












































































































































Author: Таубе П.Р. Баранова А.Г.
Tags: водоснабжение очистка воды водопотребление гигиена валеология здоровый образ жизни очистка сточных вод водоочистка
Year: 1983




II. Р. ТАУБЕ I , А. Г. БАРАНОВА
химия
И МИКРОБИОЛОГИЯ
воды
Допущено
Министерством высшего
и среднего специального образования СССР
в качестве учебника
для студентов
инженерно-строительных специальностей
вузов
МОСКВА
«ВЫСШАЯ ШКОЛА»
1983
ББК 51.21
Т12
УДК 628.1(075); (075.8)
Рецензенты:
Кафедра водоснабжения Ленинградского инженерно-стронтслыюго
института (зав. кафедрой проф. II. Ф. Федоров),
И. С. Лавров — докт. техн, наук, проф. (Ленинградский техпо/ioi и-
ческий институт им. Ленсовета)
Таубе П. Р., Баранова А. Г.
Т12 Химия и микро;Ло;югяя воды: Учебник для студентов ву-
зов.— М.: Высш, шк., 1983. — 280 с.
В пер.: 90 к.
В учебнике даются теоретические основы курегт «Химия и микробиология во-
ды», описаны физико -имачаекие основы современных методов очистки природных
сточных вод. Приводятся сведения ио общей микробиологии, выясняется роль мик-
роорганизмов в превращении органических веществ в природных условиях, а также
возможность использования микроорганизмов для оценки качества воды и контроля
за работой очистных сооружений. Описываются основные направления биохимиче-
ских процессов, протекающих при очистке питьевой и сточных вод. Изложение ве-
дется на уровне современного состояния науки.
Предназначается для студентов вузов, обучающихся по специальности «Водо-
снабжение и канализация».
т
3206000000—245
001(01) -83
ББК 51.21
6С9.3
149—83
Петр Рейнгольдович Таубе, Алевтина Гавриловна Баранова
химия и микробиология воды
И Б № 3375
гав. редакцией С. Ф. Коцдрашкова. Редактор Т. П. Федорова. Художественный ре-
дактор Т. М. Скворцова. Художник В. Я. Батищев. Мл. редакторы С. М. Ерохина,
Т. С. Костян. Технический редактор Л. А, Григорчук. Корректор С. К. Завьялова.
Изд. № Хим—671. Сдано в набор 19.01.33. Поди, в печать. 22.04.83. Формат 60 /901 'ы-
Бум. тип. № 2. Гаришург! ли;ерагурная. Печать высокая. Объем 17.5 усл. псч. л.
17,5 усл. кр.-отт. 19,64 уч.-изд. л. Тираж 15 009 экз. Зак. № 1214. Пепа 90 кои.
Издательство «Высшая школа». Москва, К-51, Неглинная ул., д. 29 14
Московская типография X? 8 Союзполиграфпрома при Государственном комитете
СССР по делам издательств, полиг^фтГи и книжной торговли, Хохловский пер.. 7.
тельство «Высшая школа», 1983
Предисловие
В Конституции СССР и специальных постановлениях ЦК КПСС
и Советского правительства обращено особое внимание на пробле-
мы охраны природы, рационального научно обоснованного, комп-
лексного использования природных ресурсов, в том числе водных.
Особенно большое внимание было уделено в решениях XXVI
съезда КПСС вопросам комплексного и планового использования
водных ресурсов, совершенствования водоохранной деятельности,
повышения эффективности работы очистных сооружений, разработ-
ки новых методов очистки воды, внедрения водооборотных систем
и создания бессточных технологических схем.
Развитие промышленности и перевод сельскохозяйственного
производства на индустриальную основу способствуют более широ-
кому применению поверхностных вод для водоснабжения. В тоже
время состав их усложняется в результате попадания в них соеди-
нений, использующихся в практической деятельности людей. Это
ведет к тому, что схема очистки таких вод становится более слож-
ной.
Необходимость надежной защиты водоемов от загрязнения
сточными водами промышленного происхождения диктует строгие
нормы допустимого содержания примесей в этих водах. Разработка
и внедрение в практику новых современных методов очистки при-
родных и сточных вод, совершенствование методов контроля за ка-
чеством воды различного назначения потребовали пересмотра из-
ложения отдельных тем курса «Химия и микробиология воды». Осо-
бое внимание было уделено описанию физико-химических основ
современных методов водоподготовки и очистки производственных
сточных вод, определению роли микроорганизмов в процессе само-
очищения водоемов и биологической очистки сточных вод.
Поскольку сведения о многих физико-химических и микробиоло-
гических процессах в общеобразовательных курсах преподносятся
в предельно сжатой, описательной форме или вообще отсутствуют,
можно считать оправданным краткое введение о свойствах воды,
растворов, дисперсных систем. Содержащиеся в тексте таблицы со-
держат минимальный справочный материал, необходимый при изу-
чении курса. Изложение дается на уровне современного состояния
науки.
Учитывая, что 'технологические аспекты процессов водоподго-
товки и очистки сточных вод' рассматриваются д. специальных кур-
3
сах, используемые аппараты и сооружения лишь упоминаются. По
мере возможности авторы старались дать необходимый объем ин-
формации с учетом уровня подготовки студентов, избегая услож-
нения за счет изложения механизма отдельных биохимических
процессов, насыщения текста незнакомыми терминами. По нашему
мнению, книга может быть использована не только студентами, по
и специалистами, работающими в области водоснабжения и кана-
лизации.
Ограниченный объем учебника и его назначение, вытекающее из
программы курса, нс позволяют подробно излагать все вопросы.
Читателям, заинтересованным в специальной литературе по узким
вопросам, мы предлагаем небольшой список наиболее авторитет-
ных монографий.
Считаем обязательным подчеркнуть, что рецензенты уделили
рукописи очень мною внимания. Нашим рецензентам—коллекти-
ву кафедры водоснабжения Ленинградского инженерно-строитель-
ного института (зав. кафедрой докт. техн, наук, проф. П. Ф. Фе-
доров) и заслуженному деятелю науки и техники, докт. техниче-
ских наук, проф. И. С. Лаврову (ЛТП им. Ленсовета) мы приносим
глубокую и искреннюю благодарность за ценные советы и крити-
ческие замечания.
Мы будем признательны читателям за и.х отзывы.
Авторы.
ВВЕДЕНИЕ
Проблема рационального использования водных ресурсов стала
одной из важнейших проблем современности. Развитие промыш-
ленности, перевод сельского хозяйства на индус триальную основу,
рост городов способствуют постоянному росту недопотребления.
Ежедневно человечество расходует до 7 млрд, т воды, что соответ-
ствует по массе общему количеству полезных ископаемых, добы-
ваемому за год. Основными потребителями воды являются хими-
ческая, нефтехимическая, целлюлозно-бумажная отрасли про-
мышленности, черная и цветная металлургия, энергетика, мелиора-
ция. Ежегодно в нашей стране на различные нужды расходуется
более 260 млрд, м3 воды. В том числе промышленность использует
более 80 млрд, м3, а коммунальное водоснабжение потребляет бо-
лее 11 млрд, м3 воды.
Почти половина потребляемого объема воды идет на орошение.
Большое количество воды необходимо для нужд животноводческих
комплексов. Возникновение новых производств обычно сопровож-
дается применением или получением реагентов — неорганических
и органических, которые часто становятся компонентами сточных
вод. Если в начале века в практической деятельности людей ис-
пользовались 54 химических элемента, то в настоящее время — бо-
лее 80. В то же время в технологические процессы включаются и
новые органические соединения, действие которых на водоемы и
качество потребляемой воды еще недостаточно изучено. Наша стра-
на имеет огромное количество источников пресной воды, но распре-
делены они неравномерно. Лишь около 15% от общего запаса при-
родных вод приходится на европейскую часть СССР, где произво-
дится около 85% всей промышленной продукции страны. Резкое
увеличение объема производственных сточных вод, наличие в них
примесей разнообразного химического состава привели к значитель-
ному загрязнению некоторых водоемов (бассейнов рек Волги, Ура-
ла и др.). Увеличение водопотребления и соответственно рост ко-
личества сбрасываемых загрязненных вод потребовали принятия
решительных мер по усилению государственного надзора за потреб-
лением^ охраной водных ресурсов, разработки комплексных меро-
приятий по их рациональному использованию и глубокого всесто-
роннего изучения проблем водного хозяйства страны.
Решение этой проблемы возможно лишь при комплексном под-
ходе. В нашей стране вопросам охраны природы, в том числе и вод-
ных ресурсов, всегда уделялось большое внимание. Основные прин-
ципы рационального использования и охраны природных ресурсов
5
были разработаны в постановлениях и декретах, принятых в пер-
вые годы Советской власти. Они были положены в основу при раз-
работке мероприятий Коммунистической партии и Советского пра-
вительства по вопросам охраны окружающей среды. В 1960 г. в
нашей стране был принят «Закон об охране природы». 10 декабря
1970 г. сессией Верховного Совета СССР были приняты «Основы
водного законодательства Союза ССР и союзных республик» ", ко-
торые определили пути решения водной проблемы. Этот законода-
тельный акт свидетельствует о том, что советское государство рас-
сматривает водные ресурсы как важнейшее народное достояние, а
проблему решения охраны и рационального использования считает
одной из самых важных задач.
Начиная с 1975 г. в Государственные планы развития народного
хозяйства включается раздел об охране природы и рациональном
использовании природных ресурсов. В период с 1970 по 1980 г. был
принят ряд постановлений ЦК КПСС и Совета Министров СССР,
направленных на предотвращение загрязнения и рациональное
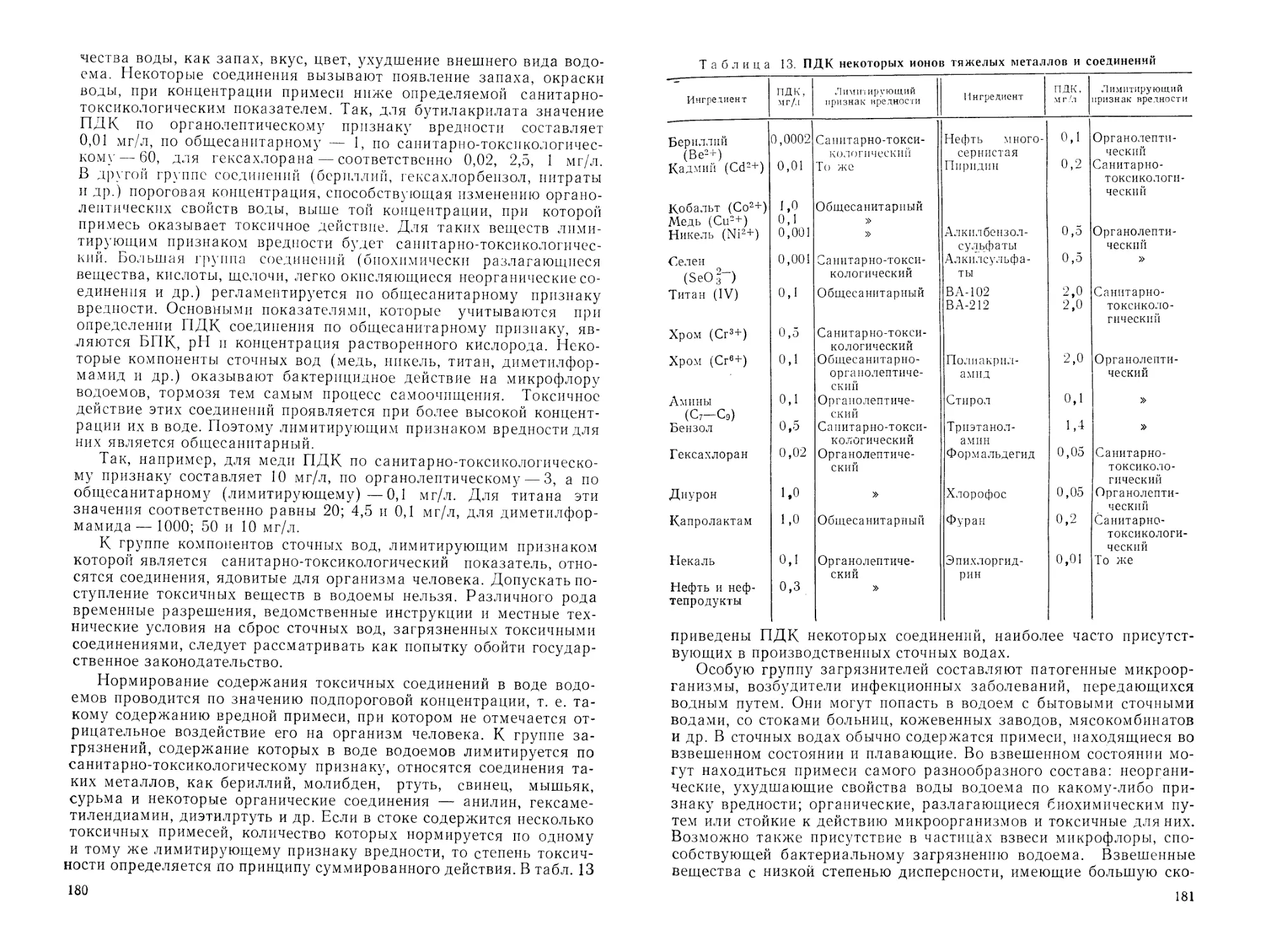
комплексное использование водных ресурсов СССР.
В «Основных направлениях экономического и социального раз-
вития СССР на 1981 —1985 годы и на период до 1990 года»*", при-
нятых на XXVI съезде КПСС, указано: «Увеличить мощности сис-
тем оборотного и повторного использования вод, разрабатывать и
внедрять на предприятиях бессточные системы водоиспользования.
Улучшить охрану водных источников, в том числе малых рек и озер,
от истощения и загрязнения».
В СССР создан Государственный комитет по гидрометеослужбе
и контролю за качеством окружающей среды. В основной закон го-
сударства — Конституцию СССР, принятую 7 октября 1977 г., вклю-
чена статья, посвященная охране и рациональному использованию
природных ресурсов (ст. 18). Все эти мероприятия продиктованы
заботой Коммунистической партии и Советского правительства о
здоровье настоящего и будущего поколений в СССР. Советский
Союз активно участвует в работе многих международных органи-
заций по охране окружающей среды. Исследования, проводимые в
научно-исследовательских институтах, высших учебных заведениях,
работниками производства способствуют внедрению новых эконо-
мичных способов очистки, повышению эффективности и интенсифи-
кации применяемых методов, позволяют сократить расход воды на
технические нужды и возвращать в производство ценные отходы.
* Сборник нормативных актов по охране природы. М., 1978, с. 214.
Основные направления экономичемского и социального развития СССР на
1981 — 1985 годы и на период до 1990 года. М„ 1981, с. 69.
РАЗДЕЛ ПЕРВЫЙ
ТЕОРЕТИЧЕСКИЕ ОСНОВЫ КУРСА
Глава I. СВОЙСТВА ВОДЫ И ВОДНЫХ РАСТВОРОВ
§ 1. Вода как химическое соединение
Рис. L Строение молекулы
воды
Природные, сточные и другие виды вод представляют собой
сложные системы, основу которых составляет химическое соедине-
ние, имеющее простую формулу Н2О. На первый взгляд, за этой
формулой стоит ничСхМ по примечательное соединение. При более
глубоком ознакомлении со строением молекулы и свойствами воды
выясняется, что опа обладает рядом уди-
вительных свойств.
Молекула воды состоит из двух ато-
мов водорода и одного атома кислорода.
Для состава воды характерно следующее
массовое содержание элементов (%): во-
дорода 11,19 и кислорода 88,81. Ядра ато-
мов в молекуле воды расположены по
углам равнобедренного треугольника, в
вершине которого находится ядро атома
кислорода. Молекула воды — плоская
угловая. В невозбужденных молекулах
воды угол НОН, образованный направле-
ниями связей кислород — водород, со-
ставляет 104°27' (для парообразного сос-
тояния) и 104°3iz (для жидкого) (рис. 1).
Межъядерное расстояние О—Н в молекулах воды равно 0,09568 им
(в газовой фазе), 0,09572 нм (в жидкой) и 0,099 нм (в фазе льда).
Колебательное, вращательное и электронное возбуждение молекул
годы вызывает изменение размеров и формы молекулы Н2О. При-
чем энергетически выгодной является одновременная деформация
угла и длины связи О—И.
Направленность химических связей и распределение электрон-
ных плотностей в молекуле воды можно объяснить электростатиче-
ским взаимодействием между атомами водорода при образовании
связей О—И и гибридизацией s- и /^-электронных орбиталей атома
кислорода. В невозбужденном атоме кислорода электронная струк-
тура 2-го слоя (2s22pz22p.v12/?y1) описывается следующим образом:
2х2-электроны образуют сферическое облако над электронным об-
лаком 1-го слоя (1s2), а плотность заряда электронов р-подуровпя
симметрично распределена в форме гантелей вдоль осей х, у, г, рас-
положенных под углом 90° относительно друг друга. Молекула
7
воды образуется в результате перекрывания оолаков s-электронов
атомов водорода с электронными облаками (2щ и 2щ;) атомов кис-
лорода. При этом максимум перекрывания наблюдается при вели-
чине угла связей 90°. ио под действием взаимного электростатиче-
ского отталкивания атомов водорода угол направления химичес-
ких связей НОН возрастает. Этому же способствует п гибридиза-
ция электронных орбиталей атома кислорода. В результате гибри-
дизации угол между валентными связями в молекуле воды стано-
вится близким к тетраэдрическому. Причем образуются две полно-
Рис. 2. Направленность электронных орбиталей (и) и схе-
ма распределения зарядов в молекуле воды (б)
стыо и две наполовину заполненные гибридные орбитали. Две из
них участвуют в образовании связи О—Н, а другие, содержащие
две неподелениые пары электронов, несимметрично удаляются от
ядра атома кислорода в сторону, противоположную связям О—Н.
Таким образом, электронное облако молекулы воды имеет вид
четырех лепестков, направленных к вершинам неправильного тет-
раэдра. В двух вершинах одной грани находятся электронные пары,
осуществляющие связь О—Н, а в двух вершинах противополож-
ной грани находятся неподелениые электронные пары атома кисло-
рода (рис. 2). Электронное облако в молекуле воды смещается к
атому кислорода, так как он имеет большую электроотрицатель-
ность. Вследствие этого вблизи ядер атомов водорода создается
избыток положительного заряда. Поэтому молекула воды поляр-
на. Полярные молекулы характеризуются электрическим моментом
диполя, который влияет на свойства веществ, состоящих из таких
молекул. Электрический момент диполя представляет собой произ-
ведение одного из зарядов на расстояние между центрами положи-
тельного и отрицательного заряда I. Для воды электрический мо-
мент диполя p = el равен 6-10~28 Кл-м. Благодаря большому зна-
чению электрического момента диполя между молекулами воды
проявляется сильное взаимодействие за счет образования водород-
ных связей между атомом кислорода одной молекулы воды и ато-
мом водорода другой.
Каждая молекула воды может образовать четыре водородных
связи: две возникают при взаимодействии неподеленных электрон-
ных пар атома кислорода с атомами водорода соседних молекул
воды и две дают атомы водорода, взаимодействующие с атомами
8
кислорода двух других молекул воды. Водородная связь возникает
в результате внедрения очень малого по размерам положительно
поляризованного атома водорода одной молекулы в электронную
оболочку отрицательно поляризованного атома кислорода другой
молекулы воды. Длина водородной связи О ... II изменяется в пре-
делах от 0,14 до 0,2 нм. Энергия ее составляет 17—33 кДж/моль.
Существованием водородных связей объясняется аномально высо-
кая температура плавления воды по сравнению с водородными
соединениями аналогов кислорода (H2S, H2Se, Н2Те). Водородные
связи и донорно-акцепторные взаимодействия способствуют возник-
новению высокой упорядоченно-
сти внутренней структуры в жид-
кой воде. Водородные связи обус-
ловливают высокое внутреннее
давление, которое способствует
появлению у воды некоторых осо-
бых свойств.
Рассмотрим структуру воды в
различных агрегатных состояни-
ях. При 0° С (273,15 К) жидкая
вода превращается в лед. Сред-
няя величина электрического мо-
мента диполя у молекулы воды в
упорядоченной структуре льда
составляет 8,58-10“28. Плотность
льда при 0° С 0,9168 г/см3.
Для воды характерно сущест-
вование нескольких полиморфных
Рис. 3. Взаимное расположение моле-
кул воды в структуре льда:
-------------химические связи,
-------------— водородные
форм льда. В зависимости от
температуры и давления, при которых происходит формирование
кристаллической фазы, различают 13 видов льда. При обычных
условиях устойчивым является лед, имеющий гексагональную
структуру. Данными рентгеноструктурного анализа подтверждено,
что в кристалле льда, имеющем молекулярную кристаллическую
решетку, каждая молекула воды тетраэдрически окружена четырь-
мя другими молекулами, образующими с ней водородные связи
(рис. 3). Подобное соединение молекул воды друг с другом спо-
собствует образованию пустот в кристаллической решетке льда. Та-
кой рыхлой структурой объясняется аномально малая плотность
воды в твердом состоянии. Свободные полости в структуре льда
способствуют образованию клатратных соединений включения кле-
точного типа. Подобные образования могут давать молекулы таких
газов, как Cl2, H2S, метан и др.
Лед обладает электропроводностью около 10-9 Ом-1-см-1 при
температуре —10° С. Электропроводность льда Бьеррум объясняет
существованием ионизационных дефектов в кристалле льда. Ионы
могут образоваться при переходе протона от одной молекулы воды
к другой по схеме 2Н2О^Н3О+ + ОН-.
Водяной пар состоит главным образом из одиночных молекул
воды, но в нем встречаются и ассоциированные молекулы (ди- и
9
тримеры). До настоящего времени едино!! теории о структуре жид-
кой возы нет. Это свидетельствует о ее чрезвычайной сложности и
недостаточной изученности. Существует несколько моделей и тео-
рий строения жидкой воды, но можно выделить два главных прин-
ципа подхода к рассмотрению самой структуры воды. ‘Эдин из них
заключается в допущении однородности структуры, другой в
представлении о смеси структурных образований.
Многие ученые придерживаются теории строения жидкой воды
и водных растворов, в основе которой лежит положение о сохра-
нении ажурной решетки льда в жидкой воде и частичном заполне-
нии пустот одиночными молекулами воды. По этой теории ближ-
няя упорядоченность молекул воды, т. с. взаимное расположение
соседних молекул воды, аналогично кристаллическому каркасу
льда, слегка нарушенному тепловым движением молекул, в пусто-
тах которого находятся одиночные молекулы воды.
О. Я. Самойловым разработана теория трансляционного дви-
жения частиц в воде, согласно которой молекулы воды льдоподоб-
ного каркаса, выйдя из состояния равновесия, совершают активи-
рованный скачок в пустоты, находятся там некоторое время, а за-
тем вновь приходят в трансляционное движение. Молекулы воды,
попавшие в пустоты, энергетически не соответствуют молекулам
каркаса. Время перехода молекул воды из одного состояния в дру-
гое составляет порядка 10~13 с.
Существуют и другие представления о структуре жидкой воды,
исходящие из однородности ее структуры. Сущность их заключает-
ся в том, что в жидкой воде сохраняются неизменными водород-
ные связи кристаллической решетки льда, но происходит их де-
формация.
Сложные взаимодействия и изменения в структуре жидкой во-
ды подтверждаются рядом температурных аномалий. Так, в интер-
вале температуры от 30 до 40° С наблюдаются изменения в свойст-
вах воды. При 35° С вода имеет наименьшую теплопроводность, а
при температуре от 0 до 35° С наблюдается наиболее резкое паде-
ние электронной поляризуемости воды под действием внешнего
поля. С этой аномалией воды связывают и ее биологическую ак-
тивность по отношению к организмам, имеющим температурный
оптимум, равный 37° С. Согласно теории Самойлова в этом интер-
вале температур происходит разрушение структурной решетки
воды.
Вторая температурная аномалия некоторых свойств жидкой во-
ды наблюдается в интервале температур от 55 до 60° С. Так, при
55° С наблюдается минимум изменения электронной поляризуемо-
сти молекул воды под действием адиабатического сжатия, отмеча-
ется минимум изменения диэлектрической проницаемости в зави-
симости от давления, значительно увеличивается звукопередача.
На особые свойства воды влияет и то, что наряду с ассоцииро-
ванными и неассоциированными молекулами в жидкой воде всегда
присутствуют ионы Н3О+ и ОН-. Так, при 18° С количество дис-
социированных молекул составляет 0,785 -10-7 моль/л, а ионное
10
произведение равно 6,16-10 1э. При 25° С ионное произведение во-
ды равно 1,04-1014 и принимается за стандартную величину.
Аномалия плотности, заключающаяся в том, что плотность льда
меньше, чем v жидкой воды, и максимум плотности около 4 С ооъ-
ясняются внутренней структурой воды. В твердом состоянии, вслед-
ствие образования тетраэдрических комплексов с рыхлой упаков-
кой, расположение молекул воды менее плотное, чем в жидкой фазе,
т. е. они занимают больший объем. При замерзании воды происхо-
дит увеличение объема примерно на 10%. При плавлении льда на-
рушается его регулярная структура и часть комплексов разруша-
ется. В воде наряду с участками, имеющими структуру, аналогич-
ную кристаллической решетке льда, появляются одиночные моле-
кулы. Нарушение регулярной структуры сопровождается повышени-
ем плотности и уменьшением объема, так как одиночные молекулы
воды заполняют полости, сохранившиеся в участках с льдоподоб-
ной структурой. С повышением температуры проявляется действие
двух факторов: теплового расширения и нарушения регулярной
структуры льда. Тепловое расширение, сопровождающееся незна-
чительным увеличением объема, связано с уменьшением упорядо-
ченности расположения молекул. При 4° С эти два фактора одинако-
вы по абсолютной величине, но противоположны по направленности
действия. При дальнейшем повышении температуры снижается
действие второго фактора, сильнее проявляется действие теплового
расширения и плотность воды уменьшается.
Тот факт, что лед легче воды, играет огромную роль в приро-
де. С наступлением морозов поверхностный слой воды в водоеме
охлаждается до температуры +4° С и как более тяжелый опуска-
ется на дно, вытесняя более теплые на поверхность. В результате
замерзание воды начинается с поверхности, а не со дна. Этому же
способствует и малая теплопроводность льда. Хорошей защитой
водоемов является и снег, покрывающий слой льда. Теплопровод-
ность снега при пл. 0,1 г/см3 соответствует теплопроводности шер-
сти, и при пл. 0,2 г/см3 — теплопроводности бумаги.
Силы межмолекулярного взаимодействия в воде обусловливают
большую величину работы, необходимой для преодоления этих сил
притяжения и перевода воды из жидкого в газообразное состояние.
Такая работа характеризуется теплотой испарения. Вода по срав-
нению с другими жидкостями имеет наибольшую теплоту испаре-
ния. Работа, необходимая для перевода вещества из твердого со-
стояния в жидкое,— теплота плавления — для воды тоже имеет
максимальное значение. При охлаждении водяного пара и при за-
мерзании воды выделяется эквивалентное количество теплоты. Вода
обладает максимальной теплоемкостью по сравнению с другими
жидкими и твердыми веществами. Эта аномалия воды имеет очень
важное значение для существования жизни на Земле. Благодаря
ей возможно возникновение огромных теплых и холодных океани-
ческих течений, сглаживающих климат теплых и холодных облас-
тей Земли. Массы воды океанов и морей служат тепловым аккуму-
лятором. Эта аномалия способствует поддержанию нормальной
11
температуры тела у теплокровных организмов при переходе из од-
ной температурной обстановки в другую. Изменение структуры во-
ды приводит к аномальному изменению теплоемкости воды в за-
висимости от температуры.
Молекулы жидкости, находящиеся в поверхностном слое, испы-
тывают притяжение со стороны молекул, лежащих в более глубо-
ких слоях и стремящихся втянуть их внутрь жидкости, что приво-
дит к возникновению поверхностного натяжения. Из всех жидкос-
тей, за исключением ртути, при обычной температуре вода имеет
максимальное поверхностное натяжение. Данное свойство воды
определяет поверхностные явления и играет важную роль в проте-
кании биохимических процессов.
Вода — единственный на Земле минерал, который может нахо-
диться одновременно в трех агрегатных состояниях. В этом состоит
еще одна аномалия воды. В жидком виде она прозрачна, аномаль-
но мало рассеивает видимый свет, поэтому бесцветна, по сильно
поглощает лучистую энергию, особенно ультрафиолетовой и инфра-
красной области спектра. Это свойство воды тоже играет важную
роль для протекания физических и биохимических процессов.
Способность многих веществ растворяться в воде и диссоцииро-
вать на ионы определяется ее высокой диэлектрической проницае-
мостью. У большинства растворителей диэлектрическая проницае-
мость находится в пределах от 10 до 50. У воды величина диэлект-
рической проницаемости максимальна и равна 81. Эта особенность
воды определяет самую большую растворяющую способность в от-
ношении веществ с полярной и ионной структурой. Аномально и
изменение вязкости воды при повышении давления. В интервале
температур от 0 до 20—30° С вязкость воды с повышением давле-
ния уменьшается.
Ниже приведены физические константы, относящиеся лишь к
одному виду воды Н216О. Но в природе встречаются и другие устой-
чивые изотопы водорода (дейтерий и тритий) и кислорода (17О,
18О). Так, на 2500 атомов 16О приходится один атом 17О и пять ато-
мов 18О. Только с учетом протия и дейтерия (тритий встречается в
природе крайне редко) можно получить следующие виды молекул
воды: Н216О; HD16O; D216O; Н217О; HD17O; D2I7O; Н218О; HD18O;
D218O.
Молекулярная масса 18,0154
Плотность жидкой воды (25° С, 101,325 кПа),
г/см3 0,9979751
Теплота образования (—Л/7 при 25° С,
101,325 кПа), кДж/моль
пар 241,9845
жидкая 286,0313
Теплота плавления (101,325 кПа), кДж/моль 6,012
Теплота испарения (101,325 кПа), кДж/моль 44,041
Удельная теплоемкость (101,325 кПа), кДж/(кгХ
ХК)
льда 2,039
жидкой воды 4,187
пара 2,039
12
Продолжение
Теплопроводность (101,325 кПа), Вт/(м-К) льда (0° С) жидкой воды (20° С) жидкой воды (45° С) пара (100° С) Скорость распространения звука при (101,325 кПа), м/с 25° С, — 2,34 0,598 0,645 0,0231 1,4963
Криоскопическая константа Эбулиоскопическая константа Вязкость (25° С, 101,325 кПа), мПа-с Температура максимальной плотности (101,325 кПа), °C с, 1,850 0,516 1,002 3,98
Диэлектрическая постоянная жидкой воды 101,325 кПа, 60 мГц) Плотность льда (0°С, 101,325 кПа), г/см3 (17° 81 0,9168
Диэлектрическая постоянная льда ( 101,325 кПа, 3000 Гц) —1° с, 79
Поверхностное натяжение (20° С, 101,325 Н/м кПа), 72,5-10-3
Наибольшее практическое значение имеет тяжелая вода D216O с
молекулярной массой 20. По свойствам тяжелая вода заметно от-
личается от обычной воды. Она кипит при 101,4° С, замерзает при
+ 3,8° С. Плотность ее при 25° С составляет 1,1042 г/см3; макси-
мальной плотностью (1,0559) она обладает при +11,2° С. Молеку-
лы D2O тоже образуют межмолекулярные дейтериевые связи, они
прочнее водородных, поэтому структура тяжелой воды более ста-
бильна. При электролизе обычной воды тяжелая вода накаплива-
ется в остатке, что и положено в основу технологии ее получения.
Растворимость солей в тяжелой воде на 10—20% ниже, чем в лег-
кой. Она угнетает жизнедеятельность растений и животных.
В природных водах содержание тяжелой воды составляет около
0,02%. Накапливание тяжелой воды происходит в условиях, спо-
собствующих испарению. Это объясняется меньшей величиной дав-
ления пара у тяжелой воды. Поэтому в морской воде ее содержится
больше, чем в пресной. Повышенное содержание тяжелой воды на-
блюдается в живых организмах и минералах. Тяжелая вода очень
гигроскопична, т. е. поглощает легкую воду и взаимодействует с
ней по схеме D2O + H2O=<=t2HDO. Вязкость тяжелой воды на 20%
выше, чем у обычной. Поэтому содержание тяжелой воды в смеси,
а следовательно, и изотопов водорода может быть определено по
вязкости.
§ 2. Понятие о системах, фазах и компонентах.
Диаграмма состояния воды
Системой называют тело или группу тел, находящихся во взаи-
модействии и мысленно обособляемых от окружающей среды. Од-
нородные системы, внутри которых не содержится поверхностей
раздела, называются гомогенными. К ним относятся растворы твер-
дых, газообразных, жидких веществ, смеси газов.
Неоднородные системы, имеющие поверхность раздела, называ-
ются гетерогенными. Гетерогенная система состоит из нескольких
фаз — гомогенных частей системы, отграниченных от остальных
частей системы псверхнсс 1ью раздела. Фазой называется совокуп-
ность всех гомсл ciiiiin частей системы, одинаковых по составу п
по физическим л химическим свойствам и oiраничепных от других
частей системы поверхностью раздела. Гомогенные системы, напри-
мер истинные растворы молекулярной и полной дисперсности, яв-
ляются однофазными. Система, состоящая из воды и льда, являет-
ся гетерогенной (двухфазной). Система из льда, жидкой воды и
водяных паров — трехфазная.
Компонентом, или составной частью системы, называют каждое
содержащееся в ней химически однородное вещество, которое после
выделения из системы может существовать в изолированном виде.
Так, в растворе хлорида натрия компонентами являются вода и
хлорид натрия. Вода в любом агрегатом состоянии содержит толь-
ко один компонент. Но числу компонентов в системе различают
одно-, двух-, трех- и т. д. компонентные системы.
Устойчивость гетерогенных систем определяется фазовыми и
химическими равновесиями. При фазовых равновесиях переход
компонентов системы из одногй фазы в другую нс сопровождается
химическим взаимодействием. Число термодинамических степеней
свободы системы при равновесии определяется числом условий, ко-
торые можно изменять в определенных пределах, не нарушая при
этом числа и видов фаз систем. Такими независимыми переменны-
ми являются концентрация, температура, давление. Равновесие в,
гетерогенных системах подчиняется закону равновесия фаз или
правилу фаз Гиббса. Для систем, в которых отсутствует химическое
взаимодействие, число термодинамических степеней свободы С в
равновесной многофазной системе равно числу компонентов К плюс
2, минус число фаз Ф:
— К —Фд-2. (1.1)
При этом допускается, что на систему оказывают влияние только
температура и давление. Число термодинамических степеней сво-
боды определяет вариантность системы: при С = 0 система инвари-
антна, т. е. данному равновесному состоянию системы соответствуют
строго определенные значения независимых переменных. При С=1
система имеет одну степень свободы (одновариантная), 1. е. можно
изменять действие одного из факторов, влияющих на равновесие-
системы. При С = 2— двухвариантная система и т. д.
Зависимость состояния системы от изменения внешних условий
можно выразить диаграммой, откладывая на осях независимые пе-
ременные. Такие диаграммы называются диаграммами состояния
или фазовыми диаграммами. Для однокомпонентной системы при
независимых переменных — температуре и давлении — число тер-
модинамических степеней свободы определяется согласно правилу
фаз:
С = 3 — Ф. (1.2)
Так как С нс может быть меньше нуля, то из уравнения (1.2) сле-
дует, что наибольшее число контактирующих фаз в однокомпонент-
14
ной системе будет равно трем. В
качестве примера рассмотрим ди-
аграмму состояния воды (рис. 4).
Из нее следует, что однофазная
система (жидкость, лед, пар)
имеет две степени свободы (двух-
вариантная), т. е. в определенных
пределах можно изменять и тем-
пературу и давление, не нарушая
равновесия. При одновременном
существовании двух фаз (жид-
кость — пар, жидкость — лед,
лед — пар) система имеет одну
степень свободы (одновариант-
ная) и при этом можно изменять
только температуру или давление,
кривых О возможно существование
4, наконец, в точке пересечения
трех фаз (Ф = 3) и число степе-
ней свободы при этом равно нулю, т. е. система инвариантная.
Точка О на этой диаграмме называется тройной точкой. Она ха-
рактеризует устойчивое одновременное существование всех трех
форм воды (твердой, жидкой и газообразной) при сохранении по-
стоянными внешних условий (температура и давление). На этой
диаграмме кривая ОА определяет устойчивость системы: жид-
кость — пар, кривая ОВ — системы лед — пар, а кривой ОС выра-
жается изменение температуры плавления льда в зависимости от
внешнего давления.
Кривыми ОА, ОВ и ОС диаграмма разделяется на три части, ко-
торые характеризуют присутствие одной фазы (жидкости, пара или
льда). Все линии пересекаются в тройной точке О, отвечающей тем-
пературе 0,01° С (0,007° С) и давлению 613 Па (4,6 мм рт. ст.). При
таких условиях находятся в равновесии все три фазы: лед^жид-
кость^пар.
§ 3. Структура водных растворов
Образование водных растворов сопровождается изменением
электрического момента диполя молекул воды, их пространствен-
ной переориентацией, изменением или разрывом водородных связей,
т. е. определенной перестройкой внутренней структуры. Если размер
молекул неэлектролита соизмерим с размером полости льдоподоб-
ного каркаса воды, то молекула внедряется в полость. Разветвлен-
ные молекулы могут занимать несколько полостей, мало изменяя
при этом структуру воды.
Большие молекулы неэлектролитов образуют в структуре воды
большие полости, аналогичные по структуре кристаллогидратам.
Энергия, необходимая для образования таких полостей, выделяет-
ся при разрыве водородных связей межи молекулами. Каждая
молекула неэлектролита может связывать большое количество мо-
лекул воды. Так, молекула метана удерживает 10—20 молекул во-
15
ды. Образование таких структур (гидратов) сопровождается выде-
лением теплоты, так как энергия взаимодействия между молеку-
лами неэлектролита и воды больше энергии взаимодействия меж-
ду молекулами воды. Способствуя разрушению структуры воды,
образование гидратов вызывает повышение температуры замерза-
ния. На этом свойстве водных растворов неэлектролитов основан
газгидратный способ опреснения морских и минерализованных вод.
Следует иметь в виду, что речь идет о замерзании растворов, содер-
жащих гидраты типа метан — вода, а не о молекулярных раство-
рах неэлектролитов, например сахара. Растворы неэлектролитов
имеют пониженную температуру замерзания по сравнению с чистой
водой.
При попадании в воду неполярных молекул достаточно больших
размеров, например молекул жидких углеводородов, происходит
разрыв водородных связей между молекулами воды, а новые связи
с растворяемым веществом не образуются. Поэтому подобные со-
единения в воде не растворяются.
Строение водных растворов электролитов будет определяться
воздействием ионов на структуру воды, которое зависит от многих
факторов. При вхождении иона в полость льдоподобного каркаса
происходит переориентация молекул воды под действием его заря-
да. Если входит катион, то результирующие электронные центры
молекул воды обращаются внутрь полости, при появлении аниона
происходит поворот протонных углов.
Определяющее значение в строении водных растворов электро-
литов имеет соотношение между величиной заряда и радиусом иона.
Крупные ионы с небольшой плотностью заряда (Rb+, К+, NO3~, I",
Вт-) вызывают резкое изменение структуры воды. Они способству-
ют разупорядочиванию структуры воды, разрыву водородных свя-
зей и увеличению подвижности молекул. Ионы с малым радиусом
и большой плотностью заряда (Cr3+, А13+, Zn2+, СО32д SO42 ) рас-
полагаются внутри полостей, не нарушая структуры, чем способ-
ствуют снижению подвижности молекул и упрочнению структуры
воды.
При образовании водных растворов электролитов снижается ве-
личина диэлектрической проницаемости, и тем заметнее, чем силь-
нее проявляется упорядочивающее действие ионов. Изменяется и
такое свойство, как текучесть: ионы Na+, SO42~ уменьшают теку-
честь раствора. Влияние ионов на структуру водного раствора зави-
сит от их концентрации. При малой концентрации в водном раство-
ре электролита могут сохраняться участки воды с ненарушенной
структурой. При больших концентрациях электролита весь объем
раствора подвержен воздействию кулоновских сил, в результате
чего вода полностью деструктурирована. Но так как действие элек-
трического заряда иона уменьшается с расстоянием, то, очевидно,
и влияние иона на структуру воды будет неодинаковым.
В водных растворах электролитов происходит гидратация ио-
нов. Процесс гидратации заключается во взаимодействии ионов
электролитов с молекулами водрц которое сопровождается как об-
16
разеванием гидратных оболочек вокруг попов, так и изменением
теплового трансляционного движения молекул воды. Если ионы
оказывают сильное воздействие на ближайшие молекулы воды,
снижая их трансляционное движение, что сопровождается упоря-
дочением структуры, то такая гидратация называется положитель-
ной. И, наоборот, р азупорядочивающие структуру ионы вызывают
повышение подвижности молекул воды, такая гидратация называ-
ется отрицательной. Она характерна для больших ионов с малым
зарядом.
Катионы связаны с молекулами воды за счет донорно-акцептор-
ного взаимодействия с неподеленной электронной парой атома кис-
лорода. Гидратная оболочка анионов формируется под действием
водородных связей. Способность к гидратации у катионов выра-
жена сильнее, чем у анионов. Сильнее гидратируются ионы с ма-
лым радиусом и большей величиной заряда.
Совместное действие катионов и анионов электролита на струк-
туру воды определяется вкладом каждого из ионов. Если преоб-
ладает действие иона с положительной гидратацией, то раствор бу-
дет более упорядоченным, чем чистая вода. Например, раствор
HNO3 является упорядоченным, так как структурирующее дейст-
вие иона Н+ преобладает над разупорядочивающим действием иона
NO3~\ А растворы KNO3, NaBr будут разупорядоченными, деструк-
турированными.
В концентрированных растворах электролитов нет свободного
растворителя — он весь входит в зоны действия ионов. Поэтому
такие растворы отличаются по свойствам от разбавленных раство-
ров. В концентрированных растворах исчезает разупорядочиваю-
щее действие ионов с отрицательной гидратацией. Растворы при
концентрации электролита выше 2 моль/л по структуре напоминают
расплавленный кристалл электролита. Если в разбавленных рас-
творах искажается структура воды ионами электролита, то кон-
центрированные растворы можно представить себе как электролит,
структура которого нарушена растворителем. Наглядным под-
тверждением взаимодействия ионов электролита с водой является
электрострикция — уменьшение (сжатие) общего объема раство-
рителя и электролита.
Образующиеся гидраты по свойствам отличаются от безводного
соединения. Иногда эту разницу в свойствах можно наблюдать ви-
зуально. Так, при растворении безводного сульфата меди вследст-
вие деформации электронной оболочки иона Си2+ появляется окра-
шенный в голубой цвет гидратированный ион [Си (4Н2О)]2+. Взаимо-
действие частиц растворенного вещества с водой может быть на-
столько сильным, что при кристаллизации они выделяются вместе
с гидратной оболочкой, образуя кристаллогидраты определенного
состава: A12(SO4)3- 18Н?О; Na2CO3- ЮН2О и др.
17
§ 4. Растворимость различных веществ в воде
Растворимость газов в воде. Газы, состоящие из полярных
молекул (аммиак, хлороводород), растворяются в воде лучше, чем
газообразные неполярные соединения (водород, метан, азот). Так,
в 1 л воды при (Г С и давлении 101,325 кПа растворяется 505 л
хлороводорода, но всего 0,002 л водорода. Растворимость некото-
рых газов в воде при 20° С и 101,325 кПа приведена ниже.
Газ О2 СО, X, С12 ХН3 H2S
Раствори-
мость га-
за, л/л 0,031 0,878 0,015 2,361 702,406 2,582
Растворимость кислорода в воде почти в два раза больше, чем
азота. Поэтому, воздух, растворенный в воде, имеет другой состав,
чем атмосферный. В сухом воздухе при 18° С объемное содержание
кислорода составляет 21,2%, а в воздухе, растворенном в воде,—
34,1%. В дистиллированной воде растворимость газов больше, чем
в водных растворах электролитов. Зависимость растворимости га-
за от концентрации растворенного электролита выражается фор-
мулой Сеченова:
\g(N0/N) = kC, (1.3)
где No и N — растворимость газа в чистой воде и в растворе элек-
тролита концентрации С; k — постоянная, зависящая от природы
газа, электролита и температуры. Так, растворимость кислорода в
воде с общим солесодержанием 40 г/кг уменьшается на 25%. Сле-
довательно, в морской воде и воде соленых озер растворенного кис-
лорода меньше, чем в речной. Растворимость газов при повышении
температуры уменьшается. Это объясняется тем, что процесс рас-
творения газов сопровождается выделением теплоты и уменьше-
нием объема. Для установления зависимости растворимости газа от
температуры используется уравнение Клапейрона — Клаузиуса:
где N1 и N2 — растворимость газа при температуре Т\ и 7V, Гисп—
молярная теплота испарения; R — универсальная газовая постоян-
ная, равная 8,31434 Дж/(моль-К). Зависимость логарифма раство-
римости газа 1пД^ от 1/Т описывается уравнением прямой. Это поз-
воляет графически устанавливать зависимость растворимости газа
в воде от температуры.
Зависимость растворимости газов от давления выражается за-
коном Генри, согласно которому количество растворенного газа
пропорционально его давлению над раствором:
С = kp, (1.5)
где С — концентрация растворенного газа; р — давление газа над
раствором; k — коэффициент пропорциональности, зависящий от
природы газа, растворителя и температуры, но не зависящий от
давления.
18
Если над раствором находится смесь газов, то растворимости
каждого из них будет пропоряпоаальна его парциальному давле-
нию над раствором (закон Генри — Дальгона). Па основании этого
закона можно вычислить растворимость различны?; газов в воде при
разных температурах. Так, при 20е С растворимость кислорода и
диоксида углерода будет выражаться произведением парциального
давления каждого из них в атмосфере на растворимость при пар-
циальном давлении 101,325 кПа.
Вследствие протекания биохимических процессов и наличия рас-
творенных соединений в воде растворимость газов несколько от-
личается от расчетных значений. Между газами, содержащимися в
воздухе и растворенными в воде водоемов, устанавливается равно-
весие. Нарушение его сопровождается изменением концентрации
растворенных газов. Если концентрация растворенного газа в воде
больше, чем следует из закона Генри —Дальтона, то он выделяется
в атмосферу до наступления равновесного состояния, и наоборот,
при концентрациях ниже равновесных происходит поглощение газа
из воздуха. Для установления состояния равновесия требуется опре-
деленное время. Перемешивание воды способствует сокраще-
нию времени, необходимого для достижения равновесного состо-
яния.
Растворимость твердых веществ в воде. Пределы растворимо-
сти твердых веществ в воде весьма широки: от 3-10 19 г, например
для HgS, до 2570 г для AgNO3 в 1 л воды. Растворимость зависит
от природы растворяемого вещества и температуры. Изменение
давления крайне незначительно влияет на растворимость твердых
веществ в воде, так как жидкости практически не сжимаются. Луч-
ше растворяются в воде те соединения, которые взаимодействуют
с водой, образуя гидраты. Растворимость твердых веществ изменя-
ется с повышением температуры различно. Так, растворимость.
KNO3 при изменении температуры от 0 до 100° С увеличивается в
18,5 раз, а растворимость NaCl — всего на 10%. Растворимость
CaSO4-2H2O, Са(ОН)2 с повышением температуры несколько/
уменьшается.
При растворении многих соединений происходит поглощение
теплоты, так как энергия, выделяющаяся в процессе гидратации,,
меньше количества энергии, необходимого для разрушения кристал-
лической решетки. Поэтому при повышении температуры раствори-
мость этих веществ в воде увеличивается. При обратном соотноше-
нии величин энергии гидратации и энергии, затрачиваемой на раз-
рушение кристаллической решетки твердого вещества, раствори-
мость соединений с повышением температуры уменьшается.
Взаимная растворимость жидкостей. Способность одной жид-
кости растворяться в другой жидкости определяется характером
химической связи в молекулах этих соединений. Полярные жидко-
сти хорошо растворяются в полярных, неполярные — в неполярных
жидкостях. Взаимная растворимость жидкостей изменяется в самых
широких пределах: от неограниченной способности растворяться
их друг в друге до практически полной нерастворимости.
1Э
Ь любых соотношениях смешиваются такие полярные жидко-
сти, как ацетон и вода. Но жидкие углеводороды (октан, декан
и др.), молекулы которых неполярны, практически не растворяют-
ся в воде. При контакте двух ограниченно растворимых друг в
друге жидкостей образуется система, состоящая из двух слоев,
каждый из которых представляет собой насыщенный при данной
температуре раствор одной жидкости в другой. Например, в систе-
ме вода — этил ацетат верхний слой будет представлять собой раст-
вор воды в этилацетате, а нижний — этилацетата в воде. Измене-
ние количества одного пли другого компонентов системы нс влияет
на равновесное распределение соединений в слоях, т. е. при посто-
янной температуре состав их остается постоянным.
При повышении температуры растворимость ограниченно раст-
воримых друг в друге жидкостей увеличивается и при определен-
ной температуре (критической температуре растворения) они стано-
вятся неограниченно растворимыми одна в другой. В некоторых
системах подобное изменение растворимости происходит и при по-
нижении температуры. Взаимная растворимость жидкостей не со-
провождается заметным изменением объема, поэтому незначитель-
но зависит от давления.
При изучении систем жидкость — жидкость Д. П. Коноваловым
было установлено, что состав пара над такой системой отличается
от состава самой смеси. В системах, состоящих из жидкостей, огра-
ниченно растворимых друг в друге, парциальное давление каждой
из них может соответствовать давлению насыщенного пара данной
жидкости из смеси, взятой отдельно. Температура кипения таких
смесей ниже, чем температура кипения каждого из компонентов.
Это так называемые азеотропные или нераздельно кипящие смеси.
В качестве примера можно рассмотреть систему фенол — вода: тем-
пература кипения фенола 181,4° С, а его смесь с водой кипит при
98,6° С.
При полном испарении одного соединения температура ки-
пения резко возрастает в результате значительного снижения
давления пара над жидкостью. Это свойство азеотропных смесей
используется для очистки производственных сточных вод от орга-
нических примесей (фенола, органических кислот) методом пере-
гонки с водяным паром.
Способы выражения концентрации растворов. Количественное
содержание компонентов в растворе характеризуется концентраци-
ей. Концентрацией раствора называется массовое или объемное со-
держание растворенного вещества в определенном объеме пли мас-
се раствора. За единицу объема часто принимают литр (дм3). На-
иболее употребительные способы выражения концентрации раст-
воров:
1) концентрация по массе показывает, сколько единиц массы
растворенного вещества содержится в определенной массе раство-
рителя. Наиболее часто пользуются процентной концентрацией. Она
показывает, сколько граммов растворенного вещества содержится в
100 г раствора;
20
2) объемная концентрация показывает, сколько единиц массы
(объема) растворенного вещества содержится в определенном объ-
еме раствора;
3) нормальная концентрация определяется числом грамм-экви-
валентов растворенного вещества в 1 л раствора;
4) молярная концентрация определяется количеством молей ра-
створенного вещества в 1 л раствора;
5) моляльная концентрация характеризуется числом молей ра-
створенного вещества, которое приходится на 1000 г растворителя.
§ 5. Свойства растворов
Между жидкой водой и паром устанавливается равновесие, ко-
торому соответствует при данной температуре определенное давле-
ние насыщенного пара. В водных растворах концентрация молекул
воды снижается и тем сильнее, чем больше содержание растворен-
ного вещества. Поэтому из фазы пара в жидкую начинают перехо-
дить молекулы воды до установления нового состояния равновесия.
Вследствие этого давление насыщенного пара над раствором будет
меньше, чем давление насыщенного пара над растворителем при
той же температуре.
Эта зависимость для разбавленных растворов выражается за-
коном Рауля (1887): относительное понижение давления насыщен-
ного пара над раствором равно отношению числа молей растворен-
ного вещества к сумме молей растворителя и растворенного веще-
ства (молярной доле растворенного вещества):
где Ро—давление насыщенного пара чистого растворителя; р —
давление насыщенного пара над раствором; п — число молей ра-
створенного вещества; N — число молей растворителя.
Понижение давления насыщенного пара над раствором вызыва-
ет изменение температуры кипения и замерзания раствора по срав-
нению с чистым растворителем. Так как давление насыщенного па-
ра над раствором ниже, чем над чистым растворителем при этой же
температуре, то растворы кипят при более высокой температуре.
Повышение температуры кипения раствора сравнительно с раство-
рителем будет тем значительнее, чем выше его концентрация. Это
соотношение, полученное Вант-Гоффом (1886), выражается урав-
нением
ЬТк--=Ет, (1.7)
где АТК — повышение температуры кипения, °C; m — моляльная
концентрация раствора; Е — эбуллиоскопическая постоянная рас-
творителя, равная для воды 0,52° С.
Понижение температуры замерзания растворов также связано с
.уменьшением давления насыщенного пара над раствором. Так как
давление пара над раствором при температуре замерзания раство-
21
рителя еще не соответствует давлению насыщенного пара над твер-
дой фазой paciF'-риц тя, то растворы замерзают при более низкой
температуре Разпспь ме кду гсмпсратурамн замерзания чистого
растворителя и paciB<pa \Г< возрастает с повышением концентра-
ции раствора. Как празпъ . из раствора вначале выкристаллизовы-
вается рас 1 ворнтель. Эю способеiвует дальнейшему понижению
температуры замерзания вследствие повышения концентрации ра-
створа. Поэтому замерзание растворов идет в некотором интервале
температур. Понижение температуры замерзания растворов также
подчиняется зависимости, выведенной Вант-Гоффом:
\Т3~~Кт, (1.8)
где АГ3 — понижение температуры замерзания, °C; т— моляльная
концентрация; /( — криоскопическая постоянная растворителя, рав-
ная для воды 1,86° С. Понижением температуры замерзания раство-
ров объясняется тот факт, что морская вода, содержащая до 3,5%
растворенных солей, замерзает при —1,9° С.
Если сосуд с раствором какого-либо вещества отделен от сосуда
с растворителем (водой) мембраной, проницаемой только для моле-
кул растворителя, то наблюдается перемещение молекул раствори-
теля через мембрану в раствор. Подобный перенос молекул воды
будет наблюдаться и тогда, когда концентрация одного из раство-
ров выше другого. Такой самопроизвольный переход молекул раст-
ворителя через полупроницаемую мембрану называется осмосом.
Движущей силой этого процесса можно считать разность концент-
раций растворителя с разных сторон мембраны. Если концентра-
ции растворов, разделенных мембраной, равны, наступает равно-
весие, характеризующееся одинаковой скоростью перехода молекул
воды в тот и другой растворы.
Гидростатическое давление, оказываемое молекулами раствори-
теля на мембрану, называется осмотическим. Осмотическое давле-
ние соответствует давлению, которое необходимо оказать на раст-
вор для достижения равновесного состояния между раствором и
растворителем, разделенными полупроницаемой мембраной. Вели-
чина осмотического давления для разбавленных растворов не за-
висит от химической природы растворителя и растворенного веще-
ства, а определяется разностью концентраций растворов, разделен-
ных мембраной. Чем больше эта разность, тем больше величина
осмотического давления. При равенстве концентраций растворов,
разделенных полупроницаемой мембраной, осмотическое давление
равно нулю. Зависимость осмотического давления от концентрации
выражается соотношением, выведенным Вант-Гоффом:
л = 1000СЯГ, (1.9)
где л — осмотическое давление, Па; С — молярная концентрация
раствора, моль/л; R — универсальная газовая постоянная
[8,314 Дж/(моль-К)]; Т — термодинамическая температура раство-
ра, К.
22
Зависимости, описываемые уравнениями (1.6) — (1-9), справед-
ливы только для растворов неэлектролитов. Это объясняется тем,
что в растворах электролитов вследствие диссоциации находится
большее число частиц. Поэтому в уравнения (1.6) — (1.9), которые
выражают зависимость свойств раствора от его концентрации, вво-
дится поправочный множитель — изотонический коэффициент I.
Он равен отношению определяемого для данного электролита осмо-
тического давления к осмотическому давлению, рассчитанному по
формуле (1.9). Изотонический коэффициент для растворов слабых
электролитов рассчитывают по формуле
i — 1 + — I), (I. 10)
где о — степень диссоциации; k—число ионов, образующихся при
диссоциации молекулы. Изотонический коэффициент выражается
отношением числа частиц, реально присутствующих в растворе, к
числу недиссоциированных молекул. При снижении концентрации
раствора величина изотонического коэффициента возрастает вслед-
ствие увеличения степени диссоциации.
§ 6. Теория сильных электролитов
Под действием полярных молекул воды при растворении элект-
ролитов происходит диссоциация их молекул на ионы. По способ-
ности к образованию ионов в водных растворах электролиты делят-
ся па ионизированные полностью, когда в растворе находятся толь-
ко ионы (сильные электролиты), и слабые, в растворах которых
наряду с ионами находятся и молекулы растворенного вещества.
Способность вещества к образованию ионов количественно выра-
жается степенью диссоциации а, представляющей собой отношение
числа молекул, распавшихся на ионы, к общему числу молекул ра-
створенного вещества. При уменьшении концентрации она возрас-
тает и в сильно разбавленных растворах приближается к 100%.
Процесс диссоциации слабых электролитов является обратимым.
Поэтому, применив закон действия масс к процессу электролити-
ческой диссоциации, получим величину константы диссоциации,
выраженную отношением числа продиссоциированных молекул к
числу недиссоциированных молекул. Так, константа диссоциации
угольной кислоты по первой ступени имеет следующее выражение:
Н2СО3^Н+ + НСОу; Ki = [Н+] [НСОу]/[Н2СО3].
При изучении растворов сильных электролитов было установ-
лено, что они не подчиняются закону действия масс. Константа дис-
социации сильных электролитов зависит от концентрации, а имен-
но при повышении концентрации она уменьшается. В 1923 г. П. Де-
бай и Э. Хюккель предложили теорию сильных электролитов, со-
гласно которой в растворах сильных электролитов находятся толь-
ко ионы. Между ионами осуществляется взаимодействие, приводя-
щее к тому, что каждый ион оказывается окруженным ионами про-
тивоположного знака (ионной сферой). Ионы, входящие в состав
23
сферы, в свою очередь, являются центрами, вокруг которых распо-
лагаются ионы противоположного знака. Ионы в растворе распре-
деляются так же, как и в кристаллах, но на значительно большем
расстоянии друг от друга. При этом их взаимное влияние ослаб-
ляется присутствием молекул растворителя. В сильно разбавлен-
ных растворах действие межионпых сил выражено незначительно
и кажущаяся степень диссоциации становится близкой к 100%. Уве-
личение концентрации раствора сопровождается усилением взаимо-
действия между ионами, что способствует уменьшению их подвиж-
ности в электрическом поле и снижением кажущейся степени
диссоциации электролита. В растворах сильных электролитов
претерпевает изменение и структура растворителя (воды) вслед-
ствие гидратации, поляризации молекул воды под действием
ионов.
При увеличении концентрации раствора подвижность ионов
снижается и поэтому растворы сильных электролитов характеризу-
ются величиной эффективной концентрации, которая меньше ис-
тинной. Она называется активной концентрацией или активностью
электролита. Между активной и истинной концентрацией сущест-
вует зависимость
а = уС, (1.11)
где а — активная концентрация, или активность, моль/'л; С — ис-
тинная концентрация раствора, моль/л; у — коэффициент актив-
ности.
Из соотношения (1.11) следует, что коэффициент активности
представляет собой отношение активности к истинной концентра-
ции. В сильно разбавленных растворах величина у приближается к
единице, а с повышением концентрации уменьшается. Использова-
ние активности позволяет проводить термодинамические расчеты
свойств растворов сильных электролитов по уравнениям, выведен-
ным для простых систем.
Если в растворе находится смесь электролитов, то электроста-
тическое действие ионов друг на друга характеризуется ионной
силой раствора /:
CiZ? + + • • • + CnZ^n
I = -----------------------, (1.12)
где Ci, С2, ..., Сп — истинные концентрации отдельных ионов;
Z2, ..., Zn — заряды ионов.
Коэффициент активности не зависит от химической природы
иона и одинаков для всех равнозарядных ионов при определенной
ионной силе раствора (табл. 1).
В водах с малым солесодержанием (до 200. мг/л) коэффициент
активности не принимается во внимание. При большей концентра-
ции растворов электролитов в расчетах необходимо учитывать ко-
эффициент активности.
24
Для растворов с ионной
силой до 0,01 коэффициент
активности иона рассчитыва-
ют по формуле
lgY = -0,5Z-Y7. (1.13)
В растворах с ионной си-
лой от 0,5 до 0,01 коэффици-
ент активности вычисляют
по формуле
0,5ZH I
Ig Y = — , , (r-14)
Формула (1.14) примени-
ма для точного расчета ко-
эффициента активности рас-
творов с концентрацией до
50 мг-экв/л.
Таблица 1. Зависимость
коэффициентов активности
для ионов с различным зарядом
от ионной силы раствора
Ионная сила рас пора Коэффициент активное in для ионов
одноза- рядных дпух за- рядных трех за- рядных
0,00 1 1 1
0,001 0,96 0,87 0,72
0,002 0,95 0,81 0,63
0,005 0,92 0,72 0,48
0,01 0,89 0,63 0,35
0,02 0,05 0,87 0,57 0,28
0,81 0,43 0,15
0,1 0,76 0,34 0,084
0,2 0,70 0,24 0,041
0,5 0,62 0,15 0,014
§ 7. Произведение растворимости
Растворимость таких веществ, как СаСО3, AgCl, FeS, CuS и
многих других, не может быть выражена обычными способами вви-
ду исключительно малой величины самой растворимости. Однако
абсолютно нерастворимых веществ нет. Для выражения раствори-
мости практически нерастворимых веществ используется условная
величина — произведение растворимости ПР.
При контакте воды с поверхностью практически нерастворимо-
го вещества, например карбоната кальция, начинается растворение
его, т. е. выход в раствор незначительного количества ионов Са2+ и
СО32ж Так как скорость растворения зависит от количества ио-
нов на поверхности, то можно допустить, что с течением времени
она не изменяется, т. е.
vi — ky. (1-15)
Наряду с растворением при повышении концентрации ионов в
растворе увеличивается скорость и2 перехода ионов из раствора на
поверхность твердой фазы. Она зависит от активности ионов, а для
сильно разбавленного раствора — от концентрации ионов в раст-
воре, и выражается уравнением
->_ • (F16)
Са^г со;
о
При определенной концентрации ионов в растворе скорости рас-
творения и осаждения становятся равными, т. е. Ui = u2- Подстав-
ляя их значения, получаем
^1 = МСа2+%02-> С-17)
откуда
*1/^2 = йСа2+ас02- = ПР- (L 18)
25
Отношение &1/&2 есть величина постоянная. Она характеризует рас-
творимость труднорастворимого электролита и называется произ-
ведением растворимости ПР. Произведение растворимости — ве-
личина постоянная и равна произведению активностей ионов в на-
сыщенном растворе труднорастворимого электролита при данной
температуре. Так, ПР карбоната кальция составляет при 20е С
5- 10 s, карбоната магния — 2-Ю-5; Mg(OH)2 — 1,2- 10““. В на-
сыщенном растворе СаСО3 существует равновесие
СаСО3^Са2+ + СО“-
твердая фаза раствор
Произведение растворимости карбоната кальция имеет следую-
щий вид:
ПР ~ а па =4,8-10—9 моаь/л. (1.19)
СаСОз Са- СО-~ ’ ' v '
Если при диссоциации электролита образуется несколько оди-
наковых ионов, то ПР равно произведению активностей ионов в
степени, соответствующей количеству образующихся ионов. Па-
пример, для М§’з(РО4)2 произведение растворимости записывается
следующим образом:
nPMg3(PO,)=-я* з-. (1.20)
Mg- pj4
Если произведение активностей ионов <ПР, то раствор являет-
ся ненасыщенным. Если оно равно ПР, то раствор будет насыщен-
ным. Если произведение активностей ионов больше ПРХ то раствор
является пересыщенным. Содержание в воде ионов Са2+, СО,2 ,
SO42 обусловливается произведением растворимости, характери-
зующим растворимость солей СаСО3, CaSO4. Зная произведение
растворимости, можно определить растворимость вещества и, на-
оборот, по величине растворимости можно рассчитать произведе-
ние растворимости. Подставив вместо активностей ионов в уравне-
ние (Е18) значения их концентраций, получим
йСа2 + = VCa2+ [Са2 4-]; дсс2_ Yco2- [СО2-]
ПРсаСОз = Yca2-1 [Са^]усоГ [СО2-].
Вследствие равенства коэффициентов активностей ионов с оди-
наковой величиной заряда можно записать:
ПРсаСОз = Y '[Са ’ ] [СО,2-], (1.21)
т. е. [Са2+] [СО* ’] = ПРСаСОз/у2.
Так как концентрации ионов [Са2+] и [СО32 ] равны, то кон-
центрация каждого из них будет соответствовать молярной кон-
центрации насыщенного раствора: \
tcas+j = [СОД] = . /-Efba. = _ Сс (1.22)
Г Y2 Y
26
В сильноразбавленных растворах, когда у~ 1, молярная концентра-
ция насыщенного раствора бинарного электролита вычисляется по
формуле
С / ПР.
(1.23)
Для вычисления концентрации насыщенного раствора электролита
в граммах на лтр нужно умножить количество молей на массу мо-
ля данного соединения.
В присутствии эюкчролпта, содержащего одноименный ион с од-
них: из ионов трудпорастворимого электролита, растворимость по-
следнего уменьшается. Это объясняется смещением равновесия
диссоциации тр\днорастворимого электролита в сторону образова-
ния нсдиссоциированных молекул, а также тем, что величина ПР,
представляющая собой произведение концентраций (активностей)
ионов, является при данной температуре постоянной величиной.
Для увеличения полноты осаждения выделяемых, из воды ионов не-
обходимо вести процесс при некотором избытке осадителя.
При введении в насыщенный раствор труднорастворимого элек-
тролита другого электролита, не содержащего с ним одноименных
ионов, наблюдается повышение растворимости. Это явление назы-
вается солевым эффектом. Он объясняется тем, что при повышении
концентрации раствора происходит снижение коэффициентов ак-
тивности, что способствует увеличению произведения концентраций
ионов труднорастворимого электролита.
§ 8. Кинетика химических реакций
Кинетика химических реакций изучает закономерности проте-
кания химических процессов во времени. Следовательно, химичес-
кая кинетика — это учение о скорости химических реакций и зави-
симости ее от различных факторов.
Скорость химической реакции измеряется изменением концент-
рации реагирующих веществ (моль/л) в единицу времени. Если
относить изменение концентрации к бесконечно малому промежут-
ку времени, то можно получить выражение истинной скорости ре-
акции v в данный момент как производной от концентрации С по
времени I:
АС
^-±-—. (1.24)
At
Если рассматривается изменение концентрации одного из исход-
ных веществ, то производная будет отрицательной величиной, так
как в процессе реакции концентрация уменьшается:
Влияние концентрации на скорость гомогенных химических ре-
акций описывается законом действия масс: при постоянной темпе-
ратуре скорость данной реакции прямо пропорциональна произве-
27
дению концентрации реагирующих веществ в степенях, равных ко-
эффициенту перед формулой данного вещества в уравнении реак-
ции. Этот закон применим для газовых реакций и реакций, проте-
кающих в сильно разбавленных растворах. Для прямой реакции
А+В = АВ закон действующих масс записывается так:
w=A[A][B], (1.26)
где v — скорость реакции; k — коэффициент пропорциональности
или константа скорости реакции; [А], [В]—концентрации реаги-
рующих веществ.
Константа скорости реакции равна скорости реакции, когда кон-
центрация каждого из реагирующих веществ равна единице. Для
реакции образования воды из водорода и кислорода
2Н2 -р Oj - 2Н2О
выражение скорости прямой реакции будет иметь вид
v - ^[Н2]2[О2].
В зависимости от количества молекул веществ, участвующих в
реакции, различают моно-, би-, тримолекулярные реакции. Наибо-
лее часто встречаются моно- и бимолекулярные реакции. Это объ-
ясняется тем, что вероятность столкновения одновременно трех
молекул чрезвычайно мала.
Важным понятием в химической кинетике является порядок ре-
акций. Он характеризуется суммой показателей степеней концент-
раций отдельных реагентов в выражении закона действия масс. Раз-
личают реакции первого, второго, третьего порядка. Порядок реак-
ции не всегда совпадает с ее молекулярностью. Большинство
химических реакций протекает в несколько стадий и скорость ре-
акции характеризуется скоростью наиболее медленно протекаю-
щей стадии. Порядок реакции будет выражаться молекулярностью
этой стадии и, как правило, отличаться от суммы коэффициентов
реакции в целом. Так, в реакциях гидролиза солей в разбавленных
водных растворах концентрация воды изменяется так незначитель-
но, что в уравнение скорости реакции она не входит, и кинетика
таких реакций будет описываться уравнениями кинетики реакций
первого порядка. Реакции разложения молекул, внутримолекуляр-
ных группировок (например, диссоциация молекулы хлора на ато-
мы) являются одномолекулярными и относятся к реакциям первого
порядка. Скорость одномолекулярной реакции выражается уравне-
нием
v=kC. (1.27)
Согласно уравнению (1.25)
dC
kC = —-----, (1.28)
dt
где k имеет размерность 1/С или
(1.29)
28
После интегрирования получаем
In С —kt —у В. (1.30)
Если начальную концентрацию (при ( = 0) принять за Со, то можно
определить постоянную интегрирования: В = 1п Со.
Отсюда
1п(С/С0) = -kt, (1.31)
C = Coe~kt. (1.32)
Уравнение (1.32) характеризует зависимость концентрации от
времени для химических реакций первого порядка. Так как в урав-
нение (Е31) концентрация вещества входит в виде отношения, то
константа скорости реакции первого порядка не будет зависеть от
размерности времени. При изменении обеих концентраций в оди-
наковое число раз интервал времени не будет изменяться, т. е. за
одни и те же промежутки времени будет реагировать одна и та же
часть взятого количества исходного вещества. Если в первую ми-
нуту прореагировала 1/100 часть от общего количества исходного
вещества, то через минуту прореагирует столько же от оставшихся
99 частей исходного вещества.
Кинетика реакции второго порядка для равенства концентра-
ций реагирующих веществ описывается уравнением
kt = (Су — C)/CqC. (1.33)
Если учитывать изменение концентрации реагирующего вещества
по времени t х=С0—С, то tlx=CQ—С. Тогда уравнение (1.33) при-
нимает вид
k — x/tC(j(C0 — х). (1.34)
В отличие от реакций первого порядка величина х в уравнении
(1.34) будет зависеть от того, в каких единицах выражаются вре-
мя и концентрация реагирующих веществ.
Влияние температуры на скорость реакции. Скорость химиче-
ской реакции обычно с повышением температуры резко возрастает.
При увеличении температуры на каждые 10° С скорость реакции
возрастает в 2—3 раза (правило Вант-Гоффа). Это объясняется
увеличением числа эффективных столкновений частиц реагирующих
веществ, т. е. в выражении скорости химической реакции измене-
ние температуры сказывается на величине константы скорости ре-
акции k. Отношение константы скорости при повышении темпера-
туры на 10° (^/+ю) к константе скорости реакции при данной тем-
пературе kt есть температурный коэффициент ую скорости реакции,
значение которого соответствует правилу Вант-Гоффа:
kt = Yio-
Значительное увеличение скорости реакции при повышении тем-
пературы объясняется увеличением числа активных молекул, т. е.
таких молекул, которые имеют энергию активации Еа, достаточную
для того, чтобы столкновение молекул закончилось их взаимодей-
29
ствием. Энергия активации — это то избыточное количество энер-
гии (по сравнению со средним), которое должна иметь молекула
в момент столкновения, чтобы оно закончилось химическим взаи-
модействием. Если энергия столкнувшихся молекул будет меньше
энергии активации, то взаимодействия между ними не произойдет.
Скорость реакции зависит от числа активных молекул, количество
которых при низкой температуре но сравнению с числом неактив-
ных молекул очень мало. Активность значительно возрастает при
повышении температуры. Активные молекулы характеризуются со-
стоянием возбуждения, вызываемого нахождением электронов на
более высоком энергетическом уровне или неустойчивым внутрен-
ним строением молекулы. Такие молекулы имеют повышенную ки-
нетическую энергию поступательного или вращательного движения.
Взаимосвязь между константой скорости реакции k и энергией
активации Ея описывается уравнением Аррениуса:
_ Е 'RT
k = Ае а , (1.35)
где А — предэкспоиенциальный множитель, связанный с вероятно-
стью и числом столкновений; е — основание логарифма; R — уни-
версальная газовая постоянная; Т — термодинамическая темпера-
тура, К.
При рассмотрении соотношений, выражающих зависимость ло-
гарифма константы от обратного значения термодинамической тем-
пературы, следует, что логарифм константы скорости реакции на-
ходится практически в линейной зависимости от обратной темпе-
ратуры.
Химическое равновесие. Многие химические реакции являются
обратимыми, т. е. течение их возможно в прямом и обратном на-
правлениях. Оба процесса совершаются одновременно и независи-
мо друг от друга, но скорость одного из них больше, чем другого.
При определенных условиях скорости прямой и обратной реакций
становятся равными, наступает момент химического равновесия.
Химическое равновесие является динамическим, подвижным.
Взаимодействие при этом не прекращается. Примером обратимой
реакции может служить реакция образования воды при темпера-
туре от 2000 до 4000° С: 2H2 + O2=«=t2H2O. Вначале взаимодействие
водорода с кислородом идет с достаточно высокой скоростью в сто-
рону образования воды. Запишем выражение скорости этого про-
цесса:
Щ = ^1[Н2]2[О2].
По мере накопления молекул воды начинает увеличиваться ско-
рость обратной реакции:
v2 = ын2ор.
В определенный момент скорости этих процессов выравниваются
(щ = ц2) и наступает момент химического равновесия:
МН2]2[О2] = £2[Н2О]2
ИЛИ
^1/^2- [Н2О]2/[Н2]2[О2] = (1.36)
30
Величина К является постоянной, так как равна отношению посто-
янных величин. Она называется константой химического равнове-
сия. Уравнение (1.36) выражает закон действия масс применитель-
но к обратимым реакциям и применимо только при сильных раз-
бавлениях. Для вычисления константы химического равновесия
растворов более высокой концентрации вместо концентраций сле-
дует пользоваться активностями. Для реакции общего вида
mA -J- + rfD
уравнение константы Ка записывается следующим образом:
Уа = 4Ч)/К>в)- С1-37)
Константа равновесия К для разбавленных растворов рассчиты-
вается по формуле
К = [СЛО^/ДАКДВ]*). (1.38)
Равновесное состояние системы может нарушаться в результате
воздействия на нее внешних факторов. Такими факторами могут
быть изменения концентрации одного из веществ системы, темпе-
ратуры и давления (для газовых систем). Переход системы, на-
ходящейся в равновесном состоянии, под влиянием изменившихся
условий в новое состояние равновесия называется смещением хи-
мического равновесия.
Смещение химического равновесия подчиняется принципу Ле
Шателье, согласно которому: если на систему, находящуюся в ус-
тойчивом равновесии, воздействовать извне, изменяя какое-нибудь
из условий, определяющих положение равновесия, тогда в системе
усиливается то из направлений процесса, течение которого ослаб-
ляет влияние произведенного воздействия, и положение равновесия
сместится в том же направлении.
При уменьшении концентрации одного из исходных веществ в
равновесной системе происходят процессы, направленные на увели-
чение концентрации этого вещества. Это достигается повышением
скорости обратной реакции до установления равновесного состоя-
ния, соответствующего изменившимся условиям. Так, при увеличе-
нии температуры система стремится уменьшить ее за счет проте-
кания эндотермической реакции. Например, такой реакцией будет
разложение воды на образующие ее водород и кислород:
2НзОд^2Н2 + 0-2 — 571 кДж
При понижении температуры в системе протекают экзотермические
реакции, т. е. в самой системе начинает выделяться теплота.
Влияние изменения давления проявляется в газовых системах,
когда количество молекул исходных веществ и продуктов реакции
неодинаково. Уменьшение давления ведет к образованию большего
числа молекул, т. е. к увеличению объема системы, а повышение
давления сопровождается уменьшением объема системы, возраста-
нием скорости реакции, в результате которой образуется меньшее
число молекул газа.
31
Принцип смещения равновесия в гомогенных системах приме-
ним п к гетерогенным системам для характеристики равновесных
состояний в отдельных фазах системы.
Возможность протекания химических реакций и условия уста-
новления равновесия. Самопроизвольное протекание химических
реакций возможно, если процесс сопровождается уменьшением
внутренней энергии системы U или переходом ее в наиболее веро-
ятное состояние, которое характеризуется большим числом возмож-
ных комбинаций взаимного расположения частиц с одинаковым
запасом энергии. Наряду с внутренней энергией для характеристи-
ки состояния системы используют термодинамическую функцию —
энтальпию II, которая включает в себя внутреннюю энергию систе-
мы и работу, совершаемую системой при переходе ее из состояния
1 в состояние 2. В процессах, протекающих без изменения объема
и температуры (изохорно-изотермических), тепловой эффект соот-
ветствует изменению внутренней энергии: Аб/=С'2—U}. Если же
процесс совершается при постоянном давлении и постоянной темпе-
ратуре (изобарно-изотермический), то тепловой эффект соответст-
вует изменению энтальпии: Д/7 = Я2—1Ц. Различие между величи-
нами внутренней энергии и энтальпии для конденсированных сред
(растворов, кристаллов) невелико.
Для экзотермических реакций ДЖО, так как система при этом
выделяет теплоту. Для эндотермических реакций Таким об-
разом, в изолированной системе при заданных условиях самопро-
извольно могут протекать экзотермические реакции. Самопроиз-
вольное протекание химических реакций возможно в направлении
наиболее вероятном, при котором состояние системы наименее упо-
рядочено. Это справедливо для изолированных систем.
Согласно второму закону термодинамики в системах могут про-
текать только те процессы, которые сопровождаются увеличением
термодинамической функции — энтропии S, а пределом соверше-
ния процесса является достижение максимального значения для
данных условий. Энтропия связана с вероятностью состояния систе-
мы соотношением 3 = &1п w, где w — число возможных комбинаций
взаимного расположения частиц с определенным запасом энергии;
k — постоянная Больцмана (отношение универсальной газовой по-
стоянной к постоянной Авогадро). Размерность энтропии Дж/
(моль-К). Изменение энтропии AS реакции можно вычислить
по разности между энтропией продуктов реакции и энтропией ис-
ходных веществ:
AS = (y«/Sz),Ip.p-(v/?/S0Hcx.B, (Е39)
где п — количество молекул каждого из веществ, участвующих в
реакции.
Термодинамические расчеты предусматривают стандартное со-
стояние вещества: концентрация — 1 моль/1000 г растворителя,
температура 25° С (298 К), давление 101,325 кПа. Стандартные
величины термодинамических функций указаны в справочных таб-
лицах.
32
Функцией, характеризующей направление и предел самопроиз-
вольного прохождения процесса в системе при постоянных значе-
ниях давления и температуры, является изобарно-изотермический
(изобарный) потенциал, или энергия Гиббса (G): G = H—TS. Из-
менение энергии Гиббса (свободной энергии) AG рассчитывают по
формуле
= SH — TSS. (1.40)
В личина T>\S соответствует той части энергии, которая не
связ .та с осуществлением химической реакции, т. е. расходуется
непр шзводительно. Таким образом, AG характеризует величину
пол< ной работы, совершаемой системой в процессе, взятую с об-
ратном знаком. Самопроизвольно могут протекать процессы, со-
провождающиеся уменьшением энергии Гиббса, т. е. при условии
AG<0. Состоянию термодинамического равновесия соответствует
минимальное значение изменения энергии Гиббса, т. е. AG = 0.
Кристаллическому состоянию соединений соответствует минимум
свободной энергии, но не наиболее вероятное состояние. При кон-
такте кристаллов с водой система переходит в менее упорядоченное
состояние (раствор), характеризующееся возрастанием энтропии.
При достижении значения энтропии, максимального для данных ус-
ловий, наступает равновесие между жидкой и твердой фазой, т. е.
образуется насыщенный раствор.
С константой равновесия К}) энергия Гиббса связана соотноше-
нием
AG = — 2,3/?rigKp. (1.41)
Изменение энергии Гиббса (для стандартного состояния) вы-
числяется по разности сумм стандартного изменения образования
конечных продуктов и начально взятых веществ:
AGO = (2nzAG)np.p - (2шАО)исх.в. (1.42)
(Значения AG0 находят по справочным таблицам.)
Изменение энергии Гиббса при образовании простых веществ
принимается равным нулю. Знак AG, а следовательно, и направ-
ленность процесса зависят от соотношения величин АЯ и TAS в
формуле (1.40). Если экзотермический процесс (АЯ<0) сопровож-
дается переходом систем в наиболее вероятное состояние, т. е. AS
имеет положительное значение, то величина AG будет отрицатель-
ной, что указывает на возможность самопроизвольного протекания
этой реакции. Если экзотермический процесс сопровождается упо-
рядочиванием системы (АЗ<0), при соотношении AH<.TNS реак-
ция самопроизвольно не пойдет несмотря на то, что процесс энер-
гетически выгоден. Протекание эндотермических реакций (АЯ>0),
сопровождающихся разупорядочиванием системы, т. е. при условии
AS>0, возможно при соотношении AH<ThS. С повышением тем-
пературы действие энтропийного фактора возрастает.
Нели процессы совершаются в системах без изменения объема
и температуры (изохорно-изотермических), то направленность про-
цесса и предел протекания характеризуют термодинамической
2—1244
33
функцией — изохорно-изотермическим (изохорным) потенциалом--
энергией Гельмгольца F. С внутренней энергией U и энтропией S
энергия Гельмгольца связана соотношением F=U—TAS. Измене-
ние энергии Гельмгольца Аг в процессе рассчитывают по уравне-
нию
AF = AC — TAS. (1.43)
Термодинамические расчеты указывают только на возможность
самопроизвольного протекания реакции при данных условиях. Ио
под действием сопротивления внешней среды, например из-за кине-
тического сопротивления, реакция может находиться в квазпршзно-
весном состоянии. Так, синтез воды из водорода и кислорода ис
идет при обычных условиях, хотя и является возможным на осно-
вании термодинамических расчетов. Такой синтез возможен с уча-
стием катализаторов.
Невозможность самопроизвольного протекания реакции, обус-
ловленная термодинамическими расчетами, вовсе не означает, что
эта реакция вообще не может быть осуществлена. Подводя энер-
гию извне (например, повышая температуру, используя электролиз,
различные виды излучений), можно вызывать протекание реакций,
не осуществляющихся самопроизвольно. Так, при нагревании, в
процессе электролиза идет разложение воды па водород и кислород.
К реакциям, не осуществимым по термодинамическим расчетам, по
протекающим за счет использования энергии солнца, относятся ре-
акции фотосинтеза в растительных организмах.
§ 9. Ионное произведение воды. Водородный показатель
Вода является малодиссоциированным соединением. Из 55,56
моля, содержащихся в 1 л воды, диссоциирует только 10“7 моля.
Вода является амфотерным электролитом. Одна молекула воды
в результате донорно-акцепторного взаимодействия является ак-
цептором протона, т. е. выполняет роль кислоты, а другая, отдавая
протон, выступает в качестве основания. В результате такого взаи-
модействия образуется ион гидроксония и гидроксид-ион:
2Н2О Н3О+ + ОН—
Свободных протонов в водных растворах не существует, но, чтобы
избежать усложнения в записи ионных уравнений, можно выразить
уравнение диссоциации воды следующим образом: Н2О^*Н+ + ОН~.
Константа диссоциации воды при 22° С составляет
а —
Кн2о = ——— — 1,8- Ю-16. (1.44)
ан2о
Приняв активность недиссоцииро&аных молекул воды за посто-
янную величину вследствие крайне незначительной степени диссо-
циации, уравнение (1.44) можно записать так
^н2ойн2о = ан+%н-- С-45)
34
Произведение z<H2oaH2o обозначается через К,с. При определен-
ной температуре Ки- является величиной постоянной:
Кда = «н+^ои_. (1.46)
Подставляя числовые значения К,, и Лн2ов (1.46), получим (25° С)
а +а = 1,8-10—16-55,56 = 1,008-10—14.
н+ он~
Произведение активностей ионов Н~ и ОН' при данной темпера-
туре есть величина постоянная и называется ионным произведением
воды !(„. Вследствие малого количества ионов Н+ и ОН' в чистой
воде вместо активностей можно взять концентрации ионов:
[Н+] [ОН-] = 10-14.
При равенстве концентраций ионов Н+ и ОН' (в чистой воде)
концентрация каждого из них составляет
[Н+] = [ОН-] = /10=14 = 10-7 моль/л.
Произведение активностей этих ионов есть постоянная величи-
на и поэтому увеличение концентрации одного из ионов сопровож-
дается соответственным уменьшением концентрации другого. Так
как попы водорода обусловливают кислотные свойства растворов,
а гпдроксид-ионы — щелочные, то при равенстве их концентраций
реакция среды раствора будет нейтральной.
При содержании ионов Н+ больше 10~7 моль/л реакция среды
будет кислой. Если же активность ионов водорода меньше 10~7, то
реакция среды будет щелочной. Значения ан+ и %н- определяются
по уравнению (1.46)
ан+ ~ ’ (1.47)
а = Kw/a+. (1.48)
ин н
Для характеристики реакции среды растворов используют не
абсолютное значение активности иона Н+, а его отрицательный де-
сятичный логарифм. Называется эта величина водородным пока-
зателем и обозначается pH:
рН=- lgaH+. (1.49)
При 25° С в нейтральном растворе pH 7, в кислых средах рН<
<7, а в щелочных рН>7. При повышении температуры степень
диссоциации воды возрастает, так как это — процесс эндотермичес-
кий (АН = 55,842 кДж/моль). Это вызывает повышение концентра-
ции ионов Н+ и одновременно ионов ОН- в чистой воде. Так, при
50° С ионное произведение воды составляет 5,474-10'14, концентра-
ции Н+ и ОН' — 2,339-10~7, pH 6,631. При понижении температу-
ры равновесие диссоциации молекул воды смещается в сторону об-
разования недиссоциированных молекул, степень диссоциации
уменьшается, что сопровождается увеличением pH, соответствую-
щего нейтральной среде. Так, при 0° С Kw= 1,139-10~15, концентра-
ция ионов Н+ и ОН~ составляет 3,38-10 8, pH 7,972.
2* 35
Отрицательный десятичный логарифм активности гидроксид-
иона рОН называется гидроксидным показателем. Зная величину
pH, можно легко вычислить рОН, так как pH + рОН= 14. Например,
при pH 3 рОН 11. Для разбавленных растворов уравнение (1.49)
принимает вид
pH = — lg [Н+]. t1-50)
Вычисление pH для растворов с концентрацией ионов ГН или
ОН больше 10 3 моль/л производится с учетом коэффициента ак-
тивности.
Для оценки качества воды важной характеристикой является
pH. У большинства поверхностных вод pH колеблется в пределах
от 6,5 до 8,5. Подземные воды иногда имеют повышенное значе-
ние pH. Кислую реакцию среды имеют рудничные воды сульфид-
ных, колчеданных месторождений, содержащие сульфат железа
(II), воды болот, в которых находится значительное количество гу-
мусовых кислот. Обычно pH природных вод данного источника яв-
ляется величиной постоянной. Постоянство pH природных вод обус-
ловлено наличием в них буферной системы, состоящей из свобод-
ной угольной кислоты и гидрокарбоиатов. Поэтому заметные
изменения pH могут быть показателем загрязнения воды бытовыми
или промышленными стоками. Дистиллированная вода, вследствие
растворения диоксида углерода из воздуха, может иметь pH до
5,7. Водородный показатель морской воды изменяется в пределах
от 8,2 до 8,5.
Знание pH необходимо для выбора правильного режима обра-
ботки воды, определения ее коррозионного действия и выбора спо-
соба обеззараживания.
Вычисление pH растворов. Расчет pH водных растворов кис-
лот, оснований, солей, сложных систем производится с учетом сте-
пени и константы диссоциации соединений, их концентрации (ак-
тивности) в растворе. В табл. 2 приведены формулы для вычисле-
ния концентрации ионов Н+ и ОН-, pH растворов соединений, наи-
более часто используемых при аналитических исследованиях.
§ 10. Гидролиз солей
При растворении в воде некоторых солей нарушается равнове-
сие диссоциации воды и реакция ср^ды раствора может стать кис-
лой или щелочной. Это объясняется тем, что ионы, возникающие
при диссоциации соли, взаимодействуют с ионами Н+ и ОН-, т. е.
растворенное вещество взаимодействует с водой. Реакция взаимо-
действия ионов растворенной соли с молекулами воды называется
гидролизом. Гидролизуются только растворимые соли и к тому же
не все. Соли, образованные сильными основаниями и сильными кис-
лотами (NaCl, KNO3, Na2SO4 и др.), гидролизу не подвергаются.
Растворы их имеют нейтральную реакцию среды. Типичные реак-
ции гидролиза:
36
Таблица 2. Расчет концентрации ионов Н+ и ОН~, pH растворов различных соединений
37
00 оо Раствор Формула для расчета 1 1римср
Слабая кислота в присутствии ее со- ли (например, СН3СООН+ + CH3COONa) [Н+] = Хкисл (£кислД>соли)> pH = рАкисл 1g /кисл/Ссоли) ИЛИ pH = рХкисл 4~ рС’кисл Р^соли ^СНзСООН =0,2; ^сНзСООХа ~ ^ещеоон ~ 1,8-105; [Н+] = 1,8-10-5= | 8.10-5.2 = 0,1 = 3,6-10-5; pH = 5 — 1g 3,6 = 5,44
Слабая щелочь в присутствии ее со- ли (например, NH4OH + NH4CI) [ОН-] = /<осн (Сосн/Ссоли)> рОН == Р^Сосн 1S (^осн/^соли) ИЛИ рОН = P-Kqch 4” рСосн Р^соли ^nh4oh = 0 4 [ОН-] = 1,8.10-5 — = 1,8-10-5.4 = J 0,1 = 7,2-10-5; рОН = 5 — 1g 7,2= 4,14; рН = 14 — 4,14 = 9,86
Соль слабой кислоты и сильной ще- лочи (например, CH3COONa) , ^Н,(/Кисл [Н+] V ‘ > F '-'соли рН = 7 + “Г р^кисл + ~2~ Ссоли’ Г, п . Р'КкиСЛ рСсОЛИ pH = 7 + 1 CcH3COONa~ °»1; ^СНзСООН = 1 ’8‘ 1()~ ;4 = /1,8.10-18= 1,35.10- 5; pH = 9 —1g 1,35 = 8,87
Р аствор Формула для расчета Пример
Соль слабого основания и сильной кислоты (например, NH4CI) [Н+]=/ Кн/°--; ' 'ХОСН 1 1 pH — 7 Р^кисл g ^соли» u т , Р^соли Р-^осн или pH—7 + 2 Cnh^ci =°»1; / 10—14-0,1 10-5 [Н+] = 1 / = = 7,4-10-6: L J |/ 1,8-10-5 1,35 pH =6 —1g 7,4 = 5,13
Соль слабой двухосновной кислоты (например, Na2CO3) " ^соли рН=7 + -у РК"исл + у 1S Ссоли, ,, „ Р^кисл — рСсоли ИЛИ pH - / + Оча2со3 = 0,1; Ссоз = 6.ю-П; Г 10-14.6-10-11 0,1 = /6.10-21 = 2,4-10-12; pH = 12 —1g 2,4 = 11,62
Кислая соль слабой двухосновной кислоты (например, NaHCO3) [н+] = у <ИСХИСЛ; Р^кисл 4" Р^кисл рп ~ - 2 Kn2co-3-1(W; Кн2со:1-- 6-10-1’; [Н+] = /з.1о-7.б-1о-и = = /18-10-18 = 4,2-10-9; pH = 9 — lg4,2 = 8,38
* Принятые обозначения: Скисл, Сосн, ^солп — молярные концентрации кислоты, основания, соли; Ккисл, Косп - константы дпссо
циации кислоты, основания; К'кисд’ ^"иисл— первая и вторая константы диссоциации слабой двухосновной кислоты; р /',, „ с;. рКосн“
показатели (отрицательный логарифм) константы диссоциации кислоты, основания; рС. с л, рС,.,, ,, ц -• пока зателп отрппа те.’1' 'И ix Л"| .трпфмоз
концентрации кислоты, соли.
1, Гидролиз солей, ооразованных сильным основанием и слаоой
кислотой, например ацетат натрия CH3COONa:
CH.COONa 4- НОН 2^2 СН3СООН + NaOH
GH3COO~ 4- Ха+ + НОН CH,;СООН + Na+ 4- ОН~
СН3СОО- -г НОН 2^2 СН3СООН 4- ОН—
Реакция протекает между анионом соли и водой. Так как обра-
зующаяся уксусная кислота — соединение малодиссоциированное,
то равновесие диссоциации воды нарушается и в растворе оказы-
вается избыток гидроксид-ионов. Реакция среды растворов солей
такого типа будет щелочной.
Соли, образованные многоосновными слабыми кислотами и
сильными основаниями, гидролизуются ступенчато. Например, гид-
ролиз карбоната натрия протекает следующим образом:
I Ступень Na2CO3 + НОН 2^2 NaHCO3 + NaOH
2Na+ 4- СО|~ + НОН 2^2 Na+ + HCOf 4- Na+ + OH~
СОз~4- НОН 2^2 НСО7 4- ОН-
Ионы НСОз- тоже могут гидролизоваться:
II ступень NaHCO3 + НОН ^2 Н2СО3 + NaOH
нсоу + НОН 2^2 Н2СО3 4- он-
Обычно гидролиз таких солей идет преимущественно по первой
стадии, т. е. является частичным. Реакция практически заканчи-
вается образованием кислой соли, так как диссоциация иона НСО3“
очень мала.
Гидролиз — процесс обратимый, поэтому равновесие гидролиза
можно сместить, вводя в раствор ионы, связывающие ОН_-ионы
(например, ионы Н+), и тем самым провести реакцию гидролиза
до образования конечных продуктов. При нагревании равновесие
гидролиза смещается вправо.
2. Гидролиз солей, образованных слабым многокислотным осно-
ванием и сильной кислотой. Пои этом оавновесие диссоциации во-
ды нарушается в результате связывания ионов ОН- катионом со-
ли и образования в растворе избытка ионов Н+. Рассмотрим это
на примере гидролиза хлорида железа (III). Гидролиз этой соли
протекает ступенчато:
I ступень FeCl3 4- НОН FeOHCl2 + НС1
Fe3+ 4- НОН FeOH2+ + Н+
II ступень FeOHCl2 + НОН Fe (ОН)2 Cl 4-НС1
FeOH2+ + НОН Fe (ОН)^ + Н+
III Ступень Fe(OH)2Cl 4-НОН Fe (ОН)3 + НС1
Fe (ОН)+ -у НОН 2^2 Fe (ОН)3 + Н +
40
Из приведенных уравнений реакций следует, что при растворе-
нии хлорида железа (III) в воде образуются основные соли, связы-
вающие гидроксид-ионы, а в растворе остается избыток ионов Н+.
обусловливающий кислую реакцию среды и тормозящий прохож-
дение II и III ступеней гидролиза. При этом гидролиз до конца
нс доходит, так как образованию гидроксида железа (III) препят-
ствует кислая среда раствора. Полный гидролиз солей такого типа
может быть осуществлен только при условии связывания ионов
(например, гидроксид-ионами).
3. Гидролиз солей, образованных слабым основанием и слабой
кислотой, например ацетата аммония:
CH3COONH4 4- ПОН СНзСООН 4- NH4OH
Равновесие гидролиза у этой соли будет смещаться вправо, так
в результате реакции образуются малодиссоциированная кислота и
слабое основание. Реакция среды в растворах солей будет опреде-
ляться абсолютной силой кислоты и основания. Соли, образованные
очень слабыми основанием и кислотой, в воде подвергаются пол-
ному гидролизу и поэтому не существуют в водных растворах. На-
пример:
АП (СО3)з 4- 6Н2О - 2А1 (ОН)3 4- 3 [Н2О 4- СО2]
угоньная кислота
Гидролиз солей представляет собой процесс равновесный, обра-
тимый и имеет динамический характер. Следовательно, к нему при-
менимы все правила и закономерности обратимых химических про-
цессов.
Способность соли к гидролизу характеризуется степенью гидро-
лиза. Степень гидролиза представляет собой отношение числа
гидролизованных молекул к общей концентрации растворенной со-
ли. Так как гидролиз процесс обратимый, то к нему применим за-
кон действия масс. Для общего случая гидролиза соли, образован-
ной сильным основанием (МеОН) и слабой одноосновной кислотой
(НА), можно записать уравнение
МеА 4- НОН = НА 4- МеОН
А- 4- НОН= НА 4-ОН—
Константа равновесия для данного процесса имеет вид
[НА] [ОН-]
[А—] [Н2О] •
(1.51)
В разбавленных растворах [Н2О] можно принять за общую кон-
центрацию молекул воды и перенести эту величину в левую часть
равенства. Произведение [Н2О]К принимается за константу гидро-
лиза Кгидр:
/<гидр= [НА] [ОН-] [А-].
(1.52)
Подставив значение [ОН ] из ионного произведения воды [О1Г ] =
= /Сг/[Н+], получим
41
/<гидр- [НА] АГ^/( [А-] [Н + ]), (1.53)
где [НА]/([А-] [Н+]) = 1//<1!А,
отсюда Агидр = ^w/A'iia- (1-54)
Подставим значение /\Гпдр в уравнение (1.52):
ЛU-ЖНА = [НА] [ОН-]/[А-]. (1.55)
Зная концентрацию соли и /(гидр, можно вычислить степень гид-
ролиза соли [3 по уравнению (1.55). Если через С обозначить мо-
лярную концентрацию соли, то число гидролизованных молекул бу-
дет [ЗС, а негидролизованных — С(1—[3). Число молекул кислоты,
получающихся при гидролизе, равно числу гидроксид-иопов. После
подстановки этих данных в уравнение (1.55) получим
Kw//Cha = C?2/(1 -?). (1.56)
Так как величина [3 всегда меньше единицы, ею можно пренебречь
и уравнение (1.56) будет иметь вид
KwlКкисл — Р'С, (1.57)
откуда
(1.58)
Константа гидролиза соли, образованной слабым основанием и
сильной кислотой, имеет вид
Кгилр = [МеОН] [Н+]/[Ме+] = КЖК0СН, (1-59 )
а степень ее гидролиза
Р = //<Ж(СКосн). (1.60)
С повышением температуры от 0 до 100° С степень гидролиза уве-
личивается. Степень гидролиза возрастает и при разбавлении ра-
створа. Чем слабее образующиеся кислота или основание, тем вы-
ше степень гидролиза.
В природных условиях возможно и взаимное усиление гидро-
лиза при растворении солей противоположных типов (например,
FeSO4 и ИагСОз). При определенном соотношении концентраций
может произойти полное гидролитическое разложение этих солей:
FeSO4 + Na2CO3 + 2Н2О = Fe (ОН)2 + Н2О + СО2 + Na2SO4
При этом не будет происходить резкого изменения кислотности или
щелочности воды.
§ 11. Буферные растворы
Многие технологические и биохимические процессы протекают
при определенном pH, постоянство которого обеспечивают буфер-
ные растворы. Буферными называются растворы, pH которых не
изменяется при прибавлении небольшого количества сильной кисло-
ты или щелочи. Буферные растворы содержат компоненты, диссо-
циирующие с образованием одноименного иона, но отличающиеся,
42
друг от друга степенью диссоциации. Основные виды буферных ра-
створов: 1) слабая кислота и ее соль: ацетатная (СН3СООН и
CH3COONa), карбонатная (Н2СО3 и NaHCO3); 2) слабое основа-
ние и его соль: аммонийная (NH4OH и NH4CI); 3) две кислые соли:
фосфатная (NaH2PO4 и Na2HPO4); 4) кислая и средняя соль: кар-
бонатная (NaHCO3 и Na2CO3). Постоянство pH буферных раство-
ров при добавлении кислоты или щелочи объясняется связыванием
вводимых ионов Н+ и ОН- компонентами буферного раствора в
малодиссоциировапные соединения. Рассмотрим это взаимодейст-
вие на примере карбонатной буферной системы (Н2СО3—NaHCO3).
Компоненты буферного раствора диссоциируют:
Н2СО3^Н + +HCOJ-; NaHCO3^±Na+ +НСО7
Гидрокарбонат натрия — сильный электролит, а диссоциация уголь-
ной кислоты (слабого электролита) подавлена в результате нали-
чия в растворе большого количества ионов НСО3~, образующихся
при диссоциации соли. При введении в этот раствор кислоты ионы
Н + будут взаимодействовать с ионами НСО3~, образуя компонент
буферного раствора — угольную кислоту: Н~ + НСО3^П2СО3.
Вследствие незначительной степени диссоциации угольной кислоты
концентрацию ионов НСО3~ в буферном растворе можно для
сильно разбавленных растворов принять равной концентрации со-
ли Сс0 лп-
Если добавить к этому буферному раствору небольшое количе-
ство сильной щелочи, то введенные ионы ОН- взаимодействуют с
тем малым количеством ионов Н+, которое образуется при диссо-
циации угольной кислоты. В результате этого взаимодействия об-
разуется малодиссоциированное соединение — вода. Снижение
концентрации ионов Н+ вызывает смещение равновесия диссоциа-
ции угольной кислоты вправо. Избыток ионов Н+ нейтрализуется
ионами ОН~, что обеспечивает постоянство pH раствора. Запишем
уравнение константы диссоциации угольной кислоты (по I ступени)
Ki = [Н+] [нсо-]/[н2со3].
Принимая во внимание незначительную степень диссоциации
угольной кислоты, можно считать концентрацию недиссоциирован-
ных молекул равной общей концентрации кислоты Скисл. После
подстановки в выражение константы диссоциации угольной кисло-
ты принятых обозначений ([НСО3~]= Ссолп’, [Н2СО3]=Скис л) ПО-
ЛуЧИМ
— [Н+] (Ссоли/СКис1).
Откуда [Н+] будет равна
[Н+]=/<1(СКис.1/Ссо.1и), (1.61)
а величина pH
pH = р/<! - 1g (СКИс.,/Сс01и). (1.62)
Так как величине! pH буферного раствора зависит не от кон-
центрации кислоты и соли, а от соотношения их концентраций, то
43
при разбавлении она остается постоянной. При сильном разбавле-
нии pH буферной смеси незначительно возрастает. Формулы для
вычисления pH буферных растворов приведены в табл. 2.
Характеристикой буферных растворов служит величина буфер-
ной емкости. Она определяется числом грамм-эквивалентов силь-
ной кислоты или щелочи, которое необходимо прибавить к 1 л бу-
ферного раствора для изменения pH на единицу. Максимальная
буферная емкость раствора соответствует содержанию компонен-
тов в эквивалентных количествах.
Кислые или щелочные стоки, попадающие в водоем, в опреде-
ленном количестве могут быть нейтрализованы карбонатной буфер-
ной системой природных вод, состоящей из свободной угольной
кислоты и гидрокарбонатов. Это же способствует поддержанию по-
стоянства pH воды при введении реагентов в процессе обработки.
В щелочных водах (при рН>8,5) буферные свойства природных
вод определяются второй карбонатной буферной системой, состоя-
щей из гидрокарбонатов и средних карбонатов (например, NaHCO3
и Na2CO3).
Поддержание оптимальной величины pH при биологической очи-
стке сточных вод, необходимой для нормального протекания про-
цессов жизнедеятельности микроорганизмов, также обеспечивается
наличием буферных систем. Так, буферные свойства иловой воды
обусловливаются присутствием в ней карбонатной, аммонийной и
фосфатной систем. Буферные растворы используются при хими-
ческом анализе воды. Например, аммонийный — при комплексоно-
метрическом определении общей жесткости, кальция, магния. Дру-
гие — для колориметрического определения pH растворов, калиб-
рования приборов (pH-метров) при потенциометрическом опреде-
лении pH.
§ 12. Активная, общая, свободная кислотность воды
Ионы водорода, обусловливающие кислотность воды, образуют-
ся при диссоциации свободных (сильных или слабых) кислот, не-
которых кислых солей (например, гидросульфата натрия), а также
вследствие гидролиза солей, образованных слабыми основаниями и
сильными кислотами. При неполной диссоциации кислоты или ча-
стичном гидролизе соли концентрация свободных ионов Н+, нахо-
дящихся в растворе, часто не соответствует концентрации водород-
ных ионов, вступающих в реакцию нейтрализации с сильным осно-
ванием. Концентрация реально присутствующих ионов Н+ в раство-
ре при данных условиях характеризуется активной кислотностью.
Активная кислотность устанавливается при колориметрическом и
потенциометрическом определении pH.
Концентрация ионов Н+, которая определяется в процессе про-
ведения реакции нейтрализации, характеризует общую кислот-
ность. Она обычно превышает активную кислотность. Только в
очень разбавленных растворах сильных кислот эти величины ока-
зываются близкими по значению. Если кислотность раствора обус-
44
ловлена присутствием свооодной угольной кислоты, то активная
кислотность будет меньше общей, что связано с очень малой сте-
пенью диссоциации этой кислоты (/\i = 3-107, Л'2 = 6• 10"11). Вве-
дение ионов ОН" при нейтрализации кислоты щелочью способст-
вует смещению равновесия диссоциации угольной кислоты в сто-
рону образования ионов Н+ и НСО3". Это связано с тем, что ионы
Н" связываются ионами ОН" в молекулы воды. При достаточном
количестве гидроксид-ионов создаются условия для полной нейт-
рализации ионов Н+ слабой кислоты: Н2СОз + 2\аОН = На2СО3 +
+ 2Н2О.
Кислотность природных вод обычно обусловлена наличием сво-
бодной угольной кислоты, а для некоторых видов вод — также гу-
миновыми и другими слабыми органическими кислотами. При на-
личии этих кислот pH воды обычно не бывает <4,5. Кислые руд-
ничные воды, содержащие сульфаты железа, марганца, вследствие
гидролиза этих солей могут иметь более низкое значение pH.
При аналитическом определении кислотности воды различают
общую и свободную кислотности. Свободная кислотность — это та
часть общей кислотности, которая вызывает снижение pH раство-
ра до 4,5 и ниже. Свободная кислотность выражается количеством
титрованного раствора сильного основания (мг-экв/л), израсходо-
ванного при нейтрализации до достижения pH 4,5.
Общая кислотность определяется количеством титрованного
раствора сильного основания (мг-экв/л), которое необходимо для
полной нейтрализации исследуемого раствора по достижении pH 8,3.
Если исследуемый раствор (вода) имеет рН>8,3, то кислотность
принимается равной нулю. Кислотность воды определяют титрова-
нием пробы исследуемой воды стандартным раствором гидроксида
или карбоната натрия. Точка эквивалентности, соответствующая
окончанию реакции нейтрализации, определяется визуально по
изменению окраски индикатора. При определении свободной кис-
лотности применяется индикатор метиловый оранжевый, а общей
кислотности — фенолфталеин. Достижение точки эквивалентности
при взаимодействии основания с сильными кислотами может быть
зафиксировано любым из кислотно-щелочных индикаторов, имею-
щих интервал перехода окраски от 4 до 9, так как уже от одной
лишней капли основания при титровании исследуемого раствора
резко возрастает pH. Активная реакция среды раствора по окон-
чании реакции будет нейтральной, так как образующаяся соль не
подвергается гидролизу. При проведении реакции нейтрализации
слабой кислоты сильным основанием активная реакция среды ра-
створа в точке эквивалентности будет щелочной, так как образую-
щаяся соль подвергается гидролизу. При проведении реакции нейт-
рализации в присутствии индикатора фенолфталеина в точке экви-
валентности (pH 8,3) могут образовываться не только средние, но
и кислые соли. Так, при определении общей кислотности природных
вод, кислотность которых обусловлена свободной угольной кисло-
той, реакция нейтрализации гидроксидом натрия заканчивается
образованием гидрокарбоната натрия: СО2 + NaOH^NaHCO3. Гид-
45
рокарОонат натрия подвергается гидролизу и реакция среягл ра-
створа становится щелочной: Nal4СО3 + Н2О^ХаОН + [Н2О + СО2].
Наличие в исследуемом растворе солей, образованных слабыми ос-
нованиями и сильными кислотами, например сульфатов железа,
алюминия, хлоридов никеля, цинка и других солей тяжелых метал-
лов, затрудняет определение точки эквивалентности вследствие гид-
ролиза этих солей. Поэтому предварительно создаются условия для
смещения реакции гидролиза вправо, для чего исследуемый раст-
вор кипятят в течение 2 мин и реакцию нейтрализации проводят в
горячем растворе. Для окрашенных, мутных, сточных вод предпоч-
тительно электрометрическое определение точки эквивалентности
(по pH).
§ 13. Активная, общая и свободная щелочность воды
Щелочность воды может быть обусловлена присутствием раство-
римых оснований, средними и кислыми солями, образованными силь-
ными основаниями и слабыми кислотами (гидрокарбонаты, кар-
бонаты, гидросиликаты, гидросульфиды и т. и.). Активная щелоч-
ность раствора характеризуется показателем рОН. Щелочность во-
ды определяется нейтрализацией соединений, обладающих щелоч-
ными свойствами, сильными кислотами. Обычно щелочность при-
родных вод обусловлена наличием гидрокарбонатов щелочных и
щелочноземельных металлов. Значение pH таких вод не превышает
8,3. Если pH воды<4,5, принято считать общую щелочность воды,
равной нулю. Для природных вод с рН<8,3 общая щелочность од-
новременно соответствует карбонатной жесткости и содержанию
ионов НСОз~ (мг-экв/л). При анализе воды определение общей ще-
лочности проводится нейтрализацией раствора сильной кислотой до
pH 4,5. Если pH исследуемого раствора >8,3, то количество силь-
ной кислоты (мг-экв/л), необходимое для снижения pH до этого
значения, характеризует свободную щелочность. Свободная щелоч-
ность обусловлена наличием свободных оснований и растворимых,
солей — карбонатов, силикатов, сульфидов. Общая щелочность во-
ды характеризуется количеством кислоты (мг-экв/л), необходимым
для снижения pH до 4,5. Если pH исследуемого раствора <8,3, то
свободная щелочность его будет равна нулю. При определении ще-
лочности, обусловленной карбонатом и гидрокарбонатом натрия,,
нейтрализацией соляной кислотой протекают реакции по уравне-
ниям:
Na2CO3 -у HCJ == NaHCO3 Д-NaCJ (pH 8,3д
NaHCO3 + HCI = H2O + CO2 щ NaCl (pH 4,5)
Точку эквивалентности определяют с помощью индикаторов фенол-
фталеина и метилового оранжевого или электрометрически по со-
ответствующим значениям pH. Зная общую т и свободную р ще-
лочности, можно рассчитать содержание карбонат- и гидрокарбо-
нат-ионов при их совместном присутствии в исследуемом растворе.
В табл. 3 приведены соотношения, по которым приближенно рас-
46
считывают содержание ионных форм при известных значениях об-
щей и свободной щелочности.
Т а б л и ц а 3. Соотношения для вычисления ионных форм по щелочности *
(). и пелс I! С’ ше : женне ..у шщеи >0 >Д!Щ)Й 41!' 'С1ЫО Г.нРжар и- на.ы, мг-зкв К:/‘бона ‘ н . ЧГ-; КВ/,' 1 ' н югнпе нежду общей и св юодной Ж С.I 'ЧНОС [ ыо 1 II 'И.щЩфСй на. ы, мг-^кв л Каубонагы, <1
р =- (J т 0 2р т 0 2 (/и — р)
2р <у //. т — 2d р ----- т 0 0
- т 0 2/?
’ Унифицированные методы анализа вод/Под род. Ю. IO. Лурье. М., 1973, с. 166.
Воды, содержащие большое количество гуминовых соединений,
имеют гуматную щелочность, которую определяют вытеснением гу-
миновых кислот из их солей действием сильной кислоты. Содержа-
ние различных форм щелочности можно вычислить по специаль-
ным номограммам.
Протолитическая теория кислот и оснований. Для характерис-
тики кислотных и основных свойств соединений может быть ис-
пользована протолитическая теория Бренстеда и электронная тео-
рия Льюиса. Согласно данным теориям кислоты и основания от-
носятся к одному классу — протолитам. Основным признаком
кислотности или основности соединения является его отношение к
протону.
Соединение, которое способно отдавать протон, т. е. быть доно-
ром, является кислотой. Основанием считается соединение, прини-
мающее протон (акцептор). Примером такого донорно-акцептор-
ного взаимодействия с переходом протона является образование
ионов Н3О+ и ОН~ из двух молекул воды. Здесь одна молекула во-
ды является кислотой, а другая — основанием. При взаимодейст-
вии кислота переходит в сопряженное с ней основание. Например,
при взаимодействии НС1 с водой:
HCI + Н2О Н3О+ Щ Cl-
кислота (1) основание (2) кислота (2) основание (1)
Отдавая молекуле воды протон, НС1 выполняет роль кислоты (1), а
молекула воды — основания (2). Образующийся ион гидроксония
становится кислотой (2), а ион С1~ — основанием (1). По теории
Бренстеда катионы поливалентных металлов в водных растворах
обладают кислотными свойствами. Они гидратируются с образо-
ванием аквакомплексов, способных давать протон. Например,
[Си (НзОф/.р^щ^Сн (Н2О) зО11]+ +Н+. При взаимодействии с аммиа-
ком вода выполняет функцию донора протона, т. е. является кисло-
той. Образующий ион NH4+ тоже обладает кислотными свойствами,
так как может отдавать протон:
NH3 + Н2О NH+ щ ОН-
основание (2) кислота (1) кислота (2) основание (1)
47
По теории Льюиса к кислотам относятся соединения, которые
являются донорами неподеленной электронной пары. По этому при-
знаку наличие протона в молекуле кислотного соединения необяза-
тельно (апротонные кислоты). Кислотами могут быть ионы и нейт-
ральные молекулы. Так, к кислотам относятся такие соединения,
как А1С13, BF3.
Недостатком теории Бренстеда является фактическое отрицание
основной реакции кислотно-основного взаимодействия — реакции
нейтрализации.
§ 14. Окислительно-восстановительные процессы
Окислительно-восстановительные процессы сопровождаются от-
тягиванием или перемещением части или всех валентных электро-
нов от ионов, атомов или молекул одного из веществ, вступающих
в реакцию, к частицам другого реагирующего вещества. В окисли-
тельно-восстановительных реакциях одновременно, взаимосвязан-
но протекают два процесса: окисление и восстановление. Окисле-
ние связано с отдачей электронов отдельными атомами или груп-
пой атомов. Восстановление заключается в присоединении элект-
ронов отдельными атомами или группой атомов. Те атомы, ионы
или молекулы, которые отдают электроны, называются восстанови-
телями. Атомы, ионы или молекулы, которые присоединяют элект-
роны, называются окислителями. В процессе окислительно-восста-
новительной реакции восстановитель окисляется, а окислитель вос-
станавливается.
В зависимости от характера химической связи (ковалентная,
ионная) в молекулах реагирующих веществ окислительно-восста-
новительная реакция может закончиться образованием соединений
с ковалентной связью различной степени полярности или, реже,
соединений с ионным типом связи. При образовании ионных соеди-
нений происходящий в процессе реакции переход электронов от
одних взаимодействующих атомов, ионов или молекул к другим,
сопровождается соответствующим изменением их электрохимичес-
кой валентности. При этом электронные облака образующихся
ионов разобщены.
Если в результате окислительно-восстановительной реакции об-
разуются ковалентные соединения за счет перекрывания электрон-
ных облаков взаимодействующих атомов, изменения валентного со-
стояния атомов практически не происходит.
Так, при образовании полярной молекулы воды из неполярных
молекул водорода и кислорода атомы водорода и кислорода не
изменяют своей валентности. В результате реакции электроны ато-
мов водорода оттягиваются в сферу электростатического притяже-
ния ядра кислорода, т. е. при этом происходит не передача элект-
ронов от одного атома к другому, а только перераспределение элек-
тронных плотностей, и пара связующих электронов оттягивается к
более электроотрицательному атому кислорода:
48
Подобное же перераспределение электронных плотностей, не со-
провождающееся полным переходом электронов, наблюдается и
при окислении и восстановлении органических соединений. Вслед-
ствие того, что электроны, образующие связь, смещены к более элек-
троотрицательному атому, в данном примере — атому кислорода,
он получает отрицательный заряд. Заряд атома, возникающий пос-
ле такого распределения электронов, называют степенью окисле-
ния. Степень окисления — это кажущийся заряд атома, который
возникает при отдаче или присоединении электронов в ионных со-
единениях или в результате притягивания или оттягивания элект-
ронных пар от одного атома к другому в молекулах полярных
соединений. При этом условно считается, что молекула состоит
только из ионов. Степень окисления может иметь положительное,
нулевое и отрицательное значения. Она вычисляется как алгебраи-
ческая сумма полярных связей. Степень окисления атомов в ионных
соединениях по величине и знаку соответствует заряду иона, а у
атомов неполярных молекул (Н2, О2 и др.) равна нулю, так как от-
сутствует одностороннее оттягивание общих пар электронов. Рас-
смотрим изменение степени окисления атома углерода при окисле-
нии щавелевой кислоты перманганатом калия. Эта реакция прово-
дится при определении перманганатной окисляемости воды по урав-
нению
2КМпО4 -у 5Н2С2О4 + 3H2SO4 = ЮСО2 + 2MnSO4 + K2SO4 + 8Н2О
Валентность атома углерода в молекуле щавелевой кислоты и
углекислого газа равна четырем, а степень окисления — соответ-
ственно + 2 и +4.
В высшей степени окисления атомы проявляют только окисли-
тельные свойства, в низшей — только восстановительные. К типич-
ным окислителям относятся хлор, фтор, кислород, перманганат ка-
лия и другие соединения, в которых содержатся атомы, имеющие
высшую степень окисления. Восстановители — металлы, водород,
сероводород и другие водородные соединения элементов, т. е. такие
соединения, в которых содержатся атомы, находящиеся в низшей
степени окисления. Соединения, в которых атомы проявляют про-
межуточную степень окисления, могут быть в зависимости от ус-
ловий проведения реакций окислителями или восстановителями
(например, сульфиты, нитриты, соли железа (II) и др.).
Эквивалент окислителя (восстановителя) равен количеству
окислителя (восстановителя), которое соответствует одному при-
соединенному (отданному) электрону в данной окислительно-вос-
49
стаиовительной реакции. Иногда окислитель и восстановитель мо-
гут находиться в одном веществе (например, в молекуле воды при
реакции разложения ее на водород и кислород). Иногда роль окис-
лителей п восстановителей выполняют атомы элементов с проме-
жуточной степенью окисления, которые входят в состав мотекулы
одного вещества. Примером такой реакции может служить полу-
чение гипохлорита натрия при взаимодействии хлора с раствором
гидроксида натрия:
о -1 +1
Cl2 + 2ХаОН NaCl + NaCIO + Н:О
Окислительно-восстановительные потенциалы. Окислители н
восстановители различаются между собой по химической актив-
ности. Сильные окислители способны к отнятию (оттягиванию)
электронов от многих восстановителей, в том числе и тех, которые
с трудом отдают электроны, т. е. слабых. Слабые окислители могут
взаимодействовать только с сильными, типичными восстановите-
лями.
В окисленной форме (MnO4_, Fe3+, С1°) степень окисления эле-
мента больше, чем в восстановленной (Mn2+, Fe2+, СП). В окис-
лительно-восстановительной реакции окисленная форма элемента
выступает в качестве окислителя, восстановленная выполняет роль
восстановителя. Чем активнее окислитель, тем меньше будут выра-
жены восстановительные свойства у восстановленной формы. На-
пример, свободный хлор является сильным окислителем, а его во-
дородное соединение будет проявлять слабые восстановительные
свойства, так как хлор имеет сильно выраженную способность к
присоединению электронов. Возможность и направленность проте-
кания окислительно-восстановительных реакций определяется ко-
личественной характеристикой химической активности окислителя
и восстановителя — окислительно-восстановительным потенциа-
лом.
Окислительно-восстановительные реакции сопровождаются из-
менением заряда или степени окисления взаимодействующих в ра-
створе ионов. Работа гальванических элементов основана на про-
текании реакций окисления —- восстановления. Так, в медно-цин-
ковом элементе осуществляется окислительно-восстановительная
реакция:
Zno + Cu2+ Cuo + Zn2+
Zn° — 2^>->Zn2+ (восстановитель)
Cu2+ -|-2e->-CuO (окислитель)
Электродные реакции окисления — восстановления протекают
на границе электрода и раствора соответствующей соли металла.
Если же вместо активных электродов использовать инертные элект-
роды (например, платиновые), которые не будут изменяться в ходе
окислительно-восстановительной реакции, а будут выполнять толь-
ко роль проводников электронов в данной реакции, то можно опре-
делить относительную величину окислительно-восстановительного
потенциала.
50
мюсилютную величину окислительно-восстановительного потен-
циала определить невозможно, поэтому его измеряют относительно
стандартного электрода, чаще всего водородного, нормальный по-
тенциал которого в реакции 21+~ + 2ещШ2 принимается равным
нулю.
При определении окислительно-восстановительного потенциала
исследуемого раствора применяют инертный (платиновый) элект-
род и стандартный электрод сравнения (каломельный, хлорсереб-
ряный, водородный и др.). Направленность протекания окислитель-
но-восстановительной реакции определяется направлением перехо-
да электронов: они будут переходить с электрода, где их количест-
во больше, на электрод, где их количество меньше. Э.д.с., равная
разности потенциалов полуэлементов, определяется компенсацион-
ным методом.
В табл. 4 приведены значения нормальных окислительно-восста-
новительных потенциалов, которые позволяют определять направ-
ление окислительно-восстановительных процессов, возможность их
протекания и одновременного существования в растворе различ-
ных соединений.
Таблица 4. Нормальные окислительно-восстановительные потенциалы, Ео
(20° С)
Окислительно-восстановительный процесс r0, В Окисли i ельно-восстановительный процесс £-0. В
Al qq А13+ 4-Зс -1,7 Fe2+ qq Fe3+ + 4-0,77
SO^~+2OH-qq SO|—+Н2О+2с -0,93 hno2 + H20qqN0~ + зн+ + 2c +0,94
S + бон- qq so|- + 3H2O + 4c —0,90 i- + 3H2o qq io~ + 6H+ + 6c -1,09'
H2 4-20H-qt2H20 + 2c -0,83 2H20qq o2 + 4H+ + 4c + 1,23
Znqq Zn2+ + 2c -0,76 Mn2+ +2H20qqMn02 + + 1,23-
Fe (OH)2 + OH- q^Fe (OH)3 + c -0,56 + 4H+ + 2c
FeqqFe2+ 4-2c —0,44 2Сгз+ +7H2oqqCr2o^- + + 1,36
Cd qq Cd2+ 4- 2c —0,40 + 14H+ +6c
Ni q±Ni2+ +2c —0,25 2Ci-qqci2 + 2c + 1,36
H202 + 2OH- qq O2 + 2H2O +2c —0,08 ci- + H20qq нею + н+ + 2c + 1,49
h2 qq 2h+ + 2c 0,00 Mn2+ 4- 4H2o qq MnOj- + + 1,51
h2s qq s + 2H+ + 2c +0,14 + 8H+ 4- 5c
с2н5он qq CH3CH0 + 2h+ + 2c +0,19 V2CI2 + H20 qq HC1O + H+ 4- c + 1,63
Cu qqcu2+ + 2c +0,337 2H2O qq H2O2 + 2H+ + 2c + 1,77
4ОН- O2 + 2H2O + 4c +0,4 o2 + H2o qq 0,3 + 2H+ + 2c +2,07
Br~ 4- бон—qqBro~+3H2o+6c +0,6 2F-qqF2 + 2c +2,85
Пользуясь нормальными окислительными потенциалами, можно
определить, какое из веществ будет в реакции окислителем (вос-
становителем), в каком направлении она пойдет и рассчитать окис-
лительно-восстановительный потенциал данного процесса. Величи-
51
на его определяется по разности окислительно-восстановительных
потенциалов окислителя и восстановителя. Например, окислитель-
но-восстановительный потенциал для реакции H2S + Cl2 = S + 2НС1
рассчитывается следующим образом. Пользуясь табл. 4, находим
нормальные окислительно-восстановительные потенциалы для пар:
СЕ^2С1~+2е, равный +1,36 В, и H2S^S + 2Н+ + 2е, равный
+ 0,14 В. Окислителем в данной реакции будет хлор, имеющий
большую величину Ео. Окислительно-восстановительный потенциал
Eh этой реакции будет равен:
£Л = ( 4- 1 .Зб^В) — ( +0,14 В)^ 1,22 В.
Положительное значение Efi указывает на возможность самопроиз-
вольного протекания окислительно-восстановительной реакции в
данном направлении. Вычисление Ео относится к стандартным ус-
ловиям проведения реакции.
Зависимость окислительно-восстановительного потенциала от
концентрации окисленной и восстановленной формы в растворе опи-
сывается формулой Нернста:
„ С 0,058 «окисл
Е = Е0 +--------1g--------
« «ВОССт
(1.63)
где Ео — нормальный окислительно-восстановительный потенциал,
В; п —- число электронов, отданных инертному электроду при про-
текании окислительно-восстановительной реакции; аОкисл — актив-
ность соединения в окисленной форме; аВосст — активность соедине-
ния в восстановленной форме. Так, для окислительно-восстанови-
,тельного процесса Cl2 + 2Fe2+=2F3+ + 2Cl~ Eh вычисляется для каж-
дой из окислительно-восстановительных пар по формулам:
О. 058 «Fe3+
1g —----
[n «Fe2+
„ 0,0о8 дс12
Eh — ^0 + 1g 2
n «Ci-
Если происходит одновременно одинаковое изменение ионных
концентраций растворов в обоих полуэлементах, то величина Е/(
будет соответствовать EQ.
Окислительная способность соединений зависит от pH в тех ре-
акциях, где одним из компонентов реакции является вода или ионы
Н+. Зависимость величины Eh от концентрации ионов Н+ можно
рассчитать по формуле Нернста:
Eh — Eq +
0,058 ,
lg
[H+]2
[H2] *
(1.64)
Так как Е0 = 0 для водородного электрода, то формула (1.64) при-
обретает вид
Eh = 0,029 lg([H+]2/[H2]). (1.65)
Отрицательный логарифм концентрации водородных ионов есть
pH, а отрицательный логарифм давления молекулярного водорода
52
обозначают через гН2. После соответствующих преобразований
уравнения (1.65) получаем
Eh = 0,029(гН2 — 2рН), (1.66)
откуда имеем
-£/г +2рН. (1.67)
- 0,029
Уравнение (1.67) позволяет охарактеризовать окислительно-вос-
становительные свойства среды: чем меньше гН2) тем легче идут
процессы восстановления. Увеличение гН2 и соответственно умень-
шение давления молекулярного водорода способствуют протеканию
окислительных процессов. Как следует из уравнения (1.67), вели-
чина Eh зависит от давления молекулярного водорода и концент-
рации ионов Н+. Поэтому она может быть использована для харак-
теристики окислительно-восстановительных условий среды только
при определенном значении pH. При равенстве молярных концент-
раций кислорода и водорода гН2~28. Для водного раствора, насы-
щенного водородом, гН2 - ().
Глава II. ПРИРОДНЫЕ ВОДЫ.
ПОКАЗАТЕЛИ КАЧЕСТВА ВОДЫ
§ 15. Круговорот воды в природе.
Мировые водные ресурсы. Водные ресурсы СССР
Воды мирового океана, атмосферы, рек, озер, подземные объ-
единяются в общее понятие — гидросфера. Отдельные составные
части гидросферы взаимосвязаны, между ними осуществляется по-
стоянный обмен, взаимодействие, т. е. в природе происходит круго-
ворот воды. Испаряющаяся с поверхности земли вода общим объ-
емохМ более 525 тыс. км3 образует атмосферные воды в форме водя-
ного пара. При охлаждении в верхних слоях атмосферы водяной
пар конденсируется в мельчайшие капли воды или кристаллы льда,
которые возвращаются на землю в форме атмосферных осадков.
Вода, испаряющаяся с поверхности морей и океанов и возвращаю-
щаяся в виде осадков вновь на поверхность воды, включается в
малый круговорот. Если же атмосферные осадки выпадают на по-
верхность суши, то часть из них оказывается непосредственно в
водах рек, озер, а большая часть в процессе фильтрации через
почву обогащается минеральными и органическими веществами,
образуя подземные воды. Вместе с поверхностным стоком они по-
ступают в воды рек, а оттуда возвращаются в океан. Вода, испа-
ряющаяся с поверхности суши, образуя атмосферные воды, в виде
осадков тоже может попасть в океан. Так замыкается большой
круговорот воды. При этом общее количество воды на Земле оста-
ется постоянным. На рис. 5 представлена схема круговорота воды.
Общее уравнение мирового водного баланса имеет вид
UQ + Uc = Оо Ос, (II. 1)
53
Рис. 5. Круговорот воды в природе:
1 — океан; 2 — поверхностный и подземный сток; 3 — бессточные области
где Uo и Uc — количество воды, испаряющейся с поверхности окса-
на и суши; Оо и О(-—количество осадков, выпадающих н’а поверх- ности океана и суши. Объем гидросферы, по современным данным,
составляет более 1,4 млрд. км3. Данные о Таблица 5. Запасы воды в различных частях гидросферы запасах воды в различных частях гидросферы приве- дены в табл. 5. Водные ресурсы вклю- чают в себя все виды вод, пригодные для использо- вания. Основная роль при
Части гидросферы Объем воды, тыс. км3 Содержание воды в раз- личных час- тях гидро- сферы, %
Мировой океан Подземные воды в том числе зона ак- тивного водообмена Ледники Озера Почвенная влага Водяные пары в атмос- фере Речные воды 1 370 323 60 000 4 000 24 000 280* 85** 14 1,2 93,96 4,12 0,27 1,65 0,019 0,006 0,001 0,0001 определении водных ре- сурсов принадлежит прес- ным водам суши, так как они наиболее доступны и постоянно возобновляют- ся. Характерной особен- ностью водных ресурсов^ является их неисчерпае- мость при рациональном использовании. Превыше- ние расхода воды в ре- зультате хозяйственной деятельности людей над
* В том числе около 5 хранилищах. ** В том числе тельных вод. тыс. км3 воды в водо- около 2 тыс. км3 ороси-
естественным восполнени-
ем ^приводит к нарушению естественного равновесия. Так, чрезмер-
ный расход подземных вод сопровождается понижением их уровня.
Для водоснабжения частично используются морские и минера-
лизованные подземные воды после соответствующей обработки.
Однако определяющей роли в водном хозяйстве они не могут иметь
так как их подготовка связана с большим расходом электроэнергии
и сложным технологическим оформлением. Запасы пресной воды
.гост) иной для использования, ограничены. Дополнительным ослож-
54
пенном является и неравномерность их распределения, что создает
напряженные условия эксплуатации водных ресурсов в отдельных
районах, особенно с высокой концентрацией промышленного про-
изводства.
По запасам водных ресурсов Советский Союз занимает второе
место после Бразилии. Среднегодовой речной сток в СССР состав-
ляет 4384 км3. О степени неравномерности распределения водных
ресурсов свидетельствуют удельные водные ресурсы для отдельных
республик и районов страны. В среднем по РСФСР они равны
30,7 тыс. м3/год на человека, в районах Дальнего Востока — 250,
Поволжья — 2,1, в Туркмении—0,5, в Молдавии — 0,2,тыс. м3/год.
В пополнении водных ресурсов важную роль играют воды озер,
общий запас воды в которых составляет примерно 26 тыс. км3.
Суммарные эксплуатационные запасы пресных и солоноватых под-
земных вод составляют около 220 км3/год. Ледники содержат око-
ло 2400 км3 воды. Общий объем ежегодно возобновляемых прес-
ных вод в СССР составляет 4634 км3, из них 4384 приходится на
речной сток, на подземные воды — 220 и наледи — 30 км3.
§ 16. Физико-химическая характеристика
природных вод
Природные воды представляют собой сложные системы, содер-
жащие растворенные вещества в виде ионов и молекул, минераль-
ные и органические соединения в форме коллоидов, суспензий и
эмульсий. В воде растворены газы, входящие в состав атмосферы,
а также вещества, образующиеся в результате жизнедеятельности
водных организмов и протекания процессов химического взаимо-
действия в самой водной среде. Формирование состава природных
вод происходит в результате взаимодействия воды с окружающей
средой — горными породами, почвой, атмосферой. При этом про-
текают процессы: а) растворение соединений; б) химическое вза-
имодействие веществ с водой и водными растворами; в) биохими-
ческие реакции; г) коллоидно-химические взаимодействия. Дейст-
вие каждого из этих процессов определяется такими условиями
взаимодействия веществ с водой, как температура, давление, гео-
логические особенности. На формирование состава поверхностных,
подземных и атмосферных вод заметно влияет усиливающаяся
практическая деятельность человека.
Изменение состава воды может наблюдаться при разбавлении,
смешении вод различного состава, изменении температуры. Раст-
ворению горных и осадочных пород способствуют химические
реакции, протекающие с образованием растворимых соединений.
Это реакции гидролиза, окисления — восстановления, карбониза-
ции, выщелачивания, ионного обмена и др. Карбонатные породы
разрушаются под действием угольной кислоты, которая переводит
труднорастворимые карбонаты в более растворимые гидрокарбо-
наты. Так, например, минерал кальцит легко выветривается под
действием угольной кислоты СаСО3 + Н2О + СО2 = Са(НСО3)2. За-
55
Таблица 6. Растворимость СаСО3 в зависимости от содержания СО2 в газовой фазе висимость растворимости карбоната кальция от со- держания СО2 приведена
Раствори- мость СаСО3, г/л Объемное содержание со2, % Примечание в табл. 6. Вещества, растворен- ные в воде, могут нахо- диться в виде молекул
0,0131 0,0634 0,1334 1,0986 0,00 0,03 0,3 100,00 При среднем содержа- нии СО2 в воздухе При среднем содержа- нии СО2 в почвенном воздухе Раствор, насыщенный СО2 при атмосферном давлении (газы, некоторые органи- ческие вещества), простых и комплексных ионов (на- пример, [UO2 (СО3)3]4~). Ионы в водных раство- рах гидратированы, они могут образовывать ак- вакомплексы типа
[Си (Н2О)3ОН]+. Возможны такие образования комплексных соеди-
нений катионов и анионов. В морской воде примерно третья часть
Са2+ и Mg2+ находится в форме недиссоципрованных сульфатных
комплексных ионов, а также образуются комплексы [XaSOJ и
лишь 8% от общего содержания сульфатов находится в форме
SO?.
Катионы могут входить в состав комплексов с органическими
соединениями. Например, широко распространенной формой содер-
жания железа в воде являются его комплексы с гуминовыми кис-
лотами, обусловливающие цветность воды. Растворимость трудно-
растворимых соединений в природных водах больше, чем в дистил-
лированной воде. Так, произведение растворимости карбоната
кальция в морской воде (при концентрации хлоридов 19 г/кг и
20° С) почти в 500 раз больше, чем в дистиллированной. В процес-
сах окисления минералов основная роль принадлежит кислороду.
Так, дисульфид железа (II) (пирит) окисляется во влажном возду-
хе по уравнению
2FeS2 + 2Н2О + 7О2 = 2FeSO4 + 2H2SO4
и дальше
4FeSO4 + 2H2SO4 + О2 = 2Fe2 (SO4)3 + 2Н2О
Принимая во внимание также и гидролиз образующейся соли
железа (III), легко объяснить причину высокой кислотности руд-
ничных вод, pH которых может быть <3. В этих водах могут рас-
творяться такие минералы, как апатит, карбонаты, сульфиды,
алюмосиликаты, бокситы и даже магнетит. При растворении апа-
тита в присутствии серной кислоты образуются фосфорная и пла-
виковая кислоты. Последняя способствует появлению в природных
водах фторидов.
Агрессивными по отношению к некоторым минералам являются
и щелочные растворы, образующиеся, например, при разложении
нефелина NaAlSiO4 под действием воды и диоксида углерода:
2NaAlSiO4 4- лгН2Э ~ СО,Na2CO34-AbO3.2SiO2-nH2O
56
Щелочные растворы вызывают растворение амфотерных окси-
дов (А12О3, ВеО) и силикатов. Вода, участвуя в реакциях гидроли-
за, обусловливает перевод в растворимые формы таких минера-
лов, как алюмосиликаты.
В формировании состава природных вод важная роль принад-
лежит процессам обмена между ионами, содержащимися в воде,
и ионами, входящими в состав алюмосиликатов почвы. В результа-
те ионного обмена происходит разделение калия и натрия. Калий
поглощается почвой, а натрий не задерживается и всегда значи-
тельно преобладает в природных водах. На формирование состава
Рис. 6. Классификация природных вод
поверхностных вод большое влияние оказывают биохимические
процессы, протекающие в живых водных организмах и при их от-
мирании. Они влияют на основной химический состав, распределе-
ние газов, содержание микроэлементов и биогенных веществ.
Многообразие природных вод по химическому составу отраже-
но в классификации В. И. Вернадского, согласно которой выделено
485 видов вод с учетом солесодержания, преобладающего иона
или группы ионов, соотношения между определенными группами
ионов, наличия специфических компонентов. По солесодержанию
(г/кг) природные воды делятся на следующие виды: 1) пресные
до 1; 2) солоноватые 1—25; 3) воды с морской соленостью 25—50;
4) соленые воды >50.
Достаточно полной классификацией природных вод по химиче-
скому составу является классификация, предложенная О. А. Алеки-
ным (рис. 6). Согласно классификации природные воды по преобла-
дающему аниону делятся на три класса: карбонатный, или гидро-
карбонатный (С), сульфатный (S) и хлоридный (С1). По
преобладающему катиону классы делятся на группы: кальциевую
(Са), магниевую (Mg) и натриевую (Ха). Группы, в свою очередь,
делятся на типы, указывающие на соотношение между ионами
(мг-экв). Первый тип характеризуется следующим соотношением:
57
Таблица 7. Изменение содержания
отдельных ионов от солесодержания
в воде
HCO3“>Ca2+ + Mg2+. Сюда отно-
сятся мягкие воды с небольшим
солесодержанием, с преобладани-
Солесо дер-
жание,
г/л (кг)
Преобладающие и> >ны
ем ионов Na+ и КС Для второго
типа выполняется соотношение
НСО3-< Са2+ + Mg2+< 11С()
-rSO42 . Состав этих вод форми-
<0,06
>2,0
>168,3
>193,9
>328,6
Са2+, SO2~, НСО>
Na+, С1-, Mg2+
SO^-, НССУГ, Na+, CI-,
Mg2-
HCO< Na+, Cl-,Mg2 L
Na+, Cl—, Mg2+
C1-, Mg2+
руется при контакте их с осадоч-
ными породами и продуктами вы-
ветривания. Это воды большинст-
ва рек, озер и подземные воды с
малым и средним солесодержани-
ем. В водах, относящихся к
третьему типу, наблюдается со-
отношение между поил ми:
HCO3~ + SO42"<Ca2~ + Mg2 или
CI~>Na+. К этому типу относятся воды морей и океанов, сил
!О-
минерализованных подземных источников. К четвертому типу от-
носятся кислые воды (НСО3~ = 0). Они могут быть только в суль-
фатном н хлоридном классах, в группах Са и Mg. По этой класси-
фикации можно кратко охарактеризовать состав воды следующей
формулой: принадлежность воды к классу указывается символом
элемента, входящего в состав преобладающего аниона. Справа,
вверху от него пишется химический символ преобладающего катио-
на, т. е. обозначается группа. Справа, внизу от символа класса рим-
ской цифрой обозначается тип. Например, С^а (вода гидрокарбо-
натного класса, группы кальция, тип II) или Sm (сульфатный
класс, группа натрия, тип III). Иногда рядом с обозначением типа
указывается солесодержание (г/кг), а у символа группы дается
общая жесткость. Когда содержание другого иона отличается от
преобладающего не больше чем на 5 мг-экв, то в формулу записы-
ваются символы двух элементов.
Концентрация основных ионов в природных водах зависит от
солесодержания. Содержание ионов лимитируется образованием
труднорастворимых соединений. Изменение содержания отдельных
ионов в воде в зависимости от солесодержания приведено в табл. 7.
§ 17. Химические ингредиенты природных вод
и их значение для оценки качества воды
Химические компоненты природных вод условно делят на пять
групп (О. А. Алехин): 1) главные ионы; 2) растворенные газы;
3) биогенные вещества; 4) микроэлементы; 5) органические ве-
щества.
Главные ионы. В природных водах установлено присутствие
более 70 химических элементов. Наиболее распространенные анио-
ны: НСО< , С1-, СО3~ , HSiCC и катионы: Na+, Са2+, Mg2+
58
К+ Гс2+. Содержание главных ионов в пресных водах составляет
90—95% от общего солесодержания.
В природных водах постоянно присутствуют ионы Са2+ и ?Bg2+,
которые обусловливают общую жесткость воды. Основной источ-
ник их поступления в воде—растворение пород, содержащих из-
вестняки, доломит, гипс, сложные алюмосиликаты. Ионы Са2+
характерны для мало- и средпемиперализоваппых вод. При повы-
шении солесодержания до 1 г/кт концентрация ионов Мц2+ увели-
чивается. В минерализованных водах они становятся преобладаю-
щими, что связано с лучшей растворимостью в воде солей магния.
В санитарно-гигиеническом отношении ионы Са2+ и Mg2+ не пред-
ставляют особой опасности, но значительная жесткость делает
воду непригодной для хозяйственно-бытовых и производственных
нужд.
Ионы Na+ и К+ встречаются почти во всех природных водах.
Соли натрия, присутствующие в водяном паре, обусловливают пе-
нистый переброс солей. Последнее делает нежелательным их при-
сутствие в воде, идущей для питания паровых котлов среднего и
высокого давления.
Гидрокарбопат-иоп часто преобладает в пресных водах. Нали-
чие его в природных водах связано с растворением карбонатных
пород под действием диоксида углерода. Ион НСО3~ устойчив в
природных водах в интервале значений pH 4,2—12. Вместе с иона-
ми Са2+ и Mg2+ он обусловливает карбонатную жесткость воды.
Для большинства природных вод содержание гидрокарбоиат-иона
(мг-экв/л) характеризует их общую щелочность.
Ионы SO4” поступают в природные воды в процессе растворе-
ния гипсовых пород, мирабилита, окисления сульфидов, серы и ор-
ганических серусодержащих веществ. Содержание сульфат-иона
лимитируется в питьевой воде; так, при концентрации более
500 мг/л у человека может проявляться расстройство деятельности
желудочно-кишечного тракта. В производственных условиях зна-
чительное содержание сульфатов в воде способствует возникнове-
нию коррозии бетона. Содержание сульфат-ионов может быть до-
статочно высоким в водах атмосферных осадков вследствие загряз-
нения воздуха промышленными выбросами.
Хлориды по общему содержанию в природных водах занимают
первое место среди анионов. Содержание их колеблется от деся-
тых долей до тысячи мг/л и более. В пресных водах их концентра-
ция обычно невелика. Они появляются в природных водах при
растворении горных пород, содержащих хлориды, выбрасываются
в большом количестве при извержении вулканов. Хлориды — по-
стоянный компонент бытовых сточных вод и стоков некоторых
производств. При концентрации хлорид-иона выше 300 мг/л у
воды появляется солоноватый привкус. Кроме того, хлориды уси-
ливают коррозию железа в воде. Повышение концентрации хло-
ридов в воде может быть косвенным показателем загрязнения
водоема сточными водами.
59
Растворенные газы. К И С Л О р О д. Изменение растворимости
кислорода в интервале температур от 0 до : 29 С при атмосферном
давлении и содержании ниже. кислорода 20,9% в воздухе приведено
Температура, °C 0 1 2 3 4 5
Растворимость СЬ, мг/л 14,66 14,23 13,84 13,34 13,13 12,80
Температура, °C 6 7 8 9 10 11
Растворимость СЦ, мг;л 12,48 12,17 11,87 11,59 11,33 11,08
Температура, °C 12 13 14 15 16 17
Растворимость Ог, мг/л 10,83 10,60 10,37 10,15 9,95 9,74
Температура, °C 18 19 20 21 22 23
Растворимость О2, мг/л 9,54 9,35 9,17 8,99 8,83 8,68
Температура, °C 24 25 26 27 28 29
Растворимость Ог, мг/л 8,53 8,38 8,22 8,04 7,92 7,63
Содержание кислорода в поверхностных водоемах определяется
поступлением его из воздуха. Оно зависит также от времени года,
глубины водоема, условий рсаэрации, жизнедеятельности водных
макро- и микроорганизмов. Снижение концентрации растворенного
кислорода может указывав на загрязнение водоема органически-
ми соединениями. В придонных слоях кислорода меньше, так какой
потребляется в процессе окисления донных отложений.
Максимум содержания растворенного кислорода наблюдается
летом в период интенсивной фотосинтетической деятельности рас-
тительных организмов. В зимний период содержание кислорода в
ьоде резко уменьшается из-за трудности реаэрации и в связи с по-
ступлением практически только подземных вод, почти не содержа-
щих кислорода. Растворенный в воде кислород придает ей осве-
жающий вкус. Благодаря высокой химической активности кислород
усиливает коррозию металлов.
Диоксид углерода. Этот газ находится в воде как в рас-
творенном виде, так и в форме угольной кислоты. Суммарное со-
держание СО2 + Н2СО3, которое определяется при анализе воды,
объединяется понятием «свободная угольная кислота». Основным
источником диоксида углерода в поверхностных водах являются
биохимические процессы распада органических веществ. Кроме
того, он попадает в поверхностные водоемы с подземными водами,
а также поглощается водой из воздуха. Большое количество диок-
сида углерода содержится в воде Мирового океана; оно в 60 раз
превышает его количество в атмосфере. Диоксид углерода необхо-
дим для обеспечения жизнедеятельности водных растительных
организмов. Режим изменения содержания диоксида углерода в
воде является противоположным аналогичному процессу для кис-
лорода. В поверхностных водах содержание диоксида углерода
обычно составляет 0,5—2, максимум 20—30 мг/л. В подземных во-
дах его концентрация выше (50 мг/л и более). Содержание диокси-
да углерода в воде зависит от солесодержапия, pH, температуры,
концентрации ионов Са2+ и характеризуется карбонатным равно-
весием. При концентрации диоксида углерода в воде выше равно-
весной проявляется агрессивное действие его по отношению к бе-
тону и железу.
60
Сероводород. В водах, не содержащих растворенного кис-
лорода, создаются условия для появления сероводорода. Он обра-
зуется в результате растворения сульфидных минералов под дей-
ствием угольной кислоты, при биохимическом разложении ссрусо-
держащих органических соединений в отсутствие кислорода,
например в донных отложениях. Так как сероводород является ток-
сичным соединением и придает воде неприятный запах, который
обнаруживается уже при концентрации его в воде более 0,3 мг/л,
то наличие H2S в питьевой воде не допускается. Кроме того, серо-
водород вызывает коррозию железа и способствует развитию серо-
бактерий. Кроме растворенного сероводорода в воде могут нахо-
диться сульфид-S2- и гидросульфид-ионы HS-, так как раствор
сероводорода проявляет свойства слабой кислоты. При pH 5—9 в
воде присутствует сероводород в равновесии с гидросульфид-ионом.
При pH<5 в воде находится только сероводород, а сульфид-ионы
появляются в щелочной среде (рН>9).
Биогенные вещества. К этой группе относят соединения, необ-
ходимые для жизнедеятельности водных организмов и образую-
щиеся ими в процессе обмена веществ. Это, в первую очередь, ми-
неральные и органические соединения азота. Органические формы
азота представлены белками и продуктами их распада. Неоргани-
ческие соединения азота (NHt, NOT , NOD могут образоваться
при разложении азотсодержащих органических соединений или
же поступают в поверхностные воды с атмосферными осадками, при
вымывании удобрений из почвы (аммонийный азот, нитраты).
Содержание и преобладание различных форм азота зависит от
условий поступления азотсодержащих соединений в воду, режима
водоема. В паводковый период наблюдается увеличение концентра-
ции органических фор?л азота вследствие смыва органических ос-
татков с поверхности почвы, летом растворимые соединения азота
потребляются водными организмами и содержание их в воде сни-
жается. Нитриты (NO?) являются промежуточной формой окисле-
ния аммонийного азота в нитраты (NOF). Их содержание в природ-
ных водах обычно невелико. Концентрация нитратов в чистых водо-
емах также оценивается сотыми, десятыми долями мг/л. Но иногда,
например при смыве удобрений, может достигать 10 мг/л и более.
Аммонийные соединения обычно содержатся в воде в малых коли-
чествах (сотые, десятые доли мг/л).
Преобладание той или иной формы содержания неорганическо-
го азота может быть использовано для определения времени, про-
шедшего с момента загрязнения водоема органическими соедине-
ниями. При свежем загрязнении в воде содержится преимущест-
венно аммонийный азот, а присутствие в воде нитратов указывает
на то, что процессы разложения органических примесей заканчи-
ваются. Наличие соединений азота в воде, используемой для
промышленного водоснабжения, нежелательно, так как они спо-
собствуют развитию микроорганизмов в трубах, градирнях, бас-
сейнах.
61
Важным биогенные ->лс'-,(-итом "илистая фосфор. Концентрация
соединений фосфора в чистых водоемах незначительна (тысячные,
сотые доли мг/л). Это — минеральные формы содержания фосфора
(ли- и гидрофосфаты Н2РО~и НР04~ ) и органические соединения.
Увеличение концентрации фосфатов до нескольких миллиграммов
в литре воды способствует массовому развитию микроорганизмов
в трубопроводах распределительной сети и водорослей в водоемах.
Соединения железа (II) содержатся только в подземных водах.
Опп поступают в воду при растворении железосодержащих пород
под действием кислот (угольной, гуминовых и др.). В поверхност-
ных водах концентрация соединений железа (III) незначительна
вследствие полного гидролиза солей. Органическая форма содер-
жания железа в воде — это сложные комплексы с гуминовыми
кислотами, имеющие коричнево-бурую окраску. При концентрации
ионов железа более 0,3 мг/л у воды появляется железистый при-
вкус, а в трубопроводах возможно развитие железобактерий. Ионы
железа наряду с ионами Са2+ и Mg2+ сообщают воде жесткость.
Соединения кремния встречаются в природных водах в форме
различных минеральных и органических соединений. Это — крем-
ниевая кислота, ее соли (гидросиликаты и силикаты), а также час-
тицы различных алюмосиликатов в коллоидном и взвешенном со-
стоянии и органические соединения кремния. Концентрация крем-
ния в природных водах обычно не превышает нескольких
миллиграммов в литре, но в водах северных рек она выше и может
достигать десятков мг/л. Соединения кремния вызывают образо-
вание на стенках теплообменной аппаратуры трудноустранимой
накипи, имеющей малую теплопроводность.
Органические вещества. Основную часть органического вещест-
ва природных вод составляют гумусовые соединения, которые об-
разуются при разложении растительных остатков. Водный гумус
содержит в основном лигнино-протеиновые соединения. В состав
его входят также углеводы, жиры и воск. Почвенный гумус вклю-
чает в себя нерастворимый гумин, перегнойные кислоты и другие
продукты распада сложных органических веществ. Перегнойные
(гумусовые) кислоты делятся на гуминовые (гуминовая и ульми-
новая) и фульвокислоты (креповая и апокреновая). Гуминовые
кислоты — высокомолекулярные соединения, продукты конденса-
ции ароматических соединений типа фенола с аминокислотами и
протеинами. Их строение еще недостаточно изучено. В зависимости
от размера молекул гуминовые соединения могут образовывать в
воде истинные, коллоидные растворы и взвеси. Гуминовые кислоты
способны, вследствие межмолекулярных взаимодействий, образо-
вывать агрегаты молекул — мицеллы. Мицеллярная масса гумино-
вых кислот составляет 3700—8270. Фульвокислоты-—высокомоле-
кулярные соединения типа оксикарбоновых кислот, содержащие
азот, с меньшим количеством углеродных атомов, чем гуминовые.
Кислотные свойства у них выражены достаточно сильно. Концентра-
ция органических веществ (водного гумуса) может достигать
50 мг/л и выше. Гуминовые кислоты составляют незначительную
.62
часть водного гумуса, основная часть его представлена фульвокис-
лотами. Цветность воды, обусловленная фульватами железа, может
быть по платипово-кобальтозой шкале 3000е и выше.
Количество органических веществ в воде характеризуется вели-
чиной окисляемостщ т. е. количеством кислорода, расходуемого на
окисление примесей сильными окислителями (КМпО4, ЩСщО;).
В чистых водах окисляемость составляет or 1 до 4 мг СЕ/л. Окисляе-
мость выше 10 мг О2/л может указываеть на загрязнение водоема
сточными водами. Воды северных областей, содержащие большое
количество гумусовых веществ, имеют повышенную окисляемость,
но безопасны в санитарном отношении.
Микроэлементы. Элементы, содержание которых в воде состав-
ляет менее 1 мг/л, относятся к группе микроэлементов. Микроэле-
менты в природных водах могут находиться в виде ионов, молекул,
коллоидных частиц, взвеси, входить в состав минеральных и орга-
нических комплексов. Алекин выделяет следующие группы микро-
элементов: 1) типичные катионы (1л+, Rb+, Cs+, Ва2+, Sr2+ и др.);
2) попы тяжелых металлов (Сп2+, Ag+, Ni24-, Cd2+ и др.); 3) амфо-
терные комплсксообразовагели (Cr, Mo, V и др.); 4) типичные
анионы (I-, F~, В г' и др.); 5) радиоактивные элементы.
Важное гигиеническое значение имеют соединения иода и фтора.
Соединения иода содержатся в поверхностных водах в количестве
нескольких тысячных долей ?vir/ri. Концентрация иода в морских
водах исчисляется сотыми долями мг/л. При недостаточно..! содер-
жании иода в питьевой воде наблюдается нарушение деятельности
щитовидной железы (эндемический зоб). Для профилактики жи-
тели районов с такой водой получают нужное количество кода с
поваренной солью. Такого рода профилактика, проводимая в со-
ветское время в Таджикистане, а позднее в Карпатах, позволила
почти полностью ликвидировать это заболевание. При содержании
иода от 0,0001 до 0,001 мг/л заболевание наблюдается у 15—30 че-
ловек из тысячи, а мри концентрации иода от 0,0014 до 0,01 мг/л —
лишь у одного из тысячи. В воде иод содержится обычно в форме
иодид-иона.
Фтор в природных водах встречается в форме фторид-иона. Его
содержание изменяется от сотых долей до десяти мг/л и более.
Концентрация фтора в природных водах СССР составляет от 0,01
до 12 мг/л. Соединения фтора активно участвуют в процессах ми-
нерализации костной ткани и зубов. При недостаточном содержа-
нии фтора в питьевой воде наблюдается кариес зубов, а при избыт-
ке развивается заболевание зубов — флюороз (пятнистая эмаль).
Воды с повышенным содержанием фтора встречаются значитель-
но реже, чем воды с малыми концентрациями.
Природные воды содержат и ионы других микроэлементов, ко-
торые могут быть естественными компонентами природных вод или
появляться в водоемах в результате практической деятельности
людей.
63.
§ 1б. основные показатели качества воды.
Требования, предъявляемые к воде хозяйственно-бытового
и промышленного водоснабжения
Выбор и оценка качества источника водоснабжения базируются
на результатах изучения его санитарного состояния. При этом
исследуются возможные источники загрязнения водоема, проводит-
ся .химический и бактериологический анализ воды, указываются
мероприятия, направленные на предотвращение загрязнения во-
доема, рассматриваются предполагаемые методы очистки воды и
дается их технико-экономическое обоснование. Объем программы
исследования водоема для определения возможности использова-
ния его в качестве источника водоснабжения определяется
ГОСТ 17.1.3.03—77 «Правила выбора и оценка качества источни-
ков централизованного хозяйственно-бытового водоснабжения».
Анализ воды проводится с соблюдением определенных правил
отбора проб и включает в себя характеристику органолептических,
химических и бактериологических показателей.
При предположении о вероятном присутствии радиоактивных
элементов, токсичных неорганических и органических соединений
проводится дополнительное исследование воды.
Отбор проб проводится для открытых водоемов с поверхности
воды и с той глубины, которая проектируется для водозабора.
Для действующего водозабора пробы воды отбираются после насо-
сов первой ступени. При анализе воды из подземных источников
пробы отбираются из того водоносного горизонта, из которого
будет идти водозабор, или из уже действующих источников, исполь-
зуемых для водоснабжения. Для получения объективной характе-
ристики свойств воды необходимо провести несколько серий ана-
лизов с учетом сезонных колебаний состава воды.
Качество воды обусловливается совокупностью растворенных
в ней минеральных и органических веществ, газов, коллоидов,
взвешенных веществ и наличием микроорганизмов. Требования к
качеству воды для хозяйственно-бытового водоснабжения предпи-
сываются государственными и международными стандартами.
В нашей стране ГОСТ на питьевую воду был впервые введен в
1937 г. Развитие промышленности, совершенствование методов
обработки воды, забота о здоровье советских людей способствова-
ли совершенствованию стандарта, определяющего качество питье-
вой воды. Действовавший с 1954 г. ГОСТ 2874—54 в связи с требо-
ванием времени был пересмотрен и с 1975 г. введен на питьевую
воду более строгий и совершенный ГОСТ 2874—73. Для этого
ГОСТа характерным является комплексный подход к оценке каче-
ства воды. Его созданию предшествовала большая работа химиков,
медиков, микробиологов, которые на основе санитарно-микробио-
логических, санитарно-токсикологических и клинико-статистических
данных установили предельно допустимые концентрации химиче-
ских соединений, уточнили уровень отдельных показателей качест-
ва воды. Использование новых реагентов при обработке воды,
64
появление в воде водоемов техногенных элементов сделало необхо-
димым регламентирование качества питьевой воды по этим пока-
зателям. Усилен и несколько изменен контроль за качеством воды
по бактериологическим показателям. Условия действия ГОСТа
определены в указаниях, которые предшествуют конкретным пока-
зателям качества воды: 1) при любом типе водоисточника, способе
обработки воды и конструктивных особенностях водопроводных
сетей свойства воды должны удовлетворять указанным в стандар-
те нормативам, обеспечивающим безопасность в эпидемиологиче-
ском отношении, безвредность химического состава и благоприят-
ные органолептические свойства воды; 2) вода, подаваемая
потребителям, должна быть защищена от случайного и системати-
ческого загрязнения путем устройства зон санитарной охраны
и герметичности водопроводной сети; 3) качество питьевой во-
ды должно определяться совокупностью ее состава и свойств
в местах поступления в водопроводную сеть и разбора воды из
наружных водоразборов и кранов внутренних водопроводных
сетей.
Показатели качества питьевой воды классифицируются по трем
группам: 1) органолептические; 2) токсичные химические соеди-
нения; 3) бактериологические.
Определение органолептических показателей не ограничивается
только интенсивностью их воздействия на органы чувств, а для
ряда соединений указаны предельно допустимые концентрации
(ПДК) их в воде, превышение которых ухудшает органолептиче-
ские свойства воды.
Оценка качества воды начинается с характеристики физических
показателей (цвет, вкус, запах, температура, прозрачность, содер-
жание взвешенных веществ).
Мутность воды. Грубодиспергированные примеси (частицы диа-
метром более 100 нм) обусловливают мутность воды. Она может
вызываться присутствием таких неорганических частиц, как песок
и глина, и некоторых органических компонентов, например про-
дуктов распада растительных и животных организмов. Мутность
присуща водам поверхностных водоемов с достаточно высокой
скоростью течения. Величина мутности зависит от характера пита-
ния водоема, свойств береговых пород, климатических и погодных
условий. Максимальные значения мутности воды наблюдаются в
предпаводковый период и во время паводка. Мутность воды дости-
гает при этом тысячи мг/л и более. Мутность воды определяют гра-
виметрическим и нефелометрическим методами. Гравиметрический
метод основан на взвешивании осадка, просушенного до постоян-
ной массы вместе с фильтром при 105° С. Осадок получают после
пропускания через фильтр определенного объема воды. Этот метод
точный, но требует много времени для выполнения. В практике
водоочистки наряду с этим используются визуальный и фотоэлек-
троколориметрический (нефелометрический) методы. Визуальный
основан на сравнении мутности исследуемой воды со стандартны-
ми образцами.
3—1244
65
Фотоэлектропефелометрический метод определения мутности
основан на способности взвешенных частиц рассеивать свет. Фик-
сация интенсивности светового потока проводится с помощью фото-
элемента. При определении мутности воды данным методом необ-
ходимо учитывать неоднородность взвешенных частиц в различных
водах и проводить корректирование стандартной шкалы, используя
гравиметрический метол. Для этого данные фотоэлектронефсломст-
рического анализа сравнивают с результатами гравиметрического
анализа.
Воды, содержащие незначительное количество взвешенных час-
тиц, прозрачны. II поэтому качество таких вод характеризуется ве-
личиной обратной мутности, — прозрачностью. Прозрачность воды
выражается высотой столба воды, через который просматривается
«крест» (толщина линий 1 мм), нанесенный черной краской на бе-
лую фарфоровую пластинку (метод определения прозрачности «по
кресту»), или определенный шрифт (определение «по шрифту»).
Вода, идущая для хозяйственно-питьевого водоснабжения, должна
иметь прозрачность «по кресту» нс менее 300 см и «по шрифту» не
менее 30 см.
Цветность воды. Гуминовые и фульвокислоты и их растворимые
соли (гуматы и фульваты железа), водоросли, могут сообщать во-
де окраску. Интенсивность окраски цветных вод характеризуется
цветностью. Цветность воды определяется по платиново-кобальто-
вой шкале и выражается в градусах. Один градус такой шкалы со-
ответствует содержанию в 1 л раствора 2,49 мг хлорплатипата
калия K2[PtCl6] и 2,018 мг хлорида кобальта СоС12-6Н2О. Опреде-
ление цветности производится колориметрическим методом. Со-
гласно ГОСТ 2874—73 цветность воды не должна превышать 20°
по платииово-кобальтовой шкале. В отдельных случаях, по согла-
сованию с органами санитарного надзора, допускается цветность
до 35°. Иногда цветность воды определяют по дихромат-кобальто-
вой шкале, приготовляемой смешением раствора, содержащего в
1 л воды 0,0875 г К2С1ДО7 и 2 г CoSO4-7H2O, 1 мл H2SO4 (пл.
1,84), и раствора, содержащего 1 мл H2SO4 (пл. 1,84) в 1 л
воды.
Запах и вкус воды. Запах и вкус природных вод обусловлены
растворенными солями, газами, органическими соединениями, об-
разующимися в процессе жизнедеятельности водных организмов.
В соответствии с происхождением запахов их делят на естествен-
ные и искусственные. Естественные запахи (рыбный, гнилостный,
болотный, плесневый и др.) возникают в результате жизнедеятель-
ности водных организмов, а также при разложении органических
веществ. Искусственные запахи (фенольный, хлорфенольный и др.)
появляются при загрязнении источников сточными водами. Соглас-
но ГОСТ 2874—73 определение запаха проводится при температу-
ре воды 20° С и при подогреве до 60° С. При этом дается качествен-
ная и количественная характеристика. Определение запаха и вкуса
производится органолептически; у воды отмечают горький, сладкий,
кислый или соленый вкус. Вес остальные вкусовые ощущения опрс-
66
деляются как привкусы. Количественно запах и вкус воды оцени-
ваются по пятибалльной шкале запахов и вкусов:
Балл 0 12 3 4 5
Интенсив- никакого очень слабый заметный отчетли- очень
ность за- слабый вый силь-
паха ный
Интенсивность запаха воды должна быть не более 2 баллов при
60° С. Вкус воды тоже нс должен быть более 2 баллов (при 20° С).
Температура воды. Температура воды зависит от местоположе-
ния источника и подвержена значительным колебаниям. Большин-
ство источников водоснабжения имеет определенный диапазон ко-
лебания температуры, обусловленный климатическими условиями.
Наиболее благоприятная температура питьевой воды 7—12° С.
Химические компоненты, вляющие на органолептические свойст-
ва воды. Химические соединения, содержащиеся в питьевой воде,
при достижении пороговой концентрации могут сильно изменять
органолептические свойства воды и ухудшать ее качество. Поэтому
ГОСТ 2874—73 предусматриваются предельно допустимые концен-
трации (ПДК) таких компонентов или регламентируется опреде-
ленный показатель качества воды. Прежде всего — общее солесо-
держание или минерализация воды. Минерализованные воды, со-
держащие более 1 г растворенных солей, имеют солоноватый или
вяжущий привкус, хуже утоляют жажду, ограниченно пригодны для
хозяйственно-бытовых нужд.
Общее содержание нелетучих минеральных и частично органиче-
ских соединений в питьевой воде характеризуется величиной сухо-
го остатка. Она определяется массой осадка, образующегося после
выпаривания и высушивания до постоянной массы (105° С) про-
фильтрованной пробы воды. При использовании этой методики
определения в полученный результат включаются и коллоидные
примеси воды, так как они не удаляются при фильтровании. Масса
сухого остатка согласно ГОСТ 2874—73 не должна превышать
1000 мг/л. Кроме этого показателя используются также и другие:
«общее содержание примесей», «растворенные вещества». Опреде-
ление общего содержания примесей отличается от определения
сухого остатка тем, что оно проводится с нефильтрованной водой,
т. е. в этот показатель включаются и взвешенные вещества. Пока-
затель «растворенные вещества» идентичен сухому остатку. Про-
каливанием высушенных осадков при 600° С определяются пока-
затели: «остаток после прокаливания» и «потери при прокалива-
нии».
Водородный показатель pH. Важным показателем чистоты и
свойств состава воды является pH. Согласно ГОСТ 2874—73 до-
пускаются колебания pH в пределах 6,5н-8,5. Отклонение pH от
этих значений указывает на нарушение стабильности воды и воз-
можное ее загрязнение.
Жесткость воды. Ионы Са2+ и Mg2+ характеризуют общую
жесткость. Умеренно жесткая вода не опасна в гигиеническом ас-
3* 67
пекте, так как с водой в организм поступает 20—30% кальция,
необходимого для поддержания в норме обмена веществ в орга-
низме. В этом отношении мягкие маломинерализовапиыс воды яв-
ляются менее желательными. Гидрокарбонатпокальциевые воды
средней жесткости наиболее приятны на вкус. Согласно ГОСТ
2874—73 общая жесткость питьевой воды должна быть не более
7 мг-экв/л.
В некоторых природных водах отмечается повышенное содер-
жание соединений железа и марганца (II). Они, нс обладая выра-
женным токсичным действием, ухудшают качество воды, придавая
ей при концентрации более 0,1—0,3 мг/л железистый привкус.
Поэтому допустимая концентрация железа в питьевой воде со-
ставляет 0,3 мг/л, а марганца-—0,1 мг/л. Постоянные компоненты
природных вод — сульфаты и хлориды при высоком содержании
также отрицательно влияют па качество воды. Так, при концентра-
ции хлоридов более 500 мг/л у воды появляется солоноватый при-
вкус. Сульфаты, если содержание их в воде превышает 500 мг/л,
угнетают некоторые функции организма и обладают слабительным
действием. В питьевой воде должно быть не более 350 мг/л хло-
ридов и 500 мг/л сульфатов.
Для обработки воды на водопроводных станциях применяются
реагенты, которые при концентрации, выше допустимой, могут от-
рицательно сказаться на качестве воды. Поэтому ГОСТ 2874—73
нормирует остаточные концентрации этих веществ в питьевой во-
де. При использовании алюминиевого коагулянта ПДК остаточно-
го алюминия составляет 0,5 мг/л. Предельно допустимая концен-
трация флокулянта-—полиакриламида 2 мг/л. При обеззаражива-
нии воды хлором концентрация свободного остаточного хлора
должна быть не менее 0,3 и не более 0,5 мг/л после 30-минутного
контакта или содержание связанного остаточного хлора должно
быть не менее 0,8 и не более 1,2 мг/л после 60-минутного контакта
хлора с водой.
Лимитируется также и содержание других обеззараживающих
реагентов. Содержание остаточного озона в воде установлено 0,1 —
0,3 мг/л, концентрация остаточного серебра — до 0,05 мг/л. Более
высокая концентрация ионов Ag+ недопустима ввиду токсичного
действия. Содержание гексаметафосфата или триполифосфата нат-
рия в воде должно быть не более 3,5 мг/л (в расчете'иа РОД ).
В природных водах, используемых для водоснабжения, иногда воз-
можно присутствие веществ, непосредственно оказывающих вред-
ное воздействие на организм. К числу таких относятся общеядови-
тыс вещества, соли тяжелых металлов, некоторые органически'?
вещества. В воде, предназначаемой для хозяйственно-бытового во-
доснабжения, установлены предельно допустимые концентрации
таких соединений.
В «Правилах по охране поверхностных- водоемов от загрязне-
ния сточными водами» приведены предельно допустимые концентра-
ции (ПДК) более 400 химических соединений. ПДК некоторых
токсических веществ приведены и в ГОСТ 2874—73 (мг/л): Ве2+
68
0,0002; Sec’+ 0,001; As3+;5+ 0,05; Pb2+ 0,1; Sr2^ 2; Mo2+ 0,5. Для ра-
диоактивных элементов радия 226Ra и стронция 90Sr установлены
соответственно допустимые уровни активности: 4,44-103 и 1,48Х
X Ю2 с-’-м ’1. Ограничивается также содержание нитратов в питье-
вой воде (в пересчете на азот) до 10 мг/л.
Предельно допустимые концентрации фтора в питьевой воде
для 1 и II климатического района 1,5 мг/л, для III-— 1,2; для IV —
0,7 мг/л. Если проводится фторирование воды, то доза фтора со-
ставляет 70—80% от ПДК для данного района.
Токсичным действием обладает и полиакриламид при содержа-
нии выше 20 мг/л. К сильнодействующим токсичным соединениям
относятся одно- и многоатомные фенолы. Наиболее часто встреча-
ется загрязнение водоема фенолом СбНзОН. При содержании фе-
нола более 0,001 мг/л начинает проявляться его токсичное дейст-
вие, обнаруживается характерный запах. Фенолы легко взаимо-
действуют1 с хлором, образуя хлорфенолы, имеющие крайне
неприятный запах. Присутствие хлорфенолов в воде даже при вы-
соком качестве воды по всем остальным показателям делает ее
непригодной для питья. Кипячение воды только усиливает хлор-
фенольный запах.
Действие некоторых токсичных соединений, одновременно при-
сутствующих в воде, может суммироваться или даже усиливаться,
несмотря на то, что содержание каждого из них, взятого в отдель-
ности, может не превышать ПДК- Возможность вредного воздейст-
вия комплекса токсичных веществ на организм учтена ГОСТ
2874—73. Если в воде содержится несколько токсичных веществ,
то сумма, выраженная в долях ПДК каждого из них, не должна
превышать единицы:
C'r ' C' ^C'n " ’
Ci, C2, ..., Cn — концентрации токсичных веществ в воде, мг/л; С/,
С2', ..., Сп —ПДК токсичных веществ в воде, мг/л.
Бактериологические показатели качества питьевой воды. Дан-
ные показатели характеризуют безвредность воды относительно
присутствия болезнетворных микроорганизмов. Общее количество
микроорганизмов — минерализаторов органических веществ, не
опасных для здоровья людей, определяется количеством колоний,
вырастающих при посеве на питательной среде 1 мл питьевой во-
ды. Общее количество таких колоний должно быть не более 100
для 1 мл воды.
Важным бактериологическим показателем является содержание
в воде бактерий группы кишечной палочки (БГКП). Количество
бактерий группы кишечной палочки (БГКП) в 1 л воды определяет
величину коли-индекса, а наименьший объем, в котором содержит-
ся одна клетка БГКП, соответствует коли-титру. Согласно ГОСТ
2874—73 коли-титр должен быть ^300, а коли-индекс ^3. Тре-
бования, предъявляемые к качеству питьевой воды в нашей стране,
превышают требования стандартов других стран.
69
Требования к воде промышленного водоснабжения. Качество
воды, используемой для промышленного водоснабжения, определя-
ется видом производства, ролью воды в технологическом процес-
се. Она может входить в состав получаемого продукта, быть тех-
нологическим сырьем, растворителем, теплоносителем и т. д.
ГОСТ 2874—73 определяет требования, которые предъявляются к
воде, используемой многими отраслями пищевой промышленности.
Рассмотрим требования, предъявляемые к качеству воды, ис-
пользуемой в промышленности. Прежде всего вода применяется
для охлаждения мартеновских и доменных печей, атомных реакто-
ров, двигателей внутреннего сгорания, конденсаторов на электро-
станциях, теплообменников на химических предприятиях и других
объектов. Основные требования к охлаждающей воде: достаточно
низкая температура, малая карбонатная жесткость, предельно ма-
лые концентрации ионов железа и сероводорода. Ограничения,
связанные с устранимой жесткостью, вызваны возможным перехо-
дом гидрокарбонатов кальция и магния при нагревании в карбона-
ты, отлагающиеся на стенках теплообменной аппаратуры.
Соединения железа, сероводород усиливают коррозию железа
в воде и вызывают образование обрастаний на внутренних поверх-
ностях труб. Поэтому содержание железа в охлаждающей воде не
должно превышать 0,1 мг/л, а сероводорода — 0,5 мг/л. Карбонат-
ная жесткость допускается до 2,8 мг-экв/л. Регламентируется
присутствие микроорганизмов, водорослей и грубодиспергирован-
ных веществ, а также веществ, вызывающих отложения в трубах.
Более строгие требования предъявляются к воде, идущей для
питания паровых котлов. Природная вода для этой цели, как пра-
вило, непригодна, и поэтому се обязательно подвергают специаль-
ной обработке. Основные требования к воде для паровых котлов:
предельно малые значения общей жесткости, растворенного кисло-
рода, минимальное солесодержанис, отсутствие взвешенных
веществ и соединений кремния. Отрицательное воздействие воды
на стенки котла связано с образованием накипи, основными компо-
нентами которой являются карбонаты и силикаты кальция и маг-
ния. Допустимая общая жесткость воды для котлов высокого дав-
ления составляет 0,017 мг-экв/л; концентрации (мкг*/л): кремние-
вой кислоты до 30 мг/л; железа до 100; кислорода до 30;
содержание масел до 0,5 мг/л. Ограничение содержания масел
связано с возможностью вспенивания воды, приводящего к пере-
носу воды с паром. Необходимость удаления кислорода обусловле-
на сильным коррозионным действием его на стенки котла.
Особенно строгие требования предъявляются к воде в некото-
рых отраслях химической и радиоэлектронной промышленности,
когда вода входит в контакт непосредственно с продуктом, на
качество которого могут вредно влиять даже небольшое содержа-
ние примесей. Вода, используемая для приготовления бетонной
смеси и строительных цементных растворов, должна иметь сухой
1 мкг (микрограмм) = 1 • 10-3 мг.
70
остаток до 5000 мг/л, рН^4. Содержание сульфатов, во избежание
сульфатной коррозии бетона, не должно превышать 2,7 г/л.
Природные и сточные воды — сложные многокомпонентные си-
стемы, содержащие примеси различного фазово-дисперсного соста-
ва. Растворимые неорганические и органические соединения обра-
зуют однофазные растворы с размерами частиц от 10 ° до Ю-'м.
Высокомолекулярные органические соединения при растворении
в воде могут образовать коллоидные растворы. В коллоидном со-
стоянии могут находиться и некоторые такие труднорастворимые
неорганические соединения, как алюмосиликаты, кремниевая кис-
лота, гидроксиды тяжелых металлов и др. Размер частиц в колло-
идных системах составляет 10~9--ИР7 м. Основная часть трудно-
растворимых неорганических и органических соединений находит-
ся в воде в форме грубодисперсных примесей, образуя суспензии
или эмульсии с размером частиц более 10~7 м.
Многообразие примесей и их различный фазово-дисперсный со-
став значительно усложняют процесс обработки воды. Л. А. Куль-
ским предложена классификация примесей воды, подлежащей
очистке, с учетом фазово-дисперсной характеристики загрязнений и
их химического состава. Примеси воды, имеющие значение для
выбора способа ее очистки, сгруппированы по признаку физико-хи-
мического состояния, которое в определенной степени зависит от
размера частиц вещества, ого фазового состава.
Все примеси воды по их отношению к дисперсионной среде де-
лятся на четыре группы (две из них образуют гетерогенные системы,
а остальные — гомогенные). К первой группе относятся не раство-
римые в воде примеси (взвеси, эмульсии, зоо- и фитопланктон) с
диаметром частиц более 10~6 м. Эти примеси, за исключением мик-
роорганизмов, образуют в воде кинетически неустойчивые системы,
т. е. они подвержены седиментации (осаждению под действием
силы тяжести). Поэтому технологические процессы, при которых
они выделяются из воды, основаны на явлении седиментации (от-
стаивание, агрегирование частиц при введении химических реаген-
тов с последующим отстаиванием); адгезии (прилипание) частиц
к поверхности твердых сорбентов, газов (флотация). Микроорга-
низмы планктона могут выделяться из воды после обработки ее
химическими реагентами и электрическим током (электрофорез).
Коллоидные примеси, вирусы, мелкие бактерии и высокомоле-
кулярные соединения входят во вторую группу. Общим свойством
для них является диаметр частиц. Так как диаметр этих частиц
очень мал, то для выделения их из воды необходимо введение хи-
мических реагентов, вызывающих агрегирование частиц (коагуля-
ция) или воздействие электрическим током (электрофорез). Важ-
ная роль при удалении из воды вирусов и мелких бактерий отводит-
ся бактерицидным реагентам (хлору, озону, перманганату калия
И др.).
Третья группа примесей воды представлена соединениями, обра-
зующими в воде молекулярные растворы (газы, органические ве-
щества, продукты жизнедеятельности водных организмов, некото-
71
рые вещества, содержащиеся в производственных сточных водах,
и др.). Эти примеси образуют гомогенные системы. Некоторые из
этих примесей могут выделяться при воздействии химических реа-
гентов (чаще всего окислителей), удаляться в процессе сорбции,
например активированным углем, физическими методами (аэриро-
ванием) .
Четвертая группа примесей представлена веществами, диссо-
циирующими в водных растворах на ионы. Удаление ионов из воды
может быть осуществлено связыванием их в труднорастворимые
(осаждение), газообразные или малодиссоциированныс соединения.
Эффективными методами для удаления таких примесей служат
газгидратный, ионообменные методы, электродиализ и др.
Таким образом, пользуясь этой классификацией, можно опре-
делить более рациональную компоновку очистных сооружений и
выбрать наиболее эффективную технологическую схему очистки
воды.
РАЗДЕЛ ВТОРОЙ
физико-химические основы очистки
ПРИРОДНЫХ и сточных вод
Глава III. УМЯГЧЕНИЕ И ОБЕССОЛИВАНИЕ ВОДЫ
§ 19. Жесткость воды
Катионы Са2+, Mg2'1' и тяжелых металлов обусловливают жест-
кость воды. Но так как в природных водах преобладают ионы Са2+
и Mg’2r, а остальные присутствуют не всегда и в незначительных
количествах, то общая жесткость природных вод может быть оха-
рактеризована суммой концентраций ионов кальция и магния, вы-
раженной в миллиграмм-эквивалентах в 1 л (мг-экв/л) или в мик-
рограмм-эквивалентах в 1 л (мкг-экв/л). Один миллиграмм-экви-
валент соответствует содержанию в воде 20,04 мг/л ионов Са2+ или
12,156 мг/л ионов Mg’24'. Такой способ выражения жесткости соот-
ветствует истинному состоянию соединений в растворе и согласу-
ется с ионной формой записи результатов анализа воды.
По величине общей жесткости природные воды делятся на сле-
дующие группы: вода очень мягкая (< 1,5 мг-экв/л), вода мягкая
(1,5—3,0 мг-экв/л), вода средней жесткости (3,0—5,4 мг-экв/л),
вода жесткая (5,4—10,7 мг-экв/л), вода очень жесткая
(>10,7 мг-экв/л).
Наиболее мягкими водами являются воды атмосферных осад-
ков, имеющие жесткость 70—100 мкг-экв/л. Жесткость подземных
вод определяется составом контактирующих с ними пород. Так,
грунтовые воды Карелии имеют очень малую жесткость (пример-
но 700 мкг-экв/л), так как они омывают труднорастворимые гра-
нитные породы, а грунтовые воды Донбасса, которые формируют-
ся под воздействием меловых и доломитовых пород, относятся к
очень жестким (около 18—20 мг-экв/л). Жесткость речных вод
зависит от климатических условий и характера питания рек.
При подземном питании жесткость воды будет больше. Наимень-
шая жесткость речной воды наблюдается в паводковый период.
В летний период жесткости речных вод зависит от количества вы-
падающих осадков. В табл. 8 приводится жесткость воды некото-
рых рек СССР.
Жесткость воды, обусловленная наличием ионов Са?+ и Mg‘2+,
принимается за общую. В воде эти ионы могут быть связаны с
различными анионами (НСОГ, SO4~ , С1~, СОз~ и др.). Соответ-
ственно этому жесткость воды, обусловленную наличием гидрокар-
бонатов и карбонатов кальция и магния, называют карбонатной
жесткостью. Сульфаты, хлориды, а также кальциевые и магниевые
соли других кислот, гидроксиды кальция и магния характеризуют
73
Таблица 8. Жесткость воды рек СССР некоторых некарбонатную жесткость. Заметно растворимый в воде гидрокарбонат каль- ция при нагревании пере- ходит в карбонат вслед- ствие нарушения углекпе-
Жесткость, мг-экв/л
Река общая карбо- натная нокар- бонат- ная каль- циевая магни- евая
Амур Волга (ниж- нее тече- ние) Днепр Дон Москва Печора Урал 1,448 6,490 5,135 5,777 4,493 3,388 8,202 1,017 3,316 4,214 4,208 2,244 2,445 4,992 0,431 3,174 0,891 1,569 0,249 0,963 3,210 1,417 4,950 3,708 4,078 3,243 2,480 6,038 0,031 1 ,540 1,427 1,690 1,250 0,908 2,164 лотиого равновесия: Са(НСОз)2^СаСО3 + + СО2 + Н2О. Карбонат кальция имеет чрезвычайно малую растворимость, равную 14,45 мг/л в дистиллиро- ванной воде при 25° С. Гидрокарбонат магния
при кипячении тоже переходит в труднорастворимые карбонат или
основной карбонат магния (MgOH)2CO3. Таким образом, при кипя-
чении жесткость воды, вызванная присутствием гидрокарбонатов
кальция и магния, как бы устраняется. Поэтому такая жесткость
называется устранимой или временной.
Следует различать понятия карбонатная и устранимая жест-
кость. При переходе НСОГ в СОз~ и при выпадении карбонатов
кальция и магния в воде остается некоторое количество ионов
Са2+, Mg2+, СОз~ , соответствующее произведению растворимости
карбоната кальция и основного карбоната магния. В присутствии
посторонних ионов растворимость этих соединений повышается.
Разность между карбонатной и устранимой жесткостью, обуслов-
ленной карбонатами кальция и магния, характеризует величину
остаточной жесткости. В некоторых природных водах наблюдается
соотношение НСОГ >Ca2+-r-M.g2+, т. е. общая щелочность превы-
шает сумму концентраций ионов Са2+ и Mg2+. Для таких вод общая
жесткость условно принимается за карбонатную, а значение некар-
бонатной не вычисляется.
§ 20. Реагентные методы умягчения воды
Снижение концентрации ионов Са2+ и Mg2+ в воде (умягчение)
может достигаться применением ряда методов (реагентных, ион-
ного обмена и термических). Выбор метода и степень умягчения
воды определяются требованиями, предъявляемыми к воде в дан-
ном технологическом процессе. Реагентные методы умягчения воды
основаны на переводе ионов Са2+ и Mg2+ в труднорастворимые
соединения (карбонаты, фосфаты и др.). Полного извлечения ионов
Са2+ и Mg2+ при использовании этих методов не происходит, так
как в растворе остается некоторое количество ионов. Концентрация
их в воде будет зависеть от величины произведения растворимости
образующихся труднорастворимых соединений.
74
Рассмотрим основные физико-химические процессы, происходя-
щие при умягчении воды методами осаждения.
Известкование воды. Сущность данного метода заключается в
переводе заметно растворимых гидрокарбонатов кальция и магния
в труднорастворимые соединения в результате взаимодействия с
известью. Гидроксид-ионы, поступающие в раствор из извести, по-
вышают pH, что вызывает' нарушение равновесия между различны-
ми формами угольной кислоты. Устойчивой формой угольной кис-
лоты в щелочной среде являются карбонат-ионы, образующие с
ионами Са2+ труднорастворимые соединения. Ионы Mg2+ в зави-
симости от pH могут выделяться из воды в форме основного кар-
боната или гидроксида. Свободная угольная кислота при взаимо-
действии с гидроксидом кальция переходит в связанную (карбонат
кальция). Повышение дозы извести (при рН>10,3) приводит к
уменьшению магниевой жесткости и замене пскарбонатной маг-
ниевой жесткости на кальциевую. Химические реакции, происходя-
щие при известковании воды, содержащей соли жесткости и сво-
бодную угольную кислоту, можно описать следующими уравне-
ниями:
Са (НСО3)2 + Са (ОН)2 = 2СаСО3| 4- 2Н2О
2Mg (НСО3)2 4-ЗСа (ОН)2 = (MgOH)2 СО3; +ЗСаСО3; 4-4Н2О(рН 9,5)
Mg(HCO3)2 +2Са(ОН).2 = Mg(OH)2| +2СаСО3| + 2Н2О(рН 10,3)
MgSO4 + Са (ОН)2 = CaSO4 + Mg (ОН)2;
СО2 4- Са(ОН)2 — СаСО3| 4- Н2О
Из приведенных выше уравнений следует, что при известкова-
нии воды снижаются карбонатная жесткость (щелочность), кон-
центрация свободной угольной кислоты и общее солесодержание.
Известкование применимо для вод с большой карбонатной жест-
костью (Жкарб>1,5 мг-экв/л), если общая щелочность меньше об-
щей жесткости воды (ЩОбщ<Жобщ). Обратное соотношение
(Щобщ>Жобщ) способствует переходу избытка гидрокарбоната
натрия в щелочной среде в карбонат. Щелочность воды нс только
не снижается, а даже несколько возрастает за счет реакции
2NaHCO3 4- Са (ОН)2 = ;СаСО3 4- Na2CO3 4- 2Н2О
Известкование воды проводится при двух режимах: если нс
предусматривается снижение магниевой жесткости, то обработка
воды ведется при pH 9,5. Для выделения ионов Mg2r и соединений
кремниевой кислоты известкование ведут при повышенной дозе
извести (pH 10,9). Первый режим известкования чаше использу-
ется при обработке природных вод, так как содержание магния в
пресных водах всегда меньше, чем кальция. При соотношении ио-
нов в воде Са2+>НСОГ практически выделяются из воды только
ионы Са2+ в форме карбоната. Если же Са2+<НСОГ , то из воды
будет удаляться и магний в форме основного карбоната.
75
Известкование воды по второму режиму позволяет связать ио-
ны Mg2+ гидроксид-ионами в труднорастворимый гидроксид маг-
ния, образующий аморфные, сильно гидратированные хлопья. При
низкой температуре обработки воды мелкие хлопья гидроксида
магния проходят через фильтр, ухулшая качество умягченной во-
ды. Кроме того, они затрудняют рост кристаллов карбоната каль-
ция на зернах контактной среды, забивают поры фильтра, снижая
тем самым эффективность работы установки. Поэтому в практике
водоподготовки известкование воды предпочтительно проводить
дозой извести, оптимальной для работы в первом режиме. Для
карбоната кальция и гидроксида магния характерно образование
пересыщенных растворов, поэтому состояние равновесия приведен-
ных химических процессов (см. с. 75) наступает не сразу, а через
длительное время.
Ниже приведены данные о растворимости гидроксида магния в
равновесных условиях и свежеполученного осадка при различной
температуре.
Температура, °C 10 20 30 40 50 60 70 80 100
Растворимость
Mg(OH)2,
мг/л 16,3 14,5 13,6 10,8 9,2 8,2 7,3 6,6 5,4
Растворимость
свежеполу-
ченного осад-
ка, мг/л 25 — 24 — 22 — 20 — 8
Из приведенных данных видно, что растворимость гидроксида
магния с повышением температуры снижается. Аналогичная зави-
симость растворимости от температуры проявляется у карбоната
и гидроксида кальция. Это позволяет добиться большей степени
умягчения воды известкованием при повышении температуры. По-
ложительное действие повышенной температуры заключается в
увеличении скорости химических реакций и ускорении процесса
кристаллизации. В практике водоподготовки процесс умягчения
идет в неравновесных условиях, поэтому степень умягчения воды
ниже, чем определенная расчетом. Так как скорость химической
реакции значительно выше скорости кристаллизации образующих-
ся соединений, то именно второй процесс и определяет время, не-
обходимое для достижения состояния равновесия. Поэтому для ин-
тенсификации обработки следует создавать благоприятные условия
для ускорения процесса образования осадка.
Для более полного умягчения воды используют следующие прие-
мы: а) повышение температуры: б) создание небольшого избытка
реагента (извести); в) перемешивание; г) введение в обрабаты-
ваемую воду веществ, способных увеличивать количество центров
кристаллизации (свежего осадка карбоната кальция и др.). На
формирование осадка отрицательно влияют органические вещест-
ва, особенно ПАВ, так как они препятствуют росту кристаллов.
При окисляемости исходной воды 75—100 мг Оз/л кристаллизация
почти отсутствует.
76
Эффективность известкования воды определяется выбором оп-
тимальной дозы извести. На основании уравнений реакций, проте-
кающих при известковании, можно ориентировочно рассчитать не-
обходимую для умягчения дозу извести (товарного продукта):
где Д1Г—доза извести (товарного продукта) в расчете на СаО,
мг/л; 28 — эквивалент СаО; Жк— карбонатная жесткость воды,
мг-экв/л; [СО2] — содержание свободной угольной кислоты, мг/л;
22 — эквивалент угольной кислоты, 0,5 — избыток извести,
мг-экв/л; — содержание оксида кальция в товарном продукте, %.
По формуле (III.1) рассчитывают дозу извести для умягчения
воды, если не требуется снижения магниевой жесткости. Для уста-
новления точной дозы извести проводится пробное умягчение воды.
Затем в этой умягченной воде определяется остаточная щелочность
титрованием пробы воды 0,1 или 0,01 н. соляной кислотой в при-
сутствии индикаторов фенолфталеина и метилового оранжевого.
Эффективным методом контроля известкования воды является
измерение pH. Наиболее полно осаждение карбоната кальция про-
ходит при величине pH, соответствующей состоянию насыщения
воды карбонатом кальция (pHs). Величину pHs рассчитывают по
формуле
РН5 = рК2 - РПРСаСОз - 1g [Са2+] - 1g Щ + 2,5 /7 + 7,6, (III.2)
где рКг, рПРСаСОз—отрицательные логарифмы константы дис-
социации угольной кислоты по 2-й ступени и прсасо3; [Са2+] —
содержание кальция, мг/л; Щ — щелочность воды, мг-экв/л; I —
ионная сила раствора.
В зависимости от условий обработки и температуры воды умяг-
чение ведут при pH на 0,3—0,5 больше, чем pHs.
Умягченная вода должна быть стабильной. Разность (Щ) меж-
ду щелочностью воды, поступающей после осветлителей на фильт-
ры (Щ]), и щелочностью профильтрованной воды (Щ2): Щ =
= Щг-Щ2 не должна превышать 0,15 мг-экв/л при температуре
воды 10—20° С и 0,1 мг-экв/л при 30—40° С.
Известково-содовый метод умягчения воды. Сущность метода
заключается в обработке воды двумя реагентами: известью и кар-
бонатом натрия (содой), вследствие чего снижается как карбонат-
ная, так и некарбонатная жесткость. Этот метод может быть исполь-
зован для вод с различным химическим составом. Уравнения хи-
мических реакций, вызывающих снижение карбонатной жесткости,
приведены при описании известкования воды. Наряду с ними про-
текают реакции по уравнениям:
CaSO4 -р Na2CO3 = СаСО3 -Р Na2SO4; СаС12 + Na2CO3 = СаСО3 -р 2NaCl
MgCl2 + Na2CO3 = MgCO3 + 2NaCl; MgCO3 -p Ca (OH)2 = Mg (OH)2 + CaCO3
MgSO4 -p Ca (OH 2 = Mg (OH)2 -4- CaSO4j MgCl2 -p Ca (CH)2 — Mg (OH)2 -p CaCl2
77
Образование осадка также сопровождается возникновением пе-
ресыщенных растворов карбоната кальция и гидроксида магния.
Поэтому приемы интенсификации процесса осаждения используют-
ся и в известково-содовом методе умягчения воды. Свободная ще-
лочность в процессе умягчения воды этим методом должна
составлять 0,3—0,5 мг-экв/л. Дозы извести рассчитывают по фор-
муле
где Ди— доза извести (товарного продукта, в расчете на СаО),
мг/л; Жв — карбонатная жесткость воды, мг-экв/л; [СО2] — содер-
жание свободной угольной кислоты, мг/л; С„ — содержание СаО
в товарной извести, %; [Mg2+] — концентрация Mg2+, мг/л; 12,16 —
миллиграмм-эквивалент магния.
Дозу СОДЫ Де (мг/л) вычисляют по формуле
Дс = 53 [Жнк Д-1 ] —,
где Жпь- — некарбонатная жесткость воды, мг-экв/л; 1 — избыток
соды, мг-экв/л; Сс — содержание безводного карбоната натрия в
товарном продукте, %; 53 — миллиграмм-эквивалент безводного
карбоната натрия. При расчете дозы соды исходя из состава кри-
сталлогидрата Na2CO3-10H2O миллиграмм-эквивалент его равен
143. Максимальная степень умягчения известково-содовым мето-
дом составляет 0,3—0,5 мг-экв/л.
Для повышения эффективности умягчения обработка воды реа-
гентами сочетается с подогревом ее до 95—98° С (термохимиче-
ский метод). Нагревание вызывает термический распад гидрокар-
бонатов:
Са (НСО3)2 = СаСО3 + СО2 + Н2О
Mg (НСО3)2 = MgCO3 + со2 + Н2О
Образующийся карбонат магния известью переводится в труд-
норастворимый гидроксид магния:
MgCO3 + Ua (ОН)2 = СаСО3 + Mg (ОН)2
Соли кальция, обусловливающие некарбонатную жесткость, при
взаимодействии с содой образуют труднорастворимый карбонат
кальция. Соли магния связываются в гидроксид магния известью
(см. с. 77). Преимущество этого метода заключается в образова-
нии крупных быстро осаждающихся хлопьев. Кроме того, доза из-
вести может быть несколько уменьшена, так как при нагревании из
воды удаляются растворенные, газы, в том числе и свободная уголь-
ная кислота. Степень умягчения воды термохимическим методом
составляет 0,2—0,3 мг-экв/л.
Иногда вместо извести для умягчения воды применяется гидро-
ксид натрия. При его использовании происходит связывание сво-
бодной угольной кислоты, снижение карбонатной жесткости и
78
уменьшение некарбонатной жесткости, обусловленной солями маг-
ния. Основные реакции, протекающие по уравнениям:
СО2 4- 2NaOH = Na2CO3 4- Н2О; Са (НСО3)2 + 2NaOH = СаСО3 4- Na2CO34- 2Н2О
Mg (НСО3)2 щ 4NaOH = Mg (ОН)2 + 2Na2CO3 + 2Н2О
MgCl2 + 2NaOH = Mg (OH)2 + 2NaCl; MgSO4 + 2NaOH --- Mg (OH), 4- Na2SO4
При взаимодействии гидроксида натрия с солями карбонатной
жесткости и свободной угольной кислотой образуется карбонат
натрия. Если в воде существует соотношение 2Жк-г[СО2]^[Са2+]4-
-4 0,5, где Жк—карбонатная жесткость воды, мг-экв/л; [СО2] — со-
держание свободной угольной кислоты, мг-экв/л; [Са2+] — содер-
жание кальция, мг-экв/л; 0,5 — избыток гидроксида натрия,
мг-экв/л; то умягчение воды достигается только введением гидро-
ксида натрия. Необходимое количество карбоната натрия образу-
ется в процессе взаимодействия гидроксида натрия с солями
карбонатной жесткости и свободной угольной кислотой. Если не-
карбонатная жесткость превышает величину карбонатной жестко-
сти, то в воду дополнительно вводят карбонат натрия. При значи-
тельной карбонатной и малой некарбонатной жесткости в воде
после умягчения образуется избыток карбоната натрия, что явля-
ется нежелательным. При повышении температуры степень гидро-
лиза карбоната натрия возрастает, что приводит к появлению в
воде избытка гидроксида натрия и агрессивной угольной кислоты:
Na2CO3 4- H2O^2NaOH 4- СО2.
Фосфатный метод умягчения воды. В этом методе в качестве
реагентов используются обычно тринатрийфосфат Na3PO4-12H2O
и динатрийфосфат Na2HPO4. Происходящие при фосфатном умяг-
чении химические реакции описываются следующими уравнениями:
ЗСаС12 4- 2Na3PO4 = Са3 ФО4)2 4- 6NaCl
3MgSO4 4- 2Na3PO4 = Mg3 (PO4)2 + 3Na2SO4
При pH обрабатываемой воды >10,5 карбонат кальция перехо-
дит в основной фосфат [Са5(ОН)4(РО4)2]. Растворимость его чрез-
вычайно мала, что позволяет снизить жесткость воды до 20 мг-экв/л.
Обычно степень фосфатного умягчения воды составляет 20—
40 мкг-экв/л. Фосфатное умягчение дает хорошие результаты при
высокой температуре воды и применяется при подготовке воды
для питания паровых котлов. Для умягчения воды при более низ-
кой температуре используется гексаметафосфат натрия (NaPO3)3
или Na2[Na4(PO3)6]. Уравнения реакций умягчения при использо-
вании этого реагента можно записать следующим образом:
2CaSO4 + Na2 [Na4 (РО3)6] = Na2 [Са2 (РО3)3] + 2Na2SO4
2MgCl2 4- Na2 [Na4 (PO3)6] = Na2 [Mg2 (PO3)3] 4- 4NaCl
Фосфатные реагенты составляют основу антинакипинов, исполь-
зующихся для обработки воды паровых котлов. В последние годы
умягчение воды методами осаждения вытесняется или дополняет-
ся более эффективным методом ионного обмена.
79
§ 21. Метод ионного обмена и его применение
при обработке воды
Реагентные методы умягчения воды имеют ряд существенных
недостатков. Они не дают достаточной степени умягчения, требуют
значительного расхода реактивов и сложны по технологическому
осуществлению. В настоящее время в практике водоподготовки стал
широко применяться ионообменный метод умягчения воды. При
этом обработка воды нс сопровождается образованием осадка, от-
падает необходимость непрерывного дозирования реагентов, дости-
гается значительная степень умягчения, уменьшается солесодсржа-
пис обработанной воды.
Сущность процесса ионного обмена. В середине XIX в. было от-
крыто свойство почв обменивать в эквивалентных количествах вхо-
дящие в их состав ионы на другие ионы, содержащиеся в почвенном
растворе. Способность к ионному обмену была позднее открыта и у
некоторых природных алюмосиликатов (глауконитов, бентонитов).
Первый искусственный минеральный ионообменный материал был
получен в начале XX в., но из-за малой механической и химической
стойкости и недостаточно высокой способности к ионному обмену он
не нашел широкого применения в практике. Несколько позднее
обработкой бурых углей серной кислотой был получен сульфоуголь,
обладающий способностью к обмену катионов. Первый полимерный
ионообменник, синтезированный Адамсом и Холмсом в 1935 г.,
положил начало большому количеству работ по синтезу новых ио-
нообменных материалов, по изучению их свойств и применению в
различных отраслях хозяйства. Наиболее широко используются
ионообменные материалы в практике подготовки природных и
очистки производственных сточных вод. Природные, искусственные
и синтетические материалы, способные к обмену входящих в их
' состав ионов на ионы контактирующего с ними раствора, называ-
ются ионитами. Иониты, содержащие подвижные катионы, способ-
ные к обмену, называются катионитами, а обменивающие анионы —
анионитами. Наибольшее практическое значение для очистки воды
имеют органические полимерные иониты, которые являются поли-
электролитами. В этих соединениях одни ионы (катионы или анио-
ны) фиксированы на углеводородной основе (матрице), а ионы
противоположного знака являются подвижными, способными к
обмену на одинаковые по знаку заряда ионы, содержащиеся в рас-
творе.
Основу синтетического ионита составляют полимерные углево-
дородные цепи с пространственной трехмерной структурой, кото-
рые содержат ионогенные (активные) группы. Активные группы
вводятся в полимер пли непосредственно при его получении, или
при последующей химической обработке неполярного (гидрофоб-
ного) полимера соединениями, содержащими будущую активную
группу ионита. Для придания полимеру трехмерной структуры
кроме основного исходного мономера в реакционную смесь вво-
дится непредельный углеводород, сшивающий линейные макромо-
80
лекулы полимера мостиками между собой. Очень часто в
качестве сшивающего реагента используется дивинилбензол
СН2 = CH—СН = СНГ В зависимости от соотношения количества
основного материала матрицы и сшивающего реагента получаются
иониты с различной степенью поперечной связанности. Если коли-
чество сшивающего реагента невелико (около 2%), то расстояния
между мостиками, соединяющими линейные макромолекулы, полу-
чаются довольно большими (более 10 нм). Иониты, имеющие такую
структуру, проницаемы для крупных ионов. Это макропористые
иониты, применяемые для удаления органических веществ из воды.
При большем содержании сшивающего реагента (8—10%) получа-
ются иониты, способные к обмену ионов средних и малых размеров.
Размер «пор» у таких ионитов колеблется от 1 до 10 нм (в среднем
2—3 нм). Такие иониты чаще всего используются в практике во-
доподготовки. Иониты с высоким содержанием сшивающего агента
(до 20%) необходимы для обмена неорганических ионов в присут-
ствии крупных органических ионов, например при обессоливании
воды, содержащей антибиотики.
Катиониты по химической природе — полимерные кислоты или
их соли. На определенных участках матрицы у них расположены
активные полярные группы, содержащие подвижные катионы во-
дорода, натрия и др. Катиониты, содержащие в качестве активной
группы сульфогруппу SO3H, являются сильнокислотными. Сульфо-
группа обладает высокой степенью диссоциации и поэтому обмен
катионов возможен у сильнокислотных катионитов в щелочной, ней-
тральной и кислой средах (при pH до 1,5). Звено сульфокатионита
КУ-2 имеет строение
—сн~сн2—сн2—сн—СН2-СН—
SO3H
Этот катионит получают совместной полимеризацией стирола
и дивипилбепзола (ДВБ) с последующей обработкой полимера кон-
центрированной серной кислотой. Иониты нерастворимы в воде,
так как их основа — углеводородная матрица — гидрофобна. Ак-
тивные полярные группы ионитов имеют большое сродство к воде,
т. е. гидрофильны. Активная группа катионитов в воде диссоцииру-
ет с образованием фиксированного на матрице аниона и подвижно-
го противоиона (катиона), который оказывается в растворе в непо-
средственной близости от активной группы. Если условно обозна-
81
чить фиксированный на матрице анион сульфокатиопита через
RSOF, то процесс диссоциации протекает по схеме R—SO3H^
^RSO?+H+.
Катиониты, содержащие карбоксильные группы — СООН, харак-
терные для карбоновых кислот, относятся к слабокислотпым. Они
способны к обмену катионов в щелочной среде (при рП>7), так
как степень диссоциации карбоксильной группы в этих условиях
возрастает. К слабокислотпым катионитам относится отечествен-
ный карбоксильный катионит КБ-4. Катиониты, содержащие раз-
личные активные группы, занимают промежуточное по.южепис меж-
ду сильно- и слабокислотными. К ним относятся сульфоугли, содер-
жащие группы •—SO3H и —СООН, катионит КУ-1, содержащий
—SO3H и —ОН. Рабочей формой катионита может быть кислотная,
когда обменивающимися ионами будут ионы Н+ (Н-катионит), пли
солевая, например натриевая, когда способным к обмену катионом
ионита будет ион Na+ (Na-катионит).
Аниониты являются полимерными основаниями или их солями.
Матрица анионита содержит активные полярные группы, способные
диссоциировать в воде с образованием подвижных анионов (ОН-,
С1~ и др.). Фиксированные на матрице катионы сообщают аниони-
ту положительный заряд. Звено анионита на основе пиридина в со-
левой хлор-форме имеет следующее строение:
С1-
—СН2—СН—СН2—СН—СН2—СН—
N+
Cl-
Диссоциация анионита в воде идет по схеме R+Q-^R+4-Cl-, где
R+ — фиксированный на матрице катион активной группы, условно
принятый за однозарядный. Высокоосновные аниониты способны к
обмену в кислой, нейтральной и щелочной средах. Низкоосновные —
только в кислой среде.
Способность ионитов к обмену ионов количественно характери-
зуется обменной емкостью, т. е. числом миллиграмм-эквивалентов
(мг-экв) ионов, обменивающихся определенной навеской сухого или
определенным объемом набухшего ионита. В зависимости от усло-
вий определения различают полную (ПОЕ), статическую (СОЕ) и
динамическую (рабочую) обменную емкость (ДОЕ, РОЕ) ионита.
Полная обменная емкость ионита характеризуется общим числом
активных групп ионита в единице объема смолы. Статическая об-
82
мениая емкость определяется числом миллиграмм-эквивалентов ио-
нов, обменивающихся при контакте раствора с определенным коли-
чеством ионита (1 г в расчете на абсолютно сухой продукт).
Основной технологической характеристикой ионитов служит ра-
бочая (динамическая) обменная емкость. Она определяется коли-
чеством ионов (мг-экв/дм3 или г-экв/м3), поглощенных определен-
ным объемом набухшего ионита в процессе фильтрования до мо-
мента появления этих ионов в растворе, выходящем из фильтра. Ра-
бочая обменная емкость зависит от природы ионита и поглощаемых
ионов, солесодержания, pH, концентрации одноименных ионов в
обрабатываемой воде, скорости фильтрования, высоты слоя иони-
та, фракционного состава, режима работы фильтра. Солевые фор-
мы катионитов имеют высокую степень диссоциации и поэтому их
активность в меньшей степени зависит от концентрации одноимен-
ного иона в растворе. Рабочая обменная емкость ионита зависит от
солесодержания. Так, например, рабочая обменная емкость катио-
нита КУ-2 при увеличении солесодержания от 1 до 15 мг-экв/л сни-
жается примерно на 15%.
Скорость обмена зависит от размера иона, величины его заря-
да и способности к гидратации. Она увеличивается с повышением
заряда иона и уменьшением степени гидратации. Рабочая обмен-
ная емкость катионитов по иону Na+ примерно в два раза меньше,
чем по ионам Са2+ или Mg2+. Аниониты имеют большую избира-
тельность к сульфат-иону по сравнению с хлорид-ионом. Рабочая
обменная емкость по сульфат-иону на 40—50% выше, чем по хло-
рид-иону. На рабочую обменную емкость влияет скорость фильтра-
ции через ионитовый фильтр. При значительной скорости фильтро-
вания воды рабочая обменная емкость заметно уменьшается. Эта
зависимость рабочей обменной емкости от скорости фильтрования
является общей для всех видов ионитов. Обычно рабочая обменная
емкость составляет около 60% от полной, но в зависимости от ре-
жима фильтрования может изменяться. Высота слоя, при которой
происходит снижение жесткости исходной воды до заданной величи-
ны, называется высотой защитного слоя ионита. На рабочую обмен-
ную емкость ионитов влияет и их фракционный состав. Чем меньше
размер зерен, тем выше скорость обмена ионов. Размер частиц ос-
новной рабочей фракции большинства марок ионитов составляет
0,5 мм.
Ионный обмен происходит эквивалентно, т. е. при равных заря-
дах число ионов ионита, перешедшее в раствор, соответствует чис-
лу ионов, поглощенных из раствора. Как и всякая химическая ре-
акция обмена, ионный обмен есть процесс обратимый и подчиня-
ется закону действия масс. Ионы, поглощенные ионитом, можно де-
сорбировать введением раствора, содержащего ион, который спосо-
бен вступить в реакцию обмена с этим ионом, т. е. отработанные
иониты можно регенерировать. Регенерация катионитов проводится
растворами кислот (для Н-катионитов) или солей. Аниониты реге-
нерируют растворами гидроксида, хлорида или карбоната натрия,
соляной кислоты. Сильнокислотные катиониты обменивают свои но-
83
пы па вес катионы, находящиеся в воде, а слабокислотные избира-
тельно поглощают катионы слабых кислот. Высокоосновные анио-
ниты способны к обмену анионов сильных и слабых кислот, а ииз-
коосиовные обменивают избирательно анионы сильных кислот.
Так как химическая реакция обмена протекает практически
мгновенно, то кинетика ионного обмена определяется скоростью
диффузии ионов через слой раствора (при концентрации его менее
0,1 г-экв/л) или скоростью диффузии ионов в фазе ионита (при боль-
шей концентрации раствора). В сильно набухшем полпстпрольном
ионите ионообменное равновесие наступает через несколько секунд.
На скорость диффузии ионов влияют размер зерен ионита, темпе-
ратура, степень поперечной связанности ионита. Таким образом,
благоприятные факторы для работы ионообменных установок: оп-
тимальный диаметр зерен ионита, достаточно высокий коэффициент
набухания ионита, большая плотность активных групп в фазе ио-
нита. Иногда для более полного использования ионита обработка
воды ведется во взвешенном слое ионита. Обработка воды на ка-
тионитах называется катионироваписм, а на анионитах — аниони-
рованием.
Основные марки синтетических ионитов отечественного произ-
водства, применяющихся при обработке воды. 1. Катионит универ-
сальный КУ-2-8 представляет собой сульфированный сополимер
стирола с 8% дивинилбензола. Это сильнокислотный сульфокатио-
нит. Выпускается в виде сферических гранул желтого или коричне-
вого цвета. Нерастворим в органических растворителях, выдержи-
вает нагревание до 120° С, устойчив к действию кислот, щелочей и
некоторых окислителей. Имеет высокую механическую прочность.
Удельный объем набухшего ионита в Н-форме до 2,9 мл/г. Полная
обменная емкость в статических условиях по 0,1 н. раствору NaOH
составляет 4,9 мг-экв/г. Применяется для умягчения, обессолива-
ния воды и для очистки производственных сточных вод от катионов
тяжелых металлов.
2. Катионит карбоксильный КБ-4П-2. Его получают сополиме-
ризацией метилметакрилата с дивинилбензолом с последующим
омылением полученного полимера щелочью. Выпускается в виде
сферических гранул белого, светло-желтого или светло-розового
цвета. Используется для тонкой очистки воды.
3. Анионит высокоосновной АВ-17. Содержит сильно диссоцииро-
ванные группы четвертичного аммониевого основания. Выпускает-
ся в виде сферических гранул светло-желтого цвета. Устойчив к
действию кислот и щелочей, органических растворителей. Удель-
ный объем набухшего ионита в ОН-форме от 2,7 до 3,3 мл/г. Пол-
ная обменная емкость в статических условиях по 0,1 н. раствору
НС1 не менее 3,5 мг-экв/г. Применяется при обессоливании воды
для удаления кремниевой кислоты и для очистки сточных вод, со-
держащих тяжелые металлы в форме анионов.
4. Анионит низкоосновной АН-22. Содержит слабоосновные ами-
ногруппы. Представляет собой сферические гранулы от светло-жел-
того до светло-коричневого цвета. Удельный объем набухшего иони-
84
та составляет от 1,8 до 2,8 мл/г. Полная обменная емкость в ста-
тических условиях по 0,1 и. раствору НС1 не менее 5,0 мг-экв/г.
Кроме указанных синтетических ионитов в практике водопод-
готовки используется сульфоуголь (катионит). Он представляет со-
бой продукт обработки бурого угля серной кислотой. По внешнему
виду это черные зерна неправильной формы с предельными разме-
рами фракций от 0,3 до 1,55 мм. Полная обменная емкость сульфо-
угля в статических условиях составляет 1,8—1,9 мг-экв./'г. Допусти-
мая температура обрабатываемой воды в нейтральной и слабокис-
лой среде 60, в слабощелочной 30—40° С. Низкая обменная емкость
и недостаточно высокая химическая и термическая устойчивость
ограничивают применение этого материала для обработки воды.
Na-Ка.ионирование. Умягчение воды в этом процессе происхо-
дит в результате замены в эквивалентном количестве ионов Са2+
и Mg2+, содержащихся в обрабатываемой воде, на ионы Na+ катио-
нита. Соли карбонатной жесткости, содержащиеся в воде, заменя-
ются соответствующими солями натрия. Метод применим для вод
с небольшой карбонатной жесткостью. Если обозначить через
R- аннон активной группы, фиксированный на матрице, то схемы
уравнений реакций обмена, протекающих при Na-катионироваиии,
можно записать следующим образом:
2NaR + СаСЬ Д: CaR2 + 2NaCl
2NaR + MgSO4 MgR2 4- Na2SO4
Na-Катионитовые фильтры регенерируют 5—10%-ным раство-
ром хлорида натрия:
CaR2 + 2NaCl Д: 2NaR + СаС12
MgR2 + 2NaCl ^±2NaR + MgCl2
Растворы, образующиеся при регенерации, представляют отход
производства. Умягченная вода имеет повышенную щелочность, по-
этому ее дополнительно обрабатывают кислотами или смешивают с
Н-катионированной водой. При высокой исходной щелочности (бо-
лее 3 мг-экв/л) и магниевой жесткости применяется комбинирован-
ный метод умягчения воды. Щелочность снижается предваритель-
ным известкованием, а затем вода подается на Na-катионитовый
фильтр. Остаточная жесткость воды, умягченной Na-катионирова-
нием, уменьшается до 0,01 мг-экв/л и ниже.
Н-Катионирование. Сущность этого метода заключается в том,
что ионы водорода Н-катионита обмениваются на содержащиеся в
воде ионы Са2+ и Mg2+. В обработанной воде появляется эквива-
лентное количество свободных кислот:
Са (НСО3)2 + 2HR CaR2 4- 2 [Н2О Щ СО2]; MgSO4 4- 2HR ^±MgR2 4- H2SO4
Mg (HCO3)2 + 2HR MgR2 4- 2 [H2O 4- CO,]; CaCl2 4- 2HR CaR2 4- 2HC1
CaSO4 4- 2HR CaR2 + H2SO4; MgCI2 + 2HR MgR2 4- 2HC1
Н-Катионирование применимо для вод с небольшой (до
1 мг-экв/л) некарбонатной жесткостью. Умягченная вода не содер-
жит карбонатов, которые заменяются свободной угольной кислотой.
85
Вследствие образования свободных кислот вода после обработки
Н-катионированием имеет кислую реакцию среды и является аг
рессивной по отношению к металлам. Для снижения избыточной
кислотности угольную кислоту удаляют физическими методами пли
нейтрализуют воду гидроксидом натрия. Для получения стабиль-
ной воды в практике водоподготовки также сочетают обработку во-
ды на Н- и Na-катиопитовых фильтрах с последующим смешением
одной и другой воды исходя из величин щелочности и жесткости.
Н-Катионитовые фильтры регенерируют 5%-ным раствором со-
ляной или 1 — 1,5%-ным раствором серной кислоты:
Cal?2 + 2НС1 2HR + СаСЬ; MgR2 + H2SO4 2HR + MgSO4
Для повышения эффективности процесса регенерации количест-
во регенеранта (хлорида натрия или кислоты) необходимо брать
в 3—3,5 раза больше, чем следует из расчета по уравнениям реак-
ций. Обычно расход серной кислоты для регенерации Н-катиони-
тового фильтра в 1,25—1,5 раза превышает количество, которое
определяется по реакциям ионного обмена.
§ 22. Обессоливание воды
Метод обработки воды, направленный па снижение общего со
лесодержания, называется обессоливанием. Различают полное и
частичное обессоливание воды. При полном обессоливании дости-
гаемое остаточное солесодержание составляет десятые или сотые
доли миллиграмма в 1 л воды. Если из воды удаляются только от-
дельные ионы для уменьшения общего солесодержаиия, то такое
обессоливание называется частичным. Полностью обессоленная во-
да необходима в теплоэнергетике, при производстве чистых хими-
ческих материалов, полупроводниковых приборов и в некоторых
других отраслях промышленности. Опреснение морской и минера-
лизованной воды для получения воды питьевого или технического
назначения представляет один из вариантов частичного обессоли-
вания.
Обессоливание воды методом ионного обмена. Метод ионного об-
мена может быть использован как для полного, так и для частич-
ного обессоливания воды. Сущность этого метода заключается в
последовательном пропускании воды через Н-катионитовый и ОН-
анионитовый фильтры. Обмен ионов Са2+ и Mg2+ на Н-катиоиито-
вом фильтре протекает так же, как и при умягчении воды. Ионы
Na+ обмениваются на ионы Н+ катионита по уравнениям:
HR + NaCl NaR -У НС1; 2HR + Na2SO4 2NaR + H2SO4
HR + NaHCOj^NaR + [CO2 + H2O]
В результате обмена образуется свободная угольная кислота
в количестве, соответствующем щелочности воды, а вместо хлори-
дов и сульфатов появляется эквивалентное количество соляной и
серной кислот. При последующем фильтровании такой воды через
ОН-анионитовый фильтр происходит обмен анионов кислот, содер-
86
жащихся в воде, на гидроксид-ионы анионита (R+ — фиксирован-
ный на матрице катион анионита, условно принимаемый за одноза-
рядный) :
ROH + НС1 RC1 -- Н2О; 2ROH у- MjSOj R>SO4 + Н2О
Таким образом после прохождения воды через Н-катионнтовый и
ОН-анионитовый фильтры происходит се обессоливание вследствие
обмена катионов на ионы водорода, а анионов на гидроксид-ионы.
Аниониты могут применяться также в карбонатной и гпдрокарбо-
Рис. 7. Схема установки для полного химического обессоливания воды:
1— катионитовый фильтр 1-й ступени; 2— аниоиитовый фильтр 1-й ступени; 3 — катиони-
товый фильтр 2-й ступени; 4 — декарбонизатор; 5 — резервный бак; 6 — аниоиитовый
фильтр 2-й ступени; 7 — катионитовый фильтр 3-й ступени; 8 — аниоиитовый фильтр 3-й
ступени
натной формах, так как при обмене сильных кислот образуется сла-
бая угольная кислота:
R2CO3 + 2HCI 2RC1 + [СО2 + Н2О]; RHCO3 + НС1 RC1 + [СО2 + Н2О]
R2CO3 + H2SO4 R2SO4 + [CO2 -у H2O]; 2RHCO3 + H2SO4 ^±R2SO4+2[CO24-H.2O]
Регенерация анионита осуществляется растворами гидроксида,
карбоната или гидрокарбоната натрия по уравнениям:
RC1 -у NaOH ROH + NaCl; R2SO4 + Na2CO3 R2CO3 -y Na2SO4
RC1 -p NaHCO3 RHCO3 + NaCl
Обессоливание воды, идущей на питание паровых котлов, про-
изводится, если суммарное содержание сульфат- и хлорид-ионов в
исходной воде не превышает 3—4 мг-экв/л. Рассмотрим схему пол-
ного химического обессоливания воды, приведенную на рис. 7. Пер-
вая ступень обессоливания представлена Н-катионитовым и ОН-
анионитовым фильтрами. На сильнокислотном Н-катионитовом
фильтре (сульфоуголь, КУ-2) происходит обмен основного количе-
ства катионов, содержащихся в воде, на эквивалентное количество
ионов водорода катионита и переход гидрокарбонатов в свободную
угольную кислоту. Регенерация фильтра производится серной или
соляной кислотой.
Аниоиитовый фильтр 1-й ступени служит для обмена анионов
сильных кислот (SOI , С1~ и др.) на гидроксид-ионы анионита.
Анионы слабых кислот при этом остаются в растворе. Загрузка
87
т----г„ njMj.Knuj,!!! iv/i atuiuninuM v7/j,c7-iшi. в начале
работы анионитового фильтра идет поглощение угольной кислоты.
Затем угольная кислота десорбируется вследствие замещения ее
анионами сильных кислот. Для регенерации анионитового фильтра
1-й ступени применяется разбавленный раствор гидроксида натрия
или отработанный раствор регенеранта после сильноосновного анио-
нитового фильтра 2-й ступени обессоливания.
Вторая ступень обессоливания воды предназначается для обме-
на катионов и анионов, оставшихся после обработки воды на фильт-
рах 1-й ступени. Регенерация Н-катионитового фильтра 2-й ступе-
ни осуществляется серной или соляной кислотами. Перед подачей
воды па анионптовый фильтр 2-й ступени производится удаление
свободной угольной кислоты физическими методами. Это необходи-
мо потому, что при обескремнивании воды на сильноосновном анпо-
нитовом фильтре 2-й ступени идет поглощение угольной кислоты.
Это уменьшает обменную емкость фильтра по кремниевой кислоте
и увеличивает расход регенерирующего раствора. Обескремнивание
воды производится на фильтре, загруженном сильпоосповным анио-
нитом АВ-17. Здесь же происходит удаление оставшихся анионов
угольной кислоты. Регенерация этого фильтра производится рас-
твором гидроксида натрия.
Третья ступень глубокого обессоливания воды представлена
Н-катионитовым и ОН-анионитовым фильтрами и служит для обмена
катионов и анионов, попадание которых в воду возможно в резуль-
тате несвоевременного отключения отработанного ионитового
фильтра. Два последних фильтра называются буферными и могут
быть заменены фильтром смешанного действия, содержащим силь-
нокислотный катионит и сильноосновной анионит. Использование
фильтров смешанного действия сопровождается трудностями их
регенерации, так как при этом возникает необходимость разделе-
ния ионитов. Этого можно избежать, применяя метод электрохими-
ческой регенерации смешанного слоя ионита.
Химически обессоленная вода содержит незначительное коли-
чество органических веществ. Часть из них, обладающая кислот-
ными или основными свойствами и способная диссоциировать, всту-
пает в реакции обмена с катионитом или анионитом. Недиссоцииру-
ющие органические вещества частично адсорбируются на поверхно-
сти зерен ионитов. Если перманганатная окисляемость исходной
воды составляет более 15—20 мг О2/л, то перед Н-катионитовым
фильтром 1-й ступени устанавливаются фильтры, загруженные ак-
тивированным углем или макропористым ионитом. Назначение
фильтров состоит в предварительной сорбции органических ве-
ществ. Применяя полностью обессоленную воду, необходимо при-
нимать меры к сохранению устойчивости ее состава во времени.
«Сверхчистая» вода имеет неустойчивое значение pH, так как в ней
отсутствует буферная система. Вследствие поглощения диоксида
углерода из воздуха она имеет слабокислую реакцию среды. При
концентрации СО2 1 мг/л вода имеет pH 5,5—5,6. В такой воде
растворяются труднорастворимые соединения: Fe(OH)3, СаСО3,
88
х\ деиивию подоонои воды устойчивы титан, луженые
медь или латунь, полиэтилен, тефлон, полипропилен.
Метод ионного обмена может быть применен и для опреснения
минерализованных вод, но большие расходы на технологический
процесс получения опресненной воды пока ограничивают его при-
менение.
Обессоливание воды методом электродиализа. Электродиализ —
это процесс переноса ионов под действием постоянного электриче-
ского тока к электродам через полупроницаемую мембрану, поме-
щенную в раствор электролита. Установка для электродиализа на-
зывается электродиализатором, который состоит из трех камер: ка-
тодной, анодной и средней — рабочей. От катодной и анодной камер
средняя отделена перегородками — мембранами. Мембраны, раз-
деляющие камеры, не пропускают коллоидные частицы, но пропус-
кают воду и ионы электролита. В средней камере находится вода,
подлежащая обессоливанию. При пропускании электрического то-
ка ионы из средней камеры переносятся к соответствующим элект-
родам. В катодную камеру поступают катионы Na+, Са2+, Н+, Mg2+
и др. Ионы водорода восстанавливаются на катоде до свободного
водорода: 2Н+ + 2(?—>-Н2.
В анодную камеру поступают анионы, содержащиеся в воде
(СИ, НСОГ, SO4” др.), и гидроксид-ионы. Гидроксид-ионы окис-
ляются на аноде с образованием кислорода, а хлорид-ионы — с об-
разованием свободного хлора:
4ОН- — 4с-*2Н2О + О2; 2CI—— 2е-+С12
В катодной и анодной камерах происходит увеличение концент-
рации растворенных веществ, а в средней камере происходит час-
тичное снижение концентрации — обессоливание. Производитель-
ность такой установки невелика вследствие дополнительного пере-
носа ионов через рабочую камеру. Повысить эффективность работы
можно при использовании активных ионитовых мембран. Такие мем-
браны обладают соответственно свойствами катионита или аниони-
та. Применение активных ионитовых мембран в электродиализе
повышает эффективность применения этого процесса для обессоли-
вания воды. На рис. 8 приведена схема трехкамерной электродиа-
лизной установки. Катодная камера отделена от камеры обессоли-
вания катионитовой мембраной, анодная камера — анионитовой.
Исходная вода подается во все камеры. В процессе работы установ-
ки в средней камере происходит обессоливание воды, а в крайних
наблюдается повышение концентрации раствора. Осуществление
процесса электродиализа с применением ионитовых мембран осно-
вано на избирательном (селективном) переносе ионов определенно-
го знака через мембрану. Анионитовая мембрана, несущая поло-
жительный заряд фиксированных на матрице катионов, избиратель-
но пропускает только анионы из раствора, отрицательно заряжен-
ная катионитовая мембрана проницаема только для катионов.
Благодаря селективной проницаемости ионитовых мембран катио-
ны из камеры обессоливания беспрепятственно проходят в катод-
89
ную камеру, а анионы — в анод-
ную. Но катионы натрия и водо-
рода из анодной камеры не могут
пройти в камеру обессоливания,
так как на их пути находится
анионитовая мембрана, практи-
чески не проницаемая для них.
Точно так же из-за наличия од-
ноименного заряда с катионито-
вой мембраной анионы (С1~
ОН-) остаются в катодной ка-
мере.
Практически в мембрану на-
ряду с обменивающимися ионами
Рис. 8. Схема трехкамерной ячейки
электродиализатора:
/ — анод, 2—анионитовая мембрана; •/—
камера обессоливания; 4 — катионитовая
мембрана; 5 — катод; 6 — катодная каме-
ра; 7 — анодная камера
проникает некоторая часть ионов
противоположного знака. Селек-
тивность мембраны определяется
числом переноса ионов. Число пе-
реноса — это та доля электриче-
ства, которая переносится через
мембрану ионами, имеющими знак заряда, противоположный заря-
ду ионов, фиксированных на матрице. Для идеального случая чис-
ло переноса ионов равно единице. Чем больше удельная обменная
емкость мембраны, тем выше ее селективность.
Использование трехкамерного электродиализатора нерентабель-
но из-за больших потерь электроэнергии при протекании электрод-
ных реакций окисления и восстановления. Значительная экономия
электроэнергии получается, если обессоливание воды осуществля-
ется в многокамерных электродиализаторах, состоящих из последо-
вательно соединенных десятков и сотен камер (рис. 9). В таких
установках создаются ячейки, отделенные друг от друга катионито-
вой и анионитовой мембранами. У катода находится катионитовая
мембрана, у анода — соответственно анионитовая. При этом коли-
чество электроэнергии, расходуемое на вторичные электродные ре-
акции, снижается с 60—70 до 1—2%. В процессе работы установки
в четных камерах получается обессоленная вода, а в нечетных об-
разуются растворы высокой концентрации.
Электродиализные установки применяются в основном для час-
тичного обессоливания морских и минерализованных вод, а также
в оборотных системах водоснабжения. Устойчивая работа установ-
ки и достигаемая степень обессоливания зависят от ионного соста-
ва воды. При большой карбонатной жесткости исходной воды на-
блюдается образование отложений карбоната кальция и гидрокси-
да магния. Особенно заметно это в катодной камере, где в процессе
электролиза создается щелочная среда. Электродиализ рациональ-
но использовать при минерализации воды от 3,5 до 35 г/кг и при
остаточном солесодержании не менее 400 мг/л.
Если в обрабатываемой воде присутствуют микроорганизмы, то
не исключена возможность образования биологических обрастаний
90
на мемиранах. для предотвращения обрастаний воду обрабатыва-
ют хлором или гипохлоритами. Содержание взвешенных веществ в
исходной воде должно быть не более 3 мг/л, железа — 0,1, бора —
0,1 мг/л, цветность воды не выше 20° С, окисляемость до 5 мгОг/л.
Отравление катионитовой мембраны возможно и в результате от-
ложения гидроксида железа. Перед поступлением воды в электро-
диализатор проводят фильтрование для удаления взвешенных час-
тиц. Электроды, применяемые в электродиализаторах, должны
Рис. 9. Схема многокамерного электродиализатора:
1 — анод; 2 — вода для промывки анода; 3 — исходная вода; 4 — вода для промывки като-
да; 5 — катод; 6 — концентрированный раствор солей; 7 — катионитовые мембраны; 8 —
анионитовые мембраны; 9— раствор гидроксида натрия; 10 — опресненная вода; // — хлор-
ная вода
иметь высокую химическую стойкость. Они изготовляются из магне-
тита, платинированного титана или графита с высокой плотностью.
Электродиализ может быть применен и для глубокого обессоли-
вания воды с общим солесодержанием 300—500 мг/л. Для уменьше-
ния электрического сопротивления межмембранный объем в каме-
ре обессоливания заполняется ионитами (катионитом и анионитом).
Отработанные иониты подвергаются электрохимической регенера-
ции. Для снижения расхода электроэнергии расстояние между мем-
бранами сокращают до 0,2—0,3 мм.
Первая электродиализная установка была введена в строй в
пос. Бекдаш на полуострове Мангышлак. В Краснодарском крае,
Туркменской ССР, Запорожской области и других районах нашей
страны работают десятки подобных опреснительных установок.
Метод гиперфильтрации (обратный осмос). Этот метод основан
на использовании осмотического переноса молекул воды через по-
лупроницаемую мембрану. Такая мембрана проницаема для мо-
91
лекул воды, но не пропускает молекулы и ионы растворенных в во-
де веществ. Явление осмоса состоит в том, что при неодинаковых
концентрациях раствора с разных сторон полупроницаемой мембра-
ны молекулы воды будут диффундировать через пес из раствора
с меньшей концентрацией в раствор с большей концентрацией. Ве-
личина возникающего осмотического давления зависит от разности
концентраций растворов по обе стороны мембраны: чем она боль-
ше, тем выше осмотическое давление. При разности концентраций
растворов, соответствующих солссодержанию морской воды
Рис. 10. Схема движения молекул воды через полупрони-
цаемую мембрану:
а—состояние равновесия; б—осмотический перенос; в—обратный
осмос; 1 — соленая вода; 2 — пресная вода; 3—мембрана
(35г/кг) и дистиллированной (10 мг/л), осмотическое давление со-
ставляет 24 -102 кПа. Обратный процесс — диффузию молекул воды
через мембрану в раствор с меньшей концентрацией можно осуще-
ствить, подавая концентрированный раствор на мембрану под дав-
лением, превышающим осмотическое (рис. 10).
Перенос молекул воды через полупроницаемую мембрану из рас-
твора с большей концентрацией в раствор с меньшей концентраци-
ей под действием внешнего давления, превышающего осмотическое
давление, называется обратным осмосом или гиперфильтрацией.
О причине обратного осмоса высказано несколько гипотез. Наибо-
лее убедительная из них состоит в том, что поры этой мембраны
проницаемы только для молекул воды. Гидратированные ионы, об-
ладающие большими размерами, проходить через поры мембраны
не могут. Движущей силой процесса обратного осмоса является
разность между прилагаемым внешним рабочим и осмотическим
давлением. Понятно, что при увеличении солесодержания исходной
воды обратный перенос молекул воды через мембрану уменьшается
вследствие повышения осмотического давления. Снижается и эф-
фект обессоливания, так как при увеличении концентрации раство-
92
ра уменьшается степень гидратации ионов, размеры их становятся
соизмеримыми с размером пор полупроницаемой мембраны.
Определяющими характеристиками полупроницаемых мембран
являются селективность, проницаемость по воде, стабильность по-
казателей во времени. Селективность мембран S характеризует сте-
пень задержания растворенных компонентов и рассчитывается по
формуле
S = (Сисх Сосг) 100/СИсх,
где Спех — концентрация растворенных солей в опресняемой воде;
СОст — остаточная концентрация солей в опресненной воде.
Селективность мембран зависит от солесодержания исходной
воды. В интервале концентраций раствора от 0,5 до 10 г/л селек-
тивность изменяется незначительно.
Проницаемость по воде определяется скоростью фильтрации
опресненной воды через единицу площади мембраны. Селектив-
ность мембран повышается при уменьшении проницаемости по во-
де. Для мембран высокого качества селективность достигает 98%.
Важным показателем качества мембран служит устойчивость
их работы во времени. Обычно срок службы мембран определяет-
ся несколькими месяцами и находится в зависимости от многих
факторов (концентрации, солевого состава опресняемой воды, ре-
жима работы установки). При сроке работы мембран больше 9 ме-
сяцев на расходы по опреснению методом гиперфильтрации почти
не влияет стоимость мембран.
Для опреснения воды методом гиперфильтрации применяются
мембраны из ацетатцеллюлозы. Они могут быть выполнены в виде
пленки, трубки, тонких полых волокон. Степень задержания раз-
личных компонентов обрабатываемой воды ацетатцеллюлозными
мембранами неодинакова: Ca2++Mg2+>SNa+ +К+; СОз ’>НСОГ>
>5О Д>С[_. Ацетатцеллюлозные мембраны имеют большую селек-
тивность по отношению к поливалентным ионам. В щелочной среде
они подвергаются гидролизу и поэтому менее устойчивы. Опти-
мальные значения pH, при которых минимально сказывается дей-
ствие гидролиза, 4,5—5,0.
Эффективность опреснения гиперфильтрацией снижается, если
исходная вода содержит ионы, способствующие образованию нерас-
творимых осадков на мембранах. Метод гиперфильтрации может
быть применен не только для опреснения природных вод, но и для
обработки воды в системах оборотного водоснабжения, для очист-
ки производственных сточных вод.
Газгидратный метод опреснения воды. Газгидратный метод
опреснения воды основан па том, что при определенной температуре
и давлении некоторые газы образуют с водой кристаллогидраты,
выпадающие в виде твердой фазы. Соединения, образующиеся в ре-
зультате внедрения молекул газа в полости кристаллической решет-
ки льда, называются газгидратами. Они относятся к так называе-
мым клатратам — нестехиометрическим соединениям включения.
Химического взаимодействия между газом и водой при образова-
93
нии газгидратов не происходит. Молекулы газа удерживаются в
фазе льда за счет проявления межмолекулярных (ван-дер-ваальсо-
вых) сил. Общая формула газгидратов М-пН2О, где М — вещест-
во (газ). Газгидраты могут иметь структуры, обозначаемые I и II.
Элементарная ячейка структуры I содержит 46 молекул воды и 8
полостей. При предельном заполнении полостей формула газгидра-
та будет 8М-46Н2О. Элементарная ячейка структуры II содержит
136 молекул воды, 8 больших и 16 малых полостей. Газгидраты
при этом образуются только в больших полостях и имеют общую
формулу 8М-136Н2О. Малые пустоты могут заполняться только
такими газами, у которых размеры атомов сравнительно невелики
(азот, кислород).
Наилучшими гидратообразующими веществами из применяв-
шихся для опреснения воды являются пропан C3Hs и фторхлорпро-
изводные углеводородов жирного ряда — фреоны. Эти вещества не-
токсичны и одновременно используются в качестве хладагентов
для поддержания температуры, необходимой для гидратообразова-
ния. Так, фреон-12 (дифтордихлорметан CF2C12), имеющий т. кип.
—23,7°, образует с водой газгидрат со структурой II. При исполь-
зовании пропана (т. кип. —42,1°С), образующего газгидрат со
структурой II, опреснение воды ведется при 2—5°С и давлении до
500 кПа. Интервалы температур и давления, при котором возможно
формирование газгидратов, зависит от солесодержания воды. Так
как образование газгидратов сопровождается выделением тепло-
ты, то в процессе кристаллизации систему необходимо охлаждать.
Для ускорения образования зародышевых кристаллов смесь газа с
водой переохлаждают. Образующиеся газгидраты удерживают на
поверхности кристаллов слой рассола, поэтому после отделения их
необходимо промывать опресненной водой.
Технологическая схема опреснительной установки газгидратным
методом включает в себя операции: получение газгидратов, отде-
ление их от рассола, очистку кристаллов от рассола, плавление газ-
гидратов и отделение пресной воды.
Дистилляционный метод. При нагревании опресняемой воды до
температуры кипения происходит интенсивное образование пара,
который почти не содержит солей. Расход теплоты на парообразо-
вание складывается из теплоты, необходимой для нагревания воды
до температуры кипения и скрытой теплоты парообразования, рав-
ной при 100° С 2445,6 кДж/кг (44,041 кДж/моль). Температура ки-
пения исходной воды зависит от общего солесодержания.
Опреснение воды методом дистилляции осуществляется па испа-
рительных установках различных конструкций с использованием
разных источников тепловой энергии, стоимость которой составля-
ет половину расходов на опреснение. Опреснение воды данным ме-
тодом осложняется образованием накипи на стенках теплообмен-
ной аппаратуры. Для предотвращения образования накипи опресня-
емую воду подвергают предварительной обработке химическими и
физическими методами. В практике опреснения воды применяются
антинакипины, механизм действия которых заключается в адсорб-
94
ции на поверхности испарителей и на зародышевых кристаллах на-
кипеобразователей. Образующиеся тонкие адсорбционные соли ан-
тинакипинов препятствуют дальнейшему росту кристаллов или
сильно замедляют его. В качестве антинакипинов могут быть ис-
пользованы некоторые поверхностно-активные вещества (ПАВ),
таннин, полиакриловая кислота и другие соединения.
Снижение концентрации накппеобразователей достигается и при
фильтровании воды через фильтр, загруженный контактной массой,
состоящей из зерен природных карбонатов. В процессе фильтрова-
ния происходит осаждение карбоната кальция и гидроксида магния
на зернах загрузки и снижение их содержания в опресняемой воде.
Разрабатываются методы предотвращения образования накипи, ос-
нованные на гндрофобизации поверхности стенок испарителей раз-
личными покрытиями.
В настоящее время проводятся работы по использованию энер-
гии ядерных установок для опреснения воды. Комплексное исполь-
зование теплоты от различных атомных реакторов позволяет про-
водить одновременно опреснение воды и получать электроэнергию.
При использовании низкопотенциального теплоносителя дистилля-
ция воды может проводиться при более низкой температуре (50—
60° С) под глубоким вакуумом (0,04-у0,08) • 102 кПа. Этот метод
применим для опреснительных установок любой производительно-
сти. В городе Шевченко работает опреснительная дистилляционная
установка, использующая энергию реактора на быстрых нейтронах,
производительностью более 100 тыс. м3/сут. В южных районах, с
большим количеством солнечных дней в году, для опреснения воды
может быть использована солнечная энергия (гелиоопреснение).
Опреснение воды вымораживанием. При замерзании минерали-
зованных вод соль и вода образуют отдельные структуры: сначала
формируются кристаллы пресного льда, смерзающиеся в агрегаты,
а на поверхности кристаллов льда и в пространстве между ними
сохраняется рассол, замерзающий при более низкой температуре.
При медленном таянии плавится сначала рассол, а затем лед. Под-
бирая оптимальные температурные режимы, можно отделить лед
от рассола. Количество опресненной воды, получаемой из 1 м3 ис-
ходного льда, зависит от общего солесодержания опресняемой во-
ды. Солесодержание исходной воды, равное 30 г/кг, позволяет полу-
чить около 0,6 м3 пресной воды. Замораживание воды может осу-
ществляться в естественных условиях, но этот способ имеет огра-
ниченное применение. Наиболее приемлемым является опреснение
воды замораживанием с применением искусственного охлаждения.
Замораживание можно осуществить при испарении воды под ваку-
умом или при контакте ее с гидрофобным хладагентом. С пониже-
нием внешнего давления температура кипения воды снижается. При
подаче воды в вакуумный замораживатель часть ее испаряется, а
около половины переходит в лед. Наиболее эффективными являют-
ся установки по опреснению воды, в которых охлаждение и замора-
живание воды осуществляется при прямом контакте ее с такими
гидрофобными нетоксичными хладагентами, как изобутан, бутан,
95
фреоны, имеющими температуру кипения ниже 0°С. Использование
их в качестве хладагентов позволяет вести опреснение воды при
давлении, близком к атмосферному. Охлаждение воды происходит
в результате испарения газов и по способу осуществления имеет
сходство с газгидратным методом.
Оценка качества опресненной и обессоленной воды. Опреснен-
ная вода по органолептическим и санитарно-химическим показате-
лям должна соответствовать требованиям, предъявляемым ГОСТом
к качеству воды. Установлены также предельно допустимые кон-
центрации некоторых элементов и определены оптимальные концен-
трации основных компонентов в опресненной воде. При употребле-
нии воды с общим солесодержанием менее 50 мг/л происходит на-
рушение водно-электролитного обмена и выведение из организма
ионов С1щ Na+, К+, Са2+. Поэтому минимальное общее солесодер-
жание для воды должно составлять 100 мг/л. Оптимальное общее
солесодержание 250 мг/л.
Таблица 9. Химический состав дистиллята
Пока- затель ста- биль- ности Щелоч- ность, мг-экв/л Жест- кость общая, мг-экв/л Соле- содер- жание, мг/л Содержание ионов, мг/л
Са2+ Mg2+ Ха + К+ СГ' 9— so2 нсо3 ^еобщ F- со2
0,3 0,01 0,1 10,0 2,0 0,3 2,0 0,7 0,5 0,4 Следы 0,4 3,5
Вода, полученная методом дистилляции, имеет очень малую ве-
личину солесодержания (10—20 мг/л), содержит мало ионов Са2+
и F-, pH ее непостоянен, так как она лишена буферных свойств.
При растворении диоксида углерода pH такой воды снижается, и
она становится агрессивной.
В табл. 9 приводится химический состав дистиллята (по данным
А. Е. Егорова и Л. С. Алексеева). В дистиллят могут попадать ор-
ганические соединения, летучие с водяным паром. В опресненной
воде могут оказаться также жизнеспособные споровые формы мик-
роорганизмов, в ней могут развиваться железо-и серобактерии. По-
этому дистиллят не может быть непосредственно использован для
хозяйственно-питьевого водоснабжения. Для получения воды, со-
ответствующей требованиям ГОСТа на питьевую воду, в дистил-
лят вводят необходимые соли (СаС12, MgSO4, NaHCO3, NaHSO4,
NaF и др.). Иногда смешивают дистиллят с морской или подзем-
ной минерализованной водой. Обычно за эталон при подготовке
питьевой воды из дистиллята берется вода Московского водопрово-
да (общее солесодержание 250 мг/л).
При опреснении воды методами гиперфильтрации и электродиа-
лиза соотношение между главными ионами в получаемой воде со-
храняется таким же, как в исходной. Мембранами предпочтительно
задерживаются ионы поливалентных металлов, галогенид-ионы, но в
малой степени задерживаются такие микроэлементы, как бор. По-
этому в опресненной воде содержание бора может превышать ПДК
96
(1 мг/л). Ацетатцеллюлозные мембраны непроницаемы для бакте-
рий, в том числе и для кишечной палочки, но не задерживают ви-
русов и бактериофагов. Поэтому определяющим фактором бакте-
риологических показателей качества воды становится наличие ви-
русов. При эксплуатации гиперфильтрационных установок для
опреснения морской воды может наблюдаться ухудшение бактерио-
логических показателей качества воды.
Опреснение воды методом электродиализа сопровождается в на-
чальный период вымыванием низкомолекулярных веществ из мем-
бран, которые ухудшают органолептические свойства получаемой
воды и способствуют появлению запаха. Поэтому рекомендуется
проводить предварительную кислотно-щелочную обработку мем-
бран.
Магнитная обработка воды. Впервые магнитная обработка воды
для предотвращения образования накипи в паровых котлах была
предложена бельгийским инженером Вермайерном (1945). Она за-
ключается в пропускании воды через магнитные поля с чередую-
щейся направленностью, которые создаются постоянными магни-
тами или электромагнитами. При этом кристаллизация соедине-
ний— накипеобразователей — происходит не на стенках теплооб-
менной аппаратуры, а в воде. Под действием омагнцченной воды на-
блюдается разрушение и ранее образовавшейся накипи. Эффект
магнитной обработки усиливается, если воду пропускать через маг-
нитное поле 3—5 раз в течение суток. Максимальное воздействие
магнитного поля наблюдается при направлении потока воды, пер-
пендикулярном магнитному полю.
Фактором, сдерживающим широкое применение магнитной обра-
ботки воды, является отсутствие строгой теории процесса, что не
позволяет заранее планировать условия и эффект обработки. Воз-
действие магнитного поля на воду связывается с тем, что часть
молекул воды, совершающих беспрерывное колебательное движе-
ние, входит с ним в резонанс. Это сопровождается возникновением
квантов энергии, вызывающих нарушение водородных связей, что
ведет к изменению структуры воды. Исследования показали, что
на свойства омагниченной воды влияют ионы растворенных ве-
ществ. Наибольший эффект магнитной обработки воды проявляет-
ся в кальциево-карбонатных водах, при этом важную роль играет
содержание диоксида углерода, растворенного в воде, который спо-
собствует образованию пересыщенных растворов карбонатов каль-
ция и магния. Эффективность процесса зависит от напряженности
магнитного поля, скорости протекания воды, химического состава и
концентраций примесей.
Для магнитной обработки воды характерно то, что между эф-
фектом обработки и напряженностью магнитного поля проявляет-
ся периодическая зависимость. Эта периодичность, обнаруживаемая
и в ряде других явлений (при электродных процессах, при измене-
нии состава жидкой фазы в суспензиях и др.), состоит в том, что
при каком-то напряжении магнитного поля обнаруживается макси-
мум воздействия на систему. Но дальнейшее увеличение напряжен-
4—1244 97
ности до некоторого предела приводит к уменьшению эффекта воз-
действия. Затем снова возникает максимум воздействия и т. п.
§ 23. Угольная кислота и ее формы содержания в воде
Диоксид углерода, растворяясь в воде, частично вступает с ней
во взаимодействие с образованием угольной кислоты. Отдельно
определить содержание диоксида углерода и угольной кислоты в
воде трудно, поэтому суммарную концентрацию этих компонентов
принимают за концентрацию свободной угольной кислоты. Так как
только около 1% растворенного диоксида углерода образует уголь-
ную кислоту, расчет содержания свободной угольной кислоты ве-
дется на диоксид углерода СО2 СВоб- Концентрация свободной
угольной кислоты в поверхностных водах определяется парциаль-
ным давлением диоксида углерода в атмосфере. Растворимость
диоксида углерода в воде, отвечающая равновесному состоянию при
атмосферном давлении, приведена ниже.
п, °C 0 5 10 15 20 25 30 35 40 45 50 60
ri2'J
Раствори-
мость СО2,
мг/л 3,35 2,77 2,32 1,97 1,69 1,45 1,25 1,11 0,97 0,86 0,76 0,58
Для достижения состояния равновесия между диоксидом угле-
рода атмосферы и растворенным в воде требуется определенное
время. Концентрация свободной угольной кислоты в поверхностных
водах иногда достигает до 10—30 мг/л. В подземных водах, особен-
но пластовых, содержание СО2 может составлять сотни миллиграм-
мов на литр.
Другой формой содержания угольной кислоты в воде являются
гидрокарбонаты, образующиеся при диссоциации угольной кисло-
ты по 1-й ступени: Н2СО3^Н+ + НСОз . Они поступают в воду так-
же в результате растворения карбонатных пород под действием
угольной кислоты:
СаСО3 + СО2 4- Н2О Са (НСО3),
MgCO3 4- СО2 + Н2О Mg (НСО3)2
Гидрокарбонаты — наиболее распространенная форма содержа-
ния угольной кислоты в природных водах при средних значениях
pH. Они обусловливают щелочность воды.
При диссоциации угольной кислоты по 2-й ступени образуют-
ся карбонат-ионы: НСО~^Н+ + СО?К Они содержатся только в ще-
лочных водах (при рН>8,4). В присутствии ионов Са2+ содержа-
ние СОз~ невелико вследствие малой растворимости карбоната
кальция. В присутствии свободной угольной кислоты растворимость
карбоната кальция возрастает в результате образования гидрокар-
бонатов. Содержание различных форм угольной кислоты в воде рас-
считывают, исходя из констант диссоциации ее по 1-й и 2-й ступе-
ням и произведения растворимости карбоната кальция. Запишем
выражение константы диссоциации угольной кислоты по 1-й сту-
пени Кь
Кг = [Н+] [НСО3-]/[Н2СО3] = 3.10-7, (Ш.З)
98
где [НСОз“] — концентрации гидрокарбонат-ионов; [Н+] — концент-
рация ионов Н+; [Н2СО3] — концентрация недиссоциированных мо-
лекул Н2СО3 и СО2 в растворе.
Диссоциация угольной кислоты по 2-й ступени /\2 описывается
уравнением
/<2=[H+J [COf-] / [НСО-], (П1.4)
где [СОз~”] — концентрация карбонат-ионов в растворе. Произведе-
ние растворимости карбоната кальция записывается следующим об-
разом:
ПРСаСО3 = аСа2 + %02-’
где аСа2+ — активность ионов Са2+ в растворе.
Содержание свободной угольной кислоты в воде при pH 54-8,3
вычисляют по формуле
_ 44Щ°
“cc>-"»6 ^юрн+0,5/7 ’ ( -S)
где '"сорсвоб— содержание свободной угольной кислоты в воде,
мг/л; Що — общая щелочность воды, мг-экв/л; I — ионная сила рас-
твора.
В природных водах устанавливается динамическое равновесие
между различными формами угольной кислоты:
Н2О + СО2 Н2СО3 Н+ + НСОу 2Н+ -р СО^
Одновременно все формы угольной кислоты в воде присутствовать
не могут. Устойчивыми системами являются следующие: 1) СО2 +
+ Н2О; 2) СО2 + НСО33) НСОГ; 4) НСОГ+СО|~; 5) СО23~ + ОН-.
Остальные системы являются неравновесными и переходят в одну
из устойчивых систем:
COf~+СО2 + Н2О = 2НСО^; НСО^ -р ОН- = СО^ + Н2О; СО2+ОН-=НСО~
Основная карбонатная система природных вод представляет со-
бой систему из свободной угольной кислоты и гидрокарбонат-иона:
Н2О + СО2 Н2СО3 Н+ + НСО^
Она обладает буферными свойствами и обеспечивает постоянство
pH природных вод. Из уравнений констант диссоциации угольной
кислоты (III.2), (Ш.З) следует, что концентрация свободной уголь-
ной кислоты находится в прямой зависимости от концентрации ио-
нов Н+, а содержание карбонат-ионов — в обратной. Таким обра-
зом, в природных водах pH зависит от соотношения различных
форм угольной кислоты. При низких значениях pH (рН<4,2) в во-
де практически присутствует только свободная угольная кислота.
При повышении pH (в интервале от 4,2 до 8,3) в воде находятся
в равновесии свободная угольная кислота и гидрокарбонаты. Со-
держание гидрокарбонат-ионов с повышением pH возрастает, а
концентрация свободной угольной кислоты уменьшается. При pH
8,3—8,4 в воде находятся практически только гидрокарбонаты
4* 99
Рис. 11. Зависимость содержания раз-
личных форм угольной кислоты от pH
(~98%). При рН>8,4 в воде
появляются карбонат-ионы, ко-
торые находятся в равновесии
с гидрокарбонатами. Если
рН>12, то карбонат-ионы ста-
новятся преобладающей фор-
мой (рис. И). Пользуясь кри-
выми, приведенными на рис. 11,
можно вычислить соотношение
содержания различных форм
угольной кислоты при опреде-
ленном значении pH (для
25° С). При увеличении общего
солесодержания кривые будут
смещаться влево. Повышение
давления вызывает смещение равновесия вправо. Увеличение тем-
пературы способствует увеличению констант диссоциации угольной
кислоты, а это приводит к смещению кривых вправо.
Угольная кислота в форме карбонат-ионов называется связан-
ной. Принято считать, что гидрокарбонаты состоят наполовину из
связанной и полусвязанной угольной кислоты, так как при разложе-
нии гидрокарбонаты дают карбонаты (связанную) и свободную
угольную кислоту (полусвязанную): 2НСОГ^СО2 + СОз~ + Н2О.
В воде, содержащей СО2Своб и гидрокарбонаты, количество связан-
ной угольной кислоты равно содержанию полусвязанной. Содержа-
ние полусвязанной угольной кислоты используется для вычисления
концентрации агрессивной угольной кислоты в воде. Если в воде
одновременно присутствуют свободная угольная кислота и гидро-
карбонаты, то в состоянии равновесия определенному содержанию
гидрокарбонат-ионов соответствует вполне определенное количест-
во свободной угольной кислоты, которую называют равновесной
угольной кислотой. Если в воде содержание свободной угольной
кислоты будет больше, чем соответствующее состоянию равнове-
сия, т. е. Са2++2НСО7<СО2 + СаСОз + Н2О, то согласно принципу
Ле Шателье часть свободной угольной кислоты будет взаимодейст-
вовать с карбонатом кальция и переводить его в более раствори-
мый гидрокарбонат. Избыток свободной угольной кислоты над рав-
новесной, растворяющий карбонат кальция, называется агрессив-
ной угольной кислотой. Часть избыточной свободной угольной кис-
лоты находится в равновесии с гидрокарбонатами, образующимися
при растворении карбонатов. Поэтому в действительности концент-
рация агрессивной угольной кислоты несколько меньше, чем общее
избыточное содержание свободной угольной кислоты над равновес-
ной.
Когда содержание в воде свободной угольной кислоты меньше
равновесного с гидрокарбонатами:
Са2+ + 2НСО“ >СО2 + СаСО3 + Н2О.
100
то равновесие смещается вправо, гидрокарбонат-ионы раз-
рушаются с образованием свободной угольной кислоты и карбокат-
ионов. Избыток карбонат-ионов взаимодействует с ионами Са2+, со-
держащимися в воде, с образованием труднорастворимого карбо-
ната кальция.
Содержание равновесной угольной кислоты [СО2]Р в состоянии
равновесия рассчитывается по формуле
[СО* = --- Г2 [НСО3-]2 V"2 [Са2+], (Ш.6)
^lnPCaCO3
где у' и у"— коэффициенты активности соответственно ионов НСС~
и Са2+; Ki и К2 — константы диссоциации угольной кислоты по 1-й
и 2-й ступеням. После логарифмирования выражения и подсчета
1gу по формуле Дебая — Хюккеля уравнение (III.6) принимает
вид
Ig [СО2]Р = pKi - рК2 + рПРСаСОз + 21g [НСС/] + 1g [Са2+] -3 /7-5,96,
(Ш.7)
где рЛь рК2 — отрицательные логарифмы констант диссоциации
угольной кислоты по 1-й и 2-й ступеням; [СО2]Р — концентрация
равновесной угольной кислоты, мг/л; рПРсаС03 — отрицательный
логарифм произведения растворимости карбоната кальция;
[НСОГ] — концентрация гидрокарбонат-ионов, мг-экв/л; [Са2+] —
концентрация ионов Са2+, мг/л; 7 — ионная сила раствора.
Для определения различных форм угольной кислоты пользуют-
ся таблицами или номограммами, построенными на основании при-
веденных формул.
Из уравнения основного карбонатного равновесия следует, что
воды с одинаковым содержанием свободной угольной кислоты, но
с различной концентрацией гидрокарбонатов будут проявлять не-
одинаковую активность по отношению к карбонату кальция. Поэто-
му маломинерализованные воды вследствие избытка свободной
угольной кислоты могут проявлять агрессивные свойства по отно-
шению к бетону. Если концентрация гидрокарбонатов больше, чем
это необходимо для осуществления равновесия со свободной уголь-
ной кислотой, т. е. вода имеет повышенную щелочность, то карбо-
натное равновесие снова становится неустойчивым, но уже из-за
избытка гидрокарбонат-ионов. Нарушением основного карбонатного
равновесия объясняется и образование карбонатной накипи при
нагревании воды. С повышением температуры растворимость диок-
сида углерода в воде уменьшается и поэтому наблюдается смеще-
ние карбонатного равновесия в направлении образования свобод-
ной угольной кислоты и карбонат-ионов при разложении гидрокар-
бонатов. С ионами Са2+ карбонат-ионы образуют карбонат каль-
ция, растворимость которого понижается с повышением темпера-
туры.
101
§ 24. Стабильность воды
Воды, в которых соблюдается основное карбонатное равнове-
сие, называются стабильными. Они не изменяют своего состава при
контакте с карбонатами, бетоном, карбонатными защитными плен-
ками. Воды, содержащие избыток свободной угольной кислоты над
равновесной, называются агрессивными. При контакте с бетоном
пли карбонатными пленками такие воды вызывают растворение
карбонатных составляющих. Агрессивное действие этих вод выра-
жается в растворении карбоната кальция и извести по уравнениям:
СаСО3 + СО2 + Н2О Са (НСО3)2
Са (ОН)2 + 2СО2 ^2 Са (НСО3)2
Воды, в которых имеется избыточное (над равновесным) содер-
жание гидрокарбонатов, т. е. повышенная щелочность, называются
нестабильными. Поскольку такие воды не насыщены свободной
угольной кислотой, карбонатное равновесие в таких водах смеща-
ется в сторону разложения гидрокарбонатов. При наличии ионов
Са2+, Mg2+ в нестабильной воде наблюдается отложение карбона-
тов на стенках аппаратуры.
Для оценки агрессивности свойств воды сопоставляют количе-
ство свободной угольной кислоты, установленное анализом, с вы-
численным по формуле (Ш.6). Определение агрессивной угольной
кислоты в воде проводится сопоставлением щелочности воды до и
после контакта ее с карбонатом кальция. При наличии в воде аг-
рессивной угольной кислоты происходит взаимодействие се с кар-
бонатом кальция, в результате чего образуется гидрокарбонат, что
повышает щелочность воды. Некоторая часть избыточной свобод-
ной угольной кислоты не является агрессивной, так как она нахо-
дится в равновесии с гидрокарбонатами, которые образуются при
растворении карбоната кальция. Так, при взаимодействии 1 мг сво-
бодной СО2 с карбонатом кальция в воду поступает 0,91 мг Са2+
и 2,77 мг НСО~. Количество агрессивной угольной кислоты можно
вычислить также по таблице исходя из содержания свободной и
полусвязанной угольной кислоты * и номограммам, составленным
по данным химического анализа воды **.
Стабильность воды оценивают по методу Ланжелье, который ос-
нован на том, что данному значению pH соответствует определен-
ное количество свободной угольной кислоты, находящейся в равно-
весии с другими ее формами. Величина pH, соответствующая рав-
новесию, называется «pH равновесного насыщения воды карбона-
том кальция» и обозначается pHs. При этом pHs вычисляют по фор-
муле
pHs = рК2- рПРсаСОз - tCa2+] - 1g Що + 2,5 /7 + 7,6, [(III.8)
где рК2 — отрицательный логарифм константы 2-й ступени диссо-
циации угольной кислоты; рПРСасо3 “ отрицательный логарифм
* П. Р. Таубе, А. Г. Баранова. Практикум по химии воды. М., 1971.
** В. А. Кляч ко, И. Э. А п е л ь ц и н. Очистка природных вод. М., 1971.
102
произведения растворимости; [Са2+]— концентрация ионов Са2+,
мг/л; Що — общая щелочность, мг-экв/л; / — ионная сила.
Характеристика свойств воды производится по «индексу насы-
щения», или «индексу Ланжелье» J, который представляет собой
разность между pH исследуемой воды и pH воды, насыщенной кар-
бонатом кальция рН4:
J = рНиссл — рН5. (HI 9)
Если J имеет положительное значение /( + ), то, следовательно,
рНИССл>рНз. При этом вода будет нестабильной. При отрицатель-
ном значении индекса Ланжелье /(—) имеется соотношение
рНИсел<рН5, т. е. вода имеет повышенную кислотность и является
агрессивной. Стабильные воды имеют соотношение рНпссл = рН.ч, и
индекс Ланжелье для них равен нулю. Практически воды счита-
ются стабильными при J— ±0,25-?0,3. Стабильность воды харак-
теризуется также показателем стабильности С (основным СОсп и
вспомогательным Свсп). Основной показатель стабильности пред-
ставляет собой отношение щелочности исследуемой воды (Щиссл)
к щелочности воды, насыщенной карбонатом кальция (Щнас):
Сосн = Шиссл/Шнас • (III. 10)
Вспомогательный показатель стабильности есть отношение pH
исходной воды (рНиссл) к pH воды, насыщенной карбонатом каль-
ция (рНнас):
Свсн — рНИссл/рНнас • (III.11)
Если С = 1, т. е. рНиссл —рНнас, то вода стабильна. При С>1 вода
нестабильная, при С<1 вода агрессивная.
Стабилизация воды, содержащей агрессивную угольную кисло-
ту, производится веществами, вызывающими повышение щелочно-
сти воды. В качестве реагентов применяются известь, гидроксид и
карбонат натрия. Реакции, протекающие при этом, описываются
уравнениями:
NaOH + СО2 = NaHCO3; Са (ОН)2 + 2СО2 = Са (НСО3)2
Na2CO3 + СО2 + Н2О = 2NaHCO3
Агрессивную воду можно сделать стабильной также путем фильт-
рования ее через слой известняка, мрамора, природного и полу-
обожженного доломита («магномассы»). Процесс обработки воды
карбонатом кальция описывается уравнением (Ш.9). При фильт-
ровании воды через слой магномассы дополнительно проходит ре-
акция взаимодействия оксида магния с угольной кислотой:
MgO 4* 2СО2 -f- Н2О — Mg (НСО3)2
Обработка нестабильных вод направлена на снижение щелочности
и заключается в обработке их кислотами (соляной, серной) или
в насыщении диоксидом углерода (рекарбонизация). Так, напри-
мер,
NaHCO3 + HCI = NaCl 4- Н2О 4- СО2
103
ria некоторых очистных сооружениях используются мраморно-
песчаные фильтры, которые позволяют получить не только освет-
ленную, но и стабильную воду.
§ 25. Коррозия железа в воде
Под коррозией металлов понимают их разрушение при взаимо-
действии с окружающей средой. В зависимости от механизма это-
го взаимодействия различают химическую, электрохимическую и
биологическую коррозии.
Химическая коррозия железа наблюдается при непосредствен-
ном контакте с кислотами и щелочами, а также с такими агрессив-
ными компонентами среды, как газы, перегретый пар, органические
растворители. В основе коррозии лежат окислительно-восстаиови-
Рис. 12. Схема коррозии
железа в воде:
1 — анодный участок; 2— про-
дукты коррозии; 3 — катодный
участок
тельные реакции с компонентом среды, сопровождающиеся образо-
ванием новых соединений, способствующих необратимому разруше-
нию металла. Многие металлы, особенно при повышенной темпера-
туре, легко окисляются кислородом. Так, при окислении железа в
зависимости от температуры могут образоваться оксиды состава:
FeO, Fe2O3, Fe3C>4. Скорость окисления железа кислородом повыша-
ется в присутствии паров воды и кислот, галогенов, диоксида серы,
диоксида углерода.
Более распространенной является электрохимическая коррозия,
проявляющаяся при контакте металлов с растворами электроли-
тов или влажным паром. Механизм этой коррозии заключается в
образовании на поверхности раздела фаз микро- и макрогальвани-
ческих элементов. В процессе работы таких элементов металл вы-
полняет, как правило, роль анода, т. е. подвергается разрушению.
Катодные участки могут быть представлены оксидами, включения-
ми шлака, сульфидов, сплавов, заклепками, сварными швами и др.
Электродный потенциал этих включений обычно выше, чем у чисто-
го железа. Поэтому в образующейся гальванической паре происхо-
дит окисление железа: Fe°—2e->Fe2+.
На рис. 12 приведена схема коррозии железа в чистой воде, воз-
никающей вследствие неравномерного снабжения поверхности же-
леза кислородом. При контакте капли воды с поверхностью желе-
за краевые участки капли быстрее насыщаются кислородом, что
способствует образованию пленки оксида железа (II), а внутри
капли из-за недостатка кислорода поверхность металла остается
чистой. Возникновение на поверхности металла участков с различ-
104
ным электродным потенциалом приводит к образованию гальвани-
ческой пары, в которой железо будет анодом, а оксидный участок —
катодом. В процессе работы гальванического элемента железо бу-
дет растворяться, посылая в воду ионы железа Fe2+. На анодном
участке оказываются в избыточном количестве электроны, которые
переходят на катодные участки, восстанавливая растворенный в во-
де кислород до гидроксид-иона (кислородная деполяризация):
О2 + 4е + 2Н2О-^4ОН-. При работе гальванического элемента у по-
верхности катодных участков в воде происходит повышение кон-
центрации гидроксид-ионов. Вследствие диффузии катионов метал-
ла и гидроксид-иона внутри капли между катодным и анодным
участками идет образование гидроксида железа (II): Fe2+ + 2OH_ =
= Fe(OH)2. Под действием растворенного в воде кислорода проис-
ходит окисление этого соединения до гидроксида железа (III):
4Fe(OH)2 + O2 + 2H2O = 4Fe(OH)3, который образует рыхлый оса-
док. При коррозии железа во влажном воздухе конечными продук-
тами являются а- и у-модификации оксид-гидроксида железа
FeO(OH)2 и Fe3O4.
Скорость коррозии определяется такими факторами, как одно-
родность металла, соотношение эффективных электродных потен-
циалов анодного и катодного участков, температура, pH, концент-
рация раствора электролита и растворенных газов; присутствие
окислителей. Усилению коррозии железа способствуют также на-
личие веществ, растворяющих продукты коррозии, перемешивание
раствора, попеременное смачивание и высушивание поверхности
металла.
При использовании в технологических процессах стабильной во-
ды на внутренних поверхностях железных труб образуется защит-
ная пленка. Для образования защитного слоя необходимы опреде-
ленные условия: наличие в воде растворенного кислорода, ионов
Са2+, гидрокарбонатов, слабощелочная реакция среды, отсутствие
агрессивной угольной кислоты и малая концентрация хлорид-ионов.
Образование защитной пленки сопровождается процессами, ана-
логичными процессу коррозии железа в чистой воде, с той лишь
разницей, что в воде присутствуют ионы, образующие труднораст-
воримые соединения с продуктами коррозии. Вследствие повышения
щелочности воды при работе гальванического элемента происходит
нарушение равновесия между ионами в растворе. Гидрокарбонат
кальция под действием гидроксид-ионов, образующихся на катоде,
переходит в труднорастворимый карбонат кальция. Ионы Fe2+, об-
разующиеся на анодном участке, под действием карбонат- и гидрок-
сид-ионов образуют труднорастворимый основной карбонат желе-
за (FeOH)2CO3. Эти соединения осаждаются на поверхности метал-
ла и при благоприятных условиях образуют плотный защитный
слой. С образованием защитного слоя электрохимические процессы
прекращаются, так как поверхность металла оказывается покрытой
плотным слоем труднорастворимых соединений.
В кислой среде на катоде восстанавливаются ионы Н+ до сво-
бодного водорода (водородная деполяризация): 2Н+ + 2е—>-Н2. По-
105
нижение pH вызывает разрушение защитной пленки, которая устой-
чива в слабощелочной среде. Повышение температуры снижает pH,
и коррозия железа усиливается. При наличии в воде растворенного
кислорода и свободной угольной кислоты коррозионный процесс
идет одновременно с кислородной и водородной деполяризацией.
Усилению коррозионных процессов способствуют ионы С1~, которые
имеют малый потенциал разряда. Присутствие ионов СН способст-
вует и образованию растворимых соединений металлов. Этим, в
частности, объясняется значительная интенсификация коррозии ме-
таллов в морской воде.
Для уменьшения коррозии паровых котлов высокого давления
применяют обессоленную воду, не содержащую кислорода и угле-
кислого газа. Обескислороживание воды производится термическим,
химическим и десорбционным методами. Для предупреждения угле-
кислотной коррозии стенок котлов в воду вводят аммиак или орга-
нические амины. Свободная угольная кислота связывается аммиа-
ком в карбонат аммония: 2УН4ОН + СО2= (NH4)2CO3 + H2O. Под-
держание слабощелочной реакции воды для питания котлов дости-
гается подщелачиванием.
§ 26. Коррозия бетона в воде
По отношению к бетону могут проявлять агрессивность различ-
ные виды природных и сточных вод. Существуют три вида разру-
шения бетона: 1) растворение и выщелачивание составных частей
бетона — обменные реакции, сопровождающиеся образованием рас-
творимых соединений; 2) реакции обмена, в результате которых
образуются в бетоне рыхлые осадки, не проявляющие вяжущих
свойств, способные вымываться во внешнюю среду; 3) реакции, при
которых образуются труднорастворимые соединения, объем кото-
рых превышает объем составных частей бетона. Классификация эта
условна. На практике встречаются виды коррозии, в которых про-
являются признаки, характерные как для первого, так и для второ-
го вида коррозии. Скорость процесса коррозии и его интенсивность
зависят как от состава и качества бетона, так и характеристики
внешней среды (химический состав, солесодержание воды, коэф-
фициент фильтрации ее через грунт, условия омывания бетона во-
дой и др.). В зависимости от химической природы коррозионного
агента различают следующие виды коррозии бетона в воде: 1) кис-
лотную; 2) углекислотную; 3) выщелачивающую; 4) магнезиаль-
ную; 5) сульфатную.
Кислотная коррозия. Составляющие цементного камня имеют
основной характер, водная вытяжка его имеет щелочную реакцию,
обусловленную гидроксидом кальция. Поэтому наиболее интенсив-
ное разрушение бетона идет при низких значениях pH воды. Гид-
роксид кальция сравнительно хорошо растворим в воде. Раствори-
мость его в дистиллированной воде при 18° С составляет 1,3 г/л.
Гидроксид-ионы, образующиеся при диссоциации, нейтрализуются
ионами Н+, что способствует его дальнейшему растворению. В при-
106
сутствии посторонних ионов, например хлорид-ионов, раствори-
мость гидроксида кальция увеличивается, что способствует более
интенсивному протеканию коррозии бетона. Сильные кислоты взаи-
модействуют с труднорастворимыми соединениями, входящими в
состав бетона (гидросиликатами, карбонатом кальция). В зависимо-
сти от растворимости образующихся в результате реакции нейтра-
лизации солей в одних случаях будет наблюдаться быстрое разру-
шение бетона, в других — медленное. Возможно и уплотнение бето-
на за счет образования труднорастворимых осадков, например фос-
фатов кальция.
Повышение кислотности воды может быть связано с поступле-
нием свободных кислот со сточными водами, растворением газов,
образующих с водой кислоты. К числу таких относятся диоксид се-
ры, сероводород, хлористый водород. Содержание в воде гидроли-
зующихся солей типа сульфатов железа, нитрата аммония также
может вызывать коррозию бетона. Величина pH воды, контактиру-
ющей с бетоном, в зависимости от конкретных условий не должна
быть ниже 5—6,3. При более кислой среде применяют кислотостой-
кие бетоны или защитные покрытия.
Углекислотная коррозия. Этот вид коррозии проявляется при на-
рушении карбонатного равновесия ввиду наличия агрессивной
угольной кислоты в воде. Равновесная угольная кислота не явля-
ется агрессивной по отношению к бетону. Она наряду с нонами
НСО - способствует образованию защитной пленки на поверхно-
сти бетона. Агрессивная угольная кислота разрушает этот защит-
ный слой. Определяющими факторами протекания углекислотной
коррозии являются концентрация СО2 в воде, условия омывания
бетона водой, плотность бетона. Бетоны, имеющие высокую плот-
ность, более устойчивы к углекислотной коррозии. Предельно до-
пустимые концентрации агрессивной угольной кислоты в воде, кон-
тактирующей с бетоном, определяются в зависимости от общего
солесодержания воды, концентрации гидрокарбонатов, условий про-
текания коррозии и составляют с учетом действующих факторов от
3 до 8,3 мг/л.
Выщелачивающая коррозия. В мягких маломинерализованных
водах содержание гидрокарбопатон незначительно и количество
свободной угольной кислоты, соответствующее равновесию с СО?
атмосферы при данной температуре, может оказаться большим, чем
это необходимо для карбонатного равновесия. Поэтому при кон-
такте с бетоном таких вод возможно растворение карбонатной
пленки. Крайне незначительная концентрация ионов Са2+ в воде
способствует растворению извести из цементного камня.
Жесткие воды обладают меньшей способностью к выщелачива-
нию ввиду значительного содержания ионов Са2ц Ионы Mg2+ при
незначительном содержании связывают гидроксид-ионы в трудно-
растворимый гидроксид магния. Мягкие воды, содержащие значи-
тельное количество гидрокарбонат-ионов, способствуют растворе-
нию карбоната кальция, если содержание свободной угольной кис-
лоты будет превышать равновесное. Минимальное содержание гид-
107
рокарбонат-ионов в воде в зависимости от условий составляет 0,4—
1,5 мг-экв/л.
Сульфатная коррозия. При значительном содержании ионов
SCV в воде происходит коррозия бетона, проявляющаяся в обра-
зовании новых соединений, обладающих значительным объемом и
способствующих образованию трещин. Чаще всего таким соедине-
нием является гидросульфоалюминат кальция («бетонная бацил-
ла»): 3CaO-Al2O3-3CaSO4-31H2O. Процесс образования гидросуль-
фоалюмината кальция при взаимодействии сульфат-ионов с четы-
рехкальциевым гидроалюминатом описывается уравнением
4СаО-А12О3- 12Н2О + 20Н2О щ 3CaSO4 -
= 3CaO-Al2O3-3CaSO4-31 Н2О + Ca (ОН)2
При содержании ионаЗО4~> 1000 мг/л выпадает CaSO4-2H2O,
так как при этом достигается его произведение растворимости в на-
сыщенном растворе Са(ОН)2. В присутствии хлоридов более
1000 мг/л сульфатная коррозия замедляется, так как растворимость
образующегося двуводного сульфата и гидросульфоалюмината
кальция увеличивается. Когда концентрация хлоридов достигает
5 г/л, коррозия полностью прекращается. Повышение стойкости бе-
тона к сульфатной коррозии достигается и уменьшением (до 5%) в
составе цемента трехкальциевого алюмината. Из-за возможности
сульфатной коррозии содержание сульфат-иона в воде для затво-
рения бетонных смесей не должно превышать 2,7 г/л.
При повышенном содержании магния в контактирующей воде
сульфатная коррозия бетона усиливается. Сульфат магния взаимо-
действует с известью с образованием рыхлого осадка гидроксида
магния и двуводного сульфата кальция по уравнению
MgSO4 + Ca(OH)2 + 2H2O = Mg(OH)2 + CaSO4-2H2O
Этот комбинированный вид коррозии бетона проявляется в морской
воде.
Магнезиальная коррозия. При содержании ионов Mg2+ в воде
более 750 мг/л наблюдается коррозия бетона, сущность которой за-
ключается в протекании реакций обмена, сопровождающихся заме-
щением ионов Са2+ в бетоне ионами Mg2+ из воды. Поскольку гид-
роксид магния имеет меньшую растворимость, чем гидроксид каль-
ция, то в порах бетона происходит образование рыхлого осадка
гидроксида магния. Снижение щелочности среды вследствие связы-
вания гидроксид-ионов создает условия для растворения гидрокси-
да магния, устойчивого при высоких значениях pH.
Сульфат магния интенсифицирует процесс сульфатной коррозии.
При высокой концентрации ионов Mg2+ и длительном контакте бе-
тона с водой возможно растворение и других составных частей бе-
тона.
Условия для возникновения коррозии бетона в воде создаются
при спуске сточных вод и смыве удобрений в поверхностные водое-
мы, при фильтровании вод, содержащих агрессивные химические
вещества через слой почвы. Химические вещества, активные по от-
108
ношению к бетону, могут содержаться также в жидкой фазе отхо-
дов животноводческих комплексов. Они могут образоваться и при
силосовании кормов с химическими добавками. Для предохранения
бетонных сооружений от воздействия воды, обладающей агрессив-
ными свойствами, используются такие мероприятия, как устройство
засыпки, содержащей карбонатные породы, отвод воды от соору-
жений и создание гидроизоляционных слоев из битумов или поли-
мерных пленок.
§ 27. Физико-химические основы удаления взвешенных
и коллоидных примесей
Сорбционные процессы. При измельчении (диспергировании) ве-
ществ происходит значительное увеличение поверхности при посто-
янном суммарном объеме всех частиц. Работа, затрачиваемая на
преодоление притяжения между частицами, переходит в энергию
поверхностного слоя, или поверхностную энергию. Чем выше сте-
пень измельчения, тем больше поверхностная энергия.
Согласно принципу минимума энергии система тел находится в
устойчивом равновесии тогда, когда свободная энергия системы
имеет наименьшую величину. Поэтому поверхностная энергия час-
тиц высокой степени дисперсности имеет тенденцию к уменьшению.
Это может быть достигнуто укрупнением частиц, связанным с
уменьшением общей поверхности или путем концентрирования на
поверхности частицы (молекул или ионов) другого вещества. Про-
цесс концентрирования растворенного или парообразного вещества
(газа) на поверхности твердого вещества или жидкости называет-
ся сорбцией. Вещество, на котором идет сорбция, называется сор-
бентом, а сорбируемое вещество — сорбатом. Сорбция, происходя-
щая на поверхности вещества, называется адсорбцией, а сопровож-
дающаяся проникновением поглощаемого вещества внутрь погло-
тителя — абсорбцией. При этом поглощенное вещество называется
сорбтивом.
Сорбция, сопровождающаяся химическим взаимодействием меж-
ду сорбентом и сорбатом, называется химической в отличие от фи-
зической сорбции, основанной на силах межмолекулярного взаимо-
действия. Химическая сорбция может ограничиваться только по-
верхностью сорбента или протекать во всем объеме вещества. Про-
цесс химической сорбции, обусловленный химическим взаимодей-
ствием, может быть необратимым, в то время как при физической
сорбции возможно отделение сорбированного вещества от сорбен-
та (десорбция). Сорбция связана с уменьшением поверхностной
энергии и поэтому протекает самопроизвольно и с выделением теп-
лоты.
С повышением температуры физическая сорбция уменьшается,
а химическая сорбция часто при этом увеличивается, но до опреде-
ленного предела. С увеличением поверхности количество сорбиро-
ванного вещества возрастает, что обусловлено возрастанием по-
верхностной энергии. Сорбция может происходить на поверхности
109
раздела между газом и твердым веществом, газом и жидкостью.,
жидкостью и твердым веществом или между двумя несмсшлваю-
щимися жидкостями.
При наличии в сорбенте групп, способных к ионному обмену,
происходит взаимодействие между ионами электролита и сорбен-
том. В результате ионы электролита сорбируются на поверхности
сорбента, а в раствор вместо них выходят ионы самого сорбента.
Так как в основе этой сорбции лежит химическая реакция ионного
обмена, то сорбция такого типа называется ионообменной сорб-
цией.
Адсорбция является обратимым процессом и в состоянии равно-
весия между количеством адсорбированного вещества и равновес-
ной его концентрацией в водном растворе устанавливается зависи-
мость, описываемая при постоянной температуре уравнением.
Фрейндлиха:
(Ш.12)
где А — адсорбция на единицу объема или массы сорбента; С —
равновесная концентрация вещества в воде; и ц — эмпирические,
константы, зависящие от температуры.
Графически эта зависимость выражается изотермой адсорбции.
Например, при сорбции фенолов активированным углем марки
КАД-иодный при концентрации фенола в воде до 0,5 мг/л и 25° С
уравнение (III.12) принимает вид А = 15,85С0’22, где А —количест-
во фенолов, сорбированных углем; С — равновесная концентра-
ция фенолов в воде, г/л.
При больших концентрациях примесей процесс сорбции описыва-
ется уравнением Лэнгмюра:
А^А^+C/iC + b), (Ш.13)
где А — количество сорбированного вещества; Аоо — предельная ад-
сорбция; С — равновесная концентрация вещества; b—постоянная,
равная той концентрации, при которой сорбция равна половине пре-
дельной сорбции. Так, для сорбции фенолов активированным углем
КАД-иодный при концентрации их в воде более 0,5 мг/л уравнение
Лэнгмюра имеет следующее выражение:
А = 18,5 + С/(0,16+С).
Способность к адсорбционному взаимодействию может быть оха-
рактеризована уменьшением свободной энергии AF при адсорбции
вещества из бесконечно разбавленных растворов. Чем больше А/7,
тем избирательнее сорбируется вещество данным сорбентом. Умень-
шение свободной энергии можно вычислить по формуле
ДД = InKa^RT, (П1.14)
где R— универсальная газовая постоянная; Т — абсолютная тем-
пература, К; Каде — константа адсорбции. Константа адсорбции
представляет собой отношение концентраций вещества в фазе сор-
бента Саде и в воде Сравн в состоянии адсорбциопного равновесия;
Уадс “ б?адс/С’равн- (III.15)
110
На адсорбционных процессах основана очистка природных и
-сточных вод. Они лежат в основе осветления и обесцвечивания во-
ды, играют большую роль в процессах самоочищения водоемов и
почвы, при биологической очистке сточных вод.
Коллоидные системы. Дисперсные системы с размером частиц
дисперсной фазы от 1 до 100 нм (10-94-10~7 м) называются кол-
лоидными. По размеру частиц они занимают промежуточное поло-
жение между грубодисперсными системами и истинными раство-
рами. Коллоидные системы являются ультрамикрогстерогенными
системами. Для них характерно наличие высокоразвитой межфаз-
ной поверхности, что в свою очередь обусловливает большой запас
свободной поверхностной энергии. Это способствует тому, что колло-
идные системы являются термодинамически неустойчивыми. В них
сильно выражено стремление к уменьшению запаса свободной энер-
гии. Реализация его возможна при уменьшении дисперсности час-
тиц в результате их укрупнения или при адсорбции на их поверхно-
сти ионов или молекул, находящихся в дисперсионной среде. Осо-
бые свойства коллоидных систем обусловлены размером частиц.
Коллоидные частицы настолько малы, что не задерживаются обыч-
ными фильтровальными материалами, не видны в обычный микро-
скоп, не оседают под действием силы тяжести. Устойчивость колло-
идных растворов со временем снижается, т. е. они подвержены
«старению».
Коллоидные растворы называют золями. В зависимости от аг-
регатного состояния дисперсионной среды выделяют аэрозоли (дис-
персионная среда — газ), гидрозоли (дисперсионная среда — вода),
органозоли (дисперсионная среда — органическая жидкость). Кол-
лоидные системы могут иметь твердую дисперсионную среду, в ко-
торых роль дисперсной фазы выполняют вещества, находящиеся в
твердом (сплавы, минералы), жидком (почвы, адсорбенты) или га-
зообразном состоянии (активированные угли, пористые материалы,
силикагель). Насколько велика межфазная поверхность в коллоид-
ных системах, можно видеть из такого примера: удельная площадь
поверхности активированных углей и силикагеля составляет
8-Ю6 и 4-106 см2/г соответственно. Частицы коллоидных систем со-
держат от 103 до 109 атомов.
По степени интенсивности взаимодействия коллоидных частиц
с дисперсионной средой различают лиофильные и лиофобные си-
стемы. Если дисперсионной средой является вода, то они называ-
ются гидрофильными и гидрофобными. В лиофильных системах
наблюдается значительное взаимодействие между дисперсионной
средой и дисперсной фазой, сопровождающееся образованием соль-
ватных (гидратных) слоев. В лиофобных системах это взаимодей-
ствие проявляется незначительно.
В зависимости от строения частиц дисперсной фазы различают
типичные коллоидные системы (суспензоиды) и растворы высоко-
молекулярных соединений (молекулярные коллоиды). Коллоидные
частицы суспензоидов состоят из агрегатов молекул не раствори-
мых в данной среде соединений. Такие системы являются гетероген-
111
ными, многофазными, так как они имеют поверхность раздела. Рас-
творы высокомолекулярных соединений (ВМС) имеют молекуляр-
ную степень дисперсности, это — устойчивые однофазные системы,
но размер макромолекул настолько велик, что становится соизмери-
мым с размером частиц типичных коллоидных растворов. Поэтому
растворы ВМС обладают рядом свойств, присущих коллоидным сис-
темам. Но в отличие от них молекулярные коллоиды образуют
устойчивые, обратимые растворы. Для них характерно наличие
сильного взаимодействия между макромолекулами растворенного
полимера и растворителем за счет образования сольватных (гидрат-
ных) слоев.
Частицы коллоидной степени дисперсности могут быть получены
измельчением твердых и жидких веществ (диспергационные мето-
ды) или объединением молекул или ионов в агрегаты (конденсаци-
онные методы). Для процессов очистки воды имеют значение глав-
ным образом конденсационные методы. Однако при формировании
состава природных вод коллоидные частицы могут образовываться
в результате самопроизвольного диспергирования контактирующих
с водой осадочных пород (алюмосиликатов). Малая концентрация
растворенных веществ, наличие органических высокомолекулярных
соединений и других примесей, экранирующих растущие кристал-
лы, способствуют образованию частиц коллоидной степени дисперс-
ности труднорастворимых соединений (гидроксидов, сульфидов тя-
желых металлов и др.), которые образуются в результате химиче-
ских реакций.
Свойства коллоидных систем. Молекулярно-кинетические свой-
ства и закономерности, описывающие законы самопроизвольного
движения молекул в растворах, могут быть использованы и для
описания свойств коллоидных систем (диффузии, осмоса). Но при
этом следует учитывать размер коллоидных частиц и их концентра-
цию в дисперсионной среде. Концентрация коллоидного раствора
характеризуется числом частиц в единице объема (частичная кон-
центрация v). Кроме того, используется грамм-частичная концент-
рация Cd, которая представляет собой отношение частичной кон-
центрации к постоянной Авогадро Na’ Cd=vlNA. Частичная (ми-
целлярная) масса коллоидной частицы Md равна произведению мас-
сы коллоидной частицы т на постоянную Авогадро:
Md = mNk. (Ш.16)
Малая грамм-частичная концентрация коллоидного раствора по
сравнению с молекулярными растворами ведет к уменьшению осмо-
тического давления. Закономерности же и механизм осмоса оста-
ются теми же самыми, только в уравнении Вант-Гоффа вместо мо-
лярной концентрации берется частичная Cd, а универсальная газо-
вая постоянная R заменяется константой Больцмана k—RfNA.
Коллоидные частицы диффундируют в дисперсионной среде,
стремясь равномерно распределиться по всему объему системы.
Диффузия обусловлена тепловым (броуновским) движением час-
тиц. Способность к диффузии измеряется коэффициентом диффу-
112
зии, который характеризует количество вещества, переносимое че-
рез площадь 1 см2. Размерность его см2-с-1 или см2-сут-1. Коэффи-
циенты диффузии в гидрозолях чрезвычайно малы. Если молекулы
в растворах проходят 1 см за несколько часов, то коллоидные час-
тицы, имеющие коэффициент диффузии 5-10~9 см2-с-1, — за три го-
да. В коллоидных растворах броуновское движение частиц дополня-
ется перемещением их под действием гравитационного поля. В гру-
бодисперсных системах наблюдается осаждение частиц под дейст-
вием силы тяжести (седиментация). Так как размеры коллоидных
частиц очень малы, то коллоидные системы являются кинетически
устойчивыми, т. е. в них не происходит осаждения дисперсной фа-
зы. Но с течением времени коллоидные частицы определенным об-
разом распределяются в объеме дисперсионной среды в зависимо-
сти от их размера. В системе устанавливается седиментационно-
диффузионное равновесие, описываемое уравнением
In (Ci/C2) = mg (h2 - ht) (p - ?q)/RTp, (III. 17)
где Ci и C2 — концентрации частиц соответственно на высоте и
/г2; т— масса частицы; р и ро — плотности частиц и дисперсионной
среды; Ад — постоянная Авогадро; g — ускорение силы тяжести.
Пользуясь уравнением (III.17), можно вычислить частичную массу
дисперсной фазы.
В зависимости от соотношения плотностей частиц дисперсной
фазы и дисперсионной среды более крупные частицы оказываются
в нижней части сосуда (при р>ро) или концентрируются в верхних
слоях (при р<ро). Потеря кинетической (седиментационной) устой-
чивости сопровождается разрушением коллоидной системы.
Оптические свойства коллоидных систем. Поскольку размер кол-
лоидных частиц меньше длин световых волн видимой части спект-
ра, то поглощенная энергия вновь испускается частицами в раз-
личных направлениях, поляризованные частицы как бы становятся
новыми источниками излучения. Рассеяние света является харак-
терным свойством коллоидных систем и подтверждает их гетероген-
ность. Интенсивность светорассеяния усиливается при наличии
крупных частиц (но с диаметром не более ’/20 длины волны света),
при коротковолновом излучении и при значительном отличии пока-
зателей преломления дисперсной фазы и дисперсионной среды. Кол-
лоидные растворы поглощают монохроматический свет, причем мак-
симум поглощения зависит от размера частиц. С уменьшением их
диаметра этот максимум смещается в коротковолновую часть
спектра.
Устойчивость коллоидных систем. Агрегативная устойчивость
коллоидных систем проявляется в постоянстве состава и сохране-
нии размеров частиц дисперсной фазы. Устойчивость коллоидных
систем характеризуется ионно-электростатическим и адсорбционно-
сольватным факторами. Коллоидные системы имеют высокоразви-
тую поверхность раздела фаз, на которой формируются слои, обла-
дающие особыми свойствами. Ионно-электростатический фактор
устойчивости коллоидных систем обусловлен одноименным зарядом
ИЗ
у коллоидных частиц. Наличие заряда подтверждается электрофо-
резом, который заключается в направленном перемещении частиц
в электрическом поле. Коллоидные растворы могут быть заряжены
положительно (золи гидроксидов металлов) или отрицательно (зо-
ли кремниевой кислоты, гуминовых кислот). Заряд коллоидной час-
тицы суспензоидов может возникать в результате избирательной
адсорбции ионов из раствора, ионизации молекул дисперсной фазы
(например, кремниевой кислоты) под действием молекул дисперси-
онной среды, адсорбции из раствора молекул, способных к диссо-
циации.
Рис. 13. Схема строения двойного электрического слоя (Д) и изменение величин
ф- и ^-потенциалов (/>):
а — потенциалопределяющие ионы; б — адсорбционный слой противоионов; в — диффузный
слой
А
Ионы, адсорбированные на поверхности нейтральной частицы
(ядра), называются потенциалопределяющими. Из ионов электро-
лита, содержащегося в окружающем растворе, адсорбируются те,
которые имеют с поверхностью одинаковую или изоморфную с ней
атомную группу. Так как на частице возникает заряд, то для дости-
жения электронейтральности из раствора к ее заряженной поверх-
ности подходят ионы со знаком заряда, противоположным заряду
потенциалопределяющих ионов (противоионы). Часть из них обра-
зует вместе с потенциалопределяющими ионами плотный адсорбци-
онный слой, расположенный в непосредственной близости от твер-
дой частицы. Остальные противоионы образуют около частицы в
растворе диффузный слой, в котором ионы находятся под действи-
ем двух сил: силы электростатического притяжения ионами проти-
воположного знака, которая удерживает их вблизи частицы, и си-
лы диффузии, стремящейся распределить их равномерно в объеме
дисперсионной среды. Так формируется двойной электрический слой
(рис. 13). Так как часть противоионов (диффузный слой) находит-
114
ся в растворе, то на границе раздела (ядро
с адсорбционным слоем — диффузный
слой) возникает скачок потенциала по срав-
нению с объемом раствора, который на-
зывается электрокинетическим или (дзе-
та)-потенциалом. Знак заряда частицы
определяется знаком заряда потенциал-
определяющих ионов.
Толщина адсорбционного слоя составля-
ет 10”7—10 ч см, а диффузного — порядка
10”4 см. Под воздействием внешних факто-
ров возможно изменение толщины диффуз-
ного слоя и количества ионов, содержащих-
ся в нем.
Таким образом, коллоидная частица име-
ет сложное строение и постоянно находится
в определенном физико-химическом взаи-
модействии с окружающей средой. Схему
строения коллоидной частицы рассмотрим на
Рис. 14. Строение колло-
идной частицы кремние-
вой кислоты
примере кремниевой
кислоты при наличии в окружающем растворе гидросиликата нат-
рия (рис. 14):
{[/иНгБЮз]• nHSiO3 -(п —• х) Na+} • xNa+
ч- ядро ->-ч- адсорбционный слой ->ч- диффузный слой
Ядро частицы составляет агрегат из т молекул кремниевой кис-
лоты. На поверхности ядра адсорбируются потенциалопределяю-
щие гидросиликат-ионы. Часть противоионов натрия (п—х) адсор-
бируется на частице и удерживается электростатическими силами
притяжения потенциалопределяющих ионов. Не полностью компен-
сированный заряд потенциалопределяющих ионов вызывает появ-
ление отрицательного заряда у частицы кремниевой кислоты.
В электрическом поле коллоидная частица вместе с адсорбцион-
ным слоем перемещается к одному полюсу, а ионы диффузного
слоя — к противоположному. Если бы коллоидная частица переме-
щалась в электрическом поле только с потенциалопределяющими
ионами, то электрокинетический потенциал должен быть равен тер-
модинамическому потенциалу фо, возникающему между поверх-
ностью частицы и раствором. Наличие в адсорбционном слое про-
тивоинов снижает величину потенциала, и электрокинетический
потенциал составляет часть полного потенциала. Если граница
диффузного слоя совпадает с границей адсорбционного (х = 0), то
электрокинетический потенциал равен нулю. Такое состояние кол-
лоидной частицы называется изоэлектрической точкой. В изоэлек-
трической точке коллоидные частицы теряют устойчивость, укруп-
няются и выпадают в осадок.
Коллоидные растворы, образованные амфотерными соединения-
ми, например гидроксидом алюминия, могут иметь различный знак
заряда в зависимости от pH. Смена заряда происходит при пере-
ходе через значение pH, соответствующее изоэлектрической точке.
115
На ^-потенциал влияет концентрация электролитов, находящихся
в растворе. При увеличении концентрации раствора ^-потенциал
уменьшается за счет перехода части ионов диффузного слоя в ад-
сорбционный. Величина (ц—х) при этом возрастает и приближа-
ется к п.
Адсорбционно-сольватный фактор агрегативной устойчивости
коллоидных систем связан с образованием на межфазной поверх-
ности слоев из молекул или ионов. Адсорбционно-сольватные слои
обладают особыми свойствами. Они имеют высокую прочность, мо-
лекулы в них взаимодействуют между собой более энергично и по-
этому менее подвижны. В их составе могут быть кристаллические
образования. Формирование таких слоев происходит в результате
определенного взаимодействия между веществом дисперсной фазы
и дисперсионной средой. Определенная ориентация молекул в сло-
ях способствует проявлению особых механических свойств, а имен-
но: повышению вязкости, упругости, сопротивления сдвигу. Разви-
тые адсорбционно-сольватные слои препятствуют сближению кол-
лоидных частиц, т. е. способствуют повышению устойчивости кол-
лоидной системы. Образование прочных адсорбционно-сольватных
слоев характерно для гидроксидов таких металлов, как алюминий,
железо и др. Согласно теории, разработанной П. А. Ребиндером
и его школой, адсорбционно-сольватные слои представляют собой
двумерные квазикристаллические структуры. Адсорбционно-соль-
ватные слои могут образовываться и крупными молекулами орга-
нических веществ. Эти ориентированные слои не разрушаются при
сближении коллоидных частиц, они создают структурно-механиче-
ский барьер, препятствующий их агрегации. Соединения, входящие
в состав адсорбционно-сольватных слоев, называются стабилиза-
торами.
Устойчивость и коагуляция коллоидных систем. Будучи термо-
динамически неустойчивыми, коллоидные системы под воздействи-
ем различных факторов могут разрушаться. Процесс соединения
коллоидных частиц друг с другом, сопровождающийся образова-
нием крупных агрегатов и разрушением коллоидной системы, на-
зывается коагуляцией. Она может происходить при введении в си-
стему электролитов, золей с противоположным знаком заряда.
Коагуляцию можно вызвать и изменением температуры, механиче-
ским воздействием, ионизирующим облучением.
Наибольшее практическое значение имеет коагуляция электро-
литами. Она начинается при определенной концентрации электро-
лита в растворе, которая называется порогом коагуляции. Коагу-
ляция электролитами протекает в соответствии со следующими пра-
вилами: 1) коагулирующее действие оказывают ионы, имеющие
знак заряда, противоположный знаку заряда потенциалопределяю-
щих ионов; 2) действие коагулирующего иона возрастает с повы-
шением заряда. Так, порог коагуляции для однозарядных ионов
составляет 25—150 ммоль/л, двухзарядных — 0,5—2, трехзаряд-
ных— 0,01—0,1 ммоль/л. Коагулирующая способность ионов с оди-
наковым знаком заряда неодинакова, сильнее она выражена у ме-
116
нее гидратированных ионов. По коагулирующей способности ионы
располагаются в лиотропные ряды, например Na+<NH4 <К+<
<M.g2+<Ca2+<Ba2+<Al3-r. На коагулирующую способность катио-
на влияет химическая природа связанного с ним аниона. Так, порог
коагуляции положительно заряженного золя гидроксида алюминия
составляет для хлорида калия 46 ммоль/л, а для сульфата калия—•
0,3 ммоль/л. Электролит, вызывающий коагуляцию коллоидного
раствора, называется коагулянтом, а коагулирующий ион — коагу-
лятором.
Коагулирующее действие ионов при коагуляции смесью элек-
тролитов изменяется неоднозначно. Если ионы имеют одинаковый
заряд и незначительно отличаются друг от друга по размеру и
степени гидратации, их коагулирующее действие суммируется. Ес-
ли ионы-коагуляторы отличаются по величине заряда, то коагу-
лирующая способность иона с меньшим зарядом подавляется по-
лизарядным (антагонизм ионов). Иногда суммарное воздействие
превышает действие каждого из них, взятого в отдельности. Подоб-
ное явление взаимного усиления называется синергизмом. При спе-
цифической адсорбции ионов, химическом взаимодействии иона-
коагулятора с ионами двойного электрического слоя приведенные
закономерности могут не всегда выполняться.
Величина порога коагуляции зависит и от природы противо-
ионов, что свидетельствует о важной роли диффузного слоя в про-
цессе коагуляции. Коагуляция сопровождается уменьшением элек-
трокинетического потенциала, а заряд поверхности может при этом
оставаться неизменным. В зависимости от концентрации коагулян-
та выделяются зоны устойчивости, медленной и быстрой коагуля-
ции. Если его концентрация ниже порога коагуляции, то скорость
коагуляции так мала, что можно считать коллоидный раствор ус-
тойчивым. При повышении концентрации коагулянта выше поро-
говой скорость коагуляции увеличивается (зона медленной коагу-
ляции). После достижения определенной концентрации и при даль-
нейшем ее повышении скорость коагуляции практически не зави-
сит от концентрации введенного электролита (зона быстрой коагу-
ляции) .
Если допустить, что коллоидные частицы имеют сферическую
форму и одинаковые размеры, то скорость быстрой коагуляции опи-
сывается уравнением скорости химической реакции 2-го порядка
(с учетом частичной концентрации). Если частицы имеют различ-
ную форму и размеры (в полидисперсных золях), скорость коагу-
ляции повышается, что связано с неодинаковой скоростью осажде-
ния скоагулировавших частиц. Вероятность столкновения их друг
с другом в таких системах возрастает (ортокинетическая коагуля-
ция) .
При введении в коллоидный раствор ионов А13+, Fe3+, сильно
поляризующих твердую поверхность, наблюдается чередование зон
коагуляции и устойчивости. Зоны устойчивости соответствуют очень
низким и достаточно высоким концентрациям электролита. После
1-й и 2-й зон устойчивости следуют зоны коагуляции. Знак заряда
117
Рис. 15. Зависимость энергии
взаимодействия между колло-
идными частицами от расстоя-
ния между ними
коллоидных частиц при переходе системы из 1-й зоны устойчивости
через зону коагуляции во 2-ю меняется на противоположный. Из-
менение знака заряда может быть обусловлено заменой потенциал-
определяющих ионов или адсорбцией противоионов на заряженной
поверхности. Чередование зон устойчивости и коагуляции в зави-
симости от количества вводимого коагулянта необходимо учитывать
при выборе оптимального количества реагента, необходимого для
обработки воды.
Механизм процесса коагуляции. Устойчивость коллоидных си-
стем зависит от соотношения электростатических сил отталкива-
ния и межмолекулярных сил притяже-
ния, действующих между частицами..
Ван-дер-ваальсовы и лондоновские си-
лы притяжения преобладают на очень
малых расстояниях и больших, когда
частицы удалены друг от друга на рас-
стояние, превышающее двойную тол-
щину диффузного слоя. На средних
расстояниях порядка эффективной тол-
щины ионной атмосферы между части-
цами может преобладать действие
электростатических сил отталкивания.
Этому способствуют достаточно высо-
кий потенциал поверхности и малая
концентрация окружающего раствора.
Если частицы находятся на таком рас-
стоянии, что их диффузные слои час-
тично перекрываются, то между ними возникают силы отталкива-
ния в результате действия одноименных полей. Диффузные слои
противоионов деформируются, происходит перераспределение
ионов в контактирующих слоях. Соединению коллоидных частиц
препятствует наличие потенциального барьера. На рис. 15 приве-
дены кривые потенциальной энергии отталкивания, энергии притя-
жения и объединенная потенциальная кривая, результирующая их
действие в зависимости от расстояния между частицами. Энергия
отталкивания считается положительной, а энергия притяжения —
отрицательной. Если высота потенциального барьера (II) и глуби-
на второго минимума (III) незначительна, частицы сближаются
между собой и коагулируют в результате ближнего взаимодействия
(I). Расстояние это составляет несколько десятых долей наномет-
ра. Агрегативная устойчивость коллоидной системы соответствует
значительной высоте потенциального барьера (II) и малой глубине
второго минимума (III). Особый вид связи между частицами на-
блюдается при достаточно большой глубине второго минимума, при
дальнем взаимодействии (расстояние около 102 нм). При этом час-
тицы образуют пары, тройники или более сложные структуры, в
которых не происходит агрегатирования частиц, т. е. дисперсность
системы не изменяется. В ней наблюдается обратимое равновесие:
золь^агрегат. Подобное состояние системы является относитель-
118
но устойчивым. Если концентрация дисперсной фазы достаточно
велика, то коллоидный раствор переходит в структурированную
систему. Это совместное взаимодействие частиц дисперсной фазы
может привести к образованию периодических коллоидных систем
(ПКС), характеризующихся образованием квазикристаллических
структур. Чем больше заряд поверхности и толщина диффузного
слоя, тем выше потенциальный барьер, который нужно преодолеть
частицам для их взаимодействия. Уменьшение высоты барьера на-
блюдается при снижении величины заряда частицы, вызванной
избирательной сорбцией ионов из раствора или при сжатии диф-
фузного слоя. Оно ведет к преобладанию сил межмолекулярного
притяжения между частицами над силами отталкивания и их
соединению, т. е. к коагуляции коллоидного раствора. Если сниже-
ние потенциального барьера происходит за счет избирательной ад-
сорбции ионов из окружающего раствора, то возникающий при
этом процесс называется нейтрализационной коагуляцией. Она со-
провождается уменьшением заряда поверхности. Если же снижение
величины заряда частицы происходит в результате сжатия диффуз-
ного слоя при повышении концентрации электролита в окружающем
растворе, то коагуляция, вызванная действием этого фактора, на-
зывается концентрационной.
Согласно физической теории устойчивости коллоидных систем
ДЛФО в области перекрывания диффузных слоев коллоидных час-
тиц вследствие перераспределения противоионов между слоями и
окружающим раствором возникают дополнительные неуравнове-
шенные электростатические силы отталкивания. Этому способству-
ет возникновение дополнительного «расклинивающего» давления в
тонком слое жидкости. В зависимости от баланса сил притяжения
и отталкивания расклинивающее давление может быть положи-
тельным, увеличивая действие сил отталкивания, или отрицатель-
ным, при котором наблюдается уменьшение слоя жидкости между
частицами. Жидкость, находящаяся в тонком слое, разделяющем
две твердые поверхности, обладает большей упругостью формы.
Действие расклинивающего давления между частицами обуслов-
лено наличием ионной атмосферы у коллоидной частицы. Чем боль-
ше размыт диффузный слой, тем сильнее проявляется действие
расклинивающего давления, тем выше устойчивость коллоидного
раствора. Е1ри введении электролита изменяется толщина диффуз-
ного слоя и пленки жидкости, разделяющей частицы. После до-
стижения порога коагуляции величина потенциального барьера
снижается настолько, что кинетическая энергия взаимодействую-
щих частиц превышает его и частицы под действием межмолеку-
лярных сил притяжения начинают сближаться, что означает на-
чало процесса коагуляции. В начале процесса коагуляции размер
образующихся агрегатов недостаточно велик и видимых измене-
ний в коллоидном растворе не наблюдается. Это период скрытой
коагуляции. Затем в результате дальнейшего укрупнения частиц
начинается образование хлопьев. При введении в разрушающийся
коллоидный раствор ВМС, имеющих макромолекулы с полярными
119
группами, наблюдается их взаимодействие с агрегатирующими час-
тицами, которое сопровождается образованием рыхлых хлопьев
(флокул). Этот процесс называется флокуляцией, а применяемое
ВМС — флокулянтом. Заканчивается процесс коагуляции седимен-
тацией— осаждением частиц под действием силы тяжести. Обра-
зующиеся осадки (коагуляты) могут быть хлопьевидными, плот-
ными, объемными, содержащими значительное количество воды,
кристаллоподобными.
Коагуляция коллоидного раствора может произойти при введе-
нии в него другого золя, имеющего противоположный заряд. Этот
процесс называется взаимной коагуляцией. Она проходит эффек-
тивно, если частичная концентрация золей примерно одинакова.
Коагуляция одного золя другим обусловлена наличием разноимен-
ного заряда у частиц.
Если коллоидные частицы соединяются непосредственно поверх-
ностями, то коагуляция будет необратимой. Если между соединя-
ющимися частицами остаются небольшие сольватные оболочки пли
двойной электрический слой, образовавшийся осадок можно снова
перевести в раствор. Такой процесс называется обратимой коагу-
ляцией. Растворы высокомолекулярных соединений (ВМС) и не-
которых органических веществ образуют на поверхности коллоид-
ных частиц прочные адсорбционно-сольватные слои, способствую-
щие повышению устойчивости коллоидного раствора, т. е. прояв-
ляют защитное действие. Стабилизирующее действие этих соеди-
нений способствует лиофилизации (гидрофилизации) дисперсных
систем: по свойствам они становятся близкими к молекулярным
коллоидам (обратимы, устойчивы). Если концентрация стабилиза-
тора недостаточна для образования адсорбционно-сольватных сло-
ев, его защитное действие резко снижается. При очистке воды со-
держащиеся в ней органические коллоиды замедляют процесс коа-
гуляции, повышая устойчивость образующихся коллоидных рас-
творов гидроксидов алюминия и железа.
Процесс коагуляции может быть обратимым, т. е. коагулят сно-
ва может переходить в золь. Этот процесс называется пептизацией
или дезагрегацией. Она осуществляется при введении растворов
электролитов или других веществ, которые называются пептизато-
рами. Так, гидроксид железа (III) легко пептизируется раствором
хлорида железа (III). Действие электролита-пептизатора заключа-
ется в том, что один из его ионов адсорбируется на поверхности
частиц осадка и сообщает им заряд, что ведет к увеличению элек-
трокинетического потенциала и расширению гидратной оболочки.
Частицы начинают отталкиваться друг от друга и переходить в рас-
твор. Пептизации подвергаются только те осадки, в которых со-
храняются гидратные оболочки и развивается двойной электриче-
ский слой. Только что полученные осадки, имеющие рыхлую
структуру, пептизируются легче. Таким образом в определенных ус-
ловиях устанавливается коллоидно-химическое равновесие: золь^
^агрегаты, в котором коллоидные частицы рассматриваются как
псевдомолекулы, а коагулят — как псевдокристалл. Часто взаимо-
120
действие между коллоидными частицами сопровождается образова-
нием структурированных систем с особыми свойствами, в которых
частицы имеют высокую степень дисперсности, например ПКС.
По теории П. А. Ребиндера существует два основных типа
структур: коагуляционные и конденсационно-кристаллические.
Коагуляционные структуры образуются в результате сцепления
коллоидных частиц под действием сил межмолекулярного воздей-
ствия в цепочки, трехмерные сетки с образованием рыхлого кар-
каса. Конденсационно-кристаллические структуры возникают в ре-
зультате реакций полимеризации и поликонденсации, при кристал-
лизации из растворов (твердение цемента). Коагуляционные струк-
туры могут быть обратимыми. Конденсационно-кристаллические
необратимы. К коагуляционным структурам относятся гели, обра-
зованные коллоидными частицами или молекулами ВМС (студ-
ни). В гидрогелях частицы дисперсной фазы, сцепляясь друг с дру-
гом, образуют трехмерную сетку, промежутки которой заполнены
водой. Близки по свойствам к гелям осадки, образующиеся при
коагуляции сильно гидратированных золей,— коагели. Их тоже рас-
сматривают как отдельный вид коагуляционных структурирован,
ных систем. В гелях дисперсионная среда неподвижна, они облада-
ют упругостью формы. Гелевую структуру имеют синтетические
ионообменные материалы и ионитовые мембраны.
Обратимость многих коагуляционных структур подтверждается
наличием перехода: золь^гель при механическом воздействии
(тиксотропия). Этим явлением обусловлено разжижение грунтов,
повышение пластичности цементного раствора под действием виб-
рации.
Поверхностно-активные вещества (ПАВ). Особую группу кол-
лоидных электролитов составляют поверхностно-активные вещества.
Это соединения, молекулы которых состоят из длинных углеводо-
родных радикалов (С8—Cis — гидрофобная часть) и небольших
полярных групп (гидрофильная часть). В сильно разбавленных
растворах они находятся в виде молекул или ионов. При увеличе-
нии числа молекул ПАВ в растворе до критической концентрации
мицеллообразования (ККМ) формируются более сложные агрега-
ты (мицеллы) за счет взаимодействия между гидрофобными участ-
ками молекул. В отличие от мицелл типичных коллоидных раство-
ров они образуются за счет взаимодействия между углеводородны-
ми радикалами молекул ПАВ и не содержат твердой фазы. В вод-
ном растворе ПАВ устанавливается равновесие: молекулы
(ионы) ^мицеллы. Поверхность мицелл гидрофилизируется в ре-
зультате ориентации в водную фазу полярных групп молекул ПАВ.
Поверхностная активность этих соединений проявляется в сни-
жении поверхностного натяжения о на границе раздела фаз в ре-
зультате концентрирования молекул ПАВ в поверхностном слое и
образования адсорбционных слоев с определенной ориентацией. На
границе раздела фаз (рис. 16) молекулы ПАВ располагаются так,
что гидрофобная часть их (радикал) обращается в сторону непо-
лярной фазы («масло»), а гидрофильная группа — в полярную (во-
121
да). Различают анионактивные, катионактивные и неионные ПАВ.
Анионактивные ПАВ (соли высших жирных, алкилсерных кислот)
диссоциируют в воде с образованием поверхностно-активного анио-
на. У катионактивных ПАВ поверхностной активностью обладает
катион. Неионные ПАВ в водных растворах находятся в недиссо-
циированном состоянии.
Способность ПАВ снижать поверхностное натяжение на грани-
це фаз и тем самым изменять свойства поверхности широко исполь-
зуется в практике. Они составляют основу моющих средств, при-
Рис. 16. Схема ориентации молекул ПАВ на границе раз-
дела фаз:
а — «масло» в воде; б — вода в «масле»
меняются в текстильной промышленности для обработки тканей,
в качестве стабилизаторов эмульсий, пенообразователей, дисперга-
торов— веществ, интенсифицирующих процесс измельчения.
Мицеллярные растворы ПАВ характеризуются солюбилизацией,
(коллоидным растворением), т. е. самопроизвольным растворени-
ем в присутствии ПАВ, обычно не растворимых в воде соединений,
например углеводородов нефти. Солюбилизация неполярных соеди-
нений происходит в результате внедрения их молекул в углеводо-
родную часть мицеллы. ПАВ, являясь незаменимыми веществами
во многих технологических процессах, в то же время затрудняют
очистку сточных вод, способствуют ухудшению качества воды в во-
доемах.
Суспензии (взвеси). Это грубодисперсные системы типа «твер-
дое вещество — жидкость». Размер частиц дисперсной фазы в них
превышает 100 нм. Дисперсная фаза в суспензиях имеет развитую
поверхность, но меньшую, чем в коллоидных системах. Так как
размеры частиц относительно велики, то суспензии — кинетически
неустойчивые системы, т. е. при отстаивании частицы дисперсной
фазы выпадают в осадок. Скорость осаждения их определяется
свойствами дисперсионной среды (плотность, вязкость) и частиц
дисперсной фазы. Если принять форму частиц за сферическую, то
скорость их осаждения и описывается законом Стокса:
2 г2(р — Po)g
(III.18)
122
где г — радиус частицы; р и р0—плотность частицы и дисперсион-
ной среды соответственно; g —ускорение в гравитационном поле;
т] — вязкость дисперсионной среды. Формула (III.18) позволяет ус-
тановить распределение частиц по размерам в данной системе, из-
меряя скорость их осаждения. Необходимые данные получают при
проведении седиментационного анализа.
Для систем, в которых дисперсионной средой служит вода, се-
диментационная устойчивость суспензий зависит также от степени
гидрофильности частиц дисперсной фазы. Если частицы смачива-
ются водой (гидрофильны), то суспензия будет более устойчивой.
Гидрофобные частицы, не смачивающиеся водой, быстро оседают
(при р>ро) или всплывают (при р<ро)- Седиментационная устой-
чивость суспензий повышается в присутствии стабилизаторов ПАВ.
Вопросы устойчивости и разрушения суспензий играют важную
роль при определении оптимальных условий выделения из воды
грубодиспергированных примесей.
Эмульсии. Дисперсные системы, состоящие из двух или более
взаимно нерастворимых жидкостей, называются эмульсиями. Ча-
ще всего приходится иметь дело с эмульсиями, в состав которых
входит вода и другая неполярная или малополярная жидкость, ус-
ловно обозначаемая термином «масло». Если дисперсной фазой
является масло, а дисперсионной средой — вода («масло в воде» —
м/в), то эмульсия называется прямой. Эмульсии типа «вода в мас-
ле» (в/м) называются обратными. Если концентрация дисперсной
фазы не превышает 0,1%, то эмульсии называются разбавленными.
Дисперсионной средой в них считается жидкость, содержащаяся в
большем количестве. Так как расстояние между каплями в таких
эмульсиях достаточно велико, то вероятность взаимодействия меж-
ду каплями мала. Но в то же время почти каждое столкновение
между ними сопровождается их слиянием, так как распределение
поверхностного заряда происходит диффузно в той и другой жид-
кости, что способствует снижению межфазного потенциала. Про-
цесс слияния капель эмульсии друг с другом называется коалес-
ценцией.
Эмульсии, содержащие от 0,1 до 74% дисперсной фазы, называ-
ются концентрированными, более 74% — высоко концентрирован-
ными. Повышение устойчивости эмульсий достигается введением
веществ, вызывающих их стабилизацию,— эмульгаторов. В качест-
ве эмульгаторов используются ПАВ и некоторые тонкоизмельчен-
ные твердые вещества. Механизм стабилизирующего действия ПАВ
заключается в образовании адсорбционно-сольватных слоев с оп-
ределенной ориентацией молекул (см. рис. 16). Действие твердых
эмульгаторов (например, бентонитовых глин, сажи и др.) заклю-
чается в формировании на межфазной границе слоя по своей струк-
туре, аналогичного коагуляционным образованиям.
ПАВ, в результате значительного снижения поверхностного на-
тяжения на межфазной границе, иногда способствуют не только
стабилизации, но и самопроизвольному эмульгированию неполяр-
ных жидкостей в воде. Они способствуют повышению как агрега-
123
тивной, так и седиментационной устойчивости эмульсий. Это свя-
зано с тем, что они понижают поверхностное натяжение и вызы-
вают увеличение дисперсности.
Эмульсии можно разрушить введением веществ — деэмульгато-
ров. Так, например, при добавлении к эмульсии ПАВ, обладающих
большей поверхностной активностью, чем эмульгатор, но не обра-
зующих защитных слоев, происходит разрушение эмульсии. Одно
и то же ПАВ в зависимости от концентрации может выполнять
для одной и той же эмульсии роль эмульгатора и деэмульгатора.
Разрушение эмульсий, образующихся в сточных водах, может быть
вызвано введением химических реагентов, например сульфата алю-
миния, а также физическими (фильтрование через пористые из-
бирательно смачиваемые материалы) и физико-химическими мето-
дами (коагуляция, флотация).
§ 28. Обработка воды коагулянтами
Природные воды являются полидисперсными системами, т. е. в
них могут содержаться одновременно частицы, размеры которых
соответствуют размерам коллоидных и грубодиспергированных час-
тиц. Освобождение воды от грубодисперсных примесей осущест-
вляется в процессе отстаивания. Коллоидные примеси при отстаи-
вании воды не удаляются.
В зависимости от характера содержащихся в воде диспергиро-
ванных частиц различают мутные и цветные воды. В мутных водах
преобладают коллоидные частицы алюмосиликатов (глинистые),
кремниевой кислоты и органоминеральных комплексов почвы. Кол-
лоидные частицы примесей природных вод, как правило, имеют
отрицательный заряд.
Основу глинистых частиц образуют гидроалюмосиликаты, кото-
рые по химической природе являются алюмокремниевыми кислота-
ми или их солями и диссоциируют с образованием сложного анио-
на и иона водорода или щелочного металла. Возникновение отри-
цательного заряда у коллоидных частиц глин обусловлено именно
данным процессом. Так как алюмокремниевые кислоты имеют ма-
лую степень диссоциации, то при снижении pH устойчивость кол-
лоидных частиц глин уменьшается. И наоборот, при повышении
щелочности воды устойчивость коллоидных частиц возрастает. Это
объясняется тем, что ионы Н+, образующиеся при диссоциации
алюмокремниевых кислот, замещаются катионами металлов, а сте-
пень диссоциации образующихся солей выше, чем соответствую-
щих кислот. В очень жестких водах коллоидные частицы глины мо-
гут иметь положительный заряд.
Гумусовые соединения, содержащиеся в воде, по своим свой-
ствам занимают промежуточное положение между коллоидными
растворами и растворами ВМС. Они более устойчивы к действию
электролитов и обладают защитными свойствами по отношению к
коллоидным растворам гидроксидов металлов. Повышая устойчи-
вость коллоидных гидроксидов, гумусовые вещества затрудняют
124
процесс очистки воды. Гумусовые соединения находятся в коллоид-
ном состоянии только при низких значениях pH. В щелочной среде
они присутствуют в виде растворимых солей.
Метод обработки воды, направленный на удаление веществ, на-
ходящихся в коллоидном состоянии с помощью химических реаген-
тов, называется коагулированием. Применяющиеся для этой цели
химические вещества называются коагулянтами. Коагулирование
воды применяется для осветления мутных и обесцвечивания цвет-
ных вод. Наряду с коллоидными примесями при коагулировании
удаляются из воды грубодисперсные частицы, а также планктон,
бактерии и вирусы. Для очистки воды применяются следующие
коагулянты: сульфат алюминия A12(SO4)3-I8H2O, сульфат желе-
за (II) FeSO4-7H2O (железный купорос), хлорид железа (III)
FeCl3-6H2O, гидроксохлорид алюминия А12(ОН)5С1, метаалюми-
нат натрия NaAlOz.
Коагулянты представляют собой соли, образованные слабыми
основаниями и сильными кислотами, поэтому в воде они подверга-
ются ступенчатому гидролизу. Например:
А1з+ + Н2О Al (ОН)2+ + Н+
А1 (ОН)2+ + Н2О^:А1 (ОН)+ + Н+
А1 (ОН)2++Н2О^±А1(ОН)з + Н +
С учетом возможности образования аквакомплексов схему гид-
ролиза соединений алюминия можно записать следующим образом:
[А1 (Н2О)б]з+ + Н2О = [А1 (Н2О)5 ОН]2+ + Н3О+
Сущность гидролиза соединений алюминия и железа сводится к
отрыву протона от гидратированного иона молекулой воды и обра-
зованию гидрооксокомплексов. Обязательным условием эффектив-
ности действия коагулянтов является полнота их гидролиза с обра-
зованием труднорастворимых гидроксидов. Растворимость гидрок-
сида алюминия при 18° С составляет 2,26-10~4, а гидроксида желе-
за (III) —2,3-10-5 мг/л. Из приведенных уравнений гидролиза сле-
дует, что для увеличения степени гидролиза необходимо связывать
образующиеся ионы Н+. Это достигается за счет гидрокарбонат-,
карбонат- или гидроксид-ионов, присутствующих в обрабатываемой
воде (щелочного резерва).
Суммарное уравнение реакции гидролиза сульфата алюминия
в присутствии гидрокарбонатов можно записать следующим обра-
зом:
А12 (SO4)3 4- ЗСа (НСО3)2 4- 6Н2О = 2А1 (ОН)3 + 3CaSO4 4- 6 [Н2О 4- CO2J
Наряду с гидроксидами алюминия и железа в воде обычно присут-
ствуют и промежуточные продукты гидролиза в форме основных
солей. Основные сульфаты алюминия практически не растворимы
в воде и образуются одновременно с гидроксидом алюминия при
рН<7,5. Образующийся гидроксид алюминия обладает амфотер-
ными свойствами. Его образование начинается при рН>4,5. При
125
рН>8,5 происходит растворение гидроксида с образованием алю-
минатов по уравнению
Л1 (ОН)з 4-3NaOH =- Na3 [Al (ОН)6]
гексагидрооксоалюминат
натрия
Для простоты изложения будем принимать, что в щелочной среде
образуется анион метаалюминиевой кислоты А1О~. Метаалюминат
натрия как соль, образованная слабой кислотой и сильным осно-
ванием, подвергается гидролизу: АЮГ+2Н2О^А1 (ОН)3 + ОН_.
На этом свойстве и основано использование его в качестве коагу-
лянта. Минимальная растворимость гидроксида алюминия наблю-
дается в интервале значений pH 6,5—7,5.
Гидроксид железа (III) имеет наименьшую растворимость при
pH 6,5. Осадок Fe(OH)3 образуется в более широком диапазоне
значений pH по сравнению с гидроксидом алюминия. Осаждение
гидроксида железа (III) начинается при рН>3. Гидроксид железа
(II) образуется при рН>7, минимальная растворимость его дости-
гается при pH 10.
Гидролиз вводимых коагулянтов заканчивается очень быстро,
в течение одной-двух минут. Гидроксиды алюминия или железа
вследствие большого разбавления образуют коллоидные растворы.
Золи гидроксидов металлов имеют положительный заряд вследст-
вие адсорбции катионов металла, образующего гидроксид, на кол-
лоидной частице. Противоионами являются анионы соли металла
(сульфат-, хлорид-ионы).
У амфотерных гидроксидов, например у гидроксида алюминия,
знак заряда коллоидного раствора зависит от pH. В кислой и
нейтральной среде золь гидроксида алюминия имеет положитель-
ный заряд. Потенциалопределяющими будут ионы А13+. В слабо-
щелочной среде (при pH 8,2) знак заряда коллоидного раствора
гидроксида алюминия становится отрицательным, так как роль по-
тенциалобразующих ионов уже выполняют алюминат-ионы или
гидроксид-ионы. Противоионами будут в таких коллоидных части-
цах катионы металлов. При низких значениях pH коллоидные час-
тицы гидроксидов металлов адсорбируют большее число катионов
(потенциалопределяющих ионов) и поэтому являются более устой-
чивыми, что объясняется меньшей полнотой гидролиза коагулянта.
Коагулирование воды — это сложный физико-химический про-
цесс, включающий в себя три стадии: 1) образование коллоидного
раствора гидроксида металла в результате полного гидролиза ко-
агулянта; 2) коагуляцию образовавшегося золя коагулянта под
действием анионов воды и его взаимодействие с коллоидными и
грубодисперсными примесями воды; 3) процесс хлопьеобразования,
завершающийся седиментацией. В процессе обработки воды необ-
ходимо поддерживать оптимальные условия для каждой стадии
процесса.
При введении коагулянта в воду происходит его диссоциация
и одновременно начинается процесс гидролиза коагулянта. Обра-
зующиеся коллоидные растворы гидроксидов алюминия или же-
126
леза (III) коагулируют под действием анионов, содержащихся в
воде. Особенно быстро проходит этот процесс при pH 6,5—7,5,
близких или соответствующих изоэлектрическому состоянию этих
золей (для гидроксида алюминия при pH 6,5—7,5; гидроксида же-
леза (Ш) при pH 5—7). Изоэлектрическая точка для коллоидных
растворов гидроксидов металлов может несколько смещаться в за-
висимости от солевого состава воды. Снижение устойчивости золей
коагулянтов происходит в результате сжатия диффузного слоя кол-
лоидных частиц. При этом часть противоионов из диффузного слоя
переходит в адсорбционный, следствием чего является уменьшение
электрокинетического потенциала (концентрационная коагуляция).
Коллоидные частицы коагулянта, полностью или частично потеряв-
шие заряд, начинают соединяться друг с другом, так как межмоле-
кулярные силы притяжения в этих условиях превышают электро-
статические силы отталкивания, обусловленные наличием одно-
именного заряда. Скоагулировавшие частицы гидроксида алюминия
или железа имеют чрезвычайно развитую поверхность и поэтому
являются прекрасными адсорбентами для коллоидных примесей,
содержащихся в воде.
Вторая стадия процесса коагуляции заключается в том, что от-
рицательно заряженные коллоидные примеси воды начинают ад-
сорбироваться коагулирующими частицами коагулянта. Снижение
или потеря заряда коллоидных частиц примесей может происхо-
дить и под действием катиона коагулянта. Если концентрации поло-
жительно заряженных золей коагулянта и коллоидных примесей
примерно одинаковы, наблюдается взаимная коагуляция. Так как
она происходит только в очень узкой зоне концентраций, то ее
можно рассматривать как частный случай процесса коагуляции.
Заряд коллоидных частиц примесей может понижаться и в ре-
зультате проникновения катионов коагулянта при сжатии диффуз-
ного слоя в адсорбционный. Иногда, например для золей кремние-
вой кислоты, возможна их специфическая адсорбция, сопровожда-
ющаяся образованием труднорастворимых соединений с потенциал-
определяющими ионами (гидроксида алюминия или железа и
кремниевой кислоты) (нейтрализационная коагуляция). Этот про-
цесс сопровождается уменьшением термодинамического потенциа-
ла коллоидных частиц примесей.
Основная роль при удалении коллоидных примесей воды отво-
дится адсорбции их растущими агрегатами скоагулировавших час-
тиц гидроксидов алюминия или железа. Максимальная адсорбция
коллоидных частиц примесей воды происходит в начальный период
формирования коагеля, т. е. в период скрытой коагуляции.
Коагуляция коллоидных частиц гидроксидов алюминия и желе-
за вызывается анионами (сульфатами, гидрокарбонатами, хлори-
дами), содержащимися в обрабатываемой воде. Коагуляция идет
с наибольшей скоростью при следующих пороговых концентрациях
анионов (г-экв/л): СК 0,07; НСОГО,005; SO1~ 0,001—0,002. В при-
родных водах концентрации этих ионов обычно меньше и поэтому
скорость коагуляции ниже.
127
Основным фактором, обусловливающим скорость процесса
коагулирования воды, является стадия хлопьеобразования. Рост
хлопьев идет в результате сорбции скоагулировавшими частицами
гидроксидов алюминия или железа коллоидных частиц, примесей
воды, укрупнения образовавшихся хлопьев за счет соединения их
друг с другом. Если в начальной стадии количество хлопьев ис-
числяется тысячами в 1 мл воды, то к концу процесса оно измеря-
ется уже единицами (5—10 в 1 мл воды). Укрупнившиеся хлопья
оседают под действием силы тяжести, увлекая за собой взвешен-
ные частицы. Чем быстрее рас-
Рис. 17. Кривая коагуляции
тут хлопья, чем больше их мас-
са и размер, тем интенсивнее
идет процесс седиментации,
тем выше степень осветле,'
воды.
При обесцвечивании воды,
когда удаляются из воды гуму-
совые соединения, имеющие
свойства гидрофильных колло-
идов, основная роль отводит-
ся специфической адсорбции
многозарядных катионов алю-
миния поверхностью сложных
высокомолекулярных гумусо-
вых кислот. В результате об-
разуются труднорастворимые комплексы алюминия. Коагуляция
гумусовых соединений алюминиевым коагулянтом является необ-
ратимой. Оптимальные значения pH обрабатываемой воды при
обесцвечивании 4,5—5,5.
Так как собственно процесс коагуляции идет очень быстро, не-
обходимо энергичное перемешивание воды после поступления коагу-
лянта. Для ускорения процесса хлопьеобразования в маломутных,
цветных водах дополнительно вводят суспензии глин для увеличе-
ния общего числа частиц. На процесс коагуляции влияет темпера-
тура обрабатываемой воды. При понижении температуры уменьша-
ется скорость теплового движения и число эффективных столкно-
вений коллоидных частиц уменьшается, поэтому устойчивость
«системы повышается.
Условия коагуляции зависят также от количества коагулянта,
введенного в воду (рис. 17). На этапе, соответствующем зоне I,
происходит обмен ионами между коагулянтом и коллоидами воды
и его гидролиз. Так как концентрация раствора коагулянта в этой
зоне ниже пороговой, то коагуляция еще не идет, поэтому мутность
воды не уменьшается (/ — зона устойчивости). При концентрации
выше пороговой идет коагуляция коллоидного раствора коагулян-
та и коллоидных примесей воды (зона коагуляции II). Чем ближе
pH к изоэлектрической точке, тем быстрее и полнее идет коагуля-
ция. Изоэлектрическая точка для гидроксида алюминия в дистил-
лированной воде соответствует pH 7,2, но под влиянием ионов,
128
содержащихся в очищаемой воде, она несколько смещается. К мо-
менту достижения изоэлектрической точки мутность воды достига-
ет минимума и некоторое время остается постоянной, что связано с
буферными свойствами гидрокарбонат-ионов, содержащихся в во-
де. После того как щелочной резерв израсходован, увеличение дозы
коагулянта вызывает усиление мутности воды. Это является след-
ствием уменьшения степени гидролиза сульфата алюминия из-за
накопления ионов в воде и прекращения коагуляции гидрокси-
да алюминия (зона устойчивости III). Дальнейшее увеличение до-
зы коагулянта приводит к перезарядке коллоидных частиц (зо-
на IV).
Количество введенного в воду коагулянта (мг/л, мг-экв/л, г/м3
и”" г-экв/м3) называется дозой коагулянта. Минимальная концен-
1'ця коагулянта, отвечающая наилучшему осветлению или обес-
цвечиванию воды, называется оптимальной дозой. Она определя-
ется опытным путем и зависит от солевого состава, жесткости, ще-
лочности воды и др. Оптимальной дозой коагулянта считается то
его минимальное количество, которое при пробном коагулировании
дает крупные хлопья и максимальную прозрачность воды через
15—20 мин. Для сульфата алюминия эта концентрация обычно
колеблется от 0,2 до 1,0 мг-экв/л (20—100 мг/л) Во время павод-
ка доза коагулянта увеличивается приблизительно на 50%. При
температуре воды ниже 4° С дозу алюминиевого коагулянта увели-
чивают почти в два раза.
Так как коагуляция коллоидных частиц гидроксидов металлов
происходит под влиянием анионов, находящихся в воде, то мягкие
воды (во время паводка) обладают плохой коагулируемостью.
Коагуляция примесей в мягких водах под действием сульфата алю-
миния происходит лучше при pH 5,7—6,6, в водах средней жест-
кости— при pH 6,6—7,2, жестких—при pH 7,2—7,6. Если требуется
снизить цветность воды, в качестве коагулянта применяется суль-
фат алюминия. Доза коагулянта для обесцвечивания воды опре-
деляется ориентировочно по формуле
Д = 4)Ц, (III.19)
где Д — доза сульфата алюминия в расчете на безводную соль,
мг/л; Ц — цветность воды по платино-кобальтовой шкале, град.
Коагулирование с подщелачиванием. При коагулировании про-
исходит снижение щелочности воды в результате взаимодействия
гидрокарбонат-ионов с ионами Н+ гидролизующегося коагулянта.
Так, при дозе сульфата алюминия 100 мг/л щелочность снижается
на 0,6 мг-экв/л, а при дозе 100 мг/л сульфата железа (II)—на
0,72 мг-экв/л. При малой щелочности и низкой температуре воды
коагулирование воды ухудшается. Для повышения эффективности
процесса при недостаточной щелочности воды проводят коагули-
рование с подщелачиванием. Вместе с коагулянтом вводят реаген-
ты, содержащие гидроксид-, карбонат-ионы, которые способствуют
повышению щелочности. Для подщелачивания обычно используют
известь (когда не ограничивается жесткость воды) и соду. Сущ-
5—1244 129
ность процессов, происходящих при подщелачивании, можно опи-
сать уравнениями реакций:
2FeCl3 у- ЗСа (ОН)2 = 2Fe (ОН)3 -г ЗСаСЬ (а)
A12(SO4)3 +3Na2CO3 4- ЗН2О = 2А1 (ОН); -у ЗХа >SO4 -ЗСО> (щ
В процессе (б) образующаяся свободная угольная кислота связы-
вается избытком карбоната натрия. Количество щелочи для подще-
лачивания определяют по формуле
Дщ = Лк-Щ + 1, (Ш.2Э)
где Дщ — доза щелочи в расчете на безводное чистое соединение
(100%-ный продукт), мг-экв/л; Дк — доза коагулянта в пересчете
на безводную соль, мг-экв/л; Щ — щелочность воды, мг-экв/л; 1 —
избыток щелочного реагента, обеспечивающий щелочной резерв во-
ды, мг-экв/л.
Для перевода дозы щелочного реагента в мг/л необходимо ум-
ножить полученный результат на эквивалент оксида кальция, рав-
ный 28, или карбоната натрия (безводного), равный 53. При не-
достаточном щелочном резерве обрабатываемой воды можно ис-
пользовать смешанный алюминиевый коагулянт, состоящий из суль-
фата алюминия и алюмината натрия. При гидролизе алюмината
натрия реакция среды становится щелочной, поэтому возможна
нейтрализация кислого раствора сульфата алюминия щелочным
раствором алюмината натрия по уравнению
A12(SO4)3 + 6NaA102 4- 12Н2О == 8А1 (ОН)3 + 3Xa2SO4
При применении алюмината натрия в качестве самостоятельного
коагулянта необходимо связывать образующийся избыток гидрок-
сид-ионов кислыми реагентами.
§ 29. Характеристика коагулянтов и флокулянтов
Характеристика коагулянтов. Коагулянты, применяемые в прак-
тике водоочистки, должны соответствовать определенным требо-
ваниям, предъявляемым к ним как реагентам и химическим про-
дуктам. Как реагент коагулянт должен обладать высокой способ-
ностью к осветлению или обесцвечиванию воды, т. е. доза его долж-
на быть небольшой. Гидролиз коагулянта, коагуляция золей гид-
роксидов металлов, хлопьеобразование и седиментация должны
протекать с высокой скоростью. При введении коагулянта pH обра-
батываемой воды не должен заметно изменяться. Как химический
продукт коагулянт должен быть дешевым, изготавливаться из до-
ступного сырья, хорошо сохраняться, быть удобным для дозиров-
ки, не проявлять сильного коррозионного действия по отношению
к железу и бетону, быстро растворяться в воде. В разработке тех-
нологии получения и применения коагулянтов большую роль сыгра-
ли исследования, проведенные во ВНИИ ВОДГЕО, Институте кол-
лоидной химии и химии воды АН УССР, Академии коммунального
130
хозяйства им. К. Д. Памфилова, Уральском научно-исследователь-
ском химическом институте.
С л ь ф а т а л ю м и н и я (сернокислый глинозем)
A12(SO4)3• 18Н2О. Он является наиболее распространенным коагу-
лянтом. Сульфат алюминия получают при обработке веществ, со-
держащих оксид алюминия (обожженные бокситы и глины, отходы
некоторых производств), серной кислотой: A12O3 + 3H2SO4 —
~A12(SO4)3 + 3H2O. Товарный продукт выпускается двух марок:
очищенный и неочищенный. При хранении сульфат алюминия сле-
живается. В очищенном содержится 40,3% безводного сульфата
алюминия (или 78,25% в пересчете на кристаллогидрат), в неочи-
щенном— 33,5 и 65,37% соответственно.
Растворимость сульфата алюминия составляет при 20° С
362 г/л. Этот коагулянт применяется для осветления и обесцвечи-
вания воды. Плотность образующихся хлопьев гидроксида алюми-
ния' 2,4. При отсутствии щелочного резерва в воде сульфат алю-
миния вызывает резкое снижение pH (до 4,5) и вода становится
агрессивной по отношению к железу и бетону.
Основной хлорид алюминия А12(ОН)5С1 • 6П2О. В на-
шей стране разработана технология производства этого коагулян-
та растворением свежеосажденного гидроксида алюминия в 0,5—
1 % растворе соляной кислоты. Он представляет собой зеленоватые
кристаллы, хорошо растворимые в воде. Этот коагулянт снижает
щелочность воды и увеличивает ее солесодержание меньше, чем
сульфат алюминия. Основной хлорид алюминия можно получать в
виде раствора, порошка, гранул.
Алюминат натрия NaA102. Его получают при взаимодей-
ствии едкого натра с оксидом или гидроксидом алюминия. Содер-
жание водорастворимого алюминия в пересчете на оксид алюминия
в техническом продукте составляет 55% • Растворимость при 20° со-
ставляет 370 г/л. Раствор имеет щелочную реакцию среды (pH 9),
поэтому для создания условий образования гидроксида алюминия
необходимо связывать избыток щелочности. При водоочистке алю-
минат натрия используется, если увеличение солесодержания за
счет анионов коагулянтов является нежелательным.
Хлорид железа (III) РеС13-6Н2О. Это соединение получают
хлорированием железной стружки при 650—750° С. Он представ-
ляет собой очень гигроскопичные красно-бурые кристаллы, хорошо
растворимые в воде. Необходимость герметизации тары создает
трудности при доставке, хранении и дозировке реагента. Коагу-
лянт применяется для осветления воды.
Сульфат железа (III) Fe2(SO4)3. Его получают при взаи-
модействии концентрированной серной кислоты с оксидом железа
(III). С водой образует кристаллогидраты различного состава. Об-
ладает довольно сильным коррозионным действием по отношению
к железу и бетону, поэтому применяется реже других коагулянтов.
Сульфат железа (II) Fe2SO4-7H2O. Он представляет со-
бой зеленоватые кристаллы, буреющие на воздухе вследствие окис-
ления Fe2+ до Fe3+. Растворимость в воде при 20° С составляет
5* 131
265 г/л. Товарный продукт выпускается двух марок, содержащих
соответственно 47 и 53% безводного сульфата железа. Для осу-
ществления коагуляции воды необходимо окислить образующийся
гидроксид железа (II) в гидроксид железа (III). С заметной ско-
ростью окисление идет при рН>7. Выпадение осадка гидроксида
железа (II) начинается при рН>8. Коагулирование сульфатом же-
леза осуществляется в щелочной среде, создаваемой известью, ко-
торую вводят в обрабатываемую воду, по уравнениям:
FeSO4 + Са(ОН)2 = Fe(OH)2
4Fe(OH)2 + О2 + 2Н2О = 4Fe(OH)3
Процесс ведется при pH не ниже 9—9,5.
Для окисления 1 мг Fe2+ необходимо 0,143 мг кислорода. Если
в обрабатываемой воде содержится 8—9 мг/л растворенного кис-
лорода, то обработку воды без дополнительного введения окисли-
теля можно проводить при дозе коагулянта до 100 мг/л (в расчете
на безводную соль). Для интенсификации процесса при недостаточ-
ном количестве растворенного кислорода в воде в качестве окисли-
теля железа (II) в воду вводят хлор. Предварительное хлорирова-
ние сульфата железа позволяет вести процесс при более низких
значениях pH (до 5,5). В присутствии гидрокарбонатов этот про-
цесс описывается следующим уравнением:
2FeSO4 4- С12 + ЗСа(НСО3)2 = 2Fe(OH)3 4- 2CaSO4 - 6СО2 + CaCi2
При коагулировании сульфатом железа (II) из воды удаляется до
50% органических примесей. Этот коагулянт применяется для ос-
ветления технических вод.
К преимуществам железных коагулянтов перед алюминиевыми
следует отнести меньшую зависимость их коагулирующей способ-
ности от солевого состава воды и величины pH, незначительное сни-
жение коагулирующих свойств при низкой температуре. Образую-
щиеся хлопья гидроксида железа имеют большую плотность (3,6),
чем гидроксида алюминия, и поэтому их осаждение идет быстрее.
Коагель гидроксида железа (III)—хороший адсорбент для орга-
нических веществ и соединений меди, мышьяка, марганца и др.
Недостаток железных коагулянтов состоит в высокой коррози-
онной активности по отношению к железу и бетону, возможности
применения их только для осветления воды. При одинаковых дозах
алюминиевого и железного коагулянтов количество образующихся
хлопьев у первого будет больше, так как его эквивалентная масса
меньше. Л. А. Кульским и сотрудниками предложен смешанный
коагулянт, который получают смешением растворов, содержащих
сульфат алюминия и хлорид железа (III) в соотношении 1 : 1. Та-
кой коагулянт проявляет положительные свойства железных коагу-
лянтов, но одновременно позволяет снизить их коррозионную ак-
тивность.
Применение флокулянтов для интенсификации процесса коагу-
лирования. Процесс коагулирования воды не отличается постоян-
ством параметров работы очистных сооружений из-за непрерывного
132
Рис. 18. Схема образования хлопь-
ев в присутствии высокомолеку-
лярного флокулянта
изменения таких показателей качества воды, как температура,
химический состав, концентрация примесей, степень их дисперсно-
сти. Для интенсификации очистки воды методом коагулирования
применяются дополнительные реагенты-флокулянты, позволяющие
ускорять процессы хлопьеобразования и осаждения. Использова-
ние флокулянтов способствует образованию прочных, быстро осе-
дающих хлопьев, что позволяет ускорить процесс обработки воды.
В настоящее время для очистки воды используются следующие
классы флокулянтов (Ю. И. Вейцер): 1) неорганические (активи-
рованная кремниевая кислота);
2) органические высокомолекуляр-
ные соединения (ВМС), полученные
переработкой природных продуктов
[альгинат натрия, крахмал, карбо-
ксиметил целлюлоза (КМЦ)]; 3) син-
тетические высокомолекулярные со-
единения (полиакриламид, полиэти-
ленимин и др.). В водных растворах
такие высокомолекулярные флоку-
лянты, как крахмал, полиоксиэти-
лен, эфиры целлюлозы, находятся в
недиссоциированном состоянии. Фло-
кулянты типа BA-2, BA-202, ВА-212,
полиэтпленимин дают активный ка-
тион (поликатион). Некоторые фло-
кулянты проявляют свойства амфо-
литов. К их числу относится гидролизованный полиакриламид. Ме-
ханизм действия флокулянтов основан на адсорбции макромолекул
флокулянта скоагулировавшими или взвешенными частицами. Так
как степень полимеризации у высокомолекулярных флокулянтов
колеблется от 1000 до 5000 звеньев, то количество активных цент-
ров, способных к адсорбции, очень велико. Поэтому одна макромо-
лекула флокулянта может адсорбироваться за счет отдельных сег-
ментов сразу несколькими твердыми частицами. Процесс флокуля-
ции включает в себя три стадии: адсорбцию, образование мостико-
подобной структуры и непосредственно флокуляцию. Образование
полимерных мостиков между макромолекулами, сорбированными
твердыми частицами, является непременным условием успешного
прохождения процесса флокуляции. Если адсорбция -макромолеку-
лы на поверхности частицы не сопровождается взаимодействием
между образующимися агрегатами, то происходит стабилизация си-
стемы (явление защиты). Макромолекулы флокулянта связывают
большое количество скоагулировавших частиц, взаимодействуют
друг с другом, образуя крупные быстро растущие хлопья (рис. 18).
Теория флокуляции, предложенная Кройтом и Трольстром, разви-
тая в работах Ла Мера, Ю. И. Вейцера, является в настоящее вре-
мя общепризнанной. Рассмотрим наиболее часто применяемые фло-
кулянты.
133
Полиакриламид (ПАА). Его получают сополимеризацией
акриламида СН2 = СН — С"' и акриловой кислоты
xNH2
СН2 = СН—СООН. Полимер содержит полиамидные (I) и кислот-
ные звенья (II):
’ — СН2 — СН —
О = С — NH>
I
- _ сн2 — СН — _
СООН
II
П1
В полимере преобладают полиамидные звенья. Кислотные звенья
в молекулах ПАА переводят в солевые (акрилатные) обработкой
известью или аммиаком. Технический продукт выпускается двух
марок: известковый, содержащий акрилат кальция, и аммиачный,
в состав которого входит акрилат аммония. ПАА представляет со-
бой белое аморфное вещество, хорошо растворимое в воде. Нали-
чие карбоксильных групп обусловливает у ПАА слабо выраженные
свойства анионного флокулянта. Наиболее эффективно примене-
ние ПАА для осветления вод с мутностью более 150 мг/л. Харак-
терно, что с повышением мутности воды флокулирующая способ-
ность ПАА возрастает. Ориентировочно доза ПАА составляет
0,05—1,5% к общему содержанию взвешенных веществ. Введение
ПАА позволяет снизить дозу коагулянта в 2—3 раза, ускорить про-
цесс осаждения хлопьев в 10—20 раз. В щелочной среде активность
ПАА резко снижается ввиду его гидролиза. Для обезвоживания
осадков используется гидролизованный полиакриламид (ГПАА),
который содержит до 20% акрилатных групп. Его получают при
обработке ПАА раствором едкого натра при 50—60°С непосред-
ственно на очистных сооружениях. Обработка природных вод, со-
держащих отрицательно заряженные коллоидные примеси, не мо-
жет быть осуществлена одним ПАА, так как он самостоятельно не
вызывает их коагуляцию, а только интенсифицирует процесс
хлопьеобразования.
Активированная кремниевая кислота. Ее получа-
ют при взаимодействии жидкого стекла (Na2O-«SiO2-mH2O) с раз-
личными веществами, вызывающими образование коллоидного рас-
твора кремниевой кислоты или труднорастворимых силикатов.
Обычно применяют неорганические кислоты, диоксид углерода, со-
ли, водные растворы которых вследствие гидролиза имеют кислую
реакцию (сульфаты аммония и алюминия), хлор и др. Схемы воз-
можных химических реакций:
Na2O-/?SiO2-/nH2O д- H2SO4~>/?SiO2-хН2О Д-Na2SC>4
Na2O-nSiO2-mH2O Д- СО2 Д- H2O-^/7SiO2-аН2О Д- Na2CO3
Na2O-/?SiO2-mH2O Д-(NH4)2SO4-^/?SiO2-A'H2O Д- Na2SO4 + 2NH4OH
Na2O-nSiO2-mH2O Д- Cl2 Д- H20>/iSi02-xH2O Д-NaCl Д-NaClO
134
Все эти процессы заканчиваются образованием оксидов крем-
ния с различной степенью гидратации. Это связано с тем, что про-
стейшие гидраты диоксида кремния (орто- и метакремниевые кис-
лоты) конденсируются с образованием поликремниевых кислот —
неорганических полимеров, которые и образуют, в зависимости от
концентрации исходных продуктов, коллоидный раствор или гель.
Обычно для получения активированной кремниевой кислоты при-
меняют 1,5—2%-ный раствор жидкого стекла (по SiO2). Через
6—8 ч золь активированной кремниевой кислоты, полученной из
такого раствора, переходит в гель. Для замедления гелеобразования
приготовленный коллоидный раствор разбавляют до 0,5%-ного (по
SiO?). Период формирования золя кремниевой кислоты до начала
перехода его в гель определяется «созреванием». Наибольшую фло-
кулирующую способность имеет созревший золь, выдержанный в
течение нескольких часов. Время созревания активированной кис-
лоты зависит от pH. Оно значительно возрастает при pH>8 и <6.
Механизм действия активированной кремниевой кислоты заклю-
чается во взаимодействии с положительно заряженными коллоид-
ными частицами коагулянтов и создании дополнительных центров
для образования хлопьев. Доза активированной кремниевой кис-
лоты при обработке воды с цветностью до 100 град, мутностью до
15 мг/л и температурой выше 5ГС составляет 2—3 мг/л. Активиро-
ванная кремниевая кислота широко используется в качестве фло-
кулянта во многих странах.
Катионный флокулянт ВА-2. Технология получения и примене-
ния его разработана в академии коммунального хозяйства
им. К. Д. Памфилова. В отличие от анионных и нсионных флоку-
лянтов катионный флокулянт ВА является и эффективным коагу-
лянтом коллоидных примесей, имеющих отрицательный заряд.
С гумусовыми соединениями, анионными ПАВ флокулянт ВА-2 об-
разует труднорастворимые комплексы. По химической природе
это — полимер, диссоциирующий в воде с образованием активного
катиона со структурой
— СН2 —СН2
СН2 —N+(CH3)3C1
При дозе ВА-2 1,0—1,5 мг/л эффект осветления воды выше, чем при
использовании сульфата алюминия. ПДК ВА-2 в воде составляет
0,5 мг/л. Этот флокулянт применяется для обработки мутных вод,
используемых для технических целей.
Катионные флокулянты ВА-212 и ВА-202, представляющие со-
бой четвертичные аммониевые соли на основе полистирола и поли-
винилтолуола, разрешены для обработки питьевой воды. ПДК та-
ких флокулянтов в воде составляет 2 мг/л.
135
§ 30. Безреагентные методы коагуляции
В практике водоочистки применяются методы удаления колло-
идных примесей воздействием на них электрическим полем. Пре-
имущества такого метода заключаются в отсутствии реагентного
хозяйства, компактности установок и возможности автоматизации
процесса. По механизму действия электрического поля на колло-
идные частицы можно выделить два направления: электрокоагуля-
цию и электрофорез.
Электрокоагуляция. Сущность метода заключается в том, что
разрушение коллоидных систем происходит под действием элек-
трофоретического перемещения частиц к соответствующему элек-
троду. При электрокоагуляции происходит поляризация ионных
сфер коллоидных частиц, коагуляция протекает при несуществен-
ном изменении электродов и среды. Электрокоагуляция не тре-
бует большой плотности тока, электроды практически не расходу-
ются. Коллоидные частицы примесей, имеющие отрицательный за-
ряд, при наложении электрического поля в результате электрофо-
реза концентрируются у анода. Этот процесс является первичным
при электрокоагуляции, так как идет он со значительной ско-
ростью. Увеличение концентрации коллоидных частиц в анодном
пространстве приводит к поляризационной коагуляции, которая,
возможно, является обратимым процессом.
Вторая разновидность процесса — электрохимическая коагуля-
ция, которая протекает при большой плотности тока и сопровож-
дается электрохимическим растворением алюминиевого или желез-
ного анода. В воду поступают ионы алюминия или железа (II), ко-
торые в результате гидролиза образуют соответствующие гидрок-
сиды. Катионы Fe2+ окисляются растворенным в воде кислородом
или хлором до Fe3+ с образованием Fe(OH)3. Последующие ста-
дии, включающие в себя концентрационную и нейтрализационную
коагуляцию коллоидных частиц, сорбцию коллоидных частиц приме-
сей скоагулировавшими гидроксидами металлов и флокуляцию
проходят по механизму, описанному при коагулировании воды.
Расход электроэнергии увеличивается при увеличении плотно-
сти тока и расстояния между электродами, снижении скорости по-
тока воды в электрокоагуляторе. Расстояние между электродами
составляет 15—20 мм, скорость потока воды должна быть не менее
0,5 м/с, плотность тока — не более 10 А/м2. Растворение 1 г алю-
миния равноценно поступлению в воду 6,35 г безводного сульфата
алюминия, а растворение 1 г железа-—1,93 г безводного хлорида
железа (III). Разработка более экономичных электролизеров, сни-
жение энергозатрат создают предпосылки для широкого внедрения
электрических методов коагуляции.
Электрофорез. В отличие от электрокоагуляции электрофорез
не сопровождается разрушением коллоидных примесей, а сводится
к их концентрированию с последующим удалением из зоны воздей-
ствия электрического поля. Электрофоретический перенос коллоид-
ных частиц осуществляется под влиянием внешнего воздействия.
136
На рис. 19 представлена электрофоретическая ячейка, в которой
электроды отделены от обрабатываемой воды токопроводящими
мембранами Л-1 и ЛЦ, не проницаемыми для коллоидных частиц.
Камера разделена фильтром Ф, через который проходят частицы
коллоидной степени дисперсности.
Вода, содержащая коллоидные при-
меси, поступает в камеру через от-
верстие 1, под давлением проходит
через фильтр в ее вторую часть и
выходит через выпускное отверстие
3. Если в камере создать электриче-
ское поле, то отрицательно заря-
женные коллоидные частицы будут
стремиться перейти в левую часть
камеры и сконцентрироваться у мем-
браны Лф отделяющей ячейку от
анода. Перемещение коллоидных ча-
стиц в направлении, противополож-
Рис. 19. Схема электрофоретиче-
ской ячейки
ном направлению течения исходного
раствора, будет возможно в том слу-
чае, если скорость электрофорети-
ческого переноса частиц будет
больше или равна скорости движения раствора Цф. В растворе,
прошедшем через ячейку, могут при этом остаться коллоидные ча-
стицы, потерявшие заряд. Концентрированный коллоидный раствор
удаляется из камеры через выпускное отверстие 2. Характерной
особенностью электрофореза является малая зависимость эффек-
тивности процесса от исходной концентрации коллоидных примесей
и основных показателей качества обрабатываемой воды.
Высокий эффект очистки воды наблюдается при использовании
комплекса электрических воздействий, который включает в себя
сочетание действия электри-
ческих полей и разрядов.
Ниже приведены данные по
очистке воды комплексом
электрических воздействий.
Электрообработка воды
при соответствующем со-
вершенствовании конструк-
ции аппаратов является од-
ним из перспективных на-
правлений в современной
технологии.
Показатель Исходная вода Обработанная J вода
Прозрачность, см 5—20 30
Мутность, мг/л 10—500 0,4
Цветность, град 30—500 9
Запах, балл 1—5 0-1
Вкус, балл 1—3 0-1
pH Окисляемость пер- манганатная, мг 6,5—7,5 6,7-7,8
О2/л 3,25 0,5—6
§ 31. Удаление из воды соединений железа
и марганца
В природных водах встречаются различные формы содержания
железа со степенью окисления +2 и +3. Это могут быть неорга-
нические и органические соединения, которые находятся в воде в
137
растворенном, коллоидном и взвешенном состояниях. Появление
железа в природных водах прежде всего связано с растворением
железосодержащих пород под действием кислот (угольной, серной,
органических) и окислителей (кислорода). Так, минерал пирит при
растворении может давать сульфат и гидрокарбонат железа (II):
FeS2 -р 2СО2 Щ 2Н2О . Fe(HCO3)2 - H2S -r S
2FeS2 4- 7O2 -p 2H2O 2FeSO4 + 2H2SO4
В присутствии кислорода соединения железа (II) неустойчивы и
окисляются с образованием соединений железа со степенью окисле-
ния + 3. Соединения железа (11), как правило, встречаются в под-
земных водах. Содержание их обычно не превышает 1 мг/л, но
может достигать нескольких десятков мг/л. Основные формы со-
держания железа (II): гидрокарбонат железа (II) и сульфат же-
леза (II). В поверхностных водах содержатся неорганические соеди-
нения железа (III) и органическая форма — гуматы железа. Соли
железа (III) в воде подвергаются гидролизу с образованием гид-
роксида железа (III), который может находиться в воде в коллоид-
ном состоянии, чему способствует наличие органических высоко-
молекулярных соединений. Обычно концентрация железа (III) в
поверхностных водах составляет сотые, реже десятые доли мг/л,
что обусловлено очень малой растворимостью гидроксида железа
(III), которая при pH 7 составляет 0,05 мг/л, а при повышении pH
она становится еще ниже. Гуматы железа имеют желто-бурую ок-
раску и обусловливают цветность природных вод. При выходе под-
земных железосодержащих вод на поверхность происходят окисле-
ние железа (II) и гидролиз с образованием гидроксида желе-
за (III):
4Fe(HCO3)2 + О2 + 2Н2О = 4Fe(OH)3 4- 8СО2
4FeSO4 + О2 + ЮН2О = 4Fe(OH)3 4- 4H2SO4
Это объясняет образование бурого осадка при стоянии прозрачной
артезианской воды и постепенное побурение на воздухе зеленова-
того налета (гидроксида железа (II)), взятого со дна родников.
Окисление железа (II) может происходить с участием железобак-
терий, использующих энергию окисления для жизнедеятельности.
Если концентрация железа выше 0,3 мг/л, то вода приобретает
неприятный железистый привкус. Отдельные отрасли производст-
ва предъявляют более строгие требования к содержанию железа
в воде, например при производстве вискозного волокна оно должно
составлять не более 0,03 мг/л, кинопленки — 0,05 мг/л.
Процесс обработки воды, проводимый для снижения содержа-
ния соединений железа, называется обезжелезиванием. Выбор ме-
тода обезжелезивания зависит от формы содержания железа в во-
де, количества растворенного кислорода, диоксида углерода, pH,
солевого состава, а также требований, предъявляемые к воде. Для
обезжелезивания подземных вод применяются различные методы:
упрощенная аэрация с последующим фильтрованием; аэрация с из-
весткованием и хлорированием; коагулирование сульфатом алюми-
138
ния с предварительным хлорированием; фильтрование воды через
загрузку из пиролюзита; катионирование воды. Для удаления же-
леза из поверхностных вод используются коагулирование, извест-
кование, окислительные методы.
Наиболее простой метод обезжелезивания воды — аэрация. Он
заключается в насыщении воды кислородом для окисления соеди-
нений железа (II) и перевода их в труднорастворимый гидроксид
железа (III). Чаще всего этот метод применяется при наличии в
воде гидрокарбоната железа (II). На окисление 1 мг железа рас-
ходуется 0,143 мг кислорода. Гидролиз соли и окисление железа
(II) могут идти одновременно или предварять друг друга в зави-
симости от количества растворенного кислорода, диоксида угле-
рода, солевого состава, pH воды. При недостаточном количестве
кислорода и в присутствии других восстановителей гидролиз пред-
шествует окислению:
Fe(HCO3)2 + 2H2O=Fe(OH)2 + 2СО2 + 2Н2О
4Fe(OH)2 + О2 + 2Н2О = 4Fe(OH)3
При окислении и гидролизе соединений железа (II) образуютя про-
межуточные продукты, например ионы FeOH+. Так, 1 мг гидроли-
зующегося железа (II) снижает щелочность воды на 0,036 мг-экв/л
и способствует образованию 1,6 мг диоксида углерода. Эффектив-
ность обезжелезивания аэрацией увеличивается при повышении
pH и увеличении количества кислорода.
Метод упрощенной аэрации для удаления неорганических соеди-
нений железа (II) при концентрации их в воде до 10 мг/л приме-
ним для вод с окисляемостью не более 6—7 мг/л, содержание ам-
монийного азота в которых не превышает 1 мг/л, а сероводорода —
0,2 мг/л. Щелочность воды должна быть не менее 2 мг-экв/л, а
рН>7. Окислительно-восстановительный потенциал обработанной
воды должен быть не менее +100 мВ, а индекс стабильности воды
7^0,05. При пропускании воды через фильтр на зернах загрузки
образуется пленка из ионов и оксидов железа (II) и (III), которая
катализирует процесс окисления и удаления железа. Если качест-
во воды не соответствует приведенным условиям, то проводится
глубокая аэрация воды или она сочетается с хлорированием или
известкованием.
При рН<7 и значительном содержании диоксида углерода про-
водится известкование. Гидроксид кальция взаимодействует с СО2,
чем повышает степень гидролиза гидрокарбоната железа и спо-
собствует его переходу в гидроксид железа (II):
СО2 + Са(ОН)2 = СаСО3 + Н2О
Fe(HCO3)2 + 2Са(ОН)2 = Fe(OH)2 + 2СаСО3 + 2Н2О
Сульфат железа (II) удаляется аэрацией воды с подщелачива-
нием:
FeSO4 + Са(ОН)2 = Fe(OH)2 + CaSO4
4Fe(OH)2 + О2 + 2Н2О = 4Fe(OH)3
139
При недостаточном количестве кислорода, необходимого для оки-
сления железа, проводят хлорирование воды или сочетают его с
известкованием. При малом содержании свободной угольной кис-
лоты иногда бывает достаточно одного хлорирования воды с под-
щелачиванием, без аэрации. Аммонийный азот замедляет процесс
окисления, так как окислительный потенциал образующегося хло-
рамина меньше, чем у свободного хлора.
При хлорировании воды с достаточным щелочным резервом
(содержащей железо (II) в форме гидрокарбоната) протекают про-
цессы, которые описываются суммарным уравнением
2Fe(HCO3)2 + С 2 + Са(НСО3)2 -- 2Fe(OH)3 4- СаС12 6СО2
Суммарное уравнение известкования и хлорирования воды, со-
держащей железо (II) в форме сульфата, можно записать сле-
дующим образом:
2FeSO4 + Cl2 I- ЗСа(ОН)2 - 2Fe(OH)3 + СаСЬ 4- 2CaSO4
Для окисления железа (II) могут быть применены перманганат
калия и диоксид марганца МпО2:
3Fe(HCO3)2 + KMnO4 + 2Н2О = 3Fe(OH)3 + МпО2 4- КНСО3 + 5СО>
Образующийся диоксид марганца МпО2 является катализатором
окисления железа (II):
4Fe(HCO3)2 4- ЗМпО2 4- ЮИ2О -> 4Fc(OH)3 4- МпО 4- Мп2О3 4- 8СО2 4- 8Н2О
Низшие оксиды марганца (МпО и Мп2О3) затем вновь окисляются
до МпО2. Обезжелезивание воды проходит также эффективно при
фильтровании воды через слой пиролюзита или кварцевого песка,
на поверхности зерен которого создается пленка из диоксида мар-
ганца.
Для удаления неорганических форм железа может быть исполь-
зован также метод ионного обмена с использованием Н-катиони-
товых фильтров. Органические формы железа удаляются коагуля-
цией и действием таких окислителей, как хлор, озон, перманганат
калия. Для обезжелезивания воды могут быть использованы желе-
зобактерии. Для вод со значительным содержанием железа эффек-
тивным методом обезжелезивания служит электрохимическая коа-
гуляция с использованием алюминиевого или стального электрода.
Применение этого метода позволяет одновременно проводить ос-
ветление, обесцвечивание и обескремнивание воды, способствует
снижению окисляемости. Для обезжелезивания воды применяется
также и напорная флотация с известкованием (для вод с содержа-
нием железа более 10 мг/л).
Марганец в природных водах обычно сопутствует железу, но в
меньшей концентрации (до 0,5 мг/л). Содержание марганца стро-
го лимитируется технологией производства искусственных волокон,
радиотехнических и текстильных товаров. ПДК марганца в питье-
вой воде составляет по органолептическому признаку 0,1 мг/л. Фор-
мы содержания марганца в воде: гидрокарбонат и сульфат марган-
140
ца (II), органические соединения. Окисление марганца кислородом
происходит при рН>9,5. Марганец (II) удаляется из воды одно-
временно с железом (II) при известковании и аэрации:
MnSO4 - Са(ОН)2 = Мп(ОН)2 + CaSO4
2Мп(ОН)2 Д- О2 + 2Н2О = 2Мп(ОН)4
Процесс завершается выпадением осадка диоксида марганца. Вме-
сто подщелачивания воды для окисления марганца (II) можно ис-
пользовать такие окислители, как перманганат калия, хлор, озон,
диоксид хлора:
3MnSO4 + 2КМ1.О4 + 2Н2О 5Mi.O2 + K2SO4 + 2H2SO4
M11SO4 + СЬ + 2Н2О = МпО2 + 2НС1 + H2SO4
M11SO4 + О3 + Н2О = МпО2 4- О2 + H2SO4
5MnSO4 Д- 2С1О2 + 6Н2О = 5МпО2 + 2НС1 + 5H2SO4
Окисление марганца (II) может осуществляться при фильтро-
вании воды через слой загрузки, активированной катализатором
МпО2, или в процессе катионирования воды на Мп-катионитовых
фильтрах, обработанных перманганатом калия. Коллоидные соеди-
нения марганца удаляются при коагулировании воды. Для пре-
дотвращения защитного действия органических высокомолекуляр-
ных веществ предварительно производится обработка воды хлором
или другими окислителями. В ГДР для удаления марганца при-
меняется биохимический метод, основанный на использовании мар-
ганецокисляющих бактерий.
§ 32. Обескремнивание воды
Содержание соединений кремния в воде ограничивается в теп-
лоэнергетике, при производстве кордной целлюлозы и искусствен-
ных волокон. Кремниевая кислота в природных водах может при-
сутствовать в виде неорганических и органических соединений.
Свободная кремниевая кислота находится в воде в форме колло-
идного раствора. Состав коллоидных частиц определяется присут-
ствием поликремниевых кислот с общей формулой xSiO2-z/H2O.
При дальнейшем изложении будем рассматривать реакции с учас-
тием метакремниевой кислоты H2SiO3 или (SiO2-H2O).
Содержание различных форм кремниевой кислоты (свободной,
гидросиликат-HSiOr и силикат-ионов SiO?C ) зависит от pH. При
рН<8 в природных водах содержится свободная кремниевая кис-
лота. В интервале pH от 8 до 11 в равновесии находятся кремние-
вая кислота и гидросиликат-ионы. Содержание соединений крем-
ниевой кислоты в природных водах колеблется от долей мг/л до
нескольких десятков и даже сотен мг/л. В поверхностных водах
содержание кремния обычно невелико, но в водах болот и рек,
имеющих болотистый тип питания, концентрация соединений крем-
ния может исчисляться несколькими десятками мг/л. Так, содержа -
141
ние соединений кремния в водах Самотлорского болотного масси-
ва достигает 60—70 мг/л (в расчете на Si).
Наиболее эффективно коллоидная кремниевая кислота удаля-
ется при коагуляции солями железа в пределах pH 4,5—7,0. При
известковании воды содержание кремниевой кислоты снижается в
результате образования труднорастворимого метасиликата каль-
ция:
Na2SiOj + Са(ОН)2 = CaSiO3 4- 2xNaOH
Существуют методы удаления кремниевой кислоты, основанные
на коагуляции ее алюминатом натрия в щелочной среде, но полно-
го обескремнивания все эти методы не дают. Наиболее распростра-
нены магнезиальные методы обескремнивания, основанные на сорб-
ции различных форм кремниевой кислоты оксидом магния и други-
ми его соединениями. С повышением температуры степень обес-
кремнивания увеличивается. Так, при 80° С содержание SiO2 сни-
жается до 0,5 мг/л, при 97° С — до 0,1 мг/л. В качестве реагентов
применяется оксид магния (каустический магнезит) пли доломити-
зировапная известь (CaO-MgO). Главные недостатки магнезиаль-
ных методов состоят в необходимости подогрева, громоздкости ап-
паратуры, точного соблюдения режима и недостаточной степени
обескремнивания.
В институте ВОДГЕО разработан метод обескремнивания, ос-
нованный на фильтровании воды через фильтр, загруженный маг-
незиальным сорбентом. Такой сорбент получается обработкой ка-
устического магнезита или хлорида магния раствором соляной кис-
лоты с последующим высушиванием (80—100° С). Такие фильтры
установлены на Череповецкой ТЭЦ. Их производительность оцени-
вается следующими показателями: снижение содержания SiO2 от
2—4 до 0,3—1,0 мг/л, кремнеемкость (по SiO2) 91 кг на 1 т сор-
бента.
Эффективно удаляется кремниевая кислота на высокоосновном
анионите АВ-17. Глубина обескремнивания может достигать 0,01 —
0,05 мг/л. Этот метод используется в схеме полного обессоливания
воды.
§ 33. Фторирование и дефторирование воды
Содержание фтора в природных водах может составлять от со-
тых долей мг/л до нескольких десятков мг/л. В воде большинства
поверхностных водоемов содержание фтора не превышает 0,5—
0,7 мг/л. В подземных минерализованных водах концентрация фто-
ра обычно выше 1 мг/л. В южных засушливых районах содержание
фтора в воде выше, чем в средней полосе и северных областях.
Так, в Казахстане в водах поверхностных водоемов концентрация
фтора достигает 10 мг/л. Основная форма содержания фтора в во-
де— фторид-ион F-. Фтор является биологически активным эле-
ментом. При недостаточном содержании фтора в воде наблюдается
кариес зубов, а при концентрации в воде фтора выше допустимой
развивается флюороз («пятнистая эмаль»).
142
При недостатке фтора в воде осуществляется ее фторирование.
В качестве реагентов используются растворимые соединения фто-
ра. Наибольшее распространение в практике фторирования воды
получили фторид натрия NaF, кремнефторид натрия Na2SiF6, гек-
сафторкремниевая кислота H2SiF6.
Технический фторид натрия — кристаллическое вещество бело-
го или светло-серого цвета с растворимостью 35—41 г/л. Раствор
фторида натрия имеет нейтральную реакцию среды. Содержание
фтора в чистом фториде натрия составляет 45,2%, а в товарном
техническом продукте — 35—40% •
Кремнефторид натрия представляет собой белое кристалличе-
ское вещество иногда с серым или желтоватым оттенком. В чистом
кремнефториде натрия содержится 60,6% фтора, в техническом —
до 55%. При изменении температуры воды от 0 до 20° С раствори-
мость повышается от 4,3 до 7,4 г/л. Раствор кремнефторида нат-
рия имеет кислую реакцию среды.
Гексафторкремниевая кислота устойчива только в растворе. Ее
транспортируют в виде 8—14%-ного раствора. Она обладает силь-
ным корродирующим действием. Содержание фтора, в пересчете
на чистую гексафторкремниевую кислоту, составляет 79% • Приме-
нение этого реагента ограничивается трудностью транспортировки
и высокой стоимостью продукта.
Так как в процессе обработки воды содержание фтора несколь-
ко снижается, то фторирование воды проводится после коагулиро-
вания перед обеззараживанием воды. В соответствии с ГОСТ
2874—73 содержание фтора в воде устанавливается в зависимости
от климатических условий. Это обусловлено тем, что поступление
фтора с водой в организм человека зависит от нормы недопотреб-
ления. В районах с высокой среднегодовой температурой оптималь-
ная концентрация фтора в питьевой воде меньше, чем в регионах
с умеренным или холодным климатом.
При концентрации фтора в воде выше-ПДК проводится дефто-
рирование (обеефторирование). Сложность обработки воды для
снижения содержания фтора заключается в том, что раствори-
мость даже труднорастворимых фторидов превышает ПДК фтора
в воде. Методы дефторирования воды основаны на способности не-
которых труднорастворимых соединений (оксидов и гидроксидов
алюминия, магния, фосфатов кальция, основных солей алюминия)
избирательно сорбировать из воды ионы F~ по механизму ионного
обмена. В зависимости от качества исходной воды фториды могут
извлекаться в процессе обработки сорбцией их свежеосажденными
хлопьями указанных сорбентов или при фильтровании воды через
слой сорбента. Последнее практикуется для подземных вод, не тре-
бующих дополнительной обработки. Эффективным сорбентом фто-
ридов является свежеосажденный гидроксид магния. При наличии
в воде фторидов проходит обмен ионов по схеме Alg(OH)2+
+ F~^MgOHF + OH_. Свежеосажденный фосфат кальция, образу-
ющийся при введении 1%-ной фосфорной кислоты в известковое
молоко, избирательно сорбирует из воды ионы F-.
143
Снижение концентрации фтора при обработке воды коагулиро-
ванием объясняется способностью гидроксида алюминия и особен-
но гидроксосульфата алюминия A1OHSO4 сорбировать ионы F~.
Для удаления из воды фтора может быть использован и основной
хлорид алюминия.
Дефторирование воды, не требующей дополнительной очистки,
осуществляется фильтрационными методами. В качестве загрузки
фильтров могут быть применены обожженный доломит, активиро-
ванный оксид алюминия, гидроксилапатит. Механизм удаления
фтора данными методами аналогичен описанному, но осуществля-
ется в динамических условиях. Наиболее распространенным сор-
бентом для этой цели служит активированный оксид алюминия.
Активация осуществляется по методике, разработанной в ВНИИ
ВОДГЕО. Она состоит в том, что товарный оксид алюминия про-
каливается в течение 3 ч при 800° С, затем обрабатывается
15%-ным раствором кальцинированной соды и снова выдержива-
ется 30 мин при 800° С. В результате такой обработки получается
активированный оксид алюминия, обладающий свойствами анио-
нита. Регенерация сорбента осуществляется 2%-ным раствором
едкого натра (с последующей нейтрализацией) или раствором суль-
фата алюминия. Если активация оксида алюминия проводится гид-
роксидом натрия, то процесс дефторирования описывается по схе-
ме ROH + F~^RF + ОНТ При использовании сульфата алюминия
активированный оксид алюминия проявляет свойства анионита в
сульфат-форме: R2SO4 + 2F_^2RF + SO4~~. Обменная емкость сор-
бента по фторид-иону уменьшается с повышением pH. Сорбцион-
ная емкость активированного оксида алюминия по фтору состав-
ляет 90—1000 г на 1 м3 загрузки фильтра. При выборе метода уда-
ления фтора из воды необходимо проводить пробное дефторирова-
ние воды.
§ 34. Удаление из воды растворенных газов
Процесс удаления растворенных газов из воды называется дега-
зацией. В воде, используемой для технологических нужд, часто яв-
ляется нежелательным присутствие таких газов, как сероводород,
кислород, диоксид углерода. Для удаления растворенных в воде
газов применяются физические и химические методы.
Согласно закону Генри — Дальтона количество газа, растворен-
ного в воде, зависит от парциального давления его над раствором.
Поэтому газы, которые практически отсутствуют в атмосфере или
содержатся в незначительном количестве (сероводород, углекислый
газ), могут быть удалены при контакте с воздухом, т. е. при аэра-
ции воды. Для газов, парциальное давление которых в воздухе до-
статочно велико, например кислорода, создаются условия для умень-
шения растворимости их в воде. Это достигается подогревом воды
при обычном давлении или осуществлением процесса в вакуумных
дегазаторах без повышения температуры. Так, сероводород из воды
обычно удаляют аэрацией. Эффективность процесса зависит от pH
144
обрабатываемой воды. При pH<5 в воде находится практически
только свободный сероводород и удаление его идет достаточно пол-
но. При аэрации без подкисления степень удаления составляет
65—70%, так как сероводород в форме гидросульфидов почти не
удаляется из воды. Применение аэрации с подкислением для удале-
ния сероводорода необходимо сочетать с последующим подщела-
чиванием воды во избежание коррозионного действия воды на ап-
паратуру. Для удаления небольшого (2—3 мг/л) избытка серово-
дорода из воды можно использовать аэрацию без подкисления (при
рН=<6,5). Остаточное содержание сероводорода в этих условиях
составляет 0,3—0,5 мг/л. При аэрации воды одновременно проис-
ходит и химическое окисление сероводорода по уравнению 2H2S +
+ O2 = 2S + 2H2O. Для удаления образующейся серы в воду после
аэрации с подкислением и последующей стабилизации вводят коа-
гулянты.
Химические методы заключаются в связывании растворенных в
воде газов в новые химические соединения. Так, для более полного
удаления молекулярного сероводорода и гидросульфидов из воды
применяются химические методы, основанные на их окислении и пе-
реводе в труднорастворимые соединения. Наибольшее распростра-
нение получил метод удаления сероводорода, основанный на дей-
ствии хлора. В зависимости от дозы хлора окисление сероводорода
может проходить с образованием свободной серы или сульфатов
по уравнениям:
H2S + С12 = 2НС1 + S
H2S + 4С12 + 4Н2О = H2SO4 -Ь 8НС1
Реакция образования сульфатов идет с малой скоростью и тре-
бует четырехкратного увеличения количества хлора, что повышает
стоимость обработки воды.
Метод аэрации может дополняться биохимическим окислением
сероводорода серобактериями в аэроокислителе или в аэротенке.
Серобактерии могут окислять сероводород до серы и сульфатов. Ис-
пользование биохимического метода окисления сероводорода встре-
чает затруднения, связанные с необходимостью нейтрализации воды
и постоянным зарастанием загрузки карбонатом кальция.
Растворенный в воде кислород удаляют в термических деаэра-
торах и вакуумных дегазаторах. Можно использовать и химические
методы, основанные на окислительных процессах с участием кисло-
рода и таких восстановителей, как диоксид серы, сульфит натрия,
гидразин или металлическое железо. При пропускании воды через
фильтры из стальной стружки кислород окисляет железо. Образую-
щийся шлам, состоящий из оксида железа (III), удаляют при про-
мывании фильтра. При обработке воды диоксидом серы, сульфитом
натрия или гидразином протекают реакции по уравнениям
2SO2 + О2 + 2Н2О = 2H2SO4
2Na2SO3 + О2 = 2Na2SO4
NH2 — NH2 + O2 = N2 + 2H2O
145
Как следует из приведенных уравнений, при обескислорожива-
нии воды диоксидом серы и сульфитом натрия наблюдается увели-
чение солесодержания. При использовании гидразина наблюдается
эффективное обескислороживание воды без изменения ее химичес-
кого состава. Обработка воды гидразином может производиться
для глубокого обескислороживания после предварительного удале-
ния кислорода физическими методами. Обескислороживание воды
гидразином является эффективным, но дорогим методом.
Диоксид углерода можно удалять из воды физическими и хи-
мическими методами. Так как парциальное давление углекислого
газа в воздухе невелико, то он удаляется аэрацией воды. Химичес-
кие методы удаления углекислого газа основаны на повышении
щелочности воды введением таких реагентов, как известь или гид-
роксид натрия:
Са(ОН)2 + 2СО2 = Са(НСО3)2, NaOH + СО2 = NaHCO3
Физические методы дегазации имеют ряд достоинств: их использо-
вание не требует реагентов, усложняющих процесс очистки; не из-
меняется солевой состав воды, улучшаются условия работы обслу-
живающего персонала. Но этими методами не всегда достигается
необходимая эффективность удаления газов. Физические методы
дегазации являются более распространенными и иногда сочетают-
ся с химическими. Химические методы позволяют получить боль-
ший эффект дегазации и поэтому используются при глубокой очи-
стке воды.
§ 35. Очистка природных вод от органических примесей
Органические вещества, образующиеся непосредственно в водо-
емах или попадающие в них из окружающей среды, часто оказыва-
ют отрицательное воздействие на свойства воды, в первую очередь
на такие органолептические показатели, как цветность, запах, вкус.
Основными естественными компонентами природных вод, обуслов-
ливающими цветность воды, являются растворимые соли железа
гумусовых кислот. Цветность природных вод может быть обуслов-
лена и присутствием дубильных веществ, которые попадают вводу
при лесосплаве. Устранить цветность, особенно значительную, не
всегда удается достаточно полно. Поэтому в практике водоочистки
иногда применяются специальные методы удаления органических
веществ, обусловливающих цветность, запахи и привкусы воды.
Основными источниками появления запаха и привкуса воды служат
соединения, выделяемые водорослями, анаэробными бактериями,
актиномицетами и другими микроорганизмами. Так, сильный зем-
листый запах имеют продукты выделения одного из видов актино-
мицетов Actinomyces coelicolor. Водоросли выделяют в воду фенол-
содержащие соединения, органические кислоты, поверхностно-ак-
тивные и другие вещества, способные ухудшить качество воды.
В результате массового отмирания водных организмов, например в
осенний период, в воду поступает большое количество продуктов
146
распада с резким запахом (меркаптаны, сероводород, фенолы, выс-
шие спирты, альдегиды, ацетон и др.)- Большое количество орга-
нических веществ попадает в водоемы и из почвы. Это могут быть
не только естественные компоненты почвы, но вещества, применяе-
мые для защиты растений от вредителей (пестициды). Они наряду
с некоторыми другими органическими примесями сточных вод обус-
ловливают возникновение запахов и привкусов искусственного про-
исхождения. Так, наличие в воде 0,72 мг/л гексахлорана С6Н6С1б
вызывает запах интенсивностью в 5 баллов. ПДК вещества 2, 4, 6-
трихлорфенола по изменению органолептических свойств воды (по-
явлению запаха) составляет 0,0004 мг/л. Для анионных поверхно-
стно-активных веществ (алкилсульфатов и алкилсульфонатов
натрия) ПДК по появлению привкуса у воды составляет 0,5 мг/л.
Методы удаления органических веществ из вод можно разде-
лить на две группы: окислительные и адсорбционные. В качестве
окислителей органических примесей природных вод используются
хлор, озон, перманганат калия, т. е. реагенты, применяемые и для
обеззараживания воды. В процессе обработки воды хлором в ос-
новном идут реакции окисления и замещения, которые при опти-
мальной дозе окислителя сопровождаются образованием соедине-
ний, не имеющих запаха, цвета и вкуса. Хлор легко окисляет аль-
дегиды, спирты, аминокислоты, действует на некоторые компонен-
ты, вызывающие цветность воды (апокренаты железа). Кренаты
железа окисляются хлором хуже. Обесцвечивание воды идет наи-
более эффективно при pH 7,5—-8,0, основная роль при этом отво-
дится хлорноватистой кислоте и гипохлорит-иону, образующимся
при гидролизе хлора в воде. Органические примеси окисляются
только тогда, когда окислительный потенциал введенного реагента
будет достаточным для протекания реакции с органическим веще-
ством. Так, применение хлора является не всегда эффективным для
окисления веществ, вызывающих запахи и привкусы воды. Коли-
чество хлора, необходимое для их окисления, выше оптимальной
дозы хлора для обеззараживания воды.
Хлорирование воды для снижения содержания органических ве-
ществ обычно проводится повышенными дозами хлора перед об-
работкой воды. Доза введенного хлора может достигать 20—30 мг/л.
Если хлорирование для улучшения органолептических свойств воды
проводится после очистки, то часто возникает необходимость де-
хлорирования воды, так как доза хлора при этом выше, чем не-
обходимо для обеззараживания воды. Для снижения цветности
воды на 3 град требуется 1 мг хлора при дозе 5 мг/л. Хлорирование
с аммонизацией эффективно лишь для предотвращения запахов
хлорфенолов. Оптимальная доза хлора выбирается по точке пере-
лома на кривой хлоропоглощения. Доза перманганата калия при
перхлорировании воды обычно составляет 0,3—0,5 мг/л.
Лучший эффект обесцвечивания и дезодорации воды дает при-
менение озона в качестве окислителя. Более высокий окислитель-
ный потенциал озона способствует энергичному окислению многих
органических веществ. Так как озон разрушает бензольные ядра,
147
то он энергично воздействует на фенолы и вещества, обусловливаю-
щие цветность воды. Озон способствует удалению запахов, возни-
кающих в результате жизнедеятельности водорослей и актиноми-
цетов. Доза озона для дезодорации воды составляет 0,5—1 мг/л, для
обесцвечивания — до 10 мг/л и выше.
В качестве окислителя органических веществ в практике водо-
очистки применяется перманганат калия. В нейтральной среде мар-
ганец (VII) восстанавливается до марганца (IV), который выде-
ляется в форме диоксида марганца (IV), труднорастворимого в
воде. При малой концентрации диоксида марганца (IV) вода вна-
чале приобретает бурую окраску и через некоторое время образу-
ются бурые хлопья, которые являются, в свою очередь, хорошими
сорбентами коллоидных примесей воды. Восстановление марганца
(VII) в марганец (IV) и окисление органических веществ осуще-
ствляется по схеме
MnOy + 2Н2О + 3e->MnO2 + 4ОН—
Перманганат калия эффективно окисляет органические вещест-
ва, обладает высоким бактерицидным действием. Перманганат ка-
лия может применяться для дезодорации воды в сочетании с хло-
ром, а иногда с активированным углом.
Адсорбционные методы извлечения из природных вод водораст-
воримых органических веществ основаны на применении активиро-
ванного угля (АУ). При обработке воды АУ в статических или ди-
намических условиях происходит снижение цветности воды, устра-
няются запахи и привкусы. Активированный уголь имеет высоко-
развитую поверхность, обусловленную наличием тонких каналов и
пор. Он является хорошим сорбентом для фенолов, спиртов, ПАВ,
продуктов жизнедеятельности водных организмов. Сорбционная ем-
кость АУ возрастает с увеличением молекулярной массы сорбируе-
мого органического вещества. Сорбционная емкость АУ в щелоч-
ной среде уменьшается. Органические вещества, хорошо диссоции-
рующие в воде, сорбируются в меньшей степени, чем неионные или
слабодиссоциированные. Обычно для дезодорации воды доза угля
составляет 10—15 мг/л при времени контакта с водой 10—20 мин.
Так как концентрации органических веществ в природных водах,
вызывающих ухудшение органолептических свойств, очень малы, то
сорбционная емкость АУ в статических условиях для этих веществ
бывает недостаточной.
Эффективно удаляются органические вещества в адсорберах со
взвешенным слоем АУ. Высокая степень удаления органических
веществ наблюдается при фильтровании воды через фильтры, за-
груженные гранулированным АУ. Однако этот способ довольно до-
рогой. Кроме того, вода, поступающая на фильтр, должна быть
предварительно подвергнута полной очистке. Для увеличения сорб-
ционной емкости АУ дезодорацию проводят перед введением хло-
ра. При повышении температуры обрабатываемой воды эффектив-
ность сорбции органических примесей АУ уменьшается.
148
Активированный уголь легко самовоспламеняется, поэтому хра-
нить его следует в герметически закрытой таре, в отдельном поме-
щении. Регенерируют АУ обработкой щелочами пли кислотами,
полная регенерация осуществляется термическим методом при про-
каливании угля при 700—800° С с ограниченным доступом кисло-
рода.
Преимущества очистки воды АУ заключаются в простоте реа-
гентного хозяйства, сохранении постоянства состава воды, высоком
эффекте дезодорации воды, возможности улавливания нежелатель-
ных органических примесей и снижении бактериального загряз-
нения.
При очистке природных вод для удаления летучих органичес-
ких соединений применяется аэрация. Продувка воздухом осущест-
вляется перед обработкой воды (преаэрация). Применение этого
метода бывает иногда достаточно для дезодорации воды и повы-
шения эффективности процесса коагулирования.
В последние годы ведется разработка новых методов обесцве-
чивания и дезодорации воды. Один из них — радиационный метод
обесцвечивания и дезодорации воды — заключается в радиолизе
воды под действием у-излучения 60Со и взаимодействии продуктов
радиолиза с органическими примесями воды. При облучении воды
в сочетании с барботажем воздухом доза излучения, необходимая
для снижения цветности воды с 50 до 20 град, составляет
1 • 104 Дж/кг (0,1 Мрад). Хлорфенольные запахи с пороговым чис-
лом 250 (запах перестает ощущаться при разбавлении в 250 раз)
снимаются той же дозой излучения без барботажа воздухом.
Доза излучения, необходимая для полного устранения землис-
того запаха, составляет 1 -102 Дж/кг. Эффект облучения не зависит
от концентрации веществ в воде, вызывающих появление запаха.
При всех достоинствах радиационного метода обесцвечивания и
дезодорации воды необходимы дальнейшие исследования по изу-
чению влияния излучения на свойства обработанной воды. Не ис-
ключена возможность образования соединений, не влияющих на
вкусовые качества воды, но токсичных.
Глава IV. ОБЕЗЗАРАЖИВАНИЕ ВОДЫ
Процесс обработки воды, проводимый для полного удаления па-
тогенных и снижения общего числа микроорганизмов, называется
обеззараживанием. Оно может осуществляться химическими и фи-
зическими методами. К химическим методам относятся хлориро-
вание, озонирование, обработка солями тяжелых металлов и др.
Обеззараживание воды физическими методами заключается в воз-
действии на микроорганизмы ультрафиолетовых лучей, ультразву-
ка, высокой температуры. В последние годы изучается возможность
использования для обеззараживания воды методов электрофореза,
электрокоагуляции, электрического разряда, комплекса электриче-
ских воздействий, гиперфильтрации, ионизирующих излучений.
149
§ 36. Хлорирование воды
Наиболее распространенным методом обеззараживания воды
является обработка газообразным хлором или его кислородными
соединениями. Обеззараживающее действие хлора проявляется в
хлорировании и окислении органических веществ, содержащихся в
воде. При обычных условиях хлор — ядовитый газ с резким запа-
хом, зеленовато-желтого цвета. При —34,6е С и нормальном давле-
нии или при повышении давления до 607,950 кПа и 15е С хлор
сжижается. Хлор в 2,45 раза тяжелее воздуха, в воде растворим
Температура, °C 0 10 20 25 сравнительно мало. Раство-
Растворимость 14,6 9,97 7,29 6,40 римость хлора в воде зави-
хлора в воде, сит от температуры,
г/л
Нормальный окислительный потенциал хлора равен 1,36 В.
Раствор хлора в воде называется хлорной водой. При темпера-
туре ниже 0° С из хлорной воды выделяются гидраты хлора общей
формулы С12-«Н2О. Наиболее вероятной формулой гидрата счи-
тается С12-8Н2О. Хлор является очень активным элементом. Он
взаимодействует почти со всеми металлами и многими неметалла-
ми. Непосредственно с кислородом хлор не соединяется, по извест-
ны его оксиды (С12О, С1О2, СЮ3, С12О7), а также кислородсодер-
жащие кислоты (НСЮ, НС1О2, НС1О3, НС1О4). В практике обезза-
раживания воды применяются свободный хлор, соли хлорноватис-
той кислоты (гипохлориты), а также диоксид хлора СЮ2. При ра-
створении хлора в воде протекает гидролиз с образованием двух
кислот: хлорноватистой и хлороводородной (соляной):
С12 + Н2О^НС1О + НС1
В щелочной среде равновесие гидролиза хлора смещается впра-
во в результате реакции нейтрализации:
НСЮ + НС1 + 2NaOHzNaCl + NaClO + 2Н2О
Хлорноватистая кислота очень слабая, выделяется из солей при
взаимодействии их даже с угольной кислотой, является сильным
окислителем. В растворах диссоциирует с образованием гипохло-
рит-иона: НСЮ+И1+ + CIO-. Константа диссоциации хлорноватис-
той кислоты при 18° С равна 3,7 • 10-8.
В зависимости от pH основными устойчивыми составляющими
водных растворов хлора являются С12, НСЮ, СЮ-. Соотношение
между этими формами соединений хлора в воде показано на рис. 20.
Кроме того, в воде могут присутствовать С12О, СЮ3, образующиеся
при разложении хлорноватистой кислоты. При pH 6,5-?8,5 в воде
содержатся преимущественно хлорноватистая кислота и гипохло-
рит-ион. Нормальные окислительные потенциалы хлорноватистой
кислоты и гипохлорит-иона составляют соответственно 1,63 и 0,4 В.
Так как окислительно-восстановительный потенциал гипохлорит-
иона меньше, то с повышением pH обеззараживающее действие
хлора снижается.
150
Механизм бактерицидного дей-
ствия хлора и его кислородсодер-
жащих соединений заключается во
взаимодействии с составными час-
тями клетки микроорганизма, в пер-
вую очередь с ферментами. Потеря
биологической активности фермен-
тов может происходить в результате
реакций окисления, хлорирования,
замещения. Изменения в структуре
ферментов ведут к нарушению об-
мена веществ в клетке микроорга-
Рпс. 20. Соотношение содержания
различных форм хлора в воде в
зависимости от pH
низма и ее отмиранию.
Количество хлора, содержащееся
в реагенте и способное вступать в
реакции взаимодействия с составны-
ми частями клеток микроорганизмов и другими примесями воды,
характеризует концентрацию активного хлора. Содержание актив-
ного хлора в соединении рассчитывается на молекулярный хлор с
учетом числа электронов, принятых окислителем при взаимодейст-
вии с иодидом калия в кислой среде:
С12 + 2KI + H2SO4 = I2 + K2SO4 + 2НС1
(а)
NaClO + 2KI + H2SO4 = I2 + K2SO4 + NaCl + Н2О (б)
NH2C1 н- 2KI 4- H2SO4 = NH4C1 + K2SO4 4- 12 (в)
Ca(CIO)2 4-4KI + 2H2SO4 = СаС12 + 2K2SO4 + 2I2 + 2H29 (r)
Один электрон, принятый окислителем, соответствует атомной
массе хлора (35,5). Из уравнений реакций следует, что в реакциях
(а) — (в) окислитель принимает по 2 электрона. Следовательно,
одному молю каждого из этих веществ соответствует 71 г актив-
ного хлора. При взаимодействии иода с гипохлоритом кальция окис-
литель принимает 4 электрона, поэтому каждому молю гипохло-
рита кальция будет соответствовать 142 г активного хлора.
Кроме хлора для обеззараживания воды применяются соли хлор-
новатистой кислоты — гипохлориты кальция и натрия и хлорная
известь, содержащая гипохлорит и хлорид кальция. Наиболее ве-
роятная формула хлорной извести Са(С1О)2-СаС12-2Н2О. Хлорную
известь получают хлорированием на холоду известкового молока
по уравнению
2Са(ОН)2 + 2С12 = Са(С1О)2 + СаС12 + 2Н2О
Хлорная известь представляет собой белый гигроскопичный по-
рошок с характерным запахом. Под действием диоксида углерода,
влаги, воздуха и света она разлагается:
Са(С1О)2 + СО2 + Н2О = СаСО3 + 2НСЮ
Окислительными свойствами обладает только активный хлор,
входящий в состав гипохлорит-иона. При растворении хлорной из-
вести происходит взаимодействие с водой с образованием хлорно-
151
ватистой кислоты. Соотношение содержания хлорноватистой кисло-
ты и гипохлорит-иона изменяется в зависимости от pH.
Качество реагента, используемого для обеззараживания воды,
определяется содержанием активного хлора. Техническая хлорная
известь должна содержать не менее 32—35% активного хлора. Вс
избежание потери активности хлорную известь транспортируют и
хранят в закрытой таре. Применяется она главным образом на не-
больших водопроводных станциях и для локального обеззаражи-
вания.
В последние годы для обеззараживания воды вместо хлорной
извести применяют гипохлорит кальция. Содержание активного
хлора в нем достигает 50—60%. Гипохлорит кальция представляет
собой порошок белого цвета. Его транспортируют и хранят в ме-
таллических барабанах. Пыль гипохлорита вызывает раздражение
слизистых оболочек дыхательных путей, глаз, кожи. Во избежание
взрыва нельзя допускать загрязнения продукта органическими со-
единениями, маслами.
В практике обеззараживания воды используется также гипохло-
рит натрия, который получают действием хлора на гидроксид нат-
рия. В Академии коммунального хозяйства им. К. Д. Памфилова
разработана конструкция электролизной установки, позволяющая
получать гипохлорит натрия непосредственно на очистных соору-
жениях. Уже начат серийный выпуск таких электролизеров. Гипо-
хлорит натрия образуется при взаимодействии продуктов электро-
лиза хлорида натрия (гидроксида натрия и хлора): 2NaOH + Cl2 =
— NaClO + NaCl + H2O. Гипохлорит натрия, вводимый в воду, гидро-
лизуется с образованием хлорноватистой кислоты: NaClO + HoO^
^NaOH + HClO.
Для обеззараживания воды на локальных установках и в по-
левых условиях применяются органические хлорамины. Это — со-
единения, в которых один или оба атома водорода в аминогруппе
—NH2 замещены атомами хлора. Различают монохлорамины
с общей формулой R—NHC1 и дихлорамины RNC12, где R — орга-
нический радикал. В практике обеззараживания воды применяют-
ся дихлорамин Б C6H5SO2NC12 и хлорамин Т СН3—С6Н4—
—SO2NClNa-2H2O. Содержание активного хлора в хлорамине Б
составляет 25,5%. Гидролиз хлораминов в воде проходит значи-
тельно медленнее, чем других хлорсодержащих реагентов, применя-
ющихся для обеззараживания, поэтому активный хлор сохраняется
в воде более длительное время. Бактерицидное действие хлорами-
нов обусловливается тем, что гидролиз их в зависимости от pH
сопровождается образованием хлорноватистой кислоты или гипо-
хлорит-иона.
В некоторых странах для обеззараживания воды применяется
диоксид хлора С1О2. Он представляет собой газ зеленовато-жел-
того цвета, с резким запахом. Растворимость диоксида хлора в
воде при парциальном давлении 13,595 кПа (100 мм рт. ст.) и 5° С
составляет 21 г/л, при 20° С—17 г/л. Диоксид хлора — соединение
нестойкое. В контакте с органическими веществами, при нагрева-
152
ншг до 60° С, электрическом разряде, на прямом солнечном свету
взрывается с образованием хлора и кислорода. В кислой среде он
устойчив, при повышении pH начинается гидролиз. Нормальный
окислительный потенциал диоксида хлора в кислой среде равен
1,5 В. Диоксид хлора получают непосредственно на водопровод-
ных очистных сооружениях из хлорита натрия ХаС1О2 и хлората
натрия NaC103.
Диоксид хлора обладает более сильным обеззараживающим
действием, чем хлор. Время контакта С1О2 с водой, необходимое для
обеззараживания, меньше, чем для хлора. Диоксид хлора окисляет
фенолы с разрушением бензольного кольца, поэтому может быть
использован для уничтожения фенольных запахов воды. Примене-
ние диоксида хлора для обеззараживания воды сдерживается не-
достаточным производством хлорита натрия.
Хлоропоглощаемость воды. Выбор оптимальной дозы хлора.
Эффект обеззараживания воды зависит от сочетания многих фак-
торов, среди которых наибольшее значение имеют биологические
особенности микроорганизмов, бактерицидное действие реагентов,
состояние водной среды; условия, в которых происходит процесс
обеззараживания. Прежде всего эффективность обеззараживания
определяется устойчивостью (резистентностью) микроорганизмов к
воздействию реагента. Большую устойчивость при обеззаражива-
нии проявляют споровые формы микроорганизмов.
Бактерицидная активность реагентов определяется окислитель-
ным потенциалом и способностью взаимодействовать с составными
частями клеток микроорганизмов. Обеззараживающее действие
реагента зависит от таких свойств обрабатываемой воды, как pH,
наличие органических и неорганических веществ, способных к окис-
лению, взвешенных и коллоидных примесей. Энергично взаимодей-
ствуют с хлором азотсодержащие органические вещества. Присут-
ствие в воде взвешенных веществ резко ухудшает процесс обезза
раживания, так как хлор адсорбируется частицами взвеси. Но в то
же время микроорганизмы, находящиеся внутри этих частиц, не
подвергаются воздействию обеззараживающего реагента. Хлор реа-
гирует и с такими неорганическими восстановителями, как сульфи-
ты, сероводород, соединения железа (II), аммиак.
Общее количество хлора, которое расходуется на взаимодейст-
вие с окисляющимися примесями, определяет хлоропоглощаемость
(хлороемкость) воды. Она зависит от свойств примесей, содержа-
щихся в обрабатываемой воде, времени контакта хлора с водой,
температуры воды и количества введенного хлора. При концентра-
ции до 1 мг/л хлоропоглощаемость изменяется пропорционально
времени контакта, при более высоких концентрациях обнаружива-
ется логарифмическая зависимость. От температуры воды хлоро-
поглощаемость зависит незначительно. Время контакта хлора с во-
дой, необходимое для обеззараживания воды, составляет 30 мин.
Количество введенного хлора должно быть таким, чтобы после
окисления примесей в воде остался некоторый избыток активного
хлора — остаточный хлор. Наличие остаточного хлора в воде по-
153'
Рис. 21. Кривые хлоропоглощаемости
воды:
1 — в отсутствие аммонийных соединений;
2—при наличии в воде аммонийных со-
единений
вышает бактерицидный эффект обеззараживания. Определение ос-
таточного хлора в воде, не подвергающейся предварительному
осветлению, проводится через каждые полчаса, а на установках с
предварительным осветлением — через каждый час.
Ориентировочные дозы хлора для отдельных видов природных
вод выбирают по окислясмости: 1) воды с окислясмостью до Юмг/л
(грунтовые, озерные и др.) имеют хлороемкость 0,6—2,5 мг/л; 2) во-
ды с окислясмостью от 10 до 20 мг/л — 2,5—4,5 мг л; 3) воды с
окислясмостью выше 20 мг/л — от 5 мг/л и выше. Хлоропо! лощае-
мость воды определяется соотно-
шением действия многих факто-
ров и не является постоянной. По-
этому она, как и оптимальная до-
за хлора, определяется экспери-
ментально с помощью графиков,
характери дующих зависимость
количества остаточного хлора от
дозы хлора (рис. 21). Кривая 1
на рис. 21 соответствует измене-
нию хлоропоглощаемости воды в
отсутствие аммиака и его соеди-
нений. Отрезок ОС па осп абс-
цисс характеризует величину
хлороемкости воды. Наличие на
кривой 2 максимума (точка Л) и
минимума (точка В) указывает на
присутствие в воде аммонийных
соединений. Хлор взаимодействует с аммиаком и его соединениями
с образованием неорганических моно- и дихлораминов. В интерва-
ле pH от 4,4 до 8 в воде могут присутствовать моно- и дихлорами-
ны, при более высоких значениях pH образуется только монохло-
рамин. Соотношение содержания моно- и дихлораминов в воде за-
висит от концентрации хлора и аммиака. При соотношении
С12 : NH3^4 : 1 в интервале pH 5—9 в воде образуется монохло-
рамин.
Уравнения реакции образования хлораминов:
NH3 + С12 = NH2C1 + НС1; NH3 + НС1О = NH2C1 -у H2O
NH3 + 2С12 = iNHClo + 2HCI
Участок ОА на кривой 2 соответствует образованию хлораминов.
В воде наряду с хлораминами содержится свободный хлор. Макси-
мум на кривой 2 (точка Л) соответствует полному связыванию ам-
миака и его соединений в хлорамины. При дальнейшем увеличении
количества хлора начинается разложение хлораминов:
NHC12 + NH2C! = N2 + 3HCI
4NH2CI 4- 3CI2 + H2O = n2 + N2O + 10HC1
Минимум на кривой 2 (точка перелома В) соответствует полному
разложению хлораминов. При дальнейшем увеличении количества
хлора наблюдается пропорциональная зависимость между количе-
154
ством введенного и остаточного хлора, и в воде находятся только
свободный хлор н продукты его гидролиза.
Хлор, входящий в состав хлорноватистой кислоты и гипохлорит-
иона, называют свободным активным хлором, а содержащийся в
хлораминах — связанным активным хлором. Количество хлора,
необходимое для разрушения образовавшихся в воде хлораминов,
должно быть примерно в 10—20 раз больше концентрации аммиака
в воле. Если хлорирование воды ведется дозой хлора, соответст-
вующей точке А на кривой 2, то в воде находится только связан-
ный (хлора.мшшый) активный хлор. При дозе хлора, которая соот-
ветствует точке перелома В пли при большей дозе в воде содер-
жится только свободный активный хлор.
Согласно ГОСТ 2874—73 концентрация свободного остаточного
хлора должна быть не более 0,5 и не менее 0,3 мг/л после 30-минут-
ного контакта хлора с водой, а концентрация связанного остаточ-
ного хлора должна быть не менее 0,8 и не более 1,2 мг/л после
часового контакта.
Методы хлорирования воды. Описанные условия образования
и разрушения неорганических хлораминов в воде лежат в основе
комбинированного метода обеззараживания — хлорирования с
аммонизацией. Этот метод применяется для вод, имеющих запахи
или привкусы. В обрабатываемую воду кроме хлора вводится ам-
миак или сульфат аммония. Если обработка аммиаком предназна-
чается для снижения интенсивности запахов исходной воды, то ам-
миак вводится до хлорирования воды. Если аммонизация прово-
дится для маскирования запаха хлора, аммиак подается в воду
после обеззараживания. Связанный остаточный хлор более дли-
тельное время сохраняется в воде, так как гидролиз хлораминов
идет значительно медленнее, чем свободного хлора. Большой опыт
по применению хлорирования с аммонизацией накоплен на водо-
проводных сооружениях Москвы и Киева.
При наличии в воде органических веществ, вызывающих запа-
хи, привкусы, используется другой комбинированный метод — хло-
рирование с манганированием. В очищаемую воду дополнительно
вводится перманганат калия, доза которого зависит от места вве-
дения его в воду. Если перманганат калия поступает в воду до ее
осветления, то доза его может достигать 1 мг/л, так как продукт
восстановления (диоксид марганца) задерживается при фильтро-
вании. Доза перманганата калия, вводимого после обработки в во-
ду, не должна превышать 0,08 мг/л. Перманганат калия является
эффективным обеззараживающим реагентом и способствует улуч-
шению органолептических свойств воды. Однако по данным неко-
торых исследований при действии перманганата калия возможно
образование токсичных продуктов деструкции органических веществ.
Для обеззараживания воды плавательных бассейнов применя-
ется хлормедный метод. Он заключается в одновременном введении
в воду хлора и сульфата меди (II). При использовании хлормед-
ного метода обеззараживание воды обеспечивается введением
3 мг/л ионов Си2+ при 0,3 мг/л свободного остаточного хлора. Иног-
155
да в практике водоподготовки для обеззараживания воды хлориро-
вание сочетается с введением солей серебра (хлорсеребряный ме-
тод) .
В зависимости от времени введения хлора в воду различают
предварительное хлорирование (прехлорирование) и постхлориро-
вание, завершающее процесс очистки воды. Прехлорирование про-
водится обычно с повышенными дозами хлора. При постхлорирова-
нии доза остаточного хлора строго регламентируется.
В зависимости от количества вводимого хлора различают нор-
мальное хлорирование и перехлорирование. При нормальном хло-
рировании для обеззараживания или при хлорировании как про-
филактическом мероприятии доза хлора регламентируется задан-
ной концентрацией остаточного хлора. Перехлорированием называ-
ется обеззараживание повышенными дозами хлора. Оно проводится
при пуске новых водопроводных сетей, для предупреждения обрас-
таний, а также для снижения интенсивности запахов и привкусов
исходной воды. При значительном загрязнении органическими ве-
ществами и непостоянстве бактериологических показателей воду
хлорируют дважды: перед очисткой и после — для обеззаражи-
вания.
Иногда необходимо осуществлять дехлорирование — удаление
избытка остаточного хлора. Химические методы дехлорирования
основаны на взаимодействии хлора с такими восстановителями, как
диоксид серы, сульфит, гидросульфит или тиосульфат натрия. Неко-
торые реакции, происходящие при дехлорировании воды, можно
описать следующими уравнениями:
SO2 + С12 + 2Н2О = 2НС1 + H2SO4
Na2SO3 + Cl2 + Н2О = Na2SO4 + 2НС1
Na2S2O3 + Cl2 + Н2О = Na2SO4 + 2НС1 + S
Расчет необходимого количества реагентов производится по
уравнениям реакций с учетом того, что сульфит натрия может быть
безводным или в форме кристаллогидрата Na2SO3-7H2O, а крис-
таллический тиосульфат имеет состав Na2S2O3• 5Н2О. Например,
при использовании 1 моль безводного сульфита натрия (126 г) свя-
зывается 1 моль хлора (71 г), т. е. на 1 мг избыточного остаточного
хлора следует вводить 1,634 мг Na2SO3.
Физико-химический метод дехлорирования воды заключается в
сорбции хлора, хлорноватистой кислоты или хлораминов активиро-
ванным углем. Сорбция соединений хлора активированным углем
сопровождается и химическим взаимодействием, в результате кото-
рого возможно восстановление хлора и хлорноватистой кислоты до
хлоридов и окисление углерода до диоксида:
2НС1О + С = 2НС1 + СО2, 2С12 + С + 2Н2О = 4НС1 + СО2
Регенерация активированного угля проводится горячим раство-
ром гидроксида натрия.
156
§ 37. Обеззараживание воды солями тяжелых металлов
Метод обеззараживания воды солями тяжелых металлов назы-
вается олигодинамией. Он основан на том, что ионы тяжелых ме-
таллов, взаимодействуя с цитоплазмой клеток микроорганизмов,
вызывают нарушения, ведущие к их гибели. В качестве обеззара-
живающих средств применяются соединения серебра и меди (II).
Поступление ионов Ag+ можно обеспечить растворением солей
серебра, контактом воды с металлическим серебром, посеребрен-
ными зернами кварца. Наиболее эффективным методом обогащения
воды серебром является электрохимическое растворение серебря-
ного анода. Преимущество соединений серебра перед остальными
обеззараживающими реагентами заключается в том, что их бактери-
цидное действие сохраняется в течение длительного времени, т. е.
они являются одновременно и консервантами. Воздействие сереб-
ра на микробиальную клетку осуществляется в два этапа. Сначала
происходит сорбция серебра на поверхности клетки, затем наблю-
дается проникновение его в клетку, что ведет к инактивации фер-
ментов. Соединения серебра вызывают у кишечной палочки лизис
цитоплазмы, повреждение нуклеотидов и отторжение содержимого
клетки от оболочки. Бактерицидное действие серебра проявляется
при концентрации его в воде более 0,04 мг/л. При концентрации се-
ребра 0,1—0,2 мг/л кишечная палочка отмирает через 40—50 мин.
Эффективность действия реагента зависит также от дозы вводимых
ионов Ag+ и времени контакта с водой. Полное обеззараживание
достигается при двухчасовом контакте. Доза вводимого серебра ко-
леблется от 0,04 до 0,2 мг/л.
На эффект обеззараживания сильно влияет присутствие ионов,
дающих с ионами серебра труднорастворимые соединения (СН,
Вг_, S2~, I- и Др.). Лучший эффект обеззараживания достигается
при обработке осветленных вод. Ионы тяжелых металлов оказыва-
ют бактерицидное действие только на вегетативные формы микро-
организмов. При повышении температуры бактерицидное действие
значительно возрастает.
§ 38. Озонирование воды
Для обеззараживания воды и улучшения ее органолептических
свойств используется озон О3, молекула которого содержит три ато-
ма кислорода. Озон — газ голубого цвета с характерным запахом,
при —115° С сжижается. Озон является неустойчивым соединением
и более сильным окислителем, чем обычный кислород. Нормальный
окислительный потенциал озона в кислой среде равен 2,07 В, в ще-
лочной — +1,24 В. Его ПДК в воздухе составляет 0,0001 мг/л. Ра-
створимость его в воде при давлении 101,325 кПа (760 мм рт. ст.)
и 20° С составляет 1,04 г/л, при 0° С — 1,42 г/л. Во влажном воз-
духе и воде озон проявляет сильное коррозионное действие по отно-
шению ко многим металлам и сплавам. Более устойчивы к действию
озона нержавеющая сталь и алюминий. Распад озона в воде со-
157
провождается образованием перекисных соединений и свободных
радикалов (ОН, НОг и др.), обладающих высокой химической ак-
тивностью. Скорость разложения озона в воде увеличивается с по-
вышением pH, температуры, солесодержания и концентрации орга-
нических примесей. В кислой среде озон более устойчив, чем в ще-
лочной. Скорость разложения озона в воде описывается уравнением
dt °3
где k — константа разложения озона, зависящая от свойств обра-
батываемой воды; С — концентрация озона. Так, для дистиллиро-
ванной воды k = 0,229, для грунтовых вод /г = 2,383, для поверхност-
ных k = 10,31-4-22,32. Чем больше окисляющихся примесей в воде,
тем выше скорость разложения озона. Повышение температуры во-
ды от 1 до 20° С увеличивает скорость распада озона в 22 раза.
Некоторые уравнения реакций, протекающих с участием свободных
радикалов, образующихся при разложении озона:
ОН -у ОН->Н2О +0; О + ОН^НО2; НО2 + НО2^Н2О - О2
Озон разлагается с выделением теплоты. При распаде 2 моль
озона с образованием кислорода выделяется 297,4 кДж. Бактери-
цидное действие озона связано с протеканием реакций окисления
(озонолиз). Вступают в реакции также свободные радикалы, обра-
зующиеся при разложении озона в воде. Озон, являясь сильным
окислителем, разрушает ароматические ядра, но не действует на
пиридиновые кольца. Полная гибель спор наблюдается только при
дозе озона 10 мг/л и выше. Кишечная палочка практически полно-
стью отмирает при дозе озона 2 мг/л. При остаточной дозе озона
0,3 мг/л инактивация вирусов на 99,99% наблюдается через 4 мин.
Бактерицидное действие заметно проявляется при концентрации
озона в воде выше 0,1 мг/л. Время контакта озона с водой, необ-
ходимое для обеззараживания, составляет 3—10 мин. Доза озона,
необходимая для обеззараживания, составляет 1—4 мг/л. Она вы-
бирается пробным озонированием воды, контроль обеззараживания
ведется по остаточному озону. Концентрация остаточного озона,
равная 0,05—0,1 мг О3/л, обеспечивает бактерицидный эффект при
озонировании очищенной воды.
Доза озона, необходимая для обеззараживания, определяется и
соотношением числа вегетативных и споровых форм и поэтому из-
меняется в зависимости от времени года. В зимний период при от-
сутствии спор при дозе озона 3—4 мг/л общее число бактерий умень-
шается на 95%, а коли-индекс — на 100%. В весенний период,
когда преобладают споровые формы, общее число бактерий при той
же дозе озона снижается только на 30—40%, а коли-индекс — на
70—80%. В весенний и летний периоды бактерицидная доза озона
составляет 4—5 мг/л. При концентрации озона выше 3 мг/л у воды
появляется ароматический запах. Озонирование воды способствует
устранению цветности, запахов и привкусов воды. Для растворен-
ных соединений при обесцвечивании расходуется 0,05—0,15 мг озо-
158
на на 1 град цветности, для коллоидных — до 0,5 мг. Озоном хо-
рошо устраняются запахи, вызываемые актиномицстами, плесне-
выми грибами, водорослями, органическими и минеральными при-
месями.
Метод обработки воды с целью уничтожения запаха называется
дезодорацией. Эффективность процесса дезодорации определяется
природой примесей и интенсивностью запаха. При озонировании
воды улучшаются вкус и цвет воды. Для предотвращения биологи-
ческого обрастания водных трактов озонирование может сочетать-
ся с предварительным хлорированием. Для повышения степени
обесцвечивания и дезодорации воды используется двойное озони-
рование.
Озон получают непосредственно на очистных сооружениях дей-
ствием тихого электрического разряда па кислород воздуха в озо-
наторах при напряжении более 10 кВ. Предварительное осушение
озонируемого воздуха необходимо для предотвращения искровых
разрядов, вызывающих увеличение расхода электроэнергии и кор-
розию аппаратуры. Полученную озоно-воздушную смесь растворя-
ют в воде, а затем дозируют в обрабатываемую воду. Выход озона
из 1 м3 воздуха достигает 18 г. Получение озона — энергоемкий
процесс, поэтому обработка воды озонированием по сравнению с
хлорированием обходится дороже.
Впервые предложение о возможности использования озона для
обеззараживания и улучшения органолептических свойств воды
внес на Русском водопроводном съезде в 1901 г. Н. П. Зимин. Он
отметил: «...озонирование, кроме уничтожения в ней (воде) бакте-
рий сообщает ей нужную бесцветность, блестящий вид и приятный
освежающий вкус, оно устраняет из воды дурной запах и не вводит
в воду никаких посторонних веществ, кроме безусловно полезного
кислорода». В 1909 г. озонирование впервые было применено для
борьбы с эпидемией холеры в Петербурге. В настоящее время озо-
нирование является перспективным методом обеззараживания во-
ды. Озонирование воды применяется во многих странах (США, Ка-
наде, Швейцарии и др.), но особое распространение оно получило
во Франции, где примерно ’/з воды, поступающей из системы цент-
рализованного водоснабжения, озонируется. В 1974—1975 гг. на
Восточной водопроводной станции Москвы введена в строй круп-
нейшая в мире озонаторная установка, которая рассчитана на об-
работку 1,2 млн. м3/сут воды. Недостаток метода заключается в от-
сутствии эффекта последействия.
§ 39. Физические методы обеззараживания воды
При водоподготовке для обеззараживания применяются два
основных физических метода: облучение воды ультрафиолетовыми
лучами и воздействие ультразвуком. Преимущества физических ме-
тодов перед химическими заключаются в отсутствии реагентного
хозяйства, возможности полной автоматизации процесса обеззара-
живания. При их использовании достигается высокая степень обез-
159»
зараживания воды. К недостаткам этих методов следует отнести от-
сутствие эффекта последействия и невозможность непрерывного
эффективного контроля процесса обеззараживания.
Обеззараживание воды ультрафиолетовым излучением. Приме-
нение ультрафиолетового излучения для обеззараживания воды ос-
новано на том, что ультрафиолетовая часть спектра, соответствую-
щая длине волн света от 200 до 300 нм, обладает сильно выражен-
ным бактерицидным действием. Эта биологически активная часть
ультрафиолетовой части спектра называется бактерицидной. Мак-
симум бактерицидного действия проявляется у излучения с длиной
волны 260 нм. Об эффективности действия излучений с различной
длиной волны света можно сделать вывод по следующим данным:
если условно принять бактерицидное действие ультрафиолетового
излучения с длиной волны 253 нм за единицу, то воздействие излу-
чения с длиной волны 365 нм будет ниже в 4000, а с длиной волны
546 нм — в 30 000 раз меньше.
Бактерицидное действие ультрафиолетового излучения вызыва-
ется протеканием фотохимических реакций, ведущих к нарушению
обмена веществ в клетках микроорганизмов, образованию биологи-
чески активных веществ и коагуляции белковых макромолекул. При
дозе облучения, вызывающей 100%-ную гибель микроорганизмов
(3000 мВт-с/м2), наблюдается уменьшение клеток в объеме, истон-
чение клеточной оболочки и отделение от нее цитоплазмы. Однако
малые дозы излучения оказывают стимулирующее действие на жиз-
недеятельность микроорганизмов.
Бактерицидный эффект обеззараживания воды ультрафиолето-
вым излучением определяется мощностью источников света, способ-
ностью воды и содержащимися в ней примесями поглощать ультра-
фиолетовое излучение, толщиной слоя воды, временем воздействия
и устойчивостью микроорганизмов к излучению. Чем выше мощ-
ность источника ультрафиолетового излучения, тем меньшее время
требуется для уничтожения микроорганизмов. Наличие в обраба-
тываемой воде взвешенных примесей и соединений, вызывающих
цветность воды, ведет к снижению бактерицидного действия ультра-
фиолетового излучения вследствие поглощения части излучения
этими компонентами воды. Поэтому данный метод обеззаражива-
ния применим для вод с высокой прозрачностью и не содержащих
веществ, обусловливающих цветность. Наиболее целесообразно
использовать этот метод для обеззараживания подземных вод, так
как они имеют меньший коэффициент поглощения по сравнению с
водами, поступающими из поверхностных источников. Контроль за
обеззараживанием воды проводится по данным бактериологичес-
кого анализа.
Время облучения, необходимое для практически полного отми-
рания микроорганизмов, возрастает с повышением количества мик-
роорганизмов в обрабатываемой воде. Споровые формы более ус-
тойчивы к излучению, чем вегетативные. Скорость отмирания мик-
роорганизмов (до степени обеззараживания 99%) описывается
уравнением скорости мономолекулярной реакции.
160
В качестве источников излучения используются ртутно-кварце-
вые лампы высокого давления и аргоно-ртутные лампы низкого дав-
ления с увиолевым стеклянным баллоном. Первая установка для
обеззараживания воды ультрафиолетовым излучением в нашей
стране была пущена в эксплуатацию в г. Уфе. В настоящее время
этот метод применяется для обеззараживания воды на многих водо-
проводных станциях, использующих в качестве водоисточника под-
земные воды. Стоимость обеззараживания подземных высокопро-
зрачных и практически бесцветных вод ультрафиолетовыми лучами
ниже, чем при хлорировании.
Обеззараживание воды ультразвуком. Впервые в нашей стране
физический метод обеззараживания воды ультразвуком был при-
менен Л. Б. Доливо-Добровольским в 1941 г. В качестве источника
ультразвуковых колебаний используются пьезокварцевые излучаю-
щие пластинки и магнитострикционные вибраторы. Под действием
ультразвука наблюдается коагуляция белковых компонентов клетки
микроорганизмов. Кроме того, при этом возрастает скорость окис-
лительных процессов. Эффективность обеззараживания зависит от
времени воздействия ультразвука на воду, интенсивности ультра-
звукового поля, частоты ультразвуковых колебаний, толщины слоя
обрабатываемой воды.
Максимальное бактерицидное действие ультразвука наблюда-
ется при частоте колебаний 500—1000 кГц. Под действием ультра-
звука происходит отмирание как вегетативных, так и споровых
форм. Этот метод не находит широкого применения в практике обез-
зараживания воды из-за ограниченной мощности генераторов ульт-
развуковых колебаний.
Глава V. СТОЧНЫЕ ВОДЫ И МЕТОДЫ ИХ ОЧИСТКИ
Сточные воды делятся на бытовые, производственные и ливне-
вые. Они отличаются друг от друга своим происхождением, соста-
вом, биологической активностью и в связи с этим — способами очи-
стки. Сравнительная характеристика бытовых и производственных
сточных вод приведена в табл. 10 (С. Н. Черкинский).
§ 40. Характеристика бытовых сточных вод
Бытовые сточные воды образуются в результате практической
деятельности и жизнедеятельности людей. При этом изменяются
физические и химические свойства воды, а также ее бактериальная
загрязненность. Химический состав бытовых вод в отличие от про-
изводственных сточных вод не подвержен значительным качествен-
ным изменениям. Данные о количестве загрязнений, которые по-
ступают в бытовые сточные воды от одного жителя в течение су-
ток при наличии централизованного водоснабжения, приведены
ниже.
6—1244
161
Таблица 10. Характеристика бытовых и производственных сточных вод
Методы обезврежи- вания । ® > 1 СО । <7__> й н О к S S к га й о Д х 0J оэ = 2 Я и г-.'Й “ О О- 5 LO и т: О' и 7; Си “ х х о и ™
Гигиеническое значение । 2 Й g S - s 0 - ° ? g га я о -- 5 I г а - ’•'п О j- m О СО 77 s я 5 О .3 2 О О Й fei Я Я ~ О о с - Я Н Q О о ш л о я =: с- 9шАооя:-2^ >_ га си о я Д?н2о:аяясз — !—|ОССяяОг- 1 , о я я я га 3
T ипичность состава свойств — о <~ч га _ О О я; ^-ага\оогаяг~2“ -ч га г: я я а СО а Е « & и ® н ffl s А Е
Токсич- ность га га J 2 , я о я 2 “ 2 ~ я и ~ “ га га 2 ~ — 7 7 3 И 5 J-4 О и
Химический состав "'S П ° К Р ® о i п 1 6 о А 2 „ га о Q- — га га га я га X — о га - ora я о.<и0ояос>20к: Нга„.^яя я Я >, Я -3 га Я ' Я Я ни га_. СО GJ 77- СО К ггГ *7 г £2 L- сп —< Ч1 5 S “ 7 £ So а - ЕН s ч § с g §
Взвешен- Реакция ные среды вещества к - 2 L 2 га ,д я ч 2 к ~ g я 2 А К- g ex s я д и д я 2 2 9 Д к 2 а га: 2 я н 2 А Л 2 га" я Ц О й азо 23оя~я22Ви Н 5 ГО Q ~ О Я Я
к § и1 я >. 2 'Я с я я о Н я и 2 ° - н а 7 га О S О W К Я О g О О я я <и Е s 2 s X g. 5 S я у
Режим выпуска К к £ £ Е к ка ° га га i 3 ? 7 О ° S ® 5 ® 2 Г3ягашоя>?£а 2 Я 2 ОягахЗоО-я'ОЗшяяя О. я о га га S га я г
Внеш- ний вид II II О ^2 эы о со . В « га Я Я =S et сх £ м а. 3 О ю Д3 ю я О Ом о
Количество ESouKES6 Е о >, д 2я^ОХ"яЯгаЯ р; а. о с с - с >, е > рякгаОяоояяя Осшчч-&яя'юд OooEo-jrarax'o
Происхождение 07 ' 1 A Д А < О 07 , 1 Q Д *Д сд со о t _ с -Q о _ н Н О й ’G* <^^0 н Н -"7 m Д О- CQ М Д >, Ч КЮ К я я £ га; « а s о О g § § g £ га g СО >-> m С— л-, ® -1 Т-О*2* S 77 о д удя s 7 2 \о о. д 5к с-> о s д \по. o«£SSg;SS£ o«^£&8§SS^S.§g-
Воды 8 а Я.5 5 2 3 £ гаГ И и £ m ® Era со га га, ш s U
Показатель Количество загрязнений наП жителя ~ г/сут
Взвешенные вещества 65
БПК5 неосветленной жидкости 54
БПК5 осветленной жидкости 35
БПКполн неосветленной жид- 75
кости
БПКполн осветленной жидко- 40
сти
Азот аммонийный 8
Фосфаты (в расчете на Р2О5), 3,3
в том числе от моющих
средств 1.6
Хлориды 9
Поверхностно-активные веще- 2,5
ства
(VJ)
Концентрацию загрязне-
ний бытовых сточных вод с
учетом нормы водоотведе-
ния определяют по формуле
а 1000
С =---------------
q
где С — концентрация при-
меси. мг/л; а — количество
загрязнений, приходящееся
на одного жителя, г/л; q —
норма водоотведения от од-
ного жителя, л/сут.
В сточных водах содер-
жатся примеси минерально-
го и органического происхождения. Минеральные соединения в бы-
товых сточных водах представлены солями аммония, фосфатами,
хлоридами, гидрокарбонатами и другими соединениями, образую-
щимися в результате жизнедеятельности человека и разложения
органических веществ. Бытовые сточные воды имеют обычно сла-
бощелочную реакцию среды (pH 7,2—7,8). Минеральные и органи-
ческие примеси в воде могут в зависимости от степени дисперсно-
сти находиться во взвешенном, коллоидном и растворенном состоя-
ниях. Соотношение между содержанием минеральных и органиче-
ских соединений (по данным С. Н. Строганова) см. в табл. 11.
При медленном движении или отстаивании бытовых сточных вод
происходит выпадение твердой фазы, содержащей от 80 до 99% во-
Таблица 11. Содержание минеральных Ды- Она включает в себя
и органических загрязнений в бытовых частицы песка, шлака, во-
локна, органические остатки
сточных водах
Распределение, °/( различного происхождения. Взвешенные органические вещества при осаждении бы- стро загнивают. Органичес- кие вещества бытовых сточ-
Загрязнения нераст- вори- мые (осадок) сус- пензии КОЛЛО- ИДЫ раство- ренные
Минеральные Органические 5 15 5 15 2 8 30 20 ных вод можно разделить на цве группы: 1) безазотис- тые, содержащие углерод, водород и кислород; 2) азот-
содержащие вещества.
Основная часть безазотистых органических примесей бытовых
сточных вод представлена углеводами и жирами. В зависимости от
сложности строения углеводов различают моно-, ди- и полисахари-
ды. Общая формула простых углеводов (моносахаридов) CnH2nOn,
сложных CnH2mOm. Из моносахаридов в сточных водах чаще дру-
гих содержатся глюкоза СеН^Ое и лактоза (молочный сахар), из
дисахаридов — сахароза СщНггОн. Сложные углеводы (полисаха-
риды) являются естественными полимерами моносахаридов и име-
ют общую формулу (С6Н10О5)п. Компонентами бытовых сточных вод
162
6*
163
являются такие полисахариды, как целлюлоза и крахмал. В отли-
чие от простых углеводов они не растворимы в воде. Целлюлоза
входит в состав оболочек растительных клеток. В желудочно-ки-
шечном тракте человека она не разлагается и не усваивается.
В сточных водах целлюлоза содержится во взвешенном состоянии,
составляя значительную часть твердой фазы. По химическому
строению она является полимером глюкозы с молекулярной массой
около 500 000. Глюкоза, образующаяся при гидролизе целлюлозы, в
отсутствие кислорода подвергается брожению, продукты которого
определяются условиями и видом микроорганизмов. При брожении
глюкозы могут образоваться этиловый, бутиловый спирты, ацетон
и некоторые органические кислоты: масляная СНз(СН2)2СООН, ук-
сусная СН3СООН, пропионовая СН3—СН2—СООН, молочная
СН3—ПН (ОН)—СООН и другие соединения.
Жиры представляют собой сложные эфиры трехатомного спир-
та — глицерина и высших карбоновых кислот. В состав их моле-
кулы могут входить остатки олеиновой, стеариновой, пальмитино-
вой и других кислот. Жиры хорошо растворимы во многих органи-
ческих растворителях. В воде в присутствии поверхностно-активных
веществ они образуют устойчивые эмульсии. Под действием фер-
ментов жиры гидролизуются с образованием глицерина и соответ-
ствующих кислот.
В бытовых сточных водах могут содержаться в небольших коли-
чествах и другие безазотистые органические соединения (кислоты,
спирты, альдегиды, кетоны и т. п.).
Азотсодержащие органические соединения представлены в быто-
вых сточных водах белками и продуктами их гидролиза — пептида-
ми и аминокислотами. Белки по химическому строению являются
естественными полимерами — продуктом конденсации аминокис-
лот. Молекулярная масса белков изменяется от десятков тысяч до
нескольких миллионов. Количество звеньев аминокислот колеблет-
ся от нескольких десятков до сотен тысяч. В образовании белков
участвуют аминокислоты различного строения с алифатическим,
ароматическим или гетероциклическим радикалами и содержащие,
кроме того, другие функциональные группы. Это обусловливает раз-
нообразие строения белковых молекул, их сложность и различную
биологическую активность. Белки, содержащие только остатки ами-
нокислот, называются протеинами. Если же в молекуле наряду с
белковыми группами содержится небелковая часть, то такие соеди-
нения называются протеидами. К протеидам относятся глико- и
мукопротеиды, которые представляют собой соединения белков с
углеводами; фосфопротеиды, содержащие фосфор; липопротеиды,
содержащие кроме белковой части липидные группы; нуклеопро-
теиды — соединения белков с нуклеиновыми кислотами. В воде бел-
ки образуют коллоидные растворы, устойчивость которых зависит от
pH, присутствия электролитов, температуры. Повышение темпера-
туры, действие ультрафиолетовых лучей, ионизирующего излуче-
ния, некоторых химических веществ способствует биологической
инактивации белков и уменьшению их растворимости в воде.
164
Широкое использование в быту синтетических моющих средств,
активной частью которых являются поверхностно-активные вещест-
ва, привело к тому, что они стали постоянным компонентом быто-
вых сточных вод. Уже при малых концентрациях ПАВ наблюдается
образование пены, что ухудшает процесс обработки жидкой фазы
сточных вод в аэротенках. Особую форму примесей бытовых сточ-
ных вод представляют микроорганизмы. Бактериальное население
бытовых сточных вод представлено микроорганизмами, живущими
в организме человека и животных, бактериями, разлагающими ор-
ганические вещества. Иногда могут присутствовать и болезнетвор-
ные формы микроорганизмов (бактерии пли вирусы). В бытовых
сточных водах встречаются яйца паразитических червей (гельмин-
тов): аскарид, остриц и др. От одного человека в сутки поступает
4,48-1012 бактериальных клеток. Общее количество микроограниз-
мов в бытовых сточных водах исчисляется десятками миллионов в
1 мл. Поэтому неочищенные бытовые сточные воды представляют
определенную опасность и в эпидемиологическом отношении.
Городские сточные воды образуются при смешении ливневых,
бытовых и предварительно обезвреженных производственных сточ-
ных вод. Количество производственных сточных вод в составе го-
родских может достигать 40—50%. Городские сточные воды, посту-
пающие на очистные сооружения биологической очистки, не долж-
ны содержать токсичные для микроорганизмов соединения в кон-
центрациях выше предельно допустимых (ПДК). Высокая плот-
ность населения, интенсивная практическая деятельность людей
приводят к образованию большого количества городских сточных
вод, которые не могут быть сброшены в водоемы без очистки. Так,
по данным Мосочиствода количество сточных вод, подлежащих
очистке на очистных сооружениях Москвы, составляло в 1972 г.
5 млн. м3/сут, а количество осадка в них — 800 т/сут (по сухому
веществу).
§ 41. Характеристика производственных сточных вод
По мере развития промышленности все большее значение при-
обретает проблема охраны водоемов от загрязнения их сточными
водами. В последние годы проблеме очистки водоемов от загрязне-
ния сточными водами уделяется большое внимание как в нашей
стране, так и за рубежом. Успешное развитие социалистической
экономики и плановое ведение хозяйства способствовало разра-
ботке единого плана охраны водоемов от загрязнения. Обобщение
данных по всей стране привело к выработке «Основ водного зако-
нодательства Союза ССР и союзных республик», принятых в декаб-
ре 1970 г. Верховным Советом СССР, в которых изложены общие
положения о порядке потребления природных вод, организации их
охраны, исключения возможности их вредного воздействия. Данным
законодательным актом предусматривается установление государ-
ственного учета и планирования использования природных вод.
165
При использовании воды водьс;.1ив для промышленных целей
должны соблюдаться технологические нормы и правила водополь-
зования. Необходимо принимать меры для уменьшения расхода во-
ды и сокращения сброса сточных вод. Сброс допускается только
тогда, когда он нс сопровождается увеличением содержания в вод-
ном объекте загрязняющих веществ свыше установленных норм и
при условии очистки водопользователем сточных вод до пределов,
установленных органами по регулированию использования и охране
вод.
В нашей стране впервые в мире вопрос охраны природы закреп-
лен законодательно в Конституции СССР (ст. 18). Только за 1976—
1978 гг. были введены в действие сооружения для очистки сточных
вод общей мощностью 25,9 млн. м3/сут сточных вод, что на 20,5%
больше, чем было намечено планом. Селенгинский и Байкальский
целлюлозно-бумажные комбинаты будут переведены на бессточные
системы водоснабжения. Выполнение мероприятий, предусмотрен-
ных Государственным планом экономического и социального разви-
тия СССР, постановлениями ЦК КПСС и Совета Министров СССР,
будет важным вкладом в дальнейшее улучшение рационального
использования водных ресурсов и их охраны.
Состав сточных вод различных промышленных предприятий за-
висит от характера производственного процесса и отличается боль-
шим разнообразием. С введением в строй новых предприятий число
их будет возрастать. В зависимости от назначения воды в процессе
производства и его характера степень загрязненности сточных вод
различна, так же как и различен характер содержащихся в ней
примесей.
По количественному содержанию примесей производственные
сточные воды делятся на загрязненные (грязные) и условно чис-
тые. К условно чистым относятся воды, имеющие такой же состав,
как и исходная вода, но отличающиеся от нее температурой. В ос-
новном, это воды, использованные для охлаждения аппаратуры, кон-
денсаторов и др. Примеси, содержащиеся в загрязненных сточных
водах, могут представлять собой продукт производства, побочные
продукты, образующиеся в результате реакции, остатки и загрязне-
ния сырья и др. В зависимости от состава примесей и специфично-
сти их действия на водоемы сточные воды делятся на следующие
группы (по А. И. Жукову):
1) воды, содержащие неорганические примеси со специфически-
ми токсичными свойствами (кислоты, щелочи, соли тяжелых ме-
таллов, сульфиды, гидроксиды металлов). В эту группу входят сто-
ки предприятий цветной и черной металлургии, гальванических
цехов, сернокислотных, азотно-туковых заводов. Они могут вызвать
изменение pH воды водоемов, коррозию бетонных и металлических
сооружений. Соли тяжелых металлов являются токсичными по от-
ношению к водным организмам и способствуют образованию «мерт-
вых» донных отложений;
2) воды, содержащие неорганические примеси, не обладающие
токсичным действием (шлак, песок, частицы пустой породы и др.).
166
К этой группе относятся сточные воды угле- и рудообогатительных
фабрик, цементных заводов и др. Примеси такого типа находятся
во взвешенном состоянии. Для водоема часто особой опасности эти
воды не представляют, но способствуют образованию донных отло-
жений, снижению прозрачности воды, что может отрицательно ска-
заться на жизнедеятельности водных организмов;
3) воды, содержащие нетоксичные органические вещества. Это —
сточные воды предприятий пищевой промышленности. Органичес-
кие примеси, содержащиеся в этих водах, разлагаются биохимичес-
ким путем. При попадании их в водоем резко возрастает окисляе-
мость, ВПК, снижается количество растворенного кислорода,
уменьшается прозрачность воды, изменяется окислительно-восста-
новительный потенциал. Нерастворимые органические примеси, осе-
дая на дно, начинают разлагаться с выделением метана, сероводо-
рода и других газов. При значительном загрязнении возникает
кислородный дефицит, нарушается гидрохимический режим водо-
ема;
4) воды, содержащие органические вещества со специфическими
токсичными свойствами (фенолы, эфиры, красители, углеводороды
и др.). К этой группе относятся сточные воды предприятий орга-
нического синтеза по производству полимеров и синтетических во-
локон, нефтеперерабатывающих предприятий и др.
Особую группу образуют сточные воды, содержащие радиоак-
тивные вещества.
§ 42. Основные примеси производственных
сточных вод
В докладе экспертов Всемирной организации здравоохранения
(ВОЗ) на VII конгрессе по водоснабжению (Женева, 1967) дается
следующая классификация видов загрязнений воды: 1) бактериаль-
ное загрязнение (бактерии, вирусы, болезнетворные микроорганиз-
мы); 2) биохимически окисляющиеся органические соединения, ко-
торые вызывают запахи и привкусы воды; 3) неорганические соли,
которые не удаляются обычными методами и могут вызвать пол-
ную непригодность воды для хозяйственно-бытовых и производст-
венных нужд; 4) неорганические соединения, являющиеся питатель-
ными веществами для растений (нитраты, фосфаты, калийные со-
ли). Они вызывают усиление роста водной растительности, способ-
ствуют «цветению» водоемов, ухудшают качество питьевой воды;
5) нефтепродукты, которые затрудняют реаэрацию водоема, оказы-
вают неблагоприятное воздействие на водные организмы, ухудша-
ют органолептические свойства воды; 6) токсичные соединения
(соли тяжелых металлов, окислители, вещества общеядовитого
действия, различные органические соединения).
В табл. 12 приведены данные об основных примесях бытовых и
производственных сточных вод с учетом интенсивности загрязне-
ния.
167
Таблица 12. Основные виды загрязнений
Загрязни ie.ni Наличие* и уровни**
бытовых нроизвэлственн >ix
цветной ме- таллургии черной метал- лургии коксохими- ческие машиномро- ительнче нефтеперера- ба I ывающие
Плавающие: твердые нефтепродукты, жиры Взвеси минеральные органические биологические Растворенные неорганические ве- щества: хлориды сульфаты фосфаты кислоты неорганические цианиды медь марганец хром свинец цинк ртуть кадмий Растворенные органические ве- щества ПАВ фенолы ацетон бутанол дибутилфталат уксусная кислота формальдегид фурфурол сероуглерод см СМ + 1 +” + 01 Ц II 1 1 1 1 1 1 I J. 1 1 1 1 1 1 1 1 5-6 4 2 0—5 До 4 3-4 0-2 2—4 0-2-3 0—1 3-4 3-4 0-3 3—4 1 + +11 1 1 1 + +”+” 1 + и 1 1 1 1 1 1 1 1 1 — см о '+ II "7 1 II 1 1 II 1 | | 1 1 1 1 1 1 1 1 |
* Знак «+» означает возможность присутствия загрязнителя в неочищенном
** Числовые обозначения соответствуют следующим уровням присутствия загрязни
5 — десятки тысяч; 6 — сотни тысяч.
Рассмотрим свойства некоторых наиболее часто встречающихся
групп загрязнений производственных сточных вод.
Нефть и нефтепродукты являются постоянными компонентами
сточных вод нефтедобывающих установок, нефтеперерабатываю-
щих, нефтехимических и машиностроительных заводов, предприя-
тий транспорта. Растворимость сырой нефти в воде составляет 5—
20 мг/л. Растворимость керосина в воде достигает 80 мг/л. В ми-
нерализованных водах растворяется 4—9 мг/л нефти. Основную
часть нефтей составляют предельные, циклические (циклопарафи-
ны, нафтены) или ароматические углеводороды. В состав нефти в
168
бытовых и производственных сточных вод
загрязнений стоков
(по видам предприятий)
—
о
О CJ <39
К о
—. • со г О о
- X с £ X X со о 5 о ~ 2 р*
— _L_
Г4 । ъ. -
1-1 Е н S о
+ + + 1
— — — — —. 3 3-4 2-3
2-4 3-4 - -- — — — 4 —
— — 3 3 4—5 3
— — — — — —г* И-
1-3 2—3 4 3—4
2-3 3 — 3 3-4 — —
— 1-2 — —• 1-3 — — 1-2
— -р- — — — — — —
— — — —- — — — —
— — + — — — —
— — — — — — — —•
— — —. — — 1 2 — —
— — — — —. — — —
-— — —р — — — —
-— — — — — — — —
— — — — — — — —
0—2 2 0—2
— — 0-2 — 0-2 — —
— — 2-3 — — — —
— — 2-3 — — — —
— — — 0—4 — — — —
— — 0—4 — — — —
- — — — 0—2 — — — —
— 2-3 — + — — — —
— — 4- —, — — — —
стоке.
телей в неочищенном стоке (мг/л):
1 — единицы;
2 — десятки; 3 — сотни; 4 — тысячи;
небольших количествах входят также соединения, содержащие се-
ру, кислород (нафтеновые кислоты, фенол), азот и смолы.
С нефтепродуктами в воду могут попасть и полициклические
ароматические углеводороды (ПАУ), например бенз-(а)-пирен, об-
ладающие канцерогенным действием. При концентрации более
0,1 мкг/'л они усваиваются морским планктоном. Образованию ус-
тойчивой эмульсии «нефть в воде» способствует присутствие в воде
поверхностно-активных веществ. Такие эмульсии образуются при
промывке танкеров, барж, резервуаров, при некоторых процессах
очистки нефти. Предельно допустимая концентрация нефти и неф-
169
тепродуктов в воде водоемов составляет 0,3 мг/л, а для много-
сернистой нефти — до 0,1 мг/л.
Фенолы являются ароматическими соединениями, в которых
гидроксидная группа связана с углеродным атомом бензольного
ядра. По количеству гидроксидных групп различают одно- и много-
атомные фенолы. Ниже приводятся формулы некоторых фенолов:
ОН
фенол
(оксибензол)
(1,2, 6-триокси-
бензол) пирен аллол
ОН
гидрохинон
(1, 4-диокеи-бензол)
Фенолы — кристаллические вещества с резким запахом и плот-
ностью, близкой к единице. Наиболее часто в производственно-сточ-
ных водах содержится фенол С6Н5ОН (карболовая кислота). Фенол
содержится в некоторых сортах нефти, образуется при химической
переработке каменного угля, является одним из важнейших полу-
продуктов промышленности органического синтеза. Крезолы полу-
чают из каменноугольной смолы. Они используются в производстве
синтетических смол и красителей. Фенолы довольно хорошо раство-
римы в воде. Растворимость фенола в воде при 20° С составляет
9,3 г/100 г. Т. пл. 41° С, т. кип. 181° С. При повышении температуры
растворимость фенола в воде возрастает. С водой он образует азео-
тропную (нераздельно-кипящую) смесь, температура кипения кото-
рой 98,6° С. Многоатомные фенолы имеют большую растворимость
в воде.
Взаимодействуя с хлором, фенол образует хлорфенолы по урав-
нению
/г-хлорфенол
Хлорфенолы имеют резкий, специфический («аптечный») запах,
который появляется в воде при концентрации фенолов более
0,001 мг/л.
В процессе получения целлюлозы из древесины по сульфитно-
му способу образуются концентрированные сульфитные щелока,
которые содержат 9—14% сухого остатка. Основными компонента-
ми сульфитных щелоков являются такие органические соединения,
как лигнин в форме кальциевых солей лигнинсульфокислот и угле-
воды (глюкоза, манноза и др.). В щелоке есть также водонераст-
воримые углеводы — целлюлоза и гемицеллюлоза. Неорганические
компоненты сульфитных щелоков представлены, в основном, суль-
170
фитом кальция, диоксидом серы. Наличие в сульфитных щелоках
углеводов обусловливает возможность биологической очистки сточ-
ных вод, но соли лигнинсульфокислот почти не разлагаются био-
химическим путем, хотя и легко подвержены действию неорганиче-
ских окислителей, например перманганата калия.
Загрязнение сточных вод сульфитными щелоками зависит от
степени утилизации их и от объема оборотных циклов водоснаб-
жения. При попадании сточных вод, содержащих сульфитные щело-
ка, в водоем наблюдается резкое изменение свойств воды: появля-
ется окраска, вспенивание, снижается pH, увеличивается содержа-
ние взвешенных веществ и начинается быстрое потребление раство-
ренного в воде кислорода. Расходование кислорода на окисление
диоксида серы, сульфитов и на биохимическое окисление углеводов
может привести к возникновению анаэробных условий в водоеме и
нарушению жизнедеятельности аэробных организмов. Наличие
углеводов в сульфитных щелоках, дефицит растворенного кислорода
в воде способствуют массовому развитию грибов (например, Lepto-
mitus) и нитчатых бактерий (Sphaerotilus natans).
Производство целлюлозы сульфатным способом заключается в
обработке древесной массы хвойных пород едким натром, сульфи-
дом или сульфатом натрия и карбонатом натрия. В черный щелок
переходят те же органические соединения, что и при сульфитном
методе, по. кроме того, образуются смоляные мыла. Среда этих
щелоков щелочная, они окрашены в темно-коричневый цвет. Чер-
ный щелок подвергается утилизации сжиганием. Органическая
часть выгорает, а неорганические вещества возвращаются в произ-
водство. Сточные воды могут загрязняться и неполностью отреге-
нерироваипыми сульфатными щелоками. Воздействие их па режим
водоема будет также проявляться в энергичном потреблении раство-
ренного кислорода для биохимического окисления углеводов. Кро-
ме того, эти стоки вызывают появление более интенсивной устойчи-
вой окраски воды, а смолисто-ароматические вещества придают
запах воде и ухудшают вкус рыбы.
В сточных водах предприятий цветной металлургии, цехов галь-
ванических покрытий и некоторых других отраслей промышленно-
сти содержатся соли тяжелых металлов. Наиболее часто в сточных
водах присутствуют медь, свинец, хром, кадмий, ртуть, цинк, ни-
кель. Тяжелые металлы в сточных водах могут содержаться в фор-
ме катионов (Си22, СН+, Cd2+, Hg2+), анионов (СгО42щ Сг^О?2-)
или комплексных ионов [Си (NH3)4]2+, [Си(CN)3]2-, [Zn(CN)4]2_.
Форма содержания тяжелых металлов в воде зависит от pH. В кис-
лой и нейтральной среде амфотерные металлы содержатся в форме
катионов (Al3+, Zn2+, СН~!), а в сильнощелочной (при рН>9) — в
форме анионов [А1(ОН)4Н, [Zn(OH)3]", [Сг(ОН)4К.
Соединения тяжелых металлов отрицательно влияют на процесс
самоочищения, вызывая угнетение жизнедеятельности аэробных
микроорганизмов. При содержании в воде всего 0,1 —10 мкг/л ртути
уже наблюдается снижение интенсивности фотосинтеза водорос-
лей. Многие верные организмы (гидробионты) обладают способно-
171
стью аккумулировать соединения тяжелых металлов. Концентрация
таких соединений в водных организмах может быть в сотни и даже
тысячи раз большей, чем в воде водоема. Токсичное действие неко-
торых тяжелых металлов может усиливаться в присутствии других.
Так, токсичность соединений никеля возрастает в присутствии сое-
динений меди (II), а ртут.ь усиливает токсичность меди. Характер-
ным свойством тяжелых металлов как загрязнителей является дли-
тельность действия. Воздействие тяжелых металлов на водоем про-
является и в нарушении газового режима, изменении буферных
свойств воды, увеличении солесодержания. Миграцию тяжелых ме-
таллов в водоемах сдерживают химические и физико-химические
процессы, способствующие самоочищению водоема от загрязните-
лей. Прежде всего это химическое взаимодействие ионов тяжелых
металлов с ингредиентами природных вод, сопровождающееся об-
разованием труднорастворимых соединений. Важная роль в этом
принадлежит гидролизу солей тяжелых металлов. Снижение кон-
центрации ионов тяжелых металлов в воде водоемов связано ч с
сорбцией их глинистыми минералами по ионообменному механизму.
Труднорастворимые соединения тяжелых металлов, попадающие
в состав донных отложений, могут быть вторичным источником за-
грязнения водоема. С изменением щелочности, pH, окислительно-
восстановительного потенциала, условий протекания процессов раз-
ложения органического вещества и других факторов может нару-
шиться равновесие между содержанием ионов тяжелых металлов в
донных отложениях и в воде, что создает возможность для частич-
ного растворения труднорастворимых соединений. От 20 до 50%
соединений тяжелых металлов, входящих в состав донных отложе-
ний, достаточно легко могут перейти в растворенное состояние.
В водоемах тяжелые металлы могут образовывать новые токсичные
соединения. Например, неорганические соединения ртути переходят
в элементорганические (метилртуть). Это указывает на необходи-
мость более полного извлечения соединений тяжелых металлов из
сточных вод. Наиболее рациональный путь для решения этой проб-
лемы — внедрение оборотного водоснабжения и безотходной тех-
нологии.
Пестициды — соединения, используемые для борьбы с сельско-
хозяйственными вредителями, болезнями растений, для уничтоже-
ния сорной растительности. Вещества, которые применяют для
борьбы с насекомыми, называются инсектицидами, с грибковыми
болезнями — фунгицидами, с сорняками — гербицидами. В послед-
ние десятилетия пестициды получили очень широкое применение
практически во всех странах. Использование пестицидов в сельском
хозяйстве позволяет значительно увеличивать выход сельскохозяй-
ственной продукции, но в то же время их использование связано с
загрязнением внешней среды этими соединениями.
Наибольшее распространение в качестве пестицидов получили
хлор- и фосфорорганические соединения. К хлорорганическим пе-
стицидам относятся гексахлоран, алдрин и др. Из фосфорорганиче-
ских соединений чаще всего используются карбофос, хлорофос, ди-
172
хлофос и др. Характерной особенностью пестицидов является их
устойчивость к биохимическому разложению. Особую опасность
представляют хлорорганические пестициды, так как в почве они
сохраняются в течение нескольких лет, например гексахлоран. Пос-
ле однократной обработки черноземной почвы хлорорганическими
соединениями через 9—12 лет в ней было обнаружено 15—35% дил-
дрина, 18—30% хлордана, 3—13% гептахлора. Пестициды, имею-
щие малую растворимость в воде, в присутствии ПАВ образуют
устойчивые дисперсии в воде. Многие из пестицидов придают воде
интенсивный запах, являются токсичными для людей и животных.
Концентрация пестицидов в водах поверхностного стока зависит от
времени года. Максимум содержания их наблюдается в период, со-
ответствующий времени обработки сельскохозяйственных угодий
пестицидами и орошения.
Обычная схема очистки питьевой воды, включающая коагулиро-
вание и хлорирование, не обеспечивает полного извлечения пести-
цидов. С учетом возможности загрязнения водоемов пестицидами
необходимо выбирать и схему очистки воды. Эффективным сорбен-
том пестицидов является активированный уголь, а окислителем —
озон.
Основные пути для предотвращения загрязнения водоемов пес-
тицидами заключаются в разработке менее токсичных препаратов
с избирательным действием, разлагающихся достаточно быстро во
внешней среде; замене химических методов борьбы с вредителями
биологическими; совершенствовании агротехнических работ, на-
правленном на сокращении количества используемых химических
препаратов, выноса загрязненных пестицидами вод в водоемы.
В сточных водах предприятий органического синтеза, по произ-
водству полимерных материалов, синтетического каучука (СК) и
некоторых других часто содержатся такие вещества, как альдегиды,
кетоны, органические кислоты, синтетические поверхностно-актив-
ные вещества (СПАВ). Альдегиды — постоянные компоненты сточ-
ных вод предприятий по производству органических кислот, феноло-
формальдегидных и карбамидных полимеров, СК. Простейшие аль-
дегиды— формальдегид (муравьиный альдегид) НСНО, ацетальде-
гид (уксусный) СН3—СНО и пропионовый альдегид СН3—СН2—
—СНО — бесцветные жидкости, смешивающиеся с водой, облада-
ют резкими специфическими запахами. В сточных водах могут со-
держаться и другие альдегиды. Из непредельных — акролеин
СН2 = СН—СНО, производное гетероциклического соединения фу-
рана — фурфурол:
СН—СН
'I н /О
СН С-С%
\/ чн
о
Ацетальдегид, пропионовый альдегид окисляются микроорганиз-
мами сравнительно легко. Фурфурол, акролеин и другие непредель-
ные альдегиды разлагаются биохимическим путем только после
173
длительной адаптации. Присутствие альдегидов в воде прежде все-
го определяется по запаху. Для ацетальдегида порог восприятия
запаха составляет всего 0,2 мг/л. Ацетальдегид легко окисляется
растворенным в воде кислородом и с участием микроорганизмов до
уксусной кислоты. При концентрации ацетальдегида 1 мг/л вели-
чина ВПК повышается на 1,3—1,5 мг Ог/л. Высокая реакционная
способность альдегидов способствует их быстрому изменению в вод-
ной среде. Так, при концентрации ацетальдегида в воде 2 мг/л про-
цесс самоочищения длится всего 3 сут. Формальдегид по сравнению
с ацетальдегидом обладает резко выраженным токсичным дей-
ствием. Предельно допустимая концентрация его в воде по токсико-
логическому признаку составляет 0,05 мг/л. Образование кислот
при окислении альдегидов способствует снижению величины pH
воды.
В производственных сточных водах многих предприятий содер-
жится ацетон. Ацетон — простейший представитель ряда кетонов,
(диметилкетон): СН3—-С — СН3Дцетон разлагается биохимическим
О
путем, поэтому его влияние на санитарный режим водоема прояв-
ляется в повышении ВПК. Подпороговая концентрация ацетона пс
изменению органолептических свойств воды составляет 40,9 мг/л,
Предельно допустимая концентрация в воде регламентируетс>!
ВПК. Более сложные кетоны разлагаются биохимическим путем
только после длительной адаптации или вовсе не разлагаются (на-
пример, метилвинилкетон).
Органические кислоты являются компонентами сточных вод
многих производств. Эти соединения влияют на свойства воды в за-
висимости от их способности к растворению, диссоциации и хими-
ческого строения. Одноосновные предельные кислоты, за исключе-
нием муравьиной, не подвергаются химическому окислению. Двух-
основные, например щавелевая, при действии окислителей легко-
окисляются до диоксида углерода и воды. Растворимость предель-
ных одноосновных кислот в воде с увеличением углеводородного
радикала быстро уменьшается. Предельные одноосновные кислоты
с числом углеродных атомов от 1 до 3 (муравьиная, уксусная, про-
пионовая) — бесцветные жидкости с резким запахом, смешиваются
с водой. Большинство кислот с числом углеродных атомов от 4 до
9 — маслянистые жидкости с неприятным запахом, ограниченно
или плохо растворимые в воде. Влияние органических кислот на ре-
жим водоема проявляется в снижении pH, появлении кислого при-
вкуса, запаха, повышении БПК. Некоторые из них проявляют ток-
сичное действие по отношению к водным организмам.
Высшие предельные одноосновные кислоты и их соли (за ис-
ключением солей щелочных металлов и аммония) в воде не раст-
воримы. Соли этих кислот называют мылами. Мыла, растворимые
в воде (натриевое, калиевое и др.), образуют полуколлоидные рас-
творы, имеющие щелочную реакцию среды. Они относятся к ани-
онным поверхностно-активным веществам (ПАВ).
174
ПАВ являются одними из основных загрязнителей водоемов.
Они могут поступать в водоем в результате недостаточно полного
удаления их на очистных сооружениях, с загрязненными грунтовы-
ми водами. По отношению их к биохимическому окислению выде-
ляют «биологически жесткие» и «биологически мягкие» ПАВ. В на-
шей стране производство и применение биологически жестких ПАВ
ограничено. Перед поступлением сточных вод, содержащих такие
соединения, на городские очистные сооружения должна проводить-
ся предварительная очистка от этого вида загрязнений. Анионные
ПАВ — алкилсульфаты R—OSO3Na легко окисляются и малоток-
сичны для микроорганизмов. При концентрации 200 мг/л не наблю-
дается отрицательного воздействия на микроорганизмы активного
ила. Легко окисляются первичные алкилбензолсульфонаты
R—so3Na с прямой цепью. Наиболее биологически жесткими
анионными ПАВ являются третичные алкилбензолсульфонаты. Так,
сульфонол НП-1 (додецилсульфонат натрия с сильно разветвлен-
ной углеводородной цепью) почти не окисляется биохимическим пу-
тем и токсичен по отношению к микроорганизмам при концентра-
ции 20 мг/л. Сульфонол НП-3 (додецилсульфонат натрия с прямой
цепью) достаточно хорошо окисляется микроорганизмами активно-
го ила даже при концентрации 100 мг/л. Алкилсульфаты окисляют-
ся быстрее и полнее, чем алкилбензолсульфонаты. Неионные ПАВ
(типа ОП-7, ОП-Ю) практически не окисляются биохимическим
.путем.
При попадании ПАВ в водоем наблюдается нарушение санитар-
ного режима вследствие пенообразования, возникновения устойчи-
вых эмульсий и суспензий труднорастворимых в воде веществ. ПАВ
влияют на органолептические свойства воды, обусловливая появле-
ние запаха, облегчают всасывание в кровь пестицидов и других
токсичных веществ уже при концентрации более 0,5 мг/л. Обычно
в городских сточных водах концентрация ПАВ колеблется от 1,5
до 10 мг/л. После очистных сооружений она снижается до 1—3 мг/л,
в водопроводной воде она изменяется от 0 до 0,1 мг/л.
ПАВ отрицательно влияют на процесс биохимической очистки:
при концентрации выше допустимой они тормозят процесс окисле-
ния органических веществ и нитрификации, вызывают интенсивное
ценообразование в аэротенках и уменьшают степень очистки сточ-
ных вод. Они способствуют образованию трудноосаждающихся
взвесей при умягчении воды методами осаждения, коагуляции. От-
рицательное воздействие ПАВ на процессы очистки воды, возмож-
ность проявления токсичного воздействия определяют необходи-
мость разработки более эффективных методов удаления их из сточ-
ных вод.
В последние годы в связи с широким применением минеральных
удобрений имеет место загрязнение водоемов соединениями таких
биогенных элементов, как азот и фосфор. Соединения азота, имею-
щие высокую растворимость, более подвержены миграции в водо-
емы. Повышение концентрации азота и фосфора в воде способству-
175
ет массовому развитию высших водных растений, микроорганизмов
планктона, в том числе и водорослей, вызывающих цветение водо-
емов. Предотвратить повышение концентрации азота и фосфора в
поверхностных водоемах можно проведением ряда мероприятий, на-
правленных на уменьшение выноса удобрений с полей.
Введение оборотных систем водоснабжения и орошение земель
вызывает увеличение общего солесодержания используемой воды.
Повышение концентрации главных ионов способствует нарушению
технологического режима, образованию отложений, ведет к засоле-
нию почвы. Поэтому снижение солесодержания отработанной воды
представляет важнейшую задачу, требующую решения.
§ 43. Основные показатели степени загрязнения
сточных вод
Анализ сточных вод. Анализ сточных вод необходим для опре-
деления возможности спуска их в водоем, метода очистки и для вы-
яснения содержания в них ценных или токсичных примесей. В отли-
чие от анализа природных вод здесь не требуется определения всех
компонентов и можно ограничиться характеристикой группы ве-
ществ, содержащих какой-либо элемент. Задачи анализа сточных
вод заключаются в составлении общей характеристики сточных вод
и их примесей, определении особо важных примесей и их концент-
рации.
Санитарно-химический анализ городских сточных вод включает
следующие определения: 1) температуры, цвета, запаха, pH; 2) сте-
пени прозрачности; 3) оседающих веществ по объему и массе;
4) взвешенных веществ и потерь при прокаливании; 5) общего со-
держания примесей, остатка их после прокаливания, потерь при
прокаливании; 6 ) азота — общего, аммонийного, нитритов, нитра-
тов; 7) окисляемости перманганатной, дихроматной [химическое
потребление кислорода (ХПК)]; 8) биохимического потребления
кислорода (БПК5, БПКполн.); 9) относительной стабильности;
10) растворенного кислорода; 11) хлоридов, свободного хлора;.
12) фосфатов; 13) фторидов; 14) специфических ингредиентов про-
изводственных сточных вод (тяжелые металлы — железо, медь,
кобальт, хром, никель, свинец, кадмий, ртуть, фенолы, цианиды,
сульфаты, синтетические ПАВ, нефтепродукты, эфирорастворимые
вещества); 15) бактериологическое исследование; 16) радиологиче-
ское исследование; 17) гельминтологический анализ.
Содержание веществ, находящихся в сточных водах в виде ио-
нов, выражается в ионной (мг/л) или эквивалентной (мг-экв/л)
формах. Полный санитарный анализ проводится на городских очи-
стных сооружениях один раз в 10 суток. Объем анализа промыш-
ленных сточных вод может быть различным, что обусловлено ха-
рактером производства.
Показатели степени загрязнения сточных вод. Предельную на-
грузку на очистные сооружения можно ориентировочно рассчитать
по данным анализа сточной воды. Характеристика ее дается после
176
определения основных показателей степени загрязнения. Опреде-
ляющую роль при выборе оптимального режима работы сооруже-
ний биохимической очистки играет степень загрязнения воды
органическими соединениями. Органические примеси сточных вод
вследствие их многообразия, сложности и трудности анализа непо-
средственно не определяются. Для характеристики степени загряз-
нения воды органическими соединениями применяются косвенные
показатели: окисляемость воды и биохимическое потребление кис-
лорода (БПК).
Окисляемость воды характеризуется количеством кислорода, эк-
вивалентным количеству расходуемого окислителя. Она выражает-
ся в миллиграммах кислорода, расходуемого для окисления ве-
ществ, содержащихся в 1 л воды. В зависимости от используемых
окислителей и полноты окисления органических веществ различают
перманганатную и дихроматную окисляемость (ХПК).
Перманганатная окисляемость характеризует количество легко-
окисляющихся органических примесей и обычно используется при
анализе природных вод. Окисление ведется перманганатом калия в
кислой среде при концентрации хлоридов до 300 мг/л. Если хлори-
дов больше, то окисление ведется в щелочной среде. Данным мето-
дом можно определять окисляемость воды без разбавления до
10 мг Ог/л. Достоверные результаты получаются не более чем при
10-кратном разбавлении, т. е. при окисляемости воды 100 мг О2/л.
Определению мешают неорганические восстановители (нитриты,
железо (II), сульфиды, сульфиты и др.).
Показателем содержания органических примесей в сточных во-
дах является величина ХПК. Окисление органических примесей
осуществляется дихроматом калия в присутствии концентрирован-
ной серной кислоты. В качестве катализатора окисления для труд-
ноокисляющихся веществ применяется сульфат серебра. При дей-
ствии дихромата калия в сильнокислой среде происходит практи-
чески полное окисление растворимых, коллоидных и нерастворимых
органических примесей. Конечные продукты окисления — диоксид
углерода, вода, аммиак, фосфаты и сульфаты. Но и в этих усло-
виях небольшая часть органических веществ остается полностью
или частично неокисленной. Степень окисления органических ве-
ществ обычно составляет 95—98%. По данным определения ХПК
можно рассчитать, зная состав органического соединения, его со-
держание в воде. И наоборот, величину ХПК можно вычислить для
определенного соединения, используя уравнение реакции его окис-
ления. Теоретическое ХПК обычно выше установленного анализом.
Практически при определении ХПК не окисляются пиридин и неко-
торые другие азотсодержащие органические соединения, а также
труднорастворимые углеводороды (бензол, нафталин, парафины).
Максимально определяемая данным методом величина окисляемо-
сти составляет 104 мг О2/л.
Степень загрязнения сточных вод выражается также количест-
вом кислорода, необходимым для окисления органических веществ
микроорганизмами в аэробных условиях. Этот показатель называ-
177
ется биохимическим потреблением кислорода (БПК). Практически
полным БПК считается количество кислорода, необходимое для
окисления органических веществ до начала процесса нитрификации
(БПКполп). В лабораторных исследованиях наряду с полным БПК
определяется биохимическое потребление кислорода в течение
5 сут — БПК5. Величина БПК5 для бытовых сточных вод составля-
ет 75—85% от БПКполи» но для городских и производственных СТОЧ-
НЫХ вод соотношение между этими величинами непостоянно, изме-
няется в широких пределах и определяется свойствами органичес-
ких примесей.
Скорость окисления органических веществ определяется их
строением и характером протекающих процессов. Количество кис-
лорода, расходуемое для окисления аммонийного азота до нитри-
тов и нитратов (нитрификации), при определении БПК не учиты-
вается. На скорость биохимического окисления органических при-
месей влияет присутствие тяжелых металлов, которые даже при не-
больших концентрациях вызывают замедление этого процесса. По-
скольку окисление органических веществ происходит под воздей-
ствием микроорганизмов, необходимо создать благоприятные усло-
вия для их жизнедеятельности. Этими условиями являются опреде-
ленная среда, оптимальная температура (20° С), отсутствие токсич-
ных и бактерицидных соединений, наличие биогенных веществ. БПК
бытовых сточных вод достигает нескольких сотен мг О2/л и зависит
от нормы водоотведения. БПК производственных сточных вод мо-
жет достигать нескольких тысяч мг/л. БПКполн в водоемах, исполь-
зующихся для хозяйственно-бытового водоснабжения, не должно
превышать 9 мг О2/л.
Разность между ХПК и БПК характеризует наличие примесей,
не окисляющихся биохимическим путем, и количество органических
веществ, идущих на построение клеток микроорганизмов. Для бы-
товых сточных вод БПКполн составляет 85—90% от ХПК. По соот-
ношению БПКполн/ХПК можно судить о возможности применения
определенного метода очистки сточных вод. Если соотношение
БПК/ХПК>0,5, то это указывает на возможность применения био-
химической очистки сточной воды; при соотношении БПК/ХПК<0,5
использование этого метода становится мало эффективным, так как
в воде содержится значительное количество биологически неокис-
ляемых веществ. Для таких сточных вод более целесообразным бу-
дет применение физико-химических или химических методов очист-
ки. Так как в процессе биохимического окисления сточных вод сни-
жается концентрация биологически окисляемых веществ, то соот-
ношение БПК/ХПК при этом уменьшается. Сточные воды, содержа-
щие биологически разлагаемые органические примеси и неоргани-
ческие восстановители, являются энергичными потребителями кис-
лорода. На окисление этих примесей идет растворенный кислород.
После полного использования растворенного кислорода окисле-
ние органических веществ может осуществляться за счет восстанов-
ления нитратов и нитритов. Если количество биохимически исполь-
зуемого кислорода достаточно для окисления органических при-
178
месей, то такие воды называются стабильными или стойкими. Ста-
бильность (стойкость) сточных вод определяется по отношению ко-
личества биохимически потребляемого кислорода к БПКполн и вы-
ражается в процентах. При наличии кислородного дефицита вода
загнивает. Относительная стабильность (стойкость) сточных вод в
лабораторной практике рассчитывается по времени обесцвечивания
метиленовой сини (в сутках).
Показатели степени загрязнения производственных сточных вод
определяются особенностями производственного процесса. Наряду с
указанными наибольшее значение имеют показатели: pH, кислот-
ность, щелочность, содержание тяжелых металлов и других токсич-
ных примесей, цветность, взвешенные вещества и плавающие при-
меси, запах воды и др.
§ 44. Условия спуска сточных вод в водоемы
Нормативные показатели, принятые в нашей стране, определяют
качество воды в водоеме независимо от состава попадающих в него
сточных вод. Это связано с тем, что критерием токсичности сточных
вод является ограничение, а то и невозможность использования во-
ды водоема для целей водоснабжения вследствие ее загрязнения.
Законодательно утвержденные предельно допустимые концентра-
ции (ПДК) различных веществ в воде водоемов обеспечивают ее
качество, исключающее отрицательное воздействие на организм
человека и затрудняющее условия водопользования. Значения ПДК
веществ-загрязнителей характеризуют состав воды у тех мест водо-
пользования, которые находятся ниже выпуска сточных вод. При
этом учитывается возможность использования водоема в качестве
водоисточника, а также его рыбохозяйственное и гигиеническое зна-
чение. При достаточном удалении места водопользования от места
выпуска сточных вод определение ПДК вредных веществ проводит-
ся с учетом разбавления воды (в расчете на минимальное) и воз-
можности устойчиво протекающего и хорошо изученного процесса
самоочищения.
Основные нормативные данные, определяющие условия спуска
сточных вод в водоемы, приведены в «Правилах охраны поверхно-
стных вод от загрязнения сточными водами». Количество соедине-
ний, для которых указаны ПДК их в воде, приближается к 500.
В настоящее время устанавливаются ПДК для многих других ве-
ществ. Методика установления ПДК включает в себя определение
предельно допустимой концентрации вещества по органолептичес-
кому, общесанитарному и санитарно-токсикологическому показате-
лям. Согласно методическому приему, предложенному С. Н. Чер-
кинским и сотр., ПДК вещества устанавливается по тому признаку
вредного действия, который характеризуется наименьшей пороговой
или подпороговой (для санитарно-токсикологического признака)
концентрацией (лимитирующий признак вредности).
Органолептический показатель вредности регламентирует кон-
центрацию вещества в результате изменения таких показателей ка-
17Р
чества воды, как запах, вкус, цвет, ухудшение внешнего вида водо-
ема. Некоторые соединения вызывают появление запаха, окраски
воды, при концентрации примеси ниже определяемой санитарно-
токсикологическим показателем. Так, для бутил акрилата значение
ПДК по органолептическому признаку вредности составляет
0,01 мг/л, по общесанитарному — 1, по санитарно-токсикологичес-
кому— 60, для гексахлорана — соответственно 0,02, 2,5, 1 мг/л.
В другой группе соединений (бериллий, гексахлорбензол, нитраты
и др.) пороговая концентрация, способствующая изменению органо-
лептических свойств воды, выше той концентрации, при которой
примесь оказывает токсичное действие. Для таких веществ лими-
тирующим признаком вредности будет санитарно-токсикологичес-
кий. Большая группа соединений (биохимически разлагающиеся
вещества, кислоты, щелочи, легко окисляющиеся неорганические со-
единения и др.) регламентируется по общесанитарному признаку
вредности. Основными показателями, которые учитываются при
определении ПДК соединения по общесанитарному признаку, яв-
ляются ВПК, pH и концентрация растворенного кислорода. Неко-
торые компоненты сточных вод (медь, никель, титан, диметилфор-
мамид и др.) оказывают бактерицидное действие на микрофлору
водоемов, тормозя тем самым процесс самоочищения. Токсичное
действие этих соединений проявляется при более высокой концент-
рации их в воде. Поэтому лимитирующим признаком вредности для
них является общесанитарный.
Так, например, для меди ПДК по санитарно-токсикологическо-
му признаку составляет 10 мг/л, по органолептическому — 3, а по
общесанитарному (лимитирующему)—0,1 мг/л. Для титана эти
значения соответственно равны 20; 4,5 и 0,1 мг/л, для диметилфор-
мамида — 1000; 50 и 10 мг/л.
К группе компонентов сточных вод, лимитирующим признаком
которой является санитарно-токсикологический показатель, отно-
сятся соединения, ядовитые для организма человека. Допускать по-
ступление токсичных веществ в водоемы нельзя. Различного рода
временные разрешения, ведомственные инструкции и местные тех-
нические условия на сброс сточных вод, загрязненных токсичными
соединениями, следует рассматривать как попытку обойти государ-
ственное законодательство.
Нормирование содержания токсичных соединений в воде водо-
емов проводится по значению подпороговой концентрации, т. е. та-
кому содержанию вредной примеси, при котором не отмечается от-
рицательное воздействие его на организм человека. К группе за-
грязнений, содержание которых в воде водоемов лимитируется по
санитарно-токсикологическому признаку, относятся соединения та-
ких металлов, как бериллий, молибден, ртуть, свинец, мышьяк,
сурьма и некоторые органические соединения — анилин, гексаме-
тилендиамин, диэтилртуть и др. Если в стоке содержится несколько
токсичных примесей, количество которых нормируется по одному
и тому же лимитирующему признаку вредности, то степень токсич-
ности определяется по принципу суммированного действия. В табл. 13
180
Таблица 13. ПДК некоторых ионов тяжелых металлов и соединений
Ингредиент ПДК, мг/л Лими-1 ирующий признак вредности Ингредиент ПДК, м г /л Лимитирующий признак вредности
Бериллий (Ве2+) Кадмий (Cd2+) Кобальт (Со2+) Медь (Си2+) 0,0002 Санитарно-токси- кологический Нефть много- сернистая 0,1 Органолепти- ческий
0,01 1,0 0,1 То же Общесанитарный » Пиридин 0,2 Санитарно- токсикологи- ческий
Никель (Ni2+) 0,001 » Алкилбензол- сульфаты 0,5 Органолепти- ческий
Селен (SeO 0,001 Санитарно-токси- кологический Алкилсульфа- ты 0,5 »
Титан (IV) Хром (Сг3+) о,1 0,5 Общесанитарный Санитарно-токси- кологический ВА-102 ВА-212 2,0 2,0 Санитарно- токсиколо- гический
Хром (Сг6+) 0,1 Общесанитарно- органолептиче- ский Полиакрил- амид 2,0 Органолепти- ческий
Амины (С7-С9) 0,1 Органолептиче- ский Стирол 0,1 »
Бензол 0,5 Санитарно-токси- кологический Триэтанол- амин 1,4 »
Гексахлоран 0,02 Органолептиче- ский Формальдегид 0,05 Санитарно- токсиколо- гический
Диурон 1,0 » Хлорофос 0,05 Органолепти- ческий
Капролактам 1 ,о Общесанитарный Фуран 0,2 Санитарно- токсикологи- ческий
Некаль Нефть и неф- тепродукты о,1 0,3 Органолептиче- ский » Эпихлоргид- рин 0,01 То же
приведены ПДК некоторых соединений, наиболее часто присутст-
вующих в производственных сточных водах.
Особую группу загрязнителей составляют патогенные микроор-
ганизмы, возбудители инфекционных заболеваний, передающихся
водным путем. Они могут попасть в водоем с бытовыми сточными
водами, со стоками больниц, кожевенных заводов, мясокомбинатов
и др. В сточных водах обычно содержатся примеси, находящиеся во
взвешенном состоянии и плавающие. Во взвешенном состоянии мо-
гут находиться примеси самого разнообразного состава: неоргани-
ческие, ухудшающие свойства воды водоема по какому-либо при-
знаку вредности; органические, разлагающиеся биохимическим пу-
тем или стойкие к действию микроорганизмов и токсичные для них.
Возможно также присутствие в частицах взвеси микрофлоры, спо-
собствующей бактериальному загрязнению водоема. Взвешенные
вещества с низкой степенью дисперсности, имеющие большую ско-
181
рость осаждения, способствуют образованию наносов в месте спус-
ка сточных вод в водоем. Поэтому запрещается сброс в водоем
сточных вод, содержащих взвешенные вещества, оседающие со
скоростью 0,4 мм/с для проточных водоемов и 0,2 мм/с для водо-
хранилищ, если даже общее их содержание не превышает установ-
ленных величин. При спуске сточных вод в водоемы, использующие-
ся для хозяйственно-питьевого водоснабжения, количество взвешен-
ных веществ должно увеличиваться не более чем на 0,25 мг/л.
Сброс сточных вод с повышенной температурой может привести
к нарушению процесса самоочищения, ухудшению кислородного
режима, угнетению жизнедеятельности гидробионтов. Поэтому в
летний период допускается повышение температуры воды водоемов
после спуска сточных вод не более чем на 3° С по сравнению со
среднемесячной температурой самого жаркого месяца за послед-
ние 10 лет.
Глава VI. ОБЗОР МЕТОДОВ ОЧИСТКИ ПРОИЗВОДСТВЕННЫХ
СТОЧНЫХ ВОД
Очистка производственных сточных вод предназначается для
удаления загрязняющих примесей, извлечения содержащихся в них
ценных веществ и обеззараживания воды перед спуском в водоем.
В зависимости от характера и степени изменения свойств при-
месей в процессе обработки различают регенеративные и деструк-
тивные методы очистки сточных вод. Удаление примесей из сточ-
ных вод при использовании регенеративных методов очистки идет
практически без изменения химического строения примесей. Эти
методы перспективны для применения в системах оборотного водо-
снабжения и для создания бессточных технологических процессов.
Регенеративные методы наиболее эффективны по технико-экономи-
ческим показателям, так как позволяют сократить расход воды, за-
траты на очистку стоков и возвратить в производство полезные
продукты. К регенеративным методам относятся экстракция, эва-
порация, ионный обмен и др. Деструктивные методы очистки сточ-
ных вод основаны на глубоком изменении химического строения
примесей, что способствует переходу их в менее сложные пли не-
токсичные соединения. К деструктивным методам очистки сточных
вод относится биологическая очистка и многие химические способы.
Механические методы очистки сточных вод рассчитаны на уда-
ление грубодиспергированных веществ. К ним относятся извлечение
крупных примесей с помощью решеток или сеток, отстаивание воды
для выделения грубодисперсных примесей, фильтрование, центри-
фугирование. Для некоторых видов сточных вод бывает достаточ-
ным применение этих методов для того, чтобы они удовлетворяли
требованиям, предъявляемым к воде, поступающей в сеть оборот-
ного водоснабжения или в водоем. Для большинства же видов про-
изводственных сточных вод необходимо дополнительное использо-
вание- других методов, позволяющих достаточно эффективно уда-
лять примеси различной природы.
182
§ 45. Физико-химические методы очистки
производственных сточных вод
Коагулирование. Этот метод используется для выделения из
сточных вод взвешенных и коллоидных примесей. Механизм про-
цесса коагулирования сточных вод аналогичен тому, который был
рассмотрен при обработке природных вод. Но переносить его меха-
нически на обработку сточных вод нельзя, так как состав примесей
сточных вод не отличается таким постоянством, как в природных.
Коагулянт необходимо выбирать так, чтобы знак заряда его кол-
лоидных частиц был противоположен знаку заряда частиц приме-
сей. Вследствие малой агрегативной устойчивости взвешенных ве-
ществ доза коагулянта для их удаления будет меньше, чем для
коагуляции коллоидных примесей. При коагулировании удаляются
примеси, обусловливающие мутность, цветность воды, снижаются
ХПК и БПК. Наряду с сульфатом алюминия в качестве коагулян-
тов широко используются хлорид железа (III), сульфат железа
(II), хлорид алюминия, основной хлорид алюминия, катионные по-
лиэлектролиты (BA-102, ВА-212 и др.). Если производственные
сточные воды не имеют достаточного щелочного резерва, необходимо
проводить коагулирование с подщелачиванием.
Перспективным методом очистки производственных сточных вод
от коллоидных и эмульгированных примесей служит электрокоагу-
ляция. Этот метод имеет ряд преимуществ перед обычным коагу-
лированием. Он относится к безреагентным методам, является бо-
лее технологичным, не требует больших производственных площа-
дей. При очистке производственных сточных вод, содержащих тон-
кодиспергированные примеси, под действием электрического поля
происходит концентрирование, коагуляция или коалесценция при-
месей, что способствует эффективному разделению фаз и очистке
воды от этого вида загрязнений. Метод электрокоагуляции оказал-
ся эффективным для очистки сточных вод от тонкодиспергирован-
ных мономеров и полимеров. Стоимость 1 м3 воды электрокоагуля-
цией в зависимости от свойств и концентрации загрязнений состав-
ляет 0,1 —10 коп, в то время как очистка воды реагентными ме-
тодами обходится в несколько раз дороже. Например, стоимость
очистки сточных вод от производства полистирола методом коагу-
ляции хлоридом магния с подщелачиванием и применением поли-
акриламида в качестве флокулянта составляет 26 коп. за 1 м3.
Электрокоагуляцию можно применять в производствах с замкну-
тым циклом водообеспечения. Но использование этого метода в
каждом конкретном случае требует предварительной отработки и
определения оптимальных условий очистки сточных вод.
Флотация. Этот метод применяется для удаления из сточных
вод эмульгированных или суспендированных примесей. Сущность
флотации заключается в образовании агрегатов «пузырек газа —
извлекаемая примесь», которые затем всплывают на поверхность.
Формирующийся на поверхности пенный слой насыщен извлекаемой
примесью и механически удаляется. При обычных видах флотации
(пневматической, напорной, пенной) газовая фаза создается пода-
183
чей воздуха в обрабатываемую воду. Воздух поступает в воду в
тонкодиспергпрованном состоянии. Диаметр пузырьков составляет
15-10-6—30-Ю-6 м.
Образование агрегатов «воздух—частица примеси» происходит в
результате концентрирования частиц примесей на границе раздела
вода — пузырек воздуха и закрепления их на поверхности пузырь-
ка вследствие межмолекулярного взаимодействия. Протекание это-
го процесса сопровождается уменьшением поверхностной энергии
пограничных слоев. Прочность образующегося агрегата зависит от
молекулярных свойств контактирующих фаз. В основе флотации
лежит избирательное смачивание, сопровождающееся уменьшением
поверхностного натяжения на границе раздела фаз. Пузырьки воз-
духа гидрофобны и поэтому гидрофобные эмульгированные части-
цы примесей вытесняют с поверхности раздела вода — пузырек воз-
духа— молекулы воды, образуя новые агрегаты частица примеси —
пузырек воздуха.
Если поверхность пузырька воздуха будет полностью покрыта
извлекаемой из воды примесью, например нефтью, то последующие
частицы будут прилипать уже на однородную поверхность. При
этом сила прилипания частиц примесей определяется уже их раз-
мером и величиной поверхностного натяжения па границе раздела
вода — частица примеси.
Для создания оптимальных условий протекания процесса фло-
тации в обрабатываемую воду вводят пенообразующие вещества,
способствующие изменению поверхностного натяжения на границе
фаз. Поверхностное натяжение воды, обрабатываемой флотацией,
должно быть не выше 60• 103—65-103 Н/м. Наиболее эффективно
флотация идет, если частицы имеют определенный размер. Для сус-
пензий этот размер частиц составляет 20—100 нм, для эмульгиро-
ванных органических веществ — 100—300 нм. Высокая суммарная
поверхность пузырьков и их полидисперсность способствуют выде-
лению из воды частиц различных размеров. Для изменения степе-
ни дисперсности частиц примесей в обрабатываемую сточную воду
вводят коагулянты. Наиболее эффективным коагулянтом, исполь-
зуемым при флотации, является хлорид железа (III). Используется
также и сульфат алюминия.
В практике очистки производственных сточных вод в послед-
ние годы стала внедряться электрофлотация. Отличие ее от обыч-
ных видов флотации заключается в том, что пузырьки тонкодис-
пергпрованного газа образуются в процессе электролиза обраба-
тываемой воды. Диаметр пузырьков газа составляет 30—40 нм. Пре-
имущества электрофлотации перед классическими видами флотации
заключаются в повышении скорости процесса, высокой степени из-
влечения примесей, особенно хлопьевидных. При электрофлотации
не обязательно введение органических реагентов-собирателей. Ши-
рокое применение этого метода ограничено высокой стоимостью
очистки. Метод флотации успешно применяется для очистки сточ-
ных вод предприятий нефтехимической, нефтеперерабатывающей,,
химической промышленности.
184
Степень очистки сточных вод данным методом составляет «о—
95% при начальной концентрации примесей 1—2 г/л. Для очистки
сточных вод сложного состава флотация сочетается с другими ме-
тодами. Флотация относится к регенеративным методам очистки
сточных вод. Флотируемые примеси извлекаются из пенного про-
дукта и возвращаются в производство. Так, на Кишиневском мясо-
комбинате методом электрофлотации из сточных вод выделяются
жиры. Электрофлотация используется на некоторых предприятиях
Белоруссии.
К флотации близок метод коалесценции, применяющийся для
выделения эмульгированных примесей. В отличие от флотации
уменьшение степени дисперсности капелек гидрофобной жидкости
основано на адсорбции их поверхностью твердого адсорбента, ко-
торый смачивается этой жидкостью лучше, чем водой. Практически
очистка воды коалесценцией осуществляется с помощью насадки,
выполняющей роль адсорбента.
В Ленинградских инженерно-строительном институте (ЛИСИ) и
технологическом институте (ЛТИ) им. Ленсовета выполнены боль-
шие исследования по электрообработке питьевых и сточных вод.
Наибольший эффект очистки воды достигается при комплексе элек-
трических воздействий (КЭВ), под которым понимается сочетание
действия электрических разрядов и полей. Этот комплексный метод
обработки воды вызывает резкое снижение агрегативной устойчи-
вости примесей воды, приводит к их коагуляции и флокуляции, поз-
воляет резко улучшить качество очищенной воды.
Экстракция. Для очистки сточных вод используется метод экс-
тракции, основанный на извлечении органических примесей с малой
растворимостью в воде с помощью органических растворителей-
экстрагентов, в которых эти соединения хорошо растворимы. Для
улучшения массообмена экстрагент дозируется в очищаемую воду
в тонкодисперсном состоянии. В результате экстракции примесь
распределяется между экстрагентом и водой. Отношение концент-
раций экстрагируемого вещества (примеси) в экстрагенте и в очи-
щенной воде при состоянии равновесия есть величина постоянная k
(коэффициент распределения):
k=C3/CK, (VI. 1)
где Сл — концентрация вещества в экстрагенте, кг/м3; Св — кон-
центрация вещества в воде, кг/м3.
Коэффициент распределения характеризует способность раство-
рителя к экстрагированию. Его величина зависит от концентрации
растворенного вещества, температуры, химической природы приме-
си и экстрагента. Поскольку коэффициент распределения зависит от
концентрации растворенного вещества, закон распределения явля-
ется приближенным и применим только для разбавленных раство-
ров. При повышении температуры коэффициент распределения
уменьшается. Это необходимо учитывать при очистке производст-
венных сточных вод. Так, при извлечении фенолов из подсмольных
вод предусматривается охлаждение.
185
1 а о лица 14. Характеристика экстрагентов,
используемых для извлечения фенола из воды
(20° С)
Экстрагенты Ко эф- фици- ент рас- преде- ления Плот- ность, г/см8 т. кип., °C Раствори- мость в поде, %
Бензол 2,2 0,88 80 Не раст-
Диизопропило- 27,8 0,725 68,7 ворим 0,2
вый эфир Этилацетат 55 0,9 77,1 7,9
Бутилацетат 51,9 0,875 125 0,7
Экстракцией удаляют
из воды фенолы, жирные
кислоты, анилин и другие
органические примеси. В
табл. 14 даны характери-
стики некоторых экстра-
гентов, применяемых для
удаления фенолов из во-
ды.
Предпочтительным бу-
дет использование того
экстрагента, который име-
ет наибольший коэффици-
ент распределения и ма-
лую растворимость в воде. Из данных табл. 14 следует, что этил-
ацетат является эффективным экстрагентом (& = 55), но он имеет
высокую растворимость в воде. Поэтому из приведенных соедине-
ний в табл. 14 наиболее целесообразно для экстракции фенолов ис-
пользовать бутилацетат.
Так как однократной экстракцией не удается достаточно полно
извлечь примеси из воды, то процесс осуществляют в несколько
ступеней. Если в одном из растворителей растворенное вещество
находится в виде ионов, а в другом — в виде молекул, то закон
распределения для бинарного электролита будет иметь вид
& б’ноиУ/'/С’мол,
(VI.2)
где Сион—концентрация растворенного вещества в растворителе,
в котором оно находится в виде ионов; Смол — концентрация веще-
ства в растворителе, в котором оно содержится в виде молекул;
Уг — коэффициент активности ионов при данной концентрации.
Методом экстрагирования удаляются органические вещества,
полнота извлечения их составляет более 90%. Эффективность экст-
рагирования зависит от растворимости вещества в воде, pH и ус-
тойчивости эмульсии. На процесс экстракции отрицательно влияют
присутствующие в воде ПАВ, так как они повышают устойчивость
образующихся эмульсий. Температура кипения экстрагента должна
быть ниже температуры кипения удаляемых примесей или их азео-
тропных смесей.
Для повышения экстрагирующего действия растворителя в
практике используются смешанные растворители. Например, раст-
воритель, содержащий 40% амилового спирта и 60% диизопропи-
лового эфира, имеет для фенолов коэффициент распределения 66,4.
После экстрагирования растворенное вещество извлекается из экст-
рагента перегонкой. Отрегенерированный экстрагент снова поступа-
ет в экстракторы, а экстрагированное вещество используется по
назначению. Экстракция относится к регенеративным методам очи-
стки сточных вод. Перспективность и широкое применение этого
метода обусловлены его высокими технико-экономическими пока-
186
зателями и возможностью использования в оборотных и бессточных
системах водоснабжения предприятий.
Пароциркуляционный метод (эвапорация). Этот метод очистки
применяется для удаления из сточных вод веществ, летучих с водя-
ным паром (фенол, анилин). Он основан на образовании некото-
рыми органическими соединениями азеотропной смеси с водяным
паром. Метод широко используется для извлечения фенолов из
сточных вод коксохимических заводов. Сущность метода заключа-
ется в том, что подлежащая очистке вода, нагретая до 100° С, посту-
пает в колонку с насадкой, которая позволяет увеличить поверхность
контакта пара с водой. Навстречу стекающей воде подается водя-
ной пар с температурой 102—103° С. При контакте пара с водой в
газовую фазу переходят вещества, летучие с водяным паром.
Распределение вещества в жидкой и газовой фазе подчиняется
закону распределения. Коэффициент распределения для водных
растворов фенола равен 2—2,5. Регенерация водяного пара осу-
ществляется в поглотительных колонках различными методами, ос-
нованными на образовании нелетучих соединений. Процесс осуще-
ствляется при повышенном давлении. Так, фенол из пара извлека-
ется обработкой его 7—15%-ным раствором едкого натра, нагре-
того до 102—-103° С. При этом образуется фенолят натрия:
СбН5ОН + ПаОН = СбН5ОПа + Н2О. Концентрация фенола в щелоч-
ном растворе составляет 10—20%. Регенерация фенолов произво-
дится диоксидом углерода:
2C6H5ONa + СО2 + Н9О = 2С6Н5ОН + Na2CO3
На работу обесфеноливающей установки отрицательно влияют
присутствующие в воде в повышенном количестве диоксид углерода,
сероводород, снижающие концентрацию циркулирующей щелочи.
В воде, подлежащей обработке данным методом, не должно содер-
жаться смол и масел, вызывающих образование отложений на по-
верхности насадки.
Метод эвапорации позволяет снижать концентрацию фенолов в
воде с 1000—2000 до 100 мг/л. Принимая во внимание, что пре-
дельное содержание фенолов в водоемах составляет 0,001 мг/л, не-
обходимо сочетать этот метод с биохимической доочисткой.
Сорбционный метод очистки производственных сточных вод.
Преимущество метода заключается в том, что он обладает высокой
эффективностью при малой концентрации примесей в воде, т. е.
когда использование других методов становится экономически не-
целесообразным или вообще невозможным. Сущность его заклю-
чается в концентрировании примесей, содержащихся в воде, на
поверхности твердого сорбента. Этот метод может быть использо-
ван как самостоятельный для глубокой очистки воды в оборотных
и бессточных системах водоснабжения.
Сорбционные методы используются для очистки сточных вод от
растворенных веществ, которые находятся в виде молекул, ионов.
При очистке производственных сточных вод методом сорбции в
зависимости от характера примесей может протекать сорбция не-
187
электролитов и слабых электролитов, ионообменная и химическая
сорбция.
Неполярные соединения, органические вещества достаточно пол-
но извлекаются неполярными сорбентами, например активирован-
ным углем. Для сорбции сильных электролитов используются та-
кие полярные сорбенты, как силикагель, ионообменные смолы. Сор-
бент может использоваться однократно или регенерироваться после
цикла сорбции. Десорбция примесей производится переводом сор-
бата в соединение, плохо сорбируемое этим сорбентом, отгонкой
или экстракцией различными растворителями.
Количество извлекаемых примесей из воды зависит от природы
сорбента и примесей, от площади поверхности сорбента, на которой
идет процесс сорбции, и условий проведения процесса (статические,
динамические). Сорбция в статических условиях заключается в
контактировании воды с определенным количеством сорбента в те-
чение некоторого времени и последующим отделением отработан-
ного сорбента путем фильтрования, отстаивания или центрифугиро-
вания. Для повышения эффективности процесса используется мно-
гоступенчатая схема, т. е. новая порция сорбента приводится в кон-
такт с уже частично очищенной водой. Сорбция в динамических ус-
ловиях является более эффективной, так как при фильтровании во-
ды через слой сорбента достигается практически полное извлечение
примесей. С экономической точки зрения сорбция в динамических
условиях предпочтительна потому, что позволяет более полно ис-
пользовать сорбент и реже проводить его регенерацию.
В практике очистки сточных вод от растворенных органических
соединений широко применяется сорбция активированным углем
(АУ). Степень извлечения загрязнений при использовании этого
метода составляет 90—99%. Эффективность сорбции органических
веществ активированным углем зависит от их химической природы
и концентрации в воде, марки активированного угля. Согласно пра-
вилу уравнивания полярностей П. А. Ребиндера активировании уг-
лем из воды в большей степени будут сорбироваться вещества,
имеющие меньшую растворимость. С повышением температуры воз-
растает скорость сорбции, но уменьшается количество сорбирован-
ного активированным углем вещества. Высокая эффективность из-
влечения органических примесей позволяет применять сорбционные
методы для доочистки сточных вод и при повторном их исполь-
зовании.
Метод ионного обмена. Этот метод используется для очистки
сточных вод, содержащих растворенные примеси в ионном виде.
Метод ионного обмена широко применяется при очистке сточных
вод, содержащих ионы тяжелых металлов, антибиотики, ПАВ, кра-
сители, органические кислоты. Преимущества этого метода перед
традиционными заключаются в возможности утилизации ценных
продуктов, содержащихся в сточных водах, сокращении объема
сбрасываемых стоков, снижении расхода реагентов, значительном
уменьшении площадей, занятых очистными установками. Использо-
вание комплексных схем позволяет не только очищать сточные воды
188
и возвращать в производство извлекаемые вещества, но и получать
обессоленную воду, проводить регенерацию рабочих растворов.
В Воронежском государственном университете разработана ионооб-
менная установка по очистке хромсодержащих сточных вод гальва-
нических цехов производительностью 2 м3/ч для промывных вод и
0,3 м3/ч для регенерации электролита. Промывные воды содержат
25—300 мг/л Cr (VI), электролит — 50—150 г/л. Применение пред-
ложенной схемы обработки сточных вод и регенерации электролита
позволило ежегодно возвращать в производство 2,5 т СгО3 в виде
раствора концентрации 30—60 г/л и получать до 9 тыс. м3 частично
обессоленной воды для промывки деталей.
Катионы тяжелых металлов обмениваются на катионитовых
фильтрах. Катиониты обычно используются в Н- или Na-форме.
Анионы тяжелых металлов СгО42-, [Cu(CN)2]~, [Au(CN)4]~ и дру-
гие обмениваются на сильноосновных анионитах, которые приме-
няются в СК- или ОН"-форме. Например:
RC1 + [Си (CN)2]- R (Си (CN)2j + Cl-
Комплексные цианиды извлекаются из воды ионитами в 1,5—
2 раза эффективнее, чем в форме ионов CN~.
На работу ионообменных фильтров отрицательно влияют взве-
шенные и коллоидные примеси, вызывающие заиливание фильтра.
Нежелательными компонентами очищаемых сточных вод являются
ПАВ, тиомочевина, фосфаты ввиду возможности образования устой-
чивых комплексных соединений с ионитом, снижающих произво-
дительность работы ионообменной установки.
Обмен крупных органических ионов, содержащихся в сточных
водах, осуществляется на макропористых ионитах. Ионный обмен
органических ионов сопровождается обычно и необменным погло-
щением молекул органического вещества, что ведет к повышению
сорбционной емкости ионита. Регенерация ионитов при извлечении
органических веществ наиболее эффективно осуществляется водно-
органическими растворами электролитов. Так, при сорбции анион-
ных ПАВ анионитом высокая степень извлечения органических
ионов и молекул при регенерации достигается при использовании
водно-диоксанового раствора хлорида натрия.
Особую группу полимерных ионообменных материалов состав-
ляют электронообменники (редокситы)—окислительно-восстано-
вительные иониты. Они используются для удаления из воды раство-
ренного кислорода, галогенов, извлечения благородных металлов,
перевода ионов металлов из одной степени окисления в другую.
Действие их основано на существовании в структуре ионита актив-
ных групп, обладающих восстановительными или окислительными
свойствами. При контакте редоксита с водой, содержащей соедине-
ния, способные к окислению или восстановлению, идут окислитель-
но-восстановительные реакции. Ионный обмен является одним из
перспективных методов очистки производственных сточных вод.
при переходе предприятий на безотходную технологию.
189
Электродиализ. Электродиализные установки с ионитовыми
мембранами, использующиеся для опреснения воды, в последние
годы стали применяться и для очистки производственных сточных
вод. Основное назначение электродиализных установок — извлече-
ние из обрабатываемой воды ионизированных примесей. Механизм
разделения примесей аналогичен тому, который был рассмотрен
при обессоливании воды. Так как при электродиализе происходит
снижение общего солесодержания обрабатываемой воды, то это де-
лает целесообразным его применение в оборотных системах водо-
снабжения. Если в обрабатываемой воде содержатся катионы ме-
таллов, образующие труднорастворимые соединения, то в промыв-
ной раствор при необходимости добавляется кислота для предот-
вращения образования осадков на поверхности мембран. В процес-
се работы установки активная реакция католита становится щелоч-
ной, а анолита — кислой. Смешением этих растворов может быть
достигнута их полная или частичная нейтрализация. Исходными
данными, которые характеризуют пригодность электродиализа для
очистки сточной воды, являются срок службы мембран и электро-
дов, расход реагентов на нужды установки, расход электроэнергии,
количество и скорость подачи воды, затраты на эксплуатацию уста-
новки. Экономически целесообразным применение электродиализа
для очистки производственных сточных вод считается в том случае,
когда извлекаемые примеси возвращаются в производство.
Гиперфильтрация (обратный осмос). Этот метод в последние
годы стал применяться как в нашей стране, так и за рубежом для
очистки производственных сточных вод от растворенных примесей
во многих отраслях промышленности. Преимущества гиперфильтра-
ции перед другими методами очистки сточных вод заключаются в
том, что этот процесс прост в эксплуатации и общие затраты элек-
троэнергии относительно невелики. Установка занимает небольшую
площадь, работа ее может быть автоматизирована. Качество очи-
щаемой воды получается настолько высоким, что она без дополни-
тельной обработки может быть направлена в водооборот. Произ-
водительность работы гиперфильтрационных установок зависит от
разности между рабочим и осмотическим давлением. При высокой
концентрации растворенных веществ рабочее давление становится
фактором, ограничивающим применение этого метода. Так, рабочее
давление при гиперфильтрации 5—10%-ных растворов солей состав-
ляет 4600—9800 кПа. Так как в кислой и щелочной средах усили-
вается гидролиз ацетатцеллюлозы, составляющей активную часть
мембран, то процесс следует проводить в интервале pH обраба-
тываемой воды от 4 до 7. С повышением температуры возрастает и
скорость гидролиза мембран, поэтому температура обрабатываемой
воды должна быть не выше 35—40° С. Рабочее давление зависит от
концентрации примесей в сточной воде. Экономически оправдан-
ным считается давление (4600—5000 кПа). Оно должно быть выше
осмотического давления образующихся концентрированных рас-
творов.
190
Метод гиперфильтрации используется для извлечения органиче-
ских веществ, для очистки сточных вод нефтеперерабатывающей
промышленности, для очистки сточных вод от ПАВ. При очистке
сточных вод может быть достигнуто снижение солесодержания на
80%.
Стоимость очистки сточных вод методом гиперфильтрации и це-
лесообразность ее применения определяются конкретными усло-
виями. С повышением производительности установки снижается
стоимость очистки. Стоимость очистки 1 м3 сточной воды колеблет-
ся от 3 до 20 коп. На больших установках она составляет 3—5 коп.
Утилизация содержащихся в сточных водах веществ и возвращение
очищенной воды в оборотную систему водоснабжения при хорошем
качестве мембран является весьма экономичным и эффективным
методом для очистки производственных сточных вод.
Наряду с гиперфильтрацией для очистки производственных сточ-
ных вод от высокомолекулярных веществ (с молекулярной массой
выше 1200) применяется метод ультрафильтрации. Обработка воды
при ультрафильтрации ведется при более низком рабочем давлении,
а используемые мембраны имеют больший (в 50—2000 раз) раз-
мер пор.
§ 46. Химические методы очистки сточных вод
В сточных водах многих производств часто содержатся высоко-
токсичные соединения: цианиды, соли тяжелых металлов, некото-
рые органические вещества. Предварительное удаление таких ве-
ществ из сточных вод является необходимым. Очистка сточных вод,
содержащих такие примеси, часто проводится с применением хими-
ческих методов. Они основаны на осуществлении химических реак-
ций, в результате которых образуются труднорастворимые соедине-
ния, происходит окисление или восстановление примесей, превра-
щение токсичных соединений в нетоксичные. Избыток щелочности
и кислотности из сточных вод удаляется методом нейтрализации.
При очистке сточных вод от солей тяжелых металлов исполь-
зуются методы осаждения. Так, ионы Zn2+, Cu2+, Ni2+ из сточных
вод можно удалить в виде труднорастворимых оснований, сульфи-
дов, карбонатов:
Cu2+ + 2ОН- = Си (ОН)2 pH 8—10
N12+ + 2ОН- = Ni (ОН]2 pH 9,5
Zn2+ + 2ОН- = Zn (ОН)2 pH 9 — 9,2
При взаимодействии данных ионов с карбонатом натрия образуют-
ся труднорастворимые основные карбонаты:
2CuSO4 + 3Na2CO3 + 2Н2О = (CuOH)2 СО3 + 2Na2SO4 + 2NaHCO3
или в ионном виде:
2Си2+ + СО|~+ 2ОН—=| (СиОН)2 СО3
2N12+ + СС)2- + 2ОН- = | (NiOH)2 СО3
3Zn2++5CO““ + 4Н2О = l Zn3 (ОН)4СО3 + 4НСО^
191
Наиболее полное осаждение попои Zn2; и Hg2; достигается при
обработке сточных вод сульфидом натрия:
Hg(NO3)2 + Na.2S — ; HgS + 2NaNO3 (ПРНу§ =1,6-10—52 моль/л)
Z11SO4 + Na2S = I ZnS + Na2SO4 (nPZns —- 7,1 • Ю—18 моль/л)
Сульфид ртути образуется в тонкодисперсном состоянии, поэтому
его коагулируют введением алюминиевого или железного коагулян-
та. В результате такой обработки содержание ртути в очищенной
воде составляет не более 0,07 мг/л. Недостатком этого метода яв-
ляется образование сероводорода при понижении pH.
Для удаления цианистых соединений из сточных вод использу-
ются реакции окисления хлором, хлорной известью и другими реа-
гентами. Окисление хлором ведется в щелочной среде, так как циа-
нид натрия гидролизуется даже в нейтральной среде, а в кислой
возможно образование еще более токсичного соединения хлорциа-
на C1CN:
NaCN + С12 + 2NaOH = NaCNO + 2NaCl + Н2О
В водных растворах цианаты гидролизуются с образованием неток-
сичных соединений:
2NaCNO + 4Н2О = 2 (NH4)2 СО3 + Na2CO3
CNO- + 2Н2О = NH + + СО'“
При окислении цианидов гипохлоритами протекающие процессы
можно выразить следующими уравнениями:
2 [Си (CN)3]2- + 7С1О- + 2ОН- + Н2О = 6CNO-+7C1- + 2Си (ОН)2 (pH 9 — 10)
Образовавшиеся цианаты окисляются гипохлоритами:
2CNO- + 3CIO- + 2Н2О = N2 + 3CI- + 2НСО“ (pH 6,5)
При очистке сточных вод восстановление примесей иногда соче-
тается с последующим окислением образовавшихся промежуточных
продуктов. Так, при очистке сточных вод предприятий по производ-
ству органических красителей и полупродуктов для разложения
нитросоединений (нитробензола, динитробензола, динитротолуола)
используется метод, который заключается в обработке металличе-
ским железом сточной воды, подкисленной серной кислотой. Нитро-
соединения выделяющимся водородом вначале восстанавливаются
до аминосоединений, которые окисляются до углекислого газа, нит-
ратов и воды.
Метод химического восстановления стойких к биохимическому
окислению азо- и нитрокрасителей, содержащихся в сточных водах
текстильной промышленности, разработан в ЛИСИ. Химизм про-
цесса аналогичен описанному. Окисление и минерализация обра-
зующихся при восстановлении аминосоединений проводится в ще-
лочной среде (pH 8—9).
При очистке сточных вод от соединений хрома (VI) на первой
стадии их восстанавливают в соли хрома (III). Для этого воду
192
обрабатывают гидросульфитом натрия NaHSO3 или сульфатом же-
леза (II):
К2СЫО7 + 6FeSO4 + 7H2SO4 = Cr2 (SC>4)j -j- 3Fe2(SO4)j + К25О4 4- 7Н2О
К2СГ2О7 4- 3NaHSO3 + IH2SO4 — Cr2 (804)3 + K2SO4 + 3XaHSO4 + 4H2O
Образующийся сульфат хрома (HI) обрабатывают гидроксидом
натрия, в результате чего получается труднорастворимый гидро-
ксид хрома:
Cr2 (SO4)3 + 6NaOH = ; 2Сг (ОН)3 + 3Na2SO4
Гидроксид хрома (III) является соединением амфотерным и может
растворяться в избытке щелочи. Поэтому при обработке воды не-
обходима строгая дозировка щелочности (pH^9).
Окислительные методы используются и для доочистки сточных
вод от органических загрязнений. В качестве окислителей могут
быть использованы хлор, озон и перманганат калия. Особенно
перспективно применение озона. Иногда при очистке сточных вод
используется электрохимический метод, основанный на протекании
окислительно-восстановительных реакций непосредственно у элек-
тродов. Например, для удаления из воды цианидов применяется
анодное окисление. Цианиды на аподе окисляются до цианатов:
CN_ + 2OH~—2с“—^СМСН + НгО. Цианаты окисляются с образова-
нием нетоксичных соединений:
2CNO- + 4011- + -> N2 + 2СО2 + 2Н2О
Для повышения эффективности процесса в очищаемую воду вво-
дятся соли, например хлорид натрия. Образующийся па аноде хлор
является дополнительным окислителем цианидов.
Химические методы, в основном, являются деструктивными.
Применение их связано с использованием реагентов, что усложня-
ет работу очистных сооружений, требует создания реагентного хо-
зяйства, точности дозировки, соблюдения мер предосторожности
при работе с этими веществами. Повышение солесодержания сточ-
ных вод при введении реагентов затрудняет повторное использова-
ние таких вод. В то же время создание новых производств, поступ-
ление в сточные воды токсичных, биохимически не разлагаемых
примесей приводит к увеличению объема сточных вод, обрабаты-
ваемых химическими методами. Если в 1975 г. из общего количест-
ва сточных вод химическими методами очищалось 6,2%, то в бли-
жайшем времени эта цифра возрастет до 8,7%, а в перспективе —
до 10,2%.
В производственных сточных водах могут содержаться свобод-
ные кислоты пли щелочи, вызывающие изменение pH, коррозию ме-
таллических и железобетонных канализационных сооружений, ги-
бель флоры и фауны водоемов. Поэтому кислые и щелочные стоки
перед спуском в водоем подвергают нейтрализации. Изменение pH
воды в водоеме при спуске вод должно быть в пределах от 0,25 до 1.
Нейтрализация кислых или щелочных вод при небольших значе-
ниях свободной кислотности или щелочности может осуществлять-
7—1244
193
ся за счет щелочности воды водоема. При поступлении кислых сто-
ков ионы Н+ взаимодействуют с гидрокарбонатам и, содержащими-
ся в воде водоема, с образованием свободной угольной кислоты:
Н+ + НСОГ = Н,0 4- СО>
Избыток гидроксид-ионов, поступающих со щелочным стоком,
связывается гидрокарбонатами кальция в труднорастворимый кар-
бонат:
Са (НСО3)2 + 2NaOH = | СаСО3 + 2Н2О + Na2CO3
При значительном содержании свободных кислот или оснований
в сточных водах производится предварительная полная или час-
тичная нейтрализация их. При нейтрализации сточных вод обычно
учитывается только свободная кислотность или щелочность.
При наличии на предприятии одновременно кислых и щелочных
стоков возможна взаимная нейтрализация стоков при их смешении
в определенных соотношениях. Зная кислотность а кислого стока
и щелочность b щелочного стока, суточный расход кислых п и ще-
лочных т вод, можно рассчитывать соотношение их, соответст-
вующее полной нейтрализации: ап = Ьт.
Для нейтрализации кислых или щелочных стоков используются
реагентный и фильтрационный методы. Реагентный метод нейтра-
лизации заключается во введении в воду нейтрализующих реаген-
тов в виде растворов. В качестве нейтрализующих реагентов для
кислых вод применяются гидроксид натрия, известь, кальциниро-
ванная сода. Самым распространенным реагентом является известь,
недостаток которой состоит в трудности осуществления автомати-
зации процесса. Обработка воды реагентными методами сопровож-
дается образованием большого количества осадка, который затем
необходимо обезвоживать и удалять.
Применение фильтрационного метода требует учета растворимо-
сти солей, образующихся при нейтрализации стоков. Так, при нейт-
рализации сточной воды, содержащей более 1,5% свободной серной
кислоты, происходит отложение труднорастворимого сульфата
кальция на зернах карбонатной загрузки. Во избежание образова-
ния осадков, забивающих фильтры, не рекомендуется использовать
доломит в качестве загрузки при содержании в воде соединений
железа (II и III). Фильтры, в которых происходит взаимодействие
примесей воды с веществом загрузки, называются мутационными.
Мутационные фильтры могут быть использованы не только для
нейтрализации сточных вод. В зависимости от химических свойств
примесей воды и загрузки на них могут протекать и другие процес-
сы, например окислительно-восстановительные.
§ 47. Сжигание органических примесей, содержащихся
в сточных водах
Для обработки промышленных отходов и высококонцентрирован-
ных производственных сточных вод химических производств, содер-
жащих органические и минеральные примеси, применяется терми-
194
ческий (огневой) метод обезвреживания. Возможность применения
этого метода определяется свойствами примесей. В табл. 15 при-
водятся данные о возможности термического обезвреживания про-
мышленных отходов различных отраслей промышленности совме-
стно с бытовыми отходами на городских установках.
Таблица 15. Классификация загрязняющих веществ различных отраслей
промышленности
Bff'U-i промышленных отходов Консистенция Воспламеняемое п> Возможность сжигания совместно с бытовыми отходами в городских уст ановках
Эфиры, спирты и другие растворители Жидкие Воспламеняю- щиеся Отсутствует
Отходы бензина, отра- ботанные масла » » Имеется
Кислоты, щелочи » Невоспламеняю- щиеся »
Шламы масел Угольные шламы Илистые » Воспламеняющие- ся Воспламеняющие- ся после подсу- шивания » Имеется при опре- деленных усло- виях
Гальванические шламы Пастообраз- ные Невоспламсияго- щиеся Отсутствует
Жиры, битуминозные от- ходы » Воспламеняющие- ся »
Отдельные виды пласт- масс, смолы (термо- пластичные) Твердые » л
Отдельные виды пласт- масс, смолы (термо- реактивные) » » Имеется при опре- деленных усло- виях
Целлюлоза, дерево, бу- мага » » Имеется
Кожа » Трудновоспламе- няющиеся Имеется при оп- ределенных ус- ловиях
Кожа, содержащая хром » То же Отсутствует
Неорганические отходы от фильтров » Невоспламеняю- щиеся • »
Концентрированные жидкие отходы, содержащие горючие ток-
сичные соединения, тоже могут обезвреживаться термическим ме-
тодом, но условия обработки будут отличаться от условий сжига-
ния бытовых отходов. Так, серьезной проблемой, возникающей при
термической обработке токсичных соединений, является очистка от-
ходящих газов.
Промышленные стоки, содержащие горючие токсичные соедине-
ния, успешно обезвреживаются огневым методом в циклонных ре-
7*
195
акторах. Сущность этого способа заключается в том, что концент-
рированные промышленные стоки в тонкодисперсном состоянии по-
даются в окислительную зону огневого факела. Вода испаряется,
горючие органические примеси сгорают с образованием нетоксич-
ных газов, а неорганические компоненты выводятся из реактора в
виде расплава. Стоимость обезвреживания промышленных стоков в
циклонных реакюрах в 1,5—2 раза ниже, чем в печах камерного п
шахтного типа.
§ 48. Очистка воды от радиоактивных изотопов
(дезактивация)
В пашен стране и во всем мире осуществляется массовое строи-
тельство п эксплуатация атомных электростанции (АЭС). Подго-
товка обогащенного урана для АЭС, работа самих атомных реак-
торов и парообразователей, переработка отходов АЭС сопряжены с
неизбежным радиоактивным загрязнением воды и образованием
сточных вод с повышенной радиоактивностью. При научных иссле-
дованиях, в промышленности, медицине широко применяются ра-
диоактивные изотопы, которые могут попадать в сточные воды, по-
этому вопрос дезактивации воды является весьма актуальным. Ра-
диоактивные вещества могут быть источниками а-, [3- или у-излуче-
ния. а-Излучепие представляет собой поток положительно
заряженных частиц. р-Излучецпе — поток электронов, у-пзлучение
имеет электромагнитную природу, которое по действию близко к
рентгеновскому и обладает наибольшей проникающей способно-
стью. Одной из характеристик радиоактивных изотопов является
период полураспада. Некоторые изотопы теряют активность очень
быстро. Например, период полураспада изотопов 16N — 6 сут, 32Р —
14,3 сут, 1311 — 8 сут и т. д. Дезактивация вод, содержащих быстро
распадающиеся изотопы, достигается при отстаивании воды в специ-
альных емкостях. Наиболее опасны радиоактивные изотопы, имею-
щие большой период полураспада: 90Sr — 19,9 года, 137Cs — 33 года,
60Со — 4,95 года и др.
В природных водах содержится до 50 радиоактивных изотопов,
многие из которых имеют большой период полураспада, по их ко-
личество крайне мало. В сточных водах радиоактивные изотопы мо-
гут быть в растворенном, коллоидном и взвешенном состоянии. При
попадании в водоем они могут переходить в донные отложения в
результате образования труднорастворимых соединений и в значи-
тельной степени аккумулироваться водными организмами, особен-
но микроорганизмами планктона. Концентрация радиоактивных
изотопов в них может в тысячи раз превышать их содержание в
воде. Такие радиоактивные изотопы, как 32Р, 90Sr, могут усваивать-
ся микроорганизмами и затем передаваться другим более высоко-
организованным организмам (рыбам, ракообразным) по трофи-
ческой цепи.
По степени активности различают низко-, средне- и высокоак-
тивные воды. Выбор метода дезактивации зависит от активности
196
обрабатываемых вод и химических свойств элемента. Высокоак-
тивные воды концентрируют упариванием, обрабатывают специаль-
ными реагентами и выдерживают в особых емкостях до полной дез-
активации. Методы дезактивации средне- и низкоактивных вод ана-
логичны используемым для обычной очистки воды, но могут иметь
ряд особенностей. Концентрация радиоактивных изотопов в воде
настолько мала, что при осаждении их не достигается произведение
растворимости образующегося соединения. Поэтому концентрация
удаляемого элемента повышается за счет введения устойчивого изо-
топа данного элемента или близкого к нему ио свойствам. Этот
прием называется соосаждением. Так, более полному удалению ме-
тодом осаждения 90Sr будет способствовать введение ионов Са2+,
а для удаления 1311 в форме иодида серебра — введение иодида
калия. Для дезактивации воды применяются методы осаждения,
коагулирование с последующим отстаиванием, метод ионного об-
мена, сорбционные методы. Например, метод контактной коагуля-
ции обеспечивает дезактивацию воды, в которой содержание 32Р в
форме фосфат-ионов в 80 раз превышает предельно допустимую
норму. Успешно удаляются коагуляцией радиоактивные изотопы,
находящиеся в коллоидном и взвешенном состоянии. Использова-
ние коагулирования с подщелачиванием позволяет снизить на 97—
98% содержание 90Sr, 141Се, 95Zr. Эффективным методом дезактива-
ции для удаления радиоактивных изотопов, находящихся в воде в
форме ионов, является метод ионного обмена. Этот способ исполь-
зуется для дезактивации отработанной воды на атомных электро-
станциях.
Дезактивация низкоактивных вод может также проводиться
биологическими методами. Дезактивация воды в процессе биоло-
гической очистки основана на способности микроорганизмов актив-
ного ила, биопленки, планктона аккумулировать радиоактивные
изотопы. Так, степень удаления плутония-239 в биофильтрах со-
ставляет 75—95%, а в аэротенках — 75—80%.
Общим недостатком всех применяемых методов является обра-
зование высокоактивных отходов (осадка, концентрата и т. п.), ко-
торые подлежат обработке или захоронению.
7*—1244
РАЗДЕЛ ТРЕТИЙ
МИКРОБИОЛОГИЯ воды
Глава VII. ОСНОВЫ ОБЩЕЙ МИКРОБИОЛОГИИ
§ 49. Предмет и задачи микробиологии.
Краткий исторический очерк развития микробиологии
как науки
Микробиология — наука о живых организмах, имеющих малые
размеры и не видимых невооруженным глазом. Задача микробио-
логии заключается в изучении строения и закономерностей разви-
тия микроорганизмов с целью выяснения роли их в процессах пре-
вращения веществ, возможности управления этими процессами.
Микроорганизмы имеют исключительно важное значение в круго-
вороте веществ в природе. Одни микроорганизмы осуществляют
распад сложных соединений в процессе разложения органических
остатков, а другие в процессе жизнедеятельности синтезируют ор-
ганические вещества из простых неорганических соединений (диок-
сида углерода, атмосферного азота и др.). Некоторые микроорга-
низмы могут вызывать болезни, а другие используются для лече-
ния ранее нс излечимых заболеваний. Микроорганизмы способст-
вуют образованию почв, под их воздействием образуются отложе-
ния некоторых полезных ископаемых (например, некоторых видов
железных и серусодержащих руд). В нашей стране создана микро-
биологическая отрасль промышленности, одной из задач которой
является получение кормовых белков из отходов нефтеперераба-
тывающих заводов.
Водная микробиология изучает строение и жизнедеятельность
микроорганизмов, находящихся в чистых и загрязненных водах,
определяет направленность и закономерности процесса самоочище-
ния, выясняет возможность использования микроорганизмов в ка-
честве индикаторов степени загрязнения воды. Особое внимание
уделяется изучению биохимических процессов, протекающих при
очистке сточных вод, влиянию микроорганизмов и продуктов их
жизнедеятельности на качество воды и работу очистных сооруже-
ний.
Микроорганизмы не представляют собой единой систематиче-
ской группы. К ним относятся одноклеточные и многоклеточные ор-
ганизмы растительного и животного происхождения: бактерии, бак-
териофаги, вирусы, некоторые водоросли и грибы, простейшие. Об-
щими отличительными признаками всех микроорганизмов является
малый размер, определяющий у них особенности высокой интенсив-
ности обмена веществ.
Морфологический этап развития микробиологии начался после
изобретения микроскопа. В дальнейшем ученые стали не только изу-
198
чать строение микроорганизмов, но и их физиологические функции.
Этому способствовали работы русского ученого М. М. Тереховского,
который впервые использовал экспериментальный метод исследо-
вания жизнедеятельности микроорганизмов. Основоположником на-
учной микробиологии является французский ученый Л. Пастер.
В своих работах он доказал, что брожение представляет собой био-
химический процесс, а его направленность определяется особенно-
стями обмена веществ микроорганизмов. Пастер впервые установил
факт развития определенных групп микроорганизмов в бескисло-
родной среде, т. е. в анаэробных условиях. Велики заслуги его и
в области медицинской микробиологии. Создание вакцины против
бешенства явилось неоценимым вкладом в медицинскую науку и
практику. Исследования Пастера имели исключительное значение
для выделения различных отраслей микробиологии.
Развитию научной микробиологии в России способствовали ра-
боты И. И. Мечникова (1845—1916). Разработанные им фагоци-
тарная теория иммунитета и учение об антагонизме микроорганиз-
мов способствовали совершенствованию методов борьбы с инфек-
ционными заболеваниями.
Большая заслуга в разработке методов изучения микроорганиз-
мов принадлежит немецкому ученому Р. Коху, который впервые по-
лучил чистые культуры, используя твердые питательные среды. Он
разработал один из способов стерилизации, описал процесс споро-
образования.
Открытие Д. И. Ивановским в конце прошлого века (1892) ви-
русов позволило объяснить причины возникновения многих заболе-
ваний человека, животных и растений. В настоящее время вирусо-
логия выделилась в самостоятельную отрасль знаний.
Важную роль в развитии научной микробиологии сыграли ра-
боты акад. Н. Ф. Гамалея по открытию особой группы микроорга-
низмов — бактериофагов, изучению изменчивости микроорганизмов.
Основы учения о хемотрофном типе питания микроорганизмов бы-
ли заложены С. Н. Виноградским. Он изучал жизнедеятельность
микроорганизмов — хемотрофов, развивающихся в почве (железо-,
серобактерий). Им был изучен процесс усвоения азота бактериями.
В формировании микологии — науки о грибах — большое значение
приобрели работы Л. С. Ценковского, изучавшего низшие грибы и
водоросли. В. Л. Омелянский изучал жизнедеятельность микроор-
ганизмов почвы и выполнил ряд исследований по изучению меха-
низма разложения целлюлозы.
Выдающихся успехов достигла микробиологическая наука в на-
стоящее время, когда созданы условия для целенаправленных на-
учных исследований. Теоретические и практические вопросы реша-
ются огромным коллективом ученых, работающих в научно-иссле-
довательских институтах. Значительная работа проводится на са-
нитарно-эпидемиологических станциях и производственных объек-
тах. Большой вклад в развитие санитарной микробиологии воды
сделан русскими и советскими учеными. Изучению микрофлоры и
фауны природных вод, выяснению закономерностей процессов са-
7** 199
моочищенпя водоемов посвящены работы Б. Б. Никитинского,
А. С. Разумова, Н. Н. Долгова, С. И. Кузнецова и других иссле-
дователей. Закономерности биохимических процессов, протекаю-
щих при очистке сточных вод, и пути их интенсификации являются
предметом работ С. Н. Строганова, Н. А. Базякиной, А. А. Имше-
нсцкого и других научных коллективов (Академия коммунального
хозяйства им. К. Д. Памфилова, ВНИИ ВОДГЕО, санитарно-ги-
гиенический институт им. Эрисмана и др.).
§ 50. Некоторые сведения по морфологии
и систематике микроорганизмов
К микроорганизмам относятся низкоорганнзованные одно- или
многоклеточные представители растительного (микрофлора) и жи-
вотного мира (микрофауна). Особую группу микроорганизмов со-
ставляют ультрамикробы (вирусы и фаги), являющиеся неклеточ-
ными формами живых организмов. Наиболее часто в природных
и сточных водах встречаются следующие группы микроорганизмов:
1) ультрамикробы (вирусы и фаги); 2) бактерии; 3) актиномице-
ты; 4) грибы; 5) водоросли; 6) простейшие; 7) коловратки.
Рассмотрим отличительные морфологические признаки основ-
ных групп микроорганизмов.
Вирус ы. Это — микроорганизмы, не имеющие клеточного
строения. Размеры структурных единиц вирусов (вирионов) колеб-
лются от 10 до 300 нм. В состав вирионов входят молекулы рибо-
нуклеиновой (РНК) или дезоксирибонуклеиновой (ДНК) кислот,
окруженные белковой оболочкой. Вирусы имеют разнообразную
форму: кубическую, сферическую, палочковидную и др. Размноже-
ние вирусов осуществляется простым делением или более сложным
путем только внутри клеток живого организма. Вирусы обладают
специфичностью действия, т. е. отдельные группы вирусов поража-
ют определенные живые организмы.
Наиболее часто в воде встречаются энтеровирусы, развивающие-
ся в пищеварительном тракте человека и животных. Вирионы энте-
ровирусов имеют малые размеры (17-—32 нм). Они состоят из мо-
лекулы РНК (20—30%) и белковой оболочки. Энтеровирусы устой-
чивы к высушиванию, могут образовывать кристаллы кубического
типа симметрии.
Особую группу вирусов представляют паразитические формы
микроорганизмов — фаги. Фаги, развивающиеся в клетках бакте-
рий, называются бактериофагами, актиномицетов — актинофаги,
грибов — микофаги, водорослей — альгофаги. Фаги имеют булаво-
видную форму (рис. 22). Утолщенная часть называется головкой,
а суженная — хвостовой. Размеры бактериофагов варьируют в пре-
делах от 50 до 100 нм. Размножение фагов осуществляется только
в живых клетках. Адсорбируясь на поверхности клетки бактерии,
фаги выделяют фермент, способствующий растворению оболочки,
после чего молекулы РНК или ДНК фагов поступают в клетку.
При этом происходит изменение обмена веществ, заключающееся
200
в том, что вместо вещества клетки идет образование вещества фа-
га. Через несколько часов оболочка клетки разрывается и во внеш-
нюю среду поступает большое количество новых фагов (порядка
нескольких сотен или даже тысяч).
Фаги поражают только определенный вид или близкие к нему
виды микроорганизмов, т. е. обладают избирательностью действия.
Это свойство бактериофагов позволяет использовать их в качестве
санитарно-показательных микроорганизмов. Для индикации опре-
деленного вида бактерий в исследуемую воду помещают небольшое
количество определенного вида
бактериофага и через некоторое
время определяют изменение чис-
ла бактериофагов в определенном
объеме жидкости — титр фага.
Возрастание этого показателя
указывает на присутствие в воде
определенного вида бактерий.
Бактериофаги встречаются в
воде водоемов, сточных водах,
почве, т. е. там, где живут сопут-
ствующие им виды микроорганиз-
мов. Бактериофаги играют опре-
деленную роль в формировании
биоценозов в природных водах.
Рис. 22. Бактериофаги в момент ата-
ки бактериальной клетки (а) и бак-
териофаги при большем увеличении
(б)
Р и к к е т с и и. Они занимают промежуточное положение меж-
ду вирусами и бактериями. Их размеры определяются десятыми
долями микрометра. Имеют шаровидную, палочковидную или ни-
тевидную форму. Риккетсии являются паразитирующими формами
живых клеток, поэтому не развиваются на искусственных питатель-
ных средах. Оболочка и цитоплазма содержат нуклеиновые кисло-
ты. Развиваясь в живом организме, риккетсии вызывают заболева-
ния— риккетсикозы (например, сыпной тиф).
Бактерии. Большинство из них является одноклеточными
формами. Размер клеток бактерий обычно варьирует в пределах от
0,4 до 10 мкм. По форме клеток бактерии делятся на группы: ша-
ровидные, палочковидные и извитые (рис. 23). Среди бактерий ша-
ровидной формы (кокков) имеются разновидности, отличающиеся
порядком расположения клеток. Если после деления клетки отхо-
дят друг от друга, то образуются одиночные кокки (микрококки).
При делении, происходящем в одном направлении, кокки могут ос-
таваться соединенными попарно, образуя диплококки или цепочки
клеток — стрептококки. При делении клеток в двух взаимно пер-
пендикулярных направлениях происходит образование групп кле-
ток, отличающихся плотной упаковкой, — сарцин. Если же деление
клеток идет беспорядочно, то образуются группы бактерий, напо-
минающие по внешнему виду виноградную гроздь,— стафиллокок-
ки. Из кокковых форм бактерий в процессе минерализации органи-
ческих веществ активно участвуют представители родов Micrococ-
cus, Sarcina, Streptococcus и др.
201
Палочковидные формы бактерий аналогично кокковым могут
быть одиночными, располагаться попарно или в виде цепочки. Па-
лочковидные формы могут иметь утолщение в различных частях
клеток. Некоторые из них образуют споры (р. Bacillus).
Извитые формы отличаются количеством витков. Бактерии,
имеющие небольшой изгиб (до витка), называются вибрионами;
с одним или несколькими (4—6) витками — спириллами; длинные
тонкие клетки с большим количеством витков — спирохетами. На-
ряду с типичными формами бактерий существуют микроорганизмы,
Рис. 23. Строение бактерий:
а, б — палочковидные; в — извитые; г — кокковые
отличающиеся от них некоторыми признаками. Это прежде всего
нитчатые формы, у которых клетки объединены общей капсулой
в тонкие длинные нити. К ним относятся некоторые виды железо-
и серобактерий. Диаметр нитей составляет 1—5 мкм. Размножение
у этих бактерий происходит делением. Молодые клетки выходят в
воду и дают затем новые нити или прикрепляются к капсуле, спо-
собствуя образованию ветвящихся колоний. Каждая нить объеди-
няет в себе несколько десятков, сотен или тысяч клеток.
В особую группу выделяются миксобактерии. На раннем этапе
развития они представляют собой овальные, заостренные к концам
клетки с обособленным ядром. После размножения клетки образу-
ют слизистые колонии (плодовые тела), в которых затем форми-
руются микроцисты. Из микроцист при прорастании вновь образу-
ются клетки миксобактерий.
Цитофаги имеют клетки удлиненной формы, тоже образуют
слизистые колонии, но без микроцист. Они участвуют в разложе-
нии сложных углеводов.
Среди бактерий много подвижных форм. Передвижение бакте-
рий осуществляется с помощью жгутиков, которые могут распола-
гаться по всей поверхности оболочки или сосредотачиваться на од-
ном конце клетки. У некоторых бактерий образуются и особые вы-
росты, роль которых окончательно не выяснена.
Бактерии проявляют свойство полиморфизма, т. е. многообразие
формы и размеров для одного и того же вида. Форма бактериаль-
ных клеток может изменяться под влиянием изменения условий
окружающей среды (температура, pH, режим питания и др.).
Бактериальная клетка имеет сложное строение. Она включает
в себя структурные элементы, необходимые для осуществления об-
202
мена веществ. Наружный слой содержимого клетки — цитоплаз-
мы — уплотнен и образует трехслойную цитоплазматическую мемб-
рану, состоящую из внутреннего липидного слоя и внешних — бел-
ковых. Цитоплазматическая мембрана непосредственно примыкает
к оболочке клетки. Она обладает избирательной проницаемостью
по отношению к различным соединениям и регулирует осмотиче-
ский перенос веществ в клетку и из нее. В цитоплазматической
мембране сосредоточены системы, катализирующие энергетические
процессы. Внешняя оболочка клетки плотная, но эластичная, что
способствует сохранению определенной формы бактерий. Иногда
эта оболочка набухает, ослизняется, образуя капсулу. В результате
соединения капсул возникают слизистые образования, содержащие
бактериальные клетки — зооглеи.
Цитоплазма представляет собой коллоидный раствор, дисперс-
ной фазой которого являются сложные белковые соединения и ве-
щества, близкие к жирам, а дисперсионной средой — вода. У неко-
торых форм бактерий в цитоплазме содержатся включения — ка-
пельки жира, серы, гликогена и др. Постоянными составляющими
бактериальных клеток являются особые выросты цитоплазматиче-
ской мембраны — мезосомы, в которых содержатся ферментные
окислительно-восстановительные системы. В этих образованиях
идут в основном процессы, связанные с дыханием бактерий. В мел-
ких включениях — рибосомах, содержащих рибонуклеиновую кис-
лоту, осуществляется биосинтез белка. Большинство видов бакте-
рий не имеет обособленного ядра. Ядерное вещество, представлен-
ное ДНК, у них не отделено от цитоплазмы и образует нуклеоид.
Транспортировка веществ, необходимых для жизнедеятельности
клетки, и отвод продуктов обмена осуществляется по особым кана-
лам и полостям, отделенным от цитоплазмы мембраной, имеющей
такое же строение, как и цитоплазматическая. Это структурное об-
разование называется эндоплазматической сетью (ретикулум).-
У некоторых палочковидных форм бактерий (бацилл, клостри-
дий) наблюдается образование спор, отличающихся высокой устой-
чивостью к воздействию факторов внешней среды. Спорообразова-
ние внутри бактериальной клетки сопровождается возникновением
участка с цитоплазмой более вязкой консистенции, который окру-
жается плотной водонепроницаемой оболочкой. Вода в спорах на-
ходится в связанном состоянии. Этим объясняется устойчивость
спор к воздействию высоких температур. Они менее восприимчивы
к действию токсичных для бактерий веществ. С наступлением бла-
гоприятных условий спора прорастает, давая бактериальную
клетку.
Размножение бактерий происходит путем деления клеток. На
твердых питательных средах бактерии при размножении образуют
колонии. Внешний вид колоний определяется видовыми особенно-
стями микроорганизмов, свойствами питательной среды и другими
факторами. Время, необходимое для завершения деления клетки,
называется временем генерации. Скорость процесса генерации за-
висит от условий внешней среды: наличия питательных веществ, pH,
203
концентрации продуктов обмена, температуры и др. При достаточ-
ном количестве питательных веществ в условиях, способствующих
отведению продуктов обмена, например в проточной воде, деление
клеток осуществляется через 15—18 мин.
Систематика бактерий проводится на основании определенных
признаков у группы микроорганизмов. Для выделения бактерий
в определенную систематическую группу необходимо тщательное
изучение морфологии, способности к перемещению, спорообразова-
нию. Отличительным признаком служит отношение клеток к окра-
шиванию. Наиболее употребительным методом дифференциации
клеток микроорганизмов является окрашивание по Граму. Сущ-
ность этого метода заключается в выяснении возможности удержи-
вания клеткой красителя (кристаллический фиолетовый — иод) при
последующей обработке препарата спиртом. Использование этого
индикаторного метода позволяет выделить две группы микроорга-
низмов: грамположительные, сохраняющие окраску; и грамотрпна-
тельныс, обесцвечивающиеся при обработке препарата спиртом.
Для отнесения микроорганизмов в определенную систематиче-
скую группу необходимо знать также особенности обмена веществ,
выяснить характерные признаки изменения внешней среды под вли-
янием жизнедеятельности микроорганизма, например образование
газов; определить способ получения энергии, отношение микроор-
ганизма к воздействию факторов внешней среды.
Наименьшей систематической единицей является вид, объеди-
няющий особей одной или нескольких разновидностей, имеющих
определенные отличительные признаки по отношению к другой бо-
лее общей группе организмов — роду. Род объединяет близкие по
морфологическим и физиологическим признакам микроорганизмы,
но имеющие некоторые особые черты, например разный цвет коло-
ний, вырастающих на питательных средах. Роды объединяются в
семейства, а семейства—в группы. Латинское название микроор-
ганизма состоит из названия рода (первое слово пишется с пропис-
ной буквы) и вида. Например, Bacteria coli. Первое слово указы-
вает на то, что кишечная палочка относится к грамотрицательным
бактериям, имеет небольшую длину клетки, не образует спор, а на-
звание вида указывает на ее естественную среду обитания.
Актиномицеты (лучистые грибы). Актиномицеты за-
нимают промежуточное положение между бактериями и грибами.
Вегетативное тело их называется мицеллием. Нити мицеллия обра-
зуют гифы диаметром 0,2—1,5 мкм. Типичные актиномицеты имеют
одноклеточный разветвленный мицеллий, не содержащий обособ-
ленных ядер. Одна часть мицеллия развивается в питательной сре-
де (субстратный мицеллий), а другая выступает над ней (воздуш-
ный мицеллий). На поверхности воздушного мицеллия образуются
спороносны. Размножаются актиномицеты делением мицеллия и
спорами. К актиномицетам близки микобактерии, представляющие
собой одиночные палочковидные клетки, иногда ветвящиеся. Спор
они не образуют. Некоторые представители актиномицетов (род
Micromonospora) не имеют воздушного мицеллия и образуют по
204
одной споре на субстратном мицеллии. иснивнии срсдии
актиномицетов является почва. Они участвуют в трансформации
многих органических веществ. Развитие их идет в присутствии кис-
лорода. Источником питания для них служат органические веще-
ства. Продукты жизнедеятельности актиномицетов, особенно в ус-
ловиях затрудненной аэрации, могут вызывать появление запахов
и привкусов у воды.
Грибы. Грибы в основном имеют многоклеточный мицеллий.
Гифы их имеют значительно больший диаметр, чем у актиномице-
тов. Подобно актиномицетам, грибы образуют ветвящийся субст-
ратный и воздушный мицеллий. У грибов наблюдается разнообра-
Рис. 24. Плесневые грибы:
а — Penicillium; б — Aspergillus; в — Mticor
зие способов размножения. Они могут размножаться простым де-
лением, почкованием, с помощью спор и половым путем. Споры у
некоторых видов образуются внутри особых образований — споран-
гиев (эндоспоры). После созревания спор оболочка спорангия раз-
рушается и они выходят во внешнюю среду. У других они форми-
руются на гифах-конидиеносцах. Такие споры называются экзоспо-
рами или конидиями. Половое размножение грибов происходит при
слиянии двух клеток, неотличимых внешне, мужской и женской.
Грибы в качестве источника питания используют органические
вещества. Наиболее часто участвуют в биохимических процес-
сах разложения органических веществ в воде и почве следующие
представители грибов: роды Aspergillus, Penicillium, дрожжи, от-
носящиеся к микроскопическим совершенным грибам (класс—ас-
комицеты); семейство мукоровых (Mucoraceae) из низших гри-
бов — фикомицетов.
Плесневые грибы (Aspergillus и Penicillium) имеют многокле-
точный мицеллий (рис. 24). Размножаются они бесполым (спора-
ми) и подовым путем. Экзоспоры вырастают на воздушных гифах,
205
имеющих на концах выросты — стеригмы. У аспергиллевых стериг-
мы находятся на овальных утолщениях, а у пеницилловых — на ки-
стевидных выростах. На стеригмах располагаются вегетативные
споры. Дрожжи (Saccharomycetes) представляют собой крупные
овальные клетки. Размножаются чаще всего почкованием, реже де-
лением и с помощью спор, которые образуются внутри клетки.
Мукоровые грибы имеют одноклеточный разветвленный мицел-
лий. Размножаются с помощью эндоспор, находящихся в сфериче-
ском спорангии. При недостатке кислорода мицеллий распадается
Рис. 25. Синезеленые (1), зеленые (2), диатомовые (3) водоросли:
а, б — р. Oscillatoria; в — Anabaena spiroides; г — Scenedesmus quadricauda; <3 — Ulothrix;
е — Diatoma vulgare; ж — Navicula
на отдельные овальные клетки (мукоровые дрожжи). Грибы класса
базидиоминетов имеют многоклеточный мицеллий. Половое размно-
жение у них происходит с помощью базидий —• больших цилиндри-
ческих клеток, образующихся после оплодотворения. На них выра-
стают четыре базидиоспоры, из которых после прорастания фор-
мируется мицеллий.
Водоросли. Наличие хлорофилла в клетках водорослей об-
условливает их способность к фотосинтезу. Различная окраска во-
дорослей объясняется тем, что наряду с хлорофиллом в их клетках
могут находиться и другие пигменты. Некоторые группы могут ис-
пользовать в качестве источника питания и органические вещества
(миксотрофные водоросли). В поверхностных водоемах и почве наи-
более часто встречаются синезеленые, зеленые и диатомовые водо-
росли (рис. 25). Синезеленые водоросли относятся к самым низко-
организованным формам. Они наиболее приспособлены к жизни в
водоемах, загрязненных органическими веществами. Многие из них
могут фиксировать молекулярный азот для биосинтеза белка. Сре-
ди синезеленых водорослей встречаются одноклеточные (Aphanizo-
menon flos aqua) и нитчатые формы (Anabaena). В их клетках в
отличие от других типов водорослей не имеется вакуолей с клеточ-
ным соком и обособленных ядер. Хлорофилл и другие пигменты (си-
ний — фикоциан, красный — фикоэритрин, оранжевый — каротин)
распределены в виде зерен в наружном слое цитоплазмы. Синезе-
леные водоросли находятся во взвешенном состоянии благодаря на-
206
личию газовых пузырьков внутри клетки. Для многих видов сине-
зеленых водорослей характерно образование слизистых оболочек,
способствующих формированию колоний.
Зеленые водоросли широко распространены в поверхностных
водоемах. Среди них встречаются одно-, многоклеточные и колони-
альные формы. Пигменты у них сосредоточены в особых образова-
ниях— хроматофорах. Размножаются они делением цитоплазмы с
образованием дочерних клеток или половым путем. Некоторые ви-
ды размножаются путем образования подвижных спор. Колонии
образуются в результате бесполого деления, при котором дочерние
клетки остаются связанными друг с другом. Клетки зеленых водо-
рослей имеют разнообразную форму (шаровидную, овальную и др.)
и содержат органоиды, характерные для клеток высших растений.
Ядро у них дифференцировано и отделено от цитоплазмы мембра-
ной. Оболочка клеток состоит из целлюлозы. В цитоплазме могут
содержаться зерна крахмала, который является продуктом фото-
синтеза. Наиболее часто в пресных водоемах встречаются из одно-
клеточных форм хлорелла (Chlorella vulgaris), хламидомонады
(Chlamidomonas), из колониальных — вольвокс (Volvox aureus),
гоипум (Gonium pectorale), из многоклеточных — улотриксовые.
Диатомовые водоросли — одноклеточные микроорганизмы,
имеющие твердые силикатные панцири. Пигменты, входящие в со-
став их клеток, имеют бурую или желтую окраску. Размножаются
они простым делением или с помощью спор. Строение кремниевого
панциря, состоящего из двух створок, является отличительным при-
знаком вида. Из диатомовых водорослей в пресных водоемах наи-
более часто встречаются астерионелла (Asterionella), синедра (Sy-
nedra), мелозира (Melosira).
Простейшие (Protozoa). К этому типу относятся одно-
клеточные животные организмы. Большинство из них имеет отно-
сительно постоянную форму клетки, чему способствует наличие
плотной, эластичной оболочки—пелликулы. Многие простейшие
способны к передвижению. Деление типа на классы основано на
особенностях строения органоидов движения. Размножение про-
стейших происходит в результате простого или множественного де-
ления или половым путем. В определенных, чаще всего в неблаго-
приятных условиях, простейшие способны к иннистированию: клет-
ки их принимают сферическую форму, окружаются двойной проч-
ной оболочкой, образуя цисту. Постоянными обитателями водоемов
являются следующие классы простейших: 1) саркодовые (Sarco-
dina); 2) жгутиковые (Mastigophora или Flagellata); 3) инфузо-
рии (Infusoria или Ciliata).
Класс саркодовых включает в себя простейших, образующих
выросты (псевдоподии) для передвижения и захвата пищи. Сюда
относятся амебы (голые и раковинные), солнечники, фораминифе-
ры и радиолярии (рис. 26). Цитоплазма у них образует два слоя:
внешний (экзоплазма) и внутренний, более темный (эндоплазма).
Амебы имеют обособленное ядро. У некоторых видов солнечников
имеется большое количество ядер (до 200). Захваченная псевдо-
207
подиями пища с небольшим количеством воды образует в клетке
пищеварительные вакуоли, где и происходит переваривание ее.
Остатки пищи выводятся из клетки. Амебы питаются бактериями,
солнечники — другими простейшими (жгутиковыми, инфузориями),
т. е. являются хищниками. В клетках простейших имеются сокра-
тительные вакуоли, которые служат для осморегуляции и выведе-
ния некоторых растворимых в воде продуктов обмена веществ.
У раковинных амеб для защиты от неблагоприятного внешнего воз-
действия имеются раковины, состоящие из органических веществ,
Рис. 26. Представители типа простейших:
а — Amoeba proteus; б — Opercularia coarctata; в — Stylonichia mytilis; г — Paramecium
caudatum; d — Vorticella
выделяемых цитоплазмой, различных включений, забираемых клет-
кой из внешней среды, кремнезема. Форма раковин разнообразная
(округлые, грушевидные, яйцевидные).
Солнечники являются постоянными обитателями пресных и мор-
ских вод. Большинство из них свободноплавающие, но некоторые
прикрепляются к субстрату. Они имеют сферическую форму и ра-
диально расположенные псевдоподии, имеющие внутри прочную
осевую нить. У некоторых видов имеется шаровидный силикатный
скелетик. Фораминиферы и радиолярии, имеющие известковые или
кремниевые скелетики, являются обитателями морей.
Жгутиковые отличаются постоянством формы и наличием тон-
ких протоплазматических выростов — жгутиков, служащих орга-
ноидами движения. Среди представителей этого класса встречают-
ся организмы с различным уровнем организации и типом питания.
Некоторые из них (вольвоксовые, хламидомонады), обладающие
фотосинтетической деятельностью, относятся к растительным орга-
низмам. Эвгленовые имеют смешанный тип питания: в условиях
освещения они получают органические вещества в процессе фото-
синтеза, а в отсутствие света переходят на питание органическими
веществами и обесцвечиваются. При этом продукты питания могут
поступать в виде растворов через пелликулу или заглатываться в
форме твердых частиц. Эвгленовые распространены в водоемах, за-
грязненных органическими веществами. Они имеют один жгутик и
хроматофоры, содержащие хлорофилл. В передней части клетки на-
208
ходится сократительная вакуоля с красным глазком (стигмой).
Оболочка у жгутиковых плотная, что обусловливает постоянство
формы клеток. Наличие определенных видов жгутиковых в водое-
мах с определенной степенью загрязнения позволяет использовать
их в качестве биологических индикаторов загрязнения воды орга-
ническими примесями. Так, в водах со значительной степенью за-
грязнения обитают мелкие жгутиконосцы (размер клетки 10—
25 мкм) рода Boclo, имеющие тело яйцевидной формы и два проти-
воположно направленных жгутика.
Инфузории имеют наиболее сложное строение из простейших
(см. рис. 26). Органоидами движения у них служат реснички. Ин-
фузории — постоянные обитатели пресных и морских вод, некото-
рые живут в почве. Это уже довольно крупные организмы. Размер
инфузории Paramecium caudatum (туфельки) составляет 0,15—
0,2 мм. Деление класса связано с расположением ресничек на по-
верхности тела. Реснички наряду с двигательными функциями мо-
гут выполнять и другую роль, создавая ток воды, облегчающий за-
глатывание пищи из воды. У некоторых инфузорий (брюхореснич-
ных) реснички соединяются в пластинки (мембранеллы) или в пуч-
ки, образуя цирры. Инфузории питаются бактериями, взвешенными
органическими примесями (детрит). Пища попадает вначале в око-
лоротовое углубление (перистом) и через ротовое отверстие в глот-
ку. Затем кусочек пищи с небольшим количеством воды переводит-
ся в эндоплазму, образуя пищеварительную вакуолю, т. е. у инфу-
зорий осуществляется внутриклеточное пищеварение. Остатки пи-
щи выводятся в определенном участке клетки. У инфузорий имеет-
ся, как правило, два ядра: большое (макронуклеус) и малое (мик-
ронуклеус). У некоторых содержится в клетке несколько ядер.
Сократительные вакуоли имеют каналы для сбора жидкости и ос-
новной резервуар, из которого жидкость выводится из клетки через
особое отверстие. Среди инфузорий встречаются свободноплаваю-
щие и прикрепленные формы. Из равноресничных инфузорий наи-
более часто встречаются Paramecium caudatum, Ciclidium glauco-
ma, Colpoda steini. Брюхоресничные инфузории имеют плоское тело
и цирры, определенным образом расположенные на поверхности
клетки. Ki ним относятся роды: Stylonichia, Aspidisca, Euplotes,
Oxytricha. С помощью цирр они перемещаются по твердому суб-
страту. Кругоресничные инфузории, как правило, ведут прикреп-
ленный образ жизни. Представители этого подкласса имеют харак-
терную форму: стебелек, закрепленный на твердой поверхности, за-
канчивающийся овальной клеткой, напоминающей колокольчик.
Реснички у них расположены на переднем крае тела, окружая око-
лоротовой диск (см. рис. 26). Особенностью кругоресничных инфу-
зорий является способность к сокращению тела или стебелька. Сре-
ди них встречаются одноклеточные (Vorticella) и колониальные
формы (Opercularia, Carchesium, Epistilis). В определенных усло-
виях у кругоресничных инфузорий наблюдается образование сво-
бодноплавающей стадии («бродяжки»), которая способствует их
расселению. Колонии их образуются в результате того, что при раз-
209
множении разделившиеся особи остаются соединенными с помощью
стебельков.
Особую группу составляют сосущие инфузории (Suctoria), ве-
дущие паразитический образ жизни. Вместо ресничек у них име-
ются особые, полые внутри щупальцы, с помощью которых они вы-
сасывают содержимое клеток. Питаются они другими простей-
шими.
Коловратки (Rotatoria). Они представляют обособлен-
ную группу животных организмов и являются родственными неко-
торым группам низкоорганизованных червей. Коловратки — много-
клеточные животные организмы, имеющие членистое строение пан-
циря. Размер их достигает до 2 мм. В передней части у них имеет-
ся ротовое отверстие округлой формы, окруженное многочислен-
ными ресничками. У коловраток уже имеется примитивная пище-
варительная система, состоящая из ротовой полости, глотки, пище-
вода, желудка и узкой кишки. Продукты обмена удаляются с по-
мощью выделительных органов. Мерцательный аппарат (реснички)
служит для создания направленного тока воды, в которой присутст-
вуют микроорганизмы (бактерии, простейшие), частицы взвешен-
ного органического вещества, составляющие пищу коловраток. Дви-
жение коловраток происходит с помощью коловращательного ап-
парата и «ноги», выступающей из панциря в задней части. Нога
используется также для прикрепления к субстрату. Коловратки ис-
пользуются в качестве индикаторных организмов при оценке ра-
боты очистных сооружений биологической очистки.
§ 51. Физиология микроорганизмов
Конструктивный и энергетический обмен. Физиология изучает
процессы, протекающие в живом организме, и их закономерности.
Современная материалистическая физиология основана на принци-
пе единства организма с окружающей средой. Взаимодействие ор-
ганизма со средой проявляется в обмене веществ и энергии (мета-
болизм). Он включает в себя два процесса: конструктивный обмен
(ассимиляция, или анаболизм) и энергетический (диссимиляция,
или катаболизм). В основе конструктивного обмена лежат биохи-
мические реакции, в процессе которых усваиваются вещества, по-
ступающие из окружающей среды, и идет создание биомассы клет-
ки. Сущность энергетического обмена заключается в разрушении
веществ, содержащихся в организме, преимущественно в результа-
те гидролитических и окислительных процессов, сопровождающих-
ся выделением энергии, необходимой для биосинтеза. Оба процесса
в клетке идут одновременно и сочетаются друг с другом. Энергия,
полученная клеткой в процессе обмена веществ, аккумулируется в
соединениях, содержащих химические связи, при разрыве которых
выделяется большое количество энергии (макроэргпческие). Часто
это соединения с фосфатными связями, например аденозинтрифос-
фат (АТФ). По мере надобности эти вещества подвергаются гид-
ролитическому распаду, сопровождающемуся выделением энергии.
210
Продукты жизнедеятельности микроорганизмов (метаоолиты;
могут выводиться из клетки или откладываться в ней, образуя раз-
личного рода включения. В благоприятных условиях обмен ве-
ществ протекает с большой скоростью. Микроорганизмы в течение
суток могут переработать такое количество питательных веществ,
которое в 20—30 раз превышает их собственную массу.
Ферменты. Биохимические реакции, составляющие основу обме-
на веществ и энергии, протекают в присутствии биологических ка-
тализаторов — ферментов (энзимов). Это соединения белковой при-
роды, повышающие скорость химических реакций в живом организ-
ме. Ферменты катализируют превращения веществ, поступающих
из окружающей среды и образующихся внутри организма. Биоло-
гические катализаторы, выделяемые во внешнюю среду и перево-
дящие сложные соединения в форму, способную усваиваться орга-
низмом, называются экзоферментами. Биохимические реакции,
протекающие внутри клетки, осуществляются при участии эндофер-
ментов. По химическому строению ферменты могут быть простыми
(протеины) или сложными (протеиды) белками. Активность фер-
ментов-протеинов обусловлена наличием активных (каталитиче-
ских) центров в белковой молекуле. Молекула ферментов-протеи-
дов включает активные группы, содержащие сложные органические
соединения или металлы (железо, кобальт и др.). Белковая часть
молекулы фермента называется фероном (носитель), а добавоч-
ная — агоном (активная группа). Отдельно ферон и агон не прояв-
ляют каталитической активности. Если агон может существовать
самостоятельно, то он называется коферментом. Коферменты обыч-
но являются промежуточными переносчиками электронов или неко-
торых атомов и атомных групп (водорода, аминогрупп и др.) от
одних соединений к другим. В состав коферментов часто входят
витамины. Так, коферменты НАД (никотинамидадениндинуклео-
тид) и НАДФ (никотинамидадениндинуклеотиддифосфат), участ-
вующие в переносе водорода, содержат витамин В5 (никотинамид).
Механизм действия ферментов связан со снижением энергии ак-
тивации взаимодействующих молекул в результате образования
ферментно-субстратного комплекса. Последовательность процес-
сов, протекающих при ферментативном катализе, можно записать
в виде схемы фермент + субстрат ферментно-субстратный комп-
лекс продукт реакции 4- фермент. Для ферментов характерно
значительное снижение энергии активации по сравнению с обыч-
ными катализаторами. Так, для разложения перекиси водорода на
кислород и воду требуется энергия активации 75,2 кДж/моль.
В присутствии катализатора (коллоидной платины) она снижается
до 50,2 кДж/моль, а фермент каталаза ее уменьшает до
8,3 кДж/моль. Действие ферментов строго специфично, т. е. они дей-
ствуют только на определенный субстрат. Активность их зависит от
pH, температуры, присутствия ряда химических соединений.
Активацию ферментов вызывают ионы Мп2+, Zn2+, Са2+, Mg2+
и некоторые органические вещества, например аскорбиновая кис-
лота. Ингибиторы тормозят их действие.
211
Ь зависимости от типа химических реакций, подвергающихся
каталитическому воздействию, выделяют следующие классы фер-
ментов: 1) гидролазы, ускоряющие процессы разложения или син-
теза, проходящие с участием воды. Так, липаза ускоряет гидролиз
жиров, сопровождающийся образованием глицерина и высших
жирных кислот; 2) оксиредуктазы катализируют окислительно-вос-
становительные реакции. Ферменты, участвующие в переносе ато-
мов водорода от одних соединений к другим, называются дегидро-
геназами; 3) трансферазы ускоряют реакции передачи атомных
групп, радикалов от одних соединений к другим. Они играют важ-
ную роль при освобождении энергии фосфатных связей; 4) лиазы
катализируют процессы распада, а иногда и синтеза органических
соединений. При реакциях разложения с их участием часто выде-
ляются простые соединения (СО2, МНз, Н2О). К этому классу, в
частности, относятся ферменты, вызывающие разложение аминокис-
лот с образованием аммиака (дезаминирование); 5) лигазы (син-
тетазы) определяют возможность синтеза сложных соединений из
простых. Ферменты этого класса играют важную роль в процессах
биосинтеза белков. Они катализируют образование связей: С — С,
С — N, С — О; 6) изомеразы способствуют осуществлению внутри-
молекулярных превращений. Некоторые из них ускоряют процессы
синтеза и распада углеводов.
Химический состав клеток микроорганизмов. Соединением, опре-
деляющим жизнеспособность клетки, является вода. Количество
воды составляет 75—90% от общей массы клетки. В ней растворе-
ны многие, необходимые для организма соединения, а биохимиче-
ские реакции протекают в водных растворах. В спорах вода нахо-
дится в связанном состоянии и содержание ее снижается до 40%.
С помощью воды осуществляется транспорт питательных веществ
в клетку и выведение из нее продуктов обмена. Она играет опре-
деленную роль и в теплорегуляции. Так, перестройка структуры во-
ды, а именно экзотермический процесс образования водородных
связей, позволяет в определенной мере снизить отрицательное воз-
действие низких температур.
К числу наиболее важных химических элементов, составляющих
основу органического вещества клеток, относятся углерод, азот, во-
дород, кислород, фосфор, сера. Органическое вещество бактерий
представлено белками, углеводами, жирами и другими группами
органических соединений. Белки — наиболее важная составная
часть живого организма. С ними связано протекание основных фи-
зиологических процессов. Белки являются пластическим материа-
лом, из которого построены клетки, могут использоваться в качест-
ве энергетического материала, особенно при неблагоприятных ус-
ловиях, входят в состав ферментов. В клетках микроорганизмов
содержится большое количество белков, отличающихся по химиче-
скому составу и строению. Они обусловливают специфичность мик-
роорганизмов и их изменчивость под воздействием окружающей
среды. В молодых клетках содержится большее количество белко-
вых соединений. Особую роль в синтезе белков выполняют нуклеи-
212
новые кислоты - рибонуклеиновая (РНК) и дезоксирибонуклеино-
вая (ДНК), обусловливающие передачу наследственных свойств
организма. Содержание белковых соединении зависит от условий
внешне!! среды и при достаточном количестве питательных ве-
ществ часть белков цитоплазмы откладывается в запас.
Углеводы являются постоянными компонентами, содержащими-
ся в клетках микроорганизмов. Качественный состав их специфи-
чен для каждого вида. В клетках бактерии количество углеводов
в зависимости от условий изменяется от 10 до 30%. Углеводы (цел-
люлоза) могут входить в состав оболочек, капсул, слизистых об-
разований. Они иногда выполняют роль запасного материала, об-
разуя включения, например гликоген. Углеводы — один из основ-
ных источников энергии.
Жиры служат запасным веществом, и количество их зависит от
наличия питательных веществ в окружающей среде. У некоторых
бактерий они выполняют защитную функцию, входя в состав кап-
сулы, не проницаемой для растворов, содержащих токсичные для
бактерий соединения. Наряду с углеводами они выполняют роль
энергетического материала.
Жироподобные соединения — липоиды, входят в состав цито-
плазмы, образуя с белком цитоплазмы липоидо-протеиновый комп-
лекс. В наружном слое цитоплазмы они образуют мембрану, кото-
рая служит для регулирования поступления соединений в клетку
из внешней среды. Липоиды и жиры образуют группу липидов. От
жиров липоиды отличаются наличием в их молекулах азота и фос-
фора.
Состав минеральных соединений, содержащихся в клетках мик-
роорганизмов, непостоянен и зависит от свойств и состава пита-
тельных веществ. Неорганические соединения составляют примерно
1/300 от массы клетки. Основными элементами являются сера, же-
лезо, хлор, кальций, натрий, калий, магний. Эти элементы участву-
ют в важнейших реакциях обмена веществ. Калий, например, иг-
рает существенную роль в углеводном обмене и синтезе органиче-
ских веществ клетки. Магний активирует деятельность некоторых
ферментов.
В состав бактериальной клетки входят также микроэлементы,
играющие чрезвычайно важную роль в регулировании обмена ве-
ществ. Это прежде всего литий, марганец, иод, кобальт, медь, цинк
и др. Они входят в состав органических соединений клетки или со-
держатся в виде растворов. Многие из этих элементов содержатся
в ферментах. Химический состав бактерий претерпевает изменения
в процессе их жизнедеятельности.
Питание микроорганизмов. Питательные вещества необходимы
живым организмам для построения структурных элементов клетки
и как источник энергии. Микроорганизмы, за исключением простей-
ших и коловраток, получают питательные вещества в виде молекул
простых веществ при всасывании их через цитоплазматическую
мембрану из окружающего раствора, так как полимерные молеку-
лы белков, полисахаридов не могут непосредственно усваиваться
213
клеткой. Под действием экзоферментов, выделяемых микроорганиз-
мами, происходит расщепление сложных соединений до простых
сахаров, аминокислот и других веществ, которые затем поступают
в клетку (внеклеточное пищеварение). Простейшие и коловратки
заглатывают пли поглощают твердые частицы пищи, которые за-
тем перевариваются внутри клетки (голозойный тин питания). Не-
которые простейшие (из жгутиковых) могут также получать пита-
тельные вещества из водных растворов всасыванием через пелли-
кулу (сапрозойный тип питания).
Транспортировка растворенных веществ через цитоплазматиче-
скую мембрану в клетку может происходить в результате разности
концентраций растворов или электрических потенциалов (для ио-
нов) по ту и другую сторону мембраны или же, в большинстве слу-
чаев, под действием особых соединений (пермеаз), содержащихся
в цитоплазматической мембране. Так, в каждой клетке кишечной
палочки содержится около 8000 молекул пермеазы, транспортирую-
щей молекулы молочного сахара — лактозы. Образующиеся в про-
цессе жизнедеятельнЪсти продукты обмена также через оболочку
выводятся в окружающую среду.
Нормальное развитие микроорганизмов наблюдается лишь тог-
да, когда в водном растворе присутствуют все элементы, необхо-
димые для построения живого вещества и осуществления жизне-
деятельности клетки. Микроорганизмы проявляют избирательность
действия по отношению к источникам питания, т. е. отдельные ви-
ды могут использовать только определенные соединения. Источни-
ками азота для биосинтеза белков микроорганизмами могут быть
аммонийные соединения, свободный азот, аминокислоты и др. Био-
генные элементы (фосфор, железо и др.) поступают обычно в виде
солей. В зависимости от природы соединений, являющихся источ-
ником углерода, выделяют следующие группы микроорганизмов:
1) автотрофы, использующие для синтеза сложных органических
соединений углекислый газ или карбонаты; 2) гетеротрофы, усваи-
вающие углерод органических соединений. Микроорганизмы, пи-
тающиеся мертвым органическим веществом, называются сапрофи-
тами. Гетеротрофы, приспособившиеся к усвоению органических
соединений, находящихся в живом организме, называются парази-
тами (паратрофами). К ним относятся вирусы, бактериофаги, бо-
лезнетворные микроорганизмы.
Микроорганизмы, осуществляющие перевод световой энергии в
энергию химических связей, называются фототрофами. Другая
группа микроорганизмов (хемотрофы) использует для биосинтеза
и поддержания жизнедеятельности клетки энергию, выделяющую-
ся при химических превращениях.
С учетом химической природы соединений — поставщиков угле-
рода — все микроорганизмы по способу получения энергии делятся
на следующие группы:
I. Фототрофы: 1) фотолитотрофы, использующие солнечную
энергию для построения живого вещества из неорганических сое-
динений (углекислого газа, воды, сероводорода). Это организмы —
214
фотосинтетики (растения, зеленые водоросли, пурпурные серобак-
терии и др.); 2) фотоорганотрофы, которые наряду с процессом
фотосинтеза способны использовать энергию химических превраще-
ний органических веществ (несерные пурпурные бактерии).
II. Хемотрофы: 1) хемолитотрофы используют энергию окисле-
ния неорганических соединений (водорода, аммиака, нитритов, же-
леза (II) и др.). К ним относятся железобактерии, нитрифицирую-
щие бактерии, бесцветные серобактерии и др. Процесс биосинтеза,
осуществляющийся за счет энергии химических превращений неор-
ганических соединений, называется хемосинтезом. О большой роли
хемолитотрофов можно судить по данным Н. А. Березиной. В при-
донных слоях Рыбинского водохранилища около 50% от общего
суточного прироста бактерий составляют хемолитотрофы. Очень
велико их значение зимой, когда в водоемах они становятся прак-
тически единственным источником питания для гетеротрофов;
2) хемоорганотрофы используют энергию химических превращений
сложных органических соединений. В эту группу входят микроорга-
низмы, осуществляющие разложение органических соединений.
При изменении внешних условий многие микроорганизмы могут
переходить на другой тип питания. Такие формы называются мик-
сотрофными. Кроме питательных веществ для нормальной жизне-
деятельности бактерий необходимо наличие особых факторов ро-
ста — витаминов. Наиболее важными из них являются витамины:
рибофлавин (В2), пиридоксин (В6), никотиновая (РР), фолиевая и
пантотеновая кислоты. Вещества —факторы роста, необходимые
для одного вида, могут образовываться в процессе жизнедеятельно-
сти другим видом микроорганизмов.
Аккумулирование энергии в клетках микроорганизмов. Обмен
веществ и энергии осуществляется в результате многих фермента-
тивных реакций, сопровождающихся выделением или поглощением
энергии. Микроорганизмы обладают способностью аккумулировать
энергию в определенных макроэргических соединениях, содержа-
щих химические связи, при разрыве которых выделяется большое
количество энергии. Одним из таких аккумуляторов энергии яв-
ляется аденозинтрифосфат (АТФ), который синтезируется из аде-
нозиндифосфата (АДФ) путем присоединения остатка фосфорной
кислоты. Синтез АТФ осуществляется за счет энергии, выделяю-
щейся при протекании ряда окислительно-восстановительных реак-
ций. Если окисление органических веществ идет при участии кис-
лорода, то процесс образования АТФ, сопряженный с ним, назы-
вается окислительным фосфорилированием. Процесс перехода АДФ
в АТФ обратим, и энергия, необходимая для обеспечения биосинте-
за, выделяется при отщеплении от молекулы АТФ фосфорной кис-
лоты. Взаимосвязь между реакциями синтеза и разложения АТФ
можно показать схематически следующим образом:
поглощение энергии
АДФ + Н„РО4 . -----------АТФ
выделение энергии
Энергия, выделяющаяся при расщеплении АТФ, расходуется также
215
на теплообразование, обеспечение активного транспорта питатель-
ных веществ через цитоплазматическую мембрану и выполнение
других функций.
У фотосинтезирующих микроорганизмов активация неорганиче-
ского фосфата и синтез АТФ происходит за счет световой энергии
(фотосинтетическое фосфорилирование). Хемолитотрофы использу-
ют энергию, выделяющуюся в окислительно-восстановительных ре-
акциях неорганических соединений (хемосинтетическое фосфорили-
рование). Таким образом обмен веществ неотделим от обмена энер-
гии: при конструктивном обмене она потребляется, а энергетиче-
ский обмен обусловливает накопление энергии.
Биологическое окисление. Типы дыхания микроорганизмов. Бро-
жение. В настоящее время понятие «биологическое окисление»
включает в себя совокупность окислительно-восстановительных ре-
акций, протекающих с участием ферментов. Эти процессы являют-
ся основным источником энергии в организме. В зависимости от на-
правленности этих реакций выделяют и различные типы дыхания.
При биологическом окислении идут окислительно-восстанови-
тельные реакции, сопровождающиеся отнятием атомов водорода от
одних соединений (доноров) и передачей его другим (акцепторам),
или реакции, связанные с переносом электронов от донора к акцеп-
тору. Эти процессы осуществляются при участии ферментов, отно-
сящихся к классу оксиредуктаз. Процессы дыхания, в которых ак-
цептором водорода или электронов является молекулярный кисло-
род, называются аэробными. Если же акцепторами будут другие
неорганические или органические соединения, то такой тип дыхания
называется анаэробным. По типу дыхания выделяют две группы
микроорганизмов: аэробы (оксибиотические формы), которым для
дыхания необходим кислород, и анаэробы (аноксибиотичсскис фор-
мы), развивающиеся в отсутствие кислорода. Между ними нет рез-
кого различия. Наряду со строгими (облигатными) аэробами и ана-
эробами есть микроорганизмы, которые могут жить в присутствии
кислорода и без него. Это микроаэрофплы, оптимум содержания
кислорода в воздухе для которых составляет 0,5—1%, и факульта-
тивные анаэробы. Так, кишечная палочка является факультатив-
ным анаэробом.
Микроорганизмы с аэробным типом дыхания встречаются как
в группе литотрофов (бактерии-нитрифпкаторы, окисляющие аммо-
нийный азот до нитритов, а затем до нитратов, железо-, серобакте-
рии), так и среди органотрофов (бактерии, окисляющие углеводы,
органические кислоты и другие органические соединения; водорос-
ли, грибы, простейшие, коловратки). При аэробном дыхании обра-
зуются конечные продукты распада — диоксид углерода и вода
(полное окисление). Если разложение органических веществ в при-
сутствии молекулярного кислорода нс идет до конца, процесс назы-
вается неполным окислением. При анаэробном дыхании разложе-
ние органического вещества часто задерживается на одной из про-
межуточных стадий. Если в окружающем растворе не содержится
питательных веществ, необходимых для получения энергии, то мик-
216
роорганизмы окисляют запасные вещества, имеющиеся в клетке
(эндогенное дыхание).
Брожением называется такой процесс, при котором фермента-
тивная окислительно-восстановительная реакция проходит в ана-
эробных условиях при участии одного соединения, а акцептором
водорода является один из продуктов реакции. Чаще всего броже-
ние наблюдается при разложении углеводов.
Брожение идет в две стадии: сначала происходит разрыв угле-
родной цепи и отнятие от молекулы окисляемого соединения (суб-
страта) атомов водорода, а затем — передача атомов водорода вос-
станавливаемому органическому соединению (акцептору), образо-
вавшемуся в ходе разложения субстрата. Эти стадии включают в
себя ряд реакций, протекающих с участием ферментов. Так, при
разложении углеводов (глюкозы, фруктозы и др.) в результате
сложных ферментативных процессов на первой стадии образуется
О
//
пировиноградная кислота: C6Hi2O6-> 2Н2 + СН3—С — СООН, из
которой на второй стадии образуются различные продукты, являю-
щиеся акцепторами водорода (масляная кислота, этиловый спирт,
ацетон и др.). Иногда она сама восстанавливается водородом в мо-
лочную кислоту (молочнокислое брожение):
СН3 — СО — СООН + 2Н2 -> 2СН3 — СН (ОН) — СООН -f- 200 кДж
Атомы водорода не передаются непосредственно от окисляемого
вещества к акцептору, а вначале они восстанавливают коферменты
анаэробных дегидрогеназ — НАД или НАДФ, образуя НАД-Н2 или
НАДФ-Н2. На второй стадии восстановленные формы этих кофер-
ментов передают, окисляясь, водород акцептору. Схематически этот
процесс можно показать следующим образом:
1-я стадия
субстрат-Н2 + НАД-> НАД-Н2 + продукт окисления (1)
2-я стадия
НАД • Н2 + акцептор -> Н2 • акцептор ,+ НАД (2)
Аэробное дыхание является сложным процессом, который вклю-
чает в себя ряд ферментативных окислительно-восстановительных
реакций, заканчивающихся передачей водорода кислороду. Пути
передачи водорода акцептоцу могут быть различными. У многих
микроорганизмов отщепление водорода от окисляемого субстрата
осуществляется первичными анаэробными дегидрогеназами (напри-
мер, НАД) по тому же механизму, что и при брожении. Затем они
передают его аэробным дегидрогеназам, в частности коферменту
ФАД (флавинадениндинуклеотид). Аэробное дыхание включает в
себя две фазы: 1) цикл реакций, в которых субстрат окисляется до
углекислого газа, а атомы водорода передаются восстанавливае-
мым соединениям; 2) передача водорода кислороду. Первая фаза
заключается в разложении пировиноградной кислоты через ряд
сложных ферментативных реакций (цикл Кребса). Суммарно для
8 1214
217
пировиноградной кислоты этот процесс можно описать уравнением
СНз — СО — СООН + 2 Н2О -> 2 СО2 + 8 Н. Во второй фазе под
действием ферментов — оксидаз водород передается кислороду. Ча-
ще всего это цитохромы — ферменты, содержащие железо. Они пе-
редают не атомы водорода, а электроны. Принимая электрон от
атома водорода, цитохромы восстанавливаются (Fe3+ переходит в
Fe2+). Последний фермент в дыхательной цепи (цитохромоксидаза)
активирует кислород и катализирует процесс образования воды
(рис. 27). Эти процессы сопряжены с синтезом АТФ.
НАД
Субстрат
+•
и
Цитохром-
оксидаза
Рис. 27. Схема окислительно-восстановительной цепи при аэробном
дыхании
При аэробном дыхании выделяется значительно больше энер-
гии, чем при анаэробном. Так, если при полном окислении молеку-
лы глюкозы образуется 38 молекул АТФ, то при брожении ее —
всего 2. Поэтому анаэробам приходится перерабатывать значитель-
но большее количество органического вещества, чем аэробам, для
получения одинакового количества энергии.
Некоторые анаэробные микроорганизмы в качестве акцептора
используют связанный кислород, входящий в состав таких соеди-
нений, как сульфаты или нитраты. Этот тип дыхания отмечается у
некоторых факультативных анаэробов, например, у бактерий — де-
нитрификаторов Pseudomonas flourescens, Paracoccus denitrificans.
В присутствии кислорода они имеют аэробное дыхание, а в бескис-
лородных средах используют в качестве акцептора кислород нит-
ратов, восстанавливая их до азота или его низших оксидов. Бакте-
рии, восстанавливающие в процессе дыхания сульфаты до серово-
дорода, являются облигатными анаэробами, например Desulfovib-
rio desulfuricans.
Закономерности роста культуры микроорганизмов. Рост и раз-
витие культуры микроорганизмов одного вида в закрытой системе,
содержащей ограниченное количество питательных веществ, будет
лимитироваться их концентрацией, а также накоплением продуктов
обмена в окружающей среде. Закономерности развития культуры
микроорганизмов одного вида описываются кривой активного роста
(рис. 28). При введении микроорганизмов в среду, содержащую
питательные вещества, вначале происходит задержка роста (lag-
фаза). Это связано с тем, что микроорганизмам необходимо опре-
деленное время для адаптации к изменившимся условиям среды.
Наличие лаг-фазы связано с изменением питания, внешних условий
218
(например, температуры, pH), наличия ингибиторов ферментатив-
ных процессов, переходом споровых форм в вегетативные и др. Зна-
ние причин, обусловливающих наличие фазы задержки роста, по-
зволяет уменьшить время, необходимое для адаптации микроорга-
низмов к данной среде. Длительность ее может быть использова-
на для характеристики влияния отдельных факторов внешней сре-
ды на жизнедеятельность микроорга-
низмов. В этой фазе прирост биомассы
идет за счет увеличения клеток мик-
роорганизмов. После адаптации начи-
нается ускоренный рост культуры за
счет возрастания скорости деления
клеток, достигающий через определен-
ное время максимума. При этом ско-
рость роста пропорциональна количе-
ству биомассы в степени (экспоненте),
Рис. 28. Фазы роста микроби-
альной культуры:
/ — лаг-фаза; II— фаза ускоренно-
го роста; III — фаза логарифмиче-
ского роста; IV— фаза замедлен-
ного роста; V— стационарная фа-
за; VI — фаза отмирания
которая является функцией времени.
В это время наблюдаются наиболее
оптимальные условия для развития
микроорганизмов, заключающиеся в
достаточном количестве питательных
веществ и при небольшом содержании
продуктов обмена в окружающей сре-
де. В логарифмической фазе скорость роста не изменяется — это
период непрерывного роста культуры. По мере расходования пита-
тельных веществ и накопления метаболитов скорость размножения
микроорганизмов постепенно снижается и фаза замедленного рос-
та переходит в стационарную, в которой количество вновь обра-
зующихся клеток примерно соответствует количеству отмирающих.
Дефицит питательных веществ и повышающаяся концентрация
продуктов обмена в окружающей среде способствуют отмиранию
клеток микроорганизмов (фаза отмирания).
Однако рост культуры нельзя рассматривать как механический
прирост или уменьшение клеток. Фазы развития культуры сопро-
вождаются физиологическими изменениями в клетках микроорга-
низмов. Молодые клетки, имеющие высокую интенсивность метабо-
лизма, постепенно стареют, что способствует снижению интенсив-
ности обменных процессов.
§ 52. Влияние факторов внешней среды
на жизнедеятельность микроорганизмов
Возможность и напряженность протекания жизненных процес-
сов у микроорганизмов определяется направленностью действия
совокупности факторов внешней среды. Они могут оказывать поло-
жительное воздействие, интенсифицируя жизнедеятельность микро-
организмов, или быть неблагоприятными для их роста и развития.
С учетом многообразия и особенностей воздействия их на жиз-
недеятельность микроорганизмов выделяют следующие группы фак-
8* 219
торов внешней среды: физические, химические и биологические.
К физическим факторам относятся следующие:
Влажность. Жизненные процессы микроорганизмов осу-
ществляются в водных растворах. В растворенном состоянии по-
ступают в клетку питательные вещества. Поэтому недостаток вла-
ги отрицательно сказывается на жизнедеятельности микроорганиз-
мов. В процессе эволюции определились группы микроорганизмов,
более или менее устойчивые к высушиванию. Наименее устойчивы
к высушиванию молодые клетки,
сохранять жизнеспособность при
кания биохимических процессов
Таблица 16. Классификация
микроорганизмов по их отношению
к температуре
споры же могут длительное время
недостатке влаги. Скорость проте-
резко снижается при понижении
влажности почвы. Процессы
аммонификации и нитрифи-
кации наиболее интенсивно
Г руины Температура, °C
минимум опти- мум макси- мум
Психрофилы Мезофилы Термофилы +12 1$ Ф 1 1 IQ О О 15-18 20—25 35-37 55—75 19-22 35—45 60—89
развитие микроорганизмов возможно
протекают при влажности,
равной 60% от общей влаго-
емкости почвы.
Температура. Мик-
роорганизмы не обладают
способностью к терморегу-
ляции и поэтому температу-
ра содержимого клетки со-
ответствует температуре
внешней среды. Нормальное
лишь в определенном интер-
вале температур, в пределах которого отмечается минимум, опти-
мум и максимум активности жизненных процессов. В зависимости
от того, при каких значениях температуры возможна жизнедея-
тельность микроорганизмов, выделяют следующие группы: 1) пси-
хрофилы, 2) мезофилы, 3) термофилы.
В табл. 16 приведены данные об интервалах температур, при
которых осуществляется жизнедеятельность микроорганизмов этих
групп.
Среди психрофилов встречаются некоторые виды бактерий, си-
незеленых водорослей, актиномицетов. У мезофилов две группы
микроорганизмов различаются по значению оптимума температур-
ного фактора. К первой группе относятся микроорганизмы, имею-
щие температурный оптимум при 20—25° С. Это—-микроорганиз-
мы, обитающие в воде, почве. Вторая группа с температурным оп-
тимумом 35—37° С представлена микроорганизмами, приспособив-
шимися к жизни в организме человека или теплокровных живот-
ных. Термофилы — микроорганизмы, развивающиеся при повышен-
ной температуре.
В природных условиях отдельные виды микроорганизмов, оби-
тающих в водной среде, могут встречаться при температурах и
—7,5° С в соляных озерах, и +99° С в термальных источниках. Диа-
томовые водоросли в большом количестве развиваются в антаркти-
ческих льдах. Наиболее чувствительны к изменению температуры
патогенные микроорганизмы. При понижении температуры наблю-
220
дается задержка развития микроорганизмов и резкое понижение
обмена веществ, но микроорганизмы сохраняют жизнедеятельность.
Некоторые бактерии выдерживают понижение температуры до
—190° С. Значительно хуже микроорганизмами переносится повы-
шение температуры. Отмирание многих вегетативных форм бакте-
рий при повышении температуры до +60° С происходит в течение
30 мин, при +70° С - через 10—15 мин, а при 100° С они гибнут в
течение нескольких секунд. Споры обладают большей устойчи-
востью к повышению температуры, они выдерживают кипячение в
течение часа и более.
Воздействие высоких температур на жизнедеятельность бакте-
рий используется для борьбы с ними. Примерами могут служить
приемы обработки материалов — пастеризация и стерилизация. Па-
стеризация заключается в нагревании продукта при 70° С в течение
30 мин. Для уничтожения всех микроорганизмов и спор применяет-
ся стерилизация, которая заключается в нагревании обеззаражи-
ваемого материала при 120° С в течение 30 мин.
Действие излучений. Микроорганизмы в процессе жиз-
недеятельности могут быть подвержены воздействию различных ви-
дов излучений. Влияние излучений на микроорганизмы зависит от
длины волны. Инфракрасное излучение оказывает тепловое воздей-
ствие на микроорганизмы. Важное экологическое значение имеет
свет для фотосинтезирующих микроорганизмов, которые не могут
развиваться в его отсутствие. Излучения определенной длины вол-
ны видимой части спектра необходимы для жизнедеятельности пиг-
ментообразующих бактерий. Изменение интенсивности освещенно-
сти сопровождается перемещением микроорганизмов, находящихся
в воде во взвешенном состоянии (фотодинамический эффект). Све-
толюбивые (фотопозитивные) виды перемещаются в верхние слои.
Фотонегативные формы характерны для донных отложений. Ульт-
рафиолетовое излучение с длиной волны от 200 до 300 нм обладает
наиболее сильно выраженным бактерицидным действием. На пря-
мом солнечном свету отмирание микробов происходит в тонком
слое воды через 20—30 мин.
Ионизирующее излучение в зависимости от его интенсивности
оказывает определенное влияние на развитие микроорганизмов. Ма-
лые дозы облучения способствуют некоторой стимуляции жизне-
деятельности микроорганизмов. Достаточно высокие дозы прекра-
щают их размножение. Большие дозы облучения вызывают отмира-
ние микроорганизмов. В присутствии кислорода эффект облучения
усиливается, что приводит к гибели большого числа микроорганиз-
мов. Этот факт можно объяснить образованием свободных радика-
лов, обладающих высокой реакционной способностью. Иницииро-
ванные ими реакции ведут к нарушению биохимических процессов
обмена веществ.
Действие электрического тока. При пропускании
слабого постоянного тока происходит электрофоретическое переме-
щение бактерий к определенному полюсу вследствие наличия у бак-
териальных клеток равномерно распределенного по поверхности за-
221
ряда (преимущественно отрицательного в нейтральных средах).
При изменении pH величина заряда изменяется, а в кислых средах
после перехода через изоэлектрическую точку бактериальные клет-
ки приобретают положительный заряд. Вследствие поляризации
при наложении электрического поля наблюдается обратимая или
необратимая коагуляция бактериальных клеток. Мощным бактери-
цидным и спороцидным фактором является электроискровой раз-
ряд и сочетание действия поля и разряда (комплекс электрических
воздействий).
Давление. Повышение давления существенно не влияет на
жизнедеятельность микроорганизмов. Они способны выдерживать
давление до 3-102 МПа и более. Вирусы и бактериофаги отмирают
при давлении порядка 6,5-102 МПа, неспорообразующие бакте-
рии— при давлении 4—5-Ю2 МПа. Споры выдерживают давление
до 2-103 МПа.
Влияние механических воздействий. Сильное дли-
тельное встряхивание вызывает отмирание бактерий, а слабое ме-
ханическое воздействие даже несколько стимулирует развитие мик-
роорганизмов. Перемешивание воды при обеззараживании ее хло-
ром повышает его бактерицидное действие. Микроорганизмы, жи-
вущие в проточной воде, обладают меньшей чувствительностью к
механическим воздействиям. Перемешивание воды способствует
процессу самоочищения водоемов, при котором происходит отмира-
ние некоторых микроорганизмов, не являющихся постоянными оби-
тателями водоема.
К химическим факторам относятся следующие:
Концентрация растворенных веществ. В процессе
эволюции микроорганизмы приспособились к различным концентра-
циям растворенных веществ в окружающей среде. Одно и то же
вещество может по-разному влиять на жизнедеятельность микро-
организмов. В малых концентрациях оно может быть стимулятором
развития микроорганизмов, а при повышении концентрации задер-
живать их рост и даже вызывать отмирание. Так, 2%-ный раствор
хлорида натрия .активизирует жизнедеятельность микроорганизмов,
а при повышении концентрации до 5—10% вызывает их отмирание.
В то же время существует ряд бактерий, которые приспособились
к жизни в водах с концентрацией хлорида натрия до 25%. Такие
микроорганизмы называются галофильными. Осмофильные дрож-
жи могут жить в насыщенном растворе сахара. Устойчивы к высо-
кой концентрации солей и некоторые виды плесневых грибов. По-
стоянство определенного химического состава клеток микроорганиз-
мов при изменении химического состава окружающей среды, напри-
мер при спуске сточных вод, обусловлено наличием в клетках меха-
низмов регуляции солевого состава. Наличие таких саморегулирую-
щихся систем обеспечивает сохранение жизнедеятельности микро-
организмов в водах с различным химическим составом.
Величина pH. Водородный показатель pH определяет не
только условия существования микроорганизмов, но и направлен-
ность биохимических процессов. Для каждого вида бактерий суще-
222
ствует оптимальная величина pH, при которой проявляется его наи-
большая биологическая активность. Влияние pH на водные микро-
организмы может быть прямым и косвенным. Прямое воздействие
заключается в изменении проницаемости клеточной оболочки. При
отклонении значения pH от оптимального происходит изменение в
обмене веществ. Влияет pH и на водно-солевой обмен микроорга-
низмов. Косвенное влияние заключается в изменении формы содер-
жания растворенных в воде веществ в зависимости от pH. Так, в
щелочной среде подавляется и даже прекращается жизнедеятель-
ность многих водорослей.из-за уменьшения растворимости соедине-
ний железа. Отклонение pH от оптимального значения ведет к по-
тере устойчивости коллоидного состояния белков цитоплазмы.
Степень воздействия влияния pH на водные микроорганизмы
зависит от их видовых особенностей. Так, один из видов коловраток
выдерживает изменение pH на 5—6 единиц. Большинство же мик-
роорганизмов более чувствительно к изменению pH. Оптимальные
значения этого фактора для них находятся в пределах от 6 до 9_
Некоторые микроорганизмы приспособились к жизни в кислых во-
дах, выдерживая понижение pH до 3,5.
В результате жизнедеятельности водных организмов в период
их интенсивного развития pH воды водоема может заметно изме-
ниться. В летний период суточные колебания pH воды под влияни-
ем фотосинтетической деятельности водных организмов могут до-
стигать 2 единиц. В дневное время происходит повышение щелоч-
ности воды из-за потребления диоксида углерода в процессе фото-
синтеза, а ночью pH воды снижается в результате насыщения.во-
ды диоксидом углерода.
Микроорганизмы обладают способностью к некоторому регули-
рованию pH. Если концентрация ионов Н+ в окружающем растворе
выше оптимальной, то они выделяют вещества, обладающие щелоч-
ными свойствами. Наоборот, повышенная щелочность окружающе-
го раствора нейтрализуется веществами, обладающими кислотными:
свойствами. Примером подобного воздействия микроорганизмов яв-
ляется значительное подкисление внешней среды в результате жиз-
недеятельности молочнокислых бактерий.
Окислительно-восстановительный потенциа л..
На нормальное развитие микроорганизмов существенно влияет на-
правление окислительно-восстановительных процессов, протекаю-
щих в окружающем растворе. Одни из них проявляют наибольшую
активность в среде, характеризующейся процессами окисления
(аэробы), другие—при преобладании процессов восстановления,
(анаэробы).
Окислительно-восстановительный потенциал Eh неочищенных
сточных вод обычно находится в пределах от 0 до —400 мВ. Чис-
тые воды пресных водоемов, содержащие достаточное количество
растворенного кислорода, имеют Е1г = 300 — 350 мВ. При положи-
тельных значениях Ел (и высоких значениях гН2) развиваются
аэробные микроорганизмы. Максимальная величина гН2, при кото-
рой возможна жизнедеятельность облигатных анаэробов, составля-
223
ег около iz. икислительно-восстановительные условия изменяются
в процессе очистки сточных вод. Восстановительные условия сточ-
ных вод способствуют развитию анаэробных форм микроорганиз-
мов. По мере того как идет разложение органических примесей, ве-
личина гН2 возрастает, в воде создаются окислительные условия,
появляется достаточное количество кислорода, что приводит к сме-
не групп микроорганизмов — замене анаэробных форм микроорга-
низмов на аэробные.
На величину окислительно-восстановительного потенциала влия-
ет присутствие неорганических соединений, способных к окислению
(железа (II), нитритов, сульфидов и др.), и pH. По наличию опре-
деленных групп микроорганизмов в воде можно сделать вывод о
характере протекающих процессов. И, наоборот, зная окислитель-
но-восстановительные условия, можно предвидеть присутствие в во-
де определенных групп микроорганизмов.
Влияние ядовитых химических соединений на
развитие микроорганизмов. Микроорганизмы очень чув-
ствительны к химическим соединениям, как к неорганическим, так
и органическим, не являющимся постоянными компонентами среды
обитания. Соли тяжелых металлов (ртути, меди, свинца, серебра
и др.) обладают сильным бактерицидным действием. Концентрация
их, достаточная для отмирания микроорганизмов, исчисляется мил-
лиграммами или даже десятыми долями миллиграмма в 1 л воды.
Вследствие малой проницаемости оболочки споры обладают мень-
шей чувствительностью по отношению к солям тяжелых металлов
и других ядовитых веществ.
Бактерицидное действие хлора, хлорной извести, озона, пер-
манганата калия и других окислителей основано на протекании
процессов окисления соединений, входящих в состав цитоплазмы
клетки. Сильным токсичным действием по отношению к микроор-
ганизмам обладают также иод, мышьяковистые и цианистые соеди-
нения. Бактерицидным действием обладают такие органические
соединения, как фенолы, низшие спирты, эфиры, формальдегид,
нафтеновые кислоты. Характер воздействия этих веществ на живое
вещество клетки заключается в их адсорбции на коллоидных час-
тицах цитоплазмы, вследствие чего нарушается нормальное про-
текание процессов обмена веществ.
Токсичность химических веществ зависит от физико-химических
свойств среды. Так, воздействие их на микроорганизмы будет раз-
личным по силе в чистой воде и в воде, загрязненной органическими
соединениями. Очень малые дозы солей марганца, кобальта, меди
и других могут стимулировать жизнедеятельность микроорганиз-
мов. Повышенная концентрация этих же соединений вызывает за-
держку или прекращение'размножения, а при дальнейшем повы-
шении концентрации происходит отмирание микроорганизмов.
Пестициды, ПАВ, многие органические соединения, содержащиеся
в сточных водах предприятий органического синтеза, также явля-
ются токсичными для микроорганизмов.
224
Биологические факторы. Различные группы организмов, объ-
единенные единой средой обитания, взаимодействуют между собой
определенным образом, образуя естественные сообщества организ-
мов— биоценозы. Развитие биоценоза направлено на повышение
стабильности системы, создание условий, при которых действие,
внешних воздействий окружающей среды будет сведено к миниму-
му. Под воздействием взаимоотношений, складывающихся в био-
ценозе, происходит определенное изменение внешней среды в сто-
рону наибольшего соответствия для жизнедеятельности организмов
этой экологической системы. В биохимическом отношении водоем
представляет собой единую экологическую систему со сложными
типами взаимоотношений между различными группами водных ор-
ганизмов (гидробионтов). Важная роль принадлежит в биоцено-
зах водоемов микроорганизмам.
Взаимные связи между различными членами биоценоза могут
быть прямыми и косвенными. Прямые связи предусматривают не-
посредственный контакт взаимодействующих организмов. Косвен-
ное влияние одних микроорганизмов биоценоза на жизнедеятель-
ность других осуществляется путем изменения физико-химических
свойств внешней среды под действием продуктов их жизнедеятель-
ности (метаболитов). В водных биоценозах косвенные связи меж-
ду организмами, проявляющиеся через выделение в воду метабо-
литов, играют важную роль. Взаимодействия между различными
группами микроорганизмов и другими живыми организмами мож-
но охарактеризовать следующими типами: 1) симбиоз, 2) метабиоз,
3) антагонизм, 4) паразитизм.
Симбиотические отношения проявляются, если одни микроорга-
низмы благоприятствуют жизнедеятельности других. Так, некото-
рые водоросли находятся в симбиотических отношениях с простей-
шими. Метабиотические взаимоотношения заключаются в том, что
вещества, выделяющиеся микроорганизмами в процессе обмена
веществ, используются другими микроорганизмами. Примером
таких взаимоотношений является деятельность бактерий — мине-
рализаторов. Органические кислоты, выделяющиеся при разложе-
нии целлюлозы целлюлозоразлагающими бактериями, служат пи-
тательными веществами для азотобактера. В свою очередь, удале-
ние продуктов обмена веществ из окружающей среды благоприят-
ствует жизнедеятельности целлюлозоразлагающих бактерий. В по-
добных взаимоотношениях находятся между собой бактерии —
аммонификаторы, нитрозо- и нитробактерии. Аммиак, выделяю-
щийся при разложении аминокислот, окисляется нитрозобактерия-
ми до нитритов, которые под влиянием нитробактерий окисляются
до нитратов. Между симбиозом и метабиотическими взаимоотноше-
ниями нельзя провести резкой границы.
Антагонистические взаимоотношения между микроорганизмами
могут проявляться в тех случаях, когда продукты обмена веществ
одних микроорганизмов являются токсичными для других. Эти
метаболиты называются антибиотиками. Примером антибиотиче-
ского действия может служить подавление жизнедеятельности гни-
225
.постных бактерий веществами, выделяемыми плесневыми грибами.
Особой формой антагонистических отношений является использо-
вание одних микроорганизмов другими в качестве пищи (хищни-
чество). Так, некоторые виды инфузорий питаются бактериями и
другими простейшими, например жгутиковыми.
Паразитизм заключается в том, что микроорганизмы, способ-
ные к питанию живым органическим веществом, проникают в орга-
низм хозяина, развиваются в нем, вызывая нарушение обмена ве-
ществ. К паразитическим формам относятся бактериофаги, болез-
нетворные бактерии, вирусы, некоторые простейшие.
В последние годы для интенсификации процессов самоочище-
ния водоемов применяют биологический способ, основанный на
использовании микроорганизмов рода Bdellovibrio, вызывающих
отмирание многих видов бактерий.
Микроорганизмы и окружающая среда. Микроорганизмы могут
существовать, только взаимодействуя с внешней средой, из кото-
рой они получают необходимые питательные вещества и в кото-
рую они выделяют продукты обмена. Жизнедеятельность микро-
организмов обусловлена внешней средой, от свойств ее зависит
направленность биохимических процессов и развитие определенных
групп микроорганизмов. В то же время микроорганизмы в процес-
се жизнедеятельности изменяют в определенной степени условия
окружающей среды, создают возможность для развития других
групп организмов. Так, в настоящее время считается, что третья
часть кислорода на земле продуцируется фитопланктоном морей.
.Микроорганизмы очень чувствительны даже к незначительным
изменениям внешней среды. Причем развитие других групп микро-
организмов наблюдается уже через несколько минут после начала
действия фактора. Примером этого может служить смена био-
ценозов при уменьшении количества растворенного кислорода в
воде.
Сложные взаимоотношения микроорганизмов с окружающей
средой можно регулировать, усиливая или ослабляя воздействие
определенных факторов. Так, при биологической очистке сточных
вод процессы ведутся таким образом, чтобы создавались опти-
мальные условия для групп микроорганизмов, участвующих в раз-
ложении органических веществ. При биологической очистке про-
изводственных сточных вод иногда приходится вводить дополни-
тельно биогенные элементы (азот, фосфор) для обеспечения нор-
мального развития микроорганизмов.
Взаимосвязь микроорганизмов является всеобщей и взаимо-
обусловленной. Микроорганизмы представляют собой наиболее
ответственное звено в круговороте элементов на Земле. В процес-
се жизнедеятельности они вступают во взаимодействие с компо-
нентами внешней среды и различными группами других организ-
мов, взаимно обусловливая существование различных экологиче-
ских групп. Взаимодействие и диалектическая взаимосвязь микро-
организмов с окружающей средой — пример осуществления
принципа всеобщей связи и взаимодействия в природе.
226
§ 53. Распространение микроорганизмов в природе
Микроорганизмы почвы. Почва — наиболее благоприятная сре-
да обитания для микроорганизмов. В ней имеется большое количе-
ство питательных веществ в форме органических остатков, необхо-
димое количество влаги, близкая к оптимальной температура.
Количественное содержание микроорганизмов в почве может под-
вергаться значительным колебаниям в зависимости от характера
и типа почв, степени аэрации, pH, времени года, удаления от по-
верхности земли и других факторов. Оно колеблется от 300 до
3000 млн. микроорганизмов в 1 г почвы. В хорошо аэрируемых
почвах содержится большое количество микроорганизмов, так как
многие из них являются аэробами. Кислые почвы менее благопри-
ятны для жизнедеятельности микроорганизмов, так как большин-
ство из микроорганизмов развивается в нейтральной или слабо-
щелочной среде.
В самом верхнем слое почвы содержится относительно мало
микроорганизмов, что объясняется проникновением солнечных лу-
чей и меньшей увлажненностью его по сравнению с более глубо-
кими слоями. Наибольшее количество микроорганизмов находится
на глубине 10—15 см, так как здесь создаются наиболее благо-
приятные условия для развития микроорганизмов. В более глубо-
ких слоях почвы количество микроорганизмов постепенно умень-
шается.
Видовой состав микрофлоры почв отличается разнообразием и
находится в прямой взаимосвязи с типом почвы. Взаимное влия-
ние микроорганизмов и состава почвы обусловливает направлен-
ность биохимических процессов и структуру поверхностного слоя
земли. Наиболее широко распространены в почве микроорганизмы-
гетеротрофы, осуществляющие разложение органических веществ.
Много в почве кокковых форм, спорообразующих палочек. В ней
всегда имеются актиномицеты, дрожжи. Автотрофные микроорга-
низмы наиболее широко представлены бактериями-нитрификатора-
мп. Кроме них могут встречаться железо- и серобактерии и различ-
ные виды микроскопических водорослей (зеленые, синезеленые,
реже — диатомовые). Из простейших наиболее распространены
саркодовые (амебы), жгутиковые и некоторые виды инфузорий.
Количество их колеблется от нескольких десятков до сотен тысяч
особей в 1 г почвы. Из грибов чаще всего встречаются роды Peni-
cillium, Aspergillus, Fusarium, Mucor. В почве могут довольно дол-
го сохраняться некоторые болезнетворные формы бактерий, особен-
но спорообразующие, например возбудитель сибирской язвы. Мик-
роорганизмы почвы участвуют в создании гумуса (перегноя), со-
став которого определяется деятельностью различных групп микро-
организмов.
Микроорганизмы воздуха. Воздух — неблагоприятная среда
для жизни микроорганизмов. Их количество в воздухе значительно*
меньше, чем в почве, и подвержено колебаниям. Микроорганизмы
попадают в воздух с пылью и мельчайшими капельками воды. На
227
жизнедеятельность микроорганизмов оказывают отрицательное
воздействие недостаточная влажность, резкие колебания темпера-
туры, солнечная радиация, отсутствие питательных веществ. Мак-
симальное количество микроорганизмов содержится в воздухе над
крупными городами в нижних слоях воздуха в теплое время года.
По мере удаления от поверхности земли количество их резко сни-
жается. На высоте 500 м в 1 см3 воздуха над Москвой содержит-
ся в среднем 2—3 бактерии, на высоте 1 км — 1,5 бактерии, а на
высоте 2 км — 0,5 бактерии. Зимой количество микроорганизмов
в воздухе значительно снижается, что связано с образованием сне-
гового покрова и понижением температуры.
Качественный состав микрофлоры воздуха не является посто-
янным и зависит от источника бактериального загрязнения. Боль-
шую часть микроорганизмов воздуха составляют различные виды
кокков, пигментообразующие бактерии, споры актиномицетов,
плесневых грибов. Наряду с безвредными микроорганизмами в
воздухе, особенно загрязненном, могут содержаться патогенные
микроорганизмы — возбудители инфекций, передающихся через
воздух (туберкулеза, гриппа и др.).
Микроорганизмы воды. Количество микроорганизмов в водной
среде определяется такими факторами, как наличие органических
веществ, содержание растворенного кислорода, температура, ха-
рактер водоема и т. и. В подземных водах и водах атмосферных
осадков содержится незначительное количество микроорганизмов.
В поверхностных водоемах численность микроорганизмов опре-
деляется степенью загрязнения воды органическими соединениями.
В воде рек она может колебаться от нескольких тысяч до миллио-
на клеток и более в 1 мл. Донный ил, содержащий большее коли-
чество питательных веществ в виде органических остатков, явля-
ется средой обитания для многих микроорганизмов. В водоемах с
замедленным стоком (водохранилищах), озерах количество микро-
организмов уменьшается при удалении от береговой линии и с
возрастанием глубины. Видовой состав микроорганизмов, живущих
в поверхностных водоемах, крайне разнообразен.
Роль микроорганизмов в круговороте углерода и азота. Первич-
ная продукция органических веществ осуществляется фотосинтези-
рующими организмами. В растениях процессы синтеза значитель-
но преобладают над процессами разложения. Органические веще-
ства растений служат пищей для животных. Они используются как
для построения клеток и тканей организма, так и для получения
энергии, необходимой для их жизнедеятельности. Но круговорот
углерода не ограничивается жизнедеятельностью растений и живот-
ных. Значительная часть органических соединений растений непри-
годна для питания животных. Огромные количества органического
углерода содержатся в отмирающих организмах. Таким образом,
эти органические соединения не смогли бы участвовать в кругово-
роте, если бы не жизнедеятельность микроорганизмов. Каждая
группа микроорганизмов разлагает определенные органические
соединения, но вследствие многообразия микроорганизмов в приро-
228
де практически подвергается разложению любое органическое со-
единение. Выделяющийся при этом диоксид углерода поступает в
биологический круговорот углерода. Таким образом, микроорганиз-
мы принимают в круговороте углерода непосредственное участие,
минерализуя огромные массы органического вещества и высвобож-
дая углерод в виде диоксида углерода.
Не меньшее значение имеет жизнедеятельность микроорганиз-
мов в круговороте азота. Растения способны усваивать азот в виде
растворимых солей. Огромное количество свободного азота атмо-
сферы для них недоступно, так же как и сложные органические
азотсодержащие соединения, содержащиеся в воде. Перевод слож-
ных белковых соединений в более простые осуществляется раз-
личными микроорганизмами в несколько этапов. Образующийся в
процессе разложения аминокислот аммиак окисляется в присут-
ствии кислорода сначала до нитритов, затем до нитратов. Часть
этих соединений используется растениями, а другая часть под
влиянием микроорганизмов восстанавливается до свободного азо-
та. Некоторые бактерии (азотфиксаторы) могут усваивать свобод-
ный азот атмосферы и переводить его в нитриты и нитраты.
Изменчивость микроорганизмов. Изменчивость является одной
из наиболее важных сторон жизни и развития микроорганизмов.
Сущность ее заключается в реакции организма на изменения, про-
исходящие во внешней среде или внутри клетки, в структуре и
функциях наследственного аппарата микроорганизма. Наследст-
венность, обеспечивающая постоянство видовых признаков, и из-
менчивость представляют собой взаимосвязанные диалектические
противоположности процесса развития организма. Именно измене-
нием и наследованием приобретаемых признаков в процессе эво-
люции произошло выделение паратрофов из группы гетеротрофов.
Микроорганизмы быстро адаптируются к изменившимся условиям
среды обитания, изменяя соответствующим образом обмен веществ.
Классическим примером адаптации микроорганизмов к воздей-
ствию внешних факторов может служить появление форм болез-
нетворных микроорганизмов, устойчивых к действию лекарствен-
ных веществ. На изменчивости микроорганизмов основано выведе-
ние микрофлоры, способной осуществлять превращения органиче-
ских веществ, которые не разлагаются обычной микрофлорой воды
или почвы. Изменения формы и функциональных особенностей
микроорганизмов могут быть вызваны действием физических,
химических пли биологических факторов. Приобретаемые микро-
организмами признаки могут быть связаны только с условиями
жизни отдельного микроорганизма и не передаваться по наслед-
ству.
Наиболее важное значение имеет наследственная (мутацион-
ная) изменчивость, обусловленная изменениями в структуре и
функциях наследственного аппарата клетки. Приобретенные приз-
наки передаются и закрепляются в следующих поколениях, давая
устойчивые видоизмененные формы микроорганизмов. Изменчи-
вость может касаться морфологических признаков или биологиче-
229
ских особенностей микроорганизма, связанных с обменом веществ.
При этом изменяется активность биохимических процессов и их
направленность.
Изучение изменчивости микроорганизмов необходимо для ин-
тенсификации биохимической очистки сточных вод, для получения
новых разновидностей микроорганизмов, обладающих нужными
свойствами. Так, например, выведены штаммы споровых бактерий,
разлагающих капролактам, — компонент сточных вод предприя-
тий по производству капрона.
Глава VIII. САНИТАРНАЯ МИКРОБИОЛОГИЯ ВОДЫ
§ 54. Биоценозы природных водоемов
Создание научной основы рационального использования ресур-
сов водоемов невозможно без изучения жизнедеятельности водных
микроорганизмов, определения их роли в процессе самоочищения
водоемов, влияния на формирование химического свойства при-
родных вод, выяснения закономерностей биохимических процессов
превращения элементов в водной среде.
Основным источником поступления микроорганизмов в поверх-
ностные пресные водоемы служит почва. Большое количество мик-
роорганизмов попадает в водоемы со сточными водами, содержа-
щими биологически разлагаемые вещества. Некоторая часть мик-
роорганизмов поступает в водоемы из воздуха.
Выделяют две экологические группы микроорганизмов: автох-
тонную и аллохтонную. Самостоятельная, первоначально сущест-
вующая микрофлора, для которой вода является естественной сре-
дой питания, называется автохтонной. Состав ее отличается ста-
бильностью и является характерным для каждого водоема. Мик-
роорганизмы, поступающие в воду извне, например со сточными
водами, образуют аллохтонную микрофлору. Эти микроорганизмы
в водоеме попадают в неблагоприятные условия, которые состоят
в отсутствии или недостатке питательных веществ, изменении тем-
пературы, pH, солевого состава, окислительно-восстановительного
потенциала и др. Поэтому микроорганизмы аллохтонной микро-
флоры спустя некоторое время после попадания в водоем отмира-
ют. Среди микроорганизмов аллохтонной микрофлоры могут быть
и патогенные формы. Между микроорганизмами автохтонной и ал-
лохтонной группы возникают антагонистические отношения, что
ускоряет отмирание несвойственных чистому водоему микроорга-
низмов. Характерным является создание в водоеме новой авто-
хтонной микрофлоры, если загрязнение определенными соединения-
ми, например нефтью, фенолами, наблюдается постоянно в тече-
ние длительного промежутка времени. Подобная же смена биоце-
нозов наблюдается и при длительном поступлении сточных вод,
содержащих тяжелые металлы.
230
Различные группы водных организмов (гидробионтов) приспо-
собились к определенным зонам обитания, образуя биоценозы в
пределах единой экологической системы (водоема). Толща воды
населена микроорганизмами планктона. Растительные формы на-
зываются фитопланктоном, а животные организмы — зоопланкто-
ном. Многие микроорганизмы зоопланктона (простейшие) облада-
ют определенной способностью к перемещению с помощью ресни-
чек, жгутиков. Коловратки совершают прыгательные движения или
плавают, используя реснички.
Поддержанию микроорганизмов во взвешенном состоянии спо-
собствует наличие у некоторых микроорганизмов в клетках газо-
вых пузырьков, как это наблюдается у раковинной амебы Arcella.
Особенно характерным для зоопланктона является перемещение
по вертикали, происходящее при изменении внешних условий (тем-
пература, освещение и др.). Размеры организмов микропланктона
колеблются от 50 мкм до 1 мм. Это — простейшие, коловратки, не-
которые водоросли и бактерии. К карликовому планктону (нанно-
планктон) относятся микроорганизмы, размер которых менее
50 мкм (бактерии, фаги, вирусы). Среди планктонных микроорга-
низмов широко представлены бактерии, вызывающие биохимиче-
ское окисление органических веществ, но встречаются и автотроф-
ные организмы. Бактерии могут вместе с водорослями, простейши-
ми развиваться на деструктурированных частицах органических
остатков (детрите).
В зимний период наблюдается минимум развития бактериаль-
ного планктона, в паводок количество бактерий значительно уве-
личивается, часто достигая максимума. Затем с развитием водо-
рослей численность их снижается, оставаясь постоянной в течение
лета, осенью во время массового отмирания фитопланктона коли-
чество бактерий резко возрастает. Причина сезонных изменений
планктона заключается в колебаниях содержания питательных ве-
ществ, необходимых для определенных групп планктона.
Крупные гидробионты, имеющие хорошо развитые органы дви-
жения (например, рыбы), называются нектоническими формами.
Микроорганизмы, развивающиеся на твердом субстрате и вызы-
вающие обрастание подводных предметов, образуют перифитон.
Обрастания могут вызываться как растительными, так и животны-
ми организмами. Из растительных форм в перифитоне преоблада-
ют водоросли.
Водные организмы, населяющие дно водоема и донные отло-
жения, называются бентосом. К бентосным формам микроорганиз-
мов относятся огромное количество бактерий, водоросли, простей-
шие, грибы и др. Особенно много бактерий, осуществляющих пре-
вращения биогенных элементов, находится в верхнем слое донных
отложений. Они могут образовывать па поверхности донных отло-
жений пленку толщиной в несколько миллиметров. Широко рас-
пространены в донных отложениях простейшие, коловратки. Мак-
симальное количество микроорганизмов в донных отложениях
наблюдается летом и осенью. На больших глубинах микробентос-
231
Таблица 17. Зависимость между
качеством воды и содержанием в ней
аэробных сапрофитных микроорганизмов
ные формы широко пред'
ставлены, в основном, бак
териями-минерализаторами.
Количество колоний, вырастающих при посеве на питательный агар 1 мл воды Опенка водоемов Процессы разложения орга- нических веществ с наи- большей напряженностью идут именно в поверхност-
~10 10—100 100— 1 000 1 000— 10 000 10 000—100 000 >100 000 Очень чистые Чистые Умеренно-загрязнен- ные Загрязненные Г рязные Очень грязные ных слоях донных отложе- ний. Классификация микро- организмов по физиологиче- ским признакам основана на их способности усваивать различные вещества из вод- ной среды в аэробных или
анаэробных условиях. Микроорганизмы различных физиологичес-
ких групп вступают между собой в сложные метабиотические или
антагонистические отношения. Основная роль в биоценозах отво-
дится группам микроорганизмов, участвующих в круговороте важ-
нейших биогенных элементов — углерода, азота, серы, фосфора.
В физиологическую группу микроорганизмов, участвующих в кру-
говороте азота, входят протеолиты, гидролизующие сложные бел-
ки до простых и аминокислот; аммонификаторы; нитрификаторы;
азотфиксирующие бактерии; денитрификаторы и другие группы
микроорганизмов. Среди этих многочисленных форм есть и облигат-
ные аэробы, например нитрификаторы, и анаэробные микроорга-
низмы.
Фотосинтезирующие формы микроорганизмов представлены в
водоемах различными группами водорослей (диатомовых, зеленых,
синезеленых). Физиологическая группа микроорганизмов, участ-
вующая в круговороте углерода, включает большое число как
аэробных, так и анаэробных форм. Основная роль принадлежит
микроорганизмам, сбраживающим (в анаэробных условиях) и
окисляющим (в аэробных) углеводы, в частности целлюлозу, ко-
торая образует основную часть органических остатков при отмира-
нии растений. В водоемах присутствуют также микроорганизмы,
окисляющие жиры, органические кислоты, углеводороды и др.
Анаэробные микроорганизмы, населяющие донные отложения, вы-
зывают разложение органических соединений и дальнейшее пре-
вращение образующихся газообразных продуктов (Н2, N2, H2S) в
другие вещества. Среди них представлены возбудители метанового,
водородного, маслянокислого и других видов брожения.
Из других физиологических групп микроорганизмов, присутст-
вующих в водоемах, следует отметить бактерии, окисляющие се-
роводород до серы или сульфатов (тионовые) и восстанавливаю-
щие (в анаэробных условиях) сульфаты до серы и даже до серо-
водорода (сульфатредуцирующие). В чистых водоемах могут со-
держаться и другие бактерии — автотрофы, например железобак-
терии.
232
Основное значение при оценке санитарного режима водоема
имеют бактерии — сапрофиты, разлагающие органическое вещест-
во. По количеству микроорганизмов сапрофитов в поверхностных
водах можно судить о санитарном состоянии водоемов (табл. 17).
§ 55. Санитарно-бактериологическая оценка
качества воды
Бактериологические показатели качества воды являются частью
исследования свойств вод любого состава, происхождения и бакте-
риальной загрязненности. Бактериологические показатели более
чувствительны при определении степени загрязнения водоема бы-
товыми сточными водами, чем результаты химического исследова-
ния. Так, по содержанию бактерий-сапрофитов можно обнаружить
загрязнение воды органическими биологическими разлагаемыми со-
единениями при разбавлении в десятки и сотни тысяч раз. Высокая
чувствительность микробиологических методов исследования име-
ет большое значение в деле охраны водной среды от загрязнения.
Основная задача санитарно-бактериологических исследований
воды заключается в получении гигиенической оценки ее качества
в отношении инфекционной опасности. Определение отдельных ви-
дов патогенных микроорганизмов требует специальных, зачастую
сложных методов микробиологического исследования, да и полу-
ченный отрицательный результат не всегда гарантирует отсутст-
вие этих форм. Поэтому в практике микробиологических исследо-
ваний применяют косвенные методы оценки качества воды по бак-
териологическим показателям. При систематических исследованиях
эпидемиологического состояния водоема в качестве санитарно-по-
казательных микроорганизмов используются наиболее характерные
представители микрофлоры кишечника человека.
Санитарно-бактериологическому исследованию подлежит вода:
а) централизованного водоснабжения и водопроводных сетей;
б) колодцев, родников; в) открытых водоемов (озер, прудов, рек);
г) сточная; д) плавательных бассейнов; е) минеральная. Его про-
изводят при выборе водоисточников, при систематическом санитар-
но-гигиеническом наблюдении за ними и по эпидемиологическим
показаниям.
Санитарно-бактериологический анализ, проводимый для оценки
качества питьевой воды, включает в себя определение двух основ-
ных показателей: 1) определение общего числа бактерий; 2) коли-
индекс или коли-титр. Методика выполнения этих исследований
регламентируется ГОСТ 18963—73.
Определение общего числа бактерий («микробное число»).
Сущность его заключается в подсчете количества колоний микро-
организмов, вырастающих в течение 24+2 ч при температуре
37+0,5° при посеве 1 мл исследуемой воды в стандартную пита-
тельную среду. В этих условиях определяются мезофильные гете-
ротрофные аэробы и факультативные анаэробы, развивающиеся на
данной питательной среде. Таким образом, показатель «общее чис-
233
ло бактерий» не соответствует действительному количеству микро-
организмов, присутствующих в воде. Например, в эту группу не
входят микроорганизмы, развивающиеся на других питательных
средах, автотрофы, облигатные анаэробы. Тем не менее этот пока-
затель определяет степень бактериального загрязнения воды.
Для оценки фекального загрязнения водоемов проводят выра-
щивание микроорганизмов при 37° С в течение 24 ч, а для контро-
ля за ходом процесса самоочищения — при 20—22° С в течение 48 ч.
Сравнительная характеристика этих определений позволяет сде-
лать вывод о степени загрязнения водоема бытовыми сточными
водами. В чистых водоемах преобладают микроорганизмы, вырас-
тающие при 20—22° С. Общее число бактерий используется для
определения эпидемиологической характеристики воды плаватель-
ных бассейнов (не более 100 в 1мл) и для оценки эффективности
биологической очистки и обеззараживания сточных вод.
Если нужно дать оценку микрофлоре воды в целом, используют
прямой микроскопический метод подсчета микроорганизмов, до-
стоинство которого заключается в более полном учете всех групп
микроорганизмов. Этот метод нельзя использовать для оценки
обеззараживания воды, так как при этом регистрируются не толь-
ко живые, но и мертвые клетки микроорганизмов. Важное значе-
ние этот метод имеет при возникновении аварийных условий в си-
стеме хозяйственно-питьевого водоснабжения, при массовом бак-
териальном загрязнении водоемов.
§ 56. Санитарно-показательные микроорганизмы
воды и почвы
В качестве санитарно-показательных организмов, применяемых
для оценки вероятности содержания в воде патогенной микрофло-
ры, используют микроорганизмы, для которых постоянной средой
обитания является кишечник человека и животных. Количествен-
ный учет микроорганизмов — индикаторов загрязнения позволяет
дать более точную оценку степени бактериального загрязнения
воды. Основные требования, предъявляемые к санитарно-показа-
тельным микроорганизмам: 1) они должны иметь общую естествен-
ную среду обитания с патогенными микроорганизмами и выделять-
ся во внешнюю среду в большом количестве; 2) во внешней среде
санитарно-показательные микроорганизмы должны по возможно-
сти равномерно распределяться и быть более устойчивыми, чем
патогенные. Они должны дольше сохраняться в воде, практически
не размножаясь, иметь большую устойчивость к воздействию раз-
личных неблагоприятных факторов, у них должна возможно в
меньшей степени проявляться изменчивость свойств и признаков;
3) методы определения санитарно-показательных микроорганиз-
мов должны быть простыми, быстрыми и иметь достаточную сте-
пень достоверности.
Из микроорганизмов естественной микрофлоры человека и жи-
вотных в наибольшей степени этим требованиям соответствуют
234
бактерии группы кишечных палочек (БГКП). Бактерии группы
кишечных палочек наряду с энтерококками, бифидобактериями
являются наиболее типичными представителями кишечной микро-
флоры человека и животных. В настоящее время принято в качест-
ве санитарно-показательных микроорганизмов использовать не
отдельные виды или роды микроорганизмов, а группы микроорга-
низмов, объединенные рядом общих признаков. Это упрощает
анализ и обеспечивает большую точность определения. К бактери-
ям группы кишечных палочек относятся грамотрицательные, не-
спорообразующие палочки, сбраживающие лактозу с образовани-
ем кислоты и газа при 37° С в течение 24—48 ч или сбраживающие
глюкозу с образованием кислоты и газа при той же температуре в
течение 24 ч и не обладающие оксидазной активностью. Целесооб-
разность использования БГКП в качестве индикатора фекального
загрязнения, особенно при анализе питьевой воды, получила науч-
ное обоснование и подтвердилась многолетней практикой санитар-
ной оценки качества воды.
Оксидазный тест используется для идентификации бактерий
семейства Enterobacteriaceae (энтеробактерии), в которое входят
БГКП, неокрашивающиеся по Граму, и вырастающие на стандарт-
ной питательной среде водные сапрофиты, не имеющие санитарно-
показательного значения. Эти группы бактерий-сапрофитов отли-
чаются от БГКП тем, что обладают ферментом — оксидазой. По-
этому при контакте колоний микроорганизмов, вырастающих при
посеве питьевой воды на стандартной питательной среде, с раство-
ром фенилендиаминовых соединений колонии микроорганизмов,
проявляющих оксидазную активность, окрашиваются в синий цвет.
Подавляющее большинство колоний, не изменивших цвета, образо-
вано БГКП, но среди них могут быть кокки, спорообразующие и
грамотрицательные палочки, не имеющие санитарно-показатель-
ного значения. Они исключаются из общего количества после пря-
мого микроскопирования и при отсутствии образования газа при
сбраживании глюкозы при 37° С. Таким образом, основным крите-
рием отнесения микроорганизмов к санитарно-показательным
БГКП является оксидазный тест и способность сбраживать глюко-
зу с образованием газа при 37° С. Если необходимо уточнить дан-
ные о происхождении, характере и давности фекального загрязне-
ния, то используют в качестве санитарно-показательного микроор-
ганизма бактерии рода Escherichia (эшерихия), наличие которых
в воде указывает на свежее загрязнение воды и увеличивает эпи-
демиологическую опасность. Предложенный учеными Чехослова-
кии и ГДР оксидазный тест был тщательно проверен и дополнен
в практике санитарно-бактериологического анализа в нашей
стране.
Предложения по уточнению методики определения БГКП были
учтены при разработке нового ГОСТа. БГКП являются основным
показателем при оценке бактериального загрязнения питьевой
воды, степени очистки и обеззараживания сточных вод, воды источ-
ников водоснабжения, морей, плавательных бассейнов.
235
Энтерококки приняты в качестве санитарно-показательных мик-
роорганизмов в стандартах некоторых стран (Великобритания,
США), а в нашей стране они используются в качестве дополни-
тельного теста при исследовании воды. Они входят в состав по-
стоянной микрофлоры кишечника человека и наряду с БГКП в
огромном количестве содержатся в бытовых сточных водах. Пре-
имущество энтерококков перед БГКП заключается в том, что они
являются более достоверным тестом на фекальное загрязнение,
устойчивы к действию бактерицидных веществ, в воде водоемов не
размножаются. Затруднения возникают из-за нечеткого определе-
ния группы энтерококков, которые должны использоваться в ка-
честве индикаторов загрязнения.
Для оценки санитарного режима водоемов может быть исполь-
зовано отношение БГКП к группе энтерококков. При загрязнении
водоема сточными водами, не подвергавшимися обеззараживанию,
БГКП преобладают над группой энтерококков. При обеззаражива-
нии сточных вод БГКП отмирают в большей степени и соотноше-
ние числа БГКП к количеству энтерококков уменьшается почти до
единицы.
Это отношение может быть использовано для контроля за
обеззараживанием сточных вод, причем анализ должен проводить-
ся не более чем через 20 ч после спуска сточных вод, так как
отдельные виды энтерококков быстро отмирают в водоеме. Комп-
лексное определение БГКП и энтерококков позволяет получить
более точные данные для оценки санитарного состояния водоема,
при выборе источника водоснабжения, установить границы и сте-
пень загрязнения водоема.
Санитарно-показательные микроорганизмы рассмотренных
групп не могут быть использованы для оценки загрязнения водо-
емов вирусами. Вирусы обладают более высокой устойчивостью,
чем патогенные бактерии, к воздействиям внешней среды и более
длительное время сохраняются в воде водоемов. Комплексные ис-
следования, проведенные в институте общей и коммунальной ги-
гиены им. А. Н. Сысина АМН СССР, показали, что в качестве са-
нитарно-показательных микроорганизмов в отношении энтеровиру-
сов можно использовать бактериофаги кишечных бактерий. Этот
тест с достаточной достоверностью может быть применен для са-
нитарной оценки качества воды рек в зонах сильного пли умерен-
ного загрязнения.
Оценка качества воды по данным санитарно-бактериологиче-
ского анализа. Для обеспечения безопасности питьевой воды в
эпидемиологическом отношении ГОСТ 2874—73 предусматривает
определение косвенных показателей: степень общего бактериаль-
ного загрязнения и содержание бактерий группы кишечной палоч-
ки (БГКП). Общее количество бактерий в неразбавленной воде
должно быть не более 100 в 1 мл. Количество БГКП, определяемое
на плотной стандартной среде с применением концентрирования
бактерий на мембранных фильтрах, должно быть не более 3 в 1 л
воды (коли-индекс). Наименьший объем воды, в котором находит-
236
ся одна кишечная палочка (коли-титр), должен быть не менее
300 мл.
К качеству воды по бактериологическим показателям в круп-
нейших городах предъявляются более строгие требования: коли-
индекс должен быть не более 2, коли-титр — не менее 500. При
наличии бактериального загрязнения воды выше допустимых норм
проводятся дополнительные исследования на присутствие бакте-
рий — показателей свежего фекального загрязнения — кишечных
палочек преимущественно Esherichia coli. Наличие их устанавли-
вается по способности ферментировать лактозу до кислоты
и газа при 43° С в присутствии ингибиторов посторонней микро-
флоры.
ГОСТ 2874—73 на питьевую воду предусматривает отсутствие
в воде микроорганизмов зоо- и фитопланктона, видимых невоору-
женным глазом. В то же время в воде, поступающей на водопро-
водные очистные сооружения, могут встречаться практически все
представители планктона водоема. Особенно большое количество
фитопланктона наблюдается в период цветения водоемов. Некото-
рые формы микроорганизмов (зеленые и диатомовые водоросли)
обладают высокой устойчивостью к действию обеззараживающих
реагентов. Поэтому для контроля за ходом эффективности процес-
са очистки и обеззараживания воды на очистных сооружениях во-
допровода проводится гидробиологический анализ воды, который
заключается в обнаружении и установлении видового и количест-
венного состава микроорганизмов, присутствующих в воде на раз-
ных этапах ее обработки. Результаты гидробиологического анали-
за выражают числом клеток микроорганизмов в 1 м3 воды. Изуче-
ние видового состава микроорганизмов планктона и ее количест-
венная характеристика позволяют проводить корректировку в про-
цессе очистки воды.
В воде поверхностных источников водоснабжения коли-индекс
не должен превышать 10 000 ни в одной из исследуемых проб. При
более сильном бактериальном загрязнении (большем значении
коли-индекса) необходимо проведение дополнительных санитарно-
бактериологических исследований для уточнения и идентификации
бактериального загрязнения: энтерококков, бактериофагов, кишеч-
ных палочек, патогенных кишечных микроорганизмов. Присутствие
Escherihia coli в количестве более 1000 в 1 л воды указывает на
свежее загрязнение водоема бытовыми сточными водами. Отноше-
ние числа БГКП к количеству Escherichia coli в такой ситуации,
как правило, меньше 10. Такой водоем не может быть использован
для целей водоснабжения, поскольку в нем создаются неблагопри-
ятные в эпидемиологическом отношении условия. На наличие фе-
кального загрязнения указывает и значительное количество энте-
рококков в пробе воды (более 1000).
Для характеристики качества воды подземных водоисточников
проводится санитарно-бактериологическое исследование на общее
содержание бактерий, БГКП и дополнительно на энтерококки, так
как они более устойчивы к действию низкой температуры и имеют
237
более длительный период выживания в подземных водах сравни-
тельно с БГКП.
Категории биологических загрязнений сточных вод и их значе-
ние для оценки эпидемиологической опасности сточных вод. Пере-
дача некоторых инфекций водным путем приобретает особую ак-
туальность вследствие всевозрастающей роли водоемов в жизни
людей. Увеличение микробиологического загрязнения водоемов
связано с поступлением большого количества сточных вод, возра-
станием водопользования, использованием их для купания и прове-
дения спортивных мероприятий. Ряд бактериальных инфекций
(брюшной тиф, дизентерия, холера, туляремия) имеют водный
путь передачи. Для вирусных инфекций (полиомиелита) тоже до-
казана возможность передачи через водную среду.
Основным источником бактериального загрязнения являются
бытовые сточные воды, стоки больниц, бань, прачечных, некоторых
видов предприятий пищевой промышленности. В сточных водах мо-
гут находиться бактерии, вирусы, бактериофаги, яйца гельминтов,
дрожжи, плесневые грибы, микроскопические водоросли, простей-
шие.
Наиболее часто водным путем передаются кишечные инфекции,
что связано с концентрацией их возбудителей в кишечнике чело-
века, массовым выведением их в водную среду и относительной
устойчивостью в воде. Хотя вода и не является благоприятной
средой обитания для патогенных форм микроорганизмов, они мо-
гут сохраняться в ней в течение нескольких суток и даже месяцев.
Так, холерный вибрион может выживать в воде в течение несколь-
ких месяцев и сохранять активность после замораживания. Осо-
бенно высокую устойчивость в сточной воде и осадке имеют ви-
русы.
При длительном нахождении в воде патогенные микроорганиз-
мы могут изменяться под влиянием условий внешней среды, но при
этом они не всегда теряют болезнетворные свойства.
В сточные воды с выделениями людей и животных попадают
яйца гельминтов. Наиболее часто присутствуют в таких водах
яйца аскарид, остриц. Биологические особенности развития обус-
ловливают деление гельминтов на две группы: геогельминты и био-
гельминты. Геогельминты развиваются во внешней среде без сме-
ны хозяина. К ним относятся аскарида, власоглав, строгилоидес,
анкилостома. Заражение человека происходит при попадании та-
кой воды в рот или путем внедрения личинок в кожный покров.
К биогельминтам относятся свиной и бычий цепни, эхинококк,
лентецы и др. В водоемах они могут использовать в качестве орга-
низмов-хозяев пресноводных рыб, раков. Количество яиц гельмин-
тов в сточных водах может исчисляться сотнями в 1 л.
Индикаторами загрязнения сточных вод чаще всего являются
бактерии-сапрофиты, способные давать колонии на стандартных
питательных средах, характеризующие наличие легко разлагаю-
щихся органических веществ, и бактерии — представители посто-'
явной микрофлоры человека и животных. Общее число бактерий
238
определяется подобно тому, как это делают при анализе питьевой
воды, только производится предварительное соответствующее раз-
бавление сточной воды. Аналогично проводится учет бактерий — по-
казателей фекального загрязнения воды (БГКП). Результаты ана-
лиза выражают в форме коли-ипдекса. Сбрасываемые в водоемы
очищенные и подвергшиеся обеззараживанию сточные воды хо-
зяйственно-питьевого или культурно-бытового водопользования
должны иметь коли-индекс не более 1000. Гельминтологический
анализ проводится для уточнения характера загрязнения и контро-
ля за эффективностью работы очистных сооружений. Дополнитель-
ные санитарно-бактериологические исследования проводятся по
специальным показаниям.
§ 57. Характер и источники загрязнения водоемов
Основным источником загрязнения водоемов являются бытовые,
производственные, сельскохозяйственные и ливневые сточные воды.
Кроме того, немаловажную роль в загрязнении водоемов играют
смывы с площади водосбора при орошении. Часть загрязнений по-
падает в водоем с подземными водами и из воздуха, при купании
людей и животных. Вклад каждого из этих источников в загрязне-
ние водоема определяется конкретными условиями. Загрязнение
водоема проявляется в нарушении биологического равновесия, в
создании которого принимают участие все микроорганизмы водо-
ема. Выделяют три фазы во взаимодействии между сточными во-
дами и водой водоема:
1) изменение физических, физико-химических свойств воды, ток-
сичное действие на гидробионты;
2) воздействие на процессы, протекающие в биоценозах во-
доема;
3) стабилизация условий в водоеме в процессе самоочищения
(Е. А. Веселов).
Интенсивность, характер и продолжительность воздействия
поступающих в водоем загрязнений на санитарный режим, хими-
ческий состав воды и условия жизнедеятельности водных организ-
мов определяется их свойствами. Так, соединения-восстановители,
легко окисляющиеся растворенным в воде кислородом, вызывают
резкое нарушение санитарного режима водоема сразу же после
спуска сточных вод. И, наоборот, некоторые биологически нераз-
лагающиеся органические соединения (хлорорганические пестици-
ды) могут обнаруживаться в воде далеко (за десятки и даже сот-
ни километров) от места их поступления в водоем. Нарушение
жизнедеятельности водных организмов, ухудшение качества воды
может вызываться соединениями, продуцируемыми некоторыми
группами микроорганизмов в самом водоеме.
Существует несколько классификаций видов загрязнений сточ-
ных вод с учетом их воздействия па микроорганизмы водоема.
Классификация видов загрязнения природных вод, предложен-
ная Л. В. Григорьевой, приведена ниже.
239
* Сюда относятся соединения, не обладающие свойствами веществ 2, 3, 4,
5-й групп.
В других классификациях кроме выделенных групп учитывают-
ся удобрения, продукты жизнедеятельности водных организмов и
взвешенные вещества. Между этими группами нет резкого раз-
граничения, их действие взаимосвязано и взаимно обусловлено.
Одни из них могут влиять и изменять свойства других, а в целом
их действие сводится к влиянию на микроорганизмы водоема.
Загрязнения, поступающие в водоем из внешних источников,
вызывают первичное загрязнение водоема. В то же время посту-
пающие в водоем соединения подвергаются трансформации, видо-
изменяются, что сопровождается изменением их свойств и степени
влияния на микроорганизмы водоема. Водные микроорганизмы,
аккумулирующие радиоактивные изотопы, включаются в пищевую
цепь и вызывают повышение концентрации радиоактивных веществ
в организмах других гидробионтов (рыб, ракообразных). После
отмирания этих гидробионтов радиоактивные элементы усваивают-
ся донными микроорганизмами или накапливаются в них. Соеди-
нения тяжелых металлов в природных водах быстро осаждаются,
вызывая загрязнение донных отложений. Но при изменении усло-
вий, например при увеличении или снижении pH, растворимость их
может увеличиваться и они вызывают вторичное загрязнение воды
водоема. Бактерии, в том числе и патогенные, попадающие в дон-
ные отложения, сохраняются в них определенное время и при пе-
ремешивании воды могут снова вымываться из иловых отложений.
Развивающиеся в большом количестве растительные и животные
организмы после отмирания также вызывают вторичное загрязне-
ние водоема, способствуя повышению содержания органического
вещества в водоеме (эвтрофикации).
Поступление биогенных элементов (азота, фосфора, калия)
240
происходит при смыве удобрений с полей, со сбрасываемыми при
орошении коллекторно-дренажными водами, с пылью, сдуваемой с
поверхности почвы. Значительное количество биогенных веществ
поступает в водоемы также со сточными водами после биологиче-
ской очистки. Соединения азота могут мигрировать в грунтовые
воды, вызывая их загрязнение. Повышение концентрации азота и
фосфора в воде водоема вызывает усиленное развитие высших
водных растений, что способствует зарастанию и засорению водо-
хранилищ, каналов и других водоемов с замедленным стоком. Для
массового развития планктона, водорослей, вызывающих цветение,
достаточно повышение концентрации фосфора в воде до нескольких
миллиграммов в литре. Изменение качества воды во время цвете-
ния и нарушение кислородного режима при массовом отмирании
фитопланктона и водной растительности способствуют ухудшению
санитарного режима водоема. В итоге этих процессов происходит
увеличение органического вещества в донных отложениях и пита-
тельных веществ в толще воды, т. е. происходит эвтрофикация во-
доема.
§ 58. Система сапробности и ее применение
для оценки степени загрязнения водоемов
Для оценки санитарного состояния водоема наряду с физико-
химическими и бактериологическими методами исследования ис-
пользуется гидробиологический метод, основанный на изучении ви-
дового и количественного состава водных организмов биоценозов.
На основании полученных данных можно сделать вывод о степени
и продолжительности загрязнения водоема и о его способности к
самоочищению.
Водоем представляет собой единую экологическую систему, по-
этому для определения санитарного состояния его изучаются груп-
пы гидробионтов, населяющие толщу воды (фито- и зоопланктон),
донные отложения (бентос) и водные организмы обрастаний (пе-
рифитон). При санитарной оценке водоема наибольшее значение
имеют организмы тех зон водоема, где с наибольшей напряжен-
ностью идут процессы самоочищения, т. е. население донных отло-
жений и обрастаний.
Биологический метод исследования водоемов основан на том,
что отдельные группы гидробионтов могут жить только при опре-
деленной степени загрязнения водоема органическими веществами.
Способность гидробионтов, обусловленную их физиологическими
особенностями, жить в воде с определенной степенью загрязнения
органическими соединениями принято называть сапробностью *.
Классификация степени загрязнения водоемов органическими
веществами в зависимости от обитания в них различных групп
организмов предложена Р. Кольквитцем и М. Марссоном. Большой
* Некоторые авторы сапробностью называют степень распада органических
веществ в загрязненных водах (Методика технологического контроля работы
очистных сооружений городской канализации. М., 1977, с. 220).
241
вклад в разработку и обоснование системы сапробности внесли
советские ученые Я. Я. Никитинский, Г. И. Долгов, А. С. Разумов.
Экологическая характеристика различной степени загрязнения во-
доемов дается по зонам сапробности. В зависимости от степени за-
грязнения воды выделяют следующие зоны сапробности: поли-
сапробную (р); а-мезосапробную (ат), |3-мезосапробную ((Зш) и
олигосапробиую (о). Иногда выделяют пятую зону — катаробпую
(/?), соответствующую самым чистым водам.
Полисапробная зона (зона сильного загрязнения) соответству-
ет свежему загрязнению водоема бытовыми сточными водами. Она
характеризуется наличием большого количества сложных белковых
соединений. Свободный кислород отсутствует, поэтому протекают
биохимические процессы, имеющие восстановительный характер.
В результате разложения органических соединений образуются
сероводород, метан, диоксид углерода, аммиак. Основное население
этой зоны представлено большим количеством бактерий. Числен-
ность их может достигать миллиона и более в 1 мл воды. Наряду
с бактериями в этой зоне развиваются бесцветные жгутиковые,
грибы, в иле находятся малощетинковые черви — тубифициды.
Общее количество видов 30—36, возможны колебания видового со-
става в зависимости от времени года.
а-Мезосапробная зона характеризуется большей степенью рас-
пада органических веществ. В воде появляется небольшое количе-
ство кислорода, который обусловливает протекание окислительно-
восстановительных процессов. В результате распада белковых со-
единений содержится большое количество аминокислот, аммиака.
Сероводород и диоксид углерода присутствуют в незначительном
количестве. В этой зоне появляются фотосинтезирующие формы
микроорганизмов. Количество бактерий исчисляется сотнями ты-
сяч в 1 мл. Широко распространены жгутиковые, грибы, инфузо-
рии. Встречаются некоторые виды синезеленых и зеленых водорос-
лей и коловратки. В иле находятся тубифициды. Общее число ви-
дов 70—76.
|3-Мезосапробная зона характеризуется наличием продуктов ми-
нерализации органических соединений — аммонийных солей, нит-
ратов, нитритов. Содержание органических веществ невелико.
В воде имеется достаточное количество растворенного кислорода,
характер протекающих процессов — окислительный. По сравнению
с предыдущими зонами резко увеличивается количество видов
(116—133) с одновременным уменьшением количества особей од-
ного вида. Количество бактерий исчисляется десятками тысяч в
1 мл. Показательным для зоны является преобладание зеленых,
диатомовых, синезеленых водорослей. Широко представлены ко-
ловратки, инфузории, низшие ракообразные, насекомые, рыбы. Эта
зона соответствует качеству воды прудов, водохранилищ со ста-
бильным санитарным режимом, рек.
Олигосапробная зона — зона чистой воды — характеризуется
окончанием окисления органических веществ, поступивших в во-
доем. Азот присутствует преимущественно в форме нитратов. Вода
242
может быть пересыщена кислородом. Разнообразен видовой состав
водных организмов (более 163). Количество бактерий невелико
(до 1000 в 1 мл).
Катаробная зона отличается крайне незначительным содержа-
нием органических веществ. К этой зоне относятся грунтовые, пла-
стовые, минеральные воды, очищенные природные воды, обрабо-
танные хлором, озоном.
Для более точного определения степени загрязнения водоемов
с учетом промышленных стоков система сапробности была уточне-
на Г. Либманном, который ввел в систему промежуточные зоны,
доводя их общее количество до семи. В классической системе сап-
робности не учитывается влияние токсичных веществ, попадающих
в водоем, на жизнедеятельность микроорганизмов — биоиндикато-
ров загрязнения, так как зоны сапробности характеризуются отно-
шением микроорганизмов к количеству растворенного кислорода.
Поэтому для оценки влияния токсичных веществ на процесс само-
очищения водоема В. И. Жадиным были предложены три шкалы
оценки загрязнения: по степени сапробности, токсобности и сапро-
токсобности. Токсобность характеризует свойство водных организ-
мов выживать в водах с различной степенью загрязнения токсич-
ными веществами. Аналогично зонам сапробности выделяются
поли-, мезо- и олиготоксобные зоны. Водоемы или их зоны, в кото-
рых невозможна жизнедеятельность водных организмов из-за вы-
сокой степени загрязнения токсичными примесями, относятся к
гипертоксобным. В. Сладочек предложил систему сапробности, в
которой выделяется 12 зон сапробности. Все эти зоны можно оха-
рактеризовать с биологических позиций (табл. 18).
§ 59. Процессы самоочищения водоемов
В результате спуска сточных вод происходит загрязнение во-
доема, проявляющееся в нарушении процессов жизнедеятельности
водных организмов и изменении качества воды. Сточные воды, по-
падая в водоем, способствуют увеличению мутности воды, измене-
нию ее химического состава, pH, уменьшению содержания раство-
ренного кислорода, снижению окислительно-восстановительного
потенциала, резкому увеличению количества микроорганизмов,
среди которых могут быть и патогенные. Нарушение нормального
режима водоема прежде всего проявляется в значительном погло-
щении растворенного кислорода. Сточные воды несут иногда по-
рядка нескольких тысяч мг/л взвешенных веществ. Производствен-
ные сточные воды часто содержат вещества, губительные для вод-
ных организмов. Бытовые сточные воды вносят в водоемы органи-
ческие соединения и являются источником патогенных микроорга-
низмов и гельминтов.
Под действием физических (разбавление, осаждение грубо-
дисперсных примесей), физико-химических (коагуляции коллои-
дов), химических (гидролиз, окислительно-восстановительные про-
цессы) и биологических процессов качество воды в водоеме спустя
243
Таблица 18. Система качества воды (система сапробности)
Категория Степень сапробности Знак зоны БПК. мгО2/.1 02. м г /л Н2.<?. м г /л Преобладающие макро-и микроорга- низмы
Катаробная . k — Разное 0
Лимносапробная Ксеносапробная X <^ ] значе- ние >8 0 Разные группы
(разбавленные сточные воды) Олигосапробная О <2,5 >6 0 гидробионтов То же
р-Мезосапробная 3 <ъ ^>4 0 Кладофоры, ры-
а-Мезосапробная а <10 0 бы, поденки и др. Стигеоклоннум, улотрикс, рыбы Тубифициды, сфе-
Полисапробная Р <50 >0,1 <0,1
Эусапробная Изосапробная i <400 .<^ 1 ротилус, осцил- лятория Цилиат — до
(концентриро- ванные сточ- ные воды) Метасапробная т <700 <1000 50 000 Флагеллят — до 20 000 Бактерий — масса Бесцветные жгу-
Гиперсапробная h <2000 <10 тиковые до 300 000 Цилиат — 0—5 Бактерий — масса Бактерий и микро-
Ультрасапробная и >120000 . <^о фита — масса Флагеллят — 0—5 Бактерий и микро-
Транссапробная Антисапробная а — — — фита — 0—5
Радиосапробная г — — — —
Криптосапробная с — — — —
некоторое время снова улучшается, т. е. происходит самоочищение
водоема. В процессе самоочищения от органических веществ ос-
новная роль принадлежит биологическим факторам. В водоемах
с проточной водой самоочищение осуществляется быстрее, чем в
водоемах с замедленным стоком. Это связано с лучшей аэрацией
воды и большей скоростью окисления органических веществ.
Так, в Волге и водохранилищах — в районах со свободным тече-
нием за сутки окисляется до 20—30% легкоокисляющихся ор-
ганических веществ, в районах с замедленным стоком—только
5-10%.
Донные отложения водоемов также подвергаются биохимиче-
скому распаду, но характер протекания этих процессов иной, чем
в толще воды. Донный ил является хорошим сорбентом органиче-
ских веществ, и на его поверхности происходят интенсивные био-
химические процессы. Выделяют три группы биоценоза дна:
1) накопители органического вещества;
244
Рис. 29. Изменение численности бак-
териального населения в процессе са-
моочищения:
1 — лето; ? — осень; 3 — зима
2) минерализаторы;
3) захоронители. В малозагрязненных водоемах население дна
представлено, в основном, минерализаторами (до 90%). В искусст-
венных каналах, облицованных щебнем, бетоном, создаются усло-
вия для развития гидробионтов-накопителей. Минерализация орга-
нических веществ в них затруднена. Условия для накопления орга-
нических веществ создаются и в водохранилищах. Так, в Учинском
водохранилище в результате биологического самоочищения в дон-
ных отложениях остается до 50% органических веществ.
С увеличением содержания органических веществ в водоеме
происходит интенсивное развитие микрофлоры, состоящей из мик-
роорганизмов-минерализаторов. Скорость и интенсивность процес-
са самоочищения находятся в
зависимости от температуры.
При температуре воды 10° С
наибольшее количество бакте-
рий обычно наблюдается через
50 ч, а при 20° С — через сут-
ки. В зависимости от химичес-
кой природы разлагающихся
веществ, присутствия токсич-
ных соединений и других фак-
торов максимум развития бак-
териальной микрофлоры может
несколько смещаться во време-
ни. На рис. 29 показано изме-
нение количества бактерий в
процессе самоочищения, проте-
кающего в реке в зависимости
от времени года.
Процесс самоочищения во-
доема при значительном загрязнении проходит через все зоны са-
пробности с соответствующей сменой биоценозов. Разложение ор-
ганических веществ в аэробных условиях осуществляется микроор-
ганизмами, окисляющими сложные органические соединения угле-
рода, азота, серы, фосфора, железа и других элементов в простые
неорганические формы. В анаэробных условиях образуются про-
дукты распада, которые могут обладать большей токсичностью, чем
исходные: например, меркаптаны, органические кислоты, сероводо-
род, метилированные производные ртути и др. Основная роль в
самоочищении водоема от органических биологически разлагаемых
веществ принадлежит бактериям. Кроме них в этом процессе участ-
вуют водоросли, грибы, простейшие.
Схема потоков энергии (кДж) в экологической системе Рыбин-
ского водохранилища, составленная на основе исследований, вы-
полненных Институтом биологии внутренних вод АН СССР, пока-
зывает характер взаимосвязи между различными группами гидро-
бионтов:
245-
Процесс биологического самоочищения водоема осуществляется
всем сообществом гидробионтов, образующих единую экологиче-
скую систему.
Загрязнение водоема токсичными примесями или любое нару-
шение целостности этой системы способствует замедлению или
полному прекращению процесса самоочищения. Активность биоло-
гического самоочищения зависит от воздействия факторов внешней
среды, но способность к самоочищению не безгранична. Фактора-
ми, определяющими протекание процесса самоочищения, являются
кислородный режим водоема, pH и окислительно-восстановитель-
ный потенциал. При концентрации кислорода менее 0,1—0,5 мг/л
изменяется направленность биохимических процессов.
Для нормальной жизнедеятельности водных организмов, участ-
вующих в процессе самоочищения, необходимо присутствие таких
биогенных элементов, как азот и фосфор. Практически полное от-
сутствие азота в воде наблюдается иногда в период массового раз-
вития водорослей, что создает неблагоприятные условия для бак-
териального окисления безазотистых органических веществ, по-
ступающих в водоем.
Роль зоопланктона в процессе самоочищения заключается в
снижении биомассы и продукции фитопланктона. Одновременно
продукты жизнедеятельности зоопланктона способствуют стимули-
рованию процесса фотосинтеза и приросту биомассы фитопланкто-
на. Таким образом, зоопланктон определенным образом регулиру-
ет количество фитопланктона в водоеме. Чем быстрее регенериру-
ются биогенные вещества, содержавшиеся в бактериях, в результа-
те жизнедеятельности животных организмов, тем более интенсивно
идет процесс самоочищения.
В процессе самоочищения важную роль играет высшая расти-
тельность. Водные растения способствуют улучшению санитарного
режима водоема, являются энергичными потребителями неоргани-
ческих форм биогенных элементов. Но при массовом их развитии
происходит засорение водоема (канала, водохранилища) и в пе-
риод отмирания наблюдается самозагрязнение водоема раститель-
ными остатками.
246
Большую работу по разложению органических веществ выпол-
няют микроорганизмы обрастаний. Роль водорослей заключается
в продуцировании кислорода и создании благоприятных условий
для микроорганизмов, окисляющих органические вещества. Кроме
того, некоторые водоросли сами используют органические соедине-
ния в качестве питательных веществ. Простейшие способствуют
удалению тонкой взвеси и коллоидных примесей, а также уничто-
жают бактерии. Количество бактерий, которое может перерабаты-
ваться простейшими, достигает 30 000 клеток для одной особи.
Коловратки, низшие ракообразные способствуют коагуляции и
осаждению взвешенных веществ. Микроорганизмы способны акку-
мулировать радиоактивные изотопы. Попадающие в водоем пести-
циды, ПАВ, соли тяжелых металлов способствуют ингибированию
процессов биологического самоочищения, а при достаточно высоких
концентрациях вызывают отмирание организмов биоценоза. Мощ-
ным биологическим фактором самоочищения водоемов и почвы яв-
ляется микроорганизм — Bdellovibrio bacteriovorus «бактериопияв-
ка», выделенный из почвы. Этот микроорганизм обладает высокой
бактерицидной активностью по отношению ко многим видам бак-
терий, в том числе и патогенным. В отличие от бактериофагов, ко-
торые тоже играют заметную роль в процессах бактериального
самоочищения водоемов, бделловибрио не обладает специфич-
ностью действия, использует для питания не только живых, но и
мертвых бактерий. Выделено несколько штаммов Bdellovibrio, из
которых все обладали литической активностью по отношению к
бактериям. Эти микроорганизмы сохраняются в речной воде до
200 сут.
В процессе самоочищения водоема некоторые легкоокисляю-
щиеся органические вещества могут подвергаться химическому
окислению. Но основная часть органических загрязнений удаляет-
ся из воды в результате биохимического окисления. Завершается
процесс самоочищения нитрификацией. Химическое окисление
продолжается несколько часов и зависит от природы органических
соединений. Прохождению этой стадии благоприятствует аэрация
воды. Одновременно с органическими веществами окисляются и
неорганические восстановители, если они присутствуют в воде.
Биохимическое окисление органических веществ длится несколь-
ко суток. Скорость его лимитируется концентрацией органических
веществ и зависит от напряженности действия факторов, оказы-
вающих влияние на жизнедеятельность организма. Если выразить
концентрацию органических примесей через БПК, то можно оха-
рактеризовать скорость разложения их через константу k. Это свя-
зано с тем, что биохимический распад основного количества
('—99%) органических соединений описывается уравнением реак-
ции первого порядка (1.32), пользуясь которым можно вычислить
константу скорости биохимического разложения органического ве-
щества k. Для расчета k пользуются методом определения БПК по
кратным срокам, т. е. через 2 и 4 или 3 и 6 сут. Подставляя значе-
ния БПК в формулу (1.32), получаем
247
t БПК2/ —ВПК, ’ v .
где t — время, сут; БПКг — ВПК для меньшего времени инкуба-
ции пробы.
На основании кинетического уравнения реакции 1-го порядка
можно вычислить также степень разложения органического веще-
ства, исходя из того, что одна и та же доля исходного вещества
разлагается за одинаковые интервалы времени.
Со сточными водами в водоем попадают и патогенные микро-
организмы. Поэтому процессы бактериального и вирусного само-
очищения водоемов имеют очень важное значение для предотвра-
щения вспышек инфекционных заболеваний. В сильно загрязнен-
ных сточных водах, особенно в присутствии токсичных соеди-
нений, патогенные микроорганизмы отмирают довольно быстро.
Время их выживания колеблется от нескольких часов до несколь-
ких суток. При разбавлении сточных вод действие неблагоприят-
ных факторов снижается. В отсутствие хорошо развитой микрофло-
ры и микрофауны в водоеме патогенные микроорганизмы могут
сохраняться в течение нескольких месяцев. Длительность выжива-
ния микроорганизмов возрастает при низкой температуре. Так, на-
пример, энтеропатогенные эшерихии в речной воде при температу-
ре от 4 до 6° С сохраняются в течение 192 сут, при 20° С—до
96 сут, при 37° С — до 68 сут.
Некоторые патогенные микроорганизмы сохраняют способность
к размножению даже в воде водоемов, образуют споры. Выделяют
следующие группы факторов, обусловливающих сроки выживания
патогенных микроорганизмов в воде: 1) биологические особенно-
сти возбудителей заболевания; 2) количество попадающих в водо-
ем микроорганизмов; 3) одновременное попадание в водоем био-
логического субстрата их естественного обитания; 4) особенности
водоема; 5) температурный фактор; 6) комплекс гидрометеорологи-
ческих факторов; 7) сопутствующая микрофлора и гидробионты
(Л. В. Григорьева).
При большом бактериальном загрязнении воды патогенными
микроорганизмами срок их выживания в водоеме увеличивается.
Этому же способствует поступление в водоем частиц биологическо-
го субстрата, на котором развиваются патогенные микроорганизмы.
Такие патогенные микроорганизмы, как возбудители дизентерии,
холерный вибрион, переносят замораживание, поэтому вспышка
инфекционных заболеваний может произойти в теплый период сле-
дующего года. В процессе самоочищения водоемов и при биологи-
ческой очистке сточных вод значительную роль играют бактерио-
фаги. Сильным бактерицидным действием обладают продукты
жизнедеятельности некоторых водорослей и грибов.
Патогенные микроорганизмы поедаются простейшими: реснит-
чатыми инфузориями, амебами, коловратками, ракообразными и
червями. После воздействия бактериофагов или других гидробион-
тов на патогенные микроорганизмы иногда наблюдается появле-
248
при рассмотрении действия биологических факторов на выживае-
мость патогенных микроорганизмов.
§ 60. Влияние микроорганизмов и продуктов
их жизнедеятельности на качество воды
Вещества, образующиеся в процессе жизнедеятельности микро-
организмов, как и сами микроорганизмы, могут стать причиной
ухудшения качества воды, особенно в водоемах с замедленным сто-
ком. Возможны также нарушения в работе гидротехнических со-
оружений. Наиболее частыми проявлениями жизнедеятельности
микроорганизмов, которые затрудняют процесс самоочищения в во-
доемах, эксплуатацию водозаборов и систем охлаждения, вызывают
изменение качества воды, являются цветение водоемов, обрастания,
появление запахов и привкусов у воды. Формирование водохрани-
лищ связано с уменьшением скорости течения воды, в результате
чего гидрохимический режим крупных водохранилищ становится
близким к режиму озер. При зарегулировании речного стока время
прохождения воды от истока до устья возрастает в 10—15 раз. Так,
в Волге до зарегулирования стока вода доходила от Рыбинска до
Волгограда в половодье за 30 сут, а в межень — за 50. После фор-
мирования каскада водохранилищ время прохождения воды на
этом участке увеличилось до 450—500 сут. Замедление водообмена
в речной системе сопровождается значительными изменениями гид-
рохимического и гидробиологического режима. Водохранилища ра-
ботают, как гигантские отстойники, поэтому в них наблюдается
концентрирование загрязнений. Поступление органических и ток-
сичных соединений, биогенных элементов способствует возникнове-
нию условий для эвтрофикации водоема, нарушению процесса са-
моочищения, зарастанию, т. е. массовому развитию высшей водной
растительности.
Для водохранилищ, как и для озер, характерным является мас-
совое развитие водорослей (цветение водоема). Это явление можно
рассматривать как новый фактор самозагрязнения водоема. В глу-
боких водоемах цветение обычно происходит в верхних слоях, в
мелководных — по всей глубине. При цветении преобладает один
или два вида микроорганизмов. Цветение длится некоторое время,
а затем пропадает. Оно может быть вызвано различными водорос-
лями. В начале весны наблюдается цветение диатомовыми водо-
рослями, при этом вода приобретает желтовато-коричневый цвет.
Наиболее распространенными диатомовыми водорослями, вызыва-
ющими цветение, являются астерионелла (Asterionella), синедра
(Synedra), мелозира (Melosira). В середине лета часто наблюдает-
ся цветение водоемов синезелеными водорослями. Характерными
представителями синезеленых водорослей, вызывающими цветение,
являются анабена (Anabaena), осциллятория (Oscillatoria), которые
придают воде голубовато-зеленый цвет и неприятный привкус и за-
пах. Обильное развитие водорослей резко ухудшает условия суще-
9—1244 249
ствования других водных организмов. Так, при цветении воды ко-
личество бактерий-сапрофитов резко снижается. При массовом от-
мирании водорослей после цветения наблюдается резкое увеличе-
ние числа бактерий-минерализаторов. Водоросли выделяют особые
антибиотические вещества, токсичные не только для бактерий, но и
для других гидробионтов, а также для домашних животных и чело-
века. Образующиеся значительные скопления водорослей вызы-
вают закупорку фильтров, что усложняет процесс очистки воды.
Цветение, вызываемое синезелеными водорослями, является осо-
бенно значительным по размерам и влиянию на режим водохрани-
лищ. Массовому развитию сине-зеленых водорослей способствует
повышение концентрации биогенных элементов, достаточно высокая
температура воды и инсоляция, ухудшение кислородного режима.
Так, например, цветение синезелеными водорослями в водохрани-
лищах Днепра и Дона продолжается 3—4 месяца и занимает по
объему 80—90% акватории. Количество биомассы водорослей дос-
тигает 60—100 г/м3. В Волжском каскаде водохранилищ цветение
наблюдается в течение 1—3 месяцев, захватывая 30—80% аквато-
рии с развитием биомассы водорослей от 4 до 33 г/м3. Основная
масса синезеленых водорослей развивается в водоеме на глубине
2—3 м. В летний период синезеленые водоросли в водохранилищах
могут составлять до 90% общей массы фитопланктона. В местах
скопления синезеленых водорослей обитают только коловратки. Во
время нагонов ветром в прибрежной зоне водохранилищ образуется
сплошная масса водорослей, в которой биомасса синезеленых водо-
рослей достигает сотен килограммов на 1 м3. Синезеленые водорос-
ли проявляют токсичные свойства, природа которых еще не изуче-
на. В 1 кт сырой массы водорослей обнаруживают до 14—15 мг фе-
нолов.
Резкое нарушение санитарного режима водоема наблюдается и
при массовом отмирании водорослей. При распаде синезеленых во-
дорослей в результате деятельности протеолитических и аммонифи-
цирующих бактерий повышается содержание аммонийного азота,
диоксида углерода, резко снижается содержание растворенного кис-
лорода, что является причиной массовой гибели рыб. При массовом
развитии водорослей затрудняется работа водозаборных сооруже-
ний, ухудшается фильтрация воды. Малые размеры клеток водоро-
слей (4—7 мкм) делают неэффективным примененье микропроце-
живания. Водоросли образуют на поверхности фильтров слизистую,
не проницаемую для воды пленку, поэтому необходима частая про-
мывка фильтров. Запахи и привкусы, появляющиеся у воды в пери-
од цветения, при применении обычной технологической схемы очи-
стки питьевой воды не устраняются.
Профилактика цветения заключается в предупреждении загряз-
нения водоема биологически разлагаемыми органическими вещест-
вами, биогенными элементами. Биологические методы борьбы с цве-
тением заключаются в создании искусственных поверхностей обрас-
тания в зоне поступления воды в водохранилище для интенсифика-
ции процессов биологического самоочищения. Практикуется также
250
создание условий для развития высшей водной растительности, яв-
ляющейся потребителем биогенных веществ.
Физические методы борьбы с цветением заключаются в искусст-
венном замутнении воды глиной, аэрации, применении всасыва-
ющих устройств для удаления водорослей. Для выделения водорос-
лей в системах технического водоснабжения, в небольших водоемах
и резервуарах возможно применение коагуляции сульфатом алю-
миния. Химические методы борьбы с цветением заключаются в об-
работке водоемов пестицидами, сульфатом меди. Токсичность этих
соединений для других водных организмов ограничивает использо-
вание их в широких масштабах. Перспективным методом борьбы с
цветением водоемов является биологический, основанный на ис-
пользовании микроорганизмов-антагонистов водорослей. Выделено
25 антагонистов синезеленых водорослей. В днепровских водохрани-
лищах выделены микроорганизмы (альгофаги), лизирующие сине-
зеленые водоросли в течение 2—6 сут. Определенную роль играет
прогнозирование времени и интенсивности цветения.
Жизнедеятельность микроорганизмов создает помехи в работе
очистных сооружений, которые состоят в появлении привкусов и
запахов у воды. Химический состав соединений, обусловливающий
появление запаха, зависит от вида микроорганизма, условий его
жизнедеятельности. Так, актиномицеты в условиях затрудненной
аэрации придают воде землистый запах. Запах воды может вызы-
ваться также массовым развитием бактерий. В зависимости от об-
разующихся метаболитов запахи могут быть также различными:
ароматический, сероводородный, плесневый, гнилостный. В период
массового развития микроорганизмов-продуцентов запахов и при-
вкусов мясо рыб также приобретает привкус. Основная роль в воз-
никновении запахов воды принадлежит аминам, органическим кис-
лотам, фенолам, эфирам, альдегидам, кетонам. Для удаления за-
пахов и привкусов, вызываемых микроорганизмами, необходимо
применение дополнительных методов очистки воды.
При массовом развитии в водохранилищах, каналах, водоводах
высшей водной растительности (макрофитов) происходит их зарас-
тание. В результате снижается пропускная способность сетей и ка-
налов, наблюдается загрязнение водоема. В зарослях создаются ус-
ловия для отложения личинок кровососущих насекомых, возника-
ют трудности при ловле рыбы. Вторичное загрязнение водоема про-
дуктами распада макрофитов сопровождается нарушением кисло-
родного режима, сменой биоценозов, возникновением благоприят-
ных условий для массового развития микроорганизмов планктона,
обрастаний. Для борьбы с зарастанием водоемов рекомендуются
различные физические методы: скашивание растений, затенение во-
доема путем посадки деревьев по берегам канала. Химические ме-
тоды борьбы с зарастаниями основаны на применении тех же сое-
динений, что и при борьбе с цветением. Биологические методы за-
ключаются в разведении таких травоядных рыб, как белый амур,
толстолобики, и животных (ондатры, нутрии).
Работе сооружений технического и питьевого водопровода нано-
9*
251
сят большой вред микроорганизмы обрастаний. Они вызывают за-
сорение и обрастание водозаборных сооружений, внутренних сте-
нок труб, вызывая их сужение. В теплообменных установках нару-
шается режим теплообмена. Попадание в воду живых или погиб-
ших микроорганизмов повышает концентрацию взвешенных ве-
ществ, появляются запахи и привкусы. Микроорганизмы обраста-
ний часто вызывают или усиливают коррозию металлов. В морских
водах, особенно теплых, обрастания развиваются быстрее и интен-
сивнее, чем в пресных. Если микроорганизмы выделяют вещества,
способные вызывать или усиливать коррозию металла, например
кислоты, то разрушение его может происходить на некотором уда-
лении от места массового развития микроорганизмов. Продукты
выделения микроорганизмов, например диоксид углерода, могут
вызывать коррозию бетона. При транспортировке сточных вод по
трубам создаются условия для развития анаэробов, например бак-
терий, восстанавливающих сульфаты, что сопровождается образо-
ванием таких коррозионных агентов, как сероводород. Некоторые
виды плесневых грибов (Penicillium, Aspergillus) и актиномицетов
вызывают коррозию натурального каучука.
На формирование обрастаний, их характер и интенсивность
влияют химический состав воды, ее температура, скорость потока,
количество растворенного кислорода, содержание питательных ве-
ществ, свойства материала, контактирующего с водой. В чистых во-
дах обрастания, чаще всего обусловлены железобактериями, серо-
бактериями, водорослями, моллюсками, губками, мшанками. При
малом количестве питательных веществ в воде микроорганизмы
планктона тоже вызывают обрастания. Теплообменная аппаратура,
гидротехнические сооружения также подвержены обрастаниям же-
лезобактериями. Присутствие сероводорода в воде создает условия
для развития серобактерий, окисляющих его до серы или сульфа-
тов. Для полисапробной зоны характерно развитие зооглейных
форм бактерий, образующих слизистые скопления.
Максимальное развитие организмов обрастаний наблюдается
при температуре, близкой к оптимальной для данной группы. Так,
некоторые виды железобактерий развиваются при низкой темпера-
туре воды. Например, для галионеллы оптимальная температура
7—10° С. Большинство же микроорганизмов обрастаний является
мезофилами с температурными оптимумами 15—20 и 35—37° С. Из-
менение температуры сопровождается сменой микроорганизмов
обрастаний. При понижении температуры в загрязненных водах
преобладают нитчатые бактерии и грибы. Летом грибы в обраста-
ниях практически отсутствуют. Быстрее всего развиваются в обра-
станиях нитчатые и зооглейные бактерии.
При использовании воды, загрязненной органическими вещест-
вами, для охлаждения промышленных установок обрастания зоо-
глейными бактериями могут развиваться в теплообменной аппара-
туре уже через 10 ч, нитчатыми — через 36 ч. Основные организмы
обрастаний: зооглейные (Zooglea ramigera), нитчатые бактерии
(Sphaerotilus natans, Cladothrix dichotoma), нитчатые формы желе-
252
зобактерий — лептотрикс (Leptothrix), кренотрикс (Crenothrix), од-
ноклеточные (гелионелла); грибы — мукор, лептомитус, фузариум,
простейшие-—жгутиковые, инфузории; мшанки и гидроидные поли-
пы; черви (нематоды и олигохеты); моллюски (дрейссена, мидия,
бптиния, вивипарус и др.); ракообразные (балянус). В водах, со-
держащих сероводород, развиваются серобактерии (Beggiatoa
alba), представляющие собой нити, плавающие в воде (рис. 30).
Эти бактерии окисляют сероводород до свободной серы, которая
откладывается в их клетках. При значительной концентрации серо-
водорода образуются пучки этих нитей, вызывающие механическую
закупорку труб. Содержание в охлаждающей воде повышенного
в
Рис. 30. Различные формы серобактерий:
а — р. Beggiatoa; б — р. Thiobacillus; в — р. Thiotrix
количества сульфатов создает предпосылки для массового развития
бактерий, восстанавливающих сульфаты до сероводорода и сульфи-
дов, которые вызывают сильную коррозию металлических труб. Об-
растания в водопроводной сети и теплообменной аппаратуре чаще
всего вызываются железобактериями. Эти бактерии относятся к ав-
тотрофам, и поэтому они живут в водах, содержащих незначитель-
ное количество органических веществ (с окисляемостью более
5 мг О2/л). Они поглощают из воды соединения железа (II) и окис-
ляют их до железа (III) (рис. 31):
4Fe (НСО3)2 + О2 + 2Н2О = 4Fe(OH)3 + 8СО2
Среди железобактерий встречаются нитчатые формы, выделя-
ющие образующийся гидроксид железа (III) в слизистую оболочку,
общую для всей нити; и одноклеточные формы, у которых гидрок-
сид железа откладывается в виде спиральных нитей вне клетки.
Образующийся гидроксид железа (III) снижает вкусовые качества
воды. При концентрации железа в воде выше 0,8—1,0 мг/л и при
благоприятных условиях развивающиеся в трубах железобактерии
могут вызвать полное зарастание внутренней поверхности. Железо-
бактерии могут развиваться и при содержании 0,1—0,3 мг/л железа
в воде, т. е. в водопроводной сети. Кроме того, они способствуют
быстрому исчезновению остаточного хлора и тем самым снижают
253
S
а.
его обеззараживающее действие. Прикрепленные водоросли не при-
носят ощутимого вреда подводным сооружениям. Значительно боль-
ший вред оказывают водоросли планктона, вызывающие во время
цветения водоема закупорку труб. Из животных наибольший вред
приносит моллюск дрсйссена, вызывающий обрастание водоприем-
ных сооружений и водоводов.
Наиболее эффективные мероприятия по борьбе с обрастаниями
заключаются в изменении нежелательных свойств воды (обезжеле-
зивание, удаление сероводорода, сниже-
ние возможности поступления органических
веществ), предотвращение цветения и мас-
сового развития высшей водной раститель-
ности в водоеме — источнике водоснабже-
ния.
Для удаления обрастаний в трубопро-
водах применяются промывка горячей
(45° С) водой и различные виды химическо-
го воздействия: хлорирование, введение ве-
ществ, вызывающих отмирание микроорга-
низмов обрастаний.
Для борьбы с биологическими обраста-
ниями, вызываемыми зооглейными, нитча-
тыми бактериями, грибами, дрейссеной, при-
меняют хлорирование воды. Развитие водо-
рослей предотвращается обработкой воды
сульфатом меди (II) (0,2—0,4 мг/л Си2+).
Она может осуществляться как профилак-
тическая мера в самом водоеме или исполь-
зуется для предотвращения обрастаний во-
дорослями в градирнях, прудах-охладите-
лях, брызгальных бассейнах. Если обрастания вызываются одно-
временно бактериями и водорослями, обработка воды сульфатом
меди (II) сочетается с хлорированием.
В воде, поступающей на водопроводные очистные сооружения,
содержатся микроорганизмы, для которых водоем является естест-
венной средой обитания (бактерии — сапрофиты и автотрофы, во-
доросли, простейшие и др.). В ней могут обнаруживаться и пато-
генные формы. Часть из них, сорбированная частицами грубодис-
персных примесей, выделяется из воды в процессе отстаивания.
Важным условием улучшения качества воды при этом является
присутствие достаточного количества растворенного кислорода.
Появление участков с кислородным дефицитом обусловливает раз-
витие анаэробных процессов разложения органических веществ, ко-
торые сопровождаются образованием соединений с неприятным за-
пахом. Предварительное хлорирование воды перед обработкой спо-
собствует снижению численности микроорганизмов. При коагули-
ровании воды микроорганизмы сорбируются растущими хлопьями
и переходят в осадок. Но и после фильтрования в воде может оста-
ваться до 10% жизнеспособных бактериальных клеток. Возмож-
Рис. 31. Различные фор-
мы железобактерий:
а — Crenothrix ferruginea;
б — Leptothrix ochraceae
254
ность присутствия патогенных бактерий и вирусов делает необходи-
мым обеззараживание воды.
Высокая эффективность задержки бактерий наблюдается в кон-
тактных осветлителях. Фильтрование воды через осветлитель сов-
мещается с контактной коагуляцией взвешенных частиц. При этом
происходит задержка микроорганизмов вместе с грубодиспергиро-
ваннымп и коллоидными примесями. В табл. 19 приведены данные
о снижении общего количества бактерий и Bacteria coli.
Таблица 19. Эффективность задержки бактерий в контактном осветлителе
Наименование объектов Периоды испытаний Эффект задержки бактерий, % Примечание
В. coli от общего количества бактерий
г. Москва Зима Весенний паводок Лето Осенний паводок 96,0 96,0 98,5 43,0 95,0 44,0 92,0 Осветлитель рабо- тал без коагу- лянта
г. Ленинград Зима Весенний паводок Лето 90,0 99,6 99,6 45,0 84,5 88,0 —
Работа фильтров медленного действия основана на образовании
биологической пленки. В период созревания фильтра на зернах за-
грузки постепенно формируется активная пленка, состоящая из ог-
ромного количества бактерий, водорослей и других микроорганиз-
мов. Фильтрование воды сопровождается освобождением ее не толь-
ко от взвешенных и коллоидных примесей, но и от микроорганиз-
мов. Обеззараживание воды после обработки—мероприятие, гаран-
тирующее от попадания в питьевую воду патогенных микроорганиз-
мов и обеспечивающее качество воды по бактериологическим пока-
зателям.
При массовом развитии диатомовых водорослей во время цвете-
ния затрудняется процесс фильтрования воды, так как они образу-
ют на поверхности фильтра слой, способствующий резкому замед-
лению скорости прохождения воды.
Глава IX. ЗНАЧЕНИЕ МИКРООРГАНИЗМОВ В ПРОЦЕССАХ
ОЧИСТКИ СТОЧНЫХ ВОД
Полная биологическая очистка сточных вод является эффектив-
ным способом предотвращения загрязнения водоемов биохимически
разлагаемыми органическими соединениями. Сущность ее заключа-
ется в разложении органических веществ, содержащихся в сточных
водах, в результате жизнедеятельности микроорганизмов. На очист-
ных сооружениях городской канализации биологической очистке
подвергаются бытовые и производственные сточные воды. Послед-
ние по мере необходимости, например при наличии токсичных сое-
динений, подвергаются предварительной обработке.
Эффективность работы станций биологической очистки опреде-
ляется подбором оптимальных условий для жизнедеятельности мик-
роорганизмов биоценозов очистных сооружений. Способность мик-
роорганизмов потреблять соединения разнообразного химического
состава и высокие адаптационные возможности к изменяющимся
условиям внешней среды позволяют очищать сточные воды от орга-
нических соединений, содержащихся в бытовых сточных водах, а
также от биохимически разлагаемых веществ, поступающих с про-
изводственными сточными водами.
Разложение органических веществ в процессе биологической
очистки может происходить в аэробных и анаэробных условиях.
Аэробные процессы обычно используются для окисления загрязне-
ний, остающихся в сточной воде после отстаивания, а именно: раст-
воренных, коллоидных и тонкодиспергированных органических при-
месей, не выделившихся при отстаивании. Окисление осуществляет-
ся аэробными микроорганизмами в естественных (биологические
пруды, поля орошения и поля фильтрации) условиях и на искусст-
венных очистных сооружениях (аэротенки, био- и аэрофильтры).
В аэротенках, окислительных прудах воспроизводятся процессы са-
моочищения, протекающие в водоемах. В биофильтрах, аэрофильт-
рах, на полях орошения и полях фильтрации воспроизводятся поч-
'венные процессы самоочищения. Эффективность удаления органи-
ческих веществ определяется технологическими особенностями
-очистных сооружений и выбором оптимальных условий для жизне-
деятельности микроорганизмов. Оптимальная нагрузка по органи-
ческим веществам, температура, pH, количество растворенного
кислорода, отсутствие токсичных примесей определяют эти ус-
ловия.
Анаэробные биохимические процессы используются для разло-
жения осадка сточных вод и иногда в качестве предварительной
ступени очистки концентрированных производственных сточных
вод. Разложение органических веществ идет с образованием мета-
на, диоксида углерода, азота, сероводорода, водорода и продуктов
неполного распада органических соединений. Этот способ обра-
ботки осадка сточных вод называется сбраживанием, которое
осуществляется в септиках, двухъярусных отстойниках и метан-
тенках.
256
§ 61. Окисление органических веществ
в аэробных условиях
Аэробные процессы разложения безазотистых органических ве-
ществ. Процессы окисления чрезвычайно разнообразны. В присут-
ствии кислорода биохимическому распаду подвергаются углеводы,
жиры, органические кислоты, углеводороды и другие соединения.
Органические вещества имеют предварительный и основной пути
окисления. Предварительное разрушение органических соединений
происходит в результате гидролиза или окисления их до таких ве-
ществ, которые затем вступают на основной путь биохимического
окисления.
Окисление целлюлозы и пектиновых веществ.
Разложение целлюлозы начинается с ферментативного гидролиза
вначале до дисахарида целлобиозы, а затем — до глюкозы:
целлюлаза
(С6Н10О5)п + 1/2 иН2О-----> 1/2 лС12Н220ц
целлобиоза
целлобиаза
1/2 лС12Н220ц + 1/2 пН2О * пС6Н120б
глюкоза
Образующаяся при окислении глюкозы в результате фермента-
тивных реакций пировиноградная кислота вступает в цикл трикар-
боновых кислот (Кребса), в результате чего выделяется диоксид
углерода, а освобождающиеся атомы водорода в конце дыхательной
цепи связываются кислородом в воду. Суммарно схему этих слож-
ных ферментативных процессов с учетом только исходного вещест-
ва и конечных продуктов можно записать следующим образом:
С6Н12О6 + 6О2 = 6СО2 + 6Н2О.
Разложение целлюлозы в аэробных условиях осуществляется
рядом бактерий, актиномицетами и грибами. Чаще всего это бакте-
рии из рода Cytophaga, а также бактерии рода Cellvibrio. Из акти-
номицетов способностью к окислению целлюлозы обладают роды
Micromonospora, Streptomyces, из грибов — Fusarium, Aspergillus и
некоторые другие. Грибы медленно разлагают целлюлозу и часто
при этом наблюдается неполное окисление с образованием лимон-
ной, щавелевой, уксусной, муравьиной и других кислот.
Пектиновые вещества, входящие в состав оболочек раститель-
ных клеток и заполняющие межклеточное пространство, по своей
химической природе являются сложными полимерными соединени-
ями. В аэробных условиях они разрушаются некоторыми плесневы-
ми грибами. Окисление пектиновых веществ тоже идет в две ста-
дии. Сначала в результате ферментативного гидролиза образуются
менее сложные водорастворимые соединения, которые затем окис-
ляются до диоксида углерода и воды.
Окисление жиров. Разложение жиров начинается с их гид-
ролиза под влиянием фермента — липазы, содержащейся во многих
257
микроорганизмах—пигментных бактериях, актиномицетах, плесне-
вых грибах родов Aspergillus и Penicillium:
СН2—О—С—Ri
6 СН,—ОН Ri—СООН
4-ЗНоО ;
СН—О—С—R2 ------___> СН—ОН - R-2—СООН
О CIH-OH R3—СООН
п U гл п п г.шцерин жирные
ЫП2—и—С—Кз кисло 1Ы
II
о
Трехатомпый спирт—глицерин быстро окисляется до диоксида уг-
лерода и воды. Высшие жирные кислоты, не растворимые в воде,
окисляются значительно труднее и медленнее. Под воздействием
микроорганизмов в аэробных условиях они окисляются до диоксида
углерода и воды:
С17Н35СООН + 26О2-> 18СО2 18Н2О
стеариновая
кислота
Окисление жирных кислот идет через стадии образования дру-
гих кислот, которые вступают в цикл Кребса. Органические кисло-
ты могут использоваться микроорганизмами в качестве источника
энергии и как материал для биосинтеза. Исключение составляет
муравьиная кислота, которая участвует только в катаболитическом
процессе.
Окисление предельных углеводородов (алка-
нов). Некоторые микроорганизмы в аэробных условиях разлагают
углеводороды с различной длиной углеродной цепи, от метана до
сложных углеводородов нефти. Группа метаноокисляющих микро-
организмов Methanomonas под действием ферментов трансформи-
рует метан в диоксид углерода и воду. Углеводороды с большим
числом углеродных атомов окисляются другими группами бактерий
по различным механизмам. Основной путь можно представить схе-
мой
+0 +0 +0 +0
R—СН3---> R—СН2ОН----> R—СНО-----> R—СООН----> С02 + Н20
спирт альдегид карбоновая
кислота
Последняя стадия заключается в преобразовании кислот в цикле
Кребса до простых продуктов, при полном окислении до диоксида
углерода и воды. Наиболее энергично окисляют алканы бактерии
Pseudomonas, микобактерии, актиномицеты. Разложение углеводо-
родов может происходить и в анаэробных условиях в присутствии
веществ, способных отдавать кислород.
Окисление ароматических углеводородов и их
производных. Многие соединения ароматического ряда, напри-
мер фенол и его гомологи, являются токсичными по отношению к
микроорганизмам. Но существуют специфические микроорганизмы
258
из групп микобактерий, бацилл, способные разлагать эти соедине-
ния при определенной концентрации их в окружающей среде. На
деятельности этих микроорганизмов основан биохимический метод
обесфеноливания воды. Наиболее полно и быстро окисление этих
соединений происходит в воде при достаточном количестве кислоро-
да. В этих условиях соединения ароматического ряда разлагаются
до диоксида углерода и воды без образования промежуточных про-
дуктов.
Поскольку все биохимические процессы проходят при участии
ферментов, то при поступлении органических веществ иного хими-
ческого состава и строения жизнедеятельность микроорганизмов
из-за токсичного действия может полностью нарушаться или же в
течение некоторого времени происходит приспособление (адапта-
ция) микроорганизмов к изменившимся условиям. Следствием это-
го является выработка новых ферментов, под действием которых
начинает разлагаться новый вид органического загрязнения. В за-
висимости от химической природы загрязнения, его концентрации,
количества микроорганизмов, скорости их размножения и других
внешних факторов период адаптации может длиться от нескольких
дней до нескольких месяцев.
Постоянными компонентами городских сточных вод являются
поверхностно-активные вещества. По отношению к биохимическому
окислению они делятся на «мягкие» и «жесткие». Жесткие ПАВ
практически не подвергаются биохимическому окислению. Способ-
ность ПАВ к биохимическому окислению определяется их химиче-
ской структурой. Легко подвергаются биохимическому окислению
анионные ПАВ— алкилсульфаты с нормальной углеводородной це-
пью. ПАВ, имеющие разветвленную углеводородную цепь, содержа-
щую бензольное ядро, и неионогенные ПАВ наиболее устойчивы к
биохимическому окислению. Способность к биохимическому окис-
лению ПАВ может быть повышена при адаптации микроорганиз-
мов, которую следует начинать с введения малых количеств ПАВ
(порядка 5 мг/л).
Процессы окисления азотсодержащих органи-
ческих веществ. Азотсодержащие органические соединения
широко распространены в природе, так как азот входит в состав
белков. Белки непосредственно не могут усваиваться микроорганиз-
мами. Однако некоторые группы микроорганизмов обладают спо-
собностью выделять во внешнюю среду протеолитические экзофер-
менты, которые гидролизуют белки с образованием соединений с
меньшей молекулярной массой, способных проникать через оболоч-
ку клетки.
ч-н2о +н20
белки----> полипептиды-» аминокислоты
Образовавшиеся аминокислоты под влиянием других микроорганиз-
мов, содержащих фермент трансаминазу, подвергаются дезамини-
рованию, т. е. разложению с выделением аммиака. Оно осуществля-
259
ется различными путями. Прямое дезаминирование:
R — СН2 — СН — СООН — R — СН=СН—COOH+NH3
I непредельная кислота
NH2
Окислительное дезаминирование:
+1 '2 О ?
R—СН—СООН---------’ R—С—COOH+NH3
I II
nh2 о
КС!ОКИСЛО!а
Восстановительное дезаминирование:
+2Н
К _ СН — СООН----> R — СН2 — СООН + NH3
nh2
Процесс разложения продуктов гидролиза белковых соединений
микроорганизмами с образованием аммиака называется аммонифи-
кацией, а микроорганизмы — аммонификаторами.
Окисление белковых соединений происходит до конца с образо-
ванием аммиака, диоксида углерода, воды. Если в белках содер-
жится сера, то в качестве промежуточных соединений образуются
еще меркаптаны (тиоспирты), а при полном распаде образуется
сероводород. Наиболее распространенные аэробные возбудители
разложения белков: Bacterium fluorescens, Bacillus subtilis, Bacil-
lus mycoides. Кроме того, разложение белковых соединений может
вызываться актиномицетами и многими грибами. Нуклеопротеиды,
содержащие нуклеиновые кислоты, связанные с аминокислотными
остатками, разлагаются с образованием углеводов — рибозы и де-
зоксирибозы, азотистых органических оснований и фосфорной кис-
лоты.
Разложение карбамида. Карбамид (мочевина) NH2—
—СО—NH2 представляет собой продукт белкового обмена у живот-
ных и человека. Под действием фермента уреазы карбамид гидро-
лизуется уробактериями с образованием аммиака и диоксида угле-
рода:
NH2 — С — NH2 + 2Н2О -> 2NH3 + СО2 + Н2О (NH4)2 СО3
Разложение карбамида осуществляется в основном аэробными мик-
роорганизмами. Образующийся карбонат аммония подвергается
гидролизу с образованием гидрокарбоната аммония и гидроксида
аммония. Наличие этих соединений обусловливает буферные свой-
ства сточной жидкости.
Н и т р и ф и к а ц и я. Аммиак, образовавшийся в результате раз-
ложения азотсодержащих органических соединений, используется в
качестве энергетического материала нитрозобактериями. Энергия,
необходимая для жизнедеятельности нитрозобактерий, получается
260
в результате ферментативных реакций окисления аммиака и солей
аммония кислородом:
2ХН3 + ЗО2 = 2HNO2 + 2Н2О; (хМН4)2СО3 + ЗО2 = 2HNO2 + СО2 + ЗН2О
Этот процесс осуществляется только в аэробных условиях. Энергия,
выделяющаяся при этом, расходуется на синтез органического ве-
щества клетки, так как нитрозобактерии — автотрофные организ-
мы. Окисление аммонийного азота начинается только после полно-
го разложения биологически разлагаемых органических примесей.
Наиболее энергичными окислителями аммиака являются бактерии
рода Nitrosomonas, представляющие собой подвижные клетки
овальной формы с длинным жгутиком.
Азотистая кислота в присутствии достаточного количества кис-
лорода окисляется другой группой микроорганизмов — нитробакте-
риями до азотной кислоты: 2HNO2 +О2 = 2HNO3. Образующиеся
кислоты нейтрализуются гидрокарбонатами, находящимися в воде
водоемов. Поэтому на первой стадии нитрификации образуются со-
ли азотистой кислоты нитриты, а на второй — соли азотной кислоты
нитраты. Этот процесс протекает при энергичной аэрации. Возбуди-
телями процесса служат бактерии рода Nitrobacter, представля-
ющие собой мелкие палочки длиной около 1 мкм, и диаметром
0,5 мкм, образующие зооглеи.
Процессы окисления аммиака и азотистой кислоты называются
нитрификацией, а бактерии — нитрифицирующими или нитрифика-
торами. Для нормального протекания процесса нитрификации необ-
ходимо определенное значение pH. Первая стадия имеет оптимум
pH 8,5, а вторая — 8,3—9,3. Образующиеся при нитрификации азо-
тистая и азотная кислоты могут вызывать разрушение подводных
бетонных сооружений.
Разложение сер у содержащих соединений. Сера
входит в состав некоторых белков. При гидролитическом распаде
белков она восстанавливается до сероводорода, который представ-
ляет собой токсичное соединение для многих групп микроорганиз-
мов. Но в водоемах и почве встречаются серобактерии, окисляющие
восстановленные соединения серы до свободной серы и сульфатов.
Эти бактерии живут при высоких концентрациях сероводорода в
окружающей среде. Сероводород для них служит источником энер-
гии для синтеза органического вещества.
Окисление сероводорода осуществляется в два этапа. Вначале
при достаточном количестве сероводорода образуется свободная
сера, которая откладывается в клетках серобактерий в виде вклю-
чений: 2H2S + O2==2H2O + 2S. Затем при снижении концентрации
сероводорода в окружающей среде идет окисление серы до сульфа-
тов: 2SH-3O2 + 2H2O = 2H2SO4. К серобактериям, окисляющим серо-
водород через образование свободной серы, относятся бесцветные и
пурпурные серобактерии. Из бесцветных серобактерий чаще всего
встречается Beggiatoa alba. Это бактерии, образующие длинные,
плавающие нити диаметром от 8 до 50 мкм и вызывающие обрас-
тания. Пурпурные серобактерии (сем. Thiorodaceae) содержат пиг-
261
мент бактериопурпурин. Они способны получать энергию путем фо-
тосинтеза. Группа тионовых бактерий окисляет сероводород
непосредственно до сульфатов. Из них наиболее часто встречается
небольшая подвижная палочка Thiobacillus thioparus.
Превращение соединений фосфора. Фосфор входит
в состав таких жизненно важных соединений, как нуклеиновые кис-
лоты, липоиды, АТФ. При разложении этих соединений освобожда-
ется фосфорная кислота, которая образует с ионами Са2 3+ и Mg2+
труднорастворимые фосфаты. Перевод их в растворимые соединения
(гидро- дигидрофосфаты) называется мобилизацией фосфатов. Он
осуществляется иод действием кислот (азотной, серной), образу-
ющихся в результате жизнедеятельности некоторых серобактерий и
нитрификаторов:
Са3(РО4)2 4-2H2SO4 = C:i(H2PO4)2 4- 2CaSO4
Са3(РО4)2 +4HNO.3 Са(Н2РО4)2 + 2Ca(NO3)2
Общая направленность аэробных процессов. Рассмотренные пу-
ти окисления основных групп органических соединений являются не-
сколько условными, так как при наличии нескольких групп окисля-
ющихся органических веществ разложение каждой из них будет за-
висеть от химической природы и концентрации других компонентов.
Общую направленность процессов аэробного окисления, осуществ-
ляемых бактериями-гетеротрофами, можно условно описать следу-
ющими схемами:
1) окисление органических веществ в процессе энергетического об-
мена:
4- (х + 0,25г/ — 0,5г)О2^л'СО2 4- 0,5г/Н2О — А//
/ у z 3 \ /у — 3 \
CxHaOzN + (х + у- - у - — \О2^хСО2 + I у- 1Н2О Щ NH3 - ДЯ
2) синтез биомассы клеток микроорганизмов в процессе конструк-
тивного обмена:
/?(C vHyOz) Д- nNH3 4- п(х 4-0,25г/ —0,5г —5)О2^-/г(С5117NO2) 4-
4- п(х —5)СО2 4-0,5п(у —4)Н2О 4- ДЯ
3) окисление органических веществ, входящих в состав клеток мик-
роорганизмов:
«(CgHyNC^) 4- 5/гО2^-5лгСО2 4- 2/гН2О 4- «ХН3 — ДЯ
Условно принятые обозначения: СхНуОг—безазотистое органиче-
ское вещество; C.v-H?yOzN — азотсодержащее соединение; СэНуКОг—
формула, выведенная на основе среднестатистического соотноше-
ния между элементами в синтезируемом клеткой веществе. Количе-
ство кислорода, необходимое для окисления органических примесей
в ферментативных реакциях конструктивного и энергетического об-
мена (см. схемы 1 и 2), характеризует полное БПК. Образующий-
ся аммиак окисляется нитрифицирующими бактериями до нитритов
и нитратов
262
После окисления органических примесей сточных вод происхо-
дит разложение содержимого клеток микроорганизмов (см. схему
3). Коэффициенты (х, у, z) и среднестатистическое соотношение
между основными элементами могут отличаться для разных групп
микроорганизмов. Поэтому эта схема весьма условна, но с помо-
щью ее можно проследить общий ход процессов преобразования
органических веществ при аэробных способах очистки сточных вод.
Первый и третий процессы являются экзотермическими, а второй
(биосинтез) — эндотермическим.
§ 62. Микроорганизмы активного ила и биопленки
Действующим началом при очистке сточных вод в аэротенках
является активный ил, представляющий собой частицы органиче-
ских веществ, населенные различными группами микроорганиз-
мов— аэробов и факультативных анаэробов. Аэрация воды способ-
ствует созданию оптимальных условий для их жизнедеятельности
и интенсификации процессов окисления органических веществ. Кро-
ме того, перемешивание воздухом способствует поддержанию ак-
тивного ила во взвешенном состоянии.
С физико-химических позиций активный ил представляет собой
структурированную коллоидную систему, обладающую высокой
сорбционной способностью. Активный ил представляет собой среду
обитания многих микроорганизмов воды и почвы, образующих
сложный биоценоз. Он получается при длительной аэрации биоло-
гической пленки биофильтров или бытовых сточных вод. Состав ак-
тивного ила определяется природой органических примесей и поэто-
му может изменяться качественно и количественно. Так, при окис-
лении фенолсодержащих сточных вод в биоценозе активного ила
преобладают псевдомонады (Pseudomonas) при общем количестве
видов микроорганизмов 48 и плотности бактериального населения
550 млн. клеток в 1 мл. А при окислении сточных вод, содержащих
анионные ПАВ (алкилсульфонаты), развиваются только десять
видов со значительным преобладанием микобактерий при общем
количестве микроорганизмов 500 000 в 1 мл.
Биоценоз активного ила при нормальной работе аэротенка
прежде всего представлен различными группами бактерий, среди
которых есть гетеротрофы и литотрофы. Численность бактерий в
активных илах составляет 108—1012 клеток на 1 мг сухого вещества.
В активном иле встречаются все морфологические группы бакте-
рий: порядок Pseudomonadineae и роды: Bacillus, Bacterium, Sar-
cina, Micrococcus и др. В нормально работающем иле содержится
небольшое количество нитчатых бактерий (сферотилюс и кладот-
рикс, рис. 32).
Бактерии в процессе очистки воды образуют слизистые скопле-
ния— зооглеи, которые характерны для хорошо сформированного
активного ила. В зооглеях преобладают кокковые и палочковидные
формы. В образовании зооглей может участвовать бактерия Zoog-
lea ramigera, образующая разветвленные зооглеи в виде лопастей.
263
основными представителями биоценозов активного ила являются
бактерии, осуществляющие биохимические процессы разложения
органических веществ. Среди них представлены все группы бакте-
рий, разлагающих безазотистые и азотсодержащие органические
соединения и недоокисленные неорганические вещества; протеоли-
ты, гидролизующие белковые соединения; аммонификаторы; бак-
терии, разлагающие углеводы, жиры, органические кислоты; нит-
рификаторы; серобактерии и др.
Кроме бактерий в активном иле развиваются грибы и актиноми-
цеты. Водоросли обычно встречаются редко и в небольшом количе-
Рис. 32. Нитчатые и зооглейные бактерии:
а — Sphaerotilus natans; б—Cladothrix dichotoma; в — Zooglea
ramigera
стве. Актиномицеты и близкие к ним микобактерии разлагают угле-
водороды, жиры, органические кислоты, углеводы. Плесневые гри-
бы участвуют в разложении углеводов, спиртов, органических кис-
лот, некоторые усваивают органические формы азота. Углеводы и
органические кислоты потребляются дрожжами. Соотношение меж-
ду различными группами микроорганизмов в активных илах приве-
дено на рис. 33.
При затрудненной аэрации или избыточном поступлении в аэро-
тенк органических загрязнений наблюдается смена биоценоза ак-
тивного ила. При длительности анаэробных условий более 15 мин
происходит значительное увеличение количества факультативных
анаэробов и подавляется жизнедеятельность аэробных форм. По-
добная смена групп микроорганизмов сопровождается образовани-
ем неоседающего ила (вспуханием). При этом в активном иле на-
блюдается массовое развитие нитчатых бактерий и грибов (Fusa-
rium) .
Микрофауна активного ила представлена разнообразными груп-
пами простейших (жгутиковыми, саркодовыми, инфузориями), ко-
ловратками, встречаются малощетинковые черви (олигохеты), ре-
же— водные клещи. Представители микрофауны очень чувствитель-
ны к концентрации органических соединений, количеству раство-
ренного кислорода, наличию токсичных соединений. Поэтому в
зависимости от изменения степени загрязнения воды в процессе
очистки или интенсивности аэрации воды в активном иле наблюда-
264
ется преобладание различных групп простейших. Так, при сильном
загрязнении воды органическими примесями (в начале процесса
очистки, при плохой работе аэротенка) в активном иле развивают-
ся в большом количестве из саркодовых мелкие амебы (например,
Amoeba limax и др.), бесцветные жгутиковые (р. Bodo, Oicomonas
и др.). Инфузории при неблагоприятных условиях из веге-
\///Л Род Pseudomonas
Р$дО1 Род Bacillus
£ I Род Bacterium
[xTwJ Род Micrococcus
| : Род Sarcina
Saccharomycetes
Рис. 33. Соотношение между различными группами микроорганизмов ак-
тивного ила
тативных форм превращаются в цисты, а при недостатке
растворенного кислорода прикрепленные формы переходят в
подвижную стадию. Наименее чувствительны к отрицатель-
ным воздействиям из равноресничных инфузорий Paramecium
caudatum, а из кругоресничных — Vorticella microstoma. При
хорошей работе аэротенка в активном иле встречается боль-
шое количество видов простейших. Особенно характерны
брюхоресничные инфузории: Aspidisca, Stylonichia, Euplotes; круго-
ресничные: Vorticella convallaria, Opercularia. Если наблюдается
дефицит питательных веществ (голодающий ил), происходит умень-
шение размеров инфузорий и они начинают инцистироваться. В ак-
тивном иле, перегруженном органическими примесями, развивают-
ся сосущие инфузории (Suctoria).
В активном иле в период завершения окисления органических
веществ и протекания процесса нитрификации развиваются в боль-
шом количестве прикрепленные инфузории (например, Carchesium),
раковинные (Arcella) и крупные голые амебы. Довольно часто
встречаются коловратки (Philodina, Notommata и др.) (рис. 34).
Водные клещи и рачки (циклопы) развиваются в голодающем иле.
Способность отдельных групп микрофауны развиваться при опреде-
265
Рис. 34. Коловратки:
а — р. Notommata;
б — р. Philodina
работы сооружения
ленных условиях используется для проведе-
ния гидробиологического анализа. При мик-
роскопировании активного ила или биоплен-
ки дается характеристика видового и коли-
чественного состава микроорганизмов, оп-
ределяется их жизнеспособность. В сочета-
нии с технологическим анализом это позво-
ляет сделать вывод о работе сооружения.
В табл. 20 приведены данные о степени раз-
вития различных групп простейших при раз-
личной работе очистных сооружений.
По результатам гидробиологического
анализа определяется режим работы соору-
жения, нагрузка по органическим вещест-
вам, устанавливается факт попадания про-
изводственных сточных вод, содержащих
токсичные вещества . При характеристике
следует учитывать интенсивность развития ин-
дикаторных форм микроорганизмов, а не отдельных видов. Анализ
кривых роста для бактерий и других микроорганизмов показывает,
какие микроорганизмы сопутствуют определенной фазе развития
бактериальной микрофлоры активного ила. Так, фаза задержки ро-
ста бактерий в активном иле сочетается с преобладанием в нем
амеб и жгутиковых. В логарифмической фазе из простейших наи-
большее развитие получают жгутиковые, увеличивается количество
свободно плавающих инфузорий. Эта фаза соответствует интенсив-
ному разложению органических примесей, но скоплений бактерий
не образуется. В фазе замедленного роста и стационарной фазе ко-
личество бактерий почти не изменяется, но идет образование хлопка
активного ила. Этой фазе соответствует максимум развития свобод-
ноплавающих инфузорий. Фаза отмирания бактерий (эндогенная)
соответствует окончанию разложения органического вещества. Чис-
ленность бактерий уменьшается в результате отмирания из-за не-
достатка питательных веществ и потребления их простейшими.
В этой фазе из простейших преобладают прикрепленные инфузо-
рии, присутствуют свободноплавающие и коловратки. Окисление
клеточного материала отмирающих бактерий идет параллельно с
процессом нитрификации (нитрифицирующий ил). Роль простей-
ших сводится к поеданию бактерий, а также к потреблению взве-
шенных веществ.
Процесс минерализации органических веществ может нарушать-
ся в присутствии токсичных соединений, ПАВ, тяжелых металлов и
др. Биологическая токсичность примесей производственных сточ-
ных вод оценивается по изменению дегидрогеназной активности.
Дегидрогеназа — окислительно-восстановительный фермент, участ-
вующий в процессе дыхания. Она очень чувствительна к действию
токсичных соединений. По степени подавления активности дегидро-
геназы у микроорганизмов активного ила или биопленки можно су-
дить о влиянии этих соединений на их жизнедеятельность. Опреде-
266
ление проводится в присутствии индикаторов, изменяющих окраску
при переходе из окисленного состояния в восстановленное под дей-
ствием дегидрогеназы.
Таблица 20. Степень развития различных групп простейших при работе
очистных сооружений_______________________________________________
Организмы Характеристика работы биологического окислителя
очень плохая плохая удовлетвори тел ьн ая (нитрификация слабая) хорошая (нитрифи- кация сильная)
Амебы Преобладают Преобладают Единичные эк- земпляры Отсутствуют
Бесцветные То же »
жгутиковые Инфузории Отсутствуют Мало Преобладают равнореснич- ные Преобладают круто- и брю- хоресничные
Коловратки Преобладают Преобладают
Способность активного ила к осаждению характеризуется ило-
вым индексом. Эта величина определяется по объему ила, образую-
щегося после 30-минутного отстаивания жидкости, содержащей
3 мг/л ила. Соотношение между объемом (мл) ила и массой сухого
вещества, содержащегося в 1 г ила, называется иловым индексом.
При хорошей работе аэротенка иловый индекс составляет 100—
120 мл/г. Повышение илового индекса до 150—200 мл/г свидетель-
ствует о нарушении работы аэротенка. Биологическая очистка счи-
тается полной, если БПКполн очищенной сточной воды составляет
менее 20 мг/л, и неполной, если БПКполн превышает 20 мг/л. Пол-
ная биологическая очистка может проводиться в двух режимах:
обычная аэрация (до начала нитрификации и минерализации ак-
тивного ила) и длительная аэрация (полное окисление), при кото-
рой частично минерализуется активный ил и идет процесс нитрифи-
кации. Понятие «полная биологическая очистка» является услов-
ным, так как часть биологически окисляющихся и неокисляющихся
в этих условиях органических соединений остается в очищенной
воде.
Биохимические процессы разложения органических веществ в
биофильтре осуществляются микроорганизмами биологической
пленки, формирующейся на зернах загрузки в период его созрева-
ния. Население биопленки представлено, в основном, теми же груп-
пами микроорганизмов, что и в биоценозе активного ила. Но в ней
более широко представлены зеленые, синезеленые и диатомовые во-
доросли, а также грибы, черви, личинки насекомых. Степень разви-
тия различных групп микроорганизмов в биофильтре определяется
составом сточных вод, условиями обработки воды. Видовой состав
биопленки подвержен также сезонным колебаниям. Водоросли раз-
виваются в верхних слоях, но диатомовые могут встречаться и в
нижних слоях загрузки. Простейшие также развиваются преимуще-
ственно в верхних слоях (в неорошаемой зоне) и в нижних. Бакте-
рии населяют все слои загрузки биофильтра. В отличие от биоцено-
267
ja активного ила в биопленке содержится большое количество ана-
эробов, иногда до 29%. В нижних слоях загрузки наблюдается
развитие большого количества червей (зона червей). Смена био-
ценозов по высоте загрузки характерна для биофильтров. В них
живут микроорганизмы, свойственные а- и p-мезосапробной зонам.
В верхних слоях живут микроорганизмы а-мезосапробной зоны:
здесь встречается большое количество нитчатых бактерий, водоро-
слей, грибов, из простейших — бесцветные жгутиковые. В средних
слоях количество этих микроорганизмов уменьшается и на смену
им приходят брюхо- и кругоресничные инфузории. Нижние слои за-
селены микроорганизмами (З-мезосапробной зоны. Бактериальное
население представлено авто- и гетеротрофами.
§ 63. Почвенные методы очистки сточных вод
Сущность данных методов заключается в фильтрации сточных
вод, содержащих органические вещества, через слой почвы. Очист-
ка воды при этом осуществляется под воздействием физических,
химических и биологических факторов.
В процессе фильтрации сточной жидкости в течение 1—2 недель
на поверхности почвы образуется активный фильтрующий слой. Он
возникает в результате задержки взвешенных и коллоидных ве-
ществ, содержащихся в сточной жидкости, и заселения этого слоя
огромным количеством аэробных микроорганизмов. Толщина ак-
тивной пленки составляет 20—40 см. В этом слое происходит накоп-
ление взвешенных, коллоидных и растворенных веществ, а также
микроорганизмов и яиц гельминтов. Благодаря чрезвычайно силь-
но развитой поверхности почвенных частиц осуществляется адсорб-
ция газов, содержащихся в сточной жидкости, в том числе необхо-
димого для жизнедеятельности микроорганизмов кислорода.
Очистка воды в определенной степени может осуществляться в
слое почвы толщиной до 1,5—2 м. В самом верхнем слое почвы за-
держиваются взвешенные вещества, яйца гельминтов, частично бак-
терии. В более глубоких слоях происходит адсорбция коллоидных
примесей и бактерий почвенными частицами, содержащими огром-
ное количество микроорганизмов, которые участвуют в разложении
органических веществ и ликвидации бактериального загрязнения.
Основную массу населения активной биологической пленки состав-
ляют бактерии-минерализаторы, осуществляющие процесс биохи-
мического окисления органических веществ.
Микроорганизмы сточных вод составляют около 1% от общего
числа почвенных микроорганизмов. Под влиянием неблагоприятных
условий и антагонистического воздействия микроорганизмов почвы
происходит почти полное отмирание патогенных форм. Задержка
патогенных микроорганизмов происходит в результате поглощения
их частицами почвы, а отмирание может вызываться антибиотиче-
скими веществами, выделяемыми почвенными микроорганизмами.
Кроме того, патогенные микроорганизмы используются простейши-
ми, коловратками и другими организмами для питания.
268
Эффективность задержки бактерий, поступающих со сточными
водами, зависит от способности их сорбироваться частицами почвы.
Количество адсорбированных бактерий зависит от их видовых осо-
бенностей и колеблется в значительных пределах (от 45 до 95%).
Степень адсорбции для кишечной палочки составляет около 94%
от общего количества, поступившего со стоком. Присутствие ПАВ в
сточных водах способствует уменьшению числа задерживаемых поч-
вой бактерий. Так, при концентрации анионного ПАВ (сульфоно-
ла) 10 мг/л расстояние, на которое переносится водой Bacteria coll,
увеличивается в 2 раза. Энтерококки и кишечные палочки при ко-
ли-индексе 105 и 10" соответственно в 1 л воды распространяются
на расстоянии от 30 до 200 см. Отмирание микроорганизмов, посту-
пающих со сточными водами, может обусловливаться в определен-
ной степени и действием таких факторов, как солнечная радиация,
неблагоприятные условия внешней среды. Прохождение сточной
жидкости через почвенный фильтрующий слой сопровождается сни-
жением общего числа микроорганизмов на 90—99,8%.
В активном слое происходит энергичное разложение азотсодер-
жащих соединений, в нем развивается количество бактерий — нит-
рификаторов, достигающее 1 млн. клеток на 1 г почвы. Содержание
азота в форме нитратов в очищенной воде может составлять 20—
25 мг/л. На глубине более 40 см окисление органических соединений
осуществляется за счет восстановления нитратов.
В процессе разложения органических веществ активно участву-
ют и дручие растительные и животные организмы биоценоза. Поч-
венные водоросли, особенно зеленые и синезеленые, являются до-
полнительным источником кислорода. Грибы используют в качест-
ве питательного материала органические соединения. Роль простей-
ших и коловраток сводится к уничтожению бактерий, переработке
иловых частиц. Черви, личинки насекомых, населяющие верхний
слой почвы, разрыхляют его и способствуют лучшей аэрации. На
полях орошения число личинок комара Chironomus pulmosus мо-
жет достигать 90 000 на 1 м2, количество поглощаемого ими органи-
ческого вещества достигает примерно 250 г/м2, из которых около
150 г минерализуется и около 100 г используется для построения
тела личинок.
Степень биохимической очистки при почвенных методах опре-
деляется количеством и качеством поступающих сточных вод и
свойствами почвы. Определяющими показателями почв служат их
проницаемость и адсорбционная способность. Наиболее благопри-
ятными в этом отношении являются супесчаные, черноземные и суг-
линистые почвы.
При избыточном количестве поступающей сточной жидкости
наблюдается возникновение анаэробных условий, что ведет к заме-
не аэробных микроорганизмов анаэробными и накоплению проме-
жуточных продуктов распада органических веществ. Нарушение
биохимического окисления может происходить и при высокой кон-
центрации неорганических и биологически неокисляемых органиче-
ских соединений.
269
Очищенная фильтрацией через слой почвы сточная вода не со-
держит яиц гельминтов. Основная масса яиц гельминтов задержи-
вается в 10-сантиметровом слое почвы, отдельные могут проникать
на глубину 30 см. Но сама почва, задерживая в значительном ко-
личестве яйца гельминтов, может явиться источником заражения
выращиваемых на ней овощей, а также обслуживающего персона-
ла. Яйца гельминтов на глубине 2 см могут сохраняться в течение
полутора лет. Подпочвенное орошение сточными водами имеет пре-
имущество перед обычными фильтрационными методами в сани-
тарном отношении. При этом не загрязняется поверхность почвы и
произрастающие растения, отсутствуют неприятные запахи вслед-
ствие поглощения газов почвой. Очищенные сточные воды характе-
ризуются следующими показателями: pH 6,5—8,5; взвешенные ве-
щества 14—70 мг/л; ХПК 20—150 мг/л; БПКб 10—40 мг/л; аммо-
нийный азот 6—70 мг/л; растворенный кислород 4—8 мг/л; сухой
остаток 400—-700 мг/л. Из приведенных данных следует, что при
биологической очистке не всегда удается получить воду, кото-
рая бы соответствовала необходимым требованиям. Иногда бывает
необходима доочистка сточных вод, прошедших биологическую очи-
стку, физико-химическими и химическими методами.
§ 64. Разложение органических веществ
в анаэробных условиях
Осадок, образующийся в первичных остойниках, и избыточный
активный ил подвергаются специальной обработке для предотвра-
щения их загнивания, улучшения структуры, уменьшения объема и
облегчения их обезвоживания. Обычно эта обработка заключается
в частичном биохимическом разложении органических веществ в
анаэробных условиях. Этот прием обработки осадка называется
сбраживанием. Рассмотрим основные направления биохимического
разложения органических веществ в анаэробных условиях. Основу
этих процессов составляют различные виды брожения.
Общее направление биохимических процессов. Б р о ж е н и е
целлюлозы. Целлюлоза устойчива к действию химических сое-
динений, не подвергается изменению в пищеварительном тракте че-
ловека, накапливается в большом количестве в почве и на дне водо-
емов. Она составляет значительную часть осадка, образующегося в
первичных отстойниках. Брожение целлюлозы начинается с фермен-
тативных реакций образования глюкозы, которая затем сбражи-
вается с образованием органических кислот, этилового спирта, ди-
оксида углерода, водорода. Разложение целлюлозы осуществляет-
ся облигатными анаэробами-бактериями, относящимися к роду
клостридий (Clostridium). Это подвижные палочки, образующие
внутри клеток крупные споры. Среди них есть мезофильные (опти-
мальная температура развития 30—35° С) и термофильные (опти-
мальная температура существования около 60° С) формы. При бро-
жении целлюлозы в зависимости от условий образуются и другие
соединения. Ниже приводятся основные продукты, получающиеся
270
при брожении целлюлозы различными видами бактерий. При мезо-
фильном брожении бактериями Clostridium omelianskii образуются
уксусная, молочная и муравьиная кислоты, этиловый спирт, СО2,
Н2; бактериями Clostridium dissolvens — масляная, уксусная и мо-
лочная кислоты, этиловый спирт, СО2, Н2; бактериями Clostridium
cellobioparum — уксусная, муравьиная и молочная кислоты, этило-
вый спирт, СО2, Н2. При термофильном брожении бактериями Clos-
tridium tcimocelhim образуются уксусная, молочная, муравьиная
кислоты, этиловый спирт, СО2, Н2. Термофильные бактерии более
полно разлагают целлюлозу, и процесс брожения идет значительно
быстрее. В табл. 21 приведены данные, характеризующие зависи-
мость между температурой, скоростью и глубиной разложения цел-
люлозы бактериями.
Таблица 21. Изменение скорости и глубины распада
целлюлозы в зависимости от температуры сбраживания
Темперачура брожения. °C Продолжитель- ность броже- ния, сут Сброжено целлюлозы, % Содержание спиртов в среде
г/л %
62 5 91,7 6,0 20,0
45 30 69,0 0,36 3,8
35 120 41,0 0,16 0,1
В качестве азотистого питания целлюлозоразлагающие бакте-
рии используют органические соединения, аммонийные соли, а неко-
торые из них способны усваивать свободный азот.
Маслянокислое брожение. Особенностью этого вида
брожения является образование масляной, уксусной кислоты, угле-
кислого газа и водорода. Оно вызывается маслянокислыми бакте-
риями (Clostridium butiricum). В качестве побочных продуктов мо-
гут получиться ацетон, бутиловый спирт. Они сбраживают простые
и сложные углеводы (целлюлозу, глюкозу, пектиновые вещества),
соли органических кислот. Схему маслянокислого брожения сум-
марно можно записать
С5Н12Об->СН3 — СН2 — СН2 — СООН + СН3СООН + СО2 + Н2
Брожение идет через стадии образования пировиноградной кислоты
с ее последующим преобразованием. Источником азота для масля--
нокислых бактерий служат пептоны, аминокислоты и аммонийные
соли, некоторые из бактерий используют также свободный азот. Уг-
леводы для них служат источником энергии и углерода. Возбудите-
ли маслянокислого брожения являются облигатными анаэробами.
Это крупные, подвижные спорообразующие палочки длиной 3—
10 мкм и диаметром 0,5—1,5 мкм. Оптимальная температура для
их развития составляет 35—37° С, предельные значения pH 6—8.
Брожение пектиновых веществ. Это брожение осуще-
ствляется бактериями и грибами, гидролизующими пектиновые ве-
271
щества с образованием менее сложных органических соединений,
которые затем сбраживаются до масляной и уксусной кислот, водо-
рода и диоксида углерода. Возбудителями брожения пектиновых
веществ являются облигатные анаэробы (Clostridium felsineum,
Clostridium pectunovorum).
Разложение жиров в анаэробных условиях также проходит
через стадию образования глицерина и жирных кислот, которые за-
тем через ряд последовательных превращений сбраживаются с об-
разованием метана и диоксида углерода. Возбудители анаэробного
разложения жиров и жироподобных соединений: Clostridium рег-
fringens, Cl. sporogenes и другие микроорганизмы. Глицерин через
стадию образования пировиноградной кислоты сбраживается с об-
разованием различных продуктов. Высшие жирные кислоты под-
вергаются сбраживанию, сопровождающемуся разрывом углерод-
ной цепи и образованием низших кислот.
Описанные ферментативные процессы составляют первую ста-
дию разложения органических соединений в анаэробных условиях,
которая называется кислым или водородным брожением. Образу-
ющиеся на первой стадии органические кислоты, спирты и другие
соединения затем подвергаются превращениям, которые заканчива-
ются образованием метана и диоксида углерода.
Метановое брожение. Метановое брожение — сложный
процесс, протекающий при участии нескольких групп микроорганиз-
мов. Направленность биохимических процессов и развитие опреде-
ленных групп метанообразующих микроорганизмов определяются
химическим составом разлагающегося субстрата, температурными
условиями и нагрузкой на сооружение по органическому веществу.
Сущность основных закономерностей метанового брожения заклю-
чается в сбраживании органических кислот, спиртов, образующихся
на первой стадии брожения, с выделением метана и диоксида уг-
лерода. Одновременно может проходить восстановление диоксида
углерода водородом до метана. Для метанового брожения харак-
терно участие воды или образование ее в процессе биохимических
реакций. Основные биохимические процессы, протекающие с обра-
зованием метана, можно выразить следующими уравнениями:
а) брожение органических (жирных) кислот
4НСООН->СН4 4- ЗСО2 4- 2Н2О
муравьиная
кислота
СН3СООН->СН4 + со2
уксусная
кислота
2СН3 — (СН2)2 — СООН + СО2 + 2Н2О->СН4 + 4СН3СООН
масляная
кислота
б) спиртов
4СН3 — ОН->ЗСН4 4- СО2 4- 2Н2О
метанол
2С2Н5ОН + СО2->СН4 + 2СН3СООН
этанол
272
4СН3 — СН — СН3 + СО>->4СН3 —- С — СН3 4- 2Н2О + СН4
I
ОН о
пропанол-2 ацетон
в) восстановление СО2 водородом
СО2 +4Н2->СН4 + 2Н2О
Около 70—75% метана образуется из уксусной кислоты, а осталь-
ное — из диоксида углерода, водорода и других соединений. Общее
эмпирическое уравнение метанового брожения с участием воды для
веществ, содержащих углерод, водород и кислород, имеет вид
Основными возбудителями метанового брожения являются сле-
дующие виды бактерий (палочковидные и кокковые формы): Met-
hanobacterium sohngenii, Methanococcus mazei, Methanosarcina met-
hanica, сбраживающие соли органических кислот; Methauobacteri-
um omelianskii, сбраживающая спирты; Methanococcus vannielii,
вызывающая восстановление диоксида углерода водородом. Мета-
новое брожение вызывается как органотрофными микроорганизма-
ми, так и литотрофами при участии фермента фередоксина, витами-
на Bi2. Коферменты НАД и НАДФ ингибируют эти реакции. Про-
цессы образования метана используются микроорганизмами в энер-
гетическом обмене. Метановое брожение идет в нейтральной или
слабощелочной среде.
Разложение азотсодержащих органических соединений в ана-
эробных условиях. Разложение белковых соединений в анаэробных
условиях заканчивается образованием таких продуктов неполного
распада, как амины, аминокислоты ароматического ряда, меркап-
таны, сероводород. Многие из образующихся веществ имеют непри-
ятный запах.
Анаэробное разложение белков вызывается спорообразующими
палочками: Bacillus putrificus, Bacillus sporogenes. Разложение
белковых соединений вызывается и факультативными анаэробами
Proteus vulgaris, Bacteria coli. Степень и интенсивность разложе-
ния белковых соединений зависит от химической структуры белка и
вида микроорганизмов. Аминокислоты, образующиеся в процессе
распада белков в анаэробных условиях, подвергаются восстанови-
тельному дезаминированию с образованием предельных органиче-
ских кислот и аммиака. Органические кислоты могут разлагаться с
•образованием метана и диоксида углерода. Продуктами аммони-
фикации в анаэробных условиях будут метан, аммиак и диоксид
углерода.
Образующийся аммиак может накапливаться в разлагающемся
субстрате или подвергаться «биологическому закреплению», т. е.
273
усваиваться микроорганизмами. При разложении белков, содержа-
щих серу, образуются тиоспирты, тиоэфиры, сероводород.
Процесс денитрификации. В анаэробных условиях нит-
раты в результате ферментативных превращений могут восстанав-
ливаться до свободного азота. Этот процесс называется денитрифи-
кацией. Денитрифицирующие бактерии широко распространены ?
водоемах и почве. Наиболее энергичные возбудители процесса де
нитрификации: Bacterium denitrificans, Pseudomonas flourescens
Восстанавливать нитраты могут некоторые серобактерии (Thioba-
cillus denitrificans). Кислород нитратов при этом идет на образова
ние сульфатов, а источником энергии является процесс окисления
элементарной серы до сульфатов. Схема этого процесса
6KNO3 + 5S + 2СаСО3 = 3K2SO4 + 2CaSO4 Д- 2СО2 Д- 3N2
§ 65. Биохимические процессы, протекающие
при разложении осадка сточных вод
Осадок сточных вод и концентрированные производственные
сточные воды с ВПК выше 5 г/л подвергаются биохимическому раз-
ложению в анаэробных условиях. Оно может происходить в соору-
жениях-септиках, представляющих собой отстойник, через который
медленно проходит сточная жидкость. В двухъярусном отстойнике
осадок отделен от проходящей сточной жидкости, его разложение
осуществляется в иловой камере. На очистных сооружениях боль-
шой производительности осадок сточных вод выделяется в первич-
ных отстойниках и вместе с избыточным активным илом подверга-
ется сбраживанию в метантенках. Интенсивность и глубина разло-
жения осадка прежде всего определяются его составом, который ко-
леблется по соотношению содержания основных органических ком-
понентой (углеводов, белков, жироподобных соединений) и неорга-
нических веществ. Обычно в осадке городских сточных вод содер-
жится 70—80% органических веществ. Так, примерный состав осад-
ка (%): белки 24, углеводы 23, жироподобные вещества до 30. Ча-
ще всего при кислом брожении осадка получаются уксусная, мас-
ляная, пропионовая кислоты. Образующиеся газы содержат диок-
сид углерода, метан, водород, сероводород. Водная фаза имеет
кислую реакцию среды (рН<5), не обладает буферными свойства-
ми, имеет резкий неприятный запах.
Для нормальной работы сооружений (двухъярусных отстойни-
ков, метантенков), в которых идет разложение осадка, необходимо
создавать условия для непременного прохождения второй стадии —
щелочного или метанового брожения. Нормальное течение процес-
са метанового брожения определяется такими факторами, как тем-
пература, время сбраживания, степень перемешивания, соотноше-
ние между количеством свежего и сброженного осадка, химический
состав осадка, pH, щелочность, концентрация летучих органиче-
ских кислот, наличие токсичных примесей, биогенных элементов,
достаточное количество микроорганизмов. Термофильное брожение
имеет ряд преимуществ перед мезофильным. К ним относятся боль-
274
шая скорость и глубина распада органических соединений, увели-
чение объема выделяющегося газа. При перегрузке метантенка све-
жим осадком создаются условия для протекания кислого брожения.
Сбраживание осадка нарушается также при поступлении в бродя-
щую масс}' веществ, токсичных для метанпродуцирующих бактерий.
В табл. 22 приведены ПДК некоторых соединений, оказывающих
отрицательное влияние на процесс сбраживания осадка.
Т а б л и ц а 22. Предельно допустимые концентрации
некоторых соединений, влияющие на процесс брожения
Соединения Концентрация, мг/л Соединения Концентрация, мг/л
допус- тимая вредная допус- тим ая вредная
Хром (VI) 3 6 Толуол 20 200
Медь (Си2+) 20 30 Амиловый спирт 50 100
Свинец (РЬ2+) 50 70 Ацетон 100 200
Cu2+ + Pb2+ 7,5- —25 25 Бензол 50 200
При термофильном брожении наблюдается полное отмирание
патогенных микроорганизмов и дегельминтизация. Яйца гельмин-
тов погибают при 49° С в течение 3 ч, а при 53° С — через час. Воз-
будители брюшного тифа, дизентерии, паратифа отмирают при тер-
мофильном брожении через несколько часов. ЛАетановые бактерии
очень чувствительны к колебаниям температуры. Поэтому в интер-
вале температур 37—42° С брожение осадка замедляется, что свя-
зано с неблагоприятным воздействием температуры, низкой для
развития термофилов и высокой — для мезофилов.
Образующийся газ содержит обычно 62—65% метана и 30—35%
углекислого газа. Сероводород при нормально протекающем ще-
лочном брожении отсутствует, так как он связывается в форме
сульфидов. Водорода содержится не более 0,5%, азота — около
2,2—2,8%. Количество газа и его состав зависят от соотношения
различных групп органических загрязнений в осадке. Так, при уве-
личении количества белков или снижении содержания жиров в
осадке выход газов уменьшается.
Метановое брожение идет в интервале pH 5,64-8,2. Оптимум
составляет 7,0—7,6. Несмотря на присутствие кислот, pH бродящей
массы изменяется очень незначительно, что связано с буферными
свойствами иловой воды, обусловленными присутствием свободной
угольной кислоты и гидрокарбонатов. Повышение щелочности вызы-
вается наличием аммонийного азота. Свободная угольная кислота,
взаимодействуя с основаниями, частично переходит в гидрокарбо-
наты, что также способствует повышению pH. Контроль за работой
метантенка ведется по качественному составу и количеству образу-
ющегося газа, осадка и по иловой жидкости. В иловой воде и сбро-
женном осадке содержится значительное количество биогенных ве-
275
ществ. При нормальном протекании метанового брожения иловая
вода должна иметь следующие показатели: pH 6,8—7,5; щелочность
40—60 мг-экв/л, жирные кислоты не более 10—12 мг-экв/л, NH4+
более 500 мг/л. Метановое брожение нарушается тогда, когда появ-
ляются условия для задержки процесса на стадии кислого броже-
ния. Этому способствует перегрузка сооружения свежим осадком,
малое количество микроорганизмов, резкое колебание температуры
и pH свежего осадка ниже оптимума, неравномерное поступление
осадка. Нарушение процесса метанового брожения проявляется в
снижении выхода газа с единицы объема поступающего осадка, из-
менении его качественного состава и уменьшения содержания ме-
тана, возрастании количества диоксида углерода и водорода и вы-
делении сероводорода. В иловой воде увеличивается содержание
органических (жирных) кислот, снижается щелочность и pH. На-
блюдается вспенивание осадка, он увеличивается в объеме, изме-
няется его цвет (из темно-серого он становится грязно-желтым), по-
является неприятный запах. Сброженный осадок содержит большое
количество соединений азота, фосфора и других биогенных элемен-
тов. Поэтому после дополнительного обезвоживания и сушки мо-
жет быть использован в качестве удобрения.
После высушивания содержание бактерий в иле снижается в
3800 раз по сравнению со сточной жидкостью, а кишечная палочка
отсутствует полностью, что указывает на безопасность ила в сани-
тарно-гигиеническом отношении.
Аэробная стабилизация осадка. В последние годы для обработ-
ки избыточного активного ила и смеси его с осадком из первичных
отстойников используется аэробная стабилизация. Она заключает-
ся в частичном окислении органических примесей, входящих в сос-
тав твердой фазы, при длительной аэрации в сооружениях типа
аэротенков. Эффективность обработки осадка зависит от состава и
свойств осадка, длительности и интенсивности аэрации, температу-
ры и других факторов. Длительность аэрации для стабилизации
активного ила составляет 7—10 сут, для осадка из первичных от-
стойников— 10—15 сут. При аэробной стабилизации происходит и
самоокисление клеточного вещества бактерий. Изменение свойств
осадка, степень разложения органических примесей при использо-
вании этого метода примерно соответствует эффекту, получаемому
при анаэробном сбраживании. Иногда процесс аэробной стабилиза-
ции называют «аэробным сбраживанием». С этим нельзя согласить-
ся, так как брожение является анаэробным процессом и не может
идти в аэробных условиях.
ЛИТЕРАТУРА
Алекин О. А. Основы гидрохимии. Л., 1970.
Апельцин И. Э., Клячко В. А. Опреснение воды. М., 1979.
Беличенко Ю. П., Полянинов Л. Я. Охрана водных ресурсов. М., 1976.
Брежнев В. И. Обеззараживание воды на городских водопроводах. М., 1970.
Возная Н. Ф. Химия воды и микробиология. М., 1979.
Вейцер Ю. И., Минц Д. М. Высокомолекулярные флокулянты в процессах
очистки воды. М., 1975.
Вольф И. В.,. Ткаченко Н. И. Химия и микробиология природных и сточных
вод. Л., 1973.
Габович Р. Д., Николадзе Г. И., Савельева Н. П. Фторирование и обесфто-
ривание питьевой воды. М., 1968.
Грановский М. Г., Лавров И. С., Смирнов О. В. Электрообработка жидко-
стей. Л., 1976.
Григорьева Л. В. Санитарная бактериология и вирусология водоемов.
М., 1975.
Голубовская Э. К. Биологические основы очистки воды. М., 1978.
Грушко Я. М. Вредные неорганические соединения в промышленных сточ-
ных водах. Л., 1979.
Доливо-Добровольский Л. Б., Кульский Л. А., Накорчевская В. Ф. Химия и
микробиология воды. Киев, 1971.
Долин П. И., Шубин В. Н., Брусенцова С. А. Радиационная очистка воды.
М„ 1973.
Жуков А. И., Монгайт И. Л., Родзиллер И. Д. Методы очистки производст-
венных сточных вод. М., 1977.
Зацепина Г. Н. Свойства и структура воды. М., 1974.
Зарубин Г. П., Овчинкин И. П. Санитарные вопросы водоснабжения и кана-
лизации. М., 1974.
Клячко В. А., Апельцин И. Э. Очистка природных вод. М., 1971.
Карелин Я. А., Жуков Д. Д., Журов В. Н., Репин Б. Н. Очистка производ-
ственных сточных вод в аэротенках. М., 1973.
Кульский Л. А. Теоретические основы и технология кондиционирования во-
ды. Киев, 1971.
Кульский Л. А., Гороновский И. Т., Когановский А. М., Шевченко М. А.
Справочник по свойствам, методам анализа и очистке воды. Киев, 1980.
Кульский Л. А., Чепцов А. С., Князькова Т. В., Кучерук Д. Д. Новые мето-
ды опреснения воды. Киев, 1974.
Кожинов В. Ф., Кожинов И. В. Озонирование воды. М., 1974.
Кастальский А. А., Минц Д. М. Подготовка воды для питьевого и промыш-
ленного водоснабжения. М., 1962.
Кузнецов Ю. В., Щебетковский В. Н., Трусов А. Г. Основы очистки воды
от радиоактивных изотопов. М., 1974.
Константинов А. С. Общая гидробиология. М., 1972.
Кузнецов С. И., Романенко В. И. Микробиологическое изучение внутренних
водоемов. М., 1963.
Львович М. И. Мировые водные ресурсы и их будущее. М., 1974.
Лукиных Н. А., Липман Б. Л., Криштул В. П. Методы доочистки сточных
вод. М., 1978.
Лурье Ю. Ю., Рыбникова А. И. Химический анализ производственных сточ-
ных вод. М., 1974.
277
Микробиология загрязненных вод/Под ред. Митчелла. М., 1974.
Мишустин Е. Н., Перцовская М. И., Горбов В. А. Санитарная микробиоло-
гия почвы. М., 1979.
Мишустин Е. Н., Емцев В. Т. Микробиология. М., 1978.
Милованов Л. В., Краснов Б. П. Методы химической очистки сточных вод
горнорудных предприятий цветной металлургии. М., 1967.
Новиков Г. В., Дударев А. Я. Санитарная охрана окружающей среды со-
временного города. Л., 1978.
Николадзе Г. И. Обезжелезивание природных и оборотных вод. М., 1978.
Очистка производственных сточных вод/Под ред. Ю. И. Турского и И. В. Фи-
липпова. Л., 1967.
Предельно допустимые концентрации вредных веществ в воздухе и воде.
Л., 1975.
Пушкарев В. В., Трофимов Д. И. Физико-химические основы очистки сточ-
ных вод от ПАВ. М„ 1975.
Поруцкий Г. В. Биохимическая очистка сточных вод органических произ-
водств. М., 1975.
Проскуряков В. А., Шмидт Л. И. Очистка сточных вод в химической про-
мышленности. Л., 1977.
Рейзин Б. Л., Стрижевский И. В., Шевелев Ф. А. Коррозия и защита ком-
мунальных водопроводов. М., 1979.
Рубенчик Л. И. Микроорганизмы — биологические индикаторы. Киев, 1972.
Роговская Ц. И. Биохимический метод очистки производственных сточных
вод. М., 1967.
Соколов В. Ф. Обеззараживание воды бактерицидными лучами. М., 1964.
Сорокин Ю. И. Роль бактерий в жизни водоемов. М., 1974.
Таубе П. Р., Баранова А. Г. Практикум по химии воды. М., 1971.
Фридрихсберг Д. А. Курс коллоидной химии. М. — Л., 1974.
Хаммер М. Технология обработки природных и сточных вод. М., 1979.
Черкинский С. Н. Санитарные условия спуска сточных вод в водоем.
М., 1971.
Чурбанова И. Н., Карюхина Т. А. Химия воды и микробиология. М., 1974.
Шевченко М. А. Физико-химическое обоснование процессов обесцвечивания
и дезодорации воды. Киев, 1973.
Оглавление
Стр.
Предисловие ............................................................ 3
Введение ............................................................... 5
Раздел первый. Теоретические основы курса .............................. 7
Глава I. Свойства воды и водных растворов............................... 7
§ 1. Вода как химическое соединение................................ 7
§ 2. Понятие о системах, фазах и компонентах. Диаграмма состояния
воды................................................................ 13
§ 3. Структура водных растворов.................................... 15
§ 4. Растворимость различных веществ в воде........................ 18
§ 5. Свойства растворов............................................ 21
§ 6. Теория сильных электролитов................................... 23
§ 7. Произведение растворимости.................................... 25
§ 8. Кинетика химических реакций................................... 27
§ 9. Ионное произведение воды. Водородный показатель............... 34
§ 10. Гидролиз солей................................................ 36
§ 11. Буферные растворы............................................. 42
§ 12. Активная, общая, свободная кислотность воды.................. 44
§ 13. Активная, общая и свободная щелочность воды.................. 46
§ 14. Окислительно-восстановительные процессы....................... 48
Глава II. Природные воды. Показатели качества воды.................. 53
§ 15. Круговорот воды в природе. Мировые водные ресурсы. Водные
ресурсы СССР........................................................ 53
§ 16. Физико-химическая характеристика природных вод............... 55
§ 17. Химические ингредиенты природных вод и их значение для оцен-
ки качества воды.............................,...................... 58
§ 18. Основные показатели качества воды. Требования, предъявляемые
к воде хозяйственно-бытового и промышленного водоснабжения 64
Раздел второй. Физико-химические основы очистки природных и сточных
вод................................................................. 73
Глава III. Умягчение и обессоливание воды.............................. 73
§ 19. Жесткость воды............................................... 73
§ 20. Реагентные методы умягчения воды............................. 74
§ 21. Метод ионного обмена и его применение при обработке воды . . 80
§ 22. Обессоливание воды............................................ 86
§ 23. Угольная кислота и ее формы содержания в воде........ 98
§ 24. Стабильность воды............................................ 102
§ 25. Коррозия железа в воде....................................... 104
§ 26. Коррозия бетона в воде...................................... 106
§ 27. Физико-химические основы удаления взвешенных и коллоидных
примесей........................................................... 109
§ 28. Обработка воды коагулянтами.................................. 124
§ 29. Характеристика коагулянтов и флокулянтов................. 130
279
Стр
§ 30. Безреагентные методы коагуляции.............................. 136
§ 31. Удаление из воды соединений железа и марганца................ 137
§ 32. Обескремнивание воды......................................... 141
§ 33. Фторирование и дефторирование воды........................... 142
§ 34. Удаление из воды растворенных газов.......................... 144
§ 35. Очистка природных вод от органических примесей............... 146
Глава IV. Обеззараживание воды.................................... 149
§ 36. Хлорирование воды............................................ 150
§ 37. Обеззараживание воды солями тяжелых металлов................. 157
§ 38. Озонирование воды............................................ 157
§ 39. Физические методы обеззараживания воды....................... 159
Глава V. Сточные воды и методы их очистки............................. 161
§ 40. Характеристика бытовых сточных вод........................... 161
§ 41. Характеристика производственных сточных вод.................. 165
§ 42. Основные примеси производственных сточных вод................ 167
§ 43. Основные показатели степени загрязнения сточных вод.......... 176
§ 44. Условия спуска сточных вод в водоемы......................... 179
Глава VI. Обзор методов очистки производственных сточных вод . . . 182
§ 45. Физико-химические методы очистки производственных сточных
вод.......................................................... 183
§ 46. Химические методы очистки сточных вод................... 191
§ 47. Сжигание органических примесей, содержащихся в сточных водах 194
§ 48. Очистка воды от радиоактивных изотопов (дезактивация) . . . 196
Раздел третий. Микробиология воды..................................... 198
Глава VII. Основы общей микробиологии................................. 198
§ 49. Предмет и задачи микробиологии. Краткий исторический очерк
развития микробиологии как науки ................................. 198
§ 50. Некоторые сведения по морфологии и систематике микроорганиз-
мов .............................................................. 200
§ 51. Физиология микроорганизмов................................... 210
§ 52. Влияние факторов внешней среды на жизнедеятельность микро-
организмов ....................................................... 219
§ 53. Распространение микроорганизмов в природе .................. 227
Глава VIII. Санитарная микробиология воды.......................... 230
§ 54. Биоценозы природных водоемов................................. 230
§ 55. Санитарно-бактериологическая оценка качества воды............ 233
§ 56. Санитарно-показательные микроорганизмы воды и почвы........ 234
§ 57. Характер и источники загрязнения водоемов.................... 239
§ 58. Система сапробности и ее применение для оценки степени за-
грязнения водоемов............................................ 241
§ 59. Процессы самоочищения водоемов.......................... 243
§ 60. Влияние микроорганизмов и продуктов их жизнедеятельности на
качество воды................................................. 249
Глава IX. Значение микроорганизмов в процессах очистки сточных вод 256
§ 61. Окисление органических веществ в аэробных условиях ..... 357
§ 62. Микроорганизмы активного ила и биопленки................ 263
§ 63. Почвенные методы очистки сточных вод.................... 268
§ 64. Разложение органических веществ в анаэробных условиях . . . 270
§ 65. Биохимические процессы, протекающие при разложении осадка
сточных вод....................................................... 274
Литература...........................................................• 277
ВЫСШЕЕ
ОБРАЗОВАНИЕ
П. Р ТАУБЕ,А.Г БАРАНОВА
химия
И МИКРОБИОЛОГИЯ
воды