Т
+ехр 7,0055 (1 - -35^
Хп Км Хо хм
0 285,4 0,562 0,438
0,04 283,9 0,540 0,420
0,10 281,7 0,546 0,394
0,1458 279,9 0,481 0,3732
Хо Гил хм хл
0 303,4 0,708 0,292
0,1 299,3 0,639 0,261
0,2 294,9 0,569 0,231
0,3 290,1 0,499 0,201
0,4 284,8 0,429 0,171
0,4811 279,9 0,372 0,1469
хм ТОЯ X, Хп
0 296,5 0,759 0,241
0,1 292,6 0,684 0,216
0,2 288,3 0,610 0,190
0,3 283,6 0,535 0,165
0,3731 279,9 0,480 0,1469
Три диаграммы плавления бинарных смесей мета-,
орто- и пара-изомеров хлорнитробензола.
1 — пара (А) + орто (В), 2 — мета (А) + пара (В),
3 — орто (А) + мета (В).
Равновесие жидкость — твердое вещество 429
пара-Изомер
(356,7 К)
орто-Изомер
(307,5 К)
построенная на допущении
мета-Изомер
(317,6 К)
Диаграмма равновесия смесей пара-, орто- и мета-
изомеров хлорнитробензола,
идеального поведения.
8.5.5. Составы и количества двух фаз. Задача со-
стоит в том, чтобы найти составы и количества каждо-
го компонента в каждой из фаз при любой
температуре — от момента начала кристаллизации до
полного отвердевания, соответствующего тройной эв-
тектике. Примем следующие обозначения: L — доля
жидкости в общей смеси; п, — число молей компонента
i в жидкости/число молей во всей смеси; пю — п, —чис-
ло молей компонента i в твердых фазах/число молей в
исходной смеси.
Этот процесс иллюстрирует рисунок 8.13, где клю-
чевые моменты процесса отвечают точкам D, Е, F и G.
Пример 8.9 демонстрирует количественные зависимости
этого процесса.
1. Момент начала кристаллизации, точка D, описы-
вается уравнением (8.54).
2. Чистые кристаллы компонента С образуются до
достижения точки F. Температуру и составы xif находят
при помощи уравнений (8.55)—(8.56а). Количество ком-
понента С, остающееся в жидкости, определяют по-
средством выражения
ncF= (па0 +nb0VcF/(\ ~ XcF), (8.57)
а количества компонентов а и b — посредством вы-
ражений:
naF ~ пао ’
nbF = пЬо-
3. Долю чистых кристаллов, которая может быть
регенерирована, рассчитывают по уравнению
nyF Па^ + ( X<-'F \
= I I . (о. 5 о)
Лс0 rtc0 yl xcF /
4. При определенной промежуточной температуре
Те, лежащей в интервале Td — Tf, состав хсе в жидкос-
ти находят при помощи уравнения (8.59)
xcE = exp[(^Smc/R)( 1 - Ттс/ТЕ)]. (8.59)
После этого посредством уравнения, аналогичного
уравнению (8.57), определяют число молей:
псЕ~ («а0+ nbQ)XcE/(\ ~ХсЕ), (8.60)
где ПаЕ = Пао И ПЬЕ = Пьо.
5. В любой точке G вдоль линии эвтектики FE4
температура и состав определяются по уравнению (8.53)
при заданном содержании того компонента, который не
осаждается (компонент А на рис. 8.12). В результате
уравнение (8.53) которое принимает следующий вид:
1 “ ха = ХЬ + хс = ехр[(ASmb/R)( 1 - ТтЬ/Г)]
+ exp[(A5wc/7?)(l - Tmc/T)], (8.61)
решается относительно Т при определенных значени-
ях Ха.
Количество жидкой фазы и количества других ком-
понентов определяются при помощи следующих
Плотность, г /мл
Рис. 8.13. Сверхкритические свойства и раство-
римость.
а — влияние давления на плотность диоксида
углерода при 40°С и давлении ниже и выше кри-
тического. Критические условия: 31,2°С, 72 бар
и 0,47 г/мл. Сверхритическая растворяющая
способность хорошо коррелируется с плот-
ностью газа-растворителя, как это показано на
рис. 8.13,5.
б — растворимость нафталина в этилене как
функция плотности растворителя [768].
в — влияние сверхкритических температур и дав-
лений этилена на растворимость нафталина в
этилене. Критические свойства этилена: 283 К и
51,6 бар [768]. Некоторые из этих данных хоро-
шо коррелируются с уравнением Ли — Кеслера
[469].
г — растворимость в сжатом этилене при 75°C
[271].
Давление, фунт/кв. дюйм
Равновесие жидкость — твердое вещество 431
уравнений:
i = ~xaG^
"cG= "сО ~ LxcG.
nbG = "bO ~ LXbG-
(8.62)
(8.63)
(8.64)
Количества компонентов, находящихся в твердой фазе,
находят суммированием:
(«cG)soIid = пс0 ~ ncG' (8.65)
(fl/>G)soIid = пЬ0 ~ nbG- (8.66)
(flaG )solid — 0- (8.67)
6. Количество жидкости, остающейся непосред-
ственно перед отвердеванием тройной эвтектики, равно
LE4 = па()/хаЕ4’ (8.68)
где эвтектический состав находят по уравнению (8.50).
8.6. Растворимость твердых веществ и
жидкостей в сверхкритических газах
Свойства сильно сжатых газов, включая растворя-
ющую способность, сходны со свойствами жидкости,
различие состоит только в том, что сильно сжатые га-
зы полностью заполняют любое ограниченное про-
странство, в которое их помещают. При умеренных
давлениях содержание конденсируемого вещества в кон-
тактирующем с ним газе определяется давлением пара
или давлением сублимации этого вещества, и содержа-
ние контактирующего вещества уменьшается, если дав-
ление системы растет. Однако при давлениях, близких
к критическому давлению газа, его растворяющая спо-
собность резко увеличивается с давлением, точно так
же, как это происходит с жидкими растворителями. Та-
кое увеличение растворимости объясняется резким
уменьшением коэффициента фугитивности газообразно-
го растворенного вещества с увеличением давления.
Указанное поведение хорошо оценивается современны-
ми уравнениями состояния. Некоторые данные, иллю-
стрирующие сказанное, приведены на рис. 8.13.
Если принять, что фаза растворенного вещества
остается чистой, баланс парциальной фугитивности рас-
творенного вещества будет определяться выражением
fiv =flL ~flL- (8.69)
После соответствующих замещений это уравнение пре-
образуется в следующее выражение:
угфгР=ф^Р^\РР)г
= ф^Р^ ехр[(К2/ЯТ)(Р - P^at)], (8.70)
которое можно разрешить относительно содержания
растворенного вещества в паровой фазе:
>’2 = (01atP|at/02^)exp[(K2/PT)(P - P|at)]. (8.71)
Знаменатель ФгР обычно доминирует над коэффициен-
том Пойнтинга, но в некоторых случаях коэффициент
парциальной фугитивности так резко падает с увеличе-
нием давления, что растворимость заметно повышает-
ся. В примере 8.11 при помощи В-усеченного вириаль-
ного уравнения анализируется растворимость некото-
рых тяжелых углеводородов в пропане. В этих случаях
даже при давлениях ниже 100 атм наивысшая раствори-
мость превышает наименьшую в 10—20 раз. Другие
примеры показаны на рис. 5.27, а также в гл. 9.
Увеличение растворимости конденсированных фаз в
газах при высоких давлениях было обнаружено более
100 лет назад, но интерес к этому явлению вновь возник
лишь недавно. В своей статье Зусе [752] описал первый
пример применения этого явления в промышленности.
Последние разработки связаны с извлечением или раз-
делением веществ, слишком нестойких для дистилля-
ции. В этих целях наиболее пригодны газы, критические
температуры которых ниже температуры воздуха. Ряд
таких газов перечислен ниже.
Газ Тс, °C Рс, бар
Диоксид углерода 31,0 73,9
Этилен 9,2 50,6
Оксид азота(1) 36,5 72,5
Этан 32,3 48,8
CCIF3 (R-13 хладагент) 28,9 38,6
CHF3 (R-23 хладагент) 25,9 48,2
Пропан 96,8 42,0
Шталь и Шильц [661] приводят в своей статье пере-
чень примерно дюжины примеров промышленного при-
менения сверхкритической экстракции. Эти же авторы
описывают использование СОг и N2O в сверхкритиче-
ском состоянии в тонкослойной хроматографии пример-
но сотни растительных лекарственных средств.
Несколько примеров применения газов в сверхкритиче-
ском состоянии в промышленности и анализе описаны
Питером и др. [552]. В обзоре литературы, выполнен-
ном Ганголи и Тодосом [295], особое внимание уделено
экстракции углем. В гл. 9 рассматриваются недавно
опубликованные материалы нескольких симпозиумов, и
там более детально обсуждается фазовое поведение в
сверхкритических условиях.
Экстрация в сверхкритических условиях, помимо ее
преимуществ для низкотемпературного выделения низ-
колетучих веществ, часто более эффективна энергети-
чески, чем обычные методы разделения, поскольку
растворимость в этом случае можно контролировать
относительно небольшим изменением давления и тем-
пературы, а изменения фазы не требуется. Одним из
примеров такого энергетически эффективного процесса
является разделение этанола и воды с применением СОг
в сверхкритическом состоянии, хотя этот процесс нахо-
дится еще на стадии пилотных разработок. Ниже мы
кратко опишем два примера промышленного примене-
ния газов в сверхкритическом состоянии и при этом
остановимся на некоторых деталях данных методов.
1. Сверхкритическая деасфальтизация остатков сы-
рой нефти после вакуумной перегонки осуществляется с
применением легких углеводородов, например пентана.
В одном конкретном случае, когда система имеет сле-
дующие характеристики: давление 39,5 атм, температу-
ра 468 К и плотность растворителя 19 фунт/фут3,
нерастворенным остается только асфальт, и его отделя-
ют от раствора отстаиванием. В результате снижения
давления до 37 атм и повышения температуры до 489 К
плотность растворителя падает до 7,7 фунт/фут3, и
нефть, и растворитель разделяются. Эти небольшие из-
Пример 8.9. Путь отвердевания тройной смеси
Чтобы улучшить отделение кристаллов в смеси м-
бензодинитрила См-СбН4 (CN)2) и jw-цианобензамида
(C6H4(CN)CON2H) состава 90/10 соответственно, в нее
добавляют л/-ксилол. Анализ процесса, поскольку коэф-
фициенты активности не известны, будет проведен при
допущении, что поведение расплава идеально. Физиче-
ские данные приведены в первой таблице.
Содержание компонентов в смеси (в мольных до-
лях) определяют по уравнениям:
Ха = ехр [6,4821(1 - 435/Т)] (м-бензодинитрил),
хь = ехр [6,5425(1 - 495/73] (м-цианобензамид),
Хс = ехр [6,1600(1 - 225,4/Т)] (м-ксилол).
Состав бинарной эвтектики находят при помощи урав-
нения (8.50):
Ха + Хь - 1 = 0, Т = 414,1, Ха = 0,7213,
хь = 0,2787.
Состав тройной эвтектики также устанавливают по
уравнению (8.50):
Ха+хь+Хс~^=д, Т = 225,30, Ха = 0,0024,
хь = 0,0004, Хс = 0,9972.
Условия начала кристаллизации определяют по уравне-
нию (8.54). Для всех исследованных здесь концентраций
л<-ксилола компонент А — это тот компонент, который
кристаллизуется чистым. Температуры исходной кри-
сталлизации находят из уравнения
Результаты расчетов приведены в таблице. Условия
осаждения вещества А соответствуют прямой, пред-
ставляющей постоянные пропорции других веществ в
исходной смеси. Рассмотрим подробно отвердевание
смеси с исходным содержанием л<-ксилола 20%, для
которой
0 = Хсо/хьо = 0,20/0,08 = 25.
Применяя уравнение (8.55), получим
(/3 + 1)х>> + Ха - 1 = 0
или
(2,5 + 1)ехр[6,5425(1 - 495/Т)]
+ ехр(6,4821(1 - 435/7)1 -1=0.
Другими словами,
Т = 386,45, Ха = 0,4429, хь = 0,1592, Хс = 0,3980.
При дальнейшем понижении температуры кристаллизу-
ются кристаллы, содержащие примеси. Путь кристал-
лизации проходит вдоль эвтектической «впадины»
веществ А а В, которая описывается уравнением (8.53).
Это уравнение можно представить как
1 - Хс = ехр[6,4821(1 - 435/Т)]
+ ехр[6,5425(1 - 495/Г)].
Чтобы найти температуры и составы, соответствую-
щие «впадине», в уравнения подставляются значения Хс
в диапазоне от условий, соответствующих моменту по-
явления кристаллов с примесями (0,3980), до момента
образования тройной эвтектики (0,9972). Результаты
показаны в третьей таблице и обозначены в виде точек
на тройной диаграмме. Ясно, что форма действитель-
ной эвтектической «впадины» (линия) отклоняется от
линейной, которая представлена в виде штриховой ли-
нии, соединяющей бинарную (Л и В) и тройную эвтек-
тику (фактически чистый л<-ксилол).
1. Термические свойства
Вещество Tf AS//R
А JW-C6H4(CN)2 435 6,4821
В JW-C6H4CNCONH2 495 6,5425
С УИ-СбН4(СН3)2 225,4 6,1600
2. Начальные и конечные условия образования чистых кристаллов
Исходная смесь В момент окончания чистой кристал-
лизации
^аО ХйО ХсО Т 1 начальная Т Ха Хь Хс
0,9 0,1 0 428,04 414,1 0,7222 0,2778 0
0,81 0,09 0,1 421,3 399,29 0,5601 0,2084 0,2315
0,72 0,08 0,2 414,0 386,45 0,4429 0,1592 0,3979
0,63 0,07 03 406,1 374,73 0,3526 0,1225 0,5249
3. Эвтектическая «впадина» между А и В
Хс Т Ха Хь Ха/ХЬ
0 414,1 0,7213 0,2787 2,5881
о,1 408,06 0,6518 0,2481 2,6274
0,2 401,49 0,5822 0,2179 2,6718
0,3 394,28 0,5120 0,1880 2,7233
0,3979 386,45 0,4429 0,1592 2,7825
0,5 377,2 0,3704 0,1296 .2,8575
0,6 366,65 0,2987 0,1012 2,9503
0,7 353,9 0,2264 0,0736 3,0742
0,8 337,3 0,1529 0,0469 3,2586
0,9 312,3 0,0783 0,0218 3,5991
0,9972 255,30 0,0024 0,0004 6,0410
Ниже суммирован состав жидкой фазы как функция
температуры на всем пути кристаллизации.
Г Хс Хь Ха
414 0,20 0,08 0,72
400 0,3093 0,1237 0,567
386,5 0,3979 0,1592 0,4429
366,7 0,6000 0,1012 0,2988
337,3 0,8000 0,0469 0,1529
225,3 0,9972 0,0004 0,0024
Взяв за основу один моль исходной смеси и испо-
льзуя уравнения материального баланса, получают ко-
личества обеих фаз и отдельных содержащихся в них
Равновесие жидкость — твердое вещество 433
компонентов. Эти данные и составы жидкой фазы при-
ведены в нижеследующей таблице.
Т L S Жидкая фаза Твердая фаза
пс Пь Па лс пь Пи
414 1 0 0,2 0,08 0,72 0 0 0
400 0,6467 0,3533 0,2 0,08 0,3667 0 0 0,3533
386,5 0,5027 0,4973 0,2 0,08 0,2227 0 0 0,4973
366,7 0,3333 0,6667 0,2 0,0337 0,0997 0 0,0463 0,6203
337,3 0,2500 0,7500 0.2 0,0018 0,0383 0 0,0683 0,6818
225,3 0,2006 0,7994 0,2 0,0001 0,0005 0 0,0799 0,7195
Например, при температуре 366,7 К и JQ = 0,6
в жидкой фазе:
в твердой фазе:
/. - пс/0,6 = 0.2/0,6 = 0,3333,
Пс - ПсО = 0,2,
пь = 0,3333(0,1012) = 0,0337,
па = 0,3333(0,2987) = 0,0997,
S = 1 - L = 0,6667,
пс = 0,2 - 0,2 = 0,
пь = 0,08 - 0,0337 = 0,0463,
А, па - 0,72 - 0,0997 = 0,6203.
Осаждение чистого компонента ,4 начинается при температуре 414,0 К и продолжается
до тех пор, пока температура не упадет до 386,5 К и мольная доля ха не станет равна
0,443. Оба компонента, А и В, кристаллизуются вместе, и кристаллизация их соответствует
.линии, связывающей бинарную (А и В) и тройную эвтектики. Второе число каждой группы
чисел, представленных па рисунке, - это молная доля компонента А(ха).
Ч-ХМ
Пример 8.10. Расчет условий образования тройной эвтектики по уравнению Вильсона
Свойства компонентов смеси
Компонент V, мл/моль Д&п тт
1 150 13 450
2 200 13 375
3 125 13 400
Параметры уравнения Вильсона
Ln = 1141,2, L2i = -364,3,
Lb = 221,5, £з1 = 144,5,
Ьгз = -281,6, £з2 = 493,5.
Ti = 342,54, xi = 0,1284, х2 = 0,5379, хз = 0,3357. Ис-
пользуя эти значения при помощи уравнений, приведен-
ных в табл. 4.7, находят коэффициенты активности, а
далее определяют Т и х, методом подстановки по
уравнению
Результаты пяти последовательных попыток, представ-
ленные ниже в виде таблицы, показывают очень близ-
кую сходимость.
Условия образо путем решения еле; S xi( = 1 = 2 ехР вания идеальной эвтектики находят дующего уравнения:
= ехр - 13 R 450 _ А Г /
+ ехр - 13 R о-х
+ ехр - 13 R 1 1 1
Попытка
1 2 3 4 5
71 1 1,5832 1,7316 1,7788 1,7932
72 1 0,1136 1,1079 1,1066 1,1062
73 1 1,1548 1,1382 1,1382 1,1311
Т 342,54 349,99 350,01 350,014 350,013
Xl 0,1284 0,0974 0,0891 0,0867 0,0860
Х2 0,5379 0,5627 0,5658 0,5665 0,5667
ХЗ 0,3337 0,3401 0,3451 0,3468 0,3473
Пример 8.11. Влияние давления на растворяющую способность пропана и на коэффициенты фугитивности тяже-
лых углеводородов
Свойства трех растворенных веществ указаны ниже
в таблице. Температура 400 К. Для оценки коэффициен-
тов парциальной фугитивности использовано вириаль-
ное уравнение Цонопулоса [696]. Во всех случаях
пропан — это компонент 1, растворенные вещества —
компонент 2. Сначала по мере повышения давления
растворимость падает, но в дальнейшем этой тенденции
противодействует влияние значительного снижения фу-
гитивности и фактора Пойнтинга, и растворимость воз-
растает.
В = у1Вц + у2В22 + 2yiy2Bi2,
1п02 = (2yiBi2 + 2у2В22 — В),
ф^Р?\РК)2
У2 = -----------
ФгР
фТ'Р?1 Г У2(Р - /?“) '
=--------ехр ------------ .
ф2Р L КГ
Графики не выходят за пределы 100 атм, поскольку
точность В-усеченного вириального уравнения недоста-
точна. Фактически точность уравнения начинает сни-
жаться при Р/Рс >0,5 Т/ Тс, что составляет около 20
атм для пропана. Однако результаты качественно вер-
ны: с увеличением давления растворимость сначала по-
нижается, а затем значительно возрастает.
Вещество Тс Рс Ус Zc w By Ps Vl
Пропан 369,8 41,9 203 0,281 0,152 -0,2069 — —
Нафталин 748,4 40,1 410 0,267 0,302 -2,746 0,0717 132,0
Гексадекан 717,0 14,0 828 0,236 0,708 -10,060 0,00437 294,1
Фенантрен 878 28,6 594 0,228 0,440 -10,369 0,00153 151,2
Пропан +
+ нафталин 526,1 40,16 0,227 -0,7044
Пропан +
+ гексан 514,9 24,55 0,430 -1,2222
Пропан +
+ фенантрен 569,8 32,67 0,246 -1,1467
Равновесие жидкость — твердое вещество 435
1000 Xрастворенное в-во $ растворенное в-во
Влияние давления на коэффициенты парциальной фуги-
тивности нафталина, гексадекана и фенантрена в пропа-
не при 400 К.
Влияние давления на растворимость нафталина, гекса-
декана и фенантрена в пропане при 400 К.
менения давления и температуры вблизи критической
точки растворителя (33,3 атм, 469,6 К) соответствуют
существенно меньшим энергетическим потребностям по
сравнению с обычной экстракцией, для которой необхо-
димо несколько более высокое отношение раствори-
тель/нефть.
2. Обеззоливание угля можно провести при помощи
растворителя, состоящего примерно на 80% из пириди-
на и на 20% из «антраценовой нефти». Критические па-
раметры пиридина: 620 К и 55,6 атм. При 617 К и 62,2
атм достигается 90%-ное обеззоливание угля. После то-
го как нерастворенный уголь и зола отделяются меха-
ническим путем от раствора, растворитель и
растворенный уголь разделяются путем изменения ус-
ловий: повышения температуры до 620 К и снижения
давления до 60 атм.
Хотя в примере 8.9 для большей наглядности при-
менялось В-усеченное вириальное уравнение, оно не
обеспечивает достаточной точности при высоких давле-
ниях в критической области. Другие уравнения дают
лучшие результаты. Соав [652] применил свою модифи-
кацию уравнения Редлиха — Квонга к равновесию меж-
ду углеводородами и твердым диоксидом углерода,
однако, чтобы результаты расчета были точными, не-
обходимо учитывать параметры бинарного взаимо-
действия. Стефан и Шабер [663] провели более
обширные исследования с использованием уравнения
Редлиха — Квонга для сверхкритических областей и
также пришли к выводу о необходимости учета пара-
метров бинарного взаимодействия, например кп =0,11
для системы диоксид углерода — алканы.
Обычно характер изменения растворимости можно
оценить при помощи уравнения (8.71) и подходящего
соотношения для коэффициента парциальной фугитив-
ности. Однако при этом можно руководствоваться и
следующими эмпирическими правилами.
1. Во многих случаях увеличение растворимости
особенно существенно при температурах, близких к кри-
тической температуре газа-растворителя. Например, как
показано на рисунке а к задаче 16, наибольшее увеличе-
ние дает СОг, критическая температура которого
(31,3°С) ближе всего температуре эксперимента (0°С).
2. Растворимость имеет тенденцию снижаться до
минимума при давлениях, близких к критическому. Рас-
творимость всех показанных на рис. 8.13, г растворен-
ных веществ минимальна вблизи давления, равного
критическому давлению этилена (744 фунт/кв. дюйм).
Между прочим, эти кривые были бы расположены го-
раздо ближе друг к другу, если бы растворимости были
выражены в мольных процентах, а не в массовых.
3. В определении растворимости главным факто-
ром, по-видимому, является плотность растворителя.
На рис. 8.13, б показано примерно линейное изменение
log(pacTBopHMocTb) от плотности в области высокого
давления. Рис. 8.13, б и в, на которых использованы
одни и те же данные, но представленные по-разному,
также показывают, что плотность достаточно хорошо
коррелирует с влиянием как температуры, так и давле-
ния. Как показано на рис. 8.13, в на примере СО2, изме-
нение самой плотности с давлением имеет ярко
выраженный нелинейный характер в области умеренно-
го и высокого давлений.
8.7. Задачи
8.1. Определите идеальную растворимость л-ксило-
ла в интервале температур 200—275 К, если известно,
что = 4090 кал/моль, Тт = 286,39 К и ДСР -
= 5,96 кал/моль • К.
8.2. Растворимость нафталина в бензоле равна
0,14 мол. доли при 0°С и 0,478 мол. доли при 45°C.
Точка плавления нафталина 80,1°С. Вычислите теплоту
плавления для каждого значения растворимости. Срав-
ните найденные значения с установленным эксперимен-
тально (4559 кал/моль).
8.3. Найдите коэффициент растворимости (6х/6Р)т,
используя уравнение Скэтчарда — Гильдебранда для ко-
эффициентов активности и следующие данные:
Т = 300 К, Vi = 50 мл/моль, V2 = 18 мл/моль, Xi =
= Х2 - 0,5, 61 - 62 = 3 (кал/мл)0,5, ДР/ = 5 мл/моль.
8.4. Для смеси SnCU(l) и Snl4(2) экспериментально
установленное значение Г2(6г - 6)2//?Т равно 3,8 при
298 К. Точка плавления и теплота плавления иодида
равны 417,7 К и 4480 кал/моль. Найдите раствори-
мость иода в этой смеси при 298 К.
8.5. Фазовая диаграмма смеси этиленгли-
коль^) + вода(В) свидетельствует о существовании мо-
лекулярного соединения ГВг и ГВг/з. Теплота плавле-
ния этиленгликоля равна 2685 кал/моль и воды 1436
кал/моль. Определите теплоту плавления двух гидра-
тов. Температуры замерзания и состав соединений ука-
заны ниже.
Вещество или эвтектика Температура замерзания, °C Содержа- ние этилен- гликоля, масс. %
Лед 0 0
Лед + ГВг -51,2 57,3
ГВ2 -49,6 61,6
ГВг + ГВг/з -63,3 75,6
ГВг/з -39,7 84,4
ГВг/з + Г -49,4 87,0
Г -12,8 100
8.6. Используйте данные, приведенные в примере
8.8, и результаты показанных в нем расчетов для сме-
сей изомеров хлорнитробензола для решения следую-
щих задач.
а. Найдите температуры начала отвердевания и
идентифицируйте вещество, которое кристаллизуется
первым, если хорто равен 0,25 или 0,50, или 0,75 для
трех значений отношения хмета/х„ара: 9,25, 0,5, 2,0.
б. Проследите для смеси состава хорто = 0,3,
хмета = 0,1, х„ара = 0,6 путь охлаждения от начала
кристаллизации до точки, при которой два вещества на-
чинают осаждаться и переходят в тройную эвтектику.
в. Определите состав жидкости и количество кри-
сталлизованного вещества при снижении температуры
до 310 К.
Равновесие жидкость — твердое вещество 437
г. Установите составы твердой и жидкой фаз и тем-
пературу, если в жидкой фазе хмета = 0,3.
8.7. а. Бензол плавится при 5,5°С, его скрытая теп-
лота плавления 2350 кал/моль; хлорбензол плавится
при -45°C, его скрытая теплота плавления 1800 кал/
моль. Эти соединения образуют простую эвтектиче-
скую систему. Принимая, что они дают идеальные сме-
си, постройте фазовую диаграмму для конденсирован-
ной системы, помечая каждую область, и определите
эвтектическую температуру и состав. {Ответ:
Т= -61 °C, хбензол = 0,263.)
б. Точки плавления и скрытые теплоты плавления
л-нитробензола (Л) и л-нитроанилина (В) имеют сле-
дующие значения:
А В
t, °C 172,8 147,5
ДА, кал/моль 6250 5135
Эти два соединения образуют простую эвтектику. По-
стройте идеальную фазовую диаграмму и сравните ее
со следующими экспериментальными результатами:
ес ХА t° С ХА
172,8 1,00 122,2 0,374
165,2 0,865 (П9,3) Эвтектика
154,0 0,694 125,2 0,294
142,0 0,551 135,4 0,176
139,0 0,525 142,3 0,084
131,3 0,444 147,5 0,000
{Ответ: Т = 118°С, хв = 0,635.)
8.8. Гептан(1) и 1,2-этандиол(3) сколько-нибудь за-
метно не смешиваются в присутствии менее 10% или
около того гексанола(2). Экспериментальные значения
коэффициента распределения: К - концентрация в фазе
гептана/концентрация в фазе этандиола, известны как
функции концентрации в фазе этандиола.
Концентрация в этандиоле, мол. % К
0,0432 1,20
0,0667 2,12
0,0932 2,43
Проверьте эти значения К, используя уравнение
NRTL при а = 0,20 и
ти = 2,9188 721 = -0,6747
723 = 0,8866 732 = 1,3954
Соответствие не будет полным, по-видимому, из-за
некоторой взаимной растворимости этих трех компоне-
нтов (DECHEMA, V/2, р. 431).
8.9. Найдите параметры уравнений Маргулеса и
Вильсона для системы нафталин + бензол исходя из эв-
тектических условий: 270,2 К и xi = 0,125.
Нафталин Бензол
Энтальпия, кал/моль 4550 2379
Тт, К 353,3 278,7
8.10. Постройте идеальную кривую отвердевания
смесей м и о-дибромбензолов.
мета орто
Энтальпия, кал/моль 13,38 12,78
Тт, °C -6,9 1,8
8.11. Антрацен и фенантрен полностью смешивае-
мы в жидкой и твердой фазах. Их теплоты плавления
равны 6898 и 4456 кал/моль соответственно. Равновес-
ные концентрации антрацена в твердой и жидкой фазах
представлены ниже в таблице.
а. Найдите соотношения каждого компонента
при каждой температуре.
б. Найдите параметры уравнения Маргулеса для
160°С и, следовательно, коэффициенты активности каж-
дого компонента.
°C XS XL
218 0 0
200 0,10 0,26
180 0,21 0,47
160 0,32 0,62
140 0,45 0,76
120 0,71 0,88
101 1 1
8.12. Найдите коэффициенты активности каждого
из компонентов смесей метанол + вода при каждой из
приведенных ниже температур плавления. Энтальпия и
температура плавления компонентов: 759,4 кал/моль и
175,4 К (метанол) и 1436 кал/моль и 273,2 К (вода).
X Т
0,0273 271,00
0,0472 268,20
0,0725 264,90
0,0994 261,50
0,1273 257,60
0,1572 253,20
0,1883 248,20
0,2215 243,20
0,2563 237,60
8.13. Даны точки замерзания смеси глицерин +
+ вода. Энтальпия плавления компонентов, кал/моль:
4421 (глицерин) и 1436 (вода) соответственно.
а. Найдите коэффициенты активности при каждой
из указанных температур.
б. Сравните идеальные кривые замерзания с приве-
денными данными.
в. Найдите параметры уравнения Вильсона из эв-
тектических условий (х = 0,28 и Т = 228,7 К) и, испо-
льзуя их, вычислите кривую замерзания.
Молярные объемы: 116,13 — глицерин, 18,02 —
вода.
X Т X Т
0,0000 273,2 0,2269 239,6
0,0213 271,6 0,2800 228,7
0,0466 268,4 0,3134 235,4
0,0774 263,7 0, 4390 254,0
0,1154 257,7 0,6378 271,6
0,1646 251,2 1.0000 291,2
8.14. Найдите параметры уравнений Маргулеса и
ван Лаара, соответствующие эвтектическим составам,
показанным ниже на двух диаграммах.
a. D- и L-Изомеры /3-бензилгидратроповой кислоты.
б. о- и л-Изомеры хлорнитробензола.
Орто Пара
Энтальпия, кал/г 28,85 31,51
Тпл, °C 32,5 83,5
Энтропии плавления оптических изомеров во всех слу-
чаях (пока не будут найдены более точные значения)
можно принять равными 13 Дж/моль-К.
8.15. Ниже указаны энтальпии и температуры плав-
ления, а также теплоемкости бифенила и 1,2-
дифенилэтана. Найдите идеальные эвтектические темпе-
ратуры, учитывая теплоемость и пренебрегая ею.
Тт, °C нт, кал/моль Ср(/.), кал/моль CP(S.), кал/моль
Бифенил 69,0 4235 63,5 54,8
1,2-Дифенилэтан 51,25 5390 74,75 67,0
8.16. Четыре представленные ниже тройные смеси
образуют простые двойные и тройные эвтектики. Оце-
ните для каждого случая параметры уравнений Вильсо-
на и NRTL исходя из данных о бинарных эвтектиках.
Используйте полученные результаты для нахождения
тройных эвтектик и сравните их с экспериментальными
данными.
Энтальпии плавления первой группы веществ опре-
делены по правилу Вальдена с ЬН/Т =14. Данные по
эвтектикам взяты из справочника Ландольта-
Бернштейна [73]. Составы выражены в массовых про-
центах.
Эвтектика Те, °C Ментол СюНгоО Фенацетин C10H13O2N Антипирин CnHei2ON2
Тройная Е 31 82,5 1,5 16
Бинарная ei 75 — 40 60
62 40 96,8 3,2 —
ез 33 84 — 16
Эвтектика Те, °C о-Нитро- анилин C6H6O2N2 ^-Нитро- анилин C6H6O2N2 л-Нитро- анилин CeHeO2N2
Тройная Е 43,3 67 20 13
Бинарная ei 52,2 75,5 24,5 —
62 56,6 83 — 17
63 89,7 — 63 37
Эвтектика Те, °C 0-Динитро- бензол C6H4O4N2 .м-Динитро- бензол C6H4O4N2 л-Динитро- бензол CeH4O4N2
Тройная Е 60 30 60 10
Бинарная 64 66 34 —
62 82 — 84 16
63 101 73,5 — 26,5
Эвтектика Те, °C 1,3-Динитро- 2,4-Динитро- 2,4,6-Три- нитротолуол С7 H5O4N3
бензол C6H4O4N2 бензол C7H4O4N3
Тройная Е 29 35,5 31 33,5
Бинарная ei 43,2 43,5 56,5 —
62 51 45,5 — 54,5
63 45,8 — 52 48
Равновесие жидкость — твердое вещество 439
Вещество Т, °C ДЯ, кал/моль ДЯ/Т
Метанол 42,5 4420 (14)
Фенацетин 134,5 5708 (14)
Антипирин 113,0 5407 (14)
о-Нитроанилин 71,2 3847 11,17
.м-Нитроанилин 147,0 5037 11,99
л-Нитроанилин 114,0 5654 14,60
о- Динитробензол 116,93 5418 13,89
.м-Динитробензол 89,7 4150 11,44
л-Динитробензол 173,5 6718 15,04
1,3-Динитробензол 89,7 4750 11,44
2,4-Динитротолуол 70,14 4752 13,84
2,4,6-Тринитротолуол 80,83 5071 14,32
8.17. Четыре указанные на графиках системы обра-
зуют молекулярные соединения.
а. Найдите энтальпии плавления молекулярных со-
единений исходя из эвтектических условий и считая сис-
тему идеальной.
б. Считая, что энтропия плавления молекулярного
соединения представляет средневзвешенную энтропию
мономеров, найдите из эвтектических условий коэффи-
циенты активности.
Вещество тт, °C ДЯя, кал/моль ЬНт/Тт
Ацетамид 81,9 3396 9,56
Уксусная кислота 16,6 2755 9,51
Тетрахлорметан -23,0 784 3,13
Коричная кислота 133,0 5402 13,30
Диоксан 11,8 3067 10,76
Фенол 40,1 2695 8,58
Пикриновая кислота 122,0 4663 11,80
Мочевина 132,6 3472 8,56
Коричная кислота
Пикриновая
кислота
140
2:1
100 мол. % 100мол.%
Уксусная
кислота
8.18. Ниже в таблице приведены характеристики
простых эвтектических систем. Указанная эвтектическая
мольная доля относится к тому соединению, которое
названо первым.
а. Найдите энтальпию плавления там, где она от-
сутствует, считая систему идеальной.
б. Найдите коэффициенты активности в тех случа-
ях, когда энтальпия плавления известна.
Мочевина
Вещество Тт дят Д5т Xi у
Пиридин -41,5 1
Формамид 2,2 1
Эвтектика -56,7 0,677
Аценафтен 94,0 1
3,5-Динитротолуол 63,5 1
Вещество Тт ДЯт Д5т Х1 у
Эвтектика 46,0 0,3042
и-Дихлорбензол 52,9 4,273 13,10
п-Хлорфенол 42,9 1
Эвтектика 27,2 0,266
Камфора 175 1
о-Нитрофенол 44,5 3,720 11,71
Эвтектика 14,5 0,518
Камфора 175 1
Пикриновая кислота 121,5 4,663 11,81
Эвтектика 70 0,693
8.19. Бинарная смесь, содержащая 90% компонента
А, имеет следующие характеристики: Та = 400,
Тъ = 475, Д5в = ДЗл = 13. Найдите долю жидкости и
ее состав при температуре, находящейся посредине ин-
тервала между температурой эвтектики и температурой
плавления осаждающегося компонента.
8.20. Ниже в таблице приведены данные для трой-
ной смеси, твердые фазы которой несмешиваемы. Най-
дите температуру, при которой начинается осаждение,
и определите, какое вещество осаждается первым.
X ASm Т 1 т У
1 0,3 13 400 2,5
2 0,5 12 425 1,2
3 °,2 13 450 4
Вещество £ к мл/моль
Хлорбензол 9,62 101,8
Толуол 8,91 106,8
Гексан 7,27 131,6
Ацетон 9,51 73,5
Метанол 14,51 40,5
Нафталин 9,90 —
8.22. Используя уравнение Соава, определите рас-
творимость фенантрена в пропане при 400 К и давлении
от 1 до 400 атм. Физические свойства этих соединений
приведены в примере 8.11.
8.23. В кристаллизаторе непрерывного действия
требуется выделить n-ксилол из его смеси с jw-ксилолом
(50 на 50) при 80°Е Выход должен составить 95% тео-
ретического, а чистота 99,5%. В этом типе кристаллиза-
тора кристаллы, выходящие из аппарата, «отмывают-
ся» от окклюдированных примесей перемещающимся
им навстречу потоком расплавленного материала почти
такого же состава. Флегмовое число в данном примере
равно 1. Данные выражены в следующих единицах:
британская тепловая единица, фунт, °F.
Вещество Т Lm СР
п-Ксилол 55,8 68,09 0,41
jw-Ксилол -54,0 46,82 0,41
Эвтектика -70,6
8.21. В примере 8.4 указана растворимость нафта-
лина в нескольких растворителях при 40°С. Температу-
ра и энтальпия плавления нафталина составляют
353,4 К и 4558 кал/моль соответственно. Определите
растворимость нафталина:
а) считая систему идеальной;
б) используя уравнение Скэтчарда — Гйльдебранда;
в) используя уравнение Скэтчарда — Гильдебранда,
модифицированное Флори — Хаггинсом.
Равновесие жидкость — твердое вещество 441
Эвтектика содержит 86% Atezno-изомера. Составьте ма-
териальный и тепловой балансы этого процесса, взяв
100 фунт исходной смеси (см. рисунок).
Примечание. Процесс полного разделения этих изо-
меров описан Далем [240]. Пентан добавляют для того,
чтобы получить эвтектики с каждым из изомеров; в
указанном процессе кристаллизация сочетается с фрак-
ционированием.
8.24. Равновесные данные для системы антрацен +
+ фенантрен получены Брэдли и Маршем [193]. Требу-
ется разделить в колонне эквимолярную смесь при
218°С на компоненты с чистотой 99%. Флегмовое чис-
ло должно быть в 1,5 раза выше его минимального
значения. Найдите флегмовое число и число теоретиче-
ских тарелок, необходимые для этого разделения.
Фенантрен
Антрацен
99% А
8.25. Ниже в таблице указаны Д//т и Ср твердых
и жидких п- и At-изомеров ксилола.
а. Постройте диаграмму Т — х.
б. Смесь, содержащая 30% n-ксилола при 80°F, за-
гружают в дистилляционную колонну и получают прак-
тически чистый п-ксилол и эвтектику. Нижнее флег-
мовое число равно 1. Составьте уравнение теплового
баланса (используйте таблицу).
8.26. Имеется смесь пара- и мета-изомеров. Пер-
вый изомер, содержание которого равно 40%, необхо-
Изомер Т or ли брит.тепл.ед. брит. тепл. ед.
фунт • моль фунт • °F
Пара, тверд. 55,9 7362 0,322 + 0,00053 Т
Пара, жидк. 0,377 + 0,00049 Т
тверд. - 54,2 4977 0,313 + 0,00055 Т
Мета, жидк. 0,389 + 0,00038 Т
пара-Изомер хр пара-Изомер
Реактор
димо выделить. Как видно из фазовой диаграммы этой
смеси (см. рисунок), эвтектика содержит 15% пара-то-
мера. После отделения кристаллов эвтектика возвраща-
ется в реакционную камеру, в которой вновь достигает-
ся равновесие; после чего смесь, содержащая 35% пара-
изомера, опять поступает в кристаллизатор. Для ско-
рости образования лера-изомера 100 моль/ч определите
состав хр смеси, поступающей в кристаллизатор, и ко-
личество рециркулирующей эвтектики, Е. Некоторые
данные показаны на схеме.
8.27. Определите эвтектические условия для систе-
мы, состоящей из двух веществ с температурами плав-
ления 320 и 380 К и теплотами плавления 3500 и 2100
кал/моль. В первом случае предположите, что данная
система является идеальной, а во втором задайтесь зна-
чениями отношений активности. Параметры уравнения
Маргулеса: Ап = 1,2 и Л21 = 1,8.
9. ДРУГИЕ АСПЕКТЫ ФАЗОВОГО РАВНОВЕСИЯ
В данной главе кратко рассмотрены фазовые равнове-
сия других типов, их более подробный анализ содер-
жится в ряде работ, на которые даны соответствующие
ссылки. Методы расчета этих равновесий разработаны
далеко не для всех систем, потому что сами расчеты
обычно довольно сложны, а промышленное применение
таких систем довольно ограничено.
9.1. Адсорбция
Твердые вещества способны селективно поглощать
газы или жидкости. Эта способность приобретает прак-
тическую важность, если удельная поверхность адсор-
бентов достигает нескольких сотен квадратных метров
на грамм. Такие адсорбенты с развитой поверхностью
представляет собой вещества, пронизанные узкими по-
рами, которые после удаления летучих соединений ста-
новятся доступными для адсорбентов. Наиболее важ-
ные адсорбенты — это активный уголь, получаемый
при частичном выгорании или пиролизе карбонизиро-
ванного органического сырья, оксид алюминия, силика-
гель, цеолиты (молекулярные сита), активируемые пу-
тем дегидратации. Исходным материалом для силикаге-
ля служит аморфный кремнезем, осажденный из
силикатов кислотами, а для цеолитов — водные алю-
мосиликатные гидрогели, которые после перекристал-
лизации превращаются в пористые кристаллы.
Степень и избирательность адсорбции из смесей зави-
сит от химической природы жидкостей и твердых ве-
ществ, а также от физических условий, определяемых
температурой и давлением. Равновесие устанавливается
между объемной жидкой фазой и адсорбированной
жидкой фазой, последнюю можно рассматривать как
существующую в двух измерениях, на свойства которой
влияет природа поверхности твердого вещества. Если
поведение фаз неидеально, эти виды фазового равнове-
сия, как и любые другие, количественно можно выра-
зить через коэффициенты фугитивности и активности.
Влияние природы поверхности твердого вещества на
адсорбцию удобнее всего рассмотреть на примере сили-
кагеля. Способность силикагеля адсорбировать углево-
дороды убывает в следующем ряду: многоядерные аро-
матические соединения > соединения бензольного ряда
> диолефицы > парафины. При адсорбции на актив-
ных углях этот порядок нарушается. На рис. 9.1 показа-
но влияние температуры и давления, а также природы
адсорбента на адсорбцию этана. Величина адсорбции
паров может быть весьма значительной — до 0,5 — 1,0
г/г адсорбента — для таких веществ, как бензол,
галоген- и кислородсодержащие органические соедине-
ния [462]. Величина адсорбции обычно увеличивается
вместе с ростом давления, убывает с повышением тем-
пературы и с уменьшением концентрации адсорбата.
При регулировании процесса адсорбции и регенерации
адсорбентов за этими переменными осуществляется
контроль.
Аналитическое разделение веществ путем адсорбции,
так называемая адсорбционная хроматография, имеет
чрезвычайно важное значение. В промышленности ад-
сорбция используется преимущественно для выделения
или удаления небольших количеств примесей, например
тяжелых углеводородов или воды из природного газа,
токсичных газов из воздуха, растворителей из воздуха
при печатании и крашении, а также биологически вред-
ных огранических веществ, например фенола, из сточ-
ных вод. Адсорбция применяется при разделении ве-
ществ сравнимой концентрации; самый наглядный при-
мер такого ее применения — разделение с
использованием цеолитов ароматических соединений и
парафинов, разделение изомеров. Некоторое время про-
мышленное разделение этана и этилена осуществлялось
в адсорбере с движущимся слоем (процесс гиперсорбции
[462]), однако в настоящее время этот процесс не испо-
льзуется. С появлением молекулярных сит, отличаю-
щихся в некоторых случаях более высокой избиратель-
ностью, вновь возник интерес к использованию адсор-
бентов для разделения таких веществ, как пропен и
пропан, а также бутен и насыщенные вещества. Энерге-
тические затраты при таком разделении меньше, чем
при классической дистилляции или экстрактивной дис-
тилляции, однако необходимость использования нестан-
дартного оборудования в большинстве случаев затруд-
няет их распространение.
9.1.1. Измерения. Несмотря на то что данные о ско-
ростях адсорбции имеют большое практическое значе-
ние, в данном случае они не представляют для нас инте-
реса. Интересующая нас информация — это равновес-
ные количества и состав фаз как функции Т, Р и
концентрации жидкого адсорбата. Получить данные о
равновесии для чистых паров или паров, смешанных с
относительно слабо адсорбируемой средой, такой как
воздух, сравнительно просто: известные количества га-
за и твердого вещества помещают в замкнутый объем
Рис. 9.1. Влияние температуры, давления и типа адсорбента на количество адсорбированного этана
(данные Union Carbide Corporation).
1 — активный уголь при 25 °C; 2 — молекулярное сито (МС) типа 4А при О °C; 3— МС типа 5А при
25 °C; 4—МС типа 4А при 25 °C; 5 — МС типа 4А при 75 °C; 6—силикагель при 25 °C; 7 — МС
типа 4А при 150 °C.
таким образом, чтобы они соприкасались; величину ад-
сорбции определяют путем взвешивания твердого веще-
ства, а составы газа и адсорбата устанавливают посред-
ством подходящего аналитического метода. Множество
данных подобного рода собрано, например, в справоч-
нике Ландольта-Бернштейна [73].
При определении адсорбционных равновесий с жид-
кими адсорбатами необходимы косвенные методы из-
мерения с тем, чтобы исключить проникновение неад-
сорбированной жидкости в поры адсорбата. Один из
таких методов предусматривает следующее: жидкость и
адсорбент помещают в раздельных открытых сосудах
в замкнутый объем постепенно образующийся пар ад-
сорбируется вплоть до достижения равновесия между
жидкой фазой, паровой фазой и фазой адсорбата. Для
достижения равновесия в таких условиях требуется, по
разным оценкам, от четырех до шести недель. Практи-
куется также удаление окклюдированного материала
центрифугированием. При использовании хроматогра-
фического метода, как, например, в работе [259], опре-
деленное количество жидкости известного состава при-
водится в равновесие с известным количеством твердо-
го вещества, помещенного в колонку. После чего через
колонку пропускают растворитель, следя за изменением
определенных свойств вытекающего потока (элюата), и
исходя из них определяют состав и количество ад-
сорбата.
Поскольку внешняя поверхность гранулы пористого
адсорбента составляет лишь незначительную часть ее
полной поверхности, единственный предел величины
адсорбции определяется объемом пор, который, как
правило, составляет 30 — 60%. Обычно такие пределы
не достигаются, если только не применяются высокое
давление, так как притяжение жидкости к твердому те-
лу уменьшается по мере последовательного накопления
молекулярных слоев на поверхности. Однако в одном
часто упоминаемом в литературе случае, а именно при
адсорбции хлора активным углем в атмосферных усло-
виях, объем адсорбата, измеряемый в тех же условиях,
превышает объем пор, что указывает на сжатие жидко-
го хлора.
9.1.2. Равновесие при адсорбции газов. Величину ад-
сорбции газов обычно выражают в виде отношения
масс г адсорбата и адсорбента как функции Т и Р. Не-
смотря на то что уравнение вида
г=/(Т,Р), (9.1)
которое было бы справедливо для любого случая, неиз-
вестно, для описания адсорбции применяется множест
во видов уравнений, параметры которых характерны
для отдельных систем. Простейшее уравнение такого
рода — уравнение Фрейндлиха, названное по имени ис-
следователя, который часто использовал его в своих ра-
ботах, имеет следующий вид:
r=fcipl>2, (9.2)
где кг, как правило, превышает единицу. Очевидно, что
этому правилу отвечают только прямолинейные участ-
ки графика, показаного на рис. 9.2. Уравнение
Ленгмюра
г= кхР/{\ + к2Р). (9-3)
Другие аспекты фазового равновесия 445
Содержание в адсорбате, мол.доли первого из названных
компонентов
Рис. 9.2. Адсорбция бинарных смесей (данные Union Carbide Corporation).
1 — этан + этилен, МС типа 4А, 25°С, 250 мм рт. ст.; 2 — то же, но при
730 мм рт. ст.; 3 — то же, но при 75°С и 730 мм рт. ст.; 4 — диоксид углерода
+ сульфид водорода, МС типа 5А, 27°С, 760 мм рт. ст.; 5 — н-пентан + н-
гексан, МС типа 5А, 100°С, 760 мм рт. ст.; 6 — этан + этилен, силикагель, 25°С,
760 мм рт. ст.; 7 — этан + этилен, углерод колумбия G, 25°C, 760 мм рт. ст.;
8 — ацетилен + этилен, МС типа 4А, 31 °C, 740 мм рт. ст.
выведено (исходя из теоретических представлений). Это
уравнение было разработано для адсорбции мономоле-
кулярного слоя, однако нашло более широкое примене-
ние Одно из достоинств уравнения Ленгмюра состоит
в том, что его можно распространить на смеси, т.е. для
компонента i можно записать
г^к^, (9.4)
где — доля поверхности, не заполненной адсорбатом.
9„ = 1/(1 + Е к{Р{). (9.5)
Суммирование выполняется для всех веществ, присут-
ствующих в смеси. Чтобы можно было, исходя из экс-
периментальных данных, оценить константы, входящие
в уравнение (9.4), удобнее всего представить это уравне-
ние в линейной форме:
P,/r, = (l + Е kiP^/ki. (9.6)
Янг и Кроуэл [145] опубликовали обзор работ, прове-
денных с применением уравнения Ленгмюра. При высо-
ких давлениях некоторого улучшения степени соответ-
ствия уравнения Ленгмюра или Фрейндлиха можно
ожидать при использовании вместо давлений фугитив-
ностей, однако количество опубликованных данных об
адсорбции при высоких давлениях, которые можно ис-
пользовать для проверки, ограниченно. Льюис и др.
[440] исследовали адсорбцию при давлениях вплоть до
20 атм, однако, поскольку их результаты представлены
только в графической форме и наименьший коэффици-
ент фугитивности составляет приблизительно 0,95, они
не могут служить подтверждением сказанного. Авторы
работы [578] получили данные об адсорбции азота при
давлениях вплоть до 15 атм, но диапазон изменения ко-
эффициентов фугитивности очень мал — от 0,999 до
1,009.
Авторы работы [740] использовали своего рода ком-
бинацию уравнений Фрейндлиха и Ленгмюра; для опи-
сания адсорбции чистых веществ они применили вы-
ражение
г = (£Р)1/л/[1 + (£Р)1/л], (9.7)
или
г
1пР= -1пК + п\п—----------, (9.8)
1 — г
а для описания адсорбции смесей выражение
1 + Е^Р,)17"/
(9.9)
В некоторых случаях эффективные параметры для сме-
си можно получить исходя из результатов измерений,
проведенных для чистых веществ, однако, как правило,
также необходимы параметры взаимодействия.
Равновесие бинарных смесей удобно представить в
виде диаграммы х—у, которая выявляет сходство ад-
сорбиционного равновесия с другими видами фазового
равновесия (рис. 9.2). Если полученные кривые в доста-
точной степени симметричны, их можно описать урав-
нением адсорбции , в котором относительная лету-
честь (а) является константой:
(1 -л) 7(1 ~*1)
71 / *1
(9.Ю)
или
1 + (а - 1 )хj '
(9.11)
Однако, как правило, а не является постоянной величи-
ной, так как зависит от состава. Эту зависимость иног-
да выражают при помощи уравнения следующего вида:
а0(1 + *1-4)
а =----------------2 ,
1 + ^2Х1 + ^Зх1
(9.12)
которое рассматривается, например, в работе [50], но
для этих констант не разработано каких-либо корреля-
ций универсального характера. Уравнение, использован-
ное в работе [259]
7
1 “7
= к\
(9.13)
считать по существу двумерной, то в уравнении энерге-
тического баланса член PdV заменяют на член irdA, ха-
рактеризующий поверхностную энергию:
dU = TdS - nd А + Е paidnai, (9.15)
где A — полная поверхность адсорбента, а тг — потен-
циал поверхности, т. & количество энергии, приходя-
щейся на единицу площади поверхности, который обыч-
но называют поверхностным давлением по аналогии с
соответствующим членом уравнения энергетического
баланса жидкой фазы. Подстрочный индекс а указывает
на фазу адсорбата. Из этого уравнения получено урав-
нение Гиббса — Дюгема для фазы адсорбата
SdT~Adn + Е naidpai = 0, (9.16)
которое при постоянной температуре записывают так:
Adn = Е naidpai. (9.17)
В состоянии равновесия химические потенциалы жидкой
фазы и фазы адсорбата равны
ра! = pfi = RTin]}. (9.18)
Следовательно, уравнение поверхностного давления
принимает следующий вид:
RT
dn = — Е naid\nfi. (9.19)
А
Поскольку посредством уравнения состояния можно
выразить зависимость парциальных фугитивностей от
давления, то, если известно экспериментально установ-
ленное отношение давления системы, величины и соста-
ва адсорбата, можно выполнить интегрирование этого
уравнения и найти поверхностное давление. Эта опера-
ция еще более упрощается, если газообразная фаза яв-
ляется идеальным раствором и можно воспользоваться
правилом Льюиса — Рендалла
Л = 7(Л- (9.20)
Более того, при умеренных давлениях фугитивность
равна давлению, что дает возможность упростить
уравнение
(A/RT)dn = Е naid\n(yiP) (9.21)
= nt d InP + E naid In7,-. (9.22)
как выяснилось, можно применять и для газовых сме-
сей, по-видимому, можно также попытаться применить
его и для жидких смесей.
9.1.3. Термодинамические основы теории адсорбции.
Фундаментальное уравнение для жидкой фазы имеет
следующий вид:
dU = TdS - PdV + Е pfidnfi, (9.14)
где подстрочный индекс f указывает на химический по-
тенциал и состав жидкой фазы. Если фазу адсорбата
Для чистого вещества последний член опускают и полу-
чают следующее выражение для определения поверх-
ностного давления:
A n/RT =
(9.23)
Если исходить из уравнения Фрейндлиха
п = кгР1/к2,
(9.24)
Другие аспекты фазового равновесия 447
интеграл равен
An/RT= кхк2Рук2, (9.25)
а в соответствии с уравнением Ленгмюра
п = кхР/(1 + к2Р), (9.26)
он составляет
Атг/RT = (кх/к2)\п(\ + к2Р). (9.27)
На практике могут потребоваться более сложные эмпи-
рические уравнения адсорбции. Например, Снайдер и
Чао [650] при анализе энтальпий адсорбции и поверх-
ностных давлений легких углеводородов применили
уравнения с пятью константами.
Если допустить, что
х( = nai/nt, (9.28)
уравнение (9.22) приобретает для бинарной смеси сле-
дующий вид:
А Х1 х2
-----dn + dlnP +—dyx+ — dy2 = 0, (9.29)
у j у2
или же только для первого компонента
А Х1~У1
drr + d\nP + dy,=0. (9.30)
мт----------------------------------------У1(1 -У1) Л1
При постоянном давлении состав жидкой фазы опреде-
ляют по уравнению
Легко видеть, что при этом устраняется необходимость
экспериментального определения состава адсорбата, ес-
ли имеются другие данные. В работе ван Несса [705]
описан метод получения таких данных, основанный на
измерении зависимости поверхностного давления от
предельной адсорбции при постоянных Т и Р для не-
скольких постоянных составов паровой фазы, однако
автор не располагал данными, которые позволили бы
ему проверить полученные результаты.
Адсорбционное равновесие смеси, представленное ав-
тором работы [499] при помощи закона Рауля, опреде-
ляется следующим выражением:
y^P^Xi/P (9.32)
гдеР* — кажущееся давление пара чистого компонента,
адсорбированного при той же температуре и поверх-
ностном давлении, что и смесь. Состав бинарной смеси,
например, определяют из такого уравнения:
X! = (P-P?)/(Pt-P5). (9.33)
Например, если уравнение поверхностного давления вы-
водят из изотерм Фрейндлиха, кажущееся давление пара
определяется как
1
Р? = — (rrA/RT)bi,
«1
1
Р? = — (nA/RT)b2,
а2
(9.34)
(9.35)
а мольная доля адсорбата при заданных давлении и
температуре составит
1
Р - — (irA/RT)b2
(9.36)
— (nA/RT)bi - — (nA/RT)b2
, °1 а2
Если молярную площадь поверхности А/п, т. е. пло-
щадь поверхности адсорбента, приходящуюся на едини-
цу количества адсорбата, можно аппроксимировать в
виде средневзвешенной суммы молярных площадей чис-
тых компонентов, то
1/и = Е Xi/rii.
(9.37)
Таким образом, полную адсорбцию смеси можно найти
в том случае, если известны величины адсорбции чис-
тых компонентов при одинаковом давлении, а мольные
доли определены по уравнению (9.36). В примере 9.1
показана корреляция поверхностных давлений чистых
веществ и их использование для прогнозирования усло-
вий адсорбционного равновесия бинарных смесей.
Неидеальность фазы адсорбата учитывается путем
введения коэффициента активности, в результате чего
закон Рауля преобразуется к следующему виду:
Л = У{Р*х{/Р.
(9.38)
Этот подход применен в работе [234], авторы которой
использовали данные об адсорбции чистых веществ и
бинарных смесей для прогнозирования равновесия в
тройных смесях. Поверхностные давления бинарных
смесей получают путем интегрирования
) «Р У1(1~ Л)
(9.39)
После чего соответствующие величины кажущихся дав-
лений пара Р* и Pi находят исходя из данных о чистых
компонентах. Коэффициенты активности рассчитывают
по уравнению (9.38) и коррелируют при помощи либо
уравнения Вильсона, либо уравнения UNIQUAC.Ha рис.
9.3 показаны полученные таким образом данные о сме-
сях этилена и пропилена. Условия равновесия в двух
тройных системах углеводородов были рассчитаны на
основе двух корреляций; мольные доли обычно не вы-
ходили за пределы 0,01. При более высоких давлениях
необходимо учитывать неидеальность паровой фазы,
Рис.9.3. Данные об адсорбционном равновесии (мол.
доли) (а) и коэффициентах активности смесей этилена
и пропилена при 20°С и 75 мм рт. ст. (б) [234].
1 — экспериментальные точки; 2,3 — расчетные дан-
ные, полученные по уравнениям UNIQUAC (2) и Вильсо-
на (5).
применяя вместо закона Рауля следующее выражение:
Ф(РУ1 = 7iP*xi,
или более полный его вариант:
4>iPyi =
(9.40)
(9.41)
однако для анализа адсорбции при высоком давлении
подобная методика пока применена не была.
9.1.4. Жидкости. Практически все исследования ад-
сорбции жидкостей проводились с использованием чис-
тых веществ или двойных смесей. В тех случаях, когда
концентрация растворенного вещества мала, систему
можно рассматривать как однокомпонентную. Для
жидких растворов и разбавленных газов в равной мере
справедливы и уравнение Фрейндлиха, и уравнение
Ленгмюра. В справочнике Ландольта-Бернштейна [73]
приведены константы уравнения Фрейндлиха примерно
для ста жидких систем, для смесей же даны только
ссылки на литературу. Адсорбция чистой жидкости
практически не зависит от давления; температура обра-
зования жидкой фазы (конденсации ) и природа жидкос-
ти являются переменными, связанными с определен-
ным адсорбентом.
Равновесие смешивающихся двойных жидких смесей
удобно представить в виде диаграммы х—х*, где х* —
адсорбат. Вид таких диаграмм зависит от природы как
адсорбента, так и жидких фаз. Сравнивая показанные
на рис. 9.4 кривые равновесия пар — жидкость и пар
— адсорбат, нетрудно проследить эту зависимость. До
сих пор еще не разработана достаточно простая мето-
дика прогнозирования диаграмм х—х* или представле-
ния их в обобщенном математическом виде. Некоторые
графики можно представить в единицах относительной
летучести, уравнение (9.10). Игл и Скотт [259] примени-
ли уравнение (9.13) для описания жидких смесей углево-
дородов; для смеси толуол + изооктан экспонента ki
варьируется в пределах от 0,628 до 0,881 в зависимости
от типа адсорбента. Данное уравнение можно перегруп-
пировать, чтобы выразить относительную летучесть
как функцию состава жидкости:
*2~1
1 — Xj
1
а = кх
(9.42)
в таком виде оно может в некоторых случаях быть
предпочтительнее уравнения (9.12), однако сравнение
этих уравнений на конкретных примерах не проводи-
лось. На рис. 9.5 и 9.6 представлены некоторые данные
по адсорбции жидких смесей.
Термодинамический анализ равновесия жидкость —
адсорбат начинают с допущения равенства химических
потенциалов или парциальных фугитивностей двух фаз:
(9.43)
Если пренебречь коэффициентом Пойнтинга, фугитив-
ность чистого компонента в левой части уравнения ста-
новится равной фугитивности насыщенных паров:
Л = Ф^, (9.44)
а чтобы определить/?, проводят такую же подстанов-
ку, как и в уравнении (9.38),
J?-p* (9.45)
где Р? — это кажущееся давление пара, соответствую-
щее поверхностному давлению смеси. После выполне-
ния обеих подстановок равновесное состояние отвечает
следующему условию:
y„0fPfci = у*Р*х*. (9.46)
Подобная методика может применяться для анализа
двух типов задач.
1. Исходя из результатов одного или всего нескольких
измерений равновесного состава жидкость — адсорбат,
можно найти параметры уравнения коэффициента ак-
тивности, которые можно использовать при расчете
равновесия адсорбции для всего диапазона составов.
Другие аспекты фазового равновесия 449
10-864
Содержание ацетона в жидкой фазе или адсорбате
Рис. 9.4. Равновесие пар — жидкость и пар — адсорбат
на древесном угле [695].
Рис. 9.5. Адсорбционное равновесие бинарной жидкости на диаграммах х — у.
1 — толуол + изооктан на силикагеле [259]; 2 — то же на древесном угле [259];
3 — дихлорэтилен + бензол на бемите [774]; 4 — то же на древесном угле [774].
мал.аили
Рис. 9.6. Адсорбция жидких смесей хлороформ + ацетон и бензол + этанол на
древесном угле [65]. На оси ординат отложено количество каждой отдельной ад-
сорбируемой жидкости, на оси абсцисс — мольная доля хлороформа (смешанно-
го с ацетоном) или мольная доля бензола (смешанного с этанолом).
Другие аспекты фазового равновесия 451
2. Параметры уравнения, используемого для вычисле-
ния коэффициентов активности, можно найти путем по-
строения регрессионных зависимостей, используя весь
интервал равновесий, поддающихся измерению.
Для выполнения подобных расчетов, помимо равно-
весных составов жидкость — адсорбат, необходимо
также располагать и рядом других данных, а именно:
коэффициентами активности жидкой фазы, поверхност-
ным давлением чистых компонентов и поверхностным
давлением смеси как функцией состава. Коэффициенты
активности жидкости можно получить посредством не-
зависимых измерений равновесия пар — жидкость, а
также при помощи метода UNIQUAC или ASOG.
При подстановке
d\nf= (V/RT)dP (9.47)
в уравнение (9.19) для чистого компонента поверхност-
поскольку и удельный объем, и величина адсорбции
жидкости по существу не зависят от давления. Для иде-
альных растворов fi = х/,, а уравнение (9.19) приводит-
ся к виду
А
—-d-n = 'L njdXnfi + 'L nidXnXj. (9.49)
Если Р и Т — постоянные величины, дифференциалы
фугитивностей чистого компонента опускают и получа-
ют выражение
А
---dir = Е n;dXnX:. (9.50)
RT 1 1
Таким образом, чтобы уравнение (9.39) соответствовало
равновесию жидкость — адсорбат, применительно к
двойной смеси его преобразуют к виду
ное давление определяется как
Атт nV
— = —Р,
RT RT
(9.48)
А М**~*1)
(77-71,)= / dx{.
RT J1 x,( 1 — x,)
(9.51)
Пример 9.1. Расчет равновесной адсорбции бинарной смеси на основе данных об адсорбции чистых компонентов
Майерс [49] применил для прогнозирования адсорб-
ции смесей диоксида углерода (1) и этилена (2) данные
об их адсорбции при различных давлениях и температу-
ре 20 °C. Величину поверхностного давления каждого из
веществ рассчитывают по уравнению (9.32):
р*
Чтобы упростить расчет, цифровые данные Майерса,
приведенные в его работе в виде небольших таблиц,
представлены в форме эмпирических уравнений:
РГ = (6,375 + 0,0656тгД/Л7)1гЛ/РТ,
Р1 = (0,500 + 0,0438itA/RT)ttA/RT.
Соотношение между равновесными составами пара и
адсорбата определяют следующим образом (давление
равно 760 мм рт. ст., температура 20 ° С).
1. Принимают такое значение irA/RT, которое в ко-
нечном итоге даст величину Xi в интервале от нуля до
единицы.
2. Находят соответствующие величины кажущегося
давления пара из уравнений (1) и (2).
3. Вычисляют мольную долю компонента 1 в фазе
адсорбата по уравнению
Xi = (760 - Р!)/(Р? - Pi)
и мольную долю паровой фазы по уравнению
У1 = Pfxi/760.
4. Подбирают такое число значений irA/RT, которое
позволяет включить в расчет весь необходимый интер-
вал составов.
Результаты расчета xi в интервале от 0 до 1 показаны
ниже в виде таблицы.
S] | i- рГ Л* Х1 У1
69,51 760,2 246,1 1,000 1,000
70 767,8 249,4 0,985 0,995
80,00 930,0 320,0 0,721 0,883
90,00 1105,4 399,4 0,511 0,743
100,00 1293,8 487,5 0,338 0,575
110,00 1495,4 584,4 0,193 0,379
120,00 1710,1 690,0 0,069 0,154
125,00 1822,3 746,1 0,013 0,031
126,20 1849,8 759,9 0,000 0,000
Величину полной адсорбции при определенном соста-
ве адсорбата находят по уравнению (9.37):
П - 1/(Х1/П1 +Х2/П2).
Величины «1 и «2 при соответствующих значениях Р?и
Р£ являются экспериментальными, их считывают с но-
мограмм, приведенных в работе Майерса [499]. Во вто-
рой таблице данного примера собраны результаты для
различных составов. Величина адсорбции измеряется в
миллилитрах (при нормальных температуре и давле-
нии) в расчете на грамм углерода.
У1 Х1 А т RT Р* Л* «1 П2 п
1 1 69,5 760 48 0 48
0,883 0,721 80 930 320 55 44 51,4
0,575 0,338 100 1294 488 64 57 59,2
0 0 126,2 0 760 0 64 64
Пример 9.2. Расчет коэффициентов активности адсорбата по данным об адсорбции бинарных жидких смесей
В этом примере коэффициенты активности адсорбата
будут определены для заданной мольной доли объем-
ной жидкой фазы (xi = 0,7). Для расчетов используются
упрощенные зависимости равновесия.
1. Рабочее давление 20 атм, A/RT = 1.
2. Давление пара Р\ - 12 атм, Рг = 20 атм; удельные
объемы определяют как Vi/RT = 0,002, Уг/РТ = 0,0012;
коэффициенты фугитивности насыщенных паров
Ф1 = Ф2= 1.
3. Величины равновесной адсорбции чистых компо-
нентов П1 = 0,5 моль/кг, «2 = 0,3 моль/кг.
4. Равновесная адсорбция смеси представлена уравне-
нием Фрейндлиха
п = пмь‘ + и(1 - Xi)^ = 0,5х?-4 + 0,3(1 - xi)0’5.
5. Параметры уравнения ван Лаара для коэффициен-
тов активности жидкой фазы А = 1,8939, В = 3,8055.
При Xi = 0,7 yi = 1,5, у2 = 3,0.
6. Равновесие между жидкостью и адсорбатом опре-
деляют по выражению
9. При заданном поверхностном давлении кажущиеся
давления пара равны
Vi/RT
Vi/RT
0,1205
002
0,1205
0,0012
= 60,3,
100,4.
10. После соответствующих подстановок в уравнение
(9.52) получаем
ф _ yi^W'x! _ 1,5(12)(0,7)
71 В*г* 60,3(0,6087)
3(2О)(О,3)
100,4(0,3913)
= 0,4582
11. Параметры уравнения ван Лаара для фазы адсор-
бата находят, используя уравнения (4.93) и (4.94):
а — (а — 1)%1
при а = 1,5 и х* = 0,6087 для Xi = 0,7.
7. Поскольку в этом случае A/RT = 1, поверхностные
давления, найденные из уравнения (9.48), соответствен-
но равны
in = (Vi/RT)P = 0,002Р = 0,04 при 20 атм,
т2 = 0,0012В = 0,024.
8. Применяя уравнение (9.51) для смеси, получаем
f И(Х1*-Х1)
тг = Tri + I ----------axi =
] Х1(1 - Xi)
'1
1
!п
------;----, ~
а - (а - 1)%1
Х1
Г 0,5xi’4 + 0,3(1 - Х1)0-5
= 0,4+ ---------------------dxi = 0,1205.
J 3-2xi
0,7
Л* = InyJ
В* = In*
X* 1П?2*1 2
1 + »
Xj 1п71
j ! !п7* 2
Х2* 1пу2
2,3080,
7,6510.
12. Составы адсорбата х* для всего диапазона соста-
вов объемной жидкой фазы xi находят по уравнению
(9.52) после его перегруппировки:
, 71 ВТ 71 12
хГ= — = —- = =----------Хь
71 Р* 71 тг/0,002
Далее определяют каждую величину Xi, соответствую-
щие поверхностные давления находят, как описано в
п. 8, коэффициент активности 7i получают через А и
В. Поскольку у* зависит от х*, заключительный этап со-
стоит в решении методом подбора относительно х* с
использованием величин Л* и В* найденных, как указа-
но в п. 11.
Рассчитанные таким образом величины (хь xf) мож-
но сопоставить с результатами решения уравнения, ука-
занного в п. 6.
Аналогичным образом, используя уравнение (9.38) и все
последующие уравнения, находят коэффициенты актив-
ности адсорбата. Коэффициент активности адсорбата
равен
7/0?atP?atxz
> (9.52)
где В,* определяют при поверхностном давлении смеси
состава х(, который находят по уравнению (9.51). Все
необходимые для такого расчета данные, по-видимому,
не были получены ни для одной из исследованных сис-
тем. В примере 9.2, который приведен выше, испо-
льзуются совершенно произвольные данные.
Другие аспекты фазового равновесия 453
9.2. Системы водных электролитов
Помимо того что вода представляет собой самое рас-
пространенное в природе соединение, она отличается от
других достаточно распространенных соединений вы-
сокой степенью молекулярной ассоциации и высокой
пробивной напряженностью. В силу первого из этих
свойств невозможно достаточно точно описать воду
при помощи приведенного уравнения состояния, а в си-
лу второго — степень диссоциации на ионы растворен-
ных в воде неорганических веществ может быть различ-
ной. Поэтому, чтобы описание фазового равновесия
водных систем было точным, следует учитывать хими-
ческое равновесие молекулярной ассоциации и ионной
диссоциации.
Возобновление исследований в данном направлении
связано с борьбой с загрязнением окружающей среды
и, в частности, с разработкой процессов переработки
угля, связанных со сбросом значительных количеств за-
грязненных сточных вод. Технология удаления SO2, H2S
и СОг абсорбцией их водными растворами достаточно
устоялась, тем не менее ее можно усовершенствовать,
особенно применительно к сложным смесям. Воздейст-
вие растворенных веществ на растворимость газов и
жидкостей и на относительную летучесть жидких сме-
сей также представляет значительный интерес для ин-
женеров-технологов .
Составить представление о современном состоянии
проблемы можно по материалам относительно недавно
прошедшего симпозиума, опубликованным в 1980 г. под
редакцией Ньюмена [14]. Ренон [586] приводит подроб-
ный обзор корреляционных методов исследования элек-
тролитического равновесия. Большинство работ, цити-
руемых в данном разделе книги, опубликованы в ука-
занном выше сборнике.
Изучая системы водных электролитов исследователи
пользуются как эмпирическими, так и теоретическими
методами. Фазовое равновесие водных систем, содержа-
щих легкие углеводороды и неорганические газы, было
в общих чертах описано Пенгом и Робинсоном [549],
применившим свое уравнение состояния в сочетании со
специальными параметрами взаимодействия бинарных
смесей с водой. Указанные параметры в очень большой
степени зависят от температуры и компонентов и варьи-
руют в интервале от — 0,7 до 0,4, для неводных двой-
ных смесей этот диапазон значительно уже — от 0 до
0,2. Результатами данного анализа, по-видимому, впол-
не можно пользоваться при решении ряда технологиче-
ских задач.
В целях анализа систем с низкой взаимной раствори-
мостью Баумгартнер и др. [14] выполнили проверку не-
скольких моделей молекулярной ассоциации воды; наи-
лучшие результаты дала комбинация 1, 2, 4, 8 и 12 вод-
ных олигомеров. Эти виды равновесия олигомеризации
совместно с кубическим уравнением состояния испо-
льзовались в ряде случаев для представления равнове-
сия пар — жидкость при давлении до 500 бар, в основу
которого было положено кубическое уравнение. Ассоци-
ация в паровой фазе была незначительной — менее 2%;
некоторые данные, полученные Баумгартнером и др.
[14] для жидкой фазы представлены ниже.
Т, ° К Содержание олигомеров, мол. доли
1 2 4 8 12
273 0,362 0,031 0,127 0,349 0,132
647(ТС) 0,866 0,082 0,052 0,0003 —
В ходе анализа остаточных энтальпий смесей пара с
углеводородами Уормалд и Коуллинг [14] рассмотрели
зависимость вириальных коэффициентов от констант
равновесия при ассоциации. В разд. 1.4 выводятся урав-
нения для подобной зависимости при димеризации. Для
нескольких первых коэффициентов эти уравнения имеют
следующий вид:
В = BQ- k2RT, (9.53)
С = Cq — (2к3 — 4k2)(RT)2, (9.54)
D = Dq~ (Зк4- 18Л2А:з + 20^)(ЯГ)3, (9.55)
и т. д. Во, Со и Do — это вириальные коэффициенты
в отсутствие ассоциации, кг — константа равновесия
при димеризации, кз — константа равновесия при три-
меризации и т. д. Однако исследователи были вынужде-
ны отказаться от непосредственного использования
этих уравнений ввиду их сложности, а также ввиду за-
висимости параметров от температуры и трудности
формулирования коэффициентов перекрестных взаимо-
действий. Взамен было выдвинуто предположение, что
остаточная энтальпия складывается из энтальпии неас-
социирующей эталонной паровой фазы и поправки на
ассоциацию. Таким образом,
AH' = АН'(В0, Cq,Dq, . . . ) + АН'(к2, к3,к4>... ).
(9.56)
Как выяснилось, эта методика предпочтительнее мето-
дики Пенга и Робинсона при рассмотрении смесей паро-
вых фаз, содержащих гептан, причем оптимальный па-
раметр бинарного взаимодействия, который они смогли
вывести, равнялся - 1,3.
Методика интерпретации воздействия ионного равно-
весия на термодинамические свойства смесей разработа-
на Питцером и др., им же выполнен обзор аналогичных
работ [557]. Основная посылка заключается в том, что
избыточную энергию Гиббса можно представить вири-
альным рядом, который включает члены, описываю-
щие взаимодействие пар и триплетов компонентов, а
также член, учитывающий бесконечное разбавление и
ионную силу /, рассматриваемую как основное свой-
ство. Таким образом,
GeK/RT = f(I) + SS XfXj + SSS XiXjXkp.ijk.
(9.57)
Член, учитывающий бесконечное разбавление /(/), явля-
ется функцией только ионной силы, и его представляют
эмпирической модификацией уравнения Дебая — Хюк-
келя. Ху также зависит от ионной силы. Соотношения,
определяющие как Ху, так и /ху*, основаны на использо-
вании измеряемых параметров для катионов и анионов
различных зарядов.
Чен и др. [219] применили эту методику для описания
взаимодействия молекул с ионами и другими молекула-
ми; при помощи шести подгоночных параметров им
удалось достаточно хорошо представить системы типа
КНСОз + К2СОз + СО2 + Н2О.
Как утверждает Ренон [586], метод Питцера имеет
ряд ограничений. 1. Вода должна оставаться основным
компонентом. 2. Зависимость параметров от темпера-
туры весьма значительна и ее характер неоднозначен.
3. Ковариантность параметров высока. 4. Каждой от-
дельной системе присущи свои специфические парамет-
ры. В целях устранения этих трудностей при разработке
модели NRTL Круз и Ренон [14], а также Чен и др. учли
как воздействие ионного равновесия на избыточную
энергию Гиббса, так и влияние «локального состава».
Хорошее соответствие экспериментальным данным для
бинарных и тройных систем было получено только при
использовании бинарных параметров. Маурер [14] вы-
полнил детальное сопоставление трех методов (в число
которых, однако, не вошел метод Питцера — NRTL)
расчета фазового равновесия летучих электролитов, та-
ких, как NH3, СОг, SO2 и H2S.
Экспериментальные данные о системах электролитов
имеют, безусловно, большое значение для разработки
методов корреляции. В библиографический обзор Вил-
хойта входит 145 наименований [14]. В том же сборнике
статей опубликованы оригинальные данные о летучести
аммиака, об удалении SO2 доломитным шламом и рас-
творами цитрата натрия.
В тех случаях, когда помимо фазового равновесия
учитывается и равновесие нескольких химических реак-
ций, расчет значительно усложняется. Наиболее прос-
тые примеры подобного рода рассмотрены в гл. 10. Ре-
нон [586] приводит основные положения программ
ЭВМ для расчета равновесия пар — жидкость при нали-
чии летучих электролитов (СОг, SO2, H2S).
9.3. Соединения и смеси неопределенного
состава
Несмотря на то что принцип соответственных состоя-
ний является краеугольным камнем прикладной термо-
динамики, для его применения необходимо располагать
данными о критических свойствах, что не всегда воз-
можно. Некоторые соединения разлагаются еще до мо-
мента достижения критической температуры, а смеси
могут состоять из многих соединений с близкими тем-
пературами кипения, состав которых может быть неиз-
вестен. Примером таких смесей могут служить нефтя-
ные фракции, каменноугольные смолы и другие природ-
ные смеси.
В результате долголетних исследований была разра-
ботана методика, позволяющая рассматривать нефтя-
ные смеси как состоящие из псевдокомпонентов со сред-
ней температурой кипения в интервале от 5 до 10 °C и
плотностью соответствующей фракции. Исходя из этих
двух основных свойств, были разработаны корреляции
для определения молекулярных масс, ацентрических ко-
эффициентов, критической температуры и критического
давления, а также пропорций ароматических, нафтено-
вых и парафиновых составляющих; некоторые из этих
свойств представлены в табл. 9.1. Поскольку эти корре-
ляции выведены на основе данных, полученных при
температуре ниже 650 °C, их не рекомендуется приме-
нять для анализа тяжелых остатков и, вероятно, про-
дуктов перегонки каменного угля, содержащих главным
образом циклические соединения. Для выполнения рас-
четов по мгновенному испарению нефтяных фракций
используется метод, основанный на уравнении Соава;
как установлено, это наиболее точный метод из числа
проанализированных Симсом и Даубертом [637], хотя
следует отметить, что результаты всех этих методов
неудовлетворительны при величине испарения ниже
примерно 20%.
Ньюман [515] осуществил проверку справедливости
ряда широко применяемых корреляций для определения
молекулярной массы, критических температуры и дав-
ления, и также давления пара некоторых циклических
соединений, содержащихся в каменноугольных смолах.
Полученные результаты говорят о том, что для каждой
фракции необходимо измерять или оценивать соотно-
шение ароматических, нафтеновых и парафиновых угле-
водородов, в противном случае при прогнозировании
давления паров жидкостей по методу Соава, Ли — Кес-
лера или Питцера — Керля можно ожидать погрешно-
стей в размере 10—15%. Результаты прогнозирования,
касающиеся твердых веществ, гораздо менее точны.
Таблица 9.1. Характеристики нефтяных фракций с узким диапазоном температур кипения
1. Ть — температура кипения при атмосферном давлении, К.
2. S — удельная масса 15/15°С.
3. М — молекулярная масса = 219,06 7*’1185'1’88ехр(0,003927г, - 3,075).
4. Рс — критическое давление, атм = 8O2,1(1O6)7V2’3125S2’3201.
5. Тс — критическая температура, К = 34,31227*’588485°’3596.
6. а) — ацентрический коэффициент = -1 + 0,18611пРс/(Тс/Ть - 1)
(Эдмистер, Petroleum Refiner 37(4): 173 — 179(1958)) [265].
Более точное уравнение для углеводородов, предложенное Ли и Кеслером, приводится в разд. 1.3.1.
7. Для фракций с узким диапазоном температур кипения различие между разными видами средних темпера-
тур кипения исчезает, и псевдокритические и критические свойства становятся тождественными.
Пп. 3—5 взяты из справочника API (Американского нефтяного института).
Другие аспекты фазового равновесия 455
Вильсон [728] приводит ряд других примеров корреля-
ций давления пара, плотностей жидкостей, поверхност-
ного натяжения и вязкости каменноугольных смол.
Авторами работы [206] разработано трехпараметри-
ческое уравнение типа уравнения Бенедикта — Уэбба —
Рубина (среди параметров отсутствуют Та Рс и ш) для
прогнозирования фазового равновесия и термодинами-
ческих свойств; к сожалению, необходимую проверку
данной методики осуществить нельзя из-за недостатка
экспериментальных данных (экспериментально установ-
лена лишь одна характеристика). Сказать что-нибудь
определенное о свойствах жидких продуктов переработ-
ки каменного угля можно будет после опубликования
справочника, работа под которым ведется вот уже не-
сколько лет при поддержке Института технологии газа
(Institute of Gas Technology, проект 8979, опубликовано
несколько годовых отчетов).
Все еще интенсивно ведется исследование нефтяных
фракций. Ярборо [736] применил методику псевдоком-
понентов, разработанную Американским нефтяным
институтом (API) для комплексного исследования фазо-
вого равновесия. В основе этой методики лежит уравне-
ние Редлиха — Квонга, в котором параметры определя-
ются как функции ацентрического коэффициента и при-
веденной температуры:
a=/e(w, Tr)R2T25/Pc, (9.58)
b=fb(t», Tr)RTc/Pc, (9.59)
причем особое внимание обращается на исходные про-
дукты и условия.
Аналогичный подход был вероятно применен и Ро-
бинсоном и др. [597], с тем чтобы сделать возможным
использование уравнения Пенга — Робинсона. Необхо-
димые для расчетов свойства определяют, проводя пе-
регонку в соответствии с американским стандартным
методом испытаний (ASTM), или же хроматографиче-
ским методом устанавливают число углеродных ато-
мов. Важные особенности этого метода содержатся в
программе для ЭВМ, разработанной Ассоциацией по
переработке газа (Gas Processors Association, lUlsa). Ме-
тодика, принятая в «Справочнике Американского не-
фтяного института», предусматривает использование
уравнения Соава. В основу ее положено представление
о псевдокомпонентах, свойства которых рассчитывают
по формулам, подобным приведенным в табл. 9.1. Па-
раметры бинарного взаимодействия с неорганическими
газами известны только для ряда соединений от н-дека-
на до триметилбензола (табл. 1.12), однако возможна
экстраполяция с известной степенью вероятности. Хро-
матографический анализ позволяет обнаруживать со-
единения, содержащие до пятидесяти атомов углерода.
Некоторые свойства соединений можно с определен-
ной степенью точности оценить методом структурных
вкладов. В литературе [80, 107, 129] описаны способы
расчета нормальных температур кипения и критических
свойств. На основе этих свойств определяют ацентриче-
ский коэффициент, используя, например, формулу
Эдмистера. Наименее точные результаты метод струк-
турных вкладов дает при оценке нормальной темпера-
туры кипения; однако если о веществе данной структу-
ры хоть что-нибудь известно, то это, как правило, све-
дения о температуре кипения при нормальных
температуре и давлении или по крайней мере давлении
пара при некоторой температуре.
В настоящее время ведется разработка уравнения со-
стояния, параметры которого можно определить непо-
средственно методом групповых вкладов. Это уравне-
ние PFGC (parameters from group contributions — пара-
метры на основе групповых вкладов) в перспективе
будет иметь особое значение при анализе компонентов
или смесей неопределенного состава; отпадет необходи-
мость в данных о критических свойствах и ацентриче-
ских коэффициентах. Содержащиеся в соединениях груп-
пы можно определять при помощи современных анали-
тических методов, например метода ЯМР13С. Опубли-
ковано несколько статей, в которых рассматриваются
перспективы такого направления ( см., например, [496]),
однако величины рассчитываемых таким образом пара-
метров будут приведены только после подтверждения
их правильности. Данный метод распространяется на
ароматические и другие типы молекул, содержащие N,
О и S, образующиеся в процессе сжижения угля, галоге-
нированные хладагенты и органические соединения. В
вышеупомянутой книге рассматривается система мета-
нол + вода + углеводороды + кислые газы; однако
для представления данной системы одинаково пригод-
ны и уравнение PFGC и уравнение Соава.
9.4. Полимеры
Фазовое равновесие полимеров играет важную роль в
ряде процессов, например в процессах 1) смешения по-
лимеров с пластификаторами, мономерами и другими
жидкостями; 2) испарения мономеров из растворов; 3)
смешения различных полимеров; 4) плавления полиме-
ров. Вплоть до настоящего времени ни для одного из
этих процессов не дано достаточно хорошего количест-
венного термодинамического описания, хотя работы в
этом направлении ведутся и сейчас. Тот факт, что поли-
меры, как правило, не подвергаются многостадийным
процессам разделения, не способствовал экономическо-
му стимулированию исследований их фазового поведе-
ния. Кроме того, решение данной проблемы в опреде-
ленной степени усложняется разнообразием возможных
состояний полимеров. Это могут быть смеси, молеку-
лярные массы компонентов которых соответствуют не-
которому определенному диапазону, и аморфные сте-
кловидные или каучукоподобные вещества, или же в за-
висимости от температуры и предыстории они могут
иметь более одной кристаллической формы. Будет
уместно процитировать замечание Бонди [190] относи-
тельно того, что его обзор литературы по термодинами-
ке фазового поведения полимеров «отражает недоста-
точный современный уровень знаний» по этому вопросу.
Исследуя смешение растворов, Флори и Хаггинс при-
шли к выводу о значимости того большого различия
в молекулярных размерах, которое характерно для мо-
номеров и полимеров. Результаты этих исследований
будут кратко описаны ниже. Удобная методика решения
указанных вопросов содержится в книгах Хаггинса [60],
где также рассмотрены и некоторые более ранние пуб-
ликации, и Чао и Гринкорна [27]. При значительном
различии молекулярных размеров компонентов смеси
энтропия смешения более точно выражается в объем-
ных, а не мольных долях. Таким образом, для смеси,
содержащей ni молей мономера и пг молей полимера,
молекулярная масса которого в т раз превышает моле-
кулярную массу мономера, используют выражение
ASmlx//? = — «1 in 0i — п2 in 02» (9.60)
вместо
ASmix//?(ideal) = — jq inxj — x2lnx2, (9.61)
Объемные доли определяют как
0! = пх/(пх 4-тп2), (9.62)
02 = тп2/(п1 4- тп2). (9.63)
Изменение энтальпии при смешении прежде всего
пропорционально массе системы, которая в свою оче-
редь пропорциональна (»i + тпг). Более того, функция,
представляющая эту энтальпию, при фх = 0 или фг = 0
должна обратиться в нуль. Простейшее уравнение, от-
ражающее эти свойства, имеет следующий вид:
Д//т1Х/7?Г = х(п1 + тп2)ф1ф2, (9.64)
где х — эмпирический параметр, названный парамет-
ром Флори, величина которого зависит от природы и
состава системы. Некоторые его численные величины
приводятся в упомянутых выше книгах Хаггинса, а так-
же Чао и Гринкорна. При температуре 25 °C в таблице
Хаггинса они варьируют от 0,14 до 0,91 примерно для
сорока данных. Параметр Флори зависит от парамет-
ров растворимости; так
X = Х0 + 77-^1 “ й2)2- (9.65)
ЛЧ 1
Объединяя уравнения для энтропии и энтальпии, по-
лучаем следующее уравнение энергии Гиббса при
смешении:
AGmix//?r= «1 in 0i 4- п2 in 02 4- х(п1 + тп2)Ф1Ф2-
(9.66)
Это уравнение легко распространить на многокомпо-
нентные смеси. Подробный вывод уравнений активнос-
ти и коэффициентов активности приведен в примере
9.3. Конечные уравнения выглядят так:
inoi = In 0i 4- (1 — \/т)ф2 4- х02> (9.67)
in у 1 = 1п(01/Х1) 4- (1 - \/т)ф2 4- х02» (9.68)
1па2 = 1п02 - (т - 1)01 4-х01- (9.69)
Мольные доли не пригодны в качестве единиц при опи-
сании полимерных смесей по той причине, что их моле-
кулярные массы велики и, как правило, точные их вели-
чины не известны, так что в итоге коэффициенты ак-
тивности лишаются своего обычного смысла. В то же
время активности, выраженные, как показано здесь, в
объемных долях, очень удобны. Условия равенства пар-
циальных фугитивностей фаз, находящихся в состоянии
равновесия, предполагает равенство активностей, если
для всех фаз используется одно и то же стандартное со-
стояние, что является аргументом в пользу применения
активностей.
На рис. 9.7 графически представлено воздействие со-
отношения молекулярных масс и параметра Флори на
активности. Изменения т в пределах 100 или около то-
го оказывают весьма незначительное влияние на актив-
ность мономера. На практике т обычно превышает
1000, и активность полимера очень мала. Это соответ-
ствует положению о том, что полимеры с высокой мо-
лекулярной массой практически нерастворимы, тем не
менее они поглощают мономеры и другие жидкости,
т. е. по отношению к веществам с низкой молекуляр-
ной массой они играют роль растворителя. Как далее
следует из этой теории, полная смешиваемость достига-
ется при величине параметра Флори ниже критической:
Хс — 0,5( 1 4- 1/т)2 — 0,5, (9.70)
а при его величине выше критической она является ча-
стичной. Исходя из зависимости х от параметров рас-
творимости и вышеупомянутого положения, можно за-
ключить следующее: чтобы смешиваемость была мак-
симальной, параметры растворимости компонентов
смеси должны быть примерно одинаковы.
Многие полимеры не смешиваются, а при смешива-
нии некоторых пар полимеров с водой образуются две
водные фазы. Например, смесь, содержащая по 8% по-
лиэтиленгликоля и декстрана, в воде образует две фазы
следующего состава: 2% полиэтиленгликоля + 20% дек-
страна и 12% полиэтиленгликоля +0,01% декстрана.
Такого рода поведение удобно для экстракции таких
ферментов, которые не экстрагируются органическими
растворителями. Регулируя концентрацию полимеров,
pH и концентрацию солей, можно получить коэффици-
енты распределения, равные пяти и более [414].
Экспериментальные методы определения активностей
для смесей полимеров включают измерение давления
пара, осмотического давления, понижения температуры
замерзания и взаимной растворимости; в то же время
измерение равновесия пар — жидкость, как правило, не
проводится в силу того, что полимеры по существу не-
летучи. Подробный анализ этих вопросов содержится,
например, в работах [54].
Выведенные выражения для активностей в принципе
можно использовать для описания растворимости, рав-
новесия плавления и равновесия пар — жидкость для
полимеров и жидкостей с низкой молекулярной массой.
Что же касается растворимости, то данная теория при-
менима, вероятно, только для разбавленных растворов.
Этот аспект данного вопроса рассмотрен Шинодой
[121]. Предпринимались также попытки распространить
эту теорию на концентрированные растворы; так, на-
пример, Бере и др. [1980] отмечают возможность испо-
льзования метода групповых вкладов UNIFAC в подоб-
ных случаях. В обзоре литературы, подготовленном
Бонди [190], приводятся ссылки на публикации, посвя-
щенные проблемам применения такого подхода для
описания равновесия плавления и равновесия пар —
жидкость, а также других видов равновесия; однако
большинство результатов неудовлетворительны.
Другие аспекты фазового равновесия 457
О 0,2 0,4 0,6 0,8
Рис. 9.7. Активности полимера (2) и мономера
(7) как функции соотношения их молекулярных
масс т и параметра Флори; если т > 100, то
в выбранном масштабе кривые inai — фг прак-
тически неразличимы.
К сожалению, выводы, сделанные в этом разделе от-
носительно термодинамики фазового равновесия поли-
меров, являются преимущественно отрицательными. В
этой области еще предстоит выполнить большую тео-
ретическую и экспериментальную работу.
9.5. Жидкие кристаллы и мицеллы
Жидкие кристаллы. Некоторые чистые жидкости (в
1978 г. их насчитывалось более 6000) и еще большее
число смесей обладают свойствами, гораздо более при-
сущими твердым кристаллам, а именно для них харак-
терны оптическая анизотропия, дифракция рентгенов-
ских лучей и ряд других определенных электрических и
термических свойств. Более того, их свойства претерпе-
вают резкие изменения при определенных температу-
рах, что свидетельствует об изменениях, происходящих
во внутренней структуре, хотя общие признаки жидко-
стей сохраняются. Например, твердый этил-и-
азоксициннамат при 140 °C внезапно превращается в
мутную жидкость, которая в свою очередь при 249 °C
также внезапно становится прозрачной; подобные изме-
нения являются воспроизводимыми и обратимыми.
Структурные фазы этого вида называются мезоморф-
ными (промежуточными формами) или жидкокристал-
лическими; по определению энтузиастов, работающих в
данной области, это — четвертое состояние материи.
Органические соединения, образующие жидкие кристал-
лы, имеют длинные и узкие молекулы с полярной груп-
пой типа —OR или — COOR у одного или обоих концов
и во многих случаях с умеренно активной группой типа
—С=С—, —C=N— или —N—О—N— в центре. Мезо-
морфизм возможен только при наличии заместителя в
ядре-положении. Молекулы стремятся располагаться
параллельно друг другу в виде скоплений или «кристал-
лов», содержащих порядка 100 000 молекул каждый.
Число входящих в жидкие кристаллы молекул, имею-
щих определенную геометрическую форму, не является
постоянным; жидкие кристаллы, статистические по при-
роде, находятся в равновесии с окружающими группами
молекул, так что размеры и форма первых могут варь-
ировать. По мере повышения температуры размеры
скоплений уменьшаются, так что в конце концов они те-
ряют способность рассеивать свет и становятся аморф-
ными жидкостями в традиционном понимании. Ввиду
ярко выраженной чувствительности к воздействию тем-
пературы жидкие кристаллы называют термотропны-
ми (зависящими от воздействия тепла) агрегатами в от-
личие от другого типа агрегатов — мицелл, структур-
ный ростав которых также зависит и от их
концентрации, в силу чего они получили название лиот-
ропных (зависящими от воздействия жидкости)
структур.
Специалисты в этой области считают удобным при-
менение весьма разветвленной классификации различ-
ных видов вероятных скоплений молекул. Смектиче-
ские (мыловидные) структуры состоят по всей длине из
параллельных пластинок толщиной в одну молекулу;
существует по меньшей мере семь подклассов смектиче-
ских структур. Нематические (нитевидные) структуры
состоят из длинных, прямых, параллельных стержней
во многих случаях с цилиндрическим поперечным сече-
нием. Холестерические структуры (название — произ-
водное от холестерола, имеющего аналогичную форму)
состоят из скрученных стержней, форма, которых иног-
да приближается к спиральной. В книге Демуса и Рих-
тера (1978) приводится 212 микрофотографий, многие
из которых выполнены в цвете, различных видов таких
веществ.
Переход от одного типа структуры к другому осу-
ществляется главным образом при изменении темпера-
туры, имеет определенный характер и является воспро-
изводимым. В табл. 9.3 указано несколько таких пере-
ходов, представлены данные о температуре и измене-
нии энтальпии и энтропии, найденные методом диффе-
ренциального термического анализа (гл. 12). Соответ-
ствующие изменения температуры можно также наблю-
дать визуально в поляризованном свете на нагреваемом
предметном столике микроскопа. Разнообразные мето-
ды наблюдений, используемые при исследовании фаз,
описаны, например, Греем и Винзором (1974).
Отвердевание смесей различного рода жидких кри-
сталлов происходит аналогично отвердеванию обычных
жидкостей. На рис. 9.8 показана фазовая диаграмма с
одной простой эвтектикой, в то время как на рис. 5.23,
е представлен эквивалент азеотропной смеси с мини-
мальной температурой кипения, образованный аморф-
ной жидкостью и жидкими кристаллами. Как установ-
лено рядом исследователей, уравнение Шредера (разд.
8.4.1) во многих случаях представляет эвтектические
температуры с точностью до одного-двух градусов
(см., например, табл. 9.2). Наибольшие изменения энер-
гии происходят во время перехода от твердой фазы к
первой жидкой кристаллической фазе; остальные фазо-
вые переходы характеризуются менее значительными
изменениями энергии. В конце данного раздела приво-
дятся прочие термические данные и корреляции.
Мицеллы. Некоторые длинные молекулы с одним по-
лярным и одним неполярным концом образуют в при-
сутствии воды и некоторых органических жидкостей так
называемые мицеллы (по-латыни крошки). Эти агрега-
ты имеют сферическую, цилиндрическую или пластин-
чатую форму, их полярные концы обращены в сторону
водной фазы, если она выполняет роль окружающей
среды. Эти структуры изображены на рис. 9.9. Если
окружающая среда является преимущественно органиче-
ской, полярные концы обращены вовнутрь; такие ми-
целлы принято называть обратными. Наиболее распро-
страненный пример мицеллярной системы — это мыло
или неионогенное поверхностно-активное вещество и
вода при наличии другого органического вещества или
без него. Мицеллы обладают интересным и полезным
свойством — они солюбилизируют органические веще-
ства, обычно не смешивающиеся с водой; этот процесс
происходит путем внедрения растворенного вещества в
состав мицеллы и изоляции его от воды полярными
концами молекул мыла.
Подобно жидким кристаллам мицеллы представляют
собой агрегаты статистической природы, состоящие в
среднем всего из нескольких сотен молекул, размеры и
форма которых варьируют в зависимости от концентра-
ции и температуры. Основное различие-между мицелла-
ми и жидкими кристаллами заключается в том, что
первые агрегаты существуют в контакте с аморфной
жидкой фазой в то время как жидкие кристаллы сами
заполняют все пространство.
В малых концентрациях поверхностно-активные ве-
щества образуют в воде истинные растворы, однако по-
сле достижения критической концентрации мицелло-
образования (ККМ) начинается образование мицелл,
продолжающееся до момента достижения некоторой су-
щественной концентрации. В большинстве случаев
ККМ весьма мала — порядка 0,01 молей для ионоген-
ных поверхностно-активных веществ типа мыла и
0,0001 молей для неиогенных поверхностно-активных
веществ типа эфира оксида этилена и алкилбензолов.
Критическую концентрацию мицеллообразования невоз-
можно выявить оптически, поскольку длина мицелл
меньше длины волны света и поскольку они сохраняют-
ся в коллоидных растворах. Прочие физические свой-
ства системы по достижении ККМ могут резко изме-
няться. Так, на рис. 9.10 показана зависимость поверх-
ностного натяжения от концентрации мыла. При
концентрации 0,07 масс. % график зависимости стано-
вится горизонтальным, по крайней мере для некоторого
дальнейшего повышения концентрации, что свидетель-
ствует о существовании при концентрации, превышаю-
щей ККМ, двух фаз — мицелл и фазы истинного рас-
твора концентрацией 0,07 масс.%.
Каждая химическая система может иметь несколько
типов мицеллярных структур в зависимости от кон-
центрации и температуры. На рис. 9.11 показана бинар-
ная мицеллярная система; в ней в зависимости от кон-
центрации и температуры может преобладать одна из
пяти различных структур: твердая, аморфная жидкая,
оптически изотропная, но по структуре жидкая, чистая
ламеллярная фаза и промежуточная (стержневидная)
фаза. Пики некоторых областей напоминают пики, со-
ответствующие образованию соединений на диаграммах
замерзания аморфных жидкостей (гл. 5 и 8). Тройная
система, изображенная на рис. 9.12, также образует раз-
Другие аспекты фазового равновесия 459
Рис. 9.8. Фазовая диаграмма л-азоксианизола (У) + п-
азоксифенетола (2), на которой показано образование
эвтектики с нематической фазой [362].
Таблица 9.2. Энтальпии и энтропии фазовых превращений ряда ди-н-алкил-п-азокси-а-
метилциннаматов [754]
о о
и и
c„h2„+1-o-c-c=ch-Q-n=n-0-ch=c=c-o-c„h2„+1
сн3 о сн3
Следует учесть, что правило Вальдена для плавления (Д5 = 13) на эти случаи не распростра-
няется.
СпН2п +1 Переход Т, °C дн, ккал/моль и AS, ;ал/моль‘К
с2н5 Твердая фаза -»• смектическая фаза А НО 7,89 20,6
Смектическая фаза А -»• нематическая фаза 125 0,26 0,65
Нематическая фаза -»• изотропная фаза 141 0,29 0,70
С3Н7 Твердая фаза-* смектическая фаза А 72 6,69 19,4
Смектическая фаза А ->• нематическая фаза 119 0,26 0,66
Нематическая фаза-> изотропная фаза 131 0,29 0,72
СбНв Твердая фаза -> смектическая фаза А Смектическая фаза А ->• изотропная 55 5,98 18,2
жидкая фаза 91 0,96 2,6
СцН23 Твердая фаза ->• смектическая фаза С 79 15,7 44,6
Смектическая фаза С -»• смектическая фаза А 83 « 0,24 « 0,71
Смектическая фаза А -* изотропная жидкая фаза 87 2,01 5,58
Мономеры Мицелла Цилиндрическая п <пзгь
мицелла (произвольно ' бксагональная
ориентированная) ^Sp^
Рис. 9.9. Последовательные стадии образования мицел-
лярных структур по мере повышения концентрации по-
верхностно-активного вещества в воде, протекающего в
направлении, указанном стрелками [169].
Рис. 9.10. Поверхностное натяжение додецилбензол-
сульфоната натрия в воде при 25 ° С [630]. Разрыв кри-
вой при концентрации ПАВ 0,07% соответствует мо-
менту достижения критической концентрации мицелло-
образования.
Другие аспекты фазового равновесия 461
350
100 90 80 70 60 50 40 30 20 10 О
Содержание лаурата натрия,°/0
Рис. 9.11. Фазовая диаграмма температура — состав
системы лаурат натрия + вода [773]. На диаграмме
изображены три вида двухфазных областей:
1. Изотропная фаза, состоящая из сферических мицелл,
которые образуют основную структурную единицу жид-
кого кристалла и размещены на объемно- или гране-
центрированной кубической упаковке внутри водного
раствора.
2. Средняя фаза, состоящая из достаточно длинных
стержнеобразных мицелл цилиндрического (иногда ква-
дратного или прямоугольного) поперечного сечения,
размещенных на гексагональной упаковке в водной сре-
де. Эта фаза сходна с нематической фазой жидких кри-
сталлов.
3. Чистая фаза, состоящая из ламеллярных мицелл с,
аналогичная смектической фазе жидких кристаллов и
отличающаяся от последней только тем, что в ней слои
разделены водным раствором.
личные чистые агрегаты и смеси агрегатов. Диаграммы
более ста подобных тройных и бинарных систем приве-
дены в книге [270]. До настоящего времени ни одна фа-
зовая диаграмма мицеллярной системы, даже простой
эвтектики, показанной на рис. 9.8, не построена на
основе термических данных.
Термодинамические свойства. Жидкие кристаллы
представляют собой более простые структурные эле-
менты, чем мицеллярные агрегаты, вследствие чего
корреляции изменений свойств, сопровождающих фазо-
вые переходы, являются более успешными. Некоторые
данные такого рода приведены в табл. 9.2. В работе,
из которой заимствованы данные этой таблицы, также
содержится множество данных об энтропиях фазового
перехода для групп химически подобных веществ с ал-
кильными цепочками различной длины, для которых в
большинстве случаев изменения энтропии последова-
тельно возрастают. О том, что уравнение Шредера ус-
пешно применяется для прогнозирования поведения ве-
ществ при отвердевании, уже упоминалось выше.
Рассматривая водные мицеллярные системы, необхо-
димо учитывать ряд факторов, усложняющих положе-
ние даже при ККМ и малых концентрациях, когда мож-
но принять, что мицеллы имеют сферическую форму.
Степень агрегирования зависит главным образом от
концентрации, температуры и ионой силы. На рис. 9.13
и в табл. 9.3 показано воздедйствие этих факторов при
ККМ, но какой-либо корреляции, пригодной для общих
расчетов, не разработано.
Некоторые исследователи обращались к проблеме
равновесия при образовании мицелл, которое записыва-
ют в виде следующего стехиометрического уравнения:
Nm (мономер) +* Л/( мицелл а), (9.71)
где N — степень агрегирования. Стандартное изменение
энергии Гиббса процесса определяется выражением
RT
ag° = —ье^ + лппс^ (9.72)
или
RT RT
ДС° = ЛТ1п(стс) + — In#- — \n(NCM). (9.73)
Что касается величин в правой стороне уравнения, то
ККМ находят из результатов измерения поверхностного
натяжения, молекулярную массу мицеллы — мембран-
ной осмометрией, а степень агрегирования соответ-
ственно — расчетом материального баланса.
Проведенный анализ [185] показал, что изменения
энергии Гиббса процесса мицеллообразования состоят
из вкладов трех фундаментальных процессов, однако
оценка каждого отдельного вклада выполнена не была.
При наличии корреляции ККМ как функции концент-
рации, температуры и ионной силы и данных об изме-
рении степени агрегирования представляется возмож-
ным записать корреляцию изменений энергии Гиббса
процесса мицеллообразования при помощи тех же пере-
менных. После этого можно получить изменение эн-
тальпии в зависимости от температуры по изохоре
Вант-Гоффа:
d(AG°/T)
4H° = ~RT2-------—-----. (9.74)
Такого рода корреляции, включающие число агрегиро-
вания, аналогичны корреляциям для изменения энергии
Гиббса реакции, выраженным через групповые вклады
составляющих.
Еще одной задачей, требующей решения, является
построение полных фазовых диаграмм, подобных пред-
ставленным на рис. 9.11 и 9.12, однако вероятность по-
лучения таких диаграмм в обозримом будущем весьма
сомнительна, поскольку даже для аморфных и неассо-
деканол
Рис. 9.12. Фазовая диаграмма тройной системы октано-
ат натрия + деканол + вода при 20°С. Структуры агре-
гатов показаны схематически: Li и Li — изотропные
фазы раствора; В, С и D — ламеллярные фазы, Е —
гексагональная фаза, F — инвертируемая гексагональ-
ная фаза. Эта выполненная Линдманом схема [446] ба-
зируется на данных оригинальной работы [270]. В по-
следующих исследованиях было показано, что В и С —
однофазные области. Переработанная диаграмма под-
готовлена Фрименом и Дэниэльсоном [293].
Рис. 9.13. Число молекул п, приходящихся на одну ми-
целлу, т. е. степень агрегирования, додецилсульфата на-
трия в 0,6 М растворе хлорида натрия как функция тем-
пературы и концентрации [233].
Другие аспекты фазового равновесия 463
Таблица 9.3. Критическая концентрация мицеллообразования (ККМ) и сте-
пень агрегирования (п) нескольких поверхностно-активных веществ с до-
бавлением и без добавления соли [553]
Поверхностно-активное вещество Раствор NaCl.M ККМ, моль/л п
Додецилсульфат натрия Вода 0,0081 80
0,02 0,00382 94
0,03 0,00309 100
0,10 0,00139 112
0,20 0,00083 118
0,40 0,00052 126
Додецил амингидрохлорид Вода 0,0131 56
0,0157 0,0104 93
0,0237 0,00925 101
0,0460 0,00723 142
Децилтриметиламмоний- Вода 0,0680 36
бромид 0,013 0,0634 38
Додецилтримегил аммоний- Вода 0,0153 50
бромид 0,013 0,0107 56
Тетрадецилтриметил- Вода 0,00302 75
аммонийбромид 0,013 0,00180 96
циированных жидкостей в областях кристаллизации
органических соединений, соединений кремния и метал-
лов, как уже было отмечено в гл. 8, это сделать не уда-
лось. Необходима разработка корреляций также и для
солюбилизации поверхностно-активными веществами.
Применительно к аморфным жидкостям в этой области
было предложено несколько корреляций, выраженных
через коэффициенты активности, полученные методом
структурных вкладов, однако в них было недостаточно
учтено влияние температуры.
Вероятно, основной причиной неудовлетворительнос-
ти результатов исследований термодинамических аспек-
тов поведения мицелл является исключительное богат-
ство этой отрасли, разнообразие составляющих и воз-
действующих на них факторов, что открывает большие
возможности сбора интересных и полезных фактов. За
этим может последовать разработка корреляций.
9.6. Смеси в сверхкритических условиях
Критические свойства многих бинарных смесей изме-
няются с достаточной регулярностью в зависимости от
состава и выражаются непрерывной кривой Р — х, про-
стирающейся от одного чистого компонента до друго-
го, над которой располагается гомогенная область, а
под ним — область жидкость + пар. Такой характер
изменения критических свойств показан, например, на
рис. 1.31 и 1.32. Однако для веществ, значительно раз-
личающихся по размерам и форме молекул, летучести
или полярности, могут наблюдаться разрывы в крити-
ческих локусах, в результате чего критические точки
чистых веществ не связываются локусом, и расщепле-
ние фаз может протекать при температурах и давлени-
ях, намного превышающих критические. Подобную
ситуацию обычно квалифицируют как равновесие
газ — газ, хотя плотности сверхкритических фаз харак-
терны для жидких фаз. Однако, как будет показано ни-
же, различие между жидкой и паровой фазами при
очень высоких давлениях не всегда явно выражено или
имеет физический смысл. Одно из проявлений воздейст-
вия высокого давления, а именно растворимость в сжа-
тых газах, было рассмотрено в разд. 8.6.
Критический локус бинарной смеси, исходящий из
критической точки вещества с меньшей летучестью, мо-
жет иметь характерную форму, зависящую от химиче-
ской природы пары. На рис. 9.14 некоторые критиче-
ские локусы имеют максимальные температуры и дав-
ления, иногда расположенные вблизи левой крити-
ческой точки, а применительно к системе Не + СН4
непрерывно расходящиеся. При достаточном повыше-
нии давления в некоторых случаях возможно схождение
кривых в одной точке, однако часто кривые заканчива-
ются в момент образования твердой фазы. Разделение
фаз в сверхкритических условиях показано на рис. 9.15
на примере смеси аммиак + азот; а на рис. 9.16 — на
примере смеси тетрафторид углерода + н-гептан.
Поведение фаз в условиях, близких к соответствую-
щим нижней критической точке чистого компонента, де-
монстрируется на примере смеси метан + гексан на
рис. 9.17; полный критических локус этой смеси дан на
рис. 9.14. В этом случае критические локусы, исходящие
из критических точек чистых компонентов, прерывают-
ся, но тем не менее совпадают на границе трехфазного
локуса. Из сопутствующих диаграмм Р — х также вид-
но, что при 193,2 К трехфазная линия все еще существу-
ет, но с повышением температуры до 198 К она преры-
вается. Несколько примеров такого типа даны в книге
[81].
Разделение фаз в сверхкритических условиях часто
связывают с тем, что смеси имеют ограниченную вза-
Температура
Рис. 9.14. Критические локусы вблизи критической точ-
ки менее летучего из компонентов смеси. Штрихи на
линиях указывают на двухфазыне области. Кривые вы-
полнены не в масштабе. Указанные давления являются
наибольшими из числа измеренных в каждом из отдель-
ных случаев.
Рис. 9.15. Ограниченная смешиваемость газообразного
аммиака и азота. Например, смесь, содержащая 57%
NH3, при температуре 100°С и давлении 2600 атм
начинает расслаиваться, в то время как смеси, содержа-
щие от 18 до 84,5% NH3, при 5000 атм являются
двухфазными. Критические свойства компонентов:
NH3 (111,3 атм, 132,6°С), N2 (33,5 атм, - 151°С) [61].
имную растворимость. Оба явления характерны для ве-
ществ, молекулы которых существенно различаются по
ряду параметров. В разд. 7.4 рассматриваются формы
областей несмешиваемости главным образом в свете
воздействия на них температуры, которое проиллю-
стрированно на рис. 5.21 ив примере 7.11. Некоторые
проявления воздействия давления показаны на рис. 5.28
и 9.18. Как видно из последнего рисунка, повышение
давления и понижение концентрации соли оказывают
сходное влияние на растворимость, однако в общем
случае влияние добавления солей не столь однозначно,
как в этом конкретном случае. Исследования, проводи-
мые в этой области, подробно излагаются Шнайдером
[618].
Как уже указывалось, выявить различие между жид-
кой и газовой фазами не всегда просто. Так, обычно
считается, что на фазовой диаграмме системы этилен
+ нитрометан (рис. 9.19, а) под выпуклым участком
критического локуса находится область смеси жидкости
и пара, однако фазы, расположенные слева от почти
вертикальных линий на рисунках 9.19, а и б, как прави-
ло, называют фазами газ + газ. Возникает вопрос, на
каком именно участке кривой жидкая фаза переходит в
газовую?
Несмотря на казалось бы необычное поведение фаз в
сверхкритических условиях, оно поддается теоретиче-
ской трактовке. Известно, что условия фазового равно-
весия газ — газ были предсказаны еще Ван-дер-Вааль-
сом, а некоторое экспериментальное подтверждение бы-
ло получено к 1900 г., однако начало современным
экспериментальным открытиям в этой области было
положено Кричевским в 1940 г. Дайтерс и Шнайдер
[245], а также Стефан и Шабер [663] применили моди-
Другие аспекты фазового равновесия 465
Рис. 9.16. а — фазовое равновесие в сверхкритических условиях смесей тетрафторида
углерода и н-гептана. Области, ограниченные параболическими кривыми и парами
линий с отмеченными на них одинаковыми температурами, являются двухфазными
[478].
б — зависимость критического локуса Рс от Тс для смеси, показанной на
рис. 9.16, а. Локус начинается в критической точке н-гептана, но не возвращается
в критическую точку тетрафторида углерода [478]. Критические свойства компонен-
тов: CF4 (37,5 бар, 227,6 К), C7Hi6 (27,4 бар, 540,2 К).
фицированное уравнение Редлиха — Квонга для описа-
ния фазового равновесия в условиях высокого давления.
Авторы работы [155] проанализировали воздействие
полярности на критические локусы. В данной области
ведется интенсивная исследовательская работа. Подроб-
ный обзор литературы по этому вопросу выполнен
Шнайдером [618], а также Роулинсоном и Свинтоном
[ИЗ].
Помимо чисто научного интереса фазовое поведение
в сверхкритических условиях имеет значение при разра-
ботке условий хранения нефти и природного газа в ре-
зервуарах, эксплуатации реакторов, хроматографическо-
го разделения веществ с малыми скоростями перемеще-
ния или с низкой термостойкостью, опреснения воды,
а также условий разделения вообще. Некоторые из этих
вопросов были рассмотрены в разд. 8.6. Основные при-
нципы и ряд примеров практического применения этих
технологических процессов описаны в недавно опубли-
кованных сборниках статей под редакцией Шнайдера и
др. [118], а также Паулайтиса и др. (1982).
Характерная особенность таких процессов состоит в
том, что давление пара и, следовательно, раствори-
мость конденсированных фаз можно значительно увели-
чить посредством повышения давления выше критиче-
ского давления газа при температуре, близкой к его
критической температуре. После достижения требуемо-
го растворения регенерация растворителя осуществляет-
ся с минимальными энергетическими затратами путем
незначительного понижения давления и в ряде случаев
одновременного незначительного понижения температу-
ры. Поскольку СОг в принципе пригоден для обработки
пищевых продуктов, значительные условия были затра-
чены на создание методики применения СОг для экс-
тракции природных продуктов, чувствительных к воз-
действию тепла. Диапазон термодинамических условий,
необходимых для проведения таких процессов, и типич-
11-864
Рис. 9.17. Диаграммы Р — Т — х системы гексан + ме-
тан вблизи критической точки метана. А — давление
паров метана; Ас — критическая точка метана, 190,6 К,
4,60 МПа; с — критические локусы; ВККТ — верхняя
критическая конечная точка, конец трехфазной линии;
НККТ — нижняя критическая конечная точка. В неко-
торых случаях критический локус от 8е может оканчи-
ваться не в НККТ, а в ВККТ, как указано в этом при-
мере [8].
ные профили температур и давлений в экстракционных
аппаратах показаны на рис. 9.20. В табл. 9.3 даны неко-
торые эксплуатационные характеристики нескольких
промышленных процессов экстракции в сверхкритиче-
ских условиях. Работа [661], из которой заимствована
данная таблица, содержит ряд правил, которыми следу-
ет руководствоваться при выборе соответствующих рас-
творителей, в ней же указаны типы веществ, экстрак-
ция которых является эффективной. Например, такие
слабополярные вещества, как углеводороды, сложные и
простые эфиры и лактоны, экстрагируются при давле-
ниях ниже 100 бар; экстракция веществ, содержащих
единичные группы с сильной полярностью, например
—ОН или — СООН, затруднена, а вещества с еще более
сильной полярностью, например сахара и аминокисло-
ты, не экстрагируются при давлениях ниже 500 бар.
Несколько примеров применения этих процессов, осу-
ществленного на практике и потенциально возможного,
приводятся Паулем и Вайзе, [94], а также Эллисом
[271]. Много внимания уделялось проблеме регенерации
жидких продуктов перегонки угля, см., например, рабо-
ты Ганголи и Тодоса [295]. О результатах работ, прове-
денных на пилотной установке с толуолом и о-крезо-
лом, сообщается в статье Мэдокса и др. [454]. Следует
однако, заметить, что четко выраженного экономиче-
ского превосходства этих процессов обнаружить не уда-
лось. Важным преимуществом растворителей в сверх-
критическом состоянии по сравнению с жидкими рас-
творителями в подобных случаях является простота
выделения первых из остатков, обусловленная относи-
тельно низкой их плотностью и высокой летучестью.
Список промышленных экстракционных процессов, в
которых в качестве растворителей используются газы в
сверхкритическом состоянии, дан в табл. 9.3.
9.7. Гйдраты
Некоторые газы образуют с водой твердые фазы, со-
храняющие стабильность при температурах, намного
превышающих температуру замерзания воды, и это их
свойство может приводить к весьма опасным послед-
ствиям. Так, при некоторых величинах температуры и
давления в газопроводах, по которым происходит
транспортировка влажного природного газа, образуют-
ся «гидратные пробки». В то же время способность га-
зов образовывать твердые гидраты используется для
опреснения морской воды пропаном. Фазовая диаграм-
ма такой системы представлена на рис. 9.21. Растворен-
ные соли и другие относительно нелетучие вещества по-
Другие аспекты фазового равновесия 467
Р, бар
Рис. 9.18. Влияние давления и концентрации растворен-
ной соли на смешиваемость изобутанола и воды [619].
Воздействие концентрации NaClO4 Н2О показано при
давлении 450 бар. Воздействие давления показано на при-
мере раствора, содержащего 5,81 г NaClCU FbO/lOO г
Н2О. В растворе, не содержащем соли, присутствует
40% изобутананола и 60% воды. Двухфазные области
ограничены параболическими кривыми.
7] К
Рис. 9.19. Диаграммы Р — Т смесей этилен + нитро-
метан (а) и этан + диметилформамид (б) [245]. Области
разделения фаз этих двух бинарных смесей расположе-
ны выше критических точек (КТ1 и КТ2 — критические
точки чистых веществ).
11
S, кДж/кг К
б
Рис. 9.20. Диапазон термодинамических эксплуатационных условий при экстракции природных продуктов СОг в
сверхкритическом состоянии (а) и профили температуры и давления в экстракционном аппарате (б) [269]. А —
подача твердых веществ в экстракционной аппарат; В — наполнение установки растворителем; С — нагревание
до температуры экстракции; D — выделение и разделение экстракта; Е — выпуск газа до давления в резервуаре;
F — отсасывание до остаточного давления; G — выпуск остаточного газа; Н — удаление отработанных твердых
веществ и экстракта; I — регенерация адсорбента.
Таблица 9.4. Промышленные процессы, в которых применяется экстракция газами в сверхкритическом состоянии
[661]
Сырье Экстрат Растворитель Условия экстракции Условия регенерации
Р, атм Т, °C
Нефть Легкие углеводороды Пропан, пропилен 100—150 100 50 бар, 100°С
Шерстяной жир Ланолин Пропан, пропилен 60—110 100—105 1 бар
Нефть Фракции углево- дородов Этилен 40—120 20 40 бар
Кофейные бобы Кофеин СО2, N2O 120—180 40—80 Адсорбция на активном угле
Табак Никотин СОг, N2O, SF6, ароматические соединения, галогенпроизводные углеводородов 65—1000 35—100 Адсорбция в докрити- ческих условиях
Хмель Смолы, а- и /3-кислоты СО2 70—400 45—50 Сверхкритические условия
Черный перец Пиперин, эфирные и жирные масла СО2 < 400 40—60 65 бар, 25—60°С, десорбирование с СО2
Гвоздика Эвгенол СО2 < 400 40—60 То же
Корица Коричный альдегид СО2 < 400 40—60 >> >,
Ваниль Ванилин СО2 < 400 40—60 » 99
Жареный кофе Ароматические СО2 < 320 —50 65 бар
соединения, глицериды,
жирные кислоты
Сброженный Ароматические СО2 < 300 —50 50—70 бар
черный чай вещества
Другие аспекты фазового равновесия 469
Пример 9.3. Уравнения активности Флори — Хаггинса
В примере представлены некоторые особенности вы-
вода уравнений для активности мономера (1) и полиме-
ра (2) из основного уравнения для энергии смешения
Гиббса, т — число молекул мономера типа 2 в полиме-
ре. См. разд. 9.4.
И]
1.0, =--------.
Д1 + тт
8. = 1пф1 ч-
9. = 1пф1 +
10. = 1пф1 +
/11 пг”] с?ф{ х(/ипг)2
— _ —---------1------------
фг ф2 d/12 (/Ji Ч- /лиг)
/?1 /?2 Ф1Ф2 у
-------------- + Х02 =
01 Ф2 п\
1-----) 02 + Х02-
. т /
тпг
2. 02 = --------
Д1 + тпг
Э01 d(j>2 0102
3. = ---------=------.
dni dni /11
301 Э02 30102
4. — — — —- ---------.
дП2 дП2 П2
5. СГа/КГ - И11пф1 + /?21n02 + x(”i + тп2)Ф1ф2 =
хтпт2
6. = П11П01 + И21П02 Ч-----------------.
nt + тпг
7. inai = (СГ"//?П =
Э/ii -
11. Inyi = 1ПС1 - 1ПХ1 =
13. 1ПО2 = 1П02 +
= 1п(ф1/Х]) +
02 + Х02-
П\ П2
01 02
0102
П2
xmnl
+------------у =
(/Ji + тпг)
14. = 1п02 - (т - 1)01 + х"*0ь
Давление, атм
6
5
3
2
1
-20246
Температура, ’С
Рис. 9.21. Фазовая диаграмма системы
пропан + вода в присутствии соли.
Pg — газообразный пропан; PL — жидкий
пропан; Н — гидрат; W — вода. Существова-
ние того или иного варианта фаз зависит от
общего количества пропана и воды в системе.
Рис. 9.22. Давления диссоциации гидратов углеводородов и воздействие концентрации метанола [32]. а — давление
диссоциации при температурах выше и ниже точки замерзания воды, б — давление диссоциации гидратов природ-
ного газа при различных концентрациях метанола.
нижают температуру плавления гидратов, как показано
на рис. 9.21 и 9.22, б. Прочие данные о давлениях дис-
социации как функции температуры приводятся на
рис. 9.22, а. Отношения этих характеристик хорошо
описываются уравнением
1пР = кх + к2/Т, (9.75)
однако, при малом температурном диапазоне, как на
указанных рисунках, выражение 1пР = к\ + к{Г практи-
чески эквивалентно приведенному выше уравнению.
Сведения об истории исследований и теории образо-
вания гидратов обобщены Дэвидсоном [242], который
приводит список из семидесяти известных гидратов.
Как установлено, эти гидраты не являются стехиомет-
рическими соединениями, а представляют собой твер-
дые растворы газа во льду. В присутствии стабилизи-
рующего газа лед образует кристаллическую структуру
с полостями примерно одинакового размера, достаточ-
но большими для того, чтобы вместить по одной моле-
куле. Важное значение имеет как химическая природа
газа, так и размер молекул; например, бутан в отличие
от изобутана не образует гидратов. Описано два основ-
ных вида кристаллической решетки; один из них, соот-
ветствующий максимальному содержанию газа, пред-
ставлен молекулярной формулой М.17 НгО в случае за-
Другие аспекты фазового равновесия 471
Концентрация этанола в парах, мол. доли (основание,
не содержащее воды)
Рис. 9.23. Равновесие гидратов в системе этан + про-
пан + вода [352].
V — пар; L\ — жидкость с большим содержанием во-
ды; Li — жидкость с большим содержанием углеводо-
рода; Н\ — гидрат структуры I; Нц — гидрат структу-
ры II.
полнения всех полостей, а другой — либо формулой
М.7-2/3 НгО, либо М.5-3/4 НгО, где М — 1 моль газа.
Тип решетки зависит от природы газа. Например, этан
образует структуру типа I, а пропан — структуру типа
II. Условия, при которых в присутствии смесей преоб-
ладает какой-либо из структурных типов, зависят от
факторов, указанных на рис. 9.23.
В соответствии с теорией, предложенной Ван-дер-Ва-
альсом, и затем развитой Платтевом (1959), доля поло-
стей, заполненных молекулами газа, выражается по-
средством уравнения, сходного с уравнением Ленгмюра,
описывающим адсорбцию,
С/Л с^ф^р
0: =---------- =-----------— , (9.76)
1 l+ZCjfj l+ZCj-yfiP’
Рис. 9.24. Диаграмма давление — состав системы
где ft и 0, — парциальная фугитивность и коэффициент
фугитивности.
Для чистого газа можно записать следующее уравне-
ние:
СфР
° ~ 1 + СфР ‘
Метод оценки коэффициентов фугитивности твердой
фазы, разработанный Пэрришем и Праузницем [541],
позволяет рассчитывать давления при диссоциации сме-
сей газов, как образующих, так и не образующих гидра-
ты. На рис. 9.24 представлена диаграмма давление —
состав в диапазоне образования гидратов для системы
пропан + метан + вода. В целях предотвращения обра-
зования гидратов в газопроводах широко применяется
впрыскивание метанола, гликоля или аммиака. В по-
следнее время было проведено изучение количественной
стороны подобных процессов. Ментен, Пэрриш и Сло-
ун (неопубликованная работа, 1982) проанализировали
эффект применения ингибиторов путем исследования их
воздействия на коэффициент активности воды. Макого-
ном [84] выполнен обзор современных методов решения
проблем, связанных с образованием гидратов газов.
9.8. Задачи
9.1. Адсорбционное равновесие системы этан + эти-
лен на силикагеле и молекулярных ситах типа 4А при
температуре 25 °C и давлении 730—760 мм рт.ст. харак-
теризуется следующими данными (заимствованы из
рис. 9.2):
метан + пропан + вода [541].
X У
S1O2 4А
1 0,275 0,470
2 0,422 0,830
3 0,563 0,902
4 0,670 0,940
5 0,752 0,962
6 0,822 0,979
7 0,880 0,985
8 0,928 0,990
9 0,968 0,995
а. Найдите средние относительные летучести для
двух групп данных, постройте соответствующие диа-
граммы х — у и определите на них положение исход-
ных данных в целях сравнения.
б. Найдите наилучшие величины параметров для каж-
дой группы данных из уравнения
Какая из корреляций наиболее подходит для этих
случаев?
9. 2. Ниже указаны величины адсорбционного равно-
весия чистых этилена и пропилена и поверхностного
давления, заимствованные из работ [234].
Давление равно 75 мм рт. ст.
моль Р, мм рт.ст х°А RT
Этилен
6,500 10~3 4,10 6,010 10-3
1,592 10-2 17,99 2,471 10“ 2
3,029 10~2 52,09 4,847 10-2
4,399 10-2 125,00 8,217 10~2
5,252 10-2 181,44 1,005 КГ1
6,056 10~2 242,32 1,169 Ю-1
6,796 10~2 309,34 1,326 10-1
7,459 10~2 384,10 1,481 Ю-1
8,060 10-2 465,00 1,630 Ю-1
8,659 10~2 546,12 1,764 10-1
9,209 10~2 632,00 1,895 10-1
9,658 10~2 728,00 2,002 Ю-1
Пропилен
1,390 10-2 0,24 1,166 10-2
3,105 10“2 2,56 3,366 10-2
5,090 10“2 12,80 1,161 10~1
7,005 10-2 30,03 1,705 Ю-1
9,095 10-2 64,90 2,356 Ю-1
1,095 10"1 122,19 3,014 10-1
1,252 10-1 209,14 3,666 Ю-1
1,377 10"1 327,70 4,268 Ю-1
1,468 10"1 463,78 4,773 Ю-1
1,575 10-1 618,47 5,217 Ю-1
1,620 10-1 713,63 5,447 10-1
а. Проверьте правильность указанных величин по-
верхностного давления.
б. Найдите диапазон равновесных составов мето-
дом Майерса и сравните с приведенными ниже экспе-
риментальными данными (представлены графически на
рис. 9.3).
Х1 У1
0,800 0,993
0,614 0,980
0,253 0,858
0,141 0,696
0,055 0,436
0,028 0,248
9.3. На основе данных [234] для адсорбированных
смесей этилена и пропилена рассчитаны коэффициенты
активности, приведенные ниже в таблице; величины ко-
эффициентов в условиях бесконечного разбавления по-
лучены путем графической экстраполяции. Исходя из
этих коэффициентов активности для бесконечного раз-
бавления, найдите параметры уравнений Маргулеса и
Висьсона, а затем из этих уравнений определите коэф-
фициенты активности при указанном ниже составе ад-
сорбата.
Х1 У1 У2
0 (0,757) 1
0,021 0,7580 1,0119
0,064 0,7637 0,9835
0,126 0,7922 0,9621
0,277 0,8407 0,8917
0,619 0,9398 0,8624
0,807 0,9991 0,7772
1 1 (0,69)
9.4. Для системы, указанной в примере 9.2, найдите
параметры уравнений ван Лаара и Маргулеса для фазы
адсорбата при составе жидкости Xi = 0,3. Другими сло-
вами, в п. 5 примите, что Xi = 0,3.
9.5. Данные об адсорбции уксусной кислоты из вод-
ного раствора древесным углем выражены через равно-
весную молярную концентрацию, отнесенную к количе-
ству адсорбированной кислоты в расчете на грамм дре-
весного угля. Проверьте эти данные при помощи
уравнений Ленгмюра и Фрейндлиха.
Молярность г/г С
0,434 0,209
0,202 0,152
0,0899 0,1083
0,0347 0,0818
0,0113 0,060
0,00333 0,037
9.6. Проверьте данные об адсорбции моноксида угле-
рода на углероде при 0 °C, используя уравнения Фрейн-
длиха и Ленгмюра.
мм рт.ст. мм (НТД)/г
100 3,38
200 6,15
Другие аспекты фазового равновесия 473
мм рт.ст. мм (НТД)/г
300 8,44
400 10,39
500 12,21
600 13,77
700 15,25
а. Для каждой из трех групп данных найдите констан-
ты уравнения Фрейндлиха, п, = а,
б.
ми,
Сравните данные для смеси с расчетными данны-
пользуясь тремя следующими правилами:
1.
2.
п = Ех,п,.
1/п = EXi/ni.
3. 1пл = ЕхДпЛ;.
9.7. Адсорбция пропилена, этилена и их смесей 2:1 на
углероде при 20°С характеризуется следующими данны-
ми (заимствованы из работы Коста и др. [234]).
Обратите внимание,
применить во всем
став паровой фазы
что ни одно из этих правил нельзя
диапазоне составов, поскольку со-
непосредственно не учитывается.
Р, мм рт.ст. и,моль/г
пропилен этилен смесь
100 0,210 0,080 0,120
200 0,245 0,110 0,165
300 0,270 0,135 0,200
400 0,285 0,155 0,220
500 0,300 0,165 0,240
600 0,310 0,180 0,255
700 0,320 0,185 0,265
10.1. Введение
Находящиеся в равновесии фазы могут содержать
такие вещества, которые при контактировании фаз, всех
или некоторых, химически взаимодействуют. В таких
случаях состояние равновесия зависит от химической
природы компонентов и их коэффициентов распределе-
ния между фазами, а также от температуры, давления
и полного состава системы. Скорость достижения хи-
мического равновесия имеет важное практическое значе-
ние. Более быстрому установлению равновесия способ-
ствуют высокие температуры, соответствующее изме-
нение давления и обычно присутствие катализаторов.
Влияние температуры часто бывает двойственным: ее
повышение ускоряет достижение равновесия, однако
при этом состав системы может оказаться нежелатель-
ным. Решая практические задачи, необходимо прежде
всего найти такие равновесные условия, которые благо-
приятствуют получению требуемого состава, а затем
изыскивать пути проведения реакции с приемлемой ско-
ростью. В данной главе будет рассмотрена только зави-
симость состава и распределения фаз при равновесии от
температуры и давления. Обсуждение начнем с теории
единичных и мультиплетных химических реакций, про-
исходящих в одной фазе, а после этого рассмотрим
многофазные процессы.
Равновесное состояние реакционных систем, как и
равновесие инертных систем, характеризуется миниму-
мом энергии Гиббса при определенных Т и Р. Таким
образом,
ntG = Z ni(G? + RTln(fi/f?) = Z ni(G? + РТ\па{).
-* минимум (10.1)
Аналогично при заданных Т и V условие равновесия, ес-
ли использовать энергию Гельмгольца, определяется
выражением
ntA = S nt (A + RT In а,) -» минимум. (10.2)
Поскольку Т и Р составляют практически более
важную группу переменных, чем Т и V, при последую-
щем обсуждении мы будем пользоваться только энерги-
ей Гиббса. Состав системы входит в эти уравнения
через парциальные фугитивности (летучести). Таким об-
разом, для газов уравнение (10.1) принимает следую-
щий вид:
ntG - 2 MG? + RT\n(yi^>iP) -> минимум. (Ю.З)
Если смесь представлена единственным стехиомет-
рическим отношением, все составы выражаются в еди-
ницах частичного превращения основного компонента £,
в этом случае минимум может быть найден исходя из
—-°-
(10.4)
Аналогично, если имеются два стехиометрических отно-
шения, равновесие выражают через две переменные £i
и £2
d(ntG) I
5ei I
J е2
d(ntG)
де2
(Ю.5)
= 0
и т. д. для любого числа стехиометрических от-
ношений.
В тех случаях, когда такой подход не позволяет вы-
явить приемлемый набор независимых стехиометриче-
ских отношений, равновесие можно непосредственно
определить из уравнения (10.1) и уравнений материаль-
ного баланса тех химических элементов, которые огра-
ничивают минимизацию, исходя из того, что содержа-
ние каждого элемента в равновесной смеси остается тем
же, что и в исходном веществе. Минимизацию энергии
Гиббса, основанную на балансе химических элементов,
наиболее легко осуществить по методу Лагранжа, кото-
рый будет описан позднее. Имеются и другие методы
проведения этой операции для сложного равновесия,
причем в некоторых конкретных случаях они могут
быть проще в исполнении, чем названный выше основ-
ной метод.
10.2. Степень «развития» химической
реакции
Стехиометрическое уравнение химической реакции,
включающее компоненты А, со стехиометрическим ко-
эффициентами Vi, можно записать как
(10.6)
где — суммы всех реагентов, а — суммы всех
продуктов. Разность между правой и левой суммами
Химическое равновесие 475
стехиометрических коэффициентов дает
Ду,- = S V,- — £ V,-.
Р R
(Ю.7)
Для заданной степени превращения одного из компоне-
нтов все присутствующие количества можно выразить
в единицах одного количества, называемого глубиной
протекания реакции
И; = ni0 ± ViE
nt - пю ( + для продуктов,
£ = ------- {
± Vi I - для реагентов.
Таким образом, для каждого компонента
+ для продуктов,
- для реагентов.
Запишем это выражение в дифференциальной форме:
dni = ± vide.
Так, например, для реакции
А\ + 2Лг —* 3^4з + 4/44
(10.11а)
эти количества определяются следующими выра-
жениями:
Дг,- = 3 + 4 - 1 ~ 2 = 4, (10.116)
Аз~ 30 Л4—Ддо Л] -^10 А2~А 20
Е — ——
1 1 4 -1 -2 (10.11В)
А1 = А10 — (10.11г)
А2 = А20 — 2е, (Ю.Пд)
Аз Л3о + Зе, (10. Не)
А д = А до + 4е. (Ю.Пж)
10.3. Уравнение равновесия
В дифференциальной форме энергия Гиббса хими-
чески реагирующей смеси
d(nG) = — nSdT + nVdP + У] Gidni
= -nSdT + nVdP + 2 Gtvide,
_ J +для продуктов,
(^-для реагентов.
(10.12)
(10.13)
Аналогично, если Т и V — независимые пере-
менные,
d(nA) = -nSdT + Pd(nV) + S divide. (10.14)
При постоянных температуре и давлении и наличии
равновесия уравнение (10.13) записывается так:
j =y.GiVi = 0 (10.15)
Эе 1 тр
\ +для продуктов,
V = <
/ - для реагентов,
или
S GiVi - 2 GiVi = 0.
Р R
(10.16)
Для каждого компонента парциальная молярная энер-
гия Гиббса зависит от энергии Гиббса этого компонента
в чистом состоянии и его фугитивности в смеси
G = G? + RT\n(fi/J?) = G? + RTlnai, (10.17)
где G? — энергия Гиббса образования вещества i из эле-
ментов. Используя подстановку уравнения (10.17), урав-
нение (10.16) можно записать в нескольких формах:
(10.8)
(Ю.9)
(10.10)
AG°= Sv Р ,-G? - S v,G? R (10.18)
= RT Sln(^//?)1''- Sln(///?)vi- _ P R (10.19)
= RT S In (a, )1'' — S In (a, )1'' _ P R (10.20)
= RT In ( Па^' ) - In ( Па^' \ P / \ R (10.21)
Разность Д6°, определяемая посредством уравнения
(10.18) — это изменение энергии Гиббса, сопровождаю-
щее реакцию между компонентами в их стандартных
состояниях.
Константа химического равновесия определяется
как
Ксе = ехр(—t±GQ /RT)
(10.22)
(10.23)
Для реакции, выраженной, например, уравнением
(10.11а),
AG = ЗСз + 4^4 — С? — 2^2 (10.24)
а3 а4
Хсе = —-у. (10.25)
°1 а2
Константы равновесия образования многих веществ
для широкого диапазона температур опубликованы в
виде таблиц Сталлом [130]. Часть этой информации по-
лучают из уравнения
В
logio^p = А + —'.
(10.26)
Значения констант А и В даны в табл. Г.1. Эта форма
температурной зависимости Кр выведена исходя из при-
емлемой средней энтальпии образования (см. вывод
уравнения (10.71)). Достаточное соответствие этого со-
отношения эксперименту подтверждает близость к рас-
четным линиям, представленным на рис. 10.6, точек,
представляющих лучшие (экспериментальные) данные
для нескольких систем, из которых наиболее исследо-
ванной является система, используемая для получения
этанола. Используя уравнение (10.26), Лафт [449] по-
строил соответствующую номограмму равновесных
данных для пятидесяти одной реакции.
Константу равновесия каждой определенной реак-
Пример 10.1. Химическое равновесие как условие минимума энергии Гиббса
Для компонентов реакции СН4 + НгО ** СО + ЗНг
энергии Гиббса и равновесные константы образования
при 800 К имеют следующие значения:
AG/ IgA"
1. СН4 2. Н2О -0,56 -48,65 0,154 13,290
3. СО -43,68 11,933
Данные константы равновесия, объединенные в
Ксе = 0,03083, будут использованы для проверки усло-
вия равновесия, найденного минимизацией энергии
Гиббса. Поведение пара считается идеальным. Для каж-
дого компонента f? - 1 и
G, = G? + RTlnfi/J? = G? + RTlnft + RTlny^P
- G? + RT lny,P.
Коэффициенты парциальной фугитивности принимают
равными единице и величины G? оценивают при f? = 1.
Мольные доли выражают через R и е,
R — отношение НгО к СГЦ в начале реакции,
е — степень превращения СН4, выраженная в моль-
ных долях,
nt — 1 + R + 2е,
У1 = (1 - e)/nt,
Уг - (R - e)/nt,
Уз = e/nt,
У4 = Зб/Лг.
Функцию энергии Гиббса смеси находят по уравнению
zirG ( G*l . D\
s = ~kt + lnj"7
= nt
InP + + Iny,
\ K-l
(1)
Строят графическую зависимость g от e для P = 1,5
при нескольких значениях R. Чтобы легче было найти
минимумы, при построении на оси ординат для каждой
кривой выбирается своя в разной степени увеличенная
(растянутая) шкала. Значения равновесного превраще-
ния, взятые из графика, сравниваются с соответствую-
щими значениями, полученными из уравнения равно-
весия,
К = 0,0308319 = ~4Р2 =------------------^£)-.р2-----------. (2)
(1 - е)(А - £)(1 + R + 2с)2
Алгебраическая минимизация уравнения (1) приводит к
уравнению (2) с ATln/f = G? + 3Cm-G?-G2. В приве-
денной ниже таблице сравниваются результаты, полу-
ченные графически и при использовании уравнения (2).
е
R графический метод расчетный метод
1 0,21 0,2076
1,5 0,26 0,2555
2 0,29 0,2976
2,5 0,33 0,3357
пени превращения е. Указаны минимумы энергии.
Химическое равновесие 477
ции получают, объединяя константы образования от-
дельных соединений. Так, в соответствии с уравнением
(10.24) имеем
1пАГсе = 3 lntf3 + 4 1пА"4 - In- 2 In^. (10.27)
10.4. Константа равновесия и стандартные
состояния
В основном численные значения константы химиче-
ского равновесия Ксе получают экспериментально, одна-
ко при нынешнем уровне знания их можно обычно
рассчитать из имеющихся в наличии данных по энергии
образования Гиббса компонентов реакции
-RTlnKce - = &H°f - TLSQf. (10.28)
Энтальпии образования измеряют калориметрически, а
абсолютные энтропии в этом уравнении рассчитывают
по измеренной теплоемкости, экстраполированной к аб-
солютному нулю, или получают из спектроскопических
данных. Такого рода информация имеется в ряде под-
робных справочников. Приведем некоторые из них.
1. Stull, Westrum, & Sinke, «Chemical Thermodynamics of
Organic Compounds» (1969). Таблицы для 917 соедине-
ний, 298—1000 К.
2. Landolt — Bornstein, П/4 (1961), IV/4a (1967), IV/4b
(1972).
3. Глушко В.П., Термодинамические свойства, 1978 и
последующие годы (всего 8 томов). М.: ВИНИТИ.
4. Texas А&М University TRC, «Selected Values of
Hydrocarbon Properties» (current, looseleaf), «Selected
Values of Chemical Compound Properties» (current,
looseleaf).
5. Chemetron Corporation, «Physical and Thermodynamic
Properties» (1969). Таблицы для 68 газов, 32—2200 F.
В примере 10.15 представлена страница из справоч-
ника Сталла.
Состояния, при которых оцениваются AG/ и ft, в
принципе произвольны, однако в целях удобства приня-
ты определенные стандарты, которые далее мы рас-
смотрим. Несколько общепринятых форм выражения
активности и соответствующих констант химических
равновесий приведены в табл. 10.1.
10.4.1 . Газы. Обычное стандартное состояние —
это газ с фугитивностью, равной единице, которая во
многих случаях является такой же, как и для реального
газа при одной атмосфере. В таких случаях константа
равновесия определяется как
К„-К)~ П?р7П?Г'. (10.29)
се J P R
Данные по ДО/ газовой фазы в справочнике Сталла
[130] и во многих других справочниках приводятся для
стандартного состояния. Вводя коэффициенты фугитив-
ности, получаем
Kce = Kf = II(0/j/P)v'7lI(0/y/P)v' (10.30)
Р R
= КфКуР^. (10.31)
Если все компоненты являются идеальными газа-
Таблица 10.1. Обобщение выражений активности и
констант химического равновесия, Ксе = Ка для реакции
VfAi & hp VjAj.
P R
рр Vi (PF): — ехр / — dP — ехр V, — СР - 1) J
Ji RT
Система Активность Константа равновесия
1. 1. Основная Основная в а Ка
2. терминах Ji/fi фугитивности Стандартное со- стояние — идеальный газ Kf/Kf.
3. при 1 атм = Чистая конден- Ф(УуР КфКуР1"'
4. сированная фаза (РЕ), Раствор конден- сированной фа- зы, стандартное состояние — K(PF)
чистое вещество yiKift/ft при 1 атм = yjXi(PF)i KyKxK(PF)
ми, коэффициенты фугитивности равны единице и кон-
станты равновесия определяются как
Ксе = Кр = КуР^.
(10.32)
Применяя правило Льюиса — Рэндалла для идеаль-
ных растворов, преобразуем уравнение (10.32) к виду
Ксе = КфКуР^.
(10.33)
Несколько примеров численных значений Кф показаны
на рис. 10.2 и 10.3. Равновесные составы нескольких ре-
акционных газообразных систем представлены как
функции температуры и давления на рис. 10.1—10.5.
10.4.2 . Чистые конденсированные фазы. Стандарт-
ное состояние — это чистое вещество при давлении в
одну атмосферу. Активность переводится в систему
давления при помощи выражения Пойнтинга
а,-Л7/? = ехр/Р RT<‘r- (10.34)
В некоторых случаях допустимо пренебречь влиянием
давления и взять за единицу активность всех конденси-
рованных фаз. Однако в примере 10.3 влияние высоких
величин давления весьма существенно.
Стандартные энергии Гиббса образования чаще все-
го даются в виде таблиц величин для газового состоя-
ния. Связь между этими значениями и значениями,
полученными для конденсированных фаз вещества А,
можно найти суммированием изменений, соответствую-
щих последовательным этапам, начиная с образования
газа из элементов и кончая образованием жидкости
(или твердого вещества) при той же самой температуре.
Пример 10.2. Различные виды выражения константы равновесия газофазной реакции А + 2В С
Давление составляет 3 атм; другие необходимые
для определения Ксе данные приведены ниже
Вещество yt it Z Р,
А 0,3 0,9 0,81 0,9
В 0,5 0,8 1,20 1,5
С 0,2 0,7 0,42 0,6
Различные способы выражения Ксе\
Ксе = К?= 0,42/(0,81)(1,2)2 = 0,3601,
Ку = 0,20/(0,3)(0,5)2 = 2,6667,
Кф = 0,7/(0,9)(0,8)2 = 1,2153,
Кр = 0,6/(0,9)(1,5)2 = 0,2963,
рД» = 3(1-1-2) =
Ксе = КуКфР^ = 2,6667(1,2153)(0,1111) = 0,3601.
Пример 10.3. Влияние давления на равновесие реакции,
Определим температуру разложения СаСОз, описы-
ваемого реакцией СаСОз <=* СаО + СОг при нескольких
давлениях,
а) принимая активности твердых веществ равными
единице,
б) применяя поправочный коэффициент Пойнтинга
к активностям.
Поскольку реакция идет при высоких температурах, ко-
эффициент фугитивности СОг берем равным единице
(см. задачу 10.9). Находим активности твердых веществ
(R = 82,05):
«СаСОз = ехр[(271,2//?Т)(Р - 1)],
«СаО = ехр[(190,7//?Г)(Р - 1)].
Соответственно константа равновесия равна
К = «СО2«СаО/оСаСОз =
= Рехр[(190,7 - 271,2)(Р - 1)/ЯТ]. (1)
Константа равновесия как функция температуры дана в
работе [59].
InA' = — 22,265/7’ + 29,26 - 1,424 InT’ + 0,0003737’ -
- 0,2076(10 “6)7’2 + 50266/7’2.
протекающей с участием твердых фаз
Эти уравнения объединяют и решают относительно
температуры методом подбора для нескольких давле-
ний; полученные результаты приведены ниже:
P, атм T, К а
корректи- ровка по Пойнтингу активность СаО СаСОз
1 1146,4 1146,4 1 1
10 1317,9 1318,5 1,016 1,023
100 1550,9 1558,9 1,160 1,235
500 1749,6 1797,1 1,940 2,567
Ясно, что при высоких давлениях коэффициент Пой-
нтинга влияет на результаты расчета температуры раз-
ложения. Давление оказывает значительное влияние на
активности, однако при наличии двух твердых веществ
их активности проявляют тенденцию к сближению, так
что влияние температуры намного меньше, чем оно бы-
ло бы в присутствии только одной конденсированной
фазы.
Пример 10.4. Энергия Гйббса образования жидкости из
Необходимые для расчета данные заимствованы из
работы [130], значения энергии Гиббса образования
жидкости (или твердого вещества) вычисляются для
сравнения из значения энергии Гйббса образования газо-
вой фазы по уравнению
Дб/(1) = AG/(g) + RT\nPs.
Jps
VLdP < 0,003. Давле-
i
ния паров указаны в приложении в табл. Г.2. Незначи-
пара
тельное различие в давлении паров можно объяснить
неполным соответствием между рассчитанными и таб-
личными значениями Дб/ (1); величины давления па-
ров, соответствующие разностям между табулирован-
ными Сталлом [130] величинами давления паров и со-
ответствующими энергиями Гиббса образования жид-
кости из пара, указаны в последней колонке таблицы.
В некоторых из приведенных примеров наблюдается до-
статочно близкое совпадение величин.
Химическое равновесие 479
Соединение 4G® (Сталл) ДС?(/) , (вычисл.) Я Ps (Сталл)
g /(S)
Пропанол -38,95 -40,79 -41,09 0,027 0,0448
Этилацетат Валериановая -78,25 -79,52 -79,47 0,128 0,117
кислота -85,37 -89,25 -90,31 2,4-(Е-4) 0,14 -(Е- 4)
Бензол 30,99 29,72 29,76 0,126 0,117
Нафталин 53,44 (48,05) 48,65 3,1 • (Е - 4) 1,1 • (Е-4)
Пример 10.5. Сравнение равновесия в жидкой и паровой фазах
Для компонентов реакции
СНзСООН + С2Н5ОН & СН3СООС2Н5 + Н2О
при 298 К энергии Гиббса образования жидкостей и га-
зов имеют следующие значения:
AGf Я (данные) Ps (Антуан)
жидкость газ
Усусная
кислота -93,06 -90,03 0,0060 0,0201
Этанол -41,62 -40,22 0,0940 0,0780
Этилацетат -79,52 -78,25 0,1171 0,1277
Вода - 56,69 - 54,64 0,0314 0,03111
4G/ - 1,5300 -2,6400
Кее 13,2488 86,3587
В этой же таблице (колонки 4 и 5) указаны давления
паров, соответствующие энергиям Гиббса образования
жидкости и пара, вычисленные по уравнению (10.38) и
по уравнению Антуана. Два значения давления паров
уксусной кислоты несколько различаются, что может
быть обусловлено молекулярной ассоциацией (см., на-
пример, данные, приведенные в задаче 1.11). Две вели-
чины Кр вычисленные исходя из энергии Гиббса и из
уравнения Антуана, имеют следующие значения: 6,5183
и 2,5301 соответственно.
Для эквимолярных количеств реагентов превраще-
ние дается уравнением
К = ?/(1 - s)2,
так что
л- К} - f0’7845, жидкая фаза, К = 13,2488,
е _ у /( + v ) _ ^0 9028> парорая фаза, к = 86,3587.
Для того чтобы реакция происходила в паровой фа-
зе при 298 К, необходимо понизить давление существен-
но ниже атмосферного.
Элементов H(gas.P —• 1); ACj(gas),
(10.35 а)
Ops
/l(gas, Р = 1) — ?l(gas, Psat); AGj = exp / VGdP,
(10.356)
H(gas, Psat) - J(liq, Psat); AG2 = 0,
(10.35b)
H(liq, Psat) - J(liq, P = 1); AG3 = exp f ' VL dP,
•^Г(1О.35г)
£ Элементов — ?4(liq, P=l); AGy(liq).
(Ю.35д)
Результирующее значение:
AGy(liq) = AG^(gas) + (VG~VL)dP. (10.36)
Если газ идеальный, а объем жидкости постоянный:
AGy(liq) = AG®(gas) +PrinPsal- VL(Psat - I).
(10.37)
При условии, что последний член в уравнении (10.37)
мал, получаем
AGj(liq) = AGj(gas) + PTlnPsat (10.38)
В примере 10.4 уравнение (10.38) проверяется в соот-
ветствии с конкретными данными. Аналогичное преоб-
разование уравнения для энтальпии дает
AtfJ(liq) = А Я? (gas) - АЯ^
dHG
дР
(10.39)
или в приближенной форме
AHy(liq) = A//JJ(gas) - АЯ^, (10.40)
где ДЯ*а* — энтальпия испарения.
(10.41)
(10.42)
Совместное решение уравнений (10.38) и (10.22) да-
ет применительно к конкретной реакции соотношение
между константами равновесия X(gas), полученными ис-
ходя из предположения, что стандартное состояние
каждого компонента — это его состояние, соответ-
ствующее фугитивности, равной единице, и A7(liq), полу-
ченными исходя из предположения, что стандартное
состояние — это состояние конденсированных фаз чис-
тых компонентов при давлении в 1 атм.
X’(gas) = tf(liq)/Caat
где
/Caat = П (P?at)v//n (P-at)v'\
Р I R
Анализ равновесных составов, используемых для
проведения одной и той же реакции в жидкой или газо-
образной фазе, выполненный в примере 10.5, обнаружи-
вает их различия. Они возникают из-за различной
степени отклонения от идеальности в обеих фазах и ха-
рактеризуются коэффициентами активности и влиянием
давления на газовую фазу.
10.4.3. Растворы конденсированных фаз. Обсужде-
ние данного вопроса будет вестись главным образом на
примере жидких растворов, но сделанные выводы бу-
дут непосредственно распространены и на твердые рас-
творы. Если уравнение состояния равно применимо и
к жидкой, и к паровой фазам, коэффициенты активнос-
ти можно вычислить так же, как это показано в разд.
4.5. За стандартное состояние принимают пар с фуги-
тивностью, равной единице, а константу равновесия на-
ходят из уравнения (10.29), как и для пара.
В общем случае тем не менее поведение фугитив-
ности конденсированных фаз следует выразить через
коэффициенты активности, принимая состояние чистых
конденсированных фаз при 1 атм за стандартное, и
определять непосредственно из эксперимента или вы-
числять исходя из молекулярной структуры. Таким
образом,
а,- = 7iX.fi/fi = YIxIexp(PlL(P - 1 )/RТ).
(10.43)
Если коэффициентом Пойнтинга можно пренебречь,
Рис. 10.1. Равновесие окисления диоксида серы SO2 кис-
лородом воздуха. Для стехиометрического уравнения
SO2 + 0,5Ог +* SO3 уравнение равновесия имеет следую-
щий вид:
е /2(1 + 2,38/? - 0,5е) г 11846,5 ,, п
------ = ехр ------!— _ Ц 256 ,
1 - е aJ Р(Я - £) I-Т + 273,2 J
где е — степень превращения SO2, Р/2 — первоначаль-
ное отношение O2/SO2, Р — давление, атм.
Давление, атм
Рис. 10.3. Химическое равновесие в синтезе метанола из водорода и монооксида углерода.
2Н2 + СО<=*СН3ОН, Хсе = К^Кр/Р, \%КР = -12,5173 + 5 090,5/Т (данные табл. Г.1).
а — зависимость коэффициента фугитивности, Кф = <£сн3он/(<£н2)2<£со [516] от давления
(мол.%) метанола; б — концентрация в условиях равновесия, вычисленная из стехиометриче-
ских соотношений водорода и монооксида углерода [230].
Рис. 10.2. Равновесие раакции синтеза аммиака.
а — зависимость коэфициента фугитивности.
КФ = 0nh3/(0n2)o>5(0h2)1,5 [58];
б — концентрация аммиака (%) в условиях равновесия,
вычисленная из стехиометрических отношений азота и
аммиака [226]
Давление, атм
12-864
Температура, °C
27 227 427 627 827 1027 7227
300 500 700 900 1100 1300 1500
Температура, К
Рис. 10.4. Равновесие между изомерами пентана (а), пен-
тена (б) и пентина (в) [601].
Химическое равновесие 483
Температура, °C
-73 +127 327 527 727 927 1127
Температура, К
Рис. 10.4 (продолжение).
константа равновесия определяется как
ксе"кукх- (10.44)
Для идеальных растворов Ку равна единице и
Ксе = Кх = П (xyvi/X (х/П (Ю.45)
Р R
В жидкофазных реакциях могут участвовать рас-
творитель и несколько растворенных веществ. Из-за
сложности отношений, связывающих коэффициенты ак-
тивности и концентрации в многокомпонентных смесях,
данные, которыми мы располагаем в настоящее время,
скудны и применение законов термодинамики раство-
ров к неидеальным реакционным системам сводится к
минимуму. Если известны точные значения энергий
Гиббса образования, экспериментальное определение
равновесных превращений даст значения Ку, соответ-
ствующие каждой равновесной смеси; различные равно-
весные составы могут быть получены для различных
исходных соотношений реагентов. При достаточном
числе таких измерений можно определить параметры
подходящего корреляционного уравнения для коэффици-
ентов активности компонентов. Так, пример 10.6 иллю-
стрирует применение метода двухпараметрического
уравнения для двухкомпонентной смеси. Минимальное
число измерений Ку требует определения соответствую-
щего числа параметров. Например, при рассмотрении
трехкомпонентной смеси в уравнение Вильсона включа-
ют шесть параметров и для проведения расчетов необ-
ходимы по крайней мере шесть равновесных составов.
10.4.4. Смешанные фазы. Если в системе одновре-
менно присутствуют и жидкая, и паровая фазы, задача
состоит в том, чтобы установить количества и составы
фаз в условиях мгновенного равновесного испарения и
химического равновесия. Для любого конкретного ве-
щества желательно принимать за стандартное одно и
то же состояние в каждой фазе, так как тогда констан-
ты химического равновесия будут численно одними и
теми же в каждой фазе. Поскольку наиболее широко из-
вестны данные по Дб/ для газов при фугитивности,
равной единице, такое стандартное состояние предпо-
чтительно для всех фаз. Если исходить из того, что в
условиях равновесия парциальные фугитивности явля-
ются одними и теми же для индивидуальных компоне-
нтов и что f? = 1, можно предложить несколько
вариантов выражений для определения активности:
ai-fiv- Ф,У<Р (10'46)
= fiL = Tixifi = YI'xI-0fPfexP( Vib(P “ PS)/PT)
= TiX^Pj(PF)i. (10.47)
Соответственно константы химического равновесия 1
12
Рис. 10.5. Равновесные составы реакции метан и вода [175]. а — влияние температуры при давлении
10 атм и Н/О = 3; б—влияние давления при температуре 1200 °F и Н/О = 3.
Давление, атм
Химическое равновесие 485
Пример 10.6. Коэффициенты активности жидкофазной реакционной смеси
Константа равновесия реакции типа А 2В, выве- денная из энергии Габбса образования, определяется как Для Xi = 0,5 коэффициенты активности даются выра- жениями 1П71 = 0,25В,
Ксе = КУКХ = 0,7. 1П72 = 0,25Л,
Результаты измерений равновесных составов, про- веденных для двух различных исходных соотношений компонентов и соответствующие вычисленные вели- чины Ку а для Xi = 0,6 1П71 = -0.032Л + 0,192В, 1п72 = 0.288Л + 0,072В. Подставляя эту пару коэффициентов активности в
Xi Ку 71 72 выражение Ку = 72/71,
0,5 1,4 (1,55) (1,72) 0,6 2,625 (1,43) (1,94) получаем два уравнения для параметров: 1 4 = (ехР 0,25Л)2 ехр 0,25В > 6о5 _ [ехр(О,288Л + 0,072В)2
Используя эти данные, можно определить параметры ехр(-0,032Л + 0,192В)
уравнения Маргулеса: In-yi = [А + 2(В - Л)Х1]Л2, 1П72 = [В + 2(А - В)х2]Х1. которые дают Л = 1,7588, В = 2,1717. Соответствую- щие коэффициенты активности показаны в таблице в скобках.
Пример 10.7. Равновесные составы одной реакции
Реакция А & В + 2С имеет следующие характе- ристики: Методом подбора получаем: £ = 0,8492 и соответ- ствующие числа молей и мольных долей:
КСе = 4,2, Кф = 0,8 (оценивается заранее), Р = 5, Па0 = 2, Пы> = 1, ПсО = 0. Соответственно па = 1,1508, уа = 0,2449, пь = 1,8492, уь = 0,3936, пс = 1,6984, ус = 0,3615.
Ку = -Кс‘- = 4’2- = 0,2100 КфР1 0,8(25) Расчетное значение К^, как правило, следует проверять после того, как найден пробный равновесный состав, и
(пьо + £)(и<-0 + 2е)2 _ (1 + е)(2е)2 (Пао - е)(пго + 2е)2 (2 - с)(3 + 2е)2 при существенном различии повторять вычисления. Однако в настоящем случае исходные данные для расче- та К^ не даны.
Пример 10.8. Неидеальная жидкофазная реакция
Для реакции Л + 2В г* С константа равновесия равна 0,5, а параметры уравнения Вильсона имеют сле- дующие значения: Кх = К/Ку = 0,5/Ку = , Ку = ус/уауъ. 4(1 - е)
Л12 - 0,6, Л21 = 1,4, Л13 = 0,7, Лз1 = 0,5, Л2з = 0,8, Л32 = 0,9. Итак, сначала принимаем, что Ку = 1, при этом £ = 0,16375. Мольные доли и коэффициенты активнос- ти табулированы. Если же Ку = 0,9085, то £ = 0,17575, при следующей попытке принимаем Ку = 0,9080.
Подстрочные индексы 1, 2 и 3 относятся к веществам Л, Ви С соответственно. Найдем равновесный состав с стехиометрическими отношениями Л и В. Уравнения для тройных коэффициентов активности даны в табл. Линейная интерполяция между попытками адекват- на и показана в нижней строчке таблицы. £ = 0,16375 £ = 0,17575 £ = 0,17580
4.7. Решение начинается с допущения, что Ку = 1, со-
ставы, полученные при таком допущении, будут испо- льзованы для обнаружения более точного значения: X 7 X 7 X
п, = 3 - 2г,
Ха = (1 - £)/Лг, А 0,3)29 1,6367 0,3112 1,6329 0,3112 В 0,6257 1,0113 0,6224 1,0102 0,6224
Хъ = 2ха, С 0,0614 1,5208 0,0664 1,5131 0,0664
Хг = £/п,. Ку 0,9085 0,9080
определяются как
Ксе = Kf =K^KyP^v, Для газов, (10.48)
= КуКхКф5 Kps ХПойнтинга для жидкости. (10.49)
Выражения, определяющие активности, можно связать,
если это даст какое-либо преимущество. Например, для
реакции А & 2В имеются четыре эквивалентные формы
(ФУ)ьР
(ФУ)а
[yx0satPsat(PF)]£
lyX0SatPSat(PP)]fl
(ФУ)ьР2
[yx0satPsat(PP)]fl
[yx0satPsat(PP)]j
(ФУ)аР
(10.50)
(10.51)
(10.52)
(10.53)
Во всех приведенных выше выражениях стандартные
состояния — это состояния с фугитивностью, равной
единице. Для многофазного равновесия (разд. 10.7)
константу равновесия Ксе обычно выражают только че-
рез фугитивности газовой фазы, и тем не менее при
формулировании уравнения мгновенного испарения, ко-
торое необходимо решать одновременно с уравнением
химического равновесия, чтобы найти общие условия
равновесия, следует использовать фугитивности обеих
фаз.
10.4.5. Компоненты, присутствующие в одной из
двух фаз. Такие компоненты по существу являются не-
летучими в реакционных условиях; они, конечно, не
принимают участие в газофазной реакции и, следова-
тельно, не включаются в газофазную Ксе. Более того,
при формулировании уравнения мгновенного испарения
считается, что у таких веществ коэффициенты фазового
равновесия равны нулю, в то время как у нераствори-
мых в жидкости газов эти коэффициенты имеют беско-
нечно большую величину. Например, рассмотрим
многофазную реакцию
Л (газ) + Р(жидкость) 2С(газ и жидкость),
в которой только компонент С присутствует в обеих
фазах. Если компонент В отсутствует в паровой фазе,
константа химического равновесия определяется следу-
ющим выражением:
Ксе (ФсУс)2?IФаУа>
(10.54)
с исчезновением А из жидкой фазы (Ка К> = 0)
уравнение испарения принимает такой вид:
zb zc
1 - Р + 1 + Р(КС- 1)
У1 = К,Х1,
Р = V/F.
(10.55)
(10.56)
(10.57)
Механизм этой реакции должен предусматривать нали-
чие межфазной поверхности, допускающей присутствие
обоих компонентов А и В и возможность их контакта.
В разд. 10.7 некоторые методы определения условий
равновесия комбинированной фазы и химического рав-
новесия рассмотрены подробно.
10.4.6. Влияние давления. Поскольку энергия обра-
зования, ДД/, определяется при фиксированном давле-
нии (обычно 1 атм) или фугитивности, равной единице,
что по существу эквивалентно давлению 1 атм, обе про-
изводные ДД° и Ксе по давлению равны нулю:
(10.58)
Однако равновесные составы газофазных реакций мо-
гут меняться под действием давления, о чем свидетель-
ствует знаменатель выражения
Ку~Ксе/КфР^
(10.59)
если Др # 0 или вследствие изменения Кф с давлением.
Последнее может быть достаточно заметным, как, на-
пример, при синтезе метанола при 300°С. Ниже пред-
ставлены некоторые результаты, полученные исходя из
данных, показанных на рис. 10.3,о.
Р, атм кф Е
уточненные @Кф=\
100 0,62 0,488 0,406
200 0,39 0,737 0,628
400 0,20 0,878 0,778
600 0,13 0,921 0,836
Эти данные получены для стехиометрических отноше-
ний реагентов.
В жидкофазных реакциях давление оказывает уме-
ренное влияние, и его можно определить или из Ку, или
из ^пойнтинга- Производная коэффициента активности
по давлению
(10.60)
обычно невелика; более того, данные редко имеются в
наличии для некоторого диапазона давлений, так что
влияние давления обычно принимается равным нулю.
Поскольку данные, позволяющие провести оценку коэф-
фициента Пойнтинга, доступны, это влияние, если оно
значительно, принимается во внимание (см. пример
10.3).
10.4.7. Влияние температуры. Вывод уравнения,
описывающего влияние температуры на константу хи-
мического равновесия, начинаем с определения
G = H~TS (10.61)
Химическое равновесие 487
или
G/T= HIT-5= Н/Т+ (dG/dT)P. (10.62)
Уравнение (10.62) является дифференциальным линей-
ным уравнением первого порядка, имеющим следующее
решение:
G
Т
Г Н
I dT + Constant.
(10.63)
Дифференцирование этого уравнения приводит к важно-
му выводу
выходов во многих важных реакциях способствуют низ-
кие температуры, что не всегда благоприятно для кине-
тики реакции. Так, при синтезе аммиака, например,
приемлемые скорости реакции достигаются только в
таких условиях, которые приводят всего лишь к 25 %-
ной степени превращения в одном цикле, что вызывает
необходимость в экстенсивном рецикле непрореагиро-
вавшего материала. В других случаях при пониженных
температурах эффективны многоступенчатые реакторы.
Когда теплота реакции практически постоянна в
определенном представляющем интерес интервале тем-
ператур, прямое интегрирование уравнения (10.65) дает
такой полезный результат:
~d(G/T) ] Я
_ дТ Т2 ’
(10.64)
(10.70)
Объединяя энергию Гиббса образования всех ре-
агентов в стехеометрическом соотношении и подставляя
в уравнение (10.64), получаем
В
1пК = А + ~ .
Т
(10.71)
d(&G°/RT) “I /д\пКсе\ ДЯ?
-----------=------------=-------—. (10.65)
дТ \ дТ J Т2
О таком линейном изменении 1пХ в зависимости от
температуры упоминается в разд. 10.3, где цитируются
некоторые данные.
Это выражение называется изохорой Вант-Гоффа
(1884 г).
Изменение стандартной энтальпии реакции, Mir,
зависит от температуры. Если в интересующем нас
температурном интервале фазовых изменений не про-
исходит,
Г т
ДН° = + / ДС0 dT,
J298 Р
где
ДСр — ^iVjCpj — £ ViCpi.
PPR
(10.66)
(10.67)
При наличии фазовых изменений в уравнении (10.66)
проводятся соответствующие изменения, что сделать
несложно. После подстановки уравнения (10.66) в урав-
нение (10.65) интегрирование дает
10.5. Равновесный состав
Рассмотрим реакцию, которая представляется един-
ственным стехиометрическим уравнением
2у.Л(. (10.72)
я р
Количества всех присутствующих веществ выражаются
через глубину протекания реакции (это понятие было
введено в разд. 10.2):
п, - ni0 \ + для продуктов, ...
£ = -------- < (10.73)
/ - для реагентов.
Общее число молей, присутствующих в какой-то мо-
мент времени, определяется выражением
(10.74)
^298
К 1
Г т
ДЯ%8 + / ДС„ dT
J29S р
dT.
а число мольных долей индивидуальных компонентов
находят как
(10.68)
Если теплота реакции ДЯ? известна при Т\, а К извест-
на при Тг (вместо обоих значений при температуре
298 К, которая обычно берется), интеграл принимает
следующий вид:
Я 1 р т 1
1П КТ1 ~ R ]т2 Т2
ДЯ^
dT dT.
(10.69)
Влияние температуры на синтез метанола исследу-
ется в примере 10.9. Ее влияние на эту реакцию, как и
на реакцию окисления диоксида серы, как показывает
рис. 10.1, весьма значительно. Получению равновесных
п,о ± У,£ | + ДЛЯ продуктов, ,1А„.
У, =----------- { (10.75)
nt / - для реагентов.
Константа равновесия выражается через мольные доли
следующим образом:
П(п(0 + v,e)v'
_________Р_______________________
к ____ _
у П (nio - Vie)vi (nt0 + £Дг)Лр
R
(10.76)
Уравнение (10.76) может быть слишком сложным, что-
бы его можно было разрешить относительно £ мето-
дом подбора, как в примере 10.7.
Для бимолекулярной реакции
A + B-C + D, (10.77)
Пример 10.9. Константа равновесия синтеза метанола как функция температуры
Реакция СО + 2Н2 ** СНзОН. Величины теплоем-
кости, энтальпии и энергии Гиббса образования при
298 К взяты из табл. Г.2.
ДЯ&8 = -48080 - (-26420) - 2(0) = -21660 кал/моль,
Ms = -38840 - (-32180) - 2(0) = -6030 кал/моль.
*298 = ехр = 26466,7,
£70 К
ДСр = Ср(СНзОН) - Ср(СО) - 2Cp(Hj)
= -15,287 + 0,01558 Т+ 6,113(Е - 6)Т2
+ 6,195(Е - 9)Т3.
Теплоту реакции вычисляем как функцию темпе-
= 8989,4 ( t - J
\ Т 298 J
-7,9651п + 0,00392(7 - 298)
+ О,5128(Е - 6)(72 - 2982)
= 0,2599(Е - 9)(73 - 2983).
(1)
Как показывает нижеследующая таблица, вычис-
ленные по уравнению (1) значения лишь удовлетвори-
тельно согласуются с результатами, приведенными
Эвеллом [278] и вычисленными по уравнению (2):
Igtf = -12,5173 + 5090,5/7 (2)
(коэффициенты взяты из табл. Г.1)
ратуры
Д/4 = -21660 + Г ACPdT
>298
= - 17862 - 15,287Т + 0.00779Т2
+ 2,О38(Е - 6)Т3 + 1,549(Е - 9)Г\
а константа равновесия как функция температуры дает-
ся уравнением
JT
298
----Г- dT
RT2
106 К
Г, °C уравнение (1) данные Эвелла [278] 1уравнение (2)
100 14,65(Е6) 10,84(Е6) 13,27(Е6)
200 22110 16950 17390
300 288 232 231
400 13,0 10,9 11,1
500 1,302 1,134 1,165
уравнение равновесия можно представить в стандарт-
ной квадратичной форме:
(*у 1 )е [-*y(wao + nbo) ^со ndol£
4- (КуПаопьо ncondo) 0. (10.78)
Реакционная система, которая представлена одно-
временно несколькими стехиометрическими уравнения-
ми, может быть выражена через глубину протекания
реакции £, для каждого уравнения и константы равнове-
сия для каждой реакции. Для того чтобы получить
уравнения, связывающие равновесие с составами, реак-
ции располагают в соответствующем порядке и допу-
скают, что они протекают последовательно; при этом
исходные количества компонентов любой определенной
реакции являются теми количествами, которые образу-
ются после достижения определенной глубины протека-
ния всех предшествующих реакций. Исключение
промежуточных количеств приводит к системе уравне-
ний, определяющих число молей всех присутствующих
веществ через начальные количества и через глубину
протекания реакций £i и £2.
Рассмотрим такой подход на примере ряда одно-
временно протекающих реакций.
2А + В & 2С, (10.79)
А + 2С& 3D. (10.80)
Запишем соответствующие выражения для глубины
протекания реакции £i и £2:
ИаО Ла1 Пс1 ПсО
£1 =--------------= пьо ~ пь =---------------
ПС1 — Пс Па — HdO
£2 = Па1 Па — - ‘ — ------
(10.81)
(10.82)
После исключения nai и пс\ из этих уравнений число
молей всех веществ, исходя из их первоначальных коли-
честв с учетом двух различных величин глубины проте-
кания реакции, составляет
Па = ПаО - 2£1 - £2, (1О.83а)
Пь = Пьо - £1, (10.836)
Пс - ПсО + 2£1 - 2£2, (1О.83в)
Tld = HdO + 3f2, (10.83г)
nt = Пю - £1. (Ю.83д)
Соответственно два уравнения равновесия имеют еле-
дующий вид: ncnt К\/КФ\ ~ „2„ р е2)> Г1анЬ г (10.84)
v nd
К2^Кф2~ „ и2~/г(е1> е2)- (10.85)
Для определения глубины протекания реакции, а следо-
вательно, и равновесного состава можно воспользовать-
Химическое равновесие 489
Рис. 10.6. Константы равновесия некоторых реакций
как функция температуры [61]. (См. также табл. Г.1 в
приложении Г.)
ся любым благоприятным методом совместного
решения нелинейных уравнений. В примере 10.10 приме-
няется графический метод решения такой задачи. Более
детально методы расчета рассматриваются в последую-
щих разделах, где также обсуждаются менее громозд-
кие способы формулирования решений для многореак-
ционного равновесия.
10.6. Множественные реакции
Даже в заведомо простых химических системах в
состоянии равновесия, кроме основных веществ, может
присутствовать большое число дополнительных компо-
нентов, образовавшихся из основных в результате раз-
личных простых или параллельных реакций. Например,
при изомеризации изопропанола в н-пропанол побоч-
ные реакции могут привести к образованию значитель-
ных количеств или следов ацетальдегида, ацетона,
водорода, диоксида углерода, воды, а возможно, и дру-
гих веществ. Пиролиз бутана, который, как можно бы-
ло бы ожидать, протекает по реакции С4Н10 С2Н4 +
+ СгНб, в действительности приводит к образованию
некоторого количества пропилена, нескольких изомеров
бутана, бутадиена, метана, водорода и даже бензола.
В стехиометрических уравнениях, которые связыва-
ют основные и побочные продукты, некоторые из по-
бочных продуктов могут оказаться комбинациями
других, однако определенное минимальное число их
всегда можно обнаружить и, исходя из этого, предста-
вить себе реакционную систему. Такие независимые сте-
хиометрические уравнения приводят к соответствую-
щим равновесным уравнениям, которые вместе с урав-
нениями материального баланса присутствующих хими-
ческих элементов составляют математические системы,
позволяющие установить равновесный состав.
Альтернативный метод обнаружения условия
равновесия — это нахождение минимума энергии Гибб-
са реакционной системы. Оба метода будут описаны де-
тально. Обзоры работ, посвященных этой теме, и
детальное описание методов приведены в монографиях
ван Зеггерна и Стори [138], Голуба и Вонки [58], Смита
и Миссена [123]. Другие ссылки даны в разд. 7.8.3.
Независимые и производные компоненты и реак-
ции. Систематический метод нахождения набора незави-
симых реакций постулированной смеси, в результате
Рис. 10.7. Схема метода релаксации или последовательно соединенных ре-
акторов для определения равновесия сложных реакций. Каждый из реакто-
ров представляет собой реактор периодического действия, и в нем дости-
гает своего равновесия реакция, обозначенная тем же номером, что и реак-
тор. Процесс начинается загрузкой исходной смеси в реактор 1, где
реакция 1 достигает стадии равновесия. После этого продукт переходит
в реактор 2, в котором равновесия достигает только реакция 2. Получен-
ный продукт переходит в реактор 3, где начинается реакция, обозначенная
номером 3. Далее продукт из реактора 3 возвращается в реактор 7, и про-
цесс продолжается до тех пор, пока состав выходящего продукта из любо-
го реактора не станет постоянным.
которых могут образоваться все компоненты реакций,
разработан Бринклеем [202] и дополнен Кандинером и
Бринклеем [384]. Первый шаг: необходимо составить
матрицу состава, в которой вещества будут распложе-
ны в виде рядов, а химические элементы — в виде коло-
нок, как в примере 10.13. Число независимых
компонентов равно рангу матрицы — самому большо-
му ненулевому определителю, получаемому из матри-
цы. Математически ранг — это максимальное число
линейных независимых рядов и колонок матрицы, см.,
например, [299]. Для нахождения ранга матрицы разра-
ботаны систематические вычислительные процедуры,
однако, если в системе присутствуют только три или че-
тыре химических элемента, его обычно легко обнару-
жить, не прибегая к вычислениям.
10.6.1. Стехиометрический метод. Метод Брин-
клея, предназначенный для вывода и совместного реше-
ния уравнений равновесия множества реакций, включает
семь шагов, как описано ниже, и этот метод применен
в примере 10.13.
1. Выбор химических компонентов, которые могут
сосуществовать в условиях равновесия.
2. Определение числа независимых компонентов.
3. Выбор ряда независимых компонентов из числа
имеющихся и идентификация оставшихся как производ-
ных компонентов.
4. Составление ряда химических уравнений, в ре-
зультате которых только из независимых компонентов
образуется отдельно каждый производный компонент.
5. Составление уравнения химического равновесия
каждой из этих реакций.
6. Составление уравнений материального баланса
для отдельных химических элементов: средневзвешен-
ная сумма элементов во всех присутствующих вещест-
вах в условиях равновесия равна их количеству в
первоначальной смеси.
7. Уравнения равновесия (п. 5) и элементного ба-
ланса (п. 6) образуют систему, которую разрешают от-
носительно состава равновесной смеси.
К некоторым из этих шагов даны некоторые по-
яснения.
1. Выбор этих компонентов, которые могут присут-
ствовать в условиях равновесия, диктуется обстоятель-
ствами и может основываться на аналогии. Лучше
ошибиться, имея слишком много компонентов, чем
слишком мало, поскольку после завершения вычислений
выяснится, что концентрации любых излишних элемен-
тов пренебрежимо малы.
2. Число независимых компонентов обычно равно
числу присутствующих химических элементов, однако
может быть и меньше, если в системе протекают реак-
ции изомеризации, полимеризации или присоединения.
Такие случаи наблюдаются при определении ранга мат-
рицы состава. Указанные вопросы обсуждаются Ари-
сом и Маем [2]. Число независимых компонентов в
одной из систем примера 10.11 меньше числа содержа-
щихся в ней химических элементов.
3. Хотя обычно возможно несколько комбинаций
независимых компонентов, численные вычисления уско-
ряются путем выбора таких компонентов, которые в ус-
ловиях равновесия могут давать наибольшие концен-
трации при наличии перечисленных ниже ограничений.
а. Компоненты должны быть стехиометрически не-
зависимы друг от друга, т. е. должна быть исключена
вероятность присутствия изомеров, полимеров или про-
дуктов присоединения.
б. В этот набор независимых компонентов должны
быть включены все исходные химические элементы.
в. Любой компонент, концентрацию которого не-
льзя выразить посредством константы химического
равновесия, например чистая конденсированная фаза с
активностью, равной единице, должен быть включен в
систему уравнений как независимый компонент.
4. Существует только один возможный путь обра-
Химическое равновесие 491
Пример 10.10. Равновесие в синтезе метанола из CQ?. и Н2 (графическое решение при Т - 350 К и Р - 500 атм)
В процессе синтеза метанола в системе одновремен-
но происходят следующие реакции:
СОг + Н2 СО + Н2О: Кх = 0,05,
l-e; 3-ei fit Г
СО + 2Н2 СНзОН: Кг = 4,458(Е - 5)
ei-e2 3-ei-2e2 ег
где £i и s2 — глубина протекания реакции. Количества
реагентов, выраженные при Н2/СО2 -Зв начале про-
цесса, равны соответственно
Соответственно уравнения константы равновесия
определяются следующими выражениями:
=
______________------------= 0,05,
(1 - £:)(3 - Ех - 2£г)
£2(4 - 2£2)2
(£1 - £z)(3 - £1 - 1£2)2Р2
= 4,458(Е - 5).
С02
Н2
СО
Н2О
СНэОН
nt
1 - £1
3 — Si — 2fi2
£i ~ £2
£1
£2
4 - 2fi2
Уравнения решаются графически. Кривые зависимости
£2 •- У(£1) построены для каждого уравнения при приня-
тых значениях £1 и найденных по методу Ньютона —
Рафсона соответствующих им значений е2. Точка пере-
сечения кривых устанавливается графически.
Пересечение при £1 = 0,463 и е2 = 0,359.
каждого производного компонента из независи-
к
зования
мых компонентов.
5. Величины константы равновесия должны быть
известны, и должны быть известны способы их опреде-
ления из энергий Гиббса образования компонентов.
7. Поскольку система уравнений обычно включает
несколько нелинейных уравнений, приходится прибегать
методам аппроксимации. Этой проблеме уделялось
много внимания в связи с многореакционным равнове-
сием, так как при этом может наблюдаться плохая схо-
димость на промежуточных стадиях последовательных
приближений корней и могут появляться отрицатель-
ные мольные доли. При этом были использованы при-
ведение уравнений к линейным системам перед или в
ходе применения метода Ньютона — Рафсона, линейное
программирование (для определения связей химических
элементов), метод крутого спуска и другие методы
оптимизации.
Если используется последовательная аппроксимация
и требуется начальная оценка состава, то такую оценку
следует проводить исходя из элементного баланса. В
примере 10.13 показано, как применять метод Ньюто-
на — Рафсона в сочетании с процедурой Бринклея.
10.6.2. Метод релаксации. После того как сформу-
лированы необходимые стехиометрические уравнения и
составлены уравнения материального баланса, следует
прежде всего установить глубину протекания реакции
отдельных компонентов. В некоторых случаях эта зада-
ча может быть решена при помощи метода релаксации,
называемого также методом последовательно соеди-
Пример 10.11. Независимые компоненты и независимые реакции
Реакция H2S + SO2. Компоненты, которые будут
присутствовать в системе в условиях равновесия, нахо-
дят из матрицы состава. Первые три строки образуют
ненулевой детерминант, таким образом, здесь присут-
ствуют три независимых компонента. Выбираем в каче-
стве таковых H2S, SO2 и НгО.
н О S
1 H2S 2 0 1
2 SO2 0 2 1
3 Н2О 2 1 0
4 Н2 2 0 0
5 S2 0 0 2
Для Н2: 2Н2О + H2S -* ЗН2 + SO2.
Для S2: 2H2S + SO2 -* 3/2S2 + 2Н2О.
Получение этанола из этилена и воды. Предпола-
гается, что при равновесии в системе присутствуют
только эти три вещества. Ранг матрицы состава 2. Вы-
бираем С2Н4 и НгО в качестве независимых компо-
нентов.
С Н О
1 С2Н4 2 4 0
2 Н2О 0 2 1
3 С2Н5ОН 2 6 1
Для С2Н5ОН: С2Н4 + НгО -► С2Н5ОН.
Ранг матрицы меньше, чем число элементов. Только
два из трех элементных балансов независимы; баланс
по водороду получают добавлением кратных балансов
по углероду и кислороду.
Чтобы найти четыре неизвестных щ, пг, пз и п, со-
вместно с уравнением равновесия используются общий
баланс и балансы по углероду и кислороду.
ненных реакторов [477], позволяющего решать уравне-
ния равновесия по-одному, серией и повторять решение
до достижения сходимости.
Авторы метода [477] применяют гипотетическую
систему, число реакторов в которой равно числу незави-
симых стехиометрических уравнений, как это изображе-
но на рис. 10.7 на примере системы для трех реакций.
Каждый реактор действует как периодический и приво-
дит к равновесию реакцию, номер которой соответству-
ет номеру реактора. Процесс начинается с загрузки
исходной смеси в первый реактор, в котором равнове-
сия достигает реакция 1. Полученный продукт перегру-
жается во второй реактор, в котором равновесия
достигает реакция 2. Далее продукт из второго реакто-
ра перегружается в третий реактор, затем в первый и
т. д. В каждый реактор поступает продукт равновесия
из предыдущего реактора, и в каждом реакторе проте-
кает только одна реакция.
Одно из преимуществ метода последовательно со-
единенных реакторов состоит в том, что каждый раз
приходится решать уравнения только с одним неиз-
вестным — глубиной протекания реакции, а это процесс
более простой, чем одновременное решение системы не-
линейных уравнений. В подходящих случаях сходимость
достигается менее чем через десять проходов через ре-
акторную систему.
Сравнение метода последовательно соединенных
реакторов с методом Ньютона — Рафсона показывает,
что последний требует начальных оценок количеств всех
компонентов и что сходимость в весьма заметной степе-
ни зависит от этих оценок. В одном из примеров приве-
денных авторами [477] процесс из семи реакций
адекватно устанавливается после восьми циклов, при
этом результаты сравнимы с полученными путем мини-
мизации энергии Гйббса.
Этот метод используется в примерах 10.14 (две ре-
акции) и 10.15 (три реакции). Кроме того, в указанных
примерах он также сравнивается с методами прямой
итерации (в первом случае) и графическим (во втором),
которыми можно пользоваться при малом числе
реакций.
10.6.3. Минимизация энергии Гйббса. Если энергия
Гиббса при данных Т и Р минимальна, что соответству-
ет равновесному состоянию системы, то это положение
может быть непосредственно применено к нахождению
равновесного соства реакционной смеси без предшеству-
ющей спецификации каких-либо стехиометрических от-
ношений. Единственная необходимая информация —
это величины энергии Гйббса образования всех химиче-
ских компонентов, которые могут присутствовать в сис-
теме в условиях равновесия, и характер изменения их
фугитивностей или коэффициентов активности, если их
нельзя принять равными единице. Этот метод отлича-
ется гибкостью по отношению к возможным компоне-
нтам, так как по завершении расчетов выясняется, что
концентрации «лишних» компонентов пренебрежимо
малы.
Указанное выше условие математически называется
условным или ограниченным минимумом, так как энер-
гия Гйббса реакционной смеси минимальна, если в усло-
виях равновесия сохраняется общее число индиви-
дуальных химических элементов, образующих содержа-
щиеся в системе химические компоненты. Возможно не-
сколько вариантов решения проблемы, а наиболее
приемлемым является метод множителей Лагранжа, ко-
торый будет использован здесь.
Множители Лагранжа — это постоянные в общем
процессе нахождения экстремума функции п пере-
менных:
/(xi, Хг...хл) “* минимум (максимум) (10.86)
с ограничениями
х2,. . . , х„) = 0 к = 1, 2, . . . , т, (10.87)
где т — дополнительные коэффициенты (т < п). Со-
гласно этому принципу, нахождение экстремума слож-
ной функции
ф = f + 2 ^кфк -* минимум (максимум) (10.88)
аналогично поиску экстремума исходной функции f.
Множители Лагранжа — это постоянные Хк.
Химическое равновесие 493
Пример 10.12. Равновесие в реакции H2S + SO2. Графическое и прямое итерационные решения
Исходная смесь содержит 35% H2S, 15% SO2 и 50%
инертного N2 при 1500 К и 0,8 атм. Предполагается, Расчет по методу
что в условиях равновесия в системе содержатся: H2S, Н2, S2, Н2О и N2. Запишем две независимые реакции и их константы равновесия: Вегштайна £1 £2
H2S Н2 + O,5S2, Ki = 0,334, 0,1947 0,0063
2H2S + SO2 ** 2НгО + 1,5S2, К2 = 30,2. 0,1083 0,0987
Выразим материальные балансы индивидуальных ком- 0,0725 0,0603 0,0566 0,1105 0,1142 0,1153
понентов через глубину протекания реакции:
1 H2S 0,35 - £1 - 2е2 0,0555 0,1157
2 Н2 £1 0,0551 0,1158
3 S2 0,5fii + 1,5£2 4 SO2 0,15 - £2 5 Н2О 2е2 6 N2 0,5 nt 1 + 0,5fii + 0,5fi2 0,0550 0,1158
и уравнения равновесия:
, ,п „ , (2€2)2[0,5(£1 + SfizlF’^O.S)0’5 0 12)
Jl ’ (0,35 - £1 - 2£2)2(0,15 - £2)(1 + 0,5fii + 0,5£2)°’5
Содержание компонентов (равновес-
ные моли), найденных
1. Эти два уравнения представлены графически сна- Компонент графи- по методу по методу
чала для определенного £1, а затем для €2, найденного чески Вегштайна релаксации
с использованием единственной переменной по методу (при-
Ньютона — Рафсона. Точка пересечения соответствует мерЮ. 13)
ei = 0,0549 и £2 = 0,1158.
2. Чтобы применить метод итерации, уравнения
равновесия преобразуют к виду H2S 0,064 0,0634 0,065
Н2 0,058 0,0550 0,055
£1 = gi(£i, £2), S2 0,200 0,2012 0,201
£2 = g2(£i, £2), SO2 0,036 0,0342 0,034
Н2О 0,228 0,2316 0,231
которые для настоящего случая определяются как N2 0,500 0,5000 0,500
0,5281(0,35 - £1 - 2£2)(1 + 0,5£i + О,5£2)0,5 Общее 1,086 1,0854 1,084
C1 (fii + 3£2)0-5
1 23,8752(0,35 - £1 - 2£2)2(0,15 - £2)(1 + 0,5fii + О,5£2)0,5 (4)
С2 Д/ (£i + 3£2)1,5
Прямая итерация предусматривает следующее: подста-
новка пары fiV’, £^0) по методу Рафсона — Ньютона —
Скэтчарда в первом уравнении дает eV’ в левой части
уравнения, последующая подстановка (eV’, £$0’) в пра-
вой части уравнения (2) дает eV’ и так до полной сходи-
мости. В настоящем примере, однако, прямая итерация
не дает достаточной сходимости, и, чтобы улучшить ее,
используют метод Вегштайна. Вычисления начинают с
того, что для €2=0 находят при помощи итерации по
Вегштайну ci из уравнения (3), подставляют найденное
значение ei в уравнение (2), находят при помощи итера-
ции по Вегштайну из этого уравнения £2 = 0,0063 и воз-
вращаются к уравнению (4) и повторяют операцию, как
это показано в таблице.
3. Метод Ньютона — Рафсона может применяться
для совместного решения уравнений. В ситуациях, похо-
жих на описываемую, обычно предпочитают найти чис-
ленные значения необходимых производных, например
Э/ _ /(1,0001€1, €2) -/(£1, с2)
Э£1 0,0001£i ’ ( ’
которые легко рассчитать на калькуляторе или компью-
тере. Поскольку в данной книге приводятся и другие
примеры использования метода Ньютона — Рафсона
для совместного решения уравнений, здесь мы более к
нему прибегать не будем.
494 Глава 10
Пересечение при ei = 0,0549, Si = 0,1158.
Приравниваемые нулю производные
/ дф \
1^7 )“° 1=1,2, (10.89)
и ограничения
Фк = О к= 1,2, . . . , т, (10.90)
составляют систему п + т уравнений, из которых xt и
Хк оцениваются при нахождении минимума (мак-
симума).
Возможен и другой подход, если процесс алгебраи-
чески возможен, т перменных можно заместить, чтобы
выразить f как функцию п - т перменных, нахождение
экстремума которой выполняется обычным образом.
Часто, однако, эта процедура более трудоемка, а если
фк математически сложно выразить, то вообще неосу-
ществима. Формальное доказательство метода множи-
телей Лагранжа можно найти в обычных курсах
высшей математики, например в «Курсе высшей мате-
матики» Смирнова [758].
В примере 10.16 рассмотрено несколько вариантов на-
хождения экстремума по методу Лагранжа и демон-
стрируются результаты, которые должны быть такими
же, что и получаемые другими, возможно, более извест-
ными методами, если таковые применимы.
Основные уравнения. Общая энергия Гиббса смеси
слагается из вкладов чистых компонентов и эффектов
смешения:
nG = Е njGi = Е n,(G? + Gi - G?)
= Е л,(Сг? + ЯГ1па,), (10.91)
или более кратко
g = nG/RT = Е + 1па(). (10.92)
Активности жидкостей выражаются через их коэффици-
енты активности
ai =fi/f*i = У/Х, = У/Л//Л, (10.93)
а в качестве стандартного принимается состояние чис-
той жидкости при давлении 1 атм. Активности газов
выражаются через их коэффициенты фугитивности
°i = Ф,У,Р = Ф{Рп,/п, (Ю.94)
фугитивность которых в стандартном состоянии равна
единице.
Поскольку коэффициенты активности и фугитив-
ности зависят от составов фаз, которые заранее неиз-
вестны, решения задач, включающие неидеальные
смеси, проводят следующим образом. Начинают с
предположения об идеальности, после чего находят со-
ставы, которые в свою очередь используют для оценки
коэффициентов активности и фугитивностей. Этот про-
цесс повторяют столько раз, сколько это необходимо
для получения желаемой точности. Так как основная
схема определения условий равновесия идеальных и не-
идеальных индивидуальных систем одна и та же, дШ
упрощения последующего обсуждения речь будет идя
только об идеальном газе.
Химическое равновесие 495
Пример 10.13. Равновесие в реакции СзН8 + SO2. Решение по методу Бринклея
Если пропан и диоксид серы реагируют при 500°С
и атмосферном давлении, можно ожидать, что в число
продуктов реакции входят СзН8, SO2, СзНб, С2Н4,
СН4, Н2О, H2S и Нг. Выводим 1) матрицу состава, 2)
подходящий набор независимых компонентов, 3) хими-
ческие уравнения для образования побочных продуктов,
4) алгебраические уравнения, из которых можно опреде-
лить состав системы в условиях равновесия.
Компонент С И О S logA>
1. С3Нб 3 6 0 0 -9,519
2. H2S 0 2 0 1 3,516
3. Н2О 0 2 1 0 13,9660
4. Н2 0 2 0 0 0
5. С3Н8 3 8 0 0 -8,030
6. С2Н4 2 4 0 0 6,788
7. СНд 1 4 0 0 0,3527
8. SO2 0 0 2 1 20,674
Детерминант, основанный на первых четырех строчках,
равен -6^0; следовательно, в системе присутствуют
четыре независимых компонента. Предположим, что
это СзНб, H2S, НгО и Нг. Запишем уравнение образо-
вания побочных продуктов:
5) СзНб + Нг = СзН8, Ks = (Т/Л)-1Л5/Л1Л4,
6) 2СзНв = ЗС2Н4, Кб = (т/л)л|/л?,
7) СзНб + ЗН2 = ЗСНд, Ку = (т/л)-1Л7/Л1Л4,
8) H2S + 2Н2О = ЗН2 + SO2; Ks = (т/п)пЬц/пгп1
9) п = ni + щ + пз + гц +
+ П$ + Лб + Пу + Л8.
Предполагая, что в исходной смеси содержится по 1
молю СзНв и SO2, составим балансы по С, Н, S и О:
1) 3 = Зл! + Эл? + 2лб + пу.
2) 8 = 6л1 + 2л2 + 2лз + 2/ц + 8лз + 4Лб + 4л?.
3) 1 = л2 + л8.
4) 2 = лз + 2л8.
Для перечисленных компонентов величины logA/ обра-
зования при 500°С табулированы в приведенной выше
матрице. Девять уравнений приведены к стандартным
формам как исходные для решения их методом Ньюто-
на — Рафсона или другим эквивалентным методом:
/1 = ЗЛ1 + Зл5 + 2лб + Пу —3 = 0,
fi = 6Л1 + 2л2 + 2лз + 2л< + 8п$ + 4лв + 4л? — 8 = 0,
/з = л2 + л8 - 1 = 0,
ft = Л3 + 2л8 —2 = 0,
/5 = ЛЛз - К5РП1П4 = 0,
/б = Рп% - Kenni = 0,
fy = ппу - КуРщп^ = 0,
ft = Pnint - Къппзпз = 0,
/9 = Л - Л1 ~ Л2 - ЛЗ - Л4 ~ Л5 - Лб - Л7 - Л8 = 0.
Допустим, что /ю — это ft при определенных на-
чальных значениях лю, л20.... Согласно методу Ньюто-
на — Рафсона, корректированные значения оценок
л7- = njo + dj, где <5у получают из системы девяти линей-
ных уравнений
/ю + S = 0’ 1 = 1» 2, ..., 9.
У_1 \ dnj J о
Вычисленные начальные значения должны удовлетво-
рять материальным балансам. Для одной из таких сис-
тем л = 3 и Лб = пу = л8 = 1. Скорость сходимости
действительно зависит от таких исходных значений.
Эта задача решена Кандинером и Бринклеем [384].
Пример 10.14. Равновесие в реакции H2S + SO2. Решение методом релаксации
Авторами работы [477] реакция, рассмотренная в
примере 10.12, решена методом релаксации. Последова-
тельность этапов решения дана ниже Предполагая, что
две реакции протекают независимо, запишем уравнения
равновесия через глубину протекания реакции:
л» для любой реакции устанавливаются при допуще-
нии, что предыдущая реакция достигает равновесия, как
это показано на циклической схеме на рис. 10.7. Реше-
ния для £1 и £2 найдены при помощи метода Вегштай-
на. Приведем уравнения для первых двух циклов:
и Г D
к - П2ПЗ I "
1 Л1 |_ Лп
(лго + £1)(лзо + O,5ei)0,5
лю - £1
0.5
0,5
0.5
Л5ЛЗ’ I р
----„'П,— I
П1П4 I Лг2
(изо + 2£г)2(лзо + 1,5сг)]
(лю - 2£г)2(л4о - е2)
пю + 0,5ei
0,334, (1)
р I °-5
---------- =30,2(2)
пю + 0,5е2 J
По мере выполнения последовательных аппроксимаций
количества пю = меняются. Исходные коли-
чества
п «4 = £1(0.5£2)°’5(0,8)0-5
’ (0,35 - £i)(l + 0,5£i)°’s ’
£1 = 0,194.
4£г(0,097 + 1,5£2)1,5(О,8)0’5
30,2 =----------------,---------------------------,
(0,156 - 2e2) (0,15 - £2)(1,097 + O,5£2)o,s
£2 = 0,066.
о 334 = (0>194 + с»)(0Л96 + 0,5£1)0>5(0,8)°-5
’ (0,023 - £i)(1,129 + 0,5£1)°-5
61 = —0,086.
30,2 - (0’132 + У(°’153 + •^>1'5<0’8)°'!0! , й - 0,033.
(0,108 - 2£2)2(0,084 - £2)(1,085 + 0,5£2)о<5
Помещенная ниже таблица — это решение, полученное
авторами работы [473].
Цикл 1 2 3 4 5 6
Реактор 1 2 3 4 5 6 7 8 9 10 11 12 12
Уравнение 1 2 1 2 1 2 1 2 1 2 1 2 2
£1 0,194 ... -0,086 -0,035 0,009 ... -0,007 ... -0,002 ...
£2 0,066 ... 0,033 0,012 0,00 ... 0,001 0,001
Исходная Парциальное
смесь, давление,
моли моли моли моли моли моли моли моли моли моли моли моли моли атм
H2S 0,35 0,156 0,023 0,108 0,044 0,080 0,056 0,069 0,061 0,065 0,063 0,064 0,063 0,0465
Н2 0 0,194 0,194 0,108 0,108 0,073 0,073 0,064 0,064 0,057 0,057 0,055 0,055 0,0406
S2 0 0,097 0,196 0,153 0,202 0,184 0,202 0,195 0,201 0,199 0,201 0,200 0,201 0,1483
SO2 0,15 0,15 0,084 0,084 0,051 0,051 0,039 0,039 0,036 0,036 0,035 0,035 0,034 0,0251
Н2О 0 0 0,132 0,132 0,197 0,197 0,221 0,221 0,228 0,228 0,230 0,230 0,231 0,1706
N2 0,50 0,50 0,50 0,50 0,50 0,50 0,50 0,50 0,50 0,50 0,50 0,500 0,50 0,3690
Сумма: 1,097 1,129 1,085 1,102 1,085 1,091 1,088 1,090 1,085 1,086 1,084 1,084 0,8001
Пример 10.15. Равновесие при диссоциации изопропанола. Решение методом релаксации
Реакция контролируется при давлении 1 атм и тем-
пературе 400 и 800 К. Предположим, что в системе при-
сутствуют следующие вещества: изопропанол,
н-пропанол, ацетон, пропионовый альдегид и водород
(нумеруются в указанном порядке), и одновременно
протекают три независимые реакции:
«ЗО-С3Н7ОН H-C3H7OH (1) (3) (1)
«30-С3Н7 ОН (СНз)2СО + Н2 (1) W (2) - (2)
«ЗО-С3Н7ОН С2Н5СНО + н2. (1) (5) (2) (3)
Константы равновесия указанных реакций имеют сле-
дующие значения:
Т, к Кг К2 К.
400 0,0640 0,0760 0,00012
800 1,9055 0,0025 0,0301
Уравнения равновесия записываются так:
Изо + £1 _ р. (4)
Пю - £1
(«40 + £2)(Л2О + £г)Р _ к (5)
(«ю - £г)(Х п»о + £г)
(«50 + £з)(«2О + £3)Р = (П10 - £з)(Х ”«> + £з) (6)
Исходное количество чистого изопропанола равно 1
молю. При 400 К достаточно 3 циклов, чтобы глубина
протекания реакции была менее 10 ~4. При 800 К для
этого необходимо 6 циклов. Представленный процесс
диссоциации йзопропанола не вполне соответствует ре-
ально происходящему, поскольку здесь не принято во
внимание, например, образование пропилена и после-
дующее его гидрирование до пропана. Однако в данном
случае задача состояла в том, чтобы на простом приме-
ре рассмотреть суть проблемы. Термодинамические
данные были взяты из книги Сталла [130].
Химическое равновесие 497
Цикл 1 2 3
Реакция 1 2 3 1 2 3 1 2 3
1. Изопропанол 1 0,9398 0,6843 0,6837 0,6992 0,6968 0,6968 0,6969 0,6969
2. Водород 0 0 0,2555 0,2561 0,2561 0,2585 0,2585 0,2585 0,2585
3. н-Пропанол 0 0,0602 0,0602 0,0602 0,0447 0,0447 0,0446 0,0446 0,0446
4. Ацетон 0 0 0,2555 0,2555 0,2555 0,2579 0,2579 0,2579 0,2579
5. Пропионовый
альдегид 0 0 0 0,0006 0,0006 0,0006 0,0006 0,0006 0,0006
т 1,00 1,00 1,2555 1,2561 1,2561 1,2585 1,2585 1,2585 1,2585
£1 0,0602 -0,0155 -0,0001
£2 0,2555 0,0024 2(106)
£з 0,0006 3(10"5) 3(10“5)
№519. Пропиловый спирт СзН8О (газ в идеальном состоянии)
кал/моль • К
ккал/моль
Т, К с? 5° (G0 - Н?98)/Г ГгО ЕГО Г1 — /7298 днЛ AG/> logKp
298 20,82 77,63 77,63 0,00 -61,55 -38,95 28,550
300 20,91 77,76 77,64 0,04 -61,58 -38,81 28,273
400 25,86 84,46 78,52 2,38 -63,11 -30,98 16,926
500 30,51 90,75 80,34 5,21 -64,40 - 22,79 9,963
600 34,56 96,68 82,57 8,47 -65,46 - 14,37 9,235
700 38,03 102,27 84,99 12,10 - 66,29 -5,79 1,807
800 41,04 107,55 87,48 16,06 -66,94 2,90 -0,791
900 43,65 112,54 89,99 20,29 -67,41 11,66 -2,831
1000 45,93 117,26 92,49 24,78 -67,73 20,47 -4,474
№520. Изопропиловый спирт СзН8О (газ в идеальном состоянии)
кал/моль • К
ккал/моль
Т, К С 0 Ср S° — (G° - H^w)/T rjO rjO Ji — /7298 дя/ AG/> logKp
298 21,21 74,07 74,07 0,00 -65,15 -41,49 30,411
300 21,31 74,21 74,08 0,04 -65,18 -41,34 30,118
400 26,78 81,09 74,98 2,45 -66,65 -33,17 18,120
500 31,89 87,64 76,86 5,40 -67,82 -24,66 10,776
600 35,76 93,81 79,18 8,78 -68,74 -15,94 5,804
700 39,21 99,58 81,68 12,53 - 69,46 -7,07 2,208
800 42,13 105,01 84,26 16,61 - 69,99 1,87 -0,511
900 44,63 110,12 86,86 20,95 -70,36 10,88 -2,642
1000 46,82 114,94 89,43 25,52 -70,58 19,93 -4,355
13-864
№546. Пропионовый альдегид СзНбО (газ в идеальном состоянии)
кал/моль • К
ккал/моль
Г, к С? S0 -(G° - Ми)/Т rjO rjO /I - /7298 дн/ ДО/ log*.
298 18,80 72,83 72,83 0,00 -45,90 -31,18 22,851
300 18,87 72,95 72,84 0,04 -45,92 -31,09 22,644
400 23,09 78,97 73,63 2,14 -47,00 -25,97 14,188
500 26,89 84,54 75,26 4,64 -47,91 -20,60 9,005
600 30,22 89,74 77,24 7,50 -48,66 -15,07 5,489
700 33,03 94,62 79,38 10,67 -49,26 -9,42 2,941
800 35,45 99,19 81,57 14,10 -49,73 -3,70' 1,011
900 37,55 103,49 83,77 17,75 - 50,08 2,08 -0,505
1000 39,27 107,54 85,95 21,59 - 50,32 7,89 -1,725
№554. Ацетон СзНбО (газ в идеальном состоянии)
кал/моль • К
ккал/моль
Г, К С? $° -(G° - Н°ы»)/Т rjO г/0 п — /7298 ЬН? ДОЛ logKp
298 17,90 70,49 70,49 0,00 -52,00 -36,58 26,811
300 17,97 70,61 70,50 0,04 - 52,02 -36,48 26,577
400 22,00 76,33 71,25 2,04 -53,20 -31,12 17,001
500 25,89 81,67 72,80 4,44 - 54,22 -25,48 11,135
600 29,34 86,70 74,70 7,20 - 55,07 -19,65 7,156
700 32,34 91,45 76,76 10,29 -55,74 -13,69 4,273
800 34,93 95,94 78,88 13,65 -56,28 -7,64 2,088
900 37,19 100,19 81,01 17,26 - 56,67 -1,54 0,373
1000 39,15 104,21 83,13 21,08 - 56,93 4,61 -1,007
Пример 10.16. Нахождение экстремума при помощи множителей Лагранжа
а. Находим экстремум f = xyz при граничном
условии
< b = x + y + z- i =0.
ф = f + \ф,
дф
= yz + X = 0,
ах
дф
- т— = xz + X = 0,
ду
дф
- Г— = ху + X = 0.
0Z
Отсюда
-X = yz = xz = ху
и соответственно
х = у = z = 1/3 и X = -1/9.
При альтернативном подходе исключаем z = 1 - х - у
и получаем
f = ху(1 - х - у) максимум.
Две производные f равны нулю при минимуме
(максимуме), таким образом,
= у(1 - 2х + у) = 0, -%- = х(1 - 2у - х) = 0,
ах ау
откуда х = у = z = 1/3, как и выше.
б. Находим экстремум f = х2 + у2 + z2 при следу-
ющих граничных условиях:
х2 у2 z2
*--^г + тп + 2г-1 = 0’
<p2=X + y + Z~l=0.
Запишем функцию Лагранжа:
ф = f + Х1Ф1 + Хгфг -♦ максимум
Химическое равновесие 499
и соответствующие производные:
4* = 2х + Xix/2 + Х2 = О,
дх
^L = 2y + Xij/3 + Х2 = О,
Ф'
4^ = 2z + X1Z/4 + Х2 = 0.
dz
Последние три уравнения можно решить относительно
у и z, используя множители Лагранжа Xi и х:
у = х(2 + Xi/2)/(2 + Xi/3),
z = х(2 + Xi/2)(2 + Х1/4).
Выразим 02 через х и множители Лагранжа:
Г 2 + \i/2 2 + Xj/2 1
L 2 + Xi/3 + 2 + Xi/4 “ ’
Для 0i запишем
Методом подбора получаем следующие результаты:
Xi = -4,333,
Х2 = -0,321,
х = 1,9277,
у = -0,5766,
z = -0,3501.
Исключение, например, у и z с целью получения функ-
ции только х и последующее дифференцирование f с
целью нахождения экстремума — операция довольно
сложная, однако в конце концов она приводит к тому
же самому результату.
в. Нахождение минимума энергии Гиббса для реак-
ции С2Нб «=* С2Нд + Н2. Запишем уравнения баланса по
углероду и водороду:
Ф1 = Л1 + Пг - 1 = 0,
02 = 3/11 + 2п2 + /1з — 3 = 0.
Запишем функцию энергии Гиббса при атмосферном
давлении, полагая газы идеальными:
g = + 1п(л,//1)].
Ki
Соответственно функция, подлежащая минимизации,
имеет следующий вид:
ф = 2 + ln(/li//l)] + Х101 + Х202.
Запишем ее производные:
-4^- = g? + ln(/i,7/i) + 2 п‘ (— _ | + 1п(/1|7л),
dnt \гц п /
- gi + ln(nt/n) + Xi + ЗХ2 = 0,
дп\
= g° + ln(/i2//i) + Xi + 2Х2 = 0,
дП2
= g3 + 1п(/1з/л) + Х2 = 0.
0/13
После исключения Xi и Х2 из последних трех уравнений,
получаем
о о о _ ДС°
g2 + gl - gl - пТ
Ki
= -In
/12/13
/71/1
Этот результат является вполне корректным. Можно
также применить и другой подход: выразить общее чис-
ло молей через глубину протекания реакции. При этом
в уравнении энергии Гиббса остается только одна пере-
менная, минимум которой можно найти довольно про-
сто. Таким образом,
g = (1 - «) (gi + In + e ($ + + 21n ! + £ j •
Минимизация этой функции дает тот же самый резуль-
тат, что и прежде.
Чтобы легче было понять суть описываемого ниже
метода, параллельно следите за ходом решения, изло-
женным в примере 10.17. В идеальном случае функция
энергии Гиббса имеет следующий вид:
g = S + 1п(л*Р/л)] = X nt[ct + 1п(т/л)], (10.95)
где
Cf-^P + lnP. (10.96)
Получаем необходимую производную
dg / 1 1 \
, = Ci + ln(wf-/«) + Еп,- I — ~ I = Ci 4- ln(/i,//i).
СП{ \ni n /
(10.97)
Запишем материальные балансы m химических эле-
ментов:
Z’;-Sau/1i = 0 J = 1. 2, . . . , m, (10.98)
для m элементов и с компонентов, где ау — число ато-
мов элемента j в молекуле i и bj — общее количество
элемента j в смеси. Ограничивающую функцию необхо-
димо миниминизировать по методу Лагранжа
f=g + 1L Xj(bj ~ S ацп{) (10.99)
= S /1,[с,- + ln(«j/«)] + S Xj(bj— 2L <lijni)-
, = 1 J~l 1-1 (10.100)
Производные по составам выглядят так:
df/d п{ = С{ + In (/1,7/1) - S aikXk = 0 i = 1, 2,..., c.
k~X (10.161)
Эти производные совместно с уравнениями материаль-
ного баланса [уравнение (10.98)] и выражением
13
2 rii = n составляют систему с + т + 1 уравнений, ко-
торые в принципе позволяют оценить л/, X* и л.
Решение уранений. Любая процедура численного
решения системы нелинейных уравнений обязательно
начинается с оценки начальных условий (в этом случае)
с + т + 1 неизвестных. Тем не менее к этим уравнени-
ям можно применить упрощенные общие методики ре-
шения, так что число неизвестных, которые необходимо
оценивать заранее, меньше чем с + т + 1, как это бу-
дет объяснено далее.
Уравнение (10.101) разрешают относительно моль-
ных долей компонентов:
у, = л,/л = ехр
т
aik^k ~ ci
k~i
(10.102)
Разделив элементные балансы, уравнение (10.98), на об-
щее число молей и проведя подстановку в них приведен-
ных выше значений у,, получаем
4. Подстановка этих мольных долей в уравнение
(10.105), которая приводит к выражению
с т
qjjу?[ 1 ~ toy? + aikX^ - q] = dj
j=l,2,...,m, (10.110)
где di = 1 и dj = 0, если у # 1.
5. Решение системы линейных зависимостей, урав-
нение (10.110), для т неизвестных исходных величин X*.
После этого из уравнения (10.105) посредством ме-
тода Ньютона — Рафсона находят уточненные значения
X*, а в дальнейшем уточненные значения мольных до-
лей находят из уравнения (10.102). В противном случае
математическая последовательность определения X* и у,
продолжается до достижения удовлетворительной схо-
димости. Для оценки сходимости отношения матери-
альных балансов уравнения (10.98) можно использовать
в форме
i =1
Ду- ехр
к =1
aik^k ~ ci
~ = 0. (10.103)
D = S 1 -
•?. aUni
(10.111)
S qij ехр
i — 1
Переменную л можно исключить из этого уравнения
при каждом значении i путем первой подстановки j = 1
в уравнение (10.103), что приводит к выражению
1 Iе
— = — S ехр[а^Хл — с,], (10.104)
л b ] ' =1
В результате подстановки полученных результатов в
уравнение (10.103) получаем
т
S aikXk -Cj = dj у = 1,2, . . . , m,
t = 1 J (10.105)
где
9/1 = 1,^ = 1, (10.106)
4ij = aij ~ ailbj/b\, dj = 0 j = 2> 3,. . . , m.
(10.107)
Уравнение (10.105) — это система m нелинейных
уравнений, которую необходимо решить численно для
т неизвестных X*.
Нахождение множителей. Для нахождения X* мы
применим метод Ньютона — Рафсона. Предваритель-
ная оценка этих величин проводится по следующей
схеме.
1. Оценка начальных величин мольных долей у',', ес-
ли какие-либо основания сделать иной выбор отсутству-
ют, все мольные доли могут быть приняты равными.
2. Разложение 1пу, в усеченный ряд Тейлора
1пу,- = 1пу? + (у, — У?)/уР- (10.108)
3. Отделение у, и замена 1пу; из уравнения (10.102)
у У = 1 — 1пу^ + Iny,) (10.109)
= у?( 1 — In у? aik$ ~ ci) I = 1,2, . . . , С.
Альтернативно разность в последующих оценках обще-
го числа молей определяется как
|л(/+1) _ л(0| < е
(10.112)
Общее число молей можно найти на каждой стадии вы-
числения из выражения
т тс
П = S ьЛ а1]У1-
У=1 J
(10.113)
Схема такого способа нахождения равновесных соста-
вов дана на рис. 10.8.
10.6.4. Сравнение трех методов. С необходимос-
тью совместного решения нелинейных уравнений прихо-
дится часто сталкиваться, и в настоящее время
разработано значительное число различных приемов,
позволяющих получать более точные результаты или
быстрее добиваться сходимости последовательных по-
пыток. Со всеми этими вопросами можно ознакомиться
более обстоятельно, если воспользоваться перечислен-
ными в начале разд. 10.6 публикациями и приведенны-
ми в них библиографическими списками. Вполне
понятно, что для решения большинства задач, в кото-
рых рассматриваются более трех реакций или более
шести — восьми веществ, желательно использовать
ЭВМ.
1. Метод релаксации (метод последовательно со-
единенных реакторов). В этом методе предусматрива-
ется определение полной стехиометрии, однако не
требуется оценивать равновесный состав. Это относи-
тельно прямой способ оценки одной неизвестной за
один цикл. Если число стехиометрических соотношений
мало, например равно шести и менее, для решения зада-
чи вполне приемлемы калькулятор или компьютер. Ре-
зультаты сравнимы с получаемыми другими методами,
однако информация об относительном количестве ма-
шинного времени, требуемого для различных методов,
отсутствует.
Химическое равновесие 501
Рис. 10.8. Определение равновесия с помощью миниза-
ции энергии Гиббса.
2. Метод совместного решения стехиометрических
уравнений (Бринклей). В нескольких варантах этого ме-
тода требуется совместное решение ряда уравнений,
число которых равно числу химических веществ плюс
единица. Применяются прямая итерация, метод Ньюто-
на — Рафсона и различные методы оптимизации. Ско-
рость и даже возможность сходимости часто в
значительной степени зависят от первоначальных оце-
нок, которые должны быть согласованы с материаль-
ными балансами химических элементов. Очевидный
метод приравнивания содержания всех компонентов к
нулю, кроме трех или четырех, которые можно ввести
в уравнение материального баланса при его рассмотре-
нии, не всегда удовлетворяет. Метод, использующий
число генераций глубины протекания всех реакций, ка-
чественно описывается Голубом и Вонкой [57].
3. Метод минимизации энергии Гйббса. Описывае-
мый здесь вариант этого метода, в котором использу-
ются множители Лагранжа, уменьшает число уравне-
ний, которые должны быть решены одновременно до
ранга состава матрицы, равного в большинстве случаев
числу присутствующих химических элементов, в свою
очередь редко превышающему 5 или 6. Метод не требу-
ет стехиометрического анализа, упрощает перечень хи-
мических компонентов, предположительно сосуществу-
ющих в условиях равновесия, и термодинамических дан-
ных; он позволяет добавлять компоненты без чрезмер-
ной перегрузки вычислений. Этот метод легко
выражается формулами с сочетанием любых двух пере-
менных Т, Р, V, Н и S как фиксированных, тогда как
в большинстве публикаций рассматриваются формулы
с фиксированными Т и Р. Минимизацию энергии Гйббса
можно осуществлять различными методами оптимиза-
ции, например, такими, как ниспадающий максимум,
линейное или выпуклое программирование, и все эти
вопросы рассматриваются в многочисленных публи-
кациях.
Предпочтительность данного метода по сравнению
с мегом Бринклея убедительно подтверждается также
следующим: 1) в первом случае приходится решать
меньшее число нелинейных уравнений и 2) решение
можно начинать с совершенно произвольного выбора
числа мольных долей.
Примеры численных решений даны в следующих
публикациях [58, 123, 384, 477, 531, 723, 746].
10.7. Объединенное химическое и фазовое
равновесие
Реакции веществ, распределенных между нескольки-
ми фазами, не относятся к числу необычных. Приведем
несколько примеров систем, используемых в промыш-
ленности, — это водные растворы кислот плюс органи-
ческие жидкости, системы, в которых проводится
гидрирование органических жидкостей, омыление вод-
ными растворами, щелочей, гидрирование этилена и
пропилена, биоферментативное расщепление органиче-
ских соединений в воде, системы, используемые в каче-
стве жидких и твердых топлив. Активности чистых
конденсированных фаз можно считать постоянными
или в незначительной степени зависящими от давления;
вычисление равновесия в таких случаях осуществляется
прямым путем. В обычной ситуации, однако, несколько
фаз представляют собой смеси. В данном разделе мы
рассмотрим системы, состоящие из двух жидких фаз и
паровой и жидкой фазы. Перейти от них к системам,
содержащим более чем две фазы, в принципе возможно,
но расчеты при этом, естественно, усложняются.
Основная схема решения многофазного химическо-
го равновесия предусматривает следующее: необходимо
представить химическое равновесие в одной фазе и за-
тем соотнести их с составами фаз при помощи отноше-
ний, описывающих равновесие фаз. Рассматривая
парожидкостные реакции, желательно выразить в виде
формул химическое равновесие в паровой фазе и связать
составы фаз с уравнением мгновенного испарения. Рас-
сматривая две жидкие фазы, целесообразнее выразить
химическое равновесие в той фазе, в которой содержат-
ся более высокие концентрации реагентов, если это
можно оценить заранее. Например, если изучаемые сис-
темы подобны используемым для нитрования аромати-
ческих жидкостей водными растворами кислот, то
заведомо известно, что реакция пойдет в первую оче-
редь в органической фазе. Во всех случаях многофазно-
го реакционного равновесия относительные количества
этих двух фаз неизвестны и оценить их нельзя.
10.7.1. Две жидкие фазы. Запишем выражения, опи-
сывающие химическое и фазовое равновесие в системе
указанного типа:
Ксе= П(а^/П(а^1 = КуКх, (10.114)
Р R
и
xt=KiXi, (10.115)
где
(10.116)
— коэффициент распределения (звездочкой обозначена
вторая фаза). Химическое равновесие сформулировано
для первой фазы. Поскольку коэффициенты активности
и коэффициенты распределения в основном зависят от
составов фаз, для успешной их оценки может понадо-
биться несколько попыток, приводящих к последова-
тельному улучшению в оценке фазовых составов.
Фазовое расслоение можно выразить общим числом
молей в каждой фазе:
Р =
nt + n*'
(10.117)
Глубина протекания реакции определяется как
щ + и* - ni0 f+ для продуктов,
£ =--------------- < ци.ыо;
±vt для реагентов.
Количество любого компонента в обеих фазах находят
по выражению
1+ для продуктов,
- для реагентов,
(10.119)
а общее количество компонентов обеих фаз рассчитыва-
ют как
nt 4- п*= nt0 4- eAv. (10.120)
Общая мольная доля
и, 4-и* nio ± ev,
=-----7~Т~- (10.121)
nt 4- п* nt0 4- eAv
В результате уравнение материального баланса между
фазами имеет следующий вид:
7/- = Дх/ + (1 -Д)х*=[Д+7Q(1 -ДЖ-, (10.122)
где /3 — доля общего количества вещества, присут-
ствующего в первой фазе. Решая уравнение относитель-
но Xi и проводя суммирование, получаем уравнение
фазового расслоения:
которое в стандартной форме выглядит следующим
образом:
"io ± £Vj__________п
- -1 + Z + еДv)[p + К({ j _ д)]
(10.124)
Если известны константа химического равновесия и
уравнение, связывающее коэффициенты активности с
составом, то остается только установить равновесие пе-
рехода вещества из одной фазы в другую, а также коли-
чества и составы обеих фаз. В этих целях можно
воспользоваться методикой, приведенной в виде блок-
диаграммы на рис. 10.9 и представленной ниже по-
этапно.
1. Задаемся значениями коэффициентов активности
в обеих фазах. После нахождения составов правиль-
ность выбора коэффициентов будет проверена и, если
соответствие окажется недостаточным, вычисления бу-
дут повторяться вплоть до получения такового.
2. Задаемся е (или 3)-
3. Находим 3 (или £) из уравнения расслоения фаз,
уравнения (10.124), и соответственно состав х, основной
фазы.
4. Оцениваем Кх, используя эти данные, и сравнива-
ем с Кх = Ксе 7 Ку.
5. Возвращаемся на стадию 2, повторяя попытки
до тех пор, пока не будет достигнуто соответствие на
стадии 4.
6. Оцениваем х* = KiXi и находим коэффициенты
активности 7, и 7* всех веществ. Сравниваем новые зна-
чения К = 7,/7* и Ку с выбранными на стадии 1 и по-
вторяем вычисления столько раз, сколько необходимо
для достижения требуемой сходимости.
В примере 10.18 этот метод применен для одного
цикла вычисления трехкомпонентной реакции, протека-
ющей в системе жидкость — жидкость.
Химическое равновесие 503
Пример 10.17. Минимизация энергии Гиббса реакции между 1 молем СН4 и 5 молями НгО при 900 К и 2 атм
Допустим, что в условиях равновесия в системе
присутствуют пять указанных ниже соединений
= ci + ln(«i/«) + Xi + 4Х2 = 0,
дП1
Соединение G/0 С Н О с = G°/RT + + 1пР
1 СН4 1,99 1 4 0 1,806
2 Н2О -47,36 0 2 1 -25,790
3 СО2 -94,58 1 0 2 -52,195
4 СО - 50,61 1 0 1 - 27,607
5 н2 0 0 2 0 0,693
= сг + ln(/i2//l) + 2Хг
О/1г
+ Хз = 0,
Запишем уравнение материальных балансов эле-
ментов:
С: 01 = 1 - «1 - «з - тц = 0,
Н: фг = 14 - 4/11 - 2/i2 - 2/is = 0,
О: 0з = 5 - /12 - 2/1з -«4=0,
п — П1 + «2 + «з + /ц + «5.
Запишем минимизируемую функцию
5 3
/ = S wt(Ci + 1п(/г,7/?)) + 2 Х*0* -♦ минимум
1 1
и ее производные
= Сз + 1п(«з/п) + Xi оПз + 2Хз = 0,
= С4 + 1п(/Ц//1) + Х1 ОПд + Хз = 0,
Э/
= С?
Э«5
+ ln(/is//i) + 2Хг = 0.
Эти девять уравнений можно решить одновременно для
шести т и трех X* при помощи метода Ньютона —
Рафсона или аналогичного ему метода. Соответствую-
щая процедура требует первоначальных оценок всех де-
вяти неизвестных. Однако для описанного в тексте
метода достаточно оценить всего пять мольных долей,
которые можно принять равными, что не ухудшит ре-
зультатов расчетов. Для рассматриваемой системы вы-
числения ведутся с использованием уравнений
(10.102)—(10.110).
\у i \ аи Ча Ci
1 2 3 1 2 3
1 1 4 0 1 -10 -5 1,806
2 0 2 1 1 2 1 -25,770
3 1 0 2 1 -14 -3 -52,195
4 1 0 1 1 -14 — 4 -27,607
5 0 2 0 1 2 0 0,693
bj 1 14 5
dj 1 0 0
Подставим уравнение (10.103)
з
fl/jtXjt = anXi + 0,2X2 + Л/3Х3
1
в уравнение (10.110) и запишем последнее для каждого
значения j.
J = 1:
<7iiji(l ~ 1ЦУ1 + auXi + 012X2 + 4/13X3 — fi)
+ ^21j2(l — 1цу2 + 4/21X1 + 022X2 + 4/23X3 — С2)
+ <7згУз(1 — 1цуз + 4/31X1 + 4/32X2 + 4/33X3 — сз)
+ <741j4(l ~ 1ПИ4 + 041X1 + 042X2 + 043X3 — С4)
+ 95175(1 - 1ЦУ5 + 051X1 + О52Х2 + О53Х3 - С5) = 1
j = 2:
Qnfifl ~ 1ЦУ1 + ОцХ1 + О12Х2 + О13Х3 — С1)
+ 4'2Л>’2(1 ~ 1ПУ2 + 021X1 + 022X2 + О23Х3 — <?2)
+ <7з2Уз(1 — 1цуз + О31Х1 + 4/32X2 + 4/33X3 - Сз)
+ <?42>'4(1 - 1ПУ4 + О41Х1 + 042X2 + 043X3 — С4)
+ <?52>'5(1 ~ 1ПУ5 + О51Х1 + 052X2 + О53Х3 — С5) = О
j = 3:
<71з.У1(1 — 1пу1 + ОцХ1 + О12Х2 + О13Х3 — ci)
+ ^23j2(l - 1ЦУ2 + 021X1 + О22Х2 + О23Х3 — Сг)
+ <7ззУз(1 — 1цуз + 4/31X1 + О32Х2 + 4/33X3 - Сз)
+ </433'4(1 ~ 1ЦУ4 + 041X1 + 042X2 + О43Х3 — С4)
+ <753У5(1 - 1ЦУ5 + 051X1 + О52Х2 + О53Х3 — С$) = О
Примем исходные мольные доли равными,
у,- = 0.2. Подставим эти значения и значения o,y, qtJ и
с, из таблицы коэффициентов в предыдущие уравнения.
Полученные выражения упростим до
0,6X1 + 1,6Х2 + 0,8Хз = -22,2204,
-7,6X1 - 6,4X2 - 8Хз = 227,5390,
-2,4X1 - 3,6X2 - 1,8Хз = 52,1794.
Решение этих трех линейных уравнений дает исходные
значения множителей Лагранжа:
Х10) = -2,0631, Х$0) = 0,2039, Х^0) = -26,6455.
Принимая эти значения за исходные, при помощи ме-
тода Ньютона — Рафсона находим, пользуясь уравнени-
ем (10.105), улучшенные значения
с / т \
S 9</«р ( S а,к^к ~ Ci) = dj, J - 1»2, з.
1=1 \*=1 /
Эти уравнения записываются для каждого значения j,
j = 1:
exp(Xi + 4X2 - Ci) + exp(2X2 + X3 - C2)
+ exp(Xi + 2Хз - C3) + exp(Xi + Хз - c4)
+ ехр(2Хг - C5) = 1,
j = 1'
- 10exp(Xi + 4X2 - Ci) + 2ехр(2Хг + Хз - сг)
- 14exp(Xi + 2Хз - C3) - 14exp(Xi + Хз - C4)
+ 2ехр(2Хг - cj) = О,
j = 3:
-5exp(Xi + 4X2 - Ci) + ехр(2Хг + Хз - Сг),
-3exp(Xi + 2Хз - сз) - 4exp(Xi + Хз - с4) = 0.
После того как решением трех предыдущих уравнений
найдены X*, посредством уравнения (10.102) определяются
уточненные значения мольных долей:
(т \
S aikXk - а I,
к = 1 /
yi = ехр(йцХ1 + йпХг + Й13Х3 — ci),
У2 — ехр(й21Х1 + 022X2 + 4/23X3 — Сг),
Уз = ехр(йз1Х1 + 4/32X2 + Л33Х3 — Сз),
у4 = exp(4/4iXi + 4Z42X2 + 4/43X3 - с4),
у5 = exp(4/siXi + 4/52X2 + 4/53X3 - с5).
С этими откорректированными значениями мольных до-
лей можно определить последующие уточненные значения
множителей Лагранжа и т. д.
10.7.2. Жидкая и паровая фазы. Поскольку энергии
Гйббса образования соединений чаще всего известны
для паровой фазы, предпочтительно составить уравне-
ние химического равновесия именно для этой фазы.
Взаимосвязь между энергиями Гиббса образования со-
единений в двух фазах исследуется в примере 10.4.
Константа химического равновесия паровой фазы опре-
деляется следующим образом:
Ксе = K-f= КфКуР^. (10.125)
Фазовое равновесие выражается посредством равновес-
ных коэффициентов парообразования:
У,
К,--------------г—-------. (10.126)
X/ ф,Р
Составляя уравнение мгновенного испарения, следует
принимать во внимание тот факт, что относительные
количества двух фаз зависят от глубины протекания ре-
акции. Количества индивидуальных веществ в обеих фа-
Химическое равновесие 505
Пример 10.18. Реакция, включающая две жидкие фазы
В исходной смеси, используемой для проведения реак-
ции 24 + В «=* С, содержатся 2 моля Л и 1 мольВ. Констан-
та равновесия Ксе равна 1,2. Предварительные расчеты
показывают, что активности и коэффициенты распределе-
ния имеют следующие приближенные значения:
7 7* к = 7/7*
4(1) 1,5 2 0,75
В(2) 2,1 1,5 1,40
С(3) 1,25 1 1,25
Используя эти значения, получаем Ку = 1,25/(1,5)2 х
х (2,1) = 0,2646. Состав первичной (основной) фазы дает-
ся выражениями
(3 - 2е)[Ц + 0,75(1 - /?)] ’
1 - £
(3 - 2е)[8 + 1,4(1 - 3)1 ’
£
(3 - 2£)№ + 1,25(1 - 0)]
Определяем наборы переменных £ и /3, глубину
протекания реакции и расслоение фаз, удовлетворяю-
щие отношению фазового равновесия
ф = /(£, 3) = -1 + Х1 + х2 + Хз = 0. (4)
Принимаем значение £, (3 находим из уравнения (4)
и результаты проверяем выражением для константы
равновесия:
Кх = хз/х?х2 = Ксе/Ку = 1,2/0,2646 = 4,5351.
(5)
Как следует из таблицы, это значение Кх получают
в том случае, если £ = 0,5637 и (3 = 0,3551. Соответ-
ствующие фазовые составы:
Xi = 0,5556,
х2 = 0,1852,
хз = 0,2592,
хГ = 0,4167,
хг = 0,2593,
хз* = 0,2340.
Коэффициенты активности, соответствующие этим
фазовым составам, пересчитываются из подходящих
уравнений, таких, как NRTL, чтобы проверить оценку,
сделанную в начале этого вычисления. Если обнаружи-
ваются значительные расхождения между новым и ста-
рым вычислениями, их следует повторять до полной
сходимости.
£ /3 Кх К„
0,2900 0,9933 1,1835 0,3132
0,3000 0,9771 1,2493 0,3306
0,4000 0,7913 2,0728 0,5485
0,5000 0,5490 3,3403 0,8838
0,5637 0,3551 4,5348 1,1999
0,6000 0,2269 5,4224 1,4348
0,6100 0,1891 5,7012 1,5085
0,6200 0,1501 5,9971 1,5868
0,6300 0,1097 6,3114 1,6700
0,6400 0,0682 6,6457 1,7585
зах определяют через глубину протекания реакций
соответствующих компонентов:
ния имеет следующий вид:
ф(£, L) = -1 + Е
л, = пю ± £vi
+ для продуктов,
- для реагентов,
ПЮ ± £Vt
L + Ki (пю + £ Д v — L)
и общее их количество в обеих фазах:
nt ~ nto £Av — V + L.
(10.127)
I +для
(^-для
продуктов
реагентов
(10.132)
(10.128)
Для точки росы L = 0 и
<Д(е, 0) = — 1 + Е
Запишем уравнения материального баланса для отдель-
ного компонента
"io ± £^
Kj(nto + eAv)
(10.133)
В то время как в точке, соответствующей температуре
начала кипения, L = то, так что
И| = Lxi + Vyi = [L + Ki(nt — L)]x{,
или
ni
Х‘~ L + Ki(nt-L) ’
(10.130)
Принимаем мольные доли равными единице:
ni
Е х, = Е ' ......— =1. (10.131)
1 L + Ki(nt- L) v
Окончательная форма уравнения мгновенного испаре-
(10.129)
и 10 ±
71 to + Kit&v
(10.134)
’/'(е. «го) = — 1 + Е
Границам двухфазной области отвечают значения е, вы-
численные при помощи этих двух уравнений. Прежде
чем анализировать двухфазный процесс, целесообразно
проверить глубину протекания однофазной реакции. За-
дача состоит в том, чтобы найти £ и расслоение фаз,
представляемые количеством жидкой фазы, L, и соот-
ветствующими фазовыми составами, которые бы удов-
летворили уравнению химического равновесия
(уравнение (10.125)). Методика вычислений при задан-
ных значениях температуры, давления и полного исход-
ного состава приведена на рис. 10.10 и описана ниже.
Рис. 10.10. Химическое равновесие в жидкой и паровой
фазах.
1. Принимаем, что обе фазы идеальны, так что
ф, = 7, = 1 и Kt = Pf/P.
2. Находим глубину протекания реакции, соответ-
ствующую точке росы и температуре начала кипения.
Эти значения будут только приблизительными, прини-
мая допущение этапа 1.
3. Выбираем значение £, лежащее в интервале, уста-
новленном на этапе 2.
4. Находим L методом подбора из уравнения мгно-
венного испарения, уравнение (10.132), и соответственно
состав жидкой фазы, х,, и состав паровой фазы,
у, = Kxi, используя значения К для идеальных фаз.
5. Оцениваем Ку и сравниваем с Ку = Kce/K^P^.
Если согласование неудовлетворительное, выбираем
другое значение £, возвращаемся к стадии 4 и повторя-
ем этот процесс до тех пор,- пока не будет получена не-
обходимая сходимость.
6. Используя значения составов, дающие удовлет-
ворительную сходимость, проводим повторное вычис-
ление 7,, <j>i, К и /Q, после чего вновь возвращаемся
к этапу 2 или 3.
Последовательные определения фазовых составов
продолжаем до согласования с подходящим критерием,
например
2 [х(* + 1) - х(/с>]2 предельное отклонение (10.135)
или до тех пор, пока не стабилизируется значение £.
Однофазные компоненты. В уравнение химическо-
го равновесия для паровой фазы нелетучие вещества не
входят, их коэффициенты фазового равновесия равны
нулю. Коэффициенты фазового равновесия нераствори-
мых газов равны бесконечности.
Идеальные системы. Если исследуемые фазы близ-
ки к идеальным, так что их фугитивности и коэфициен-
ты активности по существу равны единице для всех
составов, их коэффициенты фазового равновесия также
независимы от состава, при этом этапы 5 и 6 автомати-
чески исключаются.
В примере 10.19 рассматривается система с идеаль-
ной паровой фазой и постоянными коэффициентами фа-
зового равновесия; вычисление их для системы, в
которой степень отклонения от идеальности зависит от
состава, проводится в один цикл.
10.8. Задачи
10.1. Реакция А ЗВ имеет место при постоянной
температуре, исходной материал — чистое вещество
А. В условиях равновесия давление, соответствующее
точке росы, равно 12 атм. Давления паров чистых ком-
понентов имеют следующие значения: 73? = 20 и Рь = 9
атм. Определите величину константы равновесия Кр.
Химическое равновесие 507
Пример 10.19. Реакция, включающая жидкую и паровую фазы
Реакция А + 2В С идет при давлении 4 атм.
Константа равновесия Ксе = Кр = KyPhv - 3,5, коэффи-
циенты фазового равновесия имеют следующие значе-
ния: Ка = 5, Кь = 1 и Кс = 0,8. Используются
стехиометрические соотношения А и В. Необходимо
определить количества и составы фаз в условиях рав-
новесия.
Выразим определяемые параметры через глубину
протекания реакции £.
Л, = 1 - £,
Л|> = 2(1 - £),
Пс = £,
л, = 3 - 2fi = L + V,
п< _ п‘ _ п‘
Xi = L + KiV ~ L + Ki(nt-L) ~ L + Ki(3-2E-L) ’
У, = KiXi,
Запишем уравнение мгновенного испарения
HL, £) = -1 + S * = “1 + S L +
- 1 V* п'
+ 1 + Ki(3 - 2е - L) '
Преобразуем для решения методом итерации:
L =
0,25
5(3 - , w-e) ‘~Е а-------------
3 - 2г 0,2£ + 0,8(3 - 2е)
L находят при определенных значениях е. Полученные
результаты показаны ниже в виде таблицы. При
КСе = 3,5 глубина протекания реакции £ = 0,8429. Тем-
пературе начала кипения соответствует £ = 0,7619, а
точке росы —£ == 0,9524. Если Ксе < 1,9215, реакция
проходит только в паровой фазе, если же Ксе > 166,7,
реакция проходит только в жидкой фазе.
£ L V Уа Уь Ус Ксе
0,7619 0 1,4762 0,1613 0,3226 0,5161 1,9215
0,80 0,2800 1,1200 0,1701 0,2857 0,5442 2,4497
0,8429 0,5588 0,7554 0,1813 0,2391 0,5796 3,4968
0,85 0,6012 0,6988 0,1831 0,2308 0,5861 3,7566
0,90 0,8700 0,3300 0,1984 0,1667 0,6349 7,1979
0,95 1,0859 0,0141 0,2162 0,0909 0,6927 24,23
0,9524 1,0933 0,0019 0,0432 0,0869 0,8699 166,66
10.2. Реакция 2А & В идет при давлении 5 атм, исход-
ное количество чистого компонента А в реакторе равно
1 моль. В условиях равновесия количество жидкой фазы
составляло 0,14 моль. Равновесные коэффициенты испа-
рения компонентов имеют следующие значения: Ка = 5,
Кь = 0,5. Принимая поведение газа и раствора за иде-
альное, найдите
а) константу химического равновесия Ксе,
б) глубину протекания реакции, соответствующую
температуре начала кипения и точке росы.
10.3. Если исходное количество чистого компонента
А равно 1 молю, равновесный состав реакции в идеаль-
ной газовой фазе
А # 2В + С,
2B&D,
уа = 0,25, уа = 40. Найдите константы равновесия Ку
обеих реакций.
10.4. В условиях равновесия степень превращения (в
мольных долях) в реакции А ЗВ равна 0,30 (исходное
вещество А чистое). Соответствующее значение
Ку - 2,7. Найдите параметр симметричного уравнения
Маргулеса.
10.5. Реакция А + 2В С проходит с различными
исходными отношениями г = пьо/пао. Константа равно-
весия Кх равна 5. При каком отношении степень превра-
щения вещества А максимальна?
10.6. Для идеальной газовой реакции А ** 2В полу-
чены следующие данные: Д#298 = 3000 кал/моль А;
Д(?298 = -400 кал/моль А; ДСР = 3,5 кал/моль А К.
Давление равно 1,2 атм. Найдите температуры, при ко-
торых степени превращения вещества А равны 25, 50,
75 и 95%.
10.7. Для системы, приведенной в примере 10.7, фу-
гитивность выражается уравнением К^ = 0,6 + О,35£0,3.
Определите степень превращения и численное значение
Ч
10.8. Установите глубину протекания и равновес-
ный состав реакции разложения метанола СНзОН «=*
СО + 2Нг при температуре 200°С и давлении 5 атм.
Константа равновесия Кр равна 42 и можно принять
равным единице, поскольку давление мало.
10.9. При температуре 1800 К и давлении 450 атм
коэффициент фугитивности примерно равен 1,10 (заим-
ствовано из таблиц Ли — Кеслера). Другие необходи-
мые данные приведены в примере 10.3. Каким должно
быть давление, чтобы подавить диссоциацию СаСОз
при температуре 1800 К?
10.10. Найдите равновесный состав для реакции ди-
меризации 2А ** В при давлении 20 атм и температуре
800 К. Константа равновесия равна 1,25, вторые вири-
альные коэффициенты имеют следующие значения:
Bi = -1,802, В2 = -5,871 и Bi2 = -3,361 (куб. фут/
фунт-моль). Исходные количества веществ: пао = 2,
пьо = 0,25. А — компонент 1, В — компонент 2. Урав-
нения для определения коэффициентов фугитивности
даны в табл. 3.4.
10.11. Определите значение константы равновесия
как функции температуры для реакции диссоциации н-
пропанола на пропионовый альдегид и водород и срав-
ните их с соответствующими значениями, взятыми из
справочника Сталла цри нескольких температурах.
Данные: ДН298 = 15,650 кал/моль; Кгп =
= 2,00 (Е - 6); СР = а + ЬТ + ct2 + di*.
а Ь(Е2) с(Е6) d(E8)
н2 6,483 0,2215 -3,298 0,1826
С2НбСНО 2,800 6,244 -31,050 0,5078
С3Н7ОН 0,590 7,942 -44,310 -0,3356
К
Т данные Сталла вычислено
298 2(Е-6) 2(£-6)
500 0,1102
800 63,36
1,000 561,1
10.12. Для реакций
СНзС1(газ) + Н2О(газ“) -> СНзОН(газ) + НС1(газ),
2СН3ОН(газ) - (СН3)2О(газ) + Н2О(газ)
при 600 К константы равновесия имеют следующие зна-
чения: К\ = 0,00154 и Кг = 10,6. Покажите, что для ис-
ходной смеси метилхлорида и воды (1 моль каждого
при 600 К) равновесная парциальная степень превраще-
ния метилхлорида равна 0,0482 моль и что количество
образованного диметилового эфира равно 0,0094 моль.
10.13. При давлении 1 атм одновременно происхо-
дят следующие газофазные реакции:
24 + В & 2С, Ki =2,
А + C&D, К2 = 0,5.
В исходной смеси содержатся стехиометрические соот-
ношения А и В. Определите равновесный состав смеси
графически или по методу Ньютона — Рафсона. (От-
вет: £1 = 0,4215, £2 = 0,1452.)
10.14. Определите равновесный состав смеси, полу-
чаемой при разложении пропана в соответствии с при-
веденными ниже реакциями (1400°F, 1 атм).
СзНв ** СзНб + Н2, Кр = 7,88,
С3Н8 & С2Н4 + СНд, Кр = 755.
10.15. Ниже указаны основные реакции, происходя-
щие между водяным паром и метаном при 600°С:
СНд + Н2О ** СО + ЗН2, Кр = 0,574,
СО + Н2О & СО2 + Н2, Кр = 2,21.
Найдите равновесный состав смеси, получаемой при
смешении 1 моля метана с 5 молями водяного пара при
1 атм. Используйте метод Ньютона — Рафсона.
10.16. Найдите полную систему независимых стехи-
ометрических соотношений и их константы равновесия
для реакции диссоциации изопропанола, рассмотренной
в примере 10.15, вводя в систему три дополнительных
соединения: СзНв, СзН8 и Н2О. Равновесные константы
образования для указанных условий имеют следую-
щие значения: 400 К 800 К
.1. Изопропанол 18,120 -0,511
2. Водород
3. н-Пропанол 16,926 -0,791
4. Ацетон 17,001 2,088
5. Пропионовый
альдегид 14,188 1,011
6. Пропилен -10,175 -9,511
7. Пропан -0,651 -8,18
8. Вода 29,241 13,290
10.17. Частичное окисление метана кислородом
включает следующие реакции [716]:
СНд + 2О2 —* СО2 + 2Н2О,
СНд + СО2 ** 2СО + 2Н2,
СНд + Н2О & СО + ЗН2,
Н2О + СО & СО2 + Н2,
2СО +* СОг + С.
Считая, что в условиях равновесия в системе содержат-
ся только те соединения, которые образуются в резуль-
тате приведенных выше реакций,
а) найдите ряд независимых компонентов;
б) напишите стехиометрические уравнения образо-
вания производных компонентов и сравните с данными
уравнениями.
10.18. Водород можно получить, воздействуя на бу-
тан водяным паром при температуре 1400°F и давлении
5 атм. В равновесной реакционной смеси могут присут-
ствовать перечисленные ниже соединения.
Принимая, что R — отношение вода/бутан, со-
ставьте алгебраические уравнения, из которых можно
вычислить равновесный состав.
Соединение logipK
СН4 -1,1581
СО 10,2895
СО2 20,015
С2Н4 -6,110
С4Ню “4,40
Н2 0
Н2О 9,6453
10.19. Продукт пиролиза метана, полученный при
соотношении водяной пар/метан, равном 3:1, загружа-
ют в каталитический реактор, в котором поддерживает-
ся температура 800°F и давление 8 атм, чтобы в
результате получить углеводороды с большей молеку-
лярной массой. Предполагается, что в равновесной сме-
си содержатся следующие соединения: СО, Н2, СИ»,
С4Н10, С8Н18, Н2О, СОг, СНзОН и СН3СНО и что кон-
станты равновесия реакций образования этих соедине-
ний известны. Напишите алгебраические уравнения, из
которых можно определить равновесный состав.
Химическое равновесие 509
10.20. Два моля этилена реагируют с одним молем
гаслорода при среднем давлении и повышенной темпе-
ратуре в присутствии соответствующего катализатора.
Несколько стехиометрических уравнений, которые мо-
гут протекать в этой системе, даны ниже:
С2Н4 + ЗО2 = 2СО2 + 2Н2О,
2С2Н4 + О2 = 2С2Н4О,
2С2Н4О = НСНО + С2Н4,
6С2Н4О = 5С2Н4 + 2СО2 + 2Н2О,
2С2Н4О + О2 = 2СО2 + 2СН4,
С2Н4 + Н2О = НСНО + СН4,
СН4 + О2 = 2СО2 + 2Н2О,
2НСНО + О2 = 2НСООН,
2НСООН + О2 = СО2 + Н2О.
Определите приемлемые независимые компоненты и ре-
акции и напишите алгебраические уравнения, из кото-
рых можно установить состав равновесной смеси.
10.21. Ожидается, что система, первоначально со-
стоящая из 1 моля этилена и 1 моля воды, содержит
следующие вещества (в скобках указаны соответствую-
щие значения IgA/ при 400 К): СгН4(-22,257),
Н20(67,330), С2Н5ОН(43,528), СН3СООН(107,112),
Нг(О), СгНб(4,343). Напишите уравнения, из которых
можно определить равновесный состав а) методом
Бринклея, б) путем минимизации энергии Гйббса, при-
меняя метод Лагранжа.
10.22. Для реакции А + В +*2С известны энергии
Гиббса образования компонентов. Выразите относи-
тельную степень превращения (в долях) А через £.
а. Руководствуясь примером 10.1, выразите через е
общую энергию Гйббса реакционной смеси
g = n,G/RT = 77tS yi[G?/RT + ln(y,P)].
б. Используя метод алгебраической минимизации g,
покажите, что равновесие определяется выражением
К = [2£/(1 - £)]2,
где
-КГ\пК = 2GC° - GS - G°b.
10.23. Решите приведенные ниже задачи при помо-
щи метода Лагранжа.
a. f = х2 + у2 -* минимакс при у + 10% 1пх + 5 =
= 0.
б. f = xyz -* минимакс при (х/а)2 + (у/b)2 +
+ (z/c)2 -1=0.
в. f = cos2x + cos2y -» минимакс при у - х -
- т/4 = 0.
г. Определите равновесие реакции СОг + ЗНг **
** СНзОН + НгО для стехиометрического отношения
исходных компонентов (левая часть уравнения) и атмос-
ферного давления.
10.24. В равновесной смеси, образующейся в ре-
зультате реакции 1 моля С3Н8 с 5 молями Ог и 20 мо-
лями Nz, предположительно присутствуют следующие
вещества: СОг, Н2О, Nz, СО, Н2, Н, ОН, О, NO и О2.
а. Найдите ранг матрицы состава и соответственно
число независимых компонентов.
б. Выбрав СОг, НгО, Nz и СО в качестве независи-
мых компонентов, напишите стехиометрические уравне-
ния образования остальных соединений.
в. Напишите уравнение равновесия и балансы хими-
ческих элементов, из которых можно найти состав рав-
новесной смеси.
г. Выберите другую серию независимых компоне-
нтов и напишите соответствующие уравнения образова-
ния остальных соединений [384].
10.25. При окислении аммиака воздухом в получае-
мой равновесной смеси предположительно содержатся
NH3, NO2, NO, НгО, О2 и N2. Число молей N, Н, О,
содержащихся в исходной смеси, равно <?n, <?н и <?о со-
ответственно.
а. Проверьте, насколько NH3, НгО и О2 удовлетво-
ряют понятию независимых компонентов.
б. Составьте уравнения равновесия и материально-
го баланса, требуемые для решения этой задачи мето-
дом Бринклея.
10.26. Используйте метод релаксации для нахожде-
ния равновесных составов, получаемых в результате ре-
акций 2А& ЗВ и А + В +* С, если известно, что
па0 = 2, пьо = Псо = 0, Кг = 3, К2 = 2 и Р = 0,5.
10.27. Смесь, состоящая из азота, водорода и воды
(1, 3 и 5 частей соответственно), поддерживается при
температуре 25°С и давлении 100 мм рт. ст. до наступ-
ления химического и фазового равновесия. Условие хи-
мического равновесия выражается с помощью
уравнения Ксе = Унн3/$£ Ун* р = 125где р дано в
атмосферах. Фазовое равновесие выражено законом Ге-
нри Pi = Hix, с HN1 = 91,053 атм, НП1 = 70,658 атм,
Г/иНз ~ 0,960 атм. Давление водяного пара 25,76 мм
рт. ст. Найдите соотношения и составы двух фаз при
равновесии [759].
10.28. Газофазная димеризация 2А «=* В проходит в
присутствии нелетучего растворителя. В исходной смеси
содержится по 1 молю компонента А и растворителя.
Равновесные коэффициенты парообразования имеют
следующие значения: Ка = 5, Кь = 0,5. Константа хи-
мического равновесия Ку равна 0,080. Найдите:
а) степень превращения (в молях) вещества А *;
б) количества и составы двух фаз.
* Степень превращения должна находиться в преде-
лах 0,3—0,5.
10.29. Реакция 2А ** В проходит и в жидкой и в па-
ровой фазрх. При атмосферном давлении и температуре
350 К полная степень превращения составила 0,3. По-
стоянные уравнения ван Лаара А = 2,3026, В - 1,6094.
Другие данные:
А В
Psat, мм рт. ст. 952 86,98
0sat 0,98 0,98
ф 0,95 1,05
{PF} 1,002 0,998
Найдите
а) константу химического равновесия, V Bij
б) количества и составы двух фаз. “— “
и n2 0,044647
10.30. Для реакции 0,5 N2 + 1,5Нг «=* NH3 констан- 22 Н2 0,025626
та равновесия Лу при температуре 873 К равна 0,00156. 33 NH3 0,047731
Реакция проходит при 873 К и 200 атм со стехиометри- 12 0.033751
ческими отношениями реагентов. 14 ППИС1П1
Убедитесь в том, что степень превращения при рав- новесии равна около 0,161. 23 0,035809
Примите, что в данном случае можно применить
вириальное уравнение со следующими значениями вто-
рых вириальных коэффициентов, Вц (л/моль), вычис-
ленных по уравнению Цонопулоса.
11. ОЦЕНКА ИЗМЕНЕНИЙ ЭНТРОПИИ И ЭНТАЛЬПИИ
11.1. Основные уравнения
Анализ фазового равновесия, как правило, требует
также оценки изменений энтропии или энтальпии или
других свойств, которые могут быть вызваны измене-
ниями первичных переменных таких, как температура,
давление и состав. Например, при проектировании обо-
рудования для процессов разделения необходимы дан-
ные как о балансе энтальпии, так и об отношениях
равновесия, а при анализе эксплуатационных характери-
стик компрессоров и расширителей — данные о характе-
ре изменений энтропии.
Графики или таблицы энергетических функций, со-
ставленные на основе более или менее полных экспери-
ментальных данных, имеются всего лишь для неболь-
шого количества чистых веществ и очень немногих сме-
сей. В этот класс веществ входят легкие углеводороды,
наиболее распространенные газы и хладагенты. Менее
точные корреляции были разработаны для тяжелых
нефтяных фракций. Ввиду ограниченного объема дан-
ных, особенно для смесей, единственно возможный ме-
тод оценки изменений энергетических функций состоит
в использовании уравнений состояния для нахождения
воздействия давления и теплоемкостей идеального газа
с целью определения температурного воздействия. Дан-
ных о теплоемкости получено достаточно много, кроме
того, обычно ее величину можно с удовлетворительной
точностью оценить методом молекулярных групповых
вкладов.
Анализ отклонений энергетических функций от иде-
ального поведения в некотором отношении подобен
способу вывода уравнений для коэффициентов фугитив-
ности и коэффициентов распределения при испарении,
рассмотренному в предыдущих главах. Фундаменталь-
ные термодинамические уравнения, описывающие зави-
симость различных термодинамических свойств от Р, V,
Т, Ср, приведены в табл. 2.2 и А.З, а также в таблицах
настоящей главы.
При оценке разностей остаточных свойств требуется
выполнить интегрирование уравнений состояния. По-
скольку уравнения состояния могут быть выражены ли-
бо через давление, либо через объем, в ряде случаев
необходимо заменить независимую переменную Р на V
или наоборот. Ниже приводится список некоторых ис-
пользуемых для этого формул:
р С V1
f VdP = P2V2~Р1И-/ PdV, (П.1)
>i •'’'I
= PV-
(П.2)
i/ dP \ I / dV \ ,D
I I <*V = - I I ) dP, если интегрирование
\ ol j v I \ о 1 j p
выполняется при постоянной T,
(др\ (дт\ / „
I= -1’ «enHoe правило, (11.4)
\ 0V }т \ Or9 )V \ 01 )Р
(11.3)
(П.5)
Дифференциальные формулы, выражающие зависи-
мость энергетических функций от Р и Т или от V, мож-
но вывести из исходных определений этих функций,
чаще же представляется более удобным и, вероятно, бо-
лее надежным сделать это при помощи таблицы
Бриджмена (табл. А.7). При этом применяются следую-
щие формулы:
(дН \ (дН \
dH = ГдТ ) dT + (ар ) dP
\ ' Р \ /т
= CpdT +
dP
(П.6)
(И.7)
= CpdT+ d(PV) +
dV
(11.8)
dS =
(П.9)
RT2
= CpdT-----
(11.10)
'as \ /as \
— +— ] dP
dT J \dP J
(UH)
(1112)
(11.13)
(И.14)
(11.15)
(11.16)
(11.17) Puc- Изменения свойств в изобарических и изотер-
мических условиях.
&Mad = &МаЬ + ^Mbc + &Mcd
(11.18) чить как
dU= dH~ d(PV),
dG = dH - d(TS),
dA = dU~d(TS).
ДМ' = Mid- М. (11.22)
(П-19)
т. е. это разность величин свойства в состоянии идеаль-
(11- 20) ного газа и в реальном состоянии при одинаковых тем-
(11.21)
пературах и давлении.
Поскольку теплоемкости зависят как от Р, так и от
Т, следует с большой тщательностью выполнять оценку
воздействия изменений Т и Р на энергетические свой-
ства. В большинстве случаев теплоемкости, которые на-
зывают теплоемкостями идеального газа С1р, известны
только при атмосферных или близких к нулю давлени-
ях. Этими данными можно воспользоваться, наблюдая
последовательные изобарические и изотермические из-
менения, показанные на рис. 11.1 линией abed, сопро-
вождающие переход от состояния при Р\, 1\ до
состояния при Рг, Тг- Этим изменениям соответствуют
следующие процессы.
1. Понижение первоначального давления Pi до малой
величины Ро, при которой вещество ведет себя как иде-
альный газ при постоянной температуре Т\.
2. Интегрирование от 7\ до Т2 при постоянном дав-
лении Ро.
3. Повышение давления от Ро до Р2 при постоянной
температуре 7г.
Изменения, описанные в пп. 1 и 3, называют остаточ-
ными свойствами, или отклонениями от идеальности.
Для некоторого общего свойства М их можно обозна-
11.2. Оценка остаточных свойств
Величины остаточных свойств можно получить непо-
средственно из экспериментальных данных. Так, кало-
риметрические измерения энтальпии и теплоемкости
дают собственно остаточную энтальпию Д77' и оста-
точную энтропию ДХ'. Однако количество подобных
данных, особенно для смесей, ограничено. Некоторые
примеры экстремального воздействия давления на теп-
лоемкость показаны на рис. 1.3 и 1.4. На рис. 11.2 в
графической форме представлен ряд измеренных вели-
чин остаточных свойств этилена. В примере 11.4 экспе-
риментальные данные о зависимости PVT для азота
применены для оценки остаточных энтальпий и энтро-
пий; результаты этих вычислений сопоставляются с ре-
зультатами расчетов, выполненных на основе некото-
рых уравнений состояния.
На практике остаточные свойства находят с по-
мощью уравнений состояния, которые, конечно, основа-
ны на экспериментальных данных о функции PVT и
других данных. При этом исходят из дифференциаль-
ных уравнений, приведенных в предыдущем разделе,
при dT = 0.
Оценка изменений энтропии и энтальпии 513
Рис. 11.2. Остаточная энтальпия и энтропия этилена как
функции давления и температуры [127].
В большинстве случаев применяют уравнения состоя-
ния, выраженные через объем:
ДЯ’ = Hid - н =
V=V(P,T) (11.23)
или
RT
У = ~^Р, Т), (11.24)
а также через давление:
Р=Р(Г, Г) (11.25)
или
RT
P=~z(V,T). (11.26)
dV
дТ
dP
(11.27)
(11.28)
= (U + PV)id - (U + PV)
(11.29)
(11.30)
Единственным широко применяемым примером урав-
нения состояния, выраженным через объем, является
вириальное уравнение, а уравнение Питцера — Керля и
уравнение Ли — Кеслера могут служить примерами
уравнений состояния, выраженных через z.
Уравнения, предназначенные для определения оста-
точных энтальпий и энтропий, выводят при помощи
обеих форм уравнения состояния. Для остаточной эн-
тальпии получают следующий ряд уравнений:
= RT
(11.31)
= — RT(z - 1)-RT2
(11.32)
/4-864
Поскольку чаще всего применяются уравнения состоя-
ния, выраженные через давление, наибольший интерес
представляет уравнение (11.31).
Исходной точкой вывода выражений для остаточной
энтропии является уравнение Максвелла:
~R
_V
dP \ 1 pKidtf
— / ~~dV-
JT / J Jv V
(11.39)
(11.33)
(11.34)
Поскольку энтропия даже идеальных газов зависит от
давления и при Р -> 0 или V -» со стремится к бесконеч-
ности, остаточную энтропию нельзя получить непосред-
ственным интегрированием уравнения (11.34). Для
устранения этой трудности введем величину
RT
I/id — -
Р ’
(11.35)
которая представляет собой объем идеального газа при
давлении Р и температуре системы Т При величинах
V, превышающих Kld, поведение газа также становится
идеальным. Соответственно интегрирование уравнения
(11.34) в этом интервале дает
Kid j?
---dV.
V
(11.36)
Кроме того, для всего диапазона объемов интеграл мо-
жет быть представлен следующим образом:
r v (др \
S(~)-S(K) = - / I— J dV.
\°Т /v
(11.37)
Второй интеграл в правой части уравнения равен
л 0dR Kld RT
I ~dV = R In-----= R In—= ~7?lnz,
Jv V V PV
(11.40)
следовательно, в конечном итоге будем иметь
(П.41)
При использовании уравнения состояния, выраженно-
го через объем, исходной точкой служит другое уравне-
ние Максвелла:
(11.42)
При объединении этого уравнения с соответствующим
выражением для идеального газа получаем дифференци-
ал остаточной энтропии:
<f(Sid - S) =
(11.43)
Поскольку при Р = 0 величины Sld - S и подынтеграль-
ная функция обращаются в нуль, интегрирование дает
В целях получения окончательной подынтегральной
функции для оценки остаточной энтропии следует объ-
единить уравнения (11.36) и (11.37) со следующим урав-
нением:
AS' = Sid - S =
dP. (11.44)
(11.38)
В результате получаем
AS' = sid - 5 = S{ Kid) - S( К)
Уравнения состояния, эквивалентные уравнениям
(11.41) и (11.44), выраженные через z, обобщены в
табл. 11.1, там же даны формулы, описывающие дру-
гие остаточные свойства, выведенные путем соответ-
ствующего соединения формул для остаточной энтро-
пии и энтальпии или же аналогичным методом.
Пример 11.1. Уравнения для расчета влияния давления при Т = const на термодинамические свойства газа, описы-
ваемые уравнением состояния Клаузиуса
Уравнения, приведенные в табл. 11.1, применяются к
уравнению Клаузиуса, имеющему следующий вид:
Необходимые производные определяются следующими
выражениями:
RT _ _а_
V- b ТУ2
(1)
R
У-Ь
а
Т2У2’
(2)
Таблица 11.1. Остаточные свойства. Общие уравнения для уравнений состояния, выраженных через Р или V
Остаточное свойство, ДМ' = M'd - M, представляет
собой разность величин этого свойства в состоянии иде-
ального газа и реального газа при одинаковых темпера-
туре и давлении.
Уравнения состояния, выраженные через объем, мо-
гут иметь вид V = V(P, Т) или V = (RT/P)z(P, Т). Урав-
нения состояния, выраженные через давление, имеют
следующий вид: Р = Р(Р, Т) или Р = (RT/V)z(V, Т).
А. Для уравнений состояния, выраженных через объем,
имеем
= At/' + RT~PV= &U' + RT(1 ~z)
dz \ dV
— ) ---+ RT-PV,
dT „V
dP
dV
АЯ'
At/'
dT
dV ' ) ~ v dP (1)
\.dT p p v ( dz
= — RT2 / I —
J» \ dT
( dz \ dP
1 (2)
'P p
dV
dP
R
(3)
v
At/' = ДЯ' +PV-RT, = АЯ' — RT(\ - z),
dV
R
dT
ДЛ' = At/' - TAS',
AG' = ДЯ - TAS',
AS'
1П0 = ---
R
ЬС'р
р (d2V \
—7 ldP
\dT2 Л
( dz
2 ----
\ dT
р '-v
dP
(4)
dz
dT
dP
dP
d2z
dT2
(dP \2 у Vr 4 / ' (dp\ \ar )T
~RT (dz \ z&
- — 1 + —
. У \dT 'v v -
zRT
2
~RT (dz
dV
V2
AC'= дс;-я + (ср-с1,).
Б. Для уравнений состояния, выраженных через
давление, имеем
\H' = -
dP
dP
(15)
(16)
(17)
(18)
dT
dV-Rlnz
dz
dT
dV
— — R\nz,
(19)
(5)
(6)
(7)
(8)
(9)
dP
(10)
(20)
АЛ' = At/' - TAS',
AG" = АЯ' - TAS',
AS' АЯ'
In Ф -----------= Z — 1 — Inz —
R RT
дс;
= -RT
(21)
(22)
dV
(23)
dV /т
dT
v (d2P \
----- I dV
\dT2 Л
(24)
dz
.dT
dV
(11) (dV \2 /fdv\ cp- cv= - т — 1 / — ] p \dT )p/ \dP )T ~zR RT (dz \ -T — +— (— _ P P \dT /p 2
(12) RT Г 1 (dz \ ~ — + 1 1 P L p \dP JT. (13) Ac;= ^C'v + R-(Cp-Cv). pp (d2V \ ,,Л, = T 1 1 1 dP (14) Jo Vr2 Jp
d2z
dV
dT2
(25)
(26)
(27)
(28)
(29)
(7)
a
RT2V
(7ld - U = Hld - H - (RT - PV) = ~ (8)
Jv
oo
Jv
2a,dV =
JV2
= RT - PV +
TV TV V - b
ylid — Л = 17id—[/—7’(Sid —S)= -~+RTln
Gid - G = Hid - H - T(Sid - S) = +
(5)
(10)
dV — R\nz —
dV - .Rinz =
(6)
11.3. Специальные уравнения состояния
В табл. 11.2 содержатся формулы для расчета оста-
точных свойств, выведенные из вириального уравнения
состояния, а в табл. 11.3 приводятся формулы, получен-
ные на основе ряда других уравнений состояния. Под-
робное описание вывода вириальных форм будет
рассмотрено ниже в данном разделе. Для некоторых
уравнений состояния составлены числовые таблицы или
графики. Сравнение графиков (рис. 11.3 и 11.4) остаточ-
ных свойств, полученных из трех различных корреля-
ций второго вириального коэффициента, указывает на
существенное различие рассчитанных величин. Посколь-
ку корреляция Цонопулоса наилучшим образом описы-
вает данные о функции PVT, она была принята при
построении графиков на рис. 11.5 и 11.6.
Таблицы остаточных свойств, составленные Питце-
ром и др., содержатся, например, в книге Льюиса, Рэн-
далла и др. (1961). Эти данные представлены в
графическом виде Эдмистером [266]. Более точные дан-
ные, основанные на корреляции Ли и Кеслера, даны в
табл. Д. 15 — Д.18; эти же данные также представлены
в графической форме в справочнике Американского неф-
тяного института (API Data Book). Графики, основан-
ные на уравнении Редлиха — Квонга, были построены
Эдмистером [267]. Подобные схемы и графики удобно
использовать для проверки экспериментальных данных
по отдельным точкам, в то же время при помощи каль-
кулятора или микрокомпьютера можно легко получить
любые величины, даже используя такие сложные урав-
нения состояния, как уравнение Ли — Кеслера или Бене-
дикта — Уэбба — Рубина — Старлинга.
В большинстве случаев способ получения производ-
ных уравнений, приведенных в табл. 11.3, достаточно
прост. В качестве иллюстрации в примере 11.3 выводят-
ся формулы для расчета остаточных свойств из уравне-
ния Соава.
Вириалъное уравнение. Формулы для расчета остаточ-
ных свойств, выведенные из нескольких корреляций
второго вириального коэффициента, показаны в
Jv
~-^dV =
Т’З j/2 jV. y>
oo
(11)
RT * 2a \
v- ь t2 vy
(12)
табл. 11.2. Корреляция третьего вириального коэффици-
ента, предложенная Орби и Вера (1983), дана в
табл. 1.8. Она имеет вид уравнения Питцера:
С= (RTc/Pc)2(CW + wd1)),
(11.45)
где члены с надстрочными индексами являются функ-
циями приведенной температуры.
Исходными в данном случае являются показанные в
табл. 11.1 уравнения (2) и (5), выраженные через z. Про-
изводная имеет следующий вид:
dz \ dB' dC'
--- ) = Р + р2
дТ / dT--dT
(11.46)
Р
RT
'dB В
.dT Т
(П-47)
ДН' определяют по уравнению (7) из табл. 11.2, a AS'
определяют из следующего выражения:
dB Р
dT R
dP
Р
(11.48)
(11.49)
Оценка изменений энтропии и энтальпии 517
Таблица 11.2. Остаточные свойства, полученные из вириального уравнения
1 + B/V + C/V2 + . . . = 1 + В'Р + С'Р1 + . . .,
(1)
V = BIRT, (2)
С' = (С- B2)/(RT)2, (3)
dB' i (dB В
dT ~ RT \dT T
dC'
dT
dC dB
+-----25 —
dT dT
dC'
(5)
(6)
= PT
Используя корреляцию Эббота для коэффициента В,
получаем
2R
(7)
1.097
Hid - Н = RTcPr ~ 0.083
(И)
/ dB' Р2 dC'
+ Т Р-------+-----------
\ dT 2 dT
!?пД2льз^я корреляцию Цонопулоса для коэффициента В,
получаем
Hli — Н = R Тсрг[- 0.1445 + 0.660/7; + 0.41Ь/Т2
+ 0.0484/7? + 0.0055/7?
+ о>(—0.0637 - 0.993/7? + 1.692/7?
+ 0.072/7?)], (9)
sid_s = 7f6(°-675 + 0-722oj/r^6)- (12)
Используя корреляцию Питцера—Керля для
коэффициента В, получаем
Hid - Н= RTcPr[~(0.1445 + О.О73<о)
+ (0.660 - 0.92о>)/7у + (0.4155 + 1.5О(о)/7?
+ (0.0484 + 0.388<i) )/7? + 0.0657о)/7?],
(13)
5ld - S = —Г [0-330 + 0.277/7- + 0.036/7?
у?/ L f •
+ 0.0049/7? + о)(-0.662 + 1.269/7?
Sid - S = ЛРГ[(О.ЗЗО - 0.46(0)/7? + (0.2770 + 1.00й))/7?
+ (0.0363 + О.29(о)/7? + 0.05 84/7?]. (14)
+ 0.064/7?)].
(Ю)
где
dB / dB(0>> dB^
Тт^{КТ^1Р^\1т+ш^т
R jdB^ dB^\
~ Pc\dTr + Ш dTr J ’
точные свойства жидкостей менее значительное воз-
действие, чем на остаточные свойства газов, однако
вблизи критической точки и при довольно высоких дав-
лениях различие между жидкостью и паром становится
размытым, а их остаточные свойства соизмеримыми.
Выборка из данных, на основе которых построены гра-
(11.50) фики, представленные на рис. 11.7, а, подтверждает это
положение:
dC / (/С(°)
--(лгс/р^ —-
рг 0,4 1,0 2,0 10,0
ЬН'/ТС @ Тг = 0.9 1,48 9,94 10,29 8,74
&Н'/ТС @ТГ= 1.1 0,74 2,42 5,22 6,94
/ d&Q} dC^
(И-51)
Очевидно, что при введении третьего вириального коэф-
фициента невозможно получить простой график, подоб-
ный приведенным на рис. 11.5 или 11.6.
Жидкости. Как правило, давление оказывает на оста-
Воздействие давления определяется при помощи коэф-
фициента изотермической сжимаемости и коэффициента
термического расширения, которые для жидкостей
обычно малы. Особенно простые выражения, описыва-
ющие воздействие давления на энтальпию и энтропию
жидкостей, получают в тех случаях, если эти коэффици-
енты имеют постоянные величины, а именно:
Таблица 11.3. Остаточные свойства, полученные из некоторых уравнений состояния
Уравнение Ван-дер-Ваальса (табл. 1.3)
&H' = RT
2а b
RTV ~ (V-Ь)
AS'=-5 In [z(l - b/V)].
Уравнение Редлиха—Квонга (табл. 1.9)
ЫГ = RT
1.5а
l~z+TR^ln(l+b/V)r
(1)
(2)
(3)
AS' = -R
а
ln[z(l-b/V)]-—~rT\n(l+b/V)
IDI\1
(4)
Уравнение Соава (табл. 1.11)
Di = - = рп(оа) \/Тг/а ],, (5)
Д---= 1 — z + -(аа +P)ln(l + b/V),
RT bRT
(6)
(7)
А / D \
= l-z+- 1+— ln(l+5/z), (8)
В \ аа /
BD
= —ln(z—5) +-------ln(l+5/z). (10)
Aaa
Для чистых веществ вместо D и Е в уравнениях (9) и
(10) следует применять D, и Ei.
Уравнение Пенга—Робинсона (табл. 1.13)
Следует учесть, что между коэффициентами в опреде-
лениях ki, Ai и Bi в уравнениях Соава и Пенга-
Робинсона существует небольшое различие, а уравнения
для D и Di имеют одинаковую форму.
И' А / D \ z + 2.414В
A rt~ 1 z+ 2.828В \ + аа / ° z — 0.4145
(11)
S' BD z +2.4145
AT = ~ In(Z ~B}+ 2.8284аа 1П z - 0.4145 (12)
Для чистых веществ в уравнениях (7) — (10) следует ис-
пользовать Di вместо D.
Уравнение Бенедикта—Уэбба—Рубина (табл. 1.16)
Уравнение Бенедикта—Уэбба—Рубина—Старлинга
(табл. 1.17)
Уравнение Ли—Кеслера.
См. табл. 1.18. Подробное описание методики дано
в «Справочнике Американского нефтяного института»
(API Data Book, рр.6В1.8, 7В3.7). Приемлемым является
описанное здесь использование правил Кэя вместо бо-
лее сложных правил Ли—Кеслера.
S' D
А—-------ln[z(l -b/V)] + —— ln(l + b/V), (9)
R OKI
dH =
V- Т
dP=V(l - fiT)dP
(11.52)
= V0(l —0T)exp(—kP)dP,
(11.53)
dS = ~
(dV \
— dP = - fidP = Po0exp(-fcP) dP,
\dT Jp
(11.54)
где сжимаемость
1 /аг
k — — — I
V \dP
(11.55)
а коэффициент термического расширения
1
V \дТ
(11.56)
Полезным также представляется анализ изотерм жид-
кости на диаграммах давление — энтальпия, подобных
демонстрируемым в книге Старлинга [127]. При более
низких температурах (дН/дР)т имеет положительный
знак и незначительную величину; однако по мере при-
ближения к критической температуре величины стано-
вятся отрицательными и их абсолютные значения
намного возрастают. В примере 11.6 рассматривается
воздействие давления на ряд жидкостей.
На диаграммах, показанных на рис. 11.7, представле-
ны как жидкости, так и газы. В настоящее время наи-
лучшим образом можно определить остаточные свой-
ства неполярных жидкостей из уравнений состояния,
применимых как к жидкостям, так и к газам. Авторы
статьи [675] провели проверку шести таких методов,
охватывающую как чистые углеводороды, так и неко-
торые бинарные смеси, применительно к 1800 экспери-
ментальным данным об энтальпиях жидкостей. Как
выяснилось, уравнение Ли — Кеслера дает несколько
более точные результаты, чем уравнение Бенедикта —
Уэбба — Рубина — Старлинга, однако и более простое
уравнение состояния Соава также оказалось достаточно
надежным. Использование последнего уравнения пока-
зано в примере 11.11.
Оценка изменений энтропии и энтальпии 519
Рис. 11.3. Остаточные этальпии 77id — Н, основанные
на корреляциях второго вириального коэффициента
Питцера—Керля (7), Эббота (2) и Цонопулоса (3).
Рис. 11.4. Остаточные энтропии, полученные на основе
корреляций второго вириального коэффициента Питце-
ра—Керля (7), Эббота (2), Цонопулоса (3).
Рис. 11.5. Остаточная энтальпия, полученная на основе
вириального уравнения с корреляцией Цонопулоса. На
кривых указаны величины ацентрических коэффициен-
тов О).
Оценка изменений энтропии и энтальпии 521
Рис. 11.6. Остаточная энтропия, полученная на основе
вириального уравнения с корреляцией Цонопулоса. На
кривых указаны ацентрические коэффициенты ш.
Пример 11.2. Уравнения для остаточных свойств, полученные несколькими методами из В-усеченного вириально-
го уравнения
Уравнение
Z = PV/BT = 1 + ВР/ВТ
Используя уравнения (15) и (17), получаем
(1)
ДЯ' = ВТ - PV +
можно переписать, выразив его либо через давление,
либо через объем, либо через z, так что его можно ис-
пользовать для проверки пригодности приведенных в
табл. 11.1 уравнений для определения ЬН' и Д5'. Необ-
-ВР +
ВТ2 dB
V — В dT
ходимые для расчета производные равны
(_R dB
\dTjp р dT'
(Ьр\ = R + dB
\дТ )v V-B (V-B)2dT’
_ P ( dB B\
\dT Jp RI\dT TJ
В + ВТ dB
V-B (V—B)2dT/
= р(т~ - в\
\ dT 1
dV= (7)
(8)
(2)
0)
(4)
Уравнение (4) приводит к
Р , dB R\dP= pdB
Р + dT Р dT'
Д5'
(9)
Исходя из уравнения (5), получаем
Используемые для расчетов уравнения приведены в
табл. 11.1 под теми же номерами. При помощи уравне-
ния (1) получаем
BP Р (dB В
ВТ В \dT Т
0 L X
При помощи уравнения (19) получаем
Д5' =
Д5' =
В
Уравнение (2) дает
ДЯ' = ВТ2
Sp
о
^В
</Р=р(т^-Л).(5)
\ al 1
= Pin——
V-B
dP = р dB
Р dT'
(Ю)
Р (dB _ B\dP = J’dB _ Л
BTydT т/ Р \ dT )
(6)
В _ ВТ dB
V-B (V — В)2 dT
ВТ dB D.
+-------- P In Z =
V- В dT
dV — Plnz =
= Plnz + P^ - Plnz = P~J.
dT dT
(ID
Во всех случаях наблюдается полное соответствие.
Пример 11.3. Уравнения, используемые для определения остаточных свойств, полученные из уравнения состояния
Соава
Уравнение Соава и необходимые производные имеют
следующий вид':
р _ ВТ _ аа
~ V - b V(V+ Ь?
-Л)]'
а =
дР\ _ В _ da 1
dTjy V-b adTV(V+b)’
da _
~dT "
Используя
уравнение (17) из табл. 11.1, получаем
ДЯ' =
ВТ
V-b
аа______ВТ
V(V+ b) V-b
(1)
(2)
(3)
(4j
+ dTda 1
dT V( V + b)
dV =
(5)
I г*, da । 1 jtz
cl Г----a I---------dV =
V dT JV(V+b)
= ТГ\а~ + ь/у>-
о \ dl 1
После подстановки в уравнение (15), табл. 11.1, имеем
ДЯ' = ВТ - PV + ДЯ' =
= ВТ 1
~ )ln(l + b/V) .
bBT \ dT 1 '
Используя уравнение (19), табл. 11.1, получаем
Д5' =
Р
V
R In V - b
В da 1
V- b dTV(V+ b)
а da.
, + ьат'Л
dV-Blnz=
- Р In z =
(6)
(7)
(8)
(9)
= -P In [z(l - b/V)) + ^^ln(l + b/V) . (10)
Вывод формулы для d(aa)/dT смесей показан в
разд.11.5.
Оценка изменений энтропии и энтальпии 523
tep 11.4. Вычисление остаточных свойств непосредственно из данных по измерению объема и при помощи
иьного уравнения и уравнения Соава
Удельные объемы азота были измерены в некотором
гервале давлений и температур [247]. Эти данные
реобразуют в сжимаемости и дифференцируют, ис-
мьзуя формулу Лагранжа для неравных интервалов с
м, чтобы найти (dz/dT)p/P при О °C. После этого при
мощи уравнений (2) и (5), табл. 11.1, определяют
паточные свойства Д/Г и Д5'.
а? \
&Г)р
dP.
свойства вычисляют при О °C.
Р, атм V (мл/г) при
-25 °C 0°С 20 °C
20 35,75 39,67 42,74
40 17,65 19,72 21,33
60 11,66 13,11 14,23
80 8,694 9,828 10,70
100 6,950 7,886 8,604
200 3,645 4,139 4,524
300 2,704 3,020 3,271
400 2,287 2,510 2,693
500 2,046 2,217 2,360
600 1,890 2,032 2,146
800 1,695 1,798 1,880
1000 1,567 1,650 1,717
1100 1,516 1,591 1,654
1200 1,473 1,543 1,601
Jo 4 7
V = R |
J о
1
I Р, атм Z При (*) (*‘) Д/Г AS'
-25 °C 0°С 20 °C
20 0,9831 0,9910 0,9949 12,4389 5,8683 37,8 0,12
1 40 0,9707 0,9853 0,9930 11,8361 5,6950 — 0,23
60 0,9619 0,9825 0,9937 11,2889 5,5486 108,3 0,35
80 0,9563 0,9821 0,9963 10,6639 5,3443 — 0,46
100 0,9556 0,9850 1,0014 9,7822 5,0122 171,4 0,56
200 1,0023 1,0340 1,0531 5,4706 3,3075 — 0,97
300 1,1153 1,1317 1,1421 1,9348 1,9226 337,2 1,24
400 1,2578 1,2541 1,2538 -0,1853 1,1617 — 1,29
500 1,4065 1,3846 1,3734 -1,4009 0,7679 336,2 1,39
600 1,5592 1,5229 1,4986 -2,7006 0,2657 305,8 1,44
800 1,8644 1,7967 1,7505 -3,1086 0,2913 219,7 1,50
1000 2,1545 2,0610 1,9984 -3,4011 0,2619 123,1 1,55
1100 2,2928 2,1861 2,1176 -3,4542 0,2674 72,3 1,58
1200 2,4303 2,3128 2,2361 -3,5162 0,2650 20,6 1,61
О °C
z- 1 + 273,2^
’ 126 (, о °с
Р \дТ /р
Ю34
Экспериментальные данные, полученные для азота:
Тс = 126,2, Рс = 33,5, ы = 0,040, Т = 273,2.
Расчет с использованием уравнения Цонопулоса:
Исходя из вирального уравнения Цонопулоса, выполня-
ем подстановки в уравнения (9) и (10) (табл. 11.2):
МГ = = 7,4854/д(7’г)Р,
AS’ = §VS(7>) = 0,0593/$( 77)Р
*с
Обе величины — и Д/Г, и Д5'—линейно меняются
вместе с давлением:
Р = 0,
Р= 300.
AS'
0, Р = 0,
1,79, Р= 300.
W ~ 560,
Метод Соава (необходимые формулы см. в табл.
11.1):
R = 1,987,
а = 0,42747 Р2Т?/РС = 802,37,
b = 0,08664 RTc/Pc = 0,6485,
т = 0,48508 + 1,55171ы - 0,15613ы2 = 0,5469,
а = [1 + Jt(l - VP/Ге]2 = 0,5509,
-2-,86(1(гзк
Р /V \2
А = 0,42747а —I ~ I = 1,4998(10 " 3)Р
*с \ * /
(Р\/т \
D )( v' ) = 1,1946(10 “ 3)Р,
/с / \ * /
АВ = 1,7917(10 "6)Р2,
А - В - В2 = 0,3052(10 “3)Р - 1,4271 (10 ~6)Р2,
Z3 - Z2 + (А - В - B2)Z - АВ = 0,
b/V= — = 1,1946(10“3)—.
zRT z
Прежде всего следует разрешить уравнения относи-
тельно z для каждого давления. Далее необходимо вы-
полнить подстановки в уравнения (8) и (10) (табл. 11.3).
АН' = 542,85(1 - z) + 1420,52 In • 1 + 1’1946<10
z
AS' = 1,987 In (z - 1,1946(10’3)Р) +
+ 2,7047 In 1 +
1,1946(10-3)
Результаты расчетов, проведенных по методу Соава,
представлены ниже в виде таблицы.
AH' AS'
20 0,9952 36,31 0,1220
60 0,9933 102,61 0,3506
100 1,0016 159,19 0,5539
300 1,1532 301,25 1,1882
500 1,3822 302,73 1,4527
800 1,7541 208,44 1,6236
1000 2,0051 118,28 1,6816
На рисунке сравниваются результаты, полученные
тремя указанными меюдами.
Остаточные энтальпия и энтропия азота.
---------экспериметнальные данные,
---------- расчет с применением вириального х рав-
нения Цонопулола, о расчет по метолу
Соава.
Пример 11.4А. Вычисление остаточных энтальпий при помощи уравнений Соава и Пенга — Робинсона и таблиц
Ли — Кеслера
В примере будут использованы уравнения (9) и (И)
из табл, i 1.3, а также данные, приведенные в табл. Д. 15
и Д.1о Для чистых веществ в целях сравнения испо-
льзуются величины, взятые из графиков Американского
тефтзн'Ли института (API). Образец программы со-
гавлен для конкретных смесей этана, пропана и ч-бута-
>.а при давлении в 30 атм и температуре 478 К (400 °F).
Тля чистых компонентов и их эквимолярной смеси пи-
.лчены следующие результаты:
рассчитавтал
; уравнению
хН' (кал/г), рассчитанная
нс уравнению
<'сана 11 CHI c*.— РоЬин- сина - АРР Jill - г оава Пенга— Робин- сона
u с 9,960 0,944 5,6 5,7 6,1 6,8
I ,o99 0,872 8,9 8.5 8,8 9/.-
0 1 0.798 О 748 12,8 12,3 12.4 П. 6
0,899 -- 9,1 3,8 9,6
Для определения псевдокритически х снойщв По-
мощью уравнения Ли — Кеслера используются правила
Кэя.
Как показывает этот пример, величины, рас/ю
ные по уравнению Соава. довольно хорошо соьь.д < •г-
ся с полученными Американским нефтяным ь-т и .-ч
тем, а также вычисленными г.о уравненью .'л -
Кеслера, го же можно сказать и об уравнении Пенга -
Робинсона, хотя в последнем случае согласование е
сколько хуже. Как установили авторы статьи -б < 1 ' г, -
мерно для пяти тысяч данных результаты примеиеч-ь.
метода Соава и метода Ли — Кеслера достаточно ю,.ю
шо согласуются между собой и в среднем <„• дичаь
оз экспериментальных даины> прибди я-тащао ча
2 кал' г.
10 I RESIDUAL ENTHALPY. . A I б ю' ’
AND P-R EQNS. EQNS. 8 di 11 J „IL .
“KRESD”
20 ’ WITH SOAVE, GOSUB 42u 1C Li.A
PRINT H IN LINE 360
30 '. WITH PENG—ROBINSON, GOS>. т r
LINE 13u, PRINT Hl IN LINE л.ю
Оценка изменений энтропии и энтальпии 525
60 DIM А(3), В(3), М(3), W(3), Т1(3), Р1(3), Y(3),
А1(3)
70 N = 3
80 READ Т, Р
90 DATA 478, 30
100 MAT INPUT Y
ПО MAT READ Tl, PI, W
120 DATA 305.4, 369.8, 425.2, 48.2, 41.9, 37.5, .098,
.152, .193
130 GOSUB 420
140 А = О
150 FOR I = 1 ТО N
160 FOR J = 1 ТО N
170 А = А + Y(I) * Y(J) * (A(I) * A(J)) * .5
180 NEXT J
190 NEXT I
200 INPUT Z! Trial value
210 GOSUB 380
220 H = F/Fl
230 Z = Z - H
240 IF ABS(H/Z) < = .0001 THEN 260
250 GOTO 210
260 D = 0
270 FOR I = 1 TO N
280 FOR J = 1 TO N
290 D = D + Y(I) * Y(J) ♦ (A(I) * A(J)) * .5 * (1 +
.5 * M(I) * (T/A1(I)/T1(I) ‘ .5 + .5 * M(J) *
(T/A1(J)/T1(J)) ‘ .5)
300 NEXT J
310 NEXT I
320 H = 1.987 * T * (1 - Z + D/B * LOG(1 + B/Z))
330 Hl = 1.987 * T * (1 - Z + D/B/8 .5 *
LOG((Z + 2.414 * B)/(Z - .414 * B)))
340 PRINT “T = T; “P = ” ;P; “Y1 = Y(l);
“Y2 = Y(2); “Y3 = Y(3)
350 PRINT “Z = Z
360 PRINT “AH' = H
370 END
380 ! SR FOR Z
390 F = Z* 3 - Z* 2 + (A - В - В* 2) * Z - A * R
400 Fl = 3 ♦ Z* 2 - 2 * Z + A - В - B* 2
410 RETURN
420 ! SR FOR SOAVE
430 В = 0
440 FOR I = 1 TO N
450 M(I) = .48508 + 1.55171 * W(I) - .15613 * W(I)‘ 2
460 A1(I) = (1 + M(I) * (1 - (T/T1(I))‘ 5))' 2
470 A(I) = .42747 * A1(I) * P/P1(I) * (T1(I)/T)* 2
480 B(I) = 0.08664 * P/P1(I) * T1(I)/T
490 В = В + Y(I) * B(I)
500 NEXT I
510 RETURN
520 ! SR FOR PENG—ROBINSON
530 В = 0
540 FOR I = 1 TO N
550 M(I) - .3746 + 1.54226 * W(I) - .26992 * W(I)‘ 2
560 A1(I) = (1 + M(I) * (1 - T/T1(I))~ 5))‘ 2
570 A(I) = .45724 * A1(I) * P/P1(I) * (T1(I)/T)' 2
580 B(I) = .0778 * P/P1(I) * T1(I)/T
590 В = В + Y(I) * B(I)
600 NEXT I
610 RETURN
Тс Рс Тг Рг ^H^/RTc ЬНЫ/КГс А//', кал/г
с2 0,098 305,4 48,2 1,57 0,62 0,2811 0,0217 5,73
С3 0,152 369,8 41,9 1,29 0,72 0,4793 0,1738 8,45
д-Сд 0,198 425,2 37,5 1,12 0,80 0,789 0,458 12,78
Смесь 0,148 366,8 42,5 1,30 0,71 0,479 0,161 9,06
Пример 11.5. Численная оценка изменений свойств при Т = const при помощи уравнения состояния Ван-дер-
Ваальса
В данном примере рассматривается этан, который
при температуре 500 К подвергают сжатию от 10 до
50 атм. Константы уравнения Ван-дер-Ваальса равны
а - 5,49 и Ь = 0,064; в качестве единиц измерения вы-
браны л/моль, атм, К.
Для определения остаточных свойств, Н и S, по мето-
ду а применяются формулы из табл. 11.3, а при исполь-
зовании метода b — основные формулы, приведенные в
разд. 11.1.
Конечные значения объема и сжимаемости находят
методом проб из уравнения состояния Ван-дер-Ваальса:
Ki = 4,0325, Vi = 0,7507, zi = 0,9829 и z2 = 0,9149.
а.
2,0613 первоначально,
10,8028 окончательно.
Нг - Hi = ДЯ1 - ДЯг = -8,4175 л-атм/моль,
А„, 2а 0,08205 67 \
™ ~v-b- ‘ I
AS' = -0.082051П
(0,0027 первоначально,
0,0146 окончательно.
S2 - Si = -0,08205 In (P2/Pi) + AS/ - AS2' =
= -0,1321 + 0,0027 - 0,0146 =
= -0,1440 л • атм/моль • К.
б. Энтальпия как функция объема равна
ан\ = VЭР\ + VЭР \
дУ Jt \ду)т \дТ Jv
RTV + 2а + КГ
(V- Ь)2 V2 V - ь
Интеграл равен
Н2-Н. = -RT у --Ь-ь + \п(У- Ь)~
+ ягщк- b)
V2 = 0,7507
Vi = 4,0325
— -8,4175 л-атм/моль.
Изменение энтропии находят непосредственно следую-
щим образом:
Vz
S2 - Si =
Vi
ar Jv Vi - ь
= -0,1440 л • атм/моль • K.
Таким образом, результаты вычисления изменений эн-
тальпии и энтропии, проведенного обоими методами,
совпадают. Изменения других свойств находят путем
объединения Д/f и AS. Так,
U2-Ui=H2-Ht-{P2V2-PiVi) = -5,6275 л-атм/моль,
Л2 - Ai = U2 - Ui - T(S2 - Si) = 66,3725 л • атм/моль,
G2-Gi=H2-Hi-T(S2-Si) = 63,5825 л• атм/моль.
Рис. 11.7. Остаточные энтальпии и энтропии жидкостей
и газов. Приведеныне данные получены при критиче-
ской сжимаемости zc = 0,27; в оригинальной работе [59]
поправки даны для других величин сжимаемостей в диа-
пазоне 0,23 — 0,29.
11.4. Влияние изменений Р и Т
Изменения энергетических функций можно просто вы-
разить через теплоемкости идеального газа и остаточ-
ные свойства:
Н2~ Нг= £2С^Т+ДН\-ЛН'2, (11.57)
лт2
JT1
S2 Sj
Clpd Р2
—dT — R In~б~ + ASi - AS'2.
1
(11.58)
В последнем уравнении два члена, стоящие в правой ча-
сти уравнения, соответствуют изменению энтропии иде-
ального газа для процесса, протекающего от состояния
(Pi, Ti) до (Р2, Т2). Незначительные сложности возни-
кают в том случае, если необходимо определить изме-
нения энергий Гиббса и Гельмгольца при изменениях
как Т, так и Р; такая ситуация рассматривается в при-
мере 11.7.
Если требуется описать только изотермические изме-
нения, более простым представляется пренебречь фор-
мулами для остаточных свойств и непосредственно
выполнить интегрирование между нужными величина-
ми давления или объема. Соответствующие уравнения
Оценка изменений энтропии и энтальпии 527
Пример 11.6. Влияние давления на энтальпию и энтропию некоторых жидкостей
В этом примере мы проследим воздействие изменения
давления от 0 до 100 атм при температуре 300 К (приня-
тые единицы измерения — кал, г, К).
к=
У \дР Jt
V = Уое~кр,
АН = | Г(1 - $T)dP = Го(1
Jo
e~kpdP =
о
AS =
SVdP =
о
-kP
k
= «Ио I .
Jo
Ко, k, S и остаточные свойства имеют следующие ве-
личины:
Уо, мл/г 10Ч 10’3 АН - io’as
Уксусная 0,953 0,090 1,10 1,54 2,53
кислота
Ацетон 1,266 0,125 1,50 1,67 4,57
Бензол 1,130 0,095 1,30 1,66 3,54
Этанол 1,267 0,114 1,10 2,04 3,35
Глицерин 0,793 0,025 0,54 1,61 1,03
Метанол 1,264 0,120 1,40 1,76 4,26
Октан 1,422 0,120 0,72 2,68 2,46
Фенол 0,944 0,050 0,90 1,66 2,05
Вода 1,000 0,046 2,00 0,97 4,83
В расчете на единичную массу влияние давления прак-
тически одинаково для всех этих соединений. Все измене-
ния сравнительно малы. Влияние изменения давления в
100 атм можно приближенно считать эквивалентным
воздействию изменения температуры от 1 до 3 К.
Пример 11.7. Изменения свойств, вызванные изменениями Р и Т
Теплоемкость идеального газа С? принимаем извест-
ной как функцию температуры. Определяем изменения
свойств в соответствии с линией abed, показанной на
рис. 11.4. Величину АМ{ = М^ - Mi определяем при
(Pi, Ti), а АМ{ — при (Р2, Т2).
Запишем выражения, определяющие разность эн-
тальпий
Н2 - Hi = jp CpdT + АН{ - AHI (1)
и внутренних энергий
U2 - U1 = Н2 - Hl - (Р2К2 - P1V1) =
= \T2(dp - R)dT + AU{ - AU2. (2)
J г.
Поскольку энтропия идеального газа зависит от давле-
ния и эту зависимость также необходимо учесть при
расчете изменений энтропии, разность энтропий опреде-
ляется следующим выражением:
За стандартные условия обычно принимаются
То = 298 К и Po = 1 атм. Величины абсолютных энтро-
пий при этих условиях известны для нескольких тысяч
веществ; некоторые данные приводятся, например, в
книге Сталла и др. [130]. Необходимые оценки можно
также выполнить, пользуясь методом групповых вкла-
дов; соответствующая методика, разработанная Андер-
соном, Байером и Уотсоном, описана Хаугоеном и др.
[59] и Райдом и др. [107].
Подстановка уравнений (1) и (5) в уравнение (4) даст
требуемый результат. Аналогичным образом изменение
энергии Гельмгольца можно определить из следующего
уравнения:
А2 - Ai = U2 - Ui - (T2S2 - TiSi).
(6)
Для определения изменений функции Планка G/Т и
функции Массье А/Т требуются данные об абсолютных
энтальпиях. Так,
S2 - Si = dT - R In ~ + AS{ - ASi. (3)
I 1 Pi
Н_\
Т )z
н\
Т )i
- (S2 - Si),
(7)
Для энергии Гиббса можно записать
G2 - Gi = Нг - Hi - (T2S2 - TiSi). (4)
Энтропии в последнем уравнении должны иметь аб-
солютные значения, например,
. fT'iCj? Pi
Si = Sobs + ~dT - R In — - AS{.
J Го T po (5)
где S2 - Si дается уравнением (4), а энтальпия, напри-
мер, в состоянии 1 — следующим уравнением:
Hi = HSbs + Г’ CpdT - АН{. (8)
Численные величины абсолютных энтальпий для мно-
гих веществ приводятся, в частности, в книге Сталла
и др. [130], а также в других обзорах.
Пример 11.8. Расширение идеального газа в адиабатной турбине
Из реактора для синтеза кумола поступает смесь сле-
дующего состава: 9,64% кумола, 3,79% пропилена и
86,57% бензола; температура смеси 588,9 К, абсолют-
ное давление 600 фунт/кв. дюйм. Давление смеси необ-
ходимо понизить до атмосферного в адиабатной тур-
бине с изоэнтропийным кпд, равным 70%. В примере
будет определена температура смеси после расширения.
Остаточные свойства находят по табл. 11.2 или по
рис. 11.5 и 11.6, основываясь на вириальном уравнении
Цонопулоса. Средневзвешенные теплоемкости идеаль-
ного газа и квазикритические свойства, рассчитанные
при помощи правил Кэя, равны
С“! = -7,890 + 0,11837’— 7,515(Е-5)7’2+1,792(Е-8)7’3,
Тс = 562 К, Рс = 46,6 атм, ш = 0,221.
При начальных условиях
Тг = 1,048, Рг = 0,876,
Д5/ = 0,68(1,987-0,876) = 1,1888 кал/моль-К,
ДЯ/ = 1,032(1,987) (562-0,876) = 1009,9 кал/моль.
При изоэнтропийных условиях
f*7* >id
Si - Sj = 0 = -^dT -
J 588,9
- P In (14,7/600) - Д5/ + 1,1888.
Величину изоэнтропийной температуры находят ме-
тодом проб.
При Тг, = 465,2 Тг = 0,828, Рг = 0,0215, ДВ2' = 0,0656.
АН{ = 42,5, а величина интеграла равна -8,4923. Про-
веряя баланс, получаем
-8,4923 + 7,3699 - 0,0656 + 1,1888 = 0,0008 = 0.
Изоэнтропийное изменение энтальпии равняется
(я2 - Hi)s = f465,2 CfdT - АЩ + ДЯ/
J588,9
= -4471,5 - 42,5 + 1009,9 = -3504,1 кал/моль.
При 70%-ном кпд
0,7(-3504,1) = 9 CpdT - ДЯ2' + ДЯ/.
Попробуем величины Тг = 496,3, ТГ = 0,883:
ДЯ2' = 1,51(1,987)-(562)-(0,0215) = 36,3.
Проверяя баланс, запишем
-0,7(3504,1) = -2452,9 =
= -3426,5 - 36,3 + 1009,9 = -2452,9,
что свидетельствует о полном соответствии. Следует
отметить, что правильнее было бы выполнить оценку
вириального коэффициента, используя коэффициент пе-
рекрестных взаимодействий, а не правило Кэя, однако
в данном случае различие было бы совсем незначи-
тельным.
можно записать с помощью уравнений, выраженных че-
рез давление:
Я2-Я1
11.5. Смеси
Энтальпию или некоторое свойство М при некоторых
заданных температуре и повышенном давлении можно
найти через избыточные свойства ЛГХ или остаточные
свойства ДМ'. Рассмотрим сначала определение эн-
тальпии. Для смеси идеальных газов
ZTd = S yiH\* = S yi (Hi0 + f Tc#dT ).
\ *7Г0 /(11.61)
Исходя из определений ЕГ* и ДЯ' при заданных Р и Т
(П-59)
и
Н = Н**+Т» у(Н\Л
(11.62)
(11.60)
Данный метод упрощает, в частности, оценку измене-
ний энтропии. Результаты, получаемые при использова-
нии обоих методов, должны, безусловно, совпадать. В
примере 11.5 сопоставлены результаты, полученные
при помощи этих двух методов и уравнения Ван-дер-Ва-
альса. В примере 11.8 показано использование остаточ-
ных свойств для анализа типичной задачи, возникаю-
щей при анализе процесса в турбине.
или
Я = Я16 - AH' = S ytH\A - ДЯ' (11.63)
Очевидно, что эти два уравнения тождественны, так как
ЕР* = - ДЯ', однако в том или ином случае одно из
этих выражений может оказаться более удобным. Обе
величины оценивают из уравнений состояния; ЕР* нахо-
дят из уравнения (11.64), а - ДЯ' —при помощи урав-
нений из табл. 11.2 и 11.3.
При использовании соответствующих правил усредне-
ния свойств для смесей оба метода представляются тру-
доемкими. В задаче 11.20, например, применяется В-усе-
ченное вириальное уравнение и корреляции. Однако по-
Оценка изменений энтропии и энтальпии 529
скольку поправки энтальпии на неидеальность обычно
невелики, допустимы некоторые упрощения. Например,
метод оценки Д/f' и Д5', применяемый Американским
нефтяным институтом, основан на уравнении Ли —
Кеслера, однако для оценки некоторых псевдокритиче-
ских свойств и ацентрического коэффициента применя-
ется правило Кэя. Предполагается, что этот метод в
равной степени пригоден и при использовании других
уравнений состояния.
Аналогичным образом избыточные свойства находят
при помощи других уравнений состояния. Применяя
правило М = У]yiMj, уравнения (36) и (76) из табл. 3.2
можно преобразовать к следующему виду:
Например, при использовании уравнения Соава, для
которого уравнения для коэффициентов фугитивности
даны в табл. 3.4,
В1
1пф; = — (Z - 1)- ln(z -В)
D
А Г.В, 2 v "I / В \
+ в ~в~ ~yj(aa)ij lnl1 + 7/>
(И.71)
получаем следующую производную:
Яех = -RT2
(11.64)
(In 0,)2 ~ (In 0/)}
т2-т.
(11.72)
Gex = ЯГ(1п ф - S In 0,), (11.65)
A In 0;-
Яех - (7ех
о ex--------------
т
АГ
(11.73)
(11.66)
Если к жидкому состоянию нельзя применить ни одно
уравнение состояния, избыточные свойства выражают
через коэффициенты активности посредством уравнений
(11.67)—(11.69).
Выражение в конечных разностях (11.73) эквивалентно
производной и его находят путем подстановки двух ве-
личин температуры в уравнение (11.71); в этом уравне-
нии величины А,, В,, А, В, z, at и а зависят от тем-
пературы. Этот метод применен в примере 11.10.
На практике изменением энтальпии смешения при
давлении системы часто можно пренебречь, следова-
тельно, энтальпия смеси обращается во средневзвешен-
ную сумму энтальпий компонентов, т. е.
Яех=-ЛГ2Ех,----- , (11.67)
\ дТ 1
Н = S yiHi “ S yjHi
и
AH' ~ Z у,АН1,
(11.74)
(11.75)
Gex = RTZ xt In у/, (11.68)
+ In л
(11.69)
Степень риска подобной аппроксимации установить
трудно. Тот факт, что ее результат не согласуется с пра-
вилами усреднения свойств даже наиболее простых
уравнений состояния, может быть проиллюстрирован
на примере уравнения Ван-дер-Ваальса, для которого
остаточная энтальпия смеси определяется как
Последние три уравнения будут выведены в следующем
разделе.
Получение уравнений для производных от коэффици-
ентов парциальной фугитивности из большинства урав-
нений состояния, которые входят в уравнения (11.64)—
(11.69), может быть довольно сложным. Однако при
наличии ЭВМ всегда можно применить численный ме-
тод исходя из того, что
AH' = - RT
b
V -ь
2а
RTV
(11.76)
однако параметры выводятся из параметров компонен-
тов исходя из следующих правил:
« = (Л\7в/)2, (U.77)
b='£yibi. (11.78)
df /(1.0001 Г)-/(Г)
dT 0.0001 Т
(11.70)
Если считать энтальпии аддитивными величинами,
можно записать
причем оценку In выполняют дважды при незначи-
тельно отличающихся друг от друга температурах. Ко-
эффициент 1,0001 можно в случае необходимости
изменить.
/ Ь^ \ 2
ЫГ = RTZ У1 (7—^ )- у L у>а<-
(1179)
Sex = - R S Xj
т
1?-S64
Очевидно, что параметры а и b смеси нельзя получить
аналогичным образом из параметров для чистых ком-
понентов в уравнениях (11.76)—(11.79). Подобного рода
заключения справедливы и для более сложных уравне-
ний состояния и соответствующих им правил усредне-
ния свойств.
Если параметры уравнения состояния не зависят от
температуры, для описания ДН' и Д5' чистых веществ
пригодны те же уравнения, что и для смесей при усло-
вии использования надлежащих правил усреднения
свойств. Это заключение справедливо по отношению к
таким уравнениям состояния, как уравнения Ван-дер-Ва-
альса, Редлиха — Квонга, Бенедикта — Уэбба — Рубина
и Бенедикта — Рубина — Старлинга, а также уравнения
Ли — Кеслера; в то же время на вириальное уравнение,
уравнения Соава и Пенга — Робинсона оно не распро-
страняется.
Вириальное уравнение. Вириальные коэффициенты в
очень сильной степени зависят от температуры, что бы-
ло учтено при выводе уравнений (11.46)—(11.51). Пра-
вила усреднения свойств приведены в табл. 1.8. Для
второго вириального коэффициента справедливо сле-
дующее:
В = ЕЕ yiyjBij = Е yjBi + 2 ЕЕ yiyjBij,
l*J (11.80)
где Bij выражен при помощи корреляции типа корреля-
ции Питцера — Керля:
ви = (4°} V (11 •81)
"cij
Если / # j, сверхкритические свойства бинарных смесей
обычно оценивают по правилам Лоренца — Бертло. Не-
обходимая производная определяется как
dB y]Tci (dB^} dB^ \
dT~RZ Pci \ dT +U)i dT /
+ 2Я EE yiyj^
1 rClJ
'dB^ dstf \
ч dT + W,y dT ) ’
(11.82)
Подстановка уравнений (11.80)—(11.82) в уравнения
(11.46)—(11.51) дает уравнение, описывающее изменение
свойств. В табл. 11.2 показаны уравнения, выведенные
на основе корреляций Эббота, Цонопулоса и Питце-
ра — Керля.
Уравнения Соава и Пенга — Робинсона. Для этих
уравнений характерна одна и та же форма зависимости
параметра а от температуры
<*/= [1 +mi( ’Ж/)]2
(11.83)
и одинаковые правила усреднения свойств, применяе-
мые для комбинации параметров аа и для параметра Ь,
аа = Е Е yiyj( 1 - ку) y/(aa)i(aa)j, (11.84)
i J
Ь=Ъу^. (11.85)
Величины mi в этих уравнениях не являются тождест-
венными. Производная уравнения Соава имеет следую-
щий вид:
R 1 J(aa)
V - b У( V + b) dT ’
(11.86)
в то время как производная Пенга — Робинсона равна
дР \ R 1 d(aa)
дТ /к У-b У2 + 2ЬУ — b2 dT
(11.87)
Как следует из уравнения (11.83),
d(aa){ m, -------------
= ~ — (аа)^Тг{/а{. (11.86)
di i
Для индивидуальных членов можно записать
d _______________ d
— V(aiai)(ajaj) = у/а^а] —
(11.89)
daj dttj
а,- + а,-
1 dT J dT
(11.90)
[+ kj\/ajx/ajOtj!Tci]
(П.91)
1 _____________
= - — V(aa)i(aa)j [kjVTrJ/aj + к^Т^/а,].
(11.92)
Поскольку А, = (aa)iP/R2T2 и В = ЬР/RT, можно про-
вести следующую замену в уравнении (11.92), что даст
более простые окончательные выражения для ДН' и
Д5':
y/(aa)i(act)j = bRTy/A^AjiB.
(11.92а)
Для смеси производная равна
d(aa) 1
------= -—ЕЕ y,y;(i-fc0.)
dT 2у/Т «’ J
(11.93а)
Путем подстановки в уравнения (11.31) и (11.41) полу-
чим уравнения для определения Д/f' и Д5', которые
Оценка изменений энтропии и энтальпии 531
Пример 11.9. Расчет избыточных свойств при помощи
В примере рассматривается система этанол(1) + бен-
зол(2). Параметры растворимости и молярные объемы
компонентов смеси имеют следующие значения:
12,915 Н и 58,4 мл/моль у этанола и 9,158/7 и
89,4 мл/моль у бензола. Параметры уравнения
Вильсона Х12 и Х21 равны соответственно 1297,9 и
131,47 кал/моль.
В соответствии с уравнением Скэтчарда — Гильде-
бранда можно записать
/7ех = Xj In Ti = (61 — 5г)2[xi V\(1 — </>i)2 + ХгКгф?],
уравнений Вильсона и Скэтчарда — Гйльдебранда
Ф1 = 1/(1 + X2V2/X1V1).
Если используется метод Скэтчарда — Гильдебранда, то
предполагается, что //ех не зависит от температуры, а
5ех = 0.
Если расчет проводится по методу Вильсона, следует
воспользоваться уравнениями (9) и (10) из примера
11.11. Полученные графики указывают на низкую сте-
пень соответствия, что является вполне логичным, по-
скольку коэффициенты активности, рассчитанные на
основе этих двух уравнений, также во многом различ-
ны, как видно, например, из рис. 4.19, а.
Расчет избыточной энтальпии по
уравнениям Вильсона и Скэтчар-
да—Гильдебранда. Н*\ рассчи-
танная по уравнению Скэтчар-
да—Гильдебранда, не зависит от
температуры.
Расчет избыточной энтропии по
уравнению Вильсона. В соот-
ветствии с расчетом, проведен-
ным по уравнению Скэтчарда—
Гильдебранда, Sex = 0.
15
Пример 11.10. Расчет избыточных свойств жидких и
Эквимолярная смесь H2S (1) и н-гептана (2) при 422 К
и 5 атм находится в парообразном состоянии, а при
310,9 К и 30 атм — в жидком.
Уравнение Соава приведено в табл. 1.11, а коэффици-
енты фугитивности — в табл. 3.3 и 3.4. Избыточные
свойства Н** и Sex находим при помощи уравнений
(11.64) и (11.66), а необходимые производные — по
уравнению (11.72), используя сравнительно большую
разность температур (1%), чтобы более ярко продемон-
стрировать влияние температуры на параметры.
Как следует из данных, приведенных в табл. 1,12, па-
раметр бинарного взаимодействия = 0,0737. Кон-
станты а и Ь определяем при R = 0,08205.
Тс Рс а b
H2S 373,2 88,2 0,100 4,544 0,0301
Н-С7 540,2 27 0,351 31,103 0,1422
Смесь 14,418 0,0862
паровых смесей по уравнению Соава
Ниже приводятся сведенные в таблицу результаты
вычислений сжимаемостей и коэффициентов фугитив-
ности. Избыточные свойства получают следующим
образом.
Для паровой фазы при 422 К:
/Гх = -1,987(422)2(0,5)(-66,7 - 309,5)(Е - 6) =
= 66,6 кал/моль,
$ех = ^ + я2>/1п(ф,/ф,) =
= 66,6/422 + 1,987(0,5)(-0,0223 - 0,0312) =
= 0,105 кал/моль • К.
Для жидкой фазы при 310,9 К:
Н* = -1,987(310,9)2(0,5)(0,0158 + 0,0139) =
= -2775 кал/моль,
5ех = -2775/310,9 + 1,987(0,5)(- 0,2548 - 0,0752) =
= -9,3 кал/моль-К.
а аа 103Л 103В Z In ф In ф In(ф/ф) 106(а1пф/7)
При 422 К H2S н-С? Смесь 0,9207 1,2485 4,184 38,832 16,658 17,45 161,9 69,47 4,347 20,53 12,45 0,9868 0,8363 0,9405 -0,0132 -0,1514 -0,00907 -0,1212 -0,0223 -0,0312 -66,7 - 309,5
При 426,2 H2S H-C7 Смесь К 0,9142 1,2386 4,154 38,524 16,529 16,99 157,5 67,58 4,304 20,33 12,325 0,9872 0,8421 0,9425 .-0,0128 -0,1461 - 0,00879 -0,1215 -0,0216 -0,0246
При 310,9 H2S H-C7 Смесь К 1,1139 1,5473 5,062 48,126 20,525 233,35 2220,1 946,3 35,40 167,23 101,37 0,0582 0,1989 0,1335 - 0,2929 - 5,4491 -0,0381 -5,328 -0,2548 -0,1211 0,0158 0,0139
При 314,0 H2S H-C7 Смесь К 1,1079 1,5473 5,034 48,126 20,499 227,53 2175,1 926,5 35,05 165,58 100,37 0,0586 0,1975 0,1327 -0,2358 -5,3602 0,0108 -5,285 - 0,2466 -0,0752
приводятся в табл. 11.3. В примере 11.4А предлагается
программа для ЭВМ для расчета Д/f' смеси с использо-
ванием уравнения Соава.
Литература, посвященная данному вопросу. Дэннер
и др. (1978) предприняли в рамках исследований Амери-
канского нефтяного института комплексную проверку
нескольких уравнений состояния для прогнозирования
энтальпий чистых веществ и смесей. Результаты, полу-
ченные при применении уравнений Соава и Пенга — Ро-
бинсона, оказались хуже полученных при использовании
уравнения Ли — Кеслера, однако не уступали найден-
ным при помощи уравнения Бенедикта — Уэбба — Ру-
бина. Как показало более раннее исследование [626],
последнее уравнение дает вполне удовлетворительные
результаты. Сравнение величин избыточных энтальпий
бинарных смесей с рассчитанными по уравнению Виль-
сона показало хорошее их соответствие; авторы этого
исследования [506] исходили из допущения о квадратич-
ной зависимости экспоненциальных параметров от тем-
пературы. Авторы работы [505] установили, что метод
UNIFAC также вполне пригоден для прогнозирования
избыточных энтальпий нескольких бинарных и тройных
смесей. Один из первых обзоров методов вычисления
энтальпии опубликован Натаном в 1967 г.
Оценка изменений энтропии и энтальпии 533
11.6. Смешение жидкостей
Если ни одно из имеющихся уравнений состояния не
дает достаточно точного описания жидкой фазы, оцен-
ку энтальпий и прочих свойств таких смесей следует
осуществлять другими методами. В этом случае полез-
но обратиться к избыточным свойствам, рассмотрен-
ным в гл. 2, представляющим собой разности свойств
реальных веществ и идеальных смесей. Например, из-
быточная энтальпия определяется как
Яех = Н - S х,Я, = S х,-(Я,- - Н,) = S х, Hfx.
(11.94)
Избыточную энтальпию соотносят с другими свойства-
ми, величины которых легко определить:
меняется методика противоположного характера, за-
ключающаяся в нахождении коэффициентов активности
из установленных калориметрическими методами вели-
чин парциальных молярных энтальпий посредством
уравнения
(11.102)
В целях вывода уравнений для определения избыточ-
ных свойств пригодна любая корреляция коэффициента
активности, учитывающая влияние температуры. На-
пример, используя уравнение Скэтчарда — Гильде-
бранда, получаем
Э(Гех/Г)
d(Gex/T)
(11.95)
(11.96)
1П 5)2>
JK 1
(11.103)
д In у,-
дТ
1П У;
т
(11.104)
Для оценки избыточных энтальпий используются ре-
зультаты дилатометрических измерений избыточных
объемов, методика проведения которых детально разра-
ботана. Однако гораздо более удобным представляется
применение соотношений, в которые входит энергия
Гиббса. Избыточная энергия Гиббса выражается через
коэффициенты активности посредством следующего
уравнения:
(7ех = RTE хг-1пу(. (11.97)
Коэффициенты активности находят экспериментальным
путем посредством измерений фазового равновесия или
при помощи других методов, описанных в гл. 4. Урав-
нение избыточной энтальпии и избыточной энтропии
можно записать следующим образом:
следовательно,
Нех = RTX Xj Inу(- = S XiVjiSj - 8)2, (11.105)
и
Sex = 0.
(11.106)
Результат, полученный в уравнении (11.106), является
вполне закономерным, так как теория регулярных рас-
творов и уравнение Скэтчарда — Гильдебранда были
разработаны на основе допущения о нулевой избыточ-
ной энтропии.
Уравнения Вильсона, NRTL и UNIFAC также разрабо-
таны с учетом приближенной температурной зависи-
мости. Для параметров уравнения Вильсона
= у ехр
(11.107)
Нех = - RT2 S х.
д In у,-
дТ
(11.98)
производная равна
d^jj 8.jj^-jj
dT ~ RT2
(11.108)
Нех - Gex
5ех -----------= - R £ Xj
Т
+ In Ji
Аналогичный результат можно получить, если принять,
что Ху меняется линейно вместе с температурой. Произ-
водная коэффициента активности равна
(11.99)
Свойства смесей описываются при помощи следующих
уравнений:
d In у,-
dT
у dXjj
j ‘ dT
Н= Нех + S X/Hj,
(11.100)
5= Sex + S х,[5,+ ln(l/xz)J,
(11.101)
где свойства чистых компонентов находят в виде функ-
ций температуры и давления, как было объяснено выше
в данной главе.
В некоторых случаях, см., например, [332, 670], при-
dh-ki * V
Z^XjXkj A-kj ^xj
dT j J dT
(11.109)
Способ вывода уравнений для избыточной энтальпии и
энтропии из уравнения Вильсона для бинарной смеси
полностью показан в примере 11.11. Как уравнение
Вильсона, так и уравнение Скэтчарда — Гильдебранда
используются для численного решения в примере 11.9.
Вывод уравнений для избыточных свойств с по-
мощью уравнения NRTL выполнен автором работы
[109], результаты которой представлены в табл. 11.4.
Ренон и соавторы исходили из допущения о том, что
обе группы параметров ау и ту находятся в линейной
зависимости от температуры.
Энтальпии жидких фаз были получены по уравнению
Редлиха — Квонга Иоффе и Зудкевичем [379]. Эти авто-
ры применили модифицированную форму данного урав-
нения с параметрами, зависимыми от температуры,
однако обобщить эту температурную зависимость им
не удалось. В отчете [675] об исследованиях, предприня-
тых по инициативе Американского нефтяного институ-
та, рассматривается шесть методов, включая основан-
ные на уравнениях Соава и Бенедикта — Рубина — Уэб-
ба— Старлинга, и делается вывод относительно того,
что для описания систем углеводородов наиболее при-
годно уравнение Ли — Кеслера.
11.7. Энтальпия испарения
Изменение энтальпии, сопровождающее массообмен
между фазами, получают путем вычисления разности
энтальпий конечных состояний, которые определяют
рассмотренными-в настоящей главе методами. Для чис-
тых веществ энтальпию испарения можно найти по-
средством эмпирических методов, что представляется
гораздо менее сложным, чем применение к обеим фазам
уравнений состояния даже в тех случаях, когда это
возможно.
В основу многих из этих методов положено уравнение
Клаузиуса — Клапейрона, выражающее зависимость
давления пара и энтальпии испарения:
&HV = RT2Az(d lnP°/dT),
(11.110)
где Дг — разность сжимаемостей жидкой и паровой
фаз. Это уравнение часто соединяют с какой-либо од-
ной из множества имеющихся корреляций для давления
пара. Например, используя уравнение Антуана
1пР° = Л -В/(Т+ С),
(11.111)
получаем следующее уравнение для энтальпии ис-
парения:
= BRT2Az/(T + С)2.
(11.111а)
При этом достаточно располагать константами уравне-
ния Антуана и уравнением состояния для оценки сжима-
емостей пара и жидкости, а Дг часто принимают
равным единице.
Объединение результатов, полученных Риделем, и
следующего уравнения:
1.10937?^ (-1 + 1пРс)
Ть
0.930----~
т
л г
(11.112)
Таблица 11.4. Расчет избыточной энтальпии и энтропии с помощью уравнения NRTL [109]
XjGiJ
Gkjxk
к
Принимается, что параметры ац и Сц изменяются ли-
нейно вместе с температурой. Избыточную энтропию
находят, объединяя уравнения (6) и (7) в соответствии
со следующим:
1
5ех = -(Яех-Ех.1пУ1.), (1)
Cji = gji - gii = + CfaT- 273.15), (2)
aji = $ + a$(T~ 273.15), (3)
( СЛ С?;1 = ехр \~aJi^ b (4)
= Tji~ RT (5)
(7)
Оценка изменений энтропии и энтальпии 535
Пример 11.11. Вывод уравнений для определения избыточной энтальпии и энтропии при использовании уравнения
Вильсона для коэффициентов активности бинарных смесей
In 71 = -1п(Х1 + Л12Х2) + 0X2, (1)
In 72 = -1п(Л21Х1 + x2) - 0xi, (2)
О Л12 Л21
0 = i> Xl + A12X2 Л21Х1 + X2’ (3)
y. Aij = -~exp(-Xij/BT), (4)
dA.ij X/jAjj
_ . (51
dT RT2 ’
d@jj _ _ Xi JA12 _ х2 JA21
dT (xi + Л12Х2)2 dT (Л21Х1 + Х2)2 dT
Jin 71 X X2 dAn + Л12Х2 dT + X2 , dT (7)
dT
din 72 xi dA2i dfi - *1— dT (8)
dT Л21Х1 + x2 dT
7PX = -ВТ2 Jin 71 Jin 72 Xl + *2 dT dT (9)
Aex = /PX/7-|-jR[Xiln(Xi + Л12Х2) + Х21п(Л21Х1 +X2)] = (10)
= /rx _ T R(xi In 71 + x2 In 72). (И)
См. также уравнение (4.268).
часто дает вполне удовлетворительное уравнение, для
решения которого требуется располагать лишь данны-
ми о температуре кипения при атмосферном давлении,
критической температуре и давлении. Карруто и Кобая-
ши (1972) разработали уравнение, основанное на прин-
ципе соответственных состояний Питцера — Керля,
которое имеет следующий вид:
— =7,08(1 - т№4+ 10,95 »(1 - Л)»-456.
RTC
(11.113)
Три вышеупомянутых уравнения используются в приме-
ре 11.13, и полученные результаты сравниваются с ре-
зультатами расчета по уравнению Соава и с экспери-
ментальными данными. Результаты, полученные при
применении всех этих уравнений, за исключением урав-
нения Клаузиуса — Клапейрона — Антуана, отличаются
от вышеупомянутых величин в пределах 2%, что явля-
ется практически максимальной степенью соответствия,
полученной с помощью эмпирических методов. В книге
[585], а также [129] дано описание ряда других эмпири-
ческих выражений. Метод групповых вкладов, разрабо-
танный Шастри и др. [610], несколько отличен от
эмпирических методов. В книге Тамира и Стефана
(1983) [18] содержится подробный обзор литературы по
проблеме теплоты испарения.
11.8. Смеси углеводородов
Смеси, состоящие из большого количества углеводо-
родов, обычно принимают эквивалентными содержа-
щим меньшее число псевдокомпонентов, средние
температуры кипения и плотности которых выводят в
соответствующем отношении из их истинных темпера-
тур кипения при дистилляции. Исходя из этих свойств,
можно оценить критические свойства и молекулярные
массы компонентов и в конечном итоге, используя кор-
реляции Ли — Кеслера или другие подобные корреля-
ции, определить энтальпии.
В этих же целях часто вполне пригодны и менее тру-
доемкие методы, например подгонка корреляционных
кривых, как это показано в «Справочнике Американско-
го нефтяного института». Простая формула этого типа
предложена для жидкостей [749]:
0,3897, + 0,0004638,2
н ----------------------
/ 0,4515 \
X 10,3265 +-------- (7^/273,2 + 1)1/3 I,
(11.114)
где Н— энтальпия (кал/моль), t — температура (°C),
Ть — средняя температура кипения псевдокомпонента
(К), q — плотность (г/мл).
Гораздо более сложный метод подгонки кривой раз-
работан Карли [210] как для жидких, так и для паровых
фракций в виде функции Т, Р и характеристического
фактора; в формулу включено тридцать шесть коэффи-
циентов.
Если вычисления проводятся с использованием ЭВМ,
принято вводить величины при трех или четырех тем-
пературах, взятых из диаграмм Американского нефтя-
ного института, и использовать метод интерполяции
для нахождения промежуточных величин. При этом ча-
сто применяется либо простой полином от температу-
ры, либо уравнение следующего вида:
,/Н= кх + к2Т + к3Т2 (11.115)
11.9. Задачи
11.1. Выведите выражения для отклонений от идеаль-
ности A/f' и Д5' исходя из следующих уравнений со-
стояния:
а. Клаузиус: (Р + а/Т( V + с)2)( V - Ь) = ВТ.
б. Лоренц: (Р + a/V2)V = ВТ(1 + b/V).
в. Дитеричи: Р(У — Ь) = ВТ ехр (— a/BTV).
Пример 11.12. Расчет остаточной энтальпии и энтальпии испарения жидкого бензола по уравнению Соава
Условия проведения эксперимента: 505,9 К и
Р = 23,5 атм. Характеристики системы: Тс = 562,1 К,
Рс = 48,3 атм, ш = 0,212. Для расчета используются
формулы, приведенные в табл. 11.1; единицы измере-
ния: мл/моль, атм, К. Различные константы, связан-
ные с уравнением Соава, имеют следующие значения:
R = 82,05,
а = 18,825(Е6),
b = 82,73,
к = 0,8070,
а = 1,0845,
da
Тт
-£[1 4- £(1 - ТТ7тс)]
ТтТ
-0,00157,
+ 18’8ffiE6) [1,0845 + 505,9(0,00157)]
82,73 \
1232,4 )
18,825(Е6)(1,0845)(23,5) = 0 2785
(82,05)2(505,9)2
82,73(23,5)
82,05(505,9)
= 0,0468,
z3 - z2 + 0,2295 - 0,0130 = 0.
Величины сжимаемости жидкой и паровой фаз z соот-
ветственно равны 0,08629 (жидкая фаза) и 0,21590 и
0,69781 (паровая фаза). Для жидкой фазы:
V = zRT/P = 152,4 мл/моль,
Нлй - Н = RT(l - z) +у(“ “ W1 +Т
= 82,05(505,9)(1 - 0,08629) +
= 223,307 мл • атм/моль, 5407,8 кал/моль.
Для газовой фазы:
Z = 0,69781,
V = 1232,4 мл/моль,
Н'й - Я = 82,05(505,9)(0,30219) + 18,8^(Е6)
х [1,0845 + 505,9(0,00157)] In
= 40319,6 мл-атм/моль, 976,4 кал/моль.
Поскольку Нлй для обеих фаз имеет одинаковую величи-
ну, скрытая теплота испарения равна разности функций
отклонения:
Hv = Hv - HL = (Я* - H)L - -H)v =
= 5407,8 - 976,4 = 4431,4 кал/моль.
В «Справочнике Американского нефтяного института»
дана Н» = 4512 кал/моль, что довольно точно соот-
ветствует полученному результату. Величина
(Нлй - H)v = 998 кал/моль, приведенная в «Справочни-
ке Американского нефтяного института», также хоро-
шо соответствует результату, полученному в этом
примере.
Пример 11.13. Расчет энтальпии испарения бензола при Тг = 0,9 посредством эмпирических уравнений
Исходные данные:
Psat = 15,9008 - 2788,51/(7- 52,36)(единицы измерения:
мм рт. ст., К),
Ть = 353,3 К,
Тс = 562,1 К,
Рс = 48,3 атм,
ш = 0,212.
В соответствии с примером 11.12 разность величин
сжимаемости составляет
Z = 0,69781 - 0,08629 = 0,61152.
Уравнение Клаузиуса — Клапейрона дает
= 2788,51(1,987X505,9)W1152) = 4215 кал/моль.
(505,9 - 52,36/
Следует отметить, что без учета поправки Дг погреш-
ность результата была бы велика.
Из уравнения Риделя — Уотсона получаем
н _ 2,17(353,3)(-1 + In 48,3) Г 562,1 - 505,91°’38 _
" 0,930 - 353,3/562,1 I 562,1 - 353,3
= 4444 кал/моль.
Уравнение Каррута и Кобаяши дает
ДЯ„ = 1,987(562,1)[7,08(1 - 0,9)°’354 + 10,95(0,212)
х (1 - О,9)0,456] = 4407 кал/моль.
Вычисленная по уравнению Соава в примере 11.12
ДЯ„ = 4431. В «Справочнике Американского нефтяного
института» дана Hv = 4512 кал/моль, которой доволь-
но точно соответствуют результаты расчетов по всем
уравнениям, кроме уравнения Клаузиуса — Клапейрона.
Оценка изменений энтропии и энтальпии 537
9 Р Т
г. Бертло: ^ = ‘ + П8 р;г
11.2. Сравните отклонения Д/Г и Д£', рассчитанные
по уравнениям Редлиха — Квонга, Соава и Пенга — Ро-
бинсона для насыщенных паров толуола при 502 °F и
290 фунт/кв. дюйм. В «Справочнике Американского не-
фтяного института» дано следующее значение:
Д/Г = 22,5 брит. тепл, ед./фунт.
11.3. В сборнике DECHEMA (DECHEMA VL Equilib
Data, Part la, 221, 223) указаны следующие параметры
уравнения Маргулеса для смеси вода(1) + пропионовая
кислота(2):
Т = 60 °C: Л12 = 1,3410, Л21 = 1,9777,
Т =90 °C: Ли = 0,8593, Л21 = 1,9916.
Исходя из допущения о том, что Ап = ао + а\Т и
Л21 = bo + bi Т, найдите уравнения для избыточной эн-
тальпии и избыточной энтропии. Каковы численные ве-
личины Ях и 8ех эквимолярной смеси при 75 °C?
11.4. Уравнение для приведенного давления пара
Р°/Рс, выраженное через приведенную температуру
углеводородов, имеет следующий вид:
In Рг = С(3 - 4/Тг + 1/7?) +
+ - 64/Тг + 63).
18,744
Найдите уравнение для теплоты испарения [769].
11.5. Уравнение Ван-дер-Ваальса для н-гептана имеет
следующий вид:
(Р + 31,51/И2)( И — 0,2065) = 0,082057
(единицы измерения: атм, л/моль, К). При 500 К и
14,3 атм корни уравнения V равны 2,1483, 0,5191,
0,4080. Уравнение Антуана справедливо для давления
пара. Найдите теплоту испарения при заданной темпе-
ратуре. Общепринятая величина равна 4398 кал/моль.
11.6. Удельный объем (куб. фут/фунт) аммиака пред-
ставлен в таблице в виде функции температуры и дав-
ления. Найдите поправку на давление для
Д/Г = Н' - Н из остаточных объемов и сравните ре-
зультаты с вычисленными по вириальному уравнению
Цонопулоса и уравнению Соава. В таблице также ука-
заны истинные величины Ню — /Гоофунт/кв.дюйм
сР - с? = -{RT2B")q +
+ R (В - ТВ')2 - с + ТС' --Т2С
2
е2 +
(iCp = -(В - ТВ’) + [2В2 - 2ТВВ' - 2С + ТС' -
- (RT2/c°)(B - TB')B"]q...,
где В' = dB/dT, В" = d2B/dT2 и т. д., если исходить из
термодинамических уравнений
Ср = с% - v/dT2)pdP, цср = т(ду/дт)р - у.
11.8. Найдите избыточную энтальпию и энтропию
эквимолярной смеси этанола (1) и трихлорэтилена (2)
при давлении 1 атм. Данные заимствованы из работы
[57]:
7 = 70,8 °C,
Ли = 0,12141 }
[ параметры уравнения Вильсона,
Л21 = 0,53404 )
Vi = 58,39 мл/моль,
У1 = 89,62 мл/моль.
11.9. Корреляции избыточных энергий Гиббса и хи-
мических потенциалов смеси вода + метил этилдиамин,
C5H13N, были установлены при помощи приведенных
ниже уравнений [770]. Исходя из каждой из этих трех
корреляций, определите /Гх и 5ех при 308,15 К и
Xi = 0,25, 0,5 и 0,75.
Упрощенное уравнение
G^/RT=Ax^\ -хО + хД1 -Х!)(2х! - 1)
+ [Cj + C2(2xj - 1) + C3(2xj - 1 )xf]
rf/RT= A(1 -x02 - СД1 - 6xi + 9x2 - 4x?)
+ C2(l - 10xi + 29x? - 32x^ + 12xf)
+ C3(7xj - 46x j + 107xf - 104x? + 36x|°)
^IRT= Axj — Ci(3xj — 4xj)
+ C2(5x2 — 16x2 + 12xi)
- C3(6xJ - 35xj + 64x? - 36x}°)
\ р 7 \ V. н10 ~ Н200
10 50 100 200
100 35,07 6,843 3,304 1,520 34,4
150 38,26 7,521 3,672 1,740 24,0
200 41,45 8,185 4,021 1,935 18,3
7 К
283,15 293,15 303,15 308,15 313,15 320,15
А 1,7983 1,9409 2,0656 2,1477 2,2067 2,2647
Ci 0,01811 0,08909 0,15728 0,20630 0,24316 0,27895
с2 0,7496 0,6312 0,5597 0,535 1 0,4808 0,4527
0,1105 0,5906 0,8575 0,8690 0,8946 0,9173
11.7. Покажите, что используя вириальное уравнение
Z = 1 + Bq + Cq2 + ...,
Уравнение Вильсона: GB/RT = Xi ln(xi + Л12Х2) +
можно получить следующие уравнения для теплоемкос-
ти и коэффициента Джоуля — Томсона: + x2ln(x2 + Л21Х1)
Т, К а12 А21 aiG^)/, Дж/моль
283,15 0,2611 0,2170 35,7
293,15 0,2795 0,1292 31,9
303,15 0,2750 0,0777 25,6
308,15 0,2477 0,0615 18,7
313,15 0,2507 0,0448 11,3
320,15 0,2364 0,0340 5,5
Уравнение NRTL: Ga/RT = xiX2[A2iG2i/(xi + X2G21) +
+ A12G12/(X2 + X1G12)], где
G12 = exp (~aA2i/RT), G21 = exp (-аА2\/РТ), а Л12,
Л21 и a — подгоночные параметры.
Т, К а12 а21 а
283,15 5166,79 4922,48 0,615 3,5
293,15 6211,77 4826,83 0,568 2,8
303,15 7067,01 4812,98 0,528 4,6
308,15 7363,92 4734,57 0,503 4,1
313,15 7528,72 4651,36 0,486 5,3
320,15 7791,81 4596,28 0,467 5,3
11.10. Co скоростью 2моль/с ацетон проходит обра-
тимый цикл, состоящий из изотермического расшире-
ния (ab) от 30 до 10 атм при 1000 К и последующих
изобарического и изохорного процессов (Ьс) и (со).
Найдите изменения энтальпии, работу и тепловые эф-
фекты для процессов ab, Ьс, са и abca, используя урав-
нение Редлиха — Квонга с параметрами а = 360,5 и
b = 0,0778, выраженными в атм, К и л/моль. Теплоем-
кость (кал/моль • К) в состоянии идеального газа опре-
деляется как
С; = 1,505 + 0,06224 Т - 2,992(Е - 5)7* + 4,867(Е - 9)7*
11.11. Изоэнтропийное сжатие моноксида углерода
от 300 К и 10 атм до 50 атм протекает со скоростью
1 моль/с. Параметры уравнения Ван-дер-Ваальса, выра-
женные в атм, К, л/моль, имеют следующие значения:
а = 1,485, Ь = 0,03985. Теплоемкость идеального газа
С; = 6,79 + 0,0021 Т - 2,06(Е5)/7*
Определите температуру на выходе и изменение эн-
тальпии.
11.12. Монооксид углерода расширяется от 300 атм
при 500 К до 10 атм. Параметры уравнения Редлиха —
Квонга имеют следующие значения: а = 16,985, Ь =
= 0,0274. Использованные единицы: атм, К, л/моль.
Теплоемкость идеального газа дана в задаче 11.11.
Определите температуру на выходе.
11.13. Из чистых компонентов, температура, которых
равна 0 °C, необходимо получить эквимолярную смесь
вода + этанол при 80 °C.
а. Как изменится энтальпия системы (кал/моль), если
конечное давление по существу равно 1 атм?
б. Каким окажется дальнейшее изменение энтальпии
системы, если давление смеси повысится до 1000 атм,
а температура останется равной 80 °C?
Величины избыточной энтальпии, выраженные в виде
функции температуры, приведены ниже:
Г, °C 25 50 58 70 90 ПО
Яех, -407 -121,7 -18,5 129,2 378,5 598,7
Дж/моль
Теплоемкость этанола: 0,68 кал/моль • К; средний объ-
ем сжимаемости:
к = - 4т 4^ = 0,0001/атм,
V dp
коэффициент теплового расширения:
1 dV
= = °’ООО85/К-
Удельная масса смеси равна 0,86.
11 .14. При температуре 50 °C получают эквимоляр-
ную смесь воды и 1,4-диоксана. Определите соответ-
ствующие изменения энтальпии (каль/моль), используя:
а) уравнение Скэтчарда — Гильдебранда при
51 = 23,53, 52 = 10,13, Vi = 18 и У2 = 85,3;
б) уравнение Маргулеса при Ап = 1,7692, Л21 =
= 1,8814, допуская, что ТЧпу, = const;
в) уравнение Вильсона при Хи = 1811,85 и Х21 =
= -137,85 кал/моль;
г) уравнение NRTL при ст = 44, Agn = 825,83 и
Д#21 = 765,13 кал/моль.
11.15. Некоторое вещество описывается следую-
щим уравнением состояния: Р = 0,8277(И - 0,035) -
- 7,89/(И2 + 0,08 И - 0,0002), где единицы измерения:
атм, л/моль, К. Определите
а) энтальпию при 400 К и 30 атм относительно ее ве-
личины при 400 К и 1 атм;
б) Ср — Cv при 400 К и 30 атм.
Ниже даны некоторые решения вышеупомянутого урав-
нения состояния.
Р V (dV/dT)p
1 32,60 0,08265
15 1,898 0,006046
30 0,003758
11.16. Для некоторого газа при 500 К известны сле-
дующие данные: V = 1,0516 л/моль при Р = 10 атм и
V - 0,4132 л/моль при Р = 50 атм.
а. Найдите параметры уравнения Клаузиуса Р =
= RT/(V- b) - a/TV1.
б. Найдите изменения энтропии, внутренней энергии
и энтальпии при повышении давления от 10 до 50 атм
при температуре 500 К.
11.17. Изменение энтальпии в ходе реакции СО +
+ НгО «=* СНзОН равно - 50 880 кал/моль при 700 К и
нулевом давлении. Какова величина изменения энталь-
пии при 200 атм? Исходите из допущения о справедли-
Оценка изменений энтропии и энтальпии 539
вости уравнения Редлиха — Квонга и пренебрегите
изменением энтальпии смешения.
11.18. Покажите, что уравнение (12), приведенное в
табл. 11.3, можно записать следующим образом:
Д5'//? = -In [z(l - B/z)] + iln(l + B/z)
X , ЕУ|\Х/’(1 kij)"'/ Tri/(Xj + Trj/(Xy)].
» J
Расширьте программу, приведенную в примере 11.4А,
с тем чтобы можно было оценить Д5'.
11.19. Объясните, как влияет изменение температуры
от 300 до 600 К на ДЯ' жидкой и паровой фазы смеси,
рассматриваемой в примере 11.4А.
n4z4zjio#*nrm 'иллочуuvi чу гмопиосьил
Теоретически измерение фазового равновесия преду-
сматривает всего лишь измерение 1) давления, 2) темпе-
ратуры, 3) состава и 4) объемов фаз. Однако получить
на практике перечисленные данные достаточной точнос-
ти далеко не просто. Прежде всего необходимо убедить-
ся, что равновесие действительно существует, что тем-
пература и давление измеряются в момент существова-
ния равновесия и что отбор проб для анализа не
нарушает равновесия в существенной степени. Эти про-
блемы оказались в достаточной мере серьезными для
того, чтобы привести к разработке самого разнообраз-
ного оборудования и методик исследования нескольких
видов фазового равновесия. В этой главе будут описаны
наиболее типичные из них.
Изменение фазового состояния сопровождается изме-
нениями физических свойств, некоторые из них можно
измерить достаточно легко и с приемлемой точностью.
В ряде случаев с этой целью можно, например, прово-
дить измерение скорости звука или электропроводнос-
ти. Однако, как правило, изменение фазового состояния
устанавливается методами, относящимися к одной из
трех указанных ниже групп.
1. Визуальные методы. Пробу нагревают или охлаж-
дают в прозрачном сосуде и регистрируют резкие изме-
нения коэффициента рефракции, мутности, цвета или
образование капель или пузырьков.
2. Дилатометрический метод. Фазовые преобразова-
ния часто сопровождаются резким изменением объема
пробы. В качестве примера можно привести уретан, фа-
зовая диаграмма которого показана на рис. 5.10,6; при
1 атм удельный объем составляет 1,014 мл/г, а при
плавлении при температуре 47,9 °C объем возрастает
на 0,0599 мл/г. Как видно из табл. 12.1, фазовые пре-
вращения в трех тройных точках также сопровождают-
ся существенными изменениями удельного объема. Об-
зор многочисленных работ, посвященных дилатометри-
ческим методам, содержится, в частности, в книге
Даниэльса [30].
3. Термические методы исследования изменения тем-
пературы или теплопередачи во времени. В эту группу
входят три следующих метода.
а. Термический анализ (ТА), или метод кривых нагре-
вания или охлаждения, позволяет изучать изменение
температуры пробы во времени при приблизительно
постоянном подводе и отводе тепла.
б. Дифференциальный термический анализ (ДТА) поз-
воляет регистрировать изменение разности температур
Таблица 12.1. Изменения объема, сопровождающие фа-
зовые превращения уретана (см. рис. 5.10,6 и [73])
Давление, атм Темпера- тура, °C Фазовые превращения ДИ, мл/г
2350 66,2 I -» жидкость 0,0253
1 -► жидкость 0,0355
I - II - 0,0102
3400 25,5 I -*п - 0,00922
II - III 0,04820
I -»III 0,05742
4230 76,8 II -► жидкость 0,01840
III -► жидкость 0,06396
II - III 0,04556
пробы и эталона (который не претерпевает фазовых из-
менений) во времени в условиях подвода или отвода
тепла.
в. Дифференциальная сканирующая калориметрия
(ДСК), позволяет измерять изменение переноса энталь-
пии во времени, необходимое для поддержания одина-
ковых температур у пробы и эталонного образца. При
помощи этого метода можно определить не только
температуры, при которых происходят фазовые превра-
щения, но также и сопутствующие изменения эн-
тальпии.
Последние два метода, ДТА и ДСК, являются в выс-
шей степени точными, для их осуществления разработа-
но и выпускается промышленностью несколько типов
приборов. Эти методы применяются преимущественно
для изучения конденсированных фаз.
В этой главе не содержится описания способа прове-
дения измерений и осуществления контроля за темпера-
турой и давлением; для решения этих задач разработа-
но множество методик и специальных приборов — от
самых простых до весьма сложных. Эти вопросы под-
робно освещены в методиках лабораторных исследова-
ний, например в книге Вайсбергера и Росситера (1971),
а также Хала и др. [50]. В этих же книгах рассматрива-
ются методики измерения давления пара.
12.1. Растворимость в жидкостях
Не вдаваясь в тонкости можно сказать, что раствори-
мость определяют следующим образом: дают возмож-
Принципы экспериментальных методов исследования фазового равновесия 541
Емкость для
исследуемых
газовых проб
Бюретка- Вакуум
дозатор
Сосуд,в
котором
проводят
растворение
Бюретка с
делениями
Отбор
проб
жидкости
Уравнительный
сосуд
Жидкость под
давлением
Жидкость под
давлением
Рис. 12.1. Прибор для исследования растворения газа в жидкости и измерения количества
растворенного газа. Количество газа, вводимого в сосуд, в котором проводится растворе-
ние, определяют, измеряя начальное и конечное давление и уровень ртути в бюретке-
дозаторе. Количество газа, оставшегося нерастворенным в сосуде известного объема, опре-
деляют, устанавливая объем, занимаемый газом в уравнительном сосуде (бюретка с деле-
ниями), после выравнивания уровня ртути.
Вакуум
Газ
Раствор
Рис. 12.2. Прибор для определения количества раство-
ренного газа.
Раствор вводят в предварительно вакуумированную бю-
ретку, жидкость откачивают и газ вновь сжимают до
некоторой требуемой величины, с тем чтобы можно
было найти его массу по соответствующим делениям,
нанесенным на бюретку.
ность растворенному веществу и растворителю контак-
тировать друг с другом вплоть до достижения равнове-
сия, после чего соответствующими методами измеряют
количества фаз и их состав. Достижение равновесия
ускоряют механическим перемешиванием, встряхивани-
ем или вибрацией сосуда, либо циркуляцией одной фазы
через другую посредством перекачивания. Необходимая
длительность контактирования может составлять от
всего лишь нескольких минут до нескольких недель (для
вязких жидкостей и для труднорастворимых веществ).
Убедиться в том, что состояние равновесия достигнуто,
можно следующими способами: 1) приближаясь к этому
состоянию с двух противоположных сторон — либо от
состояния пересыщения, либо от состояния недосыще-
ния, если фактическая температура намного отличается
от требуемой; 2) периодически отбирая пробы раство-
ра, пока не будет установлено стационарное состояние.
Следует исключить возможность установления мета-
стабильного равновесия. Например, вероятность пере-
сыщения более-велика при удалении растворенного ве-
щества из раствора, чем при его растворении. Если рас-
творимость определяют, понижая температуру
раствора, следует выполнить несколько экспериментов
с различной скоростью охлаждения. При достаточно
низкой скорости охлаждения и перемешивании пересы-
щения не происходит.
Газы. Со времени проведения первых исследований
по растворимости газов, Генри (1803), Бунзен (1853) и
Оствальд (1890), используемые в этих целях методики
претерпели весьма заметные изменения. Рабочие давле-
ние и температура и, конечно, природа растворенного
вещества и растворителя, а также требуемая точность
определяют выбор методики и того или иного экспери-
ментального оборудования.
На рис. 12.1 представлена принципиальная схема при-
бора, позволяющего определять растворимость газа в
жидкости. Определенную навеску вещества-раствори-
теля помещают в сосуд, в котором проводят операцию
растворения, а затем в него подают растворяемый газ
до достижения требуемого давления и обеспечивают его
циркуляцию при помощи высокоскоростного побудите-
ля газа. Любые посторонние газы, которые могли рас-
твориться в растворителе, удаляют посредством пред-
варительного вакуумирования растворителя. В процессе
насыщения растворителя газом давление последнего
поддерживают постоянным, регулируя его подачу. По-
сле завершения насыщения определяют изменение объ-
ема в градуированной трубке и, применяя соответ-
ствующее уравнение состояния, рассчитывают количе-
ство растворенного газа. Если растворитель по своей
природе нелетуч, небольшую пробу насыщенного рас-
твора отсасывают непосредственно в бюретку вакуум-
ным насосом, как это показано на рис. 12.2. После ис-
парения газа из раствора жидкость сливают из бюрет-
ки, а оставшийся газ сжимают до атмосферного
давления с помощью ртутного столба через уравнитель-
ную воронку и фиксируют его объем.
Используемые в этом случае приборы должны иметь
достаточную термическую и механическую прочность и
должны обеспечивать возможность интенсивного пере-
мешивания, необходимого, в частности, для исследова-
ния несмешивающихся жидкостей. Большое число ра-
бот, посвященных этой теме, приводится в книге под
редакцией Дэка [29], а также в книге Вайсбергера и Рос-
ситера (1971).
Жидкости. Взаимную растворимость двух жидкостей
определяют следующим образом: необходимые количе-
ства исследуемых жидкостей, предварительно тщатель-
но взвешенные, перемешивают до достижения равнове-
сия, после этого разделяют фазы и определяют их ко-
личество и состав. В этих целях можно использовать
целый ряд аналитических методов: химический анализ,
хроматографию, рефрактометрию или денситометрию,
если измеряемые характеристики являются функциями
состава фаз. При наличии тенденции к эмульгированию
перемешивание следует выполнять осторожно, так что
анализ может потребовать нескольких часов.
Чтобы установить растворимости в некотором интер-
вале температур, приготавливают растворы известного
состава при повышенных температурах и медленно их
охлаждают, отмечая температуру, при которой проис-
ходит помутнение изотропной системы. В тех случаях,
когда точку помутнения трудно обнаружить даже с ис-
пользованием фотоэлектрических средств, в раствор
вводят микродозу красителя, растворимого главным
образом в осаждающеся фазе.
Предложенная Хиллом [346] методика позволяет ис-
ключить определение количества фаз. В соответствии с
методикой Хилла одновременно выполняют два экспе-
римента с различным соотношением двух жидкостей,
контактирующих с одной и той же паровой фазой.
В таких случаях составы двух жидкостей не зависят от
соотношений фаз, поскольку они находятся в равнове-
сии с одной и той же газовой фазой. Применяя следую-
щие условные обозначения:
т\ — масса компонента I в эксперименте 1,
тг — масса компонента 1 в эксперименте 2,
Фильтрую-
щая трубка
Отбор проб
раствора
В
Капилляр-
ная трубка
Рис. 12.3. Прибор для определения растворимости
твердого вещества.
Растворение выполняется в камере А. После заверше-
ния растворения прибор переворачивают и из камеры
В отбирают пробы отфильтрованного раствора.
х — концентрация компонента 1 в нижней фазе,
у — концентрация компонента 1 в верхней фазе,
L\ — объем нижней фазы в эксперименте 1,
Vi — объем верхней фазы в эксперименте 1,
L2 — объем нижней фазы в эксперименте 2,
V2 — объем верхней фазы в эксперименте 2,
записываем уравнения материального баланса по перво-
му компоненту:
mi = Lix + Viy,
т2 = L2x + V2y,
которые легко разрешить относительно составов фаз х
и у. Аналогичную систему уравнений записывают и ре-
шают для компонента 2 или же количество компонента
2 рассчитывают из х и у и плотностей обеих фаз.
Твердые вещества. Если высокая степень точности не
обязательна, а давление и температура близки к атмо-
сферным, для измерения растворимости твердых тел в
жидкостях пригодны достаточно простые методы и
приборы. Две фазы встряхивают вместе до достижения
равновесия, после декантации или фильтрования отби-
рают пробу жидкой фазы, которую далее анализируют
химическими методами или проводят ее испарение.
Другой широко распространенный метод заключается в
постепенном охлаждении горячего концентрированного
раствора с регистрацией температуры, при которой
происходит осаждение.
Отбор проб при предельных температурах и давлени-
ях осуществляется различными способами, некоторые
Принципы экспериментальных методов исследования фазового равновесия 543
Рис. 12.4. Схема эбулиометра.
А — куб, Б — насос Котрела, В — карман для термопа-
ры, который омывается парожидкостной смесью, пода-
ваемой насосом Котрела, Г — конденсатор, охлаждае-
мый хладагентом при - 20°С (Thomas et al., 1982).
(© Am. Chem. Soc.)
1 — стеклянные шарики размером 30 меш, припаянные
изнутри, 2 — 2-мм стержень, припаянный к дну карма-
на для термопары и пробке; 3 — защитная пробка; 4
— патрубок для подсоединения к этиленгликолевой ван-
не, 5 — змеевик диаметром 6 мм, 6 — тефлоновый за-
порный кран диаметром 8 мм, 7 — вакуумная муфта.
^6 мм (внешний
, диаметр)г18 витков
' ._Конденсатор ,
— 42 мм (внешний
32 диам.)
’ Овод пробы —
лярная трубка
— Карман для
термопары
*_6 мм (внешн.
л диаметр)
Насос
Котрела
$
f
155 см
*-Куб
•—Нихромовая
проволока
4-мм тефлоновый
запорный кран
Конденсатор
9мм (внешн.диаметр)
20 мм (внешн.диаметр)
Затвор
Спирали из стеклян-
ных шариков
.Трубка для возврата
охлажд. конденсата
6 мм (внешн.диаметр)
28мм (внешн. диаметр)
Рис. 12.5. Дифференциальный эбулиометр для измерения температур кипения разбавленных растворов.
Одну из ячеек заполняют чистым растворителем, другую — разбавленным раствором. Точность измерения разно-
стей температур составляет 0,001 К. Конструкция усовершенствованного эбулиометра показана на рис. 12.4 [260].
из них описаны Вайсбергером и Росситером [6]. Как
правило, посредством специальных приспособлений от-
бирают пробу надосадочной жидкости и помещают ее
в термостат, с тем чтобы температура пробы соответ-
ствовала требуемой для проведения анализа. Если раз-
деление фаз происходит с трудом и необходимо филь-
трование, можно использовать двухкамерный прибор
типа показанного на рис. 12.3. После насыщения рас-
твора сосуд переворачивают и раствор отфильтровыва-
ют через трубку, в то время как газ просачивается из
камеры В в камеру А по капилляру.
Если исследуемые вещества труднорастворимы, для
достижения равновесия иногда требуются дни и недели;
анализ получаемых в результате разбавленных раство-
ров может потребовать особых методов, отвечающих
особенностям конкретных систем, например, в ряде слу-
чаев измеряется электропроводность растворов. В каче-
стве общего метода исследования разбавленных систем
Митчел [487] предлагает интерферометрию.
12.2. Температура кипения и давление пара
Несмотря на простоту метода измерения температу-
ры кипения путем считывания показаний термометра,
помещенного в паровую или жидкую фазу, его исполь-
зование может привести к значительным погрешно-
стям. Поверхностное натяжение и гидростатический на-
пор вызывают перегрев, в то время как небольшие ко-
личества летучих примесей, например воды, следы
которой часто присутствуют в органических жидкостях,
снижают показания термометра в паровой фазе, кото-
рый показывает температуру конденсации смеси, а не
чистого пара.
В настоящее время при конструировании наиболее эф-
фективных эбулиометров применяется принцип Котре-
ла: вокруг термометра, находящегося в паровой фазе
(рис. 12.4 и 12.5) циркулирует смесь пузырьков и захва-
ченной жидкости. Эту основную конструкцию совер-
шенствуют при помощи устройств, предназначенных
для стока жидкости от термометра и регулирования
скорости испарения, которую в целях точного определе-
ния температуры кипения следует поддерживать в опре-
деленном интервале значений, зависящем до некоторой
степени от природы жидкости. Особенности конструк-
ций, используемых для анализа гетерогенных смесей,
изложены в книге [50].
Эбулиометры, предназначенные для функционирова-
ния в некотором интервале давлений, используют для
определения давлений пара. Давление пара при задан-
ной температуре можно определить посредством изоте-
нископа [647]. Как показано на рис. 12.6, сосуд для про-
бы и присоединенную к нему U-образную трубку ча-
стично заполняют исследуемой жидкостью. Далее
жидкость доводят до кипения, чтобы удалить из нее все
растворенные газы, и прибор термостатируют. После
этого, изменяя высоту столба ртути, регулируют давле-
ние так, чтобы началось образование пузырьков. Мо-
мент начала образования пузырьков желательно опре-
делять не визуально, а при помощи соответствующих
приборов.
Рис. 12.6. Измерение давления пара изотенископом Смита и Мензиса [647]. Баллон изотениско-
па и U-образная манометрическая трубка частично заполнены исследуемой жидкостью. Пробу
жидкости доводят до кипения, чтобы удалить весь растворенный газ, после чего температуру
понижают до требуемой величины. Наконец, внешнее давление регулируют до выравнивания
жидкости в U-образной трубке. В этот момент давление в приборе равно давлению пара веще-
ства в баллоне изотенископа.
Принципы экспериментальных методов исследования фазового равновесия 545
12.3. Равновесие пар — жидкость
Приборы с циркуляцией паровой фазы. Прибор, пред-
ложенный много лет назад Отмером (рис. 12.7) для на-
хождения равновесных составов жидкости и пара, дает
достаточно точные результаты. В сосуде А постоянно
образуется пар, который далее конденсируется и собира-
ется в приемнике В. Избыток конденсата возвращается
в куб. Когда показания термометра, размещенного в
паровой фазе, укажут на установление стационарного
режима, отбирают пробы кипящей жидкости и конден-
сата и анализируют их.
Поскольку, как указывалось при описании принципа
действия эбулиометров, показания термометра по ряду
Рис. 12.7. Прибо Отмера для определения парожидкост-
ного равновесия, использующий рециркуляцию конден-
сата до момента установления равновесия [535].
А — перегонный куб; В — паровая трубка для предот-
вращения конденсации и ректификации пара на термо-
метре; С — паровая рубашка; D — холодильник; Е —
приемник конденсата; G — присоединение к маностату;
F — затвор; J — кран для отбора пробы конденсата;
К — кран для отбора пробы жидкости; L — загрузоч-
ный кран, посредством которого в приборе можно со-
здавать избыточное давление (первоначально прибор
был предназначен для работы только при атмосферном
давлении); М — термометр; N — крепление прибора;
О — отверстие для выхода пара; Р — счетчик капель.
причин могут оказаться неточными, в современных
приборах такого типа обычно используется насос Кот-
рела. Первым из приборов такого типа был перегон-
ный куб Джиллеспи [306], показанный на рис. 12.8. По
своему действию он аналогичен перегонному кубу От-
мера, но термометр в нем омывается смесью пузырьков
и схваченной жидкости.
За многие годы в конструкцию перегонного куба для
определения равновесия пар — жидкость был внесен це-
лый ряд усовершенствований или же предпринимались
попытки их внести в целях повышения точности, упро-
щения работы с прибором, в целях приспособления его
для анализа определенных типов смесей или двухфаз-
ных жидкостей, а также для функционирования в усло-
виях предельных давлений и температур. В книге Хала
и др. [50] приводятся схемы примерно тридцати различ-
ных типов конструкций такого прибора.
Статические методы. Простой по своей сути метод
определения равновесия пар — жидкость состоит в сле-
дующем: исследуемую смесь выдерживают при таких
температуре и давлении, при которых паровая и жидкая
фазы сосуществуют, и тщательно их перемешивают до
достижения равновесия, далее отбирают пробы двух
фаз и анализируют их, стараясь не нарушать сколько-
нибудь заметным образом существующего равновесия.
Одним из затруднений, которые могут возникнуть при
низких или умеренных давлениях, является значитель-
ная масса пробы пара, необходимая для выполнения
анализа, в момент отбора которой равновесие может
заметно нарушиться. Поэтому при использовании обо-
рудования этого типа желательно применять для анали-
за хроматографию или масс-спектрометрию, так как в
этом случае объем пробы может составлять всего 1 мл
или около того.
В одной из рабочих конструкций для отбора малых
проб применяются пробковые краны, снабженные отсе-
кателями объемом 1 мл (или около этого), которые раз-
мещены на лицевой стороне крана. До момента дости-
жения равновесия клапан обращен к ячейке своей лице-
вой стороной, в нужный момент его поворачивают на
180°, так чтобы захваченная проба могла поступить в
хроматограф. При повышенных давлениях пробы по-
добного объема также могут быть достаточны для ана-
лиза при помощи других методов.
Для нахождения коэффициентов активности при бе-
сконечном разбавлении можно использовать дифферен-
циальный эбулиометр, позволяющий измерять темпера-
туры очень разбавленных растворов. В приборе, пока-
занном на рис. 12.5, один из эбулиометров содержит
чистый растворитель, другой — разбавленный раствор.
Этот прибор позволяет измерить разности температур
с точностью в пределах 0,001 К и определить воздейст-
вие концентраций порядка 0,005 мольных долей. Следу-
ет заметить, что для измерения коэффициентов актив-
ности растворителей или пар растворителей в жидко-
стях с большой удельной массой применяются методы
газовой хроматографии. Один из приборов данного ти-
па изображен на рис. 12.9. Подробный обзор такого ро-
да экспериментальных методов выполнен Летчером
[437], его дополняет работа Экерта и др. [260], опубли-
кованная позднее.
Чтобы результаты определений, проводимых при по-
вышенных давлениях или низких температурах, были
16-864
Рис. 12.8. Прибор Джиллеспи для определения паро-
жидкостного равновесия с рециркуляцией обеих фаз
[306].
А — куб; В — насос Котрела; С — термометр; D —
сепаратор; Е — трубка возврата уносимой жидкости;
F — конденсатор; G — приемник конденсата; Н — ли-
ния возврата избытка конденсата; К — нагреватель.
достаточно точными, необходимы более сложные при-
боры. Рабочие ячейки для исследования равновесия
обычно имеют окошки, позволяющие визуально опреде-
лять температуру начала кипения или точку росы. На
рис. 12.10 и 12.11 показаны два типичных варианта та-
ких приборов. Исследуемые вещества сначала помеща-
ют в дозатор при давлении, равном давлению его па-
ров, или выдавливают в дозатор ртутью. Ртуть переме-
щается по точно отградуированным трубкам, так что,
зная разность уровней ртути и плотность жидкости,
можно рассчитать массу жидкости, поступившей в до-
затор. Перемешивание осуществляется магнитной ме-
шалкой. Растворенные в жидкости газообразные приме-
си удаляют из нее в дозаторе. Необходимая температу-
ра достигается термостатированием, а необходимое
давление — поршневым насосом. После установления
равновесия для проведения анализа отбирают пробы
только жидкой фазы или обеих фаз.
Показанная на рис. 12.10 установка, разработанная
Шоттом, предназначена для изучения равновесия при
давлениях в пределах 55—70 атм. В этой установке ис-
пользуется ячейка размером 10 х 15 см. Точность изме-
рения составляет 0,4 мл/компонент, точность измере-
ния температуры находится в пределах 0,2 °C, давле-
ния — в пределах 0,8%, а точность определения состава
пара — в пределах 1—2%. Как указывает, автор, сум-
марная погрешность типичной группы измерений и ко-
эффициента активности, выведенного из наиболее точ-
ных корреляций, составляет максимум 7%, а зачастую
не превышает 5%. Некоторые детали конструкции ра-
бочих ячеек, применяемых другими исследователями
Ввод
пробы
1
Колонка
к- Термостат
[Рубашка для
I обогрева J7
'I Детектор"^
Насосы
для подачи
раствори-
теля
Баллон с
гелием
П ж Выходной
сигнал
, Счетчик
мыльных
пузырей
Ловушка для
растворителя
Выход
гелия
Рис. 12.9. Газовый хроматограф для измерения коэффи-
циентов активности бесконечного разбавления (Diner et
al., 1981).
Принципы экспериментальных методов исследования фазового равновесия 547
Баллон
с гелием
Мано-
метр
Хайзе
Вакуум
‘ Датчик
—z ♦ Датчик
Бюретка
н9
н9
Нд
Масляная'
[Рабочая
____| ячейка
Поршневой
э насос
К абсорберу
Рис. 12.10. Установка для измерения парожидкостного
равновесия при повышенных давлениях и средних тем-
пературах [620].
при давлениях до 100 бар, показаны на рис. 12.12.
Изучение зависимости давление—состав. Рабочие
ячейки для изучения равновесия статическим методом,
подобные показанным на рис. 12.10—12.12, особенно
эффективны для нахождения давления, соответствую-
щего температуре начала кипения и точке росы при ре-
гулируемой температуре и известных суммарных соста-
вах. При помощи методов, описанных в разд. 4.17.4,
исходя из результатов таких измерений, можно полу-
чить данные о парожидкостном равновесии. Данный
способ делает ненужными такие сложные и вносящие
известную неопределенность процедуры, как отбор од-
ной или обеих фаз и последующий ее анализ. И хотя,
решая вопрос о приемлемой форме корреляционных
уравнений для фугитивности и коэффициентов актив-
ности, таких, как вириальные уравнения и уравнения
Вильсона, приходится принимать определенные допу-
щения, получаемые результаты отличаются высокой
точностью.
Если давления составляют несколько десятков атмо-
сфер, можно использовать стеклянные ячейки. При
этом за образованием пузырьков или капель можно на-
блюдать визуально, но в случае необходимости можно
прибегнуть и к фотоэлектрическим методам детектиро-
вания. Число возможных вариантов конструкций и
принципов проведения статических определений весьма
велико. Исследуемые вещества подаются в рабочую
Электрический зуммер
0-13,8 МПа
Сосуд Дьюара
Электрич.
зуммер
Газовый
баллон
Ртутный
насос
Рабочая
ячейка
('—-"'JKud-
0-34,6 МПа
кость
Пар
насос
4х-^Тяга
Вакуум-насос
X
О ~ 51,8.МПа о-гтзмпа
Проба Проба
жидкой паровой
фазы
фазы
Ртутный насос
(поршневой)
Рис. 12.11. Установка для измерения парожидкостного
равновесия при повышенных давлениях и средних тем-
пературах [313].
16*
Рис. 12.13. Схемы приборов, используемых для трех основных типов термического анализа.
а — традиционный термический анализ: температура исследуемой пробы регистрируется по мере того, как меняет-
ся (с постоянной скоростью) температура печи; б — дифференциальный термический анализ; регистрируется раз-
ность температур исследуемой и эталонной проб, по мере того как меняется (с постоянной скоростью) температура
печи; в — дифференциальная сканирующая калориметрия: измеряется скорость теплопередачи к исследуемой пробе,
необходимой для поддержания нулевой разности температур между исследуемой и эталонной пробами, в то время
как температура эталонной пробы меняется с постоянной скоростью.
Принципы экспериментальных методов исследования фазового равновесия 549
ячейку для исследования равновесия путем вытеснения
ртутью или с помощью насосов. Жидкости также мож-
но загружать во взвешенных закрытых ампулах, кото-
рые вскрывают в рабочей ячейке каплей, управляемой
магнитом. Эта методика была применена авторами ра-
бот [453 , 544]. Среди многочисленных инструменталь-
ных методов, предназначенных для определения темпе-
ратуры начала кипения и точки росы, можно отметить
дифференциальный термический анализ (ДТА), который
получил очень широкое распространение при изучении
фазовых переходов; для проведения ДТА разработаны
н выпускаются промышленностью приборы различной
конструкции.
12.4. Равновесие твердое вещество —
жидкость (равновесие плавления)
I Определение температур плавления чистых веществ в
i целях их идентификации является обычной лаборатор-
j ной процедурой, для проведения которой выпускается
I множество промышленных приборов. В большинстве
* из них проба твердого вещества помещается на поверх-
ность с регулируемой температурой. Температуру плав-
ления определяют визуально при увеличении или же, ес-
; ли прибор полностью автоматизирован, при помощи
фотоэлектрических устройств.
Однако такие приборы обычно непригодны для ана-
лиза смесей, так как плавление смесей происходит в не-
котором интервале температур (исключение составляют
эвтектики), а поэтому начало и конец плавления нельзя
наблюдать сколько-нибудь точно. Более того, при ис-
следовании равновесия жидкость — твердое вещество
может возникнуть необходимость в выявлении других
типов переходов, например происходящих полностью в
твердом состоянии и приводящих к образованию меж-
молекулярных соединений.
Поскольку фазовые переходы и растворение обычно
сопровождаются существенными тепловыми эффекта-
ми, измерение последних позволит изучить изменение
фазового состояния. Если тепловые эффекты малы или
плохо поддаются толкованию в виду сложности систе-
мы, можно прибегнуть к ряду других методик, допол-
няющих или подтверждающих результаты измерений.
В зависимости от особенностей конкретной системы
можно проследить за изменениями плотности, электро-
проводности, магнитных свойств или скорости звука
или же сравнить дифракцию рентгеновских лучей.
Однако, поскольку наиболее широкое применение полу-
чили термические методы, ограничимся лишь их рас-
смотрением.
Метод кривых нагревания или охлаждения. Эти кри-
вые характеризуют изменение во времени температуры
пробы, помещенной в печь, температуру которой посте-
пенно меняют, причем лучше, чтобы эти изменения
имели линейный характер. Схема такого нагревательно-
го прибора показана на рис. 12.13,а; для охлаждения ис-
пользуется поток газа. В современном оборудовании
для термического анализа применяются небольшие про-
бы, 5—100 мг, а скорость изменения температуры мо-
жет достигать 2—10 ° С/мин.
Если происходящие фазовые изменения не сопровож-
дются сколько-нибудь существенным тепловым эффек-
том, температура пробы меняется в соответствии с
температурой печи, в противном случае на кривой из-
менения температуры наблюдается резкий излом. На
рис. 12.14,а показана гипотетическая кривая охлаждения
чистого вещества с постоянной теплоемкостью; темпе-
ратура горизонтального участка — это точка замер-
зания. Аналогичная кривая, полученная для бензола
(рис. 12.14,6), указывает на мгновенное переохлаждение
жидкой фазы. Следует заметить, что отрезок прямой,
соответствующий периоду замерзания бензола, не впол-
не горизонтален. Причем такое отклонение наблюдает-
ся даже при самом тщательном выполнении экспери-
мента и использовании самого совершенного оборудо-
вания. На графике, полученном для эвтектической
системы (рис. 12.14,6), бензойная кислота замерзает в
точке А, смесь, содержащая 20% коричной кислоты, на-
чинает замерзать в точке В, а самая низкая точка замер-
зания любой смеси — это точка образования эвтектики
(точка С). Длина горизонтальных смещений темпера-
турных кривых пропорциональна изменениям энталь-
пии фазовых переходов.
На рис. 12.14,0 и 12.15,6 показаны кривые нагревания
и охлаждения систем. При данной скорости изменений
температуры печи оба типа кривых должны быть, как
это и показано, идентичны, однако вероятность появле-
ния погрешностей более велика для кривых охлаждения
ввиду свойственной всем системам более сильной тен-
денции к переохлаждению, чем к перегреву. Соответ-
ственно при построении кривых охлаждения предпочти-
тельны пробы меньших размеров и более низкие ско-
рости изменений температуры. Процессы переходов в
твердом состоянии являются особенно вялыми, а соот-
ветствующие тепловые эффекты незначительными, поэ-
тому их определение следует проводить особенно тща-
тельно.
Для построения фазовых диаграмм сложных систем,
как показано, например, на рис. 12.15, может потребо-
ваться большое количество температурных кривых.
Однако даже при достаточном количестве таких кривых
интерпретация данных может оказаться затруднитель-
ной, вследствие чего может потребоваться использова-
ние других дополнительных методов, как это отмеча-
лось в начале данного раздела.
Дифференциальный термический анализ (ДТА). Это
один из более чувствительных, чем прямой термический
анализ, методов анализа, для проведения которого раз-
работаны и выпускаются промышленностью необходи-
мые приборы. Данный метод был разработан пример-
но в то же время, что и метод кривых нагревания и
охлаждения (Ле Шателье, 1887; Робертс-Остен, 1899),
однако в последние годы он был значительно усовер-
шенствован ввиду усложнения решаемых таким спосо-
бом задач и оживления интереса к термическим мето-
дам анализа вообще. Схема прибора для ДТА показана
на рис. 12.13,6. Прибор такого типа позволяет измерять
разность температур пробы исследуемого материала и
эталона, который не подвергается фазовым переходам
в интересующем исследователя интервале температур.
Масса пробы составляет несколько миллиграмм, темпе-
ратуру печи изменяют равномерно со скоростью 2—
10 °C. Результаты измерений изображают в виде графи-
ка зависимости разности температур от температуры эта-
лона, которая по существу является температурой печи.
Рис. 12.14. Кривые охлаждения/нагревания некоторых простых систем.
а — кривая охлаждения чистого вещества с постоянной теплоемкостью; б — кривая охлаждения для определения
температуры замерзания бензола, деления температурной шкалы составляют 0,1 °C, следовательно, погрешность
равна примерно 0,3 °C; в — изменение температуры, наблюдаемое при образовании эвтектики; г — фазовая диа-
грамма и изменение температуры системы с полностью растворимыми жидкой и твердой фазами;
д — простая эвтектическая система бензойная кислота + коричная кислота.
Рис. 12.15. Кривые охлаждения/нагревания более слож-
ных систем, а — кривые охлаждения системы, образую-
щей межмолекулярное соединение, стабильное в точке
кипения; б — перитектическая система уксусная кисло-
та + 3-диметилпиррон.
Рис. 12.16. Дифференциальный термический анализ
(ДТА) анизалдина. На полученной термограмме видны
точка плавления (?) и переход из жидкого состояния в
кристаллическое (2).
Рис. 12.17. Бинарная эвтектическая система, ее кривая охлаждения и дифференциальная термограмма при исходном
составе w.
Понижение температуры
Рис. 12.18. Гипотетическая бинарная система А + В и ее дифференциальные термограммы
при восьми различных составах [760]. (Подстрочный индекс SS — раствор твердого ве-
щества.)
Изменение энтальпии
Рис. 12.19. Фазовая диаграмма системы стиль-
бен + трифенилметан, построенная по резуль-
татам измерений, проведенных с использовани-
ем дифференциального сканирующего калори-
метра фирмы Perkin-Elmer.
а — две термограммы; наблюдаемые на каж-
дой кривой два пика соответствуют началу
плавления и эвтектической температуре, ско-
рость нагревания 5°С/мин, б — полная фазо-
вая диаграмма, построенная после нескольких
часов работы прибора.
Принципы экспериментальных методов исследования фазового равновесия 553
При наличии фазового перехода на таком графике на-
блюдается пик, как это показано на рис. 12.16 на приме-
ре анизалдина, для которого характерны два фазовых
перехода. Если полученный пик достаточно узок, а та-
кие пики обычно получают при низкой скорости измене-
ния температуры, то он легко обнаруживается и указы-
вает по существу температуру перехода. Поскольку ха-
рактеристики приборов могут сказываться на
интерпретации графиков, принято предварительно про-
водить градуировку приборов при помощи веществ,
температуры переходов которых точно известны.
Несмотря на то что форма пика на кривой, получае-
мой при проведении ДТА, зависит от скорости нагрева-
ния, размера пробы и градиента температуры, его пло-
щадь всегда пропорциональна тепловому эффекту пере-
хода. Количественные данные можно получить после
градуировки прибора по стандартным веществам.
На рис. 12.17 и 12.18 показаны кривые, полученные
при проведении дифференциального термического ана-
лиза (ДТА) простой и сравнительно сложной систем.
На некоторых кривых наблюдается до трех пиков.
Дифференциальный термический анализ (ДТА) и диф-
ференциальная сканирующая калориметрия (ДСК) ши-
роко используются для идентификации веществ и опре-
деления степени их чистоты. Термограммы многих со-
единений собраны в каталоги (например, каталоги,
составленные Сэдтлерскими исследовательскими лабо-
раториями).
Дифференциальная сканирующая калориметрия
(ДСК). В термических методах величины тепловых эф-
фектов фазовых переходов оценивают путем интегриро-
вания площади пика, учитывая также скорость нагрева-
ния. Некоторые выпускаемые промышленностью при-
боры для ДТА снабжены встроенными интеграторами,
что весьма удобно при проведении рутинных анализов.
Приборы для ДСК разработаны фирмой Perkin-Elmer
Со. (1964). Как видно из рис. 12.13,в, подвод тепла к
исследуемой пробе и эталону осуществляется раздельно.
Температура эталона изменяется равномерно с одной и
той же скоростью, в то время как подвод тепла к иссле-
дуемой пробе изменяется, с тем чтобы поддерживать
нулевую разность температур. Скорость подачи энер-
гии, требуемой для этого, регистрируется как функция
температуры пробы и опосредованная функция време-
ни, поскольку скорость нагревания также известна. Теп-
ловой эффект перехода определяют интегрированием,
для чего приборы для ДСК имеют специальные встро-
енные устройства.
Использование как ДТА, так и ДСК дает одинаковые
конечные результаты, хотя по некоторым утверждени-
ям последний метод является более точным. Термо-
граммы, полученные в стандартных условиях, являются
в высшей степени характерными; опубликованы катало-
ги обоих видов термограмм. Эти методы особенно эф-
фективны для выявления примесей в чистых веществах
и для снятия характеристик смесей и полимеров. ДСК
применяется для определения диаграмм плавления (см.,
например, рис. 12.19). Масса требуемой пробы измеря-
ется в миллиграммах, а для построения простой диа-
граммы, подобной показанной на рис. 12.19, и для
определения изменений энтальпии при переходе доста-
точно нескольких часов работы.
12.5. Обзоры экспериментальных методов
изучения фазового равновесия
Ниже дается перечень книг, содержащих информацию
по обсуждаемой проблеме, а также справка относитель-
но соответствующих журнальных статей.
X.DackM.R.J. ed. Solutions and solubilities, Part 1,
Chap. 7: The solubility of gases in liquids, 1976.
2. Hala E., Pick J., Fried V., Vilim O. Vapor-liquid equi-
librium, 1967.
3. Ze Neindre B., Vodar B. eds. Experimental thermo-
dynamics, Vol. 2: Experimental thermodynamics of non-
reacting fluds, Chap. 6: Meassurement of PVT properties
of gases and gas mixtures at low pressures; Chap. 8: PVT
relationships in gases at high pressures and high tempera-
tures; Chap. 16: Phase equilibria of liquid-gas mixtures;
Chap. 17: Liquid-solid phase equilibria, 1975.
4. McGlashan M. L. Chemical thermodynamics, Vol. 2: A
specialist periodical report, Chap. 3: Experimental
methods for studying phase behavior of mixtures at high
temperatures and pressures, 1978.
5. Pope M. I., Judd M. D. Differential thermal analysis,
1977.
6. Weissberger A., Rossiter B. W., eds., Physical methods of
chemistry, Part V: Determination of thermodynamic and
surface properties, 1971.
7. Wendlandt W. W. Thermal methods of analysis, 1976.
Приложение А
Основные формулы термодинамики
Несмотря на то что систематическими подборками
термодинамических формул, в которых не указан спо-
соб их вывода и ограничения применимости, следует
пользоваться с осторожностью, они представляют
определенную ценность для начинающих. И хотя пол-
ной подборки таких формул, по-видимому, не существу-
ет, в перечисленных ниже изданиях содержится много
таблиц, используемых при рассмотрении тех или иных
вопросов.
1. Bridgman Р. W. A Condensed Collection oL
Thermodynamic Formulas. Harvard University Press (1925).
Содержит первые и вторые производные прочих термо-
динамических свойств, выраженные через Р, V, Т, S, Ср
и их производные (см. табл. А.7).
2. Goranson R. W. Thermodynamic Relations in
Multicomponent Systems. Carnegie Institution of Wa-
shington (1930).
3. Shaw A. N. The derivation of thermodynamical
relations for a simple system. Phil. Trans. Roy. Soc.
(London), A234, 299—328 (1935).
Содержит первые и вторые производные для одноком-
понентных систем. Изложенный метод отличается
большей гибкостью, чем метод Бриджмена, поскольку
предполагает выражение любых производных через
другие свойства, помимо Р, V, Т, S и Ср. Процедуры
получения первых производных представлены также в
книге: Sherwood Т К., Reed С. Е. Mathematics in Chemi-
cal Engineering, McGraw-Hill (1939) и статье Carrol J.
Chem. Education, 42(4), 218—221 (1965).
4. Thnnell G. Relations between Intensive
Thermodynamic Quantities and their First Derivatives in a
Binary System of One Phase. W. H. Freeman and Company
(1961).
5. Sage В. H. Thermodynamics of Multicomponent
Systems. Reinhold Publishing Corporation (1965).
В приложении к книге приведены перечни формул для
систем переменной массы и состава, подобные форму-
лам Бриджмена.
6. Van Ness Н. С, Abbott М. М. Classical Thermody-
namics of Non-Electrolyte Solutions. McGraw-Hill (1982).
В тексте книги и приложениях приведены таблицы фор-
мул для смесей.
Специальные таблицы формул термодинамики содер-
жатся также в тексте настоящей книги, в частности в
гл. 2, 3, 4 и 11.
Таблица А.1. Определения и условные обозначения тер-
модинамических свойств
Термодинамическое свойство Услов- ное обоз- начение Определение
Теплота Внутренняя энергия Энтропия Энтальпия (теплосодержа- ние, функция тепла, сум- марная теплота) Функция Гельмгольца (свободная энергия и функция работы) Функция Гиббса (свобод- ная энергия, свободная энтальпия, термодинами- ческий потенциал) Объемное расширение (коэффициент объемного расширения) Изотермический объем- ный модуль упругости Адиабатический объемный модуль упругости Изотермическая сжима- емость Адиабатическая сжима- емость Теплоемкость при посто- Q и S н G В Bs кт ks U+ PV и-TS H-TS 1 V \дТ/Р / ЭР\ - Н — I \дУJ т ЛА - к( — 1 уЭК/s 1 1дУ\ V \дРJт 1 ( Э(| V \dPjs / / ds \
янном объеме Теплоемкость при посто- Cv ( — 1 = Т| — 1 XdTlv \дТ / v ( Э/Л /
янном давлении Соотношение теплоем- костей Коэффициент Джоуля Коэффициент Джоуля— Томсона (Кельвина) Функция Массье Функция Планка Ср 7 Ч М Ф 1 — 1 = т( — 1 \дТ/Р \дТ/Р Ср Cv ( дТ\ \ду)и (дт\ \дР A U - = --S Т Т G Н Т Т
Таблица А.2. Некоторые уравнения термодинамики
Приложения 555
1.
2.
3.
4.
5.
6.
7.
8.
9.
10.
11.
12.
13.
14.
15.
Первый закон термодинамики dQ = dU + dW
Второй закон термодинамики dQ - TdS
Третий закон термодинамики ljm S = 0
Закон идеального газа: РУ = RT
Закон Рауля: Р, = ytP^1
Закон Генри: Р, = кнх, или /, = ЛяХ/
Правило Льюиса и Рэндалла: / = или ф, = ф,-
Уравнение Клапейрона: — =-----
dT ТАУ
Уравнение
Уравнение
Уравнение
Уравнение
Уравнение
тг «. тг dlnP ДТ/vap
Клапейрона — Клаузиуса: ------=------- (идеальный пар)
dT RT2
Гиббса — Дюгема: Y,xtdMi
Гиббса — Гельмгольца: Т
dP
т
Гесса: изменение энтальпии реакции не зависит от пути процесса
Кирхгоффа:
ДСР
Правило Лоренца — Бертло: для параметров кубического уравнения состояния, подобного уравнению
Ван-дер-Ваальса, Va = и b = ЕуД [113].
Изохора Вант-Гоффа: (д1пКсе/дТ)р = &H°/RT2 (для химической реакции).
Таблица А.З. Основные уравнения и отношения
Фундаментальные уравнения:
nU = J{S, V, М, п2, ... ,пк)
пН = f(S, Р, Th, П2, Пк)
= n(U + РУ)
внутренняя энергия,
nA = f(T, V, Th, п2, .... Пк)
= n(U - TS)
nG = f(T Р, Th, П2, ..., Пк)
энтальпия,
энергия Гельмгольца,
энергия Гиббса.
f^n(U - TS + РУ)
Дифференциальные уравнения:
d(nU) = Td(nS) - Pd(nV) + Lindm.
d(nH) = Td(nS) + nVdP + E fj-idn t,
d(nA) = - nSdT - Pd(nV) + TL^dni,
d(nG) = - nSdT + nVdP + ’Epudni,
SPrij
Интенсивные свойства:
Уравнение Максвелла:
Равновесие химической реакции:
Keq = ехр(- &G°/RT),
/э1аКеЧ\ _ /адо°/т\
\ дТ ) \ дТ )р
Таблица А.4. Непосредственно измеряемые параметры
1.
2.
3.
Изотермическая сжимаемость: кт = -
Адиабатическая сжимаемость: к.$ = -
1 (dV\
У\дР/т
l(dV\
. . „1 (дУ\
Коэффициент термического расширения: р = — I — I
У\дТ/Р
Теплоемкость или удельная теплоемкость:
4.
5.
6.
7.
8.
9.
при постоянном давлении: Ср
(дН\ = =т(—(—
\дт/Р \дту р \дт)р\дт)
Коэффициент Джоуля — Томсона
1
~Ср
Таблица А.5. Свойства идеального газа и величины газовой постоянной, выраженные в различных единипях
pV = nRT
/дУ\ V
\дт)р Т
dU=nCvdT
dCv
дУ
I = 0
T
dT dp
dG = nCDdT- nTCD — + nRT—
p p T p
kT = l/p
kg = Cy/pCp
P= i/T
dH=nCpdT
dCp
dP
= 0,
Rk
Cp С у R, Cp ________ j , к Ср! Су
nCp dp
dS = ~rdT-nR-
T P
dT dV
dA — nC yd T nTCy nR T
Энергия Температура Моль R
фунт • кв.фут/с2 Градусы Ренкина фунт 4,969-104
фут • фунт-сила Градусы Ренкина фунт 1544
куб.фут • атм Градусы Ренкина фунт 0,7302
куб.фут(фунт-силы /
кв. дюйм) Градусы Ренкина фунт 10,73
Британская тепловая
единица Градусы Ренкина фунт 1,987
л.с./ч Градусы Ренкина фунт 7,805 10“‘
кВт ч Градусы Ренкина фунт 5,819-10“
Дж (абс.) Кельвины г 8,314
кг-м2/с2 Кельвины кг 8,314-103
кгс-м Кельвины кг 8,478-102
см3•атм Кельвины г 82,0562
кал Кельвины г 1,987
Таблица А.6. Процессы, происходящие в идеальном газе PV = RT, постоянные теплоемкости Ср и Cv, к = Cp/Cv
Ограничения
К=С
Р=С
Q = о
Обратимый адиабатический процесс
Соотношения PV Г1 = Г2 Л=Р2 Л Pl V2 Pl v\^p2 Vk2 (Kl Y Л V2 /
Соотношения РТ Л п т\ = т2 72 /p2 Г1 = \A )
Соотношения TV Д(РК) = ^(Р2-р1) Vz Ъ И П = Р(Г2- Г1) /?2 \ = РП — - 1 Vi ) = 0 12 Tx = PiPi = pPj /pj V 1 = J Г /Р \(k-D/k (-] -I L \pi / /p2 1” 1 ~ 1 . \P1 / J
АГ />2 \ (V1 \ = Г1 V’1) = 0 = Л = n i и i I T । < । । Д 1 1
Q = AtZ= СрАГ А(РК) к - 1 < II Ец 1 < II II Рг ~-RT^ И, = -ЛГ1лК = 0
W = \PdV Работа обратного процесса = 0 = Д(РК) Рт "-ЛГ1лК = - AC/= - cvA.T — RT} ’ /p2 Y*-1)/* к - 1 [ V 1 /
Приложения
U1
U1
Продолжение
Работа обратного процесса = Р(И2 - Кр
W - \VdP Работа обратного процесса = Г(ДР) = V(P2~PX) = 0
ДЯ = с^Д Т Л (РУ) к - 1 = CVA Т Д(РИ) к - 1
ДЯ = срДГ " к ~ к 1 А<РК) к — 1 = срАТ - к - ! Д(РГ)
У, -Д(РГ) fc— 1 _ -Pl И к — 1 чгч
II II 1 1 5" в II II II II 1 III £ л- Л- ** > 1 > 1 *- 1 * Й5 ~ .5 ~ - ч II -Ср др ’ />2 \(к~1)/к lU) -1 Д(РР) Г /р2 \к~"/к LW - -
= 0 ^nonfl RTl ~ к - 1 Д(РГ) к- 1 = Р1К1_ ' к - 1 ow У /р у*-о/* -] ы ч ЙГЧ
= 0 — — W = гг п kRT\ ~ к - 1 к = *-1 д< _ kPtVj к- 1 СрДР /р \(*-1)/* W -1] PV) Г /р2 у*-*)" 1
Таблица А. 7. Таблица Бриджмена для расчета первых производных термодинамических функций [26]
(dT)P= ~(dP)T= 1
/dV
(dV)P= ~(dP)v= —
\ат
Ci
(as)P=-(ap)s=^
(dV \
(dU)P= -(dP)u= Cp — P —
Vt’/p
(dH)p = ~ (dP)H = Cp
{dG)P= — (dP)G—— S
{дЛ)Р=-(дР)А = -
(dV
(dV)T= -(dT)v= - I —
[dV
(dS)p= — (dT)s = I
\dT
(dV
dV
дТ
dV
дР
(dU)s= -(dS)u = ~
VC
(dH)s = — (dS)H = —
— (dS)G — —
1
(ЭЛ)5=-(Э5)л = -
dV
4- ST----
\ dT
1
(dH)u=-(dU)H=-V
(dV
CP I
XdP
dV
dP
'd V V '
дТ
dV
VCD - ST —
\ат
dV
CP —
\ dP
f dV
CP~P[---
\dT
dV\2 '
dT
dV\2'
дТ
(dV
(dH)T=-(dT)H^-V+T^~
(dG)u=-(dU)G=~ V
dV
cp - P —
\ dT
(dG)T=-(dT)G=~ V
fdV
(dA)T=~(dT)A = P —
\dP
1
(dS)v = -(dV)s=~
dV
CP ~
\dP
'dV \2 '
.dT
'dv\2
(dV
(dU)v=-(dV)v= Cp I—
yap / \ат
’dV \2
(dV
(dH)v=-(dV)H=Cp —
\<эр / \ат
dV
dT
{dG)v=-(dV)G=~
dV
dV
dV
dV
dT
dP )T_
dT
dP
(dA)u=-(dU)A=P (Cp +S)
(dV
4- STI-
\dT
dV
dP
dV
(dG)H=-(dH)G=- V(Cp + S) + TS —
(ЭЛ)Я= ~{dH)A = -
dV
dT
dV
dT
(dV
+ pcp —
ar\2'
dT
(dV
(dA)v=-(dV)A = ~S I —
fdV
(dA)G=-(dG)A = ~S V + P
dP
/dV
-pvl —
\dT
Приложение Б. Численные методы
Среди уравнений, пригодных для изучения и пред-
ставления фазового равновесия, достаточно простых и
разрешимых аналитическим путем очень и очень немно-
го, поэтому в случае использования компьютеров следу-
ет обращаться к численным методам. Эти методы мо-
гут быть чрезвычайно сложными и могут требовать
много машинного времени, однако рассмотренные в
данном разделе наиболее простые их варианты доста-
точно экономичны с точки зрения расхода времени и
вполне приемлемы для иллюстрации излагаемого мате-
риала. Подробное описание методов, предназначенных
для специалистов, можно найти в перечисленных ниже
изданиях.
1. Gerald. Applied Numerical Analysis (1978).
2. Himmelblau. Process Analysis by Statistical Methods
(1970).
3. Lapidus. Digital Computation for Chemical Engineers
(1962).
4. Rosenbrock, Storey. Computational Techniques for
Chemical Engineers (1966).
5. Scarborough. Numerical Mathematical Analysis, 6th ed.
(1966).
6. Wblberg. Prediction Analysis (1967).
Б.1. Правила частного дифференцирования
а.Дифференциал и = и(х, у, z, ...) равен
, (ди\ /ди\ /ди \
du - I I dx +— I dy + I— ) dz.
\dx / \dy / \dz /
\ /y>z X -r / xz /xy
б. Для функции и = u(x, у)
1) цепное правило:
/ди дх \ / ду \
— — / I----- ) = ~ * 1
\<Эх ду / \ди /
' j- J ' и X ' х
2) правило перекрестного изменения порядка диф-
ференцирования :
д / ди\ д / ди\
дх \ ду /х ду \ дх /у
3) замена переменных величин, принимаемых по-
стоянными:
4) при и = const
dx =
Б.2. Нахождение корней
Задача состоит в том, чтобы найти величину х, удов-
летворяющую уравнению у = fix) = 0. При наличии
более одного корня все имеющиеся численные методы
требуют начальной оценки корня, как можно более
близкой к правильной величине.
а. Аналитический метод Ньютона — Рафсона. Если
хо — оценка корня, то улучшенная величина равна
х = х0 - kf{xo)/f'lx0),
где f (хо) — производная, а к — положительная дробь,
выбранная произвольно для улучшения сходимости; ча-
сто приемлема к = 1. Этот процесс повторяют с вновь
определяемыми величинами до получения желательной
точности.
б. Численный метод Ньютона — Рафсона. Если труд-
но найти производную аналитическим путем, можно
использовать , отношение конечных разностей. На-
пример,
/(1,0001х) -/(х)
f (х) =-----------------
0,0001х
и улучшенная оценка корня составляет
0,0001хо/(хо>
х - хо - к------------------
/(1,0001хо) - /(хо)
Производная имеет такой вид:
= (х2 - х + 1) [0,7 ехр (0,5(1 - х)2 - 1,2 ехр (0,5л2)].
dx
Коэффициент 1,0001 выбирается произвольно, его мож-
но увеличивать или уменьшать, чтобы ускорить получе-
ние сходимости или повысить точность расчетов.
в. Метод Вегштейна. Уравнение преобразуют к сле-
дующему виду:
X = fix)
Улучшенная оценка корня равна
x0/(xi) - /(хо)2
хо + /(xi) - 2/(х0) ’
где Хо — начальная оценка, a Xi = /(хо).
Далее расчет повторяют, используя улучшенное значе-
ние корня.
г. Пример. Необходимо решить следующее
уравнение:
у = - 1 + 0,7хехр(0,5(1 - х)2) + 1,2(1 - х) ехр (0,5 х2) = 0
1 - 1,2(1 - х)ехр(0,5х2)
ИЛИ % —__________________________t
0,7 ехр (0,5(1 - х)2)
Как видно из графика, это уравнение имеет два корня.
Ниже показаны три метода выполнения последователь-
ных аппроксимаций корней при трех различных началь-
ных величинах. При начальной величине 0,1 метод
Вегштейна ведет к сходимости до отрицательной вели-
чины корня, в то время как другие методы дают поло-
жительное значение корня. Во второй таблице показано
влияние начальной величины на число итераций и ко-
рень, определяемый по методу Вегштейна.
Приложения 561
| Последовательность итераций, выполненная тремя
| методами
Аналитический метод Ньютона — Рафсона Численный метод Ньютона — Рафсона Метод Вегштейна
0,9 0,9 0,9
0,7152 0,71252 0,76691
0,65784 0,65686 0,68119
0,65443 0,65458 0,65610
0,65442 0,65444 0,65443
0,65443
0,5 0,68002 0,5
0,68001 0,65580 0,73760
0,65509 0,65452 0,66818
0,65442 0,65443 0,65489
0,65442 0,65443 0,65443
0,1 0,1 0,1
1,4367 1,4368 -1,3707
1,114 0,97308 -0,49429
0,8412 0,75575 -0,47079
0,68865 0,66976
0,65562 0,65532
0,65442 0,65448
0,65443
Число итераций при использовании метода Вегштей-
на и найденная величина корня
х(°) -0.4708 0.6544
0 3
0,1 3
0,15 4
0,2 5
0,25 12
о,з 38
0,35 8
0,4 6
0,5 4
10 ! N-R SOLUTION OF Y=F(X)=0 WITH
NUMERICAL DERIVATIVE
20 SHORT X
30 INPUT X
40 PRINT X
50 GOSUB 160
60 Y1=Y
70 X=1.0001*X
80 GOSUB 160
90 Y2=Y
100 H=.0001*X*Yl/(Y2-Yl)
110 X=X/1.0001-H
120 PRINT X
130 IF ABS(H/X)<=.0001 THEN 150
140 GOTO 50
150 END
160 ! SR FOR Y=F(X)
170 Y—14-. 7*X*E XP( .5 *( 1 - X) "2)4-1,2*( 1 - X)*EXP
(.5*X"2)
180 RETURN
10.! WEGSTEIN SOL’N OF X=F(X)
20 SHORT X
30 INPUT XI
40 PRINT XI
50 X=X1
60 X2=X
70 GOSUB 160
80 X=Y1
90 GOSUB 160
100 X=(X2*Y1-X"2)/(X14-Y1-2*X)
110 IF ABS((X-Yl)/(X4-Yl))<=.0001 THEN 150
120 PRINT X
130 X1=X
140 GOTO 60
150 END
160 ! SR FOR Y1=X=F(X)
170 Y1 =( 1-1,2*( 1 ~X)*EXP(.5*X~2))/.7/EXP(.5*
(1-Xf2)
180 RETURN
10 ! N-R SOLUTION OF Y=F(X)=0 WITH
ANALYTICAL DERIVATIVE
20 SHORT X
30 INPUT X
40 PRINT X
50 GOSUB 120
60 H=Y/Y1
70 X=X-H
80 PRINT X
90 IF ABS(H/X)<=.0001 THEN 110
100 GOTO 50
110 END
120 ! SRFOR Y AND Y1
130 Y=—14-.7*X*EXP(.5*( 1—X)"2)+1,2*( 1-X)*EXP
(,5*X~2)
140 Y1=(X"2—X+1)*(.7*EXP(.5*(1—X)~2)—1.2*
EXP(.5*X"2))
150 RETURN
Б.З. Система нелинейных уравнений.
Ниже будет описан только один метод решения сис-
тем нелинейных уравнений — метод Ньютона —
Рафсона. Он имеет достаточно широкую область при-
менения, достаточно прост по своей сути и вполне при-
емлем для решения задач, рассматриваемых в данной
книге. На практике иногда может возникнуть необходи-
мость в использовании более сложных методов, для ко-
торых в библиотеках имеются машинные программы.
Некоторые из этих методов основаны на решении сис-
темы уравнений
ф,(Х1, хг, ..., Хп) = 0 i = 1, 2, ..., п
с целью нахождения минимума функции
/(хь х2, .... Хп) = Е (Ф,)2 -> minimum,
что может быть выполнено при помощи так называе-
мого метода крутого восхождения, описание которого
17-864
содержится, например, в книгах Стори (1966) и Хим-
мельблау [55]. Сравнительно сложная задача минимиза-
ции рассматривается в разд. 10.6.3.
При наличии только двух переменных иногда удобно D =
прибегнуть к графическому методу. Если имеются сле-
дующие уравнения:
D
fx fy fz
gX gy gz
hx hy hz
f(x, y) = 0,
g(x, y) = 0,
точки на графиках находят, задавая величины одной из
переменных, скажем у, и находя соответствующие вели-
чины х из каждого уравнения, например, при помощи
метода Ньютона — Рафсона для одной переменной.
Эту процедуру выполняют для ряда значений у, чтобы
получить достаточное число точек пересечения. Можно
сначала провести графическое решение и использовать
полученные величины как начальные при последующем
решении задачи численным методом, что позволит по-
лучить более точные результаты. Эта процедура ис-
польуется, в частности, в примерах 4.18, 10.10 и 10.12.
Считается, что метод решения систем линейных урав-
нений известен: при наличии лишь нескольких перемен-
ных пользуются детерминантами, а в общем случае —
матричным методом. По методу Ньютона — Рафсона
систему нелинейных уравнений с исходными перемен-
ными сводят к системе линейных уравнений, выражен-
ных через поправки к исходным переменным. Возьмем,
чтобы не усложнять задачу, систему уравнений с тремя
неизвестными;
f(x, у, z) = 0, g(x, у, z) = 0, h(x, у, z) = 0.
Если (х0, Уо, Zo) — исходные оценки корней, а (а, Ь, с)
— поправки, то улучшенные величины корней выглядят
так:
х = хо + а, У = Уо + b, z = Zo + с.
Линейные уравнения, которые следует разрешить отно-
сительно а, Ъ, и с, имеют следующий вид:
/о + afx + bfy + cfz = 0,
go + agx + bgy + cgz = 0,
h0 + ahx + bhy + chz = 0,
где, например,
fo = f(x0, yo, Zo),
fx = d//dx, полученная при (хо, Уо, Zo) и т. д.
При решении с использованием детерминантов
имеем:
При следующей подстановке исходными величинами яв-
ляются х = Хо + а, у - уо + b, z = Zo + с. Этот метод
применен в примере 4.7, а машинная программа для не-
го дана в табл. В.5.
Б. 4. Интерполяция
а. Линейная интерполяция. Решение х = /(х) = у
предусматривает две подстановки.
(Л - У1)
т =------------,
(х2 - Х1)
Oi - тх\)
Х (1 - т)
20 + 15/Г2
Пример. V = 0,2
Подстановки выполняют, используя следующие исход-
ные значения (И У) = (1, 1,1429), (1,5, 1,4375),
m = (1,4375 - 1,1429)/(1,5 - 1) = 0,5892,
V = (1,1429 - 0,5892(1))/0,4108 = 1,3479.
Правильный ответ: V = 1,3873, поэтому для повышения
точности необходимо дальнейшее уточнение исходных
данных.
б. Двойная линейная интерполяция. Даны четыре та-
булированных значения переменных zi, Z2, Z3, Zt, следу-
ет найти величину z(xy) при промежуточных значениях
основных переменных х и у.
У X] X Х2
У1 z\ z3
У z
Уз z3 z4
а =
с =
о 0 бу 6z % == ---------------------------------------------------------
Ао hy hz (х2 - Х1)(у2- Л)
D
fx fo fz
gx gO gz
hx hQ hz
fx
gx
hx
D
Лу fo
gy g0
hy h0
Пример. Найдите удельный объем перегретого пара,
исходя из приведенных в таблице величин. Результаты
линейной интерполяции показаны в распечатке, в скоб-
ках для сравнения указаны действительные значения.
Приложения 563
Т р
290 х 310
500 1,8324 1,7042
У (^)
600 2,0770 1,9360
10 READ XI,Х2, Yl, Y2, ZI, Z2, Z3, Z4
20 DATA 290,310,500,600,1.8324,2.077,1.7042,1.936
30 INPUT X,Y
40 Z=(X2-X)*(Z 1 ♦(Y2-Y)+Z2*(Y-Y1 ))+(X-Xl )*
(Z3*(Y2- Y)4-Z4*(Y-Yl))
50 Z=Z/(X2-X1)/(Y2-Y1)
60 PRINT “X=”;X;“Y=”;Y;“Z=”;Z
70 END
X = 305 Y = 570 Z = 1,90075 (1,9012)
X = 295 Y = 570 Z = 1,96933 (1,9696)
в. Интерполяция с неравными интервалами. Интерпо-
ляционный полином Лагранжа для трех точек имеет
следующий вид:
(x-xj)(x-x2) (х-х0)(х-х2)
>’0 + /1
(*0 ~ *1 )(*0 - х2) (*1 “ *0)(* 1 “ х2)
(х ~хо)(х - xj
(х2 ~*0)(*1 ~Х2)У2'
Пример. Исходя из заданных величин энтропии пара
при 875 фунт/кв. дюйм абсолютного давления при не-
скольких температурах, найдите величину энтропии при
700 ° Е
Т 540 700 800 920
S 1,4181 (1,5292) 1,5849 1,6377
Величина, полученная в результате интерполяции, равна
S = 0,2227(1,4181) + 1,1282(1,5849) - 0,3509(1,6377) =
= 1,5292.
Действительная величина, взятая из таблиц величин,
полученных для пара, составляет 1,5339.
Б. 5. Подбор кривой
а. Линеаризация. Если уравнение f(x, у) = 0 имеет две
константы, их можно легко найти, представив уравне-
ние в линейном виде:
g(x, у) = а + bh(x, j).
Примеры. 1. Вириальное уравнение z = 1 + B/V у
+ C/V2 преобразуется к виду V(z — 1) = В У C/V,
т. е. к линейному отношению между V(z — 1) и 1/И
2. Уравнения ван Лаара (разд. 4.8) представляют в
линейной форме
Vinyi = Va + \!а/в Vinyz
откуда можно найти параметры Л и В, если известны
хотя бы по два значения Inyi и Inyz.
3. Уравнение у = аехр(Ь/Т) широко применяется в
линейной форме, Inj = А + В/ Т, для представления
влияния температуры на давление пара, коэффициенты
равновесия испарения, константы химического равнове-
сия и другие свойства.
б. Метод наименьших квадратов. Наилучшие величи-
ны параметров а, Ь, ..., выражающие зависимость
группы переменных
у = /(хь х2, ..., хп, а, Ь, ...)
получают в том случае, если сумма квадратов отклоне-
ний минимальна:
Е(у, - fi)2 = minimum.
Для этого необходимо, чтобы все производные относи-
тельно неизвестных параметров были равны нулю:
=0
да дЬ дс
Помимо случая полиномиальных уравнений, дифферен-
цирование дает нелинейные уравнения для неизвестных
а, Ь, с..., относительно которых эти уравнения решают
при помощи метода Ньютона—Рафсона. Для полинома
у = а0 + aix + СгХ2 + азх3 + ... щх*
метод наименьших квадратов приводит к следующему
выражению:
Е( - yt + а0 + aix, + 02Х? + азх! + ...)2 = minimum.
Дифференцирование относительно а, дает систему
уравнений
naQ 4- OjS х{ + a^L xj + . . . = S yt,
oqL Xj + xj + a^L xj + . . . = S Xjyj,
aQ S xj + ax S xj + a2^ xj + . . . = S xjy{,
и t. д., где n — количество данных (х,, у,). Эти уравне-
ния разрешимы относительно неизвестных величин а,-
при помощи любого стандартного метода решения сис-
тем линейных уравнений.
Пример. Функция избыточной энергии Гиббса,
у = Gn/RTx(\ - xj, для системы вода + триэтил амин
известна как функция состава [777]. К данным будет
подобран квадратный полином.
X У У calc
0,05 10,2847 10,45
0,25 9,5148 9,30
0,45 9,3282 9,14
0,65 9,7665 9,96
0,85 11,3528 11,77
0,95 13,4083 13,04
Из уравнения у = а + Ьх + сх2 получаем уравнения сле-
дующего вида:
17
6а + 3,26 + 2,315d = 63,6553,
3,2a + 2,3156 + 1,853c = 35,8266,
2,315a + 1,8536 + 1,5599 = 26,9391.
В результате решения этих линейных уравнений по-
лучаем
у = 10,8896 - 9,4422х + 12.3252Х2.
Данные, приведенные в последнем столбце, рассчитаны
из этого уравнения. В указанном источнике использует-
ся более сложное коррелирующее уравнение большей
точности.
в. Нелинейный метод наименьших квадратов. В
табл. В.6. приводится программа для определения па-
раметров уравнения Вильсона и уравнения Цубоки—
Катаямы—Вильсона, которые входят в выводимые
уравнения в нелинейной форме. В работе [143] изложе-
ны основные моменты этого метода.
Б.6. Численное дифференцирование
Уравнения, используемые в этих целях, основаны
на интерполяционных формулах, выраженных через ко-
нечные разности. В работе Милна [778] дана формула
Лагранжа для нескольких интервалов. Формулы для
высших производных приведены, например, в «Спра-
вочнике Центра координационных исследований по ма-
тематическим наукам» [779]. В данном приложении для
наглядности будет использовано уравнение Лагранжа
для трех точек с неравными интервалами.
Производная интерполяционной формулы Ла-
гранжа
dy = 2х - xi - х2 уо + 2х - хр - х2 yi +
dx (хо - xi)(xb - х2) 7 (Xi - xb)(xi - хг)
(хг - хо)(х2 - Xi)
При равномерных промежутках h = Хг - Xi = Xi - хь
уб = (~3уо + 4yi - y2)/2h,
yi = (~Уо + yi)/2h,
Уг = (jo — 4yi + Зуг)/26.
Пример. Избыточные энергии Гиббса системы
вода + 1,4-диоксан имеют следующие значения [780]:
Xi 0 0,05 0,15
Gex 0 242,5 637,8
Химический потенциал равен
/и = Gex + (1 - Х1) .
Дифференцирование выполняют при помощи формулы
Лагранжа в контрольных точках и в двух других точ-
ках. Результаты представлены ниже. В последнем
столбце приведены точные величины, указанные в пер-
воисточнике.
X dG dx
ВЫЧИСЛ. исправл.
0 5,149 5,149 5,166
0.05 4,551 4,566 4,554
0.10 3,953 4,012 4,008
0.15 3,355 3,490 3,520
0.20 2,757 3,000 3,084
Кубические сплайны. Подгонку данных можно вы-
полнить, проведя кубические кривые через пары близле-
жащих точек и сгладив их в этих точках (х,, yi), путем
приравнивания их первых и вторых производных с обе-
их сторон от этих точек — со стороны х, -1 и Xi+1. Для
/-го интервала, находящегося между (х,, yi) и (xI + i,
Ji + i), кубическое уравнение имеет следующий вид:
у = а,(х - х,)3 + 6,(х - xi)2 + с,(х - х() + di.
Интервалы между абсциссами определяют по формуле
hi = Xi + i - Xi.
Коэффициенты для каждого интервала выражают через
вторые производные S< при помощи следующих
уравнений:
ai = (Si + i - Si)/6hi,
bi = Si/2,
Ci = (yI + i - yi)/hi - hi(2Si + S, + i)/6,
di = yi.
Уравнения производных имеют следующий вид:
у' = 3a,(x - xi)2 + 26,(х - Xi) + ci,
у" - 6at(x - xi) + 26,-.
Si находят путем решения системы линейных уравнений
hi - \Si - 1 + 2(hi - 1 + hi)Si + .hiSi + 1 =
= 6[(y, +1 - yi)/hi - (yi - yi-i)/hi-i]
при величине i от 2 до n - 1 и Si = Sn = 0. Для получе-
ния конечных величин в некоторых случаях использу-
ются другие процедуры, описание которых приведено,
например, в работе [43]. При равенстве интервалов
h = hi = hi +1 уравнения для S, преобразуются к следую-
щему виду:
hSi-i + 4hSi + hSi+1 = 6(yi + i + yi-i)/h.
Решение этой системы линейных уравнений является
относительно простым, поскольку матрица коэффици-
ентов в левой стороне тригональная.
Пример 2. Применение кубических сплайнов для на-
хождения химических потенциалов при наличии данных
об избыточной энергии.
Данные взяты из источника, который указан в при-
мере 1 данного раздела. Уравнения для определения Sr.
0,55г + O,1S3 = —8,470,
O,1S2 + 0,5S3 + 0,lS4 = -7,544,
0,lS3 + 0,354 = -4,212.
Приложения 565
Величины St и соответствующие значения коэффициен-
тов кубических сплайнов, а также исходные данные
представлены в табличной форме. Во второй таблице
приведены результаты вычислений производных в за-
данных и промежуточных точках, а также химические
потенциалы, рассчитанные по следующей формуле:
n = Gex + (1 - x)dGex/dx.
Результаты сравнения с величинами д, приведенными в
источнике, нельзя назвать удовлетворительными так
же, как и результаты сравнения производных в общих
точках близлежащих интервалов. Вероятно, выполяя
расчет с учетом других условий, помимо Si = S5 = О,
можно добиться большей точности подгонки. В боль-
шинстве других случаев метод кубических сплайнов го-
раздо более точен, чем другие интерполяционные
методы.
i X h S
1 0 0 0.1 0
2 0,10 454,5 0,15 -14 949
3 0,25 924,5 0,10 -9,954
4 0,35 1 112,1 0,05 -10.722
5 0,40 1 170,8 0
i a b c d
1 — — — —
2 -24 915 0 4 794 0
3 5 551 7 475 4 005 454,5
4 -1 281 4 977 2 399 924,5
5 35 740 5 361 1 063 1 112,1
* Gex dgexldx ВЫЧИСЛ. данные
0 0 4,794 4,794 5,166
0,05 242,5 4,607 4,619 4,554
0,10 454,5 4,047 4,097 4,008
0,10 — 4,005 4,059
0,15 637,8 3,299 3,442 3,520
0,20 794,1 2,676 2,935 3,084
0,25 924,5 2,137 2,527 2,693
0,25 — 2,400 2,724
0,30 1 030,2 1,892 2,355 2,342
0,35 1 112,1 1,365 2,000 2,025
0,35 — 1,063 1,803
0,40 1 170,8 795 1,648 1,738
Б. 7. Численное интегрирование
Эту операцию могут выполнять самые недорогие
калькуляторы, причем необходимая программа, как
правило, имеется, так что составлять ее приходится в
очень редких случаях. Наиболее широкое распростране-
ние получил метод Симпсона, при помощи которого
можно получить любую требуемую степень точности с
математическими функциями, используя достаточно
малые интервалы. При наличии определенных интерва-
лов желательно воспользоваться более точными фор-
мулами, выраженными через высшие разности [779].
а. Правило Симпсона
f "у dx = — (у0 + у„ + 2 22 у,; + 4 22 у, ] ,
d х0 \ even odd /
где п — четное число равных интервалов. В формуле
указаны суммы у,- с четными и нечетными надстрочны-
ми индексами.
б. Правило трапеции
рхп Д*
/ У dx - — (у0 + Уп + 2S у,).
JXQ 7.
в. Формула Лагранжа для трех неравноотстоящих
точек
р ь (Ь — а)
I ydx= б Мо>’о + А1У1 + Л2у2),
где
2(b2 + ab + а2) — 3(*j + x2)(b + а) + 6х^х2
° (*о ~ *1 )(*0 “ х1)
2(b2 + ab + а2) — 3(*о + x2)(b + а) + 6xqx2
А1 ~ ,
(%1 -х0)(*1 ~х2)
2(b2 + ab + а2) — 3(*о + х^)(Ь + а) + 6хох1
А2 = .
(Х2~х0)(*2“*1)
При а = хь и Ь = хг коэффициенты равны
2 2
2%о — 3%o%i — *0*2 + 3*1*2 — *2
Ап = ,
(*1 -Х0)(*2 ~*0)
_ (Х2~*0)2
1 (*1 -хо)(*2-х1)’
2 2
—*0 + 3*0*1 — *0*2 — 3*1*2 + 2*2
А2 (х2- Х0)(Х2- Xi)
Пример. Ниже приведены значения сжимаемости
изобутана при температуре 361 К и нескольких величи-
нах давления
P. фунт/кв.дюйм абсолют, давления 0 100 225
z 1 0,8815 0,7099
(z - 1)/P 0 -0,001185 -0,001289
Коэффициент фугитивности определяют по формуле
- 1
1пф = I dP.
J о
10 READ X0,Xl,X2,Y0,Yl,Y2A
20 DATA 0,100,225,0,-.001185,-.001289,0
30 INPUT В
40 С=В " 2+А*В+А " 2
50 А0=(2#С-3#(Х1+Х2)*(В+А)+6#Х1#Х2)/
(ХО—Х1)/(Х0—Х2)
60 А1=(2*С-3*(Х0+Х2)*(В+А)+6#Х0#Х2)/
(XI- Х0)/(Х1- Х2)
70 А2=(2*С-3#(Х0+Х1)*(В+А)+6#Х0#Х1)/
(Х2-Х0)/(Х2—XI)
80 I=(B-A)/6*(AO#YO+A1*Y1+A2#Y2)
90 F=EXP(I)
100 PRINT USING 110 ; B,F
110 IMAGE DDD,5X. DDD
120 GOTO 30
130 END
P ф
50 0.981
100 0.935
150 0.875
200 0.815
225 0.788
250 0.765
Приложение В. Программы для ЭВМ
При многократном проведении расчетов фазовых
равновесий использование ЭВМ представляется совер-
шенно необходимым. В книге по мере изложения мате-
риала приведено много коротких программ, в том
числе предназначенных для решения задач и рассматри-
ваемых в книге примеров. В данном приложении в
табл. В.1 — В. 11 показаны некоторые более длинные
программы общего характера. Они составлены на язы-
ке Бейсик для миникомпьютера НР-85.
В литературе опубликовано много алгоритмов, ис-
пользуемых при расчетах фазового равновесия; см., на-
пример, программы, приведенные в приложении Г,
заимствованные из книги Лислея [6] и статей Петерсо-
на (1978—1979). В открытой литературе помещено
очень немного действительно важных программ.
Основные программы различной степени сложности,
представляющие интерес в связи с рассматриваемой те-
мой, приведены в перечисленных ниже публикациях; все
они составлены на языке Фортран.
1. Fredenslund et. al. Vapor-Liquid Eduilibria Using
UNIFAC (1977). Определение параметров модели
UNIFAC из данных по равновесию пар — жидкость.
Определение коэффициентов фугитивности из вириаль-
ного уравнения. Коэффициенты активности, определяе-
мые с помощью уравнений UNIFAC—UNIQUAC. Пред-
сказание равновесия пар — жидкость, жидкость — жид-
кость.
2. Kojima, Tochigi Prediction of Vapor-Liquid Equili-
bria by the ASOG Method (1979). Расчет коэффи-
циентов активности исходя из молекулярной структу-
ры. Предсказание равновесия пар — жидкость при
условии идеальности паровой фазы. Программа для
определения параметров методом группового вклада из
экспериментальных данных.
3. Shahin Negahban Department of Chemical and
Petroleum Engineering, University of Kansas (1982).
Регрессия для расчета параметров по данным о равно-
весии жидкость — жидкость в двойных и тройных сис-
темах. Расчет соединительных линий для двух- и
трехкомпонентных систем с двумя жидкими фазами.
Трехкомпонентные системы с тремя жидкими фазами.
Четырехкомпонентные системы с двумя фазами. Ис-
пользование уравнений NRTL и UNIQUAC.
4. Null Phase Equilibria in Process Design (1970). Рас-
чет параметров равновесия жидкость — жидкость в
трехкомпонентных системах и двух- и трехфазных сис-
Приложения 567
темах с использованием коэффициентов активности
уравнений ван Лаара.
5. Prausnitz et al. Computer Calculations for Multi-
component Vapor-Liquid and Liquid-Liquid
Equilibria (1980). Регрессия для параметров уравнения
UNIQUAC из экспериментальных данных. Расчет вто-
рых вириальных коэффициентов, фугитивностей коэф-
фициентов активности, остаточных энтальпий и
w энтропий. Обычная процедура, применяемая для расче-
та равновесия пар — жидкость, жидкость — жидкость.
Я 6. Renon et al. Calcul sur Ordinateur des Equilibres
Я Liquide-Vapeur et Liquide-Liquide (1971). Регрессия
Я для расчета параметров уравнения NRTL из экспери-
Ш ментальных данных по равновесию пар — жидкость.
Я Обычная процедура, применяемая для расчета равнове-
я сия пар — жидкость и жидкость — жидкость.
Я 7. Smith, Missen Chemical Reaction Equilibrium
Я Analysis: Theory and Algorithms (Wiley, 1982).
Il Программы для калькулятора HP-41 С на языках Бей-
< сик и Фортран.
1 8. Sorensen Correlation of Liquid-Liquid Equilibrium
I Data, doctoral thesis, Instituttet for Kemiteknik,
1 Lyngby, Denmark (1890). Описание программы для pe-
I грессии параметров NRTL и UNIQUAC из эксперимен-
I тальных данных по равновесию жидкость — жидкость
| для трехкомпонентных систем и расчет бинодальной
j кривой для системы с двумя жидкими фазами. Рабочую
? программу, подготовленную в ее окончательном виде
М.Л. Михельсеном, можно получить в указанном инс-
титуте.
9. Sorensen, Arlt Liquid-Liquid Equilibrium Data
Collection: Ternary Systems, DECHEMA Chemistry
Data series, V-2 (1980). Бинодальные кривые для трех-
компонентных систем с двумя фазами, рассчитанные по
уравнениям NRTL и UNIQUAC.
Таблица В.1. График уравнения состояния Харменса —
Кнаппа
10 ! PLOTS OF THE HARMENS-KNAPP EOS
20 ! DATA FOR ACETONE: PC=46.4 atm,
TC=5O8.1 K, W=0.309
30 T=525
40 SCALE 0,1,5,-30.70
50 XAXIS 0,.l
60 YAXIS 1.5,10
70 YAXIS 0,10
80 READ T1,P1,W
90 DATA 508.1,46.4,.309
£100 Xl=.3211—,08* *W+.0384*W"2
0110 Bl=.1077+.76405*Xl —1.2428*X1~2+.9621*X1
~з
Qa 120 Yl= 1-3#Х1+3*ХГ2+Bl#Xl#(3-6#Xl+Bl#
Xl)
0*130 Z=B1*X1
6140 B2=.08205*Tl#Z/Pl
c!50 C2=l+(1—3*X1)/Z
160 IF T<=T1 THEN 170 ELSE 260
170 IF Al<=2 THEN 180 ELSE 220
Л180 A=.5+.27767*W+2.17225#W~2
190 B=-.022+.338#W-.854#W~2
«200 A3=(l+A#(l-(T/Tl) ,5)-B#(l-Tl/T))~2
210 GOSUB 300
220 A=.4131 + 1.1465#W
230 B=.0118
240 A3=(l+A#(l-(T/Tl) ,5)-В#(1-Т1/Т)Г2
250 GOSUB 300
260 A3=l-(.6258+1.5527*W)*LOG(T/Tl)+(. 1533+
.41 *W)*LOG(T/T1)~2
270 GOSUB 300
280 END
290 ! SR FOR X,Y
a 300 A2=A3*Yl*.08205'2*Tl~2/Pl
310 FOR X=B2+EPS TO 1.5 STEP .01
P32O Y=.08205#T/(X-B2)-A2/(X^2+B2#C2»X-
(C2- 1)*B2~2)
330 DRAW X,Y
V 340 NEXT X
350 MOVE 0,0
360 BEEP
370 RETURN
Таблица В. 2. Уравнение состояния Соава: сжимаемость
и удельный объем бинарной смеси (табл. 1.11)
10 ! PROGRAM “S-1”
20 ! SPECIFIC VOLUME OF A BINARY MIX WITH
THE SOAVE EQN. P=atm, V=liters/gmol, T=deg К
30 ! EXAMPLE DATA IN LINES 70 & 90 ARE FOR
CO2+C3H6
40 SHORT Z,V
50 OPTION BASE 1
60 DIM P(2), T(2), W(2)
70 DATA 304.2,364.9,72.9,45.45,.225,.148
80 MAT READ T,P,W
90 K=.O914
100 R=.O82O5
110 INPUT T,P,Y
120 FOR J=1 TO 2
130 A(J)=.42747*R~2*T(J)~2/P(J)*( 1+(.48508+
1.55171*W(J)-.15613*W(J)*2)*(1-(T/T(J))~5))
*2
140 B(J)=.08664»R#T(J)/P(J)
150 NEXT J
160 A=Y ~2*A( 1)+ (1 - Y) *2*A( 2)+ 2*Y*( 1 - Y)#( 1 -
K)*(A(1)*A(2))'.5
170 B=Y*B(l)+(—Y)*B(2)
180 A=A*P/R~2/T~2
190 B=B*P/R/T
200 Z=1
210 GOSUB 310
220 H=F/F1
230 Z=Z-H
240 IF ABS(H/Z)<=.0001 THEN 260
250 GOTO 210
260 V=Z*R*T/P
270 PRINT “P=”;P;“T=”;T;“Y=”;Y
280 PRINT “Z=”;Z
290 PRINT “V=”;V;“liters/gmoI”
300 END
310 ! SR FOR COMPRESSIBILITY,Z
320 F=Z"3—Z"2+(A—B—B"2)*Z—A*B
330 F1=3*Z"2—2*Z+A—В—Вл2
340 RETURN
P= 10 T=300 Y=5
Z= .91282
V = 22.2469 liters/gmol
P= 50 T=300 Y=.5
Z= .16708
V= .082253 liters/gmol
P= 25.5 T=3O3 Y=.5
Z= .75335
V = .73448 liters/gmol
Таблица В.З. Уравнение состояния Соава: коэффициен-
ты фугитивности бинарной смеси (табл. 3.4)
10 ! PROGRAM “S-2”
20 ! FUGACITY COEFFICIENTS WITH THE
SOAVE EQN. TABLE 3.4.
30 ! EXAMPLE DATA IN LINES 50 & 70 ARE FOR
CO2+C3H6
40 OPTION BASE 1
50 DIM P(2),T(2),W(2),A(2),B(2),D(2),K(2)
60 DATA 304.2,364.9,72.9,45.45,.225,.148
70 MAT READ T,P.W
80 K=.0914
90 R=.08205
100 INPUT P,T,Y1
105 PRINT “P=”;P;“T=”;T;“Y=”;Y1
110 Y2=1-Y1
120 FOR J=1 TO 2
130 K(J)= .48508+1.5 5171 #W(J)- .15163*W(Jf 2
140 A(J)=(1+K(J)*(1—(T/T(J))".5))"2
150 B(J)=.08664*R#T(J)/P(J)
160 NEXT J
170 FOR J=1 TO 2
180 D(J)=T(J)»(A( J)/P(J)) ~.5/(Y 1 *T( 1 )*(A( 1 )/P( 1)) ~
,5+Y2*T(2)*(A(2)/P(2))~.5)
190 A(J)=A(J)*.42747*R~2*T(J)~2/P(J)
200 NEXT J
210 A=Y Г2*A( 1)+ Y2~ 2*A(2)+ 2»Y 1 #Y2#( 1—K)*(A
(1)*A(2)) .5
220 A=A*P/R~2/T~2
230 C=Y1*B(1)+Y2*B(2)
240 B=C*P/R/T
250 Z=1
260 GOSUB 380
270 H=F/F1
280 Z=Z-H
290 IF ABS(H/Z)<=.0001 THEN 310
300 GOTO 260
310 FOR J=1 TO 2
320 F(J)=B(J)/C*(Z—1)—LOG(Z—B)—A/B*(2*D(J)—В
(J)/C)*LOG(1+B/Z)
330 F(J)=EXP(F(J))
340 PRINT “FUG COEFF=”;F(J)
350 NEXT J
360 PRINT “Z=”;Z
370 END
380 ! SR FOR COMPRESSIBILITY, Z
390 F=Z"3—Z~2+(A—B—B"2)*Z—A*B
400 F1=3*Z~2—2*Z+A—B—B"2
410 RETURN
P= 50 T=300 Y=5
FUG COEFF=.943593853229
FUG COEFF=1.2405 7606753
Z=.167077288477
P= 30 T=300 Y=5
FUG COEFF=.897040939721
FUG COEFF=.84238281801
Z= .675247249859
P= 10 T=300 Y=.5
FUG COEFF=.96038164773
FUG COEFF=.942323379853
Z= .912818464905
Таблица В. 4. Уравнение состояния Соава: расчет откло-
нений величин энтальпии и энтропии двухкомпонентной
смеси (табл. 11.3)
10 ! PROGRAM “S-3”
20 ! ENTHALPY & ENTROPY DEPARTURES WITH
THE SOAVE EQN. units are cal, gmol, deg К
30 ! EXAMPLE DATA IN LINES 60 & 80 ARE FOR
CO2+C3H6
40 OPTION BASE 1
50 DIM P(2), T(2), W(2)
60 DATA 304.2,364.9,72.9,45.45,.225,.148
70 MAT READ T,P,W
80 K=.O914
90 R=.08205
100 INPUT P,T,Y
IIO’FOR J=1 TO 2
120 K(J)=.48508+1.55171*W(J)—.15163*W(J)~2
130 A1(J)=.42747*R~2*T(J)~2/P(J)
140 A(J)=(1+K(J)*(1—(T/T(J))~.5))~2
150 A(J)=A(J)*A1(J)
160 B(J)=.08664*R*T(J)/P(J)
170 NEXT J
180 A1=Y~2#A 1 (1)+(1 -Yf2*A 1(2)+ 2#Y#( 1 -Y)*
(А1(1)*А1(2))Л5
190 C=Y~2*A( 1)+(1 -Yf 2*A(2)+2*Y*( 1 -Y)*( 1—K)
*(А(1)*А(2))Л.5
200 A=C*P/R~2/T~2
210 B1=Y#B(1)+(1-Y)*B(2)
220 B=B1#P/R/T
230 Z=1
Приложения 569
к) GOSUB 450
К50 H=F/F1
260 Z=Z H
270 IF ABS(H/Z)<=.0001 THEN 290
280 GOTO 240
1290 V=Z*R*T/P
1300 FOR J=1 TO 2
1310 D(J)=—(K(J)»(A1(J)*(A(J)/T/T(J)) ~.5))
1 320 NEXT J
330 Dl=—(1/2/T ~.5*(K(2)*(A1(2)*A(1)/T(2))~.5+K
(1)*(A1(1)*A(2)/T(1))~.5))
340 D=Y"2*D(1)+(1—Y)"2»D(2)+2*Y*(1—Y)»(l—
' K)*D1
350 Hl = 1,987»T»( 1 -Z)+ 1/B1 *(C-T»D)»LOG( 1+B1/
V)
360 S1 = 1.987#LOG(V/(V-B1))-1 /В 1 »D»LOG( 1+B1 /
V)
370 PRINT “P=”;P;“atm”
380 PRINT “T=”;T;“K”
390 PRINT “Y=”;Y;“mol fraction”
400 PRINT “Z=”;Z
410 PRINT “V=”;V;“liters/gmol”
420 PRINT “AH=”;Hl;“cal/gmol”
430 PRINT “AS=”;Sl;“cal/gmol/K”
440 END
450 ! SR FOR COMPRESSIBILITY, Z
460 F=Z~3-Z~2+(A-B-B~2)»Z-A»B
470 F1=3»Z~2—2*Z+A—B—B~2
480 RETURN
P = 30 atm
T= 300 К
Y = .5 mol fraction
Z= .675247249859
V = .55404036851 liters/gmol
AH = 217.212185573 cal/gmol
AS=.20582271096 cal/gmol/K
P= 10 atm
T= 300 К
Y = .5 mol fraction
Z= .912818464904
V= 2.24690265136 liters/gmol
AH = 57.9623661825 cal/gmol
AS = 4.98532833145E-2 cal/gmol/K
Таблица В. 5. Расчет параметров уравнений Вильсона и
Цубоки — Катаямы — Вильсона исходя из одного на-
бора коэффициентов активности при помощи метода
Ньютона — Рафсона (см. пример 4.7)
10 ! FINDING WILSON OR Т-К-WILSON
PARAMETERS FROM ONE SET OF ACTIVITY
COEFFS, USING THE N-R METHOD
20 ! FOR WILSON EQN, MAKE R=V2/V1=1
30 READ X,Z1,Z2,R
40 DATA .736,1.0224,1.3983,.5107
45 SHORT A,В
50 INPUT A,В
60 X2=l- X
70 C=1/(X2+X/A)-1/(X+X2/B)
80 C1=1/(X2+X/R)-1/(X+X2»R)
90 F=LOG( Z1)+LOG(( X+ X2»A)/( X+ X2»R))~ X2*
(C-Cl)
100 G=LOG(Z2)+LOG((X2+X*B)/(X2+X/R))+X*
(C-Cl)
110 Fl=A#(X2/(X+X2*A)f2
120 F2=(X2/(X2+X*B))~2
130 Gl=(X/(X+A#X2)f2
140 G2=B*(X/(X2+B#X))~2
150 D=G1»F2-F1»G2
160 H=(F»G2-G»F2)/D
170 K=(F1»G-G1»F)/D
180 PRINT USING 190 ; A,В
190 IMAGE D.DDDD,2X,D.DDDD
200 A=A+H
210 B=B+K
220 IF ABS(H/A)+ABS(K/B)>=.0001 THEN 70
230 PRINT
240 PRINT “X1=”;X
250 PRINT “A12=”;A
260 PRINT “A21=”;B
270 END
Параметры уравнения Вильсона, R = 1
.5000 .5000
.5715 .8399
.3140 1.1717
.2953 1.2602
.2924 1.2671
Xl=.736
A12=. 29235
A21=1.2672
Параметры уравнения Цубоки — Катаямы — Вильсо-
на, R = 0,5107
.5000 .5000
.7220 .8575
.3668 1.2910
.3314 1.4531
.3211 1.4782
.3210 1.4785
Х1=.736
А12=.32104
А21 = 1.4785
Таблица В.6. Расчет параметров уравнений Вильсона и
Цубоки — Катаямы — Вильсона методом нелинейной
регрессии
В качестве примера взята система вода + этанол
при 60 °C, К2/И1 = 3,244. Ниже показаны последова-
тельные аппроксимации.
10 ! “W&TKW” LEAST SQUARES NEWTON-
RAPHSON METHOD FOR WILSON AND T-K
WILSON PARAMETERS.
20 ! IN STEP #60, PLACE V=V2/V1 FOR T-K-W,
AND V=1 FOR WILSON.!
30 ! N=# OF DATA POINTS. ENTER Xl (OR X2)
AND G1 and G2 IN STEPS 80-89. CHANGE #110
IF NEEDED.
40 OPTION BASE 1
50 SHORT A,В
60 INPUT N,А,В,V
70 DIM X1(U),X2(11),G1(11),G2(11),Q(11),Q1(11),
R(11),R1(11),R2(11)
80 DATA .051,.086,.197,.375,.509,.527,.545,.808,.851,
.86,.972
81 DATA 1.028,1.079,1.161,1.366,1.591,1.608,1.621,
2.135,2.19,2.26,2.35
82 DATA 3.845,3.135,2.155,1.421,1.198,1.181,1.171,
1.022,1.015,1.01,.998
90 MAT READ X2,G1,G2
100 FOR 1=1 TO N
110 X1(I)=1-X2(I)
120 NEXT I
130 GOSUB 520
140 FOR 1=1 TO N
150 Q(I)=X1(I)*LOG(G1(I))+X2(I)*LOG(G2(I))
160 R(I)=Q(I)+Q1(I)
170 NEXT I
180 F=0
190 FOR 1=1 TO N
200 F=F+R(I)~2
210 R1(I)=X1(I)*X2(I)/(X1(I)+A*X2(I))
220 R2(I)=X1(I)*X2(I)/(B*X1(I)+X2(I))
230 NEXT I
240 S=0
250 Sl=0
260 S2=0
270 T=0
280 Tl=0
290 T2=0
300 FOR 1=1 TO N
310 S=S+R(I)#R1(I)
320 T=T +R(I)*R2(I)
330 S1=S1+R1(I)~2
340 S2=S2+R1(I)»R2(I)
350 T1=S2
360 T2=T2+R2(lf 2
370 NEXT I
380 D=S1*T2—S2*T1
390 H=(S»T2-T*T1)/D
400 K=(S1*T—T1*S)/D
403 PRINT USING 406 ; A,В
406 IMAGE D.DDDD,2X,D.DDDD
410 IF ABS(H/A)+ABS(K/B)<=.0001 THEN 460 ELSE
430
430 A=A-H
440 B=B-K
450 GOTO 130
460 PRINT “A=”;A;“B=”;B
470 END
520 FOR 1=1 TO N
530 Q1(I)=X1(I)*LOG((X1(I)+A»X2(I))/(X1(I)+V*X2
(I)))+X2(I)*LOG((X1(I)*B+X2(I))/(X1(I)/V+X2
(I)))
540 NEXT I
550 RETURN
560 END
Параметры уравнения Цубоки —Катаямы —Виль-
сона, N = 11, V2/V1 = 3,244
.2000 .2000
.6913 .5029
1.2177 .2351
1.4352 .1949
1.4608 .1890
1.4617 .1887
А= 1.4617 В=.1887
.5000 .5000
.9964 .3797
1.4144 .1703
1.4630 .1877
1.4618 .1886
1.4617 .1887
А= 1.4617 В=.18868
Параметры уравнения Вильсона, N = 11, V2/V1 = 1
.1000 .1000
.4931 .2511
.8751 .0753
.9492 .0845
.9515 .0842
.9514 .0842
^=. 95143 В=.084168
.4000 .4000
.8502 .0317
.9604 .0758
'.9514 .0841
.9514 .0842
Ч.=.95143 В=.084168
(Если в данном примере исходные величины равны 0,5
или превышают это значение, параметры уравнения
Вильсона не дают сходимости.)
Приложения 571
лица В. 7. Введение: использование метода ASOG
расчета коэффициентов активности исходя из моле-
нной структуры
Кодовые номера групп, составляющих отдельные мо-
шккулы, даны на треугольной диаграмме. Суммарное
|юо атомов, за исключением атомов водорода, в ка-
шюм-либо одном виде групп равно Vkt", исключением яв-
иются алканы, где гН2О = 1,6, рсн = 0,8 и рс = 0,5.
Манные для этого конкретного примера, т. е. смеси
[гганола (1) и бензола (2), приведены в строках 190 и
[260 программы. Параметры взаимодействия ам. вклю-
чены в программу в виде шагов 270 — 3080.
X. Группа XJ к Молекула \ i vki WK,J) S(J) vk N1(J)
CH2 (*1) ArCH (*3) OH (*6)
1 2 0 1 3 3
2 0 6 0 6 6
(Для идентификации групп пользуйтесь диаграм-
мой на следующей странице)
T= 345.0 X=0.00 X=1.00 GAM- GAM= 7.4671 1.0000
T=345.0 X= .20 X= .80 GAM= GAM= 2.6628 1.1059
T=345.0 X= .40 X= .60 GAM= GAM= 1.6235 1.7962
T=345.0 X= .60 X= .40 GAM= GAM= 1.2258 1.7962
T=345.0 X= .80 X= .20 GAM= GAM= 1.0518 2.5645
T= 345.0 X=1.00 X=0.00 GAM= GAM= 1.0000 4.0507
Таблица В.7А. Определение коэффициентов активности
исходя из молекулярной структуры при помощи метода
ASOG
10 REM *ASOG METHOD FOR ACTIVITY
COEFFICIENTS
20 REM *ENTER DATA ON 190: T, P, Q, N(K,J),
S(J), N1(J)
30 REM *ENTER GROUP NUMBERS К AND L ON
260
40 REM *INPUT MOL FR X(J) ON RUN
50 REM *T=TEMP, P=#OF GROUPS, Q=# OF
COMPOUNDS
60 SHORT A(31, 31)
70 READ T,P,Q
80 FOR K= 1 TO P
90 FOR J=1 TO Q
100 READ N(K,J)
110 NEXT J
120 NEXT К
130 FOR J=1 TO Q
140 READ S(J)
150 NEXT J
160 FOR J=1 TO Q
170 READ N1(J)
180 NEXT J
190 DATA 345,3,2,2,0,0,6,1,0,3,6,3,6
200 FORK=1 TO P
210 READ K1(K)
220 NEXT К
230 FORL=1 TO P
240 READ L1(L)
250 NEXT L
260 DATA 1,3,6,1,3,6
270 A(2,1)=EXP(-1.524+713.8/T)
280 A(3,1)=EXP(.7297-176.8/T)
290 A(4,1)=EXP(—.1842+.3/T)
300 A(5,1)=EXP(.5045-2382.3/T)
310 A(6,1)=EXP(4.7125-3060/T)
320 A(7,1)=EXP(3.5403-2282.8/T)
330 A(8,1)=EXP(—12.9277+4063.3/T)
340 A(9,1)=EXP(—1.7588+169.6/T)
350 A(10,l)=EXP(—.5097+165.7/T)
360 A(11,1)=EXP(—.4274-560/T)
370 A(12,1)=EXP(—.3699+162.6/T)
380 A(13,1)=EXP(—10.9719+4022/T)
390 A(14,1)=EXP(—.0721-264.8/T)
400 A(15,1)=EXP(— 3.7607+849.8/T)
410 A(16,1)=EXP(—12.2577+3268.8/T)
420 A(17,1)=EXP(—1.1005-346.7/T)
430 A(18,1)=EXP(,2778-274.9T)
440 A(19,1)=EXP(. 1993+2.8/T)
450 A(20,l)=EXP(—1.9291—615.1/T)
460 A(21,1)=EXP(—1.4089+228.5/T)
470 A(23,1)=EXP(—1.3346+116/T)
480 A(24,1)=EXP(.2849-151.1/T)
490 A(25,1)=EXP(—. 1134+41.1/T)
500 A(26,1)=EXP(.6926—358.5/T)
510 A(27,1)=EXP(—1.8796+482.5/T)
520 A(29,1)=EXP(—7.0135 + 1842.7/T)
530 A(30,l)=EXP(—2.0131+2.8/T)
540 A(31,1)=EXP(—,0033+6/T)
550 A(1,2)=EXP( 1.3296-995/T)
560 A(3,2)=EXP(—1.5842+247.4/T)
570 A(6,2)=EXP( 10.5763-4545.3/T)
580 A(9,2)=EXP(—1.6404+109.8/T)
590 A(12,2)=EXP(—3.4011 + 1159.1/T)
600 A(15,2)=EXP(—.7601+.4/T)
610
620
630
640
650
660
670
680
690
700
710
720
730
740
750
760
770
780
790
800
810
NQ
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
24
26
27
28
29
30
31
доступны
\26
\28
О
CS2
СН2
С-С
АгСН
СуСН
Н2О
ОН
АгОН
GOH
СО
О
СНО
СОО
СООН
нсоон
CON
CN
NH2
NH
N
ArNH2
NO2
ArNCh
Cl
CC12
CC13
CCU
ArCl
ArF
Br
CH3 то же что СН2
2
A( 16,2)=EXP(—2.1511+2.7/T)
A(17,2)=EXP(-1.6397+4.3/T)
A(18,2)=EXP(—,9317+1.9/T)
A( 19,2)=EXP(—2.7491 +2.6/T)
A(21,2)=EXP(—1.1622+80.2/T)
A(24,2)=EXP(—.8108+1.7/T)
A(25,2)=EXP(—.8444+1.1/T)
A(26,2)=EXP(—1.0915 +1.9/T)
A(l,3)=EXP(-.7457+146/T)
A(2,3)=EXP(- 5754+338.7/T)
A(4,3)=EXP(.53O1-251/T)
A(6,3)=EXP(—.5859—939.1/T)
A(7,3)=EXP(—2.0123-478/T)
A(8,3)=EXP(.9602+5.6/T)
A(9,3)=EXP(-.4021-216.8/T)
A(11,3)=EXP(-,1261+312/T)
A(12,3)=EXP(-.1541+97.5/T)
A(13,3)=EXP(—.2256—213.7/T)
A( 14,3)=EXP(—.3-232.2/T)
A( 15,3)=EXP(.8736-98/T)
A(16,3)=EXP(.864+2.3/T)
820 A(2O,3)=EXP(—2.6417+62.5/T)
830 A(21,3)=EXP(-.1225-161.1/T)
840 A(22,3)=EXP(6.783-2325.8/T)
850 A(23,3)=EXP(- 324+7.1/T)
860 A(25,3)=EXP(.2511+1/T)
870 A(26,3)=EXP(.8304—374.5/T)
880 A(27,3)=EXP(—1.5456+191.7/T)
890 A(28,3)=EXP( 145.3566—51745.1/T)
900 A(29,3)=EXP(—1.5507+253.8/T)
910 A(31,3)=EXP(- 1.37O5+279.3/T)
920 A(1,4)=EXP(. 153+2.1/T)
930 A(3,4)=EXP(-,3288+156.3/T)
940 A(6,4)=EXP(5.6308-3221.4/T)
950 A(9,4)=EXP(-2.7194+428/T)
960 A(12,4)=EXP(-,0991+2.4/T)
970 A(22,4)=EXP(—4.4362+1354.8/T)
980 A(23,4)=EXP( 1.1119-640/T)
990 A(25,4)=EXP(-,2819+3.9/T)
1000 A(26,4)=EXP(.0193—78.1/T)
1010 A(31,4)=EXP(.3599- 178.5/T)
1020 A(1,5)=EXP(—.2727-277.3/T)
Приложения 573
1030 А(6,5)=ЕХР(—5.8341 +1582.5/Т) 1040 А(8,5)=ЕХР(2.2157—450.2/Т) 1630 1640 А(25,10)=ЕХР(. 8597+. 6/Т) А(31,1О)=ЕХР(.О428+.3/Т)
1050 А(9,5)=ЕХР(.3198-91.2/Т) 1650 А(1,11)=ЕХР(2.7287-563.7/Т)
1060 А(11,5)=ЕХР(2.0292~556.8/Т) 1660 А(3,11)=ЕХР(2.2781 —14146.9/Т)
1070 А( 12,5)=ЕХР(-2.5548+659.9/Т) 1670 А(5,11)=ЕХР(—4.2492+825/Т)
1080 А(13,5)=ЕХР(—2.1113+779.7/Т) 1680 А(6,11)=ЕХР( —1.1238+2.7/Т)
1090 А(14,5)=ЕХР( 1.5229-921.5/Т) 1690 А(13,11)=ЕХР(3.32-775.3/Т)
1100 А(15,5)=ЕХР(.2189+226.8/Т) 1700 А(1,12)=ЕХР(—15.2623+515/Т)
1110 А(16,5)=ЕХР(—4.5933+1450.9/Т) 1710 А(2,12)=ЕХР(—5.8807-9/Т)
1120 А( 17,5)=ЕХР(4.4468— 1847.2/Т) 1720 А(3,12)=ЕХР(-.5812-249.3/Т)
ИЗО А(18,5)=ЕХР(—2.6892+919.5/Т) 1730 А(4,12)=ЕХР(—2.0465 +10.5/Т)
1140 А( 1,6)=ЕХР(—41.2503+7686.4/Т) 1740 А(5,12)=ЕХР(—2.4686+565.7/Т)
1150 А(2,6)=ЕХР(—.1595-248.2/Т) 1750 А(6,12)=ЕХР(.0583-455.3/Т)
1160 А(3,6)=ЕХР(2.2682-1111.5/Т) 1760 А(9,12)=ЕХР(—2.5152+489.5/Т)
1170 А(4,6)=ЕХР(— 11.9939-2231.6/Т) 1770 А( 13,12)=ЕХР(—2.132+228.5/Т)
1180 А(5,6)=ЕХР(1.4318-280.2/Т) 1780 А(23,12)=ЕХР(—23.0752+7174.8/Т)
1190 А(9,6)=ЕХР(—.3283 + 1.3/Т) 1790 А(25,12)=ЕХР(—16.8658-3637.1/Т)
1200 А(10,6)=ЕХР(.9348- 152.2/Т) 1800 А(26,12)=ЕХР(—2.8851—27.1/Т)
1210 А(11,6)=ЕХР(—.8049+4.4/Т) 1810 А(1,13)=ЕХР(9.7236-3797.5/Т)
1220 А( 12,6)=ЕХР(—.0296+2.6/Т) 3040 А(9,31 )=ЕХР(—.0999-247.9/Т)
1230 А(13,6)=ЕХР( 1.7—664.5/Т) 3050 А( 10,31)=ЕХР(. 137+2.5/Т)
1240 А( 15,6)=ЕХР(—.5965+2.2/Т) 3060 А(21,31)=ЕХР(— 1.5109+244.8/Т)
1250 А(16,6)=ЕХР(—.1045-91.9/Т) 3070 А(25,31)=ЕХР(—.2393+138.5/Т)
1260 А( 18,6)=ЕХР(5.9417-1834.8/Т) 3080 А(26,31)=ЕХР(.7392—255.8/Т)
1270 А(19,6)=ЕХР(2.6807-115.6/Т) 3090 FORK=1 ТО 31
1280 А(21,6)=ЕХР(2.8755—916.7/Т) 3100 А(К,К)=1
1290 А(23,6)==ЕХР(8.9682—2889.4/Т) 3110 NEXT К
1300 А(25,6)=ЕХР(—2.2978+605.7/Т) 3120 J=1
1310 А(26,6)=ЕХР(—9.7985+2539/Т) 3130 FOR К=1 ТО Р
1320 А(29,6)=ЕХР(.7523-589.5/Т) 3140 FORL=1 ТО Р
1330 А(30,6)=ЕХР(—.8586—421.2/Т) 3150 B(K,L)=A(K1(K),L1(L))
1340 А(31,6)=ЕХР(.9985-4056.7/Т) 3160 NEXT L
1350 А(1,7)=ЕХР(—6.5482+2719.5/Т) 3170 NEXT К
1360 А(3,7)=ЕХР(.6483-252/Т) 3180 FORJ=1 TO Q
1370 А(1,8)=ЕХР( 13.6669-5554/Т) 3190 INPUT X(J)
1380 А(3,8)=ЕХР(—7.6896—7.5/Т) 3200 NEXT J
1390 А(5,8)=ЕХР(—7.9975+2397/Т) 3210 REM *CALC OF X1(K)
1400 А( 10,8)=ЕХР(—4.2085+2478.6/Т) 3220 K=1
1410 А(1,9)=ЕХР(2.6172-865.1/Т) 3230 GOSUB 4140
1420 А(2,9)=ЕХР(.1444+40.5/Т) 3240 K=K+1
1430 А(3,9)=ЕХР(.9273-185.8/Т) 3250 IF K>=P+1 THEN 3280 ELSE 3260
1440 А(4,9)=ЕХР(3.2821 —1042.6/Т) 3260 GOSUB 4140
1450 А(5,9)=ЕХР(.0585—278.8/Т) 3270 GOTO 3240
1460 А(6,9)=ЕХР(—.7262+2.9/Т) 3280 S2=0
1470 А(10,9)=ЕХР( 1.0836+1.1/Т) 3290 FORJ=1 TO Q
1480 А(12,9)=ЕХР(—.1212+180/Т) 3300 S2=S2+X(J)»S(J)
1490 А(13,9)=ЕХР( 1.8864-543/Т) 3310 NEXT J
1500 А(16,9)=ЕХР(.О13- 152.5/Т) 3320 FOR K= 1 TO P
1510 А(21,9)=ЕХР(.0617+1.9/Т) 3330 X1(K)=S1(K)/S2
1520 А(23,9)=ЕХР(.9482-408/Т) 3340 NEXT К
1530 А(25,9)=ЕХР(.3823+3.1/Т) 3350 REM »CALC OF D(K)
1540 А(26,9)=ЕХР(.8583—252.5/Т) 3360 K=1
1550 А(31,9)=ЕХР(—.1141—88.2/Т) 2370 GOSUB 4200
1560 А(1,10)=ЕХР(—.09+32.4/Т) 3380 K=K+1
1570 А(6,10)=ЕХР(—.671 —150.8/Т) 3390 IF K>=P+1 THEN 3420 ELSE 3400
1580 А(8,10)=ЕХР(-5.3133+1673/Т) 3400 GOSUB 4200
1590 А(9,10)=ЕХР(—.8071 + 1.8224/Т) 3410 GOTO 3380
1600 А(21,10)=ЕХР(.2035+1.4/Т) 3420 REM *CALC OF C(K),G(K)
1610 А(23,10)=ЕХР( 16.7667-5200.2/Т) 3430 L=1
1620 А(24,10)=ЕХР(.5476-122.6/Т) 3440 GOSUB 4260
3450 L=L+1
3460 IF L>=P+1 THEN 3490 ELSE 3470
3470 GOSUB 4260
3480 GOTO 3450
3490 FORK=1 TO P
3500 G(K)=1-C(K)-LOG(D(K))
3510 NEXT К
3520 REM *CALC OF X2(K,J)
3530 J=1
3540 GOSUB 4320
3550 J=J+1
3560 IF J>=Q+1 THEN 3590 ELSE 3570
3570 GOSUB 4320
3580 GOTO 3550
3590 REM *CALC OF D1(K,J)
3600 J=1
3610 GOSUB 4370
3620 J=J+1
3630 IF J>=Q+1 THEN 3660 ELSE 3640
3640 GOSUB 4370
3650 GOTO 3620
3660 REM *CALC OF C1(L,J)
3670 J=1
3680 L=1
3690 GOSUB 4460
3700 L=L+1
3710 GOSUB 4460
3720 L=L+1
3730 IF L>=P+1 THEN 3760 ELSE 3740
3740 GOSUB 4460
3750 GOTO 3720
3760 J=J+1
3770 IF J>=Q+1 THEN 3780 ELSE 3680
3780 REM *CALC OF G1(K,J)
3790 FOR K=1 TO P
3800 FOR J=1 TO Q
3810 G1(K,J)=1-C1(K,J)-LOG(D1(K,J))
3820 NEXT J
3830 NEXT К
3840 REM *CALC OF G2(J)
3850 J=1
3860 G2(J)=0
3870 FOR K=1 TO P
3880 G2(J)=G2(J)+N(K,J)*(G(K)~G1(K,J))
3890 NEXT К
3900 J=J+1
3910 IF J>=Q+1 THEN 3920 ELSE 3860
3920 REM *CALC OF G3(J),G4(J)
3930 S3=0
3940 FOR J=1 TO Q
3950 S3=S3+N1(J)*X(J)
3960 NEXT J
3970 FOR J=1 TO Q
3980 R(J)=N1(J)/S3
3990 NEXT J
4000 FOR J= 1 TO Q
4010 G3(J)=1-R(J)+LOG(R(J))
4020 NEXT J
4030 FOR J=1 to Q
4040 G4(J)=EXP(G3(J)+G2(J))
4050 NEXT J
4060 REM *PRINT RESULTS
4070 PRINT USING 4110 ; T
4080 FOR J=1 TO Q
4090 PRINT USING 4120 ; X(J),G4(J)
4100 NEXT J
4110 IMAGE “T=”DDD.D
4120 IMAGE “X=”D.DD,2X,“GAM=”DDD.DDDD
4130 END
4140 REM #SR FOR S1(K)
4150 Sl(K)=0
4160 FOR J=1 TO Q
4170 S1(K)=S1(K)+X(J)*N(K,J)
4180 NEXT J
4190 RETURN
4200 REM *SR FOR D(K)
4210 D(K)=0
4220 FORL=1 TO P
4230 D(K)=D(K)+X1(L)*B(K,L)
4240 NEXT L
4250 RETURN
4260 REM *SR FOR C(K)
4270 C(L)=0
4280 FORK=1 TO P
4290 C(L)=C(L)+X1(K)*B(K,L)/D(K)
4300 NEXT К
4310 RETURN
4320 REM *SR FOR X2(K,J)
4330 FORK=1 TO P
4340 X2(K,J)=N(K,J)/S(J)
4350 NEXT К
4360 RETURN
4370 REM *SR FOR D1(K,J)
4380 K=1
4390 Dl(K,J)=0
4400 FORL=1 TO P
4410 D1(K,J)=D1(K,J)+X2(L,J)*B(K,L)
4420 NEXT L
4430 K=K+1
4440 IF K>=P+1 THEN 4450 ELSE 4400
4450 RETURN
4460 REM *SR FOR C1(L,J)
4470 C1(L,J)=O
4480 FORK=1 TO P
4490 C1(L,J)=C1(L,J)+X2(K,J)*B(K,L)/D1(K,J)
4500 NEXT К
4510 RETURN
4520 END
Таблица В. 8. Расчет пределов растворимости при помо-
щи уравнений ван Лаара и Скэтчарда — Гильдебранда.
В примере 7.5 в этих же целях используется уравнение
Маргулеса
10 ! SOLUBILITY LIMITS WITH THE VAN LAAR
EQNS “VLAAR”
20 ! SPECIFY PARAMETERS A & B, AND FIND
CORRESPONDING X & XI BY TRIAL WITH
THE N-R METHOD
30 SHORT X,X1
40 INPUT A,В
Приложения 575
50 REM *INPUT TRIAL VALUES OF X&X1
60 INPUT X,X1
70 G1=EXP(A/(1+A»X/B/(1—X))~2)
80 G2=EXP(B/(1+B*(1-X)/(A»x)f2)
90 G3=EXP(A/1+A»X1/B/(1—Xl))~2)
100 G4=EXP(B/( 1 +B*( 1 -X1 )/(A»X 1))~2)
110 Dl=—(2»A~2/(B»(1—X)~2*(1+A*X/(B*(1—X)))
3)*G1)
120 D2=2*E~2/(A*X''4(1-I-B*(1—X)/(A*X))~3)*G2
130 D3=-(2*A'2/(B*( 1-X1 )'2*( 1 +A»X 1/(B»( 1 -X1)))
3)*G3)
140 D4=2»B ~2/(A*X Г 2*( 1+B*( 1 -X1 )/(A*X 1))~3)*G4
150 M1=G1 + X*D1
160 M2=-G3-X1*D3
170 M3=—G2+(l—X)*D2
180 M4=G4-(1- X1)*D4
190 F=G1*X-G3*X1
200 G=G2»(1—X)—G4*(l—XI)
210 D=M1*M4-M2*M3
220 H=(F»M4-G*M2)/D
230 K=(G»M1-F»M3)/D
240 IF ABS(H/X)+ABS(K/X1)<= 0001 THEN 280
ELSE 250
250 X=X-H
260 X1=X1-K
270 GOTO 70
280 PRINT “A=”;A
290 PRINT “B=”; В
300 PRINT “X=”;X
310 PRINT “X»=”;X1
320 END
A= 4 A= 2
B= 2.5 B = 2.5
X= .021689 X = .28881
X* = .88404 X* =.83242
Таблица В. 9. Рассмотрение состояния равновесия двух
жидких фаз, включающих два, три или четыре компо-
нента, с использованием параметров уравнения Цубо-
ки — Катаямы — Вильсона
Параметры и соотношения объемов даны в виде
последовательностей: Ли, Ап, Ап, ..., А21, А22, А23, ...,
.... Иц, Ии, Ии, ..., Г21, V22, V23, ..., ... . Для систем
с тремя и четырьмя компонентами приводятся отдель-
ные программы. Расчет для двухкомпонентной систе-
мы выполняется при помощи программы, которая
используется в случае наличия трех компонентов.
Vij = Vj/Vi.
3. Задайте начальную величину общего состава
Zi = Z3 = 0,5, Z2 = 0 или некоторую другую величину
состава, промежуточную между значениями взаимной
растворимости двух растворителей.
4. Сходимость наступает наиболее быстро при та-
кой величине общего состава, которая дает приблизи-
тельно равное разделение фаз (F = 0,5).
5. Оптимизация сходимости достигается путем
подгонки а к некоторой величине, находящейся между
нулем и единицей, хотя в качестве исходной величины
в программах используется а - 1 (шаг 260,
F = F + аН).
6. В программе показаны последовательные ап-
проксимации ф и конечные разности (ytx, - yi'xi'), кото-
рые должны составлять примерно нуль.
Двухкомпонентная система
Задайте полную величину Ац, состоящую из девяти
элементов, и матрицы Уц, а вместо параметров третье-
го компонента (псевдокомпонента) запишите единицу.
Таким образом,
[Л] =
1
^21
1
^12
1
1
1 1 Г 1 и12 1
1 , (И= И21 1 1
1 ill
В операторе 100 программы введите Z3 = 0. Замените
Xi в операторе 120: 0,9, 0,1, 0; или оставьте их такими,
какими они записаны в программе.
Четырехкомпонентная система
1. Примите компонент 2 за растворенное вещество.
2. В качестве исходных величин примите: xi = 0,91,
хг = хз = Х4 = 0,03; X* = 0,1, хотя для улучшения схо-
димости их значениями можно варьировать.
3. За исходную величину общего состава можно
принять: Z2 = 0, Zi = Z3 = Z4 = 1/3, хотя если эта вели-
чина окажется в однофазной области, может возник-
нуть необходимость в ее подгонке.
В качестве примера взята система гексан
(1) + этанол (2) + ацетонитрил (3). Входные данные:
"1 0.0818 0.1002 0.4308 1 0.4159 0.1641 0.5677 1
"1 0.4465 0.4159
1И = 2.2400 1 0.8991
2.4909 1.1122 1
В таблице даны величины разделения фаз, F, обще-
го состава, Zi, и соответствующие значения состава для
некоторых случаев:
Трехкомпонентная система
1. Примите, что парой наиболее плохо смешиваю-
щихся компонентов являются компоненты 1 и 2.
2. В качестве исходных величин примите: Xi = 0,9,
Х2 = Хз = 0,05, х* - 0,1, хотя для улучшения сходимос-
ти этими значениями можно варьировать.
1 2 3
F= .49228
Z .5000 0.0000 .5000
X .9149 0.0000 .0851
X* .0978 0.0000 .9022
1 2 3
z F= .46986 .4500 .1000 .4500
. x .8156 .0689 .1155
X* .1260 .1275 .7465
z F= .42381 .4000 .2000 .4000
X .7194 .1364 .1442
X* .1651 .2468 .5882
z F= .33479 .3500 .3000 .3500
X .7028 .1759 .1213
X* .1724 .3625 .4651
z F= .25318 .3000 .4000 .3000
X .8012 .1397 .0591
X* .1301 .4882 .3817
Таблица В.9А. Расчет равновесных составов двух жид-
ких фаз, состоящих из двух или трех компонентов, при
помощи уравнения Цубоки — Катаямы — Вильсона
10 ! “W-14” LIQ-LIQ EQUILIB WITH T-K-WILSON
FOR 3 CPDS.
30 OPTION BASE 1
40 N=3
50 DIM A(3,3),V(3,3),X(3),X1(3),Z(3)
60 Xl(l)=.l
70 MAT READ A,V
80 DATA 1,.0818,.1002,.4308,1,.4159,.1641,.5677,1
90 DATA 1,2.24,2.491,.4465,1,1.1122,.4015,.8991,1
100 MAT INPUT Z
110 MAT READ X
120 DATA .9,.05,.05
130 F=(Z(1)-X1(1))/(X(1)-X1(1))
140 FOR 1=2 TO N
150 X1(I)=(Z(I)-X(I)*F)/(1-F)
160 NEXT I
170 GOSUB 550
180 S=0
190 T=0
200 FOR 1=1 TO N
210 K(I)=G1(I)/G(I)
220 S= S+ Z(I)*(K(I)~ 1)/(1 +(K(I)-1 )*F)
230 T=T+Z(I)*((K(I)-1)/(1+(K(I)-1)*F)) 2
240 NEXT I
250 H=S/T
260 F=F+H
270 DISP F
280 Sl=0
290 Tl=0
300 FOR 1=1 TO N
310 X1(I)=Z(I)/(1+(K(I)-1)*F)
320 IF Xl(I)<0 THEN 330 ELSE 340
330 Xl(I)=0
340 X(I)=K(I)*X1(I)
350 S1=S1+X(I)
360 T1=T1+X1(I)
370 NEXT I
380 FOR 1=1 TO 3
390 X(I)=X(I)/S1
400 X1(I)=X1(I)/T1
410 NEXT I
420 IF ABS(H/F)<=.001 THEN 430 ELSE 170
430 PRINT “F=”;F
435 PRINT
440 MAT PRINT USING 450 ; COL Z
450 IMAGE D.DDDD,2X
460 MAT PRINT USING 470 ; COL X
470 IMAGE D.DDDD,2X
480 MAT PRINT USING 490 ; COL XI
490 IMAGE D.DDDD,2X
500 FOR J=1 TO 3
510 Y(J)=G(J)*X(J)-G1(J)*X1(J)
520 DISP Y(J)
530 NEXT J
540 END
550 ! SR FOR ACT COEFFS
560 A=0
570 B=0
580 C=0
590 Al=0
600 В 1=0
610 Cl=0
620 A2=0
630 B2=0
640 C2=0
650 A3=0
660 B3=0
670 C3=0
680 FORK=1 TO N
690 A=A+X(K)*A(1,K)
700 A1=A1+X(K)#V(1,K)
710 A2=A2+X1(K)*A(I,K)
720 A3=A3+X1(K)*V(1,K)
730 B=B+X(K)*A(2,K)
740 B1=B1+X(K)*V(2,K)
750 B2=B2+X1(K)*A(2,K)
760 B3=B3+X1(K)*V(2,K)
770 C=C+X(K)*A(3,K)
780 C1=C1 + X(K)*V(3,K)
790 C2=C2+X1(K)*A(3,K)
800 C3=C3+X1(K)*V(3,K)
810 NEXT К
820 G=LOG(A1/A)+X(1)*(1/A1-1/A)+X(2)*(V(2,1)/
Bl-A(2,l)/B)
830 G( 1 )=EXP(G+ X(3)*(V(3,1 )/C 1 -A(3,1 )/C))
840 G=LOG(B 1 /В)+ X( 1 )*( V( 1,2)/A 1 -A( 1,2)/A)+ X(2)
*(1/B1- 1/B)
850 G(2)=EXP(G+X(3)*(V(3,2)/C1-A(3,2)/C))
860 G=LOG(C1/C)+X(1)*(V(1,3)/A1—A(l,3)/A)+X(2)
*(V(2,3)/B1—A(2,3)/B)
870 G(3)=EXP(G+X(3)*(1/C1~1/C))
880 G=LOG(A3/A2)+X1(1)*(1/A3-1/A2)+X1(2)*(V
(2,1)/B3-A(2,1)/B2)
890 Gl( 1 )=EXP(G+X1 (3)*(V(3,1 )/C3—A(3,1 )/C2))
900 G=LOG(B3/B2)+X1 (1 )*(V( 1,2)/A3~A( 1,2)/A2)+
Приложения 577
910 Xl(2)<l/B3- 1/В2) G1(2)=EXP(G+X1(3)*(V(3,2)/C3-A(3,2)/C2)) 470 IMAGE D.DDDD,2X 480 MAT PRINT USING 490 ; COL Xl,
920 G=LOG(C3/C2)+X1( 1 )*(¥( 1,3)/A3~A( 1,3)/А2)+ 490 IMAGE D.DDDD,2X
X1(2)*V(2,3)/B3—А(2,3)/В2) 500 FOR J=1 TO 3
930 G1(3)=EXP(G+X1(3)*(1/C3—1/С2)) 510 Y(J)=G(J)*X(J)—G1(J)*X1(J)
940 RETURN 520 DISP Y(J)
950 END 530 NEXT J
540 END
550 ! SR FOR ACT COEFFS
Таблица В. 10. Расчет равновесных составов двух жид- 560 A=0
ких фаз, состоящих из четырех компонентов, при помо- 570 B=0
щи уравнения Цубоки — Катаямы — Вильсона 580 C=0
590 D=0
10 ! “W-l 3” LIQ-LIQ EQUILIB WITH T-K-WILSON 600 A 1=0
EQN FOR 4 CPDS 610 Bl=0
20 SHORT F 620 Cl=0
30 OPTION BASE 1 630 Dl=0
40 N=4 640 A2=0
50 DIM A(4,4),V(4,4),X(4),X1(4),Z(4) 650 B2=0
60 Xl(l)=.l 660 C2=0
70 MAT READ A,V 670 D2=0
80 ! DATA FOR A, 16 ITEMS 680 A3=0
90 ! DATA FOR V, 16 ITEMS 690 B3=0
100 MAT INPUT Z 700 C3=0
ПО MAT READ X 710 D3=0
120 DATA .9, .05, .03, .02 720 FORK=1 TON
130 F=(Z(1)-X1(1))/(X(1)-X1(1)) 730 A=A+X(K)*A(1,K)
140 FOR 1=2 TO N 740 A1=A1+X(K)*V(1,K)
150 X1(I)=(Z(I)-X(I)*F)/(1-F) 750 A2=A2+X1(K)*A(1,K)
160 NEXT I 760 A3=A3+X1(K)*V(1,K)
170 GOSUB 550 770 B=B+X(K)*A(2,K)
180 S=0 780 B1=B1 + X(K)*V(2,K)
190 T=0 790 B2=B2+X1(K)*A(2,K)
200 FOR 1= 1 TO N 800 B3=B3+X1(K)*V(2,K)
210 K(I)=G1(I)/G(I) 810 C=C+X(K)*A(3,K)
220 S=S+Z(I)*(K(I)-1)/(1 +(K(I)-1 )*F) 820 C1=C1+X(K)*V(3,K)
230 T=T+Z(I)*((K(I)~ 1)/(1+(K(I)~ 1)*F))"2 830 C2=C2+X1(K)*A(3,K)
240 NEXT I 840 C3=C3+X1(K)*V(3,K)
250 H=S/T 850 D=D+X(K)*A(4,K)
260 F=F+H 860 D1=D1 + X(K)*V(4,K)
270 DISP F 870 D2=D2+X(K)*A(4,K)
280 Sl=0 880 D3=D3+X(K)*V(4,K)
290 Tl=0 890 NEXT К
300 FOR 1= 1 TO N 900 G= LOG(A1 /А)+ X( 1 )*( 1/A1 -1 /A)+ X(2)*(V(2,1)/
310 X1(I)=Z(I)/(1+(K(I)~1)*F) Bl- A(2,l)/B)
320 IF Xl(I)<0 THEN 330 ELSE 340 910 G(1)=EXP(G+X(3)*(V(3,1)/C1-A(3,1)/C)+X(4)*
330 Xl(I)=0 (V(4,1)/D1-A(4,1)/D))
340 X(I)=K(I)*X1(I) 920 G=LOG(B1/B)+X( 1 )*(V( 1,2)/A 1 ~A( 1,2)/A)+X
350 S1=S1 + X(I) (2)*(1/B1 —1/B)
360 T1=T1 + X1(I) 930 G(2)=EXP(G+X(3)*(V(3,2)/C1-A(3,2)/C)+X(4>
370 NEXT I (V(4,2)/D1-A(4,2)/D))
380 FOR 1=1 TO 3 940 G=LOG(C1/C)+X(1)»(V(1,3)/A1—A(l,3)/A)+X
390 X(I)=X(I)/S1 (2)*(V(2,3)/B1~A(2,3)/B)
400 X1(I)=X1(I)/T1 950 G(3)=EXP(G+X(3)*(1/C1~ 1/C)+X(4)*(V(4,3)/
410 NEXT I DI- A(4,3)/D))
420 IF ABS(H/F)<=.001 THEN 430 ELSE 170 960 G= LOG(D 1 /D)+X( 1 )*(V( 1,4)/A 1 -A( 1,4)/A)+ X
430 PRINT “F=”;F (2)*(V(2,4)/B1-A(2,4)/B)
440 MAT PRINT USING 450 ; COL Z 970 G(4)=EXP(G+X(3)*(V(3,4)/C1-A(3,4)/C)+X(4)*
450 IMAGE D.DDD,3X (1/D1-1/D))
460 MAT PRINT USING 470 ; COL X 980 G=LOG(A3/A2)+X1(1)*(1/A3-1/A2)+X1(2)*(V
18-864
(2,1)/B3—A(2,l)/B2)
990 G1(1)=EXP(G+X1(3)*(V(3,1)/C3-A(3,1)/C2)+
X1(4)*(V(4,1)/D3-A(4,l)/D2))
1000 G=LOG(B3/B2)+X1 (1)4 V( 1,2)/A3-A( 1,2)/A2)+
Xl(2)*(l/B3— 1/B2)
1010 G1(2)=EXP(G+X1(3)*(V(3,2)/C3-A(3,2)/C2)+
X1(4)*(V(4,2)/D3-A(4,2)/D2))
1020 G=LOG(C3/C2)+X1(1)*(V(1,3)/A3-A(1,3)/A2)+
X1(2)*(V(2,3)/B3~A(2,3)/B2)
1030 G1(3)=EXP(G+X1(3>(1/C3-1/C2)+X1(4)*(V
(4,3)/D3-A(4,3)/D2))
1040 G=LOG(D3/D2)+X1( 1 )*(V( 1,4)/A3-A( 1,4)
/A2)+X1(2)*(V(2,4)/B3-A(2,4)/B2)
1050 G1(4)=EXP(G+X1(3)*(V(3,4)/C3-A(3,4)/C2)+
Xl(4)*(l/D3—1/D2))
1060 RETURN
1070 END
Таблица B.ll. Расчет коэффициентов активности мно-
гокомпонентных смесей при помощи уравнения
Вильсона
10 ! ACT COEFF WITH WILSON EQN, “C.l 1”
20 ! ENTER X(J) ON LINE 90
30 ! ENTER MATRIX OF PARAMS, A(I,J), AS
SHOWN AT END. I IS THE ROW NO., J THE
COLUMN NO.
40 ! EXAMPLE IS OF A FOUR COMPONENT
SYSTEM. CHANGE LINES 60 AND 70 FOR
OTHER CASES
50 OPTION BASE 1
60 N=4
70 DIM X(4),A(4,4),G(4)
80 MAT READ X
90 DATA ,1,.2,.2,.5
100 MAT READ A
110 DATA 1,2.2,3.3,.44,.22,1,1.1,.11,.22,.55,1,.33,.66,
.77,.88,1
120 FORK=1 TO N
130 Gl=0
140 FOR J=1 TO N
150 G1=G1+X(J)*A(K,J)
160 NEXT J
170 G2=0
180 FOR 1=1 TO N
190 G3=0
200 FOR J=1 TO N
210 G3=G3+X(J)*A(I,J)
220 NEXT J
230 G2=G2+X(I)*A(I,K)/G3
240 NEXT I
250 G(K)=EXP(1-LOG(Gl)—G2)
260 PRINT USING 270 ; X(K),G(K)
270 IMAGE D.DDDD,4X,DD.DDDD
280 NEXT К
290 END
xi И
.1000 1.0341
.2000 1.6336
.2000 1.1395
.5000 1.4102
i X 1 2 3 4
i 1 2.2 3.3 .44
2 .22 1 1.1 .11
3 .22 .55 1 .33
4 .66 .77 .88 1
Приложение Г. Физические свойства
Данные о физических свойствах можно получить ли-
бо непосредственно из литературы и подборок стан-
дартных данных, либо их можно рассчитать, используя
соответствующие корреляции или исходя из молекуляр-
ной структуры. Описание последнего метода можно
найти в работах [25, 80, 107, 129]. По оценкам специа-
листов, ежегодно публикуется примерно 35 000 —
50 000 статей, содержащих около 2 млн. данных; следо-
вательно, для того чтобы быть в курсе всей информа-
ции, необходим машинный поиск, который выполняют
несколько систем. Ряд перечней подборок данных мож-
но найти в следующих изданиях:
1. Fratzcher W., Picht Н.Р., Bittrich H.J. The acquisition,
collection and tabulation of substance data on fluid systems
for calculations in chemical engineering. Int. Chem. Eng.
20 (1), 19 — 28 (1980).
2. Maizell R.E. How to Find Chemical Information.
Wiley (1978).
3. Mellon M.G. Cheemical Publications: Their Nature and
Use, McGraw-Hill (1982).
4. Rasmussen R, Fredenslund A. Data Banks for Chemi-
cal Engineers. Kemiingeniorgruppen (Lyngby, Demark)
(1980).
5. Torres-Marchal C. Getting chemical engineering infor-
mation from non-English-speaking countries. Chem. Eng.
88 (14), 117 — 125 (July 13, 1981).
6. Physico-Chemical Properties for Chemical Engineering.
Подготовлено Японским обществом инженеров-
химиков. Выходит практически ежегодно, начиная с
19717 г. Содержит библиографическое описание литера-
туры, рефераты теоретических статей и указания на
вид данных, содержащихся в источнике. Рассматрива-
ются данные о PVT, плотности жидкостей, давлении
паров, энтальпии испарения, теплоемкости и энталь-
пии, парожидкостном равновесии, растворимости и
теплоте растворения, вязкости, теплопроводности и
молекулярной диффузии.
Несколько банков данных поставляют за плату ин-
формацию по запросам пользователей относительно
свойств веществ. Некоторые из источников информа-
ции указаны в книге Расмуссена (см. выше), а также в
следующих изданиях:
Приложения 579
7. Peterson J.N., Chen С.-С, Evans L.B. Computer prog-
rams for chemical engineers. Chem. Eng. 145 — 154 (June
5, 1978); 69 — 82 (July 3, 1978); 79 — 86 (July 31, 1978);
107 — 115 (August 28, 1978); 167 — 173 (May 21, 1979).
Prepared in connection with the ASPEN project.
8. Hilsenrath J. Summary of on-line or interactive
physicochemical numerical data systems. National Bureau of
Standards Technical Note 1122 (1980).
9. Leesley M.E. Computer-Aided Process Plant Design.
Gulf Publishing Company (1982).
Американский институт химических инженеров пред-
лагает подобного рода услуги своим абонентам, в буду-
щем ими смогут воспользоваться все желающие.
Данные для 72 веществ, которые приводятся в насто-
ящем приложении, заимствованы главным образом из
списка данных для 468 веществ.
Другая подборка данных для 176 веществ взята из
моделирующей программы и приведена в следующем
издании:
10. Henley E.J., Seader J.D. Equilibrium-Stage Separation
Processes in Chemical Engineering. Wiley (1981).
Обширные подборки данных о фазовом равновесии
помещены в книгах (46, 68, 73, 82, 57, 124, 125]. Библи-
ография литературы о равновесных свойствах жидко-
стей опубликовали Хиза, Кидней и Мимер [4].
Параметры уравнения для расчета констант равнове-
сия образования, приведенные в табл. Г.1,
lgX„ = А + В/Т
получены методом регрессии табличных данных, ука-
занных в книге Сталла и др. [130] для температур в диа-
пазоне 298—1000 К. Данные, приведенные в табл. Г.2,
в основном взяты из таблицы, опубликованной Райдом
[107], исключение составляют лишь параметры раство-
римости: они заимствованы из других источников.
Таблица Г.1. Константы равновесия образования
IgXp = А + 1000 В/Т в диапазоне температур
298 — 1000 К
Формула Наименование А в
НС1 Хлористый водород 0,4285 4.852
Н2О Вода -2,6322 12,7401
H2S Сульфид водорода 1,1627 1,4390
H3N Аммиак -5,7596 2,5853
h4n2 Гидразин -11,8129 -4,7331
so2 Диоксид серы -0,2973 15,8165
so3 Триоксид серы -5,1933 20,9378
COC12 Фосген -2,4317 11,5237
CC14 Тетрахлорид углерода -7,0782 5,1369
co Моноксид углерода 4,7272 5,7622
co2 Диоксид углерода 0,1165 20,5658
cs2 Дисульфид углерода 7,3293 -5,5939
CHC13 Хлороформ 5,8676 5,3323
CH2O2 Муравьиная кислота -7,0563 20,5541
CH4 Метан -5,1030 4,2138
CH4O Метанол -7,7901 10,8527
C2H2 Ацетилен 2,9533 -11,8005
c2h3n Ацетонитрил -3,7480 -4,3729
C2H4 Этилен -3,6279 -2,4422
Продолжение табл. Г.1
Формула Наименование А в
С2Н4О Ацетальдегид -6,7327 9,0089
С2Н4О2 Уксусная кислота -11,1003 23,0204
с2н6 Этан -10,4383 4,8874
С2Н6О Этанол -13,0030 12,7215
с3н6 Пропилен -8,7127 -0,6258
С3Н6О Ацетон -12,6811 11,8304
С3Н8 Пропан -15,8040 6,0076
С3Н8О 1-Пропанол -18,8086 14,5270
С3Н8О Изопропанол -18,9711 14,7867
С4Н6 1,2-Бутадиен -7,4546 -8,0979
С4Н6 1,3-Бутадиен -8,1031 -5,4136
С4Н8 1-Бутен -14,1659 0,5660
СД18 t/wc-2-Бутен -14,6256 0,9963
С4Н8 щраяс-2-Бутен -14,6446 1,1448
С4н8о Метилэтилкетон -17,7890 12,9993
С4н8о2 1,4-Диоксан -25,4654 17,1068
С4н8о2 Этилацетат -21,8841 23,6913
С4Ню н-Бутан -21,1552 7,2844
С4Ню Изобутан -21,9428 7,7140
С4НюО н-Бутанол -23,7886 15,0466
С4НюО Изобутанол -23,8537 15,9293
С4НюО щре/п-Бутанол -25,5398 17,6692
С4Н10О Диэтиловый эфир -24,8400 13,8636
c5h5n Пиридин -9,9614 -6,9212
с5н8 Изопрен -13,5832 -3,5175
с5н12 н-Пентан -26,5977 8,4631
С5Н12 2-Метилбутан -26,8757 8,8811
С5Н12 2,2-Д иметилпропан -28,6731 9,4316
С5Н12О 1-Пентанол -29,2003 16,6242
с6н6 Бензол -9,5963 -3,8557
С6Н6О Фенол -12,2574 5,4163
c6h7n Анилин -15,3245 -4,0887
с6н12 Циклогексан -29,8950 7,3581
С6Н14 н-Гексан -32,0048 9,6646
С7Н6О2 Бензойная кислота -15,8402 15,7865
с7н8 Толуол -14,3874 -2,0150
С7Н16 н-Гептан -37,4102 10,8575
с8н8 Стирол -13,1807 -7,1768
С8Н10 Этилбензол -19,7509 -0,8499
С8Н18 н-Октан -42,0549 12,0549
Таблица Г. 2. Банк данных о свойствах веществ
Приведенные ниже данные взяты из приложения А
к книге Райда, Праузница и Шервуда «Свойства газов
и жидкостей» [107]; данный вариант текста подго-
товлен для CAST, Массачусетский технологический
институт, Дж. Бостоном. В скобках указаны коэффи-
циенты для пересчета в другие общепринятые
единицы.
Условные обозначения и единицы измерения
MW Молекулярная масса
TFP Нормальная точка замерзания, К
ТВ Нормальная точка кипения, К
18
тс Критическая температура, К 298 К, Н • м/кг-моль (X 2.388D - 4 =
PC Критическое давление, Н/м »* 2 = кал/моль)
(X 1/101,325 = атм). DGFORM Стандартная энергия образования соеди-
VC Критический объем, М ** 3/кг-моль нения Гиббса при 298 К. Идеальный
(=л/моль) газ, 1 атм, Н • м/кг-моль
zc Критическая сжимаемость (X 2.388D - 4 = кал/моль)
co Ацентрический коэффициент Питцера ANTI Коэффициенты уравнения Антуана для
VL Плотность жидкости при TREF, давления пара
кг/м *» 3 (г/л) ln(VP) = ANTI + ANT2/(T + ANT3) +
TREF Эталонная температура для системы + (ANT4 * T) + (ANT5 • ln(T)) +
жидкость—пар, К + (ANT6»T »» ANT7)
MUP Дипольный момент, (Н • м »» 4) »» 0,5 Давление пара выражено в Па, темпе-
(X 0.3162D25 = Дебай) ратура — в К.
CPIG1 Константы уравнения расчета теплоем- (X 1/101325 = атм)
кости идеального газа TMIN Минимальная температура для уравне-
CPIG4 CPIG1 + (CPIG2 * Т) + (CPIG3 • Т •• 2) + ния Антуана, К
+ (CPIG4 • Т •* 3) ТМАХ Максимальная температура для уравне-
(Н • м/кг-моль • К) и Т в К ния Антуана, К
(X 2.388D - 4 = кал(моль) (К) DHVB Теплота испарения для нормальной точ-
vise 1 Константа уравнения вязкости ки кипения, Н • м/кг-моль (X 2.388D -
жидкости, - 4 = кал/моль)
Н • с/м2 (X 1000 = сПз). 5 Параметр растворимости при 25 °C,
VISC 2 Константа уравнения вязкости жидкос- (кал/мл)0,5
ти, Klog(VISC) = (VISC)*((1/T)*(1/VISC)) 31 Параметр растворимости при 25 °C,
DHFORM Стандартная энтальпия образования при (мПа)0’5
АРГОН ХЛОР ХЛОРИСТЫЙ ВОДОРОД ВОДОРОД ВОДА СЕРОВОДОРОД
FORMULA AR CL2 HCL H2 H2O H2S
MW 0.39948000D+02 0.70906000D+02 0.36461000D+02 0.20160000D+01 O.18O15OOOD+O2 0.34080000D+02
TFP 0.83800000D+02 O.1722OOOOD+O3 O.159OOOOOD+O3 0.14000000D+02 O.2732OOOOD+O3 O.1876OOOOD+O3
ТВ 0.87300000D+02 O.2387OOOOD+O3 O.1881OOOOD+O3 0.20400000D+02 O.3732OOOOD+O3 0.21280000D+03
ТС O.15O8OOOOD+O3 0.41700000D+03 0.32460000D+03 0.33200000D+02 0.6473OOOOD+O3 O.3732OOOOD+O3
PC 0.48737325D+07 O.77OO7OOOD+O7 O.83O865OOD+O7 0.12969600D+07 0.22048320D+08 O.8936865OD+O7
VC 0.74900000D-01 0.12400000D+00 0.81000000D-01 0.65000000D-01 O.56OOOOOOD- 01 O.985OOOOOD- 01
ZC 0.29100000D+00 O.275OOOOOD+OO 0.24900000D+00 0.30500000D+00 0.22900000D+00 0.28400000D+00
OMEGA -0.40000000D-02 O.73OOOOOOD-01 0.12000000D+00 -22000000D+00 0.34400000D+00 0.10000000D+00
VL O.1373OOOOD+O4 O.1563OOOOD+O4 0.11930000D+04 0.71000000D+02 O.998OOOOOD+O3 0.99300000D+03
TREF O.9OOOOOOOD+O2 0.23910000D+03 O.1881OOOOD+O3 0.20000000D+02 O.293OOOOOD+O3 O.2136OOOOD+O3
MUP 0.0 0.63245552D-25 0.34785054D-24 0.0 0.56920997D-24 0.28460498D-24
CPIG1 0.20804209D+05 0.26929498D+05 0.30291498D+05 0.27143024D+05 0.32242547D+05 0.31941097D+05
CPIG2 -0.32112756D-01 0.33837718D+02 -0.72012960D+01 0.92737620D+01 0.19238346D+01 0.14364911D+01
CPIG3 0.51665112D—04 -0.38690219D-01 0.12459917D-01 -0.13808066D-01 0.10554923D-01 0.24321121D-01
CPIG4 0.0 0.15470226D- 04 -0.38979108D-05 0.76450968D-05 -0.35964612D-05 —0.11764908D—04
VISC1 O.1O757OOOD+O3 O.19196OOOD+O3 O.37278OOOD+O3 0.13820000D+02 O.65825OOOD+O3 O.34279OOOD+O3
VISC2 0.22268146D+02 0.46662679D+02 0.85850678D+02 0.24838213D+01 0.12362298D+03 0.67601597D+02
DHFORM 0.0 0.0 -0.92360808D+08 0.0 -0.24199704D+09 -0.20180376D+08
DGFORM 0.0 0.0 -O.95333436D+O8 0.0 -0.22876675D+09 -O.33O7572OD+O8
ANTI -0.73078490D+04 —0.36143310D+04 -0.64603120D+04 -0.51752110D+04 -0.31397400D+02 —0.93201210D+02
ANT2 0.17633770D+06 0.19292300D+06 0.28430820D+06 0.38036580D+05 -0.20463660D+04 —0.82474460D+00
ANT3 0.39162290D+02 0.10600010D+03 0.82323240D+02 0.82040620D+01 -0.75402240D+02 —0.18128770D+03
ANT4 -0.63849410D+01 —0.10226150D+01 -0.23612360D+01 -O.3O3O7O2OD+O2 -0.12054280D-01 -0.55504840D-01
ANT5 0.14404790D+04 0.60082960D+03 0.11128860D+04 0.14664860D+04 O.9165751OD+O1 0.21720190D+02
ANT6 0.58241780D-02 0.3616337OD-O3 0.10312470D-02 0.12734970D+00 0.4879195OD-17 0.66817760D-10
ANT7 0.20000000D+01 0.20000000D+01 0.20000000D+01 0.20000000D+01 0.60000000D+01 0.40000000D+01
TMIN O.838OOOOOD+O2 O.1722OOOOD+O3 O.159OOOOOD+O3 0.15660000D+02 O.2732OOOOD+O3 O.1876OOOOD+O3
TMAX O.15O8OOOOD+O3 0.41700000D+03 O.3246OOOOD+O3 O.332OOOOOD+O2 O.6473OOOOD+O3 O.3732OOOOD+O3
DHVB 0.65314080D+07 0.20431584D+08 0.16161048D+08 0.90434880D+06 0.40683136D+08 0.18673128D+08
5 5.33 8.708 7.0110 23.53 8.8
51 10.91 17.818 14.346 48.15 18.0
Приложения 581
АММИАК ГИДРАЗИН ГЕЛИЙ-4
FORMULA H3N H4N2 HE(4)
MW O.17O31OOOD+O2 0.32045000D+02 O.4OO3OOOOD+O1
TFP O.1954OOOOD+O3 O.2747OOOOD+O3 0.0
ТВ O.2397OOOOD+O3 O.3867OOOOD+O3 0.42100000D+01
TC O.4O56OOOOD+O3 O.653OOOOOD+O3 O.519OOOOOD+O1
PC 0.1127747 3D+O8 0.14692125D+08 0.22696800D+06
VC O.725OOOOOD+O1 O.961OOOOOD- 01 O.573OOOOOD-01
zc 0.24200000D+00 0.26000000D+00 0.30100000D+00
OMEGA 0.25000000D+00 0.32800000D+00 —O.387OOOOOD+OO
VL O.639OOOOOD+O3 0.10080000D+04 0.123OOOOOD+O3
TREF O.2732OOOOD+O3 O.293OOOOOD+O3 0.43000000D+01
MUP 0.47434164D-24 0.94868328D-24 0.0
CPIG1 0.27314683D+05 0.9767 8044D+04 0.0
CPIG2 0.23831266D+02 0.18945270D+03 0.0
CPIG3 0.17073770D—01 -0.16571354D+00 0.0
CPIG4 -0.11848644D-04 0.6024805 2D-04 0.0
VISC1 0.34904000D+03 0.52498000D+03 0.0
VISC2 0.69012222D+02 0.10926159D+03 0.0
DHFORM -0.45719856D+08 0.95249700D+08 0.0
DGFORM —0.16161048D+08 0.15863785D+09 0.0
ANTI -0.36891000D+02 —0.81.937220D+04 0.17144170D+02
ANT2 -0.80343580D+03 0.67364210D+06 -0.33732900D+02
ANT3 -0.66161870D+02 0.17465370D+03 O.179OOOOOD+O1
ANT4 -0.20785260D-01 -0.12760300D+01 0.0
ANT5 0.10587760D+02 0.12518600D+04 0.0
ANT6 0.77121770D-16 0.27092930D-03 0.0
ANT7 0.60000000D+01 0.20000000D+01 0.0
TMIN 0.19540000D+03 0.35915OOOD+O3 0.30000000D+01
TMAX 0.40560000D+03 O.653OOOOOD+O3 0.50000000D+01
DHVB 0.23362344D+08 0.44798760D+08 0.92109600D+05
5 12.408 18.11
Si 25.389 37.06
АЗОТ КИСЛОРОД ДИОКСИД СЕРЫ
N2 02 O2S
0.28013OOOD+O2 O.31999OOOD+O2 0.64063000D+02
O.633OOOOOD+O2 0.54400000D+02 0.19770000D+03
O.774OOOOOD+O2 0.90200000D+02 0.26300000D+03
O.1262OOOOD+O3 0.15460000D+03 0.43080000D+03
0.33943875D+07 0.5045 9850D+07 O.7883O85OD+O7
O.895OOOOOD-01 0.73400000D-01 0.12200000D+00
0.29000000D+00 0.28800000D+00 0.26800000D+00
0.40000000D-01 0.21000000D-01 0.25100000D+00
0.80400000D+03 0.11490000D+04 0.14550000D+04
O.781OOOOOD+O2 0.90000000D+02 0.26300000D+03
0.0 0.0 0.50596442D-24
0.31149792D+05 0.28105988D+05 0.23852200D+05
-0.13565232D+02 -0.36801972D-02 0.66988800D+02
0.26795520D-01 0.17458956D-01 -0.49613580D-01
-0.11681172D-04 -0.10651219D- 04 0.13280530D-04
O.9O3OOOOOD+O2 0.85680000D+02 O.39785OOOD+O3
0.18216343D+02 0.18371721D+02 0.81046903D+02
0.0 0.0 -0.29705346D+09
0.0 0.0 -0.30036103D+09
-0.19496430D+04 -0.14079980D+04 -0.18537110D+02
0.47772050D+05 0.39737770D+05 -0.15622620D+04
0.39737620D+02 0.48151280D+02 -0.45780700D+02
—0.13499100D+01 -0.79895110D+00 -0.17722480D-01
0.38108330D+03 0.26744380D+03 0.75143910D+01
0.13866070D-07 0.49293240D-08 0.35731280D-13
0.40000000D+01 0.40000000D+01 0.50000000D+01
0.63300000D+02 0.61840000D+02 O.1977OOOOD+O3
0.12620000D+03 0.15460000D+03 O.43O8OOOOD+O3
0.55810044D+07 0.68244840D+07 0.24932394D+08
4.40 4.0 6.0-
9.00 8.18 12.28
ТРИОКСИД СЕРЫ ФОСГЕН ТЕТРАХЛО- МОНОКСИД УГЛЕРОДА диоксид ДИСУЛЬФИД
РИД УГЛЕРОДА УГЛЕРОДА УГЛЕРОДА
FORMULA O3S CCL20 CCL4 со СО2 CS2
MW O.8OO58OOOD+O2 0.98916OOOD+O2 0.15382300D+03 0.28010000D+02 0.44010000D+02 0.76131000D+02
TFP O.29OOOOOOD+O3 O.145OOOOOD+O3 O.25OOOOOOD+O3 0.68100000D+02 0.21660000D+03 O.1613OOOOD+O3
ТВ O.318OOOOOD+O3 O.28O8OOOOD+O3 O.3497OOOOD+O3 O.817OOOOOD+O2 O.1947OOOOD+O3 O.3194OOOOD+O3
ТС 0.49100000D+03 O.455OOOOOD+O3 O.5564OOOOD+O3 0.1329OOOOD+O3 0.30420000D+03 O.552OOOOOD+O3
PC 0.82073250D+07 O.56742OOOD+O7 0.45596250D+07 0.34957125D+07 0.73764600D+07 O.79O335OOD+O7
VC 0.13000000D+00 0.19000000D+00 0.27600000D+00 O.931OOOOOD-01 0.94000000D-01 0.17000000D+00
ZC O.26OOOOOOD+OO O.28OOOOOOD+OO 0.27200000D+00 O.295OOOOOD+OO 0.27400000D+00 0.29300000D+00
OMEGA 0.41000000D+00 0.20400000D+00 0.19400000D+00 0.49000000D-01 O.225OOOOOD+OO 0.11500000D+00
VL 0.17800000D+04 0.13810000D+04 O.1584OOOOD+O4 O.8O3OOOOOD+O3 O.777OOOOOD+O3 0.12930000D+04
TREF O.318OOOOOD4-O3 O.293OOOOOD+O3 0.29800000D+03 O.81OOOOOOD+O2 O.293OOOOOD+O3 O.273OOOOOD+O3
MUP 0.0 0.34785054D-24 0.0 0.31622776D-25 0.0 0.0
CPIG1 0.45904D+5 0.28089241D+05 0.40716630D+05 0.30869276D+05 0.19795190D+05 0.27444474D+05
CPIG2 0.5240D+2 0.13607100D+03 0.20486012D+03 -0.12853476D+02 0.73436472D+02 0.81265788D+02
CPIG3 -0.2731D-1 -0.13736891D+00 -0.22696643D+00 0.27892462D-01 -0.56019384D-01 -0.76660308D-01
CPIG4 0.5560D-5 0.50702148D-04 0.88425216D-04 -0.12715312D-04 0.17153320D-04 0.26728531D-04
VISC1 O.13728OOOD+O4 0.0 0.54015OOOD+O3 0.94060000D+02 O.578O8OOOD+O3 O.274O8OOOD+O3
VISC2 0.18691687D+03 0.0 0.11120589D+03 0.19104228D+02 0.94446586D+02 0.62734410D+02
DHFORM -0.39552700D4-09 -0.22106304D+09 —O.1OO4832OD+O9 —0.11061526D+09 -0.39376854D+09 0.11714666D+09
DGFORM -0.37061554D+09 —0.20691166D+09 -0.58280256D+08 -0.13736891D+09 -0.39464777D+09 0.66946932D+08
ANTI 0.79498460D+05 0.20649270D+02 —0.3421OO8OD+O2 —0.30121770D+04 -0.52840260D+04 -0.18102110D+04
ANT2 -0.51465900D+07 —0.21673100D+04 -0.11587170D+04 0.62507530D+05 0.30915310D+06 0.14750340D+06
ANT3 0.12721910D+03 —0.4315OOOOD+O2 -0.87403690D+02 0.33139810D+02 0.11114140D+03 0.17363370D+03
ANT4 0.16615650D+02 0.0 - O.135OOO3OD-01 -0.33345090D+01 —O.1OO9929OD+O1 -0.23123160D+00
ANT5 -0.12628050D+05 0.0 0.93655890D+01 0.61935790D+03 0.85020940D+03 0.27678890D+03
ANT6 -0.44255980D-02 0.0 0.10799190D-16 0.37100650D-02 0.18866410D-14 0.31644950D-10
ANT7 0.20000000D4-01 0.0 0.60000000D+01 0.20000000D+01 0.60000000D+01 0.40000000D+01
TMIN 0.29000000D+03 O.213OOOOOD+O3 O.25OOOOOOD+O3 O.681OOOOOD+O2 O.16731OOOD+O3 0.22080000D+03
TMAX O.491OOOOOD+O3 O.341OOOOOD+O3 O.5564OOOOD+O3 O.1329OOOOD+O3 O.3O42OOOOD+O3 O.552OOOOOD+O3
DHVB 0.40678949D+08 0.24409044D+08 0.30019356D+08 0.60457392D+07 0.17165880D+08 0.26753652D+08
5 15.329 7.1200 9.338 3.1300 7.1200 9.8640
51 31.366 14.5691 19.108 6.4047 14.5691 20.1839
Приложения 583
ХЛОРОФОРМ МУРАВЬИНАЯ КИСЛОТА МЕТАН МЕТАНОЛ АЦЕТИЛЕН ВИНИЛХЛОРИД
FORMULA CHCL3 CH202 CH4 СН40 С2Н2 C2H3CL
MW 0.11937800D+03 0.46025000D+02 0.16043000D+02 0.32042000D+02 0.2603 8000D+02 0.62499000D+02
TFP 0.20960000D+03 0.28150000D+03 0.90700000D+02 O.1755OOOOD+O3 0.19240000D+03 0.11940000D+03
ТВ O.3343OOOOD+O3 O.3738OOOOD+O3 0.1117OOOOD+O3 O.3378OOOOD+O3 0.18920000D+03 0.25980000D+03
TC O.5364OOOOD+O3 O.58OOOOOOD+O3 0.19060000D+03 O.5126OOOOD+O3 O.3O83OOOOD+O3 0.42970000D+03
PC 0.547155OOD+O7 0.0 0.46001550D+07 0.80958675D+07 0.61402950D+07 0.56032725D+07
VC 0.23900000D+00 0.0 0.99000000D-01 0.11800000D+00 0.11300000D+00 0.16900000D+00
ZC 0.29300000D+00 0.0 O.288OOOOOD+OO 0.22400000D+00 0.27100000D+00 0.26500000D+00
OMEGA 0.21600000D+00 0.0 0.80000000D-02 0.55900000D+00 0.18400000D+00 0.12200000D+00
VL 0.14890000D+04 0.12260000D+04 0.42500000D+03 O.791OOOOOD+O3 O.615OOOOOD+O3 0.96900000D+03
TREF O.293OOOOOD+O3 0.28800000D+03 0.1117OOOOD+O3 O.293OOOOOD+O3 0.189OOOOOD+O3 O.259OOOOOD+O3
MUP 0.34785054D-24 0.47434164D-24 0.0 0.53758719D-24 0.0 0.47434164D-24
CPIG1 0.24002924D+05 0.11714666D+05 0.19250906D+05 0.21151714D+05 0.26820641D+05 0.59494428D+04
CPIG2 0.18932710D+03 0.13577792D+03 0.52125660D+02 0.70924392D+02 0.75781080D+02 0.20192936D+03
CPIG3 -0.184093 60D+00 0.84112812D-01 0.11974248D-01 0.25870237D-01 -0.50074128D-01 -0.15361369D+00
CPIG4 0.66570120D-04 0.20167816D-04 —0.11316920D—04 -0.28516295D-04 0.14122076D-04 0.47729520D-04
VISC1 0.39481000D+03 0.72935000D+03 0.11414000D+03 O.5553OOOOD+O3 0.0 0.27690000D+03
VISC2 0.8579723 8D+02 O.13921O37D+O3 0.22912330D+02 0.10823454D+03 0.0 0.59450112D+02
DHFORM —0.10132056D+09 0.37886353D+09 -0.74901852D+08 -0.20130134D+09 0.22688269D+09 0.35169120D+08
DGFORM —0.68579784D+08 0.35123065D+09 -0.50869620D+08 —0.16261531D+09 0.20934000D+09 0.51539508D+08
ANTI -0.26501960D+02 0.21880970D+02 -0.18436630D+04 0.43744470D+02 -0.14634450D+03 0.38131010D+04
ANT2 -0.14830630D+04 O.359958OOD+O4 O.62O333OOD+O5 -0.46749420D+04 0.12143980D- 01 -0.25799530D+06
ANT3 -0.6775 7450D+02 O.26O9OOOOD+O2 0.58912410D+02 -0.27120530D+02 -0.19209530D+03 0.12676080D+03
ANT4 -0.12913810D- 01 0.0 -0.80123750D+00 -0.68086370D-03 —0.18888080D+00 0.61228460D+00
ANT5 O.8239852OD+O1 0.0 0.335193OOD+O3 —0.29141350D+01 0.35883170D+02 -0.59201660D+03
ANT6 O.1352358OD-16 0.0 0.24853960D-08 0.17842010D-16 0.15316000D- 03 -0.95496840D-07
ANT7 0.60000000D+01 0.0 0.40000000D+01 0.60000000DT01 0.20000000D+01 0.30000000D+01
TMIN 0.21456000D+03 0.27100000D+03 O.9O7OOOOOD+O2 0.20504000D+03 0.19240000D+03 0.23633500D+03
TMAX O.5364OOOOD+O3 O.4O9OOOOOD+O3 0.19060000D+03 0.51260000D+03 O.3O83OOOOD+O3 0.42970000D+03
DHVB 0.29726280D+08 0.21938832D+08 0.81851940D+07 0.35277977D+08 0.16956540D+08 0.20640924D+08
5 9.236 12.1 5.680 14.510 5.329
18.899 24.8 11.623 29.691 10.904
АЦЕТОНИТРИЛ ЭТИЛЕН АЦЕТАЛЬДЕГИД УКСУСНАЯ КИСЛОТА ЭТАН ЭТАНОЛ
FORMULA C2H3N C2H4 C2H4O С2Н4О2 С2Н6 С2Н6О
MW 0.41053000D+02 0.28054000D+02 0.44054000D+02 0.60052000D+02 O.3OO7OOOOD+O2 0.46069000D+02
TFP O.2293OOOOD+O3 0.10400000D+03 0.15020000D+03 O.2898OOOOD+O3 O.899OOOOOD+O2 0.15910000D+03
ТВ O.3548OOOOD+O3 0.16940000D+03 O.2936OOOOD+O3 0.39110000D+03 O.1845OOOOD+O3 0.35150000D+03
ТС O.548OOOOOD+O3 0.28240000D+03 0.46100000D+03 O.5944OOOOD+O3 O.3O54OOOOD+O3 0.51620000D+03
PC 0.48332025D+07 O.5O358525D+O7 0.55728750D+07 0.57856575D+07 0.48838650D+07 0.63834750D+07
VC O.173OOOOOD+OO 0.12900000D+00 0.15400000D+00 0.17100000D+00 0.14800000D+00 0.16700000D+00
zc O.184OOOOOD+OO O.276OOOOOD+OO 0.22000000D+00 0.20000000D+00 O.285OOOOOD+OO 0.24800000D+00
OMEGA O.321OOOOOD+OO O.85OOOOOOD-O1 O.3O3OOOOOD+OO 0.45400000D+00 0.98000000D-01 0.63500000D+00
VL O.782OOOOOD+O3 O.577OOOOOD+O3 O.778OOOOOD+O3 0.10490000D+04 O.548OOOOOD+O3 0.78900000D+03
TREF O.293OOOOOD+O3 O.163OOOOOD+O3 O.293OOOOOD+O3 O.293OOOOOD+O3 O.183OOOOOD+O3 0.29300000D+03
MUP 0.11067972D-23 0.0 0.79056940D-24 0.41109609D-24 0.0 0.53758719D-24
CPIG1 0.20481826D+05 0.38058012D+04 0.77162724D+04 0.48399408D+04 0.54093456D+04 0.90141804D+04
CPIG2 0.11961688D+03 0.15658632D+03 0.18225140D+03 0.25485052D+03 0.17810647D+03 0.214071O8D+O3
CPIG3 -0.44924364D-01 -0.83484792D-01 - O.1OO65O67D+OO —0.17530132D+00 -0.69375276D-01 -0.83903472D-01
CPIG4 0.32029020D-05 0.17551066D-04 0.23801958D-04 0.49487976D-04 0.87127308D-05 0.13732704D-05
VISC1 0.33491000D+03 0.16898000D+03 O.3687OOOOD+O3 O.6OO94OOOD+O3 O.1566OOOOD+O3 0.68664000D+03
VISC2 0.72894789D+02 0.35212913D+02 0.75058843D+02 0.12109599D+03 0.33760263D4-02 0.12999361D+03
DHFORM 0.87922800D+08 O.52335OOOD+O8 -0.16646717D+09 -0.43513412D+09 -0.84740832D+08 -0.23496322D+09
DGFORM 0.10567483D+09 0.68161104D+08 —0.13339145D+09 -0.37693760D+09 -0.32950116D+08 -0.16839310D+09
ANTI —O.3OO5728OD+O4 -0.17884590D+04 0.21140870D+02 -0.14271780D+03 -0.20410290D+04 -0.75760900D+02
ANT2 0.25039050D+06 0.77818890D+05 —0.24651500D+04 0.22656960D+00 0.94395410D+05 -0.31006470D+04
ANT3 0.17508420D+03 0.83119340D+02 -0.37150000D+02 -O.33O9145OD+O3 0.88739680D+02 -0.40500640D+02
ANT4 -0.36956270D+00 -0.57861500D+00 0.0 -0.46303860D-01 -0.60049O8OD+OO -0.88140770D-01
ANT5 0.45560340D+03 0.30942770D+03 0.0 0.28820290D+02 0.34802740D+03 0.20812080D+02
ANT6 0.46555470D-10 0.36372090D-06 0.0 0.13776960D-10 0.31743870D-06 0.50453330D-04
ANT7 0.40000000D+01 0.30000000D+01 0.0 0.40000000D+01 0.30000000D+01 0.20000000D+01
TMIN 0.22930000D+03 0.11296OOOD+O3 0.21000000D+03 0.32692000D+03 O.12216OOOD+O3 0.20648000D+03
TMAX 0.54800000D+03 0.28240000D+03 O.32OOOOOOD+O3 0.59440000D+03 O.3O54OOOOD+O3 O.5162OOOOD+O3
DHVB 0.31401000D+08 0.13552672D+08 0.25748820D+08 0.23697288D+08 0.14716602D+08 0.38769768D+08
3 12.049 5.801 9.844 10.051 6.050 12.915
51 24.655 11.870 20.143 20.567 12.380 26.427
Приложения 585
ПРОПИЛЕН АЦЕТОН ПРОПИОНОВАЯ КИСЛОТА МЕТИЛАЦЕТАТ ПРОПАН 1-ПРОПАНОЛ
FORMULA C3H6 C3H6O C3H6O2 C3H6O2 C3H8 C3H8O
MW 0.42081000D+02 O.58O8OOOOD+O2 0.74080000D+02 O.74O8OOOOD+O2 0.44097000D+02 0.60096000D+02
TFP O.879OOOOOD+O2 O.1782OOOOD+O3 O.2525OOOOD+O3 O.175OOOOOD+O3 O.855OOOOOD+O2 O.1469OOOOD+O3
ТВ O.2254OOOOD+O3 O.3294OOOOD+O3 0.41400000D+03 O.33O1OOOOD+O3 0.23110000D+03 O.37O4OOOOD+O3
TC O.365OOOOOD+O3 0.50810000D+03 O.612OOOOOD+O3 O.5O68OOOOD+O3 O.3698OOOOD+O3 O.5367OOOOD+O3
PC 0.46204200D+07 0.470148OOD+O7 O.537O225OD+O7 0.46913475D+07 0.4245 5175D+O7 0.51675750D+07
VC 0.18100000D+00 0.20900000D+00 O.23OOOOOOD+OO 0.22800000D+00 0.20300000D+00 0.21850000D+00
ZC O.275OOOOOD+OO O.232OOOOOD+OO 0.24200000D+00 0.25400000D+00 0.28100000D+00 0.25300000D+00
OMEGA 0.14800000D+00 0.30900000D+00 O.536OOOOOD+OO 0.32400000D+00 O.152OOOOOD+OO 0.62400000D+00
VL O.612OOOOOD+O3 O.79OOOOOOD+O3 O.993OOOOOD+O3 O.934OOOOOD+O3 O.582OOOOOD+O3 O.8O4OOOOOD+O3
TREF O.223OOOOOD+O3 O.293OOOOOD+O3 O.293OOOOOD+O3 O.293OOOOOD+O3 O.231OOOOOD+O3 O.293OOOOOD+O3
MUP 0.12649110D-24 O.917O6O5OD-24 0.47434164D-24 0.53758719D-24 0.0 O.53758719D-24
CPIG1 0.37095048D+04 0.63011340D+04 0.56689272D+04 O.1655O42OD+O5 -0.42244812D+04 0.24702120D+04
CPIG2 0.23454454D+03 0.2605 8643D+03 O.36889895D+O3 0.2245 38O8D+O3 0.30626442D+03 0.332515 66D+O3
CPIG3 —0.11-601623D+00 -0.12526906D+00 -0.28646086D+00 -0.43417116D-01 - O.15863785D+OO -0.18551711D+00
CPIG4 0.22047689D-04 O.2O377156D-O4 0.98766612D-04 0.29144315D-04 O.3214625OD-O4 0.42956568D-04
VISC1 O.27384OOOD+O3 O.36725OOOD+O3 O.535O4OOOD+O3 0.40862000D+03 O.22267OOOD+O3 O.951O4OOOD+O3
VISC2 0.53901514D+02 0.77291732D+02 0.11175727D+O3 0.84706479D+02 0.47690488D+02 0.16116547D+03
DHFORM 0.20431584D+08 —0.21771360D+09 -0.45544010D+09 -O.4O972O25D+O9 O.1O391638D+O9 -0.25656710D+09
DGFORM O.6276O132D+O8 -0.15315314D+09 -0.36956884D+09 0.0 -0.23487948D+08 -0.16190356D+09
ANTI -0.2495 7960D+04 0.21544070D+02 0.15382540D+04 0.115045 9OD+O3 -0.21284520D+04 O.3982635OD+O3
ANT2 0.11799810D+06 -0.29404600D+04 -0.12824810D+06 -0.77691770D+04 0.1171OO8OD+O6 -0.22098850D+05
ANT3 O.94O7885OD+O2 - O.3593OOOOD+O2 O.1425641OD+O3 O.1O52171OD+O2 O.1O98733OD+O3 0.28134120D+02
ANT4 - O.8528422OD+OO 0.0 O.1688537OD+OO 0.11846230D-01 -0.49471660D+00 0.55274220D-01
ANT5 O.42674O5OD+O3 0.0 -O.2266232OD+O3 -0.14594400D+02 O.35O6767OD+O3 - O.5955OO9OD+O2
ANT6 O.3578286OD-O3 0.0 - O.852O342OD-08 0.13296100D-16 0.17788010D-06 O.33O3832OD-O5
ANT7 0.20000000D+01 0.0 0.30000000D+01 0.60000000D+01 0.30000000D+01 0.20000000D+01
TMIN 0.14600000D+03 O.241OOOOOD+O3 O.3366OOOOD+O3 0.20272OOOD+O3 O.14792OOOD+O3 O.21468OOOD+O3
TMAX O.365OOOOOD+O3 O.35OOOOOOD+O3 O.612OOOOOD+O3 O.5O68OOOOD+O3 O.3698OOOOD+O3 O.5367OOOOD+O3
DHVB 0.18421920D+08 0.29140128D+08 0.32238360D+08 0.30144960D+08 0.18786172D+08 0.41784264D+08
6 6.208 9.566 12.385 9.014 6.400 12.050
<51 12.703 19.574 25.342 18.445 13.096 24.657
ИЗОПРОПИЛОВЫЙ СПИРТ ГЛИЦЕРИН 1,2-БУТАДИЕН 1,3-БУТАЦИЕН 1-БУТЕН цис-2-БУТЕН
FORMULA C3H8O C3H8O3 C4H6 C4H6 C4H8 C4H8
MW 0.60096000D+02 O.92O95OOOD+O2 0.54092000D+02 0.54092000D+02 O.561O8OOOD+O2 O.561O8OOOD+O2
TFP O.1847OOOOD+O3 0.29100000D+03 O.137OOOOOD+O3 O.1643OOOOD+O3 O.878OOOOOD+O2 0.13430000D+03
ТВ O.3554OOOOD+O3 O.563OOOOOD+O3 0.28400000D+03 O.2687OOOOD+O3 O.2669OOOOD+O3 O.2769OOOOD+O3
ТС O.5O83OOOOD+O3 O.726OOOOOD+O3 0.443 7OOOOD+O3 O.425OOOOOD+O3 0.41960000D+03 O.4356OOOOD+O3
PC O.4762275OD+O7 O.668745OOD+O7 0.44988300D+07 0.43265775D+07 0.40226025D+07 O.42O49875D+O7
VC 0.22000000D+00 O.255OOOOOD+OO 0.21900000D+00 0.22100000D+00 0.24000000D+00 0.23400000D+00
ZC 0.24800000D+00 0.28000000D+00 0.26700000D+00 0.27000000D+00 O.277OOOOOD+OO 0.27200000D+00
OMEGA 0.72400000D+00 0.0 0.25500000D+00 O.195OOOOOD+OO O.187OOOOOD+OO 0.20200000D+00
VL O.786OOOOOD+O3 0.12610000D+04 O.652OOOOOD+O3 O.621OOOOOD+O3 O.595OOOOOD+O3 O.621OOOOOD+O3
TREF O.293OOOOOD+O3 0.29300000D+03 O.293OOOOOD+O3 O.293OOOOOD+O3 O.293OOOOOD+O3 O.293OOOOOD+O3
MUP O.53758719D-24 0.94868328D-24 0.12649110D-24 0.0 0.94868328D-25 0.94868328D-25
CPIG1 0.32426766D+05 0.84238416D+04 0.11199690D+05 -0.16872804D+04 -O.2993562OD+O4 0.43961400D+03
CPIG2 O.18848974D+O3 0.44421948D+03 0.27235134D+O3 0.34185222D+03 0.35319845D+O3 O.29533687D+O3
CPIG3 0.64058040D-01 - O.315894O6D+OO -0.14683108D+00 -0.23400025D+00 -0.19904047D+00 -0.10178111D+00
CPIG4 -0.92612016D-04 O.9378432OD-O4 0.30890210D-04 0.63346284D-04 0.44631288D-04 -O.6154596OD-O6
VISC1 0.11397OOOD+O4 O.33371OOOD+O4 0.0 0.3005 9OOOD+O3 O.2563OOOOD+O3 0.26894000D+03
VISC2 O.1747O193D+O3 O.29743861D+O3 0.0 O.62O7OO56D+O2 0.54674549D+02 0.56842739D+02
DHFORM -O.2726O255D+O9 -O.58531464D+O9 0.16232224D+09 0.11023844D+09 -0.12560400D+06 -0.69919560D+07
DGFORM - O.1735OO99D+O9 0.0 O.19857992D+O9 O.15O76667D+O9 O.71343O72D+O8 O.659OO232D+O8
ANTI 0.235 8567OD+O2 0.2213197OD+O2 0.20996670D+02 O.2O66547OD+O2 -0.26389800D+04 -0.23513930D+04
ANT2 -0.36402000D+04 -0.44870400D+04 -0.23972600D+04 -0.21426600D+04 0.14105410D+06 0.12905430D+06
ANT3 - O.5354OOOOD+O2 -0.14020000D+03 -0.30880000D+02 - O.343OOOOOD+O2 O.1O92926OD+O3 O.114O957OD+O3
ANT4 0.0 0.0 0.0 0.0 -0.76149810D+00 - O.6587477OD+OO
ANT5 0.0 0.0 0.0 0.0 0.44014500D+03 O.39O7181OD+O3
ANT6 0.0 0.0 0.0 0.0 O.2774342OD-O3 O.2347O37OD-O3
ANT7 0.0 0.0 0.0 0.0 0.20000000D+01 0.20000000D+01
TMIN O.273OOOOOD+O3 0.44000000D+03 O.245OOOOOD+O3 O.215OOOOOD+O3 O.16784OOOD+O3 0.17424000D+03
TMAX O.374OOOOOD+O3 0.60000000D+03 O.3O5OOOOOD+O3 O.29OOOOOOD+O3 0.41960000D+03 O.4356OOOOD+O3
DHVB 0.39858336D+08 0.61127280D+08 0.24283440D+08 0.22483116D+08 0.2193045 8D+08 0.23362344D+08
11.572 17.69 7.950 6.940 6.766 6.760
<*1 23.679 36.20 16.267 14.201 13.845 13.832
Приложения 587
МЕТИЛЭТИЛКЕТОН ТЕТРАГИДРОФУРАН 1,4-ДИОКСАН ЭТИЛАЦЕТАТ н-БУТАН ИЗОБУТАН
FORMULA C4H8O C4H8O C4H8O2 C4H8O2 C4H1O C4H1O
MW 0.72107000D+02 0.72107000D+02 0.88107000D+02 0.88107000D+02 0.58124000D+02 0.58124000D+02
TFP O.1865OOOOD+O3 0.16470000D+03 0.28500000D+03 0.18960000D+03 0.13480000D+03 0.1136OOOOD+O3
ТВ 0.35280000D+03 O.3391OOOOD+O3 0.37450000D+03 O.35O3OOOOD+O3 O.2727OOOOD+O3 O.2613OOOOD+O3
TC O.5356OOOOD+O3 0.54020000D+03 O.587OOOOOD+O3 0.52320000D+03 O.4252OOOOD+O3 0.40810000D+03
PC 0.41543250D+07 0.51878400D+07 0.52081050D+07 O.383OO85OD+O7 0.37996875D+07 O.36477OOOD+O7
VC 0.26700000D+00 0.22400000D+00 0.23800000D+00 0.28600000D+00 O.255OOOOOD+OO O.263OOOOOD+OO
ZC 0.24900000D+00 0.25900000D+00 0.25400000D+00 0.25200000D+00 0.27400000D+00 O.283OOOOOD+OO
OMEGA 0.32900000D+00 0.0 0.28800000D+00 O.363OOOOOD+OO O.193OOOOOD+OO O.176OOOOOD+OO
VL O.8O5OOOOOD+O3 O.889OOOOOD+O3 0.10330000D+04 0.90100000D+03 0.57900000D+03 O.557OOOOOD+O3
TREF O.293OOOOOD+O3 O.293OOOOOD+O3 0.29300000D+03 0.29300000D+03 0.29300000D+03 O.293OOOOOD+O3
MUP 0.10435516D-23 0.53758719D-24 0.12649110D-24 0.60083274D-24 0.0 0.31622776D-25
CPIG1 0.10944295D+05 -0.19104368D+05 - -0.53574293D+05 0.72347904D+04 0.94872888D+04 -0.13900176D+04
CPIG2 O.35591987D+O3 0.51623244D+03 0.59871240D+03 0.40716630D+03 0.33130148D+03 O.384725O5D+O3
CPIG3 -0.18999698D+00 -0.41315342D+00 -0.40850608D+00 -0.2091725 3D+00 —0.11082460D+00 —0.1845 960ID+00
CPIG4 0.39196822D-04 0.14540756D-03 0.10621912D-03 0.28545602D-04 -0.28219032D-05 0.28951722D-04
VISC1 0.42384000D+03 0.41979000D+03 0.66036000D+03 0.4273 8OOOD+O3 0.26584000D+03 0.30251000D+03
VISC2 0.87760659D+02 0.88991097D+02 O.1285O773D+O3 0.88826671D+02 0.5 7054241D+02 O.63321324D+O2
DHFORM -0.23852200D+09 -0.18434480D+09 - -0.31526604D+09 -0.44321465D+09 -0.12623202D+09 -O.1346O562D+O9
DGFORM -0.14616119D+09 0.0 -0.18091163D+09 -0.32761710D+09 - 0.17165880D+08 -0.20892132D+08
ANTI -0.78357580D+04 0.20999670D+02 0.21025470D+02 0.48610870D+02 -0.20775660D+04 -0.22805130D+04
ANT2 0.53123170D+06 -0.27683800D+04 - -0.29668800D+04 -0.82999240D+04 0.13037520D+06 0.1370518OD+O6
ANT3 O.1386953OD+O3 -0.46900000D+02 - -0.62150000D+02 0.28708980D+02 0.1296717OD+O3 0.12196710D+03
ANT4 -0.15758780D+01 0.0 0.0 -0.47696360D-01 -0.4067 327OD+OO —0.46894860D+00
ANT5 0.12395040D+04 0.0 0.0 -0.46184520D+00 0.33409570D+03 0.36914510D+03
ANT6 0.41455620D-03 0.0 0.0 0.34456160D-04 0.1113578OD—06 O.137195OOD-06
ANT7 0.20000000D+01 0.0 0.0 0.20000000D+01 0.30000000D+01 0.30000000D+01
TMIN 0.26780000D+03 0.27000000D+03 O.275OOOOOD+O3 0.20928000D+03 O.17OO8OOOD+O3 O.16324OOOD+O3
TMAX O.5356OOOOD+O3 O.37OOOOOOD+O3 0.41000000D+03 O.5232OOOOD+O3 0.42520000D+03 0.40810000D+03
DHVB 0.31233528D+08 0.29600676D+08 0.36383292D+08 0.32238360D+08 0.22407754D+08 O.2131O812D+O8
8 9.199 9.10 10.13 8.974 6.634 6.730
Si 18.823 18.62 20.73 18.363 13.575 13.771
н-БУТАНОЛ 2-БУТАНОЛ ИЗОБУТАНОЛ трет-Б УТИЛОВЫЙ СПИРТ ДИЭТИЛОВЫЙ ЭФИР ДИЭТИЛЕНГЛИКОЛЬ
FORMULA С4НЮО С4НЮО С4НЮО С4НЮО С4НЮО С4НЮОЗ
MW 0.74123OOOD+O2 0.74123000D+02 0.74123000D+02 0.74123000D+02 0.74123000D+02 0.10612200D+03
TFP O.1839OOOOD+O3 O.1585OOOOD+O3 0.16520000D+03 0.29880000D+03 0.15690000D+03 0.26500000D+03
ТВ 0.39090000D+03 O.3727OOOOD+O3 O.381OOOOOD+O3 O.3556OOOOD+O3 O.3O77OOOOD+O3 0.51900000D+03
ТС 0.56290000D+03 O.536OOOOOD+O3 0.54770000D+03 O.5O62OOOOD+O3 0.46670000D+03 O.681OOOOOD+O3-
PC 0.44177700D+07 0.41948550D+07 0.42961800D+07 0.39719400D+07 0.36375675D+07 0.46609500D+07
VC 0.27400000D+00 0.26800000D+00 O.273OOOOOD+OO O.275OOOOOD+OO 0.28000000D+00 O.316OOOOOD+OO
ZC 0.25900000D+00 0.25200000D+00 O.257OOOOOD+OO 0.25900000D+00 0.26200000D+00 0.26000000D+00
OMEGA 0.59000000D+00 0.57600000D+00 O.588OOOOOD+OO 0.61800000D+00 0.28100000D+00 0.0
VL O.81OOOOOOD+O3 0.80700000D+03 O.8O2OOOOOD+O3 O.787OOOOOD4-O3 O.713OOOOOD+O3 0.11160000D+04
TREF 0.29300000D+03 0.29300000D+03 O.293OOOOOD+O3 0.29300000D+03 O.293OOOOOD+O3 0.29300000D+03
MUP 0.56920997D-24 0.53758719D-24 0.53758719D-24 0.53758719D-24 0.41109609D—24 0.0
CPIG1 0.32657040D+04 0.57526632D+04 —0.77078988D+04 —0.48612935D+05 0.21423856D+05 0.73059660D+05
CPIG2 0.41801011D+03 0.42454152D+03 0.46892160D+03 0.71719884D+03 0.33586510D+03 0.34608089D+03
CPIG3 -0.22416127D+00 —0.23282795D+00 - O.28838678D4-OO —0.70840656D+00 - -O.1O353956D+OO -0.14678921D+00
CPIG4 0.46850292D-04 0.47729520D-04 0.72306036D-04 0.29198743D-03 - -0.93574980D-05 0.18463788D-04
VISC1 0.9845 4000D+03 0.14417000D+04 0.11991000D+04 0.97210000D+03 0.35314000D+03 0.19430000D+04
VISC2 0.16726246D+03 O.19617583D+O3 0.18483874D+03 0.17129030D+03 0.72767733D+02 0.24155823D+03
DHFORM -0.27486342D+09 -0.29282479D+09 -0.28340449D+09 -0.31262836D+09 - -0.25238030D+09 -0.57149820D+09
DGFORM -0.15089227D+09 -0.16772321D+09 -0.16743013D+09 -0.17777153D+09 - -0.12242203D+09 0.0
ANTI 0.22108770D+02 0.22102970D+02 0.21763970D+02 0.21747570D+02 0.98234300D+01 0.21925370D+02
ANT2 -0.31370200D+04 -0.30260300D+04 -0.28747344D+04 -0.26582900D+04 - 0.25462230D+04 -0.41225200D+04
ANT3 -0.94430000D+02 - O.8665OOOOD4-O2 -O.1OO3OOOOD+O3 -0.95500000D+02 - -0.35442790D4-02 -0.12250000D+03
ANT4 0.0 0.0 0.0 0.0 -0.68873780D-02 0.0
ANT5 0.0 0.0 0.0 0.0 0.22952660D+01 0.0
ANT6 0.0 0.0 0.0 0.0 0.28640650D-16 0.0
ANT7 0.0 0.0 0.0 0.0 0.60000000D+01 0.0
TMIN O.288OOOOOD+O3 0.29800000D+03 0.29300000D+03 0.29300000D+03 0.18668000D+03 0.40200000D+03
TMAX 0.40400000D+03 O.393OOOOOD+O3 O.388OOOOOD+O3 O.376OOOOOD+O3 0.46670000D+03 0.56000000D+03
DHVB 0.43124040D+08 0.408213OOD+O8 0.42077340D+08 0.39062844D+08 0.26711784D+08 0.57233556D+08
11.440 11.08 10.949 10.316 7.544 13.551
<^1 23.409 22.67 22.404 21.109 15.437 27.728
Приложения 589
ПИРИДИН н-ПЕНТАН 2-МЕТИЛБУТАН 2,2-ДИМЕТИЛПРОПАН 1-ПЕНТАНОЛ БЕНЗОЛ
FORMULA C5H5N C5H12 C5H12 C5H12 C5H12O C6H6
MW O.791O2OOOD+O2 0.72151000D+02 0.72151000D+02 0.72151000D+02 0.88150000D+02 0.78114000D+02
TFP 0.23150000D+03 0.14340000D+03 0.1133OOOOD+O3 O.2566OOOOD+O3 O.195OOOOOD+O3 O.2787OOOOD+O3
ТВ O.3885OOOOD+O3 0.30920000D+03 0.30100000D+03 O.2826OOOOD+O3 0.41100000D+03 O.3533OOOOD+O3
TC 0.62000000EH03 O.4696OOOOD+O3 0.46040000D+03 O.4338OOOOD+O3 0.58600000D + 03 O.5621OOOOD+O3
PC 0.56336700D+07 0.33741225D+07 0.33842550D+07 0.32018700D+07 0.38503500D + 07 0.48939975D+07
VC 0.25400000D+00 0.30400000D+00 O.3O6OOOOOD+OO O.3O3OOOOOD+OO 0.32600000D + 00 O.259OOOOOD+OO
ZC O.277OOOOOD+OO 0.26200000D+00 0.27100000D+00 0.26900000D+00 0.26000000D + 00 O.271OOOOOD+OO
OMEGA 0.24000000D+00 O.251OOOOOD+OO O.227OOOOOD+OO 0.19700000D+00 O.58OOOOOOD+OO 0.21200000D+00
VL O.983OOOOOD+O3 0.62600000D+03 0.62000000D+03 O.591OOOOOD+O3 O.815OOOOOD+O3 O.885OOOOOD+O3
TREF O.293OOOOOD+O3 O.293OOOOOD+O3 O.293OOOOOD+O3 O.293OOOOOD+O3 O.293OOOOOD+O3 O.289OOOOOD+O3
MUP 0.72732385D-24 0.0 0.31622776D-25 0.0 0.53758719D-24 0.0
CPIG1 0.39791347D+O5 -0.3625 7688D+04 -0.95249700D+04 -0.16592288D+05 0.38686032D+04 -0.33917267D+05
CPIG2 0.49278636D+03 0.48734352D+03 0.50660280D+03 0.55516968D+03 0.50450940D+03 0.47436444D+03
CPIG3 -0.355 79426D+00 -0.25 803248D+00 -0.27293749D+00 -0.33063160D+00 - -O.26393587D+OO -0.30170081D+00
CPIG4 0.1004413 3D—03 0.53046756D-04 0.57233556D-04 0.76325364D- 04 0.51204564D-04 0.71301204D—04
VISC1 O.6185OOOOD+O3 O.31366OOOD+O3 O.36732OOOD+O3 O.35554OOOD+O3 0.11511000D+04 O.54564OOOD+O3
VISC2 0.12077243D+03 0.6646925 7D+02 0.74699240D+02 0.73905376D+02 0.18293405D+03 0.10791118D+03
DHFORM 0.14025780D+09 -0.14653800D+09 -0.15457666D+09 -0.16609036D+09 - -0.2989375 2D+09 0.82982376D+08
DGFORM O.19O33193D+O9 - O.83736OOOD+O7 -0.14821272D+08 -0.15239952D+08 - -0.14611932D+09 0.12974893D+09
ANTI 0.20983770D+02 —0.31144360D+02 -0.25965910D+04 -0.53893750D+04 0.21419770D+02 -0.37675460D+02
ANT2 -0.30951300D+04 -0.12065 390D+04 0.15024030D+06 0.31478200D+06 - -0.30268900D+04 -0.10975230D+04
ANT3 - 0.6115OOOOD+O2 -0.64139850D+02 0.12090470D+03 0.11741390D+03 - -O.1O5OOOOOD+O3 -0.93532510D+02
ANT4 0.0 -0.16082770D-01 -0.67404480D+00 -0.13554940D+01 0.0 -0.14047730D-01
ANT5 0.0 0.91627870D+01 0.42657630D+03 0.87916190D+03 0.0 0.99482220D+01
ANT6 0.0 0.31313440D-16 0.22501220D-03 0.43847210D-03 0.0 0.10416500D-16
ANT7 0.0 0.60000000D+01 0.20000000D+01 0.20000000D+01 0.0 0.60000000D+01
TMIN O.285OOOOOD+O3 0.18784000D+O3 O.I8416000D+03 O.2566OOOOD+O3 0.31000000D+03 O.2787OOOOD+O3
TMAX O.425OOOOOD+O3 0.46960000D+03 0.46040000D+03 O.4338OOOOD+O3 O.411OOOOOD+O3 0.56210000D+03
DHVB 0.35169120D+08 0.25790688D+08 0.24702120D+08 0.22767818D+08 0.44380080D+08 0.30781354D+08
10.62 7.020 7.020 7.020 11.12 9.158
<*1 21.73 14.364 14.364 14.364 22.75 18.739
ФЕНОЛ
АНИЛИН
ЦИКЛОГЕКСАН МЕТИЛИЗОБУТИЛКЕТОН БЕНЗОЙНАЯ КИСЛОТА ТОЛУОЛ
FORMULA MW TFP ТВ C6H6O 0.94113OOOD+O2 O.314OOOOOD+O3 O.455OOOOOD+O3 C6H7N 0.93129000D+02 O.267OOOOOD4-O3 O.4575OOOOD+O3 C6H12 0.84162000D+02 O.2797OOOOD+O3 O.3539OOOOD+O3 C6H12O 0.10016100D+03 O.189OOOOOD+O3 O.3896OOOOD+O3 C7H6O2 0.12212400D+03 O.3956OOOOD+O3 O.523OOOOOD+O3 C7H8 0.92141000D+02 O.178OOOOOD+O3 O.3838OOOOD+O3
ТС 0.69420000D+03 O.699OOOOOD+O3 O.5534OOOOD+O3 O.571OOOOOD+O3 O.752OOOOOD+O3 0.5917OOOOD+O3
PC 0.61301625D+07 O.53O943OOD+O7 0.40732650D+07 0.32727975D+07 0.45596250D+07 0.41137950D+07
VC 0.22900000D+00 O.27OOOOOOD+OO 0.30800000D+00 O.371OOOOOD+OO O.341OOOOOD+OO O.316OOOOOD+OO
ZC 0.24000000D+00 0.24700000D+00 O.273OOOOOD+OO 0.26000000D+00 O.25OOOOOOD+OO 0.26400000D+00
OMEGA 0.44000000D+00 O.382OOOOOD+OO O.213OOOOOD+OO 0.40000000D+00 0.62000000D+00 O.257OOOOOD+OO
VL O.1O59OOOOD+O4 0.10220000D+04 0.77900000D+03 O.8O1OOOOOD+O3 0.10750000D+04 O.867OOOOOD+O3
TREF O.313OOOOOD+O3 O.293OOOOOD+O3 0.29300000D+03 0.29300000D+03 0.40300000D+03 O.293OOOOOD+O3
MUP 0.50596442D-24 0.50596442D-24 0.94868328D-25 0.88543773D-24 0.53758719D-24 0.12649110D-24
CPIG1 -0.35843195D+05 -0.40515664D+05 -0.54541444D+05 0.38937240D+04 -0.51292487D+05 -0.24354616D+05
CPIG2 0.59829372D+03 0.63848700D+03 0.61127280D+03 0.56563668D+03 0.62927604D+03 0.51246432D+03
CPIG3 -0.4827 3804D+00 - O.5133O168D+OO -0.25233844D+00 - O.3318O39OD+OO -0.42370416D+00 -0.2765 3814D+00
CPIG4 0.15269260D-03 O.163327O7D-O3 0.13213541D—04 0.82312488D-04 0.10621912D-03 0.49111164D-04
VISC1 0.14055000D+04 0.10746000D+04 O.65362OOOD+O3 O.47365OOOD+O3 0.26176000D+04 0.467 33OOOD+O3
VISC2 0.20675411D+03 0.17885216D+03 0.12456186D+03 0.98093576D+02 0.27794845D+03 0.96736798D+02
DHFORM -0.96422004D+08 0.86917968D+08 -0.12321752D+09 -0.28403251D+09 -0.29039645D+09 0.50032260D+08
DGFORM -0.32908248D+08 0.16680211D+09 0.31777812D+08 0.0 -0.21055417D+09 0.12208709D+09
ANTI 0.21054660D+03 -O.19266O3OD+O3 -0.16959900D+03 0.20609270D+02 0.22056170D+02 -0.22847860D+02
ANT2 -O.2255542OD+O5 -O.7556426OD+O2 -0.13416300D+00 -0.28936600D+04 -0.41907000D+04 -0.18741860D+04
ANT3 0.74616710D+02 -0.24438150D+03 -0.27659790D+03 - O.7O75OOOOD+O2 - O.1252OOOOD+O3 -0.72921440D+02
ANT4 -0.10845870D-01 -0.90804580D-01 -0.10627640D+00 0.0 0.0 -0.10836630D-01
ANT5 -0.25260010D+02 O.39O5296OD+O2 0.36245390D+02 0.0 0.0 0.74851470D+01
ANT6 0.14980730D-04 0.32747950D-04 0.48013040D-04 0.0 0.0 0.75672290D-17
ANT7 0.20000000D+01 0.20000000D+01 0.20000000D+01 0.0 0.0 0.60000000D+01
TMIN O.314OOOOOD+O3 0.38445000D+03 0.27970000D+03 O.285OOOOOD+O3 O.4O5OOOOOD+O3 O.23668OOOD+O3
TMAX 0.69420000D+03 O.699OOOOOD+O3 O.5534OOOOD+O3 O.425OOOOOD+O3 O.56OOOOOOD+O3 0.5917OOOOD+O3
DHVB 0.45636120D+08 0.41868000D+08 0.29977488D+08 0.35587800D+08 0.50660280D+08 0.33201324D+08
8 12.106 11.461 8.193 8.58 8.914
24.772 23.452 16.765 17.557 18.240
Приложения 591
СТИРОЛ о-КСИЛОЛ м-КСИЛОЛ п-КСИЛОЛ ЭТИЛБЕНЗОЛ h-OKTAH
FORMULA C8H8 C8H10 C8H10 C8H10 C8H10 C8H18
MW 0.104152OOD+O3 O.1O6168OOD+O3 0.10616800D+03 0.10616800D+03 0.10616800D+03 0.11423200D+03
TFP 0.2425 0000D+03 0.24800000D+03 0.225 3OOOOD+O3 0.28640000D+03 0.17820000D+03 0.21640000D+03
ТВ 0.41830000D+03 0.41760000D+03 0.41230000D+03 0.4115OOOOD+O3 0.40930000D+03 O.3988OOOOD+O3
TC 0.64700000D+03 O.63O2OOOOD+O3 0.61700000D+03 0.61620000D+03 0.61710000D+03 O.5688OOOOD+O3
PC 0.39922050D+07 0.37287600D+07 0.35463750D+07 0.35159775D+07 0.36071700D+07 0.24824625D+07
VC 0.0 0.36900000D+00 0.37600000D+00 0.37900000D+00 0.37400000D+00 0.49200000D+00
ZC 0.0 0.26300000D+00 0.26000000D+00 0.26000000D+00 0.26300000D+00 O.259OOOOOD+OO
OMEGA 0.25700000D+00 0.31400000D+00 O.331OOOOOD+OO 0.32400000D+00 0.30100000D+00 0.39400000D+00
VL 0.90600000D+03 O.88OOOOOOD+O3 0.86400000D+03 0.86100000D+03 0.86700000D+03 O.7O3OOOOOD+O3
TREF 0.29300000D+03 0.29300000D+03 0.29300000D+03 0.29300000D+03 0.29300000D+03 0.29300000D+03
MUP 0.31622776D-25 0.15811388D-24 0.94868328D-25 0.31622776D-25 0.12649110D-24 0.0
CPIG1 -0.28248340D+05 -0.15851225D+05 -0.29165249D+05 -0.25091492D+05 -0.43098919D+05 -0.60959808D+04
CPIG2 0.61587828D+03 0.59620032D+03 0.62969472D+03 0.60415524D+03 0.70715052D4-03 0.77120856D+03
CPIG3 -0.40230961D+00 -0.34432243D+00 -0.37471860D+00 -0.33737234D+00 -0.48106332D+00 -0.41951736D+00
CPIG4 0.99352764D-04 0.75278664D-04 0.84782700D-04 0.68202972D-04 0.13008388D-03 0.88550820D-04
VISC1 0.52864000D+03 0.51354000D+03 0.45342000D+03 0.47516000D+03 0.47282000D+03 O.4737OOOOD+O3
VISC2 0.10765617D+03 0.10594135D+O3 0.95195399D+02 0.98626941D+02 0.98720249D+02 0.97031344D+02
DHFORM 0.14745910D+09 0.19008072D+08 0.17249616D+08 0.17961372D+08 0.29810016D+08 -0.20858638D+09
DGFORM 0.21394548D+09 0.12217082D+09 0.11894699D+09 0.12120786D+09 O.13O67OO3D+O9 0.1641225 6D+08
ANTI 0.20912070D+02 0.80811040D+01 -0.32447180D+04 -0.43637890D+04 —0.12931300D+02 —0.1173785OD+O3
ANT2 -0.33285700D+04 -0.34240540D+04 0.24302390D+06 0.33397640D+06 -0.33902620D+04' —0.15641240D+04
ANT3 -0.63720000D+02 -0.50815350D+02 0.16336200D+03 0.16450780D+03 -0.36508840D+02 -0.60739230D+02
ANT4 0.0 -0.52463410D-02 - O.5884763OD+OO -0.76178230D+00 —0.15539180D—01 -0.86680810D-01
ANT5 0.0 0.24766890D+01 O.5O6855OOD+O3 0.67750020D+03 0.66026580D+01 0.26971140D+02
ANT6 0.0 0.48165370D-17 0.14444320D-03 0.18044380D-03 0.71440690D-11 0.41398210D-04
ANT7 0.0 0.60000000D+01 0.20000000D+01 0.20000000D+01 0.40000000D+01 0.20000000D+01
TMIN O.3O5OOOOOD+O3 0.25208000D+03 0.24680000D+03 0.28640000D+03 0.24684000D+03 0.2275 2OOOD+O3
TMAX 0.46000000D+03 O.63O2OOOOD+O3 0.61700000D+03 0.61620000D+03 0.61710000D+03 O.5688OOOOD+O3
DHVB 0.36843840D+08 0.36843840D+08 0.36383292D+08 0.36006480D+08 O.355878OOD+O8 0.34436430D+08
6 9.211 8.987 8.818 8.769 8.783 7.551
<51 18.848 18.389 18.044 17.943 17.972 15.451
Приложение Д. Параметры и корреляции
Таблицы настоящего приложения являются дополни
тельным материалом к гл. 1, 3, 4 и И.
Табл. Д.1. Величины Z(0) (уравнение Ли — Кеслера)
Табл. Д.2. Величины Z(1) (уравнение Ли — Кеслера)
Табл. Д.З. Параметры бинарного взаимодействия
Табл. Д.4. Константы уравнений состояния
Табл. Д.5. Значения констант уравнения Бенедикта — Уэбба — Рубина
Табл. Д.6. Значения [In (J7p)](0) (уравнение Ли — Кес- лера)
Табл. Д.7. Значения [In (/7р)]0) (уравнение Ли — Кес- лера)
Табл. Д.8. Некоторые параметры уравнения Виль- сона
Табл. Д.9. Некоторые параметры уравнения Мар- гулеса
Табл. Д.10. Некоторые параметры уравнения ван Лаара
Табл. Д.11. Параметры объема и площади поверхнос- ти групп уравнения UNIFAC для расчета парожидкостного равновесия
Табл. Д.12. Параметры взаимодействия уравнения
UNIFAC для расчета парожидкостного
равновесия
Табл. ДЛЗ. Параметры объема и площади поверхнос-
ти групп уравнения UNIFAC для расчета
равновесия жидкость — жидкость
Табл. Д.14. Параметры взаимодействия групп уравне-
ния UNIFAC для расчета равновесия жид-
кость — жидкость
Н° — Н (0)
Табл. Д.15. Значения РТс (уравнение Ли —
Кеслера)
Н° - Н (1)
Табл. Д.16. Значения РТс (уравнение Ли —
Кеслера)
Табл. Д.17. Значения 1 ' ° ДЗ | (0) (уравнение Ли —
Кеслера)
Г 5° - 51 (О
Табл. Д.18. Значения R (уравнение Ли —
Кеслера)
Таблица Д.1. Значения Z(0) (уравнение Ли — Кеслера)
PR
TR 0.010 0.050 0.100 0.200 0.400 0.600 0.800 1.000 1.200 1.500 2.000 3.000 5.000 7.000 10. 000
0.30 0.0029 0.0145 0.0290 0.0579 0.1158 0.1737 0.2315 0.2892 0.3470 0.4335 0.5775 0.8648 1.4366 2.0048 2.8507
0.35 0.0026 0.0130 0.0261 0.0522 0.1043 0.1564 0.2084 0.2604 0.3123 0.3901 0.5195 0.7775 1.2902 1.7987 2.5539
0.40 0.0024 0.0119 0.0239 0.0477 0.0953 0.1429 0.1904 0.2379 0.2853 0.3563 0.4744 0.7095 1.1758 1.6373 2.3211
0.45 0.0022 0.0110 0.0221 0.0442' 0.0882 0.1322 0.1762 0.2200 0.2638 0.3294 0.4384 0.6551 1.0841 1.5077 2.1338
0.50 0.0021 0.0103 0.0207 0.0413 0.0825 0.1236 0.1647 0.2056 0.2465 0.3077 0.4092 0.6110 1.0094 1.4017 1.9801
0.55 0.9804 0.0098 0.0195 0.0390 0.0778 0.1166 0.1553 0.1939 0.2323 0.2899 0.3853 0.5747 0.9475 1.3137 1.8520
0.60 0.9849 0.0093 0.0186 0.0371 0.0741 0.1109 0.1476 0.1842 0.2207 0.2753 0.3657 0.5446 0.8959 1.2398 1.7440
0.65 0.9881 0.9377 0.0178 0.0356 0.0710 0.1063 0.1415 0.1765 0.2113 0.2634 0.3495 0.5197 0.8526 1.1773 1.6519
0.70 0.9904 0.9504 0.8958 0.0344 0.0687 0.1027 0.1366 0.1703 0.2038 0.2538 0.3364 0.4991 0.8161 1.1241 1.5729
0.75 0.9922 0.9598 0.9165 0.0336 0.0670 0.1001 0.1330 0.1656 0.1981 0.2464 0.3260 0.4823 0.7854 1.0787 1.5047
0.80 0.9935 0.9669 0.9319 0.8539 0.0661 0.0985 0.1307 0.1626 0.1942 0.2411 0.3182 0.4690 0.7598 1.0400 1.4456
0.85 0.9946 0.9725 0.9436 0.8810 0.0661 0.0983 0.1301 0.1614 0.1924 0.2382 0.3132 0.4591 0.7388 1.0071 1.3943
0.90 0.9954 0.9768 0.9528 0.9015 0.7800 0.1006 0.1321 0.1630 0.1935 0.2383 0.3114 0.4527 0.7220 0.9793 1.3496
0.93 0.9959 0.9790 0.9573 0.9115 0.8059 0.6635 0.1359 0.1664 0.1963 0.2405 0.3122 0.4507 0.7138 0.9648 1.3257
0.95 0.9961 0.9803 0.9600 0.9174 0.8206 0.6967 0.1410 0.1705 0.1998 0.2432 0.3138 0.4501 0.7092 0.9561 1.3108
0.97 0.9963 0.9815 0.9625 0.9227 0.8338 0.7240 0.5580 0.1779 0.2055 0.2474 0.3164 0.4504 0.7052 0.9480 1.2968
0.98 0.9965 0.9821 0.9637 0.9253 0.8398 0.7360 0.5887 0.1844 0.2097 0.2503 0.3182 0.4508 0.7035 0.9442 1.2901
0.99 0.9966 0.9826 0.9648 0.9277 0.8455 0.7471 0.6138 0.1959 0.2154 0.2538 0.3204 0.4514 0.7018 0.9406 1.2835
1.00 0.9967 0.9832 0.9659 0.9300 0.8509 0.7574 0.6353 0.2901 0.2237 0.2583 0.3229 0.4522 0.7004 0.9372 1.2772
1.01 0.9968 0.9837 0.9669 0.9322 0.8561 0.7671 0.6542 0.4648 0.2370 0.2640 0.3260 0.4533 0.6991 0.9339 1.2710
PR
TR 0.010 0.050 0.100 0.200 0.400 0.600 0.800 1.000 1.200 1.500 2.000 3.000 5.000 7.000 10.000
1.02 0.9969 0.9842 0.9679 0.9343 0.8610 0.7761 0.6710 0.5146 0.2629 0.2715 0.3297 0.4547 0.6980 0.9307 1.2650
1.05 0.9971 0.9855 0.9707 0.9401 0.8743 0.8002 0.7130 0.6026 0.4437 0.3131 0.3452 0.4604 0.6956 0.9222 1.2481
1.10 0.9975 0.9874 0.9747 0.9485 0.8930 0.8323 0.7649 0.6880 0.5984 0.4580 0.3953 0.4770 0.6950 0.9110 1.2232
1.15 0.9978 0.9891 0.9780 0.9554 0.9081 0.8576 0.8032 0.7443 0.6803 0.5798 0.4760 0.5042 0.6987 0.9033 1.2021
1.20 0.9981 0.9904 0.9808 0.9611 0.9205 0.8779 0.8330 0.7858 0.7363 0.6605 0.5605 0.5425 0.7069 0.8990 1.1844
1.30 0.9985 0.9926 0.9852 0.9702 0.9396 0.9083 0.8764 0.8438 0.8111 0.7624 0.6908 0.6344 0.7358 0.8998 1.1580
1.40 0.9988 0.9942 0.9884 0.9768 0.9534 0.9298 0.9062 0.8827 0.8595 0.8256 0.7753 0.7202 0.7761 0.9112 1.1419
1.50 0.9991 0.9954 0.9909 0.9818 0.9636 0.9456 0.9278 0.9103 0.8933 0.8689 0.8328 0.7887 0.8200 0.9297 1.1339
1.60 0.9993 0.9964 0.9928 0.9856 0.9714 0.9575 0.9439 0.9308 0.9180 0.9000 0.8738 0.8410 0.8617 0.9518 1.1320
1.70 0.9994 0.9971 0.9943 0.9886 0.9775 0.9667 0.9563 0.9463 0.9367 0.9234 0.9043 0.8809 0.8984 0.9745 1.1343
1.80 0.9995 0.9977 0.9955 0.9910 0.9823 0.9739 0.9659 0.9583 0.9511 0.9413 0.9275 0.9118 0.9297 0.9961 1.1391
1.90 0.9996 0.9982 0.9964 0.9929 0.9861 0.9796 0.9735 0.9678 0.9624 0.9552 0.9456 0.9359 0.9557 1.0157 1.1452
2.00 0.9997 0.9986 0.9972 0.9944 0.9892 0.9842 0.9796 0.9754 0.9715 0.9664 0.9599 0.9550 0.9772 1.0328 1.1516
2.20 0.9998 0.9992 0.9983 0.9967 0.9937 0.9910 0.9886 0.9865 0.9847 0.9826 0.9806 0.9827 1.0094 1.0600 1.1635
2.40 0.9999 0.9996 0.9991 0.9983 0.9969 0.9957 0.9948 0.9941 0.9936 0.9935 0.9945 1.0011 1.0313 1.0793 1.1728
2.60 1.0000 0.9998 0.9997 0.9994 0.9991 0.9990 0.9990 0.9993 0.9998 1.0010 1.0040 1.0137 1.0463 1.0926 1.1792
2.80 1.0000 1.0000 1.0001 1.0002 1.0007 1.0013 1.0021 1.0031 1.0042 1.0063 1.0106 1.0223 1.0565 1.1016 1.1830
3.00 1.0000 1.0002 1.0004 1.0008 1.0018 1.0030 1.0043 1.0057 1.0074 1.0101 1.0153 1.0284 1.0635 1.1075 1.1848
3.50 1.0001 1.0004 1.0008 1.0017 1.0035 1.0055 1.0075 1.0097 1.0120 1.0156 1.0221 1.0368 1.0723 1.1138 1.1834
4.00 1.0001 1.0005 1.0010 1.0021 1.0043 1.0066 1.0090 1.0115 1.0140 1.0179 1.0249 1.0401 1.0747 1.1136 1.1773
Приложения 595
Таблица Д.2. Значения Z(1) (уравнение Ли — Кеслера)
PR
Тг 0.010 0.050 0.100 0.200 0.400 0.600 0.800 1.000 1.200 1.500 2.000 3.000 5.000 7.000 10. 000
0.30 -0.0008 -0.0040 -0.0081 -0.0161 -0.0323 -0.0484 -0.0645 -0.0806 -0.0966 -0.1207 -0.1608 -0.2407 -0.3996 -0.5572 -0.7915
0.35 -0.0009 -0.0046 -0.0093 -0.0185 -0.0370 -0.0554 -0.0738 -0.0921 -0.1105 -0.1379 -0.1834 -0.2738 -0.4523 -0.6279 -0.8863
0.40 -0.0010 -0.0048 -0.0095 -0.0190 -0.0380 -0.0570 -0.0758 -0.0946 -0.1134 -0.1414 -0.1879 -0.2799 -0.4603 -0.6365 -0.8936
0.45 -0.0009 -0.0047 -0.0094 -0.0187 -0.0374 -0.0560 -0.0745 -0.0929 -0.1113 -0.1387 -0.1840 -0.2734 -0.4475 -0.6162 -0.8606
0.50 -0.0009 -0.0045 -0.0090 -0.0181 -0.0360 -0.0539 -0.0716 -0.0893 -0.1069 -0.1330 -0.1762 -0.2611 -0.4253 -0.5831 -0.8099
0.55 -0.0314 -0.0043 -0.0086 -0.0172 -0.0343 -0.0513 -0.0682 -0.0849 -0.1015 -0.1263 -0.1669 -0.2465 -0.3991 -0.5446 -0.7521
0.60 -0.0205 -0.0041 -0.0082 -0.0164 -0.0326 -0.0487 -0.0646 -0.0803 -0.0960 -0.1192 -0.1572 -0.2312 -0.3718 -0.5047 -0.6928
0.65 -0.0137 -0.0772 -0.0078 -0.0156 -0.0309 -0.0461 -0.0611 -0.0759 -0.0906 -0.1122 -0.1476 -0.2160 -0.3447 -0.4653 -0.6346
0.70 -0.0093 -0.0507 -0.1161 -0.0148 -0.0294 -0.0438 -0.0579 -0.0718 -0.0855 -0.1057 -0.1385 -0.2013 -0.3184 -0.4270 -0.5785
0.75 -0.0064 -0.0339 -0.0744 -0.0143 -0.0282 -0.0417 -0.0550 -0.0681 -0.0808 -0.0996 -0.1298 -0.1872 -0.2929 -0.3901 -0.5250
0.80 -0.0044 -0.0228 -0.0487 -0.1160 -0.0272 -0.0401 -0.0526 -0.0648 -0.0767 -0.0940 -0.1217 -0.1736 -0.2682 -0.3545 -0.4740
0.85 -0.0029 -0.0152 -0.0319 -0.0715 -0.0268 -0.0391 -0.0509 -0.0622 -0.0731 -0.0888 -0.1138 -0.1602 -0.2439 -0.3201 -0.4254
0.90 -0.0019 -0.0099 -0.0205 -0.0442 -0.1118 -0.0396 -0.0503 -0.0604 -0.0701 -0.0840 -0.1059 -0.1463 -0.2195 -0.2862 -0.3788
0.93 -0.0015 -0.0075 -0.0154 -0.0326 -0.0763 -0.1662 -0.0514 -0.0602 -0.0687 -0.0810 -0.1007 -0.1374 -0.2045 -0.2661 -0.3516
0.95 -0.0012 -0.0062 -0.0126 -0.0262 -0.0589 -0.1110 -0.0540 -0.0607 -0.0678 -0.0788 -0.0967 -0.1310 -0.1943 -0.2526 -0.3339
0.97 -0.0010 -0.0050 -0.0101 -0.0208 -0.0450 -0.0770 -0.1647 -0.0623 -0.0669 -0.0759 -0.0921 -0.1240 -0.1837 -0.2391 -0.3163
0.98 -0.0009 -0.0044 -0.0090 -0.0184 -0.0390 -0.0641 -0.1100 -0.0641 -0.0661 -0.0740 -0.0893 -0.1202 -0.1783 -0.2322 -0.3075
0.99 -0.0008 -0.0039 -0.0079 -0.0161 -0.0335 -0.0531 -0.0796 -0.0680 -0.0646 -0.0715 -0.0861 -0.1162 -0.1728 -0.2254 -0.2989
1.00 -0.0007 -0.0034 -0.0069 -0.0140 -0.0285 -0.0435 -0.0588 -0.0879 -0.0609 -0.0678 -0.0824 -0.1118 -0.1672 -0.2185 -0.2902
1.01 -0.0006 -0.0030 -0.0060 -0.0120 -0.0240 -0.0351 -0.0429 -0.0223 -0.0473 -0.0621 -0.0778 -0.1072 -0.1615 -0.2116 -0.2816
Тг PR
0.010 0.050 0.100 0.200 0.400 0.600 0.800 1.000 1.200 1.500 2.000 3.000 5.000 7.000 70.000
1.02 -0.0005 -0.0026 -0.0051 -0.0102 -0.0198 -0.0277 -О.ОЗОЗ -0.0062 -0.0227 -0.0524 -0.0722 -0.1021 -0.1556 -0.2047 -0.2731
1.05 -0.0003 -0.0015 -0.0029 -0.0054 -0.0092 -0.0097 -0.0032 -0.0220 -0.1059 -0.0451 -0.0432 -0.0838 -0.1370 -0.1835 -0.2476
1.10 -0.0000 0.0000 0.0001 0.0007 0.0038 0.0106 0.0236 0.0476 0.0897 0.1630 0.0698 -0.0373 -0.1021 -0.1469 -0.2056
1.15 0.0002 0.0011 0.0023 0.0052 0.0127 0.0237 0.0396 0.0625 0.0943 0.1548 0.1667 0.0332 -0.0611 -0.1084 -0.1642
1.20 0.0004 0.0019 0.0039 0.0084 0.0190 0.0326 0.0499 0.0719 0.0991 0.1477 0.1990 0.1095 -0.0141 -0.0678 -0.1231
1.30 0.0006 0.0030 0.0061 0.0125 0.0267 0.0429 0.0612 0.0819 0.1048 0.1420 0.1991 0.2079 0.0875 0.0176 -0.0423
1.40 0.0007 0.0036 0.0072 0.0147 0.0306 0.0477 0.0661 0.0857 0.1063 0.1383 0.1894 0.2397 0.1737 0.1008 0.0350
1.50 0.0008 0.0039 0.0078 0.0158 0.0323 0.0497 0.0677 0.0864 0.1055 0.1345 0.1806 0.2433 0.2309 0.1717 0.1058
1.60 0.0008 0.0040 0.0080 0.0162 О.ОЗЗО 0.0501 0.0677 0.0855 0.1035 0.1303 0.1729 0.2381 0.2631 0.2255 0.1673
1.70 0.0008 0.0040 0.0081 0.0163 0.0329 0.0497 0.0667 0.0838 0.1008 0.1259 0.1658 0.2305 0.2788 0.2628 0.2179
1.80 0.0008 0.0040 0.0081 0.0162 0.0325 0.0488 0.0652 0.0816 0.0978 0.1216 0.1593 0.2224 0.2846 0.2871 0.2576
1.90 0.0008 0.0040 0.0079 0.0159 0.0318 0.0477 0.0635 0.0792 0.0947 0.1173 0.1532 0.2144 0.2848 0.3017 0.2876
2.00 0.0008 0.0039 0.0078 0.0155 0.0310 0.0464 0.0617 0.0767 0.0916 0.1133 0.1476 0.2069 0.2819 0.3097 0.3096
2.20 0.0007 0.0037 0.0074 0.0147 0.0293 0.0437 0.0579 0.0719 0.0857 0.1057 0.1374 0.1932 0.2720 0.3135 0.3355
2.40 0.0007 0.0035 0.0070 0.0139 0.0276 0.0411 0.0544 0.0675 0.0803 0.0989 0.1285 0.1812 0.2602 0.3089 0.3459
2.60 0.0007 О.ООЗЗ 0.0066 0.0131 0.0260 0.0387 0.0512 0.0634 0.0754 0.0929 0.1207 0.1706 0.2484 0.3009 0.3475
2.80 0.0006 0.0031 0.0062 0.0124 0.0245 0.0365 0.0483 0.0598 0.0711 0.0876 0.1138 0.1613 0.2372 0.2915 0.3443
3.00 0.0006 0.0029 0.0059 0.0117 0.0232 0.0345 0.0456 0.0565 0.0672 0.0828 0.1076 0.1529 0.2268 0.2817 0.3385
3.50 0.0005 0.0026 0.0052 0.0103 0.0204 О.ОЗОЗ 0.0401 0.0497 0.0591 0.0728 0.0949 0.1356 0.2042 0.2584 0.3194
4.00 0.0005 0.0023 0.0046 0.0091 0.0182 0.0270 0.0357 0.0443 0.0527 0.0651 0.0849 0.1219 0.1857 0.2378 0.2994
Приложения
Таблица Д.З. Параметры бинарного взаимодействия кц для расчета псевдокритических температур компонентов,
Гец = (1 - kn)(TciTc2)°’5, а также перекрестных коэффициентов уравнения Редлиха — Квонга, уравнения Пенга — Робинсона и уравне-
ния Соава или Вц вириального уравнения. Полностью формулы приведены в табл. 1.5. Величины в скобках получены путем интерполя-
ции или при помощи оценок [99]
Система Система fcuxio2 Система кп х 102
(1) (2) AruXlO2 (1) (2) (1) (2)
Метан Этилен 1 н-Пентан Изопентан 0 Азот Метан 3
Этан 1 (или изо- н-Гексан 0 Этилен 4
Пропилен 2 пентан) Циклогексан 0 Этан 5
Пропан 2 н-Гептан 0 Пропилен (7)
н-Бутан 4 н-Октан 0 Пропан (9)
Изобутан 4 Бензол (1) н-Бутан 12
н-Пентан 6 Толуол (1)
Изопентан 6 Аргон Метан 2
н-Гексан 8 н-Г ексан н-Гептан 0 Этилен 3
Циклогексан 8 (или цикло- н-Октан 0 Этан 3
н-Гептан 10 гексан) Бензол (1) Кислород 1
н-Октан (12) Толуол 1 Азот 0
Бензол (8)
Толуол (8) н-Гептан н-Октан 0 Тетрафтор- Метан 7
Нафталин 14 Бензол (1) метан Азот 2
Толуол (1) Гелий (16 ± 2)
Этилен Этан 0
(или этан) Пропилен 0 н-Октан Бензол (1) Водород Азот 0
Пропан 0 Толуол (1) Аргон 0
н-Бутан 1 Метан 3
Изобутан 1 Бензол Толуол (0) Этан (5)
н-Пентан 2 Пропан (7)
Изопентан 2 Диоксид Метан (5 ± 2) н-Бутан (8)
н-Гексан 3 углерода Этилен 6 Изобутан (8)
Циклогексан 3 Этан
н-Г ептан 4 Пропилен
н-Октан (5) Пропан
Бензол 3 н-Бутан
Толуол (3) Изобутан
Нафталин 8 н-Пентан
Изопентан
Пропилен Пропан 0 Нафталин
(или пропан) н-Бутан 0
Изобутан 0 Сероводород Метан
н-Пентан 1 Этилен
Изопентан 0 Этан
н-Гексан (1) Пропилен
Циклогексан (1) Пропан
н-Г ептан (2) н-Бутан
н-Октан (3) Изобутан
Бензол 2 н-Пентан
Толуол (2) Изопентан
Диоксид
углерода
н-Бутан Изобутан 0
(или н-Пентан 0 Ацетилен Метан
изобутан) н-Изопентан 0 Этилен
н-Гексан 0 Этан
Циклогексан 0 Пропилен
н-Гептан 0 Пропан
н-Октан (1) н-Бутан
Бензол (1) Изобутан
Толуол (1) н-Пентан
Изопентан
8 н-Пентан (9)
10 Изопентан (9)
11 ± 1 н-Г есан 10
16 ± 2
(16 ± 2) Гелий Азот 16
(18 ± 2) Аргон 5 ± 1
(18 ± 2) Метан (46)
24
Неон Метан 28
5 ± 1 Криптон 20 ± 2
(5 ± 1)
6 Криптон Метан 1
(7)
8
(9)
(9)
10
(Ю)
8
(5)
6
8
7
9
(Ю)
(Ю)
(П)
(И)
Приложения 599
Таблица Д.4. Константы некоторых уравнений состояния для ряда веществ
Уравнение Ван-дер-Ваальса: (Р + a/V2)(V — b) = RT, а = 27R2T2/64PC, b = RTC/3PC.
Уравнение Бертло: (Р + a/TV2)(V - b) = RT, а = 21R2T%/64PC, b = 9RTC/\28PC.
Уравнение Дитеричи: P(V — b) = RTехр(—а/RTV), а = 4R2T2/e2, b = RTc/e2Pc, е = 2.718.
RT р у 8
Уравнение Битти — Бриджмена: Р~ „
V И V3 V
Rc
Р = RTBq — Aq-
RcBq
Y = - RTBQb + AQa -
RBpbc
T2
Единицы: P, атм, V, л/моль, Т, К, R = 0,08206.
Газ т 1 с Рс Ван-дер-Ваальс Бертло Дитеричи
а b а b а b
Н2 33.2 12.8 0.24463 0.02661 8.1217 0.01497 0.3139 0.02881
Не 5.19 2.24 .034161 0.023766 0.1773 0.013369 0.04383 0.025731
Аг 150.8 48.1 1.3431 0.032159 202.54 0.018089 1.7235 0.034818
n2 126.2 33.5 1.3506 0.03864 170.45 0.02174 1.7331 0.04184
О2 154.6 49.8 1.3634 0.03184 210.78 0.01791 1.7496 0.03448
со2 304.2 72.8 3.60111 0.04286 1098.5 0.02411 4.6337 0.04641
сн4 190.6 45.4 2.2732 0.04306 433.27 0.02422 2.9169 0.04662
С2Н5ОН 516.2 63.0 12.016 0.08405 6202.7 0.04728 15.418 0.09100
с6н6 562.1 , 48.3 18.583 0.11937 10446.0 0.06715 23.845 0.12924
Битти — Бриджмен
Газ Ао а Во b 104С
Не 0.0216 0.05984 0.01400 0 0.004
Ne 0.2125 0.2196 0.02060 0 0.101
Аг 1.2907 0.02328 0.03931 0 5.99
Н2 0.1975 -0.00506 0.02096 -0.04359 0.050
n2 1.3445 0.02617 0.05046 -0.00691 4.20
О2 1.4911 0.02562 0.04624 0.004208 4.80
Air 1.3012 0.01931 0.04611 -0.01101 4.34
со2 5.0065 0.07132 0.10476 0.07235 66.00
сн4 2.2769 0.01855 0.05587 -0.01587 12.83
(С2Н5)2О 31.278 0.12426 0.45446 0.11954 33.33
Приложения 601
Таблица Д.5. Значения констант уравнения Бенедикта — Уэбба — Рубина (Р, атм, V, л/моль, Т, К, R = 0,08206).
Таблица заимствована из книги Голуба и Вонки «Химическое равновесие в газообразных системах», Reidel, 1976.
Впервые напечатана в работе Новака, Малиевского, Матуса и Собра «Газы и газовые смеси, поведение и термоди-
намические свойства», Академия Наук ЧССР, 1972
Вещество А0 so Со а Ь
Водород3 9.7319 X 10“ 2 1.8041 X 10“ 2 3.8914Х 102 -9.2211 X Ю~3 1.7976 X 10“4
Азот 1.1925 0.0458 5.8891 X 103 0.0149 1.98154 X 10“3
Азот 0.872086 2.81066 X 10“ 2 7.81375 X 103 3.12319 X 10“ 2 3.2351 X 10“3
Кислород 1.4988 4.6524 X 10-2 3.8617 X 103 -4.0507 X 10“ 2 -2.7963 X 10“4
СО 1.34122 5.45425 X 10-2 8.562 X 103 3.665 X 10“2 2.6316 X 10“3
СО 1.03115 0.040 1.124 X 104 3.665 X 10“2 2.6316 X 10“3
со2 2.7374 4.9909 X 10“ 2 1.38564 X 105 1.3681 X 10-1 7.2105 X 10“3
со2 2.51604 4.48842 X 10-2 1.474405 X 105 1.3681 X 10-1 4.12381 X 10“3
С02а 2.7634 4.5628 X 10“ 2 1.1333 X 105 5.1689 X 10“ 2 3.0819 X 10“3
S02a 7.08538 0.10896 4.43966 X 105 6.87046 X 10“2 1.93727 X 10“3
so2 2.12042 2.61817 X 10-2 7.93840 X 105 0.844680 1.46531 X 10“2
N2Oa 3.0868 5.1953 X 10“ 2 1.2725 X 105 0.10946 3.7755 X 10“3
H2S 3.10377 3.48471 X 10-2 1.9721 X 105 0.144984 4.42477 X 10“3
NH3 3.78928 5.16461 X 10~2 1.78567 X 105 0.10354 7.19561 X 10“4
CH3C1 4.56359 5.07705 X 10-2 5.83918 X 105 0.180052 5.19665 X 10“3
Метан 1.8550 4.2600 X Ю-2 2.257 X Ю4 0.0494 3.38004 X 10“3
Метан 1.79894 4.54625 X 10-2 3.18382 X 104 0.04352 2.52033 X 10“3
Этан 4.15556 6.27724 X 10-2 1.79592 X 105 0.34516 1.1122 X 10“2
Этилен 3.33958 5.56833 X 10-2 1.31140 X 105 0.259 0.00860
Ацетилен 1.5307 5.5851 X 10~3 2.1586 X 105 -0.10001 -3.7810 X 10“5
Пропан 6.87225 9.7313 X Ю-2 5.08256 X 105 0.9477 0.0225
Пропен 6.11220 8.50647 X Ю-2 4.39182 X 105 7.74056 X Ю-1 1.87059 X Ю-2
Пропин3 5.10806 6.9779 X 10“ 2 6.40624 X 105 0.69714 1.4832 X 10“2
Изобутан 10.23264 1.37544 X 10-1 8.49943 X 105 1.93763 4.24352 X 10“2
Бутан 10.0847 1.24361 X 10-1 0.99283 X 106 1.88231 3.99983 X 10“2
1-Бутен3 9.05497 0.116019 9.27248 X 105 1.68197 3.4815 X 10“2
2-Бутен, цис 9.82266 0.121971 1.0719 X 106 1.91732 3.8444 X 10“2
1,3-Бутадиен 7.41998 9.5452 X 10“2 1.03999 X 106 1.39146 2.8002 X 10“ 2
Изобутилен 8.95325 1.16025 X 10-1 9.2728 X 105 1.6927 3.48156 X 10“2
Пентан 12.1794 0.156751 2.12121 X 106 4.0748 6.6812 X 10“2
Изопентан 12.7959 0.160053 1.74632 X 106 3.7562 6.6812 X 10“ 2
Неопентан3 14.9413 0.19534 1.07186 X 106 2.72334 5.71607 X 10“2
Неопентан3 7.06955 5.17798 X 10~2 1.62085 X 106 2.06202 4.62003 X 10“2
1 -Пентен 11.05352 1.27921 X 10-1 1.38870 X 106 2.262816 4.2286 X 10“ 2
Г ексан 14.4373 1.77813 X Ю-1 3.31935 X 106 7.11671 1.09131 X Ю-1
Гептан 17.5206 1.99005 X 10-1 4.75574 X 106 10.36475 1.51954Х Ю-1
Нонан -41.456199 -9.64946 ХЮ-1 2.75136 X 106 37.17914 6.04989 X Ю-1
Декан -19.38795 -9.46923 ХЮ-1 3.43152 X 106 59.87797 9.86288 X Ю-1
а Константы рассчитаны при помощи типовой программы на отделении физической химии Института химической техноло-
гии, Прага, с использованием данных, заимствованных из литературы.
Продолжение табл. Д.5
Вещество с а У Диапазон приложимости ^шах, аТМ
Т, °C до dr
Водород® -2,4613 X 102 -0.34215 X 10"5 1.89 X 10"3 (0)-(150) 2.5 2500
Азот 5.48064 X 102 2.91545 X 10"4 7.5 X 10"3 (—163)—(200) 1.25 600
Азот 5.47364 X 102 7.093 X 10~5 4.5 X 10~3 (—170)—(100) 2.0
Кислород -2.0376 X 102 8.641 X 10"6 3.59 X 10“3 (—110)—(125) 0.8
СО 1.04 X 103 1.350 X Ю"4 0.006 (—140)—(—25) 1000
СО 1.04 X 103 1.350 X Ю"4 0.006 (—25)—(200) 1000
СО2 1.49183 X 104 8.4658 X 10"5 5.393 X 10"3 (Ю)—(150) 700
СО2 1.49183 X 104 8.4658 X 10“5 5.253 X 10“3 (150)—(250) 700
СО2а 7.0672 X 103 1.1271 X 10~4 4.94 X 10"3 (0)—(275) 2.1 700
SO2a 5.85038 X 104 5.86479 X 10"4 8.687 X 10"3 (Ю)—(250) 2.0 200
SO2 1.13356 X 105 7.1951 X 10"5 5.923 X 10“3 (Ю)—(250) 2.0 200
N2Oa 1.3794 X 104 9.377 X 10"5 5.301 X 10“3 (—30)—(150) 2.0 200
H2S 1.87032 X 104 7.0316 X 10"5 4.555 X 10~3 (5)-(170) 2.2 700
NH3 1.57536 X 102 4.651890 X 10"6 1.980 X 10~2 (0)-(300) 1.5 1100
CH3C1 6.87309 X 104 4.13840 X 10~4 1.131 X 10“2 (40)—(220) 2.1 300
Метан 2.545 X 103 1.24359 X 10~4 0.006 (—70)—(200) 1.8 400
Метан 3.5878 X 103 3.30 X 10“4 1.05 X 10"2 (0)_(350) 1.8 400
Этан 3.2767 X 104 2.43389 X 10"4 1.18 X 10"2 (0)—(275) 1.6 300
Этилен 2.112Х 104 1.78 X 10“4 9.23 ХЮ"3 (0)-(200) 1.6 300
Ацетилен 6.0162 X 103 -5.549 X 10“5 7.14 X Ю“3 (20)—(250) 1.6 150
Пропан 1.29 X 105 6.07175 X 10"4 0.022 (100)—(275) 1.75
Пропен 1.02611 X 105 4.55696 X 10~4 1.829 X 10" 2 (25)—(300) 1.45
Пропин® 1.09855 X 105 2.7363 X 10~4 1.245 X 10“2 (50)—(200) 300
Изобутан 2.8601 X 105 1.07408 X 10~3 0.034 (100)—(240) 1.8
Бутан 3.1640 X 105 1.10132 X 10”3 3.4 X 10"2 (150)—(300) 1.8
1-Бутен® 2.7493 X 105 9.1084 X 10“4 2.96 X 10"2 (150)—(250) 250
2-Бутен, цис 3.33972 X 105 1.05693 X 10~3 3.27 X 10"2
1,3-Бутадиен 2.45052 X 105 7.09881 X 10“4 2.35 X 10"2
Изобутилен 2.7492 X 105 9.10889 X 10"4 2.96 X 10“2 (150)—(275) 1.8
Пентан 8.2417 X 105 1.810Х 10"3 4.75 X 10"2 (140)—(280) 1.5 200
Изопентан 0.695 X 106 1.70 X 10"3 4.63 X 10~2 (130)—(280) 1.5 200
Неопентан® 4.73969 X 105 2.24898 X 10"3 5.352 X 10“2 (160)—(275) 2.1 250
Неопентан® 4.31017Х 105 2.51254 X 10"3 5.342 X 10"2 (30)—(200) 1.7 70
1-Пентен 4.53779 X 105 1.219208 X 10"3 3.595 X 10"2
Гексан 1.51276 X 106 2.81086 X 10"3 6.668 X 10~2 (275)—(350) 1.8
Гептан 2.47 X 106 4.35611 X 10"3 9 X 10"2 (275)—(350) 1.8
Нонан 2.516085 X 106 3.230516 X 10"3 12.23 X 10~2 (40)—(250) 700
Декан 7.822297 X 106 4.35394 X 10"3 15.3 X 10"2 (40)—(250) 700
а Константы рассчитаны при помощи типовой программы на отделении физической химии Института химической техноло-
гии, Прага, с использованием данных, заимствованных из литературы.
Таблица Д.6. Значения [log( f/p)}(0) (уравнение Ли — Кеслера)
PR
TR 0.010 0.050 0.100 0.200 0.400 0.600 0.800 1.000 1.200 1.500 2.000 3.000 5.000 7.000 10.000
0.30 -3.708 -4.402 -4.696 -4.985 -5.261 -5.412 -5.512 -5.584 -5.638 -5.697 -5.759 -5.810 -5.782 -5.679 -5.461
0.35 -2.471 -3.166 -3.461 -3.751 -4.029 -4.183 -4.285 -4.359 -4.416 -4.479 -4.547 -4.611 -4.608 -4.530 -4.352
0.40 -1.566 -2.261 -2.557 -2.848 -3.128 -3.283 -3.387 -3.463 -3.522 -3.588 -3.661 -3.735 -3.752 -3.694 -3.545
0.45 -0.879 -1.575 -1.871 -2.162 -2.444 -2.601 -2.707 -2.785 -2.845 -2.913 -2.990 -3.071 -3.104 -3.063 -2.938
0.50 -0.344 -1.040 -1.336 -1.628 -1.912 -2.070 -2.177 -2.256 -2.317 -2.387 -2.468 -2.555 -2.601 -2.572 -2.468
0.55 -0.008 -0.614 -0.911 -1.204 -1.488 -1.647 -1.755 -1.835 -1.897 -1.969 -2.052 -2.145 -2.201 -2.183 -2.096
0.60 -0.007 -0.269 -0.566 -0.859 -1.144 -1.304 -1.413 -1.494 -1.557 -1.630 -1.715 -1.812 -1.878 -1.869 -1.795
0.65 -0.005 -0.026 -0.283 -0.576 -0.862 -1.023 -1.132 -1.214 -1.278 -1.352 -1.439 -1.539 -1.612 -1.611 -1.549
0.70 -0.004 -0.021 -0.043 -0.341 -0.627 -0.789 -0.899 -0.981 -1.045 -1.120 -1.208 -1.312 -1.391 -1.396 -1.344
0.75 -0.003 -0.017 -0.035 -0.144 -0.430 -0.592 -0.703 -0.785 -0.850 -0.925 -1.015 -1.121 -1.204 -1.215 -1.172
0.80 -0.003 -0.014 -0.029 -0.059 -0.264 -0.426 -0.537 -0.619 -0.685 -0.760 -0.851 -0.958 -1.046 -1.062 -1.026
0.85 -0.002 -0.012 -0.024 -0.049 -0.123 -0.285 -0.396 -0.479 -0.544 -0.620 -0.711 -0.819 -0.911 -0.930 -0.901
0.90 -0.002 -0.010 -0.020 -0.041 -0.086 -0.166 -0.276 -0.359 -0.424 -0.500 -0.591 -0.700 -0.794 -0.817 -0.793
0.93 -0.002 -0.009 -0.018 -0.037 -0.077 -0.122 -0.214 -0.296 -0.361 -0.437 -0.527 -0.637 -0.732 -0.756 -0.735
0.95 -0.002 -0.008 -0.017 -0.035 -0.072 -0.113 -0.176 -0.258 -0.322 -0.398 -0.488 -0.598 -0.693 -0.719 -0.699
0.97 -0.002 -0.008 -0.016 -0.033 -0.067 -0.105 -0.148 -0.223 -0.287 -0.362 -0.452 -0.561 -0.657 -0.683 -0.665
0.98 -0.002 -0.008 -0.016 -0.032 -0.065 -0.101 -0.142 -0.206 -0.270 -0.344 -0.434 -0.543 -0.639 -0.666 -0.649
0.99 -0.001 -0.007 -0.015 -0.031 -0.063 -0.098 -0.137 -0.191 -0.254 -0.328 -0.417 -0.526 -0.622 -0.649 -0.633
1.00 -0.001 -0.007 -0.015 -0.030 -0.061 -0.095 -0.132 -0.176 -0.238 -0.312 -0.401 -0.509 -0.605 -0.633 -0.617
1.01 -0.001 -0.007 -0.014 -0.029 -0.059 -0.091 -0.127 -0.168 -0.224 -0.297 -0.385 -0.493 -0.589 -0.617 -0.602
1.02 -0.001 -0.007 -0.014 -0.028 -0.057 -0.088 -0.122 -0.161 -0.210 -0.282 -0.370 -0.477 -0.573 -0.601 -0.588
1.05 -0.001 -0.006 -0.013 -0.025 -0.052 -0.080 -0.110 -0.143 -0.180 -0.242 -0.327 -0.433 -0.529 -0.557 -0.546
1.10 -0.001 -0.005 -0.011 -0.022 -0.045 -0.069 -0.093 -0.120 -0.148 -0.193 -0.267 -0.368 -0.462 -0.491 -0.482
1.15 -0.001 -0.005 -0.009 -0.019 -0.039 -0.059 -0.080 -0.102 -0.125 -0.160 -0.220 -0.312 -0.403 -0.433 -0.426
1.20 -0.001 -0.004 -0.008 -0.017 -0.034 -0.051 -0.069 -0.088 -0.106 -0.135 -0.184 -0.266 -0.352 -0.382 -0.377
1.30 -0.001 -0.003 -0.006 -0.013 -0.026 -0.039 -0.052 -0.066 -0.080 -0.100 -0.134 -0.195 -0.269 -0.296 -0.293
1.40 -0.001 -0.003 -0.005 -0.010 -0.020 -о.озо -0.040 -0.051 -0.061 -0.076 -0.101 -0.146 -0.205 -0.229 -0.226
1.50 -0.000 -0.002 -0.004 -0.008 -0.016 -0.024 -0.032 -0.039 -0.047 -0.059 -0.077 -0.111 -0.157 -0.176 -0.173
1.60 -0.000 -0.002 -о.ооз -0.006 -0.012 -0.019 -0.025 -0.031 -0.037 -0.046 -0.060 -0.085 -0.120 -0.135 -0.129
1.70 -0.000 -0.001 -0.002 -0.005 -0.010 -0.015 -0.020 -0.024 -0.029 -0.036 -0.046 -0.065 -0.092 -0.102 -0.094
Приложения 603
Продолжение табл. Д.6
PR
TR 0.010 0.050 0.100 0.200 0.400 0.600 0.800 1.000 1.200 1.500 2.000 3.000 5.000 7.000 10.000
1.80 -0.000 -0.001 -0.002 -0.004 -0.008 -0.012 -0.015 -0.019 -0.023 -0.028 -0.036 -0.050 -0.069 -0.075 -0.066
1.90 -0.000 -0.001 -0.002 -о.ооз -0.006 -0.009 -0.012 -0.015 -0.018 -0.022 -0.028 -0.038 -0.052 -0.054 -0.043
2.00 -0.000 -0.001 -0.001 -0.002 -0.005 -0.007 -0.009 -0.012 -0.014 -0.017 -0.021 -0.029 -0.037 -0.037 -0.024
2.20 -0.000 -0.000 -0.001 -0.001 -о.ооз -0.004 -0.005 -0.007 -0.008 -0.009 -0.012 -0.015 -0.017 -0.012 0.004
2.40 -0.000 -0.000 -0.000 -0.001 -0.001 -0.002 -о.ооз -о.ооз -0.004 -0.004 -0.005 -0.006 -0.003 0.005 0.024
2.60 -0.000 -0.000 -0.000 -0.000 -0.000 -0.001 -0.001 -0.001 -0.001 -0.001 -0.001 0.001 0.007 0.017 0.037
2.80 0.000 0.000 0.000 0.000 0.000 0.000 0.001 0.001 0.001 0.002 0.003 0.005 0.014 0.025 0.046
3.00 0.000 0.000 0.000 0.000 0.001 0.001 0.002 0.002 0.003 0.003 0.005 0.009 0.018 0.031 0.053
3.50 0.000 0.000 0.000 0.001 0.001 0.002 0.003 0.004 0.005 0.006 0.008 0.013 0.025 0.038 0.061
4.00 0.000 0.000 0.000 0.001 0.002 0.003 0.004 0.005 0.006 0.007 0.010 0.016 0.028 0.041 0.064
Таблица Д. 7. Значения [log(///?)](1) (уравнение Ли — Кеслера)
PR
TR 0.010 0.050 0.100 0.200 0.400 0.600 0.800 1.000 1.200 1.500 2.000 3.000 5.000 7.000 10. 000
0.30 -8.778 -8.779 -8.781 -8.785 -8.790 -8.797 -8.804 -8.811 -8.818 -8.828 -8.845 -8.880 -8.953 -9.022 -9.126
0.35 -6.528 -6.530 -6.532 -6.536 -6.544 -6.551 -6.559 -6.567 -6.575 -6.587 -6.606 -6.645 -6.723 -6.800 -6.919
0.40 -4.912 -4.914 -4.916 -4.919 -4.929 -4.937 -4.945 -4.954 -4.962 -4.974 -4.995 -5.035 -5.115 -5.195 -5.312
0.45 -3.726 -3.728 -3.730 -3.734 -3.742 -3.750 -3.758 -3.766 -3.774 -3.786 -3.806 -3.845 -3.923 -4.001 -4.114
0.50 -2.838 -2.839 -2.841 -2.845 -2.853 -2.861 -2.869 -2.877 -2.884 -2.896 -2.915 -2.953 -3.027 -3.101 -3.208
0.55 -0.013 -2.163 -2.165 —2.169 -2.177 -2.184 -2.192 -2.199 -2.207 -2.218 -2.236 -2.273 -2.342 -2.410 -2.510
0.60 -0.009 -1.644 -1.646 -1.650 -1.657 -1.664 -1.671 -1.677 -1.684 -1.695 -1.712 -1.747 -1.812 -1.875 -1.967
0.65 -0.006 -0.031 -1.242 -1.245 -1.252 -1.258 -1.265 -1.271 -1.278 -1.287 -1.304 -1.336 -1.397 -1.456 -1.539
0.70 -0.004 -0.021 -0.044 -0.927 -0.934 -0.940 -0.946 -0.952 -0.958 -0.967 -0.983 -1.013 -1.070 -1.124 -1.201
0.75 -0.003 -0.014 -0.030 -0.675 -0.682 -0.688 -0.694 -0.700 -0.705 -0.714 -0.728 -0.756 -0.809 -0.858 -0.929
Продолжение табл. Д. 7
0.80 -0.002 -0.010 -0.020 -0.043 -0.481 -0.487 -0.493 -0.499 -0.504 -0.512 -0.526 -0.551 -0.600 -0.645 -0.709
0.85 -0.001 -0.006 -0.013 -0.028 -0.321 -0.327 -0.332 -0.338 -0.343 -0.351 -0.364 -0.388 -0.432 -0.473 -0.530
0.90 -0.001 -0.004 -0.009 -0.018 -0.039 -0.199 -0.204 -0.210 -0.215 -0.222 -0.234 -0.256 -0.296 -0.333 -0.384
0.93 -0.001 -0.003 -0.007 -0.013 -0.029 -0.048 -0.141 -0.146 -0.151 -0.158 -0.170 -0.190 -0.228 -0.262 -0.310
0.95 -0.001 -о.ооз -0.005 -0.011 -0.023 -0.037 -0.103 -0.108 -0.114 -0.121 -0.132 -0.151 -0.187 -0.220 -0.265
0.97 -0.000 -0.002 -0.004 -0.009 -0.018 -0.029 -0.042 -0.075 -0.080 -0.087 -0.097 -0.116 -0.149 -0.180 -0.223
0.98 -0.000 -0.002 -0.004 -0.008 -0.016 -0.025 -0.035 -0.059 -0.064 -0.071 -0.081 -0.099 -0.132 -0.162 -0.203
0.99 -0.000 -0.002 -о.ооз -0.007 -0.014 -0.021 -0.030 -0.044 -0.050 -0.056 -0.066 -0.084 -0.115 -0.144 -0.184
1.00 -0.000 -0.001 -о.ооз -0.006 -0.012 -0.018 -0.025 -0.031 -0.036 -0.042 -0.052 -0.069 -0.099 -0.127 -0.166
1.01 -0.000 -0.001 -о.ооз -0.005 -0.010 -0.016 -0.021 -0.024 -0.024 -0.030 -0.038 -0.054 -0.084 -0.111 -0.149
1.02 -0.000 -0.001 -0.002 -0.004 -0.009 -0.013 -0.017 -0.019 -0.015 -0.018 -0.026 -0.041 -0.069 -0.095 -0.132
1.05 -0.000 -0.001 -0.001 -0.002 -0.005 -0.006 -0.007 -0.007 -0.002 0.008 0.007 -0.005 -0.029 -0.052 -0.085
1.10 -0.000 -0.000 0.000 0.000 0.001 0.002 0.004 0.007 0.012 0.025 0*041 0.042 0.026 0.008 -0.019
1.15 0.000 0.000 0.001 0.002 0.005 0.008 0.011 0.016 0.022 0.034 0.056 0.074 0.069 0.057 0.036
1.20 0.000 0.001 0.002 0.003 0.007 0.012 0.017 0.023 0.029 0.041 0.064 0.093 0.102 0.096 0.081
1.30 0.000 0.001 0.003 0.005 0.011 0.017 0.023 0.030 0.038 0.049 0.071 0.109 0.142 0.150 0.148
1.40 0.000 0.002 0.003 0.006 0.013 0.020 0.027 0.034 0.041 0.053 0.074 0.112 0.161 0.181 0.191
1.50 0.000 0.002 0.003 0.007 0.014 0.021 0.028 0.036 0.043 0.055 0.074 0.112 0.167 0.197 0.218
1.60 0.000 0.002 0.003 0.007 0.014 0.021 0.029 0.036 0.043 0.055 0.074 0.110 0.167 0.204 0.234
1.70 0.000 0.002 0.004 0.007 0.014 0.021 0.029 0.036 0.043 0.054 0.072 0.107 0.165 0.205 0.242
1.80 0.000 0.002 0.003 0.007 0.014 0.021 0.028 0.035 0.042 0.053 0.070 0.104 0.161 0.203 0.246
1.90 0.000 0.002 0.003 0.007 0.014 0.021 0.028 0.034 0.041 0.052 0.068 0.101 0.157 0.200 0.246
2.00 0.000 0.002 0.003 0.007 0.013 0.020 0.027 0.034 0.040 0.050 0.066 0.097 0.152 0.196 0.244
2.20 0.000 0.002 0.003 0.006 0.013 0.019 0.025 0.032 0.038 0.047 0.062 0.091 0.143 0.186 0.236
2.40 0.000 0.002 0.003 0.006 0.012 0.018 0.024 0.030 0.036 0.044 0.058 0.086 0.134 0.176 0.227
2.60 0.000 0.001 0.003 0.006 0.011 0.017 0.023 0.028 0.034 0.042 0.055 0.080 0.127 0.167 0.217
2.80 0.000 0.004 0.003 0.005 0.011 0.016 0.021 0.027 0.032 0.039 0.052 0.076 0.120 0.158 0.208
3.00 0.000 0.001 0.003 0.005 0.010 0.015 0.020 0.025 0.030 0.037 0.049 0.072 0.114 0.151 0.199
3.50 0.000 0.001 0.002 0.004 0.009 0.013 0.018 0.022 0.026 0.033 0.043 0.063 0.101 0.134 0.179
4.00 0.000 0.001 0.002 0.004 0.008 0.012 0.016 0.020 0.023 0.029 0.038 0.057 0.090 0.121 0.163
Примечание. Эта информация представлена в виде графиков в «Справочнике Американского нефтяного института», рис. 7G1.4, 7G1.5,
7G1.6 и 7G1.7.
Приложения 605
Компоненты Х12 — Хп, кал/моль Х12 - Х22, кал/моль Р, мм рт. ст. т,
1 2
Ацетон Бензол 494.92 -167.91 760
Тетрахлорметан 651.76 -12.67 760
Хлороформ -72.20 -332.23 760
2,3-Диметилбутан 948.29 234.96 760
Этанол 38.17 418.96 760
Метанол -214.95 664.08 760
-203.03 666.99 55
н-Пентан 996.75 262.74 760
2-Пропанол 127.43 284.99 760
429.17 53.40 55
Вода 439.64 1405.49 760
Ацетонитрил Вода 694.08 1610.07 760
Бензол Ацетон -167.91 494.93 760
1-Бутанол 160.12 817.67 760
Т етрахл орметан -103.41 204.82 760
Хлороформ 141.62 -204.22 760
Циклогексан 187.23 80.02 760
Циклопентан 266.56 -24.18 760
Этанол 131.47 1297.90 760
н-Гептан 99.35 292.94 760
73.63 364.63 75
н-Гексан 173.93 169.92 760
Метанол 153.86 1620.36 760
Метил ацетат 229.25 -23.84 760
Метилциклогексан -4.15 360.92 760
Метилциклопентан 161.44 97.33 760
1-Пропанол -73.91 1370.32 760
67.14 1222.07 75
2-Пропанол 160.53 1007.94 760
272.35 1066.93 500
1-Бутанол Бензол 817.67 160.12 760
Толуол 887.80 104.68 760
Бутил- Этилциклогексан 643.51 636.11 400
целлозольв н-Октан 1070.54 298.62 400
Тетрахлор- Ацетон -12.67 651.76 760
метан Бензол 204.82 -103.41 760
2-Пропанол 111.11 1232.94 760
Целлозольв Этилбензол 755.77 121.89 760
н-Гексан 834.86 656.23 760
Гексен-1 370.05 705.47 760
н-Октан 989.04 622.77 760
Хлороформ Ацетон -332.23 -72.20 760
Бензол -204.22 141.62 760
2,3-Диметилбутан 213.88 223.69 760
Этилацетат -367.50 -92.50 760
Метанол -373.30 1703.68 760
Метилацетат -451.09 113.24 760
Метилэтилкетон -231.61 -235.12 760
Циклогексан Бензол 80.02 187.23 760
Этанол 303.42 2151.01 760
Метилацетат 345.11 691.65 760
2-Пропанол 69.02 1734.12 760
223.13 1590.51 500
Толуол -414.68 909.36 760
Циклопентан Бензол -24.18 266.56 760
2,3-Диметил- Ацетон 234.96 948.29 760
бутан Хлороформ 223.69 213.88 760
Метанол 449.08 2771.85 760
Продолжение табл. Д.8
Приложения 607
Компоненты Х12 — Х11, кал/моль Х12 — Х22, кал/моль р, мм рт. ст. Т, °C
1 2
1,4-Диоксан н-Гексан 806.80 164.58 760
Гексен-1 495.19 176.39 760
Этанол Ацетон 418.96 38.17 760
Бензол 1297.90 131.47 760
Циклогексан 2151.01 303.42 760
Этилацетат 844.69 -178.81 760
822.03 -62.43 40
744.81 -52.14 60
н-Гептан 2096.50 617.57 760
н-Гексан 2281.99 283.63 760
Метанол -511.39 598.44 760
Метилциклопентан 2221.47 161.53 760
Толуол 1238.70 251.93 756
Вода 382.30 955.45 760
Этилацетат Хлороформ -92.50 -367.50 760
Этанол -178.81 844.69 760
-62.43 822.03 40
-52.14 744.81 60
Метанол -200.36 985.69 760 60
-316.92 1203.57 40
20.32 866.15 50
-173.45 1030.15 60
1-Пропанол -198.72 661.24 760
42.39 558.40 40
-25.10 519.67 60
2-Пропанол 60.99 289.68 760
39.77 664.42 40
45.72 488.11 60
Этилбензол Целлозольв 121.89 755.77 760
Этилциклогексан 396.01 -240.92 400
Г ексиленгликоль 52.43 1601.04 400
н-Октан 304.31 -134.87 760
Этилцикло- Бутилцеллозольв 636.11 643.51 400
гексан Этилбензол -240.92 396.01 400
Г ексиленгликоль 76.95 3592.40 400
н-Г ептан Бензол 292.94 99.35 760
364.63 73.63 75
Этанол 617.57 2096.50 760
1-Пропанол 316.22 1353.98 75
н-Гексан Бензол 169.92 173.93 760
Целлозольв 656.23 834.86 760
1,4-Диоксан 164.58 806.80 760
Этанол 283.63 2281.99 760
Гексан-1 415.18 -279.86 760
Метилциклопентан 272.09 -175.70 760
1-Пропанол 834.85 812.66 760
1,2,3 -Т рихлорпропан 116.39 1106.54 760
Гексан-1 Целлозольв 705.47 370.05 760
1,4-Диоксан 176.39 495.19 760
н-Гексан -279.86 415.18 760
1,2,3-Трихлорпропан 156.93 570.31 760
Гексиленгликоль Этилбензол 1601.04 52.43 400
Этилциклогексан 3592.40 76.95 400
Метанол Ацетон 664.08 -214.95 760
666.99 -203.03 55
Бензол 1620.36 153.86 760
Хлороформ 1703.68 -373.30 760
2,3-Диметилбутан 2771.85 449.08 760
Продолжение табл. Д.8
Компоненты X12 — Xu, кал/моль X12 “ ^22, кал/моль P, мм рт. CT. T, °C
1 2
Methanol (continued) Ethanol 598.44 -511.39 760
Ethyl acetate 985.69 -200.36 760
1203.57 -316.92 40
866.15 20.32 50
1030.15 -173.45 60
Methyl acetate 834.06 -78.81 760
2-Propanol 88.02 -30.19 760
Water 205.30 482.16 760
Methyl acetate Benzene -23.84 229.25 760
Chloroform 113.24 -451.09 760
Cyclohexane 691.65 345.11 760
Methanol -78.81 834.06 760
Methylcyclohexane Benzene 360.92 -4.15 760
2-Propanol 209.75 1831.76 500
Methylcyclopentane Benzene 97.33 161.44 760
Ethanol 161.53 2221.47 760
n-Hexane -175.70 272.09 760
Toluene -451.92 957.61 760
Methyl-ethyl-ketone Chloroform -235.12 -231.61 760
n-Octane Butyl cellosolve 298.62 1070.54 400
Cellosolve 622.77 989.04 760
Ethylbenzene -134.87 304.31 760
2-Propanol 422.41 1391.09 400
n-Pentane Acetone 262.74 996.75 760
1-Propanol Benzene 1370.32 -73.91 760
1222.07 67.14 75
Ethyl acetate 661.24 -198.72 760
558.40 42.39 40
519.67 -25.10 60
n-Heptane 1353.98 316.22 75
n-Hexane 812.66 834.85 760
Water 1015.80 1284.61 760
1942.36 1144.00 40
1051.44 1188.52 60
2-Propanol Acetone 284.99 127.43 760
53.40 429.17 55
Benzene 1007.94 160.53 760
1066.93 272.35 500
Carbon tetrachloride 1232.94 111.11 760
Cyclohexane 1734.12 69.02 760
1590.51 223.13 500
Ethyl acetate 289.68 60.99 760
664.42 39.77 40
418.11 45.72 60
Methanol -30.19 88.02 760
Methylcyclohexane 1831.76 209.75 500
n-Octane 1391.09 422.41 400
2,2,4-T rimethylpentane 1231.69 183.12 760
Toluene 1-Butanol 104.68 887.80 760
Cyclohexane 909.36 -414.68 760
Ethanol 251.93 1238.70 756
Methylcyclopentane 957.61 -452.92 760
1,2,3-Trichloropropane n-Hexane 1106.54 116.93 760
Hexene-1 570.31 156.39 760
2,2,4-T rimethylpentane 2-Propanol 183.12 1231.69 760
water Acetone 1405.49 439.64 760
Acetonitrile 1610.07 694.08 760
Продолжение табл. Д.8
Приложения 609
Компоненты Х12 — Хп, кал/моль Х12 - Х22, кал/моль Р, мм рт. ст. Т, °C
1 2
2,2,4-Три- Этанол 955.45 382.30 760
метилпентан Метанол 482.61 205.30 760
1-Пропанол 1284.61 1015.80 760
1144.00 1942.36 40
1188.52 1051.44 60
Таблица Д.9. Некоторые параметры уравнения Маргулеса (Холмс и ван Винкл, 1970) (© Am. Chem. Soc.)
Компоненты
1 2 А12 ^21 Р, мм Т, °C рт.ст.
Ацетон Бензол 0.2012 0.1533 760
Т етрахл орметан 0.3874 0.3282 760
Хлороформ -0.3051 -0.2676 760
2,3-Диметилбутан 0.6345 0.6358 760
Этанол 0.2569 0.2870 760
Метанол 0.2634 0.2798 760
0.2762 0.2877 55
н-Пентан 0.7386 0.6329 760
2-Пропанол 0.2152 0.2688 760
0.3154 0.2428 55
Вода 0.9709 0.5576 760
Ацетонитрил Вода 1.0489 0.8231 760
Бензол Ацетон 0.1533 0.2012 760
1-Бутанол 0.3449 0.5651 760
Т етрахлорметан 0.0359 0.0488 760
Хлороформ -0.0824 -0.0532 760
Циклогексан 0.1462 0.1640 760
Циклопентан 0.1634 0.1290 760
Этанол 0.5718 0.7883 760
н-Гептан 0.0842 0.1899 760
0.0953 0.2088 75
н-Гексан 0.1430 0.2010 760
Метанол 0.7494 0.8923 760
Метил ацетат 0.1219 0.0939 760
Метилциклогексан 0.0760 0.1760 760
Метил циклопентан 0.1342 0.1606 760
1-Пропанол 0.3251 0.7332 760
0.4303 0.7286 75
2-Пропанол 0.4523 0.6551 760
0.5392 0.7527 500
1-Бутанол Бензол 0.5651 0.3449 760
Толуол 0.5340 0.3699 760
Бутил- Этилциклогексан 0.5814 0.5784 400
целлозольв н-Октан 0.6903 0.5227 400
Тетрахлор- Ацетон 0.3282 0.3874 760
метан Бензол 0.0488 0.0359 760
2-Пропанол 0.4763 0.7656 760
Целлозольв Этилбензол 0.4379 0.3750 760
н-Гексан 0.6633 0.7183 760
Гексен-1 0.4228 0.5818 760
н-Октан 0.6117 0.7467 760
20-864
Компоненты
1 2 А12 Л21 Р, мм рт ст. Т, °C
Хлороформ Ацетон -0.2676 -0.3051 760
Бензол -0.0532 -0.0824 760
2,3 -Диметилбутан 0.1637 0.2677 760
Этилацетат -0.2726 -0.4275 760
Метанол 0.3702 0.7767 760
Метилацетат -0.2112 -0.3270 760
Метилэтилкетон -0.2938 -0.3507 760
Циклогексан Бензол 0.1640 0.1462 760
Этанол 0.7743 1.0699 760
Метилацетат 0.5789 0.5313 760
2-Пропанол 0.5006 0.9539 760
0.5883 0.9795 500
Толуол 0.0689 0.1563 760
Циклопентан Бензол 0.1290 0.1634 760
2,3-Диметил- Ацетон 0.6358 0.6345 760
бутан Хлороформ 0.2677 0.1637 760
Метанол 1.1265 1.5255 760
1,4-Диоксан н-Гексан 0.5230 0.4857 760
Гексен-1 0.3577 0.3755 760
Этанол Ацетон 0.2870 0.2569 760
Бензол 0.7883 0.5718 760
Циклогексан 1.0699 0.7743 760
Этилацетат 0.3925 0.3313 760
0.4816 0.4130 40
0.4080 0.3849 60
н-Г ептан 1.0806 1.0226 760
н-Гексан 1.1738 0.8337 760
Метанол 0.0081 0.0189 760
Метилциклопентан 1.1965 0.7065 760
Толуол 0.7066 0.6933 756
Вода 0.6848 0.3781 760
Этилацетат Хлороформ -0.4275 -0.2726 760
Этанол 0.3313 0.3925 760
0.4130 0.4816 40
0.3849 0.4080 60
Метанол 0.4463 0.4229 760
0.4213 0.5626 40
0.5324 0.4482 50
0.4737 0.4767 60
1-Пропанол 0.1982 0.2849 760
0.3412 0.3923 40
0.2749 0.3094 60
2-Пропанол 0.2112 0.1961 760
0.3717 0.4573 40
0.3037 0.3256 60
Этилбензол Целлозольв 0.3750 0.4379 760
Этилциклогексан 0.0800 0.0626 400
Г ексиленгликоль 0.3105 0.7358 400
н-Октан 0.0889 0.0903 760
Этилцикло- Бутилцеллозольв 0.5784 0.5814 400
гексан Этилбензол 0.0626 0.0800 400
Г ексиленгликоль 0.4293 0.9448 400
н-Гептан Бензол 0.1899 0.0842 760
0.2088 0.0953 75
Этанол 1.0226 1.0806 760
1-Пропанол 0.7719 0.7548 75
н-Гексан Бензол 0.2010 0.1430 760
Целлозольв 0.7183 0.6633 760
Продолжение табл. Д.9
Приложения 611
Компоненты
1 2 а12 л21 Р, мм рт ст. Т, °
н-Гексан (continued) 1,4-Диоксан 0.4857 0.5238 760
Этанол 0.8337 1.1738 760
Гексен-1 0.0283 0.0078 760
Метилциклопентан 0.0188 0.0014 760
1-Пропанол 0.8511 0.5763 760
1,2,3-Трихлорпропан 0.4298 0.6916 760
Гексен-1 Целлозольв 0.5818 0.4228 760
1,4-Диоксан 0.3755 0.3577 760
н-Гексан 0.0078 0.0283 760
1,2,3 -Т рихл орпропан 0.3382 0.4307 760
Гексилен- Этилбензол 0.7358 0.3105 400
гликоль Этилциклогексан 0.9448 0.4293 400
Метанол Ацетон 0.2798 0.2634 760
0.2877 0.2762 55
Бензол 0.8923 0.7494 760
Хлороформ 0.7767 0.3702 760
2,3-Диметилбутан 1.5255 1.1265 760
Этанол 0.0189 0.0081 760
Этилацетат 0.4229 0.4463 760
0.5626 0.4213 40
0.4482 0.5324 50
0.4767 0.4737 60
Метилацетат 0.4393 0.4261 760
2-Пропанол -0.0326 -0.0329 760
Вода 0.3794 0.2211 760
Метилацетат Бензол 0.0939 0.1219 760
Хлороформ -0.3270 -0.2112 760
Циклогексан 0.5313 0.5789 760
Метанол 0.4261 0.4393 760
Метилцикло- Бензол 0.1760 0.0760 760
гексан 2-Пропанол 0.6785 1.0343 500
Метилцикло- Бензол 0.1606 0.1342 760
пентан Этанол 0.7065 1.1965 760
н-Гексан 0.0014 0.0188 760
Толуол 0.0694 0.1627 760
Метилэтил- Хлороформ -0.3507 -0.2938 760
кетон Бутилцеллозольв 0.5227 0.6903 400
н-Октан Целлозольв 0.7467 0.6117 760
Этилбензол 0.0903 0.0889 760
2-Пропанол 0.8524 0.8044 400
н-Пентан Ацетон 0.6329 0.7386 760
1 -Пропанол Бензол 0.7332 0.3251 760
0.7286 0.4303 75
Этилацетат 0.2849 0.1982 760
0.3923 0.3412 40
0.3094 0.2749 60
н-Гептан 0.7548 0.7719 75
н-Гексан 0.5763 0.8511 760
Вода 1.0536 0.4393 760
1.0748 0.4507 40
1.0825 0.4653 60
2-Пропанол Ацетон 0.2688 0.2152 760
0.2428 0.3154 55
Бензол 0.6551 0.4523 760
0.7527 0.5392 500
Т етрахлорметан 0.7656 0.4763 760
Циклогексан 0.9539 0.5006 760
0.9795 0.5883 500
20'
Компоненты
1 2 а12 Л21 Р, мм рт ст. Т, °
2-Пропанол Этилацетат 0.1961 0.2112 760
0.4573 0.3717 40
0.3256 0.3037 60
Метанол -0.0329 -0.0326 760
Метилциклогексан 1.0343 0.6785 500
н-Окт-ан 0.8044 0.8524 400
2,2,4-Триметилпентан 0.6601 0.6924 760
Толуол 1-Бутанол 0.3699 0.5340 760
Циклогексан 0.1563 0.0689 760
Этанол 0.6933 0.7066 756
Метилциклопентан 0.1627 0.0694 760
1,2,3-Три- н-Гексан 0.6916 0.4298 760
хлорпропан Гексен-1 0.4307 0.3382 760
2,2,4-Триме- 2-Пропанол 0.6924 0.6601 760
тилпентан Ацетон 0.5576 0.9709 760
Вода Ацетонитрил 0.8231 1.0489 760
Этанол 0.3781 0.6848 760
Метанол 0.2211 0.3794 760
1-Пропанол 0.4393 1.0536 760
0.4507 1.0748 40
0.4653 1.0825 60
Таблица Ц.10. Некоторые параметры уравнения ван Лаара (Холмс и ван Винкл, 1970)
(© Am. Chem. Soc.)
Компоненты
1 2 л12 Л21 Р, мм рт.ст. Т, °C
Ацетон Бензол 0.2039 0.1563 760
Т етрахл орметан 0.3889 0.3301 760
Хлороформ -0.3045 -0.2709 760
2,3-Диметилбутан 0.6345 0.6358 760
Этанол 0.2574 0.2879 760
Метанол 0.2635 0.2801 760
0.2763 0.2878 55
н-Пентан 0.7403 0.6364 760
2-Пропанол 0.2186 0.2690 760
0.3158 0.2495 55
Вода 0.9972 0.6105 760
Ацетонитрил Вода 1.0680 0.8207 760
Бензол Ацетон 0.1563 0.2039 760
1-Бутанол 0.3594 0.5865 760
Т етрахл орметан 0.0360 0.0509 760
Хлороформ -0.0858 -0.0556 760
Циклогексан 0.1466 0.1646 760
Циклопентан 0.1655 0.1302 760
Этанол 0.5804 0.7969 760
н-Гептан 0.0985 0.2135 760
0.1072 0.2361 75
н-Гексан 0.1457 0.2063 760
Метанол 0.7518 0.8975 760
Метил ацетат 0.1292 0.0919 760
Метилциклогексан 0.0910 0.1901 760
Метилциклопентан 0.1360 0.1605 760
1-Пропанол 0.3772 0.7703 760
Продолжение табл. Д.10
Приложения 613
Компоненты
1 2 А12 Р, мм рт Т, °C
А21 СТ.
Бензол 0.7564
0.4508 75
2-Пропанол 0.4638 0.6723 760
0.5455 0.7716 500
1-Бутанол Бензол 0.5865 0.3594 760
Толуол 0.5430 0.3841 760
Бутилцелло- Этилциклогексан 0.5814 0.5784 400
зольв н-Октан 0.6967 0.5318 400
Тетрахлор- Ацетон 0.3301 0.3889 760
метан Бензол 0.0509 0.0360 760
2-Пропанол 0.4918 0.7868 760
Целлозольв Этилбензол 0.4402 0.3762 760
н-Гексан 0.6629 0.7206 760
Гексен-1 0.4367 0.5860 760
н-Октан 0.6158 0.7507 760
Хлороформ Ацетон -0.2709 -0.3045 760
Бензол -0.0556 -0.0858 760
2,3-Диметилбутан 0.1736 0.2790 760
Этилацетат -0.2868 -0.4478 760
Метанол 0.4104 0.8263 760
Метилацетат -0.2249 -0.3343 760
Метилэтилкетон -0.2990 -0.3486 760
Циклогексан Бензол 0.1646 0.1466 760
Этанол 0.7811 1.1031 760
Метилацетат 0.5799 0.5317 760
2-Пропанол 0.5322 1.0162 760
0.6156 1.0158 500
Толуол 0.0702 0.2578 760
Циклопентан Бензол 0.1302 0.1655 760
2,3-Диметил- Ацетон 0.6358 0.6345 760
бутан Хлороформ 0.2790 0.1736 760
Метанол 1.1276 1.5408 760
1,4-Диоксан н-Гексан 0.5260 0.4850 760
Гексен-1 0.3578 0.3757 760
Этанол Ацетон 0.2879 0.2574 760
Бензол 0.7969 0.5804 760
Циклогексан 1.1031 0.7811 760
Этилацетат 0.3972 0.3311 760
0.4833 0.4151 40
0.4093 0.3842 60
н-Г ептан 1.0832 1.0208 760
н-Гексан 1.2005 0.8422 760
Метанол 0.0088 0.0254 760
Метилциклопентан 1.2330 0.7332 760
Толуол 0.7067 0.6932 756
Вода 0.7292 0.4104 760
Этилацетат Хлороформ -0.4478 -0.2868 760
Этанол 0.3311 0.3972 760
0.4151 0.4833 40
0.3842 0.4093 60
Метанол 0.4470 0.4227 760
0.4278 0.5741 40
0.5399 0.4476 50
0.4736 0.4768 60
1-Пропанол 0.2051 0.2893 760
0.3444 0.3913 40
0.2762 0.3092 60
2-Пропанол 0.2113 0.1964 760
0.3747 0.4604 40
Компоненты
1 2 А12 А21 Р, мм рт ст. Т, °(
Метилацетат 2-Пропанол 0.3039 0.3261 60
Этилбензол Целлозольв 0.3762 0.4402 760
Этилциклогексан 0.0821 0.0628 400
Гексил енгликоль 0.3719 0.8383 400
н-Октан 0.0890 0.0902 760
Этилцикло- Бутилцеллозольв 0.5784 0.5814 400
гексан Этилбензол 0.0628 0.0821 400
Г ексиленгликоль 0.4770 1.1219 400
н-Гептан Бензол 0.2135 0.0985 760
0.2361 0.1072 75
Этанол 1.0208 1.0832 760
1-Пропанол 0.7719 0.7550 75
н-Гексан Бензол 0.2063 0.1457 760
Целлозольв 0.7206 0.6629 760
1,4-Диоксан 0.4850 0.5260 760
Этанол 0.8422 1.2005 760
Гексен-1 0.0393 0.0114 760
Метилциклопентан 0.0023 0.0226 760
1-Пропанол 0.8734 0.5952 760
1,2,3-Трихлорпропан 0.4520 0.7257 760
Гексен-1 Целлозольв 0.5860 0.4367 760
1,4-Диоксан 0.3757 0.3578 760
н-Гексан 0.0114 0.0393 760
1,2,3-Трихлорпропан 0.3419 0.4372 760
Гексилен- Этилбензол 0.8383 0.3719 400
гликоль Этилциклогексан 1.1219 0.4770 400
Метанол Ацетон 0.2801 0.2635 760
0.2878 0.2763 55
Бензол 0.8975 0.7518 760
Хлороформ 0.8263 0.4104 760
2,3-Диметилбутан 1.5408 1.1276 760
Этанол 0.0254 0.0088 760
Этилацетат 0.4227 0.4470 760
0.5741 0.4278 40
0.4476 0.5399 50
0.4768 0.4736 60
Метил ацетат 0.4394 0.4262 760
2-Пропанол -0.0325 -0.0329 760
Вода 0.3861 0.2439 760
Метил ацетат Бензол 0.0919 0.1292 760
Хлороформ -0.3343 -0.2249 760
Циклогексан 0.5317 0.5799 760
Метанол 0.4262 0.4394 760
Метилцикло- Бензол 0.1901 0.0910 760
гексан 2-Пропанол 0.6886 1.0659 500
Метилцикло- Бензол 0.1605 0.1360 760
пентан Этанол 0.7332 1.2330 760
н-Гексан 0.0226 0.0023 760
Толуол 0.0717 0.2475 760
Метилэтил- Хлороформ -0.3486 -0.2990 760
кетон Бутилцеллозольв 0.5318 0.6967 400
н-Октан Целлозольв 0.7507 0.6158 760
Этилбензол 0.0902 0.0890 760
2-Пропанол 0.8535 0.8043 400
н-Пентан Ацетон 0.6364 0.7403 760
1-Пропанол Бензол 0.7703 0.3772 760
0.7564 0.4508 75
Продолжение табл. Д.10
Приложения 615
Компоненты
1 2 А12 а21 Р, мм рт ст. Т, °C
1-Пропанол Этилацетат 0.2893 0.2051 760
0.3913 0.3444 40
0.3092 0.2762 60
н-Г ептан 0.7550 0.7719 75
н-Г ексан 0.5952 0.8734 760
Вода 1.1433 0.5037 760
1.2315 0.5305 40
1.1879 0.5224 60
2-Пропанол Ацетон 0.2690 0.2186 760
0.2495 0.3158 55
Бензол 0.6723 0.4638 760
0.7716 0.5455 500
Тетрахл орметан 0.7868 0.4918 760
Циклогексан 1.0162 0.5322 760
1.0158 0.6156 500
Этилацетат 0.1964 0.2113 760
0.4604 0.3747 40
0.3261 0.3039 60
Метанол -0.0329 -0.0325 760
Метилциклогексан 1.0659 0.6886 500
н-Октан 0.8043 0.8535 400
2,2,4-Триметилпентан 0.6603 0.6927 760
Толуол 1-Бутанол 0.3841 0.5430 760
Циклогексан 0.2578 0.0702 760
Этанол 0.6932 0.7067 756
Метилциклопентан 0.2475 0.0717 760
1,2,3-Трихлор- н-Гексан 0.7257 0.4520 760
пропан Гексен-1 0.4372 0.3419 760
2,2,4-Триметил- 2-Пропанол 0.6927 0.6603 760
пентан Ацетон 0.6105 0.9972 760
Вода Ацетонитрил 0.8207 1.0680 760
Этанол 0.4104 0.7292 760
Метанол 0.2439 0.3861 760
1-Пропанол 0.5037 1.1433 760
0.5305 1.2315 40
0.5224 1.1879 60
Таблица Д.11. Параметры объема и площади поверхности групп уравнения UNIFAC для расчета парожидкостного
равновесия [308] (© Am. Chem. Soc.)
Основная группа Подгруппа No. Rk Qk
1 сн3 1 0.9011 0.848 Гексан: 2 СН3, 4 СН2
“СН2” сн2 2 0.6744 0.540 2-Метилпропан: 3 СН3, 1 СН
сн 3 0.4469 0.228 2,2-Диметилпропан: 4 СН3, 1 С
с 4 0.2195 0.000
2 сн2=сн 5 1.3454 1.176 1-Гексен: 1 СН3, 3 СН2, 1 СН2=СН
“С=С” сн=сн 6 1.1167 0.867 2-Г ексен: 2 СН3, 2 СН2, 1 СН=СН
сн2=с 7 1.1173 0.988 2-Метил-1 -бутен: 2 СН3, 1 СН2, 1 СН2=С
сн=с 8 0.8886 0.676 2-Метил-2-бутен: 3 СН3, 1 СН=С
с=с 9 0.6605 0.485 2,3-Диметил-2-бутен: 4 СН3, 1 С=С
3 АСН 10 0.5313 0.400 Бензол: 6 АСН
“АСН” АС 11 0.3652 0.120 Стирол: 1 СН2=СН, 5 АСН, 1 АС
4 АССН3 12 1.2663 0.968 Толуол: 5 АСН, 1 АССН3
“АССН2” ассн2 13 1.0396 0.660 Этилбензол: 1 СН3, 5 АСН, 1 АССН2
АССН 14 0.8121 0.348 Кумол: 2 СН3, 5 АСН, 1 АССН
5 ОН 15 1.000 1.200 2-Пропанол: 2 СН3, 1 СН, 1 ОН
основная подгруппа № я*
группа
“ОН” 6 “СН3ОН” CH3OH 16 1.4311 1.432 Метанол: 1 СН3ОН
7 H2O 17 0.92 1.40 Вода: 1 Н2О
“Н2О” 8 “АСОН” ACOH 18 0.8952 0.680 Фенол: 5 АСН, 1 АСОН
9 CH3CO 19 1.6724 1.488 Группа кетонов — 1 СН3, 1 СН2, 1 СН3СО
“СН2СО” CH2CO 20 1.4457 1.180 2-й углерод; 2-бутанон: Группа кетонов — любой другой углерод; 2 СН3, 1 СН2, 1 СН2СО
10 CHO 21 0.9980 0.948 3-пентанон: ацетальдегид: 1 СН3, 1 СНО
“СНО” 11 CH3COO 22 1.9031 1.728 Бутилацетат: 1 СН3, 3 СН2, 1 СН3СОО
“ССОО” CH2COO 23 1.6764 1.420 Бутилпропаноат: 2 СН3, 3 СН2, 1 СН2СОО
12 “НСОО” HCOO 24 1.2420 1.188 Этилформиат: 1 СН3, 1 СН2, 1 НСОО
13 CH3O 25 1.1450 1.088 Диметиловый эфир: 1 СН3, 1 СН3О
“СН2О” CH2O 26 0.9183 0.780 Диэтиловый эфир: 2 СН3, 1 СН2, 1 СН2О
CH—0 27 0.6908 0.468 Диизопропиловый эфир: 4 СН3, 1 СН, 1 СН—О
fch2o 28 0.9183 1.1 Тетрагидрофуран: 3 СН2, 1 fch2o
14 ch3nh2 29 1.5959 1.544 Метиламин: 1 ch3nh2
“CNH2” ch2nh2 30 1.3692 1.236 Пропил амин: 1 СН3, 1 СН2, 1 ch2nh2
chnh2 31 1.1417 0.924 Изопропиламин: 2 СНз, 1 CHNH2
15 CH3NH 32 1.4337 1.244 Диметиламин: 1 СН3, 1 CH3NH
“CNH” ch2nh 33 1.2070 0.936 Диэтил амин: 2 СН3, 1 СН2, 1 CH2NH
CHNH 34 0.9795 0.624 Диизопропил амин: 4 СН3, 1 СН, 1 CHNH
16 “(C)3N” CH3N 35 1.1865 0.940 Триметиламин: 2 СН3, 1 CH3N
ch2n 36 0.9597 0.632 Триэтил амин: 3 СН3, 2 СН2, 1 CH2N
17 “ACNH2” acnh2 37 1.0600 0.816 Анилин: 5 АСН, 1 ACNH2
18 c5h5n 38 2.9993 2.113 Пиридин: 1 C5H5N
“pyridine” C5H4N 39 2.8332 1.833 З-Метилпиридин: 1 СН3, 1 C5H4N
C5H3N 40 2.667 1.553 2,3-Диметилпиридин: 2 СНз, 1 С5НзМ
19 CH3CN 41 1.8701 1.724 Ацетонитрил: 1 CH3CN
“CCN” ch2cn 42 1.6434 1.416 Пропионитрил: 1 СН3, 1 CH^N
20 COOH 43 1.3013 1.224 Уксусная кислота: 1 СН3, 1 соон
“COOH” HCOOH 44 1.5280 1.532 Муравьиная кислота: 1 нсоон
21 CH^l 45 1.4654 1.264 1-Хлорбутан: 1 СН3, 2 сн2, 1 СН2С1
“CC1” CHC1 46 1.2380 0.952 2-Хлорпропан: 2 СН3, 1 СНС1
CC1 47 1.0060 0.724 2-Хлор-2-метилпропан: 3 СН3, 1 СС1
22 CH2C12 48 2.2564 1.988 Дихлорметан: 1 СН2С12
“CC12” CHC12 49 2.0606 1.684 1,1-Дихлорэтан: 1 СНз, 1 СНС12
CC12 50 1.8016 1.448 2,2-Дихлорпропан: 2 СН3, 1 СС12
23 CHC13 51 2.8700 2.410 Хлороформ: 1 СНС13
“CC13” CC13 52 2.6401 2.184 1,1,1 -Трихл орэтан: 1 СН3, 1 СС13
24 CC14 53 3.3900 2.910 Тетрахл орметан: 1 ссц
“CC14” 25 “ACC1” ACC1 54 1.1562 0.844 Хлорбензол: 5 АСН, 1 АСС1
26 CH3NO2 55 2.0086 1.868 Нитрометан: 1 CH3NO2
“CNO2” ch2no2 56 1.7818 1.560 1-Нитропропан: 1 СНз, 1 СН2, 1 CH2NO2
chno2 57 1.5544 1.248 2-Нитропропан: 2 СН3, 1 CHNO2
27 acno2 58 1.4199 1.104 Нитробензол: 5 АСН, 1 ACNO2
“ACNO2” 28 “CS2” cs2 59 2.057 1.65 Дисульфид углерода 1 cs2
29 CH3SH 60 1.8770 1.676 Метантиол: 1 CH3SH
“CH3SH” CH2SH 61 1.6510 1.368 Этантиол: 1 СН3, 1 CH2SH
Продолжение табл. Д.11
Приложения 617
Основная Подгруппа № Rk группа G*
30
“furfural” furfural 62 3.1680 2.481 Фурфураль: 1 furfural
31 “DOH” 32 (СН2ОН)263 2.4088 2.248 1,2-Этандиол: 1(СН2ОН)2
«« J55 33 I 64 1.2640 0.992 1-Иодэтан: 1 СН3, 1 СН2, 1 I
“Вг” Br 65 0.9492 0.832 1-Бромэтан: 1 СН3, 1 СН2) 1 Вг
34 CH=C 66 1.2920 1.088 1-Гексин: 1 СН3, 3 СН2, 1 сн=с
“С=С” C=C 67 1.0613 0.784 2-Гексин: 2 СН3, 2 СН2, I С=С
35 Me2SO 68 2.8266 2.472 Диметил сульфоксид: 1 Me2SO
“Me2SO” 36 ACRY 69 2.3144 2.052 Акрилонитрил: 1 ACRY
“ACRY” 37 C1(C=C) 70 0.7910 0.724 Трихлорэтилен: 1 СН=С, 3 С1(С= =С)
“С1СС” 38 ACF 71 0.6948 0.524 Г ексафторбензол: 6 ACF
“ACF” 39 “DMF” DMF-1 72 3.0856 2.736 Диметилформамид: 1 DMF-1
40 “CF2” DMF-2 CF3 73 74 2.6322 1.4060 2.120 1.380 Диэтилформамид: Перфторгексан: 2 СН3, 1 DMF-2 2 CF,, 4 CF,
cf2 CF 75 76 1.0105 0.6150 0.920 0.460 Перфторметилциклогексан: 1 СН3, 5 СН2, 1 CF
Таблица Д.12. Параметры взаимодействия уравнения UNIFAC для расчета парожидкостного равновесия [308]
(© Am. Chem. Soc.)
1 2 3 4 5 6 7 8
1 сн2 0.0 -200.0 61.13 76.50 986.5 697.2 1318.0 1333.0
2 С=С 2520.0 0.0 340.7 4102.0 693.9 1509.0 634.2 547.4
3 АСН -11.12 -94.78 0.0 167.0 636.1 637.3 903.8 1329.0
4 АССН2 -69.70 -269.7 -146.8 0.0 803.2 603.2 5695.0 884.9
5 ОН 156.4 8694.0 89.60 25.82 0.0 -137.1 353.5 -259.7
6 СН3ОН 16.51 -52.39 -50.00 -44.50 249.1 0.0 -181.0 -101.7
7 Н2О 300.0 692.7 362.3 377.6 -229.1 289.6 0.0 324.5
8 АСОН 275.8 1665.0 25.34 244.2 -451.6 -265.2 -601.8 0.0
9 СН2СО 26.76 -82.92 140.1 365.8 164.5 108.7 472.5 -133.1
10 СНО 505.7 n.a. n.a. n.a. -404.8 -340.2 232.7 n.a.
11 ССОО 114.8 269.3 85.84 -170.0 245.4 249.6 10000.0 -36.72
12 НСОО 90.49 91.65 n.a. n.a. 191.2 155.7 n.a. n.a.
13 СН2О 83.36 76.44 52.13 65.69 237.7 339.7 -314.7 n.a.
14 CNH2 -30.48 79.40 -44.85 n.a. -164.0 -481.7 -330.4 n.a.
15 CNH 65.33 -41.32 -22.31 223.0 -150.0 -500.4 -448.2 n.a.
16 (C)3N -83.98 -188.0 -223.9 109.9 28.60 -406.8 -598.8 n.a.
17 ACNH2 5339.0 n.a. 650.4 979.8 529.0 5.182 -339.5 n.a.
18 pyridine -101.6 n.a. 31.87 49.80 -132.3 -378.2 -332.9 -341.6
19 CCN 24.82 34.78 -22.97 -138.4 185.4 157.8 242.8 n.a.
20 COOH 315.3 349.2 62.32 268.2 -151.0 1020.0 -66.17 n.a.
21 CC1 91.46 -24.36 4.680 122.9 562.2 529.0 698.2 n.a.
22 CC12 34.01 -52.71 121.3 n.a. 747.7 669.9 708.7 n.a
23 CC13 36.70 -185.1 288.5 33.61 742.1 649.1 826.7 n.a.
24 CC14 -78.45 -293.7 -4.700 134.7 856.3 860.1 1201.0 10000.
25 ACC1 -141.3 -203.2 -237.7 375.5 246.9 661.6 920.4 n.a.
26 CNO2 -32.69 -49.92 10.38 -97.05 341.7 252.6 417.9 n.a.
Продолжение табл. Д.12
1 2 3 4 5 6 7 8
27 ACNO2 5541.0 n.a. 1824.0 -127.8 561.6 n.a. 360.7 n.a.
28 CS2 -52.65 16.62 21.50 40.68 823.5 914.2 1081.0 n.a.
29 CH3SH -7.481 n.a. 28.41 n.a. 461.6 382.8 n.a. n.a.
30 furfural -25.31 n.a. 157.3 404.3 521.6 n.a. 23.48 n.a.
31 DOH 140.0 n.a. 221.4 150.6 267.6 n.a. 0.0 838.4
32 I 128.0 n.a. 58.68 n.a. 501.3 n.a. n.a. n.a.
33 Br -31.52 n.a. 155.6 291.1 721.9 n.a. n.a. n.a.
34 C=C -72.88 -184.4 n.a. n.a. n.a. n.a. n.a. n.a.
35 Me2SO 50.49 n.a. -2.504 -143.2 -25.87 695.0 -240.0 n.a.
36 ACRY -165.9 n.a. n.a. n.a. n.a. n.a. 386.6 n.a.
37 C1CC 41.90 -3.167 -75.67 n.a. 640.9 726.7 n.a. n.a.
38 ACF -5.132 n.a. -237.2 -157.3 649.7 645.9 n.a. n.a.
39 DMF -31.95 37.70 -133.9 -240.2 64.16 172.2 -287.1 n.a.
40 CF2 147.3 n.a. n.a. n.a. n.a. n.a. n.a. n.a.
9 10 11 12 13 14 15 16
1 CH2 476.4 677.0 232.1 741.4 251.5 391.5 255.7 206.6
2 C=C 524.5 n.a. 71.23 468.7 289.3 396.0 273.6 658.8
3 ACH 25.77 n.a. 5.994 n.a. 32.14 161.7 122.8 90.49
4 ACCH2 -52.10 n.a. 5688.0 n.a. 213.1 n.a. -49.29 23.50
5 OH 84.00 441.8 101.1 193.1 28.06 83.02 42.70 -323.0
6 CH3OH 23.39 306.4 -10.72 193.4 -180.6 359.3 266.0 53.90
7 H2O -195.4 -257.3 14.42 n.a. 540.5 48.89 168.0 304.0
8 ACOH -356.1 n.a. -449.4 n.a. n.a. n.a. n.a. n.a.
9 CH2CO 0.0 -37.36 -213.7 n.a. 5.202 n.a. n.a. n.a.
ЮСНО 128.0 0.0 n.a. n.a. 304.1 n.a. n.a. n.a.
11 CCOO 372.2 n.a. 0.0 372.9 -235.7 n.a. -73.50 n.a.
12 HCOO n.a. n.a. -261.1 0.0 n.a. n.a. n.a. n.a.
13 CH2O 52.38 -7.838 461.3 n.a. 0.0 n.a. 141.7 n.a.
14 CNH2 n.a. n.a. n.a. n.a. n.a. 0.0 63.72 -41.11
15 CNH n.a. n.a. 136.0 n.a. -49.30 108.8 0.0 -189.2
16 (C)3N n.a. n.a. n.a. n.a. n.a. 38.89 865.9 0.0
17 ACNH2 18 byridine- -399.1 n.a. n.a. n.a. n.a. n.a. n.a. n.a.
-51.54 n.a. n.a. n.a. n.a. n.a. n.a. n.a.
1TCCN -287.5 n.a. -266.6 n.a. n.a. n.a. n.a. n.a.
20 COOH -297.8 n.a. -256.3 312.5 -338.5 n.a. n.a. n.a.
21 CC1 286.3 -47.51 n.a. n.a. 225.4 n.a. n.a. n.a.
22 CC12 423.2 n.a. -132.9 n.a. -197.7 n.a. n.a. -141.4
23 CC13 552.1 n.a. 176.5 488.9 -20.93 n.a. n.a. -293.7
24 CC14 372.0 n.a. 129.5 n.a. 113.9 261.1 91.13 -126.0
25 ACC1 128.1 n.a. -246.3 n.a. n.a. 203.5 -108.4 1088.0
26 CNO2 -142.6 n.a. n.a. n.a. -94.49 n.a. n.a. n.a.
27 ACNO2 n.a. n.a. n.a. n.a. n.a. n.a. n.a. n.a.
28 CS2 303.7 n.a. 243.8 n.a. 112.4 n.a. n.a. n.a.
29 CH3SH 160.6 n.a. n.a. 239.8 63.71 106.7 n.a. n.a.
30 furfural 317.5 n.a. -146.3 n.a. n.a. n.a. n.a. n.a.
31 DOH n.a. n.a. 152.0 n.a. 9.207 n.a. n.a. n.a.
32 I 138.0 n.a. 21.92 n.a. 476.6 n.a. n.a. n.a.
33 Br -142.6 n.a. n.a. n.a. 736.4 n.a. n.a. n.a.
34 C=C 443.6 n.a. n.a. n.a. n.a. n.a. n.a. n.a.
35 MejSO 110.4 n.a. 41.57 n.a. -122.1 n.a. n.a. n.a.
36 ACRY n.a. n.a. n.a. n.a. n.a. n.a. n.a. n.a.
37 C1CC -8.671 n.a. -18.87 n.a. -209.3 n.a. n.a. n.a.
38 ACF n.a. n.a. n.a. n.a. n.a. n.a. n.a. n.a.
39 DMF 97.04 n.a. n.a. n.a. -158.2 n.a. n.a. n.a.
4O.CF2 n.a. n.a. n.a. n.a. n.a. n.a. n.a. n.a.
Продолжение табл. Д.1.
Приложения 619
17 18 19 20 21 22 23 24
1 сн2 1245.0 287.7 597.0 663.5 35.93 53.76 24.90 104.3
2 С=С n.a. n.a. 405.9 730.4 99.61 337.1 4584.0 5831.0
3 АСН 668.2 -4.449 212.5 537.4 -18.81 -144.4 -231.9 3.000
4 АССН2 764.7 52.80 6096.0 603.8 -114.1 n.a. -12.14 -141.3
5 ОН -348.2 170.0 6.712 199.0 75.62 -112.1 -98.12 143.1
6 СН3ОН 335.5 580.5 36.23 -289.5 -38.32 -102.5 -139.4 -67.80
7 Н2О 213.0 459.0 112.6 -14.09 325.4 370.4 353.7 497.5
8 АСОН n.a. -305.5 n.a. n.a. n.a. n.a. n.a. 1827.0
9 СН2СО 937.9 165.1 481.7 669.4 -191.7 -284.0 -354.6 -39.20
10 СНО n.a. n.a. n.a. n.a. 751.9 n.a. n.a. n.a.
11ССОО n.a. n.a. 494.6 660.2 n.a. 108.9 -209.7 54.47
12 НСОО n.a. n.a. n.a. -356.3 n.a. n.a. -287.2 n.a.
13 СН2О n.a. n.a. n.a. 664.6 301.1 137.8 -154.3 47.67
14 CNH2 n.a. n.a. n.a. n.a. n.a. n.a. n.a. -99.81
15 CNH n.a. n.a. n.a. n.a. n.a. n.a. n.a. 71.23
16 (C)3N n.a. n.a. n.a. n.a. n.a. -73.85 -352.9 -8.283
17 ACNH2 0.0 n.a. -216.8 n.a. n.a. n.a. n.a. 8455.0
18 pyridine n.a. 0.0 -169.7 -153.7 n.a. -351.6 -114.7 -165.1
19 CCN 617.1 134.3 0.0 n.a. n.a. n.a. -15.62 -54.86
20 COOH n.a. -313.5 n.a. 0.0 44.42 -183.4 76.75 212.7
21 CC1 n.a. n.a. n.a. 326.4 0.0 108.3 249.2 62.42
22 CC12 n.a. 587.3 n.a. 1821.0 -84.53 0.0 0.0 56.33
23 CCi3 n.a. 18.98 74.04 1346.0 -157.1 0.0 0.0 -30.10
24 CC14 1301.0 309.2 492.0 689.0 11.80 17.97 51.90 0.0
25 ACC1 323.3 n.a. 356.9 n.a. -314.9 n.a. n.a. -255.4
26 CNO2 n.a. n.a. n.a. n.a. n.a. n.a. n.a. -34.68
27 ACNO2 5250.0 n.a. n.a. n.a. n.a. n.a. n.a. 514.6
28 CS2 n.a. n.a. 335.7 n.a. -73.09 n.a. -26.06 -60.71
29 CH3SH n.a. n.a. 125.7 n.a. -27.94 n.a. n.a. n.a.
30 furfural n.a. n.a. n.a. n.a. n.a. n.a. 48.48 -133.1
31 DOH 164.4 n.a. n.a. n.a. n.a. n.a. n.a. n.a.
32 I n.a. n.a. n.a. n.a. n.a. -40.82 21.76 48.49
33 Br n.a. n.a. n.a. n.a. 1169.0 n.a. n.a. 225.8
34 C=C n.a. n.a. 329.1 n.a. n.a. n.a. n.a. n.a.
35 Me2SO n.a. n.a. n.a. n.a. n.a. -215.0 -343.6 -58.43
36 ACRY n.a. n.a. -42.31 n.a. n.a. n.a. n.a. n.a.
37 C1CC n.a. n.a. 298.4 2344.0 201.7 n.a. 85.32 143.2
38 ACF n.a. n.a. n.a. n.a. n.a. n.a. n.a. -124.6
39 DMF 335.6 n.a. n.a. n.a. n.a. n.a. n.a. -186.7
40 CF2 n.a. n.a. n.a. n.a. n.a. n.a. n.a. n.a.
25 26 27 28 29 30 31 32
1 CH2 321.5 661.5 543.0 153.6 184.4 354.5 3025.0 335.8
2 C=C 959.7 542.1 n.a. 76.30 n.a. n.a. n.a. n.a.
3 ACH 538.2 168.0 194.9 52.07 -10.43 -64.69 210.4 113.3
4 ACCH2 -126.9 3629.0 4448.0 -9.451 n.a. -20.36 4975.0 n.a.
5 OH 287.8 61.11 157.1 477.0 147.5 -120.5 -318.9 313.5
6 CH3OH 17.12 75.14 n.a. -31.09 37.84 n.a. n.a. n.a.
7 H2O 678.2 220.6 399.5 887.1 n.a. 188.0 0.0 n.a.
8 ACOH n.a. n.a. n.a. n.a. n.a. n.a. -687.1 n.a.
9 CH2CO 174.5 137.5 n.a. 216.1 -46.28 -163.7 n.a. 53.59
10 CHO n.a. n.a. n.a. n.a. n.a. n.a. n.a. n.a.
11 CCOO 629.0 n.a. n.a. 183.0 n.a. 202.3 -101.7 148.3
12 HCOO n.a. n.a. n.a. n.a. 4.339 n.a. n.a. n.a.
13 CH2O n.a. 95.18 n.a. 140.9 -8.538 n.a. -20.11 -149.5
14 CNH2 68.81 n.a. n.a. n.a. -70.14 n.a. n.a. n.a.
Продолжение табл. Д.12
25 26 27 28 29 30 31 32
15 CNH 4350.0 n.a. n.a. n.a. n.a. n.a. n.a. n.a.
16 (C)3N -86.36 n.a. n.a. n.a. n.a. n.a. n.a. n.a.
17 ACNH2 699.1 n.a. -62.73 n.a. n.a. n.a. 125.3 n.a.
18 pyridine n.a. n.a. n.a. n.a. n.a. n.a. n.a. n.a.
19 CCN 52.31 n.a. n.a. 230.9 21.37 n.a. n.a. n.a.
20 COOH n.a. n.a. n.a. n.a. n.a. n.a. n.a. n.a.
21 CC1 464.4 n.a. n.a. 450.1 59.02 n.a. n.a. n.a.
22 CC12 n.a. n.a. n.a. n.a. n.a. n.a. n.a. 177.6
23 CC13 n.a. n.a. n.a. 116.6 n.a. -64.38 n.a. 86.40
24 CC14 475.8 490.9 534.7 132.2 n.a. 546.7 n.a. 247.8
25 ACC1 0.0 -154.5 n.a. n.a. n.a. n.a. n.a. n.a.
26 CNO2 794.4 0.0 533.2 n.a. n.a. n.a. 139.8 304.3
27 ACNO2 n.a. -85.12 0.0 n.a. n.a. n.a. n.a. n.a.
28 CS2 n.a. n.a. n.a. 0.0 n.a. n.a. n.a. n.a.
29 CH3SH n.a. n.a. n.a. n.a. 0.0 n.a. n.a. n.a.
30 furfural n.a. n.a. n.a. n.a. n.a. 0.0 n.a. n.a.
31 DOH n.a. 481.3 n.a. n.a. n.a. n.a. 0.0 n.a.
32 I n.a. 64.28 n.a. n.a. n.a. n.a. n.a. 0.0
33 Br 224.0 125.3 n.a. n.a. n.a. n.a. n.a. n.a.
34 CsC n.a. 174.4 n.a. n.a. n.a. n.a. n.a. n.a.
35 Me2SO n.a. n.a. n.a. n.a. 85.70 n.a. 535.8 n.a.
36 ACRY n.a. n.a. n.a. n.a. n.a. n.a. n.a. n.a.
37 C1CC n.a. 313.8 n.a. 167.9 n.a. n.a. n.a. n.a.
38 ACF n.a. n.a. n.a. n.a. n.a. n.a. n.a. n.a.
39 DMF n.a. n.a. n.a. n.a. -71.00 n.a. -191.7 n.a.
40 CF2 n.a. n.a. n.a. n.a. n.a. n.a. n.a. n.a.
33 34 35 36 37 38 39 40
1 CH2 479.5 298.9 526.5 689.0 -0.505 125.8 485.3 -2.859
2 C=C n.a. 523.6 n.a. n.a. 237.3 n.a. 320.4 n.a.
3 ACH -13.59 n.a. 169.9 n.a. 69.11 389.3 245.6 n.a.
4 ACCH2 -171.3 n.a. 4284.0 n.a. n.a. 101.4 5629.0 n.a.
5 OH 133.4 n.a. -202.1 n.a. 253.9 44.78 -143.9 n.a.
6 CH3OH n.a. n.a. -399.3 n.a. -21.22 -48.25 -172.4 n.a.
7 H2O n.a. n.a. -139.0 160.8 n.a. n.a. 319.0 n.a.
8 ACOH n.a. n.a. n.a. n.a. n.a. n.a. n.a. n.a.
9 CH2CO 245.2 -246.6 -44.58 n.a. -44.42 n.a. -61.70 n. a.
ЮСНО n.a. n.a. n.a. n.a. n.a. n.a. n.a. n.a.
11 CCOO n.a. n.a. 52.08 n.a. -23.30 n.a. n.a. n.a.
12 HCOO n.a. n.a. n.a. n.a. n.a. n.a. n.a. n.a.
13 CH2O -202.3 n.a. 172.1 n.a. 145.6 n.a. 254.8 n.a.
14 CNH2 n.a. n.a. n.a. n.a. n.a. n.a. n.a. n.a.
15 CNH n.a. n.a. n.a. n.a. n.a. n.a. n.a. n.a.
16 (C)3N n.a. n.a. n.a. n.a. n.a. n.a. n. a. n.a.
17 ACNH2 n.a. n.a. n.a. n.a. n.a. n.a. -293.1 n.a.
18 pyridine n.a. n.a. n.a. n.a. n.a. n.a. n.a. n.a.
19 CCN n.a. -203.0 n.a. 81.57 -19.14 n.a. n.a. n.a.
20 COOH n.a. n.a. n.a. n.a. -90.87 n.a. n.a. n.a.
21 CC1 -125.9 n.a. n.a. n.a. -58.77 n.a. n.a. n.a.
22 CC12 n.a. n.a. 215.0 n.a. n.a. n.a. n.a. n.a.
23 CC13 n.a. n.a. 363.7 n.a. -79.54 n.a. n.a. n.a.
24 CC14 41.94 n.a. 337.7 n.a. -86.85 215.2 498.6 n.a.
25 ACC1 -60.70 n.a. n.a. n.a. n.a. n.a. n.a. n.a.
26 CNO2 10.17 -27.70 n.a. n.a. 48.40 n.a. n.a. n. a.
27 ACNO2 n.a. n.a. n.a. n.a. n.a. n.a. n.a. n.a.
28 CS2 n.a. n.a. n.a. n.a. -47.37 n.a. n.a. n.a.
29 CH3SH n.a. n.a. 31.66 n.a. n.a. n.a. 78.92 n.a.
30 furfural n.a. n.a. n.a. n.a. n.a. n.a. n.a. n.a.
31 DOH n.a. n.a. -417.2 n.a. n.a. n.a. 302.2 n.a.
32 I n.a. n.a. n.a. n.a. n.a. n.a. n.a. n.a.
33 Br 0.0 n.a. n.a. n.a. n.a. n.a. n.a. n.a.
34 C=C n.a. 0.0 n.a. n.a. n.a. n.a. -119.8 n.a.
Продолжение табл. Д.12
Приложения 621
33 34 35 36 37 38 39 40
35 Me2SO n.a. n.a. 0.0 n.a. n.a. n.a. -97.71 n.a.
36 ACRY n.a. n.a. n.a. 0.0 n.a. n.a. n.a. n.a.
37 С1СС n.a. n.a. n.a. n.a. 0.0 n.a. n.a. n.a.
38 ACF n.a. n.a. n.a. n.a. n.a. 0.0 n.a. n.a.
39 DMF n.a. 6.699 136.6 n.a. n.a. n.a. 0.0 n.a.
40 CF2 n.a. n.a. n.a. n.a. n.a. n.a. n.a. 0.0
п. а. отсутствует.
Параметры уравнения UNIFAC для взаимодействия с
группой АСОН
Основная группа m вт, АСОН, к aacoh, m, К
CH2 1333.0 275.8
C=C 547.4 1665.0
ACH 1329.0 25.34
acch2 884.9 244.2
OH -259.7 -451.6
CH3OH -101.7 -265.2
H2O 324.5 -601.8
CH2CO -133.1 -356.1
ccoo -36.72 -449.4
pyridine -341.6 -305.5
CC14 10000.0 1827.0
DOH 838.4 -687.1
Параметры взаимодействия групп уравнения UNIFAC
для существующих групп атп, К (ранее обозначаемых
п. а. — «отсутствует»)
Основная группа m Основная группа и Omn> К Опт» К
C=C CCOO 71.23 269.3
c=c HCOO 468.7 91.65
c=c ACC1 959.7 -203.2
ACH CC12 -144.4 121.3
acch2 (C)3N 23.50 109.9
acch2 acno2 4448.0 -127.8
OH acno2 157.1 561.6
CH3OH (C)3N 53.90 -406.8
CH3OH acnh2 335.5 5.182
CH3OH ACC1 17.12 661.6
CH2CO acnh2 937.9 -399.1
CH2CO pyridine 165.1 -51.54
CH2CO ACC1 174.5 128.1
CHO CH2O 304.1 -7.838
ccoo DOH -101.7 152.0
HCOO CC13 -287.2 488.9
CH2O DOH -20.11 9.207
CH2O Br -202.3 736.4
cnh2 (C)3N -41.11 38.99
cnh2 CCI4 -99.81 261.1
CNH (C)3N -189.2 865.9
acnh2 CCN -216.8 617.1
acnh2 ACC1 699.1 323.3
pyridine COOH -153.7 -313.5
pyridine CC12 -351.6 587.3
pyridine CC14 -165.1 309.2
CCN ACC1 52.31 356.9
main group m main group n amn> & anm> К
COOH CC13 76.75 1346.0
CC1 ACC1 464.4 -314.9
cno2 ACNO2 533.2 -85.12
cno2 I 304.3 64.28
H2O DOH 0.0 0.0
OH CH3SH 147.5“ 461.6
а В работе [641] величина этого параметра указана неверно.
Параметры UNIFAC для группового новых групп взаимодействия уравнения
Основная группа т Основная группа п Отп» Опт» К
сн2 С=С 298.9 -72.88
с=с с=с 523.6 -184.4
СН2СО с^с -246.6 443.6
CCN с^с -203.0 329.1
cno2 с^с -27.70 174.4
сн2 Me2SO 526.5 50.49
АСН Me2SO 169.9 -2.504
ассн2 Me2SO 4284. -143.2
он Me2SO -202.1 -25.87
СН3ОН Me2SO -399.3 695.0
Н2О Me2SO -139.0 -240.0
СН2СО Me2SO -44.58 110.4
ссоо Me2SO 52.08 41.57
СН2О Me2SO 172.1 -122.1
СС12 Me2SO 215.0 -215.0
СС13 Me2SO 363.7 -343.6
СС14 Me2SO 337.7 -58.43
CH3SH Me2SO 31.66 85.70
DOH Me2SO —417.2 535.8
сн2 ACRY 689.0 -165.9
Н2О ACRY 160.8 386.6
CCN ACRY 81.57 -42.31
сн2 C1CC -0.505 41.90
с=с C1CC 237.3 -3.167
АСН C1CC 69.11 -75.67
он C1CC 253.9 640.9
СН3ОН C1CC -21.22 726.7
СН2СО C1CC -44.42 -8.671
ссоо C1CC -23.30 -18.87
СН2О C1CC 145.6 -209.3
CCN C1CC -19.14 298.4
соон C1CC -90.87 2344.
Параметры группового взаимодействия уравнения
UNIFAC для новых групп
Основная группа Основная группа атп, К апт, R
т n
СС1 CICC -58.77 201.7
СС13 CICC 85.32
СС14 CICC -86.85 143.2
cno2 CICC 48.40 313.8
cs2 CICC -47.37 167.9
сн2 ACF 125.8 -5.132
АСН ACF 389.3 -237.2
ассн2 ACF 101.4 -157.3
он ACF 44.78 649.7
сн3он ACF -48.25 645.9
СС14 ACF 215.2 -124.6
сн2 DMF 485.3 -31.95
с=с DMF 320.4 37.70
АСН DMF 245.6 -133.9
ассн2 DMF 5629.0 -240.2
он DMF -143.9 64.16
сн3он DMF -172.4 172.2
Н2О DMF 319.0 -287.1
СН2СО DMF -61.70 97.04
СН2О DMF 254.8 -158.2
acnh2 DMF -293.1 335.6
СС14 DMF 498.6 -186.7
CH3SH DMF 78.92 -71.00
DOH DMF 302.2 -191.7
с=с DMF -119.8 6.699
Me2SO DMF -97.71 136.6
сн2 cf2 -2.859 147.3
сн2
с=с
АСН
ассн2
он
СН3ОН
Н2О
АСОН
СН2СО
СНО
ССОО
НСОО
СН2О
cnh2
CNH
(C)3N
acnh2
PYRIDINE
CCN
COOH
CCI
CCI2
CCI3
CC14
ACC1
CNO2
acno2
cs2
CH3SH
FURFURAL
DOH
B|
c c
DMSO
ACRY
CICC
ACF
DMF
CF;
[5
ОПРЕДЕЛЯЕМЫЕ ПАРАМЕТРЫ ЕЗ
в
10
1»«и
12
13
5
6
«в»1
3^S
1
1
20.
3
Яв»
20|
21
22
23
«я»
25
2в|
27
2в
29
30
31
32
33
34
35
Зв
37
Зв
39
401
«»«
я«1и
Таблица Д.13. Параметры объема и площади поверхности групп для расчета равновесия жидкость — жидкость
уравнения UNIFAC [456] (© Am. Chem. Soc.)
Основная группа Подгруппа No. Rk Qk
1 “СН2” CH3 CH2 1 2 0.9011 0.6744 0.848 0.540 Бутан: 2 CH3, 2 CH2
CH 3 0.4469 0.228 2-Метилпропан: 3 CH3, 1 CH
c 4 0.2195 0.000 2,2-Диметилпропан:4 CH3, 1 C
2“С=С” CH2=CH 5 1.3454 1.176 1-Гексен: 1 CH3, 3 CH2, 1 CH2=CH
CH=CH 6 1.1167 0.867 2-Гексен: 2 CH3> 2 CH2, 1 CH=CH
CH=C 7 0.8886 0.676 2-Метил-2-бутен: 3 CH3, 1 CH=C
CH2=C 8 1.1173 0.988 2-Метил-1 -бутен: 2 CH3, 1 CH2, 1 СН2=С
3 “АСН” ACH 9 0.5313 0.400 Бензол: 6 АСН
AC 10 0.3652 0.120 Стирол:! СН2=СН, 5 АСН, 1 АС
4 “АССН2” ACCH3 11 1.2663 0.968 Толуол:5 АСН, 1 АССН3
acch2 12 1.0396 0.660 Этилбензол: 1 СН3, 5 АСН, 1 АССН2
ACCH 13 0.8121 0.348 Кумол: 2 СН3, 5 АСН, 1 АССН
5 “ОН” OH 14 1.0000 1.200 2-Бутанол:2 СН3, 1 СН2, 1 CH, 1 ОН
6 Pl 15 3.2499 3.128 1-Пропанол:1 pi
7 P2 16 3.2491 3.124 2-Пропанол: 1 Р2
8 H2O 17 0.92 1.40 Вода: 1 Н2О
9 ACOH 18 0.8952 0.680 Фенол: 5 АСН, 1 АСОН
10 “СН2СО” CH3CO 19 1.6724 1.488 Группа кетонов — 2-й углерод: 2-бутанон: 1 СН3, 1 СН2, 1 СН3СО
CH2CO 20 1.4457 1.180 Группа кетонов — любой другой углерод;
3-пентанон: 2 СН3, 1 СН2, 1 СН2СО
11 CHO 21 0.9980 0.948 Ацетальдегид: СН3, 1 СНО
12 furfural 22 3.168 2.484 1 Фурфураль:
13 “СООН” COOH 23 1.3013 1.224 Уксусная кислота: 1 СН3, 1 СООН
HCOOH 24 1.5280 1.532 Муравьиная кислота: 1 НСООН
14 “СООС” CH3COO 25 1.9031 1.728 Бутилацетат: 1 СН3, 3 СН2, 1 СН3СОО
СНгСОО 26 1.6764 1.420 Бутилпропаноат:2 СН3, 3 СН2, 1 СН2СОО
15 “СН2О” CH3O 27 1.1450 1.088 Диметиловый эфир: 1 СН3, 1 СН3О
CH2O 28 0.9183 0.780 Диэтиловый эфир: 2 СН3, 1 СН2, 1 СН2О
CH-O 29 0.6908 0.468 Диизопропиловый эфир: 4 СН3, 1 UH, 1 СН—О
fch2o 30 0.9183 1.1 Тетрагидрофуран: 3 СН2, 1 FCH2O
16 “СС1” CH2C1 31 1.4654 1.264 1-Хлорбутан: 1 СН3, 2 СН2, 1 СН2С1
CHC1 32 1.2380 0.952 2-Хлорпропан: 2 СН3, 1 СНС1
CC1 33 1.0060 0.724 2-Хлор-2-метилпропан: 3 СН3, 1 СС1
17 “СС12” CH2C12 34 2.2564 1.988 Дихлорметан: 1 СН2С12
CHC12 35 2.0606 1.684 1,1-Дихлорэтан 1 СН3, 1 СНС12
CC12 36 1.8016 1.448 2,2-Дихлорпропан: 2 СН3, 1 СС12
18 “СС13” CHC13 37 2.8700 2.410 Хлороформ:] CHC13
CC13 38 2.6401 2.184 1,1,1-Трихлорэтан: 1 СН3, 1 СС13
19 CC14 39 3.3900 2.910 Тетрахлорметан: 1 ССЦ
20 ACC1 40 1.1562 0.844 Хлорбензол: 5 АСН, 1 АСС1
21 “CCN” CH3CN 41 1.8701 1.724 Ацетонитрил: 1 CH3CN
ch2cn 42 1.6434 1.416 Пропионитрил: 1 СН3, 1 CH2CN
22 acnh2 43 1.0600 0.816 Анилин:5 АСН, 1 ACNH2
23 “CNO2” ch3no2 44 2.0086 1.868 Нитрометан; 1 CH3NO2
ch2no2 45 1.7818 1.560 1-Нитропропан: 1 СН3, 1 СН2, 1 CHNO2
chno2 46 1.5544 1.248 2-Нитропропан:2 CH3, 1 CHNO2
24 acno2 47 1.4199 1.104 Нитробензол:5 АСН, 1 ACNO2
25 “DOH” (CH2OH)2 48 2.4088 2.248 1,2-Этандиол: 1 (СН2ОН)2
26 “DEOH” (HOCH,CH2)2O 49 4.0013 3.568 Диэтиленгликоль :1 (НОСН2СН2)2О
27 “pyridine” C5H5N 50 2.9993 2.113 Пиридин: C5H5N
C5H4N 51 2.8332 1.833 3-Метилпиридин:1 СН3, 1 C5H4N
C5H3N 52 2.667 1.553 2,3-Диметилпиридин:2 СН3, 1 C5H3N
28 “ТСЕ” CC12=CHC1 53 3.3092 2.860 Трихлорэтилен: 1 СС12=СНС1
29 “MFA” HCONHCH3 54 2.4317 2.192 Мети л формамид: 1 HCONHCH3
30 “DMFA” HCON(CH3)2 55 3.0856 2.736 Диметил формамид: 1 HCON(CH3)2
31 “TMS” (CH2)4SO2 56 4.0358 3.20 Тетраметиленсульфон: 1 (CH2)4SO2
32 “DMSO” (CH2)2SO 57 2.8266 2.472 Диметилсульфоксид: 1 (CH2)2SO
Таблица Д. 14. Параметры взаимодействия групп уравнения UNIFAC для расчета равновесия жидкость — жидкость
(надстрочный индекс 1 = Р1 или Р2 получен на основе обобщенных данных; надстрочный индекс 2 — это пара-
метр, рассчитанный из данных о парожидкостном равновесии) [456] (© Am. Chem. Soc.)
1 CH2 2 C=C 3 ACH 4 acch2 5 OH 6 Pl 7 P2 8 H2O
1 сн2 0 74.54 -114.8 -115.7 644.6 329.6 310.7 1300.0
2 С=С 292.3 0 340.72 4102.02 724.4 1731.0 1731.0 896.0
3 АСН 156.5 —94.782 0 167.02 703.9 511.5 577.3 859.4
4 АССН2 104.4 -269.72 -146.82 0 4000.0 136.6 906.8 5695.0
5 ОН 328.2 470.7 -9.210 1.270 0 937.3 991.3 28.73
6 Р1 -136.7 -135.7 -223.0 -162.6 -281.1 0 0 -61.29
7 Р2 -131.9 -135.7 -252.0 -273.6 -268.8 0 0 5.890
8 Н2О 342.4 220.6 372.8 203.7 -122.4 247.0 104.9 0
9 АСОН -159.8 -473.2 -470.4 -63.15 -547.2 -547.2 -595.9
10 СН2СО 66.56 306.1 -78.31 -73.87 216.0 401.71 -127.61 634.8
11 СНО 146.1 517.0 -75.30 223.2 -431.3 643.41 231.41 623.7
12 FURF 14.78 -10.44 -184.9 444.7 -94.64 732.3 211.6
13 СООН 1744.0 -48.52 75.49 147.3 118.4 728.71 349.11 652.3
14 СООС -320.1 485.6 114.82 -170.02 180.6 -76.64 -152.8 385.9
15 СН2О 1571.0 76.442 52.132 65.692 137.1 -218.1 -218.1 212.8
16 СС1 73.80 -24.362 4.6802 122.92 455.1 351.5 351.5 770.0
17 СС12 27.90 -52.712 669.2 -186.11 -401.61 740.4
18 СС13 21.23 -185.12 288.52 33.612 418.4 -465.7 -465.7 793.2
19 СС14 89.97 -293.702 -4.702 134.72 713.5 -260.3 512.2 1205.0
20 АСС1 -59.06 777.8 -47.13 1989.0 390.7
21 CCN 29.08 34.782 56.41 -53.29 2011.0 63.48
22 ACNH2 175.8 -218.9 -15.41 529.02 -239.8
23 CNO2 94.34 375.4 113.6 -97.05 483.8 264.7 264.7 13.32
24 ACNO2 193.6 7.180 -127.1 332.6 439.9
25 DOH 108.5 247.3 453.4 -289.3 -424.3
26 DEOH 81.49 -50.71 -30.28 -99.56
27 PYR -128.8 -225.3 -124.6 -319.2 203.0
28 TCE 147.3 837.9 1153.0
29 MFA -11.91 176.7 -80.48 -311.0
30 DMFA 14.91 132.1 -17.78 -262.6
31 TMS 67.84 42.73 59.16 26.59 1.110
32 DMSO 36.42 60.82 29.77 55.97
Продолжение табл. Д.14
Приложения 625
9 ACOH 10 CH2CO 11 CHO 12 FURF 13 COOH 14 COOC 15 CH2O 16 CCl
1 СН2 2255.0 472.6 158.1 383.0 139.4 972.4 662.1 42.14
2 С=С 343.7 -214.7 1647.0 -577.5 289.32 99.612
3 АСН 1649.0 593.7 362.3 31.14 461.8 6.02 32.142 -18.812
4 АССН2 292.6 916.7 1218.0 715.6 339.1 5688.02 213.12 -114.12
5 ОН -195.5 67.07 1409.0 -140.3 -104.0 195.6 262.5 62.05
6 Р1 -153.2 -47.411 -344.11 299.3 244.41 19.57 1970.0 -166.4
7 Р2 -153.2 353.81 -338.61 -241.8 -57.981 487.1 1970.0 -166.4
8 Н2О 344.5 -171.8 -349.9 66.95 -465.7 -6.320 64.42 315.9
9 АСОН 0 -825.7 -898.3
10 СН2СО -568.0 0 -37.362 120.3 1247.0 258.7 5.2022 1000.0
11 СНО 128.02 0 1724.0 0.750 -245.8 751.82
12 FURF 48.93 -311.6 0 1919.0 57.70
13 СООН -101.3 1051.0 -115.7 0 -117.6 -96.62 19.77
14 СООС -337.3 58.84 1090.0 -46.13 1417.0 0 -235.72
15 СН2О 52.382 1402.0 461.32 0 301.12
16 СС1 483.9 -47.512 337.1 225.42 0
17 СС12 550.6 808.8 437.7 -132.92 -197.72 -21.35
18 СС13 342.2 203.1 370.4 176.52 -20.932 -157.12
19 СС14 1616.02 550.0 70.14 438.1 129.52 113.92 И.802
20 ААС1 190.5 1349.0 -246.32
21 CCN -349.2 2.410
22 ACNH2 -860.3 857.7 681.4
23 CNO2 377.0 152.4 —94.492
24 ACNO2 -230.4 211.6
25 DOH 523.O2 82.77 -75.23 -1707.0 29.86
26 DEOH
27 PYR —222.72 -201.9
28 TCE 417.4 123.2 639.7
29 MFA
30 DMFA -281.9
31 TMS
32 DMSO
17 18 19 20 21 22 23 24
CCl2 CCl3 CCl4 AC Cl CCN acnh2 cno2 acno2
1 CH2 -243.9 7.5 -5.550 924.8 696.8 902.2 556.7 575.7
2 C=C 337.12 4583.02 5831.O2 405.92 425.7
3 ACH -231.92 3OOO.O2 -878.1 29.13 1.640 -1.770 -11.19
4 ACCH2 -12.142 -141.32 -107.3 1208.0 689.6 3629.02 -175.6
5 OH 272.2 -61.57 -41.75 -597.1 -189.3 -348.22 -30.70 -159.0
6 Pl 128.61 1544.0 224.6 150.8
7 P2 5O7.81 1544.0 -207.0 150.8
8 H2O 370.7 356.8 502.9 -97.27 198.3 -109.8 1539.0 32.92
9 ACOH 4894.02 -851.6 -16.13
10 CH2CO -301.0 12.01 -10.88 902.6 430.6 1010.0 400.0 -328.6
11 CHO
12 FURF -347.9 -249.3 61.59
13 COOH 16700 48.15 43.83 874.3 942.2 446.3
14 COOC 108.92 -209.72 54.572 629.02 -149.2
15 CH2O 137.82 -154.32 47.672 95.182
16 CC1 110.5 249.22 62.422
17 CC12 0 56.332
18 CC13 0 -30.102 70.042 -75.50
19 CC14 17.972 51.902 0 475.82 492.02 13O2.O2 490.92 534.72
20 ACC1 -255.42 0 346.2 -154.52
21 CCN -15.622 —54.862 -465.2 0
22 ACNH2 -S216.3 8455.02 0 179.9
23 CNO2 -34.682 794.42 0
21-864
Продолжение табл. Д.14
17 18 19 20 21 22 23 24 CCl2 CCl3 CCl4 ACCl CCN ACNH2 CNO2 ACNO2
24 25 26 27 28 29 30 31 32 ACNO2 514.62 175.8 0 DOH -241.7 164.42 481.32 -246.0 DEOH PYR -114.72 -906.5 -169.72 -944.9 TCE MFA DMFA TMS DMSO
25 DOH 26 DEOH 27 PYR 28 TCE 29 MFA 30 DMFA 31 TMS 32 DMSO
1 CH2 527.5 269.2 -300.0 -63.6 928.3 331.0 561.4 956.5
2 C=C 500.7 115.4 784.4 265.4
3 ACH 358.9 363.5 -578.2 364.2 -58.10 21.97 84.16
4 ACCH2 337.7 1023.0 -390.7 238.0 132.2
5 OH 536.6 53.37 183.3 -44.44
6 Pl
7 P2
8 H2O -269.2 -873.6 1429.0 -364.2 -117.4 18.41
9 ACOH -538.6 -637.32
10 CH2CO 211.6 148.0
11 CHO
12 FURF -278.2 -208.4 -13.91 173.8
13 COOH 572.7 -2.160
14 COOC 343.1
15 CH2O
16 CC1
17 CC12
18 CC13 18.982
19 CC14
20 ACCl 124.8 -387.7
21 CCN 134.3
22 ACNH2 125.32 924.5
23 CNO2 139.82
24 ACNO2 963.0
25 DOH 0
26 DEOH 0
27 PYR 0
28 TCE 0
29 MFA 0
30 DMFA 0
31 TMS 0
32 DMSO 0
(О)
(уравнение Ли — Кеслера)
Таблица Д.15. Значения
ТГ - Н
RTC
PR
TR 0.010 0.050 0.100 0.200 0.400 0.600 0.800 1.000 1.200 1.500 2.000 3.000 5.000 7.000 10.000
0.30 6.045 6.043 6.040 6.034 6.022 6.011 5.999 5.987 5.975 5.957 5.927 5.868 5.748 5.628 5.446
0.35 5.906 5.904 5.901 5.895 5.882 5.870 5.858 5.845 5.833 5.814 5.783 5.721 5.595 5.469 5.278
0.40 5.763 5.761 5.757 5.751 5.738 5.726 5.713 5.700 5.687 5.668 5.636 5.572 5.442 5.311 5.113
0.45 5.615 5.612 5.609 5.603 5.590 5.577 5.564 5.551 5.538 5.519 5.486 5.421 5.288 5.154 4.950
0.50 5.465 5.463 5.459 5.453 5.440 5.427 5.414 5.401 5.388 5.369 5.336 5.270 5.135 4.999 4.791
0.55 0.032 5.312 5.309 5.303 5.290 5.278 5.265 5.252 5.239 5.220 5.187 5.121 4.986 4.849 4.638
0.60 0.027 5.162 5.159 5.153 5.141 5.129 5.116 5.104 5.091 5.073 5.041 4.976 4.842 4.704 4.492
0.65 0.023 0.118 5.008 5.002 4.991 4.980 4.968 4.956 4.945 4.927 4.896 4.833 4.702 4.565 4.353
0.70 0.020 0.101 0.213 4.848 4.838 4.828 4.818 4.808 4.797 4.781 4.752 4.693 4.566 4.432 4.221
0.75 0.017 0.088 0.183 4.687 4.679 4.672 4.664 4.655 4.646 4.632 4.607 4.554 4.434 4.303 4.095
0.80 0.015 0.078 0.160 0.345 4.507 4.504 4.499 4.494 4.488 4.478 4.459 4.413 4.303 4.178 3.974
0.85 0.014 0.069 0.141 0.300 4.309 4.313 4.316 4.316 4.316 4.312 4.302 4.269 4.173 4.056 3.857
0.90 0.012 0.062 0.126 0.264 0.596 4.074 4.094 4.108 4.118 4.127 4.132 4.119 4.043 3.935 3.744
0.93 0.011 0.058 0.118 0.246 0.545 0.960 3.920 3.953 3.976 4.000 4.020 4.024 3.963 3.863 3.678
0.95 0.011 0.056 0.113 0.235 0.516 0.885 3.763 3.825 3.865 3.904 3.940 3.958 3.910 3.815 3.634
0.97 0.011 0.054 0.109 0.225 0.490 0.824 1.356 3.658 3.732 3.796 3.853 3.890 3.856 3.767 3.591
0.98 0.010 0.053 0.107 0.221 0.478 0.797 1.273 3.544 3.652 3.736 3.806 3.854 3.829 3.743 3.569
0.99 0.010 0.052 0.105 0.216 0.466 0.773 1.206 3.376 3.558 3.670 3.758 3.818 3.801 3.719 3.548
1.00 0.010 0.051 0.103 0.212 0.455 0.750 1.151 2.584 3.441 3.598 3.706 3.782 3.774 3.695 3.526
1.01 0.010 0.050 0.101 0.208 0.445 0.728 1.102 1.796 3.283 3.516 3.652 3.744 3.746 3.671 3.505
Приложения
Продолжение табл. Д.15
PR
TR 0.010 0.050 0.100 0.200 0.400 0.600 0.800 1.000 1.200 1.500 2.000 3.000 5.000 7.000 10.000
1.02 0.010 0.049 0.099 0.203 0.434 0.708 1.060 1.627 3.039 3.422 3.595 3.705 3.718 3.647 3.484
1.05 0.009 0.046 0.094 0.192 0.407 0.654 0.955 1.359 2.034 3.030 3.398 3.583 3.632 3.575 3.420
1.10 0.008 0.042 0.086 0.175 0.367 0.581 0.827 1.120 1.487 2.203 2.965 3.353 3.484 3.453 3.315
1.15 0.008 0.039 0.079 0.160 0.334 0.523 0.732 0.968 1.239 1.719 2.479 3.091 3.329 3.329 3.211
1.20 0.007 0.036 0.073 0.148 0.305 0.474 0.657 0.857 1.076 1.443 2.079 2.807 3.166 3.202 3.107
1.30 0.006 0.031 0.063 0.127 0.259 0.399 0.545 0.698 0.860 1.116 1.560 2.274 2.825 2.942 2.899
1.40 0.005 0.027 0.055 0.110 0.224 0.341 0.463 0.588 0.716 0.915 1.253 1.857 2.486 2.679 2.692
1.50 0.005 0.024 0.048 0.097 0.196 0.297 0.400 0.505 0.611 0.774 1.046 1.549 2.175 2.421 2.486
1.60 0.004 0.021 0.043 0.086 0.173 0.261 0.350 0.440 0.531 0.667 0.894 1.318 1.904 2.177 2.285
1.70 0.004 0.019 0.038 0.076 0.153 0.231 0.309 0.387 0.466 0.583 0.777 1.139 1.672 1.953 2.091
1.80 0.003 0.017 0.034 0.068 0.137 0.206 0.275 0.344 0.413 0.515 0.683 0.996 1.476 1.751 1.908
1.90 0.003 0.015 0.031 0.062 0.123 0.185 0.246 0.307 0.368 0.458 0.606 0.880 1.309 1.571 1.736
2.00 0.003 0.014 0.028 0.056 0.111 0.167 0.222 0.276 0.330 0.411 0.541 0.782 1.167 1.411 1.577
2.20 0.002 0.012 0.023 0.046 0.092 0.137 0.182 0.226 0.269 0.334 0.437 0.629 0.937 1.143 1.295
2.40 0.002 0.010 0.019 0.038 0.076 0.114 0.150 0.187 0.222 0.275 0.359 0.513 0.761 0.929 1.058
2.60 0.002 0.008 0.016 0.032 0.064 0.095 0.125 0.155 0.185 0.228 0.297 0.422 0.621 0.756 0.858
2.80 0.001 0.007 0.014 0.027 0.054 0.080 0.105 0.130 0.154 0.190 0.246 0.348 0.508 0.614 0.689
3.00 0.001 0.006 0.011 0.023 0.045 0.067 0.088 0.109 0.129 0.159 0.205 0.288 0.415 0.495 0.545
3.50 0.001 0.004 0.007 0.015 0.029 0.043 0.056 0.069 0.081 0.099 0.127 0.174 0.239 0.270 0.264
4.00 0.000 0.002 0.005 0.009 0.017 0.026 0.033 0.041 0.048 0.058 0.072 0.095 0.116 0.110 0.061
(уравнение Ли — Кеслера)
Таблица Д.16. Значения
Н° - Н (1)
RTC
PR
TR 0.010 0.050 0.100 0.200 0.400 0.600 0.800 1.000 1.200 1.500 2.000 3.000 5.000 7.000 10.000
0.30 11.098 11.096 11.095 11.091 11.083 11.076 11.069 11.062 11.055 11.044 11.027 10.992 10.935 10.872 10.781
0.35 10.656 10.655 10.654 10.653 10.650 10.646 10.643 10.640 10.637 10.632 10.624 10.609 10.581 10.554 10.529
0.40 10.121 10.121 10.121 10.120 10.121 10.121 10.121 10.121 10.121 10.121 10.122 10.123 10.128 10.135 10.150
0.45 9.515 9.515 9.516 9.517 9.519 9.521 9.523 9.525 9.527 9.531 9.537 9.549 9.576 9.611 9.663
0.50 8.868 8.869 8.870 8.872 8.876 8.880 8.884 8.888 8.892 8.899 8.909 8.932 8.978 9.030 9.111
0.55 0.080 8.211 8.212 8.215 8.221 8.226 8.232 8.238 8.243 8.252 8.267 8.298 8.360 8.425 8.531
0.60 0.059 7.568 7.570 7.573 7.579 7.585 7.591 7.596 7.603 7.614 7.632 7.669 7.745 7.824 7.950
0.65 0.045 0.247 6.949 6.952 6.959 6.966 6.973 6.980 6.987 6.997 7.017 7.059 7.147 7.239 7.381
0.70 0.034 0.185 0.415 6.360 6.367 6.373 6.381 6.388 6.395 6.407 6.429 6.475 6.574 6.677 6.837
0.75 0.027 0.142 0.306 5.796 5.802 5.809 5.816 5.824 5.832 5.845 5.868 5.918 6.027 6.142 6.318
0.80 0.021 0.110 0.234 0.542 5.266 5.271 5.278 5.285 5.293 5.306 5.330 5.385 5.506 5.632 5.824
0.85 0.017 0.087 0.182 0.401 4.753 4.754 4.758 4.763 4.771 4.784 4.810 4.872 5.008 5.149 5.358
0.90 0.014 0.070 0.144 0.308 0.751 4.254 4.248 4.249 4.255 4.268 4.298 4.371 4.530 4.688 4.916
0.93 0.012 0.061 0.126 0.265 0.612 1.236 3.942 3.934 3.937 3.951 3.987 4.073 4.251 4.422 4.662
0.95 0.011 0.056 0.115 0.241 0.542 0.994 3.737 3.712 3.713 3.730 3.773 3.873 4.068 4.248 4.497
0.97 0.010 0.052 0.105 0.219 0.483 0.837 1.616 3.470 3.467 3.492 3.551 3.670 3.885 4.077 4.336
0.98 0.010 0.050 0.101 0.209 0.457 0.776 1.324 3.332 3.327 3.363 3.434 3.568 3.795 3.992 4.257
0.99 0.009 0.048 0.097 0.200 0.433 0.722 . 1.154 3.164 3.164 3.223 3.313 3.464 3.705 3.909 4.178
1.00 0.009 0.046 0.093 0.191 0.410 0.675 1.034 2.471 2.952 3.065 3.186 3.358 3.615 3.825 4.100
1.01 0.009 0.044 0.089 0.183 0.389 0.632 0.940 1.375 2.595 2.880 3.051 3.251 3.525 3.742 4.023
Приложения 629
Продолжение табл. Д.16
77? PR
0.010 0.050 0.100 0.200 0.400 0.600 0.800 1.000 1.200 1.500 2.000 3.000 5.000 7.000 10.000
1.02 0.008 0.042 0.085 0.175 0.370 0.594 0.863 1.180 1.723 2.650 2.906 3.142 3.435 3.661 3.947
1.05 0.007 0.037 0.075 0.153 0.318 0.498 0.691 0.877 0.878 1.496 2.381 2.800 3.167 3.418 3.722
1.10 0.006 0.030 0.061 0.123 0.251 0.381 0.507 0.617 0.673 0.617 1.261 2.167 2.720 3.023 3.362
1.15 0.005 0.025 0.050 0.099 0.199 0.296 0.385 0.459 0.503 0.487 0.604 1.497 2.275 2.641 3.019
1.20 0.004 0.020 0.040 0.080 0.158 0.232 0.297 0.349 0.381 0.381 0.361 0.934 1.840 2.273 2.692
1.30 0.003 0.013 0.026 0.052 0.100 0.142 0.177 0.203 0.218 0.218 0.178 0.300 1.066 1.592 2.086
1.40 0.002 0.008 0.016 0.032 0.060 0.083 0.100 0.111 0.115 0.108 0.070 0.044 0.504 1.012 1.547
1.50 0.001 0.005 0.009 0.018 0.032 0.042 0.048 0.049 0.046 0.032 -0.008 -0.078 0.142 0.556 1.080
1.60 0.000 0.002 0.004 0.007 0.012 0.013 0.011 0.005 -0.004 -0.023 -0.065 -0.151 -0.082 0.217 0.689
1.70 0.000 0.000 0.000 -0.000 -0.003 -0.009 -0.017 -0.027 -0.040 -0.063 -0.109 -0.202 -0.223 -0.028 0.369
1.80 -0.000 -0.001 -0.003 -0.006 -0.015 -0.025 -0.037 -0.051 -0.067 -0.094 -0.143 -0.241 -0.317 -0.203 0.112
1.90 -0.001 -0.003 -0.005 -0.011 -0.023 -0.037 -0.053 -0.070 -0.088 -0.117 -0.169 -0.271 -0.381 -0.330 -0.092
2.00 -0.001 -0.003 -0.007 -0.015 -0.030 -0.047 -0.065 -0.085 -0.105 -0.136 -0.190 -0.295 -0.428 -0.424 -0.255
2.20 -0.001 -0.005 -0.010 -0.020 -0.040 -0.062 -0.083 -0.106 -0.128 -0.163 -0.221 -0.331 -0.493 -0.551 -0.489
2.40 -0.001 -0.006 -0.012 -0.023 -0.047 -0.071 -0.095 -0.120 -0.144 -0.181 -0.242 -0.356 -0.535 -0.631 -0.645
2.60 -0.001 -0.006 -0.013 -0.026 -0.052 -0.078 -0.104 -0.130 -0.156 -0.194 -0.257 -0.376 -0.567 -0.687 -0.754
2.80 -0.001 -0.007 -0.014 -0.028 -0.055 -0.082 -0.110 -0.137 -0.164 -0.204 -0.269 -0.391 -0.591 -0.729 -0.836
3.00 -0.001 -0.007 -0.014 -0.029 -0.058 -0.086 -0.114 -0.142 -0.170 -0.211 -0.278 -0.403 -0.611 -0.763 -0.899
3.50 -0.002 -0.008 -0.016 -0.031 -0.062 -0.092 -0.122 -0.152 -0.181 -0.224 -0.294 -0.425 -0.650 -0.827 -1.015
4.00 -0.002 -0.008 -0.016 -0.032 -0.064 -0.096 -0.127 -0.158 -0.188 -0.233 -0.306 -0.442 -0.680 -0.874 -1.097
Примечание. Эта информация представлена в виде графиков в «Справочнике Американского нефтяного института», рис. 7В3.4
Таблица Д.17. Значения
5° _ 5 I (°)
—-— I (уравнение Ли — Кеслера)
I
PR
TR 0.010 0.050 0.100 0.200 0.400 0.600 0.800 1.000 1.200 1.500 2.000 3.000 5.000 7.000 10.000
0.30 11.614 10.008 9.319 8.635 7.961 7.574 7.304 7.099 6.935 6.740 6.497 6.182 5.847 5.683 5.578
0.35 11.185 9.579 8.890 8.205 7.529 7.140 6.869 6.663 6.497 6.299 6.052 5.728 5.376 5.194 5.060
0.40 10.802 9.196 8.506 7.821 7.144 6.755 6.483 6.275 6.109 5.909 5.660 5.330 4.967 4.772 4.619
0.45 10.453 8.847 8.157 7.472 6.794 6.404 6.132 5.924 5.757 5.557 5.306 4.974 4.603 4.401 4.234
0.50 10.137 8.531 7.841 7.156 6.479 6.089 5.816 5.608 5.441 5.240 4.989 4.656 4.282 4.074 3.899
0.55 0.038 8.245 7.555 6.870 6.193 5.803 , 5.531 5.324 5.157 4.956 4.706 4.373 3.998 3.788 3.607
0.60 0.029 7.983 7.294 6.610 5.933 5.544 5.273 5.066 4.900 4.700 4.451 4.120 3.747 3.537 3.353
0.65 0.023 0.122 7.052 6.368 5.694 5.306 5.036 4.830 4.665 4.467 4.220 3.892 3.523 3.315 3.131
0.70 0.018 0.096 0.206 6.140 5.467 5.082 4.814 4.610 4.446 4.250 4.007 3.684 3.322 3.117 2.935
0.75 0.015 0.078 0.164 5.917 5.248 4.866 4.600 4.399 4.238 4.045 3.807 3.491 3.138 2.939 2.761
0.80 0.013 0.064 0.134 0.294 5.026 4.649 4.388 4.191 4.034 3.846 3.615 3.310 2.970 2.777 2.605
0.85 0.011 0.054 0.111 0.239 4.785 4.418 4.166 3.976 3.825 3.646 3.425 3.135 2.812 2.629 2.463
0.90 0.009 0.046 0.094 0.199 0.463 4.145 3.912 3.738 3.599 3.434 3.231 2.964 2.663 2.491 2.334
0.93 0.008 0.042 0.085 0.179 0.408 0.750 3.723 3.569 3.444 3.295 3.108 2.860 2.577 2.412 2.262
0.95 0.008 0.039 0.080 0.168 0.377 0.671 3.556 3.433 3.326 3.193 3.023 2.790 2.520 2.362 2.215
0.97 0.007 0.037 0.075 0.157 0.350 0.607 1.056 3.259 3.188 3.081 2.932 2.719 2.463 2.312 2.170
0.98 0.007 0.036 0.073 0.153 0.337 0.580 0.971 3.142 3.106 3.019 2.884 2.682 2.436 2.287 2.148
0.99 0.007 0.035 0.071 0.148 0.326 0.555 0.903 2.972 3.010 2.953 2.835 2.646 2.408 2.263 2.126
1.00 0.007 0.034 0.069 0.144 0.315 0.532 0.847 2.178 2.893 2.879 2.784 2.609 2.380 2.239 2.105
1.01 0.007 0.033 0.067 0.139 0.304 0.510 0.799 1.391 2.736 2.798 2.730 2.571 2.352 2.215 2.083
Приложения 63
Продолжение табл. Д.17
PR
TR 0.010 0.050 0.100 0.200 0.400 0.600 0.800 1.000 1.200 1.500 2.000 3.000 5.000 7.000 10. 000
1.02 0.006 0.032 0.065 0.135 0.294 0.491 0.757 1.225 2.495 2.706 2.673 2.533 2.325 2.191 2.062
1.05 0.006 0.030 0.060 0.124 0.267 0.439 0.656 0.965 1.523 2.328 2.483 2.415 2.242 2.121 2.001
1.10 0.005 0.026 0.053 0.108 0.230 0.371 0.537 0.742 1.012 1.557 2.081 2.202 2.104 2.007 1.903
1.15 0.005 0.023 0.047 0.096 0.201 0.319 0.452 0.607 0.790 1.126 1.649 1.968 1.966 1.897 1.810
1.20 0.004 0.021 0.042 0.085 0.177 0.277 0.389 0.512 0.651 0.890 1.308 1.727 1.827 1.789 1.722
1.30 0.003 0.017 0.033 0.068 0.140 0.217 0.298 0.385 0.478 0.628 0.891 1.299 1.554 1.581 1.556
1.40 0.003 0.014 0.027 0.056 0.114 0.174 0.237 0.303 0.372 0.478 0.663 0.990 1.303 1.386 1.402
1.50 0.002 0.011 0.023 0.046 0.094 0.143 0.194 0.246 0.299 0.381 0.520 0.777 1.088 1.208 1.260
1.60 0.002 0.010 0.019 0.039 0.079 0.120 0.162 0.204 0.247 0.312 0.421 0.628 0.913 1.050 1.130
1.70 0.002 0.008 0.017 0.033 0.067 0.102 0.137 0.172 0.208 0.261 0.350 0.519 0.773 0.915 1.013
1.80 0.001 0.007 0.014 0.029 0.058 0.088 0.117 0.147 0.177 0.222 0.296 0.438 0.661 0.799 0.908
1.90 0.001 0.006 0.013 0.025 0.051 0.076 0.102 0.127 0.153 0.191 0.255 0.375 0.570 0.702 0.815
2.00 0.001 0.006 0.011 0.022 0.044 0.067 0.089 0.111 0.134 0.167 0.221 0.325 0.497 0.620 0.733
2.20 0.001 0.004 0.009 0.018 0.035 0.053 0.070 0.087 0.105 0.130 0.172 0.251 0.388 0.492 0.599
2.40 0.001 0.004 0.007 0.014 0.028 0.042 0.056 0.070 0.084 0.104 0.138 0.201 0.311 0.399 0.496
2.60 0.001 0.003 0.006 0.012 0.023 0.035 0.046 0.058 0.069 0.086 0.113 0.164 0.255 0.329 0.416
2.80 0.000 0.002 0.005 0.010 0.020 0.029 0.039 0.048 0.058 0.072 0.094 0.137 0.213 0.277 0.353
3.00 0.000 0.002 0.004 0.008 0.017 0.025 0.033 0.041 0.049 0.061 0.080 0.116 0.181 0.236 0.303
3.50 0.000 0.001 0.003 0.006 0.012 0.017 0.023 0.029 0.034 0.042 0.056 0.081 0.126 0.166 0.216
4.00 0.000 0.001 0.002 0.004 0.009 0.013 0.017 0.021 0.025 0.031 0.041 0.059 0.093 0.123 0.162
Таблица Д.18. Значения S° - S (1) (уравнение Ли — Кеслера)
R
PR
TR 0.010 0.050 0.100 0.200 0.400 0.600 0.800 1.000 1.200 1.500 2.000 3.000 5.000 7.000 10. 000
0.30 16.782 16.774 16.764 16.744 16.705 16.665 16.626 16.586 16.547 16.488 16.390 16.195 15.837 15.468 14.925
0.35 15.413 15.408 15.401 15.387 15.359 15.333 15.305 15.278 15.251 15.211 15.144 15.011 14.751 14.496 14.153
0.40 13.990 13.986 13.981 13.972 13.953 13.934 13.915 13.896 13.877 13.849 13.803 13.714 13.541 13.376 13.144
0.45 12.564 12.561 12.558 12.551 12.537 12.523 12.509 12.496 12.482 12.462 12.430 12.367 12.248 12.145 11.999
0.50 11.202 11.200 11.197 11.192 11.182 11.172 11.162 11.153 11.143 11.129 11.107 11.063 10.985 10.920 10.836
0.55 0.115 9.948 9.946 9.942 9.935 9.928 9.921 9.914 9.907 9.897 9.882 9.853 9.806 9.769 9.732
0.60 0.078 8.828 8.826 8.823 8.817 8.811 8.806 8.799 8.794 8.787 8.777 8.760 8.736 8.723 8.720
0.65 0.055 0.309 7.832 7.829 7.824 7.819 7.815 7.810 7.807 7.801 7.794 7.784 7.779 7.785 7.811
0.70 0.040 0.216 0.491 6.951 6.945 6.941 6.937 6.933 6.930 6.926 6.922 6.919 6.929 6.952 7.002
0.75 0.029 0.156 0.340 6.173 6.167 6.162 6.158 6.155 6.152 6.149 6.147 6.149 6.174 6.213 6.285
0.80 0.022 0.116 0.246 0.578 5.475 5.468 5.462 5.458 5.455 5.453 5.452 5.461 5.501 5.555 5.648
0.85 0.017 0.088 0.183 0.408 4.853 4.841 4.832 4.826 4.822 4.820 4.822 4.839 4.898 4.969 5.082
0.90 0.013 0.068 0.140 0.301 0.744 4.269 4.249 4.238 4.232 4.230 4.236 4.267 4.351 4.442 4.578
0.93 0.011 0.058 0.120 0.254 0.593 1.219 3.914 3.894 3.885 3.884 3.896 3.941 4.046 4.151 4.300
0.95 0.010 0.053 0.109 0.228 0.517 0.961 3.697 3.658 3.647 3.648 3.669 3.728 3.851 3.966 4.125
0.97 0.010 0.048 0.099 0.206 0.456 0.797 1.570 3.406 3.391 3.401 3.437 3.517 3.661 3.788 3.957
0.98 0.009 0.046 0.094 0.196 0.429 0.734 1.270 3.264 3.247 3.268 3.318 3.412 3.569 3.701 3.875
0.99 0.009 0.044 0.090 0.186 0.405 0.680 1.098 3.093 3.082 3.126 3.195 3.306 3.477 3.616 3.796
1.00 0.008 0.042 0.086 0.177 0.382 0.632 0.977 2.399 2.868 2.967 3.067 3.200 3.387 3.532 3.717
1.01 0.008 0.040 0.082 0.169 0.361 0.590 0.883 1.306 2.513 2.784 2.933 3.094 3.297 3.450 3.640
Приложения 633
Продолжение табл. Д.18
PR
TR 0.010 0.050 0.100 0.200 0.400 0.600 0.800 1.000 1.200 1.500 2.000 3.000 5.000 7.000 10.000
1.02 0.008 0.039 0.078 0.161 0.342 0.552 0.807 1.113 1.655 2.557 2.790 2.986 3.209 3.369 3.565
1.05 0.007 0.034 0.069 0.140 0.292 0.460 0.642 0.820 0.831 1.443 2.283 2.655 2.949 3.134 3.348
1.10 0.005 0.028 0.055 0.112 0.229 0.350 0.470 0.577 0.640 0.618 1.241 2.067 2.534 2.767 3.013
0.15 0.005 0.023 0.045 0.091 0.183 0.275 0.361 0.437 0.489 0.502 0.654 1.471 2.138 2.428 2.708
1.20 0.004 0.019 0.037 0.075 0.149 0.220 0.286 0.343 0.385 0.412 0.447 0.991 1.767 2.115 2.430
1.30 0.003 0.013 0.026 0.052 0.102 0.148 0.190 0.226 0.254 0.282 0.300 0.481 1.147 1.569 1.944
1.40 0.002 0.010 0.019 0.037 0.072 0.104 0.133 0.158 0.178 0.200 0.220 0.290 0.730 1.138 1.544
1.50 0.001 0.007 0.014 0.027 0.053 0.076 0.097 0.115 0.130 0.147 0.166 0.206 0.479 0.823 1.222
1.60 0.001 0.005 0.011 0.021 0.040 0.057 0.073 0.086 0.098 0.112 0.129 0.159 0.334 0.604 0.969
1.70 0.001 0.004 0.008 0.016 0.031 0.044 0.056 0.067 0.076 0.087 0.102 0.127 0.248 0.456 0.775
1.80 0.001 0.003 0.006 0.013 0.024 0.035 0.044 0.053 0.060 0.070 0.083 0.105 0.195 0.355 0.628
1.90 0.001 0.003 0.005 0.010 0.019 0.028 0.036 0.043 0.049 0.057 0.069 0.089 0.160 0.286 0.518
2.00 0.000 0.002 0.004 0.008 0.016 0.023 0.029 0.035 0.040 0.048 0.058 0.077 0.136 0.238 0.434
2.20 0.000 0.001 0.003 0.006 0.011 0.016 0.021 0.025 0.029 0.035 0.043 0.060 0.105 0.178 0.322
2.40 0.000 0.001 0.002 0.004 0.008 0.012 0.015 0.019 0.022 0.027 0.034 0.048 0.086 0.143 0.254
2.60 0.000 0.001 0.002 0.003 0.006 0.009 0.012 0.015 0.018 0.021 0.028 0.041 0.074 0.120 0.210
2.80 0.000 0.001 0.001 0.003 0.005 0.008 0.010 0.012 0.014 0.018 0.023 0.035 0.065 0.104 0.180
3.00 0.000 0.001 0.001 0.002 0.004 0.006 0.008 0.010 0.012 0.015 0.020 0.031 0.058 0.093 0.158
3.50 0.000 0.000 0.001 0.001 0.003 0.004 0.006 0.007 0.009 0.011 0.015 0.024 0.046 0.073 0.122
4.00 0.000 0.000 0.001 0.001 0.002 0.003 0.005 0.006 0.007 0.009 0.012 0.020 0.038 0.060 0.100
Примечание. Эта информация представлена в виде графиков в «Справочнике Американского нефтяного института», рис. 7F1.4
и 7F1.5.
Приложения 635
Приложение Е
Используемые единицы и их эквиваленты
Несмотря на повсеместно наблюдаемую тенденцию
использования Международной системы единиц, боль-
шинство данных до сих пор записывается в старых еди-
ницах, о которых поэтому необходимо иметь
отчетливое представление. Тем более, что такие усто-
явшиеся единицы, как, например, атмосфера или мил-
лиметры ртутного столба, будут, вероятно,
применяться еще долго. Международная система еди-
ниц весьма удобна тем, что ее легко можно распростра-
нять на новые понятия и применять в широком
диапазоне величин.
Так, например, если принять, что красота воспетой
Гомером Елены — это единица красоты, то ее можно
определить как количество красоты, способной приве-
сти в движение 1000 кораблей. Однако эта единица
слишком громоздка, и поэтому гораздо удобнее поль-
зоваться такой единицей красоты, как микро-Елена, т.
е. количество красоты, способное привести в движение
гребную шлюпку. Эта единица логически определима и
вполне удобна.
Ниже указаны наиболее полезные литературные ис-
точники, содержащие таблицы эквивалентов различных
единиц.
1. Eshbach O.W., Sounders М. (eds) Handbook of
Engineering Fundamentals. Wiley (1975).
2. Mullin J.W. Recent development in the changeover to
the international system of units. The Chemical
Engineer, 353 (October 1971).
3. SI for AIChE. Publication
Таблица E.l. Эквиваленты единиц
Множители
микро = 10(Е - 6)
милли = 10(Е — 3)
санти = 10(Е - 2)
деци = 10(Е - 1)
кило = 10(ЕЗ)
мега = 10(Е6)
Длина
1 фут = 0,3048 м = 30,48 см -- 304,8 мм
Объем
1 фут3 = 0,0283 м3 = 7,481 американских галло
нов
1 м3 = 35,34 фут3 = 1000 л
Объем газа в стандартных условиях
22,414 л/моль при 0 °C и 1 атм
359,05 фут3/моль при 32 °F и 1 атм
Давление
1 атм = 760 мм рт. ст. = 101,325 Н/м2 =
= 1,01325 бар = 10,33 кг/м2
= 14,696 фунт-сила/дюйм2 =
= 2116,2 фунт-сила/фут2
1 бар = 10 0000 Н/м2
1 Па = 1 Н/м2
Энергия, работа и теплота
1 брит. тепл. ед. = 252,16 кал = 1055,06 Дж =
= 0,2930 Вт • ч = 10,41 л • атм
1 л.с. • ч = 0,7457 кВт • ч = 778 куб. фунт-сила
= 2545 кал
1 кал= 4,1868 Дж
1 Дж= 1 Нм=1 Вт-с = 0,2388 кал
= 0,000948 брит. тепл. ед.
Мощность
1 фунт-сила/с = 0,0018182 л.с. = 1,356 Вт =
= 0,0012856 брит. тепл. ед./с = 0,3238 кал/с
1 В = 1 Дж/с =1 Нм/с
Температура
К (Кельвин) = 100 °C + 273,16
= [°F (градус Фаренгейта; -г
+ 459,6]/1,8 = °R (градус Ренкина)/1,8
1 °R = 1,8 К = F + 459,6
1 °C = (F - 32)/1,8
Разность температур
1 °C = 1 К = 1,8 °R = 1,8 °F
Теплоемкость и энтропия
1 кал/г • К = 4,1868 Дж/г • К
= брит. тепл, ед./фунт • °R
Удельная энергия
1 кал/г = 4,1868 Дж/г = 1,8 брит. тепл, ед./фунт
Объемный расход
1 фут3/с = 0,028316 м3/с = 28,316 л/с
Гравитационная постоянная
gc = 1 кг-масса/Н • с2
= 1 г • см/дин • с2
= 9,806 кг-масса/кг-сила • с2
= 32,174 фунт-масса/фунт-сила • с2
Универсальная газовая постоянная
См. табл. А.5
Масса
1 фунт = 0,4536 кг
1 кг = 2,2046 фунт
Плотность
1 фунт/фут3 = 16,018 кг/м3
1 г/см3 = 62,43 фунт/фут3
Сила
1 фунт-сила = 0,4536 кг-сила = 4,448 Н
ЛИТЕРАТУРА
Сборники статей и библиографические указатели
1. “Phase Equilibrium and Fluid Properties in the Chemical
Industry”. Proceedings, Second International Conference,
Berlin, March 17, 1980, European Federation of Chemical
Engineers (EFCE), DECHEMA, 1980.
2. Chao К. C. Applied Thermodynamics, Based on a sympo-
sium sponsored by Industrial and Engineering Chemistry,
June 12—14, 1967, American Chemical Society Publica-
tions, 1968.
3. Chao К. C, Robinson R. L. “Equations of State in En-
gineering and Research”. Advances in Chemistry Series
182. American Chemical Society, 1979.
4. Hiza M. J., Kidnay A. J., Miller R. C Equilibrium Proper-
ties of Fluid Mixtures. 1. A Bibliography of Data on
Fluids of Cryogenic Interest. 2. A Bibliography of Ex-
perimental Data on Selected Fluids. Plenum Press, 1975,
1982.
5. Kehiaian H. V., Zwolinski B. J. (eds). International Data
Series. Series A: Selected Data on Mixtures. Series B:
Thermodynamic Properties of Aqueous Organic Systems.
Thermodynamics Research Center, Texas A and M Univer-
sity, 1971-data.
6. Leesley M. E. (ed.). Computer-Aided Process Plant De-
sign. Gulf Publishing Company, 1982.
7. Le Neindre B, Vodar B. (eds). Experimental Ther-
modynamics II. Experimental Thermodynamics of Non-
reacting Fluids. International Union of Pure and Applied
Chemistry. Butterworths, 1975.
8. McGlashan M. L. (ed.). Chemical Thermodynamics: A
Specialist Periodical Report. Vol. I (1973). Vol. II (1978).
The Chemical Society, London.
9. Mah R. S. H., Seider W. D. Foundations of Computer-
Aided Process Design. Vols. I and II. American Institute
of Chemical Engineers, 1980.
10. Mittal K. L. (ed.). Micellization, Solubilization and
Microemulsions, Vols. 1 and 2. Plenum Press, 1977.
11. Mittal K. L. (ed.). Solution Chemistry of Surfactants,
Vols. 1 and 2 Plenum Press, 1977.
12. Mittal K. L„ FendlerE. J. (ed.). Solution Behavior of Sur-
factants. Vols 1 and 2. Plenum Press, 1982.
13. Newman S. A. (ed.). Chemical Engineering Ther-
modynamics. From the Second World Congress of Chemi-
cal Engineering, Montreal, Oct. 4—9, 1981. Ann Arbor
Science, 1982.
14. Newman S. A., Barner H. E., Klein M., Sandler S. I. (eds).
“Thermodynamics of Aqueous Systems with Industrial
Applications”. ACS Symposium Series 133. American
Chemical Society, 1980.
15. PaulaitisM. E., Penninger J. M. L., Gray R. D., Jr., David-
son P (eds). Chemical Engineering at Supercritical Fluid
Conditions. Ann Arbor Science, 1983.
16. Skinner H. A. (ed.). International Review of Science:
Physical Chemistry. Series 2, Vol. 10. Thermochemistry
and Thermodynamics. Butterworths, 1975.
17. Storvick T. S., Sandler S. I. (eds). “Phase Equilibria and
Fluid Properties in the Chemical Industry”, ACS Symposi-
um Series 60. American Chemical Society, 1977.
18. Tamir A., Stephan K. Heats of Phase Change of Pure
Components and Mixtures: A Literature Guide. Elsevier,
1983.
19. Wichterle I., Linek J., Hala E. Vapor-Liquid Equilibrium
Bibliography and Supplements 1, 2 and 3. Elsevier 1973,
1976, 1979, 1982.
20. Wishiak J., Tamir A. Liquid-Liquid Equilibria and Extrac-
tion (1980). Mixing and Excess Thermodynamic Proper-
ties: A Literature Source Book and Supplement (1978,
1982). Phase Diagrams: A Literature Source Book, Parts
A and В (1981). Elsevier.
Книги
21. Bevington P. R. Data Reduction and Error Analysis for
the Physical Science, McGraw-Hill, 1969.
22. Blasdale W. C. Equilibria in Saturated Salt Solutions.
Reinhold, 1927.
23. Blinder S. M. Advanced Physical Chemistry Macmillan,
1969.
24. Bowden S. T. The Phase Rule and Phase Reactions. Mac-
millan, 1938.
25. Bretsznajder S. Prediction of Transport and other Physical
Properties of Fluids, Pergamon 1971; Wlasnosci Gazow i
Cieczy. Wydawnictwa Naukowo-Techniczne (Warsaw),
1962.
26. Bridgman P. W. Condensed Collection of Thermodynam-
ic Formulas. Harvard University Press, 1926.
27. Chao К. C, Greenkorn R. A. Thermodynamics of Fluids:
An Introduction to Equilibrium Theory. Marcel Dekker,
1975.
28. Christensen J. J., Hanks R. W., Izatt R. M. Handbook of
Heats of Mixing, Wiley, 1982.
29. Dack M. R. J. (ed.) Solutions and Solubilities, Parts I and
II. Technique of Organic Chemistry, Vol. 8, A. Weissberg-
er (ed.). Wiley-Interscience, 1976.
Литература 637
30. Daniels Т С. Thermal Analysis, Wiley, 1973.
31. Daubert T. E., Graboski M. S., Danner R. P. Documenta-
tion of the Basis for Selection of the Contents of
Chapter 8 — Vapor-Liquid Equilibrium К-Values in Tech-
nical Data Book — Petroleum Refining, No. 8—78,
American Petroleum Institute, 1978.
32. Deaton W. M., Frost E. M., Jr. Gas Hydrates and Their
Relation to the Operation of Natural-Gas Pipe Lines. U.S.
Bureau of Mines Monograph 8, 1946.
33. Dickerson R. E. Molecular Thermodynamics. Benjamin,
1969.
34. Doring R., Knapp H., Oellrich L. R., Plocker U J., Praus-
nitz J. M. Vapor-Liquid Equilibria for Mixtures of Low-
Boiling Substances, DECHEMA, 1982.
35. Durell С. V. Homogeneous Coordinates. G. Bell and Sons,
1961.
36. Dymond J. H, Smith E. B. The Virial Coefficients of Pure
Gases and Mixtures: A Critical Compilation. Clarendon
Press, Oxford, 1980.
37. Edmister W. C. Applied Hydrocarbon Thermodynamics,
Vols. I and II. Gulf Publishing Company, 1974, 1984.
38. Ferguson F. D., Jones T. K. The Phase Rule. Butterworths,
1966.
39. Francis A. W. Liquid-Liquid Equilibriums. Wiley-Inter-
science, 1963.
40. Fredenslund A., Gmehling J., Rasmussen P. Vapor-Liquid
Equilibria Using UNIFAC. Elsevier, 1977.
41. Fried V., Натека H. E, Blukis U Physical Chemistry
Macmillan, 1977.
42. Gaskell D. R. Introduction to Metallurgical Ther-
modynamics. McGraw-Hill, 1973.
43. Gerald C F. Applied Numerical Analysis. Addison-Wesley,
1978.
44. Герасимов Я. (ред.). Физическая химия. — М.: Хи-
мия, 1970.
45. Glasstone S. Textbook of Physical Chemistry. Van Nos-
trand 1946.
46. Gmehling J., Onken U Vapor-Liquid Equilibrium Data
Collection. DECHEMA Chemistry Data Series, Vol. 1,
DECHEMA 1977 ff.
47. Guggenheim E. A. Thermodynamics, 7th ed. North-Holl
and, 1977.
48. Haase R., Thermodynamik der Mischphasen, Springer,
1956.
49. Haase R., Schonert H., Solid-Liquid Equilibrium, Perga-
mon, 1969.
50. Hala E., Pick J., Fried V., Vilim O. Vapor-Liquid
Equilibrium. Pergamon, 1967.
51. Henley E. J., Rosen E. M. Material and Energy Balance
Computations. Wiley, 1969.
52. Henley E. J., Seader J. D Equilibrium-Stage Separation
Operations in Chemical Engineering, Wiley, 1981.
53. Hildebrand J. H, PrausnitzJ. M., Scott R. L. Regular and
Related Solutions. Van Nostrand, 1970.
54. Hildebrand J. H, Scott R. L, The Solubility of Non-
electrolytes. Dover, 1964.
55. Himmelblau D. M. Process Analysis by Statistical
Methods. Wiley, 1970.
56. Himmelblau D. M., Brady В L., McKetta J. J., Jr. Survey
of Solubility Diagrams for Tfernary and Quaternary Liquid
Systems. Special Publication No. 30, Bureau of Engineer-
ing Research, University of Texas, Austin, 1959.
57. Hirata M., Ohe S., Nagahama K., Computer-Aided Data
Book of Vapor-Liquid Equilibria. Elsevier, 1976.
58. Holub R., Vonka P. Chemical Equilibrium of Gaseous
Systems. Reidel, 1976.
59. Hougen O. A., Watson К. M., Ragatz R. A. Chemical
Process Principles. Part II. Thermodynamics. Wiley, 1959.
60. Huggins M. L., Physical Chemistry of High Polymers.
Wiley, 1958.
61 Карапешьянц M. X. Химическая термодинамика. —
M.: Химия, 1975.
62. Kaufman L., Bernstein H. Computer Calculation of Phase
Diagrams. Academic Press, 1970.
63. Keenan J. H„ Keyes F. G., Hill P. G., Moore J. G. Steam
Tables, English Units (1969), SI Units (1978), Wiley.
64. King M. B. Phase Equilibrium in Mixtures. Pergamon,
1969.
65. Kipling J. J. Adsorption from Solutions of Nonelectro-
lytes. Academic Press, 1965.
66. Klotz L. M., Rosenberg R. M. Chemical Thermodynamics
Basic Theory und Methods. Benjamin-Cummings, 1972.
67. Коган В. Б. Гетерогенные равновесия. —Л.: Химия,
1968.
68. Коган В. Б., Фридман В. М., Кафа ров В. В. Парожид-
костное равновесие. — М.: Наука, 1966.
69. Kofler L. Thermomicromethoden zur Kennzeichnung or-
ganischer Stoffe und Stoffgemische. Verlag Chemie, 1954.
70. Kojima K., Tochigi K. Prediction of Vapor-Liquid
Equilibria by the ASOG Method. Elsevier, 1979.
71. Kubashewski O., Alcock С. В Metallurgical Thermo-
chemistry, 5th ed. Pergamon Press, 1979.
72. Kubo R. Thermodynamics, an Advanced Course with
Problems and Solutions, North-Holland, 1968.
73. Landolt-Bornstein, Numerical Data and Functional Rela-
tionships in Science and Tbchnology II/2a: Vapor-Liquid
Equilibria and Osmotic Phenomena (1960); II/2b: Solu-
tion Equilibria (1962); II/2c: Solution Equilibria (1964);
II/3: Melt Equilibria and Surface Phenomena (1956);
IV/4cl: Absorption of Gases, Low Vapor Pressure Liquids
(76); IV/4c2: Absorption of Gases in High Vapor Pressure
Liquids (in preparation); NS IV/2: Heats of Mixing and
Solution (1976); NS IV/3: Thermodynamic Equilibria, of
Boiling Mixtures (1975). Springer Verlag, 1950ff.
74. Lapidus L. Digital Computation for Chemical Engineers.
MgGraw-Hill 1962.
75. Larkin J. V. (ed.). Selected Data on Mixtures. Internation-
al Data Series B, Thermodynamic Properties of Aqueous
Organic Systems. Engineering Sciences Data Unit. Ltd.,
1978ff.
76. Lipka J., Graphical and Mechanical Computation. Wiley,
1918.
77. Loney S. L. The Elements of Coordinate Geometry,
Part II. Trilinear Coordinates, etc. Macmillan, 1923.
78. Lupis С. H. P., Chemical Thermodynamics of Materials.
North Holland, 1983.
79. Lydersen A. L., Greenkorn R. A., Hougen O. A. General-
ized Thermodynamic Properties of Pure Fluids. University
of Wisconsin Eng. Exp. Station, Report 4 (October), 1955.
80. Lyman W. J., Reehl W. F, Rosenblatt D. H. Handbook of
Chemical Property Estimation Methods: Environmental
Behavior of Organic Compounds. McGraw-Hill, 1982.
81. McGlashan M. L. Chemical Thermodynamics. Academic
Press, 1979.
82. Mackenzie R. C (ed.). Differential Thermal Analysis,
Vol. 2: Applications. Academic Press, 1972.
83. Maczynski A. (ed.). Verified Vapor-Liquid Equilibrium
Data: Thermodynamical Data for Technology, several
parts. PWN-Polish Scientific Publishers, 1976ff.
84. Makogon Yu. F. Hydrates of Natural Gas, Penn Well,
1981.
85. Mason E. A., Spurting T. H. The Virial Equation of State.
International Encyclopedia of Physical Chemistry and
Chemical Physics, Topic‘10, Vol. 2. Pergamon Press, 1969.
86. Modell M., Reid R. C. Thermodynamics and its Applica-
tions Prentice-Hall, 1983.
87. Nielsen L. E. Predicting the Properties of Mixtures. Mar-
cel Dekker, 1978.
88. Null H. R. Phase Equilibrium in Process Design. Wiley-
Interscience, 1970.
89. Nyvlt J. Solid-Liquid Phase Equilibria. Elsevier, 1977.
90. Ohe S. Computer-Aided Data Book of Vapor Pressure,
Data Publishing Company, Tokyo, 1976.
91. Opfell J. B„ Pings C. J., Sage В. H. Equation of State for
Hydrocarbons. Monograph on API Research Project 37,
American Petroleum Institute, 1959.
92. Partington J. R. An Advanced Treatise on Physical •
Chemistry. Vol. I: Fundamental Principles and Properties
of Gases; Vol. II: The Properties of Liquids. Longmans,
1950, 1951.
93. Partington J. R., Shilling W. G. The Specific Heat of
Gases E. Benn, 1924.
94. Paul R. E M., Wise W. S. Principles of Gas Extraction.
Mills and Boon Ltd., 1971.
95. Pickering S. E Relations between the Temperatures, Pres-
sures and Densities of Gases. Circular No. 279, U. S.
Bureau of Standards, 1925.
96. Pope M. I., Judd M. D. Differential Thermal Analysis: A
Guide to the Technique and its Applications. Heyden,
1977.
97. Prausnitz J. M. Molecular Thermodynamics of Fluid
Phase Equilibria. Prentice-Hall, 1969.
98. Prausnitz J. M., Anderson T, Grens E., Eckert C,
Hsieh R., O’Connell J. Computer Calculations for Mul-
ticomponent Vapor-Liquid and Liquid-Liquid Equilibria.
Prentice Hall, 1980.
99. Prausnitz J. M., Chueh P. L. Computer Calculations for
High Pressure Vapor Liquid Equilibria. Prentice-Hall,
1968.
100. Prausnitz J. M., Eckert C. A., Orye R. V, O’Con-
nell J. P., Computer Calculations for Multicomponents
Vapor Liquid Equilibria. Prentice-Hall, 1967.
101. Priestley E. B, Wojtowicz P J., Sheng P. Introduction to
Liquid Crystals. Plenum Press, 1975.
102. Prigogine I., Defay R. Chemical Thermodynamics. Long-
mans, 1954.
103. Prince A. Alloy Phase Equilibria. Elsevier, 1966.
104. Rasmussen R, Fredenslund A. Data Banks for Chemical
Engineers. Kemiigeniorgruppen, Lyngby, Denmark,
1980.
105. Redlich O. Thermodynamics Fundamentals and Applica-
tions. Elsevier, 1978.
106. Reed T. M., Gubbins К. E. Applied Statistical Mechan-
ics: Thermodynamic and Transport Properties of Fluids.
McGraw-Hill, 1973.
107. Reid R. C, Prausnitz J. M., Sherwood T. K. The Proper-
ties of Gases and Liquids. McGraw-Hill, 1958, 1966,
1977.
108. Reisman A. Phase Equilibria, Basic Principles, Applica-
tions. Experimental Techniques, Academic Press, 1970.
109. Renon H, Asselineau L., Cohen G., Raimbault C Calcul
sur Ordinateur des Equilibres Liquide-Vapeur et Liquide-
Liquide. Editions Technip, 1971.
110. Ricci J. E. The Phase Rule and Heterogeneous Equilibri-
um. Van Nostrand, 1951.
111. Rosenbrock H. H, Storey C. Computational Techniques
for Chemical Engineers. Pergamon Press, 1966.
112. Rowlinson J. S. The Perfect Gas. International Ency-
clopedia of Physical Chemistry and Chemical Physics,
Topic 10, Vol. 5. Pergamon Press, 1963.
113. Rowlinson J. S., Swinton F. L. Liquids and Liquid Mix-
tures, 3rd ed. Butterworths, 1982.
114. Sage В H. Thermodynamics of Multicomponent Sys-
tems. Reinhold Publishing Company, 1965.
115. Sage В H, Lacey W. N. Volumetric and Phase Behavior
of Hydrocarbons. Stanford University Press, 1939.
116. Salzer H. E„ Richards С. H, Arsham I. Tables for the
Solution of Cubic Equations. McGraw-Hill, 1958.
117. Scarborough J. B. Numerical Mathematical Analysis.
Johns Hopkins University Press, 1966.
118. Schneider G. M., Stahl E., Wilke G. Extraction with Su-
percritical Gases. Verlag Chemie, 1980.
119. Sherwood T K. A Course in Process Design. MIT Press,
1963.
120. Sherwood T K., Redd С. E. Applied Mathematics in
Chemical Engineering. McGraw-Hill, 1939.
121. Shinoda K. Principles of Solution and Solubility. Marcel
Dekker, 1978.
122. Smith J. M., Van Ness H. C. Introduction to Chemical
Engineering Thermodynamics. McGraw-Hill, 1975.
123. Smith W. R., Missen D. W. Chemical Reaction Analysis:
Theory and Algorithms, Wiley, 1982.
124. Sorensen J. M. Correlation of Liquid-Liquid Equilibri-
um Data. Instituttet for Kemiteknik, Danmarks Tekniske
Hojskole, 1980.
125. Sorensen J. M., Arlt W. Liquid-Liquid Equilibrium Data
Collection, 3 parts. DECHEMA, 1979, 1980.
126. Stanley H. E. Introduction to Phase Transitions and Crit-
ical Phenomena. Clarendon Press, Oxford, 1971.
127. Starling К. E. Fluid Thermodynamic Properties for Light
Petroleum Systems. Gulf Publishing Company, 1973.
128. Stephen H, Stephen T, Silcock H. Solubilities of Inor-
ganic and Organic Compounds, 7 parts. Pergamon, 1979.
129. Sterbacek A., Biskup B, Tausk P. Calculation of Proper-
ties Using Corresponding States Methods. Elsevier, 1979.
130. Stull D. R., Westrum E. E, Sinke G. C. The Chemical
Thermodynamics of Organic Compounds. Wiley, 1969.
131. Tabor D. Gases, Liquids and Solids. Cambridge Universi-
ty Press, 1979.
132. Tamas E, Pal I. Phase Equilibrium Spatial Diagrams, II-
liffe Books, 1970.
133. Teeple J. E. Industrial Development of Searles Lake
Brines. Reinhold. 1929.
134. Temperley H. N. V., Trevena D. H. Liquids and their
Properties. Ellis Horwood Ltd., 1978.
135. Treybal R. Liquid Extraction. McGraw-Hill, 1963.
136. Вукалович M. П., Новиков И. И. Уравнения состоя-
ния реальных газов. — М.: Госэнергоиздат, 1948.
Литература 639
137. Van Ness H. C, Abbott М. М. Classical Thermodynam-
ics of Non-Electrolyte Solutions with Applications to
Phase Equilibria. McGraw-Hill, 1982.
138. Van Zeggeren E, Storey S. H The Computation of
Chemical Equilibria. Cambridge University Press, 1970.
139. Vogel R. Heterogeneous Equilibria (in German).
Akademische Verlagsgeseilschaft, 1959.
140. Wendlandt W. W., Thermal Methods of Analysis. Wiley,
1974.
141. West D. R. F. Ternary Equilibrium Diagrams, 2nd ed.
Chapman and Hall, 1982.
142. Wolberg J. R. Prediction Analysis. Van Nostrand, 1967.
143. Yaws C. L. Physical Properties: A Guide to the Physical,
Thermodynamic and Transport Properties of Industrially
Important Chemical Compounds, McGraw-Hill, 1977.
144, Еремин E. H. Основы химической кинетики. — M.:
Высшая школа, 1976.
145. Young D. М., Growell A. D. Physical Adsorption of
Gases. Butterworths, 1962.
146. Zernike J. Chemical Phase Theory, Kluwer, Deventer,
Holland, 1955.
Журнальные статьи
147. Abbott M. M. Cubic Equiations of State (review).
AIChE Journal, 19, 596—601, (1973).
148. Abbott M. M. Cubic Equations of State: An Interpretive
Review, К. C. Chao and R. L. Robinson (eds.). Equations
of State in Engineering and Research. 47—70, Advances
in Chemistry Series 182, Am. Chem. Soc. 1979.
149. Abbott M. M„ Floess J. K., Walsh G. E, Jr., Van Ness
H. C. Vapor-liquid equilibrium. Part IV: Reduction of
P-x data for ternary systems. AIChE Journal, 21, 72—76
(1975).
150. Abbott M. M., Van Ness H. C. Vapour-liquid equilibri-
um. Part III: Data reduction with precise expressions for
Ge, AIChE Journal, 21, 62—71 (1975).
151. Abbott M. M., Van Ness H. C. An extension of Barker’s
method for reduction of VIE data. Fluid Phase
Equilibria, 1, 3—11 (1977).
152. Abrams D. S., Prausnitz J- M. Statistical Thermodynam-
ics of liquid mixtures: A new expression for the excess
Gibbs energy of partly or completely miscible systems.
AIChE Journal, 21, 116—128 (1975).
153. Abrams D. S., Seneci E, Chueh P. L., Prausnitz J. M.,
Thermodynamics of multicomponent liquid mixtures
containing subcritical and supercritical components. Ind.
Eng. Chem. Fundamen., 14, 52—54 (1975).
154. Adler S. R, Spencer С. E, Ozkardesh H., Kuo С. M. In-
dustrial uses of equations of state: A state of the art
review, T. S. Storvick and S. I. Sandler (eds.). Phase
equilibria and fluid properties in the chemical industry,
150—199. ACS Symposium Series 60, Am. Chem. Soc.
(1977).
155. Alwani Z„ Schneider G. M. Fluid mixtures at high pres-
sure: Phase Separation and critical phenomena in binary
mixtures of a polar compound with supercritical carbon
dixode, ethane and ethene up to 1000 bar. Ber Bunsenges
Physik Chemie, 80, 1310—1315 (1976).
156. Ambrose P. The correlation and estimation of vapor
pressures. Proceedings of the NPL Conference: Chemical
Thermodynamic Data on Fluids and Fluid Mixtures. IPC
Science and Technology Press (1979).
157. Ambrose D„ Counsell, Davenport J. Chem. Ther-
modynamics, 2, 283 (1970).
158. Anderson T. E, Prausnitz J- M. Computational methods
for high-pressure phase equilibria and other fluid-phase
properties using a partition function. 1: Pure fluids;
2: Mixtures; Ind. Eng. Chem. Process Des. Dev., 19, 1—4
(1980).
159. Anderson T E, Prausnitz J. M. Application of the
UNIQUAC equation to calculation of multicomponents
phase equilibria. 1: Vapor-liquid equilibria; 2: Liquid-
liquid equilibria. Ind. Eng. Chem. Process Des. Dev., 17,
552—567 (1978).
160. Antezane F. J., Cheh J. Y. Component fugacities in
hydrogen-ammonia-propane mixtures. 1: The fugacity of
hydrogen; 2: The fugacity of ammonia. Ind. Eng. Chem.
Fundamen., 14, 224—232 (1975); 15, 96—99 (1976).
161. Arich G., KikicL, Allessi P. The liquid-liquid equilibrium
for activity coefficient determination. Chem. Eng.
Science, 30, 187—191 (1975).
162. Аристович В. Ю., Степанова Э. И. Расчетное опре-
деление многокомпонентных азеотопов по данным
бинарных систем. Журн. прикл. хим., 43, 10, 2192—
2197 (1970).
163. Ashraf Е A., Vera J. Н. A simplified group method anal-
ysis. Fluid Phase Equilibria, 4, 211—228 (1980).
164. Asselineau L„ Bogdanic G., Vidal J. Calculation of ther-
modynamic properties and vaport-liquid equilibria of
refrigerants. Chem. Eng. Science, 33, 1269—1276 (1978).
165. Asselineau L., Bogdanic G., Vidal J. A versatile al-
gorithm for calculating vapor-liquid equilibria. Fluid
Phase Equilibria, 3, 273—290 (1979).
166. Аветян В. С., Карп Л. Е., Петлюк Ф. В., Серафи-
мов Л. А. «Математическое описание фазового рав-
новесия в полиазеотропных смесях», Журн. физ.
хим., 52, 6, 1425—1427 (1970).
167. Bae Н. К, Nagahama К., Hirata М. Measurement and
correlation of high pressure vapor-liquid equalibria for
the systems ethylene + 1-butene and ethylene + propy-
lene. J. Chem. Eng. Japan, 14, 1—6 (1981).
168. Baker L. E., Luks K. D. Critical Point and saturation
pressure calculations for multicomponent systems. Paper
SPE 7478, presented at 53rd Annual Fall Technical Con-
ference of Society of Petroleum Engineers of Al ME, held
in Houston, Texas, October 1—3, 1978.
169. Bansal V. K., Shah D. O. Micellar solutions for improved
oil recovery. Mittal K. L. (ed.). Solution Chemistry of
Surfactants, 1, 87—113 (1977).
170. Barieau R. E. The calculation of fugacities from volu-
metric and phase equilibrium data by means of the
Gibbs-Duhem equation. Ind. Eng. Chem. Fundamen.,
10, 428—433 (1971).
171. Barker J. A. Determination of activity coefficients from
total pressure measurements. Australian J. Chem., 6,
207—210 (1953).
172. Barner H. E., Adler S. B. Low temperature B-W-R appli-
cations. Hydrocarbon Processing, 47, (10), 150—156
(1968).
173. Barner H. E., Adler S. B. Three-parameter formulation
of the Joffe equation of state. Ind. Eng. Chem. Funda-
men., 9, 521—530 (1970).
174. Barner H. E., Adler S. B. Calculation of solution
nonideality from binary T-x data. Ind. Eng. Chem.
Process Des. Dev., 12, 71—75 (1973).
175. Baron R. E., Porter J. H, Hammond О. H. Chemical
Equilibria in Carbon-Hydrogen-Oxygen Systems. MIT
Press (1975).
176. Barton A. F. Solubility parameters. Chem. Reviews, 75,
73—753 (1975).
177. Barton P., Holland R. E., McCormick R. H. Correlation
of vapor-liquid equilibria of Сз—Cs hydrocarbons using
solubility parameters. Ind. Eng. Chem. Process Des.
Dev., 13, 378—383 (1974).
178. Beattie J. A., Stockmayer W. H. The thermodynamics
and statistical mechanics of real gases. Treatise on Physi-
cal Chemistry Vol. 2, Ihylor H. S. and Glasstone S. (eds.),
187—352. Van Nostrand, 1951.
179. Belton G. R., Fruehan R. J. The determination of activi-
ties by mass spectrometry. I: The metallic systems iron-
nickel and iron-cobalt. J. Phys. Chem., 71, 1403—1409
(1967).
180. Bender E. Equations of state for ethylene and propylene.
Cryogenics, 15 (11), 667—673 (1975).
181. Benedict M., Webb G. B., Rubin L. C. An empirical equa-
tion for thermodynamic properties of light hydrocarbons
and their mixtures. J. Chem. Physics, 8, 334—345 (1940);
10, 747—758 (1942).
182. Benedict M., Webb G. B., Rubin L. C. An empirical equa-
tion for thermodynamic properties of light hydrocarbons
and their mixtures: Fugacities and liquid-vapor
equilibria. Chem. Eng. Progress, 47 (8), 419 (1951); 47
(9), 449—454 (1951).
183. Beret S., Prausnitz J. M. Perturbed hard-chain theory:
An equation of state for fluids containing small or large
molecules. AIChE Journal, 21, 1123—1132 (1975).
184. Beutier D., Renon H Representation of NH3-H2S-H2O,
NH3-CO2-H2O and NH3SO2-H2O vapor-liquid
equlibria. Ind. Eng. Chem. Process Des. Dev., 17,
220—230 (1978).
185. Birdi K. S. Thermodynamics of micelle formation, Mittal
K. L. (ed.). Solution Chemistry of Surfactants, 1,
151—169 (1977).
186. Bishnoi P. R., Miranda R. D., Robinson D. В BWR Ap-
plied to NG/SNG needs. Hydrocarbon Processing, 54,
(11), 197—201 (1974).
187. Bishnoi P. R., Robinson D. В New mixing rules for the
BWR parameters to predict mixture properties. Canad.
J. Chem. Eng., 50, 101—107 (1972).
188. Bishnoi P. R., Robinson D. В Mixing rules improve BWR
use. Hydrocarbon Processing, 51 (11), 152—156 (1972).
189. Black C. Phase equilibria in binary and multicomponent
systems: Modified van Laar-type equation. Ind. Eng.
Chem., 50, 403—412 (1958).
190. Bondi A. Polymer equilibria, Storvick T. S. and San-
dler S. I. (eds.). Phase Equilibria and Fluid Properties in
the Chemical Industry. ACS Symposium Series, 60, 118—
140 (1977).
191. Boston J. E, Mathias P. M. Phase equilibria in a third
generation process simulator. Phase Equilibria and Fluid
properties in the Chemical Industry, 2nd Int. Conf., Ber-
lin, March 17—21, 1980, EFCE DECHEMA 1980.
192. Boublik T Progress in statistical thermodynamics ap-
plied to fluid phase. Fluid Phase Equilibria, I, 37—87
(1977).
193. Bradley G., Marsh J. K. The system anthracene-phenan-
threne. J. Chem. Soc. (London), 136—138, 650—652
(1933).
194. Brancker A. V., Hunter T. G., Nash A. W. The quater-
nary system acetic acid-chloroform-acetone-water at
25C. J. Phys. Chem., 683—698 (1940).
195. Brandani V. Use of infinite-dilution activity coefficients
for predicting azeotrope formation at constant tempera-
ture and partial miscibility in binary liquid mixtures. Ind.
Eng. Chem. Fundamen., 13, 154—156 (1974); 14, 73
(1975).
196. Brandani V., Di Giacomo G., Foscolo P. U. Isothermal
vapor-liquid equilibria for the water-formaldehyde sys-
tem: A predictive thermodynamic model. Ind. Eng.
Chem. Process Des. Dev., 19, 179—185 (1980).
197. Brandani V., Prausnitz J. M. Thermodynamics of gas
solubility in liquid solvents and solvent mixtures. Fluid
Phase Equilibria, 7, 259—274 (1981).
198. Breedveld G. J. E, Prausnitz J. M. Thermodynamic
properties of supercritical fluids and their mixtures at
very high pressures. AIChE Journal, 19, 783—796 (1973).
199. Brelvi S. W. Fugacities of supercritical hydrogen, helium,
nitrogen and methane in binary liquid mixturs. Ind. Eng.
Chem. Process Des. Dev., 19, 80—84 (1980).
200. Brielles J., Dedit A., Lallemand M., Le Neindre B„
Lerou Y„ Vermese J, Vidal D. Equation of state of gases
at high pressures and low or moderate temperatures,
Neindre B. Le and Vodar B. (eds.). Experimental Ther-
modynamics. Vol. 2, 347—383, Butterworths, 1975.
201. Бриль Ж. А., Мозжухин А. С., Петлюк Ф. В., Сера-
фимов Л. А. «Математическое моделирование при
помощи ЭВМ равновесия жидкость — жидкость —
пар многокомпонентных смесей», Журн. физ. хим.,
47, ч. I, 2614—2617, ч. II, 2714—2717, ч. III
2771—2773, ч. IV 2774—2775 (1973).
202. Brinkley S. R. Note on the conditions of equilibrium for
systems of many constituents. J. Chem. Physics, 14,
563—564 (1946).
203. Brinkman N. D., Tao L. C., Weber J. H. The calculation
of the parameters for the Wilson equation for a ternary
system. Ind. Eng. Chem. Fundamen., 13, 156—157
(1974).
204. Brown G. G., Souders M., Jr., Smith R. L. Pressure-
volume-temperature relations of paraffin hydrocarbons.
Ind. Eng. Chem., 24, 513—515 (1931).
205. Bruin S. Activity coefficient relations in miscible and
partially miscible multicomponent systems. Ind. Eng.
Chem. Fundamen., 9, 305—314 (1970).
206. Brule M. R., Lin С. T, Lee L. L., Starling К. E. Mul-
tiparameter corresponding-states correlation of coal-
fluid thermodynamic properties. AIChE Journal, 28,
616—625 (1982).
207. Byer S. M., Gibbs R. E„ Van Ness H C. Vapor-liquid
equilibrium. II: Correlations from P-x data for 15 sys-
tems. AIChE Journal, 19, 245—252 (1973).
208. Буданцева Л. С, Лестева T M. «Метод расчета со-
става паровой фазы по концентрационной зависимо-
сти температур кипения в бинарных системах»,
Журн. физ. хим., 50, 8, 1687—1690 (1976).
209. Cajander В. С., Hipkin Н G., Lenoir J. М. Prediction of
equilibrium ratios from nomograms of improved accura-
cy. J. Chem. Eng. Data, 5, 251—259 (1960).
210. Carli A. A correlation for enthalpy of petroleum frac-
tions. Chemical Processing, 87—88 (April 1974).
211. Carlson H. C, Colburn A. P. Vapor-liquid equilibria of
nonideal solutions. Ind. Eng. Chem., 34, 581—589
(1942).
Литература 641
212. Carnahan N. E, Starling К. E. Equation of state for
nonattracting spheres. J. Chem. Physics, 51, 635—636
(1969).
213. Carnahan N. E, Starling К. E. Intermolecular repulsions
and the equation of state for fluids. AIChE Journal, 18,
1184—1189 (1972).
214. Carniti P, Cori L., Ragaini V. A critical analysis of the
Hand and Othmer-Tobias correlations. Fluid Phase
Equilibria, 2, 39—47 (1978).
215. Carroll B. On the use of Jacobians in thermodynamics.
J. Chem. Education, 42 (4), 218—221 (1965).
216. Castillo J., Grossmann I. E. Computation of phase and
chemical equilibria. Computers and Chemical Engineer-
ing, 5, 99—108 (1981).
217. Chang S.-D, Yu C.-Y. A generalized virial equation of
state and its application to vapor-liquid equilibria at low
temperatures. Advances in Cryogenic Engineering, 17,
255—268 (1972).
218. Chao К. C, Seader J. D. A general correlation of vapor-
liquid equilibria in hydrocarbon mixtures. AIChE Jour-
nal, 7, 598—605 (1961).
219. Chen С. C, Britt H. I., Boston J. E, Evans L. B. Two new
activity coefficient models for the vapor-liquid equilibri-
um of electrolyte systems. In: Newman S. A. (ed.), Ther-
modynamics of Aqueous Systems with Industrial
Applications. ACS Symposium Series 133, Am. Chem.
Soc. (1980).
220. Cheng S.-L, Chan T. L. Estimation of Wilson parameters
by non-linear regression. The Chemical Engineering
Journal, 4, 282—286 (1972).
221. Chiu J. Visual observation in differential thermal analy-
sis. Analytical Chemistry, 35, 933—934 (1963).
222. Christiansen L. J., Michelsen M. L., Fredenslund A. Suc-
cessive approximation in distillation calculations using
the Soave-Redlich-Kwong equation of state. 12th Sympo-
sium on Computer Applications in Chemical Engineer-
ing (1979) (CACE).
223. Chueh P. L., Prausnitz J. M. Vapor-liquid equilibria at
high pressures: Vapor-phase fugacity coefficients in non-
polar and quantum-gas mixtures. Ind. Eng. Chem. Fun-
damen., 6, 492—498 (1967).
224. Chueh P L„ Prausnitz J M. Calculation of high-pressure
vapor-liquid equilibria. Ind. Eng. Chem., 60, 34—52
(1968).
225. Chung W. K., Haman S. E. M„ Lu B. C-Y. A modified
Redlich-Kwong equation of state capable of representing
the liquid state. Ind. Eng. Chem., Fundamen., 16,
494—495 (1977).
226. Clark E G., Koppany C. R. Method tests V/L correla-
tions. Hydrocarbon Processing, 57 (11), 282—286 (1978).
227. Clark P. E., Clow A., Easter brook E. K., Haendler H. M.,
IddlesH. A. Ternary systems with ammonium nitrate and
guanidine nitrate. J. Phys, and Colloid Chemistry, 53,
1009—1015 (1949).
228. Colburn A. P In: Chemical Engineers Handbook, 528
(1950).
229. Collatz L. In: Handbuch der Physik, 2, 353 (1955).
230. Comings E. W. High Pressure Technology, McGraw-Hill,
1956.
231. Cooper H. W., Goldfrank J. C. B-W-R constants and new
correlations. Hydrocarbon Processing, 46, (12), 141—146
(1967).
232. Cope J. Q., Lewis W. K., Weber H. C. Generalized ther-
modynamic properties of higher hydrocarbon vapors.
Ind. Eng. Chem., 28, 887—892 (1931).
233. Corti M., Degiorgio V. Investigation of aggregation
phenomena in aqueous sodium dodecyl sulfate solutions
at high NaCl concentration by quasielastic light scatter-
ing. In K. L. Mittal (ed.), Solution Chemistry of Surfac-
tants, Vol. 1, 377—390. Plenum Press (1979).
234. Costa E., Sotelo J. L., Calleja G., Marron C. Adsorption
of binary and ternary hydrocarbon gas mixtures on acti-
vated carbon: Experimental determination and theoreti-
cal prediction of the ternary equilibrium data. AIChE
Journal, 27, 5—12 (1981).
235. Coward L, Gale S. E, Webb D. R. Process engineering
calculations with equatiolns of state. Trans. I. Chem. E.,
56, 19—27 (1978).
236. Coward I., Webb D. R. Computational difficulties with
the Chao and Seader correlation. Computers and Chemi-
cal Engineering, 2, 177—187 (1978).
237. Cox J. D, Lawrenson I. J. The PVT behavior of single
gases, M. L. McGlashan (ed.). Chemical Thermodynam-
ics, Vol. 1, A Specialist Periodical Report, 162—203. The
Chemical Society, London, 1973.
238. Cragoe C. S. in: International Critical Tables, III, 228
(1928).
239. Cysewski G. R., Prausnitz J- M. Estimation of gas solu-
bilities in polar and nonpolar solvents. Ind. Eng. Chem.,
Fundamen., 15 304—309 (1976).
240. Dale G..H. in: Encyclopedia of Chemical Processing and
Design, 13, 464, Dekker (1981).
241. Danner R.P, Nicoletti M. P, Al-Ammer R. S. Determina-
tion of gas mixture adsorption equilibria by the tracer-
pulse technique. Chem. Eng. Science, 35, 2129—2133
(1980).
242. Davidson D. W. Clathrate hydrates. Water, a Comprehen-
sive Treatise, edited by F. Franks, Vol. 2, Chap. 3, Plenum
Press, 1973.
243. Deam J. R„ Maddox R. N. How to figure three-phase
flash. Hydrocarbon Processing, 48, (7), 163—164 (1969).
244. Deaton W. M., Frost E. M. Gas Hydrates. U. S. В. M.
Monograph 8 (1946).
245. Deiters U, Schneider G. M. Fluid mixtures at high pres-
sures: Computer calculations of the phase equilibria and
the critical phenomena in fluid binary mixtures from the
Redich-Kwong equation of state. Ber Bunsenges Physik
Chemie, 80, 1316—1321 (1976).
246. de Ligny C. L., Van der Veen N. G., Van Houwellingen
J. C. Correlation and prediction of solubility and entropy
of solution of gases in liquids by means of factor analy-
sis. Ind. Eng. Chem. Fundamen., 15, 336—340 (1976).
247. Deming W. E., Shupe L. E. Some physical properties of
compressed gases. I. Nitrogen, Physical Review, Second
Series, 37, 638—654 (1931).
248. De Santis R., Gironi E, Marrelli L. Vapor-liquid
equilibrium from a hard-sphere equation of state. Ind.
Eng. Chem. Fundamen., 15, 183—189 (1976).
249. De Santis R., Grande B. An equation for predicting third
virial coefficients of nonpolar gases. AIChE Journal, 25,
931—938 (1979).
250. De Santis R., Marrelli L., Muscetta P. N. Liquid-liquid
equilibria in water-aliphatic alcohol system in the
presence of sodium chloride. The Chem. Eng. Journal,
11, 207—214 (1976).
251. Dincer S., Bonner D. C, Elefritz R. A. Vapor-liquid
equilibria in the benzene-polybutadiene-cyclohexane sys-
tem. Ind. Eng. Chem. Fundamen., 16, 54—59 (1979).
252. Djordjevic B. D., Mihajlov A. N., Grozdanic D. K.,
Tasic A. Z., Horvath A. L. Applicability of the Redlich-
Kwong equation of state and its modifications to polar
gases. Chem. Eng. Science, 32, 1103—1107 (1977).
253. Djordjevic В D., Mihajlov-Dudukovic A. N., Tasic A. Z.
Correlation of second virial coefficients of polar gases
by Redlich-Kwong equation of state. AIChE Journal, 26,
858—862 (1980).
354. Домнина E. В., Захаров H. Д. «Термодинамический
метод предсказания критических параметров трой-
ных систем», Журн. физ. хим., 52, 1427—1429 (1978).
255. Donohue М. D., Prausnitz J М. Statistical ther-
modynamics of solutions in natural gas and petroleum
refining Research Report RR-26 (1977), Gas Processors
Association, 1812 First Place, TUlsa, Oklahoma 74103.
AIChE Journal, 24, 849—860 (1978).
256. Douslin D. R. The pressure, volume and temperature
properties of fluids, International Review of Science,
Physical Chemistry. Series 2, Vol. 10: Thermochemistry
and Thermodynamics, Skinner H. A. (ed.), 191—246
(1975).
257. Dowling G. R„ Todd W. G. Comparing vapor-liquid
equilibrium correlations. Chemical Engineering, 80 (6),
115—120 (March 19, 1973).
258. Drahos J., Wichterle I., Hala E. A generalized method
for calculation and prediction of vapor-liquid equilibria
at high pressures. Fluid Phase Equilibria, 1, 173—184
(1978).
259. Eagle S„ Scott J. W. Liquid phase adsorption equilibria
and kinetics. Ind. Eng. Chem., 42, 1287—1294 (1950).
260. Eckert C. A., Newman В A., Nicolaides G. L.,
Long T. C. Measurement and application of limiting ac-
tivity coefficients. AIChE Journal, 27, 33—40 (1981).
261. Eckert C. A., Thomas E. R., Johnston К. P. Non-
electrolyte solutions: State of the art review. Proc. 2nd
International Conf, on Phase Equilibria and Fluid
Properties in the Chemical Industry. West Berlin, March
1980, DECHEMA.
262. Eckert C. A., Thomas E. R., Newman В A., Nicolaid-
es G. L. Limiting activity coefficients from differential
ebulliometry. J. Chem. Eng. Data, 27, 233—240 (1982).
263. Edmister W. C. Pseudo-critical for mixtures. Petroleum
Refiner, 27 (4), 134—142 (1948).
264. Edmister W. C. Convergence correction to vapor-liquid
equilibrium ratios. Pet. Refiner, 28 (9), 95—102 (Septem-
ber 1949).
265. Edmister W. C. Compressibility factors and equations of
state. Petroleum Refiner, 37 (4), 173—179 (1958).
266. Edmister W. C. Isothermal pressure corrections to the en-
thalpy and entropy. Hydrocarbon Processing, 46 (4),
165—170 (1967).
267. Edmister W. C. Compressibility factors and fugacity
coefficients from the Redlich-Kwong equation of state.
Hydrocarbon Processing, 47 (9), 239—244 (1968).
268. Egan C. J., Luthy R. V. Separation of xylenes: Selective
solid compound formation with carbon tetrachloride,
Ind. Eng. Chem., 47, 250—253 (1955).
269. Eggers R., Tschiersch R. Development and design of
plant for high-pressure extraction of natural products. In:
Schneider G. M., Stahl E. and Wilke G. (eds.), Extraction
with Supercritical Gases, 176—188, Verlag Chemie
(1980).
270. Ekwall P. Composition, properties and structures of li-
quid crystalline phases in systems of amphilic com-
pounds, Brown G. H. (ed.). Advances in Liquid Crystals,
Vol. 1, 1—142 (1975).
271. Ellis S. R. M. Vapour phase extraction processes. British
Chem. Eng., 16, 358—361 (1971).
272. Ellis S. R. M., Jonah D. A. Prediction of activity coeffi-
cients at infinite dilution. Chem. Eng. Science, 17,
971—976 (1962).
273. Elwell R. H, Welch L. M. Rectification in ternary sys-
tems containing azeotropes. Ind. Eng. Chem., 37,
1224—1231 (1945).
274. Elshayal I. M., Lu В G-Y. Prediction of vapor-liquid
equilibria by means of a modified Clausius equation of
state. Canad. J. Chem. Eng., 51, 76—81 (1973).
275. Elshayal I. M., Lu B. C.-Y. Measurement of total pres-
sures for ethylene-propane mixtures. Canad. J. Cheip.
Eng., 52, 83—87 (1975).
276. Epstein L. F. Redlich-Kwong equation of state: Exact
critical constant relations. Chem. Eng. Science, 31, 87
(1976).
277. EveleinK. A., Moore R. G. Prediction of phase equilibria
in sour natural gas systems using the Soave-Redlich-
Kwong equation of state. Ind. Eng. Chem. Process Des.
Dev., 18, 618—624 (1979).
278. Ewell R. H, Harrison J. M., Berg L. Azeotropic distilla-
tion. Ind. Eng. Chem., 36, 871—875 (1944).
279. Ezekwe J. Ph. D. Thesis, University of Kansas (1982).
280. Fair J. Sorption processes. Chem. Eng., 76 (14), 90—110
(July 14, 1969).
281. Ferrell R. A. in: Gores W. S. and Chilton F. Conference
on Fluctuations in Superconductors, Stanford Research
Institute (1968).
282. Findlay R. A., Weedman J. A. Separation and purifica-
tion by crystallization. Advances in Petroleum Chemistry
and Refining, Vol. 1. 119—208 (1958).
283. Fishtine 5. H Estimates of saturated fluid densities and
critical constants. Ind. Eng. Chem. Fundamen., 2,
148—155 (1963).
284. Francis A. W. Ternary systems of liquid carbon dioxide
J. Phys. Chem., 50, 1099—1114 (1954).
285. Francis A. W. Ternary systems with three separate bino-
dal curves. J. Phys. Chem., 60, 20—27 (1956).
286. Fredenslund A., Gmehling J., Michelsen M. L., Rasmus-
sen P, Prausnits J. M. Computerized design of mul-
ticomponent distillation columns using the UNIFAC
group contribution method for calculation of activity
coefficients. Ind. Eng. Chem. Process Des. Dev., 16,
450—462 (1977).
287. Fredenslund A., Jensen T, Magnussen T, Michel-
sen M. L., Rasmussen P. Phase equilibria, flash and dis-
tillation calculations. Computer-Aided Process Plant
Design, edited by M. E. Leesley, Gulf Publishing Compa-
ny (1982).
288. Fredenslund A., Jones R. L., Prausnitz J- M. Groupcon-
tribution estimation of activity coefficients in nonideal
liquid mixtures. AIChE Journal, 21, 1086—1098 (1975).
289. Fredenslund A., Michelsen M. L., Sorensen J. M. Liquid-
liquid equilibrium calculations using activity coefficient
models. Proceedings, 2nd International Conference, Ber-
Литература 643
lin, March, 21, 1980, EFCE, DECHEMA, 433—444.
290. Fredenslund A., Rasmussen P., Michelsen M. L. Recent
progress in the computation of equilibrium ratios. Sym-
posium on Distillation, AIChE, 87th National Meeting,
Boston, August 19—22, 1979.
291. Fredenslund A., Rasmussen R., Michelsen M. L. Recent
progress in the computation of equilibrium ratios. Chem.
Eng. Commun., 4, 485—500 (1980).
292. Fredenslund A., Rasmussen P., Mollerup J., Ther-
mophysical and transport properties for chemical process
design. Foundations of Computer-Aided Chemical
Process Design, Mah R. S. H. and Seider W. D. (eds.),
Vol. 2, 1—29 (1980).
293. Friman R., Daniellson I. Lamellar mesophase with high
contents of water — X-ray investigations of sodium
octanoate-decanol-water system. J. Colloid Interface Sci.,
86, 501—514 (1982).
294. Fuller G. G. A modified Redlich-Kwong equation of state
capable of representing the liquid state. Ind. Eng. Chem.
Fundamen., 15, 254—257 (1976).
295. Gangoli N., Thodos G. Liquid fuels and chemical feed-
stocks from coal by supercritical gase extraction: A
review. Ind. Eng. Chem. Prod. Res., 16, 208—216 (1977).
296. Gas Processors Association. GPA Peng-Robinson Pro-
grams, including new C7 + Subroutine. Gas Processors
Association, 1812 First Place, 15 East Fifth Street, Tulsa,
Oklahoma 74103.
297. Gautam R., Seider W. D. Computation of phase and
chemical equilibrium. I: Local and constrained minima
in Gibbs free energy; II: Phase splitting; III: Electrolytic
solutions. AIChE Journal, 25, 991—1015 (1979).
298. George B., Brown L. P., Farmer С. H, But hod R, Man-
ning F. S. Computation of multicomponents, multiphase
equilibrium. Ind. Eng. Chem. Process Des. Dev., 15,
372—377 (1976).
299. Gere J. M„ Weaver W. Matrix Algebra for Engineers. Van
Nostrand, 1965.
300. Germann F. E. E., Germann E. P. Line coordinate
representation of solubility curves. Ind. Eng. Chem., 36,
93—96 (1944).
301. Ghosh S. K., Chopra S. J. Activity coefficients from the
Wilson equation. Ind. Eng. Chem. Process Des. Dev., 14,
304—308 (1975).
302. Gibbs R. E., Van Ness H. C. Vapor-liquid equalibria
from total-pressure measurements: A new apparatus.
Ind. Eng. Chem. Fundamen., 11, 410—413 (1972).
303. Gibbons R. M. The equation of state of neon between
and 70 K. Cryogenics, 9 (8), 251—260 (1969).
304. Gibbons R. M. Equations for the second virial coeffi-
cient of polar molecules. Cryogenics, 14 (7), 399—403
(1974).
305. Gibbons R. M., Coulthurst J. R., Farrell D., Gough D.
Gillett. Industrial uses of thermodynamic data. Proceed-
ings, NPL Conference Chemical Thermodynamic Data
on Fluids and Mixtures. IPC Science and Technology
Press (1979).
306. Gillespie D. T. Vapor-liquid equilibrium still for miscible
liquids. Ind. Eng. Chem. Analytical Ed., 18, 575—577
(1946).
307. Gmehling J. G., Anderson T. E, Prausnitz J- M. Solid-
liquid equilibria using UNIFAC. Ind. Eng. Chem. Funda-
men., 17, 269—273 (1978).
308. Gmehling J., Rasmussen P., Fredenslund F. Vapor-liquid
equilibria by UNIFAC group contribution revision and
extension. 2. Ind. Eng. Chem. Process Des. Dev., 21,
118—127 (1982).
309. Gmelhling J., Onken U Calculation of activity coeffi-
cients from structural group contributions. Int. Chem.,
Eng., 19, 566—570 (1979).
310. Goldwasser S. R. Basis for calculating equilibrium gas
composition on a digital computer. Ind. Eng. Chem., 51,
595—596 (1959).
311. Gomez-Nieto M., Thodos G. Benedict-Webb-Rubin
parameters for ethanol. Canad. J. Chem. Eng., 54,
438—440 (1976).
312. Gomez-Nieto M., Thodos G. Generalized vapor pressure
equation for nonpolar substances. Ind. Eng. Fundamen.,
17, 45—52 (1978).
313. Gomez-Nieto M„ Thodos G. Vapor-liquid equilibrium
behavior for the propane-acetone system at elevated pres-
sures. Chem. Eng. Science, 33, 1589—1595 (1978).
314. Gothard E A., Codrea Ciobanu M. E, Breban D. G.,
Bucur С. I., Sorescu G. V. Predicting the parameters in
the Wilson equations for activity coefficients in binary
hydrocarbon systems. Ind. Eng. Chem. Process Des.
Dev., 15, 333—337 (1976).
315. Graboski M. S., Daubert T E. A modified Soave equa-
tion of state for phase equilibrium calculations.
1: Hydrocarbon systems, Ind. Eng. Chem. Process Des.
Dev., 17, 443—448 (1978). 2: Systems containing CO2,
H2S, N2 and CO. Ibid, 448—454 (1978). 3: Systems con-
taining hydrogen. Ibid, 18, 300—306 (1979).
316. Gray R. D. Industrial experience in applying the Redlich-
Kwong equation to vapor-liquid equilibria. Chao К. C.
and Robinson R. L. (eds.); Equations of state in en-
gineering and research, 253—270. Advances in Chemistry
Series 182. Am. Chem. Soc. 1979.
317. Gray R. D. Case studies of difficult industrial problems:
Computation of phase equilibria in the critical region.
Proc., 2nd Int. Conf., Berlin, March 21, 1980, EFCE,
DECHEMA, 919—928.
318. Grayson H. G., Streed C. W. Vapor-liquid equilibrium
for high temperature, high pressure hydrogen-hydro-
carbon mixtures. Sixth World Petroleum Congress
Proceedings, Sect. Ill, 233—245, (1963).
319. Griffiths R. B, Wheeler J. C. Critical points in multicom-
ponent systems. Physical Review, A2, 1047—1063 (1970).
320. Grossmann W. E., Davidson J. Computation of restricted
chemical equilibria. Computers and Chem. Eng., 6,
181—184 (1982).
321. Guerreri G. Vapour-liquid equilibria in non-polar mul-
ticomponent systems. British Chemical Engineering, 15,
927—931 (1970).
322. Gunn R. D. Corresponding state: Theoretical develop-
ment for mixtures. AIChE Journal, 18, 183—188 (1972).
323. Hadden S. T, Grayson H. G. New charts for hydrocar-
bon vapor-liquid equilibria. Hydrocarbon Processing, 40
(9), 207—218 (1961).
324. Hala E. Vapor-liquid equilibrium in the critical region:
Concentration limits of binary systems. Ind. Eng. Chem.
Fundamen. 14, 136—137 (1975).
325. Halm R. L., Stiel L. I. A fourth parameter for the vapor
pressure and entropy of vaporization of polar fluids.
AIChE Journal, 13, 351—355 (1967).
326. Hammick D. L„ Hills G. M., Howard J. The composition
of the compounds of picryl chloride and of a o-trinitro-
benzene and benzene. J. Chem. Soc. London, 1530—1532
(1932).
327. Hamam S. E., Chung W. K., Elshayal I. M., Lu R C.-Y.
Generalized temperature-dependent parameters of the
Redlich-Kwong equation of state for vapor-liquid
equilibrium calculations. Ind. Eng. Chem. Process Des.
Dev., 16, 51—59 (1977).
328. Handa Y. P., Benson G. R. Volume changes on mixing
two liquids: A review of the experimental techniques and
the literature data. Fluid Phase Equilibria, 3, 185—249
(1979).
329. Hankinson R. W., Coker T. A., Thomson H. G. Get ac-
curate LNG densites with COSTALD. Hydrocarbon
Processing, 61(4), 207—208 (1982).
330. Hankinson R. W, Langfitt B. D., Tassios D. P. A single
parameter equation for the prediction of multicompo-
nent vapor-liquid data from binary isobaric data. Canad.
J. Chem. Eng., 50, 511—514 (1972).
331. Hankinson R. W„ Thomson G. H. A new correlation for
saturated densities of liquids and their mixtures. AIChE
Journal, 25, 653—663 (1979).
332. Hansk R. W., O’Neill T K., Christensen J. J. The predic-
tion of vapor-liquid equilibrium from heat of mixing for
binary hydrocarbon-alcohol mixtures. Ind. Eng. Process
Des. Dev., 18, 408—414 (1979).
333. Hansen G., Beerbower A. Solubility parameters. Ency-
clopedia of Chemical Technology, Supplement (1971).
334. Harmens A., Knapp H Three-parameter cubic equation
of state for normal substances. Ind. Eng. Fundamen., 19,
291—294 (1980).
335. Hayden J. G., O’Connell J. P. A generalized method for
predicting second virial coefficients. Ind. Eng. Chem.
Process Des. Dev., 14, 209—216 (1975).
336. Hecht G., Holste C. Limitations of density calculations
with reduced equations of state (in German). Chem.
Technik, 17, 518—524 (1965).
337. Helpinstill J. G., Van Winkle M. Prediction of infinite di-
lution activity coefficients for polar-polar binary sys-
tems. Ind. Eng. Chem. Process Des. Dev., 7, 213—220
(1968).
338. Herington E. E G. Tests for consistency of experimental
isobaric vapor-liquid equilibrium data. Institute of
Petroleum Journal, 37, 457—470 (1951); symmetrical-
area tests for the examination of the reliability of vapour-
liquid equilibrium data. Inst. Chem. Engineers (Lon-
don), International Symposium on Distillation, Septem-
ber 8—10, 1969, 3, 17—24.
339. Herskovitz Kf., Gottlieb M. UNIFAC group contribution
method for silicone compounds. Ind. Eng. Chem.
Process Des. Dev., 20, 407—409 (1981).
340. Heidemann R. A. Predict three-phase equilibria.
Hydrocarbon Processing, 53 (11), 167—170 (1974).
341. Heidemann R. A. Three-phase equilibria using equations
of state. AIChE Journal, 20, 847—855 (1974).
342. Heidemann R. A. Use of infinite-dilution activity coeffi-
cients for predicting azeotrope formation at constant
temperature and partial miscibility in binary liquid mix-
tures. Ind. Eng. Chem. Fundamen., 14, 72—73 (1975).
343. Heidemann R. A., Khalil A. M. The calculation of criti-
cal points. AIChE Journal, 26, 769—779 (1980).
344. Hildebrand J. H, Lamoreaux R. H Solubility of gases
in liquids: Fact and theory. Ind. Eng. Chem. Fundamen.,
13, 110—115 (1974).
345. Hildebrand J. H, Sharma J. N. The activities of molten
alloys of thallium with tin and lead. JACS, 462—471
(1929).
346. Hill A. E. The mutual solubility of liquids. I. Mutual
solubility of ethyl ether and water. II. The solubility of
water in benzene. JACS, 45, 1143—1155 (1923).
347. Hiranuma M. A new expression similar to the three-
parameter Wilson equation. Ind. Eng. Chem. Funda-
men., 13, 219—222 (1974).
348. Hiranuma M. The contribution of non-neighbor pairs to
free energy of mixing. J. Chem. Eng. Japan, 8, 69—77
(1975).
349. Hiza M. J., Duncan A. G. A correlation for the predic-
tion of interaction energy parameters for mixtures of
small molecules. AIChE Jornal, 16, 733—737 (1970).
350. Hlavaty K. Correlation of the binodal curve in a ternary
liquid mixture with one pair of immiscible liquids. Coll.
Czech. Chem. Commun., 37, 4005—4007 (1972).
351. Hlavaty K. Semirational method for the construction of
equations of state for liquids and gases. Coll. Czech.
Chem. Commun., 39, 2927—2934 (1974).
352. Holder G. H, Hand J. H Multiple-phase equilibria in
hydrates from methane,' ethane, propane and water.
AIChE Journal, 28, 440—447 (1982).
353. Holland R. E. Correlation of vapor-liquid equilibria us-
ing solubility parameters. Ph. D. thesis, Pennsylvania
State University (1971).
354. Holmes M. J., Van Winkle M. Prediction of ternary
vapor-liquid equilibria from binary data. Ind. Eng.
Chem., 62, 21—31 (1970).
355. Holste J. C, Watson M. Q., Bellomy M. T, Eubank P. T,
Hall K. R. Determination of interaction second virial
coefficients: He-CO? system. AIChE Journal, 26,
954—964 (1980).
356. Horry S. E., Prausnitz J. M. Molecular thermodynamics
of monolayer gas adsorption on homogeneous and hete-
rogeneous solid surfaces. AIChE Symposium Series, 74,
3—9 (1967).
357. Норке S. W. Application of equations of state in Exxon’s
production operations. ACS Symposium Series, 60,
221—223 (1977).
358. Horvath A. L. Redlich-Kwong equation of state: review
for chemical engineering calculations. Chem. Eng.
Science, 29, 1334—1340 (1974).
359. Horvath C., Lin H.-J. A simple three-parameter equation
of state with critical compressibility-factor correlation.
Canad. J. Chem. Eng., 55, 450—456 (1977).
360. Hoy K. L. New values of the solubility parameters from
vapor pressure data. J. Paint Technology, 42, No. 541,
76—118 (1970).
361. Hrynakowski K, Szmyt M. Solid-liquid equilibria in sys-
tems in which incongruent melting binary compounds
occur. Ill (in German). Z. Physik Chem., 175A, 83—98
(1935).
362. Hsu E. C-H, Johnson J. F. Prediction of eutectic tem-
peratures, compositions and phase diagrams for binary
mesophase systems. Mol. Cryst. Liq. Cryst., 27, 95—104
(1974).
363. Hulme D S., Raynes E. P. Eutectic mixtures of nematic
4'-substituted 4-cyanobiphenyls. J. C. S. Chem. Comm.,
98—99 (1974).
364. Huron M. J. Use of the Soave equation and of the stabili-
ty conditions for calculating the critical points of mix-
Литература 645
tures. Chem. Eng. Science, 31, 837—839 (1976).
365. Huron M. J., Dufour G. N., Vidal J., Vapour-liquid
equilibrium and critical locus curve calculations with the
Soave equation for hydrocarbon systems with carbon di-
oxide and hydrogen sulfide. Fluid Phase Equilibria, 1,
247—265 (1978).
366. Huron M. J., Vidal J. New mixing rules in simple equa-
tions of state for representing vapour-liquid equilibria of
strongly non-ideal mixtures. Fluid Phase Equilibria, 3,
255—271 (1979).
367. Hutchins R. A. Liquid phase adsorption — Maximizing
performance. Chem. Eng., 87 (4), 101—110 (February 25,
1980).
368. Inoue M., Azumi K., Suzuki N. A new vapor pressure
assembly for static vapor-liquid equilibrium. Ind. Eng.
Chem. Fundamen., 14, 312—314 (1975).
369. Ishikawa T, Chung W. K., Lu B. C.-Y. A cubic perturbed,
hard sphere equation of state for thermodynamic proper-
ties and vapor-liquid equilibrium calculations. AIChE
Journal, 26, 372—378 (1980).
370. JaneckeE. The system НгО + CO2 + NH3. Z. fur Elek-
trochem., 35, 716—727 (1929).
371. Jelinek J., Hlavacek V. Compute boiling points faster
Hydrocarbon Processing, 50 (8), 135—136 (1971).
372. Jenkins J. D., Gibson-Robinson M. Vapour-liquid
equilibrium in systems with association in both phases:
The simultaneous evaluation of liquid phase models and
thermodynamic consistency testing for the system acetic
acid-toulene. Chem. Eng. Science, 32, 931—938 (1977).
373. Joffe J. Fugacities in gas mixtures. Ind. Eng. Chem., 40,
1738—1741, 2439—2442 (1948).
374. Joffe J. Combining rules for the third parameter in the
pseudocritical method for mixtures. Ind. Eng. Chem.
Fundamen., 10, 532—533 (1971).
375. Joffe J. Vapour-liquid equilibria by the pseudocritical
method. Ind. Eng. Chem. Fundamen., 15, 298—303
(1976).
376. Joffe J. Vapor-liquid equilibria and densities with the
Martin equation of state. Ind. Eng. Chem. Process Des.
Dev., 20, 168—172 (1981).
377. Joffe J., Joseph H, Tassios D. Vapor-liquid equilibria
with a modified Martin equation. Newman S. A. (ed.).
Chemical Engineering Thermodynamics. 211—220
(1982), Ann. Arbor Science.
378. Joffe J., Zudkevitch D. Prediction of critical properties
of mixtures: Regorous procedure for binary mixtures.
AIChE Symposium Series, 81, 43—51 (1967).
379. Joffe J., Zudkevitch D. Prediction of liquid-phase enthal-
pies with the Redlich-Kwong equation of state. Ind. Eng.
Chem. Fundamen., 9, 545—548 (1970).
380. Johnson D. W., Colver С. P. Mixture properties by com-
puter Hydrocarbon Processing, 47 (12), 79—83 (1968); 48
(1), 127—133 (1969).
381. Jolls K. R., Willers G. P. Computer generated phase dia-
grams for ethylene and propylene. Cryogenics, 18 (6),
329—336 (1978).
382. Jolls K. R., Willers G. P, Jenson L. D. The computer
generation of thermodynamic phase diagrams. Trans, of
the Computers in Education Division of ASEE, 8 (10)
(1976).
383. Kalra H, Robinson D. R. Vapor-liquid equilibrium in a
six-component simulated sour natural gas system at
subambient temperatures. Fluid Phase Equilibria, 3,
133—144 (1979).
384. Kandiner H. J., Brinkley S. R. Jr. Calculation of complex
equilibrium relations. Ind. Eng. Chem., 42, 850—855,
1526 (1950).
385. Kappallo W., LundN., Schaffier K. Intermolecular forces
between equal and different molecules from the virial
coefficient. Z. phys. Chem. (Frankfurt), 37 (3,4)
196—209 (1963).
386. Kata K., Nagahama K., HirataM. Generalized interaction
parameters for the Peng-Robinson equation of state: car-
bon dioxide-n-paraffin binary systems. Fluid Phase
Equilibria, 7, 219—231 (1981).
387. Kato M., Chung W. K, Lu В C.-Y. Modified parameters
for the Redlich-Kwong equation of state. Canad. J.
Chem., Eng., 54, 441—445 (1976).
388. Kato M., Chung W. K., Lu В C.-Y. Binary interaction
coefficients of the Redlich-Kwong equation of state.
Chem. Eng. Science, 31, 773—776 (1976).
389. Katz D. L., Firoozabadi A. Predicting phase behavior of
condensate/crude oil systems using methane interaction
coefficients. J. Pet. Tech. 1649—1655 (November 1978).
390. KatzD. L., KurataF. Retrograde condensation. Ind. Eng.
Chem., 32, 817—827 (1940).
391. Kaul В. K., Prausnitz J. M. Second virial coefficients of
gas mixtures containing simple fluids and heavy
hydrocarbons. Ind. Eng. Chem. Fundamen., 16,
335—339 (1977).
392. Kay W. B. Density of hydrocarbon gases and vapors at
high temperature and pressure. Ind. Eng. Chem., 28,
1014—1019 (1936).
393. Kay W. R Liquid vapor phase equilibrium relations in
the ethane-n-heptane system. Ind. Eng. Chem., 30,
459—465 (1938).
394. Kehiaian H. V, Grolier J. P. E,, Kechavarz M. R., Ben-
son G. C, Kiyohara O., Handa Y. P. Thermodynamic
properties of binary mixtures containing ketones. VII:
Analysis of the properties of n-alkanone + n-alkane, and
л-alkanone+n-alkanone mixtures in terms of a quasi-
chemical group contribution model. Fluid Phase
Equilibria, 7, 95—120 (1981).
395. Kesler M. G., Lee В. I. On the development of an equa-
tion of state for vapor-liquid equilibrium calculations.
ACS Symposium Series, 60, 236—240 (1977).
396. Kesler M. G., Lee В. I., Benzing D. W., Gruz A. Method
improves convergence pressure predictions. Hydrocarbon
Processing, 48 (6), 177—179 (1969).
397. Kesler M. G., Lee R I., Sandler S. I. A third parameter
for use in generalized thermodynamic correlations. Ind.
Eng. Chem. Fundamen., 18, 49—54 (1979).
398. KikicL, Alessi P. Liquid-liquid equilibrium for the activi-
ty coefficient determination: Effect of mutual solubility
between binary systems and auxiliary solvents. Canad.
J. Chem. Eng., 55, 78—81 (1977).
399. Kikic I., Allessi P, Rasmussen P, Fredenslund A. On the
combinatorial part of the UNIQUAC and UNIFAC
models Canad. J. Chem. Eng., 58, 253—258 (1980).
400. King M. R, Al-Najjar J., Kassim K. The solubilities of
carbon dioxide, hydrogen sulfide and propane in some
normal alkane solvents. I: Experimental determinations
in the range 15—70 C and comparison with ideal solu-
tion values; II: Correlation of data at 25 °C in terms of
solubility parameters and regular solution theory. Chem.
Eng. Science, 32, 1241—1252 (1977).
401. KingM. M., Al-Najjar H. S. H, AUK. Integration of the
Jonah-King equation to predict gas solubilities over a
range of temperatures when the solubility at one tem-
peratures is known. Chem. Eng. Science, 34, 1080—1082
(1979).
402. Knobler С. M. Volumetric properties of gaseous mix-
tures, McGlashan M. L. (ed.). Chemical Thermodyna-
mics, Vol. 2, a Specialist Periodical Report, 199—237.
The Chemical Society, London (1978).
403. Knox W. G., Hess M., Jones G. E, Smith H. B. The hy-
drate process. Chem. Eng. Progress, 57 (2), 66—71 (1961).
404. Kobayashi R., Katz D. L. Vapor-liquid equilibria for bi-
nary hydrocarbon-water systems. Ind. Eng. Chem., 45,
440—451 (1953).
405. Коган В. Б., Ципарис И. Н. «Определение парожид-
костного равновесия для тройных систем с химиче-
ским взаимодействием между компонентами»,
Журн. прикл. хим., 41, 10, 2517—2521 (1968).
406. Kohman G. Т., Andrews D. И. Solubility relations in iso-
metric organic compounds. V: The construction of the
ideal ternary solubility diagram and its use in analysis.
J. Phys. Chem, 29, 1317—1324 (1925).
407. Koningsveld R. Thermodynamics of polymer solutions.
Int. Chem. Eng., 19, 420—429 (1979).
408. Konstam A. H, Feairheller W. R. Jr. Calculation of solu-
bility parameters of polar compounds. AIChE Journal,
16, 837—840 (1970).
409. Krolikowski T. S. Industrial view of the state of the art
in phase equilibria, Storvick T. S. and Sandler S. I. (eds.);
62—86 ACS Symposium Series 60. American Chemical
Society (1977).
410. Kruis A., in: Landolt-Bornstein IV/4c/5 (1976).
411. Krumins A. E., Rastogi A. K, Rusak M. E., Tassios D.
Prediction of binary vapor-liquid equilibrium from one-
parameter equations. Canad. J. Chem. Eng., 58,
663—669 (1980).
412. Ku P. S., Dodge В. E Compressibility of the binary sys-
tems, helium+nitrogen and carbon dioxide+ethylene.
J. Chem. Eng. Data, 12, 158—164 (1967).
413. Kudchaker A. P. Thermodynamic properties of Refriger-
ant 500, Chao К. C. and Robinson R. L. (eds.) Equations
of state in engineerin research. Advances in Chemistry
Series 182, 305—322. Am. Chem. Soc. (1979).
414. Kula M.-R. Extraction and purification of enzymes using
aqueous two-phase systems. In: Applied Biochemistry
and Bioengineering Wingard L. B. et al. (eds.), Vol. 2,
71—195, Academic Press (1979).
415. Kummel R., Wilde G. Osmotic and activity coefficients
in the ternary system water-calcium choloride-urea at
298.15 K. Fluid Phase Equilibria, 2, 215—223 (1978).
416. Kurnik R. T, Holla S. J., Reid R. C. Solubility of solids
in supercritical carbon dioxide and ethylene. J. Chem.
Eng. Data, 26, 47—51 (1981).
417. Лабинов С. Д., Бойко H. В., Соломин H. К. «Новая
методика определения констант уравнения В—W—R
для газовых смесей», Журн. физ. хим., 41, 3, 618—621
(1967).
418. Ladurelli A. J., Eon С. Н, Guiochon G. Fallibilities in-
herent in the Wilson equation applied to systems having
a negative excess Gibbs energy. Ind. Eng. Chem. Funda-
men., 14, 191—195 (1975).
419. Lahiri A. K. Multicomponent multiphase equilibria.
Fluid Phase Equilibria, 3, 113—121 (1979).
420. Lambert J. D., Roberts G. A. H, Rowlinson J. S., Wilkin-
son V. J. Second virial coefficients of organic vapors.
Proc. Roy. Soc. London, 196A, 113—125 (1949).
421. Langmuir I. Third Colloid Symposium Monograph.
Reinhold (1925).
422. Lapina R. P. Predict water thermo properties with
BWRS. Hydrocarbon Processing, 54 (2), 115—118 (1975).
423. Larrinaga L. Graphically determining the Wilson
parameters. Chem. Eng., 88 (7), 87—91 (April 7, 1981).
424. Leach M. J. An approach to multiphase vapor-liquid
equilibria. Chemical Engineering, 84 (10), 137—140 (May
23, 1977).
425. Lee В. I., Kesler M. G. A generalized thermodynamic
correlation based on three-parameter corresponding
states. AIChE Journal, 21, 510—527 (1975).
426. Lee H. H, Warner J. C. The ternary system diphenyl-
diphenylamine benzophenone. J. Am. Chem. Soc., 55,
4474—4477 (1933).
427. Lee H. H, Warner J. C. The system biphenyl-bibenzyl-
naphthalene. Nearly ideal binary and ternary systems. J.
Am. Chem. Soc., 57, 318—321 (1935).
428. Lee S-М., Eubank P. T., Hall K. R. Truncation errors as-
sociated with the virial equation. Fluid Phase Equilibria,
1, 219—224 (1978).
429. Leesley M. E., Heyen G. The dynamic approximation
method of handling vapor-liquid equilibrium data in
computer calculations for chemical processes. In:
Computer-aided Process Plant Desing, Leesley M. E.
(ed.). Gulf Publishing Company (1982).
430. Legret D., Richon D., Renon H. Vapor liquid equilibria
up to 100 MPa: A new apparatus. AIChE Journal, 27,
203—207 (1981).
431. Iceland T. W. Equations of state for phase equilibrium
computations: Present capabilities and future trends.
Proceedings, 2nd International Conference, Berlin,
March 21, 1980, EFCE, DECHEMA, 283—334.
432. Leland T. W., Chappelear P. S. The corresponding states
principle: A review of current theory and practice. Ind.
Eng. Chem., 60, 15—43 (1968).
433. Leland T. W., Mueller W. H. Applying the theory of cor-
responding states to multicomponent systems. Ind. Eng.
Chem., 51, 597—600 (1959).
43 4.1-enoir J. M. Predict К values at low temperatures.
Hydrocarbon Processing, 48 (9), 167—172; 48 (11),
121—124 (1969).
435. Lenoir J. M., Koppany C. R. Need equilibrium ratios?
Do it right. Hydrocarbon Processing, 46 (11), 249—252
(1967).
436. I-enoir J. M., White G. A. Predicting convergence pres-
sure. Petroleum Refiner, 37 (3), 173—181 (1958).
437. Letcher T. M. Activity coefficients at inifinite dilution
from gas-liquid chromatography. In: Chemical Ther-
modynamics, McGlashan M. L. (ed.). Vol. 2 A Specialist
Periodical Report, 46—70. The Chemical Society, Lon-
don (1978).
438. Levelt-Sengers J. M. H. Critical behavior in fluids. In:
High Pressure Technology, Vol. II, Spain I. L. and
J. Paauwe (eds.) Dekker (1977).
439. Levelt-Sengers J. M. H. Critical exponents at the turn of
the century. Physica, 82A, 319—351 (1976).
440. Lewis W. K., Gilliland E. R., Chertow B., Cadogan W. P.
Литература 647
Adsorption equilibria: Hydrocarbon mixtures. Ind. Eng.
Chem., 42, 1319—1332 (1950).
441. Leyendecker W. R„ Gunn R. D. Prediction of compo-
nents fugacities and related properties of mixtures.
AIChE Journal, 18, 188—193 (1972).
442. Lin C-Т., Daubert T. E. Prediction of the fugacity coeffi-
cients of nonpolar hydrocarbon systems from equations
of state. Ind. Eng. Chem. Process Des. Dev., 17,
544—549 (1978).
443. Lin С. T, Daubert T. E. Prediction of partial molar
volume from the Lee-Kesler equation of state. AIChE
Journal, 25, 365—367 (1979).
444. Lin C.-Т., Daubert T. E. Estimation of partial molar
volume and fugacity coefficients in mixtures from the
Soave and Reng-Robinson equations of state. Ind. Eng.
Chem. Process Des. Dev., 19, 51—59 (1980).
445. Lin C.-J, Норке S. W. Application of the BWRS equa-
tion to methane, ethane, propane and nitrogen systems.
AIChE Symposium Series, 140, 37—47 (1974).
446. Lindman R, Lindblom G., Wennerstrom H, Gustav-
vson H. Ionic interactions in amphilic systems studies by
NMR. K. L. Mittal (editor), Solution Chemistry of Sur-
factants, Vol. 1, 195—227 (1977).
447. Lu B. C.-Y., Yu P., Sugie A. H. Prediction of vapor-liquid-
liquid equalibria by means of a modified regula falsi.
Chem. Eng. Science, 29, 321—326 (1974).
448. Lucas K. Calculation of properties of gases and luquids
by the molecular theory — state of knowledge and appli-
cation in chemical engineering. Ind. Chem. Eng., 18,
408—416 (1978).
449. Luft N. W. Heats of reaction. Chem. Eng., 64 (11),
235—237 (1957).
450. Luna J. L. E, de Castro F. B. Evaluations of various
modifications of the Redlich-Kwong equation. Ind.
Chem. Eng., 18, 611—626 (1978).
451. Lydersen A. L., Greenkorn R. A., Hougen O. A. General-
ized thermodynamic properties of pure fluids. Univ. Wis-
consin Eng. Exp. Sta. Rept. (October 4, 1955) (The data
are reproduced by Hougen et al. Thermodynamics, 1959).
452. McCarty R. D. A modified Benedict-Webb-Rubin equa-
tion of state for methane using recent experimental data.
Cryogenics, 14 (5), 276—280 (1974).
453. McGlashan M. L., Willamson A. G. Thermodynamics of
mixtures of «-hexane and «-hexadecane, Part 2: Vapor
pressures and activity coefficients. Trans. Faraday Soc.,
57, 588—600 (1961).
454. Maddocks R. R., Gibson 1, Williams D. F. Supercritical
extraction of coal. Chem. Eng. Progress, 75 (6), 49—55
(1979).
455. Maffiolo G., Vidal J., Asselineau L. Vapor-liquid
equilibrium constants of hydrocarbon mixtures at
moderate pressures (in French). Chem. Eng. Science, 30,
625—630 (1975).
456. Magnussen T, Rasmussen P., Fredenslund A. UNIFAC
parameter table for prediction of liquid-liquid equalibria.
Ind. Eng. Chem. Process Des. Dev., 20, 331—339 (1981).
457. Magnussen T., Sorensen J. M., Rasmussen R, Fredens-
lund A. Liquid-liquid equilibrium data: Prediction. Fluid
Phase Equilibria 4, 151—163 (1980).
458. Maher P. J., Smith B. D. Infinite dilution activity coeffi-
cient values from total pressure VLE data: Effect of
equation of state used. Ind. Eng. Chem. Fundamen., 18,
354—357 (1979).
459. Mair B. J., West haver J. W., Rossini E D. Theoretical
analysis of fractionating processes of adsorption. Ind.
Eng. Chem., 42, 1279—1286 (1950).
460. Malbrunot P. PVT relationships in gases at high pres-
sures and high temperatures, Le Neindre B., Vodar B.
(eds.), Experimental Thermodynamics, Vol. II,
383—420. Butterworths (1975).
461. Mandell L., Ekwal P. The three-components system sodi-
um caprylate-decanol-water. The phase equilibria at
20 C. Acta Polytechnica Scandinavica, Series 74, I—III
(1968).
462. Mantell C. L. Adsorption 162, McGraw-Hill (1951).
463. Mapstone G. E. Tie-line interpolation and checking.
British Chem. Eng., 15, 778—779 (1970).
464. Marina J. M., Tassios D. P. Effective local compositions
in phase equilibrium correlations. Ind. Eng. Chem.
Process Des. Dev., 12, 67—71 (1973).
465. Martin J. J. Equations of state. Ind. Eng. Chem., 59,
34—52 (1967).
466. Martin J. J. Cubic equations of state — which? Ind. Eng.
Chem. Fundamen., 18, 81—97 (1979); 19, 128—129
(1980).
467. Martinez-Ortiz J. A., Manley D. B. Direct solution of the
isothermal Gibbs-Duhem equation by an iterative
method for binary systems. AIChE Journal, 23,
393—395 (1977).
468. Martinez-Ortiz J. A., Manley D. R Direct solution of the
isothermal Gibbs-Duhem equation for multicomponents
systems. Ind. Chem. Process Des. Dev., 17, 246—351
(1978).
469. Masuoka H., Tawaraya R„ Saito S. Calculation of solid-
liquid equilibria using the modified BWR equation of
state of Lee and Kesler. J. Chem. Eng. Japan, 12,
257—262 (1979).
470. Maurer G., Prausnitz J. M. On the derivation and exten-
sion of the UNIQUAC equation. Fluid Phase Equilibria,
2, 9—99 (1978).
471. Mauri C. Unified procedure for solving multiphase-
multicomponent vapor-liquid equilibrium calculation.
Ind. Eng. Chem., Process Des. Dev., 19, 482—489 (1980).
472. Mauser H. Phase diagrams: Their determination and use
(in German). Ullmann, Enzyklopadie der technischen
Chemie, II/l. Verlag Chemie (1972).
473. Maxwell J. C. On the dynamical evidence of the molecu-
lar constitution of matter, Nature, 11, 357—359,
374—377 (1975).
474. Mecke R. Forces in Liquids (in German). Z. Elek-
trochem., 52, 269 (1948).
475. MedaniM. S., Hasan M. A. Phase equilibria calculations
with a modified Redlich-Kwong equation of state.
Canad. J. Chem. Eng., 56, 251—256 (1978).
476. Medir M., Giralt E Correlation of activity coefficients
of hydrocarbons in water at infinite dilution with
molecular parameters. AIChE Journal, 28, 341—343
(1982).
477. Meissner H. R, Kusik C. L., Dalzell W. H. Equilibrium
composition with multiple reactions. Ind. Eng. Chem.
Fundamen., 8, 659—665 (1969).
478. Mendonca J. M. M., Bett К. E. Phase equilibria in binary
mixtures of carbon tetrafluoride and «-alkanes at high
pressure. Newman S. A. (ed.), Chemical Engineering
Thermodynamics, 117—130. Ann Arbor Science (1982).
479. Mentzer R. A., Greenkorn R. A., Chao K.-C. Principle of
corresponding states and vapor-liquid equilibria of
molecular fluids and their mixtures with light gases. Ind.
Eng. Chem. Process Des. Dev., 20, 240—252 (1981).
480. Michels A., Blaisse B., Michels C. The isotherms of CO2
in the neighborhood of the critical point and round the
coexistence lines. Proc. Roy. Soc., A160, 358—375 (1937).
481. Michelsen M. L. Calculation of phase envelopes and cri-
tical points for multicomponent mixtures. Fluid Phase
Equilibria, 4, 1—10 (1980).
482. Miller D. G. Estimating vapor pressures — a comparison
of equations. Ind. Eng. Chem., 56, 46—57 (1964).
483. Miller D. G. Joule-Thomson Inversion Curve, Cor-
responding States and Simpler Equations of State. Ind.
Eng. Chem. Fundamen., 9, 585—589 (1970).
484. Mills M. B., Wills M. J., Bhirud V. L. The calculation of
density by the BWRS equation of state in process simula-
tion contexts. AIChE Journal, 26, 902—910 (1980).
485. Miller R. C., HizaM. J. Experimental molar volumes for
some LNG-related saturated liquid mixtures. Fluid Phase
Equilibria, 2, 49—57 (1978).
486. Миндович E. П. «Термический анализ и рентгено-
структурное исследование аддитивных соединений
пикриновой кислоты с циклическими углеводорода-
ми», Журн. физ. хим., 30, 5, 1082 (1956).
487. Mitchell S. A method for determining the solubility of
sparingly soluble substances. J. Chem. Soc. (London),
1333—1336 (1926).
488. Miyahara K., Sadotomo H., Kitamura K. Evaluation of
the Wilson parameters by nomographs. J. Chem. Eng.
Japan, 3, 157—160 (1970).
489. Modell M. Criteria of criticality. Storvick T. S. and San-
dler S. I. (eds.), Phase equilibria and fluid properties in
chemical engineering, 369—368. ACS Symposium Series
60. Am. Chem. Soc. (1977).
490. Moldover M. R., Gallagher J. S. Critical points of mix-
tures: An analogy with pure fluids. AIChE Journal, 24,
267—278 (1978).
491. Mollerup J. Thermodynamic properties from cor-
responding state theory. Fluid Phase Equilibria, 4, 11—34
(1980).
492. Mollerup J. A note on excess Gibbs energy models, equa-
tions of state and local composition concept. Fluid Phase
Equilibria, 7, 121—138 (1981).
493. Morgan M. S., Reid R. C. Generalized partial quantities
from Pitzer’s expansion and pseudocritical rules. AIChE
Journal, 16, 889—890 (1970).
494. Morimi J., Nakanishi K. Use of the Wilson equation to
calculate solid-liquid phase equilibria in binary and ter-
nary systems. Fluid Phase Equilibria, 1, 153—160 (1977).
495. Morris J. P, Zellars G. R. Vapor pressure of liquid cop-
per and activities in liquid Fe-Cu alloys. Trans. AIME,
206, 1086—1090 (1956).
496. Moshfegian M., Shariat A., Erbar J. H. Application of
the PEGC-MES equation of state to synthetic and natu-
ral gas systems. Newman S. A. (ed.), Thermodynamics of
aqueous systems with industrial applications, ACS Sym-
posium Series, 133, 333—360 (1980).
497. Muir R. E, Howat C. S. III. Predicting solid-liquid
equilibrium data from vapor-liquid data. Chemical En-
gineering, 89 (4), 89—92 (February 22, 1982).
498. Mundis C. J., Yarborough L., Robinson R. L. Jr. Vapori-
zation equilibrium ratios for CO2 and H2S in paraffinic,
naphthenic and aromatic solvents. Ind. Eng. Chem.
Process Des. Dev., 16, 254—259 (1977).
499. Myers A. L. Adsorption of gas mixtures: Thermodyna-
mic approach. Ind. Eng. Chem., 60 (5), 45—49 (1968).
500. Myers A. L., Prausnitz J- M. Thermodynamics of mixed-
gas adsorption. AIChE Journal, 11, 121—127 (1965).
501. Nagata I., Katoh K. Effective UNIQUAC equation in
phase equilibrium calculation. Fluid Phase Equilibria, 5,
225—244 (1980).
502. Nagata I., Nagashima M„ Ogura M. A comment on an
extended form of the Wilson equation to correlation of
partially miscible systems. J. Chem. Eng. Japan, 8,
406—408 (1975).
503. Nagata I., Nagashima M. Correlation and prediction of
vapor-liquid and liquid-liquid equilibria. J. Chem. Eng.
Japan, 9, 6—11 (1976).
504. Nagata I., Ohta T. Calculation of high-pressure phase
equilibrium from total pressure-liquid composition data.
Ind. Eng. Chem. Process Des. Dev., 15, 211—215 (1976).
505. Nagata I., Ohta T. Prediction of the excess enthalpies of
mixing for mixtures using the UNIFAC method. Chem.
Eng. Science, 33, 177—182 (1978).
506. Nagata I., Yamada T. Correlation and prediction of ex-
cess thermodynamic functions of strongly nonideal li-
quid mixtures. Ind. Eng. Chem. Process Des. Dev., 13,
47—53 (1974).
507. Nagata I., Yasuda S. Fugacity coefficients in binary mix-
tures containing acetic acid. J. Chem. Eng. Japan, 8,
398—400 (1975).
508. Nagata I., Yasuda S. On the Carnahan-Starling equation
of state. J. Chem. Eng. Japan, 10, 64—65 (1977).
509. Nakamura R., Breedveld G. J. E, Prausnitz J. M. Ther-
modynamic properties of gas mixtures containing com-
mon polar and nonpolar components. Ind. Eng. Chem.
Process. Des. Dev., 15, 557—564 (1976).
510. Nakanishi K., Wada H., Touhara H. Thermodynamic ex-
cess functions of methanol+piperidine solutions at
298.15. Fourth Int. Conf, on Chem. Thermodynamics
(August 26, 1975), Montpellier, France, 169—175.
511. Nath J. Acentric factor and the heats of vaporization for
unassociated polar and nonpolar organic liquids. Ind.
Eng. Chem. Fundamen., 18, 297—298 (1979).
512. Nath J., Das S. S., Yadav a M. L. On the choice of acen-
tric factor. Ind. Eng. Chem. Fundamen., 15, 223—225
(1976).
513. Nathan D. I. Prediction of mixture enthalpies. British
Chem., Eng. 12 (2), 223—226, (1967).
514. Nelder J. A., Mead R., A simplex method for function
minimization. Computer Journal, 7, 308—313 (1964).
515. Newman S. A., Correlations evaluated for coal-tar li-
quids. Hydrocarbon Processing, 60 (12), 133—142 (1981).
516. Newton R. FL, Dodge R E Ind. Eng. Chem., 27, 577
(1935).
517. Nghiem L. X., Aziz K. A robust iterative method for
flash calculations using the Soave-Redlich-Kwong or the
Peng-Robinson equation of state. Paper No. SPE 8285,
presented at 54th annual Fall Technical Conference of the
Society of Petroleum Engineers, Las Vegas (September
23—26, 1979).
518. Nghiem L. X., Fong D. K., Aziz K- Compositional
modelling with an equation of state. Paper No. 9306,
presented at 55th Annual Fall Technical Conference of
Литература 649
the Society of Petroleum Engineers, Dallas (September
21—24, 1980).
519. Niedzwiecki J. L., Springer R. D., Wolfe R. G. Multicom-
ponent distillation in the presence of free water. Chem.
Eng. Progress, 76 (4), 57—58 (1980).
520. Nishiumi H. Thermodynamic property prediction of Сю
to C20 paraffins and their mixtures by the generalized
BWR equation of state. J. Chem. Eng. Japan, 13, 74—76
(1980).
521. Nishiumi H. An improved generalized BWR equation of
state with three polar parameters applicable to polar sub-
stances. J. Chem. Eng. Japan, 13, 178—183 (1980).
522. Hishiumi H., Robinson D. B. Compressibility factor of
polar substances bases on a four-parameter correspond-
ing states principle. J. Chem. Eng. Japan, 14, 259—266
(1981).
523. Nishiumi H., Saito S. Correlation of the binary interac-
tion parameter of the midified generalized BWR equa-
tion of state J. Chem. Eng. Japan, 10, 176—180 (1977).
524. Nitta T, Katayama T. A new interpretation of the Wil-
son equation as a short-cut form of the associated solu-
tion theory J. Chem. Eng. Japan, 7, 381—382 (1974).
525. Null H. R. Application of the Wilson equation to solid-
liquid equilibria. AIChE symposium Series, 63, 52—56
(1967).
526. Ochi K, Hiraba S., Kojima K. Prediction of solid-
equilibrium using ASOG. J. Chem. Eng. Japan, 15,
59—61 (1982).
527. Ochi K., Lu B. C.-Y. Determination and correlation of bi-
nary vapor-liquid equilibrium data. Fluid Phase
Equilibria, 1, 185—200 (1978).
528. O’Connell J. P. Thermodynamics of gas solubility.
Proceedings. 2nd International Conference, Berlin
(March 21, 1980), EFCE, DECHEMA, 445—456.
529. Oellrich L., Plocker U, Prausnitz J. M., Knapp H.
Methods for calculation of phase equilibria and enthal-
pies with the aid of equations of state (in German).
Chem. Ing. Tech., 49, 955—965 (1977).
530. Oishi T, Prausnitz J. M. Estimation of solvent activities
in polymer solutions using a group-contribution method.
Ind. Eng. Chem. Process Des. Dev., 17, 333—339 (1978).
531. Oliver R. C, Stephanou S. E., Baier R. W. Calculating
free energy minization. Chem. Eng., 69 (4), 121—128
(February 19, 1962).
532. OryeR. V. Prediction and correlation of phase equilibria
and thermal properties with the BWR equation of state.
Ind. Eng. Chem. Process Des. Dev., 8, 579—588 (1969).
533. Orye R. V., Prausnitz J. M. Multicomponent equilibria
with the Wilson equation. Ind. Eng. Chem., 57, 18—26
(1965).
534. Osborn A. How to calculate three-phase flash vaporiza-
tion. Chemical Engineering, 71 (26), 97—100 (December
21, 1964).
535. OthmerD. E Composition of vapors from boiling binary
solutions. Ind. Eng. Chem., 20, 743—746 (1928).
536. Othmer D. E Correlating physical and chemical data for
chemical engineering use. Proceedings, 11th International
Congress of Pure and Applied Chemistry, London
(1947).
537. Ott J. B, Coates J. R., Hall H. T. Jr. Comparison of
equations of state in effectively describing PVT relations.
J. Chem. Education, 48, 515—517 (1971).
538. Otto J. Equations of state (in German). Handbuch der
Experimental Physik, Bd, 8/2, W. Wien and F. Harms
(eds.), 207—246. Akademische Verlagsgesellschaft, 1929.
539. Ozkardesh H., TarakadR. R., Adler S. В Single-set treat-
ment of all available binary system VLE data leads to
simplified correlation procedures. Ind. Eng. Chem. Fun-
damen., 17, 206—209 (1978).
540. Palmer D. A. Predicting equilibrium relationships for
maverick mixtures. Chem. Eng. (1975), 80—85 (June 9,
1975).
541. Parrish W. R., Prausnitz J. M. Dissociation pressures of
gas hydrates formed by gas mixtures. Ind. Eng. Chem.
Process Des. Dev., 11, 26—35 (1972).
542. Passut C. A., Danner R. P. Development of a four-
parameter corresponding states method: Vapor pressure
prediction. AIChE Symposium Series, 140, 30—36
(1974).
543. Павлов С. Ю., Карпачева Л. Л., Боснер M. E., Кир-
нос А. Б. «Предсказание коэффициентов активности
на основе параметров взаимодействия структурных
групп», Журн. физ. хим., 52, 4, 887—889 (1978).
544. Pemberton R. С, Mash С. J. Thermodynamic properties
of aqueous non-electrolyte mixtures. II: Vapour pressures
and excess Gibbs energies for water + ethanol at 303.15
to 363.15 determined by an accurate static method. J.
Chem. Thermodynamics, 10, 867—888 (1978).
545. Peng D-Y, Robinson D. B. A new two-constant equation
of state. Ind. Eng. Chem. Fundamen., 15, 59—64 (1976).
546. Peng D.-Y., Robinson D. В Two and three phase equilibri-
um calculations for systems containing water. Canad. J.
Chem. Eng., 54, 595—599 (1976).
547. Peng D.-Y., Robinson D. B. A rigorous method for
predicting the critical properties of multicomponent sys-
tems from an equation of state. AIChE Journal, 23,
137—144 (1977).
548. Peng D.-Y, Robinson D. В Calculation of three-phase
solid-liquid-vapor-equilibrium. Chao К. C. and Robin-
son R. L. (eds.). Equations of state in engineering and
research, 185—196, Advances in Chemistry Series 182,
Am. Chem. Soc. (1979).
549. Peng D.-Y., Robinson D. В Two- and three-phase
equilibrium calculations for coal gasification and related
proceses. Newman S. A. (ed.). Thermodynamics of aque-
ous systems with industrial applications ACS Symposium
Series, 133, 393—414 (1980).
550. Pesult D. R. Binary interaction constants for mixtures
with a wide range in component properties. Ind. Eng.
Chem. Fundamen., 17, 235—242 (1978).
551. Peter S. Thermodynamics of multicomponent systems as
a basis for physico-chemical separation processes. Int.
Chem.. Eng., 19, 410—419 (1979).
552. Peter S„ Brunner G., Riha R. High pressure phase
equilibria and their technical application (in German).
Chem. Ing. Technik, 46, 623—668 (1974).
553. Phillips J. N. The erngetics of micelle formation. Trans.
Faraday Soc., 51, 561—569 (1955).
554. Pistorius C. W. F.T. Melting points and volume changes
upon melting. Neindre B. Le and Vodar B. (eds.), Ex-
perimental Thermodynamics, Vol. II, 803—834, Butter-
worths, 1975.
555. Pitzer K. S., Curl R. F. et al. Volumetric and ther-
modynamic properties of fluid — enthalphy, free energy
and entropy Ind. Eng. Chem., 50, 265—274 (1958). (Cm.
также JACS, 77, 3427, 3433, 79, 2369.)
23-864
556. Pitzer К. S. Origin of the acentric factor. Storvick T. S.
and Sandler S. I. (eds). Phase equilibria and fluid proper-
ties in the chemical industry, 1—10. ACS Symposium Ser-
ies 60, Am. Chem. Soc. (1977).
557. Pitser K. S. Thermodynamics of aqueous electrolytes at
various temperatures, pressures and compositions. New-
man S. A. (ed.), Thermodynamics of Aqueous Systems
with Industrial Applications, ACS Symposium Series
133, Am. Chem. Soc. (1980).
558. Pitzer K. S. Thermodynamics of electrolyte solutions
over the entire miscibility range. Newman S. A. (ed.),
Chemical Engineering Thermodynamics, 309—322
(1982). Ann Arbor Science.
559. Pitzer K. S. Theory: Ion interaction approach. Pyt-
kowicz R. (ed.), Activity Coefficients in Electrolyte Solu-
tions, Vol. 1, 157—208, CRC Press (1979).
560. Plocker U, Knapp H, Prausnitz J. M. Calculation of
high pressure vapor-liquid equilibria from a correspond-
ing states correlation with emphasis on symmetric mix-
tures. Ind. Eng. Chem. Process Des. Dev., 17, 324—332
(1978).
561. Poling В E., Grens E. A. II, Prausnitz J. M. Ther-
modynamic properties from a cubic equation of state:
Avoiding trivial roots and spurious derivatives. Ind. Eng.
Chem. Process Des. Dev., 20, 127—130 (1981).
562. Pollin A. G., Fried V., Yorizane M. The prediction of ex-
cess enthalphy and excess Gibbs energy from volumetric
data. J. Chem. Eng. Japan, 11, 326—327 (1978).
563. Potts A. D., Davidson D. W. Ethanol hydrate. J. Phys.
Chem., 69 (3), 996—1000 (1965).
564. Prausnitz J- M. Solubility thermodynamics in chemical
engineering J. Phys. Chem., 66, 640—645 (1962).
565. Prausnitz J- M. State of the art review of phase
equilibria. Storvick T. S. and Sandler S. I. (eds.), Phase
equilibria and fluid properties in the chemical industry,
11—62, ACS Symposium Series, 60, Am. Chem. Soc.
(1977).
566. Prausnitz J. M. Practical applications of molecular ther-
modynamics for calculating phase equilibria. Interna-
tional Chemical Engineering, 19, 401—409 (1979).
567. Prausnitz J. M. State of the art review of phase
equilibria. Proceedings, 2nd International Conference,
Berlin (March 21, 1980). EFCE, DECHEMA, 231—282.
568. Prausnitz J- M. Calculation of phase equilibria for sepa-
ration operations. Trans. I. Chem. E., 59, 3—16 (1981).
569. Prausnitz J. M., Gunn R. D. Volumetric properties of
nonpolar gaseous mixtures. AIChE Journal, 4, 430—435
(1958).
570. Prausnitz J- M., Gunn R. D. Pseudocritical constants
from volumetric data for gas mixtures. AIChE Journal,
4, 494 (1958).
571. Prausnitz J- M., Shair F. H. A thermodynamic correla-
tion of gas solubilities. AIChE Journal, 7, 682—687
(1961).
572. Prausnitz J. M., Targovnik J. H. Salt effects in aqueous
vapor-liquid equilibria. Ind. Eng. Chem. Chem. and
Eng. Data Series, 3, 234—239 (1958).
573. Prins A. Mixtures of liquid crystals in binary systems.
Z. Physik. Chem., 67, 689—723 (1909).
574. Rachford H. H, Rice J. D. Procedure for use of electron-
ic digital computers in calculating flash vaporization
hydrocarbon equilibrium. Petrol. Technol., Sect. 1, p. 19,
Sect. 2, p. 3 (October 1952).
575. Rackett H G. Equation of state for saturated liquids. J.
Chem. Eng. Data, 15, 514—517 (1970).
576. Raimondi L. A modified Redlich-Kwong equation of
state for vapour-liquid equilibrium calculations. Chem.
Eng. Science, 35, 1269—1275 (1980).
577. Rasmussen R, Fredenslund A. Prediction of separation
factors using group contribution methods: A review.
Separation and Purification Methods, 7 (2), 147—182
(1978).
578. Ray G. C., Box E. O. Adsorption of gases on activated
charcoal. Ind. Eng. Chem., 42, 1315—1318 (1950).
579. Rea H. E. Effect of pressure and temperature on the li-
quid densities of pure hydrocarbons. J. Chem. Eng. Data,
18, 227—230 (1973).
580. Redlich O. On the three-parameter representation of the
equation of state. Ind. Eng. Chem. Fundamen., 14,
257—260 (1975).
581. Redlich O., Kwong J. N. S. On the thermodynamics of
solutions: V: An equation of state. Fugacities of gaseous
solutions. Chem. Review, 44, 233—244 (1949).
582. Redlich O., Ngo V В. T. An improved equation of state.
Ind. Eng. Chem. Fundamen., 9, 287—290 (1970).
583. Ree E H, Hoover W. G. Seventh virial coefficients of
hard spheres and hard disks. J. Chem. Physics, 46, 4181—
4197 (1967).
584. Reich R., Ziegler W. T., Rogers К A. Adsorption of
methane, ethane and ethylene gases and their binary and
ternary mixtures and carbon dioxide on activated carbon
at 212—301 К and pressures to 35 atmospheres. Ind. Eng.
Chem., Process Des. Dev., 19, 336—344 (1980).
585. ReidR. C., BeegleB L. Critical point criteria in Legendre
transform notation. AIChE Journal, 23, 726—732
(1977).
586. Renon H. Deviations from ideality in electrolyte solu-
tions. Mah R. S. H. and Seider W. D. (eds.). Foundations
of Computer-Aided Chemical Process Design, Vol. 2,
53—83 (1980) AIChE.
587. Renon H, Prausnitz J- M. Local compositions in ther-
modynamic excess functions for liquid mixtures. AIChE
Journal, 14, 135—144 (1968).
588. Renon H, Prausnitz J. M. Estimation of parameters for
the NRTL equation for excess Gibbs energies of strongly
non-ideal liquid mixtures. Ind. Eng. Chem. Process. Des.
Dev., 8, 413—419 (1969).
589. Rheinboldt H, Kircheisen M. Melting diagrams of sys-
tems with miscibility gaps: Picric acid+triphenyl-
methane, dinitrotoluene+urea, phenylenediamine+tri-
phenylmethane. J. Fur Praktische Chem., 112, 187—195
(1926).
590. Richon D., Antoine P., Renon H. Infinite dilution activity
coefficients of linear and branched alkanes from Ci to
C9 in n-hexadecane by inert gas stripping. Ind. Eng.
Chem. Process Des. Dev., 19, 144—147 (1980).
591. Richter H. R. D., Burniside В. M. A general programme
for producing pressure-enthalphy diagrams. J. Meeh.
Science, 17 (1), 31—39 (1975).
592. Righter W. M., Hall К R. Optimal truncation of the viri-
al equation. AIChE Journal, 21, 406—407 (1975).
593. Rizzi A., Huber J. E K. Comparative calculations of ac-
tivity coefficients in binary liquid mixtures at infinite di-
lution using the “solution of groups” model. Ind. Eng.
Chem. Process Des. Dev., 20, 204—210 (1981).
594. Robinson D. B, Peng D.-Y., Ng H.-J. Applications of the
Литература 651
Peng-Robinson equation of state. Storvick T. S. and San-
dler S. I. (eds.), Phase Equilibria and Fluid Properties is
the Chemical Industry, ACS Symposium Series 60 (1977).
595. Robinson D. R, Peng D.-Y. The use of equations of state
in multiphase equilibrium calculations. Proceedings, 2nd
International Conference, Berlin (March 21, 1980),
EFCE, DECHEMA, 335—354.
596. Robinson D. R, Peng D.-Y., Ng H.-J. Applications of the
Peng-Robinson equation of state. ACS Symposium Ser-
ies, 60, 200—220 (1977).
597. Robinson D. R., Peng D.-Y., Ng H.-J. Capability of the
Peng-Robinson programs. Part 1: VIE and critical
property calculations Hydrocarbon Processing, 57 (4),
95—98 (1978).
598. Robinson R. L., Chao К. C. A correlation of vaporiza-
tion equilibrium ratios for gas processing streams. Ind.
Eng. Chem. Process Des. Dev., 10, 221—229 (1971).
599. Robinson R. L., Chao K. C. A correlation of vaporiza-
tion equilibrium ratios for gas processing systems. Ind.
Eng. Chem. Process Des. Dev., 10, 221—229 (1979).
600. Ross R. A., Seider W. D. Simulation of three-phase distil-
lation towers. Computers and Chemical Engineering, 5,
7—20 (1980).
601. Rossini F. D. in: Science of Petroleum, V/1, Oxford, 1950.
602. Rowlinson J. S. The properties of real gases. Encyclope-
dia of Physics. Vol. XII, Flugge S. (ed.). Springer-Verlag,
1958.
603. Rowlinson J. S. Legacy of van der Waals. Nature,
244—417 (August 17, 1973).
604. Rudman P. S., in: Herman H. (ed.), Advances in Materi-
als Research 4, 147—194, Interscience (1970).
605. Russell R. A. Non-ideal liquid activity coefficients Erdol
and Kohle, 29, 407—409 (1976).
606. Сабылин И. И., Полозов А. Г., Клионский А. Б.
«Методика расчета равновесия жидкость— жид-
кость — пар в трехкомпонентных системах», Журн.
прикл. хим., 52, 2, 308—312 (1979).
607. Sanderson R. V., Chien Н. Н. Y. Simultaneous chemical
and phase equilibrium calculation. Ind. Eng. Chem.
Process Des. Dev., 12, 81—85 (1973).
608. Santacesaris E. Measurement of activity coefficients at
infinite dilution by stripping and detention time
methods. Fluid Phase Equilibria, 3, 167—176 (1979).
609. Sarashina E., Nohka J., Arai Y., Saito S. Correlation of
critical locus for binary mixtures by the BWR equation
J. Chem. Eng. Japan, 7, 219—222 (1974).
610. Sastri S. R. S., Rao M. V. R., Reddy K. A., Doraiswa-
my L. K. A generalized method for estimating the latent
heat of organic compounds. Brit. Chem. Eng., 14 (7),
959—963 (1969).
611. Saville G. Measurement of PVT properties of gases and
gas mixtures at low pressures. Le Neindre B. and Vodar B.
(eds.). Experimental Thermodynamics. Vol. II, 321—346.
Butterworths (1975).
612. Sayegh S. G., Vera J. H. Model — free methods for
vapor-liquid equilibria calculations. Chem. Eng. Science,
35, 2247—2256 (1980).
613. Sayegh S. G., Vera J. H, Ratcliff G. A. Vapor-liquid
equilibria for the ternary system n-heptane/n-pro-
panol/lchlorobutane and its constituent binaries at
298.15 K. Canad. Chem. Eng., 57, 513—519 (1979).
614. Scatchard G., Hamer W. J. The application of equations
for chemical potentials of partially miscible solutions.
JACS, 57, 1805—1809 (1935). The application of equa-
tions for chemical potentials to equilibrium between solid
solution and liquid solution. JACS, 75, 1809—1811
(1935).
615. Scheller W. A., Narashmha Rao S. V. Isothermal vapor-
liquid equilibrium data for system heptane-2-pentanone
at 90 C. J. Chem. Eng. Data, 18, 223—225 (1973).
616. Schmidt G., Wenzel H. A modified van der Waals type
of equation of state. Chem. Eng. Science, 35, 1503—1512
(1980).
617. Schneider G. M. Phase equilibria of liquid and gaseous
mixtures at high pressures. Le Neindre B. and B. Vodar
(eds.), Experimental Thermodynamics, Vol. II,
787—802. Butterworths (1975).
618. Schneider G. M. High pressure phase diagrams and criti-
cal properties of fluid mixtures. McGlashan M. L. (ed.),
Chemical Thermodynamics, Vol. 2: A Specialist Periodi-
cal Report, 105—146. The Chemical Society, London
(1978).
619. Schneider G. M., Russo C. Effect of pressure on miscibi-
lities of liquids. V. Effect of salts on miscibilities of
propanol + water, 2-butanol + N20 and pyridine + water
up to 6000 bar, Ber Bunsengesellschaft, 70, 1008—1014
(1966).
620. Schotte W. Collection of phase equilibrium data for
separation technology. Ind. Eng. Chem. Process Des.
Dev., 19, 432—439 (1980).
621. Schreiber L. R., Eckert C. A. Use of infinite dilution ac-
tivity coefficients with Wilson’s equation. Ind. Eng.
Process Des. Dev., 10, 572—576 (1971).
622. Schreinemakers F. A. H. Equilibria in the system
water + phenol+acetone. Z. Physik. Chem., 33, 78—98
(1900).
623. Schulte H. W„ Grenzheuser P., Gmehling J. Application
of a modified Wilson Equation to liquid mixtures show-
ing phase splitting. Fluid Phase Equilibria, 4, 185—196
(1980).
624. Schweitzer O. R., Wales С. E. Phase Equilibria, Part 1:
Phase rule and equilibria relations: Part 2: Equilibria in
one-components systems. Chemical Engineering 117—
120 (May 27, 1963); 111—114 (June 24, 1963).
625. Scott R. L. Liquid State. Physical Chemistry. VIIIA, edit-
ed by Eyring, Henderson and Yost, 1—83, Academic
Press, 1971.
626. Sehgal I. J. S., Yesavage V. E, Mather A. E., Powers J. E.
Enthalphy predictions tested by data. Hydrocarbon
Processing, 47 (8), 137—143 (1968).
627. Seider W. D., Gautam R., White C W. Computation of
phase and chemical equilibrium: A review. R. G. Squires
& G. V. Reklaitis (eds.), Computer Applications to
Chemical Engineering, 115—134, ACS Symposium Series
124, Am. Chem. Soc. (1980).
628. Seider W. D., Gautam R., White C W. III. Computation
of phase and chemical equilibrium: A review. Founda-
tions of Computer-Aided Chemical Process Design,
115—134, AIChE (1980).
629. Sengers J. V, Levelt-Sengers A. The critical state. Chem.
and Eng. News, 104—118 (June 10, 1968).
630. Shah D. O., Chan K. S., Giordano R. M. The effect of
dissolved oils and alcohols on the CMC of synthetic and
petroleium sulfonates. Mittal K. L. (ed.), Solution
Chemistry of Surfactants, Vol. 1, 391—406 (1979).
Plenum.
23
631. Shah К. К., Thodos G. A comparison of equations of
state. Ind. Eng. Chem., 57, 30—37 (1965).
632. Shah M. K., Bishnoi P. R. Multistage multicomponent
separation calculations using thermodynamic properties
evaluated by the SRK/PR equation of state. Canad. J.
Chem. Eng., 56, 478—486 (1978).
633. Shah P. N., Yaws C. L. Densities of liquids. Chem. Eng.,
83 (21), 131—133 (October 25, 1976).
634. Shaw A. N. The derivation of thermodynamical relations
for a simple system. Phil. Trans. Roy. Soc. (London),
A234, 299—328 (1935).
635. Signer R., Arm H., Daeniker H. Vapor pressures, densi-
ties, thermodynamic mixing functions and refractive in-
dexes of the binary systems water-tetrahydrofuran and
water-diethylether at 25 C (in German). Helv. Chim.
Acta., 52, 2347—2351 (1969).
636. Silverman N„ Tassios D. The number of roots in the Wil-
son equation and its effects on vapor-liquid equilibrium
calculations. Ind. Eng. Chem. Process Des. Dev., 16,
13—20 (1977).
637. Sim W. J., Daubert T. E. Prediction of vapor-liquid
equilibria of undefined mixtures. Ind. Eng. Chem.
Process Des. Dev., 19, 386—393 (1980).
638. Simnick J. J., Lin H. M., Chao К. C. The BACK equation
of state and phase equilibria in pure fluids and mixtures.
Chao К. C. and R. L. Robinson (eds.), Equations of state
in engineering and research, 209—234, Advances in
Chemistry Series 182. Am. Chem. Soc. (1979).
639. Simonet R., Behar E. A modified Redlich-Kwong equa-
tion of state for accurately representing pure component
data. Chem. Eng. Science, 31, 37—43 (1976).
640. Simonetty J., Yee D, Tassios D. Prediction and correla-
tion of liquid liquid-equilibria. Ind. Eng. Chem. Process
Des. Dev., 21, 174—180 (1982).
641. Skjold-Jorgensen S., Kolbe B, Gmehling J., Rasmussen P.
Vapor-liquid equilibria by UNIFAC group contribution:
Revision and extension. Ind. Eng. Chem. Process Des.
Dev., 18, 714—722 (1979).
642. Skjold-Jorgensen S., Rasmussen R, Fredenslund A. On
the temperature dependence of the UNIQUAC/UNIFAC
models Chem. Eng. Science, 35, 2389—2403 (1980).
643. Skjold-Jorgensen S., Rasmussen R, Fredenslund A. On
the concentration dependence of the UNIQUAC/
UNIFAC models, Chem. Eng. Science, 37, 99—111
(1982).
644. Skolnik H. Effect of pressure in azeotropy. Ind. Eng.
Chem., 43, 172—276 (1951).
645. Sloan E. D., Jr., Mullins J. C. Nonideality of binary ab-
sorbed mixtures of benzene and Freon-11 on highly
graphitized carbon at 298.15 K. Ind. Eng. Chem. Funda-
men., 14, 347—355 (1975).
646. Slocum E. W. Multipurpose high-pressure phase-
equilibrium apparatus. Ind. Eng. Chem. Fundamen., 14,
126—128 (1975).
647. Smith A., Menzies A. W. C. Studies in vapor pressure.
JACS, 897—914 (1910).
648. Smith B. D. Simplified calculation of chemical equilibria
in hydrocarbon systems containing isomers. AIChE Jour-
nal, 5, 26—28 (1959).
649. Smith W. R. The computation of chemical equilibrium
in complex systems: Review. Ind. Eng. Chem. Fundamen.
19, 1—10 (1980).
650. Snyder С. E, Chao К. C. Heat of adsorption of light
hydrocarbons and their mixtures on activated carbon.
Ind. Eng. Chem. Fundamen., 9, 437—443 (1970).
651. Soave G. Equilibrium constants from a modified
Redlich-Kwong equation of state. Chem. Eng. Science,
27, 1197—1203 (1972).
652. Soave G. S. Application of the Redlich-Kwong equation
of state to solid-liquid equilibria calculations. Chem.
Eng. Science, 33, 225—229 (1979).
653. Soave G. S. Application of a cubic equation of state to
vapour-liquid equilibria of’ systems containing polar
compounds I. Chem. E. Symposium Series, No. 56,
1.2/1—1.2/16 (1979).
654. Soave G. Rigorous and simplified procedures for deter-
mining the pure components parameters in the Redlich
Kwong-soave equation of state. Chem. Eng. Science, 35,
1725—1729 (1980).
655. Sokolov В. I. Equation of state of a saturated liquid. Rus-
sian J. Phys. Chem. 50, 668—669 (1976).
656. Sood S. K, Haselden G. G. Corrections to Co for the
Benedict-Webb-Rubin equation. AIChE Journal, 16,
891—892 (1970).
657. Sorensen J., Magnussen T, Rasmussen R, Fredenslund A.
Liquid-liquid equilibrium data: Their retrieval, correlation
and prediction. Fluid Phase Equilibria, 2,297—308 (1979);
3, 47—82 (1979); 4, 47—82 (1979); 4, 151—163 (1980).
658. Spear R. R., Robinson R. L., Chao К. C. Critical states
of mixtures and equations of state. Ind. Eng. Chem. Fun-
damen., 8, 2—8 (1969).
659. Spear R. R., Robinson R. L., Chao К. C. Critical states
of ternary mixtures and equations of state. Ind. Eng.
Chem. Fundamen., 10, 588—592 (1971).
660. Spencer С E, Danner R. P. Prediction of bubble-point
density of mixtures. J. Chem. Eng. Data, 18, 230—234
(1973).
661. Stahl E., Schilz W. Extraction with supercritical gases in
thin-layer chromatography: Applicability to natural sub-
stances (in German). Chem. Ing. Technik, 48, 773—778
(1976).
662. Stein E R, Miller E. J. Extension of the Hayden-
O’Connel correlation to the second virial coefficients of
some hydrogen-bonding mixtures. Ind. Eng. Chem.
Process Des. Dev., 19, 123—138 (1980).
663. Stephan K, SchaberK. Phase equilibria for vapour phase
extraction processes. Ger. Chem. Eng., 2, 38—45 (1979).
664. Stiel L. I. A generalized theorem of corresponding states
for the thermodynamic properties of non-polar and po-
lar fluids. Chem. Eng. Science, 27, 2109—2115 (1972).
665. Stookey D. J., Smith B. D. Prediction of excess free ener-
gy from excess enthalpy and excess volume data for
hydrocarbon mixtures. Ind. Eng. Chem. Process Des.
Dev., 12, 372—376 (1973).
666. Su G.-J., Chang C.-H. Generalized equation of state for
real gases. Ind. Eng. Chem., 38, 802—806 (1946).
667. Su G.-J, Viswanath D. S. Generalized Benedict-Welb-
Rubin equation of state for real gases. AIChE Journal,
11, 205—207 (1965).
668. Sugi H„ Katayama T. Ternary liquid-liquid and miscible
binary vapor-liquid equlibrium data for the two systems
n-hexane +ethanol+acetonitrile and water + acetonitri-
le+ethyl acetate. J. Chem. Eng. Japan, 11, 167—172
(1978).
669. Sutton T. L., MacGregor J. F. The analysis and design
of binary vapour-liquid equilibrium experiments.
Литература 653
I: Parameter estimation and consistency tests; II: The de-
sign of experiments. Canad. J. Chem. Eng., 55, 602—613
(1977).
670. Tai T. B., Ramalho R. S., Kaliaguine S. Application of
Wilson’s equation to the determination of vapor-liquid
equilibrium data and heats of mixing for nonideal solu-
tions. Canad. J. Chem. Eng., 50, 771—776 (1972).
671. TajblD. G., Kanofsky J. S„ Braband J M. UOP’s OLEX
process: New applications. Energy Processing/Canada,
61—63 (May — June, 1980).
672. Takeo M., Nishii K., Nitta T, Katayama T. Isothermal
vapor-liquid equilibria for two binary mixtures of hep-
tane with 2-butanone and 4-methyl-2-pentanone meas-
ured by a dynamic still with a pressure regulation. Fluid
Phase Equilibria, 3, 123—131 (1979).
673. Tamir A., Wisniak J. Vapor equilibrium in associating
systems (water-formic acid-propionic acid). Ind. Eng.
Chem. Fundamen., 15, 274—280 (1976).
674. Tang Y. P. On the solution of BWR compressibility.
Canad. J. Chem. Eng., 48, 726—727 (1970).
675. Tarakad R. R., Danner R. P. A comparison of enthalpy
prediction methods. AIChE Journal, 22, 409—411
(1976).
676. Tarakad R. R., Danner D. P. An improved corresponding
states method for polar fluids: Correlation of second
virial coefficients. AIChE Journal, 23, 685—695 (1977).
677. Tarakad R. R., Spencer С. E, Adler S. B. A comparison
of eight equations of state to predict gas phase density
and fugacity. Ind. Eng. Chem. Process Des. Dev., 18,
726—739 (1979).
678. Tassios D. P. Infinite dilution relative volatilities through
gas-liquid chromatography. Ind. Eng. Chem. Process
Des. Dev., 11, 43—46 (1972).
679. Tassios D Limitations in correlating strongly nonideal
binary systems with the NRTL and LEMF equations.
Ind. Eng. Chem. Process Des., Dev. 15, 574—578 (1976).
680. Tassios D. The number of roots in the NRTL and LEMF
equations and the effect on their performance. Ind. Eng.
Chem. Process Des. Dev., 18, 182—186 (1979).
681. Taylor S. L., Reed T. M., III. Virial coefficients and criti-
cal properties of perfluorohexanes. AIChE Journal, 16,
738—741 (1970).
682. Tee L. S., Gotoh S., Stewart W. E. Molecular parameters
for normal fluids, The Lennard-Jones and Kihara poten-
tials, IEC Fundamen., 5, 356—357 (1966).
683. Teja A., Sandler S. I. (on Part II). A corresponding states
equation for saturated liquid densities. I: Application to
LNG; II: Application to calculation of swelling factors
of СОг-crude oil systems. AIChE Journal, 26, 337—345
(1980).
684. Teja A. S., Sandler S. I., Patel N. C. A generalization of
the corresponding states principle using two nonspherical
reference fluids. The Chem. Eng. J. 21, 21—28 (1981).
685. Thompson P. A. An equation for liquid-vapor saturation
densities as a function of pressure. Chao К. C. and
Robinson R. L. (eds.). Equation of state in engineering
and research, 365—384, Advances in Chemistry Series
182. Am. Chem. Soc. 1979.
686. Thomson G. H, Brobst K. R., Hankinson R. W. An im-
proved correlation for densities of compressed liquids
and liquid mixtures. AIChE Journal, 28, 671—676
(1982).
687. Tinoco-Garcia L., Cano-Dominguez J. A new technique
for solving multi-phase equilibria. Unpublished, Institu-
te Mexicano del Petroleo (1979).
688. Tochigi K, Kojima K. The determination of group Wil-
son parameters to activity coefficients by ebulliometer J.
Chem. Eng. Japan, 9, 267—273 (1976).
689. Tochigi K., Hiraga M., Kojima K. Prediction of liquid-
liquid equilibria for ternary systems by the ASOG
method. J. Chem. Eng. Japan, 13, 159—162 (1980).
690. Tochigi K, Lu В C.-Y., Ochi K., Kojima K. On the tem-
perature dependence of ASOG parameters for VLE cal-
culations. AIChE ’Journal, 27, 1022—1024 (1981).
691. Torres-Marchal C. Graphical design for ternary distilla-
tion systems. Chemical Engineering, 88 (21), 134—155
(October 19, 1981).
692. Tryhorn F. G., Wyatt W. F. Adsorption. I: Adsorption by
coconut charcoal from alcohol-benzene and acetone-
benzene mixtures. Trans. Faraday Soc., 21, 399—405
(1925).
693. Tryhorn E G., Wyatt W. E Adsorption. II: The adsorp-
tion by a coconut charcoal of saturated vapours of some
pure liquids. Trans. Faraday Soc., 22, 134—145 (1926).
694. Tryhorn E G., Wyatt W. E Adsorption. Ill: Stages in the
adsorption by a coconut charcal from vapour mixtures
of alcohol and benzene, and of acetone and benzene.
Trans. Faraday Soc., 23, 139—145 (1927).
695. Tryhorn E G., Wyatt W. F. Adsorption. IV: Adsorption
by coconut charcoal from binary mixtures of saturated
vapours: The systems methyl alcohol-benzene, ethyl
alcohol-benzene, л-propyl alcohol-benzene and n-buthyl
alcohol-benzene. Trans. Faraday Soc., 24, 36—47 (1928).
696. Tsonopoulos C. An empirical correlation of second virial
coefficients. AIChE Journal, 20, 263—272 (1974).
697. Tsonopoulos C. Second virial cross-coefficients: Correla-
tion and prediction of ky. Chao К. C. and Robin-
son R. L. (eds.). Equations of state in engineering and
research, 143—162. Advances in Chemistry Series 182.
Am. Chem. Soc. (1979).
698. Tsonopoulos C., Prausnitz J. M. Equations of state: A
review for engineering applications. Cryogenics, 9 (10),
315—327 (1969).
699. Tsuboka T, Katayama T. Modified Wilson equation for
vapor-liquid and liquid-liquid equilibria. J. Chem; Eng.
Japan, 8, 181—187 (1975).
700. Tsuboka T, Katayama T Correlations based on local
fraction model between new excess Gibbs energy equa-
tions. J. Chem. Eng. Japan, 8, 404—406 (1975).
701. Unno Y., Hoshino D., Nagahama K., Hirata M. Predic-
tion of solid-liquid equilibria from vapor-liquid equilibri-
um data using the solution of groups model. J. Chem.
Eng. Japan, 12, 81—85 (1979).
702. van der Waals J. H. On the continuity of the gaseous and
liquid, state. Dissertation, Leiden (1973). Physical
Memoirs, English translation by Threlfall and Adair,
Physical Society, 1, iii 333 (1890).
703. van der Waals J. H„ Plateeuw J. C. Clathrate solutions.
Advances Chem. Physics, II, 1—57. Interscience Publish-
ers (1959).
704. Van Laar J. J. The vapor pressure of binary mixtures. Z.
Physik Chem., 72, 723—751 (1910). On the theory of
vapor pressures of binary mixtures. Z. Physik Chem., 83,
599—608 (1913).
705. Van Ness H. C. Adsorption of gases on solids: Review
of the role of thermodynamics. Ind. Eng. Chem. Funda-
men., 8, 464—473 (1969).
706. Van Ness H. C. On integration of the coexistence equa-
tion for binary vapor-liquid equilibrium. AIChE Jour-
nal, 16, 18—22 (1970).
707. Van Ness H. C On use of constant-pressure activity
coefficients. Ind. Eng. Chem. Fundamen., 18, 431—433
(1979).
708. Van Ness H. C, Abbott M. M. Vapor-liquid equilibrium.
Part VI: Standard state fugacities for supercritical com-
ponents. AIChE Journal, 25, 645—655 (1979).
709. Van Ness H. C, Byer S. M., Gibbs R. E. Vapor-liquid
equilibrium. I: An appraisal of data reduction methods.
AIChE Journal, 19, 238—244 (1973).
710. Van Ness H. C, Pedersen E, Rasmussen P. Vapor-liquid
equilibrium. V: Data reduction by maximum likelihood.
AIChE Journal, 24, 1055—1062 (1978).
711. Varhegyi G., Eon С H. Calculation of the free energy
parameters from ternary liquid-liquid equilibrium data.
Ind. Eng. Chem. Fundamen., 16, 182—185 (1977).
712. Vera J. H., Prausnitz J. M. Generalized van der Waals
theory for dense fluids. The Chemical Engineering Jour-
nal, 3, 1—13 (1972).
713. Verhille R. P. Effects of equilibrium data correlating
equations on the design of continuous rectification
columns for binary separations. I. Chem. E. Symposium
Series, No. 56. 1.3/1—1.3/23 (1979).
714. Vidal J. Mixing rules and excess properties in cubic equa-
tions of state. Chem. Eng. Science, 33, 789—791 (1978).
715. Вийроя А. К., Каллас Ю. И., Сийрде Э. К. Исполь-
зование метода UNIFAC при нахождении фазовых
равновесий в многокомпонентных системах, Журн.
прикл. хим., 51, 11, 2471—2474 (1973).
716. Viswanath D. S., Su G.-J. Generalized PVT behaviour of
gases. AIChE Journal, 11, 202—204 (1965).
717. Wagner W. New vapor pressure measurements for argon
and nitrogen and a new method for establishing rational
vapor pressure equations. Cryogenics, 470—482 (August
1973).
718. Wales С. E. Phase equilibria. Ill: Behavior of one-
component systems; IV: Equilibria in two-components
systems; V: Phase equilibria in binary systems. Chemical
Engineering, 141—144 (July 22, 1963); 167—174 (August
19, 1963); 187—192 (September 16, 1963).
719. Weller H., Schuberth H., Leibniz E. Vapor-liquid
equilibrium of the system phenol/n-butylacetate/water at
44.4 C (in German). J. Prakl. Chemie, Series 4, 21,
234—249 (1963).
720. Wellman P, Katell S. How pressure and temperature af-
fect steam-methane reforming. Hydrocarbon Processing,
42 (6), 135—137 (1963).
721. Wenzel H., Rupp W. Calculation of phase equilibria in
systems containing water and supercritical components.
Chem. Eng. Science, 33, 683—687 (1978).
722. White M. G., Chao К. C. Principle of corresponding
states of liquid solutions. Ind. Eng. Chem. Fundamen.,
14, 166—171 (1975).
723. White W. B., Johnson S. M., Dantzig G. В Chemical
equilibrium in complex mixtures. J. Chem. Physics, 28,
751—755 (1958).
724. Wichterle I. High pressure vapour-liquid equilibrium: A
review. Fluid Phase Equilibria, 1, 161—172 (1977); 1,
225—245 (1978); 1, 305—316 (1978); 2, 59—78 (1978); 2,
143—159 (1978).
725. Williamson A. G. Phase equilibria of two-components
systems and multicomponent systems. Neindre B. Le. and
Vodar B. (eds.), Experimental Thermodynamics, II,
749—786. Butterworths (1975).
726. Wilson G. M. Vapor-liquid equilibrium. XI: A new ex-
pression for the excess free energy of mixing. J. Am.
Chem. Soc., 86, 127—130 (1964).
727. Wilson G. M. Areas of research on activity coefficients
from group contributions at the Thermochemical Insti-
tute. Storvick T. S. and Sandler S. I. (eds.), Phase
equilibria and fluid properties in chemical engineering,
429—444, ACS Symposium Series 60. Am. Chem. Soc.
(1977).
728. Wilson G. M. Thermophysical and transport properties
of synthetic fuel systems at extreme temperatures and
pressures. Mah R. S. H. and Seider W. D. (eds.), Founda-
tions of Computer-Aided Chemical Process Design,,
Vol. II, 31—51 (1980). AIChE.
729. Wilson G. M., Deal С. H. Activity coefficients and
molecular structure. Ind. Eng. Chem. Fundamen., 1,
20—23 (1962).
730. Winnick J., Kong J. Excess volumes of mixtures contain-
ing polar liquids. Ind. Eng. Chem. Fundamen., 13,
292—293 (1974).
731. Witonsky R. J., Miller J. G. The second virial coefficient
of the helium-nitrogen system from 175 to 475 C, JACS,
85, 282—286 (1963).
732. Wohl A. Investigations on equations of state. Z. physik
Chem., 87, 1—39 (1914); 99, 207—241 (1921).
733. Wojtowicz P J. in: Priestley et al. (eds.), Introduction to
Liquid Crystals 333—350, Plenum Press (1975).
734. Yamada T. An improved generalized equation of state.
AIChE Journal, 19, 286—291 (1973).
735. Yamada T., Gunn R. D. Saturated liquid molar volumes:
The Rackett equation. J. Chem. Eng. Data, 18, 234—235
(1973).
736. Yarborough L. Application of a generalized equation of
state to petroleum reservoir fluids, Chao К. C. and
Robinson R. L. (eds.), Ewuations of state in engineering
and research, 385—440, Advances in Chemistry'Series
182, Am. Chem. Soc. 1979. (
737. Yen L. C, Alexander R. E. Estimation of vapor and li-
quid enthalpies. AIChE Journal, 11, 334—339 (1965).
738. Yeo К. O., Christian S. D. A thermodynamic method for
predicting solubilities of solutes in nonpolar solvents.
Ind. Eng. Chem. Fundamen., 13, 196—198 (1974).
739. Yokoyama C, Arai K, Saito S. in: Newman (ed.) Semi-
empirical equation of state for polar substances on the
basis of perturbation theory, Chemical Engineering
Thermodynamics, Ann Arbor Science (1983).
740. Yon С M., TUrnock P. H. Multicomponent adsorption
equilibria on molecular sieves. AIChE Symposium Ser-
ies, 117, 75—83 (1971).
741. Yorizane M., Miyano Y. A generalized correlation for
Henry’s constants in nonpolar binary systems. AIChE
Journal, 24, 181—186 (1978).
742. Young C L. Experimental methods for studying phase
behavior of mixtures at high temperatures and pressures.
McGlashan M. L. (ed.), Chemical Thermodynamics.
Литература 655
II: A Specialist Periodical Report, 71—104, The Chemical
Society, London (1977).
743. Yu W. C, Lee H. M., Ligon R. M. Predict high pressure
properties. Hydrocarbon Processing, 61 (1), 171—178
(1982).
744. Закс И. А., Крупоткин И. Л. «Использование эле-
ментов диаграмм жидкостных равновесий», Журн.
прикл. хим., 51, 2125—2128 (1978).
745. Zarkarian J. A., Anderson Е Е., Boyd J. A., Prausnitz
J. М. UNIFAC parameters from gas-liquid chromato-
graphic data. Ind. Eng. Chem. Process Des. Dev., 18,
657—661 (1979).
746. Zeleznik F. J., Gordon S. Calculation of complex chemi-
cal equilibria. Ind. Eng. Chem., 60, 27—57 (1968).
747. Zellars G. R., Payne S. L., Morris J. P., Kipp R. L. The
activities of iron and nickel in liquid Fe-Ni alloys. Trans.
AIME, 215, 181—192 (1959).
748. Zellner M. G., Claitor L. C., Prausnitz J. M. Prediction
of vapor-liquid equilibria and ethalpies of mixtures at
low temperatures. Ind. Eng. Chem. Fundamen., 9,
549—564 (1970).
749. Жванецкий И. В., Платонов В. М. «Аналитические
уравнения для расчета энтальпии и теплоемкости
жидких нефтепродуктов», Нефтехимия, XIV, №5,51
(1970).
750. Zhvanetskii I. В., Platonov V. М. Calculation of phase
equilibrium constants for petroleum cuts. Int. Chem.
Eng., 18, 84—85 (1978).
751. Zief M., Wilcox W. R. (eds.). Chap. 2: Phase diagrams,
Wblten G. M., Wilcox W. R. Chap. 11: Column crystalli-
zation, Albertins R., Gates W. C, Powers J. E. Chap. 16:
Phillips fractional-solidification process, McKay D. L.
Fractional Solidification, Vol. 1, Marcel Dekker (1967).
752. Zhuse T. P. Compressed hydrocarbon gases as a solvent.
Petroleum (London), 298—300 (August 1960).
753. Zudkevitch D, Joffe J. Correlation and prediction of
vapor-liquid equilibria with Redlich-Kwong equation of
state. AIChE Journal, 16, 112—119 (1970).
754. Barral Johnson Liquid Crystals and Plastic Crystals,
Gray and Johnson, eds, 1974, v. 2, pp. 254—306.
755. Glanville, Sage, Lacey Ind. End. Chem., 42, 508 (1952).
756. NGPSA Engineering Data Book, 1976
757. Заимствовано из Edmister, Lee Applied Hydrocarbon
Thermodynamics, v. 1, 1983.
758. Смирнов Курс высшей математики, т. I. — М.: Физ-
матгиз, 1964.
759. Sandler Chemical and Engineering Thermodynamics,
1977.
760. Hunter, Brown. Ind. Eng. Chem., 39, 1343 (1947).
761. Smittenberg, Hoog, Henkes. J. Am. Chem. Soc., 60, 17
(1938).
762. Cady G. H. J. Am. Chem. Soc., 56, 1431 (1934).
763. Platto Z. Physik. Chem., 59, 383 (1907).
764. Sisler Davidson, Lyon, JACS, 66, 1888 (1944).
765. Andreus J. Phys. Chem., 29, 1041 (1925).
766. Labowitz, Arents Physical Problems and Solutions
(1969).
767. Sillen et al. Problems in Physical Chemistry (1952).
768. Цеканская и др. Журн. физ. хим., 38, 2166 (1964).
769. Zia, Thodos Can. J. Chem. Eng., 52, 530 (1974), Hein-
rich et al. Int. Chem. Eng., 20, 77 (1980).
770. Int. Data Series, Series B, 120, 121 (July 31, 1980).
771. Handbook of Tables for Applied Engineering Science,
1973, p. 77.
772. Kennard S. M. S., McCuster P. A. J. Am. Chem. Soc., 70,
3375 (1948).
773. Hudson Z. Physik Chem., 47, 113 (1904).
774. Kipling Proceedings of the Second International Con-
gress of Surface Activity, 111, 462 (1957).
775. Mackenzie, ed., Differential Thermal Analysic, v. 2,
1972.
776. Weber, Meissner Thermodynamics for Chemical En-
gineers, Wiley, 1957
777. Int. Data Series, Prop. Aq. Solutions, No. 110, June 3,
1980.
778. Milne Numerical Calculus, 1949.
779. CRC Handbook of Mathematical Sciences, 5th ed., 1978.
780. Int. Data Series, Thermo. Prop. Aq. Org. Systems,
No. 97, 1979.
Авогадро
число 89, 94
Адиабатическое расширение 143
Аддитивность
давлений
— парциальных 10
объемов парциальных 10
Адсорбент 443
Адсорбция 443
равновесная
— смеси 446
------ бинарной 451
чистых веществ 447
Азеотропность 221
Азеотропные смеси 228, 232, 247,
262, 265
двойные 229
многокомпонентные 229
тройные 229
Активности 139, 244
коэффициент 40, 135, 176, 179,
191, 218, 223, 244
растворов электролитов 176
химическая 27
Амагата закон И
Аморфная жидкость см. Жидкость
аморфная
Анализ дифференциальный термиче-
ский 548, 549, 551, 553
Баланс
материальный 254, 319, 345, 379,
393, 427, 503
тепловой 441
энергетический 117
энтальпийный 239, 325
Бертло уравнение 23, 25, 67
Битти — Бриджмена уравнение
состояния 25, 46, 51, 70, 74
Бинарная эвтектика см. Эвтектика
бинарная
Бесконечное разбавление 218, 229,
247
Ван-дер-Ваальса уравнение
состояния 19, 22, 46, 60, 93, 112,
147
Ван Лаара уравнение 180, 181, 182,
187, 211, 236, 246, 248, 368, 375,
509
Вильсона уравнение 44, 181, 182,
184, 185, 194, 198, 210, 213, 215,
236, 241, 245, 248, 361, 367, 533,
537
Водородная связь 35, 222
Газ
легкий 110
— носитель 220
метастабильный 23
сверхкритический 431
Газы
идеальные 18, 88, 133, 149, 282,
479
неидеальные 18
реальные 92, 149
Газовая хроматография 220
Гйббса
правило фаз 250
энергия см. Энергия Гйббса
Пгббса — Гельмгольца уравнение 142
Гйббса — Дюгема уравнение 117,
135, 136, 146, 248
Гомогенная жидкость см. Жидкость
гомогенная
Давление
влияние на
-----равновесие жидкость —
твердая фаза 276
-----фугитивность 162
----- состав
-------смесей 268
----------азеотропных 227
------- эвтектики 283
диссоциации 470
избыточное 176
конвергентное 313, 314, 317, 318,
323
критическое 38
— многокомпонентных смесей 313
насыщения 21, 69, 11, 167, 310
насыщенного газа 167
насыщенной жидкости 85
осмотическое 176, 244
пара 18, 159, 248, 311, 413
Давление пара бинарной смеси 354
— водяного 509
— жидкости 163
— растворителя 219
---- чистого 178
— твердого вещества 163
парциальное 133, 244, 300
приведенное 159
равновесия 137
разложения 163
смеси 245, 285
системы 219
Дальтона закон 11
Диаграммы фазовые 421, 424, 441
бинарные 344
давление — состав 228, 471
двухкоординатные 250, 265, 288,
297
Гельмгольца 137
жидкость — твердое вещество 271
изотермическая
— кипения 228
нерастворимые твердые фазы 267
плавления 269, 297, 428
плоскостные 252, 254, 282
пространственные 137, 259, 296
— трехмерные 250, 252
равновесия 429
распределения 261
состав — давление 396
состав — температура 342
состав — температура — давле-
ние 254
стереоскопические 250
температура — состав 228, 375
термодинамические 71, 139
треугольные 284
трехкомпонентные 256, 291, 385,
387, 406, 428
трехкоординатная 288, 297
фазовые 352, 461, 462, 464
четырехкомпонентной смеси 259
Р—Т 260, 467
Р—Т—х 466
Р— х 248, 258, 275, 344, 393
Р— V—Т 262
Предметный указатель 657
Диаграммы фазовые Т—х 248, 258,
264, 344, 417, 424
х—у 262, 264, 340, 341, 344, 348,
394, 450
Дилатометр 135
Димеризация 49
Дистиллат 299
Дистилляция
многокомпонентная 311
Дросселлирование 113
Жидкость
аморфная 277
гомогенная 271, 273
метастабильная 23
насыщенная 94
переохлажденная 163, 252, 253,
409
смешивающаяся 269
Законы см. также Правила
Амагата 39
Бойля И
Гей-Люссака И
Генри 118, 160, 244, 257, 337
Клапейрона 11
Льюиса — Рендалла 118, 157, 168
идеальный газов 10, 24
Рауля 118, 156, 158, 169, 244, 257,
265, 354, 447
термодинамический 136
Шарля 11
Замерзание 176
Затвердевание 269
Зона
жидкости
— метастабильной переохлажден-
ной 252
жидкость — твердая фаза 274
двухфазная 252
однофазная 253
перегретого пара 252
твердой фазы 252
Идеальные газы 10, 24
Идеальные растворы 132, 157
Изобара 254, 262, 285
Изоплета 252, 261, 262, 264
Изотенископ 544
Изотерма 262, 285
идеального газа 18
критическая 22, 69
сверхкритическая 22
Фрейндлиха 447
экспериментальная 20
5-образная 21
Испарение
адиабатическое 324
изоэнтропное 322
мгновенное 108
равновесное 322, 483
расчет 380
Калориметрия дифференциальная
сканирующая 540, 553
Коннодаль 254
Конденсация 103
тройной смеси 324
чистых веществ 266
Константа(ы)
Антуана 212
Вильсона 212
Генри 161, 244, 310
критического состояния 89
интегрирования 144
Маргулеса 212
равновесия 36, 165, 284, 477, 487,
489, 496, 507
— димеризации 112, 453
— химического 475, 483, 502, 507
уравнения
— Редлиха — Кистера 192
— NRTL 212
— UNIQUAC 212
фазового равновесия 310
химического равновесия 35
Коэффициент(ы)
активности 40, 135, 176, 179, 191,
218, 223, 244, 311, 340, 381, 395,
411, 412, 414, 419, 485, 533
— адсорбата 452
— бесконечного разбавления 192,
197, 546
— бинарных смесей 535
— расчет 227, 235
— тройной смеси 346
ацентрический 11, 24, 29, 32, 59,
76, 84, 454, 455, 529
бинарного взаимодействия 82, 222
вириальный 31, 45, 142, 519
давления энтальпии 113
Джоуля — Томсона 113, 114, 118,
141, 537
изотермической сжимаемости 52,
517
отклонения 106
относительной фугитивности 133
парциальной фугитивности 82, 309
перекрестного взаимодействия 41
Пойнтинга 170, 227, 233, 309, 310,
315, 333, 338, 431, 448, 478, 480
полярности 83
равновесия
— испарения 107
распределения 310, 315, 321, 325,
410
растворимости 436
регрессионный 386
сжимаемости 29, 79, 92
смеси 45
стехиометрический 138, 474, 475
теплового расширения 24, 97, 113,
517
фазового распределения 178
фугитивности 32, 63, 68, 78, 80,
135, 144, 227, 309, 333, 481
— смесей 146
Коэффициент(ы) фугитивности чис-
тых веществ 145
— эмпирический 44
Кривые
бинодальная 256, 291, 388, 402,
424
замерзания 283
затвердевания 267
идеальная 281
испарения 253
кипения смесей 229
конденсации 327
коэффициентов активности 248
нагревания 411, 549, 550, 551
насыщения 57, 58, 61
охлаждения 299, 550, 551
плавления 253, 267, 270
равновесия 295, 418
— истинные 344
распределительная 284, 288, 289,
291
сублимации 253
температура — состав 267
энергии Гиббса 140, 360
— смеси 359
— смешения 356, 362, 363, 364,
365
---бинарных смесей 363
Критерий
Полинга 113
статистический 246
Кристалл жидкий 457
Кристаллизация 264, 298
Критические
давление 31, 96, 99
плотность 96
свойства 22, 23 , 95
— тройных смесей 101
сжимаемость 11, 67, 95
температура 31, 96, 99
— растворения 269
точка 9, 62, 102, 288
— растворения 269, 271
Летучесть относительная 221, 232,
446
Линия
кристаллизации 270
ликвидуса 229, 248
отвердения 274
пара 229, 248
соединительная 289
солидуса 269
равновесных фаз жидкость —
жидкость 289
Локус критический 464, 466
Метастабильные условия 137
Метод(ы)
Бриджмена 119
Бринклея 495, 501
Вегштайна 54
градиентов 196
групповых вкладов 207
Метод дилатометрический 236, 540
итерационный 232, 507
калориметрический 135, 236, 533
Кардано 22, 28
Карлсона и Кольберна 368
Лагранжа 494
наименьших квадратов 42, 236
Ньютона — Рафсона 28, 70, 197,
198, 227, 235, 316, 327, 492, 493,
500, 508
отсекаемых отрезков касательной
153
Питцера 44, 47
Питцера — Керля 38
проб и ошибок 204
редакции 491, 493, 495, 496, 500,
509
рецикла 490
симплексный 196, 227
скорейшего спуска 228
спектрального анализа 88
статистические 186, 545
стехиометрический 490
термический 540
хроматографический 220
Чао — Сидера 222, 311, 312, 348
Шоу 141
API 314, 323
ASOG 44, 211, 224, 227, 418, 419
UNIFAC 44, 226, 227, 413, 418, 419
Многокомпонентные смеси 208
Моделирование
жидкой фазы 109
паровой фазы 109
Молекулярная
ассоциация 35
структура 44
Молекулярный кластер 195
Момент дипольный 31, 34
Моногидрат 261
Насыщение 61
Ионограмма параметров уравнения
Вильсона 196
Неполярный компонент 222
Область
гомогенная 294
гетерогенная 290
двухфазная 252, 287 , 288 , 290, 291,
355, 357, 464, 505
метастабильного состояния 115,
355
многофазная 293
множественных корней 214, 215
нестабильная 355
ограниченной растворимости 272
однофазная 288, 290, 357
перегрева 170
переохлаждения 170
растворимости 288
стабильная 355
сверхкритическая 170, 436
трехфазная 254, 288, 290, 385
Объем
атомный 14
избыточный 127, 236, 242
критический 22
молекулярный 47, 295
молярный 182, 189, 222, 409
насыщения 21, 27
насыщенной жидкости 69, 85
остаточный 18
парциальный молярный 141
приведенный 12, 13
сжатой жидкости 85
сжижения 102
смеси 222
удельный 21, 86
чистого растворителя 177
эффективный молекулярный 21
Осмотическое давление 176, 177,
242, 244
водного раствора 242
крови 242
Пар водяной 300
Параметр(ы)
бинарного взаимодействия 24, 41,
59, 62, 63, 72, 210
взаимодействия 224
— между компонентами 183
корреляционный 45
критический 52
множественные 186
непроизвольности 183
объема 208
перекрестных взаимодействий 24,
53, 62
поверхности 208
растворимости 83, 197, 213, 221,
222
сжимаемости 78
уравнения
— ван Лаара 186, 234, 244, 345,
438
— Вильсона 186, 195, 198, 199,
237, 437
— Маргулеса 192, 199, 238 , 245,
438
— NRTL 206, 409
электростатический 27
энергетический 34
Перитектика 252, 271, 273
Плавление
инконгруэнтное 252
конгруэнтное 252
Плотность
молярная 96
насыщения 97
насыщенной жидкости 83, 85
насыщенных паров 111
сжатой жидкости 83, 85
Поверхность
энергия Гельмгольца — темпера-
тура — объем 122
Поверхность энергия Гйббса — дав-
ление — температура 122
энтальпия — энтропия — давле-
ние 122
Полиморфизм 260
Полиморфные превращения 250
Полярность 34, 158
Постоянная (ые) см. также Кон-
станты, Коэффициенты,
Числа
Больцмана 34, 88, 94
Генри 404
газовая 22
— универсальная 23
равновесия 163
Потенциал
бинарного взаимодействия 89
бинарной системы 90
жесткой сферы 90, 92
ионизационный 34
Кихары 90
Леннарда-Джонса 89, 91
межмолекулярного взаимодейст-
вия 45, 108
Сазерленда 90
химический 102, 123, 137
Правило(а) см. также Законы
аддитивности 22, 24
Кэя 37, 58, 102, 529
Ладурелли 213, 218
Лоренца — Бертло 24, 59, 333,
530
Льюиса — Рендалла 446
Максвелла 111
подобия размерностей 27
Праузница — ГУнна 39
рычага 252, 253, 255
Симпсона 138
Тассио 219
усреднения свойств 40, 154
— псевдокритических 24
фаз Гйббса 250, 252, 257
Приведенные
давление 25, 78
— пара 30
объем 25
плотность 71
температура 25, 65, 78
уравнение состояния 25
Принцип(ы)
Ле-Шателье 264
Максвелла 27
независимого действия 223
соответственных состояний 11, 23,
25, 71
Процесс см. также Реакции
димеризации 115
периктектический 270, 274
обратимый
— адиабатический 113
осмотический 137
Работа 117
Предметный указатель 659
Работа изотермического процесса
219
обратимого процесса 117
Равновесие(я)
адсорбционное 448
бинарных смесей 446
двухкомпонентных систем 269
диффузионное 137
гидростатическое 137
жидкость — адсорбат 448
жидкость — жидкость 8, 202, 207,
208, 210, 211, 227, 233, 261, 352,
383, 384
жидкость — пар 57, 242, 406
жидкость — расплав 8
жидкость — твердая фаза 264,
276, 295
жидкость — твердое вещество
252, 255 , 260, 411, 424
интермолекулярного соединения
264
испарения 64
метастабильное 384
многофазных систем 399
пар — жидкость 71, 108, 202, 209,
211, 220, 233, 252, 255, 261, 262,
284, 309, 418, 449, 547
— тройной смеси 248
пар — жидкость — жидкость 299,
392, 394
пар — жидкость — твердая фаза
263, 400
пар — твердое вещество 252
плавления 406, 411
растворения 406
реакции 481
стабильное 384
температурное 137
трехфазное 393
фазовое 8, 29, 137, 227, 289, 291,
293, 295, 297, 501, 504, 509
— в сверхкритических условиях
339
химическое 474, 476, 481, 506
Равновесный состав
смеси 143
фаз 291
Разбавление
бесконечное 199, 200, 204, 211
псевдокритическое 199
Распределение Максвелла — Боль-
цмана 88
Растворение газа 541
Растворимость
бинарных смесей 381
газов 170, 222, 338, 391
— в жидкостях 337
жидкостей 229, 388
идеальная 408, 412
массовая 244
ограниченная 257, 272
— жидких фаз 272
— твердых фаз 273
Растворимость ограниченная
взаимная
— жидкостей 264
— твердого вещества 406, 542
— твердых растворов 264
смеси 221
твердых веществ 170
Растворитель 176, 409
Раствор (ы)
ассоциированный 195
атермический 157
водный 300
жидкий 160
идеальные см. Идеальные
растворы
концентрированный 178, 456
критический 353
разбавленный 178
твердый 160, 252, 273 , 274, 277
регулярный 157, 221
Рауля закон 169, 265
Реактор периодического действия
490
Реакция
бимолекулярная 487
газофазная 486
глубина протекания 502, 506, 507
жидкофазная 483
независимая 258
сложная многокомпонентная 138
химическая 138
Режим
адиабатический 117
изотермический 219
Свойства
аддитивные 132
жидкой фазы 180
идеальной смеси 132
избыточные 31, 127, 529, 532
— расчет 175
криогенные 108
критические 27, 58, 162
общие 123
остаточные 22, 511, 512, 517
парциальные молярные 123, 124
псевдокритические 24, 81
приведенные 23
реальной смеси 132
специфические 123
термодинамические 34, 461, 511,
514
физические 34
чистых компонентов 221
электростатические 27
экстенсивные 132
Сжимаемость 400
газа 165
жидкости 9, 334
изотермическая 97, 113
кажущаяся 36
критическая 23
Сжимаемость парциальная молярная
129
паровой фазы 334
расчет 50, 84
Система (ы)
бинарная 262, 394
— эвтектическая 551
водные органические 206
водных электролитов 453
двухкомпонентные 128, 261
жидкость — жидкость 209, 264,
288, 366, 384, 392
замкнутая 117
идеальная 506
конденсированные 260, 295
меритектические 274, 279
мицеллярные 290
многокомпонентные 210, 424
нераздельнокипящие 265
однокомпонентная 252, 260
открытая 117
пар — жидкость 309, 380, 392
пар — жидкость — жидкость 397
перитектические 274, 279, 551
проточная открытая 117
с взаиморастворимыми жидкими
фазами 266
— взаиморастворимыми тверды-
ми фазами 264
— конгруэнтной тройной компо-
зицией 295
— несмешивающимися жидкостя-
ми 295
— перитектической тройной ком-
позицией 295
реакционная 488
твердая фаза — жидкость 251
трехкомпонентная 253, 285, 393
тройные 104, 288, 295, 403
четырехкомпонентная 257, 387
— двухфазная 386
Смешиваемость 290
жидкостей 222
жидких смесей 221
фаз 420
Смесь(и)
азеотропные 83
бинарные 104, 138, 209, 367, 403,
417, 440
внутренняя энергия 132
газообразная 115
двухкомпонентная 49, 136, 194,
229, 392
жидкие 352
идеальная 155, 248
многокомпонентные 197, 208, 233,
379
неводные 206
неполярные 197
полярные 197
трехкомпонентные 39, 209, 379
Смесь тройная 256, 397, 425, 432
четырехкомпонентная 126, 155,
209, 381
эквимолярная 37, 347
смешиваемость 353
Соединения
бинарные 251
метастабильные 296
интермолекулярные 252, 257, 259,
262, 267, 269, 274, 276, 278, 296,
297
твердые 251
тройные
251
Состав
азеотропных смесей 233
жидкость — адсорбат 448
жидкой фазы 231, 325
многокомпонентных смесей 161
молекулярный 271
паровой фазы 230, 235, 284, 289,
325
равновесный 352, 371, 378, 401,
485, 487, 507, 508
— парожидкостный 286
— смеси пар — жидкость 218, 219
тройных систем 252
фракционный 379
эвтектический 417, 423
Состояние
газообразное 133
докритическое 24
идеальное 131
метастабильное 388
неравновесное 118
неустойчивое 137
перегретое 57
равновесное 118
сверхкритическое 275
стабильное 388
стандартное 118
твердое 269
термодинамическое 135
Стабильность термодинамическая
353
Стандартные
состояние 159, 169
— газа 159
— отклонение 195
— растворенного вещества 159
— растворителя 159, 160
условия 112, 140
Степень
ассоциации 35
димеризации 49
превращения 36, 475, 476, 507,
508, 509
— относительная 140
развития химической реакции 474
растворимости 295
Стехиометрические соединения 251
Структура(ы)
нематическая 458
Структура холестерическая 458
ячеистая 204
Сумма
мольных долей 24
избыточных энергий Гиббса 209
Температура
влияние
— на коэффициенты активности
207, 236, 237
-----параметры
-----растворимости 223, 245
-------уравнения Вильсона 239
-------уравнения Ван Лаара
239, 240
-----состав смесей 267
замерзания 176, 177, 243
— морской воды 243
изоэнтропийная 528
испарения 310
кипения 31, 85, 228, 230, 244, 262,
284, 454, 544
— смеси 218
— разбавленных растворов 543
криогенная 71, 85
критическая 34, 162, 454
— растворения 354, 375
насыщения 243
начала кипения 329, 507
— расчет 346
нормальная 31
пара 326
плавления 34, 160, 251, 417, 420,
428
— твердого вещества 159
— эвтектики 276
----- тройной 428
приведенная 17, 146
равновесия 406
растворения 276, 290, 381
— критическая 382
сверхкритическая 106, 310
субкритическая 106
характеристическая 17
эвтектическая 298, 300
Теорема
однородных функций 132
Эйлера 128
Теоретическая тарелка 248
Теория регулярных растворов 222
Теплоемкость
идеальных газов 18, 512, 527, 538
избыточная 127
удельная 107
— воды 243
— льда 243
Теплота
испарения 97, 98, 537
плавления 160, 243, 437
смешения 135
сублимации 160
Термодинамика статистическая 29
Термодинамические поверхности 122
реакции 487
свойства 120
функции 88, 117, 223
— парожидкостного равновесия
102
характеристики 45
Термограмма 552
Точка
азеотропы 227
Бойля 113
замерзания 160, 176, 177, 178, 437
инверсии Джоуля-Томсона 113
кипения 87, 176, 316
— смеси 299
кристаллизации 260
критическая 56, 252, 430, 436
метастабильная твердая 253
начала кипения 326, 392, 394, 398
перитектическая 279, 297
плавления 34, 160, 162, 270, 272,
296, 406, 551
— нестабильная 278
растворимости 288
росы 103, 310, 318, 323, 324, 326,
329, 330, 392, 394, 398, 507
— расчет 336
седловидная 228
тройная 160, 170, 251, 252, 253,
259, 407
эвтектическая 298, 422, 423 , 427
Третий закон термодинамики 169
Уравнения см. также Уравнения со-
стояния
Антуана 15, 35, 311, 534
Бенедикта — Уэбба — Рубина 9,
11, 21, 41, 46, 69, 70, 72, 75,
112, 147, 311, 518
Бенедикта — Уэбба — Рубина —
Старлинга 9, 41, 73, 76, 106,
147, 310, 516, 518
Бертло 23, 25, 67
Блэка 248
бинарные 128
Больцмана 15
ван Лаара 180, 181, 182, 187, 211,
236, 246, 248, 368, 375, 509
Вильсона 44, 181, 182, 184, 185,
194, 198, 210, 213, 215, 236, 241,
245, 248, 361, 367, 418, 420, 421,
422, 423, 533, 537
вириальное 11, 70, 516
возмущенной жесткой сферы 107
В-усеченное вириальное 141, 233,
313, 434, 522
Гиббса — Гельмгольца 142
Гиббса — Дюгема 117, 135, 136,
146, 248
Гугенгейма 114
Дитеричи 23, 25
испарения 320, 486
Предметный указатель 661
Уравнения испарения мгновенного
507
идеального газа 74
Клапейрона 29
Клаузиуса 514
кубическое 21, 50
Ленгмюра 445 , 446
Ли — Кеслера 35, 71, 79, 80, 86,
147, 418, 513, 518, 524, 534
Ли — Кеслера — Плекера 9
Максвелла 514
Маргулеса 180, 181, 182, 187, 211,
235, 236, 340, 361, 366, 367, 374,
375, 422, 538
Мартина 51
материального баланса 393
многопараметрическое 186
Накамуры — Бридвельта — Пра-
узница 106
Нишиуми 70
парообразования 326
Пенга — Робинсона 51, 62, 64,
65, 66, 147, 281, 310, 315, 518,
524, 530
Питцера 247
Плекера — Ли — Кеслера 310
полиномиальное 27
приведенное 21, 53
равновесия 475, 480, 488, 496
растворимости 407
регрессионное 242
Редлиха — Квонга 11, 40, 52, 53,
58, 60, 65, 93, 104, 112, 147,
311, 335, 436, 516, 518, 530
Редлиха — Кистера 187, 192, 246
Ренона 204
симметричное 181, 182, 185, 235,
367
Скэтчарда — Гильдебранда 181,
182, 184, 185, 191, 202, 211, 213,
236, 246, 311, 338, 339, 390, 407,
409, 531, 533
Скэтчарда — Гильдебранда —
Флори — Хаггинса 184, 191,
202, 221, 246, 313, 332
Скэтчарда и Хамера 187
Старлинга 70
Старлинга — Хана 41
стехиометрическое 259
Соава 41, 51, 59, 61, 65, 66, 112,
147, 310, 311, 315, 334, 522, 524,
529, 530
термодинамическое 170
трансцендентное 195
универсальное
— квазихимическое 207
Флори и Хаггинса 221, 223, 407,
469
Фрейндлиха 445, 446, 452
фугитивности 22
Фуллера 63
фундаментальное 121
Хана — Старлинга 76, 104
Уравнения Харменса — Кнаппа 11,
61, 68
Цонопулоса 112, 167, 510, 523
Цубоки — Катаямы — Вильсона
181, 182, 184, 185, 202, 246, 248,
361, 364, 365, 367, 374, 375
Шредера 297
Эббота 39
Эд мистера 35
Эллиса и Джонса 245
энергетического баланса 118
ASOG 387, 388, 451
LEMF 384
NRTL 180, 181, 184, 185, 204, 205,
210, 211, 216, 217, 236, 361, 367,
384, 420, 423, 533
UNIQUAC 180, 181, 184, 185, 205,
236, 248, 384, 388, 451
UNIFAC 387, 388, 533
Уравнения состояния 14
Битти — Бриджмена 25, 46, 51,
70, 74
Бойля 14
Ван-дер-Ваальса 19, 22, 46, 60, 93,
112, 147, 518, 525, 530
Гей-Люссака 11, 14
Готро и Коутса 219
Дальтона 10
Джоуля — Томсона 14
Клаузиуса 14, 25, 67, 74, 111, 409
Планка 17
Смолуховского 16
Условия
изобарические 512
изотермические 512
изоэнтропийные 528
Фаза
жидкая 178, 227, 251, 286, 287,
288, 294, 309, 429, 433, 459, 479,
504
— изотропная 459
— расслоение 233
изотропная 459, 461
конденсированная 133, 159, 275,
477
метастабильная 260
— паровая 253, 309
нематическая 459
обогащенная 298
обедненная 298
паровая 20, 108, 227, 286, 287,
479, 504
— неидеальность 219, 227, 232
пересыщенная 384
сжиженная 281
смектическая 459
смешанная 483
твердая 252, 274, 275 , 280, 294,
433, 459
Фазовые
диаграммы см. Диаграммы
фазовые
Фазовый переход 298
равновесие 64, 138, 227, 250, 289,
291, 293, 295
Фугитивность
газов 165, 169, 477, 486
жидкостей 160, 165, 311
зависимость от давления 162, 163
конденсированной фазы 159, 170
методы расчета 159, 160
насыщенной жидкости 162
насыщенного пара 162
относительная 315
паровой фазы 159, 165, 170, 400
парциальная 77, 144, 227, 406, 431
— молярная 124
перегретой жидкости 339
смеси 144, 166
— тройной 166
твердого вещества 160, 163
чистых веществ 145
Функции
избыточная 135
— энергии Гиббса 248
Лагранжа 498
Масьё 132, 527
однородная 128
отклонения 78, 83
Планка 132, 136, 527
потенциальная 89, 94
приведенной температуры 56
разделения 113
распределения 119
термодинамическая 135
целевая 186, 195
энергетическая 137
Характеристики
чистой жидкости 176
Химические
потенциал 176, 257
превращение 269
соединение 271
состав 298
Хроматография адсорбция 443
Хроматографическая колонка 220
Числа см. также Константы, Коэф-
фициенты, Постоянные
Авогадро 89, 94
координационное 183, 204, 208,
236
Прандтля 97, 99
степеней свободы системы 257
фаз 293
флегмовое 440
Шредера уравнение 297, 417, 420,
425
Эбулиометр 543, 545
дифференциальный 543
Эбулиоскопия дифференциальная 221
Эбулиоскопическое измерение 220
Эвтектика 252, 269, 271, 273
бинарная 267, 425, 426, 438
Эвтектика твердофазная 279
тройная 259, 260, 295, 296, 424,
433, 436, 438
Эвтектические
смесь 271, 273
точка 255
Эквимолярная
смесь 247
— двухкомпонентная 244
— тройная 111, 244
Энергия
взаимодействия молекул 208
внутренняя 88, ИЗ, 117, 118, 221
— испарения 208, 247
вращательная 88
Гельмгольца 87, 88, 117, 526
Гйббса
— бинарной смеси 355, 358
— избыточная 40, 127, 135, 171,
172, 180, 191, 204, 245, 246, 404
— минимизация 344, 378, 398,
492, 501, 503
— образования 140, 475
Энергия парциальная молярная 102,
123, 166
— смеси 140, 344, 476, 492
— смешения 353, 358, 361
— чистых веществ 204, 345, 398
испарения 222
квантованная 88
колебательная 88
молекул 88
образования 399
потенциальная 88
равновесная 137
характеристическая 34
Энтальпия
жидкости 107
замерзания 178
— чистой жидкости 178
идеального газа 131
избыточная 127, 142, 172, 241,
531, 533, 534
— парциальная 237
— эквимолярных смесей 245
изменение 121
Энтальпия испарения 178, 247, 479,
534, 536
— воды 242
кристаллизации 178
молярная 243
— парциальная 533
отклонения 108
остаточная 32, 73, 512, 513, 519,
524
— жидкостей 526
плавления 407, 409, 414, 420, 438
смешения 456
Энтропия
идеального газа 68, ИЗ, 527
избыточная 135, 172, 221, 531, 534
общая 133
остаточная 73, 513, 514, 519, 520,
521
— жидкостей 526
плавления 407, 420
смешения 415
— идеального газа 133
Якобиан 104
ОГЛАВЛЕНИЕ
6. Равновесие в системах пар — жидкость ....
6.1. Паровая и жидкая фазы ................
6.2. Константа фазового равновесия ........
6.3. Равновесия с К, не зависимыми от состава
6.4. Равновесия с К, зависящими от состава ..
6.5. Растворимость газов в жидкостях ......
6.6. Бинарные диаграммы равновесия пар —
жидкость ..................................
6.7. Минимизация энергии Гйббса ...........
6.8. Задачи ...............................
7. Равновесия жидкость — жидкость ...........
7.1. Жидкие смеси .........................
7.2. Фазовые диаграммы ....................
7.3. Смешиваемость и термодинамическая ста-
бильность .................................
7.4. Бинарные смеси .......................
7.5. Трех- и многокомпонентные смеси ......
7.6. Прогнозирование условий равновесия в сис-
темах жидкость — жидкость с применением
уравнений UNIFAC или ASOG .................
7.7. Растворимость жидкостей ..............
7.8. Равновесие пар — жидкость — жидкость ...
7.9. Равновесие в системе пар — жидкость —
твердое вещество ..........................
7.10. Задачи ..............................
8. Равновесие жидкость — твердое вещество ..
8.1. Введение .............................
8.2. Растворимость твердых веществ в жидко-
сти .......................................
8.3. Равновесие плавления .................
8.4. Бинарные смеси .......................
8.5. Многокомпонентные системы ............
8.6. Растворимость твердых веществ и жидко-
стей в сверхкритических газах .............
8.7. Задачи ...............................
9. Другие аспекты фазового равновесия ......
9.1. Адсорбция ............................
9.2. Системы водных электролитов ..........
9.3. Соединения и смеси неопределенного соста-
ва ........................................
9.4. Полимеры .............................
9.5. Жидкие кристаллы и мицеллы ...........
9.6. Смеси в сверхкритических условиях ....
9.7. Гидраты ..............................
9.8. Задачи ...............................
309
309
310
316
337
344
344
347
352
352
352
353
367
379
387
388
392
400
401
406
406
406
411
417
424
431
436
443
443
453
454
455
457
463
466
471
10. Химическое равновесие ................. 474
10.1. Введение ........................... 474
10.2. Степень «развития» химической реакции . 474
10.3. Уравнение равновесия ............... 475
10.4. Константа равновесия и стандартные со-
стояния ................................. 477
10.5. Равновесный состав ................. 487
10.6. Множественные реакции .............. 489
10.7. Объединенное химическое и фазовое равно-
весие ................................... 501
10.8. Задачи ............................. 506
11. Оценка изменений энтропии и энтальпии .. 511
11.1. Основные уравнения ................. 511
11.2. Оценка остаточных свойств .......... 512
11.3. Специальные уравнения состояния .... 516
11.4. Влияние изменений Р и Т ............ 526
11.5. Смеси .............................. 528
11.6. Смешение жидкостей ................. 533
11.7. Энтальпия испарения ................ 534
11.8. Смеси углеводородов ................ 535
11.9. Задачи ............................. 535
12. Принципы экспериментальных методов иссле-
дования фазового равновесия ................ 540
12.1. Растворимость в жидкостях .......... 540
12.2. Температура кипения и давление пара .... 544
12.3. Равновесие пар — жидкость .......... 545
12.4. Равновесие твердое вещество — жидкость
(равновесие плавления) .................. 549
12.5. Обзоры экспериментальных методов изуче-
ния фазового равновесия ................. 553
Приложения ................................. 554
Приложение А. Основные формулы термодина-
мики ..................................... 554
Приложение Б. Численные методы .......... 560
Приложение В. Программы для ЭВМ ......... 566
Приложение Г. Физические свойства ........ 578
Приложение Д. Параметры и корреляции .. 593
Приложение Е. Используемые единицы и их эк-
виваленты ................................ 635
Приложение Ж. Литература ............... 636
Предметный указатель ....................... 656
УВАЖАЕМЫЙ ЧИТАТЕЛЬ!
Ваши замечания о содержании книги, ее оформлении,
качестве перевода и другие просим присылать по
адресу: 129820, Москва, И-110, ГСП, 1-й Рижский
пер., д. 2, издательство «Мир».
Научное издание
Стенли М. Уэйлес
ФАЗОВЫЕ РАВНОВЕСИЯ В ХИМИЧЕСКОЙ ТЕХНОЛОГИИ
В 2-х частях
2
Заведующий редакцией академик О.А. Реутов.
Зам. зав. редакцией З.Ф. Ходецкая. Старший научный редактор Р.И. Краснова.
Младший редактор И.С. Ермилова. Мл. научный редактор И.И. Землячева. Художник
Ю.С. Урманчеев. Художественный редактор М.Н. Кузьмина. Технический редактор
Н.И. Борисова. Корректоры В.А. Валуева, Р.Л. Вибке.
ИБ № 6583
Подписано к печати 13.06.89.
Формат 84 х 108 Ив. Бумага офсетная № 1.
Гарнитура тайме. Печать офсетная, (фотоофсет).
Объем 11,25 бум. л. Усл. печ. л. 37,8.
Усл. кр.-отт. 64,66. Уч.-изд. л. 40,89.
Изд. № 3/5747. Тираж 3200 экз. Зак.864.
Цена 6 р. 20 к.
Издательство «Мир» В/О «Совэкспорткнига» Государственного комитета СССР
по делам издательств, полиграфии и книжной торговли.
129820, ГСП, Москва И-110, 1-й Рижский пер., 2.
Набрано в Межиздательском фотонаборном центре издательства «Мир»
Можайский полиграфкомбинат В/О «Совэкспорткнига»
Государственного комитета СССР по делам издательств,
полиграфии и книжной торговли.
143200, Можайск, ул. Мир, 93.