/









































































































































































































































































































































































Text
Е. Д. Бабенков
очистка воды
КОАГУЛЯНТАМИ
ИЗДАТЕЛЬСТВО -НАУКА-
АКАДЕМИЯ НАУК СССР
ОРДЕНА ТРУДОВОГО КРАСНОГО ЗНАМЕНИ
ИНСТИТУТ ФИЗИЧЕСКОЙ ХИМИИ
Е. Д. Бабенков
ОЧИСТКА ВОДЫ
КОАГУЛЯНТАМИ
ИЗДАТЕЛЬСТВО «НАУКА»
МОСКВА 1977
УДК 628.162.5 + 628.335',628.344/.347
Очистка воды коагулянтами. Бабенков Е. Д. М., «Нау¬
ка», 1977, стр. 356
Книга посвящена очистке природной воды и сточных жид¬
костей гидролизующимися коагулянтами. Дан систематизиро¬
ванный обзор исследований в этой области. Приведены необ¬
ходимые сведения по теории коагуляции. Рассмотрены меха¬
низм коагуляции и электрокоагуляцин минеральных и органи¬
ческих примесей воды, факторы, влияющие на эффективность
процесса и качество очищенной воды, методы интенсификации
коагуляции, возможность удаления растворенных примесей и
микроэлементов, вопросы совмещения коагуляции с другими
методами водоподготовки. Дано обоснование расчета опти¬
мальной дозы коагулянта.
Монография рассчитана на научных работников и инже¬
неров, занятых вопросами подготовки воды для хозяйственно¬
питьевых и технических целей, а также вопросами очистки
промышленных и бытовых стоков. Может рассматриваться
как дополнительное учебное пособие для студентов и аспиран¬
тов.
Таблиц 24. Иллюстраций 83. Библ. 1847 назв.
Ответственные редакторы:
доктор технических наук
Е. Ф. КУРГАЕВ,
кандидат физико-математических наук
В. М. МУЛЛЕР
20503-123
Б 055(02-77) 96-7'
© Издательство «Наука», 1977 г
ПРЕДИСЛОВИЕ
Обработка коагулянтами — самый распространенный метод
очистки воды от грубодисперсных и коллоидных загрязнений.
Масштабы применения метода коагуляции увеличились в послед¬
ние годы и, судя по прогнозам, будут продолжать увеличиваться.
Поэтому актуален поиск путей к усовершенствованию этого мето¬
да — повышению скорости формирования и отделения коагули¬
рованных взвесей в осадок.
Известно, что базой для изысканий, направленных на разви¬
тие и усовершенствование какого-либо метода или процесса,
должна являться четко выработанная теоретическая платформа.
Сегодняшняя практика коагулирования основана, главным об¬
разом, на эмпирических выводах. В пособиях по водоподготовке
теоретические аспекты коагуляции рассматриваются лишь в
общих чертах, а со времени появления монографий JI. А. Куль-
ского и др. «Физико-химические основы очистки воды коагуля¬
цией» (1950) и О. И. Мартыновой «Коагуляция при водоподго¬
товке» (1951) прошло более 25 лет.
Между тем результаты именно теоретических исследований
в области фундаментальных наук, в частности физической и кол¬
лоидной химии, заставляют критически пересмотреть принятые
взгляды на механизм коагуляционных взаимодействий, позволя¬
ют создать модель процесса, близкую к процессу реальному,
наметить новые пути повышения технологической эффективности
коагулирования.
Благодаря работам физико-химиков, в первую очередь совет¬
ских, созданы и развиты новая физическая теория коагуляции
коллоидов, теория формирования коагуляционных структур,
химия комплексных и высокомолекулярных соединений. Полу¬
чила развитие теория флокуляции, изменились взгляды на роль
механического перемешивания воды, обработанной реагентами,
обнаружено и объяснено большое значение специфических хими¬
ческих реакций в ходе агломерации и структурообразования.
Некоторые из результатов теоретических исследований успеш¬
но применены в водоподготовке. Они помогли уяснить влияние
pH среды на структурные особенности коагулятов, подобрать
условия для более полного проявления интенсифицирующего
действия магнитного и ультразвукового полей, установить связи
между оптимальной дозой коагулянта и катионообменной емко-
4
ПРЕДИСЛОВИЕ
стыо минеральных примесей воды, между величиной электроки-
нетического потенциала взвесей и их способностью к осаждению,
ввести в практику очистки воды новые флокулянты, усовершен¬
ствовать технику коагуляционных исследований.
При решении большой группы дальнейших задач ощущается
потребность в монографии, в которой было бы раскрыто совре¬
менное состояние вопроса, систематизированы и обсуждены ре¬
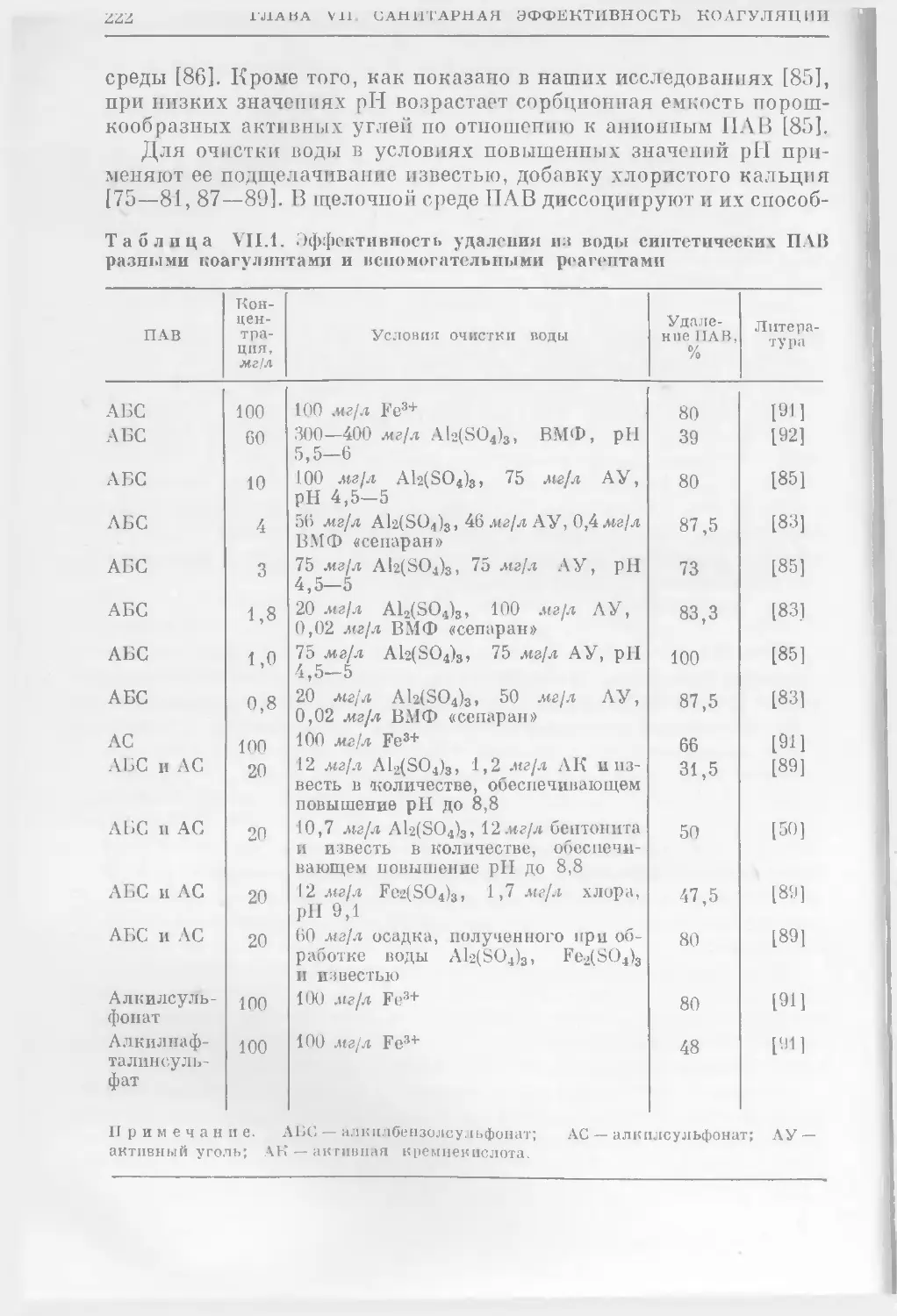
зультаты новейших исследований, увязаны теоретические поло¬
жения и практический опыт, выделены наиболее важные аспекты
проблемы. Это тем более необходимо, что интенсификация работы
водоочистных сооружений развивается, главным образом, в нап¬
равлении усовершенствования реагентной обработки воды.
Книга, предлагаемая читателям, явилась плодом обработки
и критического анализа результатов большого числа отечествен¬
ных и зарубежных работ, а также собственных исследований
автора. В ней рассмотрены физические и коллоидно-химические
процессы, протекающие при очистке коагулянтами природных и
сточных вод, кинетика и эффективность коагуляции, методы оп¬
тимизации отдельных стадий, вопросы рационального совмещения
коагулирования с другими методами очистки. Анализируя от¬
дельные стадии процесса коагуляции водных примесей, автор
пришел к выводу, что многие из наблюдаемых явлений могут най¬
ти удовлетворительное объяснение, если их рассматривать как
результат проявления дальнодействующих молекулярных сил,
существование которых предсказано физической теорией коагу¬
ляции и в последние годы подтверждено экспериментально. На
основе этого вывода развиты представления об оптимальной дозе
коагулянта и ее связи с качественными параметрами обрабаты¬
ваемой воды.
В изложении материала выбран обычный порядок — от об¬
суждения существа элементарных процессов к выводу общих
закономерностей, от теории к эксперименту и практике.
В первой и по существу вводной главе обсуждаются важней¬
шие свойства водных дисперсных систем и явлений, имеющих
место на границе раздела жидкой и твердой фаз. Даны основные
сведения о природе сил взаимодействия дисперсных частиц с ок¬
ружающей их средой, без 'которых невозможно дальнейшее из¬
ложение закономерностей процесса коагуляции.
Вторая глава содержит характеристику загрязнений природ¬
ной воды. Особое внимание уделено составу и физико-химическим
свойствам дисперсных примесей.
В третьей главе описаны виды коагулянтов и закономерности
их гидролиза, состав продуктов гидролиза,5 формирование и
свойства коагуляционных структур.
Четвертая глава посвящена рассмотрению взаимодействия
частиц, коагулирующих в присутствии электролитов и под влия¬
ПРЕДИСЛОВИЕ
5
нием различных физических факторов. Изложена теория коагу¬
ляции лиофобных золей.
В пятой главе, посвященной кинетике коагуляции дисперсных
частиц электролитами и гидролизующимися коагулянтами, об¬
суждаются условия перехода медленной коагуляции в быструю и
особенности коагуляции частиц в движущейся среде, анализиру¬
ется роль перемешивания воды в ходе формирования коагулиро¬
ванной взвеси.
В шестой главе рассмотрен механизм взаимодействия продук¬
тов гидролиза коагулянтов с примесями воды и на его основе —
один из главных вопросов водоподготовки: определение оптималь¬
ной дозы коагулянта для воды разного состава. Обсуждаются
методы коагулирования и выделения коагулированной взвеси в
осадок.
В седьмой главе описаны сфера применения минеральных ко¬
агулянтов, санитарная эффективность коагуляции и электрокоа¬
гуляции как при самостоятельном использовании коагулянтов,
так и в сочетании с другими реагентами.
Восьмая глава содержит подробный анализ существующих ме¬
тодов интенсификации коагуляции: реагентных (за исключением
флокуляции) и безреагентных. Флокуляции полиэлектролитами,
требующей более детального изложения, посвящена самостоятель¬
ная девятая глава. В ней приводятся сведения о видах флокулян-
тов, механизме и эффективности их действия.
Последняя, десятая глава знакомит с примерами успешного
использования коагулирования в некоторых специфических про¬
цессах водоподготовки — при удалении кальция, магния, желе¬
за, марганца, кремния, фтора и сероводорода; при очистке бы¬
товых и промышленных стоков и обработке осадков.
Ставя своей целью рассмотрение проблемы с общих позиций,
автор не включил сведения о технических средствах, используе¬
мых при проведении коагулирования, методах расчета и констру¬
ирования сооружений для разделения суспензий. Эти сведения
имеются в специальной литературе, известной широкому кругу
специалистов. Из-за ограниченного объема в книгу не вошли раз¬
делы о технике проведения коагуляционных исследований и спо¬
собах регенерации осадков. Зато подробно проанализированы
сущность отдельных способов и приемов коагулирования, специ¬
фика очистки воды от таких «новых» видов загрязнений, как
пестициды, синтетические детергенты, фосфаты. При этом неяс¬
ные моменты, особые явления, не укладывающиеся пока в рам¬
ки теории,автор старался не обходить молчанием, а, напротив,
сосредоточить на них внимание читателей.
Возможно, некоторые выводы носят полемический характер,
требуют уточнения. Это естественно, так как предлагаемая книга
является первой попыткой систематического изложения Целого
6
ПРЕДИСЛОВИЕ
комплекса вопросов, относящихся к очистке воды коагулян¬
тами.
Автор благодарит доктора технических наук профессора
Е. Ф. Кургаева, поддержавшего саму идею написания моногра¬
фии и давшего ряд ценных советов после просмотра рукописи.
Автор выражает также признательность члену-корреспонден-
ту АН СССР Б. В. Дерягину, академику АН УССР JI. А. Кульско-
му, кандидату физико-математических наук В. М. Муллеру и
кандидату химических наук И. Т. Гороновскому за чрезвычайно
полезные критические замечания по рукописи, которые автор
учел в процессе ее доработки.
ВВЕДЕНИЕ
Коагуляция — это процесс укрупнения дисперсных частиц за
счет их взаимодействия и объединения в агрегаты. Вещества, спо¬
собные вызвать коагуляцию частиц, называют в общем случае
коагуляторами, а в водоподготовке — коагулянтами или гидро¬
лизующимися коагулянтами. Последние не только вызывают коа¬
гуляцию частиц загрязнений, но и образуют, гидролизуясь, мало¬
растворимые продукты, способные объединяться в крупные хлопья.
Важная особенность коагулирования состоит в том, что при
правильной организации процесса, помимо решения основной
технологической задачи — очистки воды от грубодисперсных и
коллоидных загрязнений, с его помощью можно добиться замет¬
ного удаления некоторых истинно растворенных примесей.
История применения коагулянтов для очистки воды, по-ви¬
димому, берет начало в Древнем Египте, где еще в XVI в. до на¬
шей эры в качестве коагулянта использовали сок сладкого мин¬
даля [1]. Коагулирующие свойства алюмокалиевых квасцов были
известны египтянам, римлянам, грекам [2]. В Европе квасцы
начали применять только в середине XVIII в. и относились к
ним довольно долго с опасениями, о чем свидетельствует запись
Дельвига в первом русском руководстве по водоснабжению:
«Нельзя не осуждать всякого очищения, которое вводит в химиче¬
ский состав воды новое вещество, прежде в ней не заключающееся.
По этой причине, а равно и по ценности квасцов способ очищения
ими весьма редок и может быть употреблен только при малом
количестве воды» [3].
Первые эксперименты по использованию сернокислого алю¬
миния проведены в 1827 г. [4]. В 1884 г. американец Хайят полу¬
чил патент на очистку воды фильтрами с предварительной обра¬
боткой воды сернокислым алюминием [5]. В 1865—1872 гг. ре¬
комендованы для практического использования соли окисного
железа [6], в 1887 г. запатентовано карманное фильтрующее уст¬
ройство с предварительным выделением в осадок гидроокиси же¬
леза [7]. Систематическое использование сульфатов алюминия и
железа в практике очистки воды началось в 80—90-х годах прошло¬
го столетия [6, 8]. В это же время проведено изучение техноло¬
гических возможностей коагулирования, опубликованы сообще¬
ния об использовании коагулянтов при очистке торфяных и
сточных вод [9, 10].
8
ВВЕДЕНИЕ
По данным Пискунова [11], в России приоритет в очистке
воды коагулянтами принадлежит И. О. Плятсу. В 1886 г. Зем-
бицкий [12] предложил использовать для подготовки питьевой
воды хлорное железо, а первые исследования эффективности
этого коагулянта провел в конце века Бунге [13]. К этому времени
полностью или частично 29 городских водопроводов работали на
воде поверхностных источников, а на семи из них (города Гель¬
сингфорс, Тифлис, Нижний Новгород, Двинск, Новочеркасск,
Нахичевань, Владимир) применяли коагулирование [14].
В начале XX в. в практику водоснабжения входят скорые
фильтры, работающие с применением коагулирования. Опыт их
эксплуатации указывает на высокую обеззараживающую способ¬
ность коагулянтов. В 1912 г. в качестве коагулянта начинают
применять хлорированный железный купорос [15], в 1929 г.—
алюминат натрия [4], в 1950-х годах — оксихлорид алюминия
[16, 17].
В СССР в 1920-х годах применялся почти исключительно не¬
очищенный сульфат алюминия. Использование железосодержащих
коагулянтов началось после того, как по инициативе Академии
коммунального хозяйства им. К. Д. Памфилова в Днепропетров¬
ске, Куйбышеве, Челябинске и Свердловске были успешно про¬
ведены исследования по очистке воды с помощью железного
купороса.
В 1937 г. Д. Бэйлис предложил использовать в качестве фло-
кулянта активную кремнекислоту, а начиная с 1950-х годов в
практику водоочистки прочно входят органические флокулянты.
В последующие годы применение коагулянтов и флокулянтов
в области водоподготовки непрерывно увеличивалось. Это обус¬
ловлено несколькими причинами:
1) ростом водопотребления, происходящим в основном за счет
поверхностных источников;
2) ухудшением качества воды в этих источниках;
3) повышением требований к качеству очищенных природных
и сточных вод.
На заре развития централизованного водоснабжения, в 50-х
годах прошлого века расход воды на каждого жителя европей¬
ских столиц составлял 15—50 л/сут. К концу века он вырос до
100—150 л/сут (Лондон, Санкт-Петербург, Париж) [3, 18]. Глав¬
ным источником водоснабжения были подземные воды, но по ме¬
ре роста водопотребления все чаще становилось необходимым
использовать речную и озерную воду. Так, к 1910 г. число водо¬
проводов России, использующих поверхностные источники, сос¬
тавило 55% от общего их числа [14].
За годы Советской власти число городов с централизованным
водоснабжением увеличилось в нашей стране в 30 раз. Новый
перспективный план развития водного хозяйства предусматривает
ВВЕДЕНИЕ
9
увеличение водопотребления населением к 1980 г. до 101 млн. м3/
/сут. Это означает, что среднесуточный расход воды только на
хозяйственно-питьевые нужды одного горожанина возрастет до
360 л. В некоторых крупных городах (Москва, Ленинград, Киев,
Волгоград и др.) запланированная норма водопотребления уже
достигнута [19].
К расходу воды на хозяйственно-питьевые цели нужно доба¬
вить сильно выросшее водопотребление промышленностью, осо¬
бенно новыми ее отраслями. Например, на производство 1 т про¬
дукции в химической промышленности расход воды (в м3) состав¬
ляет [20]:
К 1980 г. потребление воды промышленностью СССР возрас¬
тет до 419 млн. м3/сутп.
С увеличением общего водопотребления доля воды, забирае¬
мой в разных странах из открытых водоемов, возросла до 70—95%
Потребление воды из открытых источников продолжает уве¬
личиваться по двум причинам:
1. Не хватает пресных подземных вод (в СССР запасы таких
вод примерно в 20 раз меньше запасов поверхностных) [24].
2. Неисчерпаемые запасы воды Мирового океана остаются фак¬
тически неиспользованными: суммарная суточная производи¬
тельность всех действующих на земном шаре опреснительных
установок составила в 1968 г. 800 тыс.м3, в 1970 г.— 1114 тыс. м3,
а, согласно прогнозам, к концу 70-х годов не достигнет и
4 млн. ж3 [25, 26]. Если опреснение морской воды будет развивать¬
ся более высокими темпами и удастся резко удешевить его, за¬
дача водоснабжения больших территорий не будет решена пол¬
ностью из-за необходимости подачи опресненной воды на значи¬
тельные расстояния.
Являясь источниками водоснабжения, реки, озера и водохра¬
нилища одновременно служат коллекторами сточных вод, коли¬
чество которых растет пропорционально водопотреблению. Био¬
логическая жизнь водоемов, их способность к самоочищению
нарушается присутствием нефти и масел, фенола, синтетических
детергентов, пестицидов, целого ряда микроэлементов, анализ
содержания в воде которых сильно затруднен. Известны много¬
численные примеры, когда загрязнение воды в разных районах
нашей планеты достигало катастрофических размеров. В резуль¬
тате гидротехнического строительства усилилось развитие водо¬
Искусственный шелк, капрон, нитрон
Канролактам
Фотохимические товары
Химико-фармацевтические товары
550-750
3500-4500
6900-9000
1900-17 000
[21-23].
10
ВВЕДЕНИЕ
рослей и планктона и, как следствие, появились неприятные за¬
пахи и привкусы воды.
На территории СССР и других стран введены в действие но¬
вые законы о водопользовании и охране водных ресурсов. Строго
регламентировано качество сбросных вод, максимально исполь¬
зуется оборотное водоснабжение, запрещено или ограничено про¬
изводство медленноокисляющихся веществ. Борьба за чистоту
водоемов уже дала результаты. Но использование природных вод
все же невозможно без их глубокой физико-химической очистки,
в которой коагулированию принадлежит ведущая роль.
Расширение сферы применения коагулянтов происходит и в
связи с более высокими требованиями к качеству питьевой воды.
Если примерно до 1880 г. при оценке питьевых качеств воды ис¬
ходили из самых общих физико-химических показателей, а позд¬
нее — из биологических [27], то теперь дополнительно нормиро¬
вано содержание многих микроэлементов. Хотя I Международный
конгресс в Амстердаме рекомендовал использовать для питьевых
целей воду с содержанием взвешенных примесей Не более 0,5 мг/л,
некоторые исследователи полагают, что величина предельно
допустимых концентраций этих примесей должна быть много
ниже: установлена прямая связь между качеством воды и числом
сердечно-сосудистых заболеваний [28].
В соответствии с действующими нормативами по некоторым
показателям к качеству питьевой воды предъявляют такие же и
даже более высокие требования, чем к химически чистым реак¬
тивам. В международных [29] и европейских [30] стандартах ука¬
заны предельно допустимые концентрации некоторых веществ
(в мг/л): синтетические детергенты — 0,2, свинец — 0,1, медь,
марганец, мышьяк, сероводород, хром и цианиды — 0,05, кад¬
мий и селен — 0,01, фенолы — 0,001. Рекомендуются следую¬
щие пороговые концентрации фосфорорганических инсектицидов
(по органолептическому признаку, в мг/л): хлорофос и карбофос —
0,05, тиофос — 0,03, метафос — 0,02 [31]. С I1 января 1975 г.
в СССР введен в действие новый государственный стандарт на
питьевую воду (ГОСТ 2874-73), в котором по сравнению с ранее
действующими стандартами повышены требования к мутности
воды и концентрации микроэлементов. Так, например, содержа¬
ние селена в питьевой воде не должно превышать 0,001, а берил¬
лия — 0,0002 мг/л [32].
В ряде отраслей народного хозяйства особые требования предъ¬
являются к качеству технической воды. На паросиловых уста¬
новках эти требования касаются жесткости воды, содержания
кислорода и свободной углекислоты, кремнекислоты и фосфатов.
В воде, предназначенной для некоторых химических производств,
а также для заводнения нефтяных пластов, содержание железа
не должно превышать 0,05—0,2 мг/л.
ЛИТЕРАТУРА
11
Наконец, отметим, что за последние годы значительно расши¬
рилось применение коагулянтов при очистке промышленных и
бытовых сточных вод, при обработке осадков. Если раньше
хозяйственно-бытовые стоки проходили почти исключительно
механическую и биохимическую обработки, теперь в дополнение
к ним, в особенности для нормализации работы перегруженных
очистных сооружений [33], успешно применяют коагулирование.
Введены в строй первые установки, на которых методы биологи¬
ческой очистки полностью заменены физико-химическими мето¬
дами, в том числе коагулированием [34].
Совершенствование приемов и методов реагентной обработки
воды было бы невозможным без теории коагуляционных и фло-
куляционных явлений, в развитии которой выдающаяся роль
принадлежит советским ученым Н. П. Пескову, Н. А. Фуксу,
П. А. Ребиндеру, Б. В. Дерягину и другим.
В плане развития идей коллоидной и физической химии при¬
менительно к процессам очистки природных и сточных вод над
повышением эффективности коагулирования успешно работали
и работают в СССР И. Э. Апельцин, Ю. И. Вейцер, С. А. Возне¬
сенский, И. Т. Гороновский, И. Ф. Ефремов, В. А. Клячко,
B. П. Криштул, Л. А. Кульский, Е. Ф. Кургаев, И. С. Лавров,
Д. М. Минц, В. Т. Турчинович, С. А. Шуберт, С. В. Яковлев и
многие другие; за границей — Е. Биин, А. Блэк, Г. Гудзон,
C. Кавамура, В. Ля Мэр, У. Ланжелье, Е. Матиевич, С. Мэтт¬
сон, С. Мацкрле, М. Нода, Ч. О’Мелиа, Р. Пакхам, Т. Рэддик,
В. Стамм и другие.
Л итература
1. М. N. Baker. The Quest for Pure Water. N. Y., American Water Works
Association, 1948.
2. M. Frison. Techn. sanit. et municip., 48, 158 (1953).
3. Руководство к устройству водопроводов, составленное бароном Дель¬
вигом, полковником корпуса инженеров путей сообщения. М., Типо¬
графия Ф. Готье, 1856, стр. 6 раздела «Способы очищения воды».
4. Manual of British Water Engineering Practice. Cambridge, Heffer and
Sons Ltd., 1961.
5. A. S. Hyatt. Пат. США 293740 (1884).
6.С. А. Вознесенский. Физико-химические процессы очистки воды. М.,
Госстройиздат, 1934, стр. 8.
7. К. Е. Oehler. Уот Wasser 1963, Bd. 30. Weinheim/Bergstr., 1964, S. 127.
8. J. E. Singley, A. P. Black. J. Amer. Water Works Assoc., 59, 1549 (1967).
9. P. T. Austen, F. A. Wilber. Annual Beport of the State Geologist of New
Jersey, 1884, p. 141.
10. G. W. Fuller. Report of the Investigations into the Purification of the
Ohio River Water at Louisville. N. Y., D. Van Nostrand, 1898.
И. П. И. Пискунов. Водоснабжение фабрик и заводов. М., Госстройиздат.
1951, стр. 44.
12. Ф. К. Зембиикий. Очистка воды химическим способом для питья. СПб.,
1886.
12
ВВЕДЕНИЕ
13. Н. Л. Бунге. Обесцвечивание днепровской воды химическим способом
(сообщение на II Русском водопроводном съезде). СПб., изд. Постоянного
бюро русских водопроводных съездов, 1895.
14. Ф. А. Данилов. Водопроводы русских городов. СПб., изд. Постоянного
бюро русских водопроводных съездов, 1911, стр. 168.
15. Е. V. Bull. Proc. Inst. Water Supply Assoc. N. Y., 1912, p. 119.
16. T. Stones. J. and Proc. Inst. Sewage Purific., N 4, 350 (1955).
17. Англ. пат. 823082 (1959).
18. В. E. Тимонов. Водоснабжение и водостоки, вып. 1. СПб., Типография
Ю. Н. Эрлих, 1899, стр. 128.
19. Л. А. Кулъский, В. В. Даль. Проблемы чистой воды. Киев, «Наукова
думка», 1974, стр. 106.
20. Е. 3. Аграноник, II. Д. Бондарев, В. П. Иванов и др. Водоснабжение
и канализация предприятий химической промышленности. JL, Стройиз-
дат, 1967, стр. 130.
21. Сб. «V Международный конгресс по водоснабжению». Под ред. Ф. А. Ше¬
велева. М., Изд-во МКХ РСФСР, 1963, стр. 154.
22. С. М. Драчев. Водоснабжение и санитарная техника, № 9, 40 (1969).
23. В. И. Лялъко. Вечно живая вода. Киев, «Наукова думка», 1972, стр. 98.
24. В. Р. Лозанский. Сб. «Водоснабжение, канализация, гидротехнические
сооружения», вып. 13. Киев, «Вуд1вельник», 1971, стр. 58.
25. В. А. Клячко. Водоснабжение и санитарная техника, № 8, 35 (1969).
26. С. М. Wong. Proc. Seminary Nucl. Power held in Bombay. Bombay,
1970, p. 29.
27. Н. А. Кашкаров. Современные способы очистки воды. Томск, Типо¬
литография Сибирского товарищества печатного дела, 1912, стр. 37.
28. И. Е. Элъпинер. Природа, № 7, 23 (1966).
29. Международные стандарты питьевой воды. Женева, изд. Всемирной ор¬
ганизации здравоохранения, 1973, стр. 37.
30. Европейские стандарты питьевой воды. Женева, изд. Всемирной орга¬
низации здравоохранения, 1972, стр. 35.
31. С. II. Черкинский, К. И. Акулов, Г. Н. Красовский. Гигиена и санитария,
№ 4, 17 (1966).
32. С. Н. Черкинский. Технология очистки питьевой воды и санитарно-ги¬
гиенические требования к ее качеству (материалы семинара). Изд. Моск.
Дома научно-техн. пропаганды им. Ф. Э. Дзержинского, 1974, стр. 20.
33. К. Klantsch. Gas-Wasser-Abwasser, 53, 301 (1973).
34. Environ. Sci. and Technol., 7, 804 (1973).
Глава I
ФЕНОМЕНОЛОГИЯ ВОДНЫХ ДИСПЕРСИИ
I. 1. СТЕПЕНЬ ДИСПЕРСНОСТИ И ФОРМА ЧАСТИЦ
Система называется дисперсной, если она включает дискрет¬
ные образования (дисперсную фазу), распределенные в массе ос¬
новного несущего вещества (дисперсионной среды). По размеру
частиц дисперсной фазы (степени дисперсности) дисперсные сис¬
темы делят на три группы [1, стр. 15]:
1. Грубодисперсные системы. Определяющий размер частиц
(для сферических — диаметр, для кубических—длина ребра,
для вытянутых — эквивалентный диаметр) более 0,1 мкм. К та¬
ким системам относят суспензии, эмульсии.
2. Коллоидные системы. Размер частиц 0,1 мкм — 1 нм.
Частицы включают от нескольких единиц до сотен миллионов моле¬
кул вещества. Коллоидные системы называют также золями,
а в случае, когда дисперсионной средой является вода,— гидро¬
золями.
3. Истинные растворы. Размер частиц не превышает 1 нм, т. е.
соответствует размерам отдельных молекул или ионов.
Особую, четвертую группу составляют растворы высокомоле¬
кулярных веществ (ВМВ), в которых отдельные макромолекулы
могут объединяться в «пачки», достигающие размеров коллоид¬
ных частиц.
В данной классификации верхний предел дисперсности кол¬
лоидных систем (1 нм) обусловлен тем, что при дальнейшем дроб¬
лении вещества мы переходим уже от агрегатов молекул к от¬
дельным молекулам, нижний предел дисперсности (0,1 мкм) свя¬
зан, как будет показано ниже, с резким снижением интенсивности
теплового движения частиц.
Классификация носит условный характер. Грубодисперсные
и коллоидные системы, в которых существует физическая грани¬
ца раздела фаз, в отличие от истинных (гомогенных) растворов
являются гетерогенными, т. е. многофазными (в простейшем слу¬
чае — двухфазными). Причем суспензии с частицами размером
порядка нескольких микрометров проявляют свойства, схожие
с коллоидными системами, и их часто объединяют под общим на¬
званием микрогетерогенных систем.
С повышением степени дисперсности частиц площадь поверх¬
ности раздела фаз резко возрастает. По величине удельной по¬
верхности, подсчитываемой как частное от деления суммарной
площади поверхности частиц на общий объем или вес дисперсной
14
ГЛАВА I. ФЕНОМЕНОЛОГИЯ ВОДНЫХ ДИСПЕРСИЙ
фазы, коллоидные системы занимают особое положение среди дис¬
персных систем: в истинных растворах поверхность раздела в
обычном смысле отсутствует, а удельная поверхность грубодис¬
персных систем невелика. Поэтому поведение коллоидных раст¬
воров в гораздо большей мере, чем каких-либо других дисперс¬
ных систем, определяется поверхностными явлениями.
Природные и сточные воды, как и большинство дисперсных
систем, содержат частицы разного размера. Такие системы в от¬
личие от монодисперсных, т. е. имеющих частицы одинакового
размера, называются полидисперсными. Важнейшей характери¬
стикой полидисперсных систем является функция распределения
частиц по размеру, показывающая отношение числа частиц с дан¬
ным размером к общему-числу частиц в системе.
По форме, имеющей очень большое значение для коагуляцион¬
ных взаимодействий и структурообразования, коллоидные и гру¬
бодисперсные частицы могут быть изодиаметрическими и анизо-
диаметрическими. Размеры первых во всех трех измерениях
приблизительно равны. К ним относятся частицы сферические,
кубические. Анизодиаметрические частицы вытянуты в одном или
двух измерениях, они могут быть дискообразными, игольчатыми,
палочковидными. Эллипсоидальные частицы занимают некоторое
промежуточное положение.
I. 2. МОЛЕКУЛЯРНО-КИНЕТИЧЕСКИЕ СВОЙСТВА
Тепловое (броуновское) движение. Частицы дисперсной фазы
испытывают удары молекул дисперсионной среды, находящихся
в непрерывном и хаотическом тепловом движении, и вследствие
этого сами перемещаются в пространстве. Перемещение является
результатом усредненного действия всех ударов и происходит со
скоростью, гораздо меньшей, чем скорость движения молекул.
При этом мелкие частицы перемещаются в различных направле¬
ниях. Частицы крупные (3—5 мкм) обладают большей массой, а
вероятность взаимной компенсации ударов с разных сторон у
них возрастает. Поэтому они совершают лишь небольшие колеба¬
тельные движения со скоростью долей миллиметра в секунду.
Частицы диаметром более 5 мкм практически не подвержены
броуновскому движению. Средняя величина смещения частицы
АХ за определенный промежуток времени Ат определяется урав¬
нением Эйнштейна:
где R — газовая постоянная; Т — абсолютная температура;
N — число Авогадро; т] — динамическая вязкость среды; г —
радиус частиц.
Дт
Зят]г ’
(1.1)
1.2. МОЛЕКУЛЯРНО-КИНЕТИЧЕСКИЕ СВОЙСТВА
15
Диффузия. Под влиянием теплового движения происходит
самопроизвольное выравнивание концентрации частиц по объему
раствора — их диффузия. Скорость диффузии подчиняется закону
Фика:
-Ат = DS {Ас/АХ)Дт, (1.2)
где Ат — количество продиффундировавшего вещества; D —
коэффициент диффузии; S — площадь, через которую происходит
диффузия; Ас/АХ — градиент концентрации вещества.
Знак минус перед Ат указывает на то, что процесс идет в нап¬
равлении уменьшения концентрации вещества.
Из уравнения (1.2) становится понятным, что коэффициент
диффузии численно равен количеству вещества, продиффунди-
ровавшему за единицу времени через единицу площади сечения,
нормального к направлению диффузии, при градиенте концент¬
рации, равном единице. Между коэффициентом D и средним квад¬
ратичным перемещением частицы за время Ат существует простая
связь, подтверждаемая результатами экспериментов:
D = АХ2/2 Ах. (1.3)
Из уравнений (1.1) и (1.3) получаем выражение Эйнштейна
для коэффициента диффузии сферических частиц:
n^RT 1 п/л
N бят]г ' ( )
Это уравнение имеет важное значение для практических рас¬
четов, так как по найденным экспериментально значениям D
позволяет определить размер частиц. Для несферических частиц
выражение блтпг заменяется более сложным, и величина коэффи¬
циента D в этом случае меньше, чем для сферических частиц той
же массы.
Для молекул и ионов неорганических веществ в водной среде
величина D равна приблизительно 1 ■ 10_6 см2/сек, а для коллоид¬
ных частиц она ниже на 2—4 порядка. Скорость диффузии мак¬
ромолекул высокомолекулярных веществ при одинаковых ус¬
ловиях близка к скорости диффузии коллоидных частиц.
Вязкость. Течение дисперсных систем отличается от течения
истинных растворов, поскольку частицы твердой фазы искрив¬
ляют пути движения отдельных молекул жидкости. Как следст¬
вие у дисперсных систем наблюдается ранняя турбулентность
при меньших, чем для истинных растворов, числах Рейнольдса.
Частицы твердого вещества сужают также пространство, занятое
жидкостью, и увеличивают градиент скорости в поперечном се¬
чении потока. Поэтому вязкость дисперсной системы всегда выше
вязкости среды.
16
ГЛАВА I. ФЕНОМЕНОЛОГИЯ ВОДНЫХ ДИСПЕРСИЙ
Вязкость разбавленного коллоидного раствора т], согласно
статистическим расчетам, выполненным Эйнштейном, может быть
описана уравнением
где 1|0 — вязкость дисперсионной среды; с0 — объемная концент¬
рация дисперсной фазы.
Уравнение (1.5) справедливо для случая, когда частицы шаро¬
образны и не взаимодействуют друг с другом. Объемная концент¬
рация частиц, покрытых гидратным слоем толщиной hr, вычисля¬
ется по уравнению [2]
где г — радиус.
Для частиц анизодиаметрических вязкость системы всегда
выше, чем определенная по уравнению (1.5), так как жидкость,
попадающая в объем, образующийся вокруг таких частиц при их
вращении, становится как бы связанной с частицей, увеличивает
ее объем [1, стр. 336].
В случае эллипсоидных частиц
где А и В — коэффициенты, зависящие от степени асимметрии
частиц [3, стр. 46].
Имеющиеся экспериментальные данные по вязкости суспен¬
зий, образующихся в процессе очистки воды гидролизующимися
коагулянтами, подтверждают справедливость соотношения (1.5).
С увеличением скорости потока вязкость систем, содержащих
анизодиаметрические частицы, уменьшается, так как последние
ориентируются вдоль потока и их вращение затрудняется. Связь
между логарифмом вязкости и температурой среды в простейшем
случае близка к линейной, но часто нарушается из-за влияния
температуры на агрегативную устойчивость частиц. С увеличе¬
нием концентрации дисперсной фазы вязкость системы возрас¬
тает и уже не может быть описана уравнением (1.5). Причины
этого заключаются как в изменении гидродинамических условий,
так и в проявлении сил притяжения или отталкивания между
частицами.
Вязкость золей, содержащих заряженные частицы, зависит,
кроме всего прочего, от степени дисперсности частиц, величины
их заряда. По Смолуховскому [4, стр. 486]
где — удельная электропроводность; е — диэлектрическая
постоянная; £ — электрокинетический потенциал двойного элект¬
рического слоя (см. ниже).
г] = т|0(1 + 2,5 с0),
(1.5)
Со = с0 (1 + 3hT/r),
(1.6)
Г] = rio (1 + Ас0 + Вс02),
(1.7)
(1.8)
1.2. МОЛЕКУЛЯРНО-КИНЕТИЧЕСКИЕ СВОЙСТВА
17
С увеличением степени дисперсности частиц и уменьшением
абсолютной величины их заряда уравнение (1.8) переходит в
уравнение (1.5).
В одной из экспериментальных работ [5] показано на примере
суспензий угля и других минералов, что при с0 < 0,2 распреде¬
ление частиц по размерам на величину вязкости не влияет, тогда
как при с0 = 0,2—0,4 с увеличением размера частиц вязкость
возрастает. В другой работе [6] установлено, что гидрофобиза-
ция стеклянных и кварцевых частиц за счет их покрытия силико¬
новой смазкой приводит к уменьшению эффективного объема час¬
тиц и величины их заряда и тем самым понижает вязкость суспен¬
зий.
Вязкость растворов ВМВ значительно выше вязкости колло¬
идных растворов той же концентрации, что объясняется способ¬
ностью макромолекул к образованию ассоциатов. С молекулярным
весом (MB) полимера вязкость связана соотношением
ц = К [МВ]«, (1.9)
где К — коэффициент, постоянный для раствора данного ВМВ в
данном растворителе; а — коэффициент, зависящий от формы
и гибкости макромолекул.
С увеличением скоростного градиента вязкость растворов ВМВ
вследствие разрушения ассоциатов и ориентации вытянутых моле¬
кул вдоль потока (как это имеет место в коллоидных растворах с
анизодиаметрическими частицами) уменьшается. Добавка веществ,
способных влиять на взаимодействие коллоидов и макромоле¬
кул, изменяет вязкость дисперсных систем.
Кинетическая устойчивость. Существует два типа устойчиво¬
сти дисперсных систем: агрегативная и кинетическая.
Агрегативная устойчивость — это способность системы сохра¬
нять степень дисперсности своих частиц. Она объясняется нали¬
чием вокруг частиц двойного электрического слоя ионов и соль¬
ватных оболочек и будет рассмотрена ниже.
Под кинетической (седиментационной) устойчивостью пони¬
мают способность частиц противостоять силе тяжести. Каждая
частица дисперсной фазы подвержена действию двух противопо¬
ложных сил: силы тяжести, вызывающей седиментацию, и силы
диффузии, препятствующей ей. Соотношение сил зависит от сте¬
пени дисперсности частиц: чем крупнее частицы, тем меньше они
подвержены броуновскому движению и тем интенсивнее седимен-
тируют.
Суспензии, в которых диффузия отсутствует, являются кине¬
тически неустойчивыми системами: их частицы осаждаются в
течение сравнительно небольшого отрезка времени. К абсолютно
кинетически устойчивым системам относятся истинные растворы.
Коллоидные растворы составляют промежуточный тип систем.
18
1 ЛАВА I. ФЕНОМЕНОЛОГИЯ ВОДНЫХ ДИСПЕРСИЙ
V, m/vac
JO1
5
3
г
10°
S
3
г
/а
10'z / 3 S №'7 2 J S 70°
Рис. 1.1. Зависимость v от d при
разных значениях yt (температу¬
ра воды 10° С)
В этих растворах в результате
взаимодействия между силами тя¬
жести и диффузии устанавливает¬
ся равновесие, характеризующееся
постепенным уменьшением кон¬
центрации частиц в направлении
снизу вверх.
Чем меньше частицы, тем мед¬
леннее они опускаются под дей¬
ствием силы тяжести и тем боль¬
ший срок требуется для достиже-
ния равновесия. Установлению
равновесия мешают колебания тем¬
пературы в системе, толчки и со¬
трясения. Поэтому практически
все золи обнаруживают одинако¬
вую концентрацию дисперсной фа¬
зы по высоте слоя.
Растворы ВМВ обладают, как
правило, высокой кинетической
устойчивостью, что связано с малой
плотностью растворенного веще¬
ства.
Для подсчета скорости и осаж¬
дения сферических частиц, не под¬
верженных тепловому движению,
применимо известное уравнение
Стокса:
где g — ускорение силы тяжести;
рт и рв — плотности соответствен¬
но вещества дисперсной фазы и
воды; d — диаметр частиц.
Если осаждающиеся частицы
многокомпонентны, величина рт
определяется с учетом плотности и
относительного объема каждого
компонента.
С увеличением температуры от
5 до 30° С вязкость воды умень¬
шается вдвое, а плотность — лишь
на 2%. Поэтому повышение тем¬
пературы воды способствует седи¬
ментации частиц, не подвер-
1.3. АДСОРБЦИЯ НА ПОВЕРХНОСТИ РАЗДЕЛА
19
женных или мало подверженных тепловому движению. Если
вещество легче воды (например, эмульсия масла), величина
Рт — Рв в уравнении (1.10) имеет отрицательный знак, вместо
осаждения будет происходить всплывание частиц.
За меру устойчивости супензий обычно принимают величину,
обратную скорости осаждения ее частиц. В водоподготовке спо¬
собность к седиментации часто характеризуют величиной так на¬
зываемой «гидравлической крупности» — скорости падения частиц
в воде при температуре 10° С. Зависимость «гидравлической круп¬
ности» v от размера d и объемного веса ут частиц взвеси показана
на рис. 1.1.
Формула Стокса справедлива для идеальных систем. Факти¬
ческие скорости осаждения всегда отличаются от значений и,
подсчитанных по этой формуле, вследствие теплового движения
частиц, их неправильной формы и гидратации, объединения их в
агрегаты. В зависимости от объемной концентрации частиц или
их агрегатов (с0) изменяется соотношение между истинной (v) и
расчетной (и) скоростями осаждения:
v = и (I — с0)п, (1.11)
где п ~ 5.
В монодисперсных системах все частицы оседают с одинаковой
скоростью, поэтому вес накапливающегося осадка Р пропорцио¬
нален времени осаждения т (рис. 1.2, а). Для полидисперсных
систем прямая зависимость между Рит нарушается (рис. 1.2, б).
Кривые, показанные на рис. 1.2, носят название седиментацион-
ных и являются важнейшей характеристикой фракционного сос¬
тава дисперсной фазы.
Прежде чем перейти к обсуждению причин агрегативной устой¬
чивости дисперсных систем, рассмотрим в самых общих чертах
процессы, происходящие на границе раздела контактирующих
сред и определяющие, как будет видно ниже, поведение частиц
в ходе коагуляции.
3. АДСОРБЦИЯ НА ПОВЕРХНОСТИ РАЗДЕЛА
Поверхность частиц дисперсной фазы обладает свободной энер¬
гией, существование которой можно объяснить следующим об¬
разом [7]. Молекулы, атомы или ионы, находящиеся на поверх¬
ности раздела фаз, не равноценны тем же молекулам, атомам и
ионам, находящимся внутри каждой фазы. Внутри фазы моле¬
кулы окружены себе подобными и их силовое поле насыщено
симметрично. Поле молекул, лежащих на поверхности, асиммет¬
рично: часть его находится вне фазы и не насыщена. Эта ненасы-
щенность и является источником свободной энергии.
20
ГЛАВА I. ФЕНОМЕНОЛОГИЯ ВОДНЫХ ДИСПЕРСИЙ
Под действием поверхностных сил происходит изменение
концентрации компонентов в поверхностном слое по сравнению с
объемной фазой, т. е. протекает процесс адсорбции (сорбции).
Адсорбция может быть положительной, если энергия взаимодей¬
ствия растворенного вещества с молекулами, находящимися на
поверхности адсорбента, выше, чем с молекулами растворителя,
и отрицательной, когда наблюдается обратное явление. В даль¬
нейшем мы будем говорить в основном о положительной адсорб¬
ции, т. е. сгущении вещества на поверхности раздела фаз. В слу¬
чае неэлектролитов сорбируются молекулы вещества, в случае
электролитов — их ионы.
В процессах очистки природных вод важное значение имеют
и молекулярная, и ионная сорбции. '
Помимо своей главной задачи — извлечения из воды нежела¬
тельных примесей — адсорбирующее вещество (адсорбент) вы¬
полняет функции катализатора, так как молекулярные и ионные
реакции на поверхности раздела протекают обычно значитель¬
но быстрее, чем в объеме среды. Это объясняется увеличением
концентраций молекул и ионов, их ориентацией, ослаблением
связей между отдельными атомами.
Различают два основных вида адсорбции: физическую и хи¬
мическую. К силам, обусловливающим физическую адсорбцию,
относят молекулярные взаимодействия: 1) молекул с постоян¬
ным диполем (ориентационный эффект)-, 2) молекул с индуциро¬
ванным диполем (индукционный эффект)', 3) неполярных моле¬
кул (дисперсионный эффект), а также 4) силы, обусловливающие
водородную связь [1, стр. 851. Исследования последних лет привели
к выводу, что одной из важнейших составляющих адсорбционных
сил являются так называемые силы изображения, появление кото¬
рых связано с различием диэлектрических проницаемостей веще¬
ства дисперсных частиц и окружающей среды.
Физическая адсорбция протекает самопроизвольно и всегда
обратима. Количество вещества, адсорбированного на данном
участке поверхности в данный момент времени, определяется не
только перечисленными силами взаимодействия, но и силами де¬
сорбции, возникающими в результате теплового движения частиц.
Причем для каждой концентрации адсорбирующегося вещества
(адсорбтива) и для каждой температуры среды существует сос¬
тояние адсорбционного равновесия.
Силы, обусловливающие химическую адсорбцию (хемосорб¬
цию),— специфически валентные. В отличие от физической адсорб¬
ции хемосорбция обычно необратима. С повышением температуры
среды хемосорбция, требующая значительной энергии активации,
возрастает. Соединения, образующиеся при хемосорбции на по¬
верхности раздела фаз, нельзя рассматривать как новое вещество,
так как, несмотря на возникновение химических связей, поверх-
1.3. АДСОРБЦИЯ НА ПОВЕРХНОСТИ РАЗДЕЛА
21
Рис. 1.3. Изотерма адсорбции вещества из раствора
Рис. 1.4. Характер влияния температуры среды t на форму изотермы адсорб¬
ции (t1 > t2 > t3)
иостные атомы адсорбента продолжают сохранять связь с осталь¬
ными его атомами.
Провести резкую границу между физической и химической ад¬
сорбцией во многих случаях довольно трудно: адсорбция одних
и тех же веществ на одном и том же адсорбенте в одних условиях
может иметь физический, в других — химический характер.
В частности, повышение температуры снижает физическую, но
увеличивает химическую адсорбцию.
Расчеты показывают, что энергия притяжения молекул к по¬
верхности убывает обратно пропорционально третьей степени их
расстояния от поверхности и, следовательно, с увеличением это¬
го расстояния должна быстро уменьшаться. Однако, по резуль¬
татам экспериментов, толщина слоя адсорбированных молекул
может достигать значительной величины — нескольких тысяч
ангстрем [8, стр. 124]. Это указывает на дальнодействие адсорб¬
ционных сил, которое не нашло пока четкого теоретического
обоснования.
Количество адсорбированного вещества выражают по избытку
его, отнесенному к 1 см2 поверхности адсорбента или к 1 г адсор¬
бента. В первом случае это количество обозначается Г (молъ/см2),
во втором — V(моль/г). При постоянной температуре между ко¬
личеством физически адсорбированного вещества Г и его равно¬
весной концентрацией ср в объеме раствора существует динамиче¬
ское равновесие, описываемое так называемой изотермой адсорб¬
ции. В зависимости от природы сорбента и сорбтива, а также ус¬
ловий проведения сорбции изотермы имеют разную форму. При
хемосорбции на изотермах могут появляться ступени и резкие
изломы.
Одна из наиболее часто встречающихся изотерм (лэнгмюров-
ского типа) показана на рис. 1.3. Она может быть разделена на три
участка. Прямолинейный участок I характеризует такое состоя¬
22
ГЛАВА I. ФЕНОМЕНОЛОГИЯ ВОДНЫХ ДИСПЕРСИЙ
ние системы, когда поверхность адсорбента еще в значительной
степени свободна. Здесь величина Г пропорциональна ср. Второй
прямолинейный участок III, почти горизонтальный, соответствует
большим ср и характеризует адсорбцию в условиях, когда поверх¬
ность адсорбента почти полностью насыщена адсорбтивом. Ве¬
личина Г уже практически не зависит от ср. Криволинейный
участок II отвечает промежуточной стадии.
С повышением температуры адсорбция, отвечающая состоя¬
нию равновесия, уменьшается. Однако конечное значение коли¬
чества адсорбированного вещества (Г,*) не меняется: как показа¬
но на рис. 1.4, концы изотерм адсорбции при разных температу¬
рах постепенно сближаются.
С известным приближением, довольно часто допустимым в
практических расчетах, для описания изотермы адсорбции из
растворов можно использовать эмпирическое уравнение Фрейнд¬
лиха:
Г = Вср/п, (1.12)
в котором (3 — коэффициент, равный по своему физическому
смыслу величине адсорбции при ср = 1 молъ/л, а п — константа,
значение которой при адсорбции из водных растворов находится
в пределах 2—10.
Для случая экспоненциального распределения активных
центров уравнение (1.12) нашло теоретическое подтверждение
[3, стр. 85].
К настоящему времени предложено множество различных тео¬
рий адсорбции. Давать их обзор не входит в наши задачи, тем
более, что это уже сделано другими исследователями [1, 8]. От¬
метим лишь, что ни одна из существующих теорий (в том числе
теория БЭТ, наиболее полно учитывающая разные стороны про¬
цесса) не является универсальной вследствие специфического
характера адсорбции в разных условиях. Для решения практи¬
ческих задач, связанных с очисткой воды от дисперсных и истин¬
но растворенных примесей, нам вполне достаточно воспользовать¬
ся представлениями Лэнгмюра о мономолекулярной адсорбции,
сохраняющими свое значение до настоящего времени.
В своей теории, развитой еще в 1915 г., Лэнгмюр исходил из
следующих предположений.
1. Адсорбция происходит не на всей поверхности сорбента,
а лишь на отдельных ее активных участках — ребрах, выступах.
2. Каждый активный участок, адсорбируя молекулу адсорб-
тива, становится уже не способным к дальнейшей адсорбции. Та¬
ким образом, на поверхности адсорбента образуется лишь моно-
молекулярный слой адсорбтива.
3. Адсорбированные молекулы удерживаются на активных
участках только в течение определенного времени. В результате
1.3. АДСОРБЦИЯ НА ПОВЕРХНОСТИ РАЗДЕЛА
23
флуктуации (непрерывного колебания) энергии молекулы могут
оторваться от этих участков, и их место занимают новые моле¬
кулы.
4. Взаимодействием между адсорбированными молекулами
можно пренебречь.
На основе этих предположений Лэнгмюр дал простую зави¬
симость 1 величины молекулярной адсорбции от равновесной
концентрации адсорбтива:
Г = ГэоТ^-, (1.13)
где Гоо — максимальная адсорбция; К — константа адсорбцион¬
ного равновесия.
Аналогичное выражение может быть получено исходя из зако¬
на действия масс в любой гетерогенной химической реакции.
Как будет ясно из последующего изложения, использование вы¬
ражения (1.13) позволяет интерпретировать результаты некото¬
рых исследований по очистке воды гидролизующимися коагу¬
лянтами.
На степень адсорбции сильное влияние оказывают свойства
адсорбента, адсорбтива и среды и, в частности, интенсивность
поля действующих молекулярных сил — полярность. В качестве
количественной характеристики полярности твердых частиц,
погруженных в жидкость, используют величину удельной сво¬
бодной энергии на поверхности частиц — поверхностное натяже¬
ние.
В соответствии с правилом Ребиндера адсорбция веществ бу¬
дет происходить, если полярность их лежит между полярностью
среды и адсорбента. Следовательно, чем больше разность поляр¬
ностей между растворяемым веществом и раствором, т. е. чем ме¬
нее растворимо вещество, тем лучше оно будет адсорбироваться.
Действительно, неполярные гидрофобные вещества (в частности,
активный уголь) хорошо адсорбируют поверхностно-активные ве¬
щества, что широко используется в водоподготовке. Помимо по¬
лярности, важную роль играют другие параметры. С увеличением
молекулярного веса адсорбтива адсорбция возрастает. Этим
объясняется, в частности, хорошая адсорбция красителей.
Вещества пористые и с шероховатой поверхностью адсорбиру¬
ют сильнее. Поэтому аморфные адсорбенты всегда эффективнее
кристаллических. Чем уже поры адсорбента и чем крупнее моле¬
кулы адсорбтива, тем меньше и медленнее адсорбция. Для ком¬
пенсации недостаточной скорости диффузии и ускорения наступ¬
ления адсорбционного равновесия часто применяют перемеши¬
вание жидкости.
1 Вывод уравнения Лэнгмюра приводится в учебниках коллоидной и физи¬
ческой химии.
24
ГЛАВА I. ФЕНОМЕНОЛОГИЯ ВОДНЫХ ДИСПЕРСИЙ
Адсорбция электролитов, имеющая наиболее важное значение
в водных растворах, резко отличается от молекулярной адсорб¬
ции. Участки поверхности адсорбента, несущие заряд, как пра¬
вило, адсорбируют противоположно заряженные ионы, а из ио¬
нов разной валентности сильнее адсорбируются многовалентные.
Сказывается и влияние природы ионов. Так, из ионов одинаковой
валентности лучше адсорбируются ионы большего радиуса: они
сильнее поляризуются и обладают меньшей гидратацией, что уве¬
личивает силы их притяжения к поверхности. По способности
адсорбироваться ионы могут быть расположены в следующие ря¬
ды, называемые лиотропными:
Анионы SOf" < Cl" < Br- < N0“ < J" < CNS~ < < 0H~
Катионы Na+ < K.+ < Mg2+ < Ca2+ < Ba2+ < Al3+ < Fe3+ < < H+
Первостепенное практическое значение при коагулировании
водных примесей, умягчении и обессоливании воды имеет обмен¬
ная адсорбция, в ходе которой адсорбент, поглощая опреде¬
ленное количество каких-либо ионов, выделяет одновременно
в раствор эквивалентное количество других ионов того же знака,
вытесненных с поверхности. Происходит обмен ионов.
Обменная адсорбция обладает следующими особенностями:
1) к обмену способны только определенные ионы;
2) адсорбция не всегда обратима;
3) адсорбция протекает медленнее, чем адсорбция неэлектро¬
литов;
4) при.обменной адсорбции может происходить изменение pH
среды, когда обмениваемыми ионами являются Н+ или ОН'.
Адсорбция коллоидов 2, по Пескову [9], тем больше, чем выше
степень их дисперсности и меньше устойчивость. Поскольку с
уменьшением степени дисперсности частиц устойчивость их сни¬
жается, кривая, изображающая зависимость адсорбции коллои¬
дов от степени дисперсности, проходит через максимум. Скорость
адсорбции практически всегда определяется скоростью диффузии,
и часто 90—95% адсорбтива связывается адсорбентом в течение
первых двух-трех секунд.
Кривые скорости адсорбции при разных температурах t имеют
вид, показанный на рис. 1.5, и описываются уравнением
dTJdx = К (Гр - Гт), (1.14)
где К — коэффициент; Гр — количество адсорбированного ве¬
щества, отвечающее установлению адсорбционного равновесия в
данных условиях.
Выражение (1.14) можно использовать для приближенных
расчетов скорости адсорбции истинно растворенных веществ на
2 Правильнее, по-видимому, говорить об адагуляции коллоидов (см. ниже).
1.3. АДСОРБЦИЯ НА ПОВЕРХНОСТИ РАЗДЕЛА
25
Г
'‘'/777? V7,
О
Рис. 1.5. Характер влияния температуры среды t на скорость адсорбции
вещества из раствора (tx > i2)
Рис. 1.6. Капля воды на несмачиваемой (а) и смачиваемой (б) поверхностях
грубодисперсной фазе, возникающей в ходе гидролиза коагу¬
лянтов.
С повышением температуры скорость адсорбции возрастает,
но одновременно, вследствие увеличения интенсивности теплового
движения, уменьшается величина адсорбции, отвечающая равно¬
весному состоянию системы. Поэтому кривые для температур tx
и t2 < £i, как показано на рис 1.5, пересекаются.
Говоря о свойствах активного угля как сорбента, мы употре¬
били термин «гидрофобное вещество». По степени прочности связи
между дисперсной фазой и дисперсионной средой все дисперсные
системы делят на лиофобные и лиофильные или для случая, когда
дисперсионной средой является вода,— на гидрофобные и гидро¬
фильные.
На гидрофильных поверхностях преобладают ненасыщенные
атомные, ионные или полярные связи, что и обусловливает взаи¬
модействие поверхностей с молекулами воды или их ассоциата-
ми. На гидрофобных поверхностях преобладают насыщенные
связи, потому они слабо взаимодействуют с водой. Степень гид-
рофильности различных материалов определяют чаще всего ве¬
личиной краевого угла смачивания 0 водой поверхности материа¬
лов (рис. 1.6),' принимая условно, что при 0 < 90° поверхность
хорошо смачивается водой и ее можно считать гидрофильной;
при 0 > 90й поверхность гидрофобна [10, стр. 11].
По Ребиндеру [И], критерием для оценки гидрофобности или
гидрофильности дисперсных частиц является величина поверхно¬
стного натяжения, которая может быть больше или меньше не¬
которого граничного значения (ат), определяемого кинетической
энергией теплового движения частиц:
ат =_ fiRT/Nr2,
(1.15)
26
ГЛАВА I. ФЕНОМЕНОЛОГИЯ ВОДНЫХ ДИСПЕРСИЙ
где р — безразмерный множитель, равный примерно 30; г — сред¬
ний размер частиц, участвующих в броуновском движении, рав¬
ный приблизительно 10"6 см.
При комнатной температуре величина ат составляет около
0.1 эрг-см~2. Гидрофобные системы характеризуются высоким
поверхностным натяжением (а ат), а потому мало гидрати¬
рованы и обладают резко выраженной границей раздела фаз. Сте¬
пень их дисперсности зависит от условий образования и стаби¬
лизации. Гидрофильные системы, напротив, обладают малым по¬
верхностным натяжением (о < от) и проявляют сродство с во¬
дой. Четкая граница раздела фаз отсутствует, а дисперсность
частиц не является случайной величиной, зависящей от условии
образования и стабилизации, а определяется природой обеих фаз.
Нужно, однако, иметь в виду, что использование величины от
в качестве критерия гидрофильности системы затруднено: усло¬
вию о < от отвечает лишь особая немногочисленная группа сис¬
тем, занимающих по существу промежуточное положение между
коллоидными и истинными растворами [12]. Правильнее говорить,
что лиофильность или лиофобность зависят от природы сил, дей¬
ствующих на поверхности частиц: обусловлены ли эти силы элект¬
рическим взаимодействием ионных атмосфер, окружающих кол¬
лоидные частицы, или они имеют неионную природу [13]. Во вся¬
ком случае четкой границы между гидрофильными и гидрофоб¬
ными системами провести нельзя. К гидрофобным системам отно¬
сят золи металлов, к гидрофильным — желатину, агар-агар, крах¬
мал. Промежуточный тип систем составляют золи кремнекислоты,
гидроокиси железа, алюминия, хрома и других металлов. Степень
гидрофильности этих систем зависит от pH среды [14].
1. 4. АГРЕГАТИВНАЯ УСТОЙЧИВОСТЬ
Важнейшая особенность лиофобных золей и суспензий, определя¬
ющая всю сумму наблюдаемых поверхностных явлений, состоит
в существовании двойного электрического слоя ионов и скачка по¬
тенциала на границе раздела фаз. Причинами возникновения
двойного электрического слоя являются разница в диэлектри¬
ческих свойствах материала среды и дисперсной фазы, специфи¬
ческие молекулярные силы, обусловливающие избирательную
адсорбцию ионов из раствора, или ионизация поверхностных мо¬
лекул вещества самой дисперсной фазы.
Распределение ионов вблизи поверхности раздела определя¬
ется действием противоположно направленных сил: сил моле¬
кулярного притяжения, удерживающих ионы у поверхности, сил
электростатического притяжения или отталкивания и диффузион¬
ных сил, стремящихся выровнять концентрацию ионов в объеме
среды.
1.4. АГРЕГАТИВНАЯ УСТОЙЧИВОСТЬ
27
Строение двойного слоя можно представить схемой, показан¬
ной на рис. 1.7. Ионы, находящиеся в избытке на поверхности
(называемые потенциалобразующими ионами), компенсируются
противоионами. Часть ионов удерживается поверхностью на
сравнительно близком расстоянии, порядка нескольких ангст¬
рем, образуя так называемый плотный ионный слой. Остальные
ионы образуют внешний диффузный слой, в котором в упорядо¬
ченное распределение ионов вмешиваются силы теплового дви¬
жения. Под действием диффузии этот слой оказывается как бы
«размытым» в направлении от поверхности раздела в глубину
раствора. Заряд частицы обусловлен избытком ионов какого-
либо одного знака в структурной геометрической единице слоя.
Ионы, находящиеся в избытке, являются, таким образом, потен-
циалопределяющими. Помимо ионов, в образовании двойного слоя
участвуют молекулы, содержащие электрические диполи.
Теоретически диффузный слой ионов распространяется внутрь
среды на неограниченно большое расстояние. Поэтому употреб¬
ляемое в коллоидной химии понятие «толщина» двойного слоя
носит весьма условный характер. В теории сильных электролитов
«толщина» двойного слоя определяется величиной дебаевского
радиуса гд, связанной с концентрацией и валентностью ионов
симметричного электролита 3 следующей зависимостью:
в которой к — константа Больцмана; Т — абсолютная темпера¬
тура; е — заряд электрона; п — число ионов в 1 см3 раствора;
?,i — валентность ионов.
При комнатной температуре
где с — концентрация электролита (в моль!л).
Согласно выражению (1.16'), величина rD в зависимости от
концентрации электролита может изменяться на несколько по¬
рядков — от нескольких ангстрем (концентрированные растворы)
до сотен микрометров (разбавленные растворы).
Рассмотрим в качестве примеров строение частиц золей гид¬
роокисей железа и алюминия. По мнению большинства исследо¬
вателей (Песков, Кройт, Паули, Думанский и др.), потенциал-
определяющими ионами золя гидроокиси железа, полученного
при гидролизе FeCl3, могут быть катионы Fe3+, FeO+ или Fe2022+.
(1.16)
(1.16')
3 Симметричным называется такой электролит, в котором ионы противо¬
положного знака обладают одинаковой валентностью,
28
ГЛАВА I. ФЕНОМЕНОЛОГИЯ ВОДНЫХ ДИСПЕРСИЙ
Рис. 1.7. Схема строения двойного электрического слоя на твердой по¬
верхности
I — плотный слой; II — диффузный слой; М—N — граница поверхности; т—п—гра¬
ница между плотным и диффузным слоями
Рис. 1.8. Схема строения мицеллы гидроокиси железа
I •— плотный слой; II — диффузный слой
Каргин и др. [15—18] пришли к выводу, что потенциалопре-
деляющим может быть катион водорода. Для этого случая строе¬
ние частицы золя показано на рис. 1.8.
Частицу с окружающим ее плотным слоем ионов называют
гранулой, а с двойным слоем — мицеллой. Построение мицеллы
представляют следующим образом 4:
1) на поверхности Fe(OH)a адсорбируются ионы Н+;
2) противоионы С1“, связанные в плотном слое, уменьшают
заряд частицы, но не нейтрализуют его полностью. Знак заряда
гранулы определяется ионами Н+, находящимися в избытке;
3) противоионы С1~, имеющиеся в диффузном слое, нейтрали¬
зуют положительный заряд гранулы и делают мицеллу нейтраль¬
ной.
Структуру мицеллы Fe(OH)3 можно представить формулой
{[Ге (ОН)3]т-геН+ (п — х) С1_}+жС1_.
или
{[Fe (ОН)3]т • reFeOCl, FeO+ (п - ж) С1-}+а;С1-.
Для мицеллы гидроокиси алюминия, образованной при гид¬
ролизе A12(S04)3, можно написать по аналогии
{[ А1 (OFP3] rn • nH+V-2 (п - х) S0l~}+l/2xS02-
4 В действительности строение гранул гораздо сложнее. Из-за неоднород¬
ности материала самого ядра, шероховатости поверхности, неравномер¬
ности распределения адсорбционных центров и наличия дефектов и микро¬
примесей в кристаллах поверхность раздела электрически неоднородна,
1.4. АГРЕГАТИВНАЯ УСТОЙЧИВОСТЬ
29
или в предположении, что потенциалопределяющим ионом явля¬
ется А13+
{[ А1 (ОН)3] т • nAl3+V-2 (» - *)SO|-}+3M S02-.
Как и в случае Fe(OH)3, мнения о строении мицеллы гидро¬
окиси алюминия расходятся. В частности, по Халлу [19], потен¬
циалопределяющим может быть катион Al6(OH)JJ. Дальнейшее
изложение (см. гл. III) покажет, что вследствие полимеризации
продуктов гидролиза А13+ и Fe3+ и их способности образовывать
со многими ионами комплексные соединения приведенные выше
формулы гидрозолей носят весьма условный характер.
Полный скачок потенциала ф0 между твердой частицей и точ¬
кой в глубине раствора называют поверхностным или термодина¬
мическим потенциалом (рис. 1.9). В плотном слое, толщина d
которого может быть определена как расстояние от центра тяже¬
сти зарядов внутренней обкладки поверхности до плоскости, про¬
ходящей через центры ближайших к поверхности противоионов
(так называемой плоскости наибольшего приближения [8, стр.
197]), падение потенциала имеет линейный характер, а в диффуз¬
ном слое из-за существования объемного заряда эта линейность
нарушается.
Потенциал плоскости наибольшего приближения фх является
важнейшей характеристикой двойного слоя, но его величина не
может быть измерена экспериментально. Информацию о величине
Ф, можно получить лишь путем измерения другой, близкой к ф!
величины — электрокинетического потенциала t. Этот потен¬
циал, называемый обычно дзета-потенциалом (ДП), определяется
как потенциал плоскости скольжения фаз при их относитель¬
ном перемещении, вызванном внешними силами. Точное положе¬
ние плоскости (а, может быть, и слоя) скольжения неизвестно
[20], но во всяком случае для разбавленного электролита (како¬
вым является природная вода), когда дебаевский радиус очень
велик и падение потенциала происходит по пологой кривой (см.
рис. IV.2), можно считать ДП примерно равным потенциалу пло¬
скости наибольшего приближения фх.
Величина ДП может быть найдена, например, из результатов
микроэлектрофоретических измерений:
£ = Апци/еН, (1-17)
где и — средняя скорость электрофоретического переноса час¬
тицы; Н — градиент потенциала внешнего электрического поля.
Величина ДП частицы определяет ее способность к коагуля¬
ции и во многих случаях является основным критерием полноты
протекания процесса. Как видно из рис. 1.10, величина ДП за¬
висит от толщины диффузного слоя ионов. Чем сильнее размыт
30
ГЛАВА I. ФЕНОМЕНОЛОГИЯ ВОДНЫХ ДИСПЕРСИЙ
Рис. 1.9. Падение потенциала в двойном электрическом слое
Рис. 1.10. Влияние концентрации индифферентного электролита на тол¬
щину двойного слоя и величину электрокинетического (£) потенциала (кон¬
центрация электролита возрастает от кривой 3 к кривой 1)
диффузный слой, тем выше ДП, и, наоборот, при предельном сжа¬
тии диффузного слоя значение ДП приближается к нулю.В изо-
электрическом состоянии, когда дзета-потенциал равен нулю,
строение мицелл можно представить формулами
{[Fe(OH)3]m.«H+.nCl-}°,
{[H2Si03]m-«Si0f-.2reH+}'> .
В зависимости от степени сродства вещества дисперсной фазы
с окружающими его электролитами последние делятся на индиф¬
ферентные и неиндифферентные. Потенциалопределящие ионы
неиндифферентных электролитов способны повышать термодина¬
мический потенциал частиц. С другой стороны, ионы противопо¬
ложного знака, находящиеся с ними в паре, могут вызывать сжа¬
тие диффузного слоя и понижать тем самым ДП. При малых кон¬
центрациях неиндифферентного электролита обычно проявляется
первая тенденция, происходит увеличение ф0 и £. При больших
концентрациях на первый план выступает роль сопутствующих
ионов: происходит снижение ДП вплоть до перемены знака на
противоположный [1, стр. 292].
Индифферентные электролиты не влияют на величину ф0, но
сжимают двойной слой, уменьшая ДП.
С увеличением адсорбируемости противоионов дзета-потен-
циал снижается интенсивнее. Без труда проникают к поверхности
твердой фазы ионы Н+, имеющие по сравнению с прочими очень
малые размеры. Ионы гидроксила адсорбируются легко благода¬
ря своему большому дипольному моменту, поэтому pH среды ока¬
зывает сильное влияние на величину и знак ДП.
1.4. АГРЕГАТИВНАЯ УСТОЙЧИВОСТЬ
31
Кислые вещества (например, кремнекислота или танин) при¬
обретают в воде отрицательный заряд. Амфотерные соединения
(белки, гидроокиси некоторых металлов, почвенные коллоиды)
могут быть заряжены отрицательно или положительно. В природ¬
ной воде глинистые и кварцевые частицы имеют обычно отрица¬
тельный заряд, а гидроокиси алюминия или железа, образую¬
щиеся в ходе гидролиза коагулянтов,— положительный.
Снижение pH увеличивает положительный заряд золей, за¬
ряженных положительно, и уменьшает отрицательный заряд зо¬
лей, заряженных отрицательно. Особенно сильно влияние pH
среды на гидрозоли А1(ОН)3 и Fe(OH)3, являющиеся амфотерными
соединениями. В слабокислой среде (pH 4,5—6,5) избыток водо¬
родных ионов подавляет ионизацию гидроокисей по кислотному
типу и они ведут себя как основания:
А1 (ОН) з t; А1 (ОН)^ + ОН".
Потенциалопределяющим ионом будет А1(ОН)2+, и гидроокись
поэтому заряжается положительно.
В средах с pH < 4,5 потенциалопределяющими становятся
катионы алюминия:
А1 (ОН)о1' + ОН- ;=? А13+ + ЗОН-.
Величина положительного заряда возрастает.
В слабощелочной среде (pH 7,5—8) ионизация по основному
типу невозможна из-за избытка ОН~-ионов:
А1 (ОН)3 П А1 (0Н)20- + Н+.
Потенциалобразующим будет ион А1(0Н)20~, и частица приобре¬
тает отрицательный заряд.
При более высоких pH
А1 (0Н)20- + Н+ AlOf + Н+ + Н20.
В чистой воде изоэлектрическое состояние золя соответствует
определенному значению pH, называемому pH изоэлектрической
точки и обозначаемому рНг. В растворах электролитов рНг в
присутствии катионов смещается в щелочную сторону, а в при¬
сутствии анионов — в кислую [21]. Влияние на ДП температуры
среды также двояко. С повышением температуры, с одной стороны,
возрастает интенсивность теплового движения противоионов и
может произойти увеличение толщины диффузного слоя и ДП;
с другой стороны, уменьшится адсорбция потенциалопределяющих
ионов, что приведет к падению ДП.
Частицы тонкодисперсных суспензий с единым компактным
ядром, например частицы глин, построены по такому же принци¬
пу, как и частицы золей, а изменение их потенциалов подчиня¬
ется тем же закономерностям [7, стр. 244].
32
ГЛАВА I. ФЕНОМЕНОЛОГИЯ ВОДНЫХ ДИСПЕРСИЙ
\
\
ч
\
\
ч
Если частицы суспензии — агрегаты, состоящие из нескольких
более мелких частиц и не имеющие общего заряда, то их поверх¬
ностные свойства определяются частными свойствами каждой
мелкой частицы. Причем снижение ДП лишь некоторой доли
таких частиц не означает еще потери устойчивости агрегата в
целом.
Между двумя частицами одинаковой природы в дисперсных си¬
стемах действуют молекулярные силы притяжения (силы Ван-дер-
Ваальса) и электростатические силы отталкивания. Чтобы
оценить количественно взаимодействие частиц, нужно знать харак¬
тер изменения величины этих сил с расстоянием, их дальнодей¬
ствие.
Впервые объяснение агрегативной устойчивости дисперсных
систем и их коагуляции с количественным учетом суммарной
энергии взаимодействия частиц было дано Дерягиным, а затем
более детально Дерягиным и Ландау. Несколько позднее этот же
подход к проблемам устойчивости и коагуляции осуществили
Фервей и Овербек. Поэтому теория взаимодействия и коагуля¬
ции дисперсных частиц получила название теории Дерягина —
Ландау—Фервея—Овербека или сокращенно ДЛФО.
Характер изменения энергии притяжения и отталкивания с
увеличением расстояния между частицами показан на рис. 1.11.
Результирующая потенциальная кривая построена путем геомет¬
рического сложения ординат потенциальных кривых притяжения
и отталкивания и показывает, что на больших расстояниях дол¬
жны преобладать силы молекулярного притяжения. Это обуслов¬
лено тем, что силы притяжения и силы отталкивания убывают
по мере удаления от поверхности частицы по разным законам:
силы притяжения — обратно пропорционально расстоянию, а
силы отталкивания — по экспоненциальному закону. По этой
же причине на средних расстояниях, соответствующих толщине
ионных оболочек частиц, могут превалировать силы отталкивания,
на меньших расстояниях (порядка 10 А) — силы притяжения и,
О
Рис. 1.11. Потенциальная кри
вая частицы золя
1 — энергия отталкивания;
2 — энергия притяжения;
3 — результирующая кривая
/
1.4. АГРЕГАТИВНАЯ УСТОЙЧИВОСТЬ
33
наконец, на очень малых расстояниях — вновь силы отталкива¬
ния 5.
Как показано на рис. 1.11, на потенциальной кривой взаимо¬
действия частиц имеется два энергетических минимума (первичный
и вторичный) и максимум, называемый барьером отталкива¬
ния. Взаимодействие частиц определяется высотой барьера оттал¬
кивания, глубиной энергетических минимумов и энергией соуда¬
ряющихся частиц. Возможны следующие случаи.
1. Энергия столкнувшихся частиц недостаточна для преодо¬
ления барьера отталкивания, а глубина вторичного минимума
недостаточна для того, чтобы удержать частицы вместе. Коагуля¬
ции не происходит.
2. Энергия частиц достаточна для преодоления барьера оттал¬
кивания, а глубина первичного минимума достаточна для того,
чтобы удержать частицы вместе. Происходит коагуляция за счет
ближнего взаимодействия.
3. Энергия частиц недостаточна для преодоления барьера от¬
талкивания, но глубина вторичного минимума достаточна для
удержания частиц вместе. Происходит коагуляция за счет даль¬
него взаимодействия частиц. Зонтаг и Штренге [22, стр. 10] не¬
правильно называют этот последний случай коагуляции «флоку-
ляцией».
Мартынов и Муллер [23] показали, что коагуляция за счет
дальнего взаимодействия частиц может иметь место при до¬
статочно высоком значении ф0-потенциала и низкой валентности
противоионов. Для дисперсий с относительно крупными частица¬
ми характерно наличие глубокого вторичного минимума и, сле¬
довательно, «дальняя» коагуляция наиболее вероятна. Эксперимен¬
тальные данные, которые мы приведем в следующих главах, убе¬
ждают в том, что фиксация сильно гидратированных частиц про¬
дуктов гидролиза алюминия и железа происходит преимущест¬
венно во вторичном энергетическом минимуме 6. Рассмотрим силы
взаимного притяжения и отталкивания частиц.
Сила взаимного притяжения двух сферических частиц радиу¬
сом г, находящихся на произвольном растоянии В друг от друга
(рис. 1.12), по уравнению Гамакера [4, стр. 377] составляет
® В учебниках и руководствах по водоснабжению при описании потенциаль¬
ной кривой взаимодействия частиц, как правило, допускают серьезные
ошибки.
6 Фридрихсберг [8, стр. 259] высказывает предположение, что частицы гидро¬
окисей алюминия, железа, марганца и кремния вообще не могут сблизиться
на расстояния, достаточные для коагуляции в первичном энергетическом
минимуме.
_ 4Г2
№
■]
(1.18)
2 Е. Д. Бабенков
34
ГЛАВА I. ФЁНОМЁНОЛОГИЯ! ЙОДЙЫХ ДИСПЕРСИЙ
М
Я
Рис. 1.12. Схема взаимодействия частиц
Рис. 1.13. Схема строения адсорбционного гидратного слоя у твердой по¬
верхности (М — N — граница поверхности)
где А — константа Гамакера, слагающаяся из отдельных кон¬
стант, характеризующих свойства частиц дисперсной фазы и сре¬
ды. Она равна приблизительно 1-10~14—1-10~12 эрг [22, стр. 55].
При малых расстояниях между поверхностями частиц зави¬
симость (1.18) принимает вид
Энергия отталкивания двух сферических частиц для случая,
когда Н0 <^; г, определяется уравнением Дерягина—Ландау:
Согласно уравнениям (1.18) и (1.19), силы притяжения и от¬
талкивания пропорциональны радиусу частиц в первой степени.
Следовательно, концентрация электролита, определяющая равен¬
ство Fnp = For, не зависит от размера частиц. Этот вывод, одна¬
ко, не всегда подтверждается экспериментом [22, стр. 53]. Как по¬
казали исследования [24, 25], при одной и той же концентрации
золя порог его коагуляции (см. гл. IV) по мере понижения сте¬
пени дисперсности частиц проходит через минимум.
Во многих случаях агрегативная устойчивость коллоидных
систем объясняется не только существованием электростатических
сил отталкивания, но и другими факторами [26—28]. Первый из
них — сольватация (гидратация) частиц, т. е. образование на
их поверхности оболочек из молекул дисперсионной среды.
Из схемы строения гидратного слоя у поверхности твердого
тела, показанной на рис. 1.13, видно, что по мере приближения
к границе раздела упорядоченность расположения диполей воды
возрастает. За счет строгой ориентации диполей и прочного сцеп¬
^пр = — Аг/12Н0
(1.18')
(1.19)
1.4. АГРЕГАТИВНАЯ УСТОЙЧИВОСТЬ
35
ления их между собой гидратные слои обладают повышенной вяз¬
костью и слабой растворяющей способностью [29, стр. 22]. По
мнению Ребиндера, способность сольватных оболочек противо¬
действовать слипанию частиц заключается, главным образом, в
наличии у оболочек определенных механических свойств. Деря¬
гин объясняет эту способность возникновением положительного
расклинивающего давления 7.
Влияние гидратации на устойчивость коллоидов изучено недо¬
статочно, однако ясно, что оно неодинаково для золей гидрофиль¬
ных и гидрофобных. Некоторая гидратация последних объясня¬
ется гидратацией адсорбированных ионов или молекул. После
снижения заряда гидратные оболочки уже не препятствуют сли¬
панию частиц. Коллоиды с гидрофильной поверхностью сохраня¬
ют устойчивость и после устранения заряда. Следовательно, на¬
личие гидратных оболочек является для них более важным фак¬
тором стабилизации, чем двойной электрический слой [26].
Вторым дополнительным фактором агрегативной устойчиво¬
сти частиц является структурно-механический. Его действие про¬
является тогда, когда поверхность частиц покрыта слоем молекул
стабилизатора, обладающим структурной вязкостью и механи¬
ческой прочностью. Структурно-механический фактор стабилиза¬
ции обусловливается, главным образом, адсорбционными слоями
ориентированных поверхностно-активных и высокомолекулярных
веществ [22, 31, 32]. По мнению некоторых исследователей [33,
34], причиной стабилизирующего действия являются не собст¬
венно ПАВ и ВМВ, а наступающая вследствие их адсорбции гид-
рофилизация поверхности. Таким образом, действие второго
дополнительного фактора стабилизации сводится к действию
первого.
Теоретические и экспериментальные исследования разных авто¬
ров показывают, что стабилизация частиц дисперсной фазы насту¬
пает при толщине структурных оболочек от нескольких единиц
до нескольких десятков ангстрем.
Агрегативная устойчивость суспензий зависит от тех же фак¬
торов, что и устойчивость золей. Лишь для анизодиаметрических
частиц суспензий большое значение приобретают, кроме рассмот¬
ренных, дипольные взаимодействия. В частности, минимальная
устойчивость вытянутых частиц палыгорскита (средняя длина
7 Согласно современной теории устойчивости, в понятие расклинивающего
давления вкладывается широкий смысл. При сближении лиофобных частиц
расклинивающее давление определяется суммой двух независимых слагае¬
мых: ван-дер-ваалъсового взаимодействия молекул прослойки и смежных
фаз и ионно-электростатического взаимодействия. Если частицы одинаковы
по природе, то, как показано выше, первое слагаемое всегда отрицательно,
а второе — всегда положительно. При -сближении лиофильных частиц
учитывается третья слагающая расклинивающего давления, обусловленная
перекрытием граничных слоев жидкости [30].
2*
36
ГЛАВА I. ФЕНОМЕНОЛОГИЯ ВОДНЫХ ДИСПЕРСИЙ
250 нм) имеет место при значении ДП, равном не нулю, а при¬
близительно 30 мв, что совпадает с условием минимальной элект¬
рической поляризуемости частиц [35].
Для количественной оценки агрегативной устойчивости сус¬
пензий в зависимости от их концентрации используют ряд ме¬
тодов: изучение скорости осаждения частиц с получением кривых
распределения по размерам, определение скорости накопления
осадков, исследование их структурно-механических свойств [36]
и другие методы, подробно рассматриваемые в руководствах по
коллоидной и физической химии.
Агрегативную устойчивость растворов ВМВ следует, по-види¬
мому, рассматривать, как устойчивость гомогенных растворов.
Но вследствие больших размеров молекул и их способности об¬
разовывать ассоциаты устойчивость ВМВ может быть обусловле¬
на также двойным электрическим слоем или сольватными оболоч¬
ками. Потеря электрического заряда, по данным Воюцкого [1,
стр. 465], лишь незначительно сказывается на степени устойчи¬
вости ВМВ, что говорит об их лиофильности.
Добавление электролита способствует дегидратации макромо¬
лекул ВМВ, степень которой определяется не столько зарядом
добавленных ионов, сколько их дегидратирующей способностью
[8, стр. 316]. Этот процесс, получивший название высаливания,
резко отличается от коагуляции лиофобных золей и требует го¬
раздо больших (на 3—5 порядков) концентраций электролита.
Высаливание можно рассматривать как следствие «конкуренции»
ионов и макромолекул за молекулы воды. В результате высали¬
вания возникают хлопьевидные образования макромолекул.
Л итература
1. С. С. Воюцкий. Курс коллоидной химии. М., «Химия», 1975.
2. Р. Т. Марченко. Физическая и коллоидная химия. М., «Высшая школа»,
1965.
3. А. Г. Пасынский. Коллоидная химия. М., «Высшая школа», 1968.
4. Г. Р. Кройт. Наука о коллоидах, т. 1. Необратимые системы. М., ИЛ,
1955
5. Т. С. Rao. Chem. Age India, 21, N 1, 76 (1970).
6. D. N. Saraf, S. D. Khullar. Canad. J. Chem. Engrs, 53, 449 (1975).
7. H. П. Песков, E. М. Александрова-Прейс. Курс коллоидной химии.
М.— JL, Госхимиздат, 1948.
8. Д. А. Фридрихсберг. Курс коллоидной химии. JI., «Химия», 1974.
9. Н. П. Песков. Физико-химические основы коллоидной науки. М.— Л.,
Госхимиздат, 1935.
10. А. Д. Зимон. Адгезия жидкости и смачивание. М., «Химия», 1974.
11. П. А. Ребиндер. Коллоидн. ж., 20, 527 (1958).
12. Ю. М. Глазман. Коллоидн. ж., 24, 275 (1962).
13. Б. В. Дерягин. Сб. «Исследования в области поверхностных сил». М.,
Изд-во АН СССР, 1961, стр. 21.
14. В. Н. Крестинская, Н. И. Муратова. Коллоидн. ж., 9, 43 (1947).
Г. ЛИТЕРАТУРА
37
15. В. А. Каргин, В. В. Киселев. ЖФХ, 11, 461 (1938).
16. В. А. Каргин, Г. В. Климовицкая. Там же, стр. 467.
17. В. Ф. Устъ-Каикинцев. Коллоидн. ж., 15, 394 (1953).
18. М. Ф. Талина. Изв. вузов. Химия и хим. технол., 11, 613 (1968).
19. Е. S. Hall. J. Appl. Chem., 15, 197 (1965).
20. С. С. Духин, Б. В. Дерягин. Электрофорез. М., «Наука», 1976, стр. 23.
21. М. R. Buydens. Techn. sanit. et municip., 40, 139 (1953).
22. Г. Зонтаг, К. Штренге. Коагуляция и устойчивость дисперсных систем.
JI., «Химия», 1973.
23. Г. А. Мартынов, В. М. Муллер. Сб. «Исследования в области поверх¬
ностных сил». М., «Наука», 1972, стр. 7.
24. Б. В. Барбой, В. М. Барбой и др. Коллоидн. ж., 31, 660 (1969).
25. Б. В. Барбой, Ю. М. Глазман. Коллоидн. ж., 32, 10 (1970).
26. С. С. Воюцкий. Коллоидн. ж., 23, 353 (1961).
27. Ю. М. Глазман, Д. Н. Стражеско, Е. Ф. Жельвис и др. Коллоидн. ж.,
21, 263 (1959).
28. Н. D. Napper. Sci. Progress, 55, N 217, 91 (1967).
29. В. А. Глембоцкий, В. И. Классен. Флотация. М., «Недра», 1973.
30. Б. В. Дерягин. Сб. «Успехи коллоидной химии». М., «Наука», 1973,
стр. 30.
31. Сб. «Исследования в области поверхностных явлений». Под ред. П. А. Ре¬
биндера. М.— JI., ОНТИ, 1936, стр. 7.
32. Сб. «Исследования по физико-химии технических суспензий». Под ред.
П. А. Ребиндера. М.— Л., ОНТИ, 1933, стр. 9.
33. Ю. М. Глазман, И. П. Сапон. Сб. «Исследования в области поверхност¬
ных сил». М., «Наука», 1964, стр. 258.
34. И. П. Сапон, Ю. М. Глазман. Коллоидн. ж., 20, 274 (1958).
35. С. П. Стойлов, И. Б. Петканшин. Докл. Болг. АН, 24, 487 (1971).
36. JI. И. Эделъман, Д. С. Соминский. Коллоидн. ж., 21, 126 (1959).
Глава II
ХАРАКТЕРИСТИКА ЗАГРЯЗНЕНИЙ
ПРИРОДНЫХ ВОД
Состав воды источников водоснабжения определяется геологи¬
ческими, гидрогеологическими и климатическими условиями, а
также плотностью населения, хозяйственной деятельностью че¬
ловека, его культурой. Поскольку метод коагуляции использу¬
ется главным образом при очистке вод поверхностных источников,
обсуждая качественные показатели природной воды, мы будем
иметь в виду прежде всего воду рек, озер и водохранилищ.
Вещества минерального и органического происхождения при¬
сутствуют в воде во всех видах дисперсного состояния. В грубо¬
дисперсном (взвешенном) состоянии находятся глинистые, квар¬
цевые, известковые и гипсовые частицы, ряд веществ животного и
растительного происхождения; в коллоидном — частицы глин,
соединения кремния и железа, сера, продукты жизнедеятельно¬
сти и распада микроорганизмов, гуминовые вещества; в истинно
растворенном — газы, неорганические соли щелочных, щелочно¬
земельных и тяжелых металлов, ряд органических соединений,
а также бром, иод и другие.
В классификации Кульского [1], истинно растворенные при¬
меси воды подразделены по характеру их поведения в процессах
очистки воды на молекулярно растворенные (газы и органические
соединения с малодиссоциированными группами) и ионно раство¬
ренные (главным образом, минеральные соли). Характерные
свойства последних — ярко выраженная способность к гидра¬
тации и заметное влияние на структуру и физические свойства
воды.
Количество минеральных солей в водоисточниках зависит от
способа их питания и колеблется в широких пределах: от 30
(поверхностное питание) до 2000 мг/л (подземное питание). Со¬
левой состав вод определяется преимущественно содержанием
ионов НСО^, SO4'. Са2+, Mg2+, Na+. По преобладающему аниону
воды рек делят на три класса: гидрокарбонатные (Волга, Днепр
и др.), сульфатные (Дон, Сев. Донец и др.) и хлоридные (Ишим,
Эмба и др.). В свою очередь каждый класс по преобладающему
катиону подразделяют на кальциевую, магниевую и натриевую
группы [2].
Концентрация органических веществ также неодинакова: от
2 до 200 мг/л. Большие концентрации характерны для рек, в бас¬
сейне которых имеются болота. Наличие разнообразных питатель¬
ii.i. ФИЗИКО-ХИМИЧЕСКИЕ ПОКАЗАТЕЛ к КАЧЕСТВА воды
39
ных веществ создает благоприятные условия для развития рас¬
тительных и животных организмов. Поэтому воды поверхностных
источников богаты микрофлорой и микрофауной.
Вся сумма процессов, происходящих в водах рек, озер и водо¬
хранилищ, рассматривается как совокупность самозагрязнения и
самоочищения. Самозагрязнение состоит во взмучивании воды и
накоплении в ней органических веществ. Самоочищение включает
следующие процессы: 1) осаждение грубодисперсных примесей и
естественно скоагулировавших коллоидов, 2) окисление органи¬
ческих веществ и неустойчивых минеральных примесей, 3) нейт¬
рализация загрязнений.
В озерах круговорот веществ в ходе самоочищения направ¬
лен, главным образом, на захоронение в иле, а в реках — на ми¬
нерализацию [3]. Большую роль в самоочищении водоемов
играет способность поверхностного слоя воды аккумулировать
молекулы веществ, понижающих поверхностное натяжение: бел¬
ков, жиров, соединений фосфора, марганца, железа [4].
Существенным отличием наземных водоисточников от подзем¬
ных является непостоянство состава воды в течение года. В зави¬
симости от интенсивности паводков и дождей, протекающих фи¬
зико-химических и биологических процессов состав воды изменя¬
ется. Так, во время паводка происходит разбавление воды источ¬
ника талыми водами с одновременным увеличением содержания
механических примесей. При благоприятном температурном ре¬
жиме в воде озер и водохранилищ происходит массовое развитие
планктона — цветение.
II. 1. ФИЗИКО-ХИМИЧЕСКИЕ ПОКАЗАТЕЛИ КАЧЕСТВА ВОДЫ
Запах и вкус воды обусловливаются присутствием природных
примесей и промышленных загрязнений. Из минеральных при¬
родных веществ запах и вкус свойственны, например, сероводо¬
роду, соединениям железа и марганца, сульфатам, хлоридам.
Запах и вкус воды биологического происхождения (травяной,
болотный, земляной, рыбный и др.) являются следствием жизне¬
деятельности и отмирания высших водных растений (рдест, рого¬
листник, ряска, стрелолист и др.), лучистых грибков, плесеней,
водорослей, некоторых форм бактерий [5].
Из промышленных загрязнений, сбрасываемых в водоемы,
запах и вкус свойственны ароматическим и алифатическим угле¬
водородам, спиртам, фенолам, кетонам, альдегидами др. Наимень¬
шими пороговыми концентрациями (по запаху) обладают фенолы,
некоторые хлорированные углеводороды, пестициды [5, 6].
С бытовыми сточными водами в источники попадают синтетические
детергенты, обладающие неприятным запахом и вкусом при кон¬
центрациях 0,2—0,3 мг/л [7]. Запах и вкус воды, содержащей
40 ГЛАВА II. ХАРАКТЕРИСТИКА ЗАГРЯЗНЕНИЙ ПРИРОДНЫХ ВОД
алкилбензолсульфонаты и фенолы, усиливаются при обработке
воды хлором. Так, хлорфенолы ощущаются в воде при концент¬
рации 0,01 мг/л [8].
Интенсивность запаха и вкуса непостоянна в течение года и с
повышением температуры воды увеличивается.
Мутность. Прозрачность. Примеси, находящиеся в дисперс¬
ном состоянии, обусловливают мутность воды. Величина мут¬
ности определяется интенсивностью светорассеяния и пропор¬
циональна весовой концентрации взвешенных веществ.
С изменением степени дисперсности взвеси мутность меняется
неоднозначно: при увеличении размеров частиц (начиная от мо¬
лекулярных) интенсивность светорассеяния сначала возрастает,
а затем постепенно уменьшается. Максимальное светорассеяние
соответствует коллоидной степени дисперсности. Высокой мут¬
ностью обладают воды среднеазиатских рек. Реки северных и
центральных областей СССР несут гораздо меньше взвесей. Не¬
которые реки Сибири (например, Ангара) обладают ничтожно
малой мутностью. Мутность речных вод минимальна зимой и
максимальна во время паводков. Цветение водохранилищ приво¬
дит к повышению мутности воды летом.
Определение концентрации взвешенных веществ осуществля¬
ется либо весовым способом (в мг/л), либо с помощью фотоэлектро¬
колориметра (в единицах оптической плотности, пересчитываемых
на мг/л). Более простыми, но менее точными являются косвенные
методы, основанные на визуальном анализе мутности или проз¬
рачности воды.
Цветность воды, ее окраска объясняются наличием гуминовых
соединений, танина, солей железа, окрашенных отходов произ¬
водства. Цветность свойственна в основном источникам с болот¬
ным питанием. В зависимости от состава окрашивающих веществ
вода может приобрести желтоватый, бурый, зеленоватый и дру¬
гие цвета [9]. Нужно иметь в виду, что цветность воды не опреде¬
ляется общим содержанием в ней органических веществ, посколь¬
ку часть последних не имеет окраски. Так, например, при об¬
работке коагулянтом волжской воды цветность уменьшалась на
63%, а содержание органических веществ — лишь на 39%. Для
окской воды эти показатели были равны соответственно 47 и 22%
[10, стр. 30].
В течение года цветность воды меняется иногда очень резко.
Колебания цветности могут происходить в такт с колебаниями
мутности или независимо от них (например во время цветения
водорослей).
Количественно цветность выражают в условных градусах плати-
но-кобальтовой шкалы. По данным Лебедевой [11], среднее коли¬
чество природных органических веществ, обусловливающее один
градус цветности, составляет 0,27 мг/л. В большинстве водоемов
II.1.ФИЗИКО-ХИМИЧЕСКИЕ ПОКАЗАТЕЛИ КАЧЕСТВА ВОДЫ
41
величина цветности воды находится в пределах 20—60 град, но
в небольших реках с заболоченными берегами может достигать
1400 град.
Иногда для характеристики органических веществ, обуслов¬
ливающих цветность, рекомендуют использовать так называе¬
мый коэффициент цветности, равный частному от деления цвет¬
ности воды (в град) на ее окисляемость [12, стр. 14]. Значения
этого коэффициента для большинства природных вод находятся
в пределах 1—8.
Сухой остаток. Суммарное количество минеральных и органи¬
ческих примесей, содержащихся в воде в истинно растворенном
и коллоидном состоянии, характеризуют величиной сухого остат¬
ка. Величина его определяется взвешиванием остатка после вы¬
паривания некоторого объема предварительно профильтрованной
воды и выражается в миллиграммах на литр.
По Шкробу и Вихреву [13], величина сухого остатка речной
воды колеблется в пределах от 60 до 1000 мг/л и для большинства
рек составляет 150—400 мг/л. Часть сухого остатка, которая
удаляется при прокаливании, дает ориентировочное представле¬
ние о содержании в воде органических веществ.
Ионная сила раствора. Электропроводность. Важнейшей фи¬
зико-химической характеристикой солевого состава обрабаты¬
ваемой воды является ионная сила раствора (7Р), определяемая
как полусумма произведений концентрации сг каждого из при¬
сутствующих ионов на величину его заряда zt в квадрате
Для условно пресных вод с общим содержанием солей до
1 г/л справедливо эмпирическое выражение
в котором Рс — общее солесодержание воды (в мг/л), принимае¬
мое равным величине сухого остатка после прокаливания.
Важным показателем общей концентрации растворенных со¬
лей является электропроводность воды. Значения ионной силы
раствора и электропроводности во многих случаях могут быть
связаны с результатами очистки воды коагулянтами.
Показатель концентрации водородных ионов (pH). Вода яв¬
ляется слабым электролитом и диссоциирует лишь в незначитель¬
ной степени:
(H-I)
/ р = 2,2-10 -6Р0,
(П-2)
H2OiiOH- + Н+.
Ионы водорода существуют в виде иона гидроксония (Н30+),
но для простоты обозначим их Н+. Отрицательный логарифм ак¬
тивности водородных ионов (pH) является мерой кислотности
42 ГЛАВА II. ХАРАКТЕРИСТИКА ЗАГРЯЗНЕНИЙ ПРИРОДНЫХ ВОД
Рис. 11.1. Зависимость соотношений (а) углекислых соединений от pH среды
Рис. II.2. Зависимость pH воды от температуры
воды. В природных водах величина pH определяется количеством
растворенного углекислого газа (рис. II.1), гидролизом солей
железа, присутствием гуминовых кислот или промышленных
отходов. Обычно величина pH наземных вод находится в пределах
6,5—8,5 и более или менее постоянна в течение года. Это обстоя¬
тельство объясняется наличием буферной системы:
Н2С03 ^ НСО^ + Н+.
Прибавление к воде некоторых количеств щелочи или кислоты
мало влияет на величину pH: при добавлении щелочи часть Н+-
ионов в приведенной реакции связывается в малодиссоциирован-
ную воду, а диссоциация новых количеств Н2С03 восстанавли¬
вает первоначальную концентрацию водородных ионов; при до¬
бавлении кислоты избыточные водородные катионы связываются
анионами НСОз в малодиссоциированную угольную кислоту.
Повышение температуры сопровождается увеличением ионного
произведения воды и некоторым снижением величины pH, как
это видно из графика на рис. II.2 (подробнее см. гл. III). В тех
случаях, когда в водоеме лимитирующим фактором развития
фитопланктона является концентрация С02, pH воды в течение
суток может колебаться в пределах нескольких десятых единицы
[14].
Жесткость воды определяется суммой содержащихся в ней
ионов кальция и магния, поступающих в воду при растворении
частиц известняка, гипса, доломита и других минералов и коле¬
блется в пределах от 1—2 до 6—8 мг-экв!л.
Щелочность воды обусловлена присутствием в ней бикарбо-
натных и карбонатных ионов и почти всегда соответствует вели¬
чине карбонатной жесткости. Воды рек, например, обладают
щелочностью 0,5—4 мг-экв/л.
(1.1. ФиЗиКО-ХиМиЧЁСКЙЕ ПОКАЗАТЕЛИ КАЧЕСТВА воды
43
Окисляемость воды — количество кислорода (в мг/л), потреб¬
ное для окисления веществ, присутствующих в ней,— обуслов¬
ливается в основном наличием органических веществ и лишь в
незначительной степени — быстроокисляющихся соединений же¬
леза, сероводорода, нитритов. Ее величина используется для
косвенной количественной характеристики концентрации орга¬
нических загрязнений.
В большинстве рек СССР окисляемость воды находится в пре¬
делах 5—10 мгО^/л. Окисляемость озерных и водохранилищных
вод достигает 10—16 мг02/л. Вода некоторых рек, особенно тех, в
бассейнах которых имеются болота, может иметь более высокую
окисляемость — до 60 мгО^л.
Растворенные газы. В растворенном виде в воде находятся
кислород, азот, углекислый газ, сероводород и иногда метан.
Растворимость газа сгз зависит от его природы, парциального
давления ра и температуры воды:
сгз = К рп, (II.3)
где К — коэффициент пропорциональности, равный раствори¬
мости газа при данной температуре и давлении 1 ат.
Значения сгз для перечисленных газов приведены в табл. II.1.
Таблица II.1. Растворимость газов (мг/л) при парциальном
давлении 1 ат [15, стр. 52]
Газ
Температура воды,
ас
0
5
10
15
20
25
30
40
Кислород
69,5
60,7
53,7
48
43,4
39,3
35,9
30,8
Азот
29,1
—
22,8
—
18,9
—
17,0
13,8
Углекислый газ
3350
2770
2310
1970
1690
1450
1260
970
Сероводород
7070
6000
5110
4410
3950
3380
2980
2360
Обогащение воды кислородом происходит из воздуха и в ре¬
зультате процессов фотосинтеза водной растительности. Ряд про¬
цессов — окисление, дыхание организмов, гниение и другие —
снижают содержание растворенного кислорода. В открытых источ¬
никах содержится до 15 мг02/л.
Наличие в воде растворенного азота объясняется его поступле¬
нием из воздуха и некоторыми биохимическими процессами: раз¬
ложением органических веществ, бактериальным восстановлением
азотистых соединений. Поскольку воздух является главным по¬
ставщиком азота, а последний обладает большим парциальным дав
44 ГЛАВА II. ХАРАКТЕРИСТИКА ЗАГРЯЗНЕНИЙ ПРИРОДНЫХ ВОД
лением, чем кислород, растворенный азот обычно содержится в
больших количествах, чем кислород.
Источник появления в воде углекислого газа, помимо возду¬
ха,— био- и геохимические процессы. К факторам, приводящим
к снижению содержания С02, относятся фотосинтез, растворение
карбонатных пород с образованием бикарбонатов и др. Концен¬
трация С02 в поверхностных водах колеблется от 0,5 до 2,0 мг/л.
Присутствие сероводорода связано с процессами восстановле¬
ния и разложения некоторых минеральных веществ (серный кол¬
чедан, гипс) и гниением органических остатков. Вследствие легкой
окисляемости в поверхностных водоемах сероводород находится
в ничтожных количествах.
Сульфаты и хлориды. Соли серной и соляной кислот встре¬
чаются почти во всех природных водах, что обусловлено широкой
распространенностью легко размываемых осадочных пород, окис¬
лительными процессами, а иногда — загрязненностью воды сто¬
ками. Концентрация сульфатов и хлоридов в значительной сте¬
пени предопределяет эффективность очистки воды коагулянтами.
Содержание SO4” в поверхностных водах колеблется в преде¬
лах 60—100 мг/л. Концентрация С1_ достигает иногда нескольких
сот миллиграммов на литр и изменяется в очень широких пределах.
Железо и марганец. Железо может содержаться в составе ор¬
ганоминеральных комплексов, обладающих достаточно высокой
растворимостью или находящихся в коллоидном состоянии. В ре¬
ках, загрязненных шахтными водами и стоками травильных це¬
хов, часто содержится железный купорос, постепенно окисляю¬
щийся. Если в воде присутствует сероводород, может образо¬
вываться тонкодисперсная взвесь FeS, придающая воде черную
окраску. Содержание железа в воде достигает в некоторых слу¬
чаях 3—5 мг/л.
Марганец содержится в виде гидратированных ионов или ком¬
плексов с органическими соединениями в истинно растворенном
и коллоидном состоянии. В водоемах, загрязненных сточными во¬
дами, концентрация марганца может достигать 4 мг/л.
Фтор поступает в водоемы вследствие растворения некоторых
природных минералов или со сточными водами. Концентрация
фтора в воде обычно не превышает 0,3—0,4 мг/л, но иногда дости¬
гает 12 мг/л.
Соединення кремневой кислоты (кремнекислоты) находятся
в воде в коллоидном и истинно растворенном состоянии. Гидрат¬
ные формы кремневой кислоты — метакремневая кислота
H2Si03 (Si02-H20), ортокремневая кислота H4Si04(Si02-2Н20)
и другие поликремневые кислоты — малорастворимы и образуют
коллоидные растворы. В зависимости от pH воды в коллоидном
состоянии может находиться от 3 до 20% общего количества крем¬
некислоты [15, стр. 51].
П.2. ПРИМЕСИ ЕСТЕСТВЕННОГО ПРОИСХОЖДЕНИЯ
45
Прежде чем перейти к обсуждению свойств основных видов
загрязнений (удаляемых коагуляцией) — грубодисперсных и
коллоидных примесей, приведем краткие сведения о влиянии ион¬
но-растворенных веществ на структурные особенности воды.
Согласно современным представлениям, каждая полярная мо¬
лекула воды связана прочной водородной связью с четырьмя дру¬
гими молекулами и образует ажурную тетраэдрическую структур¬
ную сетку [16, стр. 259]. По-видимому, ионы растворенного веще¬
ства, имеющие малый радиус, размещаются в пустотах такой
сетки и слабо влияют на структуру воды (гидрофобное внедрение).
Ионы большого радиуса могут замещать водные молекулы в струк¬
турных узлах (гидрофильное внедрение). В результате взаимодей-
стия ионов растворенного вещества с молекулами воды изменяют¬
ся термодинамические свойства воды, нарушается ее упорядочен¬
ная структура в ближайшем окружении (увеличение энтропии)
или, наоборот, происходит стабилизация структуры (уменьше¬
ние энтропии). Напомним, что энтропия определяется как отно¬
шение количества тепла, полученного или отданного системой, к ее
абсолютной температуре.
Действие ионов зависит не только от их размеров, но и от за¬
ряда, строения, электронной конфигурации. Преобладание разу-
порядочивающего действия на структуру воды характерно в основ¬
ном для однозарядных ионов, таких, как Cl-, J~, Вг", К+ и др.,
преобладание упорядочивающего действия — для многозарядных и
многоатомных ионов: РО4-, SO4-, Al3+, Mg2+ и др. Соответствен¬
но первые называются ионами с отрицательной гидратацией,
вторые — с положительной.
Количественной характеристикой действия ионов на структуру
воды является суммарное изменение энтропии. Расчетные значе¬
ния этой характеристики в энтропийных единицах (кал! г-град К)
для некоторых ионов приводим по Крестову [17]:
К+ +8,2' НС03- -6,0 S042~ -30,2 Р04з- -85,9
Cl" +1,5 Н+ -13,7 Са2+ -31,6 А13+ -94,6
Na+ —1,3 ОН" —20,8 Fe2+ —49,4 Fe3+ —94,6
Из приведенных данных видно, что катионы А13+ и Fe3+ ока¬
зывают по сравнению с другими ионами наибольшее упорядочи¬
вающее действие на структуру воды.
II. 2. ПРИМЕСИ ЕСТЕСТВЕННОГО ПРОИСХОЖДЕНИЯ
II. 2.1. Минеральные примеси
Распределение частиц минеральных примесей по размерам зави¬
сит от скорости течения воды и вида подстилающих грунтов. Ча¬
стицы, вымываемые из почв, находятся в основном в грубодисперс-
46 ГЛЛВЛ ii. XAPAKTI'PИСТИНА ЗАГРЯЗНЕНИЙ ПРИРОДНЫХ ВОД
ном состоянии, однако содержание коллоидной фракции может
достигать 50% (глинистые почвы) [18, стр. 351J. Характеристика
дисперсного состава минеральных примесей приведена в табл. II.2.
Таблица II.2. Дисперсный состав минеральных примесей воды
Примесь
Размер ча¬
стиц, мкм
Удельная
поверхность,
мг1г
Литература
Кварцевый песок
100-1000
[15, стр. 9]
Ил
5-50
—
[15, стр. 9]
Глина (без указания минерального
состава)
Минералы
0,1-5
100-1000
[19, 20]
каолинит
0,3-4
10-150
[19—26]
бентонит (монтмориллонит)
0,05-0,3
100-750
[24—27]
гидрослюда
0,1-2
100-150
[25, 26, 28]
вермикулит
1,5-2
470-670
[25, 26]
Не подлежит сомнению, что эффективность очистки воды коа¬
гулянтами в значительной мере определяется фазово-дисперсной ха¬
рактеристикой примесей и их специфическими адсорбционными
свойствами. В соответствии с принятой классификацией взвешен¬
ные вещества условно делят на три основные фракции: пелитовую
(< 10 мкм), алевритовую (от 10 до 100 мкм) и псаммитовую
(^> 100 мкм). Для большинства рек существует тесная связь
между фракционным и минералогическим составом взвешенных
веществ и их общим весовым содержанием. Чем больше взвеси
несет река, тем больший процент составляет пелитовая фракция
взвеси. В частности, в период паводков доля этой фракции возра¬
стает до 70—80%. Доля пелитовой фракции растет также вниз по
течению реки, так как увеличивается концентрация глинистых
частиц и уменьшается концентрация кварцевых. В табл. II.3
даны сведения по дисперсному и минеральному составу взвесей
некоторых рек СССР [20].
В пелитовой фракции преобладают гидрослюда и монтморил¬
лониты, входящие в состав многих почв и являющиеся составной
частью бентонитовых и отбеливающих (гумбрин, асканит) глин.
Примерный состав монтмориллонитов — Al2(OH)2[Si4O10] ■ пН20.
Группа гидрослюд представлена главным образом иллитом, вер¬
микулитом, глауконитом. Группа каолинитов охватывает мине¬
ралы состава Al203-2Si02-2H20 [29].
Таблица II.3 Дисперсный и минеральный состав взвесей некоторых рек СССР
Дисперсный состав
(вес. %) при размере
частиц, мкм
<10
10—100
>100
Река и место отбора
проб
Концен¬
трация
взвеси,
мг/л
Минеральный состав, вес. %
кварц
гидро¬
слюда
монт¬
морил¬
лонит
монтмо¬
риллонит
и гидро¬
слюда
монтмо¬
риллонит
и каоли¬
нит
хлорит
каль¬
цит
прочие
минералы
Аму-Дарья (Чатлы)
2670
80
20
39
35
Амур (Комсомольск)
31
30
55
15
50
25
Днестр (Берислав)
120
40
55
5
18
65
Дон (Калач)
15,6
35
50
15
35
25
Енисей (Красноярск)
149
15
65
20
57
15
Колыма (Средне-Ко-
лымск)
Кубань (Темрюк)
306
55
35
10
45
25
1300
47
50
3
40
—
Кура (Сальйны)
3230
25
75
—
40
—
Обь (Барнаул)
392
30
60
10
57
15
Риони (Поти)
770
60
40
—
43
—
-Сура (устье)
387
80
20
—
35
—
Терек (ст. Каргалин-
•ская)
1900
50
40
10
30
Урал (Гурьев)
698
75
20
5
30
10
20
20
42
24
55
38
25
20
6
—
—
15
—
—
17
—
10
10
10
—
18
10
10
10
18
18
15
7
12
—
16
15
10
8
—
—
10
20
16
9
15
15
2
II.2. ПРИМЕСИ ЕСТЕСТВЕННОГО ПРОИСХОЖДЕНИЯ
48
ГЛАВА II. ХАРАКТЕРИСТИКА ЗАГРЯЗНЕНИЙ ПРИРОДНЫХ ВОД
В чистом виде глины почти не встречаются. Они содержат в ка¬
честве примесей кварцевый песок, слюду, полевой шпат, биотит,
мусковит, рудные и другие минералы, отнесенные к «прочим»
в последней графе табл. II.3.
По данным исследователей, в реках Англии в составе дисперсных
примесей преобладают каолиниты [30], в реках США — монтмо¬
риллониты [31].
Частицы минеральных примесей имеют самую разную фор¬
му, но в основном анизодиаметричны. Например, глинистые ча¬
стицы имеют вид чешуек (табличек), у которых длина и ширина
значительно превышают толщину (у монтмориллонитов — в де¬
сятки и сотни раз) [21, 32]. В химический состав глин входят окис¬
лы кремния, алюминия, железа, титана, кальция и другие, но ос¬
новными являются первые три компонента. Их процентное со¬
держание в разных глинах показано в табл. II.4.
Таблица II.4. Содержание основных компонентов в минералах
Минерал
Содержание компонентов, вес. %
Литература
S1O2
А120з
F62O3
Бентонит (монтморил¬
лонит)
51,0-63,8
18,0-26,6
1,3-16,0
[25, 33-37]
Каолинит
46,9
32,2
1,3
[25]
Вермикулит
35,5
10,5
5,6
[25]
Гидрослюда
54,6
13,4
8,3
[25]
Глинистые частицы можно рассматривать как своего рода ам-
фотерные соединения, включающие кислый (Si02) и основной
(Ме203) компоненты. Общий заряд частиц и pH их изоэлектриче-
ской точки могут быть связаны с соотношением Si02 : Ме203.
Как показано в работах [35, 36], с уменьшением размеров частиц
это соотношение уменьшается.
Всестороннее исследование свойств глинистых минералов как
сорбентов, выполненное Овчаренко и др. [25, 26], показало, что
по структурным свойствам их целесообразно подразделить на
три группы:
1) слоистые минералы с расширяющейся структурой (монтмо¬
риллонит, вермикулит);
2) слоисто-ленточные минералы (палыгорскит);
3) слоистые минералы с жесткой структурой (пирофиллит,
гидрослюда, тальк, каолинит).
Минералы первой группы являются микропористыми, причем
объем их пор в ходе сорбции увеличивается. Ко второй группе от¬
II.2. ПРИМЕСИ ЕСТЕСТВЕННОГО ПРОИСХОЖДЕНИЯ
49
носятся минералы, обладающие меньшей внутренней пористостью.
Минералы третьей группы характеризуются наличием лишь внеш¬
ней адсорбирующей поверхности, и поэтому их адсорбционная
способность связана главным образом со степенью дисперсности.
Коллоидно-химические свойства минеральных загрязнений
зависят не только от их структуры и химического состава, но и от
свойств поверхностных слоев. Часть катионов из числа тех, ко¬
торыми почвы обогащают воду, адсорбируется на частицах за¬
грязнений. Катионы Са2+, Н+, К+, Na+, NH4 и др. полностью за¬
нимают адсорбционные участки частиц, и всякая дальнейшая ад¬
сорбция носит катионообменный характер. По Гриму [21, стр. 49],
способность глин к катионному обмену объясняется тремя об¬
стоятельствами:
1. Разрушением связей по краям кремнезем-глиноземных
структурных единиц, которое должно быть сбалансировано ад¬
сорбирующимися катионами. Количество разрушенных связей,
а следовательно, и обменная способность глин увеличивается
с ростом степени дисперсности частиц.
2. Замещением в кристаллической решетке четырехвалентного
кремния катионами с более низкой валентностью— алюминия или
магния. У монтмориллонита и вермикулита такими замещениями
обусловлено около 80% их общего катионного обмена.
3. Замещением водорода краевых гидроксильных групп таки¬
ми катионами, которые в свою очередь способны к обмену.
Установлено, что в тех глинах, где катионный обмен вызван
нарушением связей, обменные катионы сосредоточены по краям
чешуек, а в тех, где обменные реакции связаны с замещением в
кристаллической решетке, обменные катионы расположены глав¬
ным образом на поверхности базальных плоскостей. По энергии
поглощения глинами катионы располагаются в ряд [37]:
Fe3+ > А13+ > Н+ > Ва2+ > Са2+ > Mg2+ > NH+ > К+ > Na+ > Li+.
Общее количество способных к обмену катионов, приходя¬
щееся на 100 г минерала, называется его катионообменной ем¬
костью (Ev) и выражается в мг-экв/100 г. Катионообменная ем¬
кость взвесей, содержащихся в обрабатываемой воде, влияет на
расход коагулянта и является важным показателем технологиче¬
ских свойств воды. Значения Ек, определенные для некоторых ми¬
неральных веществ, приведены в табл. II.5.
Величина Ек зависит от степени дисперсности частиц. Напри¬
мер, для каолина зависимость Ек от степени дисперсности частиц
иллюстрируют данные, приведенные в табл. .11.6.
Обменные катионы влияют на заряд частиц минеральных за¬
грязнений, ориентацию кристаллов и степень агрегации частиц,
пористость осадков. Замена Na+ на Са2+, как правило, приводит
50 ГЛАВА XI. ХАРАКТЕРИСТИКА ЗАГРЯЗНЕНИЙ ПРИРОДНЫХ ВОД
Таблица II.5. Катионообменная емкость почв, донпых отложении
и минералов
Почвы, донные отложения и минералы
Вк,
мг-эк е/100 г
Литература
Почвы
подзолистые
6-8
[18, стр. 3521
черноземпые
40-60
[18, стр. 352]
торфяники
60-100
[18, стр. 352]
Донные отложения
р. Дамодар
12,5
[38]
р. Гудзон
5,4-24,5
[38]
р. Делавэр
9,5-85,8
[38, 39]
Минералы
кварц
1
[38]
биотит
3
[40]
пирофиллит
5
[38]
каолинит, диккит
0,5-25,5
[21, 23, 24, 29,36 , 38,
40, 41]
гидрослюда, палыгорскит, хло¬
3—70
121, 29, 40-42]
рит
бентонит (монтмориллонит)
25—150
[21, 23 , 24 , 36 , 40—43]
вермикулит, волконскоит
80—170
[21, 29, 40, 41]
Примечание. В донных отложениях от 1 до 24% Ек приходится на долю орга¬
нических веществ [39].
Таблица II.6. Катионообменная емкость каолина
(Наименова¬
ние каолина
Удельная
поверхность
частиц,
Ж2/8
Ек.
.мг-экв/100 г
Наименова¬
ние каолина
Удельная
поверх¬
ность
частиц,
м2/г
Ек,
мг-экв/100 г
Береговский
8,4
0,5
Турбовский
i6,i
4,5
Глуховецкий
11,3
1,0
Дубровский
31,9
12,5
Просяновскпй
14,8
2,0
Глуховской
97,6
25,5
к коагуляции частиц [44]. Пористость осадка каолинитовой сус¬
пензии понижается в присутствии катионов в ряду [45]:
Mg2+ > Са2+> Na+ > Li+.
Примерно в такой же последовательности катионы влияют на
теплоту смачивания водой глуховского каолина [46J:
Ga21- > Mg2+ > Cu2+ > Na+ > К+ > Li+,
Й.2. ПРЙМЁСЙ ЁСТЁСТ?ВЁННОГО ЙРОЙСХОЙДЁНЙЯ
51
Особенно велико влияние на коллоидно-химические свойства глин
катионов Al3+, Н+, NH£, Со2+, Mg2+, Th4+ [47—49].
Важное для водоочистки явление, позволяющее объяснить
влияние добавок известкового молока на процессы структуро-
образования, отмечено в работах Овчаренко и др. [45, 50]: при
смешении глинистых суспензий с гидроокисью кальция на поверх¬
ности частиц глин формируется слой гидросиликата кальция,
обеспечивающий упрочение поверхностных контактов и повы¬
шение прочности коагуляционных структур. По способности к
этому явлению минералы располагаются в ряд: палыгорскит
монтмориллонит каолинит.
Частицы почвы способны поглощать не только катионы, но и
многовалентные анионы, например РО|_. Поскольку процесс ад¬
сорбции РО|~ необратим, можно предполагать хемосорбцию и обра¬
зование комплексных соединений. fcv'-i
При ионообменной сорбции органических красителей на монт¬
мориллоните свойства последнего резко меняются. Вследствие
гидрофобизации поверхности происходит коагуляция частиц,
причем степень изменения гидратации глины зависит от глубины
протекания катионного обмена между неорганическими катиона¬
ми глины и органическими катионами красителя [51].
Ионизацией материала частиц и адсорбцией на их поверхно¬
сти ионов, присутствующих в воде (в частности, ионов Н+ и ОН"),
объясняются еще два важных физических явления — суспензи¬
онный эффект и поверхностная проводимость. Суспензионный эф¬
фект состоит в том, что значения pH суспензии или золя и ультра¬
фильтрата той же суспензии (золя) неодинаковы. Для суспензий
разных минералов разница в величинах pH (АрН) составляет 0,03—
0,19 единицы и увеличивается с ростом концентрации и степени
дисперсности частиц. «Направление» суспензионного эффекта
(знак АрН) совпадает со знаком заряда частиц минералов [52, 53].
Наличие поверхностной проводимости связано с возникнове¬
нием добавочной электропроводности, обусловленной избытком
ионов вблизи поверхности раздела. Поверхностная электропро¬
водность значительно выше, чем электропроводность компонентов
(около 1,5-10-6 ом-1-см'1 для воды и около 1-10-11 ом-1-СМ"1
для глины). Например, 1%-ная суспензия глины в дистиллиро¬
ванной воде обладает электропроводностью 1-10~4 ом^-см'1 [54].
От природы адсорбированных ионов и величины Ек зависит
количество воды, связанной с поверхностью частиц. По способ¬
ности удерживать воду катионы и анионы располагаются в ряды
[55]:
Na+>Ca2+ >Н+>А13+;
он- > СО2- > СН3СОО- > > Cl-.
52 ГЛАВА II. ХАРАКТЕРИСТИКА ЗАГРЯЗНЕНИЙ ПРИРОДНЫХ ВОД
При низких температурах способность воды смачивать неко¬
торые минералы снижается. Это особенно заметно при использо¬
вании талой воды. Так, например, краевой угол смачивания све¬
жеталой водой типично гидрофильного кварца достигает 60°.
Можно предположить, что в этих условиях вследствие упорядо¬
ченности структуры воды энергия диполей направлена в основном
на взаимодействие друг с другом, а связь воды с поверхностью
минералов ослабевает [56, стр. 25]. В присутствии электролитов
заметная гидрофобизация кварцевых частиц наступает лишь при
высоких концентрациях электролитов — более 1 г-экв/л [57].
Значения дзета-потенциала (ДП) кварцевых и глинистых ча¬
стиц в поверхностных водах с величиной pH порядка 6,5—7,5
находятся обычно в пределах —5 -т 40 мв [35, 36, 52, 58] и опре¬
деляются соотношением кислых и основных компонентов. Напри¬
мер, при pH ~ 6,3—6,5 величина ДП глинистых частиц с соот¬
ношением Si02 : Ме203 ~ 3 равна —26 мв, а частиц с соотноше¬
нием Si02: Ме203 ~ 1 — лишь —16 мв [35]х.
Ниже приводятся значения ДП (в мв) частиц разных минера¬
лов в дистиллированной воде, измеренные методом потенциала
протекания [52]:
Имеются экспериментальные данные о том, что в многокомпо¬
нентных смесях частиц разной природы налипание одних частиц на
другие вызывает соответствующее изменение ДП [60]. Изменение
температуры в пределах 1—20° С практически не влияет на ДП ча¬
стиц бентонита и каолинита в дистиллированной воде [61].
Влияние pH среды на величину ДП не всегда носит однознач¬
ный характер. Так, при обработке глинистых суспензий щелочью
может произойти сначала повышение, а затем — снижение ДП ча¬
стиц вплоть до перезарядки. Повышение потенциала объясняется,
вероятно, замещением противоионов А13+ диффузного слоя на ка¬
тион щелочи и увеличением (вследствие этого) толщины слоя.
Когда же катионов А13+ остается мало, они уже не препятствуют
вхождению катионов щелочных металлов в адсорбционный слой:
толщина диффузного слоя уменьшается и величина ДП падает
Существенное влияние на ДП минеральных коллоидов оказы¬
вает присутствие в воде ПАВ, ВМВ и активного хлора [58]. Ну-
Магнезит
Пирофиллит
Гшхс
Мусковит
+9,3 Каолинит —14,5
—5,5 Биотит —15,3
—9,2 Бентонит —18,9
—10,6 Нефелин —31,0
[62].
1 По современным представлениям [59], заряд частиц кремнеземов в области
значений pH 5—8 обусловлен диссоциацией молекул воды, связанных
координационно с поверхностными атомами кремния.
II.2. ПРИМЕСИ ЕСТЕСТВЕННОГО ПРОИСХОЖДЕНИЯ
53
Рис. II.3. Седиментационная
кривая взвеси природной воды
Р — вес выпавшего осадка;
т •— длительность отстаивания
левому заряду глинистых и кварцевых частиц соответствуют зна¬
чения pH 2—5 [63—65].
Удельный вес (в г/см3) разных минералов изменяется в довольно
широких пределах [66]:
Известняк 2,2—2,4 Кварц 2,65—2,75
Гипс 2,15—2,75 Доломит 2,8—3,0
Глины 2,44—2,53 Гетит 4,0—4,3
Однако, если учесть, что главным компонентом взвешенных
примесей является глина, для ориентировочных расчетов можно
принять некоторую среднюю величину удельного веса р ~
— 2,5 г/см3. Согласно расчетам по уравнению Стокса, при р =
= 2,5 г/см3 гидравлическая крупность частиц диаметром 100 мкм рав¬
на 10 м/час, а частиц диаметром 1 мкм — 0,001 м/час. Разница
очень велика. Если к тому же принять во внимание, что на осаж¬
дение частиц размером менее 50 мкм заметное влияние оказывает
тепловое движение молекул среды [67], станет еще более очевид¬
ной большая разница в скорости осаждения псаммитовых и пели-
товых фракций минеральных примесей. Поэтому кривая седимен¬
тации дисперсных загрязнений природной воды сильно выгнута,
а верхний ее участок почти параллелен оси абсцисс (рис. 11.3).
Для технологической оценки гранулометрического состава
взвеси по кривой седиментации устанавливают ее показатель
осаждаемости. Он выражается дробью, в числителе которой весо¬
вое количество взвеси (в процентах к первоначальному), выпа¬
дающей в осадок за определенное время тх, а в знаменателе — ко¬
личество взвеси, выпадающей в осадок за время х2 [68].
Более точные сведения о фракционном составе взвеси можно по¬
лучить, построив кривую распределения частиц по размеру (см.
гл. I)2.
2 Методика построения кривых распределения описана в руководствах по
коллоидной химии.
54 ГЛАВА II. характеристика загрязнений ПРИРОДНЫХ вод
II. 2.2. Органические примеси
Органические примеси воды представлены, главным образом,
высокомолекулярными веществами: высшими полисахаридами и
белками (протеинами). К первым относятся крахмал — продукт
фотосинтеза растений и целлюлоза — главная составная часть
растений (клетчатка).
Органические примеси, обусловливающие цветность воды 3,
находятся в коллоидном и истинно растворенном состоянии.
Коллоидная фракция имеет высокую степень дисперсности. На¬
пример, при фильтровании цветной воды только 5% окрашиваю¬
щих веществ задерживалось на фильтре с размером пор 0,45 мкм,
и 13% — на фильтре с размером пор 10 нм [69J.
Вымываемый водами гумус, содержание которого в почвах до¬
стигает 75%, представляет собой сложный комплекс органиче¬
ских соединений — продуктов физико-химических и биологи¬
ческих процессов превращения остатков растительного происхож¬
дения. Удельный вес гумуса равен приблизительно 1,4 г/см3 [70].
Гумусовые вещества являются продуктом конденсации арома¬
тических соединений фенольного типа с аминокислотами и про¬
теинами [71]. Они сходны по строению и свойствам, но отличаются
молекулярным весом и соотношением функциональных групп
[72]. Удельная поверхность частиц почвенного гумуса составляет
в среднем 1900 м2/г [73], катионообменная емкость достигает не¬
скольких сот миллиграмм-эквивалентов на литр. Гуминовые ве¬
щества составляют от 45 до 90% почвенного гумуса [12, 74] и
представлены кислотами и их солями. В коллоидном состоянии
находится лишь часть из них.
Коллоидная фракция торфяной воды состоит преимущественно
из частиц размером 0,5—1 мкм, имеющих форму, близкую к сфери¬
ческой, и не обладающих заметной пористостью [75J. По Гапону
[19], удельная поверхность таких частиц достигает 6000 м2/г.
Гуминовые кислоты по данным электронно-микроскопического
анализа представлены мельчайшими сферическими частицами,
склонными к объединению в цепные агрегаты [76, 77]. Согласно
расчетам Джуриона [78], размер ассоциатов гуминовых кислот
составляет ~ 50 А. По своему составу гуминовые кислоты —
сложные ВМВ, в молекулах которых имеются ароматические
кольца изо- и гетероциклического строения, а также карбок¬
сильные и фенольные группы [12, 79]. Большинство авторов под^
черкивает карбоксильный характер гуминовых кислот [80—82],
однако единого мнения об их природе и строении до сих пор не
3 Деление окрашивающих воду веществ на коллоидные и истинно растворен¬
ные весьма условно: под «коллоидной» фракцией этих веществ часто подра¬
зумевают ассоциаты макромолекул ВМВ, проявляющие поверхностные
свойства.
II.2. ПРИМЕСИ ЕСТЕСТВЕННОГО ПРОИСХОЖДЕНИЯ
55
выработано. Это объясняется не только большим разнообразием
кислот, но и аналитическими трудностями.
По данным Блэка и Кристмана [81], эквивалентный вес гуми¬
новых кислот составляет 68—240. Их элементарный состав сле¬
дующий: 52—62% углерода; 30—39% кислорода; 2,9—5,4% во¬
дорода; около 4% азота [12, 83]. Молекулярный вес гуминовых
кислот колеблется в пределах от 300—400 до нескольких тысяч
[81, 84, 85], мицеллярный вес, по данным Когановского [86, 87J,
достигает 3700—8270. Растворы гуминовых кислот обладают
сильно выраженным кислотным характером. Они осаждаются ка¬
тионами Са2+, Со2+, Ni2+ [88] и Mg2+ [74, 89]. Этим объясняется
меньшая окраска жестких вод.
Помимо гуминовых кислот, среди окрашивающих соединений
воды выделяют коллоидные и истинно растворенные фулъвокис-
лоты (креновые и апокреновые кислоты), обладающие повышен¬
ной устойчивостью к действию сорбентов и окислителей. В фуль-
вокислотах содержание углерода ниже, а водорода несколько вы¬
ше, чем в гуминовых кислотах. Однако как объяснить различие
в поведении гуминовых и фульвокислотных фракций в процессе
коагуляции — различием химического состава и строения или раз¬
мером частиц — пока не вполне ясно. Изучение строения этих
компонентов методами ИК-спектроскопии и рентгеноструктур¬
ного анализа привело Гончарову и др. [901 к выводу о разном
строении функциональных групп в молекулах. Хроматографически
выделено четыре вида фульвокислот, имеющих разное строение
[91]. Отмечается более высокая степень дисперсности фульвокис¬
лот по сравнению с гуминовыми кислотами [92].
Соотношение коллоидной и истинно растворенной фракций
окрашивающих веществ в разных источниках неодинаково и ко¬
леблется в течение года. При исследовании гумуса озер Эстонии
обнаружено, что по мере роста общего содержания в воде орга¬
нических соединений увеличивается концентрация коллоидных
окрашивающих веществ. В озерах у верховных болот окраши¬
вающие вещества представлены в основном гуминовыми кисло¬
тами, а в озерах у низовых болот— фульвокислотами [93]. В Волге,
Днепре, Доне, Припяти, в'Клязьминском водохранилище на долю
фульвокислот приходится от 69 до 97 % общей цветности; из них
20—50%—истинно растворенные фульвокислоты [12, 941.
Гуминовые вещества гидрофильны [95, 961, их агрегативная
устойчивость объясняется наличием развитых гидратных оболо¬
чек. Наиболее гидрофилен молодой гумус черноземных почв [97].
Действие электролитов на гуминовые вещества сводится к образо¬
ванию пористых ассоциатов размером от 16~до 50 мкм [75, 86].
Часто образуются комплексные ассоциатыг гуминовой кислоты
с железом и другими металлами [71, 79, 84,г85]. С ростом концен¬
трации электролитов вероятность ассоциации возрастает.
56
ГЛАВА И. ХАРАКТЕРИСТИКА ЗАГРЯЗНЕНИЙ ПРИРОДНЫХ ВОД
Согласно наблюдениям Таран и др. [981, при хлорировании
воды, содержащей гуминовые вещества, на кривой зависимости
скорости реакции от pH имеются два максимума: первый отвечает
наибольшему обесцвечиванию за счет окисления гидроксильных
групп до карбоксильных, второй — окислению аминных групп
гуминовых кислот. Наиболее полное обесцвечивание хлором
воды, содержащей гуминовые кислоты, происходит в нейтраль¬
ной среде.
Коллоидные окрашивающие вещества несут чаще всего отри¬
цательный заряд, достигающий максимума при pH около 8, а их
изоэлектрическая точка лежит в области значений pH 3,5—4,5 [64].
В отдельную группу органических веществ, обусловливающих,
в частности, цветность воды, выделяют галловую кислоту и та¬
нины, относящиеся к классу дубильных. По данным Васенкина
[99], содержание дубильных веществ составляет в коре березы
2,4—10%, ели — 7—12%, ивы — 10—12%, лиственницы—10—
15%, сосны — 2,2—4,2%. Богаты таниновой кислотой корни ду¬
ба и ивы, мхи, папоротники, зеленые водоросли [69].
По своему строению дубильные вещества не являются индиви¬
дуальными химическими соединениями, а представляют собой
комплекс веществ, для которых общим является наличие аромати¬
ческих колец с несколькими оксигруппами. Молекулярный вес
дубильных веществ достигает нескольких тысяч, а изоэлектри¬
ческая точка соответствует pH ~ 6 [100]. Источником появления
в воде кислых полисахаридов являются опавшие листья [101].
Органические вещества, придающие воде запах и вкус, пред¬
ставляют собой сложные смеси ароматических углеводородов и
кислородсодержащих соединений (спиртов, альдегидов, кетонов,
сложных эфиров). Они хорошо сорбируются на активных углях
и разрушаются сильными окислителями [102, 103]. В их составе
обнаружены разные летучие и нелетучие кислоты: сиреневая, ва¬
нилиновая, масляная, уксусная, муравьиная, пропионовая, пи-
ровиноградная, янтарная, молочная, щавелевая, винная и др.
[104—106]. Содержание этих кислот в воде поверхностных водо¬
источников СССР составляет, по данным Хоменко и Гончаровой
[106], 3-10-2 — 15,7-10~2 мг-экв/л.
Коллоидные частицы органических веществ, как правило, об¬
ладают ярко выраженными гидрофильными свойствами, что объ¬
ясняется наличием в составе многих из них полярных групп
(ОН_,СОз“, HC03~, SOl-, РОГ), прочно связанных с дипольными
молекулами воды. Такие коллоидные частицы представляют со¬
бой рыхлые агрегаты цепных молекул, обильно насыщенные во¬
дой. Особенно велика гидрофильность белков и их производных.
Белковую молекулу можно схематически представить в виде
NH2 — R-COOH.
11.2. ПРИМЕСИ естественного ПРОИСХОЖДЕНИЯ
57
В нейтральной среде могут образовываться двойственные (ам-
фотерные) ионы
NH+-R - СОО-.
В кислой и щелочной среде существуют равновесия
NH2 —R—СООН + НС1 ^NH+ — R — СООН + С1-,
NH2 — R — COOH + NaOH г? NH2 — R - COO" + Na+ + H20.
В первом случае потенциалопределяющими группами являются
—NH3+, во втором —СОО".
Заряд белковых частиц обусловлен диссоциацией ионоген¬
ных групп или адсорбцией ионов, находящихся в растворе. В изо-
электрической точке большинство белков находится в недиссоци-
ированном состоянии, а количества анионов и катионов, адсорби¬
рованных из раствора, равны. Поскольку изоэлектрическое
состояние большинства белков отвечает значениям pH < 7, в при¬
родных водах они заряжены отрицательно. Впрочем, как и для
обычных коллоидов, на величину pH изоэлекарической точки бел¬
ков сильное влияние оказывает солевой состав воды.
Вследствие высокой гидрофильности коагуляция белков про¬
текает медленно, однако в изоэлектрическом состоянии белки мож¬
но скоагулировать полностью [107]. Значительное снижение
устойчивости белков, как и многих других гидрофильных веществ,
достигается уменьшением количества полярных групп путем об¬
работки воды хлором, озоном и другими окислителями.
В группу биологических примесей входят простейшие расти¬
тельные и животные организмы, находящиеся во взвешенном со¬
стоянии (планктон) или прикрепленные ко дну водоемов (бентос).
Применительно к задаче коагулирования нас будет интересовать
более всего фитопланктон, быстро развивающийся в озерах и во¬
дохранилищах в теплые периоды года и создающий большие труд¬
ности при очистке воды.
Характерной особенностью многих водохранилищ является
весеннее и осеннее диатомовое и летнее синезеленое цветение,
т. е. массовое развитие водорослей [108, 109]. В период вегетации
диатомового планктона преобладают следующие виды водорослей:
Asterionella (звездчатой формы, объемом 420—500 мкм3), Diatoma
и Fragilaria .(ленточной формы, объемом 320 мкм3), Melosira
(1100—1300 мкм3). В. период цветения синезеленых водорослей
преобладают виды Aphanizomenon (60 мкм3), Microcystis (14 мкм3),
АпаЪаепа (90 мкм3) [110]. Среди синезеленых водорослей суще¬
ствуют как планктонные, так и бентосные формы. Некоторые из
них (АпаЪаепа circinalis, Aphanizomenon flocs-aquae) выделяют
в воду вещества, придающие привкусы и запахи [111].
t ЛАВА tl. ХАРАКТЕРИСТИКА ЗАГРЯЙЙЁЙЙЙ ПРЙРОДНЬГХ Й6Д
Отдельные клетки водорослей объединены в колонии, имеющие
иногда общую слизистую оболочку. Число клеток в 1 мл воды до¬
стигает сотен тысяч. В присутствии фосфатов и других биогенных
веществ развитие водорослей ускоряется 1110, 112].
Помимо обычных для поверхностных вод сапрофитовых (непа¬
тогенных) форм бактерий, в воде могут присутствовать попавшие
со стоками патогенные формы — бактерии дизентерии, брюшного
тифа, холеры, туляремии. Большинство бактерий погибает в ре¬
зультате процессов самоочищения через 3—4 дня, но отдельные
особи сохраняются гораздо более длительное время. По данным
Григорьевой [113], сроки выживаемости кишечной палочки, воз¬
будителей брюшного тифа, дизентерии, туляремии, чумы, полио¬
миелита исчисляются десятками суток, а спор сибиреязвенных
палочек — годами.
Бактерии используют для питания углерод неорганических
веществ, органические соединения, живой белок. Нарушение ус¬
ловий питания посредством изменения pH, температуры и кисло¬
родного режима приводит к подавлению развития бактерий и их
отмиранию [11, стр. 273].
По степени биологического загрязнения водоемы делят на три
основные категории: полисапробную (наиболее загрязненная во¬
да), мезосапробную и олигосапробную. Число колоний бактерий
в 1 мл воды в этих зонах соответственно составляет до 1 • 10е, до
1 • 10Б и до 1 • Ю3. В воде озер и водохранилищ содержится обычно
до 1,5- Ю3, в воде рек — до 1,25 • 105 колоний бактерий в 1 мл.
Особо важным показателем санитарного состояния водоема
считают концентрацию в воде кишечной палочки (В. Coli), посколь¬
ку ее присутствие говорит о загрязненности воды фекальными
стоками.
Бактерии делят по форме на шаровидные — кокки (1—2 мкм),
палочкообразные — бациллы и собственно бактерии (1—4 мкм) и
спиралъно-извитые — спиреллы, спирохеты, вибрионы (1—20 мкм).
В состав бактерий входит 1—4% жиров, 8—14% белков и 80—
85% воды. В микроколичествах содержатся фосфор, калий,
кальций, магний, железо и другие элементы [114 (стр. 267), 115].
Вирусы не обладают клеточной структурой и имеют размер 10—
100 нм [115, стр. 248].
Колонии бактерий и вирусов (биоколлоиды) состоят из сотен
и тысяч клеток и отличаются периодичностью строения. Толщина
водных прослоек между клетками 1—3 мкм, т. е. соизмерима
с размерами бактериальных клеток [116, стр. 114].
Планктон и биоколлоиды обладают большой поверхностью
раздела и несут электрический заряд. В частности, по данным
Бернгардта [117], ДП водорослей Oscillatoria rubescens составляет
—8 13 мв. Некоторое снижение заряда достигается хлори¬
рованием воды. Например, смесь бактерий (9 вес. ч. В. Coli +
II.2. ПРИМЕСИ ЕСТЕСТВЕННОГО ПРОИСХОЖДЕНИЯ
59
+4 вес. ч. В. Subtilis +1 вес. я. В. Proteus Mirabilis) общей концен¬
трацией 2,5-104 колоний в 1 мл имела ДП от —27 до —71 мв
(в среднем —50 мв). После 24-часового контакта этой смеси с хло¬
ром (2 мг/л остаточного свободного хлора) среднее значение ДП
снизилось до —34 мв [118].
II. 2.3. Органо-минеральные ассоциаты и комплексы
Выше указывалось на способность гуминовых кислот образовы¬
вать комплексные соединения с катионами металлов. Этой же осо¬
бенностью обладают дубильные и некоторые другие органиче¬
ские вещества. Причем многие комплексы (особенно с окислами
железа) имеют интенсивную окраску [68 (стр. 43), 119, 120].
При одновременном присутствии в воде частиц минеральных
примесей и органических веществ в результате адсорбции уста¬
навливается определенное равновесие, зависящее от концентрации
и поверхности примесей. Адсорбция на глинах органических кра¬
сителей, метакриловой, олеиновой и а-аминовой кислот описы¬
вается изотермами Лэнгмюра или Фрейндлиха [121,122], но наблю¬
далась также и форма изотермы, характерная для хемосорбции
[123-125].
Количественная оценка адсорбции органических веществ (осо-
. бенно ПАВ и ВМВ) на механических примесях имеет важное зна¬
чение, поскольку дает возможность судить о состоянии поверх¬
ности, оценить способность частиц к коагуляции.
Исследования, проведенные с водой из р. Миссури, показали,
что с минеральными частицами взвеси ассоциировано около 60%
органических веществ [1261. С увеличением степени гидрофиль¬
ности минеральных взвесей возрастает количество органических
примесей, удаляемых коагуляцией, что указывает на более высо¬
кую адсорбционную способность гидрофильных минералов. С дру¬
гой стороны, коллоидные частицы и макромолекулы органических
веществ могут проявлять по отношению к частицам минералов
защитное действие и затруднять их коагуляцию [119,127]. Гумино¬
вые вещества проявляют защитное действие по отношению к поч¬
венным суспензиям, золям кремнекислоты, гидроокисям алюми¬
ния и железа. Этим объясняется, в частности, наличие растворен¬
ного железа в водах, богатых кислородом \128]. Максимальная
адсорбция гуминовых веществ имеет место при низких значениях
pH воды, когда их ДП близок к нулю (рис. II.4).
Ионогенные и неионогенные ПАВ, адсорбируясь на глинах
в небольших количествах, вызывают их гидрофобизацию [129,
130], сопровождающуюся объединением в агрегаты [131]. Однако
с увеличением концентрации ПАВ начинают проявлять защит¬
ное действие (подробнее см. раздел II.3.1).
Хорошо адсорбируются на минеральных частицах бактерии и
вирусы. Причем адсорбция разных микроорганизмов носит ярко
60 ГЛАВА II. ХАРАКТЕРИСТИКА ЗАГРЯЗНЕНИЙ ПРИРОДНЫХ ВОД
Рис. 11.4. Зависимость величины адсорбции (Г) гуминовых веществ на
частицах глины от pH воды [10]
Рис. 11.5. Процентное распределение (р) дисперсных частиц загрязнений
природной воды по величине ДП
выраженный селективный характер и протекает с неодинаковой
скоростью [132]. Поскольку ДП клеток бактерий имеет высоко¬
отрицательные значения, сорбцию их на глинах можно проследить
по росту средней величины ДП глинистых частиц в присутствии
бактерий [133]:
Объект исследования ДП, мв
Суспензия глины —16
То же и В. Coli (4-108 колоний ъ I мл) —24
То же и В. Proteus (6 -105 колоний в 1 мл) _34
Можно, таким образом, прийти к выводу, что наличие в по¬
верхностных водах дисперсных и истинно растворенных веществ
разной природы со своими индивидуальными свойствами и зарядом
позволяет говорить лишь о некоторых средних значениях ДП.
Действительно, как показано на рис. II.5 и в табл. II.7, наблю¬
дается большой разброс значений ДП примесей в источниках во¬
доснабжения США.
В речных и озерных водах величина ДП частиц загрязнений
находится обычно в пределах —15 25мв, но если они загряз¬
нены дополнительно сточными водами, содержащими органиче¬
ские примеси, ДП достигает —50 и даже —60 мв. Это отражается
и на средней величине заряда частиц. Биин [133] приводит сле¬
дующие результаты замеров средних значений ДП частиц за¬
грязнений речной воды, смешанной в разных пропорциях с хозяй¬
ственно-бытовыми сточными водами:
Содержание сточных вод в речной воде, % 0 10 25 50
Средние значения ДП, мв —21 —23,7 —27 —29
II.3. ПРИМЕСИ, ПОСТУПАЮЩИЕ СО СТОЧНЫМИ ВОДАМИ
61
Таблица II.7. Значения ДП частиц загрязнений в источниках
водоснабжения США
ДП, мв
Источник водоснабжения
минималь¬
ный
максималь¬
ный
средний
Литература
р. Делавэр
-13
-33
-21,5
[118, 134]
р. Потомак
—16
-21
-19
[32]
Водохранилища в ок¬
рестностях Вашингтона
—14
-27
—
[32]
р. Гудзон
-7
-28
-17
[135]
В периоды паводков, когда источники водоснабжения попол¬
няются чистыми водами, значения ДП дисперсных загрязнений
понижаются. Таким образом, заряд частиц примесей может слу¬
жить косвенной характеристикой степени загрязненности воды.
Кавамура [136] предложил использовать в качестве интегрального
показателя характера и количества дисперсных загрязнений сум¬
марный заряд примесей в единице объема воды, определяемый
методом так называемого «коллоидного титрования».
II. 3. ПРИМЕСИ, ПОСТУПАЮЩИЕ СО СТОЧНЫМИ ВОДАМИ
С хозяйственно-бытовыми и производственными сточными водами,
в том числе со стоками с промплощадок, в водоемы попадают бел¬
ки, жиры, масла, нефть и нефтепродукты, красители, смолы, ду¬
бильные вещества, моющие средства и многие другие загрязнения.
С полей вымываются удобрения и пестициды — средства борь¬
бы с вредителями сельскохозяйственных культур. Поэтому в водах
открытых источников водоснабжения в разных концентрациях
содержатся фактически любые химические элементы, в том числе
такие вредные для здоровья, как свинец, цинк, олово, хром, медь.
Не имея целью дать полный обзор состава загрязнений, попа¬
дающих со сточными водами, и полагая, что свойства биологиче¬
ских примесей достаточно подробно рассмотрены в предыдущем
разделе этой главы, остановимся лишь на некоторых видах загряз¬
нений, отличительными признаками которых являются: широкая
распространенность, особенно в последние годы; токсические свой¬
ства; трудное отделение при очистке сточных вод; медленное
окисление и разложение в открытых водоемах; мешающее дей¬
ствие, оказываемое на процессы очистки воды, в том числе на коа¬
гуляцию; '.способность быть индикаторами глубины очистки воды
от отдельных 'элементов.
62 ' ГЛАВА II. ХАРАКТЕРИСТИКА ЗАГРЯЗНЕНИЙ ПРИРОДНЫХ ВОД
К загрязнениям с перечисленными особенностями, называемым
иногда микрозагрязнениями, мы отнесем синтетические поверх¬
ностно-активные вещества, фосфаты, пестициды и радиоактивные
примеси. Рассмотрим их свойства.
II. 3.1. Синтетические поверхностно-активные вещества
К поверхностно-активным веществам (ПАВ), о которых неодно¬
кратно упоминалось выше, относятся соединения, обладающие спо¬
собностью при адсорбции на поверхности раздела значительно
понижать поверхностную энергию. Эта способность объясняется
наличием в структуре молекул, помимо неполярной углеводород¬
ной части, асимметрично расположенных полярных групп атомов.
Полярные группы, такие, как СОО-, ОН”, POj”, ориентируются
на границе раздела фаз преимущественно в сторону более поляр¬
ной фазы (воды), в то время как углеводородные участки стре¬
мятся приблизиться к менее полярной фазе.
Характер действия ПАВ определяется числом и расположением
в молекуле полярных групп, а также степенью полярности (гид¬
рофильности) последних. Молекулы, имеющие полярную груп¬
пу на конце, образуют на поверхности раздела вертикально ори¬
ентированный адсорбционный слой. Наличие такого слоя способ¬
ствует несмачиванию поверхности водой, т. е. ее гидрофобизации.
В тех же случаях, когда полярные группы расположены в середине
молекулярной цепи или по ее концам, адсорбированные молекулы
как бы стелются по поверхности (горизонтальная ориентация)
и придают ей меньшую гидрофобность.
Полярные группы в воде ионизируются. Если заряд ионов от¬
рицательный, вещество называют анионоактивным (или анион¬
ным), если положительный — катионоактивным (или катион¬
ным). Первое адсорбируется главным образом на положительно
заряженных участках поверхности, второе — на отрицательно
заряженных. ПАВ с полярными группами, не несущими заряда
или несущими очень слабый заряд (—ОН, —СО и др.), называют
неионогенными, или неионными. За счет водородных связей они
присоединяют молекулы воды и поверхность раздела гидрофили-
зуется. Важнейшей особенностью ПАВ с достаточно большим
углеводородным радикалом и сильной полярной группой является
их способность при некоторой критической концентрации (ККМ)
образовывать мицеллы — скопления множества ориентированных
молекул. Такие ПАВ называют коллоидными. Так же как и при
адсорбции на поверхности, молекулы коллоидных ПАВ в мицел¬
лах ориентированы полярными группами в сторону более поляр¬
ной фазы (рис. II.6).
К поверхностно-активным веществам относится Оолыная часть
рассмотренных в предыдущем разделе органических кислот и их
lli. примеси, Поступающие со сточныМй водами
63
солей, спирты, эфиры, белки. Наиболее обширную группу искус¬
ственных 1IAB составляют вещества, входящие в состав современ¬
ных моющих средств — синтетических детергентов (СД), все
более вытесняющих из производства жировые мыла. Широкое ис¬
пользование СД в народном хозяйстве связано с их высокой тех¬
нологической эффективностью, не зависящей от жесткости и ще¬
лочности воды, и сравнительно дешевым способом получения_—
из продуктов нефте- и углехимического синтеза. ПАВ, входящие
в состав СД, могут быть анионными, катионными, амфотерными
и неионогенными. Наибольшее применение получили анионные
ПАВ: алкилсульфаты, алкилсульфонаты, алкилбензолсульфо-
наты, алкиларилсульфонаты и другие.
Характерной особенностью синтетических ПАВ является их
слабая окисляемость. Время, в течение которого концентрация
анионных ПАВ в воде водоемов снижается на 50% за счет биохими¬
ческого разложения, составляет, по Лукиных [137, стр. 19], от
нескольких суток до десятков и даже сотен суток в зависимости
от химического строения. Период полураспада алкилбензолсуль-
фоната в речной воде равен примерно 15 суткам [138]. По спо¬
собности к окислению ПАВ делят на биохимически «мягкие» (ал¬
килсульфаты, линейные алкилбензолсульфонаты, эфиры, амиды
жирных кислот, алкилсульфонаты) и «жесткие» (алкилбензол¬
сульфонаты, алкилфеноксиполиэтиленгликоли) [139].
Средняя концентрация синтетических ПАВ в бытовых сточ¬
ных водах — 16 мг/л [137, стр. 25]. При очистке сточных вод сте¬
пень удаления ПАВ находится в пределах 13—88%, составляя
в среднем 45% для «жестких» и 80% для «мягких» ПАВ [139—
141]. Все это обусловливает поступление ПАВ в водоемы 4.
Поведение в воде синтетических ПАВ носит тот же характер,
что и природных. Мицеллы низших членов ряда (п ~ 10) алкил-
бензолсульфоната натрия (CnH2n+1C6H4S03Na) — небольшие,
сильно гидратированные, сферической формы; мицеллы высших
членов ряда (ге^>10) — сравнительно крупные и асимметричные.
Сорбция ПАВ (деканолсульфонат и додецилсульфат натрия)
/7олярные группы
Рис. II.6. Схема строения ми¬
целлы ПАВ
4 Подробный обзор о загрязнении водоемов СД дан в работе [142].
64 ГЛАВА II. ХАРАКТЕРИСТИКА ЗАГРЯЗНЕНИЙ ПРИРОДНЫХ ВОД
на частицах монтмориллонита протекает с образованием связи
между анионом ПАВ и катионом кальция глины [143]. Под дей¬
ствием как ионогенных, так и неионогенных ПАВ происходит пеп-
тизация суспензии монтмориллонита [144]. На золь гидроокиси
железа, по данным Мегаро и Кондо [1451, ПАВ в зависимости от
концентрации могут оказывать коагулирующее или пептизирую-
щее действие.
Исследование воздействия неионогенных ПАВ — эфиров це-
тилового спирта и полиэтиленгликоля — на заряд частиц золей
показало, что в присутствии ПАВ ДП положительно и отрица¬
тельно заряженных частиц снижается тем быстрее, чем длиннее
цепи ПАВ [146]. Можно предположить, что снижение ДП является
результатом происходящего при адсорбции ПАВ сдвига границы
скольжения фаз в глубину раствора.
II. 3.2. ФОСФАТЫ
Помимо основного инградиента (ПАВ), с моющими средствами
в источники водоснабжения поступают конденсированные фос¬
фаты: пиро- и трифосфаты. Например, в природные воды Калифор¬
нии 20—40% общего фосфора попадает с синтетическими детер¬
гентами [147].
В воде конденсированные фосфаты гидролизуются с образо¬
ванием ортофосфатов. Гядролиз пирофосфата протекает по урав¬
нению
Na4P207 + Н20 = 2Na„HP04.
Гидролиз трифосфата проходит в две стадии:
Na6P3O10 + Н20 = Na3HP20, + Na2HP04,
Na3HP20, + H20 =Na2HP04+NaH2P04.
На скорость гидролиза сильное влияние оказывают pH, тем¬
пература и солевой состав воды [148,149]. Концентрация фосфатов
(в диапазоне 1—50 мг/л) мало влияет на скорость их гидролиза
[149]. Ниже приведены данные из работы [148], характеризующие
зависимость периодов полураспада фосфатов (в сутках) от темпе¬
ратуры воды (при исходной концентрации фосфатов 20 мг/л):
5° С 20° С 35° С
Пирофосфат 220 120 45
Трифосфат 240 60 20
При удалении фосфатов из сточных вод на канализационных
сооружениях возникают серьезные трудности. Применение реа-
гентных методов целесообразно лишь при очистке не более чем на
90%, так как более глубокое удаление фосфатов связано с резким
И.З. ПРИМЕСИ, ПОСТУПАЮЩИЕ со СТОЧНЫМИ ВОДАМИ
65
увеличением расхода реагентов [150, стр. 105]. Поэтому, а также
вследствие медленного гидролитического распада фосфаты при¬
сутствуют в открытых водоемах. Не исключается их концентрация
до 1 мг/л [134].
С катионами металлов фосфаты образуют комплексные соеди¬
нения. В частности, возможна реакция
Na6P3O10 + Са2+ Na3 1СаР3Ою] + 2Na+
и, как следствие, ускорение гидролитического распада фосфатов
в жестких водах [148]. Кислая среда и присутствие бактерий также
способствуют ускорению гидролиза [151, 152].
Сорбция полифосфатов и метафосфатов на частицах глины, са¬
жи, окислов металлов, пигментов приводит к росту заряда частиц
[134, 153] и сопровождается пептизацией. Так, по данным Биина
и др. [134], ДП частиц глинистой суспензии, равный в обычных
условиях —13,5 мв, после добавления 1 мг/л гексаметафосфата
увеличился до —33 мв. Пептизирующее действие фосфатов может
быть объяснено связыванием ими катионов многовалентных ме¬
таллов, выступающих в роли коагулирующих ионов для отрица¬
тельно заряженных частиц.
Вредное влияние фосфатов на качество воды в источниках во¬
доснабжения состоит также в их стимулирующем влиянии на раз¬
витие водорослей [154, 155] и фитопланктона [156].
I. 3.3. ПЕСТИЦИДЫ
Наибольшее распространение получили хлорорганические (геп¬
тахлор, алдрин, дилдрин, эндрин, ДДТ, гексахлоран) и фосфор-
органические (карбофос, хлорофос, фосфамид) пестициды. Их
пороговые концентрации, ухудшающие органолептические свой¬
ства воды, чрезвычайно малы: 0,01 —0,001 мг/л [150 (стр. 94), 157,
158]. Попадая в организм, пестициды воздействуют на нервную
и сердечно-сосудистую системы, на функцию некоторых органов
[159—191]. Поэтому вода, предназначенная для питьевых целей,
должна быть подвергнута глубокой очистке.
Окисление пестицидов протекает чрезвычайно медленно. Ис¬
следования показали, что за восемь недель концентрация в воде
эндрина, дилдрина, гептохлорэпоксида, ДДТ, ДДД, ДДЕ, азо-
дрина и ВНС не претерпевает заметных изменений [162]. Гидролиз
паратиона, входящего в состав фосфорорганических пестицидов,
заканчивается через 30—40 суток [163].
Большинство пестицидов хорошо адсорбируется на частицах
глин [164]. Сорбция дилдрина возрастает при понижении pH среды
от 10 до 6. Температура воды и присутствие разных органических
веществ мало влияют на величину адсорбции [165]. Инсектициды
3 Е. Д. Бабенков
66 ГЛАВА И. ХАРАКТЕРИСТИКА ЗАГРЯЗНЕНИЙ ПРИРОДНЫХ ВОД
легко аккумулируются живыми организмами: через 24 часа кон¬
центрация ДДТ в дафниях превышает концентрацию в воде в 16—
23 тыс. раз [166].
И. 3.4. РАДИОАКТИВНЫЕ ВЕЩЕСТВА
Обогащение поверхностных вод радиоактивными веществами про¬
исходит вследствие выщелачивания и растворения радиоактивных
минералов, при добыче и промывке руд, при загрязнении источни¬
ков сточными водами атомных реакторов, предприятий по изго¬
товлению и регенерации тепловыделяющих элементов, медицин¬
ских учреждений. В источниках водоснабжения могут присут¬
ствовать изотопы: 10Ве, 14С, 24Na, 27Mg, 40К, 46Са, 87Rb, 89Sr, 90Sr,
91Y, 95Nb, 106Ru, 131J, 137Cs, 140Ba, 144Ce, 220Rn, 226Ra, 232U и другие
[167 (стр. 9), 168].
Фазово-дисперсное состояние радиоизотопов зависит от их спо¬
собности образовывать малорастворимые продукты гидролиза и
комплексные соединения с ионами, содержащимися в воде. Боль¬
шая часть изотопов, как правило, адсорбируется на механических
примесях воды. Например, в водоемах США на долю взвешенных
частиц приходится 50—60% общей (5-активности воды [169].
Имеются, однако, и другие примеры: в воде Рейна лишь 1% ра¬
диоактивных веществ связан с дисперсной фазой, остальные же
присутствуют в растворенном виде [170].
Распределение радиоактивности между водой, грунтом и био¬
массой водоемов в сильной степени зависит от состава изотопов
и pH среды [171—173]. В иле может накапливаться до 90% актив¬
ности [167, стр. 38]. Значительная часть радиоактивных веществ
поглощается водорослями, планктоном, моллюсками, рыбой [174,
175]. Особенно заметна тенденция к накоплению в планктонных ор¬
ганизмах и на взвешенных частицах ила изотопов 32P,46Sc, 64Cu,
65Zn,67As, 137Cs [167 (стр. 38),176]. В живых организмах концентри¬
руются в основном растворимые соединения, в осадках — нера¬
створимые [136].
Некоторые радиоактивные вещества могут образовывать в воде
самостоятельные радиоколлоиды, формирование которых не свя¬
зано с адсорбцией на посторонней фазе [177, 178].
Литература
1. Л. А. Кульский. Теоретическое обоснование технологии очистки воды.
Киев, «Наукова думка», 1968, стр. 18.
2. О. А. Алекин. Основы гидрохимии. JI., Гидрометеоиздат, 1970.
3. В. И. Жадин. Труды Всесоюзного гидробиологического общества,
т. 14. М., Изд-во АН СССР, 1963, стр. 9.
4. С. М. Драчев, А. А. Былинкина, И. Н. Сосунова. Там же, стр. 66.
5. В. А. Реберг. Водоснабжение и санитарная техника, № 3, 4 (1941).
II. ЛИТЕРАТУРА
67
6. JI. А. Кулъский, М. А. Шевченко, Е. М. Калинийчук. Методы улучше¬
ния запаха и вкуса питьевой воды. М., Изд-во МКХ РСФСР, 1961,
стр. 21.
7. Е. Paulson. Water and Sewage Works, 110, 216 (1963).
8. Сб. «VIII Международный конгресс по водоснабжению». Под ред.
Ф. А. Шевелева. М., Стройиздат, 1970, стр. 39.
9. Н. С. Попова. Водоснабжение и санитарная техника, № 9, 7 (1958).
10. Л. А. Кулъский. Основы технологии кондиционирования воды. Киев,
Изд-во АН УССР, 1963.
11. Н. С. Лебедева. Автореферат канд. дисс. М., ВНИИ ВОДГЕО, 1954.
12. М. А. Шевченко. Физико-химическое обоснование процессов обесцве¬
чивания и дезодорации воды. Киев, «Наукова думка», 1973.
13. М. С. Шкроб, В. Ф. Вихрев. Водоподготовка. М.— Л., «Энергия», 1966,
стр. 410.
14. L. G. Rich, J. F. Andrews, Т. М. Keinath. Water and Sewage Works, 119,
126 (1972).
15. В. А. Клячко, И. Э. Апельцин. Подготовка воды для промышленного
и городского водоснабжения. М., Стройиздат, 1962.
16. Д. Эйзенберг, В. Кауцман. Структура и свойства воды. Л., Гидрометео-
издат, 1975, стр. 259.
17. Г. А. Крестов. Термодинамика ионных процессов в растворах. Л.,
«Химия», 1973, стр. 191.
18. С. А. Балезин, Б. В. Ерофеев, Н. И. Подобаев. Основы физической и кол¬
лоидной химии. М., «Просвещение», 1975.
19. Е. Н. Гапон. Коллоидн. ж., 9, 29 (1947).
20. А. А. Иванова, Г. С. Коновалов. Гидрохимические материалы, т. 55. Л.,
Гидрометеоиздат, 1971, стр. 79.
21. Р. Э. Грим. Минералогия и практическое использование глин. М.,
«Мир», 1967.
22. В. N. Rolfe. US Geol. Survey. Profess. Paper, N 340-D, 334 (1962).
23. Ю. И. Тарасевич, Ф. Д. Овчаренко. Коллоидн. ж., 31, 753 (1969).
24. R. A. Baker, Luh Ming-Dean. Water Res., 5, 839 (1971).
25. Ф. Д. Овчаренко, Ю. И. Тарасевич, Ф. А. Велик и др. Коллоидн. ж., 35,
467 (1973).
26. Ф. Д. Овчаренко, Ю. И. Тарасевич. Там же, стр. 867.
27. Ю. И. Тарасевич, В. М. Балицкая и др. Коллоидн. ж., 30, 889 (1968).
28. D. G. Edwards, А. М. Posner et al. Nature (Engl.), 206, N 4980, 168 (1965).
29. В. С. Комаров. Адсорбционно-структурные, физико-химические и ката¬
литические свойства глин Белоруссии. Минск, «Наука и техника»,
1970, стр. 5.
30. R. F. Packham. J. Colloid Sci., 20, 92 (1965).
31. А. P. Black, Ching-lin Chen. J. Amer. Water Works Assoc., 57, 354
(1965).
32. R. L. Williams. Ibid., p. 801.
33. Ф. Кржиль. Исследование и оценка технических адсорбционных ве¬
ществ. Харьков—Киев, «Кокс i хем1я», 1933.
34. М. И. Слюсарева, Л. С. Частникова. Сб. «Адсорбционные свойства
природных и синтетических сорбентов». Ташкент, «ФАН», 1969, стр. 22.
35. Когё ёсуй, № 49, 1 (1962); РЖХим., 17И198-17И205 (1963).
36. Т. Абдуллаев, Г. И. Николадзе, Б. М. Еремин. Пуск и наладка очист¬
ных сооружений (материалы семинара). Изд. Моск. Дома научно-
техн. пропаганды им. Ф. Э. Дзержинского, 1971, стр. 25.
37. И. В. Кононов. Водоснабжение и канализация, вып. 1 (21). М., изд.
ЦБНТИ МЖКХ РСФСР, 1972, стр. 1.
38. М. N. Rao, С. A. Sastry. J. Inst. Engrs (India). Public Health Engng
Div. 49, 141 (1969).
39. S. J. Toth, A. N. Ott. Environ. Sci. and Technol., 4, 935 (1970).
3*
68 ГЛАВА II. ХАРАКТЕРИСТИКА ЗАГРЯЗНЕНИЙ ПРИРОДНЫХ ВОД
40. Н. Loffler. Wasserwirtschaft—Wassertechnik, 17, 11 (1967).
41. Е. М. Сергеев. Грунтоведение. Изд-во МГУ, 1971.
42. В. Pilipovich, А. P. Black, F. A. Eidsness et al. J. Amer. Water Works
Assoc., 50, 1467 (1958).
43. A. P. Black, S. A. Hannah. J. Amer. Water Works Assoc., 53, 438 (1961).
44. B. G. Laffer, О. M. Posner, J. P. Quirk. J. Colloid and Interface Sci., 30,
355 (1969).
45. Ф. Д. Овчаренко. Сб. «Успехи коллоидной химии». М., «Наука», 1973,
стр. 67.
46. Ю. И. Тарасевич, И. А. Грибина. Коллоидн. ж., 32, 577 (1970).
47. N. К. Mitra, В. Sandilya. Indian Ceram., 13, N 11, 257 (1969).
48. И. И. Марциан, В. М. Балицкая. Сб. «Глины, их минералогия, свойства
и практическое значение». М., «Наука», 1970, стр. 101.
49. Ш. Батталова, Н. Д. Пак. Там же, стр. 135.
50. Э. Г. Агабалъянц, А. А. Говоров, Э. В. Маркина. Там же, стр. 151.
51. Н. В. Пшеничная, М. В. Эйриш. Исследование и использование глин
и глинистых материалов (материалы симпозиума в г. Алма-Ате
16—21 сент. 1968 г.). Алма-Ата, «Наука», 1970, стр. 249.
52. Ю. М. Чернобережский, Н. Г. Алексеева. Коллоидн. ж., 28, 609 (1966).
53. Ю. М. Чернобережский, С. Н. Зубкова, С. Д. Усанова. Коллоидн. ж., 27
780 (1965).
54. В. Б. Черногоренко. Коллоидн. ж., 22, 730 (1960).
55. Г. В. Куколев, Я. М. Сыркин. Коллоидн. ж., 17, 90 (1955).
56. В. А. Глембоцкий, В. И. Классен. Флотация. М., «Недра», 1973, стр. 25.
57. Г. С. Стрелъцин. Пятая Всесоюзная конференция по коллоидной химии
(тезисы докладов). М., Изд-во АН СССР, 1962, стр. 281.
58. А. Кооритс, У. Пальм. Уч. зап. Тартуского ун-та, 265, 61 (1970).
59. В. Ф. Киселев. Сб. «Электронные явления в адсорбции и катализе на
полупроводниках». М., «Мир», 1969, стр. 164.
60. Е. Е. Бибик, Е. А. Соколова, И. С. Лавров. Коллоидн. ж., 32, 301 (1970).
61. М. F. Mohtadi, Р. N. Rao. Water Res., 7, 747 (1973).
62. Е. Т. Ускова. Коллоидн. ж., 23, 204 (1961).
63. J. М. Cases, P. Ch. Touret, D. Vestier. С. r. Acad. Sci., C272, 728 (1971).
64. А. А. Кастальский, Д. М. Минц. Подготовка воды для питьевого и
промышленного водоснабжения. М., «Высшая школа», 1962, стр. 58.
65. А. М. Годен. Флотация. М., Госгортехиздат, 1959.
66. А. М. Когановский. Коллоидн. ж., 11, 417 (1949).
67. Б. М. Левин. Труды Московского института инженеров транспорта,
вып. 164. М., «Высшая школа», 1963, стр. 173.
68. В. А. Клячко, И. Э. Апелъцин. Очистка природных вод. М., Стройиздат,
1971, стр. 137.
69. А. P. Black, D. G. Williams. J. Amer. Water Works Assoc., 53, 585 (1961).
70. И. П. Антипов-Каратаев. Современные методы исследования физико¬
химических свойств почв, вып. 1. Руководство для полевых и лабора¬
торных исследований почв. М.— JL, Изд-во АН СССР, 1945, стр. 5.
71. М. М. Кононова. Проблемы почвенного гумуса и современные задачи
его изучения. М., Изд-во АН СССР, 1951.
72. R. F. Packham. Proc. Soc. Water Treat. Exam., 13, 316 (1964).
73. E. H. Гапон. Коллоидн. ж., 9, 329 (1947).
74. О. И. Мартынова. Коагуляция при водоподготовке. М.— JL, Госэнер-
гоиздат, 1951, стр. 6.
75. М. П. Воларович, Н. В. Чураев. Коллоидп. ж., 16, 241 (1954).
76. Т. А. Кухаренко. Природа, № 5, 92 (1953).
77. Т. А. Кухаренко. Труды Института горючих ископаемых, вып. 5. М.,
Изд-во АН СССР, 1955, стр. И.
78. R. Jurion. Bull. Soc. chim. France, N 6, 2622 (1968).
79. W. L. Lamar. US Geol. Survey. Profess. Paper, N 600-D, 24 (1968),
II. ЛИТЕРАТУРА
69
80. R. F. Christman. Trend Engng Univ. Washington, 16, N 4, 10 (1964).
81. J. Shapiro. Limnol. and Oceanogr., 2, 161 (1957).
82. A. P. Black, R. F. Christman. J. Amer. Water Works Assoc., 55, 897
(1963).
83. Ю. И. Вейцер, P. М. Стерина. Научные труды АКХ им. К. Д. Пам¬
филова, вып. 52. Водоснабжение, № 5. М., ОНТИ, 1969, стр. 27.
84. М. Ghassemi, R. F. Christman. Limnol. and Oceanogr., 13, 583 (1968).
85. М. М. Кононова. Органическое вещество почвы. М., Изд-во АН СССР,
1963.
86. А. М. Когановский, Т. М. Ровинская. Коллоидн. ж., 17, 83 (1955).
87. А. М. Когановский. Коллоидн. ж., 24, 34 (1962).
88. Р. А. Идрисова, А. И. Мун и др. Труды Института химических наук,
вып. 25. Алма-Ата, «Наука», 1969, стр. 74.
89. A. Otsuki, R. G. Wetzel. Limnol. and Oceanogr., 18, 490 (1973).
90. Т. О. Гончарова, В. А. Криулькова и др. Гидрохимические материалы,
т. 48. JL, Гидрометеоиздат, 1968, стр. 112.
91. Б. Г. Мурзаков. ЖПХ, № 6, 1373 (1969).
92. В. В. Пономарееа. Почвоведение, № 12, 714 (1947).
93. Н. Simm. Arch. Hydrobiol., 66, 273 (1969).
94. Д. М. Минц, Т. К. Сиденко, 3. С. Ващенко и др. Сб. «Санитарная
техника». М., Изд-во МКХ РСФСР, 1954, стр. 33.
95. П. А. Ребиндер. Труды Третьей Всесоюзной конференции по коллоид¬
ной химии. М., Изд-во АН СССР, 1956, стр. 7.
96. П. А. Ребиндер. Коллоидн. ж., 20, 527 (1958).
97. JI. А. Кулъский, А. М. Когановский, И. Т. Гороновский и др. Физико¬
химические основы очистки воды коагуляцией. Киев, Изд-во ДН УССР,
1950, стр. 50.
98. П. Н. Таран, Р. С. Касъянчук и др. Сб. «Физико-химическая механика
и лиофильность дисперсных систем», вып. 2. Киев, «Наукова думка»,
1971, стр. 297.
99. В. С. Васенкин. Технология экстрактивных веществ дерева. М., Гослес-
техиздат, 1944.
100. S. Kawamura. Water and Sewage Works, 114, 282 (1967). .
101. W. A. De Long, M. Schnitzer. Soil Sci. Soc. Amer. Proc., 19, 360 (1955).
102. E. M. Middleton, A. A. Reosn. Ind. and Engng Chem., 48, 268 (1956).
103. JI. А. Кулъский, М. А. Шевченко, В. П. Чупова. Гигиена и санитария,
№ 5, 16 (1957).
104. R. F. Christman, М. Ghassemi. J. Amer. Water Works Assoc., 58, 723
(1966).
105. И. А. Гончарова. Гидрохимические материалы, т. 52. JI., Гидрометео¬
издат, 1969, стр. 17.
106. А. Н. Хоменко, И. А. Гончарова. Гидрохимические материалы, т. 55.
JI., Гидрометеоиздат, 1971, стр. 32.
107. N. Zdenek. Wasserwirtschaft—Wassertechnik, 7, 432 (1957).
108. Е. С. Велъмина. Качество подготовки воды (материалы семинара), сб.
№ 2. Изд. Моск. Дома научно-техн. пропаганды им. Ф. Э. Дзержинского,
1967, стр. 25.
109. D. Е. Francisco, Ch. М. Weiss. J. Water Pollut. Contr. Fed., 45, 480
(1973).
110. А. А. Гуревич. Пресноводные водоросли. М., «Просвещение», 1966.
111. J. К. Silvey, D. E. Henley, J. T. Wyatt. J. Amer. Water Works Assoc.,
64, 35 (1972).
112. С. M. Palmer. J. Amer. Water Works Assoc., 52, 897 (1960).
113. JI. В. Григорьева. Санитарная бактериология и вирусология водоемов.
М., «Медицина», 1975, стр. 71.
114. Н. Ф. Возная. Химия воды и микробиология. М..[«Высшая школа», 1967,
115. М. Фробишер. Основы микробиологии. М., «Мир», 1965.
70 ГЛАВА II. ХАРАКТЕРИСТИКА ЗАГРЯЗНЕНИЙ ПРИРОДНЫХ ВОД
116. И. Ф. Ефремов. Периодические коллоидные структуры. JI., «Химия»,
1971, стр. 114.
117. Н. Bernhardt. Yom Wasser 1965, Bd. 32. Weinheim, 1966, S. 193.
118. E. L. Bean, S. J. Campbell, F. R. Anspach. J. Amer. Water Works Assoc.,
56, 214 (1964).
119. W. Stumm, J. J. Morgan. J. Amer. Water Works Assoc., 54, 971 (1962).
120. Сб. «Коагулянты для очистки питьевой воды». Под ред. В. Т. Турчино-
вича. М., Изд-во МКХ РСФСР, 1948, стр. 16.
121. К. М. Махкамова, Г. М. Вирская, К. С. Ахмедов. Сб. «Адсорбционные
свойства природных и синтетических сорбентов». Ташкент, «Фан»,
1969, стр. 134.
122. Н. С. Курбанова, Э. А. Арипов, К. С. Ахмедов. Там же, стр. 123.
123. М. Б. Бакиева, Н. Ю. Насырова, Э. А. Арипов и др. Там же, стр. 97.
124. W. Bodenheimer, L. Heller. Clay Minerals, 7, 167 (1967).
125. V. Hofmann, I. Dammler. Chimia, 23, 476 (1969).
126. H. N. Myrick, D. W. Pyckman. J. Amer. Water Works Assoc., 55, 783
(1963).
127. М. E. Шишниашвили, A. Л. Бацанадзе, JI. H. Одилавадзе. Пятая Все¬
союзная конференция по коллоидной химии (тезисы докладов). М.,
Изд-во АН СССР, 1962, стр. 45.
128. В. А. Клячко. Сб. «Исследования по водоподготовке». М., Стройиздат,
1956, стр. 107.
129. В. П. Руди, Н. К- Альбота. Пятая Всесоюзная конференция по коллоид¬
ной химии (тезисы докладов). М., Изд-во АН СССР, 1962, стр. 279.
130. А. К. Мискарли, В. Я. Землянская. Коллоидн. ж., 28, 696 (1966).
131. G. W. Kalb, R. В. Curry. Clays and Clay Minerals, 17, 47 (1969).
132. G. Bitton. Water Res., 9, 473 (1975).
133. E. L. Bean. Bull. Engng and Archit., N 56, 46 (1966).
134. E. L. Bean, S. J. Campbell, F. R. Anspach. Water Works and Wastes
Engng, 2, N 11, 47 (1965).
135. Т. M. Riddick. J. Amer. Water Works Assoc., 53, 1007 (1961).
136. S. Kawamura, G. P. Hanna. Ind. Water Engng, 4, 21 (1967).
137. H. А. Лукиных. Очистка сточных вод, содержащих синтетические
поверхностно-активные вещества. М., Стройиздат, 1972.
138. С. N. Sawyer. Sewage and Ind. Waters, 30, 757 (1958).
139. H. А. Лукиных, Б. Л. Липман и др. Водоснабжение и канализация,
вып. 3. М., изд. ЦБНТИ МКХ РСФСР, 1964, стр. 97.
140. Н. А. Лукиных. Научные труды АКХ Им. К. Д. Памфилова, вып. 63.
Городская канализация, № 5. М.— JI., ОНТИ, 1970, стр. 3.
141. Water and Waste Treatment, 10, N 4, 194 (1964).
142. С. H. Wayman. Progress Industrial Microbiology, v. 10. Edinburgh—
London, 1971, p. 219.
143. II. Scott. Kolloid. Z. und Z. Polymere, 219, 42 {1967).
144. H. Scott. J. Colloid and Interface Sci., 26, 133 (1968).
145. K. Megaro, T. Kondo. J. Chem. Soc. Japan. Pure Chem. Sect., 76, 642
(1955).
146. Ю. М. Глазман, Г. М. Кабыш. Коллоидн. ж., 31, 27 (1969).
147. D. Jenkins, W. J. Kaufman, P. Me Gauhey et al. Water Res., 7, 265
(1973).
148. S. S. Russel, М. C. Jesse, W. Graham. J. Amer. Water Works Assoc., 48,
55 (1956).
149. E. Bullock, M. J. Newlands. Fisheries Research Board of Canada Halifax,
Aug., 1969, p. 23.
150. Сб. «IX Международный конгресс по водоснабжению». Под ред.
Ф. А. Шевелева. М., Изд-во МЖКХ РСФСР, 1973.
151. А. Шварц, Д. Перри, Д. Берч. Поверхностно-активные вещества и мою¬
щие средства. М., ИЛ, I960,
It. литература
И
152. N. L. Clesceri, F. G. Lee. Internet. J. Air and Water Pollut., 9, 723 (1965).
153. F. Tokiwa, T. Imamura. J. Amer. Oil Chem. Soc., 46, 280 (1969).
154. D. Matulova. Vodni hospod., 14, 377 (1964).
155. Т. E. Maloney. J. Water Pollut. Contr. Fed., 38, 38 (1966).
156. C. N. Sawyer. J. Amer. Water Works Assoc., 57, 1431 (1965).
157. К. E. Quentin, E. Husehenbeth. Vom Wasser 1968, Bd. 35. Weinheim,
1969, S 76.
158. J. Zuezak. Rocz. Panst._ zakl. hig., 22, 113 (1971).
159. H. P. Nicholson, J. R. Thoman. Mosquito News, 24, 169 (1964).
160. Г. А. Цатурова, JI. Г. Милокостова и др. Гигиена и санитария, № 9,
94 (1968).
161. А. П. Пулатов. Сб. «Гидрогеология и инженерная геология аридной
зоны СССР», вып. 9. Ташкент, «Фан», 1969, стр. 45.
162. /. W. Eichelberger, J. J. Lichtenberg. Environ. Sci. and Technol., 5, 541
(1971).
163. К. E. Quentin, G. W. Philipp. Vom Wasser 1969, Bd. 36. Weinheim/
Bergstr, 1970, S. 54.
164. Huang Ju-Chang, Liao Cheng-Sun. J. Sanit. Eng. Div. Proc. ASCE,
N 5, 1057 (1970).
165. Huang Ju-Chang. J. Water Pollut. Contr. Fed., 43, 1739 (1971).
166. D. G. Crosly, R. K. Tucker. Environ. Sci. and Technol., 5, 714 (1971).
167. IO. В. Кузнецов, В. H. Щебетковский, А. Г. Трусов. Основы дезакти¬
вации воды. М., Атомиздат, 1968.
168. А. А. Хоникевич. Очистка радиоактивно-загрязненных вод. М., Атом¬
издат, 1968, стр. 71.
169. L. R. Setter et al. J. Amer. Water Works Assoc., 51, 1377 (1959).
170. H. Schneider, W. Block. Gas- und Wasserfach, 109, 1410 (1968).
171. E. H. Тимофеева-Расовская и др. Труды Института биологии Уральского
филиала АН СССР, т. 22. Свердловск, 1962, .стр. 49.
172. М. Эйзенбад. Радиоактивность внешней среды. М., Атомиздат, 1967.
173. А. В. Каншина. Труды ВНИИ ВОДГЕО, вып. 15. М., 1967, стр. 54.
174. J. Davis. J. Amer. Water Works Assoc., 50, 1505 (1958).
175. R. Foster. Sewage and Ind. Wastes, 31, 1409 (1959).
176. А. С. Смирнов. ЖВХО им. Д. М. ^Менделеева, 5, 651 (1960).
177. И. Е. Старик, Н. И. Алексеенко, II. Г. Роговская. Изв. АН СССР, ОХН,
1956, 755.
178. И. Е. Старик, Н. Г. Роговская. Ж. неорг. химии, 1, 598 (1968).
Глава III
КОАГУЛЯНТЫ И ИХ ГИДРОЛИЗ
III. 1. виды КОАГУЛЯНТОВ
Обработка воды коагулированием производится добавлением
к ней минеральных солей с гидролизующимися катионами, анод¬
ным растворением металлов или простым изменением pH среды,
если в обрабатываемой воде (сточной жидкости) уже содержатся
в достаточном количестве катионы, способные образовывать при
гидролизе малорастворимые соединения.
В практике очистки питьевых и сточных вод в качестве коагу¬
лянтов обычно используют соли алюминия, соли железа или их
смеси в разных пропорциях [1—76]. В редких случаях находят
применение соли магния [77—79], цинка [77, 80, 81] и титана [82,
83].
III. 1.1. Соли алюминия
Виды и состав коагулянтов на основе солей алюминия приведены
в табл. II 1.1. Наибольшее применение получил сернокислый алю¬
миний (сульфат алюминия), который производят обработкой H2S04
сырой или обожженной глины (каолин, бокситы, нефелин и др.)
Таблица II1.1. Алюминийсодержащие коагулянты
Содержание, вес. %
Коагулянт
Формула
A1.0,
нераствори¬
мых примесей
Сульфат алюминия
неочищенный
Al2(S04),.18H20
>9
>
<23
очищенный
Al2(S04)8-18H20
>13,5
<1
Al2(S04V14H20
17-19
—
AI2(S04)8-14H20
28,5
3,1
Алюминат натрия
NaAKte
45-55
6-8
Полихлорид (оксихло-
Al2(OH)nCl(e_n)
40-44
—
рид) алюминия
Квасцы
алюмокалиевые
KA1(S04)*.12H20
10,2-10,7
0,04-0,2
аммиачные
N H4A1(S04)2 • 12H20
11,0-11,2
III.1. ВИДЫ КОАГУЛЯНТОВ
73
с последующей фильтрацией раствора, упаркой и кристаллиза¬
цией. Описаны также способы получения сульфата алюминия
путем кипячения алюминиевой стружки [1], анодного растворе¬
ния алюминиевых листов в H2S04 [2], обработкой отходов металла
кислотами и щелочами [3].
В США в 1930-х годах многие муниципальные управления
считали экономически выгодным приготовление сернокислого
алюминия на местах с использованием в качестве сырья бокситов
{4]. Эта тенденция продолжала сохраняться и позднее [5].
В СССР в 1924—1941 гг. делались попытки использования
в качестве сырья для производства сернокислого алюминия отхо¬
дов промышленности — золы и шлаков. Однако обилие инертных
примесей в полученных таким способом коагулянтах не позволило
рекомендовать их для широкого применения [6, стр. 44].
Исходными продуктами для получения алюмината натрия и
оксихлорида алюминия служат свежеосажденная гидроокись или
окись алюминия. Первый коагулянт получают растворением этих
продуктов в разбавленной щелочи (NaOH), второй — в разбав¬
ленной соляной кислоте. Иногда для получения Al2(0H)nCl6-n
используют хлористый алюминий, являющийся отходом алюми¬
ниевой промышленности [7] или полученный посредством анод¬
ного растворения алюминия в растворе поваренной соли [8].
В Японии предложен способ получения оксихлорида алюминия
непосредственно из глины, кальцинированной при температуре
600° С [9].
При использовании в качестве электролита соляной кислоты
и проведении анодного растворения алюминия до соотношения
А1 : С1 ~ 2 : 1 можно получить оксихлорид алюминия, затрачивая
около 1 кет-час электроэнергии в пересчете на 1 кг готового про¬
дукта [10]. В зависимости от принятого способа и режима произ¬
водства оксихлорида число п в формуле Al2(OH)flCl6-n может ме¬
няться от 1 до 5, а основность, т. е. доля гидроксильных групп от
общего числа анионов, находиться в пределах 60—80%.
Интересный способ производства алюминийсодержащих коа¬
гулянтов в двухкамерном диафрагменном электролизере предложен
в 1968 г. Мотомурой [11]. В обе камеры непрерывно подается ра¬
створ NaCl. Под действием постоянного тока в анодной камере об¬
разуется А1С13, имеющий кислую реакцию, а в катодной камере —
NaAl02, обладающий щелочной реакцией. Перед дозировкой в об¬
рабатываемую воду продукты анодной и катодной камер смеши¬
вают в определенной пропорции, обеспечивающей требуемую ве¬
личину pH. В качестве варианта вместо поваренной соли воз¬
можно использование морской воды. Разработана технология из¬
готовления смешанных хлорид-сульфатных коагулянтов [12, 13].
Помимо коагулянтов, перечисленных в табл. III.1, для обра¬
ботки природных вод и, в особенности, сточных жидкостей пред¬
74
ГЛАВА III. КОАГУЛЯНТЫ И ИХ ГИДРОЛИЗ
ложены различные глины, алюминийсодержащие отходы произ¬
водства, пасты и смеси:
Литература
Шламовые отходы производства A12(S04)3, содержащие крем-
некислоту [14]
Коагулянты, полученные взаимодействием основных хло¬
ридов алюминия со щелочными реагентами [15]
Сернокислый алюминий с добавкой активированного угля
и силикатных материалов [1, 16
(стр. 210), 17]
Смесь сульфата алюминия с алюминатом натрия и.добавкой
гидрофильных веществ типа крахмала [18]
Основной сульфат алюминия, получаемый смешением
A12(S04)3 с основным хлоридом алюминия и последующей
нейтрализацией щелочью [19,20]
Смесь алюмината натрия с тонкодисперсными остатками от
выщелачивания боксита щелочью [21]
Смесь, полученная обработкой пирофиллита сильной кис¬
лотой с последующим добавлением Na2C03, NaA102, а так¬
же глин, сульфатов многовалентных металлов, растительной
вытяжки [22]
Травильные растворы . [23, 24]
Смесь, образующаяся при выщелачивании глины серной
кислотой и нейтрализации избыточной кислотности обож¬
женной глиной [25]
Паста, полученная осаждением солей алюминия из сточных
вод аммиаком [26]
Основные соли алюминия, активированные многовалент¬
ными анионами кислот (H2S04, H3P04, H2Si03) [27]
Продукт обработки глин соляной и азотной кислотами с
последующей термообработкой [28]
Продукт, полученный в результате смешения многооснов¬
ного хлористого алюминия с сульфатами, сульфитами и
фосфатами алюминия, железа, магния, кальция и других
металлов [29]
Алюминийсодержащие коагулянты изготовляют и поставляют
на водоочистные сооружения в виде кусков и плит, порошка, гранул,
а также в виде желе и растворов, содержащих от 7 до 32% А1203 [30
(стр. 68), 31—35]. Применение жидких коагулянтов во многих слу¬
чаях дает неоспоримые экономические преимущества [5, 36, 37].
III. 1.2. Соли железа
Виды и состав коагулянтов на основе солей железа приведены
в табл. III.2. Наиболее распространено хлорное железо, которое
получают обычно обработкой хлором железного лома, иногда непо¬
средственно на водоочистных станциях [38—40]. Используются
также методы анодного растворения железа в растворах поварен¬
ной соли или серной кислоты [2, 41—43].
Ш.1. ВИДЫ КОАГУЛЯНТОВ
75
Таблица III.2. Железосодержащие коагулянты
Содержание, вес. %
Каогулянт
Формула
Fe20;i
нерастворимых
примесей
Хлорное железо
FeCl3-6H20
>95
Железный купорос
FeS04-7H20
>47
<i
Хлорированный железный ку¬
Fe2(S04)3 + FeC'3
—
—
порос
Сернокислая окись железа
Fea(S04)3-2H20
68—76
<40
Железный купорос получают из растворов, образующихся
при травлении металла. Применение аэрации дает возможность
получить коагулирующие растворы с концентрацией FeSG4 по¬
рядка 20% [44]. Предполагается, что под действием кислорода
воздуха формируются соли вида Fe4(OH)10SO4, обладающие силь¬
ным коагулирующим действием [45].
Первые исследования по применению железного купороса про¬
ведены в СССР в 1935 г. на Саратовском водопроводе, а в 1940 г.
этот коагулянт был рекомендован для широкого использования
[46].
Исследования, проведенные в Академии коммунального хо¬
зяйства, показали возможность использования для очистки воды
травильных растворов металлообрабатывающих заводов, содер¬
жащих до 78% FeS04-7H20 [6, стр. 67].
Недостатком железного купороса является необходимость
иметь для перевода двухвалентного железа в трехвалентное вы¬
сокий щелочной резерв или применять предварительное хлори¬
рование его растворов. Самостоятельное применение рекомендо¬
вано [47] лишь при pH воды более 9.
Хлорированный железный купорос получают непосредственно
на водоочистных станциях путем обработки FeS04 хлором
6FeS04 +3 Cl2 2FeCl3 + 2Fe2(S04)3.
Теоретический расход хлора в приведенной реакции составляет
0,128 г на 1 г FeS04-7H20, а практически он достигает 0,16—
0,20 г [16, стр. 220]: часть хлора расходуется, по-видимому, на
окисление органических примесей воды и образование более
сложных соединений с железом (гл. VII). Ряд других способов
получения хлорированного железного купороса разработан в Япо¬
нии [48, 49].
Проверка действия хлорированного купороса, проводившаяся
на водопроводах разных городов СССР в 1939—1940 гг., дала
удовлетворительные результаты [50]. Однако большая потреб¬
76
ГЛАВА III. КОАГУЛЯНТЫ И ИХ ГИДРОЛИЗ
ность в хлоре препятствуют широкому распространению этого
коагулянта. За рубежом положительные результаты при доста¬
точно хороших экономических показателях получены в США,
Англии, Бельгии, Голландии [51—53].
Сернокислое окисное железо производят путем обработки окиси
железа серной кислотой. В качестве сырья часто используют пи-
ритные (колчеданные) огарки — отходы сернокислотного произ¬
водства [54, 55]. Известен способ получения Fe2(S04)3 из отходов
сернокислотного производства двуокиси титана, содержащих
31—35% Fe203. Полученный коагулянт не уступает по техноло¬
гическим свойствам сернокислому железу из пиритных огарков
[56]. При очистке сточных вод железосодержащие отходы произ¬
водства двуокиси титана могут как коагулянт иметь самостоятель¬
ное значение [57, 58]. Описан также способ приготовления
Fe2(S04)3 обработкой раствора FeS04 сернистым газом [59].
В СССР первые опыты по проверке коагулирующего действия
Fe2(S04)3 проведены в 1918 г. В 1941—1944 гг. испытания были
продолжены и дали положительные результаты. Однако отмечено,
что Fe2(S04)3 требует добавления к воде извести и хлора и медлен¬
нее гидролизуется, чем другие железосодержащие коагулянты.
Помимо перечисленных железосодержащих солей, запатентовано
и описано несколько способов получения гидроокисей железа
путем пропуска воды через слой железной стружки [60—64].
Для повышения скорости растворения металла обрабатываемую
воду подкисляют [65, 66] или используют вместо стружки сталь¬
ные опилки [67]. Для интенсификации окисления двухвалентного
железа применяют аэрацию [68].
III. 1.3. Смеси солей алюминия и железа
Смеси солей алюминия и железа приготавливают заранее (в ра¬
створных баках) или в самой обрабатываемой воде — за счет одно¬
временного или последовательного дозирования их растворов.
Существуют способы производства смешанных коагулянтов кис¬
лотной обработкой шлака, золы и глин, содержащих заметные ко¬
личества железа и алюминия. Некоторые коагулирующие смеси
солей алюминия и железа перечислены ниже:
Смесь A12(S04)3, FeS04 и хлора [69]
Коагулянт Ferric-floc, содержащий 14% Fe„03, 4% А1203 и 1,5%
ТЮ2 [70]
Смесь A12(S04)3 и Fe2(S04)3, полученная обработкой серной
кислотой золы каменных углей и содержащая 20,8—29,3%
А1203 и 6,7-13,7% Fe203 [71]
Кислые растворы, образующиеся при обработке минеральными
кислотами монтмориллонитовых глин и содержащие около
16 г/л А13+ и около 6 г/л двух- и трехвалентного железа ... [72]
111.2. ГИДРОЛИЗ КОАГУЛЯНТОВ
77
Смесь отработанной кислоты, гидроокиси алюминия, бокситов
и глины [73]
Смесь солей алюминия и железа с каменноугольной золой . . [74]
Коагулянт, полученный обработкой шлака растворами НС1 и
H2S04, содержащий соли алюминия, железа и активную крем¬
некисл оту [75]
Смесь, полученная в результате обжига некоторых измельчен¬
ных минералов (силиката, феррита, алюмината, алюмосиликата,
алюмоферрита) [76]
III. 2. ГИДРОЛИЗ КОАГУЛЯНТОВ
III. 2.1. Реакции гидролиза
Реакцию гидролиза катионов металла Ме+ представляют обычно
в общем виде:
Ме+ + Н20 ^ МеОН + Н+.
Из реакции видно, что скорость гидролиза Fr пропорциональ¬
на молярным концентрациям катионов металла и воды или (по¬
скольку величину [Н20] в данной реакции можно считать постоян¬
ной) только концентрации [Ме+], т. е. дозе коагулянта:
Vr — Kv [Ме+], (III.1)
где Кг — константа скорости гидролиза.
С повышением температуры воды на каждые 10° С константа
скорости гидролиза, как и других химических реакций, возра¬
стает по закону Вант-Гоффа в 2—4 раза. Скорость гидролиза
солей алюминия и железа возрастает при добавлении к воде гид¬
роокисей этих металлов [84 (стр. 101), 85 (стр. 50)]. Каталитиче¬
ское влияние гидроокисей описывается уравнением
KJK2 = (cjc2)n, (III.2)
в котором и К2 — константы скорости гидролиза при концен¬
трациях катализатора сх и с2, а п — показатель степени, равный
1,3-1,6 [86].
При очистке воды мы имеем дело с относительно малыми кон¬
центрациями катионов металлов, а их гидролиз протекает практи¬
чески мгновенно.
Из уравнения реакции гидролиза в соответствии с законом
действия масс следует
[МеОН][Н+] (Ш 3)
Лг 1Ме+] ’ v '
т. е. чем больше Кг, тем сильнее равновесие смещается в сторону
образования МеОН, Н+ и гидролиз протекает полнее. Если теперь
78
ГЛАВА III. КОАГУЛЯНТЫ И ИХ ГИДРОЛИЗ
числитель и знаменатель умножить на величину [ОН ], то полу¬
чим
Kr = Kw/Kk, (III.4)
где Kw — [Н+ЫОН-]— ионное произведение воды, а Кк =
= [Ме+]- [ОН~]/[МеОН] — константа диссоциации гидроокиси ме¬
талла.
Из выражения (II 1.4) следует важный для водоподготовки вы¬
вод о том, что металлы, образующие гидроокиси с меньшей кон¬
стантой диссоциации, подвержены большему гидролизу.
Степень диссоциации гидроокисей выражают обычно произве¬
дением растворимости Lv —Кк [МеОН]. Значения Lv при темпе¬
ратуре 18° С для А1(ОН)3 равны 1,9• 10-33, для Fe(OH)3 — 3,8-
•10"38, для Fe(OH)2 — 4,8-10"16 [87]. Следовательно, гидролиз ка¬
тионов трехвалентного железа протекает полнее, чем гидролиз
катионов алюминия и двухвалентного железа.
Катионы металлов сильно гидратированы, и мы уже говорили
о том, что их соединения с водой можно рассматривать как ком¬
плексные, содержащие воду в качестве лигандов. Реакции гидро¬
лиза таких аквакомплексов можно представить как ступенчатое
отщепление протонов от молекул воды [88—90]. Например, для
А1(Н20)Г:
А1 (Н20)3+ й [А1 (Н20)6(0Н)Р+ + Н+,
[А1 (Н20)5(0Н)]г+ [А1(Н20)4(0Н)2]+ + Н+,
[А1(Н20)4(0Н)2]+ [А1 (Н20)з(0Н)з1 + Н+,
[А1 (Н20)з(0Н)3] ^ [А1 (НаО)2(ОН)4Г + Н+.
Продукты реакций гидролиза — гидроксокомплексы (окси-
гидраты) 1 и малорастворимые основные соли, о составе и свойст¬
вах которых пойдет речь в разделе III.2.2.
Значения pH, при которых продукты гидролиза выделяются
в осадок, зависят от целого ряда физико-химических условий [91].
Рассмотрим случаи образования гидроокисей и основных солей.
Для гидроокиси трехвалентного катиона металла
Lp = я,ме3Н—Ь За30н-, (III.5)
где а — активность ионов в растворе. Выразив величину а0н- че¬
рез ионное произведение воды и активность ионов водорода,
получим
«он- = К\у/ян+,
Z/p = аме3+ + 3 (/Cw/^H+)3,
1 Употребляемый нами и другими авторами термин «гидроокись металла»
в значении продукта гидролиза солей алюминия и железа — лишь дань
сложившейся традиции.
III.2. ГИДРОЛИЗ КОАГУЛЯНТОВ
79
^WaMe3+ + 3
flH+= 7J, ’
p
pH = — lgaH+= 4-lgbp—lg^w—-|-lgaMe3+ + 3. (III.6)
Из уравнения (III.6) видно, что увеличение концентрации соли
и связанное с ним повышение активности катиона металла сдви¬
гают pH гидратообразования в сторону меньших значений. При¬
сутствие в растворе посторонних веществ, приводящее к изме¬
нению его ионной силы, также должно влиять на pH гидрато¬
образования, так как при этом будет меняться средний коэффици¬
ент активности. С помощью выражения (III.6) можно определить
зависимость концентрации негидролизовавшихся катионов от
величины pH среды:
Ln. Me (ОН)3
[Ме3+] = • 10зрН . (III.7)
w
Если теперь представить основную соль, образующуюся при
гидролизе металла, общей суммой х молекул нормальной соли и у
молекул гидроокиси
х (Mev+ Av_) у [Ме(ОН)3],
то рассуждения, аналогичные приведенным выше, позволят вы¬
вести зависимость
РН = lgLp - *v ++ у- lg aMe + 3 - Ig aA - zA - Ig Kw,
(III. 8)
в которой z\ — валентность анионов.
В соответствии с формулой (III.8) pH образования основных
солей зависит не только от активности катиона металла, но и от
активности присутствующих анионов а\. Пользуясь выражениями
(III.6) и (III.8), можем определить, какой из двух продуктов —
основная соль или гидроокись — будет выпадать в осадок при
заданном значении pH.
Благодаря наличию буферной емкости, определяемой углекис¬
лотным равновесием, величина pH воды не претерпевает заметных
изменений до тех пор, пока эта емкость не будет исчерпана. Пол¬
ный гидролиз катионов алюминия и железа происходит только
при достаточной буферной емкости воды. По наблюдениям Хей-
манна [85, стр. 52], гидролитическое равновесие зависит от сте¬
пени дисперсности продуктов гидролиза железа, причем грубо'
дисперсная фаза находится в равновесии с меньшими концентра¬
циями FeCl3, чем тонкодисперсная.
80
ГЛАВА III. КОАГУЛЯНТЫ И ИХ ГИДРОЛИЗ
В табл. II 1.3 приведены расчетные и экспериментально найден¬
ные значения pH осаждения из растворов малорастворимых про¬
дуктов гидролиза алюминия и железа.
Т аб л и ц а III.3. Значения pH осаждения и растворения продуктов
гидролиза алюминия и железа
Катион
Начало осажде¬
ния
Полное
осаждение
Начало растворения
Литература
А13+
3,3-4,0
5,2
7,8
[92]
3,0
7,0
9,0
[93]
3,9
4,8
—
[94]
4,4
—
7,8
[95]
2,5-3,0
6,0
~10,0
[96]
4,5
5,5—7,0
8,0
[30, стр. 142]
Fe3+
1,5-2,3
4,1
14,0
[92]
2,2
3,2
. —
[94]
—
3,5
—
[30, стр. 142]
Fe2+
6,5-7,5
9,7
13,5
[92]
9,5
—
[30, стр. 142]
Величина pH, отвечающая минимальной растворимости про¬
дуктов гидролиза коагулянтов, зависит, как это ясно из всего пре¬
дыдущего материала, от аниона исходной соли. Так, для FeCl3
она равна 7,0, для Fe2(S04)3 — 6,2, для FeS04 — 9,5, а для смеси
Fe2(S04)3 и FeCI3 — 6,5 [97]. По данным некоторых авторов [98
(стр. 16), 99], минимальная растворимость продуктов гидролиза
соответствует их изоэлектрическому состоянию.
III. 2.2. Состав продуктов гидролиза
Когда мы будем рассматривать закономерности коагуляции золей
электролитами (гл. IV) и в качестве примера—коагуляцию гидро¬
золей алюминия и железа, мы увидим, что многие наблюдаемые
явления объясняются специфическими химическими взаимодей¬
ствиями.
До 30-х годов большинство исследователей придерживалось
мнения, что процесс коагуляции А1(ОН)3 и Fe(OH)3 целиком опре¬
деляется обменной адсорбцией ионов-стабилизаторов на ионы до¬
бавленного электролита. Причем подразумевалось, что ионный
обмен происходит в эквивалентных количествах. Дальнейшие
работы выявили, однако, частые нарушения эквивалентности
обмена, это дало основание предположить важную роль химиче¬
ских взаимодействий между молекулами и ионами твердой и жид-
III.2» ГИДРОЛИЗ КОАГУЛЯНТОВ
81
кой фаз. Для подтверждения этой роли необходимо было выра¬
ботать более четкое представление о строении золей гидроокисей
металлов.
Схемы строения мицелл А1(ОН)3 и Fe(OH)3, приведенные в
гл. I, отражают действительный состав двойного слоя лишь в са¬
мом общем виде и призваны иллюстрировать влияние сжатия слоя
на устойчивость частиц. Уже в 1920-х годах Бриттон [100],Матт-
сон [101], а также Рабинович и др. [102—105], Песков [106]
и другие указывали, что на поверхности частиц гидроокисей про¬
исходит образование малорастворимых химических соединений
с иными, отличными от гидроокисей свойствами.
В 1937 г. Каргин и др. [107,108] пришли к выводу, что адсорб¬
ционные процессы при коагуляции гидрозолей определяются пре¬
имущественно не обменом ионов, а образованием малорастворимых
основных солей. По их данным, стабилизирующим электролитом
золя А1(ОН)3 является соединение А1203-НС1, которое, взаимо¬
действуя с коагулирующим электролитом (например, Na2S04),
образует хорошо адсорбирующуюся соль — в данном случае ок-
сисульфат A1203-H2S04. Степень адсорбции определяется кон¬
центрацией и растворимостью соли и меняется с изменением pH.
Уменьшение содержания S04~-hohob в осадке при понижении pH
объясняется увеличением растворимости оксисульфата, а при
повышении pH — малым содержанием в интермицеллярной жид¬
кости А1203-НС1. Область существования малорастворимых со¬
лей соответствует значениям pH среды 3—6,5.
Позднее было установлено [109], что адсорбционные и электро¬
химические свойства гидроокиси алюминия могут практически
целиком определяться сульфатными, фосфатными и силикатными
основными солями, растворимость которых в воде составляет
1-10'4 — 1 • 10“6 г-экв/л.
При добавлении к воде сернокислого алюминия наиболее ве¬
роятно образование оксисульфатов. Это подтверждается резуль¬
татами экспериментов. В частности, в процессе титрования ра¬
створа A12(S04)3 щелочью раствор становится нейтральным при
меньшем расходе щелочи, чем нужно для образования А1(ОН)3,
а полученный осадок содержит БО^-ионы [110].
Образование оксисульфатов рассматривается также как обмен
на ионы SO|~ гидроксильных ионов диффузного слоя А1(ОН)3
[111]. Эта точка зрения подтверждается повышением pH среды
в процессе образования A1203-H2S04 [111, 112].
На рис. III.1 приведена диаграмма примерного распределения
разных продуктов гидролиза сернокислого алюминия в широком
диапазоне pH обрабатываемой воды [113].
Большая часть приведенных для А1(ОН)3 сведений относится
й к золю гидроокиси железа. По одним данным [114, 115], при до-
82
ГЛАВА III. КОАГУЛЯНТЫ И ИХ ГИДРОЛИЗ
Рис. III.1. Процентное распре¬
деление (р) продуктов гидролиза
A12(S04)3 в зависимости от pH
воды
Рис. III.2. Схема реакций гид¬
ролиза алюминия
бавлении электролитов в этих зо¬
лях наряду с ионообменными про¬
цессами происходит дополнитель¬
ная адсорбция соляной кислоты,
присутствующей в интермицелляр-
ной жидкости. В результате ад¬
сорбции эквивалентность обмена
нарушается. По другим данным
[116], сорбция на Fe(OH)3 одно-и
двухвалентных анионов (Cl“, S04~,
СгОГ) подчиняется закономерно¬
сти, предсказываемой теорией
ДЛФО (см. гл. IV), носит ионооб¬
менный характер и уменьшается
с ростом pH. Нарушение эквива¬
лентности обмена и хемосорбция
обнаружены лишь в присутствии
анионов Н2РС>4.
Среди работ, посвященных гид-
ратообразованию, особое место за¬
нимает обзорная работа Стамма и
Моргана [99], которая заставила
обратить внимание на большое зна¬
чение химических взаимодействий
в процессе коагуляции. Показано,
что в результате конденсации про¬
стых продуктов гидролиза в усло¬
виях пересыщения раствора воз¬
никают полиядерные гидроксид-
ные соединения, которые обладают
гораздо более сильной коагули¬
рующей способностью, чем катио¬
ны АР+ и Fe3+ [99, 117, 118].
В табл. III.4 и на схеме рис. III.2,
по данным ряда авторов [99, 119,
120], показаны некоторые реакции
гидролиза и соответствующие им
константы равновесия Кг при 25°С.
Предполагается [121], что воз¬
растание величины pH среды (от
4 до 7) способствует увеличению
степени полимеризации гидроксо-
комплексов алюминия по схеме
А1 (ОН)+ Ale (ОН)°+ - Ali„ (ОН)|+
-А121 (011)12+_Л154(ОН){484+.
HI.2. ГИДРОЛИЗ КОАГУЛЯНТОВ
83
Таблица Ш.4. Реакции гидролиза катионов алюминия и железа
и константы их равновесия
Реакция
ig Kr
А13+ + Н20 ^ [А10Н]2+ + Н+
-5,03
2А13+ + 2НгО ^ [А12(ОН)2]4+ + 2Н+
-6,27
А13+ + ЗШО ^ А1(ОН)3 + ЗН+
—9,1
А1(ОН)з + НаО ^ [А1(ОН)4]-+Н+
—12,74
8А13+ + 20Н20 ^ [А18(ОН)2о14+ + 20Н+
6А13+ + 15Н20 ^ [А16(ОН)16]3+ + 15Н+
[А1(0Н)]2+ + Н20 “2 [А1(ОН)2]+ + Н+
[Fe(H20)3l3+ + Н20 ^ [Fe(H20)60H]2+ + Н30+
-2,17
[FeOH]2+ + HaO ^ [Fe(OH)2]+ + H+
-4,7
Fe3+ + ЗН2О ^ Fe(OH)3 -f ЗН+
—6,0
Fe(OH'3 + HaO ^ [Fe(OH)4]- + H+
-18,5
2Fe3+ + ЗН2О ^ [Fe2(OH)3]3+ + 3H+
—
2Fe3+ + 5H20 ^ [Fe2(OH)5l+ + 5H4-
—
3Fe3+ + 2H20 ^ [Fe3(OH)2]7+ + 2H+
—
3Fe3+ -j- 4H20 ^ [Fe3(OH)4p++4H+
-5,77
При этом заряд полимеризованного иона постепенно повышается
(от _|_i до -(-18), а отношение величины заряда к числу атомов алю¬
миния уменьшается от 1 до 73. К наиболее вероятным формам сое¬
динений алюминия относят А12(ОН)5 [122], Al4(OH)g+ [123, 124],
А17(ОН)£ и А113(ОН)м [117, 125], А118(ОН)49 [126]; а к формам сое¬
динений железа — Fe3(OH)4+ [117]. Эти формы можно рассматри¬
вать как промежуточное звено между простыми ионами и поли¬
электролитами .
Как отмечалось, о степени гидролиза металла и преимущест¬
венном составе продуктов гидролиза при данном значении pH
можно судить по результатам титрования растворов солей ще¬
лочью. Методика такого титрования и расчета количества (Шп¬
ионов, связанных с атомом алюминия, описана.в работах [99, 120,
127]. Авторы отмечают, что путем титрования можно получить
более достоверные сведения, чем исходя из трудноопределяемых
констант равновесия в реакциях гидролиза.
На рис. III.3 показаны результаты определения соотношения
между отдельными формами продуктов гидролиза алюминия
и железа при их исходных концентрациях, близких к условиям
очистки воды. Из диаграмм видно, что в зоне pH 5—7,5 системы
пересыщены в отношении нерастворимых продуктов, в то время
как растворимые примеси содержатся в ничтожных количествах.
84
ГЛАВА III. КОАГУЛЯНТЫ И ЙХ ГИДРОЛИЗ
Рис. III.3. Расчетные концентрации продуктов гидролиза алюминия (а)
и железа (б) в водных растворах в зависимости от pH воды [117]
Сходные результаты приведены другими исследователями [128—
131]. Несколько позднее построены диаграммы процентного со¬
держания разных форм трехвалентного железа, показанные на
рис. III.4 и III.5. В 1971 г. Бабушкин и др. [134] составили диа¬
грамму полей устойчивости Fe3+ при разных значениях pH и раз¬
ных концентрациях катионов железа.
Таблица III.5. Реакции комплексообразования катионов алюминия
и железа и константы их равповесия [99]
Реакция
lgKr
А1з+ + ро43- ^ А1Р04
22,0
А13+ + Р20,“- ^ [А1Р20,Г
—
А13+ + F- ^ [A1F]2+
6,13
А13+ + 3(ацетат)~ [А1(ацетат)3]
—
А13+ + 3(оксалат)2~ ^ [А1(оксалат)3]3_
16,3
А13++салицилат2- ^ [А1(салицилат)]+
14,0
Fe3+ + Р043- ^ FeP04
21,9
Fe3++ НР042- ^ [FeHP04]+
8,4 •
Fe3++ Р20,4- ^ [КеР20,]-
Fe3+ + F" ^ [FeF]2+
5,17
Fe3++ ацетат" ^ [Ре(ацетат)]2+
—
Fe3+-)- З(оксалат)2- [Ге(оксалат)3]3~
23,9
Fe3+ фенолат- [Ге(фенолат)]2+
8,2
Fe3++ цитрат4- ^ [Ее(цитрат)]-
25,0
Fe3++ салицилат2- ^ [Ре(салицилат)]+
15,82
Ш.2. ГИДРОЛИЗ КОАГУЛЯНТОВ
85
S
Л
1т$
Fe3*J
ион);
Ft(0H)j
Fe ОН2*
FelDH)3
щ
Р,Л
Рис. III.4. Процентное распределение (р)
продуктов гидролиза трехвалентного железа
в водных растворах в зависимости от pH „п
воды [132]
Молярная концентрация: а — 1 -10—3; б — ЫО-4;
в — 1-10-»
Рис. III.5. Процентное распределение (р)
продуктов гидролиза трехвалентного железа
в зависимости от pH воды [133]
Для катионов А13+ и Fe3+ характерна сильная тенденция к об¬
разованию соединений не только с ионами гидроксила, но и с дру¬
гими анионами и ионизированными группами гидрофильных ор¬
ганических веществ: фосфатными, сульфатными, карбоксильными
и другими. Помимо малорастворимых продуктов, показанных на
рис. III.1, предполагается формирование [A1(H20)60H]S04
(с произведением растворимости 1-10"28), [А1 (H20)4(0H)2]2S04
(с произведением растворимости 1-10~23), AIe(0H)12(S04)3 [135,
136]. Константы равновесия некоторых реакций комплексообра-
зования для температуры воды 25° С приведены в табл. III.5.
Описано образование катионами железа и алюминия ком¬
плексов с анионами органических кислот [137—139]. В общем
случае любые комплексообразующие ионы, присутствующие
в воде, можно рассматривать как конкурирующие с ОН~-груп-
86
ГЛАВА III. КОАГУЛЯНТЫ И ИХ ГИДРОЛИЗ
пами за координационные участки оксигидратов. При pH 7
находящиеся в достаточно высоких концентрациях SO4-- и
С1_-ионы могут вытеснить гидроксильные ионы и занять их места
[99]. Вероятность «координационного внедрения» анионов опре¬
деляется их концентрацией и валентностью, а также значением
pH окружающей среды. Как будет показано в гл. VI, в присут¬
ствии конкурирующих анионов удовлетворительная коагуляция
продуктов гидролиза А13+ и Fe3+ протекает при более низких
величинах pH, чем в их отсутствии, поскольку для удовлетворения
координационных требований катионов металлов и обра¬
зования малорастворимых соединений требуется меньшее коли¬
чество ионов гидроксила. Исключение составляют лишь те анио¬
ны (например, оксалаты и салицилаты), которые с катионами ме¬
талла образуют растворимые комплексы. В этом случае pH опти¬
мальной коагуляции смещается в сторону более высоких значе¬
ний.
Значения pH изоэлектрической точки гидроокиси алюминия
в воде, не содержащей посторонних ионов, составляют, по данным
разных авторов, 7,6—8,2, а в присутствии анионов уменьшаются
на 1-1,5 [6 (стр. 12), 97, 140-143].
III. 3. КОАГУЛЯЦИОННЫЕ СТРУКТУРЫ II ИХ СВОЙСТВА
В коллоидной химии различают два типа структур дисперсного
материала: первичные и вторичные. Первичная структура — это
структура отдельных частиц золей и суспензий. С ней связаны
электрические свойства частиц, их сольватация, молекулярно¬
кинетические свойства дисперсий. Вторичные структуры возни¬
кают вследствие взаимодействия отдельных частиц. От них зави¬
сят объем и механические свойства осадков. Вторичные струк¬
туры часто называют сверхмицеллярными [144, 145].
У гидрофобных коллоидов можно довольно отчетливо диффе¬
ренцировать свойства, связанные с типом структуры, у гидро¬
фильных такая дифференциация затруднена.
Образующиеся в пересыщенной системе зародыши малораство¬
римых веществ могут укрупняться за счет роста индивидуальных
частиц твердой фазы (кристаллизация) или путем коагуляцион¬
ных взаимодействий с формированием пространственных агре¬
гатов. Поэтому сверхмицеллярные структуры в свою очередь
делят на конденсационно-кристаллизационные и коагуляционные
[146, 147]. Схема построения последних показана на рис. III.6.
Для образования коагуляционной структуры, помимо астаби-
лизации первичных частиц, необходимо превысить некоторую кри¬
тическую концентрацию твердой фазы в растворе. В присутствии
посторонней поверхности раздела зародышеобразование и струк-
турообразование протекают интенсивнее, с меньшими энергоза-
III.3. КОАГУЛЯЦИОННЫЕ СТРУКТУРЫ И ИХ СВОЙСТВА
87
Рис. II 1.6. Схема коагуля¬
ционной структуры
1 — частицы дисперсной фазы;
2 — астабилизированные участки
поверхности;
3 — участки поверхности, сохра¬
нившие устойчивость;
4 — полости, заполненные водой
тратами и при более низких пересыщениях (см. также гл. V).
Положительную роль играют флуктуации содержания твердой
фазы.
В этой главе мы рассмотрим структурообразование и физиче¬
ские свойства «чистых» коагулятов 2 — гидроокисей и основных
солей алюминия и железа,— не касаясь пока свойств коагулиро¬
ванной взвеси, которая включает в себя дополнительно механиче¬
ские примеси и другие загрязнения воды. На них мы остано¬
вимся в гл. VI.
Имеются сведения [84 (стр. 139), 148] (подтвержденные виско-
зиметрическими исследованиями) о том, что структурообразова¬
ние в гидрозолях алюминия и железа может рассматриваться как
образование на первом этапе сплошной пространственной сетки
и ее разрыв под влиянием гидродинамических воздействий и про¬
цессов старения на втором этапе. В результате разрыва обра¬
зуются микрохлопья, укрупняющиеся в ходе дальнейшего объ¬
единения.
На скорость структурообразования большое влияние оказы¬
вают размер и форма исходных частиц. Плавное перемешивание
и повышение температуры способствуют ускорению образования
структур, так как увеличивают вероятность преодоления оста¬
точного энергетического барьера между частицами.
Частицы взаимодействуют участками, на которых произошло
наиболее полное устранение причин их агрегативной устойчиво¬
сти [147, 149 (стр. 316)]. Поэтому свойства структур в первую
очередь определяются степенью астабилизации частиц. При не¬
полной астабилизации структуры образуются рыхлые, а их внут¬
ренние полости заполнены дисперсионной средой [150, стр. 226].
Образованию рыхлой структуры способствуют высокая дисперс¬
ность и анизодиаметрия частиц [145, 147, 151]. Гели А1(ОН)3,
Fe(OH)3, суспензии глин с удлиненными частицами-чешуйками
образуют рыхлые коагуляционные структуры.
2 Коагулятами в коллоидной химии называют продукты коагуляции, агре¬
гаты скоагулированных частиц.
88
ГЛАВА III. КОАГУЛЯНТЫ И ИХ ГИДРОЛИЗ
Рис. III.7. Зависимость максимального размера (d) агрегатов в коллоидном
растворе А1(ОН)3 от концентрации NaCl [145]
Рис. III.8. Зависимость седиментационного объема суспензий каолина от
концентрации КС1 [145]
Размер частиц (мкм): 1 — 1,5—4,0; 2 — 2,5—2,6 (п — отношение объема осадка к объему
исходной суспензии)
При понижении устойчивости частиц, когда лиофобные участки
занимают существенную часть их поверхности, формируются от¬
носительно компактные структуры. Поэтому в экспериментах
(рис. III.7) можно наблюдать, что с увеличением концентрации
электролита размер хлопьев коагулята сначала, как правило,
возрастает, а затем уменьшается. Нужно, однако, иметь в виду,
что во многих исследованных случаях увеличение плотности коа¬
гуляционных структур может быть удовлетворительно объяс¬
нено с точки зрения образования новых соединений со своими ин¬
дивидуальными структурными свойствами.
Среди других факторов, влияющих на величину агрегатов ча¬
стиц, важное значение имеет степень пересыщения раствора по
отношению к осаждаемому веществу — чрезмерно большие дозы
коагулянта вызывают уменьшение размеров хлопьев.
Ориентация и плотность упаковки частиц в коагуляционных
структурах могут быть неодинаковыми в разных участках. Уста¬
новлено, в частности, что в золях Fe(OH)3 имеется по крайней мере
две группы частиц, отличающихся по механическим свойствам
[152]. Это явление связано, вероятно, со способностью анизодиа-
метрических частиц Fe(OH)3 к построению тактоидов — агрегатов
с ясно выраженной периодичностью расположения первичных ча¬
стиц [153].
В зависимости от плотности упаковки частиц меняется седи-
ментационный объем скоагулированной твердой фазы. Частицы
устойчивые свободно скользят в осадке одна по поверхности
другой и потому занимают в конце концов такое взаимное поло¬
жение, которое соответствует минимуму потенциальной энергии.
Ш.З. КОАГУЛЯЦИОННЫЕ СТРУКТУРЫ и ИХ СВОЙСТВА
89
Осадки образуются плотные. Неполностью астабилизированные
взвеси сохраняют в коагуляционной структуре то случайное вза¬
имное положение, в котором они соприкоснулись, и их седимента-
ционный объем велик. Например, объемная концентрация твердой
фазы в осадке агрегативно устойчивой суспензии кварца достигает
54%, а в осадке неустойчивой суспензии — лишь 7% [154,
стр. 495].
Как видно из графика на рис. III.8, на примере каолинитовой
суспензии кривые зависимости объема коагуляционных структур
от концентрации коагулирующего электролита обнаруживают
максимум. Повышение валентности и степени гидратации коагу¬
лирующих ионов приводит к увеличению объема осадка [154
(стр. 496), 155].
Гели менее чувствительны к свойствам электролитов, чем золи
и суспензии, поскольку, помимо сил молекулярного притяжения
и остаточных сил электростатического отталкивания, на струк¬
турные свойства гелей влияют другие факторы. В условиях не¬
больших концентраций катионов металла, имеющих место при
добавлении коагулянтов к обрабатываемой воде, на структуру
продуктов гидролиза сильное влияние оказывает ориентация ча¬
стиц в агрегатах. Причем, так как с увеличением числа гидро¬
ксильных групп, связанных с катионом металла, скорость ориента¬
ции понижается, гидроокиси двухвалентных металлов имеют
обычно кристаллическую структуру, а гидроокиси трехвалент¬
ных — аморфную [84 (стр. 105), 119 (стр. 10)]. На аморфный Ха¬
рактер свежеосажденных гидроокисей алюминия и железа указы¬
вают результаты многих исследований [156—164]. Со временем,
хотя гидратные оболочки и препятствуют упорядочению струк¬
туры, происходит постепенная кристаллизация гидроокисей [145,
163, 164].
Исследуя механизм формирования гидроокиси алюминия, Бе-
рестнева и др. [158] показали, что первоначально образуются
очень мелкие аморфные шарики, размером около 0,2 мкм, но скоро
происходит их перегруппировка, слипание и возникают ячеистые
структуры. В узлах ячеек начинается кристаллизация. Скорость
кристаллизации зависит от способа получения и диализации гид¬
роокисей. По-видимому, процесс кристаллизации начинается
сразу же после образования А1(ОН)3, но может быть обнаружен
аналитическими методами лишь через существенный промежуток
времени, измеряемый сутками и неделями. Так, Истомина и др.
[165] показали, что кристаллизация А1(ОН)3 в растворе A12(S04)3
обнаруживается только через 4—5 недель после образования.
Рентгенографическим, электронно-микроскопическим и элек¬
тронно-графическим методами анализа установлено, что гели
А1(ОН)3 вначале имеют структуру бемита, которая переходит
постепенно в структуру гидраргиллита [166, стр. 15]. Этот порядок
00
ГЛАВА III. КОАГУЛЯНТЫ И ИХ ГИДРОЛИЗ
перехода одной формы гидроокиси в другую подтвержден ИК-
спектроскопическими исследованиями [164]. Однако медленно
происходящие структурные перестройки влияют лишь на свой¬
ства осадков гидроокисей, хранящихся длительное время, и сле¬
дует согласиться с Халлом [121] в том, что единственной формой
гидроокиси алюминия, вызывающей флокуляцию частиц загряз¬
нений, является аморфная форма.
По данным Смита и др. [167], существуют три формы мало¬
растворимых продуктов гидролиза алюминия: 1) мономерные
соединения А13+ с ОН_-группами; 2) полиядерные образования,
включающие от 20 до 400 атомов алюминия; и 3) микрокристалли¬
ческие частицы. Все три формы одновременно присутствуют в геле,
причем со временем вторая форма переходит в третью. Спустя не¬
сколько недель третья форма приобретает структуру гиббсита.
При наличии затравки в виде ранее сформированной гидро¬
окиси А1(ОН)3 кристаллизация в осадках развивается быстрее,
о чем свидетельствует, в частности, улучшение их фильтрацион¬
ных свойств.
Сведения, приводимые разными авторами о структуре гидро¬
окиси железа, противоречивы. По одним данным [119, стр. 51],
кристаллизация Fe(OH)3 начинается только через несколько не¬
дель, по другим — сразу же [168] или через 30 мин с момента
образования [161]. Анализы обнаруживают структуру Fe(OH)3,
сходную с гетитом [84, стр. 105] или гематитом [169]. Недавние
исследования Репиной и др. [170] показали, что ядра мицелл
свежеосаждепной гидроокиси железа имеют неустойчивую поли¬
мерную структуру, которая в результате дегидратации и оксо-
ляции постепенно распадается на отдельные молекулы Fea03-
•7гН20. Распад приводит к сильному пересыщению объема первич¬
ных частиц по концентрации таких молекул и благоприятствует
кристаллизации.
На скорость кристаллизации А](ОН)3 и Fe(OH)3 сильно вли¬
яет значение pH среды. По Кандыкину [162], переход аморфной
гидроокиси алюминия в кристаллическую начинается при pH
^>7,09, причем кристаллическая форма бемита обнаруживается при
pH 8,5 через 2—3 суток, а при pH 9,3 — уже через 3—4 часа после
образования А1(ОН)3.
С точки зрения влияния структурных особенностей А1(ОН)3
и Fe(OH)3 на степень очистки воды коагулянтами нас будут в пер¬
вую очередь интересовать такие свойства коагуляционных струк¬
тур, как площадь их поверхности, гидратация, степень подвер¬
женности старению и пептизации, тиксотропная обратимость,
прочность, адсорбционная способность. Обсудим эти свойства
в перечисленном порядке.
Площадь поверхности. Ермоленко и Левина [159—161] уста¬
новили, что при pH 4,2—8,0 частицы гидроокиси алюминия имеют
Ш.З. КОАГУЛЯЦИОННЫЕ СТРУКТУРЫ И ИХ СВОЙСТВА
91
в основном размер 2—2,5 мкм, но наряду с ними содержатся части¬
цы размером 4—6 и 10—30 мкм. С понижением температуры от 20
до 5° С количество мелких частиц уменьшается, а крупных воз¬
растает. При pH около 8 доля мелких частиц больше, чем при
pH 4,2—6,2. По Кандыкину [162],при pH 8,5—9,3 в гидроокиси алю¬
миния преобладают частицы величиной 0,01—0,05 мкм. В гелях
Fe(OH)3 первичные частицы имеют размер 10—30 мкм [160, 161].
По мере кристаллизации размер частиц гидроокисей умень¬
шается. Уменьшается и количество воды в ячейках. Изменение
удельной поверхности А1(ОН)3 и Fe(OH)3 (s0, м2/г) можно просле¬
дить по данным результатов седиментационного анализа [161]:
В момент фор¬
1 После
Через
Через
мирования
диализа
5 час.
24 часа
85,2
25,6
113,4
116,9
22,1
20,5
27,7
37,7
Использование для определения удельной поверхности гелей
других методов привело к иным результатам. Так, расчеты, вы¬
полненные на основе данных по адсорбционной способности, дают
значения s0 от 200 до 1000 м2/г [161, 171]. Согласно измерениям
Буайе и др. [172], удельная поверхность свежеприготовленного
геля А1(ОН)3 составляет 450—550 м2/г и понижается со временем
до нескольких квадратных метров на грамм. По Пионтковской
и др. [173], величина s„ равна 46 м2/г и не изменяется со временем.
На величину s0 гидроокисей сильное влияние оказывают вид
и концентрация анионов, способных к «координационному вне¬
дрению), в особенности концентрация ОН"-ионов. В частности,
увеличение pH среды от 8,5 до 9,3 приводит к росту s0 А1(ОН)3
с 228 до 264 м2/г [162]. Для гелей Fe(OH)3, полученных осаждением
из раствора нитрата железа, в щелочной среде s0 = 320 м2/г,
а в кислой — лишь около 100 м2/г [174].
Несмотря на расхождение в количественной оценке удельной
поверхности, можно сделать вывод о высокой пористости свеже-
сформированных аморфных гелей и сохранении ими развитой внут¬
ренней поверхности по крайней мере в течение первых часов. Это
позволяет предположить, что фиксация первичных частиц в гелях
происходит на сравнительно больших расстояниях за счет дально-
действующих сил притяжения.
Гидратация. Согласно формулировке, предложенной Кресто¬
вым [1751, под гидратацией ионов и молекул в общем случае по¬
нимается совокупность изменений, связанных с образованием ион¬
ного (молекулярного) раствора в воде. При гидратации происхо¬
дит нейтрализация и перераспределение заряда ионов по окружа¬
ющим их молекулам воды.
Различают «ближнюю» и «дальнюю» гидратацию. Первая харак¬
теризует взаимодействие ионов с ближайшими молекулами воды.
92
ГЛАВА III. КОАГУЛЯНТЫ И ИХ ГИДРОЛИЗ
Водные молекулы прочно связаны с ионами растворенного веще¬
ства и участвуют с ними как одно целое в броуновском движении.
Второй вид гидратации объясняется взаимодействием ионов с мо¬
лекулами воды, не входящими в ближайшее окружение. Молеку¬
лы воды испытывают лишь незначительное влияние ионных
полей без потери поступательных степеней свободы. Толщина
«ближнего» гидратного слоя имеет молекулярные размеры,
а число молекул воды, удерживаемых ионами в этом слое, назы¬
вается координационным числом (или лигандностъю) и является
важной количественной характеристикой гидратации. Лиганд-
ность ионов зависит от их природы, концентрации электролита,
температуры и некоторых других условий.
По результатам геометрических расчетов в зависимости от
того, расположены ли ионы в узлах или пустотах ячеистой струк¬
туры воды, лигандность ионов в «ближней» гидратации равна 4
или 6. С уменьшением концентрации электролита вероятность
размещения ионов в пустотах каркаса воды уменьшается. Поэтому
в разбавленных растворах лигандность ионов минимальна.
Водонасыщенность продуктов гидролиза коагулянтов зависит
от их состава и структуры. Благодаря большому заряду и относи¬
тельно малому радиусу, катионы А13+ и Fe3+ сильно гидратиро¬
ваны. Причем по экспериментальным данным степень гидратации
Fes+ заметно ниже, чем А13+. Это объясняют большим радиусом
катиона железа и высокой скоростью полимеризации Fe(OH)3.
Присоединенные к катионам гидроксильные группы также связы¬
вают большие количества воды.
Особенно большие количества воды гидроокиси алюминия и
железа включают в начальный момент структурообразования,
когда в дополнение к химической гидратации происходит механи¬
ческий захват воды [150 (стр. 226), 176 (стр. 59)]. В этот момент
количество воды в ячейках в сотни раз превышает содержание твер¬
дой фазы. По данным Запрометова и Вирской [155], степень струк¬
турно-механической гидратации Fe(OH)3, подсчитанная как от¬
ношение объема включенной воды FB к объему твердого вещества
FT, достигает 600—900. Согласно экспериментам Кургаева [176,
стр. 60], отношение VJVT в неуплотненных осадках составляет
для А1(ОН)3 — 1210, Fe(OH)3 — 790, Fe(OH)2 — 115 и для смеси
А1(ОН)3 и Fe(OH)3 — 675. Захваченная вода прочно удерживается
в капиллярах гелей и может быть удалена лишь при нагревании
до 140-170° С [177, 178].
г Степень гидратации гелей зависит от скорости их формирования.
По данным Рюммеля [1791, чем ниже скорость формирования, тем
меньше воды захватывает гель. Если рассматривать процесс струк¬
турообразования как ориентированную агрегацию частиц, уве¬
личение водонасыщенности при больших скоростях образования
твердой фазы можно объяснить запаздыванием ориентации.
Ш.З. КОАГУЛЯЦИОННЫЕ СТРУКТУРЫ И ИХ СВОЙСТВА
93
Компактность, плотность хлопьев коагулята выражают вели¬
чиной их объемного веса, которая по данным разных авторов на¬
ходится в пределах 1,0012—1,028 г/см3 [176 (стр. 60), 180, 181].
Гидратация и структурная плотность гелей зависят, как и удель¬
ная поверхность, от анионного состава воды. Продукты гидролиза
солей алюминия в кислой среде образуют крупные, но более рых¬
лые хлопья, чем в щелочной, где структурные свойства опреде¬
ляются присутствующими в больших количествах ОН-- и НС03-
ионами [119 (стр. 36), 182]. Изменение структуры хлопьев влечет
за собой изменение скорости их осаждения.
Значения pH, при которых формируются наиболее плотные,
быстро осаждающиеся хлопья коагулята, являются важнейшей
технологической характеристикой используемых коагулянтов.
Для продуктов гидролиза сернокислого алюминия они находятся
в пределах 5,5—7,5 [176 (стр. 10), 181, 183, 184], для продуктов
гидролиза солей железа — в пределах 6—7 [6 (стр. 14), 84 (стр.
104), 119 (стр. 52), 185] и 8—9,5 [6 (стр. 14), 119 (стр. 51), 176
(стр. 10), 185]. По Чалому [166, стр. 23], при осаждении продуктов
гидролиза солей алюминия и железа из сернокислых солей рас¬
твором NaOH наименьшему объему осадка соответствует соотно¬
шение [ОН-]: [Ме3+] от 2,5 до 3; при этом образуются соли
Al2(SO4)3-10Al(OH)3 или Fe(0H)S04-4Fe(0H)3.
Регулируя pH и солевой состав воды, можно в известной сте¬
пени изменять плотность, оптимизировать условия образования
и осаждения хлопьев. Исследования, проведенные в ЦНИИ МПС,
позволили сделать вывод о возможности активного воздействия
на структуру осадка А1(ОН)3 путем его промывки растворами
кислот и оснований [182, 186]. Существенного увеличения плот¬
ности осадка гидроокиси алюминия можно достигнуть, если
через гидроокись, сформированную в избытке щелочи, пропускать
углекислый газ [157].
Старение гелей. Всю сумму процессов, происходящих в коа¬
гулятах после их образования, часто объединяют общим термином
«старение». По Пескову [150, стр. 363], старение гелей — это
самопроизвольно протекающий процесс, направленный в сторону
увеличения пассивности системы в отношении поверхностных
явлений. Но часто старение рассматривается и в более общем
смысле — как переход системы из неравновесного состояния в рав¬
новесное [187]. Так или иначе в результате старения, независимо
от того, происходит ли кристаллизация или нет, хаотическое рас¬
положение частиц в агрегатах заменяется ориентированным, чис¬
ло контактов между частицами возрастает вследствие их стрем¬
ления занять такое взаимное положение, при котором потен¬
циальная энергия системы минимальна [149 (стр. 490), 188].
Старение гелей сопровождается отделением части интермицел-
лярной (капиллярной) жидкости и постепенной утратой первона¬
94
ГЛАВА III. КОАГУЛЯНТЫ И ИХ ГИДРОЛИЗ
чальных поверхностных свойств [149 (стр. 490), 189]. Повышение
температуры и концентрации электролитов, увеличение pH среды
ускоряют старение [162, 190]. При наличии в растворе некоторых
органических веществ, например, нефтепродуктов, старение за¬
медляется [191], что, по-видимому, объясняется ухудшением усло¬
вий массообмена и кристаллизации [192, 1931.
Тиксотропия. Следующее важное свойство коагуляционных
структур — тиксотропная обратимость, или тиксотропия, под
которым понимается способность структур восстанавливать связи
после их механического разрушения. Кройт [154, стр. 505] пред¬
лагает рассматривать тиксотропию как одну из форм коагуляции.
Тиксотропия определяется способностью частиц к «дальней»
агрегации — взаимодействию во вторичном энергетическом ми¬
нимуме. Как уже указывалось, при неполной астабилизации
частиц в местах их контакта сохраняются водные прослойки
с отрицательным расклинивающим давлением. Толщина этих
прослоек равна приблизительно 0,1—0,3 мкм [144, 145, 191, 194,
195]. Прослойки снижают прочность структуры, но, обладая сма¬
зочной способностью, придают ей пластичность, эластичность
[146, 196]. Чем тоньше прослойка, тем менее прочна структура,
но тем полнее происходит ее восстановление после разрушения
[191].
Тиксотропия особенно велика в гелях с анизодиаметрическими
частицами [154 (стр. 505), 197]. Хлопья гидроокисей алюминия и
железа довольно быстро восстанавливают свою первоначальную
структуру. На скорость восстановления влияют pH среды и раз¬
мер первичных частиц [191]. Повышение температуры и медленное
перемешивание способствуют тиксотропному восстановлению.
Строение хлопьев при этом упорядочивается, плотность и проч¬
ность возрастают [84 (стр. 141), 198].
Свежеосажденные коагуляты солей алюминия и железа могут
быть без ущерба подвергнуты перемешиванию с довольно высоким
скоростным градиентом. Однако по мере старения тиксотропные
свойства гелей ухудшаются.
Пептизация. Под действием ионов, способных адсорбироваться
на поверхности частиц коагуляционной структуры, последняя
может разрушиться. Этот процесс, обратный коагуляции, носит
название пептизации.
Для того чтобы произошла пептизация, необходимо нли уве¬
личить заряд частиц до значения, превышающего критическое,
или гидрофилизировать их поверхность. Свежие осадки, в которых
процесс кристаллизации еще не получил развития, пептизируются
лучше, чем постаревшие. Перемешивание, встряхивание и повы¬
шение температуры ускоряют пептизацию. Хорошими пептиза-
торами гидроокисей алюминия и железа являются кислоты, ще¬
лочи, ПАВ [149 (стр. 255), 199].
Ш.З. КОАГУЛЯЦИОННЫЕ СТРУКТУРЫ И ИХ СВОЙСТВА
95
Согласно правилу Оствальда — Бузага, с увеличением коли¬
чества осадка его пептизируемое количество сначала растет, а за¬
тем падает до нуля. Это объясняется тем, что по мере возраста¬
ния массы осадка при постоянном количестве пептизатора доля
последнего, адсорбируемая единицей поверхности осадка, будет
уменьшаться. Из правила вытекает важное следствие о том, что
путем добавления к золю избытка осадка, из которого он получен
пептизацией, можно скоагулировать этот золь.
Прочность. Прочность коагуляционных структур оксигидра-
тов алюминия и железа зависит от количества и вида содержащихся
в воде электролитов и степени дисперсности структурообразую¬
щих частиц. Регулируя концентрацию коагулирующих или ста¬
билизирующих ионов, добавляя твердый наполнитель, подвергая
эти структуры разным физическим воздействиям, можно в извест¬
ных пределах изменять и их прочность.
Анализ результатов экспериментальных данных разных ис¬
следователей приводит к выводу, что прочность хлопьев А1(ОН)3
и Fe(OH)3, так же как и многих других агломератов [200], не зави¬
сит от прочности первичных частиц, а определяется прочностью
контактов между ними и числом контактов в единице объема агло¬
мерата. Число контактов и их распределение в структурной сетке
агломерата зависят от размеров и формы первичных частиц и их
ориентации. Что же касается прочности каждого контакта, то она
в основном определяется силами адгезии. И прежде чем перейти
к количественной оценке прочности коагулятов, рассмотрим крат¬
ко закономерности адгезии.
Под адгезией в широком смысле слова понимают взаимодей¬
ствие макрочастиц с твердой поверхностью. Взаимодействие час¬
тиц друг с другом — частный вид адгезии — получил название
аутогезии [201].
Сила адгезии зависит от кривизны взаимодействующих поверх¬
ностей, величины свободной энергии на границе раздела жидкой и
твердой фаз, а также температуры среды [201—204]:
где Fa — сила адгезии; и е'2 — кривизны поверхностей; Н„ —
расстояние между поверхностями.
Для аутогезии двух сферических частиц радиусом г1 и г2
(III.10)
где а — поверхностная энергия на границе раздела фаз.
Если радиус одной частицы во много раз больше радиуса
другой (переход от аутогезии к адгезии), уравнение (III.10)
96
ГЛАВА III. КОАГУЛЯНТЫ И ИХ ГИДРОЛИЗ
принимает вид
Fa = 4 nor, (111.10')
где г — радиус малой частицы.
По уравнению (III.10') с увеличением г сила адгезии линейно
возрастает. Однако в реальных условиях силам адгезии проти¬
востоят силы тяжести и инерции частиц, пропорциональные г3
и возрастающие с увеличением г быстрее, чем силы адгезии. Сле¬
довательно, силы адгезии выступают на первый план при наличии
в системе мелких частиц.
Далее нужно иметь в виду, что значения Fa, рассчитанные по
уравнению (III.10') для аэрозолей, дают отклонения от экспери¬
ментальных данных на 2—3 порядка. По мнению Фукса [205],
отклонение связано с тем, что под символом г в этом уравнении
должен выступать не радиус частиц, а радиус субмикроскопиче-
ских выступов, по которым происходит контактирование, т. е.
должна учитываться шероховатость поверхности.
В воздушной среде сила адгезионного взаимодействия слага¬
ется из электрических, молекулярных и капиллярных сил [201,
стр. 172]. В жидкой среде капиллярные и электрические силы не
проявляются, а молекулярные силы заметно экранируются жид¬
кой прослойкой. Кроме того, жидкая прослойка обусловливает
появление дополнительных сил отталкивания. Взаимодействие
сил притяжения и отталкивания с учетом толщины зазора между
поверхностями рассмотрено в гл. I. Характер этого взаимодей¬
ствия определяет адгезию как фиксацию частиц во вторичном
энергетическом минимуме.
Мацкрле и др. [206] предприняли попытку рассчитать (правда,
без учета расклинивающего давления) значения константы моле¬
кулярного притяжения А в уравнении (1.18') для адгезии и ауто¬
гезии частиц А1(ОН)3 и Fe(OH)3 к различным поверхностям в вод¬
ной среде. Они получили следующие средние значения А:
Поверхность
Частица
А-10»2, эрг
Поверхность Частица
Л-101*, эрг
А1(ОН)3
А1(ОН)3
1,26
СаС03 А1(ОН)3
1,40
Fe(0H)3
1,43
Fe(OH)3
1,72.
Fe(OH)3
А1(ОН)3
1,43
Уголь А1(ОН)3
1,70
Fe(OH)3
1,88
Fe(OH)s
1,73
Si02
А1(ОН)3
1,27
Fe(OH)3
1,43
Из результатов расчетов видно, что гидроокись железа обладает
лучшими адгезионными свойствами, чем гидроокись алюминия.
Рассмотрение результатов экспериментов по выявлению влия¬
ния на силы адгезии физико-химических свойств среды и поверх¬
ностей приводит к следующим обобщениям.
III.3. КОАГУЛЯЦИОННЫЕ СТРУКТУРЫ И ИХ СВОЙСТВА
97
1. Рост заряда частиц, увеличение ионной силы раствора и
прогрессирующее во времени утоньшение водных прослоек вызы¬
вают увеличение сил адгезии [201, 207—210]. На основании этого
сделан вывод [211, 212], что наилучшая адгезия гидроокисей алю¬
миния и железа на отрицательно заряженной поверхности кварце¬
вого песка будет иметь место в высокоминерализованной воде и
в начальный момент гидролиза, когда в окружающей среде при¬
сутствуют катионы А13+ и Fe3+.
2. При понижении температуры воды (от 40 до 0Ь С) адгезия
уменьшается [201, стр. 202], что, по всей вероятности, связано
с увеличением гидратации частиц и плотности воды в прослойках.
По данным Кургаева [176, стр. 64], для гидроокиси алюминия со¬
отношение коэффициентов а, учитывающих интенсивность слипа¬
ния первичных частиц, при температурах воды t и 20ь С описыва¬
ется эмпирическим уравнением
а, = 0,5- + 0,025г. (III.И)
3. С уменьшением краевого угла смачивания поверхностей
(гидрофилизация) адгезия уменьшается [201, стр. 205].
4. Шероховатость поверхности по одним данным [213] усили¬
вает адгезию, по другим [201, стр. 208] уменьшает ее. По-видимому,
расхождение результатов связано с разным масштабом шерохо¬
ватости: малые частицы на грубошероховатой поверхности имеют
большую площадь соприкосновения, чем крупные частицы на
мелкошероховатой поверхности.
5. В присутствии ПАВ и комплексообразующих веществ (гек¬
саметафосфат и трифосфат натрия) адгезия снижается [201,
стр. 199].
6. В кислой среде (pH 5—7) адгезия продуктов гидролиза сер¬
нокислого алюминия [186] и стеклянных частиц [201, стр. 194]
выше, чем в щелочной (pH 7—8).
7. Чрезмерно интенсивное перемешивание приводит к отрыву
частиц от макроповерхностей. Поэтому кривая зависимости адге¬
зии гидратированных частиц от интенсивности перемешивания
имеет максимум. Дэвис [214] предложил уравнение для описания
этого эффекта и показал его справедливость на примере гидрооки¬
си железа.
Достаточно концентрированные гели обладают механической ,
прочностью, которую можно оценить экспериментально и охарак¬
теризовать предельным напряжением сдвига, модулем упругости,
периодом релаксации [215].
Для оценки механической прочности гелей по предельному
напряжению сдвига применяют в основном два метода: танген¬
циального смещения пластинки (метод Вейлера — Ребиндера)
и закручивания цилиндра (метод Шведова). Эти методы исполь¬
зованы для оценки механических характеристик А1(ОН)я и Fe(OH)3
4 Е. Д. Бабенков
98
ГЛАВА' III. КОАГУЛЯНТЫ И ИХ ГИДРОЛИЗ
Тетеркиным и Лебедевой [181, 216], Барышниковой [217], Мити¬
ным [218, 219], Радвинским и Пучковым [220, 221]. Гороновский
и др. [222—224] определяли предельное напряжение сдвига коа¬
гулирующих систем по началу качения шарика [225].
Кургаев [176 (стр. 52), 226, 227], исходя из структурно-механи¬
ческих свойств коагулятов, разработал и успешно применил метод
определения критической величины давления, соответствующе¬
го пределу прочности хлопьев на сжатие.
Для разных коагулятов исследователями [181, 216, 218, 220,
222—224, 228] получены следующие значения предельного напря¬
жения сдвига (в мг/см2):
А1(ОН)3 5-11
Fe(OH)3 6-18
80% А1(ОН)3 +20%Са(ОН)2 16
Критические величины давления (в мг/см2) составляют по Кур-
гаеву [176, стр. 60]:
А1(ОН)3 7
Fe(OH)3 22
Fe(OH)2 26
50% А1(ОН)3 + 50% Fe(OH)3 14
Механическое диспергирование осадка Fe(OH)3 с последующим
его тиксотропным восстановлением способствует увеличению кри¬
тического давления от 22 до 25 мг/см2 [176, стр. 60].
Существенным пробелом в исследованиях является отсутствие
попытки установить влияние на прочность коагуляционных
структур их возраста, а также температуры и величины pH
среды.
Адсорбционная способность. В рапних работах адсорбцию
на частицах золя и гелях малорастворимых продуктов гидролиза
алюминия и железа рассматривали почти исключительно как
замену анионов во внешней обкладке двойного слоя анионами
солей, присутствующих в воде. Дальнейшие исследования [108]
продемонстрировали важную роль химических взаимодействий.
Установлено, что адсорбция истинно растворенных веществ
на гидроокисях описывается изотермами Лэнгмюра и Фрейндли¬
ха, причем вначале может иметь место хемосорбция, а затем поли-
слойная адсорбция [229, 230]. В кислой среде адсорбируются
преимущественно анионы. Сорбция катионов Си2+, Zn2+, Ni2+,
Ми2+ на гидроокисях А1(ОН)3 и Fe(OH)a начинается соответст¬
венно при значениях pH выше чем 4, 5, 6 и 8 [231].
Изучению сорбционной способности и емкости А1(ОН)3 и
Fe(OH)3 по отношению к высокомолекулярным веществам посвящен
ряд работ. Когановский [232—234], исследуя адсорбцию гумино¬
вых веществ и красителей, установил, что изотермы носят ступен-
III.3. КОАГУЛЯЦИОННЫЕ СТРУКТУРЫ II ИХ СВОЙСТВА
99
ДПл мВ
40
О
Рис. II 1.9. Зависимость ДП
гидроокиси железа от величи¬
ны удельной сорбции (Г0) кон¬
го красного [232]
чатый характер с резкими увеличениями количества адсорбиро¬
ванного вещества. Ступенчатый характер изотерм автор объясняет
внезапными увеличениями доступности внутренней поверхности
пор сорбента вследствие пептизации гидроокисей. Он показал
далее, что сорбируемость веществ тем выше, чем сильнее ассоци¬
ированы ионы, образующие мицеллу, а между величиной удель¬
ной сорбции красителя и дзета-потенциалом гидроокиси железа
существует почти линейная связь (рис. III.9).
В начальный момент образования золей наблюдалось резкое
падение адсорбции. При pH около 7,2 это падение происходило
в течение первых 0,5—1 мин для Fe(OH)3 и 3—10 мин для А1(ОН)а
[234]. Наступающая затем коагуляция не уменьшала адсорбции,
что, по мнению Когановского, объясняется свободным доступом
сорбтива к внутренней поверхности крупнопористых гидрооки¬
сей. Повышенную адсорбционную емкость золей в момент гидро¬
лиза автор связывает с максимальной концентрацией в этот
момент катионов А13+ и Fe3+, являющихся по отношению к отрица¬
тельно заряженным частицам сорбтива коагулирующими. Началь¬
ная сорбционная способность А1(ОН)3 выше, чем Fe(OH)3, и
меньше снижается во времени [98, стр. 61].
Ермоленко и др. [159—161, 173] на основе результатов экспе¬
риментов пришли к выводу, что уменьшение сорбции во времени
связано с изменением пористой структуры гидрогелей в резуль¬
тате кристаллизации — несмотря на постоянство размера первич¬
ных частиц сорбционная способность А1(ОН)3 с течением времени
резко снижалась (табл. III.6).
Адсорбция красителей зависит от условий формирования гид¬
роокисей. При недостатке щелочи сорбируются преимущественно
кислые красители [141]. Например, азолитин связывается гидро¬
окисью алюминия при pH ~ 6,3 [160].
Коагуляты гидроокисей, полученные в среде, содержащей
НС03- и СГ-анионы, обладали по отношению к гуминовым вещест¬
вам большей адсорбционной емкостью (примерно в 2 раза), чем
полученные в среде НС03- и ЭО^-анионов [156]. Это можно объяс¬
нить разной пористостью коагулятов [235, стр. 130] и более силь-
4
100
ГЛАВА III. КОАГУЛЯНТЫ И ИХ ГИДРОЛИЗ
Таблица III.6. Адсорбция красителей (вес. %)
на геле А1(ОН)3 в зависимости от его возраста [159]
Возраст геля, мин
Азолитмин
Ализариновый
желтый
'0
78,0
54,4
5
24,6
—
10
—
45,0
30
23,6
17,2
60
23,6
—
ной конкуренцией SO4- (по сравнению с СГ) с гуминовыми веще¬
ствами за адсорбционные участки гидроокисей.
При небольших добавках к А1(0Н)3 гидратов, обладающих
крупнопористой структурой, адсорбционная емкость гидроокиси
возрастает, при добавках мелкопористых гидратов — уменьша¬
ется [236]. С увеличением степени гидратации А1(ОН)3 и Fe(OH)3
их адсорбционная емкость увеличивается [224, 237].
Интересно также следующее наблюдение: для гидроокиси
алюминия, полученной осаждением при малых скоростях переме¬
шивания, характерна меньшая удельная сорбция и большая ее
зависимость от температуры, чем для той же гидроокиси, получен¬
ной при высоких скоростях перемешивания [124].
Удельная сорбция гуминовых веществ на гидроокиси алюми¬
ния составляет в зависимости от принятых исходных условий от
1—1,4 [98, стр. 59] до 16 мг [238] на \ мг А13+. По данным Горо-
новского [235, стр. 118], 1 мг-экв А1(ОН)3 снижает цветность воды
на 30—100 град.
Литература
1. S. Constantlnescu, G. Zavoianu et al. Пат. CPP 42550 (1964).
2. Я. Д. Рапопорт. Автореферат канд. дисс. М., АКХ им. К. Д. Памфи¬
лова, 1970.
3. Ch. P. Roddy. Water Works and Wastes Engng, 9, N 12, 34 (1972).
4. S. T. Powell, L. V. Carpenter, L. R. Setter et al. Chem. and Metallurg.
Engng, N 8, 481 (1939).
5. JI. H. Фальковская. Научные труды АКХ им. К. Д. Памфилова, вып. 22.
Водоснабжение, № 3. М.— JI., ОНТИ, 1963, стр. 158.
6. Сб. «Коагулянты для очистки воды». Под ред. В. Т. Турчиновича. М.,
Изд-во МКХ РСФСР, 1948.
7. М. Zdybiewska, D. Cymbalko. Przem. chem., 47, 232 (1968).
8. С. Симада. Яп. пат. 6386 (1959).
9. С. Такесита, К. И гуси, II. Аракава и др. Нихон кагаку кайси, № 8,
1606 (1973); РЖХим., 4И321 (1974).
10. Франц. пат. 1398820 (1965).
И. И. Мотомура. Яп. пат. 38110 (1972); 37588 (1972).
ITI. ЛИТЕРАТУРА
101
12. W. Becher, J. Massonne. Пат. ФРГ 2107970 (1975).
13. Т. Эмбу, С. Яманака. Мидзусёри гидзюцу, 16, 77 (1975); РЖХим.,
7И534 (1976).
14. /. Kucharski, J. Kepinski et al. Пат. ПНР 43643 (1961).
15. А. А. Фурман, 3. А. Орловский и др. Авт. свид. СССР 125192 (1959);
Бюлл. изобр., № 24 (1959).
16. Г. Бэббит, Дж. Доланд. Водоснабжение. М., Госстройиздат, 1958.
17. М. Prison. Techn. sanit. et municip., 48, 158 (1953).
18. H. J. Horvitz. Пат. США 3082173 (1963).
19. P. Фукумори. Яп. пат. 49-40552 (1974).
20. Э. А. Левицкий, В. Н. Максимов. Авт. свид. СССР 146302 (1961); Бюлл.
изобр., № 8 (1962).
21. А. О. Walker. Пат. США 3023169 (1962).
22. О. М. Bacon. Пат. США 3350303 (1967); 3350304 (1967); 3446742 (1969).
23. Пат. ФРГ 2318653 (1974).
24. Й. Морита. Яп. пат. 49-40553 (1974).
25. S. Lerczynski, J. Kucharski. Przem. chem., 44, 458 (1965).
26. /. Kucharski, A. Mazaraki et al. Пат. ПНР 41043 (1958).
27. Y. Aiba, T. Furumori. Пат. США 3544476 (1970).
28. С. И. Савчук, Ю. П. Лобанов, Н. М. Восвилов и др. Авт. свид. СССР
359237 (1971); Бюлл. изобр., № 35 (1972).
29. С. Бан, С. Хатано, Т. Кобаяси. Яп. пат. 19242 (1972); Й. Мурата.
Яп. пат. 49-40792 (1974); Р. Фукумори. Яп. пат. 49-40551 (1974).
30. В. А. Клячко, И. Э. Апельцин. Подготовка воды для промышленного
и городского водоснабжения. М., Стройиздат, 1962.
31. М. Lane. Water and Sewage Works, 106, 339 (1959).
32. R. W. OcKershausen. Water and Sewage Works, 105, 61 (1958).
33. K. A. Melkersson. Mod. Kemi, N 2, 75 (1968).
34. В. Т. Турчинович. Улучшение качества воды. М.— JI., Стройиздат,
1940, стр. 43.
35. А. А. Кастальский, Д. М. Минц. Подготовка воды для питьевого и
промышленного водоснабжения. М., «Высшая школа», 1962, стр. 68.
36. .Т. Amer. Water Works Assoc., 49, 1315 (1957).
37. J. Amer. Water Works Assoc., 50, 1259 (1958).
38. Л. А. Кулъский. Обезвреживание и очистка воды хлором. М., Изд-во
МКХ РСФСР, 1947, стр. 348.
39. Л. А. Кулъский, М. Н. Булава, И. Т. Гороновский и др. Проектирова¬
ние и расчет очистных сооружений водопроводов. Киев, «Буд1вельник»,
1972, стр. 68.
40. Г. Г. Руденко, И. Т. Гороновский. Удаление примесей из природных
вод на водопроводных станциях. Киев, «Буд1вельник», 1976, стр. 88.
41. М. А. Шевченко. Укр. хим. ж., 17, 252 (1951).
42. М. А. Шевченко. Там же, стр. 427.
43. Л. А. Кулъский. Рационализация технологии очистки питьевых вод.
Киев, Изд-во АН СССР, 1951.
44. Е. С. Абазаев. Водоснабжение и санитарная техника, № 5, 8 (1959).
45. /. С. Rhodes. Пат. США 3347787 (1967).
46. Водоснабжение и санитарная техника, № 3, 62 (1941).
47. J, С. Schrippers. Tijdschr. drinkwatervoorz en afvalwaterbehandel,
N 14, 320 (1971).
48. Т. Ябу. Яп. пат. 14241 (1963).
49. Т. Сакакура, М. Мацухаси. Яп. пат. 19421 (1964).
50. М. К. Игнатов, Г. С. Лейбович. Водоснабжение и санитарная техника,
№ 3, 57 (1941).
51. Water and Sewage Works, 116, 198 (1969).
52. L. L. Hedgepeth, N. C. Olsen, W. C. Olsen. J. Amer. Water Works Assoc.,
20, 467 (1928).
102
ГЛАВА III. КОАГУЛЯНТЫ И ИХ ГИДРОЛИЗ
53. А. С. Коган. Вопросы водоснабжения и санитарной техники в Бельгии.
М., Изд-во МКХ РСФСР, 1960, стр. 78.
54. П. В. Мозжухин. Гигиена и санитария, № 1, 2 (1944).
55. В. Радуич, Я. Белышсва. Жилищно коммунальное хозяйство, № И,
19 (1957).
56. Г. Г. Ломакина, С. П. Сукач, А. И. Зелъцерман и др. Сб. «Водоснаб¬
жение, канализация, гидротехнические сооружения», вып. 15. Киев,
«Буд1вельннк», 1972, стр.4.
57. Ж. И. Антончук, С. П. Сукач. Труды ВНИИ ВОДГЕО, вып. 25. М.,
1970, стр. 69.
58. И. А. Макаров, А. Д. Стром и др. Авт. свид. СССР 244949 (1967);
Бюлл. изобр., № 18 (1969).
59. М. С. Schwartz. J. Amer. Water Works Assoc., 30, 551 (1938).
60. W. Husmann. Osterr. Wasserwirtschaft, 13, 4 (1961).
61. T. Niwa, S. Ozaki. Пат. США 3349921 (1967).
62. N. Matsumoto, II. Arimitsu. Пат. США 3461067 (1969).
63. X. Кавасима, К. Нисикава и др. Осака когё дайгаку киё. Рико-хен.
А13, № 2, 103 (1969); РЖХим., 3H523 (1970).
64. К. Reinhardt. Пат. ФРГ 1258808 (1968).
65. С. М. Шифрин, И. Г. Краснобородъко. Прогрессивные методы очистки
природных и сточных вод (материалы научно-технической конферен¬
ции), сб. № 2. Изд. Моск. Дома научно техн. пропаганды им.
Ф. Э. Дзержинского, 1971, стр. 5.
66. P. J. Gaughan, J. К. Brown. Пат. США 3575853 (1971).
67. Е. A. Zdansky. Швейц. пат. 335085 (1959).
68. I. Т. Fucik, U. Meyer. Австрийск. пат. 322461 (1975).
69. J. Kepinski, Н. Adamski. Zesz. nauk. Politechn. szezecinsk., N 34, 45 (1964).
70. H. Beckhaus. Wochenbl. Papierfabr., 97, 291 (1969).
71. /. Ganczarczyk. Gaz, woda i techn. sanit., 32, 92 (1958).
72. Пат. ФРГ 1933035 (1971).
73. Ю. Сугавара, Т. Ямада и др. Яп. пат. 91 (1967).
74. W. С. Harsh. Пат. США 3446731 (1969).
75. V. Cocheci, R. Taubert et al. Bull, stiint. §i tehn. Inst, politehn. Timisoara,
12, N 1, 83 (1967).
76. K. Kdwert. Швед. пат. 347954 (1972).
77. E. D. Сапп. Пат. США 3377271 (1968).
78. С. G. Thompson, J. F. Singley, A. P. Black. J. Amer. Water Works Assoc.,
64, 11 (1972).
79. Environ. Sci. and Technol., 7, 304 (1973).
80. К. Тоёда, К. Окада и др. Яп. пат. 27226 (1969).
31. R. R. Seiler. Швейц. пат. 479490 (1969).
82. X. Накамура. Яп. пат. 12733 (1960).
83. X. Накамура. Яп. пат. 12733 (1960).
84. Л. А. Кулъский. Основы технологии кондиционирования воды. Киев,
Изд-во АН УССР, 1967.
85. С. А. Вознесенский. Физико-химические процессы очистки воды. М.—Л.,
Госстройиздат, 1934.
86. G. Н. Bredig. Anorganische Fermente. Leipzig, 1901, S. 62.
87. И. Т. Гороновский, Ю. П. Назаренко, E. Ф. Некряч. Краткий справоч¬
ник по химии. Киев, «Наукова думка», 1974, стр. 346.
88. А. Поллинг. Общая химия. М., «Мир», 1974, стр. 439.
89. Л. А. Кулъский. Теоретическое обоснование технологии очистки воды.
Киев, «Наукова думка», 1968, стр. 29.
90. Сб. «Химия координационных соединений». Под ред. Дж. Бейлара.
М., ИЛ, 1960.
91. Я. И. Герасимов, В. П. Древинг, Е. II. Еремин, и др. Курс физической
химии, т. II. М., «Химия», 1973, стр. 451.
XII. ЛИТЕРАТУРА
103
92. Ю. Ю. Лурье. Справочник по аналитической химии. М., «Химия»,
1971, стр. 248. «■
93. И. И. Вадюхин. Водоснабжение и санитарная техника, № 10, 35 (1939).
94. II. И. Блок. Качественный химический анализ. М., Госхимиздат, 1952.
95. И. И. Жуков. Избранные труды. М., Изд-во АН СССР, 1952, стр. 234.
96. Л. Кертман. Курс качественного анализа. М., Госхимиздат, 1937.
97. М. R. Buydens. Techn. sanit. et municip., 40, 139 (1953).
98. Л. А. Кулъский, А. М. Когановский, И. Т. Гороновский и др. Физико¬
химические основы очистки воды коагуляцией. Киев, Изд-во АН УССР,
1950.
99. W. Stumm, J. Morgan. J, Amer. Water Works Assoc., 54, 971 (1962).
100. II. Britton. J. Chem. Soc., 127, 2120 (1925).
101. S. Mattson. J. Phys. Chem., 32, 1532 (1928).
102. А. И. Рабинович. ЖФХ, 1, 469 (1930).
103. А. И. Рабинович, В. А. Каргин. Там же, стр. 65.
104. А. И. Рабинович, В. А. Каргин. Е. В. Федиман. ЖФХ, 2, 63 (1931).
105. А. И. Рабинович, Е. В. Федиман. Там же, стр. 336.
106. Н. П. Песков. Физико-химические основы коллоидной науки. М.—JI.,
Госхимиздат, 1935.
107. В. А. Каргин, Н. А. Оганджанова. ЖФХ, 10, 782 (1937).
108. 3. Я. Берестнева, В. А. Каргин. Там же, стр. 593.
109. М. Е. Шишниашвили, В. А. Каргин, А. Л. Бацанадзе. ЖФХ, 21, 391
(1947).
110. X. Г. С. Бриттон. Водородные ионы. Определение и значение их в тео¬
ретической и прикладной химии. Л., Химтеорет, 1936.
111. Б. П. Никольский, В. И. Парамонова. ЖФХ, 2, 5 (1931).
112. Б. А. Скопинцев. Водоснабжение и санитарная техника, № 3, 57 (1938).
113. В. А. Клячко, И. Э. Апелъцин. Очистка природных вод. М., Стройиздат,
1971, стр. ИЗ.
114. В. А. Каргин, В. В. Киселева. ЖФХ, 11, 461 (1938).
115. В. А. Каргин, Г. В. Климовицкая. Там же, стр. 467.
116. Ю. М. Глазман, А. А. Баран, В. М. Барбой и др. Коллоидн. ж., 27,
513 (1965).
117. W. Stumm, Ch. R. O'Melia. J. Amer. Water Works Assoc., 60, 514 (1968).
118. A. S. Teot, S. L. Daniels. Environ. Sci. and Technol., 3, 825 (1969).
119. О. И. Мартынова. Коагуляция при водоподготовке. М.— Л., Гос-
энергоиздат, 1951.
120. /. Е. Singley, А. P. Black. J. Amer. Water Works Assoc., 59, 1549 (1967).
121. Y. Inoue, A. Osugi, Y. Kanagi et al. J. Chem. Soc. Japan. Pure Chem.
Sect., 82, 310 (1964).
122. E. S. Hall. J. Appl. Chem. 15, 197 (1965); E. S. Hall, R. F. Packham.
J. Amer. Water Works Assoc., 57, 1149 (1965).
123. T. Okura, K. Goto et al. Mem. Fac. Engng Hokkaido Univ., 11, 25 (1960).
124. E. Matijevic, G. F. Janayer, M. Kerker. J. Colloid Sci., 19, 333 (1964).
125. J. Amer. Water Works Assoc., 63, 99 (1971).
126. G. Biederman. Svensk kem. tidskr. 76, 362 (1964).
127. A. P. Black, D. G. Williams. J. Amer. Water Works Assoc., 59, 1549
(1967).
128. E. Matijevic, G. E. Janayer. J. Colloid and Interface Sci., 21, 197 (1966).
129. М. C. Fyerstenay, P. Somasyndaran, D. W. Fyerstenay. Trans. Inst.
Mining and Metallurgy, 74, 381 (1965).
130. A. P. Black, Ching-Cin Chen. J. Amer. Water Works Assoc., 59, 1173
(1967).
131. К. M. Yao. Water and Sewage Works, 114, 295 (1967).
132. J. E. Singley, J. H. Sullivan. J. Amer. Water Works Assoc., 61, 190
(1969).
133. Л. H. Mалашевич, С. А. Левина и др. Коллоидн. ж., 31, 543 (1969).
104
ГЛАВА III. КОАГУЛЯНТЫ И ИХ ГИДРОЛИЗ
134. В. И. Бабушкин, Ю. Н. Зенченко, К. А. Токарь и др. Сб. «Водоснабже¬
ние, канализация, гидротехнические сооружения», вып. 13. Киев,
«Будгвельник», 1971, стр. 13.
135. В. И. Малое. XXX Научно-техническая конференция МИСИ им.
В. В. Куйбышева (тезисы докладов). Изд. МИСИ им. В. В. Куйбышева,
1971, стр. 20.
136. В. С. Сажин, А. К. Заполъский, Н. Н. Захарова. ЖПХ, № 7, 1420
(1968).
137. Д. Ф. Васильев. Коллоидн. ж., И, 377 (1949).
138. Б. Н. Хрусталев, В. В. Пушкарев и др. Коллоидн. ж., 30, 286 (1968).
139. Н. Bernhardt, A. Wilhelms. Vom Wasser 1971, Bd. 38. Weinheim, 1971,
S. 215.
140. Т. E. Larson, A. M. Buswell. Ind. and Engng Chem., 32, 132 (1940).
141. И. Т. Гороновский. Канд. дисс. Киев, ИОНХ АН УССР, 1955.
142. А. P. Black, S. A. Hannah. J. Amer. Water Works Assoc., 53, 438 (1961).
143. Т. T. Collins, H. L. Davis B. W. Rowland. Paper Trade J., 113, 94 (1941).
144. А. И. Рабинерсон. Проблемы коллоидной химии. JI., Химтеорет, 1937.
145. Г. И. Фукс. Колоидн. ж., 12, 228 (1950).
146. П. А. Ребиндер. Совещание по вязкости жидкостей и коллоидных рас¬
творов, т. I. М., Изд-во АН СССР, 1941, стр. 361.
147. П. А. Ребиндер. Труды Третьей Всесоюзной конференции по коллоид¬
ной химии. М., Изд-во АН СССР, 1956, стр. 7.
148. Н. R. Hay. Water and Sewage Works, 93, 6 (1946).
149. С. С. Воюцкий. Курс коллоидной химии. М., «Химия», 1975.
150. Н. П. Песков, Е. М. Александрова-Прейс. Курс коллоидной химии.
М.— Л., Госхимиздат, 1948.
151. И. С. Артемов. Коллоидн. ж., 9, 225 (1947).
152. А. И. Рабинерсон, Г. И. Фукс. Труды Ленинградского отделения Все¬
союзного института удобрений, агропочвоведения и агротехники. Л.,
1933, стр. 22.
153. И. Ф. Ефремов. Периодические коллоидные структуры. Л., «Химия»,
1971, стр. 14.
154. Г. Р. Кройт. Наука о коллоидах, т. I. М., ИЛ, 1955.
155. Б. Г. Запрометов, Г. М. Вирская. Коллоидн. ж., 10, 339 (1948).
156. И. Т. Гороновский. Сб. «Исследования по водоподготовке». М., Гос¬
стройиздат, 1956, стр. 5.
157. И. М. Колътгоф, Е. Б. Сендэл. Количественный анализ. М.— Л., Гос¬
химиздат, 1941.
158. 3. Я. Берестнева, Т. А. Корецкая, В. А. Каргин. Коллоидн. ж., 13,
323 (1951).
159. Н. Ф. Ермоленко, С. А. Левина. Изв. АН БССР, № 1, 107 (1954).
160. Н. Ф. Ермоленко, С. А. Левина. Труды Третьей Всесоюзной конферен¬
ции по коллоидной химии., Изд-во АН СССР, 1956, стр. 276.
161. С. А. Левина, Н. Ф. Ермоленко. Коллоидн. ж., 17, 287 (1955).
162. Ю. М. Кандыкин. Коллоидн. ж., 26, 318 (1964).
163. Е. R. Landa, R. G. Gast. Clays and Clay Minerals, 21, 121 (1973).
164. T. Sato. Z. anorg. allgem. Chem., 391, 69 (1972).
165. И. А. Истомина, 3. Я. Берестнева, В. А. Каргин. Коллоидн. ж., 21,
574 (1959).
166. В. П. Чалый. Гидроокиси металлов. Киев, «Наукова думка», 1972.
167. R. W. Smith, J.D. Нет. US. Geol. Survey. Water-Supply Paper,
№ 1827-D, 51 (1972).
168. М. B. Weiser. Ind. and Engng Chem., 32, 1487 (1940).
169. K. W. Towe, W. F. Bradley. J. Colloid and Interface Sci., 24, 384 (1967).
170. H. С. Репина, H. Ф. Ермоленко, М. Д. Эфрос и др. Коллоидн. ж., 36,
585 (1974).
171. Z. anorg. allgem. Chem., 273, 227 (1953).
III. ЛИТЕРАТУРА
105
172. G. С. Bye, J. G. Robinson. Chem. and Ind., № 15, 612 (1963).
173. М. А. Пионтковская, Я. В. Жигайло, JI. А. Еременко и др. Коллоидн.
ж., 21, 347 (1959).
174. P. Becker, R. Baro. J. chim. phys. et phys.-chim. biol., 67, 195 (1970).
175. Г. А. Крестов. Термодинамика ионных процессов в растворах. JL, «Хи¬
мия», 1973, стр. 106.
176. Е. Ф. Кургаев. Основы теории и расчета осветлителей. М., Госстрой¬
издат, 1962.
177. П. В. Руфимский. Коллоидн. ж., 21, 351 (1959).
178. II. И. Исколъдский. Химия растворов алюминиевой промышленности.
М.— Л., ОНТИ НКТП, 1937.
179. W. Rummel. Wasserwirtschaft—Wassertechnik, 10, 121 (1960).
180. Н. И. Беляев. Метеорология и гидрология, № 5, 6 (1940).
181. Н. С. Лебедева. Автореферат канд. дисс. М., ВНИИ ВОДГЕО, 1954.
182. Е. Д. Бабенков. Водоснабжение и санитарная техника, № 10, 24 (1965).
183. R. F. Packham. J. Appl. Chem., 12, 556 (1962).
184. И. Bernhardt. Vom Wasser 1965, Bd. 32. Weinheim, 1966, S. 193.
185. R. K. Ham, R. F. Christman. J. Sanit. Engng Div. Proc. ASCE, № 3,
481 (1969).
186.JE-Д. Бабенков. Сб. «Методы очистки и контроля качества воды». М.,
«Транспорт», 1966, стр. 15.
187. G. Langhammer. Wiss. Z. Karl-Marx-Univ. Leipzig. Math, naturwiss.
Beihe, 19, 399 (1970).
188. P. Э. Нейман, P. И. Сухорукова. Коллоидн. ж., 12, 363 (1950).
189. А. Я. Зворыкин, Ф. М. Перельман. Коллоидн. ж., 9, 37 (1947).
190. Н. Krischner, К. Torkar, R. Hoffmann. Monatsh. Chem., 98, 2348 (1967).
191. М. И. Медведев, А. Г. Шаблина. Сб. «Нефтепереработка и нефтехимия»,
вып. 9. Киев, «Буд1вельник», 1973, стр. 50.
192. Г. Фрейндлих. Тиксотропия. М.— Л., Госхимиздат, 1939.
193. S. L. Нет, Е. J. Russo, S. М. Bahai et al. J. Pharm. Sci., 59, 317 (1970).
194. М. П. Воларович, Д. М. Толстой. ЖФХ, 4, 815 (1933).
195. И. Н. Антипов-Каратаев. Руководство для полевых и лабораторных
исследований почв, т. 4. Современные методы исследования физико¬
химических свойств почв, вып. 1. М.— Л., Изд-во АН СССР, 1945,
стр. 86.
196. Г. И. Фукс. Пятая Всесоюзная конференция по коллоидной химии
(тезисы докладов). М., Изд-во АН СССР, 1962, стр. 33.
197. В. Питтель, А. Рабинерсон. Коллоидн. ж., 4, № 5 (1938).
198. Е.Н. Тетеркин. Сб. «Исследования по водоподготовке». М., Стройиздат,
1955, стр. 17.
199. 10. С. Зуев. Коллоидн. ж., 12, 114 (1950).
200. И. Г. Бажал, О. Д. Куриленко. Переконденсация в дисперсных си¬
стемах. Киев, «Наукова думка», 1975, стр. 14.
201. А. Д. Зимон. Адгезия пыли и порошков. М., «Химия», 1976.
202. Б. В. Дерягин, II. А. Кротова. Адгезия. М., Изд-во АН СССР, 1949.
203. Н. А. Кротова, Ю. М. Кириллова. Труды Третьей Всесоюзной конфе¬
ренции по коллоидной химии. М., Изд-во АН СССР, 1956, стр. 329.
204. Б. В. Дерягин. Труды Моск. Дома ученых, вып. 2. М., Изд-во АН
СССР, 1937, стр. 75.
205. Н. А. Фукс. Механика аэрозолей. М., Изд-во АН СССР, 1955, стр. 317.
206. V. Mackrle, S. Mackrle. Adhese vefiltracnim lozi. Praha, 1959.
207. Б. В. Дерягин, А. Д. Зимон. Коллоидн. ж., 23, 544 (1961).
208. II. К. Адам. Физика и химия поверхностей. М.— Л., Гостехиздат, 1947.
209. А. Д. Малкина, Б. В. Дерягин. Коллоидн. ж., 12, 431 (1950.)
210. Б. Тремийон. Разделение на ионообменных смолах. М., «Мир», 1967.
211. Л. А. Кулъский, М. И. Медведев. Сб. «Санитарная техника», вып. 10.
Киев, «Вуд1вельник», 1971, стр. 147.
1{>6
ГЛАВА III. КОАГУЛЯНТЫ И ИХ ГИДРОЛИЗ
212. Г. И. Пинская. Сб. «Научные основы технологии очистки воды». Киев,
«Наукова думка», 1973, стр. 30.
213. В. В. Morgan. Monthly Bull. Brit. Coal Utilisation Bes. Assoc., 25,
125 (1961).
214. A. G. Davies. J. Colloid and Interface Sci., 28, 48 (1968).
215. С. Я. Вейлер, П. А. Ребиндер. ДАН СССР, 19, 5 (1948).
216. E. H. Тетеркин, Н. С. Лебедева. Физические и технологические свой¬
ства взвесей, образующихся при обработке воды. М., Стройиздат,
1962.
217. Т. И. Барышникова. Труды инж.-строит. фак-та Челяб. политехи, ин-та,
вып. 3, 3 (1959).
218. Б. А. Митин. Коллоидн. ж., 28, 852 (1966).
219. Б. А. Митин. Зав. лаб., 28, 1259 (1962).
220. М. Б. Радвинский, А. И. Пучков. Сб. «Водоснабжение, канализация,
гидротехнические сооружения», вып. 13. Киев, «Буд1вельник»,
1971, стр. 38.
221. М. Б. Радвинский, А. И. Пучков. ЖПХ, 46, 204 (1973).
222. И. Т. Гороновский. Зав. лаб., 19, 940 (1953).
223. И. Т. Гороновский, А. М. Волошинова. Коллоидн. ж., 16, 333 (1954).
224. А. Б. Заварило, Э. В. Галингер, И. Т. Гороновский. Сб. «Физико-хими-
ческая механика и лиофильность дисперсных систем», т. 2. Киев, «Нау¬
кова думка», 1971, стр. 273.
225. Ю. С. Зуев. Коллоидн. ж., 12, 36 (1950).
226. Е. Ф. Кургаев. Коллоидн. ж., 19, 72 (1957).
227. Е. Ф. Кургаев. Методы определения физических параметров контактной
среды в осветлителе. Информационное письмо № 411. М., изд. ЦНИИ
МПС, 1957.
228. Ю. М. Климов, Б. А. Митин. Коллоидн. ж., 32, 458 (1970).
229. Л. А. Кулъский, А. М. Когановский. ЖПХ, 17, 599 (1944).
230. J. G. Paterson, Т. Salman. Trans. Inst. Mining and Metallurgy, 79, 91,
(1970).
231. Г. А. Соломин, Т. О. Гончарова и др. Гидрохимические материалы, т. 46.
JI., Гидрометеоиздат, 1968, стр. 150.
232. А. М. Когановский. Коллоидн. ж., 11, 237 (1949).
233. А. М. Когановский. Там же, стр. 417.
234. А. М. Когановский. Коллоидн. ж., 13, 283 (1951).
235. И. Т. Гороновский. Физико-химическое обоснование автоматизации
технологических процессов обработки воды. Киев, «Наукова думка»,
1975.
236. С. А. Левина, Н. Ф. Ермоленко. Сб. «Ионообмен и сорбция из растворов».
Минск, Изд-во АН БССР, 1963, стр. 100.
237. W. Stumm, С. P. Huang, S. R. Jenkins. Croat, chem. acta, 42, 223 (1970).
238. W. Rummel. Fortschr. Wasserchem., № 1, 87 (1964).
Глава iV
ЗАКОНОМЕРНОСТИ ВЗАИМОДЕЙСТВИЯ
КОАГУЛИРУЮЩИХ ЧАСТИЦ
IV. 1. КОАГУЛЯЦИЯ ЗОЛЕЙ ЭЛЕКТРОЛИТАМИ
Из представлений о факторах агрегативной устойчивости дисперс¬
ных систем вытекает, что коагуляция может наступить в резуль¬
тате протекания разных физических и химических процессов.
Для очистки воды наибольшее практическое значение имеет коа¬
гуляция электролитами, когда снижение (исчезновение) энерге¬
тического барьера, препятствующего слипанию частиц, происхо¬
дит за счет повышения концентрации электролитов.
Из-за относительно малых концентраций электролитов в прес¬
новодных источниках водоснабжения дисперсные примеси воды
не могут быть скоагулированы естественным образом в той степе¬
ни, которая оказалась бы достаточной для их отделения в осадок.
Такие возможности возникают лишь при увеличении минерали¬
зации воды в несколько раз, например, при смешении речной и
морской вод в дельтах рек [1]. Поэтому закономерности коагуля¬
ции мы будем рассматривать главным образом на примере продук¬
тов гидролиза коагулянтов, для успешной коагуляции которых
содержание электролитов в природной воде оказывается достаточ¬
ным.
Из строения мицелл ясно, что коагулирующим действием обла¬
дают только те ионы, заряд которых противоположен заряду час¬
тиц,— так называемые коагулирующие ионы. Для золей с отри¬
цательно заряженными частицами коагулирующими являются
катионы, для золей с положительно заряженными частицами —
анионы.
Чтобы вызвать коагуляцию золя, необходимо превысить неко¬
торую максимальную концентрацию ионов-коагуляторов — порог
коагуляции. Согласно экспериментальным данным, величина порога
зависит от концентрации золя и некоторых других условий. На¬
пример, чем выше концентрация золей А1(ОН)3 и Fe(OH)3, тем
выше и порог их коагуляции [2, стр. 120] 1. Теоретически обосно¬
вано уменьшение пороговой концентрации с увеличением радиуса
частиц коллоидной системы при значениях дзета-потенциала
<^25 мв [3]. Поэтому значение порога коагуляции служит лишь
относительной характеристикой эффективности коагулирующих
ионов по отношению к данному золю.
1 Это объясняется явлениями катионного обмена (см. раздел II.2.1).
108
ГЛАВА IV. ВЗАИМОДЕЙСТВИЕ КОАГУЛИРУЮЩИХ ЧАСТИЦ
Согласно правилу Шульце—Гарди, обоснованному теоретиче¬
ски Дерягиным, коагулирующая сила иона тем больше, чем выше
его валентность. По экспериментальным данным, собранным Овер-
беком [2, стр. 432], соотношение средних пороговых концентраций
одно-, двух-, трех- и четырехвалентных анионов для золя А1(ОН)3
составляет 1 : 0,012 : 0,0015 : 0,0010, а одно- и двухвалентных
анионов для золя Fe(OH)3 — 1 : 0,018. По другим сведениям 14]
коагулирующие концентрации одно-, двух- и трехвалентных ани¬
онов для золя А1(ОН)3 относятся как 1 : 0,013 : 0,0017. На соот¬
ношение влияет диэлектрическая проницаемость среды [5].
Коагулирующая сила ионов одинаковой валентности зависит
от их радиуса и степени гидратации. По возрастанию коагулирую¬
щих свойств ионы одинаковой валентности можно расположить
в лиотропные ряды. В частности, для золя гидроокиси железа по
Думанскому [61:
СН3СОО- > CNS- > Cl" > ОН- > N0“ > CIO” > J-;
по Крлеже [7]: .
СН3СОО~> NO“> С1~,
SO2" > оксалат2- > НРО2- > SiO2^
[Fe (CN)s]3- > цитрат3- > P03_j
[Fe(CN)6]4->P20*-
По мнению Руцкова [8], последовательность расположения ионов
в лиотропных рядах объясняется разным их влиянием на диспер¬
сионную среду (сжимающее или разрыхляющее действие), по
мнению Майтака [9] — разной способностью ионов проходить
сквозь потенциальный барьер двойного слоя посредством так
называемого туннельного> эффекта.
Поскольку заряд частиц связан с адсорбцией ионов, коагули¬
рующая способность последних определяется, в частности, их
адсорбируемостью. Рассматривая в этом аспекте действие коагу¬
лирующих ионов, авторы работы [10] установили:
1) коагулирующее действие ионов возрастает с увеличением
адсорбируемости;
2) снижение заряда частиц золя тем сильнее, чем меньше кон¬
центрация электролита, при которой достигается требуемая сте¬
пень адсорбции коагулирующих ионов.
Высокой адсорбируемостью объясняется, например, сильное
коагулирующее действие некоторых органических ионов.
На коагулирующую способность влияют не только свойства
коагулирующего, но и сопутствующего иона, т. е. иона, заряжен¬
ного одноименно с частицами дисперсной фазы. Это объясняют
адсорбцией сопутствующих ионов на поверхности частиц, меж-
IV. 1. КОАГУЛЯЦИЯ ЗОЛЕЙ ЭЛЕКТРОЛИТАМИ
109
ионными взаимодействиями или взаимодействием сопутствующих
ионов с молекулами дисперсионной среды [11].
При коагуляции гидрозоля А1(ОН)3 хлоридными ионами с раз¬
ными сопутствующими катионами выявлен следующий лиотроп¬
ный ряд [6]:
NH4C1> KCl>NaCl.
Увеличение концентрации электролита сверх пороговой
(в 2—10 раз) приводит к существенному (до 70%) возрастанию
адсорбции коагулирующих ионов [12].
Коагуляция наступает обычно не в изоэлектрической точке,
а после снижения заряда частиц до некоторой критической вели¬
чины. Причем адсорбция коагулирующих ионов не превышает
к этому моменту половины предельной величины адсорбции [10].
Значение дзета-потенциала, соответствующее критической вели¬
чине заряда частицы (ДПкр), по одним данным, не зависит от вида
электролита и составляет в среднем +30 мв (см. таблицу), по дру¬
гим — такая зависимость существует.
Критические значения дзета-потенциала частиц
F(‘203 при коагуляции разными электролитами
[13, стр. 289]
Электролит
Концентрация электролита,
мг-экд/л
ДГ^кр» мв
КС1
100,0
+34
NaOH
7,5
+31
K.,S04
6,6
+32
К2Сг04
6,5
+32
K3Fe(CN)e
0,65
+30
Приведенные в таблице значения ДПК„ совпадают со значением,
найденным Мантрой [14].
Величина ДПкр гидрозоля А1(ОН)3, по данным Рэддика,
находится в пределах +15 -=■—[-20 мв [15], а согласно эксперимен¬
там Кульского ь др. [16], она составляет при коагуляции хлори¬
дами +75 мв, сульфатами +25 мв, бикарбонатами +9 мв.
Нельзя, однако, забывать о том, что ДП и потенциал плоскости
наибольшего приближения (срх) — понятия не тождественные
(см. гл. I), и величина ДП не является точной мерой степени
устойчивости частиц. К тому же, как показали расчеты [17],
приблизительное постоянство значения, отвечающего потере час¬
тицей агрегативной устойчивости, должно сохраняться не для
потенциала ф4, а для произведения его критической величины на
валентность коагулирующего иона.
1,10
ГЛАВА IV. ВЗАИМОДЕЙСТВИЕ КОАГУЛИРУЮЩИХ ЧАСТИЦ
IV. 1.1. Физическая теория коагуляции
В нашу задачу не входит обсуждение многочисленных теорий
коагуляции, развитых различными исследователями в конце про¬
шлого века — начале нынешнего. Они представляют лишь исто¬
рический интерес. В настоящее время общепринята физическая
теория коагуляции лиофобных золей Дерягина — Ландау —
Фервея — Овербека [18—22], в которой степень устойчивости
системы определяется из баланса молекулярных и электростатиче¬
ских сил (см. гл. I). Хотя детальная разработка этой теории еще
не завершена, она, благодаря принципиально верной трактовке
роли поверхностных сил разной природы, позволила объяснить
целый ряд коллоидно-химических явлений.
Физическая теория различает два типа коагуляции:
1) концентрационную, при которой потеря устойчивости частиц
связана со сжатием двойного слоя;
2) нейтрализационную, когда наряду со сжатием двойного
слоя уменьшается потенциал срх.
Концентрационная коагуляция характерна для сильно заря¬
женных частиц и высоких концентраций электролита. Чтобы
объяснить механизм концентрационной коагуляции, нужно пом¬
нить, что чем выше потенциал фх, тем сильнее противоионы притя¬
гиваются к поверхности частиц и своим присутствием экранируют
рост электрического поля. Поэтому при высоких значениях ф!
силы электростатического отталкивания между частицами не
возрастают безгранично, а стремятся к некоторому конечному
пределу. Этот предел достигается при фх более 250 мв. Отсюда
следует, что взаимодействие частиц с высоким ф1-потепциалом не
зависит от величины этого потенциала, а определяется только кон¬
центрацией и зарядом противоионов.
По мере увеличения концентрации электролита величина ДП
снижается, а фх практически сохраняет свое значение (рис. IV. 1).
Соответствие между степенью устойчивости и ДП отсутствует.
Если концентрация электролита мала, на результирующей кривой
энергии взаимодействия частиц (см. рис. 1.11) имеется высокий
энергетический барьер. По мере повышения концентрации элек¬
тролита происходит сжатие двойного слоя и радиус действия сил
электростатического отталкивания постепенно уменьшается до
области, где уже достаточно велики силы притяжения и может
произойти коагуляция частиц.
Теория дает следующую величину порога концентрационной
коагуляции:
гЦкТу
<1УЛ>
где СК — константа, слабо зависящая от отношения зарядов ка¬
тиона и аниона электролита; 8 — диэлектрическая проницаемость
IV. 1. КОАГУЛЯЦИЯ ЗОЛЕЙ ЭЛЕКТРОЛИТАМИ
111
м
гп т'
ft
N л п' X
N
л
Рис. IV.1. Взаимосвязь между фгпотенциалом и ДП для сильно заряжен¬
ной частицы (концентрационная коагуляция)
Рис. IV.2. Взаимосвязь между фгпотенциалом и ДП для слабо заряженной
частицы (нейтрализационная коагуляция)
раствора; А — константа, характеризующая молекулярное при¬
тяжение частиц; е — заряд электрона; ?г — валентность противо-
иона.
Из уравнения (IV.1) видно, что порог коагуляции не зависит
от и обратно пропорционален шестой степени валентности
противоионов. Для одно-, двух-, трех- и четырехвалентных ионов
соотношение порогов коагуляции будет равно
1 : 0,016: 0,0013: 0,00024.
Указанное соотношение теоретически обосновывает и подтвер¬
ждает правило Шульце — Гарди. Найденная количественная про¬
порция между пороговыми концентрациями коагулирующих ионов
разной валентности совпадает со многими экспериментальными
данными [23, 24]. Нужно, однако, иметь в виду, что связь между
у и г,, выраженная уравнением (IV.1), представляет предельный
случай. Теория показывает, что при относительно низких значе¬
ниях ДП показатель степени у Z; может снизиться до двух.
Нейтрализационная коагуляция характерна для слабо заря¬
женных частиц. Потеря агрегативной устойчивости обусловлена
ростом адсорбции противоионов и снижением потенциала диф¬
фузного слоя фх.
При невысоких концентрациях электролита, когда толщина
диффузного слоя велика, значения ф! и ДП близки (рис. IV.2).
Поэтому значение ДП при нейтрализационной коагуляции доста¬
точно надежно характеризует степень устойчивости золя.
или
112
ГЛАВА IV. ВЗАИМОДЕЙСТВИЕ КОАГУЛИРУЮЩИХ ЧАСТИЦ
Согласно теории Дерягина, критическая величина потенциала
связана с условиями нейтрализационной коагуляции соотноше¬
нием
где Сн — константа; % — величина, обратная толщине диффуз¬
ного слоя.
Для решения задачи водоочистки важно иметь ответ на вопрос:
в каких случаях преобладает концентрационная и в каких — ней¬
трализационная коагуляция? Мы уже отмечали, что одновалентные
коагулирующие ионы производят относительно слабое сни¬
жение заряда частиц, в то время как двух- и трехвалентные спо¬
собны снизить его в несколько раз. Поэтому коагуляция много¬
валентными ионами проходит, как правило, по нейтрализацион-
ному механизму, а одновалентными — по концентрационному
Результаты исследований последних лет приводят, однако,
к выводу, что даже в случае многовалентных коагулирующих
ионов нарушение агрегативной устойчивости гидрофобных золей
никогда не протекает чисто нейтрализационно. Хотя адсорб¬
ционные явления, приводящие к нейтрализации заряда частиц,
во всех исследованных случаях имели весьма существенное значе¬
ние, важная роль принадлежала и сжатию двойного слоя [26—
IV. 1.2. Специфические явления
В коллоидной химии накоплен богатый экспериментальный мате¬
риал, относящийся к тем или иным частным вопросам коагуляции.
Привлекая его в выборочном порядке, можно обосновать или
опровергнуть фактически любые, даже противоречивые теорети¬
ческие представления. Поэтому к экспериментальному фонду
следует относиться с большой осторожностью.
Наиболее серьезные ошибки в эксперименте возникают из-за
использования авторами различных нестандартных способов при¬
готовления и очистки (диализации) золей, методов физического
и физико-химического анализа. В частности, степень агрегатив¬
ной устойчивости золя Fe(OH)3, часто являющегося объектом
исследования при изучении механизма коагуляции, зависит от
способа его приготовления [30]. Результаты определения ДП
дисперсных частиц связаны со способом измерения их электро¬
форетической подвижности.
Вместе с тем среди обилия экспериментальных данных часто
проступают закономерности обобщающего характера, не уклады¬
вающиеся в рамки привычных теоретических представлений, но
(IV.2)
[17, 25 (стр. 122)].
29].
IV. 1. КОАГУЛЯЦИЯ ЗОЛЕЙ ЭЛЕКТРОЛИТАМИ
ИЗ
Рис. IV.3. Схема чередования
зон устойчивости и неустой¬
чивости отрицательно заря¬
женных частиц золя при
возрастании концентраций (с)
многовалентных катионов
достаточно тщательно проверенные разными исследователями.
Это — особые аномальные или специфические явления, не нашед¬
шие пока отражения в теоретических разработках.
В сфере коагуляции такие специфические явления делятся, по
Воюцкому [13, стр. 300], на четыре основные группы: 1) явления
неправильных рядов; 2) антагонизм и синергизм ионов; 3) привы¬
кание; 4) коллоидная защита и сенсибилизация. В той или иной
форме все названные явления имеют место при очистке воды коагу¬
лянтами и потому заслуживают внимательного рассмотрения.
Явление неправильных рядов состоит в том, что при постепен¬
ном добавлении к золю электролита происходит чередование зон
устойчивости и неустойчивости частиц: сначала наблюдается сни¬
жение устойчивости и коагуляция частиц, затем устойчивость
возрастает и вновь понижается (рис. IV.3). Такое явление вызы¬
вают многовалентные катионы Fe3 + и А13+, большие ионы органи¬
ческих красителей и алкалоидов.
Одно из объяснений явления неправильных рядов состоит
в следующем. При малых количествах электролита многовалент¬
ных коагулирующих ионов недостаточно, чтобы снизить ДП и
довести его значение до критического. С увеличением концентра¬
ции электролит начинает проявлять коагулирующее действие
(зона /, рис. IV.3). Значение ДП в этот момент находится в преде¬
лах между — ДПкр и + ДПкр. При дальнейшем росте концен¬
трации многовалентных ионов ДП частиц сначала вновь стано¬
вится выше критического (зона//), а затем уменьшается (зона III).
Происходит концентрационная коагуляция золя.
Явление неправильных рядов может быть также объяснено
действием не самих коагулирующих ионов, а продуктов их гид¬
ролиза — гидроокисей и основных солей. Действительно, в кислой
среде, когда гидролиз А13+ и Fe3+ не происходит, даже весьма
высокие концентрации катионов не способны вызвать перезарядку
золей [31].
114
ГЛАВА IV. ВЗАИМОДЕЙСТВИЕ КОАГУЛИРУЮЩИХ ЧАСТИЦ
В слабокислой и слабощелочной средах формируются продукты
гидролиза с высоким положительным зарядом и сильной способ¬
ностью к перезарядке частиц золя. Наконец, в области высоких
pH формирующиеся гидроксиды несут отрицательный заряд и
перезарядка частиц не происходит.
Правильность точки зрения на роль гидролиза катионов алю¬
миния и железа подтверждается результатами исследований по
коагуляции водных примесей, на которых мы подробно остановим¬
ся в гл. VI и VII, а также тем обстоятельством, что перезарядка
частиц золя может быть вызвана добавлением и двухвалентных
коагулирующих ионов при условии, что они гидролизуются с об¬
разованием малорастворимых соединений [13, стр. 301].
Антагонизм и синергизм. Поскольку в системе с жидкой дис¬
персионной средой присутствуют ионы-стабилизаторы, процесс
коагуляции при добавке электролита даже в простейшем случае
происходит под влиянием двух видов ионов. Если же средой яв¬
ляется природная вода, содержащая разные электролиты, то при¬
ходится говорить о действии на дисперсную фазу смеси многих
ионов.
В коагулирующем действии смеси электролитов различают
три явления: аддитивность — когда электролиты действуют на
золь независимо один от другого; антагонизм — электролиты как
бы противодействуют один другому и для коагуляции золя их
требуется больше, чем при аддитивном действии; синергизм —
электролиты как бы помогают друг другу и для коагуляции их
требуется меньше, чем в случае аддитивности.
По данным работ [32, 33], отклонения в действии электроли¬
тов от аддитивности в ту или иную сторону (антагонизм и синер¬
гизм) являются следствием либо взаимного электростатического
влияния коагулирующих ионов в объеме раствора и в электриче¬
ском поле коллоидных частиц (для сильно заряженных золей),
либо конкуренцией коагулирующих ионов за адсорбционные
участки поверхности раздела. В некоторых случаях антагонизм
электролитов может объясняться химическим взаимодействием
ионов, в результате которого образуются комплексы с более сла¬
бым коагулирующим действием. В качестве примера такого слу¬
чая приводят коагуляцию золей AgJ и AgBr солями алюминия
[34].
Иногда добавление к золю некоторых количеств электролита,
меньших, чем пороговые, приводит к его застудневанию — обра¬
зованию пространственной сетчатой структуры. Это явление
характерно для золей с анизодиаметрическими частицами и объяс¬
няется тем, что, будучи не полностью астабилнзированными, эти
частицы одними участками своей поверхности сцепляются, дру¬
гими — отталкиваются [2, стр. 405]. Необходимые для застудне¬
вания концентрации электролитов (в ммоль/л) уменьшаются с уве¬
IV.i. КОАГУЛЯЦИЯ ЗОЛЕЙ ЭЛЕКТРОЛИТАМИ Ц5
личением валентности коагулирующих ионов, что может быть
показано на примере золя окиси алюминия:
NaCl 77 К2(СОО)2 0,36
КС1 80 K3Fe(CN)6 0,10
K2SO4 0,28 K4Fe(CN)e 0,08
Привыкание. При постепенном добавлении электролита час¬
тицы золей иногда теряют устойчивость при концентрациях элек¬
тролита, меньших или больших, чем требуется для потери их ус¬
тойчивости при единовременном вводе электролита. Эти явления
получили названия соответственно отрицательного или положи¬
тельного привыкания и объясняются разными авторами по-раз¬
ному.
По Глазману [35], причина отрицательного привыкания
заключается в том, что при постепенном добавлении электролита
каждая его последующая порция действует уже на частично де¬
стабилизированный золь. Согласно же Крестинской [13, стр. 303],
отрицательное привыкание связано с химическими изменениями
в системе. Она допускает, в частности, что при постепенном при¬
бавлении соляной кислоты к золю гидроокиси железа в результате
образования FeCl3 происходит уменьшение количества дисперс¬
ной фазы и одновременное увеличение количества стабилизатора.
Положительное привыкание чаще всего объясняют тем, что
частицы, постепенно выпадающие в осадок, выводят из раствора
адсорбированные коагулирующие ионы. Однако положительное
привыкание золя Fe(OH)3 можно связать и с более высокой сте¬
пенью гидратации частиц при постепенном добавлении электро¬
лита [36].
Защита и сенсибилизация. Как указывалось в гл. I, при до¬
бавлении к дисперсной системе достаточно больших количеств
ВМВ и ПАВ последние образуют на поверхности частиц адсорб¬
ционные слои, которые гидрофилизируют золь, повышают его
устойчивость. Это явление носит название коллоидной защиты.
Коллоидную защиту способны вызывать щелочные мыла и де¬
тергенты, спирты, белки, полисахариды с большим количеством
полярных групп и другие вещества. Защитное действие зависит
от природы и концентрации защитного вещества и золя, степени
дисперсности частиц золя, pH среды, наличия разных примесей.
Наибольшим защитным действием обладают вещества с большим
молекулярным весом и заряженные одноименно с частицами золя
[37]. По данным Овербека, количества разных гидрофильных ве¬
ществ (в мг), способные предотвратить коагуляцию 10 мл золя
Fe(OH)3 при добавлении 1 мл 10%-ного раствора NaCl, следую¬
щие: желатина — 5, яичный альбумин — 15, декстрин — 20,
гуммиарабик — 25, сапонин — 115 [2, стр. 441]. Защитное дей¬
116
ГЛАВА IV. ВЗАИМОДЕЙСТВИЕ КОАГУЛИРУЮЩИХ .ЧАСТИЦ
ствие ПАВ возрастает с увеличением длины неполярной цепи
в их молекулах [38].
Защищенные частицы золей имеют примерно ту же величину
ДП, что и макромолекулы ВМВ. По современным представлениям
коллоидная защита является следствием возникновения или уси¬
ления разных факторов стабилизации: гидратации поверхности
частиц [13 (стр. 305), 39, 40], увеличения ф1-потенциала частиц
[13 (стр. 305), 41], взаимного отталкивания гибких макромолекул,
адсорбированных на частицах золя [42]. Адсорбция защитного
вещества на поверхности частиц продуктов гидролиза многова¬
лентных металлов может сопровождаться химическим взаимодей¬
ствием с образованием в поверхностных слоях гидрофильных ком¬
плексов [43, 44].
Добавление к золю небольших количеств ВМВ, которые не
обеспечивают полное покрытие частиц золя, вызывает явление,
противоположное защите — сенсибилизацию, т. е. повышение
чувствительности золя к действию электролитов [13 (стр. 306),
41, 45]. Явлением сенсибилизации объясняется, в частности, быст¬
рая коагуляция суспензий угля и глин при добавлении крахмала
и извести [2, стр. 442], увеличение сорбции Fe(OH)3 на зернах гра¬
фита в присутствии желатины [44].
Сенсибилизирующее действие ВМВ проявляется независимо от
знака заряда частиц золей, как это видно из данных [6], приведен¬
ных ниже:
Золи Знак заряда Сенсибилизаторы
АЮН)3 -)- Желатина
Fe(OH)3 Крахмал, сапонин, альбумин, гликоген
SiC>2 — Желатина
Каолин — Альбумин
Красители Танин
» _ »
Если макромолекулы сенсибилизатора несут заряд, разно¬
именный с зарядом коллоидных частиц, сенсибилизацию объясняют
как одну из форм взаимной коагуляции коллоидов, а в случае
одноименных зарядов — объединением частиц в агрегаты за счет
возникновения полимерных мостов, т. е. флокуляцией.
Предполагается, что защитное или сенсибилизирующее дей¬
ствие неионогенных 1IAB на частицы кварца и глины связано
с ориентацией макромолекул на поверхности частиц в зависимости
от соотношения в ПАВ гидрофильных и гидрофобных групп [46].
Выполненные недавно Еременко и др. [47] исследования сорбции
неионогенного ВМВ (полиоксиэтилена) на поверхности кварцевых
частиц показали, что действие полимера на величину ДП этих
частиц можно объяснить уменьшением степени диссоциации си-
ланольных групп или молекул воды, связанных с атомами крем¬
IV.2. КОАГУЛЯЦИЯ ЗОЛЕЙ БЕЗ ДОБАВЛЕНИЯ ЭЛЕКТРОЛИТОВ
117
ния координационно, и перемещением положения «плоскости
скольжения» в глубину жидкой фазы.
Исследования по изучению влияния различных красителей на
гидрозоль А1(ОН)3показали, что повышение концентрации основ¬
ных красителей (метиленовый голубой, малахитовый зеленый)
вызывает сначала повышение, затем падение устойчивости золя.
При повышении же концентрации кислых красителей (ализарин,
сульфокислота, метилоранж) устойчивость частиц А1(ОН)3 растет
во всем диапазоне концентраций красителя.
Рассмотренные выше закономерности и специфические явления
относятся главным образом к электролитной коагуляции частиц,
однородных по природе и близких по размеру (гомокоагуляция).
При обработке коагулянтами природной воды, вследствие разно¬
родности состава присутствующих загрязнений и применения
разных технологических приемов для ускоренного отделения коа¬
гулированной взвеси в осадок, имеют место гетерокоагуляция
и гетероадакоагуляция 2.
Коагуляционное взаимодействие между частицами, отличаю¬
щимися по природе, имеет место в основном в том случае, когда
эти частицы несут разноименные заряды. Поэтому такое взаимо¬
действие рассматривали вначале как результат взаимной нейтра¬
лизации зарядов. Однако позже был доказан факт коагуляции
частиц, разнородных по природе, но заряженных одноименно.
Теория взаимодействия разнородных частиц разработана Деря¬
гиным. Согласно этой теории, силовой барьер, возникающий между
заряженными разнородными частицами, зависит только от вели¬
чины заряда частицы, заряженной слабее. Поэтому при появлении
в растворе развитой посторонней поверхности с ДП, близким
к нулю, произойдет коагуляция всей системы в целом, несмотря
на то, что частицы исходного золя останутся устойчивыми в отно¬
шении слипания между собой. Согласно представлениям Пескова
[48], одной из причин потери устойчивости дисперсными системами
в присутствии чужеродной поверхности является адсорбция
стабилизатора на поверхности.
Гетерокоагуляция и гетероадакоагуляция имеют важнейшее
значение в процессе контактного осветления воды.
IV. 2. КОАГУЛЯЦИЯ ЗОЛЕЙ
БЕЗ ДОБАВЛЕНИЯ ЭЛЕКТРОЛИТОВ
Частичная или полная потеря устойчивости золей и суспензий
может быть вызвана не только путем добавления к ним электро¬
литов, но и физическим воздействием интенсивного перемешивания,
2 Гетерокоагуляция — коагуляция частиц разной природы; гетероадако¬
агуляция (гетероадагуляция) — коагуляция частиц на макроповерхно-
стях отличной от частиц природы.
118
ГЛАВА IV. ВЗАИМОДЁЙСТВИЁ КОАГУЛИРУЮЩИХ ЧАСТИЦ
нагревания, замораживания, ультрафиолетового и ионизирующего
излучений, ультразвука, электрического и магнитного полей.
Хотя при очистке воды эти физические методы коагуляции не
имеют пока самостоятельного значения, а применяются только в до¬
полнение к основному — химическому методу, тем не менее оии
позволяют ощутимо улучшить (ускорить) очистку и заслуживают
подробного рассмотрения. Надо, однако, иметь в виду недостаточ¬
ную изученность механизма физических методов астабилизации
золей и чрезвычайно слабую освещенность их в литературе.
Перемешивание, нагревание. Коагуляция в результате интен¬
сивного механического перемешивания золей и суспензий обус¬
ловлена временным нарушением адсорбционного баланса стаби¬
лизатора у поверхности частиц и снижением вследствие этого их
агрегативной устойчивости. Нагревание среды вызывает увеличе¬
ние интенсивности броуновского движения, частичную десорб¬
цию стабилизатора, уменьшение степени гидратации частиц [13
(стр. 311), 49 (стр. 220)].
Замораживание. Коагуляция гидрозолей и гидрогелей под
действием замораживания объясняется постепенным вымерзанием
воды, увеличением вследствие этого концентрации электролитов
в поверхностных слоях частиц, сжатием двойного слоя и сниже¬
нием устойчивости [6, 50—54]. Вымерзание воды может протекать
одновременно по двум механизмам: за счет пленочной диффузии
молекул из слоя коагулята к поверхности ранее образовавшегося
льда и вследствие возникновения центров кристаллизации воды
внутри самого слоя [55]. Частицы, слипшиеся при замораживании,
образуют коагуляционные структуры, сохраняющиеся при отта-
ивани .
На коагулирующее действие замораживания сильное влияние
оказывают состав электролита и температурный режим замора¬
живания. Вольхин и др. [50, 56] показали на примере осажденной
гидроокиси железа, что если температура замораживания (t3)
ниже температуры образования электролитом криогидрата (tK),
электролит затвердевает в прослойках между кристаллами льда
с образованием эвтектики. При этом независимо от исходной кон¬
центрации электролита происходит глубокое промораживание
осадка Fe(OH)3 и его объем после оттаивания заметно уменьшается.
Если же t3 tK, в прослойках между кристаллами льда электро¬
лит достигает такой концентрации, при которой при данной тем¬
пературе вымораживание больше не происходит, сжатие Fe(OH)3
прекращается, а объем осадка после оттаивания тем выше, чем
больше исходная концентрация электролита.
Быстрое замораживание гораздо менее эффективно, чем мед¬
ленное, так как в этом случае вода застывает в виде сплошной
массы и концентрирования электролита вблизи поверхности частиц
не происходит.
IV.2. КОАГУЛЯЦИЯ ЗОЛЕЙ БЕЗ ДОБАВЛЕНИЯ ЭЛЕКТРОЛИТОВ
119
Наложение электрического поля. При наложении на дисперс¬
ные системы электрического поля происходит ориентация частиц
вдоль силовых линий поля и последующая их агрегация.
Объединение частиц в поле переменного тока исследовали
подробно в 1941 г. Кройт и Фогель [57]. Они установили, что боль¬
шое значение имеют поляризационные силы, деформация и раз¬
рушение ионных оболочек мицелл. Относительную длительность
существования агрегатов после прекращения воздействия электри¬
ческого поля авторы объяснили развитием рекристаллизацион-
ных процессов в местах контакта частиц.
Изучение воздействия на суспензии глин поля постоянного
тока [58] показало, что степень ускорения седиментации частиц
под влиянием поля зависит от напряженности электрического
поля и гидратации глинистых частиц. Оптимальным условиям
воздействия поля отвечают значения ДП, близкие к ДПкр. В 50-х
годах ряд работ по структурообразованию дисперсных систем
в поле постоянного тока опубликовали Френкель, Гиндин и др.
[59]. По Гороновскому [60], коагуляция частиц гидроокисей ме¬
таллов в поле постоянного тока связана со следующими эффектами:
электрофоретическим переносом частиц и их последующим разря¬
жением на электродах; возникновением высоких концентраций
коагулирующих ионов; коагулирующим действием ионов, перехо¬
дящих в раствор с электродов; взаимной коагуляцией дисперсных
частиц с частицами, перезарядившимися на электродах.
Согласно современным представлениям, основной причиной
агрегации частиц в электрическом поле следует считать прира¬
щение энергии взаимодействия частиц за счет поляризационной
составляющей [61 (стр. 68), 62, 63]. Поляризуемость частиц объяс¬
няется либо свойствами их материала (поляризация вследствие
разных диэлектрических проницаемостей твердого вещества и
дисперсионной среды), либо перераспределением ионов двойного
слоя и появлением ионной составляющей дипольного момента,
обусловливающей взаимодействие частиц на сравнительно больших
расстояниях, в 2—3 раза превышающих размеры частиц
[64].
Возникающие агрегаты ориентируются своей длинной осью
вдоль силовых линий электрического поля и имеют форму цепочек
длиной 150—200 мкм [65, 66].
Поляризационное взаимодействие вносит ощутимый вклад
в процесс объединения частиц в агрегаты уже при напряженно¬
стях электрического поля в несколько вольт на 1 см [67]. Интен¬
сивность образования агрегатов растет с увеличением концен¬
трации суспензии, напряженности поля и с уменьшением (до
некоторого предела) частоты переменного тока [66]. Вследствие
агрегации частиц повышается вероятность их взаимодействия с
другими частицами [68, 69].
120
ГЛАВА IV. ВЗАИМОДЕЙСТВИЕ КОАГУЛИРУЮЩИХ ЧАСТИЦ
Коагуляция дисперсий может быть вызвана не только дей¬
ствием переменного и постоянного электрических полей, но и
электрического разряда малой мощности. Электрический разряд,
по данным Кожемякина и др. [70, 71], действует в широком диа¬
пазоне концентраций суспензий и коагулирует устойчивые сус¬
пензии глины и кварцевого песка с образованием быстро оседаю¬
щих агрегатов размером до 2 мм. Снижение устойчивости частиц
объясняется поляризацией ионных сфер или материала частиц.
Наложение магннтного поля. Несмотря на большой объем
исследований, проведенных по магнитной обработке дисперсных
систем, до сих пор не выработано единой точки зрения на меха¬
низм воздействия магнитного поля. Само же воздействие носит
большей частью избирательный характер, проявляется по-раз-
ному и сильно зависит от напряженности поля.
Известно, что молекулы воды обладают определенной магнит¬
ной восприимчивостью и под влиянием магнитного поля способны
изменять свой магнитный момент. Об изменении свойств воды,
подвергнутой магнитной обработке (ее электропроводности, вяз¬
кости), свидетельствуют работы многих авторов [72—78]. Эти
изменения связывают со структурными явлениями, разобщением
существующих [79, 80] или возникновением новых [74] ассоциатов
молекул, в результате чего смачиваемость твердых поверхностей
уменьшается [81, 82].
По данным некоторых авторов, в растворах электролитов под
действием магнитного поля происходит дегидратация ионов и
возникают ионные ассоциаты [83—85]. Причем предполагается
также переход ионов на более высокую ступень ионизации [86].
Эти явления в свою очередь вызывают уменьшение гидратации
и ДП коллоидных и суспендированных частиц, изменение степени
взаимного сцепления агрегатов в осадках, облегчают образование
зародышей новой фазы [79, 82, 83, 87—90]. Последнее обстоятель¬
ство используется для борьбы с накипеобразованием в котлах.
При наличии в воде солей, способных к гидролизу, увеличение
диэлектрической проницаемости вследствие ориентационной и
упругой поляризации молекул воды приводит, по мнению Бата¬
лина [78], к ускорению гидролиза и образованию центров конден¬
сации новой фазы.
Магнитная обработка водных растворов и суспензий далеко не
всегда, однако, приводит к желаемым результатам [91, 92]. Мно¬
гое зависит от режима обработки, напряженности поля, свойств
материала твердой фазы, электролитного состава воды. Наиболее
устойчивые результаты достигаются при воздействии магнитного
поля на частицы ферромагнетиков. Структурообразование в вод¬
ных суспензиях железа и окиси железа описано Гервеем [93].
Лавров и др. [61, 63] подробно изучили влияние магнитного поля
(напряженностью до 1600 а/см) на скорость коагуляции и осажде-
IV.2. КОАГУЛЯЦИЯ ЗОЛЕЙ БЕЗ ДОБАВЛЕНИЯ ЭЛЕКТРОЛИТОВ
121
иия частиц магнетита в воде и установили с помощью прямых
микроскопических наблюдений факт ориентации частиц вдоль
силовых линий. По данным Зубарева [94], наложение магнитного
поля на броуновское движение анизодиаметрических частиц при¬
водит к их ориентации длинной осью- перпендикулярно силовым
линиям. Ориентация сопровождается взаимодействием частиц
на дальних расстояниях и возникновением периодических колло¬
идных структур [61, стр. 82].
В ряде работ изучалось действие магнитного поля на водные
суспензии глин. Осипов [95], исследуя структурирование суспен¬
зий монтмориллонита, каолинита и гидрослюды, объяснил замет¬
ное увеличение вязкости этих суспензий при циклическом намаг¬
ничивании присутствием в глинах ферромагнетика Fe203. Действи¬
тельно, монтмориллонит и гидрослюда, в которых содержание
Fe203 достигает 4%, реагировали на действие магнитного поля
сильнее, чем каолинит, включающий лишь 0,6—1,1% Fe203.
Как выяснилось в последнее время, эффективность магнитной
обработки воды в борьбе с накипеобразованием сильно зависит
от содержания в воде соединений железа. Наилучшие результаты
получены при оптимальной концентрации железа, величина ко¬
торой зависит от pH [96].
На существенную роль ориентационного эффекта в дополнение
к дегидратирующему действию магнитного поля указывают
Круглицкий и др. [96—98], исследовавшие структурообразование
высококонцентрированных суспензий монтмориллонита и каоли¬
нита. Однако вряд ли можно считать, что при коагуляции глини¬
стых примесей ориентационному эффекту принадлежит ведущая
роль. Это показывают, в частности, опыты, в которых авторы
сначала подвергали магнитной обработке дистиллированную воду
и лишь затем добавляли к ней тонкоизмельченные каолин или
глинистый сланец; предварительное омагничивание воды интен¬
сифицировало агрегацию и седиментацию частиц [99]. Высокая
технологическая эффективность предварительно проведенной
магнитной обработки воды показана Шаховым и другими на при¬
мере коагуляции гидроокисей алюминия и железа (см. гл. VIII).
Воздействие ультразвуком. Коагулирующее действие ультра¬
звука на дисперсные системы объясняют чаще всего увеличением
интенсивности диффузии частиц и их скоплением в местах пучно¬
стей звуковых волн. Как и в случае механического перемешива¬
ния, увеличение диффузии вызывает нарушение сферической сим¬
метрии двойного слоя частиц и возникновение дипольного момента
[100].
Егоров [101, 102] установил, что при действии ультразвука на
суспензию кварца с поверхности твердой фазы десорбируются
потенциалопределяющие ионы и причиной коагуляции частиц
является не только их направленное движение, но и изменение
122
ГЛАВА IV. ВЗАИМОДЕЙСТВИЕ КОАГУЛИРУЮЩИХ ЧАСТИЦ
поверхностного потенциала ф0. Десорбция потенциалопределяю-
щих ионов подтверждена увеличением электропроводности дис¬
персионной среды после осаждения частиц кварца и некоторым
снижением ее pH.
Способность к скоплению в местах пучностей звуковых волн
зависит от плотности и размера частиц. Чем крупнее частицы или
чем выше их плотность по сравнению с плотностью дисперсион¬
ной среды, тем выше вероятность скопления. В водных дисперсиях
заметного скопления мелких коллоидных частиц не происходит.
Эффективность ультразвука особенно велика при коагуляции
аэрозолей, где разница в плотностях частиц и среды особенно
велика [65, 100].
Иногда коагулирующее действие ультразвука связывают с воз¬
никновением кавитации — образованием и последующим замыка¬
нием пустот в жидкости, формирующихся вследствие высоких
разрывающих напряжений. Однако опыт показывает, что коа¬
гулирующее действие ультразвук оказывает при интенсивностях
озвучивания 0,3—0,5 вт/см2, тогда как для возбуждения кавита¬
ции в воде необходима интенсивность от 2—3 до нескольких тысяч
ватт на квадратный сантиметр. По-видимому, эффект состоит
в том, что мелкие частицы в ультразвуковом поле докавитацион-
ной мощности увлекаются водой и, попадая в зону действия сил
притяжения крупных частиц, взаимодействуют с ними.
Отмечают возможную роль некоторых частных последствий
ультразвуковой обработки, таких, как дезагрегация ВМВ и хими¬
ческое изменение их макромолекул [103], снижение pH воды, обра¬
зование Н202 [104].
С увеличением до некоторого предела интенсивности и дли¬
тельности озвучивания коагулирующее действие ультразвука
усиливается, но затем может произойти диспергирование образо¬
вавшихся агрегатов [105—107]. Согласно наблюдениям Круглиц-
кого и др. [108], при ультраозвучивании водных суспензий као¬
линита в течение одной минуты происходит разрыв связей между
дисперсной фазой и дисперсионной средой и разрушение первич¬
ных агрегатов суспензий. Затем начинается образование более
Компактных и прочных агрегатов, которые после нескольких минут
озвучивания также разрушаются. Возникает эластичная коагуля¬
ционная структура с упорядоченным строением сетки. По данным
Лычникова и др. [109], максимальный коагуляционный эффект
действия ультразвука на суспензию иллитовой глины наблюда¬
ется при определенном соотношении размеров частиц и длины
волны и может быть объяснен увеличением; вероятности взаимо¬
действия частиц в первичном энергетическом минимуме.
Коагулирующее действие ультразвука зависит также от час¬
тоты и амплитуды колебаний.
Ионизирующие излучения. Возможность применения иони¬
IV. ЛИТЕРАТУРА
123
зирующих излучений для коагуляции дисперсий широко изуча¬
ется, однако единое мнение о механизме действия излучений до
сих пор отсутствует. Исследования влияния ионизирующей ради¬
ации на дзета-потенциал золей железа, меди, серебра и золота
показали, что радиация вызывает изменение ДП частиц золей и
соответственно — изменение порога их коагуляции хлористым
кальцием. Степень изменения ДП зависит от размера частиц,
природы золей и дозы радиации [110].
По данным Нанобашвили и др. [111], у-облучение золей, в том
числе гидрозоля Fe(OH)3, влияет на их электропроводность, pH,
оптическую плотность, но при наличии в растворе органических
защитных веществ (агар-агара, желатины) таких изменений не
происходит.
Облучение золя гидроокиси алюминия нейтронами и у-лучами
усиливает адсорбцию на А1(ОН)3 фосфатов, но ухудшает адсорб¬
цию метиленового голубого [112]. Облучение сопровождается
потерей части воды и глубокими структурными изменениями гид¬
роокиси [113]. По данным Грюна [114], у-облучение не влияет на
устойчивость золя А1203, но вызывает коагуляцию частиц Fe203.
Предполагается [115, 116], что первичными радиолитическими
продуктами при у-облучении гидрозолей являются гидратирован¬
ный электрон и свободный радикал ОН. Электроны нейтрализуют
Н+-ионы, стабилизирующие мицеллы (например, Се(ОН)4), что
приводит к снижению их стабильности и коагуляции.
Помимо прямого физико-химического действия на двойной слой
коллоидных частиц, сопровождающегося уменьшением ДП, облу¬
чение повышает скорость коагуляции за счет разрушения неко¬
торых трудноокисляющихся органических веществ [117—121].
Скорость осаждения частиц порошков минералов, примешанных
к предварительно обработанной ^-излучателями дистиллирован¬
ной воде, увеличилась для флюорита в 3—4, кальцита — в 2—3,
угля и боксита — в 1,5—2 раза [122].
Литерат ура
1. Ф. В. Чухров. Коллоиды в земной коре. М., Изд-во АН СССР, 1955.
2. Г. Р. Кройт. Наука о коллоидах, т. 1. Необратимые системы. М., ИЛ,
1955.
3. В. М. Муллер, Б. В. Дерягин. ДАН СССР, 176, 1111 (1967).
4. С. А. Вознесенский. Физико-химические процессы очистки воды. М.— Л.,
Стройиздат, 1934.
5. К. Chandra, S. Ghosh. Rev. Phys. Chem. Japan, 33, 31 (1963).
6. А. В. Думанский. Учение о коллоидах. М., Госхимиздат, 1948.
7. F. Krleza. Glasnik hem i technol. (BiH), 15, 77 (1967).
8. А. П. Руцков. Коллоидн. ж., 15, 288 (1953).
9. Г. П. Майтак. Коллоидн. ж., 28, 689 (1966).
10. В. М. Барбой, Ю. М. Глазман, И. М. Дыкман. Пятая Всесоюзная кон¬
ференция по коллоидной химии (тезисы докладов). М., Изд-во АН СССР,
1962, стр. 39.
124
ГЛАВА IV. ВЗАИМОДЕЙСТВИЕ КОАГУЛИРУЮЩИХ ЧАСТИЦ
11. С. Т. Телешов. Там же, стр. 43.
12. А. А. Баран, Ю. М. Глазман, Д. Н. Стражеско. Коллоидн. ж., 26,
278 (1964).
13. С. С. Воюцкий. Курс коллоидной химии. М., «Химия», 1975.
14. N. Maitra. J. Indian Chem. Soc., 44, 521 (1966).
15. Т. M. Riddick. J. Amer. Water Works Assoc., 53, 1007 (1961).
16. Л. А. Кулъский, А. М. Когановский, И. Т. Гороновский и др. Физико¬
химические основы очистки воды коагуляцией. Киев, Изд-во АН УССР,
1950, стр. 39.
17. Ю. М. Глазман. Пятая Всесоюзная конференция по коллоидной химии
(тезисы докладов). М., Изд-во АН СССР, 1962, стр. 30.
18. Б. В. Дерягин. Изв. АН СССР, ОХН, 1937, 1153.
19. Б. В. Дерягин. Коллоидн. ж., 6, 291 (1940).
20. Б. В. Дерягин. Коллоидн. ж., 7, 285 (1941).
21. Б. В. Дерягин, JI. Д. Ландау. ЖЭТФ, 15, 662 (1945).
22. В. V. Derjaguin, L. D. Landau. Acta Phys. Chim. URSS, 14, 633 (1941).
23. H. М. Кудрявцева. Автореферат канд. дисс. М., ИФХ АН СССР, 1963.
24. A. S. Teot. Ann. New York Acad. Sci., 155, 593 (1969).
25. А. Г. Пасынский. Коллоидная химия. М., «Высшая школа», 1968.
26. В. М. Барбой, 10. М. Глазман. Сб. «Исследования в области поверхност¬
ных сил». М., «Наука», 1967, стр. 207.
27. Е. Ф. Желъвис, Ю. М. Глазман. Коллоидн. ж., 29, 196 (1967).
28. В. М. Барбой. Коллоидн. ж., 27, 643 (1965).
29. В. М. Барбой, Ю. М. Глазман, И. М. Дыкман. Коллоидн. ж., 24,
382 (1962).
30. К. С. Nand, S. Ghosh. Proc. Nat. Acad. Sci. India, A34, 78 (1964).
31. E. Matijevic, G. E. Janauer, M. Kerker. J. Colloid. Sci., 19, 333(1964).
32. Ю. М. Глазман, И. М. Дыкман, Е. А. Стрельцова. Коллоидн. ж., 20,
149 (1958).
33. Ю. М. Глазман, И. М. Дыкман. Коллоидн. ж., 18, 13 (1956).
34. L. J. Stryker, Е. Matijevic. J. Phys. Chem., 73, 1484 (1969).
35. Ю. М. Глазман, Б. Э. Тартаковская. Коллоидн. ж., 11, 299 (1949).
36. 10. А. Клячко, II. П. Кондратюк. Зав. лаб., № 8, 901 (1941).
37. С. А. Балезин, Б. Ф. Ерофеев, II. И. Подобаев. Основы физической и
коллоидной химии. М., «Просвещение», 1975, стр. 362.
38. М. Е. Краснокутская, Ю. М. Глазман. Сб. «Исследования в области
поверхностных сил». М., «Наука», 1967, стр. 219.
39. Y. М. Glazman. Disc. Faraday Soc., 4, 255 (1966).
40. Ю. М. Глазман, И. П. Сапон. Сб. «Исследования в области поверх¬
ностных сил». М., «Наука», 1964, стр. 258.
41. А. В. Думанский. ЖФХ, 1, 481 (1930).
42. С. С. Воюцкий. Коллоидн. ж., 23, 353 (1961).
43. М. Е. Шишниашвили, А. Л. Бацанадзе, Л. Н. Одилавадзе. Пятая Все¬
союзная конференция по коллоидной химии (тезисы докладов). М.,
Изд-во АН СССР, 1962, стр. 45.
44. Л. Д. Скрылев, Л. Н. Калитина. Коллоидн. ж., 27, 876 (1965).
45. R. Н. Ottewill, A. Watanable. Kolloid Z., 170, 38 (1960).
46. И. II. Кравченко, Б. И. Генкина. Коллоидн. ж., 29, 206 (1967).
47. Б. В. Еременко, Б. Э. Платонов, И. А. У сков и др. Коллоидн. ж., 36,
250 (1974).
48. Н. П. Песков. ЖРХО, 49, 1 (1917).
49. Н. П. Песков, Е. М. Александрова-Прейс. Курс коллоидной химии.
М.— «П., Госхимиздат, 1948.
50. В. В. Вольхин, В. Л. Золотавин. Коллоидн. ж., 23, 134 (1961).
51. В. Л. Золотавин, В. В. Волъхин. Там же, стр. 276.
52. В. Л. Золотавин, В. В. Волъхин. Коллоидн. ж., 22, 305 (1960).
IV. ЛИТЕРАТУРА
125
53. В. В. Волъхин. Сборник научных трудов Пермского политехнического
института, вып. 71, 47 (1970).
54. Е. И. Пономарев, В. В. Волъхин, В. Л. Золотавин. Там же, стр. 64.
55. В. В. Волъхин, Е. И. Пономарев, В. Л. Золотавин. Коллоидн. ж., 35,
144 (1973).
56. В. М. Любарский. Водоснабжение и канализация, вып. 14. М., изд.
ЦБТИ МЖКХ, 1969, стр. 67.
57. R. Kruyt, G. G. Vogel. Kolloid. Z., 95, 2 (1941).
58. Б. Г. Запрометов, К. С. Ахмедов. Коллоидн. ж., 7, 563 (1941).
59. Л. Г. Гиндин, А. Е. Волъпян. Успехи химии, 37, 130 (1968).
60. И. Т. Гороновский. Канд. дисс. Киев, ИОНХ АН УССР, 1955.
61. И. Ф. Ефремов. Периодические коллоидные структуры. JT., «Химия»,
1971.
62. М. Г. Грановский, И. С. Лавров, О. В. Смирнов. Электрообработка жид¬
костей. JI., «Химия», 1976, стр. 30.
63. В. М. Корнилов, И. С. Лавров, О. В. Смирнов и др. ЖПХ, 48, 357
(1975).
64. А. Е. Ковалев, И. С. Лавров. Коллоидн. ж., 32, 63 (1970).
65. II. А. Фукс. Механика аэрозолей. М., Изд-во АН СССР, 1955.
66. Т. А. Воробьева, И. Н. Влодавец. Коллоидн. ж., 31, 668 (1969).
67. О. Г. Усьяров, И. С. Лавров и др. Коллоидн. ж., 28, 596 (1966).
68. Е. Е. Бибик, И. С. Лавров и др. Коллоидн. ж., 30, 494 (1968).
69. И. С. Лавров. Коллоидн. ж., 23, 423 (1961).
70. В. Г. Кожемякин, И. С. Лавров, О. В. Смирнов и др. ЖПХ, 42, 1903
(1969).
71. В. Г. Кожемякин, И. С. Лавров и др. Санитарная техника. Доклады
XXVII научной конференции ЛИСИ (1—9 февр. 1969). Л., 1969, стр. 14.
72. В. И. Миненко, С. М. Петров, М. Н. Минц. Магнитная обработка во¬
ды. Харьк. кн. изд-во, 1962.
73. II. К. Лопарев. О физических безреагентных методах водоподготовки.
Изд. Ленингр. Дома научно-техн. пропаганды, 1959.
74. О. И. Молоканов, А. П. Сальников, В. И. Осенев. Сб. «Вопросы теории
и практики магнитной обработки воды и водных систем». М., изд. ЦНИИ
информ. и техн.-эконом. исслед. цвет, металлургии, 1971, стр. 87.
75. В. И. Классен. Там же, стр. 5.
76. Э. В. Миллер, В. И. Классен, А. Д. Кущенко. Там же, стр. 59.
77. Д. Ф. Файзуллаев, С. Джурабеков и др. ДАН УзбССР, № 8, 13 (1968).
78. Б. С. Баталин. Сборник научных трудов Пермского политехнического
института, вып. 108, 116 (1972).
79. Е. Ф. Тебенихин, Б. Т. Гусев. Сб. «Водоподготовка и внутрикотловые про¬
цессы», вып. 2. М.— Л., Госэнергоиздат, 1963, стр. 41.
80. С. А. Брунс, В. И. Классен, А. К. Коньшина. Коллоидн. ж., 28, 153
(1966).
81. С. В. Щербакова. Труды Иркутск, политехи, ин-та, вып. 46, 23 (1969).
82. В. И. Классен, В. И. Литовко, Э. И. Русская. Коллоидн. ж., 33, 366 (1971).
83. А. И. Шахов, С. С. Душкин. Сб. «Коммунальное хозяйство», вып. 1.
Киев, «Буд1вельник», 1964, стр. 102.
84. О. И. Мартынова, Е. Ф. Тебенихин, Б. Т. Гусев. ЖПХ, 41, 2782 (1968).
85. В. С. Духанин, Н. Г. Ключников. ЖПХ, 46, 1204 (1973).
86. D. С. Avampato. Пат. США 3511776 (1970).
87. А. И. Шахов, С. С. Душкин. Сб. «Энергетика и электротехническая про¬
мышленность», № 1 (17). Киев, 1964, стр. 54.
88. А. И. Шахов, С. С. Душкин. Сб. «Наука и техника в городском хозяй¬
стве», вып. 5. Киев, «Буд1вельник», 1966, стр. 147.
89. В. И. Классен, В. И. Литовко, Э. II. Русская. Сб. «Вопросы теории и
практики магнитной обработки воды и водных систем». М., изд. ЦНИИ
информ. и техн.-эконом. исслед. цвет, металлургии, 1971, стр. 203.
126
ГЛАВА IV. ВЗАИМОДЕЙСТВИЕ КОАГУЛИРУЮЩИХ ЧАСТИЦ
90. А. И. Шахов, А. В. Ширяева, А. С. Аветисов. Изв. вузов. Энергетика,
№ 12, 112 (1966).
91. К. Szakall, М. Nyilassy. Hidrol. Kozl., № 10, 477 (1965).
92. Г. Г. Вудрова, А. Д. Сорокин, П. И. Подрезов. Уголь Украины, № 6,
51 (1972).
93. Е. N. Harvey. J. Colloid Sci., 8, 543 (1953).
94. В. А. Зубарев. ЖФХ, 46, 2635 (1972).
95. Ю. В. Осипов. Коллоидн. ж., 28, 713 (1966).
96. В. А. Кишиневский, Е. Ф. Тебенихин. Труды Моск. энергетического
ин-та, вып. 238, 89 (1975).
97. В. И. Оробченко, Ф. Д. Овчаренко, Н. Н. Круглицкий. Коллоидн. ж.,
29, 836 (1967).
98. Н. Н. Круглицкий, В. И. Оробченко и др. ДАН СССР, 179, 151 (1968).
99. В. И. Классен, Ю. 3. Зиновьев. Коллоидн. ж., 29, 758 (1967).
100. Л. Бергман. Ультразвук и его применение в науке и технике. М., ИЛ,
1957.
101. П. А. Егоров. Коллоидн. ж., 30, 363 (1968).
102. П. А. Егоров. Коллоидн. ж., 33, 834 (1971).
103. И. И. Жуков. Избранные труды. М., Изд-во АН СССР, 1952, стр. 479.
104. А. Г. Лифшиц. Труды ЦНИИ хлопчатобумажной промышленности,
часть 4. М., «Легкая индустрия», 1971, стр. 81.
105. В. П. Рюмин. Автореферат канд. дисс. МХТИ им. Д. И. Менделеева,
1964.
106. В. В. Минченко, С. П. Ничипоренко, Н. Н. Круглицкий. Сб. «Физико¬
химическая механика и лиофильность дисперсных систем». Киев,
«Наукова думка», 1968, стр. 151.
107. М. П. Воларович, И. И. Лиштван и др. Коллоидн. ж., 30, 677 (1968).
108. II. Н. Круглицкий, В. В. Симуров и др. ДАН СССР, 164, 861 (1965).
109. Д. С. Лычников, Г. А. Мартынов. ДАН СССР, 167, 855 (1966).
110. W. М. Smith, О. J. Ledbetter. Water and Sewage Works, 114, 496 (1967).
111. E. М. Нанобашвили, И. Г. Бахтадзе. Пятая Всесоюзная конференция
по коллоидной химии (тезисы докладов). М., Изд-во АН СССР, 1962,
стр. 201.
112. L. Jacimovic, S. Velicovic, N. Ajdacic. Radiochim. acta, 12, 49 (1971).
113. В. H. Крюков, С. И. Кузнецов и др. ЖПХ, 44, 909 (1971).
114. W. N. Grune. Water and Sewage Works, 112, 262 (1965).
115. M. Haissinsky. J. chim. phys. et phys.-chim. biol., 68, 545 (1971).
116. D. S. Ballantine. Isotop. and Radiat. Technol., 8, 415 (1971).
117. F. D. Miraldi, E. J. Morgan. Пат. США 3603788 (1971).
118. M. Gerrard. Isotop. and Radiat. Technol., 8, 429 (1974).
119. D. S. Ballantine. Ibid., p. 445.
120. D. S. Ballantine, L. A. Miller. Chem. EngngProgr. Symp. Ser., 66, 187
(1970).
121. D. D. Woodbridge, L. A. Mann, W. R. Garrett. Bull. Environ. Contam.
and Toxicol., 7, 80 (1972).
122. В. И. Классен, II. Г. Малышева и др. Коллоидн. ж., 31, 628 (1969).
Глава V
КИНЕТИКА КОАГУЛЯЦИИ
Важнейшие условия успешного протекания хлопьеобразования —
создание оптимальных гидродинамических условий смешения и
более или менее длительного перемешивания коагулянта с обра¬
батываемой водой. Первый этап смешения иногда называют быст¬
рым или скорым перемешиванием, второй — медленным или
флокуляционным.
В учебниках и руководствах по очистке воды часто указывают,
что основное назначение быстрого перемешивания коагулянта
с водой — как можно более полное и быстрое распределение реа¬
гента в массе воды. Как свидетельствуют многочисленные наблю¬
дения, роль быстрого перемешивания на этом далеко не исчерпы¬
вается. В гетерогенном процессе, каковым является зародыше-
образование и формирование частиц малорастворимых продуктов
гидролиза коагулянта, характер и интенсивность перемешивания
накладывают сильный отпечаток на физические свойства образу¬
ющихся впоследствии хлопьев коагулированной взвеси [1—4].
Поэтому быстрое перемешивание следует рассматривать прежде
всего как первый и очень важный этап образования новой фазы.
Приведем несколько примеров:
1) для достижения одного и того же эффекта осветления с по¬
мощью растворов коагулянтов разной концентрации (от 0,05
до 20%) требуется разная интенсивность и длительность смешения
этих растворов с водой [5—8];
2) правильный выбор режима смешения приводит к экономии
коагулянта [6, 9];
3) результаты коагулирования зависят от промежутка времени
между моментом ввода коагулянта и началом быстрого переме¬
шивания [6, 10].
Заметное влияние режима последующего медленного переме¬
шивания объясняется сильной зависимостью скорости коагуляции
от числа столкновений коагулирующих частиц. Коэффициент
молекулярной диффузии в жидкости невелик, и скорость гетеро¬
генных реакций, имеющих промышленное значение, в большой
мере определяется гидродинамическими условиями. Уже при
самых незначительных скоростях потока перенос вещества начи¬
нает преобладать над молекулярной диффузией. Однако в лами¬
нарном потоке механизм переноса остается таким же, как и в не¬
подвижной среде [11]. При турбулентном режиме движения пере¬
128
ГЛАВА V. КИНЕТИКА КОАГУЛЯЦИИ
нос массы происходит вследствие беспорядочного перемещения
малых объемов жидкости и коэффициент турбулентной диффузии
превосходит коэффициент молекулярной диффузии в тысячи раз.
V. 1. ПОНЯТИЕ О БЫСТРОЙ И МЕДЛЕННОЙ КОАГУЛЯЦИИ
В общем случае скорость коагуляции Системы зависит от концен¬
трации электролита, определяющей степень устойчивости частиц
и эффективность их взаимных столкновений. Эта зависимость
выражается графиком, показанным на рис. V.I. При малых кон¬
центрациях электролита с эффективность соударений частиц -ф,
т. е. отношение числа столкновений, окончившихся слипанием,
к общему числу столкновений, близка-к нулю. По мере роста с
скорость коагуляции v увеличивается, однако еще не все столкно¬
вения эффективны (Ocilx^l). Такую коагуляцию называют
медленной [12]. При я|з = 1 энергетический барьер между сталки¬
вающимися частицами исчезает и наступает быстрая коагуляция —
все столкновения частиц заканчиваются образованием агрегатов.
Зависимость v = / (с) теоретически и экспериментально иссле¬
довали Глазман и др. [13], Бхаттачария и Кумар [14, 15], Шри-
вастава и Эброль [16] и другие. Наибольшее практическое приме¬
нение получило эмпирическое уравнение
c-a = ^Ti> (V-1)
дающее в диапазоне исследованных концентраций с электролита
линейную связь между с и длительностью периода времени т,
прошедшего от момента добавления электролита до момента за¬
вершения коагуляции (а, т и п — константы). Проверка этого
уравнения на примере коагуляции золя А1(ОН)3 хлоридом, суль¬
фатом и фосфатом натрия дала положительные результаты [17].
Согласно теории Смолуховского, скорость быстрой коагуляции
золя зависит от начальной концентрации частиц п0, интенсивно¬
сти их броуновского движения и радиуса действия сил притя¬
жения. Для скорости увеличения числа пх агрегатов частиц Смо-
луховский получил следующее выражение, соответствующее ре¬
акции второго порядка:
dnjdx =» К (п0 - пх)2, (V.2)
в котором К — константа скорости коагуляции.
Обозначив п0 — пх через пт — количество частиц в единице
объема воды по прошествии времени т, получим
dnjnl = —Kdr, (V.3)
или после интегрирования в пределах от п0 до пх и от 0 до т:
• (V-4)
V.l. ПОНЯТИЕ О БЫСТРОЙ И МЕДЛЕННОЙ КОАГУЛЯЦИИ
129
Рис. V.I. Зависимость скоро¬
сти коагуляции (v) от концен¬
трации электролита (с)
В уравнениях (V.2) — (V.4) константа скорости коагуляции
определяется выражением
К ж 4nDR ж 8nDr, (V.5)
в котором D — коэффициент диффузии одиночных частиц; г —
радиус частиц; R — расстояние, на которое должны приблизиться
частицы, чтобы произошло их объединение (предполагается ра¬
венство R ж 2г, т. е. существование короткодействующих сил при¬
тяжения).
Чтобы исключить из уравнения (V.5) размер частиц, Смолу
ховский ввел новое понятие — время коагуляции Ti/2, в течение
которого количество частиц в единице объема золя уменьшается
вдвое. Уравнение (V.4) приняло вид:
=
по
гмг* (V'6>
Теория Смолуховского рассматривает одновременное столк¬
новение лишь двух частиц и не учитывает возможность коллек¬
тивных взаимодействий, возрастающую с увеличением исходной
концентрации частиц и объема образующихся агрегатов [18, 19
(стр. 393)]. Тем не менее она находит экспериментальное подтвер¬
ждение [20 (стр. 262), 21, 22].
Для описания медленной коагуляции Смолуховский ввел
в уравнение (V. 6) коэффициент эффективности сближений (столк¬
новений) частиц:
п — 52 (V.6')
l+lJ)(T/Т1 j/2)
Фукс [23] дал позднее теоретическое истолкование коэффициента
т|з, объяснив медленную коагуляцию аэрозолей как следствие
остаточного энергетического барьера, высота которого тем больше,
чем крупнее агрегаты частиц. Дерягин [24—27] успешно перенес
эти представления на область лиофобных золей.
Приведенные выше рассуждения относятся к монодисперсным
системам. Вместе с тем еще до появления работ Смолуховского
Вигнер [28] и другие исследователи отмечали, что в реальных
5 Е. Д. Бабенков
130
ГЛАВА V. КИНЕТИКА КОАГУЛЯЦИИ
полидисперсных — системах коагуляция протекает быстрее: круп¬
ные частицы способствуют коагуляции более мелких. Вигнер впер¬
вые дал определение двум видам или двум стадиям коагуляции:
перикинетической, протекающей при броуновском движении час¬
тиц, и ортокинетической, в которой транспортировка частиц
должна обеспечиваться искусственным перемешиванием, так как
увеличившиеся размеры не позволяют частицам участвовать
в тепловом движении.
Частным случаем ортокинетической коагуляции является
гравитационная коагуляция, протекающая вследствие разницы
в скоростях осаждения частиц полидисперсной системы. В ре¬
зультате гравитационной коагуляции образуются дождевые капли
и снежные хлопья. Немаловажное значение она имеет и в процес¬
сах разделения суспензий отстаиванием (см. гл. VI).
В 20-х годах Мюллер [19, стр. 401], применив теорию Смолу¬
ховского к полидисперсным золям, объяснил наблюдавшийся
в экспериментах автокаталитический характер протекания коа¬
гуляции. В отличие от монодисперсной системы, где вероятность
столкновения частиц учитывается произведением ADR [уравнение
(V.5)], в системе из двух видов частиц, заметно отличающихся по
своим размерам и г2, эта вероятность пропорциональна,по Мюл¬
леру, величине
[4 + (Yгг/г2 — Yг2/гх)2] DR,
а изменение числа частиц в ходе коагуляции описывается урав¬
нением
N* + ~ 1 + т/тт [4 + (ф*п + 1) (1 + т/Г1/2)ф -1] ’ (V’7)
в котором пt — число мелких частиц и агрегатов, образуемых
только мелкими частицами в единице объема золя ко времени т;
N, — число крупных частиц и агрегатов, включающих, по край¬
ней мере, одну крупную частицу в единице объема золя к этому
же времени; Ф = + 1)/2kr — фактор Мюллера, определяющий
степень бидисперсности системы; kT = rx/r2; кп = NJn0-, N0 и
п0 — начальная концентрация соответственно крупных и мелких
частиц.
Результаты расчетов показывают, что выражение в квадрат¬
ных скобках уравнения (V.7) приводит к существенному отклоне¬
нию от уравнения Смолуховского при значениях кт и кп более
десяти.
Экспериментальная проверка теории Мюллера дала положи¬
тельные результаты [19 (стр. 402), 29]. В таблице приведены зна¬
чения констант коагуляции частиц бидисперсных аэрозолей, под¬
считанные Фуксом [20, стр. 266] по уравнениям Мюллера.
V.l. ПОНЯТИЕ о быстрой и медленной коагуляции
131
Константы скорости коагуляции (хЮ10 см3/сек) частиц радиусом \
[П, мм
г2, мм
10-*
10“5
Ю-*
ю-»
10-2
10-6
4,5
10-6
90
12
10"4
5 • 103
80
5,2
10-3
7,7 -104
840
16
3,2
10-2
8 • 105
8,5 -103
140
9,6
3,0
Из результатов расчетов видно, какое большое влияние на
скорость коагуляции системы оказывает распределение частиц
по размерам. Забегая несколько вперед, отметим, что особенно
велика роль степени полидисперсности системы при коагуляции
в условиях турбулентного перемешивания. Так, Самыгин и др.
[30] установили, что скорость объединения относительно мелких
(7—11 мкм) частиц с более крупными (~ 60 мкм) в турбулентном
потоке может быть на несколько порядков выше, чем скорость
объединения только мелких. Авторы объясняют это различием
в механизме встречи частиц — инерционном в случае полидис-
персной системы и диффузионном в случае монодисперсной.
Для того чтобы правильно предсказать ход коагуляции, не¬
обходимо знать исходное распределение частиц по размерам и
решить задачу об изменении этого распределения во времени.
Общее интегральное уравнение процесса необратимой коагу¬
ляции с учетом распределения частиц по размерам имеет следую¬
щий вид:
у
д? (^’Т) = ^ Л (со, F — со) р (со, т) р (F — со, т) dco —
О
со
— р(У,т)^ Л(7,со)р(со, r)dco, (V.8)
о
где р (F, т) — плотность распределения частиц системы относи¬
тельно их объема V; A (F, со) — коэффициент, выражающий за¬
висимость частоты столкновений частиц от их объемов V и со,
а также от конкретных условий протекания коагуляции.
При броуновском движении
при перемешивании
А"'р (F, со) - АОр (F, со) + (V + Зсо'/^'/. + Зсо’^з + со),
5*
132
ГЛАВА V. КИНЕТИКА КОАГУЛЯЦИИ
где к — константа Больцмана; Т — абсолютная температура сре¬
ды; 11 — динамическая вязкость среды; G — скоростной градиент.
При броуновской коагуляции частиц, мало отличающихся по
размерам, ядро уравнения — коэффициент A (V, со) — может быть
принято постоянным и уравнение (V.8) решается довольно просто.
В случае же коагуляции в турбулентном потоке решение уравне¬
ния сильно затруднено.
Вместе с тем в очистке воды нас в первую очередь интересует
коагуляция в турбулентном потоке, как имеющая наиболее важ¬
ное практическое значение. Знание закономерностей такой коагу¬
ляции позволяет ответить на вопросы: каково распределение по
размерам хлопьев коагулированной взвеси после завершения
коагуляции; когда и с какой интенсивностью следует перемеши¬
вать воду с раствором коагулянта, чтобы добиться наилучших
результатов очистки; какие требования должны быть предъявлены
к конструкции смесительных бассейнов и сооружений для удале¬
ния взвеси в осадок.
Прежде чем перейти к рассмотрению этих сложных вопросов,
необходимо, хотя бы в первом приближении, оценить величину
такого критического размера коагулирующих частиц, который
соответствует переходу перикинетической стадии коагуляции
в ортокинетическую.
V. 2. КОАГУЛЯЦИЯ В ДВИЖУЩЕЙСЯ СРЕДЕ
V. 2.1. Ламинарный поток
Представим себе две частицы 1 и 2, движущиеся в ламинарном
потоке жидкости вдоль плоской стенки в направлении оси х\
причем скорость частицы 2 равна v, а в направлении z существует
скоростной градиент dv/dz. Центр частицы 2 поместим в начало
координат.
По Смолуховскому, взаимодействие частиц 1 и 2 произойдет,
если расстояние между их центрами уменьшится до величины
радиуса сферы притяжения /?12 = + г2 (рис. V.2). Число час¬
тиц, достигших за единицу времени поверхности шарового слоя
радиусом /?12, будет равно произведению расхода потока q в сфере
притяжения на число частиц 1 в единице объема жидкости (гах)
dq — z(dv/dz) 2 (В.\г — z2)l/‘dz. (V.9)
Здесь zdv/dz — относительная скорость потока на расстоянии z
от оси х, a dA =2 (i?i2 — z2)‘/2dz — площадь, через которую про¬
ходит поток (рис. У.З).
V.2. КОАГУЛЯЦИЯ В ДВИЖУЩЕЙСЯ СРЕДЕ
133
Рис. V.2. Модель взаимодей¬
ствия коагулирующих частиц
(по Смолуховскому)
Рис. V.3. Модель взаимодействия частиц в ламинарном потоке в простран¬
стве (а) и на плоскости (б)|
Число взаимодействий частиц 1 с центральной частицей 2 за
единицу времени в единице объема жидкости составит
Ft 12
Ni — Arii z (dv/dz) (R2^ —z^'I’dz, (V.10)
o
или после замены производной dv/dz на некоторый средний по
времени скоростной градиент dv/dz
~~ Ni = у П! Jdvjdz)R\i. (V.10')
Для п2 центральных частиц, подставив в уравнение (V.10')
7?i2 = (dx + d2)/2, будем иметь
A^i2 = -g- пгпг (dy d-if dv/dz. (V.ll)
Обозначив dv/dz = G, получим уравнение Смолуховского для коа¬
гуляции бидисперсного золя в ламинарном потоке:
N* ™ = i n1n2G(d1 + dtf. (V.ll')
134
ГЛАВА V. КИНЕТИКА КОАГУЛЯЦИИ
Для монодисперсного золя
ДГлам _ A. n2Gd3.
(V.12)
Сравнение уравнения (V.12) с выражением (V.5) для константы
скорости броуновской коагуляции показывает, что соотношение
числа актов коагуляции в ламинарном потоке и в неподвижной
среде
Если коагулирующие частицы невелики, произведение DR
можно заменить (согласно уравнению Стокса — Эйнштейна) на
Согласно расчетам, при температуре 20° С равенство ЛГлам = NSp
выдерживается, если диаметр частиц составляет 2-iQ-3/G'!> мм.
Для значения скоростного градиента G = 1 сек-1 в ламинар¬
ном потоке величина R равна приблизительно 2 мкм. Это означает,
что при размере частиц около 1 мкм ортокинетическая коагуляция
начинает преобладать над перикинетической. Порядок величины
переходного (критического) размера частиц согласуется с экспе¬
риментальными данными.
V. 2.2. Турбулентный поток
Выражение (V.12) для случая коагуляции в турбулентном потоке,
строго говоря, не применимо, поскольку величина скоростного
градиента не может в полной мере охарактеризовать структуру
потока. В то же время экспериментальная проверка применимости
этого уравнения для турбулентного режима во многих случаях
дала удовлетворительные результаты. Это объясняется тем, что
величина скоростного градиента и размер турбулентных пульса¬
ций, ответственных за столкновение частиц, определяются одними
и теми же параметрами — механической энергией, поглощенной
единицей массы жидкости, и вязкостью последней.
В соответствии со схемой, показанной на рис.'V.4, при условии
равновесия некоторого выделенного объема жидкости между си¬
лами давления р и силами трения / существует зависимость [31]:
pAyAz + [/ + (df/dz) Az] АхАу = fAxAy + [р + (dp/dx) Azl-
ДГлам/ДГбр = GR3/SDnR.
(V.13)
2A; ТУЗят]. Тогда
Тулам/ДГбр = цсГП3/2кТ.
(V.13')
• AyAz.
(V.14)
Энергия, затраченная на перемещение выделенного объема
жидкости за единицу времени, 'равна произведению момента вра¬
щения (JAxAy) Az на угловую скорость dv/dz. Следовательно,
V.2. КОАГУЛЯЦИЯ в движущейся СРЁДЁ
135
Рис. V.4. Схема, показываю¬
щая соотношение между сила¬
ми давления (р) и силами тре¬
ния (/), действующими на эле¬
ментарный объем жидкости в
турбулентном потоке
удельная затрата энергии на единицу объема жидкости составляет
W = = fdv/dz. (V.15)
Поскольку dv/dz = G, а / = г\dv/dz (г] — динамическая вяз¬
кость жидкости), то
W = т] (dv/dzу = т]G2, (V.15')
или в окончательном виде
G = У W/ц = У~гф, (V.16)
где ех — диссипация энергии за единицу времени, отнесенная
к единице массы среды; v — кинематическая вязкость среды.
Выражение (V.16) позволяет количественно оценить степень
турбулизации потока и используется в дальнейшем для расчета
критерия скорости коагуляции водных примесей. Инженерные
методы определения величины удельных энергетических затрат
при использовании перемешивающих устройств разного типа изла¬
гаются в работах [6, 32—35].
Виганд и Франкенбергер [36] одними из первых получили
выражение для учета влияния на коагуляцию турбулентного
перемешивания. Однако они исходили из неверного предположе¬
ния, что градиент скорости, наблюдаемый на больших расстоя¬
ниях, сохраняет свое значение вплоть до расстояний, сравнимых
с размером коагулирующих частиц.
В 40-х годах влияние турбулентного перемешивания на кине¬
тику коагуляции исследовали Пшенай-Северин [37, 38], Туниц-
кий [39], Таверовский [40], использовавшие целый ряд допущений
и приближений, но правильный подход к рассматриваемому во¬
просу стал возможен только с позиций теории изотропной турбу¬
лентности Колмогорова [41]. В этой теории турбулентное течение
рассматривается как результат наложения на основную (среднюю)
скорость течения жидкости спектра непрерывных пульсаций ско¬
ростей разного масштаба. Кинетическая энергия крупномасштаб¬
ных пульсаций, возникающих при отрыве вихрей от поверхности
136
ГЛАВА V. КИНЕТИКА КОАГУЛЯЦИИ
обтекаемых предметов, передается постепенно пульсациям сред¬
него и мелкого масштаба. Масштаб пульсаций X, имеющих ско¬
рость vx, связан с числом Рейнольдса соотношением
Re>. == Xvyjv.
Когда размер пульсаций уменьшается до некоторой критиче¬
ской величины Я,0, то под влиянием сил вязкости их энергия начи¬
нает убывать значительно быстрее. Происходит диссипация энер¬
гии — переход в теплоту. Критическая величина пульсаций
носит название внутреннего масштаба турбулентности и опре¬
деляется, с одной стороны, удельной диссипацией механической
энергии, с другой — вязкостью перемешиваемой среды.
Метод Колмогорова получил развитие в теории турбулентной
коагуляции [42]. Хотя эта теория разрабатывалась для аэрозо¬
лей, принятый в ней подход к определению влияния масштаба
турбулентных пульсаций на закономерности коагуляции может
быть в принципе использован и для гидрозолей. Поэтому на этой
теории мы остановимся подробнее. Автор исходил из следующих
предположений:
1) во всех практически встречающихся случаях перемешива¬
ния аэрозолей размер и радиус коагуляции частиц мал по сравне¬
нию с внутренним масштабом турбулентности;
2) происходит полное увлечение частиц турбулентными пуль¬
сациями того масштаба, который в основном ответствен за столк¬
новения частиц.
Стационарная диффузия к поверхности сферы описывается
уравнением
в котором / — поток частиц на 1 см2 поверхности сферы коагуля¬
ции; — эффективный коэффициент диффузии, равный сумме
коэффициентов броуновской и турбулентной диффузий.
За относительное перемещение двух частиц, находящихся на
расстоянии I друг от друга, ответственны пульсации масштаба
I, так как при I обе частицы переносятся как единое
целое, а при X < I пульсации имеют малую интенсивность и ими
можно пренебречь. Поэтому перемещение частиц можно охаракте¬
ризовать коэффициентом диффузии £)тур6 = %v\.
Согласно методу Колмогорова,
где а и р - постоянные числовые множители, определяемые экс¬
перимента л ьно.
4лг2/ = 4яО*г2 ,
(V.17)
при X Х0 Ятурб = ае}'3 Х4/3,
при X^.'ko DTyp6 = рУe1/v V,
(V.18)
(V.19)
V.2. КОАГУЛЯЦИЯ В ДВИЖУЩЕЙСЯ СРЕДЕ
137
Наиболее интенсивные пульсации масштаба X Х0 выравни¬
вают концентрацию частиц аэрозоля в объеме среды, а пульсации
масштаба X < Я0 определяют взаимодействия частиц при сближе¬
нии. Поэтому расстояния I < Х0 вносят основной вклад в диффу¬
зионное сопротивление сближающихся частиц и ход процесса
определяется уравнением (V.19), по которому величина DTурб
пропорциональна X2.
Если радиус коагуляции R удовлетворяет неравенству
Уъ/у1Р>Р,
то £)турб > D и в уравнении (V.17) Z)* можно заменить на ,Отурб.
Тогда после интегрирования (V.17) получаем следующее выраже¬
ние для потока частиц:
j = р Уъф Rn0. (V.20)
Поскольку полный поток частиц на поверхность сферы радиу¬
сом R равен 4nR2j, число столкновений частиц в единице объема
за 1 сек с учетом выражения (V.20) составляет
ДГтурб = 4я[1R3 Уъф nl (V.21)
Из уравнения (V.21) видно, что величина iVTyp6, как и величина
дглам в уравнении' (V.12), пропорциональна радиусу коагуляции
R в третьей степени. Кроме того, она возрастает пропорционально
У 8] или скорости потока в степени 3/2. Сравнение значений iVTypR
и jV6p = AnRDnl дает соотношение
ЛГтурб/Л/бр = y^fiR*/D. (V.22)
Согласно выражению (V.22) величина критического размера
частиц, при которой соблюдается равенство iVTyp6 = Л7’бр, лежит
в диапазоне 5—10 мкм. Примерно тот же порядок величины кри¬
тического размера частиц (1,6—4,6 мкм) дают расчеты, выполнен¬
ные Тамбо [43] при условии равенства энергии частиц, подвер¬
женных турбулентному перемешиванию
£тУРб ^ ^Tn{el/\)Rb
и броуновскому перемещению
£бр = kT
Из результатов приведенных расчетов следует, что уже при
размерах агрегатов порядка нескольких микрометров броуновская
коагуляция резко замедляется, а ламинарная и турбулентная —
ускоряются. Критический размер агрегатов частиц соответствует
наиболее трудным транспортным условиям процесса.
138
ГЛАВА V. КИНЕТИКА КОАГУЛЯЦИИ
Недавно Абрахамсон [44] провел теоретическое исследование
случая коагуляции в турбулентном потоке относительно крупных
частиц (размером более 100 мкм), для которых скорости переме¬
щения как по величине, так и по направлению могут быть приняты
независимыми. Исходя из нормального распределения скоростей
и введя в расчеты среднеквадратичные скорости двух коагули¬
рующих частиц U\ и U\, он получил уравнение!
Минимальный размер частиц dmtn, при котором соблюдается спра¬
ведливость предположения о независимости скоростей частиц,
Абрахамсон определил из соотношения
где рв и рт — соответственно плотность воды и материала частиц.
Прежде чем перейти к обсуждению применимости изложенных
соображений к кинетике коагуляции гидролизующимися коагу¬
лянтами, коснемся кратко вопроса влияния [формы частиц на ско¬
рость коагуляции. j i | ) ]
Известно, что анизодиаметрические частицы коагулируют
быстрее, чем изодиаметрические. По Мюллеру, объяснение со¬
стоит в том, что из-за вращения (под действием броуновского дви¬
жения) анизодиаметрические частицы обладают большим диамет¬
ром столкновений и большей константой диффузии, чем изодиа¬
метрические.
) Приняв упрощенно форму поглощающей поверхности вытяну¬
той частицы за эллипсоид с соотношением размеров осей а, Мюл¬
лер показал, что отношение константы скорости коагуляции таких
частиц к константе скорости коагуляции сферических частиц
составляет приблизительно In 2а. Это означает, что при а = 10
скорость коагуляции анизодиаметрических частиц превышает
скорость коагуляции сферических частиц в уравнении Смолухов¬
ского в 3 раза. Такой порядок увеличения скорости коагуляции
с изменением формы частиц подтвержден подробным исследова¬
нием аэрозолей [45, 46]. Экспериментальная проверка уравнения
Смолуховского, модифицированного с учетом вращения частиц,
дала положительные результаты [47]. ) 1
It..
V. 3. КОАГУЛЯЦИЯ ГИДРОЛИЗУЮЩИМИСЯ КОАГУЛЯНТАМИ
С УЧЕТОМ РАЗРУШЕНИЯ АГРЕГАТОВ
Коагуляция гидролизующимися коагулянтами обладает в кинети¬
ческом отношении целым рядом особенностей, вносящих сущест¬
венные коррективы в конечный результат и потому требующих
учета. Перечислим эти особенности.
ДГТур<5 = У VI + 01
(V.23)
dmin= 15^-^-^,
Б1 РТ
(V.24)
V.3. КОАГУЛЯЦИЯ ГИДРОЛИЗУЮЩИМИСЯ КОАГУЛЯНТАМИ
139
1. В коагуляции участвуют как частицы загрязнений, так и
частицы новой дисперсной фазы, образующейся в ходе гидролиза
коагулянтов.
2. Скорость образования новой фазы является функцией ско¬
ростей собственно химической реакции и диффузии.
3. Размер формирующихся хлопьевидных агрегатов в тысячи
раз превосходит размеры элементарных частиц.
4. Сформированные хлопья коагулированной взвеси по
своим физическим свойствам резко отличаются от тех идеальных
агрегатов, образование которых предполагается при теоретиче¬
ском рассмотрении кинетики коагуляции.
Исследования хлопьеобразования при очистке воды разви¬
вались в двух направлениях. В одних случаях осуществлялся
чисто инженерный подход и усилия исследователей были направ¬
лены на выявление оптимальных условий процесса и критериев
перемешивания. К таким работам можно отнести, например,
работу Биина [48], в которой в качестве безразмерного критерия
работы перемешивающего устройства рекомендуется использовать
отношение количества воды, вовлекаемой в перемешивание ло¬
пастями мешалки за единицу времени, к общему расходу воды
через камеру, предназначенную для перемешивания.
В других случаях производились попытки установить на осно¬
ве уравнений Смолуховского связь между длительностью хлопье¬
образования, остаточной мутностью обработанной воды и струк¬
турой потока. Здесь исследователи встретились с серьезными
трудностями, во-первых, потому что в реальных условиях водо¬
очистки наряду с собственно коагуляцией происходит формирова¬
ние новых количеств твердой фазы при одновременном отделении
части хлопьев в осадок; во-вторых, неясно, до какого предель¬
ного размера частицы образующейся дисперсной фазы следует
рассматривать как элементарные, возникшие вследствие пересыще¬
ния раствора и участвующие в дальнейшей коагуляции в качестве
первичных частиц; в-третьих, число первичных частиц должно быть
установлено с учетом кинетики зародышеобразования малораство¬
римых продуктов гидролиза коагулянтов.
Как известно, образование зародышей твердой фазы в пере¬
сыщенном растворе происходит вследствие объединения молекул
в стабильные ассоциаты такого размера, при котором силы при¬
тяжения, действующие между молекулами, преобладают над си¬
лами, стремящимися их разобщить. Число молекул в кристалли¬
ческих зародышах в зависимости от типа их соединения в решетке
может находиться в пределах от 4 до 60 [49, стр. 41]. Зародыше-
образованию предшествует так называемый период индукции,
длительность которого соответствует времени, необходимому
для накопления критических концентраций вещества [50,
стр. 51].
140
ГЛАВА V. КИНЕТИКА КОАГУЛЯЦИИ
Скорость образования зародышей выражается уравнением
v3 = K3exp{-E3/RT), (V.25)
в котором К3 — константа скорости зародышеобразования, а
E3 = jnr2a (V.26)
есть минимальная энергия, необходимая для образования заро¬
дыша.
В присутствии посторонней твердой фазы, которая всегда име¬
ется в обрабатываемой воде, зародышеобразование ускоряется,
что объясняют или уменьшением работы зародышеобразования
на поверхности раздела фаз, или увеличением статистической
вероятности сорбции молекул на этой поверхности.
Перемешивание среды в области небольших пересыщений спо¬
собствует зародышеобразованию вследствие повышения вероят¬
ности объединения молекул в стабильные ассоциаты, причем по¬
вышение вероятности может быть следствием изменения транс¬
портных условий или, как полагает Нывлт [49, стр. 47],— сжатия
отдельных участков жидкости.
Скорость дальнейшей кристаллизации г>кр является функцией
удельной поверхности твердых частиц s0 и величины относитель¬
ного пересыщения раствора Ас [51]:
so Ас
укр= (l/A^p) + (1/D) - (V.27)
где Ккр и D — соответственно константы кристаллизации и диф¬
фузии.
Исследования гидролиза солей, применяемых в качестве коа¬
гулянтов, показывают, что в условиях водоподготовки гидролиз
и полимеризация продуктов гидролиза (отвечающие стадиям ин¬
дукции и зародышеобразования) протекают практически мгно¬
венно. Так, по данным, собранным Ханом и Стаммом [52],
константа скорости гидролиза катионов алюминия равна приблизи¬
тельно 1-1010 сек-1, константа полимеризации продуктов гидроли¬
за— 1 -102—1 • 103 сек-1. Следовательно, наибольший практиче¬
ский интерес в отношении интенсификации процесса для нашего
случая представляет последующая «коагуляционная» стадия
хлопьеобразования.
В 1964 г. Фаир и Джеммал [53], полагая, что размеры частиц,
участвующих в коагуляции, ограничены, с одной стороны, усло¬
виями перикинетической коагуляции (статически устойчивый
диаметр), а с другой — кинетической стабильностью агрегатов,
связанной с интенсивностью перемешивания, рассчитали по
системе уравнений Смолуховского изменение числа частиц во
V.3. КОАГУЛЯЦИЯ ГИДРОЛИЗУЮЩИМИСЯ КОАГУЛЯНТАМИ
141
времени и пришли к заключению, что произведение величины
скоростного градиента на начальную счетную концентрацию частиц
однозначно описывает процесс коагуляции.
Дальнейшее применение теории Смолуховского к очистке воды
гидролизующимися коагулянтами мы находим у Гудзона [54, 55],
который попытался представить про¬
цесс очистки как «исчезновение» час¬
тиц загрязнений (dt < 10 мкм) за счет
их «поглощения» хлопьями, образо¬
ванными продуктами гидролиза коа¬
гулянта (df ж 2 • 103 мкм). Пренебре¬
гая размером частиц загрязнений, он
представил в соответствии с уравне¬
нием (V.12) скорость уменьшения
числа щ первичных частиц в виде
— drii/dx — -g- ^n-jifGd3f. (V.28)
Поскольку объемная концентра¬
ция хлопьев в воде предполагается
постоянной и составляет
с0 = ^ЯЙ;3/6, (V.29)
вместо выражения (V.28) получаем
— dtii/dx = c0ni (V.30)
и после интегрирования
njn0 = exp (—tyc^Gx/n). (V.31)
Согласно уравнению (V.31), отношение числа частиц загряз¬
нений (гат), оставшихся в воде по прошествии времени т, к перво¬
начальному числу частиц (тг0) зависит от произведения G-c0, т. е.
при G = const определяется целиком свойствами объемных продук¬
тов гидролиза коагулянта.
Для проверки выражения (V.31) Гудзон представляет объем¬
ную концентрацию с0 в виде частного от деления количества коа¬
гулянта, добавленного к единице объема обрабатываемой воды а,
на количество коагулянта, содержащееся (в виде продуктов гидро¬
лиза) в объеме сформированных хлопьев. Эксперименты, проведен¬
ные на воде р. Детройт при разных значениях G, а и т, позволили
выявить зависимость г]зс0 = / (а). Из графика, показанного на
рис. V.5, видно, что эта зависимость при G < 27 сек*1 близка
к линейной, а увеличение G приводит к уменьшению г|зс0. Если
разделить мнение автора о постоянстве эффективности столкно¬
Рис. V.5. Зависимость между
дозой коагулянта (а) и пара¬
метром г|зс0 при разных зна¬
чениях скоростного градиен¬
та (G)
142
ГЛАВА V. КИНЕТИКА КОАГУЛЯЦИИ
вений г|з при любых значениях G, то уменьшение произведения гр с0
с ростом G можно объяснить снижением с0, т. е. уплотнением
хлопьев.
Слабыми местами в рассуждениях Гудзона являются представ¬
ления о механизме коагуляции как о «поглощении» загрязне¬
ний хлопьями коагулята и предположение о постоянстве зна¬
чений я|).
Харрис и др. [56] при изучении ортокинетической коагуляции
водных примесей применили более правильный подход, считая,
что в результате дестабилизации частиц загрязнений образуются
микрохлопья, имеющие примерно одинаковый размер. Если каж¬
дый из микрохлопьев включает в себя i первичных частиц, то его
объем определяется выражением
Fi== Tnr\ = Tnirb (v-32)
в котором Г; и Г\ — соответственно радиусы микрохлопьев и пер¬
вичных частиц.
Тогда объемная концентрация хлопьев определяется следую¬
щим образом:
оо оо
со = ^ 4 nrirei=4 яг1 Xiirii' (v.33)
i=l i=l
где nt — численная концентрация хлопьев.
Скорость убывания первичных частиц при г|з = 1 можно пред¬
ставить в виде
'*■»>«?.«• <v-34>
i=l
где р — максимально возможное число первичных частиц в каж¬
дом из хлопьев (подразумевается, что при столкновении хлопьев,
содержащих р первичных частиц, с первичными частицами ско¬
рость образования новых агрегатов компенсирована скоростью
обратного процесса — отделения от агрегатов первичных частиц
под действием касательных напряжений);
Hti — радиус столкновения первичных частиц с хлопьями,
содержащими i таких частиц,
Rn = Ri+R,^ агг (i1/. + 1), (V.35)
где а — отношение радиуса столкновения частицы к ее истинному
радиусу.
V.3. КОАГУЛЯЦИЯ ГИДРОЛИЗУЮЩИМИСЯ КОАГУЛЯНТАМИ
143
Комбинацией уравнений (V.33)—(V.35) с заменой в (V.35)
верхнего предела на р получим выражение
может рассматриваться как мера степени, с которой распределе¬
ние по размеру хлопьев отличается от распределения по размеру
первичных частиц. Максимальное значение 8Р равно 8 и дости¬
гается, когда присутствуют только первичные частицы. По мере
роста числа хлопьев, состоящих из р первичных частиц, величина
8Р приближается к нулю.
Работу камеры хлопьеобразования можно представить как
действие многих последовательно соединенных элементарных
реакторов, общий объем которых равен объему реальной камеры
(ячейковая модель). Применение уравнения (V.36) к группе,
состоящей из т таких реакторов с общей продолжительностью х
пребывания воды в них, дает выражение
где пх — численная концентрация частиц, не коагулированных
ко времени т; К = ч|ш3с0/ппй — экспериментально определяемая
константа скорости коагуляции; Е — экспериментально опреде¬
ляемый параметр, являющийся функцией гт.
Использовав ту же экспериментальную технику, что и Гудзон,
авторы [56] провели исследования по коагуляции суспензии као¬
лина, содержащей 47% частиц менее 1 мкм и 89% частиц менее
10 мкм. В качестве коагулянта применен сернокислый алюминий
в дозе 25 мг/л. Число элементарных реакторов т составляло 1,
Обработку результатов экспериментов проводили в следующем
порядке.
1. Строили графики зависимости п0/пТ от величины G (при
п0 = const, т = const), от величины т (при G = const, п0 = const)
и от величины (при 6-т = const).
(V.36)
в котором параметр
2Х
i=l
(V.37)
(V.38)
которое при т 1 принимает вид
njnz = exp (KEGn0т),
(V.39)
2 и 4.
144
ГЛАВА V. КИНЕТИКА КОАГУЛЯЦИИ
Рис. У.6. Зависимость степени коагуляции (п0/пх) частиц от параметра Е,
характеризующего эффективность столкновений частиц при разном числе m
элементарных реакторов
Рис. V.7. Зависимость степени коагуляции (n0/nz) частиц от параметра
Gn0T при разном числе т элементарных реакторов
2. Определяли зависимость п0/пх от произведения КЕ и путем
экстраполяции до значения ra0/wT = 1 (при котором Е = 8) на¬
ходили величину К.
3. По величине К определяли зависимость п0/пх = / (Е)
при разных значениях т (рис. V.6).
4. Используя график рис. V.6, рассчитывали по уравнению
(V.39) значения п0/пг, соответствующие разным величинам про¬
изведения СгП0х при разном числе элементарных реакторов
(рис. V.7).
Результаты экспериментов показывают, что с ростом Сит
произведение Etyc0 уменьшается. Если допустить постоянство if)c0,
то это уменьшение можно объяснить, по Харрису, изменением
распределения агрегатов по размеру.
Выводы Гудзона и Харриса с сотрудниками совпадают в том,
что уравнение Смолуховского для градиентной коагуляции
в принципе может быть применено к случаю гидролизующихся
коагулянтов [57], но причины уменьшения произведения
с ростом произведения Gx они видят разные: по Гудзону, про¬
исходит уменьшение объемной концентрации хлопьев за счет их
уплотнения, по Харрису — перераспределение хлопьев по раз¬
меру. Результаты экспериментальных исследований указывают
на необходимость учета обоих явлений.
V.3. КОАГУЛЯЦИЯ ГИДРОЛИЗУЮЩИМИСЯ КОАГУЛЯНТАМИ 145
Безразмерное произведение Gx, или, как его обычно записы¬
вают GT, известно под названием критерия перемешивания
Кэмпа, поскольку Кэмп первым предложил использовать его для
анализа гидродинамических условий хлопьеобразования. Мы уви¬
дим далее, что это произведение, действительно, довольно тесно
может быть связано со скоростью формирования хлопьев, но толь¬
ко при условии обработки коагулянтом воды одного и того же
состава.
С изменением качества исходной воды оптимальные значения
GT сильно отклоняются от некоторой средней величины. По¬
этому критерий Кэмпа должен быть дополнен третьим сомножи¬
телем, отражающим влияние концентрации дисперсной фазы.
Как вытекает ив результатов работ [53—58], им может явиться
счетная концентрация частиц загрязнений в обрабатываемой
воде п0 или объемная концентрация хлопьев коагулированной
взвеси с0 (см. рис. У.5 и Y.7). Экспериментальные доказательства
возможности использования произведения c0GT в качестве кри¬
терия условий очистки воды гидролизующимися коагулянтами
приведены в работе [59].
Одним из главных вопросов, возникающих при попытке при¬
менения уравнений Смолуховского к процессу очистки воды
гидролизующимися коагулянтами,— это вопрос о правомерности
использования величины скоростного градиента для характери¬
стики турбулентного потока. Выражения (V.12) и (V.21) для
числа актов коагуляции в ламинарном и турбулентном потоках
отличаются множителем, характеризующим гидродинамический
режим. Равенство Улам = 7VTyp6 наступает при
G ж Зяр |/"iL «~ DTyi>6. (V.40)
Аргаман и Кауфман [60] сделали попытку объяснить приме¬
нимость уравнения (V.17) для ламинарного потока к случаю
перемешивания на основе статистической теории турбулентности,
исходя из предположения, что частота столкновений частиц опре¬
деляется пульсациями масштаба X Х0. Это предположение выте¬
кает из реальных условий протекания процесса хлопьеобразова-
ния’в воде, когда размер образующихся агрегатов значительно
превышает размер агрегатов в аэрозоле.
Для коэффициента турбулентной диффузии авторы [60] путем
качественного анализа связи между спектром кинетической энер¬
гии, размером частиц и их диффузией, использовав ряд допуще¬
ний, получили выражение
£)тУрб _ кт jfr2, (V.41)
в котором Кт — некоторый размерный коэффициент, учитываю¬
щий влияние на скорость диффузии масштаба турбулентных пуль¬
146
ГЛАВА V. КИНЕТИКА КОАГУЛЯЦИИ
саций; й2 — квадрат скорости флуктуаций — функция спектра
энергии и частоты пульсаций; г/ — радиус хлопьев.
Для числа столкновений первичных частиц с хлопьями, ис¬
пользуя (У.41) и рассматривая процесс коагуляции как погло¬
щение частиц хлопьями, получаем
УТ7рб = 4лКтй2я0«/Г/. (V.42)
Уравнение (V.42) соответствует уравнению Смолуховского
при замене К^й2 на G. Правомерность такой замены подтверждена
результатами экспериментов, согласно которым между й2 и 6г
существует прямая пропорциональность.
Изучение природы остаточной мутйости воды после проведения
коагуляции и отделения в осадок хлопьев в отстойниках или на
фильтрах привело некоторых исследователей к выводам, что оста¬
точная мутность обусловлена «некоагулируемостью» отдельных
видов загрязнений вследствие их специфических свойств. К таким
свойствам относят высокую агрегативную устойчивость частиц
и их малую массу, не позволяющую частицам при столкновениях
войти в достаточно прочный контакт друг с другом или с продук¬
тами гидролиза коагулянта [61]. Однако, как можно проследить
по многочисленным производственным данным, почти всегда
между количеством выносимых с очистных сооружений механи¬
ческих примесей и остаточным содержанием алюминия или же¬
леза наблюдается пропорция, близкая к соотношению исходных
количеств этих компонентов на входе потока в водоочистное
устройство. Поэтому правильнее предположить, что остаточная
мутность является следствием разрушения хлопьев коагули¬
рованной взвеси.
Для однородной дисперсной системы, содержащей щ агре¬
гатов частиц сорта к (где к — число первичных частиц в агрегате),
уравнение кинетики коагуляции с учетом распада образующихся
агрегатов можно составить исходя из следующих соображений [62].
Число слипаний агрегатов сортов i и / в один агрегат сорта
i + / равно ацгг^П], а число распадов агрегатов i + 7 на агре¬
гаты i и / — bijTiirij (ац и Ьц — соответственно коэффициенты
агрегации и распада). Тогда элементарный поток размеров агре¬
гатов из состояний г и / в состояние г + / запишется в виде
fa — aijtiitij — bijtiitij, (V-43)
а уравнение баланса числа агрегатов — в виде
dnic/dx = (т) — Fk^ (т), (V.44)
к—1
где х — время; к — 1,2,3,..., оо; ^ (т) — полный
i=i
поток всех возможных состояний агрегатов с i < к в состояние к',
V.3. КОАГУЛЯЦИЯ ГИДРОЛИЗУЮЩИМИСЯ КОАГУЛЯНТАМИ
147
f/Bs
/
\
Рис. V.8. Зависимость доли
(\|)) эффективных столкновений
частиц от концентрации катио¬
нов алюминия при разных зна¬
чениях pH
4 — 5,0
1 — 5,75;
2 — 5,5;
3 — 5,25;
О
5 . Я 15 го
'10\тУ7*/г
//£, г (т) — полный поток из состояния к во все состоя¬
ния с i к.
Вследствие разрушения хлопьев эффективность столкновения
частиц, учитываемая в расчетах параметром 1|з, как бы уменьша¬
ется, а сам процесс коагуляции замедляется и при определенных
условиях прекращается полностью. Результаты экспериментов,
проведенных Ханом и Стаммом [52], показали, что величина г|з
зависит от отношения дозы коагулянта к концентрации взвешен¬
ных примесей воды и от pH среды, т. е. от параметров, определяю¬
щих прочность хлопьев. Как видно из рисунка Y.8, максимальные
значения i]) соответствуют примерно одной и той же дозе коагу¬
лянта и имеют тенденцию увеличиваться с ростом pH.
Результаты оценки степени разрушения агрегатов частиц за¬
висят прежде всего от того, каким силам отведена роль ответствен¬
ных за разрушения.
Расчеты Фукса [20, стр. 309] для аэрозоля, в которых автор
в качестве сил, ответственных за разрушение элементарного
агрегата из двух сферических частиц радиусом г, принял центро¬
бежные силы, показали, что силы отрыва преобладают над силами
адгезионного взаимодействия при условии
Поскольку удельная поверхностная энергия частиц о равна
нескольким десяткам эргов на квадратный сантиметр, согласно
расчетам, отрыв частиц произойдет при г менее 100 А. Таким об¬
разом, можно полагать, что в реальной дисперсной системе цен¬
тробежные силы отрыва не внесут изменений в теоретическое
значение константы скорости коагуляции.
(V.45)
148
ГЛАВА V. КИНЕТИКА КОАГУЛЯЦИИ
В ламинарном потоке жидкости хлопья коагулированной
взвеси совершают вращательное движение, которое вызывает,
по-видимому, лишь некоторое перераспределение частиц внутри
хлопьев^ без разрушения коагуляционной структуры [63].
( Согласно расчетам Бензе [64], в воде зависимость критической
величины скоростного градиента Glfp, при котором наступает
Рис. V.9. Схема разрушаю¬
щего действия на агрегаты час¬
тиц турбулентных пульсаций
масштаба X.
равенство сил притяжения и отрыва, от размера частиц выражает¬
ся соотношением
GK р — 3- гЯ2
(V.46)
где А и Н0 — уже известные параметры (см. гл. I).
В турбулентном потоке на хлопья коагулированной взвеси
действуют мощные касательные усилия, производящие тем боль¬
шие разрушения, чем крупнее хлопья. Именно эти усилия боль¬
шинство исследователей считает причиной разрушения хлопьев.
Энштейн и Пантелят [65] определили максимальный размер
хлопьев в турбулентном потоке, считая, что разрушение их про¬
исходит за счет ударных сил, возникающих вследствие турбулент¬
ных пульсаций масштаба X. Исходя из предположения, что равно¬
действующие этих сил проходят через центры тяжести каждой
половины агрегата (рис. V.9), авторы установили следующую
связь максимального размера хлопьев с их прочностью на сдвиг
рс и скоростью потока v:
1 / рс \3/2
<1Гх>кф-3(-) -
(V.47)
где Кф — коэффициент, зависящий от формы агрегата; рв — плот¬
ность воды.
Эксперименты с суспензиями, флокулированными полиакрил¬
амидом, подтвердили обратную пропорциональность размера
з „ /Рс\312
„V* ~ I/ / « V
хлопьев величине гг, однако произведение
зависит
от свойств суспендированных частиц.
Дифференциальные уравнения, описывающие процесс разруше¬
ния агрегатов под действием гидродинамических сил с учетом
V.3. КОАГУЛЯЦИЯ ГИДРОЛИЗУЮЩИМИСЯ КОАГУЛЯНТАМИ
149
характера распределения разрушающих усилий, получены в ра¬
боте [66].
В 1965 г. Хэм и Кристман [67] высказали предположение, что
разрушение хлопьев в потоке воды обусловлено не касательными
силами среза, а взаимными столкновениями. Такая трактовка
явления предполагает, что роль скоростного градиента сводится
лишь к изменению числа соударений хлопьев, а разрушающее
действие потока полностью учитывается величиной коэффициен¬
та я|). Это не всегда соответствует экспериментальным данным.
Разрушающее действие потока, определяемое как скорость
появления в воде первичных частиц, оторвавшихся с поверхности
хлопьев, может быть связано с гидродинамическими условиями
перемешивания соотношением [60, 68]
dn1
г f
КвП) — G, (V.48)
dr г*
где пх и nf — концентрации в воде соответственно первичных ча¬
стиц и хлопьев; гг и г/ — размер первичных частиц и хлопьев;
К в — константа скорости разрушения хлопьев.
Общее изменение концентрации первичных частиц с учетом
разрушения хлопьев составляет [68, 69]
по 1 + кАот
- 1 + KBG2T ' <V-49)
В этом выражении п\ и щ — концентрации первичных частиц
в воде на входе и на выходе ее из камеры хлопьеобразования;
Т — длительность пребывания воды в камере; К а — константа
скорости образования хлопьев.
Проверка уравнения (Y.49) показала соответствие теоретиче¬
ских предпосылок экспериментальным данным. Несколько позже
Паркер и др. [70] уточнили это выражение, показав, что при лю¬
бом из двух возможных способов разрушения — отрыве единич¬
ных элементарных частиц от хлопьев или разрушении хлопьев на
более или менее равные части — справедливо выражение
и? 1 + КGT
~ = ГГ... А в— (V.49')
\ l + KB(cJn1)GmT v '
в котором св — весовая концентрация твердой фазы в воде;
m — показатель степени, равный 2 или 4 в зависимости от способа
разрушения.
Опыт эксплуатации водоочистных сооружений свидетельствует
о том, что при любых исходных условиях минимальной остаточной
мутности воды после отстаивания соответствуют вполне опреде¬
ленные оптимальные значения скоростного градиента, созданного
150
ГЛАВА V. КИНЕТИКА КОАГУЛЯЦИИ
Рис. V.10. Зависимость эф¬
фекта осветления воды трех
разных источников водо¬
снабжения от величины
скоростного градиента G,
созданной при перемеши¬
вании воды после ее обра¬
ботки коагулянтом
при перемешивании воды в камерах хлопьеобразования. Причем
для каждого заданного значения G устанавливается равновесие
между вероятностью образования хлопьев и их средним разме¬
ром [64]. По данным Гриффита и Уильямса [71], оптималь¬
ные значения скоростного градиента составляют 22—45 сек'1
(рис. V.10); по данным других исследователей — 20—74 сек-1
[32], 30—60 сек'1 [72], а при добавлении полиэлектролита —
50—175 сек^1 [73]. В опытах Цуноды [74] на примере коагуляции
суспензии каолина показано, что при увеличении расхода энергии
на перемешивание (в кем ■ сек'1-м'3) в 2 раза константа скорости
уменьшения мутности суспензии (в мин'1) в ходе последующего
отстаивания возрастает примерно на 25%.
Л итерат ура
1. В. И. Козлов. Сборник трудов ГИСИ им. В. П. Чкалова, вып. 25. Горьк.
кн. изд-во, 1956, стр. 160.
2. Е. Д. Бабенков. Сб. «Методы очистки и контроля качества воды». М.,
«Транспорт», 1966, стр. 15.
3. J. W. Moffett. J. Amer. Water Works Assoc., 60, 1255 (1968).
4. В. Г. Печников, Е. Г. Хайлов. Сб. «Технология очистки питьевой воды
г. Москвы». М., Стройиздат, 1974, стр. 88.
5. W. W. Nicols. Water Works and Sewerage, 88, 325 (1941).
6. J. D. Griffith, R. G. Williams. J. Amer. Water Works Assoc., 64, 825 (1972).
7. В. А. Ромоданова. Сб. «Научные основы технологии очистки воды». Киев,
«Наукова думка», 1973, стр. 30.
8. S. Kawamura. J. Amer. Water Works Assoc., 65, 417 (1973).
9. R. A. Trelies, D. J. Bengolea, A. G. Poccard. J. Amer. Water Works Assoc.,
32, 742 (1940).
10. M. R. Buydens. Techn. sanit. et municip., 48, 139 (1953).
11. В. Г. Левич. Физико-химическая гидродинамика. М., Изд-во АН СССР,
1952.
12. С. С. Воюцкий. Курс коллоидной химии. М., «Химия», 1975.
13. Ю. М. Глазман и др. Коллоидн. ж., 15, 161 (1953).
14. А. К. Bhattacharya, R. Kumar. J. Colloid. Sci., 10, 55 (1955).
15. A. K. Bhattacharya, R. Kumar. J. Colloid Sci., 11, 171 (1956).
16. R. S. Srivastava, I. P. Abrol. Kolloid. Z. und. Z. Polymere, 222, 61 (1968).
17. A. Marciniak-Fabianowa, W. Wojciak. Prace Komisji Matematyczno-
przyrodniczej, 7, № 10, 53 (1960).
18. E. Ф. Желъвис, Ю. М. Глазман. Коллоидн. ж., 29, 196 (1967).
V. ЛИТЕРАТУРА
151
19. Г. Р. Кройт. Наука о коллоидах, т. 1. Необратимые системы. М., ИЛ,
1955.
20. Н. А. Фукс. Механика аэрозолей. М., Изд-во АН СССР, 1955.
21. Н. М. Кудрявцева. Автореферат канд. дисс. М., ИФХ АН СССР, 1963.
22. Ю. М. Глазман. Коллоидн. ж., 24, 275 (1962).
23. N. A. Fuks. Z. Phys., 89, 736 (1934).
24. Б. В. Дерягин. Изв. АН СССР, ОХН, 1937, 1153.
25. В. V. Derjaguin. Acta Physicochim. URSS, 10, 333 (1939).
26. Б. В. Дерягин. Коллоидн. ж., 6, 291 (1940).
27. В. V. Derjaguin. Trans. Faraday Soc., 36, 203 (1940).
28. G. Wiegner. Kolloid. Z., 8, 227 (1911).
29. B. A. Matthews, С. T. Rhodes. J. Colloid and Interface Sci., 32, 332 (1970).
30. В. Д. Самыгин, А. А. Барский и др. Коллоидн. ж., 30, 581 (1968).
31. Т. R. Camp, Р. С. Stein. J. Boston Soc. Civil Engrs, 30, 219 (1943).
32. T. R. Camp. Trans. Amer. Soc. Civil Engrs, 120, 1 (1955).
33. T. R. Camp. J. Amer. Water Works Assoc., 60, 656 (1968).
34. A. G. Bhole, S. M. Dhabadgaonkar, S. G. Tarnekar. J. Inst. Engrs (India).
Public Health Engng Div., 55, 37 (1975).
35. /. Lai Ruey, H. E. Hudson, J. E. Singley. J. Amer. Water Works Assoc.,
67, 553 (1975).
36. A. Wigand, E. Frankenberger. Phys. Z., 31, 204 (1930).
37. С. В. Пшенай-Северин. Автореф. канд. дисс. М., Геофизический ин-т
АН СССР 1953
38. С. В. Пшенай-Северин. ДАН СССР, 94, 865 (1954).
39. Н. Н. Туницкий. ЖФХ, 20, 1137 (1946).
40. Е. Н. Таверовский. Сб. «Новые идеи в области изучения аэрозолей». М.,
Изд-во АН СССР, 1949, стр. 108.
41. А. Н. Колмогоров. ДАН СССР, 30, 299 (1941).
42. В. Г. Левич. ДАН СССР, 99, 809 (1954).
43. N. Tambo. Mem. Fac. Engng Hokkaido Univ., 11, 585 (1965).
44. J. Abrahamson. Chem. Engng Sci., 30, 1371 (1975).
45. И. С. Артемов. Коллоидн. ж., 9, 225 (1947).
46. И. С. Артемов. Сб. «Новые идеи в области изучения аэрозолей». М.,
Изд-во АН СССР, 1949, стр. 68.
47. D. N. Sutherland, I. Goodarz-Nia. Chem. Engng Sci., 26, 2071 (1971).
48. E. L. Bean. Water Works Engng, 106, 33 (1953).
49. Я. Нывлт. Кристаллизация из растворов. М., «Химия», 1974.
50. Б. Дельмон. Кинетика гетерогенных реакций. М., «Мир», 1972.
51. D. G. Stevenson. J. Inst. Water Engrs, 21, 119 (1967).
52. H. H. Hahn, W. Stumm. J. Colloid and Interface Sci., 28, 134 (1968).
53. G. M. Fair, R. S. Gemmell. J. Colloid Sci., 19, 360 (1964).
54. H. E. Hudson. J. Amer. Water Works Assoc., 57, 885 (1965).
55. H. E. Hudson. J. New Engl. Water Works Assoc., 80, 232 (1966). ^
56. H. S. Harris, W. J. Kaufman, R. B. Krone. J. Sanit. Engng Div. Proc.
ASCE, № 6, 95 (1966).
57. J. Sanit. Engng Div. Proc. ASCE, № 3, 155 (1972).
58. E. Д. Бабенков. Оптимальная доза коагулянта при очистке воды. М.,
«Транспорт», 1974.
59. /. Soucek, J. Sindelar. The Use of a Dimensionless Criterion in the Charac¬
terization of Flocculation. Praha—Podbaba, 1967.
60. Y. Argaman, W. J. Kaufman. J. Sanit. Engng Div. Proc. ASCE, № 2,
223 (1970).
61. R. L. Williams. J. Amer. Water Works Assoc., 57, 801 (1965).
62. Г. А. Мартынов, В. М. Муллер. ДАН СССР, 207, 1161 (1972).
63. D. J. Thomas. J. Amer. Inst. Chem. Engrs, 10, 517 (1964).
64. F. Benze. Dechema Monographien, 59, 29 (1968).
65. С. И, Эпштейн, Г. С. Пантелят. Сб. «Вопросы технологии обработки во¬
152
ГЛАВА V. КИНЕТИКА КОАГУЛЯЦИИ
ды промышленного и питьевого водоснабжения», № 2. Киев, «Буд1вель-
ник», 1969, стр. 96.
66. R. Kamal, I. Patterson. Canad. J. Chem. Engrs, 52, 707 (1974).
67. R. K. Ham, R. F. Cristman. J. Sanit. Engng Div. Proc. ASCE, № 3, 481
(1965).
68. Y. Argaman, W. J. Kaufman. Sert Report, № 68—5. Univ. Calif., July
1968.
69. Y. Argaman. J. Amer. Water Works Assoc., 63, 775 (1971).
70. D. S. Parker, W. J. Kaufman, D. Jenkins. J. Sanit. Engng Div. Proc.
ASCE, № 1, 79 (1972).
71. /. D. Griffith, R. G. Williams. .T. Amer. Water Works Assoc., 64, 825 (1972).
72. H. Lengweller, W. Buser. Helv chim. acta, 44, 796 (1961).
73. J. D. Walker. J. Amer. Water Works Assoc., 60, 321 (1968).
74. С. Цунода. Суйдо кёкай дзасси, № 408, 22 (1968); РЖХим., 17И318 (1969).
Глава VI
ОПТИМАЛЬНАЯ ДОЗА КОАГУЛЯНТА
В коагуляции под действием электролитов, основанной на класси¬
ческой модели двойного электрического слоя, подразумевается
независимость оптимальной дозы коагулятора от концентрации
дисперсной фазы. При обработке воды гидролизующимися коагу¬
лянтами между концентрацией примесей и потребностью в коагу¬
лянтах имеется вполне определенная связь. Это различие объяс¬
няется важной особенностью процесса коагуляции гидролизую¬
щимися коагулянтами — возникновением новой дисперсной фазы
со своими специфическими свойствами. К тому же, как мы видели,
электрокинетический потенциал сильно гидратированных дисперс¬
ных примесей природной воды обычно имеет величину ниже кри¬
тической (~30 мв) и, следовательно, не может в полной мере
характеризовать степень их агрегативной устойчивости.
К таким системам физическая теория коагуляции лиофобных
золей может быть применена только в том случае, если учтено
влияние третьей составляющей расклинивающего давления, обу¬
словленной перекрытием граничных слоев и изученной пока не¬
достаточно. Вместе с тем расчет оптимальной дозы коагулянта
возможен лишь тогда, когда достоверна модель процесса коагуля¬
ции, известен характер взаимной фиксации частиц продуктов
гидролиза и примесей воды при разном их количественном соот¬
ношении, иначе говоря, когда раскрыт механизм формирования
коагулированной взвеси.
О механизме коагуляции и построении хлопьев коагулирован¬
ной взвеси часто высказываются мнения весьма неопределенные
и противоречивые [1—9]. Современного исследователя вряд ли
может удовлетворить объяснение процесса как «механический
захват» загрязнений осаждающимися хлопьями или как след¬
ствие появления в системе новой кинетически неустойчивой фазы.
Такие представления, даже подкрепленные ссылками на физиче¬
скую теорию гетерокоагуляции, не отражают в полной мере всей
совокупности явлений, так как не учитывают ни химической сто¬
роны процесса — хемосорбции, образования малорастворимых
гидроксокомплексов, их полимеризации и кристаллизации; пи
кинетических особенностей системы, проявляющихся особенно
ярко в различии закономерностей осветления и обесцвечивания
воды. Поэтому прежде чем говорить об оптимальной дозе коагу¬
лянта и методах ее расчета, проведем краткий анализ мнений,
154
ГЛАВА VI. ОПТИМАЛЬНАЯ ДОЗА КОАГУЛЯНТА
высказываемых о механизме формирования коагулированной
взвеси, и сопоставим сделанные выводы с результатами экспери¬
ментов .
VI. 1. МЕХАНИЗМ ФОРМИРОВАНИЯ КОАГУЛИРОВАННОЙ ВЗВЕСИ
VI. 1.1. Осветление воды
В первых работах по осветлению воды гидролизующимися коагу¬
лянтами большинство исследователей разделяло точку зрения
Миллера [10—13] на главенствующую роль нейтрализации частиц
суспензий катионами А13+ и Fe3+ [14, 15]. В 1948 г., обобщив и
сопоставив экспериментальные материалы, Блэк [1] предложил
следующую гипотезу трехстадийного процесса:
а) катионы А13+ и Fe3+ нейтрализуют заряд частиц взвеси;
б) образуются положительно заряженные микрохлопья, так¬
же способные нейтрализовать заряд частиц;
в) микрохлопья объединяются в хлопья, различимые невоору¬
женным глазом.
Следующим шагом вперед явились количественные исследова¬
ния Ланжелье и др. [16, 17], в которых установлено, что при
коагуляции минеральных примесей возможны два случая.
1. При высокой катионообменной емкости взвеси (Ек—
~ 250 мг-экв/л) астабилизации и удовлетворительная коагуля¬
ция частиц наступают под действием лишь одних катионов А13+.
М); 2. При умеренных значениях Ек (~50 мг-экв/л), характерных
для поверхностных вод, осветление протекает удовлетворительно
только в присутствии дополнительного связующего материала
(«binder alum»), а потребность в коагулянте складывается из
количества, пропорционального величине Ек, и количества, не¬
обходимого для накопления связующего материала.
По-видимому, несовершенство техники эксперимента (опреде¬
ление обменной емкости взвеси и оптимальной дозы коагулянта)
не позволило Ланжелье проверить выводы на природных водах
разного состава. К тому же методика исследований не преду¬
сматривала поддержания постоянства значений pH среды, что,
безусловно, оказывало влияние и на химический состав продуктов
гидролиза алюминия и на величину Ек.
Взгляды Блэка и Ланжелье не совпадают полностью, но оба
автора первостепенное значение в снижении заряда минеральных
частиц придают катионам алюминия. Позднее, когда были приме¬
нены стандартные методы химических анализов и определения
электрофоретической подвижности частиц, подтвердилась точка
зрения Мэттсона [18—20], Каргина и др. [21] о сильном нейтра¬
лизующем действии продуктов гидролиза алюминия. Получены
VI.1. МЕХАНИЗМ ФОРМИРОВАНИЯ КОАГУЛИРОВАННОЙ ВЗВЕСИ 155
новые сведения [2, 3, 22, 23], которые можно систематизировать
следующим образом.
1. Продукты гидролиза алюминия более эффективны в отно¬
шении астабилизации глинистых частиц, чем катионы А13+ и Н+.
2. По мере увеличения дозы A12(S04)3 дзета-потенциал (ДП)
частиц сначала резко снижается, а после достижения изоэлектри-
ческой точки его изменение становится незначительным. Коагуля¬
ция протекает наилучшим образом при ДП <0 и требует соли
алюминия в 6—8 раз меньше, чем это необходимо для достижения
ДП = о.
3. Суспензии глин с малой обменной емкостью (Ек = 10—
80 мг-экв/л) коагулируют при меньших дозах коагулянта, чем
суспензии с высокой обменной емкостью частиц (Е„ = 270—
340 мг-экв/л), но в обоих случаях доза A12(S04)3, требующаяся для
достижения ДП = 0 (в мг-экв/л), во много раз превосходила 1
величину Ек. Для глин с высокой обменной способностью суще¬
ствует связь между Ек и потребностью в коагулянте, для глин
с низкой обменной способностью такой связи не обнаружено.
4. Наилучшее осветление воды происходит при pH 7,5—-8,5,
т. е. в зоне изоэлектрической точки гидроокиси алюминия (см.
гл. III), когда последняя имеет минимальную растворимость.
5. При pH ниже 4,5 удовлетворительно протекающая коагу¬
ляция объясняется действием катионов алюминия 2.
6. Во многих опытах связь между остаточной мутностью и
электрофоретической подвижностью частиц не обнаружена.
В исследованиях с сернокислым железом [26] показано, что
величина Ек замутнителя на дозу коагулянта не влияет.
Эксперименты с натуральными взвесями разных рек США [41
в основных чертах подтвердили перечисленные выводы, но зона
оптимума pH для A12(S04)3 оказалась смещенной в более кислую
среду. При обработке воды р. Сакраменто обнаружено несколько
pH-зон коагуляции при отрицательных или нулевых значениях
ДП. Наиболее широкая из них (6,5—7,5) лежит в области уско¬
ренного формирования малорастворимых продуктов гидролиза и,
так же как и эта область, расширяется с увеличением дозы коагу¬
лянта.
В 1962 г. Мацкрле [27] подверг сомнению гипотезу о сущест¬
венной роли катионного обмена и выдвинул на первый план физи¬
ческие условия протекания процесса. Перикинетическую фазу
коагуляции он рассматривает как комплекс процессов гидролиза
коагулянта, кристаллизации продуктов гидролиза с образованием
положительно заряженных микрочастиц, компенсации отрица-
1 По предположению авторов избыточное количество коагулянта является
функцией щелочности воды.
2 По [24] — при pH < 4, по [25] — при pH < 3.
156
ГЛАВА VI. ОПТИМАЛЬНАЯ ДОЗА КОАГУЛЯНТА
-7
О
а, аг а3
а
Рис. VI.1. Характер зависимости остаточной мутности (М0) воды от дозы
коагулянта (а)
Рис. VI.2. Расположение зон коагуляции и стабильного состояния золя
A1(N03)3 в координатах: концентрация золя — pH [31]
тельного заряда примесей, взаимного объединения дестабилизи¬
рованных частиц в микрохлопья.
Допуская, что исчезновение энергетического барьера в системе
является следствием снижения поверхностного (ср0) потенциала,
автор объясняет наблюдаемую в экспериментах зависимость оста¬
точной мутности воды от дозы коагулянта (рис. VI. 1) следующим
образом. При а < ах увеличение М0 с ростом а вызвано образова¬
нием в растворе большого количества микрокристаллов. По мере
накопления кристаллов энергетический барьер отталкивания
между частицами загрязнений снижается, и в точке перелома
кривой наступает быстрая коагуляция. С ростом дозы коагулянта
от а1 до а2 остаточная мутность уменьшается, а ср0 достигает зна¬
чения, близкого к нулю, и сохраняет его вплоть до а ~ а3.
При а~^> а3 потенциал становится положительным и происходит
рестабилизация частиц загрязнений, сопровождающаяся ухуд¬
шением и прекращением коагуляции.
Теория Мацкрле при всей ее стройности не учитывает многих
физических и химических аспектов, в том числе важной роли
адсорбции в механизме коагуляции [28].
Совершенно иной точки зрения придерживается Пакхам [29.,
30]. Он установил, что ход коагуляции слабо связан с зарядом
частиц и ионной силой раствора и практически целиком опреде¬
ляется скоростью образования и количеством малорастворимых
коагулятов. Последние обволакивают («enmeshmenU) частицы
взвеси и, объединяясь, образуют агрегаты, способные к осажде¬
нию. Влияние pH на хлопьеобразование сводится к влиянию кон¬
центрации Н+- и ОН~-ионов на состав и структуру продуктов
гидролиза.
VI. 1. МЕХАНИЗМ ФОРМИРОВАНИЯ КОАГУЛИРОВАННОЙ ВЗВЕСИ 157
В более поздней работе Блэк и Чииг Лин-чен [25] разделили
точку зрения Пакхама на механизм коагуляции в рН-зоне 7—8,5.
На примере разбавленной суспензии каолинита они показали, что
наиболее интенсивное снижение отрицательного заряда глинистых
частиц происходит при pH ~ 5 и объясняется специфической
адсорбцией полимерных продуктов гидролиза алюминия. Катио¬
ны алюминия (при pH ~ 3) и гидроокись (при pH ~ 8) не способ¬
ны переменить знак заряда частиц на противоположный, и коагу-
лация в первом случае обусловливается сжатием двойного слоя,
а во втором — опутыванием частиц по Пакхаму.
Полученные результаты согласуются с выводами Матиевича
и др. [31, 32], сделанными при обработке азотнокислым алюминием
золя AgJ с отрицательно заряженными частицами. На рис. VI.2,
где в координатах pH — концентрация A1(N03)3 показано распо¬
ложение зон коагуляции и стабильного состояния частиц золя,
наклон линии, отделяющей одну зону от другой, определяется соот¬
ношением [А13+] : [ОН-] в продуктах гидролиза коагулянта.
Анализ выражения Штерна для полной стандартной энергии
адсорбции, а также обработка большого числа эксперименталь¬
ных данных [8, 32—36] приводят к заключению, что в случае
гидролизующихся коагулянтов доля участия химической энергии
в акте стабилизации может быть выше, чем электростатической.
Причем сорбирующиеся соединения способны вызвать коагуля¬
цию золя в значительно меньших концентрациях, чем несорбирую-
щиеся.
Гораздо более сильная сорбционная способность гидроксо-
комплексов по сравнению с катионами А13+ и Fe3+ связана, вероят¬
но, с большей их гидрофильностью и усилением ковалентных
связей между атомами металла и специфическими участками
поверхности твердой фазы [8, 24, 37]. Как установил Маккензи [38]
в опытах по коагуляции суспензии кварца хлорным железом,
водородные мостики, возникающие между ОН-группами гидро-
ксокомплексов и кварцевых частиц, обеспечивают прочную связь,
не нарушаемую перемешиванием. С увеличением pH среды, когда
основным продуктом гидролиза становится гидроокись железа,
сорбирующаяся на частицах чисто физически, прочность связей
понижается.
Таким образом, можно предположить, как это и делают неко¬
торые исследователи [39—41], существование флокуляции, выз¬
ванной полимерными комплексами алюминия и железа, наподобие
флокуляции высокомолекулярными соединениями. Для увязки
«флокуляционного» механизма с электростатическими явлениями
приходится постулировать [7], что из-за большого многообразия
продуктов гидролиза в обрабатываемой воде даже ири'рН ~ рН;
присутствуют заметные количества положительно заряженных
продуктов. Они легко сорбируются на частицах глины, снижая
158
ГЛАВА VI. ОПТИМАЛЬНАЯ ДОЗА КОАГУЛЯНТА
их заряд, а преобладающие нейтральные фракции обеспечивают
хлопьеобразование.
При такой трактовке процесса коагуляции становится очевид¬
ным, что с увеличением концентрации или заряда частиц загряз¬
нений созданию оптимальных условий будет способствовать уве¬
личение доли и повышение заряда положительно заряженных
продуктов гидролиза. Действительно, как показано, например,
Кимом и др. [42], концентрированные суспензии глин лучше коа¬
гулируют при pH < рН;.
Интересный подход к рассмотрению механизма коагуляции
с позиций растворимости и структурной прочности продуктов гид¬
ролиза алюминия осуществил Халл [43]. В основе его гипотезы
лежит предположение, что растворимость гидроокисных соеди¬
нений, максимальная в первоначальный момент, понижается
вследствие перегруппировки в более стабильные кристаллические
формы, образования полиядерных формаций и взаимодействия
с поверхностью дисперсной фазы.
При малой площади дисперсной фазы S (маломутные воды
с относительно крупными частицами), когда соотношение [А13+]:*5
велико, формируются наиболее растворимые продукты, простран¬
ственная решетка которых не имеет перекрестных связей и пото¬
му легко разрушается. При малых соотношениях [А13+] : S
(мутные воды с тонкодисперсной взвесью) образуются менее рас¬
творимые и более прочные агрегаты. Форма выделяющихся про¬
дуктов гидролиза, а следовательно, и структурная прочность
зависят от pH среды и могут быть связаны с величиной ДП (см.
ниже). По мере уменьшения соотношения [Al3+J : S (переход от
грубодисперсной к тонкодисперсной суспензии) изоэлектрическое
состояние хлопьев наступает при более низких значениях pH.
Существенный недостаток почти всех приведенных рассужде¬
ний состоит в том, что они не затрагивают кинетической стороны
процесса: с изменением численной концентрации частиц дисперс¬
ной фазы и режима перемешивания воды меняется роль различных
факторов в механизме коагуляции.
Вернемся к работам Пакхама. В экспериментах с минеральны¬
ми суспензиями Пакхам показал [301, что для большинства из
них (иллит, кварц, кальцит, каолин и др.) при исходном содержа¬
нии 50 мг/л оптимальная зона коагуляции не зависит от вида ми¬
нерала и лежит в области pH 6,8—7,8. Однако с увеличением сте¬
пени дисперсности частиц (монтмориллонит, 50 мг/л) или концен¬
трации взвеси (каолин, 500 мг/л) область оптимума pH расши¬
ряется за счет более низких значений и составляет 4,5—7,5. Этот
результат совпадает с данными Халла, но объяснен по-другому.
По мнению Пакхама, системы с большим числом частиц в единице
объема раствора ведут себя~как типичные лпофобньте золи, чув¬
ствительные к изменению концентрации электролитов; в разбав-
VI. 1. МЕХАНИЗМ ФОРМИРОВАНИЯ КОАГУЛИРОВАННОЙ ВЗВЕСИ 159
Рис. VI.3. Зависимость дозы
сернокислого алюминия (а50),
необходимой для 50%-ного ос¬
ветления суспензий бентонита
и каолинита, от Ек- суспензий
при разных значениях pH
воды (значения pH показаны
на кривых) [42]
ленных же системах скорость коагуляции ^контролируется^ рас¬
стоянием между частицами. Для обозначения^первого вида коагу¬
ляции Пакхам предлагает использовать термин Ван Ольфена [44]
«edge-to-face» — коагуляция выступами поверхности; второй вид
коагуляции он называет «sweep» — подметающей. Количественной
оценки условий перехода от одного вида коагуляции к другому
Пакхам не дает. Этот пробел попытались восполнить другие
исследователи.
Ким и др. [42] в 1965 г. тщательно изучили влияние на дозу
коагулянта площади поверхности частиц и катионообменной ем¬
кости минеральных примесей. В качестве параметра, определяю¬
щего потребность в коагулянте, авторы, как и Пакхам, исполь¬
зовали его дозу а50, необходимую для 50%-ного удаления взве¬
шенных веществ 3.
Из данных по исследованию коагуляции при содержании взве¬
си в воде от 10 до 200мг/л (рис. VI.3) видно, что для разных глин —
бентонита и каолинита — связь lg а50—lg Ек выражена сход¬
ными кривыми с чередованием отрицательного наклона (малая
мутность) с положительным (большая мутность). При Ек ^
^ 10 мг-экв/л уменьшению дозы коагулянта способствовали зна¬
чения pH, близкие к рНг гидроокиси алюминия, а при Ек >
^> 10 мг-экв/л — более низкие pH (~6,5). Весовые концентрации
суспензий, соответствующие перемене знака наклона кривых
(около 100 мг/л для каолинита и 50 мг/л для бентонита), зависели,
по-видимому, от размера глинистых частиц: более низкие концен¬
трации характерны для относительно мелких частиц бентонита.
Ранее к выводу об увеличении удельного расхода коагулянта
3 См. раздел VI.2.1.
160
ГЛАВА VI. ОПТИМАЛЬНАЯ ДОЗА КОАГУЛЯНТА
(на 1 мг/л дисперсной фазы) с уменьшением размера частиц за¬
грязнений пришла Лебедева [451.
Для высокомутных вод зависимость удельной потребности
в коагулянте от степени дисперсности взвеси носит противополож¬
ный характер [46, 47], причем оптимальные значения pH с умень¬
шением Ек суспензий возрастают, составляя для бентонита — 6,6,
каолинита — 7,5, профиллита — 8,3, кварца — 8,0 [47].
Таким образом, каждый из рассмотренных параметров
(Ек и размер частиц) не определяет оптимальных условий коа¬
гуляции однозначно и их влияние должно рассматриваться
в совокупности. Действительно, даже характер сорбции продук¬
тов гидролиза коагулянта на частицах глины определяется при¬
родой частиц: в случае каолина они адсорбируются преимущест¬
венно на ребрах частиц, а в случае монтмориллонита — на ба¬
зальных поверхностях [48]. Характер сорбции оказывает влия¬
ние на скорость коагуляции, которая при одних и тех же условиях
убывает в ряду суспензий: каолинитовая монтмориллонито-
вая кварцевая [49].
Стамм и О’Мелиа [8, 40] на примере кварцевой суспензии по¬
казали, что при малых концентрациях твердой фазы стехиометри-
ческого соотношения между площадью поверхности частиц и не¬
обходимой дозой коагулянта не существует. Они объяснили это
обстоятельство или слишком малыми возможностями системы
в отношении локализации пересыщения раствора (фактор пересы¬
щения), или необходимостью иметь дополнительное количество
твердой фазы для увеличения числа столкновений частиц (кине¬
тический фактор). Второе предположение представляется более
правомерным.
Рассмотрим имеющиеся материалы по взаимной связи опти¬
мальных условий протекания процесса (доза коагулянта, pH)
с ДП частиц. Под оптимальной будем понимать такую минималь¬
ную дозу коагулянта, при которой после отделения в осадок коа¬
гулированной взвеси (в течение заданного промежутка времени)
достигается наименьшая остаточная мутность воды.
В серии работ Блэка и сотрудников с искусственно приготов¬
ленными глинистыми суспензиями (65—300 мг/л) зависимость
между остаточной мутностью и конечными значениями ДП хлопь¬
ев обнаружена лишь для монтмориллонита. Опыты, проведенные
с водой различных источников водоснабжения, показали, что
при коагулировании сульфатом алюминия наименьшая остаточ¬
ная мутность имеет место при ДП порядка 10 мв [50, 51]. Однако
результаты оказались хорошо воспроизводимыми лишь в лабора¬
тории. В производственных условиях на связь ДП с дозой коагу¬
лянта сильное влияние, помимо pH среды, оказывало присутствие
органических примесей [52]. Кроме того, как известно [531, при
значениях ДП порядка 5 мв результаты микроэлектрофоретиче-
VI.i. МЁХАНиЗМ ФОРМИРОВАНИЯ КОАРУЙИРОЙАЙЙОЙ ВЗВЁСЙ 1б1
ских измерений практически не воспроизводятся — наблюдается
сильный разброс опытных точек, а если частицы загрязнений
оказываются полностью покрытыми продуктами гидролиза, на¬
ходящимися в изоэлектрическом состоянии, то дальнейшее уве¬
личение дозы коагулянта («overdose») не влияет на подвижность
хлопьев [26].
Попытки обосновать связь величины ДП коагулированной
взвеси с условиями коагуляции предпринимали Тамбо [54],
Халл [431, Шульц и Старке [53] и другие. Тамбо, исходя из основ¬
ных положений физической теории коагуляции лиофобных золей
и уравнения Дебая—Гюккеля, подсчитал, что в условиях водо¬
очистки критические значения ДП хлопьев должны находиться
в пределах 8—12 мв, причем по мере повышения минерализации
воды эти значения уменьшаются.
На практике удовлетворительная связь между дозой коагу¬
лянта и величиной ДП достигается лишь при неизменных исход¬
ных условиях, и потому дзета-потенциал как критерий скорости
и полноты протекания коагуляции может быть использован
только при одном и том же составе воды в источнике [55, 56].
Между наклоном кривых ДП — pH и формой осаждающихся
продуктов гидролиза алюминия существует довольно четкая
взаимосвязь [43J, описываемая уравнением
или с учетом произведения растворимости /р гидроксокомплексов,
адсорбирующихся на поверхности частиц загрязнений,
Уравнения (VI.1) и (VI.2) не отражают влияния общей мине¬
рализации и ионного состава воды, которое проявляется, во-пер¬
вых, в буферных свойствах системы, во-вторых — в химическом
составе, структурных особенностях и заряде частиц коагулятов.
Рассмотрение.этого влияния даст дополнительные сведения о за¬
кономерностях построения коагулированной взвеси.
Для вод с малым солесодержанием зоны оптимума pH коагу¬
ляции обычно уже, чем для вод с большим солесодержанием [30,
57 (стр. 138), 58 (стр. 45)], но, по-видимому, гораздо большее зна¬
чение имеет концентрация отдельных ионов [59 (стр. 107), 60, 61].
Особенно велика роль анионов, в том числе входящих в состав
коагулянтов. Анионы, способные к образованию малораство¬
римых комплексов, смещают положение изоэлектрической точки
продуктов гидролиза в сторону низких значений pH [30, 62, 63].
По данным Кульского [59, стр. 106], скорость коагуляции
гидроокисей алюминия и железа достигает максимума при кон¬
центрациях SO4-, С1~ и НСОз, во много раз превышающих кон-
ДП = const — 0,028 pH,
(VI.1)
ДП = const — 0,028 pH — 0,056 lg Zp.
(VI.2)
6 Е. Д. Бабенков
162
ГЛАВА VI. ОПТИМАЛЬНАЯ ДОЗА КОАГУЛЯНТА
Рис. VI.4. Зависимость дли¬
тельности (т) коагулирования
воды алюминатом натрия от pH
воды при разных концентра¬
циях ионов SO®- (мг/л)
1 — 25; 2 — 125
дентрацию анионов в природных водах. Исследования влияния
анионов в концентрациях, близких к реальным [59 (стр. 107),
64—69], привели к следующим выводам:
1) наибольшая скорость коагуляции продуктов гидролиза
FeCl3 и А1С13 имеет место при увеличенных концентрациях НС03
и С1--ионов, а продуктов гидролиза Fe2(S04)3 и A12(S04)3— при
приблизительно одинаковых концентрациях S04-, НС03 и С1-;
2) анионный состав сильнее влияет на соли алюминия, чем
на соли железа;
3) зоны оптимальных концентраций анионов расширяются
при использовании смесей солей алюминия и железа.
Наиболее сильным коагулирующим действием из рассматри¬
ваемых анионов обладают сульфаты. Они расширяют оптималь¬
ные зоны pH [30, 58, 65, 70], как это показано на рис. VI.4, и
уменьшают потребность в коагулянтах: алюминате натрия, хлори¬
дах алюминия и железа. Улучшение коагуляции с увеличе¬
нием концентрации S04- при pH < 6,5 Клячко и Апельцин [57,
стр. 138] объясняют тем, что сульфаты являются противоионами
по отношению к образующимся в кислой среде положительно
заряженным продуктам гидролиза. В действительности все об¬
стоит несколько сложнее.
Измерения электрофоретической подвижности хлопьев вы¬
явили понижение рН; коагулятов алюминия примерно на единицу
от добавления 36 мг-экв/л S04- [62]. Аналогично сульфатам, но
еще сильнее действуют фосфаты (см. гл. VII). Все это говорит
в пользу «химической» концепции влияния анионов, т. е. образова¬
ния малорастворимых солей. Но обращает на себя внимание и то
обстоятельство, что стимулирующее действие анионов на ско¬
рость хлопьеобразования при добавке к воде сернокислого алю¬
миния изменяется примерно в том же ряду, в котором эти анионы
влияют на суммарное изменение энтропии воды [30, 59 (стр. 108),
71]:
РО^- > SO®- > Cl- > нсо-
VI. 1. МЕХАНИЗМ ФОРМИРОВАНИЯ КОАГУЛИРОВАННОЙ ВЗВЕСИ 163
На контактную коагуляцию соединений алюминия в зернистом
слое наиболее сильное влияние оказывают бикарбонаты, действие
же сульфатов незначительно [72, 73]. Хлориды, по одним данным,
стимулируют коагуляцию, действуя аналогично сульфатам (в кон¬
центрациях, в 8 раз больших) [30], по другим — ингибируют про¬
цесс, затрудняя хлопьеобразование при низких значениях pH
[59 (стр. 108), 64].
Катионный состав воды, по мнению большинства исследовате¬
лей [59 (стр. 106), 63, 74, 75], мало влияет на ход коагуляции.
По данным Кульского и Гороновского [76, стр. 52], замена в обра¬
батываемой воде натриевых солей на кальциевые и магниевые
приводит только к некоторому снижению величины pH вследствие
различия в силе оснований. Однако несомненно влияние катионов
на устойчивость дисперсных загрязнений. Не исключается их
участие и в реакциях комплексообразования.
Влияние катионов на глинистые суспензии при pH < 7
уменьшается в ряду Са2+ Mg2+ Na+, а при pH > 7 — в ряду
Mg2+> Са2+> Na+ [71]. Для нормального хода коагуляции
солями железа рекомендуется суммарная концентрация кальцие¬
вых и магниевых ионов не ниже 1—1,5 мг-экв/л [77, стр. 141].
На основании имеющихся материалов можно допустить, что
при обработке йоды в зоне pH < рНг, когда продукты гидролиза
коагулянта имеют положительный заряд, ускорению коагуляции
будет способствовать присутствие солей с одновалентными катио¬
нами и многовалентными анионами (например, Na3P04), а в зоне
pH рНг — присутствие солей с многовалентными катионами
и одновалентными анионами (например, СаС12) [71]. Однако экспе¬
риментальных данных для подтверждения этой гипотезы пока
недостаточно.
VI. 1.2. Обесцвечивание воды
Известно, что обработкой воды коагулянтами можно устранить
до 80—90% веществ, обусловливающих цветность, но механизм
обесцвечивания еще не вполне ясен. Кульский и Когановский
в ряде работ предложили гипотезу сорбционного механизма обес¬
цвечивания воды. Согласно этой гипотезе, обесцвечивание насту¬
пает в результате сорбции окрашивающих веществ на поверхности
продуктов гидролиза коагулянта. Использование уравнения
Лэнгмюра для описания связи между величиной исходной цветно¬
сти Ц и дозой коагулянта а, требующейся для снижения цветности
до 20 градусов, в предположении о сорбционном механизме обес¬
цвечивания дает выражение
Г = ц~20 = г 20
А + 20
6*
164
ГЛАВА VI. ОПТИМАЛЬНАЯ ДОЗА КОАГУЛЯНТА
по которому при любых значениях Ц Г<х> = const и А = const,
между Ц и а существует линейная зависимость. В действитель¬
ности, как показывают исследования и производственный опыт,
соотношение а = / (Ц) в большинстве случаев описывается сте¬
пенной зависимостью, а количество окрашивающих веществ,
удаляемых коагуляцией, составляет лишь небольшую долю от
полной адсорбционной способности формирующихся продуктов
гидролиза.
За рубежом одна группа исследователей [3, 23, 31, 32, 78, 791
поддерживает и развивает гипотезу Севилла [80] и Миллера
[10—13], согласно которой обесцвечивание объясняется главным
образом взаимной коагуляцией противоположно заряженных кол¬
лоидов цветности и продуктов гидролиза коагулянта. Представи¬
тели другой группы [7, 39, 81—85] ведущую роль в обесцвечива¬
нии отводят химическому взаимодействию между продуктами
гидролиза катионов металлов и карбоксильными группами ани¬
онных окрашивающих веществ, приводящему к образованию ком¬
плексов. Эту гипотезу предложил Холл в 1914 г. [86], но по-
настоящему она привлекла внимание исследователей после ряда
работ, опубликованных в шестидесятых годах [7, 39, 84]. Ответ
на вопрос, какая из этих двух гипотез верна, затруднителен преж¬
де всего из-за большого разнообразия и недостаточной изучен¬
ности природы окрашивающих соединений.
Кульский [64] показал, что при небольшой цветности воды
(30—40 град) коагуляция продуктов гидролиза алюминия несколь¬
ко ускоряется, а при большой (250 град) — замедляется. Наблю¬
даемое явление он объяснил сенсибилизирующим действием
гуминовых веществ в первом случае и защитным — во втором.
Влияние цветности воды на скорость хлопьеобразования отмечали
и другие исследователи [87]. В Японии выдан патент на способ
ускорения коагуляции путем добавления к воде небольших ко¬
личеств гуматов — продуктов кислотной обработки каменного
угля [88]. Все эти сведения указывают на необходимость различать
действие коагулянта на окрашивающие вещества и действие
окрашивающих веществ на формирование хлопьев.
Усовершенствование техники эксперимента дало возможность
получить новые материалы, которые можно систематизировать
следующим образом.
А. Максимальная степень обесцвечивания воды как в лабора¬
торных, так и в производственных условиях имеет место при зна¬
чениях ДП ~ 0 (рис. VI.5) [23, 51, 60]. Однако эффект, достигае¬
мый при снижении ДП за счет добавления кислоты, не равно¬
ценен эффекту, достигаемому в присутствии катионов коагулян¬
та [83].
Б. Между исходной цветностью воды и потребностью в коа¬
гулянте часто обнаруживается связь, близкая к линейной [39, 78,
VI. 1. МЕХАНИЗМ ФОРМИРОВАНИЯ КОАГУЛИРОВАННОЙ ВЗВЕСИ 105
4 а, г/галл
Рис. VI.5. Зависимость конечной цветности воды Цк (а) и электрофоретиче¬
ской подвижности и (б) хлопьев коагулированной взвеси, образовавшейся
при обработке воды сульфатом железа, от pH
1 — без добавления к воде СаС12; 2 — после добавления к воде 1 мг-экв/л СаС12
Рис. VI.6. Зависимость между цветностью исходной воды (Ци) и минималь¬
ной дозой Fe2(S04)3, необходимой для обесцвечивания воды до заданного
уровня [78]
89—911 4. Примером может служить рис. VI.6, где показаны ко¬
личества «реагирующих» веществ: железа и окрашивающих сое¬
динений (по весовому эквиваленту фульвокислотной фракции).
Авторы [45, 921 отмечают, что для снижения цветности воды на
один градус требуется от 0,85 до 4,5 мг/л химически чистого сер¬
нокислого алюминия. Наряду с этим имеются многочисленные
свидетельства отсутствия стехиометрического соотношения [45,
46, 77, 91, 93-951.
В. Наилучшее обесцвечивание достигается в узкой области
оптимальных значений pH, которая определяется видом коагу¬
лянта, щелочностью воды, концентрацией и свойствами окраши¬
вающих веществ. Оптимальные значения pH для сернокислого
алюминия составляют 4,5—6,2 [13, 23, 39, 82, 94, 96—991, для
хлорного железа — 3,5—5 [23, 39, 82, 83, 100J. Они совпадают
с наилучшими условиями формирования высокозаряженных
гидроксокомплексов.
По мере увеличения исходной цветности (рис. VI.7) и тем¬
пературы (рис. VI.12) воды оптимум pH смещается в сторону вы¬
соких концентраций водородных ионов [39, 60, 78, 98] и оптими-
В некоторых работах фиксировали потребность в коагулянте для удаления
вэщэств цвзгногти лишь на 50%.
166
ГЛАВА VI. ОПТИМАЛЬНАЯ ДОЗА КОАГУЛЯНТА
ст,мг/л
Рис. VI.7. Зависимость оптимальных значений pH обесцвечивания воды
коагулянтом от цветности исходной воды (Ци) [78]
Рис. VI.8. Взаимосвязь менаду остаточной концентрацией (сг) гуминовой
кислоты в воде и ДП коагулированной взвеси [39]
зация pH приобретает более важное значение, особенно для
фульвокислот [82, 101].
Г. Дубильные вещества и фульвокислоты удаляются хуже,
чем гуминовые вещества и требуют более низких значений pH
[39, 102 (стр. 13), 103, 104].
Д. Обесцвечивание завершается в первые минуты после добав¬
ления коагулянта к воде. Дальнейшие процессы хлопьеобра-
зования, седиментации и фильтрации не дают дополнительного
удаления веществ цветности [97, 105]. В отличие от вод, содер¬
жащих минеральные примеси, результаты обесцвечивания в ла¬
бораторных и производственных условиях совпадают [97]. Все эти
факты говорят в пользу «химической» гипотезы обесцвечивания.
Е. Влияние солевого состава проявляется по-разному и может
быть объяснено конкуренцией анионов с веществами цветности
за координационные участки гидроксокомплексов алюминия и
железа.
С увеличением концентрации НСОз-ионов сверх некоторого
предельного значения потребность в коагулянте возрастает [95,
105, 106]. Рост концентрации сульфатов также дает отрицательный
эффект [60, 83, 105], который может быть компенсирован
полностью или частично добавлением эквивалентных количеств Са2+
и Mg2+. Присутствие этих катионов расширяет зону оптимальных
pH в сторону высоких значений и повышает глубину обесцвечи¬
вания [60, 82, 83]. Причем обращает на себя внимание то, что по
своему стимулирующему действию катионы располагаются в тот
же ряд, что и при коагуляции глинистых суспензий (pH > 7,5) :
Mg2+> Са2+> Na+ [60]. Изменение концентрации Na+ и Cl-
мало влияет на остаточную цветность воды [83, 105].
VI.2. ВЫВОР ДОЗЫ КОАГУЛЯНТА
167
Как показали Халл и Пакхам [391, между логарифмом остаточ¬
ной концентрации гуминовых веществ и величиной ДП в широком
диапазоне значений pH (4—8) существует линейная зависимость,
позволяющая считать справедливым равенство
ДП = conFt - 28 lg [Х-], (VI.3)
в котором [Х“] — концентрация ионогенных групп окрашиваю¬
щих веществ, главным образом карбоксильных. В общем случае
глубина обесцвечивания определяется степенью диссоциации
карбоксильных групп и растворимостью гидроксидных комплек¬
сов, а оптимум значений pH (рис. VI.8) — соотношением между
числом диссоциированных карбоксильных групп и количеством
малорастворимых продуктов гидролиза коагулянта. Некоторое
несоответствие значений экспериментально найденных оптималь¬
ных pH и рассчитанных с помощью уравнения (VI.6) может быть
объяснено или неравновесным состоянием системы, или погреш¬
ностями расчета, связанными с недоучетом влияния других пара¬
метров.
Прогрессивное увеличение потребности в коагулянте при pH,
превышающих оптимальные значения, обусловлено, вероятно,
ионизацией новых кислотных групп (предположительно феноль¬
ных) и образованием растворимых А1-хелатных комплексов
[39, 81].
VI. 2. ВЫБОР ДОЗЫ КОАГУЛЯНТА
При недостатке коагулянта плохие результаты очистки воды
объясняют чаще всего неполной астабилизацией частиц загряз¬
нений, при избытке — новой стабилизацией (рестабилизацией)
частиц вследствие их перезарядки. В обоих случаях коагуляция
протекает вяло, обработанная вода опалесцирует, содержит
в заметных количествах остаточные алюминий и железо. Оптими¬
зация режима коагулирования составляет центральную техноло¬
гическую задачу.
Сведения о связи оптимальной дозы коагулянта а с качествен¬
ными параметрами воды противоречивы. При достаточно сильном
загрязнении воды оказываются справедливыми эмпирические за¬
висимости
а = А (св)П1, а = к (Ц)%
в которых св и Ц — соответственно содержание взвешенных
примесей в воде и ее цветность.
Для значений св, меньших 100 мг/л, характерных почти для
всех источников водоснабжения страны большую часть года,
экспериментальных данных о зависимости а = /х (св)П1 очень
мало, а строительные нормы и правила [46] никаких рекоменда¬
{68
ГЛАВА Vi. ОПТИМАЛЬНАЯ доЗа НоаГуляйТА
ций по выбору дозы коагулянта при малом содержании взвеси
в воде не дают.
Исследования направлены, главным образом, на обработку
методами линейного и нелинейного регрессионного анализа много¬
летних данных по разным источникам водоснабжения с последую¬
щим выводом эмпирических зависимостей [107, 108, 109 (стр. 121)].
Делаются также попытки получения обобщенной теоретической
зависимости дозы коагулянта от показателей качества воды,
поскольку использование такой зависимости в автоматической
системе управления очисткой воды может за счет снижения
расхода коагулянта дать существенный экономический эффект.
На одной из таких попыток, осуществленной автором [110, 111],
мы остановимся подробнее.
Предложенное выше (см. стр. 160) определение оптимальной
дозы коагулянта носит технологический характер. В отличие от
абстрактной «теоретической» потребности в коагулянте, которая
по мере приближения к нулю концентрации загрязнений также
приближается к нулю, реальная технологически оптимальная
доза коагулянта стремится к некоторому пределу (а -> а0), вели¬
чина которого зависит от задач очистки и принципа работы водо¬
подготовительного устройства. Приведем два примера.
При удалении из маломутной воды небольших количеств
фтора на осветлителях со взвешенным осадком «теоретическая»
потребность в гидроокиси алюминия, подсчитанная из соображе¬
ний сорбции фтора, может оказаться значительно ниже фактиче¬
ской потребности, т. е. потребности с учетом поддержания стабиль¬
ных параметров контактной среды в осветлителе. При очистке
маломутной воды на контактных осветлителях потребуются мень¬
шие дозы коагулянта, чем при очистке в отстойниках, поскольку
при контактной коагуляции необходимость в дополнительном
количестве продуктов гидролиза для «сшивания» микрохлопьев
мала или вовсе отсутствует.
Таким образом, технологически оптимальная доза коагулянта
(которую в дальнейшем мы будем называть просто оптимальной)
зависит от условий отделения взвеси в осадок, а разница между
ней и дозой ч<теоретической» характеризует степень совершенства
водоочистного аппарата и является резервом экономии коагу¬
лянта.
Как указывалось, для оценки результатов экспериментов Пак¬
хам [29, 30] ввел понятие дозы коагулянта, обеспечивающей
50%-ное удаление загрязнений - аъо. Кроме Пакхама этот пока¬
затель использовали в работе Ким и сотр. [42], Халл [43] и другие.
Хорошую воспроизводимость дозы а&0 можно объяснить харак¬
тером влияния постепенно возрастающих дозировок коагулянта
на величину остаточной мутности воды (см. рис. VI.1): значе¬
ние а50 соответствует крутому спаду кривой М0 — / (а). Однако,
VI.2. ВЫБОР ДОЗЫ КОАГУЛЯНТА
169
но нашему мнению, доза аъ0 не отражает практической потребно¬
сти в коагулянте, так как отношение а50 : аопт в зависимости
от условий коагуляции может иметь разные, сильно отличающиеся
значения.
Гораздо более объективным показателем является доза, тре¬
бующаяся для очистки воды до приемлемой степени (90—95%).
Удовлетворительная воспроизводимость ее при использовании
фотоколориметрического метода измерения оптической плотности
пробы во времени показана ранее [110, 112].
По-видимому, правильный подход к расчету оптимальной
дозы коагулянта может быть осуществлен, если исходить из на¬
личия дальнодействующих сил притяжения между частицами,
существование которых предсказано теорией ДЛФО и подтвер¬
ждено многочисленными экспериментами. Действительно, очень
многие свойства коагулятов, такие, как адгезионная способность,
тиксотропия, приблизительное постоянство удельной поверхности
во времени, относительно малая прочность и ее зависимость
от вероятности коллективных взаимодействий частиц, могут быть
удовлетворительно интерпретированы лишь с учетом взаимодей¬
ствия частиц во вторичном энергетическом минимуме. О прояв¬
лении дальнодействующих сил говорит и тот факт, что все те
меры физического воздействия, которые увеличивают вероятность
дальнего взаимодействия частиц, способствуют ускорению коагу¬
ляции (см. гл. IV).
Характер дальнего взаимодействия отличается от характера
ближнего взаимодействия. Во-первых, первичные частицы со¬
храняют свою индивидуальность; во-вторых, из-за сравнительно
небольшой глубины вторичного энергетического минимума агре¬
гаты довольно легко распадаются под действием внешних сил;
в-третьих, несмотря на наличие гидратных оболочек, частицы
могут вновь объединяться в агрегаты [33, стр. 254].
При использовании концепции о дальнодействующих силах
притяжения для теоретического обоснования оптимальной дозы
коагулянта целесообразно, как и в предыдущем разделе, от¬
дельно рассмотреть процессы осветления и обесцвечивания воды.
VI. 2.1. Осветление воды
Анализ экспериментальных материалов, выполненный в преды¬
дущих разделах, показывает, что необходимыми и достаточными
условиями успешного осветления воды являются: 1) обволакива¬
ние частиц взвеси продуктами гидролиза коагулянта для обеспе¬
чения их слипания при столкновениях; 2) накопление достаточно
большого количества твердой фазы для построения хлопьев коагу¬
лированной взвеси, отвечающих по своим свойствам технологиче¬
ским требованиям, «Обволакивание» — термин, заимствованный
170
ГЛАВА VI. ОПТИМАЛЬНАЯ ДОЗА КОАГУЛЯНТА
у Пакхама, рассматривается здесь как закрепление и последую¬
щее наращивание на поверхности частиц продуктов гидролиза
коагулянта. В отличие от флокуляции полиэлектролитами частицы
взвеси вследствие большой толщины окружающих их гидроксид-
ных оболочек полностью утрачивают индивидуальные свойства.
В соответствии с принятыми допущениями можно рассма¬
тривать оптимальную дозу коагулянта как складывающуюся из
двух частей: аи обеспечивающей обволакивание взвеси продук¬
тами гидролиза, и а2, дающей дополнительное количество твердой
фазы для удовлетворения кинетических требований процесса.
Не исключено, что контакты между частицами загрязнений
(в частности, лиофобных) и продуктами гидролиза вследствие
разнородности их материала и разноименности зарядов осущест¬
вляется за счет коагуляции в первичном энергетическом миниму¬
ме. Такой контакт отличается высокой прочностью. Однако моно-
слойное покрытие частиц загрязнений продуктами гидролиза
коагулянта не обеспечивает критической массы хлопьев, достаточ¬
ной для их быстрого осаждения. Требуется дополнительный
«строительный материал» на формирование вокруг одной или не¬
скольких частиц загрязнений «облака» продуктов гидролиза.
Сильно гидратированные и одинаковые по природе частицы скоа-
гулированы в этом «облаке» в области вторичного энергетического
минимума и потому могут отрываться и прилипать вновь. Действи¬
тельно, отрыв и прилипание можно наблюдать экспериментально.
Дополнительное количество твердой фазы, не связанной не¬
посредственно с поверхностью частиц загрязнений, является ре¬
зервом, за счет которого происходит быстрое восстановление раз¬
рушающихся в потоке воды агрегатов астабилизированных частиц.
Можно допустить, что между коагулятами, связанными и не свя¬
занными с поверхностью частиц загрязнений, происходит посто¬
янно эквивалентный обмен, характеризующийся некоторой средне¬
статистической константой равновесия: разрушенные хлопья за счет
резерва коагулята в течение определенного практически прием¬
лемого промежутка времени полностью восстанавливаются. В от¬
личие от гипотез Стамма—О’Мелиа [40] и Пакхама [29, 30] в нашей
гипотезе подчеркивается определяющая роль кинетики коагуля¬
ции. Это дает возможность разграничить условия коагуляции
при малых и больших концентрациях взвеси.
Установившийся режим, в котором между продуктами гидроли¬
за, связанными с дисперсными примесями воды и присутствую¬
щими в виде резервного «строительного» материала, наступило
равновесие, формально сходен с адсорбцией. Обозначим через аадс
потребность в коагулянте для формирования продуктов гидролиза,
адсорбировавшихся на поверхности дисперсных частиц, а через
яРв — для формирования равновесного количества коагулята.
Тогда применение уравнения Лэнгмюра [40] дает для оптимальной
VI.2. ВЫБОР ДОЗЫ КОАГУЛЯНТА
171
дозы коагулянта
аопт — аадс + Ярв — ^ ^ _^g ^ [1 — ksl оо (1 — 0р)],
(VI.4)
где 0Р — степень покрытия коагулятом поверхности частиц
взвеси; s — площадь поверхности частиц; к константа равно¬
весия; Гоо — максимальная величина адсорбции.
При аадс арв (хорошая сорбция) уравнение (VI.4) прини¬
мает вид
т. е. между а0Пт и s существует линейная связь.
При аадс с арв (плохая сорбция)
т. е. аопт не зависит от s.
Таким образом, при больших значениях Г^, к и s или при
0Р -> 0 обнаруживается линейная связь между дозой коагулянта
и площадью поверхности частиц, а при малых значениях Гоо, к
и s или при 0Р —>-1 не обнаруживается никакой связи* Этот
вывод не отражает результатов экспериментов, согласно которым
в области высоких концентраций взвеси линейная связь между
а0пт и s существует лишь в логарифмических координатах,
а в области низких концентраций взвеси зависимость дозы коа¬
гулянта от s сохраняется, хотя и меняется ее характер. Кроме того,
для характеристики процесса коагуляции гидролизующимися
коагулянтами вряд ли целесообразно использовать параметры Г^
и 0Р, физический смысл которых в данном случае весьма сомни¬
телен.
Чтобы исключить из расчетов параметры Г* и 0Р и осущест¬
вить хотя бы приблизительный подход к определению величины
оптимальной дозы коагулянта, воспользуемся уравнением Фрейнд¬
лиха, описывающим изотерму адсорбции с достаточной для прак¬
тических расчетов точностью даже в тех случаях, когда толщина
слоя адсорбированного вещества велика. Для дозы ах можно на¬
писать:
где ку - коэффициент, определяющий астабилизирующие свойства
продуктов гидролиза коагулянта; п - показатель степени, за¬
висящий от химических условий коагуляции.
После замены равновесной концентрации продуктов гидролиза
арв на а2 будем иметь следующее выражение:
(VI.5)
d\/S = kiOpg,
(VI.7)
^опт — kxsai -j- ci%.
(VI.8)
172
ГЛАВА VI. ОПТИМАЛЬНАЯ ДОЗА КОАГУЛЯНТА
Величина а2, с одной стороны, зависит от особенностей работы
водоочистного устройства, которые определяют критическую массу
хлопьев, соответствующую их успешному отделению в осадок,
и минимально допустимую скорость восстановления этой массы,
с другой стороны — от количества хлопьев в единице объема
среды. Чем больше это количество, тем быстрее восстанавливаются
хлопья и, следовательно, тем меньше требуется равновесного
коагулята. Поскольку количество хлопьев в единице объема среды
определяется числом столкновений коагулирующих частиц, кото¬
рое в свою очередь пропорционально квадрату их концентрации,
величину аг можно представить в виде
а2 = k2/(Na + NBy, (VI.9)
где к2 - коэффициент, учитывающий кинетические особенности
протекания процесса коагуляции; Na - концентрация частиц
продуктов гидролиза коагулянта; NB - концентрация частиц
загрязнений.
При отсутствии загрязнений в воде, когда NB = 0, уравнение
(VI.9) преобразуется в выражение
а2 = а0 = kJNl, (VI.9')
где к2 - коэффициент, характеризующий кинетические условия
коагуляции только частиц продуктов гидролиза.
Из уравнений (VI.8) и (VI.9) получаем
Яопт = а1 а2 — k\S [к2 (Уа -j- NB) “]1/u -(- &2 (Va-fiVe) 2 —
= K[s (Va + NB)~V" + k2 (Va + NBy\ (VI.10)
где Kv = /cx/cf.
Анализ уравнения (VI.10) затруднен, поскольку требует
экспериментального определения параметров s, Na, и NB■ Упро¬
стим анализ путем замены этих параметров на соответствующие
весовые концентрации загрязнений св и продуктов гидролиза
коагулянтов са. Такую замену можно сделать при условии по¬
стоянства размера и плотности частиц загрязнений и продуктов
гидролиза коагулянта.
Обозначив произведения отдельных коэффициентов, входящих
в оба члена правой части выражения (VI.10), через Кг и К2, полу¬
чим
«опт = KlCa (ca + kcB)~2ln + К2 (са + kcBy2. (VI.И)
Коэффициент к характеризует среднестатистическое соотно¬
шение размеров частиц загрязнений воды и продуктов гидролиза
коагулянта на характерном этапе построения коагуляционной
структуры. При одной и той же степени дисперсности первичных
V1.H. ВЫБОР ДОЗЫ КОАГУЛЯНТА
173
Рис. VI.9. Зависимость дозы
коагулянта (а) от содержания
взвеси в воде (св)
частиц коагулята, которая определяется в основном величиной pH
среды [113, 114], коэффициент к зависит только от дисперсного
состава загрязнений.
Уравнение (VI.11) отражает совокупное влияние основных
условий протекания коагуляции на величину оптимальной дозы
коагулянта.
Оптимальная доза коагулянта, вычисленная по уравнению
(VI.11), может быть представлена суммой двух слагаемых
(рис. VI.9), одно из которых (аг) возрастает, а другое (а2) умень¬
шается с ростом ев. Кривая а = f (св) носит V-образный характер
с минимумом, соответствующим некоторой критической концен¬
трации взвеси скр. Наличие минимума объясняется тем, что при
св < сКр рост св приводит к более медленному возрастанию а1г
чем уменьшению а2, а при св скр величина а2 быстро становится
пренебрежимо малой и оптимальная доза почти целиком опреде¬
ляется величиной аг. Таким образом, при св < скр узким местом
коагуляции является недостаточное число столкновений частиц,
а при св скр — малая вероятность их слипания.
Опытом эксплуатации водоочистных сооружений установлено,
что существенное улучшение кинетики коагуляции наступает
обычно при св = 10—50 мг/л, эти значения можно считать крити¬
ческими. Наличие минимума на кривой аопт = / (св), представ¬
ленной на рис. VI.9, было выявлено ранее в экспериментах по
частичному осветлению суспензий [42]. Уменьшение аопт с уве¬
личением концентрации взвеси до 63,3—150 мг/л показано в рабо¬
тах [25, 41, 115, 116]. Однако результаты этих экспериментов
не нашли теоретического обоснования. Отсутствие сведений
о критических величинах концентрации взвеси скр в других пуб¬
ликациях объясняется использованием методик визуального под¬
бора оптимальных доз коагулянта, которые в области св < скр
дают плохо воспроизводимые результаты.
Оценка численных значений коэффициентов, входящих в урав¬
нение (VI.11), выполненная нами ранее [110J, показала, что
при концентрации взвеси св < скр это уравнение может быть
174
глава VI. оптимальная Доза коагулянта
заменено более простым
а0пт = Кс?, (VI.12)
в котором показатель степени тп может находиться в пределах от О
до 0,8.
Обработка результатов экспериментов многих авторов
(рис. VI.10) приводит к выводу, что в случае св ^> 100 —200 мг/л
все они описываются уравнением (VI.12) при значениях К =
= 1,0—10,0 и тп — 0,28—0,65. Значения коэффициентов зави¬
сят от вида использованного коагулянта, условий водоочистки и
поставленных технологических задач (см. таблицу).
Значения коэффициентов в уравнении (VI.12) но данным разных авторов
Коэффициенты
Литература
Коэффициенты
Литература
К
m
К
m
4,5
0,43
[46]
10,0
0,39
[119]
3,1
0,42
[46]
8,9
0,41
[120]
3,4
0,44
[45]
3,0
0,33
[57]
6,9
0,34
[117]
1,0
0,58-0,65
[121]
1,3
0,53
[117]
1,3
0,28
[122]
3,7
0,49
[118]
Экспериментальная проверка уравнения (VI.11), проведенная
для суспензий монтмориллонитовой и каолинитовой глин в ши¬
роком диапазоне значений св при разной интенсивности перемеши¬
вания коагулянта с водой, а также разных значениях pH, щелоч¬
ности и температуры воды [110—113], подтвердила его справедли¬
вость. Для обоих замутнителей обнаружено (рис. VI. 11) предска¬
занное теоретически существование минимума на кривых а0пт =
= / (св) в области са = 30—50 мг/л. Значения коэффициентов,
входящих в уравнение, составляли в зависимости от условий экс¬
периментов: Кх = 1,08—1,60; К2 = (1,53—5,2)-105; п = 2,2—
9,5; к = 0,9—2,3.
Особый интерес представляют экспериментальные данные о
связи оптимальной дозы коагулянта со степенью дисперсности ми¬
неральных частиц. Исследователи давно отмечали, что повышение
степени дисперсности частиц взвеси вызывает прирост расхода
коагулянта [123, стр. 112]. Это вполне объяснимо, если учесть,
например [124], что адсорбция продуктов гидролиза алюминия
на образцах мусковита и биотита с размером частиц d < 0,08 мкм
примерно в 10 раз выше, чем на образцах этих же минералов с раз¬
мером частиц 2—5 мкм. Попытка количественной оценки явления
VI.2. ВЫБОР ДОЗЫ КОАГУЛЯНТА
175
а, тг/м
Рис. VI.10. Зависимость дозы безводного сернокислого алюминия (а) от св
по экспериментальным данным разных авторов
1 и 2 — по [46] (соответственно для тонкодисперсной и грубодисперсной взвеси); 3—
по [45]; 4 и 5 — по [117] (соответственно для отстойников и осветлителей); б — по [118];
7 — по [119]
Рис. VI.11. Зависимость а от св для воды, замутненной монтмориллонитом
(кривая 1) и каолинитом (кривая 2)
позволила установить эмпирическую зависимость [4U].
а0Пт = Kssm, (VI. 13)
где Ks и т — константы, характеризующие условия сорбции.
Согласно исследованиям других авторов, вместо площади по¬
верхности частиц в правую часть уравнения может быть подстав¬
лена величина катионообменной емкости взвеси [42] или суммар¬
ный заряд частиц, определенный методом «коллоидного титрова¬
ния» [125]. При этом степенная форма зависимости сохраняется.
Поскольку эта форма зависимости не нашла теоретического обос¬
нования, уравнение (VI. 13) применимо только для тех частных
случаев, когда очищается вода одинакового состава.
Согласно предложенной гипотезе, в области высоких весовых
концентраций взвеси, т. е. при св сКр, процесс коагуляции ха¬
рактеризуется недостаточно высокой вероятностью слипания час¬
тиц. Повысить ее можно путем увеличения степени покрытия час¬
тиц продуктами гидролиза коагулянта. Чем мельче взвесь, тем
больше потребуется коагулянта. Для вод с очень большой мут¬
ностью расстояния между поверхностями соседних частиц соизме¬
римы с толщиной гидрооксидных оболочек. Поэтому можно пред¬
положить, что увеличение св таких вод будет способствовать умень¬
шению потребности в коагулянте. Действительно, подобная зако¬
номерность обнаружена при обработке коагулянтом воды р. Вахш,
содержащей около 8 г/л взвешенных примесей [126].
176
ГЛАВА VI. ОПТИМАЛЬНАЯ ДОЗА КОАГУЛЯНТА
При св < скр улучшению коагуляции способствует повышение
вероятности столкновений частиц. Чем меньше размеры частиц,
тем больше вероятность их столкновений. Поэтому для тонкодис¬
персных взвесей в области с < сКр должны требоваться меньшие
дозы коагулянта.
Уравнение (VI. 10) позволяет количественно оценить влияние
степени дисперсности частиц загрязнений на величину оптималь¬
ной дозы коагулянта при очистке воды с разной мутностью [110].
Правда, такая оценка может быть лишь весьма приблизительной,
поскольку большое значение имеет не только средний размер час¬
тиц, но и характер распределения частиц по размерам: скорость
объединения мелких частиц (10—20 мкм) с более крупными (40—
60 мкм) в турбулентном потоке может превосходить скорость объ¬
единения частиц одинакового размера в сотни раз [127]. Тем не ме¬
нее опытные данные согласуются с предполагаемым характером
влияния степени дисперсности взвеси на величину аопт- увеличе¬
ние размера частиц в зоне св скр способствует уменьшению
потребности в коагулянте, а в зоне св < скр — увеличению. Зна¬
чения сКр с увеличением степени дисперсности взвеси несколько
снижаются [110].
С понижением температуры во всем диапазоне исследуемых
концентраций замутнителей (до 400 мг/л) зафиксировано увеличе¬
ние потребности в коагулянте. Особенно заметно отрицательное
влияние низких температур при св < скр [110].
Из обзорных материалов, изложенных в предыдущих главах,
ясно, что при низких температурах и недостаточно интенсивном
перемешивании среды вялая коагуляция является следствием за¬
медленного теплового движения молекул и повышенной вязкости
среды, увеличения гидратации коагулирующих частиц, уменьше¬
ния адгезионных сил и прочности хлопьев, уменьшения числа
взаимных столкновений частиц. Другой существенный момент —
влияние температуры на оптимальные значения pH. По упрощен¬
ной формуле [60]
-pH =-g-!g ffw-lgPe, (VI.14)
в которой K-w — ионное произведение, а рв — плотность воды;
увеличение температуры от 0 до 50° С дает понижение pH с 7,47
до 6,63. Соответственно уменьшаются оптимальные значения pH
(рис. VI.12).
Некоторые авторы [128] полагают, что влияние температуры
на этом заканчивается, и если коагулирование проводить с надле¬
жащей корректировкой pH, то доза коагулянта не зависит от тем¬
пературы воды. Действительно, во многих случаях подщелачива-
ние холодной воды позволяет улучшить коагуляцию и уменьшить
расход коагулянта [58 (стр. 55), 129], но не менее сильцое оптимц-
VI.2. ВЫБОР ДОЗЫ КОАГУЛЯНТА
177
Рис. VI. 12. Зависимость оп¬
тимальных значений pH обес¬
цвечивания воды сульфатом
железа от ее температуры (<)
[60]
зирующее действие оказывает добавление замутнителей (см. гл.
VIII), улучшающее транспортные условия протекания процесса.
Поэтому в действии температуры нужно различать химическую и
физическую стороны.
От температуры воды зависит скорость формирования коагу¬
лированной взвеси и конечный размер хлопьев [130]. Причем даже
интенсивное перемешивание не в состоянии компенсировать отри¬
цательное влияние низких температур. В качестве оптимальных
температур при использовании A12(S04)3 называют 25—30° С [131,
стр. 44], 28—30° С [99], 36—40° С [132, стр. 70]. При температуре
выше 30° С возможно ухудшение коагуляции Fe203 вследствие
чрезмерно интенсивного броуновского движения молекул [133].
Влияние режима перемешивания воды с раствором коагулянта
также проявляется двояко. Как показали наши исследования
[110, 134—136], перемешивание с умеренной интенсивностью
(GT = 1,2-105) позволяет ускорить хлопьеобразование, увели¬
чить плотность коагулированной взвеси и снизить на 10—20%
расход коагулянта. Последнее обстоятельство отмечено и други¬
ми исследователями [137 (стр. 231), 138, 139]. Однако увеличение
критерия Кэмпа до 4,1-10® приводило к замедлению формирова¬
ния хлопьев во всем диапазоне принятых св, что объяснено нару¬
шением при высоких скоростных градиентах тиксотропной обра¬
тимости коагуляционных структур (подробнее о влиянии переме¬
шивания см. гл. VIII).
Воздействие температуры и условий перемешивания коагулян¬
та с водой на величину я0пт в уравнении (VI. 11) учитывается изме¬
нением параметров К2 и са.
Для оценки влияния pH среды на оптимальную дозу коагулянта
обозначим через ас дозу коагулянта, обеспечивающую накопление
критической концентрации продуктов гидролиза, необходимой для
формирования и осаждения хлопьев (соответствующее pH — рНс);
через аРн — дозу коагулянта, требующуюся для достижения оптима¬
льного значения рН(рН0); через рНи — pH исходной воды. Рассмо¬
трим пример, когда используется кислотный коагулянт, а щелочной
178
ГЛАВА VI. ОПТИМАЛЬНАЯ ДОЗА КОАГУЛЯНТА
резерв исходной воды достаточен для полного его гидролиза. Воз¬
можны два случая.
Первый случай: рН0 < pH,,. При арн ас по мере увеличения
дозы коагулянта критическая концентрация хлопьеобразования
достигается раньше, чем значение рН0 (рН0 < рНс <; рНи). Для
полной и быстрой коагуляции требуется или увеличить дозу коа¬
гулянта до достижения рН0, или добавить к обрабатываемой воде
эквивалентное количество кислоты акпся, которое может быть вы¬
ражено функцией: / (рНи — рН0) — / (ас).
При арн < ас по мере увеличения дозы коагулянта рН„ дос¬
тигается раньше, чем в воде накапливается критическая концент¬
рация продуктов гидролиза (рНс < рН0 << рНи). Необходима до¬
за щелочного реагента ащел == / (яс) — / (рНи — рН0).
Второй случай: рН0 рНи. При использовании кислотного
коагулянта (рНс < рНи < рН0) доза щелочи находится из выра¬
жения / (ас) + / (pHо — рНи). Функция / (ас) определяется с
учетом буферных свойств системы.
Аналогичные выражения могут быть составлены для щелочных
коагулянтов.
Эффективность каждого коагулянта определяется, таким обра¬
зом, соотношением в нем кислотных (щелочных) и связующих
свойств в. Если значения рНс и рН0 не совпадают, создание опти¬
мальных условий коагуляции с одновременной экономией коагу¬
лянта требует корректировки pH воды.
[Случай, когда рН0 < рНс <; рНи, к водам с малой мутностью
и цветностью не подходит, поскольку для них, как правило,
рН0 ж рНг, а рН; рНи. Он более реален при обработке мутных
вод, содержащих мелкодисперсную взвесь с большой катионооб¬
менной емкостью, или цветных вод с достаточным щелочным ре¬
зервом, для которых рН0 < рНг. Чтобы уменьшить потребность
в кислоте при обработке, в частности, мутных вод с небольшой
цветностью, стремятся использовать более кислые соли железа
с одновалентными сопутствующими ионами (например, FeCl3).
Эти ионы не входят в состав гидрооксидных комплексов и не сме¬
щают вследствие этого рНг в сторону более низких значений [7].
?Для компенсации отрицательного действия SO^-hohob при ис¬
пользовании сернокислых коагулянтов полезно увеличить жест¬
кость воды добавлением, например, хлорида кальция. Эти способы
находят применение при обработке питьевых и сточных вод.
!*Для вод с малой мутностью и высоким pH, когда число столк¬
новений коагулйрующих частиц недостаточно и его необходимо
увеличить за счет малорастворимых продуктов гидролиза, харак¬
терен случай, когда рНс < рН0 <; рНи. Вместо добавки щелочи
5 В этом, вероятно, заключена одна из причин хороших технологических
свойств смешанных коагулянтов,
VI.2. ВЫБОР ДОЗЫ КОАГУЛЯНТА
179
а, мг/л
Рис. VI.13. Зависимость а от св при разных конечных значениях pH воды
(рНк), замутненной монтмориллонитом (а) и каолинитом (б)
1 — рНк нерегулируемое; 2 — рНк ~5; 3 — рНк^7,5
может оказаться рациональным применение алюмината натрия
или оксихлорида алюминия. Целесообразность использования этих
коагулянтов подтверждена испытаниями на московском водопро¬
воде. Из щелочей следует добавлять такие, катион которых не свя¬
зывал бы SO^-ионы и не препятствовал бы уменьшению pH, на¬
пример, NaOH, NaHC03. Чем выше жесткость воды, тем выше ее
рН0 и, следовательно, тем больше требуется щелочи.
Анализ рассмотренных примеров дает возможность сделать
вывод, что подщелачивать целесообразно маломутные воды с низ¬
кой цветностью, а подкислять — мутные и высокоцветные. Эти при-
мерынеисчерпывают, однако, всех возможных в практике случаев.
Например, маломутная, но загрязненная промышленными отхо¬
дами вода р. Потомак при pH ~ 6 требовала примерно в два раза
меньше сернокислого алюминия, чем при pH ж рНг [50]. Кроме
того, как показано в наших ранних исследованиях [114], pH сре¬
ды сильно влияет на физические параметры коагулированной взве¬
си и, следовательно, на эффективность действия водоочистного
устройства.
Для искусственно приготовленных глинистых суспензий эко¬
номия коагулянта достигалась при малых св подщелачиваиием, а
при больших — иодкислением среды (рис. VI. 13). Подщелачива-
ние приводило к увеличению показателя степени п и уменьшению
коэффициента Кх в уравнении (VI.11).
Известно, что величина остаточной щелочности воды во мно¬
гих случаях более надежный критерий для оценки результатов
коагулирования, чем значения pH [140]; причем изменение ис¬
ходной щелочности может вызвать как увеличение, так и умень¬
шение а0пт [65, 73, 95, 141—143]. Одни авторы с целью экономии
коагулянта рекомендуют проводить лишь частичную нейтрализа-
180
ГЛАВА VI. ОПТИМАЛЬНАЯ ДОЗА КОАГУЛЯНТА
Pfy
Рис. VI.14. Влияние конеч¬
ной расчетной концентрации
НСО” в воде на pH изоэлек-
трической точки продуктов
гидролиза соли алюминия (pHj)
цию кислотности [143, 144], другие советуют придерживаться
такого режима коагулирования, при котором общая щелочность
воды уменьшается на 40% [125].
Многообразие рекомендаций по поддержанию величины оста¬
точной щелочности в каждом частном случае коагулирования
позволяет предположить, что роль НСОз-ионов не исчерпывается
нейтрализацией водородных катионов — продукта гидролиза коа¬
гулянта. Однако в соответствии с действующими нормами [46]
дозу щелочного реагента назначают всегда из расчета поддержа¬
ния некоторого щелочного резерва, величина которого (1 мг-жв/л)
установлена эмпирически и является оптимальной только для
группы вод определенного состава.
[Обработка литературных данных и результаты собственных
исследований автора позволяют высказать ряд новых соображе¬
ний о роли щелочности в коагуляционном процессе. Они бази¬
руются на следующих экспериментальных выводах.
1. Скорость коагуляции механических примесей зависит фак¬
тически целиком от свойств малорастворимых продуктов гидроли¬
за коагулянта.
2. Бикарбонаты обладают способностью координироваться при
катионах А13+, заметно смещая положение изоэлектрической точки
в сторону более низких значений pH (рис. VI. 14).
3. Образующиеся при гидролизе катионы водорода легко ад¬
сорбируются на поверхности грубо дисперсных и коллоидных при¬
месей воды и могут снижать их заряд 6.
Опыты по подбору доз A]2(S04)3 для воды с разной щелочно¬
стью [110] показали, что увеличение щелочности вызывает прирост
дозы коагулянта при св 150 мг/л, т. е. в зоне, где скорость коа¬
гуляции определяется вероятностью слипания частиц. При св
•^150 мг/л увеличение щелочности или не влияло на дозу A12(S04)3,
в Высокую адсорбируемость катионов водорода объясняют их небольшими
размерами и сильной поляризующей способностью.
Vi.2. ВЫБОР ДОЗЫ КОАГУЛЯНТА
181
или способствовало ее снижению. Результаты согласуются с дан¬
ными Стамма, О’Мелиа [8] и Коростелевой [73].
В ходе экспериментов наблюдали также следующие явления.
1. При щелочности воды более 1,0—1,5 мг-экв/л снижение ще¬
лочности от добавления коагулянта соответствует расчетному,
вытекающему из хода реакции
При меньшей щелочности и особенно при большом количестве ор¬
ганических и минеральных загрязнений снижение щелочности
от добавления коагулянта в несколько раз меньше расчетного.
2. Замена в опытах сернокислого алюминия хлорным вызыва¬
ет необходимость поддержания более высокого щелочного резер¬
ва.
3. Структура и свойства хлопьев коагулированной взвеси за¬
висят от последовательности добавления к воде коагулянта и ще¬
лочи.
^Некоторые из этих явлений наблюдались и другими авторами.
Сопоставление экспериментальных данных приводит к выводу,
что величина щелочности воды активно влияет на глубину гидро¬
лиза коагулянта, определяя состав, заряд и структуру формирую¬
щихся продуктов. По-видимому, весь комплекс реакций, проте¬
кающих ири гидролизе соли алюминия, можно представить схе¬
мой
Схема включает, помимо собственно гидролиза, три реакции:
1) нейтрализацию катионов водорода (Н+ + НСОз); 2) формиро¬
вание гидроксид-бикарбонатных соединений алюминия [А1(ОН)п -|-
+ НС03]; 3) образование основных солей алюминия с други¬
ми анионами воды [А1(ОН)3 + А"]. Для определенных условий
очистки воды в системе этих реакций существует свое динамиче¬
ское равновесие.
В случае достаточно высоких pH и щелочности исходной воды,
когда при реакции гидролиза коагулянта не испытывается ни не¬
достатка в ОН“-ионах, пи затруднений в отводе из сферы реакции
катионов Н+, разница АЩ между щелочностью исходной (Щи)
и конечной (Щк) близка к расчетной. Однако это еще не означает,
что гидролиз прошел до образования А1(ОН)3: если Щк больше
некоторой критической величины, НСОз-ионы, наряду с другими
анионами воды, могут активно конкурировать с ОН“-группами
за координационные участки А13+. При этом образуются соедине¬
ния с меньшим значением рНг.
А12 (S04)3 + 6НСО- = 2А1 (ОН)3 + 3SQ2- + бСО^ .
А13+ + Н20
Al (OH)rt<—А”
\
Н+ + НСОз
182
ГЛАВА VI. ОПТИМАЛЬНАЯ ДОЗА КОАГУЛЯНТА
■ Для маломутных вод, как уже отмечалось, наилучшая коагу¬
ляция происходит при условии рН0 » рНь и сдвиг рНг в сто¬
рону низких значений способствует уменьшению расхода коагу¬
лянта. Так как ионы SO4 и С1~ в концентрациях, обычных для
коагулированной воды, не могут конкурировать с ОН'-ионами
при высоких pH, желательно иметь избыток бикарбонатов. Нали¬
чие бикарбонатов при очистке маломутных вод, кроме того, способ¬
ствует образованию объемистых хлопьев, что компенсирует в из¬
вестной мере недостаточное число частиц. Чем меньше в воде анио¬
нов, конкурирующх с ОН-, тем более высокие значения Щк тре¬
буются для снижения значений pH,-. Этим объясняется необходи¬
мость повышения щелочности при очистке маломинерализованных
вод и при использовании хлорных коагулянтов вместо сернокис¬
лых.
При очистке цветных, а также мутных вод, содержащих высо¬
коустойчивую взвесь, наиболее эффективны положительно заря¬
женные продукты гидролиза (рН0 -< рНг). Присутствие большого
количества анионов нежелательно. Величина Щк должна быть
невысокой и тем меньше, чем выше концентрация других анионов.
Отсюда вытекают необходимость уменьшения Щк с увеличением
цветности и высокий эффект обесцвечивания воды электрокоагу¬
лированием (см. гл. VII).
При низких pH на ход коагуляции и на условие равновесия
трех рассмотренных реакций особый отпечаток накладывает на¬
личие взвешенных веществ в воде. Если реакция НСО3 + Н+ зат¬
руднена, степень гидролиза алюминия уменьшается. Взвесь, на¬
ходящаяся в воде или добавленная в качестве замутнителя, сор¬
бирует избыточные катионы водорода, выполняя как бы функцию
дополнительной щелочности. Причем гидролиз А13+ проходит фак¬
тически глубже, чем это следует из равенства ДЩ = Щи — Щк.
Смещением равновесия в рассмотренных реакциях можно объ¬
яснить «коагулирующее» действие воздуха, а также влияние на
коагуляцию механического перемешивания коагулированной во¬
ды, способов и порядка добавления к воде коагулянта и подщела¬
чивающих реагентов.
Приведенные сведения позволяют говорить об оптимальных
значениях остаточной щелочности коагулированной воды, под¬
держивание которых составляет важную технологическую задачу,
способствуя снижению затрат коагулянта и улучшению качества
очищенной воды.
VI. 2.2. Обесцвечивание воды
Зависимость оптимальной дозы коагулянта от цветности воды но¬
сит менее сложный характер, чем ее зависимость от содержания
взвешенных примесей. Уравнения физической адсорбции краси¬
VI.2. ВЫБОР ДОЗЫ КОАГУЛЯНТА
183
телей на пористых сорбентах для описания этой зависимости не¬
пригодны. Рекомендуется использовать следующие эмпирические
формулы:
аопт ВД*, согласно [46, 57 (стр. 144)];
«опт = &аЩи + ^зЦ% согласно [106];
а0Пт = #4Щи, согласно [145];
а0Пт= + КвСз*, согласно [95];
аопт = ЩиЦГ*
или
«опт = Ка + -j- KWYЦь согласно [109, стр. 121]. (VI.20)
В этих уравнениях Кип — эмпирические коэффициенты; Cs —
величина суммарного заряда коллоидных окрашивающих веществ,
определяемая «коллоидным» титрованием [95]; Ц, = Ц — 20 —
цветность, подлежащая устранению в ходе коагулирования воды.
Уравнения, кроме исходной цветности и щелочности, не учи¬
тывают никаких других показателей качества воды, область их
применения ограничивается теми частными условиями, для кото¬
рых они составлены.
При расчете дозы коагулянта необходимо учитывать природу
окрашивающих веществ и механизм их удаления. Устранение истин¬
но растворенной фракции цветности связано с образованием гид-
роксокомплексов, о чем свидетельствует наблюдаемая в некоторых
случаях линейная зависимость: аопт = / (Ц) [82]. Коллоидные ве¬
щества цветности удаляются, по-видимому, по тому же механизму,
что и минеральные взвеси. Если это так, то расчет дозы коагулян¬
та может быть выполнен по формуле (VI. 11), записанной в следую¬
щем виде:
ак = di -f- = K\SK (ca -j- &Цк) -2ra К2 (ca -j- Щк) 2, (VI.21)
где Цк — цветность, обусловленная присутствием коллоидных
веществ, в град.
Анализ результатов экспериментов, представленных в виде
кривых а0пт = / (Ц) [110], показывает, что для цветных вод кри¬
тические величины цветности, отвечающие минимальной дозе коа¬
гулянта, или совсем отсутствуют, или выражены гораздо слабее,
чем для замутненных вод. Это можно объяснить гидрофильностью
и малой массой окрашивающих коллоидов, вследствие чего ки¬
нетические особенности коагулирующей системы определяются
почти исключительно концентрацией продуктов гидролиза коа¬
гулянта. Доза коагулянта при Ц < ЦКр по мере роста цветности
уменьшается медленнее, чем но мере роста мутности.
(VI.15)
(VI.16)
(VI.17)
(VI.18)
(VI. 19)
184
ГЛАВА VI. ОПТИМАЛЬНАЯ ДОЗА КОАГУЛЯНТА
а, лгг/л
Рис. VI. 15. Зависимость а от
Ци при разных значениях ще¬
лочности исходной воды (Щи,
мг-экв/л) и конечных pH (рНк)
1 — Щи = 2,1, рНк нерегулируе¬
мое; 2 — 1ЦИ = 1,2, рНк нерегули¬
руемое; 3 — Щи = 2,1, рНк —5,0
"50 750 250
Поскольку влияние состава обрабатываемой воды на значения
параметров в уравнении (VI.21) должно носить тот же характер,
что и в процессе осветления, зависимость аопт = / (Ц) для дос¬
таточно высоких значений Цк можно представить в виде
ак^ад1г2/л^Я[(1-е')ЦнГ, (VI.22)
где 0' = Цр/Ц — доля цветности, обусловленная истинно раство¬
ренными веществами.
Если реакция истинно растворенных веществ цветности с про¬
дуктами гидролиза коагулянта не осложнена никакими специфи¬
ческими поверхностными явлениями, связь между количествами
прореагировавших компонентов будет линейной:
ар = ЯЦр = Я0'Ц. (VI.23)
Выражения (VI.22) и (VI.23) объединяют приведенные выше
эмпирические зависимости. Общую потребность в коагулянте
нельзя, однако, представить как сумму ак и ар, так как продукты
гидролиза, образовавшиеся при добавлении к воде дозы ак, бу¬
дут реагировать с истинно растворенной фракцией цветности. При
этом дозы ак и ар перекрывают одна другую и фактическая потреб¬
ность в коагулянте будет определяться большей из них. С увели¬
чением доли истинно растворенных веществ цветности вероятность
линейной взаимосвязи а и Ц повышается, но дать точную коли¬
чественную оценку влияния 0' на оптимальную дозу коагулянта
пока не представляется возможным из-за отсутствия достоверных
сведений о дисперсном составе и химических свойствах веществ
цветности. Можно лишь оценить роль параметров качества воды.
Результаты опытов по подбору оптимальных доз сернокислого
алюминия, необходимых для снижения цветности воды до 10—20
град и доведения ее оптической плотности до 0,1 единицы, показа¬
ны на рис. VI. 15. Кривые, соединяющие экспериментальные точки,
могут быть описаны уравнением а — /Щ™ с разными значениями
коэффициентов К пт. Первая кривая — уравнением а = 8,4 Ц°>48;
Vi.2. ВЫВОР ДОЗЫ КОАГУЛЯНТА
185
вторая кривая — а — 5,6 Ц°>51; третья кривая — а = 1,32 Ц0-82.
Анализ результатов опытов позволил сделать следующие выводы.
1. Увеличение щелочности исходной воды от 1,2 до 2,1 мг-гкв/л
вызывает почти пропорциональное увеличение дозы коагулянта.
Причем значение показателя степени существенно не изменяется.
2. Добавление соляной кислоты с целью доведения конечного
значения pH воды до пяти приводит к уменьшению потребности в
сульфате алюминия почти во всем диапазоне принятой цветности.
Оно сопровождается увеличением показателя степени тп до 0,82,
что свидетельствует о приближении характера зависимости меж¬
ду йопт и Ц к линейному и служит подтверждением результатов,
полученных ранее [78].
Уравнения (VI. 11), (VI.22) и (VI.23) относятся к случаям об¬
работки или только мутных, или только цветных вод. При одно¬
временном присутствии в воде взвешенных и окрашивающих ве¬
ществ оптимальные условия удаления каждого компонента гид¬
ролизующимися коагулянтами не совпадают, особенно в отноше¬
нии значений pH среды: в области значений pH, оптимальных для
обесцвечивания, удаление минеральных примесей, как правило,
задерживается. Поэтому дозу коагулянта приходится назначать,
руководствуясь каким-либо одним показателем (остаточной мут¬
ностью или цветностью).
Согласно экспериментам, в обоих случаях фактическая доза
коагулянта отличается от расчетной. Величина ошибки зависит
от индивидуальных свойств загрязнений и их концентрации. Так,
добавление к цветной воде относительно небольших количеств
каолинита (50 мг/л) оказывает весьма слабое действие на глубину
обесцвечивания коагулянтами, но интенсифицирует процесс хлопье-
образования [39]. Большие же концентрации замутнителя спо¬
собствуют обесцвечиванию воды,- но могут привести к увеличению
потребности в коагулянте. Что касается окрашивающих соедине¬
ний, они, как уже отмечалось, в высоких концентрациях способ¬
ны стабилизировать минеральную взвесь.
Факт сильного влияния загрязнений, включенных в структуру
«чистых» коагулятов, на их физические свойства требует коррек¬
тировки нашей оценки этих свойств, выполненной в гл. III. Такая
корректировка поможет уяснить и конкретизировать зависимость
расхода коагулянта от качества обрабатываемой воды. Кроме то¬
го, подчеркивая «технологический» характер самого понятия «оп¬
тимальная доза коагулянта», мы уже указывали на важную роль
способа, выбранного для отделения коагулированной взвеси в
осадок. Поэтому следующий раздел посвящен рассмотрению
свойств коагулированной взвеси и способов ее выделения в осадок.
В нем мы попытаемся показать, что представления, положенные в
основу расчета оптимальной дозы коагулянта, в принципе спра¬
ведливы для любого выделения коагулированной взвеси из воды.
186
глава vi. оптимальная Доза коагулянта
VI. 3. СВОЙСТВА КОАГУЛИРОВАННОЙ ВЗВЕСИ
И СПОСОБЫ ЕЕ ВЫДЕЛЕНИЯ В ОСАДОК
VI. 3.1. Свойства коагулированной взвеси
По физическим свойствам коагулированная взвесь существенно
отличается от «чистых» коагулятов.
Размер хлопьев определяется соотношением между молеку¬
лярными силами, удерживающими частицы вместе, и гидродина¬
мическими силами отрыва, стремящимися разрушить агрегаты.
При оптимальной дозе коагулянта размер хлопьев обычно макси¬
мален. Уменьшение размера при недостатке или избытке коагу¬
лянта объясняется, по-видимому, отклонением числа связей меж¬
ду частицами от оптимального. По данным Хэма и Кристмана [41],
увеличение длительности перемешивания воды с раствором хлор¬
ного железа вызывает смещение pH образования наиболее круп¬
ных хлопьев в сторону больших значений.
Размер хлопьев, как правило, лежит в пределах 0,5—3 мм.
Меньшие значения относятся к обработке цветных вод. При ис¬
пользовании солей железа хлопья образуются более крупные.
Для характеристики крупности хлопьев предложено [117, стр. 56]
пользоваться понятием эквивалентного диаметра, подсчитываемо¬
го по величине скорости свободного осаждения v из уравнения
где 7Х — объемный вес хлопьев; Кф — коэффициент формы хлопь¬
ев; v — кинематическая вязкость воды.
Результаты расчетов d3KB по уравнению (VI.24) с использова¬
нием опытных значений v и ух указывают на уменьшение размера
хлоиьев с увеличением мутности и цветности воды.
Гидратация коагулированной взвеси с увеличением концентра¬
ции минеральных частиц и окрашивающих веществ заметно умень¬
шается. При обработке сернокислым алюминием воды, содержа¬
щей 1 г!л механических примесей, степень структурно-механиче¬
ской гидратации взвеси, подсчитанная по методике Кургаева, сни¬
жается в 15—30 раз [117, (стр. 60), 119]. Однако и в этом случае
содержание воды в хлопьях достигает 85% [146].
Объемный вес хлопцев определяют с учетом относительных
объемов, плотности воды и плотности твердой фазы, включенных
в структуру
(VI.24)
= 7в + §т (Yt — Yb),
(VI. 25)
где Yb и Yt — объемный вес воды и твердой фазы; 8Т — объем
твердого вещества в единице объема хлопьев.
VI.3. СВОЙСТВА КОАГУЛИРОВАННОЙ ВЗВЕСИ
187
Результаты экспериментальных определений ух сильно зави¬
сят от принятой методики. По методике, основанной на наблюде¬
ниях за уплотнением коагулированной взвеси [117 (стр. 52), 147],
для гидроокиси алюминия величина ух составляет 1,0012 г/смя
и возрастает с увеличением количества дисперсных загрязнений в
обрабатываемой воде до 1,042 г/см3. Определения с помощью пик¬
нометра дают значения от 1,016 до 1,18 г/см3 [45, 148].
Накопленные экспериментальные данные не отражают, к со¬
жалению, следующих обстоятельств.
1. Влияния pH на структурную плотность коагулированной
взвеси, особенно заметного при очистке маломутных вод.
2. Взаимосвязи ух с условиями перемешивания: если при зна¬
чениях скоростного градиента G 60 сек-1 объемный вес взвеси
практически не зависит от величины G [149], то при возрастании
G от 80 до 1000 сек'1 содержание твердого вещества в объеме хлопь¬
ев увеличивается примерно в 5 раз, а водонасыщенность структу¬
ры снижается соответственно с 96,5 до 89,2% [146].
3. Уменьшения ух из-за наличия газовой составляющей.
Объемная концентрация коагулированной взвеси с0 определя¬
ется содержанием воды в хлопьях. По результатам фильтрации она
составляет в обычных условиях коагулирования 0,025—0,1%
[150, 151].
Согласно результатам экспериментов, величина с0 прямо про¬
порциональна дозе коагулянта и обратно пропорциональна ско¬
ростному градиенту, создаваемому при перемешивании воды [139].
Использование счетчика Коултера дало возможность Свифту и
Фрейнландеру [152, 153] показать на примере коагуляции элект¬
ролитами разных коллоидных систем справедливость зависимости:
2,3 lg /л/'t
С°= кС, (Т2 —тх)’ (VL26)
где Nx и No — число частиц в единице объема воды’в’моменты време¬
ни TjHT2; к— константа, равнаятр Е/2п (г|) — коэффициент, харак¬
теризующий эффективность столкновений коагулирующих частиц;
Е — параметр, учитывающий распределение частиц но размеру).
Для гидролизующихся коагулянтов, в отличие от исследова¬
ний [152, 153], в которых авторы имели дело с изменяющимися
размерами и числом частиц при относительном постоянстве с0,
водонасыщенность хлопьев по мере их роста возрастает. Поэтому
величина с0 в течение периода хлопьеобразования определяется
значениями G их [146]. Значение скоростного градиента опреде¬
ляет предельный размер хлопьев, и после того как он достигнут,
перемешивание мало влияет на с0 (рис. VI. 16). Распределение
хлопьев по размеру также достигает постоянства. Таким образом,
использование уравнения (VI. 26) ограничено длительностью пе¬
риода роста хлопьев.
188
ГЛАВА VI. ОПТИМАЛЬНАЯ ДОЗА КОАГУЛЯНТА
Из экспериментальных данных видно (рис. VI. 17), что за счет
интенсификации перемешивания можно добиться существенного
увеличения плотности хлопьев (в 5—6 раз). Нужно, однако, опа¬
саться нарушения тиксотропных свойств коагулированной взвеси.
Газонасыщенность хлопьев является следствием выделения га¬
зов из самой воды, а также проведения процессов, связанных с
Рис. VI.16. Общий характер
зависимости размера хлопьев
коагулированной взвеси (d) от
величины скоростного градиен¬
та (G), создаваемого при переме¬
шивании воды после ее обработки
коагулянтом
^ Г
обогащением воды воздухом (флотация, аэрация, озонирование)
или другими газами (коагуляция, электрокоагуляция). Основными
источниками появления газовых пузырьков являются подсосы
воздуха в сальниках насосов и трубопроводах, подогрев воды с
последующим снижением Давления на входе в очистные сооруже¬
ния. При практикуемом обычно на ТЭЦ подогреве воды до 25—30° С
растворимость воздуха резко снижается и в холодное время года
из одного литра обрабатываемой воды может перейти в дисперсное
состояние до 10—13 см3 воздуха. На необходимость учета газовы-
деления в технологических расчетах указывают Кургаев и др. [154].
Включение газовых пузырьков в хлопья коагулированной
взвеси объясняется преимущественным выделением"пузырьков на
поверхности твердой фазы. Ориентировочная оценка соотноше¬
ния количеств газа, выделившихся на твердой поверхности и в
объеме воды, может быть выполнена посредством термодинамиче¬
ского анализа [155, стр. 178].
На образование пузырьков в объеме воды затрачивается работа
Wn = Wt + W2 + W3, (VI. 27)
где W1 — 4nr2om-v — работа образования новой поверхности
4
раздела; W2 = -jnr3p —работа разрыва сплошности воды; И^з =
= ^nrspB—работа заполнения образовавшейся полости водяным
паром (г — радиус пузырька; р — давление внутри пузырька;
рв — упругость водяного пара).
Работа образования пузырька на твердой поверхности опреде¬
ляется выражением
Wт — 510ж_г -f- SjOjK-r 51<3Х_Ж W2 Н~ W3 >
(VI.28)
VI.3. СВОЙСТВА КОАГУЛИРОВАННОЙ ВЗВЕСИ
189
о
40 80 7Z0 в,г/7в.&
Рис. VI.17. Зависимость объемной концентрации (с0) хлопьев от величины
скоростного градиента (G), создаваемого при перемешивании воды после ее
обработки коагулянтом [146]
Рис. VI.18. Соотношение величин работ, затрачиваемых на образование
пузырьков в объеме раствора {Wn) и на твердой поверхности (WT), в зависи¬
мости от краевого угла (0) смачивания водой твердой поверхности
в котором % — площадь поверхности пузырька; s2 — площадь по¬
верхности контакта пузырька с твердым телом; а — поверхност¬
ное натяжение на границе раздела фаз.
Значением W2 + W3 в обоих уравнениях можно пренебречь
ввиду его малости по сравнению с остальными членами. Тогда, по¬
лагая, что пузырек имеет форму шарового сегмента, получаем:
„ 0
l+tg2-o-
WJWт = з — - - (VI .29)
у 3tg2— + i
где 0 — краевой угол смачивания водой твердой поверхности.
Соотношение WJWt всегда больше единицы (рис. VI. 18). Об¬
разование пузырьков на твердой поверхности всегда требует мень¬
ших энергозатрат, чем образование в объеме воды. С увеличением
0, т. е. с уменьшением гидрофильности поверхности, вероятность
образования пузырьков на поверхности возрастает. Для минера¬
лов, присутствующих в виде взвешенных примесей в очищенной
воде, значения 0 составляют обычно от 60 до 85°, следовательно,
вероятность выделения воздуха на поверхности примесей на 15—
30% выше, чем в объеме воды.
Помимо выделения газов на поверхности твердой фазы, газо-
насыщенность хлопьев может быть следствием столкновений твер¬
дых частиц с пузырьками, выделившимися в объеме воды. Если
энергия столкновения достаточна для преодоления сопротивления
жидкостных пленок, такие пузырьки фиксируются на поверхнос¬
ти. Фиксации способствуют разноименные электрические заряды
пузырьков и твердых частиц [156, стр. 295].
190
ГЛАВА VI. ОПТИМАЛЬНАЯ ДОЗА КОАГУЛЯНТА
Рис. VI. 19. Действие разрушающих сил на агломераты с симметрично (а)
и несимметрично (б) упакованными частицами
Прочность связи пузырька с поверхностью может быть уста¬
новлена из условия равновесия между силой тяжести частиц и си¬
лой их гидростатического подъема. Согласно расчетам, выполнен¬
ным на основе результатов исследований Кабанова и Фрумкина
[157], чем гидрофобнее поверхность, тем прочнее обеспечивается
закрепление пузырька на ней.
Не исключается также механическое защемление пузырьков
во внутренних полостях коагулированной взвеси при построении
хлопьев.
Газонаполнение хлопьев, отчетливо наблюдаемое в лаборатор¬
ных опытах, приводит как бы к уменьшению их объемного веса
и должно учитываться в выражении (VI.25) введением газовой
составляющей:
7х = Ув + 6Т (7т — 7в) + 8Г (7Г — 7в). (VI. 30)
Как показали наши исследования, в результате диспергирова¬
ния первоначально возникших коагуляционных структур можно
добиться существенного снижения содержания газа в хлопьях [158].
Прочность хлопьев, как и всякого агломерата, зависит от гра¬
нулометрического состава образующихся частиц и пластичности
[158]. Агломераты частиц, неоднородных по размеру, прочнее. Это
объясняется тем, что группу симметрично упакованных частиц
одинакового размера можно разрушить по четким плоскостям
кливажа; при неодинаковой величине частиц эти плоскости смы¬
каются (рис. VI.19) и прочность агрегата возрастает.
Водные прослойки в местах соприкосновения частиц способ¬
ствуют такой их ориентации, при которой хлопья оказывают по¬
току жидкости минимальное сопротивление. Но эти же прослой¬
ки ослабляют взаимодействие частиц.
Прочность хлопьев коагулированной взвеси рассматривают
обычно как способность взвеси сопротивляться действию сил сдви¬
га, вызванных наличием гидравлического градиента в потоке во¬
VI.3. СВОЙСТВА КОАГУЛИРОВАННОЙ ВЗВЕСИ
191
ды. Поскольку прочность хлопьев настолько мала, что не подда¬
ется прямому измерению, применяют разные косвенные методы
оценки, основанные главным образом на изучении взаимосвязи
между интенсивностью перемешивания и размером хлопьев. Раз¬
мер хлопьев фиксируют посредством фото- и киносъемок [159],
турбидиметрически [139, 160], с помощью электронных счетчиков
[41, 152, 161]. Особенно большими возможностями обладает при¬
меняющийся все чаще метод Коултера, при котором взвешенные
частицы при прохождении через дюзу определенного размера вы¬
зывают увеличение сопротивления электролита, фиксируемое им¬
пульсным изменением силы тока на выходе электрической схемы;
величина импульса пропорциональна размеру частиц.
В 1938 г. Гудзон [162] предложил для оценки прочности хлопь¬
ев использовать так называемый индекс прочности
где АН — минимальные потери напора в загрузке, при которых
происходит проскок взвеси в фильтрат; d — эквивалентный диа¬
метр зерен загрузки; L — высота ее слоя.
Более обоснованный подход к оценке прочности хлопьев коа¬
гулированной взвеси может быть сделан с позиций теории Минца,
рассматривающей фильтрацию как результат процессов отрыва
и прилипания дисперсных частиц. Методика определения пара¬
метров, характеризующих прочность взвеси, хорошо освоена [163].
По мнению Хэма [43], прочность хлопьев определяется кон¬
центрацией атомов металла (алюминия или железа), связанных
через ОН'-группы в пространственную решетку. Концентрация
атомов в свою очередь зависит от растворимости продуктов гидро¬
лиза и, следовательно, от pH среды. По мере сдвига изоэлектриче-
ской точки от pH 8 к pH 5 растворимость продуктов гидролиза па¬
дает и прочность хлопьев возрастает. Установлено также [41, 161],
что отклонение от оптимума pH приводит к заметному снижению
прочности хлопьев.
Между величиной скоростного градиента и касательным нап¬
ряжением сдвига тс в перемешиваемой среде существует прибли¬
зительное соотношение [164]:
в котором г] — вязкость среды. Минимальная величина G, вызы¬
вающая разбивание хлопьев, равна примерно 100 сек-1. Поэтому
при г| = 1-10_2?гз расчет по уравнению (VI.32) дает величину
предельного касательного напряжения тпр, равную 2,7 дин/'см2.
По измерениям Барышниковой [148], предельное напряжение
сдвига осадков, образующихся при очистке воды коагулянтами,
составляет от 1,5 до 6 мг/см2. Измеренные Кургаевым [117, стр. 60]
i = A Hd/L,
(VI.31)
тс = г}G,
(VI.32)
192
ГЛАВА VI. ОПТИМАЛЬНАЯ ДОЙА КОАГУЛЯНТА
критические давления, приводящие к разрушению каркаса гидро¬
окисей алюминия и железа, имеют тот же порядок — соответст¬
венно 7 и 22 мг/см2. Осадки по прочности располагаются в ряд [45]:
А1 (ОН)з + глина > Ге(ОН)з > А1 (ОН)з > А1(ОН)з +
+ окрашивающие вещества.
£ ^Одни авторы указывают на увеличение прочности хлопьев с
ростом соотношения в них минеральных примесей и продуктов
гидролиза коагулянта [161], другие [41] — на уменьшение. Столь
же противоречивые мнения высказываются по поводу влияния
на прочность коагулированной взвеси предварительного диспер¬
гирования осадка [177 (стр. 60), 148]. Эти расхождения в значи¬
тельной мере связаны с методическими погрешностями: недоуче¬
том условий коагуляции и возраста исследуемых осадков.
Если же исходить из предположения, высказанного в начале
этой главы, о том, что фиксация продуктов гидролиза на поверх¬
ности частиц дисперсных примесей воды осуществляется за счет
сил притяжения ближнего действия, а взаимодействие частиц
продуктов гидролиза друг с другом — за счет дальнодействующих
сил, то можно сделать вывод об увеличении прочности хлопьев с
увеличением количества примесей: во-первых, из-за увеличения
числа контактов в первичном энергетическом минимуме, во-вто¬
рых, вследствие увеличения глубины вторичного энергетического
минимума при коллективном взаимодействии частиц [165].
VI. 3.2. Способы выделения коагулированной взвеси
в осадок
Работа сооружений, предназначенных для выделения коагулиро^
ванной взвеси в осадок, основана главным образом на двух прин¬
ципах — осаждении под действием сил тяжести и прилипании
к зернам фильтрующихся загрузок. Осаждение осуществляется или
из горизонтального потока воды (отстойники), или из вертикаль¬
ного потока, псевдоожижающего твердую фазу продуктов гидро¬
лиза (осветлители). Отделение взвеси фильтрацией производят
при движении воды сверху вниз (фильтры) или снизу вверх (кон¬
тактные осветлители). Центробежные силы при выделении коагу¬
лированной взвеси используются редко.
Мы дадим в этом разделе краткую характеристику трех основ¬
ных способов отделения коагулированной взвеси с точки зрения
тех требований, которые должны быть предъявлены к свойствам
взвеси для завершения осветления воды в возможно более корот¬
кие сроки и при наименьшем расходе коагулянта. Вопросы кон¬
структивного характера, приемы эксплуатации очистных устройств
здесь не рассматриваются, поскольку они подробно освещены в
литературе [57, 59, 77, 102, 117, 137, 166—168].
V1.3. СВОЙСТВА КОАГУЛИРОВАННОЙ ВЗВЕСИ
193
VI. 3.2.1. Отстаивание
Скорость осаждения астабилизированных частиц зависит от тем¬
пературы воды и их размера, формы, плотности, состояния по¬
верхности. На частицу массой т, находящуюся на некотором
расстоянии от центра гравитационного поля, действуют сила тя¬
жести mg, сила подъема т (Рв/Рч)^ и сила трения, препятствую¬
щая осаждению tydR/dx, где g — ускорение силы тяжести; рв и
Рч — соответственно плотность воды и частицы; г|) — коэффици¬
ент сопротивления; R — расстояние между центром частицы и
центром гравитационного поля.
Из баланса сил
—tydRldx + mg — т (pB/p4)g = О
следует
и = dR/dx = (1 - рв/рч), (VI.33)
или для сферических частиц диаметром d
(VI. 33')
При значениях чисел Рейнольдса Re = (udpB)/x\, меньших чем
2, которые характерны для мелких медленно осаждающихся час¬
тиц, гр ~ 24/Re и выражение (VI.33') принимает вид формулы
Стокса:
1 „ Рч-Рв ™
и — ~То~ ё » •
18 ° т)
При больших числах Рейнольдса (2 < Re < 500)
г|з ~ 18,5/Re0-6.
Наконец, при Re^> 500 коэффициент сопротивления уже не зави¬
сит от величины Re и становится равным приблизительно 0,4
[77, стр. 134].
Для коэффициента сопротивления, помимо приведенных, су¬
ществуют другие эмпирические выражения. Одно из них, полу¬
ченное при обработке многочисленных экспериментальных данных
на ЭВМ и справедливое в интервале значений Re от близких к ну¬
лю до 1000, опубликовано Танакой и др. [169]:
lg у = a (lgRe)2 + Ъ (lgRe) + С, (VI.34)
где а, Ъ и С — коэффициенты.
В проточном горизонтальном отстойнике направление и вели¬
чина скорости движения хлопьев определяются путем векторного
сложения трех составляющих (рис. VI.20): горизонтальной ско-
7 Е. Д. Бабенков
194
ГЛАВА VI. ОПТИМАЛЬНАЯ ДОЗА КОАГУЛЯНТА
V
Рис. VI.20. Скорость движе¬
ния частицы, осаждающейся
в горизонтальном отстойнике
U-(o
рости потока V, вертикальной скорости осаждения, подсчитывае¬
мой по уравнению (VI.33), и взвешивающей скорости со, обуслов¬
ленной турбулентными пульсациями. При числах Re, имеющих
обычно место в горизонтальных отстойниках (300—1900) [170],
со ~ 0,035 V. Подсчитывая и, нельзя забывать о возможном вклю¬
чении в хлопья газовых пузырьков.
Для агрегативно устойчивых суспензий между высотой столба
жидкости и длительностью осаждения взвеси существует прямая
пропорциональность. Эффект осветления воды, содержащей та¬
кую взвесь, в горизонтальных отстойниках практически не зави¬
сит от их глубины. При осаждении коагулированной взвеси, вслед¬
ствие значительной гравитационной коагуляции, с увеличением
высоты слоя воды эффект осветления возрастает. По данным Вей-
цера и Колобовой [171], для достижения одинакового эффекта ос¬
ветления воды в колонках разной высоты h требуется разное вре¬
мя х, которое связано с h соотношением
где п — коэффициент гравитационной коагуляции, равный для
случая обработки воды сернокислым алюминием 0,37—0,75 [172] 7.
Для хлопьев коагулированной взвеси необходимо также учи¬
тывать их форму, вводя поправку в коэффициент сопротивления.
Поскольку в разных гидравлических условиях форма хлопьев
проявляет себя неодинаково, Нечаев [173] ввел в расчет скорости
осаждения так называемый динамический коэффициент формы.
Этот коэффициент равен отношению скорости осаждения равнове¬
ликого по объему шара к скорости осаждения частицы произволь¬
ной формы той же плотности. На основе результатов эксперимен¬
тов даны следующие значения динамического коэффициента фор¬
мы в разных гидравлических условиях [174]:
Кф'" = 0,95 ф-»-5» — ламинарный режим;
Кфвр = 0,75 ф-0’78 Аг °'06 — переходный режим;
А'фУрб = 1,35 ф-1,41— турбулентный режим.
7 Это выражение формально сходно с уравнением (И 1.2) каталитического
ускорения гидролиза в присутствии ранее образовавшейся твердой фазы.
(VI. 35)
vi.з. свойства коагулированной взвеси
195
Здесь коэффициент сферичности ф = 4,9 F2,3/s; V — объем части¬
цы; s — площадь поверхности частицы; Аг — критерий Архи¬
меда.
На результаты отстаивания взвеси сильное влияние оказыва¬
ют: равномерность распределения и сбора воды в отстойниках;
перепад температур, обусловленный колебаниями температуры
воды в источнике водоснабжения или происходящими в осадке
биохимическими процессами; сужение сечения отстойников нако¬
пившимся осадком; наличие завихрений и водоворотов вокруг
выступов и колонн.
По данным Демуры и др. [175], на характер движения воды в
отстойнике большое влияние оказывает сам процесс осаждения
взвеси. В результате переноса осаждающимися хлопьями воды
возникает придонное течение, которое может быть зафиксировано
с помощью микротермистеров. Это течение увеличивает транспор¬
тирующую способность основного потока и приводит к ухудше¬
нию остаивания.
Основной характеристикой степени гидравлического совершен¬
ства отстойника принято считать коэффициент его объемного ис¬
пользования (Коб), равный отношению среднего фактического вре¬
мени пребывания воды к расчетному. По данным, собранным Бе¬
резой [176], значения К0о находятся в пределах 0,41—0,81. При
температуре воды, близкой к нулю, возможно уменьшение коэф¬
фициента до 0,384 [177]. Еще меньшие значения К0g имеют место
при особенно неблагоприятном перепаде температур — когда в
отстойник поступает более теплая вода [178].
Заслуживает внимания комплексный метод оценки гидравлики
отстойников, учитывающий режим течения, поперечную цирку¬
ляцию воды, наличие «мертвых» зон [179]. Для определения гид¬
равлических характеристик отстойников используют минераль¬
ные соли, органические красители, люминесцирующие и радиоак¬
тивные вещества.
Улучшения гидравлических условий отстаивания достигают
устройством продольных и поперечных (дырчатых) перегородок,
отжимных щитов, водосливов, своевременным удалением осадка,
рассосредоточенным отбором отстоенной воды и другими инженер¬
но-техническими мероприятиями [57 (стр. 181), 166 (стр. 115),
167 (стр. 138)].
Помимо длительности отстаивания, эффект задержания коа¬
гулированной взвеси зависит от мутности исходной воды. При
обработке маломутной воды в горизонтальных отстойниках Север¬
ной водопроводной станции г. Москвы в среднем в течение года
задерживалось 75% взвеси [180]. Чем выше концентрация взве¬
шенных веществ, включая твердую фазу продуктов гидролиза коа¬
гулянтов, тем выше процент отделенной взвеси. Однако остаточ¬
ная мутность, как правило, возрастает.
7*
196
ГЛАВА VI. ОПТИМАЛЬНАЯ ДОЗА КОАГУЛЯНТА
Иногда дополнительный эффект осветления имеет место в
двухэтажных отстойниках с последовательным движением воды
сначала по первому, затем по второму этажу и с поворотом потока
в вертикальной плоскости. Эффект объясняется образованием в
зоне поворота взвешенного слоя хлопьев коагулированной взве¬
си [181].
Важным шагом в усовершенствовании процесса отделения коа¬
гулированной взвеси явилось внедрение тонкослойного отстаи¬
вания в наклонных каналах или трубках. Этот метод отстаивапия
предложен в 1904 г. Хазеном [182] и получил развитие в работах
Кэмпа [183] и других исследователей [184—187]. В результате
уменьшения глубины слоя осветляемой воды, улучшения гидрав¬
лических условий отстаивания и отделения осадка метод дает воз¬
можность увеличить коэффициент объемного использования от¬
стойников до 0,80—0,85 [188] и значительно сократить длительность
пребывания воды в них.
Одна из серьезных причин ухудшения работы отстойников —
разбивание хлопьев на входе воды — может быть устранена или
сведена к минимуму оборудованием отстойников встроенными ка¬
мерами хлопьеобразования. В качестве примера эффективности
такого решения служат отстойники оренбургского водопровода,
на которых достигнуто увеличение производительности на 60%
и несколько улучшено качество осветленной воды [189]. Постепен¬
ное увеличение объема встроенных камер привело к созданию так
называемых зашламленных отстойников, действующих наподо¬
бие осветлителей со взвешенным осадком [190].
VI. 3.2.2. Осветление во взвешенном слое осадка
В осветлителях со взвешенным осадком осуществлен так называе¬
мый шламоконтактный способ очистки, обладающий по сравнению
с отстаиванием следующими преимуществами.
1. Ускоряется процесс хлопьеобразования за счет каталитиче¬
ского влияния ранее сформированной взвеси и увеличения интен¬
сивности массообмена.
2. Улучшаются гидравлические условия очистки. Только бла¬
годаря наличию взвешенного слоя хлопьев коэффициент объем¬
ного использования осветлителей возрастает на 15—25% [117,
стр. 51].
3. Снижается расход коагулянта вследствие более полного
использования адсорбционных свойств продуктов гидролиза.
Уровень взвешенного слоя хлопьев остается стабильным при
условии, если скорость восходящего потока превосходит скорость
начала псевдоожижения и не превышает критической скорости
уноса хлопьев. Эта скорость, являющаяся функцией некоторой
средней скорости осаждения хлопьевидной взвеси, определяет
VI.a. CBUilCiHA йиАХ УЛИ^иЬАНИОИ ВЗВЕСИ
197
производительность осветлителей и является их основным расчет¬
ным параметром.
При свободном осаждении частиц скорость седиментации за¬
висит от вязкости воды и индивидуальных свойств каждой части¬
цы. В случае осветления во взвешенном слое имеет место стеснен¬
ное осаждение хлопьев, скорость которого меньше скорости сво¬
бодного осаждения и зависит от объемной концентрации взвеси.
Благодаря этой зависимости достигается стабильное положение
уровня взвеси в довольно широком диапазоне скоростей восходя¬
щего потока воды.
Результаты экспериментов показывают, как мы отмечали уже
в I главе, что скорости свободного и и стесненного v осаждения
связаны зависимостью
где с0 — объемная концентрация взвеси, доли единицы; п — пока¬
затель степени, зависящий от числа Рейнольдса и формы ча¬
стиц.
Согласно результатам исследований Кургаева, главной при¬
чиной уменьшения скорости осаждения с ростом с0 является по¬
явление «кажущейся» вязкости, обусловленной турбулентными
пульсациями [117 (стр. 24), 191]. Для соотношения «кажущейся»
(г|к) и молекулярной (г)) вязкостей в диапазоне с0 = 0,07—0,6
дано следующее выражение:
При значениях с0, равных 0,15 и меньше, для практических
расчетов рекомендовано уравнение
Еще несколько эмпирических выражений, предлагаемых для
описания связи v = / (и, с0) приводит в своем обзоре Миллер [192].
На основе обзора сделан вывод, что в случае суспензий с полидис-
персными частицами, подверженными флокуляции, величина v
лежит в пределах между скоростями свободного осаждения мел¬
ких и крупных частиц.
Арикава [193] предлагает для скорости стесненного осаждения
частиц уравнение, связывающее ее не только с величиной с0, но
и с максимальным значепием объемной концентрации твердой фа¬
зы в осажденной суспензии с,пах, которая, как известно, определя¬
ется величиной остаточного энергетического барьера, т. е. степенью
(VI.36)
(VI.37)
v = и (1 — 3,5 с0).
(VI. 38)
198
ГЛАВА VI. ОПТИМАЛЬНАЯ ДОЗА коагулиптд
астабилизации частиц:
и (I с0)~
А + В (c0lcmax)n - (VI-39)
где А, В и п — константы.
Бонд [194] при выводе уравнения для скорости стесненного
осаждения взвеси установил, что найденное им соотношение
v = и (1 - 2,78#) (VI.40)
справедливо лишь для стационарного взвешенного слоя. Для ус¬
ловий очистки воды в осветлителях, когда новая некоагулирован-
ная взвесь непрерывно вводится в слой, а коагулированная взвесь
отводится, выражение (VI.40), по мнению Бонда, должно быть до¬
полнено вторым членом — некоторой фиктивной скоростью 1>ф,
характеризующей формирование новых хлопьев [195, 196]:
v' = и (1-2,78 с'*) + (VI.40')
Уф = vjs, (VI.41)
где vK — скорость образования коагулированной взвеси, см3!сек;
s — площадь поверхности образовавшейся взвеси, см2.
Введение параметра v$ имеет следующий физический смысл:
при некоторых (достаточно высоких) скоростях образования коа¬
гулированной взвеси объемная концентрация взвеси становится
более высокой при тех же значениях v. Параметр Уф может быть
увеличен за счет рециркуляции осадка.
Степень очистки воды (критерий сепарации Кс) связан, по Кур-
гаеву, с концентрацией коагулированной взвеси в контактной сре¬
де осветлителя следующей зависимостью:
Кс = af>Lcl (VI.42)
в которой аир — эмпирические коэффициенты, учитывающие
влияние на эффект осветления температуры и физико-химических
свойств хлопьев; L — высота взвешенного слоя хлопьев.
С остаточным содержанием взвеси в очищенной воде М„ кри¬
терий сепарации связан соотношением [117, стр. 72]
М0 = 260/Кс. (VI. 43)
Рассмотрение процесса осветления с точки зрения вероятнос¬
ти задержания первичных частиц взвешенным слоем хлопьев да¬
ет следующее выражение для эффекта сепарации [77 (стр. 178), 197
198]:
Мх/Мж = в»*, (VI.44)
где Ма и Мх — содержание некоагулированных взвешенных при¬
месей соответственно в исходной воде и в воде, прошедшей через
VI.3. СВОЙСТВА КОАГУЛИРОВАННОЙ ВЗВЕСИ
199
Рис. VI.21. Зависимость ос¬
таточной мутности (М0) освет¬
ленной воды от скорости вос¬
ходящего потока воды (v)
в осветлителе со взвешенным
осадком [199]
М$,мг/л
слой взвешенных хлопьев толщиной х; Ъ — коэффициент, учиты¬
вающий условия сепарации.
Линейная связь между эффективностью очистки и высотой
взвешенного слоя сохраняется, согласно экспериментам, лишь
до некоторого значения L, после превышения которого остаточная
мутность воды уже не зависит от высоты слоя. Некоторые авторы
объясняют это недостаточно полным задержанием первичных час¬
тиц [57 (стр. 247), 197]. Однако более верным (на основании све¬
дений по влиянию перемешивания, приведенных в VIII главе)
представляется вынос обломков хлопьев из контактной среды.
На справедливость этого предположения указывает факт увели¬
чения М0 с ростом скорости восходящего потока воды (рис. VI.21).
Поскольку скорости свободного и стесненного осаждения час¬
тиц растут пропорционально разнице объемных весов хлопьев и
воды [уравнение (VI.33')], даже незначительное повышение рч
приводит к заметному увеличению пир: например, при увеличе¬
нии рч от 1,002 до 1,006 г/см3 скорость и возрастает в 3 раза. При
используемых на практике скоростях восходящего потока воды
(0,7—1,4 мм/сек) объемная концентрация взвеси в контактной
среде находится в пределах 0,05—0,10, а весовая концентрация при
обработке маломутной воды составляет 0,2—0,5 г/л, при обработ¬
ке мутной — 0,5—6 г/л.
В ходе реагентного умягчения воды допустимые скорости подъ¬
ема воды значительно выше и сильно зависят от соотношения ко¬
личеств Mg(OH)2 и СаС03 в формирующейся взвеси. При возрас¬
тании относительного содержания гидроокиси (начиная с 1 до
45%) скорость потока должна быть снижена с 2,7 до 1,5 мм/сек
[200].
В случае небольшой мутности очищаемой воды процесс освет¬
ления можно интенсифицировать путем добавления во взвешенный
слой мелкого кварцевого песка (d ~ 120 мкм) [201] и дробленого
магнетита (d ~ 250 мкм) [202]. Отмечаются преимущества рецир¬
куляции осадка и пульсационной подачи воды в осветлитель [203—
200
ГЛАВА VI. ОПТИМАЛЬНАЯ ДОЗА КОАГУЛЯНТА
209]. Сконструирован так называемый суперпульсатор — аппарат,
в котором пульсационная подача воды совмещена с тонкослойным
отстаиванием [210].
VI. 3.2.3. Фильтрование
Термин «контактная коагуляция» удачно характеризует сумму
явлений, происходящих при пропускании коагулированной воды
через слои зернистых материалов. Это относится в особенности к
контактным осветлителям и фильтрам, действующим в режиме
прямоточной коагуляции, когда смешение коагулянта с водой про¬
изводится непосредственно перед входом воды в загрузку. Однако
и на фильтрах, предназначенных для доочистки воды после пер¬
вой ступени обработки, также имеет место контактная коагуля¬
ция.
Коагуляция в контакте с посторонней твердой поверхностью
отличается от коагуляции в объеме воды следующими особеннос¬
тями: 1) протекает быстрее; 2) требует меньших доз коагулянта;
3) менее чувствительна к температуре воды; 4) протекает удовлет¬
ворительно даже при малой мутности и низком щелочном резер¬
ве исходной воды. Перечисленные особенности связаны с тем, что
для выделения взвеси из воды достаточным условием является
лишь предварительная астабилизация частиц.
Центральный вопрос механизма фильтрации — это вопрос о
природе, дальнодействии и величине сил притяжения, действую¬
щих между разнородными поверхностями коагулированной взвеси
и зерен загрузки. Современные представления о механизме фильт¬
рации сводятся к следующему.
На поверхности зерен- фильтрующего материала возникает
двойной электрический слой, толщина которого определяется ус¬
ловиями, рассмотренными в гл. I, и достигает 100—1000 А [211,
212]. При движении воды относительно зерен появляется допол¬
нительный потенциал протекания. Под действием молекулярных
сил адгезии, проявляющихся на расстоянии до 1 мкм от поверх¬
ности зерен [213], происходит прилипание частиц взвеси. Интен¬
сивность прилипания во много раз выше взаимного сцепления од¬
нородных частиц и зависит от площади поверхности зерен в еди¬
нице объема материала.
Прилипание частиц стабильных суспензий во многом определя¬
ется их зарядом. Экспериментально установлено, что глубина
проникновения в фильтрующий слой частиц бентонита [214], монт¬
мориллонита [215], кварца, корунда, гидроокиси магния [216]
связана с величиной их ДП. Взаимодействие примесей с зернами
песка и полистирола происходит в соответствии с законами элект¬
ростатики [217, 218]. Но не подлежит сомнению также немалая
VI.3. СВОЙСТВА КОАГУЛИРОВАННОЙ ВЗВЕСИ
201
роль в фиксации частиц утоныиения гидратных слоев на взаимо¬
действующих поверхностях.
В случае, когда в дисперсную систему добавлен гидролизую¬
щийся коагулянт, электростатические эффекты имеют меньшее
значение [214, 219]. Во всяком случае результаты исследований
показывают, что поверхность зерен загрузки быстро обволакива¬
ется продуктами гидролиза и приобретает их положительный за¬
ряд [63, 220]. Наилучшему задержанию песчаными фильтрами коа¬
гулированной взвеси отвечают значения pH среды, равные или
ниже семи [221, 222]. В щелочной среде фильтры работают гораздо
хуже [114, 223]. Но данным Кульского и Медведева [224], наибо¬
лее интенсивное прилипание имеет место при значениях дзета-
потенциала частиц взвеси около —2 мв.
Образование гидроокисных пленок на поверхности фильтрую¬
щих материалов начинается с закрепления небольших первичных
агрегатов [225], затем происходит постепенное наращивание про¬
дуктов гидролиза. По мере накопления осадка интенсивность при¬
липания взвеси к зернам каждого слоя загрузки уменьшается.
Все это, как и при построении хлопьев в объеме, говорит о прояв¬
лении дальнодействующих сил притяжения.
Согласно теории фильтрации, разработанной Минцем [226],
основной причиной, обусловливающей уменьшение интенсивности
задержания взвеси, является разрушение и перенос хлопьев в по¬
следующие слои загрузки. Причем предельное насыщение норово¬
го пространства всегда заметно меньше единицы [227, 228].
Изменение потерь напора в процессе фильтрации, выраженное
как отношение удельных потерь напора в данный момент времени
ix к удельным потерям напора чистого фильтрующего слоя г0,
связано с величиной предельной плотности насыщения порового
пространства
где т'0 — начальная пористость загрузки фильтра; Ат = т'0 —
— т' — изменение пористости в ходе фильтрации.
Показатель степени п зависит от свойств частиц взвеси и фильт¬
рующего материала и составляет, по данным Митина [229], для
суспензий, обработанных A12(S04)3 и полиакриламидом, 2,0—3,3.
Чем ближе к месту входа воды в загрузку вводится раствор коа¬
гулянта, тем большей адгезионной способностью и прочностью
обладает накапливающаяся в порах взвесь.
В соответствии с теорией фильтрации малоконцентрированных
суспензий [226, стр. 75] уменьшение концентрации веществ Ас
в элементарном слое загрузки с единичной площадью поперечно¬
Рн = Ат'/т'0
зависимостью
(VI. 45)
Uh = 1/(1 - Р»)п,
(VI. 46)
202
ГЛАВА VI. ОПТИМАЛЬНАЯ ДОЗА КОАГУЛЯНТА
го сечения (рис. VI.22) описывается уравнением
Ас = — (са — сх) = — ^ Ах, (VI.47)
в котором градиент концентрации дс/дх выражен частной произ¬
водной, так как концентрация частиц в каждом сечении зависит
Рис. VI.22. Схема процесса
фильтрации воды через зер¬
нистую загрузку
от двух переменных — расстояния от поверхности загрузки х и
продолжительности фильтрации т.
Весовое количество вещества, задерживаемое слоем Ах, равно
AQ = vAc = -v^Ax, (VI.48)
где v — скорость фильтрации.
Количество отложений в слое составляет рхАх, а скорость их
накопления за единицу времени
AQ-^^Ax, (VI.49)
где pi— плотность насыщения пор осадком в единице объема заг¬
рузки за единицу времени.
Из выражений (VI.48) и (VI.49) получаем уравнение баланса
вещества
g — v£. (VI.50)
В уравнение (VI.50) входят две независимые переменные —
с и рг. Поэтому для описания процесса Минц использует второе
выражение
Ас = Ас! — Ас2, (VI.51)
в котором Ас, — уменьшение концентрации частиц за счет их при¬
липания; Ас., — увеличение концентрации частиц за счет их от¬
рыва.
VI.3. СВОЙСТВА КОАГУЛИРОВАННОЙ ВЗВЕСИ
203
Уменьшение концентрации частиц прямо пропорционально
средней концентрации частиц в элементарном слое толщиной Ах:
где Ъ' — параметр, характеризующий величину сил адгезии и
аутогезии.
Увеличение концентрации частиц прямо пропорционально ко¬
личеству накопившегося в слое к данному моменту времени осад¬
ка и обратно пропорционально v:
где а — параметр, характеризующий величину гидродинамиче¬
ских сил отрыва частиц.
или после дифференцирования с учетом выражения (VI.50)
Аналогичным путем может быть получено уравнение для плот¬
ности насыщения:
При интегрировании уравнений (VI.55) и (VI.56) Минц допус¬
кает однородность состава загрузки и монодисперсность осветляе¬
мой суспензии. Интегральные формы уравнений (VI.55) и (VI.56)
показывают, что кинетика процесса фильтрации определяется
двумя безразмерными критериями X' и Т'
Одинаковым значениям X' и Т' соответствуют одинаковые зна¬
чения с/си и pJPni где си — концентрация коагулиров'анной взве¬
си в исходной воде, а Рн — ужэ известная нам величина предель¬
ной плотности насыщения норового пространства.
Графики изменения концентрации суспензий во времени при
их фильтрации через слой загрузки разной толщины показывают
близкое соответствие теории экспериментальным данным. Имею¬
щиеся расхождения связаны в основном с полидисперсностью взве¬
сей, неоднородностью загрузок, проскоком в фильтрат труднокоа-
гулируемых частиц. Отмеченное в экспериментах более интенсив¬
ное задержание загрязнений в начале фильтроцикла связано, по
А сх = Ъ'сАх,
(VI. 52)
(VI.53)
Из уравнений (VI.47), (VI.51) — (VI.53) получаем
(VI. 54)
(VI.55)
(VI. 56)
= /(X', Т'), Х' = Ъ’х, Т' = а'т.
(VI.57)
204
ГЛАВА VI. ОПТИМАЛЬНАЯ ДОЗА КОАГУЛЯНТА
мнению Минца, с образованием на поверхности зерен пленок про¬
дуктов гидролиза.
Зависимость между критериями фильтрации X' и Т' описыва¬
ется уравнением
в котором К ш Х0 — коэффициенты, значения которых зависят
только от требуемого эффекта осветления воды С!Сп. На основе
выражения (VI.58) получена следующая формула для длительности
защитного действия загрузки т3:
Соотношение параметров Ъ' и а , определяемых эксперимен¬
тально и зависящих от режима фильтрации, свойств взвеси и ма¬
териала загрузки, имеет размерность скорости и характеризует
скорость проникновения взвеси в глубину загрузки.
Опыт эксплуатации фильтровальных сооружений позволяет
сделать вывод, что очистка воды до уровня, предусмотренного
ГОСТом на питьевую воду, достигается при скоростях фильтра¬
ции на фильтрах 8—12 м/час, на контактных осветлителях — 5—
6 м/час. Проблема интенсификации работы фильтровальных соо¬
ружений решается за счет 1) увеличения крупности зерен загруз¬
ки с одновременным увеличением высоты слоя, 2) применения
фильтрации в направлении убывания крупности зерен, 3) исполь¬
зования новых фильтрующих материалов. Помимо широко
применяемых материалов — кварцевого песка, антрацита и керам¬
зита — все большее распространение находят из материалов есте¬
ственного происхождения горелые породы, доменные и вулкани¬
ческие шлаки, гранат, пиролюзит, магнетит, аглопорит, шунгизит,
ильменит; из искусственных — графит, капрон, полистирол, по¬
ливиниловая и полиамидная смолы. Иногда материалы естествен¬
ного происхождения подвергают специальной обработке (силико¬
ном, смолами, окислами железа, полиэлектролитами).
Дробленые фильтрующие материалы обладают, как правило,
большей грязеемкостью, чем гранулированные. Это их преимуще¬
ство почти всегда объясняют большей пористостью. Не подлежит,
однако, сомнению и важная роль дефектов кристаллической ре¬
шетки, возникающих при дроблении и увеличивающих энергети¬
ческую неоднородность поверхности зерен. Известно, например,
что свободная энергия, приходящаяся на единицу площади раз¬
дробленных зерен кварца, гораздо выше, чем нераздробленных, и
зависит, кроме того, от выбранного способа дробления.
Правильная оценка технологических свойств фильтрующих
материалов предусматривает также учет объема застойных зон,
X' = КТ' + Х0,
(VI.59)
VI.3. СВОЙСТВА КОАГУЛИРОВАННОЙ ВЗВЕСИ
205
гидратации поверхности зерен и возможности выделения на ней
газовых пузырьков [230—232].
При коагулировании вод с относительно"небольшой исходной
мутностью и умеренной цветностью наиболее экономичным про¬
цессом выделения в осадок взвеси является прямоточная обработ¬
ка воды в фильтрующем слое с добавлением коагулянта непосред¬
ственно перед загрузкой [233]. За границей этот метод получил
название «microfloc» [234—237]. При скоростях фильтрации от 4
до 25—50 м/час прямоточная коагуляция дает возможность сни¬
зить расход коагулянта на 25—70% [236, 238—240]. Одним из
вариантов применения метода является использование коагулянтов
при очистке воды на каркасно-пленочных фильтрах [241, 242].
Прямоточная коагуляция испытывает большие затруднения,
если исходная вода содержит планктон, водоросли, нефтепродук¬
ты. Эти затруднения связаны с быстрой закупоркой пор первых
же слоев фильтрующей загрузки и интенсивным ростом потерь
напора. Часть водорослей проскакивает в фильтрат, по-видимо-
му, из-за высокого остаточного энергетического барьера [63].
Для отделения мешающих дисперсных примесей, а также в тех
случаях, когда содержание взвешенных веществ в источнике во¬
доснабжения периодически или постоянно превышает среднюю
величину, на которую рассчитаны сооружения одноступенчатой
очистки (100—200 мг/л), целесообразно предварительное отделе¬
ние части примесей. Таким путем можно достигнуть значительно¬
го снижения грязевой нагрузки на фильтры и контактные освет¬
лители и расширить сферу их применения за счет источников с
более мутной водой.
Необходимость в ряде случаев предварительного отделения
части примесей в равной степени относится и к работающим с при¬
менением коагулянтов сооружениям первой ступени очистки. Хотя
экономически обоснованные рекомендации по предельно допус¬
тимому содержанию взвеси перед этими сооружениями отсутству¬
ют, можно все же полагать, что для отстойников они лежат в диа¬
пазоне 2—4 г/л, для осветлителей со взвешенным осадком —
1—2 г/л.
Для частичного осветления воды и отделения водорослей и
планктона применяют отстаивание, обработку воды в гидроцикло¬
нах, на барабанных сетках и микрофильтрах. Как правило, по¬
мимо снижения нагрузки на основные очистные сооружения, дос¬
тигается заметная экономия коагулянтов. Однако нужно иметь
в виду, что в результате предварительного осветления удаляются
наиболее крупные фракции взвешенных веществ — более 15—
20 мкм. Это приводит к уменьшению степени полидисперсности
взвеси без существенного изменения площади ее поверхности в
единице объема воды и может вызвать ухудшение кинетических
условий коагуляции.
206
ГЛАВА VI. ОПТИМАЛЬНАЯ ДОЗА КОАГУЛЯНТА
Для предварительного осветления водохранилищной воды в
Калифорнии (США) после дождей и штормов применяется распы¬
ление коагулянта по зеркалу воды с лодок [243]. При определении
степени предварительного осветления мутных вод (или, наоборот,
замутнения маломутных) с учетом потребности в коагулянте сле¬
дует использовать выражение (VI.12).
Ли т ера т ура
1. А. P. Black. Water and Sewage Works, 95, 142 (1948).
2. A. P. Black, S. A. Hannah. J. Amer. Water Works Assoc., 53, 438 (1961).
3. A. P. Black, D. G. Williams. Ibid., p. 589.
4. A. P. Black, Ching-lin Chen. J. Amer. Water Works Assoc., 57, 354 (1965).
5. Ю. И. Вейцер. ЖВХО им. Д. И. Менделеева, 5, 628 (1960).
6. Ch. R. O’Melia. Water Quality Improvement by Physical and Chemical
Processes. Austin—London, Univ. Texas Press, 1970, p. 219.
7. W. Stumm, J. J. Morgan. J. Amer. Water Works Assoc., 54, 971 (1962).
8. W. Stumm, Ch. R. O'Melia. J. Amer. Water Works Assoc., 60, 514 (1968).
9. G. Oskam. Tijdschr. drinkwatervoorz en afvalwaterbenhandel, 2,120 (1969).
10. L. B. Miller. Publ. Health Repts, 38, 1995 (1923).
11. L. B. Miller. Publ. Health Repts, 40, 351 (1925).
12. L. B. Miller. Ibid., p. 1413.
13. L. B. Miller. Ibid., p. 1472.
14. E. Bartow, A. P. Black, W. E. Sanbury. Ind. and Engng Chem., 25, 898
(1933).
15. A. P. Black. J. Amer. Water Works Assoc., 26, 1713 (1934).
16. W. F. Langelier, II. E. Ludwig. J. Amer. Water Works Assoc., 41, 163
(1949).
17. W. F. Langelier, F. L. Harvey, G. L. Russel. J. Sanit. Engng Div. Proc.
ASCE, № 2, 118 (1952).
18. S. Mattson. Kolloidchem. Beihefte, 14, 227 (1922).
19. S. Mattson. J. Phys. Chem., 32, 1532 (1928).
20. S. Mattson. Soil Sci., 32, 343 (1931).
21. 3. Я. Берестнева, В. А. Каргин. ЖФХ, 10, .593 (1937).
22. /. В. Pilipovich, А. P. Black, F. A. Eidsness et al. J. Amer. Water Works
Assoc., 50, 1467 (1958).
23. A. P. Black. Water and Sewage Works, 108, 192 (1961).
24. A. S. Teot. Ann. New York Acad. Sci., 155, 593 (1969).
25. A. P. Black, Ching-lin Chen. J. Amer. Water Works Assoc., 59, 1173 (1967).
26. A. P. Black, J.V. Walters. J. Amer. Water Works Assoc., 56, 99 (1964).
27. S. Mackrle. J. Sanit. Engng Div. Proc. ASCE, № 3, 1 (1962).
28. /. Sanit. Engng Div. Proc. ASCE, № 6, 117 (1962); № 1, 75 (1963).
29. R. F. Packham. J. Appl. Chem., 12, 556, 564 (1962).
30. R. F. Packham. J. Colloid Sci., 20, 81 (1965).
31. E. Matijevic, F. E. Janaver, M. Kerker. J. Colloid Sci., 19, 333 (1964).
32. E. Matijevic, F. E. Janaver. J. Colloid and Interface Sci., 21, 197 (1966).
33. Д. А. Фридрихсберг. Курс коллоидной химии. JI., «Химия», 1974.
34. Е. Matijevic, М. В. Abramson, R.H.Ottewill et al. J. Phys. Chem., 65,
1724 (1961).
35. М. C. Fuerstenay, P. Somasundaran, D. W. Fuerstenay. Trans. Inst. Mi¬
ning and Metallurgy, 74, 381 (1965).
36. W. Stumm, C. P. Huang, S. R. Jenkins. Croat, chem. acta, 42, 223 (1970).
37. К. K. Jain, K. Li, R. R. Rothfus. J. Environ. Engng Div. Proc. ASCE,
№ 4, 411 (1973).
VI. ЛИТЕРАТУРА
207
38. М. W. Mackenzie. Trans. Soc. Mining Engrs ATME, 235, 82 (1966).
39. E. S. Hall, R. F. Packham. J. Amer. WaterWorks Assoc., 57, 1149 (1965).
40. Ch. R. O'Melia, W. Stumm. J. Colloid and Interface Sci., 23, 437 (1967).
41. R. K. Ham, R. F. Christman. J. Sanit. Engng Div. Proc. ASCE, № 3,
481 (1969).
42. W. Kim, H. F. Ludwig, W. D. Bishop. J. Amer. Water Works Assoc.,
57, 327 (1965).
43. E. S. Hall. Disc. Faraday Soc., 42, 197 (1966).
44. H. van Olphen. Disc. Faraday Soc., 11, 82 (1951).
45. II. С. Лебедева. Автореферат канд. дисс. М., ВНИИ ВОДГЕО, 1954.
46. Строительные нормы и правила, часть II, гл. 31. М., Стройиздат, 1976.
47. М. N. Rao, С. A. Sastry. J. Inst. Engrs (India.) Public Health Engng
Div., 49, 141 (1969).
48. /. L. Me Atee, L. M. Wells. J. Colloid and Interface Sci., 24, 203 (1967).
49. H. Hahn, U. Neis. Vom Wasser 1972, Bd. 39. Weinheim, 1972, S. 91.
50. R. L. Williams. J. Amer. Water Works Assoc., 57, 801 (1965).
51. E. L. Bean, S. J. Campbell, F. R. Anspach. J. Amer. Water Works Assoc.,
56, 214 (1964).
52. S. Hall. J. Appl. Chem., 15, 197 (1965).
53. G. Schulz, W. Starke. Acta hydrochim. et hydrobiol., 1, 199 (1973).
54. N. Tambo. Mem. Fac. Engng Hokkaido Univ., 11, 585 (1965).
55. E. Д. Бабенков. Водоснабжение и санитарная техника, № 1, 7 (1969).
56. V. S. Gupta, S. К. Bhattachariya et al. J. Amer. Water Works Assoc.,
67, 21 (1975).
57. В. А. Клячко, И. Э. Апелъцин. Подготовка воды для промышленного и
городского водоснабжения. М., Стройиздат, 1962.
58. О. И. Мартынова. Коагуляция при водоподготовке. М.— JI., Гос-
энергоиздат, 1951.
59. Л. А. Кулъский. Основы технологии кондиционирования воды. Киев,
Изд-во АН УССР, 1963.
60. /. S. Moulding, R. Н. Harris. J. Amer. Water Works Assoc., 60, 460
(1968).
61. G. P. Hanna, A. J. Rubin. J. Amer. Water Works Assoc., 62, 315 (1970).
62. S. P. Marion, A. W. Thomas. J. Colloid. Sci., 1, 221 (1946).
63. H. Bernhardt. Vom Wasser 1965, Bd. 32. Weinheim, 1966, S. 193.
64. Л. А. Кулъакий. Рационализация технологии очистки питьевых вод.
Киев, Изд-во АН УССР, 1951.
65. И. Т. Гороновский. Сб. «Исследования по водоподготовке». М., Гос¬
стройиздат, 1956, стр. 5.
66. Л. А. Кулъский, И. Т. Гороновский. Укр. хим. ж., 15, 419 (1949).
67. Л. А. Кулъский, И. Т. Гороновский, М. И. Когановская. Укр. хим. ж.,
16, 231 (1950).
68. Л. А. Кулъский, М. А. Шевченко, А. М. Когановский. Там же, стр. 64.
69. Л. А. Кулъский, А. М. Когановский. Укр. хим. ж., 18, 197 (1952).
70. P. D. Haney. J. Hydraulics Div. Proc. ASCE, № 4, 103 (1956).
71. Clarification of Drinking Water. Library of Congress. Washington, 1948.
72. P. П. Коростелева, Г. П. Чайковский. Труды Хабаровского ин-та же-
лезнодор. транспорта, вып. 41, 266 (1970).
73. Р. П. Коростелева. Автореферат канд. дисс. МИСИ им. В. В. Куйбы¬
шева, 1972.
74. И. И. Жуков. Избранные труды. М., Изд-во АН СССР, 1952, стр. 234.
75. R. F. Packham. Proc. Soc. Water Treat. Exam., 12, 15 (1963).
76. Л. А. Кулъский, В. Ф. Накорчевская, В. А. Слипченко. Активная
кремнекислота и проблема качества воды. Киев, «Наукова думка»,
1969.
77. В. А. Клячко, И. Э. Апелъцин. Очистка природных вод. М., Стройиздат,
1971.
208
ГЛАВА VI. ОПТИМАЛЬНАЯ ДОЗА КОАГУЛЯНТА
78. А. P. Black, J. Е. Singley, G. P. Whittle et al. J. Amer. Water Work
Assoc., 55, 1347 (1963).
79. С. A. Вознесенский. Физико-химические процессы очистки воды. М.— Л.,
Стройиздат, 1934.
80. Т. Saville. J. New Engl. Water. Works Assoc., 31, 78 (1917).
81. J. Shapiro. J. Amer. Water Works Assoc., 56, 1062 (1964).
82. J. Amer. Water Works Assoc., 59, 1023 (1967).
83. /. E. Singley, J. S. Maulding, R. H. Harris. Water Works and Wastes
Engng, 2, № 3, 52 (1965).
84. H. Ф. Ермоленко, С. А. Левина. Труды Третьей Всесоюзной конферен¬
ции по коллоидной химии. М., Изд-во АН СССР, 1956, стр. 276.
85. R. F. Packham. Proc. Soc. Water Treat. Exam., 13, 316 (1964).
86. F. E. Hale. Ind. and Engng Chem., 6, 632 (1914).
87. Т. О. Гончарова, H. И. Кужекова и др. Гидрохимические материалы,
т. 48. JI., Гидрометеоиздат, 1968, стр. 103.
88. Д. Дэхара, К. Тага. Яп. пат. 11260 (1967).
89. А. P. Black, R. F. Christman. J. Amer. Water Works Assoc., 55, 897
(1963).
90. J. E. Singley, A. P. Black. J. Amer. Water Works Assoc., 59, 1549 (1967).
91. W. Rummel. Fortschr. Wasserchem., N 1, 87 (1964).
92. Г. Г. Руденко. Сб. «Санитарная техника, водоснабжение и канализация»,
вып. 4. Киев, «Буд1вельник», 1966, стр. 3.
93. Z. Novak. Wasserwirtschaft—Wassertechnik, 7, 432 (1957).
94. М. И. Гоштейн, А. И. Руденко. Труды научно-исслед. ин-тов Свердл.
облздравотдела, сб. № 6. Свердл. обл. изд-во, 1936, стр. 142.
95. S. Kawamura, G. Р. Наппа, К. S. Shumate. J. Amer. Water Works
Assoc., 59, 1003 (1967).
96. J. Troedsson. Svensk papperstidn., 41, 203 (1938).
97. D. J. Miller, J. T. West. Water and Water Engng, 70, 291 (1966).
98. R. W. Ockershausen. Pulp and Paper Mag. Canada, 69, 29 (1968).
99. H. П. Белоусов. Энергетик, № 1, 7 (1973).
100. L. L. Hedgepeth, N. C. Olsen, W. C. Olsen. J. Amer. Water Works Assoc.,
20, 467 (1928).
101. J. Amer. Water Works Assoc., 62, 311 (1970).
102. Л. А. Кулъский, E. М. Калинийчук. Кондиционирование питьевой во¬
ды. М., Стройиздат, 1964.
103. Г. Л. Грановская, А. Д. Семенов. Труды НИИ хроматографии Воро¬
нежского ун-та, вып. 2, 83 (1968).
104. Ю. И. Вейцер, Л. Н. Паскуцкая, 3. А. Колобова и др. Научные труды
АКХ им. К. Д. Памфилова, вып. 8. Водоснабжение, № 2. М.— JL,
ОНТИ, 1961, стр. 3.
105. Л. 3. Казанцева. Автореферат канд. дисс. Челяб. политехнич. ин-т, 1967.
106. Г. Г. Руденко. Сб. «Санитарная техника, водоснабжение и канализация»,
вып. 2. Киев, «Буд[вельник», 1965, стр. 5.
107. Г. В. Вычегжанина, М. С. Винарский. Труды ВНИИ ВОДГЕО, вып. 53
М., 1975, стр. 132.
108. С. Н. Еленин, М. И. Карлинская, Г. В. Михайлова. Водоснабжение и
санитарная техника, № 2, 5 (1972).
109. И. Т. Гороновский. Физико-химическое обоснование автоматизации
технологических процессов обработки воды. Киев, «Наукова думка»,
1975.
110. Е. Д. Бабенков. Оптимальная доза коагулянта при очистке воды. М.,
«Транспорт», 1974.
111. Е. Д. Бабенков. ЖПХ, 48, 2166 (1975).
112. Е. Д. Бабенков, Э. И. Кирсанова, В. Е. Г уртовенко. Труды ЦНИИ
МПС, вып. 468. М., «Транспорт», 1972, стр. 69.
ИЗ. Е. Д. Бабенков. Вестник ВНИИ железнодор. транспорта, № 4, 45 (1966).
VI. ЛИТЕРАТУРА
209
114. Е. Д. Бабенков. Водоснабжение и санитарная техника, № 10, 24 (1965).
115. /. С. Baderia, С. L. Toshniwal. J. Inst. Engrs (India). Public Health
Engng Div., 49, 103 (1969).
116. Л. А. Кулъский, JI. И. Панченко, Ю. И. Козловский и др. Сб. «Водо-
подготовка и очистка промышленных стоков», вып. 9. Киев, «Наукова
думка», 1972, стр. 78.
117. Е. Ф.Кургаев. Основы теории и расчета осветлителей. М., Госстройиздат,
1962.
118. А. И. Егоров. Сб. «Исследования по водоподготовке». М., Госстройиздат,
1956, стр. 53.
119. С. М. Джафаров. Автореферат канд. дисс. М., ВНИИ ВОДГЕО, 1969.
120. В. А. Борисов, Г. В. Голубь, И. Ф. Чуприн. Водоснабжение и санитар¬
ная техника, № 6, 4 (1969).
121. D. Georgescu, I. Fazekas. Studii protect. §i apur. apel, 6, 453 (1965).
122. V. Cocheci, I. Horescu, I. Pirvu. Bui. stiint. §i tehn. Inst, politehn. Ti¬
misoara, 7, 249 (1962).
123. II. А. Кашкаров. Современные способы очистки воды. Томск, Типо¬
литография Сибирского товарищества печатного дела, 1912, стр. 112.
124. Р. М. Huang, L. М. Kozak. Nature (Engl.), 228, № 5276, 1084 (1970).
125. S. Kawamura, G. P. Hanna. Ind. Water Engng, 4, 21 (1967).
126. JI. Г. Петрова. Труды ВНИИ ВОДГЕО, вып. 16. М., 1967, стр. 10.
127. Б. В. Дерягин, В. Д. Самыгин, А. К. Лившиц. Коллоидн. ж., 26, 179
(1964).
128. М. F. Mohtadi, Р. N. Rao. Water Bes., 7, 747 (1973).
129. A. Choinacki. Gospod. wodna, 28, 49 (1968).
130. Б. А. Скопинцев, М. Т. Голубева. Водоснабжение и санитарная техни¬
ка, № 10, 45 (1938).
131. Обработка воды на тепловых электростанциях. Под ред. В. А. Го-
лубцова. М., «Энергия», 1971.
132. Л. А. Кулъский, А. М. Когановский, И. Т. Гороновский и др., Физико¬
химические основы очистки воды коагуляцией. Киев, Изд-во АН УССР,
1950.
133. S. J. Doctor, К. P. Soni, I. М. Bhatt. Indian J. Chem., 9, 77 (1971).
134. E. Д. Бабенков. Сб. «Методы очистки и контроля качества воды». М.,
«Транспорт», 1966, стр. 15.
135. Е. Д. Бабенков. Канд. дисс. МИСИ им. В. В. Куйбышева, 1966.
136. Е. Д. Бабенков. Материалы Третьей научно-технической конференции
молодых исследователей. М., изд. ЦНИИ МПС, 1967, стр. 6.
137. Г. Бэббит, Дж. Доланд. Водоснабжение. М., Госстройиздат, 1958.
138. П. Е. Богомазов. Смеситель со спиральной струей внутрь. Ростов-на-
Дону, 1934.
139. Н. Е. Hudson. J. Amer. Water Works Assoc., 57, 885 (1965).
140. Л. А. Кулъский, И. Т. Гороновский и др. Авт. свид. СССР 286616
(1968); Бюлл. изобр., № 34 (1970).
141. Г. П. Рыхликов. ЖПХ, 35, 221 (1962).
142. Г. П. Рыхликов. Научные труды Омского мед. ин-та, вып. 69, 45 (1966).
143. Э. К. Сийрде, И. И. Сутт. Авт. свид. СССР 276811 (1969); Бюлл. изобр.,
№ 23 (1970).
144. R. R. Wright, W. L. Booe. Water and Sewage Works, 118, 88 (1971).
145. J. Soutek. Sb. Vysoke skoly chem.-technol. Praze. Odd. Fak. technol.
paliv a vody, 1, 37 (1957).
146. T. R. Camp. J. Amer. Water Works Assoc., 60, 656 (1968).
147. E. Ф. Кургаев. Коллоидн. ж., 19, 72 (1957).
148. Т. И. Барышникова. Канд. дисс. Свердловск, Уральск, политехи, ин-т,
1954.
149. A. I. Lagvannar, R. S. Gemmel. J. Amer. Water Works Assoc., 60, 1040
(1968).
210
ГЛАВА VI. ОПТИМАЛЬНАЯ ДОЗА КОАГУЛЯНТА
150. Т. R. Camp. J. Sanit. Engng Div. Proc. ASCE, N 2, 1 (1964).
151. T. R. Camp. J. Sanit. Engng Div. Proc. ASCE, N 2, 55 (1965).
152. D. L. Switf, S. K. Friedlander. J. Colloid Sci., 19, 621 (1964).
153. D. L. Swift, S. K. Friedlander. J. Colloid Sci., 20, 1070 (1965).
154. E. Ф. Кургаев, В. H. Фрог, Э. И. Кирсанова и др. Теплоэнергетика,
№ 4, 48 (1973).
155. В. А. Глембоцкий, В. И. Классен. Флотация. М., «Недра», 1973.
156. А. Д. Зимон. Адгезия жидкости и смачивание. М., «Химия», 1974.
157. Б. В. Кабанов, А. Н. Фрумкин. ЖФХ, 4, 538 (1933).
158. С. Ludwig. Chem. Engng, 61, 156 (1954).
159. G. M. Fair, R. S. Gemmel. J. Colloid Sci., 19, 360 (1964).
160. II. S. Harris, W. J. Kaufman, R. B. Krone. J. Sanit. Engng Div. Proc.
ASCE, № 6, 95 (1966).
161. S. A. Hannah, J. M. Cohen et al. J. Amer. Water Works Assoc., 59, 843
(1967).
162. H. E. Hudson. J. Amer. Water Works Assoc., 40, 868 (1948).
163. B. 3. Мельцер, В. П. Криштул, В. М. Трескунов. Сб. «Очистка и обез¬
зараживание питьевой воды». М., изд. ЦБНТИ МЖКХ РСФСР, 1972,
стр. 13.
164. F. Benze. Dechema Monographien, 59, 29 (1968).
165. И. Ф. Ефремов. Периодические коллоидные структуры. JL, «Химия»,
1971, стр. 61.
166. В. Т. Турчинович. Улучшение качества воды. М.— JL, Стройиздат, 1940.
167. Е. Д. Бабенков, М. М. Блувштейн. Пособие по пуску, наладке и эк¬
сплуатации очистных сооружений водопровода. М., Стройиздат, 1968.
168. И. Т. Гороновский, Г. Г. Руденко. Эксплуатация станций подготовки
хозяйственно-питьевой воды. Киев, «Буд1вельник», 1975.
169. Z. Tanaka, К. Iinoya. J. Chem. Engng Japan, 3, 261 (1970).
170. А. И. Береза, JI. И. Высоцкий. Научные труды Саратовского поли¬
техи. ин-та, вып. 29, 61 (1968).
171. Ю. И. Вейцер, 3. А. Колобова. Научные труды АКХ им. К. Д. Памфи¬
лова, вып. 1. Водоснабжение. М., ОНТИ, 1960, стр. 56.
172. 3. А. Колобова. Научные труды АКХ им. К. Д. Памфилова, вып. 30.
Водоснабжение, № 4, М.— Л., ОНТИ, 1964, стр. 84.
173. А. П. Нечаев. Труды ВНИИ ВОДГЕО, вып. 25. М., 1970, стр. 5.
174. А. П. Нечаев. Труды ВНИИ ВОДГЕО, вып. 40, ч. 1. М., 1972 (1973),
стр. 3.
175. М. В. Демура, Е. В. Юрков. Сб. «Санитарная техника», вып. 10. Киев,
«Буд1вельник», 1971, стр. 130.
176. А. И. Береза. Вопросы гидравлики горизонтальных отстойников. Изд.
Саратовского ун-та, 1966.
177. А. А. Сурин, 3. Я. Городищер. Сборник научных работ ЛНИИ комму¬
нального хозяйства, вып. 1. Водоснабжение. Л., 1950, стр. 3.
178. 3. Я. Городищер. Там же, стр. 16.
179. М. Rebhun, Y. Argaman. J. Sanit. Engng Div. Proc. ASCE, N° 5, 37
(1965).
180. IO. Б. Багоцкий, E. В. Сизова. Водоснабжение и санитарная техника,
JV« 4, 6 (1956).
181. М. М. Блувштейн, Е. Д. Бабенков. Опыт эксплуатации двухэтажного
горизонтального отстойника. М., Изд-во МКХ РСФСР, 1961.
182. A. Hazen. Trans. Amer. Soc. Civil Engrs, 53, 45 (1904).
183. T. R. Camp. Trans. Amer. Soc. Civil Engrs, 111, 895 (1946).
184. G. Culp, S. Hanzen et al. J. Amer. Water Works Assoc., 60, 681 (1968).
185. В. М. Корабельников. Автореферат канд. дисс. М., АКХ им. К. Д. Пам¬
филова, 1973.
186. К. Танака. Сангё кикай, № 257, 34 (1972); РЖХим., 15И79 (1972).
187. А. P. Livingston. Water and Sewage Works, 116, 119 (1969).
VI. ЛИТЕРАТУРА
211
188. А. И. Жуков, В. Г. Пономарев и др. Прогрессивные методы очистки
природных и сточных вод (материалы конференции), сб. № 2. М., Изд.
МИСИ им. В. В. Куйбышева, 1971, стр. 82.
189. И. М. Миркис. Технология обработки питьевых вод (материалы семи¬
нара), сб. № 1. Изд. Моск. Дома научно-техн. пропаганды им. Ф. Э. Дзер¬
жинского, 1961, стр. 66.
190. И. М. Миркис. Водоснабжение и санитарная техника, № 12, 18 (1965).
191. Е. Ф. Кургаев. Инженерно-физический ж., 15, № 1, 79 (1968).
192. D. G. Miller. Water and Water Engng, 68, 52 (1964).
193. Ю. Арикава. Сэнтан, 17, № 5, 332 (1967); РЖХим., 15Б1488 (1968).
194. A. W. Bond. J. Inst. Water Engrs, 15, 494 (1961).
195. A. W. Bond. Civil Engng Trans. Inst. Engrs Austral., 7, № 2, 141 (1965)
196. A. W. Bond. J. Inst. Water Engrs, 20, 477 (1966).
197. 3. В. Чернова. Научные труды АКХ им. К. Д. Памфилова, вып. 8.
Водоснабжение, № 2, М.— Л., ОНТИ, 1961, стр. 98.
198. I. Tesarik, J. Vostrc.il. Water Res., 9, 887 (1975).
199. V. Mackrle, V. Mican et al. Wasserwirtschaft—Wassertechnik, 9, 22 (1959).
200. М. H. Безенфлейш. Теплоэнергетика, № 3, 24 (1960).
201. А. И. Шахов, С. С. Душкин. Водоснабжение и санитарная техника, № 12,
1 (1971).
202. G. Gomella. Франц. пат. 1501912 (1967).
203. A. A. Kalinske, F. Asce. Civil Engng, № 12, 60 (1960).
204. M. Robinson. Water and Water Engng, 68, 96 (1964).
205. H. Kittner. Wasserwirtschaft—Wassertechnik, 10, 105 (1960).
206. W. Rummel. Ibid., S. 114.
207. V. Mican, I. Tesarik. Ibid., S. 117.
208. H. D. Dehne. Пат. США 3390776 (1968).
209. /. H. Duff. Пат. США 3473665 (1969).
210. С. L. Walton. Effluent and Water Treat. J., 14, 2 (1974).
211. W. Dallman. Freiberg. Forschungsh., A, N 510, 7 (1972).
212. W. Dallman. Freiberg. Forschungsh., A, N 513, 45 (1972).
213. Г. И. Дистлер, С. A. Кобзарева. Сб. «Исследования в области поверх¬
ностных сил». М., «Наука», 1967, стр. 97.
214. С. V. Smith. J. New Engl. Water Works Assoc., 81, 170 (1967).
215. R. M. Jorden. J. Amer. Water Works Assoc., 55, 771 (1963).
216. H. Baldanf, R. Helfricht, U. Weise. Neue Bergbautechn., 2, 533 (1972).
217. А. М. Фоминых, 3. E. Гальперин и др. Изв. вузов. Строительство и ар¬
хитектура, № 12, 110 (1968).
218. Ю. И. Вейцер, 3. А. Колобова, Г. М. Сафонова. Научные труды АКХ
им. К. Д. Памфилова, вып. 97. Водоснабжение. М., ОНТИ, 1974, стр. 33.
219. О. Синогара, Т. Уеда. Суйдо кекай дзасси, № 492, 9 (1975); РЖХим.,
ЗИ421 (1976).
220. R. J. Hunter, А. Е. Alexander. J. Colloid Sci., 18, 846 (1963).
221. D. G. Miller, J. T. West. Water and Water Engng, 70, 291 (1966).
222. H. E. Hudson. J. New Engl. Water Works Assoc., 80, 232 (1966).
223. JI. А. Кулъский, М. И. Медведев. Сб. «Санитарная техника», вып. 10.
Киев, «Буд1вельник», 1971, стр. 147.
224. JI. А. Кулъский, М. И. Медведев. Сб. «Новая аппаратура и автоматиче¬
ские устройства по технологии обработки воды», вып. 9. Киев, «Науко¬
ва думка», 1972, стр. 63.
225. Е. М. Казаков, Г. А. Китаев, С. Г. Мокрушин. Коллоидн. ж., 22, 23
(1960).
226. Д. М. Минц. Теоретические основы технологии очистки воды. М.,
Стройиздат, 1964.
227. Б. А. Митин. Сб. «Процессы фильтрования при очистке природных
и сточных вод». Челябинск, Южно-Уральское кн. изд-во, 1965, стр. 39.
212
ГЛАВА VI. ОПТИМАЛЬНАЯ ДОЗА КОАГУЛЯНТА
228. В. 3. Мельцер. Научно-теоретическая конференция аспирантов АКХ
им. К. Д. Памфилова (тезисы докладов). М., ОНТИ, 1970, стр. 21.
229. Б. А. Митин. Сб. «Процессы фильтрования при очистке природных
и сточных вод». Челябинск, Южно-Уральское кн. изд-во, 1965, стр. 27.
230. В. 3. Мельцер. Научные труды АКХ им. К. Д. Памфилова, вып. 98.
Водоснабжение. М., ОНТИ, 1973, стр. 97.
231. В. А. Жужиков. Фильтрование. Теория и практика разделения суспен¬
зий. М., «Химия», 1971, стр. 173.
232. Е. Ф. Кургаев, Г. В. Шерстобитов, 9. И. Кирсанова, Е. В. Демченко.
Водоснабжение и санитарная техника, № 7, 9 (1976).
233. Г. Г. Руденко. Сб. «Санитарная техника, водоснабжение и канализа¬
ция», вып. 7. Киев, «Буд1в ельник», 1968, стр. 131.
234. А. II. Rice, W. R. Conley. Tappi, N 1, А167 (1964).
235. В. Price. Amer. City, N 2, 80 (1962).
236. Т. V. Garel. Water Works and Wastes Engng, 2, N 12, 34 (1965).
237. K. Kato. Japan Chem. Quart., 2, N 4, 46 (1966).
238. Г. Д. Павлов. Водоснабжение и санитарная техника, № 6, 4 (1970).
239. А. Я. Кыйв, 9. Р. Каар и др. Труды Таллинского политехи, ин-та, А,
№ 298, 89 (1970).
240. Е. А. Горбачев, Н. Ф. Санкин. Труды ГИСИ им. В. П. Чкалова, вып.
58. Горьк. кн. изд-во, 1971, стр. 102.
241. G. R. Bell. Proc. Intemat. Water Conf., 22, 169 (1961).
242. D. E. Burns, E. R. Baumann et al. J. Amer. Water Works Assoc., 62, 121
(1970).
243. W. R. Ree. J. Amer. Water Works Assoc., 55, 275 (1963).
Глава VII
САНИТАРНАЯ ЭФФЕКТИВНОСТЬ КОАГУЛЯЦИИ
VII. 1. СФЕРА ПРИМЕНЕНИЯ И СВОЙСТВА КОАГУЛЯНТОВ
Сфера применения коагулянтов определяется их составом и физи¬
ко-химическими свойствами, стоимостью, требованиями, предъяв¬
ляемыми к устройствам для приготовления и дозирования их рас¬
творов. Рассмотрим сферу и масштабы применения разных видов
коагулянтов в том же порядке, что и при их описании в гл. III.
VII. 1.1. Соли алюминия
Сернокислый алюминий используют для очистки мутных и цвет¬
ных вод: очищенный — при высокой мутности, неочищенный или
содержащий в качестве инградиентов глины и силикатные мате¬
риалы — при низкой мутности воды. Этот коагулянт эффективен
в диапазоне значений pH 5—7,5, причем чем выше жесткость воды
и ниже ее цветность, тем выше оптимальные значения pH среды
[1 (стр. 139), 2,3 (стр. 45)]. Относительно низкая стоимость, хо¬
рошая растворимость, отсутствие особых требований к обращению
с сухим и растворенным продуктом сделали сульфат алюминия
наиболее распространенным коагулянтом.
Установлено, что сульфат алюминия обладает большей эффек¬
тивностью, чем соли железа, при удалении из воды дубильных [4]
и гуминовых веществ [5]. Попутно с обесцвечиванием воды проис¬
ходит заметное снижение ее окисляемости. Например, при обра¬
ботке воды с исходной цветностью 30—45 град дозой A12(S04)3,
равной 100 мг/л, в области значений pH 6—6,5 происходило сни¬
жение цветности на 89 %, а перманганатной окисляемости — на
67% [6].
Отмечено [7], что сульфат алюминия с повышенным содержа¬
нием А1203 (28,5%) отличается от обычного коагулянта лучшей
обесцвечивающей способностью, которая может быть объяснена
иными структурно-химическими свойствами продуктов гидроли¬
за. В случаях недостаточного щелочного резерва воды или необхо¬
димости повышения значений pH требуется нейтрализация кис¬
лотности коагулянта, которая достигается добавлением щелоч¬
ных реагентов. Теоретически на нейтрализацию 100 мг A12(S04)3-
•18Н20 расходуется 0,9 мг-экв НС03. Практически же удельная
потребность в щелочах определяется конкретными химическими
условиями коагуляции и находится в пределах от 0,47 (для высо¬
коцветных вод) до 0,7—0^82 мг-экв НС03 [8, стр. 216].
214
ГЛАВА VII. САНИТАРНАЯ ЭФФЕКТИВНОСТЬ КОАГУЛЯЦИИ
Применение «черного» коагулянта — сернокислого алюминия
с добавкой активного угля — рекомендовано для вод невысокой
мутности при наличии органических веществ, обусловливающих
запахи и привкусы.
Алюминат натрия в отличие от сульфата алюминия — щелоч¬
ной реагент, и его использование позволяет получить воду с пока¬
зателем стабильности, близким к единице. В некоторых случаях
замена сернокислого алюминия алюминатом натрия дает экономи¬
ческие преимущества [9].
Алюминат натрия гидролизуется при pH ' 10,5 [10], а при
pH 9,3—9,8 образует быстроосаждающиеся хлопья [И]. Однако
оптимальным условиям очистки воды отвечают значения pH 4,5—
8,0 [12, 13]. Для нейтрализации избыточной щелочности необхо¬
димо добавление кислоты. В технологии очистки воды для под-
кисления иногда используют дымовые газы. Реакция нейтрализа¬
ции проходит по уравнению:
2NaA102 + С02 + 3H20 2А1 (ОН)3 + Na2C03.
В большинстве описанных в литературе случаев при низкой
щелочности обрабатываемой воды нейтрализация достигается за
счет совместного использования алюмината натрия и сульфата
алюминия [14—16]. Самостоятельное употребление алюмината нат¬
рия вследствие довольно высокой его стоимости ограничивается
случаями низких значений pH обрабатываемой воды.
Сульфат алюминия и алюминат натрия дозируют в обрабаты¬
ваемую воду в соотношениях 1 : 10—1 : 20, руководствуясь при
этом достижением оптимальных значений pH. Реакция нейтрали¬
зации протекает по уравнению
6NaA102 + Al2 (S04)3 + 12Н20 ^ 8А1 (ОН) 3 + 3Na2S04.
Совместное применение NaA102 и AI2(S04)3 дает возможность
повысить эффект осветления и обесцвечивания воды, увеличить
плотность и скорость осаждения хлопьев коагулированной взве¬
си, расширить зону оптимума pH, уменьшить отрицательное влия¬
ние низких температур [1 (стр. 142), 8 (стр. 210), 16, 17].
Обработка по данному способу воды р. Делавэр показала, что
оптимальным условиям коагуляции соответствуют более высокие
отрицательные значения дзета-потенциала коагулированной взве¬
си (—6 -= 10 мв) по сравнению с обработкой воды одним только
сернокислым алюминием (—5 ч 8 мв) [18]. Это позволяет не¬
сколько снизить общий расход коагулянта.
Оксихлорнд алюминия в сравнении с сульфатом алюминия име¬
ет следующие технологические преимущества [19—29].
1. При хранении не слеживается и не теряет коагулирующих
свойств. Имеет примерно в 3 раза более высокое содержание во¬
VII.1. СФЕРА ПРИМЕНЕНИЯ И СВОЙСТВА КОАГУЛЯНТОВ
215
дорастворимого алюминия. Растворы оксихлорида не требуют ан¬
тикоррозийной защиты аппаратуры.
2. Оксихлорид алюминия обладает меньшей кислотностью и
потому пригоден для очистки вод с небольшим щелочным резер¬
вом. Например, в реакциях
2А12 (ОН)6С1 + Са tHC03)2->4Al (0Н)8 + СаС12 + 2С02,
2А12 (S04)3 + ССа (НС03)2 -> 4А1 (ОН)3 + 6CaS04 + 12С02
снижение щелочности от оксихлорида алюминия в 6 раз меньше,
чем от эквивалентного (по А1203) количества сульфата алюминия.
3. Расширяется зона оптимума pH главным образом в сторону
низких значений.
4. При использовании оксихлорида алюминия солесодержание
воды увеличивается меньше, чем при использовании AI2(S04)3.
Это важно для воды, предназначенной для нужд теплоэнергетики,
производства целлюлозы и искусственного волокна.
5. Ускоряются хлопьеобразование и осаждение коагулиро¬
ванной взвеси. При очистке малоцветных вод (преимущественно
мутных и минерализованных) уменьшается расход коагулянта.
6. Уменьшается количество остаточного алюминия в обрабо¬
танной воде.
Совместное применение оксихлорида и сульфата алюминия поз¬
воляет, по данным Левицкого и Максимова [30], снизить общий
расход коагулянта по А1203 на 10—15%.
Аммиачные квасцы предназначены для обработки хлорирован¬
ной воды с целью уменьшения запаха хлора за счет образования
хлораминов.
VII. 1.2. Соли железа
К преимуществам солей железа по сравнению с солями алюминия
относят [3 (стр. 38), 8 (стр. 217), 31—34]:
1. Лучшее их действие при низких температурах обрабатывае¬
мой воды.
2. Более широкую зону оптимальных значений pH среды.
3. Большую прочность и гидравлическую крупность хлопьев,
лучшие тиксотропные свойства.
4. Применимость для вод с более широким диапазоном солево¬
го состава.
5. Способность устранять запахи и привкусы, обусловленные
присутствием сероводорода, удалять марганец, сорбировать сое¬
динения меди и мышьяка, катализировать окисление фенола, са¬
хара и других органических соединений.
Некоторые авторы указывают на меньший расход солей же¬
леза по сравнению с сульфатом алюминия [33, 35, 36], а также на
их лучшую обесцвечивающую способность [34, 35].
216
ГЛАВА VII. САНИТАРНАЯ ЭФФЕКТИВНОСТЬ КОАГУЛЯЦИИ
Среди недостатков солей железа отмечают [31—33, 37 (стр.
ИЗ), 38-43]:
1. Образование при реакции катионов железа с некоторыми
органическими соединениями (см. гл. II) сильно окрашенных
растворимых комплексов.
2. Сильные кислотные свойства, корродирующее действие на
аппаратуру.
3. Менее развитую поверхность хлопьев.
4. Необходимость добавления извести или хлора для окисле¬
ния Fe2+ при использовании солей закисного железа. В противном
случае хлопьеобразование сильно замедляется (например, при
содержании 52% Fe2+ в смеси с Fe3+ скорость протекания коагу¬
ляции примерно в 2,5 раза ниже, чем при отсутствии Fe2+).
Наилучшая коагуляция примесей воды солями железа имеет
место при pH 3,5—6,5 или 8,0—11,0. Существование двух зон оп¬
тимума pH может быть объяснено в соответствии с диаграммой
состояния железа в растворе (см. гл. III) сильной нейтрализующей
способностью катионов железа в кислой среде и хорошими адсорб¬
ционными и адгезионными свойствами гидроокиси железа в зоне
высоких значений pH. Обесцвечивание протекает наилучшим об¬
разом в зоне pH 3,5—5,0 [34, 43]. Замена солей алюминия солями
железа позволяет достигнуть оптимума pH при меньших затратах
коагулянта. Необходимость в подщелачивании отпадает и может
даже возникнуть потребность в подкислителях [44].
Результаты проведенных исследований указывают на преиму¬
щество солей железа при очистке мутных жестких вод с высокими
значениями pH (например, паводочных вод) [40, 45—47], а также
при обработке стоков [16, 48]. Это объясняется их способностью
хорошо осветлять воду при более низкой остаточной щелочности
и катализировать окисление органических примесей растворенным
кислородом. Примесь титана не препятствует использованию со¬
лей железа, поскольку в ходе коагуляции концентрация Ti4+
в воде уменьшается от значений, близких к 1 мг/л, до 0,04—0,085
мг/л [49, 50].
При коагулировании солями закисного железа главная тех¬
нологическая задача состоит в заблаговременном переводе двух¬
валентного железа в трехвалентное. Окисление Fea+ кислородом,
содержащимся в воде, происходит медленно. Его скорость зависит
от величины pH среды и достигает приемлемой для практических
целей величины лишь при pH > 8 (рис. VII.1). Для интенсифика¬
ции процесса окисления прибегают к подщелачиванию воды, ее
перемешиванию, аэрации, обработке хлором.
Необходимость подщелачивания сильно усложняет процесс.
К тому же добавленная щелочь может затруднять обесцвечивание
воды. Поэтому FeS04 используют в качестве коагулянта главным
VII.1. СФЕРА ПРИМЕНЕНИЯ И СВОЙСТВА КОАГУЛЯНТОВ
217
Рис. VII.1. Зависимость времени окисления (т0) двухвалентного железа
кислородом от pH воды
Рис. VII.2. Зависимость степени окисления двухвалентного железа от про¬
должительности (т) аэрации воды с интенсивностью подачи воздуха 8 л!мин
[52]
образом при реагентном умягчении воды известью или содой
(см. гл. X).
В замкнутой системе (без доступа воздуха) Fe2+ окисляется
примерно в 4 раза медленнее, чем в открытой [51]. Скорость окис¬
ления двухвалентного железа при аэрации воды с интенсивностью
подачи воздуха 8 л/мин может быть примерно определена по экс¬
периментальному графику Рюммеля [52], показанному на рис.
VII.2. Из графика видно, что для полного окисления 550 мг Fe2+
требуется проводить аэрацию в течение 14 мин. В ходе аэрации
происходит временное снижение pH воды (от 8,05 до 7,35), которое
Рюммель объясняет тем, что реакция гидролиза Fe2+ с выделени¬
ем водорода протекает быстрее, чем нейтрализация Н+ добавлен¬
ной известью. После завершения гидролиза величина pH повыша¬
ется.
Для ускорения окисления железа Сато [53] предлагает прово¬
дить аэрацию воды в присутствии активного угля, утверждая,
что такой способ дает хорошие результаты даже при pH ~ 2.
Скорость окисления Fe2+ повышается при фильтрации воды через
кварцевый песок, причем ранее образовавшееся трехвалентное
железо катализирует процесс. Эффективность действия песка воз¬
растает с увеличением жесткости исходной воды [54].
Если исходить из предположения, что в присутствии хлора
реакция окисления проходит по уравнению
2Fe2+ + С12 -» 2Fe3+ + 2С1~,
то расход хлора на 1 мг Fe2+ составляет 0,63 мг. Фактически же
количество затрачиваемого хлора зависит от целого ряда уело-
218
ГЛАВА VII. САНИТАРНАЯ ЭФФЕКТИВНОСТЬ КОАГУЛЯЦИИ
вий — величины pH воды, дозы хлорсодержащего реагента, соле¬
вого состава воды. Исследования, проведенные в ЦНИИ МПС, по¬
казали, что удельные затраты хлора на окисление 1 мг двухвалент¬
ного железа, полученного электролитическим путем, находятся
в пределах от 0,42 до 1,05 мг. Несовпадение этих затрат с расчет¬
ными объясняется формированием сложных гидроксокомплексов.
По предположению Конокшиоли и др. [55], в процессе хлорирова¬
ния образуются димеры, в которых железо выступает в четырех¬
валентной форме
ОН
Подробные исследования, выполненные в 1971 г. Сысоевой
[56], позволяют объяснить зависимость скорости окисления Fe2+
хлором от величины pH среды образованием на промежуточной
стадии реакции гипотетического радикала С12Н по схеме
Fe2+ + Cl2 ^ Fe3+ + Cljf (К1г tfj,
Cl” + Н+ -» С12Н (A’j),
Fe2+ + Cl2H->Fe3+ + 2Cl- + H+ (К3).
Путем использования метода квазистационарных состояний
получено следующее выражение для скорости уменьшения катио¬
нов Fe2+ при хлорировании:
Аналогичным уравнением описывается скорость окисления железа
кислородом [57].
Выразив величину [Fe3+] через произведение растворимости
гидроокиси железа или основной соли [Fe2S04(0H)4], Сысоева
и др. получили уравнение зависимости константы скорости реак¬
ции от величины pH среды и доказали его соответствие результа¬
там ранее проведенных экспериментов [58].
Исследованиями, проведенными в Академии коммунального
хозяйства им. К. Д. Памфилова [59], установлено, что снижение
щелочности воды после добавления к ней железного купороса за¬
висит от дозы введенного хлора. В частности, от добавления
100 мг/л FeS04 при дозе хлора 13 мг/л щелочность снижалась на
1,1 мг-экв/л, а при дозе 20 мг/л — на 1,4 мг-экв/л. На основании
этих результатов сделан вывод о необходимости при обработке
\/
ОН
d. [Fe2+]
dx
VII. 1. СФЕРА lIFHMJinrjxaifx/x jrx
воды хлорированным железным купоросом иметь щелочной ре-
зерв порядка 1,5 мг-экв/л.
Использование хлорированного железного купороса при обес¬
цвечивании воды дает лучшие результаты по сравнению с другими
солями железа. Однако в такие сезоны года, когда цветность ис¬
ходной воды невысока, экономически выгоднее хлор заменять из¬
вестью.
VII. 1.3. Смеси солей алюминия и железа.
Соли магния
Почти во всех случаях применения смесей солей алюминия и же¬
леза достигнуты лучшие результаты очистки воды, чем при раз¬
дельном использовании солей алюминия и железа. Наблюдаемое
при этом расширение зоны оптимальных значений pH можно объяс¬
нить большим разнообразием продуктов гидролиза со своими ин¬
дивидуальными свойствами, а ускоренное осаждение хлопьев —
изменением структуры коагулята за счет более плотной упаковки
частиц. Однако экспериментальные данные, которые позволяли
бы с достаточными основаниями подтвердить или, наоборот, опро¬
вергнуть эти предположения, отсутствуют.
Оптимальными соотношениями количеств A12(S04)3 и FeCl3
при обработке вод разного состава считают от 1 : 1 до 1 : 2 [37,
60—62]. Отмечается [40,45], что преимущества смешанного коа¬
гулянта проявляются наиболее сильно при очистке холодных вод:
увеличивается степень очистки, исчезает опалесценция обрабо¬
танной воды, наблюдаемая при использовании одного лишь серно¬
кислого алюминия. В случае применения A12(S04)3 вместе с же¬
лезным купоросом (с добавкой извести) есть опасность сузить зо¬
ну оптимума значений pH, поскольку при pH < 7,3 возрастает
концентрация в воде остаточного железа, а при pH 8— оста¬
точного алюминия.
Соли магния применяют при значениях pH среды 10,9—11,2,
когда образуется гидроокись магния с произведением раствори¬
мости ~6-10~10 [63]. Гидроокись магния формируется в плотные,
быстро осаждающиеся хлопья, а отработанный осадок легко реге¬
нерируется [64]. Сообщается [65j, что после успешных испытаний
на опытной установке в г. Монтгомери (США) приступили к ис¬
пользованию в промышленных масштабах карбоната магния с его
последующей регенерацией из осадка путем обработки углекис¬
лым газом. Образующийся при этом бикарбонат магния легко
растворяется и извлекается из фильтрата в виде MgC03.
При очистке сточных вод, содержащих поливиниловый спирт,
применение гидроокиси магния в качестве коагулянта дало луч¬
шие результаты, чем применение солей алюминия и железа [66].
VII. 2. ЭФФЕКТИВНОСТЬ УДАЛЕНИЯ ВРЕДНЫХ ПРИМЕСЕЙ
В предыдущей главе процесс коагуляции рассматривался на при¬
мерах осветления и обесцвечивания воды, т. е. удаления из нее
основной массы минеральных и органических загрязнений естест¬
венного происхождения. С точки зрения санитарной эффективно¬
сти коагуляции и надежности этого метода по гигиеническим пока¬
зателям нас в первую очередь будет интересовать степень очистки
воды от вредных для здоровья микрозагрязнений и биологичес¬
ких примесей.
Ориентируясь на степень значимости коагуляции в комплексе
водоподготовительных процессов, мы сочли целесообразным не¬
сколько изменить последовательность рассмотрения отдельных
видов загрязнений, принятую в гл. II, и начать изложение с наи¬
более важной для коагуляции задачи — удаления истинно рас¬
творенных микрозагрязнений, поступающих в источники водо¬
снабжения со сточными водами.
VII. 2.1. Удаление микрозагрязнений
VII. 2.1.1. Синтетические поверхностно-активные вещества
Очистка вод от синтетических ПАВ — весьма серьезная проблема.
Хлорирование дозами, применяемыми обычно в практике водо¬
снабжения, не эффективно, а при большой дозе (36 мг/л) одного из
самых сильных окислителей — двуокиси хлора — концентрация,
например, алкилбензолсульфоната (АБС) и алкил сульфата (АС)
снижается с 20 только до 12 мг/л [67]. При фильтрации воды на
медленных песчаных фильтрах концентрация синтетических де¬
тергентов (СД) уменьшается лишь на 13—50%. Коагулирование
воды, хотя и не избавляет ее полностью от детергентов, является
важнейшим барьерным мероприятием против их появления в пить¬
евой воде в недопустимых концентрациях.
Исследования по коагулированию воды, содержащей синтети¬
ческие ПАВ, развивались в двух направлениях: выявления дей¬
ствия ПАВ на скорость коагуляции и определения эффективности
коагулянтов в отношении ПАВ. Обнаруженное в экспериментах
замедление коагуляции примесей воды объясняли вначале исклю¬
чительно действием основного моющего компонента. Позднее
установили [68, 69], что, по крайней мере, частично оно может
быть отнесено на счет присутствующих в моющих средствах фос¬
фатов. В чистом виде лишь два типа АБС из числа исследованных
замедляли коагуляцию при концентрации их 8—20 мг/л; осталь¬
ные не оказывали влияния даже при содержании более 25 мг/л.
По данным Коэна и др. [70], мешающее действие додецилбен-
золсульфоната натрия на коагуляцию примесей воды сульфатом
&
Рис. VI 1.3. Зависимость сте- ^
пени удаления АБС от pH воды
при ее обработке сернокислым
алюминием (200 мг/л)
железа обнаруживается при концентрации ПАВ 8 мг/л; мешающее
действие лаурилсульфата — при концентрации 4 мг/л. Для устра¬
нения этого действия пришлось увеличить дозу коагулянта.
Изучение влияния АБС на работу фильтров после предвари¬
тельной обработки воды сульфатом алюминия привело к заключе¬
нию о больших потерях напора в присутствии ПАВ [71, 72]. Это
можно объяснить возрастанием адгезионных свойств, прочности
и объема коагуляционных структур. Результаты более обстоятель¬
ных опытов, поставленных в 1965 г. с учетом влияния на работу
фильтра величины pH среды [73], привели к выводу о возможно-
ти образования малорастворимого комплекса
[А1в(ОН)„]з+-АБС.
При значении pH 7,0 расхождения в приросте потерь напо¬
ра на опытном и контрольном фильтрах были невелики, и большая
часть АБС проходила в фильтрат. С понижением pH комплексообра-
зование протекало интенсивнее, хлопья становились крупнее
и образовывались даже при pH 4,8, чего не наблюдалось в от¬
сутствие АБС. Наилучшим условиям удаления АБС при обработ¬
ке воды сульфатом алюминия (дозами 15—50 мг/л) и последующим
отстаиванием соответствовали величины pH 4,5—4,8 (рис. VII.3).
Сильное влияние величины pH среды на степень удаления ПАВ
коагуляцией отмечали и другие исследователи, причем одни из
них считают, что оптимум pH лежит в области низких значе¬
ний (3,6—5,0) [74—76], другие — в области высоких (11—12)
[75-81].
Удаление СД при низких значениях pH осуществляют с помо¬
щью кислых солей алюминия и железа, доводя в некоторых слу¬
чаях дозу коагулянта (например, при очистке стоков прачечных)
до нескольких граммов на литр [75], а также используя подкислите-
ли [82], активный уголь [75,83—85], флокулянты [74,831.
Очистка в кислой среде основана на реакциях комплексообра-
зования между катионами алюминия и железа и анионными груп¬
пами ПАВ. Ее результаты могут быть предусмотрены зарапее па
основе зависимости состава продуктов гидролиза от величины pH
ziii:
ГЛАВА Vii. иАНИТАРИАЯ ЭФФЕКТИВНОСТЬ КОАГУЛЯЦИИ
среды [86]. Кроме того, как показано в наших исследованиях [85],
при низких значениях pH возрастает сорбционная емкость порош¬
кообразных активных углей по отношению к анионным ПАВ [85].
Для очистки воды в условиях повышенных значений pH при¬
меняют ее подщелачивание известью, добавку хлористого кальция
[75—81, 87—89]. В щелочной среде ПАВ диссоциируют и их способ-
Та блица VII.1. Эффективность удаления из воды синтетических ПАВ
разными коагулянтами и вспомогательными реагентами
ПАВ
Кон-
цен-
т ра¬
ция,
мг/л
Условия очистки воды
Удале¬
ние ПАВ,
%
Литера¬
тура
АБС
100
100 мг/л Fe3+
80
[91]
АБС
60
300—400 мг/л Al2(S04)3, ВМФ, pH
5,5-6
39
[92]
АБС
10
100 мг/л Al2(S04)3, 75 мг/л АУ,
pH 4,5-5
80
[85]
АБС
4
56 мг/л Ak(S04)3, 46 мг/л АУ, 0,4 мг/л
ВМФ «сепаран»
87,5
[83]
АБС
3
75 мг/л Al2(S04)3, 75 мг/л АУ, pH
4,5—5
73
[85]
АБС
1,8
20 мг/л A12(S04)3, 100 мг/л АУ,
0,02 мг/л ВМФ «сепаран»
83,3
[83]
АБС
1,0
75 мг/л Al2(S04)3, 75 мг/л АУ, pH
4,5—5
100
[85]
АБС
0,8
20 мг/л Al2(S04)3, 50 мг/л АУ,
0,02 мг/л ВМФ «сепаран»
87,5
[83]
АС
100
100 мг/л Fe3+
66
[91]
АБС и АС
20
12 мг/л A12(S04)3, 1,2 мг/л АК и из¬
весть в количестве, обеспечивающем
повышение pH до 8,8
31,5
[89]
АБС и АС
20
10,7 мг/л A12(S04)3, 12мг/л бентонита
и известь в количестве, обеспечи¬
вающем повышение pH до 8,8
50
[50]
АБС и АС
20
12 мг/л Fe2(S04)3, 1,7 мг/л хлора,
pH 9,1
47,5
[89]
АБС и АС
20
60 мг/л осадка, полученного при об¬
работке воды Al2(S04)3, Fe2(S04)3
и известью
80
[89]
Алкилсуль¬
фонат
100
100 мг/л Fe3+
80
[91]
Алкилнаф-
талинсуль-
фат
100
100 мг/л Fe3+
48
[91]
Примечание. АБС — алкилбензолсульфонат; АС — алкилсульфонат; АУ —
активный уголь; АК — активная кремнекислота.
VII.2. ЭФФЕКТИВНОСТЬ УДАЛЕНИЯ ВРЕДНЫХ ПРИМЕСЕЙ
223
ность вступать в химическую реакцию с продуктами гидролиза
коагулянта возрастает [77,78]. При этом важная роль принадле-
яжт катионному составу воды. Добавление натриевых солей го¬
раздо менее эффективно, чем кальциевых, что объясняется боль¬
шей активностью алюмо-кальциевых солей в реакции с анионами
ПАВ.
По методу, разработанному Лурье и Антиповой [77—81], сме¬
шение соли алюминия с известковым молоком при повышенных
значениях pH приводит к формированию алюмината кальция
{[Са (A102)2]m ■ гсСа2+ (2п — х) ОН-}* (ОН“),
который при взаимодействии с ПАВ обменивает гидроксильные
группы на анионы. Для приготовления алюмината кальция ре¬
комендованы А1С13 и A12(S04)3. Смешение реагентов с известковым
молоком может производиться заблаговременно или в обрабаты¬
ваемой воде.
Наряду с концепцией о прямом химическом взаимодействии
ПАВ с добавленными к воде соединениями, нельзя не учитывать
физической адсорбции. Показано, в частности, что продукты гид¬
ролиза коагулянта одинаково эффективны как при удалении ани¬
онных, так и неионогенных ПАВ, а закономерности удаления АС
и АБС могут быть описаны уравнением физической адсорбции [67].
Возможность использования выражения Фрейндлиха для коли¬
чественной оценки сорбции моющего средства ОП-7 на осадках
А1(ОН)3, Fe(OH)3 и Mg(OH)2 при pH 6 доказана Хрусталевым
и др. [90]. В табл. VII.1 приведены результаты очистки воды от не¬
которых синтетических ПАВ разными коагулянтами.
Из приведенных в табл. VII. 1 данных видно, что обработка
воды солями алюминия и железа позволяет уменьшить концентра¬
цию синтетических ПАВ на 31,5—100%. Преимущество солей же¬
леза, на которое указывают некоторые исследователи [92, 93],
в приведенных примерах не обнаруживается.
VII. 2.1.2. Фосфаты
На помехи коагуляции, вызываемые присутствием фосфатов в во¬
де, указывают многие исследователи [68, 89, 94—98]. Морган
и Энгельбрехт [96], изучая влияние фосфатов на осветление воды
солями железа и алюминия, пришли к заключению, что с увели¬
чением концентрации трифосфата и пирофосфата от 0,5 до 2,0 мг/л
(по Р2Ов) коагуляция постепенно ухудшается. Мешающее действие
фосфатов авторы оценили по соотношению коэффициентов удаления
взвешенных веществ из воды в присутствии фосфатов и в их от¬
сутствие. Для A12(S04)3 это соотношение оказалось равным в сред¬
нем 0,85-0,95, для FeCl3 -0,91-0,97, для Fe2(S04)3 —0,94—
0,99. С увеличением доз коагулянтов и жесткости воды степень по¬
224
ГЛАВА VII. САНИТАРНАЯ ЭФФЕКТИВНОСТЬ КОАГУЛЯЦИИ
мехи уменьшается, с понижением температуры — возрастает
[96, 981.
Для достижения того же эффекта осветления доза коагулянта
в присутствии фосфатов должна быть повышена. Уменьшение по¬
требности в коагулянте с увеличением жесткости воды можно объ¬
яснить снижением заряда твердой фазы, образованием фосфатно¬
кальциевых комплексов или обоими этими обстоятельствами.
Пороговые концентрации (мг/л) различных фосфатов, при кото¬
рых становится заметным ухудшение коагуляции водных примесей
сульфатом алюминия, приведены ниже [68J:
Доза А13+, мг/л Триполифосфат Пирофосфат Ортофосфат
10 0,5 1,0 6,0
20 2,0 2,0 16,0
Эти данные согласуются с результатами исследований Рао [97]
по коагулированию солями алюминия воды, содержащей 0,5—
5 мг/л полифосфатов и 2—10 мг/л ортофосфатов. Рао установил
также, что пробы воды, содержащей 1—3 мг/л полифосфата и ме¬
тафосфата, требовали для достижения той же степени осветления
увеличения доз сульфата алюминия в 2,3—3,7 раза. При обработ¬
ке воды р. Делавэр, содержащей 0,16 мг/л метафосфата, можно
было обойтись в 1,5 раза меньшими дозами A12(S04)3, чем при об¬
работке воды ее притока, содержащей 1,46 мг/л метафосфата [18].
Тем не менее считается [99], что реагентные методы удаления фос¬
фатов наиболее дешевы и надежны.
Характерно влияние фосфатов на структурные и седиментаци-
онные свойства коагулированной взвеси: хлопья становятся более
крупными и рыхлыми. Но на заранее сформированные хлопья
гидроокисей фосфаты оказывают слабое действие [95]. Видимо,
поэтому, используя фракционированное коагулирование, общую
дозу сернокислого алюминия, требующуюся для эффективного
удаления полифосфатов, можно снизить на 25% [97]. Для повы¬
шения скорости осаждения хлопьев рекомендуются добавки фло-
кулянтов, главным образом катионных полиэлектролитов.
При обработке воды солями железа с добавкой известкового
молока коагуляция в присутствии фосфатов протекает несколько
лучше, чем при обработке солями алюминия [97]. Что касается
технологической эффективности разных коагулянтов в отношении
удаления фосфатов, то она зависит от конкретных условий очистки
воды. Одни авторы отдают предпочтение алюминийсодержащим
коагулянтам, указывая на меньшую потребность в них (примерно
в 1,5 раза) по сравнению с железосодержащими [101, 102]; другие
[103] придерживаются противоположной точки зрения; наконец,
по данным работы [104] одинаковые молярные дозировки А13+
и Fe3+ соответственно в виде хлорированного железного купороса
VII.2. ЭФФЕКТИВНОСТЬ УДАЛЕНИЯ ВРЕДНЫХ ПРИМЕСЕЙ
225
Рис. VI 1.4. Зависимость оп- 5
тимальных значений pH коагу- 5^
лирования воды сернокислым §
алюминием от содержания в sT
ней ортофосфатов различной ^
концентрации [100] (мг-экв/л):
1 — 0; г — 0,05; 3 — 0,5; 4 — 5 .
и сульфата алюминия обеспечивают одинаковую степень удаления
фосфатов.
Согласно экспериментальным данным, при очистке от фосфатов
сточных вод (в диапазоне исходных концентраций POJ3 4—60 мг/л)
оптимальные соотношения [Ме3+] ; [Р] лежат в пределах от 1 : 1
до 6 : 1, причем большие соотношения имеют место при меньших
исходных концентрациях фосфатов [102, 105—116].
Вопрос об оптимальных дозах коагулянтов тесно переплетает¬
ся с вопросом о механизме удаления фосфатов. Отсутствие чисто
стехиометрических соотношений заставляет предполагать протека¬
ние специфических реакций и, так же как и в случае ПАВ, мнения
исследователей разделяются: одни считают, что преобладают хими¬
ческие взаимодействия, другие — физическая адсорбция. Под¬
робные исследования, выполненные Хенриксеном [103, 117], при¬
водят к выводу об образовании труднорастворимых соединений.
Об этом же свидетельствует тот факт, что под влиянием возраста¬
ющих концентраций фосфатов оптимальные значения pH коагуля¬
ции (как и в присутствии сульфатов) смещаются в сторону более
низких значений (рис. VII.4). Однако при исходной концентра¬
ции фосфатов порядка 6—12 мг/л имеет место и физическая адсорб¬
ция их на продуктах гидролиза коагулянта, которая может быть
описана уравнением Лэнгмюра.
По данным работ Попеля [118, 119], количество удаленных ко¬
агуляцией фосфатов (в %) может быть подсчитано по уравнению
р = Кх + К2 lg с + К3 lg [Р] + Я4рН, (VII.1)
в котором Кх, К2, кз и А’4 — коэффициенты; с — молярная кон¬
центрация добавленных катионов Al3+ или Fe3+; [PI — исходная
концентрация фосфатов. Автор указывает,*что для достижения
р = 90—95% необходимо, чтобы концентрация с была равна
2—6 ммолъ/л.
При использовании Л12(304)з’'оптималыше значения pH для
удаления фосфатов из сточных вод лежат в интервале 6,0—7,1;
при использовании Fe2(S04)3 — в интервале 4,7—7,4. Помимо кор¬
ректировки значений pH среды и добавления ВМФ, для повыше-
8 Е. Д. Бабенков
226
ГЛАВА VII. САНИТАРНАЯ ЭФФЕКТИВНОСТЬ КОАГУЛЯЦИИ
ния эффективности удаления фосфатов коагуляцией предлагают
добавлять к воде анионные ПАВ, образующие с фосфатами ком¬
плексы [120]; использовать смесь растворов NaOH,. А1(ОН)3 и крах¬
мала [121 ]; вводить различные глинистые сорбенты, например вер¬
микулит, обработанный солями железа и алюминия [122]; добав¬
лять ионообменные продукты [123]. Результаты лабораторных
и производственных опытов указывают на высокую эффективность
электрокоагулирования [124—126], а также коагулирующих рас¬
творов, полученных путем обработки отходов металла кислотами
и щелочами [127].
По мнению большинства исследователей, применение коагу¬
лянтов при очистке сточных вод от фосфатов не оказывает влияния
на последующий процесс метанового сбраживания осадка [102, 106,
128], однако объем осадка возрастает в 1,5—2 раза [102, 104].
VII. 2.1.3. Пестициды
Сведений об эффективности применяемых способов очистки воды
от пестицидов очень мало. Это обусловлено прежде всего трудно¬
стями определения малых концентраций ядохимикатов. Однако
имеющихся данных достаточно для того, чтобы сделать вывод
о довольно высокой эффективности коагулирования по сравнению
с другими методами обработки воды, например окислительными.
Как показали Робек и др. [129], двукратное хлорирование воды
и добавление перманганата калия не дают результатов. Лишь озон
в высоких концентрациях (35—38 мг/л) снижает содержание пес¬
тицидов примерно наполовину. В то же время применение коагу¬
ляции с последующим фильтрованием воды обеспечило уменьше¬
ние концентрации линдана (гексахлорана) на 10, алдрина — на 35,
дилдрина — на 55, бутоксиэтилового эфира — на 65, паратиона —
на 80 и ДДТ — на 98%. Сходные результаты по перечисленным
пестицидам получены в другой работе [130].
Степень удаления гексахлорана и симазина коагуляцией, по
данным Шевченко и др. [131], растет с увеличением дозы A^SOJg
от 50 до 200 мг/л, а при дальнейшем увеличении дозы (до 500 мг/л)
не изменяется, составляя соответственно 80 и 10%. Согласно экс¬
периментам Коша и др. [132], удаление ДДТ и ДДЕ коагуляцией
протекает тем успешнее, чем больше исходная вода содержит орга¬
нических примесей.
Эффективность коагуляции может быть повышена добавлением
к воде активных углей и природных сорбентов. При дозах уголь¬
ного порошка порядка 10—20 мг/л степень удаления хлороргани-
ческих пестицидов возрастала до 90% [129]. Кульский и др. [133]
установили, что величина сорбции из сточных вод ДДТ на угле
марки КАД-иодный достигает 0,8% от веса угля.
VII.2. ЭФФЕКТИВНОСТЬ УДАЛЕНИЯ ВРЕДНЫХ ПРИМЕСЕЙ
227
VII. 2.1,4. Радиоактивные вещества
Степень дезактивации коагулированием определяется изотопным
составом радиоактивных веществ (РВ) и их состоянием в растворе.
Если РВ адсорбированы на механических примесях или сами на¬
ходятся в коллоидно-дисперсном состоянии, достигается устойчи¬
вая дезактивация. Степень извлечения РВ определяется в этом
случае глубиной осветления воды. По отношению к истинно раство¬
ренным РВ коагулянты гораздо менее эффективны. Однако при¬
сутствие в обрабатываемой воде дисперсных примесей, а также ис¬
кусственное ее замутнение позволяют получить по многим изото¬
пам неплохие результаты, делают коагуляцию одним из основных
методов дезактивации.
Исследования по дезактивации воды коагулированием, прово¬
димые при исходной радиоактивности примесей от 1 • 10-5 до 1 •
• 10~9 кюри!л и дозах коагулянтов 10—500 мг/л, показывают, что
в ходе двухступенчатой очистки активность воды снижается на 70—
90%. Причем РВ, ассоциированные со взвесью, удаляются на
97—100%. На 90—98% удаляются изотопы элементов, способных
гидролизоваться с образованием малорастворимых соединений
ниобия, церия, иттрия, циркония, празеодима, неодима и др. Кон¬
центрация остальных элементов уменьшается лишь на 10—60%.
К трудноудаляемым коагулированием изотопам относятся 89Sr,
90Sr, 131J, 137Cs, 140Ba [37 (стр. 397), 134—137]. Особенно плохо
удаляется 131J, образующий с другими ионами высокорастворимые
соединения.
Поскольку связывание растворенных РВ происходит за счет
реакций комплексообразования и адсорбции на продуктах гидро¬
лиза коагулянта, удаление РВ носит избирательный характер.
С помощью сернокислого алюминия удалялось, например, около
половины содержащегося в воде стронция и рутения и лишь
20% цезия. Радиоактивный фосфор снимался на 96,8% [138].
При дозах сульфатов алюминия и железа 180 мг/л достигнуто
практически полное устранение (3-активных грубодисперсных при¬
месей, а также устранение на 60% истинно растворенных РВ.
По эффективности дезактивации некоторые авторы отдают пред¬
почтение железосодержащим коагулянтам [139, 140], что связано,
по-видимому, с образованием малорастворимых продуктов гидро¬
лиза железа в области более высоких, чем для алюминия, значе¬
ний pH. При pH 10 с помощью сульфата железа (100 мг/л) 90Sr
удалялся примерно на 30% [141]. Комацу [142], исследуя дезакти¬
вацию воды, содержащей смесь изотопов стронция и иттрия, выя¬
вил преимущества гидроокиси титана по сравнению с солями алю¬
миния и железа.
Все те меры, которые способствуют улучшению очистки воды
от взвешенных примесей, водорослей и планктона (применение
8*
228
ГЛАВА VII. САНИТАРНАЯ ЭФФЕКТИВНОСТЬ КОАГУЛЯЦИИ
окислителей, полиэлектролитов и др.), дают положительные ре¬
зультаты и в отношении дезактивации. Добавление флокулянтов
(аэрофлок-548, S-3000, RD-4053 и др.) позволило снизить опти¬
мальные значения pH при коагулировании воды хлорным желе¬
зом с 11,5 до 7—8 и сохранить при этом высокий процент сниже¬
ния р- и у-активности —85—95 [142,143]. Указывается, что иногда
к очищенной воде полезно добавлять для соосаждения некоторое
количество стабильных элементов [144, 145].
Поскольку многие изотопы гидролизуются в щелочной среде
(pH 8—11), подщелачивание воды улучшает очистку. Так, с помо¬
щью сульфатов железа и алюминия при pH 4,4 удаляется от 3
до 30% радиоактивного рутения, а при pH 7,5—8,4 от 74 до 98%
[143]. По этой же причине высокой дезактивирующей способно¬
стью обладают карбонатная и фосфатная коагуляции. Известь ис¬
пользуется преимущественно вместе с солями алюминия или желе¬
за для поддержания повышенных значений pH, фосфаты могут
применяться и самостоятельно.
С помощью солей КН2Р04 и Na3P04 (100—200 мг/л) изотопы
91Y, 95Zr 144Се удаляются на 99,2—99,8% [145]. На хорошую дез¬
активирующую способность фосфатов в отношении 90Sr указывают
результаты экспериментов Бернгардта и Гартмана [146], в от¬
ношении продуктов деления урана — Мархарта [137]. Самостоя¬
тельного применения фосфатная коагуляция при очистке поверх¬
ностных вод не находит, но использование фосфатов совместно
с солями алюминия и железа, а также с известковым молоком за¬
метно повышает степень устранения радиоактивных примесей [143,
144, 147 (стр. 13)].
Наибольший эффект в удалении из воды РВ имеет место при
фракционированном добавлении коагулянта [141, 148, 149]. Один
из путей повышения эффекта состоит в использовании электрокоа¬
гуляции. Заметного улучшения дезактивации можно достичь вне¬
сением в обрабатываемую воду в дополнение к коагулянтам мел¬
кодисперсных минеральных сорбентов. Результаты применения
этого метода представлены в табл. VI.2.
Для очистки сточных вод от РВ в качестве сорбентов рекомен¬
дованы иллит, джибсит, лимонит [150]. Использование тонкоиз-
мельченных глинистых сланцев позволяет повысить степень дез¬
активации воды коагулированием, отстаиванием и фильтрацией
с 60—85 до 90—98%. Изотопы стронция, иттрия, цезия, бария,
церия и празеодима обработкой воды полиэлектролитами (лайтрон-
886 и сепаран-2610) с предварительной добавкой измельченных
глинистых сланцев удаляются на 65,5—100% [151].
По данным Орловой и др. [152], хорошо сорбирует изотопы из
воды моренный суглинок: при дозе его 100 мг/л активность воды,
содержащей радиоизотопы церия, празеодима, рутения, ниобия,
кальция, бария, цезия, циркония и бария, снижается на 98%.
VII.2. ЭФФЕКТИВНОСТЬ УДАЛЕНИЯ ВРЕДНЫХ ПРИМЕСЕЙ
229
Таблица VII.2. Стенень дезактивации воды (%) при обработке
глиной 1143]
Изотоп
Концентрация глины, мг'л
100
750
5000
«Sc
53
91,7
96,9
89Sr
2-12
14-22
49-52
91Y
22-49
56-70
93,6
116Cd
3
30
64
137Cs
38
87
98
140Ba и 140La
41
58
85
144Ce
70-80
86
185\^
0
4
49
В присутствии бентонита улучшается очистка воды коагулянтами
от 32Р [37J. Оптимальные дозы глин в ряде случаев очень высоки —
до 6 г/л, а длительность достижения адсорбционного равновесия
равна приблизительно 1,5 час [153].
VII. 2.1.5. Прочие примеси
При удалении из воды мышьяка наиболее эффективны железосо¬
держащие коагулянты. Если исходная концентрация As не превы¬
шает 3 мг/л, достаточна доза коагулянта 40 мг/л (по Fe203), чтобы
снизить ее до норм качества питьевой воды [154]. Доливо-Добро-
вольским наилучшие результаты достигнуты при использовании
Fe2(S04)3 и извести с поддержанием значений pH обрабатываемой
воды в пределах 7,0—9,5 [155]. Из других солей железа для уда¬
ления мышьяка рекомендованы хлорное железо [156] и хлориро¬
ванный железный купорос [157]. Практически полная очистка
воды с исходной концентрацией As до 2 мг/л достигается при дозе
FeCI3 32 мг/л [158].
Использование при очистке городских сточных вод сернокисло¬
го алюминия и извести, помимо удаления мышьяка, позволяет до¬
биться значительного снижения концентрации меди, свинца, хро¬
ма, никеля, кадмия, ртути. На степень удаления ртути сильное
влияние оказывает мутность воды (с увеличением мутности эффек¬
тивность действия сульфата железа возрастает), причем оптималь¬
ные значения pH составляют 10,7—11,4. Неорганическая ртуть
отделяется успешнее, чем органическая [159]. Наиболее широки¬
ми возможностями обладает способ удаления ртути путем коагу¬
ляции коллоидных и грубодисперсных частиц сульфида ртути,
230
ГЛАВА VII. САНИТАРНАЯ ЭФФЕКТИВНОСТЬ КОАГУЛЯЦИИ
образующихся при добавлении к воде сернистого натрия. Этот
способ позволяет снизить концентрацию ртути на 99%.
По данным авторов работы [160], концентрации металлов в реч¬
ной воде после ее обработки коагулянтами, осветления и фильтро¬
вания на песчаных фильтрах снижаются в среднем: железа и мар¬
ганца на 65%, никеля и меди — на 50%, свинца и хрома — на
30%. По данным других авторов [161], после обработки речной во¬
ды известью и солями железа, 6-часового отстаивания и фильтра¬
ции через песчаные и сорбционные фильтры пятивалентный мышь¬
як удаляется на 98%, кадмий — на 95%, ртуть и трехвалентный
мышьяк — на 60—90%, а барий, радий и селен — менее чем на
60%.
Указывается [162], что соосаждение молибдена продуктами
гидролиза железа протекает успешнее, чем продуктами гидролиза
алюминия. При значениях pH 3—4 и соотношении концентраций
железа и молибдена более 100 возможно удаление молибдена из
сточных вод на 80—90%. Степень извлечения из обрабатываемой
воды капролактама в зависимости от его исходной концентрации
составляет 9,2—21,8% [163].
VII. 2.2. Удаление биологических прммесей
VII. 2.2.1. Планктон и водоросли
В период массового развития планктона и водорослей при обработ¬
ке воды коагулянтами на водоочистных станциях возникают боль¬
шие затруднения. Присутствие биологических организмов тормо¬
зит коагуляцию, требует существенного увеличения доз коагу¬
лянтов, иногда — в 2—3 раза [164—169].
Эффективность коагулирования в отношении планктона и во¬
дорослей зависит от их исходной концентрации. Применение коа¬
гулирования для обработки воды на Северной станции московско¬
го водопровода (мутность 7,2 мг/л, цветность 40 град, содержание
планктона 1900 клеток в 1 мл) дозой сернокислого алюминия
3,9 мг/л (по А1203) позволило после 1,5—2-часового отстаивания
снизить концентрацию планктона на 64,6%. При этом произошло
попутное уменьшение мутности воды на 62,5% и цветности на 50%
[165]. Использование коагулирования для очистки воды от диато¬
мовых водорослей в двуступенчатой схеме очистки привело к сни¬
жению концентрации водорослей на 73—83%: около 24% клеток
задерживалось отстаиванием, остальные — фильтрацией [170].
В 1968 г. проведены подробные исследования по коагулирова¬
нию сульфатом алюминия воды, содержащей водоросли Synedra,
Fragilaria, Spirogyra, Nitischia, Navicula, Anabena, Oscillatoria,
Phormidium, Desmidium, Hydrodictyon, Pinnilaria, Vlotrix, Ankis-
trodesmus, Cyclotella [171].
VII.2. ЭФФЕКТИВНОСТЬ УДАЛЕНИЯ ВРЕДНЫХ ПРИМЕСЕк
231
Авторы установили, что снижение содержания водорослей в во¬
де происходило на 81—91,5%, в то время как количество бактерий
уменьшалось на 98—100%, а взвешенных веществ — на 60—
74,5 %. По отношению к каждому виду загрязнений обнаружен чет¬
ко выраженный оптимум дозы коагулянта.
В работе [170] отмечаются преимущества хлорного железа по
сравнению с сульфатом алюминия.
Повышения степени удаления водорослей и планктона удается
добиться за счет применения органических высокомолекулярных
флокулянтов [168, 172—174] и активной кремнекислоты [175].
Действие флокулянтов, как и в случае суспензий, состоит в обра¬
зовании мостовых связей и укрупнении хлопьев коагулированной
взвеси. Оптимальные дозы полиэлектролитов определяются фазой
развития водорослей: при переходе линейной фазы роста в лога¬
рифмическую и стационарную они возрастают примерно в 3 раза.
Скорость флокуляции достигает максимума при 50%-ной степени
покрытия поверхности клеток макромолекулами. В области низ¬
ких pH стимулирующее действие катионных флокулянтов возрас¬
тает, что, по-видимому, связано со снижением заряда клеток и
и электростатических сил их взаимного отталкивания [173].
Помимо флокулянтов, для улучшения удаления планктона и во¬
дорослей коагуляцией применяют хлорирование воды, добавляют
к ней известковое молоко или активный уголь.
Для эффективной борьбы с водорослями Oscillatoria rubescens
при их массовом развитии (до 100 тыс. клеток в 1 мл) рекомендует¬
ся двухстадийное коагулирование воды с применением полиэлек¬
тролита по схёме: 1) сернокислый алюминий—сода—хлор; 2) сер¬
нокислый алюминий — флокулянт. Как показали испытания,
первоначальная величина дзета-потенциала клеток (—24 мв) умень¬
шалась при обработке воды по этому способу после первой стадии
обработки до —19 мв, после второй — до —10 мв [168].
При очистке воды от водорослей (исходная концентрация до
50 тыс. клеток в 1 мл) по схеме «микрофильтрация—коагулирова¬
ние (с добавлением активного угля)—хлорирование—отстаива¬
ние — известкование—фильтрация» выявлено заметное стимули¬
рующее действие извести. Тем не менее, фильтрат содержал от 50
до 100 клеток водорослей в 1 мл воды [176].
VII. 2.2.2. Бактерии и вирусы
Бактерии. Дезинфицирующие реагенты, даже такой универсаль¬
ный реагент, как хлор, не всегда обеспечивают надежное обезза¬
раживание воды [177]. Гарантированного обеззараживания можно
достигнуть, если бактерии не только разрушены окислителями,
но и отделены от очищенной воды [178 (стр. 292)]. Поэтому коагу¬
лирование следует рассматривать как способ, дополняющий дей¬
ствие дезинфектантов.
232
ГЛАВА VII. САНИТАРНАЯ ЭФФЕКТИВНОСТЬ КОАГУЛЯЦИИ
Эффективность обеззараживания воды коагулянтами зависит
от полноты протекания процессов хлопьеобразования и последую¬
щего отделения хлопьев. Больше всего сведений накоплено по
обеззараживанию от кишечной палочки (В. Coli), содержание ко¬
торой в воде уже долгое время считается показателем биологичес¬
кой загрязненности воды. Из этих сведений видно, что коагули¬
рование воды с последующим отстаиванием (без добавления окисли¬
телей) способно очистить воду от В. Coli на 80—95%. Примерно
такой же процент обеззараживания (70—90%) достигается по об¬
щему числу бактерий в воде. Концентрация микроорганизмов
в осадке канализационных станций уменьшается после обработки
его хлорным железом в 10®—104 раз [179].
По данным Стритера [8], между содержанием бактерий в ис¬
ходной воде си и в воде, подвергнутой коагулированию и отстаи¬
ванию с0С, существует эмпирическое соотношение
Если длительность отстаивания Т выражена в часах, коэффициен¬
ты С и и равны соответственно 0,57 и 0,88.
На существование связи между степенью обеззараживания во¬
ды от бактерий и длительностью отстаивания коагулированной
взвеси, а также мутностью исходной и очищенной воды указывают
результаты работ [180—184].
С увеличением дозы коагулянта эффект обеззараживания воз¬
растает. Так, согласно экспериментам, увеличение дозы A12(S04)3-
•14 Н20 от 15 до 50 и 100 мг!л вызвало повышение степени обезза¬
раживания воды от В. Coli соответственно от 82 до 95 и 99,5%
[185]; увеличение дозы Al2(S04)a от 15 до 25 мг!л — от 75 до 99%
Степень обеззараживания воды коагулянтами во многих слу¬
чаях находится в прямой зависимости от степени удаления взве¬
шенных веществ. Поэтому остаточная мутность воды после обра¬
ботки коагулянтами может служить ориентировочным показателем
надежности обеззараживания. По свидетельству Дюрена [187],
при очистке воды от бактерий Streptococcus faecalis и Escherichia
Coli в присутствии замутнителей (кварц и бентонит), между про¬
центным удалением бактерий (так же как и глин) Y и требуемой
дозой коагулянта (сульфата алюминия) а существует зависимость
в которой m и С' — эмпирические константы.
Бадериа и др. [180] дают следующую эмпирическую зависи¬
мость между процентным удалением из воды В. Coli Y и взвешен¬
ных примесей X (рис. VII.5):
(VII. 2)
[186].
Yam = С',
(VI 1.3)
Y = 2.98 X -170.
(VI 1.4)
VII.2. ЭФФЕКТИВНОСТЬ УДАЛЕНИЯ ВРЕДНЫХ ПРИМЕСЕЙ
233
Рис. VII.5. Соотношение ме¬
жду величинами процентного
удаления из воды взвешенных
примесей (X) и В. Coli (У)
в результате коагулирования
[180]
Рис. VII.6. Зависимость про¬
центного удаления (р) взвешен¬
ных веществ (кривые 1) и
бактерий (кривые 2) от дозы
коагулянта при мутности ис¬
ходной воды 30 (а) и 150 мг/л
(б) [180]
90
70
50
— -
1
*
в
* ' /
в
•
н -Л
7
•
у
•
•
f
.
•
А
1
50
70 90 Л
10‘
\В
X 6
№
-а—
"*'* S
ч
)/
/V
^ V
У
. - ..
ео
80
ГОО
W
/г
^ /
— -
— —. ^
Ь
50
80
Оптимальные дозы коагулянта для удаления взвешенных ве¬
ществ и бактерий разные. На основе результатов тщательных ис¬
следований по обеззараживанию воды двух источников, содержа¬
щей от 30 до 150 мг/л минеральной взвеси и разные количества
В. Coli (210, 460 и 2400 клеток в 1 мл установлено, что кривые, опи¬
сывающие связь между процентным удалением взвеси (св) и бак¬
терий (Y) и дозой коагулянта, имеют вид парабол (рис. VII.6),
на которых точки максимума для св и У не совпадают как в гори¬
зонтальном, так и в вертикальном направлении. Кривые описы¬
ваются уравнением (VII.3) при следующих значениях коэффици¬
ентов: для удаления взвеси С = 2,029, т. = 0,39; для удаления
бактерий С = 2,033, т = 0,57 [180].
Разная степень удаления механических примесей и бактерий
обусловлена, вероятно, различиями в коагуляции мутных и мало¬
мутных вод (см. гл. VI). Это предположение подтверждается тем
обстоятельством, что при меньшем содержании примесей (30 и
и 50 мг/л) доза сульфата алюминия, обеспечивающая максималь¬
ное обеззараживание воды (см. рис. VII.6), ниже, чем доза, приво¬
дящая к наибольшему удалению примесей; при большей величине
св (135—150 мг/л) наблюдается обратное явление.
234
ГЛАВА VII. САНИТАРНАЯ ЭФФЕКТИВНОСТЬ КОАГУЛЯЦИИ
По-видимому, основной причиной удаления микроорганизмов
в ходе коагуляции является их адсорбция на продуктах гидроли¬
за коагулянта, хотя и не исключается возникновение мостовых
связей между катионами А13+ или Fe3+ и линейными полимерными
субстанциями бактерий и вирусов [181]; наиболее прочная связь
между продуктами гидролиза и бактериями достигается в кислой
среде, о чем свидетельствуют, в частности, данные Рубина и др. [188].
Вирусы могут быть удалены коагуляцией с последующим от¬
стаиванием и фильтрованием воды на 98—99,99%. Причем основ¬
ная часть вирусов удаляется после коагуляции и отстаивания.
Например, эксперименты по доочистке сточных вод показали, что
после обработки сернокислым алюминием и отстаивания процент
удаления вирусов составил 99,85, а после фильтрации —99,99
[189]. Примерно такой же эффект достигается при умягчении во¬
ды, обладающей высокой магнезиальной жесткостью. Но в этом
случае положительные результаты объясняются пе столько фор¬
мированием малорастворимых продуктов гидролиза магния,
сколько высокими значениями pH среды. Например, показано
[190], что при увеличении pH от 10,8 до 11,2 инактивация вирусов
возрастает от 50 до 98,5%. Предполагается, что это связано с де¬
натурацией протеиновых оболочек и разрушением вирусов в ще¬
лочной среде [191].
Многие авторы полагают, что в основе механизма удаления ви¬
русов коагуляцией лежит процесс комплексообразования трехва¬
лентных металлов с ионизированными группами протеинов [192 —
195]. Вместе с тем показано, что для описания процесса ассоциа¬
ции вирусов с продуктами гидролиза коагулянтов применимо
уравнение адсорбции Фрейндлиха [192].
С увеличением исходной концентрации вирусов, а также тре¬
буемой степени их удаления доза коагулянта возрастает [192,
196—198]. Присутствие посторонних органических веществ, ка/и¬
онов Са2+ и Mg2+ в одних случаях приводит к положительным [198,
199], в других — к отрицательным результатам [194, 195, 200].
Нужно иметь в виду отмеченную в исследованиях избиратель¬
ность действия коагуляции по отношению к вирусам. Например,
вирус иолиомиэлита при коагулировании воды, ее отстаивании
и фильтрации через диатомитовые фильтры удаляется очень пло¬
хо [2011; для удаления из воды бактериофага F-2 (на 99%) требо¬
валось сернокислого алюминия в 2 раза меньше, чем хлорного же¬
леза [202]. Кроме того, после коагуляции вирусы остаются живы¬
ми, и если не произведено своевременное удаление осадка из очист¬
ных устройств, существует опасность вторичного заражения воды
[191, 195, 200].
Оптимальные значения pH удаления коагуляцией бактериофа¬
га Т-4 составляли 5,1—5,5, бактериофага М-2—5,9—6,1, вирусов
коксаки и бактериальных —5,5—5,6 [185, 192, 195].
VII.2. ЭФФЕКТИВНОСТЬ УДАЛЕНИЯ ВРЕДНЫХ ПРИМЕСЕЙ
235
Удаление вирусов происходит обычно пропорционально удале¬
нию взвешенных примесей [195, 199], что объясняется их сорбцией
на минеральных частицах. Известно, например, что на части¬
цах суспензии бентонита сорбируется свыше 99% вирусов ящура
и коксаки [1771. И хотя условия максимального удаления вирусов
и взвеси (так же как и в случае бактерий) не всегда совпадают, оста¬
точная мутность воды может служить показателем санитарной
надежности очистки [178].
Эффективность обеззараживания воды коагулянтами повы¬
шается при добавлении активной кремнекислоты [196, 198] и орга¬
нических высокомолекулярных флокулянтов [195, 199, 203]. Ни¬
же приведены результаты удаления (в %) бактериофага Т-2 ири
исходном его содержании 106—107 микробных тел в 1 мл с помо¬
щью как одного сернокислого алюминия, так и в сочетании с ани¬
онным (геркулес-СМС), катионным (нэльколит-605) и неионоген¬
ным (нэльколит-110) флокулянтами (дозой 1 мг/л) [199]:
A.12(S04)3) Без ВМФ Геркулес-СМС Нэльколит-605 Нэльколит-110
Из приведенных данных видно, что при самостоятельном ис¬
пользовании (без коагулянта) анионный и неионогенный полиэлек¬
тролиты действовали слабее, чем катионный, а в сочетании с
A12(S04)3 их действие усиливалось. Однако степень удаления бак¬
териофага при совмещении коагуляции с флокуляцией оставалась
примерно такой же как и при одном только коагулировании.
По данным авторов [195], применение катионных флокулянтов
позволяет увеличить степень очистки воды от микроорганизмов
(бактериофаги Т-4 и М-2). Этот результат объясняется, по всей ве¬
роятности, более высоким содержанием в исследованной воде ме¬
ханических примесей, благодаря которому эффективность действия
ВМФ проявляется отчетливее. В подтверждение этого вывода мож¬
но сослаться на работу Карлсона и др. [203], в которой авторы до¬
казывают возможность существования сложных глинисто-поли-
мерно-вирусных комплексов.
Обработка воды солями алюминия и железа, в особенности сер¬
нокислым закисным железом,— эффективный способ очистки воды
от ботулинического токсина. Применение полиакриламида и ка¬
тионного флокулянта ВА-2 дает хорошие результаты только в соче'
тании с гидролизующимися коагулянтами 1204, 205].
мг/л
о
5 56-58
40 98
до 40
39-52
96
84-94
80-94
98
до 37
60-73
96 ■
236
ГЛАВА VII. САНИТАРНАЯ ЭФФЕКТИВНОСТЬ КОАГУЛЯЦИИ
VII. 3. ЭФФЕКТИВНОСТЬ КОАГУЛЯЦИИ В СОЧЕТАНИИ С ДРУГИМИ
МЕТОДАМИ ВОДОПОДГОТОВКИ
VII. 3.1. Обработка окислителями
Хлор и другие окислители разрушают гидрофильные органичес¬
кие соединения, стабилизирующие дисперсные примеси воды, и об¬
легчают условия протекания коагуляции. Особенно эффективно
применение окислителей при обработке цветных вод.
В зависимости от состава органических примесей и дозы окис¬
лителя расход коагулянта моя^ет быть в большей или меньшей сте¬
пени снижен. Иногда он сокращается в 2,5 [205] и даже в 4 раза
[206]. Увеличивается гидравлическая крупность хлопьев коагу¬
лированной взвеси, ускоряется осветление воды [207, стр. 12].
Кроме того, предварительная обработка воды окислителями поз¬
воляет поддерживать водоочистные сооружения в лучшем сани¬
тарном состоянии, предотвращать вторичное заражение воды ми¬
кроорганизмами и загнивание осадка и тем самым увеличивать
длительность работы отстойников между чистками. В присутствии
хлора гели гидроокиси алюминия, накопленные в загрузке кон¬
тактных осветлителей, дольше сохраняют адсорбционные свойства
при прекращении подачи коагулянта [208, 209].
Выбор окислителя зависит от местных условий и задач водо¬
очистки. Наиболее распространено предварительное хлорирова¬
ние газообразным хлором или хлорной известью. При низких тем¬
пературах, а также при наличии в источнике водоснабжения тру-
дноокисляющихся ароматических веществ эффективны озон и пер¬
манганат калия [210].
При очистке цветных вод величину дозы окислителя подбира¬
ют, ориентируясь на рациональную глубину обесцвечивания.
Удельный расход хлора для снижения цветности на один градус,
по данным Восточной водопроводной станции г. Москвы [211],
уменьшается с увеличением цветности исходной воды и составля¬
ет при цветности более 50 град 0,17—0,20 мг/л (рис. VII.7). Рас¬
ход безводного сернокислого алюминия составляет около 1,5 мг/л
на градус цветности [212].
Так как эффективность хлорирования увеличивается с ростом
дозы хлора, на некоторых водоочистных станциях применяют
обработку воды повышенными дозами хлора —5—10 мг/л [211,
213, 214]. Такой режим (перехлорирование) обеспечивает разру¬
шение особо стойких органо-минеральных (в частности, железо¬
органических) комплексов, однако вода на выходе из очистных со¬
оружений может содержать недопустимо высокие концентрации
остаточного хлора и требовать дехлорирования. В этих случаях
в качестве коагулянтов целесообразно применение солей закисно¬
го железа, если реакция Fe2+ с оставшимися после хлорирования
органическими веществами не дает окрашенных соединений.
VII.3. СОЧЕТАНИЕ С ДРУГИМИ МЕТОДАМИ ВОДОПОДГОТОВКИ
237
Рис. YII.7. Зависимость ко¬
личества хлора, необходимого
для устранения одного граду¬
са цветности, от цветности ис¬
ходной воды Ци (по данным
Восточной водопроводной стан¬
ции г. Москвы)
С lz , мг/уг
В качестве способа рационального совмещения перехлорирова-
ния воды и обработки ее коагулянтом заслуживает внимания пре¬
дложение Руденко [214] о раздельной обработке воды в отстойни¬
ках: добавление коагулянта и хлора (в обычно принимаемой дозе)
в один поток воды, а известкового молока и хлора (в повышен¬
ной дозе) — в другой.
Перехлорирование или предварительное хлорирование воды
обычными дозами стремятся осуществить в таких точках техноло¬
гической схемы, чтобы промежуток времени между вводом хлора
и коагулянта был максимальным: на насосной станции Первого
подъема и даже в водозаборном ковше. При проведении хлориро¬
вания в ковше Восточной водопроводной станции г. Москвы обна¬
ружен важный сопутствующий эффект —20—40%-ное освобожде¬
ние воды от взвешенных примесей [211].
Исследованиями установлено, что в большинстве случаев дей¬
ствие окислителей проявляется в снижении ДП дисперсных при¬
месей. Однако известны случаи, когда наблюдался противополож¬
ный эффект. Например, при хлорировании воды р. Потомак, заг¬
рязненной стоками, ДП частиц взвеси вырос от —20 до —23 мв
[215, 216]. Причину такого явления авторы видят в неоднород¬
ности состава частиц, полагая, что гидрофильные, трудно окисля¬
ющиеся частицы с большой величиной ДП окружены оболочками
из относительно легко окисляющихся соединений. Кульский
объяснил обнаруженное им повышение устойчивости каолиновой
суспензии в присутствии хлора стабилизирующим действием хло¬
ра на соединения алюминия и железа, входящие в состав глинис¬
тых частиц. Отрицательное влияние хлора на удаление коагули¬
рованием синезеленых водорослей Microcystis связывают с разру¬
шением водорослевых колоний [211].
В зависимости от принятых условий обработки воды 1 мг!л
озона снижает цветность на 3—12 град [217—219] и, так же как
и хлор, может или астабилизировать частицы дисперсной фазы,
вызывая их коагуляцию, или приводить к образованию высоко¬
устойчивых систем (например, при доочистке сточных вод).
238
ГЛАВА VII. САНИТАРНАЯ ЭФФЕКТИВНОСТЬ КОАГУЛЯЦИИ
Добавление КМп04 приводит к разрушению органических
комплексов железа и марганца, трудно окисляемых хлором. В хо¬
де реакции происходит некоторое, как правило, полезное для коа¬
гуляции повышение pH. Кроме того, улучшение коагуляции обу¬
словливается образованием малорастворимой гидроокиси марган¬
ца [210, 220].
Следует отметить еще одно важное и интересное обстоятель¬
ство. Известно, что при сильном бактериальном заражении воды
успешному протеканию процесса обеззараживания ее хлором и
другими окислителями способствует перемешивание воды с вы¬
соким скоростным градиентом. Механизм этого явления до сих
пор не раскрыт. Во всяком случае объяснить его с точки зрения
увеличения диффузии молекул окислителя к поверхности микро¬
организмов и через их липоидные оболочки довольно трудно. На
наш взгляд, объяснение нужно искать в образовании периодичес¬
ких коллоидных структур — биоколлоидов, о которых шла речь
в гл. II. Под действием интенсивного перемешивания возможна
перегруппировка отдельных клеток биоколлоидов и, как следст¬
вие, увеличение доступности внутренних клеток для дезинфици¬
рующего действия окислителей. Так или иначе при совместном
использовании окислителей и коагулянтов этот эффект надо учи¬
тывать: интенсивное перемешивание, требующееся для повышения
эффекта обеззараживания, может привести к необратимому раз¬
биванию хлопьев и потому должно быть прекращено до наступле¬
ния стадии хлопьеобразования.
В приведенных выше примерах речь шла о вводе окислителей,
предшествующем коагуляции. Это наиболее распространенный
прием. В некоторых случаях, однако (например, для отделения
основной массы загрязнений и снижения хлоропоглощаемости во¬
ды), целесообразно предварительное коагулирование. И тогда пе¬
ред технологами возник важны ! вопрос об эффективности окисле¬
ния загрязнений, покрытых оболочками продуктов гидролиза ко¬
агулянта. Ответ на этот вопрос требует проведения специальных
исследований.
VII. 3.2. Обработка активным углем
Обработка природных и сточных вод активным углем (АУ) приме¬
няется для удаления природных веществ, обусловливающих за¬
пах, цветность, вкус и окисляемость воды, фенолов, масел, нефте¬
продуктов, пестицидов и синтетических ПАВ. Обработку осущест¬
вляют либо добавлением к воде угольного порошка (от 2—3 до
100—150 мг/л), либо фильтрацией воды через слой гранулирован¬
ного АУ. Первый способ обладает преимуществами в отношении
скорости достижения сорбционного равновесия и может быть ис¬
пользован при обработке воды, не подвергнутой предварительной
VII.3. СОЧЕТАНИЕ С ДРУГИМИ МЕТОДАМИ ВОДОПОДГОТОВКИ
очистке. Его применение связано, однако, с необходимостью от¬
деления отработанного угля в осадок.
До настоящего времени коагулирование с последующим освет¬
лением воды является наиболее распространенным способом отде¬
ления угольного порошка. Продукты гидролиза коагулянта и сор¬
бент дополняют друг друга в технологическом отношении: присут¬
ствие коагулянта позволяет удалить из воды дисперсные примеси,
плохо снимаемые АУ; присутствие сорбента — избавиться от орга¬
нических веществ, затрудняющих коагуляцию. Но в то же время
катионы А13+ и Fe3+, вводимые с коагулянтами, могут конкуриро¬
вать с удаляемыми веществами за адсорбционные участки АУ,
а гидроокисные оболочки — уменьшать доступность для адсорб¬
тива внутренней поверхности угольных пор, экранировать их.
С другой стороны, включение пористых частиц АУ в хлопья
коагулированной взвеси вызывает изменение размера., формы
и объемного веса хлопьев, скорости их осаждения. Степень поме¬
хи зависит от дозы АУ и принятого способа осветления воды. Так,
при дозировании АУ в количествах более 50 мг/л перед фильтро¬
вальными сооружениями работа фильтрующих слоев заметно
ухудшилась, потребовалось добавление флокулянта. При пере¬
ходе от «мокрого» способа дозирования угольного порошка (в ви¬
де водной суспензии) к «сухому» длительность фильтроцикла со¬
кратилась на 13—14% [221]. Осветлители со взвешенным осадком
при дозах АУ около 150 мг/л работали неустойчиво даже при ско¬
рости восходящего потока 0,5 мм/сек [222].
Исследования, выполненные Когановским и Петренко [223],
показали, что во взвешенном слое, образованном продуктами гид¬
ролиза алюминия или железа и частицами АУ, размер агрегатов
частиц зависит от соотношения количеств гидроокисей и угля
(табл. VГ1.3). С увеличением дозы АУ агрегаты становились более
симметричными, но степень расширения взвешенного слоя не из¬
менялась.
Таблица VII.3. Размеры агрегатов (.нк.и), состоящих из продуктов
гидролиза алюминия или железа и частиц АУ [223]
Продукт
Содержание АУ в хлопьях коагулированной взчеси, вес. %
гидролиза
0
60
80
95
100
А1(ОН)3
Fe(OH)3
175
189
63,2
100
80
119
Примечание. Размеры агрегатов частиц определены при скорости потока воды
во взвешенном слое хлопьев, равной 2,04 мм!сек.
240
ГЛАВА VII. САНИТАРНАЯ ЭФФЕКТИВНОСТЬ КОАГУЛЯЦИИ
Рис. VI 1.8. Доза Al8'1", не¬
обходимая для частичного
осветления угольных сус¬
пензий разной концентра¬
ции [224]
1 — 50%-;
2 - 80%-ное осветление
Рис. VI 1.9. Доза А13+, не¬
обходимая для коагуля¬
ции угольных суспензий
разной концентрации
с, л;г/л
¥
10‘
8
6
4
в hV'mrM 70
У
/
j
г
-Г
/
1
>
1
1Z
К\?*лгг/л
Взаимодействие продуктов гидролиза коагулянта с частицами
угля носит специфический характер, определяющийся: высокими
отрицательными значениями ДП частиц АУ (около 40 мв) [18, 215]; ’
сильно развитой поверхностью частиц АУ (400—800 м~/г); резким
увеличением вероятности химического взаимодействия между ка¬
тионами Ме3+ и молекулами адсорбтива вследствие повышенных
их концентраций на поверхности угля; высокой сорбционной ем¬
костью АУ по отношению к катионам водорода, образующимся
в ходе гидролиза коагулянта.
ат,мг/м
100
60\
40
3^»
1
25 SO
7S А У ,мг/л
Рис. VII.10. Зависимость удельной сорбции (Г0) метиленовоготголубого на
активном угле от весового соотношепия (а) доз коагулянта и угля
Рис. VII.11. Влияние добавок АУ на величину оптимальной дозы серно¬
кислого алюминия (аопт) при обработке воды с разным содержанием взвешен¬
ных примесей (мг/л):
1 — 2; 2 — 25; 3 — 100
VII.3. СОЧЕТАНИЕ С ДРУГИМИ МЕТОДАМИ ВОДОПОДГОТОВКИ
241
В опытах Леттермана и др. [224] по коагуляции сульфатом
алюминия угольных суспензий определены оптимальные дозы коа¬
гулянта, необходимые для достижения заданного (50 или 80%)
эффекта осветления и минимальной мутности отстоенной воды.
Как видно из рис. VII.8 и VII.9, первая доза при концентрации
угольной суспензии 100—200 мг/л имеет минимум, вторая — мо¬
нотонно возрастает с увеличением концентрации суспензии. Про¬
является тот же характер зависимости доз коагулянта от содержа¬
ния взвеси, что и для глинистых суспензий (см. рис. VI.3). Мини¬
мальная остаточная мутность воды соответствовала определенным
значениям щелочности. Увеличение щелочности от 0,6 до 4,0 мг-
экв/л привело к^смещению_оптималыюго_ значения pH от 7,4 до
6,8. ц
Ввод АУ после добавления коагулянта в описываемых исследо¬
ваниях дал отрицательные результаты: остаточная мутность воды
возросла. При обработке активным углем (5 мг/л) воды с неболь¬
шим содержанием взвеси (10 мг/л) дозу сернокислого алю¬
миния удалось снизить, а формирование хлопьев протекало
быстрее.
Существенный пробел в работе Леттермана и сотрудников —
отсутствие попытки определить экранирующее влияние продуктов
гидролиза коагулянта на сорбционную способность АУ — вос¬
полнен исследованиями, проведенными в ЦНИИ МПС [225—227].
Установлено, что в присутствии продуктов гидролиза A12(S04)3
величина сорбции макромолекул метиленового голубого на уг¬
лях марок КАД и ОУ заметно уменьшается. Экранирующее дей¬
ствие продуктов гидролиза уменьшалось с увеличением интенсив¬
ности перемешивания воды и при значениях скоростного градиен¬
та около 800 секГ1 становилось мало заметным: по-видимому, под
действием касательных усилий среза гидроокисные оболочки раз¬
рушались и не препятствовали проникновению макромолекул к по¬
верхности угля. Высокие значения скоростного градиента, тре¬
бующиеся для устранения экранирующего действия продуктов
гидролиза, свидетельствуют о прочной связи продуктов гидролиза
с поверхностью угольных частиц и подтверждают предположение
(см. гл. VI) о вероятности взаимодействия разнородных частиц
в первичном энергетическом минимуме.
С увеличением степени покрытия угольных частиц гидроокис-
ными соединениями сорбция метиленового голубого прогрессивно
уменьшалась, но при соотношении доз коагулянта и АУ, равном
2 : 1, достигала постоянного значения (рис. VII.10). На основе
полученных результатов рекомендован следующий режим обра¬
ботки воды:
1) добавление угольного порошка и перемешивание его с водой
в течение 15—20 мин при величине скоростного градиента
800 секГ1\
242
ГЛАВА VII. САНИТАРНАЯ ЭФФЕКТИВНОСТЬ КОАГУЛЯЦИИ
2) добавление раствора коагулянта за 1 мин до окончания бы¬
строго перемешивания;
3) перемешивание в течение 5 мин при величине скоростного
градиента 100 сек"1.
На рис. VII. 11 представлены результаты опытов по выявлению
оптимальных доз коагулянта (а), требующихся для осветления
в течение 40 мин воды, обработанной углем марки КАД по реко¬
мендованному режиму. При очистке маломутной воды (содержа¬
ние взвешенных веществ около 2 мг/л) добавление АУ в количест¬
вах 10,25 и 50 мг/л привело к экономии коагулянта, что может
быть объяснено стимулирующим действием угольного порошка
как замутнителя. Расход сернокислого алюминия уменьшился
примерно на 10%.
Интенсификация коагуляционного процесса оказалась особен¬
но заметной при низких значениях щелочности обрабатываемой
воды —0,3—0,4 мг-экв/л. Недостаток щелочного резерва компен¬
сировался, по-видимому, сорбцией водородных катионов на части¬
цах АУ. В остальных исследованных вариантах мутности исход¬
ной воды (25 и 100 мг/л) добавки угля вызвали увеличение расхода
коагулянта. При обычно применяемых в практике дозах АУ —
не более 25 мг/л — следует ожидать увеличения оптимальных доз
A12(S04)3 на 5 —10%.
При фильтрации воды, обработанной коагулянтом, через слой
гранулированного АУ коагулированная взвесь задерживается
в первых по ходу движения воды слоях угля и ее экранирующее
влияние на сорбцию истинно растворенных веществ невелико
[228].
VII. 3.3. Флотация
Если в обрабатываемой жидкости в заметных концентрациях при¬
сутствуют эмульгированные и поверхностно-активные примеси —
нефтепродукты, жиры, масла, синтетические детергенты — ра¬
циональным методом выделения загрязнений из воды может стать
их флотация пузырьками воздуха или других газов. Применяют
флотацию главным образом при очистке производственных сточ¬
ных вод. Возможности использования этого метода в питьевом во¬
доснабжении пока изучаются в лабораторных условиях.
При флотации подъемная сила газовых пузырьков, оценка ко¬
торой выполнена в гл. VI, призвана (в отличие от седиментацион-
ного метода разделения суспензий) играть положительную роль,
а сам процесс объединения частиц загрязнений можно рассматри¬
вать как частный случай гетерокоагуляции, где величина и знак
заряда частиц взвешенных примесей, свойства адсорбционных
слоев на поверхности раздела фаз имеют очень важное значение.
В зависимости от давления, под которым находится жидкость
в момент добавления газа, флотация может быть безнапорной или
VII. 3. СОЧЕТАНИЕ С ДРУГИМИ МЕТОДАМИ ВОДОПОДГОТОВКИ
243
напорной. Безнапорную флотацию осуществляют при атмосферном
давлении за счет диспергирования газа в объеме жидкости с помо¬
щью специальных распылителей или пористых перегородок. В на¬
порной флотации (микрофлотации) жидкость сначала насыщают
газом под давлением в несколько атмосфер, а затем перепускают
в открытую камеру, где газ переходит из растворенного состоя¬
ния в дисперсное, выделяясь по всему объему камеры в виде мель¬
чайших (100—200 мкм) пузырьков.
Эффективность напорной флотации вследствие малых размеров
и чрезвычайно большой поверхности пузырьков, как правило,
выше, чем безнапорной. Насыщать воздухом можно всю обраба¬
тываемую воду (прямая флотация) или только ее часть с последую¬
щим смешением с остальной частью воды (рециркуляционная фло¬
тация). Этот второй способ при добавлении коагулянтов наиболее
предпочтителен, так как предотвращает разбивание хлопьев коа¬
гулированной взвеси [229].
В электрофлотации в качестве флотационных используются
газы, выделяющиеся на поверхности электродов в результате
электрохимического процесса. О высокой эффективности этого
метода флотации свидетельствуют многочисленные примеры его
использования, в том числе — с добавкой минеральных коагулян¬
тов или катионов А13+ и Fe3+, перешедших в обрабатываемую воду
при анодном растворении металлов. Некоторое интенсифицирую¬
щее действие оказывает сопутствующее электрофлотации наложе¬
ние электрического поля [230].
Роль коагулянтов, как и других «собирателей» флотации, со¬
стоит, во-первых, в подготовке поверхности твердых частиц к за¬
креплению на газовых пузырьках, во-вторых — в укрупнении
частиц. Успешнее всего флотируются частицы размером 20 —
100 мкм, что объясняется наибольшей вероятностью их закрепле¬
ния на поверхности пузырьков [231].
Опыт эксплуатации флотаторов свидетельствует о высокой эф¬
фективности коагулирования при очистке вод от нефти и масел.
Степень очистки достигает 90—99%, причем лучшие результаты
соответствуют высоким значениям pH среды (9—10), при которых
эмульгированные примеси гидрофилизируются [229].
Содержание мылов в сточных водах прачечных можно снизить
методом флотации при добавлении 600 мг/л сернокислого алюминия
на 77%, а при добавлении 500 мг/л хлорного железа — на 97%
[232, стр. 96]. Использование серной кислоты и сульфата алюми¬
ния позволило удалить 40% синтетических ПАВ [84].
Описаны случаи успешного применения коагулянтов при фло¬
тационной очистке сточных вод целлюлозно-бумажной промышлен¬
ности [233], заводов по переработке рыбы, боен и свиноферм [234,
235], производства лаков и смол [236, 237]. Методами флотации
удается в значительной степени удалить из воды бактерии, гумино-
244
ГЛАВА VII. САНИТАРНАЯ ЭФФЕКТИВНОСТЬ КОАГУЛЯЦИИ
вые кислоты и планктон [237, 238]. Добавление к цветной воде
в дополнение к основному «собирателю»— катионоактивному аце-
татамину канифоли — сернокислого алюминия (дозой 50—
100 мг/л) позволило снизить расход «собирателя» и увеличить сте¬
пень удаления гуминовых кислот методом флотации до 95%
[239].
[Степень очистки зависит от соотношения между величиной
окисляемости воды и количеством примесей, подлежащих удале¬
нию [240]. Это соотношение определяет, по-видимому, состояние
поверхности дисперсных частиц, их гидрофилыюсть. Оптималь¬
ные значения pH при удалении разных примесей, по данным Кас¬
селя и др. [237], следующие: бактерий —3,5, кварцевых частиц —
4,5—6,5, гуминовой кислоты —6,9—9,5, полистирольных лаков —
8—11. Сточные воды, загрязненные латексом, очищаются при до¬
бавлении сернокислого алюминия наиболее успешно при pH 3—7.
При высоких оптимальных значениях pH среды используют
соли железа (или анодно растворенное железо), а в остальных слу¬
чаях предпочтение отдают алюминиевым коагулянтам, образую¬
щим легкофлотируемые продукты. Результаты экспериментов [2411
по очистке бытовых сточных вод напорной флотацией, проведен¬
ные с добавлением серпокислого алюминия и анионного флокулян-
та, позволили рекомендовать этот метод для обработки стоков в те
сезоны года, когда сооружения биологической очистки перегруже¬
ны и не справляются с нагрузкой.
[Для осуществления флотации с одновременной обработкой во¬
ды коагулянтами предложены разнообразные технологические схе¬
мы [228, 232, 242—246]. Некоторые из них предусматривают реге¬
нерацию коагулянта [245, 246].
VII. 4. ЭЛЕКТРОКОАГУЛЯЦИЯ
В основе электрокоагуляции лежит процесс анодного растворе¬
ния металлов под действием постоянного электрического тока.
Перешедшие в раствор катионы А13+или Fe2+ гидролизуются и слу¬
жат активными коагулянтами для дисперсных примесей. Сопут¬
ствующее анодному растворению наложение на коагулирующую
систему электрического поля и связанный с ним электрофорез час¬
тиц играют подчиненную роль [247].
Количество железа, растворяющегося в единицу времени, про¬
порционально силе рабочего тока и достигает 95—98% от теорети¬
ческого количества, определяемого законами Фарадея. Выход
алюминия может превышать расчетную величину, что одни иссле¬
дователи объясняют дополнительным растворением катода (за
счет высоких концентраций щелочи в прикатодном слое), другие —
образованием промежуточных форм катионов алюминия с валент¬
ностью порядка единицы.
VII.4. ЭЛЕКТРОКОАГУЛЯЦИЯ
245
В отличие от обычного коагулирования солями железа и алю¬
миния при электрокоагуляции вода не обогащается анионами SO]-,
С1“ и другими, что сказывается благоприятно на обработке вод, со¬
держащих растворенные загрязнения. Однако в ходе электролиза
воды или водных растворов солей происходит выделение значи¬
тельных количеств газов (водорода на катоде и кислорода на ано¬
де), пузырьки которых вызывают флотацию хлопьев. Вероятность
флотации, резко замедляющей осая^дение хлопьев в отстойниках
или осветлителях, увеличивается с уменьшением содержания взве¬
си в исходной воде.
При электрокоагуляции, в отличие от обработки воды кислыми
солями алюминия и железа, происходит некоторое повышение pH
среды, благодаря которому в отдельных случаях отпадает необхо¬
димость в добавке подщелачивающих реагентов.
Электрохимическое коагулирование практикуется не только
для выделения из воды твердых дисперсных примесей, но и эмуль¬
гированных веществ, а также растворенных газов (кислород, серо¬
водород, хлор), фенолов, радиоактивных и поверхностно-активных
веществ 1. Кроме того, как отмечено в гл. III, анодным растворе¬
нием металлов в растворах поваренной соли, соляной и серной кис¬
лот получают хлориды и сульфаты алюминия и железа, используе¬
мые в качестве коагулирующих растворов. В связи с этим исследо¬
вания по анодному растворению металлов, начатые еще в конце
прошлого века, расширяются.
Компактность и простота эксплуатации электрокоагуляторов
делают метод электрохимической коагуляции особенно пригодным
при обработке малых объемов воды, например на судах морского
и речного флота [248].
Основной элемент электрокоагулятора — набор железных или
алюминиевых пластин, в зазорах между которыми протекает обра¬
батываемая вода или раствор электролита. Для борьбы с пасси¬
вацией металлов (с целью снижения затрат электроэнергии), а
а также для равномерного износа электродных пластин периоди¬
чески производят смену их полярности.
Удельные затраты электроэнергии могут быть снижены за
счет уменьшения расстояния между электродами и плотности тока,
увеличения электропроводности раствора и скорости его движения
в межэлектродных зазорах. Однако при значительном содержании
в обрабатываемой воде взвешенных примесей (особенно в присут¬
ствии ПАВ, образующих устойчивые иены) даже при достаточно
высоких скоростях протекания воды в межэлектродных зазорах
и ширине последних 10—15 мм может произойти закупорка. По¬
этому такую воду необходимо подвергать предварительной очистке.
1 Примеры использования электрокоагулирования для решения разных
технологических задач приведены в гл. VIII и X.
246
ГЛАВА VII. САНИТАРНАЯ ЭФФЕКТИВНОСТЬ КОАГУЛЯЦИИ
Рис. VII.12. Распределение
электрического тока в ваннах
с биполярными электродами
1 — корпус ванны;
2 — монополярные электроды;
3 — биполярные электроды
Подключение пластин электродного пакета к источнику тока
может быть параллельным или последовательным. В первом слу¬
чае все электроды действуют как монополярные, во втором — про¬
межуточные пластины работают биполярно. Последовательное под¬
ключение имеет ряд важных преимуществ перед параллельным:
оно позволяет работать при меньших токах и использовать малога¬
баритные выпрямители. Однако в ваннах с биполярно подключен¬
ными электродами (рис. VII.12) возникают паразитные токи (токи
утечки), не участвующие в полезной работе. Соотношение между
рабочим Iр и паразитным /п токами может быть подсчитано из ба¬
ланса напряжения:
и (tfo + гр/р) = U0 + nr„In, г (VII.5)
где U0 — потенциал разложения электролита, т. е. наименьшее
значение э.д.с., необходимое для начала электролиза; гр и гп —
сопротивление слоев электролита на участках протекания соответ¬
ственно рабочего и паразитного токов; п — число элементарных
электролизных ячеек, образуемых электродами.
Помимо электрокоагуляторов с пластинчатыми электродами,
делаются попытки применить для целей очистки воды коагулято¬
ры со стружечными электродами — отходом металлообработки.
Преимущество металлической стружки состоит в большей площа¬
ди поверхности. Но нарушение электроконтактов в местах сопри¬
косновения стружек и закупоривание пор продуктами гидролиза
приводят к быстрому нарастанию сопротивления. В электрокоагу¬
ляторе со стружечным анодом, помимо паразитного тока и рабоче¬
го тока, вызывающего многократную поляризацию внутренних
слоев стружки, возникает транзитный ток, который проходит по
металлу стружки, вызывая лишь однократную его поляризацию.
Исследования, проведенные автором [249], показали, что воз¬
растание напряжения на клеммах стружечного электролизера свя¬
зано главным образом с нарушением контакта между стружечным
пакетом и токоподводящей анодной пластиной, а также газонапол-
нением пор. Установлено, что использование стружки как бипо¬
лярно действующего электрода, не соприкасающегося с токопод¬
VII.4. ЭЛЕКТРОКОАГУЛЯЦИЯ
247
водящими пластинами, а также разделение стружки на отдельные
слои перфорированными прокладками из диэлектрика (с протоком
воды через них) позволяют в значительной степени стабилизиро¬
вать напряжение и добиться некоторого снижения энергетических
затрат [250].
Один грамм растворенного алюминия эквивалентен 6,35 г без¬
водного сернокислого алюминия, а один грамм железа —1,93 г
безводного хлорного железа; однако фактическая потребность
в электролитически приготовленных коагулянтах и коагулянтах-
солях можем сильно отличаться от рассчитанной, если исходить
из условий эквивалентности. Кроме того, необходимо иметь в виду,
что железо растворяется в двухвалентной форме, и скорость осаж¬
дения продуктов гидролиза железа сильно зависит от степени его
окисления. Поскольку при pH воды ниже 7,0 катионы Fe2+ окис¬
ляются медленно, приходится прибегать к подщелачиванию воды
или обработке ее хлором 2.
Потребность в больших количествах вспомогательных реаген¬
тов может сделать процесс электрокоагуляции с растворением же¬
леза неэкономичным [247]. В этом случае выгоднее использовать
флотацию или электрофлотацию. На одной из водоочистных
станций Англии из-за больших расходов электроэнергии приш¬
лось отказаться от способа электрокоагуляции с железными элек¬
тродами в пользу простого хлорирования металлолома [251].
Некоторые новые возможности снижения энергетических затрат
при анодном растворении железа открывает добавление к обрабаты¬
ваемой годе или сточной жидкости поваренной соли, которая вы¬
полняет функцию депассиватора и увеличивает электропроводность
еоды. Отмечено, что с ростом концентрации NaCl несколько воз¬
растает и выход металла по току [252].
Проведенные в ИОНХ АН УССР технологические испытания
смеси растворов FeCl3 и FeCl2, полученных при электролизе смеси
растворов FeCl2 и поваренной соли [42, 252] показали, что ухудше¬
ние осаждения продуктов гидролиза становится существенным,
если соотношение Fe2+ : Fe3+ превышает 1 : 1 [42]. Таким образом,
содержание довольно больших количеств Fe2+ можно считать все
же допустимым.
Скорость осаждения продуктов гидролиза железа, полученного
электролитически, сильно зависит от мутности исходной воды.
По наблюдениям автора в незамутненной водопроводной воде до¬
статочно интенсивное хлопьеобразование имеет место лишь при
концентрации общего железа не менее 30 мг/л. Оптимальные дозы
растворенного железа и алюминия колеблются в широких преде¬
лах: от 10—25 мг/л А13+ при обезжелезивании воды [253] до
400 мг/л Fe3+ при обработке жиросодержащих стоков [254]. При
2 См. раздел VII. 1.
248
глава vii. санитарная эффективность коагуляции
очистке природных вод обычно достаточно 30—60 мг/л железа
[42].
Технологическая эффективность и преимущество электрокоа¬
гуляции перед коагуляцией солями не подлежит сомнению при
обработке природных вод, содержащих большие количества орга¬
нических веществ. Они объясняются высокой активностью катио¬
нов А13+ и Fe3+, отсутствием дополнительных количеств анионов
и, как следствие,— большей вероятностью образования металло¬
органических комплексов.
Электрокоагуляция лучше удаляет фульвокислоты [255, 256],
дает возможность достигнуть более глубокого обесцвечивания во¬
ды, снижения ее окисляемости [257—259]. Методом электрокоагу-
ляции хорошо удаляются шестивалентный хром [260, 261], кремне-
кислота [262] и другие растворенные вещества, перечисленные вы¬
ше. Эффективное удаление хрома при использовании железных
анодов объясняется протеканием прямой химической реакции вос¬
становления шестивалентного хрома до трехвалентного и после¬
дующим образованием гидрата:
6Fe2+ + Na2Cr207 + 19Н20 6Fe (0№3 + 2Сг (ОН)з + 2NaOH + 6Н2.
Согласно уравнению реакции, для удаления 1 вес. ч. Cf6+ тре¬
буются 3 вес. ч. Fe2+. Отмечаемое в опытах уменьшение потреб¬
ности Fe2+ связано с протеканием: сопутствующих реакций.
Важные сведения о технологических свойствах коагулирую¬
щих растворов, полученных путем анодного растворения алюми¬
ния и железа в растворах серной кислоты и поваренной соли, при¬
ведены Рапопортом [257].
Л итература
1. В. А. Клячко, И. Э. Апелъцин. Подготовка воды для промышленного
и городского водоснабжения. М., Стройиздат, 1962.
2. И. И. Жуков. Избранные труды. М., Изд-во АН СССР, 1952, стр. 237.
3. О. И. Мартынова. Коагуляция при водоподготовке. М., Госэнерго-
издат, 1951.
4. М. А. Шевченко, Р. С. Касъянчук, Укр. хим. ж., 30, 1103 (1964).
5. М. И. Гоштейн, А. И. Руденко. Труды научно-исслед. ин-тов Свердл.
облздравотдела, сб. № 6. Свердл. обл. изд-во, 1936, стр. 142.
6. О. Н. Шемякина. Труды ВНИИ ВОДГЕО, вып. 29. М., 1971, стр. И.
7. д. Б. Гитис, Н. Ш. Сафиуллин, Е. Ф. Дубрава и др. Водоснабжение и
санитарная техника, № 8, 9 (1971).
8. Г. Бэббит, Дж. Доланд. Водоснабжение. М., Стройиздат, 1958.
9. W. A. Boiler. Water and Wastes Engng, 11, N 12, 21 (1974).
10. T. Stones. J. Proc. Inst. Sewage Purific., N 2, 120 (1955).
И. М. E. Rew. J. Amer. Water Works Assoc., 52, 852 (1960).
12. C. O. Bown, D. J. Rose. Canad. Pulp and Paper Ind., 11, N 1, 41 (1958).
13. E. J. Bonner. Works Engng and Factory Serv., N 10, 24 (1970).
14. П. И. Васильева. ЖПХ, 3, 207 (1930).
15. Л. А. Кулъский, А. М. Когановский, И. Т. Гороновский и др. Физико¬
VII. ЛИТЕРАТУРА
249
химические основы очистки воды коагуляцией. Киев, Изд-во АН УССР,
1950.
16. М. Lane. Water and Sewage Works, 106, 339 (1959).
17. M.Adhikari, S. K. Gupta, B.Banerjce. J. Indian Chem. Soc., 51, 891
(1974).
18. E. L. Bean, S. J. Campbell, F. R. Ansspach. Water and Wastes Engng,
2, N 11, 47 (1965).
19. С. А. Шуберт, В.П.Криштул, В. JI. Драгинский и др. Водоснабже¬
ние и канализация, вып. 2 (22). М., изд. ЦБТИ МЖКХ РСФСР, 1972,
стр. 1.
20. Ю. В. Багоцкий, Т. А. Дмитриева, 3. М. Елдашева и др. Водоснабже¬
ние и канализация, вып. 1 (15). М., изд. ЦБТИ МЖКХ РСФСР, 1965,
стр. 15.
21. Ю. Б. Багоцкий, Т. А. Дмитриева и др. Городское хозяйство Москвы,
№ 2, 26 (1964).
22. В. А. Клячко. Водоснабжение и санитарная техника, № 7, 13 (1962).
23. 10. А. Бардин, Е. С. Шалашова. Водоснабжение и санитарная техника,
№ 6, 27 (1965).
24. JI. Н. Паскуцкая. Повышение технической и экономической эффектив¬
ности работы водопроводно-канализационных сооружений (тезисы
докладов и сообщений на Всесоюзной научно-технической конференции
в Новосибирске 2—5 июля 1968 г.). М., 1968, стр. 64.
25. С. М. Джафаров. Труды Бакинского филиала ВНИИ ВОДГЕО, вып. 5,
26 (1970).
26. Б. М. Щипачев. Водоснабжение и санитарная техника, № 10, 13 (1962).
27. Я. Б. Лазаревский. Труды Азерб. НИИ водных проблем, вып. 2. Баку,
1971, стр. 221.
28. Look (Japan), 16, N 190, И (1972).
29. С. М. Джафаров. За технический прогресс, № 7, 35 (1971).
30. Э. А. Левицкий, В. Н. Максимов. Авт. свид. СССР 146302 (1961); Бюлл.
изобр., № 8 (1962).
31. Сб. «Коагулянты для очистки питьевой воды». Под ред. В. Т. Турчино-
вича. М., Изд-во МКХ РСФСР, 1948, стр. 15.
32. М. R. Buydens. Techn. sanit. et municip., 40, 139 (1953).
33. В.А.Бедрин. Изв. вузов. Строительство и архитектура, № 8, 113
(1959).
34. А. P. Black, J. Е. Singley, G. R. Whittle et al. J. Amer. Water Work,;
Assoc., 55, 1347 (1963).
35. E. С. Абазаев. Водоснабжение и санитарная техника, № 5, 8 (1959).
36. I. Fazekas, D. Georgescu et al. Studii aliment, apa, 2, 133 (1965).
37. Л. А. Кулъский. Основы технологии кондиционирования воды. Киев,
Изд-во АН УССР, 1963.
38. В. Е. Тимонов. Водоснабжение и водостоки, вып. II. СПб., 1900.
39. А. Васильев. Жилищно-коммунальное хозяйство, № 6, 25 (1957).
40. М. Ф. Булычев. Санитарная техника. Доклады на XXIII научной кон¬
ференции Ленинградских инженерно-строительных институтов (ян¬
варь, 1965). Л., 1965, стр. 54.
41. Е. С. Абазаев. Водоснабжение и санитарная техника, № 6, 4, (1956).
42. Л. А. Кулъский, М. А. Шевченко. Укр. хим. ж., 17, 239 (1951).
43. D. G. Miller, J. Т. West. Water and Water Engng, 70, 342 (1966).
44. E. E. Драхлин. Водоснабжение и санитарная техника, № 1, 5 (1972).
45. М. М. Блувштейн, Е. Д. Бабенков. Опыт эксплуатации двухэтажного
горизонтального отстойника. М., Изд-во МКХ РСФСР, 1962.
46. Г. П. Рыхликов. Научные труды Омск, сельскохоз. ин-та, вып. 74,
106 (1970).
47. Н. В. Веселова. Гигиена и санитария, № 9, 44 (1950).
48. С. G. Hyde. J. Amer. Water Works Assoc., 27, 631 (1935).
250
ГЛАВА VII. САНИТАРНАЯ ЭФФЕКТИВНОСТЬ КОАГУЛЯЦИИ
49. Г. Г. Ломакина, Л. Н. Семенова, Ж. И. Антончук. Сб. «Водоснабже¬
ние, канализация, гидротехнические сооружения», вып. 15. Киев,
«Буд1вельник», 1972, стр. 8.
50. Г. Г. Ломакина, С. П. Сукач, А. И. Зелъцерман и др. Там же, стр. 4.
51. S. Kolaczkowski, P. Surawski. Fortschr. Wasserchem., N 1, 196 (1964).
52. W. Rummel. Wasserwirtschaft — Wassertechnik, 10, 121 (1960).
53. И. Camo. Яп. пат. 49-26657 (1974).
54. A. Hult. Effluent and Water Treat. J., 13, 209 (1973).
55. T. J. Conocchioli, E. J. Hamilton, N. Sutin. J. Amer. Chem. Soc., 87,
926 (1965).
56. В. В. Сысоева. ЖПХ, 44, 2558 (1971).
57. В. В. Сысоева, А. Л. Ротинян. Там же, стр. 254.
58. В. В. Сысоева, Г. Н. Доброхотов, И. А. Строева и др. ЖПХ, 41, 1946
(1966).
59. М. К. Игнатов, Г. С. Лейбович. Водоснабжение и санитарная техника,
№ 3, 57 (1941).
60. Е. Ф. Кургаев. Коллоидн. ж., 19, 72 (1957).
61. Л. А. Кулъский. Рационализация технологии очистки питьевых вод.
Киев, Изд-во АН УССР, 1951.
62. Ф. Т. Добрынин. Водоснабжение и санитарная техника, № 5, 50 (1941).
63. В. П. Чалый. Гидроокиси металлов. Киев, «Наукова думка», 1972.
64. С. G. Thompson, J. Е. Singley, А. P. Black. J. Amer. Water Works
Assoc., 64, 11 (1972).
65. Environ. Sci. and Technol., 7, 304 (1973).
66. Л. И. Шмидт. ЖПХ, 46, 1763 (1973).
67. J. С. Vaughn, R. F. Falkenthal, R. W. Schmidt. J. Amer. Water Works
Assoc., 48, 30 (1956).
68. S. S. Russell, М. C. Jesse, W. Graham. Ibid., p. 55.
69. H. А. Лукиных. Очистка сточных вод, содержащих синтетические по¬
верхностно-активные вещества. М., Стройиздат, 1972, стр. 76.
70. J. М. Cohen, G. A. Rourke et al. J. Amer. WaterWorkS Assoc., 51, 1255
(1959).
71. H. S. Lawrence, D. G. Charles. J. Amer. Water Works Assoc., 48, 45
(1956).
72. L. H. Sanjord, C. D. Gates. Ibid., p. 40.
73. N. R. Sedlander, C. D. Gates. Ind. Engng Chem. Proc. Design and De¬
velop., 4, N 1, 55 (1965).
74. Франц. пат. 1424872 (1966).
75. J. Chudoba, P. Fitter. Sb. Vysoke skoly chem.-technol. Praze. Odd. Fak.
technol. paliv a vody, 9, 123 (1966).
76. B. L. Rosenthal, J. E. O'Brien et al. Water and Water Engng, 67, 352
(1963).
77. Ю. Ю. Лурье, П. С. Антипова. Труды ВНИИ ВОДГЕО, вып. 14. М.,
1966, стр. 43.
78. Ю. Ю. Лурье, П. С. Антипова. Научные труды АКХ им. К. Д. Пам¬
филова, вып. 63. Городская канализация, № 5. М., ОНТИ, 1970,
стр. 133.
79. 10. Ю. Лурье, П. С. Антипова. Водоснабжение и санитарная техника,
№ И, 18 (1966).
80. Ю. Ю. Лурье, П. С. Антипова. Текстильная промышленность, № 2,
13 (1968).
81. 10. 10. Лурье, П. С. Антипова. ЖВХО им. Д. И. Менделеева, 12, 671
(1967).
82. 11. А. Лукиных, Э. С. Разумовский и др. Научные труды АКХ им.
К. Д. Памфилова, вып. 63. Городская канализация, № 5. М., ОНТИ,
1970, стр. 77.
83. А. II. Rice, W. R. Conley. Пат. США 3252899 (1965).
VII. ЛИТЕРАТУРА
251
84. А. И. Цветкова, Г. П. Щетинина. Научные труды АКХ им.
К. Д. Памфилова, вып. 63. Городская канализация, № 5. М., ОНТИ,
1970, стр. 121.
85. Е. Д. Бабенков, М. В. Грехова. Водоснабжение и санитарная техника,
№ 6, 21 (1968).
86. В. В. Пушкарев, Д. И. Трофимов. Физико-химические особенности очи¬
стки сточных вод от ПАВ. М., «Химия», 1975, стр. 61.
87. V. Kresta, М. Koubik. Chem. prumysl., 14, 287 (1964).
88. D. L. Morall. Англ. пат. 987144 (1963).
89. R. S. Smith, J. M. Cohen, G. Walton. J. Amer. Water Works Assoc., 48,
60 (1956).
90. Б. H. Хрусталев, В. В. Пушкарев, II. В. Драницына. Коллоидн. ж.,
29, 283 (1967).
91. W. Husmann. Osterr. Wasserwirtschaft, 13, 4 (1961).
92. F. М. Кabaine, S. Rohrsetzer, E. Wolfram. Magyar kem. folyoirat., 76, N 4,
166 (1970).
93. V. V. Volk, M. L. Jackson. J. Water Pollut. Contr. Fed., 40, 205 (1968).
94. А. М. Когановский. Коллоидн. ж., 24, 34 (1962).
95. S. A. Hannah, J. M. Cohen, G. G. Robeck. J. Amer. Water Works Assoc.
59, 843 (1967).
96. J. J. Morgan, R. S. Engelbrecht. J. Amer. Water Works Assoc., 52, 1303
(1960).
97. M. N. Rao. J. Inst. Engrs (India). Public Health Engng Div., 48, 115
(1968).
98. C. N. Sawyer. J. Amer. Water Works Assoc., 57, 143 (1965).
99. H. Marson. Effluent and Water Treat. J., 11, 441 (1971).
100. R. F. Packham. J. Colloid Sci., 20, 81 (1965).
101. M. Maulaz. Gas-Wasser-Abwasser, 52, 81 (1972).
102. W. A. Eberhardt, J.B. Nesbitt. J. Water Pollut. Contr. Fed., 40, 1239
(1968).
103. A. Henriksen. Schweiz. Z. Hydrol., 24, N 2, 253 (1962).
104. A. Aregger. Gas-Wasser-Abwasser, 52, 78, (1972).
105. E.A. Thomas, H.Rai. Gas-Wasser-Abwasser, 50, 179 (1970).
106. E. L. Johnson, J. H. Beeghly et al. Public Works, 100, N 11, 66 (1969).
107. L. Ulmgren. J. Water Pollut. Contr. Fed., 47, 696 (1975).
108. F. M. Middleton. Water Bes., 6, 475 (1972).
109. J. A. Meginess, R. D. Ilarriger. Water and Wastes Engng, 10, N 3, 35
(1973).
110. G. E. Galonian, D. В. Aulenbach. J. Water Pollut. Contr. Fed., 45, 8
(1973).
111. М. C. Mulbarger, D. G. Shifflett. Chem. Engng Progr. Symp. Ser., 67,
107 (1971).
112. A. Shindala. Water and Sewage Works, 119, 66 (1972).
113. A. Shindala. Ibid., p. 50.
114. A. B. Hais, J. B. Stamberg, D. F. Bishop. Chem. Engng Progr. Symp.
Ser., 68, 35 (1972).
115. D. Cole, P. Tveite. Public Works, 103, N 10, 86 (1972).
116. B. Ericsson. Water Res., 7, 227 (1973).
117. A. Henriksen. Schweiz. Z. Hydrol., 25, N 2, 380 (1965).
118. J. Popel. Stuttgart. Ber. Siedlungswasserwirtsch., N 16, 199 (1966).
119. J. Popel. Vodni hospod., 18, 291 (1968).
120. /. Block. Пат. США 3583909 (1971).
121. W. С. Steigerwald. Пат. США 3607742 (1971).
122. A. S. Patil, J. W. Icraus, J. Block. Пат. США 3677939 (1972).
123. R. Rober, K. P. Dougherty. Chem. Engng Progr. Symp. Ser., 69, 241
(1973).
124. E. Dobolyi. Water Res., 7, 329 (1973).
252
ГЛАВА VII. САНИТАРНАЯ ЭФФЕКТИВНОСТЬ КОАГУЛЯЦИИ
125. L. A. Campbell, A. J. Horton. Water and Polut. Contr. (Canada), 110,
N 3, 28 (1972).
126. E. Dobolyi. Hidrol. kozl., N 3, 117 (1973).
127. Ch. P. Roddy. Water and Wastes Engng, 9, N 12, 34 (1972).
128. E. F. Barth, М. B. Ettinger. Пат США 3480144 (1968).
129. G. G. Robeck, K. A. Dostal et al. J. Amer. Water Works Assoc., 57, 181
(1965).
130. E.A. Sigworth. Ibid., p. 8.
131. М. А. Шевченко, П. В. Марченко, П. Н. Таран и др. Водоснабжение
и санитарная техника, № 10, 31 (1974); Очистка питьевых и сточных вод
от ядохимикатов. Киев, «Бу/ивельник», 1975.
132. R. Koch, К. Strobel. Z. ges. Hyd., 20, 442 (1974).
133. JI. А. Кулъский, A. Г. Шабалина. Сб. «Химическая промышленность
Украины», вып. 1 (31). Киев, 1967, стр. 30.
134. К. А. Большаков и др. Труды Второй Международной конференции по
мирному использованию атомной энергии. Женева, 1958. Доклады
советских ученых, т. 4. М., Атомиздат, 1959, стр. 211.
135. L. Media. Ingeneria sanit., 7, N 6, 176 (1959).
136. Н. Bernhardt, Н. Hartman et al. Vom Wasser 1963, Bd. 30. Weinheim/
Bergstr., 1964, S. 104.
137. H. Marchart. Osterr. Chem. Ztg, 65, N 12, 373 (1964).
138. Г. В. Якимов. Очистка воды и сточной жидкости от радиоактивных изо¬
топов. М., Изд-во МКХ РСФСР, 1961, стр. 47.
139. О. J. Stevenson. Appl. microbiol., 13, N 10, 383 (1960).
140. P. S. Bull, J. V. Evans, R. J. Knight, J. Appl. Chem. and Biotechnol.,
25,801 (1975).
141. И. А. Соболев и др. Изотопы в СССР, № 13, 41 (1969).
142. Т. Komatsu. J. Water Pollut. Contr. Fed., 39, 123 (1967).
143. А. С. Смирнов. ЖВХО им. Д. И. Менделеева, 5, 651 (I960).
144. Nucl. Engng., 2, N 19, 413 (1957).
145. R. A. Lauderdal. Ind. and Engng Chem., 43, 1538 (1951).
146. H. Bernhardt, II. Hartmann et al. Vom Wasser 1964, Bd. 31. Weinheim/
/Bergstr., 1965, S. 85.
147. А. С. Коган. Вопросы водоснабжения и санитарной техники в Бельгии.
М., Изд-во МКХ РСФСР, 1960.
148. П. Ф. Долгих. ЖПХ, 35, 995 (1962).
149. В. Л. Золотавин и др. Радиохимия, 13, 164 (1971).
150. Т. Tamura. Proc. Internat. Clay Conf. Jerusalem, 1, 425 (1966).
151. M. Lacy, M. De Laguna. Ind. and Engng Ch«m., 50, 1193 (1958).
152. E. И. Орлова, Т. П. Горбатенкова и др. Гигиена и санитария, № 2,
50 (1965).
153. С. L. Sumrall, Е. J. Middlebrooks. J. Amer. Water Works Assoc., 60, 485
(1968).
154. А. И. Эттингер, 3. А. Кологриева. Сборник научных работ ЛНИИ ком¬
мунального хозяйства, вып. 1. Водоснабжение. Л., 1950, стр. 65.
155. В. В. Доливо-Доброволъский. Научные доклады высшей школы. Ме¬
таллургия, № 3, 104 (1958).
156. J. Hollo, I. Szebenyi et al. Period. Polytechn. Chem. Engng, 12, N 3, 283
(1968). '
157. K. Kermer. Пат. ГДР 69089 (1970).
158. Y. S. Shen. J. Amer. Water Works Assoc., 65, 543 (1973).
159. G. S. Logsdon, J. M. Symons. Ibid., p. 554.
160. G. M. Zemansky. J. Amer. Water Works Asfoc., 66, 606 (1974).
161. T. J. Sorg, G. S. Logsdon, J. M. Simons. Z. Wasser- und Abwasser-
Forsch., 8, 149,(1975).
162. G. R. Le Gendre, D. D. Runnells. Environ. Sci. and Technol., 9, 744
(1975).
VII. ЛИТЕРАТУРА
253
163. К. О. Ласточкина. Гигиена и санитария, № 11, 109 (1972).
164. Ю. Б. Багоцкий. Труды ВНИИ ВОДГЕО, вып. 8. М., 1964, стр. 12.
165. Е. С. Велъмина. Качество подготовки воды (материалы семинара), сб.
№ 2. Изд. Моск. Дома научно-технической пропаганды им. Ф. Э. Дзер¬
жинского, 1967, стр. 25.
166. Е. С. Велъмина. Городское хозяйство Москвы, № 4, 25 (1959).
167. К. В. Clarke. J. Inst. Water Engrs, 15, 233 (1961).
168. H. Bernhardt. Arch. Hydrobiol., 61, N 3, 311 (1965).
169. J. McKendrick, R. K. Williams. J. Water Pollut. Contr. Fed., 68, 523
(1969).
170. L. Kable, R. Keil et al. Hydrobiologia, 34, 575 (1969).
171. P. K. Khanna, С. L. Toshniwal. Univ. Boorkee Bes. J., 10, 63 (1968).
172. M. G. Me Garry. J. Water Pollut. Contr. Fed., 42, 191 (1970).
173. M. W. Tenney, W. E. Echelberger et al. Appl. microbiol., 18, 965 (1965).
174. В. М. Любарский, В. М. Трескунов, Р. М. Стерина. Научные труды
АКХ им. К. Д. Памфилова, вып. 98. М., ОНТИ, 1973, стр. 86.
175. J. R. Baylis, А. Е. Mrva. J. Amer. Water Works Assoc., 55, 1536 (1963).
176. R. R. Speedy, N. B. Fisher et al. J. Amer. Water Works Assoc., 61, 289
(1969).
177. Сб. «Использование адгезионных и адсорбционных процессов для уда¬
ления из воды взвесей и микроорганизмов». Под ред. JI. А. Кульского
и М. Н. Ротмистрова. Киев, «Наукова думка», 1973.
178. В. А. Клячко, И. Э. Апелъцин. Очистка природных вод. М., Строй¬
издат, 1973.
179. Е. Н. Kampelmacher, J. L. Noorle. Tijdschr. drinkwatervoorz en af-
valwaterbehandel, 3, 604 (1970).
180. J. C. Baderia, C. L. Toshniwal. J. Inst. Engrs (India). Public Health
Engng Div., 49, 103 (1969).
181. P. V. Panicker, P. M. Vagi, N. V.Rao. Environ. Health, 6, 176 (1964).
182. С. H. Черкинский, H. FI. Трахтман. Обеззараживание питьевой воды.
М., Медгиз, 1962, стр. 40.
183. R. P. Mathur. Environ. Health, 7, 191 (1965).
184. Н. Л. Козлова, И. П. Осадчих, X. М. Тартаковская и др. Водоснабже¬
ние, вып. 1 (5). М., изд. ЦБТИ МКХ РСФСР, 1965, стр. 3.
185. R. W. Ockershausen. Pulp and Paper Mag. Canada, 69, 29 (1968).
186. S. L. Chang, R. E. Stevenson, A. R. Bryant et al. Amer. J. Public He¬
alth, 48, 51 (1958).
187. F. A. Duuren. Water and Water Engng, 71, 360, 414, 454 (1967).
188. A. J. Rubin, S. S. Lackey. J. Amer. Water Works Assoc., 60, 1156 (1968).
189. H. W. Waif, R. S. Safferman, A. R. Nixson. J. Amer. Water Works
Assoc., 66, 526 (1964).
190. G. Berg, R. B. Dean, D. R. Dahling. J. Amer. Water Works Assoc., 60,
193 (1968).
191. O. J. Sproul. J. Amer. Water Works Assoc., 64, 31 (1972).
192. S. L. Chang, P. C. Isaac, N. Baine. Amer. J. Hyg., 57, 253 (1953).
193. P. W. Kabler, S. L. Chang, N. A. Clarke et al. Proc. 5th Sanit. Engrs Conf.
Univ. Illinois Urbana, 1963.
194. S. L. Chang,\ R. E. Stevenson, A. R. Bryant et al. Amer. J. Public He¬
alth, 48,1159 (1958).
195. G. G. Robeck, N. A. Clarke, K. A. Dostal. J. Amer. Water Works Assoc.,
54, 1275,(1962).
196. G. G. Robeck, N. A. Clarke, K. A. Dostal. Bull. Engng and Archit., N 52,
40 (1962).
197. G. Berg. J. New Engl. WaterWorks Assoc., 78, 79 (1964).
198. V. Frankova, R. Cervenka et al. Ceskosl. hyg., 9, 489 (1964).
199. R. T. Thorup, F. P. Nixon, D. F. Wentworth et al. J. Amer. Water
Works Assoc., 62, 97 (1970).
254
ГЛАВА VII. САНИТАРНАЯ ЭФФЕКТИВНОСТЬ КОАГУЛЯЦИИ
200. J. F. Manwaring, М. Chandhuri, R. S. Engelbrecht. J. Amer. Water
Works Assoc., 63, 248 (1971).
201. H. J. Carlson, G. M. Ridenour, C. F. McKhann. Amer. J. Public Health,
32, 1256 (1942).
202. J. R. Neefe, J. R. Raty, J. O. Reinhold et al. Amer. J. Public Health,
37, 365 (1947).
203. G. F. Carlson, F. E. Woodward, D. F. Wentworth et al. J. Water Pollut.
Contr. Fed., 40, 89 (1968).
204. JI. И. Коробейникова, H. Ф. Соколова, R. А. Рябченко. Научные труды
НИИ вакцин и сывороток, вып. 24. М., 1975, стр. 25.
205. Е. R. Штанников, А. А. Шиндряев. Гигиена и санитария, № 10, 18
(1971).
206. F. P. Adams. J. Amer. Water Works Assoc., 25, 1086 (1933).
207. С. A. Ro3HeceucKuU. Физико-химические процессы очистки воды. М.— JT.,
Стройиздат, 1934.
208. 3. Я. Городищер. Труды Поволжской конференции. Горький, изд.
Научн-техн. об-ва санитарной техники и городского хозяйства, 1957,
стр. 107.
209. Ю. И. Колодный. Опыт работы контактных осветлителей с безгравий-
ной загрузкой. Горьк. кн. изд-во, 1963.
210. Е. Ф. Золотова. Качество подготовки питьевой воды (материалы семи¬
нара), сб. № 2. Изд. Моск. Дома научно-техн. пропаганды им.
Ф. Э. Дзержинского, 1967, стр. 17.
211. Е. С. Шалашова, Л. П. Казакова, 3. С. Фейгина и др. Водоснабжение
и канализация, вып. 8. М., изд. ЦБТИ МКХ РСФСР, 1966, стр. 5.
212. R. И. Маркизов.,Очистка воды коагулянтами. М., Изд-во МКХ РСФСР,
1954, стр. 14.
213. Л. С. Круглов. Водоснабжение и канализация, вып. 1 (15). М., изд.
ЦБТИ МКХ РСФСР, 1965, стр. 10.
214. Г. Г. Руденко. Сб. «Коммунальное хозяйство», вып. 1. Киев, «Буд1вель-
ник», 1964, стр. 117.
215. Е. L. Rean, S. J. Campbel, F. R. Anspach. J. Amer. Water Works As¬
soc., 56, 214 (1964).
216. R. L. Williams. J. Amer. Water Works Assoc., 57, 801 (1965).
217. R. Ф. Кожинов. Установки для озонирования воды. М., Стройиздат,
1968, ,стр. 39.
218. Л. А. Кулъский, М. А. Шевченко. Водоснабжение и санитарная техника,
№ 3, 10 (1960).
219. R. С. Шалашова. Жилищно-коммунальное хозяйство, № 6, 18 (1960).
220. А. P. Rlack, A. Smith. J. Amer. Water Works Assoc., 54, 926 (1962).
221. Л. А. Кулъский, С. P. Заякин. Сб. «Санитарная техника», вып. 8. Киев,
«Буд1вельник», 1969, стр. 125.
222. R. Л. Криштул, С. R. Гуслистов. Водоснабжение и санитарная тех¬
ника, № 8, 25 (1963).
223. А. М. Когановский, R. Г. Петренко. Коллоидн. ж., 16, 184 (1954).
224. R. D. Letterman, J. Е. Quon, R. S. Gemmell. J. Amer. Water Works
Assoc., 62, 652 (1970).
225. Т. П. Лимонова, E. Д. Бабенков. Наладка и пути повышения эффектив¬
ности работы водопроводных и канализационных сооружений (материа¬
лы семинара). Изд. Моск. Дома научно-техн. пропаганды им.
Ф. Э. Дзержинского, 1975, стр. 73.
226. Е. Д. Бабенков, Т. П. Лимонова. Технология очистки питьевой воды
и санитарно-гигиенические требования к ее качеству (материалы се¬
минара). Изд. Моск. Дома научно-техн. пропаганды им. Ф. Э. Дзер¬
жинского, 1974, стр. 89.
227. Е. Д. Бабенков, Т. П. Лимонова. Водоснабжение и санитарная тех¬
ника, № И, 14 (1975),
VII. ЛИТЕРАТУРА
255
228. А. Е. Sylvia. Water and Sewage Works, 122, 66 (1975).
229. И. И. Караваев, Н. Ф. Резник. Водоснабжение и санитарная техника.
№ 5, 18 (1970).
230. О. A. Clemens, J. V. Ziemba. Food Engng, 43, N 8, 47 (1971).
231. В. А. Глембоцкий, В. И. Классен. Флотация. М., «Недра», 1973, стр. 188.
232. А. И. Мацнев. Очистка сточных вод флотацией. Киев, «Буд1вельник»,
1976.
233. J. Dollfus, /- L. Burgand. Houille blanche, 22, 411 (1967).
234. L. R. Vuuren, G. J. Stander et al. Water Res., 2, 177 (1968).
235. R. И. Кречетов, 3. Д. Вахчиванжи. Труды Кишиневского политехи,
ин-та, вып. 26, 101 (1971).
236. П. Михайлов, А. Шулъман и др. Сб. «Горючие сланцы», № 6. Таллин,
изд. Республ. Дома научно-техн. пропаганды, 1970, стр. 19.
237. Е. A. Cassell, Е. Matijevic, F. J. Mangreavite et al. J. Amer. Inst. Chem.
Engrs, 17, 1486 (1971).
238. G. G. Cillie, L. R. Vuuren et al. Advances Water Pollution Research,
v. 2. Washington, 1967 p. 1.
239. Л. Д. Скрылев, A. H. Пурич, Ю. С. Городенцев. ЖПХ, 48, 685 (1975).
240. О. А. Иванов, 3. Г. Беляева. Сб. «Транспорт и хранение нефти, нефте¬
продуктов», вып. 12. М., изд. Министерства нефт. промышленности,
1970, стр. 29.
241. L. Vandevenne, A. Dubois. Tribune CEBEDEAU, 28, 159 (1975).
242. I. P. Mail. Пат. США 3418236 (1969).
243. /. R. Lage. Швейц. пат. 468938 (1972).
244. Б. М. Матов, Н. Г. Матова. Авт. свид. СССР 299463 (1969); Бюлл.
изобр., № 12 (1971).
245. Б. М. Матов. Электронная обработка металлов, № 3, 80 (1966).
246. Е. М. Калинийчук, И. А. Макаров, И. И. Василенко и др. Химия и
технология топлив и масел, № 10, 23 (1972).
247. Е. Е. Гришина. Труды ВНИИ ВОДГЕО, вып. 14. М., 1966, стр. 29.
248. L. Lazorko. Prod. Engng, 41, N 21, 36 (1970).
249. Е. Д. Бабенков, М. В. Грехова. Труды ЦНИИ МПС, вып. 468. М., «Тран¬
спорт», 1972, стр. 38.
250. Е. Д. Бабенков, М. В. Грехова. Авт. свид.'СССР 414200 (1972); Бюлл.
изобр., № 5 (1974).
251. N. J.-Pugh. J. Inst. Water Engrs, И, 17 (1957).
252. М. А. Шевченко. Укр. хим. ж., 17, 252 (1951).
253. П. П. Строкач. Сб. «Санитарная техника», вып. 12. Киев, «ByfliBenb-
ник», 1972, стр. 165.
254. А. П. Мартынова. Труды Харьк. ин-та инженеров железнодор. тран-
порта, вып. 75, 42 (1965).
255. 3. Я. Ярославский. Канд. дисс. М., Всесоюзный и.-и. ин-т гидротех¬
ники и мелиорации, 1964.
256. А. Н. Кривчун, П. С. Цыганков. Изв. вузов. Пищевая технология, № 6,
51 (1974).
257. Я. Д. Рапопорт. Автореферат канд. дисс. М., АКХ им. К. Д. Памфи¬
лова, 1970.
258. А. Н. Кривчун, П. С. Цыганков. Фермент, и спирт, промышленность,
№ 5, 16 (1973).
259. В. А. Борисов, Г. В. Голубь и др. Водоснабжение и санитарная техни¬
ка, № 6, 4 (1969).
260. В. Г. Ревенко, А. А. Мамаков. Электронная обработка материалов, № 1,
55 (1976).
261. Е. I. Onstott, W. S. Gregory, Е. F. Thode. Environ. Sci. and Technol.,
7, 333 (1973).
262. П. П. Строкач, В. А. Слипченко. Сб. «Научные основы технологии очи¬
стки воды». Киев, «Наукова думка», 1973, стр. 11.
Глава VIII
ИНТЕНСИФИКАЦИЯ КОАГУЛЯЦИИ
На каком бы принципе не была основана работа сооружений,
предназначенных для выделения коагулированной взвеси из воды,—
— седиментации, фильтрации, центрифугировании или флота¬
ции — главными свойствами, определяющими скорость и полноту
отделения взвеси в осадок, являются структурная прочность и
и плотность хлопьев, их адгезионная способность и тиксотропная
обратимость. Для улучшения этих параметров особенно жела¬
тельного при обработке маломутных холодных вод, в практике
водоочистки выработаны различные приемы и методы. Они позво¬
ляют, как правило, не только улучшить технологические свойства
взвесей, но и ускорить их формирование — интенсифицировать
процесс коагуляции.
Применяемые методы улучшения очистки воды коагулянтами
целесообразно подразделить на два больших класса [1]. К перво¬
му классу следует отнести методы, связанные с внесением в обра¬
батываемую воду дополнительных реагентов:
1) флокулянтов; 2) окислителей; 3) регуляторов величины pH
воды; 4) минеральных замутнителей.
Ко второму классу следует отнести методы, не требующие ис¬
пользования дополнительных реагентов:
1) перемешивание воды, обработанной коагулянтами;
2) осуществление наиболее рациональных способов добавле¬
ния коагулянтов к воде;
3) рециркуляцию коагулированной взвеси через зону ввода
новых порций коагулянта;
4) совмещение коагуляции гидролизующимися коагулянтами
с физическими методами коагуляции — обработкой воды в маг¬
нитном и электрическом поле, ультразвуком и т. д.
В дальнейшем методы интенсификации коагуляции, отнесен¬
ные к первому классу, будем называть реагентными, а методы, от¬
несенные ко второму классу,— безреагентными.
VIII. 1. РЕАГЕНТНЫЕ МЕТОДЫ
Поскольку флокулянтам и флокуляции дисперсных систем посвя¬
щена гл. IX, а действие окислителей подробно рассматривалось
в гл. VII, в этом разделе мы остановимся на регулировании вели¬
чины pH воды и применении минеральных замутнителей.
Vllt.l. РЕАГЕНТНЫЕ МЕТОДЫ
257
VIII. 1.1. Регулирование величины pH воды
Необходимость корректировки величины pH воды в ходе коагуля¬
ции обоснована в гл. VI. Ниже рассматриваются свойства и ре¬
жим добавления щелочных и кислотных реагентов, а также дости¬
гаемые при этом технологические эффекты.
Наиболее широкое применение получили щелочные реагенты,
а среди них — известь, получаемая обжигом при температуре
900-^1200° С известняков, мела и доломитов. Помимо окиси каль¬
ция, в состав извести входят карбонат кальция, окись магния, при¬
меси из глины и песка. В зависимости от содержания окиси маг¬
ния известь делят на кальциевую « 7 % MgO) и магнезиальную
0 7% MgO). Чаще всего известь используют в гашеном виде.
Образующаяся при гашении известь-пушонка содержит до 67%
СаО и MgO. Реже применяют измельченный карбонат кальция
[2,3], негашеную известь [4—6], кальцинированную соду и едкий
натр [7, стр. 75], бикарбонат натрия [2]; при очистке сточных вод —
отходы цементного производства и шлаки, содержащие СаО [8].
Из-за малой растворимости гашеную известь дозируют чаще всего
в виде известкового молока, содержащего до 15% СаО, но иногда
используют и насыщенные растворы (0,12—0,13% СаО).
При выборе щелочного реагента рекомендуется учитывать ко¬
личество углекислоты, выделяющейся в реакции нейтрализации
водородных катионов:
при добавлении соды
111
Н+ -f- ~2~ СОз —» ~2~ СС>2 + ~2~ Н2О,
при добавлении извести или едкого натра
Н++ ОН--► Н20.
Как видно из приведенных реакций, применение Na2C03 и
СаС03 дает дополнительные, количества углекислоты, что ока¬
зывает влияние и на величину pH среды и на осаждаемость коагу¬
лированной взвеси.
Известковое молоко, содержащее большое количество твердой
фазы, а также молотые или гранулированные карбонат и окись
кальция следует использовать при очистке маломутных вод, когда
добавление дисперсных материалов способствует ускорению коа¬
гуляции. Рекомендуемое в последние годы применение извести
в негашеном виде объясняется стремлением избавиться от потерь
активной части при гашении, которые достигают 20%, и меньшей
потребностью СаО по сравнению с Са(ОН)2 (примерно на 25%)
при решении одной и той же технологической задачи [5]. Ращук
и др. [6] показали, что при использовании молотой строительной
извести для подщелачивания железосодержащих сточных вод, по.
9 Е. д. Бабенков
ГЛАВА VIII. ИНТЕНСИФИКАЦИЯ КОАГУЛЯЦИИ
мимо ускорения коагуляции и осаждения хлопьев, происходит
значительное (в 2—4 раза) сокращение объема осадка.
Как сообщалось при рассмотрении способов удаления синтети¬
ческих ПАВ, взаимодействие СаО с продуктами гидролиза коагу¬
лянта приводит к образованию алюмината кальция, хорошо сор¬
бирующего истинно растворенные примеси. Хотя вопрос о составе
и фазовых превращениях системы Ме(ОН)3 + Са(ОН)2 еще во мно¬
гом неясен 1 [9], можно все же утверждать, что кальциевые щелоч¬
ные реагенты обладают несомненным преимуществом перед натри¬
евыми. Возможно, это связано с меньшей растворимостью соеди¬
нений кальция (например, CaS04) и их иными структурными свой¬
ствами. По данным Медведева и Шабалиной [12], гели гидроокиси
алюминия в кальциевой форме подвержены менее интенсивному
старению, чем те же гели, выделенные в натриевой форме.
Рэддик и др. [3] предложили карбонат кальция заблаговремен¬
но добавлять в рабочий раствор A12(S04)3 в пропорции, обеспечи¬
вающей получение зародышевых хлопьев алюмината кальция,
и этой смесью обрабатывать воду. На способ получения железо¬
кальциевых коагулирующих растворов путем смешения раствора
железного купороса с известковым молоком и последующего оки¬
сления смеси кислородом воздуха выдан патент в Бельгии [13].
В большинстве же случаев коагулянт и щелочной реагент дози¬
руют порознь. Выбор точки ввода щелочного реагента и проме¬
жутка времени между добавлением коагулянта и щелочи произво¬
дят в зависимости от местных условий.
Если преследуется цель создания в воде дополнительного ще¬
лочного резерва и улучшения коагулирования за счет механичес¬
ких добавок, подщелачивающие реагенты вводят перед коагулян¬
том. Например, на Новосибирском водопроводе (р. Обь) наилуч¬
шая коагуляция имела место при добавлении к воде сначала из¬
весткового молока, затем (через 2 мин) — хлорного железа и,
спустя 10—15 сек — сернокислого алюминия [14]. По сведениям
Хэма и Кристмана [15], корректировка значений pH до того, как
к воде добавлен коагулянт, дает лучшие результаты в отношении
размера и прочности образующихся хлопьев. Имеются, однако,
примеры успешного применения обратного порядка ввода реаген¬
тов: при обработке воды р. Иртыш лучшие результаты достигнуты
в случае, когда добавление известкового молока производилось че¬
рез 0,5—2 мин после ввода коагулянта [16].
Лебедева [17] в исследованиях с FeS04 пришла к выводу, что
если целью подщелачивания является повышение значений pH
воды не более чем до 8, щелочной реагент следует вводить одновре-
1 По Робертсу [10], продукт реакции — гексагональный гидроалюминат
кальция состава 4Са0,А1203-19Н20. Свойства систем А1С13 + Са(ОН)2 и
A12(S04)3 + Са(ОН)2 и формы состояния в них алюминия рассмотрены
Цукамото [111.
VIII.1. РЕАГЕНТНЫЕ МЕТОДЫ
259
менно с коагулянтом. Если же требуются большие значения pH
(например, в процессе реагентного умягчения воды), щелочной ре¬
агент лучше добавлять к воде заблаговременно, выдерживая раз¬
рыв во времени 7—10 мин. Причина изменения порядка добавле¬
ния реагентов состоит в более длительном формировании в воде
карбоната кальция по сравнению с продуктами гидролиза железа.
Особенно важное значение порядок добавления реагентов име¬
ет при обработке цветных вод, так как гидроксильные ионы могут
во-первых, стабилизировать окрашивающие вещества, во-вторых,
конкурировать с ними за координационные участки гидроксокомп-
лексов (см. гл. II, VI). Новак [18] при очистке умеренно цветных
вод рекомендует добавлять известь после окончания перикинети-
ческой стадии коагуляции, но указывает, что в случае высокоцвет¬
ных мягких вод «возврат цветности», вызванный рестабилизацией
гуминовых веществ, может произойти даже тогда, когда Са(ОН)2
введен после завершения ортокинетической стадии.
Турчинович [19] предлагает проводить известкование воды пос¬
ле осаждения из обесцвечиваемой воды основной массы хлопьев
коагулированной взвеси, а, согласно экспериментам Дэана и Ди¬
кинсона [20], промежуток времени (в интервале от 0 до 10 мин)
между вводом A12(S04)3 и извести при обработке маломутных цвет¬
ных вод не оказывает влияния на результаты очистки. Указывается
на преимущества пропуска коагулированной воды перед отстаи¬
ванием через камеру, заполненную зернами известняка или каль¬
цита [21].
При использовании для обработки цветных вод солей железа
на режим подщелачивания накладываются дополнительные огра¬
ничения. Исследования Блэка и др. [22] показали, что в цветной
воде, обработанной сульфатом я^елеза в области оптимальных для
устранения цветности значений pH, 50—80% общего железа пред¬
ставлено двухвалентной формой. Перевод этой формы в трехва¬
лентную требует доведения pH до 8,3, но при этом происходит
«возврат цветности», объясняемый переходом окрашенных соеди¬
нений двухвалентного железа в еще более окрашенные соединения
трехвалентного железа. Следовательно, величина pH воды, об¬
работанной щелочью, в этом случае не должна превышать 8,3.
Из кислотных реагентов для корректировки значений pH при¬
меняют почти исключительно серную кислоту, подчеркивая ее пре¬
имущества при обработке вод с малой буферной емкостью, реже —
соляную и угольную кислоты [23, 24 (стр. 217), 25, 26]. Клячко
и Шемякина [26] с целью снижения pH до оптимальных значений
рекомендуют подвергать воду предварительному Н-катионирова-
нию.
Применение подкислителей особенно целесообразно при обра¬
ботке цветной воды, так как приводит к заметному повышению эф¬
фекта обесцвечивания и экономии коагулянта [27]. Подкисление
9*
260
ГЛАВА VIII. ИНТЕНСИФИКАЦИЯ КОАГУЛЯЦИИ
воды позволяет увеличить глубину очистки ее от ПАВ, микроорга¬
низмов, кремнекислоты и некоторых других примесей. Иногда при
очистке некоторых видов сточных вод целесообразно проводить
двойную корректировку значений pH среды с последовательным
вводом кислотного и щелочного реагентов [28].
Предложен способ кислотной активации самих растворов коа¬
гулянтов за счет добавления к ним серной, фосфорной или кремние¬
вой кислот [29]. Указывается, что скорость седиментации коагу¬
лированной взвеси возрастает после такой активации в 20 раз.
VIII. 1.2. Применение минеральных замутнителей
При обычно используемых дозах коагулянтов степень пересыще¬
ния воды малорастворимыми продуктами гидролиза соответствует
метастабильной зоне, где энергия пересыщения может оказаться
недостаточной для возникновения зародышей твердой фазы. По¬
этому внесение искусственных замутнителей, частицы которых
играют роль дополнительных центров конденсации продуктов гид¬
ролиза, способствует ускорению коагуляции примесей при очист¬
ке маломутных вод. Замутнение резко усиливает флокулирующее
действие полиэлектролитов.
Следующая задача, которая может быть успешно решена при
замутнении обрабатываемой воды — утяжеление хлопьев коагу¬
лированной взвеси, увеличение их гидравлической крупности.
Если замутняющее вещество способно, кроме того, сорбировать ра¬
створенные примеси, возрастает глубина очистки воды. Наконец,
если частицы замутнителя сорбируют ионы, определяющие сте¬
пень устойчивости золей, то облегчаются условия коагуляции 2.
Частным случаем такой сорбции является поглощение глинами ка¬
тионов водорода, образующихся при гидролизе коагулянтов. Оно
особенно важно при обработке вод с низким щелочным резервом
[30].
Из перечисленных задач вытекают требования, предъявляемые
к замутнителям — высокая степень дисперсности, достаточно
большая сорбционная емкость по отношению к нежелательным при¬
месям, наличие заряда, противоположного по знаку заряду про¬
дуктов гидролиза коагулянтов. Наиболее распространенный замут-
нитель — глины: монтмориллонит (бентонит), палыгорскит и др.
Под действием электролитов, содержащихся в воде, отрицательно
заряженные частицы глин коагулируют, и потому некоторые гли¬
нистые минералы могут употребляться даже в качестве самостоя¬
тельных коагулянтов, например монтмориллонит в Na-форме, на¬
2 Это вытекает из правила Оствальда—Бузага и объясняется уменьшением
среднего количества стабилизатора на единицу площади поверхности дис¬
персной фазы.
VIII. 1. РЕАГЕНТНЫЕ МЕТОДЫ
261
зываемый аскангелем, использовался (совместно с известью) для
очистки вод Закавказья [31].
К преимуществам глин как замутнителей относят [32]: ^эффек¬
тивность при любых значениях pH; 2) отсутствие необходимости
в дополнительных реагентах; 3) отсутствие влияния на органо¬
лептические показатели качества воды.
Из глинистых замутнителей чаще всего применяется бентонит.
Эффективность его действия зависит от размеров частиц [33, 34].
Для улучшения сорбционных свойств бентонит перед употребле¬
нием рекомендуют обрабатывать едким натром [35] или (в случае
малой жесткости исходной воды) известью — присутствие ионов
Са2+ способствует снижению агрегативной устойчивости частиц
замутнителя и их коагуляции. При флотации бентонит может ис¬
пользоваться как флотореагент [36, 37].
В сочетании с коагулянтами и флокулянтами глины и глинис¬
тые сланцы дают хорошие результаты при очистке воды от радио¬
активных веществ, фосфатов, ПАВ, микроорганизмов (см. гл. VII).
Достигается экономия коагулянтов. Оптимальные дозы глин ле¬
жат в пределах от нескольких десятков до нескольких сот мил¬
лиграммов на литр.
Второй по значению замутнитель — карбонат кальция, облег¬
чающий хлопьеобразование не только за счет создания центров
конденсации твердой фазы, но и за счет повышения pH, особенно
важного для маломинерализованных вод, а также для формирова¬
ния железо- и алюмокальциевых комплексов [38—42]. Карбонат
кальция (кальцит) дозируют в виде тонкодисперсного порошка
в количествах от 12—18 до 240 мг/л. Рэддик [43] считает оптималь¬
ным такое соотношение между дозами кальцита и сернокислого
алюминия, при котором каждый из хлопьев содержит около 50
кристалликов СаС03.
В качестве замутнителей рекомендованы также сульфаты, фос¬
фаты и хроматы бария [42—44], окиси алюминия, хрома, железа
и кобальта, карбонаты и хроматы свинца, фосфаты хрома [44],
измельченный магнетит [45], летучие золы [46], мелкий кварцевый
песок [47, 48], портланд-цемент [49], металлическая пыль [50],
силикатсодержащие отходы производства [51], кизельгур, активи¬
рованный серной кислотой [52], молотый антрацит [53] и др.
Из числа перечисленных заслуживают особого внимания за-
мутнители, предназначенные для многократного использования.
Измельченный антрацит (размер частиц около 500 мкм, удельная
поверхность 92 см21г) рекомендуется добавлять во взвешенный слой
осветлителей, откуда он вместе с отработанной коагулированной
взвесью непрерывно отделяется, затем очищается в гидроциклоне
и возвращается в осветлители [53].
Предлагаемый Шиманским и др. [45] способ многократного
использования магнетитового порошка (с размером частиц менее
262
ГЛАВА VIII. ИНТЕНСИФИКАЦИЯ КОАГУЛЯЦИИ
60 мкм) предусматривает отделение магнетита от осадка коагули¬
рованной взвеси в магнитном поле. При использовании стальной
пыли имеется в виду дополнительная возможность ускорения
хлопьеобразования за счет магнитной коагуляции [50].
VIII. 2. БЕЗРЕАГЕНТНЫЕ МЕТОДЫ
VIII. 2.1. Перемешивание воды
В практике очистки воды коагулянтами различают два режима
перемешивания: с большой интенсивностью (быстрое перемеши¬
вание) и с малой (медленное перемешивание). В первом режиме ра¬
ботают устройства, предназначенные для смешения растворов
реагентов с водой, во втором — камеры хлопьеобразования.
Анализ материалов, приведенных в гл. V, показывает, что до
тех пор, пока основная масса участвующих в коагуляции частиц
не достигнет размера 5—10 мкм, перемешивание воды, каким бы
интенсивным оно ни было, не в состоянии улучшить транспортные
условия коагуляции. На первом этапе коагулирования гораздо
большее значение имеет создание таких исходных условий, ко¬
торые оказали бы благоприятное влияние на ход дальнейшего
хлопьеобразования. Помимо равномерного распределения коа¬
гулянта в объеме воды, интенсифицирующее действие быстрого
перемешивания состоит в его влиянии на численную концентрацию
зародышевых частиц коагулированной взвеси, распределение этих
частиц по размеру и характер их взаимной фиксации в агрегатах.
Исследования, проведенные автором [30, 54], позволили уста¬
новить, что повышение интенсивности смешения воды с раство¬
ром коагулянта вплоть до значений G, вызывающих дисперги¬
рование коагуляционных структур в момент их возникновения
(период скрытой коагуляции), приводит к увеличению плотности
структур, отделению углекислоты и ускорению седиментации об¬
разующихся хлопьев. Возможность создания благоприятных ки¬
нетических условий хлопьеобразования за счет увеличения сте¬
пени дисперсности частиц коагулята интенсивным перемешива¬
нием обрабатываемой воды показана Печниковым и Хайловым
[55]. Но, как мы уже знаем, чрезмерно интенсивное и длительное
перемешивание воды может привести к необратимому разбиванию
микрохлопьев, резкому ухудшению адгезионных свойств взвеси
и замедлению последующего хлопьеобразования. Поэтому интен¬
сивность быстрого перемешивания должна быть ограничена. На
практике в смесительных устройствах значения скоростного гра¬
диента поддерживают обычно в пределах 300—350 сект1 [56], а
перемешивание осуществляют в течение 1—2 мин. Увеличение
G до 500—1000 секГ1 позволяет сократить период быстрого пере¬
V11I.2. ЁЕЗРЕАГЁНТНЫЕ МЕТОДЫ
мешивания до 10—30 сек [7 (стр. 94), 30, 54]. Соответственно мо¬
жет быть уменьшен рабочий объем смесителя или увеличена его
производительность.
Технологический эффект, достигаемый при перемешивании,
зависит от выбранного способа перемешивания. Применяют, в
основном, два типа смесителей. Работа смесителей первого типа,
называемых иногда гидравлическими, основана на использовании
кинетической энергии потока самой обрабатываемой воды; работа
смесителей второго типа, называемых механическими,— на ис¬
пользовании средств принудительного перемешивания. И в том,
и в другом случае перемешивание достигается за счет появления
градиента скоростей при обтекании потоком воды механических
препятствий. Поэтому оба эти способа можно объединить под об¬
щим названием механического перемешивания.
Часто перемешивание воды с раствором коагулянта осущест¬
вляют путем воздушного барботажа. Этот способ — пневматичес¬
кое перемешивание — обладает важными особенностями. Наконец,
иногда пользуются комбинированными смесительными устройст¬
вами, в которых совмещены разные приемы перемешивания:
эжекторная рециркуляция и воздушный барботаж, насосная ре¬
циркуляция и перемешивание в дырчатых перегородках [57, 58]
и др.
Ниже мы дадим краткую характеристику эффективности двух
основных способов перемешивания — механического и пневма¬
тического.
VIII. 2.1.1. Механическое перемешивание
Выбор типа смесителя зависит от вида коагулянта и вспомогатель¬
ных реагентов, производительности очистных сооружений, рас¬
полагаемого напора, стоимости электроэнергии и ряда других ус¬
ловий, влияние которых подробно обсуждается в специальной ли¬
тературе [7, 19, 24]. Здесь нам хотелось бы подчеркнуть лишь сле¬
дующее важное обстоятельство. Как бы ни была совершенна си¬
стема турбулизации воды в объеме смесительного пространства,
однородность условий перемешивания может быть достигнута
только тогда, когда за время пребывания в смесителе вся вода ус¬
певает однократно или многократно проходить через определен¬
ную зону наиболее интенсивного перемешивания, т. е. подвергает¬
ся рециркуляции 3.
Наряду с созданием однородных условий перемешивания, си¬
стема рециркуляции обеспечивает качественно новый эффект,
поскольку вследствие каталитического влияния ранее образовав¬
3 Идея рециркуляции коагулируемой воды через зону впода попых порций
коагулянта принадлежит Кургаеву.
264
ГЛАВА VIII. ИНТЕНСИФИКАЦИЯ КОАГУЛЯЦИИ
шейся твердой фазы облегчаются формирование и рост зародышей
коагулированной взвеси [58]. Кроме того, наличие внутренней ре¬
циркуляции приводит к заметному увеличению фактического вре¬
мени пребывания воды в смесителе. Для необратимой реакции соот¬
ношение времени пребывания воды в реакторе с рециркуляцией
и без нее может быть подсчитано приблизительно по уравнениям
Никитина [59].
Хорошие результаты дает способ смешения воды с раствором
коагулянта во взвешенном слое зернистого материала, в котором
интенсивный массообмен и гетерогенный катализ сочетаются с ре¬
циркуляцией ранее образовавшихся малорастворимых продуктов
гидролиза.
Камеры хлопьеобразования, в которых осуществляется мед¬
ленное перемешивание, рассчитываются на время пребывания во¬
ды 20—60 мин и так же, как и смесители, делятся на гидравличес¬
кие, механические, барботажные и комбинированные [7 (стр. 164),
60]. Стремление обеспечить такие гидравлические условия пере¬
тока обрабатываемой воды из камер хлопьеобразования в очист¬
ные сооружения, при которых сформированные хлопья не под¬
вергались бы разрушениям, привело к идее создания встроенных
камер, т. е. совмещенных в едином блоке с очистными устройст¬
вами.
Скорость движения воды в камерах хлопьеобразования, отве¬
чающая минимальной остаточной мутности воды, лежит в преде¬
лах 0,12—0,7 м!сек [61, 62 (стр. 136), 63, 64]. Для характеристики
режима работы камер чаще всего пользуются критерием Кэмпа
GT, который, как показывает практика, должен составлять
1-104—2-105. Большие значения G относятся к водам с высоким
содержанием грубодисперсных примесей; большие значения Т —
к маломутным водам [17]. Довольно широкий диапазон оптималь¬
ных значений GT и зависимость критерия от качества исходной
воды подтверждают предположения о необходимости учета чис¬
ленной и объемной концентрации дисперсной фазы, в том числе
продуктов гидролиза коагулянта. По-видимому, после создания
более надежных способов регистрации этих параметров будут по¬
лучены дополнительные материалы для выбора оптимального ре¬
жима хлопьеобразования.
Преимущества, достигаемые при использовании медленного
перемешивания, отмечает подавляющее большинство исследова¬
телей, особенно при малой мутности и низкой температуре воды
в источнике [85]. Успешно применяется рекомендованное Лан-
желье перемешивание с постепенно снижающейся интенсивностью
[66, 67]. Однако известны случаи, когда перемешивание, осуще¬
ствляемое во время хлопьеобразования, не дало никаких преиму¬
ществ. Например, применение механических мешалок со ступен¬
чато уменьшающейся скоростью вращения на Северной водопро¬
VIII.2. БЕЗРЕАГЕНТНЫЕ МЕТОДЫ
265
водной станции г. Москвы не дало улучшения качества отстоенной
воды. Это, возможно, связано с низким содержанием взвеси в ис¬
ходной воде или разбиванием хлопьев на входе в отстойник, в ре¬
зультате чего эффект, достигнутый перемешиванием, оказался
экранированным. Подобные примеры требуют тщательного ана¬
лиза.
В зарубежных конструкциях смесительных камер преимуще¬
ственное распространение получили лопастные мешалки, и много¬
численные публикации свидетельствуют об их высокой эффектив¬
ности. К числу преимуществ механического перемешивания перед
гидравлическим относят обеспечение лучшего качества осветлен¬
ной воды, возможность экономии до 40% коагулянта, гибкое ре¬
гулирование интенсивности, малые потери напора [24 (стр. 231),
68]. В СССР камеры хлопьеобразования с механическим переме¬
шиванием пока не получили распространения, хотя результаты
исследований [54, 55] позволяют надеяться на их успешное при¬
менение на многих источниках водоснабжения.
Увеличение интенсивности перемешивания, легко достигае¬
мое при механическом способе, открывает возможности для суще¬
ственного сокращения длительности перемешивания. Об этом сви¬
детельствуют результаты, полученные при двухэтапном переме¬
шивании коагулированной воды с практически одинаковыми зна¬
чениями критерия Кэмпа: 48 000 для первого и 45 000 для второго
этапа [69]. При сохранении постоянства условий перемешивания
на втором этапе (G = 50 сект1, Т = 900 сек) длительность первого
этапа перемешивания без ущерба для качества отстоенной воды
можно было уменьшить с 16 до 4 мин за счет соответствующего
увеличения G от 50 до 200 сект1.
VIII. 2.1.2. Пневматическое перемешивание
Еще в 20—30-х годах исследователи придавали значение важному
свойству способа пневматического перемешивания воды, обрабо¬
танной коагулянтами — отделению заметных количеств углекис¬
лоты [70, 71].
В 1954 г. Камбозу [72] на примере коагулирования мутных вод
(около 750 мг/л взвешенных веществ) показал, что 5—7-минутная
аэрация воды после добавления сернокислого алюминия дает
возможность в 3—4 раза сократить потребность в коагулянте, в
4 раза ускорить формирование и осаждение хлопьев, избавиться
от опалесценции очищенной воды. Высокую эффективность аэ¬
рации автор объяснил отделением углекислоты, выделяющейся во
время гидролиза A12(S04)3.
Позднее Егоров и др. [73—75] в ходе лабораторных исследова¬
ний пневматического перемешивания коагулируемой воды уста¬
новили, что для достижения наилучших результатов очистки воз-
266
ГЛАВА VIII. ИНТЕНСИФИКАЦИЯ КОАГУЛЯЦИИ
дух должен вводиться обязательно в момент образования микро¬
хлопьев коагулированной взвеси 4, а пузырьки его должны быть
как можно более мелкими. При расходе воздуха в размере 10—
20% от количества обрабатываемой воды оказалось возможным
уменьшить расход коагулянта в 1,5—2,5 раза. Производственные
испытания, осуществленные на высокомутных водах (с содержа¬
нием взвеси 900 и 1200 мг/л) подтвердили результаты лаборатор¬
ных опытов. Обработку воды воздухом рекомендовано проводить
в течение 8—10 [сек.
В других исследованиях, предпринятых в 1930 г. Жуковым
[76] по определению оптимальных условий очистки невской воды,
отмечено, что при пневматическом перемешивании ускорение ко¬
агуляции наступало раньше чем происходило заметное повышение
pH среды, связанное с отделением углекислоты. Следовательно,
ускорение коагуляции не могло быть объяснено только десорбци¬
ей С02.
Анализ возможной роли разных факторов в улучшении коагуля¬
ции примесей воды при аэрации, а также результаты исследований
[77], выполненных нами ранее, приводят к выводу, что достигае¬
мый эффект обусловлен одновременным протеканием по крайней
мере четырех процессов:
1) специфического механического перемешивания воды пузырь¬
ками воздуха;
2) образования зародышей твердой фазы продуктов гидролиза
на поверхности газовых пузырьков;
3) выделения из воды избыточных количеств углекислого газа;
4) окисления кислородом, выделяющимся из воздуха, органи¬
ческих примесей.
Скорость всплывания пузырьков и связанная с ней интенсив¬
ность перемешивания среды определяются величиной подъемной
силы
где d — диаметр пузырька; рв и рг — плотность воды и газа;
•ф — коэффициент сопротивления; va — скорость подъема пу¬
зырька.
В реальных условиях из-за наличия вихревых потоков исполь¬
зование уравнений (VIII.1) и (VIII.2) может внести ошибку в рас¬
* В лабораторных опытах разрыв во времени между вводом коагулянта и
началом аэрации состарил 15—25 сек.
FПОД ~~Q~ (Рв Рг)
(VIII.1)
и испытываемым сопротивлением
(VIII.2)
VIII.2. БЁЗРЁАГЁНТНЫЕ МЕТОДЫ
267
четы va. Целесообразно пользоваться следующими выражениями
[78]:
для пузырьков размером до 0,4 и 0,9—1 мм
1 70 ё (Рв ~ Рг) /TfTTI 04
Vn = "36" ч (VIII.3)
для пузырьков размером 0,4—0,9 мм
va = 26,4 d; (VIII.4)
для пузырьков размером более 1 мм
vn ~ 26—30 см/сек.
При распылении воздуха через пористые перегородки размер
образующихся пузырьков равен 2,5—3,0 мм, и скорость их подъ¬
ема может быть принята для расчетов в пределах 30—35 см/сек.
Согласно расчету, выполненному нами в соответствии с рекомен¬
дациями Кэмпа [79], максимальный скоростной градиент вблизи
поверхности поднимающегося с такой скоростью пузырька имеет
порядок 2-103 сек'1.
Роль пузырьков воздуха как центров хлопьеобразования в
коагулирующей системе объясняется, в первую очередь, налипа¬
нием твердых частиц вследствие самопроизвольного стремления
системы к уменьшению потенциальной энергии. Работа закрепле¬
ния твердых частиц на поверхности пузырьков определяется по¬
верхностным натяжением, краевым углом смачивания и размера¬
ми (кривизной поверхности) пузырьков (см. гл. VI). Причем ра¬
бота закрепления большой частицы на малом пузырьке воздуха
уменьшается с уменьшением размера частицы [78, 80].
Процесс образования аэрофлокул и кинетику коагуляции гид¬
розолей на поверхности газовых пузырьков исследовали Штарк
[81], Геллер и Петерс [82, 83]. Установлено, в частности, что рез¬
кое увеличение скорости коагуляции золя гетита [a-FeOOH]
с ростом поверхности раздела вода — воздух является следствием
поверхностной коагуляции. Исходя из предположения, что сгу¬
щение частиц золя на поверхности пузырьков подчиняется урав¬
нению адсорбции Лэнгмюра, авторы [82, 83] получили выражение
для описания кинетики поверхностной коагуляции и подтверди¬
ли его справедливость экспериментально.
В фиксации твердых частиц на поверхности газовых пузырь¬
ков важное значение принадлежит отрицательному заряду по¬
верхности, обусловленному ионными парами в. Величина заряда
меняется в зависимости от размеров пузырька и анионного состава
воды. В присутствии кислот величина заряда смещается в сторо¬
6 Анионы преобладают со стороны газовой фазы, катионы — в диффузном
слое со стороны водной среды [80].
268
ГЛАВА VIII. ИНТЕНСИФИКАЦИЯ КОАГУЛЯЦИИ
ну положительных значений, в присутствии щелочей — в сторону
отрицательных. Особенно сильное действие оказывают адсорби¬
рующиеся органические вещества.
При подъеме пузырьков, помимо их прямого соприкосновения
с частицами твердой фазы, происходит подсос частиц в кормовую
зону пузырьков. Как показывает киносъемка, сталкиваясь с пу¬
зырьками, частицы скользят по их поверхности вниз и закрепляют¬
ся в нижней полусфере. На захват частиц пузырьками воздуха
заметное влияние оказывает электрическое поле, возникающее
вокруг пузырьков при их подъеме [84—86].
Для описания десорбции углекислоты из воды при продувке
воздуха предложены следующие выражения [87—89]:
в которых с о и ст — концентрация углекислоты соответственно
в начальный момент и ко времени т; cs — концентрация насы¬
щения (растворимость углекислоты в данных условиях); s — пло¬
щадь поверхности контакта воды с воздухом; Дсср — средняя
движущая сила процесса десорбции, зависящая главным образом
от исходной концентрации С02 и условий аэрации; V — объем
воды, подвергаемой аэрации; К — коэффициент десорбции, имею¬
щий в уравнениях (VIII.5) — (VIII.7) несколько разный физичес¬
кий смысл и разную размерность.
По приведенным выражениям можно проследить влияние усло¬
вий аэрации на конечные результаты. Изменение температуры ока¬
зывает двойное действие: уменьшает параметр с5, в результате
чего возрастает движущая сила десорбции (сх — cs), и увеличи¬
вает скорость газопереноса вследствие снижения сопротивления
пленки и увеличения коэффициента диффузии.
По данным Хатчисона и Шервуда [90], коэффициент десорбции
углекислоты К2 в уравнении (VIII.6) с увеличением скорости пе¬
ремешивания в 10 раз возрастает примерно в 5 раз. Скорость де¬
сорбции наиболее велика в первые мгновения после образования
пузырька, когда пленка имеет минимальную толщину.
Шафер [89] проверил выражение (VIII.7) при аэрации пузырь¬
ками воздуха размером 2—8 мм. Величина коэффициента Ка
зависела от типа аппарата и интенсивности перемешивания воды.
Она возрастала с увеличением поверхности контакта воды с возду¬
хом и температуры. Экспериментально найденная зависимость
C-с == / (т) описывалась кривой, лежащей на графике между расчетны¬
ми кривыми для ламинарного и развитого турбулентного режимов.
С-{ С о - (7ср,
(VIII.5)
(VIII.6)
= — К3г,
(VIII.7)
VIII.2. ЕЕЗРЕАГЕНТНЫЁ МЕТОДЫ
269
Наши исследования [77] по пневматическому перемешиванию
воды после добавления коагулянта показали, что десорбция угле¬
кислоты в этих условиях подчиняется несколько иным закономер¬
ностям. Чем меньше углекислоты содержала исходная вода до
смешения с раствором сернокислого алюминия и чем ниже была
доза коагулянта, тем успешнее протекал процесс десорбции С02.
Полнота отделения углекислоты определялась не столько дви¬
жущей силой десорбции (с0 — cs), сколько отношением количест¬
ва поданного воздуха к количеству углекислоты в воде.
Этот на первый взгляд неожиданный результат может быть объ¬
яснен следующим образом. В ходе аэрации воды, обработанной
коагулянтом, вследствие отделения С02 гидролиз катионов А13+
протекает полнее и образуется дополнительное количество угле¬
кислоты. Чем больше доза коагулянта, тем меньше ощущаются
результаты аэрации: наиболее заметное увеличение pH в ходе
аэрации (0,4 единицы) соответствовало в описываемых опытах до¬
зе A12(S04)3, равной 50 мг/л. Таким образом, роль аэрации воды
в момент гидролиза коагулянта сводится к отделению свободной
углекислоты; смещению равновесия реакции
НСО; + Н+ s: С02 + Н20
вправо; углублению реакции гидролиза; уменьшению количества
бикарбонатов, связанных с катионами алюминия химическими
силами (см. гл. VI); смещению вследствие этого рН{ продуктов
гидролиза в сторону более высоких значений. Прямыми резуль¬
татами аэрации являются перестройка структуры хлопьев коагу¬
лированной взвеси и улучшение их гидравлической крупности.
С изложенной точки зрения на механизм действия аэрации нахо¬
дят объяснение результаты очистки невской воды [76].
В проведенных исследованиях обнаружено важное отличие
пневматического перемешивания от механического: несмотря на
приблизительно такую же интенсивность перемешивания (по ве¬
личине скоростного градиента), длительная аэрация воды, обра¬
ботанной коагулянтом (до 10 мин), не приводила к нарушению
тиксотропных свойств коагулированной взвеси. Увеличение плот¬
ности взвеси оказалось тем заметнее, чем длительнее была аэра¬
ция и короче промежуток времени от момента ввода коагулянта
до начала аэрации. При расходе воздуха в размере 20% от коли¬
чества обрабатываемой маломутной (2—3 мг/л) воды потребность
в сульфате алюминия удалось снизить на 30% [1].
Еще большая экономия коагулянта (на 50%) достигнута при
аэрации маломутной воды (13 мг/л) Котляровым [91]. Применение
пневматического перемешивания воды с использованием для рас¬
пыления воздуха пористых труб позволило снизить содержание
углекислоты в воде на 66 % и увеличить производительность ос¬
270
ГЛАВА VIII. ИНТЕНСИФИКАЦИЯ КОАГУЛЯЦИИ
ветлителей Сыктывкарской ТЭЦ [92]. Хорошие технологические
результаты удалось получить при совмещении аэрации воды с
рециркуляцией коагулированной взвеси [57, 58, 93, 94].
VIII. 2.2. Подбор рационального режима
добавления коагулянтов к воде
Места ввода коагулянтов на водоочистных станциях определяются
принятой технологической схемой, качеством исходной воды,
необходимостью добавления щелочей или кислот, флокулянтов и
сорбентов. В двухступенчатой технологической схеме раствор
коагулянта добавляют к воде обычно непрерывно одной порцией
в начало или середину смесителя, в водораспределительные уст¬
ройства осветлителей со взвешенным осадком, в камеры хлопье¬
образования; в одноступенчатой — в непосредственной близости
от входа воды в загрузку фильтровальных сооружений.
Подбор наиболее рациональных способов добавления коагу¬
лянтов основан на максимальном использовании концентрацион¬
ных и каталитических эффектов, с одной стороны, и лучших гид¬
родинамических условий перемешивания — с другой.
VIII. 2.2.1. Фракционированное коагулирование
Фракционированное коагулирование 6 предусматривает добавле¬
ние расчетного количества коагулянта к воде не одной, а двумя
или несколькими последовательными порциями. Обработку воды
разными последовательно добавляемыми коагулянтами можно так¬
же рассматривать как фракционированное коагулирование.
Технологический эффект, достигаемый при фракционировании
дозы коагулянта, почти всегда объясняют с кинетической точки
зрения — образованием в результате гидролиза первых порций
коагулянта твердой фазы, выступающей в роли центров хлопье¬
образования при гидролизе последующих порций коагулянта [62
(стр. 11), 95]. Как известно, скорость налипания мелких частиц
на крупные может в несколько раз превышать скорость взаимной
коагуляции мелких частиц (см. гл. V). Большое значение, несом¬
ненно, имеет также «омолаживание» взвеси, полученной в резуль¬
тате гидролиза предшествующих порций коагулянта, т. е. процесс
обновления ее активности в отношении поверхностных явлений —
сорбции и адгезии [1, 54].
Впервые фракционированное коагулирование применено, по-
видимому, в Аргентине [95], где на примере A12(S04)3, FeCl3 и
FeS04 показана возможность снижения остаточного содержания
взвеси в очищенной воде в 1,5—2 раза. Наилучшие результаты
6 Иногда этот способ коагулирования называют «дробным», или «частичным».
VIII.2. БЕЗРЕАГЕНТНЫЕ МЕТОДЫ
271
достигнуты при работе на смешанном коагулянте с дозировкой
сначала двух частей сернокислого алюминия, а затем одной части
хлорного железа. Установлено, что оптимальный промежуток вре¬
мени между вводом фракций коагулянта равен 90—120 сек. Эко¬
номия коагулянта составила 17%.
Опыт применения фракционированного коагулирования на
одной из водоочистных станций Англии [96] показал, что при обес¬
цвечивании воды первая фракция коагулянта не должна превы¬
шать половины общей дозы. Такой режим приводит к формирова¬
нию на первом этапе мелких, медленно осаждающихся хлопьев
гидроокиси алюминия, которые, по мнению исследователей, вслед¬
ствие развитой поверхности раздела хорошо сорбируют окраши¬
вающие примеси. Расход коагулянта по сравнению с обычным спо¬
собом ввода уменьшился.
Исследование фракционированного коагулирования продол¬
жено автором [1,54]. При обработке воды с разным содержанием
взвеси (Мп) сернокислым алюминием найдено, что этот метод дает
возможность на 6—20% увеличить плотность коагулированной
взвеси. Степень уплотнения возрастала с увеличением относитель¬
ного содержания продуктов гидролиза коагулянта в хлопьях (см.
таблицу).
Влияние фракционированного коагулирования на плотность
коагулированной взвеси [54]
Ма, мг/л
Дозы Al2(S04)a,
мг/л
Условный
объемный
вес коагу¬
лирован¬
ной
взвеси,
мг/см3
Ми, мг/л
Дозы Ab(S04)3,
мг/л
Условный
объемный
вес коагу¬
лирован¬
ной
взвеси,
мг/см*
/1-я
фрак¬
ция
2-я
фрак¬
ция
1-я
фрак¬
ция
2-я
фрак¬
ция
0
200
1,85
50
200
3,12
0
150
50
2,22
50
150
50
3,30
25
200
—
2,62
100
200
—
5,95
25
150
50
2,92
100
150
50
6,42
Примечание. Условный объемный вес взвеси определен по результатам 3-х ча¬
сового отстаивания по методике Кургаева [62].
Дополнительного уплотнения взвеси удалось добиться при дис¬
пергировании продуктов гидролиза, образовавшихся после добав¬
ления первой фракции коагулянта, интенсивным механическим
перемешиванием. Однако для такого режима особенно важным бы¬
ло правильное распределение (в зависимости от длительности и ин¬
тенсивности диспергирования) общей дозы коагулянта на фракции,
272
ГЛАВА VIII. ИНТЕНСИФИКАЦИЯ КОАГУЛЯЦИИ
Чем сильнее разрушались коагуляционные структуры, возникшие
в результате ввода первой фракции коагулянта, тем большей дол¬
жна была быть доля второй фракции для восстановления тиксо-
тропных и поверхностных свойств хлопьев. В то же время степень
уплотнения коагулированной взвеси росла с увеличением первой
фракции дозы коагулянта. В условиях проведенных эксперимен¬
тов оптимальный режим коагулирования, позволяющий добиться
увеличения плотности коагулированной взвеси в 1,5—2 раза (без
ущерба для ее поверхностных свойств), соответствовал разделению
дозы коагулянта на две равные части и длительности диспергиро¬
вания 30—60 сек [54].
VIII. 2.2.2. Концентрированное коагулирование
Концентрированное коагулирование 7 заключается в дозирова¬
нии всего потребного количества коагулянта лишь в часть обраба¬
тываемой воды. После тщательного смешения с раствором коагу¬
лянта поток обработанной воды объединяют (обычно в начале ка¬
мер хлопьеобразования) с потоком остальной — некоагулирован-
ной воды. Этот способ, широко используемый в химической про¬
мышленности для ускорения протекания гетерогенных реакций,
для очистки воды был впервые применен на Восточной станции
московского водопровода в 1947 г. [97].
Сардановский и Казакова [98] указывают на следующие пре¬
имущества метода концентрированного коагулирования:
1) распределение всего коагулянта только в части воды создает
условия для ускоренного хлопьеобразования;
2) после смешения с необработанной водой хлопья, сформиро¬
ванные в условиях повышенной концентрации коагулянта, хо¬
рошо удаляют водные примеси.
При обработке методом концентрированного коагулирования
волжской воды достигнуто дополнительное уменьшение ее мут¬
ности и цветности, снижено содержание остаточного алюминия.
Наилучшие результаты достигнуты при отношении расхода обра¬
батываемой коагулянтом воды к расходу остальной воды 1 :1,5,
но авторы [98] рекомендуют для каждого конкретного случая
это соотношение подбирать опытным путем, исходя из соображе¬
ний, чтобы доза коагулянта, отнесенная к обрабатываемой части
потока, не была ниже оптимальной по хлопьеобразованию и выше
предела емкости щелочного резерва. Применение концентрирован¬
ного коагулирования позволило уменьшить расход сернокислого
алюминия на 20—30% [98, 99].
Метод концентрированного коагулирования модифицирован
Дризом [100, 101]. В небольшую часть (около 1%) очищаемой во¬
7 Концентрированное коагулирование называют иногда «раздельным», но
этот термин неудачен, так как не отражает существа способа.
VIII.2. БЕЗРЕАГЕНТНЫЕ МЕТОДЫ
273
ды вводится все потребное количество коагулянта и известковое
молоко в количестве, обеспечивающем получение pH воды около
4,5. После часового пребывания в резервуаре-реакторе получен¬
ный раствор смешивается с основным потоком обрабатываемой
воды. В отстойниках образуются крупные и плотные хлопья,
эффект осветления воды возрастает. Наблюдаемый эффект Дриз
объяснил формированием труднорастворимых основных солей алю¬
миния типа Al (0H)S04 со своими индивидуальными свойствами,
но не подлежит сомнению и существенная роль образующихся алю-
мо-кальциевых комплексов.
Метод концентрированного коагулирования не всегда дает
положительные результаты [56]. Поэтому можно предполагать,
что в его основе лежат также специфические явления, обусловлен¬
ные свойствами обрабатываемой воды и загрязнений. По-видимо¬
му, в условиях низких значений pH могут формироваться разви¬
тые полимерные структуры, на которые дальнейшее повышение
pH (за счет смешения с основным потоком воды) не оказывает
заметного действия.
VIII. 2.2.3. Прерывистое (периодическое) коагулирование
Этот вид коагулирования основан на более полном использовании
свойств продуктов гидролиза коагулянта при их избытке. Он со¬
держит элементы метода концентрированного коагулирования
и состоит в чередовании периодов подачи в обрабатываемую воду
увеличенных доз коагулянта с периодами полного прекращения
коагулирования [102].
Применение метода дает возможность значительно сократить
потребность в коагулянте, увеличить длительность фильтрацион¬
ных циклов на фильтрах и контактных осветлителях. Так, при об¬
работке маломутной воды по двухступенчатой схеме за счет со¬
вмещения концентрированного коагулирования с периодическим
достигнута экономия коагулянта на 30—40% [97, 99]. Рекомен¬
дуемая Бардиным [99] длительность периода «коагулирования»
равна 1—3 час, а соотношение длительностей «коагулирования»
и «некоагулирования» — от 3 : 1 до 0,3 : 1.
По данным Котлярова [91], прерывистое коагулирование во¬
ды, содержащей 13 мг/л взвешенных веществ, при одинаковой дли¬
тельности периодов «коагулирования» и «некоагулирования» —
0,5; 1 и 2 часа — позволяет при прямоточном фильтровании сни¬
зить дозу сульфата алюминия примерно в 2 раза. Еще более вы¬
сокая экономия коагулянта (50—80%) получена при одноступен¬
чатой очистке амурской воды [103].
Казанцевой [104, 105] и Вильсону [106] удалось с помощью
периодического коагулирования повысить степень обесцвечивания
воды. Применив этот метод в процессе прямоточного фильтрования,
274
ГЛАВА VIII. ИНТЕНСИФИКАЦИЯ КОАГУЛЯЦИИ
Казанцева установила, что улучшенное отделение окрашивающих
примесей обусловлено более низкими значениями pH в период
«коагулирования». Наилучший с точки зрения обесцвечивания
осадок коагулированной взвеси формировался в порах загрузки
контактного осветлителя при pH среды 6,0—6,5 [104]. В периоды
«некоагулирования» исходная вода по мере прохождения через за¬
грузку обогащается сульфатными ионами, ее щелочность и ве¬
личина pH уменьшаются, что свидетельствует о замене в продук¬
тах гидролиза анионов SO4- на ОН" и НСО3.
Уплотнение осадка в порах загрузки уменьшает его обесцве¬
чивающую способность. Эффективность метода прерывистого коа¬
гулирования понижается также при увеличении цветности обраба¬
тываемой воды [104].
VIII. 2.3. Рециркуляция осадка коагулированной взвеси
Обработка воды с возвратом части отработанного осадка в зону
ввода новых порций коагулянта в ряде случаев привела к зна¬
чительной (до 30%) экономии коагулянта и ускорению осаждения
коагулированной взвеси в отстойниках и осветлителях. Наблю¬
даемые эффекты являются следствием более полного использова¬
ния свойств коагулянта, ускоренного формирования хлопьев
в контакте с ранее выделенным осадком.
При известково-содовом умягчении воды преимущества ре¬
циркуляции осадка очевидны. Во всех описанных примерах ее
использования достигнуты положительные результаты [107—
109]. Плотность осадков в ходе рециркуляции возрастала в не¬
сколько раз.
Взвеси, образующиеся при коагулировании воды, обладают ма¬
лой прочностью и гораздо хуже, чем осадки, полученные при
умягчении, сохраняют свою поверхностную активность. Поэтому
попытки применить рециркуляцию таких взвесей не всегда за¬
канчивались успешно. Бесспорно только, что этот метод помогает
произвести зарядку осветлителей со взвешенным осадком. О целе¬
сообразности рециркуляции осадков в остальных случаях мнения
специалистов расходятся.
Существуют два способа рециркуляции — по внутреннему
и наружному контуру. Рециркуляция по внутреннему контуру
предусматривает возврат образовавшихся хлопьев в зону добав¬
ления раствора реагента без вывода их из рециркуляционной ем¬
кости. По этому принципу работают осветлители со взвешенным
осадком в США, ГДР, Франции и некоторых других странах.
Рециркуляция по наружному контуру включает отвод шламовой
жидкости из камер хлопьеобразования, отстойников, осветлите¬
лей или фильтров, частичное отстаивание ее (иногда с добавлением
реагентов), обеззараживание и возврат осадка в смеситель. От
VIII.2. БЕЗРЕАГЕНТНЫЕ МЕТОДЫ
275
выбранного способа рециркуляции, а также от использованной
для этой цели аппаратуры (насосы, эжекторы, эрлифты, турбин¬
ные и пропеллерные мешалки) зависит достигнутый технологи¬
ческий эффект.
Калинске [110] на основе опыта эксплуатации осветлителей
с рециркуляционными устройствами рекомендует поддерживать
расход рециркулирующей шламовой жидкости в пределах 3—5-
кратной производительности осветлителя, а максимальную ско¬
рость рециркуляции — не более 1,5 м/сек.
Для рециркуляции по наружному контуру Кингерли [111],
руководствуясь результатами исследований на Днепровском во¬
допроводе г. Киева, предлагает использовать отработанную коа¬
гулированную взвесь в дозах 20—40 мг/л; Блувштейн [112] по
результатам пусконаладочных работ на Владимирском водопро¬
воде — в дозах 150—200 мг/л; Мизра и др. [113] — в дозах 100—
400 мг/л. Паоло и др. [114] советуют к обрабатываемой воде добав¬
лять шламовую жидкость в количестве 0,6—5%, а Беляев и Ко¬
лодный [115] — в количестве 10% от расхода воды. Добавление
осадка к воде рекомендуется осуществлять перед вводом коагу¬
лянта, но иногда для перекачки осадка используют те же насо¬
сы, что и для дозирования раствора коагулянта. Известен также
способ предварительного добавления раствора коагулянта к
осадку [116].
Метод рециркуляции осадка используют главным образом для
интенсификации коагулирования маломутных вод, но имеются и
примеры, когда с помощью этого метода достигнуто заметное (око¬
ло 25%) ускорение осветления воды, содержащей до 20 г/л тонко¬
дисперсных примесей [113, 117].
Преимущества рециркуляции при обработке цветных вод и по¬
лучаемую экономию коагулянта объясняют обычно более полным
использованием сорбционных свойств продуктов гидролиза коа¬
гулянтов по отношению к органическим соединениям [112, 118,
119]. Как полагает, однако, Гадек [120], сорбционная емкость коа¬
гулятов алюминия и железа исчерпывается уже за первый цикл
коагулирования, а при рециркуляции осадка может наступить
десорбция органических веществ и ухудшиться качество очищен¬
ной воды.
Биин [121] высказал опасение в отношении вторичного загряз¬
нения воды бактериями. Опыт эксплуатации систем рециркуля¬
ции осадка и повторного использования сточных вод от промывки
отстойников и фильтров не дает оснований для этих опасений —
при наличии предварйтельного хлорирования вторичного загряз¬
нения воды не происходит [112, 118, 122].
Экономическая и технологическая эффективность рециркуля¬
ции осадка водопроводных станций подтверждена результатами
исследований Глена и др. [123].
276
ГЛАВА VIII. ИНТЕНСИФИКАЦИЯ КОАГУЛЯЦИИ
VIII. 2.4. Физические методы интенсификации
Вопросы, связанные с механизмом действия физических (без-
электролитных) методов коагуляции на дисперсные системы, об¬
суждаются в гл. II. Этот раздел посвящен оценке технологических
эффектов, достигаемых при совмещении указанных методов с об¬
работкой гидролизующимися коагулянтами.
VIII. 2.4.1. Наложение элетрического поля
Эффекты, наблюдаемые при наложении электрического поля на
дисперсную систему, коагулирующую под действием электролитов,
иногда трудно отделить от побочных процессов, сопровождающих
электролиз,— анодного растворения металла, выделения газов
и др. Тем не менее с полной очевидностью можно говорить об ин¬
тенсифицирующем действии — сильном или слабом — электри¬
ческого поля на коагуляцию дисперсных примесей воды и продук¬
тов гидролиза коагулянтов. По данным Кульского и др. [124,
125], это действие, названное «коагуляцией под током», проявляет¬
ся даже при очень малом напряжении на электродах (меньшем на¬
пряжения разложения воды).
Предпринятая нами попытка выделить наиболее важные для
практики результаты исследований по изучению действия электри¬
ческого поля как интенсификатора коагуляции привела к следую¬
щим выводам.
1. В поле постоянного тока ускоряются процессы формирова¬
ния и осаждения коагулированной взвеси, полученной при обра¬
ботке сульфатом алюминия мутных вод [126]; повышается степень
очистки воды от органических и неорганических примесей фильт¬
рованием [127, 128]; улучшается отделение эмульгированных жи¬
ров [129] и водорослей [130, 131]. С увеличением концентрации
взвешенных веществ и ростом напряженности электрического поля
эффективность обработки воды повышается [126, 130, 132].
2. Газы, выделяющиеся на электродах, облегчают условия
очистки воды за счет окисления органических примесей [133].
3. В электрическом поле постоянного тока ускоряется обезво¬
живание осадков сточных вод. Рекомендуемая плотность тока со¬
ставляет 0,01—0,02 а/м2, напряжение на электродах —50—60 в
[134, 135].
4. При использовании переменного тока промышленной час¬
тоты положительные результаты достигнуты в случае обработки
сильно минерализованных (до 6 г/л) мутных (до 3 г/л) шахтных сто¬
ков. Потребность в коагулянте снизилась на 30—40%.
Для снижения расхода энергии и получения более стабильных
результатов рекомендован способ обработки воды в электрическом
поле последовательно в несколько этапов с постепенным увеличе¬
VIII.2. БЕЗРЕАГЕНТНЫЕ МЕТОДЫ
277
нием напряженности поля от одного этапа к другому [136]. Пред¬
ложены электролизеры и фильтры с загрузкой из электропровод¬
ных материалов — активного угля и магнетита [137 —142].
VIII. 2.4.2. Наложение магнитного поля
Наиболее подробные исследования по изучению технологических
возможностей магнитной обработки в процессе коагулирования
воды гидролизующимися коагулянтами и подбору рационального
режима обработки выполнены Шаховым и сотр. [143—152]. Вы¬
воды, вытекающие из этих работ, можно систематизировать сле¬
дующим образом.
1. В отсутствие гидролизующегося коагулянта наибольший
эффект выделения взвеси из воды с общим солесодержанием 500—
600 мг/л и щелочностью 5,8—6,0 мг-экв/л после ее обработки в 12-
контурном магнитном генераторе (с чередованием полярности кон¬
туров) достигнут при напряженности магнитного поля около
400 а/см в течение 0,6 сек.
2. Совместное осаждение карбоната кальция и гидроокиси
магния, образующихся в ходе известкового умягчения воды, уско¬
ряется после проведения магнитной обработки воды примерно
в 1,5 раза. Оптимальные условия обработки: напряженность поля
200—300 а/см, скорость движения воды в рабочем зазоре генера¬
тора 0,5—1,0 м/сек.
3. Омагничивание изменяет свойства продуктов гидролиза
алюминия и железа:
а) уменьшаются структурно-механическая гидратация и дзе¬
та-потенциал частиц; максимальной степени снижения этих пара¬
метров у золя гидроокиси алюминия соответствует напряженность
поля 560 а/см;
б) на 30—40% возрастает сорбционная емкость продуктов гид¬
ролиза Коагулянтов по отношению к гуминовым веществам: для
Fe(OH)3 — с 284 до 379 мг/г (после обработки в поле напряжен ¬
ностью 480а/см), для А1(ОН)3 — с 348 до 494 мг/г (после обработ¬
ки в поле напряженностью 400 а/см.)
4. При очистке вод, содержащих минеральные взвеси, магнит¬
ная обработка дает следующие преимущества:
а) вызывает увеличение плотности и гидравлической крупности
хлопьев коагулированной взвеси;
б) позволяет повысить производительность отстойников и ос¬
ветлителей со взвешенным осадком;
в) приводит к некоторому снижению мутности осветленной во¬
ды.
Независимо от исходного содержания взвеси в обрабатываемой
воде (в пределах от нескольких до 250 мг/л), наилучшие резуль¬
таты достигаются при напряженности магнитного поля 80 а/см.
278
ГЛАВА VIII. ИНТЕНСИФИКАЦИЯ КОАГУЛЯЦИИ
Концентрация взвеси в контактной среде осветлителя возрастает
в 1,7—2,5 раза, объемный вес (по пикнометру) — с 1,027 до
1,043 г!см?. Наиболее высокий эффект дало омагничивание воды
с повышенной щелочностью и повышенным содержанием мине¬
ральных примесей. При концентрации примесей менее 250 мг/л
магнитная обработка не улучшила осветления.
5. Омагничивание цветной воды позволило увеличить объем¬
ный вес коагулированной взвеси (по пикнометру) с 1,023 до
1,041 г/см3 в случае Fe(OH)3 и с 1,019 до 1,035 г/см3 в случае
А1(ОН)3.
6. Для интенсификации коагуляционного процесса рекомен¬
дован следующий режим:
а) омагничивание воды за 10—60 сек до ввода раствора коагу¬
лянта;
б) скорость воды в рабочем зазоре магнитного генератора рав¬
на 1 м/сек;
б) количество знакопеременных магнитных контуров в гене¬
раторе равна 4—6;
г) длительность омагничивания 0,6—1 сек.
7. Расход электроэнергии при магнитной обработке составляет
5—8 вгп'Час на 1 м3/час производительности водоочистной уста¬
новки.
8. Возможно омагничивание лишь части (например, половины)
обрабатываемой воды с последующим смешением ее (до ввода коа¬
гулянта) с остальной водой.
9. Интенсифицировать коагуляцию можно путем магнитной
обработки раствора коагулянта. Эффект активации раствора за¬
висит от напряженности магнитного поля.
В экспериментах, проводимых другими исследователями па¬
раллельно с работами Шахова и сотрудников, получены противо¬
речивые результаты. По данным Щукиной [153], магнитная обра¬
ботка не оказывает никакого влияния на коагуляцию суспензий
каолина (200 —6000 мг/л) сернокислым алюминием и хлорным же¬
лезом. Зато, как показал Дроздов [154], обработка воды в поли-
градиентном магнитном поле в присутствии FeCl3 и извести привела
к резкому ускорению хлопьеобразования. Бартник и др. [155,
156] получили наилучшие результаты при омагничивании воды
после образования зародышевых хлопьев коагулированной взве¬
си и объяснили эти результаты взаимным притяжением разно¬
именно заряженных участков хлопьев.
В 1968—1969 гг. в ЦНИИ МПС проведены исследования [157]
по магнитной обработке воды, содеря<ащей разные количества за-
мутнителя (25—250 мг/л) и коагулированной разными дозами
A12(S04)3 и РеС13 при одинаковых конечных значениях pH среды
(6,4—6,7). Установлено, что интенсифицирующее действие маг¬
нитного поля достигает максимума при очистке вод средней мут¬
VIII.2. БЕЗРЕАГЕНТНЫЕ МЕТОДЫ
279
ности. Процессы формирования и осаждения коагулированной
взвеси заканчивались в 1,5—2 раза быстрее, чем без омагничива-
ния, оптическая плотность осветленной воды возросла в 1,5 —
2,5 раза. Достигнута существенная экономия коагулянта — в
2—3 раза. Оптимальная напряженность магнитного поля в этих
экспериментах независимо от исходных условий составляла при¬
близительно 400 а/см.
Большой интерес представляет магнитная обработка суспен¬
зий, содержащих ферромагнитные примеси: стальную пыль,
Fe304, 6-FeOOH, Y-Fe203 [158—160]. При очистке сточных вод
мартеновских печей и конвертеров этот способ позволяет при на¬
пряженности магнитного поля 800—1200 а/см повысить в 1,5 ра¬
за производительность очистных сооружений и снизить в 2—10 раз
остаточные концентрации примесей. Магнитное коагулирование
сточных вод газоочисток сталеплавильного и доменного производ¬
ства оказалось эффективным при концентрации твердых приме¬
сей не менее 1 г/л и проходило наиболее успешно в полиградиент-
ном поле. Разработаны разные типы магнитных коагуляторов на
электрических и постоянных магнитах [161—163]. Магнитное по¬
ле оказывает также интенсифицирующее действие на процесс ос¬
ветления сточных вод, содержащих гидроокись железа [164].
VIII. 2.4.3. Воздействие ультразвуком
Имеются примеры успешного использования коагулирующего
действия ультразвука (УЗ) при обработке водных суспензий кар¬
боната кальция, кварца и угля, красителей, волокон целлюло¬
зы, золя двуокиси марганца [165—169]. Оптимальные частоты ле¬
жат в диапазоне от нескольких килогерц до 1 мгц.
В некоторых случаях хорошие результаты достигаются при ис¬
пользовании в помощь гидролизующимся коагулянтам низкоча¬
стотных (50 гц) механических колебаний. При этом происходит
ускорение седиментации частиц каолиновой суспензии пример¬
но в 2 раза [170].
Как и при наложении электрического и магнитного полей,
применение УЗ не всегда дает положительные результаты. На¬
пример, озвучивание с частотой 1 мгц каолинитовой суспензии, об¬
работанной сульфатом алюминия, привело к дополнительному
снижению остаточной мутности суспензии в 2,5—3,2 раза [171].
В других экспериментах озвучивание воды (0,5 и 8 кгц), содержа¬
щей коагулированную взвесь, дало те же результаты, что и про¬
стое механическое перемешивание, произведенное в оптимальном
режиме [172]. По-видимому, при выборе параметров ультразву¬
ковой обработки необходимо иметь в виду диспергирующее дей¬
ствие УЗ на водные примеси. Рюмин [166] показал, что озвучива¬
ние суспензии красителя (частота колебаний 23,7 кгц, длитель¬
280
ГЛАВА VIII. ИНТЕНСИФИКАЦИЯ КОАГУЛЯЦИИ
ность озвучивания 3 мин) уменьшает количество не только мелких,
но и крупных частиц, а содержание частиц средних размеров воз¬
растает. О разрушении некоторой доли крупных частиц полидис-
персной системы под действием УЗ свидетельствуют материалы,
собранные Бергманом [167].
В результате разрушающего действия УЗ погибают некоторые
бактерии, зоопланктон и водоросли [165, 173 (стр. 241)], разру¬
шаются фенолы [174]. Это содействует успешной коагуляции. Од¬
нако разрушение механических примесей, уменьшающее степень
полидисперсности суспензий, может оказать отрицательное влия¬
ние. Нужно также иметь в виду, что под действием УЗ из воды вы¬
деляются пузырьки газов, которые могут оказаться защемленны¬
ми в хлопьях коагулированной взвеси и вызвать их флотацию.
Исследования, выполненные во ВНИИ ВОДГЕО по ультра¬
звуковой коагуляции различных суспензий, привели к заключе¬
нию, что наилучшие результаты имеют место при относительно
низких частотах УЗ — 8 и 18 кгц\ с увеличением интенсивности
УЗ скорость коагуляции возрастает; оптимальная продолжитель¬
ность озвучивания составляет 1 —3 мин\ существует оптимальная
концентрация твердой фазы, при которой коагулирующее дейст¬
вие УЗ максимально; в условиях проведенных экспериментов она
находится в пределах 5—12 вес. % [175].
Как показывают эксперименты, коагулирующее действие УЗ
определяется не только его частотой и мощностью, но также ам¬
плитудой колебаний, направлением распространения звуковых
волн, свойствами суспендированных частиц. В случае ферромаг¬
нитных материалов — Fe(OH)2,Fe(OH)3 и Fe304 — ультраозву¬
чивание с частотой 1 мгц приводит к многократному увеличению
скорости осаждения твердой фазы [176].
Предложены многочисленные способы практического приме¬
нения УЗ на водоочистных сооружениях [177—179]. Все они пре¬
дусматривают обработку воды после добавления раствора коагу¬
лянта.
VIII. 2.4.4. Ионизирующее облучение
За последние годы по радиационной обработке воды накоплен зна¬
чительный экспериментальный материал, касающийся, главным
образом, очистки сточных вод. Из этих материалов видно, что дей¬
ствие ионизирующих у-, р- и рентгеновских излучений преимуще¬
ственно сводится к интенсификации окисления растворенным в
воде кислородом органических и минеральных примесей, в ре¬
зультате чего имеют место следующие явления [180—192]: обез¬
зараживание воды; разложение синтетических ПАВ, фенолов,
цианидов и других медленно окисляющихся соединений; дезодо¬
рация воды; ускорение осаждения взвешенных примесей; обес-
VIII. ЛИТЕГАТУРА
281
цвечивание воды; улучшение обезвоживания осадков. Все перечи¬
сленные процессы облегчают обработку воды гидролизующимися
коагулянтами, расширяют сферу их применения. В качестве ис¬
точников излучения используют большей частью 60Со и 137Cs,
реже 90Sr, 204Т1, 147Рш, 152Еи и др. Как правило, для достижения
перечисленных технологических эффектов достаточны дозы об¬
лучения 1*104—1•10е рад.
По данным некоторых авторов, эффективность облучения зо¬
лей не зависит от знака заряда частиц и ряда других исходных ус¬
ловий [193]. Однако положительные результаты и их воспроизво¬
димость достигаются все же не всегда. Описаны даже случаи,
когда радиационная обработка приводила к стабилизации золей
[180, 186]. В качестве наиболее эффективного средства для интен¬
сификации действия ионизирующих излучений хорошо себя за¬
рекомендовало в экспериментах пневматическое перемешивание,
обогащающее воду кислородом [180].
Большого внимания заслуживают результаты экспериментов
Брегвадзе и др. [194, 195] по 7-облучению (мощностью 800 рад/сек
и дозой 3—5 Мрад) растворов сернокислого алюминия. Авторы
указывают, что после такого облучения раствор коагулянта акти¬
вируется и скорость хлопьеобразования возрастает в 2—3 раза.
Эффект, вероятно, состоит в воздействии радиации на структуру
ионных и молекулярных ассоциатов [196].
Облучение не вызывает ядерных превращений в растворах коа¬
гулянтов и образцах вод и, следовательно, безопасно [188, 195].
Литература
1. Е. Д. Бабенков. Автореферат канд. дисс. МИСИ им. В. В. Куйбышева,
1966.
2. R. С. Hoather. Water and Water Engng, 65, 525 (1961).
3. Т. M. Riddick, N. Lindsay et al. Water and Sewage Works, 105, 15 (1958).
4. A. A. Hirsch. J. Amer. Water Works Assoc., 54 1531 (1962).
5. Water and Wastes Engng, 7, N 5, 52 (1970).
6. 11. JI. Ращук, В. А. Шипунов. Изв. вузов. Строительство и архитекту¬
ра, № 7, 152 (1971).
7. В. А. Клячко, И. Э. Апелъцин. Подготовка воды для промышленного
и городского водоснабжения. М., Стройиздат, 1962.
8. Н. Л. Ращук, А. Л. Рабинович. Водоснабжение и санитарная техника,
№ 7, 29 (1966).
9. В. М. Колбасов, Г. В. Топилъский, Ю. М. Бугпт. ЖПХ, 43, 1291 (1970).
10. М. X. Roberts. J. Appl. Chem., 7, 543 (1957).
11. К. Цукамото. Ёсуйто Хайсуй, 15, 477 (1973); РЖХим., 23И382 (1973).
12. М. И. Медведев, А. Г. Шабалина. Сб. «Нефтепереработка и нефтехимия»,
вып. 9. Киев, «Наукова думка», 1973, стр. 50.
13. Бельг. пат. 641033 (1969).
14. Е. С. Абазаев. Водоснабжение и санитарная техника, № 6, 4 (1956).
15. R. К. Ham, R. F. Christman. J. Sanit. Engng Div. Proc. ASCE, N 3, 481
(1969).
282
ГЛАВА Vllt. ИНТЕНСИФИКАЦИЯ КОАГУЛЯЦИИ
16. Г. П. Рыхликов. Научные труды Омского сельскохоз. ин-та, вып. 82,
86 (1971).
17. Н. С. Лебедева. Автореферат канд. дисс. М., ВНИИ ВОДГЕО, 1954.
18. Z. Nov&k. Wasserwirtschaft—Wassertechnik, 7, 432 (1957).
19. В. Т. Турчинович. Улучшение качества воды. М.— Л., Стройиздат,
1940, стр. 41.
20. J. С. Dean, I. L. Dickinson. Appita, 24, 118 (1970).
21. S. P. Hansen, Ch. W. Botsford et al. Пат. США 3482695 (1969).
22. A. P. Black, J. E. Singley, G. P. Whittle et al. J. Amer. Water Works
Assoc., 55, 1347 (1963).
23. S. T. Powell, L. V. Carpenter, L. R. Setter et al. Chem. and Metallurg.
Engng, N 8, 481 (1939).
24 Г. Бэббит, Дж. Доланд. Водоснабжение. М., Стройиздат, 1958.
25. Water and Water Engng, 66, 416 (1962).
26. В. А. Клячко, О. Н. Шемякина. Авт. свид. СССР 325215 (1967); Бюлл.
изобр., № 3 (1972).
27. Е. Е. Драхлин. Водоснабжение и санитарная техника, № 1, 5 (1972).
28. F. Baer, К. Xylander. Пат. ФРГ 1517730 (1975).
29. Y. Aiba, Т. Furumori. Пат. США 3544476 (1970).
30. Е. Д. Бабенков. Оптимальная доза коагулянта при очистке воды. М.,
«Транспорт», 1974.
31. Сб. «Коагулянты для очистки питьевой воды». Под ред. В. Т. Турчи-
новича. М., Изд-во МКХ РСФСР, 1948, стр. 17.
32. М. Frison. Techn. sanit. et municip., 48, 158 (1953).
33. M. R. Buydens. Ibid., p. 139.
34. А. С. Коган. Интенсификация работы городского водопровода. М.,
Изд-во МКХ РСФСР, 1955, стр. 88.
35. S. Kalandra, J. Jandl et al. Пат. ЧССР 125459 (1965).
36. R. В. Crieves, С. J. Crandall. Water and Sewage Works, 113, 432 (1966).
37. R. B. Crieves, S. M. Schwartz. J. Amer. Water Works Assoc., 58, 1129
(1966).
38. G. H. Spaulding, H. N. Lowe, R. P. Shmitt. J. Amer. Water Works
Assoc., 43, 793 (1951).
39. J. Kelus. Gaz, woda i techn. sanit., 34, 19 (1960).
40. Water Works Engng, 113, 712 (1960).
41. H. N. Lowe. Water and Sewage Works, 104, 99 (1957).
42. Т. M. Riddick. Пат. США 3097163 (1963).
43. Т. M. Riddick. J. Amer. Water Works Assoc., 53, 1007 (1961).
44. R. Richards. Пат. США 3575854 (1971).
45. L. Szymanski, J. Grejner. Пат. ПНР 47884 (1967).
46. J. D. Eye, Т. K. Basu. J. Water Pollut. Contr. Fed., 42, 121 (1970).
47. А. И. Шахов, С. С. Душкин. Водоснабжение и санитарная техника,
№ 12, 1 (1971).
48. W. Meins. Kommunalwirtschaft, 6, 216 (1975).
49. W. Farnham. Пат. США 2964466 (1960).
50. Water and Sewage Works, 118, 12 (1971).
51. I. D. Ionescu, S. Constantinescu et al. Bull. Inst, politechn. Iasi, 16,
2/187 (1970).
52. К. Моридзаки, М. Ватанабэ. Яп. пат. 49-49069 (1974).
53. Франц. пат. 2071027 (1971).
54. Е. Д. Бабенков. Сб. «Методы очистки и контроля качества воды». М.,
«Транспорт», 1966, стр. 15.
55. В. Г. Печников, Е. Г. Хайлов. Сб. «Технология очистки питьевой воды
г. Москвы». М., Стройиздат, 1974, стр. 88.
56. J. D.[Griffith, R. G. Williams. J. Amer. Water Works Assoc., 64, 825 (1972).
57. E. Ф. Кургаев, E. Д. Бабенков, В. E. Гуртовенко И др. Авт. свид.
СССР 182602 (1965); Бюлл. изобр., № 11 (1966).
VIII. ЛИТЕРАТУРА
283
58. Е. Д. Бабенков, Д. С. Щербаков. Водоснабжение и санитарная техника,
№ 5, 7 (1967).
59. А. К. Никитин. Теоретические основы хим. технологии, 4, 700 (1970).
60. L. Vrale, R. М. Jorden. J. Amer. Water Works Assoc., 63, 52 (1971).
61. А. И. Пацюков. Сборник научных трудов ГИСИ им. В. П. Чкалова,
вып. 58. Горьк. кн. изд-во, 1971, стр. 106.
62. Е. Ф. Кургаев. Основы теории и расчета осветлителей. М., Стройиздат,
1962.
63. Е. Н. Тетеркин, Н. С. Лебедева. Физические и технологические свой¬
ства взвесей, образующихся при обработке воды. М., Стройиздат, 1952.
64. В. И. Маркизов. Очистка воды коагулянтами. М., Изд-во МКХ РСФСР,
1954, стр. 12.
65. Я. Л. Файншиль. Автореферат канд. дисс. М., ВНИИ ВОДГЕО, 1970.
66. G. J. Hazey. WaterWorks Engng, 116, 195 (1963).
67. G. A. Harris, R. J. Ringwood. Public Works, 101, N 1, 65 (1970).
68. G. E. Wilcomb. J. Amer. Water Works Assoc., 24, 1416 (1932).
69. R. J. Те Kippe, R. K. Ham. J. Amer. Water Works Assoc., 63, 439 (1971).
70. П. E. Богомазов. Смеситель со спиральной струей внутрь. Ростов-на-
Дону, 1934.
71. М. Pirnie. Engng News-Becord, 90, 883, (1923).
72. G. Cambosu. Instituto d’igiene Dell’universita di Parma. Parma, 1954.
73. А. И. Егоров, И. С. Морозова, Р. В. Поспелова. Труды ВНИИ ВОДГЕО,
вып. 15. М., 1967, стр. 90.
74. И. С. Морозова. Труды ВНИИ ВОДГЕО, вып. 25. М., 1970, стр. 64.
75. А. И. Егоров, И. С. Морозова. Труды ВНИИ ВОДГЕО, вып. 48. М.,
1975, стр. 16.
76. И. И. Жуков. Избранные труды. М., Изд-во АН СССР, 1952, стр. 234.
77. Е. Д. Бабенков. Вестник ВНИИ железнодор. транспорта, № 4, 45 (1966).
78. В. И. Классен, В. А. Мокроусов. Введение в теорию флотации. М.,
Металлургиздат, 1953.
79. Т. R. Camp. Trans. Amer. Soc. Civil Engrs, 120, 1 (1955).
80. А. М. Годэн. Флотация. М., Госгортехиздат, 1959,
81. Н. М. Stark. J. Amer. Chein. Soc., 52, 1930 (1930).
82. W. Heller, J. Peters. J. Colloid and Interface Sci., 32, 592 (1970).
83. J. Peters, W. Heller. J. Colloid and Interface Sci., 33, 578 (1970).
84. Б. R. Дерягин. Пятая Всесоюзная конференция по коллоидной химии (те¬
зисы докладов). М., Изд-во АН СССР, 1962, стр. 14.
85. Б. В. Дерягин, С. С. Духин. Коллоидн. ж., 21, 37 (1959).
86. С. С. Духин, Б. В. Дерягин. Коллоидн. ж., 22, 587 (1960).
87. А. А. Кастальский. Проектирование устройств для удаления из воды
растворенных газов в процессе водоподготовки. М., Госстройиздат, 1957.
88. P. Haney. J. Amer. Water Works Assoc., 50, N 9 (1958).
89. W. Schafer. Vom Wasser 1967, Bd. 34. Weinheim, 1968, S. 443.
90. М. H. Hatchinson, Т. K. Sherwood. Engng Chem., 29, 836 (1937).
91. А. С. Котляров. Сб. «Вопросы технологии обработки воды промышлен¬
ного и питьевого водоснабжения», вып. II. Киев, «Буд1вельник», 1969,
стр. 86.
92. Е. Ф. Кургаев, Б. Н. Фрог, Э. И. Кирсанова, Т. П. Лимонова. Тепло¬
энергетика, № 4, 48 (1973).
93. Т. М. Riddick. Пат. США 3075645 (1958).
94.- Е. Д. Бабенков. Водоснабжение и канализация, вып. 11. М., изд.
ЦБТИ МКХ РСФСР, 1967, стр. 74.
95. R. A. Trelles, D. J. Bengolea, A. G. Poccard. J. Amer. Water Works
Assoc., 32, 742 (1940).
96. N. J. Pugh. J. Inst. Water Engrs, 11, 17 (1957).
97. Ю. А. Бардин. Труды Поволжской конференции. Горький, изд. научно-
техн. об-ва санитарной техники и городского хозяйства, 1957, стр. 85.
284
ГЛАВА VIII. ИНТЕНСИФИКАЦИЯ КОАГУЛЯЦИИ
98. А. Н. Сардановский, Л. П. Казакова. Водоснабжение и санитарная тех¬
ника, № 4, 9 (1955).
99. Ю. А. Бардин. Научные труды АКХ им. К. Д. Памфилова, вып. 52.
Водоснабжение, № 5. М., ОНТИ, 1969, стр. 56.
100. И. А. Дриз. Водоснабжение и санитарная техника, № 12, 10 (1958).
101. И. А. Дриз. Водоснабжение и санитарная техника, № 1, 39 (1969).
102. Д. Бонев. Гидротехника и мелиорация, № 1, 13 (1966).
103. Г. И. Воловик. Труды Хабаровск, ин-та инженеров железнодор. тран¬
спорта, вып. 40, 225 (1971).
104. Л. 3. Казанцева. Автореферат канд. дисс. Челябинский политехи, ин-т,
1967.
105. Т. И. Барышникова, Л. 3. Казанцева. Изв. вузов. Строительство и архи¬
тектура, № 3, 87 (1965).
106. A. L. Wilson. J. Appl. Chem., 10, 377 (I960).
107. Е. Ф. Кургаев. Паровозное и вагонное хозяйство, № 2, 27 (1941)
108. F. V. Frissora. Ind. and Water Engng, 4, 26 (1967).
109. Ch. H. Lawrance. J. Amer. Water Works Assoc., 55, 177 (1963).
110. A. A. Kalinske. Civil Engng, N 12, 60 (1960).
111. А. Д. Кенгерли. Уч. зап. Азерб. политехи, ин-та. Серия X, № 1 (13).
Баку, 1969, стр. 133.
112. М. М. Блувштейн. Повышение эффективности работы очистных соору¬
жений водопровода. М., Стройиздат, 1971, стр. 120.
113. N. D. Misra, G. J. Padma Rao. J. Indian Pulp and Paper, 22, 411 (1968).
114. P. E. Paolo, B. J. Schwartz, L. K. Wang. Water and Sewage Works,
119, 123 (1972).
115. И. И. Беляев, Ю. И. Колодный. Гигиена и санитария, № 1, 108
(1968).
116. Z. Novak, М. Karpisek. Vodni hospod., 15, 309 (1965).
117. М. Adhikari, S. К. Gupta, R. D. Biswas. Bes. and Ind., 19, 9 (1974).
118. А. Д. Кенгерли. За технический прогресс, № 4, 38 (1970).
119. Л. А. Кулъский, М. В. Демура. Использование промывной воды фильт¬
ров и осадков отстойников в технологическом процессе очистки воды.
Киев, изд. Научно-технического общества санитарной техники и город¬
ского хозяйства, 1958.
120. J. Hddek. Voda, 35, N 12, 392 (1956).
121. Е. L. Bean. Bull. Engng and Archit., N 56, 46 (1966).
122. Ю. Б. Багоцкий, А. А. Борзакова. Водоснабжение и санитарная техни¬
ка, № 9, 8 (1962).
123. R. W. Glenn, J. F. Judkins, J. Morgan. J. Amer. Water Works Assoc.,
65, 414 (1973).
124. Л. А. Кулъский. Теоретические основы и технология кондициониро¬
вания воды. Киев, «Наукова думка», 1971, стр. 137.
125. И.\Т. Гороновский. Автореферат канд. дисс. Киев, ИОНХ АН УССР,
1955.
126. Т. Кубота, Ф. Такэи и др. Яп. пат. 3506 (1965).
127. К. Кубо. Яп. пат. 47-28596 (1972).
128. И. С. Лавров, В. И. Барабанов, Р. А. Окунев. Труды Ленингр. инж.-
строит. ин-та, вып. 75, 54 (1973).
129. J. P. Hess, L. Clegg. Англ. пат. 859417 (1961).
130. В. И. Журавлев. Сб. «Научные основы технологии очистки воды». Киев,
«Наукова думка», 1973, стр. 18.
131. Е. С. Мацкевич, Л. А. Кулъский, В. И. Журавлев. Авт. свид. СССР
371172 (1969); Бюлл. изобр., № 12 (1973).
132. В. P. Gdcsine. Magyar kem. folyoirat, 69, N 5, 191 (1963).
133. J. G. Noble. Process Biochem., 8, 19 (1973).
134. Л. E. Волков, И. С. Лавров, В. Н. Пономарева и др. Труды Ленингр.
инж.-строит, ин-та, вып. 75, 105 (1973).
VIII. ЛИТЕРАТУРА
285
135. Н. К. Антоневич, Л. Е. Волков, И. С. Лавров и др. Там же, стр. 112.
136. И. А. Веребрюсов, И. С. Лавров, Г. А. Пузырев и др. Авт. свид. СССР
346237 (1970); Бюлл. изобр., № 23 (1972).
137. Ю. В. Кузьменко. Авт. свид. СССР 230079 (1967); Бюлл. изобр., № 34
(1968).
138. P. Lutsch. Wasser, Luft und Betrieb, 12, 702 (1968).
139. H. А. Масленников. Научные труды АКХ им. К. Д. Памфилова, вып. 20.
Городская канализация, № 2. М.— Л., ОНТИ, 1963, стр. 97.
140. Е. В. Гребеневич. Научно-теоретическая конференция аспирантов
АКХ им. К. Д. Памфилова. М., ОНТИ, 1966, стр. 20.
141. A. S. King. Пат. США 3801482 (1974).
142. 3. Я. Ярославский, В. М. Корнилов, И. С. Лавров и др. ЖПХ, 48, 650
(1975).
143. А. И. Шахов, А. Н. Музыченко и др. Водоснабжение и санитарная тех¬
ника, № 11, 6 (1963).
144. А. И. Шахов, А. В. Ширяева, А. С. Аветисов. Изв. вузов. Энергетика,
№ 12, 112 (1966).
145. А. И. Шахов, А. С. Аветисов. Сб. «Водоснабжение, канализация, гид¬
ротехнические сооружения», вып. 13. Киев, «Буд1велышк», 1971, стр. 17.
146. А. И. Шахов, С. С. Душкин. Сб. «Коммунальное хозяйство», вып. 1.
Киев, «Буд1вельник», 1964, стр. 102.
147. А. И. Шахов, С. С. Душкин. Сб. «Вопросы технологии обработки воды
промышленного и питьевого водоснабжения», вып. 1. Киев, «Буд1вель-
ник», 1969, стр. 53.
148. А. И. Шахов, С. С. Душкин. Там же, стр. 48.
149. А. И. Шахов, С. С. Душкин. Сб. «Наука и техника в городском хозяй¬
стве», вып. 5. Киев, «Буд1вельник», 1966, стр. 147.
150. А. И. Шахов, С. С. Душкин. Сб. «Энергетика и электротехническая
промышленность», вып. 1 (17). Киев, изд. Научно-техн. об-ва энерго¬
промышленности УССР, 1964, стр. 54.
151. А. И. Шахов. Прогрессивные методы очистки природных и сточных
вод (материалы научно-технической конференции), сб. № 2. М., изд.
МИСИ им. В. В. Куйбышева, 1971, стр. 107.
152. А. И. Шахов, С. С. Душкин. Сб. «Санитарная техника», вып. 11. Киев,
«Буд1вельник», 1971, стр. 130.
153. Л. Н. Щукина. Труды ВНИИ ВОДГЕО, вып. 40. ч. 1. М., 1972 (1973),
стр. 145.
154. Ф. П. Дроздов. Изв. вузов. Энергетика, № 7, 60 (1973).
155. J. A. Bartnik. Iron and Steel Engrs, 46, N 3, 106 (1969).
156. J. A. Bartnik, A. F. San Miguel. Англ. пат. 1240970 (1971).
157. E. Д. Бабенков, Э. И. Кирсанова, В. Е. Гуртовенко. Труды ЦНИИ МПС,
вып. 468. М., «Транспорт», 1972, стр. 69.
158. Б. Т. Гусев, Е. А. Леонтьев. Доклады научно-технической конференции
по итогам научно-исследовательских работ за 1968—1969 гг. Московско¬
го энергетического института. Секция теплотехническая. М., 1969, стр.[[66.
159. J. Gega, J. Wrobel. Zesz. nauk AGH, N 356, 197 (1972).
160. М. Сува, Я. Сузуки, С. Сузукияр$. Когё ёсуй, № 176, 42 (1973); РЖХим.,
4И61 (1974).
161. Г. М. Левин, Г. С. Пантелят, К. Ш. Шустер и др. Научные труды Все¬
союзного научно-исследовательского и проектного института по очистке
и технологии газов, сточных вод и использованию вторичных энерго¬
ресурсов предприятий черной металлургии, вып. 15. Харьков, 1972,
стр. 88.
162. Я. А. Карелин, О. С. Хабаров. Водоснабжение и санитарная техника,
№ 12, 13 (1969).
163. В. А. Холодный, И. Н. Ожиганов. Водоснабжение и санитарная техника,
№ 3, 1 (1970).
286
ГЛАВА VIII. ИНТЕНСИФИКАЦИЯ КОАГУЛЯЦИИ
164. Я. Сузуки, С. Сузуки, Т. Фуйи. Когё ёсуй, № 196, 50 (1975); РЖХим.,
18И41 (1975).
165. Л. Н. Фалъковская. Водоснабжение и канализация (новая техника жи¬
лищно-коммунального хозяйства), серия IV, вып. 3. М., Стройиздат,
1964, стр. 47.
166. В. П. Рюмин. Автореферат канд. дисс. МХТИ им. Д. И. Менделеева,
1964.
167. Л. Бергман. Ультразвук и его применение в науке и технике. М., ИЛ,
1957.
168. Т. П. Дорош, В. В. Сало, П. А. Егоров и др. Сб. «Обогащение полезных
ископаемых», вып. 8. Киев, «Технша», 1971, стр. 88.
169. S. Prakash, О. Prakash. Kolloid Z. und Z. Polymere, 188, 136 (1963).’
170. E. K. Obiakor, R. L. Whitmore. Nature (Engl.), 205, N 4969, 381 (1965).
171. Л. А. Кулъский, H. H. Биневская. Сб. «Физико-химическая механика
и лиофильность дисперсных систем». Киев, «Наукова думка», 1968, стр.
стр. 291.
172. С. Я. Стародубцев. Водоснабжение и санитарная техника, № 3, 4 (1942).
173. С. Н. Черкинский, Н. II. Трахтман. Обеззараживание питьевой воды
М., Медгиз, 1962.
174. Г. С. Топилъский, П. А. Егоров и др. Укр. хим. ж., 34, 853 (1968).
175. В. Е. Завьялов, В. Д. Петрушкин. Труды ВНИИ ВОДГЕО, вып. 40, ч. 1.
М., 1972 (1973), стр. 141.
176. Л. В. Юрьева, Э. Е. Элик, С. Ш. Гохман. Труды Уральск, лесотехн. ин-
та, вып. 31, 123 (1973).
177. М. Posa, I. Neagu. Пат. СРР 52355 (1970).
178. J. Klose, J. Schmidt et al. Пат. ГДР 75277 (1970).
179. Франц. пат. 1242377 (1960).
180. П. И. Долин, В. Н. Шубин, С. А. Брусенцева. Радиационная очистка
воды. М., «Наука», 1973.
181. A. I. Mytelka. Пат. США 3553089 (1971).
182. В. L. Lenz, Е. S. Robbins et al. Pulp and Paper Mag. Canada, 72, 75 (1971).
183. D. S. Balantine, L. A. Miller. Chem. Engng Progr. Symp. Ser., 66, 187
(1970).
184. D. D. Woodbridge, L. A. Mann, W. R. Garret. Bull. Environ. Contam. and
Toxicol., 7, N 2—3, 80 (1972).
185. A. H. Vajdic. J. Amer. Water Works Assoc., 63, 459 (1971).
186. R. H. Andrews, M. R. Fielding. Isotop. and Badiat. Technol., 8, 452 (1971).
187. F. D. Miraldi, E. J. Morgan. Пат. США 3603788 (1971).
188. M. Gerrard. Isotop. and Badiat. Technol., 8, 429 (1971).
189. D. S. Balantine. Ibid., p. 415.
190. H. R. Spragg. Пат. США 3677935 (1972).
191. D. M. Compton, S. J. Black et al. Nucl. News (USA), 13, N 9, 58 (1970).
192. И. Э. Апелъцин, В. А. Клячко, E. Ф. Золотова и др. Водоснабжение и са¬
нитарная техника, № 5, 8 (1973).
193. Е. М. Нанобашвили, Н. А. Бах. Сборник работ по радиационной химии.
М., Изд-во АН СССР, 1955, стр. 123.
194. У. Д. Брегвадзе, Г. С. Чубинидзе. Доклады научно-технической конферен¬
ции по использованию ионизирующих излучений в народном хозяйстве,
вып. 3. Тула, Приокское кн. изд-во, 1970, стр. 137.
195. У. Д. Брегвадзе. Сб. «Радиационная обработка пищевых продуктов».
М., Атомиздат, 1971, стр. 178.
196. М. М. Крыжановский, Ю. В. Ласточкин, В. Е. Миронов. ЖПХ, 42, 929
(1969).
Глава IX
ФЛОКУЛЯЦИЯ И ФЛОКУЛЯНТЫ
Под флокуляцией в коллоидной химии понимают агрегацию
частиц, в которой в дополнение (а иногда взамен) к непосредствен¬
ному контакту частиц происходит их взаимодействие через моле¬
кулы адсорбированного флокулянта [1, 2]. В этом состоит суще¬
ственное отличие процесса флокуляции от коагуляции.
Флокуляция является наиболее эффективным способом интенси¬
фикации очистки воды гидролизующимися коагулянтами. Приме¬
няемые в практике водоподготовки флокулянты — это неоргани¬
ческие или органические высокомолекулярные соединения. Из
неорганических флокулянтов широко используется активная
кремне кислота, из органических—разного рода природные и син¬
тетические ВМВ. По сложившейся традиции высокомолекулярны¬
ми флокулянтами (ВМФ) называют обычно только органические
продукты, но надо иметь в виду, что и кремнекислота в процессе
приготовления образует полимерные соединения.
IX. 1. АКТИВНАЯ КРЕМНЕКИСЛОТА
Хотя свойства геля кремнезема были известны еще в прошлом
веке, первооткрывателями флокулирующего действия растворов
нейтрализованных кремнеземов по праву считают Бейлиса и не¬
зависимо от него Грефа и Шуорма [3, 4 , 5 (стр. 6)], доказавших эф¬
фективность этих растворов в производственных масштабах. «Вы¬
сокая эффективность, дешевое и недефицитное сырье, полная без¬
вредность золей кремнекислоты для здоровья человека,— пишет
Кульекий [6, стр. 46],— обеспечивают им особое место среди фло¬
кулянтов, используемых при подготовке воды».
В СССР первые исследования по активации и использованию
активной кремнекислоты (АК) проводились Будриным [7], Позд¬
няковой [8], затем Кульским, Слипченко, Накорчевской и др. [6,
9—24], Первовым 125, 26], Гервицем [27—30] и др. За рубежом по¬
добные работы проводили Блэк [31], Крист [32], Келюс [33] и др.
Имеются сведения о применении АК на некоторых крупных водо¬
проводах США, Англии, Канады, ФРГ и ГДР, Швеции, Испании,
Финляндии, Норвегии, Японии [12, 19 (стр. 122), 33]. В СССР
промышленное использование АК наложено на пяти водопроводах
[19 (стр. 108), 30].
288
ГЛАВА IX. ФЛОКУЛЯЦИЯ И ФЛОКУЛЯНТЫ
IX. 1.1. Методы приготовления (активации)
Обычно в качестве сырья для производства АК применяют жид¬
кое стекло — водный раствор силиката натрия, содержащий 22,9—
39% Si02 и 8,6—14,6% Na20. Предлагается также использовать
силикат кальция и.трисиликат магния [34].
Формирование золей АК, способных флокулировать гидрооки¬
си металлов и взвешенные примеси воды, достигается нейтрализа¬
цией щелочности растворов силикатов путем кислотной обработ¬
ки. При этом кремнезем выделяется (конденсируется) в виде золя
монокремниевой кислоты, который, полимеризуясь со временем,
образует гель. Процесс состоит в дегидратации молекул и форми¬
ровании силоксановых связей
ОН ОН
I I
2Si (ОН)* -» НО—Si—О—Si—ОН + Н20
I I
ОН он
При этом значительная часть силанольных групп [Si(OH)n] со¬
храняется [35].
Помимо реагентов, обусловливающих только понижение pH
растворов силикатов — кислот и их ангидридов, для активации
используют соли, которые способны образовывать при реакции
малорастворимые соединения. К ним относятся кислые алюминий-
и железосодержащие коагулянты, алюминат натрия, оксихлорид
алюминия и др. Реакции активации разными реагентами приведены
в работах [19 (стр. 16), 36, 37 (стр. 149)].
Наиболее перспективными активаторами считают хлор, сер¬
ную кислоту, сульфат алюминия, кремнефтористый натрий. Спо¬
соб и режим активации сильно влияют на физико-химические и
технологические свойства готового продукта и могут быть разными
в зависимости от условий водообработки. Основными показателя¬
ми при оценке активации кислыми реагентами являются степень
нейтрализации щелочности жидкого стекла и конечное значение
pH золя.
При использовании минеральных кислот оптимальная степень
нейтрализации составляет 80—85% [3,9 (стр. 118), 19 (стр. 130),
25], при использовании A12(S04)3 — 70—75% [25]. Гервиц [30]
рекомендует поддерживать соотношение Si02 : А1203 равным
6:1, утверждая, что образующиеся при этом алюмосиликаты об¬
ладают наилучшими флокулирующими свойствами. В случае
применения хлора соотношение С12 : Na20 следует поддерживать
в пределах 0,75 : 1—1,5 : 1 [12,19 (стр. 140), 37 (стр. 310)]. Гра¬
фик связи степени нейтрализации с величиной pH раствора жид¬
кого стекла представлен на рис. IX.1.
IX. 1. АКТИВНАЯ КРЕМНЕКИСЛОТА
289
pH
10
Рис. IX.1. Зависимость сте¬
пени нейтрализации (ан) 2%-
ного раствора (по Si02) жид- g
кого стекла разными реаген¬
тами от pH среды [36]
Реагенты: S
1 — NaHC03 или С02;
2 — Cl2, НС1 или H2S04;
3 — ai2(S04)s ^
Концентрацию исходного раствора жидкого стекла по Si02
поддерживают в пределах 0,5—3,5% и чаще всего принимают
равной 1,5—2% [9 (стр. 119), 12,19 (стр. 153), 25,29, 38 (стр. 309),
39, 40, 41 (стр. 80), 42—44]. По завершении перемешивания ис¬
ходного раствора с активатором должен быть выдержан период
так называемого вызревания золя — 0,5—2 час. Он составляет
20—80% от времени полного застудневания и образования геля
[12, 45].
Наибольшая скорость полимеризации золя наблюдается при
значениях pH 5—8. С повышением температуры раствора от 0 до
50° С застудневание ускоряется примерно в 5 раз. Немалое значе¬
ние имеет режим перемешивания [5 (стр. 21), 30]. Скорость застуд¬
невания увеличивается при повышении концентрации ионов в во¬
де, используемой для приготовления раствора, особенно катионов
Са2+, Mg2+ и Fe3+ [45]. Однако, как показано некоторыми исследо¬
вателями [12, 19 (стр. 145)], при использовании жесткой воды фло-
кирующие свойства АК ухудшаются. Это, по-видимому, связано
с образованием малорастворимых силикатов.
С повышением концентрации SiOa полимеризация ускоряется.
Регулировать процесс можно изменением степени нейтрализации
раствора силиката натрия и выбором соответствующего актива¬
тора. При использовании для активации хлора период вызревания
сокращается до 20—40 мин [12, 19 (стр. 144)].
Созревший золь разбавляют водой до концентрации по Si02
0,5—0,75% [12, 19 (стр. 173), 38 (стр. 309), 41 Гетр. 82), 45] и ис¬
пользуют в качестве рабочего раствора в течение 8—10 час. В Япо¬
нии разработан метод приготовления АК, способной храниться
в разбавленном виде в течение месяца [46].
В последние годы с целью снижения затрат на реагентное обо¬
рудование и использование АК в момент ее наибольшей активно¬
сти периодическое (порционное) приготовление АК заменяют не¬
прерывным [12, 22, 25, 30, 45, 47, 48]. Обзоры по соответствующе¬
му оборудованию даны в работах [19, 37]. Предложены способы
10 Е. Д. Бабенков
290
ГЛАВА IX. ФЛОКУЛЯЦИЯ И ФЛОКУЛЯНТЫ
получения А К путем активации жидкого стекла непосредственно
в трубопроводе, по которому АК подается на очистные сооруже¬
ния [22, 49]. Кулъский и Слипченко для интенсификации актива¬
ции и улучшения технологических свойств АК рекомендуют хло-
ро-воздушную смесь [16] и электрохимический способ [21].
IX. 1.2. Свойства и механизм действия
Частицы АК имеют сферическую форму и размер от 10 до 1500 А
[19 (стр. 15), 27]. Их изоэлектрическая точка лежит в области зна¬
чений pH 4—5. Поэтому при величинах pH, обычно имеющих ме¬
сто после нейтрализации силиката, частицы несут отрицательный
заряд. Некоторая доля продуктов нейтрализации находится, по-
видимому, в истинно растворенном состоянии [50]. Естественно
предполагать, что в основе флокулирующего действия АК лежит
взаимная коагуляция противоположно заряженных золей [31, 51,
52]. Но нельзя оставлять без внимания и важную роль частиц АК
как центров конденсации продуктов гидролиза коагулянта [32].
Ланжелье и Людвиг [53] положительное влияние добавок АК
объясняют повышением ионообменной емкости дисперсной фазы.
Хотя разноименность зарядов АК и продуктов гидролиза алю¬
миния и железа имеет, несомненно, важное значение, объяснить
действие АК полностью взаимной коагуляцией невозможно. Со¬
гласно экспериментальным данным, улучшение коагуляции проис¬
ходит в широких пределах соотношения Si02 : Ме3+. По мнению
Кульского и др. [9 (стр. 120), 19 (стр. 22)], наиболее вероятно воз¬
действие АК на компактность и прочность вторичных коагуляцион¬
ных структур за счет возникновения разветвленных кремнекисло¬
родных связей.
Подчиненная роль разноименности зарядов подтверждается
и тем обстоятельством, что наилучшие результаты очистки воды
достигаются, если АК добавлена после завершения гидролиза
коагулянта и образования первичных частиц твердой фазы. В не¬
давно проведенных исследованиях [54] установлено, что в раство¬
рах, содержащих Als+ и Si02, образуется аморфный продукт, от¬
вечающий по составу формуле Al2Si205(0H)4.
IX. 1.3. Опыт применения и эффективность
Активная кремнекислота применяется для интенсификации коа¬
гуляции при очистке не только природных, но и некоторых видов
сточных вод 1. В отдельных случаях, например, при удалении из
1 Примеры использования АК для обработки стоков разного состава приве¬
дены в гл. X.
IX.1. АКТИВНАЯ КРЕМНЕКИСЛОТА
291
воды грубодисперсных примесей [55] или железа [19 (стр. 106)]
возможно употребление АК в качестве самостоятельного коагулян¬
та. В присутствии АК происходит ускорение окисления двухвалент¬
ного железа и образование железо-кремниевых комплексов [56].
Совместное применение АК и гидролизующихся коагулянтов
дает следующие преимущества.
1. Увеличивается плотность коагулированной взвеси [57],
ускоряется хлопьеобразование и осаждение хлопьев [9 (стр. 122),
20, 33, 41].
2. Достигается более стабильная работа осветлителей со взве¬
шенным осадком [9 (стр. 120]), сокращается длительность их за¬
рядки [25, 58]. Становится возможным повышение производитель¬
ности отстойников [19 (стр. 58), 59] и вихревых камер хлопьеобра¬
зования [60].
3. При обработке малоцветных вод достигается снижение пот¬
ребности в коагулянтах на 10—40% [10, 19 (стр. 108), 20, 33,
61, 62].
4. Возрастает глубина очистки воды за счет лучшего отделения
в осадок коагулированной взвеси и удаления примесей, плохо
удаляемых коагулянтами (например, кремнекислоты [13, 14,
19 (стр. 105), 20, 25, 33, 58].
5. При низкой исходной щелочности воды добавление АК поз¬
воляет нормализовать процесс коагуляции без добавления подще¬
лачивающих реагентов [19 (стр. 45), 20, 33, 63].
6. Улучшаются адгезионные свойства коагулированной взве¬
си, повышается ее прочность. Это дает возможность повысить
грязеемкость фильтров [15, 17, 18, 20].
7. Применение АК особенно эффективно при низких темпера¬
турах обрабатываемой воды [7, 9 (стр. 120), 10, 19, (стр. 54), 20,
64, 65].
8. Расширяется область оптимальных значений pH [19 (стр. 40),
20, 33].
9. Остаточный кремний в обработанной воде практически от¬
сутствует [19 (стр. 64), 25, 58].
По некоторым данным АК мало эффективна в сочетании Гс
солями железа [33, 66, 67], хотя описаны и примеры успешного
совместного использования этих реагентов [44, 68, 69].
При очистке воды от взвешенных примесей относительное сни¬
жение мутности в результате добавления АК возрастает с увели¬
чением дозы флокулянта (до некоторого предела), а также с рос¬
том концентрации и степени дисперсности примесей. Оптималь¬
ная доза АК находится в пределах 5—10% от дозы сернокислого
алюминия. Большие дозы требуются при очистке воды, содержа¬
щих высокодисперсные примеси [19, стр. 32]. Для активации
рекомендуются сернокислый алюминий, серная кислота, крем¬
нефтористый натрий.
10*
292
ГЛАВА IX. ФЛОКУЛЯЦИЯ И ФЛОКУЛЯНТЫ
pH
Мо, мг/л
Рис. IX.2. Зависимость времени (т) вызревания геля АК от pH среды при
обработке жидкого стекла хлором [44]
Рис. IX.3. Зависимость остаточной мутности (А/0) воды от процентного
соотношения (а) доз АК и сернокислого алюминия
Исходная мутность воды — 8 мг/л\ исходная цветность — 75 град', доза коагулянта —
40 мг/л [19]
Имеется много примеров успешного применения АК при очи¬
стке цветных вод [5 (стр. 164), 63, 70—72]. Ларионов и др. [55]
показали, что самостоятельное использование АК в больших до¬
зах (60 мг/л) приводит к некоторому снижению цветности (на 10 —
20%), которое, по их мнению, объясняемся соосаждением гумино-
вых веществ частицами золя флокулянга. Однако, по данным Куль-
ского, АК не обладает связующими свойствами по отношению к
веществам цветности.
При использовании совместно с коагулянтом АК не только не
способствует обесцвечиванию воды, но, напротив, экранирует дей¬
ствие продуктов гидролиза, затрудняя удаление гуминовых ве¬
V 7
I о
-/
-г
а
VV
\\
/
к
к
J # №.,/нг/л
С==
А
V
\
А
Рис. IX.4. Влияние порядка добавления к воде сернокислого^ алюминия
и АК на электрофоретическую подвижность (и) коагулированной взвеси (а)
н степень (р) удаления взвеси из воды (б)
Сплошные кривые — добавление АК перед коагулянтом; пунктирные — добавление
АК после коагулянта; 1 — 15, 2 — 3 мг/л АК
IX.1. АКТИВНАЯ КРЕМНЕКИСЛОТА
293
ществ. Отмечены даже случаи, когда цветность обработанной воды
превышала исходную [10, 36]. Поэтому в случае цветных вод АК
следует использовать только как средство для ускорения осажде¬
ния коагулированной взвеси, снижения остаточной мутности во¬
ды. Из активаторов лучшие результаты дают хлор и хлоро-воз-
душная смесь [10, 16, 19 (стр. 148), 2?, 44] (за счет избыточного ко¬
личества хлора улучшается обесцвечивание воды). Рекомендован
также сернокислый алюминий [10]. Зависимость скорости вызре¬
вания АК в присутствии хлора от степени нейтрализации 2%-
ного раствора Si02 показана на рис. IX.2.
При очистке окрашенных вод небольшой мутности диапазон
эффективных доз АК ограничивается 20% от дозы коагулянта.
Чаще всего применяют дозы в размере 10% от дозы коагулянта
(рис. IX.3).
Большинство исследователей указывает, что обработка воды
A12(S04)3 с добавкой АК идет наиболее успешно в диапазоне значе¬
ний pH 5,5—7,5, причем применение АК расширяет область оп¬
тимума pH и диапазон концентрации анионов, в котором коагу¬
ляция протекает успешно. В жестких водах эффективность дей¬
ствия АК выше, чем в мягких, а зона оптимальных pH, как и
в случае одного только коагулянта, смещена в сторону более
низких значений.
По данным Института коллоидной химии и химии воды АН
УССР, применение АК при обработке маломутной цветной воды
(Днепровская водопроводная станция г. Киева) позволило сни¬
зить затраты на реагенты на 18—21% и уменьшить в 1,5—2 раза
мутность отстоенной воды. При очистке мутной воды, хотя расход
коагулянта и удалось снизить на 25%, эксплуатационные затра¬
ты несколько возросли [62].
При обработке цветных вод АК следует вводить после коагу¬
лянта с разрывом во времени от 10 сек до 3 мин [10, 19 (стр. 25),
20, 36, 64, 73]. Большие интервалы времени относятся к случаю
очистки холодных вод. Низкий эффект очистки воды при добавле¬
нии АК до ввода коагулянта объясняют или экранированием крем-
некислотой продуктов гидролиза коагулянта и снижением вслед¬
ствие этого их сорбционной способности по отношению к окраши¬
вающим соединениям, или способностью частиц кремнекислоты
связывать катионы алюминия, в результате чего дисперсные за¬
грязнения оказываются неполностью астабилизированными. Наи¬
более благоприятный момент для ввода АК наступает после до¬
стижения сорбционного равновесия между веществами цветности
и продуктами гидролиза коагулянта.
О порядке добавления реагентов при обработке малоцветных,
но мутных вод мнения исследователей расходятся. Согласно экс¬
периментам Блэка и др. [74], порядок добавления АК и A12(S04)3
к воде, замутненной каолином, не имеет значения (рис. IX.4).
294
ГЛАВА IX. ФЛОКУЛЯЦИЯ И ФЛОКУЛЯНТЫ
Некоторые специалисты отдают предпочтение предварительному
вводу АК. Миркис установил, что такой порядок ввода реагентов
благоприятен при осветлении речной воды, содержащей 120—
1500 мг/л взвешенных примесей [62]. Этот же порядок добавления
реагентов принят на водоочистной станции г. Эйпен (Бельгия)
[41, стр. 78] и при очистке стоков красильных производств [75].
При очистке днепровской воды АК рекомендуют вводить через
15—120 сек после добавления раствора сернокислого алюминия.
Такого же мнения придерживаются японские специалисты, иссле¬
довавшие очистку сильно загрязненной воды р. Энкачавы хлори¬
рованным железным купоросом и АК [44].
Перед фильтровальными сооружениями, работающими по прин¬
ципу контактной коагуляции, добавлять АК следует после сме¬
шения коагулянта с водой — на входе воды в загрузку [19
(стр. 25), 24] или в толщу самого фильтрующего слоя [76].
IX. 2. ОРГАНИЧЕСКИЕ ВЫСОКОМОЛЕКУЛЯРНЫЕ
ФЛОКУЛЯНТЫ
Известно, что еще в XX в. до н. э. в Индии для очистки воды при¬
меняли органические вещества некоторых растений. Тем не менее,
флокулирующее действие органических полиэлектролитов впер¬
вые описано в 1951 г. [77], а систематическое изучение их техно¬
логической эффективности начато в 1952 г. В 1967 г. в США ре¬
комендовано для практического использования 9 флокулянтов
разного состава [78]; в 1969 г. это число выросло до 18 [79], в
1970 г.— до 79 [80], а в 1971 — до 134 [81]. Большинство из ре¬
комендованных флокулянтов—органические полиэлектролиты.
Применение органических высокомолекулярных флокулян¬
тов (ВМФ) позволяет резко ускорить образование и осаждение
хлопьев коагулированной взвеси, сократить потребность в коа¬
гулянтах и увеличить санитарно-гигиеническую надежность ра¬
боты водоочистных сооружений. Помимо очистки природных вод,
в которой ВМФ используются, как правило, совместно с коагу¬
лянтами, они находят широкое применение при обработке сточных
вод и осадков различных промышленных производств. Подробные
обзоры по применению ВМФ даны в работах [3, 36, 82—88].
Полиэлектролиты — продукты полимеризации или поликон¬
денсации отдельных малых молекул в макромолекулы. Молекуляр¬
ные цепочки достигают длины более 1 мкм и могут быть жест¬
кими (например, у полисахаридов) или гибкими. Гибкость обус¬
ловлена вращением отдельных участков цепи вокруг одинарных
валентных связей атомов углерода. При некоторых условиях, на¬
пример, при повышении температуры, минимальной потенциальной
анергии гибких макромолекул отвечает клубкообразная форма.
iX.2. ОРГАНИЧЕСКИЕ ВЫСОКОМОЛЕКУЛЯРНЫЕ ФЛОКУЛЯНТЫ
295
В общем случае конфигурация макромолекул зависит от ион¬
ной силы раствора и числа заряженных участков полимерной це¬
пи. С увеличением степени ионизации макромолекул их цепи вы¬
тягиваются. Добавление к раствору полимера противоионов при¬
водит к уменьшению длины макромолекулярных цепей и соответ¬
ствующему снижению вязкости раствора [88]. Это, по-видимому,
связано с сжатием двойных электрических слоев на одноименно
заряженных участках (сегментах) и ослабеванием их взаимного
отталкивания 2.
Согласно одной из первых теорий, при адсорбции макромоле¬
кулы контактируют с поверхностью твердой фазы лишь частью
сегментов, остальная их часть простирается в глубь раствора [89,
90]. Позднее установлено, что характер размещения полимерных
цепочек на твердой поверхности зависит от энергии взаимодейст¬
вия макромолекул с поверхностью. Для описания адсорбции поли¬
меров предложено выражение [91, 92]
fi = Ка, (IX.1)
Рс (1-9) Рс
где рс — число сегментов макромолекулы, адсорбционно закре¬
пленных на поверхности; 0 — доля поверхности сорбента, покры¬
тая сегментами; а — активность полимера в данном растворе;
К — константа.
Нетрудно заметить, что при рс = 1 уравнение (IX.1) перехо¬
дит в уравнение Лэнгмюра.
IX. 2.1. Виды и состав флокулянтов
Флокулянты, используемые в водоподготовке, представляют со¬
бой природные и синтетические водорастворимые линейные поли¬
меры анионного, катионного, амфотерного и неионогенного типов.
В качестве природных флокулянтов наибольшее распростра¬
нение получили высшие полисахариды — крахмал, целлюлоза
и их производные. В зависимости от соотношения количеств ами¬
лозы и амилопектина молекулярный вес полисахаридов колеб¬
лется в пределах от нескольких сот тысяч до нескольких
миллионов. Флокулянты со слабо выраженными анионными свойст¬
вами получают, главным образом, щелочной обработкой карто¬
фельного или кукурузного крахмала [77, 93]. Другими способами
обработки можно получить продукты с сильно выраженными ани¬
онными и катионными свойствами [5 (стр. 26)j.
2 Другие важные свойства макромолекул были рассмотрены в гл. I и II.
296
ГЛАВА IX. ФЛОКУЛЯЦИЯ И ФЛОКУЛЯНТЫ
Структурная формула крахмала:
н он
н
[ с с
\ /I
\ / ОН
с
о—
/ \ н
\|
с
/ \
/
\
о
с
•о
н
СН2ОН
Длина макромолекул достигает 4000—8000 А при поперечном раз¬
мере 3—7,5 А.
На основе крахмала выпускают под разными коммерческими
названиями флокулянты в ПНР (Р-26, L-М и др.), Франции
(NH-19, като, флокгель и др.), ЧССР (флокал), США (супергель,
виспрофлок, гамако и др.), Японии и других странах [94—100].
Исследованы флокирующие свойства крахмала, обработанного
акриламидом, карбамилэтилированного крахмала и продукта
реакции диальдегидного крахмала с полиакриламидом [101], крах¬
мала, смешанного с глицерином [102] и обработанного хлорбу-
тенилтриметиламмонийхлоридом [103].
Целлюлозу получают путем обработки древесины или других
растительных материалов щелочью или монохлоруксусной кис¬
лотой [76, 96, 104—107]. Поскольку короткие фракции целлюлозы
(п ~ 40) нерастворимы в воде, под разными названиями (СМС,
КМЦ, флокулес, сольвитоза, НЕС, целлюфикс и др.) используют¬
ся метилцеллюлоза, карбоксилметилцеллюлоза и оксиэтилцеллю-
лоза, обладающие анионными свойствами. Их молекулярный вес
составляет от нескольких десятков до сотен тысяч.
Альгинатсодержащие флокулянты получают путем щелочной
обработки морских водорослей и применяют в практике очистки
воды с 1957 г. [108—111]. Структурная формула полиальгинатов
та же, что у крахмала и целлюлозы, но группы СН2ОН, СН2ОСН3
и др. заменены группой COONa. Полиальгинаты обладают анион¬
ными свойствами.
Во Франции, Японии и других странах разработано несколь¬
ко способов изготовления порошкообразных полиальгинатов или
их растворов, содержащих до 90 и даже до 100% очищенного
альгината натрия [107, 112—115]. В зависимости от задач водоочи¬
стки степень нейтрализации альгиновой кислоты (содой или едким
натром) в коммерческом продукте меняют в широких пределах
[116—118]. Альгинатсодержащие флокулянты выпускаются ино¬
странными фирмами под разными названиями (келкзоль, келджип,
велгум и др.) с молекулярным весом от 20 до 200 тысяч.
IX.2. ОРГАНИЧЕСКИЕ ВЫСОКОМОЛЕКУЛЯРНЫЕ ФЛОКУЛЯНТЫ
297
Помимо перечисленных природных флокулянтов, при очистке
воды находят ограниченное применение белковые дрожжи, тол¬
ченые семена некоторых деревьев, агар-агар, протеин и нуклеин-
содержащие вещества, выделяемые из отходов пищевой промыш¬
ленности, а также различные органические смеси [107, 119—123].
Под названием гуартек рекомендован к использованию в качест¬
ве неионогенного флокулянта экстракт семян бобового растения
Cyamopsic psoraliades [83, 108].
Синтетические флокулянты получили гораздо более широкое
применение, чем природные, во-первых, потому что они обладают
лучшими флокуляционными свойствами; во-вторых, их производ¬
ство (из продуктов переработки угля и нефти), как правило, свя¬
зано с меньшими затратами. Синтетические флокулянты по хи¬
мическому составу делят обычно на четыре группы: полиэтилен и
его производные, полиамиды, полиамины, полиакрилы. К пятой
группе можно отнести сополимеры представителей первых четы¬
рех групп.
Наибольшее применение получили полиакрилат натрия, поли¬
акриламиды и их сополимеры. Структурная формула полиакри¬
лата натрия:
г-СН2—СН--1
I
СО
I
CNa
Флокулянт является продуктом гидролиза полиакрилонитри-
лов. Это анионный полимер с молекулярным весом от нескольких
тысяч до нескольких миллионов [5, 35, 82]. Коммерческие назва¬
ния: седипур-А, крилиум-9, HPAN и др.
Полиакриламид (ПАА) получают полимеризацией растворов
моноакриламида в присутствии эквимолярной смеси тиосульфата
натрия и перхлората аммония [104]. Его структурная формула:
-СН2—СН-
I
СО
I
NH2
Молекулярный вес ПАА зависит от его чистоты и метода полиме¬
ризации. В результате диссоциации образуются отрицательно за¬
ряженные группы —СОО~ и положительно заряженные группы
=Ш$.
Наилучшими флокулирующими свойствами обладает частично
гидролизоватгаый полиакриламид, в котором некоторая доля амид¬
ных групп заменена карбоксильными [124 (стр. 116), 125, 126].
298
ГЛАВА IX. ФЛОКУЛЯЦИЯ И ФЛОКУЛЯНТЫ
Это объясняется тем, что в негидролизованном ПАА молекулы
лишь слегка вытянуты, а амидные группы диссоциированы слабо.
По мере протекания гидролиза молекулы полимера удлиняются,
а отрицательно заряженные карбоксильные группы придают им
отрицательный заряд. Полиакриламид, гидролизованный на 67%,
обладает ярко выраженными анионными свойствами.
Из сополимеров высокой флокулирующей способностью обла¬
дают продукты взаимодействия неионизированных веществ — ак-
рилонитрила, акриламида, винилового спирта, винилацетата и
других с электролитами — солями акриловой и малеиновой кис¬
лот [104].
В Советском Союзе производство ПАА разработано Институ¬
том высокомолекулярных соединений АН СССР совместно с ВНИИ
Галургии [84]. Технический продукт под разными названиями
(АМФ, ПАНГ и др.) имеет молекулярный вес 5,2—5,6 млн. [127].
Токсического действия ПАА в тех концентрациях, в которых фло-
кулянт может присутствовать в питьевой воде, не обнаружено
[128-132].
Полиакриламидами являются и наиболее эффективные зарубеж¬
ные флокулянты: сепаран-2610, РАМ-100, суперфлок-16, магни-
флок, седипур, полифлок [36, 96, 124 (стр. 6)] и многие другие.
Американские, немецкие, голландские, французские и итальян¬
ские фирмы выпускают ПАА в порошкообразном, гранулирован¬
ном или гелеобразном виде.
К синтетическим ВМФ, обладающим высокой флокулирующей
способностью, относятся также поливиниловый спирт и его произ¬
водные [107, 133]; сополимеры эфиров уксусной кислоты с малеи-
новым ангидридом [36, 82]; полиэлектролиты, содержащие в бо¬
ковой цепи алкоксильные группы [134]; продукты реакции тетра¬
метил оксипропандиамина с дихлорэтилэфиром [135]; сополимеры
мочевины и формальдегида с сильнокислотными радикалами [136];
хлористо-водородные соли полиметилвинилпиридина [137]; поли-
диамилдиметиламмонийхлорид [138]; азогетероциклические фос-
фоновые кислоты [139] и др. Однако эти флокулянты не получили
пока широкого распространения.
В 60-х годах в МХТЙ им. Д. И. Менделеева разработана тех¬
нология синтеза катионных ВМФ серии ВА — поливинилбензил-
триметиламмония и поливинилбензилпиридиния с молекулярным
весом от 26 до 270 тысяч и обменной емкостью поглощения около
5 мг-экв/г [140—142]. Эти флокулянты успешно прошли испытания
и, по всей вероятности, получат применение на водоочистных стан¬
циях.
Исследователи [143—154] предлагают использовать смеси ор¬
ганических флокулянтов со щелочными реагентами, а также гли¬
нами, угольной золой, молотым кварцем и другими наполните¬
лями.
IX.2. ОРГАНИЧЕСКИЕ ВЫСОКОМОЛЕКУЛЯРНЫЕ ФЛОКУЛЯНТЫ
299
IX. 2.2. Свойства и механизм действия
Флокуляция в водоподготовительных процессах рассматривается
как «сшивание» микрохлопьев — вторичных коагуляционных
структур, их объединение в более крупные и тяжелые агрегаты
(флокулы).
Обсуждая в предыдущих главах явления защиты и сенсибилиза¬
ции, а также свойства высокомолекулярных веществ, мы уже ка¬
сались отчасти сложного механизма взаимодействия макромоле¬
кул с поверхностью твердой фазы и отмечали последствия этого
взаимодействия. Флокуляция с помощью ВМФ может протекать,
по крайней мере, по трем механизмам.
1. Сжатие двойного слоя, снижение агрегативной устойчиво¬
сти частиц и, как следствие, объединение частиц под действием
молекулярных сил притяжения [154]. Допускается обмен простых
анионов дисперсной фазы на анионные группы полимеров [155].
2. Химическое взаимодействие макромолекул с веществами,
входящими в состав природной воды и ее взвесей, или предвари¬
тельно добавленными ионами гидролизующихся коагулянтов.
Возможно комплексообразование [156, 157].
3. Формирование мостиков полимера между отдельными ча¬
стицами твердой фазы вследствие закрепления молекулярных це¬
почек на поверхности разных частиц [155, 158—160].
Первые два механизма предполагают, что молекулярный вес
ВМФ в ходе флокуляции не играет существенной роли, хотя и мо¬
жет определять некоторые параметры процесса. Однако сильное
влияние молекулярного веса, подчеркиваемое многими исследова¬
телями (см. ниже), говорит в пользу мостовой модели флокуляции.
Электростатическим явлениям — смещению дзета-потенциала ча¬
стиц твердой фазы в сторону больших положительных значений
при использовании катионных ВМФ и в сторону больших отри¬
цательных значений при использовании анионных ВМФ — при¬
надлежит, вероятно, подчиненная роль.
Согласно мостовой модели [161] флокуляция состоит, во-пер-
вых, в закреплении концов макромолекул на поверхности частиц
и, во-вторых,— в адсорбции простертых в глубину раствора сег¬
ментов на вакантных участках соседних частиц. Предполагается
[88, 104, 162—166] существование следующих закономерностей:
1) оптимальные условия флокуляции достигаются при дозах
ВМФ, обеспечивающих покрытие доступных участков поверхно¬
сти твердых частиц;
2) пересыщение поверхности частиц молекулами полимера
приводит к ухудшению флокуляции, поскольку в этом случае
свободные концы макромолекул могут адсорбироваться на той же
поверхности, образуя петли, и число мостовых связей между со¬
седними частицами уменьшается;
300
ГЛАВА IX. ФЛОКУЛЯЦИЯ И ФЛОКУЛЯНТЫ
3) при интенсивном перемешивании, способном разрушить по¬
лимерные связи, происходит разобщение сфокулированных час¬
тиц; если доза ВМФ меньше оптимальной, то мостовые связи сла¬
бее противостоят касательным усилиям среза;
4) между оптимальной дозой полимера и площадью, доступной
для адсорбции поверхности частиц дисперсной фазы, существует
линейная зависимость.
Возникновение полимерных мостиков имеет место даже тогда,
когда флокулирующие частицы заряжены противоположно. При¬
чем снижение заряда частиц в зависимости от характера сил, от¬
ветственных за адсорбцию ВМФ, может иметь большее или мень¬
шее значение [166].
Результаты ультрамикроскопических, электронно-микроско-
пических и рентгеноскопических исследований [161, 167, 168]
подтверждают предположение о том, что первопричиной флоку-
лирующего действия ВМФ является адсорбционное закрепление
концов макромолекул на твердых поверхностях. При этом катион¬
ные флокулянты адсорбируются преимущественно по катионооб¬
менному механизму на межслойной и наружной поверхностях гли¬
нистых частиц, а анионные — на положительно заряженных уча¬
стках [161, 169, 170]. Большое значение принадлежит образованию
водородных связей между недиссоциированной группой поли¬
мера и гидроксильными группами или атомами водорода на по¬
верхности алюмосиликатов [104, 124 (стр. 117), 155, 171].
Скорость и величина адсорбции полимеров зависят от свойств
их макромолекул. Вытянутые макромолекулы адсорбируются
лучше, и в этом состоит одно из главных преимуществ синтетиче¬
ских флокулянтов над природными.
Большое значение имеет гибкость макромолекул, плотность
их зарядов и характер взаимодействия с поверхностью адсорбента.
У полимеров, слабо взаимодействующих с поверхностью, число
адсорбировавшихся сегментов пропорционально квадратному кор¬
ню из молекулярного веса; у сильно взаимодействующих полиме¬
ров — молекулярному весу [91, 92]. По данным Зильберберга [172,
173], длина простирающихся в глубь раствора концов макромо¬
лекул определяется не столько их молекулярным весом, сколько
гибкостью.
На характер сорбции ВМФ сильное влияние оказывает приро¬
да дисперсной фазы и наличие в растворе молекул, способных
к конкурирующей адсорбции. Хотя сорбция полимерных моле¬
кул на минералах удовлетворительно описывается уравнением
Лэнгмюра, величина ее для разных минералов может сильно от¬
личаться. Так, поливиниловый спирт адсорбируется на частицах
каолина в количествах, в десятки раз меньших, чем на частицах
бентонита [174].
Существенную роль играет концентрация ВМФ и твердой фа¬
IX.2. ОРГАНИЧЕСКИЕ ВЫСОКОМОЛЕКУЛЯРНЫЕ ФЛОКУЛЯНТЫ
301
зы в растворе. Изучение сорбции полиакрилата гуанидина на ча¬
стицах бентонита, каолина и кварца позволило установить, что
изотермы адсорбции не проявляют тенденции к насыщению.
Авторы [175] объяснили это изменением формы макромолекул
при повышении концентрации полимера. С увеличением концент¬
рации суспензии число мостовых связей возрастает, по-видимому,
вследствие уменьшения расстояний между отдельными частица¬
ми [88].
Концентрации полиэлектролитов, необходимые для начала
флокуляции суспендированных минеральных частиц, намного
ниже по сравнению с концентрациями простых электролитов,
вызывающими начало коагуляции этих же частиц (табл. IX.1).
Таблица IX.1 Пороговые концентрации катионов, вызывающие
коагуляцию (флокуляцшо) минеральных частиц [161]
Катионы и катион¬
ные полиэлектро-
литы
Валент¬
ность
Пороговые
концентра¬
ции, мг-экв/л
Катионы и катион¬
ные полиэлектро¬
литы
Валент¬
ность
Пороговые
концентра¬
ции, мг-экв/л
Натрий
Кальций
Алюминий
+i
+2
+3
1,3-2,3
0,43-0,5
0,4-0,6
Диметиламино-
этилметакрилат
Вини лбу тил пир 11-
ДИН
—
0,005-0,02
0,005—0,01
Измерение дзета-потенциала макромолекул катионных ВМФ
отдельно и в смеси с коллоидными растворами показало, что
заряд смеси близок к заряду макромолекул [176]. Это свидетель¬
ствует об образовании на поверхности коллоидных частиц до¬
вольно толстых оболочек полимера. С ростом концентрации ВМФ
отрицательный заряд частиц кварца постепенно уменьшался,
а затем наступала перезарядка частиц [177].
Увеличение катионной активности поливинилпиридина при
приблизительном постоянстве величины молекулярного веса поз¬
волило в несколько раз снизить оптимальную дозу флокулянта,
но степень астабилизации отрицательно заряженных частиц
суспензии возросла при этом незначительно [88]. С другой сто¬
роны, как показал Диксон [178], существенное уменьшение
оптимальной дозы катионного ВМФ достигается при возрастании
его молекулярного веса от 189 до 1000; дальнейшее увеличение
молекулярного веса (от 1000 до 32000) приводит лишь к незна¬
чительному сокращению потребности во флокулянте.
Сопоставление результатов воздействия рассмотренных фак¬
торов на флокуляционный процесс приводит к выводу, что даже
в случае обработки суспензий катионными ВМФ нейтрализацион-
302
ГЛАВА IX. ФЛОКУЛЯЦИЯ И ФЛОКУЛЯНТЫ
ная коагуляция играет второстепенную роль. Во всяком случае,
кинетика флокуляции определяется числом полимерных мости¬
ков, возникающих между отдельными частицами.
Флокулирующее действие неэлектролитов и анионных поли¬
электролитов проявляется только в том случае, когда молекулы
их могут приблизиться на расстояние, достаточное для проявле¬
ния сил сцепления. По мнению Вейцера [84], это условие соблю¬
дается для суспензий с относительно крупными частицами. По¬
этому такие частицы могут быть сфлокулированы анионными
ВМФ без добавления минеральных коагулянтов. В случае тонко¬
дисперсных взвесей требуется предварительное снижение устой¬
чивости частиц, поскольку сами анионные ВМФ, имеющие одно¬
именный с частицами заряд, не в состоянии вызвать флокуляции.
Эта точка зрения на характер влияния размера частиц в ходе
флокуляции подтверждается результатами исследований Гор-
ловского и Хейнмана [179, 180].
Экспериментальные данные, приведенные в табл. IX.2 по
статье Вейцера и др. [181], указывают на существенное различие
в действии ПАА на дзета-потенциал (ДП) отрицательно заря¬
женных частиц гидрослюды (иллита) и положительно заряжен¬
ных частиц гидроокиси железа без предварительного добавления
к суспензиям низкомолекулярных электролитов.
Таблица IX.2. ДП частиц иллита и Fe(OH)3 при разных дозах ПАА
Доза ПАА,
мг/л
ДП (мз) частиц суспензий
Доза ПАА,
мв/л
ДП (мв) частиц суспензий
иллита
Fe(OH)3
иллита
Fe<OII)3
0
-32,3
+57,6
50
-31,3
+30,2
5
-34,5
—
100
-32,8
+ 8,2
15
-32,6
—
150
—
0
25
I
DO
00
+39,5
По мере снижения ДП флокуляция частиц Fe(OH)3 усили¬
вается, но наибольшей скорости она достигает лишь после добав¬
ления электролита (Na2S04), вызывающего дополнительную аста-
билизацию частиц 3. Для флокуляции иллитовой суспензии до¬
бавление электролита оказалось необходимым условием. Сильные
флокулирующие свойства анионных ВМФ проявляются для сус¬
пензий кальцита и флюорита с положительно заряженными час¬
тицами [124 (стр. 118)].
3 Влияние электролитов на флокулирующие свойства 11А А рассмотрено
в работах Вирской и цр. [182, 183].
XX.2. ОРГАНИЧЕСКИЕ ВЫСОКОМОЛЕКУЛЯРНЫЕ ФЛОКУЛЯНТЫ
303
Установлено, что полиэлектролиты, в молекулах которых
имеются нейтральные или отрицательно заряженные группы,
обладают лучшим флокулирующим действием. Причем в акте
сорбции принимают участие нейтральные амидные и гидроксиль¬
ные группы. Анионные группы, по-видимому, препятствуют лишь
свертыванию макромолекул в клубки, расширяя сферу действия
полимера [104].
Исследователи установили также, что анионный полиэлектро¬
лит (ПАА) действует на лиофобные золи с отрицательно или поло¬
жительно заряженными частицами наподобие любого высоко¬
молекулярного соединения: сенсибилизируя их при сравнительно
небольших дозах и стабилизируя при больших. Добавление
ПАА перед вводом электролита привело к защите золей. Элек-
тронно-микроскопическое изучение процесса стабилизации пока¬
зало, что ее причина состоит в образовании пространственных
сетчатых флокуляционных структур, в которых частицы связаны
нитеобразными молекулярными пачками [184].
Отмечено также [185], что при использований анионных поли¬
электролитов образование мостиков и прочность их закрепления
на поверхности частиц сильно зависят от образования комплек¬
сов из катионов и функциональных групп полимеров.
Установленное экспериментально улучшение флокуляции в
присутствии катионов Са2+ может быть объяснено реакцией ком-
плексообразования и возникновением более прочных мостовых
связей [36, 162, 165, 185]. Фосфаты, хорошо сорбирующиеся на
твердых поверхностях, оказывают противоположное действие —
ингибируют астабилизацию частиц поле электролитами даже в
том случае, когда они добавлены в раствор после добавления
флокулянта [186]. Характер взаимодействия флокулянтов с гу-
миновыми веществами и фульвокислотами определяется, в основ¬
ном, строением ВМФ и их силой как оснований [187].
По данным Грота и др. [188], добавление к суспензии кварца
органических веществ, способных образовывать водородные свя¬
зи, ухудшает процесс флокуляции полиакриламидом, по-види¬
мому, вследствие конкуренции этих веществ с ITAA за образова¬
ние водородных связей с силанольными группами на поверхности
кварца.
Действие ПАА, как и многих других флокулянтов, зависит
от величины pH среды [124 (стр. 118), 189]. В кислой среде карбо¬
ксильные группы ПАА не диссоциируют и ДП макромолекул
близок к нулю. Флокуляция идет за счет образования водород¬
ных связей между амидными и карбоксильными группами поли¬
мера и молекулами, находящимися на поверхности взвешенных
примесей. В нейтральной и щелочной средах происходит сильная
диссоциация карбоксильных групп, и действие ПАА в этих усло¬
виях связано, вероятно, с понижением заряда частиц загрязнений
304 ГЛАВА IX. ФЛОКУЛЯЦИЯ и ФЛОКУЛЯНТЫ
вследствие адсорбции на них полимера. На флокулирующее дей¬
ствие производных крахмала величина pH оказывает меньшее
действие, чем на полиакриламид [101].
Дополнительные сведения, подтверждающие важное значение
мостовых связей, мы находим в описании результатов эксперимен¬
тов по определению фильтруемости сфлокулированных осадков
и изучению влияния интенсивности перемешивания воды на фло-
куляцию. В опытах по фильтрации [190, 191] показано, что по
мере увеличения дозы ВМФ (синтетических и природных, катион¬
ных и анионных) удельное сопротивление осадков уменьшается,
а скорость фильтрации соответственно возрастает (пропорцио¬
нально дозе ВМФ). Причина состоит в образовании простран¬
ственной сетки и гидрофобизации поверхности частиц под дей¬
ствием адсорбировавшихся полимеров. Под давлением флокулы
проявляют пластические свойства.
Вязкие силы среза, возникающие в жидкости вблизи поверх¬
ности флокулирующих частиц при перемешивании, могут вызвать
уменьшение адсорбции флокулянта, разрушение полимерных
мостиков, перераспределение частиц внутри агрегатов, изменение
конфигурации адсорбированных молекулярных цепей [192, 193].
Биркнер и др. [185] на примере суспензии латекса показали,
что агрегаты частиц, образовавшиеся под действием одного только
простого электролита (NaCl), разрушаются гораздо быстрее, чем
флокулы, сформированные в присутствии полиэлектролита; при¬
чем во втором случае нарушение связей между частицами, обра¬
зующими флокулу, наступает при перемешивании в результате
разрыва макромолекулярных цепочек.
IX. 2.3. Кинетнка флокуляции
Во многих работах, в частности [165, 185, 194], показано, что
скорость сорбции полимеров на частицах водных суспензий очень
велика, и кинетика флокуляции практически полностью опре¬
деляется скоростью построения флокул. Однако использование
уравнения Смолуховского для скорости уменьшения числа пер¬
вичных частиц в ходе агрегации не представляется правомерным,
так как флокуляция обладает целым рядом важных особенно¬
стей, проявляющихся в механизме встречи и объединения час¬
тиц, в воздействии физико-химических условий протекания про¬
цесса на конфигурацию макромолекул.
Горловский и Хейнман [179] показали, что вероятность фло¬
куляции W$ полиэлектролитом двух частиц может быть описана
уравнением
== g—sjs<) ^2 — g~S2/so^si/s2 — 2si/so^
(IX.2)
IX.2. ОРГАНИЧЕСКИЕ ВЫСОКОМОЛЕКУЛЯРНЫЕ ФЛОКУЛЯНТЫ
305
в котором символом s обозначены средние площади: — участка
контакта при столкновении частиц; s2 — поперечного сечения
ассоциата молекул флокулянта; s0 — поверхности частиц в одном
ассоциате.
При выводе уравнения авторы исходили из следующих пред¬
положений:
1) флокуляция частиц происходит за счет образования поли¬
мерных мостиков из ассоциатов молекул флокулянта, причем
частицы слипаются тогда, когда на каком-либо из соприкасаю¬
щихся участков поверхности закреплен хотя бы один ассоциат;.
2) если частицы сталкиваются участками, на каждом из кото-
рых закреплено хотя бы по одному ассоциату, флокуляции не
происходит;
3) столкновения частиц участками поверхности, свободными
от ВМФ, не приводят к их объединению.
Другие авторы [160, 190, 195] для описания кинетики флоку¬
ляции использовали уравнение Смолуховского, модифицирован'
ное фактором 0Р (1 — 0Р), в котором 0Р — степень покрытия
поверхности частиц полиэлектролитом:
dNJdx = KN,ер (1 - 0Р). (1Х.З)
Согласно выражению (IX.3), максимальный эффект флоку¬
ляции достигается при покрытии поверхности частиц флокулян-
том наполовину. Однако это далеко не всегда подтверждается
экспериментами. Например, оптимальные условия флокуляции
кварцевой суспензии достигаются при 0Р = 0,33, а оптимальные
условия флокуляции глин — при 0Р = 0,09 — 0,22 [165, 192].
По-видимому, должны учитываться другие, дополнительные усло¬
вия процесса и в первую очередь — молекулярный вес поли¬
электролита.
При использовании ВМФ с большим молекулярным весом
определяющим параметром флокуляции может оказаться ско¬
рость диффузии макромолекул. Тогда кинетика процесса будет
определяться интенсивностью перемешивания. Действительно, как
установили Биркнер и Морган [185], с увеличением скоростного
градиента в потоке суспензии латекса, обработанной ВМФ, от
11 до 70—80 сек'1 скорость флокуляции возрастает. Уменьшение
числа первичных частиц в ходе флокуляции подчинялось урав¬
нению первого порядка только при оптимальных дозировках
ВМФ в течение 10-минутного отрезка времени от начала флоку¬
ляции. В последующем линейная связь между константой ско¬
рости флокуляции и величиной скоростного градиента не выдер¬
живалась вследствие разрушения агрегатов гидродинамическими
силами.
Некоторые ученые, например Акерс [196], допускают, что
иногда контролирующей стадией флокуляции является адсорб-
l/411 Е. Д. Бабенков
306
ГЛАВА IX. ФЛОКУЛЯЦИЯ И ФЛОКУЛЯНТЫ
ция полимера, призванная обеспечить достаточно длинные его
петли на поверхности частиц и требующая более длительного
периода времени, чем последующее объединение частиц в агре¬
гаты.
Положительное влияние увеличения молекулярного веса на
скорость флокуляции суспензий отмечают многие исследовате-
Рис. IX.5. Схема флокирую-
щего действия ВМФ по Уол¬
лесу [201]
Ли [125, 197, 198], хотя имеются исключения, например, для
суспензий с волокнистыми частицами [199]. Для описания зави¬
симости между скоростью флокуляции и молекулярным весом
(MB) или длиной молекулярных цепей (10) ПАА предложены
выражения [200]:
lg (?i - V2) = 0,66 lg MB + lg 3,73к + 10-4, (IX.4)
lg (Vx - Vt) = 1,19 lg l'J‘ + 5,17, (IX.5)
в которых Vi и V2 — скорости флокуляции суспензии соответ¬
ственно с добавкой и без добавки ПАА; к — константа.
Интересная попытка теоретического обоснования влияния
MB полиэлектролита на скорость и эффективность флокуляции
предпринята Уоллесом [201]. Он, как и многие другие исследова¬
тели, исходил из предположения, что в определенный момент
времени макромолекулы ВМФ одним своим концом закреплены
на поверхности частиц твердой фазы, тогда как другой остается
свободным и простирается на расстояние h', пропорциональное
величине MB, в глубь раствора (рис. IX.5). Для числа столкно¬
вений N частиц в единицу времени, полагая, что коэффициент
диффузии частиц после адсорбции ВМФ остается неизменным,
а эффективный радиус взаимодействия возрастает, Уоллес полу¬
чил выражение
6 (2г + Л')2 D
(R — 2г — /г')4 ’
где г — радиус первичных частиц; D — коэффициент диффузии;
R — расстояние между центрами двух флокул ирующих частиц.
Величина h', определенная расчетами по значениям MB раз¬
ных флокулянтов, колеблется в пределах 1 —16 мкм, т. е. в де¬
сятки раз превышает размер дисперсных примесей воды. В урав¬
нении (IX.6) между параметрами h' и г обнаруживается взаимо¬
связи приводящая к выводу о том^ что* когда размеры флокул
N =
(IX.6)
IX.2. ОРГАНИЧЕСКИЕ ВЫСОКОМОЛЕКУЛЯРНЫЕ ФЛОКУЛЯНТЫ
307
становятся в 10—100 раз больше длины полимерных цепей,
последние перестают быть эффективным средством флокуляции.
Далее, согласно расчетам, при концентрации суспензии
0,01 —10% удвоение MB полиэлектролита приведет в результате
роста ti к увеличению начальной скорости флокуляции на 1 —
2 порядка. Наконец, из уравнения (IX.6) следует, что интенси¬
фицирующее действие добавленного флокулянта проявляется не
сразу, Когда число столкновений частиц и так достаточно велико
(вследствие больших значений D), а спустя некоторое время.
IX. 2.4. Опыт применения и эффективность
При оптимальном' соотношении между дозой ВМФ и площадью
поверхности дисперсной фазы возникают прочные хлопья (фло¬
кулы). Предельное напряжение сдвига осадка, образующегося
в результате обработки флокулянтом гидроокисей алюминия и
железа, по мере увеличения дозы флокулянта сначала увеличи¬
вается, а затем, достигнув максимума, снижается [202, 203].
Существование максимума объясняется образованием наиболее
прочных форм связи между полимером и твердой поверхно¬
стью [181].
Уплотнение осадка, содержащего ВМФ, в первые часы проте¬
кает быстрее, чем осадка обычной коагулированной взвеси, но
конечная весовая концентрация оказывается примерно одина¬
ковой [127]. В некоторых работах утверждается, что в присут¬
ствии ВМФ возрастает плотность хлопьев, однако эксперимен¬
тальные данные не приводятся. Взвеси, полученные с добавкой
ВМФ, обладают также лучшей тиксотропией [204].
Анионные ВМФ, как правило, обеспечивают высокий техно¬
логический эффект только в сочетании с гидролизующимися
коагулянтами. Наиболее полные сведения о свойствах анионных
ВМФ имеются по полиакриламидам, применение которых в СССР
в производственных масштабах началось в 1960 г. [205]. Накоп¬
ленный в нашей стране и за рубежом опыт использования ПАА
позволяет сделать следующие выводы.
1. ПАА в небольших дозировках позволяет повысить степень
осветления воды, производительность отстойников, осветлителей
со взвешенным осадком, фильтров и контактных осветлителей;
улучшить технологические свойства коагулированной взвеси при
отделении в осадок отработанного угольного порошка и других
сорбентов [127]; увеличить степень обесцвечивания [206—208] и
обеззараживания [209] воды.
2. Эффективность действия ПАА возрастает с увеличением
концентрации взвеси в воде [84, 205].
3. При очистке мутных вод, особенно вод, содержащих грубо-
дисперсную взвесь, применение ПАА дает возможность снизить
потребность в коагулянте в 2—3 раза [84, 127, 208, 210].
11 * Е. Д. Бабенков
308
ГЛАВА IX. ФЛОКУЛЯЦИЯ И ФЛОКУЛЯНТЫ
4. ПАА не удаляет окрашивающих соединений [58, 130, 204,
211]. Отмеченное в ряде случаев улучшение обесцвечивания воды
объясняется, по-видимому, так же как и при использовании актив¬
ной кремнекислоты, лучшими физическими характеристиками
хлопьев. При обработке цветных вод добавление ПАА не позво¬
ляет уменьшить расход коагулянта [127].
5. Дозирование ПАА перед фильтровальными сооружениями
приводит к увеличению длительности защитного действия загруз¬
ки [84, 204, 205, 212]. Одновременно возрастают потери напора,
что связывают с повышением прочности и адгезии хлопьев. Появ¬
ляется возможность увеличить крупность загрузки [201, 202,
213]. Требуется повышать интенсивность и длительность промыв¬
ки загрузки после окончания фильтроцикла [214, 215].
6. В одних случаях применение ПАА дает высокий экономи¬
ческий эффект [216, 217], в других — себестоимость очистки
воды несколько возрастает [214]. Расчеты показывают, что ис¬
пользование ПАА перед осветлителями со взвешенным осадком
экономически оправдано при дозе флокулянта не более 0,5 мг/л
или при больших дозах, но при условии одновременного сниже¬
ния расхода коагулянта примерно на 10 мг от каждого милли¬
грамма добавленного ПАА. Применение ПАА перед фильтрами
в количестве 0,015—0,05 мг/л экономически оправдано, если
производительность фильтров возрастает при этом соответственно
на 10—60% [218]. В присутствии ПАА возрастают оптимальные
значения критерия перемешивания Кэмпа [219].
Сходные результаты обеспечивают и другие анионные ВМФ.
Наиболее полные сведения имеются по сапарану и нэлько. Луч¬
шими условиями применения этих ВМФ считают такие, при
которых ДП образовавшихся в результате коагуляции хлопьев
имеет небольшую отрицательную величину [74, 220]. В этих
условиях использование сепарана NP-10 или нэлько-614 в дозах
1 мг/л обеспечивает уменьшение потребности в сернокислом алю¬
минии на 10—15 мг/л.
Хорошие результаты дала проверка эффективности аэрофлока,
седомакса, седипура, виспрофлока и некоторых других анионных
флокулянтов, выпускаемых зарубежными фирмами [221—225].
Для улучшения отделения органических примесей полиэлектро¬
литы рекомендуется применять совместно с глинами [207, 226].
Катионные ВМФ применяются самостоятельно или совместно
с минеральными коагулянтами. В СССР дана положительная
санитарно-токсикологическая оценка флокулянтам ВА-2, ВА-3,
ВА-2Т, ВА-102 и ВА-212 [140, 227—229]. По их технологической
эффективности можно привести следующие сведения.
1. Флокулянты серии ВА обеспечивают очистку мутных вод
без применения минеральных коагулянтов [140, 142]. По данным
лаборатории Уфимского водопровода, 1 мг/л ВА-2 заменяет
IX.2. ОРГАНИЧЕСКИЕ ВЫСОКОМОЛЕКУЛЯРНЫЕ ФЛОКУЛЯНТЫ
309
примерно 6 мг/л А1203 [230]. Использование ВА перед осветли¬
телями со взвешенным осадком позволяет повысить концентрацию
твердого вещества в контактной среде [230—233].
2. Флокулянты серии ВА не изменяют значений pH и щелоч¬
ности обрабатываемой воды [234].
3. При обработке цветных вод происходит значительное уве¬
личение их мутности и окисляемости. Цветность воды по одним
данным [140] возрастает, по другим [142] — несколько снижается
(на 7—10 град от 1 мг/л введенного флокулянта).
Ъ величение мутности воды, по-видимому, является следст¬
вием образования малорастворимых соединений при взаимодей¬
ствии между карбоксильными и фенольными группами гуми-
новых веществ и основными группами флокулянтов серии ВА.
При некоторой дозе полимера, близкой к оптимальной, происхо¬
дит полное связывание гуминовых кислот, причем эта доза зна¬
чительно ниже необходимой для нейтрализации кислот. Такое
несовпадение доз объясняют глобулярной формой ассоциатов
гуминовых веществ и невозможностью соприкосновения макро¬
молекул с кислотными группами, расположенными внутри ассо¬
циатов [140, 141, 235].
При фильтровании цветной воды, обработанной флокулян-
тами ВА, прозрачность ее возрастает, а цветность и окисляемость
несколько снижаются [232—234]. Добавление этих флокулянтов
к предварительно коагулированной цветной воде не влияет на
качественные показатели отстоенной и профильтрованной воды.
Сделан вывод о возможности самостоятельного применения фло¬
кулянтов серии ВА для очистки цветных вод лишь в том случае,
если технологическая схема предусматривает последующее филь¬
трование [140].
4. При обработке воды ВА-2 очень важно — находятся ли
молекулы полимера в развернутом или свернутом виде, что зави¬
сит от химического состава воды и режима перемешивания. В ла¬
минарном потоке, по данным Кульского и др. [236], флокуляция
протекает быстрее, чем в турбулентном.
5. При осветлении концентрированных суспензий, например
сточных вод драг [237], флокулянты ВА часто оказываются эффек¬
тивнее полиакриламида, применяемого как самостоятельно, так
и в сочетании с сернокислым алюминием.
Сходным образом ведут себя другие известные катионные поли¬
электролиты. Испытания, проведенные в АКХ им. К. Д. Пам¬
филова, показали, что один из наиболее распространенных ВМФ —
нэлько-600 — обладает примерно такими же свойствами, как и
ВА [140]. Из другого источника [105] известно, что нэлько-600
малоэффективен в низкощелочных, слабоминерализованных во¬
дах, но может надеяшо осветлять воду, содержащую водоросли
и триполифосфат натрия.
1]**
310
ГЛАВА 1Х.|,ФЛОКУЛЯЦИЯ И ФЛОКУЛЯНТЫ
Рлс. IX.6. Влияние pH на электрофоретическую подвижность (и) коагули¬
рованной взвеси (пунктирная линия и на конечную цветность (Цк) воды
(сплошная линия) при обработке воды ВМФ дозой 60 мг/л [241]
Рис. IX.7. Влияние дозы ВМФ на электрофоретическую подвижность (и)
коагулированной взвеси (пунктирная линия) и на конечную цветность (Цк)
воды (сплошная линия)
2 — обработка воды_50 мг/л A12(S04)3 при pH 4,85; 2 — обработка воды 30 мг/л Fe2(S04)3
при pH 3,55
При использовании катионных ВМФ в качестве добавок к гид¬
ролизующимся коагулянтам оптимальная величина ДП хлопьев
близка к нулю [238]. На водоочистной станции в Уотерфорде
(США), где исходная вода сильно загрязнена органическими
веществами сточных вод, во избежание перерасхода сернокислого»
алюминия и чрезмерного подкисления воды принят следующий
режим реагентной обработки [239, 240]: добавление сернокислого
алюминия с доведением величины ДП хлопьев до —7 -г- —10 мв,
затем ввод катионного ВМФ (0,5—4 мг/л) и создание величи¬
ны ДП, близкой к нулю.
Блэк и др. [241] показали экспериментально, что наилучшее
обесцвечивание воды при самостоятельном применении катион¬
ных полиэлектролитов происходит в области значений pH 4—5,
когда флокулы имеют небольшой положительный заряд или на¬
ходятся в изоэлектрической точке (рис. IX.6). При использовании
ВМФ совместно с коагулянтом увеличение дозы полимера вызы¬
вало снижение отрицательного заряда хлопьев и даже перемену
его знака на противоположный (рис. IX.7). Однако дополни¬
тельного удаления веществ цветности не происходило.
Применение неионогенных ВМФ в сочетании с гидролизую¬
щимися коагулянтами дает, как правило, меньший эффект, чем
IX.2. ОРГАНИЧЕСКИЕ ВЫСОКОМОЛЕКУЛЯРНЫЕ ФЛОКУЛЯНТЫ
311
применение анионных и катионных. Особенно это относится к
случаю маломутных [242, 243] и цветных [211] вод.
Природные полиэлектролиты, хотя и уступают во многих
случаях по своему интенсифицирующему действию синтетиче¬
ским ВМФ, могут быть успешно использованы при обработке
наиболее «трудного) типа вод — вод, богатых органическими
загрязнениями. Применение крахмала оказалось эффективным в со¬
четании с железо- и алюминий содержащими коагулянтами при низ¬
ких температурах воды [76, 77, 244]. Однако недостаток крахмала
как флокулянта состоит в значительных остаточных его концен¬
трациях в очищенной воде [94]. Использование гидроксилэтил-
целлюлозы (НЕС) и карбоксилметилцеллюлозы (СМС) позволяет
снизить расход коагулянта и расширить зону оптимальных зна¬
чений pH [76].
Альгинатсодержащие флокулянты рекомендуется применять
при очистке цветных вод с низким щелочным резервом и величи¬
ной pH менее 6,8, подвергая предварительно альгиновую кислоту
нейтрализации до значений pH 4,4—6,5. Если же очищаемая
вода не содержит больших количеств органических веществ,
нейтрализацию кислоты следует проводить до величины pH
7,2—8,2 [116, 117]. По данным Акадзавы [245], 1 мг альгината
натрия эквивалентен по действию примерно 4 мг активной кремне¬
кислоты. С увеличением мутности исходной воды экономия коа¬
гулянта при использовании альгината натрия (0,2 мг/л) возра¬
стает [107].
В качестве добавок, способных сорбировать органические
загрязнения и улучшать коагуляцию, хорошо проявили себя
натуральные растительные масла в концентрации около 3% от
веса твердой фазы [246].
Из приведенных материалов ясно, что наилучшая флокуляция
имеет место при определенном соотношении между концентра¬
цией золя или суспензии и дозой ВМФ. Это соотношение зависит
от степени устойчивости дисперсной системы и свойств полиэлек¬
тролитов. Ограничение дозы ВМФ снизу диктуется желанием
достигнуть наилучших результатов очистки, ограничение сверху
обусловлено защитным действием чрезмерно больших концентра¬
ций флокулянтов [84, 162, 204].
В исследованиях, посвященных изучению влияния длины
молекулярных цепочек полимеров на их оптимальную дозировку,
обнаружено, что ВМФ с короткими цепочками макромолекул
имеют более узкую зону оптимальных доз по сравнению с ВМФ,
имеющими Длинные цепочки [186]. Этот результат может быть
объяснен более полной адсорбцией короткоцепочечных поли¬
меров и связанным с ней быстрым насыщением поверхности.
С увеличением в коагулирующей системе соотношения между
концентрациями природных взвешенных примесей и продуктов
312
ГЛАВА IX. ФЛОКУЛЯЦИЯ И ФЛОКУЛЯНТЫ
гидролиза коагулянта потребность в анионных флокулянтах
возрастает. По-видимому, для связывания гидроксидных соеди¬
нений требуется больше полимера, чем для связывания глини¬
стых частиц [130, 161, 212].
Обычно оптимальные дозы синтетических анионных ВМФ
лежат в пределах 0,1—2 мг/л или 0,1—2% от веса твердой фазы
загрязнений. Большие значения относятся к случаю дозирова¬
ния перед отстойниками, меньшие — перед осветлителями со
взвешенным осадком. При добавлении перед фильтрами и кон¬
тактными осветлителями доза ВМФ может быть уменьшена до
0,01—0,5 мг/л.
Оптимальные дозы катионных ВМФ при самостоятельном
применении составляют 0,5—4 мг/л, при совмещении с коагу¬
лянтами — 0,1—0,3 мг/л. Указывается, что для первого случая
потребность во флокулянте прямо пропорциональна обменной
емкости глинистых и гумусовых примесей и обратно пропорцио¬
нальна собственной обменной емкости [247]. Для вод, содержа¬
щих примерно от 10 до 100 мг/л взвешенных веществ и имеющих
цветность 50—100 град, доза флокулянта ВА перед фильтро¬
вальными сооружениями определяется только величиной цвет¬
ности [1401.
Альгинат натрия применяют в дозировках 0,1—1 мг/л [245,
248J, производные крахмала — до 25—50 мг/л [98], поливини¬
ловый спирт — до 1 г/л [133].
В периоды года, когда содержание взвеси в обрабатываемой
воде превышает 50 мг/л, полиакриламид и другие анионные ВМФ
выгоднее добавлять перед очистными сооружениями первой
ступени; при меньшем содержании взвеси — перед фильтра¬
ми [216, 217, 249]. В последнем случае, а также на станциях
контактного осветления флокулянты следует вводить непосред¬
ственно перед входом воды в загрузку: полнее используются
свойства полимеров и обеспечивается формирование более проч¬
ных коагуляционных структур 1107, 130, 212].
Промежуток времени между моментами добавления к воде
коагулянта и флокулянта подбирается таким образом, чтобы,
с одной стороны, обеспечивалось равномерное распределение
раствора коагулянта в объеме воды и успевали образоваться
микрохлопья коагулированной взвеси; а с другой стороны —
чтобы не произошло существенного уменьшения численной кон¬
центрации частиц (за счет агломерации) и ухудшения поверхно¬
стных свойств коагулированной взвеси (в результате старения).
Обычно этот промежуток времени находится в пределах 1 —
4 мин [250]. Чем ниже мутность и температура воды и выше ее
цветность, тем длительнее должен быть разрыв во времени между
вводом коагулянта и флокулянта.
Необходимо учитывать также режим обеззараживания воды.
IX. ЛИТЕРАТУРА
313
Если ввод ПАА предшествует добавлению хлора, степень обезза¬
раживания воды может понизиться [209]. В случае предваритель¬
ного хлорирования могут ухудшиться технологические свойства
катионного ВМФ [240]. Первое явление можно объяснить экра-
нируюцим действием макромолекул ВМФ на процесс обезвре¬
живания биологических объектов, второе — деструкцией макро¬
молекул под воздействием окислителя.
Описаны случаи, когда хорошие результаты очистки достиг¬
нуты при последовательной обработке воды катионными и анион¬
ными ВМФ [244, 251] и совмещении органических ВМФ с актив¬
ной кремнекислотой [19 (стр. 107), 251, 252]. Альгинатсодержа¬
щие ВМФ рекомендуется добавлять к воде перед вводом коагу¬
лянта [107, 253].
Литература
1. П. А. Ребиндер, М. Е. Липец, М. М. Римская и др. Физико-химия фло¬
тационных процессов. М.—JL, Металлургиздат, 1933.
2. V. К. La Мег. J. Colloid Sci., 19, 291 (1964).
3. J. R. Ryalis. J. Amer. Water Works Assoc., 29, 1355 (1937).
4. A. V. Graf, W. B. Schworm. Water Works Engng, 90, 1514 (1937).
5. Ю. И. Вейцер, Д. М. Минц. Высокомолекулярные флокулянты в про¬
цессах очистки воды. М., Стройиздат, 1975.
6. Л. А. Кулъский. Теоретическое обоснование технологии очистки воды.
Киев, «Наукова думка», 1968.
7. Р. Н. Будрин. Водоснабжение и санитарная техника, № 5, 16 (1940).
8. С. И. Позднякова. Там же, стр. 24.
9. Л. А. Кулъский. Основы технологии кондиционирования воды. Киев,
Изд-во АН УССР, 1963.
10. Л. А. Кулъский, М. А. Шевченко, Г. Ю. Лемпке. Водоснабжение и сани¬
тарная техника, № И, 24 (1956).
11. Л. А. Кулъский, В. А. Слипченко, В. Ф. Накорчевская. Укр. хим. ж., 30,
108 (1964).
12. В. А. Слип'енко. Автореферат канд. дисс. Киев, Институт коллоидной
химии и химии воды АН УССР, 1968.
13. В. Ф. Накорчевская, Л. А. Кулъский. Водоснабжение и санитарная тех¬
ника, № 1, 1 (1970).
14. Л. А. Кулъский, В. А. Слипченко, В. Ф. Накорчевская и др. MicbKe гос-
подарство Украши, № 4, 21 (1966).
15. В. Ф. Накорчевская, Л. А. Кулъский. Укр. хим. ж., 35, 958 (1969).
16. Л. А. Кулъский, В. А. Слипченко, В. Ф. Накорчевская. Авт. свид. СССР
176834 (1963). Бюлл. изобр., № 23 (1965).
17. Л. А. Кулъский, В. Ф. Накорчевская. ДАН УССР, Б, 1015 (1968).
18. В. Ф. Накорчевская, Л. А. Кулъский. Водоснабжение и санитарная тех¬
ника, № 9, 6 (1969).
19. Л. А. Кулъский, В. Ф. Накорчевская, В. А. Слипченко. Активная крем-
некислота и проблема качества воды. Киев, «Наукова думка», 1969.
20. В. Ф. Накорчевская. Автореферат, канд. дисс. Киев, Институт коллоид¬
ной химии и химии воды АН УССР, 1968.
21. В. А. Слипченко, Л. А. Кулъский. Водоснабжение и санитарная техника,
№ 7, 7 (1968).
22. В. А. Слипченко, Е. М. Шелякова. Сб. «Водоподготовка и очистка про¬
мышленных стоков», вып. 9. Киев, «Наукова думка», 1972, стр. 52.
314
ГЛАВА IX. ФЛОКУЛЯЦИЯ И ФЛОКУЛЯНТЫ
23. В. А. Слипченко. Сб. «Новая аппаратура и автоматические устройства
по технологии обработки воды». Киев, «Наукова думка», 1972, стр. 15.
24. Сб. «Использование адгезионных и адсорбционных процессов для уда¬
ления из воды взвесей и микроорганизмов». Под ред. JI. А. Кульского
и М. Н. Ротмистрова. Киев «Наукова думка», 1973.
25. Г. Г. Первое. Труды ВНИИ ВОДГЕО, вып. 8. М., 1964, стр. 3.
26. Г. Г. Первое. Автореферат канд. дисс. М., ВНИИ ВОДГЕО, 1964.
27. Э. И. Гервиц. Научно-теоретическая конференция аспирантов АКХ им.
К. Д. Памфилова. М., ОНТИ, 1966, стр. 24.
28. Э. И. Гервиц. Научно-теоретическая конференция аспирантов АКХ им.
К. Д. Памфилова. М., ОНТИ, 1969, стр. 12.
29. Э. И. Гервиц. Прогрессивные методы очистки природных и сточных вод
(материалы научно-технической конференции), сб. № 1. М., изд. МИСИ
им. В. В. Куйбышева, 1971, стр. 97.
30. Э. П. Гервиц. Автореферат канд. дисс. М., АКХ им. К. Д. Памфилова,
1974.
31. С. A. Black. J. Amer. Water Works Assoc., 45, 1101 (1953).
32. W. Christ. Wasserwirtschaft—Wassertechnik, 10, 99 (1960).
33. J. Kelus. Gaz, woda i techn. sanit., 34, 19 (1960).
34. Water Works Engng, 113, 712 (1960).
35. Б. М. Мацюк. Коллоидн. ж., 36, 800 (1974).
36. Ю. И. Вейцер. ЖВХО им. Д. И. Менделеева, 5, 628 (1960).
37. В. А. Клячко, И. Э. Апельцин. Подготовка воды для промышленного и
и городского водоснабжения. М., Стройиздат, 1962.
38. П. Гамер, Д. Джексон, И. Серстон. Очистка воды для промышленных
предприятий. М., Стройиздат, 1968.
39. R. С. Merril. Ind. and Engng Chem., 40, 1355 (1948).
40. A. E. Griffin. J. Amer. Water Works Assoc., 46, 571 (1954).
41. А. С. Коган. Вопросы водоснабжения и санитарной техники в Бельгии.
М., Изд-во МКХ РСФСР, 1960.
42. /. D. Walker. Water and Water Engng, 59, 534 (1955).
43. M. H. Cubberley. Пат. США 2935482 (1960).
44. К. Иситоби. Когё ёсуй, № 24, 34 (1960); РЖХим., 22И251 (1961).
45. F. В. Jackson. Water and Water Engng, 62, 437 (1958).
46. Т. Исибаси, К. Сомено. Суйдо кёкай дзасси, № 447, 23 (1971); РЖХим.;
16И285 (1972).
47. F. Smith. Англ. пат. 1399598 (1975).
48. Ю. И. Вейцер, Э. И. Гервиц, М. С. Басин и др. Авт. свид. СССР 332052
(1970); Бюлл. изобр., № 10 (1972).
49. К. Н. Albrecht, F. Landauer et al. Пат. ГДР 65240 (1970).
50. 3. Я. Берестнева, Г. А. Корецкая, В. А. Каргин. Коллоидн. ж., 11, 369
(1949).
51. Н. R. Hay. J. Amer. Water Works Assoc., 36, 626 (1944).
52. A. Denne. Wasser, Luft und Betrieb, 5, 123 (1961).
53. W. F. Langelier, H. F. Ludwig. J. Amer. Water Works Assoc., 41, 163
(1949).
54. J. D. Нет., С. E. Roberson, C. J. Lind et al. US Geol. Survey. Water-Supply
Paper, N 1827-E, 31 (1973).
55. Ю. В. Ларионов, Б. А. Скопинцев. Биология внутренних вод. Информ.
бюлл., № 15. JI., «Наука», 1972, стр. 63.
56. J. Е. Schenk, W. J. Weber. J. Amer. Water Works Assoc., 60, 199 (1968).
57. H. С. Лебедева. Автореферат канд. дисс. М., ВНИИ ВОДГЕО, 1954.
58. Г. Г. Первое. Научные сообщения ВНИИ ВОДГЕО. Водоснабжение.
М., 1960, стр. 23.
59. N. J. Pugh. J. Inst. Water Engrs, И, 17 (1957).
60. Я. Л. Файншиль. Автореферат канд. дисс. М., ВНИИ ВОДГЕО, 1970.
61. Т. Тояма. Кагаку дайгаку сюхо, № 1, 10 (1959); РЖХим., 69968 (1960).
IX. ЛИТЕРАТУРА
315
62. И. М. Миркис. Водоснабжение и санитарная техника, № 8, 35 (1967)
63. Н. R. Hay. Water and Sewage Works, 100, 107 (1953).
64. Применение силиката натрия при улучшении качества воды. Информа¬
ционное письмо АКХ им. К. Д. Памфилова, № 38. М., ОНТИ, 1954.
65. Т. М. Riddick. J. Amer. WaterWorks Assoc., 52, 502 (1960).
66. Г. Бэббит, Дж. Доланд. Водоснабжение. М., Стройиздат, 1958.
67. М. Г. Литвиненко. Труды Харьк. научн. мед. об-ва, вып. 12, 54 (1958).
68. W. Christ, L. Scholz. Wasserwirtschaft—Wassertechnik, 8, 361 (1958).
69. A. Choinacki. Gospod. wodna, 28, 49 (1968).
70. K. Doink. Zellstoff und Papier, 13, N 11, 331 (1964).
71. R. A. Pepper. J. Inst. Water Engrs, 15, 47 (1961).
72. A. G. Strom. Austral. Civil Engng, 9, N 7, 27 (1968).
73. S. Kawamura. J. Amer. Water Works Assoc., 65, 417 (1973).
74. A. P. Rlack, S. A. Hannah. J. Amer. Water Works Assoc., 53, 438 (1961).
75. E. Peyron. Teintex, 32, 419 (1967).
76. R. Ф. Накорчевская, JI. А. Кулъский. Авт. свид. СССР 453356 (1974);
Бюлл. изобр., № 2 (1975).
77. С. Е. Johnson. Ind. and Engng Chem., 43, 1080 (1951).
78. J. Amer. Water Works Assoc., 50, 462 (1958).
79. J. Amer. Water Works Assoc., 51, 574 (1959).
80. F. Russo, R. L. Carr. Water and Sewage Works, 117, Reference number,
72 (1970).
81. J. Amer. Water Works Assoc., 63, 388 (1971).
82. H. I. Fiedler. Wasserwirtschaft—Wassertechnik, 8, 561 (1958).
83. R. Oliver et al. Mechanisation, 24, N 3, 47 (1960).
84. Ю. И. Вейцер, Л. H. Паскуцкая. Научные труды АКХ им. К. Д. Памфи¬
лова, вып. 1. Водоснабжение. М., ОНТИ, 1961, стр. 104.
85. Л. И. Гюнтер, Ю. И. Вейцер. Научные труды АКХ им. К. Д. Памфило¬
ва, вып. 20. Городская канализация, № 2. М.—JL, ОНТИ, 1963, стр. 114.
86. /. Reuter. Techn. eau, N 267, 25 (1969).
87. A. L. Kowa, M. Swiderska-Broz. Pr. nauk. Inst. inz. chem. i uzzad. ciep.,
N 14, 231 (1972).
88. J. Amer. Water Works Assoc., 63, 99 (1971).
89. С. С. Воюцкий. Растворы высокомолекулярных соединений. М., Госхим-
издат, 1960, стр. 42.
90. Е. Jenckel, В. Rumbach. Z. Elektrochem., 55, 612 (1951).
91. R. Simha, H. L. Frisch, F. R. Eirich. J. Phys. Chem., 58, 584 (1953).
92. H. L. Frisch, R. Simha. J. Phys. Chem., 58, 507 (1954).
93. P. Kepa. Химия и технология крахмала. М., Пищепромиздат, 1956.
94. М. Boruch, A. Sroczynski. Przem. spozywczy, 21, N 10, 17 (1967).
95. J. Kucharski, A. Moniuszko. Oczyszczanie wod i sciekow przemyslowych
metoda koagulacji. Warszawa, Wydawn. nauk.-techn., 1967.
96. A. Klein. Ind. chim., 55, N 612, 301 (1968).
97. B. P. Vogh, J. E. Warrington et al. J. Amer. Water Works Assoc., 61, 276
(1969).
98. A. D. Nevers. Пат. США 3157594 (1964).
99. H. Канаяма, К. Аяма и др. Яп. пат. 3288 (1967).
100. X. Судзуки, С. Танака. Яп. пат. 19523 (1972).
101. М. Инано. Когё кагаку дзасси, 72, 2298 (1969); РЖХим., 10Б1677
(1970).
102. М. Канагава. Яп. пат. 49-40793 (1974).
103. W. G. Hunt, R. J. Belz. Пат. США 3835114 (1974).
104. A. S. Michaels. Ind. and Engng Chem., 46, 1485 (1954).
105. J. M. Cohen, G. A. Rourke, R. L. Woodward. J. Amer. Water Works As¬
soc., 50, 463 (1958).
106. А. Б. Здановский, E. И. JIяховская. ЖПХ, 44, 510 (1971).
107. Когё ёсуй, № 49, 1 (1962); РЖХим., 17И198-17И205 (1963).
316
ГЛАВА IX. ФЛОКУЛЯЦИЯ И ФЛОКУЛЯНТЫ
108. L. Е. Petterson, I. W. Opie. Ind. and Engng Chem., 50, 1013 (1958).
109. H. Maass. Alginsaure und Alginate. Heidelberg, «Chemie und Technik»,
1959.
110. С. Ф. Кузькин, В. П. Небера. Бюлл. цветной металлургии, № 13/90. М.,
изд. Центр, ин-та информации цветной металлургии, 1957.
111. В. Чемпен. Морские водоросли и их использование. М., ИЛ, 1953.
112. Франц. пат. 1351561 (1963).
113. G. Desbos. Франц. пат. 1570527 (1969).
114. G. Desbos. Швец. пат. 488629 (1970).
115. G. Desbos. Пат. США 3627680 (1971).
116. G. Desbos, J. Sibony, G. Thomas et al. Франц. пат. 2032724 (1970).
117. G. Desbos, J. Sibony, G. Thomas et al. Франц. пат. 2032725 (1970).
118. Франц. пат. 2133266 (1972).
119. И. А. Якубович. Цветные металлы, № 12, 9 (1957).
120. N. Y. Dhekane, G. В. Ambawane, S. D. Pagar et al. J. Inst. Engrs (India).
Public Health Engng Div., 52, 10 (1972).
121. М. H. Yueh. Пат. США 3681283 (1972).
122. С. Gomella. L’eau, 37, N 4, 57 (1950).
123. R. Schwarz, T. Hennig. Пат. ФРГ 973320 (1960).
124. М. H. Савицкая, Ю. Д. Холодова. Полиакриламид. Киев, «Техшка», 1969.
125. Е. Е. Сегалоеа, 3. Д. Туловская и др. Коллоидн. ж., 30, 128 (1968).
126. А. М. Фоминых, О. Г. Гириков. Изв. вузов. Строительство и архитекту¬
ра, № 7, 140 (1970).
127. Ю. И. Вейцер, В. Л. Криштул. Условия производственного применения
ПАА в технике очистки питьевой воды. Информационное письмо АКХ
им. К. Д. Памфилова, № 12 (173). М., ОНТИ, 1962.
128. W. Weindel, Н. Sontheimer. GWF. Wasser/Abwasser, 112, N 2, 76 (1971).
129. Л. А. Рахманона. Гигиена и санитария, № 12, 20 (1964).
130. Л. Н. Паскуцкая. Научные труды АКХ им. К. Д. Памфилова, вып. 22.
Водоснабжение, № 3. М.—Л., ОНТИ, 1963, стр. 37.
131. В. А. Клячко, И. Э. Апельцин. Очистка природных вод. М., Стройиздат,
1971, стр. 122.
132. Ю. И. Вейцер, 3. А. Колобова. Научные труды АКХ им. К. Д. Памфило¬
ва, вып. 8. Водоснабжение, № 2. М.—Л., ОНТИ, 1961, стр. 38.
133. R. Cervenka. Пат. ЧССР 123933 (1967).
134. J. W. Rysnar. Пат. США 3492225 (1970).
135. Е. Witt. Пат. США 3632507 (1972).
136. М. Сугихара. Яп. пат. 20167 (1970).
137. Л. И. Габриелова, А. К. Лившиц. Авт. свид. СССР 122449 (1959); Бюлл.
изобр., № 18 (1959).
138. J. P. Kleler. Water and Wastes Engng, 6, N 6, 42 (1969).
139. D. Redmore. Пат. США 3859211 (1975).
140. Ю. И. Вейцер, Р. М. Стерина. Водоснабжение и санитарная техника,
№ 9, 14 (1965).
141. 10. И. Вейцер, Р. М. Стерина. Научные труды АКХ им. К. Д. Памфи¬
лова, вып. 52. Водоснабжение, № 5. М., ОНТИ, 1969, стр. 27.
142. Р. М. Стерина. Научно-теоретическая конференция аспирантов АКХ
им. К. Д. Памфилова. М., ОНТИ, 1966, стр. 29.
143. Water and Sewage Works, 118, Reference number, 185 (1971).
144. D. S. Blaisdell, R. E. Klaaset al. Пат. США 3142638 (1963).
145. W. Rebhun, N. Narkis et al. Water Res., 3, 344 (1969).
146. J. Green. Нат. США 3130167 (1964).
147. W. Grunert. Пат. ГДР 43118 (1966).
148. L. Demeter, B. Galgoczi et al. Англ. пат. 1081096 (1967).
149. W. L. Denman, Ch. M. Hwa. Англ. пат. 1054677 (1967).
150. Ph. В. Reilly. Proc. 27th Internat. Water Conf. Pittsburgh, nov., 1966
(Engrs Soc. West. Pennsylvania). Pittsburgt, 1966, p. 171.
IX. ЛИТЕРАТУРА
317
151. О. М. Bacon. Пат. США 3515666 (1970).
152. J. J. Н гоп as. Пат. США 3066095 (1962).
153. J. Green. Пат. США 3276998 (1966).
154. 7. Geberiigi. Пат. ФРГ 2332512 (1975).
155. A. S. Michaels, О. Morelos. Ind. and Engng Chem., 47, 1801 (1955).
156. D. Guttman, T. Higachi. J. Amer. Pharm. Assoc., 45, 659 (1956).
157. T. Higachi, R. Kuramoto. J. Amer. Pharm. Assoc., 43, 393 (1954).
158. T. W. Healy, V. K. La Mer. J. Phys. Chem., 66, 1835 (1962).
159. W. F. Linke, R. B. Booth. Trans. Amer. Inst. Mining and Metallurg. Engrs,
217, 364 (1960).
160. К. M. Yao. Water and Sewage Works, 114, 295 (1967).
161. R. A. Ruehrwein, D. W. Ward. Soil Sci., 73, 485 (1952).
162. V. K. La Mer et al. J. Colloid Sci., 11, 704, 710, 716, 720 (1956); 12, 230,
566 (1957); 13, 589 (1958).
163. V. K. La Mer, T. W. Healy. Rev. Pure and Appl. Chem., 13, 112 (1963).
164. J. С. Капе, V. K. La Mer, H. D. Linford. J. Phys. Chem., 67, 1977 (1963).
165. A. P. Black, F. B. Birkner, J. J. Morgan. J. Amer. Water Works Assoc.,
57, 1547 (1965).
166. W. Stumm, Ch. R. O'Melia. J. Amer. Water Works Assoc., 60, 514 (1968).
167. Э. М. Натансон. Коллоидн. ж., 20, 556 (1958).
168. E. W. Fischer. Kolloid Z., 160, 120 (1958).
169. A. D. Me Laren. J. Phys. Chem., 58, 129-(1954).
170. A. D. Me Laren, G. H. Peterson, I. Barshad. Soil Sci. Soc. Amer. Proc.,
22, 239 (1958).
171. R. M. Holmes, S. J. Toth. Soil Sci., 84, 479 (1957).
172. A. Silberberg. J. Phys. Chem., 66, 1872 (1962).
173. A. Silberberg. Ibid., p. 1884.
174. JI. E. Кузнецова, H. H. Серб-Сербина. Коллоидн. ж., 30, 853 (1968).
175. В. А. Введенская, Н. Е. Огнева, В. В. Коршак и др. Труды МХТИ им.
Д. И. Менделеева, вып. 66, 173 (1970).
176. Н. Е. Ries. Nature (Engl.), 226, N 5240, 72 (1970).
177. Н. Е. Ries, В. L. Meyers. Science, N 3835, 1449 (1968).
178. J. К. Dixon et al. J. Colloid Sci., 23, 465 (1968).
179. С. И. Г орловский, В. Я. Хайнман. Пятая Всесоюзная конференция по
коллоидной химии (тезисы докладов). М., Изд-во АН СССР, 1962,
стр. 200.
180. С. И. Горловский, В. Я. Хейнман. Обогащение руд, № 4, 8 (1961).
181. 10. И. Вейцер, 3. А. Колобова, Р. М. Стерина. Научные труды АКХ
им. К. Д. Памфилова, вып. 22. Водоснабжение, № 3. М.—JI., ОНТИ,
1963, стр. 19.
182. Г. М. Вирская, М. М. Арефьева, М. Мансуроваи др. Сб. «Структурообра-
зование в дисперсных системах в присутствии полиэлектролитов». Таш¬
кент, «Фан», 1970, стр. 56.
183. Л. Д. Шин, Г. М. Вирская, К. С. Ахмедов. Там же, стр. 62.
184. Г. Р. Бочкарев, Ю. П. Коврижных. Коллоидн. ж., 31, 334 (1969).
185. F. В. Birkner, J. J. Morgan. J. Amer. Water Works Assoc., 60, 175 (1968).
186. B. P. Werkentin, R. D. Miller. Soil Sci., 85, 14 (1958).
187. Г. Л. Грановская, Л. А. Семенова, Л. Б. Зубакова и др. Сб. «Теория и
и практика сорбционных процессов», вып. 7. Изд. Воронежского ун-та,
1972, стр. 106.
188. О. Griot, J. A. Kitchener. Trans. Faraday Soc., 61, 1026 (1965).
189. F. R. Rirkner, J. K. Edzwald. J. Amer. Water Works Assoc., 61, 645 (1969).
190. V. K. La Mer. Disc. Faraday Soc., 42, 248 (1966).
191. Токё когё сикэнсё хококу, 56, 274 (1970); РЖХим., 19Б1831 (1971).
192. А. М. Kragh, W. R. Langston. .Т. Colloid Sci., 17, 101 (1962).
193. J. Amer. Water Works Assoc., 62, 311 (1970).
194. A. P. Black, F. B. Birkner, J. J. Morgan. J. Colloid Sci., 21, 626 (1966).
318
ГЛАВА IX. ФЛОКУЛЯЦИЯ И ФЛОКУЛЯНТЫ
195. P. G. Saffman, J. S. Turner. J. Fluid Mechanics, 1, 16 (1956).
196. R. J. Akers. Filtr. and Separ., 9, 423 (1972).
197. H. J. Fiedler. Zucker, 16, N 10, 273 (1957).
198. R. C. Tilton, J. Murphy, J. K. Divon. Water Res., 6, 155 (1972).
199. Т.'И. Погарская, E. E. Сегалова, Т. К. Брукус и др. ЖПХ, 41, 2630 (1968).
200. J. Chem. Soc. Japan. Pure Chem. Sect., 82, 1297 (1961).
201. W. E. Walles. J. Colloid and Interface Sci., 27, 797 (1968).
202. Б. А. Мишин. Коллоидн. ж., 28, 852 (1966).
203. M. Б. Радвинский, А. И. Пучков. Сб. «Водоснабжение, канализация, гид¬
ротехнические сооружения», вып. 13. Киев, «Буд1вельник», 1971, стр. 38.
204. Ю. И. Вейцер, Л. Н. Паскуцкая, 3. А. Колобова и др. Научные труды
АКХ им. К. Д. Памфилова, вып. 8. Водоснабжение, № 2. М.—Л.,
ОНТИ, 1961, стр. 3.
205. Ю. И. Вейцер, 3. Я. Городищер, В. П. Криштул и др. Научные труды
АКХ им. К. Д. Памфилова, вып. 1. Водоснабжение. М., ОНТИ, 1960,
стр. 104.
206. А. P. Black. J. Amer. WaterWorks Assoc., 51, 1545 (1959).
207. С. P. Blackley. Public Works, 90, 2 (1959).
208. H. Т. Лубочников, E. П. Правдин. Научные труды АКХ
им. К. Д. Памфилова, вып. 52. Водоснабжение, № 5. М., ОНТИ, 1969,
стр. 53.
209. П. Ф. Соколова, И. И. Троицкий и др. Труды Центр, н.-и. дезинфекц.
ин-та, вып. 19, 95 (1970).
210. Б. Л. Вахлер. Опыт эксплуатации систем промышленного и коммуналь¬
ного водоснабжения (материалы семинара). Изд. Моск. Дома научно-
технич. пропаганды им. Ф. Э. Дзержинского, 1966, стр. 62, 96.
211. J. Е. Singley, J. S. Maulding et al. Water Works and Wastes Engng, 2,
N 3, 52 (1965).
212. 10. И. Колодный. Опыт работы контактных осветлителей с безгравийной
загрузкой. Горьк. кн. изд-во, 1963.
213. W. R. Conley, P. W. Pitman. J. Amer. Water Works Assoc., 52, 1319
(1960).
214. М. М. Блувштейн, В. М. Корабельников. Водоснабжение и канализация,
вып. 2 (6). М., изд. ЦБТИ МКХ РСФСР, 1965, стр. 3.
215. В. И. Козлов, Ю. И. Колодный. Труды ГИСИ им. В. П. Чкалова, вып. 51,
Горьк. кн. изд-во, 1968, стр. 26.
216. И. Н. Осипова, В. Ф. Милованов и др. Научные труды АКХ
им. К. Д. Памфилова, вып. 30. Водоснабжение, 4. М.—Л., ОНТИ, 1964,
стр. 47.
217. Г. А. Орлов. Там же, стр. 62.
218. Г. А. Орлов. Научно-теоретическая конференция аспирантов АКХ им.
К. Д. Памфилова. М., ОНТИ, 1963, стр. 22.
219. Ю. И. Вейцер, Г. Н. Луценко, А. И. Цветкова. Водоснабжение и санитар¬
ная техника, № 9, 2 (1975).
220. Е. L. Bean, S. J. Campbell, F. R. Anspach. J. Amer. Water Works Assoc.,
56, 214 (1964).
221. S. Rozgai. Kemija u industrji, 14, N 3, 134 (1965).
222. J. Chattopadhyay, A. K. Chakravarti et al. Chem. Ind. News, 10, N 3—4,
124 (1960).
223. Англ. пат. 981963 (1960).
224. South. Power and Ind., 80, N 10, 42 (1962)-.
225. I. Tesarik, J. Vostrcil. Tribune CEBEDEAU, 21, 7 (1968).
226. J. R. Fanselow. Пат. США 3617561 (1971).
227. М. Б. Трахтман. Гигиена и санитария, № 9, 15 (1969).
228. Б. Р. Битвицкая. Гигиена и санитария, № 5, 13 (1969).
229. Б. Р. Битвицкая. Научные труды АКХ им. К. Д. Памфилова, вып. 76.
Водоснабжение, № 7. М., ОНТИ, 1970, стр. 84.
IX. ЛИТЕРАТУРА
319
230. В. Л. Драгинский, Н. Т. Лубочников и др. Там же, стр. 10.
231. И. Н. Синицина. Там же, стр. 35.
232. Л. Н. Паскуцкая, В. Л. Драгинский. Водоснабжение и санитарная тех¬
ника, № 1, 15 (1970).
233. Л. II. Паскуцкая, В. Л. Драгинский. Сб. «Ионообменные материалы в на¬
уке и технике» (материалы семинара). Изд. Моск. Дома научно-техн.
пропаганды им. Ф. Э. Дзержинского, 1969, стр. 183.
234. В. Л. Драгинский. Научные труды АКХ им. К. Д. Памфилова, вып. 52.
Водоснабжение, № 5. М., ОНТИ, 1969, стр. 39.
235. Ю. И. Вейцер, Р. М. Стерина. Сб. «Ионообменные материалы в науке
и технике» (материалы семинара). Изд. Моск. Дома научно-техн. пропа¬
ганды им. Ф. Э. Дзержинского, 1969, стр. 187.
236. Л. А. Кулъский, Л. И. Панченко, В. А. Зайцева. Сб. «Водоподготовка
и очистка промышленных стоков», вып. 9.' Киев, «Наукова думка», 1972,
стр. 30.
237. Ф. Э. Молочникова, Е. И. Силина. Колыма, № 10, 14 (1968).
238. R. L. Williams. J. Amer. Water Works Assoc., 57, 801 (1965).
239. Т. M. Riddick. Chem. Engng, 68, 141 (1961).
240. Т. M. Riddick. J. Amer. Water Works Assoc., 53, 1007 (1961).
241. A. P. Rlack, D. G. Williams. Ibid., p. 589.
242. E. L. Bean, S. J. Campbell, F. R. Anspach. Water Works and Wastes
Engng, 2, N 11, 47 (1965).
243. E. L. Rean. Bull. Engng and Archit., N 56, 46 (1966).
244. А. К. Лившиц, Л. И. Габриелова. Горный журнал, № 2, 67 (1960).
245. А. Акадзава. Суйдо кёкай дзасси, № 268, 69 (1957); РЖХим., 39088
(1959).
246. П. I. Waterman, S. Sijderins. Пат. США 2761563 (1958).
247. Ю. И. Вейцер, Г. С. Колесников и др. Сб. «Ионный обмен и иониты». JL,
«Наука», 1970, стр. 262.
248. С. Кавамура. Суйдо кёкай дзасси, 12, № 303 (1959); РЖХим., 73868
(1960).
249. Ю. Б. Багоцкий, 3. Б. Чернова и др. Научные труды АКХ им. К. Д. Пам¬
филова, вып. 30. Водоснабжение, № 5. М., ОНТИ, 1969, стр. 46.
250. Технические указания на применение полиакриламида ПАА для очист¬
ки питьевых вод на городских водопроводах. М., ОНТИ АКХ
им. К. Д. Памфилова, 1969.
251. /. Т. Rurke, М. Т. Dajani et al. Пат. США 3377274 (1968).
252. Т. М. Riddick. Bull. Engng and Archit., N 56, 37 (1966).
253. S. Kawamura. J. Amer. Water Works Assoc., 65, 417 (1973).
Глава X
КОАГУЛЯЦИЯ В СПЕЦИФИЧЕСКИХ ПРОЦЕССАХ
водоподготовки
X. 1. УДАЛЕНИЕ ИСТИННО РАСТВОРЕННЫХ ПРИМЕСЕЙ
Кальций и магний. Известно, что химические реакции умягче¬
ния воды протекают практически мгновенно, но для формиро¬
вания хорошо осаждающихся агломератов требуется довольно
длительное время. Основные продукты известкового и известково¬
содового умягчения — карбонат кальция и гидроокись магния.
Возможно также образование сложных комплексных соединений.
Продукты реакции постепенно из коллоидного состояния
переходят в грубодисперсное: карбонат кальция — за счет
процессов кристаллизации, гидроокись магния — за счет коагуля¬
ционного структурообразования. Частицы СаС03 имеют симмет¬
ричную форму, характерную для конденсационно-кристаллиза-
ционных структур. Они обладают высокой плотностью, прочно¬
стью и не способны к тиксотропии. Коагуляционные структуры
Mg(OH)2, состоящие из первичных частиц размером около 100 А,
сильно гидратированы и обладают гораздо меньшей плотностью
[1, стр. 124]. По данным Лебедевой [2], объемный вес свежеоса-
жденной гидроокиси магния, определенный с помощью пикно¬
метра, равен 1,075 г!см3, т. е. примерно тот же, что у гидрооки¬
сей железа и алюминия. Согласно измерениям Кургаева
[3, стр. 60], объемный вес взвесей, образованных СаС03 и Mg(OH)2
в разной пропорции, составляет (г/см3):
100% Mg(OH)2 1,0026 15% Mg(OH)2+85% СаСОз 1,072
100% СаС03 1,26 25% Mg(OH)2 + 75% СаС03 1,028
С увеличением исходной концентрации золя Mg(OH)2 плот¬
ность коагулята возрастает [4].
В условиях формирования малорастворимой фазы при умяг¬
чении воды ведущая роль принадлежит значению pH среды, тем¬
пературе, интенсивности перемешивания. Оптимальные вели¬
чины pH выделения карбоната кальция лежат в пределах 9—10,
выделения гидроокиси магния — 10—11 [2, 3 (стр. 20)].
С повышением температуры и интенсивности перемешивания
воды скорость образования твердой фазы растет. С понижением
температуры процесс выделения и коагуляции Mg(OH)2 проте¬
кает медленнее, чем кристаллизация СаС03, твердая фаза про¬
дуктов формируется раздельно и технологические свойства взвеси
ухудшаются. В этом случае повышением интенсивности пере¬
Х.1. УДАЛЕНИЕ ИСТИННО РАСТВОРЕННЫХ ПРИМЕСЕЙ
321
мешивания воды трудно компенсировать отрицательное влияние
низких температур, поскольку оно может привести к разруше¬
нию структуры Mg(OH)2. При температурах 25—30° С скорости
образования СаС03 и Mg(OH)2 примерно одинаковы, и создаются
наиболее благоприятные условия формирования грубодисперсной
взвеси. При температуре выше 40° С из-за быстрого выпадения
в осадок основной массы шлама процесс роста структурирован¬
ных систем замедляется [1, 5 (стр. 44)].
Специфика совместного выделения твердой фазы продуктов
реакции умягчения состоит также в том, что гидроокись магния
задерживает кристаллизацию СаС03, причем с увеличением отно¬
сительного содержания Mg(OH)2 взвесь все более приобретает
свойства непрочных коагуляционных структур [6]. Условиям
формирования взвеси с наилучшими седиментационными харак¬
теристиками отвечает соотношение Mg(OH)2: СаС03 = 0,1—0,35
[3, стр. 69]. По мнению Кургаева, это связано с проявлением
максимальных сил сцепления частиц в агрегатах.
Помимо реакций совместного выделения СаС03 и Mg(OH)2,
происходит взаимодействие их с примесями, обусловливающими
мутность и цветность воды. Вследствие малых количеств гидро¬
окись магния не может быть эффективным коагулянтом для этих
примесей. Возникает необходимость в добавлении к воде солей
алюминия и железа.
Дозы коагулянтов зависят от степени устойчивости взвеси.
Изучение электрофоретической подвижности частиц СаС03 и
Mg(OH)2 в области значений pH, оптимальных для известково¬
содового умягчения, показало [71, что они имеют разноименные
заряды: СаС03 — отрицательный, Mg(OH)2 — положительный.
Отрицательный заряд СаС03 обусловлен преимущественным пе¬
реходом в раствор катионов Са2+ вследствие их большей по срав¬
нению с С03“ теплотой .гидратации [8, стр. 20J. С увеличением
магнезиальной жесткости отрицательный дзета-потенциал частиц
твердой фазы постепенно приближается к нулю и может даже
перемениться на положительный [91. Таким образом, величина
заряда и степень устойчивости частиц твердой фазы определяются
при умягчении соотношением [Mg(OH)2] : [СаС03]. По-видимому,
минимальная устойчивость отвечает приведенным выше соотно¬
шениям 0,1—0,35.
Добавление коагулянта перед вводом извести и соды способ¬
ствует увеличению скорости формирования малорастворимых
продуктов за счет устранения защитных коллоидов [5 (стр. 97),
10 (стр. 347)]. В системе Mg—A12(S04)3—Н20 возможно образова¬
ние сложных гетерополиядерных аквакомплексов, например,
следующего состава [11]:
[Mg (Н20)в12 [Al (H20)eh [SO4 (Н20)3]5.
322
Глава х. коагуляция в процессах водоподготовки
Длительность осаждения хлопьев сокращается на 60—70%. На
15—20% уменьшается остаточная жесткость воды.
При обработке вод с малой жесткостью и щелочностью коагули¬
рование может оказаться полезным для увеличения в известко¬
ванной воде остаточной кальциевой жесткости с целью углуб¬
ления дальнейшей декарбонизации. Коагулирование предлагается
использовать при умягчении не только пресных, но и соленых
(морских) вод [12].
Из коагулянтов при умягчении воды могут, естественно,
использоваться лишь те, которые образуют малорастворимые
соединения в области значений pH 9—И: NaA102, FeS04, FeCl3,
Fe2(S04)3 [13, 14J. Алюминат натрия действует не только как коа¬
гулянт, но и как подщелачивающий реагент, позволяющий сни¬
зить расход извести. Кроме того, способность продуктов гидроли¬
за NaA102 удалять из воды магний связана, по-видимому, с обра¬
зованием основных алюминатов магния [15, стр. 34].
При низкой магнезиальной жесткости (< 0,6 мг-экв/л) 'алю¬
минат натрия малоэффективен, хотя коагуляцию можно улуч¬
шить добавкой сернокислого магния или фосфата натрия. Однако
применять NaA102 экономически выгоднее все же при обработке
вод с высокой жесткостью [15 (стр. 34), 16]. В остальных случаях,
в особенности, если умягчаемая вода содержит много взвешенных
примесей, эффективны более дешевые соли железа. Их оптималь¬
ная доза (по безводному продукту) может быть определена в пер¬
вом приближении по эмпирическому соотношению [10, стр. 3431
®опт = 3 }/ Св , (Х.1)
в котором св — содержание взвешенных веществ в умягченной
воде.
Иногда используют смеси алюмината натрия с солями железа
[17, стр. 210]. Наилучшие результаты достигаются при коагули¬
ровании в две ступени: перед вводом извести и соды и после не¬
го [18, 19]. В дополнение к коагулянтам применяют активную
кремнекислоту [15 (стр. 35), 20, 21], органические полиэлектро¬
литы [22]. Ускорение кристаллизации СаС03 достигается исполь¬
зованием в качестве затравки мелкого песка, молотых карбо¬
ната кальция и окиси магния [23, 24]. Целесообразна рециркуля¬
ция осадка.
Железо и марганец. При наличии в воде (например, артезиан¬
ской) достаточно больших количеств двухвалентного железа
задача его удаления сводится к окислению до трехвалентного
посредством аэрации и подщелачивания известью х. Количе¬
ства образующихся при этом продуктов окисления и гидролиза
железа хватает для достижения критической концентрации и
1 Об окислении Fe2+ см. гл. VII.
Х.1. УДАЛЕНИЕ ИСТИННО РАСТВОРЕННЫХ ПРИМЕСЕЙ
323
проведения коагуляции в практически приемлемый промежуток
времени. Если же содержание растворенного железа не превы¬
шает 30 мг/л, воду приходится дополнительно обрабатывать
коагулянтом 2.
Как мы уже отмечали в гл. II, в поверхностных водах железо
содержится в виде истинно растворенных или коллоидных ком¬
плексов с органическими веществами и серой. Если преобладает
коллоидная фракция, для очистки воды, как правило, оказы¬
вается достаточным лишь добавление определенного количества
коагулянта. В случае же, когда основная масса железа пред¬
ставлена растворенными соединениями, необходимыми стано¬
вятся подщелачивание воды известью, добавление окислителей.
По мнению Кульского [26], соли железа и их смеси с солями
алюминия в процессе обезжелезивания обладают преимущества¬
ми перед сернокислым алюминием вследствие большей сорбцион¬
ной способности продуктов гидролиза железа. По-видимому,
немалая роль принадлежит и каталитическим эффектам. Однако
на практике предпочтение чаще всего отдают более легким в обра¬
щении солям алюминия. Совместное использование хлорного
железа и алюмината натрия дает возможность удалить из воды
не только железо и марганец, но и кремнекислоту. Оптимальное
соотношение NaA102 : FeCl3 равно 1 : 1 [26, стр. 93].
Иидзима и др. [27] предлагают для интенсификации процесса
обезжелезивания добавлять в избыточном количестве известковое
молоко, а затем, отделив образовавшуюся гидроокись железа
фильтрованием, нейтрализовывать избыток щелочи добавлением
солей алюминия. Эффект заключается в образовании алюминатов
кальция.
Результаты сравнительных испытаний разных методов реагеит-
ной обработки воды, содержащей небольшие количества железа
(5—6,5 мг/л), с последующим отделением хлопьев в осветлителе
со взвешенным осадком представлены в табл. Х.1.
В Институте коллоидной химии и химии воды АН УССР раз¬
работан метод удаления железа пропусканием воды через взве¬
шенный слой высокодисперсных частиц мела и гидроокиси алю¬
миния при малых скоростях восходящего потока. По мнению
авторов, в основе метода лежит реакция
4СаС03 + 4FeSCU + 02 + 6Н20 = 4Fe (ОН)3 + 4CaSOi + 4С02,
но, по-видимому, нельзя исключать и роли формирующихся в
этих условиях алюмо-кальциевых комплексов. При соотношении
А1(ОН)3 : СаС03, равном 1 : 1, указанным методом можно удалить
95—98% железа [26].
2 От небольших количеств растворенного железа можно освободиться филь¬
трационным способом, основанным на каталитическом действии кварце¬
вого песка и ранее образовавшихся продуктов гидролиза железа [25].
324
ГЛАВА X. КОАГУЛЯЦИЯ В ПРОЦЕССАХ ВОДОПОДГОТОВКИ
Таблица Х.1. Степень обезжелезивания воды разными реагентами [28J
Реагент
Доза реагента в отдельных сериях экспериментов, мг/л
СаО
30-35
30-35
30-35
30-35
30-35
30-35
30-35
FeCl3
—
2-10
—
4,0
—
—
—
FeS04
—
—
—
—
—
5-10
С1г
—
—
0,5
—
0,5-2
2-4
2-4
СаС03
—
—
—
—
—
10
—
АК
—
—
4,0
2,0
—
—
—
Остаточное содер¬
жание железа, мг/л
3,9
1,8
1,0
2,3—3,0
<3,5
1,4
1,1—1,4
Достаточно полное окисление двухвалентного марганца в
трех- и четырехвалентный с образованием гидроокисей Мп(ОН)3
и Мп(ОН)4 происходит в зоне значений pH 9,5—10,5 [10, стр. 429].
Процесс протекает в несколько стадий с образованием промежу¬
точного соединения МпООН [29]. Так же, как и при обработке
железосодержащих вод, может оказаться полезной добавка окис¬
лителей.
Описан способ удаления марганца (при его исходной кон¬
центрации в воде около 1 мг/л) путем добавления в качестве акти¬
ватора КМп04 и последующей обработкой сернокислым алюми¬
нием [30]. В этом случае коагулянт следует добавлять к воде
через 5—10 мин после введения активатора [31, стр. 95]. Чехо¬
словацкие исследователи [32] в дополнение к КМп04 рекомендуют
добавлять МпС12 и MnS04 и проводить коагуляцию при pH 6,2—
6,5. Этим способом содержание марганца может быть снижено с
1,5 до 0,15 мг/л.
Фтор. В процессах обесфторивания воды используются сорб¬
ционные свойства продуктов гидролиза коагулянтов и способ¬
ность катионов А13+ к комплексообразованию. Главные реагенты
при хемосорбционном обесфторивании — гидроокиси и основные
соли алюминия и магния. Их применение дает преимущества по
сравнению с активным углем, активной окисью алюминия, фос¬
фатами [33].
Уже в первых исследованиях по сорбции фтора на продуктах
гидролиза алюминия, проведенных в 1934 г. [34], обнаружено,
что потребность в сернокислом алюминии связана с количеством
удаляемого фтора стехиометрически, причем для удаления 1 г F
требовалось 40—45 г A12(S04)3. Сделано предположение об об¬
разовании малорастворимых фтор-алюмшшевых комплексов.
Х.1. УДАЛЕНИЕ ИСТИННО РАСТВОРЕННЫХ ПРИМЕСЕЙ
325
Позднее, однако, показано, что удельный расход A12(S04)3
сильно зависит от режима обесфторивания, в особенности от ве¬
личины pH среды, и может значительно превышать 40—45 г/г.
В частности, по данным Лурье и др. [35], для снижения концен¬
трации фтора от 10 до 1—1,5 мг/л при pH > 6,5 требуется до
120 г сульфата алюминия на 1 г удаленного фтора, а при pH ~ 4,
т. е. в зоне формирования основных солей и полимерных комплек¬
сов, удельная потребность в коагулянте снижается до 30—40 г/г.
С уменьшением исходного содержания фтора удельный расход
A12(S04)3 возрастает.
Из работы Кульпа и Штальтенберга [33], посвященной изуче¬
нию разных режимов обесфторивания воды сернокислым алюми¬
нием, вытекает следующее.
1. Для снижения концентрации фтора с 3,6 до 1,0 мг/л тре¬
буется 140—170 мг/л безводного сернокислого алюминия.
2. Наилучшее обесфторивание достигается при определенных
гидравлических условиях: интенсивное перемешивание воды с
раствором коагулянта в течение 2 мин, а затем — медленное
перемешивание, предотвращающее выпадение хлопьев в осадок.
Химическое равновесие в системе наступает через 30 мин после
прекращения быстрого перемешивания.
3. В период медленного перемешивания pH следует поддер¬
живать равным приблизительно 5, а при отделении хлопьев в
осадок — повышать pH добавлением щелочи до значений, со¬
ответствующих оптимальным условиям осаждения.
4. Фракционированный ввод коагулянта 5—6 отдельными пор¬
циями позволяет сократить потребность в коагулянте примерно
на 20%.
5. Применение в качестве вспомогательных средств коагуля¬
ции глин, гидроксилэтилцеллюлозы и активной кремнекислоты,
а также рециркуляция отработанного осадка способствуют ус¬
корению собственно коагуляции, но не дают ощутимых резуль¬
татов в отношении обесфторивания.
Дальнейшие исследования, проведенные Клячко [10 (стр. 447),
36], в основном подтвердили эти выводы: процесс обесфторивания
сернокислым алюминием протекал наилучшим образом при pH
4,8—5,5, а удельный расход A12(S04)3 зависел от величины за¬
данной остаточной концентрации фтора (см. рисунок).
Приобесфторивании воды солями алюминия возможны разные тех¬
нологические решения. Деревянко и др. [37] рекомендуют двухсту¬
пенчатое коагулирование с очисткой воды на осветлителях со
взвешенным осадком. После обработки воды, содержащей 4,5 мг/л
фтора, сернокислым алюминием (85 мг/л на первой и 40 мг/л на
второй ступени) остаточные концентрации фтора составляли 0,8-—
1,2 мг/л. Большая часть фтора (60—70%) удалялась после пер¬
вой ступени. Кузякин и Когановский [38] описали способ обес-
12 Е. Д. Бабенков
326
ГЛАВА X. КОАГУЛЯЦИЯ В ПРОЦЕССАХ ВОДОПОДГОТОВКИ
Зависимость остаточной кон¬
центрации фтора (Сф) в воде
от отношения (а) дозы серно¬
кислого алюминия к исходной
концентрации фтора
фторивания сточных вод сульфатом алюминия с добавкой поли¬
акриламида.
Следует иметь в виду, что заметное снижение концентрации
фтора в речной воде происходит при обычном режиме реагентной
обработки — дозами коагулянта, предназначенными для освет¬
ления воды. Например, при очистке иртышской воды содержание
фтора уменьшалось с 0,11—0,15 до 0,07—0,1 мг/л [39]. Замена
сульфата алюминия на оксихлорид позволяет расширить область
оптимальных pH среды в сторону более высоких значений [40].
Ориентировочная оценка обесфторивающей способности разных
коагулянтов сделана в работе [41].
Помимо своего основного назначения — связывать анионы фто¬
ра, алюминийсодержащие коагулянты можно использовать для
регенерации активной окиси алюминия, применяемой при обес-
фторивании в виде фильтрующей загрузки. Удаление фтора окисью
алюминия рассматривается как ионообменный процесс, а регене¬
рация ее раствором A12(S04)3 — как образование комплексов
Al—F с одновременной нейтрализацией избыточной щелочности
[31 (стр. 108), 42]. Обесфторивание воды в ходе известково-содо-
вого умягчения объясняли первоначально сорбцией фтора на
хлопьях Mg(OH)2 [43, 44]. Позднее доказано образование мало¬
растворимого оксифторида магния [45].
На основе экспериментов и производственного опыта уста¬
новлена следующая зависимость количества удаленного фтора
от концентрации магния в умягчаемой воде [43]:
Foe = F„(1 — 0,07 /[М£Г), (Х.2)
где FH и Foc — концентрации фтора соответственно в исходной
и осветленной воде, мг/л\ [Mg] — концентрация магния в исход¬
ной воде, мг/л.
В исследованиях, выполненных во ВНИИ ВОДГЕО, выраже¬
ние (Х.2) нашло экспериментальное подтверждение при отде¬
лении твердой фазы отстаиванием. При использовании осветли¬
телей со взвешенным осадком на удаление 1 г фтора расходова¬
Х.1. УДАЛЕНИЕ ИСТИННО РАСТВОРЕННЫХ ПРИМЕСЕЙ
327
лось 20—30 г Mg, т. е. примерно в 2 раза меньше, чем это следу¬
ет из соотношения (Х.2). Установлено также что обесфторивание
протекает наиболее интенсивно в зоне pH 9,4—10,5, оптималь¬
ных для выделения Mg(OH)2, и завершается в основном за 8—
10 мин. Температура воды не оказывает существенного влияния
на длительность и глубину обесфторивания [31 (стр. 136), 36].
В случае недостаточной магнезиальной жесткости воды полезны
добавки сульфата или хлорида магния [10, стр. 449].
Кремний. Принято считать, что удаление продуктами гидро¬
лиза алюминия, железа и магния коллоидной фракции кремне¬
кислоты вызвано взаимной коагуляцией золей, а удаление истин¬
но растворенной фракции — сорбцией [10, стр. 431]. Однако не
подлежит сомнению и важная роль чисто химических взаимодей¬
ствий.
Обескремнивание солями алюминия происходит наилучшим
образом в области pH 7,1—7,6 [46, стр. 643]. Поэтому при исполь¬
зовании A12(S04)3 воду, как правило, требуется подщелачивать.
Большие количества SO|”-hohob, вводимых с раствором сернокис¬
лого алюминия, могут затруднять удаление кремневой кислоты
вследствие конкуренции с ней за координационные участки гид-
роксокомплексов. В этом отношении лучшие результаты дает
использование алюмината натрия (с подкислением воды до оп¬
тимума pH). На удаление 1 г кремнекислоты расходуется 10—15 г
NaA102. При значениях pH 8,5—9,5 кремнекислота успешно уда¬
ляется солями железа. Удельный расход последних не превыша¬
ет 5—8 г на 1 г удаленной кремнекислоты. Результаты исследо¬
ваний Схенка и Вебера [47] указывают на возможность формиро¬
вания комплексов Fe2+—Si02.
Соли алюминия и железа могут быть заменены свежеосажден-
ными гидроокисями металлов, действующими эффективно в до¬
зах 100—200 мг/л при pH 7,5—8,5. Целесообразно применение
электрокоагуляции [15, 48].
В условиях магнезиального умягчения воды хорошо сорбиру¬
ет кремнекислоту гидроокись магния. Оптимальные величины pH
находятся в пределах 10—10,5: при pH < 10 ухудшение удаления
кремнекислоты связано с недостаточной ее диссоциацией, при
pH > 10,5 — с уменьшением сорбционной емкости Mg(OH)2. До¬
бавление коагулянтов способствует не только быстрому отделе¬
нию Mg(OH)2 в осадок, но и уменьшению остаточных концентра¬
ций кремния. Дополнительного обескремнивания на 10—35%
можно достигнуть добавлением к воде 5—15 мг/л активной кремне¬
кислоты [49, 50].
Сероводород. В работах советских ученых [51, 52] получил
развитие метод удаления сероводорода, предусматривающий ис¬
пользование солей железа. По этому методу в результате прямого
химического взаимодействия гидроокиси железа с сероводородом
12*
328
ГЛАВА X. КОАГУЛЯЦИЯ В ПРОЦЕССАХ ВОДОПОДГОТОВКИ
в щелочной среде
2Fe (ОН)8 + 3H2S = Fe2S3 + 6Н20
1
и в кислой среде
2Fe (ОН)з + 3H2S = 2_FeS + 6Н20 + S
I
образуются сульфиды железа. Они малорастворимы и быстро
выпадают в осадок. Гидроокись железа может быть регенерирова¬
на аэрацией:
2Fe2S3 + 6Н20 + 302 = 4Fe (ОН)3 + 6S,
4FeS + 6Н20 + ЗОа = 4Fe (OH)s + 4S.
Стехиометрически на реакцию с 1 г H2S расходуется 0,23 г
FeS04 или 0,45 г FeCl3. Однако фактическая потребность в коа¬
гулянтах, как показали исследования, проведенные во ВНИИ
ВОДГЕО [10 (стр. 481), 52], может в несколько раз превышать
теоретическую.
Сотрудниками Ленинградского НИИ АКХ им. К. Д. Памфи¬
лова предложен способ удаления сероводорода основным хлори¬
дом алюминия при значениях1 pH среды 7,5—8,5 [53]. С целью
экономии коагулянта процесс удаления H2S рекомендуется осу¬
ществлять в осветлителях со взвешенным осадком.
X. 2. ОЧИСТКА СТОЧНЫХ ВОД
X. 2.1. Бытовые и промышленно-бытовые
сточные воды
Несмотря на то, что способ очистки воды коагулянтами из¬
вестен с древних времен, его использование при обработке быто¬
вых и промышленно-бытовых сточных вод до недавнего времени
сильно ограничивалось. Это связано, во-первых, с тем, что кри¬
териями оценки санитарной надежности очистки являлись почти
исключительно биологические показатели. Во-вторых, исследова¬
тели указывали на большую потребность в коагулянтах, а также
на ряд технических и экономических причин ограничения исполь¬
зования коагуляции, а именно:
1. Трудность дозировки коагулянтов из-за резких колебаний
состава сточных вод.
2. Плохие условия отделения в осадок коагулированной взве¬
си вследствие колебания температуры и расхода воды.
3. Резкое увеличение объема осадка с одновременным умень¬
шением возможности его утилизации.
Х.2. ОЧИСТКА СТОЧНЫХ ВОД
329
По вопросу совмещения химического коагулирования с
биологическими методами очистки мнения исследователей расхо¬
дятся. Одни исследователи утверждают, что при попадании про¬
дуктов гидролиза коагулянта в анаэробный сбраживатель про¬
цесс сбраживания нарушается вследствие обволакивания биомассы
и создания гидроокисных оболочек, препятствующих про¬
никновению кислорода; по мнению других, присутствие в осадках
сточных вод гидроокисей не ухудшает процессов сбраживания,
обезвоживания и отделения газов. Очевидно, «экранирующее»
действие гидроокисных оболочек определяется видом коагулянта
и его дозой.
Из числа упомянутых причин ограниченного использования
коагулянтов при очистке бытовых и промышленно-бытовых сточ¬
ных вод первая и вторая причины могут быть в принципе устра¬
нены за счет внедрения автоматического дозирования коагулянтов
по качественным показателям стоков, устройства резервуаров-
усреднителей для выравнивания расхода и температуры воды,
применения тонкослойного отстаивания и проведения ряда дру¬
гих мероприятий. Повышенный расход коагулянтов может быть
в известной степени компенсирован (с экономической точки зре¬
ния) использованием дешевых отходов промышленного производ¬
ства и высокомолекулярных флокулянтов.
Наибольшие трудности связаны, по-видимому, со значитель¬
ным увеличением объема осадков. Если в отсутствие коагуляции
объем осадка составляет 0,4—0,6% от объема обработанной сточ¬
ной жидкости, то при коагуляции этот показатель возрастает до
2,3-2,5% [54, 55]. ЙЗ?,
Вместе с тем нельзя не учитывать весьма существенные пре¬
имущества, которыми способы физико-химической 'обработки
воды, и, в частности, коагулирование, обладают перед традици¬
онными методами биологической очистки бытовых и промышленно¬
бытовых стоков.
1. Сокращение длительности очистки.
2. Меньшая площадь очистных сооружений.
3. Более полное удаление из сточной жидкости трудноокисля-
ющихся примесей (фосфатов, синтетических ПАВ и др.) и микро¬
элементов .
4. Почти полная независимость результатов очистки от сезо¬
на года.
5. Меньшее изменение величины pH сточной жидкости.
6. Меньшая загниваемость осадков.
7. Неподверженность процесса очистки влиянию токсических
веществ.
8. Лучшие возможности для автоматизации процесса очистки.
Опыт применения коагуляции при очистке бытовых и промыш¬
ленно-бытовых сточных вод показывает, что по эффективности
330
ГЛАВА X. КОАГУЛЯЦИЯ В ПРОЦЕССАХ ВОДОПОДГОТОВКИ
этот метод не уступает неполной биологической очистке в аэро-
тенках: происходит снижение биохимической потребности в кис¬
лороде (ВПК) на 50—75%, окисляемости — на 70—80%, взве¬
шенных примесей — на 30—65%; задерживается часть бактерий.
Наилучший эффект дает использование железосодержащих коа¬
гулянтов [54—58]. Для подщелачивания применяется известь.
Оптимальные дозы коагулянтов находятся в пределах 50—
200 мг!л по безводному продукту и могут быть снижены на 20—
30% за счет рециркуляции коагулированной взвеси [56] и добав¬
ления полиэлектролитов [56, 59]. Процесс коагуляции может
быть совмещен с обработкой воды активным углем [60].
Указывается [56, 61], что метод коагуляции вполне конкурент¬
носпособен в сравнении с методами биологической очистки сточ¬
ных вод, а при использовании в качестве коагулянтов отходов
производства стоимость его может оказаться ниже. На станции
очистки сточных вод округа Маттабассетт (США) отказались от
биохимического метода в пользу обработки вод хлорным желе¬
зом и известью из-за сильного изменения величины pH в резуль¬
тате биохимической очистки [58]. На другой станции [59] обра¬
ботку воды коагулянтами считают целесообразной зимой и в пе¬
риод паводков, когда биологическая очистка ухудшается вслед¬
ствие недостаточного количества субстрата.
В соответствии с программой перспективного внедрения новых
технологических решений в области очистки сточных вод Управ¬
ление но охране окружающей среды США предполагает широкое
внедрение коагуляции для удаления фосфатов [61]. Уже теперь
коагуляция как способ очистки воды от фосфатов используется
примерно на 300 пунктах обработки сточных вод [62]. В перспек¬
тивном плане развития канализации реагентным методам очист¬
ки отведена доминирующая роль [63].
Описанные примеры использования коагулянтов относятся к
сточным водам, подвергнутым лишь простейшей предваритель¬
ной обработке — механической очистке от крупных примееей.
В последние годы коагулирование все более широко применяет¬
ся и как способ доочистки (вторичной или третичной очистки)
стоков после проведения биохимической обработки. В этих слу¬
чаях наряду с солями железа находят применение соли алюминия
и их смеси с солями железа [64—73], в качестве флокулянтов —
полиэлектролиты [66, 68, 72] и активная кремнекислота [68].
Для улучшения отделения грубодисперсных примесей рекомен¬
дуют перед добавлением коагулянтов пропускать через сточную!
воду топочные газы [65], используют рециркуляцию осадка [71].
При доочистке сточных вод наряду с удалением механических
примесей и снижением химической потребности в кислороде (ХПК)
уменьшаются концентрации фосфатов, азота, синтетических де¬
тергентов и многих других примесей. Оптимальные значения
Х.2. ОЧИСТКА СТОЧНЫХ ВОД
331
pH при использовании солей окисного железа составляют 4—6,, 5,
а область критических значений Д11 коагулированной взвеси
-15 -г- +10 мв [74].
X. 2.2. Промышленные сточные воды
Очистка коагулянтами промышленных сточных вод имеет много
характерных особенностей, обусловленных разнообразием их фи-
зико-химического состава, резкими колебаниями во времени кон¬
центрации загрязнений, температуры и расхода сточных вод. Со-
лесодержание и величина pH вод могут быть самыми разными и
определять выбор коагулянта и вспомогательных реагентов. В
ряде случаев, как уже отмечалось, открываются возможности
использования в качестве коагулянтов гидролизующихся соеди¬
нений, присутствующих в сточных водах или образующихся в
результате смешения стоков разных цехов (производств).
Главная трудность удаления из промышленных сточных вод
дисперсных загрязнений состоит в том, что эти загрязнения чаще
всего обладают совершенно разными коллоидно-химическими
свойствами. Например, тяжелые шламы, формирующиеся при
очистке коагулянтами стоков металлообрабатывающей или уг¬
леобогатительной промышленности, быстро выпадают в осадок,
в то время как эмульгированные примеси легче удалить флота¬
цией. Изменения pH среды, вызванные добавлением коагулянтов
и вспомогательных реагентов, могут оказать благоприятное влия¬
ние на поведение одних примесей, но вызвать стабилизацию дру¬
гих.
В этом разделе мы попытаемся дать обзор наиболее характер¬
ных примеров использования коагуляции для очистки промыш¬
ленных сточных вод разного типа.
X. 2.2.1. Нефте- и маслосодержащие сточные воды
После нефтеловушек, действующих без применения коагулянтов,
остаточное содержание нефти и нефтепродуктов составляет 50—
250 мг/л [75]. Коагулирование солями алюминия и железа поз¬
воляет снизить его в 2—3 раза [75—79], причем лучшие результа¬
ты дает флотация. Оптимальные дозы коагулянтов лежат в пре¬
делах 25—100 мг/л [76—80], но иногда превышают и 300 мг/л [81].
Результаты очистки зависят от исходной концентрации нефти
в сточных водах, величины pH среды, содержания взвешенных
примесей и степени их дисперсности. Регулировка pH произво¬
дится обычно добавлением известкового молока [75, 81]. Удель¬
ный расход A12(S04)3 колеблется от 1,1 до 4 г на 1 г удаленных
нефтепродуктов [76, 77, 79, 81].
Из солей железа особенно широкое применение находит за-
кисное сернокислое железо [75, 80, 82—86]. Отмечается, что эф¬
332
ГЛАВА X. КОАГУЛЯЦИЯ В ПРОЦЕССАХ ВОДОПОДГОТОВКИ
фект очистки во многом зависит от выбранного для окисления
двухвалентного железа щелочного реагента и солевого состава
воды [84].
Хорошие результаты достигаются при использовании элект¬
рофлотации с растворимыми анодами. Так, на Горьковском неф-
темаслозаводе концентрацию нефтепродуктов в электрофлота¬
торе со стальными анодами удалось снизить с 376 до 23 мг/л при
удельном расходе электроэнергии 0,6—0,9 кет-час/м3 или 2,4—
3,6 вт-час на 1 г удаленной нефти [77]. По данным Герасимова
[87], в процессе электрофлотации с растворимыми анодами расхо¬
дуется около 0,25 г железа на удаление 1 г нефти. Железо для
электрофлотации рекомендуется использовать в закисном виде,
так как хлопья Fe(OH)2 легче флотируются [88]. В качестве фло¬
кулянтов применяют органические полиэлектролиты [89 (стр. 66),
90] и активную кремнекислоту [77, 91].
Имеются сведения о высокой технологической эффективности
гидролизующихся коагулянтов при очистке сточных вод от мазу¬
та [92, 93] и масел [77, 94—99]. Применение флотации с добавкой
сернокислого алюминия и серной кислоты дало возможность сни¬
зить концентрацию масел на одном из заводов компании Джене-
рал Моторе на 99% [99]. Электрофлотация в присутствии хлор¬
ного железа и сернокислого алюминия обеспечила при затратах
электроэнергии 1,06 квт-час/м3 уменьшение содержания масла
с 200—1200 до 5—15 мг/л [95]. В качестве оптимальных рекомен¬
дованы значения pH < 7 [90, 99].
В результате тщательных исследований очистки маслосодер¬
жащих стоков металлообрабатывающего завода установлено, что
из числа многих испытанных коагулянтов (в комбинации с из¬
вестью, кислотой и ВМФ) лучшие результаты дает совместное
применение FeS04 и Са(ОН)2 в щелочной среде [100].
Интенсифицировать отделение масел можно предварительным
центрифугированием [98], добавкой тонкодисперсной окиси же¬
леза [97], активной кремнекислоты [101], полиэлектролитов [90,
96], торфа [102].
Высокая эффективность коагулирования делает этот способ
очистки нефте- и маслосодержащих вод одним из наиболее перс¬
пективных.
X. 2.2.2. Сточные воды предприятии
целлюлозно-бумажной промышленности
Из коагулянтов наиболее широкое применение находит серно¬
кислый алюминий. Реже используют хлорное железо [103—107],
железный купорос [108—110], оксихлорид алюминия [111]. До¬
зы коагулянтов колеблются в пределах от 35 до 600 мг/л [112,
ИЗ], составляя в среднем 80—200 мг/л. Для корректировки зна¬
чений pH почти исключительно используется известь [103, 107,
Х.2. ОЧИСТКА СТОЧНЫХ ВОД
333
109, 112, 114]. Замена извести содой приводит к ухудшению ос¬
ветления сточных вод [103].
Оптимальные значения pH для солей алюминия и окисных
солей железа составляют 5—6 [110, 115, 116], но при отсутствии
органических коллоидов (например, красящих веществ) эти зна¬
чения не выходят за границы области, обычной для природных
вод. Обработка стоков железпым купоросом наиболее эффектив¬
на при pH 9—9,5 [110]. В результате коагулирования происхо¬
дит снижение концентрации грубодисперсных примесей, краси¬
телей, уменьшение окисляемости и ВПК. По некоторым показа¬
телям степень очистки сточных вод коагулянтами превышает
степень очистки биологическими методами [107].
Для обработки сульфатных стоков рекомендована электро¬
коагуляция с растворимым алюминиевым анодом и добавкой со¬
лей трехвалентного железа [106] или предварительной обработ¬
кой воды щелочью [117]. В качестве сорбентов и замутнителей
предложено использовать бентонит и другие глины [103, 117],
каменноугольную золу [118]; в качестве флокулянтов — актив¬
ную кремнекислоту [107, ИЗ, 119, 120], катионные и анионные
полиэлектролиты [ИЗ, 118, 120—124], карбоксилметилцеллюло-
зу [124].
Примером коагуляции целлюлозы и других органических за¬
грязнений без добавления гидролизующихся коагулянтов может
служить способ смешения вискозных и кислых вод в соотношении,
обеспечивающем поддержание pH ~ 4 [125]. В результате нейт¬
рализации едкого натра, являющегося стабилизатором соедине¬
ний целлюлозы, происходят коагуляция и осаждение примесей.
Коагуляция ускоряется при добавлении сульфата натрия.
Для улучшения отделения органических соединений и более
глубокого осветления стоков производства сульфатной целлюло¬
зы используется метод двухстадийного коагулирования серно¬
кислым алюминием [126]. На первой стадии за счет ввода коагу¬
лянта pH понижают примерно до 4. Происходит формирование
алюминийорганических комплексов. На второй стадии добавле¬
нием известкового молока повышают pH до 5,4, что приводит к
образованию хлопьев коагулированной взвеси и их выпадению
в осадок.
Имеется тенденция к расширению использования коагуляции
при очистке сточных вод предприятий целлюлозно-бумажной про¬
мышленности [127].
X. 2.2.3. Сточные воды, содержащие красящие
и дубильные вещества
Помимо сернокислого алюминия, применяют закисные и окисные
соли железа [128—133], хлористый алюминий [131], глины [134].
В зависимости от состава и исходной концентрации красителей
334
ГЛАВА X. КОАГУЛЯЦИЯ В ПРОЦЕССАХ ВОДОПОДГОТОВКИ
и дубильных веществ расход коагулянта меняется от 200—300
до 800—1000 мг/л [129, 130, 135—137], но в отдельных случаях
(например, при очень больших концентрациях загрязнений) до¬
стигает 2000 мг/л [138]. Коагуляцию солями алюминия и трехва¬
лентного железа желательно осуществлять при относительно низ¬
ких значениях pH, порядка 4—6, коагуляцию солями двухвалент¬
ного я?елеза — при pH ~ 9—10 [137, 139]. Для интенсификации
процесса коагуляции рекомендуется использовать минеральные
сорбенты [140], высокомолекулярные флокулянты [129, 135—137,
139, 141] и окислители [128]. Достигается снижение ВПК и ХПК
сточных вод на 40—70% [129, 137, 142], окисляемости — на 50—
70% [130, 135].
Хорошие результаты при очистке сточных вод, содержащих
красители на основе алкильно-эфирных смол, достигнуты приме¬
нением FeS04 или A12(S04)3 (300—400 мг/л), извести и флокулян¬
та Р-26. Окисляемость при этом снизилась на 80—90% [143].
Таблица Х.2. Примеры использования разных реагентов
при обработке сточных вод
Виды сточных вод
Реагенты
Литера¬
тура
Сточные воды, содержащие
хром
Соли двух- и трехвалентного же¬
леза
[163]
цианиды
FeS04
[164]
фенол и формальдегид
Соли алюминия и железа
[165]
пекаль
То же
[166, 1671
латекс
A1-2(S04)3, Fe2'4S04)3
1168]
Бентонит
[169]
Оксихлорид алюминия и ПАА
[170]
тр инитротолуо л
FeCl3, A12(S04)3
[171]
многоядерные ароматиче¬
AU(S04'3, сажа, известь
[172]
ские углеводороды
Сточные воды производств
поливинилхлорида
A12(S04)3, FeCl3, ПАА
[173, 174]
полимеризационных пласт¬
A12(S04)3, А К
[175]
масс
синтетического спирта
FeS04, A12(S04)3, и шесть
[176]
желатины
FeCl3, A12(S04)3
[177,178]
искусственного волокна
FeS04
[1791
кинопленки
A12(S04)3
[180]
Х.З. ОБРАБОТКА ОСАДКОВ
335
X. 2.2.4. Прочие сточные воды
Имеются многочисленные примеры эффективного использования
солей алюминия и железа с добавкой и без добавки извести и дру¬
гих вспомогательных средств при очистке стоков от первичной
обработки пищевых продуктов, а также от продуктов, образую¬
щихся в результате их переработки: от промывки зерна [144]
и сахарной свеклы [145], предприятий плодоовощной промышлен¬
ности [146—148], животноводческих ферм [149, 150], боен [151],
макаронных фабрик [152], заводов по производству пива [153],
вина [154, 155] и ликеро-водочных изделий [156].
Большое распространение метод очистки сточных вод коагу¬
лянтами получил на горнообогатительных и металлургических
предприятиях [125, 157—162], в различных отраслях химической
и текстильной промышленности. Несколько наиболее интересных
примеров удачного использования коагуляции для очистки сточ¬
ных вод разных производств приведены в табл. X. 2.
Применение электрокоагуляции со стальными или алюминие¬
выми электродами рекомендовано для очистки локальных стоков
предприятий машиностроительной и химической промышленнос¬
ти, содержащих эмульгированные примеси, хром и некоторые
виды полимеров.
X. 3. ОБРАБОТКА ОСАДКОВ
X. 3.1. Коагуляция замораживанием
Обработка осадков водопроводных и канализационных очистных
станций путем вымораживания воды — один из видов «физичес¬
кой» коагуляции, цель которого состоит в снижении остаточного
заряда структурообразующих частиц [181] и количественном пе¬
рераспределении различных форм связи воды с твердым веществом
с переводом части связанной воды в свободное состояние [182].
Вымораживание воды является эффективным способом уплот¬
нения осадков, повышения скоростей их осаждения после оттаи¬
вания, резкого увеличения производительности вакуум-фильт-
ров [182—184]. Как показали исследования, проведенные во
ВНИИ ВОДГЕО [185, стр. 81], замораживанием при температуре
—5 -j 40° С удается снизить влажность осадка А1(ОН)3 с 97
до 40—58% через трое суток и до 25—40% — через пятеро суток.
Применение замораживания осадка на водопроводной станции
округа Монро (США) [1861 в течение зимы дало возможность уве¬
личить содержание сухого вещества в нем от 3,5 до 17,5%.
Температура, которую нужно поддерживать при обработке
осадков замораживанием, зависит от размера пор осадков: чем
меньше поры, тем более низкие температуры требуются для дос¬
336
ГЛАВА X, КОАГУЛЯЦИЯ В ПРОЦЕССАХ ВОДОПОДГОТОВКП
тижения одного и того же эффекта [182, 187]. Максимальное раз¬
рушение первоначальной структуры осадка и его уплотнение име¬
ют место при медленном замораживании [188]. Существенное зна¬
чение имеют время пребывания осадка в оттаявшем состоянии и
режим перемешивания осадка после оттаивания [189].
Искусственное замораживание — дорогостоящий процесс
[185, 190]. Поэтому, к сожалению, приходится констатировать,
что замораживание может дать ощутимый экономический эффект
лишь тогда, когда оно происходит естественным путем. Это огра¬
ничивает сферу применения метода.
Анализ преимуществ и недостатков метода замораживания,
а также влияния условий замораживания на конечные физичес¬
кие параметры осадков дан в работах [191—193].
X. 3.2. Обработка коагулянтами и флокулянтами
Замораживание дает ощутимые и устойчивые результаты в отно¬
шении осадков коагулированной взвеси, образующейся при очист¬
ке природных вод. В осадках сточных вод в больших количест¬
вах присутствуют органические вещества, которые могут препятст¬
вовать процессу коагуляции при замораживании, точнее — его
необратимости. Такие осадки перед замораживанием или осуществ¬
лением какого-либо другого способа обезвоживания (на ваку¬
ум-фильтрах, пресс-фильтрах и т. д.) обрабатывают коагулянта¬
ми, известью, флокулянтами, хлором. Предпочтение отдают хлор¬
ному железу [178, 194—200], хлорированному или нехлорирован-
ному железному купоросу [196, 201, 202], сернокислому окисному
железу [178, 203]. Из алюминийсодержащих коагулянтов чаще
всего используют A12(S04)3.
В результате экспериментальных исследований установлено,
что больший эффект дает обработка коагулянтами сырого (несбро-
женного) осадка, поскольку степень дисперсности первичных
частиц ниже, чем частиц сброженного осадка, и потребность в
коагулянтах уменьшается [201]. Присутствие в осадках соеди¬
нений железа, хрома, меди и кислот приводит к экономии коагу¬
лянтов; присутствие нефтепродуктов, масел и жиров — к их пе¬
рерасходу [195, 204].
Дозы коагулянтов возрастают также с увеличением концент¬
рации твердого вещества в осадке, его щелочности и промежутка
времени, прошедшего от момента образования осадка до момента
ввода коагулянта [196]. С целью сокращения расхода коагулян¬
та осадок рекомендуется перед обработкой коагулянтом промыть
очищенной водой [196, 201]. Расход коагулянта находится обыч¬
но в пределах от 1,5—2 до 8,5—10% от веса сухого вещества осад¬
ка [195, 199, 204, 205], хотя имеются сведения и о более высоких
дозах: в частности, исследования, проведенные в Англии [201],
X. ЛИТЕРАТУРА
337
выявили необходимость применения хлорированного железного
купороса в количестве 14—18% от веса сухого вещества осадка.
Туровский [206] предлагает определять потребность в коагу¬
лянте при обработке осадка перед вакуум-фильтрацией с учетом
его влажности, щелочности и удельного сопротивления. Добав¬
ление извести в количестве 0,6—25% [204, 206] от веса сухого
вещества (по СаО) приводит к значительному (на 30—50%) сокра¬
щению расхода коагулянтов.
По данным Теннея и др. [207], оптимальные значения pH сре¬
ды при обработке осадков сточных вод перед вакуум-фильтрами
составляют: для солей железа 6—7, для солей алюминия 4,5—
5,5. При совместном применении хлорного железа и извести ре¬
комендуется сначала добавлять коагулянт, затем — известь, а
смешение осадка с реагентами осуществлять в ершовых смесите¬
лях, поддерживая pH среды выше 9 [195, 196, 201]. Оценка фильт¬
рационной способности активного ила (после биохимической очи¬
стки стоков производства древесно-волокнистых плит), обрабо¬
танного FeS04, СаО, A12(S04)3, FeCl3 и золой, показала, что наи¬
более обезвоженный осадок получается при использовании FeCl3
и СаО [200].
С целью экономии реагентов Туровский [197] предложил сна¬
чала обрабатывать осадок хлорным железом и сразу же подвер¬
гать его уплотнению (для снижения влажности до 94%), а затем
добавлять известь в количестве 10—12% от веса сухого вещества
осадка.
Самостоятельно или в сочетании с гидролизующимися коагу¬
лянтами применяются катионные и анионные полиэлектролиты:
сепаран, нэлько-600, аэрофлок [194], виниловый полимер [208],
ПАА [203], аквафлок-410 и -415 [209], праэстол-114, -2935 и -444К
[210], седипур [211].
Л итератора
1. В. П. Чалый. Гидроокиси металлов. Киев, «Наукова думка», 1972.
2. Н. С. Лебедева. Автореферат канд. дисс. М., ВНИИ ВОДГЕО, 1954.
3. Е. Ф. Кургаев. Основы теории и расчета осветлителей. М., Стройиздат,
1962.
4. S. В. Kaningo, Р. К. De, V. P. Ват. Indian J. Technol., 8, 23 (1970).
5. Обработка воды на тепловых электростанциях. Под ред. В. А. Голубдова.
М., «Энергия», 1966.
6. Е. Ф. Кургаев. Коллоидн. ж., 19, 72 (1957).
7. А. P. Black, R. F. Christman. J. Amer. Water Works Assoc., 53, 737 (1961).
8. О. H. Григоров. Электрокинетические явления. Л., Изд-во ЛГУ, 1973,
стр. 20.
9. F. Z. Saleeb, II. S. Hanna. Маджаллат аль-кимия ли-льджумхурийа
аль-арабийа аль-муттахида, 12, 229 (1969); РЖХим., 5Б1707 (1971).
10. В. А. Клячко, И. Э. Апвлъцин. Очистка природных вод. М., Стройиздат,
1971.
И. А. С. Мошинский, В. П. Чибизов. ЖПХ, 48, 2186 (1975).
338
ГЛАВА X. КОАГУЛЯЦИЯ В ПРОЦЕССАХ ВОДОПОДГОТОВКИ
12. В. В. А шанин, Б. Н. Голубков и др. Водоснабжение и санитарная тех¬
ника, № 3, 4 (1968).
13. В. М. Квятковский. ЖВХО им. Д. И. Менделеева, 5, 637 (1960).
14. В. С. Бутлер. Энергетика за рубежом. Сборник переводов под ред.
М. С. Шкроба, вып. 4. М.—Л., Госэнергоиздат, 1957, стр. 14.
15. П. Гамер, Д. Джексон, И. Серстон. Очистка воды для промышленных
предприятий. М., Стройиздат, 1968.
16. /. S. Hess. Amer. City, 75, N 8, 161 (1960).
17. Г. Бэббит, Дж. Доланд. Водоснабжение. М., Стройиздат, 1958.
18. А. И. Егоров. Сб. «Исследования по водоподготовке». М., Стройиздат,
1956, стр. 53.
19. Д. Н. Смирнов. Водоснабжение и санитарная техника, № 7, 33 (1956).
20. R. S. Stout, В. S. Shuey. Amer. City, 80, N 5, 108 (1965).
21. С. R. Henry. J. Amer. Water Works Assoc., 50, 61 (1958).
22. W. L. Denman, Ch. M. Hwa. Англ. пат. 1054677 (1967).
23. A. Treanor, J. Ignatius. Англ. пат. 1061209 (1967).
24. A. R. Le Compte. Пат. США 3262877 (1966).
25. А. М. Перлина, 3. Я. Городищер, Я. С. Едалин. Научные труды АКХ
им. К. Д. Памфилова, вып. 30. Водоснабжение, № 4. М.—Л., ОНТИ,
1964, стр. 3.
26. JI. А. Кулъский. Основы технологии кондиционирования воды. Киев,
Изд-во АН УССР, 1963.
27. Д. Иидзима, II. Като. Яп. пат. 1313 (1965).
28. W. Christ. Wasserwirtschaft—Wassertechnik, 10, 99 (1960).
29. М. A. Kessick, J. J. Morgan. Environ. Sci. and Technol., 9, 157 (1975).
30. J. A. bevitr. J. New Engl. Water Works Assoc., 80, 19 (1966).
31. E. Ф. Золотова, Г. IO. Acc. Очистка воды от железа, фтора, марганца
и сероводорода. М., Стройиздат, 1975.
32. A. G. Curev, D. Zubcenko. Пат. ЧССР 116575 (1965).
33. R. L. Culp, Н. A. Stoltenberg. J. Amer. Water Works Assoc., 50, 423 (1958).
34. С. S. Roruff. Ind. and Engng Chem., 26, 69 (1934).
35. Ю. Ю. Лурье, А. И. Рыбникова, 3. В. Николаева. Очистка воды от фто¬
ра. М., Стройиздат, 1952.
36. В. А. Клячко. Сб. «Исследования по водоподготовке». М., Стройиздат,
1959, стр. 4.
37. А. Деревянко, II. Масальская. Жилищно-коммунальное хозяйство, № 10,
11 (1960).
38. Е. Б. Кузякин, А. М. Когановский. Цветные металлы, № 12, 40 (1962).
39. Г. П. Рыхликов. Научные труды Омского мед. ин-та, вып. 70, 27 (1965).
40. Э. А. Левицкий, Я. Б. Лазовский. Авт. свид. СССР 261996 (1967); Бюлл.
изобр., № 5 (1970).
41. W. Pawlita. Рг. nauk Inst. inz. ochr. srodow PWr, N 27, 145 (1974).
42. E. A. Savinell, A. P. Black. J. Amer. Water Works Assoc., 50, 33 (1958).
43. R. D. Scott, A. E. Kimbery et al. J. Amer. Water Works Assoc., 29, 9
(1937.)
44. C. S. Finkbeiner. Water Works Engng, 91, 990 (1938).
45. A. S. Zettlemoyer, E. A. Zettlemoyer, W. C. Wolker. J. Amer. Chem. Soc.,
69, 1312 (1947).
46. В. А. Клячко, И. Э. Апельцин. Подготовка воды для промышленного
и городского водоснабжения. М., Стройиздат, 1962.
47. J. Е. Shenk, W. J. Weber. J. Amer. Water Works Assoc., 60, 199 (1968).
48. П. П. Строкач, В. А. Слипченко, Л. А. Кулъский. Электронная обработ¬
ка материалов, № 5, 57 (1975).
49. L. L. Klinger. Tappi, 37, 891 (1954).
50. Л. А. Кулъский, В. Ф. Накорчевская В. А. Слипченко. Активная крем-
некислота и проблема качества воды. Киев, «Наукова думка», 1969,
стр. 108.
X. ЛИТЕРАТУРА
339
51. Д. П. Козырев, И. Г. Купцов. Удаление сероводорода из артезианских вод
посредством гидратов окислов железа с последующей их регенерацией.
М., Гостоптехиздат, 1946.
52. И. Э. Апельцин, Е. Ф. Золотова, Е. С. Перемыслова. Сб. «Исследования
по водоподготовке». М., Стройиздат, 1959, стр. 143.
53. Л. Б. Лазовский, В. М. Щипачев, С. В. Эльковский. Авт. свид. СССР
414199 (1971); Бюлл. изобр., № 5 (1974).
54. М. Л. Ожервлъвва. Труды ГИСИ им. В. П. Чкалова, вып. 31. Горьк. кн.
изд-во, 1959, стр. 3.
55. Л. В. Политковская. Труды ГИСИ им. В. П. Чкалова, вып. 25. Горьк.
кн. изд-во, 1956, стр. 191.
56. G. Е. Weismantel. Chem. Engng, 78, 98 (1971).
57. Е. Hokervall. Effluent and Water Treat. J., 11, 551 (1971).
58. J. R. Srymanski. Amer. City, 85, N 2, 72 (1970).
59. K. Peck. Water and Wastes Engng, 8, N 11, 30 (1971).
60. J. R. Stukenberg. J. Water Pollut. Contr. Fed., 47, 338 (1975).
61. М. Ямасаки, Т. Асано. Мицзусёри гидзюцу, 14, 1065 (1973); РЖХим.,
10И360 (1974).
62. R. W. Ockershausen. Water and Sewage Works, 112, 80 (1975).
63. A. C. Gross. Environ. Sci. and Technol., 8, 414 (1974).
64. А. И. Жуков. Водоснабжение и канализация, вып. 2. М., изд. ЦБТИ
МКХ РСФСР, 1963, стр. 63.
65. X. Йокоути. Яп. пат. 28173 (1964).
66. А. Н. Rice. Англ. пат. 1083343 (1967).
67. G. Culp, S. Hansen. Amer. City, 82, N 6, 96 (1967).
68. H. J. Graeser, P. D. Haney. Water and Wastes Engng, 5, N 12, 34 (1968).
69. H. R. Gustafson. Пат. США 3408289 (1968).
70. R. Sosewitz, V. W. Bacon. Civil Engng, N 11, 34 (1969).
71. J. L. Rose. Chem. Engng Progr. Symp. Ser., 67, 63 (1971).
72. F. P. Sebastian. Ibid., p. 410.
73. С. М. Драчев. Водоснабжение и санитарная техника, № 9, 40 (1969).
74. W. М. McLellon, Т. М. Keinath, Chao Chia Chen. J. Water Pollut. Contr.
Fed., 44, 77 (1972).
75. И. Л. Монгайт. Водоснабжение и санитарная техника, № 1, 23 (1955).
76. И. Л. Монгайт, И. Д. Родзиллер. Водоснабжение и санитарная техни¬
ка, № 7, 19 (1957).
77. А. И. Мацнев. Очистка сточных вод флотацией. Киев, «Бугцвелытик»,
1976.
78. W. Templeton. Wastes Engng, 27, 12 (1956).
79. R. N. Simonsen. Hydrocarbon Process and Petrol Befiner, 41, N 5, 145
(1962).
80. J. Cornelissen, W. Steck. Erdol und Kohle, 14, 742 (1961).
81. Я. А. Карелин. Водоснабжение и санитарная техника, № 5, 32 (1956).
82. Е. М. Калинийчук, И. А. Макаров, И. И. Василенко и др. Химия и тех¬
нология топлив и масел, № 10, 23 (1972).
83. И. А. Макаров, Е. М. Калинийчук, И. И. Василенко и др. Сб. «Нефте¬
переработка и нефтехимия», вып. 7. Киев, «Наукова думка», 1972, стр.
стр. 56.
84. Е. М. Калинийчук, И. И. Василенко, Н. А. Суховерхова. Труды Всесоюз¬
ного объединения «Нефтехим», вып. 5. М., ОНТИ, 1973, стр. 321.
85. J. Strankmuller, К. Н. Mangold et al. Wasserwirtschaft—Wassertechnik,
10, 379 (1960).
86. D. Heinz, D. Radeck et al. Wiss. und Fortschr., 20, 242 (1970).
87. И. R. Герасимов. Труды Уфимского нефт. ин-та, вып. 3, 29 (1960).
88. Б. М. Матов, П. М. Степанов и др. Коллоидн. ж., 32, 91 (1970).
89. Л. А. Кулъский, Е. М. Калинийчук. Кондиционирование воды. .4., Строй¬
издат, 1964.
340
ГЛАВА X. КОАГУЛЯЦИЯ В ПРОЦЕССАХ ВОДОПОДГОТОВКИ
90. A. Gasser. Австрийск. пат. 299832 (1972).
91. Б. А. Миткалев, Э. Г. Иоакимис и др. Труды Башк. НИИ по переработ¬
ке нефти, вып. 1. Уфа, 1959, стр. 216.
92. Н. Ф. Резник, И. И. Караваев и др. Путь и путевое хозяйство, № 7, 19
(1963).
93. О. А. Иванов, 3. Г. Беляева. Сб. «Транспорт и хранение нефти и нефте¬
продуктов», № 12. М., 1970, стр. 29.
94. Е. Baer. Electroplat. and Metal Finish., 18, N 6, 201 (1965).
95. W. Brecht. ETR Eisenbahntechn. Rundschau, 20, 167 (1971).
96. R. Unterhansberg, W. Shaper. Wasser, Luft und Betrieb, 13, 392 (1969).
97. X. Судзуки. Яп. пат. 9137 (1958).
98. И. Л. Монгайт, С. В. Захарьина. Авт. свид. СССР 349644 (1970); Бюлл.
изобр., № 26 (1972).
99. Water and Wastes Engng, 10, N 3, 13 (1973).
100. D. R. Woods, M. W. Slezak. J. Water Pollut. Contr. Fed., 45, 2136 (1973).
101. A. R. Middleton. Water and Sewage Works, 110, 251 (1963).
102. Water and Pollut. Contr. (Canada), 112, N 8, 18 (1974).
103. T. P. Quirk. Wastes Engng, 31, 82 (1960).
104. J. Polis. Papir a celul., 19, 9 (1964).
105. S. E. Smith, R. F. Christman. J. Water Pollut. Contr. Fed., 41, 222 (1969).
106. Т. Гото, H. Унохара и др. Камипа гикёси, 25, № 2, 59 (1971); РЖХим.,
23И539 (1971).
107. V. Mican, J. Tesafik. Wasserwirtschaft—Wassertechnik, 10, 117 (1960).
108. С. Skalicky, P. Sochdn. Papir a celul., 14, 273 (1959).
109. A. Munteanu, E. Cute. Studii protect, si epur. apel., 7, 265 (1966).
110. A. Munteanu, E. Cute. Tribure CEBEDEAU, 20, 122 (1967).
111. A. H. Онохин. Труды Ленингр. технология, ин-та целлюл.-бумажной
промышленности, вып. 20, 202 (1967).
112. Е. S. Packard. Paper Trade J., 145, 27 (19б1).
113. R. P. Logan, T. W. Lesperance. Proc. 8th Annual Pacific. Northwest In-
dustr. Waste Conf. Washington, 1957, p. 207.
114. А. А. Пряникова. Химическая переработка древесины. Реферативная
информация, № 17. М., Министерство химической промышленности,
1967, стр. 10.
115. R. R. Fuller. Пат. США 3627679 (1971).
116. М. Б. Беров, Л. И. Шмидт, R. М. Шапченко. Бумажная промышлен¬
ность, № 2, 6 (1973).
117. D. О. Herer, F. Е. Woodard. Tappi, 59, 134 (1976).
118. А. И. Попов. Авт. свид. СССР 252943 (1966); Бюлл. изобр., № 29 (1969).
119. М. Gafitanu, V. Marculescu et al. Studii protect, si epur. apeal., 7, N 1,
339 (1966).
120. W. T. Rattershill. Pulp and Paper Mag. Canada, 69, 49 (1968).
121. А. Ониси. Камипа гикёси, 25, № 2, 79 (1971); РЖХим., 23И540 (1971).
122. J. Bebin. Papeterie, 93, 1067 (1971).
123. J. R. Roydston. Forest Prod. .Т., 21, N 9, 58 (1971).
124. Л. И. Гюнтер, Ю. И. Рейцер. Научные труды АКХ им. К. Д. Памфило¬
ва, вып. 20. Городская канализация, № 2. М.—Л., ОНТИ, 1963,
стр. 114.
125. Г. И. Фишман, А. А. Литвак. Водоснабжение и очистка сточных вод
предприятий химических волокон. М., «Химия», 1971.
126. J. Relin, P. Boulenger, J. С. Bourdelot. Advances Water Pollut. Bes. Proc.
6 (,lInternat. Conf., Jerusalem, 1972. Oxford, 1973, p. 595.
127. J. Zielinski. Prz. pap., 29, N 9, 320 (1973).
128. S. Perkowski. Przegl. skorrany, 13, N 10, 241 (1958).
129. O. J. Spoul, P. F. Atkins et al. J. Water Pollut. Contr. Fed., 38,508(1966).
130. R. A. Prabhakara, D. D. Dasnurkar. J. Inst. Engrs (India). Public Health
Engng Div., 49, 134 (1969).
X. ЛИТЕРАТУРА
341
131. В. Mejstrik, F. Sillinger et al. Пат. ЧССР 92431 (1959).
132. X. Иида, М. Эндо. Когё кагаку дзасси, 70, № 3, 403 (1967); РЖХим.,
17И329 (1967).
133. X. Иида, Н. Кувабара, М. Эндо. Токё когё сюкэнсё хококу, 59, № 2,
63 (1964); РЖХим., 17И223 (1965).
134. Т. Иосида. Яп. пат. 27170 (1965).
135. W. Schawrz. Osterr.-Abwasser-Rundschau, 11, N 4, 67 (1966).
136. Melliand. Textiller, 53, N 2, 230 (1972).
137. В. Villiermet, P. M. Geliy. Technicuir, 7, N 2, 31 (1973).
138. И. Папазов, С. Карцев. Труды НИИ водоснабжения, канализации и са¬
нитарной техники, вып. 8. София, 1973, стр. 51.
139. У. Маэда, К. Сюго, К. Ишикава п др. Нихон кагаку кайси, № 6, 1117
(1974); РЖХим., 1И584 (1975).
140. Ch. Н. Mobius, Т. Н. Giinther. Wochenbl. Papierfabr., 102, 559 (1974).
141. II. Куроки. Кагаку, 28, № И, 1017 (1973); РЯ{Хим., 10И436 (1974).
142. И. Папазов, А. Николов. Труды НИИ водоснабжения, канализации и са¬
нитарной техники, вып. 8. София, 1973, стр. 69.
143. P. Pietraszek, М. Suchnicka. Gospod. wodna, 33, 263 (1973).
144. Н. Kloss, N. Losakiewicz, T. Masztakowska. Prz. zloz.-mlyn., 17, N 1,
20 (1973).
145. H. J. Fiedler. Zucker, 10, N 12, 273 (1957).
146. T. Wolski. Przet.-wor. owoc.-warz. i Koncentr., 2, N 4, 122 (1958).
147. B. A. Sard. Chem. and Ind., N 49, 2051 (1962).
148. Ch. H, Gockel. Vom Wasser 1970, Bd. 37. Weinheim, 1971, S. 216.
149. D. Spremo. Пат. СФРЮ 24863 (1965).
150. A. В. Wheatland, В. J. Borne. Chem. and Ind., N 9, 357 (1964).
151. М. C. Dart. Process Biochem., N 5, 11 (1974).
152. X. Има. Ню фудо индасутори, 14, № 12, 36 (1972); РЖХим., 13И377
(1973).
153. К. P. Dietrich. Brauweit, 106, N 44, 818 (1966).
154. Н.-Е. Klotter, Е. Hantge. Уот Wasser 1965, Bd. 32. Weinheim, 1966,
S. 310.
155. И. С. Лавров, Н. И. Рукобратский, О. В. Смирнов и др. Виноделие и ви¬
ноградарство, № 1, 51 (1973).
156. Й. Йосидзава, Ю. Исикава, Ц. Судзуки. Нихон дзёдзо кёкай дзасси, 67,
1059 (1972); РЖХим., 13И381 (1973).
157. И. И. Шабунин, В. А. Седова и др. Ученые записки ЦНИИ оловян. про¬
мышленности, № 1. Новосибирск, Зап.-Сиб. кн. изд-во, 1966, стр. 18.
158. С. А. Костин, А. Т. Казаков и др. Научные труды Кузнецкого научно-
исследовательского и проектно-конструкторского ин-та углеобогащения.
Прокопьевск, 1972.
159. Ф. Э. Молочникова, Е. И. Силина. Колыма, № 7, 10 (1965).
160. И. М. Соболева, С. В. Пелътихин и др. Сб. «Охрана водоемов и методы
очистки воды». Киев, 1962, стр. 114.
161. В. В. Найденко. Тезисы докладов конференции по методам очистки газо¬
вых выбросов и промстоков от вредных веществ. Дзержинск, 1967,
стр. 159.
162. Б. П. Краснов, Л. В. Милованов и др. Цветные металлы, № 3, 8 (1959),
163. D. A. Wilms, F. F. Notebaert, A. A. Van Haute. Advances Water Pollut.
Res. Proc. 6th Internat. Conf., Jerusalem, 1972. Oxford, 1973, p. 615.
164. Б. И. Коробицин. Сб. «Современные способы очистки сточных вод цехов
гальванопокрытий машиностроительных предприятий». Новосибирск,
1968, стр. 140.
165. G. Chqdrynski, J. Kucharski et al. Пат. ПНР 56613 (1968).
166. М. II. Бочкарев, В. П. Соколов. Авт. свид. СССР 173141 (1964); Бюлл.
изобр., № 14 (1965).
167. A. L. Kowal, Z. Krutul. Gaz, woda i techn. sanit., 40, 191 (1966).
342
ГЛАВА X. КОАГУЛЯЦИЯ В ПРОЦЕССАХ ВОДОПОДГОТОВКИ
168. Е. 3. Аграноник, Н. Д. Бондарев, В. П. Иванов и др. Водоснабжение
и канализация предприятий химической промышленности. Л., Стройиз¬
дат, 1967.
169. Е. Hechler. Wasser, Luft und Betrieb, 12, 762 (1968).
170. Я. Намида, М. Идзуми. Яп. пат. 49-32268 (1974).
171. V. Solin, К. Buridnek. Sb. Vysoke skoly chem.-technol. Praze. Odd. Fak.
technol. paliv a vody, 2, 237 (1958).
172. P. С. Водяник, В. А. Ливке. Авт. свид. СССР 215111 (1966); Бюлл. изобр.,
№ 12 (1968).
173. В. П. Соколов. Хим. промышленность, № 1, 33 (1967).
174. В. П. Соколов. Тезисы докладов конференции по методам очистки газо¬
вых выбросов и промстоков от вредных веществ. Дзержинск, 1967,
стр. 80.
175. Б. И. Иванов, Н. Н. Никитина. Сб. «Очистка сточных вод топливной
промышленности и нефтехимического синтеза». М., «Химия», 1965,
стр. 68.
176. И. Л. Монгайт, С. И. Конобеев. Водоснабжение и санитарная техника,
№ 3, 20 (1955).
177. F. Jursik. Sb. Vysoke skoly chem.-technol. Praze. Odd. Fak. technol.
paliv a vody, 5, 45 (1961—1962).
178. H. G. Scholz. Adhasion, 11, N 1,7 (1967).
179. R. D. Sadow. Ind. Water and Wastes, 6, N 2, 42 (1961).
180. G. J. Mohanrao, K. P. Krishnamoorthi et al. Water Bes., 1, 181 (1967).
181. V. V. Volk, M. L. Jackson. J. Water Pollut. Contr. Fed., 40,205 (1968).
182. R. М. Любарский.Водоснабжение и канализация, вып. 14. М., изд. ЦБТИ
МКХ РСФСР, 1969, стр. 67.
183. G. S. Logsdon, Е. Edgerley. J. Amer. Water Works Assoc., 63, 734 (1971).
184. P. F. Mahoney, W. J. Duensing. J. Amer. Water Works Assoc., 64, 665
(1972).
185. Сб. «Коагулянты для очистки питьевой воды». Под ред. В. Т. Турчино-
вича. М., Изд-во МКХ РСФСР, 1948.
186. S. L. Bushop, G. P. Fulton. Public Works, 99, N 6, 94 (1968).
187. A. A. Giessen, J. G. Rensen. J. Inorg. and Nucl. Chem., 30, 1739 (1968).
188. Ph. Messerschmidt. Luft- und Kaltetechn, 8, 82 (1972).
189. В. M. Любарский. Научно-теоретическая конференция аспирантов АКХ
им. К. Д. Памфилова. М., ОНТИ, 1969, стр. 15.
190. R. М. Любарский. Научно-теоретическая конференция аспирантов АКХ
им. К. Д. Памфилова. М., ОНТИ, 1967, стр. 39.
191. D. Вепп, P. W. Doe. Filtr. and Separ., 6, 383 (1969).
192. E. И. Пономарев, В. В. Вольхин, В. Л. Золотавин. Научные труды Перм¬
ского политехи, ин-та, № 71, 64 (1970).
193. В. М. Любарский. Водоснабжение и канализация, вып. 14. М., изд.
ЦБТИ МКХ РСФСР, 1969, стр. 67.
194. К. Bargman. Sewage and Ind. Wast., 30, 1079 (1958).
195. H. А. Лукиных, И. С. Туровский. Основные принципы технологического
расчета установок по механическому обезвоживанию осадков сточных
вод на барабанных вакуум-фильтрах. Информационное письмо АКХ им.
К. Д. Памфилова, № 1 (162). М., ОНТИ, 1962.
196. И. С. Туровский. Научные труды АКХ им. К. Д. Памфилова, вып. 20.
Городская канализация, № 2. М., ОНТИ, 1963, стр. 55.
197. И. С. Туровский. Авт. свид. СССР 252214 (1968); Бюлл. изобр., № 28
(1969).
198. Water and Sewage Works, 116, 434 (1969).
199. P. О. Richter. Process Biochem., 6, 21 (1971).
200. S. Bachanek. Pr. Inst. gosp. wod., 7, 57 (1972).
201. И. С. Туровский. Водоснабжение и канализация, вып. 2. М., изд. ЦБТИ
МКХ РСФСР, 1963, стр. 91.
X. ЛИТЕРАТУРА
343
202. Contract J., 219, N 4603, 170 (1967).
203. R. Ellerkes. Англ. пат. 1188394 (1970).
204. Л. В. Замошин. Водоснабжение и канализация, вып. 8. М., изд. ЦБТИ
МКХ РСФСР, 1966, стр. 66.
205. P. S. Ashman, P. F. Roberts. Water Pollut. Contr. (Gr. Brit.), 69, 638
(1970).
206. H. Д. Николаева. Бумажная промышленность, № 8, 10 (1969).
207. M. W. Tenney, W. F. Echelberger et al. J. Water Pollut. Contr. Fed., 42,
1 (1970).
208. S. Mogelnicki, E. M. Gatza, Пат. США 3409546 (1968).
209. Water and Sewage Works, 116, 361 (1969).
210. H. Reckerath, G. Wilgand. Aufbereit.-Techn., 11, 229 (1970).
211. O. Tabasaran. Water Res., 5, 61 (1971).
ЗАКЛЮЧЕНИЕ
Материалы монографии свидетельствуют о широком применении
коагулянтов прежде всего в области очистки природных вод.
В системах хозяйственно-питьевого и промышленного водоснаб¬
жения процесс коагуляции, осуществляемый в головной части
очистных сооружений, дает возможность отделить от обрабатывае¬
мой воды основную часть дисперсных загрязнений и некоторую
долю истинно растворенных веществ, обусловливающих мутность
и цветность воды. Этим самым облегчаются условия дальнейшей
более глубокой очистки воды.
Технический прогресс влечет за собой не только увеличение
водопотребления и повышение требований к качеству обработки
природной воды, но и необходимость повторного использования
сточных вод промышленности и теплоэнергетики. В решении этой
задачи методу коагуляции принадлежит, безусловно, очень важ¬
ная роль: коагуляция как самостоятельный способ очистки сточ¬
ных вод или как способ их доочистки (после обработки биологи¬
ческими методами) начинает широко применяться в практике.
Важная особенность метода коагуляции состоит в том, что
он позволяет резко снизить содержание в обрабатываемой воде
многих веществ, вредных для здоровья, таких, как бактерии и
вирусы, пестициды, синтетические детергенты, радиоактивные при¬
меси. Поэтому коагулирование следует рассматривать как важное
«барьерное» мероприятие в борьбе за оздоровление окружающей
среды.
Результаты анализа современного состояния научных и про¬
изводственных достижений в области очистки воды коагулянтами
показывают, что благодаря заметным успехам, достигнутым при
разработке теоретических основ процесса коагуляции, а также
при решении частных задач, имеющих прикладное значение, ме¬
тод коагуляции и его техническое оформление получили в послед¬
ние годы значительное развитие. Причем в разработке целого
ряда важнейших проблем, создании теории коллоидно-химичес¬
ких и поверхностных явлений, структурообразования, флокуля¬
ции и отделения коагулированной взвеси в осадок наша страна
опережает другие развитые страны.
Ведущая роль в решении перечисленных проблем принадлежит
крупным научным коллективам Института физической химии
АН СССР, Института коллоидной химии и химии воды АН УССР,
ЗАКЛЮЧЕНИЕ
345
кафедр коллоидной химии Московского и Ленинградского универ¬
ситетов, Научно-исследовательского института коммунального во¬
доснабжения и очистки воды при Академии коммунального хо¬
зяйства им. К. Д. Памфилова, Всесоюзного научно-исследователь-
ского института водоснабжения, канализации, гидротехники и
инженерной гидрогеологии (ВОДГЕО), а также целого ряда от¬
раслевых научно-исследовательских и учебных институтов.
Характерной тенденцией развития исследований является бо¬
лее тесное взаимодействие науки с практикой: сфера деятель¬
ности научных организаций расширилась за счет разработки
вопросов, имеющих прикладное значение; заметно увеличилась
роль науки на производстве, окрепла экспериментальная база
пунктов хозяйственно-питьевого и промышленного водоснабже¬
ния, повысилась квалификация специалистов-технологов по очи¬
стке воды. Во многих крупных городах на производственных
объектах водоснабжения и канализации теперь проводится пла¬
номерная научная работа.
Имеющийся опыт соединения науки о дисперсных системах
с практикой водоподготовки уже дал заметные результаты. Вмес¬
те с тем еще многие задачи, особенно задачи, требующие комплекс¬
ного подхода к решению, а также учета реальных масштабов про¬
изводства, остаются не решенными. Научный подход к проблеме
коагулирования примесей природных и сточных вод требует глу¬
бокого знания как свойств загрязнений самих вод, так физико¬
химической сущности протекающих при коагуляции процессов.
Проведенный нами анализ дает возможность выделить наи¬
более важные задачи, стоящие перед исследователями.
1. Развитие теории коагуляции частиц во вторичном энерге¬
тическом минимуме. Знание закономерностей коагуляции частиц
за счет сил дальнего взаимодействия позволит объяснить влия¬
ние толщины и структурных свойств гидратных оболочек на проч¬
ность формирующихся агрегатов; рассчитать величину сил, от¬
ветственных за прилипание хлопьев к материалам фильтрующих
загрузок; выявить такие условия коагуляции, при которых ближ¬
няя и дальняя агрегации частиц будут проявляться в наибольшей
степени.
2. Изучение механизма и кинетики очистки воды коагулян¬
тами с учетом коллективных взаимодействий частиц и распада
образующихся агрегатов. Результаты изучения позволят произ¬
вести обоснованный выбор режима перемешивания обрабатывае¬
мой воды, установить оптимальные промежутки времени меж¬
ду моментами ввода коагулянта и вспомогательных реагентов,
уточнить конструктивные параметры смесителей и камер хлопье¬
образования.
3. Дальнейшее исследование зависимости оптимальной дозы
коагулянта от качественных параметров обрабатываемой воды и
346
ЗАКЛЮЧЕНИЕ
условий коагулирования. Научно обоснованный подход к расче¬
ту оптимальных доз коагулянтов даст возможность экономно
расходовать коагулянты, планировать их производство и поступ¬
ление на водоочистные станции.
4. Совершенствование существующих и разработка новых ме¬
тодов интенсификации коагуляции и электрокоагуляции с исполь¬
зованием флокулянтов, сорбентов и замутнителей, с привлече¬
нием методов физического воздействия на скорость коагуляции
и структурные свойства коагулятов.
5. Изучение химического состава и структуры продуктов гид¬
ролиза коагулянтов, их способности к образованию комплексных
химических соединений с примесями воды и полимерных форма¬
ций.
6. Дальнейшее изучение природы загрязнений воды, в осо¬
бенности свойств веществ, обусловливающих цветность, и «новых»
видов загрязнений, поступающих в источники со сточными водами.
К кругу практически важных задач, не рассмотренных в мо¬
нографии, следует также отнести задачи, связанные с обработ¬
кой и утилизацией шламов, регенерацией коагулянтов, усовер¬
шенствованием инструментальных методов контроля за ходом
коагуляции.
На основании изложенного, можно констатировать, что поиск
путей к повышению интенсивности, экономической и технологи¬
ческой эффективности процесса коагуляции примесей природных
и сточных вод еще далеко не завершен, но основные направления
поиска уже выбраны. Не вызывает сомнения, что эти направле¬
ния в ближайшие годы получат широкое развитие.
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Агрегативная устойчивость 17, 26 сл.
Адгезия 95 сл.
Адсорбент 20
Адсорбтив 20
Адсорбции
изотерма 21
скорость 24 сл.
Адсорбция
ионная 20
молекулярная 20
обменная 24
окрашивающих веществ 98 сл.,
163 сл.
физическая 20
химическая 20 сл.
электролитов 24
Альгинат натрия 296, 311 сл.
Активная кремнекислота (см. кремне-
киелота активная)
Активный уголь 74, 214, 217, 238 сл.
Алюминат натрия 72 сл., 214
Алюминий сернокислый 72 сл., 213 сл.
Алюминия гидроокись 77 сл., 116 сл.,
123, 154 сл., 181
Алюминия оксихлорид 72 сл., 204 сл.
Антагонизм ионов 114
Астабилизация 87, 154 сл.
Бактерии
свойства 58
удаление из воды 231 сл.
Барьер отталкивания 33
Белки 56 сл.
Биологические примеси воды 57 сл.
Броуновское движение молекул 14
Взвесь коагулированная
газонасыщенность 188
гидратация 186
объемная концентрация 187 сл.
объемный вес 186 сл.
прочность 190 сл.
размер хлопьев 186
Вирусы 58 сл., 231 сл.
Вода природная
активная реакция 41 сл.
взвешенные примеси 38, 45 сл.
вкус и запах 39
жесткость 42
ионный состав 41
минеральные примеси 45 сл.
мутность 40
окисляемость 43
органические примеси 54 сл.
органо-минеральные ассоциагы
59 сл.
прозрачность 40
растворенные примеси 41 сл., 161 сл.
сухой остаток 41
цветность 40
щелочность 42
электропроводность 41
Водоросли 57 сл., 230 сл.
Высокомолекулярные вещества
защитное действие 115 сл.
свойства 13, 17, 36, 287 сл.
Высокомолекулярные флокулянты
(см. флокулянты)
Вязкость
дисперсных систем 15 сл.
кажущаяся 197
растворов высокомолекулярных
веществ 17
Гели
адгезионные свойства 95 сл.
адсорбционные свойства 98 сл.
гидратация 91
кристаллизация 89
пептизация 94
прочность 95 сл.
размер частиц 90 сл.
старение 93
структура 86 сл.
тиксотропия 94
удельная поверхность 91
Гетероадакоагуляция 117
Гетерогенная система 13
Гетерокоагуляция 117
Гидратация
ионов 45,91
структурно-механическая 34
Гидролиз 77 сл.
Гидрофильность 25 сл.
348
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Гидрофобность 25 сл.
Гомокоагуляция 117
Градиент скоростной 133, 135, 262 сл.
Гранула 28
Грубодисперсные примеси воды 13,
38 сл.
Гуминовые вещества 54 сл.
Давление расклинивающее 35
Двойной электрический слой
сжатие 30, 110 сл.
строение 26 сл.
Дестабилизация коллоидов 30,110 сл.
Детергенты синтетические
свойства 63 сл.
удаление из воды 220 сл.
Дзета-потенциал 29, 52, 109 сл., 160
сл.
Дисперсная система 17
Дисперсная фаза 13
Диффузия
молекулярная 15
турбулентная 136
Диффузный слой ионов 27
Дубильные вещества 56, 166, 333 сл.
Железа гидроокись 78 сл.
Железо сернокислое 74 сл., 215 сл.
Железо хлорное 74 сл., 215 сл.
Замутнение воды 260 сл.
Замутнители 260 сл.
Зародышеобразование 139 сл.
Заряд частиц 27 сл.
Защита коллоидная 115 сл.
Золи гидроокиси
алюминия 28 сл., 31, 33, 78, 90 сл.,
107 сл., 116 сл., 123,154 сл., 181
железа 27 сл., 30, 33, 78 сл., 88,
90 сл., 107 сл., 112, 115 сл., 123,
154 сл.
магния 320 сл.
марганца 33
Изотерма адсорбции 21
Изоэлектрическая точка
продуктов гидролиза коагулянта
86, 155, 161 сл., 177 сл.
частиц взвеси 53
Изоэлектрическое состояние 30, 53,
86, 161
Ионный обмен 24, 49
Ионы
гидратация 45
коагулирующие 107
потенциалобразующие 27
потенциалопределяющие 27
Каолин (каолинит) 46 сл., 72, 158 сл.,
174 сл., 260
Катионообменная емкость 49 сл.,
154 сл.
Квасцы 72 сл., 215
Кинетика
адсорбции 19 сл.
коагуляции 127 сл.
Кинетическая устойчивость 17
Коагулирование
концентрированное 272 сл.
прерывистое (периодическое) 273
фракционированное 270 сл.
Коагулированная взвесь (см. взвесь
коагулированная)
Коагулирующее действие ионов 107 сл.
Коагулянты гидролизующиеся
алюминийсодержащие 72сл.,213 сл.
виды 72 сл.
железосодержащие 74 сл., 215 сл.
оптимальная доза 160, 167 сл.
смешанные 76 сл., 219 сл.
состав 72 сл.
Коагулят 87
Коагулятор 7
Коагуляции
кинетика 127 сл.
методы интенсификации 256 сл.
ортокинетическая стадия 130
перикинетическая стадия 130, 155
скорость 128 сл.
теория 32, 110 сл.
Коагуляция коллоидов
без добавления электролитов 117 сл.
быстрая 128 сл.
в ламинарном потоке 132 сл.
в магнитном поле 120 сл., 277 сл.
в турбулентном потоке 134 сл.
в электрическом поле 119 сл., 276
взаимная 117
гидролизующимися коагулянтами
138 сл.
замораживанием 118, 335 сл.
изменением температуры 118
ионизирующим облучением 122,
280 сл.
контактная 200 сл.
концентрационная 110 сл.
медленная 128 сл.
нейтрализационная 110 сл.
перемешиванием 118
ультразвуком 121, 279 сл.
электролитами 107 сл.
Коллоидные частицы
анизодиаметрические 14, 87
заряд 26 сл.
изодиаметрические 14
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
349
размер 13
Коэффициент объемного использова¬
ния 195
Коэффициент формы 194
Коэффициент цветности 41
Краевой угол смачивания 25, 189
Крахмал 295 сл., 311
Кремнекислота 44, 327 сл.
Кремнекислоты активная
добавление к воде 293 сл.
оптимальная доза 293
приготовление 288 сл.
свойства 290
эффективность 290 сл.
Кривая
потенциальная взаимодействия ча¬
стиц 32
седиментации 19, 53
Критерий
Архимеда 194
Кэмпа 145, 177, 265, 267
Рейнольдса 193
Крупность взвеси гидравлическая 19
Лиотропные ряды 24, 108
Лиофильность 25
Лиофобность 25
Магнитная обработка воды 120 сл.,
277 сл.
Масла 331 сл.
Масштаб турбулентности 136
Методы интенсификации коагуляции
276 сл.
Механизм
коагуляции гидролизующимися
коагулянтами 153 сл.
обесцвечивания воды 163 сл.
осветления воды 154 сл.
Микрофлотация
Минеральные примеси воды
катионообменная емкость (способ¬
ность) 49 сл.
размер частиц 46
свойства 46 сл.
состав 46 сл.
форма частиц 48
Мицелла 28, 62
Молекулярное притяжение 32 сл.
Монтмориллонит (бентонит) 46, 76,
159, 174 сл., 260
Мутность 40
Натяжение поверхностное 23
Нефть и нефтепродукты 331 сл.
Обезжелезивание воды 322 сл.
Обескремнивание воды 327 сл.
Объемная концентрация примесей
93, 187
Окислители 236 сл.
Оптимальная доза коагулянта 153,
167 сл.
Оптимальная температура 176 сл.
Оптимальные значения pH 160, 165,
177 сл.
Органические примеси воды
гуминовые вещества 54 сл.
гуминовые кислоты 54 сл.
дубильные вещества 56
фульвокислоты 55 сл., 166
Осадки
коагуляция замораживанием 335 сл.
обработка коагулянтами и флоку¬
лянтами 336 сл.
рециркуляция 274 сл.
Осаждение частиц
свободное 197
стесненное 197
Осветление воды 154 сл., 169 сл., 192
сл.
Отстаивание 193 сл.
Очистка воды коагулянтами от
бактерий 231 сл.
вирусов 231 сл.
водорослей 230 сл.
дубильных веществ 333 сл.
железа 322 сл.
кальция 320 сл.
красящих веществ 333 сл.
кремния 327 сл.
магния 320 сл.
марганца 322 сл.
масел 331 сл.
мышьяка 229
нефти и нефтепродуктов 331 сл.
пестицидов 226
планктона 230 сл.
поверхностно-активных веществ
220 сл.
радиоактивных веществ 227 сл.
ртути 229 сл.
сероводорода 327 сл.
фосфатов 223 сл.
фтора 324 сл.
Пептизация 64, 94
Перезарядка 113
Перемешивание воды
быстрое 127
гидравлическое 263
медленное 127
механическое 263 сл.
пневматическое 182, 263, 265
350
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Пестициды 65 сл., 226
Планктон 57, 230 сл.
Плотность
гелей 93
коагулированной взвеси 187 сл.
Плотный слой ионов 27
Поверхностная проводимость 51
Поверхностно-активные вещества
62 сл., 220
Поверхностное натяжение 23
Показатель осаждаемости взвеси 53
Полиакриламид 297 сл.
Полимеризация продуктов гидроли¬
за коагулянта 82
Полисахариды 295 сл.
Полиэлектролиты 294 сл.
Полярность 23
Порог коагуляции 107
Потенциал
двойного электрического слоя 29
поверхностный (термодинамичес¬
кий) 29
протекания 200
электрокинетический 29, 52,109 сл.,
160 сл.
Правило Ребиндера 23
Правило Шульце—Гарди 108
Предельное напряжение сдвига 97 сл.,
190 сл.
Привыкание 115
Продукты гидролиза
алюминия 80 сл.
железа 80 сл.
Равновесие
адсорбционное 20
реакций гидролиза 77 сл.
Радиоактивные вещества 66, 227 сл.
Радиус дебаевский 27
Растворимость газов в воде 43
Рециркуляция осадка 274 сл.
Ряды лиотропные 24, 108
Седиментационная кривая 19, 53
Седиментационная устойчивость 17
Седиментация 17
Сенсибилизация 115
Силы взаимодействия частиц
молекулярные 32
электростатические 32
Синергизм 114 сл.
Система дисперсная 17
Скоростной градиент 133, 135, 262 сл.
Слой
двойной электрический 26 сл.
диффузный 27
Смачивание 25, 189
Сольватация 34, 45, 91
Старение гелей 93
Структура
аморфная 89 сл.
вторичная 86
коагуляционная 86
конденсационно-кристаллизацион¬
ная 86
первичная 86
Суспензионный эффект 51
Теория
Дерягина (ДЛФО) 32, 110 сл.
Минца 201 сл.
Смолуховского 128 сл.
Тепловое движение молекул 14
Тиксотропия 94
Уголь активный 74, 214, 217, 238 сл.
Удельная поверхность
гидроокисей 91
дисперсных примесей воды 46
Ультразвуковая обработка воды
121 сл., 279 сл.
Умягчение воды 320 сл.
Уравнение
Лэнгмюра 22 сл., 170
Смолуховского 128 сл.
Стокса 18, 193
Эйнштейна 15
Устойчивость
агрегативная 17, 26 сл.
высокомолекулярных веществ 18
кинетическая 17
коллоидов 17 сл.
суспензий 19
Физическая теория коагуляции 32,
110 сл.
Фильтрация (фильтрование) 200 сл.
Флокулянты
анионные 295, 307
катионные 295, 308
неионогенные 295, 310 сл.
оптимальная доза 312 сл.
природные 295 сл., 311
синтетические 297 сл.
эффективность 231, 307
Флокуляции
кинетика 304 сл.
механизм 299 сл.
Флокуляция 116, 287 сл.
Флотация 242 сл.
Фосфаты 64 сл., 223 сл.
Хемосорбция 20
Хлопья коагулированной взвеси
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
351
газонасыщенность 188
коэффициент формы 194
образование 153 сл.
прочность 190
размер 186
Цветность 40 сл.
Шульце—Гарди правило 108
Электрокинетический потенциал 29,
52, 109 сл., 160 сл.
Электрокоагуляция 244 сл.
Электролиты
индифферентные 30
неиндифферентные 30
Электропроводность воды 41
Электрофлотация 243
Энергетический минимум
вторичный 33
первичный 33
Энтропия 45
Явление неправильных рядов 113 сл
ОГЛАВЛЕНИЕ
ПРЕДИСЛОВИЕ 3
ВВЕДЕНИЕ 7
Глава I
ФЕНОМЕНОЛОГИЯ ВОДНЫХ ДИСПЕРСИЙ 13
1.1. Степень дисперсности и форма частиц 13
1.2. Молекулярно-кинетические свойства . 14
1.3. Адсорбция на поверхности раздела 19
1.4. Агрегативная устойчивость 26
Литература 36
Глава II
ХАРАКТЕРИСТИКА ЗАГРЯЗНЕНИЙ ПРИРОДНЫХ ВОД .... 38
11.1. Физико-химические показатели качества воды 39
11.2. Примеси естественного происхождения 45
11.2.1. Минеральные примеси . 45
11.2.2. Органические примеси 54
11.2.3. Органо-минеральные ассоциаты и комплексы 59
11.3. Примеси, поступающие со сточными водами 61
11.3.1. Синтетические поверхностно-активные вещества 62
11.3.2. Фосфаты 64
11.3.3. Пестициды . 65
11.3.4. Радиоактивные вещества . 66
Литература 66
Глава III
КОАГУЛЯНТЫ И ИХ ГИДРОЛИЗ 72
111.1. Виды коагулянтов 72
II 1.1.1. Соли алюминия 72
111.1.2. Соли железа 74
111.1.3. Смеси солей алюминия и железа 76
111.2. Гидролиз коагулянтов 77
111.2.1. Реакции гидролиза 77
111.2.2. Состав продуктов гидролиза 80
111.3. Коагуляционные структуры и их свойства 86
Литература 100
ОГЛАВЛЕНИЕ
353
Глава IV
ЗАКОНОМЕРНОСТИ ВЗАИМОДЕЙСТВИЯ КОАГУЛИРУЮЩИХ
ЧАСТИЦ 107
IV. 1. Коагуляция золей электролитами 107
IV.1.1. Физическая теория коагуляции 110
IV.1.2. Специфические явления 112
IV.2. Коагуляция золей без добавления электролитов 117
Литература . . 123
Глава V
КИНЕТИКА КОАГУЛЯЦИИ 127
V.I. Понятие о быстрой и медленной коагуляции 128
V.2. Коагуляция в движущейся среде 132
V.2.I. Ламинарный поток 132
V.2.2. Турбулентный поток 134
V.3. Коагуляция гидролизующимися коагулянтами с учетом
разрушения агрегатов 138
Литература 150
Глава VI
ОПТИМАЛЬНАЯ ДОЗА КОАГУЛЯНТА 153
VI. 1. Механизм формирования коагулированной взвеси 154
VI.1.1. Осветление воды 154
VI. 1.2. Обесцвечивание воды 163
VI.2. Выбор дозы коагулянта 167
VI.2.1. Осветление воды 169
VI.2.2. Обесцвечивание воды 182
VI.3. Свойства коагулированной взвеси и способы ее выделения в
осадок 186
VI.3.1. Свойства коагулированной взвеси 186
VI.3.2. Способы выделения коагулированной взвеси в осадок. 192
VI.3.2.1. Отстаивание 193
VI.3.2.2. Осветление во взвешенном слое осадка .... 196
VI.3.2.3. Фильтрование 200
Литература 206
Глава VII
САНИТАРНАЯ ЭФФЕКТИВНОСТЬ КОАГУЛЯЦИИ 213
VII.1. Сфера применения и свойства коагулянтов 213
VII.1.1. Соли алюминия 213
VII.1.2. Соли железа 215
VII.1.3. Смеси солей алюминия и железа. Соли магния . 219
VII.2. Эффективность удаления вредных примесей 220
VII.2.1. Удаление микрозагрязнений 220
354
ОГЛАВЛЕНИЕ
VII.2.1.1. Синтетические поверхностно-активные вещества . 220
VII.2.1.2. Фосфаты 223
VII.2.1.3. Пестициды 226
VII.2.1.4. Радиоактивные вещества 227
VII.2.1.5. Прочие примеси 229
VII.2.2. Удаление биологических примесей 230
VII.2.2.1. Планктон и водоросли . 230
VII.2.2.2. Бактерии и вирусы 231
VII.3. Эффективность коагуляции в сочетании с другими методами
водоподготовки 236
VII.3.1. Обработка окислителями 236
VII.3.2. Обработка активным углем 238
VII.3.3. Флотация 242
VII.4. Электрокоагуляция 244
Литература 248
Глава VIII
ИНТЕНСИФИКАЦИЯ КОАГУЛЯЦИИ 256
VIII.1. Реагентные методы 256
VIII.1.1. Регулирование величины pH воды 257
VIII.1.2. Применение минеральных замутнителей 260
VIII.2. Безреагентные методы 262
VIII.2.1. Перемешивание воды '262
VIII.2.1.1. Механическое перемешивание 263
VIII.2.1.2. Пневматическое перемешивание 265
VIII.2.2. Подбор рационального режима добавления коагу¬
лянтов к воде -г . . . 270
VIII.2.2.1. Фракционированное кеагулирование 270
VIII.2.2.2. Концентрированное коагулирование 272
VIII.2.2.3. Прерывистое (периодическое) коагулирование . . 273
VIII.2.3. Рециркуляция осадка коагулированной взвеси . . . 274
VIII.2.4. Физические методы интенсификации 276
VIII.2.4.1. Наложение электрического поля 276
VIII.2.4.2. Наложение магнитного поля . . . . • 277
VIII.2.4.3. Воздействие ультразвуком 279
VIII.2.4.4. Ионизирующее облучение 280
Литература 281
Глава IX
ФЛОКУЛЯЦИЯ И ФЛОКУЛЯНТЫ 287
IX.1. Активная кремнекислота 287
IX.1.1. Методы приготовления (активации) 288
IX.1.2. Свойства и механизм действия 290
IX.1.3. Опыт применения и эффективность 290
ОГЛАВЛЕНИЕ
355
IX.2. Органические высокомолекулярные флокулянты 294
IX.2.1. Виды и состав флокулянтов 295
IX.2.2. Свойства и механизм действия 299
IX.2.3. Кинетика флокуляции 304
IX.2.4. Опыт применения и эффективность 307
Литература 313
Глава X
КОАГУЛЯЦИЯ В СПЕЦИФИЧЕСКИХ ПРОЦЕССАХ ВОДОПОД-
ГОТОВКИ 320
X.1. Удаление истинно растворенных примесей 320
Х.2. Очистка сточных вод 328
X.2.1. Бытовые и промышленно-бытовые сточные воды . . 328
Х.2.2. Промышленные сточные воды 331
Х.2.2.1. Нефте- и маслосодержащие сточные воды 331
Х.2.2.2. Сточные воды предприятий целлюлозно-бумажной
промышленности 332
Х.2.2.3. Сточные воды, содержащие красящие и дубильные
вещества 333
Х.2.2.4. Прочие сточные воды 335
Х.З. Обработка осадков . 335
Х.3.1. Коагуляция замораживанием 335
Х.З.2. Обработка коагулянтами и флокулянтами . ... 336
Литература 337
ЗАКЛЮЧЕНИЕ 344
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ 347
Евгений Дмитриевич Бабенков
ОЧИСТКА ВОДЫ КОАГУЛЯНТАМИ
Утверждено к печати
Ордена Трудового Красного Знамени
Институтом физической химии
Академии наук СССР
Редактор Б. Г. Аристов
Редакторы издательства В. М. Орлов и Л. Г. Чокалова
Художник Г. А. Астафьева
Художественный редактор С. А. Литвак
Технический редактор Р. Г. Грузинова
Корректоры И. А. Талалай, В. С. Федечкина
Сдано в набор 10/XIX 1976 г.
Подписано к печати 28/II 1977 г.
Формат 60х90Чм. Бумага типографская № 2.
Уел. печ. л. 22,26. Уч.-изд. л. 24,2.
Тираж 6600. Т-03772. Тип. зак. 1496
Цена 1 р. 71 к.
Издательство «Наука»
103717 ГСП, Москва, К-62, Подсосенский пер., д. 21
2-я типография издательства «Наука»
121099, Москва, Г-99, Шубинский пер., 10
ИСПРАВЛЕНИЯ И ОПЕЧАТКИ
Стр.
Строка
Напечатано
Должно быть
102
15 СН.
12733
12735
192
И св.
177
117
192
15 сн.
фильтрующихся
фильтрующих
228
13 сн.
VI
VII
296
7 св.
после формулы
флокирующие
флокулирующие
Е. Д. Бабенков