

























































































































































































































































Author: Ковальчук В.П. Васильев В.Г. Бойко Л.В. Зосимов В.Д.
Tags: почвоведение почвенные исследования история исторические науки растения ботаника
ISBN: 966-95049-4-5
Year: 2010




В.П. Ковальчук, В.Г. Васильев,
Л.В. Бойко, В.Д. Зосимов
МЕТОДОВ ИССЛЕДОВАНИЯ
Ш ПОЧВ И РАСТЕНИЙ
Институт сахарной свеклы НААНУ
В.П. Ковальчук, В.Г. Васильев,
JI.B. Бойко, В.Д. Зосимов.
СБОРНИК
МЕТОДОВ ИССЛЕДОВАНИЯ
ПОЧВ И РАСТЕНИЙ
Киев-2010
УДК 631.42
УДК 631.8:001.8
ISBN 966-95049-4-5
Ковальчук В.П., Васильев В.Г, Бойко Л.В., Зосимов В.Д, Сборник
методов исследования почв и растений. — К.: Труд-ГриПол - XXI
вк, 2010. - 252 с.
Изложенные в книге «Сборник методов исследования почв и
растений» методы анализа почв, растений и продукции растениеводства
рассмотрены и одобрены Ученым Советом института сахарной свеклы
НААНУ. Они апробированы в Институте и его сети на протяжении многих
лет и рекомендуются для проведения аналитических исследований в опыт¬
но-селекционной сети Института и координируемых учреждениях.
В книге изложены методы и технология проведения анализов почв,
растений и продукции растениеводства в легкодоступной форме, что
облегчает, ускоряет и упрощает работу аналитика.
Сборник подготовили сотрудники Института кандидаты с.-х. наук
В.П.Ковальчук, В.Г.Васильев и Киевского ОДПТЦ «Облдержродючгсть»
Л.В.Бойко, В.Д.Зосимов.
В проверке и апробации изложенных в сборнике методов участвовали:
к.б.н. К.А.Маковецкий, старший научный сотрудник Е.И.Костенко, научный
сотрудник И.И.Бойко и специалист А.П.Николаенко, за что авторы
сборника выражают им благодарность.
Ответственный за выпуск - доктор с.-х наук, академик Н.В.Роик.
Рецензент: кандидат с.-х наук А.В. Стуненко.
Сборник издан при содействии ОАО КОДЛ «Агросервис».
Председатель правления В.П. Иващук.
ISBN 966-95049-4-5 ^ „
© В.П.Ковальчук, В.Г. Васильев
Л.В.Бойко, В.Д. Зосимов
Издательство «XXI BiK» - Труд-ГриПол
Киев - 2010
Ученому почвоведу-агрохимику, профессору
И.И. Канивцу посвящается
СОДЕРЖАНИЕ
Стр.
I. ПРЕДИСЛОВИЕ 8
II. АНАЛИЗ РАСТЕНИЙ 10
Отбор и подготовка образцов к анализу 10
Определение абсолютно сухого вещества и гигроскопической
влаги в растительном материале 16
Определение сырой золы 19
Определение азота, фосфора, калия, натрия, кальция, магния в одной навеске 21
Определение фосфора в растворе золы 24
Определение калия и натрия в растворе золы 27
Определение азота в растворе золы с отгонкой на приборе Сереньева 29
Определение азота в растворе золы колориметрическим методом 31
Определение кальция и магния в растворе золы 34
Различные формы азота в растениях 37
Определение белкового и небелкового азота по методу Барнштейна 37
Определение содержания азота в растениях фотометрическим
методом с реактивом Несслера 40
Определение нитратов в растениях 42
Определение содержания небелкового азота в сахарной свекле
фотометричным методом с трихлоруксусной кислотой
(метод Тонкаля, Наумовой, Васильева) 45
Определение марганца, цинка, железа, меди и бора в одной навеске 47
Озоление растительного материала 48
Определение марганца, меди, цинка, железа в растворе золы
атомноабсорбционным методом 50
Определение бора в растворе золы колориметрическим методом с
азометином Н 53
Определение молибдена колориметрическим методом с роданистым
калием 56
Определение сахаристости и кондуктометрической золы в корнеплодах
свеклы в одной навеске 59
Определение доброкачественности (чистоты) очищенного нормального
сока сахарной свеклы методом Силина 63
Определение пектиновых веществ по пектату кальция 67
Определение пектиновых веществ объемным методом (по С.Я.Раик) 69
Определение крахмала поляриметрическим методом 70
Определение крахмала в картофеле и других крахмалонакопителях 71
Приближенное определение крахмала в мякоти плодов и в корнеплодах 71
Определение клетчатки 72
Определение клетчатки по модифицированному методу Кюршнера
и Хафера 73
Определение клетчатки по Х.Н.Починку 74
Определение органических кислот 77
Органические кислоты и методы их определения 77
Определение лимонной кислоты 77
Определение яблочной кислоты 79
-5-
Определение щавелевой кислоты 82
Определение пировиноградной кислоты 84
Определение суммы органических кислот (по Х.Н.Починку) 85
Определение общего содержания липидов (масел).(по Ермакову ) 87
Определение масла по массе (весу) сухого обезжиренного остатка 90
Метод определения жирнокислого состава масла и эруковой кислоты 91
Газохроматографический метод определения жирнокислого состава
масла семян рапса и сурепицы 91
Фотоколориметрический метод определения общего содержания
глюкозинолатов в семенах рапса и сурепицы палладиевым реактивом 92
III АНАЛИЗ ПОЧВЫ 94
Отбор и подготовка почвы к анализу 94
Определение гумуса 95
Определение гумуса по методу Тюрина в модификации ЦИНАО 97
Групповой и фракционный состав гумуса 101
Метод определения группового и фракционного состава гумуса по
Тюрину в модификации Пономаревой 101
Пирофосфатный метод определения группового состава гумуса 107
Определение общего азота 110
Определение нитрификационной способности почвы (метод Кравкова) 112
Определение гидролизуемого азота по Корнфильду 114
Определение гидролизуемого азота методом Тюрина и Кононовой 115
Определение содержания минеральных форм азота в почве 117
Определение аммония 120
Определение нитратов и обменного аммония в почвах
методом ННЦ ИПА 122
Определение нитратов ионометрическим методом 126
Определение подвижных форм фосфора и калия в почвах 130
Метод Кирсанова 131
Метод Чирикова 133
Метод Мачигина 135
Определение содержания обменного калия в почве методом Масловой 137
Метод определения гидролитической кислотности почв 139
Определение pH, обменной кислотности, подвижного алюминия,
марганца, обменного кальция, магния и серы в одной навеске 141
Определение pH и обменной кислотности 141
Определение подвижного алюминия 143
Определение обменного кальция 144
Определение обменного магния 145
Атомно-абсорбционное определение кальция и магния 146
Определение подвижной серы по методу ЦИНАО 150
Определение суммы поглощенных оснований 153
Определение емкости поглощения почв методом Бобко-Алешина в
модификации ЦИНАО 154
Анализ водной вытяжки 156
Определение сухого и прокаленного остатков водной вытяжки 157
Определение щелочности водной вытяжки 158
Определение сульфат-ионов в водной вытяжке 160
-6-
Определение хлор-ионов в водной вытяжке 163
Определение катионного состава водной вытяжки (Са, Mg, К, Na) 164
Определение подвижных марганца, меди, цинка и кобальта в одной
вытяжке атомно-абсорбционным методом 165
Извлечение подвижных Mn, Zn, Си, Со 166
Определение марганца 167
Определение цинка в некарбонатных почвах 168
Определение цинка в карбонатных почвах 169
Определение меди и кобальта 171
Определение подвижного бора 172
Определение содержания молибдена в почве 174
Определение интенсивности выделения С02 из почвы 177
Определение ферментативной активности почв 178
Активность инвертазы 178
Активность целлюлазы 181
Активность протеаз 183
Активность уреазы 185
Активность фосфатазы 187
Активность пероксидаз 188
Активность полифенолоксидаз 189
Активность нитратредуктазы 192
IV. ПРИБОРЫ И ПРИНЦИП ИХ РАБОТЫ 194
Прибор Сереньева для определения азота 194
Приборы для определения содержания растворимой золы методом
кондуктометрии 195
Кондуктометр типа ОК-Ю2 196
Переносной кондуктометр типа №571 197
Кондуктометр-золомер КЛЗ-1 198
Приборы для определения pH 200
рН-метр pH-121 200
рН-метр ПЛП-64 201
Иономер И-123 202
Фотометр фотоэлектрический КФК-3-01-«ЗОМЗ» 202
Спектрофотометр С-115 212
Спектрофотометр Квант-2 АТ 214
Весы-влагомер 216
V. ТЕХНИКА БЕЗОПАСНОСТИ
ПРИ ПРОВЕДЕПИИ ЛАБОРАТОРНЫХ РАБОТ 217
Правила пользования и работа с химическими реактивами 217
Меры предосторожности при работе со стеклянной посудой 217
Правила пользования газом 217
Правила пользования электрооборудованием 218
Оказание первой помощи 218
VI. ПРИЛОЖЕНИЕ 221
ЛИТЕРАТУРА 249
-7-
1. ПРЕДИСЛОВИЕ
Решение вопросов, связанных с повышением плодородия почв и улуч¬
шением качества производимой на них сельскохозяйственной продукции,
невозможно без проведения глубоких разносторонних аналитических и тех¬
нологических исследований. Ведь от методически и организационно пра¬
вильной их постановки, достоверной информативности широкого круга
ученых и практиков во многом зависит результативность дальнейших на¬
учных разработок в области оптимизации земледельческих приемов возде¬
лывания сельскохозяйственных культур, создании перспективных селек¬
ционных материалов, улучшении качества получаемой продукции.
С интенсификацией сельскохозяйственного производства в научных ис¬
следованиях значительно возрастают требования к объемам и качеству ана¬
литической информации, что не только предполагает внедрение в практи¬
ку лабораторных работ высокоточных и высокопроизводительных методов
исследования, но и усовершенствование измерительной техники и
приборов.
Используя богатейший научный материал из области аналитических
исследований почв, растений и продукции растениеводства, можно кон¬
статировать, что агрохимическая наука в этом плане имеет большие нара¬
ботки, а именно: разработано много методов, применяя которые можно
дать всестороннюю информацию о почве, растении и получаемой продук¬
ции.
Но на практике, к сожалению, по разным причинам не всегда можно
использовать самые современные методы анализа из-за дороговизны при¬
боров, посуды, реактивов. В большей степени это касается небольших
лабораторий, пользующихся старыми приборами и оборудованием, поэто¬
му мы оставили их описание и параллельно дали описание некоторых но¬
вых приборов, используемых в лабораториях.
Принимая это во внимание, мы включили в «Сборник методов иссле¬
дования почв и растений» разные методы анализа, но с полной технологи¬
ческой схемой их выполнения, где аналитик может выбрать для своих ис¬
следований (анализов) тот или иной метод, не нарушая при этом общих
требований к получению аналитической информации.
Многие из описанных методов уже сертифицированы Госстандартом,
иные проходят этот процесс, но в конечном результате все они должны
заканчиваться математической обработкой и соответствовать единым
оценочным требованиям.
Используемые и применяемые в процессе исследований методические
указания, посуда, фильтры и реактивы должны в полной мере соответ¬
ствовать официальным стандартам. При необходимости могут быть исполь-
-8-
чованы также другие аналогичные источники или эквивалентные им по
ишчимости официальные методические указания или рекомендации.
Составителями сборника преследовалась цель подать материал так, что¬
бы в одной книге пользователь мог получить полную информацию о порядке
и последовательности проведения анализа, начиная от отбора и подготовки
образца к анализу и до обработки полученных результатов, не теряя время
на поиск технологических приемов и всего, с чем связано выполнение
самого анализа. С этой целью составлены графические технологические
карты с описанием всех составных проведения анализа.
В „Приложении" указаны описание кислот, их удельная масса, нор¬
мальность растворов, атомные веса элементов, пересчетные коэффициенты,
данные о буферных смесях, сведения о количестве исходного вещества для
приготовления процентных растворов кислот, аммиака, составе и свой¬
стве солей и другие.
Обобщая пожелания аналитиков, иметь под рукой единый сборник наи¬
более часто используемых методов химического и технологического ана¬
лиза почв, растений и продукции их переработки, мы задались целью сгруп¬
пировать эти методы по соответствующим направлениям и подготовили
такой сборник, не претендуя на всеобъемлющее изложение (обобщение)
всех методов проведения анализов, а ограничились лишь теми, которые
чаще других используются в сети института и некоторых других научных
учреждениях, проводящих соответствующие исследования и анализы.
2. АНАЛИЗ РАСТЕНИЙ
2.1 ОТБОР И ПОДГОТОВКА ОБРАЗЦОВ К АНАЛИЗУ
2.1.1. Общие положения
Необходимым условием для правильной химической характеристики семян, зер¬
на, плодов, корней, листьев, стеблей и т.п. по малой навеске является правильное
взятие пробы и подготовка ее к анализу. Этому вопросу должно уделяться особое
внимание.
В средние образцы должно войти как можно большее количество растений или
их частей, но в то же время пробы не должны быть слишком громоздкими, чтобы не
осложнять их транспортировку. Поэтому по каждой культуре или группе культур уста¬
навливаются определенные параметры образцов, дающие возможность охарактери¬
зовать их по наиболее важным показателям химического состава, а в ряде случаев и
по технологическим качествам.
При отборе образцов во всех вариантах опыта необходимо соблюдать одинако¬
вую высоту среза растений, однотипность обрезки ботвы.
Взятие образцов не должно проводиться во время дождя, полива или сразу пос¬
ле них, а также при росе на листьях. Отбор их необходимо проводить в утренние
часы, до наступления жары или в конце дня (всегда в одно и то же время).
Каждый образец должен быть снабжен четко заполненной этикеткой.
Все операции по отбору, а в случае надобности и фиксации образцов, должны
быть проведены в один и тот же день по всем вариантам опыта. Для этого должна
быть проведена соответствующая подготовительная работа, дающая возможность ус¬
корить отбор образцов: заготовлены этикетки, пронумерованы и взвешены мешки
под образцы, проверена исправность весов, подготовлены опытные делянки или ис¬
следуемые участки.
2.1.2. Подготовка к отбору образцов
За два-три дня до уборки урожая выделяют учетные площади делянок, убирают
растения с защитных полос; при выпадении или сильном повреждении растений на
учетной площади делают выключки и уточняют учетные площадки.
В опытах с зерновыми, зернобобовыми культурами и травами выключки делают при
выпадении растений в рядках на отрезках более 50см; в опытах с культурами широкоряд¬
ного посева (сахарная свекла, картофель, подсолнечник, кукуруза и др.) - при выпадении
подряд трех и более гнезд при гнездовом посеве.
В выключки необходимо вводить соседние с выпавшими растения: по одному в опытах
с пропашными культурами и на полосе 25-30см в опытах с растениями узко- рядного
посева (зерновые, травы и т.д.).
Делянки, где выключки составляют более 50% учетной площади, выбраковывают.
Также исключают делянки, более 30% учетной площади которых выбраковано, если они
по урожайности значительно отличаются от остальных делянок варианта.
Для отбора образцов основной и побочной продукции заранее заготавливают из
фанеры, картона или плотной бумаги этикетки, на которых проставляют простым каран¬
дашом или шариковой ручкой номер или название опыта, культуру, вид отбираемой
продукции, вариант, повторение, номер делянки, дату взятия образца. Остальные сведения
(сорт, предшественник, фаза развития, вносимые удобрения, содержание вариантов опыта)
должны быть записаны в реестре и регистрационном журнале.
- 10-
Этикетки готовят по количеству намечаемых образцов (количество делянок х число
видов продукции).
Подготавливают также необходимое количество больших мешков (для 20-40-корне-
вых проб сахарной свеклы и других крупных образцов), малых мешочков из плотной
ткани - для образцов зерна, семян, соломы и др.
Для сохранения образцов зеленой массы, корнеплодов сахарной свеклы и др. от
подсыхания лучше использовать полиэтиленовые мешки.
При необходимости отбора образцов в начале вегетации, когда растения еще неболь¬
шие, перед выходом в поле подготавливают листы пергаментной бумаги, по размеру
достаточные для заворачивания образцов. Все необходимые сведения записывают на
внешней стороне бумажного листа. Количество их равно числу предполагаемых проб.
2.1.3. Исходный образец
Исходный образец в полевых опытах отбирают отдельно с каждого повторения,
что дает возможность провести математическую обработку, либо смешанный из двух
и более несмежных повторностей, в зависимости от программы каждого опыта.
Следует учитывать, что представительный по химическому составу образец (ко¬
эффициент репрезентативности близок к 100%) может быть получен из проб, взятых
с двух несмежных или всех повторностей варианта.
Исходный образец с крупноделяночных опытов отбирается небольшими пучками
растений, расположенных по диагонали делянки на равном друг от друга расстоянии,
или с нескольких пробных площадок, выделенных на учетной делянке. При ограничен¬
ном числе повторений для отбора образцов можно использовать защитные полосы.
Отбор образцов с малых делянок проводится, главным образом, в селекционной
практике. Индивидуальную изменчивость сельскохозяйственных культур в селекци¬
онной практике изучают на отдельных растениях, находящихся в одинаковых усло¬
виях произрастания. Поэтому для взятия проб выбирают наиболее выравненные од¬
нородные участки, на делянках которых берут через определенные промежутки не
менее 10 индивидуальных исходных образцов (отдельных растений) одновременно
со стандартным сортом (стандартом), высеваемым через 1-10 опытных делянок, в
зависимости от выравненности рельефа и плодородия участка.
Для исходных смешанных образцов корнеплодов с малых делянок берут с каждо¬
го варианта не менее 10 растений. При отборе зерновых, зернобобовых и масличных
культур используют весь урожай соответствующего вида продукции с данной делянки
(зерно, семена, солома, зеленая масса).
В вегетационных и лизиметрических опытах в сосуды высевают завышенное в
2-3 раза количество семян. После получения укоренившихся всходов проводят 1-3
прорывки, приурочивая последние к определенным фазам развития растений. Со¬
единяют вырванные растения всех повторностей одного варианта и, тщательно очи¬
стив их от почвы, сушат и измельчают. При уборке урожая поступают так же, как и
при отборе проб на малых делянках.
2.1.4. Средняя проба
Исходные образцы, доставленные в лабораторию, разделяют на отдельные орга¬
ны: корни, ботву, семена, зерно, солому и т.д., взвешивают, определяют весовое соот¬
ношение. Затем грубо измельчают вегетативную массу ножницами или соломорез¬
кой, крупные семена - на лабораторной электрической мельнице, хорошо перемеши¬
вают и отбирают квартованием среднюю пробу.
-11 -
Отдельную пробу сырого материала отбирают сразу же для определения содер¬
жания абсолютно сухого вещества, сахаристости и проведения некоторых других ана¬
лизов.
При подготовке к химическому анализу образцы, разложенные тонким слоем,
высушивают при комнатной температуре или с подогревом до температуры не более
50-60°С до ломкого состояния.
В биохимическом анализе проводят предварительную фиксацию образцов для
инактивации растительных ферментов и предотвращения развития микробиологи¬
ческих процессов. С этой целью свежеотобранные образцы помещают в предвари¬
тельно нагретый до 120-130°С сушильный шкаф в открытых бумажных или картон¬
ных коробках или лотках (при этом температура сразу опускается до 100-105°С) и
фиксируют 20-30мин. при t - 105°С, а затем досушивают, как указано выше.
В таком состоянии образцы сохраняются в лаборатории необходимый период
времени.
2.1.5. Аналитическая проба
Из среднего образца, хорошо перемешав его, отбирают методом квартования
аналитическую пробу массой 50-150г воздушно-сухого вещества в зависимости от
предполагаемого набора анализов и размалывают его.
Измельчают материал до размера частиц 0,5-1,0мм. Более тонкий помол
необходим при употреблении небольших навесок (менее 200мг).
Хранятся образцы в течение 10 месяцев в проветриваемом помещении в закрытых
картонных или металлических коробках (бюксах).
При взятии навесок аналитическую пробу еще раз тщательно перемешивают, чтобы
исключить расслоение частиц по размерам и массе.
Образцы одного вида продукции (например, корни или зерно) со всех вариантов
одного повторения опыта должны поступать на анализ одновременно.
Таковы общие положения условий отбора растительных образцов в различных
по назначению и задачам опытах.
2.1.6. Специфика отбора и подготовка проб отдельных культур
2.1.6.1. Сахарная свекла
Отбор проб и подготовка к анализу растений сахарной свеклы или ее отдельных
частей (органов) проводится, в зависимости от целей исследования, в различные
периоды вегетации и за 2-3 дня до уборки урожая. Для взятия исходных образцов
растений в полевых опытах отступают от смежной делянки 1м, или 2-3 рядка,
протягивают по диагонали делянки шнур и отбирают на пяти рядках по 4 растения,
начиная с места пересечения шнура с рядками. В зависимости от ширины делянки
места для отбора проб намечают через каждые 1-3 рядка. То же выполняют и по второй
диагонали. Всего отбирают 40 растений, выкапывают лопатой, освобождают от земли,
складывают во влажный мешок с этикеткой и держат в тени с целью предохранения
их от высыхания. Окончательную очистку корнеплодов и взвешивание всего растения,
отделение ботвы и учет массы корнеплода проводят в лаборатории. Массу ботвы
определяют по разности. Точность взвешивания при прорывке - 0,1 г, при уборке -
0,1кг.
При отборе проб перед уборкой результаты взвешивания массы корнеплодов и
ботвы приплюсовывают к общему урожаю с делянки.
- 12-
Если для анализа необходимы лишь листья, их отбирают, как и общие пробы по
диагонали через каждые 3-4 рядка с четырех растений. Разделив листья целого
растения, не выкапывая его, на 4 примерно равные части, снимают одну четвертую
часть листьев всех порядков, заворачивают во влажную фильтровальную бумагу, кладут
этикетку и направляют в лабораторию, где готовят их к анализу
Растения, отобранные для анализа, разделяют на органы: листовые пластинки
(жилки не удаляют), черешки, корнеплоды.
Отдельно взятые листовые пластинки или черешки используют для анализов в
свежем или сухом виде. Измельчение необходимо проводить острым ножом, так как
резка тупым ножом приводит к значительным потерям растворимых веществ вместе
с выжатым соком.
Масса свежего растительного материала, подготовленного для биохимического
анализа, должна составлять 250-300г, сухого - для химического анализа - 25-50г.
Аналитическую пробу готовят из сухой средней пробы.
Корнеплоды, отобранные для анализа (единичные корнеплоды или групповая
проба), осторожно моют, не оставляя в воде, чтобы не было потерь сахара, вытирают
насухо или проветривают на воздухе. Хранить корнеплоды до анализа можно в течении
5-7 дней немытыми в полиэтиленовых мешках при t = 1-5°С, что не приводит к
заметным изменениям в содержании сахара в них.
Корнеплод измельчают на мезгообразователе или ручной терке, вырезая по всей
длине, от головки до хвостика, сегмент, достигающий его сердцевины. Мелкие
корнеплоды следует измельчать полностью.
Полученную мезгу хорошо перемешивают в чашке, часть отбирают для проведения
анализа в свежем материале (сахаристость, кондуктометрическая зола, калий, натрий
и т.д.), остальную фиксируют и сушат или консервируют для последующих анализов.
При необходимости сохранения корнеплодов для дальнейшей селекционной
работы (поверхность сегмента) обрабатывают цементом или золой.
2.1.6.2. Зерновые культуры
Отбор проб зерновых и зернобобовых культур (кроме кукурузы) проводят в период
вегетации по фазам развития. Образцы отбирают в четырех местах делянки по 25см с
двух смежных рядков, что составляет 2 погонных метра на делянку.
Взятые образцы освобождают от почвы легким встряхиванием и заворачивают в
бумагу. В помещении пробу разбирают, подсчитывают число растений и отдельных
стеблей, отделяют надземную массу от корней, учитывают массу сухого и сырого ве¬
щества.
Растения, разделенные на отдельные органы, фиксируют, высушивают при 60-
70°С и размалывают для анализа. Анализы следует проводить раздельно в листьях,
стеблях, колосьях и корнях, так как отдельные органы в сухом виде имеют различный
удельный вес и средняя проба из целого растения не будет отражать действительного
соотношения его частей, а результаты анализа - истинного содержания определяе¬
мых элементов питания.
Для взятия образцов в период уборки вырывают все растения с корнями с двух
соседних рядков на четырех площадках размером по 0,25м2 (с длиной рядков х,см =
2500/2ш, где Ш - ширина междурядий, см), расположенных по одной из диагоналей
делянки на равных расстояниях друг от друга.
Корни обрезают, а надземную часть растений, стараясь не засорить ее землей,
кладут в заранее взвешенный мешок с этикеткой, укладывая их колосьями в одну
- 13-
сторону, взвешивают с точностью до Юг и сушат под навесом в условиях хорошего
проветривания. Обмолот каждого образца проводят вручную, не вынимая растений
из мешка, или небольшими ручными молотилками с тщательно последующей очисткой.
Взвешивают отдельно зерно и солому. Средний образец отбирают массой 2кг зерна и
не менее 500г соломы с половой. Если масса среднего образца зерна будет больше
2кг, его уменьшают квартованием. Чтобы отобрать 500г соломы с половой исходные
пробы ее предварительно режут соломорезкой или ножницами на куски размером 2-
Зсм и тщательно перемешивают.
При уборке урожая комбайном средний образец зерна отбирают при высыпании
его из бункера. При этом набирают не менее 10 выемок, пересекая механическим
пробоотборником или специальным ковшом струю зерна при высыпании через равные
промежутки времени.
Для отбора образцов соломы с половой используют пробные снопы, которые берут
в различных местах рядков, срезая пучки растений вручную на той же высоте, на какой
они срезаются комбайном. Последние отбирают при широкорядном посеве с одного
рядка, при ленточном - с двух соседних, чтобы вошли растения как среднего так и
крайнего рядка ленты; при обычном рядковом посеве с шириной междурядий 15см -
также с двух рядков, чтобы вошли растения, семена которых высеяны на различную
глубину передним и задним сошниками; при узкорядном - с трех соседних рядков.
При отборе индивидуальных проб зерновых, зернобобовых и масличных культур
зерно с обмолоченного растения (без включения зерна с подгонов и зеленых бобов
верхних ярусов), очищенное от примесей, рассыпают на бумаге, подравнивают в
квадратную фигурку с помощью угольника и делят на 4 части по диагонали (метод
квартования). Зерно из противоположных треугольников собирают для анализа.
Половина зерна с одного растения вполне отражает весь урожай этого растения.
Образцы доставляют в лабораторию в матерчатых мешочках. Масса пробы зерна
должна составлять 800-1500г в зависимости от предполагаемого набора определений
(учитывая технологический анализ), семян - 200г, зеленой массы - до 300г.
Перед размолом пробу зерна очищают от посторонних включений - комочков
земли, соломы, сорняков и семян других растений - и подсушивают в термостате при
температуре 60-70°С. Размол зерна для определения питательных веществ ведется
на электромельницах типа «Пируэт». Для массовых анализов принята тонина помола
в 1мм. Размолотые пробы перемешивают и помещают в те же картонные коробки
(или бюксы), в которых они сушились.
Таким же путем проводится подготовка остальных частей растений (соломы,
листьев и т.д.).
Для технологического анализа размол зерна проводят на специальных зерновых
мельницах.
2.1.63. Кукуруза
Анализ растений кукурузы при изучении роста и развития ее в зависимости от условий
выращивания лучше начинать с фазы интенсивного роста - образования 5-6 листьев,
выбрасывания метелки, молочно-восковой и полной спелости. В опытах с кукурузой
трудно отобрать пробы, которые отражали бы все особенности растений, так как даже в
пределах одной делянки растения сильно варьируют по высоте, массе, химическому составу
и другим показателям.
Образцы отбирают по одной из диагоналей учетной делянки в 10 гнездах. В пробу
берут все растения из намеченного гнезда, чтобы правильно учесть влияние площади
питания, приходящейся в среднем на каждое растение делянки.
- 14-
Взятые образцы в лаборатории разделяют на отдельные органы - листья, стебли (с
листовыми влагалищами), початки без обертки (обертки присоединяются к листьям), а в
фазу полной спелости - листья, стебли, стержни початков и зерно. После взвешивания
всех органов составляют для каждого из них средний образец.
Пробу зерна с початков отдельных растений кукурузы отбирают после обмолота и
перемешивают. Для этого достаточно взять 1/5 или 1/10 часть урожая зерна, но не менее 25г.
Каждый лист растения делят на две части по центральной жилке и берут по 1/2 в
пробу, сюда же берут по 1/2 листка обертки. Стебель делят по диаметру на четыре части и
в образец берут 1/4 часть по его длине. Проводят грубое измельчение образцов и при
необходимости уменьшают их квартованием. После подсушивания до ломкого состояния
образец размалывают до тонины помола в 1мм.
2.1.6.4. Картофель
Образцы картофеля отбирают по диагонали учетной делянки, пропустив два
защитных рядка, по 10 кустов на двух несмежных повторениях в периоды бутонизации,
цветения и далее до уборки через каждые 10 дней.
Кусты тщательно выкапывают, отряхивают от земли, клубни отделяют от
столонов, очищают от почвы и взвешивают урожай с 10 кустов. Учитывая массу ботвы,
очищенной от столонов.
После взвешивания всего образца отбирают исходные образцы клубней и ботвы
массой до 1кг. В первый срок отбора проб, когда масса клубней еще небольшая, берут
1/4 урожая с 10 кустов, но не сортируя клубни по размерам. Позже, когда величина
клубней увеличивается, массу клубней сортируют на 3 фракции (мелкие, средние и
крупные). Каждую фракцию отдельно взвешивают и вычисляют процентное
соотношение ее к массе всех клубней. Согласно этому соотношению составляют пробу
массой 1кг.
Для составления индивидуальных образцов растения картофеля, где клубней много,
делят пополам так, чтобы в каждую половину вошли в одинаковых соотношениях крупные,
средние и мелкие клубни. Одну из этих проб используют для химического анализа. Для
изучения зеленой массы берут 1/4 надземной части растений (ботвы, черешков, листовых
пластинок, стеблей) с одинаковым соотношением отдельных органов.
Для получения среднего образца в лаборатории отобранные клубни разрезают
вдоль (через вершину и пуповинную часть) на 4 части и берут 1/4 в смешанный образец.
Затем их режут на ломтики и сушат в сушильном шкафу при температуре не выше
60°С до ломкости. По окончании сушки пробу измельчают.
Ботву грубо измельчают острым ножом, хорошо перемешав, отбирают среднюю
пробу квартованием, сушат и измельчают для анализа.
2.1.6.5. Лен-долгунец
Образцы льна для химического анализа в основные фазы роста и развития
(«елочка», быстрый рост, бутонизация, цветение, зеленая спелость и ранняя желтая
спелость) отбирают с учетной делянки, которую обходят вокруг по защитной дорожке
и через определенное число шагов выдергивают по 2-3 растения на расстоянии
вытянутой руки, всего 100-200 растений (в зависимости от размера делянки).
Из отобранных растений готовят прямо в поле смешанный образец, удаляя
корневую часть на уровне семядольных листьев. Образцы с этикетками раскладывают
на чистые листы бумаги и сушат в хорошо проветриваемом крытом помещении.
За 2-3 дня до уборки отбор проб проводят на четырех площадках размером 0,25м2
- 15-
по одной из диагоналей учетной делянки. На этих площадках, длина которых вычислена
в зависимости от ширины междурядий (длина = 2500:2111, где Ш - ширина междурядий),
вырывают все растения с двух смежных рядков. После обмолота полученных снопов
отбирают образцы семян и соломки по 150-300г.
Подсушенные до воздушно-сухого состояния пробы измельчают сначала на стебле-
резке или ножницами (соломку), а затем на мельничках типа «Пируэт». Уменьшение
средней пробы соломки проводится после грубого измельчения и перемешивания
путем квартования.
2.1.6.6. Подсолнечник
Отбор образцов подсолнечника во время вегетации проводится в фазе 1-2 пар
настоящих листьев, перед или во время прорывки в количестве 25-50 растений с учетом
делянки, а в фазе образования корзинки, цветения и созревания - по 10 растений с делянки.
За 10-15 дней до уборки намечают, привязав к ним ленты, на делянках по 10
типичный растений, размещенных вдоль одной из диагоналей, и организуют
систематический сбор засохших листьев этих растений.
Отобранные свежие образцы после взвешивания разделяют на отдельные органы,
устанавливают весовое соотношение их в сыром и воздушно-сухом состоянии.
Вымолоченные корзинки делят на 4 части и j каждой в измельченном виде берут
на среднюю пробу. Листья, черешки, стебли после высушивания измельчают и
отбирают квартованием среднюю пробу.
В семенах подсолнечника разных сортов относительный процент кожуры
значительно варьирует, а химический состав зародышей с эндоспермом также
существенно различается, поэтому часто анализируют только освобожденные от
оболочек семена.
Масличные семена измельчают перед анализом и хранят недолго, предохраняя
от света и окисления (лучше всего в алюминиевых бюксах).
2.2. ОПРЕДЕЛЕНИЕ АБСОЛЮТНО СУХОГО ВЕЩЕСТВА
И ГИГРОСКОПИЧЕСКОЙ ВЛАГИ
В РАСТИТЕЛЬНОМ МАТЕРИАЛЕ
2.2.1. Принцип метода
Абсолютно сухое вещество в растительном материале определяется весовым ме¬
тодом после высушивания его в сушильном шкафу при температуре 100-105°С до
постоянной массы. Показатель гигроскопической влаги получают по разности веса
воздушно сухого и абсолютно сухого вещества.
Определение абсолютно сухого вещества и гигроскопической влаги может про¬
водиться в воздушно-сухом (1) и сыром (2) материале. Кроме общепринятого, можно
использовать экспресс-методы (3).
2.2.2. Технологическое оборудование
Весы типа BJIKT-500.
Бюксы стеклянные или алюминиевые.
Эксикатор с водопоглощающим веществом.
Сушильный шкаф.
Палочки стеклянные оплавленные.
Прибор Чижовой. Пакеты из фильтровальной бумаги.
- 16-
2.2.3. Реактивы
1. Кальций хлористый прокаленный.
2. Кислота соляная, разбавленная дистиллированной водой в соотношении 1:1.
3. Песок очищенный и прокаленный. Белый речной песок отмучивают в стеклянной
посуде водопроводной водой, заливают соляной кислотой, разбавленной 1:1, хорошо
перемешивают и оставляют на ночь закрытым. После этого песок промывают водопро¬
водной водой до исчезновения кислой реакции (проба на лакмус), затем дистиллирован¬
ной и высушивают. Промытый песок просеивают через сито с диаметром ячеек 1-1,5мм
и прокаливают для удаления органических веществ. Хранят в закрытой банке.
2.2.4. Ход анализа
1. Чистые стеклянные или алюминиевые бюксы просушивают в течение ЗОмин. в
сушильном шкафу при t = 105°С, охлаждают в эксикаторе и взвешивают вместе с крыш¬
ками на аналитических весах или весах типа BJIKT. Из подготовленной аналитической
пробы во взвешенные бюксы берут навески (в двухкратной повторности) размолотого
для анализа воздушно-сухого материала-2,5-5,Ог. Открытые бюксы вместе с крышками
помещают в сушильный шкаф, нагрев которого отрегулирован на 105°С, и сушат в тече¬
ние 4-6 часов (в зависимости от анализируемого продукта). Затем закрывают их крыш¬
ками, охлаждают в эксикаторе до комнатной температуры и взвешивают на тех же ве¬
сах. После этого дополнительно сушат еще 2 часа, охлаждают и взвешивают. При раз¬
нице в массе бюксов после первого и второго высушивания не более 0,02 г высушива¬
ние считается законченным.
2. Содержание абсолютно сухого вещества в свежем растительном материале (кор¬
неплоды сахарной свеклы, плоды, жом и др.) определяется следующим образом.
Из средней пробы измельченного образца после тщательного перемешивания бе¬
рут на весах типа BJIKT не менее двух навесок по Юг с точностью до 0,01 г в предва¬
рительно высушенные и взвешенные бюксы.
Если влажность материала очень высокая, в бюксы перед взятием навески до¬
бавляют 5г чистого песка и стеклянную палочку, которую кладут по диагонали бюкса,
чтоб не мешала закрывать его крышкой (масса песка и палочки учитывается при та¬
рировании бюксов).
Хорошо перемешанный с навеской песок делает ее более рыхлой и ускоряет вы¬
сушивание. В противном случае на поверхности навески образуется плотная корка, а
внутри вещество долго остается сырым.
Сушка ведется в два этапа. Сначала бюксы со снятыми крышками (без крышек)
помещают в сушильный шкаф с хорошей вентиляцией и выдерживают при темпера¬
туре 50-60°С до хрупкого состояния содержимого. После этого их сушат еще 3-4 часа
при 105°С. Охлажденные в эксикаторе бюксы взвешивают, вновь сушат в течение 1
часа при температуре 100°С, снова охлаждают и взвешивают. Если разница между
двумя взвешиваниями превышает 0,02г, сушат еще полчаса, снова охлаждают и взве¬
шивают. В том случае, когда при высушивании масса начинает увеличиваться (за счет
процессов окисления), высушивание прекращают и в расчет берут самое меньшее
показание.
При резком повышении температуры сверх 105°С начинается обугливание и по¬
теря массы. Легко окисляющиеся вещества (семена льна, конопли, жмых, корне- и
клубнеплоды, барда и др..) нельзя сушить даже при такой температуре. Это вызывает
потерю углекислого газа, аммиака и других летучих соединений. Такие материалы
сушат в вакууме.
- 17-
3. Кроме обычных, применяемых в лабораторных, существуют еще методы быст¬
рого определения абсолютно сухого вещества в растительных материалах.
Основным экспресс-методом определения влажности и сухого вещества семян сель¬
скохозяйственных культур является высушивание навески размолотого материала в су¬
шильном шкафу в соответствии с режимами, указанными в таблице 1.
Таблица 1.
№
Семена культур
Температура
высушивания,
°С
Продолжи¬
тельность
высушивания,
мин.
Пшеницы, гороха, ржи, тритикале, ячменя, овса,
гречихи, вики
150
20
Зерновых и зернобобовых (кроме указанных в
пункте 1), люпина, эспарцета, подсолнечника,
арахиса, клещевины, сои
130
40
Овощных (кроме гороха, фасоли, бобов), бахчевых,
кормовых трав и корнеплодов, медоносных трав,
льна, юнопли, горчицы, кенафа
130
60
Табака, махорки
130
20
Масличных (кроме указанных в п.2 и 3),
эфиромасличных, технических (кроме указанных в
п.З и 4) и лекарственных
105
300
Сахарной свеклы
130
60
Примечание: семена табака и махорки с влажностью выше 12% высушивают в течении 30
мин.
Перед взятием навесок измельченный материал тщательно перемешивают в банке,
в которой хранится образец, затем отбирают из разных мест две навески по 5г в пред¬
варительно взвешенные бюксы. Бюксы ставят в сушильный шкаф, отрегулированный
и разогретый до нужной температуры. Время высушивания считается с момента уста¬
новления указанной в табл. 1 температуры в загруженном бюксами шкафу. Высушенные
бюксы с навесками переносят в эксикатор, охлаждают примерно ЗОмин. и взвешивают
с точностью до 0,01г.
Для быстрого определения сухих веществ в сыром, прессованном и сушеном жоме
и других растительных материалах применяется прибор Чижовой. Навеску сырого ма¬
териала около Зг (взвешивание ведут с точностью до 0,01 г) помещают в высушенных и
взвешенных пакетах из фильтровальной бумаги в прибор и сушат 15-20мин. при тем¬
пературе 140-145°С. После высушивания пакеты охлаждают в эксикаторе и взвешивают.
2.2.5. Обработка результатов.
Содержание абсолютно сухого вещества и гигроскопической влаги при опреде¬
лении любым из приведенных методов рассчитывают по формулам:
СД,% =
_ (а2 - а) 100
ГДл,%=(о‘ аг)10°
ще СВ - абсолютно сухое вещество;
- 18
ГВл - гигроскопическая влага;
а - масса бюкса, г;
я, - масса бюкса с навеской до высушивания, г;
а2 - масса бюкса с высушенной навеской, г;
100 - коэффициент пересчета в проценты.
Разница между параллельными определениями не должна превышать 0,3% (абс.).
2.2.6. Технологическая карта
2.3. ОПРЕДЕЛЕНИЕ СЫРОЙ ЗОЛЫ
2.3.1. Принцип метода
Метод определения золы основан на сжигании органического вещества при сво¬
бодном доступе воздуха. При сжигании углерод, водород и частично кислород улету¬
чиваются в виде углекислоты и паров и остаются только минеральные элементы в
виде окисных соединений.
«Сырой» золу называют потому, что в ее составе могут быть механические при¬
меси (песок), несгоревшие частицы угля.
2.3.2. Технологическое оборудование
Весы аналитические.
Муфельная печь.
Тигли фарфоровые №3-4.
Эксикатор.
2.3.3. Ход анализа
Чистые фарфоровые тигли прокаливают в муфельной печи 1-2 часа. Тигли долж¬
ны быть пронумерованы, для чего используют раствор хлорного железа (FeC13 6Н20,
0,5% водный раствор).
После охлаждения в эксикаторе тигли взвешивают на аналитических весах и в
них берут навески воздушно-сухой пробы 2-5г
Пробу укладывают рыхло для более полного и быстрого озоления. Тигли ставят в
холодную муфельную печь и поднимают температуру до 200°С. Начало озоления про¬
водят медленно для того, чтобы исключить возможное разбрасывание мелких частиц
пробы. Эту операцию можно проводить и на электрической плитке в вытяжном шка¬
фу. Через 50-60мин. после того, как проба перестала дымить, температуру в муфеле
- 19-
поднимают до 525-550°С (темно-красное каление) и сжигают обуглившуюся массу до
тех пор пока зола не приобретет светло-серый цвет. Иногда зола имеет красно-бурый
или зеленоватый оттенок из-за наличия окислов железа и марганца. Для полного
озоления пробы обычно достаточно 5-6 часов. По окончании прокаливания тигли с
золой охлаждают вначале в выключенной муфельной печи, а затем в эксикаторе и
взвешивают на аналитических весах.
В случае, если в золе остались несгоревшие обуглившиеся частицы, тигли охлаж¬
дают, прибавляют в их несколько капель горячей дистиллированной воды или 2-3
капли 3% раствора перекиси водорода и снова осторожно прокаливают в муфельной
печи. После такой обработки остатки угля быстро сгорают.
Определение сырой золы можно проводить также при озолении материала для
определения микроэлементов. При этом взвешивание тиглей (пустых, с раститель¬
ным материалом и с золой) проводится на аналитических весах.
2.3.4. Обработка результатов
Содержание сырой золы рассчитывают по формуле:
в. (а-в)Л0 О
Сырая зола, % = ,
н
ще а- масса тигля с сырой золой, г;
в - масса тигля, г;
н - навеска, г;
100 - коэффициент пересчета в проценты.
Анализ считается проведенным с достаточной точностью, если отклонение повтор¬
ного анализа от среднего (принимаемого за 100%, рассчитанного по данным основного
и повторного) не превышает допустимых расхождений, приведенных в табл. 2.
Таблица 2.
Содержание сырой
золы, %
Допустимые отклонения от среднего при повторных определениях
(в отн. %)
в одной лаборатории
в разных лабораториях
1,0- 10,0
5,0
10,0
Свыше 10,0
5,0
10,0
Пример: содержание сырой золы, по данным двух определений, составляет со¬
ответственно 1,020 и 1,120%.
1,020 + 1,120
Среднее содержание золы = = 1,070%
Отклонение каждого единичного определения от среднего равно:
1.07- 1,02 = 0,05
1.07- 1,12 = -0,05 абс.%,
что составляет± ?z05100 = ±4,6отн.%
1,07
По данным таблицы, для этой концентрации считается допустимой относитель¬
ная ошибка ± 5%, что в абсолютной величине составляет 0,054% золы, а разница в
данном примере ± 0,05%, или относительная ошибка ± 4,6%, что ниже допустимой.
Такой результат считается достоверным.
-20-
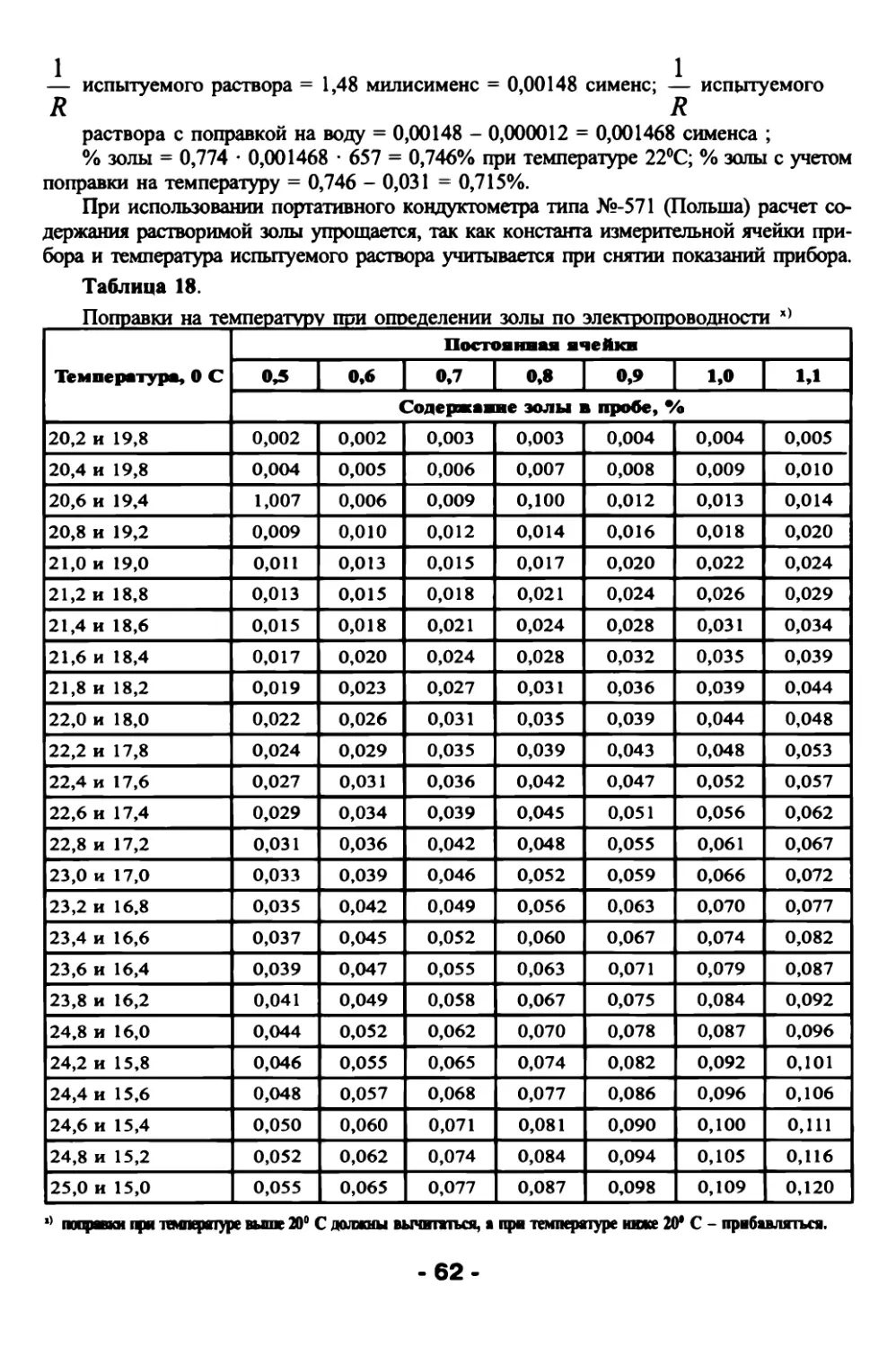
2.3.5. Технологическая карта
Зольный состав растений подвержен значительным колебаниям в зависимости
от условий произрастания растений (влияние климата, почв, внесенных удобрений
и т.д.). В табл. 3 приведены примерные данные общего содержания золы в различных
органах растений.
Таблица 3. Содержание сырой золы в различных органах сельскохозяйственных
растений (в % к сухому веществу).
Зерио:
в
1
1
клубнеплоды:
озимой пшениц ы
1,6-2,0
свеклы
0,5-1,1
озимой ржи
1,7-2,1
картофеля
1,0-1,1
овса
1,9-3,0
Солома:
кукурузы
1,5-1,7
пшеничная
4,8-6,6
ячменя
2,3-2,6
ржаная
4,1-6,1
гречихи
1,1-2,7
овсяная
5,0-5,8
Семена:
гречишная
6,0-6,5
подсолнечника
1,8-4,9
Сено:
клевера
3,0-3,8
клеверное
5,0-5,5
хлопчатника
3,2-4,5
тимофеевки
4,0-6,0
гороха
2,6-3,1
Ботва и стебли:
льна
3,9-8,7
свеклы
2,7-3,5
кукурузы
5,0-5,5
подсолнечника
2,0-3,3
картофеля
3,0-5,0
2.4. ОПРЕДЕЛЕНИЕ АЗОТА, ФОСФОРА, КАЛИЯ, НАТРИЯ,
КАЛЬЦИЯ, МАГНИЯ В ОДНОЙ НАВЕСКЕ
2.4.1. Озоление растительного материала
2.4.1.1. Принцип метода
Подготовка растительных материалов к определению зольных элементов и азота
основана на «мокром» озолении органического вещества концентрированной серной
кислотой в присутствии перекиси водорода.
Для органического азота эту реакцию (на примере аланина) можно выразить урав¬
нением:
- 21 -
2CH3CHNH2COOH + 13H,SOd
(NH4)2S04 + 6C02+ 16H20 + 12SO,
Следует отметить, что при таком способе озоления не учитывается азот нитра¬
тов, нитритов и некоторых гетероциклических соединений.
Фосфор органических соединений (липидов, нуклеиновых кислот, фитина и др.)
гидролизуется до фосфатов.
В результате озоления калий, натрий, кальций, магний и другие зольные элемен¬
ты связываются в виде сернокислых и частично фосфорнокислых солей.
Полученный раствор после разбавления дистиллированной водой является ис¬
ходным для определения азота и зольных элементов.
2.4.1J. Технологическое оборудование
Весы аналитические или торсионные.
Весы типа ВЛКТ.
Электрические плитки.
Песчаные бани.
Мерные колбы или пробирки на 50, 75 или 100мл из термостойкого стекла.
Пипетка с грушей или капельница для закапывания 30% раствора перекиси
водорода.
Дозаторы для крепких кислот на 5 и 10мл.
2.4.13. Реактивы
Кислота серная х.ч., концентрированная.
Перекись водорода, 30% водный раствор (пергидроль).
2.4.1.4. Ход анализа
Навеску 0,2-0,4г абсолютно сухой пробы, что удобно для расчетов, взвешивают
на торсионных весах и помещают в колбы Кьельдаля или термостойкие пробирки
на 50-100мл, заливают 5-10мл концентрированной H2S04 и 0,2(0,5)мл. пергидроля.
Содержимое колб осторожно перемешивают, ставят на холодную песчаную баню и
медленно нагревают. При озолении образцов с высоким содержанием сахаров (мезга
сахарной свеклы и др.) часто в начале нагревания в колбах происходит сильное вспе¬
нивание. В этом случае лучше начальный нагрев вести без перекиси водорода до
появления в колбах белого дымка. Если же вспенивание все равно имеет место, следует
колбы снять с плитки и по стенке осторожно, не встряхивая их, закапать 2-3 капли
спирта. Пробитый плотный слой пены оседает, после этого озоление продолжают
обычным порядком. Иногда эту процедуру приходится проводить дважды. Озоление
ведут при температуре 270-350°С. Через 30-40мин. колбы снимают с нагревательного
прибора, слегка охлаждают, добавляют сначала 3-5, а в последующие разы 2-3 капли
Н202 и продолжают озоление до полного обесцвечивания раствора. После последнего
добавления Н202 колбы нагревают еще в течении 15-20 мин. до полного разложения
избытка Н.02.
После охлаждения раствор осторожно разбавляют дистиллированной водой при
постоянном помешивании. Затем повторно охлаждают, переносят в мерные колбы
на 200-250мл, доводят до метки и перемешивают. Раствор служит исходным для оп¬
ределения азота, фосфора, калия, натрия, кальция и магния. Определение их лучше
проводить в следующем порядке: фосфор, калий, натрий, азот, кальций, магний. Это
связано с тем, что на дне колбы после озоления всегда имеется небольшой осадок
кремнекислоты. Отбор аликвот для определения фосфора, калия и натрия лучше вести
-22-
Таблица 4. Содержание азота и зольных элементов в различных органах рас¬
тений (% на абсолютно сухое вещество).
Растения | Азот | Фосфор | Калий | Натрий | Кальций | Магний
Сахарная свекла
Молодое растение
3,0-6,0
0,45-1,1
4,2-8,3
2,0-4,0
0,7-1,5
0,9-1,2
Корнеплоды (мезга)
0,5-1,6
0,45-0,7
0,4-1,8
0,03-0,1
0,2-1,0
0,1-0,4
Листья
1,5-5,0
0,2-0,9
2,5-6,3
1,4-3,0
1,0-2,5
0,6-0,7
Семена
1,5-2,5
0,1-0,4
0,6-2,1
-
Собственно семя
3,0-5,0
0,9-1,3
0,8-1,7
0,06-0,2
0,2-0,5
0,7-1,0
Околоплодник
1,0-2,0
0,1-0,4
1,7-3,0
0,3-0,7
0,7-1,7
0,8-1,5
Озимая пшеница
Зерно
1,5-3,5
0,2-0,9
0,2-0,8
-
0,03-0,05
0,1-0,2
Солома
0,2-1,0
0,02-0,03
0,2-0,8
0,08-0,1
0,12-0,25
-
Корневые остатки
0,5-1,5
0,05-0,2
0,1-0,2
-
-
-
Пожнивные остатки
0,3-1,0
0,02-0,2
0,1-1,2
-
-
-
Ячмень
Зерно
1,5-2,0
0,2-0,6
0,2-0,6
-
0,04-0,06
-
Солома
0,3-1,0
0,01-0,02
0,2-1,6
-
0,15-0,30
-
Овес
Зерно
1,5-3,0
0,2-0,6
0,4-0,6
-
0,05-0,2
0,1-0,2
Солома
0,6-0,9
0,05-0,06
-
-
0,25-0,40
-
Горох
Зерно
2,5-4,5
0,2-0,9
0,1-1,2
-
-
-
Солома
0,5-1,5
0,02-0,05
0,2-0,8
-
1,2-1,5
0,4-0,6
Корневые остатки
1,5-2,0
0,05-0,2
0,4-1,2
-
-
Пожнивные остатки
0,5-1,5
0,05-0,15
0,2-0,8
-
-
-
Кукуруза
Зерно
1,5-3,0
0,2-0,5
0,2-0,6
-
0,01-0,03
0,1-0,2
Стебли
0,5-1,0
0,1-0,5
0,8-2,0
-
-
Корневые остатки
0,5-1,5
0,05-0,2
0,8-1,2
-
-
-
при большом слое надосадочной жидкости, т.к. попавший в раствор осадок мешает
колориметрическому и спектральному определению этих элементов и не влияет на
определение азота по Сереньеву.
Одновременно проводят холостое определение через все стадии анализа, кроме
взятия навески анализируемого вещества.
Примерное содержание минеральных элементов и азота в некоторых культурах
зерно-свекловичного севооборота приведено в таблице 4.
2.4.1.5. Технологическая карта
-23-
2.4.2. Определение фосфора в растворе золы
2.4.2.1. Принцип метода
Для определения фосфора использован колориметрический метод, основанный
на способности ортофосфатов давать с молибденовой кислотой при определенной
концентрации водорода соли фосфорномолибденовой гетерополикислоты
Н7[Р(М0207)> • Н20.
При восстановлении части входящего в его состав шестивалентного молибдена
до пятивалентного образуется молибденовая синь, точный состав которой еще не
установлен. Интенсивность окраски раствора пропорциональна концентрации
фосфора при таких условиях реакции, когда восстанавливается лишь связанный с
фосфором молибден, а остальное его количество находится в виде избытка молибда-
та аммония.
Устойчивость получаемого окрашенного комплекса зависит от реакции среды,
концентрации молибдена и его соотношения с суммарной кислотностью раствора и
применяемого восстановителя.
Как показали исследования Мещерякова, для кислых испытуемых растворов, ка¬
кими являются растительный минерализат и почвенные вытяжки (концентрация Н-
ионов 0,06-0,14мг-экв/мл.), стабильность результатов без их предварительной нейт¬
рализации практически достигается применением молибденовых реактивов, обес¬
печивающих в колориметрированных растворах концентрацию Н-ионов 0,36-0,46мг-
экв/мл., а соотношение водорода к MQ03 равное 21.
Лучшим восстановителем для массового анализа в настоящее время можно счи¬
тать предложенную Мэрфи и Райли аскорбиновую кислоту при стабилизации окраски
сурьмяновиннокислым калием.
Определению мешают: трехвалентное железо в концентрации >0,04мг на 1мл
окрашенного раствора, окись кремния >0,8мг/мл., органические кислоты, гумусовые
вещества, фтор, мышьяк (Гинзбург, 1958).
Окраска развивается в течение Юмин. без нагревания и устойчива более суток.
2Л2.2. Технологическое оборудование
Весы аналитические и типа BJ1KT.
Фотоэлектроколориметр.
Пипетка мерная с резиновой грушей, поршневая либо шприц-дозатор на 1мл.
Дозатор на 19мл.
Цилиндры мерные на 500мл.
Пробирки или конические колбы на 40-50мл.
Колбы мерные на 1000, 500, 200мл.
2.4.2.3. Реактивы
1. Окрашивающий раствор.
Реактив А. 6г молибденовокислого аммония растворяют в 200мл дистиллиро¬
ванной воды. 0,1454г сурьмяновиннокислого калия растворяют в 100мл воды. Оба
реактива растворяются при слабом нагревании. Охлаждают, вливают в мерную колбу
емкостью 1л, доливают 500мл 5н. H2S04 (реактив 2) и доводят до метки дистиллиро¬
ванной водой. Хранят в темной склянке.
Реактив Б. 0,887г аскорбиновой кислоты растворяют в 168мл реактива А и дово¬
дят объем до 1л дистиллированной водой. Реактив Б готовят в день работы. В течение
2-3 дней при хранении в холодильнике он сохраняет свои качества.
-24-
2. 5 н. H.SOr 140мл концентрированной серной кислоты вливают в дистиллиро¬
ванную воду и после охлаждения объем доводят до 1л.
3. Образиовые растворы для определения фосфора.
Исходный образцовый раствор А ,393г химически чистого перекристаллизован-
ного КН2Р04 растворяют в дистиллированной воде и доводят до метки в мерной
колбе на 1л. В 1мл исходного образцового раствора содержится 1мг фосфора. Этот
раствор является исходным для приготовления шкалы.
Однозамещенный фосфорнокислый калий KHjPC^ часто бывает загрязнен двух-
и трехзамещенным фосфатом калия. Проверить чистоту его можно при помощи ин¬
дикаторов. Так, раствор КН2Р04 имеет кислую реакцию (pH 4,3) и окрашивает метил-
рот в красно-оранжевый цвет; К2НР04 • ЗН20 с pH 9,6 окрашивает фенолфталеин в
интенсивно малиновый, а тимолфталеин в светло-синий; KjP04 с pH 13,2 легко узнать
по пурпурной окраске фенолфталеина и интенсивно синей тимолфталеина. Для очи¬
стки от примесей КН2Р04 промывают декантацией 5-6 раз этиловым спиртом, а затем
перекристаллизовывают 1-2 раза из горячего водного раствора. На растворение ЮОг
КН2Р04 требуется 190мл горячей воды. При охлаждении (чашку с горячим раствором
помещают в снег или воду со льдом) выпадает до 60г перекристаллизованной соли.
Выпавший осадок отфильтровывают под разрежением и сушат сначала между листа¬
ми фильтровальной бумаги, а затем 2-3 часа в сушильном шкафу при температуре
110°С. Сухую безводную соль хранят в банке с притертой крышкой.
Шкала образцовых растворов. В мерные колбы вместимостью 500мл наливают
дистиллированную воду, добавляют 25мл концентрированной серной кислоты, пе¬
ремешивают, затем вливают указанные в таблице 5 количества исходного образцово¬
го раствора КН2Р04 и доводят водой до метки.
По оптической плотности образцовых растворов строят калибровочный график:
на горизонтальной оси которого наносят значения концентрации фосфора в 1мл ра¬
створа или в % на абсолютно сухое вещество, на вертикальной - оптическую плот¬
ность раствора.
Таблица 5.
Номера
Показатели^^!^^
1
2
3
4
5
6
7
8
Объем образцового
раствора, мл.
0
1
2
4
6
8
10
16
Содержание Р в мг в
1 мл, в % на абс.суже
вещество
0
0,002
0,004
0,008
0,012
0,016
0,020
0,032
При н=0,2 г
V= 50 мл
0
0,05
0,1
0,2
0,3
0,4
0,5
0,8
При н=0,4 г
V = 50 мл
0
0,025
0,05
0,1
0,15
0,2
0,25
0,4
2.4.2.4. Ход анализа
Из исходного раствора, не взмучивая осадка кремнекислоты, берут пипеткой по
1,0мл жидкости в пробирки. Добавляют дозатором 19,0мл реактива Б, хорошо пере¬
мешивают и через 1час колориметрируют, используя светло-красный светофильтр с
-25-
максимумом пропускания 620нм и кюветы с толщиной поглощающего слоя 10-20мм.
Окраска устойчива в течении 24 часов.
Одновременно окрашивают шкалу образцовых растворов. Для этого отбирают
по 1мл каждого образцового раствора, (табл.5) доливают 19мл реактива Б, переме¬
шивают и через 1 час колориметрируют вместе с испытуемыми растворами.
Сравнение всех растворов - шкалы, холостого и испытуемых - ведут с нулевым
раствором шкалы.
2.4.2.5. Обработка результатов
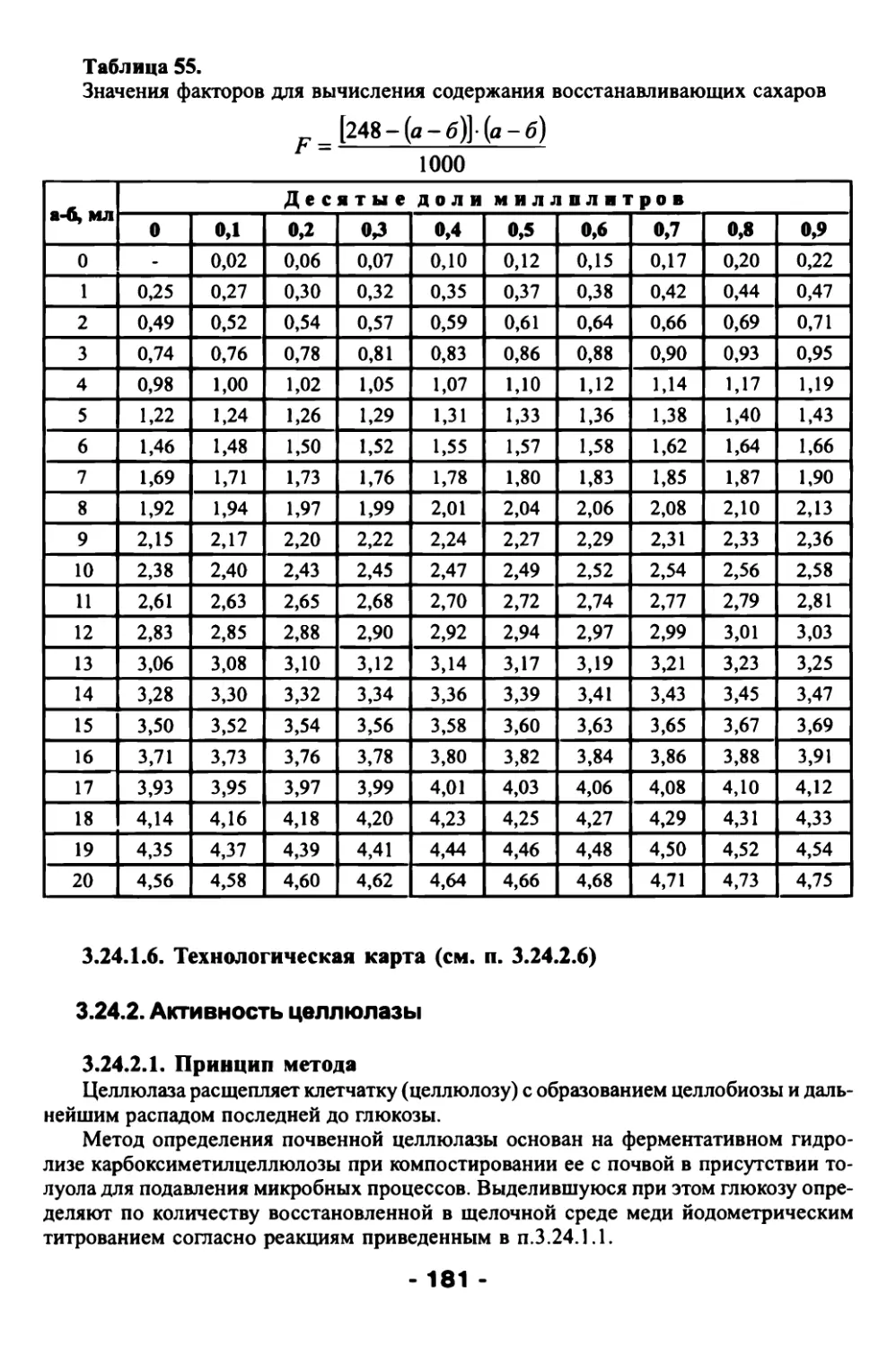
По графику находят содержание фосфора в аликвоте: вычисление проводят в %
на абсолютно сухое вещество по формуле:
гУ'Аа-О-у т
н• />1000
где V - объем исходного раствора в мл. (50мл);
а - количество мг Р в 1мл колориметрируемого раствора по графику;
в - количество мг Р в 1мл холостого раствора по графику;
р - количество мл. исходного раствора, взятого для колориметрирования (1мл);
н- навеска, г;
100 - коэффициент пересчета в проценты;
1000 - коэффициент пересчета мг в г.
При работе с постоянными навесками материала (н=0,2 или 0,4г абсолютно сухо¬
го вещества) и доведении испытуемого раствора до одинакового объема (У=50мл) сле¬
дует при построении калибровочного графика на горизонтальной оси откладывать
концентрацию фосфора сразу в % на абсолютно сухое вещество. Это значительно ус¬
коряет обработку полученных результатов.
Анализ считается проведенным с достаточной точностью, если отклонения ре¬
зультатов повторных определений от среднего не превышают указанные в таблице 6.
Пример расчета см. п.2.3.4.
Таблица 6.
Содержание фосфора, %
Допустимые отклонения от среднего при повторных
определениях (в отн.%)
в одной лаборатории
в разных лабораториях
0,10 - 0,2
10,0
16,0
0,20-0,5
8,0
12,0
0,51-3,0
6,0
10,0
Свыше 3,0
5,0
8,0
2.4.2.6. Технологи чес кая карта
-26-
2.4.3. ОПРЕДЕЛЕНИЕ КАЛИЯ И НАТРИЯ В РАСТВОРЕ ЗОЛЫ
2.43.1. Принцип метода
Количественное определение калия и натрия на пламенном фотометре основано
на зависимости между интенсивностью излучения в пламене возбуждаемых атомов
калия и натрия и концентрацией их в растворе. При определении калия используют
спектральные линии 766,5 и 769,9нм; натрия - 589нм.
2.432. Технологическое оборудование
Весы аналитические.
Пламенный фотометр.
Пипетка мерная с резиновой грушей или поршневая, либо шприц-дозатор на 1мл.
Дозатор на 9мл.
Пробирки или стаканчики на 20-50мл.
Колбы мерные на 500 или 1000мл.
2.43.3. Реактивы
1 .Образиовые растворы для определения калия.
Исходный образцовый раствор. 1,912г перекристализованного КС1 растворяют в
литровой мерной колбе и доводят дистиллированной водой до метки. Раствор содер¬
жит 1мг калия в 1мл и служит исходным для приготовления шкалы образцовых ра¬
створов.
Шкала образцовых растворов. В мерные колбы емкостью 500мл наливают не¬
много дистиллированной воды, добавляют 25мл концентрированной серной кислоты,
необходимые количества (мл.) исходного раствора КС1, указанные в таблице 7, дово¬
дят дистиллированной водой до метки и тщательно перемешивают.
2. Образиовые растворы на натрий.
Исходный образцовый раствор. 2,542г NaCl растворяют в литровой мерной кол¬
бе и доводят до метки дистиллированной водой. В 1мл исходного образцового раство¬
ра содержится 1мг Na.
Шкала образцовых растворов. В мерные колбы на 500мл наливают немного дис¬
тиллированной воды, добавляют 25мл концентрированной серной кислоты, необхо¬
димые количества (мл.) исходного образцового раствора NaCl, указанные в таблице 8,
доводят водой до метки и тщательно перемешивают.
2.43.4. Ход анализа
В пробирки или стаканчики набирают образцовые растворы шкалы для определе¬
ния калия или натрия. Нуль гальванометра устанавливают по нулевому раствору шка¬
лы, применяя при этом необходимый светофильтр (для калия или натрия). Проверяют
испытуемые растворы и при необходимости разбавляют так, чтобы при фотометриро-
вании на пламенном фотометре показания их не превышали показаний шкалы.
Чаще всего употребляется разведение 1 до 10мл дистиллированной водой, для
чего используются пипетка на 1мл и дозатор на 9мл. Реже растворы просматриваются
без разведения.
Фотометрируют образцовые растворы шкалы, а затем испытуемые растворы.
Для контроля стабильности работы фотометра после каждых 10 растворов прово¬
дят повторный отсчет 1-2 образцовых растворов, близких по содержанию калия (или
натрия) к испытуемым. При незначительном изменении показаний гальванометра про-
- 27-
Таблица 7.
ЧччШмер> колб
Показатели
0
1
2
3
4
5
б
7
8
9
10
Объем исходного
раствора КС1, мл.
0
1
2
4
5
8
10
15
20
30
40
Концентрация калия
в мг/мл. в % на
абс.сухое вещество:
0
0,002
0,004
0,008
0,010
0,016
0,020
0,030
0,040
0,060
0,080
при н=0,2г У=50мл
без разведения
0
0,05
0,1
0.2
0,25
0,4
0,5
0,75
1.0
1,5
2,0
при н=0,2г У=50мл
разведение 1/10
0
0,5
1,0
2,0
2,5
4,0
5,0
7,5
10,0
15,0
20,0
при н=0,4г У=50мл
разведение 1/10
0
0,25
0,5
1,0
1,25
2,0
2,5
3,75
5.0
7,5
10,0
Таблица 8.
^Номера колб
Показатели —^
0
1
2
3
4
5
б
7
8
9
10
Объем исходного
образцового раствора
NaCl, мл.
0
0,5
1
3
6
9
12
18
24
30
40
Концентрация натрия
в мг/мл.,в % на
абс.сухое вещество:
0
0,001
0,002
0,006
0,012
0,018
0.024
0,036
0,048
0,060
0,080
при н=0,2г У=50мл
без разведения
0
0,025
0,05
0,15
0,3
0,45
0,6
0,9
1,2
1,5
2,0
при н=0,2г У=50мл
разведение 1/10
0
0,25
0,5
1,5
3,0
4,5
6,0
9,0
12.0
15,0
20,0
при н=0,4г У=50мл
разведение 1/10
0
0,125
0,25
0,75
1,5
2,25
3,0
4,5
6,0
7,5
10.0
изводят регулировку при помощи диафрагмы. При значительных отклонениях -
заново просматривают всю шкалу и десять предыдущих испытуемых растворов.
По окончании фотометрирования строят калибровочный график, где на оси абс¬
цисс откладывают содержание в образцовых растворах шкалы калия (или натрия) в мг
на 1 мл раствора, на оси ординат - показания шкалы гальванометра.
2.43.5. Обработка результатов
Расчет проводят по формуле
K(Na),% =
СУЛ 00
«1000 ’
-28-
где С- количество калия (натрия) в 1 мл раствора, найдено данное по графику, мг;
V - объем исходного раствора, мл.;
100 - коэффициент пересчета в проценты;
н - навеска, г;
1000 - коэффициент пересчета мг в г.
Пример вычисления содержания калия. Навеска абсолютно сухого вещества
0,2г;; объем исходного раствора - 50мл; отсчет по шкале гальванометра - 25, что по
графику соответствует 0,04мг калия в 1мл; разбавление 1 до 10.
0,04.50 100 10
0,2 1000 1
Для наиболее типичных в практике лаборатории случаев в таблицах 7 и 8 пред¬
ставлены также данные для построения калибровочного графика в % калия или на¬
трия на абсолютно сухое вещество образца.
В таблице 9 приведены допустимые отклонения результатов при повторных опре¬
делениях калия.
Таблица 9.
Содержание калия,
%
Допустимые отклонения от среднего при повторных
определениях (в отн. %)
в одной лаборатории
в разных лабораториях
0,5 - 1,00
17,0
25,0
1,0- 3,0
9,0
13,0
3,0 - 5,0
7,0
10,0
Свыше 5,0
5,0
7,0
2.43.6. Технологическая карта
2.4.4. ОПРЕДЕЛЕНИЕ АЗОТА В РАСТВОРЕ ЗОЛЫ С ОТГОНКОЙ
НА ПРИБОРЕ СЕРЕНЬЕВА
2.4.4.1. Принцип метода
Определение азота в растворах после мокрого озоления проводится путем отгон¬
ки аммиака под вакуумом на приборе Сереньева. Применяемый в приборе титрант
содержит избыток борной кислоты и титрованное количество серной кислоты. Это
дает возможность одновременно поглощать и оттитровывать выделяющийся аммиак.
Реакции идут согласно уравнениям
5NH4OH + Н3В03 + H.S04 = (ЫНДВОз + (NH4)2S04 + 5Н.0
2(NH4)3B03 + 3H2S04 = 3 (NH 4 )2 S04 + 2Н3В03.
2.4.4.2. Технологическое оборудование
Весы аналитические и типа BJ1KT.
Приборы Сереньева.
Пипетка Мора на 20 мл и с делениями на 10 мл.
-29-
Цилиндры мерные на 10, 20, 25 мл.
Колбы мерные на 100, 1000 мл.
Стаканы на 800 или 1000 мл.
Стеклянные бутыли на 10 и 20 л.
2.4.43. Реактивы
1. 1% раствор лимонной кислоты - электролит для реактора. На 1л раствора бе¬
рут 10 г лимонной кислоты.
2. Титрант 0.02 н. или 0.05 н. по серной кислоте. Расчет дан на Юл титрованного
раствора.
Фиксаналы серной кислоты (2 или 5 ампул на Юл) переводят дистиллированной
водой в мерную колбу (на 1-2л), туда же вливают 200мл 0,1% спиртового раствора
индикатора метилрота (в кислотной форме), доводят содержимое колбы до метки ди¬
стиллированной водой и выливают в основной 10-литровый баллон.
Растворяют в подогретой до 60-80°С дистиллированной воде 40г борной кисло¬
ты. Остывший раствор количественно переносят в мерную колбу (1-2л), туда же вли¬
вают 100мл 0,1% спиртового раствора индикатора метиленблау, доводят до метки ди¬
стиллированной водой и переливают в основной баллон. Доливают недостающее (до
Юл) количество дистиллированной водой и хорошо перемешивают. Проверяют титр
кислоты путем отгонки на приборе Сереньева Юмл точно 0,02 н. (NH4)2S04 и титро¬
вания его приготовленным раствором.
3. 0.02 н. раствор (NH4):SQ4 готовится из химически чистой хорошо высушен¬
ной при 60-70°С соли. На 1л раствора берут на аналитических весах 1,3216г соли.
4. 30-35% раствор едкого натра с добавкой CuSO (куприт натрия). Раство¬
ряют NaOH (350г на 1л дистиллированной воды), и CuS04 • 5Н20 (1,2г в 30-50мл
дистиллированной воды при нагревании). Оба раствора охлаждают. В полученную
щелочь осторожно, по каплям, при энергичном помешивании вливают раствор
купороса, избегая образования черного осадка. Перемешивание продолжают до
получения синего раствора.
2.4.4.4. Ход анализа
Аликвоту исходного раствора (10-25мл в зависимости от содержания азота в ана¬
лизируемом материале) переносят в реакторную камеру прибора Сереньева. Туда же
заливают раствор щелочи с сернокислой медью до изменения окраски раствора. Реак¬
цию отгонки аммиака проводят под вакуумом в течение 5 мин. при слабом кипении
электролита в рубашке реактора. (Подробное описание работы прибора Сереньева см.
в п. 4.1).
Связывание выделяющегося аммиака проводится в барботере, в который перед
началом реакции заливают 1,5-2,0мл титрованного раствора, там же ведут и дальней¬
шее титрование до стойкой границы перехода окраски раствора от зеленой до лило¬
вой.
Постоянство такой окраски в течение 10-15 сек. свидетельствует о том, что анализ
окончен, в противном случае необходимо добавить 1-2 или больше капель титранта.
После окончания реакции отключают вакуум трехходовым краном, сливают из
реактора и барботера жидкость, бывшую в реакции, и готовят новую пробу.
Отгонку холостой пробы проводят методом добавок, т.е. в реактор к предыдущей
пробе доливают аликвоту холостой и продолжают отгонку, а отсчеты на бюретке про¬
водят не от нуля, а от предыдущего замера.
-30-
2.4.4.5. Обработка результатов
Расчет результатов ведут по формуле:
хго/ (a-e) V N 0.014 100 ,
TV, % =
р:н
где а - количество серной кислоты, пошедшей на титрование испытуемого раствора, мл.;
в - количество серной кислоты, пошедшей на титрование холостого раствора, мл.;
0,014 - количество азота (г), связанного 1 мл 1,0 н. раствора серной кислоты;
100 - коэффициент пересчета в проценты;
V - объем исходного раствора, мл.;
р - аликвота, взятая для отгонки, мл.;
н- навеска, г;
N - нормальность раствора серной кислоты.
Пример расчета. Для «мокрого» озоления взята абсолютно сухая навеска 0,200г.
После сжигания объем раствора доведен до 50мл. Для отгонки взято 20 мл; на титро¬
вание пошло 13,5мл 0,02 н. H2S04. На холостое определение пошло 0,3мл. Содержа¬
ние общего азота будет равно
N а, (13,5-0,3) 50 0,02 0,014 100
,/0 20 0,2
Допустимые расхождения результатов повторных определений азота приведены в
таблице 10.
Таблица 10.
Содержание азота, %
Допустимые отклонения от среднего при новторных
определениях (в отн. %)
в одной лаборатории
в разных лабораториях
0,2 - 0,5
13
20
0,51 - 1,0
8
11
1,01 - 3,0
5
6
Свыше 3,0
3
4
2.4.4.6. Технологическая карта
2.4.5. ОПРЕДЕЛЕНИЕ АЗОТА В РАСТВОРЕ ЗОЛЫ
КОЛОРИМЕТРИЧЕСКИМ МЕТОДОМ
2.4.5.1. Принцип метода
Колориметрическое определение азота основано на получении, при взаимодей¬
ствии аммония с активным хлором и салицилатом натрия, окрашенного в голубой цвет
индофенольного соединения.
В качестве источника активного хлора используется гипохлорид, в качестве ката¬
лизатора - нитропруссид натрия.
Зеленый цвет колориметрируемого раствора обусловлен, в основном, смешени¬
ем голубой индофенольной окраски с желтой окраской применяемых реактивов.
-31 -
Реакция протекает согласно уравнению
ОН
C00Na + (NH4)2S04 + 6Na0Cl + 8Na0H -Na2S04 +
гипохлорит
салицилат натрия
+ 10H20 + 4Na2C03 + 6NaCl + 2NaO ~^ ^>~ N ^> = 0.
натриевая соль индофенола
Влияние двух- и трехвалентных катионов устраняется введением трилона Б.
2.4.52. Технологическое оборудование
Фотоэлектроколориметр.
Весы типа BJIKT и аналитические.
Колбы конические или пробирки на 50мл.
Шприц-дозатор на 0,5мл или пипетка с грушей на 1мл.
Колбы мерные на 1000мл.
Дозаторы на 1;18мл.
2.4.5.3. Реактивы
1. Окрашивающий раствор
Исходный окрашивающий раствор. 57г салицилата натрия, 16,7г калия-натрия
виннокислого и 27г гидроокиси натрия растворяют в 700мл дистиллированной воды,
кипятят раствор в течение 20 мин. для удаления загрязнения аммонием. После охлаж¬
дения в полученный раствор добавляют 0,4г нитропруссида натрия и доводят объем
до 1л дистиллированной водой без аммиака. Хранится реактив в темной склянке в
холодильнике в течение месяца.
Рабочий окрашивающий раствор. Исходный окрашивающий раствор разбавляют
водой из расчета 1:8 и растворяют в нем 4мг NaOH и 2г трилона Б на 1л конечного
раствора. NaOH добавляется для нейтрализации серной кислоты испытуемого раствора.
Реактив готовят в день проведения анализа.
2. Восстановитель
Исходный раствор гипохлорита натрия. В стакане перемешивают 150г хлорной
извести (технической) с 250мл дистиллированной воды. В другом стакане 105г без¬
водного углекислого натрия растворяют в 250мл дистиллированной воды. Оба раствора
сливают при постоянном перемешивании. Масса сначала густеет, затем разжижается.
Полученную суспензию оставляют на 1-2 суток для отстаивания, затем прозрачную
жидкость сливают и фильтруют.
В полученном реактиве содержится 6-10% активного хлора. Точную его концент¬
рацию определяют следующим образом: 1мл прозрачного фильтрата разбавляют 50
мл воды, прибавляют 2г йодистого калия и 10мл 1 н. раствора НС1. Образовавшийся
иод оттитровывают 0,1 н. раствором тиосульфата натрия, приготовленного из фикса-
нала, до исчезновения вишневой окраски.
Расчет концентрации активного хлора проводится по формуле:
С1, %= а-0.00355 -100,
-32-
где а - количество мл. 0,1 н. тиосульфата натрия, пошедшего на титрование;
0,00355 - 1 мл 0,1 н. раствора тиосульфата натрия соответствует 0,00355г хлора;
100 - коэффициент перевода в проценты.
Например, на титрование 1мл исходного раствора гипохлорита натрия пошло
19,8мл 0,1 н. раствора Na2S203, следовательно, концентрация активного хлора равна
19,8- 0,00355- 100 = 7,029%.
Раствор хранится в холодильнике в склянке из темного стекла в течение года.
Рабочий раствор гипохлорита натрия. Исходный раствор гипохлорита натрия
разбавляют дистиллированной водой до концентрации 0,125% и используют для ана¬
лиза в течение дня.
Пример расчета при разведении. Исходный раствор гипохлорита имеет концент¬
рацию хлора 7,03%. Для получения 100мл 0,125% раствора нужно взять исходного - х:
0,125 100
х = — = 1,78 мл
7,03
3. Образиовые растворы для определения азота.
Исходный образцовый раствор. 1,9 Юг хлористого аммония растворяют в дис¬
тиллированной воде и доводят объем до 1л. Раствор содержит 0,5мг азота в 1 мл.
Рабочая шкала образцовых растворов. В мерные колбы на 50мл наливают объе¬
мы исходного образцового раствора, указанные в таблице 11, приливают по 5мл кон¬
центрированной H2S04 и перемешивают. После охлаждения колбы объем раствора
доводят дистиллированной водой до метки и снова перемешивают.
Раствором сравнения служит нулевой раствор шкалы.
Таблица 11.
Номера колб
Показатели —
1
2
3
4
5
б
7
8
Объем исходного образцового раствора,мл.
0
2
4
6
8
10
12
14
Концентрация N, мг/мл.
0
0,02
0,04
0,06
0,08
0,10
0,12
0,14
4. 1 н. НС1 82мл концентрированной НС1 (плотность 1,19г/см3) доводят до 1л
дистиллированной водой.
5. 01 н. раствор тиосульфата натрия (гипосульфита) готовят из фиксанала.
6. Безаммиачная вода. Проводят испытание на содержание аммиака в дистилли¬
рованной воде. В пробирку наливают около 5мл этой воды и прибавляют каплю реак¬
тива Несслера. При появлении светло-желтой окраски вода непригодна для анализа и
ее следует очистить от следов аммиака. Для этого в колбу наливают 3-5л воды, подще¬
лачивают ее 0,5-1г щелочи и кипятят для удаления аммиака до тех пор, пока испарится
до 1/4 ее первоначального объема. Кипячение необходимо проводить в свободном
от аммиака помещении. По охлаждении воду сливают в бутыль и закрывают проб¬
кой. Перед употреблением снова делают пробу с реактивом Несслера. Дезаммини-
рование воды можно проводить как указано в п.3.8.3.2.
2.4.S.4. Ход анализа
Для определения азота в пробирки или конические колбы вместимостью 50мл
-33-
отбирают шприцем-дозатором или градуированной пипеткой по 0,5мл анализиру¬
емых растворов и растворов шкалы. Приливают к ним дозатором 18,5мл рабочего
окрашивающего раствора и 1мл рабочего раствора гипохлорита натрия. Хорошо
перемешивают после добавления каждого реактива и оставляют на 1 час для разви¬
тия окраски. Одновременно проводят холостое определение.
Колориметрируют растворы в кювете с толщиной просвечиваемого слоя 10мм при
длине волны 655 нм (красный светофильтр) не ранее чем через час и не позднее чем
через 2,5 часа после добавления раствора гипохлорита натрия.
2.4.5.5. Обработка результатов
Содержание азота в анализируемом материале определяют по калибровочному
графику, на горизонтальной оси которого откладывают концентрацию азота в мг/мл.,
а на вертикальной - оптическую плотность.
Расчет содержания азота ведется по формуле:
(.-.)-к.1М
/7 «1000
где а - количество азота в мг/мл. анализируемого раствора, найденное по графику,
в - количество азота в мг/мл. холостого раствора, по графику;
V - общий объем вытяжки, мл.;
р - аликвота, взятая для окрашивания, мл.;
н- навеска, г;
100 - коэффициент для пересчета в проценты;
1000 - коэффициент для перевода мг в г.
При очень высокой концентрации азота его определение можно проводить из
растворов, подготовленных для определения калия, где обычно проводится разбавле¬
ние в 10 раз, или брать для анализа в пробирки градуированной пипеткой 0,1 -0,4мл
испытуемого раствора, добавляя до 0,5мл дистиллированной водой, а затем доливать
необходимые реактивы.
2.4.5.6. Технологическая карта
2.4.6. ОПРЕДЕЛЕНИЕ КАЛЬЦИЯ И МАГНИЯ В РАСТВОРЕ ЗОЛЫ
2.4.6.1. Принцип метода
Трилонометрическое определение кальция и магния основано на образовании ус¬
тойчивых в щелочной среде комплексных соединений этих элементов с трилоном Б.
Реакция, на примере кальция, предоставлена следующим уравнением:
^СН,COONa СН COONa
N < N«C
! ^снсоон | ^СНХОО
СН, 2 СН.
' +CaSO. Са + H.SO.
СН9 СН_
! ^СН,СООН ' ^СН,СОО
N < N <"
Х СН COONa ^ СН COONa
-34-
Как видно из приведенного уравнения, выделяющаяся кислота может смещать
реакцию раствора ниже интервала, в котором кальций (или магний) дает устойчи¬
вые комплексы с трилоном, что ведет к искажению результатов. Поэтому в анализе
для нейтрализации выделившегося водородного иона используют буферные ра¬
створы или избыток щелочи.
Определению магния в почвах, водах и растениях мешают ионы меди, марганца,
железа и алюминия, особенно при анализе кислотных вытяжек.
Для связывания меди в устойчивый комплекс используют диэтилдитиокарбамат
натрия. Чтобы удержать марганец в виде двухвалентного иона, вводят в раствор вос¬
становитель - солянокислый гидроксиламин.
Чтобы снизить концентрацию мешающих ионов, аликвотную часть раствора пе¬
ред титрованием сильно разбавляют дистиллированной водой (в 2-10 раз), что не ме¬
шает титрованию.
Конечную точку титрования кальция устанавливают по изменению окраски ме¬
талл индикатора флуорексона при pH 13. Магний определяют по разности результатов
титрования суммы кальция и магния по хромогену черному и титрования кальция по
флуорексону.
2.4.6i. Технологическое оборудование
Весы типа BJ1KT.
Колбы конические на 250мл.
Пипетка или шприц-дозатор на 10 и 20мл.
Бюретка на 25 и 50мл.
Капельница.
Колбы мерные на 100мл; 1000мл.
2.4.6.3. Реактивы
1. 0.02 н. раствор трилона Б. 3,72г соли растворяют в дистиллированной воде,
доводят до метки в мерной колбе на 1л и фильтруют.
Для установления нормальности раствора трилона Б берут 10мл 0,01н. раствора
сернокислого магния, приготовленного из фиксанала, добавляют 100мл дистиллиро¬
ванной воды, 5мл аммиачного буферного раствора, 5-7 капель индикатора хромогена
черного и титруют трилоном Б при интенсивном перемешивании раствора до перехо¬
да фиолетовой окраски в голубую.
Если нет фиксанала, берут 1,230г MgS04 • 7Н20 или 1,020г MgCl2 • 6Н20 и раство¬
ряют в дистиллированной воде в мерной колбе на 1л. Полученный 0,01н. раствор
соли магния используют для определения нормальности раствора трилона Б так,
как это указано выше.
Расчет нормальности трилона Б проводят по формуле:
нормальность трилона Б;
нормальность раствора соли магния;
количество трилона Б, пошедшего на титрование, мл.;
количество раствора соли магния, взятое для титрования, мл.
2. Флуорексон. Юг хлористого калия и 0,1 г флуорексона тщательно растирают в
ступке. Хранят в темном сухом месте в склянке с притертой пробкой.
где N
Nx
а -
X
а
-35-
3. Малахитовый зеленый. 0,2г индикатора растворяют в 100мл дистиллирован¬
ной воды.
4. Хромоген черный. 5 г индикатора растирают с 95г NaCl. Хранят в склянке из
темного стекла с притертой пробкой.
5. 20% раствор КОН. 200г едкого кали растворяют в 1л дистиллированной воды
(работу желательно вести в вытяжном шкафу).
6. Буферный раствор. 20г NH4C1 растворяют в воде, добавляют 100мл 25% раствора
NH4OH, хорошо перемешивают и дистиллированной водой доводят объем до 1л.
7. Лимоннокислый натрий.
8. Гидроксиламин солянокислый 5%.
9. Диэтилдитиокарбамат натрия.
2.4.6.4. Ход анализа
Определение кальиия. Аликвотную часть испытуемого раствора (10-20мл) разбав¬
ляют дистиллированной водой до 75-100мл в конической колбе, вносят 1-2 капли 0,2%
водного раствора малахитового зеленого (раствор окрашивается в голубовато-зеле¬
ный цвет) и прибавляют 20% раствор КОН до обесцвечивания, а затем избыток - 10мл
этого раствора. Вносят на кончике ножа лимоннокислый натрий, гидроксиламин и
индикатор флуорексон. После прибавления каждого раствор перемешивают. Раствор
становится желтоватым, с интенсивно зеленой флуоресценцией. Титруют 0,02н. ра¬
створом трилона Б до оранжево-розовой окраски при резком уменьшении зеленой
флуоресценции. Внесение в раствор малахитового зеленого и особенно щелочи про¬
водится непосредственно перед титрованием, а не заранее. В качестве «свидетеля»
используют 100мл дистиллированной воды, в которую добавляют в тех же количе¬
ствах вышеуказанные реактивы.
Определение магния. Такую же аликвотную часть раствора, как и на определение
кальция (10-20мл), помещают в коническую колбу емкостью 250мл. В раствор броса¬
ют кусочек красной бумажки Конго или индикаторной универсальной бумаги и по
каплям добавляют 10% КОн, чтобы нейтрализовать кислоту до pH 5,2. Вносят 2-3
кристаллика диэтилдитиокарбамата натрия, 1-2мл восстановителя солянокислого гид¬
роксил амина (5%). После этого добавляют 50мл дистиллированной воды, 20мл хло-
ридно-аммиачного буфера, чтобы обеспечить щелочную среду, соответствующую pH
10, при которой происходит извлечение магния трилоном Б из комплексного соедине¬
ния магния с металлиндикатором. В последнюю очередь добавляют на кончике
ножа 10-15г металлоиндикатора хромогена черного и титруют сумму (Са + Mg) 0,02н.
раствором трилона Б до перехода окраски из вишневой в сине-голубую.
2.4.6.5. Обработка результатов
Содержание кальция и магния рассчитывают по формулам:
Са 0/ (а - в) • iV • К • 100 • 0,02
рн
Mg 0/, (с-а)-АТ-К-100-0,012^
рн
ще а - количество трилона Б, пошедшее на титрование Са, мл.;
в - количество трилона Б, пошедшее на титрование холостой пробы, мл.;
-36-
с - количество трилона Б, пошедшее на титрование суммы (Са + Mg), мл.;
N - нормальность трилона Б;
V- исходный объем анализируемого раствора, мл.;
100 - коэффициент пересчета в проценты;
р - количество исходного анализируемого раствора, взятое для титрования, мл.;
н - навеска, г;
0,02 - г-экв Са;
0,012- г-экв Mg.
2.4.6.6. Технологические карты
Определение кальция.
2.5. РАЗЛИЧНЫЕ ФОРМЫ АЗОТА В РАСТЕНИЯХ
2.5.1. ОПРЕДЕЛЕНИЕ БЕЛКОВОГО И НЕБЕЛКОВОГО АЗОТА
ПО МЕТОДУ БАРНШТЕЙНА
2.5.1.1. Принцип метода
Метод основан на количественном определении азота белков, осаждаемых ще¬
лочным раствором сернокислой меди для отделения от других соединений, и небел¬
кового азота, оставшегося в растворе, путем мокрого озоления с последующей дис¬
тилляцией на приборе Сереньева.
2.5.1.2. Технологическое оборудование
Весы типа BJIKT.
-37-
Электро- или газовая печь.
Водяная баня или термостат.
Ступки фарфоровые.
Колбы конические или стаканы на 250мл.
Дозаторы на 20, 25 и 50мл.
Воронки стеклянные d = 7см.
Палочки стеклянные.
Колбы Кьельдаля на 100- 150мл.
Колбы мерные на 100 и 1000мл.
Пипетка или шприц-дозатор на 25 и 50мл.
2.5.1.3. Реактивы
1. 6% раствор сернокислой меди. 60г CuS04 • 5Н20 растворяют и доводят объем
до 1 л дистиллированной водой.
2. 1.25% раствор едкого натра. 12,5г NaOH растворяют и доводят объем до 1л
дистиллированной водой.
3. 1% раствдр хлористого бария. Юг BaCI^ растворяют и доводят объем до 1л
дистиллированной водой.
4. Серная кислота кониентрированная (плотность 1,84 г/см3).
5. Смешанный катализатор. Смешивают 400г сернокислого калия, 100г серно¬
кислой меди, 2г селена, ЗОг марганцовокислого калия и хорошо растирают в тонкий
порошок (работу проводить в вытяжном шкафу) в фарфоровой ступке.
6. Фенолсерная кислота. Растворяют 40г чистого фенола (C6f]OH) в концент¬
рированной серной кислоте и доводят той же кислотой до 1л.
1. 1% раствор лимонной кислоты.
8. Титрант 0.02н. по серной кислоте (см.п.2.4.4.3.).
9. Раствор куприта натрия (см. п. 2.4.4.3.).
2.5.1.4. Ход анализа
Навеску сырого вещества (5г) растирают с песком в ступке и смывают 50мл дис¬
тиллированной водой в коническую колбу или стакан на 250мл. При анализе воздуш¬
но-сухого материала (зерно, семена) тонко размолотую навеску (около 0,5г) помеща¬
ют сразу в колбу или стакан на 250мл, заливают 50мл горячей воды. Суспензию дово¬
дят до кипения (если вещество богато крахмалом, во избежание клейстеризации огра¬
ничиваются 10-минутным нагреванием на водяной бане при температуре 40-50°С). Не
охлаждая содержимого колб, прибавляют 25мл 6% раствора сернокислой меди и при
постоянном помешивании стеклянной палочкой небольшими порциями приливают
25 мл 1,25% раствора едкого натра. Образовавшаяся основная соль сернокислой меди
- CuS04 ■ Cu(OH)2 и является осадителем белков. Осадку дают отстояться не менее 2
часов (лучше оставить на ночь).
После отстаивания надосадочную жидкость сливают по стеклянной палочке на
предварительно промытый кипятком фильтр, осадок промывают в колбе несколько
раз (декантацией) горячей водой, затем количественно переносят на фильтр и продол¬
жают промывать теплой водой до отсутствия следов ионов S04 (проба с хлористым
барием). При правильном промывании объем фильтрата обычно не превышает 400-
500мл. Промытый фильтр с осадком подсушивают в термостате при 50-60°С до тех
пор, пока он не будет легко извлекаться из воронки, после чего фильтр с осадком
аккуратно сворачивают и переносят в колбу Кьельдаля емкостью 150мл.
-38-
Затем в колбы наливают 20мл концентрированной H2S04, добавляют 0,1-0,2г
смешанного катализатора и, неплотно закрыв горлышко колбы стеклянной пробкой
или маленькой воронкой, сжигают образец. Сжигание ведут при слабом кипении ра¬
створа до полного обесцвечивания содержимого колбы. После окончания озоления,
охлаждения и доведения до определенного объема в аликвотной части или во всей
пробе проводят определение азота на приборе Сереньева. (Описание работы прибора
Сереньева см. в разделе 4.1). Параллельно ведут холостое определение, сжигая про¬
мытый и высушенный фильтр.
Для определения общего содержания небелкового азота используют фильтрат,
полученный при отмывке белка. Его количество доводят до определенного объема
(500мл), хорошо перемешивают и, в зависимости от предполагаемого количества
небелкового азота, отбирают для анализа] или S часть общего объема в колбы Кьель-
даля. К пробе приливают 1 мл концентрированной серной кислоты (чтобы не улету¬
чился аммиак) и кипятят содержимое колбы до тех пор, пока объем жидкости в ней
не уменьшится до 10- 15мл., затем добавляют 3-5мл концентрированной серной
кислоты, смешанный катализатор и проводят озоление вытяжки до полного
обесцвечивания. Аммиак, образовавшийся при озолении небелковых азотосодержащих
веществ, отгоняют на приборе Сереньева.
Если в исследуемом образце можно ожидать заметное содержание нитратного
азота, то его озоление ведут с фенолсерной кислотой, т.к. при озолении с одной серной
кислотой без восстановления нитратов азот нитратов не будет учтен.
2.5.1.5. Обработка результатов
Количество белкового и небелкового азота вычисляют так же, как при определе¬
нии общего азота по Кьельдалю (см.п.2.4.4.5).
Содержание белка в % рассчитывают по формуле:
Белок, -/о = («-«>^0>014100-6,25
рн
где а - количество серной кислоты, пошедшее на титрование испытуемого
раствора, мл.;
в - количество серной кислоты, пошедшее на титрование холостого
раствора, мл.;
N - нормальность раствора серной кислоты;
0,014 - количество азота (г), связанного с 1,0 мл 1,0 н. раствора серной кислоты;
100 - коэффициент пересчета в проценты;
н - навеска, г;
6,25 - коэффициент пересчета на белок;
V- объем исходного раствора, мл.;
р - аликвота, взятая для отгонки, мл.
Коэффициент пересчета на белок 6,25 не одинаков для разных культур, для семян
некоторых растений в соответствии с содержанием азота в их белках пользуются дру¬
гими коэффициентами, а именно: для пшеницы, ржи, овса он равен 5,7; для люпина,
подсолнечника, льна, конопли - 5,5; гречихи, кукурузы, фасоли - 6,00.
Общее содержание небелкового азота можно определить и как разность между
содержанием общего и белкового азота в двух параллельно взятых и проанализиро¬
ванных одновременно пробах исследуемого вещества.
Определение белкового и небелкового азота при оценке качества растительных
продуктов играет важную роль.
Содержание белка в семенах и зерне различных сельскохозяйственных культур
приведено в таблице 12.
-39-
Таблица 12.
Культура
Содержание белка в % на сухое вещество
Пшеница
12-24
Ячмень
10-18
Овес
9-19
Люпин
27-34
Горох
22-38
Лен
18-34
Подсолнечник
13-19
2.5.1.6. Технологическая карта
2.5.2.0ПРЕДЕЛЕНИЕ СОДЕРЖАНИЯ АЗОТА В РАСТЕНИЯХ
ФОТОМЕТРИЧЕСКИМ МЕТОДОМ С РЕАКТИВОМ НЕССЛЕРА
2.5.2.1.Принцип метода
После мокрого озоления растительного материала все соединения азота преобра¬
зуются в аммиачную форму При взаимодействии аммонийного азота с реактивом Нес¬
слера образуется йодид меркураммония, окрашенный в желтый цвет разных оттенков в
-40-
соответствии с концентрацией аммонийного азота.
(NH)2S04 + 4K2[HqJ4) + 8КОН = 2NH2JHqO + K2S04 + 4KJ + 6^0
2.5.2.2.Tехнологическое оборудование
Фогаэтаарскоторимяр
Пипетки на 5 - 10мл.
Мерные колбы на 100мл; 500мл.
Бюретка на 25 - 50мл.
2.5.2.3.Реактивы
1.Основной образиовый раствор сульфата аммония.235Кт (NH4)2S04 растворя¬
ют в дистиллированной воде без аммиака и в мерной колбе доводят объем до 500мл. В
1мл раствора содержится 1мг аммонийного азота.
2.Образиовый рабочий раствор сульфата аммония. 25мл основного раствора
(NH4)2S04 переносят пипеткой в мерную колбу на 500мл и доводят до метки дистил¬
лированной водой не содержащей аммиак. В 1мл такого раствора содержится 0,05мг
азота. 50мл этого раствора переносят пипеткой в мерную колбу на 500мл и дистилли¬
рованной водой (без аммиака) доводят объем раствора до метки. В 1мл такого образ¬
цового раствора содержится 0,005мг азота.
3. Реактив Несслера.
4. 50%-ный раствор сегнетовой соли.
5. 10%-ный раствор NaOH.
6. Фенолфталеин.
2.5.2.4.Ход анализа
1-5-10мл раствора после мокрого озоления растительного материала перено¬
сят пипеткой в мерную колбу на 100мл. Содержание аммиачного азота в колбе
емкостью 100мл не должен превышать 0,5 - 0,6мг.
Избыток кислоты в растворе нейтрализуют 10%-ным раствором NaOH, прове¬
ряя окончание нейтрализации лакмусовой бумагой. Бумага должна приобрести
слабый синий оттенок. В том случае, если на нейтрализацию отдельно взятой про¬
бы вытяжки объемом 5мл затрачивается не более 2мл 10%-ного раствора NaOH,
нейтрализацию раствора можно не проводить. После нейтрализации раствор в кол¬
бе доводят дистиллированной водой, не содержащей аммиак, до 80-90мл и тщатель¬
но перемешивают. Доливают в колбу из бюретки 2мл 50%-ного раствора сегнето¬
вой соли и снова раствор в колбе перемешивают. Затем приливают 2мл реактива
Несслера, доводят водой до метки, перемешивают и через 10 мин. фотометрируют.
Для определения концентрации аммиака в растворе строится калибровочный
график с учетом оптической плотности окрашенного раствора. Для построения
калибровочного графика в пронумерованные колбы емкостью 100мл последова¬
тельно из бюретки приливают 0; 2; 4; 6; 8; 10; 15; 20 и 25мл образцового раствора
(NH4)2S04. Объем жидкости во всех колбах доводят дистиллированной водой (не
содержащей аммиак) до 90мл, раствор перемешивают, приливают 2мл 50%-ного
раствора сегнетовой соли и 2мл реактива Несслера, естественно перемешивая со¬
держимое колбы после добавления каждого реактива, содержимое колб до метки
доводят дистиллированной водой и тщательно перемешивают. Раствор первой кол¬
бы, содержащей все реактивы, кроме аммонийного азота, принимается за раствор
сравнения.
Фотоэлектроколориметр калибруется (настраивается) по раствору сравнения.
Содержание общего азота (N) в растительном материале, в % на сухое вещество,
рассчитывается по формуле:
-41 -
а 100 100
” w (l00-y)’
где a - количество азота, определенное по калибровочному графику, мг;
т - расчетная масса растительной навески, мг;
100 - коэффициент для пересчета в %;
у - содержание гигроскопической влаги в растительном анализируемом
материале, %.
2.5.2.5.Технологическая карта
2.5.3 ОПРЕДЕЛЕНИЕ НИТРАТОВ В РАСТЕНИЯХ
2.53.1. Принцип метода
Нитраты извлекают из растительного материала 1% уксусной кислотой. Метод
основан на восстановлении нитратов до нитритов гидразином с последующим оп¬
ределением их колориметрическим б-нафтиламинным методом (см.п.3.8.2.).
В пробе определяется сумма нитратного и нитритного азота, однако содержание
последнего очень низкое и находится в пределах ошибки анализа.
2.53.2. Технологическое оборудование
Весы типа ВЛКТ.
Фотоэлектроколориметр.
Ступки фарфоровые d = 10-12см.
Колбы мерные на 50 или 100, 500, 1000мл.
Колбы конические на 100- 150мл.
Воронки d = 5-7см.
Стеклянные палочки.
Стаканы на 100-150мл.
Пипетка с грушей или шприц-дозатор на 5мл.
Дозаторы на 5, 10, 25мл.
2.5.33. Реактивы
1. 0.5% раствор пирофосфата Na. 5г натрия пирофосфорнокислого и 6г NaOH
растворяют в дистиллированной воде и доводят объем до 1л. Хранится длительное время.
2. Раствор катализатора. 2,5г CuS04 • 5Н20 растворяют в дистиллированной воде и
-42-
доводят объем до 1л. Раствор сохраняется длительное время.
3. Восстановитель.
Запасной раствор восстановителя. 13,8г гидразина сернокислого растворяют в
дистиллированной воде и доводят объем до 1л в мерной колбе. Раствор хранится 6 месяцев.
Рабочий восстанавливающий раствор. 6мл раствора катализатора и 200мл раствора
восстановителя переносят в мерную колбу емкостью 1л, доводят до метки дистиллирован¬
ной водой и перемешивают. Раствор хранится в течение 1 недели в склянке из темного
стекла.
4. Окрашивающий раствор.
Запасной окрашивающий раствор. К 500мл дистиллированной воды приливают 200мл
ортофосфорной кислоты и растворяют 2г б-нафтиламина (1 -нафтиламина) или N - (1-
нафтил) - этилендиамин гидрохлорида и доводят в мерной колбе до объема 2л
дистиллированной водой. Раствор хранится в холодильнике в склянке из темного стекла
не более 1 месяца.
Рабочий окрашивающий раствор. В 200мл запасного окрашивающего раствора
растворяют 1г сульфаниламида (стрептоцида), прибавляют 0,2г трилона Б, предваритель¬
но растворенного в дистиллированной воде и доводят объем до 1л.
5. Сульфаниламид. В качестве реактива можно использовать аптечный препарат
стрептоцид белый, нерастворимый (порошкообразный, без наполнителя).
6. Образиовые растворы для определения нитратов.
Исходный образцовый раствор. 0,7220г х.ч. азотнокислого калия, высушенного при
100- 105°С до постоянного веса, взвешивают на аналитических весах, растворяют в
дистиллированной воде, доводят в мерной колбе на 1л до метки и перемешивают.
Полученный раствор содержит 0,1мг нитратного азота (или 0,443мг нитратов) в 1мл.
Рабочая шкала образцовых растворов. В мерные колбы емкостью 500мл наливают
немного дистиллированной воды и добавляют 2,5мл ледяной уксусной кислоты, вливают
исходный образцовый раствор в количествах, указанных в таблице 13, доводят до метки
дистиллированной водой и хорошо перемешивают.
Из полученных образцовых растворов берут аликвоты, равные объему испытуемых
растворов, окрашивают их аналогично испытуемым и затем колориметрируют.
Калибровочный график строят, откладывая на горизонтальной оси концентрацию
нитратного азота в мг/мл., на вертикальной - оптическую плотность растворов.
Таблица 13.
^ ^^Номера колб
Показате л и
1
2
3
4
5
б
7
8
9
объем исходного
образцового раствора, мл.
0
2
5,0
10,0
15,0
20,0
30,0
40,0
60.0
содержание N-N03 в
мг/мл.,
0
0,0004
0,001
0,002
0,003
0,004
0,006
0,008
0,012
в % на сырое вещество
0
0,001
0,0025
0,005
0,0075
0,010
0,015
0,020
0,030
в мг/кг сырого вещества
0
10
25
50
75
100
150
200
300
2.53.4. Ход анализа
Из средней пробы отвешивают 4г свежего растительного материала или до 1г воз¬
душно-сухого (в навеске должно быть 10-60мкг нитратного азота). Одновременно берут
-43-
навеску для определения абсолютно сухого вещества. Навеску для анализа переносят в
фарфоровую ступку, растирают с 1 % уксусной кислотой, затем смывают тем же раствором
растертую навеску в мерную колбу емкостью 100мл и доводят до метки. Жидкость
фильтруют через сухой беззольный фильтр (белая лента) в стакан.
В конические колбы отбирают пипеткой 5 мл фильтрата, добавляют дозатором
10мл 0,5% раствора пирофосфата натрия, 10мл рабочего восстанавливающего раствора
и хорошо перемешивают после добавления каждого реактива.
Через 10 мин. приливают дозатором 25мл рабочего окрашивающего раствора,
снова перемешивают и оставляют на 15 мин. для полного развития окраски.
Обычно желтая окраска вытяжки исчезает при добавлении последнего раствора.
В каждой партии ставится холостой опыт: 5мл любой из определяемых вытяжек
+10мл пирофосфата натрия +10мл рабочего восстанавливающего раствора +25мл ор-
тофосфорной кислоты, разбавленной в 50 раз, что соответствует рабочему окрашива¬
ющему раствору без сульфаниламида и а-нафтиламина.
Колориметрируют растворы в кювете с толщиной просвечиваемого слоя 1см при
желто-зеленом светофильтре (545нм), если используется N-( 1 -нафтил)-этилендиа-
мин дигидрохлорид, и при зеленом светофильтре (520 нм), если используется а-на-
фтиламин, не ранее чем через 15 мин. и не позднее чем через 1,5 часа после прибав¬
ления окрашивающего раствора.
Сравнение всех растворов шкалы, испытуемых и холостого определения ведут с
нулевым раствором шкалы.
2.5.3.5. Обработка результатов
Содержание нитратного азота рассчитывают в процентах или мг/кг растительной
массы (сырой или абсолютно сухой) по формулам:
С V 100
N-NO,, % = , (1)
«1000
CV 100
N-NO,, мг/кг = , (2)
н
растительной массы
где С - количество нитратного азота в 1 мл раствора;
найденного по графику, мг;
V - объем исходного раствора, мл.;
100 - коэффициент пересчета в проценты (1);
н - навеска, г;
1000 - коэффициент перевода мг в г (1);
1000 - коэффициент пересчета на 1кг растений (2).
Пример вычисления. Навеска сырых листьев 4г (0,66г абсолютно сухого ве¬
щества); объем исходного раствора - 100мл; отсчет по шкале колориметра - 9,0, что
по графику соответствует 0,003мг/мл.
0,003 100 100 |||М„С0/
N-NO,, % = А/г. 1 ааа ” U,Ul)75 /о на сырое в-во (0,0454% на абс.сухое в-во)
4(U,oo)*lUUU
0,003 100 1000 _^с
N-NO., мг/кг = -75 мг/кг сырого в-ва (454 мг/кг абс.сухого в-ва)
* 4(0,оо)
растительной массы
-44-
2.53.6. Технологическая карта
Взятие навесок
весы типа ВЛКТ, шпатель, ступки
фарфоровые
Приведение к заданному объему
колбы мерные на 100мл, воронки
(1=5см, палочки стеклянные
колбы на 100мл, пипетка или
шприц-дозатор на 5мл
Окрашивание нш>б
дозатор на 25мл
Измельчение растительного мате¬
риала
ступка с пестиком
Фильтрование
стаканы на 100-150мл, воронки d=
5см, фильтры
РрсстанрвлсниЕ цитратов
дозаторы на 10мл (2шт.)
Колоцуметрдрование
ФЭК (зеленый светофильтр)
2.5.4. ОПРЕДЕЛЕНИЕ СОДЕРЖАНИЯ НЕБЕЛКОВОГО АЗОТА
В САХАРНОЙ СВЕКЛЕ ФОТОМЕТРИЧНЫМ МЕТОДОМ
С ТРИХЛОРУКСУСНОЙ КИСЛОТОЙ.
(Метод Е.А.Тонкаля, Е.П.Наумовой, В.Г.Васильева)
2.5.4.1. Принцип метода
При определении небелковых форм азота в растениях в качестве осадителя ис¬
пользуется трихлоруксусная кислота.
2.5.4.2. Технологическое оборудование
Мельница для измельчения растительных образцов.
Весы типа ВЛКТ, торсионные или аналитические.
Фотоэлектроколориметр.
2.5.43. Реактивы
1. Серная кислота-HSOf кониентрированная, уд.вес 1.84г/смК
2. Раствор трихлоруксусной кислоты (25%). 250г трихлоруксусной кислоты
(СС13СООН) растворяют в 1 л мерной колбе в дистиллированной воде и доводят
объем раствора до метки.
4. Раствор едкого натрия (5%). 50г NaOH растворяют в дистиллированной воде
(без углекислоты) и объем раствора доводят до 1 литра.
5. Реактив Несслера.
6. Стандартный раствор сернокислого аммония (NH}g04. 0,2358г (NH^SC^
х.ч. растворяют в мерной колбе на 0,5л в воде без аммиака. 100мл этого раствора
доводят в мерной колбе на 0,5л водой до метки. В 1мл раствора содержится 0,02мг N.
7. Раствор сегнетовой соли (40 - 50%). 40 - 50г сегнетовой соли (KNaC4H406 •
4Н20) растворяют в дистиллированной воде и доводят объем раствора до 100мл.
8. Индикатор 0,2% раствор тимолблау или фенолфталеина.
Из существующих осадителей трихлоруксусная кислота (ТХУК) является наилуч¬
шим осадителем белка. Осаждение белков гидратом окиси меди, применяемое при
-45-
анализе растений сахарной свеклы по методу Барнштейна дает завышенную величину
белкового азота за счет осаждения полипептидов и части аминокислот.
Осаждение белков ТХУК с дальнейшим применением ускоренного сжигания с
перекисью водорода и определением азота колориметрически дает возможность сокра¬
тить продолжительность анализа в два раза, уменьшить расход реактивов и повысить
точность результатов.
2Л4.4. Ход анализа
Навеска с содержанием 30-50мг азота (свежих листьев 3-5г; корнеплодов - 5-
10г; фиксированных сухих образцов - листьев 0,25г; корнеплодов - 0,5г) растирается
в ступке и переносится в колбу Эрленмейера емкостью 100-150мл и заливается 40мл
дистиллированной воды. При определении азота в сухой массе растений измельчен¬
ная навеска заливается 40мл дистиллированной воды и настаивается в течение 30
мин. при периодическом взбалтывании. Колбы закрываются стеклянными пробками
(можно использовать пробки от колб Кьельдаля ).
Содержимое колб медленно нагревают до кипения и к теплому раствору добав¬
ляют при взбалтывании 10мл 25% раствора ТХУК. Эту кислоту берут с таким расче¬
том, чтобы в растворе, где осаждается белок, ее концентрация составляла 5%. Раствор
оставляют в спокойном состоянии до следующего дня.
После отстаивания жидкость фильтруют и 20-30мл фильтрата переносят в колбу
Кьельдаля, добавляют 5мл концентрированной H2S04 Содержимое колб хорошо взбал¬
тывают, подогревают до полного испарения влаги, о чем свидетельствует появление
в них белого дыма. Затем колбы снимают с огня и слегка охлаждают, по каплям вносят
перекись водорода (4-6 капель) до посветления жидкости и вновь нагревают на огне.
Последующее добавление перекиси водорода (по 2-3 капли) проводят через каждые
15-20 минут, как при добавлении первой порции перекиси водорода, до полного
сжигания.
После охлаждения содержимое колб полностью переносят в мерную колбу емко¬
стью 200-250мл, охлаждают и доводят дистиллированной водой до метки, хорошо
взбалтывают. Для колориметрирования берут (в зависимости от содержания в образце
азота) 5-10мл раствора и помещают в мерную колбу емкостью 100мл. Параллельно
столько же раствора помещают в коническую колбу для проведения предварительной
нейтрализации, добавляют 1-2 капли тимолблау и титруют 3-6% раствором NaOH
до перехода розовой окраски в желтую. При отсутствии тимолблау можно
пользоваться фенолфталеином. Однако в этом случае необходимо от объема щелочи,
израсходованной при титровании, исключить 0,1-0,2мл, как вступившую в реакцию
в избыточном количестве.
Количество щелочи, израсходованной на предварительную нейтрализацию, при¬
бавляют в мерную колбу для получения нейтральной реакции. Затем добавляют по
1мл 40-50% раствор сегнетовой соли и по 2мл реактива Несслера. После добавления
каждого из этих реактивов содержимое колб хорошо взбалтывают и доводят до метки
дистиллированной водой, перемешивают и через 15 мин. колориметрируют при си¬
нем свете светофильтра. Для подсчета количества азота пользуются калибровочной
кривой (график), построенной по стандартному раствору, приготовленному из
(NH«)2S04.
Кроме опыта с навеской испытуемого вещества, необходимо проведение конт¬
рольного опыта на чистоту реактивов. Нулевая точка прибора устанавливается по
контрольному образцу.
-46-
где X - процент азота на абсолютно сухое вещество;
а - количество азота, найденное по калибровочному графику, мг;
б - навеска абсолютно сухого вещества, г;
в - объем, в котором растворена навеска + количество ТХУкислоты,
взятое для осаждения белка, мг;
г - количество фильтрата, взятое для сжигания, мг;
д - объем, в котором растворена навеска после сжигания, мл;
е - количество раствора, взятое для колориметрирования, мл.
При определении небелкового азота, когда растения содержат значительное
количество нитратного азота, сжигание образцов необходимо проводить не серной
кислотой, а 4% раствором фенолсерной кислоты, т.к. при сжигании только серной
кислотой нитратный азот, имеющийся в растении, не будет определен и показатели
небелкового азота будут занижены.
2.5.4.5. Технологическая карта
2.6. ОПРЕДЕЛЕНИЕ МАРГАНЦА, ЦИНКА, ЖЕЛЕЗА, МЕДИ И БОРА
В ОДНОЙ НАВЕСКЕ
2.6.1. ОБЩИЕ ПОЛОЖЕНИЯ
Определение микроэлементов в растениях можно разделить на два основных этапа:
1) минерализация анализируемого материала и получение раствора золы;
2) определение микроэлементов в растворах золы.
Из известных методов минерализации растительного материала наиболее удобен
сухой способ озоления.
Для определения микроэлементов в растворах золы применяются колориметри¬
ческие и спектрофотометрические методы.
При определении микроэлементов возможны искажения получаемых результатов
за счет загрязнения анализируемого вещества в процессе анализа, так называемый
«фон», который зависит от чистоты применяемых в работе реактивов, посуды и дру¬
гих факторов. Уровень и колебания «фона» определяют предел обнаружения элемен¬
та и точность анализа. Поэтому к качеству воды и реактивов, чистоте посуды и
рабочего места предъявляются повышенные требования.
Для определения меди, цинка, кобальта используется химическая посуда из тер¬
мостойкого боросиликатного стекла типа «Пирекс», ПТТ или ПТ-ТУ. Для определе¬
ния бора пользуются посудой из безборного стекла марок ДГ-29, ХУ КЛП, С-90 или
кварца. Для хранения реактивов при определении бора можно использовать склянки
из зеленовато-голубого стекла.
При проведении операций, не требующих нагревания, для хранения воды и ре¬
активов, за исключением концентрированных (более 50%) кислот и органических ра¬
створителей, лучше использовать полиэтиленовую посуду.
Особенно важно для каждого микроэлемента применять отдельную посуду и до¬
зирующие устройства, т.к. использование последних в различных анализах может
повлечь за собой загрязнение их анализируемым элементом и искажение результатов.
Многие анализы несовместимы не только в отношении посуды, но даже помещения.
Например, использование КМп04 при определении бора в почве может внести искажения
при проведении в том же помещении анализа по определению марганца и т.д.
Для снятия фона холостой опыт (включающий все стадии анализа, кроме взятия
навески) необходимо проводить в каждой партии анализа не менее чем в 2-кратной
повторности и средний результат вычитать из результата анализов испытуемых проб.
Если результаты холостого опыта очень высокие, методом исключения находят
источник загрязнения и устраняют его. Очистка реактивов очень трудоемка, поэтому
ее следует проводить только в случае крайней необходимости.
При определении микроэлементов широко используются органические раство¬
рители для экстракции комплексных соединений из водных растворов. Эту операцию
проводят в делительных воронках (лучше всего), цилиндрах, пробирках или склянках
с хорошо притертыми пробками, чтобы не пользоваться смазкой, растворяющейся в
органических растворителях. При работе с цилиндрами или пробирками для отбора
экстракта используют шприцы или пипетки с оттянутыми до капилляра кончиком и
грушей для создания разрежения.
Для очистки фильтров от следов микроэлементов их промывают не менее 5 раз
чистой НС1, разбавленной бидистиллированной водой (1:100), а затем хорошо отмы¬
вают от кислоты бидистиллированной водой и сушат при температуре 95°С.
Посуду новую моют содовым или мыльным раствором, затем водой и заливают
разбавленной НС1 (около 10%) на 10-12 часов. После этого промывают 4-5 раз водо¬
проводной и 3 раза дистиллированной или бидистиллированной водой.
Посуду, бывшую в работе, очищают разбавленной НС1 и хорошо промывают дис¬
тиллированной, а затем бидистиллированной водой.
2.6.2. ОЗОЛЕНИЕ РАСТИТЕЛЬНОГО МАТЕРИАЛА
2.6.2.1. Принцип метода
Оэоление проводится сухим способом при t = 500°С в течение 4 часов. Золу обраба¬
тывают соляной кислотой для переведения окислов и карбонатов элементов, содержа¬
щихся в ней, в хлориды. Полученный раствор служит для определения бора, марганца,
цинка, железа, кобальта, меди. В этом же растворе можно определять кальций и магний.
При озолении растительного материала для анализа на микроэлементы можно
одновременно определять процент сырой золы. Для этого взвешивание растительной
массы и полученной из нее золы (в тиглях) проводят на аналитических весах до пере¬
ведения последней в раствор и рассчитывают процент золы.
2.622. Технологическое оборудование
Весы типа ВЖТ-500 или аналитические.
Печь муфельная.
Плитка электрическая с закрытой спиралью.
Тигли фарфоровые на 30-50мл.
-48-
Колбы или пробирки мерные на 50мл.
Дозаторы на 2,5 и 5 мл или бюретки на 25 или 50мл
Палочки стеклянные.
Стекла часовые.
Воронки стеклянные d=5 или 7см.
Промывалка.
2.6.2.3. Реактивы
1. Зн. НС1. 246мл концентрированной х.ч. НС1 плотность 1,19г/мл разбавляют до
1л бидистиллированной водой.
2. 0.75 Н.НС1. 62мл концентрированной х.ч.НС1 (плотность 1,19г/мл) разбавляют
бидистиллированной водой до 1л.
3. Кониентрированная х.ч. HNOr
4. Вода бидистиллированная.
2.6.2.4. Ход анализа
5г воздушно-сухого размолотого растительного материала взвешивают на весах
типа BJIKT-500 с погрешностью не более 0,01 г, помещают в фарфоровый тигль и ста¬
вят его в холодную муфельную печь, постепенно повышают температуру до 300°С и
выдерживают при этой температуре до прекращения выделения дыма. Затем доводят
температуру до 500°С (темно-красного каления) и озоляют пробу в течение 4 часов.
После охлаждения золу в тиглях смачивают дистиллированной водой, приливают к
ней 5мл Зн. НС1, накрывают тигль часовым стеклом и выдерживают на электрической
плитке, нагретой до ~ 100°С, в течение 30 минут. После охлаждения тигля нижнюю
поверхность часового стекла обмывают над ним небольшим количеством воды, за¬
тем, с помощью стеклянной палочки, не фильтруя, содержимое тигля, переносят че¬
рез воронку в мерную колбу или пробирку емкостью 50мл. Тигель, палочку и воронку
смывают водой, стараясь как можно полнее перенести частицы не растворившегося
остатка, доводят до метки, хорошо перемешивают и дают раствору отстояться (лучше
на ночь). После отстаивания над осадочную жидкость используют для определения
микроэлементов. Допускается центрифугирование или фильтрование раствора через
беззольный фильтр, проверенный на отсутствие определяемого элемента.
Одновременно в каждой партии образцов ведут в двукратной повторности хо¬
лостую пробу.
2.6.2.5. Технологическая карта
-49-
2.6.3. ОПРЕДЕЛЕНИЕ МАРГАНЦА, МЕДИ, ЦИНКА, ЖЕЛЕЗА
В РАСТВОРЕ ЗОЛЫ АТОМНО-АБСОРБЦИОННЫМ МЕТОДОМ
2.63.1. Принцип метода
В основу атомно-абсорбционного метода положено явление селективного погло¬
щения световой энергии свободными атомами определяемых элементов, образующихся
при введении их растворов в воздушно-ацетиленовое или воздушно-пропан-бутано-
вое пламя.
Количественное определение элементов осуществляется по градуировочным
графикам, полученным путем фотометрирования образцовых растворов.
Для устранения химических помех при определении марганца и железа в про-
пан-бутановом пламени в растворы золы предварительно добавляют стронций до кон¬
центрации 5 и 1 мг/мл..
Используют аналитические линии марганца - 279,5; меди - 324,7; цинка - 213,9
и железа - 248,Знм.
Техника работы на атомно-абсорбционном спектрофотометре описана в пп.4.6.*
. * В настоящее время существует множество атомно-абсорбционных приборов, вы¬
пускаемых в различных странах. Мы же приводим описание только некоторых из них.
2.63.2. Технологическое оборудование
Установка для атомно-абсорбционного анализа: спектрофотометр, лампы с по¬
лым катодом ЛСП-1 для определения марганца, меди, цинка и железа, ацетилен или
пропан-бутан (баллоны), компрессор.
Пипетки градуированные на 5, 1-20 и 50мл.
Дозаторы на 5 - 9мл.
Пробирки мерные на 10 или 20мл.
Колбы мерные на 100, 500, 1000мл.
2.6.3.3. Реактивы
1. Соляная кислота. хм. - 3 и 0.75н.. см. п. 2.6.2.3.
2. Образиовые растворы для определения марганиа.
Исходный образцовый раствор (раствор А).
4,388г сернокислого марганца MnS04 • 5Н20 растворяют в бидистиллированной
воде и доводят объем до 1л. Полученный раствор содержит 1 мг/мл марганца. Ра¬
створ хранят 1 год.
Рабочий образцовый раствор (индивидуальный или смешанный) - раствор Б.
В мерную колбу емкостью 500мл наливают 50мл раствора А, доводят до метки
0,75н. НС1 и перемешивают. Приготовление смешанного раствора Б см. табл. 14. Ра¬
створ содержит 100мкг/мл. марганца. Хранится раствор до 3 месяцев.
Таблица 14.
Исходные образцовые
растворы - растворы А
Объем раствора А,
мл.
Концентрация элемента в рабочем
образцовом (индивидуальном или
смешанном растворе) - растворе Б, мкг/мл
На марганец
50
100
На медь
25
50
На цинк
10
20
На железо
100
200
Примечание. Смешанный рабочий образцовый раствор готовится в мерной колбе на 500mjl
-50-
Шкалу образцовых растворов (индивидуальных или смешанных) готовят в мер¬
ных колбах емкостью 100мл, куда помещают указанные в таблице 15 количества ра¬
створа Б, доводят объемы до метки 0,75н. НС1 и хорошо перемешивают. Готовят шка¬
лу в день проведения анализов.
3. Образиовые растворы для определения меди.
Исходный образцовый раствор (раствор А).
3,929г сернокислой меди CuS04* 5Н20 растворяют в бидистиллированной воде,
содержащей 1мл концентрированной серной кислоты, и доводят объем до 1л. Полу¬
ченный раствор содержит 1 мг/мл. меди. Сохраняется он в течение года.
Рабочий образцовый раствор (раствор Б). В мерную колбу емкостью 500мл на¬
ливают 25мл раствора А, доводят до метки 0,75н. НС1 и хорошо перемешивают. (При¬
готовление смешанного раствора Б см. табл.. 14). Содержание меди в растворе Б -
50мкг/мл.. Срок хранения до 3 месяцев.
Шкалу образцовых растворов (индивидуальных или смешанных) готовят в мер¬
ных колбах емкостью 100мл в день проведения анализа. Отмеривают указанные в таб¬
лице 15 количества раствора Б, доводят до метки 0,75н. НС1 и хорошо перемешивают.
Таблица 15.
Номера
колб
Объем рабочего
образцового
раствора -
раствор Б, мл.
Концентрация
элементов в образцовом
растворе шкалы, мкг/мл.
Содержание элементов в
анализируемом
материале в мг/кг сухого
вещества
Мп
Zn
Си
Fe
Мп
Zn
Си
Fe
1
0
0
0
0
0
0
0
0
0
2
2
2
0,4
1,0
4
20
4
10
40
3
5
5
1,0
2,5
10
50
10
25
100
4
10
10
2,0
5,0
20
100
20
50
200
5
20
20
4,0
10
40
200
40
100
400
Примечание. Смешанную шкалу образцовых растворов готовят в мерных колбах на 100мл.
4. Образиовые растворы для определения иинка.
Исходный образцовый раствор (раствор Л). 1г металлического цинка смачивают
бидистиллятом, растворяют в 7-10мл разбавленной НС1 (1:1) и доводят объем дистил¬
лированной водой до 1л. Полученный раствор содержит 1 мг/мл. цинка. Срок хране¬
ния раствора 1 год.
Рабочий образцовый раствор (индивидуальный или смешанный) - раствор Б. В
мерную колбу емкостью 500мл отмеривают 10мл раствора А, доводят до метки 0,75н.
НС1 и хорошо перемешивают. (Приготовление смешанного раствора Б см. табл.. 14).
Содержание цинка в растворе Б - 20мкг/мл.
Шкалу образцовых растворов (индивидуальных или смешанных) готовят в день
проведения анализа в мерных колбах емкостью 100мл. Отмеривают указанные в таб¬
лице 15 количества раствора Б, доводят до метки 0,75н.НС1 и хорошо перемешивают.
5. Образиовые растворы для определения железа
Исходный образцовый раствор (раствор А).
8,635г сульфата аммония железа FeNH4(S04)2 • 12 Н20 растворяют в бидистилли¬
рованной воде, содержащей 2,5мл концентрированной серной кислоты, и доводят
объем раствора водой до 1л. Раствор хранится в течение года.
- 51 -
Рабочий образцовый раствор (индивидуальный или смешанный) - раствор Б.
В мерную колбу емкостью 500мл отмеривают 100мл раствора А, доводят до
метки 0,75н. соляной кислотой и хорошо перемешивают. (Приготовление смешан¬
ного раствора Б см. табл. 14). Содержание железа в растворе 200 мкг/мл.. Срок
хранения до 3 месяцев.
Шкалу образцовых растворов (индивидуальных или смешанных) готовят в день
проведения анализа. В мерные колбы емкостью 100 мл отмеривают указанные в таб¬
лице 15 количества раствора Б, доводят до метки 0,75 н. НС1 и хорошо перемешивают.
6. Раствор хлористого строниия 30,4г SrCl2<H20 растворяют в 500мл бидистил-
лированной воды, приливают 82мл концентрированной НС1 и доводят водой объем до 1л.
2.63.4. Ход анализа
Для определения марганца настраивают атомно-абсорбционный спектрофотометр
в соответствии с инструкциями, прилагаемыми к прибору и лампе. Регулировкой по¬
ложения горелки относительно просвечивающего луча и распылителя устанавливают
размах шкалы, вводя в пламя 1-й и 5-й растворы сравнения.
При использовании пламени ацетилен-воздух при установившемся стабильном
режиме работы прибора в пламя вводят нулевой раствор шкалы и устанавливают на¬
чало отсчета (0 оптической плотности). Вводят в пламя остальные растворы шкалы в
порядке возрастания концентрации марганца в них и анализируемые растворы, запи¬
сывая соответствующие им значения оптической плотности. Через каждые десять ис¬
пытуемых растворов проверяют нулевой и один из растворов шкалы для контроля за
градуировкой прибора. Во время работы между испытуемыми растворами в пламя
вводят воду для промывки горелки.
Определение меди, цинка и железа проводят аналогично, настраивая атомно-аб¬
сорбционный спектрофотометр на соответствующий элемент.
При использовании пламени пропан-бутан-воздух определение меди и цинка идет
аналогично описанному для воздушно-ацетиленового пламени. В анализе на марга¬
нец и железо в растворы золы и образцовой шкалы вводят 1% хлористый стронций в
соотношении 1:1 для марганца и 4:1 для железа.
Анализ всех определяемых элементов ведут последовательно, прогревая одновре¬
менно их лампы в блоке предварительного прогревания ламп с полым катодом - БП-15.
2.63.5. Обработка результатов
По результатам фотометрирования образцовых растворов строят калибровочные
графики, откладывая на горизонтальной оси содержание определяемого элемента в
мкг/мл образцовых растворов или в мг/кг анализируемого материала, а на вертикаль¬
ной оси - отсчет измерительного прибора. По графику находят содержание соответ¬
ствующего элемента и вычитают из него результат холостого определения.
Расчет ведут по формуле
(a-e)V 100
Mn(Cu, Zn, Fe), % = , (1)
или и-1000000
(а-в)ГЮОО
Mn (Си, Zn, Fe), мг/кг = —— , (2)
растительной массы Н • 1UUU
где а - содержание Mn (Си, Zn, Fe) в испьпуемом растворе по графику, мкг/мл.;
в - содержание Mn (Си, Zn, Fe) холостом растворе по графику, мкг/мл.;
-52-
V - общий объем испытуемого раствора, мл.;
н - навеска, г;
100 - коэффициент пересчета в проценты (1);
1000000 - коэффициент перевода мкг в г (1);
1000 - коэффициент пересчета на 1кг воздушно-сухого материала (2 - числитель);
1000 - коэффициент перевода мкг в мг (2 - знаменатель).
При расчетах содержания Мп и Fe, определяемых в пропан-бутановом пламени,
необходимо учитывать разведение раствора за счет введения раствора SrCl2.
Для удобства обработки данных при постоянной навеске 5г исследуемого матери¬
ала и общем объеме раствора золы 50мл можно строить калибровочный график, от¬
кладывая на горизонтальной оси содержание микроэлементов сразу в мг/кг сухого
вещества (табл.. 15).
При высоком содержании Mn (Си, Zn, Fe) раствор золы разводят в 5-10 раз 0,75н.
HCI. Например, в случае высокого содержания марганца в пробирку берут 2мл ра¬
створа золы, 8мл 0,75н. HCI (при ацетилен-воздушном пламени) и 2мл раствора золы,
8мл 0,75н. HCI и 10мл SrCl2 (при пропан-бутан-воздушном пламени) и перемешива¬
ют. Найденное по калибровочному графику содержание марганца увеличивают соот¬
ветственно в 5 и 10 раз.
Допустимые отклонения результатов при повторных определениях составляют в
одной лаборатории 10%, в разных - 15%.
2.63.6. Технологическая карта
2.6.4. ОПРЕДЕЛЕНИЕ БОРА В РАСТВОРЕ ЗОЛЫ
КОЛОРИМЕТРИЧЕСКИМ МЕТОДОМ С АЗОМЕТИНОМ Н
2.6.4.1. Принцип метода
Метод основан на образовании окрашенного в желтый цвет комплексного соеди¬
нения бора с азометином Н в водных растворах при pH = 5,2 и измерении их оптичес¬
кой плотности на фотоэлектроколориметре. Мешающее влияние меди, алюминия и
железа устраняют связыванием их в комплексе трилоном Б.
2.6.4.2. Технологическое оборудование
Весы аналитические и типа ВЛКТ-500.
Фотоэлектроколориметр.
-53-
Пробирки из безборного стекла d= 15-20мм с притертыми или полиэтиленовыми
пробками.
Шприц-дозатор или пипетка с грушей на 1мл.
Воронка Бюхнера и колба Бунзена.
Дозаторы на 4 и 5мл.
Бюретка на 25мл.
Пипетка градуированная на 10мл.
Колбы конические на 1 и 2л.
Колбы мерные на 50, 100, 1000мл.
Стаканы на 100-150мл.
2.6.4.3. Реактивы
1. Разбавленная серная кислота. К 4 частям дистиллированной воды добавляют
осторожно, при постоянном помешивании, 1 часть концентрированной H2S04.
2. Буферный маскирующий раствор. 500г уксусно-кислого аммония и 10г трило-
на Б растворяют в дистиллированной воде и доводят объем до 1л. Полученный ра¬
створ подкисляют разбавленной (1:4) H2S04 до получения pH 5,2 ±0,2. Реакцию
проверяют стеклянным электродом.
3. Азометин Н. 0.9% раствор. 0,9г азометина Н и 2г аскорбиновой кислоты ра¬
створяют в 20-30мл дистиллированной воды при осторожном нагревании на водяной
бане при 90-100°С. После полного растворения реактивов полученный раствор пере¬
ливают в мерную колбу емкостью 100мл и доводят до метки дистиллированной водой,
при необходимости фильтруют. Хранят раствор в холодильнике до двух недель. Если
при хранении раствор помутнеет, перед использованием его следует снова подогреть
на водяной бане до полного растворения.
Азометин Н получают из мононатриевой соли Н-кислоты (1 -амино-8-нафтол-3,6
дисульфокислоты мононатриевая соль - CJ0H8O7NS2Na*15H2O) и салицилового аль¬
дегида С7Н602. Растворяют 18г мононатриевой соли Н-кислоты в 1л дистиллирован¬
ной воды при осторожном нагревании и фильтруют раствор в коническую колбу
емкостью 2-3л. Теплый раствор нейтрализуют 10% КОН (-30 мл) до pH 7 по универ¬
сальной индикаторной бумаге, прибавляют по каплям концентрированную НС1(~4
мл), непрерывно перемешивая раствор до получения pH около 2. Затем к раствору
приливают 20мл салицилальдегида и встряхивают в течение часа на механическом
встряхивателе. Колбу с раствором оставляют до полного выделения азометина Н -
на 12-16 часов. Осадок фильтруют на воронке Бюхнера, промывают 5-7 раз этило¬
вым спиртом порциями по 10-15мл и сушат 3 часа при температуре 100°С. Полу¬
ченное вещество оранжевого цвета хранят в банке темного стекла с хорошо притер¬
той пробкой. Свойства реактива сохраняются в течение продолжительного времени.
4. Смешанный окрашивающий реактив. Смешивают растворы азометина Н и мас¬
кирующего буферного в отношении 1:1. Смесь готовят в день проведения анализа.
5. Образиовые растворы для определения бора.
Исходный образцовый раствор. Навеску борной кислоты 0,572г растворяют в дис¬
тиллированной воде и объем доводят до 1л. Полученный раствор содержит 100мкг/мл
(0,0001 г/мл) бора.
Рабочий образцовый раствор. 25мл исходного образцового раствора бора разбавля¬
ют в мерной колбе до 100мл 0,75н. НС1. Полученный раствор содержит 25мкг/мл бора.
Шкалу образцовых растворов готовят в мерных колбах емкостью 50мл, куда на¬
ливают объемы рабочего стандартного раствора, указанные в таблице 16, и доводят
до метки 0,75н.НС1.
-54-
Таблица 16.
" — Номера колб
Показатели
1
2
3
4
5
6
7
Объем рабочего раствора В, мл.
0
1
2
4
6
8
10
Содержание В в образцовом растворе шкалы, мкг/мл
0
0,5
1
2
3
4
5
Содержание В в анали-зируемом материале, мкг/кг
0
5
10
20
30
40
50
2.6.4.4. Ход анализа
Помещают в пробирки по 1мл прозрачного раствора золы и образцовых раство¬
ров шкалы, приливают по 5мл дистиллированной воды и 4мл смешанного окрашива¬
ющего реактива, перемешивают и оставляют на 2 часа для развития окраски.
При очень низком содержании бора в образцах аликвоту можно увеличить до 5мл,
добавляя столько дистиллированной воды, чтобы в сумме аликвота + вода = 6мл.
Оптическую плотность растворов измеряют на фотоэлектроколориметре при длине
волны и 20нм (темно-синий светофильтр) в кювете с толщиной просвечиваемого слоя 2см
в сравнении с нулевым раствором шкалы. По результатам колориметрирования образцо¬
вых растворов шкалы строят калибровочный график, откладывая на оси абсцисс содержа¬
ние бора в мкг/мл. раствора, а на оси ординат - значение оптической плотности.
2.6.4.5. Обработка результатов
Расчет ведется по формулам:
(fl-e)K-lOO
В, % = , (1)
или и* р\000000
в, (!-!>• Г (2)
н • р 1000
растительной .массы
где а -концентрация В в испытуемом растворе по графику, мкг/мл.;
в - концентрация В в холостом растворе по графику, мкг/мл.;
V - общий объем испытуемого раствора, мл.;
100 - коэффициент пересчета в проценты (1);
р - количество исходного раствора, взятое для колориметрирования, мл.;
н- навеска, г;
1000000 - коэффициент перевода мкг в г (1);
1000 - коэффициент пересчета на 1 кг материала (2 - числитель);
1000 - коэффициент перевода мкг в мг (2 - знаменатель).
Для удобства обработки данных при постоянной навеске исследуемого матери¬
ала и общем объеме раствора 50мл можно в калибровочном графике сразу отклады¬
вать содержание бора в мг/кг сухого вещества (табл. 16).
2.6.4.6. Технологическая карта
-55-
2.7. ОПРЕДЕЛЕНИЕ МОЛИБДЕНА
КОЛОРИМЕТРИЧЕСКИМ МЕТОДОМ С РОДАНИСТЫМ КАЛИЕМ
2.7.1. Принцип метода
Определение молибдена в растительном материале проводится в отдельной на¬
веске после сухого озоления. Для более полного окисления органических веществ
проводится дополнительная обработка азотной кислотой и повторное прокаливание в
муфеле. Прокаленный остаток растворяют при нагревании в соляной кислоте и опре¬
деляют Мо колориметрическим методом (см.3.14.1).
2.7.2. Технологическое оборудование
Весы аналитические и типа BJIKT-500.
Фотоэлектроколориметр.
Пипетки или шприц-дозатор на 25 мл.
Дозаторы на 1, 2, 2,5, 5, 8, 10, 15 мл.
Пипетки градуированные на 5, 10 мл.
Пипетка с оттянутым носиком на 10 мл.
Капельница.
Воронки делительные или цилиндры с пришлифованными пробками
на 50-100мл с дополнительной меткой на 30мл.
Колбы мерные на 50, 250 и 1000мл.
2.7.3. Реактивы
1. 20% раствор НС1. 492мл концентрированной х.ч. НС1 приливают к дистилли¬
рованной воде и разбавляют до 1л.
2. 6.5% раствор НС1. 330мл 20% НС1 разбавляют дистиллированной водой до 1л.
3. 1% раствор железо-аммонийных квасиов. Юг квасцов растворяют при нагре¬
вании в 20% НС1. После охлаждения объем доводят до 1л 20% НС1. Раствор содержит
около 1мг/мл. железа.
4. 10% раствор роданистого калия. Юг реактива растворяют в дистиллирован¬
ной воде, разбавляют до 100мл и фильтруют.
5. 20% раствор двухлористого олова. 20г реактива растворяют в 20мл 20% НС1.
Накрывают стакан с раствором часовым стеклом и кипятят в течение 10 мин. После
охлаждения раствор разбавляют дистиллированной водой до 100мл.
6. Изоамиловый или н-бутиловый спирт, ч.д.а.
7. Этиловый спирт.
8. Промывной раствор. Смешивают 6 мл 20% двухлористого олова и 94мл 6,5%
соляной кислоты.
9. Образиовые растворы для определения молибдена.
Исходный образцовый раствор. 0,184г молибденовокислого аммония -
(NH4)6Mo7 • 024 • 4Н20, взвешенного с погрешностью не более 0,001г, растворяют
в дистиллированной воде и доводят объем до 1л. Полученный раствор содержит 0,1 г
мг/мл молибдена.
Рабочий образцовый раствор. Отбирают градуированной пипеткой 2,5мл исход¬
ного образцового раствора молибдена и разбавляют дистиллированной водой в мер¬
ной колбе до 250мл. Полученный раствор содержит 1мкг/мл. молибдена.
Шкалу образцовых растворов_ готовят в делительных воронках или цилиндрах,
куда наливают по 1мл раствора железо-аммонийных квасцов, указанные в таблице
-56-
17 объемы рабочего образцового раствора молибдена, по 10мл 20% НС1 и дистил¬
лированную воду до общего объема 30мл (до метки). Дальше с ними проводят те
же операции, что и при анализе раствора золы.
Таблица 17.
' '—— Номер делительной воронки
Показатели ■—
1
2
3
4
5
6
7
Объем рабочего раствора Мо, мл.
0
0,5
1
2
3
4
5
Количество Мо в экстракте изоамилового спирта , мкг
0
0,5
1
2
3
4
5
Содержание Мо в анализируемом материале, мг/кг
0
0,1
0,2
0,4
0,6
0,8
1,0
2.7.4. Ход анализа
Юг воздушно-сухого растительного материала взвешивают в фарфоровых тиглях
на весах типа BJIKT-500 (в случае одновременного определения процента золы взве¬
шивание ведут на аналитических весах), ставят в холодную муфельную печь, посте¬
пенно повышают температуру до 300°С и выдерживают при этой температуре до пре¬
кращения выделения дыма. Доводят температуру до 500°С (темно-красного каления)
и озоляют еще в течение 4 часов. После охлаждения золу смачивают 5 мл дистиллиро¬
ванной воды, приливают 2мл концентрированной HN03 и выпаривают содержимое
тигля на электрической плитке с закрытой спиралью досуха. Затем тигль ставят в
холодный муфель, повышают температуру до 500°С и прокаливают остаток в течение
15мин. Приливают к прокаленному остатку 5мл 6н. НС1 и выпаривают на плитке до¬
суха, не допуская прокаливания остатка.
Сухой остаток растворяют при нагревании в 20мл 6н. НС1 и фильтруют в мерную
колбу емкостью 50мл. Стенки тигля и фильтр на воронке обмывают малыми порциями
дистиллированной воды, доводят раствор в колбе до метки дистиллированной водой
и хорошо перемешивают.
Одновременно в каждой партии образцов ведут в двухкратной повторности холо¬
стую пробу.
В делительную воронку или цилиндр с пришлифованной пробкой помещают 25мл
раствора золы, добавляют 1мл железо-аммонийных квасцов и разбавляют дистилли¬
рованной водой до 30мл (на делительной воронке предварительно отмечают объем
30мл). Затем к растворам золы и стандартным растворам шкалы приливают дозатора¬
ми 2,5мл 10% роданида калия и 2мл 20% двухлористого олова, перемешивая содержи¬
мое воронки или цилиндра после прибавления каждого реактива.
После обесцвечивания окраски роданида железа приливают 15мл изоамилового
или н-бутилового спирта и встряхивают воронку в течение 2мин. После того как фазы
полностью расслоятся, отбрасывают нижний водный слой, оставив около 0,5мл его в
воронке с экстрактом. Удаление его из цилиндра проводят шприцем или пипеткой с
грушей с хорошо оттянутым носиком. Экстракт обрабатывают промывным раство¬
ром, который готовят перед промывкой. Добавляют по Юмл промывного раствора в
делительную воронку с экстрактом и встряхивают ее в течение 1 мин.
После разделения фаз, водный слой полностью удаляют (в делительной воронке
или пипеткой с оттянутым носиком), отбирают верхний окрашенный слой в цилиндре и
переносят его в кювету колориметра с толщиной просвечиваемого слоя Зсм. Добавляют
в кювету 5 капель этилового спирта, перемешивают экстракт стеклянной палочкой и
оставляют на 5мин. для оседания капелек воды. Окраска устойчива в течение ЗОмин.
-57-
Оптическую плотность прозрачного экстракта, окрашенного в оранжевый
цвет, измеряют в сравнении с изоамиловым или н-бутиловым спиртом на фото¬
электроколориметре при длине волны 470нм (синий светофильтр).
По результатам фотометрирования образцовых растворов строят калибровоч¬
ный график, откладывая на горизонтальной оси содержание молибдена в мкг/мл. или
в мг/кг воздушно-сухош материала, соответствующее образцовому раствору, а на вер¬
тикальной оси - значение оптической плотности.
Содержание молибдена в анализируемом материале находят по калибровочно¬
му графику и вычитают из него результат холостого определения.
2.7.5. Обработка результатов
Расчет ведется по формулам:
м,,о/о = (£-«)• (,)
р-н-1000000
или (a-e)-V 1000
Мо, мг/кг = - , (2)
растительной массы р * И ' 1000
где а - количество Мо в испытуемом экстракте по графику, мкг;
в - количество Мо в экстракте из холостого раствора по графику, мкг;
V- общий объем испьпуемого раствора, мл.;
р - количество испьпуемого раствора, взятое для экстрагирования Мо, мл.;
н - навеска, г;
100 - коэффициент пересчета в проценты (1);;
1000000 - коэффициент перевода мкг в г (1);
1000- коэффициент пересчета на 1 кг воздушносухого материала
(2 - числитель);
1000 - коэффициент перевода мкг в мг (2 - знаменатель).
Для удобства обработки данных при постоянной навеске 1 Ог исследуемого мате¬
риала, общем объеме - 50мл и отборе раствора для экстракции - 25мл можно строить
калибровочный график, откладывая на горизонтальной оси содержание Мо сразу в
мг/кг сухого вещества (табл. 17).
2.7.6. Технологическая карта
-58-
2.8. ОПРЕДЕЛЕНИЕ САХАРИСТОСТИ
И КОНДУКТОМЕТРИЧЕСКОЙ ЗОЛЫ В КОРНЕПЛОДАХ
СВЕКЛЫ В ОДНОЙ НАВЕСКЕ
2.8.1. Принцип метода
Метод основан на получении водной вытяжки из мезги корнеплодов сахарной свек¬
лы в последовательном определении в ней золы (путем кондуктометрического опре¬
деления электропроводности раствора и пересчета на золу) и содержания сахара по¬
ляриметрическим методом.
2.8.2. Технологическое оборудование
Кондуктометр.
Поляриметр.
Весы типа BJIKT-500.
Автоматический дозатор или автоматическая пипетка на 178, 89, или 44,5 мл.
Стаканы на 250-300 мл или 10-позиционные кассеты.
Воронки пластмассовые.
Фильтры тонкие d= 15-18см.
Пипетка градуированная или дозатор на 1мл.
2.8.3. Реактивы
1. Маточный раствор свиниового уксуса. ЗООг уксуснокислого свинца Рв(СН3(ПЦ2
• ЗН20 и 100г окиси свинца РвО (свинцовый глет) растирают в ступке до образования
однородной массы. Пересыпают в коническую колбу на 1л, добавляют 50мл дистилли¬
рованной воды и ставят на водяную баню на 4 часа, помешивая через каждые 15мин.
Затем содержимое колбы доводят до 1л и отстаивают в течение суток. Можно вместо
выдерживания на бане колбу помещать на сутки в термостат при температуре 45°С.
Маточный раствор свинцового уксуса должен иметь сильно щелочную реакцию и
плотность 1,23-1,24г/см3, но не давать окрашивания с фенолфталеином.
2. Вместо маточного раствора можно пользоваться сухим осветлителем. кото¬
рый приобретают в магазинах или готовят следующим образом. Измельчают в мелкий
порошок ЗООг уксуснокислого свинца и ЮОг свинцового глета, хорошо перемешива¬
ют их и нагревают в течение 3-4 часов на водяной бане. В течение этого времени глет
полностью обесцвечивается или имеет слабо-розовый цвет. Затем его хорошо измель¬
чают и просеивают.
Плотность сухого осветлителя 1,5-1,7.
2мл обычного свинцового уксуса соответствует 0,35г сухого осветлителя. Для за¬
мены свинцового уксуса сухим осветлителем при определении сахаристости свеклы
его следует брать в количестве 1г на пробу.
При работе его нужно измельчать, хранить в склянке с притертой пробкой.
-59-
Внимание! Р астирать, расплавлять и просеивать соли свинца при приготов¬
лении маточного раствора или сухого осветлителя необходимо в вытяжном шкафу
или пользуясь респиратором.
3. Раствор сахарозы для проверки поляриметра. Готовят один или два раствора
сахарозы и просматривают их в поляризационной трубке на 400мм. Навески раство¬
ряют в 200мл дистиллированной воды.
Навеска сахарозы, г Показания поляриметра,
% сахара
26
100
6,5
25
5,2
20
3,9
15
2,6
10
2.8.4. Ход анализа
Корнеплод свеклы тщательно очищают от земли во избежание искажений показа¬
ний кондуктометра.
Мезгу получают при помощи сверла или сегментной терки. На квадратные листки
пергамента или кальки размером 5 х 5см берут в 2-3-кратной повторности навески (13
или 6,5г) хорошо перемешанной мезги, смывают ее дистиллированной водой из авто¬
матической пипетки (емкостью 89 или 44,5мл) в стаканчики, хорошо перемешивают
стеклянной палочкой и оставляют на полчаса, перемешивая еще не менее 3 раз в тече¬
ние этого времени.
При использовании автоматического дозатора берут среднюю порцию мезги ло¬
жечкой на квадратик кальки и помещают на чашку весов дозатора. Дозатором, соеди¬
ненным с баллоном дистиллированной воды, автоматически отмеривают соответству¬
ющий данной массе мезги объем воды.
Получасовое стояние стаканов с полученной смесью можно заменить 6-минут¬
ным взбалтыванием на ротаторе.
В полученной смеси проводят определение содержания растворимой золы кон-
дуктометрическим методом. (Работа на кондуктометре описана в п.4.2). Электрод кон¬
дуктометра опускают в стаканчик со смесью и по шкале прибора снимают показания
электропроводности раствора. Перед следующим определением электрод обмывают
смесью следующей пробы или высушивают его фильтровальной бумагой.
В стаканчик, где определялась кондуктометрическая зола, добавляют 1мл маточ¬
ного раствора свинцового уксуса или меркой 1г сухого осветлителя, хорошо переме¬
шивают стеклянной палочкой и оставляют на 3-5мин. для осветления. Затем содержи¬
мое стаканчика фильтруют в чистую сухую посуду и поляризуют в трубке длиной 400мл.
Показания сахариметра выражены в процентах содержания сахара в свекле.
Проверку поляриметра проводят перед работой по образцовым растворам сахарозы.
2.8.4.1. Использование нетоксичных осветлителей
при определении сахара в свекле
1. Гидросульфат алюминия АI (OH)SO^ х.ч. или ч.д.а.
Приготовление маточного раствора. Взвешивают ЮОг A1(0H)S04, растворя¬
ют дистиллированной водой, переливают в мерную колбу на 1000мл. Доводят объем
до метки, перемешивают и переливают в бутыль для хранения.
Приготовление рабочего раствора. Цилиндром отмеряют 25мл маточного раство¬
-60-
ра, переносят в мерную колбу на 1000мл, доводят дистиллированной водой до мет¬
ки, перемешивают.
2.Алюминий хлористый Л1С13 6НО. х.ч. или ч.д.а.
Приготовление маточного раствора.
Взвешивают 180г А1С1Э * 6Н20, растворяют дистиллированной водой и вливают в
мерную колбу на 1000мл. Объем раствора доводят дистиллированной водой до
метки, перемешивают и переливают в бутыль для хранения.
Приготовление рабочего раствора. Цилиндром отмеряют 22,1мл маточного ра¬
створа, переносят в мерную колбу объемом 1000мл и доводят дистиллированной во¬
дой до метки, перемешивают. Раствор используют для анализа.
3. Алюминий сернокислый (сульфат алюминия) Al/SOJ^ • 18ПХ): х.ч. или ч.д.а.
Приготовление маточного раствора. Взвешивают 240г A12(S04)3 • 18Н20, растворя¬
ют дистиллированной водой и переливают в мерную колбу на 1000мл. Раствор доводят
дистиллированной водой до метки, перемешивают и переливают в бутыль для хранения.
Приготовление рабочего раствора. Цилиндром отмеривают 20мл маточного
раствора, переносят в мерную колбу на 1000мл. Объем доводят дистиллированной
водой до метки и перемешивают. Раствор используется для анализа.
2.8.4.2. Ход анализа
См. п.2.8.4. К навеске мезги 13 или 6,5г, взвешенной с точностью до 0,1г и пере¬
несенной в дигестионный стакан, приливается из бюретки или дозатором соответ¬
ственно 89,1 или 44,5мл рабочего раствора свинцового уксуса или вышеуказанных
осветителей. Хорошо несколько раз перемешивается и отстаивается 20-30мин. Затем
проба фильтруется в чистый сухой стакан через бумажный фильтр, первые порции
фильтрата отбрасываются, а полученный прозрачный фильтрат поляризуется в кюве¬
те 400 или трубке длиной 400мм. Полученный показатель - выражение содержания
сахара в свекле в % при холодной дигестии.
2.8.5. Обработка результатов
При расчете кондуктометрической золы вводится поправка на электропроводность
воды, которая применяется для дегерирования мезги свеклы. С этой целью перед на¬
чалом работы проверяют электропроводность воды и вычитают ее значение из заме¬
ров электропроводности испытуемых растворов. Полученная электропроводность яв¬
ляется исходной величиной при расчете содержания кондуктрометрической золы, ко¬
торое определяется по следующей формуле:
_ „ 1
% золы к массе свеклы — ^ • — 657,
где С - проверенная константа измерительной ячейки прибора;
1
— - электропроводность испытуемого раствора в сименсах, за вычетом
R электропроводности воды;
657 - эмпирический коэффициент перевода электропроводности в золу
для нормального раствора дигерата.
Измерение электропроводности желательно проводить при температуре смеси
близкой к 20°С. При отклонении от указанной температуры производится корректи¬
ровка количества золы (табл. 18). 1
Пример расчета. С = 0,774; — воды = 12 микросименс = 0,000012 сименс;
-61 -
— испытуемого раствора = 1,48 милисименс = 0,00148 сименс; — испытуемого
R R
раствора с поправкой на воду = 0,00148 - 0,000012 = 0,001468 сименса ;
% золы = 0,774 • 0,001468 • 657 = 0,746% при температуре 22°С; % золы с учетом
поправки на температуру = 0,746 - 0,031 = 0,715%.
При использовании портативного кондуктометра типа №-571 (Польша) расчет со¬
держания растворимой золы упрощается, так как константа измерительной ячейки при¬
бора и температура испытуемого раствора учитывается при снятии показаний прибора.
Таблица 18.
Поправки на температуру при определении золы по электропроводности х)
Температура, 0 С
Постоянная я*
«ейки
0,5
0,6
0,7 |
1 о.» 1
| 0,9
1,0
1,1
Содержание золы в пробе, %
20,2 и 19,8
0,002
0,002
0,003
0,003
0,004
0,004
0,005
20,4 и 19,8
0,004
0,005
0,006
0,007
0,008
0,009
0,010
20,6 и 19,4
1,007
0,006
0,009
0,100
0,012
0,013
0,014
20,8 и 19,2
0,009
0,010
0,012
0,014
0,016
0,018
0,020
21,0 и 19,0
0,011
0,013
0,015
0,017
0,020
0,022
0,024
21,2 и 18,8
0,013
0,015
0,018
0,021
0,024
0,026
0,029
21,4 и 18,6
0,015
0,018
0,021
0,024
0,028
0,031
0,034
21,6 и 18,4
0,017
0,020
0,024
0,028
0,032
0,035
0,039
21,8 и 18,2
0,019
0,023
0,027
0,031
0,036
0,039
0,044
22,0 и 18,0
0,022
0,026
0,031
0,035
0,039
0,044
0,048
22,2 и 17,8
0,024
0,029
0,035
0,039
0,043
0,048
0,053
22,4 и 17,6
0,027
0,031
0,036
0,042
0,047
0,052
0,057
22,6 и 17,4
0,029
0,034
0,039
0,045
0,051
0,056
0,062
22,8 и 17,2
0,031
0,036
0,042
0,048
0,055
0,061
0,067
23,0 и 17,0
0,033
0,039
0,046
0,052
0,059
0,066
0,072
23,2 и 16,8
0,035
0,042
0,049
0,056
0,063
0,070
0,077
23,4 и 16,6
0,037
0,045
0,052
0,060
0,067
0,074
0,082
23,6 и 16,4
0,039
0,047
0,055
0,063
0,071
0,079
0,087
23,8 и 16,2
0,041
0,049
0,058
0,067
0,075
0,084
0,092
24,8 и 16,0
0,044
0,052
0,062
0,070
0,078
0,087
0,096
24,2 и 15,8
0,046
0,055
0,065
0,074
0,082
0,092
0,101
24,4 и 15,6
0,048
0,057
0,068
0,077
0,086
0,096
0,106
24,6 и 15,4
0,050
0,060
0,071
0,081
0,090
0,100
0,111
24,8 и 15,2
0,052
0,062
0,074
0,084
0,094
0,105
0,116
25,0 и 15,0
0,055
0,065
0,077
0,087
0,098
0,109
0,120
1} попран» (фя лмкрпуре выше 20° С должны вычитаться, а при температуре ниже 20* С - прибавляться.
-62-
Имея показатели содержания кондуктометрической золы в корнеплоде и са¬
харистости, выраженные в % к массе свеклы, получают расчетные величины, ха¬
рактеризующие технологические качества свеклы (потери сахара в мелласе, выход
сахара, выход мелласы, коэффициент завода и МБ-фактор):
1. Потери сахара в мелласе (ПМ) в % к массе свеклы
ПМ = 3,76 • Р,
где 3,76 - переводной коэффициент;
Р - содержание кондукгрометрической золы, %.
2. Расчетный выход сахара (Б) к массе свеклы
Б = Д - 0,9 - ПМ,
где Д - сахаристость свеклы, %;
0,9 - потери сахара в производстве до мелласы в % к массе свеклы.
3. Коэффициент завода (КЗ) - количество сахара, которое будет получено из 100кг
сахара, содержащегося в свекле,
Б
КЗ = — • 100,
4. Выход мелласы (М)
М = 7,5 • Р
где 7,5 - эмпирический коэффициент.
5. Величина МБ-фактора (количество мелласы, приходящейся на 100кг вырабо¬
танного белого сахара) М
МБ = —100,
Б
где М - расчетный выход мелласы в % к массе свеклы.
2.8.6. Технологическая карта
Получение мезги
мезгообразователь
(групповой или инди¬
видуальный)
Взятие навески, получение дигепата
автоматический дозатор или весы типа BJ1KT-500
и автоматическая пипетка кратная 44,5 мл; стака¬
ны на 250-300мл; 10-позиционные кассеты
Взбалтывание раствора
ротатор
Определение золы
кондуктометр
Осветление лигерята
пипетка или дозатор
на 1 мл
Фильтрование
воронки пластмассовые, фильтры, стаканы или
10-позиционные кассеты
Определение гдтяря
поляриметр
2.9. ОПРЕДЕЛЕНИЕ ДОБРОКАЧЕСТВЕННОСТИ (ЧИСТОТЫ)
ОЧИЩЕННОГО НОРМАЛЬНОГО СОКА САХАРНОЙ СВЕКЛЫ
МЕТОДОМ СИЛИНА
2.9.1. Принцип метода
Сок, выжатый из средней измельченной пробы корнеплодов, с помощью пресса,
-63-
очищают известковым молоком и нейтрализуют углекислым газом. В очищенном
нормальном соке содержание сухого вещества определяется рефрактометром, со¬
держание сахара - поляриметрическим методом. По данным содержания сухого ве¬
щества и сахара в очищенном соке определяют доброкачественность (чистоту) сока.
2.9.2. Технологическое оборудование
Весы типа ВЖТ.
Ручной пресс.
Поляриметр-сахариметр.
Рефрактометр.
Цилиндр на 100мл.
Пипетки на 100, 25,10, 5, 2мл.
Колбы на 150-200мл.
Стаканы на 100мл.
2.9.3. Реактивы
1. Известковое молоко. 5г СаО растворить в 100мл дистиллированной воды, пе¬
ред использованием взбалтывать.
2. 1н. раствор уксусной кислоты. 60мл ледяной уксусной кислоты плотностью
1,05г/см3 довести дистиллированной водой до 1 литра.
3. Аиетат свиниа. 600т ацетата свинца (РЬ(СН300)2 * ЗН20) растирают в ступке
с 200г оксида свинца (РЬО) до образования однородной массы. Пересыпают в кони¬
ческую колбу на 2л, добавляют 100мл дистиллированной воды и помещают на кипя¬
щую водяную баню и нагревают колбу с обратным холодильником 6-8 часов до про¬
светления смеси и перехода красновато-желтого цвета в белый или бледно-розовый.
Перемешивают содержимое колбы через каждые 20-30мин. Затем содержимое колбы
охлаждают и доводят дистиллированной водой до метки. Дают отстояться в течение
суток в темном месте до просветления.
Раствор свинцового уксуса должен иметь сильную щелочную реакцию и плот¬
ность 1,23-1,24г/см3, реагировать на лакмус как сильная щелочь, но не давать окраши¬
вания с фенолфталеином.
Или способ приготовления со ссылкой на п.2.8.3.
4.Фенолфталеиновая бумага. 3,5г лимонной кислоты и 1,5г фенолфталеина, пере¬
носят в мерную колбу на 1л, растворяют в 50% этиловом спирте и доводят объем до
метки. Этим раствором пропитывают фильтровальную бумагу, высушивают на воздухе
и разрезают на полоски. Показатель щелочности должен соответствовать 0,07% СаО.
2.9.4. Ход анализа
Для анализа отбирают не менее 40 корнеплодов. Целесообразнее корнеплоды ото¬
брать непосредственно перед уборкой урожая. С этой целью натягивают шнур попе¬
рек рядков или по диагонали делянки и отбирают только те корнеплоды, которые на¬
ходятся непосредственно под шнуром. Если шнур непосредственно на растение не
попадает, то берется ближний к шнуру корнеплод и всегда с одной и той же стороны.
Из корнеплодов средней пробы (40 штук) вырезаются сегменты вдоль вертикаль¬
ной оси (V4 - V8 часть корнеплода) таким образом чтобы в лабораторную пробу вошли
все части корнеплода (головка, шейка, собственно корнеплод и хвостовая часть). Про¬
бу измельчают на механической терке и тщательно перемешивают. Огбирают 1 кг мезги-
кашки, заворачивают в сухую салфетку из плотной фильтрующей ткани и выжима¬
-64-
ют сок ручным винтовым прессом. Из каждой пробы сок выжимается при одина¬
ковом количестве оборотов рукоятки пресса. Пену из сока удаляют с помощью
часового стекла или ложки. Для удаления пузырьков воздуха сок энергично пере¬
мешивается стеклянной палочкой и оставляется в покое на 30-40 минут.
Для очистки в два стакана набирают пипеткой или цилиндром по 100мл сока и
нагревают до кипения. В нагретый сок приливают в два приема по 10мл известковог о
молока. Первый раз 10мл вливается медленно по каплям на протяжении 2 минут при
энергичном помешивании сока, а вторые 10мл известкового молока приливают быст¬
ро, энергично перемешивая раствор. Затем сок снова нагревают до кипения и быстро
фильтруют в горячем состоянии в колбу емкостью 150-200мл.
Фильтрат после охлаждения в той же колбе насыщают углекислым газом до нейт¬
ральной реакции, проверяя конец нейтрализации фенолфталеиновой бумажкой. Кап¬
ля сока, нанесенная на фенол(|ггалеиновую бумажку стеклянной палочкой должна да¬
вать слабо-розовое, но не малинового цвета окрашивание. Пробу проводят в конце
нейтрализации, когда раствор в колбе помутнеет вследствие образования карбоната
кальция.
Раствор с осадком карбоната кальция медленно кипятят 5 минут для разложения
гидрокарбонатов. Уровень раствора в колбе фиксируется восковым карандашом. Пос¬
ле окончания кипячения уровень жидкости в колбе доводится до метки дистиллиро¬
ванной водой. Затем раствор быстро фильтруют в горячем состоянии через складча¬
тый фильтр. В очищенном таким образом соке определяется содержание сухих ве¬
ществ и сахара.
Определение сухого вещества. Сухое вещество в соке определяется рефракто¬
метрически.
2.9.5. Технологическая карта
Очистка сока
стаканы, цилиндр на
100мл, пипетки на 10 мл
Нагрев и (Ьильтрование
Насыщение Фильтрата СО
эл.плитка, колбы на 150,^ фенолфталеиновая бумага,
200мл, воронки, фильтры стекл.палочки
Кипячение и фильтрование
эл.плитка, складчатый фильтр
Определение сухого вещества
рефрактометр
2.9.6.0пределение сахара в соке
В оттарированный стакан или фарфоровую чашку отвешивают 26г сока. Вначале
пипеткой вливают 25мл сока, а затем по каплям добавляют сок до 26г. затем переносят
сок в мерную колбу на 100мл, смывая чашку или стакан водой до 3/4 объема колбы. К
соку приливают 1-2- капли фенолфталеина и по каплям 1н раствор уксусной кислоты
с целью нейтрализации до исчезновения малинового цвета окрашивания. После этого
приливают 2мл свинцового уксуса, доводят объем раствора водой до метки, хорошо
перемешивают и сразу же фильтруют. Прозрачный фильтрат поляризуют с помощью
сахариметра в кювете на 200мм. Одно деление сахариметра при этих условиях соот¬
ветствует 1% сахара.
- 65 -
2.9.7. Технологическая карта.
Взятие навесок
Взбалтывание
стаканы, колбы на 100мл,
пипетки на 25мл
ШШ
ротатор
Фильтрование
воронки, фильтры,
стаканы
Поляоиза-
шш
сахариметр
2.9.8. Обработка результатов
Чистота очищенного нормального сока (ч) вычисляется по формуле, %:
а 100
4 = ,
с
где а - содержание сахара в соке, %;
с - содержание сухого вещества в соке, %;
100 - коэффициент для пересчета в %.
По данным чистоты сока и содержания сахара в корнеплодах рассчитывается
ожидаемый технологический выход сахара (т) по формуле, %
Т=(Д-0,9)(1-^^ т ) ;
ч
где Д - содержание сахара в корнеплодах, %;
0,9 - потери сахара в производстве от массы корнеплодов;
ч - чистота сока;
т - мелясообразуклций коэффициент.
Мелясообразующий коэффициент в лабораторных условиях определяется по
содержанию калийно-натриевой золы в соке или по чистоте мелассы по формуле
д
т= 100—^ ’
где Д - чистота мелассы.
Если Д мелассы равна 58,0, то в 100кг сухого вещества ее содержится 58,0кг
сахара и 100 - 58,0 = 42,0кг несахаров, т.е. мелассообразующий коэффициент равен
58.0 _ ,
2.9.10. Дигестионный сок и свекла
Нормальную (26,026г) или полунормальную (13,013г) навеску свекловичной кашки
дигерируют с водой при 80°, как при определении сахара в свекле, но не прибавляя
свинцового уксуса; по охлаждении доливают до метки 100мл и фильтруют. Получаем
дигестионный сок свеклы. Определяем его электропроводность \, кондуктометри-
ческим методом. Зная \ можем определить содержание золы (углекислой) в дигес-
тионном соке в процентах по весу его, умножая электропроводность на 98. Количе¬
ство золы равно \* 98%. Это в том случае, когда для получения дигестионного сока
брали 26,026г свеклы на 100мл.
Если же было взято 13,013 г свеклы на 100 мл, то количество золы равно Х84,5%.
ГЬ Адигестионного сока можно определить также количество золы в процентах по
весу свеклы; оно при навеске 26,026 г будет А.373 %, а при навеске 13,013 г - X* 657 %.
-66-
2.9.11. Технологическая карта определения
электропроводности в соке
2.10. ОПРЕДЕЛЕНИЕ ПЕКТИНОВЫХ ВЕЩЕСТВ
ПО ПЕКТАТУ КАЛЬЦИЯ
2.10.1. Принцип метода
Этот метод основан на переведении различных пектиновых веществ в раствор,
превращении их в пектиновую кислоту, осаждении последней в виде кальциевой соли
и учете ее весовым способом.
В зависимости от цели исследования можно определять отдельно растворимый
пектин и протопектин или учитывать сумму пектиновых веществ. Ход анализа остает¬
ся одним и тем же, с тем лишь отличием, что в первом случае осаждают и учитывают
пектиновую кислоту отдельно в двух экстрактах, а во втором - экстракты соединяют и
определяют сумму пектиновых веществ.
2.10.2. Технологическое оборудование
Сушильный шкаф.
Центрифуга.
Механическая качалка.
Мерные и конические колбы на 250мл.
2.10.3. Реактивы
1. 0,3н. раствор соляной кислоты.
2. 1% раствор лимоннокислого аммония.
3. 0,4% раствор едкого натра.
4. 6% раствор уксусной кислоты.
5. 0,5% и 11,1% растворы хлористого кальция.
2.10.4. Ход анализа.
Раздельное определение пектиновых веществ. Навеску 25г свежего материала
тщательно растирают с измельченным стеклом в ступке до совершенно однородной
массы и трижды извлекают сахара спиртом.* Остаток заливают в конической колбе
100мл воды, нагретой до 45°С, выдерживают при этой температуре 30 минут (на водя¬
ной бане). Лучше остаток после извлечения поместить в центрифужную пробирку на
100 мл, прилить воды, нагретой до 40-45° и, закрыв резиновой пробкой, энергично
взбалтывать пробирку на электрической качалке или руками 15-20 минут. Через 20
минут пробирки уравновешивают и центрифугируют, жидкость сливают через бумаж¬
ный складчатый фильтр в мерную колбу на 250мл. Остаток в центрифужной пробирке
еще дважды декантируют водой (75 и 60мл) и все экстракты, соединенные в мерной
колбе, доводят до метки (если раствор мутный, его еще раз центрифугируют и фильт¬
руют). Таким образом получают вытяжку №1 воднорастворимых пектинов.
-67-
Затем весь остаток из центрифужных пробирок с 50мл 0,3н. раствора соляной
кислоты переносят в коническую колбу. Закрывают колбу пробкой с вертикально по¬
ставленным обратным холодильником и нагревают 30 мин. на кипящей водяной бане.
После этого жидкость фильтруют через складчатый фильтр в мерную колбу на 500мл,
остаток несколько раз промывают на фильтре горячей водой, сливая в ту же колбу.
Затем фильтр вместе с остатком переносят палочкой в экстракционную колбу, залива¬
ют 50-70мл 1% раствора лимоннокислого аммония и также нагревают в кипящей бане
30 мин. Экстракт фильтруют в ту же мерную колбу, промывают горячей водой и после
охлаждения доводят до метки. Полученная вытяжка №2 содержит нерастворимый в
воде протопектин и пектиновую кислоту.
В дальнейшем весь ход анализа в обеих вытяжках совершенно одинаков.
*В навесках свежего материала без предварительной обработки спиртом и отделения спиртоне¬
растворимого остатка можно определять пектин только при массовых анализах. Во всех остальных
случаях обязательно полное извлечение сахаров (3-кратное: 1 раз 95%-ным и дважды - 80%-ным
спиртом), затем промывание ацетоном и подсушивание на воздухе.
Омыление пектиновых веществ. К 50 или 100мл вытяжки №1, в зависимости от
разбавления и от ожидаемого количества пектина прибавляют 50 или 100мл 0,4%-
ного едкого натра и оставляют на ночь при комнатной температуре. Утром раствор
подкисляют тем же объемом (50 или 100мл) нормальной уксусной кислоты и осажда¬
ют пектиновую кислоту 50 (или 100)мл. 11,1%-ного раствора хлористого кальция.
Полученный осадок пектата кальция отфильтровывают через заранее высушенный до
постоянного веса фильтр. Затем осадок на фильтре промывают 0,5%-ным раствором
хлористого кальция, после этого многократно промывают холодной дистиллирован¬
ной водой для удаления хлористого кальция (проверяют пробой на хлор с азотнокис¬
лым серебром). Под конец промывают несколько раз горячей водой для удаления со¬
лей. Фильтр с осадком переносят (пинцетом) в бюкс и сушат до постоянного веса при
100-105°. Вес осадка, полученный из разности между весом стакана и осадком пекти¬
на после сушки и весом их же без пектина (до фильтрования осадка), умножают на
0,9235 и тем самым переводят на пектиновую кислоту. Вес осадка пектата не должен
превышать 0,03г. Если осадка больше, то его не удается высушить, и результаты полу¬
чаются чрезмерно повышенными. Учитывают разбавление и вычисляют процентное
соотношение пектина.
С вытяжкой №2 протопектина поступают совершенно также, но ее нейтрализуют
едким натром до прибавления щелочи, потребной для омыления.
Определение пектинов длительно, хотя и несложно. Целесообразно проводить
одновременно анализ трех образцов, если растворимый пектин учитывают отдельно,
или 6 образцов, если определяют только общее количество пектиновых веществ. За
день до анализа подготовляют складчатые фильтры из плотной фильтровальной бума¬
ги, на краях фильтров надписывают простым карандашом тот же номер, который име¬
ет и бюкс. Фильтры должны быть предварительно высушены до постоянного веса при
105° (6-8 часов сушки). Фильтры с осадком пектата кальция также высушивают до
постоянного веса при 105°. Если вес осадка пектата превышает 0,03г анализ надо по¬
вторить, взяв, например, вместо 100 мл вытяжки 50мл.
Пример вычисления. Навеска сырой моркови 25г, водная вытяжка доведена до
250мл. Для определения взяты пробы по 50мл, вес осадков пектата кальция в них
0,030г и 0,026г, в среднем 0,028г. В навеске 25г содержится 0,1405г пектата кальция,
или 0,562% на сырой вес и 4,95% на сухой вес (при содержании сухого вещества 11,8%).
Эту величину умножают на 0,9235 для перевода пектата кальция на пектиновую кис¬
лоту: 4,59 • 0,9235 = 4,24%. Аналогично вычисляют протопектин.
-68-
2.10.4.1. Технологическая карта
2.10.5. ОПРЕДЕЛЕНИЕ ПЕКТИНОВЫХ ВЕЩЕСТВ
ОБЪЕМНЫМ МЕТОДОМ (по С.Я.Раик)
2.10.5.1. Ход анализа
Измельченную навеску 10-15г свежего материала заливают горячим этиловым
спиртом (из расчета получения конечной концентрации спирта 80-82%) и нагревают
на кипящей водяной бане (с воздушным холодильником) 20-30 минут для извлечения
сахаров, затем фильтруют через бумажный фильтр в мерную колбу. Извлечение спир¬
том повторяют 3-4 раза для полного удаления сахаров. Фильтр вместе с остатком под¬
сушивают при 50° до удаления спирта (по запаху). Затем остаток вместе с фильтром
заливают в колбу 50мл воды, нагретой до 45°, и при этой температуре экстрагируют
воднорастворимый пектин на водяной бане в течение часа. Отфильтровывают в мер¬
ную колбу на 200мл, промывают водой и, охладив, доводят объем до метки. Для из¬
влечения нерастворимых фракций пектина остаток переносят в экстракционную кол¬
бу, заливают 50мл 0,3н. НС1 и нагревают полчаса в кипящей водяной бане с обратным
воздушным холодильником. Фильтруют в мерную колбу на 200-250мл и промывают 2-
3 раза горячей водой. Фильтр вместе с осадком возвращают в ту же колбу, перенося
количественно посредством 50мл 1%-ного раствора лимоннокислого аммония, и ста¬
вят в кипящую водяную баню на полчаса. Фильтруют в ту же мерную колбу, где нахо¬
дится фильтрат от соляно-кислой вытяжки, промывают горячей водой, после охлаж¬
дения доводят до метки. Из обоих экстрактов берут по 2 пробы по 50мл в конические
колбы на 250мл. Затем для омыления воднорастворимого пектина приливают в каж¬
дую колбу по 50мл 0,1н. раствора NaOH, а в экстракт нерастворимого пектина - еще
дополнительно столько щелочи, сколько требуется для нейтрализации соляной кисло¬
ты (установить отдельно). Последние остатки эфирных связей опыляются с трудом.
Это омыление необходимо проводить не менее 3-4 часов. Раствор удобно оставлять
на ночь, потому что все последующие операции, включая растворение осадка, следует
закончить в один день. После омыления во все пробы добавляют по 50мл 1н. раствора
уксусной кислоты и через несколько минут - по 50мл 5%-ного раствора медного купо¬
роса. Раствор с осадком фильтруют спустя 30-40 минут через беззольный фильтр (бе¬
лая или красная лента). Осадок тщательно промывают горячей водой до исчезновения
в промывной воде реакции на медь.
Затем осадок вместе с фильтром помещают в колбу для титрования, заливают 30-
40мл горячей воды и добавляют несколько капель аммиака. Раствор приобретает си¬
неватый оттенок из-за окраски комплекса аммиака та меди. Затем приливают 8-10мл
-69-
2н. раствора H2S04, добавляют 5г KI и титруют из микробюретки раствором 0,01 и.
гипосульфита в присутствии свежеприготовленного крахмала. Восстановление необ¬
ходимо проводить в слабокислой среде, так как высокая кислотность приводит к за¬
метному окислению ионов йода кислородом воздуха Однако при слишком малой кис¬
лотности реакция 2Си + 4Г = 2CuI + 12 сильно замедляется и конечная точка титрова¬
ния становится неясной - йодкрахмальная окраска после окончания титрования очень
быстро появляется вновь, что указывает на возобновление выделения йода.
Вычисление проводят по формуле:
V К Т100
% Си = .
н
где v - число куб.сантиметров Na2S203, пошедшее на титрование;
К - поправочный коэффициент к титру гипосульфита;
Т - титр Na2S203 (по меди равен 0,06357 • N);
и - навеска в титруемом объеме (учетное разведение).
При К= 0,01, Т= 0,0006357. Пектат-Са (%) = (% Си х 6,5).
2.11. ОПРЕДЕЛЕНИЕ КРАХМАЛА
ПОЛЯРИМЕТРИЧЕСКИМ МЕТОДОМ
2.11.1. Принцип метода
состоит в гидролизе крахмала и определения в гидролизате угла вращения. Удель¬
ное вращение [6]D гидролизатов крахмала, полученного из семян различных культур
при концентрации крахмала в растворе около 2 % (как показали исследования Н.А.-
Бондаренко и В.А.Смирнова), имеют близкие значения [6]D, равные 181.
2.11.2. Технологическое оборудование
Мерные колбы на 50 или 100мл.
Поляриметр (сахариметр) СУ-5.
Торсионные весы на 5г.
Штатив или приспособление для встряхивания мерных колб.
2.11.3. Реактизы
1. 1%-ный и 5%-ный растворы НС1;
2.5%-ный раствор фосфорновольфрамовой кислоты или раствор железистоси¬
неродистого цинка (последний готовят из 30%-ного раствора сернокислого цинка и
15%-ного раствора желтой кровяной соли; при осаждении белков и других коллоидов
вначале приливают 1-2мл раствора сернокислого цинка, а затем, после встряхивания,
прибавляют столько же раствора желтой кровяной соли).
2.11.4. Ход определения крахмала в семенах.
В основе анализа лежит метод Эверса. Отвешивают навески по 3±0,001г тонко
измельченных семян и пересыпают в мерные колбы на 100мл, приливают по 12,5мл
1%-ной соляной кислоты. Хорошо перемешав, приливают еще 12,5мл этого же ра¬
створа и колбы нагревают в бурно кипящей водяной бане в течение 15 минут. Во
время кипячения содержимое следует систематически перемешивать, для чего при
проведении серии анализов удобно пользоваться установкой для экстракции и
гидролиза. Сначала содержимое колбы загустевает за счет клейстеризации крахмала,
-70-
а затем разжижается. По истечении времени гидролиза приливают 30мл воды и
колбам дают остыть. Затем приливают 5мл раствора 5%-ной фосфорновольфрамовой
кислоты и после взбалтывания доводят объем водой до метки. На этом этапе анализ
можно прервать на несколько часов или оставить в холодильнике до утра.
Экстракт перед определением вращения фильтруют в сухую колбу. Прозрачный бес¬
цветный фильтрат наливают в поляризационную трубку (обращать внимание на ее дли¬
ну) и определяют угол вращения в поляриметре. Условия гидролиза крахмала (навески,
разбавление, кипение бани, взбалтывание) должны быть одинаковыми для всех проб.
Содержание крахмала в процентах (х) вычисляют по формуле:
100 а >100 -0,3465
1811-н
где а- угол вращения, найденный при отсчете в поляриметре
(если поляриметр с прямой шкалой, то полученные данные
переводят в градусы круговой шкалы);
181 - фактор для крахмала, соответствующий [6]D;
L - длина трубки (в дм);
н - навеска;
100 - объем экстракта.
Пример вычисления. Навеска муки риса была 3,0±0,001г, угол вращения (б)
по сахариметру СУ-5 = 35,8°, L трубки = 2. После подставления значений в формулу
получают содержание крахмала в процентах (68%).
2.11.4.1. Технологическая карта.
2.11.5. ОПРЕДЕЛЕНИЕ КРАХМАЛА В КАРТОФЕЛЕ
И ДРУГИХ КРАХМАЛОНАКОПИТЕЛЯХ
Наряду с крахмалом в клубнях картофеля содержится до 1% сахаров, поэтому при
точных анализах сахар следует извлечь концентрированным спиртом, а остаток упот¬
ребить для определения крахмала. Сахар определяют одним из соответствующих ме¬
тодов. Для сравнительной оценки сортов в зрелых клубнях картофеля крахмал опре¬
деляют часто вместе с сахаром.
Из отобранной и размельченной на измельчителе тканей средней пробы клубней
отвешивают 2 навески по 15±0,01г и растирают дополнительно в ступке с 5мл 5%-ной
соляной кислоты до однородной массы, затем переносят в мерную колбу на 100мл и
добавляют 25 мл 1%-ной соляной кислоты, одновременно ополаскивая ступку. После
этого колбы укрепляют в штативе и погружают в кипящую водяную баню на 15 минут.
В дальнейшем поступают так, как указано в прописи определения крахмала в муке семян.
2.11.6. ПРИБЛИЖЕННОЕ ОПРЕДЕЛЕНИЕ КРАХМАЛА
В МЯКОТИ ПЛОДОВ И В КОРНЕПЛОДАХ
После размельчения проб в мясорубке или кухонном комбайне отвешивают на¬
-71 -
вески по 10±0,01г мякоти тыквы (для петрушки, сельдерея, брюквы навески следует
увеличить), помещают в сосуд измельчителя, приливают туда же 9мл воды и раз¬
мельчают в течение 5 минут с перерывами. Полученную суспензию переносят в колбу
на 200мл для клейстеризации крахмала, кипятят 5 минут с обратным холодильником.
После охлаждения набирают 5 мл «болтушки» в пробирку и прибавляют туда 3 капли
0,3%-ного раствора йода. Интенсивность появившейся окраски зависит от содержа¬
ния и состава крахмала, а также от количества раствора йода и взвешенных частиц
мякоти. Ввиду этого необходимо сохранять единообразие в приемах анализа. Образ¬
цы, содержащие в свежей мякоти около 1% крахмала, дают светло-голубую окраску,
2% - светло-синюю, 2,5% - синее окрашивание, 3,5% - темно-синее окрашивание.
Окраска образцов, содержащих более 4,5% крахмала, трудно различима на глаз. Для
приближенного количественного определения следует составить шкалу, пользуясь
навеской (Юг) корнеплодов, не содержащих крахмала (морковь, свекла сахарная), к
которой добавляют препарат крахмала из такого расчета, чтобы иметь шкалу, соот¬
ветствующую содержанию крахмала на сырой вес от 1 до 7%, с интервалом в 0,5%.
Этот метод может быть использован для отбора корнеплодов на повышение крахма¬
листости.
2.12. ОПРЕДЕЛЕНИЕ КЛЕТЧАТКИ
2.12.1.1. Определение клетчатки по Кюршнеру и Гапеку
Проводимая модификация может быть рекомендована в качестве универсального
метода. Последний основан на окислении, разрушении и растворении различных хи¬
мических соединений, входящих в состав растений, смесью уксусной и азотной кислот.
При этом клетчатка практически не растворяется, отфильтровывается и взвешивается.
2.12.1.2.Технологическое оборудование
Колбы на 100-120мл с притертыми холодильными трубками.
Тигли Гуча или стеклянные тигли с пористым дном №2.
Песчаная баня.
2.12.1.3. Реактивы
1. Смесь по объему (1:10) азотной кислоты (плоти. 1,4г/см3) и 80%-ной уксусной кислоты.
2. Этиловый эфир и спирт.
2.12.1.4. Ход анализа
Отвешивают навески около 1±0,0002г крупно измельченных семян в колбы на
120мл, приливают 40мл смеси кислот и, закрыв колбу обратным холодильником, на¬
гревают на песчаной бане в течение часа. Затем отфильтровывают белый, слегка кре¬
мовый осадок через стеклянный тигель №2 или тигель Гуча с асбестовым фильтром.
Если осадок после отсасывания экстракта кремового цвета, то его промывают 1-2
раза горячей 0,2М спиртовой щелочью. После этого несколько раз промывают не¬
большими порциями дистиллированной воды и под конец - 10мл спирта с эфиром.
При этом следят, чтобы растворители хорошо отмыли стенки тиглей (при анали¬
зе масличных семян на них может остаться маслянистая жидкость). Тигли с чисто
белым осадком высушивают до постоянного веса при 105°.
При анализе кормовых и овощных растений навески сухого материала увеличи¬
вают до 1,5г и заливают 50мл смеси кислот и также нагревают в течение часа.
Процентное содержание клетчатки (х) вычисляют по формуле:
Ов. -в) 100
х =
н
-72-
ще: в - вес сухого (без осадка) тигля;
в;- вес тигля с сухим осадком;
н - навеска.
Если для определения клетчатки использован воздушно-сухой материал, то дела¬
ют пересчет на сухое вещество. Этим методом при серийных анализах один лаборант
выполняет 12 определений. А.В. Петербургский (1968) описывает модификацию мето¬
да Кюршнера и Ганека, по которой навеску 1 г кормовых растений нагревают на водяной
бане от 1 до 2 часов с 30мл смеси кислот (5 объемов азотной и 100 объемов 80%-ной
уксусной). Далее фильтруют, промывают водой, спиртом и эфиром. Следует отметить,
что здесь экономически нецелесообразно увеличивать расход уксусной кислоты.
Примечание. При использовании асбеста для приготовления фильтров его кипятят в смеси азот¬
ной и уксусной кислот (1:10) и затем промывают водой. После этого его употребляют для приготовле¬
ния фильтра.
2.12.1.5. Технологическая карта
2.12.2.0ПРЕДЕЛЕНИЕ КЛЕТЧАТКИ ПО МОДИФИЦИРОВАННОМУ
МЕТОДУ КЮРШНЕРА И ХАФЕРА
2.12.2.1. Принцип метода
Метод основан на окислительном разрушении и растворении различных веществ
(в том числе сопутствующих клетчатке) растворами азотной кислоты в спирте и спир¬
товой щелочи. Особенно ценен метод для определения клетчатки в различных веге¬
тативных органах, богатых гемицеллюлозами и лигнином.
2.12.2.2. Технологическое оборудование
Баня водяная.
Колбы с обратными холодильниками для экстракции и гидролиза.
Колбы конические на 200мл.
Тигли Гуча или стеклянные с пористой пластинкой №2.
2.12.2.3. Реактивы
1. Этиловый или метиловый спирт.
2. Смесь этилового спирта (4 объема) с 1 объемом азотной кислоты (плотн.1,4).
3. 0, ЗМ спиртовой раствор щелочи.
4. Эфир этиловый.
2.12.2.4. Ход анализа
Отвешивают по 1-3±0,001г из средней пробы крупно измельченных сухих веге¬
тативных частей растений в колбы на 200 - 250мл (величина навесок зависит от
содержания клетчатки в анализируемом материале).
В колбы с навесками приливают по 30 или 50мл смеси спирта и азотной кислоты.
Смесь готовят для серии образцов, приливая к спирту азотную кислоту при размеши¬
вании. Колбы с обратными холодильниками ставят на кипящую водяную баню на один
-73-
час. Колбы в воду не погружают, и кипение должно быть равномерным. Через час
коричнево-желтый раствор отстаивают и сливают через стаканчик с пористым дном.
Для обеспечения быстроты фильтрования, на дно стаканчика помещается слой высу¬
шенного и взвешенного песка (или мелко толченого стекла). При фильтровании следят
за тем, чтобы на фильтре был слой жидкости. Если анализируемый материал богат
белковыми веществами или содержит много сопутствующих клетчатке веществ, то
целесообразно, не перенося осадка из колбы, добавить еще реагирующей смеси и по¬
вторить нагревание в течение 30 минут с обратным холодильником на той же бане.
Жидкость приобретает лимонно-желтый или слабо-коричневый цвет (в зависимости
от оставшихся примесей). Когда жидкость вторично слита (через тот же стаканчик),
остаток в колбе промывают небольшими порциями крепкого спирта.
К промытому осадку в колбе прибавляют 50мл 0,ЗМ спиртового раствора щело¬
чи и нагревают вновь в течение часа на водяной бане при хорошем кипении или на
песочной бане. При отсасывании щелочного раствора белый осадок клетчатки пере¬
носят на фильтр того же тигля, промывают 2 раза порциями по 10мл горячей воды, а
затем 1-2 раза спиртом (спиртовые растворы собирают и отгоняют для следующих
анализов). Затем стеклянные тигли или тигли Гуча сушат до постоянного веса при
105° и после охлаждения взвешивают.
В некоторых случаях тиши необходимо промыть после спирта эфиром. При не¬
обходимости экономии спирта можно использовать водный раствор щелочи, но от-
фильтровывание водных растворов иногда очень осложняется.
Вычисление результатов производят по формуле:
(в,-в) 100
X 9
н
ще: в - вес сухого (без осадка) тигля;
ву- вес тигля с сухим осадком;
н - навеска.
2.12.2.5. Технологическая карта
2.12.3. ОПРЕДЕЛЕНИЕ КЛЕТЧАТКИ ПО X. Н. ПОЧИНКУ
Клетчатка - высокомолекулярный полисахарид (С6Н|0О5)п, образующий главную
составную часть оболочек растительных клеток. Чистая клетчатка - белое вещество без
вкуса и запаха, волокнистого строения. Она не растворима в воде, разбавленных
кислотах и щелочах, а также в органических растворителях. Кроме клетчатки в состав
клеточных стенок входят гемицеллюлозы, лигнин а также в малых количествах смолы,
жиры, дубильные и красящие вещества. При определении клетчатки указанные веще¬
ства необходимо удалить с целью получения ее в чистом виде.
-74-
2.123.1. Принцип метода
Исследуемое вещество обрабатывают смесью уксусной и азотной кислот при нагрева¬
нии. При этом растворяется межклеточное вещество и клетчатка распадается на отдельные
волокна. Происходит также удаление лигнина, гемицеллюлоз и других веществ. Идет гидро¬
лиз крахмала, пентозанов и др. веществ. После удаления примесей промыванием водой
клетчатка окисляется бихромагом калия в серно-кислой среде до углекислоты и воды:
С6Н]0О5 + 4^0^ + 16H2S04 = 6С02 + 4Cr,(S04)3 + 4K,S04 + 21 1^0
Избыток бихромата определяется титрованием солью Мора или йодометрически.
С6Нп05
Грамм-эквивалент клетчатки —^24— Равен 6,75.
2.123.2. Технологическое оборудование
Сушильный шкаф.
Лабораторная мельница.
Сито с отверстиями 1 мм2
Аналитические весы.
Центрифуга с пробирками на 15млсо стеклянной шариковой пробкой.
Стеклянные палочки.
Колбы конические на 250мл.
Бюретка на 25мл
Пипетка на 25мл.
Фарфоровые ступки с пестиком.
2.123.3. Реактивы
1. Смесьазотншиуксуснойкисшп.}^ 100мл 80%-ной уксусной кислшы берут 10мл азотной
кислоты (плотн. 1,4) хорошо перемешивают и сохраняют в склянке с притертой
стеклянной пробкой.
2. 0,5н. раствор бихромата калия в серной кислоте. 25г ±0,01 г KjCrjO., раство¬
ряют в 250мл дистиллированной воды в колбе емкостью не менее 2-х литров и по¬
степенно при охлаждении приливают 800мл концентрированной H2S04 (плотн. 1,84).
Тщательно перемешивают и после охлаждения переливают в склянку с притертой
стеклянной пробкой. Хранят в темном месте.
3. 0,1н. раствор соли Мора. 40г соли Мора растворяют в дистиллированной воде,
приливают 20мл концентрированной серной кислоты, разбавляют водой до 1л и пере¬
мешивают. Раствор готовят на неделю, проверив титр в день определения. Нормаль¬
ность раствора соли Мора устанавливают по 0,1н. раствору бихромата калия. Для это¬
го набирают пипеткой 25мл 0,1н. раствора К?Сг207, переносят в колбу для титрования,
прибавляют 5мл концентрированной серной кислоты, 3-5 капель раствора фенилант-
раниловой кислоты и титруют раствором соли Мора до перехода окраски из вишнево¬
фиолетовой в зеленую. Нормальность раствора соли Мора высчитывают по формуле:
25 ОД
к =
а
где К -нормальность раствора соли Мора
а - объем раствора соли Мора, затраченный на титрование бихромата
калия (в мл).
4.0,2% раствор фенилантраниловой кислоты. 0,1г фенил антраниловой кислоты (С6Н5
- NH - С6Н4СООН), растирают в ступке с 5мл 0,1 н. раствора NaOH, переносят раствор в
мерную колбу или цилиндр на 50мл, споласкивают (в цилиндр) ступку дистиллированной
водой. Раствор доводят до 50мл и перемешивают. Хранят в темной склянке.
-75-
2.123.4. Ход анализа
Исследуемый материал сушат, измельчают и просеивают через сито с отверстия¬
ми 1 мм2. Из полученного порошка берут навеску с таким расчетом, чтобы в ней нахо¬
дилось от 3 до 25мг чистой клетчатки (0,05г листьев и стеблей, 0,4г зерна). Навеску
взвешивают на аналитических весах, переносят в центрифужную пробирку и прили¬
вают смесь уксусной и азотной кислот. При навеске менее 0,1г берут 5мл смеси, а при
навеске выше 0,1 г берут 10мл смесь уксусной и азотной кислот. Пробирку закрывают
стеклянной шариковой пробкой с палочкой и помещают в кипящую воду на 25 минут,
периодически размешивая. После этого центрифугируют, прозрачный раствор осто¬
рожно сливают по палочке насколько это возможно, а к остатку приливают 10мл ди¬
стиллированной воды, перемешивают, центрифугируют и раствор сливают. Промы¬
вают водой 3 раза порциями по 10мл. К промытому осадку клетчатки приливают
пипеткой 10мл 0,5н. раствора бихромата калия в серной кислоте, перемешивая
стеклянной палочкой. Пробирку помещают в кипящую воду на 10 минут, периоди¬
чески помешивая содержимое. После этого охлаждают и раствор выливают в колбу
для титрования емкостью 250мл, смывая остаток 10-15мл дистиллированной воды.
Раствор охлаждают, прибавляют 5 капель 0,2%-ного раствора фенилантраниловой
кислоты и титруют 0,1н. раствором соли Мора до перехода вишнево-фиолетовой ок¬
раски в зеленую. Отдельно титруют 10мл 0,5н. раствора бихромата калия, разбавив
его 15мл дистиллированной воды и охладив. Можно остаток бихромата определить
йодометрическим методом так, как при определении крахмала. На основании полу¬
ченных результатов находят содержание клетчатки по формуле:
0,675 к (а-в)
х = ,
н
ще jc - содержание клетчатки, %;
к - нормальность титрованного раствора соли Мора;
а - объем 0,1 н. раствора соли Мора, затраченный при контрольном
титровании 10см3 бихромата, мл;
в - объем 0,1 н. раствора соли Мора, затраченный при определении
клетчатки, мл;
н - навеска исследуемого вещества, г;
0,675 - нормальный титр клетчатки, умноженный на сто.
2.123.5. Технологическая карта.
-76-
2.13. ОПРЕДЕЛЕНИЕ ОРГАНИЧЕСКИХ КИСЛОТ
2.13.1. ОРГАНИЧЕСКИЕ КИСЛОТЫ
И МЕТОДЫ ИХ ОПРЕДЕЛЕНИЯ
Органические кислоты растений разнообразны по своему строению, и некоторые
из них широко распространены в различных органах. Ряд кислот под влиянием фер¬
ментов находится во взаимных превращениях (цикл трикарбоновых кислот; взаимное
превращение щавелевой, глноксиловой и гликолевой кислот).
Органические кислоты являются продуктами превращения углеводов; при синте¬
зе белков они образуют углеродные скелеты аминокислот. Количественное содержа¬
ние органических кислот в растениях подвергается суточным и сезонным изменени¬
ям. Установлено, что различные виды и сорта растений имеют различие и по содержа¬
нию в них отдельных органических кислот.
Органическими кислотами, представляющими большой практический интерес,
являются лимонная, яблочная, винная, щавелевая, янтарная и др.. Извлечение этих
кислот из растений основано на хорошей растворимости их в воде, спирте и эфире.
Наиболее удобным способом для выделения органических кислот с целью последова¬
тельного количественного и качественного их изучения является экстракция этило¬
вым эфиром растительных веществ, подкисленных минеральными кислотами. Каче¬
ственное и количественное определение органических кислот основано не только на
рациональном использовании различных их химических свойств, но и на неодинако¬
вой подвижности их при хроматографировании на колонках, бумаге и в тонких слоях.
2.13.2. ОПРЕДЕЛЕНИЕ ЛИМОННОЙ КИСЛОТЫ
2.13.2.1. Принцип метода
Определение лимонной кислоты в плодах или листьях основано на окислении
лимонной кислоты перманганатом до ацетондикарбоновой кислоты и бромировании
этого продукта до образования пентабромацетона. Последний определяют весовым
или объемным методом.
2.13.2.2. Технологическое оборудование
Весы типа ВЛКТ.
Центрифуга.
Аппарат Сокслета.
Эксикатор.
Водяная баня.
Ступка с пестиком.
Мерный цилиндр на 250мл.
Мерные колбы на 200мл.
Пипетки градуированные на 5, 10, 20, 50мл.
Стеклянный тигель с пористым дном №2.
Бюретка.
2.13.2.3. Реактивы
1. 20%-ная серная кислота.
2. 20%-ный раствор бромистого калия.
-77-
3. 5%-ный раствор перманганата.
4. Насыщенный раствор сернокислого железа (40г в 100мл воды, подкисленной
серной кислотой).
5. 0,1 н. раствор едкого натра или едкого кали.
6. 5%-ный раствор фосфорновольфрамовой кислоты.
2.13.2.4. Ход анализа
Извлечение лимонной кислоты из сухих плодов, листьев. Навеску от 5 до 10 ±0,01 г
(в зависимости от количества лимонной кислоты в них), тщательно растирают в ступке
с добавлением толченого стекла, переносят в мерный цилиндр на 250мл и доводят
водой до 100мл. Затем прибавляют 15мл 20%-ной серной кислоты (1: 8). Оставляют
на 15-30 минут и прибавляют воды до 200мл. смесь хорошо встряхивают и оставляют
на 2 часа; во время настаивания неоднократно взбалтывают, после этого прибавляют
5-10мл 5%-ной фосфорновольфрамовой или метафосфорной кислоты, хорошо пере¬
мешивают и центрифугируют или фильтруют через сухой фильтр в сухую посуду.
Хорошим растворителем лимонной кислоты является ацетон (в 100 частях ра¬
створителя 4 части кислоты). Извлечение органических кислот ацетоном из сухого
материала проводят в аппаратах Сокслета. Ацетоновые вытяжки упрощают дальней¬
шие процедуры.
Окисление (Ьильтрата. Берут 50мл прозрачного фильтрата в колбу на 200мл и
добавляют 5мл 30%-ного раствора бромистого калия, 10мл разведенной серной кис¬
лоты (1:1) и после перемешивания быстро добавляют 20мл 5%-ного раствора пер¬
манганата. Смесь оставляют на 10 минут под вытяжкой, периодически взбалтывая
без подогревания, затем охлаждают колбу до температуры 10°. Если появившийся
вначале бурый осадок двуокиси марганца исчезает, то определение надо повторить с
прибавлением большего количества раствора перманганата.
Избыток окислителя удаляют прибавлением 20мл насыщенного раствора закис-
ного сернокислого железа до полного обесцвечивания. Смесь с образовавшимся в
этих условиях пентабромацетоном ставят в воду со льдом или в холодильник. На
этом этапе определение можно прервать до утра. За это время мутный раствор
постепенно осветляется, а осадок уплотняется на дне колбы.
Затем сливают жидкость с осадка декантацией через стеклянный тигель с порис¬
тым дном №2. Фильтр с осадком тщательно промывают до нейтральной реакции по
метилоранжу. Осадок пентабромацетона должен быть чисто белым или слабо окра¬
шенным.
Тигель с осадком высушивают в эксикаторе над серной кислотой, взвешивают и,
зная вес сухого тигля, по разности узнают вес осадка пентабромацетона (СНВг2 - СО
- СВг3), 1мг которого соответствует 0,483мг лимонной кислоты.
Количество пентабромацетона можно определить и объемным методом. В
этом случае промытый осадок с фильтра переносят в колбу на 200мл, растворяя в
20мл спирта, затем добавляют 50мл 0,1н. раствора щелочи. Колбу ставят на водяную
баню и нагревают 30 минут при температуре не выше 90° для полного растворения
осадка. Затем раствор охлаждают и избыток щелочи оттитровывают 0,1н. серной
кислотой в присутствии метилрота до светло-розового цвета.
Количество лимонной кислоты в процентах (х) вычисляют по формуле:
я-0,483 К 100
х = ,
н
-78-
ще: а - количество мл 0,1н. щелочи, связанной
в реакции с пентабромацетоном;
0,483 - количество мг лимонной кислоты, соответствующее
1мл 0,1 и. щелочи;
К - коэффициент разбавления (отношение объема экстракта к объему,
взятому для окисления);
н - навеска материала.
2.13.2.5. Технологическая карта
2.13.3. ОПРЕДЕЛЕНИЕ ЯБЛОЧНОЙ КИСЛОТЫ
2.133.1. Определение яблочной кислоты методом Ньючера и Викери
2.13.3.1.1. Принцип метода. Этот метод основан на получении бормопроизвод-
ного яблочной кислоты, которое перегоняют с водяным паром в раствор парадинитро-
фенилгидразина. В результате реакции с 2,4-динитрофенилгидразином выпадает нера¬
створимый в воде оранжево-желтый осадок - озазона. Последний растворяют в
пиридине и, прибавляя раствор щелочи, получают синее окрашивание. Интенсивность
последнего определяют на спектрофотометре или колориметре сравнением интенсив¬
ности окраски опытных растворов со стандартным раствором яблочной кислоты.
2.13.3.1.2. Технологическое оборудование
Весы типа BJIKT.
Фотоэлетроколориметр КФК-3 или микроколориметр.
Аппарат Сокслета.
Установка для перегонки продуктов окисления яблочной кислоты с водяным
паром на шлифах.
Пробирки с отводной трубкой.
Мерные колбы на 250, 100 и 50мл.
Стеклянный тигель с пористым дном №4.
Бюретки.
Колбы Кьельдаля.
2.13.3.1.3. Реактивы
1. Стандартный раствор яблочной кислоты.
2. Растворы серной кислоты 1:4 и 1:1.
3. 1 н. раствор бромистого калия (11,9г на 1л дистиллированной воды).
4. 1,5 н. раствор перманганата калия (47,4г на 1л).
-79-
5. 3%-ная перекись водорода.
6. Активированный уголь.
7. Петролейный эфир с температурой кипения 35-60°.
8. 5 н. раствор едкого натра (20г на 1л дистиллированной воды).
9. 2,4-динитрофенилгидразин.
10. Пиридин с температурой кипения 116°.
11. Этиловый (серный) эфир - очищенный и свежеперегнанный (см. приложение).
2.13.Э.1.4. Ход анализа
Извлечение кислот. Навеску 5±0,01 г сухого, тщательно измельченного материала
растирают в ступке с 4мл раствора серной кислоты (1:4) и оставляют стоять на ночь
(для перевода органических кислот в свободную форму). На следующий день добавляют
для обезвоживания 5г безводного сернокислого натрия и, тщательно растерев,
оставляют стоять на ночь. После этого сухой порошок анализируемого материала
количественно переносят в бумажный патрон и извлекают кислоты 20-кратным
объемом эфира, настаивая в широкогорлой колбе в течение суток. Затем патрон с
материалом помещают в экстрактор аппарата Сокслета, сливая эфирный настой туда
же, и продолжают извлечение в течение 20-24 часов. После этого эфирный экстракт
переливают целиком в делительную воронку на 250мл и взбалтывают с 100мл
дистиллированной воды (20-30 минут) до полного извлечения органических кислот.
Эфир при этом обесцвечивается, а водная вытяжка окрашивается, так как в воду, кроме
органических кислот, переходят пигменты и некоторые смолы. Водный слой из
делительной воронки сливают в мерную колбу на 250мл и доводят водой до метки.
Из этого исходного раствора берут в случае необходимости отдельно пробу для
определения лимонной кислоты.
Для определения яблочной кислоты берут 50мл этой водной вытяжки, осторож¬
но нейтрализуют 5 н. раствором едкого натра с фенолфталеином и количественно
переносят в мерную колбу на 100мл, куда добавляют 0,2г активированного животного
или древесного угля и доводят водой до метки. Сильно встряхивают 2 минуты и затем
фильтруют в сухую коническую колбочку.
Окисление яблочной и лимонной кислот. В конические колбы на 100мл берут по
25мл фильтрата и прибавляют по Змл серной кислоты (1:1), 2мл нормального раство¬
ра бромистого калия, а затем колбу ставят на водяную баню при температуре не выше
20-22° на 10 минут. После этого приливают 5мл 1,5н. раствора перманганата при
частом взбалтывании (реакцию следует проводить в вытяжном шкафу, так как при
этом выделяются пары брома). После окончания реакции колбу охлаждают до 5-10° и
добавляют по каплям охлажденной 3%-ной перекиси водорода до полного обесцве¬
чивания раствора. Избыток перекиси водорода разрушают, приливая по каплям слабый
раствор перманганата до появления бледно-розовой окраски.
Удаление пентабромацетона. При наличии в анализируемом материале лимон¬
ной кислоты она выпадает в виде пентабромацетона (мелкий белый осадок). Для
удаления последнего смесь обрабатывают петролейным эфиром. Для этого окислен¬
ную смесь вместе с осадком пентабромацетона переносят в делительную воронку на
100-200мл. Остатки в колбе смывают петролейным эфиром и также переносят в де¬
лительную воронку. Смесь взбалтывают в делительной воронке 5-6 раз, добавляя при
этом каждый раз по 5-10мл петролейного эфира. Промывание петролейным эфиром
заканчивают, когда осадок пентабромацетона полностью будет удален из раствора.
Всего для промывания должно быть использовано ЗО-бОмл петролейного эфира (в
зависимости от величины осадка).
Перегонка водяным паром. Промытый водный раствор, освобожденный от пен¬
табромацетона, переливают в колбу Кьельдаля и соединяют все части установки для
перегонки. (Колбу Кьельдаля во время перегонки тщательно отепляют или слегка по¬
догревают во избежание конденсации водяного пара). Остатки петролейного эфира
-80-
удаляют продуванием «грушей». Трубку холодильника погружают в раствор 2,4-ди-
нитрофенилгидразина , 25-30мл которого наливают в коническую колбу на 100мл.
Отгонку ведут при сильном нагреве до тех пор, пока в колбе останется 5мл. Воду
через холодильник пропускают только первые 5-10 минут, затем приток воды пре¬
кращают и перегонку продолжают. В приемнике образуется оранжево-желтый оса¬
док озазона - продукт реакции динитрофенилгидразина с окисленной яблочной кис¬
лотой.
После отгонки с паром приемник с оранжевым осадком отсоединяют от холо¬
дильника, предварительно обмыв трубку струей воды от приставших частиц осад¬
ка. Колбу с осадком охлаждают до комнатной температуры и осадок тщательно от¬
фильтровывают через стеклянный тигель с пористым дном (№4). Затем несколько
раз промывают водой, каждый раз ополаскивая приемник для полного переноса
оранжево-желтого осадка в тигель. Тигель с осадком сушат в течение 2-3 часов при
100-110°.
Растворение оранжево-желтого осадка. Сухой осадок в тигле растворяют горя¬
чим пиридином, отсасывая в пробирку с отводной трубкой (всю дальнейшую работу,
связанную с пиридином, лучше производить в вытяжном шкафу с хорошей тягой).
Пиридин нагревают до кипения, накрыв колбу часовым стеклом. В тигель с осадком
приливают 5-6мл горячего пиридина и отсасывают, повторяя эту процедуру 3 раза.
Затем пиридиновый раствор из пробирки переносят в мерную колбу на 25мл и туда же
смывают остатки пиридинового раствора в пробирке несколькими куб. сантиметрами
горячего пиридина. Раствор в мерной колбе охлаждают, доводят до метки пиридином
и перемешивают. Из этого объема берут 2-5мл раствора озазона в мерную колбу на
100мл, приливают 50мл воды и 10мл 5н. раствора едкого натра. Сразу же появляется
голубая или синяя окраска в зависимости от концентрации, которая не исчезает в тече¬
ние двух часов. Колбу доводят до метки и перемешивают.
Колориметрирование. Раствор с голубой или синей окраской фотометрируют,
применяя светофильтр, пропускающий свет с длиной волны 570нм. Результаты опре¬
делений находят по составленной калибровочной кривой или при использовании
колориметра, сравнивая со стандартным раствором яблочной кислоты.
При измерении колориметром концентрацию яблочной кислоты в процентах (х)
вычисляют по формуле:
К в а 100
х =
в\ н
ще К - концентрация яблочной кислоты в стандартном растворе;
в - толщина слоя стандартного раствора в колориметре при равной
интенсивности окраски;
в/ - толщина слоя исследуемого раствора в колориметре;
а - разбавление;
н - навеска.
При определении яблочной кислоты в свежем растительном материале не¬
обходимо к мезге или отжатому соку прибавить столько 4н. серной кислоты, чтобы
показатель pH соответствовал около 0,6-0,9, и затем обезводить сернокислым натрием.
Навеска и разбавление должны быть рассчитаны на содержание яблочной кислоты в
пробе от 0,2 до 2,5мг.
Пример вычисления. 5±0,005г сухих ягод черной смородины обработаны этиловым эфиром.
Из этого экстраета кислоты переведены в водную вытяжку и объем доведен до 250мл. Отсюда взято 50мл
для окисления. Из этого объема яблочная кислота переведена в производное с динитрофенилгидразином.
Желтый осадок - озазона растворен в 25мл пиридина, из этого объема взято 5мл, разбавлено до 100мл и
окрашенный раствор сравнен в колориметре со стандартным раствором яблочной кислоты, в котором
содержится 2мг яблочной кислоты. Толщина слоя стандартного раствора (в/) - 30мм, толщина
исследуемого раствора (в) - 32,5мм, следовательно:
-81 -
0,002.30.25.100-250
32,5-5-50-5
Приготовление раствора 2,4-динитрофенилгидразина. В фарфоровой чашке 5г этого
реактива, растворяют постепенно небольшими порциями (50мл) соляной кислоты (200мл
обычной кислоты на 800мл дистиллированной воды); затем смесь кипятят 1-2 минуты,
хорошо помешивая; после охлаждения раствор разбавляют водой до 1мл и фильтруют
через плотный фильтр (фильтрование возобновляют перед употреблением).
Приготовление стандартного раствора яблочной кислоты. Яблочную кислоту
дважды перекристаллизовывают из водного раствора и высушивают до постоянного
веса в вакуум-эксикаторе надсерной кислотой. Навеску 200мг яблочной кислоты ра¬
створяют в мерной колбе на 100мл нормальным раствором серной кислоты и дово¬
дят до метки. В растворе кислоты реактив сохраняется долгое время.
Из этого раствора берут 5мл, что соответствует 10мг яблочной кислоты, в колбу
на 100мл приливают 2мл н. раствора бромистого калия, охлаждают до 20° и добавля¬
ют 5мл 1,5н. раствора перманганата. После охлаждения реакционной смеси до 10°
обесцвечивают 3%-ным раствором перекиси водорода. Затем окисленную смесь пе¬
регоняют в колбу, содержащую 25мл 2,4-динитрофенилгидразина, полученный осадок
озазона отфильтровывают, промывают и растворяют в горячем пиридине, доведя
объем в мерной колбе до 25мл. Отсюда берут 5мл, или 2мг.
2133.1.5. Технологическая канта.
2.13.4. ОПРЕДЕЛЕНИЕ ЩАВЕЛЕВОЙ КИСЛОТЫ
2.13.4.1. Принцип метода
Метод количественного определения основан на осаждении щавелевой кислоты
в виде щавелевокислого кальция, почти нерастворимого в холодной воде.
2.13.4.2. Технологическое оборудование
Весы типа ВЛКТ.
Колбы мерные на 50, 200, 250 и 500 мл.
Пипетки градуированные на 10, 50 мл.
Бюретка.
Гомогенизатор.
-82-
2.13.43. Реактивы
1. Реактив для осаждения щавелевой кислоты (к раствору 25г хлористого каль¬
ция в небольшом количестве воды, помещенному в мерную колбу на 500мл, доливают
до метки 50%-ную уксусную кислоту и раствор ЗЗОг кристаллического уксуснокисло¬
го натрия в 300мл воды; оба раствора хорошо перемешивают, реактив выдерживают
48 часов при температуре 3-7° и фильтруют).
2 Борная кислота.
3. 0,1н. раствор перманганата калия.
4. 1%-ный раствор AgNOy
2.13.4.4. Ход анализа
Для определения свободной и связанной в виде кальциевой соли щавелевой кис¬
лоты используют экстракт, полученный из свежего материала. Если определяют в су¬
хом материале, то 5±0,01г сухих листьев растирают со стеклянным песком с 5мл 10%-
ной серной кислоты и небольшим количеством спирта. Затем все переносят в мерный
цилиндр или колбу и доливают водой до 250-500мл, встряхивают и оставляют на не¬
сколько часов или на ночь. Свободную щавелевую кислоту определяют в водных вы¬
тяжках, полученных без подкисления.
Для приготовления вытяжки применяют следующие способы.
1. Из предварительно измельченной средней пробы берут навеску, которую допол¬
нительно измельчают или в измельчителе тканей (гомогенизатор), или в фарфоровой
ступке. В первом случае навеску помещают в стакан гомогенизатора, заливают нагретой
предварительно до 80° водой и экстрагируют в течение 10 минут. Затем новыми
порциями воды количественно переносят содержимое стакана в мерную колбу и по
остывании доводят до метки.
2. При отсутствии гомогенизатора в фарфоровой чашке (известного веса) отвеши¬
вают навеску 25±0,01г из тщательно перемешанной непосредственно перед взвешива¬
нием средней пробы. Навеску количественно переносят в фарфоровую ступку, смывая
туда же из чашки остатки навески дистиллированной водой, и тщательно растирают с
2-Зг толченого стекла до однородной кашицеобразной массы (небольшие кусочки
тканей, например, кожицы плодов томатов, слив и т.д., должны быть растерты).
Растертую массу количественно переносят в коническую колбу на 250мл, заливают 150-
200мл горячей дистиллированной воды и нагревают в течение часа на водяной бане
при 75-80°. Через час колбу снимают с бани и переносят содержимое ее в мерную колбу
на 500мл (удобно переносить при помощи воронки со срезанным концом), ополаски¬
вая несколько раз колбу. После охлаждения вытяжки объем ее доводят до метки и,
перемешав тщательно содержимое, фильтруют вытяжку в любую другую колбу
Для осаждения щавелевой кислоты в колбы на 200мл берут по 50мл вытяжки, при¬
бавляют до щелочной реакции аммиак и 1-2г борной кислоты (последнюю прибавляют
для затруднения осаждения солей винной кислоты и ее оптических изомеров). Затем
прибавляют 10мл реактива для осаждения щавелевой кислоты и оставляют на 48 часов
при температуре не выше 7°. Выделившийся осадок щавелевокислого кальция отфильт¬
ровывают и промывают горячей водой до отрицательной реакции на хлор (отсутствие
осадка от прибавления нескольких капель 1%-ного раствора азотнокислого серебра). В
дальнейшем щавелевую кислоту определяют по одному из следующих способов.
1. Осадок щавелевокислого кальция смывают с фильтра водой из промывалки в колбу
и сразу же приливают горячую 10%-ную серную кислоту через тот же фильтр, чтобы
растворить следы оксалата. Осадок в колбе при помешивании переходит в раствор; при
медленном растворении осадка колбу с жидкостью нагревают. Фильтр промывают горячей
-83-
водой и раствор титруют 0,1н. раствором перманганата до появления розовой окраски.
Зная, сколько мл перманганата затрачено на титрование взятой пробы и зная титр
перманганата по щавелевой кислоте, узнают количество кислоты (в г).
Процент щавелевой кислоты (х) находят по формуле:
а в-100
х =
в, ♦«
ще: в - общий объем вытяжки;
а - щавелевая кислота в пробе вытяжки;
в, - объем пробы;
н - навеска опытного образца.
2. Осадок щавелевокислого кальция вместе с фильтром переносят в колбу, ра¬
створяют в точном объеме титрованного раствора H2S04 и избыток кислоты оттит-
ровывают 0,1н. раствором щелочи (1мл 0,1 н. К^04 равен 4,5мг щавелевой кислоты).
3. При больших осадках щавелевокислого кальция щавелевая кислота может быть
определена весовым путем по окиси кальция после озоления (1мг окиси кальция со¬
ответствует 1,605мг безводной щавелевой кислоты).
2.13.4.5. Технологическая карта
2.13.5. ОПРЕДЕЛЕНИЕ ПИРОВИНОГРАДНОЙ КИСЛОТЫ
2.13.5.1. Принцип метода
Пировиноградная кислота (CI^ - СО -СООН) в кислой среде реагирует с кислыми
сернистокислыми солями (KHS03 или NaHS03), давая бисульфитное соединение
СН3 - СО - СООН + NaHS03 — СН3С(ОН) - (0S02Na) -СООН.
Избыток прибавленного бисульфита связывают иодом:
NaHS03 +12 + Н20 — NaHS04 + 2HI.
Соединение пировиноградной кислоты с бисульфитом разлагают и бисульфит
оттитровывают иодом.
2.13.5.2. Технологическое оборудование
Микробюретка (или бюретка) с делениями 0,025мл.
Мерные колбы на 200 и 300мл.
Пипетки градуированные на 5, 10, 20мл.
Конические колбы на 100мл.
-84-
2.13.5.3. Реактивы
1. 1%-ный раствор бисульфита натрия (или калия.) Хранят 2-3 дня.
2. 0,1 н. и 0,01 н. растворы йода.
3. Двууглекислый натрий.
4. 1%-ный раствор крахмала.
5. 5-10%-ный раствор фосфорновольфрамовой кислоты.
2.13.5.4. Ход анализа
В коническую колбу наливают 20-25мл вытяжки и осаждают белки прибавлением
нескольких капель раствора фосфорновольфрамовой кислоты. К 5-10мл вытяжки, из
которой осажден белок фосфорновольфрамовой кислотой, добавляют Змл 1%-ного
раствора бисульфита натрия; содержимое колбы перемешивают и оставляют на 30
минут. Затем избыток бисульфита удаляют сначала 0,1н. раствором, а затем 0,01н.
раствором йода.
Соединение пировиноградной кислоты с бисульфитом разрушают добавлением 1г
сухого бикарбоната натрия и выделившийся бисульфит оттитровывают 0,01 н. раство¬
ром йода (1мл 0,01 н. раствора йода соответствует 0,44мг пировиноградной кислоты).
В присутствии больших количеств аминокислот метод не дает верных результа¬
тов. В таких случаях необходимо экстракт молочной кислоты до окисления пропус¬
тить через колонку с ионообменной смолой.
2.13.53. Технологическая карта
2.14, ОПРЕДЕЛЕНИЕ СУММЫ ОРГАНИЧЕСКИХ КИСЛОТ
(ПО X. Н. ПОЧИНКУ)
2.14.1. Принцип метода
В растениях органические кислоты находятся в свободном состоянии или в виде
растворимых и нерастворимых в воде солей. Их извлекают раствором азотной кислоты в
70%-ном этиловом спирте. При этом пектиновые вещества и другие коллоиды остаются в
осадке. В полученном растворе азотную кислоту нейтрализуют едким натром, а органичес¬
кие кислоты осаждают уксуснокислым свинцом. Осаждение идет в 70%-ном растворе
этилового спирта, подкисленного уксусной кислотой для предотвращения образования
основных солей. В 70%-ном спирте свинцовые соли органических кислот практически
нерастворимы. После отделения осадка центрифугированием и промывания 70%-ным
спиртом для удаления избытка свинца осадок обрабатывается карбонатом натрия. При этом
образуется нерастворимый в воде углекислый свинец, а органические кислоты и пигменты
-85-
в виде натриевых солей переходят в раствор:
РЬА + Na2C05 = РЬС03 + Na^ , где А - анион кислоты.
После центрифугирования и удаления раствора осадок растворяется в соляной кислоте
РЬС03 + 2НС1 = РЬС12 + С02 + Н20.
В растворе находится эквивалентное кислотам количество ионов свинца, которое
определяется комплексометрическим методом. Для этого добавляют в избытке три-
лон Б, который реагирует с ионами свинца, образуя прочное комплексное соединение:
РЬС12 + Na2H2C..Hi20:N2 + 2NH4OH = Na-PbC, Д203Ы2 + 2NH4C1 + 2H20.
Остаток трилона Б титруют сернокислым магнием:
Na2H2C10H12O3N2 + MgS04 + 2NH4OH = Na2MgC10H12O}N2 + (NH^SO, + 2H.O.
Суммарное количество ди- и трикарбоновых кислот выражают через яблочную
или лимонную кислоту. Грамм-эквивалент яблочной кислоты, = 67 , а лимонной
384 - 24
кислоты = 64,04.
6
2.14.2. Технологическое оборудование
Аналитические весы.
Фарфоровая ступка.
Центрифуга с набором пробирок.
Стаканы на 50мл.
Стеклянные палочки.
2
2.14.3. Реактивы
1. 70%-ный раствор этилового спирта. К 100мл 95%-ного этилового спирта при¬
бавить 39мл дистиллированной воды и перемешать.
2. 20%-ный раствор уксуснокислого свинца. 20г уксуснокислого свинца
РЬ(СН3СОО)2 • ЗН20, растворяют в 80мл дистиллированной воды, добавляют 1мл
ледяной уксусной кислоты и разбавляют такой же водой до 100мл.
3. 2н.раствор азотной кислоты. 127мл азотной кислоты (плотн. 1,42) разбавляют
дистиллированной водой до 1л и перемешивают.
4. 0,1н. раствор соляной кислоты. 1н. раствор соляной кислоты разбавляют в 10
раз дистиллированной водой.
5. 0,02н. стандартный раствор сернокислого магния. 2,4650г MgS04 • 7^0 ра¬
створяют в дистиллированной воде, приливают 5мл серной кислоты (1:9), раствор
переносят в мерную колбу на 1л., доводят водой до метки и тщательно перемешивают.
6. 2н. раствор едкого натра. 80г NaOH ч.д.а. растворяют в 1л дистиллированной
воды, нагревают до кипения и кипятят 30 минут, затем охлаждают и хранят в хорошо
закрытой склянке. Этот раствор можно также приготовить из 20%-нош без аммиачно¬
го раствора едкого натра разбавлением в 2,5 раза безаммиачной водой.
7. Аммиачный буферный раствор pH 10. 55г хлористого аммония х.ч. растворяют
в 350мл 25%-ного раствора аммиака и разбавляют дистиллированной водой до 1л.
8. Индикатор хромоген черный ЕТ-100. 0,2г индикатора черного и 20г хлористого
натрия х.ч. высыпают в ступку и тщательно растирают. Сохраняют в темной склянке.
50мг этой смеси всыпают в колбу при титровании.
9. 0,1н. раствора трилона Б. 18,60г высушенной при 80°С соли Na2H2C]0Hl2OgN2*
•2Н20 растворяют в дистиллированной воде и разбавляют до 1л водой.
2.14.4. Ход анализа
Взвешивают такое количество растительного материала, чтобы в нем находилось
-86-
от 5 до 30мг суммы кислот (1-2г свежего или 0,3-0,5г сухого вещества), и навеску
растирают в ступке сначала с 0,5г силикагеля или песка, а затем с 1мг 2н. раствора
азотной кислоты и 5мл 70%-ного этилового спирта. Растертую массу переносят в
центрифужную пробирку, смывая остаток 10мл 70%-ного спирта. Тщательно переме¬
шивают в течение 15-20 минут при частом перемешивании. Затем центрифугируют,
раствор сливают в стакан на 50мл, а к осадку прибавляют 5мл 70%-ного спирта и
настаивают 5 минут при частом перемешивании. После этого центрифугируют, ра¬
створ сливают в стакан, а осадок промывают еще один раз 5мл 95%-ного этилового
спирта, добавляя центрифугат к раствору в стакане. Для нейтрализации азотной кис¬
лоты прибавляют одну каплю раствора фенолфталеина и добавляют по каплям при
перемешивании 2н. раствор NaOH до появления розово-красной окраски индикато¬
ра. В том случае, когда исследуемый раствор окрашен, то для нейтрализации азотной
кислоты прибавляют 1мл 2н. раствора едкого натра. После нейтрализации азотной
кислоты добавляют 0,3мл ледяной уксусной кислоты и 1 мл 20%-ного раствора уксус¬
нокислого свинца, перемешивают и оставляют на 30 минут для выделения осадка.
Затем центрифугируют, раствор сливают в склянку с отработанным спиртом, а ста¬
кан и осадок, находящийся в центрифужной пробирке, промывают 2 раза 70%-ным
спиртом порциями по 5мл. При этом весь осадок переносят в пробирку. К промыто¬
му осадку в пробирке приливают 10мл 1%-ного раствора углекислого натрия и пере¬
мешивают стеклянной палочкой в течение 5 минут, пока разложится весь осадок и
свинец перейдет в углекислую соль, которую затем отделяют центрифугированием.
Прозрачный раствор отбрасывают, а к осадку приливают 10мл 0,1н. раствора соляной
кислоты, перемешивают и выливают в колбу для титрования на 100мл, смывая остаток
дистиллированной водой. В полученном растворе свинец определяют комплексомет¬
рическим методом. Для этого добавляют Змл аммиачного буферного раствора, 5мл 0,1 н.
раствора трилона Б, 50мг индикатора хромогена черного ЕТ-100 и титруют 0,02н.
раствором сернокислого магния до перехода сине-зеленой окраски в винно-красную.
Отдельно титруют 5 мл 0,1 н. раствора трилона Б 0,02 н. сернокислым магнием.
Из полученных данных вычисляют сумму кислот по формуле:
6,7 •(а-в)-к
х = ,
н
где х - содержание суммы кислот, %, в расчете на яблочную кислоту;
а - объем 0,02 н. раствора сернокислого магния, затраченный на
5 мл 0,1 н. раствора трилона Б, мл.;
в - объем 0,02 н. раствора сернокислого магния, затраченный при
определении кислот, мл.;
к - нормальность раствора сернокислого магния;
н - навеска исследуемого вещества, г;
6,7 - нормальный титр яблочной кислоты, умноженный на 100 для
пересчета в проценты.
2.15. ОПРЕДЕЛЕНИЕ ОБЩЕГО СОДЕРЖАНИЯ ЛИПИДОВ (МАСЕЛ)
(По А.И. Ермакову и сотрудников)
2.15.1. Принцип метода
Методы количественного определения масла в семенах основаны на способности
его растворяться в некоторых органических растворителях. В семенах и других расти¬
-87-
тельных объектах содержится ряд веществ, которые полностью или частично переходят
в раствор вместе с маслом. Эфирно-этиловый, или петролейный экстракт семян после
удаления растворителя называют «сырым жиром». Процент масла в измельченных семе¬
нах может быть определен по массе (весу) «сырого жира», извлеченного из определен¬
ной навески, или по массе обезжиренной сухой навески. Большая часть «сырого жира» -
липидов семян является тришициридами с примесью сопутствующих веществ.
Определение сырого жира в семенах по массе (весу) извлеченного масла.
При анализе семян с небольшим содержанием масла, а также травянистых материалов
этот метод дает более достоверные результаты, чем метод обезжиренных остатков
2.15.2.Технологическое оборудование
1. Аппарат Сокслета на 100-200мл (объем рабочей части экстрактора).
2. Бюксы для материала.
3. Эксикаторы - большой для колб и средний для исследуемого материала.
4. Фильтровальная бумага (плотная).
2.15.3.Реактивы
1. Эфир этиловый (серный) или петролейный (точка кипения 40-60°).
2.15.4.Аппарат Сокслета и работа с ним.
Этот аппарат используют для извлечения различных веществ эфиром или други¬
ми растворителями. Он состоит из шарикового или спирального обратного холодиль¬
ника, экстрактора и колбы. Колба сообщается с экстрактором трубками. Все три части
соединяются между собой шлифами. Объемы экстрактора и колб находятся в соответ¬
ствии один к другому (100, 200, 400мл). Эти аппараты монтируют батареями по 4-6шт
в ряд или по кругу (баня на 6 гнезд). Растворитель нагревают воздушными или водя¬
ными банями.
Пары растворителя, образующиеся в колбе в результате кипения, поднимаются по
трубке экстрактора в холодильник и сгустившись в жидкость, стекают в экстрактор. В
экстрактор помещают материал, из которого производится извлечение. Нагрев регу¬
лируют по скорости наполнения экстрактора растворителем. Если нагрев недостато¬
чен для того, чтобы пары поднялись в холодильник при высококипящих жидкостях,
прибегают к утеплению асбестовым шнуром паропроводных трубок экстрактора, на¬
чиная от шлифа, и опускают колбу внутрь нагревательного прибора.
Как только уровень растворителя в экстракторе поднимается, раствор с извлечен¬
ным маслом стечет в колбу. После этого весь процесс повторяется снова. Иногда пос¬
ле первого стекания растворитель не наполняет экстрактор (не доходит до верхнего
конца сифона) и непрерывно стекает. Это объясняется дефектом сифона: узким диа¬
метром трубки с перехватом, вследствие чего из сифона жидкость не вытекает полно¬
стью. Растворитель не должен сильно кипеть, в противном случае он не успевает ох¬
ладиться в холодильнике и улетучивается. В результате этого правильное действие
сифона также нарушается.
Ток воды, проходящий через холодильники, должен быть отрегулирован так, что¬
бы не было отпотевания холодильников и стекания воды на шлифы. Если этого нельзя
избежать, сделав перетяжку из бечевки на трубке холодильника так, чтобы концы бе¬
чевки свисали по наружной стенке экстрактора.
Необходимо, чтобы все шлифы были чистыми , иначе их во время работы «заест».
При работе с огнеопасными жидкостями не следует оставлять аппараты Со¬
кслета без присмотра.
-88-
2.15.5. Ход анализа
1. Берут 2 навески из каждого измельченного материала от 3 до 12± 0,005г. в зависимости от
содержания в нем масла. Так, при масличносш материала до 10% употребляют 10-12г, а при
масличности 50-60% достаточно взял» 1 -2г.
При единичных анализах для извлечения масла применяют воздушно-сухой мате¬
риал, в котором одновременно определяют содержание воды. При массовых анализах
удобнее производить определения в сухом материале. Для этой цели сушат цельные,
не измельченные семена и после измельчения снова подсушивают. Навески помещают
в пакетики или патроны из плотной фильтровальной бумаги.
2. Каждый пакетик помещают в экстрактор аппарата Сокслета. Во взвешенные
колбочки этого аппарата наливают 2/3 - 3/4 объема сухого, чистого, без перекисей эти¬
лового эфира. (Для разрушения перекисей эфир промывают 5%-ным раствором же¬
лезного купороса, затем сушат и перегоняют с несколькими кристаллами железного
купороса.). Затем колбу соединяют со своим экстрактором через притертый шлиф и,
установив колбу в гнездо нагревательного прибора, соединяют экстрактор с обратным
холодильником. Нагревание и кипение эфира регулируют так, чтобы за час происхо¬
дило 3-4 сливания.
Для полной экстракции достаточно 12 часов. Полнота извлечения масла зависит
от тонины помола материала. (Иногда необходимо навески растирать в ступке со стек¬
лянным порошком, после чего все количественно переносят в патрон, а ступку проти¬
рают ватой, смоченной эфиром, и тоже помещают в патрон). Если в процессе сушки
материала был допущен перегрев, то это ведет к окислению части масла (особенно
масел льна, ляллеманции, конопли). Окисленная часть масла трудно растворяется,
вследствие чего результаты определения будут занижены.
Конец экстракции устанавливается практическим путем - по времени работы ап¬
парата при определенном числе сливаний экстракта, с учетом величины навески мате¬
риала и ее измельчения.
3. После окончания экстракции аппарат разбирают, сливают остаток раствора
из экстрактора в колбу. Из колбы с раствором масла отгоняют растворитель на
установке для перегонки или другим способом. Затем эту колбу с маслом продува¬
ют резиновой грушей для удаления паров эфира и сушат около часа в термостате
при 100-105° до постоянного веса. По разности между массой (весом) колбы с мас¬
лом и без масла узнают массу (вес) масла, которое было извлечено из навески.
Содержание масла в процентах (х) вычисляют по формуле:
(я-я,)-100
х — >
н
где а - масса (вес) колбы с маслом;
а} - тоже без масла;
и - навеска.
Достоверность и совпадаемость результатов зависят от степени измельчения ма¬
териала, на что обращают особое внимание.
Нередко при измельчении в ступке нескольких граммов мелких семян (мака, гор¬
чицы, кунжута и др.) часть их остается цельными или плохо раздробленными. Это
является главной причиной заниженных результатов.
Положительные свойства этилового (серного) эфира заключаются в низкой тем¬
пературе кипения. К недостаткам серного эфира как растворителя при определениях
масла относится его способность не только разлагать воду (до 8%), но и ряд веществ.
Петролейный эфир обладает более высокими точками кипения, но не извле¬
кает воды и хуже извлекает различные сопутствующие маслу вещества.
-89-
2.16. ОПРЕДЕЛЕНИЕ МАСЛА ПО МАССЕ (ВЕСУ)
СУХОГО ОБЕЗЖИРЕННОГО ОСТАТКА
На опытных станциях наиболее часто применяется вариант этого метода в мо¬
дификации С.В.Рушковского, рассчитанный на массовые анализы семян для оцен¬
ки селекционного материала.
2.16.1.Технологическое оборудование
в дополнение к вышеприведенному:
Банки с притертыми пробками на 1-1,5л для настаивания, желательно широкогорлые;
Стакан с притертой пробкой диаметром 4,5см, высотой 7,5см дня взвешивания пакетов.
2.16.2.Реактивы
1. Петролейный эфир с точкой кипения 70°-или авиабензин.
2.16.3.Ход анализа
Две навески по 1г измельченных, высушенных или воздушно-сухих семян (в после¬
днем случае одновременно в отдельных навесках определяют количество воды) пересы¬
пают в отдельные пакеты, предварительно высушенные и пронумерованные. (Навески
можно помещать и в предварительно высушенные пакеты, затем подсушить в сушиль¬
ном шкафу в течение 1,5-2 часов при 100-105° и после охлаждения взвесить). Пакеты с
навесками по 10-12 шт. помещают в марлевые мешочки, опускают в банку с притертой
пробкой и настаивают с авиабензином или с петролейным эфиром в течение двух суток
(сменяя бензин 2-3 раза). После такого настаивания мешочек с частично обезжиренными
пакетами помещают в патрон аппарата Сокслета и извлекают в течение 2-4 часов остатки
масла этиловым эфиром, причем эфир в экстракторе должен сливаться 4 раза в час.
После экстракции пакеты извлекают из банки, помещают в широкий кристаллиза¬
тор и дают под тягой испариться растворителю, затем их помещают в стаканчики с
притертыми крышками и сушат в течение 2-3 часов при 100-105° в сушильном шкафу.
После охлаждения в эксикаторе их взвешивают. (Желтые или коричневые пятна на паке¬
тах после их сушки, происходящие в результате окисления масла, указывают на его
неполное извлечение). Взвешивание выполняют быстро, на торсионных весах. Разница
в весе пакета с навеской до экстракции и после экстракции должна показывать вес
(содержание) масла. Поэтому плохо высушенные перед экстракцией пакет и навеска
дают завышенные значения в содержании масла, что является постоянным источником
ошибок в этом анализе. Преимущество этого метода состоит в более экономном расхо¬
де растворителя на каждое определение и в большей производительности.
Приведенный метод дает вполне удовлетворительные результаты при анализе
различных растительных объектов, если учитывать извлечение воды в процессе
анализа. При массовых анализах высокомасличных семян (40-70%) расхождение в
содержании масла между парными определениями допустимо до 1%.
Повышение производительности методов зависит от возможности механизации и
распределения каждой операции метода между отдельными работниками. Быстрота
взвешивания на торсионных весах избавляет от взвешивания в стаканах с притертыми
пробками и уменьшает ошибки, происходящие за счет гигроскопичности.
Пример вычисления. Навеска сухих измельченных семян горчицы вместе с паке¬
тиком равна 1,588г, масса сухого пакетика - 0,474г, масса пакетика после обезжири¬
вания - 1,118г. Следовательно, масса семян была 1,114г, а масла - 0,470г. Таким
образом, процент масла (*) в сухих семенах равен:
х=М70Л00 %
1,114
-90-
2.16.4. Технологическая карта
2.17. МЕТОД ОПРЕДЕЛЕНИЯ ЖИРНОКИСЛОГО
СОСТАВА МАСЛА И ЭРУКОВОЙ КИСЛОТЫ
2.17.1. ГАЗОХРОМАТОГРАФИЧЕСКИЙ МЕТОД
ОПРЕДЕЛЕНИЯ ЖИРНОКИСЛОГО СОСТАВА
МАСЛА СЕМЯН РАПСА И СУРЕПИЦЫ
2.17.1.1. Принцип метода
Метод основан на быстром получении из масла метиловых эфиров высших
жирных кислот при помощи 5%-ного раствора металлического натрия в метаноле в
среде неполярного углеводородного растворителя с последующим их разделением
газожидкостной хроматографией.
2.17.1. 2. Технологическое оборудование
Г азовый хроматограф.
Лабораторный пресс.
Пробирки.
Пипетки на 1, 10мл.
Микрошприц МШ-10.
Стеклянные воронки.
Бумажные фильтры.
2.17.1.3. Реактивы
Гексан.
5%-ный раствор металлического натрия в метаноле.
2.17.1. 4. Ход определения
Масло из семян извлекают выдавливанием на лабораторном прессе при давле¬
нии 150атм. Выступившее вокруг поршня масло отсасывают микропипеткой, две
капли помещают в пробирку с 2мл гексана и добавляют 0,2мл
5%-ного раствора металлического натрия в метаноле. Пробирку встряхивают в
-91 -
течение 1мин., затем оставляют на 2-3 мин при комнатной температуре и фильтруют
через бумажный фильтр. К фильтрату прибавляют 5мл дистиллированной воды и встря¬
хивают смесь. Верхнюю фазу используют для хроматографического анализа.
Разделение метиловых эфиров высших жирных кислот проводят на газовом
хроматографе. Необходимо иметь также детектор пламенно-ионизационный, колонку
стеклянную длиной 250мм с внутренним диаметром Змм, наполненную 15%-ным
полиэтиленгликольсукцинатом (вид II) на Парохроме 1,0,125-1,315мм (газ-носитель-
азот, скорость пропускания через колонку - 30-50мл/мин, температура термостата -
176°С, детектора - 220°С, скорость ленты-самописца - 0,15 или 0,30см/мин).
Микрошприцем МШ-10 пробу вносят в испаритель в количестве 0,8-1,4мкл.
При анализе масла семян рапса и сурепицы пики основных жирных кислот на
хроматографе по мере их выхода располагаются в следующем порядке: пальмитино¬
вая кислота (16:0), пальмитолеиновая кислота (16:1), стеариновая кислота (18:0), оле¬
иновая кислота (18:1), линолевая кислота (18:2), линоленовая кислота (18:3), эйкозе-
новая кислота (20:1), эруковая кислота (22:1).
Процентное содержание жирных кислот в исследуемой пробе определяют мето¬
дом внутренней нормализации по отношению площади пиков отдельных кислот к
суммарной площади всех пиков. Площади рассчитывают путем умножения высоты
пика на его ширину, измеренную на половине высоты.
Время анализа одной пробы - 40-50 мин.
2.17.2. ФОТОКОЛОРИМЕТРИЧЕСКИЙ МЕТОД ОПРЕДЕЛЕНИЯ
ОБЩЕГО СОДЕРЖАНИЯ ГЛЮКОЗИНОЛАТОВ В СЕМЕНАХ
РАПСА И СУРЕПИЦЫ ПАЛЛАДИЕВЫМ РЕАКТИВОМ
2.17. 2.1. Принцип метода
Метод основан на колориметрическом определении палладиевым реактивом
алкенилглюкозинолатов (глюконапина, глюкобрассинапина, прогойтрина), содер¬
жащихся в водном экстракте семян.
2. 17. 2. 2. Технологическое оборудование
Мерные колбы на 50мл.
Пипетки градуированные на 1 и 2мл.
Водяная баня.
Фотоэлектроколориметр.
Лабораторная мельница.
2.17. 2. 3. Реактивы
30%-ный раствор сульфата цинка.
15%-ный раствор гексацианоферрата (II) калия.
0,02 М раствор тетрахлорид (II) палладата натрия.
2.17. 2. 4. Ход анализа
Навеску (1г) тщательно измельченных семян рапса (сурепицы) помещают в кол¬
бочку на 50мл и оставляют на 5-7мин на водяной бане с температурой не ниже 90° С,
после чего добавляют 30мл кипящей воды (дистиллированной) и продолжают вы¬
держивать при той же температуре 20мин. После охлаждения прибавляют по 1мл
30%-ного раствора сульфата цинка и 15%-ного раствора гексацианоферрата (II) калия,
-92-
взбалтывают, доводят водой до метки и еще раз тщательно перемешивают. После
отстаивания или фильтрации к 0,5мл осветленного раствора добавляют 1,5мл 0,02М
водного раствора тетрахлорида (II) палладата натрия, перемешивают и через 1 Омин
снимают эстинцию при длине волны 440нм против холостой пробы на фотоэлектро¬
колориметре с использованием кювет толщиной Змм.
Процентное содержание глюкозинолатов в семенах рапса и сурепицы вычисля¬
ют путем умножения показаний фотоколориметра на коэффициент 7,62.
Один аналитик анализирует за день 20 проб.
Для получения Na2PdCl4 (тетрахлорида (II) палладата натрия) эквимолярные ко¬
личества концентрированных водных растворов хлорида палладия и хлорида натрия
сливают в фарфоровую чашку, интенсивно перемешивают и медленно упаривают на
водяной бане.
После высушивания в эксикаторе (24 ч) светло-коричневый порошок сохраняют
без доступа влаги. Для работы используют свежеприготовленный раствор тетрахло¬
рида (II) палладата натрия.
2.17. 2. 5.Технологическая карта
-93-
3. АНАЛИЗ ПОЧВЫ
«Полное и всестороннее изучение почвы
и происходящих в ней процессов требует
приложения системы различных методов».
А.А. Роде
3.1. ОТБОР И ПОДГОТОВКА ПОЧВЫ К АНАЛИЗУ
3.1.1. Отбор образцов
Правильность методики и техника ее проведения является важнейшим услови¬
ем достоверности полученных в химическом почвенном анализе данных. Отбор и
подготовка образцов почв к анализу осуществляется в соответствии с существую¬
щими стандартами.
При проведении крупномасштабных агрохимических обследований почв и аг¬
рохимической паспортизации земель сельскохозяйственного назначения отбор об¬
разцов почвы проводится согласно «Методики сплошного почвенно-агрохими¬
ческого мониторинга сельскохозяйственных угодий Украины».
Основные требования к отбору:
1) пробы почвы со всех сравниваемых опытных делянок должны отбираться с
одинакового числа точек, количество которых зависит от пестроты почвенного по¬
крова и задач исследования;
2) масса индивидуальных почвенных проб при составлении смешанного образ¬
ца должна быть примерно одинаковой;
3) отбор, а в случае необходимости и фиксация образцов должны быть проведе¬
ны в один день по всем сравниваемым вариантам опыта одного повторения. Для
этого следует выполнить соответствующую предварительную работу, дающую воз¬
можность ускорить отбор образцов - подготовить этикетки, пронумеровать мешки
для образцов;
4) отбор образцов нельзя проводить во время дождя или сразу после него;
5) отбор следует проводить в утренние часы, всегда в одно и то же время;
6) каждый образец должен быть снабжен четко заполненной этикеткой, в кото¬
рой указывается название опыта, место его проведения, номер варианта и повторе¬
ния, горизонт почвы, дата взятия образца.
Подробные сведения - название и место расположения опыта, содержание ва¬
риантов, возделываемая культура или номер варианта и поля, повторность, почвен¬
ная разность и т.д. - должны быть записаны в реестре.
Вопрос о количестве повторностей при отборе образцов решается конкретно
при разработке программы и методики проведения исследований.
Образцы сохраняются в лаборатории в течение года в закрытых картонных или
металлических коробках (бюксах).
Исходные и последующие за ротацию образцы почвы хранятся весь период веде¬
ния полевого опыта в стеклянных либо металлических плотно закупоренных емкостях.
Огёор образцов почвы в поле для анализов должен проводиться с 6-10 точек варианта
(в зависимости от микрокомплексности почвенного покрова). Средний образец отбира¬
ется методом квартования и немедленно доводится до воздушно-сухого состояния.
Сушку целесообразно проводить в хорошо вентилируемом помещении при
подогреве до 40°С с помощью калориферных устройств, избегая воздействия пря¬
мых солнечных лучей.
Образцы поступают в лабораторию в высушенном состоянии в стандартных
маркированных коробках для хранения с указанием даты отбора, номера образца,
его характеристики и сопровождаются общей ведомостью. Ведомость должна со¬
-94-
держать сведения о типе почв, глубине отбора, названии и содержании опыта, пе¬
речня определений (определении подвижных NPK, микроэлементов, форм кислот¬
ности, состава почвенного поглощающего комплекса и т.д.).
3.1.2. Подготовка почвы
Для общего агрохимического анализа ведется следующим образом. Отбирают пин¬
цетом посторонние включения (корни, насекомых, гальку и прочее) и новообразова¬
ния (ортштейны, известковые отложения и т.д.), полностью измельчают и одновремен¬
но просеивают почву через сито с диаметром ячеек 2мм на размолочных машинах.
Из размолотого образца методом квартования отбирают пробы для анализов,
требующих более тонкого помола: для определения содержания гумуса и азота -
около 5 г почвы; емкости поглощения, содержания карбонатов или гипса - около
50г; для валового химического анализа - примерно 30г.
Пробу почвы для определения гумуса, азота, карбонатов, гипса и для валового
анализа очищают от корешков, пользуясь лупой и эбонитовой палочкой, а затем полно¬
стью растирают в размолочной машине с агатовой ступкой марки КМ-1 и просеивают
через сито с диаметром ячеек 0,25мм. Хранят в пакетах из кальки или пергамента.
Для определения емкости поглощения почву растирают в ступке резиновым
пестиком и просеивают через сито 1мм.
Целесообразно отбор почвенных образцов в поле вести непосредственно в мар¬
кированные коробки, в них же проводить сушку почвы и возвращать после размо¬
ла. Это исключает операцию по пересыпке образцов из одной тары в другую, необ¬
ходимость ее дополнительной маркировки, что требует значительных затрат рабо¬
чего времени и служит источником ошибок.
Для определения нитратного азота, закисного железа и ферментативной актив¬
ности почв образцы в полиэтиленовых мешках доставляют в лабораторию в день
отбора при естественной влажности. Их можно хранить в таком состоянии до 5
суток при температуре не более +5° С.
3.2. ОПРЕДЕЛЕНИЕ ГУМУСА
3.2.1. Принцип метода
Метод основан на окислении перегноя почвы бихроматом калия по уравнению:
ЗСорг. + 2К2Сг207 + 8H2S04 = 2Cr2(S04)3 + 2K,S04 + 8Н20 + 3C02.
Избыток бихромата в растворе после окисления перегноя титруют солью Мора:
К.Сг.О^ 7H2S04 + 6FeS04 = Cr2(S04)3 + 3Te2(SOJ3 + 7H20 .
3.2.2. Технологическое оборудование
Весы торсионные.
Газовая или электропечь.
Дозатор для крепких кислот на 10мл.
Колбы конические на 100мл.
Бюретка на 25 или 50мл.
Капельница.
Воронки d = 3-5см.
Часы песочные на 5 мин.
3.2.3. Реактивы
1. 0,4 н. К2Сг207. 40г тонко измельченной соли К2Сг207 растворяют в 500-600мл
дистиллированной воды, фильтруют через бумажный фильтр, доводят дистиллиро¬
-95-
ванной водой объем до 1л и переливают в термостойкую посуду емкостью 2,5-5л. К
этому раствору небольшими порциями, при осторожном перемешивании, прили¬
вают 1л концентрированной H2S04, накрывают часовым стеклом и оставляют до
полного охлаждения. Раствор хранят в темной склянке с притертой пробкой.
2. 0,2 к раствор сот Мора. 80г (NH4)2S04 • FeS04 • 6Н20 заливают 700-800мл 1н.
H2S04 (20мл концентрированной Н^Ю4 на 1л воды), растворяют, фильтруют и дово¬
дят до 1л дистиллированной водой.
Нормальность устанавливают по 0,1 и. раствору КМп04 . Для этого в коническую
колбу на 150мл отмеряют 10мл раствора соли Мора, приливают 1мл концентрированной
H;S04, 30мл дистиллированной воды и титруют 0,1н. раствором КМп04 до слабо-розо¬
вой окраски, не исчезающей в течение 1 мин. Нормальность вычисляют по формуле:
N V
V,
где N1 - нормальность соли Мора;
VI - объем раствора соли Мора, мл.;
N2 - нормальность раствора КМп04;
V2 - объем раствора КМп04 , мл.
3. 0,1 н. раствор КМп04 . 3,2г КМп04 переносят в бутыль темного цвета, прили¬
вают 1л дистиллированной воды, перемешивают (чтобы раствор не соприкасался с
пробкой) и оставляют стоять 10-14 дней. За время отстаивания на дно выпадает
осадок Мп02 . Отстоявшуюся жидкость сифонируют.
Нормальность раствора КМп04 устанавливают по 0,1н. раствору щавелевой
кислоты, приготовленной из фиксанала. Для этого берут 20мл раствора щавелевой
кислоты в колбы на 150мл, приливают 10мл разбавленной H2S04 (1:3), нагревают до
температуры 75° С и титруют раствором перманганата калия до появления стойкой
слабо-розовой окраски.
Нормальность вычисляют по формуле:
V,
где N1 - нормальность раствора КМп04;
VI - объем раствора КМп04, пошедший на титрование, мл.;
N2 - нормальность раствора Н2С204;
V2 - объем раствора Н2С204, мл.
4. Раствор фенилантраниловой кислоты. 0,2г кислоты растворяют в 100мл
0,2% раствора Na2C03.
5. Прокаленная пемза, песок или лесс.
3.2.4. Ход анализа
Из тщательно подготовленного образца почвы (см.п. 3.1.2.) берут навеску 0,1-0,5г, в
зависимости от предполагаемого содержания углерода, и переносят в сухую коничес¬
кую колбу емкостью 100мл, приливают 10мл 0,4н. раствора К2Сг207 , закрывают колбу
воронкой диаметром 3-5см и в течение 5 мин. (песочные часы) проводят медленное
кипячение при температуре 140-180° С. По окончании кипячения обмывают горло
колбы и воронку 10-20мл дистиллированной воды, прибавляют 5-6 капель 0,2% ра¬
створа фенилантраниловой кислоты и титруют солью Мора до перехода окраски в
зеленую.
Параллельно ведут холостое определение, добавляя вместо почвы прокаленный песок.
3.2.5. Обработка результатов
Расчет проводят по формуле:
-96-
™ _ (a-e) N-0.003 100
С /О — »
н
где С - содержание гумуса в почве, выраженное в углероде, %;
N - нормальность соли Мора;
а - объем соли Мора, пошедший на титрование холостой пробы, мл.;
в - объем соли Мора, пошедший на титрование пробы, мл.;
н - навеска почвы, г;
0,003 - г-экв углерода.
Для пересчета на гумус результат умножают на 1,724.
Допустимые расхождения результатов приведены в таблице 19.
Таблица 19.
Содержание гумуса,
%
Допустимые отклонения от среднего при повторных
определениях (в отн. %)
в одной лаборатории
в разных лабораториях
0.1 - 0,5
12
18
0,5 - 1,0
8
11
1.0 - 1,5
5
8
1,5 - 2,0
4
6
2,0 - 5,0
4
5
Свыше 5,0
3
4
3.2.6. Технологическая карта.
Взятие навесок
->
Добавление оаствопа бихромата
весы торсионные, шпатель, колбы
конические на 100-150мл.
цилиндр мерный или дозатор для
крепких кислот на 10мл, воронки d
= 3-5см
Кипячение
-
Титрование
бюретка на 25-50мп, капельница
электропечь с закрытой спи-рал ью,
прокладка листового асбеста, часы
песочные на 5мин.
З.2.А. ОПРЕДЕЛЕНИЕ ГУМУСА ПО МЕТОДУ ТЮРИНА
В МОДИФИКАЦИИ ЦИНАО
32.1Л- Метод отбора проб
Образцы почвы доводят до воздушно-сухого состояния, измельчают и просеи¬
вают через сито с круглыми отверстиями диаметром 1-2мм. Корни и неразложив-
шиеся растительные остатки оставшиеся на сите, удаляют. Образцы хранят в короб¬
ках или пакетах. Из размолотой почвы отбирают пробу массой 3-5г для тонкого
измельчения. При отборе пробы из коробки почву тщательно перемешивают на
всю глубину коробки. Из пакетов почву высыпают на ровную поверхность, распре¬
деляют слоем не более 1см и отбирают пробу не менее чем из пяти мест. Видимые
невооруженным глазом неразложившиеся корни и растительные остатки удаляют.
Отобранную пробу измельчают и просеивают через плетеное сито с размером
-97-
ячеек 0,25мм. Оставшиеся на сите частицы почвы также измельчают и полностью
просеивают через сито.
Для тонкого измельчения используют ступки и измельчительные устройства из
фарфора, стали и других твердых материалов.
Измельченную пробу тщательно перемешивают, распределяют тонким слоем на
ровной поверхности и отбирают пробу для анализа не менее чем из пяти мест.
32.2Л* Аппаратура и материалы
Фотоэлектроколориметр.
Баня водяная на 50 - 100 пробирок.
Весы лабораторные 2-го класса точности с наибольшим пределом взвешивания 200г
и 4-го класса точности с наибольшим пределом взвешивания 500г.
Весы торсионные.
Дозаторы с погрешностью дозировки не более 1%.
Бюретки емкостью 25 и 100мл 2-го класса точности.
Груша резиновая со стеклянной трубкой или устройство для барбатации.
Ступка фарфоровая с пестиком.
Палочки стеклянные длиной 30см.
Посуда мерная лабораторная 2-го класса точности.
Колбы конические емкостью 250мл.
Пробирки стеклянные термостойкие емкостью 50 мл.
Штативы для пробирок.
Бумага фильтровальная.
3.23.А. Реактивы
Аммоний-железо (II) сернокислый (соль Мора), х.ч. или ч.д.а.,
или железо (II) сернокислое, 7-водное ч.д.а.
Калия гидроокись, х.ч., ч.д.а. или ч.
Калий двухромовокислыйу х.ч. или ч.д.а.
Калий марганцовокислый, 0,1 н., стандарт-титр.
Натрий сернистокислый, ч.д.а. или натрий сернистокислый, 7-водный, ч.д.а.
Кислота серная, х.ч. или ч.д.а., концентрированная и раствор
с концентрацией 0,5моль/дм3.
Вода дистиллированная.
З.2.4.А. Подготовка к анализу
Приготовление хромовой смеси. 40г тонко измельченного двухромовокислого
калия, взвешенного с погрешностью не более 0,1 г, растворяют в дистиллированной
воде, доводя объем до 1000мл, тщательно перемешивают и переливают в колбу из
термостойкого стекла вместимостью 3000мл.
К полученному раствору осторожно приливают небольшими порциями по 100мл,
с интервалом в 10 - 15 мин, 1000мл серной кислоты. Колбу с раствором накрывают
стеклянной воронкой и оставляют до полного охлаждения. Затем раствор переливают
в склянку с притертой пробкой.
Раствор хранят неограниченно долго.
Приготовление раствора восстановителя. 40г соли Мора или 27,8г 7-водного
сернокислого железа (II), взвешенного с погрешностью не более 0,1 г, растворяют в
700мл раствора серной кислоты с концентрацией 0,5моль/дм3. Раствор фильтруют через
двойной складчатый фильтр в мерную колбу, доводят объем до 1000мл дистиллиро¬
ванной водой и тщательно перемешивают.
-98-
Концентрацию раствора проверяют каждые 3 дня по раствору марганцовокисло¬
го калия, приготовленному из стандарт-титра. Для титрования в три конические кол¬
бы наливают из бюретки по 10мл раствора соли Мора или раствора 7-водного серно-
кислога железа (II), приливают по 1мл концентрированной серной кислоты, 50мл
дистиллированной воды и оттитровывают раствором марганцовокислого калия до
слабо-розовой окраски, не исчезающей в течение 1 мин. Для вычисления коэффици¬
ента поправки используют среднее арифметическое результатов трех титрований.
Коэффициент поправки (К) вычисляют по формуле:
V
где У1 - объем раствора марганцовокислого калия, израсходованного
на титрование,мл;
V - объем раствора соли Мора или 7-водного сернокислого железа (II),
взятый дня титрования,мл.
Раствор хранят в бутыли из темного стекла, к которой с помощью сифона присое¬
диняют бюретку. Для предохранения раствора от окисления кислородом воздуха к бу¬
тыли присоединяют склянку Тищенко с щелочным раствором сернистокислого натрия.
Приготовление щелочного раствора сернистокислого натрия. 40г безводного
сернистокислого натрия или 80г 7-водного сернистокислого натрия взвешивают с
погрешностью не более 0,1 г и растворяют в 700мл дистиллированной воды.
Юг гидроокиси калия, взвешенного с погрешностью не более 0,1 г, растворяют в
300мл дистиллированной воды. Полученные растворы смешивают.
32.5Л. Проведение анализа
Окисление гумуса почвы. Массу пробы почвы для анализа определяют исходя из
предполагаемого содержания гумуса в почве.
Масса пробы для анализа почв составляет:
с содержанием гумуса более 1% - 50 - 100мг;
-«- « - « 4-7% - 100 - 200мг;
-«- « - « 2-4% - 250 - 350мг;
- «- « - « до 2% - 500 - 700мг.
Пробы почвы, взвешенные с погрешностью не более 1мг, помещают в пробир¬
ки, установленные в штативы. В пробирки с анализируемыми пробами и в десять
чистых пробирок для приготовления растворов сравнения приливают дозатором
или из бюретки по 10мл хромовой смеси и помещают в них стеклянные палочки.
Почву с хромовой смесью тщательно перемешивают. Затем штативы с пробирками
погружают в кипящую водяную баню и выдерживают в ней в течение 1ч с момента
закипания воды в бане после погружения пробирок. Уровень хромовой смеси в пробир¬
ках должен быть на 2 - Зсм ниже уровня воды в бане. Содержимое пробирок перемеши¬
вают стеклянными палочками через каждые 20мин. По истечении 1ч штативы с пробир¬
ками вынимают и погружают в водяную баню с холодной водой. После охлаждения в
пробирки с пробами почвы приливают дозатором по 40мл дистиллированной воды, а в
пробирки для приготовления растворов сравнения приливают раствор восстановителя,
приготовленный по п.3.2.4.А, и дистиллированную воду в объемах, указанных в табл.20.
Затем из пробирок вынимают стеклянные палочки и тщательно перемешивают содер¬
жимое барбатацией воздуха, нагнетаемого резиновой грушей через стеклянную трубку.
Растворы оставляют для оседания почвенных частиц и полного осветления.
Определение гумуса. Растворы сравнения и испытуемые растворы фотометриру-
ют через 12-18 часов в кювете с толщиной просвечиваемого слоя 10-20мм относитель¬
но раствора сравнения №1 при длине волны 590нм или используя оранжево-красный
светофильтр с максимумом пропускания в области 560-600нм. Раствор в кювету фото¬
электроколориметра переносят осторожно, не взмучивая осадка на дне пробирки.
Таблица 20.
Характеристика
раствора
Номер раствора сравнения
Объем дистиллированной воды, мл
40
38
36
32
30
25
20
15
10
Объем раствора восстановителя, мл
10
15
20
25
30
Масса гумуса, соответствую 1щя
объему восстановителя в растворе
сравнения, мг
1,03
2,07
4,14
5,17
7,76
10,3
12,9
15,5
3.2.6А. Обработка результатов
По результатам фотометрирования растворов сравнения строят градуировочный
график. На оси абсцисс откладывают массу гумуса в миллиграммах, соответствую¬
щую объему восстановителя в растворах сравнения, а по оси ординат - соответствую¬
щие им показания прибора. Пользуясь градуировочным графиком, по результатам ана¬
лиза определяют массу гумуса в анализируемых пробах.
Массовую долю гумуса (X) в процентах вычисляют по формуле
х.±1~т
т
где
А - масса гумуса в анализируемой пробе, найденная по графику, мг;
К - коэффициент поправки концентрации восстановителя;
т - масса пробы почвы, мг;
100 - коэффициент пересчета в проценты.
За результат анализа принимают значение единичного определения гумуса.
Результат анализа округляют до сотых долей процента - при содержании гумуса
до 10% и до десятых долей процента - при содержании гумуса более 10%.
Допускаемые отклонения от аттестованного значения стандартного образца при
доверительной вероятности Р=0,95 или от среднего арифметического при повторных
анализах указаны в таблице 21.
Таблица 21.
Массовая доля гумуса в почве, %
Допускаемые отклонения, %
в одной лаборатории
в разных лабораториях
До 2
20
30
Св.2 >5
20
20
>5
10
12
3.2.7А. Технологическая карта
- 100-
3.3. ГРУППОВОЙ И ФРАКЦИОННЫЙ СОСТАВ ГУМУСА
3.3.1. Общие положения
Фракционирование гумуса проводят по растворимости его в щелочных и кислот¬
ных экстрагентах и выделении в каждой фракции основных групп органического ве¬
щества - гуминовых и фульвокислот.
Такой подход позволяет изучить формы и прочность закрепления гумусовых ве¬
ществ почвой.
3.3.2. Метод определения группового и
фракционного состава гумуса по Тюрину
в модификации Пономаревой
33.2.1. Принцип метода
Метод дает наиболее полную дифференциацию по формам связи гумуса с мине¬
ральной частью почвы. В анализе предусматривается выделение следующих фракций:
фракция 1 - растворимая в 0,1н. NaOH - гумусовые кислоты (гуминовые и фуль-
вокислоты ), свободные и связанные с подвижными полуторными окислами;
фракция 1а - растворимая в 0,1н. H2S04 при декальцировании почвы - фульво-
кислоты, свободные и связанные с подвижными полуторными окислами (агрес¬
сивные фульвокислоты);
фракция 2 - растворимая (после декальцирования) в 0,1 н. NaOH - гумусовые
кислоты (гуминовые и фульвокислоты), связанные в основном с кальцием;
фракция 3 - растворимая в 0,02н. NaOH при 6-часовом нагревании - гумусовые
кислоты (гуминовые и фульвокислоты), связанные с устойчивыми полуторными окис¬
лами и слоистыми минералами.
33.2.2. Технологическое оборудование
Весы типа ВЛКТ.
Фотоэлектроколориметр.
Газовая или электропечь.
Водяная баня.
Колбы конические на 100 и 300мл.
Колбы плоскодонные из термостойкого стекла с повышенным тубусом на 100мл.
Воронки d = 9см.
Пипетка или шприц-дозатор на 10, 15, 25мл.
33.2.3. Реактивы
1. 0,1н. NaOH. 6,3мл 16н. NaOH доводят до 1л дистиллированной водой, не содер¬
жащей С02. Для этого воду кипятят, упаривая 1/3 часть объема.
2. 0,02 н. NaOH. 200мл 0,1н. NaOH разбавляют дистиллированной водой не со¬
держащей С02 и доводят объем до 1л.
- 101 -
3. 20% NaOH. 20 г NaOH растворяют в 100мл дистиллированной воды.
4. 1,0н. H2S04. 20мл концентрированной H2S04 осторожно вливают в стакан с
дистиллированной водой и доводят объем до 1л.
5. 0,1н. H2S04. 2,8мл концентрированной H2S04 или содержимое фиксанала ра¬
створяют в дистиллированной воде и доводят объем до 1л.
6. Прокаленная пемза, песок или лесс.
7. Насыщенный раствор Na2S04. 180г соли растворяют в 1л дистиллированной воды.
8. Растворы для определения содержания углерода (см. п.3.2.3.).
9. Растворы для определения содержания азота (см. п.3.4.3.).
10. Мурексид.
33.2.4. Ход анализа
3.3.2.4.1. Извлечение гумусовых веществ 0,1 н. раствором NaOH - фракция 1.
Берут навеску в зависимости от содержания гумуса в почве
2,5г при гумусированности почвы 10%
5-Юг _**_ Ю-3%
Ю-15г — 3-0,5%
20г _ *♦ — 0,5%
Пробу переносят в коническую колбу на 300-400мл, заливают 200мл 0,1н. NaOH,
закрывают пробкой и оставляют на 24 часа. Затем прибавляют 50мл насыщенного
раствора Na2S04, хорошо перемешивают, отстаивают , повторно взбалтывают и
фильтруют через плотный фильтр (синяя лента).
а) Определение содержания Собщ и 14общ. во фракции
В коническую колбу емкостью 100мл берут Ю-50мл фильтрата, прибавляют не¬
много прокаленного песка или пемзы, выпаривают на водяной бане и определяют орга¬
нический углерод по методу Тюрина (см. п.3.2.4.).
Для определения содержания азота такую же аликвоту вытяжки как и для углеро¬
да переносят в плоскодонную колбу, нейтрализуют 1н. H2S04 до выпадения полутор¬
ных окислов и выпаривают на водяной бане. Сухой остаток заливают Змл концентри¬
рованной H2S04. Далее анализ ведут как описано в п.3.4.4.
б) Определение С и N гуминовых кислот
50мл вытяжки переносят в стакан на 100мл, прибавляют 5мл 2,Он. раствора H2S04,
перемешивают, накр'ывают часовым стеклом, нагревают на водяной бане в течение
ЗОмин. при температуре 80°С и фильтруют через беззольный фильтр диаметром 7см
(белая лента). Осадок на фильтре промывают 2-3 раза 0,1н. раствором H2S04 . Затем
воронку с фильтром ставят на стакан, в котором проводилось осаждение, растворяют
осадок на фильтре горячим 0,1н. раствором NaOH и доводят объем до 100мл.
Для определения С и N берут в зависимости от окраски раствора аликвоту 15-
25мл соответственно в коническую и плоскодонную колбы на 100мл. Последующий
анализ проводят как указано в пункте „а”.
3.3.2.4.2. Извлечение органических веществ 091н. HJS04 (декальцирование
почвы)- фракция 1а. Навеску почвы 2,5-20,От, равную навеске при выделении фрак¬
ции 1, переносят в колбу на 300мл, приливают 200мл 0,1н. H2S04 , перемешивают,
отстаивают 24 часа. Затем фильтруют в колбу на 500мл, промывают почву на филь¬
тре 0,1 н. раствором H2S04 до исчезновения реакции на Са. Для проверки набирают
в пробирку 1мл промывных вод, прибавляют 1мл 20% NaOH и несколько кристал¬
лов мурексида. Сиреневый цвет раствора указывает на отсутствие кальция. В каче¬
стве раствора сравнения используют дистиллированную воду, окрашенную в ще¬
лочной среде этим индикатором.
- 102-
После полной промывки Са доводит объем вытяжки с промывными водами до
500-1000мл.
Для определения углерода 100мл вытяжки нейтрализуют сухой содой до появле¬
ния мути, прибавляют прокаленный песок, выпаривают на водяной бане. В сухом ос¬
татке определяют углерод по методу Тюрина (см. п.3.2.4.).
Анализ на азот ведут после выпаривания 100мл вытяжки и озоления сухого остат¬
ка в концентрированной H2S04 (см. п.3.4.4.)
В этой же вытяжке можно также определять Са, Fe, А1.
Следует отметить, что данная вытяжка неустойчива к микробному разложению,
поэтому анализ ее следует проводить в течение 2-3 дней.
3.3.2.4.3. Извлечение органических веществ 0,1н. NaOH после декальцирования
почвы - фракция 2. Влажный остаток почвы после декальцирования смывают с фильт¬
ра 0,1н. NaOH в колбу, где проводилось декальцирование, доводят объем до 200мл 0,1 н.
NaOH, закрывают пробкой и выдерживают 24 часа. Затем прибавляют 50мл насыщен¬
ного раствора Na.S04, перемешивают и через 15мин. фильтруют через плотно при¬
гнанный к воронке бумажный фильтр. Почву на фильтре промывают 2% раствором
Na2S04 до полного осветления промывных вод, доводят объем фильтрата до 500мл и
определяют:
а) содержание С и N во фракции как показано в п.3.3.2.4.1а;
б) содержание С и N гуминовых кислот согласно п.3.3.2.4.16;
в) оптическую плотность гуминовых кислот, для чего аликвотную часть вытяжки
гуминовых кислот фракции 2 с известным количеством углерода разбавляют дистил¬
лированной водой до содержания углерода 10мг/л раствора - для черноземов и серых
лесных почв и до 70мг/л раствора - для подзолистых и бурых лесных почв.
Измерение оптической плотности раствора производят на фотоколориметре с синим
светофильтром (длина волны 430нм) в кювете с толщиной просвечиваемого слоя 1см ион
на спектрофотометре. Индекс оптической плотности (Ес) вычисляют по формуле:
Есмг/л = ^ -,
с
где Д - оптическая плотность раствора;
С - концентрация углерода в вытяжке;
п - степень разбавления.
33.2.4*4. Извлечение органических веществ 0,02н. NaOH - фракция 3. Остаток
почвы после предыдущей щелочной вытяжки в сыром состоянии смывают в ту же
колбу горячим раствором 0,02H.NaOH и доводят объем 0,02н. щелочью до 200мл.
Колбы накрывают часовым стеклом и ставят в водяную баню на 6 часов, а затем
оставляют на 16-18 часов. После отстаивания добавляют 50мл насыщенного раство¬
ра Na2S04, фильтруют, промывают почву на фильтре 2% раствором Na2S04 до пол¬
ного обесцвечивания промывных вод и доводят объем до 500мл.
Определяют:
а) содержание С и N во фракции как показано в п.3.3.2.4.1а;
б) содержание С и N гуминовых кислот согласно п.3.3.2.4.16.
33.2.5. Обработка результатов
Расчеты содержания углерода и азота во всех фракциях гумусовых веществ про¬
водят по формулам:
_ (a-e) N 0.003 100
- 103-
где Сфр. - содержание углерода фракции в % к почве;
N - нормальность соли Мора;
а - объем соли Мра, пошедший на титрование холостой пробы, мл.;
в - объем соли Мора, пошедший на титрование пробы, мл.;
н — аликвотная навеска почвы, г;
0,003 - г-экв углерода.
. с#-100
СФР- = .
общ.
где сфр. - содержание углерода фракции в % к Собщ. почвы;
Сфр. - содержание углерода фракции в % к почве;
Собщ. - общее содержание углерода в почве, %.
(а-в) 0,02-/Г-14-100
*•“ «1000
где Мфр. - содержание азота в % к почве;
(а - в) - количество титранта, пошедшего на титрование пробы,
без холостого, мл.;
0,02 - нормальность титранта;
К - поправка к титру;
14 - мг-экв азота;
н - аликвотная навеска почвы, г;
1000 - коэффициент пересчета на г-экв.
Расчеты группового и фракционного состава гумуса проводят по прилагаемым
ниже формам 1 и 2.
Форма 1. Содержание углерода гумусовых веществ в почвенных вытяжках (в
буквенном выражении).
Показа¬
тели
Собщ
почвы,
%
ОД H.NaOH
вытяжка,
фракция 1
0,1 и. H2S04
вытяжка
фракция 1а
ОД H.NaOH
вытяжка после
декалыдирования
фракция 2
0,02 и. NaOH
вытяжка,
фракция 3
Собщ| Сгк| Сфр
Собщ
Собщ[ Сгк
Сфр
Собщ| Сгк
Сфр
Содержание углерода
% к
почве
Н
А
В
А-В
Е
А1
В1
А1-В1
А2
В2
А2-В2
% к С
общ
почвы
100
а
в
а - в
е
а1
в1
а1 - в!
а2
в2
а2 - в2
Форма 2. 1 рупповой состав гумуса (С фракций к С общ. почвы, %)
(рассчитывается на основании данных формы I)
Собщ.
почвы,
%
Сгк
Сфр
С негидро¬
лизуемого
остатка
Сгк
Сфр
(
ракции
сумма
фракции
сумма
1
2
3
1а
1
2
3
100
в
в1 - в
в2
Угк
е
(а-в)-е
(а1-в1)- (а-в)-е
а2-в2
Уфк
100-
(Упс+Уфк)
Угк
Уфк
Примечание; аналогично проводятся количества азота в составе гумуса.
- 104-
105-
3.4J.6. Технологическая карта.
Фракция I.
Взятие навесок
весы типа BJIKT, шпатель,
колбы конические на 300мл
Добавление 0.1 H.NaOH
цилиндр или дозатор на
200мл, пробки
Отстаивание
дозатор на 50мл
Фильтрация
колбы на 300мл, воронки (^см,
фильтры (синяя лента)
J
Определение углерода общего
Отбор аликвот
колбы конические на 100мл,
пипетка или шприц-дозатор
Добавление раствора
Кипячение
-►
Выпаривание
водяная баня
-►
бихромата
цилиндр или дозатор
для конц.кислот на
-►
эл.печь с закрытой
спиралью, асбестовая
прокладка, часы пе¬
-
Титрование
бюретка на 25мл,
капельница
на 10- 15мл, шпатель
10мл, воронки d=5cM
сочные
Определение углерода и азота гумииовых кислот
Отбор аликвот
стаканы на 100мл,
дозаторы на 5,50мл
Отстаивание
водяная баня,
часовое стекло
Фильтпаиия
колбы конические на 100мл,
воронки d-7cM, фильтры (бе¬
лая лента), промывалка
Растворение осадка
исходные стаканы на
100мл, промывалка
Доведение до
заданного объема
цилиндр на 100мл
Определение азотаобщего
Отбор аликвот
колбы плоскодонные
на 100мл, пипетка
или шприц-дозатор
на 10-20мл, шпатель
IMmaz
—►
лизания
—►
капельница
Выпаривание
водяная баня
Лобаштенме
KQHU.H.SOi
разливочное устр-во
для конц. кислот
Озоление
газовая или эл.
печь, вытяжной
шкаф, капельни¬
ца
Перегонка и
титрование
прибор Серенье-
ва, часы песоч¬
ные
3.4.2.6. Технологическая карта (продолжение)
Фракция 1а.
Взятие навесок
весы типа ВЛКТ, колбы
конические на 300мл,
шпатель
Отмывка
колбы на 500мл,ворон¬
ки ё=11см, цилиндр на
200мл, фильтры
Фракция 2
фильтрат
Определение углерода
Определение азота
осадок
t
фильтрат
Перенос обратно
Повеление по заданного
Фильтрация
в колбы на 300мл
объема
колбы на 500мл,
колба на 300мл,
нромывалка
—►
цилиндры на 200мл, до¬
затор на 50мл, пробки
воронки ё=9см,
фильтры
фильтрат
Фракция 3
осадок
► Определение углерода общего
]
►| Определение углерода гуминовых кислот |
Определение азота гуминовых кислот
►I Определение азота общего
*
фильтрат
^ Определение углерода общего
ТТрпрнлр л^пятнп
Доведение по за¬
Остывание
Фильтряттия
1 XwUVtlvL UUL/dlnU
в колбы на 300мл
данного объема
\/У X ШаДЦЩУ
водяная баня,
колбы на 500мл.
^ Определение углерода гуминовых кислот
колбы на 300мл,
промывалка
►
цилиндр на 200мл,
дозатор на 50мл
►
часовое
стекло
►
воронки с!=9см,
фильтры
Определение азота гуминовых кислот
фильтрат
^1 Определение азота общего
3.3.3. ПИРОФОСФАТНЫЙ МЕТОД ОПРЕДЕЛЕНИЯ
ГРУППОВОГО СОСТАВА ГУМУСА
333.1. Принцип метода
Ускоренный метод определения состава гумуса исключает длительный процесс
декальцирования почвы и многократные обработки ее 0,1н. раствором NaOH.
При обработке почвы смесью Na4P207 + NaOH (pH 12,5) происходит наиболее
полное замещение кальция, железа и алюминия на натрий с образованием раствори¬
мых гуматов и фульватов натрия и нерастворимых фосфорнокислых солей соответ¬
ствующих катионов.
Реакции идут по схеме: ,
[R(COO)4],Ca2 + Na4P.O, -►R(COONa4) + f Ca^O,
[R(COO)4]3Fe4 + 3Na4P207—►3R(COONa4) + i Fe4(P207),
[R(COO)J,Al4 + 3Na4P207—►3R(COONa4) + | A14(P207),
Разделение фракций гумусовых веществ, связанных с кальцием и с не силикатны¬
ми формами железа (и алюминия), проводят путем дополнительного определения в
отдельной навеске количества гумусовых веществ, извлекаемых из недекальцирован-
ной почвы 0,1н. раствором NaOH (см. п.3.3.2.4.1.).
Для черноземных почв этот метод и определение группового состава гумуса по
Тюрину в модификации Пономаревой дают близкие результаты.
3332. Технологическое оборудование
Весы типа ВЛКТ.
Газовая или электропечь.
Центрифуга.
Водяная баня.
Колбы конические на 100 и 250мл.
Колбы плоскодонные из термостойкого стекла с высоким тубусом на 100мл.
Дозаторы на 100мл.
Мерные колбы на 100мл.
Воронки.
Пипетки на 10, 15, 25мл.
33.3.3. Реактивы
1. Смесь 0,1н. пирофосфата натрия с 0,1н. NaOH. 6,3мл 16н. NaOH и 44,6г
Na4PJO? • 10Н20 растворяют в дистиллированной воде, не содержащей С02
(см.п.3.8.3.2.), и доводят объем до 1л. Раствор должен иметь pH = 12,5. Готовят в день
проведения анализа.
2. 1н. H2S04. 28мл концентрированной H2S04 осторожно вливают в стакан с
дистиллированной водой и доводят объем до 1л.
3. 0,05н. H2SOr Растворяют 1,4мл концентрированной H2S04 в дистиллирован¬
ной воде и доводят объем до 1л.
4. 0,05н. NaOH. 2г NaOH растворяют в дистиллированной воде и доводят объем до 1л.
5. Растворы для определения содержания углерода см. п.3.2.3.
6. Растворы для определения содержания азота см. п.3.4.3.
7. Прокаленная пемза, песок или лесс.
333.4. Ход анализа
3.3.3.4.1. Определение содержания С и N в исходной почве (см. пп.3.2.4., 3.4.4).
3.3.3.4.2. Извлечение гумусовых веществ из почвы пирофосфатом No.
Навеску почвы 5г, просеянную через сито с диаметром ячеек 1мм, переносят в
коническую колбу на 250мл, заливают 100мл свежеприготовленной смеси 0,1н. пиро¬
- 107-
фосфата натрия с 0,1н. NaOH, закрывают колбу резиновой пробкой и оставляют на
16 часов. Затем содержимое колбы взбалтывают и фильтруют через плотный бу¬
мажный фильтр (синяя лента) либо центрифугируют.
3.3.3.4.3. Определение содержания органического углерода и азота в вытяжке
В коническую колбу емкостью 100мл берут аликвоту фильтрата в количестве
10- 15мл, добавляют прокаленный песок и выпаривают на водяной бане. Затем про¬
водят определение углерода по Тюрину (см. п.3.2.4.).
Для определения азота в плоскодонные колбы берут аналогичную аликвоту вы¬
тяжки, нейтрализуют 1н. H2S04 до появления мути и выпаривают на водяной бане.
Остаток сжигают в Змл концентрированной H2S04 и определяют азот (см. п. 3.4.4.).
3.3.3.4.4. Определение содержания С и N гуминовых кислот
25-40мл вытяжки переносят в стакан емкостью 100мл, куда по каплям при пере¬
мешивании прибавляют для коагуляции геля гуминовых кислот 1н. H2S04 до появле¬
ния мути (pH = 1,3-1,5). Содержимое стакана перемешивают, накрывают часовым
стеклом и нагревают на водяной бане в течение ЗОмин. при температуре 80°С, после
чего оставляют на 16-18 часов до полного осаждения геля.
Отфильтровывают через ткяный фильтр диамегром 7см, осадок промывают 0,05н. H2S04,
затем растворяют на фильтре горячим раствором 0,05н. NaOH и доводят объем до 100мл.
Для определения С и N берут аликвоты по 15-20мл, дальнейшее определение см. п.3.3.2.4.
3.3.3.4.5. Определение количества С и N, переходящих в раствор при обра¬
ботке почвы 0,1 н. H2S04
Навеску почвы 5г помещают в коническую колбу емкостью 250мл, заливают 200мл
0,1н. H2S04, тщательно перемешивают и оставляют на 16-18 часов. После отстаива¬
ния фильтруют через фильтр с белой лентой, почву на фильтре промывают 0,1н.
H2S04 и доводят объем фильтрата до 500мл.
Для определения С в коническую колбу на 100мл берут 50мл фильтрата, нейтра¬
лизуют сухой солью Na2C03 до появления мути, добавляют прокаленный песок,
выпаривают, дальнейшие операции см. п.3.2.4.
При определении N в плоскодонную колбу на 100мл берут 50мл фильтрата, выпа¬
ривают, озоляют в Змл концентрированной H2S04, дальнейшие операции см. п.3.4.4.
3333. Обработка результатов
Расчеты проводят по формулам (см. п.3.3.2.5.). Групповой состав гумуса рассчи¬
тывают по формам 3 и 4.
Форма 3. Содержание углерода гумусовых веществ в почвенных вытяжках
(в буквенном выражении).
Показателя
Собвдпочвы, %
Пнрофосфат + NaOH
ОДн. NaOH
Собщ. | Сгк
Сфк
Собщ
Сгк
Сфк
Содержание углерода
в % к почве,
Н
А
В
А-В
А1
В1
А1-В1
в % к Собщ. почвы
100
а
в
а-в
а1
в1
а1-в1
Форма 4. Групповой состав гумуса (С фракции к Собщ. почвы, % рассчиты¬
вается на основании данных формы 3).
Общее
содержание
углерода в
почве, %
Сгк
Сфк
Состатка
Сен
Сфк
свободные п
несиликатпые R203
связанные
с Са
сумма
100
в1
в - в1
в
а-в
100-а
в/а-в
- 108-
- 109-
3.43.6. Технологическая карта
Определение азота см.п. 3.3.2.
Определение гумуса см.п.3.2.6
Взятие аликвот
цилиндр или шприц-доза-
тор на 25-40мл, стаканы на
100мл
Осаждение гуминовых
кислот
капельница, часовое стек-
ло, водяная баня, часы
Фильтрование и промывание осадка
колбы на 250мл, воронки д=7см, филь¬
тры с синей лентой, промывалка
Растворение осадка
стаканы для осажде¬
ния гуминовых кис¬
лот
Доведение до заданного
объема
цилиндр на 100мл
Отбор аликвот
пипетка или шприц-дозатор на 10-20мл, колбы ко¬
нические на 100мл (для гумуса), колбы плоскодон-
ные на 100мл (для азота)
Выпаривание
водяная баня
3.4. ОПРЕДЕЛЕНИЕ ОБЩЕГО АЗОТА
3.4.1. Принцип метода
Метод основан на разложении органических веществ почвы (для перевода азота в
аммонийную форму) концентрированной серной кислотой в присутствии перекиси
водорода путем мокрого озоления (см. п. 2.4.1.4.).
Содержание аммонийного азота в растворе после сжигания определяют отгонкой
его на приборе Сереньева (см. п.2.4.4.4.).
3.4.2. Технологическое оборудование
Приборы Сереньева.
Весы торсионные, аналитические типа ВЛКТ.
Газовая или электропечь.
Колбы Кьельдаля или плоскодонные тугоплавкие колбы с высоким тубусом на 100мл.
Стеклянные пробки полые внутри.
Дозирующее устройство для крепких кислот.
Капельница.
Пипетки Мора на 20мл, с делениями на 10мл.
Стаканы на 800 или 1000мл.
Баллоны на 10 и 20л.
3.4.3. Реактивы
1. 1%раствор лимонной кислоты. Растворяют лимонную кислоту из расчета 1г
на 100мл ее водного раствора.
2. Титрант 0,02н. по серной кислоте. Содержимое двух фиксаналов 0,1н. Н S04
переводят дистиллированной водой в мерную колбу емкостью 1-2л, добавляют 200мл
0,1% спиртового раствора индикатора метилрота, доводят дистиллированной водой
до метки и сливают в основной бутыль.
В той же колбе растворяют в дистиллированной воде при 60°С 40г борной кисло¬
ты, добавляют 100мл 0,1% спиртового раствора метиленблау, доводят до метки и сли¬
вают в основной бутыль.
Доводят объем раствора в бутыле до Юл.
Титр проверяют по 0,02н. раствору NH4C1 или (NH4)7SO .
3. 0,02н. NHJCI или (NHJ£Or 1,0698г NH4C1 или 1,3216г (NH4)2SO , взятых на
аналитических весах, растворяют в дистиллированной воде и доводят объем до 1л.
4. Раствор куприта Na в 30% щелочи. Растворяют CuS04 • 5Н20 (1Дг в мини¬
мальном количестве дистиллированной воды - 30-50мл) и NaOH (35Or на 1000мл ди¬
стиллированной воды) и охлаждают до комнатной температуры. В полученную ще¬
лочь осторожно, по каплям, при энергичном взбалтывании вливают раствор купоро¬
са, избегая образования черного осадка. Перемешивание продолжают до получения
синего раствора куприта натрия.
3.4.4. Ход анализа
Навеску 0,5г для дерново-подзолистых и оподзоленных почв и 0,2-0,Зг для черно¬
земов (в зависимости от содержания в них азота), взятую на торсионных весах, пере¬
носят в колбу или пробирку для сжигания, заливают 5мл концентрированной H2S04,
выдерживают 10-12 часов, а затем нагревают до слабого кипения. Озоление проводят
в течение 4-7 дней (в зависимости от состава почвы). К концу озоления раствор не¬
сколько раз обрабатывают 2-3 каплями Н202. После последнего введения перекиси
продолжают нагрев еще 2-3 часа, затем раствор охлаждают и доводят дистиллирован¬
ной водой до объема 50мл.
-110-
40мл анализируемого раствора переводят в реактор прибора Сереньева, за¬
ливают при включенном вакууме щелочным раствором куприта натрия до окра¬
шивания смеси в голубой или грязно-бурый цвет и отгоняют в течение 5мин.
Титрование ведут до устойчивой в течение 10-15сек. фиолетовой окраски реак¬
тивной смеси в барбатере прибора Сереньева. Холостую пробу определяют мето¬
дом добавок (см. п. 2.4.Д.4.). Работа прибора Сереньева описана в разделе 4.1.
3.4.5. Обработка результатов
Расчет проводят по формуле:
(g-«)-0,02-JM4-100 (а-в)-0,028-#
и-1000 ~ н
где N - содержание азота в анализируемой почве, %;
а - количество титранта, пошедшего на титрование пробы, мл.;
в - количество титранта, пошедшего на холостое титрование, мл.;
0,02 - нормальность титранта;
К - поправка к титру;
14 - мг-экв азота;
н — аликвотная навеска, соответствующая 40 мл раствора, г.
Допустимые расхождения повторных определений приведены в таблице 22.
Таблица 22.
Содержание азота, %
Допустимые отклонения от среднего при повторных
определениях (в отн. %)
в одной лаборатории
в разных лабораториях
0,01-0,05
9
14
0,05-0,1
7
11
0,1-0,2
5
8
Свыше 0,2
4
7
3.4.6. Технологическая карта
Ддбавление кони. Нд80д
разливочное устройство либо доза¬
тор для агрессивных жидкостей на
5 мл
Доведение до заданного объема
колба мерная на 50мл
Перегонка и ТДТРРШИК
прибор Сереньева, часы песочные
-111 -
3.5. ОПРЕДЕЛЕНИЕ НИТРИФИКАЦИОННОЙ
СПОСОБНОСТИ ПОЧВЫ (Метод Кравкова)
В стакан для компостирования, на дно которого насыпан дренаж (в виде битого
стекла), вставляется стеклянная трубочка для улучшения аэрации почвы. Масса стака¬
на, дренажа и стеклянной трубочки составляет тару.
Можно использовать взвешенные плоскодонные колбы объемом 300 - 350мл.
ЮОг воздушно-сухой почвы переносят через воронку в стакан или колбу, почва
предварительно просеивается через сито с отверстиями 1 - 2мм, и доводят влажность
почвы до 60% капиллярной влагоемкости, которая предварительно определяется в
лаборатории.
Количество воды, необходимой для доведения влажности почвы до 60% капил¬
лярной влагоемкости вычисляется следующим образом: допустим, что капиллярная
влагоемкость взятой почвы составляет 40% (в пересчете на воздушно-сухую почву),
тогда 60% капиллярной влагоемкости составляет 40*60/100=24%, тогда к ЮОг почвы
необходимо прилить 24мл воды. Если влажность взятой почвы составляет 5,6%, то к
100г почвы необходимо прилить 18,4мл воды (24мл - 5,6мл).
Из каждого образца почвы берут параллельно две навески - одну для компостиро¬
вания, другую для определения исходного содержания нитратного азота.
Почву, взятую для компостирования распределяют по дну ровным слоем и увлаж¬
няют дистиллированной водой до 60% капиллярной влажности, медленно приливая
ее по стенке колбы или стакана. Когда вода попадает прямо на почву, может образо¬
ваться корка, что резко нарушает условия аэрации и приведет к неправильным резуль¬
татам.
В массовых анализах приливают в среднем следующее количество воды к ЮОг
почвы:
- дерново-подзолистые песчаные и супесчаные 15 мл
- тяжелосуглинистые, темно -серые лесные, светло-каштановые,
обыкновенные черноземы и сероземы 20мл
- типичные мощные и оподзоленные черноземы, лугово-черноземы 25мл
- красноземы 30мл.
Стаканы, колбы с навесками почвы накрывают фильтровальной бумагой, а бу¬
тылки закрывают ватным тампоном и помещают в термостат где выдерживают 7 -
14 суток при температуре 28°С. Затем их вынимают и приливают по 100мл раствора
0,05% K2S04, после чего при помощи элекгромешалки перемешивают суспензию
Змин. Вытяжку отфильтровывают через складчатые фильтры из простой фильтро¬
вальной бумаги. Таким же образом готовят вытяжки из навесок исходных почв (без
компостирования) для определения начального содержания нитратного азота.
В фарфоровые чашки для выпаривания отбирают по 5 или 10мл фильтрата при
компостировании почв и 50мл вытяжки из исходных почв и выпаривают на водяной
электрической бане.
Одновременно выпаривают образцовые растворы в объеме 2-5 - 10 - 15 — 20 —
30мл. Чашки снимают с бани, когда еще на дне остается несколько капель жидкости,
которая испарится с горячей поверхности после снятия чашки с водяной бани.
В чашки с сухим остатком приливают дозатором по 1мл дисульфофеноловой кис¬
лоты и тщательно размешивают ее по дну и стенкам чашки закругленным концом стек¬
лянных палочек. Через Юмин. прибавляют 15мл дистиллированной воды и нейтрали¬
зуют жидкость 20%-ным NaOH при помешивании до установления неисчезающего
- 112-
желтого окрашивания. Окрашенные растворы переносят в мерные колбы на 50мл.,
чашки ополаскивают водой, сливая ее в те же колбы и объем жидкости в колбах дово¬
дят водой до метки. Если окраска испытуемого раствора интенсивнее наиболее окра¬
шенного образцового раствора, то определенную его часть переносят в колбу на 50мл
и разбавляют водой до метки, учитывая затем разбавление при вычислении результата
определения.
Колориметрирование нитратов проводят в тот же день на ФЭКе с синим свето¬
фильтром.
Содержание нитратов в почве вычисляют по калибровочной кривой.
Таблица 23. Приготовление шкалы образцовых растворов для калибровочной
кривой (0,002мг азота в 1мл)
Объем образцового
раствора нитрата, мл
Количество азота в
образцовом растворе, мг
Количество азота на 1кг
почвы, мг*
2
0,004
2
5
0,010
5
10
0,020
10
20
0,040
20
30
0,060
30
* Содержание азота дано для случая, когда при определении берется 10 мл вытяжки.
Таблица 24.
Группировка почв по содержанию
нитратов (степень обеспеченности в мг
на ЮОг почвы)
Низкая - 0,8 - 1,5 хорошая - 3,0 - 6,0
Средняя - 1,5 - 3,0 высокая - > 6,0.
Нитрификационная способность почвы (X) в мг N03 или N-N03 на ЮОг почвы
вычисляют по формуле: X = Х2 -
где Х2 - содержание N03 или N-N03 в почве после компостирования, мг на 100г почвы;
X, - содержание N03 или N-N03 в почве до компостирования, мг на 100г почвы.
Нитриф икациопны е
способности почвы
Содержание NO,
мг па ЮОг почвы
очень низкая
<0,5
низкая
0,5 - 0,8
средняя
©
00
1
повышенная
1,5 - 3,0
высокая
3,0 - 6,0
очень высокая
>6,0
3.5.1. Технологическая карта
-113-
3.6. ОПРЕДЕЛЕНИЕ ГИДРОЛИЗУЕМОГО АЗОТА
ПО КОРНФИЛЬДУ
3.6.1. Принцип метода
Метод основан на щелочном гидролизе органических азотных соединений почвы
при воздействии на нее 1н. раствора NaOH в течение 2 суток при температуре 26°С.
Выделившийся аммиак поглощается борной кислотой и определяется прямым титро¬
ванием серной кислотой.
Реакции идут согласно уравнениям:
3NH.OH + Н3ВО, = (NHJ, В03 + ЗН20
2(NH4)3 ВОэ + 3H2S04 = З^Н^ S04 + 2Н3ВО?.
3.6.2. Технологическое оборудование
Весы типа ВЛКТ.
Термостат.
Чашки Конвея.
Пипетки на 2 и 5мл.
Бюретка на 25мл.
Капельница.
3.6.3. Реактивы
1. 1н. NaOH. 40г реактива растворяют в дистиллированной воде и доводят объем до 1л.
2. 2%раствор борной кислоты. 2г реактива растворяют в 100мл дистиллирован¬
ной воды.
3. Индикатор Гроака. 0,2г метилового красного растворяют в 100мл этилового
спирта; 0,1 г метилового голубого растворяют в 100 мл этилового спирта. Полученные
растворы смешивают. Раствор хранят в темной склянке.
4. 0,01н. H2S04. Фиксанал серной кислоты растворяют в дистиллированной воде
и доводят объем до 1л в мерной колбе. 100мл этого раствора разбавляют дистиллиро¬
ванной водой до 1л в мерной колбе.
3.6.4. Ход анализа
2г почвы помещают в одно из внешних отделений чашки Конвея. В другую часть
внешнего отделения чашки приливают 5мл 1,0н. раствора NaOH. Во внутреннюю
емкость чашки вводят 2мл 2% раствора борной кислоты. Чашку закрывают крышкой
с пришлифованными краями и в течение 20-30сек. осторожно вращают для того, что¬
бы раствор щелочи смешался с почвой. Затем чашки помещают в термостат на 48
часов при температуре 26°С. По истечении указанного срока содержимое внутреннего
отделения чашки титруют 0,01н. раствором H2S04 в присутствии индикатора Гроа¬
ка до перехода окраски раствора с зеленого в фиолетовый.
- 114-
3.6.5. Обработка результатов
Расчет проводят по формуле:
1Г а 0,01 14 100
N, = .
н
где Na - содержание азота, мг/100г почвы;
а - объем 0,01 и. H2S04, пошедшей на титрование, мл.;
0,01 - нормальность H2S04;
14 - мг-экв азота;
н- навеска почвы, г.
3.6.6. Технологическая карта
3.7. ОПРЕДЕЛЕНИЕ ГИДРОЛИЗУЕМОГО АЗОТА МЕТОДОМ
ТЮРИНА И КОНОНОВОЙ
3.7.1 .Принцип метода
Метод основан на гидролизе почвы 0,5н. раствором серной кислоты при комнат¬
ной температуре и последующем определении общего количества азота, переходяще¬
го в вытяжку. При этом обычно учитывается как минеральный азот (аммиак и нитра¬
ты), так и водорастворимые и гидролизуемые формы органических соединений азота
в виде аминокислот, амидов, некоторых групп белковых веществ.
Реакции проходят согласно уравнениям
21^0, + 8H2S04 = 2 К, S04 + 2 Cr2 (S04)3 + 8Н20 I 302
RNH СНСООН +H„S04 > NH, + С02 + S02 + Н20
2 nh; + h2so4 = (Niy2 so4
H2S04 + Fe = FeS04 + H.
H2S04 + Zn = ZnS04 + Й2
2NH, + H2S04=(NH4)2S04
3.7.2.Технологическое оборудование
Весы типа BJ1KT.
Прибор Кьельдаля.
Бюретки 25мл, 100мл.
Пипетки 25, 50мл.
Мерный цилиндр 50мл.
Капельница.
3.7.3.Реактивы
1. Смесь иинковой пыли и восстановленного железа. 9:1.
2 0.5н. раствор серной кислоты. 14мл серной кислоты плотностью 1,84г/см3
растворяют в дистиллированной воде и доводят объем в мерной колбе до 1л.
- 115-
3. 0.02н. раствор серной кислоты. Фиксанал серной кислоты растворяют в
дистиллированной воде и доводят объем до 1л в мерной колбе. 200мл этого раство¬
ра разбавляют дистиллированной водой до 1л в мерной колбе.
4. 10% раствор бихромата калия. 10г реактива растворить в дистиллированной
воде и довести объем до 100мл в мерной колбе.
5. Индикатор метиловый красный. 0,2г реактива растворить в 100мл 60% этило¬
вого спирта.
6. 50% раствор NaOH. 50г реактива растворить в дистиллированной воде и дове¬
сти объем в мерной колбе до 100мл.
3.7.4. Ход анализа
20г почвы, из которой удалены корневые остатки растений, перенести в колбу
емкостью 250 - 300мл и прилить 100мл 0,5н раствора серной кислоты. После 3 ми¬
нутного взбалтывания раствор оставляют в покое при комнатной температуре на
протяжении 16-18 часов. Затем раствор фильтруют через сухой беззольный фильтр.
50мл фильтрата переносят в коническую колбу емкостью 150 - 200мл и добавляют
0,5г смеси цинковой пыли и восстановленного железа и кипятят на электрической
плитке до полного растворения смеси железа и цинка. В процессе нагревания и
кипячения колбы с раствором должны быть накрыты воронками.
После охлаждения раствора в колбы приливают 5мл H2S04 плотностью 1,84г/см3 и
выпаривают жидкость до появления белых паров и жидкости бурого цвета. Снова
охлаждают колбы и приливают 2мл 20% раствора хромовой кислоты или 2,5мл 10%
раствора бихромата калия. Накрывают колбы воронками и на протяжении Юмин.
кипятят при слабом нагревании до появления зеленого оттенка раствора. Если ра¬
створ приобретает зеленую окраску сразу же после добавления хромовой и серной
кислот, то анализ проводят повторно, уменьшая навеску почвы или добавляют в
колбу 1 - 2мл хромовой кислоты и двойное количество серной кислоты.
В приемную колбу прибора Кьельдаля вливают 20мл 0,02н. раствора H2SO^ 3
капли индикатора метилового красного и опускают трубку холодильника в раствор.
В отгонную колбу количественно переносят испытуемый раствор, вносят гранулу
цинка, мерным цилиндром осторожно по стенке колбы приливают 20мл 50% ра¬
створа щелочи. При этом верхняя часть горловины колбы не должна смачиваться
щелочью. Реакция раствора должна быть щелочной, о чем свидетельствует измене¬
ние цвета окрашивания раствора и появления бурого цвета осадка. После добавле¬
ния к раствору щелочи отгонная колба должна быть немедленно присоединена к
каплеуловителю. В процессе дистиллирования необходимо следить за тем, чтобы в
патрубок холодильника не засасывался раствор из приемной колбы. Продолжи¬
тельность отгонки 30 - бОмин. Считается, на протяжении 15 - 20мин. кипения ра¬
створа из отгонной колбы дистиллируется 75 - 85% общего содержания аммиака.
После отгонки 2/3 объема жидкости проверяют полноту отгонки аммиака с
помощью реактива Несслера. С этой целью в одну из двух пробирок из трубки
холодильника (после ополаскивания дистиллированной водой) набирают 1 - 2мл
дистиллята и приливают 1 -2 капли реактива Несслера. Во вторую пробирку налива¬
ют эквивалентное количество дистиллированной воды и реактива Несслера. Визу¬
ально сравнивают окраску жидкостей в пробирках. Отсутствие желтоватого цвета
окрашивания свидетельствует об окончании отгонки аммиака.
По окончании отгонки конец трубки холодильника ополаскивают водой в при¬
емную колбу и титруют 0,02н. раствором NaOH. Определяют количество 0,02н. ра¬
створа H2S04 затраченное на связывание аммиака. Перед титрованием рекомендуется
раствор прокипятить для удаления угольной кислоты.
-116-
Для определения содержания азота в реактивах необходимо провести конт¬
рольный опыт в аналогичных условиях и количестве реактивов за исключением
анализируемой почвы. Если титры H2S04 и NaOH не соответствуют точной моляр¬
ной концентрации, необходимо вводить поправку к титру.
3.7.5.0бработка результатов
Содержание легкогидролизуемого азота (N), в миллиграммах на 100г почвы рас¬
считывают по формуле: . ч
xr (F-F.V 0.28 100
N = - 1- ,
т
где V - объем точно 0,02н. раствора H2S04 взятой для поглощения аммиака^иг
V 1 - объем точно 0,02н. раствора NaOH, затраченного на титрование остатаа
H2S04 в исследугмом растворе, мл;
0,28 - количество азота, соответствующее 1мл 0,02н. р-ра H2S04, мг,
100 - коэффициент для пересчета в %;
т - расчетная масса почвы (взятая на определение), г.
При наличии азота в реактивах от его содержания в вытяжке вычитается его со¬
держание в реактивах.
3.7.6.Технологическая карта
3.8. ОПРЕДЕЛЕНИЕ СОДЕРЖАНИЯ
МИНЕРАЛЬНЫХ ФОРМ АЗОТА В ПОЧВЕ
3.8.1. Принцип метода
Нитраты и обменный аммоний извлекаются из почвы 1н. раствором КС’1 при соот¬
ношении почва :раствор 1: 2,5, одноминутном взбалтывании и настаивании в течение
18-20 часов. Определение содержания ионов в вытяжке проводят колориметрически.
-117-
Для окрашивания нитратов использован а-нафтиламиновый метод. При этом
нитраты восстанавливают до нитритов в щелочной среде гидразином в присутствии
меди как катализатора. Выделяющийся в реакции молекулярный азот обеспечивает
консервацию образовавшихся нитритов:
При взаимодействии нитритов с сульфаниламидом и а-нафтил амином в кис¬
лой среде образуется окрашенное в красно-розовый цвет азосоединение:
Для определения аммония использована реакция индофенольной зелени, образу¬
ющейся в щелочной среде при взаимодействии NH4 с гипохлоритом и салицилатом
натрия согласно уравнению:
Влияние двух- и трехвалентных катионов при окрашивании нитратов и аммония
устраняется введением трилона Б.
Определение нитратного и аммонийного азота в почве может проводиться в све-
жеотобранных образцах с естественной влажностью, а также в сухих образцах, про¬
сеянных через сито с диаметром ячеек 2мм.
3.8.2. Определение нитратов
3.8.2.1. Технологическое оборудование
Фотоэлектроколориметр.
Весы типа ВЛКТ.
Кассеты 10-позиционные.
Встряхиватель для 10-позиционных кассет.
Дозаторы на 10, 25, 50 мл.
Воронки d=7cM (желательно пластмассовые).
Пипетка с грушей или шприц-дозатор на 5мл.
3.8.2.2. Реактивы
1. 0,5% раствор пирофосфата натрия. 5г натрия пирофосфорнокислого и 6 г
NaOH растворяют в дистиллированной воде и доводят объем до 1л.
2. Раствор катализатора. 2,5г CuS04 • 5Н20 растворяют в дистиллированной
воде и доводят объем до 1л. Раствор сохраняется длительное время.
3. Восстановитель
Исходный раствор восстановителя. 13,8г гидразина сернокислого растворяют в ди¬
стиллированной воде и доводят объем до 1л в мерной колбе. Раствор хранится 6 меся¬
цев.
Рабочий восстанавливающий раствор. 6мл раствора катализатора и 200мл ра¬
створа восстановителя переносят в мерную колбу емкостью 1л, доводят до метки
дистиллированной водой и перемешивают. Раствор хранится в течение 1 недели в
склянке из темного стекла.
4. Окрашивающий раство.
Запасной окрашивающий раствор. К 500мл дистиллированной воды приливают
200мл ортофосфорной кислоты, растворяют 2г а-нафтиламина (1 -нафтиламина)
или ^(1-нафтил)-этилендиамин дигидрохлорида и доводят в мерной колбе объем
2NaNO + N,Hd + Na J\07 + 4NaOH —> 2 NaNO, + N. + Na,SO, + 2Na4P04 + 5H,0
Л 2 4 4 2 7 2224 34 2
+ NH4C1 + 3NaOCl + 4NaOH > 4NaCl + 2Na2CO} +
- 118-
до 2л дистиллированной водой. Раствор хранится в холодильнике в склянке из тем¬
ного стекла не более 1 месяца.
Рабочий окрашивающий раствор. В 200мл запасного окрашивающего раство¬
ра растворяют 1г сульфаниламида (стрептоцида), прибавляют 0,2г трилона Б, пред¬
варительно растворенного в дистиллированной воде, и доводят водой объем до 1л.
5. Сульфаниламид
В качестве реактива можно использовать аптечный препарат - стрептоцид бе¬
лый нерастворимый.
6. Образцовые растворы для определения нитратов
Исходный образности раствор. 0,722г калия азотнокислого, высушенного при
температуре 100-105°С до постоянной массы, взвешивают с погрешностью не более
0,001 г, растворяют в 1н. КС1 и доводят им до метки в мерной колбе на 1л. Раствор
содержит 0,1мг М-М03 в 1мл.
Рабочая шкала образцовых растворов. В мерные колбы вместимостью 250мл вли¬
вают количества исходною образцового раствора, указанные в таблице 25, и доводят
до метки раствором 1н. КС1.
Таблица 2S.
' "—-— Номера колб
Показатели
1
2
Э
4
5
6
7
8
Объем исходного образцового
раствора, мл.
0
2,5
5,0
7,5
10,0
15,0
20,0
30,0
Содержание N-N03
в мг/л раствора
0
1,0
2,0
3,0
4,0
6,0
8,0
12,0
в мг/кг почвы
0
2,5
5,0
7,5
10,0
15,0
20,0
30,0
Из полученных образцовых растворов берут аликвоты равные по объему пробам
почвенных вытяжек, окрашивают их так же, как при анализе вытяжек, а затем колори¬
метр ируют. Калибровочный график строят, откладывая на оси абсцисс содержание
азота N-N03 мг/кг почвы, на оси ординат - оптическую плотность.
3.8.23. Ход анализа
20г почвы заливают 50мл 1 н. раствора КС1 , перемешивают в течение 1мин. и
оставляют на 18-20 часов. На следующий день суспензию взбалтывают и фильтруют.
К 5мл вытяжки добавляют 10мл 0,5% раствора пирофосфата натрия, перемеши¬
вают, прибавляют 10мл рабочего восстанавливающего раствора и снова перемешива¬
ют. Через 10 мин. приливают 25мл рабочего окрашивающего раствора, перемешива¬
ют и оставляют на 15мин. до полного развития окраски.
Колориметрируют в кювете с толщиной просвечиваемого слоя 1см при 545нм
(желто-зеленый светофильтр), если используется N-( 1 -нафтил)-этилендиамин дигид¬
рохлорид и при 520нм (зеленый светофильтр), если используется а- нафшламин,
не ранее чем через 15мин. и не позднее чем через 1,5 часа после прибавления
окрашивающего раствора.
Аналогично проводят окрашивание шкалы образцовых растворов.
3.8.2.4. Обработка результатов
Содержание азота в анализируемых почвах находят непосредственно в мг N-
N03 на 1кг почвы по калибровочному графику.
-119-
Допустимые отклонения результатов определения приведены в таблице 26.
Таблица 26.
Содержание нитратов,
мг/кг почвы
Допустимые отклонения от среднего при повторных
определениях (в отн. %)
в одной лаборатории
в разных лабораториях
5 - 10
17
25
10-20
14
20
Свыше 20
12
18
3.8.2.5. Технологическая карта
3.8.3. Определение аммония
3.8.3.1. Технологическое оборудование
Фотоэлектроколориметр.
Весы типа ВЛКТ и аналитические.
Кассеты 10-позиционные.
Встряхиватель для 10-позиционных кассет.
Воронки d=7 см (желательно пластмассовые).
Дозаторы на 1, 18 и 50мл.
Пипетка или автоматическая пипетка на 50мл.
Колбы конические на 50мл.
Колбы мерные на 1000мл.
3.8J.2. Реактивы
1. Окрашивающий раствор
Исходный окрашивающий раствор. 56,7г салициловокислого натрия, 16,7г калия-на¬
трия виннокислого и 26,7г NaOH растворяют в 700мл дистиллированной воды и дезами¬
нируют раствор до удалений загрязнений аммонием. После охлаждения раствора в нем
растворяют 0,4г нитропруссида натрия и доводят объем до 1л безаммиачной дистиллиро¬
ванной водой. Реактив хранят в темной склянке в холодильнике в течение 1 месяца.
Рабочий окрашивающий раствор. Исходный окрашивающий раствор разбавляют
безаммиачной дистиллированной водой в соотношении 1 : 8 и растворяют в нем трилон
Б - 2г на 1л конечного раствора. Реактив готовят в день проведения анализа.
- 120-
2. Восстановитель
Исходный раствор гипохлорита натрия. 150г хлорной извести перемешивают с 255мл
дистиллированной воды. В другой емкости растворяют 105г Na2C03 в 255 мл дистилли¬
рованной воды. Затем оба раствора сливают вместе при постоянном перемешивании.
Масса сначала густеет, затем разжижается. Полученную суспензию оставляют на 1-2
суток для отстаивания, затем надосадочную жидкость сливают и фильтруют.
Полученный реактив имеет концентрацию активного хлора 6-10%. Для точного
определения его концентрации 1мл реактива разбавляют дистиллированной водой до
50мл, прибавляют 2г KJ и 10мл 1н. НС1. Образовавшийся йод оттитровывают 0,1н.
раствором гипосульфита (1мл 0,1н. раствора гипосульфита соответствует 0,00355г
активного хлора).
Расчет концентрации активного хлора в исходном растворе гипохлорита прово¬
дят по формуле:
С = а • 0,00355 • 100,
где С - концентрация активного хлора, %;
а - количество мл. 0,1н. раствора гипосульфита, пошедшего на титрование;
0,00355 - титр гипосульфита по активному хлору;
100 - коэффициент пересчета в проценты.
Раствор хранится в склянке из темного стекла до 1 года.
Рабочий раствор гипохлорита натрия. Исходный раствор гипохлорита натрия раз¬
бавляют до концентрации 0,125% дистиллированной водой. Раствор готовят в день
проведения анализа.
Реактивы для определения концентрации активного хлора в исходном растворе, а
также примеры расчета его концентрации и разбавления см. пп.2.4.5.3.
3. Образцовая шкала для определения аммиачного азота
Исходный образцовый раствор. 0,382г NH4C1 взвешивают с погрешностью не
более 0,001 г, растворяют в мерной колбе на 1л, доводят до метки 1н. раствором КС1.
Полученный раствор содержит 0,1мг N-NH4 в 1мл.
Рабочая шкала образцовых растворов. В мерные колбы емкостью 250мл налива¬
ют количества исходного образцового раствора, указанные в таблице 27, доводят до
метки 1н. раствором КС1.
Таблица 27.
Номера колб
Показатели —
1
2
3
4
5
б
7
8
Объем исходного образцового раствора, мл.
0
5
10
20
30
40
50
60
Содержание N-NH,
в мг/л раствора
0
2
4
8
12
16
20
24
в мг/кг почвы
0
5
10
20
30
40
50
60
Окрашивание и колориметрирование рабочей шкалы образцовых растворов про¬
водят так же, как описано в анализе для почвенных вытяжек.
Калибровочный график строят, откладывая на оси абсцисс содержание N-NH4 в
мг/кг почвы, на оси ординат - оптическую плотность.
4. Безаммиачная вода. Для приготовления указанных выше реактивов без амми¬
ачную воду готовят следующим образом. Добавляют к 5л дистиллированной воды
0,5-1,0г щелочи, нагревают до кипения и дезаминируют в течение 5-10мин. проточ¬
ной струей воздуха, используя для этих целей вакуум-насос или водоструйный насос.
Удаление аммиака можно проводить также путем длительного кипячения раствора.
- 121 -
3.833. Ход анализа
Из вытяжек, полученных для определения нитратов и аммония в почве, отбирают
1,0мл, добавляют 18мл рабочего окрашивающего реактива и 1,0мл рабочего раствора
гипохлорита натрия. Растворы перемешивают после каждого дозирования реагентов
и оставляют на 1 час для развития окраски.
Колориметрируют в кювете с толщиной просвечиваемого слоя 1см при 655нм
(красный фильтр) не ранее чем через 1 час и не позднее чем через 2,5 часа после
прибавления рабочего раствора гипохлорита натрия.
3.83.4. Обработка результатов
Содержание аммонийного азота в анализируемых почвах находят по калибровоч¬
ному графику непосредственно в мг N-NH4 на 1кг почвы.
3.83.S. Технологическая карта
3.9. ОПРЕДЕЛЕНИЕ НИТРАТОВ
И ОБМЕННОГО АММОНИЯ В ПОЧВАХ МЕТОДОМ ННЦ ИПА
3.9.1. Принцип метода
Определение проводится в почвах природного и нарушенного сложения. Допус¬
тимое отклонение анализа при массовой концентрации обменного аммония 20мг/100
г в диапазоне от 0,1 до 1мг/100г; при 15мг/100г в диапазоне от 1 до Змг/ЮОг; при
10мг/100г в диапазоне не более Змг/ЮОг.
Допустимое отклонение концентрации нитратов в почве при двух параллельных
при концентрации 20мг/100г в диапазоне от 0,01мг/100г; при концентрации 15мг/100г
в диапазоне более 1мг/100г.
Извлечение нитратов (N-N03) и обменного аммония (N-N04) в почве проводится
1% раствором K2S04 в одной навеске и далее определение обменного аммония с реак¬
тивом Несслера и нитратов спектрометрическим методом в модификации Цыганка.
Пробы почвы анализируют при естественной влажности, но не более чем че¬
рез 5 часов после их отбора или доводят до воздушно-сухого состояния путем под¬
сушивания при температуре до 40°С. Допускается хранение почвенных образцов в
состоянии природной влажности не более 2 суток при температуре от +1 до +5°С
после чего они должны быть проанализированы или высушены.
Образцы почвы в воздушно-сухом состоянии измельчают, пропускают через сито
с диаметром отверстий 1мм и хранят в пакетах или коробках.Пробы почвы для
- 122 -
анализа в состоянии естественной влажности высыпают на ровную поверхность,
тщательно перемешивают, разравнивают слоем не более 1см и отбирают ее не
менее чем из 10 мест в количестве 30г.Аналогично отбирают пробу для определе¬
ния влажности весом 10 - 15г.
3.9.2. Реактивы
1. V% раствор сернокислого калия. 1г реактива, взвешенного с погрешностью не
более 0,01 г, растворить в 100мл дистиллированной воды в мерной колбе (экстраги¬
рующий раствор).
2. Раствор сегнетовой соли. 50г соли калия-натрия виннокислого, взвешенного с
погрешностью не более 0,1г, растворяют в мерной колбе на 100мл и доводят до метки
дистиллированной водой не содержащей аммиак. При наличии в воде иона аммония
раствор кипятят до его отсутствия на реакцию с реактивом Несслера. Раствор разбав¬
ляют безаммиачной водой до исходного объема и повторяют пробу на аммоний.
3. Запасной раствор кониентраиии N-NQ1 0. мг/1мл или 100мг/1л .Готовят из хи¬
мически чистого перекристаллизованного KN03. Реактив перед взятием навески про¬
сушивают при 105°С до постоянного веса, охлаждают в эксикаторе. Навеску 0,7216г
реактива, взвешенную с погрешностью не более 0,001 г растворяют в экстрагирую¬
щем растворе (п.1) и доводят объем раствора в мерной колбе до 1л.
4. Растворы сравнения для определения N-NOyB мерные колбы емкостью 500мл
переносят объемы раствора, указанные в табл.28, сегнетовой соли 50% концентрации (п.2).
Таблица 28.
Характеристика раствора
Номер раствора сравнения
1
2
3
4
5
объем раствора, приготовленного по п.З, мл
25
20
10
100*
50*
концентрация N-NO, в растворах сравнения, мг/л
5
4
2
1
0,5
Раствор готовится из раствора №1.
5. Запасной раствор кониентраиии N-NHJQ.1 мг/мл или 100мг/1л Готовится из
х.ч. перекристаллизованного аммония хлористого. Реактив перед взятием навески
просушивают при 50°С до постоянного веса, затем охлаждают в эксикаторе. Навеску
0,3820г аммония хлористого взвешивают с погрешностью не более 0,001 г, растворя¬
ют в дистиллированной воде и доводят объем раствора в мерной колбе до 1л.
6. Рабочий раствор кониентраиии N-NH' 0.01 мг/мл. Готовят 10-кратным раз¬
бавлением запасного раствора (п.5).
7. Растворы сравнения для определения N-NHi В мерные колбы емкостью 50 мл
берутся объемы раствора указанные в табл.29 и приготовленного согласно п.6. Объе¬
мы растворов в колбах доводят до метки дистиллированной водой и перемешивают.
Таблица 29.
Характеристика раствора
Номер раствора сравнения
1
2
3
4
5
объем раствора, приготовленного по п.6, мл
1
2,5
5,0
7,5
10
юнцентрация N-N04 в растворах сравнения, мг/мл
0,0002
0,0005
0,0010
0,0015
0,0020
Раствор сравнения используют для градуировки ФЭКа в день проведения анали¬
за. Окрашивание растворов сравнения проводят аналогично окрашиванию вытя¬
жек, которые анализируются одновременно с ними.
- 123-
3.9.3. Ход анализа
Пробы почвы, ЗОг взвешенные с погрешностью не более 0,1 г пересыпают в
емкости вставленные в 10-позиционные кассеты или конические колбы. К пробам
почвы приливают дозатором или цилиндром по 150мл экстрагирующего раствора
1% K2S04. Колбы с раствором и навеской почвы взбалтывают на ротаторе 1 час и
фильтруют или взбалтывают 15мин. и оставляют на ночь. Затем суспензии взбалты¬
вают вручную и фильтруют через фильтры отмытые от аммиака. Их используют
при определении N-N03 и N-NH4
Определение N-NQ3 После фильтрования в вытяжках на спектрофотометре
определяют их оптическую плотность. Сначала измерение проводится при длине
волны 220 нм, а затем при 240нм.
Определение N-NHr После фильтрования определяют какое количество вытяж¬
ки необходимо для анализа. Для этого в пробирку вливают 5мл фильтрата, прибавля¬
ют по 2 капли сегнетовой соли и раствора Несслера, взбалтывают. Если аммония так
много, что появляется бурый осадок, то для анализа берется 10мл фильтрата. В том
случае, если раствор приобретает красноватый оттенок - от 15 до 25мл, а если желто¬
го - от 30 до 40мл. Необходимое количество фильтрата отмеряют пипеткой и перено¬
сят в мерную колбу емкостью 50мл, приливают дистиллированную воду до объема
40мл, перемешивают, приливают 2мл сегнетовой соли, 2мл реактива Несслера и до¬
водят дистиллированной водой до метки. Взбалтывают и колориметрируют на ФЭКе
с синим светофильтром или на спектрофотометре при длине волны 440нм. Обяза¬
тельно проведение параллельно холостого анализа на чистоту реактивов.
3.9.4. Обработка результатов
По результатам спектрофотометрии растворов сравнения строится градуировоч¬
ный график. На оси абсцисс откладывается концентрация N-N03 мг/1л, а на оси орди¬
нат - соответствующую ей оптическую плотность, обусловленную нитратами, кото¬
рая определяется по формуле:.
Д„= Д*,- Дд-1.5 ,
где Дн - оптическая плотность обусловленная поглощением нитратов;
Д^ - оптическая плотность всей вытяжки, которая определена при
ч длине волны 220нм;
Дд - оптическая плотность всей вытяжки, которая определялась при
длине волны 240нм;
1,5 - коэффициент пересчета оптической плотности при переходе от
длины волны 240нм к рабочей 220нм.
Массовая концентрация N-N03 в почве определяется непосредственно по градуи¬
ровочному графику и пересчитывается по формуле
д-К-100
1 ” т 1000 ’
где Сj - массовая концентрация N-N03, мг/100г почвы;
а - концентрация N-N03, мг/1л;
т - масса почвы, г;
V - объем 1% раствора K2S04, взятого для приготовления вытяжки, мл;
100 - коэффициент для пересчета на ЮОг почвы;
1000 - коэффициент для пересчета мл на 1л.
- 124-
Расчет концентрации N-NH4 в мг/100 г почвы. По результатам фотометрии
растворов сравнения строится градуировочный график. На оси абсцисс откладыва¬
ется содержание N-NH4 в растворах сравнения, мг/мл, на оси ординат - соответ¬
ствующие им показания ФЭКа. По градуировочному графику находится массовая
концентрация N-NH4, мг/мл. Пересчет массовой концентрации N-NH4 проводится
по формуле „ _a-v- 50 100
2" wF,
где С2 - массовая концентрация N-NH4 ,мг/100г почвы;
а - количество аммония, найденное по калибровочному графику, мг/мл;
т - масса почвы, г;
V - объем 1% K2S04, взятого для приготовления вытяжки, мл;
Vt - объем вытяжки, взятый для окрашивания, мл;
50 - объем мерной колбы, в которой проводится окрашивание, мл;
100 - коэффициент для пересчета на 100г почвы.
Если результаты измерений N-N03 или N-NH4 выходят за границы градуиро¬
вочного графика, то определение повторяют уменьшая объем вытяжки для анализа.
При анализе проб в состоянии природной влажности результаты анализа пере¬
считывают на абсолютно сухую почву, умножая на коэффициент гигроскопической
влаги по формуле: i лл
К. —
~~ 100 —'
ще К- коэффициент гигроскопической влаги;
100 - коэффициент пересчета в %;
W - массовая доля влаги в почве.
Массовую часть влаги в почве в % определяют высушиванием пробы в термо¬
стате при 100 (±5)°С на протяжении 6 часов и вычисляют по формуле
т1 - т2
ще m - масса пробы с бюксом до высушивания, г;
nij - масса пробы с бюксом после высушивания, г;
- масса пустого бюкса, г;
100 - коэффициент пересчета в %.
Массовая часть азота в пересчете на абсолютно сухую почву определяется по
формуле:
Х, = Х К ,
ще Xj - массовая часть азота в абсолютно сухой почве, мг/100г почвы;
X - массовая часть азота во влажной почве, мг/100г почвы;
К - коэффициент гигроскопической влаги.
3.9.5. Технологическая карта
- 125-
3.10. ОПРЕДЕЛЕНИЕ АЗОТА НИТРАТОВ
ИОНОМЕТРИЧЕСКИМ МЕТОДОМ
3.10.1. Метод отбора проб
Пробы почвы анализируют в состоянии естественной влажности, но не более
чем через 5ч после их отбора или доводят до воздушно-сухого состояния путем
подсушивания при температуре до 40°С. Допускается хранение проб в состоянии
естественной влажности не более 2 суток при температуре 1- 5°С, после чего они
должны быть проанализированы или высушены.
Пробы в воздушно-сухом состоянии измельчают, пропускают через сито с круг¬
лыми отверстиями диаметром 1-2мм и помещают в коробки или пакеты. Пробу на
анализ из коробки отбирают шпателем или ложкой, предварительно перемешав почву
на всю глубину коробки. Из пакетов пробу высыпают на ровную поверхность, тща¬
тельно перемешивают, распределяют слоем не более 1см и отбирают не менее чем из
пяти точек пробу для анализа массой 20,0г.
Пробы в состоянии естественной влажности тщательно перемешивают, распре¬
деляют слоем толщиной не более 1см на ровной поверхности и отбирают не менее
чем из десяти точек пробу для анализа массой 20,0г. Аналогично отбирают для опре¬
деления влажности пробу массой 5-Юг.
3.10.2.Аппаратурам материалы
Иономер или рН-метр милливольтметр с погрешностью измерений не более
5мВ.
Электрод нитратный ионоселективный типа ЭИМ-I, ЭИМ-Н,ЭМ-NO3-0,1 или
электрод, имеющий такие же технические и метрологические характеристики.
Электрод сравнения хлорсеребряный насыщенный образцовый 2-го разряда
или электрод, имеющий такие же технические и метрологические характеристики.
Мешалка лабораторная электромеханическая или встряхиватель с возвратно¬
поступательным движением с частотой колебаний не менее 75мин
Термостат с автоматической регулировкой температуры в пределах (105±5)°С.
Весы лабораторные 2-го класса точности с наибольшим пределом взвешивания
200г и 4-го класса точности с наибольшим пределом взвешивания 500г.
Весы квадрантные с устройством пропорционального дозирования ВКПД-40г
с погрешностью дозирования не более 2%.
- 126-
Дозаторы с погрешностью дозирования не более 1% или цилиндры исполнения
1 или 2 вместимостью 50мл.
Колбы мерные исполнения 1 или 2, 2-го класса точности, вместимостью 1000мл.
Пипетки и бюретки исполнения 1,2,3,4,5 2-го класса точности.
Стаканы химические, исполнения 1 или 2, вместимостью 50мл.
Кассеты 10-позиционные с технологическими емкостями или колбы конические
исполнения 1 или 2, вместимостью 250мл.
Бумага фильтровальная.
3.10.3. Реактивы
Квасцы алюмокалиевые, ч.д.а.
Калий азотнокислый, х.ч.
Калий хлористый, х.ч.
Вода дистиллированная.
Калий сернокислый, ч.д.а., раствор концентрации с(1/2*К2804)=1маль/дм3 (1 н.).
3.10.4. Подготовка к анализу.
Приготовление раствора алюмокалиевых квасиов с массовой долей 1%. Готовят
из расчета 10г алюмокалиевых квасцов, взвешенных с погрешностью не более 0,1 г на
1000мл раствора.
Приготовление растворов сравнения
а). Приготовление раствора кониентраиии с (NO :-)=0Амалъ/дм1 (рС^т =7). 10,11 г
азотнокислого калия, высушенного до постоянной массы при температуре (105±5)°С,
взвешивают с погрешностью не более 0,01 г, помещают в мерную колбу вместимос¬
тью 1000мл и растворяют в экстрагирующем растворе, доводя объем до метки.
Раствор хранят в склянке с притертой пробкой не более 1 года. При появлении
мути или осадка раствор заменяют свежеприготовленным.
б). Приготовление раствора кониентраиии с (NO}Ч=0.01моль/дм3 (рС:.т =2). Го¬
товят 10-кратным разбавлением раствора, приготовленного по п.а), экстрагирующим
раствором в день проведения анализа.
в)Приготовление раствора кониентраиии с (NO^)=0.001 моль/дм3 (оС..П1 ж3). Го¬
товят 10-кратным разбавлением раствора, приготовленного по п. б), экстрагирующим
раствором в день проведения анализа.
г)Приготовление раствора кониентраиии с (N0^0.0001моль/дм3 (z)CNm =4).
Готовят 10-кратным разбавлением раствора, приготовленного по п. в) экстрагиру¬
ющим раствором в день проведения анализа.
Приготпйпрлие гщ&прктгюднаго раствора. 10,11 г азотнокислого калия и 0,37г хло¬
ристого калия, взвешенных с погрешностью не более 0,01 г, помещают в мерную колбу
вместимостью 1000см3 и растворяют в дистиллированной воде, доводя объем до метки.
Раствор хранят в склянке с притертой пробкой не более 1 года. При появлении
мути или осадка раствор заменяют свежеприготовленным.
Подготовка электродов к работе. Новый нитратный ионоселективный электрод
тщательно промывают дистиллированной водой и ополаскивают приэлектродным ра¬
створом и выдерживают в течение 24 ч в растворе концентрации с (Ы03*)=0.1моль/
да3. После этого электрод помещают на 10мин в дистиллированную воду, промокают
фильтровальной бумагой и проверяют его функцию, используя растворы сравнения. В
диапазоне от 2 до 4 единиц рС^3 электрод должен иметь линейную функцию с накло¬
ном (5б£3)мВ на единицу pCN03 Если характеристика отличается от заданной, элект-
род непригоден для работы. В перерывах между работой электрод хранят в раство¬
ре концентрации с (Ыо3)^=0.1моль/дм3.
- 127-
Электрод сравнения готовят к работе в соответствии с инструкцией завода-
изготовителя. В перерывах между работой электрод хранят в дистиллированной
воде. Первичная и периодическая поверка электрода - согласно инструкции.
3.10.5.Проведение анализа
Пробы почвы массой 20,От взвешивают с погрешностью не более 0,1 г и помеща¬
ют в технологические емкости или конические колбы емкостью 250 мл. К пробам
приливают по 50мл экстрагирующего раствора. Пробу с раствором перемешива¬
ют на электромеханической мешалке или встряхивателе в течение 3 мин. Получен¬
ные суспензии используют для определения нитратов.
При использовании весов с устройством пропорционального дозирования экстра¬
гента допускается отбор пробы массой 15,0-25,0г.Допускается пропорциональное из¬
менение массы пробы почвы и объема экстрагирующего раствора при погрешности
дозирования не более 2 %.
Определение нитратов. Перед измерением нитратный ионоселективный элект¬
род тщательно ополаскивают дистиллированной водой и выдерживают его в дистил¬
лированной воде в течение 10 мин.
При непосредственном определении pCN03 равными 4 и 2, используя для контроля
раствор с pCN03 равное 3 . При этом отклонения значений pCNQ3 не должны превышать
0,02 единицы pCN03 от номинального значения контрольного раствора сравнения.
При измерении в милливольтах электродную пару погружают в растворы сравне¬
ния, начиная с меньшей концентрации и определяют ЭДС.
После градуировки прибора электроды тщательно ополаскивают дистиллирован¬
ной водой, промокают фильтровальной бумагой и приступают к определению нитра¬
тов в суспензиях.
Перед измерениями суспензии взбалтывают. Электродную пару погружают в сус¬
пензию и считывают показания прибора не ранее, чем через 1 мин после прекращения
заметного дрейфа показаний прибора.
Настройку прибора проверяют по растворам сравнения не менее трех раз в тече¬
ние рабочего дня, используя каждый раз свежие порции растворов сравнения. Темпе¬
ратура анализируемых вытяжек и растворов сравнения должна быть одинаковой.
3.10.6. Обработка результатов
При непосредственном измерении pCN03 массовую долю азота нитратов в почве в
миллионных долях определяют с помощью таблицы пересчета по величине pCN03.
Таблица 30. Пересчет рСту в массовую долю азота нитратов в почве, млн1 (мг
на 1кг почвы)
P^NOJ
Сотые доли рСч<
0900
0,01
0,02
0,03
0,04
0,05
0,06
0,07
0,08
0,09
2,5
109
107
105
102
100
97,7
95,5
93,3
91,2
89,1
2,6
87,1
85,1
83,2
81,3
79,4
77,6
75,9
74,1
72,4
70,8
2,7
69,2
67,6
66,1
64,6
63,1
61,7
60,3
58,9
57,5
56,2
2,8
55,0
53,7
52,5
51,3
50,1
49,0
47,9
46,8
45,7
44,7
2,9
43,6
42,7
41,7
40,7
39,8
38,9
38,0
37,2
36,3
35,5
3,0
34,7
33,9
33,1
32,4
31,6
30,9
30,2
29,5
28,8
28,2
- 128-
Продолжение таблицы 30
Р
Сотые доли pCNm
0,00
0,01
0,02
0,03
0,04
0,05
0,06
0,07
0,08
0,09
3,1
27,5
26,9
26,3
25,7
25,1
24,6
24,0
23,4
22,9
22,4
3,2
21,9
21,4
20,9
20,4
20,0
19,5
19,1
18,6
18,2
17,8
з„з
17,4
17,0
16,1
16,2
15,9
15,5
15,1
14,8
14,5
14,1
3,4
13,8
13,5
13,2
12,9
12,6
12,3
12,0
11,8
11,5
11,2
3,5
11,0
10,7
10,5
10,2
10,0
9,80
9,60
9,30
9,10
8,90
3,6
8,70
8,50
8,30
8,10
7,90
7,80
7,60
7,40
7,20
7,10
3,7
6,90
6,80
6,60
6,50
6,30
6,20
6,00
5,90
5,80
5,60
3,8
5,50
5,40
5,20
5,10
5,00
4,90
4,80
4,70
4,60
4,50
3,9
4,40
4,30
4,20
4,10
4,00
3,90
3,80
3,70
3,60
3,50
4,0
3,50
3,40
3,30
3,20
3,20
3,10
3,00
3,00
2,90
2,80
При измерении в милливольтах по результатам определения ЭДС в растворах
сравнения строят градуировочный график в линейном масштабе. На оси абсцисс
откладывают значения pCNQ3 раствора сравнения, а на оси ординат - соответствую¬
щие им показания прибора (мВ). По градуировочному графику определяют значе¬
ния pCND3 анализируемых суспензий.
Массовую долю азота нитратов в миллионных долях в почве определяют по вели¬
чине pCN03 с помощью таблицы.
При построении градуировочного графика масштаб выбирают такой, чтобы дли¬
на оси абсцисс была равна 20см, а оси ординат - 15-20см. Построение градуировоч¬
ного графика проводят по результатам единичных определений.
При анализе проб в состоянии естественной влажности, результат анализа пере¬
считывают на сухое состояние, умножая на коэффициенты и К2, учитывающие мас¬
совую долю влаги в почве и увеличение объема экстрагирующего раствора, взаимо¬
действующего с анализируемой пробой, за счет содержащейся в почве влаги, которые
вычисляют по формулам: jqq
к' ~ 100-W’
где W - массовая доля влаги в анализируемой почве, %;
к _ 100 250
А2 “ тжГ ИЛИ Л, =
100--? 250-fT
2.5
где W - массовая доля влаги в анализируемой почве, %;
2,5 - соотношение массы пробы почвы и объема экстрагирующего
раствора.
Массовую долю влаги в почве (W) в процентах определяют высушиванием пробы
в термостате при температуре (105±5)°С в течение 6 часов и вычисляют по формуле:
W = ^L.m,
т — ш.
- 129 -
где т - масса пробы с бюксом до высушивания, г;
т1 - масса пробы с бюксом после высушивания, г;
т2 - масса пустого бюкса, г;
100 - коэффициент пересчета в проценты.
Массовую долю азота нитратов в пересчете на сухую почву (Xt) в миллионных
долях вычисляют по формуле
Х{=ХКхК29
где X - массовая доля азота нитратов во влажной почве, млн'1.
За результат анализа принимают значение единичного определения. Результат ана¬
лиза выражают в миллионных долях (мг на 1кг почвы) с округлением до первого деся¬
тичного знака.
Допускаемые относительные отклонения при двусторонней доверительной веро¬
ятности Р=0,95 от среднего арифметического результатов повторных анализов при
выборочном статистическом контроле составляют:
20% - при массовой доле азота нитратов в почве до Юмлн-1 ;
15% - св. Юмлн1.
3.10.7. Технологическая карта
3.11. ОПРЕДЕЛЕНИЕ ПОДВИЖНЫХ ФОРМ
ФОСФОРА И КАЛИЯ В ПОЧВАХ
3.11.1. Принцип методов
Методы основаны на извлечении подвижных форм фосфора и калия различными
экстрагентами в зависимости от генезиса почв:
- для дерново-подзолистых и оподзоленных почв - 0,2н. раствором НС1 при соот¬
ношении почва : раствор - 1:5, времени перемешивания суспензии 1мин. и отстаива¬
ния в течение 15 мин. (метод Кирсанова);
- для черноземных не карбонатных почв - 0,5н. раствором СН3СООН при соот¬
ношении почва : раствор - 1 : 25, времени встряхивания 1 час и отстаивания в течение
18-20 часов (метод Чирикова);
- для карбонатных почв - 1% раствором (NH4)2C03 при соотношении почва :
раствор - 1 : 20, времени встряхивания 5 мин. и отстаивания в течение 18-20 часов
при температуре 25 ± 2°С (метод Мачигина).
Определение фосфора в вытяжке калориметрическое - по интенсивности окрас¬
ки молибденовой сини. В качестве восстановителя молибдена используется аскорби¬
новая кислота в присутствии сурьмы как стабилизатора реакции (см.п.2.4.2.1.).
Калий в вытяжке определяется методом пламенной фотометрии (см. п.2.4.3.1.).
Анализ вытяжки на содержание фосфора и калия проводят либо на отдельных
приборах, либо на поточных линиях типа «Мединген» и др.
- 130-
3.11.2. Метод Кирсанова
3.11.2.1. Технологическое оборудование
Фотоэлектроколориметр.
Пламенный фотометр.
Весы типа BJTKT и аналитические.
Кассеты 10-позиционные.
Встряхиватель для 10-позиционных кассет.
Воронки d= 7см (желательно пластмассовые).
Дозаторы на 1 и 19мл.
Пипетка или шприц-дозатор на 1мл.
3.11.2.2. Реактивы
1. 0,2н. НС1. Концентрированную кислоту вливают в дистиллированную воду из
расчета 16мл на 1л раствора. Концентрацию приготовленной кислоты проверяют тит¬
рованием раствором едкого натра в присутствии фенолфталеина. Для анализа допус¬
тима нормальность кислоты от 0,19 до 0,21н.
2. 5н. H2S04. Концентрированную серную кислоту вливают в дистиллирован¬
ную воду из расчета 140мл на 1л раствора.
3. Окрашивающий раствор
Реактив А. 6г молибденовокислого аммония растворяют в 200мл дистиллирован¬
ной воды. 0,145г сурмяновиннокислого калия растворяют в 100мл дистиллированной
воды. Оба раствора готовят при слабом нагревании. Охлажденные растворы прилива¬
ют к 500мл 5н. раствора серной кислоты и доводят объем дистиллированной водой до
1л. Хранят в темной склянке.
Реактив Б. 0,887г аскорбиновой кислоты растворяют в 168мл раствора А и доводят
до объема 1л дистиллированной водой. Раствор готовят в день проведения анализа.
4. Образцовые растворы для определения фосфора
Исходный образцовый раствор. 0,192г химически чистого одно замещенного фос¬
фата калия растворяют в 0,2н. растворе НС1, доводят объем до 1л. Полученный ра¬
створ содержит 0,1мг P2Q в 1мл. (Очистку реактива см. п.2.4.2.3.).
Рабочая тикала образцовых растворов. В мерные колбы на 500мл отбирают коли¬
чества исходного раствора КН2Р04, указанные в таблице 31. Объем доводят до 500мл
0,2н. НС1.
Таблица 31.
^ Номера колб
Показатели ^
1
2
3
4
5
6
7
8
9
10
11
Объем исходного раствора, мл.
0
5
15
25
50
75
100
125
150
200
250
Содержание Р205
в мг/л раствора
0
1
3
5
10
15
20
25
30
40
50
в мг/кг почвы
0
5
15
25
50
75
100
125
150
200
250
Окрашивание образцовых растворов для калибровки фотоэлектроколориметра
проводится аналогично окрашиванию анализируемых вытяжек.
Калибровочный график строят, откладывая на оси абсцисс содержание P2Q в
мг/кг почвы, на оси ординат - оптическую плотность соответствующих образцо¬
вых растворов рабочей шкалы.
5. Образцовые растворы для определения калия
- 131 -
Исходный образцовый раствор. 0,792г хлористого калия растворяют в 0,2н. ра¬
створе НС1 и доводят им объем раствора в мерной колбе до 1л. Полученный раствор
содержит 0,5мг К^О в 1мл.
Рабочая шкала образцовых растворов. В мерные колбы емкостью 250мл отбира¬
ют количества исходного раствора КС1, указанные в таблице 32.
Таблица 32.
" ' _ Номера колб
Показатели
1
2
3
4
5
6
7
8
9
10
Объем исходного раствора, мл.
0
1
2
4
6
10
20
40
60
80
Содержание К20,мг/кг почвы
0
10
20
40
60
100
200
400
600
800
Растворы доводят до метки 0,2н. НС1 и используют для построения калибровоч¬
ного графика, для чего откладывают на оси абсцисс содержание К20 в мг/кг почвы,
на оси ординат - оптическую плотность соответствующих образцовых растворов ра¬
бочей шкалы.
3.11.23. Ход анализа
10г почвы заливают 50мл 0,2н. НС1, взбалтывают в течение 1 мин., затем отстаи¬
вают 15 мин. и фильтруют. Экстракцию фосфора и калия из почвы проводят при тем¬
пературе 18-20°С.
Для определения фосфора отбирают 1мл фильтрата, приливают к пробам по 19мл
раствора Б и перемешивают. Через 10 мин. приступают к определению P2Q на
фотоэлектроколориметре при красном светофильтре.
Калий определяют на пламенном фотометре, непосредственно распыляя вытяжку
в пламя. Используют светофильтр, пропускающий аналитические линии калия 766,5
и 769,9нм.
Образцовые растворы шкалы отбирают и окрашивают так же, как и почвенные
вытяжки.
Холостое определение проводят параллельно с испытываемыми вытяжками и
вычитают его значение из результата.
3.11.2.4. Обработка результатов
Содержание P2Q и К20 в мг/кг почвы определяют по графикам, построенным
на основании шкалы образцовых растворов. Допустимые расхождения результатов
повторных определений от среднего приведены в таблице 33.
Таблица 33.
Содержание элементов
питания в почве, мг/кг
Допустимые отклонения от среднего при повторном
определении, %
в одной лаборатории |
| в разных лабораториях
р2о5
до 30
20
35
от 30 до 60
15
25
свыше 60
15
20
К,О
до 80
15
20
от 80 до 120
15
15
от 120 до 170
10
15
свыше 170
10
10
- 132-
3.11.2.5. Технологическая карта (см. п.3.11.3.5.)
3.11.3. МЕТОД ЧИРИКОВА
3.113.1. Технологическое оборудование
Фотоэлектроколориметр
Пламенный фотометр
Весы типа ВЛКТ и аналитические
Кассеты 10-позиционные.
Встряхиватель для 10-позиционных кассет.
Воронки d=7cM.
Дозатор на 18мл.
Пипетка или шприц-дозатор на 2мл.
3.11.3.2. Реактивы
1. 0,5н. CHJCOOH. 28,5мл концентрированной (ледяной) уксусной кислоты вли¬
вают в дистиллированную воду и доводят объем до 1л. Концентрацию проверяют тит¬
рованием раствором едкого натра в присутствии фенолфталеина. Допустимые кон¬
центрации растворов уксусной кислоты - 0,49-0,51н.
2. Окрашивающий раствор
Реактив А (см. п.3.11.2.2.).
Реактив Б. 0,948г аскорбиновой кислоты растворяют в 178мл реактива А, доводят
объем до 1л дистиллированной водой. Раствор готовят в день проведения анализа.
3. Образцовые растворы для определения фосфора
Исходный образцовый раствор. 0,192г одно замещенного фосфата калия раство¬
ряют в 0,5 н. уксусной кислоты, доводят объем до 1л. Исходный раствор содержит
0,1мг Р20 в 1мл. (Очистку реактива см. п.2.4.2.3.).
Рабочая шкала образцовых растворов. В мерные колбы емкостью 500мл отбира¬
ют количества исходного раствора КН2Р04, указанные в таблице 34.
Растворы доводят до 500мл 0,5н. уксусной кислотой. Окрашивание образцовых ра¬
створов для построения калибровочной шкалы проводят аналогично окрашиванию ана¬
лизируемых почвенных вытяжек. Калибровочный график строят, откладывая на оси абс¬
цисс содержание P2Q в мг/кг почвы, на оси ординат - оптическую плотность растворов.
Таблица 34.
Номера колб
Показатели '
1
2
3
4
5
б
7
8
9
Объем исходного образцового раствора, мл.
0
4
10
15
20
30
40
50
60
Содержание P?Os в мг/л раствора,
0
0,8
2
3
4
6
8
10
12
в мг/кг почвы
0
20
50
75
100
150
200
250
300
Объем исходного раствора КС), мл.
0
0,5
1
2
4
8
12
16
-
Содержание К20, мг/кг почвы
0
25
50
100
200
400
600
800
-
5. Образцовые растворы для определения калия
Исходный образцовый раствор. 0,792г хлористого калия растворяют 0,5н. уксус¬
ной кислотой, доводят объем до 1л. Раствор содержит 0,5мг К20 в 1мл.
- 133-
Рабочая шкала образцовых растворов. В мерные колбы емкостью 250мл отби¬
рают количества исходного раствора КС1, указанные в таблице 34.
Растворы доводят до объема 250мл 0,5н. уксусной кислотой. Калибровочный гра¬
фик строят, откладывая на оси абсцисс содержание К20 в мг/кг почвы, на оси
ординат - оптическую плотность раствора.
3.1133. Ход анализа
4г почвы заливают 100мл 0,5н. уксусной кислоты. Взбалтывают в течение 1 часа и
затем оставляют на 18-20 часов. На следующий день суспензию фильтруют. Берут 2мл
фильтрата, прибавляют 18мл реактива Б и перемешивают. Через 10 мин. приступают к
определению на красном светофильтре. Окраска растворов устойчива в течение су¬
ток. В оставшемся фильтрате определяют калий на пламенном фотометре.
Параллельно ведут холостое определение.
3.113.4. Обработка результатов
Содержание фосфора и калия в анализируемых почвах находят по графику непосред¬
ственно в мг P2Q и К20 на 1кг почвы и вычитают результат холостого определения.
Допустимые расхождения результатов при повторных определениях приведены в
таблице 35.
Таблица 3S.
Содержание элементов
питания в ночве, мг/кг
Допустимые отклонения от среднего при повторных
определениях (в отн. %)
в одной лаборатории
в разных лабораториях
P2Os до 50
20
20
свыше 50
15
20
К20 до 100
15
20
свыше 100
10
15
3.113.5. Технологическая карта
Взятие навесбк
Добавление экстоагента
весы типа BJTKT, шпатель, кассе¬
ты 10-позиционные
-►
дозатор на 50 мл
Взбалтывание
Филынование
встряхиватель
кассеты, воронки d=7cM, фильтры
1
▼
Фотометоиоование
Отбор аликвот
пламенный фотометр (свето¬
фильтр на калий)
->
колбы на 50мл, шприц-дозатор на
2мл,(по Чирикову), на 1 мл (по
Кирсанову)
Окрашивание проб
дозатор на 18мл(по Чирикову), на
19мл (по Кирсанову)
-►
ФЭК (красный светофильтр)
- 134-
3.11.4. МЕТОД МАЧИГИНА
3.11.4.1. Технологическое оборудование
Весы типа ВЖТ и аналитические.
Фотоколориметр.
Пламенный фотометр.
Термостат.
Кассеты 10-позиционные.
Встряхиватель для 10-позиционных кассет.
Воронки d=5 и 7 см.
Дозаторы на 35 и 75мл.
Пипетка или шприц-дозатор на 15мл.
3.11.4.2. Реактивы
1. 1% углекислый аммоний. Растворяют соль в дистиллированной воде из расчета
10г на 1л .
Раствор должен иметь pH 9,0 и допустимую концентрацию углекислого аммония
0,198-0Д08н. Последнюю устанавливают титрованием 0,1н. НС1 по метилоранжу.
При pH ниже 9,0 доводят крепким аммиаком до pH 9,0, а затем устанавливают
нормальность на основании следующих расчетов. Допустим, концентрация углекис¬
лого аммония при растворении 10г (ЫН4)2ССК в 1л воды равна 0,150н., а после дове¬
дения pH этого раствора до 9,0-0,180н. Чтобы достичь 0,208н концентрации, необхо¬
димо повысить ее на 0,028н. (0,208 н.-0,180 н.). Поскольку растворение 10г соли в 1л
воды дает 0,150н. раствор, то для увеличения нормальности на 0,028 н. следует на 1л
раствора добавить 1,87г (NH4)2C03 согласно уравнению:
0,028-10 10„
X = — = 1,87г.
0,15
Для проверки концентрации проводят повторное титрование.
Если же раствор углекислого аммония имеет pH 9,0, проверяют его нормальность
и доводят до заданной как описано выше.
2. 30% H2S04 (раствор 1). 165мл концентрированной H2S04 вливают в 835мл
дистиллированной воды.
3. Марганцовокислый калий (раствор II). 17,5г КМп04 растворяют в 1л дистил¬
лированной воды.
4. Смесь растворов: раствор II с раствором I смешивают в соотношении 1:2,5.
Готовят в день проведения анализа.
5. Окрашивающий раствор
Реактив А (см. п.3.11.2.2.).
Реактив Б. 1,17г аскорбиновой кислоты при неокрашенных почвенных вытяжках
и 2,33г при окрашенных растворяют в 222мл реактива А и доводят объем до 1л дис¬
тиллированной водой. Реактив готовят в день проведения анализа.
6. Образцовые растворы для определения фосфора.
Исходный образцовый раствор для определения ФосФооа. 0,192г КН2Р04 ра¬
створяют в 1% растворе углекислого аммония и доводят объем до 1л. Раствор содер¬
жит 0,1мг P2Q в 1 мл и является исходным для приготовления рабочей шкалы.
(Очистку реактива см. п.2.4.2.3.).
Рабочая шкапа образцовых растворов. В мерные колбы емкостью 500мл отбира¬
ют количества исходного раствора КН2Р04, указанные в таблице 36. растворы дово¬
дят до метки 1% (NH4)2C03 .
- 135-
Таблица 36.
' Номера колб
Показатели
1
2
3
4
5
6
7
8
9
Объем исходного образцового раствора, мл.
0
2,5
5
7,5
10
12,5
15
20
25
Содержание Р005, мг/кг почвы
0
10
20
30
40
50
60
80
100
Окрашивание образцовых растворов для калибровки фотоколориметра проводит¬
ся аналогично окрашиванию анализируемых почвенных вытяжек. Калибровочный
график строят, откладывая на оси абсцисс содержание P2Q в мг/кг почвы, на оси
ординат - оптическую плотность раствора.
7. Образцовые растворы для определения калия
Исходный образцовый раствор. 0,792г КС1 растворяют в 1% растворе углекисло¬
го аммония, доводят им объем до 1л. Раствор содержит 0,5мг К20 в 1мл.
Рабочая шкала образцовых растворов. В мерные колбы на 500мл отбирают коли¬
чества исходного раствора, указанные в таблице 37.
Таблица 37.
■— Номера колб
Показатели ■——
1
2
3
4
5
6
7
8
9
10
Объем исходного раствора КС1, мл.
0
0,5
1
2
3
5
10
20
30
40
Содержание К70, мг/кг почвы
0
10
20
40
60
100
200
400
600
800
Растворы доводят до объема 500мл 1% углекислым аммонием. Калибровочный
график строят, откладывая на оси абсцисс содержание К20 в мг/кг почвы, на оси
ординат - оптическую плотность раствора.
3.11.43. Ход анализа
5г почвы заливают 100мл 1% углекислого аммония, взбалтывают в течение 5 мин.,
выдерживают в термостате в течение 18-20 часов при температуре 25±2°С, а затем
отфильтровывают. 15 мл фильтрата переносят в коническую колбу, приливают по 2мл
смеси серной кислоты и марганцовокислого калия, закрывают воронкой и кипятят в
течение 2 мин. с момента закипания. После охлаждения приливают 35мл реактива Б,
содержащего 2,3Зг аскорбиновой кислоты, и перемешивают.
Колориметрируют полученный раствор при красном светофильтре через 10 мин.
после добавления реактива Б.
Если вытяжки не окрашены, определение проводят следующим образом. К 15мл
фильтрата приливают 35мл реактива Б, содержащего 1,17г аскорбиновой кислоты и
колориметрируют как указано выше.
В оставшемся фильтрате определяют калий на пламенном фотометре.
Параллельно ведут холостое определение.
3.11.4.4. Обработка результатов
Содержание фосфора и калия в анализируемых почвах находят по графику непос¬
редственно в мг P2Q и К20 на 1кг почвы и вычитают результат холостого определе¬
ния.
Допустимые расхождения результатов при повторных определениях приведены в
таблице 38.
- 136-
Таблица 38.
Содержание элементов
питания в почве, мг/кг
Допустимые отклонения от среднего (в отн. %)
в одной лаборатории
в разных лабораториях
Р?05 до 15
35
40
15-30
25
30
свыше 30
20
20
К20 до 200
10
20
свыше 200
10
15
3.11.4.5. Технологическая карта
3.12. ОПРЕДЕЛЕНИЕ СОДЕРЖАНИЯ ОБМЕННОГО КАЛИЯ
В ПОЧВЕ МЕТОДОМ МАСЛОВОЙ
3.12.1. Принцип метода
Метод рекомендуется для определения обменного калия в некарбонатных почвах.
Извлечение обменного калия из почвы 1н. раствором CH3COONH4 при соотношении
почвы и раствора 1:10.
К+ nh+4
[ППК] Са2+ + CH3COONH4 —> [ППК] NH+4
К+ Са2+
К* + CH,COONH.
- 137-
После часового взаимодействия почвы и раствора ацетата аммония количе¬
ство калия определяется в вытяжке с помощью пламенного фотометра.
3.12.2. Технологическое оборудование
Пламенный фотометр.
Весы типа ВЖТ.
Мерный цилиндр на 50 мл.
Мерные колбы на 250 мл.
3.12.3. Реактивы
1. 1н. раствор аиетат аммония. 77г CH3COONH4, взвешенные с погрешностью
не более 0,1 г, растворяют в дистиллированной воде и доводят объем 1н раствором
CH3COONH4 до 1л в мерной колбе. pH раствора должен соответствовать 6,8-7,0. При
необходимости до указанных параметров раствор доводят уксусной кислотой или ам¬
миаком.
2. Исходный образцовый раствор. 0,73г КС1 взвешивают с погрешностью не
более 0,001 г, растворяют в мерной колбе на 1л и доводят объем раствором CH3COONH4
до метки. В 1мл раствора содержится 0,5мг К20
3.12.4. Ход анализа
5г почвы, взвешенной с погрешность не более 0,1 г , переносят в колбу емкостью
200-250мл, приливают 50мл 1н. раствора CH3COONH4 и взбалтывают на ротаторе 1
час. Суспензию фильтруют и определяют в фильтрате содержание калия с помощью
пламенного фотометра.
Для построения калибровочного графика готовят шкалу образцовых растворов. В
мерные колбы вместимостью 250мл приливают соответствующее таблице 39 количе¬
ство исходного образцового раствора КС1, доводят объем до метки 1 н. раствором
CH3COONH4.
Таблица 39.
Номер образцового раствора
1
2
3
4
5
6
7
8
Объем исходного раствора КС1,мл
0
1,0
2,5
5,0
7,5
10,0
15,0
20,0
Содержание калия соответствующее мг К20
в 1 кг почвы
0
20
50
100
150
200
300
400
Содержание калия (К^О) в мг на 1кг почвы вычисляют по формуле:
a^uuu
2 т•1000
где а - количество К20 , определенное по калибровочному графику, мг;
V - объем фильтрата, мл;
т - масса навески почвы, г;
1000 - коэффициент для пересчета количества мг К20 на 1мл раствора.
На оси абсцисс кроме концентрации калия (мг К20) можно указать содержание
калия в мг К20 на 1000г (1кг) почвы.
Обеспеченность растений обменным калием указана таблице 40.
- 138-
Таблица 40. Обеспеченность растений обменным калием, мг KjO на 1000г
почвы (по методу Масловой).
Обеспеченность растений
Содержание калия
Очень низкая
<50
Низкая
50 - 100
Средняя
100- 150
Повышенная
150 - 200
Высокая
200 - 300
Очень высокая
>300
3.12.5. Технологическая карта
3.13. МЕТОД ОПРЕДЕЛЕНИЯ ГИДРОЛИТИЧЕСКОЙ
КИСЛОТНОСТИ ПОЧВ
3.13.1. Принцип метода
Метод основан на вытеснении поглощенного водорода из почвы 1н. раствором
CH3COONa при соотношении почва : раствор - 1:2,5 с последующим рН-метричес-
ким определением кислотности суспензии.
3.13.2. Технологическое оборудование
Весы типа BJIKT.
рН-метр.
Встряхиватель для кассет или магнитная мешалка.
Кассеты 10-позиционные.
Дозатор на 50мл.
3.13.3. Реактивы
1. 1н. раствор CH3COONa. 136г СНЗССХЖа • ЗН20 растворяют в дистиллиро¬
ванной воде и доводят объем до 1л.
Раствор должен иметь pH 8,3-8,4. Если раствор имеет рН<8,3, его подщелачивают
10% раствором NaOH, при рН>8,4 - подкисляют раствором СН3СООН. Раствор
хранится не более 3 дней.
2. Буферные растворы для рН-метрии с pH 4,01; 6,86; 9,18 (см.п.4.3.4.).
- 139-
3.13.4. Ход анализа
20г почвы заливают 50мл 1н. раствора CH3COONa (pH 8,3-8,4), взбалтывают в
течение 1 мин., отстаивают 18-20 часов, повторно перемешивают 1мин. и измеряют
pH суспензии. Показания pH-метра снимают с точностью до 0,01. Электроды при пе¬
реносе из одной почвенной суспензии в другую не обмывают, а слегка промокают
фильтровальной бумагой.
Перед началом работы проводят настройку рН-метра по буферным растворам с
pH 4,01; 6,86; 9,18. Во время работы настройку прибора периодически контролируют
по буферному раствору с pH 6,86.
Порядок работы на рН-метрах подробно описан в п.4.3.1.
3.13.5. Обработка результатов
Величину гидролитической кислотности почвы в мг-экв/100г почвы находят по
величине pH, пользуясь таблицей 41.
Допустимые отклонения от среднею значения при повторных определениях гид¬
ролитической кислотности в почве в одной лаборатории 12% или ±0,05 pH, для раз¬
ных лабораторий 25% или ±0,1 pH.
Таблица 41.
pH сотые доля
pH
0,00
0,01
0,02
0,03
0,04
1 0,05 |
0,06
0,07
0,08
1 0,09
Гидролитическая кислотность, мг-экв/100 г почвы
6,0
17,3
16,9
16,6
16,2
15,8
15,5
15,2
14,9
14,5
14,2
6,1
13,9
13,6
13,3
13,1
12,8
12,5
12,2
12,0
11,7
11,5
6,2
11,2
11,0
10,8
10,5
10,3
10,1
9,84
9,64
9,44
9,23
6,3
9,04
8,83
8,65
8,45
8,25
8,11
7,92
7,76
7,59
7,41
6,4
7,28
7,11
6,97
6,81
6,69
6,53
6,38
6,25
6,11
5,98
6,5
5,85
5,73
5,61
5,48
5,37
5,25
5,14
5,03
4,92
4,82
6,6
4,71
4,61
4,52
4,42
4,32
4,23
4,14
4,05
3,96
3,82
6,7
3,79
3,71
3,63
3,56
3,48
3,40
3,33
3,26
3,19
здз
6,8
3,05
2,99
2,92
2,86
2,80
2,74
2,68
2,62
2,57
2,52
6,9
2,46
2,41
2,35
2,31
2,25
2,21
2,16
2,11
2,07
2,02
7,0
1,98
1,94
1,90
1,86
1,82
1,78
1,74
1,70
1,67
1,63
7,1
1,60
1,56
1,53
1,50
1,46
1,43
1,40
1,37
1,34
1,31
7,2
1,28
1,26
1,23
1,20
1,18
1,15
1,13
1,10
1,08
1,06
7,3
1,03
1,01
0,99
0,97
0,95
0,93
0,91
0,89
0,87
0,85
7,4
0,83
0,81
0,80
0,78
0,76
0,75
0,73
0,72
0,70
0,68
7,5
0,67
0,66
0,64
0,63
0,61
0,60
0,59
0,58
0,56
0,55
7,6
0,54
0,53
0,52
0,51
0,49
0,48
0,47
0,46
0,45
0,44
7,7
0,43
0,43
0,42
0,41
0,40
0,39
0,38
0,37
0,37
0,36
7,8
0,35
0,34
0,33
0,33
0,32
0,31
0,31
0,30
0,29
0,29
7,9
0,28
0,28
0,27
0,26
0,26
0,25
0,25
0,24
0,24
0,23
8,0
менее 0,23
- 140-
3.13.6. Технологическая карта
3.14. ОПРЕДЕЛЕНИЕ pH, ОБМЕННОЙ КИСЛОТНОСТИ,
ПОДВИЖНОГО АЛЮМИНИЯ, МАРГАНЦА,
ОБМЕННОГО КАЛЬЦИЯ, МАГНИЯ И СЕРЫ В ОДНОЙ НАВЕСКЕ
3.14.1. Принцип метода
Определение основано на извлечении обменных катионов из почвы 1 н. КС1 при
отношении почвы к раствору 1 :2,5. Для анализа почвенной вытяжки применяются
следующие методы:
потенциометрический - измерение pH;
фотоколориметрический - определение А1, Мп;
потенциометрическое титрование - определение обменной кислотности;
трилонометрическое титрование - определение Са, Mg.
3.14.2. Определение pH и обменной кислотности
3.14.2.1. Технологическое оборудование
Весы типа BJIKT.
рН-метр
Встряхиватель для 1 Оппозиционных кассет и магнитная мешалка.
Кассеты 10-позиционные.
Дозатор на 75мл.
Воронки d = 9см (желательно пластмассовые).
Стаканы на 100мл.
3.14.2.2. Реактивы
1. 1н. раствор KCl. 75г КС1 растворяют в дистиллированной воде и доводят объем
до 1л.Полученный раствор должен иметь pH = 5,6-6,0. Если pH меньше 5,6, требуемое
значение устанавливают прибавлением к раствору 10 % КОН, а при pH больше 6,0 -
10% HCL.
2. Буферные растворы для настройки рН-метра (см. п.4.3.4.).
3. 0,1 н. NaOH . 4г NaOH растворяют в дистиллированной воде и доводят объем
раствора до 1л. Устанавливают титр раствора по 0,1н. фиксанальному раствору
H2S04 по фенолфталеину.
4. 1% раствор фенолфталеина. 1г фенолфталеина растворяют в 100мл 60% эти¬
лового спирта.
3.14.23. Ход анализа
ЗОг почвы заливают 75мл 1н. раствора КС1 и перемешивают в течение 1 мин. В
полученной суспензии замеряют pH, для чего погружают в суспензию стеклянный
электрод и электрод сравнения рН-метра и через 1,5-2 мин. замеряют pH. Более про¬
изводительно вести определение на 10-позиционном рН-метре.
- 141 -
Настройку рН-метра проводят по буферным растворам с pH = 4,01; 6,86; 9,18.
Порядок работы на рН-метрах подробно изложен в п.4.3.1.
После определения pH суспензию фильтруют.
Для определения обменной кислотности 25мл фильтрата переносят в стакан ем¬
костью 100мл, устанавливают на магнитную мешалку, помещают в него «магнитик»,
погружают в раствор электродную пару и кончик бюретки, заполненной ОДн. раство¬
ром NaOH. Включают магнитную мешалку, устанавливают на блоке автоматического
титрования значение эквивалентной точки рН=8,2 и время выдерживания 30 сек. Руч¬
ку «виды работ » ставят в рабочее положение, начиная тем самым титрование. Титро¬
вание проходит автоматически, конец титрования определяют по загоранию сигналь¬
ной лампочки.
3.14.2.4. Обработка результатов
Результаты определения pH почвенных вытяжек фиксируют непосредственно
по шкале прибора.
Допустимые расхождения результатов при повторных определениях в одной ла¬
боратории 0,2 единицы pH, в разных лабораториях - 0,3 единицы pH.
Показатель обменной кислотности определяют по формуле:
К = а • N • 10,
где К - обменная кислотность, мг-экв/100г почвы;
а - объем раствора щелочи, пошедшего на титрование, мг;
N - нормальность раствора щелочи;
10 - коэффициент пересчета на ЮОг почвы.
Допустимые расхождения результатов при повторных определениях обменной
кислотности приведены в таблице 42.
Таблица 42.
Показатели
Пределы значений обменной кислотности, мг-экв/100 г ночвы
менее ОД
ОД-4,5
0,5-1,0
1,0-3,0
более 3,0
Допустимые отклонения в
% от среднего значения
50
20
15
10
5
3.14.2.5 Технологическая карта
весы типа BJIKT, кассеты 10-пози-
ционные, шпатель
-►
Добавление 1 н. KCI
дозатор на 75 мл
Взбалтыватель
встряхиватель или магнитная
мешалка
-
ЗдшрлН
рН-метр, термометр
Фильтрование
Титрование
стаканы на 100 мл, воронки d=7-
9см, фильтры (белая лента)
-►
рН-метр, термометр, магнитная
мешалка, бюретка на 25 мл.
- 142-
3.14.3. Определение подвижного алюминия
3.143.1. Принцип метода
Метод основан на образовании окрашенного комплекса алюминия с хромазуро-
лом и фотоколориметрировании раствора. Влияние железа устраняется восстановле¬
нием его до двухвалентного состояния аскорбиновой кислотой. Для создания слабо
кислой реакции (pH 5,8) используется ацетатный буферный раствор.
3.143.2. Технологическое оборудование
Кассеты 10-позиционные.
Дозаторы на 25мл.
Пипетка или автоматическая пипетка на 1мл.
Фотоэлектроколориметр.
3.143.3. Реактивы
1. 0,02% раствор аскорбиновой кислоты. 0,2г реактива растворяют в 1л дистил¬
лированной воды. Раствор готовят в день проведения анализа.
2. Окрашивающие растворы
Запасной окрашивающий раствор. 326г CH3COONa растворяют примерно в
800мл дистиллированной воды, прибавляют 5мл ледяной уксусной кислоты, в полу¬
ченной буферной смеси растворяют 1г хромазурола и доводят объем раствора до
1л дистиллированной водой. После отстаивания в течение 1 дня раствор фильтруют.
Хранят в склянке из темного стекла в течение 1 месяца.
Рабочий окрашичяюптий раствор. Разбавляют 8 объемов запасного окрашивающе¬
го раствора 92 объемами дистиллированной воды. Готовят в день проведения анализа.
3. Образцовые растворы на алюминий
Исходный образцовый раствор. 0,450г металлического алюминия взвешивают с
погрешностью не более 0,001 г помещают в мерную колбу на 500мл, заливают 10мл
НС1 1 : 1 и после прекращения бурного выделения пузырьков водорода нагревают на
водяной бане до полного растворения алюминия. Охлаждают и доводят дистиллиро¬
ванной водой до метки. Полученный раствор содержит 0,1мг-экв/мл. алюминия.
Рабочая шкала образцовых растворов. Рабочий образцовый раствор готовят пу¬
тем разбавления исходного в 10 раз 1н. КС1. Данный раствор содержит 0,01мг-экв/мг
алюминия, что соответствует 2,5мг-экв алюминия на 100г почвы.
В мерные колбы на 100мл приливают количества рабочего образцового раствора,
указанные в таблице 43, и доводят объемы до меток 1н. КС1.
Из полученных образцовых растворов берут аликвоты и проводят окрашивание
так же, как и в анализируемых почвенных вытяжках. Калибровочный график строят,
откладывая на оси абсцисс содержание А1 в мг-экв/100г почвы, на оси ординат - опти¬
ческую плотность.
Таблица 43.
~ Номера колб
Показатели
1
2
3
4
5
6
7
8
9
Объем рабочего образцового раствора, мл.
0
2
4
6
8
12
16
20
24
Содержание А1, мг-экв на 100 г почвы
0
0,05
0,10
0,15
0,20
0,30
0,40
0,50
0,60
- 143-
3.143.4. Ход анализа
К 1мл 1 н. КС1 - вытяжки (см.п.З.Л.2.3.) приливают 25мл 0,02% раствора аскор¬
биновой кислоты, перемешивают, прибавляют 25мл рабочего окрашивающего раствора,
снова перемешивают.
Фотоколориметрирование проводят в кювете с толщиной просвечиваемого слоя
1см при 545 нм (желто-зеленый светофильтр) не ранее чем через 10 мин. и не позднее
чем через 30 мин. после окрашивания. Оптическую плотность измеряют относитель¬
но «нулевого» раствора образцовой шкалы.
3.143.5. Обработка результатов
Содержание подвижного алюминия в анализируемой почве находят по калибро¬
вочному графику непосредственно в мг-экв А1/100г почвы.
Допустимые расхождения результатов при повторных определениях в одной ла¬
боратории - 15%, в разных лабораториях - 20%.
3.143.6. Технологическая карта
3.14.4. Определение обменного кальция
3.14.4.1. Принцип метода
Метод основан на комплексонометрическом титровании кальция при pH = 13 три-
лоном Б с использованием флуорексона в качестве индикатора (см. п.2.4.6.1.).
Влияние марганца устраняют восстановлением его гидроксил амином, меди - свя¬
зыванием ее диэтилдитиокарбаматом.
3.14.4.2. Технологическое оборудование
Весы типа BJIKT.
Колбы конические на 250мл.
Пипетки или шприц-дозатор на 10, 25мл.
Дозаторы на 15 и 100мл.
Капельница.
Бюретка на 25мл.
3.14.4.3. Реактивы
1. 0,05н. трипон Б 9,3г реактива растворяют в дистиллированной воде и доводят
- 144-
объем до 1л. Нормальность устанавливают по 0,05н. раствору СаС12.
2. 0,05н. СаС12. 2,502г СаС03, высушенного до постоянной массы, помещают в
мерную колбу на 1л, прибавляют 10мл НС1 (1 : 1). После растворения навески объем
доводят до метки дистиллированной водой.
3. Флуорексон. Смешивают флуорексон с КС1 или KN03 в соотношении 1:100.
Хранят в темной банке с притертой пробкой.
4. 5% гидроксиламин солянокислый.
5. 20% раствор КОН.
3.14.4.4. Ход анализа
10мл КС1 -вытяжки (см. п.3.111.2.3.) при анализе тяжелых почв, 25мл - при анали¬
зе легких, а также почв с pH меньше 4,5 берут в колбы на 250мл, разбавляют дистил¬
лированной водой примерно до 100мл, прибавляют 0,5мл 5% гидроксил амина, 15мл
20% КОН, 2-3 кристаллика диэтилдитикарбамата Na, 30-50мг флуорексона и титру¬
ют 0,05н. трилоном Б до перехода окраски от интенсивной зеленой флуоресценции
до оранжево-розовой. Проводят также титрование холостой пробы.
3.14.45. Обработка результатов
Расчет проводят по формуле
„ (a-e)N 100
С а = ,
н
где С а - содержание обменного кальция в почве, мг-экв/100г почвы;
а - количество трилона Б, пошедшего на титрование
анализируемой пробы, мл.;
в - количество трилона Б, пошедшего на титрование холостой пробы, мл.;
N - нормальность трилона Б;
н - аликвотная навеска, г.
3.14.4.6. Технологическая карта (см. П.3.14.5.6Л
3.14.5. Определение обменного магния
3.145.1. Принцип метода
Определяют комплексонометрически сумму (Са + Mg) при pH 10 (см. п.2.4.6.1.),
магний вычисляют по разности между содержанием (Са + Mg) и Са в растворе.
3.14.5.2. Технологическое оборудование ,
Весы типа BJTKT.
Колбы конические на 250мл.
Пипетки на 10 и 25мл.
Дозаторы на 5 и 50мл.
Капельница.
Бюретка на 25мл.
3.14.5.3. Реактивы
1. 0,05 н. трипон Б (см. п.3.14.4.3.).
2. Хромоген черный. 5г индикатора растирают с 95г NaCl. Хранят в склянке из
темного стекла с притертой пробкой.
- 145 -
3. Хлоридно-аммиачный буфер. 20г NH4C1 растворяют в 100мл дистиллирован¬
ной воды, приливают 100мл 25% раствора аммиака и доводят объем до 1л дистилли¬
рованной водой.
4.5% гидроксиламин.
5. Диэтилдитиокарбамат натрия.
3.14.5.4. Ход анализа
10мл КС1 -вытяжки (см.п.3.8.2.3.) при анализе тяжелых почв и 25мл - при анали¬
зе легких почв берут в коническую колбу на 250мл, добавляют соответственно 50-
25мл дистиллированной воды, 5мл хлоридно-аммиачного буфера, 0,5мл 5% гидрокси-
ламина, 2-3 кристаллика диэтилдитиокарбамата Na, 10-15мг хромогена черного и тит¬
руют сумму (Са + Mg) 0,05н. раствором трилона Б до перехода окраски из вишневой в
сине-голубую.
3.14.5.5. Обработка результатов
Содержание обменного магния в почве расчитывают по формуле:
(с —«)•*•!<»
н
где Mg - содержание обменного магния в почве, мг-экв/100г почвы;
с - объем трилона Б, пошедшего на титрование суммы (Са + Mg), мл.;
а - объем трилона Б, пошедшего на титрование Са, мл.;
N - нормальность трилона Б;
н - аликвотная навеска, г.
3.14.5.6. Технологическая карта.
3.15. АТОМНО-АБСОРБЦИОННОЕ ОПРЕДЕЛЕНИЕ
КАЛЬЦИЯ И МАГНИЯ
3.15.1. Сущность метода
3 аключается в извлечении обменного кальция и обменного (подвижного) маг-
- 146-
пия из почвы раствором хлористого калия и последующем измерении поглоще¬
ния света свободными атомами определяемых элементов, образующимися з пла¬
мени при введении в него анализируемого раствора. Для устранения влияния
сопутствующих элементов, образующих с кальцием или магнием труднодиссо-
циируемые в пламени соединения, в атомизируемые растворы вводят избыток
стронция.
Определение кальция проводят по аналитической линии 422,7 нм, магния - 285,2 нм.
3.15.2. Метод отбора проб (см. п.3.1 - стр 94)
3.15.3.Аппаратурам материалы
Атомно-абсорбционный спектрофотометр с лампами с полным катодом для оп¬
ределения кальция и магния (допускается использование газовой смеси состава про-
пан-бутан-воздух и ацетилен-воздух).
Весы лабораторные 2-го класса точности с наибольшим пределом взвешивания 200г
и 4-го класса точности с наибольшим пределом взвешивания 500г.
Дозаторы с погрешностью дозирования не более 1% или пипетки и бюретки 2-го
класса точности.
Посуда мерная лабораторная 2-го класса точности.
Стаканы химические вместимостью 100мл2.
Бумага фильтровальная.
3.15.4. Реактивы
Кислота соляная, х.ч. или ч.д.а., концентрированная и раствор с массовой до¬
лей 25%.
Калий хлористый , х.ч. или ч.д.а., раствор концентрации с (КС1) - 1 моль/дм3 (1н.).
Кальций углекислый.
Окись магния.
Вода дистиллированная.
3.15.5. Подготовка к анализу
Приготовление запасного раствора стронция массой кониентраиии 20мг/
60,8г 6-водного хлористого стронция, взвешенного с погрешностью не более
0,1 г, растворяют в 600мл дистиллированной воды, приливают 250мл концентриро¬
ванной соляной кислоты, дистиллированной водой доводят объем раствора до
1000мл.
Раствор хранят в склянке с притертой пробкой не более 1 года.
Приготовление рабочего раствора строниия. 1 объем запасного раствора хлори¬
стого стронция смешивают с 8 объемами дистиллированной воды.
Раствор готовят в день проведения анализа.
Приготовление раствора калъиия кониентраиии с l/2SCa2L = О.бмоль/дмл
(0. бн.) 30,02г углекислого кальция, высушенного до постоянной массы при темпе¬
ратуре 100-105°С, взвешивают с погрешностью не более 0,01г, помещают в мерную
колбу вместимостью 1000мл, приливают 120мл раствора соляной кислоты с массо¬
вой долей 25% и после полного растворения навески приливают 500мл дистиллиро¬
ванной воды. В раствор добавляют 75г хлористого калия, взвешенного с погрешно¬
стью не более 0,1 г, дистиллированной водой доводят объем до метки и тщательно
перемешивают.
- 147-
Раствор хранят в склянке с притертой пробкой не более 1 года.
Приготовление раствора магния кониентраиии с 2молъ/дм1 (0.2н
4,031г окиси магния, прокаленной до постоянной массы при температуре 500°С, взве¬
шивают с погрешностью не более 0,001 г, помещают в мерную колбу вместимостью
1000мл, приливают 80мл раствора соляной кислоты с массовой долей 25% и после
полного растворения навески приливают 500мл дистиллированной воды. Добавляют
в раствор 75г хлористого калия, взвешенного с погрешностью не более 0,1 г, дистилли¬
рованной водой доводят объем до метки и тщательно перемешивают.
Раствор хранят в склянке с притертой пробкой не более 1 года.
Допускается приготовление растворов кальция концентрации с (1/2Са2+) =
0,6моль/дм3 (0,6н.) и магния концентрации с (l/2Mg2+)=0^MOJib/flM3(0,2H.) в общей
мерной колбе вместимостью 1000мл. Масса хлористого калия, добавляемого в ра¬
створ, должна при этом составлять 75г.
Приготовление раствора кальиия кониентраиии с (\ ИСа^-ОАЗмоль/дм1
<0.15 н.) 250мл раствора кальция концентрации с (1/2Са2+) = 0,6 моль/дм3 (0,6н.)
помещают в мерную колбу вместимостью 1000мл и раствором хлористого калия
концентрации 1 моль/дм3 доводят объем до метки.
Раствор хранят в склянке с притертой пробкой не более 1 года.
Приготовление раствора магния кониентраиии с П/2М%2±)=0.05молъ/дм1
(0.05н.). 250мл раствора магния концентрации с (l/2Mg2+)=0,2Manb/flM3(0,2H.) поме¬
щают в мерную колбу вместимостью 1000мл и раствором хлористого калия кон¬
центрации 1 моль/дм3 доводят объем до метки.
Раствор хранят в склянке с притертой пробкой не более 3 месяцев.
Допускается приготовление растворов кальция концентрации с (1/2Са2+)=0,15
мопь41м3(0,15 н.) и магния концентрации с (l/2Mg2+)=0,05мoль/дм3(0,05н.). в общей
мерной колбе вместимостью 1000мл.
Приготовление растворов сравнения
Приготовление растворов сравнения для анализа проб почвенных горизонтов, не
насыщенных основаниями, а также легких по механическому составу.
В мерные колбы вместимостью 250мл помещают указанные в табл.44 объемы
растворов кальция концентрации с (1/2Са2*)=0,15моль/дм3(0,15н.) и магния концент¬
рации с (l/2Mg2+)H),05MoW(aM3(0,05H.). Объемы растворов в колбах доводят до метки
раствором хлористого калия концентрации 1 моль/дм3 и тщательно перемешивают.
Таблица 44.
Характеристика растворов
Номер раствора сравнения
1
2
3
4
5
6
7
8
Объем растворов кальция концентрации с
(1/2Са2+)=0,15 моль/дм3 и магния
концентрации с (l/2Mg**) =0,05моль/дм3
0
2
4
8
12
16
20
24
Концентрация кальция с (1/2Са2>)
в растворе сравнения, моль/дм3
0
1,2
2,4
4,8
7,2
9,6
12,0
14,4
в пересчете на 100г почвы, ммоль
0
0,3
0,6
1,2
1,8
2,4
3,0
3,6
Концентрация магния с (l^Mg2*)
в растворе сравнения, с моль/дм3
0
0,4
0,8
1,6
2,4
3,2
4,0
4,8
в пересчете на 100г почвы, ммоль
0
од
0,2
0,4
0,6
0,8
1,0
1,2
- 148-
Приготовление растворов сравнения для анализа проб почвенных горизон¬
тов. насыщенных основаниями. В мерные колбы вместимостью 250мл помещают
указанные в таблице 44 объемы растворов кальция концентрации с (1/2Са2+) =
О,6моль/дм3 (0,6н.) и магния концентрации с (1/2Л^2+)=0,2маль/дм3(0,2н.).
Объемы растворов в колбах доводят до метки раствором хлористого калия кон¬
центрации 1моль/дм3.
Растворы сравнения для определения кальция и магния допускается готовить в
общих колбах и раздельно.
Растворы сравнения используют для градуировки атомно-абсорбционного спект¬
рофотометра в день проведения анализа.
Таблица 45.
Характеристика растворов
Номер раствора сравнения
1
2
3
4
5
6
7
8
Объем растворов кальция концентрации с
(1/2Са2+)=0,6 моль/дм3 и магния концентрации
с (1/2М£+) =0,2 моль/дм3, см3
0
5
10
20
30
40
50
60
Концентрация кальция с (1/2Са2+)
в растворе сравнения, моль/дм3
0
12
24
48
72
96
120
144
в пересчете на 100г почвы, ммоль
0
3
6
12
18
24
30
36
Концентрация магния с (1/2М^+)
в растворе сравнения, с моль/дм3
0
4
8
16
24
32
40
48
в пересчете на ЮОг почвы, ммоль
0
1
2
4
6
8
10
12
3.15.6. Проведение анализа
Приготовление вытяжки из почвы. Для анализа используют фильтраты вытяжек,
приготовленных согласно п 3.ф.2.3
Определение кальиия и магния с использованием газовой смеси состава пропан-
бутан-воздух. При анализе проб почвенных горизонтов, не насыщенных основания¬
ми, в химические стаканы отбирают по 5 мл фильтратов и растворов сравнения и при¬
ливают по 50мл рабочего раствора стронция.
При анализе проб почвенных горизонтов, насыщенных основаниями, в химичес¬
кие стаканы дозатором или пипеткой отбирают по 2мл фильтратов и растворов срав¬
нения и приливают по 50мл рабочего раствора стронция.
Полученные растворы вводят в пламя и измеряют поглощение света по аналити¬
ческой линии 422,7нм при определении кальция и по аналитической линии 285,2 ни
при определении магния.
При определении магния наконечник горелки устанавливают под углом 30° к лучу
с полым катодом.
n~r„ArKWfr ггпшт ]{ |<гттГ итлпьяпаамиеи лаяойай rupcu состава аиети-
лен-воздух. При анализе проб почвенных горизонтов, не насыщенных основаниями,
отбирают по 1мл фильтратов и растворов сравнения и приливают по 50мл рабочего
раствора стронция.
При анализе почвенных горизонтов, насыщенных основаниями, отбирают по 0,2мл
фильтратов и растворов сравнения и приливают по 50мл рабочего раствора стронция.
- 149 -
Полученные растворы вводят в пламя и измеряют поглощение света по аналити¬
ческой линии 422,7 нм при определении кальция и 285,2 нм при определении магния.
Допускается пропорциональное изменение объемов проб анализируемых вы¬
тяжек, растворов сравнения и рабочего раствора хлористого стронция при погреш¬
ности дозирования не более 1%.
3.15.7. Обработка результатов
По результатам фотометрирования растворов сравнения строят градуировочный
график. На оси абсцисс откладывают концентрации кальция или магния в растворах
сравнения в пересчете в миллимоли в 100г почвы, а по оси ординат - соответствую¬
щие им показания атомно-абсорбционного спектрофотометра.
Количество эквивалентов кальция или магния в анализируемой почве находят не¬
посредственно по градуировочному графику и вычитают из него результат холос¬
того опыта. Если результат определения выходит за пределы градуировочного гра¬
фика, определение повторяют, предварительно разбавив фильтрат раствором хло¬
ристого калия концентрации 1 моль/дм3. Результат, найденный по графику, увеличи¬
вают во столько раз, во сколько был разбавлен фильтрат.
За результат анализа принимают значение единичного определения кальция или
магния.
Результат анализа выражают в миллимолях в 100г почвы с округлением до перво¬
го десятичного знака.
При проведении массовых анализов вместо построения градуировочного графи¬
ка допускается градуирование шкалы прибора по растворам сравнения в день прове¬
дения анализа.
Допускаемые относительные отклонения от среднего арифметического результа¬
тов повторных анализов при выборочном статистическом контроле при доверитель¬
ной вероятности Р=0,95 составляют:
25% - дня количества эквивалентов кальция до 1 ммоль в 100г почвы, 12,5% - св.1
до 5ммоль в 100г почвы, 10% - св.5 ммоль в 100г почвы;
30 % - для количества эквивалентов магния до 0,2 ммоль в ЮОг почвы, 15% -
св.0,2 до 2 ммоль в ЮОг почвы, 10% - св. 2 ммоль в ЮОг почвы.
3.16. ОПРЕДЕЛЕНИЕ ПОДВИЖНОЙ СЕРЫ
ПО МЕТОДУ ЦИНАО
3.16.1. Аппаратура и материалы
Фотоэлектроколориметр.
Баня водяная.
Весы лабораторные 2-го класса точности с наибольшим пределом взвешивания 200г
и 4-го класса точности с наибольшим пределом взвешивания 500г.
Дозаторы с погрешностью дозирования не более 1% или пипетки и бюретки 2-го
класса точности.
Посуда мерная лабораторная 2-го класса точности.
Пробирки стеклянные вместимостью 50мл.
Бумага фильтровальная.
3.16.2. Реактивы
Кислота соляная, раствор концентрации с (НС1) = 1 моль/дм3 (1а)
Калий хлористый, раствор концентрации с (КС1) = 1 моль/дм3 (1н.)
- 150-
Натрия гидроокись, х.ч. или ч.д.а., раствор массовой концентрации 5г/дм3.
Натрий сернокислый безводный, х.ч.
Барий хлористый 2-водный, х.ч. или ч.д.а.
Крахмал растворимый.
Соль динатриевая этилендиамин-М,М,№,Мг-тетрауксусной кислоты 2-водную
(трилон Б), х.ч. или ч.д.а.
Вода дистиллированная.
3.16.3. Подготовка к анализу
3.163.1.Приготовление осаждающего раствора
20г двуводного хлористого бария, взвешенного с погрешностью не более 0,1 г,
помещают в стакан из термостойкого стекла вместимостью 1000мл, приливают при¬
мерно 800мл дистиллированной воды и 60мл раствора соляной кислоты концентра¬
ции 1 моль/дм3. Стакан помещают на кипящую водяную баню. В горячий раствор до¬
бавляют 5г крахмала, взвешенного с погрешностью не более 0,1 г и предварительно
разведенного небольшим количеством дистиллированной воды. Смесь нагревают на
водяной бане при непрерывном помешивании до получения прозрачного раствора.
Затем раствор охлаждают, переносят в мерную колбу вместимостью 1000мл, доводят
дистиллированной водой объем раствора до метки и тщательно перемешивают.
Раствор хранят в склянке с притертой пробкой в холодильнике не более недели.
Перед использованием раствор фильтруют через бумажный фильтр.
3.163^Приготовление раствора серы массовой концентрации 01мг/см3
0,443г сернокислого натрия, высушенного до постоянной массы при температуре
100-105°С, взвешивают с погрешностью не более 0,001 г, помещают в мерную колбу
вместимостью 1000мл, растворяют в растворе хлористого калия концентрации 1 моль/
да3, доводя объем раствора до метки и тщательно перемешивают.
Раствор хранят в склянке с притертой пробкой в холодильнике не более 3 месяцев.
ЗЛбЗЗ.Пригоговление растворов сравнения
В мерные колбы вместимостью 250мл помещают указанные в таблице 46 объе¬
мы раствора серы массовой концентрации 0,1мг/см3. Объемы растворов доводят
до метки раствором хлористого калия концентрации 1 моль/дм3 и тщательно пере¬
мешивают.
Таблица 46.
Характеристика раствора
Номер раствора сравнения
1
2
3
4
5
6
7
8
Объем раствора серы, см3
0
2
4
8
12
16
20
24
Концентрация серы
в растворе сравнения, мг/дм3
0
0,8
1,6
3,2
4,8
6,4
8,0
9,6
в пересчете на массовую долю в почве, млн 1
0
2
4
8
12
16
20
24
Растворы хранят в склянках с притертыми пробками не более 1 мес.
Растворы сравнения используют для градуировки фотоэлектроколориметра в
день проведения анализа.
- 151 -
3.16.3.4Лриготовление моющего раствора
5г трилона Б, взвешенного с погрешностью не более 0,1 г, растворяют в 1000мл
раствора гидроокиси натрия массовой концентрации 5г/дм3.
3.16.4. Проведение анализа
Приготовление вытяжки из почвы (см п. 3.1$.2.3.).
Для анализа используют фильтраты вытяжек, приготовленных.
Определение серы. В пробирки отбирают по 15мл фильтратов и растворов сравне¬
ния. К пробам приливают по 15мл осаждающего раствора и тщательно перемешивают.
Взвеси не ранее чем через 10 мин. после прибавления осаждающего раствора
фотометрируют в кювете с толщиной просвечиваемого слоя 5см относительно раствора
сравнения №1 при длине волны 520нм или используя светофильтр с максимумом про¬
пускания в области 500-540нм. Перед помещением в кювету фотоэлектроколоримет¬
ра взвесь необходимо перемешать. Взвесь оптически устойчива в течение 7 часов.
Допускается пропорциональное изменение объемов проб анализируемых вытяжек, ра¬
створов сравнения и осаждающего раствора при погрешности дозирования не более 1%.
Кюветы фотоэлектроколориметра в пробирки после работы помещают в моющий
раствор на 1 час.
3.16.5. Обработка результатов
По результатам фотометрирования растворов сравнения строят градуировоч¬
ный график. На оси абсцисс откладывают концентрации серы в растворах сравне¬
ния в пересчете на массовую долю в почве (млн-1), а на оси ординат - соответству¬
ющие показания фотоэлектроколориметра.
Массовую долю серы в анализируемой почве определяют непосредственно по
градуировочному графику и вычитают из него результат холостого опыта.
Если результат анализа выходит за пределы градуировочного графика, определе¬
ние повторяют, предварительно разбавив фильтрат раствором хлоритсого калия кон¬
центрации 1 моль/дм3. Результат, найденный по графику, увеличивают во столько
раз, во сколько был разбавлен фильтрат.
При проведении массовых анализов вместо построения градуировочного графи¬
ка допускается градуирование шкалы прибора по растворам сравнения в день прове¬
дения анализа.
За результат анализа принимают значение единичного определения серы.
Результат анализа выражают в миллионных долях с округлением до первого деся¬
тичного знака.
Допускаемые относительные отклонения от среднего арифметического результа¬
тов повторных анализов при выборочном статистическом контроле при доверитель¬
ной вероятности Р=0,95 составляет 35% при массовой доле серы в почве до 2*5 млн1,
15% - св. 2.5 до 5 млн*1, 10 % - св. 5 млн-1.
3.16.6. Технологическая карта
-152-
3.17. ОПРЕДЕЛЕНИЕ СУММЫ ПОГЛОЩЕННЫХ ОСНОВАНИЙ
3.17.1. Принцип метода
Метод основан на вытеснении поглощенных оснований из почвы 0,1 н. раствором
НС1 при соотношении почва : раствор -1:5.
Для почв, богатых основаниями (S > 15мг-экв/100 г почвы), это соотношение рас¬
ширяют до 1 : 10.
Метод пригоден для анализа безкарбонатных почв.
3.17.2. Технологическое оборудование
Электроплитка с регулируемым нагревом.
Воронки d = 3 и 7см.
Кассеты 10-позиционные.
Колбы плоскодонные на 100мл.
Капельница.
Пипетка на 50мл.
Бюретка на 25мл.
Дозатор на 100мл.
3.17.3. Реактивы
1. 0,1н. НС1 . Раствор готовят из фиксанала, разбавляя его содержимое дистил¬
лированной водой в 1л мерной колбе.
2. 1,0% раствор фенолфталеина. 1г фенолфталеина растворяют в 100мл 70% эти¬
лового спирта.
3. 0,1н. NaOH (КОН). 4 г NaOH или 5,6г КОН растворяют в дистиллированной
воде и доводят объем до 1л. Титр щелочи устанавливают по 0,1н. НС1.
3.17.4. Ход анализа
10-20г почвы (в зависимости от содержания поглощенных оснований) заливают
100мл 0,1н. НС1, взбалтывают в течение часа и оставляют на 24 часа. Затем взбалты¬
вают и фильтруют через сухой беззольный фильтр. 50мл фильтрата кипятят Змин.,
добавляют 3-4 капли фенолфталеина и в горячем состоянии титруют 0,1н. раствором
щелочи до неисчезающей в течение 1мин. слабо розовой окраски. Параллельно ведут
холостое титрование.
3.17.5. Обработка результатов
Расчет проводят по формуле:
вв_(и-в)-0,1-^-100
— >
н
где S - сумма поглощенных оснований, мг-экв/100г почвы;
а - объем 0,1 н. NaOH, пошедшей на титрование холостой пробы, мл.;
в - объем 0,1 н. NaOH, пошедшей на титрование анализируемой пробы, мл.;
0,1 - нормальность NaOH;
К - поправка к титру 0,1 н. NaOH;
н - аликвотная навеска, г.
- 153-
3.17.6. Технологическая карта
3.18. ОПРЕДЕЛЕНИЕ ЕМКОСТИ ПОГЛОЩЕНИЯ ПОЧВ
МЕТОДОМ БОБКО-АСКИНАЗИ-АЛЕШИНА
В МОДИФИКАЦИИ ЦИНАО
3.18.1. Принцип метода
Сумма всех катионов, способных к обмену, определяется путем замещения всех
обменных катионов одним катионом, не содержащимся в почве и последующего оп¬
ределения его количества. Обменные катионы вытесняются из почвы раствором ВаС12
и затем определяется количество бария (Ва), поглощенного из почвы.
Са2+
[ППК] Mq2* + 3 ВаС12 <— > ЗВа2+ + СаСК + MqCl2 + 2НС1
2Н+
H2S04 + 2NaOH = Na2S04 + 2Н20
3.18.2. Технологическое оборудование
Весы типа BJTKT.
Мерный цилиндр на 50, 100мл.
Стеклянные палочки.
Промывалка.
pH метр.
Пипетки 20мл.
Бюретка 50мл.
Химический стакан на 50мл.
3.18.3. Реактивы
1. 1 н. раствор хлорида бария буферный с рН=6.5. 6,1г ВаС12 • 2Н20 и 6,8г
(CH3OOQ2Ba растворяют в 1л мерной колбе дистиллированной водой и доводят pH
раствора до 6,5 путем добавления растворов гидрата окиси бария или уксусной кисло¬
ты. Затем объем раствора доводят водой до метки.
2. 0.05 н. раствор серной кислоты. Содержимое фиксанала ОДн. H2S04 доводят в
мерной колбе дистиллированной водой до 1л, затем полученный раствор разбавляют
в два раза.
- 154-
3. 0.1 н. раствор NaOH. 4 г NaOH растворяют в дистиллированной воде и дово¬
дят объем раствора до 1л. Устанавливают титр раствора по 0,1н.фиксанальному ра¬
створу H2S04 по фенолфталеину.
4. Раствор соляной кислоты (1 : 250; 1 : 60).
5. Фенолфталеин.
Анализ некарбонатных почв
3.18.4. Ход анализа
Навеску почвы 2,5г, предварительно просеянную через сито с отверстиями 1мм и
взвешенную с погрешностью до 0,05г, переносят в стеклянный стакан емкостью 50мл.
К почве приливают 25-30мл буферного раствора ВаС12 и тщательно перемешивают. С
помощью стеклянной палочки почву из стакана вместе с фильтратом переносят на
фильтр. С помощью промывалки остатки почвы из стакана тщательно смывают на
фильтр буферным раствором ВаС12. Для полного насыщения барием приливают на
почву 10- 15 мл буферного раствора ВаС12. Каждую последующую порцию раствора
приливают на почву после того, как полностью профильтруется предшествующая.
Насыщение почвы барием продолжают до тех пор пока pH фильтрата не будет соот¬
ветствовать 6,5, т.е.исходного буферного раствора. Для полного замещения обменных
катионов почвы барием необходимо следующее количество раствора: для легких почв
- 150-200мл, средне- и тежело-суглинистых - 200-250мл.
После окончания насыщением бария почву на фильтре промывают один раз дис¬
тиллированной водой и оставляют на ночь. Сухой фильтр с почвой переносят в хими¬
ческий стакан емкостью 250-300мл, приливают 100мл 0,05н. раствора H2S04, взбал¬
тывают 5 мин. и фильтруют. 20мл фильтрата переносят в химический стакан емкос¬
тью 100мл и титруют 0,1н. раствором NaOH с 2 каплями фенолфталеина до слабо¬
розового окрашивания. Параллельно проводят контрольное титрование, 20мл 0,05н.
раствора H2S04 в стакане емкостью 100мл титруют 0,1н. раствором NaOH с двумя
каплями фенолфталеина до появления слабо-розового окрашивания.
Анализ карбонатных, загипсованных и засоленных почв
3.18.5. Ход анализа
Из почв, предварительно удаляют карбонаты, гипс и другие соли. С этой целью
навеску 2,5г переносят в стакан емкостью 50мл и декантируют раствором HCI (1 :
250). Периодически на почву приливают небольшое количество кислоты. После от¬
ставания жидкость сливают на фильтр. Почва обрабатывается кислотой до тех пор
пока в фильтрате не появится специфическая реакция на ионы кальция.
При наличии в почве гипса или карбонатов ее сначала 2-3 раза обрабатывают ра¬
створом HCI (1 : 60) до прекращения выделения пузырьков С02 и далее до появления
реакции на ионы кальция.
Для реакции на ионы кальция 50мл фильтрата последних порций нейтрализуют
0,1н. раствором NaOH, приливают несколько миллилитров насыщенного раствора
оксалата аммония, кипятят 2 - Змин. и отстаивают 20мин. отсутствие осадка оксалата
кальция свидетельствует об отсутствии в растворе ионов кальция.
Затем почву полностью переносят на фильтр, насыщают барием, как указано ра¬
нее, и анализируют как почву не содержащую карбонатов.
Емкость поглощения почвы в миллиграмм-эквивалентах на 100г почвы рассчиты¬
вается по формуле:
- 155-
Е =
(а-«)• Я 100 100
V т
где а - количество 0,1 н. раствора NaOH, пошедшее на титрование 20мл
0,05н раствора H2S04, мл;
в количество 0,1н. раствора NaOH, пошедшее на титрование 20мл
фильтрата (после взаимодействия 0,05н. R2S04 с почвой, насыщенной
барием), мл;
Н- нормальность раствора NaOH, мг-экв/мл;
100- объем 0,05н.раствора . H2S04, взятое для замещения поглощенного
почвой бария, мл;
100 - коэффициент для пересчета результатов анализа на 100г почвы;
т - навеска почвы, г;
V - объем фильтрата, взятый для титрования, мл.
3.18.6. Технологическая карта
Подготовка к анализу
просеивание почвы, весы типа ВЛКТ,
шпатель, стканы на 50 мл
Добавление экстрагента
дозатор на 50 мл, мерный цилиндр на
50мл
Нзбалтывание шеое-мепптанирЪ о-иа
Смывание почвы на фильтр
и насыщение бапием
Стеклянная палочка
промывалка, пипетка 10-15мл,
фильтры, стаканы 300-350мл
ЩМШЫШса почвц!
->
Взбалтывание суспензии
пипетки на 100мл, мер-ные цилинд¬
ры на 100мл, стаканы на 250-300мл
Фильтрование
стеклянные палочки, хим.стаканы
150-200мл, воронки d=7-9cM,
фильтры, промывалка
-►
Титоование
пипетки на 20мл, стаканы на 100мл,
бюретка на 20-50 мл
3.19. АНАЛИЗ ВОДНОЙ ВЫТЯЖКИ
Для изучения химического состава засоленных почв и динамики легкодоступных
питательных веществ широко используется анализ водной вытяжки. В составе ее оп¬
ределяют плотный и прокаленный остаток, щелочность нормальных карбонатов и
бикарбонатов, содержание G , SO 4,"NO 3,’NO 2', Са ;*Mg “, Na‘, К* и др. ионов.
3.19.1. Получение водной вытяжки
3.19.1.1. Принцип метода
Метод основан на извлечении легкорастворимых солей из почвы водой (отноше¬
ние почвы к воде 1 : 5) при 3-минутном взбалтывании и фильтровании.
3.19.1.2. Технологическое оборудование
Кассеты 10-позиционные.
- 156-
Встряхиватель.
Воронки d = 9см.
Дозатор на 150мл.
3.19.13. Ход анализа
ЗОг воздушно-сухой почвы переносят в кассеты, заливают 150мл свежеперегнан-
ной дистиллированной воды, взбалтывают 3 мин. и фильтруют через складчатый
фильтр, перенося на него как можно больше почвы. Первые мутные порции фильтра¬
та перефильтровывают.
3.19.1.4. Технологическая карта
3.19.2. Определение сухого и прокаленного остатков водной
вытяжки
3.19.2.1. Принцип метода
Метод основан на высушивании вытяжки на водяной бане и прокаливании в му¬
фельной печи при t = 600°С. Первое дает представление об общем содержании в почве
растворимых в воде органических и минеральных соединений, а второе - только ми¬
неральных солей и характеризует степень минерализации водной вытяжки.
3.19.2.2. Технологическое оборудование
Аналитические весы.
Сушильный шкаф.
Водяная баня.
Муфельная печь.
Кассеты 10-позиционные или колбы на 250мл.
Эксикатор.
Фарфоровые чашки d = 7см.
Шприц-дозатор или цилиндр на 25 или 50мл.
Воронки d = 9см.
3.19.23. Ход анализа
25-50мл водной вытяжки помещают в предварительно взвешенные фарфоровые
чашки и ставят на водяную баню для выпаривания. По окончанию выпаривания чаш¬
ки с остатком высушивают в сушильном шкафу при t=105°C в течение 3 часов, охлаж¬
дают в эксикаторе и взвешивают на аналитических весах. Затем чашки прокаливают в
муфельной печи при t=600°C. Первое прокаливание проводят 10-15мин. с момента
достижения указанной температуры. Если за это время остаток не побелеет, чашки
- 157-
охлаждают и смачивают содержимое несколькими каплями дистиллированном воды,
затем подсушивают остаток на водяной бане и снова ставят в муфельную печь. После
полного сгорания органических веществ чашки с остатком охлаждают в эксикаторе и
взвешивают.
3.19.2.4. Обработка результатов
Расчеты ведут по формулам:
0 =
•100
где
я-
ае
н -
100
и
сухой остаток, %;
масса сухого остатка, г;
аликвотная навеска, г;
- коэффициент пересчета на 100 г почвы.
^ Д л юо
0п=-—,
Н
где О - прокаленный остаток, %;
ап - масса прокаленного остатка, г;
н - аликвотная навеска, г;
100 - коэффициент пересчета на 100г почвы.
Для большинства почв количество сухого остатка должно примерно равняться
сумме ионов в процентах. Допустимые расхождения 5%. В торфяниках и почвах со¬
лонцеватого ряда сумма ионов в % соответствует величине прокаленного остатка.
3.19.2.5. Технологическая карта
3.19.3. Определение щелочности водной вытяжки
3.193.1. Принцип метода
Карбонатную и бикарбонатную щелочность водной вытяжки определяют в од¬
ной пробе титрованием сначала СО *3‘ в присутствии фенолфталеина, затем НСО '3 с
индикатором метилоранжем. Реакции идут согласно уравнениям
- 158-
со; + н = нсо;
нсо; + н = н2о + со2
3.193.2. Технологическое оборудование
Потенциометр ЛПМ-60 М или рН-340, ЭВ-74, pH-150, AI-123.
Магнитная мешанка.
Колбы на 150 - 200 мл.
Пипетка или шприц-дозатор на 25мл.
Капельницы - 2 шт.
Бюретка на 25мл.
Стаканы на 100мл.
3.1933. Реактивы
1. 1% раствор фенолфталеина. 1г индикатора растворяют в 100мл 70% спирта.
2. 0,05% раствор метилоранжа. 50мг индикатора растворяют в 100мл дистилли¬
рованной воды.
3. 0,02н. раствор HjSOr Содержимое фиксанала 0,1н. раствора H2S04 доводят
дистиллированной водой до 1л, затем полученный раствор разбавляют в 5 раз водой.
3.193.4. Ход анализа
Опряяаттеттие С03 ."В колбы с 25мл фильтрата прибавляют по 2-3 капли фенолф¬
талеина и титруют пробы 0,02н. раствором H2S04 до обесцвечивания раствора.
Опрмгапрнйе НСОд. *К пробам, в которых определяли ион С03 ,* прибавляют 3-4
капли метилоранжа и титруют 0,02н. раствором H2S04 до перехода желтой окраски
в оранжевую с розовым оттенком.
Потенциометрическое определение СР3~и Н003*. В темно окрашенных водных
вытяжках при анализе солонцеватых и торфяных почв титрование щелочности следу¬
ет проводить потенциометрически.
В стаканы берут 20 мл водной вытяжки, помещают в нее «магнитик» и ставят
стаканы на магнитную мешалку. Затем в раствор погружают электроды и определяют
pH вытяжки. Если pH вытяжки меньше 8,3, нормальных карбонатов в растворе нет и
определять можно только щелочность бикарбонатов, титруя вытяжку 0,02 н. раство¬
ром H2S04 до pH 4,4. В вытяжках с pH выше 8,3 определяют оба вида щелочности. В
этом случае вытяжку сначала титруют 0,02 н. раствором H2S04 до pH 8,3 и фиксиру¬
ют расход кислоты, а затем продолжают титрование до pH 4,4.
Рассчитывают содержание карбонатов и бикарбонатов так же, как и при обычном
титровании.
3.193.5. Обработка результатов
Расчет проводят по формулам
.._д,-ЛМ00-2
О)
н
(а2 -2а,)-ТУ-100
(2)
н
- 159-
где С03” - содержание воднораствсримого карбонат-иона в почве,
мг-экв/100 г почвы (1);
НСО3 - аедзжаниево/дюрастворимсго^жщзбаиг-ионавпочве,
мг-экв/100 г почвы (2);
а1 - объем раствора H2S04 ,пошЕящего№ПИ1рование
пофенолфгакинумл.;
а2 - объем раствора H2S04 , пошедшего наттрованиеп
метилоранжу, мл.;
N - нормальность раствора H2S04;
н - аликвотная навеска, г;
100 - коэффициент пересчета на 100 г почвы;
2 - коэффициент пересчета на общее количество нормальных
карбонатов.
Дляпересжгавпрсадетыталученнуювеличину содержания НС03 (мг-жв/1 ООгпочвы) умножают
на0,061,аСЮ3 -на0,03.
3.193.6. Т ехнол огическая карта
3.19.4. Определение сульфат-ионов в водной вытяжке
3.19.4.1. Принцип метода
Метод основан на осаждении сульфат-иона барием и титровании избытка после¬
днего трилором Б с использованием флуорексона в качестве индикатора. Комплексо¬
метрическое определение S04* возможно в том случае, когда концентрация сульфа¬
тов в растворе обеспечивает выпадение осадка BaS04 , что соответствует 1-2мг S04
в объеме пробы. Максимальное содержание S04 в титруемой пробе не должно
превышать 20 мг. Чтобы обеспечить полноту осаждения сульфатов в водных вытяж¬
ках сильно засоленных почв, требуется большое разбавление - в 50-100 раз.
3.19.4.2. Технологическое оборудование
Аналитические весы.
Электроплитка.
Шпатель.
Колбы на 250 мл.
Пипетка или шприц-дозатор на 2, 5-10 мл.
Дозаторы на 5, 10, 15 мл.
Пробирки на 20 мл.
Капельница.
Бюретка на 25 мл.
- 160-
3.19.43. Реактивы
1 .HCl (I : 1).
2. Бумага конго красный.
3. 0,2% раствор малахитового зеленого. 200мг индикатора растворяют в 100мл
горячей дистиллированной воды или спирта.
4. 0,01Мраствор ВаС1у 2,07г ВаС12 растворяют в 1л дистиллированной воды.
5. 5Мраствор КОН. 280г КОН растворяют в 1л дистиллированной воды.
6. Флуорексон. Смешивают индикатор с КС1 или KN03 в соотношении 1 : 100.
Хранят в темной склянке с притертой пробкой.
7. 0,01 Мраствор трилона Б. 3,722г трилона Б растворяют в дистиллированной
воде, доводят объем до 1л и фильтруют. Нормальность устанавливают по 0,05 н. ра¬
створу СаС12 (см. п.3.14.4.3.).
8. Исходный образцовый раствор для определения S04 . *0,181 г K2S04 или 0.335г
Na2S04 • 10Н20 растворяют в дистиллированной воде и доводят объем до 1л. Полу¬
ченный раствор содержит 0,1мг S04’ в 1мл.
3.19.4.4. Ход анализа
Качественная проба. В пробирку берут 2мл водной вытяжки и доводят дистил¬
лированной водой объем до 10мл. Готовят шкалу образцовых растворов, для чего
отбирают в ряд пробирок указанных в таблице 47 объемы исходного образцового
раствора и доводят до 10мл водой. Затем во все пробирки добавляют по 2 капли НС1
(1 : 1) и 0,5мл 10% раствора ВаС12 . После добавления каждого реактива содержимое
пробирок перемешивают. Сравнивают выпавшие осадки водной вытяжки и шкалы.
В зависимости от величины осадка в испытуемой вытяжке устанавливают объем
аликвоты для анализа, как показано в таблице 47.
Таблица 47.
_ Номера пробирок
Показатели ' ^
1
2
3
Объем исходного образцового раствора, мл.
1
5
10
Содержание S04", мг в 10мл (шкала)
0,1
0,5
1,0
Содержание S04" в пробе, мг/мл.
0,05
0,25
0,5
Объем аликвоты, который следует брать в анализ, мл.
50
10
5
Содержание S04" в аликвоте, мг
2,5
2,5
2,5
Количественное определение. 5-50мл водной вытяжки (объем установлен на ос¬
новании качественной пробы) помещают в колбы на 250мл, добавляют соответствен¬
но 100-50мл воды, бросают кусочек бумажки конго и приливают по каплям НС1 (1:1)
до перехода окраски бумажки в сине-фиолетовый цвет. Нагревают раствор до кипения
и прибавляют 10-15мл 0,01М раствора ВаС12 для осаждения сульфат-ионов. Оставля¬
ют раствор стоять в течение 1-2 часов или до следующего дня. Затем добавляют 1-2
капли малахитового зеленого и небольшими порциями 5 М раствора КОН до обесцве¬
чивания малахитового зеленого и избыток КОН - 2мл.
В раствор шпателем добавляют флуорексон (30-50мг) и титруют 0,01М раство¬
ром трилона Б до появления оранжево-розовой окраски и снижения зеленой флуорес¬
ценции.
Поскольку в этом анализе одновременно с SO” титруется и Са,‘ параллельно
- 161 -
устанавливают объем трилона Б, пошедшего на его определение. Отдельно опреде¬
ляют объем трилона Б, пошедшего на титрование раствора ВаС12, взятого для осаж¬
дения сульфат-ионов.
3.19.4.5. Обработка результатов
Расчет проводят по формуле:
„ \а-{б-в)\ К- 0,00096 • 100
so;1 — з
где SO/1 - количество воднорастворимого сульфат-иона в почве, %;
а - объем трилона Б, пошедшего на титрование раствора ВаС12,
взятого для осаждения сульфат-ионов, мл.;
б - объем трилона Б, пошедшего на титрование избытка Ва" и Са”, мл.;
в - объем трилона Б, пошедшего на титрование иона Са1',
присутствующего в вытяжке, мл.;
н - аликвотная навеска, г;
0,00096 - титр 0,01 М раствора трилона Б по S04" ;
К - поправка к титру;
100 - коэффициент пересчета на 100г почвы.
Для пересчета в мг-экв/100 г почвы содержания S04" (%) следует разделить на 0,048.
3.19.4.6. Технологическая карта
Качественная проба.
Взятие аликвот
Добавлен и е
Осаждение
Водная
проб и шкалы
НО и НО
сульфатов
вытяжка
см.п.3.11.1.
пробирки,
шприц-дозатор
на 2 мл
-►
пипетка на 10
мл, капельница
пипетка или
шприц-дозатор
на 0,5 мл
Количественный анализ
Визуальное колоримет-
рцовмие
образцовая шкала
Взятие аликвот
Полкисление
TlamoBfluuP
Осаедение
шприц-дозатор
на 5-50 мл
оаствооа
бюретка,
индикатор
ПШ рсНапИС
электро-плитка
сульфатов
дозатор на 15 мл
Отстаивание
Подшелачивание
Титрование
оаствооа
раствора
индикатор, бюретка
индикатор, шпатель,
бюретка
часы
- 162-
3.19.5. Определение хлор-ионов в водной вытяжке
3.19*5.1. Принцип метода
метод основан на взаимодействии ионов хлора и серебра с образованием белого
осадка. В присутствии хрома иона как индикатора в точке эквивалентности идет осаж¬
дение красно-бурого хромата серебра. Реакции протекают согласно уравнениям
Ag’ + СГ = |AgCl
2 Ag’ + CrO/’= |Ag2Cr04
В присутствии карбонатов образуется также Ag2C03, поэтому титрование
следует вести в пробах, в которых оттитрована щелочность.
3.19.5.2. Технологическое оборудование
Колбы с пробами после определения щелочности.
Пипетка или дозатор на 1 и 50 мл.
Бюретка на 25-50 мл.
3.19.5.3. Реактивы
1. 10% раствор К7СгОА. 10г соли растворяют в 100мл дистиллированной воды.
2. 0,02 н. раствор AgNOv 3,4г соли растворяют в дистиллированной воде и
доводят объем до 1л. Титр проверяют по 0,1н. раствору NaCL, приготовленному из
фиксанала, а в качестве индикатора используют 10% раствор К2Сг04.
3.19.5.4. Ход анализа
В пробу, в которой оттитрована щелочность, добавляют 30-50мл дистиллирован¬
ной воды, 1мл 10% раствора К2Сг04 и титруют 0,02н. AgN03 до появления красно-
бурого осадка.
3.19.5.5. Обработка результатов
Расчет содержания СГ проводят по формуле:
Q* N 'Т Л 00
сг= ,
н
где СГ - содержание в почве хлора, мг-экв/100 г почвы;
а - объем раствора AgN03 , пошедшего на титрование анализируемой
пробы, мл.,
N - нормальность раствора AgN03 ;
Т - попрака к титру раствора AgN03;
н - аликвотная навеска, г;
100 - коэффициент пересчета на ЮОг почвы.
Для пересчета процента содержания хлора полученную величину умножают на 0,035.
3.19.5.6. Технологическая карта
- 163-
3.19.6. Определение катионного состава водной вытяжке
(Са\ Mg”, К’, Na’)
3.19.6.1. Принцип метода
Определение Са” и Mg” проводят трилонометрически. Описание метода см.
пп.3.14.4. и 3.14.5.
Концентрацию ионов натрия и калия в водной вытяжке определяют на пламенном
фотометре. При этом их содержание должно быть в пределах от 1 до ЮОмг/л. Если
водные вытяжки более концентрированы и окрашены, их следует разбавить.
3.19.6.2. Технологическое оборудование
Пламенный фотометр.
Колбы мерные на 1000мл.
Бюретка на 50мл.
3.19.6.3. Реактивы
1. Образцовые растворы для определения Na и К
Исходный образцовый раствор. Готовят 0,1 н. раствор NaCl и КС1 . Для этого
содержимое двух фиксаналов (на NaCl и на КС1) переводят в мерную колбу на 1 л и
доводят до метки дистиллированной водой.
Рабочая шкала образцовых растворов. В мерные колбы на 1л отбирают количе¬
ство исходного раствора, указанное в таблице 48, и доводят до метки дистиллирован¬
ной водой.
При построении калибровочного графика откладывают на оси абсцисс содержа¬
ние Na (К) в % или мг-экв/100г почвы, на оси ординат - оптическую плотность соот¬
ветствующих образцовых растворов шкалы.
Таблица 48.
^ Номера колб
Показатели .
1
2
3
4
5
б
7
8
Объем 0,1 н. раствора NaCl
(КС!), мл.
5
10
20
30
40
60
80
100
Содержание Na (К},мг-экв. на
100 г почвы
0,25
0,5
1,0
1,5
2,0
3,0
4,0
5,0
Na, %
0,00575
0,0115
0,023
0,0345
0,046
0,069
0,092
0,115
К,%
0,00975
0,0195
0,039
0,0585
0,078
0,117
0,156
0,195
3.19.6.4. Ход анализа
Непосредственно в водной вытяжке, а затем в образцовых растворах шкалы опре¬
деляют натрий на пламенном фотометре при светофильтре с длиной волны 589 нм.
После окончания этих замеров используя светофильтр с длиной волны 766 нм и
определяют калий в тех же вытяжках и образцовых растворах шкалы.
3.19.6.5. Обработка результатов
Содержание водорастворимого Na и К находят по графику непосредственно в
мг-экв/100 г почвы или процентах.
- 164-
Расчеты содержания Са” и Mg” в мг-экв на 100г почвы приведены в пп. 3.14.4.5.
и 3.14.5.5. Для пересчета в % полученную величину содержания Са” (мг-экв/100 г
почвы) умножают на 0,0204, a Mg” - на 0,01216.
Допустимые расхождения между сумой катионов и анионов в мг-экв/ЮОг почвы
пс должны превышать 5 %.
3.19.6.6. Технологическая карта.
Водная вытяжка
Фпггпмегшшование на Na’ ГК’)
(см. п. 3.19.1)
пламенный фотометр
3.20. ОПРЕДЕЛЕНИЕ ПОДВИЖНЫХ МАРГАНЦА» МЕДИ,
ЦИНКА И КОБАЛЬТА В ОДНОЙ ВЫТЯЖКЕ
АТОМНО-АБСОРБЦИОННЫМ МЕТОДОМ
3.20.1. Принцип метода
Подвижные формы Mn, Zn, Си и Со извлекают из не карбонатных и карбонатных
почв ацетатным буферным раствором с pH = 4,8 при соотношении почва : раствор - 1 : 10
и времени взаимодействия 1 час.
Определение их концентрации в почвенной вытяжке атомно-абсорбционным ме¬
тодом основано на измерении поглощения света свободными атомами этих микро¬
элементов, образующихся в пламени при введении в него анализируемых раство¬
ров и растворов сравнения (см. п.2.6.3.1.).
Марганец и цинк определяют путем непосредственного введения в воздушно-аци-
тиленовое пламя вытяжки из почвы. Соответственно используют аналитическую ли¬
нию марганца 279,5 нм и цинка - 213,8 нм.
При использовании воздушно-пропан-бутанового пламени цинк определяют ана¬
логично, а к анализируемым растворам на марганец для устранения химических по¬
мех вводят спектральный буферный раствор кальция.
В карбонатных почвах органические вещества, переходящие в вытяжку, предва¬
рительно окисляют. Для устранения помех, связанных с неселективным поглощением
микроэлементов (Са, Mg, Na, Al, Fe), цинк связывают в комплекс с диэтилдитиокарба-
матом и экстрагируют из водного раствора изоамилацетатом.
Аналогичную подготовку к атомно-абсорбционному определению необходимо
проводить при анализе на медь и кобальт как для карбонатных, так и для не карбонат¬
ных почв. При этом решается также задача концентрирования этих элементов до уровня
разрешающей способности прибора (табл..48). Используют аналитические линии меди
324,7 нм и кобальта - 240,7 нм.
Использование диэтилдитиокарбамата Na как реагента связано с тем, что он дает
устойчивые комплексы с цинком, медью и кобальтом, хорошо растворимые в ряде
органических растворителей. Количество реагента устанавливают в зависимости от
объема взятой в анализ вытяжки, так чтобы конечная концентрация его в водной фазе
равнялась 0,001 М.
При выборе органического растворителя (изоамилацетата) предъявляются следу¬
ющие требования: растворитель не должен давать большого поглощения или излуче¬
ния света, а также нарушать стабильность пламени; должен иметь низкую раствори¬
мость в воде и обеспечивать высокую растворимость и устойчивость экстрагируемых
комплексов.
Общие требования к анализам на микроэлементы изложены в п.2.6.1.
- 165-
3.20.2. Извлечение подвижных Mnv Zn, Си, Со
3.20.2.1. Технологическое оборудование
Набор ареометров.
Колбы на 500 мл.
Воронки d = 10см.
Цилиндр мерный или дозатор на 250мл.
3.20.2.2. Реактивы
1. Ацетатный буферный раствор с pH =4,8. Измеряют ареометром плотность
аммиака и уксусной кислоты, из которых готовят раствор, и по таблицам находят их
концентрации в весовых процентах. Объемы реактивов, необходимые для приготов¬
ления буферного раствора, вычисляют по формулам:
для аммиака 25 • 0 907
у — 75
Ч ’
для уксусной кислоты 98 • 1.0549
Vч — 108 ,
C2d,
где VI и V2 - искомые объемы аммиака и уксусной кислоты, мл ;
С1 - фактическая концентрация аммиака в весовых процентах;
dl - соответствующая ей плотность, г/см 3 ;
25 - заданная концентрация аммиака в весовых процентах;
0,907 - соответствующая ей плотность, г/см3;
75 - объем 25 % аммиака, необходимый для приготовления буферного
раствора, мл.;
С2 - фактическая концентрация уксусной кислоты, %;
d2 - соответствующая ей плотность, г/см3 ;
98 - заданная концентрация уксусной кислоты, %;
1,0549 - соответствующая ей плотность, г/см3 ;
108 - объем 98 % уксусной кислоты, необходимой для приготовления
буферного раствора, мл..
Расчетные количества уксусной кислоты и аммиака приливают к 500-600мл
бидистиллированной воды и доводят объем бидистиллятом до 1л.
3.20.23. Ход анализа
25г почвы переносят в колбу на 500мл, заливают 250мл ацетатного буферного
раствора с pH = 4,8, взбалтывают в течение 1 часа и фильтруют через складчатый
фильтр (белая лента) d = 15см, отмытый от следов микроэлементов (см.п.2.6.1.). Пер¬
вую мутную порцию отбрасывают. Параллельно ведут холостую пробу.
3.20.2.4. Технологическая карт»
- 166-
3.20.3. Определение марганца
3.203.1. Технологическое оборудование
Установка для атомно-абсорбционного анализа (см.п.2.6.32.), лампа с полым ка¬
тодом для определения Мп.
Весы аналитические.
Колбы мерные на 100, 1000 мл.
Пипетки градуированные на 5, 20 мл.
Дозатор на 5 мл.
3.203.2. Реактивы
1. Ацетатный буферный раствор с pH - 4,8 (см.п.3.20.2.2.).
2. Образцовые растворы для определения марганца
Исходный образцовый раствор - раствор А (см.п.2.6.3.3.),содержит 1мг/мл. марганца.
Рабочий образцовый раствор - раствор Б. В мерную колбу на 100мл берут 9,1 мл
раствора А и доводят до метки ацетатным буферным раствором с pH = 4,8. раствор
готовят в день проведения анализа, содержит 1СЮмкг/мл. Мп.
Шкала образцовых растворов. В мерные колбы на 100 мл переносят указанные в
таблице 49, объемы рабочего стандартного раствора марганца и доводят до метки
ацетатным буферным раствором с pH = 4,8.
Таблица 49.
— ЦумйрпДратопйлга раСТВОра
Показатели ‘—
1
2
3
4
5
6
7
Объем раствора Б, мл.
0
1
2
5
10
15
20
Концентрация Мп в образцовом растворе шкалы,
мкг/мл
0
1
2
5
10
15
20
Содержание Мпв почве, мг/кг
0
10
20
50
100
150
200
3. Раствор СаС12 , содержащий в 1мл 1,25мг кальция. 3,125г карбоната каль¬
ция растворяют в небольшом количестве дистиллированной воды, содержащей 22мл
концентрированной НС1, и, когда навеска полностью растворится, доводят объем до
1л водой.
3.2033. Ход анализа
Настраивают атомно-абсорбционный спектрофотометр на определение марган¬
ца. Устанавливают размах шкапы по 1-у и 7-у образцовым растворам. Почвенная вы¬
тяжка, см.п.3.20.2.3.
Фотометрируют шкалу образцовых растворов и почвенные вьггяжки. После каждых
десяти измерений проверяют настройку прибора по 1-у и 7-у образцовш раствора!.
При использовании пропан-бутан-воздушного пламени пробу и растворы шкалы
разбавляют вдвое раствором СаС12. Фотометрирование проводят на атомно-абсор-
бционном спектрофотометре, как указано выше.
3.203.4. Обработка результатов
Строят калибровочный график, откладывая на оси абсцисс содержание Мп в мг/100г
почвы, а на оси ординат - соответствующие ему показания измерительного прибора.
- 167-
По калибровочному графику находят содержание марганца в анализируемой
пробе почвы. Фиксируют результат холостого определения.
Допустимые расхождения результатов повторного определения от среднеарифме¬
тического в одной лаборатории - 20%, в разных лабораториях - 30%.
3.20.3.5. Технологическая карта
3.20.4. Определение цинка в некарбонатных почвах
3.20.4.1. Технологическое оборудование
Установка для атомно-абсорбционного анализа (см.п.2.6.3.2.), лампа с полым
катодом для определения цинка.
Весы аналитические.
Колбы мерные на 50, 100, 1000 мл.
Пипетки градуированные на 5, 10 мл.
3.20.4.2. Реактивы
1. Ацетатный буферный раствор с pH - 4,8 (см.п.3.20.2.2.).
2. Образцовые растворы для определения цинка.
Исхайыый образцовый раствор - раствор А. 0,1 ООг ± 0,001 металлического цинка
помещают в мерную колбу на 1000мл и растворяют в небольшом количестве билис-
тиллированной воды (30-50мл), содержащей 10мл 20% НС1, и доводят бидистилля¬
том объем до 1л. Раствор содержит 100мкг/мл..
Рабочий образиовый раствор - раствор Б. В мерную колбу на 100мл переносят
5мл раствора А и доводят до метки ацетатным буфером. Раствор содержит 5мкг/мл.
Zn. Готовят его в день проведения анализа.
Шкапа образцовых растворов. В мерные колбы на 50мл берут указанные в табли¬
це 50 объемы раствора Б и доводят до метки ацетатным буфером.
Почвенная вытяжка, см.п.3.20.2.3.
Таблица 50.
Показатели
Номера колб
1
2
3
4
5
б
Объем раствора Б, мл.
0
1
~
5
7
10
Концентрация Zn в образцовом растворе шкалы, мкг/мл.
0
0,1
0,3
0,5
0,7
1,0
Содержание Zn в почве, мг/кг
0
1
3
5
7
10
- 168-
3.20.43. Ход анализа
Настраивают атомно-абсорбционный спектрофотометр на определение цинка.
Определение проводят по аналитической линии 213,8 нм. Размах шкалы устанавлива¬
ют по первому и шестому образцовым растворам, фотометрируют всю шкалу
образцовых растворов, а затем вытяжки из почв.
После каждых 10 измерений корректируют настройку прибора по первому и ше¬
стому образцовым растворам.
3.20.4.4. Обработка результатов
Калибровочный график строят, откладывая на оси абсцисс содержание цинка в
мг/кг почвы, соответствующее образцовому раствору, а на оси ординат - отсчет изме¬
рительного прибора. Находят по калибровочному графику содержание цинка в пробе
и вычитают результат холостого определения.
3.20.4.5. Технологическая карта (см. П.3.20.5.5Л
3.20.5.0пределение цинка в карбонатных почвах
3.20.5.1. Технологическое оборудование
Установка для атомно-абсорбционного анализа (см.п.2.6.3.2.). Лампа с полым ка¬
тодом на Zn.
Электропечь с закрытой спиралью и регулятором нагрева.
Весы аналитические.
Чашки фарфоровые на 50мл.
Цилиндр мерный на 50мл.
Дозаторы на 5, 10, 20 и на 2мл для крепких кислот.
Воронки делительные на 100мл.
Колбы мерные на 50, 100, 1000мл.
Пробирки с притертой пробкой.
Пипетки градуированные на 1 и 10мл.
3.20.5.2. Реактивы
1. 0,4% HCL 10мл концентрированной НС1 х.ч. или 20мл 20% НС1 перегнанной
разбавляют до 1 л бидистиллированной водой.
2. Ацетатный буферный раствор с pH 5,8. 128мл ледяной уксусной кислоты и 150 мл
25% аммиака приливают к 500-600мл бидистиллированной воды и доводят объем до 1л.
3. 0,2% раствор диэтилдитиокарбамата натрия. 0,2г реактива растворяют в
100мл бидистиллированной воды.
4. Изоамиловый эфир уксусной кислоты (изоамилацетат), ч.
5. Образцовые растворы для определения цинка.
Исходный образцовый раствор - раствор А (см.п.3.20.4.2.).
Рабочий образцовый раствор - раствор Б. 1мл раствора А помещают в мерную
колбу на 100мл и доводят до метки 0,4% НС1. Раствор содержит 1мкг/мл. цинка. Гото¬
вят его в день проведения анализа.
Таблица 51.
Номера колб
Показатели ~ —
1
2
3
4
5
Объем раствора Б, мл.
0
5
10
15
20
Концентрация Zn в образцовом растворе шкалы, мкг/мл.
0
0,1
0,2
0,3
0,4
Содержание Zn в почве, мг/кг
0
0,5
1,0
1,5
2,0
- 169-
Щкпла образиовых растворов. В мерные колбы на 50мл переносят указанные
в таблице 51 объемы раствора Б и доводят до метки 0,4% НС1.
3.20.53. Ход анализа
50мл вытяжки (см.п.3.20.2.3.) выпаривают в фарфоровой чашке до получения влаж¬
ного остатка. К остатку приливают 2мл концентрированной HN03 и 2мл H2Q и выпари¬
вают до влажных солей. После такой же повторной обработки остаток выпаривают досу¬
ха, растворяют при нагревании в 20мл 0,4% НС1 и фильтруют в делительную воронку на
100мл. Предварительно в воронке должен быть отмечен объем 25мл. Фильтр на воронке
промывают несколько раз небольшими порциями 0,4% НС1 и доводят объем до 25мл.
Приливают к фильтрату 10мл ацетатного буферного раствора с pH 5,8 и 5мл 0,2% раство¬
ра диэтилдитиокарбамата натрия, перемешивают после каждого прибавления реактива.
Затем добавляют 10мл изоамилацетата, встряхивают 1мин. и после разделения фаз отбра¬
сывают водный слой, а экстракт сливают в пробирку с притертой пробкой.
Настраивают атомно-абсорбционный спектрофотометр. Определение цинка
проводят по аналитической линии 213,8нм. Вводят в пламя изоамилацетат и регули¬
руют расход горючего таким образом, чтобы получить пламя с четко очерченным
внутренним конусом, не отрывающееся при прекращении подачи эфира.
Размах шкалы устанавливают по чистому изоамилацетату и экстракту из шестого
образцового раствора. Затем фотометрируют экстракты из шкалы образцовых раство¬
ров и анализируемых почвенных вытяжек. После каждых 10 измерений проводят кор¬
ректировку работы прибора, вводя в пламя чистый изоамилацеат и экстракт шестого
образцового раствора.
3.20.5.4. Обработка результатов
Строят калибровочный график, откладывая на оси абсцисс содержание цинка в
мг/кг почвы, соответствующее образцовому раствору, а на оси ординат - показания
измерительного прибора.
Содержание цинка в почве находят по калибровочному графику, вычитая из него
результат холостого определения.
3.20.5.5. Технологическая карта
в некарбонатных почвах
Растворение и промывка осадка
Дозатор на 20мл,воронки дели¬
тельные на 100мл, воронки d=7cM,
фильтры
I
Концентрирование и экстракция
дозаторы на 5, 10мл, пробирки с
притертой пробкой
- 170-
3.20.6. Определение меди и кобальта
3.20.6.1. Технологическое оборудование
Установка для атомно-абсорбционного анализа (см.п.2.6.3.2.), лампы с полным
катодом для определения Си и Со.
Весы аналитические.
Воронки делительные на 200-250мл.
Цилиндры мерные на 100 мл.
Пипетки градуированные на 1, 5, 10мл.
Пробирки с притертыми пробками на 10-20мл.
Колбы мерные на 100, 500, 1000 мл.
Дозаторы на 5 и 10мл.
3.20.6.2. Реактивы
1. Ацетатный буферный раствор с pH 4,8 (см.п.3.20.2.2.)
2. 0,5% раствор диэтилдшпиокарбамата натрия. 5г реактива растворяют в 100мл
бидистиллированной воды. Реактив готовят в день проведения анализа.
3. Изоамиловый эфир уксусной кислоты (изоамилацетат), ч.
4. Образцовые растворы для определения меди и кобальта.
Исходный образцовый раствор на медь - раствор А. 1,965г CuS04 * 5Н20,
взвешенного с погрешностью не более 0,001 г, растворяют в бидистиляте, содержа¬
щем 10мл 20% НС1, переносят в мерную колбу на 500мл и доводят объем до метки.
Раствор содержит 1мг/мл Си.
Исходный образцовый раствор на кобальт - раствор А. 0,477г CoS04 • 7Н20,
взвешенного с погрешностью не более 0,001 г растворяют в бидистилляте, подкисля¬
ют раствор 1мл концентрированной H2S04 и доводят объем до 1л в мерной колбе.
Раствор содержит 100 мкг/мл. Со.
Рабочий смешанный образцовый раствор - раствор Б. В мерную колбу на 1л
переносят 1мл раствора А на медь и 5мл раствора А на кобальт и доводят ацетатным
буферным раствором с pH 4,8 до метки. Полученный раствор содержит 1 мкг/мл Си и
0,5мкг/мл. Со, готовят его в день проведения анализа.
Шкала образцовых растворов. В мерные колбы на 1000мл вводят указанные в
таблице 52 объемы рабочего смешанного раствора меди и кобальта и доводят ацетат¬
ным буфером с pH 4,8 до метки.
Таблица S2.
—^Номера колб
Показатели "
1
2
3
4
5
б
7
Объем раствора Б, мл.
0
10
20
40
60
80
100
Концентрация в образцовом растворе шкалы, мкг/мл
Си
0
0,01
0,02
0,04
0,06
0,08
0,10
Со
0
0,005
0,01
0,02
0,03
0,04
0,05
Содержание в почве, мг/кг
Си
0
0,1
0,2
0,4
0,6
0,8
1,0
Со
0
0,05
0,1
0,2
0,3
0,4
0,5
- 171 -
3.20.63. Ход анализа
В делительную воронку на 200-250мл переносят 100мл вытяжки или образцового
раствора, приливают 10мл 0,5% раствора диэтилдитиокарбамата натрия, перемеши¬
вают, приливают 5 мл изоамилацетата и встряхивают 1 мин. После разделения фаз
нижний водный слой сливают, а экстракт собирают в пробирку с притертой пробкой.
Настраивают атомно-абсорбционный спектрофотометр на определение Си.
Вводят в пламя изоамилацетат и регулируют расход газа таким образом , чтобы
получить не отрывающееся, с четким очертанием внутреннего конуса пламя. Уста¬
навливают размах шкалы по чистому изоамилацетату и экстракту седьмого образ¬
цового раствора. Фотометрируют экстракты шкалы образцовых растворов, а затем
почвенных вытяжек, проверяя настройку прибора после каждых десяти измерений
по чистому изоамилацетату и экстракту седьмого образцового раствора.
Закончив анализ проб на содержание меди, настраивают прибор на определение
кобальта. Устанавливают размах шкалы по чистому изоамилацетату и экстракту седь¬
мого образцового раствора, фотометрируют шкалу образцовых растворов и экстрак¬
ты почвенных вытяжек. Проверку и корректировку прибора проводят так же, как и
при определении меди.
3.20.6.4. Обработка результатов
Строят калибровочный график, откладывая на оси абсцисс содержание кобальта
или меди в мг/кг почвы, соответствующее образцовому раствору, а на оси ординат -
показание прибора. По калибровочным графикам находят содержание Си и Со в по¬
чве и вычитают результат холостого определения.
3.20.6.5. Технологическая карта
3.21. ОПРЕДЕЛЕНИЕ ПОДВИЖНОГО БОРА
3.21.1. Принцип метода
Метод основан на извлечении бора из почвы горячей водой, содержащей сернокис¬
лый магний для коагуляции почвенных коллоидов, при отношении почвы к воде 1 : 5 и
последующем его калориметрическом определении.
Для окрашивания использована реакция образования в водной среде (pH 5,2) ком¬
плекса бора с азометином Н желтого цвета. Окраску органических веществ вытяжки
устраняют окислением их перманганатом. Влияние меди, железа и алюминия снима¬
ют связыванием их в комплексы с трилоном Б. Необходимое для образования азоме-
тинового комплекса бора pH раствора поддерживают ацетатным буферным раство¬
ром. Общие требования к анализу на бор изложены в п.5.5.
- 172-
3.21.2. Технологическое оборудование
Весы типа BJIKT и аналитические.
Фотоэлектроколориметр или спектроколориметр.
Баня водяная.
Кассеты 10-позиционные.
Колбы конические на 250мл из безборного стекла.
Воронки.
Дозатор на 50мл.
Пробирки из безборного стекла или полиэтиленовые на 10 или 20мл с пробками.
Пипетки градуированные на 1и 10мл.
Дозаторы на 0,5 и 4мл, шприц-дозатор на 5мл.
Колбы мерные на 50, 100 и 1000мл.
3.21.3. Реактивы
1. 0,1% раствор MgSOr 1г реактива, х.ч., растворяют в дистиллированной воде
и доводят объем до 1л.
2. HJS04 (1: 4) - см.п.2.6.4.3.
3. Буферный маскирующий раствор (см.п.2.6.4.3.).
4. 0,9% раствор азометина Н (см.п.2.6.4.3.).
5. Смешанный окрашивающий раствор (см.п.2.6.4.3.).
6. Окисляющий раствор. Смешивают раствор серной кислоты (1: 4) и 0,7% ра¬
створ марганцовокислого калия в соотношении 1:1. Готовят раствор в день проведе¬
ния анализа.
7. 10% раствор аскорбиновой кислоты. 20г реактива растворяют в 200мл дистил¬
лированной воды. Раствор хранят в холодильнике не более 1 недели.
8. 10% раствор КОН. 20г реактива растворяют в 200мл дистиллированной воды.
9. Образцовые растворы для определения бора
Исходный образцовый раствор (см.п.2.6.4.3.).
Рабочий образцовый раствор (см.п.2.6.4.3.).
Щкяпя образцовых растворов. В мерные колбы на 50мл берут указанные в табли¬
це 53 объемы рабочего стандартного раствора и доводят до метки дистиллирован¬
ной водой. Готовят шкалу в день проведения анализа.
Таблица §3.
' — Номера колб
Показатели
1
2
3
4
5
6
Объем рабочего раствора бора, мл.
0
1
2
4
6
8
Количество В, мкг/мл.
0
0,1
0,2
0,4
0,6
0,8
Содержание В в почве,мг/кг
0
0,5
1
2
3
4
3.21.4. Ход анализа
Пробу почвы массой 10г взвешивают с погрешностью не более 0,1г и помещают в
коническую колбу на 250мл. К пробе приливают 50мл 0,1% раствора сернокислого
магния. В колбу вставляют маленькую воронку в качестве обратного холодильника и
умеренно кипятят суспензию на плитке в течение 5 мин. Затем снимают ее с плитки,
суспензию перемешивают и фильтруют в горячем состоянии через складчатый
фильтр. Первую порцию фильтрата отбрасывают. 5мл вытяжки переносят в про¬
- 173-
бирку, прибавляют 0,5мл окисляющего реактива, перемешивают и выдерживают в
водяной бане 10мин. при t=90-100° С. После охлаждения раствора приливают 0,5мл
10% аскорбиновой кислоты для растворения двуокиси марганца, а затем 4 мл сме¬
шанного окрашивающего реактива, перемешивают и оставляют на 2 часа.
Аналогично проводят окрашивание растворов образцовой шкалы.
Фотометрируют растворы при длине волны 440 нм (синий светофильтр) в кюве¬
тах с толщиной просвечиваемого слоя 2см относительно нулевого раствора шкалы.
Одновременно ставят холостой опыт, проводя его через все стадии анализа, ис¬
ключая взятие пробы почвы.
3.21.5. Обработка результатов
По результатам фотометрирования строят калибровочный график, откладывая на
оси абсцисс содержание бора в почве в мг/кг, соответствующее образцовому раствору,
а на оси ординат - значение оптической плотности этого раствора.
3.21.6. Технологическая карта
Добавление 0.1% р-оа MnSO
дозатор на 50 мл
ФнЛИРЭЦЩ
воронки d=9cM, фильтры с синей
лентой
Окисление органических веществ
пипетка на 1 мл или дозатор на
0,5 мл, водяная баня
Колориметрирование
ФЭК (синий светофильтр) кювета
-2см
3.22. ОПРЕДЕЛЕНИЕ СОДЕРЖАНИЯ МОЛИБДЕНА В ПОЧВЕ
3.22.1. Принцип метода
Метод основан на извлечении молибдена из почвы оксалатным буфером с pH 3,3
при отношении почвы к раствору 1:10 и взбалтывании в течение 1 часа. Содержание
молибдена в вытяжке определяют колориметрически, для чего использована реакция
образования окрашенного комплекса пятивалентного молибдена с роданидом щелоч¬
ного металла. Для концентрирования этого комплекса проводят экстракцию его изоа-
миловым или н-бутиловым спиртом. Молибден восстанавливают до пятивалентного
состояния двухлористым оловом, которое одновременно восстанавливает Fe(CNS)3
до бесцветного Fe(CNS)2 . Для более полного устранения окраски трехвалентного
роданистого железа и некоторых других элементов экстракт роданит-молибденово-
го комплекса промывают раствором SnCl2.
- 174-
Влияние железа на интенсивность окраски роданида молибдена компенсируют
и ведением железа в шкалу образцовых растворов.
3.22.2. Технологическое оборудование
Весы типа ВЛКТ и аналитические.
ФЭК.
Ротатор или встряхиватель.
Печь муфельная.
Плита электрическая с закрытой спиралью и регулятором нагрева.
Кассеты 10-позиционные.
Колбы конические на 300 и 500мл.
Колбы мерные на 100, 1000мл.
Цилиндр или шприц-дозатор на 100 и 150мл.
Дозатор на 2, 5, 8, 10мл.
Стаканы из термостойкого стекла на 100мл.
Воронки делительные на 100мл.
Пипетка градуированная на 5мл.
Воронки d= 5 и 9см.
3.22.3. Реактивы
1. Оксалатный буферный раствор с pH 3,3. 25г щавелевокислого аммония и
12,6г щавелевой кислоты растворяют в горячей дистиллированной воде и после
охлаждения доводят до объема 1л.
2. 20% раствор HCL 492мл НС1, х.ч. или ч.д.а., (плотность 1,19г/см3) приливают
к дистиллированной воде и доводят объем до 1л.
3. 6,5% раствор HCL 330мл 20% НС1 разбавляют дистиллированной водой до
1л.
4. 10% раствор KCNS. Юг реактива, х.ч. или ч.д.а., растворяют в дистиллиро¬
ванной воде, доводят объем до 100мл и фильтруют.
5. 20% раствор SnClr 20г реактива, ч.д.а., растворяют в 20мл 20% НС1, накрыва¬
ют часовым стеклом и кипятят раствор в течение 10 мин., затем охлаждают и раз¬
бавляют водой до 100мл.
6. 1% раствор железо-аммонийных квасцов. Юг реактива, х.ч. или ч.д.а., ра¬
створяют при нагревании в 20% НС1 и после охлаждения доводят этой кислотой
объем до 1л. Раствор содержит 1 мг/мл. железа.
7. Образцовые растворы для определения молибдена
Исходный образцовый раствор. 0,184г ± 0,001 г молибденовокислого аммония,
х.ч., растворяют в воде и доводят объем в мерной колбе до 1л. Раствор содержит
0,1 мг/мл. молибдена, хранится не более 1 года.
Рабочий стандартный раствор молибдена. 2,5мл исходного образцового раство¬
ра молибдена переносят в мерную колбу на 250мл и доводят дистиллированной
водой до метки. Раствор содержит 0,1мкг/мл. молибдена. Готовят в день проведе¬
ния анализа.
Шкаля образцовых растворов. В делительную воронку берут по 5мл раствора
железо-аммонийных квасцов и указанные в таблице 54 объемы рабочего стандар¬
тного раствора молибдена. Приливают по 5мл 20% НС1 и дистиллированную воду
до объема 25мл (до метки на делительной воронке). Далее поступают как и при
анализе вытяжки (см. ход анализа п.3.22.4).
- 175-
Таблица 54.
Номер делительной вороики
Показатели
1
2
3
4
5
б
7
Объем рабочего р-ра Мо, мл.
0
0,5
1,0
2,0
3,0
4,0
5,0
Содержание Мо в почве, мг/кг
0
0,05
0,1
0,2
о,з
0,4
0,5
Промывной раствор. Сливают 6 мл 20% раствора SnCl2 и 94 мл 6,5% НС1.
Раствор готовят непосредственно перед промывкой экстракта роданида молибдена.
3.22.4. Ход анализа
15г почвы помещают в коническую колбу на 500мл, приливают 150мл оксалатно-
го буферного раствора и взбалтывают в течение 1 часа. Вытяжку фильтруют через
складчатый фильтр. Первую мутную порцию фильтрата отбрасывают.
100мл вытяжки переносят в стакан из термостойкого стекла и выпаривают на плите
досуха. Стакан оставляют на плите еще 30 мин. для полного обезвоживания остатка и
частичной возгонки оксалатов, помещают в холодный муфель и прокаливают остаток
в течение 4 часов, постепенно повышая температуру до 500°С. Прокаленный остаток
растворяют при нагревании в 10 и 20% НС1 и фильтруют в делительную воронку с
отметкой 25мл. Промывают фильтр 4-5 раз дистиллированной водой, доводя объем
раствора до метки. Затем к раствору в воронке приливают 2,0мл 10% KCNS и 2,0мл
20% SnCl2, перемешивая после прибавления каждого реактива. После обесцвечива¬
ния окраски роданида железа прибавляют 8 мл изоамилового или н-бутилового спир¬
та и встряхивают воронку в течение 2 мин. Дают фазам полностью расслоиться, ниж¬
ний водный слой отбрасывают, оставив около 0,5мл его в воронке с экстрактом. Экст¬
ракт заливают 10мл промывного раствора и встряхивают воронку в течение 1мин.
После разделения фаз водный слой полностью сливают и отбрасывают, экстракт пере¬
носят в кювету колориметра, добавляют 5 капель этилового спирта, перемешивают
экстракт стеклянной палочкой и оставляют на 5мин. для оседания капелек воды. Ок¬
раска экстракта не изменяется в течение ЗОмин.
Оптическую плотность прозрачного экстракта, окрашенного в оранжевый цвет,
измеряют относительно изоамилового или н-бутилового спирта на спектроколориметре
«Спекол» при длине волны 470нм, используя приставку ЕК-5 и кюветы с толщиной
просвечиваемого слоя 5см, или на ФЭКе при синем светофильтре в кювете с толщи¬
ной просвечиваемого слоя Зсм. Параллельно измеряют оптическую плотность раствора
контрольного опыта, которая не должна превышать 0,05 единицы.
Если оптическая плотность раствора контрольного опыта больше указанной ве¬
личины, то это означает загрязнение молибденом одного или нескольких используе¬
мых реактивов.
Для окраски образцовой шкалы берут по 25мл каждого раствора сравнения и про¬
водят анализ аналогично описанному для почвенных проб.
3.22.5. Обработка результатов
По результатам фотометрирования строят калибровочный график, откладывая на
оси абсцисс содержание Мо в почве в мг/кг, соответствующее образцовому раствору, а
на оси ординат - значение оптической плотности этого раствора.
Допустимые отклонения повторного определения от среднеарифметического зна¬
чения не должны превышать 30%.
- 176-
3.22.6. Технологическая карта
3.23. ОПРЕДЕЛЕНИЕ ИНТЕНСИВНОСТИ
ВЫДЕЛЕНИЯ С02 ИЗ ПОЧВЫ
3.23.1. Принцип метода
Метод основан на поглощении С02, выделяемой из почвы, раствором щелочи в
замкнутом пространстве и последующем осаждении в форме ВаСОг Реакции идут
согласно уравнениям:
CD, + 2NaOH = Na2C03 + HzO
Na2C03 + ВаСЦ = ВаС03 + 2NaCl.
Количество С02 учитывают по разности между исходным и конечным содер¬
жанием щелочи в растворе.
3.23.2. Технологическое оборудование
Сосуд-изолятор - ведро или кристаллизатор, окрашенные снаружи в белый цвет.
Чашки Петри.
Пипетки на 10, 20мл.
Бюретка на 25мл.
Капельница.
3.23.3. Реактивы
1. 0,1 и NaOH. 4г реактива растворяют в дистиллированной воде и доводят объем до i л.
2. 0,1н НС1 или HSO . Фиксанал 0,1н. НС1 или H2S04 растворяют в дистиллиро¬
ванной воде и доводят объем до 1л.
- 177-
3. Фенолфталеин. 1г фенолфталеина растворяют в 100мл 96% этилового спирта.
4. 10% BaCL. Юг BaCL растворяют в 100мл дистиллированной воды.
3.23.4. Ход анализа
Под сосуд-изолятор ставится чашка Петри с 10-20мл 0,1н. раствора щелочи.
Сосуд-изолятор заглубляют на 1-1,5см в землю и плотно присыпают почвой наруж¬
ные стенки во избежание попадания воздуха снаружи.
После экспозиции, которая устанавливается в зависимости от интенсивности
выделения СО, из почвы (1-3 часа), сосуд-изолятор снимают, добавляют в щелочь
10мл ВаС12 10% и титруют 0,1 н. НС1 или H2S04 по фенолфталеину. Титрование
производится непосредственно в поле в чашках Петри. Одновременно определяют
содержание С02 в воздухе сосуда-изолятора, для чего чашку со щелочью ставят под
сосуд-изолятор на закрытую полиэтиленовой пленкой поверхность почвы.
При сравнении нескольких опытных делянок определения нужно проводить од¬
новременно на всех сравниваемых вариантах в одни и те же часы (желательно с 9 до
13 часов). Повторность 3-5-кратная.
3.23.5. Обработка результатов
Количество выделившийся из почвы С02 определяют по формуле:
, (а- в) -N 22 10000
А = ,
t S
где А - количество C02,miAi2 в час;
а - количество 0,1 н. НС1, израсходованной на титрование щелочи на
кошроле, мл.;
в - количество 0,1 н. НС1, израсходованной на титрование щелочи на
испытуемых вариантах, мл.;
22 - мг-экв С02;
t - время экспозиции (часы);
S - площадь почвы под сосудом-изолятором (см2);
10000 - пересчет на 1 м2 площади;
N - нормальность НС1 или H2S04.
3.23.6. Технологическая карта
3.24. ОПРЕДЕЛЕНИЕ ФЕРМЕНТАТИВНОЙ АКТИВНОСТИ ПОЧВ
3.24.1. Активность инвертазы
Инвертаза катализирует гидролиз дисахаров (сахарозы, раффинозы, генциано-
зы и других) до моносахаров.
3.24.1.1. Принцип метода
Определение почвенной инвертазы основано на компостировании почвы с ра-
- 178-
створом сахарозы в качестве субстрата в присутствии толуола, подавляющего микро¬
биологическую деятельность почвы. Активность фермента учитывается по количе¬
ству глюкозы, образовавшейся при ферментативном гидролизе сахарозы. Для этой
цели использована реакция восстановления глюкозой двухвалентной меди до одно¬
валентной, протекающая в щелочной среде.
СН2ОН
I
НО — С — н
н —с —он
+ гсисон^ —> но — с — н + Си,о + н.о.
н — с — он
I
с = о
ч
СН2ОН
I
НО —
I
с —н
н —
с —он
но —
с —н
н —
с —он
о
II
II
_и
\
и
Образовавшуюся закись меди определяют йодометрически. Реакции идут согласно
приведенным ниже уравнениям:
KJ03 + 5KJ + 3H.S04 = 3J2 + 3K.SO, + 3H20.
Выделившийся йод в присутствии щавелевой кислоты взаимодействует с заки¬
сью меди:
Cu20 + J2 + Н2С204 = CuC204 + CuJ2 + Н20.
Избыток йода оттитровывают раствором гипосульфита:
2Na,S А + = Na,S40. + 2NaJ.
2 2 3 l 2 4 6
Полной пропорциональности между количеством восстанавливающих сахаров
и закисью меди не наблюдается, поэтому для расчетов используется эмпирическая
формула, выведенная из экспериментальных данных.
3.24.1.2. Технологическое оборудование
Весы типа BJIKT.
Термостат.
Водяная баня.
Колбы на 150мл с пробками.
Дозаторы на 5, 10, 20, 100мл.
Пипетки или шприц-дозатор на 1-5мл.
Бюретки на 25мл.
Воронки d= 7см.
Капельница с грушей.
3.24.1.3. Реактивы
/. Медно-щелочной реактив. 75г Na2C03 безводный растворяют примерно в
500мл дистиллированной воды и постепенно прибавляют к нему раствор, приготов¬
ленный из 12г CuS04*5H20 и 21,5г винной кислоты, растворенных в 400мл дистил¬
лированной воды. К полученному раствору прибавляют 0,89г КЮ3, и 8г KJ, дово¬
дят объем водой до 1л, закрывают воронкой, нагревают и держат в течение 15мин. в
кипящей водяной бане, а затем отстаивают 16-18 часов и сифонируют.
2 Смесь щавелевой и серной кислот. Растворяют в 800мл дистиллированной воды
60г щавелевой кислоты и прибавляют 70мл концентрированной серной кислоты. Ра¬
створ охлаждают и доводят дистиллированной водой объем до 1л.
- 179-
3. 0.01н. раствор гипосульфита. 25г Na2S203-5H20 растворяют в дистиллиро¬
ванной воде, в которой предварительно растворено 2г NajCOj ,и доводят объем до
1л. Полученный 0,1н раствор гипосульфита разбавляют в 10 раз. Титр устанавлива¬
ют по 0,1 н фиксанальному раствору К2Сг207. Для этого к 2мл 0,1н раствора К2Сг207
прибавляют 5мл 20% раствора KJ, 10мл 10% НС1 или H2S04 и титруют гипосульфи¬
том в присутствии 1мл 0,5% раствора крахмала.
4. 5% раствор сахарозы. 5 г сахарозы растворяют в 100мл дистиллированной воды.
5. 1н. раствор KSP4 или КС1 . 174г K2S04mm 74,5г КС1 растворяют в воде и
объем доводят до 1л. ~ ~
6.Нитратно-фосфатный буфер pH 5.0.71,8г Na2HP04 • 12НгО растворяют в ди¬
стиллированной воде и доводят объем до 1л, 21,008г лимонной кислоты растворяют
в дистиллированной воде и доводят объем до 1л. Растворы сливают в соотношении 10,3 :
9,7. Хранят в склянках с притертыми пробками, предварительно обработав 3-5 кап¬
лями толуола при t = +5°С.
3.24.1.4. Ход анализа
К навеске почвы 5г приливают 0,5мл толуола, прибавляют 10мл цитратно-фос-
фатного буфера с pH 5,0 и 10 мл 5% раствора сахарозы. Колбы закрывают пробками,
встряхивают и компостируют в термостате 48 часов при температуре 37°С.
После компостирования содержимое доводят до объема 200мл 1н. раствором
К2804или КС1. К аликвотной части фильтрата (1-5 мл) прибавляют 5мл медно-ще¬
лочного реактива, перемешивают, накрывают воронкой, выдерживают в кипящей во¬
дяной бане 15 мин. и быстро охлаждают под струей холодной воды. Затем прилива¬
ют 5мл смеси щавелевой и серной кислот, выделившийся йод оттитровывают 0,01 н.
раствором гипосульфита в присутствии крахмала.
Параллельно ведут контрольное определение, для чего к 5мл медно-щелочного
реактива после 15-минутного нагревания на водяной бане и охлаждения прибавля¬
ют 5 мл смеси кислот и оттитровывают 0,01 н. раствором гипосульфита.
3.24.1.5. Обработка результатов
Расчеты ведут по формуле:
Л [248 - (а - б)] - (д - б\ 100 - Г
1000 н
или
. F-100-Г г |248-(а-б)](а-б)
л — "Р" • F= Ш
ще А - активность инвертазы в мг глюкозы на ЮОг почвы;
а - количество гипосульфита 0,01 н., пошедшего на титрование
контрольного раствора, мл;
б - количество гипосульфита 0,01 н., пошедшего на титрование
исследуемого раствора, мл;
н - аликвотная навеска, г;
248 - эмпирический пересчетный коэффициент;
Т - поправка к титру гипосульфита.
Расчетные значения F приведены в таблице 55.
Допустимая ошибка определения ±5% от средней арифметической двух повтор¬
ных определений.
- 180-
Таблица 55.
Значения факторов для вычисления содержания восстанавливающих сахаров
г [248-{а-б)]-{а-б)
1000
а-б, мл
Десятые доли миллплятров
0
ОД
0,2
0,3
0,4
0,5
0,6
0,7
0,8
0,9
0
-
0,02
0,06
0,07
0,10
0,12
0,15
0,17
0,20
0,22
1
0,25
0,27
0,30
0,32
0,35
0,37
0,38
0,42
0,44
0,47
2
0,49
0,52
0,54
0,57
0,59
0,61
0,64
0,66
0,69
0,71
3
0,74
0,76
0,78
0,81
0,83
0,86
0,88
0,90
0,93
0,95
4
0,98
1,00
1,02
1,05
1,07
1,10
1,12
1,14
1,17
1,19
5
1,22
1,24
1,26
1,29
1,31
1,33
1,36
1,38
1,40
1,43
6
1,46
1,48
1,50
1,52
1,55
1,57
1,58
1,62
1,64
1,66
7
1,69
1,71
1,73
1,76
1,78
1,80
1,83
1,85
1,87
1,90
8
1,92
1,94
1,97
1,99
2,01
2,04
2,06
2,08
2,10
2,13
9
2,15
2,17
2,20
2,22
2,24
2,27
2,29
2,31
2,33
2,36
10
2,38
2,40
2,43
2,45
2,47
2,49
2,52
2,54
2,56
2,58
11
2,61
2,63
2,65
2,68
2,70
2,72
2,74
2,77
2,79
2,81
12
2,83
2,85
2,88
2,90
2,92
2,94
2,97
2,99
3,01
3,03
13
3,06
3,08
3,10
3,12
3,14
3,17
3,19
3,21
3,23
3,25
14
3,28
3,30
3,32
3,34
3,36
3,39
3,41
3,43
3,45
3,47
15
3,50
3,52
3,54
3,56
3,58
3,60
3,63
3,65
3,67
3,69
16
3,71
3,73
3,76
3,78
3,80
3,82
3,84
3,86
3,88
3,91
17
3,93
3,95
3,97
3,99
4,01
4,03
4,06
4,08
4,10
4,12
18
4,14
4,16
4,18
4,20
4,23
4,25
4,27
4,29
4,31
4,33
19
4,35
4,37
4,39
4,41
4,44
4,46
4,48
4,50
4,52
4,54
20
4,56
4,58
4,60
4,62
4,64
4,66
4,68
4,71
4,73
4,75
3.24.1.6. Технологическая карта (см. п. 3.24.2.6)
3.24.2. Активность целлюлазы
3.24.2.1. Принцип метода
Целлюлаза расщепляет клетчатку (целлюлозу) с образованием целлобиозы и даль¬
нейшим распадом последней до глюкозы.
Метод определения почвенной целлюлазы основан на ферментативном гидро¬
лизе карбоксиметилцеллюлозы при компостировании ее с почвой в присутствии то¬
луола для подавления микробных процессов. Выделившуюся при этом глюкозу опре¬
деляют по количеству восстановленной в щелочной среде меди йодометрическим
титрованием согласно реакциям приведенным в п.3.24.1.1.
- 181 -
3.24.2.2. Технологическое оборудование
Весы типа ВЛКТ.
Термостат.
Водяная баня.
Колбы на 150 мл с пробками.
Дозаторы на 5, 20 и 50мл.
Пипетка или шприц-дозатор на 20мл.
Воронки d=7cM.
Бюретка на 25мл.
Капельница с грушей.
3.24.2.3. Реактивы
1. Фосфатный буфер pH 6.8. 11,876г Na^IPO^HjO растворяют в дистиллиро¬
ванной воде и доводят объем до 1л; 9,078г КН2Р04 растворяют также в воде и дово¬
дят объем до 1л. Растворы сливают в соотношении 1:1. Хранят в холодильнике, пред¬
варительно обработав 3-5 каплями толуола.
2.1%КМ11.1г карбоксиметилцеллюлозы растворяют в 100мл дистиллированной воды.
Остальные реактивы см. п. 3.24.1.3.
3.24.2.4. Ход анализа
20г почвы помещают в колбу, обрабатывают 0,5мл толуола, прибавляют 10мл 1%
КМЦ и 10мл фосфатного буфера с pH 6,8. Колбы закрывают пробками и взбалтывают.
Компостирование проводят в течение 7 дней при температуре 37°С в термостате.
Затем в колбу с почвенной суспензией заливают 20мл 1н. раствора K2S04miH КС1
и фильтруют. К 20мл фильтрата прибавляют 5мл медно-щелочного реактива и выдер¬
живают в кипящей водяной бане в течение 15 мин., затем быстро охлаждают под стру¬
ей воды. В жидкость добавляют 5мл смеси серной и щавелевой кислот и титруют
выделившийся йод 0,01н. раствором гипосульфита в присутствии крахмала.
Параллельно ведут контрольное определение, для чего к 5мл медно-щелочного
реактива после 15-минутного нагревания на водяной бане и быстрого охлаждения
прибавляют 5мл смеси кислот и оттитровывают 0,01н. раствором гипосульфита.
3.24.2.5. Обработка результатов
Активность фермента рассчитывают по формуле:
Л [248 - {а - б)]- (а - б)-1000 • Т
1000-н
ще А - активность целлюлозы в мкг глюкозы на 1 г почвы;
а - количество 0,01 н. гипосульфита, пошедшего на титрование контрольного
раствора, мл;
б - количество 0,01 н. гипосульфита, пошедшего на титрование
исследуемого раствора, мл;
н - аликвотная навеска, г;
Т- поправка к титру гипосульфита;
248 - эмпирический пересчетный коэффициент;
или ^ _ F ’ ЮОО • Т где ^ _ |248 — (а — б)]- (а — б)
н ’ 1000
значения которого приведены в таблице 55.
Допустимая ошибка - ±5% от средней арифметической двух повторных определений.
- 182-
3.24.2.6. Технологическая карта
Взятие навесок
весы типа ВЛКТ,шпатель,емкости
на 150мл с пробками
Др^ацление сц£си_ластдодов
субстрата и буфера
дозатор на 20 мл
Д р£авл дди e_p-g_a КС1
дозатор на 100мл (для инвертазы);
на 20мл(для целлюлазы)
Отбор аликвот
колбы на 50 или 100мл,пипетки с
грушей на 1,2 или 5мл (для инвер¬
тазы),на 20 мл(для целлюлазы)
Добавление толуола
капельница с грушей
Т ермостатирование
термостат при t= 37°С
Фил ьтрование
колбы на 150мл,воронки d=7cM,
фильтры с черной лентой d=llcM
Добавление медного реактива
дозатор на 5 мл (высокой точности)
Нагрев
водяная баня
Добавление реактива
дозатор на 5 мл
Iитрование
бюретка на 25мл,
капельница для крахмала
3.24.3. Активность протеаз
Протеазы - группа ферментов, катализирующих гидролитическое расщепление
белков и промежуточных продуктов их распада до аминокислот.
3.24.3.1. Принцип метода
Метод определения почвенных протеаз основан на компостировании почвы с же¬
латином в качестве субстрата в присутствии толуола как антисептика. Образовавший¬
ся при ферментативном гидролизе аминный азот определяют медным способом, сущ¬
ность которого состоит в том, что аминокислоты реагируют со свежеосажденным гид¬
ратом окиси меди с образованием растворимого медного комплекса.
/О
Cu(OH). + 2NH2 - R - С > Cu(NH2RCOO)2 + 2Н20.
хОН
Содержание меди учитывают колориметрически с использованием реакции об¬
разования ее диэтилдитиокарбаматного комплекса, окрашенного в желтый цвет.
Линейная зависимость между количеством аминокислот и оптической плотнос¬
тью растворов сохраняется в пределах от 1 до ЗОмкг аминного азота.
3.24.3.2. Технологическое оборудование
Весы типа ВЛКТ.
Термостат.
Фотоэлектроколориметр.
Колбы с пробками на 50 и 100-150мл.
Пробирки ё=15мм.
Дозаторы на 5,10,20 и 25мл.
- 183-
Воронки d=5 и 7см.
Шприц-дозатор на 1-2 и 10мл.
Капельница с грушей.
3.24.3.3. Реактивы
1. Фосфорный буфер pH 5.6. 11,876г Na2HP04*2H20 растворяют в дистиллиро¬
ванной воде и доводят объем до 1л; 9,078г КН2Р04 растворяют в дистиллированной
воде и доводят объем до 1л. Растворы сливают в соотношении 0,5:9,5.
2. 1% раствор желатина. 1г желатина растворяют в 100мл теплой дистиллиро¬
ванной воды.
3. Суспензия (Ьосфата меди •
а) СиС12 • 2НгО - 27,Зг на 1л дистиллированной воды;
б) Na2HP04 • 2Н20 - 64,Зг растворяют в 1000мл дистиллированной воды, содер¬
жащей 7,2г NaOH.;
в) боратный буфер - 57,2г буры растворяют в 1,5л дистиллированной воды, при¬
бавляют 100мл 1н. НС1 и доводят объем до 2л.
Растворы сливают в соотношении 1 а : 26 : 2в.
4. 2% раствор диэтилдитиокарбамата натрия. 2г соли растворяют в 100мл
дистиллированной воды.
5. Исходный обоазиовый раствор для определения аминного азота. 0,134г CuCl^
• 2Н20 растворяют в дистиллированной воде и доводят объем до 1л. 1мл раствора
содержит 0,05мг Си.
3.24.3.4. Ход анализа
Юг почвы переносят в колбу, обрабатывают 1мл толуола, заливают 50мл 1% ра¬
створа желатина и 5мл фосфатного буфера pH 5,6, закрывают пробкой и взбалтыва¬
ют. Компостирование проводят при температуре 37°С в течение 72 часов в термо¬
стате. Суспензию фильтруют через фильтр с белой лентой. К 10мл фильтрата прибав¬
ляют 10мл суспензии фосфата меди, встряхивают и отстаивают в течение 30 мин.
Затем фильтруют через фильтр с синей лентой. 1мл фильтрата переносят в пробирку
на 20мл, доливают 10-20мл дистиллированной воды и прибавляют 0,1 мл 2% диэ¬
тилдитиокарбамата натрия.
Фотометрировацие окрашенного в желтый цвет раствора проводят при синем
или зеленом светофильтре.
3.24.3.5. Обработка результатов
Активность фермента рассчитывают по формуле:
, 0,0022 а * 100
Л = ,
н
где А - активность фермента в мг аминного азота на 100г почвы;
а - показания по графику в мг/мл Си;
0,0022 - коэффициент пересчета на аминный азот;
н - аликвотная навеска.
При построении графика берут 0,1мл, 0,2мл и т.п. до 2мл раствора СиС12, содер¬
жащего в 1мл 0,05мг Си, доливают 10мл дистиллированной воды и приливают 0,1мл
2% диэтилдитиокарбамата натрия.
Допустимая ошибка - ±8% от средней арифметической двух повторных опреде¬
лений.
- 184-
3.243.6. Технологическая карта
Взятие навесок
-►
Добавление толуола
весы типа BJIKT, шпатель, емкос¬
ти на 150мл с пробками
капельница с грушей
Добавление смеси р-ров
Термостатирование
желатина и буфера
термостат при t=37° С
дозатор на 25 мл
Фильтаование
-►
Отбор аликвот
колбы на 100мл, ворон-ки d=7cM,
фильтры с бе-лой лентой d=llcM
пипетки Мора или шприц-дозатор
на 10мл, колбы на 100 или 50мл
Добавление суспензии Досфата
-►
колбы на 50- 100мл, во- ронки
d=5cM, фильтры с синей лентой
меди
дозатор на 10 мл
Отбоо аликвот
->
Окпашивание
пробирки на 20 или 30мл,
пипетка с грушей или шприц-до¬
затор на 1-2мл
дозатор на 10 или 20мл, пипетка
на 1 мл или капельница
Коллриметрирование
ФЭК (зеленый светофильтр)
3.24.4. Активность уреазы
Уреаза катализирует реакцию гидролитического разложения мочевины на амми¬
ак и углекислоту.
3.24.4.1. Принцип метода
Определение почвенной уреазы основано на компостировании почвы с мочеви¬
ной в качестве субстрата в присутствии толуола как антисептика. Выделившийся в
результате ферментативной реакции аммоний определяется колориметрическим ме¬
тодом индофенольной зелени (см.п.3.8.1.)
3.24.4.2. Технологическое оборудование
Весы типа BJIKT и аналитические.
Термостат.
Фотоэлектроколориметр.
Колбы с пробками на 50 и 100-150мл.
Дозаторы на 1, 10, 18, 20 и 100мл.
Воронки ё=5см.
Шприц-дозатор на 1мл.
Капельница с грушей.
3.24.4.3. Реактивы
/. Фосфатный буфер pH 6.8. 11,876г Na2HP04 • 2Н20 растворяют в дистиллиро¬
ванной воде и доводят объем до 1л.
- 185-
9,078г КН 2РО 4 растворяют в дистиллированной воде и доводят объем до 1л.
Оба раствора сливают в соотношении 1:1. Хранят в холодильнике, обработав 3-5 кап¬
лями толуола.
2 10% раствор мочевины. Юг мочевины растворяют в дистиллированной воде
и доводят объем до 100мл.
3. Рабочий окрашивающий раствор (см.п.3.8.3.2.).
4. Рабочий раствор гипохлорита натрия (см.п.3.8.3.2.).
3.24.4.4. Ход анализа
5г почвы обрабатывают 0,5 мл толуола, заливают 10мл 10% раствора мочевины и
10мл фосфатного буфера pH 6,8, закрывают пробкой и взбалтывают. Компостируют в
течение 24 часов в термостате при температуре 37°С. Затем прибавляют 100мл 1н.
K2S04wih КС1 , взбалтывают и отфильтровывают. Берут 1мл фильтрата, добавляют
18мл рабочего окрашивающего раствора и 1мл рабочего раствора гипохлорита натрия.
Растворы перемешивают после каждого дозирования реагентов и оставляют на 1 час
для развития окраски. Колориметрируют при 655нм - красный светофильтр.
Параллельно ведут окрашивание рабочей шкалы образцовых растворов (см.п.3.8.3.3.)
3.24.4.5. Обработка результатов
Активность фермента рассчитывают по формуле:
а 100 _ а
~ н-1000 ~10V
ще А- активность фермента, учитываемая по количеству в мг N-NH4 на
100 г почвы;
а - показания по графику, мг/л N-NH4;
н - аликвотная навеска, г.
Допустимая ошибка определения - ±5 %.
3.24.4.6. Технологическая карта
- 186-
3.24.5. Активность фосфатазы
Фосфатаза катализирует отщепление остатков фосфорной кислоты от органофос¬
фатов (фитина, нуклеиновых кислот и др.).
3.24.5.1. Принцип метода
Метод основан на компостировании почвы с Са-глицерофосфатом в присутствии
толуола как антисептика. Выделившиеся в результате ферментативного гидролиза мине¬
ральные фосфаты определяют колориметрически в виде молибденовой сини.
3.24.5.2. Технологическое оборудование
Весы типа ВЛКТ.
Фотоэлектроколориметр.
Термостат.
Колбы с пробками на 50 и 100-150мл.
Дозаторы на 18, 20 и 50мл.
Воронки ё=7см.
Шприц-дозатор на 2мл.
Капельница с грушей.
3J4.5J. Реактивы
1. Аиетатный 6v<ben pH 6.2. Смешать 1н. растворы СН3СООН и CH3COONa в соот¬
ношении 1:32 или к 1000мл 1н. CH3COONa (82г), прибавить 1,9мл уксусной кислоты.
2. 0.5% Са-?лииррп(Ьос(Ьат. 0,5г Са-глицерофосфата растворяют в 100мл ацетат¬
ного буфера.
3. Растворы для определения фосфатов (см.п.3.11.2.2.).
3.24.5.4. Ход анализа
5г почвы обрабатывают 0,5мл толуола, заливают 20мл 0,5% Са-глицерофосфата
в ацетатном буфере pH 6,2, закрывают пробкой и взбалтывают Компостируют в течение
48 часов в термостате при температуре 37° С. Добавляют 50мл 1н. . К^С^или КС1 ,
взбалтывают, осадок отфильтровывают.
К 2мл фильтрата приливают 18 мл реактива Б и колориметрируют через 10 мин.
при красном светофильтре. Образцовые растворы шкалы отбирают и окрашивают
так же, как и анализируемые пробы. Параллельно проводят холостое определение.
3.24.5.5. Обработка результатов
Активность фосфатазы рассчитывают по формуле
а 100 _ а
"h-IOOO-IOV
где С - активность фосфатазы, учитываемая по количеству мг Р205 на
ЮОг почвы;
а - показания по графику, мг/л Р205;
н - аликвотная навеска, г.
Допустимая ошибка определения - ±5 %.
- 187-
3.24.5.6. Технологическая карта
Взятие навесок
весы типа ВЛКТ, шпатель,
емкости на 150мл с пробками
Добавление смеси р-ров
субстрат0 и
дозатор на 20 мл
Добавление р-па KCI
дозатор на 50 мл
и квот
колбы на 50мл, пипетка с грушей
или шприц-дозатор на 2 мл
Колориметрироваиие
ФЭК (красный светофильтр)
Добавление толуола
капельница с грушей
ТерМОСТ«т»црпаии1>
термостат при t=37° С
Фильтрование
колбы на 100мл, воронки d=7cM,
фильтры d= 11см
проП
дозаторы на 18 мл
3.24.6. Активность пероксидаз
Пероксидазы катализируют окисление органических веществ почв (фенолов, ами¬
нов, некоторых гетероциклических соединений) за счет кислорода перекиси водорода и
органических перекисей. Большое распространение в почве имеют пероксидазные
реакции, в которых в качестве перекиси используют хиноны - «структурные фрагменты»
гумусовых веществ, т.е. пероксидазы участвуют в процессах разложения гумуса.
3.24.6.1. Принцип метода
Метод основан на способности гваякола окисляться перекисью водорода в при¬
сутствии пероксидаз до тетрагваякохиноза, окрашенного в красно-коричневый цвет.
Активность фермента учитывается по интенсивности его окраски.
Реакция идет согласно приведенному уравнению
^ОН , О • С6Н3(ОСН3) • С6Н3(ОСН3) • о
4С6Н + 4Н202 =1 i + 8Н.0.
Х>СН3 О • С6Н3(ОСНз) • С6Н3(ОСН3) • о
3.24.6.2. Технологическое оборудование
Весы типа ВЛКТ и аналитические.
Термостат.
Фотоэлектроколориметр.
Встряхиватель.
Конические колбы на 100 и 250мл.
Дозаторы на 1 и 75мл.
Пробирки с меткой на 10мл.
Воронки d=7cM.
3.24.6.3. Реактивы
L 0.3% гваякол. 1,5мл гваякола вливают в 300мл дистиллированной воды и ра¬
створяют при сильном встряхивании.
- 188-
2. 0.5н.Нр.. 0,5мл перегидроля растворяют в 150мл дистиллированной воды.
3. Стандартный раствор AgNO'. 0,1968г AgN03 растворяют в дистиллирован-
иой воде в мерной колбе на 250мл. 1мл этого раствора содержит 0,5мг Ag.
4. Построение капибповочной кривой. В серию пробирок набирают от 0,1 до 6мл
стандартного раствора AgN03H доводят дистиллированной водой объем до 10мл,
прибавляют по 1мл 0,3% раствора гваякола, перемешивают, а затем по 1 мл 3%
раствора персульфата аммония. Через 15 минут измеряют интенсивность окраски при
зеленом светофильтре. На основании полученных измерений строят калибровочную
кривую.
5. O.Sh.NHjNO^ 40г соли растворяют в дистиллированной воде и доводят объем до 1л.
3.24.6.4. Ход анализа
50г почвы заливают 100мл 0,5н. раствора NH4N03, взбалтывают в течение 30
мин. и фильтруют через фильтр с синей лентой. К 10мл фильтрата прибавляют 1мл
0,3% гваякола и 1мл 0,05н. Н202, перемешивают, а затем компостируют в течение 1
часа в термостате.
Колориметрируют при зеленом светофильтре.
3.24.6.S. Обработка результатов
Расчет проводят формуле
А =
С 2,53 100
где А - активность пероксидазы, выраженная в мкмолях гваякола;
С - показания по графику, мг/мл Ag;
н - аликвотная навеска, г.;
2,53 - пересчет в мк моли гваякола;
Допустимая ошибка определения - ±8 %.
3.24.6.6. Технологическая карта
Взятие навесок
весы типа BJIKT, шпатель,
емкости на 250мл с пробками
Нзбалтмвание
встряхиватель или ротатор
Отбор аликвот
пробирки с меткой на 10 мл
Термпстатирование
термостат при t—30° С
Добавление экстрагента
дозатор на 75 мл
Фильтрование
колбы на 100мл, воронки ё=7см,
фильтры с синей лентой d=15 см
Окрашивание нроб
2 дозатора на 1 мл
КплприМРтудрлййНИР
ФЭК (зеленый светофильтр)
3.24.7. Активность полифенолоксидаз
Полифенолоксидазы участвуют в превращении органических соединений аромати¬
ческого ряда в компоненты гумуса. Они катализируют окисление фенолов до хинонов. В
соответствующих условиях при гетерополиконденсации хинонов с аминокислотами и
полипептидами идет новообразование первичных гумусовых соединений.
- 189-
3.24.7.1. Принцип метода
Метод основан на способности почвенных полифенолоксидаз катализировать
реакцию окисления дифенолов кислородом воздуха. При pH 6,0 пирокатехин (о-ди-
фенол) быстро превращается в о-хинон:
Образовавшийся в этой ферментативной реакции ортохинон восстанавли¬
вается аскорбиновой кислотой:
■о-
,0 сн,-сн-сн-с = с-с = о
* ♦ I I II
он он он он
сн2 - сн
I !
он он
сн-с-с”с =
II II
о о
он
Реакция протекает при попеременном многократном окислении пирокатехина
почвенной полифенолоксидазой и восстановлении его аскорбиновой кислотой. Коли-
чество окисленной при этом аскорбиновой кислоты определяют йодометрически.
3.24.7.2. Технологическое оборудование
Весы типа ВЛКТ и аналитические.
Встряхиватель.
Термостат.
Колбы на 150 и 250мл.
Дозаторы на 1, 2,3, 5, 20 и 75мл.
Воронки ё=7см.
Бюретка на 25мл.
Капельница.
3.24.7.3. Реактивы
1. Раствор пирокатехина 0.2%. Отвешивают на технических весах 0,2г пирока¬
техина и растворяют в 100 мл дистиллированной воды.
2. Раствор аскорбиновой кислоты 0.4%. Отвешивают 0,4г аскорбиновой кисло¬
ты, растворяют в дистиллированной воде и доводят объем до 100мл.
3. Раствор йодата калия 0,01 н^. 0,3566г KJ03 растворяют дистиллированной во¬
дой в мерной колбе на 1л. прибавляют 5мл 1н. раствора NaOH и 2г KJ и доводят
водой до метки. Раствор хранят в темной склянке.
4. Раствор ортофосфорной кислоты 5%. 30мл Н3Р04 разбавляют дистиллиро¬
ванной водой до 1л.
5. 0.5н. NHjNOj 40г соли растворяют в дистиллированной воде и доводят объем до 1л.
- 190-
6. Фосфатный 6vd)ev pH 6.4. 1,36г КН2Р04 растворяют в безуглекислой воде,
добавляют 2,5мл 1н. раствора NaOH, разбавляют такой же водой до 200мл и хранят в
плотно закрытой склянке.
7. 0.5% раствор крахмала. 0,^г крахмала заваривают в 100мл дистиллированной воды.
3.24.7.4. Ход анализа
50г почвы заливают 100мл 0,5н. раствора . NH4N03, взбалтывают в течение 30
мин. и фильтруют через фильтр с синей лентой.
К 20мл фильтрата прибавляют 1мл фосфатного буфера с pH 6,4; 2мл 0,4% раствора
аскорбиновой кислоты и взбалтывают, затем прибавляют Змл 0,2% раствора пирокате¬
хина, повторно взбалтывают и компостируют в течение 1 часа при температуре 30° С в
термостате. После компостирования в раствор добавляют 5мл 5% ортофосфорной или
метафосфорной кислоты, 1мл 0,5% раствора крахмала и титруют 0,01Н КЮ3 до появле¬
ния неисчезающей синей окраски. Параллельно ведут холостое определение.
3.24.7.5. Обработка результатов
Активность полифенолоксидаз вычисляют по формуле:
, (а -б)-100
А — ,
н
где А - активность полифенолоксидаз в мл 0,01 н. раствора КЮ3,пошедшего
на титрование, на 100 г почвы;
а - объем КЮ3 0,01 н., пошедшего на титрование холостой пробы, мл;
б - объем KJ03 0,01 н., пошедшего на титрование исследуемой пробы, мл;
н - аликвотная навеска, г.
Допустимая ошибка определения - ± 8 %.
3.24.7.6. Технологическая карта
Примечание: посуда сугубо персональная.
- 191 -
3.24.8. Активность нитратредуктазы
Нитратредуктаза катализирует первую стадию протекающего в почве процесса
денитрификации - восстановления нитратов до нитритов.
3.24.8.1. Принцип метода
Определение активности фермента основано на компостировании почвы с ра¬
створом азотнокислого калия в присутствии глюкозы в качестве донатора водорода и
углекислого кальция для поддержания нейтральной реакции среды. Количество
нитритов, накопившихся в результате ферментативного восстановления нитратов,
учитывают анафтиламинным методом согласно реакции, описанной в п. 3.8.1.
3.24.8.2. Технологическое оборудование
Весы типа ВЛКТ.
Анаэростат с вакуумным насосом.
Термостат.
Фотоэлектроколориметр.
Конические колбы на 150мл.
Дозаторы на 1, 20, 25 и 50мл.
Шприц-дозатор на 5 мл.
Воронки ё=7см.
3.24.8.3. Реактивы
1.1% раствор KNOy 1г соли растворяют в дистиллированной воде и доводят
объем до 100мл.
2. 1% рйствор глюкозы. 1г глюкозы растворяют в дистиллированной воде и до¬
водят объем до 100мл.
3. Окрашивающий раствор (см.п.3.8.2.2.).
4. Исходный образиовый раствор. 0,6071 г KN02 растворяют в дистиллирован¬
ной воде, переносят в мерную колбу на 1л и доводят водой до метки. 1 мл раствора
содержит 0,1мг N - N02.
5. Образиовые растворы шкалы. В мерные колбы на 250мл вносят приведенные
в таблице 56 количества исходного образцового раствора KN02, доводят водой до
метки и добавляют 5-6 капель толуола. Растворы хранят до 2 месяцев.
Таблица S6.
. Номера колб
Показатели
1
2
3
4
5
6
7
8
Объем исходного образцового растворгцмл
0
2,5
5,0
7,5
10,0
15,0
20,0
30,0
Содержание N - N0,
мг/л раствора
0
1,0
2,0
3,0
4,0
6,0
8,0
12,0
мг/кг почвы
0
2,5
5,0
7,5
10,0
15,0
20,0
30,0
3.24.8.4. Ход анализа
5г почвы помещают в колбы, добавляют 20мг углекислого кальция и 1мл 1% ра¬
створа KN03, перемешивают, прибавляют 1мл 1% раствора глюкозы и снова переме¬
шивают. Колбы ставят в анаэростат, из которого воздух эвакуируют до разрежения 10-
12мм рт. ст., и компостируют в термостате при 30° С в течение 24 часов. Затем в колбы
- 192-
добавляют 50мл 1н. раствора КС1 и фильтруют. К 5мл фильтрата прибавляют 20мл
дистиллированной воды и 25мл рабочего окрашивающего раствора, перемешивают и
оставляют на 15 мин. до полного развития окраски. Колориметрируют в кювете с тол¬
щиной просвечиваемого слоя 1см при 520нм (зеленый светофильтр).
Аналогично проводят отбор, окрашивание и колориметрирование образцовых
растворов шкалы.
3.24.8.5. Обработка результатов
По результатам колориметрирования растворов образцовой шкалы строят калиб¬
ровочный график, на оси абсцисс которого откладывают количество мг N02 в мл
раствора, а на оси ординат - его оптическую плотность.
По показаниям оптической плотности испытуемых растворов определяют по гра¬
фику концентрацию в них N02.
Расчеты проводят по формуле^
d • 100
~ « 1000 ?
где А - активность фермента, учитываемая по количеству в мг N02 на
100 г почвы;
а - показания по графику, мг/л N02;
н - аликвотная навеска, г.
допустимая ошибка определения - ±5 %.
3.24.8.6. Технологическая карта
Взятие навесок
весы типа ВЛКТ, шпатель,
колбы на 150мл с пробками
Создание вакуума
анаэростат или эксикатор
с краном, вакуумный насос
Фильтрование
воронки d =7см,
фильтры d =11 см
Прибавление окрашивающего
ряртипря
дозаторы на 20 и 25 мл
Добавление реактивов
2 дозатора на 1 мл, шпатель
Добавление р-оа КС1
дозатор на 50 мл
Отбор аликвот
пипетка с грушей или шприц-
дозатор на 5 мл
Колориметрирование
ФЭК (зеленый светофильтр)
- 193-
4. ПРИБОРЫ И ПРИНЦИП ИХ РАБОТЫ
4.1. ПРИБОР СЕРЕНЬЕВА ДЛЯ ОПРЕДЕЛЕНИЯ АЗОТА
4.1.1. Принцип действия
Прибор предназначен для быстрого макро-и микроопределения аммонийного азо¬
та в озоленных серной кислотой растительных и почвенных образцах, а также в раз¬
личных почвенных вытяжках и грунтовых водах.
Конструкция прибора обеспечивает принудительную отгонку аммиака под ваку¬
умом и титрование его серной кислотой в присутствии борной при совмещении опе¬
раций отгона и титрования.
4.1.2. Устройство прибора Сереньева
Прибор состоит из следующих основных блоков: 1) сосуд реакционный; 2) бар-
ботер; 3) сосуд со щелочью; 4) сосуд с раствором титранта; 5) вакуум-насос или
водоструйный насос; 6) трансформатор с плавным регулированием напряжения.
Реакционный сосуд представляет собой удлиненный цилиндр, в который вмон¬
тированы: а) воронка с барбатажной трубкой, зауженной книзу и доходящей до дна
сосуда; б) спускной кран; в) газоотводная трубка; г) термостатирующая рубашка; д)
выходные отверстия из термостатирующей рубашки с титановыми электродами; е)
выходное отверстие из термостати¬
рующей рубашки для заливки
раствора электролита и выхода пара.
Барботер состоит из следующих
деталей: а) удлиненного суженного на
конус сосуда; б) спускного крана; в)
трехходового крана соединенного с
вакуумным насосом; г) барбатажной
трубки для приема аммиака; д) гор¬
ловины с пробкой для бюретки; е) ру¬
башка водяного охлаждения.
Вакуумный насос. Рабочее давле¬
ние около 0,08-0,1 кг/см2 может быть
обеспечено водоструйным насосом,
вакуум-насосом, отсасывателем ме¬
дицинским марки ЭОХП и другими
вакуумным оборудованием.
Трансформатор. Для создания
рабочего напряжения 100-110V мо¬
гут быть использованы трансформа¬
торы типа ЛАТР, светорегуляторы на
300W, реостаты. При одновременной
работе двух приборов Сереньева воз¬
можно их последовательное подклю¬
чение к одному трансформатору. Ра¬
бочее напряжение в этом случае 220-
Общий вид прибора Сереньева. 250V.
- 194-
4.1.3. Подготовка к работе
1. Заполняют рубашку реакционного сосуда 1% водным раствором лимонной кис¬
лоты (электролит) примерно на 3/4 ее объема.
2. Перекрывают спускные краны.
3. Подключают электроды через ЛАТР в сеть и устанавливают напряжение 200-
220V для закипания раствора электролита. Затем необходимо снизить напряжение до
I 10V,t.k. в течение всего периода работы кипение должно быть очень слабым.
4. Подключают водяное охлаждение к рубашке барботера.
5. Включают вакуум-насос, и трехходовой кран переводят в положение «вакуум»,
чем создают направленный поток воздуха от реактора к барботеру.
6. Набирают в бюретку раствор титранта.
4.1.4. Порядок работы
1. Заливают из бюретки в барботер 1,5-2 мл титранта.
2. Анализируемый раствор либо его аликвотную часть (10-20 мл в зависимости
от содержания в нем азота) количественно переводят через воронку в реактор.
3. Вводят в воронку через реактор раствор щелочи до перехода окраски реакционной
смеси в голубой или бурый цвет (сильно щелочная среда), смывают воронку водой.
Выделившийся аммиак направленным потоком воздуха принудительно пере -
гоняется в барботер, где по мере поступления связывается с титрантом.
4. Титрование ведут до стойкого в течение 1 мин. перехода окраски с зеленой до
фиолетовой от 1-2 капель титранта. Продолжительность отгонки - 5 мин.
5. По окончании титрования переводят трехходовой кран барботера в положе¬
ние «атмосферное давление» и выводят реакционные смеси из барботера и реактора
через спускные краны. После выполнения указанных в п.5 операций прибор готов
для последующих определений азота.
6. По окончании работы на приборе проводят промывку реактора под вакуумом
водой. Отключение выполняют в следующем порядке: а) сливают промывные воды;
б) выключают вакуумный насос; в) отключают от сети JIATP; г) перекрывают воду,
идущую к холодильнику.
7. Во избежание порчи спускного крана реактора от взаимодействия остатков щелочи
со стеклом необходимо после выключения прибора кран вынуть и хранить отдельно.
4.1.5. Меры безопасности
1. Прибор в процессе эксплуатации должен быть надежно заземлен.
2. Запрещается оставлять без присмотра работающий прибор.
3. Электроды должны быть прочно закреплены в вертикальном положении.
4. Уровень электролита необходимо поддерживать постоянным. Доливать до от¬
меченного уровня следует дистиллированной водой при выключенном из сети при¬
бора. Отверстие для выхода пара из нагревательной рубашки реактора должно быть
постоянно открыто.
Электролит следует полностью менять один раз в месяц.
4-2. ПРИБОРЫ ДЛЯ ОПРЕДЕЛЕНИЯ СОДЕРЖАНИЯ РАСТВОРИМОЙ
ЗОЛЫ МЕТОДОМ КОНДУКТОМЕТРИИ
4.2.1. Принцип действия
кондуктометров состоит в определении электропроводности растворов или сус¬
пензий и расчете по этому показателю содержания сырой (растворимой) золы.
Для определения содержания растворимой золы применяют кондуктометры КМ-
1А, ОК-Ю2 (Венгрия), Шандера-Пишдола (бывшая Чехословакия), КМ-2 (ГДР),
портативный - №571 (Польша).
- 195-
4.2.2. Кондуктометр типа ОК-Ю2
4.2.2.1. Настройка прибора
Перед работой прибор прогревают в течение 15 мин., включив в сеть с напряже¬
нием 220V, проводят его настройку, поверку константы измерительной ячейки и пос¬
ле этого приступают к измерению электропроводности испытуемых растворов.
При включении в сеть на панели прибора загорается красная лампочка. Пере¬
ключатель «MEASURE» - «CAL» устанавливаю в положение «CAL», а переключа¬
тель пределов измерения в положение 500 миллисименс. Потенциометром
«CALYBRATE» стрелку прибора устанавливают на деление, отмеченное красным тре¬
угольником. Затем переводят переключатель в положение 150 микросименс. Стрелка
прибора при этом должна оставаться в прежнем положении (на красном треугольни¬
ке). При отклонении стрелку возвращают на красный треугольник при помощи по¬
тенциометра «CAL», расположенного на задней панели прибора. Такую регулировку
проводят несколько раз для большей точности работы прибора.
4.2.2.2. Проверка константы измерительной ячейки
Константа измерительной ячейки вводится в расчет содержания кондуктометри-
ческой золы испытуемого раствора и должна иметь показания в пределах 0,6-0,8.
Проверку ее следует проводить периодически 1 раз в 8-10 месяцев.
Для установки постоянной измерительной ячейки следует провести калибровку
прибора по 1,0; 0,1; 0,01 н. растворам хлористого калия.
Переключатель «MEASURE» - «CAL» ставят в положение «MEASURE». Штекер
колоколообразного электрода вставляют в гнездо с надписью «MEASURE». Для про¬
верки константы измерительной ячейки замеряют электропроводность стандартных
растворов хлористого калия. При измерениях раствор должен покрывать все три пла¬
тиновых кольца колоколообразного электрода. Переключатель пределов измерения
ставят на такую ступень, при которой стрелка прибора будет находиться в средней
части шкалы измерения. Если переключатель диапазонов находится в положении 5,
50, 500, то показание электропроводности снимают по верхней шкале, в положении
1,5; 15 или 150 - по нижней шкале.
Показания электропроводности в мили- или микросименсах переводят в симен¬
сы (1 милисименд равен 10 3 сименса, а 1 микросименс 10 ^сименса).
Постоянную прибора находят по формуле С =
ще С - постоянная измерительной ячейки;
К - удельная электропроводность стандартного раствора хлористого
калия (см.таблицу 57);
* - электропроводность такого же раствора хлористого калия, снятая по
1 / к шкале данного кондуктометра в сименсах.
Таблица 57. Удельная электропроводность раствора КС1
Температура, 0 С
1,0 и.
ОД н.
0,01 н.
15
0,09252
0,01048
0,001147
16
0,09441
0,01072
0,001173
17
0,09631
0,01095
0,001199
- 196-
Температура, 0 С
1,0 D.
ОД D.
0,01 н.
18
0,09822
0,01119
0,001225
19
0,10014
0,01143
0,001251
20
0,10207
0,01167
0,001278
21
0,10400
0,01191
0,001305
22
0,10594
0,01215
0,001332
23
0,10789
0,011239
0,001359
24
0,10984
0,01264
0,001386
25
0,11180
0,01288
0,001413
Пример расчета. Электропроводность 0,1 н. раствора КС1 равна 16,0 милиси-
менс, или 16,0 х 10 ° = 0,016 сименс при температуре 23° С. Удельную электропро¬
водность находим по таблице 47, в данном случае К = 0,01239.
^ 0,01239
01аодд с = ~тьо= '
4.2.2.3. Проведение измерений см. п. 2.8.4.
4.2.3. Переносной кондуктометр типа № 571
Прибор может использоваться как в лабораториях, так и в полевых условиях,
работает на 4-х батареях «крона».
Допустимая ошибка измерений - не более 2 %.
В зависимости от геометрии электрода ячейка имеет различную «постоянную» (С).
4.2.3.1. Подготовка прибора к работе
1. Устанавливают в кондуктометре 4 батареи «крона».
2. Подключают измерительную ячейку к гнезду ввода.
3. Устанавливают корректором «ноль прибора.
4. Проверяют работу батарей. С этой целью ставят переключатель вида работ в
позицию «ВАТ». Стрелка при этом должна сместиться в черную зону шкалы.
5. Ручку компенсации температуры выводят в положение 25° С.
6. Переключатель вида работ ставят в позицию «Reg».
7. Ручкой «Reg К» переводят стрелку шкалы на величину, равную постоянной
«К» ячейки данного кондуктометра (по паспорту прибора).
На диапазонах 10, 30^S cm*1. При измерении в диапазоне lmS cm1 постоянную «К»
устанавливают при выключенной ячейке или при других позициях переключателя.
4.2.3.2. Проведение измерений
1. Переключатель компенсации температуры устанавливают на температуру, рав¬
ную температуре исследуемых растворов.
2. Погружают электроды в замеряемые растворы так, чтобы расстояние от них до
дна сосуда, в котором измеряется электропроводность, было не менее 2 см.
3. Замеряют электропроводность воды при положении переключателя диапазо¬
нов измерений - 100.
4. Замеряют электропроводность испытуемых растворов в соответствующем ди¬
апазоне измерений.
- 197-
5. Кондуктометр должен включаться только во время измерения - это увеличива¬
ет срок использования батарей.
6. По окончании измерений переключатель вида работ следует устновить в пози¬
ции «WYL».
7. При ежедневной работе на кондуктометре электрод кондуктометра следует хра¬
нить погруженным в чистую воду.
4.2.4. Кондуктометр-золомер KJ13-1
Кондуктометр по исполнению соответствует категории УХЛ и категории разме¬
щения 4.1; 4.2 ГОСТ 15150-69 и предназначен для измерения массовой доли золы в
сахаре, удельной электрической проводимости и температуры жидких сред в сахар¬
ной и других областях промышленности.
4.2.4.1. Контролируемая среда
- 28%-ный водный раствор сахара -песка или сахара- рафинада;
- 5%-ный водный раствор сахара -сырца;
- нетоксичные и неагрессивные жидкие среды.
4.2.4.2. Диапазон измерения
массовой доли золы в процентах к сухим веществам от 0.0018% до 0.9%.
4.2.4.3. Диапазон измерения
удельной электрической проводимости от 1мкСМ/см до 15000мкСм/см.
4.2.4.4. Диапазон измерения
температуры контролируемой среды от 5° до 50° С.
4.2.4.5. Пределы допустимого
значения основной абсолютной погрешности при измерении
- массовой доли золы в сахаре ± (0,0005 + 0,03х)%, где х - измеренное значение
содержания золы;
- удельной электрической проводимости ±( 1+0,02х)мкСм/см, где х - измеренное
значение удельной электрической проводимости;
- температуры ± 0,2° С.
4.2.4.6. Кондуктометр обеспечивает
- регистрацию результатов измерения на индикаторе;
-автоматическую температурную компенсацию показаний, приведенную к 20° С при
изменении температуры контролируемой среды в диапазоне температур от 15° до 25° С,
- автоматизированную калибровку;
- диагностику технического состояния.
4.2.4.7. Цена единицы
наименьшего разряда цифрового индикатора :
- при измерении массовой доли золы - 0,01%;
- при измерении удельной электрической проводимости - 0,01мкСм/смч0,001х;
- при измерении температуры - 0,01°С.
4.2.4.8. Длительность одного измерения
- не более 2 мин.
- 198-
4JL4J). Устройство, подготовка к работе и принцип работы
Кондуктометр представляет собой измерительное устройство, состоящее из пер-
иичного преобразователя (ПП) и измерительного преобразователя (ИП), обеспечива¬
ющее автоматическое измерение массовой доли золы в сахаре, измерение удельной
электрической проводимости и температуры жидких сред с выдачей результатов из¬
мерения на цифровой индикатор.
Функциональная схема кондуктометра состоит из 15 узлов, каждый из которых
ныполняет определенную заданную функцию.
4.2.4.10. Подготовка к работе
заключается в проверке общего состояния прибора, в подключении к электросе¬
ти и подключении заземления. Затем подключается первичный преобразователь (ПП)
к разъему (на задней панели прибора) и нажать кнопку «СЕТЬ», а на цифровом инди¬
каторе высвечивается значение константы кондуктрометрической ячейки, соответ¬
ствующее значению указанному в приложении, т.е.0,722 с технологическим номером
№222.
Прибор готов к работе.
4.2.4.11. Порядок работы при введении поправки на дистиллят
Убедившись, что первичный преобразователь (ПП) находится вне контролиру¬
емой среды (в воздухе) и сухой, нажать на кнопку калибровки (в зависимости от
режима - один, два раза) до появления надписи «Погрешность ИЗМ. ЦЕПИ» и
величина погрешности не должна превышать ±0,3%. После введения поправки на
дистиллят, первичный преобразователь помещается в сосуд с дистиллированной
водой, а кнопкой «УЭП» выбирается измерение .УЭП при текущей температуре
«УЭП»=х.хх Т=ххх, после чего на цифровом индикаторе высвечивается измерен¬
ное значение удельной Электрической проводимости дистиллированной воды, ис¬
пользуемой для приготовления сахарных растворов и это значение не должно пре¬
вышать 10мкСм/см.
После нажатия на кнопку «ЗАПИСЬ» прибор сам переключается в режим изме¬
рения зольности, а значение электропроводности дистиллята, используемое для вы¬
числения поправки, должно высветиться в правом верхнем углу индикатора в режи¬
ме измерения зольности.
4J.4.12. Порядок работы в рабочем режиме
Поместив первичный преобразователь в заранее приготовленный для контроля
раствор производится выбор рабочего режима нажатием соответствующей кнопки
1. «ЗОЛА 28%» - измерение процентного содержания золы в 28%-ном раство¬
ре сахара-псска или сахара-рафинада
«ЗОЛА 5%» -измерение процентного содержания золы в 5%-ном растворе сахара-
сырца
2. «УЭП» «Т» - измерение удельной электропроводимости растворов и температуры.
3. «УЭП» при 20°С Kt = 0,0™ или
«УЭП» при 25°С Kt = 0,0ХХ\
Это данные удельной электропроводности приведенной к 20^С или к 25°С.
Измерение температуры приведения и температурного коэффициента производится
кнопками «—>», «I». Трижды нажав кнопку «—>», выберите ввода константы, и далее
кнопками «—>», «I» изменяются константа и температура приведения.
Если маркер не вывести из поля индикатора, то Kt не запоминается, а сохраняет-
- 199-
с я до отключения прибора от сети. При следующем включении установятся предыду¬
щие установки.
Значение измеряемых величин высвечивается на индикаторе.
ПРИМЕЧАНИЯ:
1. Перед каждым измерением первичный преобразователь промывается 2-3 раза
контролируемым раствором.
2. При погружении кондуктрометрической ячейки контролируемый раствор
должен полностью покрыть кольцевые электроды, отверстия в верхней части стек¬
лянного корпуса ячейки должны находиться вне контролируемого раствора.
3. После проведения каждого измерения тщательно промывается 2-3 раза пер¬
вичный преобразователь дистиллированной водой.
4. Сахарные растворы необходимо готовить в соответствии с ДСТУ 2317 или
ГОСТ 12574-67.
5. Если измеряемая величина превышает верхнее значение диапазона измере¬
ния прибора, на цифровом индикаторе высвечивается сообщение об этом.
4.3. ПРИБОРЫ ДЛЯ ОПРЕДЕЛЕНИЯ pH
4.3.1. Принцип действия
приборов основан на измерении электродвижущей силы (ЭДС), которая возни¬
кает в цепи с платиновым электродом, насыщенным водородом, и зависит от кон¬
центрации водородных ионов в испытываемой среде.
В агрохимических лабораториях массовых анализов рН-метры используются для
определения pH почвенных вытяжек, гидролитической кислотности поточным ме¬
тодом, а также при приготовлении растворов.
Наиболее пригодным для массовых анализов является 10-позиционный рН-метр
типа ПЛП (СССР), для одиночных определений pH растворов используются рН-мет-
ры типа рН-метр-милливольтметр pH-121 (СССР), ОР-204/1 (Венгрия) и другие, ко¬
торые обеспечивают точность определений не менее ±0,05 pH.
4.3.2. рН-метр рН-121
Для определения pH используют основной электрод ЭСЛ-43-07 и вспомогатель¬
ный электрод сравнения ЭВП-1МЗ.
432Л. Подготовка прибора к работе
1. Нажать кнопку «0, t».
2. Включить прибор в сеть 220 V.
3. Прогреть прибор в течение 30 мин.
432.2. Порядок работы
1. Переключатель термокомпенсации устанавливают в положение «руч» или
«авто» в зависимости от выбранного режима измерений. При ручном регулировании
нажимаю!' кнопку «-1-14», ручкой ТЕМПЕРАТУРА РАСТВОРА выставляют по верх¬
ней шкале прибора температуру контролируемого раствора.
2. Погружают электроды в контролируемый раствор на 4-6 мм от дна. Время ус¬
тановления показаний - 3 мин.
3. Нажимают кнопки «ДИАПАЗОНА ИЗМЕРЕНИЯ,» «pH», а при включении вто¬
рой ячейки и кнопку «ИЗМ».
4. По соответствующим буферным растворам сначала ручкой КАЛИБРОВКА ус¬
-200-
танавливают стрелку прибора на pH 1,68, а затем ручкой КРУТИЗНА - на pH 9,22,
повторяя это несколько раз.
5. При замене контролируемого раствора и по окончании измерения должна быть
нажата кнопка «О, t».
6. По окончании работы с прибором электроды для измерения pH должны оста¬
ваться погруженными в воду или 0,1 М раствор НС1.
4.3.3. рН-метр ПЛП-64
Прибор отличается большой производительностью, т.к. проводится одновремен¬
ное определение pH в 10 анализируемых образцах.
Перед работой рН-метр проверяют и настраивают по буферным растворам. Для
этого все 10 пар электродов погружают в буферный раствор с pH 4,01. Через 3 мин.-
носле погружения подключают каждую пару электродов к измерительному блоку и
записывают показания. Если они отличаются от теоретического значения буферного
раствора более чем на 0,05 pH, необходимо произвести настройку с помощью уста¬
новленных потенциометров, расположенных на измерительном блоке. Для доступа
к потенциометрам нужно снять планку с номерами ключей, отвинтив два винта. Ре¬
гулировка проводится вращением отверткой осей потенциометров, находящихся
против соответствующих ключей, до максимального совпадения показаний прибора
со значением pH буферного раствора.
Затем электрод вынимают из буферного раствора, обмывают дистиллированной
водой и помещают в буферный раствор с pH 9,18. Через 3 мин. записывают показа¬
ния каждой электродной пары рН-метров.
При отклонении показаний всех электродных пар более чем на 0,1 pH одновре¬
менно в большую или меньшую сторону прибор настраивают корректором РАЗМАХ
ШКАЛЫ, расположенным на левой боковой панели измерительного блока. Для этого
берут одну электродную пару, погружают в буферный раствор с pH 9,18 и корректо¬
ром РАЗМАХ ШКАЛЫ устанавливают стрелку прибора на значении буферного ра¬
створа. Затем электродную пару вынимают из буферного раствора, обмывают дис¬
тиллированной водой и помещают в буферный раствор с pH 4,01. Корректором
НАЧАЛО ШКАЛЫ, расположенным также на боковой левой панели, стрелку прибора
устанавливают на значение буферного раствора. После этого повторно проверяют
показания прибора по буферному раствору с pH 9,18.
После того как будут найдены положения переменных сопротивлений НАЧАЛО
ШКАЛЫ И РАЗМАХ ШКАЛЫ, при которых ошибки измерений в буферных раство¬
рах 4,01 и 9,18 pH не превышает 0,1 pH, нужно снова проверить показания всех элек¬
тродных пар по буферному раствору с pH 4,01 и, если нужно, довести стрелку изме¬
рительного прибора до значения буферного раствора установочными потенциомет¬
рами для каждой электродной пары.
Для измерения pH кассету с суспензиями почв устанавливают на подъемный сто¬
лик рН-метра, и, поднимая столик, погружают электроды в суспензии. После погру¬
жения выдерживают в течение 1,5 мин., и, подключая последовательно каждую пару
электродов к измерительному блоку прибора соответствующим ключом, записывают
значение pH. Электроды при переносе из одной кассеты с суспензиями в другую ди¬
стиллированной водой не обмывают.
4.3.4. Буферные растворы для рН-метрии
Буферные растворы готовятся из реактивов квалификации «стандарт-титры» для
рН-метрии согласно ТУ, рассчитанных на 1000 мл буферного раствора.
-201 -
pH 1,68 (при t = 15-20°)
pH 3,56 (при t= 15-25°)
pH 6,90 (при t= 15°)
pH 6,88 (при t = 20°)
pH 6,86 (при t = 25°)
pH 9,27 (npnt= 15°)
pH 9,22 (при t = 20°)
pH 9,18 (при t = 25°)
Для приготовления буферных растворов применяется дистиллированная вода,
предварительно прокипяченная в течение 30-40 мин. для удаления углекислоты.
4.3.5. Меры безопасности
1. рН-метры в процессе эксплуатации должны быть надежно заземлены. Не разре¬
шается работать с прибором, у которого не светится глазок индикации включения.
2. Во время профилактических работ и ремонта прибор должен быть отключен от сети.
4.4. ИОНОМЕР И-123
Прибор предназначен для измерения pH и рХ растворов, температуры раство¬
ров, ОВП потенциалов, и концентрации ионов.
Прибор эксплуатируется в следующих условиях:
- температура окружающей среды 10-30° С
- относительная влажность воздуха до 80%
- напряжение переменного тока от 198 до 242 В частота 50 Гц
Характеристика прибора.
Гранично допустимые абсолютные ошибки при определении pH в условиях гра¬
дуировки прибора
- буферными растворами 1-го разряда - 0,01
- буферными растворами 2-го разряда - 0,02.
Гранично допустимые абсолютные ошибки при определении рХ - 0,02.
Гранично допустимые абсолютные ошибки при определении ОВП - 0,5 мВ.
Гранично допустимые абсолютные ошибки при определении температуры - 0,5°С.
Время измерения не более 5 мин.
Время непрерывной работы не более 8 часов.
4.5. ФОТОМЕТР ФОТОЭЛЕКТРИЧЕСКИЙ КФК-3-01-«ЗОМЗ»
4.5.1 Назначение
Фотометр предназначен для измерения спектрального коэффициента направлен¬
ного пропускания (СКНП), оптической плотности и скорости изменения оптической
плотности прозрачных жидкостных растворов, а также для определения концентра¬
ции веществ в растворах после предварительной градуировки фотометра.
По условиям эксплуатации в части воздействия климатических факторов внеш¬
ней среды фотометр относится к исполнению УХЛ категории 4.2 по ГОСТ 15150-69
и условиями работы его являются:
- температура окружающей среды, +10 -35° С
- относительная влажность воздуха, 65 ± 15%
- напряжение питающей сети, 220 ± 22В
- частота питающей сети, 50 ± 0,5 Гц.
0,05М тетраоксалат калия
Насыщенный раствор тартрата калия -
0,025М однозамещенный фосфат калия -
0,025М двузамещенный фосфат натрия -
0,01М тетраборат натрия
-202-
.5.2.Технические данные
- Диапазон длин волн, 315 - 990 нм,
- Выделяемый спектральный интервал не более 5 нм.
Диапазон измерений:
для СКНП 1 - 99
оптической плотности, Б 0,004 - 2
Диапазон показаний:
- коэффициента пропускания, % 0,1-100
-оптической плотности, Б 0-3
- концентрации, единиц концентрации 0,001 - 9999
Предел допускаемого значения основной абсолютной погрешности при измере¬
нии СКНП, % 0,5
Отклонения от линейности при измерении оптической плотности должно быть
не более:
- в диапазоне от 0,004 до 0,200 Б, Б(абс.) 0,004
- в диапазоне от 0,201 до 2,000 Б, %(отн.) 6
Предел допускаемой основной абсолютной погрешности установки длины вол¬
ны, нм 3
Предел допускаемого значения среднего квадратического отклонения случайной
составляющей основной абсолютной погрешности должен быть не более:
- при измерении СКНП, % 0,15;
-при измерении оптической плотности, Б 0,003.
Время установления рабочего режима, мин., не более 30
Рабочая длина кювет, мм 1,3,5, Ю, 20,30,50,100
Микропроцессорная система обеспечивает выполнение следующих задач:
-учет времени выхода на рабочий режим с подачей звукового сигнала с отобра¬
жением текущего времени на индикаторе;
-автоматическое измерение и учет сигнала при неосвещенном фотоприемнике;
- градуировка;
- измерение оптической плотности;
- измерение СКНП;
- измерение концентрации по фактору;
- измерение концентрации концентрации при градуировке по одному или шести
стандартным растворам;
- измерение скорости изменения оптической плотности с возможностью про¬
смотра хода реакции и выбора линейного участка;
-ввод и хранение в памяти коэффициента факторизации. После выключения со¬
храняется в памяти МПС;
- ввод и хранение в памяти концентрации стандартных растворов. После вык¬
лючения сохраняется в памяти МПС;
- измерение и хранение в памяти значений оптических плотностей стандартных
растворов. После выключения сохраняется в памяти МПС;
- диалог с оператором и отображает ошибки оператора и дает их анализ;
- выход на внешнюю ЭВМ или принтер.
4.5.3. Устройство и принцип работы
Внешний вид фотометра представлен на рис.А, В (А - вид спереди; В - вид
сзади), где обозначено:
-203-
2 5 14 3
Рис.А Вид фотаметра спереди
4
3 2 1 8 6
Р и с. В В ид ([хягыъмезпра сзади
Рис.А. 1 -металлическое основание на котором закреплены узлы прибора; 2 - ко¬
жух; 3 - съемная крышка юоветного отделения; 4 - рычаг для ввода в световой пучок
кювет с исследуемым раствором; 5 - ручка для поворота дифракционной решетки и
установки требуемой длины волн.
Рис. В. 1- розетка подключения блока проточной кюветы и зажим защитного
заземления; 2 - держатель плавной вставки 1 А; 3 - шнур с вилкой для подключения к
электрическому питанию; 4 - выключатель сетевого напряжения; 5 - крышка кювет-
ного отделения; 6 - съемная крышка для доступа к лампе; 7 - микропроцессорный
блок; 8 - разъем для подключения ЭВМ.
Принцип действия фотометра основан на сравнении потока излучения Фо, про¬
шедшего через «холостую пробу» (растворитель или контрольный раствор, по отно¬
шению к которому производится измерение) и потока излучения Ф, прошедшего че¬
рез исследуемый раствор.
Потоки излучения Фо и Ф фотоприемником преобразуются в электрические сиг¬
налы Uo, U и Ut (Ut - сигнал при неосвещенном фотоприемнике), которые обраба¬
тываются встроенной микро-ЭВМ и представляются на индикаторе в виде коэффи¬
циента пропускания, оптической плотности, скорости изменения оптической плот¬
ности, концентрации.
При измерений коэффициента пропускания ф рассчитывается формула
где U, Uo, Ut - электрические сигналы, пропорциональные потоку излучения,
прошедшему через исследуемый раствор, «холостую пробу»,
темновому току соответственно.
При измерении оптической плотности D рассчитывается формула
При измерении скорости изменения оптической плотности А рассчитывается формула
где (D2- D,) - разность значений оптической плотности за интервал времени t;
т= — х100% =
0)
(2)
-204-
t - интервал времени в минутах, задаваемый с клавиатуры; t может принимать
значения 1, 2, 3, ...9 мин.
При измерении концентрации по фактору Сф рассчитывается формула
Uo-UT
U-Uт ’
где F - значение коэффициента факторизации; определяется потребителем и вво¬
дится в память с клавиатуры; может принимать значения от 0,001 до 9999.
При измерении концентрации по стандарту С рассчитывается формула
Сф = F xD = F х lg-
(4)
C = ^SB-y.D = ^-S!L\g———.
Д.. Д.. и-UT
(5)
гае С
значение концентрации стандартного раствора; вводится в
память с помощью клавиатуры;
Dct- значение оптической плотности стандартного раствора, измеряется
на фотометре и водится в память микро-ЭВМ;
D - значение оптической плотности измеряемого раствора.
При измерении концентрации по шести стандартным растворам Сх рассчитывает¬
ся формула
С -С
^"ст(л+1) ст(п)
ст(и+1) ^ст(я)
(6)
где п=1,2,—,6;
С и Г
СТ<п)"
D , D
X (
стСп))
^ст<п ♦ I) >0.0^ ,
значения концентрации n-ого и (п+1)-ого стандартного
раствора соответственно;
, значения оптической плотности исследуемого, п-ого
и (п+1)-ого стандартного растворов соответственно.
Здесь нить лампы (1) изображается конденсором (2) в плоскости
входной щели Д1, заполняя ее светом. Далее входная щель изобража¬
ется вогнутой дифракционной решеткой (4) и вогнутым зеркалом (5) в
плоскости выходной щели Д2. вращая дифракционную
решетку вокруг оси, параллельной ее штрихам, выде¬
ляют выходной щелью излучение в узких спектраль¬
ных интервалах в диапазоне от 325 до 990 нм. Объек¬
тив (7,8) изображает с увеличением выходную щель
перед линзой (10). Линза (10) создает в плоскости фо¬
топриемника (11) световое пятно.
Для уменьшения рассеянного излучения
при работе в диапазоне длин волн 315-400 нм после
входной щели автоматически вводится (а затем авто¬
матически выводит-
10
\
h-
ся) светофильтр (3).
В юоветное от¬
деление (между
объективом 7,8 и
линзой 10) устанав¬
ливаются прямоу¬
гольные кюветы (9).
-205-
Рис.З Схема электрическая структурная
Электрическая структурная схема (рис.З) состоит из преобразователя VD опти¬
ческого излучения в электрический сигнал, микро-ЭВМ А2, преобразователя угла по¬
ворота дифракционной решетки в напряжение АЗ вместе с датчиком угла поворота,
стабилизатора напряжения осветителя А4, где
EL - осветитель
VD - преобразователь оптического излучения
А2 - микро-ЭВМ
АЗ - датчик угла поворота дифракционной решетки
А4 - стабилизатор напряжения осветителя.
1.0. Работа на приборе
Работа на фотометре должна производиться в чистом помещении, свободном от
пыли, паров кислот и щелочей. Вблизи фотометра не должны располагаться громоз¬
дкие изделия, создающие неудобства в работе оператора.
Все регулировочные работы, связанные с проникновением в корпус фотометра к
токоведущим частям, замена неисправных деталей, разъединение и подключение
штепсельных разъемов, должны производиться после отсоединения фотометра от сети.
Включается фотометр в сеть через электрошнур. Розетка у потребителя должна
быть подсоединена к заземляющей шине.
1.0. Подготовка фотометра к работе
Установить фотометр на рабочем месте. Следить за тем, чтобы на фотометр не
попадали прямые солнечные лучи.
Тумблер «СЕТЬ» установить в выключенном положении. Закрыть крышку кю-
ветного отделения.
Подсоединить фотометр к сети 220 В, 50 Гц. Включить тумблер «СЕТЬ».
Подготовка фотометра к работе осуществляется в автоматическом режиме:
- на индикаторе отображается символ завода-изготовителя «ОАО ЗОМЗ», сооб¬
щение «ПРОГРЕВ ПРИБОРА» и показания таймера;
- по истечении 2,5 мин на индикаторе отображается надпись - шифр фотометра
«КФК-3-01»;
- по истечении 5 мин автоматически учитывается «нулевой отсчет», включается
-206-
источник излучения; на индикаторе отображается значение длины волны в нм, над¬
пись «ПРОГРЕВ ЛАМПЫ» и показания таймера;
- по истечении 10 мин фотометр выдает звуковой сигнал готовности к работе и
на индикаторе отображается надпись
«ГОТОВ К РАБОТЕ ВВЕДИТЕ РЕЖИМ».
Для сокращения времени подготовки фотометра к работе при закрытой крышке
кюветного отделения после включения тумблера «СЕТЬ» последовательно два раза с
интервалом 20-30 с нажать клавишу «D». После звукового сигнала и появления над¬
писи «ГОТОВ К РАБОТЕ ВВЕДИТЕ РЕЖИМ» фотометр готов к работе. При этом
погрешность измерений не нормируется.
В модификации фотометра с одним четырехстрочным индикатором реализована
возможность переучета «нулевого» сигнала в процессе работы. Для этого нажатием
клавиши «D» («С») выбрать режим «ф - коэффициент пропускания».
При закрытой крышке кюветного отделения нажать клавишу «#», на индикаторе
должно отобразиться сообщение «Градуировка», затем
«ИЗМЕРЕНИЕ ф = 100±0,2 %».
Открыть крышку кюветного отделения, нажать клавишу «0».
На индикаторе отобразиться сообщение «ИЗМЕРЕНИЕ ф=0,0±0,1%».
Если значение «0,0» отобразилось с большим отклонением, повторно нажать кла¬
вишу «0».
При проведении указанной операции для исключения влияния внешней засвет¬
ки рекомендуется накрыть фотометр (кюветное отделение) черной тканью.
Для отключения звукового сигнала нажать клавишу «D».
1.1. Порядок работы
Для установления рабочего режима и обеспечения стабильной работы фотометр
необходимо выдержать не менее 30 минут с момента включения.
При работе в диапазоне длин волн 315-450 нм перед измерениями фотометр не¬
обходимо выдерживать не менее 5 минут при закрытой крышке кюветного отделе¬
ния.
1.2. Измерение коэффициента пропускания или оптической плотности.
1.3. Ручкой установки длин волн установить необходимую по роду измерений
длину волны.
\Л. Установить в кюветное отделение кюветы с «холостой пробой» и исследуе¬
мым раствором. Кювету с «холостой пробой» установить в дальнее гнездо юовето-
держателя, а кювету с исследуемым раствором - в ближнее гнездо. (О выборе рабочей
длины кюветы см. ниже).
Ручку перемещения кювет установить в крайнее левое положение, при этом в
сетевой пучок вводится кювета с «холостой пробой».
Закрыть крышку кюветного отделения.
1.5.Клавишей выбора режима «D» («С») выбрать режим измерения «ф - КОЭФ¬
ФИЦИЕНТ ПРОПУСКАНИЯ» («А - ОПТИЧЕСКАЯ ПЛОТНОСТЬ»). Нажать клави¬
шу «#». На индикаторе должно отобразиться «ГРАДУИРОВКА», через 3-5 с данная
надпись исчезает и вместо нее отображается «ИЗМЕРЕНИЕ», ф = 100±0,2 %» («А=
0,000±0,002»). Если значение «100» («0,000») отобразилось с большим отклонением,
повторно нажать клавишу «#».
1.6. Ручку перемещения кювет установить вправо до упора. При этом в световой
пучок вводится кювета с исследуемым раствором. На индикаторе отображается зна¬
чение коэффициента пропускания в % (оптической плотности в Б) исследуемого ра¬
створа.
1.7. Операции по п. 1.4 - 1.6 повторить три раза. Значение коэффициента пропус¬
- 207-
кания (оптической плотности) исследуемого раствора определяется как среднее ариф¬
метическое из полученных отсчетов.
1.8. Измерение концентрации вещества в растворе по фактору.
Для измерения концентрации вещества в растворе по фактору необходимо предвари¬
тельно выполнить ряд подготовительных операций в следующей последовательности:
- выбрать длину волны измерений;
- выбрать кювету;
- построить градуировочный график и определить значение коэффициента фак¬
торизации;
- ввести значение F в память МПС;
- измерить концентрацию вещества.
1.9. Выбор длины волны.
По методике п.п. 1.0.- 1.6. измерить оптические плотности исследуемого раство¬
ра в диапазоне длин волн поглощения дацным раствором.
Построить график зависимости оптической плотности данного раствора от дли¬
ны волны излучения, откладывая по горизонтальной оси значения длин волн в нм,
по вертикальной - измеренные значения оптической плотности в Б.
Выбрать такой участок, где выполняются следующие условия:
- оптическая плотность имеет максимальную величину;
- ход кривой примерно параллелен горизонтальной оси, т.е. оптическая плот¬
ность слабо зависит от длины волны.
Длина волны, соответствующая этому участку, выбирается для измерения. Если вто¬
рое условие не выполняется, то рабочая длина волны выбирается по первому условию.
2.0. Выбор кюветы
Погрешность измерения оптической плотности зависит от измеряемой величи¬
ны и достигает минимума при оптической плотности 0,4 Б. поэтому при работе на
фотометре рекомендуется путем соответствующего выбора длины рабочего слоя -
рабочей длины кюветы - работать вблизи указанного значения оптической плотнос¬
ти, т.е. в диапазоне от 0,3 до 0,6 Б.
2.1. Построение градуировочного графика и определение коэффициента факторизации.
Приготовить ряд растворов исследуемого вещества с известными концентраци¬
ями, охватывающими область возможных изменений концентраций.
По методике п.п. 1.0. - 1.6. для выбранной длины волны измерить оптические
плотности всех растворов и построить градуировочный график, откладывая по гори¬
зонтальной оси известные концентрации, а по вертикальной - соответствующие им
измеренные значения оптической плотности.
По градуировочному графику для среднего значения концентрации С определить
значение оптической плотности D.
Рассчитывать коэффициент факторизации F по формуле
2.2. Измерение концентрации по фактору.
Подготовить фотометр к работе согласно п. 1.0. F —
Выполнить операции по п. 1.З., 1.4. D
Нажатием клавиши «D» («С») выбрать режим измерений концентрации по фактору.
При этом на индикаторе должно отобразиться «Сф- КОНЦЕНТРАЦИЯ ПО ФАКТОРУ».
Нажать клавишу «В». На индикаторе отображается надпись «ВВЕДИТЕ: Кф = 0.000»
При этом курсор находится в первом разряде значения Кф.
При помощи нажатий клавиши «В», перемещающей курсор вправо, либо «А»,
перемещающей курсор влево, и нажатия соответствующей цифровой клавиши ввес¬
ти в память МПС значения коэффициента факторизации Кф
Если при наборе была допущена ошибка, установить курсор в нужном разряде и
нажать соответствующую цифровую клавишу.
-208-
Нажать клавишу «D» («С»). Выбрать режим «С+-КОНЦЕНТРАЦИЯ ПО ФАКТОРУ».
Нажать клавишу «#». На нижнем индикаторе отображается надпись
«ГРАДУИРОВКА».
По окончании градуировки на индикаторе отображается
«ИЗМЕРЕНИЕ: С =0.000±0.002»
Если значение «0.000» отобразилось с большим отклонением, повторно нажать
клавишу «#».
Ручку перемещения кювет установить в крайне правое положение, при этом в
световой пучок вводится кювета с исследуемым раствором. На индикаторе отобра¬
жается отсчет, соответствующий концентрации исследуемого раствора.
Операцию повторить три раза. Определить концентрацию исследуемого раствора
как среднее арифметическое из полученных отсчетов.
Далее, заменяя кюветы с исследуемыми растворами, аналогично определяйте их
концентрации.
2.3. Измерение концентрации вещества в растворе по стандарту.
2.3.1. Измерение концентрации вещества в растворе по одному стандартному ра¬
створу.
Подготовить фотометр к работе согласно п. 1.0.
Выполнить операции по п.п. 1.2, 1.3, при этом в дальнее гнездо кю вето держате¬
ля установить кювету с «холостой пробой», а в ближнее гнездо - кювету со стандар¬
тным раствором.
Нажатием клавиши «D» («С») выбрать режим измерения концентрации по одно¬
му стандартному раствору, при этом на индикаторе отображается надпись
«С, -КОНЦЕНТРАЦИЯ ПО 1 СТ. Р-РУ».
Нажать клавишу «В», при этом на индикаторе отображается надпись
«А =0.000 С =0.000»
ст ст
По методике 2.3 вести в память МПС значение концентрации стандартного ра¬
створа. Введенное значение концентрации стандартного раствора отображается на
индикаторе, т.е. «Сст=Х.ХХХ».
Измерить оптическую плотность стандартного раствора. Для этого нажать клави¬
шу «#» - градуировка по «холостой пробе» (ручка перемещения кювет в крайнем левом
положении). На индикаторе отображается надпись «ГРАДУИРОВКА». По окончании
градуировки на нижнем индикаторе отображается:
«А =Х.ХХХ С =Х.ХХХ».
ст ст
Повторно нажать клавишу «D». МПС фотометра выходит на режим измерения
концентрации по одному стандартному раствору с отображением на индикаторе «С ,
-КОНЦЕНТРАЦИЯ ПО 1 СТ. Р-РУ».
Кювету со стандартным раствором заменить на кювету с исследуемым раство¬
ром. Ручку перемещения кювет перевести в крайнее левое положение - в световой
пучок вводится кювета с «холостой пробой». Нажать клавишу «#». На индикаторе
отображается «ГРАДУИРОВКА». По окончании градуировки - надпись
«ИЗМЕРЕНИЕ Сы=0.000±0.002».
Если значение «0.000» отобразилось с большим отклонением, повторно нажать
клавишу «#».
Ручку перемещения кювет перевести в крайнее правое положение - в световой
пучок вводится кювета с исследуемым раствором. На индикаторе отображается зна¬
чение концентрации исследуемого раствора «ИЗМЕРЕНИЕ Сс|=Х.ХХХ».
Операцию повторить три раза. Определить концентрацию исследуемого раствора
как среднее арифметическое из полученных отсчетов.
-209-
Далее, заменяя кюветы с исследуемыми растворами, аналогично определяйте их
концентрации.
3.4. Измерение концентрации по шести стандартным растворам.
Измерение концентрации вещества по шести стандартным растворам проводится,
как правило, при нелинейной зависимости концентрации от оптической плотности.
Подготовить фотометр к работе согласно п. 1.0.
Выполнить операции по п.п. 1.3, 1.4, при этом в дальнее гнездо кюветодержате-
ля установить кювету с «холостой пробой», а в ближнее - с первым стандартным
раствором. Ручку перемещения кювет установить в крайнее левое положение - в све¬
товой пучок вводится кювета с «холостой пробой».
Нажатием клавиши «D» («С») выбрать режим измерения по шести стандартным ра¬
створам с отображением на индикаторе надписи
«С^ - КОНЦЕНТРАЦИЯ ПО 6 СТ.Р-РАМ».
Нажать клавишу «В» - на нижнем индикаторе отображается надпись
«А^.000 Сс11=0.000»
Если в память МПС предварительно были введены значения указанных пара¬
метров, то при нажатии клавиши «В» их значения и отображаются на индикаторе.
По методике п.2.3 ввести в память МПС значение концентрации первого стан¬
дартного раствора. При этом на индикаторе отображается вводимое значение кон¬
центрации «С^Х.ХХХ»
Нажать клавишу «#», на индикаторе отображается надпись «ГРАДУИРОВКА».
По окончании градуировки на индикаторе отображается надпись
«ИЗМЕРЕНИЕ - 1 СТ.Р. Аст = 0.000».
Ручку перемещения кювет установить в крайнее правое положение - в световой
пучок вводится кювета с первым стандартным раствором. На индикаторе отобража¬
ется измеренное значение оптической плотности первого стандартного раствора А
=х.ххх»
Нажать клавишу «D». На индикаторе отображается значение оптической плотно¬
сти и введенное значение концентрации первого стандартного раствора
«А =Х.ХХХ С =Х.ХХХ».
См См
Градуировка по первому стандартному раствору завершена.
Кювету с первым стандартным раствором заменить на кювету со вторым стан¬
дартным раствором. Ручку перемещения кювет установить в крайнее левое положе¬
ние - в световой пучок вводится кювета с «холостой пробой».
Нажать клавишу «D» На нижнем индикаторе отображается
«А =О.ОООС =0.000»
СТ2 ст2
Аналогично провести градуировку по второму, третьему, четвертому, пятому и
шестому стандартным растворам.
Кювету с последним стандартным раствором заменить на кювету с исследуе¬
мым раствором. Ручку перемещения кювет перевести в крайнее левое положение - в
световой пучок вводится кювета с «холостой пробой».
Нажать клавишу «#»,на индикаторе отображается надпись «ГРАДУИРОВКА». По
окончании градуировки на индикаторе отображается
«ИЗМЕРЕНИЕ С =0.000 ±0.002».
Если значение «0.000» отобразилось с большим отклонением, повторно нажать
клавишу «#».
Ручку перемещения кювет перевести в крайнее правое положение - в световой
пучок вводится кювета с исследуемым раствором. На индикаторе отображается зна¬
чение концентрации исследуемого раствора «ИЗМЕРЕНИЕ Ссб = Х.ХХХ».
Операцию повторить три раза. Концентрацию исследуемого раствора опреде¬
лять как среднее арифметическое из полученных отсчетов.
-210-
Далее, заменяя кюветы с исследуемыми растворами, аналогично определяйте их
концентрации.
Примечания.
1. Если концентрация исследуемого раствора удовлетворяет условию
С > С > С
ст.тт исс. ст.тах.
то на индикаторе отобразится «ВНЕ ДИАПАЗОНА», что говорит о неправиль¬
ном выборе стандартных растворов.
2. Веденные значения концентраций стандартных растворов и
соответствующие им измеренные оптические плотности сохраняются в памяти
МПС после выключения фотометра.
Вывод на индикатор введенных ранее значений концентраций Сст1 - Сст6 и соот¬
ветствующих им измеренных оптических плотностей Аст] - Аст6 производится с помо¬
щью нажатия клавиши «В» и последовательного нажатия клавиши «D».
3. Градуировку по шести стандартным растворам можно проводить в любой
последовательности, а не только от меньшей концентрации к большей.
2.4. Определение скорости изменения оптической плотности раствора.
Подготовить фотометр к работе согласно п. 1.0.
Выполнить операции по п.п. 1.3, 1.4.
Выбрать режим измерения активности последовательным нажатием клавиши
«D»(«C»). При этом на индикаторе отображается надпись «КИНЕТИКА».
Нажать клавишу «А» - на индикаторе отображается надпись
«ВВЕДИТЕ: t кинетики = X мин»
Ввести время t мин проведения кинетических измерений, для этого нажать одну из
цифровых клавиш. Время t мин может принимать значения 1, 2, 3,4, 5,6, 7, 8, 9 минут.
Нажать клавишу «D» - на индикаторе отображается надпись «КИНЕТИКА», од¬
новременно время t мин вводится в память.
Нажать клавишу «В» - на индикаторе отображается надпись
«А = Х.ХХХ К = Х.ХХХ»
АКТИВ.
Нажать клавишу «А» - на индикаторе во второй строке в первом разряде появля¬
ется курсор. Ввести значение априорного коэффициента К нажатием цифровых кла¬
виш. При этом положение «запятой» определяется нажатием клавиши «*».
Примечание. Если значение К введено неверно, нажатием клавиш «А» и «В» установи¬
те курсор в нужном разряде и нажатием соответствующей цифровой клавиши введите
требуемое значение.
Нажать клавишу «D» - на индикаторе отображается надпись
«Л»™» = Х.ХХХ К = Х.ХХХ»
При этом курсор в нижней строке исчезает. Ввод «К» завершен.
Нажать клавишу «D» - на нижнем индикаторе отображается надпись «КИНЕТИ¬
КА» - фотометр готов к измерению активности.
Нажать клавишу «#» - на индикаторе отображается надпись «ГРАДУИРОВКА».-
Через 3 - 5с данная надпись исчезает - на индикаторе отображается
«Нажмите - * А = 0.000.± 0.002».
Градуировка завершена.
Установить ручку перемещения кювет в крайнее правое положение - в световой
пучок вводится кювета с исследуемым раствором.
Нажать клавишу «*» - фотометр производит измерение активности с отображе¬
нием результатов хода реакции через временной интервал 15 сек. при этом на ниж¬
нем индикаторе отображается:
- на верхней строке - текущее время реакции;
- на нижней строке - активность за временной интервал dt = 15 с
«t = XX:XX dA/dt = Х.ХХХ».
-211 -
По истечении времени t мин измерение активности завершается и на инди¬
каторе отображается измеренное значение активности и введенное значение апри¬
орного коэффициента К «Амгп|в = Х.ХХХ К = Х.ХХХ».
Примечания.
1. Результаты измерения активности за временной интервал dt=15 с вводятся в па¬
мять и могут быть выведены на нижний индикатор по окончании заданного времени t мин
измерения активности с целью определения линейного участка реакции. Просмотр осуще¬
ствляется последовательным нажатием клавиши «В» (вперед) или клавиши «А» (назад).
Начало линейного участка отмечается нажатием клавиши «1», при этом пред¬
варительно на верхнюю строку нижнего индикатора устанавливают «точку» начала
линейного участка. На индикаторе отображается надпись
«dA/dt [XX]'I Х.ХХХ
dA/dt[XX] = Х.ХХХ”
Конец линейного участка отмечается нажатием клавиши «2», при этом предва¬
рительно на нижнюю строку нижнего индикатора устанавливают «точку» конца
линейного участка. На индикаторе отображается надпись
«dA/dt [XX]' =Х.ХХХ
dA/dt[XX] IX.XXX".
Затем последовательным нажатием клавиши «В» выйти на режим определения
активности на линейном участке с отображением на нижнем индикаторе надписи
«А = Х.ХХХ К = Х.ХХХ».
актив.
2. Активность А определяется по формуле
it
где Ai - оптическая плотность исследуемого раствора в конце реакции
Ао - оптическая плотность исследуемого раствора в начале реакции
t - время реакции
К - априорный коэффициент
i - порядковый номер временного интервала.
3. введенные значения временного интервала t мин и априорного коэффициента
К сохраняются в памяти микрорпроцессорной системы и при отключении фотомет¬
ра от электрической сети.
2.5 работа с внешней ЭВМ и принтером.
Связь осуществляется по стандарту RS232C.
Программное обеспечение разрабатывается пользователем.
Протокол обмена связи КФК-3-01-«ЗОМЗ» (модификация с двумя ЖК дисплеями).
- скорость обмена -9600 бод
- длина передаваемых данных -8 бит
- контроль четности -отсутствует
- количество стоповых бит -1.
Связь осуществляется по нуль-модемному (трехпроводному) соединению.
4.6. СПЕКТРОФОТОМЕТР С -115
Спектрофотометр С-115 может работать в трех атомно-абсорбционных режимах:
- режим измерения атомного поглощения;
- режим измерения неатомного поглощения;
- режим измерения атомного поглощения с компенсацией неатомного поглощения.
- 212 -
Для установки режима измерения атомного поглощения необходимо нажать кла¬
вишу ИМП-2 и включить лампу с полым катодом.
Для установки режима измерения неатомного поглощения необходимо нажать
клавишу ИМП-2 и включить дейтериевую лампу. Лампу с полым катодом при этом не
включать.
Для установки режима измерения атомного поглощения компенсацией неатом¬
ного поглощения необходимо нажать клавишу ИМП-2 и включить лампу с полым
катодом нажав клавишу ЛПК и дейтериевую лампу нажав клавишу ДДС.
Во всех трех режимах работы можно получать отсчет в единицах оптической
плотности, поглощении, концентрации.
Для выбор шкалы измерений необходимо нажать соответствующие клавиши:
Д - отсчет в миллибеллах, шкала оптической плотности;
1 -Т - отсчет в единицах поглощения, %;
С А - отсчет в единицах концентрации.
Измерение концентрации реализуется при условии линейной зависимости оп¬
тической плотности от концентрации.
Рис.4 Спектрофотометр С-И5
4.6.1. Подготовка спектрофотометра С-115 к работе
1. Установить лампу с полым катодом.
2. Включение спектрофотометра в сеть.
Убедиться перед включением спектрофотометра в сеть, чтго ручки регулировки
тока лампы с полым катодом ЛПК дейтериевой лампы ДДС и фотоэлектронного ум¬
ножителя находятся в крайнем левом положении, а их клавиши находятся в положе¬
нии выключено.
Включить кнопку «Сеть».
Включить кнопку «ЛПК», переместить ручку регулировки тока лампы с полым
-213-
катодом вправо, установить ток по шкале микроамперметра в пределах от 2 до 25 та
(от 4 до 50 делений шкалы). При этом необходимо помнить, что включение и выклю¬
чение лампы с полым катодом и дейтериевой лампы следует всегда производить при
нулевом токе (ручки регулировки тока находятся в крайнем левом положении). Нару¬
шение этого правила может привести к преждевременному выходу из строя ламп.
Помещая лампу вдоль оптической оси добиться чтобы сфокусированное изобра¬
жение полого катода находилось на уровне середины отсека атомизатора или сере¬
дины насадки горелки.
Добиться, вращая регулировочные винты держателя лампы с полым катодом,
симметричного относительно центра заполнения выходного окна отсека атомизато¬
ра спектрофотометра излучением лампы.
Окончательная юстировка лампы с полым катодом осуществляется после настрой¬
ки спектрофотометра на максимум линии излучения.
3.Настройка монохроматора.
3.1. Включить спектрофотометр и установить требуемый источник излучения.
3.2. Установить ручкой регулировки ФЭУ напряжение питания ФЭУ в пределах
800-1500 V (от 40 до 75 делений на шкале микроамперметра ФЭУ).
3.3. Включить кнопку «Щель» и установить размер щели - 0,1 мм.
3.4. Нажать клавиши «ИМП 1» «1-Т», установить однолучевой абсорбционный
режим работы спектрофотометра.
3.5. Перевести тумблер быстрого сканирования спектра в положение «Умень¬
шить» или «Увеличить» в зависимости от показания счетчика длины волн.
3.6. Перевести не доходя (1-2 нм) тумблер длины волн быстрого сканирования в
нейтральное положение.
3.7. Используя кнопки шагового сканирования «Уменьшить» или «Увеличить» и
наблюдая за показаниями индикатора ЛПК добиться максимальной длины светового
шнура на индикаторе.
3.8. Если в процессе настройки индикатор ЛПК зашкаливает, то необходимо
уменьшить напряжение питания на ФЭУ. Максимально допустимые показания
индикатора ЛПК должны составлять от 5 до 10 делений шкалы индикатора ЛПК.
3.9. Добиться, вращая котировочные винты на держателе лампы с полным като¬
дом максимальной длины светового шнура на индикаторе ЛПК. Бели в процессе на¬
стройки индикатор ЛПК зашкаливает, то необходимо уменьшить напряжение питания
на ФЭУ.
Следует погнить, что при высокой чувствительности спектрофотометра имеет место
высокий уровень шумов прибора, поэтому необходимо в процессе настройки прибора
оптимизировать соотношение чувствительности прибора с уровнем шумов.
4.6.2. Спектрофотометр Квант-2АТ
Спектрофотометр Квант-2АТ относится к разряду современных аналитических
комплексов работающего с компьютерным программным обеспечением Кант-2АТ.
К персоналу, работающего с комплексом Квант-2АТ, предъявляются требования
к знанию компьютера, правил техники безопасности, правил противопожарной бе¬
зопасности.
Комплекс состоит из измерительного блока (прибора) и компьютера с программ¬
ным обеспечением. Ручная настройка прибора не предусмотрена. Предварительно
проводятся калибровки по измеряемым элементам. Все настройки и установки запи¬
сываются в память компьютера и при включении они передаются в измерительный
блок, который в свою очередь, является исполнительным устройством с системой
- 214-
датчиков и сервоприводов. В приборе одновременно могут быть установлены шесть
измерительных ламп на разные элементы. На каждый элемент может быть несколько
различных калибровок, например, на разные диапазоны измерений, на разные объек¬
ты измерений - почва, вода, растения. Калибровки сделаны с привязкой к значениям
концентраций, поэтому результаты измерений мы получаем сразу в окне программы.
1. Включить компрессор для подачи воздуха. Открыть подачу газа (баллон или
система газоснабжения). Открыть предохранительный вентиль воздушной системы.
2. Снять защитные чехлы с прибора и компьютера. Подключить электропитание.
Включить прибор - клавиша «Сеть» и компьютер.
3. На рабочем столе запускаем ярлык «Квант-2 А». Ждем около 2 минут для тести¬
рования системы и ее автонастройки (прибор возвращается в нулевое состояние). В
окне программы внизу загораются зеленые индикаторы: «Прибор», «Воздух», «Гид¬
розатвор». Если индикаторы не горят или происходит сбой, то закрываем программу:
на самом приборе нажимаем клавишу «Сброс» и запускаем программу повторно.
4. Выбираем окно «Газовая система». Поджигаем пламя, нажимаем клавишу «Под¬
жечь пламя». Пламя поджигается и контролируется прибором автоматически. Внизу
в окне загорается индикатор «Пламя». Оптимальный показатель расхода пламени 6-8
мл в минуту.
5. Нажимаем управляющую клавишу «Методика», выбираем измеряемый «Эле¬
мент», выбираем рабочий график. Подтверждаем свой выбор «Ок». Ждем около 2
минут, пока прибор не настроится.
6. Проводим проверку калибровки. Нажимаем «Калибровка», выбираем «Уточ-
Рис.5 Спектрофотометр Квант-2 АТ
-215-
нить». В окне вводим концентрацию раствора, который будет измерять, нажимаем
«Уточняющая концентрация». В этом же окне нажимаем «Измерение», в новом окне
проводим «Обнуление», потом «Измерение». Система померяет и введет поправоч¬
ный коэффициент. Закрываем все окна. Разрешается продолжать измерения, если по¬
правочный коэффициент находится в пределах 0,9-1,1.
7. Проводим сами измерения. Нажимаем «измерение», выбираем «Добавить
образец». В этом же окне нажимаем «Измерение», в новом окне проводим «Обнуле¬
ние», потом «Измерение».Далее действуем по схеме «Добавить образец» - «Обнуле¬
ние» - «Измерение». После всех измерений результаты можно сохранить.
8. Если нужно измерить следующий элемент, то повторяем пункты 6-8 для дру¬
гого элемента.
9. После всех измерений закрываем окно «Измерение», гасим пламя «Погасить
пламя», закрываем программу «Квант-2А», выключаем прибор, компьютер, закрыва¬
ем предохранительный вентиль воздушной системы. Выключаем компрессор, закры¬
ваем подачу газа.
Во время эксплуатации могут возникнуть следующие проблемы:
1. Если прибор при включении не настраивается, всплывает окно с предуп¬
реждением о невозможности настроить или установить параметры, тухнем пламя -
тогда закрываем все окна, закрываем программу «Квант-2А», на приборе нажимаем
клавишу «Сброс» и запускаем повторно.
2. Если забился распылитель (измерения равны нулю, пламя не окрашивается
в красный цвет) - то выкручиваем трубку, с комплекта ЗИП берем специальную иголку
и прочищаем его. Вкручиваем трубку обратно.
3. Прибор работает в режиме сравнения образца с дистиллированной водой и
с каждым следующим измерением дистиллированная вода загрязняется. Поэтому же¬
лательно ее менять после каждых 10 измерений. Если показатели близки к нулю или
с отрицательным значением (Си, Zn, Pb) - то заменяем дистиллированную воду. Зак¬
рываем окно «Измерение». С правой стороны выбираем окно «ЛД Фотоприемник»,
нажимаем «Балансировать». После балансировки можно возвращаться к измерениям.
4.7. ВЕСЫ-ВЛАГОМЕР
Весы-влагомер - это прецизионный, лабораторный измерительный прибор, ко¬
торый предназначен для быстрого измерения содержания воды (влажности) в образ¬
цах разных материалов.
Весы-влагомер представляют собой прецизионные лабораторные весы и соеди¬
ненную с ними камеру сушки, которая обеспечивает стабильную температуру высу¬
шивания во время измерения. При такой конструкции метод измерения относитель¬
ной влажности отличается от известных традиционных методов.
Принцип действия весов -влагомера сводится к:
- взвешиванию исследуемого образца перед высушиванием, во время высушива¬
ния и после высушивания;
- автоматическое окончание измерения при полном испарении воды из образца
(высушивание до сталой массы), или после окончания определенного времени высу¬
шивания ;
- расчет результатов высушивания образцов для принятого метода работы;
- передача полученных данных к печатному устройству или компьютеру, если
есть такая необходимость.
Нагревание и стабилизацию весов-влагомера необходимо провести после пер¬
вого подключения в сеть.
-216-
5. ТЕХНИКА БЕЗОПАСНОСТИ
ПРИ ПРОВЕДЕНИИ ЛАБОРАТОРНЫХ РАБОТ
В целях предупреждения несчастных случаев в лабораториях необходимо раци¬
онально организовать рабочие места сотрудников, строго соблюдать санитарные
условия труда и требования технической и пожарной безопасности.
5.1. Правила пользования и работа с химическими реактивами
1. Все химические реактивы должны храниться в соответствующей упаковке и
иметь ярлыки с обозначением содержимого.
2. Концентрированные кислоты хранятся в стеклянных емкостях, помещенных в
прочные деревянные корзины, заполненные стружкой.
3. При разливе концентрированных кислот следует применять сифоны, оборудо¬
ванные ручными насосами, либо разливочные устройства.
4. Переливание «дымящих» кислот и аммиака производится в помещениях, снаб¬
женных вытяжными устройствами.
5. Работа с плавиковой кислотой проводится обязательно в полиэтиленовой, пла¬
тиновой или парафинированной изнутри стеклянной посуде.
6. При проведении массовых анализов с применением хлорной кислоты следует не
реже одного раза в неделю промывать водой внутренние стенки вытяжного шкафа.
7. Растворение гидроокисей натрия и калия проводится в фарфоровых стаканах
при постоянном перемешивании.
8. Все работы, связанные с разливом крепких растворов кислот и щелочей, сле¬
дует выполнять в прорезиненных фартуках, защитных очках.
9. Смешивание и разбавление веществ, сопровождающееся выделением тепла,
следует выполнять в термостойкой или фарфоровой посуде.
10. Пролитые кислоты и щелочи разбавляют большим количеством воды, затем
нейтрализуют кислоты, посыпая мелом или содой до прекращения вскипания. Обра¬
зующуюся массу убирают, смывая водой.
5.2. Меры предосторожности при работе со стеклянной посудой
1. Посуду, предназначенную для нагревания кислот до высокой температуры, сле¬
дует до использования прокипятить в течение 10 мин. в концентрированном раство¬
ре хлористого натрия.
2. Все виды термической и механической обработки стекла необходимо прово¬
дить в защитных очках.
3. При открывании притертых пробок в случае их залипания следует постучать
вокруг пробки деревянным бруском, а если не поможет, осторожно нагреть горлови¬
ну склянки. В зависимости от содержимого склянки можно в зазор между пробкой и
горловиной ввести несколько капель этилового спирта или уксусной кислоты.
4. При надевании на стеклянные трубки резиновых шлангов необходимо пра¬
вильно подобрать диаметр последних и концы смочить водой.
5.3. Правила пользования газом
1. При работе с газом должна быть включена вентиляция.
-217-
2. Газовые горелки должны содержаться в чистоте и порядке.
3. Герметичность газовой сети следует поверять, используя мыльный раствор.
4. при обнаружении запаха газа необходимо принять меры к устранению неисп¬
равностей, выключить нагревательные приборы и проветрить помещение.
5. Баллоны со сжиженным газом должны находиться вне помещения в специ¬
альных запираемых металлических ящиках.
5.4. Правила пользования электрооборудованием
1. Для обесточивания всех помещений лаборатории должен быть общий щит и
рубильник.
2. Все нагревательные приборы должны иметь постоянное место с достаточной
теплоизоляцией.
3. Работы с муфелем и печью для выпечки хлебцов необходимо проводить в бре¬
зентовых перчатках.
4. При пользовании электроэнергией запрещается:
а) работать с неисправным электрооборудованием;
б) вскрывать электрические щитки и магнитные пускатели;
в) держать легковоспламеняющиеся и летучие жидкости вблизи нагреватель¬
ных приборов;
г) пользоваться для подключения проводниками с поврежденной изоляцией,
без штепселей, а также самодельными предохранителями;
д) работать с незаземленным электрооборудованием.
Дежурный сотрудник обязан проверить в конце рабочего дня: перекрыты ли га¬
зопроводы и выключен ли рубильник щита энергопитания.
5.5. Оказание первой помощи
5.5.1. При поражении электротоком
1. Необходимо немедленно обесточить пострадавшего, отключив рубильник, или
перерубив провода инструментом (кусачками, топором и др.), ручки которого имеют
надежную изоляцию.
2. Если нельзя отключить установку, необходимо освободить пострадавшего от
электропроводки при помощи сухой палки или резиновых перчаток. При освобожде¬
нии пострадавшего нужно действовать одной рукой.
3. При всех случаях поражения электротоком следует немедленно вызвать меди¬
цинскую помощь. До прихода мед работника необходимо делать следующее:
а) если пострадавший в сознании, но до этого был в обмороке или продол¬
жительное время находился под током, ему необходимо обеспечить полный покой до
прибытия врача и дальнейшее наблюдение в течение 2-3 часов. А в случае невоз¬
можности быстро вызвать врача - срочно доставить пострадавшего в лечебное уч¬
реждение при помощи транспортных средств или носилок;
б) при отсутствии сознания, но сохранившемся дыхании пострадавшего нуж¬
но уложить ровно, расстегнуть одежду, создать приток свежего воздуха, давать нюхать
нашатырный спирт, обрызгивать лицо водой, растирать и согревать тело. Если пост¬
радавший плохо дышит - очень редко и судорожно, следует делать искусственное
дыхание.
-218-
5.5.2. При отравлениях
1. При отравлении газом удалить пострадавшего из опасной зоны, освободить от
стесняющей одежды, остерегаясь охлаждения тела. При остановке дыхания необхо¬
димо делать искусственное дыхание.
2. При попадании ядовитого вещества в органы пищеварения вызвать рвоту, а
затем дать средства, нейтрализующее действие яда (см.п.5.5.5.).
5.5.3. При ожогах
1. При термических ожогах необходимо применять следующие меры:
а) в случае воспламенения одежды на работнике необходимо накрыть его
плотным одеялом или облить водой;
б) при ожогах первой степени обожженное место можно присыпать двууглекис¬
лым натрием (питьевой содой), картофельным крахмалом, тальком или сделать примочки
из свежеприготовленных растворов питьевой соды (2 % раствор), марганцовокислого калия
(5 % раствор), лучшим средством для примочек является 96 % этиловый спирт;
в) при ожогах второй и третьей степени разрешается делать только примочки
из раствора марганцовокислого калия и наложить сухую асептическую повязку. Ле¬
чение таких ожогов проводит медицинский работник.
2. При химических ожогах кислоту или щелочь, попавшие на кожу, необходимо
удалить ватным тампоном, промыть пораженное место обильным количеством воды
и обработать нейтрализующими веществами:
а) при ожогах кислотами (соляной, серной, азотной) - 2 % раствором карбо¬
ната аммония или бикарбоната натрия;
б) при ожогах щелочами - 2 % раствором уксусной кислоты (или 1 % раство¬
ром соляной кислоты);
в) при ожоге плавиковой кислотой промыть большим количеством воды и
наложить компресс с пастой, приготовленной из гидрата окиси магния.
3. Очень опасны химические ожоги для глаз. Попадание в глаза органических
растворителей очень болезненно, поражение свинцом, ртутью, окисью углерода, ам¬
миаком приводит к ухудшению зрения, а перекись водорода может вызвать полную
потерю зрения. Во избежание поражения глаз при работе с перечисленными соеди¬
нениями необходимо пользоваться защитными очками.
Попавшие в глаза брызги удаляют путем промывания обильным количеством воды.
5.5.4. При ранениях и порезах
1. При незначительных порезах рану промывают раствором марганцовокисло¬
го калия и смазывают края раствором йода. Для дезинфекции применяют 3 % ра¬
створ перекиси водорода, а затем место ранения перевязывают стерильным бин¬
том.
2. При глубоких порезах и сильном кровотечении для быстрой остановки кро¬
ви накладывают жгут и направляют в лечебное учреждение (жгут нельзя держать
более полутора часов во избежание омертвения ткани).
3. При порезах стеклом необходимо промыть рану под сильной струей воды,
обработать края раны перекисью водорода, забинтовать. Дальнейшую помощь ока¬
зывает врач. Не рекомендуется самому удалять глубоко проникшие осколки стекла,
так как это может привести к увеличению поверхности ранения.
-219-
Вещества,
вызывающие отравления, и применяемые противоядия
№
п/п
Вещества,
вызывающие
отравления
Противоядия
1
Кислоты
Прополоскать рот водой или 5 % раствором NaHC03,
пить молою, суспензию окиси магния (15г MgQ на 100
мл воды), известковую воду (100мл известювой воды
разбавить водой до 1 л), жидюе мучное тесто.
2
Щавелевая кислота
Вызвать рвоту. Дать известювую воду (приготовление
см.п.1), касторовое масло.
3.
Пары азотной кислоты и
окислы азота
Поюй. Дать 2г норсульфазола или других
сульфаниламидных препаратов. Вдыхание кислорода.
4.
Аммиак
Дать в большом количестве воду подкисленную уксусом
или лимонным соком, вызвать рвоту. Давать
растительное масло, молоко, яичный белок. Вынеси на
свежий воздух.
5.
Соли бария
Вызвать рвоту, дать слабительное - MgS04, Na2S04.
6.
Медь и ее соли
Промыть желудок раствором перманганата калия
(1:1000). Внутрь принимать 1 % р-р КМп04 по 1
ст.ложке через каждые 5 мин. или водную суспензию
окиси магния (15 г MgQ на 100 мл воды).
7.
Мышьяк, сурьма, цинк
или ртуть
Вызвать рвоту Дать слабительное (сернокислый
магний, сернокислый натрий), после чего давать через
каждые 10-15 мин.болтушку до прекращения рвоты. (С
остав болтушки: 100г сернокислого окисного железа
растворить в 300 мл воды и в него влить 20г окиси
магния, растертого в 300мл воды).Лучше принять 1
флакон свежеприготовленного противоядия С
трижижевского: 1,25г бикарбоната калия, 0,1 г едюго
натра, 0,38г сернокислого магния, 0,5-0,7г сероводорода
в 100 мл раствора.
8.
Соединение олова
Вызвать рвоту. Дать суспензию окиси магния в воде,
растительное масло.
9.
Перманганат калия
Промыть желудок, асторовое масло. Внутрь
крахмальный клейстер, танин, уголь. Полоскать рот
раствором хлората калия, амфора.
10
Соединение серебра
Дать большое юличество раствора хлористого натрия
или противоядия Стрижижевсюго (см.п.7).
И.
Натрий фтористый
Дать известювую воду или разбавленный (2 %) раствор
хлористого кальция.
- 220 -
6. ПРИЛОЖЕНИЯ
Таблица 1.
Атомные веса некоторых элементов (данные Международной
комиссии по определению атомных весов на 1973год).
Элемент
Обозначение
АгомныЙ вес
Элемент
Обозначение
Атомный вес
Азот
N
14,0067
Мышьяк
As
74,9216
Алюминий
А1
26,98154
Натрий
Na
22,98977
Барий
Ва
137,34
Никель
Ni
58,70
Бор
В
10,81
Олово
Sn
118,69
Бром
Вг
79,904
Платина
Pt
195,09
Ванадий
V
50,9414
Ртуть
Hg
200,59
Водород
Н
1,0079
Рубидий
Rb
854678
Вольфрам
W
183,85
Свинец
Pb
207,2
Железо
Fe
55,847
Селеп
So
78,96
Золото
Au
196,9665
Сера
S
32,06
Иод
J
126,9045
Серебро
Ag
107,868
Калий
К
39,098
Стронций
Sr
87,62
Кальций
Са
40,08
Титан
Ti
47,90
Кислород
О
15,9994
Ушерод
С
12,011
Кобальт
Со
58,9332
Уран
и
238,029
Кремний
Si
28,086
Фосфор
p
30,97376
Литий
Li
6,941
Фтор
F
18,99840
Магний
Mg
24,305
Хлор
Cl
35,453
Марганец
Mn
54,9380
Хром
Cr
51,996
Медь
Си
63,546
Цезий
Cs
132,9054
Молибден
Mo
95,94
Цинк
Zn
65,38
Таблица 2.
Пересчетные коэффициенты для перевода процентного содержания
ионов в миллиграмм-эквиваленты.
Ион
Эквивале¬
нтный
вес
Милли¬
грамм-
эквивалент
Коэффициент
Д=1000
экввес
(для пересчета
% в мг-экв)
Ион
Эквивале-
нтный
вес
Милли¬
грамм-
эквивалент
Коэффициент
Л ж 1000
1 ж экввес
(для пересчета
% в мг-экв)
H+
1,00
0,001
992,1
а
35,46
0,035
28,2
К+
39,10
0,039
25,6
N0,
62,00
0,062
16,1
Na+
23,00
0,023
43,5
N0,
46,00
0,046
21,7
ш:
18,04
0,018
55,4
so42-
48,00
0,048
20,8
-221 -
Продолжение табл.2
Са2*
20,00
0,020
49,9
нсо,
61,01
0,061
16,4
Mg2*
12,16
0,012
82,2
СО,2
30,00
0,030
33,3
Fe2*
27,92
0,028
35,8
РОд3
31,67
0,032
31,6
Fe3*
18,61
0,019
53,7
S2-
16,03
0,016
62,4
АР
9,00
0,009
11,2
38,03
0,038
26,3
Mn2+
27,46
0,027
36,4
Пример пересчета с использованием коэффициента А. содержание поглощенного маг¬
ния равно 0,04%. Для перевода в миллиграмм-эквиваленты процентное содержание умножают
на коэффициент А и получают 0,04 • 82,2 = 3,288. округленно эту величину принимают равной
3,29 мг-экв.
Таблица 3.
Аналитические множители.
Найдено
Ищут
Пересчетный
юэффициент
(множитель)
Найдено
Ипуг
Пересчетный
юэффициент
(множитель)
AgCl
Cl
0,247
со2
СаО
1,274
AgCl
AgNO,
1,185
со2
CaCO,
2,274
А1А
A1
0,529
со2
Na7CO,
2,409
А1А
AIPO,
2,393
со,
NaHCO,
1,909
A1A
A»(SOA
3,356
СаСО,
С
0,119
BaSO,
so4
0,411
СаСО,
co2
0,439
BaS04
so3
0,343
СаСО,
CO,
0,599
BaSO,
s
0,137
СаСО,
Ca
0,400
BaSO.
H,sot
0,420
СаСО,
CaS04
1,3,60
BaSO,
Ba
0,588
СаСО,
CaSO>2RO
1,720
BaS04
CaS04
0,583
CaSO,
Ca
0,294
BaS04
CaS0,-2H,0
0,737
CaSO,
CaO
0,412
Г^мус
co2
0,579
CaSO,
CaCO,
0,735
Гумус
co2
2,123
Fe
Fe2Oe
1,430
С
co2
3,664
Fe
FeO
1,286
С
Гумус
1,724
Fe
FeCr
2,269
со,
Гумус
0,471
Fe
FeCls
2,904
со,
с
0,272
Fe
FeS047H,0
4,978
сог
СО,
1,364
Fe
Fe(SOJ,
3,580
со2
Са
0,911
Fe,0,
Fe
0,699
ре1°з
FeO
0,900
N
NO,
4,427
Fe,0,
FePO,
1,889
N
n7o.
3,356
Fe,0,
FeCls
2,031
N
NaNO,
6,069
Fe,0,
Fe,(S04X
2,504
N04
N
0,226
-222-
Продолжение табл.З
Найдено
Ищут
Пересчетный
коэффициент
(множитель)
Найдено
Ищут
Пересчетный
коэффициент
(множитель)
Н,0
H
0,111
NO.
na
0,871
н,о
0
0,888
NO,
NaNO,
1,371
КС1
к
0,524
NA
N
0,259
КС1
K20
0,632
NA
NO,
1,148
к
0,449
NA
NaNO,
1,574
k2so4
K20
0,541
nh4
N
0,776
К,SO,
KC1
0,856
nh4
NO,
3,437
Mg,p,o,
Mg
0,218
nh4
na
2,994
Mg,p,o,
MgO
0,362
nh4
NaNOj
4,712
Mg,P,o,
MgCO,
0,757
nh4
NH,
0,944
MgS04
Mg
0,202
NaCl
Na
0,393
MgS04
MgO
0,335
NaCl
Na,0
0,530
Mgso4
MgCO,
0,700
NaCl
Na,CO,
0,907
MgCO,
MgfHCO,),
1,735
NaCl
NaHCO,
1,437
Mg,PA
p
0,279
Na,S04
Na
0,323
Mg,P70,
po4
0,853
NaHCO,
Na2C03
0,630
Mfep,o,
0,638
Na,CO,
NaHCO,
1,585
Mn
MnO
1,291
NaHCO,
Na
0,274
Mn
MnO,
1,582
Na
NaHCO,
3,653
Mn
KMnO.
2,876
Si07
Si
0,467
MgSO,
Mn
0,363
SK)2
SiO,
1,266
MgS04
MnO
0,469
SK)2
Si04
1,533
MgSO,
MnO,
1,575
SK^
Si,07
1,400
Таблица 4.
Количество кислот и аммиака для приготовления их процентных растворов (в мл
на 1 л раствора)*.
Исходное
в-во
Удельный вес исх.
в-ва при 15°С
Весовой процент
исх. в-ва
25%-
ный
20%-
ный
10%-
ный
5%-
ный
2%-
ный
1%-
ный
НС1
1,19
37,23
634,8
496, В
236,4
115,2
45,5
22,6
H.SO,
1,84
95,6
167,7
129,9
60,6
29,3
11,5
5,6
HNO,
1,40
65,6
313,0
243,6
115,0
56,0
22,0
10,8
СН^СООН
1,05
99,5
247,8
196,7
97,1
48,2
19,2
9,0
nh4oh
0,91
25,0
1000,0
814,0
422,0
215,4
87,2
43,7
*Е.В.Аринушкина. руководство по химическому анализу почв. 2-е изд. Изд-во МГУ, 1970.
-223-
Таблица 5.
Удельный вес и процентное содержание уксусной кислоты при 15° С*.
Удельный
вес
СН3СООН,
%
Удельный
вес
СН3СООН,
%
Удельный
вес
СН3СООН,
%
Удельный
вес
СН3СООН,
%
1,0000
1
1,0363
26
1,0623
51
1,0747
76
1,0020
2
1,0375
27
1,0636
52
1,0748
77
1,0037
3
1,0388
28
1,0638
53
1,0748
78
1,0052
4
1,0400
29
1,0646
54
1,0748
79
1,0070
5
1,0412
30
1,0653
55
1,0748
80
1,0083
6
1,0424
31
1,0660
56
1,0747
81
1,0098
7
1,0436
32
1,0666
57
1,0746
82
1,0113
8
1,0447
33
1,0673
58
1,0744
83
1,0127
9
1,0459
34
1,0679
59
1,0742
84
1,0140
10
1,0470
35
1,0685
60
1,0739
85
1,0157
11
1,0481
36
1,0691
61
1,0736
86
1,0171
12
1,0492
37
1,0697
62
1,0731
87
1,0185
13
1,0502
38
1,0702
63
1,0726
88
1,0200
14
1,0513
39
1,0707
64
1,0720
89
1,0214
15
1,0523
40
1,0712
65
1,0713
90
1,0228
16
1,0533
41
1,0717
66
1,0705
91
1,0242
17
1,0543
42
1,0721
67
1,0696
92
1,0256
18
1,0552
43
1,0725
68
1,0686
93
1,0270
19
1,0562
44
1,0729
69
1,0674
94
1,0280
20
1,0571
45
1,0733
70
1,0660
95
1,0298
21
1,0580
46
1,0737
71
1,0644
96
1,0311
22
1,0589
47
1,0740
72
1,0625
97
1,0324
'23
1,0598
48
1,0742
73
1,0604
98
1,0337
24
1,0607
49
1,0744
74
1,0580
99
1,0350
25
1,0615
50
1,0746
75
1,0553
100
*удельный вес уксусной кислоты в пределах 1,0553-1,0748 соответствует двум ра¬
створам с различным процентным содержанием СН}СООН. Пользоваться таблицей в этом
случае можно лишь при условии проведения пробы на разбавление: если удельный вес уксус¬
ной кислоты при разбавлении водой увеличивается - содержание СН3СООН больше 77%,
если удельный вес уменьшается - содержание CHJCOOH меньше 77%.
Таблица 6.
Удельный вес и содержание H,SQ4 водных растворов серной кислоты при 15° С.
Удельный
вес
Содержание H,S04
Удельный
вес
Содержание HjS04
Удельный
вес
Содержание H,S04
в 100 г
р-ра
в 1000мл
р-ра
в 100 г
р-ра
в 1000мл
р-ра
в 100 г
р-ра
в 1000мл
р-ра
1,310
40,35
529
1,530
62,53
957
1,750
81,56
1427
-224-
Продолжение табл.6
Удельный
вес
Содержание H,S04
Удельный
вес
Содержание H,SOd
Удельный
вес
Содержание H,S04
в 100 г
в 1000мл
в 100 г
в 1000мл
в 100 г
в 1000мл
р-ра
р-ра
р-ра
р-ра
р-ра
р-ра
1,320
41,50
548
1,540
53,43
977
1,760
82,44
1451
1,330
42,66
567
1,550
64,26
996
1,770
83,51
1478
1,340
43,74
586
1,560
65,20
1017
1,780
84,50
1504
1,350
44,82
605
1,570
66,09
1038
1,790
85,70
1534
1,360
45,88
624
1,580
66,95
1058
1,800
86,92
1564
1,370
46,94.
643
1,590
67,83
1078
1,805
87,60
1581
1,380
48,00
662
1,600
68,70
1099
1,810
88,30
1598
1,390
49,06
682
1,610
69,56
1120
1,815
89,16
1618
1,400
50,11
702
1,620
70,42
1141
1,820
90,05
1639
1,410
51,15
721
1,630
71,27
1162
1,822
90,40
1647
1,420
52,15
740
1,640
72,12
1182
1824
90,80
1656
1,430
53,11
759
1,650
72,96
1204
1,826
91,25
1666
1,440
54,07
779
1,660
73,81
1225
1,828
91,70
1676
1,450
55,03
798
1,670
74,66
1246
1,830
92,10
1685
1,460
55,97
817
1,680
75,50
1268
1,832
92,70
1698
1,470
56,90
837
1,690
76,38
1289
1,834
93,25
1710
1,480
57,83
856
1,700
77,17
1312
1,836
93,80
1722
1,490
58,74
876
1,710
78,04
1334
1,838
94,60
1739
1,500
59,70
896
1,720
, 7892
1357
1,840
95,60
1759
1,510
60,65
916
1,730
79,80
1381
1,520
61,59
936
1,740
80,68
1,404
Таблица 7.
Удельный вес и содержание HN03 водных растворов азотной кислоты при 15° С.
Удельный
вес
Содержание HNO,
Удельный
вес
Содержание HNO,
Удельный
вес
Содержание HNO,
в ЮОг
р-ра
в 1000мл
р-ра
в 100 г
р-ра
в 1000мл
р-ра
в 100 г
р-ра
в 1000мл
р-ра
1,000
0,10
1
1,140
23,81
266
1,280
44,41
568
1,005
1,00
10
1,145
24,08
276
1,285
45,18
581
1,010
1,90
19
1,150
24,84
286
1,290
45,95
593
1,015
2,80
28
1,155
25,60
296
1,295
46,72
605
1,020
3,70
38
1,160
26,36
306
1,300
47,49
617
1,025
4,60
47
1,165
27,12
316
1,305
48,26
630
1,030
5,50
57
1,170
27,88
326
1,310
49,07
643
1,035
6,38
66
1,175
28,63
336
1,315
49,89
656
-225-
Продолжение табл.7
Удельный
вес
Содержание HN04
Удельный
вес
Содержание HNO,
Удельный
вес
Содержание HN04
в 100 г
р-ра
в 1000мл
р-ра
в 100 г
р-ра
в 1000мл
р-ра
в 100 г
р-ра
в 1000мл
р-ра
1,040
7,26
75
1,180
29,38
347
1,320
50,71
669
1,045
8,13
85
1,185
30,13
357
1,325
51,53
683
1,050
8,99
94
1,190
30,99
367
1,330
52,37
697
1,055
9,84
104
1,195
31,62
378
1,3325
52,80
704
1,060
10,68
113
1,200
32,36
388
1,335
53,22
710
1,065
11,51
123
1,205
33,09
399
1,340
54,07
725
1,070
12,33
132
1,210
33,82
409
1,345
54,93
739
1,075
13,15
141
1,215
34,55
420
1,350
55,79
753
1,080
13,95
151
1,220
35,28
430
1,355
56,66
768
1,085
14,74
160
1,225
36,03
441
1,360
57,57
783
1,090
15,53
169
1,230
36,78
452
1,365
58,48
798
1,095
16,32
179
1,235
37,53
463
1,370
59,39
814
1,100
17,11
188
1,240
38,29
475
1,375
60,30
829
1,105
17,89
198
1,245
39,05
486
1,380
.61,27
846
1,1Ю
18,67
207
1,250
39,82
498
1,3833
61,92
857
1,115
19,45
217
1,255
40,58
509
1,385
62,24
862
1,120
20,23
227
1,260
41,34
521
1,390
63,23
879
1,125
21,00
236
1,265
42,10
533
1,395
64,25
896
1,130
21,77
246
1,270
42,87
544
1,400
65,30
914
1,135
22,54
256
1,275
43,64
556
-
-
-
Таблица 8.
Удельный вес и содержание НС1 водных растворов соляной кислоты нри 15° С.
Удельный
вес
Содержание НС1
Удельный
вес
Содержание НС1
Удельный
вес
Содержание НС1
в 100 г
р-ра
в 1000мл
р-ра
в 100 г
р-ра
в 1000мл
р-ра
в 100 г
р-ра
в 1000мл
р-ра
1,000
0,16
1,6
1,075
15,16
163
1,145
28,61
328
1,005
1,15
12
1,080
16,15
174
1,150
29,57
340
1,010
2,14
22
1,085
17,13
186
1,152
29,95
345
1,015
3,12
32
1,090
18,11
197
1,155
30,55
353
1,020
4,13
42
1,095
19,06
209
1,160
31,52
366
1,025
5,15
53
1,100
20,01
220
1,163
32,10
373
1,030
6,15
63
1,105
20,97
232
1,165
32,49
379
1,035
7,15
74
1,110
21,92
243
1,170
33,46
391
1,040
8,16
85
1,115
22,86
255
1,171
33,65
394
-226-
Продолжение табл.8
Удельный
вес
Содержание НС 1
Удельный
вес
Содержание НС1
Удельный
вес
Содержание IICI
в 100 г
в 1000мл
в 100 г
в 1000мл
в 100 г
в 1000мл
р-ра
р-ра
р-ра
р-ра
р-ра
р-ра
1,045
9,16
96
1,120
23,82
267
1,175
34,42
404
1,050
10,17
107
1,125
24,78
279
1,180
35,39
418
1,055
11,18
118
1,130
25,75
291
1,185
36,31
430
1,060
12,19
129
1,135
26,70
302
1,190
37,23
443
1,065
13,19
150
1,140
27,66
315
1,195
38,16
456
1,070
14,17
152
1,1425
28,14
321
1,200
39,11
469
Таблица 9.
Удельный вес и содержание NH3 водных растворов аммиака при 15° С.
Удельный
вес
Содержание NH3
Удельный
вес
Содержание NR,
Удельный
вес
Содержание
в 100 г
р-ра
в 1000мл
р-ра
в 100 г
р-ра
в 1000мл
р-ра
в 100 г
р-ра
в 1000мл
р-ра
1,000
0,00
0,0
0,958
10,47
100,2
0,920
21,75
199,9
0,998
0,45
4,5
0,954
11,60
110,6
0,914
23,68
216,2
0,996
0,91
9,1
0,952
12,17
115,8
0,910
24,99
227,2
0,994
1,37
13,6
0,950
12,74
120,9
0,908
25,65
232,7
0,992
1,84
18,2
0,948
13,31
126,1
0,906
26,31
238,2
0,990
2,31
22,8
0,946
13,88
131,2
0,904
26,98
243,7
0,986
3,30
32,5
0,944
14,46
136,4
0,902
27,65
249,2
0,982
4,30
42,2
0,942
15,04
141,4
0,900
28,33
254,7
0,980
4,80
47,0
0,940
15,63
146,8
0,898
29,01
260,3
0,974
6,30
61,3
0,938
16,22
152,0
0,894
30,37
271,3
0,970
7,31
70,8
0,936
16,82
157,3
0,892
31,05
276,7
0,968
7,82
75,6
0,934
17,42
162,6
0,890
31,75
283,3
0,964
8,84
85,1
0,930
18,64
173,2
0,886
33,25
294,3
0,962
9,37
90,1
0,926
19,87
183,8
0,884
34,10
301,2
0,960
9,91
95,1
0,922
21,12
194,6
0,882
34,95
308,0
Таблица 10.
Концентрация наиболее употребляемых растворов кислот и аммиака.
Наименование
Удельный вес
при 200 С
Массовая доля,
%
Нормальность
раствора
Аммиак концентрированный
0,907
25,0
13,4
разбавленный
0,957
10,0
6,0
разбавленный
0,977
5,0
3,0
Азотная кислота концентрированная
1,400
67,0
15,0
-227-
Продолжение табл. 10
Наименование
Удельный вес
при 200 С
Массовая доля,
%
Нормальность
раствора
разбавленная
1,115
20,0
3,5
разбавленная
1,054
10,0
1,7
Серная кислота концентрированная
1,834
95,0
36,0
разбавленная
1,178
25,0
6,0
разбавленная
1,032
5,0
1,2
Соляная кислота юнцентрированная
1,184
37,0
12,0
разбавленная
1,098
20,0
6,0
разбавленная
1,047
10,0
3,0
Уксусная кислота ледяная
1,050
100,0
17,5
разбавленная
1,013
10,0
1,7
разбавленная
1,005
5,0
0,9
Фосфорная кислота концентриров.
1,700
85,0
14,7
Фтористоводородная к-та концентр.
1,146
46,6
26,3
Хлорная кислота (150 С)
1,540
60,0
9,2
- 228-
-229-
Таблица И.
Количество исходных веществ для приготовления 1 л титрованного раствора разной нормальности.
Исходное химически
чистое вещество
Молеку¬
лярная
масса
Эквива¬
лентная
масса
1 н.
0,5 н.
0,2 н.
0,1 н.
0,05 н.
0,02 н.
0,01 н.
Вещества для установления
титра указанных растворов и
их эквивалентная масса для
данной реакции
Н^О,. d 1.84
98,08
49,04
28 мл
14 мл
5,6 мл
2,8 мл
1,4 мл
0,56 мл
0,28 мл
Бура Na2B40? (перекристал-
лизованная при t 600 С);
190,7
HCI d 1,19
36,46
36,46
82 мл
41 мл
16,4 мл
8,2 мл
4,1 мл
1,64 мл
0,82 мл
njc2o4-2Hfl
126,07
63,4
-
-
-
6,3 г
3,15 г
1,26 г
0,63 г
Титрованный раствор
КМп04; 31,61
KMn04 в кислой среде
158,03
31,61
-
-
-
3,16 г
1,58 г
0,63 г
0,32 г
Щавелевокислый натрий
Na С,0 ; 67,01
2 2 4’ *
NaOH
40,00
40,00
40,0 г
20,Ог
8,0 г
4,0 г
2.0 г
0,80 г
0,40 г
Янтарная кислота Н6С404;
59,04
КОН
956,11
56,11
56,11 г
28,06 г
11,2 г
5,60 г
2,8 г
1,12 г
1,56 г
Ва(0Н),-8Н,0
315,50
157,75
157,75г
78,88 г
31,54 г
15,77 г
7,88 г
3,15 г
1,58 г
AgN03
169,89
169,89
-
-
-
17,00 г
8,50 г
3,40 г
1,70 г
Хлористый натрий NaCl;
58,45
Na2S203-5H20
248.21
248,21
-
-
-
24,80 г
12,40 г
5,00 г
2,50 г
Двухромовокислый калий
К5Сг707;49,04
FeS04(NH4)2S04*6H20
(соль Мора)
392,16
392.16
-
-
78,40 г
39,20 г
19,60 г
7,84 г
3,92 г
Титрованный раствор К20;
31,61
K2Cr207
291,22
19,04
-
-
9,18 г
1,90 г
2,45 г
0,98 г
0,49 г
Соль Мора; 392,16
Трилон Б
372,25
186,12
-
-
-
18,61 г
9,30 г
3,72 г
1,86 г
Сернокислый магний
MgSO^^O; 123,25
Таблица 12.
Количество исходных веществ для приготовления процентных растворов кислот
и аммиака, мг/л процентного раствора.
Исходное
вещество
Удельный вес
исходного в-ва при 15°
Массовая доля
исходного в-ва, %
25 %
20%
10%
2 %
1 %
НС1
1,19
37,23
634,8
496,8
236,4
45,5
22,6
H7so4
1,84
95,60
167,7
129,0
60,6
11,5
5,6
HNO,
1,40
65,6
313,0
243,6
115,0
22,0
10,8
СН,СООН
1,05
99,5
247,8
196,7
97,1
19,2
9,0
nh4oh
0,9125
25,0
1000,0
814,0
422,0
87,2
43,7
Таблица 13.
Буферные смеси для работы с ферментами. Смесь Серенсена II.
Состав цитратной смеси
pH
Состав цитратной смеси
pH
цитрат*,
мл
0,1 н.
НС1, мл
0,1 н.
NaOH, мл
цитрат*,
мл
0,1 н.
НС1 мл
0,1 н.
NaOH, мл
10,00
-
1,04
7,00
3,00
0,50
4,45
1,00
9,00
-
1,17
8,00
2,00
1,00
4,65
2,00
8,00
-
1,42
9,00
1,00
2,00
4,83
3,33
6,67
-
2,27
10,00
0,00
4,00
4,96
4,00
6,00
-
2,97
9,50
-
4,50
5,02
4,50
5,50
-
3,36
9,00
-
4,75
5,11
4,75
5,25
-
3,53
8,00
-
5,31
5,00
5,00
-
3,69
7,00
-
5,57
5,50
4,50
-
3,95
6,00
-
5,97
6,00
4,00
-
4,16
5,50
-
6,33
5,25
-
6,68
*221,008 г кристаллической лимонной кислоты в 200 мл свободного от углекислоты
нормального раствору NaOH с разбавлением водой до 1 л.
Таблица 14.
Буферные смеси для работы с ферментами. Смесь Серенсена II.
Состав фос<
этной смеси
pH
Состав фос(
отной смеси
рн
1/15 М* Na2HPQ4
1/15 М** К^РО,
1/15 М* Na0HPO,
1/15 М** КН2Р04
0,10
9,90
4,94
6,00
4,00
6,98
0,25
9,75
5,29
7,00
3,00
7,17
0,50
9,50
5,59
8,00
2,00
7,38
1,00
9,00
5,91
9,00
1,00
7,73
2,00
8,00
6,24
9,50
0,50
8,04
3,00
7,00
6,47
9,75
0,25
8,34
4,00
6,00
6,64
9,90
0,10
8,67
5,00
5,00
6,81
10,00
0,00
9,18
*1/15 MNafiPO 4 - 11,876 г в 1л воды. ** 1/15 М КН2Р04 - 9,078 г в 1 л воды.
-230-
Таблица 15.
Приготовление процентных растворов разбавлением крепких или смешанных ра¬
створов разной концентрации (правило креста).
С - заданная концентрация раствора
С, - концентрация исходного (более крепкого) раствора
С2 - концентрация слабого процентного раствора или воды, если разведение
проводится водой.
А, = Сх - С2 искомое количество объемов воды или процентного раствора мень¬
шей концентрации, необходимое для разбавления.
А2 = С - С2 искомое количество объемов раствора большей процентной кон¬
центрации.
Например: для приготовления 5% ZnS04 из 45% раствора необходимо взять 5 объе¬
мов этого раствора и смешать с 40 объемами дистиллированной воды.
Таблица 16.
Приготовление титрованных растворов кислот.
Кислота
Нормальность приготовляемого раствора
2,0
1,0
0,5
0.2
0,1
0,05 | 0,02
0,01
Плотность
Взять нижеследующее юличество (мл)юнцентрирован-
ной кислоты и довесит в мерной юлбе дистиллированной
водой до 1 л
Соляная
1,19
164
82
41
16,4
8,2
4,1
1,64
0,82
Серная
1,84
58
28
14
5,6
2,8
1,4
0,56
0,28
Азотная
1,38
148
74
37
14,8
7,4
3,7
1,48
0,74
Ортофосфорная
1,72
118х
59
29,5
11,8
5,9
2,95
1,18
0,59
59хх
29,5
11,8
2,95
1,18
0,59
0,30
Уксусная ледяная
1,05
120
60
30
12
6,0
3,0
1,2
0,6
х Эквивалентная масса НЗР04 при титровании по метиловому оранжевому соответствует 98,04;
хх При титровании по фенолфталеину эквивалентная масса равна 49,02.
Таблица 17.
Допустимые отклонения объемов мерной посуды.
Емкость, мл.
Отклонения, ±мл
Емкость, мл.
Отклонения, ±мл
Колбы
Пипетки, бюретки
Колбы
Пипетки, бюретки
2
-
0,01
100
0,08
0,08
5
-
0,01
200
0,10
0,10
10
-
0,02
500
0,15
-
25
0,03
0,03
1000
0,30
-
50
0,05
0,05
2000
0,50
-
-231 -
Таблица 18
Изменение массы воды в объеме 1000 мл (правильно
откалиброванной посуды) при разных температурах.
°с
Масса, г
°С
Масса, г
°С
Масса, г
15
997,94
20
997,18
25
996,14
16
997,01
21
997,00
26
995,90
17
997,67
22
996,80
27
995,65
18
997,51
23
996,58
28
995,40
19
997,35
24
996,36
29
995,14
Таблица 19.
Важнейшие индикаторы.
1. Кислотно-основные
Индикатор
Концентрация,
%
Раство¬
ритель
Интервал
перехода, pH
Окраска индикатора
Метиловый
оранжевый
0,1
Вода
3,4-4,4
Красная-оранжево-желтая
Конго красный
(юнгорот)
0,1
Вода
3,0-5,2
Сине-фиолетовая-красная
Метиловый красный
(метил рот)
0,2
0,1
Спирт
80%
4,4-6,2
Красная-желтая
Тимоловый синий
I переход
II переход
0,1
Спирт
20%
8,0-9,6
Красная-желтая
Желтая-синяя
Малахитовый
зеленый
0,1
Вода
11,5-13,2
Зел ен ая-бесцв етная
Фенолфталеин
0,1
1,0
Спирт
95%
7,4-10,0
Бесцветная-малиновая
2. Комплексометрические
Индикатор и
определяемые ионы
Приготовление
индикатора
pH
титруемого
р-ра
Окраска раствора индикатора
Мурексид
(Са2+, Си2*, Na2+)
Сухая смесь
индикатора с NaCl
в отношении 1:100
9,0-11,0
Фиолетовая - красная
Флуорексон
(Са2+, Си2*, Si^Ba2* .Мп2*,
Co2+,Fe3+)
Сухая смесь
индикатора с КС1 в
отошении 1:100
13,2
Розовая со слабой
фпуоресценцией-ярю-зеленая
флуорес-пирукищя
Эриохром черный Т -
хромоген черный
(Zrr+,Mn2+,Cd2\Pb2+,Hq2+,
М^+,Са2+)
Сухая смесь
индикатора с NaCl
в отношении 1:100
6,0-11,0
Синяя -винно-красная
-232-
3. Окислительно-восстановительные
Метиленовый голубой
0,1-0,2г растворить
в 100 мл 60
%спирта
—
Синяя - бесцветная
N - фени лантрани ловая
кислота
0,2 г растворить в
100мл 0,2 %
Na,CO,
—
Красно-фиолетовая -
бесцветная
Таблица 20.
Характеристика известковых материалов.
Название
материала
Форма
извести в
материале
Содержание
карбоната в
пересчете на
СаСО„ %
Влажность,
%
Способ получения
Мел
СаСОэ
90-100
ДО 10
размол природных
залежей
Известняковая
мука
СаСО,
75-100
10-15
размол твердых
извести яюв
Доломитовая
мука
СаСОэ +
MgCO,
85-98
10-15
размол доломити-
зированных известняков
Мергель
СаСОэ +до5%
MgCO,
до 20
25-35
природные залежи
Негашеная и
гашеная известь
СаО
Са(ОН)2
135
обжиг и последующее
гашение
Известняковые
ту4ь!
СаСОэ
75-96
до 50
природные залежи
Торфотуфы
СаСО,
до 80
10-70
природные залежи
Дефекат
N - 0.5 %
P,Os - 0.5-0.7%
fc,o - 0.1-1%
СаС03
Са(ОН)2
до 60-80
до 40 в
сыром
виде
отход сахарного
производства
Доменные
шлаки
силикаты
кальция и
магния
до 85
до 8
отходы выплавки чугуна
Цементная пыль
СаС03 + СаО
до 80
до 2
отходы цементных
заводов
Таблица 21.
Группировка почв за величиной кислотности и щелочности.
Степень кислотности или щелочности
Класс
Цвет на картограмме
Величина pH
Очень сильнокислые
9
Темно-зеленый
<4,0
Сильнокислые
8
Темно-красный
4,1-4,5
Среднекислые
7
Светло-красный
4,6-50
Слабокислые
6
Оранжевый
5,1-5,5
Близкие к нейтральним
5
Желтый
5,6-6,0
-233-
Продолжение табл.21
Степень кислотности или щелочности
Класс
Цвет на картограмме
Величина pH
Нейтральные
4
Светло-зеленый
6,1-7,0
С л або щелочи ы е
3
Голубой
7,1-7,5
Среднещелочные
2
Синий
7,6-8,0
Сильнощелочные
1
Фиолетовый юричневый
8,1-8,5 >8,5
Таблица 22.
Классификация почв за содержанием обменных кальция и магния.
Содержание элементов
Класс
Цвет на картограмме
Са
Mg
мг-экв/100 г почвы
Очень низкое
1
голубой
<2,5
<0,5
Низкое
2
Синий
2,6-5,0
0,6-1,0
Среднее
3
Светло-зеленый
5,1-10,0
1,1-2,0
Повышенное
4
Зеленый
10,1-15,0
2,1-3,0
Высоюе
5
Темно-синий
15,1-20,0
3,1-4,0
Очень высоюе
6
Темн о-зеленый
>20,0
>4,0
Таблица 23.
Классификация почв за суммой поглощенных оснований..
Сумма (S)
Класс
Цвет на картограмме
S, мг-экв/100г почвы
Очень низкая
1
Розовый
<5,0
Низкая
2
Темно-розовый
5,1-10,0
Средняя
3
Красный
10,1-15,0
Повышенная
4
Голубой
15,1-20,0
Высокая
5
Сиреневый
20,1-30,0
Очень высокая
6
Фиолетовый
>30.0
Таблица 24.
Классификация почв по степени насыщения основаниями.
Степень насыщения основами (V)
Класс
Цвет на картограмме
V, %
Очень низкая
1
Светло-оранжевый
<30,0
Низкая
2
Оранжевый
30,1-50,0
Средняя
3
Розовый
50,1-70,0
Повышенная
4
Красный
70,1-90,0
Высокая
5
Красно-юричневый
>90,0
-234-
Таблица 25.
Классификация почв за содержанием аммонийного и
нитратного азота, в мг на кг почвы.
Содержание азота
Класс
Цвет на картограмме
N-N03
n-nh4
N-NO3+ N-NH,
Очень низюе
1
Красный
< 5,0
<5,0
< 10,0
Низкое
2
Оранжевый
5,1-8,0
5,1-8,0
10,0-15,0
Среднее
3
Желтый
8,1-10,0
8,1-10,0
15,1-20,0
Повышенное
4
Зеленый
10,1-15,0
10,1-15,0
20,1-30,0
Высокое
5
Голубой
15,1-30,0
15,1-30,0
30,1-60,0
Очень высоюе
6
Синий
>30,0
>30,0
>60,0
Таблица 26.
Классификация почв за содержанием гумуса
согласно ДСТУ 4362:2004.
Содержание гумуса
Класс
Цвет на картограмме
Показатель
Очень низкое
1
Красный
< 1,1
Низюе
2
Оранжевый
1,1-2,0
Среднее
3
Желтый
2,1-3,0
Повышенное
4
Зеленый
3,1-4,0
Высокое
5
Голубой
4,1-5,0
Очень высоюе
6
Синий
> 5,0
Таблица 27.
Классификация почв за содержанием гумуса по Тюрину.
Содержание гумуса
Класс
Цвет на картограмме
Показатель
Очень низюе
1
Красный
<2,0
Низюе
2
Оранжевый
2,1-4,0
Среднее
3
Желтый
4,1-6,0
Повышенное
4
Зеленый
6,1-8,0
Высоюе
5
Голубой
8,1-10,0
Очень высоюе
6
Синий
> 10,0
Таблица 28.
Группирование почв по гидролитической кислотности.
Степень кислотности
Класс
Цвет на картограмме
Гидролитическая кислотность
Очень сильнокислая
6
Фиолетовый
>6,0
Сильнокислая
5
Сиреневый
5,1-6,0
Кислая
4
Красный
4,1-5,0
Среднекислая
3
Розовый
3,1-4,0
Слабокислая
2
Оранжевый
2,1-3,0
Нейтральная
1
св.-оранжевый
<2,0
-235-
Таблица 29.
Классификация почв за содержанием подвижного фосфора*
Содержание
подвижного
фосфора
Класс
Цвет на
картограмме
Содержание подвижного фосфора в мг на кг почвы
по Кирсанову
по
Чириюву
по
Мачигину
по
Риму
мин.почвы
торфов ища
Оч. низюе
1
Красный
<25
0-50
<20
< 10
<50
Низюе
2
Оранжевый
26-50
50-100
21-50
1Ы5
51-70
Среднее
3
Желтый
51-100
100-200
51-100
16-30
71-140
Повышенное
4
Зеленый
101-150
200-400
101-150
31-45
>140
Высокое
5
Голубой
151-250
400-600
151-200
46-60
-
Оч.высоюе
6
Синий
>250
>600
>200
>60
-
Примечание: у торфовищах с кислой реакцией содержание подвижного фосфо¬
ра определяется по методу Кирсанова, ас щелочной - по методу Энгера-Рима.
Таблица 30.
Классификация почв за содержанием обменного калия.
Содержание
обменного
калия
Класс
Цвет на
картограмме
Содержание подвижного фосфора в мг на кг почвы
по Кирсанову
по
Чириюву
по
Мачигину
по
Эгнеру
Риму
мин.почвы
Т0рф0ВИ1Щ
Оч. низюе
1
расный
<40
0-80
<20
<50
-
Низюе
2
Оранжевый
41-80
80-160
21-40
51-100
<70
Среднее
3
Желтый
81-120
160-240
41-80
101-200
71-140
Повышенное
4
Зеленый
121-170
240-340
81-120
201-300
>140
Высоюе
5
Голубой
171-250
340-600
121-180
301-400
-
Оч.высоюе
6
Синий
>600
>180
>400
-
Таблица 31.
Классификация почв за содержанием легкогидролизованного азота
по Корнфнльду, в мг на кг почвы.
Содержание азота
Класс
Цвет на
картограмме
Показатель по
орнфнльду
Показатель по
Тюрину-Кононовой
Очень низюе
1
Красный
< 100
<30
Низюе
2
Оранжевый
101-150
31-40
Среднее
3
Желтый
151-200
41-50
Повышенное
4
Травяной
>200
51-70
Высоюе
5
Зеленый
71-100
Очень высоюе
6
Темно-зеленый
>100
-236-
Таблица 32.
Группировка почв по содержанию подвижных форм азота
(обобщенные данные) ДСТУ 4Э62.БЗ.
Группа
Степень
обеспеченности
Содержание N, мг на 1кг почвы
Нитрфикаци-
онная
способность
по Кравыову
Минеральный
азот (сумма
нитратного и
аммиачного)
гидролизованный.
гидролизован.
Тюрин и Кононова
Корнфильд
1
Очень низкая
<30
< 100
<5
< 10
2
Низкая
31-40
101-150
6-8
11-15
3
Средняя
41-50
151-200
9-15
16-24
4
Повышенная
51-70
>200
16-30
25-30
5
Высокая
71-100
31-60
31-35
6
Очень высокая
>100
>60
>35
Таблица 33.
Группировка почв по содержанию подвижного фосфора и обменного калия
(обобщенные данные) ДСТУ 4362 Б.4, Б.5.
Группа
Содержание в
почве
мг на 1 кг почвы
Кирсанов
Чириков
Мачигин
рл
к2°
к2о
РА
к2°
1
Низкое
<50
< 80
<50
<40
<15
< 100
2
Среднее
51-100
81-120
51-100
41-80
16-30
101-200
3
Повышенное
101-150
121-170
101-150
81-120
31-45
201-300
4
Высокое
151-250
171-250
151-200
121-180
46-60
301-400
5
Оч. высокое
>250
>250
>200
>180
>60
>400
Таблица 34.
Классификация почв за содержанием обменной серы.
Содержание серы
Класс
Цвет на картограмме
Показатель
Низкое
2
Оранжевый
<6,0
Среднее
3
Желтый
6,1-12,0
Высокое
5
Голубой
> 12,0
-237-
Таблица 35.
Определение содержания микроэлементов в почве и их градации.
Класс
Название микрорэлементов
В
Мо
Мп
Со
Zn
Си
обеспеченности
вытяжка
н,о
вытяжка
буфер рН-3,3
вытяжка
0,1 и H,S04
вытяжка 1н
HNO,
вытяжка 1н
КС1
вытяжка 1н
НС1
Очень низкий
<0,15
<0,05
< 15
<0,5
<0,3
<0,7
Низкий
0,16-0,22
0,051-0,07
16-20
0,51-0,70
0,31-0,50
0,71-1,00
Средний
0,23-0,33
0,071-0,100
21-30
0,71-1,00
0,51-0,70
1,01-1,50
Повышенный
0,34-0,50
0,101-0,150
31-45
1,01-1,50
0,71-1,00
1,51-2,20
Высокий
0,51-0,70
0,151-0,220
46-70
1,51-2,20
1,01-1,50
2,21-3,30
Очень высокий
0,71-1,00
0,221-0,330
71-100
2,21-3,30
1,51-2,20
3,31-5,00
Рис. - Доступность элементов минерального питания в зависимости от величи¬
ны обменной кислотности (рНКС1) .
-238-
Таблица 36.
Группирование почв за содержанием подвижных форм микроэлементов,
которые определяются в вытяжке ацетатно-аммонийного буферного раствора
(pH 4,8) мг/кг почвы.
Эле¬
мен¬
ты
гдк
Группы и градации почв за содержанием микроэлементов
1
2
3
4
5
6
7
8
Очень
низкий
Низкий
Средний
Повы¬
шенный.
Высокий
Оч.
высокий
Мп
0-63
<5
5-7
7-10
10-15
15-20
20-30
30-45
>45
Zn
0-8,7
<1
1-1,5
1,5-2,0
2-3
3-5
5-7
7-10
>10
Си
0-2,8
<0,1
0,1-0,15
0,16-0,2
0,2-0,3
0,3-0,5
0,5-0,7
0,7-1,0
>1,0
Со
0-1,8
<0,07
0,07-0,1
0,1-0,15
0,15-0,2
0,2-0,3
0,3-0,5
0,5-0,7
>0,7
Таблица 38.
Потребность сельскохозяйственных культур в микроэлементах
(по данным различных научных учреждений).
Культура
В
Си
Fe
Мп
Zn
Мо
Люцерна
Высокая
Высокая
Средняя
Средняя
Низкая
Высокая
Клевер
Средняя
Средняя
Средняя
Средняя
Низкая
-
Кукуруза
Средняя
Средняя
Средняя
Средняя
Высокая
Низкая
Сорго
Низкая
средняя
Высокая
Высокая
Высокая
-
Соя
Низкая
Низкая
Высокая
Высокая
Средняя
Высокая
Пшеница
Низкая
Высокая
Низкая
Высокая
Низкая
-
Ячмень
Низкая
Высокая
Низкая
Средняя
Низкая
Низкая
Горох
-
-
-
Высокая
-
Средняя
Подсолнечник
Высокая
Средняя
-
Средняя
Средняя
-
Сахарная свекла
Высокая
Средняя
Средняя
Высокая
Средняя
Средняя
Рапс
Высокая
Низкая
-
Высокая
Низкая
Высокая
Лен
Средняя
Высокая
-
-
Высокая
-
Томаты
Средняя
Средняя
Средняя
Средняя
Средняя
-
Огурцы
-
Средняя
-
Высокая
-
-
Лук репчатый
Высокая
Средняя
Высокая
Средняя
Средняя
Высокая
Капуста белокачан.
Высокая
Средняя
-
Высокая
Средняя
Высокая
Капуста цветная
Высокая
Средняя
-
Средняя
Низкая
Высокая
Морковь
Высокая
Высокая
-
Высокая
-
-
Картофель
Средняя
-
-
Средняя
Средняя
-
Винотрад
Высокая
Средняя
Высокая
Высокая
Высокая
-
Яблоня
Высокая
Средняя
Высокая
Высокая
Высокая
-
-239-
Таблица 39.
Оптимальные интервалы величин обменной кислотности ночвы
(рНсодеме) для развития основных сельскохозяйственных культур.
Культура
Величина рН._,
Культура
Величина pH. ,
Озимая пшеница
6,3-7,5
Томаты
6,3-7,0
Ярая пшеница
6,0-7,5
Морковь
5,6-7,0
Жито
5,0-7,7
Столовая свекла
7,0-7,5
Кукуруза
6,0-7,5
Лук
6,4-7,9
Ячмень
6,0-7,5
Редис
5,8-7,3
Овес
5,0-7,5
Шпинат
6,0-7,5
Просо
5,5-7,5
Салат
6,0-7,0
Гречка
4,7-7,5
Конопля
7,1-7,4
Репак
5,8-7,0
Мак
6,8-7,2
Рис
4,0-6,0
Сахарная свекла
7,0-7,5
Подсолнух
6,0-7,0
Картофель
5,0-6,0
Горох
6,0-7,0
Смородина
5,5-6,9
Фасоль
6,4-7,1
Крыжовник
4,5-4,8
Вика
5,7-6,5
Малина
5,8-6,5
Люцерна
7,0-8,0
Клубника
5,0-6,0
Люпин
4,6-6,0
Виноград
6,0-7,5
Соя
6,0-7,5
Яблоня
6,5-7,2
Конюшина
6,0-7,0
Груша
5,8-7,2
Тимофеевка
4,5-7,6
Вишня
6,7-7,2
Лен
5,5-6,5
Персик
6,0-7,5
Капуста
7,0-7,4
Абрикос
6,0-6,7
Огурцы
6,4-7,5
-240-
Таблица 40.
Основные методические характеристики атомно-абсорбционного анализа
(М.Э.Брицке).
Элемент
Длина
вошы
аналитичес¬
кой /ЦНИИ,
нм
Тип
пламени
Характеристи¬
ческая
концентрация,
мкг/мл
Диапазон
оптима/ъных
содержаний,
мкг/мл
Предела обнаружения
двухлучевой
прибор
однолучевой
прибор
ЭЛЕМЕНТЫ 1 ГРУППЫ
Щелочные металлы
Литий
670,78
Ацетилен -
воздух
0,04
0,5-5,0
0,0006
0,005
323,26
9,4
Натрий
589,00
Ацетилен -
воздух
0,015
0,3-3,0
0,002
0,002
589,59
330,23
-II-
330,30
-II-
2,8
Калий
769,90
-//-
0,12
2,0-20
766,49
Пропан -
воздух
0,05
1,0-10
0,005
0,05
404,72
Водород-
воздух
25,0
404,41
Рубцдий
794,76
-0-
0,4
4,0-40
780,02
0,05
2С20
0,005
0,1
421,56
47,0
420,18
24,0
Цезий
852,11
-II-
0,5
8С80
0,05
0,1
455,54
-//-
42,0
Подгруппа меди
Медь
327,4
Ацетилен -
воздух
0,2
324,75
0,1
2С20
0,002
0,01
249,22
7,2
222,57
1,5
216,51
0,6
Серебро
338,29
Ацетилен -
воздух
0,11
328,07
0,06
2С20
0,002
0,01
Золото
267,6
-II-
0,36
242,8
-II-
0,20
2С20
0,02
0,05
ЭЛЕМЕНТЫ II ГРУППЫ
Берилтй, магний и щвлочноземегъные металлы
Берилшй
234,86
Ацетилен -
воздух
0,03
0,5-5
0,001
0,01
-241 -
Продолжение табл. 40
Элемекг
Длина
волны
аналитичес¬
кой линии,
нм
Тип пламени
Характеристи-
ческая
концентрация,
мкг/мл
Диапазон
огттима/ъных
содержаний,
мкг/мл
Предегы обнаружения
двухлучевой
прибор
однолучевой
прибор
Магний
285,21
-II-
0,007
0,1-2
0,0001
0,001
202,58
-II-
0,17
Ка/ъций
422,67
-II-
0,07
1С10
0,001
0,01
Ацетилен -
дшитроксид
Стронций
460,73
Ацетилен -
воздух
0,15
2С20
0,01
0,03
Ацетилен -
динитроксцд
Барий
553,55
Ацетилен-
динитроксид
0,3
5-100
0,02
0,08
350,11
5,0
Подгруппа цинка
Цинк
213,86
Ацетилен -
воздух
0,0150
0,2-3
0,002
0,02
Кадмий
228,8
-II-
0,25
0,5-5
0,001
0,01
Ртуть
253,65
Беспламен-
ная
атомизация
-
ЭЛЕМЕНТЫ III ГРУППЫ
Подгруппа скандия и лантаноиды
402,37
Ацетилен -
ДИНИфОКСИД
0,9
402,04
1,2
391,18
0,65
10-100
0,1
0,5
390,75
0,65
Иттрий
414,29
Ацетилен -
динитроксвд
2,8
412,83
2,4
410,24
2,0
50-200
0,3
0,7
407,74
2,2
Европий
459,4
-II-
0,6
5-100
0,04
0,1
Диспро¬
зий
421,17
-II-
0,8
10-200
0,2
0,7
419,49
0,9
418,68
1,0
404,6
1,3
-242-
Продолжение табл.40
Элемент
Д/ина
ВОЛНЫ
аналитичес¬
кой /ЦНИИ,
нм
Тип пламени
Характеристи-
ческая
концентрация,
мкг/мл
Диапазон
оптима/ъных
содержаний,
мкг/мл
Пределы обнаружения
двухлучевой
прибор
однолучевой
прибор
Го/ъмий
416,3
-//-
2,6
20-200
0,1
0,5
410,38
1,5
405,39
2,0
Эрбий
415,11
Ацетилен-
ДИНИфОКСИД
2,4
400,8
0,9
10-200
0,1
0,5
389,27
4,5
386,28
2,4
Туллий
410,58
-II-
1,0
20-200
409,42
1,3
374,41
1,2
371,79
0,7
20-200
0,2
0,6
Иттербий
398,8
-II-
0,2
1С20
0,04
0,1
346,44
Ацетилен-
воздух
0,7
246,45
0,9
Аломиний и элементы подгруппы галгия
Аломи-
ний
396,15
Ацетиле н-
динитроксед
1,6
10-150
0,03
0,2
309,27
1,0
309,28
Галлий
294,42
Ацетилен-
воздух
294,36
287,42
2,5
20-200
0,1
0,5
Индий
451,13
-II-
2,7
410,48
2,6
325,61
0,9
303,94
0,9
10С50
0,05
0,3
Талгий
377,57
-II-
1,4
276,79
0,5
5-100
0,03
0,2
ЭЛЕМЕНТЫ IV, V И VI ГРУПП
Кремний
251,61
Ацетилен-
ДИНИфОКСИД
2,0
20-200
0,08
0,3
Титан
365,35
-II-
2,0
20-200
0,09
0,4
364,27
2,2
-243-
Продолжение табл.40
Элемент
Дтна
во/иы
аналитичес¬
кой ЛИНИ4,
НМ
Тип пламени
Характеристи¬
ческая
концентрация,
мкг/мл
Диапазон
оптимальных
содержаний,
мкг/мл
Предел»! обнаружения
двухлучевой
прибор
однолучевой
прибор
Олово
224,60
Водород-
воздух
3,0
20-200
0,02
0,3
Ацетилен-
воздух
Свинец
283,31
Ацетилен-
воздух
0,5
4-40
0,02
0,07
216,99
0,2
Ванадий
318,31
Ацетилен-
динитроксод
1,7
10-100
0,06
0,2
318,40
318,54
Сурьма
217,58
Ацетилен-
воздух
0,5
10-100
0,61
0,5
231,15
0,9
Висмут
222,82
0,9
10-100
223,06
0,4
10-100
0,04
0,1
Хром
357,87
Ацетилен-
воздух
0,1
1-20
0,003
0,03
359,35
0,2
Молибден
313,26
Ацетилен-
динитроксцд
0,5
10-100
0,03
0,12
ЭЛЕМЕНТЫ VIIИ VIII ГРУПП
Марганец и элементы семейства железа
Марганец
279,48
Ацетилен-
воздух
0,05
1-10
0,002
0,01
279,83
0,07
280,11
0,10
Железо
248)32
-//-
0,10
2-20
0,01
0,03
248,82
0,17
252,74
0,46
302,06
0,37
302,11
Кобал>т
240,72
-//-
0,15
241,16
0,27
242,49
0,18
243,58
0,44
252,14
0,30
Никель
232,00
0,10
2-20
0,01
0,03
341,48
0,35
352,45
0,33
-244-
Продолжение табл.40
Элемент
Д^на
воты
аналтнес-
кой линии,
нм
Тип пламени
Характеристи¬
ческая
концентрация,
мкг/мл
Диапазон
оптимагъных
содержаний,
мкг/мл
Пределы обнаружения
двухлучевой
прибор
однолучевой
прибор
Платиновые металлы
Рутений
249,89
Ацетилен-
воздух
0,5
5-50
0,3
-
Родий
343,49
-//-
0,5
4-40
0,03
0,3
Палладий
244,79
-//-
0,2
2-20
0,02
0,2
Платина
265,94
-//-
2,5
20-200
0,1
-
Атомно-абсорбционный спеюрохимический анализ М. Э. Брицке М. «Химия» 1982
-245-
ОЧИСТКА ДИЭТИЛОВОГО (СЕРНОГО) ЭФИРА
Серный эфир - (С2Н5 - О - С2Н5), молекулярный вес 74,08, температура кипения
35°, плотность 0,714. Эфир чрезвычайно летуч и легко воспламеняется. Распростра¬
няясь в воздухе, пары его дают опасные, взрывчатые, гремучие смеси, поэтому он
является веществом чрезвычайно огнеопасным.
Использованный серный эфир бывает загрязнен главным образом органически¬
ми веществами и содержит воду (100г серного эфира могут поглотить 7,5 г воды).
При длительном стоянии продажный эфир содержит перекиси, которые опасны при
перегонке. Для очистки эфир перегоняют с дефлегматором. Перед перегонкой его
обязательно промывают 5%-ным раствором железного купороса. Промывание с целью
удаления спирта, ацетона и разрушения перекисных соединений в эфире проводят
(0,2 объема раствора от объема эфира) 2-3 раза, подкисляя серной кислотой до слабо
кислой реакции и добавляя железный купорос, чтобы разрушить перекиси, которые
могут образоваться в неочищенном эвире и при перегонках могут вызвать взрыв. Эфир,
содержащий перекиси, обладает резким запахом. На каждый куб. дециметр этилового
эфира берут 10 г кристаллического железного купороса. Затем отгоняют, собирая
фракции, кипящие при 33-37°; остальные фракции собирают отдельно и употребляют
для дальнейшего фракционирования.
Фракции эфира, кипящие при 33-37°, обезвоживают гранулированным свежепро-
сушенным при 100-105° или прокаленным при 725° плавленным хлористым кальци¬
ем, для чего в склянку на 5-7 дм3 наливают 3-4 дм4 перегнанного эфира и добавляют
хлористый кальций. Гранулированный хлористый кальций берут в количестве 1/5
объема от взятого для просушки эфира, а плавленный - в меньшем количестве.
Склянку с эфиром и хлористым кальцием оставляют на ночь после чего произво¬
дят отгонку с дефлегматором. Собирают фракции эфира, кипящие при 34-36°, приемник
плотно соединяют с аллонжем холодильника и снабжают трубкой, наполненной
хлористым кальцием, для устранения поглощения эфиром воды из воздуха.
Примесь спирта, содержащегося в эфире, может быть удалена одним из следую¬
щих двух способов.
1. Эфир многократно промывают небольшими порциями воды. Этот способ
довольно легко и быстро приводит к цели, но недостатком его является значительная
потеря эфира вследствие его заметной растворимости в воде.
2. Спирт в эфире окисляют до уксусного альдегида или уксусной кислоты, кото¬
рые затем удаляют в виде альдегидной смолы или уксуснокислого натрия. В склянку
с эфиром прибавляют небольшое количество тщательно растертого в порошок мар¬
ганцовокислого калия и 1-2 куска (Юг) едкого натра. Через несколько часов поверх¬
ность кусков едкого натра покрывается бурым осадком альдегидной смолы. Опера¬
цию эту повторяют до тех пор, пока через 12 часов больше не будет наблюдаться
выделение окрашенной смолы на едком натре. Эфир сливают в чистую сухую колбу,
сушат над хлористым кальцием в течение 24 часов и перегоняют с дефлегматором.
Для получения абсолютного эфира обезвоженный эфир сушат несколько дней над
свежеплавленным хлористым кальцием, которого берут 15-20 % от веса жидкости,
после чего быстро отфильтровывают через складчатый фильтр в сухую склянку, куда
кладут затем натрий в виде проволоки или небольших кусочков, очищенных от окиси.
-246-
Пока продолжается выделение водорода, склянку закрывают пробкой со вставленной
в нее хлор кальциевой трубкой, на которую для уменьшения испарения надевают
короткую, вытянутую в капилляр стеклянную трубку.
-247-
Агрохимические методы исследования почв. - М., Наука, 1975.
Азот в земледелии нечерноземной полосы . - Л., Колос, 1973.
Азот в почвообразовании и земледелии. Труды X Международного конгресса
почвоведов. Т.9. - М., 1974.
Аринушкина Е.В. Руководство по химическому анализу почв. - Изд-во Москов¬
ского университета, 1970.
Агрох1м1чний ан&шз. /За ред.. М.М. Городнього / К. 2005.
Баланс азота удобрений в дерновоподзолистых почвах (по данным опытов с J5N).
- Бюллетень ВИУА, 1976, №25.
Варюшкина Н.М., Мельникова М.Н. Методические указания по проведению
исследований с изотопом азота 15N и определению элементов питания лизиметри¬
ческих водах. М., 1978.
Городний Н.М., Мельничук С.Д. и др. Практическая биохимия и управление
качеством продукции растениеводства. Киев, «Аристей», 2006.
Инструкция по химико-техническому контролю и учету сахарного производства.
- Киев, 1970.
Инструкция для лабораторий Государственной агрохимической службы по ана¬
лизам кормов. - М., ЦИНАО, 1978.
Инструкция по эксплуатации прибора для определения аммонийного азота (при¬
бор Сереньева). - М., 1978.
Лисовал А.П. и др. Агрохимия, Киев, 1984.
Методические указания по анализу почв, кормов, растений и удобрений. - М.,
ЦИНАО, 1976.
Методические указания по колориметрическому определению подвижных форм
микроэлементов в почвах. - М., ЦИНАО, 1977.
Методические указания по атомно-абсорбционному определению микроэлемен¬
тов в вытяжках из почв и в растворах золы, кормов и растений. М., ЦИНАО, 1977.
Методические указания по определению технологических качеств сахарной свек¬
лы. Киев, ВНИС, ВНИИСП, 1978.
Методические указания по проведению аналитических исследований. - Киев,
ВНИС, 1979.
Методические указания по организации контроля качества растительных и по¬
чвенных анализов в системе ВНИС. - Киев, ВНИС, 1982.
Методические указания по определению тяжелых металлов в почвах и продук¬
ции растениеводства. - М., 1989.
«Методика сплошного почвенно-агрохимического мониторинга сельскохозяй¬
ственных угодий Украины» К. 1994 г.
Методические указания «Оценка селекционного материала рапса и сурепицы на
содержание эруковой кислоты и глюкозиколатов». М. 1988 г.
Методы биохимического исследования растений под ред. А.И.Ермакова - Л., «Ко¬
лос», 1972.
Методика агрох1м1чного обстеження тепличних грунта i субстрата та особли-
BocTi застосування добрив. К. 2005.
-248-
Методические указания по проведению исследований н длительных опытах с
удобрениями. Сб. ВИУА, ч.Э. Анализ растений, М., 1985.
Методика определения состава и свойств почв. Сб. Кн.1., Харьков, 2004. УААН.
Национальный научный центр «Институт почвоведения и агрохимии им. Соколовс¬
кого».
Нацюнальний стандарт Укражи. Насшня Ыльськогосподарських культур. Методи
виэначення якостг ДСТУ 4138-2002. К. 2003.
Попович Л.П. Ф1эико-х1м1чш дослшження продукци i рослинництва. К., 1993.
Починок Х.Н. Методы биохимического анализа растений. 1C, «Наумова думка», 1976.
Система методов исследования в почвоведении. Изд.„Наука", Новосибирск, 1971.
Современные методы химического анализа почв и растений (методические ука¬
зания). К. 1984.
Силин П.М., Силина Н.П. Химический контроль свеклосахарного производства.
М., «Пищепромиэдат», 1960.
ОПЕЧАТКИ
Страница
, Пункт, строка, абзац, квадрат тех
карты, графа таблицы, формула
Напечатано
Должно быть
5
24 строка сверху
атомноабсорбционным
атомно-абсорбционным
16
в п 2 16 6,9 сверху
J
Уа
19
в п 2 3 3,2 сверху
FeC13 • 6Н20
FeCl3 • 6Н20
24
в п. 2 4 2 1 ,4 сверху
4 абзац, 6 строка сверху ‘
Н7[Р(Мо207)б]П-Н20
МоОз
НтРЧМо^Мп • Н20
МоОз
39
в п 2 5 1 4 2 абзац, 4 строка сверху
j или S
% ИЛИ */з
43
в п 2 5i3 3 ,11 строка сверху
табл 13,4сгрока
2г б-нафгиламина
N-N03
2г a-нафгиламина - • *
N-N03 |
44
в п 2 5 3 4,5 абзац, 3 строка
а-нафтиламина
а-нафтиламина ’ ;
49
в п 2 6 2 3,1строка
5 строка
плотность 1,19 г/мл
хч HN03
плотность 1,19 t/cmj -
х ч HN03, плотность 1 ,40Wcmj ’
59
в п 2 8 3,1 строка
5 строка сверху
Р.(СНзС00Н)2-ЗН20
РвО
1,5-1,7
PbfCHjCOOH), • ЗН20 ' '
РЮ
1, 5-1/7Г/СМ3
69
в п 2 10 5 1, строка 16 снизу
опыляются
омыляются
78
в п 2.13.2.4 ,14 строка
30%-ного
20%-ного *"
82
в п 2 13 3 14, 4 строка сверху
8 строка сверху
до 1 мл
надсерной кислотой
до 1 л р
над серной кислотой*
118
в п 3 8 22,4 строка снизу
окрашивающий раство
окрашивающий раствор -
135
«в п 3 114 2,15 строка снизу
раствор П с раствором I
раствор I с растворомгП,
139
в п 3 13 3 ,1 строка
СНЗСООЫа. 136г
CH3COONa -ЗН20
CHjCOONcl 136г * ■
Cft3C00Na-3H20
141
в п 3 14 2 2,4сгрока сверху
HCL
НС1
142
в п 3 14 2 5 , квадрат 3
взбалтыватель
встряхиватель
147
в п 3 15 3,9сгрока сверху
100м л 2
100мл
151
впЗ 1632
0lMr/CMJ
0,1мг/см^
152
в п 3 16 4, 1,2 строки сверху
вытяжек, приготовленных
вытяжек, приготовленных?*
(смпзвгз) 14
154
в п 3 17 6,2 квадрат
добавление 1 н КС!
добавление 0,1 н НС)
161
впЗ 1943 п 8
для определения
S04 *0,118г K2S04
для определения SO 4 0,181 г
K2S04
172
в п 3.21 1 последняя сгрока
вп 55
вп 26
174
в п 3 216, рамка 2
MnS04
MgS04
189
вп 3 24 6 4 , 4 строка
в термостате
в термостате при t=30°C
191
в п 3 24 7 4,2 строка снизу
ОДИН КЮз
0,01н KJO3
192,193
В п.З 24.8 3 , 3 24 8 5
no2
N03
220
табл,графа 3 п 5
п 6
строка 4,5 сверху п 7
строка 8,9 сверху п 7
MgS04, Na2S04
KMn04
С остав
С триж ижевского
MgS04, Na2S04
KMn04
Состав
Стрижижевского
221
табл 1, графа 4,5
Селеп, So
Селен, Se
227-228
табл 10, строка 2, графа 2
(+ продолжение)
при 200С
при 20,0°С
231
табл 15, строка7
Ai=Cj -С2
Aj=Ci — С
232
табл 19, графа 4, строка 2 снизу
флуорес-п ирующая
флуоресцирующая
233-239
В названиях таблиц
21,22,23,25,26,27,29,30,31,34,36
за (величиной,
содержанием, суммой)
по (величине, содержанию,
сумме)
236
табл 29, графа 5
графа 8
в примечании под таблицей
табл 30, графа 3
табл 31, графа 4
торфовища
по Риму
у торфовищах
расный
Показатель по орнфильду
торфяники
по Эгнеру-Риму
в торфяниках
Красный
Показатель по Корнфильду
237
табл 32, графа 3
графа 4
Тюрин и Кононова
Корнфильд
по Тюрину- Коновой
по Корнфильду
240
табл 39, 1 колонка
ярая пшеница, жито, гречка
репак, подсолнух,
конюшина
яровая пшеница, рожь, гречиха
рапс, подсолнечник
клевер
248
литература, строка 11
питания лизиметрических
питания в лизиметрических
238
Рис Доступность элементов
читать „Таблица 37" Рис
Доступность элементов
Научно-практическое издание
КОВАЛЬЧУК Владимир Павлович
ВАСИЛЬЕВ Валерий Гаврилович
БОЙКО Леонид Васильевич
ЗОСИМОВ Владимир Дмитриевич
СБОРНИК
МЕТОДОВ ИССЛЕДОВАНИЯ
ПОЧВ И РАСТЕНИЙ
Ответственный за выпуск - доктор сельскохозяйственных наук,
академик Н.В. РОИК
Орнпнал-макет: подготовлено в ТзОВ
«ТРУД-ГриПол»
Дизайн, верстка: К.О. Ягольник
Пщп. до друку 16.03.2010. Формат 60x84 1/16. Пап1р офсет. №1. Офс. друк.
Гарн. Times New Roman.
Наклад 300 прим. Замовлення №41
Друк: ТОВ "Лазурит-Полнраф",
01042, м. Киш, вул. Леваневського, 8/7
1нстнтут цукрових буряк!в НААНУ, 03141, м. КиЬ-141, вул Клтчна, 25
тел. 275-50-00, 270-44-19.
Х1М1ЧНИХ ЕЛЕМЕНТ1В Д.1.
1ЕР10ДРВД
ii
ш
IV
I
У
п
I/I
I
11
Hydrog
ВОДЕ
I
1, 00794
ii
lemum
ВОДЕНЬ
Li
LlMnurn
л I ri й
3
$,&41
Natrium
HATPiri
5
К
Kiilliin
К А Л I
19
39,0963
b
63,546
С u
Cuprum
Mi дь
%7
65,467®
Rubldltifi
ЛдТй
4
9,0121$
iU
ВегШгит
ВЕРИЛ 1И
12
24,305
Mu
Macineslum
МАГМ 1 Й
20
4 5,078
С а
Calcium. ^
кдльц1й
III
IV
Borum
BOP
5
10.S1 f
\i
13
26,90 f 54
Aluminium
a jfi ю м IИIЙ
21
44,95691
Sc
Scandium,
£КлпД|Й
30
В5лЗЙ
Zwkum
циИК
С а
Gallium
ГАЛ1Й
31
69,723
Sr
38
87,*2
39
8fi,goss
atr&ntium _
yjinum
6
12,0H
Cerboneum
ByГЛЕЦЬ
Si
Sllicium
КРЕМН1Й
14
28,0855
22
47.88
I !
1 itomutti
ТИТАН
32
72.59
(. с
Germanium
ГЕРМАН(й
4«
91,224
Zirfcofuufi
V
N
rjiii
A30
7
14t0O67
iNiiro^emum
V
15
30,97376
foosphofum
ФОСФОР
M
SO, 9415
Vanadium
ВАНД1И
As
33
Arsemcurri
МйШ'ЯК
41
№,8064
Nh
VI
О
Q*ygenium
КИСЧИЬ
*
15.994
U>
o 32,066
Sulfur
CiPKA
24
51,9961
( r
Chromium
XPOWl
Se
34
78,96
Seiemum
СЕЛЕН
94
ШЬс1ап1«)1п
Л I БД £ Hi
VII
(II)
О
18,993403
Ftorum
ФТОР
Cl
17
35,453
СЫогцт
ХЛОР
25
54,9380 y|
M&rg&num
МАРГАИЕЦЬ
vr«
He
Г£Я(Й
2
4.002602
10
20л179
Neon
НЕОН
A i
Argon
АРГОИ
IK
39,948
26
55,047
Fe
Ferry rn
ЗАЛ 13 О
Br
.9^4
бготшп
iVe)
127, ere
tellurium
теллур
\v
Wqltrm\hm
ВОЛЬФРАМ
84
20tf|
Polot* чип
ПОЛОЙЙ
I Ti
Technetium
TEXHЕ4*л
Kr
Kryplcrt
КРИПТОН
36
аз.во
•44
10 no?
flut
1Ы
i
fodum
ЙСД
53
126:9045
7^
106,207
И
ftfceruif
PEH
4
m
At
85
1210J
A Galium
АСТАТ
!07
106
12631 IJnh;
UnnflosKviufP j '^n
У*та Л гёкС« W У ннШс <Ш Vi Й
lins
RO
R207
HR
Xe
S4
131.2»
Xenon
ксенон
76
190,2
I •
!Ca
символ
елемента
поряд ковой I
20# •J HD^ei
40,076
I 8&i« —
4EZ—
назва
елемен та
атоадна
~ маса
с:о
Cobattuitt
кегдлы
2К
од.69
Ni
Ni&COtUm
н г к аль
j - s-елементи
- рчзлемент!
•I*
102,«Э&$| *
к»
RLc
№
и
46
106,42
I’d
Pa'ieon
ПДЛАД
- d-елементи
- f-елементи
Rn
RaOOn
РАДОН
Osmium
ОСМ1Я
86
[222]
I (Ж
,26&J Шо
11
1^2,2::
Ir
iddiiiir.
1р|Д1й
7«
iO'j 08 , >
f
Platinum
ПЛн.ИИА
tt*9
12661 Une
L'rv iH< >* tiU П • [ ШЙиДОкгт
jftmiffOK?)й[ ШнЛКшй
Dun
Unnunnthun'
УНМУ*»п1/||и|
RO
190
Thrjriym
to pm
91
am p.|
n
гшьш и
ЪРАШ
4J ^
№7) I\p
8$.№Я
P"51 Am
ШШ'РМЩШ
06
mr' Cm
Curium
«0?»
97
t№) Rk
&ЕРКЛШ
•>h
{feu
кдМвт
um
Г” Fm
i P'OtfoiWt}
[ ФЕРМ1.Й
■ip;
r^' Md
ытмептЫ
102
Ябв,*0Й9^^
яомлдо
WJ« £ ^
лоуртш
M
1244 j
Pll
платой!
Nil
f2«3M Jg
ЁМШТЕЙИ
»*
.