/






















































































Author: Негина В.Р. Козырева Э.А.
Tags: неорганическая химия ядерная техника ракетная техника
Year: 1978




Text
АНАЛИЗ БОРА, ЕГО СОЕДИНЕНИЙ И ПРЕССКОМПОЗИЦИЙ (ПРАКТИЧЕСКОЕ РУКОВОДСТВО)
Под общей редакцией кандидата технических наук В. Р. Негиной
Москва • Атомиздат • 1978
УДК 546.27
Анализ бора, его соединений и пресскомпози-ций (практическое руководство). М., Атомиздат, 1978, с. 92 . Авт.: Негина B.F., Козырева Э.А., Дегтярева О.Ф. и др.
Изложены методики анализа бора, борида титана и пресскомпозиций на основе бора или карбида бора с полипропиленом. Описаны методики переведения вещества в раствор сплавлением с пер- (пиро)-сульфатом калия или кислотным разложением, определения основного вещества (бора, титана, углерода, водорода), примесей (кислорода, азота, углерода, фтора, хлора, серы) и 22-32 металлических примесей с применением тит-риметрических, элементорганических, потенциометрических, спектральных, пламенноспектрометрических и других методов анализа.
Рис. 17. Табл. 9. Список литературы 134 наименования.
А
20502-111
034(01)-78
-111-78
Атомиздат*
1978
ВВЕДЕНИЕ
Бор и его соединения в настоящее время находят все большее применение в различных областях науки, техники, промышленности.
В основном бор применяют в металлургии для синтеза жароупорных и химически стойких боридов, а также в качестве легирующей добавки в различных коррсзионно- и жаростойких сплавах.
Возрастает применение бора и его сплавов в ядерной энергетике в качестве замедлителя ядерных процессов и при изготовлении экранов для поглощения нейтронов. В данном случае большое сечение захвата нейтронов бором хорошо сочетается с его высокой термо- и химической стойкостью.
Кроме того, бор и его соединения применяют в пиротехнике, ракетной технике, для борирования деталей и изготовления керметов. Перспективно применение бора в полупроводниковой технике.
физические и механические свойства бора, которые обусловливают его использование, зависят от химической чистоты, кристаллической структуры, что определяется различными условиями технологического изготовления, качеством исходных компонентов.
Промышленность предъявляет высокие требования к качеству бора и его тугоплавких соединений. Поэтому введен контроль по содержанию основного вещества и примесей.
В данном руководстве описаны методики анализа кристаллического бора, карбида бора, борида титана, пресскомпози— ций, составленных на их основе с полимерными материалами.
Эти методики разработаны вновь или усовершенствованы в соответствии с предъявляемыми требованиями.
Для переведения в раствор бора и его тугоплавких соединений предложено сплавление их с К28208 и NaHSO4
3
(взамен сплавления с содой или спекания с окислами кальция и бария). Этот метод имеет много преимуществ: исключает использование платиновой посуды, допускает сплавление образцов пробы массой до 50-100 мг, образующийся прозрачный плав облегчает контроль за полнотой сплавления и т.д.
Для определения нитридного азота предложен прибор, позволяющий переводить пробы в раствор сплавлением со щелочами в замкнутой системе с дальнейшим определением его по методу Кьельдаля.
Для определения фтора предложено переводить пробу в раствор сплавлением с содой, отделяя большое количество натрия осаждением на смоле КУ-2 в статических условиях.
Для определения углерода, водорода, примесей серы й хлора в пресскомпозициях для разрушения навески использованы методы сожжения в кислороде. Для определения углерода и водорода подобраны низкотемпературный плавень ^2°5 и Режим сожжения. Для определения серы и хлора - сожжение в быстром токе кислорода (до ~ 2 л/мин) с улавливанием летучих примесей в поглотительном растворе.
Разработан прямой метод спектрального определения 32 элементов в карбиде бора и боре . с чувствительностью 3 • _ 1*10*"2% и 29 элементов в пресскомпозициях с
чувствительностью 1 • 10~4 - 3 •10’"^%. Методом фракционной дистилляции определены 22 примеси с чувствительностью 1- 10’^ - 5*10~3% в бориде титана.
Г Л А В A 1
ОПРЕДЕЛЕНИЕ ОСНОВНОГО ВЕЩЕСТВА
В КРИСТАЛЛИЧЕСКОМ БОРЕ, ЕГО ТУГОПЛАВКИХ СОЕДИНЕНИЯХ И ПРЕССКОМПОЗИЦИЯХ
1.1. ПЕРЕВЕДЕНИЕ ВЕЩЕСТВА В РАСТВОР
Кристаллический бор представляет собой порошок черного цвета с металлическим блеском. физико-химические свойства бора в значительной степени зависят от его чистоты, степени кристалличности и дисперсности. Так, кристаллический бор от аморфного отличается большей реакционной устойчивостью даже при сравнительно высокой температуре.
Кристаллический бор стоек к воздействию кислот, а перекись водорода, персульфат аммония и перманганат калия медленно окисляют его. Обычно для переведения в раствор кристаллического бора применяют метод сплавления с карбонатами щелочных металлов (КЫаСО3, Ма2СОз) или с пер— (пиро)-сульфатом калия(к2^0£,|Т,2,3| • Сплавление проводят в платиновых тигля; . .хк-орые при этом значительно изнашиваются, особенно из-за примесей в анализируемом веществе. Сплавление с пер-(пиро) -сульфатом проводят в кварцевой колбе с обратным холодильником.
Наиболее устойчивой фазой в системе Ti-В является т;вх[46], Соляная и плавиковая кислоты на борид титана не действуют, серная кислота медленно реагирует с боридом титана только при нагревании. Борид титана ‘растворяют в
смеси азотной кислоты и перекиси водорода или серной и
азотной кислот, а также разлагают расплавленными щелочами, карбонатами и бисульфатами щелочных металлов, РЬ202> Na2O2. Для разложения борида титана используют также метод спекания с СаО, МдО и ВаСО3 [ 4-6] . Универсальный метод переведения в раствор боридов разработан Блюменталем [7]. Метод основан на сплавлении в платиновом тигле боридов с 10-кратным избытком карбонатов натрия и калия с добавкой 0,1-0,2 г NaN03[8]. Бориды титана, циркония и
5
железа могут быть переведены в раствор также сплавлением с бисульфатом калия в кварцевом тигле. После переведения боридов в раствор металлы отделяют осаждением щелочью, содой или Ва(0Н)2*
о Карбидам бора приписывают различный химический состав, начиная с ВС до В^С- Основным соединением системы В-С считают В. С илиBJL,Соединение В4С имеет большую стойкость к агрессивным средам, на него не действуют концентрированные и разбавленные кислоты и их смеси как на холоде, так и при нагревании [9,]. Смеси серной и азотной, плавиковой и азотной кислот действуют очень слабо [10-13]. Карбид бора стоек также к воздействию разбавленных и концентрированных растворов щелочей, их смесей с окислителями. Однако отношение карбида бора к окислителям зависит от его фазового состава. Поэтому универсальными методами переведения В4С в раствор являются метод сплавления и метод спекания с карбонатами щелочных металлов при 800-900°С [14-17]. .Сплавление или спекание проводят в железных или никелевых тиглях в присутствии перекиси натрия или карбоната бария [6-13]. Однако при этом возможно загрязнение анализируемой пробы веществом тигля. При спекании с карбонатом бария металлические примеси при последующем выщелачивании выпадают в осадок, с которым может теряться некоторое количество борной кислоты..
Данные по переведению в раствор пресскомпозиций на основе кристаллического бора, карбида бора и полимерных материалов в литературе отсутствуют.
В работе [18| было предложено сплавлять кристаллический бор, его тугоплавкие соединения и пресскомпозиции в муфельной печи в кварцевом тигле с пер- (пиро) -сульфатом калия при 700°С. При этом не происходит потерь бора и анализируемые вещества не загрязняются. Растворение борида титана и бора может быть проведено в окислительной смеси состава серная кислота + азотная кислота + пиросульфат калия при нагревании. При сплавлении с пер- (пиро)-сульфатом калия анализ проводится в два раза быстрее по сравнению со сплавлением с KNaCO3. Сплавление с персульфатом калия протекает значительно быстрее (15-30 мин) если к нему добавить гидросульфат натрия в отношении 4:1. При сплавлении образуется прозрачный жидкий плав, позволяющий визуально определить полноту сплавления [134].
6
Методики переведения вещества в раствор
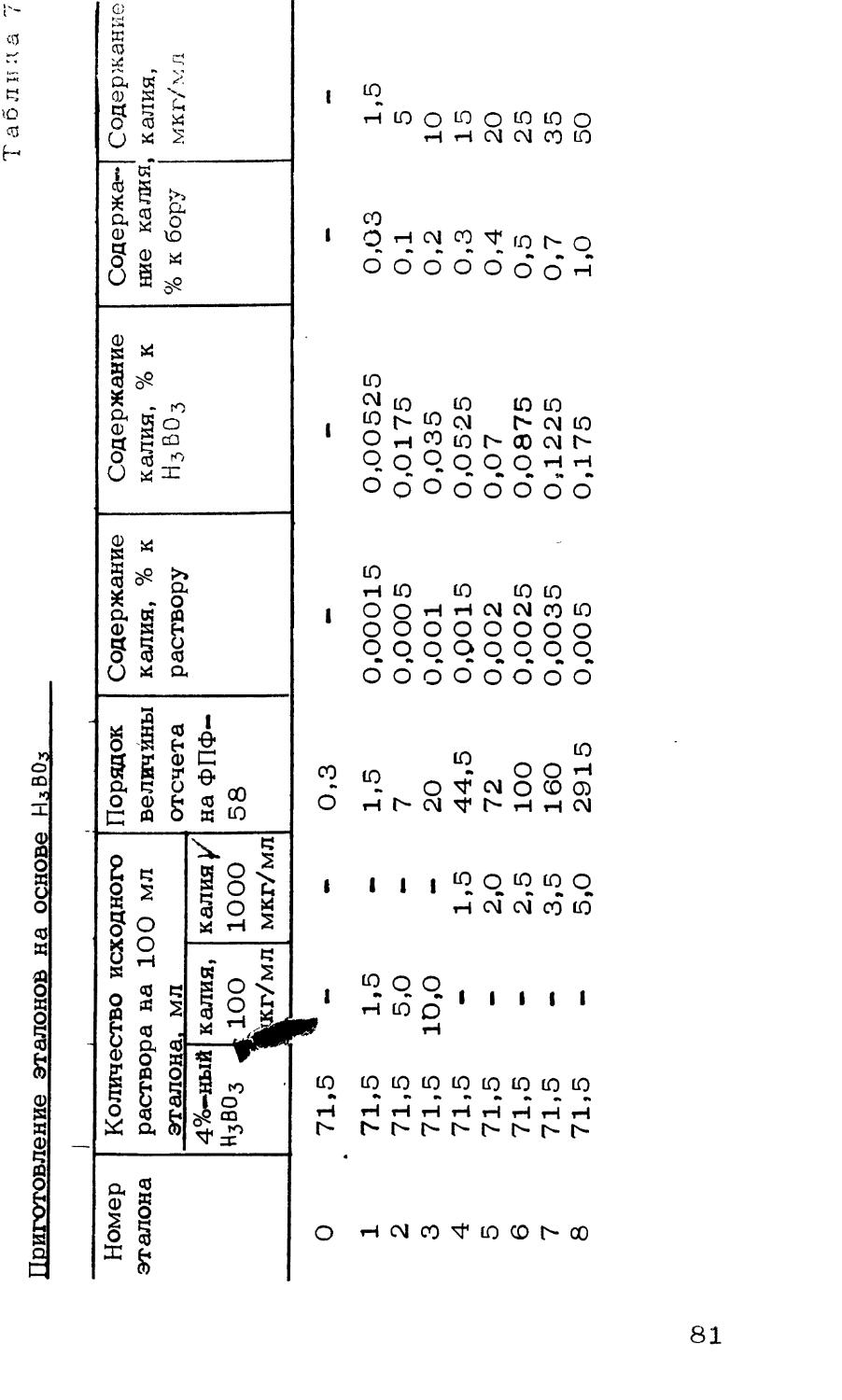
1; Сплавление с пер- (пиро) - сульфатом калия.
Методика основана на сплавлении пробы с пер— (пиро)— сульфатом калия при 700-750°С в течение 1-1,5 ч.
Методика применима для переведения в раствор кристаллического бора, борида титана и пресскомпозиций состава бор + полипропилен; карбид бора + полипропилен; 3 бор + бакелит.
Отбор проб. Для анализа берут навеску вещества с размером частиц до 250 мк: для кристаллического бора 50 мг;
для карбида бора 100 мг;
для пресскомпозиций состава бор кристаллический + полипропилен; ВдС + полипропи-
лен; бор кристаллический + бакелит 100 мг;
для борида титана
200 мг
(навески взвешиваются с точностью до 0,0002 г).
Если частицы вещества имеют размер больше указанного, то они предварительно размалываются или растираются в ступке, изготовленной из карбида бора.
Применяемые реактивы
Персульфат калия Пиросульфат калия Серная кислота
ГОСТ 4146-65, чда.
ГОСТ 7172-65, чда.
ГОСТ 4204-66, чда
Н 2 3 О 4 ч
(2 н,. раствор).
Ход' анализа. Сплавление проводят в кварцевом тигле, который покрывают кварцевой крышкой с небольшим отверстием посредине. Навеску высыпают на дно тигля и засыпают сверху 3,5 г персульфата калия. Тигель закрывают крышкой и ставят в холодный муфель. Температуру поднимают до 400°С при открытом, затем до ~ 700-750^С закрытом муфеле и при этой температуре выдерживают навеску час-полтора. Плав кристаллического бора, карбида бора и пресскомпозиций на их основе выщелачивают горячей водой. Плав борида титана выщелачивают 2 н. Н2ЗО4. Затем растворы переносят в мерную колбу на 250 мл и доводят до метки дистиллированной водой, в случае борида титана - 2н. H2SO4.
2) Сплавление со смесью персульфата калия и гидросульфата натрия.
7
Методика основана на сплавлении пробы с персульфатом калия и гидросульфатом натрия при температуре 700°С.
** с Методика применима для переведения в раствор кристаллического бора, £арбида_бора, борида титана и пресскомпо-зиций состава бор + полипропилен; карбид бора + полипропилен; бор + бакелит.
' ' Отбор проб. Для анализа берут навеску вещества, взвешенную с точностью до 0,0002 г с размером частиц для кристаллического бора, TiB2 и пресскомпозиций до 250 мк, карбида бора до 8-10 мм:
для кристаллического бора 50 мг;
для карбида бора 100 мг;
для пресскомпозиций на основе кристаллического бора или карбида бора 100 мг;
для борида титана 200 мг.
Если вещества имеют размер частиц больше указанных, то они предварительно размалываются или растираются в ступке, изготовленной из карбида бора (для ВдС) или из агата ( для TiB2) . _
z Применяемые реактивы
Персульфат калия К2320 8 ч ГОСТ 4146-65, чда. Кислый сернокислый натрий (гидросульфат натрия) NaHS04? МРТУ 6-09-5231-68, чда.
Серная кислота HgSO^ , ГОСТ 4204-66, чда (2 н. раствор).
Ход анализа. Сплавление проводят в кварцевом тигле, который покрывают кварцевой крышкой с небольшим отверстием посредине . Навеску высыпают на дно тигля и засыпают сверху 2 г NaHS04, затем 8 г К2320з • Тигель закрывают крышкой и ставят в холодный муфель. Температуру поднимают до 400°C при открытом, затем до 700-750°С закрытом муфеле и при этой температуре выдерживают навеску в течение 30 мин. Плав кристаллического бора, карбида бора и пресскомпозиций на их основе выщелачивают горячей водой. Плав борида титана выщелачивают 2 н. H2SO4. Растворы переносят в мерную колбу на 250 мл и доводят до метки дистиллированной водой, в случае борида титана -2 н. H2SO4 .
3) Кислотное разложение смесью серная кислота + азотная кислота + персульфат калия. '
Метод основан на растворении пробы в смеси H2SQ4 + + HN О3 + К 2520^ при кипячении в течение 2 ч.
Методика применима для переведения в раствор борида титана, кристаллического бора.
8
Отбсрр проб. Для анализа берут навеску вещества с точностью 0,0002 г, с размером частиц до 250 мк.
Применяемые реактивы
Пиросульфат калия K2S2O7 , ГОСТ 7172-65, чда.
Серная кислота н2304 , ГОСТ 4204-66, чда.
Азотная кислота HN03 , ГОСТ 4461-67, чда.
Ход анализа. Растворение пробы проводят в кварцевой колбе, подсоединенной к обратному холодильнику. Навеску высыпают в колбу, куда предварительно насыпано 10-15 г K^S207, и приливают 8-20 мл серной и 3 мл азотной кислот. Колбу подсоединяют к обратному холодильнику и реакционную смесь кипятят до полного растворения навески. Раствор количественно переносят в мерную колбу на 500 мл, охлаждают и доводят до метки дистиллированной водой. Раствор выдерживают в течение 2 ч.
1.2. ОПРЕДЕЛЕНИЕ ОБЩЕГО БОРА
Бор относится к группе кислотообразующих элементов, и поэтому большинство методик его определения основано на титровании борной кислоты.
Для определения бора в кристаллическом боре, карбиде бора, бориде титана после их переведения в раствор используется метод алкалиметрического титрования образующейся борной кислоты.
Борная кислота является слабой неорганической кислотой, образующей с многоатомными спиртами более сильные комплексные кислоты, которые могут быть оттитрованы раствором NaOH при рН=7^-10. Для повышения кислотности растворов борной кислоты используют маннит, глицерин, фруктозу, инвертный сахар, глюкозу, пропиле н-г ли коль, глюконат кальция и др. [19-21] . Для определения борной кислоты удобнее применять маннит в сухом виде (1г на 10 мл 0,1 н. раствора борной кислоты), т.к. при этом не происходит разбавление анализируемого раствора, как и при использовании глицерина, глюкоза же при определении борной кислоты дает заниженные результаты [22,23].
Необходимым условием титрования борной кислоты является отсутствие СС>2> ионов аммония и фтора, легко гидролизующихся или взаимодействующих с маннитом металлов ( Fe, Ti, Al, РЬ , Ni, Zn, Co , Cu, Mo, Zr, Ta , Or, W, V , Hg, Re, As , Sb j Ge, Те) [24-29] .
Определение конечной точки титрования осуществляется либо индикатором, обычно фенолфталеином, либо прибором.
9
Определение борной кислоты с индикацией эквивалентной точки потенциометрическим методом может быть проведено в присутствии ионов легкогидролизуюшихся металлов связыванием их в прочный комплекс. Так, алюминий рекомендуется связывать в цитратный комплекс [2б! , для комплексообразования титана, хрома, циркония используют органические кислоты (щавелевая, винная, лимонная; [14] . Титан
маскируется перекисью водорода или комплексеном Ш [29-36]. Многие мешающие катионы: Fe+ Со+\ Nih2, Cu+2, РЬ+2. А1+3, Cd+2,Ca+2, Mg +2, Bat?, РЗЗ - могут быть • замаскированы добавлением комплексона Ш [29,30,32-36] . Таким образом, потенциометрическое титрование имеет преимущества перед титрованием с визуальной индикацией точки эквивалентности. С помощью потенциометрического титрования можно определить бор в окрашенных и очень разбавленных растворах, содержащих 0,01-0,1 мкг бора [ 37]. Потенциометрическое титрование позволяет устранить влияние слабых кислот и оснований, которые титруются при рН=4т9. Основы метода потенциометрического титрования изложены в работах [26,27,39,40].
Методики определения
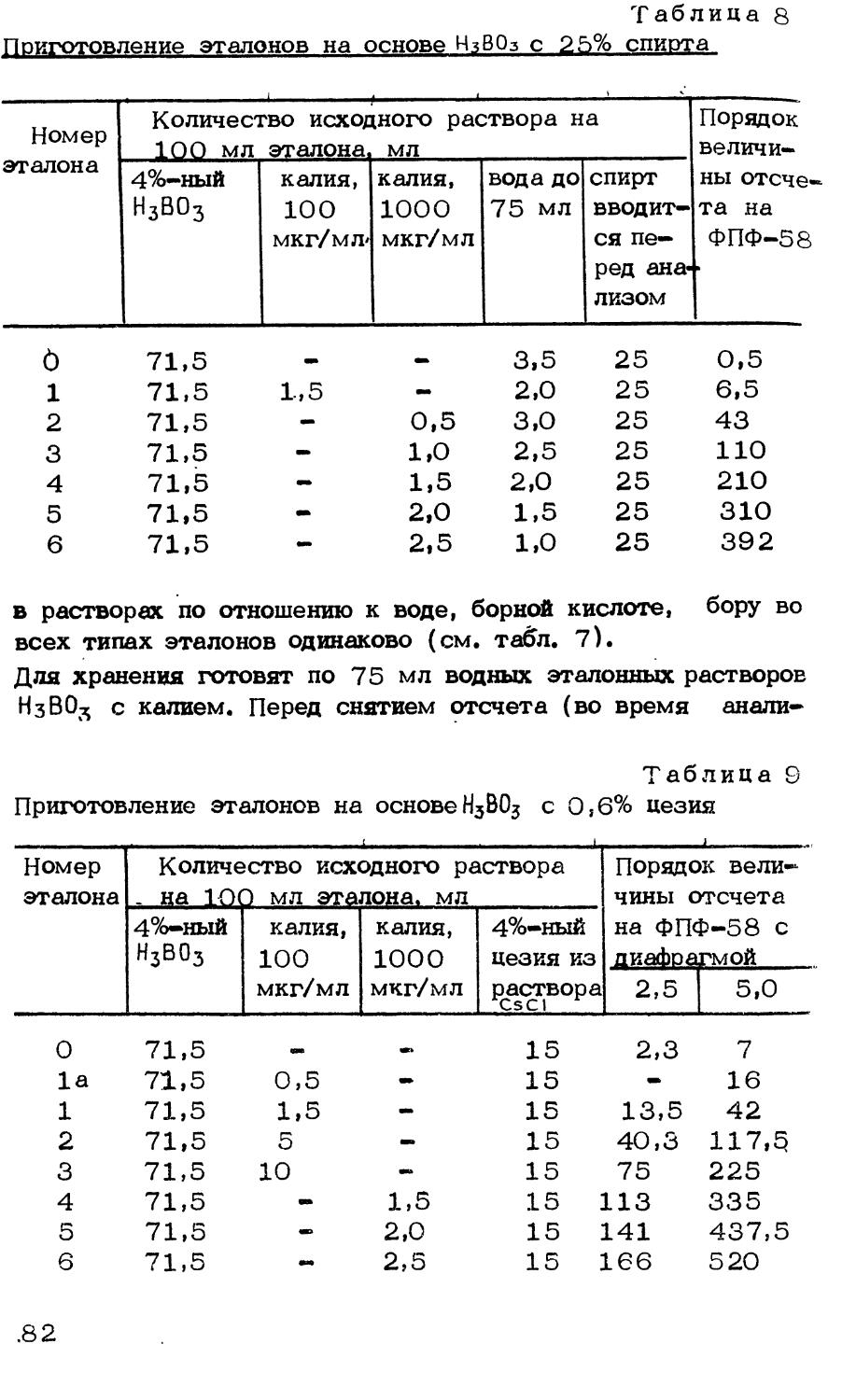
1) Алкалиметрическое определение с визуальной индикацией.
Алкалиметрическое титрование бора основано на титровании борной кислоты раствором щелочи. Алкалиметрическое титрование бора с визуальной индикацией точки эквивалентности может быть использовано Для определения бора в виде борной кислоты при анализе кристаллического бора, карбида бора и пресскомпозиций на их основе состава кристаллический бор + полипропилен; карбид бора + полипропилен; бор + бакелит. Определение проводят в средах:
а) окислительная смесь состава 15г пиросульфата калия, 3 мл азотной кислоты ( d = 1,38), 8 мл серной кислоты ( d = 1,84). Чувствительность определения 4 мг бора. Относительная погрешность метода 0,7%;
б) раствор плава с персульфатом калия или пиросульфатом калия в воде. Чувствительность определения 3* 5 мг бора.
Относительная погрешность метода 0,6%;
10
в) раствор плава со смесью ^2^2°2 ьЧаН30д в
воде. Чувствительность определения 2 мг бора. Относительная погрешность 0,5%.
Отбор проб. Для анализа отбирают аликвоту раствора объемом 100 мл для кристаллического бора и 50 мл для карбида бора и пресскомпозиций на их основе. В аликвоте должно содержаться не менее 10-15 мг бора. Перед отбором аликвоты анализируемый раствор должен быть выдержан при комнатной температуре 2 ч со времени приготовления.
Применяемые реактивы
Маннит CH20H(CH0H)4CH20H , ГОСТ 8321, чда.
Едкий натр NaOH , ГОСТ 4328-66, чда (0,1 н.,
6 н. раствор, 30%-ный раствор).
Соляная кислота НС! ? ГОСТ 3118-67, чда (0,1 н. раствор).
Фенолфталеин С2ОН14О4 , ГОСТ 5850-51 (1%-ный спиртовой раствор).
Метиловый красный (СНз)2НСбН4М “NCgH/jCOOH , ГОСТ 5853-51 (0,2%-ный раствор).
Борная кислота Н3ВО3 , ГОСТ 9656-61, хч. Стандартный раствор борной кислоты.
Раствор приготовляют из борного ангидрида, получаемого прокаливанием борной кислоты. Прокаливание проводят в платиновой чашке при 800°С в течение 45-60 мин до полного исчезновения пузырьков в расплаве. Затем чашку вынимают и осторожно касаются дном холодной воды, предварительно налитой в фарфоровый сосуд. При резком охлаждении плав растрескивается и отстает от чашки. Кусочки борного ангидрида помещают в сухой чистый бюкс и ставят в эксикатор для охлаждения. Для приготовления 500 мл 0,1 н. раствора борной кислоты отбирают 1,7410 г В20з с точностью 0,0002 г.
Ход анализа. Титрование проводят следующим образом: к аликвоте анализируемого раствора добавляют несколько капель индикатора метилового красного. Раствор нейтрализуют сначала 30%-ным NaOH, затем 0,1 н. раствором NaOH, далее слегка подкисляют 0,1 н. НС! и снова нейтрализуют 0,1 н. NaOH. Нейтрализацию раствора повторяют до гех пор, пока от одной капли 0,1 п. NaOH цвет анализируемого раствора не изменится от красного до соломенно-желтого. В- строго нейтральный раствор добавляют 1 г манвига, 10-
11
12 капель фенолфталеина и титруют 0,1 н. Na ОН до слабо-розовой окраски. Для проверки конца титрования к раствору добавляют еще 0,5-1 г маннита, 3-5 капель фенолфталеина, и если от одной капли шелочи раствор окрасится в слабо—розовый цвет, то титрование заканчивают, в противоположном случае повторно добавляют маннит, и раствор вновь титруют щелочью-.
Количественное содержание бора во взятой навеске рассчитывают по формуле:
где V - количество 0,1 н. Na ОН, пошедшее на титрование, мл; /7 - объем анализируемого раствора, мл; В - объем аликвоты раствора, взятой на титрование, мл; п - навеска анализируемого вещества, мг; Т - титр 0,1 н. NaOH, устанавливается в условиях рабочей пробы по стандартному раствору борной кислоты. Для этого отбирают 20 мл 0,1 н. стандартного раствора борней кислоты в коническую кслбу для титрования. Добавляют те же реактивы, как в рабочем опыте, и в том же количестве. Титрование проводят подобно описанному на с. И.
2) Алкалиметрическое определение с потенциометрической индикацией.
Алкалиметрическое определение бора с потенциометрической индикацией используют при анализе борида титана, кристаллического бора, карбида бора и пресскомпозиций на их основе.
Титрование проводят на приборе типа ЛПУ-01 или рН-262 при рН=6,9 со стеклянным и насыщенным каломельным электродами.
Определение проводят в средах:
а) раствор плава с пер- X пиро) -сульфатом калия или со смесью K2s208+NaHS04 в 2 н.H2SO4. Чувствительность определения 3 мг бора. Относительная погрешность 0,4%;
б) раствор плава с пер- ( пиро) - сульфатом калия в воде. Чувствительность определения 3-4 мг бора. Относительная погрешность 0,4%;
в) раствор плава со смесью K2S208 +NaHSOzj в воде. Чувствительность определения 2 мг бора. Относительная погрешность 0,5%;
12
г) окислительная смесь состава 10-15 г К23207 , 3 мл HNO3, 8-20 мл H2SO4 . Чувствительность определения 4 мг бора. Относительная погрешность 0,7%.
Отбор проб. Для анализа отбирают аликвоту раствора объемом 50 мл с содержанием бора не менее 5-15 мг. При анализе борида титана на содержание бора к раствору добавляют 1 мл 30%-нсй перекиси водорода.
Применяемые реактивы см. на с. 1.1
Ход анализа. Титрование проводят согласно инструкции, прилагаемой к потенциометру. В стакан с анализируемым раствором опускают электроды, подсоединенные к прибору. Раствор нейтрализуют до рН=6,9, вначале 6 н., азатем 0,1 н. раствором N а О Н.
После нейтрализации к жидкости прибавляют маннит( ~ 1 г), затем раствор тщательно перемешивают и титруют 0,1 н. раствором Na ОН до pH = 6,9.
Процентное содержание бора рассчитывают по формуле:
„ fo ч T-V-Я -100
где I/ - количество 0,1 н. NaOH , пошедшее на титрование, мл; /7 - объем анализируемого раствора, мл; В -объем аликвоты раствора, взятой на титрование, мл; /г -навеска анализируемого вещества, мг; Т - титр 0,1 н. NaOH, устанавливается потенциометрическим титрованием в условиях рабочей пробы по стандартному раствору борной кислоты (см. с. 11,12).
3) Определение бора в присутствии примесей металлов: Ti , Fe , Mg , Al , Zr , Cu , Pb, S n , Zn , Co , Ni , Ca , W , Si .
Бор в виде борной кислоты может быть определен в присутствии целого ряда металлических примесей, таких^ как Ti , Fe, Mg,AI,Zr,Cu,Pb, Sn , Zn , Co , Ni , Ca W , Si, обычным алкалиметрическим титрованием с потенциометрической индикацией. Мешающее влияние примесей металлов устраняют добавлением комплексообразователей (5%-ного
раствора лимонной кислоты, винной кислоты или перекиси во-. Г д
дорода). Погрешность определения 5 мг бора в присутствии 35 мг примесей металлов не превышает 1%. Метод применим для определения микроколичеств бора. При титровании 0,2 мг бора в присутствии трехкратного количества титана относительная погрешность определения составляет 3%.
13
Применяемые реактивы см. на с. 11
Отбор проб. Для анализа отбирают аликвоту раствора, содержащую не менее 5 мг бора в виде борной кислоты (см. с. 1J.
Ход анализа. К раствору добавляют 10 мл 5%—ной лимонной кислоты или 1—5 мл 30%-ной перекиси водорода и раствор нейтрализуют вначале 6 н. NaOH , а затем 0,1 н. раствором NaOH до pH = 6,9. Титрование бора проводят, как описано на с. 11.
1.3. ОПРЕДЕЛЕНИЕ ТИТАНА В БОРИДЕ ТИТАНА
Анализ борида титана, выпускаемого по ТУ 15-66, предусматривает определение титана весовым методом после осаждения купфероном. Осадок купфероната титана прокаливается до постоянной массы. Эта операция довольно длительная. Определение титана значительно ускоряется при использовании метода комплексонометрического титрования. Поскольку ион Т|,2*+ легко гидролизуется и медленно реагирует с трилоном Б, применяется обратное титрование перо— ксидного комплекса [Ti0(H202H2+ [129], который образу-
ется при добавлении к сернокислому раствору борида титана перекиси водорода. В качестве титрантов при обратном титровании титана используют растворы цинка с эриохромом черным Т в качестве индикатора, если ванадия с дифенил-карбазоном, растворы железа с салициловой кислотой, висмута с ксиленоловым оранжевым, соли меди с ПАН и др. [130]. Чаше всего в качестве индикатора используют ксиленоловый оранжевый [131-133].
Комплексонометрическое титрование титана применяется при анализе различных веществ, однако обычно требуется его предварительное отделение с помощью экстракции или ионного обмена. Экстракцию обычно осуществляют купфероном. При определении титана в присутствии алюминия, железа и магния используют иногда метод вытеснения трилона из комплекса с титаном. При этом титан связывается в фосфат, выпадающий в осадок. Однако в данном случае соосаждение AI и Fe вредит точности определения титана, и избыток фосфа т-ионов может вызвать разложение комплексонатов мешающих элементов [130].
14
Методики определения
1) Комплексонометрическое определение.
Обратное комплексонометрическое определение титана с ксиленоловым оранжевым в качестве индикатора рекомендуется проводить при отсутствии в растворе ионов металлов, образующих с ксиленоловым оранжевым устойчивые окрашенные комплексы ( Fe , Al , Zn , Mg и др).
Определение титана в бориде титана проводят в средах: а) раствор плава с пер- (пиро) - сульфатом калия или со смесью2 н. серной кислоте. Чувствительность определения 7 мг. Относительная погрешность метода 0,5%;
б) окиалительная смесь состава 15 r^^Oz^ 3 млН^О^ 20 мл н2S0z,. Чувствительность определения 7—8 мг. Относительная погрешность 0,7%.
Применяемые реактивы
Перекись водорода Н202 , ГОСТ 10920-64, хч (30%-ная). Трилон Б Na2^10^2^16Г>8 > ГОСТ 10652-63, чда (0,1 н. раствор).
Соляная кислота HCI , ГОСТ 3118-67, чда.
Ксиленоловый оранжевый, МРТУ 6-09-2503-65, чда(0,5%-ный водный раствор).
Растворяют 500 мг индикатора в 80 мл дистиллг /ванной воды, добавляют 2 мл концентрированной соляной кислоты, раствор перемешивают и прибавляют 2%-ный ЫЩОН по каплям до изменения окраски раствора в сиренево-йиш-невую. Полученный раствор доводят до объема . 100 мл дис-* тиллированной водой.
Аммиак водный NH^OH, ГОСТ 3760-64, чда (2%-ный раствор) .
Уксуснокислый натрий CR^COONa , ГОСТ 199-68, чда.
Уксусная кислота СН3СООН , ГОСТ 61-69, хч.
Буферная смесь (рН=5,5) HN03-
Растворяют в воде 540 г уксуснокислого натрия и доводят до 1 л дистиллированной водой. Полученный раствор смешивают* с 500 мл 1 н. уксусной кислоты (28,6 мл). Сульфат цинка ZnSOzj, ГОСТ 4174-69, хч (0,1 н. раствор). Катионит КУ-2.
Катионит КУ-2 отмывают дистиллированной водой от мелкой фракции и замачивают сначала в воде (1 сут), затем в 6 н. HCI (1 сут). Далее смолу обрабатывают на водяной бане с
15
6 н. НС1 7 которую постоянно меняют на свежую, до получения бесцветных сливов. Смолу отмывают водой до нейтральной реакции по метилоранжу и используют для анализа.
Отбор проб. Для анализа отбирают аликвоту анализируемого раствора с содержанием титана не менее Юг-15 мг.
Ход анализа. Анализируемый раствор упаривают до появления паров SO3 и охлаждают. К аликвоте добавляют 50мл дистиллированной воды, 10 капель 30%-ной перекиси водорода, 110 мл дистиллированной воды. Таким образом создается среда - 0,3 н. H2SO4. Далее к раствору добавляют 25 мл 0,1 н. раствора трилона Б и выдерживают 10-15 мин при комнатной температуре. После этого добавляют 10 капель ксиленолового оранжевого и нетрализуют аммиаком, сначала концентрированным, а затем разбавленным 2%-ным NH4OH до перехода цвета ицдикатора из желтого в сиреневато-фиолетовый .
К раствору добавляют 10 мл буферной смеси с рН=5,5 (цвет раствора желтый), разбавляют водой до 400 мл, прибавляют 2-3 капли ицдикатора и титруют раствором 0,1 н. сульфата цинка до перехода окраски индикатора из желтой в красную от одной капли титрующего раствора.
Количество титана рассчитывают по формуле.
_ 2,395 Ч И-СУ2) » 100
п-В ’
где 2,395 - количество титана, соответствующее 1мл 0,1 н. раствора трилона Б, мг; Щ - количество 0,1 н. раствора трилона Б, взятое в избытке по отношению к количеству титана, мл ( И/ = 25 мл); 1/2 - количество 0,1 н. раствора сульфата цинка, пошедшее на титрование избыточного количества трилона Б, мл; С - коэффициент объемного соотношения между 0,1 н. растворами трилона Б и сульфата цинка; А - объем анализируемого раствора, мл; В - объем аликвоты, мл; п - навеска, мг; К - поправочный коэффициент, найденный экспериментально.
При разложении борида титана окислительной смесью К = = 1,01;; при сплавлении с пер- (пиро)-сульфатом калия К = = 0,014; при сплавлении со смесью K2S208 + NaHSO^ К = 1,0.
При содержании в TiB2 примеси железа выше 0,1% рекомендуется вводить в формулу расчета содержания титана поправку на железо, предварительно определив его сульфосалициловым методом [85].
16
Для определения коэффициента объемного соотношения отбирают 25 мл 0,1 н. раствора трилона Б, 20 мл буферного раствора, добавляют 300—400 мл дистиллированной воды, 6-7 капель ксиленолового оранжевого и титруют 0,1 н. раствором сульфата цинка до перехода окраски из желтой в сиренево-красную.
Коэффициент объемного соотношения определяют по формуле
где И/ - объем 0,1 н. раствора трилона Б, мл; *" объем 0,1 н. раствора сульфата цинка, необходимый для титрования, мл.
2) Определение титана в присутствии примесей металлов.
Определение титана в присутствии мешающих примесей металлов Al, Си, Pb , Ni,Zn , Со, Мп, Са, составляющих в сумме не >2%, проводится с отделением пероксидного комплекса титана на ионообменной колонке ( -Н = 40 см, d = 1 см) со смолой КУ-2 в Н-форме. Определение титана проводят после сплавления борида титана со смесью K2S20g-i-NaH504 из 2 н* серной кислоты. Чувствительность определения 8 мг титана. Относительная погрешность 0,6%.
Применяемые реактивы см. на с. 15
Отбор проб. Для анализа отбирают аликвоту анализируемого раствора с содержанием титана не менее 10-15 мг (см. с. 16).
Ход анализа, К аликвоте раствора добавляют ~ 0,5 мл 30%-ного раствора перекиси водорода и 100 мл дистиллированной воды. Раствор пропускают через смолу КУ-2 в Н-форме (скорость пропускания ~ 30 капель в минуту). После осаждения пероксидного комплекса титана на колонке смола промывается 100 мл дистиллированной воды. Со смолы пероксидный комплекс титана смывается 200 мл 1 н. Н2ЗСЦ,ив растворе определяется титан методом обратного комплексонометрического титрования (см. с. 16).
1.4. ОПРЕДЕЛЕНИЕ ВОДОРОДА И УГЛЕРОДА в цтесскомпозициях
Все существующие методы определения углерода и водорода из одной навески органического вещества основаны на сожжении его до С02 и Н20 с последующим определением
17
этих соединений. Навеску сжигают в кварцевой трубке в токе кислорода в присутствии твердых окислителей или катализаторов.
М.О. Коршун, В.А. Климовой и другими авторакш [41 -44] предложен метод скоростного сожжения в быстром токе кислорода в пустой трубке с предвгфительным пиролитическим разложением вещества.
Как известно, большую трудность представляет окисление веществ с повышенной термической стойкостью, веществ, содержащих элементы, которые способны после озоления образовывать или карбиды, или стекловидную пленку на углероде, или термически устойчивые карбонаты. К таким веществам относятся пресском позиции, состоящие из карбида бора с полипропиленом (ПКБ) и бора с полипропиленом (БСП). В литературе не имеется сведений по элементному анализу этих материалов.
Г Карбид бора является тугоплавким, трудноежигаемым соединением ( fnJ1 = 2450°С) [1, 13]. Известно, что начинает окисляться в токе кислорода при 500°С, однако даже при 1000 С без катализатора он полностью не окисляется [45].
При сожжении карбида бора и бора образуется окись В2О3.
При сожжении пресскомпозиции ПКБ наряду с В20з может образовываться уголь, покрытый стекловидной пленкой, которая не окисляется при темепературе 2000°С.
При сожжении пресскомпозиции БСП дополнительно может образоваться карбвд бора. Образующаяся окись бора является летучим соединением, которое оседает на стенках сжигатель-ной трубки и в поглотительных аппаратах, что приводит к коррозии 'аппаратуры. При разработке методики анализа пресскомпозиций были учтены такие особенности материалов.
Для полноты разложения предложено применять в качестве плавня-катализатора V2^5 , которая позволяет количественно окислить навеску анализируемого материала уже при t = 900т-9 50°С. Для доокисления продуктов разложения используется температура зоны окисления 700°С при токе кислорода 40 мл/мин.
Для предотвращения улетучивания В20з из стаканчика пиролитического разложения навеска вещества засыпается дробленым кварцем.
18
Методики определения
В основу определения углерода и водорода положен метод предварительного пиролитического разложения вещества с последующим окислением продуктов пиролиза.
Анализируемое вещество подвергают пиролизу в кварцевом стаканчике при температуре 90 0-9 50°C в присутствии °5 • Выделяющиеся при этом продукты разложения до-окисляются в пустой кварцевой трубке при 700 С в токе кислорода. Конечные продукты окисления Н20 иСО^ поглощаются ангидроном и аскаритом.
Метод предназначен. для элементорганического анализа пресскомпозиций В^С + полипропилен и В + полипропилен на углерод и водород.
Относительная погрешность определения углерода 1,5— 2%. Относительная погрешность определения водорода 1%.
Применяемые реактивы
Ангидрон И9(0104)2 т МРТУ 6-09-3856—67.
Аскарит - едкий натр NaOH, препарированный на асбесте, ДАРТУ 6-09-659 2-70.
Силикагель КСК, фракция 0,5-1 мм, ГОСТ 39 56-54.
Кислород баллонный, ГОСТ 5583-58, высший сорт.
Вакуумная смазка типа Рамзая.
Глицерин CjHgOg, ГОСТ 6259-52.
Стеклянная вата.
Янтарная кислота с4Иб04, ГОСТ 6341-52, или сахароза ci2H22°fu ГОСТ 5833-54.
Едкий натр NaOH, ГОСТ 4328-66.
Азотная кислота HNO3, ГОСТ 4461-67.
Пятиокись ванадия V2°5 t прокаленная в тигле в муфельной печи при t ~ 700°С в течение 1 ч, МРТУ 6-09-6504—70.
Материалы и оборудование
Кварцевая трубка для сожжения ( I = 450 мм, =
наруЖн = 13т15 мм).
Поглотительные аппараты Прегля для С02 и Н20» Реометр для определения скорости потока кислорода. Кварцевый стаканчик для пиролитического разложения навески.
колонки типа Фрезениуса для поглотителей С02 и Н20 в' системе очистки кислорода.
19
Печи:
а) в зоне окисления - МА-2/20; б) для пиролиза - МА-Г/61.
Лабораторный автотрансформатор ЛАТР 1.
Термопара хромель-алюмель с милливольтметром МПП-154М на 1000°С.
Микрошпатель для взятия навески.
Кислородный редуктор.
Крючок для извлечения стаканчика из трубки. Микровесы типа ВЛМ-20г-М.
Трубки вакуумные: 2x4 мм; 3x4 мм; 7 х7 мм,
ТУ 38-105881-75.
Эксикатор с прокаленным CaCl^-
Подставка для поглотительных аппаратов.
Блок для терм ост атиров ания поглотительных аппаратов. Дробленый кварц.
Битый кварц с размером зерен 0,5-1 мм заливают 20%-ным раствором МаОН^ нагревают до кипения, сливают щелочь, промывают водой до нейтральной реакции, затем повторяют ту же операцию с HNO3 (1:1), зерна сушат, прокаливают в течение 1 ч при 900°С и охлаждают.
Отбор проб. Для проведения анализа отбирают микрошпателем в кварцевый стаканчик среднюю пробу (0,01-0,015 г) и взвешивают на микровесах с точностью до 0,00001 г. Навеску засыпают VzPd в три-четыре раза больше объема взятой навески, перемешивают и засыпают на 2/3 стаканчика кварцем).
Ход анализа. Установку для разложения органического вещества монтируют? в соответствии с рис. 1*. Через систему пропускают кислород со скоростью 45—50 мл/мин. Скорость потока контролируют по реометру. Одновременно включают электропечи (6,7) и нагревают до 700 и 900°С соответственно. Кварцевую трубку прокаливают передвижением печи (7) по всей длине в токе кислорода в течение 40-60 мин. Проверяют "фон* установки. Для этого предварительно взвешенные поглотители присоединяют к установке и в течение 30 мин пропускают кислород со скоростью 40 мл/мин при нагретых печах, после чего поглотители снова взвешивают.
U - трубку используют, если анализируемый материал содержит азот.
20
\ рис. 1. Схема установки для определения углерода и | водорода:
j 1 - реометр; 2 - поглотительный аппарат Прегля с ас-V каритом; з - U -образная трубка для поглощения окис-iV/лов азота; 4 - поглотительный аппарат Прегля с ангид-Сроном; 5 - асбестовый экран; 6 - печь в зоне окисле-У ния; 7 - печь пиролиза; 8 - кварцевый стаканчик; 9 -h кварцевая трубка для сожжения; 10 — загрузочный шлиф; f 11 “ система для очистки кислорода от СС^и Н20> 12 " € кислородный баллон М
Перед каждым взвешиванием поглотители слегка проти-^.ают замшей или марлей и термостатируют в течение 5 мин весовой комнате. Установку считают готовой к анализу, £сли изменение массы поглотителей (фон) не превышает (J,О 5-0,1 мг.
Перед рабочим анализом проводят контрольное сожжен че Салонного вещества с определенным содержанием углерс ха 'Я водорода.
Стаканчик с навеской вносят через загрузочный шлиф в трубку для сожжения при отсоединенных поглотительных аппаратах и непрекращающемся токе кислорода. Располагают ^открытым концом в сторону печи окисления на расстоянии >8—12 см от нее. После загрузки в течение 5-10 мин то-Лом кислорода из системы удаляют воздух, попавший в труб-ЛУ при загрузке. Затем присоединяют к установке предварительно взвешенные поглотительные аппараты, проверяют по реометру скорость кислорода и начинают сожжение.
Печь надвигают на ту часть трубки, где находится открытый Конец стаканчика, и перемещают постепенно в течение 10-15 мин вдоль него против тока кислорода так, чтобы ве
21
ществе рассчитывают по формуле: 4'с(%) = 0,2729 • ,(g'g')10°—
где 0с -.содержание углерода в поглотителя с аскаритом, мг;
щество или продукты разложения сгорали в самом стаканчике.
По окончании разложения навески печь держат на стаканчике до полного выжигания налета угля на его стенках. Если оставшийся в стаканчике уголь не удается выжечь таким образом, отодвигают печь (7) на 1-2 мин, дают стаканчику остыть (при этом он заполняется кислородом) и повторяют нагревание. Такую операцию можно проделать два-три раза, после чего печь продвигают вперед до шлифа и в обратном направлении до печи окисления. Время пиролиза и прокаливания трубки - 30 мин.
После сожжения вещества поглотители отсоединяют от трубки и термостатируют перед взвешиванием в течение 5 мин в весовой комнате. По окончании работы трубку для сожжения охлаждают и герметизируют стеклянной заглушкой.
Наряду с сожжением анализируемого вещества проводят контрольное сожжение с катализатором V2O5 для установления холостой пробы в условиях рабочего анализа.
Расчет результатов анализа. Содержание углерода в ве-
4
веществе, %; а - привес - привес поглотителя при
холостом опыте, мг; п - навеска вещества, мг; 0,2729 -коэффициент пересчета с двуокиси углерода на углерод.
Содержание водорода рассчитывают по формуле:
£н(%) = 0,1119
(a -af)W0 а
где /?н - содержание водорода в исследуемом веществе, %; а - привес поглотителя с ангидроном,мг; а1 - привес поглотителя при холостом опыте, мг; л - навеска вещества, мг; 0,1119 - коэффициент пересчета с воды на водород.
J Л .3 h:
ОПРЕДЕЛЕНИЕ ПРИМЕСЕЙ
В КРИСТАЛЛИЧЕСКОМ БОРЕ, ЕГО ТУГОПЛАВКИХ СОЕДИНЕНИЯХ И ПРЕССКОМПОЗИЦИЯХ
2.1. ОПРЕДЕЛЕНИЕ КИСЛОРОДА
Бор с кислородом образует соединения различной степени ркисленности, наиболее распространенными из которых являются В205 и H5BOj [ 14, 46-48]. В литературе имеются также самые противоречивые данные о физико-химических действах л формулах субокислов [46-49] . Наиболее изученными субикислами являются В202 и В^О [50-52].
При прохмышленном получении карбида бора в качестве исходных веществ используются борная кислота и сажа:
4Н3ВО3 * 7С = В 4 С б СО + б Н2С •
Процесс протекает при 1900-2200°С в атмосфере аргона, при этом возможны следующие реакции с образованием окислов бора:
2 н-3во3 —>- в2о3 + зн2о ;
В 5 + 5 С О--->• 2 В - 3 СО 2 )
4 В + С ---э- В4С ;
в2оз +• с —з 2 о 2 -СО j
В2о3 - 16В —- з в6о . В основу получения борида титана по двум технологиям [59] положены реакции:
TiO2 - 4В---^TiB2- 2ВО ; (1)
2Т(02 + В4С + ЗС = 2ТiВ2 -4С0 ; (2)
С-В2О3-----СО - В202* < 3)
23
Образование окисных соединений бора возможно за счет реакций (1) - (3). Борный ангидрид в технологическом процессе используется в качестве присадки для снижения содержания в бориде титана свободного углерода. Субокисел В202 является неустойчивым соединением [ 50] и может разлагаться на воздухе.
Для определения кислорода в боре и его соединениях наиболее универсальным является метод выкуум—плавления. Метод достаточно чувствителен (0,01%) и точен (относительная погрешность 10%). Этим методом можно определить все содержание кислорода, независимо от того, в какой. форме он находится. Однако он требует сложного аппаратурного оснащения [53, 60, 61] .
Метод определения окислов бора и борной кислоты, основанный на их растворимости в воде, с последующим определением бора в водной фазе колориметрическим [1, 54] или потенциометрическим [55] методами значительно проще. Этот метод является косвенным, расчет количества кислорода ведут по содержанию бора в водной фазе. Большим недостатком является допущение, что кислород находится только в виде В20з , т.е. не учитывается возможное присутствие субокислов бора.
Ниже предлагается метод определения кислорода в боре и его тугоплавких соединениях с применением реактива Фишера [56-58] .
Преимущество метода. К. Фишера в том, что кроме B20j и Н3ВО3 можно определить кислород, содержащийся в виде адсорбированной влаги, а также в виде субокислов, которые взаимодействуют с этилцеллозольЬом с выделением воды (например ; В202 ).
Определение общего кислорода методом К. Фишера
Кислородсодержащие примеси в боре - борная кислота, борный ангидрид и В2О2 - взаимодействуют с реактивом Фишера с образованием воды. Избыток реактива Фишера определяют при помощи титрования стандартным раствором воды в этилцеллозольве;
а) н3во3+зсн3он---В(ОСН3)3 + ЗН2О ;
(C5H5N)2SO2 + I2 + 2H20 +2C5H5N
24
—«-(C5H5N)2 -H2S04 + 2C5H5N-Hi;
S) B205 + 6CH30H—► 2B(.0CH3)3 + 3H20;
* B02 12 H 2® *“5 H 5 N
—-(C5H5N)2 H2S0Z| + 2C5H5N-Hl .
!
В данной методике вместо метанола для приготовления реактива Фишера применяют этилделлозольв с2н5-о-сн2он .
* Метод пригоден для определения кислородсодержащих примесей в кристаллическом и аморфном боре, карбиде бора
? И бориде титана. Относительная погрешность определения --составляет 3% при содержании кислорода 0,5 - 1%.
Применяемые реактивы
ВЭтилделлозольв с2Н50-о~сн2он, ГОСТ 8313-60, марка "к". Ц|1ирвдин технический c5h5n, ГОСТ 2747-44.
Пиридин хранят под слоем гидрида кальция. Перед ис-ЙВюльзованием его сливают декантацией и фильтруют через {лажный фильтр в атмосфере с относительной влажностью более 40%. В том случае, если пиридин поставляется в нежной упаковке объемом не более одного литра, он не эбует дополнительной осушки.
д металлический 12, ГОСТ 4159-64.
Металлический раздробленный иод хранится в эксикаторе хлористым кальцием.
рная кислота HgSOz, , ГОСТ 4204-66.
дал атрий сернистокислый безводный N&2SO3, ГОСТ 195-66. рБром Вг2, ГОСТ 4109-64.
Ж Уксуснокислый натрий (трехводный) Na(CHjCOO) ЗН2О, ||МРТУ 6-09-2082-65 осч 2.
Приготовление реактива Фишера, Для приготовления 1 л ®реактива Фишера берут 425 мл этилцеллозольва, 340 мл
^пиридина и 85 мл пиридина, насыщенного сернистым газом |*х( насыщение пиридина сернистым газом проводят из расчета ^>40 г сернистого газа на 500 мл пиридина).
К смеси добавляют 2-3 г иода. После полного растворе-ния иода проводят бромирование смеси до появления красно-
Ватого цвета свободного иода: х ‘Г.
•21 + Вг2=12 + 2Вг.
25
2
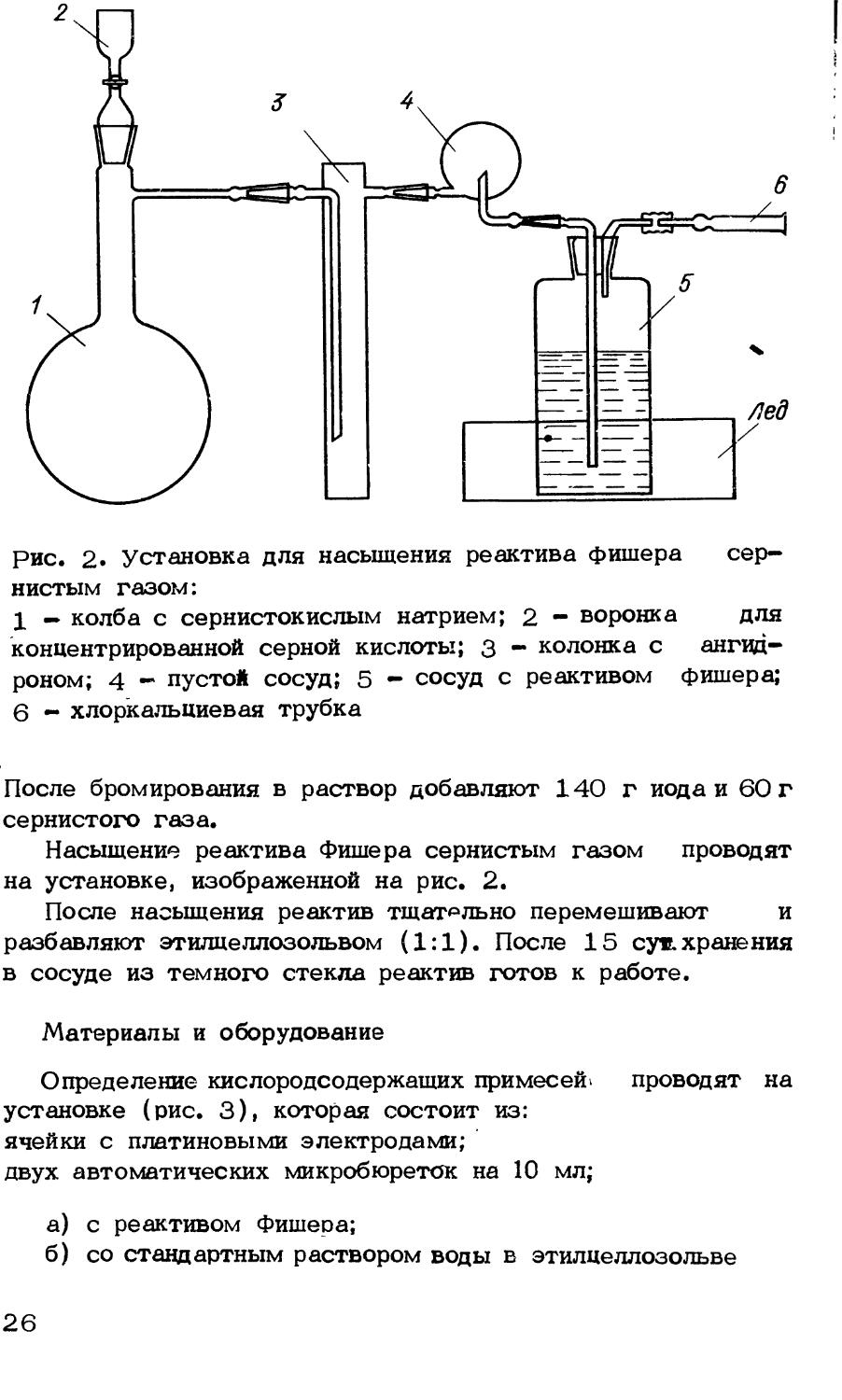
рис. 2» Установка для насыщения реактива фишера сернистым газом:
1 — колба с сернистокислым натрием; 2 - воронка для концентрированной серной кислоты; 3 - колонка с ангидроном; 4 - пустой сосуд; 5 - сосуд с реактивом фишера;
6 - хлоркальциевая трубка
После бромирования в раствор добавляют 140 г иода и 60 г сернистого газа.
Насыщение реактива Фишера сернистым газом проводят на установке, изображенной на рис. 2.
После насыщения реактив тщательно перемешивают и разбавляют этилцеллозольвом (1:1). После 15 сут. хранения в сосуде из темного стекла реактив готов к работе.
Материалы и оборудование
Определение кислородсодержащих примесей> проводят на установке (рис. 3), которая состоит из: ячейки с платиновыми электродами;
двух автоматических микробюреток на 10 мл;
а) с реактивом Фишера;
б) со стандартным раствором воды в этилцеллозольве
26
Рис. 3. Установка для определения кислорода методом фишера
для амперометрического титрования
для осушки воздуха;
эй мешалки ММ-2, МРТУ 42-1503-62;
•циевых трубок.
27
1
2
Рис. 4- Ячейка для титрования
Ячейка (рис. 4) состоит из цилиндра объемом 150 мл с пятью вводами; 1 и 2 - стандартные шлифы, плавно соединяющиеся с ячейкой через резиновые или каучуковые шланги. К этим вводам подсоединяют автоматические микробюретки, на носики которых напаяны соответственно стандартные шлифы. Носики бюреток опущены в ячейку на 3-5 мм от ее верхней стенки. В отверстие (4) вставляют электроды (3), запаянные в стеклянные трубочки диаметром 5 мм. Электроды вставляют в резиновую пробку, которая плотно входит в отверстие ячейки. Ввод (5) снабжен резиновой пробкой. В резиновую пробку вставлен крючок из жесткой проволоки. Крючок вставляют в пробку на вакуумной смазке, что позволяет свободно перемещать его вверх и вниз. В ячейку помещают размешиватель (6), изготовленный из железного стержня, запаянного в стекло. Размешиватель приводится в движение магнитной мешалкой. В шлиф (7) вставляют пробоотборник (8). Платиновые электроды (3) изготовляют из платиновой проволоки диаметром 0,5 мм, короткие обрезки которой припаивают к медной проволоке. Медцый провод служит для подсоединения электродов к схеме микроамперометрического титрования. Электроды впаивают в стеклянные трубки так, чтобы не было трещин в месте спая. Двухходовой кран (9) служит для слива отработанного реактива Фишера из ячейки.
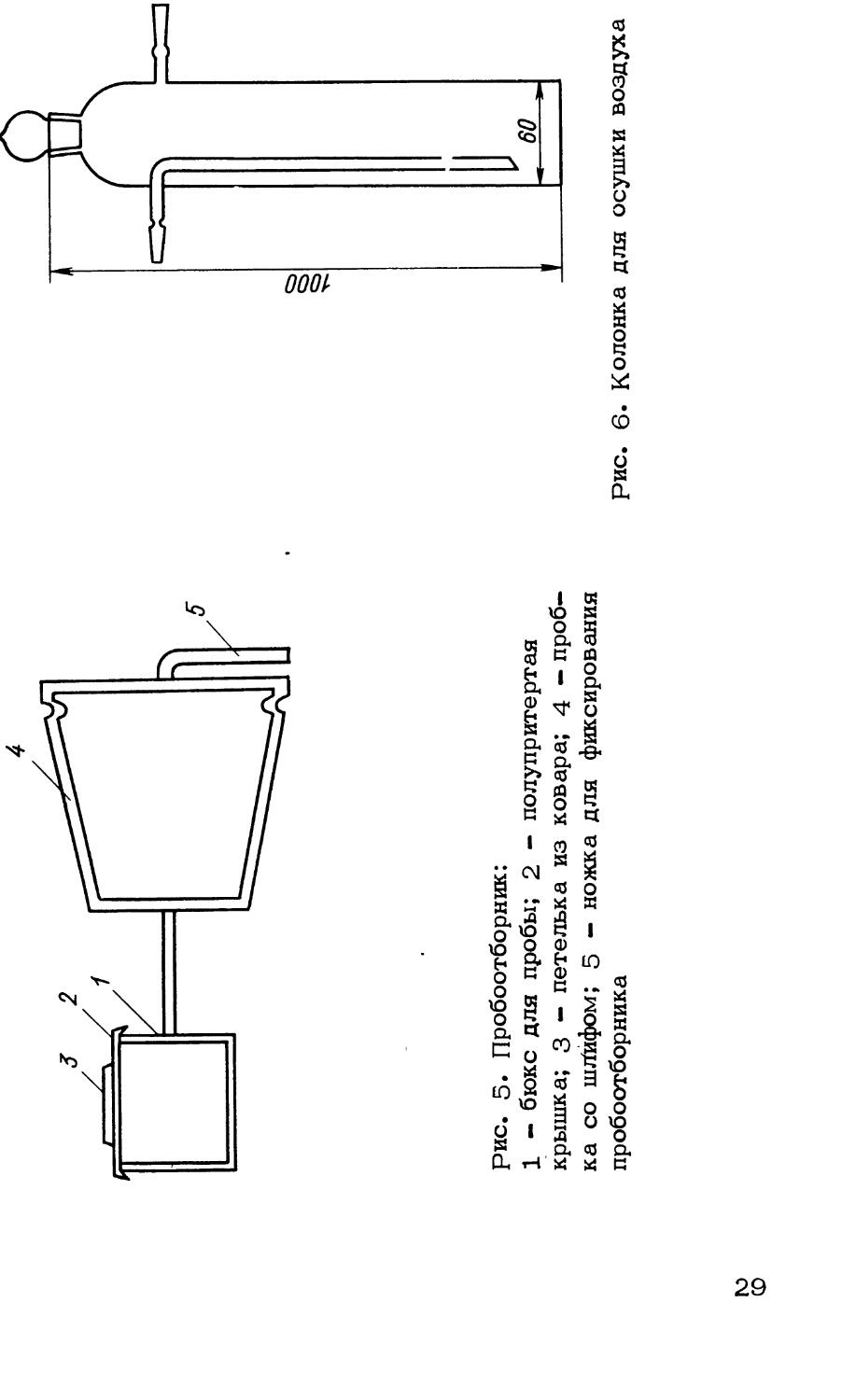
Пробоотборник показан на рис. 5.
28
Рис. 5. Пробоотборник:
1 - бюкс для пробы; 2 - полупритертая крышка; 3 — петелька из ковара; 4 — пробка со шЯифом; 5 - ножка для фиксирования пробоотборника
рис. 6- Колонка для осушки воздуха
го
со
рис. 7* Схема амь . /метрического титрования:
Б - сухой элемент (Зслоя) U = 1,45 8j/?} - сопротивление типа МЛТ-2, 100 . кОм; - проволочное сопротивление ПЭ-7, 5,5 кОм; М - микроамперметр М 240, 50 мкд;Вк -переключатель тумблера ТП-1-2; Я~ ячейке для титрования с платиновыми электродами
Автоматические микробюретки снабжены хлоркальциевыми трубками, заполняются нагнетанием. Воздух при этом осушается в колонке длиной 1 м (рис, 6), заполненной последовательно хлористым кальцием на одну четверть, ангидроном на две четверти и фосфорным ангидридом на одну четверть.
Схема амперометрического титрования изображена на рис. 7.
Определение титра реактива Фишера
Титр реактива Фишера устанавливают по стандартному раствору воды в осушенном эти л целлозольве. Осушку этил-целлозольва проводят на колонке, заполненной молекулярными ситами марки 4А (рис. 8). Длина колонки - 400 мм, диаметр - 25 мм, скорость пропускания этилцеллозольва -60 капель в минуту.
Навеску воды берут на аналитических весах в сухую мерную колбу на 500 мл из расчета 1-0,5 мг на 1 мл этилцеллозольва и доводят до метки осушенным этилцеллозоль-вом.
Количество стандартного раствора воды в этилцеллозоль-ве при титровании должно быть равно или не более чем в два раза меньше количества реактива Фишера. Титр раствора воды устойчив в течение двух недель.
30
8. установка для осушки
этилделло-
1,4 - хлоркальциевые трубки; 2 - воронка с притертой крышкой; 3 - колонка с молекулярными ситами; 5 - приемник
Титрование реактива Фишера раствором воды в этилдел-юзольве проводят в ячейке (см. рис. 4). £ Перед титрованием проб подготавливают
платиновые
Электроды. Для этого в ячейку наливают реактив Фишера до Погружения в него электродов. Включают магнитную мешалку. При помощи регулируемого сопротивления подбирают та-стрелка середины
напряжение, чтобы перед началом титрования оамперметра отклонялась приблизительно до
Реактив Фишера оттитровывают стандартным раствором Воды в этилделлозольве до установки стрелки микроамперметра на нуль, отработанный реактив Фишера сливают.
? Эту операцию повторяют дважды. Напряжение, подаваемое к Электродам, не снимают в течение определения холостых и рабочих проб. Для установления титра реактива Фишера в Ячейку наливают 10 мл реактива и титруют стандартным раствором воды в этилделлозольве.
31
Титрование вначале ведут со скоростью подачи раствора 1 -0,5 мл а интервалом 3-4 с. Как только стрелка начнет отклоняться, порции стандартного раствора воды умень-шают до 0,3 - 0,2 мл. У нулевой точки раствор прибавля-ют по каплям с интервалом 5—7 с.
Титр реактива Фишера (в миллиграммах воды) рассчитывают по формуле
т _____гхУ_____i
” 500 (10-СИ?
где п - навеска воды, взятая для приготовления стандартного раствора воды' в этилцеллозольве, мг; И J количество стандартного раствора воды в этилцеллозольве, пошедшее на титрование 10 мл реактива Фишера; С - коэффициент объемного соотношения реактива Фишера к осушенному раствору этилцеллозольва, из которого приготовлен стандартный раствор воды.
Если титр реактива Фишера окажется больше 1 мг/мл, то его следует погасить добавлением воды, необходимое количество которой рассчитывают по формуле:
Ио ,
где Тп - титр приготовленного реактива Фишера, мг/мл;
7Н - титр реактива Фишера, который необходимо получить, т.е. Тн должен быть равен 1-0,5 мг/мл; Ио -объем приготовленного реактива Фишера, мл. После гашения повторно определяют титр реактива Фишера. Проверку титра реактива Фишера можно проводить также и по соли (CH3coo)Na-3H2O .Навеску тригидрата ацетата натрия (9-10мг) отбирают в маленький стаканчик и выбрасывают в ячейку. Добавляют 10 мл реактива Фишера, перемешивают 15 мин и титруют стандартным раствором воды в этилцеллозольве, как описано в рабочем опыте (см. с. 31).
Титр реактива Фишера в миллиграммах воды вычисляют по формуле:
т 0,397т?
Г = —-—--- )
где К-10-10 у', п. - навеска тригидрата ацетата натрия, мг; 0,397 отношение количества Н20 в молекуле ацетата к его молекулярному весу; к - количество реактива Фишера, связанное водой в навеске ацетата натрия, мл; Vj - количество
32
стандартного раствора воды в этилнеллозольве, пошедшее на титрование в этом опыте, мл; И - количество стандартного раствора воды в этилпеллозольве, пошедшее на титрование 10 мл реактива Фишера, мл; 10 - количество реактива Фишера, мл.
Титр реактива Фишера со временем меняется, поэтому его необходимо проверять через 2-3 дня.
Отбор проб. Навеску анализируемого вещества 0,4-1,0 г отбирают в бюкс пробоотборника в атмосфере с влажностью не более 0,2 г/м^.
Ход анализа. Пробоотборник вставляют в шлиф ячейки и включают схему амперометрического титрования.В ячейку из микробюретки наливают 10 мл реактива Фишера, включают магнитную мешалку. При этом вся влага из воздуха ячейки поглощается реактивом Фишера. Стрелка микроамперметра отклоняется до середины шкалы. Из другой микробюретки добавляют стандартный раствор воды в этилделлозольве до падения стрелки микроамперметра до нуля. Отработанный реактив Фишера сливают через кран (9) (см. рис. 4). В ячейку снова наливают 10 мл реактива Фишера. Раствор перемешивают в течение 30 мин и оттитровывают стандартным раствором воды в этилделлозольве.
Пошедшее на титрование количество стандартного раствора воды в этилделлозольве является величиной холостого опыта. Раствор сливают через кран (9). Ячейка готова к рабочему опыту. При помощи крючка (*£) снимают крышку с бюкса пробоотборника и поворотом пробоотборника навеску бора сбрасывают в ячейку. Наливают 10 мл реактива Фишера, включают магнитную мешалку и проводят перемешивание пробы в течение 30 мин. Стрелка микроамперметра при этом отклоняется до середины шкалы. Титрование избытка реактива Фишера проводят стандартным раствором воды в этилделлозольве до падения стрелки микроамперметра на нуль.
Расчет содержания кислорода в навеске бора проводят по формуле
м __<</--0 т 0.8» <°» , -°2 V П
где 10 - количество реактива Фишера, мл; V - количество стандартного раствора воды в этилделлозольве, пошедшее на "холостую* пробу, мл; И, - количество стандартного рас
33
твора, пошедшее на рабочую пробу, мл; п - навеска анализируемого вещества, мг; Т - титр реактива Фишера^ мг/мл; 0,89 - содержание кислорода в молекуле воды.
2.2. ОПРЕДЕЛЕНИЕ АЗОТА
Азот в кристаллическом боре находится в основном в виде нитрида бора BN. Однако в литературе [46, 47] имеются указания на существование других нитридов, таких,как
, В3(Ыз)з ’ B3N3 •
Нитрид бора BN - соединение устойчивое по отношению к воздействию различных химических реагентов. Поэтому особую трудность представляет переведение его в раствор.
Для определения азота в тугоплавких соединениях, в том числе и кристаллическом боре, наиболее универсальным является метод вакуум-плавления [20, 53] . Этот метод обладает достаточной чувствительностью (0,01%) и точностью (относительная погрешность определения 10%), требует сложной аппаратуры.
Особое место занимают химические методы анализа [1, 13, 62, 63] , при которых металл растворяется в подходящем неорганическом растворителе (кислоте, перекиси водорода), а азот вместе с выделяющимся водородом образует аммонийные соли. Аммонийные соли разлагают щелочью с выделением аммиака. Образовавшийся аммиак отгоняют паром и поглощают серной кислотой, избыток которой оттит-ровывают едким натром.. Этот метод получил название метода Кьельдаля.
В процессе синтеза кристаллический бор подвергается высокотемпературной обработке при 2000°С, в результате которой сильно повышается устойчивость как самого кристаллического бора, так и присутствующей примеси нитрида бора, и они плохо растворяются в кислотах.
Методика определения
Предлагаемый метод основан на сплавлении навески кристаллического бора с твердой щелочью (КОЧ или Na ОН) при температуре 300-700°С с образованием растворимых боратов.
Азот, находящийся в виде нитрида бора, реагирует с твердой щелочью при температуре 300 С с выделением аммиака по реакции
34
Z6N - 6 KLH B2C--5 f-3K20 +2NH з .
Выделившийся аммиак отгоняют паром из водного раствора боратов по методу Кьельдаля и улавливают серной кислотой, избыток которой оттитровывают едким натром. По смешанному индикатору определяют конец титрования.
Относительная погрешность определения при содержании азота 0,1 - 1% составляет 5%. Методика применяется для анализа кристаллического бора.
Применяемые реактивы
Едкое кали КОН , ГОСТ 4203-65.
/Едкий натр NaOH, ГОСТ 4328-66, 0,1 н. раствор. Для ; приготовления 0,1 н. раствора Na ан навеску (4 г) переносят в мерную колбу на один литр и растворяют в воде. "После охлаждения раствора объем его доводят до метки водой.
^Серная кислота Н2504 - фиксанал, МРТУ 6-09-1678-64. Растворы 0,01 н. серной кислоты и щелочи готовят разбавь Гением 0,1 н. раствора.
фетиловый красный (сн3)2нс6н4№КСьн^соонл ГОСТ 5853-51. Иетиленовый голубой G16H18cin3s* зн2о , МРТУ 6-09-6045-69. Смешанный индикатор. Готовят два раствора. Первый раствор-0,3% метиловый красный в этиловом спирте. Второй раствор - 0,1% метиленовый голубой в воде. 50 мл первого раствора смешивают с 10 мл второго раствора.
Спирт этиловый ректификат С2Н50Н , ТУ-19П-39-69.
Для приготовления всех растворов используется дважды перегнанная вода. Все реактивы должны иметь марку хч 1ли чда.
! Материалы и оборудование
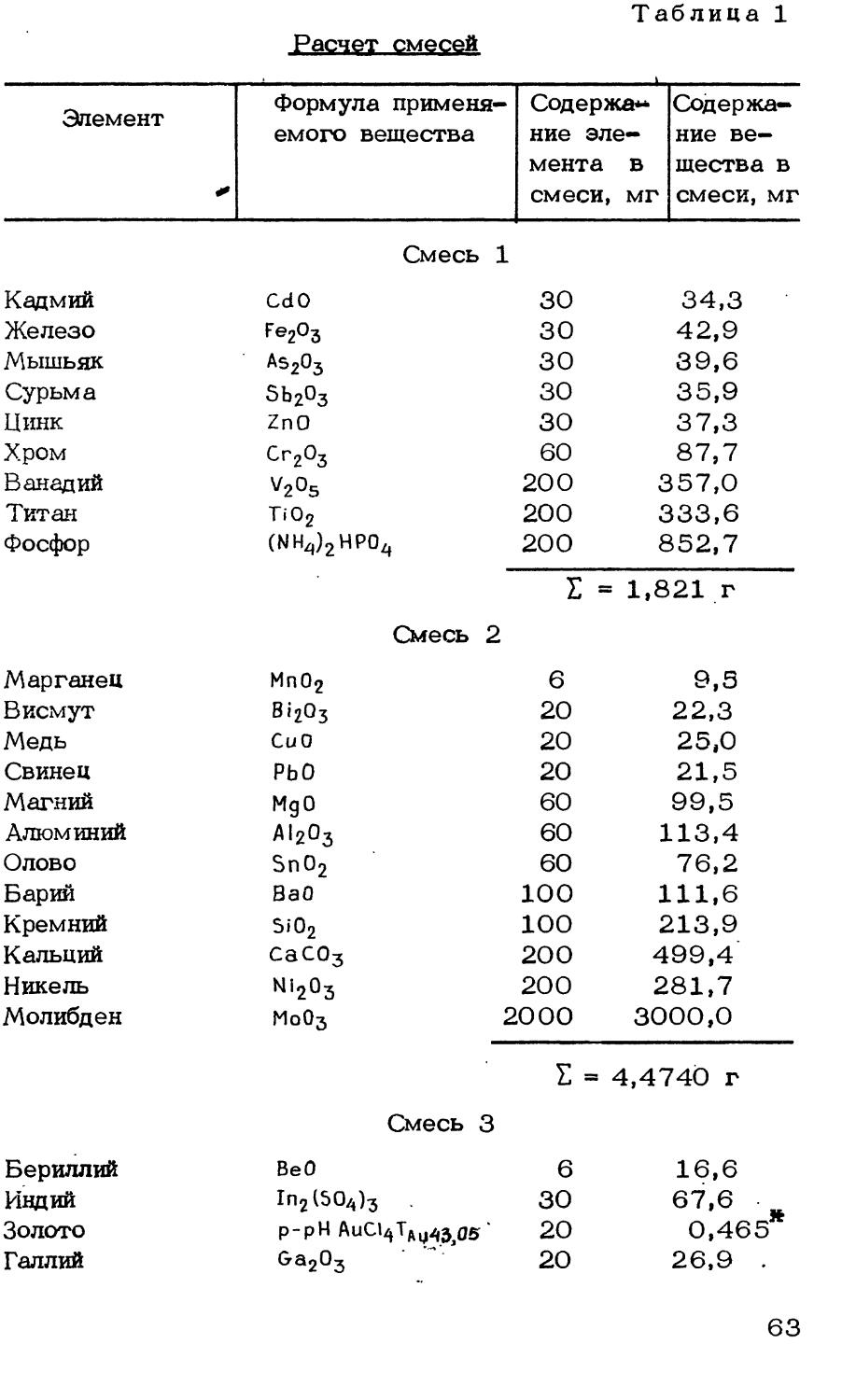
~ Установка для определения азота (рис. 9).
> Отбор пробы. Навески кристаллического бора 0,05 г и | едкого кали 4-5 г взвешивают на часовых стеклах. Затем 2 г едкого кали пересыпают в кварцевый стаканчик, тудг Г же помещают навескуг кристаллического бора и засыпают его оставшимся количеством едкого кали.
I- Взвешивание бора проводят на аналитических весах с £ точностью до 0,0002 г.
< Ход анализа. Перед началом работы реакционную пробир-ку и приемник тщательно промывают,, а затем весь аппарат » пропаривают в течение 15-20 мин. Отгон в приемнике прове-; 3<‘
г
Рис. 9. Установка для определения азота:
1 - колба - парообразователь; 2 - трехходовой кран; 3^ - реакционная пробирка ( d — 26т28 мм, I = ЮО мм); 4 - кварцевый стаканчик; 5 - трубчатая вертикальная печь ( d = 50 мм, I - 160 мм); 6 -шлиф-холодильник;, 7 - каплеуловитель; 8 - холодильник; 9 - приемник на 250 мл
ряют на содержание аммиака. Кварцевый стаканчик с навеской помещают в реакционную пробирку. Шлиф пробирки смазывают вакуумной смазкой, хорошо притирают к холодильнику и привязывают проволочкой за крючки. На реакционную пробирку надвигают заранее нагретую до 110-120°С электропечь и одновременно присоединяют приемник с 20 мл
36
0,01 н. раствора серной кислоты. Кран поворачивают так, чтобы колба соединялась с воздухом.
Сплавление бора проводят в течение 3,0 ч при различной температуре:
110-120°С - 30 мин;
300 - 30 мин;
550 - 1 ч;
650 - 40 мин;
700 - 20 мин.
После сплавления электропечь охлаждают до 100-140°С. Одновременно включают электроплитку для нагревания воды в парообразователе. Воду в парообразователе нагревают до бурного кипения, затем поворотом крана пускают пар в реакционный аппарат. Плав в стаканчике растворяется, и аммиак вместе с паром отгоняется в приемник. При отгоне аммиака температуру в печи поддерживают 200-250°C. Указанная температура необходима для того, чтобы набрать в приемник не менее 200 мл отгона.
По окончании отгона аммиака электропечь отключают, и отгон в приемнике титруют 0,01 н. раствором едкого натра со смешанным индикатором. Параллельно с рабочим опытом ставят холостой опыт в тех же условиях т
Содержание азота рассчитывают по следующей формуле
где V - количество 0,01 н. раствора Na ОН, пошедшее на титрование холостой пробы, мл; У, - количество 0,01н. раствора щелочи, пошедшее на титрование рабочей пробы, мл; п - навеска анализируемого вег’ >ства, г; К - коэффициент пересчета?
С 14-0,01
К 1000 1
С - коэффициент объемного соотношения между 0,01 н. раствором H2SD4 и 0,01 н. раствором Na ОН, 14 - атомная масса азота.
2.3. ОПРЕДЕЛЕНИЕ УГЛЕРОДА
В основном все методы определения углерода в различных материалах сводятся к сожжению навесок этих веществ с плавнями (медью, свинцом, оловом, окислами свинца, мед^
37
ванадия и др.) или без них в токе кислорода при 1ООО -13 50°С. Количество образовавшейся двуокиси углерода определяется затем различными методами: газообъемным, весовым , объемно- баритовым, потенциометрическим*газохро-матографическим. Газообъемный метод определения имеет недостаточную точность и применяется в качестве экспрессного метода. При содержании углерода в образцах до 4% сжигают навески до 1 г, используя при этом эвдиометры со- сетствующей емкости. В последнее время в заводских ; .гЧ'иаториях., широко применяется автоматический прибор Л; 29.Принцип действия прибора основан на количественном определении образовавшейся при сжигании пробы двуокиси углерода методом автомэтического кулонометрического титрования Г 64] .
Для определения углерода менее 1% используется объемно-баритовый метод, основанный на поглощении двуокиси углерода титрованным раствором Ва(0Н)2. Избыток гидроокиси бария оттитровывается уксусной или янтарной кислотой по фенолфталеину, При использовании соляной кислоты в раствор Ва(ОН)2 перед титрованием добавляется 0,5 мл 10%-ного раствора BaCI^ для предотвращения частичной диссоциации ВаСОз в нейтральной среде [65].
Для определения малых количеств углерода применяется кондуктометрический метод, впервые предложенный Брунсом [бб] и впоследствии использованный другими авторами [67, 68, 69]. После некоторого усовершенствования [70 ] этот метод был использован для определения малых количеств углерода (8-10 мкг) в навесках веществ 0,01-0,02г.
Для уменьшения продолжительности анализа применяют потенциометрический метод [64, 71 - 76].
В последнее время используются высокочастотные печи [72] , преимуществом которых является быстрота нагрева.
Интересным является колориметрическое определение углерода [77, 78] .
Применяемые реактивы
Спирт этиловый ректификованный С2Н50Н,ТУ-19П-39-69.
Перекись водорода Н202 , ГОСТ 10929-64, хч, чда.
Едкий натр МаОН, ГОСТ 4328-66, хч, чда.
Вода дистиллированная Н20 , ГОСТ 6709-72.
Гидрат окиси бария Ва(он)2, ГОСТ 4107-69/(0,1 и 0,01 н.
38
растворы). Для приготовления 0,1 н. раствора Ва(0Н)2берут навески 16-20 гВа(0Н)2-8Н20 и 50 г ВаС12 и растворяют в 1 л дистиллированной кипяченой воды.
Для приготовления 0,01 н. раствора Ba(QH)z навески соответственно уменьшают в 10 раз.
Образовавшемуся в растворе осадку карбоната бария дают полностью отстояться, после чего раствор используют для работы. Хранят раствор без доступа С02 длительное время. Для работы часть раствора отфильтровывают ' в атмосфере без СОв сосуд автоматической бюретки, которую сверху закрывают хлоркальциевой трубкой, наполненной аска-ритом. Раствор применяют для объемно-баритового метода.
Приготовление титрованного раствора гидрата окиси бария. Для приготовления 0,01 н. раствора 1,5 г BaCDH^-S^O растворяют в литре дистиллированной воды, предварительно кипятят в течение 2 ч. Раствор закрывают пробкой и оставляют на 2-3 дня. Титр раствора устанавливают по 0,01 н. раствору НС1, приготовленному из фиксанала в присутствии фенолфталеина. Хранят раствор без доступа 002 в сосуде автоматической бюретки, которую сверху закрывают хлоркальциевой трубкой, заполненной аскаритом. При определении примеси углерода до 0,5% используют 0,01 н. раствор, при содержании углерода > 0,5% - 0,1 н. раствор. Раствор применяют для метода с потенциометрической индикацией. Барий хлористый Вас 12-2^0, ГОСТ 4108-65. Для i риготовле-ния электролита 10 г ВаС12-2Н2о растворяют в 900 мл дистиллированной кипяченой воды. К раствору добавляют 5 мл этилового спирта, 5 мл 3%-ного раствора перекиси водорода, доводят до метки кипяченой водой и ' тщательно перемешивают.
Приготовление титрованного раствора хлористого бария. Для"Т1рйготовления 0,01 н. раствора 1,2 г8аС12-2Н20 растворяют в 900 мл свежекипяченой дистиллированной воды, добавляют 0,4 г Na он. Раствор доводят до метки, тщательно перемешивают, помещают в герметичный сосуд, соединенный с бюреткой для титрования. Титр раствора устанавливают по 0,01 н. раствору соляной кислоты, приготовленному из фиксанала в присутствии фенолфталеина. При определении примеси углерода до 0,5% используют 0,01 н. раствор, при содержании углерода 0,5% - 0,1 н. раствор. Раствор применяется для метода с потенциометрической индикацией.
39
Кислота соляная HCI, ГОСТ 3118-67, чда,(0,1 и 0,01 н. растворы), фиксанал, МРТУ 6-09-1678-64.
Окись меди гранулированная Си О, ГОСТ 4468-48, прокаленная в течение 3 ч при 700-800°С.
Окись меди порошкообразная СиО т ГОСТ 4469-^48, прокаленная в течение 3 ч при 700-800°С.
Магний хлорнокислый, безводный Мд(СЮ4)2 , МРТУ 6-09-3856-67, чда, высушенный в вакууме при 150-200°С в течение 3 ч.
Аскарит, МРТУ 6-09-6592-70, чда.
Кислота серная H2SOz| , ГОСТ 4204-66, чда.
Калий марганцевокислый КИпО^, ГОСТ 4527-65, чда.
Фенолфталеин С^о^О^ ? ГОСТ 5850-51. Для приготовления 0,5%-ного раствора 0,2 г фенолфталеина растворяют в 25 мл этилового спирта, добавляют 15 мл воды и перемешивают.
Хромовый ангидрид Сг03, ГОСТ 3776-68, чда.
Калий двухромовокислый К2Сг2о7, ГОСТ 4220-65, хч,чда. Лента никелевая толщиной 0,3 мм, шириной 51—300 мм, мягкая или полутвердая НП4, НП2, ГОСТ 2170-62, ТУ 14-7-95-65, ТУ 14-7-14-65.
Пятиокись ванадия , МРТУ 6-09-6594-70, чда.
Материалы и оборудование
Установки для определения углерода (рис. 10, 11). Микробюретки с автоматической установкой нуля, содержащие титрованный раствор гидрата окиси бария или соляной кислоты.
Лабораторный pH-метр типа ЛПУ-01 для измерения изменения потенциала, настроенного, как милливольтметр с диапазоном измерения +400 - О, мВ.
Отбор проб. Для ацидиметрического определения с визуальной индикацией (объемно-баритовый метод) навески проб отбирают на воздухе в кварцевые или никелевые лодочки. Лодочки предварительно моют, сушат и прокаливают в токе кислорода при 800-900°С в течение 2 ч.
Навески .0,015-0,020 г для карбида бора; 0,1-0,3 г для бора, борида титана и других материалов (в зависимости от содержания примеси углерода) отбирают с точностью до 0,0002 г.
40
Для алкалиметрического определения с потенциометрической индикацией навески исследуемого вещества 0,1-0,3 г в зависимости от содержания в нем углерода, отбирают на воздухе в фарфоровые лодочки, предварительно^ прокаленные в токе кислорода при 1200-1350°С, засыпают 0,5 -1,0 г окиси меди.
1) Ацидиметрическое определение с визуальной, индикацией (объемно-баритовый метод).
Метод основан на сожжении навески с плавнем в токе кислорода при 1000-1200 С в установке (рис. 10). Образующийся углекислый газ поглощается раствором гидрата окиси бария, избыток которого титруют соляной кислотой с фенолфталеином:
Ва(ОН)2+С02--ВаС03 + Н20;
Ва(ОН)2 + 2HCI —*-ВаС12 + 2Н20.
Относительная погрешность метода составляет 3-5% для содержания углерода до 0,5% и 1% для содержания углерода свыше 0,5%.
Отбор проб (см. с. 40).
Ход анализа. Проведение контрольного опыта с плавнем.
Для определения углерода в карбиде бора, бороде титана и боре используют в качестве плавня окись меди или пяти-окись ванадия. Перед опытом окись меди прокаливают в течение 3 ч при 700-800°С в муфельной печи. Навеску прокаленной окиси меди или пятиокиси ванадия 0,5-1,0 г отбирают в кварцевую лодочку и взвешивают на технических весах с погрешностью +0,08 г. В трубку для сожжения предварительно вставляют прокаленный в муфельной печи при 1000°С никелевый патрон, в патрон вводят лодочку с навеской окиси меди и проводят контрольный опыт в таких же условиях, что и рабочий (см. с.43). По окончании опыта замеряют общее количество соляной кислоты , пошедшее на титрование во всех ловушках.
'Фон' установки (в миллилитрах соляной кислоты) рассчитывают по формуле:
где к - суммарное количество Ва(он)2 вушках, мл; С - коэффициент объемного растворов НО и Ва(ОН)2; — суммарное
во всех ло-соотношения количество
41
рис. 10. Установка для определения углерода объемно-баритовым методом:
1 - кислородный баллон с редуктором; 2 - сосуд Тищенко С 4%-ным раствором КИпСЦ в 20%-ном КОН \ з - сосуд Тищенко с 5%-ным раствором в концентри-
рованной H^SOzi; 4 - кварцевая трубка с СиО; 5, 14» 15 -трубчатые печи; 6 - пришлифованный колпачок; 7, 10 -
колонки с ангидридом; 8, 9 - колонки с аскаритом; 11 -пришлифованный колпачок: 12 - двухходовой кран; 13 -
кварцевая трубка с никелевым патроном ( d = 16 т 20мм
I '= 600т800 мм); 16 - сосуд Тищенко с 5%-ным раствором К2СГ2О7 в концентрированной H2SO4 или$ -образной трубки с хромовым ангидридом; 17, 18, 19 - ловушки, наполненные раствором гидрата окиси бария; 20 — сосуд Тищенко с концентрированной
HCI пошедшее на титрование раствора Ва(0Н)2 во всех ловушках, мл.
При навеске СиО или 1/2 05> равной 1г, V должно быть не более 1,2 мл, а расхождения между определениями не должны превышать +0,0 5 мл. Для проверки правильности работы установки периодически проводят сожжение стали (стандартного образца) с определенным содержанием углерода.
Определение коэффициента объемного соотношения .растворов окиси бария и соляной кислоты. Ловушки для поглощения СО2 присоединяют к системе в токе кислорода
(рис. 10). Систему предварительно продувают сухим кисло
42
родом в течение 1 ч с печами (5, 15), нагретыми до 500 , 800°С соответственно.
В токе кислорода каждую ловушку заполняют 10 мл раствора гидрата окиси бария и закрывают пробкой со шлифом. После этого в токе кислорода раствор гидрата окиси бария оттитровывают раствором соляной кислоты по фенолфталеину. Титрование раствора в ловушках проводят в обратном порядке.
Коэффициент объемного соотношения С рассчитывают по формуле:
где I/ - объем раствора HCI , пошедший на титрование, мл; Vf - объем раствора Ва(0Н)2 ? взятый для опыта, мл. Желательно, чтобы С составляло 1 + 0,03.
Проведение определения. Анализируемую пробу вещества засыпают 0,5-1 г прокаленной' окиси меди или V2Q5’
Печи 5 и 15 нагревают до 500-800°С соответственно.
Лодочку с навеской осторожно в токе кислорода вносят в трубку и помещают в никелевый патрон при помощи’*' кварцевой трубки диаметром 13 мм, подсоединяют пустые поглотители и продувают установку кислородом в течение *1 ч. Затем каждую ловушку в токе кислорода заполняют по ходу газа раствором гидрата окиси бария (10 мл) и закрывают пробкой со шлифом. На кварцевую трубку с навеской надвигают электрическую печь 14. Постепенно в течение 40-60 мин температуру печи 14 поднимают до 1200°С и выдерживают 15 мин.
Затем в течение 20-30 мин температуру понижают до 1000°С, после чего печь отключают за 30 мин до титрования, а установку продолжают продувать кислородом до конца титрования.
Углерод, содержащийся в пробе, сгорает при большом избытке кислорода до углекислого газа и поглощается гидратом окиси бария. Избыточное количество гидрата окиси бария оттитровывают соляной кислотой в присутствии 1-2 капель фе-> нолфталеина. Титрование проводят в обратном порядке: замеряют общее количество соляной кислоты, пошедшее на титрование гидрата окиси бария во всех поглотителях и рассчитывают содержание углерода по формуле
43
N9[(V1C~Vt')- V] 100 n 1000
где - суммарное количество Ва(он)2 во всех ловуш-
ках, взятое для поглощения углекислого газа, мл; С - коэффициент объемного соотношения растворов HCI и Ва(0Н)2‘>
1/у - количество раствора HCI , пошедшее на титрование избытка Валону, мл; V - "фон" установки с плавнем, мл HCI ; Npci - нормальность раствора НО; Эс - грамм-эквивалент углерода (3=6); п. - навеска продукта, г. t
Содержание углерода можно также рассчитать по формуле
100 п 1000
где 1/2 - суммарное количество HCI , пошедшее на титрование Ba(OH)£j во всех ловушках в контрольном опыте, мл; п * ^hci » Эс 7 “ обозначения соответствуют предыдущей
формуле.
Примечание, Данной формулой можно пользоваться, если концентрации. Ва(0Н)2 и нс 1 постоянны в контрольном и рабочем опытах.
Алкалиметрическое определение,,^.. ^охеВД^ометричес-кой индикацией после скоростного сржж^щр пообыЛ Принцип метода' заключается в следующем: двуокись углерода, выделяющаяся после сожжения навески вещества в высокотемпературной печи при 1350-1400°С, улавливается поглотительным раствором с определенным значением pH. Изменение pH раствора фиксируется чувствительным потенциометром.
Для сожжения образцов использовалась печь с силито-выми нагревателями, изготовленная по чертежам ЦНИИЧер-мет (г. Москва).
В печь вставлены четыре фарфоровые трубки. Одна трубка служит для очистки кислорода, вторая - для сожжения анализируемых проб и две запасные. Запасные трубки позволяют переключать систему в ходе анализа.
Данный метод является экспресс-методом - на проведение одного анализа затрачивается 10-15 мин. Чувствитель-
44
Рис. Ц. установка для определения углерода потенциометрическим методом:
1 - кислородный баллон; 2 - редуктор; з - сосуд Тищенко с 4%-ным раствором KMnOzj в 20%-ном растворе КОН; 4 - сосуд Тищенко с концентрированной ~ H^SOzj;
5 - фарфоровая трубка ( d = 21 т 26 мм, I = 750т 850 мм); 6 - колонка с ангидроном; 7 -* колонка с аска-ритом; 8 - фарфоровая трубка для сожжения; 9 - си-литовая печь; 10 - силитовые нагреватели; 11 - сосуд с 5%-ным раствором К2Сг2О7 в концентрированной H2SO4 (или хромовым ангидридом, перемешанным с битым стеклом); 12 - трехходовой кран; 13 - трубка со стеклянной ватой; 14 - потенциометрическая ячейка; 15 - микробюретка с автоматической установкой нуля;
16 - потенциометр
ность определения - 0,06 мг углерода в анализируемой навеске образца. Относительная погрешность метода, определенная на стандартных образцах стали, составляет 3% и не изменяется в широком диапазоне концентраций углерода (0,06-0,006%).
Отбор проб (см. с.41).,
Ход анализа. Подготовка установки к работе (рис. 11). Силитовую. печь нагревают до 1000°С, пускают в систему кислород и продувают установку в течение 2 ч со скоростью 100 мл/мин. Затем температуру печи поднимают до 13 50°C. Соединяют потенциометрическую ячейку с фарфоровой трубкой для сожжения и пропускают кислород еще
45
Рис. 12. Потенциометрическая ячейка:
1 - стеклянный фильтр № 2;
2 - электролит; 3 - платиновый электрод (катод); 4 -крышка из органического стекла; 5 - каломельный электрод
15 мин. В потенциометрическую ячейку (рис. 12) наливают 100-120 мл электролита и пропускают кислород с такой скоростью, чтобы жидкость в сосуде энергично перемешивалась с образованием слоя пены 4-5 мм у стенок сосуда.
Дальнейшие операции проводят в токе кислорода. Потенциометрическую ячейку закрывают крышкой из органического стекла с четырьмя отверстиями. Через одно отверстие вводят платиновый электрод-катод, через другое - каломе— левый электрод сравнения - анод. К электродам присоединяют pH-метр и фиксируют положение стрелки гальванометра. В третье отверстие крышки вставляют микробюретку и по каплям прибавляют титрованный раствор едкого гидрата окиси бария и хлористого бария до установления pH—9,9. Значение потенциала при этом pH составляет 100 мВ, которое и принимают за первоначальное значение. Через четвертое отверстие выходит избыток кислорода в атмосферу.
» Проверка работы установки по стандартному образцу
стали. Для проверки правильности работы установки используют стандартные образцы стали с определенным содержа
46
нием углерода. Сожжение стандартного образца проводят периодически один раз в месяц и обязательно при сборке установки или после замены каких-либо деталей (фарфоровые трубки, колонки с поглотителями и др.).
Для сожжения используют образцы, аналогичные по со* держанию углерода анализируемым образцам, с тем же плавнем и в том же режиме.
Проведение контрольного опыта. Перед проведением контрольного опыта с плавнем определяют "фон* самой установки. С этой целью нагревают печь до температуры, как при проведении рабочего опыта, и в течение 10 мин продувают кислород. Титруют раствор в потенциометрической ячейке. Установку считают пригодной к работе, если на титрование затрачено не более 0,2 мл 0,01 н. раствора гидрата окиси бария.
Для проведения контрольного опыта с плавнем берут окись меди в фарфоровую лодочку, прокаленную в токе кислорода при 1200°С, в количестве 0,5-1 г.
Лодочку с окисью меди вводят при помощи свальной спицы с крючком на конце в трубку для сожжения, нагретою до 13 50°C, и сжигают в течение 10 мин. Замеряют количество титрованного раствора гидрата окиси бария, пошедшее на титрование контрольного опыта И.
При количестве окиси меди 0,5-1 г объем 0,01 н. раствора гидрата окиси бария, затраченного на титрование, не должен превышать 0,8 мл.
Анализ исследуемого вещества. Лодочку с навеской помещают в фарфоровую трубку для сожжения. Печь предварительно нагревают до 1350°С. Трубку закрывают резиновой пробкой со стеклянной трубкой, соединяют таким образом с кислородным баллоном и потенциометрической ячейкой, следят при этом за показаниями стрелки pH-метра. В первый момент поступления кислорода в потенциометрическую ячейку стрелка должна оставаться в первоначальном положении. По мере сгорания навески, выделения углекислого газа и его реакции с Ва(0Ю2рНраствора изменяется, следовательно, изменяется потенциал электрода, в результате чего стрелка pH-метра начинает отклоняться влево.
Сразу же добавляют титрованный раствор гидрата окиси бария и, таким образом, стрелка постепенно возвращается в первоначальное положение. В процессе сжигания пробы и поглощения углекислого газа в потенциометрической ячейке
47
стречку pH-метра все время удерживают в первоначальном положении путем добавления в ячейку титрованного раствора гидрата окиси бария. Титрованный раствор гидрата окиси бария прибавляют осторожно по каплям.
Титрование считают законченным, когда стрелка рН-мет-ра встанет неподвижно в первоначальное положение.
Если стрелка pH-метра встала в положение ниже первоначального, это указывает на то, что раствор перетитрован, и анализ считают испорченным.
Содержание углерода в анализируемом продукте рассчитывают по формуле *
Qc(%) =
/V, (l/f - У) 100
п tOQC
где /V - нормальность титрованного раствора едкого бария; 9С - грамм-эквивалент углерода; - объем титрованного раствора едкого бария, пошедший на титрование рабочей пробы, мл; V - объем титрованного раствора едкого бария, пошедший на титрование контрольной пробы, мл;
п - навеска образца.
2.4. ОПРЕДЕЛЕНИЕ ФТОРА, ХЛОРА, СЕРЫ
Для определения фтора в различных материалах используются известные методы Штарка [79], Берцелиуса [80] и их разнообразные видоизменения. Все аналитические методы отделения фтора основаны на ограниченной растворимости неорганических фторидов, летучести тетрафторида кремния или трифторида бора, устойчивости фторидных комплексов с алюминием, цикорием, железом, торием и титаном.
Для определения фтора используются весовые, объемные, колориметрические, нефелометрические, потенциометрические и амперометрические методы.
В настоящее время весовые методы используются лишь в качестве контрольных [81]. Наиболее распространенными из них считаются:
1) осаждение фтора хлоридом кальция в нейтральной или щелочной среде;
2) осаждение смешанного фторхлорида свинца из Нейтрального или слабокислого раствора;
48
3) определение в виде K^Si F6 •,
4) определение в виде В i F 3 .
Титриметрические методы применимы для растворов, содержащих фтор после выделения его из пробы. При содержании фтора >0,010 г погрешность составляет 0,5%гпри содержании фтора 0,00 5-0,0000 5 г - 10-20%.
Большинство этих методов основано на связывании фтора с различными металлами в виде с лабоионизованных соединений» В качестве титрантов используются растворы солей тория, циркония и других металлов. Конечная точка титрования фиксируется по возникновению окраски или изменению кислотности раствора.
Колориметрические методы определения фтора основаны на его способности разрушать окрашенные соединения Zr,Th Т i , Fe с цветными реагентами. При этом ионы фтора образуют бесцветные фториды. Изменение окраски фиксируется визуально или фотоколориметрически. В последнее время предложен метод прямого определения фтора [82] по тройному комплексу: фтор-РЗЭ - ализарин-комплексон, который окрашен в красно-фиолетовый цвет.
Кроме химических методов используются также радиохимические методы. Эти методы позволяют определять фтор непосредственно в исходных соединениях и характеризуются высокой чувствительностью и быстротой. Однако эти методы имеют ограниченное применение из-за сложности аппаратуры.
Во всех случаях при определении фтора необходимо предварительно отделение его от мешающих элементов. Для отделения применяются различные методы:
1) осалодение труднорастворимых соединений;
2) дистилляция фтористого водорода из сульфатных, фосфатных или хлоридных растворов [81, 83-85] с водяным паром;
3) дистилляция фтора кислотами без пропускании водяного пара [8б];
4) пирогидролиз;
5) хроматографические методы [86].
В литературе отсутствуют какие-либо указания по определению примеси фтора в кристаллическом боре. Наибольшая трудность в данном случае заключается в переведении кристаллического бора в раствор.
49
Для переведения кристаллического бора в раствор рекомендуется сплавление с содой, поташем, пиро- и персульфатом калия. В результате проведения серии опытов обнаружено, что для последующего определения фтора наиболее целесообразно проводить сплавление с Na2CO3 или NaКСОз . Большой избыток ионов натрия мешает определению фтора, поэтому необходимо предусмотреть отделение фтора
от натрия. Для этой цели можно использовать ионообменные смолы (как катиониты, так и аниониты). С анионитных смол фторид-ионы элюируются 0,05 М раствором хлористого аммония с pH = 8,8. После отделения от мешающих элементов фтор в растворе может быть определен любым из перечисленных выше методов.
В литературе имеется много методов определения летучих примесей серы и хлора в неорганических веществах, органических соединениях и полимерах.
Применяемые в аналитической химии методы концентрирования микропримесей: экстрация, осаждение, ионный обмен-не могут быть применены к определению летучих примесей серы и хлора в пресскомпозициях, так как требуется предварительное разрушение органической части самого вещества. Разрушение органической части проводят в замкнутых системах при высокой температуре, чаще всего в атмосфере кислорода или водорода, а выделяющиеся летучие примеси улавливают подходящим поглотителем.
К таким методам относятся многочисленные варианты сожжения в трубке, в колбе с кислородом, сплавление со щелочами, сожжение в кислородной бомбе и т.п.
Конечное определение летучих примесей после их улавливания, как правило, не представляет особого труда и решается обычными методами аналитической химии.
В настоящее время имеется целый ряд руководств по определению микропримесей в нефтях и нефтепродуктах [87, 88], природных объектах [89], биологических материалах [90-92] и т.п.
Данные по определению летучих примесей серы и хлора в пресскомпозициях на основе карбида бора с полипропиленом и кристаллического бора с полипропиленом в литературе отсутствуют.
За основу метода определения летучих примесей серы и хлора была взята методика разрушения полиэтилена сожжением в кварцевой трубке в быстром токе кислорода (2 — 4 л/мин) с предварительной деструкцией [93] .
50
Методики определения
1) Определение фтора.
Метод основан на реакции ионов фтора с солями тория с образованием нерастворимого фторвда тория. Избыток тория при титровании образует с ализарином красно-фиолетовый лак. Погрешность метода - 2-4% при содержаний в анализируемом растворе 1 мг фтора.
Метод применим для определения фтора в кристаллическом боре после его сплавления с содой.
Применяемые реактивы
Натрий углекислый Na2CQ3, безводный^ ГОСТ 83-63, хч, чда.
Кальций хлористый caci2> ГОСТ 4161-67, чда, насыщенный раствор. _
Кислота соляная HCI , ГОСТ 3118-67, хч, чда‘(б и 0,1 н. растворы^
Торий азотнокислый Th(№3)4, ТУ МХП У 266-51, чда. Приготовление раствора азотнокислого тория. 0,2 г. азотнокислого торияTh(ND3)4 растворяют в 0,8 л дистиллированной воды.
К раствору добавляют 50 мл 0,1 н. НС1, доводят до 1 л дистиллированной водой и тщательно перемешивают.
Натрий фтористый NaF •> , ГОСТ 4463-66, чда.
Катионная смола КУ-2. Подготовка катионита марки КУ-2. Катионит замачивают на сутки в 6 н. HCI, после чего отмывают дистиллированной водой до нейтральной реакции по метилоранжу.
Аморфный бор В, ТУ 6-02-585-70.
Ализариновый красный S, МРТУ 6-09-1485-64, чда. Приготовление ализаринового красного 5. Навеску индикатора 0,1 г растворяют в 10 мл 0,1 н. расавора NaOH и доводят дистиллированной водой до 100 мл.
Установление титра раствора Фтористого натрия. Берут навеску фтористого натрия'(0,04*0,04 5 гди взвешивают на аналитических весах с точностью до 0,0002 г. ' Навеску растворяют в мерной колбе объемом 1 л, доводят до метки дистиллированной водой, перемешивают. Подготовленный раствор переносят в полиэтиленовую посуду с плотно закрывающейся крышкой. Титр раствора рассчитывают по навеске NaF .
Установление титра раствора азотнокислого тория_____по
Фтору. В пять стаканов на 200 мл отбирают 1-5 мл титрирован-51
ного раствора NaF. Раствор доводят дистиллированной водой до 50 мл, добавляют 1 мл насыщенного раствора СаС1я> 1 мл раствора ализаринового красного 3 и по каплям 0,1 н. HCI до перехода сиреневой окраски в желтую, а затем еще 2,5 мл.
Одновременно готовят контрольную пробу, куда добавляют все используемые при анализе реактивы, за исключением MaF. В контрольную пробу прибавляют 3 мл раствора азотнокислого тория.
Рабочий раствор тщательно перемешивают и через 2-3 мин титруют раствором азотнокислого тория до цвета контрольной пробы.
Титр раствора Th по фтору рассчитывают по формуле
Т=a/V ,
где а - количество фтора, взятое в пробу для титрования, мг; И - объем раствора ThfNOj^, пошедший на титрование, мл.
Примечание, Титрование раствором Thпроводят из микробюретки с ценой деления 0,02 мл. Титры рабочих растворов проверяют через 2-3 мес.
Отбор проб. Навески проб 0,05-0,07 г'^ взвешенные с точностью 0,0002 г^ отбирают на воздухе на часовое стекло, на другое часовое стекло отбирают 6 г соды, часть ёе отсыпают в платиновый тигель, чтобы образовался слой толщиной не менее 0,5 см. Навеску анализируемого вещества переносят в платиновый тигель, засыпают оставшейся содой и осторожно перемешивают.
Ход анализа. Навеску кристаллического бора, смешанную с содой в платиновом тигле, закрывают крышкой и сплавляют в муфеле при 900-950°С в течение 2-2,5 ч.
Одновременно готовят контрольную пробу "свидетель". Для этого отбирают 0,05-0,07 г аморфного бора, не содержащего фтора, смешивают с 6 г соды в платиновом тигле и сплавляют, аналогично анализируемой навеске.
Плавы рабочей и контрольной проб охлаждают, выщелачивают горячей дистиллированной водой 3-4 порциями по 10-15 мл.
Растворы фильтруют в мерные колбы на 50 мл через фильтр "синяя лента", доводят до метки дистиллированной водой.
52
В стаканы на 100 мл помещают подготовленную смолу КУ-2 приблизительно на 1/5 объема стакана и приливают аликвоту 20 мл анализируемого раствора. Растворы в стаканах периодически перемешивают до прекращения выделения пузырьков углекислого газа. Затем растворы отфильтровывают в стаканы на 200 мл через фильтр "белая лента". Смолу три-пять раз отмывают декантацией дистиллированной водой по 5-7 мл, после чего ее переносят на фильтр и промывают еще 5-10 мл воды. Общий объем раствора (как рабочего, так и контрольного) не должен превышать 50-70мл.
К растворам прибавляют 1 мл насыщенного раствора 1 мл раствора ализаринового красного S и по каплям 0,1 н. раствор HCI до перехода окраски в желтую, азатем еще 2,5 мл. *
В раствор контрольной пробы "свидетель" вносят те же реактивы, что и в рабочий раствор, и 3 мл титрованного раствора Th (NO3)4 •
Рабочий раствор титруют из микробюретки азотнокислым торием до перехода окраски от желтой к розовой до сравнения с окраской "свидетеля".
Содержание примеси фтора рассчитывают по формуле
&F(%) =
(Vf-V)T А п В
100
где - объем раствора азотнокислого тория, пошедший на титрование рабочей пробы, мл; ¥ — объем раствора азотнокислого тория, добавленный в контрольную пробу для приготовления "свидетеля", мл; Т — титр раствора азотнокислого тория по фтору, мг/мл; В - объем аликвот анализируемого раствора, мл; А - общий объем анализируемого , раствора, мл; п - навеска, мг.
2) Определение серы и хлора
Метод основан на сожжении с предварительной деструкцией вещества в пустой кварцевой трубке в быстром токе кислорода ( 2 л/мин) и улавливании летучих примесей в , растворе соды. Сера и хлор определяются из аликвот раствора титрованием азотнокислым свинцом и азотнокислой ртутью соответственно.
£ Определяемый предел для серы - 3,2‘10"“4%, для хлора-Г 7*10"^% (при навеске 2 г).
? Относительная погрешность при оС = 0,95 для се м 10%, для хлора - 5%.
&
г
Методика применима для определения летучих примесей серы и хлора в пресскомпозициях на основе карбида бора с полипропиленом (ПКБ) и бора кристаллического с полипропиленом (БСП).
Применяемые реактивы
Кали едкое КОН, ГОСТ 4203-65, хч, 30%-ный раствор. Углекислый натрий ГОСТ 83-63, хч, 0,05 н. раствор. Свинец азотнокислый РЬ(М03)2, МРТУ 6-09-23 70-65, хч, 0,01 н. раствор.
Азотная кислота HN03, ГОСТ 4461-67, хч, 0,0 2 н. раствор. Гидроокись аммония NH^OH , ГОСТ 3760-64, хч, 0,02 н. раствор.
Мочевина NH^CONH^ , ГОСТ 6691-67, чда, 1%-ный водный раствор.
Ацетон СН3СОСН3, ГОСТ 2603-63, перегнанный, чда.
Дитизон C6H5NHNHC(==C')N я NC6h6 , ГОСТ 10165-62, чда, 0,05%-ный ацетоновый раствор (свежеприготовленный).
Уксусная кислота СН3С00М, ГОСТ 61-69, 20%-ный раствор. Бромфеноловый синий, ТУ МГУХП 271-59, 0,1%-ный раствор в 20 %-ном спирте.
Дымящая азотная кислота HN03 .
Этиловый спирт с2н5он , ТУ 19П-39-69.
Азотнокислая ртуть Hg(но3)2, ГОСТ 4520-68, хч, 0,01 н. раствор.
Натр едкий NaOH, ГОСТ 4328-66, хч, 0,2 щ раствор. Бромфеноловый синий с19нюВг/)056 , ТУ МГУХП 271-59, 0,025%-ный раствор, готовится из 0,1%-ного раствора.
Дифе нилк ар базой, ГОСТ 2636—51, 2%-ный спиртовой
раствор.
Допускается применение реактивов других ГОСТов (с наименьшим содержанием хлоридов).
Материалы и оборудование
Установка для сожжения (рис. 13).
Автоматическая микробюретка емкостью 10 мл с ценой деления 0,02 мл. Химические стаканы на 100 мл.
Мерные колбы на 50 мл.
Отбор пробы. Навеска (1-2 г) отбирается с точностью 0,0002 г в алундовый тигель.
54
Рис. 13 . Установка для сожжения:
1 - баллон с кислородом; 2 - реометр РДС; 3 - очистительная система для кислорода (сосуды*Тищенко с 30%-ным раствором КОН и колонка Фрезениуса с аскаритом); 4 - предохранительный сосуд; 5,6,12 - шлифты; 7 - отросток для деструкции; 8 - алундовый тигель; 9 - печь типа ТГ-О2; 10 печь типа Т 40/600; И - кварцевая трубка; 13,14 - абсор -беры
Ход анализа. Сожжение анализируемых материалов проводят на установке (рис. 15).
Электропечь Т—40/600 включают через лабораторный автотрансформатор (ЛАТР) в сеть и в течение 1 ч нагревают до 900-1000°С.
Затем кварцевую трубку (рис. 14) помещают в электропечь Т-40/600. Отросток для деструкции одновременно помещают в электропечь ТГ-0 2. Алундовый тигель с навеской ПКБ или БСП помещают через шлиф в отросток для дест-
1 - кварцевый отросток; 2 - патрубок; 3 - пористый кварцевый фильтр № 2
55
диняют к трубке через шлиф. Систему продувают кислородом в течение 5 мин со скоростью 2 л/мин по реометру. Кислород выводят через резиновый шланг в вытяжной шкаф. В абсорберы (рис. 15) наливают поглотительные растворы 15 мл 0,005 н. Na2co3l абсорберы присоединяют к установке.
Печь ТГ-0 2 включают в сеть через ЛАТР и постепенно нагревают от 20 до 950°С в течение 2 ч, после чего печь выключают, сожжение заканчивают.
Дожигание продуктов разложения на стенках трубки проводят передвижением ее в печи в течение 10 мин. После окончания сожжения от системы отключают ток кислорода, выключают печь. Абсорберы отсоединяют от установки и поглотительные растворы из них вместе с промывными водами количественно переносят в мерную колбу на 50 мл.
56
Раствор доводят до метки бидистиллированной водой.
Перед проведением сожжения ПКБ и БСП в новой трубке или с новым баллоном кислорода проводят ^фоновый" опыт в тех же условиях.
Содержание серы и хлора в этих опытах не должно превышать содержания их в реактивах, В случае превышения этого количества проводят дополнительную очистку кислорода и трубки.
Для определения серы аликвоту рабочего раствора 25 мл, взятую в стакан на 100 мл, упаривают на кипящий водяной бане с 1—2 каплями дымяТпей азотной кислоты для окисления 50£~ в SOj'. Сухой остаток растворяют в 1-2 мл бидистиллированной воды и упаривают досуха. К остатку добавляют 1 мл раствора мочевины и вновь проводят упаривание досуха. К сухому остатку добавляют 4 мл бидистиллированной воды, раствор нейтрализуют по бромфеноловому синему 0,02 н. раствором ИЩОМ или 0,02 н. HNO3 до оливково-зеленой окраски, добавляют 1 мл 20%-ной уксусной кислоты, 25 мл ацетона и 20 капель раствора дитизона.
Раствор титруют 0,01 н. раствором РЬ(К103)2до изменения цвета индикатора от изумрудно-зеленого через темно-серый до неисчезающего вишнево-красного. Титрование проводят по каплям с тщательным перемешиванием стеклянной ;палочкой.
' Установку титра раствора Pb(NO3)2 проводят по 0,01 н. H2SD4 с теми же реактивами и в тех же условиях, как и
.определение серы из рабочих проб.
Содержание серы вычисляют по формуле
" ,
Где V - количество 0,01 н. раствора Pb(N0j)2, пошедшее на •титрование рабочей пробы, мл; - количество 0,01 н. раствора Pb(NO3)21 пошедшее на титрование холостой пробы, мл; 0,16 - пересчетный коэффициент на серу при 0,01 н. растворе РЬ (К103)2? п * навеска, мг; 1//п - часть рабочего раствора.
Перед каэвдым определением. проводится холостой опыт на содержание серы в реактивах.
Для определения хлора к аликвоте рабочего раствора 10-25 мл добавляют одну каплю индикатора бромфенолового синего (0,025%), раствор нейтрализуют 0,2 н. НЫО3 до желтого цвета, добавляют одну каплю 0,2 н. Na ОН и кипя-
57
тят 1 мин для удаления С02- После охлаждения до комнатной температуры к раствору добавляют 2 мл 0,2 н. HNO3 и 5 капель дифенилкарбазона ♦ #
Раствор титруют 0,01 н. НдСИОз'Ь до получения розовофиолетовой окраски. Титрование рекомендуется проводить со *свидетелем". В качестве "свидетеля* используется "холостая" проба на реактивы. Установку титра раствора Hg(N03>; проводят по 0,01 н. Naci с теми же реактивами и в тех же условиях, как и определение хлора из рабочих проб.
Содержание хлора вычисляют по формуле
п (о/}. (^^1) /о^5 т юо
И - количество 0,01 н. раствора Hg(N03)2? пошедшее на титрование рабочей пробы, мл; У1 г- количество 0,01 н. раствора Hg(N03)2, пошедшее на титрование -холостой пробы; мл; 0,3545 - пересчетный коэффициент на хлор при 0,01 н« HglNOsV, п * навеска, мг; \!пг - часть рабочего раствора.
Одновременно проводят холостой опыт на содержание хлоридов в реактивах.
2.5. ОПРЕДЕЛЕНИЕ МЕТАЛЛИЧЕСКИХ ПР^УГ-^Й СПЕКТРАЛЬНЫМ МЕТОДОМ
Известны многие спектральные методики анализа бора и его соединений на содержание примесей.
Прямое сжигание бора в источнике света используется редко вследствие появления в спектре сильного фона за счет излучения окислов бора, образующихся в разряде [102,105, 109] . Для уменьшения этого эффекта возбуждение спектра проводят в атмосфере азота [94], аргона [108] или используют спектрографы высокой дисперсии [106, 107]. Это позволяет определять в боре, карбиде и нитриде бора примеси с чувствительностью 5*10*4 _ 1*ю*^% и даже (1 -4) 10*5 [108].
Обычно же для определения малых примесей в боре (л< 10*3 _ п. jq-4%) применяют концентрирование их путем удаления бора в виде борноэтилового или борнометило-вого эфира* [95, 101, 104, 109], борного ангидрида [96], хлористого' [ 98] , или фтористого бора [99, lOCj. Концентрат примесей анализируют на основе сульфата калия [95J ,
58
спектрально чистого порошка никеля [96, 97], хлористого серебра [99*] и т.п. Анализ кристаллического бора на Al ,Мд, Si, Fe проводят с чувствительностью 5’10"^%. В никелевом концентрате определяют Mg, Fe,Mn, Si, Cu,Zn,Pb,3n, Sb,Bi с чувствительностью 5е 10"^ - 2*10"“^%. Концентрат на основе хлорида серебра анализируют с чувствительностью 5*10~5% для№ и Та и 1.10""^% для Мп и По.
Методы, предусматривающие предварительное концентрирование примесей [95 - 101] , применяют для определения ограниченного числа элементов и требуют много времени для проведения анализам Применение распыления аэрозоля раствора Н3ВО3, получаемого с помощью ультразвука, не дало увеличения чувствительности больше чем п» 10"^% [110].
В известных нам работах не описаны спектральные методы контроля чистоты борвда титана и смесей кристаллического бора или карбида бора с полипропиленом.
Основная трудность, возникающая при работе с боридом титана, - это окисление пробы, которое начинается уже при 900-1000°С [111] . Температура пробы в электроде
( > 1200°С) достаточна для образования окисных соединений бора, создающих сильный фон в спектрах.
При сжигании смесей полипропилена с бором и карбидом бора благодаря быстрому сгоранию органического вещества до летучих продуктов наблюдается нестабильность возбуждения спектров (выброс пробы во время экспозиции) и связанное с этим занижение интенсивности линий некоторых элементов, которые увлекаются из пробы отходящими газами и быстро уводятся из зоны разряда.
В процессе разработки методов анализа было изучено влияние добавок угольного порошка, ^носителей*, формы электродов и других факторов на стабильность горения дуги, ^интенсивность возбуждения бора к примесей. Проведенные. j наблюдения дали возможность подобрать сравнительно прос- I тые и надежные методики контроля чистоты бора, карбида \ бора, борида титана и пресскомпозиций на содержание боль- / того числа примесей прямыми спектральными методами анализа. Так, метод анализа бора предусматривает определение Hg, As, Р, которые ранее в боре не определялись, кроме того,. чувствительность определения Sb,cdrzn, Со, С г и др. значительно повышена по сравнению с известными ^прямыми методами анализа бора [94, 10 7-111] . Метод не требует защитной атмосферы инертного газа и спектрографа j
59
большой дисперсии. Анализы выполняют на спектрографах средней дисперсии отечественного производства.
Методики определения
1) Определение 32 элементов в боре [103] и карбиде бора.
Анализ бора проводят методом трех эталонов. Эталоны готовят на основе аморфного бора. В качестве носителя используют углекислый литий (1% Li добавляют к массе пробы). Эталоны и пробы разбавляют в отношении 1:1 угольным порошком и сжигают в дуге постоянного или переменного тока. Спектры фотографируют одновременно на двух спектрографах с кварцевой и стеклянной оптикой.
Чувствительность определения Зв10~5 - 1*10"^%. Относительная погрешность определения 6-20%.
Анализ кристаллического бора и карбида бора по сравнению с анализом аморфного бора имеет небольшие особенности, которые объясняются различной дисперсностью порошков и наличием кристаллической структуры у кристаллического бора и карбида бора, а также различием плотностей анализируемых материалов. По насыпному весу (весу пробы в объеме кратера электрода) пробы кристаллического бора тяжелее аморфного бора в 1,16 раза, а пробы карбида бора - в 1,33 раза. Градуировочные графики строятся по эта-зонам, приготовленным на основе более легкого аморфного бора. Поэтому результаты для карбида бора и кристаллического бора, полученные по этим графикам, необходимо уменьшить на соответствующий коэффициент, равный отношению насыпных весов проб и эталонов. А так как плотность проб от партии к партии меняется, то коэффициент пересчета необходимо уточнять для каждой партии. Правильность результатов при анализе карбида бора и кристаллического бора по эталонам, приготовленным на аморфном боре, проверена путем сравнения данных, полученных параллельно атомноабсорбционным, пламенно-фотометрическим, химическим и разработанным спектральным методами.
Отбор проб. Отбор проб для анализа, взвешивание, очистку от поверхностных загрязнений проводят на воздухе в условиях защиты от возможных атмосферных и других внешних загрязнений.
Навески для анализа (0,5-1 г) взвешивают на часовом стекле на аналитических весах с точностью 0,0002 г.
60
В отобранные пробы к 1 г дисперсного бора или; карбида бора добавляют 1 г угольного порошка и 0,0 53 г углекислого лития.
Материал компактных проб перед отбором измельчают. Для первичного дробления куски вещества завертывают в несколько слоев полиэтиленовой пленки и разбивают. Размер частиц вещества после первичного дробления не должен превышать ^1 мм. Тонкое измельчение раздробленного материала осуществляют истиранием его в ступках из карбида бора.
К измельченной пробе добавляют уголь и углекислый литий. Смеси перемешивают в плексигласовой ступке в течение 10-20 мин.
Применяемые реактивы
Применяют реактивы квалификации чда, хч, осч, спч.
Алюминий окись А120з,ТУ О33 69-65.
Барий окись ВаО, МРТУ 6-09-4530-67.
Бериллий окись ВеОтВТУ МХП 3420-52.
Бор аморфный В, ТУ 6-02-585-70.
Ванадий окись V2O5, МРТУ 6-09-2031-64.
Висмут окись В»203> МРТУ 6-09-2012-64.
Галлий окисьйа2°з » МРТУ 6-09-5183-68.
Железо окись Fe203, МРТУ 6-09-2388-65.
Золото металлическое ’99,99, ГОСТ 7222-54.
Индий сернокислый Ir^CSOz^j, МРТУ 6-09-5492-68.
Кадмий окись Cdo, МРТУ 6-09-20 29-64.
Кальций углекислый Сасо3? МРТУ 6-09-34-62.
Калий углекислый К2созг МРТУ 6-09-1354-64.
Кобальт окись1 cqzo31 ТУ 20П-26-69.
Кремний окись S1O2., МРТУ 6-09-3267-66.
Литий углекислый и2со3^ МРТУ 6-09-4843-67.
Магний окись MgQ, МРТУ 6-09-1546-64.
Марганец окись Мп02, МРТУ 6-09-3353-66.
Медь окись СиО, МРТУ 6-09-3866-67.
Молибден окись Мо03, МРТУ 6-09-3901-67.
Мышьяк окись As2O3, ВТУ ВНИИ ХФ.
Натрий углекислый Na2C03, МРТУ 6-09-2819-66.
Никель окись NiiQ3, МРТУ 6-09-3958-67.
Олово окись SnO2j МРТУ 6-09-254-63.
Палладий хлористый PdCl2, МРТУ-6-09-1964-64.
Ртуть окись НдО, МРТУ 6-09-331-63.
61
Рубидий азотнокислый 1?ЬЫОз7 МРТУ 6-09-2720-65.
Свинец окись РЬО, МРТУ 6-09-994-64-Сурьма окись 5Ьг.0з; МРТУ 6-09—6704-70.
Таллий окись T003, МРТУ 6-09-4876-67.
Титан окись Ti02?МРТУ 6-09-5083-68.
Угольный порошок^ РОСЧ-7-4
Угли спектральные диаметром 6 мм, безборные^
ТУ 16-538.240-74.
Фосфорнокислый аммоний двузамещенный (N н2 н POz, 7 ГОСТ 3772-64.
Хром окись Cr20j. МРТУ 6-09-6250-69.
Цинк окись ZnO| МРТУ 6-09-1604-64.
Материалы и оборудование
Спектрограф ИСП-22 или любой другой с кварцевой оптикой.
Спектрограф ИСП-51 с камерой F = 270.
Генератор дуги переменного тока ДГ-2.
Генератор дуги постоянного тока 120 В, 20 А (применяют в случае проведения анализа в дуге постоянного тока).
Микрофотометр МФ-2 или МФ-4*
Фотопластинки спектральные^. ТИП JU, репродукционные штриховые сверхконтрастные, панхром, инфра-760.
Пленочный фильтр пропускаемостью 35%.
Ступки плексигласовые^агатовые и из карбида бора диаметром 90-120 мм.
Ход анализа. Эталоны готовят на основе аморфного бора. Примеси в основу вводят из окислов^из термически неустойчивых солей определяемых элементов, кроме Au и Pd7 которые добавляют из солянокислых растворов. Для уменьшения погрешности при взвешивании предварительно готовят четыре смеси примесей. Расчет смесей производят с учетом чувствительности анализа (см. табл. 4) и заданной концентрации примесей в эталонах (см. табл. 3)
Смеси элементов растирают по 30-40 мин в агатовой ступке. Из смесей примесей готовят основную смесь. Нап-римеру'для эталонов состава, указанного в табл. 3, смешивают 0,3642 г смеси 1; 0,2 г смеси 4; 0,0895 г смеси 2; 0,0400 г смеси 3. Получают 0,6937 г основной смеси. Угольный порошок добавляют в эталоны и пробы в отношении 1:1, углекислый литий используют в качестве носителя, его вводят в количестве 1% И к массе пробы.
Расчет смесей
Таблица 1
X
Элемент * Формула применяемого вещества Содержание элемента в смеси, мг Содержание вещества в смеси, мг
Смесь 1
Кадмий CdO 30 34,3
Железо Fe2°3 30 42,9
Мышьяк As203 30 39,6
Сурьма ЗЬгОз 30 35,9
Цинк ZnO 30 37,3
Хром Cr2O3 60 87,7
Ванадий V205 200 357,0
Титан Ti 02 200 333,6
Фосфор (NH4)2HP04 200 852,7
E = 1,821 г
Смесь 2
Марганец MnO2 6 9,5
Висмут ВЦО3 20 22,3
Медь CuO 20 25,0
Свинец PbO 20 21,5
Магний MgO 60 99,5
Алюминий AI203 60 113,4
Олово SnO2 60 76,2
Барий BaO 100 111,6
Кремний SiO2 100 213,9
Кальций caco3 200 499,4
Никель Ni203 200 281,7
Молибден M0O3 2000 3000,0
E = 4,4740 г
Смесь 3
Бериллий BeO 6 16,6
Индий In2(5O/4)3 30 67,6 де,
Золото р-pH АиСЦТАц4^05 ' 20 0,465
Галлий Ga203 20 26,9 .
63
Продолжение табл. 1
Элемент Формула применяемого вещества Содержание элемента в смеси, мг Содержание вещества в смеси, мг
Палладий р-р PdCl2TpdWi3jMr/Hn 60 1,69*
Таллий Т1203 100 111,7
Кобальт С°2О3 200 281,4
Ртуть НдО 300 323,9
Бор В до 2 г
Смесь 4
Натрий Na 2^0 10 23,1
Калий К2СО5 20 35,3
Рубидий JbND3 200 345,2
Бор В до 1 г
В миллилитрах.
Первым приготовляют 1У эталон без угля и лития (в 10 г этого эталона содержится 0,5203 г основной смеси и 9,4797 г бора). Разбавлением эталона основой и угольным порошком, содержащим литий (на 10 г угля берут 0,5324 rLi2.CO3), получают все необходимые эталоны (табл. 2). Эталоны растирают в плексигласовой ступке в течение 10-30 мин.
Приготовление эталонов
Таблица 2
№ эталона Эталон 1У без угля и лития, г Основа - бор, г Угольный порошок + Li2CO г
1 0,2 5,800 6,3192
П 0,6 5,400 6,3192
Ш 2,0 4,000 6,3192
1У 6,0 — 6,3192
64
Содержание элементов в эталонах, масс, %
Элементы 1 П Ш iy
Be , Мп 3 • io-5 9.-10"5 3-10“4 9.-10"4
Pb, Си, Bi, Ga, Au 1,-IO"4 3rl0“4 l:10"3 3tl0-3
In l,5:10“4 4,5.-10“4 : 1.5.-10"3 4,5-Ю"3
Sn, Al, Mg , Pd 3:10~4 9:10“4 з.-10-з 9-Ю"3
S.i,'Ba,TI, Na 5-10"4 i.Sficr3 5лЮ"3 1,5,-IO-2
Ni ,Ca,Co,К 1.-Ю”3 _ 3.-Ю"3 1:10”2 3:10"2
Zn,Sb, As, Fe,Cd, Ho 1,5 • 10 4,5-Ю"3 1.5-10"2 4,5.tl0"2
Cr 3,O-lCTd Э^Ю"3 3-10-2 9;10”2
P,Ti,V, Mo,Rb 1 . Ю-3 3,-Ю"3 i.-ier1 ЗП0-1
Возбуждение спектра эталонов и проб можно производить как в дуге переменного, так и в дуге постоянного тока.
В качестве электродов используют угольные стержни диаметром 6 мм. Нижний электрод имеет кратер глубиной 5 мм, диаметром 3,8 мм. Верхний электрод для дуги постоянного тока затачивают на конус с диаметром площадки 2 мм, для дуги переменного тока верхний электрод имеет форму ” зонтика* (рис. 16).
Рис. 16. уголные электроды (а) и стальной стержень (б) для уплотнения проб в электродах
Анализируемые пробы ( ~ 70 мг) засыпают в кратер электродов и уплотняют стальным стержнем с иглообразным выступом на конце (диаметр иглы у основания - 1 мм, высота — 5 мм).
Возбуждение спектра в дуге переменного тока производят от генератора ДГ-2, Сила тока в начале экспозиции -6 А, через 4-5 с самопроизвольно возрастает до 7А. Экспозиция на ИСП-51 - 25 с, на ИСП-22 - 60 с. Через 30 с область спектра > 2950^ (на ИСП-22) перекрывают затемненной, шкалой длин волн, для чего поворотный механизм шкалы соединяют со стопорным винтом. Приспособление для перекрытия спектра удобно использовать со спектрографом ИСП-22. При работе на ИСП-28 и ИСП-30 вместо перекрывания спектра можно применять пластинки меньшей чувствительности.
Возбуждение спектра в дуге постоянного тока производят при силе тока 7А - 15 с и 12А - 35 с. Изменение тока осуществляют с помощью переключения сопротивления в цепи дуги. Общее время экспозиции на спектрографе ИСП-51 - 25 с. Область спектра J> 2950 Д перекрывают на ИСП-22 шкалой длин волн через 25 с.
Спектры фотографируют одновременно на двух спектрографах ИСП-22 и ИСП-51 с камерой F = 270, оптические оси которых располагают под прямым углом друг к другу. Ширина щели спектрографа ИСП-51 - 0,008 мм, ИСП-22 -0,010 мм (и 0,015 мм в дуге переменного тока).
Свет от источника возбуждения фокусируют на щели спектрографов с помощью линз с промежуточной диафрагмой. Размер прорези диафрагмы у спектрографа ИСП-22 - 1,5х х25 мм, у ИСП-51 - 0,5x25 мм [103, 112].
Электроды устанавливают по их теневой проекции на диафрагму, расстояние между электродами в аналитическом промежутке - 1,5 мм. Регистрацию спектра на спектрографе ИСП-22 производят с помощью фотопластинок спектральных (тип Ш) до области 2450А и репродукционных штриховых сверхконтрастных в длинноволновой области спектра. Кассету спектрографа ИСП-51 заряжают фотопластинками панхром и инфра-760 на области 6800-8000 X.
Линию натрия ослабляют пленочным фильтром пропускае-мостью 34-35%, который укрепляют в прорези бленды в кассетной части спектрографа ИСП-51.
Каждый эталон или пробу фотографируют по 3-4 раза.
По аналитическим линиям (см. табл. 4) строят градуи-
66
Таблица 4 Аналитические линии концентраций определения примесей в боре
Элементы ^Аналити-ческие линии, А Пределы определяемых концентраций в
дуге постоянного тока, % дуге переменного тока, %
_ 9 —4 —2
Цинк Zn 1 2138,56 ЗгЮ^-ЫО 5:10 -1-10
Znl 3345,57 1,5:10~3-3-10“2 1,5:10"3-3*10“2
Кадмий П2265,02 1,5:10 -1,5’10 5:10 -5*10
Cd I 2288,02 Сурьма 12311,47 Sb —4 -2 1,5 • 10 -1,5:10 -4 -2 6-10 -3:10 -4 -2 1:10 -1:10 _ о _О 1:10 -5:10
Сурьма I 2598,05 Sb -4 -2 5:10 -5:10 _ о _о 1:10 -5:10
Барий ВаГТ4934,02 Мышьяк! 2349,84 As -4 -2 5:10 -1,5:10 —2 —1 5:10 -1:10 _ о _9 1:10 -3:10 - i.s.’io^-r.io”1
Бериллий I 2348,61 ве — 5 —4 1,5:10 -5:10 1,5:1о"5-1:Ю"3
Кобальт I 2407,25 Со -4 -2 5:10 -5:10 —2 —1 1:10 -1:10
ЗолотоАиТ 2427,95 Кремни й5®6! 2514,38 Si 7?10-5-7г10“3 —2 —9 1:10 -3-.10 7:10-5-5.-10-3 _ о _9 1:10 -3:10
-4-2
Ртуть Hg 1 2536,52 5:10 -5.*10 ФосфорР 1 2553,28 б-Ю^-б^ю”1
Марта-нец Мп П2593,73 3:10 -3:10‘
—2 —9
1-10 -5’10
-2 -1
5*10 -5*10
LkA&IO"3
3OxxFe 12599,57 1,5:10"3-5-10"2 1,5:10”3-5-10 _ —4 —2 —2 —9
Магний 12782,97 3:10 -3:10 1:10 -5*10
Мд
67
Продолжение табл. 4
Элементы Аналити-ческие о линии, А Пределы определяемых концентраций в
дуге постоянного тока, % дуге переменного тока, %
Свинец 12833,07 РЬ lilO"4-!; ю"2 5;10"5-3:10“3
ОловоSn I 2839,99 5.-10“5-3:10"3 -4 -2 1,5-10 -1:10
ХромСг П2843.25 1:1о“3-з:1о“2 1,5.-10“3-1-.10-1
О 14254,35 I.-IO^-I-.IO"1 1:10~3-1:10-1
Галлий 1 2874,24 &а 5.'10-5-3:10~3 5,-1О“5-3-1О“3
Индий In 13039,36 шо^-з.-ю"3 i-io^-s.-io"3
НикельЮ 130 50,82 1:10"3-2-10“2 l.SilO^-S'.lO”2
Алюми-
ний А1 13082,16 ItlO^-S’lO”3 —4 _ Q 3:10 -5:10
Й1Т3961.53 1-10-4-1-10~2 —4 -2 2:10 -3-10
Висмут 13067,72 1:10”4-2:10'"3 1-.10“4-1:ю"3
Титан Ti П 3088,03 -2 -1 1:10 -3;10 -2 -1 1:10 -1’10
Ванадий 13185,40 -2 —1 1:10 -3:10 -2 -1 3:10 -3:10
V Молибден I 3170,3 5 3:10~2-3,,10"1 -2 -1 1:10 <1:10
Мо Палладий I 3242,70 -4 -2 3:10 -1:10 -4 -2 3:10 -Г. 10
Pd Медь**Си1 3273,96 .4 —Ч 1:10 -1:10 l.-io^-l-.io"3
Каш3цийХХТ4226,73 -4 -2 3:10 -1’10 -4 -2 3:10 -1*. 10
ТаллийТ! 15350,46 -4 -2 5:10 -2’10 -4 -2 5:10 -2*10
—4 —9 Натрий Na I 5889, 95 5:10 -5/10 1- io^-1-io-i
Калий К I 7664,90 1: Ю”3-!-10-1 1:10 °-1-10
Рубидий Bbl 7800,23 -2 -1 1-10 -3-10 -2 -1 3:10 -3:10
Линия кадмия Cd I 2288,0 2 используется при отсутствии мышьяка.
Чувствительность определения этих элементов занижена из-за присутствия их в основе.
68
ровочные графики в координатах S-LgC,AS-lgC или LgJ-LgC, LgJi- Lg 32-LgC.
При анализе по относительной интенсивности в качестве линий сравнения используют линии лития ои бора;Li j 2475 , 29А, Li I 2562, 54A, В f*2088, 8 ЗА, а также фон у аналитических линий.
Условия приготовления проб и съемки спектров для кристаллического бора и карбида бора сохраняют такими же, как и для аморфного бора, т.к. экспериментально . установлено^ что время выгорания примесей из проб кристаллического бора и карбида бора, смешанных с угольным порошком (1:1) с добавлением 1% Li к массе пробы (в ввде 1л2С03\ и эталонов на основе аморфного бора одинаково: основная масса примесей выгорает в первые 30-40 с экспозиции (общая экспозиция - 60 с).
2) Определение 22 элементов в бориде титана [112].
Анализ борида титана проводят методом фракционной дистилляции с ^носителем* в дуге переменного тока.
Пробы и эталоны разбавляют угольным порошком в отношение^ 10:1. Элементы сравнения - кобальт и галлий (1 х «10 % каждого к массе пробы), 'носитель* - К2СОз (0,6%
калия к массе пробы) добавляют вместе с угольным порошком.
Чувствительность анализа 1*10 -1-'10~ %. Относи-
тельная погрешность определения 10-20%.
Отбор проб. Отбор проб проводят аналогично описанному на с.60, 61 .
Для приготовления пробы к 1 г дисперсного борида титана добавляют 0,1113 г угольной смеси и тщательно перемешивают в плексигласовой ступке в течение 2П лн
Угольную смесь готовят заранее следующим образом: 5 г угля смачивают 0,25 мл раствора C0SO4 (ТСо =20мг/мл) и 0,44 мл раствора (Т&а =11,43 мг/мл). Смесь
просушивают при 100°С в течение 1 ч, добавляют 0,530 г K2GO3 (300 мг К ) и хорошо перемешивают. Эту же смесь используют для приготовления эталонов.
Применяемые реактивы
Применяют реактивы, перечисленные на с.61. Исключают соли Au , In , Pd , Hg , РЧЬ ,Т1 , примеси которых в бориде титана не определяют.
69
ЛЛатериалы и оборудование
Для проведения анализа используют материалы и оборудование указанные на с. 62. Генератор дуги постоянного тока для анализа Ti В2 не применяют. Используют спектральные фотопластинки тип Ш, тип I, панхром.
Ход анализа, В к^естве основы для приготовления эталонов используют одну из наиболее чистых проб борида титана или готовят из чистых мелкодисперсных частиц титана и бора сжиганием смеси их в определенных условиях. Примеси в эталоны вводят в виде окисей или термически неустойчивых солей методом, описанным на с. 62 Правильность результатов, полученных этим методом, подтверждается путем сравнения данных анализа одних и тех же проб из порошков и растворов [112].
Для нижних электродов используют угольные стержни, заточенные в форме '‘'рюмки* (диаметр кратера - 3,9 мм, высота - 4,5 мм; диаметр ножки - 2,4 мм, высота - 3 мм). Верхний электрод имеет форму *зонтика* (диаметр площадки - 6 мм, толщина - 1,3 мм; диаметр ножки - 2,4 мм, длина - 7 мм), Высота Электродов - 30 мм (рис. 17).
Пробы и эталоны сжигают в дуге переменного тока в режиме *спекания*. В момент зажигания сила тока равна 8 А, через 1-2 с она самопроизвольно поднимается до 10 А и остается такой до конца экспозиции (45 с для ИСП-22 и 13 с для ИСП-51). Участок спектра с >2900 д на ИСП-22 через 15 с после начала горения дуги перекрывают затемненной шкалой длин волн.
Кассету кварцевого спектрографа заряжают спектральными фотопластинками тип Щ до 2700 А и тип 1 на длин-
Рис. 17. Угольные электроды для анализа борида титана
70
новолновую область спектра. По аналитическим линиям (табл. 5) строят градуировочные графики в координатах s-lgC, AS-LgC. При анализе по относительной интенсивности используют линии ^сравнения Со 12521,36 А, J 3159, 67 А, Со J 3449,47 А (для железа, «никеля, хрома, меди) и Ga
2450,07 А, ба 12719,65 А (для алюминия, кремния, кальция, магния, свинца, олова).
Таблица 5
Аналитические линии и интервалы определения примесей в бориде титана
Определяемый элемент Аналитические линии, А Пределы определяемых концентраций,%
Фосфор Р 12136,20 3-ю“3-6.‘1о“2
Р 12149,11 —Ч —2 3:10 -6.-10
&1 Si 1 2211,75 -3 -2
Кремний 5-10 -5:10
Si I 2435,16 1,10“3„5:10“2
Sfcl Алюминий Al I 2263,45 l!10~2-2?10"'1
Al 1 2367,06 l:10“3-5.-10“2
Al 13082,16 _4 -2 5;:10 -1:10
Кадмий Cd П2265.02 3-.10"l-3:10"?
Cd I 2288.02*3 —4 —2 l;10 -3.-10
«9 Никель Ni 1 2311,0 . 1:10 -1:10
Ni 1 3050,82 MO^-riO”1
Сурьма Sb I 2311,47 -4 -2 •3tl0 -1-10
Барий Ba П 2335,27 2:10~3-4-10“2
Ba П 4934,09 -3 -2 1:10 -4:10
Бериллий Be I 2348,61 IrlQ-^-S.’lO"4
Мышьяк As 1 2349,84 -3 -1 3:10-1:10
As 1 2456,53 -2 -1 l;10 -l’.lO
xiz *1 Железо Fe П 2382,04 ЗгЮ^-З.-Ю”1
Fe П 2410,52 2.-10’‘2-5,_-10“1
71
Продолжение табл. 5
Определяемый элемент Аналитические о линии, А Пределы определяемых концентраций, %
аь9 Олово Sn 2429,50 -4 -2 5:10 -1:10
-4 -3
Sn I 3175,02 1-ДО -3:10
Марганец Мп “2576,10 —4 — Ч 1:10 -5:10
Мп 2595,76 —Ч — 9 2:10 -5:10
it 9 Хром Сг 22677,16 s-io^-i.stio”1
аь1 Магний Mg I 2781,42 S.-lQ^-ltlo”1
Мд 13096,90 —4 —2 3-10 -2:10
кл *1 Медь Си 1 2824,37 3:10"3-1- IO-1
Си 13273,96 I-IO^-S’IO-3
Свинец РЬ 1 2833,07 -4 -2 2:10 ^-1:10
Висмут Bi 1 2897,98 4:10-1:10
Bi 13067,72 l-.lO^-SllO”3
Кальций Са 113179,33 2:io"3_i-.io-1
Са П3181.28 -2 -1 2:io -здо
Са П3968.47 -4 -2 3-io -2;io
Ванадий V 13183,98 l-10"2-l: IO"1
Цинк Zn 14810,53 5-10-3_5;10“2
Натрий* Na I 5889,95 Q 9 1-10 -2-10
Литий Li I 6707,84 З-Ю^-б-Ю”3
«^Слабые линии Si, Д1 , Fe , Мп , Си, Са, Мд рекомен-
дуют при.лспользовании основы, сильно загрязненной этими
элементами.
*2 Li-
Чувствительность определения N i,Sn,с г, Na несколько занижена за счет загрязнений основы.
j^3 _ °
Линидо Cd 1 2288,02 А используют при отсутствии
мышьяка в пробл-х-, о
*4 Линия As 1о2349,84 А нечетко разрешается с линией Ti 2349,94 А, но ее можно использовать для визуальной оценки при содержании мышьяка в пробах <1 *107?>.
72
3) Определение 29 элементов в пресскомпозициях.
Пресскомпозиции ВСП и ПКБ ~ смеси кристаллического бора или карбида бора с полипропиленом - анализируют прямым спектральным методом в дуге переменного тока.
Пробы и эталоны разбавляют угольным порошком в отношении 1-.2 и добавляют литий (Li^COj) в количестве 1% от массы пробы.
Чувствительность определений составляет 5*10" —
3 • 10 . Относительная погрешность анализа 10-20%.
Методика разработана для анализа мелкодисперсных смесей состава 30% бора или карбида бора и 20% полипропилена.
Возможен анализ смесей, состоящих, например, из 85% бора и 15% полипропилена. В подобных случаях пробы предварительно разбавляют чистым мелкодисперсным полипропиленом до 20%-ного содержания последнего. При получении результатов необходимо вводить коэффициент пересчета, учитывающий разбавление пробы.
Отбор проб. Отбор проб проводят аналогично описанному на с. 60.
Для приготовления пробы к 0,5 г дисперсной пресскомпозиции добавляют 1,027 г смеси угля с литием и тщательно перемешивают в плексигласовой ступке в течение 20 мин.
Смесь угля с литием'готовят заранее следующим образом: 12 г угольного порошка тщательно перемешивают с 0,324 г Li2CO3 в плексигласовой ступке в течение 30-40 мин.
Применяемые реактивы
Применяют реактивы, перечисленные на с. 61. Исключают соли Be , Ga , Аы, In , Р4 , .
Добавляют следующие реактивы:
Аммиак водный NH^OH , МРТУ 6-09-3266-66.
Бензол для криоскопии СбН6, ГОСТ 5955-68.
Вольфрам окись И/03/МРТУ 6-09-4676-67.
Полипропилену ТУ 38-3040-73.
Полистирол блочный Д, ГОСТ 9440-60 (зольность ^3*10~3%).
Серебро хлористое ДдСЬ МРТУ 6-09-5322-68.
Теллур металлический Te,T-U
73
Материалы и оборудование
Для проведения анализа используют материалы и оборудование, указанные на с. 62., Генератор дуги постоянного тока для анализа пресс композиций не применяют.
Используют фотопластинки: спектральные тип Щ^УФШ-З, панхром и инфра-760.
Ход анализа, В качестве основы для приготовления эталонов используют аморфный бор и мелкодисперсный полипропилен. Примеси в эталоны вводят в виде окисей или термически неустойчивых солей методом, описанным на с. 62.Серебро добавляют из раствора AgCI в 18%-ном аммиаке, тел-лур-из раствора, приготовленного из металлического Те (если отсутствует окись теллура).
Для электродов используют угольные стержни диаметром 6 мм, длиной 30 мм. Нижний электрод имеет кратер диаметром 3,8 мм, глубиной 6 мм. Верхний электрод - формы "зонтик" с толщиной площадки 1 мм и ножкой диаметром 2 мм и длиной 7 мм (ем. рис, 17),
Анализируемые эталоны и пробы ( ^76 мг) засыпают в кратер электрода, уплотняют стальным стержнем с иглообразным выступом на конце (диаметр^ иглы у основания -1 мм, длина - 2 мм) и сверху закапывают двумя каплями 2%-ного раствора полистирола в бензоле (вторая капля наносится после высыхания первой).
Возбуждение спектров производят в дуге переменного тока. Сила тока в начале экспозиции - 8 А, через 2-3 с самопроизвольно возрастает до 9 А.
Время экспозиции для ИСП-^Л - 20 с, для ИСП-22 -60 с. Область спектра > 2460 А перекрывают шкалой длин волн через 25 с.
По аналитическим линиям (табл. 6)строят градуировочные графики в координатах s-lgC*, ДЗ-lgC. При анализе по относительной плотности почернения используют фон около аналитических линий.
Насыпные массы эталонов и проб несколько отличаются из-за различий в плотности аморфного и кристаллического бора и карбида бора1. Необходимо следить за насыпными массами и при выдаче результатов полученную по графикам концентрацию примеси в пробе уменьшать на коэффициент пересчета.
74
Т а бли ца 6
Аналитические линии и концентрационные пределы определения примесей в пресскомпозициях БСП и ПКБ
Определяемый элемент Аналитические линии, А Пределы определения, %
Цинк Zn 12138,56 _ Q П 1-10 -3:10
Фосфор Р 12149,11 Q _1 5Ц0 -1:10
Висмут Bi 12230,61 1UO"3-3:1O-2
* Медь Bi 12897,98 Cui 2227,78 Q 2;10 -3:10 гдо^-здо-2
Кадмий Cd 12265,02 ЗДО^-бсЮ"2
Сурьма SbI 2311,47 РДО^-З-Ю-2
Барий ВаП 2335,27 1:10“3-1-Д0"2
Мышьяк ВаП 4934,09 As 12349,84 1:Д0”3-3;10”2 _ О о 3:10 -6:10
Алюминий* Al I 2367,02 -2 -1 3:10 -3:10
Т еллур Al 12263,45 Tj 12385,76 1-ДО"2-!-.!©"1 UlO^-l-.lO"2
Кобальт Co 12424,93 —4 -2 4:10-1:10
Олово Sn 12429,49 -4 -2 5:10 -3:10
Кремний Si 12435,16 —ч —1 3;10 -1-Д0
Железо* Si I 2124,15 Fe I 2472,91 -2 -1 1:10 -3:10 _1 4:10 -3:10
Ртуть Fen 2404,88 Hg 12536,52 пдо^-здо-1 5:Ю"3-6:Ю“2
Марганец МпП2576,10 ГЛ 0^-2:10-3
Хром Мп П 2933,06 Cr П2677.16 2-10^-2-ДО-2 2:10-3-6-Д О-2
Cr 14254,35 1:10"3-3-10~2
75
Продолжение табл. 6
О пред еляемый элемент Аналитические линии, А Пределы определения, %
Магний Мд 12782,97 2:io-3-b.io-1
Свинец РЬ 12833,07 —4 — Q 2‘10 -6-.10
Вольфрам W Т 2896,45 -2 -1 3:10 -3;Ю
Титан Ti П 3088,03 -2 -1 1*.Ю -3*10
Никель Ni 1 3050,82 2Д0"3-2:10"2
Молибден Mo I 3170,35 -2 -1 3:10 -3:10
Ванадий V 13183,98 -2 -1 1-Д0 -3:10
Серебро Ад 13280,68 SjlO^-lUO”3
ч Кальций Са 14226,73 -Ч -1 3:10 -1J0
Таллий Са П 3179,33 TI 15350,46 3:10 -1:10 1:.ю"3-1:1о"2
Натрий Na Т 5889,95 —Я —1 5-10 -1x10
Калий К 17664,90 1чо"3-з-.1о-2
Использование более сильных линий меди, алюминия, железа затруднено из-за загрязнения основы1.
2.6. ОПРЕДЕЛЕНИЕ КАЛИЯ
ПЛАМЕННО-ФОТОМЕТРИЧЕСКИМ МЕТОДОМ
Пламенно-фотометрический метод получил широкое применение в аналитической химии. Имеется ряд обзоров и монографий [113-116] по использованию его для анализа щелочных, щелочноземельных металлов и других элементов. Чувствительность анализа этим методом в значительной степени зависит от вида применяемой аппаратуры. На отечественных приборах максимальная чувствительность (0,001-0,18. мкг/мл К) достигнута в случае применения . спектрографа ИСП-51 ° фотоэлектрической приставкой ФЭП-1 [117] и фотометра на основе монохроматора УМ-2 [127].
76
Гораздо меньше чувствительность при использовании фотометров со светофильтрами [113, 118-120], хотя они и значительно дешевле. Так, чувствительность определения калия на фотометре ФПФ-58 не превышает 0,6 мкг/мл.
Пламенную фотометрию используют в основном для определения элементов в водах различного происхождения.
Определению примесей в чистых металлах обычно предшествует обогащение пробы: удаление основы или извлечение примесей. Иногда удается получить довольно высокую чувствительность (до 0,001-0,01%) без предварительного концентрирования примесей. Это достигают применением фотометров высокой дисперсии, выполненных на основе призменных приборов [116, 121-124, 126] или, в меньшей степени, добавлением щелочных металлов в пробы для подавления ионизации атомов и разбавлением проб органическими растворителями, понижающими вязкость и поверхностное натяжение растворов [125]. В первом случае чувствительность растет за счет монохроматизации выделяемого излучения при условии одновременного использования высокоэффективных усилителей, во втором - за счет повышения концентрации атомов в пламени, увеличения скорости распыления, уменьшения размера капель, т.е. более эффективного использования раствора*. В работе [126], выполненной на призменном фотометре, изложена методика определения калия в аморфном боре. Однако авторы не сообщают данных по влиянию состава анализируемых растворов борной кислоты и примесей других щелочных металлов на интенсивность излучения калия. Без этого невозможно проводить определение калия с достаточной точностью, особенно в горячем воздушно-ацетйленовом пламени.
Методики определения
Применяют пламенно-фотометрический метод определения калия с использованием ацетилено-воздушного пламени и пламенного фотометра ФПФ-58 [128]. Пробу бора (50-100 мг) растворяют в азотной кислоте или в смеси азотной и серной кислот (кристаллический бор и карбид бора). Раствор упаривают, остаток обрабатывают водой для удаления кислот. Анализу подвергают водный раствор борной кислоты. Повышение чувствительности определения достигают введением в растворы 0,6% цезия или 25% спирта. При использовании водных и спиртовых растворов без
7
добавки цезия получают завышенные результаты в случае загрязнения проб другими щелочными металлами. Для водных растворов проб суммарное содержание щелочных металлов (кроме калия) не должно превышать 0,2%, для спиртовых растворов - 0,01% к массе бора. Введение 0,6% цезия в растворы эталонов и проб устраняет влияние других щелочных примесей на интенсивность излучения калия.
Чувствительность метода 0,5-5 мкг/мл, что соответствует определению 0,01-0,1% калия в боре из растворов 0,5% по бору,(или 2,86% по Н3ВО3). Относительная погрешность определения * 2-3%.
Методику используют для анализа калия в аморфномт кристаллическом боре и в мелкодисперсном порошке карбида бора после переведения их в раствор.
.Применяемые реактивы
Применяют реактивы квалификации чда, хч, осч, спч.
Азотная кислота HNO3 ( d = 1,34 г/см^); МРТУ-6-03-171-65.
Борная кислота НзВ0з,МРТУ 6-09-785-63.
Вода дистиллированная, бидистиллированная, МРТУ 6-09-688-63
Калий азотнокислый KN031 МРТУ 6-09-2044-64.
Серная кислота H2SO4 ( d = 1,828 г/см^), ГОСТ 14262-69.
Цезий хлористый CsCI, МРТУ 6-09-4550-67.
Этиловый спирт С2Н5ОН , ТУ 19 П-39-69.
Отбор проб. Отбор проб проводят на воздухе с применением защиты от возможных внешних загрязнений методом, описанным на с. 60 .
Навеску 50 г аморфного бора растворяют после слабого подогревания в разбавленной (1:4) 4N03, раствор упаривают на электроплитке при постоянном перемешивании. Полученный остаток обрабатывают 2-3 раза в 5-10 мл дистиллированной воды и раствор упаривают досуха.
Навеску кристаллического бора 50 мг помещают в сухую кварцевую колбу на 300-500 мл, куда наливают 1,5 мл концентрированной HNO3 и 4 мл концентрированной H^SOzi • Растворение проводят при постепенном нагревании смеси до кипения. В этих условиях кристаллический бор растворяется, если образующиеся пары NO окисляются до N20z| и возвращаются в реакционную смесь. Поэтому для растворения выбирают колбу с длинным и узким горлом ( d = 10т*12мм
78
h. = 1404460 мм), которое охлаждают во время реакции влажным марлевым полотном (или используют обратный холодильник). Растворение длится 30-60 мин.
Мелкодисперсный карбид бора В^С растворяют в кипящей смеси серной и азотной кислот значительно медленнее. На 50 мг BzjC берут 3 мл НМ03 и 8 мл H2SO4 . После прекращения выделения красно-бурых паров (спустя ~ 60 мин после начала реакции) в охлажденную смесь добавляют еще 3 мл HN03 и растворение продолжают еще 60-90 мин. Если навеска растворяется неполностью, добавляют еще 3 мл ныо£.
Раствор кристаллического бора или карбида бора переносят в кварцевую чашку, упаривают и прокаливают в муфеле при постепенном нагревании до 350-400 С. После прекращения выделения паров H^SOzj в чашку два-три раза добавляют дистиллированную воду по 5-10 мл и раствор снова упаривают и прокаливают.
Сухой остаток, полученный от проб аморфного, ристал-лического бора или карбида бора, растворяют в минимальном объеме бцдистиллированной воды и переносят в мерную колбу на 10 мл. В зависимости от дальнейшего хода определения анализируемый раствор готовят одним из следующих трех способов:
1. Раствор в мерной колбе доводят до метки бцдистил-лированной водой. Получают раствор 0,5% по бору.
2. В мерную колбу добавляют 2,5 мл этилового спирта и воду до метки. Получают 25%-ный спиртовый раствор, 0,5% по бору.
3. В мерную колбу добавляют 1,5 мл 4% по цезию рас-TBopaCsCI и воду до метки. Получают раствор 0,6% по цезию и 0,5% по бору.
Аппаратура
Фотоэлектрический пламенный фотометр, модель ФПФ-58. Муфельная печь МП-2У.
Кварцевая чашка диаметром 50—75 мм.
Электроплитка, ГОСТ 306-58.
Баллон с растворенным ацетиленом, ГОСТ 5948-60 и ГОСТ 5457-60.
Кристаллический карбвд бора в кипящей смеси серной и азотной кислот за 2-3 ч растворяется на ~ 6%.
79
Редуктор ацетиленовый РД-2АМ.
Редуктор давления воздуха РДВ-5-СТУ 101-451-63. Затвор водяной для ацетилена ЭСП-7-62.
Рукава резинотканевые, тип Г, ГОСТ 8318-57.
Посуда мерная и колбы с пробками для приготовления и хранения растворов.
Ход анализа. Эталоны представляют собой 0,5% по бору растворы борной кислоты в воде (2,86% Н3ВО3 ) с добавкой или без добавки соли цезия или спирта и с различным содержанием калия (от 0,5 мкг/мл до 50 мкг/мл). Желательно работать на эталонах, содержащих 0,6% цезия, в этом случае чувствительность определения максимальна (табл.7, 8, 9) и отсутствует влияние примесей других щелочных элементов на интенсивность излучения калия. Растворы с 25% спирта дают хорошую чувствительность, но являются неустойчивыми, так как спирт быстро испаряется; рекомендуется эталонные растворы готовить в виде водных, а спирт добавлять непосредственно перед анализом. Эталонные растворы без добавки цезия и спирта дают минимальные отсчеты, поэтому их лучше использовать при содержании калия в боре больше 0,1%. Необходимо также учитывать тот факт, что присутствие в пробе бора примеси натрия или других щелочных металлов увеличивает величину отсчета для калия. На 25%-ных спиртовых растворах влияние щелочных металлов проявляется с концентрации их больше 0,01%, на водных без цезия с концентрации больше 0,2% по отношению к массе бора. Работать на водных эталонах без Н3ВО3 нельзя, так как присутствие борной кислоты в растворах проб снижает величину отсчета по калию.
В качестве исходных растворов для приготовления эталонов используют:
1. 4%-ный раствор борной кислоты: 40 г Н3В03 растворяют при слабом нагревании в 800 мл бвдистиллирован-ной воды. Раствор переносят в мерную колбу на 1 л и доводят до метки бвдистиллированной водой.
2. Растворы KNO3 в воде: 1000 мкг/мл и 100 мкг/мл калия. Раствор 1000 мкг/мл калия получают
растворением 258,5 мгКМ03 в воде в мерной колбе на 100 мл. Из этого раствора разбавлением его в 10 раз го-трвят растврр калия 100 мкг/мл.
3. 4% по цезию раствор CsCl: 5,067 г CsCI растворяют в бвдистиллированной воде в мерной колбе на 100 мд.
Исходные растворы для получения эталонов трех типов сливают в соответствии с табл. 7, 8, 9. Содержание калия 80
Таб ли да
Приготовление эталонов на основе НзВ03__
Номер эталона Количество исходного раствора на 100 мл эталона, мл Порядок величины отсчета на ФПФ-58 Содержание калия, % к раствору Содержание калия, % к H5BO3 Содержа-[ Содержание ние калия, калия,
% к бору мкг/мл
4%—ный| Н3В03 1 калия, ДОО ккг/мл калия/ 1000 мкг/мл
0 71,5 1 — 0,3 — —‘ — —
1 71,5 1,5 — 1,5 0,00015 0,00525 0,03 1,5
2 71,5 5,0 — 7 0,0005 0,0175 0,1 5
3 71,5 10,0 — 20 0,001 0,035 0,2 10
4 71,5 — 1,5 44,5 0,0015 0,0525 0,3 15
5 71,5 — 2,0 72 0,002 0,07 0,4 20
6 71,5 — 2,5 100 0,0025 0,0875 0,5 25
7 71,5 — 3,5 160 0,0035 , 0,1225 0,7 35
8 71,5 — 5,0 2915 0,00 5 0,175 1,0 50
00 м
Таблица 8
Приготовление эталонов на основе НзВОз с 25% спирта
Номер эталона Количество исходного раствора на 1ОО мл эталона, мл Порядок величины отсчета на ФПФ-58
4%-ный Н3В03 калия, 100 мкг/ МЛ' калия, 1000 мкг/мл вода до 75 мл спирт вводится перед анализом
0 71,5 * — 3,5 25 0,5
1 71,5 1,5 — 2,0 25 6,5
2 71,5 — 0,5 3,0 25 43
3 71,5 — 1,0 2,5 25 110
4 71,5 — 1,5 2,0 25 210
5 71,5 — 2,0 1,5 25 310
6 71,5 — 2,5 1,0 25 392
в растворах по отношению к воде, борной кислоте, бору во
всех типах эталонов одинаково (см. таол. 7).
Для хранения готовят по 75 мл водных эталонных растворов
НзВ0я с : калием. Перед снятием отсчета (во время анали-
Таблица 9
Приготовление эталонов на основе Н3ВО3 с 0,6% цезия
л............................... ............................................... .. 1-_________________________________________________i
Номер эталона Количество исходного раствора _ на 100 мл эталона, мл Порядок величины отсчета на ФПФ-58 с диафрагмой
4%-ный Н3В03 калия, 100 мкг/мл калия, 1000 мкг/мл 4%-ный цезия из раствора
2,5 5,0
0 71,5 15 2,3 7
1а 71,5 0,5 — 15 — 16
1 71,5 1,5 — 15 13,5 42
2 71,5 5 — 15 40,3 117,5
3 71,5 10 — 15 75 225
4 71,5 от 1,5 15 : L13 335
5 71,5 2,0 15 : L41 437,5
6 71,5 2,5 15 166 520
.82
за) к части этих растворов добавляют спирт, например на 7,5 мл раствора добавляют 2,5 мл спирта.
Величины отсчетов в табл. 8, 9 получены при диафрагме основного плеча, равной 5.
В эталоне № 1а содержание калия по отношению к бору равно 0,01%.
Холостой опыт (эталон Ns О) можно не учитывать, так как величина отсчета в этом случае (как было установлено дополнительно) определяется в основном чистотой раствора цезия (который вводят и в эталоны, и в пробы) и, возможно, фоновым излучением растворов борной кислоты.
Анализ проводят на фотоэлектрическом пламенном фотометре ФПФ-58, раствор сжигают в ацетилено-воздушном пламени при давлении ацетилена 160 мм вод.ст. и воздуха 0,40 мм рт.ст. Раздвижную диафрагму основного плеча раскрывают полностью до деления 5,0 и реже, для растворов с цезием - до 2,5. Подачу ацетилена в горелку из баллона, вынесенного за пределы здания, производят через два ацетиленовых редуктора, соединенных 9-метровым резинотканевым шлангом, и редуктор давления воздуха РДВ-5. Один редуктор укрепляют на баллоне, второй - перед водяным затвором. Водяной затвор предохраняет систему от обратного удара взрывной волны, возникающей, если скорость истечения газа становится ниже скорости распространения пламени. Водяной затвор устанавливают непосредственно перед редуктором РДВ-5, который используют вместо недостаточно надежно работающего вентиля ацетиленовой горелки, вмонтированного в корпус фотометра.
Воздух в горелку нагнетают с помощью воздушного компрессора через воздушный объем. Компрессор (КВМ-8) и воздушный объем поставляют в комплекте с фотометром ФПФ-58.
Для проведения анализа необходимо минимум 5 мл раствора. Перед снятием отсчетов с микроамперметра обязательно контролируют с помощью редуктора РДВ-5 постоянство давар^я ацетилена в горючей смеси, так как от этого в основном зависит воспроизводимость полученных результатов. За исключением момента измерения, микроамперметр должен постоянно находиться в арретированном состоянии.
Определение калия в пробе проводят по калибровочным прямым, которые строят в координатах: содержание калия' в
33
боре (%) - величина отсчета по микроамперметру. График прямолинейный и проходит через начало координат при диафрагме 5,0 только для эталонов, приготовленных на борной кислоте с добавкой 0,6% цезия.
Проведение измерений. Через распылитель в пламя го« релки вводят дистиллированную воду, при этом указатель микроамперметра устанавливают с помощью потенциометра на нуль. Вводят эталонные растворы в пламя горелки, Перед введением каждого эталонного раствора обязательно устанавливают указатель микроамперметра на нуль по дистиллированной воде или ( для спиртовых растворов) - по 25%-ному раствору спирта в воде.
Вводят растворы проб в пламя горелки и записывают отклонение указателя микроамперметра. Отсчеты по микроамперметру для эталонов и проб снимают 2-3 раза до получения совпадающих результатов. При распылении анализируемого раствора борной кислоты в пламя горелки наблюдается уменьшение величины отсчета, если раствор вводят продолжительное время, отсчеты снимают недостаточно быстро или когда систему плохо промывают водой после предыдущего раствора.
Содержание калия находят по калиброванным прямым построенным в координатах: величина отсчета - содержали* калия в эталонах (% к массе бора).
СПИСОК ЛИТЕРАТУРЫ
1. Васильева М.Г, и др. Анализ бора и его неорганических соединений. М., Атомиздат, 1965.
2. Eberle A.R., Pinte L.J., Lerner M,Wt "Analyt.Chem1*, 1964, v. 36, N 3, p. 674.
3. Eberle A.R<>, Lerner M.W. '’AnalytChem.’1, 1964, “v. 36, N 7, p. 1282.
4. Модылевская К.Д.. Лютая М.Д., Назарчук Т.Н, Информационное письмо ИМСС АН УССР, 1960, № 285.
5. Васильева М.Г, "Журн. аналит. х'имии", 1962, т. 17, № 4, с. 530, 531.
6. Кугай Л.Н., Назарчук Т.Н, "Заводск. лаборатория",
1971, т. 37, № 9, с. 1053.
7. Blumenthal Н. ’’Analyt.Chem.”, 1951, v. 23, N 7, р. 992.
8. Лютая. М.Л.., Назарчук Т.Н., Модылевская К.Д, Авт. свид. № 142805, кл. 42.-Бюл. изобр. 722364/23 от 20.03.61-
9. Ridhway R. ’’Trans. Amer. Electrochem. See.”, 1934, V. 66,”p. 117.
10. Назарчук Т.Н, "Журн. аналит. химии", 1959, № 4,
с. 2665.
11. Миклашевский А.И, "Заводск. лаборатория", 1938,№ 7, с. 168.
12. Шафран И.Г, "Журн. прикл. химии", 1938, т. 2, с. 56.
13. Самсонов Г.В, и др. Анализ тугоплавких соединений. М., Металлургиздат, 1962.
14. Котляр Е.Е, Назарчук Т.Н, Химические свойства и методы анализа тугоплавких соединений (Ин-т проблем материаловедения АН УССР). Киев, "Наукова думка", 1969.
15. Рысс .,И<ГА1.-СДУЦкац МгМл.. гДокл. АН СССР', 1947, т. 57, с. 689.
1 6. Марковский Л.Я, "Журн. прикл. химии", 1960, т. 33, с. 570.
17. Модылевская К.Д., Самсонов Г.В. "Укр. хим. журн.", 1959, т. 25, № 1.
18. Негина В.Р.. Козырева Э.А.. Соколова М.А. и др. "Заводок. лаборатория", 1970, т. 36, № 5, с. 5^.3.
85
19. Эристави Д.И., Броучек Ф.И. Аналитические методы определения бора. Тбилиси, "Медниереба", 1965.
20. Немо друк А. А., Кара лова З.Н. Аналитическая химия бора. М., "Наука", 1964.
21. Яковлев П.Я., Козина Г.В, "Заводск. лаборатория4',
1963, т. 29, № 8, с. 920.
22. Михеев В.И, и др, "Журн. неорганич. химии", 1957 2, с. 1249.
23. Йемодрук А.А., Маноле Б.А. Аналитическая химия. Т. 1 Аналитическая химия бора и его соединений. xvj., иэд. ВИНИТИ, 1973, с. 10.
24. Родионова М.А., Луконина З.Н.. Мун А.И. и др. "Изв, АН КазССР. Сер. хим.", 1974, № 5, с. 10*15.
25. Родионова-М.АГГ Косенко Т.П, "Материалы Д-й научной конференции молодых ученых АН КазССР1", 1970, с. 246*248.
26. Яковлев П.Я., Козина Г.В, Новые методы испытаний металлов. - "Труды ЦНИИЧермет", 1962, т.24, с. 179.
27. Яковлев П.Я., Козина Г.В, *3аводск. лаборатория", 1960, т. 26, № 12, с. 1342.
28. Пятницкий И.В, "Укр. хим. журн.", 1964, т. 30, № 3, с. 311.
29. Негина В.Р., Козырева Э.А, "Заводск. лаборатория", 1968, т. 34, с. 278.
30. Щербаков В.Г, и др, "Труды семинара по жаростойким материалам АН УССР", 1961, вып. 6, с. 52.
31. Patrovsky V. "Talanta", 1963, v. 10.
32. Хииро Кадзуо,nBull. Gevt. Industr. Res. Inst.",* 1962, v. 13, N 3, p. 198.
33. pfibil, Wunsch L. "Chem. listy", 1951, v. 45, p.337.
34. Доморев A.B., Подчайнова B.H, Современные методы химического и спектрального анализа материалов. Сб. обзоров и методик. М., "Металлургия", 1967.
35. Tereshko J. "Analyt. Chem.1’, 1963, v. 35, p.157.
36. Кугай Л,Н,« Назарчук Т.Н. "Журн. аналит. химии", 196JL т. 16, с. 205.
37. Csapo J. е.а. "Z. analyt. Chem.", 1956. Bd 151 S. 273.
38. Woodward C., RjKmian H.N. ”Soc. Analyt. Chem." ^-973, v. 11 , p. o3(J^^Analyt. Pci. моногр. № 1)
39. Дейкова "Докл. ГСХА", 1965, вып. ЮЗ.
86 .
40. Юнг Г.В, Инструментальные методы химического анализа. М., Госхимиздат, 1960.
41. Климова В.А. Основные микрометоды анализа органических соединений. М., 'Химия", 1967.
42. Коршун М.О.. Климова В.А. 'Журн. аналит. химии", 1949, т. 4, № 5, с. 292.
43. Климова В. А.. Коршун М.О. "Журн. аналит. химии", 1951, т. 6, № 2, с. 230.
44. Гельман Н.Э., Шевелева Н.С, 'Журн. аналит. химии", 1965, т. 20, № 6, с. 719.
45. Печентковская Л.Р., Назарчук Т.Н, В кн.: Химические свойства и методы анализа тугоплавких соединений. Киев, "наукева думка", 1969, с. 26—32-
46. Самсонов г.В., Серебрякова Т.Н., Неронов В.А, Бориды. М., ’ Атомиздат, 1975»
47. Самсонов г.В., марковский л.я. и др. Бор, его соединения и сплавы. Киев, "Наукова думка", i960.
48. Hall Н„ Compton L. "J. Inorg. and Nucl. Chem.", 1965, v. 4, p. 2213.
49. Марковский Л.Я., Белов м.П. 'журн. прикл. химии', 1967, т. 40, №,4. с.727-734.
50. S[rai France e.'ai'Sapere". 1965, v. 41, p.381-385.
51. Kanda F.A. "J. Amer. Chem. Soc", 1965, v. 78, p. 1509.
52. Wilberg E. "Z. anorg. und allgem. Chem.", 1930, Bd 191, S. 2.
53. Туровцева 3.M., Кунин л.а. Анализ газов в металлах,. Изд-во АН СССР, 1959.
54, Hakoila Erkki Y, е.а. "Analyt. Chem.", 1972,v. 44, p. 1857.
55. Scher Aharon e.a. "Israel J. Chem.", 1972, v. 10, p. 693.
56. Митчел пж., Смит П. "Акваметрия", Изд-во иностр.лит. 1952, с. 259.
57. Eberius Е„ Bounes Н. "Z. Angew chem.", 1959, v. 5, р. 168.
58. Thompson R. "Chem. in Britain", 1971, v. 7, N 4, p. 140.
59. МРТУ 6-09-267-63; ТУ-15-ббч
60. Booth Е», Bryant F, e.a, "Analyst", 1957, 82. p. 50.
61. Ray Sinha. "J. Chem. Soc.", 1946, p. 322.
87
62. КОТЛЯР R.F., Кучай Л.Н. Информационное письмом 21 Киев, Изд-во дн СССР, I960.
63. Миненков Н.И., Назарчук Т.Н._ М., "Mei.аллургия", "порошковая металлургия", 1965, № 6.
64. Яковлев П.Я., Яковлева Е.Ф., Оржеховская А.И. Определение углерода в металлах. М., "Металлургия", 19 /
65, Belcher е.а. Anal. Chim. Asta, 1958, v.l9,pe,-^ .
66. Брунс Б, и др, "Журн. аналит. химии", 1947, т. 2. вып. 5, с. 294.
67. Bennet E.L. е.а. ’’Analitical Chem1’, 1950, v. 22, N р. 445.
68. Брунс Б., Брауде Г, "Журн. аналит. химии", 1949, вып. 5, с. 316.
69• Koch W., Malissa Н. ’’Arch. EisenhOt-tenwesen’ 1958, Bd 29, N 9, S. 543.
70. Крюков П.А., Рейнгартен P.B, "Журн. аналит. химии 1955, т. ю, № 1, с. 51.
71. Qelsen W. е.а. ’’Arch. Eisenhlittenmesen”, 1951, . Bd 22, N 7/8, S. 225-229.
7 2. Fischer Vfc Bastins M, ’’Metall”. 1960, v. 14, b 1 p. 429.
73. Натансон К .to, и др, "Заводск. лаборатория", 19 70,т.зс, № 9, с. 1033-1036.
74. Яковлев П.Я., Оржеховская А.И, Новые методы испытания металлов (ЦНИИЧермет), М-, Металлургиздат. 1963, вып. 31, с. 144.
75. Быковская to.И, "Журн. аналит. химии", 1962, т. 17, вып. 5, с. 607.
76. Яковлев П.Я., Оржеховская А.И, Новые методы испытания металлов (ЦНИИЧермет}; М«, "Металлургия", 1964, вып. 37, с. 7»
77. Otake W. ” Light Metalls”, 1958, v. 8, N 2, p. 59.
78. Mukaewaki Kimio, Bunsaki Kagaku. ’’Analyst J.”, 1960, v. 9, N 5, p. 427.
79. Stark G.” Z. anorg. und allgem. Chem’!, 1911 Bd 70, S. 173.
80. Гилл§браадт .В^Ф Додде ль.. др, Практическое ру-
ководство по неорганическому анализу. Мм Госхимиздат, 1960, с. 933.
81. Николаев Н.С., Суворова С.Н, и др. Аналитическая химия фтора. Мм "Наука", 1970.
88
82. Попов В.А», Руденко Э.И.< Яковлев ГПЯ, "Заводск. лаборатория", 1972, № 8, с. 908.
83о Эльвинг Ф», Хортон Ч,. Уиллард X, Фтор и его соединения. М.» Изд-во иностр, лит. 1956, с. 45-178.
84. Лайтинен Г.А, Химический анализ. М., "Химия", 1966, с. 243.
85. Щарло Г, Методы аналитической химии. М.» "Хими*", 1965, с. 885.
86. Киселева F.K. Анализ фторсодержащих соединеш^, Мм 'Химия", 1966.
87. Milner O.J. Analysis of Petroleum for Trace Ele-ments. Oxford, 1963.
88. Кюрегян O.K, Эмиссионный спектральный анализ нефтепродуктов. М., "Химия", 19 68.
89. Астафьева В.В.Методы анализа минерального сырья "Аппетиты", 1971, с. 14-17.
90. Асатиани В.С, Биохимическая фотометрия. М., Изд-Bt АН СССР, 1957.
91. Определение микроэлементов в биологических объект тах. Под ред. А.Р. Вильдма. Рига, "Зинатне", 1968.
9 2. Филов В.А, Определение ядохимикатов в биологических субстратах. М.-Л.» "Наука", 1964.
93. Негина В.Р., Крашенинникова ЕЛЬ, Михайлина А.Т.идр, "Журн. аналит. химии", 1967, т. 22, № 10, с. 1552-1558.
94. Швангирадзе Р.Р., Мозговая Г.Д., Щ^тцдина Э.В.
У Журн. аналитич. химия", 1962, т. 17, выл. 1, с. 94.
95. Забияко В,И. "Заводск. лаборатория", 1964, т. 30, № с. 1084-1085.
96. Глуховецкая Н.П.. Лернер Л.А. "Заводок, лаборатория", 1964, т. 30, № 9, с. 1082-1083.
97. Гегечкори Н.М.. Шварц /КМ. 'Заводок, лаборатория", 1953, т. 19, № 5, с. 580.
98. Winslow Е., Liebhafsky Н.11 J. Amer. Chem. Soc 1942, v. 64, N 2725.
и
99. Тарасевич Н.И., Железнова А.А. "Журн. аналит. химии", 1963, № 11, с. 1345.
100. Бондаренко Л.С.. Сотникова Н.П., Пейзулаев щ.И.и др. Методы анализа веществ высокой чистоты, м.» "Наука", 1965, с. 483-486.
101. Попков К.К., Кудрявцев А.С, Методы анализа веществ высокой чистоты. М., "Наука", 1965, с. 486-487.
89
10 2. Semour Lader, ” Appl. Spectroscopy”, 1958, v. 3 p. 85, *
103. Дегтярева О.Ф., Островская М._Ф_. "Журн. аналит. химии", 1967, т. 22, вып. 12, с. 1863.
104. Kim Guk Hong, Li Yong Sik, Kim Gwang Sung "Пунсок хвахак", 1971, v. 9, N 3, p. 111-116.
105. Забияко В.И., Булычева И.Б, "Труды Уральск. НИХИ", 1964, вып. 11, с. 82.
106. Dixit R.M., Venka Т., Bramanian R. ”Govt India Atom. Energy Conmis.”, 1971, p. 564.
107. Hobdt G. “Appl. Spectroscopy”, 1962, v. 16, p. 396.
108. Кравченко Л.Ф, "Заводск. лаборатория", 1969, т. 35, № 6, с. 681.
109. Pichat Philipps. ”Bull. Soc. chim. France”, 1967, v. 7, p. 2606.
110. Тарасевич H.H., Железнова А.А., Абдулаев А.АЛВестн. Моск, ун-та. Сер. П Химия", 1971, т. 12, № 5, с. 593.
111. Самсонов Г.В., Уманский Н.С, Твердые соединения тугоплавких металлов. М., Металлургиздат, 1957.
112. Дегтярева О.Ф., Синидина Л.Г.. Барихина Т.А. "Журн. аналит. химии", 1973, т. 28, вып. 6, с.1164-1168.
ИЗ. Полуэктов Н.С, Методы анализа по фотометрии пламени. М., Госхимиздат, 1967.
114. Бурриель-Марти Ф., Рамирес-Муноос X, фотометрия пламени. М., Изд-во иностр, лит. 1962.
115. Heermann R., Alkemade С. Th. "J.Plammenpho-tometrie”, 2 neubearb. Aufl. Berlin - Gottingen -Heidelberg, Springer, 1960, VIII. S. 394, 88-DM.
116. Полуэктов Н.С, Экспрессные методы анализа при помощи фотометрии пламени в цветной металлургии. М., Металлургиздат, 1958.
117 Лебедев В.И.. Вайнштейн Э.Е, "Журн. аналит. химии^, 1961, т. 16, вып. 2, с. 124.
118. Боровик-Романова Т.Ф, Спектрально-аналитическое определение щелочных и щелечноземельных элементов. М., ИЭД-во АН СССР, 1956.
119. Бонч-Осмоловская К.С, "Уч. зап. ин-та геол. Арктики. региональная геология", 1964, вып. 4, с. 252.
90
120. Todor Dumitru N, ’’Dari Sceamci Sedint. Com. geol. RPR”, 1962-1963 (1964), 50, part. 1,317-333e
121. Голубева И, А, В кн.: Проблемы большой металлургии и физической химии новых сплавов. М., 'Наука*, 1965, с. 304.
122. Журавлев Г.И., Гаврилова И.А, 'Журн. аналит.химии', 1964, т. 19, вып. 1, с. 54.
123. Kramer Н., Pinto L.T. ’’Analyt. chim. asta”, 1965, v. 33, N 4, p. 438.
124. Золотавин B.A.. Букреев Ю.Ф., Толтов Л.К, г_ НДР?.
'Журн. прикл. спектроскопии', 1965, вып. 5, с. 461.
125. Sajo-Fesze М., Keng Q.H., Bellomo A. ”Atti Soc. pelorit sei. fis. mat. enatur”, 1964, v. 10, N. 3, p. 255.
126. Васильева М.Г., Лалыкина B.M., Михарашвили H.A, и Анализ бора и его неорганических соединений.’М.» "'М'омиздат, 1955.
127. Полуэктов Н.С., Никонова М.П., Виткун Р.А, - 'Журн. аналит. химии', 1958, т. 13, выд. 1, с. 48.
128. Дегтярева О.Ф., Шевченко П.П, 'Журн. аналит. химии', 1968, т. 23, вып. Ю, с. 1560.
129. Бабко A.K.i Волкова А.И., Лисиченок С, Л, 'Журн. не-орган. химии', 1966, т. Ц, № 3, с. 478.
130. Шварденбах Г., Флашка Г, Комплексонометрическое титрование! М., 'Химия', 1970.
131. Елинсон С.В., Победила Л.И, 'Заводск. лаборатория', 1963, т. 29, № 2, с. 139.
132. Lassner Е., Schart R. ’’Taianta”, 1960, v. 7, N -1-2, p. 12-17.
133. Lieber W, ”Z. analyt. Chem.”, 1960, Bd 177, Hf.6, S.429.
134. Балакшина A.B., Соколова M.A., 'Заводск. лаборатория', 1976, № 2, с. 152.
91
ОГЛАВЛЕНИЕ
Введение........................*.............. 3
Глава 1* Определение основного вещества в кристаллическом боре, его тугоплавких соединениях и пресскомпозидиях . . • • 5
1.1. Переведение вещества в раствор 5 1.2. Определение общего бора....... g
1.3. Определение титана в бориде титана .............................. 14
1.4. Определение водорода и углерода в пресскомпозидиях..................17
Глава 2. Определение примесей в кристаллическом боре, его тугоплавких соединениях и пресскомпозидиях..........................23
2.1. Определение кислорода..........23
2.2. Определение азота............. 34
2-3- Определение углерода ....... 37 2.4. Определение фтора, хлора, серы . 48 2.5. Определение металлических примесей спектральным методом . . 58 2-6. Определение калия пламенно-фотометрическим методом..............76
Список литературы .............................85
ИВ № 20502
Негина Валентина Романовна, Козырева Элеонора Анатольевна, Дегтярева Ольга Федоровна, Балакшина Ариадна Васильевна, Зотикова Галина Анатольевна, Михайлина Александра Тимофеевич Орликова Евгения Гавриловна, Синидина Людмила Григорьевна
АНАЛИЗ БОРА, ЕГО СОЕДИНЕНИЙ И ПРЕСС КОМПОЗИЦИЙ (практическое руководство)
Редактор Т. Молоканова
Художественный редактор А. Кирьянов
ХудожникА. Ординарцев
Технический редактор И.Тимофеева
Корректор 3. Авдюшева
Сдано в набор 17.1.1978 г. Подписано к печати 19/УП-1978г.
Т-12956 Формат бумаги 60x90 1/16. Бумага офсетная № 2
Усл.печ.л. 5,75. Уч.-изд.л. 5,2. Тираж 1250 экз. Цена 30 коп. ' Зак. изд. 75076. Зак. тип. 2^60
Атомиздат, Москва, К-31, ул. Жданова, 5/7
АНАЛИЗ БОРА,
ЕГО СОЕДИНЕНИИ И ПРЕСС-HOM ПОЗИЦИИ
АТОМИЗДАТ