
































































































































































































































































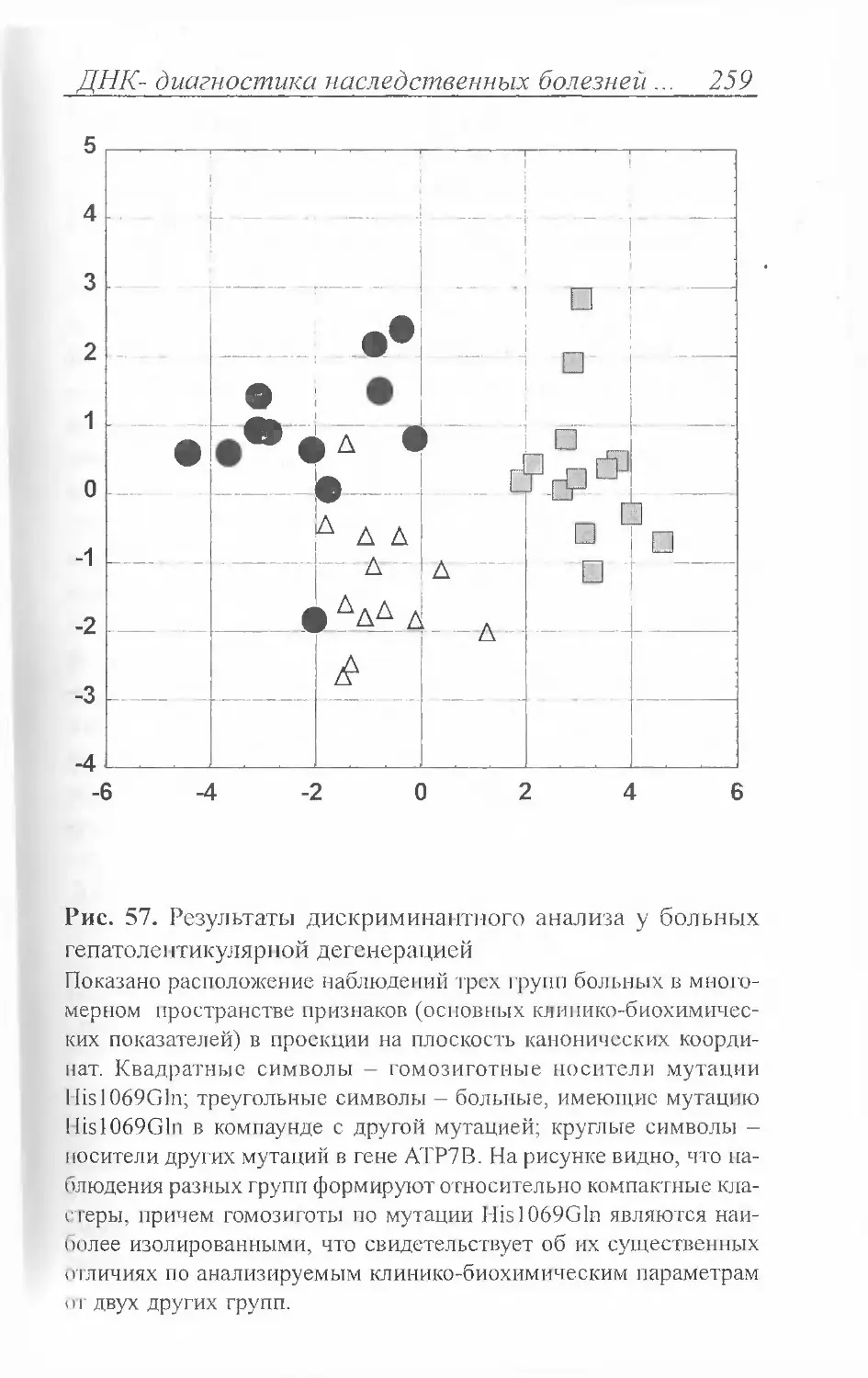




























































































































































































































































































































Author: Иллариошкин С.Н. Иванова-Смоленская И.А. Маркова Е.Д.
Tags: нервная система невропатология неврология общая генетика общая цитогенетика иммуногенетика эволюционное учение видообразование филогенез алкоголизм как болезнь нейрохирургия психиатрия медицина генетика
ISBN: 5-89481-100-7
Year: 2002




С.Н. Иллариошкин
И.А, Иванова-Смоленская
Е.Д. МарковаДНК-диагностика
и медико-генетическое
консультирование
в неврологииМЕДИЦИНСКОЕ ИНФОРМАЦИОННОЕ АГЕНТСТВО
С.Н. Иллариошкин,И.А. Иванова-Смоленская,
Е.Д. МарковаДНК-Д И АГНОСТИКА
И МЕДИКО¬
ГЕНЕТИЧЕСКОЕ
КО НСУЛ ЬТИ РОВАН И Е
В НЕВРОЛОГИИМЕДИЦИНСКОЕ ИНФОРМАЦИОННОЕ АГЕНТСТВО
МОСКВА-2002
УДК 616.8-07:575.113
ББК 56.12
И 44Иллариошкин С.Н.И 44 ДНК-диагностика и медико-генетическое консультирова-
ние в неврологии / С.Н. Иллариошкин, И.А. Иванова-
Смоленская, Е.Д. Маркова — М.: Медицинское информацион¬
ное агентство, 2002. — 591 с.: ил.15ВЫ 5-89481-100-7Монография посвящена современным возможностям ДНК-
диагностики и основанного на ней медико-генетического консульти¬
рования при наследственных заболеваниях нервной системы.
Авторами обощен большой собственный опыт в данной области
исследований в сопоставлении с результатами аналогичных работ,
полученными в ведущих лабораториях мира. Представлены сведения
об организации генетического материала в клетке, типах мутаций,
методах ДНК-анализа, геномной классификации монотонных
заболеваний нервной системы и принципах их генодиагностики,
подходах к ДНК-анализу мскотороых мульгифакториальпых
болезней мозга, а также принципах профилактики наследственных
заболеваний нервной системы в отягощенных семьях. Приложения
включают сведения о современной номенклатуре мутаций человека,
впервые обойденный в отечественной литературе детальный каталог
генов наследственных заболеваний нервной системы (по состоянию
на конец 2001 года), а также словарь генетических терминов.Для неврологов, психиатров, генетиков.УДК 616.8-07:575.113
ББК 56.12©С.Н. Иллариошкин,И.А. Иванова-Смоленская,Е.Д. Маркова, 2002
© Оформление “Медицинское
информационное агентство”, 2002Все права защищены. Никакая часть
данной книги не может быть воспроиз¬
ведена в какой бы то ни было форме
без предварительного разрешения
18В!Ч 5-89481-100-7 владельцев авторских прав.
Оглавление 3ОглавлениеИИ'ДИСЛОВИЕ 7ВВЕДЕНИЕ 11I ПАВА 1. Общие принципы генодиагностики 191.1. Строение молекулы ДНК / РНК и организациягенетического материала в клетке 191.2. Основы генеалогического анализа в неврологии 361.2.1. Наследственные факторы в патогенезе
неврологических заболеваний 361.2.2. Типы наследования моногенных болезней.Принципы сбора генеалогической информации .. 391.3. Характеристика основных типов мутаций,
вызывающих наследственные заболевания
нервной системы 601.4. Методы ДНК-диагностики 741.4.1. Прямая ДНК-диагностика 751.4.2. Косвенная ДНК-диагностика 951.5. Стратегия генетического картированияи его роль в идентификации новых геновнаследственных заболеваний 101ГЛАВА 2. Проблема генетической гетерогенности
и классификации наследственныхзаболеваний нервной системы 112ГЛАВА 3. ДНК-диагностика наследственныхмоногенных болезней нервной системы 134Г1. Наследственные нервно-мышечные болезни 1343.1.1. Прогрессирующие мышечные дистрофии 1343.1.1.1. Х-сцепленные НМД Дюшенна и Бекера
(дистрофинопатии) 1353.1.1.2. Аутосомные формы конечностно-
поясных ПМД 1443.1.1.3. Лице-лопаточно-плечевые ПМД 1553.1.1.4. Миотоническая дистрофия 1583.1.1.5. Другие формы ПМД 1623.1.2. Врожденные миопатии 1693.1.3. Наследственные невропатии 1733.1.3.1. Демиелинизирующие моторно¬
сенсорные невропатии (НМСН тип I)... 1763.1.3.2. Аксональные моторно-сенсорные
невропатии (НМСН тип II) 189
4Оглавление3.1.3.3. Моторные, сенсорные и сенсорно¬
вегетативные невропатии 1913.1.4. Спинальные амиотрофии и другие
наследственные болезни мотонейрона 1933.1.4.1. Проксимальные спинальные
амиотрофии детского возраста 1943.1.4.2. Спинально-бульбарная амиогрофия
Кеннеди (болезнь Кеннеди) 2003.1.4.3. Семейный боковойамиотрофический склероз 2023.1.5. Нервно-мышечные каналопатии 2053.2. Наследственные заболевания с преимуществен¬ным вовлечением экстрапирамидной системы ..2103.2.1. Хорея Гентингтона и другие формы
наследственной хореи 2103.2.2. Наследственные дистонии 2253.2.2.1. Торсионная дистония 2263.2.2.2. Пароксизмальная дистония 2433.2.2.3. Другие формы наследственной
дистонии 2453.2.3. Гепатолентикулярная дегенерация 2503.2.4. Эссенциальный тремор и семейный
паркинсонизм 2613.2.4.1. Эссенциальный тремор 2623.2.4.2. Семейные формы первичного
паркинсонизма 2653.3. Наследственные заболевания с преимущест¬венным вовлечением координагорных систем
(наследственные атаксии) 2823.3.1. Аутосомно-доминантные
спиноцеребеллярные атаксии 2833.3.1.1. Клинико-генетическая характе¬
ристика аутосомно-доминантныхатаксий 2833.3.1.2. ДНК-диагностика аутосомно-
доминантных атаксий 2983.3.2. Болезнь Фридрейха 3053.3.3. Другие формы наследственных атаксий 3173.3.3.1. Атаксия, обусловленнаяизолированным дефицитом
витамина Е 317
Оглавление53.3.3.2. Эпизодические атаксии 3193.3.3.3. Атакеия-телеангиэктазия 3243.3.3.4. Врожденная гипоплазия мозжечка 3283.4. Наследственные заболеванияс преимущественным вовлечением
пирамидной системы (наследственные
спастические параплегии) 3303.4.1. Аутосомно-доминантныеспастические параплегии 3313.4.2. Рецессивные спастические параплегии 3363.5. Наследственные заболеванияс преимущественным вовлечением
когнитивной сферы(наследственные деменции) 3433.5.1. Семейные формы болезни Альцгеймера 3433.5.2. Прионные болезни 3503.5.3. Другие формы наследственных деменций .... 3603.5.3.1. Лобно-височная деменция с
паркинсонизмом 3603.5.3.2. Деменция при семейной церебральной
амилоидной ангиопатиибританского типа 3623.5.3.3. Семейные деменции, ассоциированныес хромосомой 3 3633.5.3.4. Болезнь Гентингтона 3653.6.11аследственные формы эпилепсий 3663.6.1. Идиопатические наследственныеэпилепсии 3663.6.1.1. Каналопатии 3673.6.1.2. Другие кандидатные локусы 3743.6.2. Симптоматические наследственные
эпилепсии: синдром прогрессирующей
миоклонус-эпилепсии 3803.6.2.1. Миоклонус-эпилепсия
Унферрихта-Лундборга 3 803.6.2.2. Миоклонус-эпилепсия с рваными
красными волокнами(синдром МЕИНН) 3843.6.2.3. Наследственные болезни накопления .... 3843.7. Факоматозы 3893.7.1. Нейрофиброматоз 389
6Оглавление3.7.1.1. Нейрофиброматоз 1-го типа 3903.7.1.2. Нейрофиброматоз 2-го типа 3943.72. Туберозный склероз 3983.7.3. Синдром Гиппеля-Линдау 4033.7.4. Атаксия-телеангиэктазия 4073.8. Митохондриальные энцефаломиопатии 4073.8.1. Молекулярные основы
митохондриальных болезней 4073.8.2. Клинико-генетическая характеристика
митохондриальных энцефаломиопатий 4133.8.2.1. Заболевания,обусловленные мутациями
митохондриальной ДНК 4153.8.2.2. Заболевания, обусловленные мутациями
ядерной ДНК и нарушением
межгеномных взаимодействий 4233.8.3. ДНК-диагностика митохондриальных
энцефаломиопатий 429ГЛАВА 4. Молекулярно-генетический анализ
наиболее распространенных
мультифакториальныхзаболеваний нервной системы 4384.1. Анализ генетических ассоциаций 4404.2. Иммуногенетический анализ
мультифакториальных заболеванийнервной системы 4534.3. Молекулярное тестирование
при онкологическихзаболеваниях нервной системы 458ГЛАВА 5. Современные научные, этические
и организационные аспекты медико¬
генетического консультирования
в неврологии (Иллариошкин С.Н.,
Иванова-Смоленская И.А,Маркова Е.Д., Клюшников С.А.) 465ПРИЛОЖЕНИЯ1. Сводный каталог генов наследственныхзаболеваний нервной системы 4982. Номенклатура генных мутаций человека 5293. Словарь специальных генетическихтерминов, использованных в тексте 535БИБЛИОГРАФИЧЕСКИЙ УКАЗАТЕЛЬ 547
ПредисловиеНа протяжении всего XX в. генетика человека и
медицина постоянно развивались в теснейшем взаимо¬
действии. Из всех разделов медицины наибольшее вни¬
мание к генетике проявляли неврологи, а наследствен¬
ные болезни нервной системы были постоянно в фоку¬
се генетики. Многие общегенетические закономернос¬
ти были открыты при изучении именно этой группы
болезней.Отечественные неврологи A.B. Кожевников,H.М. Бехтерев, С.Н. Давиденков, Н.В. Коновалов,
P.A. Ткачев, Л.О. Бадалян и другие внесли огромный
вклад в развитие учения о наследственных болезнях не¬
рвной системы: описаны новые нозологические формы,
разработаны принципы дифференциальной диагности¬
ки для многих сходных болезней, разработаны методы
печения на основе расшифровки патогенеза, объяснены
генетические причины клинического полиморфизма,
сформулирована концепция генетической гетерогеннос¬
ти наследственных болезней, обосновано медико-гене-I ическое консультирование семей с наследственной па¬
тологией.Термин «нейрогенетика» давно вошел в широ¬
кую медицинскую лексику. Уже в 20-3 0-х годах среди
неврологов твердо сформировалось понимание значе¬
ния менделизма для теоретической и практической не¬
врологии. Именно на основе тонкого использования
клинико-генеалогического метода была произведена
«инвентаризация» наследственных болезней нервной
системы, найдены диагностические критерии сходных
нозологических форм. Однако к 70-м годам XX в. на¬
ступило «насыщение» генетического направления в
8Предисловиеневрологии. Разрешающие возможности клинико-гене¬
алогического, близнецового, популяционного методов
были уже полностью исчерпаны. Дальнейшее углублен¬
ное изучение этих болезней шло уже с применением
биохимических, электронно-микроскопических, гисто¬
химических, электрофизиологических методов.Новый импульс для развития нейрогенетики
дали методы генной инженерии. Успехи молекулярной
генетики человека привели к идентификации генов,
мутации в которых вызывают болезни нервной систе¬
мы, их клонированию, расшифровке первичных про¬
дуктов нормальных и мутантных аллелей. Все это в
корне изменило базовые принципы классификации и
диагностики наследственных заболеваний нервной си¬
стемы, разработки методов их лечения. Справедливос¬
ти ради следует напомнить, что подобную ситуацию
предвидел выдающийся отечественный невролог и ге¬
нетик С.Н. Давиденков. В 1925 году он подчеркнул, что
рациональная классификация наследственных болезней
должна быть «каталогом генов, а не фенотипических
различий». И далее он писал: «Этот полный каталог
генов еще не может быть сделан в настоящее время, и
его приходится ожидать от будущих исследователей».
Каталог генов такого рода, составленный В.А. Макю?-
юсиком через 40 лет, выдержал с 1966 года 12 изданий
(с дополнениями в каждом) и сейчас представлен в Ин¬
тернет под названием OMIM (Online Mendelian
Inheritance in Man).Современному врачу-неврологу и научному со¬
труднику необходимо знать генетические основы на¬
следственных болезней нервной системы не в меньшей
мере, чем морфологические и физиологические зако¬
номерности. В последние годы на русском языке опуб¬
ликованы полезные книги по клиническим и генети-
Предисловие9чсским аспектам наследственных болезней нервной
системы (Болезни нервной системы. Руководство для
ирачей в 2-х томах / под ред. Н.Н.Яхно и др. М.: Меди¬
цина, 1995; Наследственные болезни нервной системы.
Руководство для врачей / под ред. Ю.Е. Вельтищева иII,А.Темина. М.: Медицина, 1998; Молекулярная невро-
погия, авторы - В.Н. Горбунова, Е.А. Савельева-Васи-III,ева, В.В. Красильников. СПб.: Интермедика, 2000).
()днако накопление генетических знаний идет с такой
огромной скоростью, что требуется пересмотр концеп¬
ций или пополнение сведений при изложении фунда¬
ментальных и прикладных основ генетики болезней
нервной системы. В такой работе очень важно найти
баланс между клиническим и молекулярно-генетичес¬
ким представлением каждой нозологической формы. За
»гу трудную работу взялись С.Н. Иллариошкин,II.A. Иванова-Смоленская и Е.Д. Маркова под углом зре¬
ния ДНК-диагностики и медико-генетического консуль¬
тирования. Выполнили они эту работу успешно со всех
точек зрения, хотя писать подобного рода книги очень
трудно, поскольку «молекулярная грамота» должна
органично вписываться в клиническую генетику болез¬
ней. Это удалось авторам безукоризненно, с моей точ¬
ки зрения, потому что они обладают преемственным
опытом неврологов-генетиков (от Н.В. Коновалова,
Г.Д. Ткачева) и не жалели сил в последние годы для
организации работы молекулярно-генетической лабора¬
тории. Эта лаборатория стала одной из лучших по ди¬
агностике болезней нервной системы.В доступной для врача форме изложены общие
принципы генодиагностики, куда очень органично
иставлен раздел о генетическом анализе в неврологии.11оследнее очень важно, потому что современная диаг¬
ностика наследственных болезней обязательно должна
10Предисловиесочетать в себе глубокий генеалогический анализ и
высочайшую молекулярно-генетическую технику.В монографии представлены современные све¬
дения о природе генов и их мутаций, вызывающих бо¬
лезни нервной системы. Эта глава будет интересна не
только для врачей, но и для генетиков.Последовательно и достаточно объемно в соот¬
ветствии с новой генетической классификацией изло¬
жена молекулярная генетика и ДНК-диагностика на¬
следственных моногенных болезней нервной системы.
Данный раздел монографии (глава 3) можно рассмат¬
ривать как хороший справочник по молекулярным ос¬
новам наследственных неврологических болезней и
методам их диагностики.Впервые сделана попытка изложить молекуляр¬
но-генетические подходы к анализу наиболее распрос¬
траненных мультифакториальных заболеваний нервной
системы. Это очень важный раздел в молекулярной не¬
врологии, который должен интенсивно разрабатываться.Книга С.Н. Иллариошкина, И.А. Ивановой-Смо¬
ленской, Е.Д. Марковой «ДНК-диагностика и медико¬
генетическое консультирование в неврологии» очень
нужна в наше время широкому кругу специалистов (вра¬
чей, генетиков, преподавателей), поскольку далеко не у
каждого есть возможность получить объективную ин¬
формацию в хорошо систематизированном виде.Академик РАМН
ВведениеОдной из наиболее характерных современной
медицины на рубеже столетий является интенсивное
развитие молекулярной генетики и методов ДНК-анали-
за. Это явилось результатом серии фундаментальных
открытий и разработки ряда новаторских технологий в
соответствующих областях биологической науки, а так¬
же успешной реализацией Международной программы
«Геном человека», имевшей целью идентификацию всего
человеческого генома. Обществу еще только предстоит
оценить и в полной мере осмыслить грандиозное значе¬
ние полной расшифровки наследственного материала
человека, но уже сегодня это событие по своим общете¬
оретическим и практическим последствиям приравни¬
вается к открытию электричества и атомной энергии или
выходу человечества в космос [Киселев Л.Л., 2000]. На¬
копленная совокупность знаний позволяет говорить о
переходе медицинской науки и практики на принципи¬
ально новый, молекулярный уровень изучения патоло-I ии человека - уровень, связанный с анализом патоло-
. ■//ческой анатомии генома человека [Пузырев В.П., Сте¬
панов В.А., 1997; МсКшюк V., АтЬе^ег I., 1993]. Ука¬
чанный подход знаменуется не только глубоким пости¬
жением молекулярных основ наследственных заболева-
пий, но и разработкой надежных методов их генодиаг¬
ностики, активной профилактики и генной терапии
| Веаис1е1 А., 1999]. Более того, именно на основе техно-
погий молекулярной генетики и молекулярной биоло¬
гии возможен прорыв в решении таких ключевых про¬
пнем современной медицины, как онкогенез, сердечно¬
сосудистые заболевания, сахарный диабет, старение и др.В настоящее время не вызывает сомнений тот
факт, что ориентация в области «молекулярной медици¬
12Введениены», знание ее потенциала, основных принципов и ме¬
тодов исследования являются абсолютно необходимы¬
ми для врача любой специальности, вступающего в
XXI век.Одним из разделов клинической медицины, наи¬
более ярко демонстрирующим современные возможно¬
сти молекулярной генетики, является неврология [Ива-
нова-Смоленская И.А., 1996; Еуаш О., 1998]. Это объяс¬
няется исключительной сложностью генетической орга¬
низации функций мозга, высокой степенью дифферен-
цировки и «энергетической ранимости» нейронов и, как
следствие - их частым вовлечением в патологический
процесс при наследственно обусловленных системных
заболеваниях, метаболических расстройствах и повреж¬
дениях структурных белков клетки. Исследования в об¬
ласти молекулярной генетики представляют особую ак¬
туальность в связи с высоким удельным весом нейроге-
редитарных заболеваний в общей структуре неврологи¬
ческой патологии, глубокой инвалидизацией больных с
прогрессирующей психической и физической дезадап¬
тацией, а также фатальным течением этих тяжелых и в
большинстве случаев неизлечимых страданий.Разнообразные методы ДНК-анализа заняли свое
прочное место в исследовательском арсенале клиничес¬
кой неврологии с начала 90-х годов XX столетия. К на¬
стоящему времени известно уже более 300 наследствен¬
ных заболеваний нервной системы, для которых полно¬
стью расшифрованы первичные молекулярные дефекты
или установлена хромосомная локализация мутантных
генов. К числу таких заболеваний относятся большое
число форм прогрессирующих мышечных дистрофий и
врожденных миопатий, наследственные спиноцеребел-
лярные атаксии, наследственные спастические парапле¬
гии, спинальные амиотрофии. наследственные моторные
Введение13: I сенсорные невропатии, наиболее частые наследствен-
11 ые заболевания экстрапирамидной системы (хорея Ген-
Iннгтона, эссенциальный тремор, торсионная дистония,
болезнь Вильсона-Коновалова), митохондриальные эн-I к-фаломиопатии и др. По существу, на фоне этих откры¬
тий происходит коренное преобразование идеологии и
методических возможностей нейрогенетики и ряда дру¬
гих разделов клинической неврологии. Наиболее ярким
отражением достигнутого прогресса стало появление
новейших и надежных методов диагностики, основан¬
ных на ДНК-тестировании и идентификации конкрет-II ых генетических дефектов, лежащих в основе болезни.’ )то, в свою очередь, повлекло за собой изменение базо-111,IX принципов систематизации наследственных заболе¬
ваний нервной системы и пересмотр всей номенклату¬
ры этих заболеваний. Существенно изменились прин-
11,ипы медико-генетического консультирования: благодаря
ДНК-диагностике у неврологов появилась возможность
раннего выявления носителей мутантного гена и в ряде
случаев проведения превентивной терапии на пресимп-
томатической стадии болезни. Более того, проведение
пренатальной ДНК-диагностики позволяет в настоящее
время достоверно оценивать генетический статус плода11 при необходимости рекомендовать прерывание бере-
менности, способствуя профилактике повторных случа¬
ев заболевания в отягощенных семьях. Новейшей стра¬
ницей профилактики наследственной неврологической
патологии является разработка методов преимплантаци-
онной ДНК-диагностики (см. главу 5).Следует особенно отметить, что успехи «молеку-
пярной медицины» относятся не только к узкой областиI [«следственных моногенных заболеваний нервной сис¬
темы. Напротив, они имеют широкое общеклиническое
шачение и чрезвычайно важны для понимания патоге¬
14Введениенеза наиболее частых мультифакториальных болезней
мозга. При этом моногенные болезни выступают в каче¬
стве своеобразных моделей для изучения близких к ним
спорадических (наиболее частых) форм соответствую¬
щих заболеваний. Как пример можно привести опыт
изучения болезней Паркинсона и Альцгеймера. Как из¬
вестно, идиопатическим паркинсонизмом страдают не
менее 1% лиц старше 60 лет, а болезнь Альцгеймера яв¬
ляется ведущей причиной деменции в современном об¬
ществе. В последние годы было показано, что не менее
10% всех случаев болезней Паркинсона и Альцгеймера
носят наследственный характер и обусловлены повреж¬
дениями нескольких недавно открытых ядерных генов,
имеющих отношение к процессам нейронального транс¬
порта и внутриклеточного протеолиза нейрональных
белков. Поскольку ключевые звенья патогенеза наслед¬
ственных и спорадических случаев болезней Паркинсо¬
на и Альцгеймера могут быть весьма сходными, даль¬
нейшее изучение молекулярной основы семейных форм
будет способствовать раскрытию интимных патогенети¬
ческих механизмов спорадических случаев этих заболе¬
ваний, представляющих собой актуальнейшую пробле¬
му современной неврологии. Аналогичным образом,
известные на сегодня наследственные формы эпилепсии,
цереброваскулярных заболеваний, бокового амиотрофи¬
ческого склероза и т.д. служат своеобразным «мости¬
ком», позволяющим на основе молекулярных техноло¬
гий подходить к изучению патогенеза этих широко рас¬
пространенных заболеваний нервной системы. Все это
дает надежду на разработку новых эффективных мето¬
дов их лечения и профилактики.Таким образом, достигнутый прогресс в данной
области исследований изменяет само мышление невро¬
логов и диктует настоятельную необходимость более
Введение15активного внедрения достижений молекулярной генети¬
ки в повседневную практику. Между тем, до настоящего
времени приходится констатировать существование зна¬
чительных пробелов в преподавании основ молекуляр¬
ной генетики и молекулярной биологии в медицинских
вузах нашей страны. Клинические неврологи плохо зна¬
комы с современными возможностями молекулярной
медицины даже в рамках своей основной специальнос¬
ти. Несмотря на то, что количество публикаций, посвя¬
щенных анализу генов наследственных болезней нервной
системы, носит характер «информационного взрыва»,
подавляющее число этих работ опубликовано в новей¬
ших зарубежных журналах, в большинстве случаев не¬
доступных для практического врача. Это тем более обид¬
но, поскольку в настоящее время в России существует
практическая возможность генодиагностики основных
наиболее распространенных наследственных заболева¬
ний нервной системы. Такая диагностика проводится
на базе специализированных лабораторий нескольких
ведущих научных центров страны, среди которых в пер¬
вую очередь можно назвать НИИ неврологии РАМН
(Москва), Институт молекулярной генетики РАН (Мос¬
ква), Медико-генетический научный центр РАМН (Мос¬
ква), Институт акушерства и гинекологии им. Д.О. Отта
РАМН (Санкт-Петербург), Уфимский научный центр
РАН (Уфа), НИИ медицинской генетики Сибирского
отделения РАМН (Томск).В отечественной литературе в последние годы
были опубликованы отдельные обзоры и монографии,
посвященные тем или иным вопросам молекулярной
диагностики и генотерапии наследственных заболеваний.
Выход ряда из этих книг, хотя и весьма ограниченным
тиражом [Шишкин С.С., Калинин В.Н., 1992;
Пузырев В.П., Степанов В.А., 1997; Горбунова В.Н.,
16ВведениеБаранов B.C., 1997; Горбунова В.Н., 1999], стал замет¬
ным событием для нашей страны и позволяет рекомен¬
довать их в качестве незаменимых пособий для врачей
любых специальностей. Однако подавляющая часть до¬
ступной для отечественного читателя литературы по дан¬
ной проблеме носит либо общий ознакомительный ха¬
рактер, либо адресована главным образом врачам-гене-
тикам и специалистам в области молекулярной биоло¬
гии. Принимая во внимание ту особую роль, которую
играют нейрогенетика и нейронауки в общем прогрессе
современной молекулярной медицины, следует признать
назревшую необходимость появления отдельной моно¬
графии, посвященной теоретическим и практическим
аспектам ДНК-диагностики в неврологии и ориентиро¬
ванной на практических врачей-неврологов. Такая кни¬
га перед вами, читатель.В настоящей монографии авторами предпринята
попытка обобщения уникального опыта в области ДНК-
диагностики и медико-генетического консультирования
при наследственных заболеваниях нервной системы, на¬
копленного в нейрогенетическом отделении НИИ невро¬
логии РАМН. Нейрогенетическое отделение Института,
созданное в 1964 году Романом Александровичем Тка¬
чевым, стало первым в мире специализированным кли¬
ническим отделением данного профиля, из недр которо¬
го выросла ведущая школа нейрогенетики в нашей стра¬
не. Данное отделение продолжило традиции изучения
наследственных и дегенеративных заболеваний нервной
системы, заложенные основателем Института невроло¬
гии, выдающимся отечественным неврологом Никола¬
ем Васильевичем Коноваловым. Приоритетный вкладН.В. Коновалова в изучение одного из тяжелейших на¬
следственных заболеваний нервной системы - гепато-
лентикулярной дегенерации - признан во всем мире и
Введение17по праву отражен в общепринятом названии данной бо¬
лезни (болезнь Вильсона-Коновалова). За 35-летнюю ис¬
торию существования нейрогенетического отделения
Института неврологии под его наблюдением находились
свыше 1500 семей, отягощенных различными моноген-
ными наследственными заболеваниями центральной и
периферической нервной системы. Сотрудниками отде¬
ления описан ряд новых форм наследственных невроло¬
гических заболеваний, детально изучены вопросы фе¬
нотипического полиморфизма и биохимических звень¬
ев патогенеза и на этой основе разработаны новые мето¬
ды диагностики и лечения этих тяжелых страданий. В
1994 году при отделении была создана специализиро¬
ванная ДНК-лаборатория, что ознаменовало собой на¬
чало нового этапа в изучении наследственных заболева¬
ний нервной системы, связанного с внедрением наибо¬
лее современных молекулярно-генетических технологий.
На протяжении последних 5 лет работа отделения в об¬
ласти ДНК-диагностики, картирования генов и клини¬
ко-молекулярных сопоставлений проводилась в тесном
сотрудничестве с рядом ведущих российских и зарубеж¬
ных научных центров. Авторы хотели бы выразить осо¬
бую благодарность за плодотворную совместную работу
коллективу сотрудников отдела молекулярных основ ге¬
нетики человека Института молекулярной генетики РАН
(руководитель отдела Лауреат Государственной премии
РФ, доктор биологических наук, профессор С.А. Лим-
борская, ведущий научный сотрудник кандидат биоло¬
гических наук П.А. Сломинский). Важным для станов¬
ления данного направления исследований стало для нас
сотрудничество с молекулярным центром Гарвардского
университета в Бостоне (профессор Х.О. Breakefield), не¬
врологическим отделом Колумбийского университета в
Нью-Йорке (профессора М. Brin, S. Faim), отделением
18Введениеневрологии Института мозга Университета г. Ниигаты в
Японии (профессор S. Tsuji), молекулярной лаборатори¬
ей Отдела патологии нейронов Национального Инсти¬
тута Здоровья и Медицинских Исследований в Париже
(профессор A. Brice).Разумеется, освоение принципов и идеологии
ДНК-диагностики невозможно без твердых базовых зна¬
ний об основных закономерностях строения генома и
молекулярных основах рассматриваемых групп заболе¬
ваний человека. В связи с этим в настоящей моногра¬
фии представлен ряд обзорных разделов, посвященных
общим принципам генеалогического анализа, организа¬
ции генетического материала клетки, основным мето¬
дам ДНК-диагностики, геномной классификации наслед¬
ственных болезней нервной системы. В частных разде¬
лах монографии обобщаются современные представле¬
ния о молекулярной генетике наиболее распространен¬
ных наследственных болезней нервной системы, для
которых разработаны методы ДНК-диагностики.Приложение включает сводный каталог генов
нейрогередитарных заболеваний (по состоянию на конец
2001 года), современную номенклатуру мутаций, а также
краткий словарь генетических терминов, существенно
облегчающий восприятие материала. Таким образом,
предлагаемая монография, помимо общего знакомства с
проблемой ДНК-диагностики, может в определенном
смысле служить руководством по молекулярной
неврологии для практических неврологов и врачей других
специальностей.Издание настоящей книги, впервые обобщающей
результаты исследований в новой бурно развивающейся
области неврологии, является вкладом ученых НИИ не¬
врологии РАМН в недавно завершившуюся Программу
«Десятилетие мозга» (1990-2000 гг.).
ГЛАВА 1ОБЩИЕ ПРИНЦИПЫ
ГЕНОДИАГНОСТИКИ1.1. СТРОЕНИЕ МОЛЕКУЛЫ ДНК/РНК
И ОРГАНИЗАЦИЯ ГЕНЕТИЧЕСКОГО
МАТЕРИАЛА В КЛЕТКЕМолекулярной основой наследственности у всех
прокариот и эукариот является особый класс биоорга-
нических веществ - нуклеиновые кислоты, подразделя¬
ющиеся по своему химическому составу и биологичес¬
кой роли на дезоксирибонуклеиновые кислоты (ДНК) и
рибонуклеиновые кислоты (РНК).Оба типа нуклеиновых кислот представляют со¬
бой нитевидные молекулы, состоящие из отдельных
структурных единиц - нуклеотидов, соединенных в мно¬
гозвеньевую полинуклеотидную цепь. Каждый нуклео¬
тид состоит из следующих трех химически различных
частей: I) остатков 5-углеродного сахара - дезоксирибо-
зы (в ДНК) и рибозы (в РНК), образующих «остов» по-
линуклеотидной нити; 2) четырех азотистых оснований-
аденина (А), гуанина (в), цитозина (С) и тимина (Т) (в
молекуле РНК последнее основание заменено на
урацил и), причем каждое азотистое основание ковален¬
тно соединено с первым атомом углерода сахара посред¬
ством гликозидной связи; 3) фосфатной группы, соеди¬
няющей соседние нуклеотиды в единую цепь посред¬
ством формирования фосфодиэфирных связей между 5’-
атомом углерода одного сахара и 3 ’-атомом углерода дру¬
гого. Первичная структура полинуклеотидных цепей
ДНК и РНК представлена на рис. 1.
20 Глава 15'-конецН \ /НЦитозинЦ ГуанинТимин-о-р=оІ01З'-конец5'-конецІ-о-р=о/пNДценинЦитозин"0-Р=0Урацилі-0~р=0І01З’-конецДНК РНКРис. 1. Первичная структура ДНК и РНК
Общие принципы генодиагностики 21Запись генетической информации осуществляет¬
ся линейно от 5’-конца к 3’-концу молекулы нуклеино¬
вой кислоты. В состав одной такой молекулы может вхо¬
дить до многих миллионов нуклеотидов.В клетке молекулы ДНК существуют в виде спи-
рализованной двойной цепи (двойной спирали), нити ко¬
торой антипараллельны, т.е. имеют противоположную
ориентацию. Двойная цепь ДНК образуется благодаря
слабым водородным связям между комплементарными
основаниями: аденин строго комплементарен тимину. а
цитозин - гуанину (рис. 2).5’ 3’> ПОЛНЫЙ виток
двойной спирали
ДНК (3,4 нм)водородная связьсахарофосфатный
остов5’Рис. 2. Двойная спираль ДНК
22Глава 1При определенных условиях указанные водород¬
ные связи могут разрываться, приводя к появлению од¬
ноцепочечных молекул (денатурация ДНК), а в даль¬
нейшем образовываться вновь между теми же компле¬
ментарными участками (ренатурация, или гибридиза¬
ция ДНК). В процессе гибридизации происходит точное
восстановление исходной двойной спирали ДНК. Имен¬
но наличие комплементарности обеспечивает как точ¬
ность самовоспроизводства ДНК в каждом цикле кле¬
точного деления (этот процесс носит название реплика¬
ция), так и восстановление нарушенного нуклеотидного
состава молекулы ДНК. В связи с комплементарностью
нуклеотидов в составе двойной спирали длину молеку¬
лы ДНК принято выражать в парах оснований (п.о.), а
также тысячах пар оснований (килобазы, кб) и милли¬
онах пар оснований (мегабазы, мб). В состав ДНК чело¬
века как биологического вида входит около 3 миллиар¬
дов п.о.Направленный синтез молекулы ДНК в клетке
осуществляется особым ферментом - ДНК-полимеразой.
Этот процесс предполагает «расплетение» двойной спи¬
рали на участке синтеза и образование особой белково¬
нуклеиновой структуры—репликационной вилки; посте¬
пенное продвижение репликационной вилки вдоль двой¬
ной спирали сопровождается последовательным присо¬
единением к вновь образуемой цепи оснований, комп¬
лементарных однонитевой ДНК-матрице (синтез расту¬
щей цепи ДНК всегда протекает строго в направлении
от 5’ к 3’). Комплементарный синтез ДНК требует при¬
сутствия в среде отдельных «кирпичиков» для удлине¬
ния растущей молекулы - четырех видов молекул дезок-
сирибонуклеотид-трифосфатов (с1АТР, сПТР, с!СТР и
сЮТР). Весь процесс инициируется особыми затравка¬
ми - праймерами, представляющими собой короткие
Общие принципы генодиагностики 23олигонуклеотидные молекулы, комплементарные опре¬
деленному стартовому участку ДНК-матрицы.Дискретной единицей наследственности у высших
организмов является ген. Совокупность всех генов оп¬
ределенного биологического вида определяется терми¬
ном геном (иногда данный термин относится к полной
генетической системе отдельной клетки или конкретно¬
го организма). Ген в своем наиболее практическом по-11 имании представляет собой строго определенный уча¬
сток молекулы ДНК, последовательность которого зак¬
лючает в себе всю информацию, необходимую для син¬
теза молекулы белка или РНК. Генетическая информа-
I щя зашифрована посредством универсального для всех
живых организмов генетического кода, представляюще¬
го собой набор нуклеотидных триплетов - кодонов. Каж¬
дый такой триплет (т.е. каждая последовательность из 3
нуклеотидов) кодирует синтез одной, строго определен¬
ной аминокислоты в составе белка (таблица 1).Считывание кодонов в процессе передачи гене¬
тической информации происходит последовательно
(I финцип линейности генетического кода), и любой нук¬
леотид может входить в состав только одного кодона
(принцип неперекрываемости генетического кода). Ге¬
нетический код является вырожденным, т.е. допускает
кодирование каждой из 20 аминокислот несколькими
возможными комбинациями триплетов (всего таких ком¬
бинаций может быть 64). Расшифровка точной последо¬
вательности нуклеотидов определенного информацион¬
ного участка гена позволяет однозначно идентифициро¬
вать последовательность аминокислот в составе соответ¬
ствующего полипептидного участка белка и его размер.
11олный гаплоидный геном человека (т.е. кодируемый
одной смысловой нитью ДНК) включает, ориентировочно,
около 30 ООО—40 ООО генов.
Таблица 1Триплетный генетический код (на уровне молекул РНК)ПервоеоснованиеВтороеоснованиеиСАGиUUU фенилаланин
UUC фенилаланин
UUA лейцин
UUG лейцинUCU серии
UCC серин
UCA серин
UCG серинUAU тирозин
UAC тирозин
UAA стоп-кодон
UAG стоп -кодонUGU цистеин
UGC цистеин
UGA стоп-кодон
UGG триптофанСCUU лейцин
CUC лейцин
CUA лейцин
CUG лейцинCCU пролин
ССС пролин
ССА пролин
CCG пролинCAU гистидин
CAC гистидин
САА глутамин
CAG глутаминCGU аргинин
CGC аргинин
CGA аргинин
CGG аргининАAUU изолейцин
AUC изолейцин
AUA изолейцин
AUG метионинACU треонин
АСС треонин
АСА треонин
ACG треонинAAU аспарагин
AAC аспарагин
ААА лизин
AAG лизинAGU серин
AGC серин
AGA аргинин
AGG аргининGGUU валин
GUC валин
GUA валин
GUG валинGCU аланин
GCC аланин
GCA аланин
GCG аланинGAU аспарагиновая кислота
GAC аспарагиновая кислота
GAA глутаминовая кислота
GAG глутаминовая кислотаGGU глицин
GGC глицин
GGA глицин
GGG глицинbo■fe<Глава 1
Общие принципы генодиагностики 25Гены человека и других высших организмов име¬
ют чрезвычайно сложную структурно-функциональную
организацию и содержат различные по своей биологи¬
ческой роли нуклеотидные участки. Одни из них (экзо-
ны) являются относительно короткими, представляют
собой кодирующие последовательности и определяют
аминокислотный состав белков; другие участки гена (ин-
троны) являются обычно значительно более протяжен¬
ными и не несут непосредственной информационной
нагрузки. Окончательная роль интронов до настоящего
времени не установлена; предполагается, что они могут
иметь отношение к регуляции экспрессии генов и конт¬
ролю тонких механизмов «считывания» генетической
информации. В состав генов входят также особые регу¬
ляторные участки (промоторы, энхансеры, различные
сигнальные последовательности), обеспечивающие ини¬
циацию, интенсивность и определенную временную пос¬
ледовательность процессов нуклеотидного синтеза на
ДНК-матрице, а также модификацию промежуточных
полинуклеотидных продуктов. По ориентировочным
оценкам, собственно кодирующие последовательности
ДНК составляют не более 3-10% всего генома человека.В любой клетке организма содержится полный
набор генов, однако лишь небольшая их часть является
функционально активной в каждой конкретной ткани,
т.е. экспрессируется. Под экспрессией гена понимают
реализацию записанной в нем генетической информа¬
ции, приводящую к синтезу первичных молекулярных
продуктов гена - РНК и белка. Именно временная и тка¬
невая избирательность экспрессии генов определяет спе¬
цифику дифференцировки и функционирования различ¬
ных органов, тканей и клеток организма в онтогенезе.Процесс передачи генетической информации оп¬
ределяется так называемой центральной догмой моле¬
кулярной биологии: ДНК—»РНК—>белок. Согласно совре-
26Глава 1интрон 1ДНК (экспрессирующийся ген)транскрипцияпервичный РНК-транскрипт
(преРНК)аса.сплаисингмолекула РНК
без интроновинтрон 2концевые модификации
(полиаденилирование,
"кэпирование”)кДНКI/ /
# /
# //У '& I$&зрелая мРНК [сарі — г I Л у\ааатрансляцияпервичная полипептидная оооосооосоососоосоо
молекуласозревание белка
(гликозилирование,
конформационные
изменения и т.д.)зрелый функционально
активный белокРис. 3. Передача генетической информации в клетке
Общие принципы генодиагностики 27менным представлениям, путь от гена к белку является
весьма сложным и распадается не несколько самостоя¬
тельных этапов, схематично показанных на рис. 3.На первом этапе происходит «переписывание»
нуклеотидной последовательности гена путем синтеза
комплементарной ему молекулы РНК (;транскрипция).
Транскрипция направляется ферментом РНК-полимера-
юй и ведет к образованию в ядре клетки молекул пер¬
вичного РНК-транскрипта (преРНК). Молекула преРНК
представляет собой точный слепок ДНК-матрицы транс¬
крибируемого гена. Синтезированная преРНК проходит
стадию созревания (процессинг): эта стадия включает в
себя как модификацию концевых участков цепи (способ¬
ствующую стабилизации молекулы), так и удаление изI юрвичного РНК-транскрипта некодирующих участков-
нитронов.Процесс «вырезания» интронов, который носит
название сплайсинг, является важнейшим звеном созре-
кания преРНК и приводит к тому, что в составе РНК
остаются лишь последовательно «сшитые» друг с дру¬
гом смысловые участки, комплементарные экзонам гена.
Ключевую сигнальную роль в осуществлении сплайсинга
играют определенные нуклеотидные последовательнос¬
ти, фланкирующие каждый из экзонов (так называемые
сайты сплайсинга); при локализации мутаций в сайтах
сплайсинга может происходить нарушение интимных
механизмов удаления интронов из состава преРНК и как
результат - синтез аномального по структуре пептида.
()бразующаяся после вырезания интронов зрелая РНК
носит название информационной, или матричной
(мРНК); по своей длине мРНК во много раз короче са¬
мого транскрибируемого гена и его первичного РНК-
гранскрипта.Следующий этап передачи генетической инфор¬
мации происходит в цитоплазме. Он заключается в сбор¬
28Глава 1ке на рибосомах молекул белка по матрице мРНК (про¬
цесс трансляции). Аминокислоты транспортируются к
рибосомам особым классом молекул - транспортными
РНК (тРНК). Каждая тРНК отвечает за транспортиров¬
ку строго определенной аминокислоты, причем эта спе¬
цифичность определяется наличием в составе тРНК уни¬
кальной 3-нуклеотидной последовательности, называе¬
мой антикодоном. По мере продвижения рибосомы
вдоль молекулы мРНК антикодоны различных тРНК,
несущих «свою» аминокислоту, последовательно распоз¬
наются комплементарными им кодонами мРНК. В ре¬
зультате этого происходит последовательное присоеди¬
нение «нужных» аминокислот к растущей полипептид-
ной цепи. Процесс трансляции инициируется триплетом
AUG, кодирующим аминокислоту метионин. Таким об¬
разом, метиониновый кодон в составе РНК открывает
рамку считывания генетической информации; как было
указано, это считывание происходит в соответствии с
правилом «один триплет - одна аминокислота». Сигна¬
лом окончания трансляции служит один из трех особых
кодонов (UAA, UAG или UGA), получивших название
стоп-кодоны (нонсенс-кодоны); распознавание стоп-ко¬
дона на рибосоме прекращает синтез полипептидной
цепи.По окончании трансляции первичная полипептид-
ная молекула претерпевает определенные посттрансля-
ционные модификации, превращаясь в функционально
зрелый продукт. «Дозревание» белка происходит, как
правило, в соответствующих органеллах клетки.Существует принципиальная возможность друго¬
го пути передачи генетической информации (т.е. в на¬
правлении, обратном положениям центральной догмы)
- от молекулы РНК к молекуле ДНК. В естественных
условиях такой путь свойственен некоторым РНК-содер-
жащим вирусам, в то время как у высших организмов
Общие принципы генодиагностики 29обратная транскрипция мРНК в ДНК возможна in vitro с
участием особого фермента-об/>шгшой транскриптазы.
В результате образуется искусственная молекула ДНК,
не содержащая интронов и полностью комплементар¬
ная мРНК-матрице (так называемая комплементарная
ДНК, или кДНК) (рис. 3, пунктирная стрелка). Посколь¬
ку молекула кДНК представляет собой, образно говоря,
концентрированный кодирующий продукт исходного
гена, она является весьма ценным материалом при про¬
ведении различных молекулярно-генетических исследо¬
ваний (в том числе с целью генодиагностики и
гемотерапии).ДНК человека в основной своей части упакована
и ядрах клеток в форме компактных структур - хромо¬
сом. Каждая хромосома представляет собой одну гиган¬
тскую гиперспирализованную молекулу ДНК, образую-I цую сложный комплекс с особыми ядерными белками -I истонами. Во всех ядерных соматических клетках на¬
бор хромосом парный (по одному от каждого родителя),
и половых - одинарный. В норме у человека 46 хромо¬
сом представлены 23 парами, из которых 22 являются
неполовыми (аутосомы) и 1 пара-половые хромосомы
(X и Y). Набор половых хромосом детерминируют пол
индивидуума (XY - мужской, XX - женский).В составе каждой хромосомы можно выделить
длинное и короткое плечо, разделяемые центральной
перетяжкой - центромерой. Хромосомы не идентичны
по своим размерам и строению (соотношение длинного
п короткого плеча). При использовании специальной
дифференциальной окраски хромосомы приобретают
поперечно-исчерченный вид, который является их «ин¬
дивидуальным портретом», позволяющим идентифици¬
ровать каждую из хромосом при световой микроскопии.
) 1,анная картина хромосом определяется их нуклеотид-
п ым составом, а также соотношением эухроматина и
30Глава 1гетерохроматина (эухроматин - нуклеопротеидный ком¬
плекс с низкой плотностью упаковки, содержащий ос¬
новное число активно транскрибируемых генов; гетерох¬
роматин - конденсированная часть нуклеопротеиндно-
го комплекса, содержит в основном некодирующие вы¬
сокоповторяющиеся последовательности ДНК). В каче¬
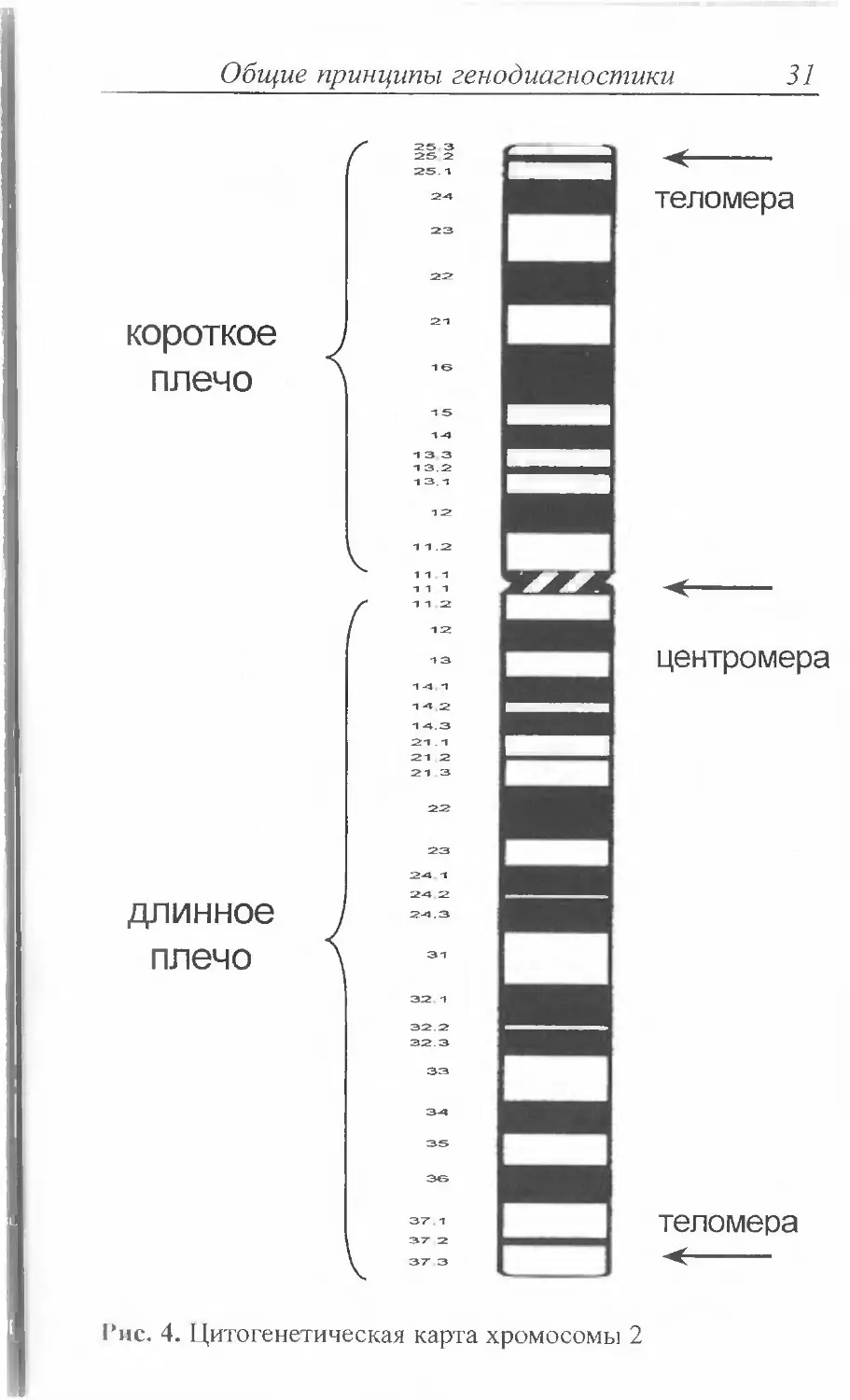
стве примера на рис. 4 представлена цитогенетическая
карта 2-й хромосомы (эта хромосома является одной из
наиболее «значимых» применительно к нейрогенетичес-
ким заболеваниям). В соответствии с действующей меж¬
дународной цитогенетической номенклатурой (ISCN,
1978) каждый участок хромосомы обозначается симво¬
лом, отражающим плечо хромосомы (р - короткое, q -
длинное), номер сегмента по направлению от центроме¬
ры к концевому участку хромосомы (теломере), далее
внутри каждого сегмента - номер полосы, а также иног¬
да-номер более мелких субъединиц в составе полос. Так,
символ 2р 13.1 подразумевает участок, расположенный
в 1-й субъединице 3-й полосы 1-го сегмента короткого
плеча хромосомы 2. Центромерные и наиболее терми¬
нальные теломерные участки каждого плеча хромосомы
обозначаются, соответственно, символами сеп и ter. На¬
пример, обозначение 17pter указывает на наиболее дис¬
тальный терминальный участок короткого плеча 17-й
хромосомы.Деление соматической клетки и ее ядра (митоз)
сопровождается сложными многофазными трансформа¬
циями хромосом (рис. 5): 1) в процессе митоза происхо¬
дит удвоение каждой хромосомы на основе комплемен¬
тарной репликации молекулы ДНК с образованием двух
сестринских нитевидных копий (хроматид), соединен¬
ных в области центромеры; 2) в последующем сестрин¬
ские хроматиды разъединяются и эквивалентно распре¬
деляются по ядрам дочерних клеток.
Общие принципы генодиагностики 31короткоеплечог{1 51 -41 3.313.21 з. 1теломераVГ11.211.1
1 1 111.212
1 3
1-4.1
1-4.2
1-4.3
21 . 1
21 .2
21 3центромерадлинноеплечо232-4.124.22-4.332.232.33-435V37. 1
37.2
37-.3теломера1*ис. 4. Цитогенетическая карта хромосомы 2
32Глава 1Рис. 5. Схема митозаМеханизм эквивалентного расхождения генетического материала
по дочерним клеткам показан на примере 2 пар хромосомРис. 6. Схема мейозаМеханизм образования четырёх гаплоидных клеток показан на
примере 2 пар хромосом.
Общие принципы генодиагностики 33В результате в делящихся соматических клетках
поддерживается идентичность хромосомного набора и
iепетического материала. Отдельно следует сказать о
| к-йронах - высокодифференцированных постмитотичес-
к их клетках, не претерпевающих клеточных делений на
протяжении жизни. Компенсаторные возможности ней¬
ронов в ответ на действие повреждающих факторов ог-I ». и шчиваются внутриклеточной регенерацией и репара-I щей ДНК в неделящемся ядре, чем в значительной сте¬
пени обусловлена специфика нейропатологических про-I i.cccob наследственной и ненаследственной природы.Совершенно иной тип деления - мейоз - харак¬
терен для половых клеток. Главной особенностью мей-
оча являются два последовательных деления клетки-II редшественника и ее ядра, в то время как хромосомы
удваиваются лишь однажды. Схематично механизм мей-
оча выглядит следующим образом (рис. 6): 1) в первом
делении мейоза дочерние клетки получают из каждой
хромосомной пары по одной гомологичной хромосоме,
( •< »стоящей из удвоенных сестринских хроматид (посколь¬
ку при этом число хромосом в дочерних клетках умень-
ш; 1стся вдвое, данное деление является редукционным);) но втором делении сестринские хроматиды разъеди¬
няются и эквивалентно расходятся по образующимся
чрелым половым клеткам - гаметам. В результате число
х ромосом в гаметах оказывается вдвое меньшим по срав¬
нению с исходной родительской клеткой. После слия¬
ния ядер половых клеток при оплодотворении зигота
получает стандартный двойной набор хромосом. Дан¬
ный механизм обеспечивает постоянство числа хромо¬
сом у разных поколений организмов, размножающихся
половым путем.Важнейшей биологической ролью мейоза явля¬
ется обеспечение генетического разнообразия особей в
34Глава 1результате «перемешивания» отцовских и материнских
генов в гамете. Это достигается двумя путями. Во-пер¬
вых, как показано на рис. 6, в первом делении мейоза
распределение отцовских и материнских хромосом по
дочерним клеткам происходит случайным образом, в
результате чего гаметы несут различные комбинации
родительских хромосом. Второй фундаментальный ме¬
ханизм поддержания генетического разнообразия заслу¬
живает того, чтобы быть разобранным более подробно,
поскольку он имеет прямое отношение к теме настоя¬
щей монографии - ДНК-диагностике. Данный механизм
схематично показан на рис. 7.А Б ВЖ жжжРис. 7. Кроссинговер между гомологичными хромосомами
в мейозеА. Спаривание гомологичных хромосом. Б. Образование хиазмы и
обмен гомологичными участками хромосом. В. Образующиеся в
результате кроссинговера рекомбинантные хромосомы.
Общие принципы генодиагностики 351} начальной фазе первого деления мейоза гомологич¬
ные хромосомы располагаются друг напротив друга и
спариваются, образуя одну или несколько зон контакта
(хиазм) между отдельными несестринскими хроматида-
ми (рис. 7 А, Б). Далее пара хроматид, образовавшая хи¬
азму, обменивается участками ДНК - процесс, носящий,
название кроссинговер. В результате кроссинговера об¬
разуются рекомбинантные хромосомы, состоящие из уча¬
стков, имеющих происхождение от разных родительс¬
ких линий (рис.7В). По завершении мейоза рекомбинан¬
тные хромосомы разойдутся по разным гаметам. Таким
образом, кроссинговер представляет собой частный слу¬
чай генетической рекомбинации - процесса перераспре¬
деления генетического материала родителей при пере¬
даче потомству. Важным следствием кроссинговера ста¬
новится создание новой комбинации генов у потомков
при соединении родительских гамет. Поскольку при ре¬
комбинации происходит обмен генетического материа¬
ла между отцовской и материнской хромосомами, этот
феномен всегда должен приниматься во внимание при
анализе наследования хромосом в процессе проведения
косвенной ДНК-диагностики и расчете генетического
сцепления (см. далее).В клетке имеется около 5% ДНК, не входящей в
состав хромосом ядра и локализованной в митохондри¬
ях. Митохондриальная ДНК (мтДНК) представляет со¬
бой короткую двуцепочечную кольцевую молекулу, ко¬
торая состоит из 16 569 п.о. и содержит 37 генов, коди¬
рующих синтез некоторых видов рибосомальной и транс¬
портной РНК, а также небольшого числа полипептидов
дыхательной цепи митохондрий. Особенностями мтДНК
являются: 1) отсутствие некодирующих областей - нит¬
ронов; 2) некоторые отличия генетического кода по срав¬
нению с ядерной ДНК; 3) более простая организация и
36Глава 1отсутствие связи с белками-гистоиами; 4) несовершен¬
ство системы репарации ДНК. Два последних фактора
лежат в основе того, что повреждаемость (темп мутиро¬
вания) мтДНК на порядок выше, чем ядерной ДНК. Одна
митохондрия (а их число в клетке может достигать мно¬
гих десятков и сотен) обычно содержит несколько ко¬
пий мтДНК - в среднем около 5. Поэтому популяция
молекул мтДНК в любой клетке и ткани является весь¬
ма значительной. Каждая молекула мтДНК реплициру¬
ется самостоятельно, и при делении клетки различные
молекулы мтДНК вместе с митохондриями в случайном
порядке переходят в цитоплазму дочерних клеток. Та¬
ким образом, клетки и ткани могут содержать как мтДНК
одного вида (гомоплазмия), так и комбинацию нормаль¬
ной и мутантной мтДНК в различных соотношениях (ге-
тероплазмия). Набор митохондрий в зиготе и, следова¬
тельно, во всех клетках организма имеет исключитель¬
но материнское происхождение - из цитоплазмы яйцек¬
летки, поэтому мтДНК всегда наследуется по материнс¬
кой линии. Мутации мтДНК лежат в основе особого
класса заболеваний - митохондриальных цитопатий
(см. главу 3, раздел 3.8).1.2. Основы генеалогического анализа
в неврологии1.2.1. Наследственные факторы в патогенезеневрологических заболеванийГенетические механизмы играют одну из ключе¬
вых ролей в развитии патологического процесса при за¬
болеваниях нервной системы, при этом конкретный
вклад наследственных факторов при той или иной нозо¬
логической форме может быть различным. С этой точки
зрения среди всего многообразия неврологической па-
Общие принципы генодиагностики 37гологии можно выделить три большие группы заболе¬
ваний.К первой группе относятся моногенные наслед¬
ственные заболевания - они патогенетически обуслов¬
лены мутациями в одном «главном гене», повреждение
которого имеет определяющий эффект для развития бо¬
лезни. Наследование указанного генетического дефекта
приводит к повторному появлению («выщеплению»)
патологического фенотипа в рамках конкретной семьи.
Результатом таких мутаций обычно является функцио¬
нально значимый дефект фермента, рецептора, структур¬
ного белка клетки или транспортной молекулы, что и
лежит в основе четкой причинно-следственной взаимо¬
связи между повреждением гена и развитием болезни.
Наследование моногенных болезней в родословной под¬
чиняется строгим закономерностям, отражающим харак¬
тер сегрегации генетического дефекта в ряду поколений.
Такие болезни иногда принято называть менделирующи-
ии - в честь выдающегося чешского исследователя-экс-
периментатора Грегора Менделя, сформулировавшего в
1866 г. основные общебиологические законы наслед¬
ственности (менделевские законы). При наследственных
менделирующих заболеваниях роль основного генети¬
ческого локуса в этиопатогенезе болезни является веду¬
щей, однако фенотипическая экспрессия мутации может
в определенной степени модифицироваться действием
ряда других экзогенных и эндогенных факторов (усло¬
вия среды, характер питания, особенности энергетичес¬
кого метаболизма организма и отдельных тканей, дей¬
ствие генов-модификаторов и т.д.).Вторая группа включает большое число заболе¬
ваний, определяемых как полигенные, или мультифак-
ториальные. В их патогенезе ведущее значение имеет
действие ряда взаимосвязанных биохимических, имму¬
38Глава 1нологических и иных механизмов, контролируемых раз¬
личными генетическим локусами. При этом конкретные
варианты одного или нескольких основных генов не яв¬
ляются абсолютно «фатальными» и лишь формируют
определенный «генетический фон», специфический для
каждого индивидуума и определяющий особенности вза¬
имодействия организма со средой. В результате в семье
наследуется не сама болезнь, а предрасположенность к
развитию определенного патологического состояния,
которая может реализоваться лишь в конкретных небла¬
гоприятных природно-средовых и социальных услови¬
ях. Данные болезни относят к болезням предрасположен¬
ности. Лица с наследственной предрасположенностью
имеют повышенный (по сравнению с общепопуляцион¬
ным) риск развития соответствующего заболевания, од¬
нако степень этого риска определяется сложным взаи¬
модействием совокупности генетических и средовых
факторов. Наследование мультифакториальных заболе¬
ваний не подчиняется строгим менделевским законам,
но в отягощенных семьях может наблюдаться накопле¬
ние повторных случаев болезни, отражающее действие
общих для членов семьи факторов риска [Бочков Н.П.,
1997]. К заболеваниям с несомненной наследственной
предрасположенностью относятся шизофрения, болезнь
Паркинсона, гипертоническая болезнь, сахарный диабет
и др. В ряду полигенных мультифакториальных болез¬
ней особую группу составляют онкологические заболе¬
вания, в развитии которых, как убедительно показано в
последние годы, ведущее значение имеют повреждения
ряда генов в соматических клетках организма [Гарькав-
цева Р.Ф., Гарькавцев И.В., 1999].Наконец, существует и третья группа заболева¬
ний нервной системы, в этиологии которых основное
значение принадлежит различным экзогенным факторам
Общие принципы генодиагностики 39(черепно-мозговая травма, нейроинфекции, интоксика¬
ции и т.д.). При этих заболеваниях роль генотипа огра¬
ничена регулированием восприимчивости организма,
>ффективности иммунного ответа и характера адапта¬
ционно-компенсаторных реакций в ответ на внешнее
воздействие.В настоящей монографии основное внимание бу¬
дет уделено заболеваниям из первых двух групп (главы 3
и 4), для которых роль наследственности наиболее зна¬
чима и методы генодиагностики получили наибольшее
развитие.1.2.2. Типы наследования моногенных болезней.Принципы сбора генеалогической информацииТип наследования является важнейшей и посто¬
янной характеристикой любого моногенного заболева¬
ния; он отражает функциональную значимость соответ¬
ствующего мутантного гена, его хромосомную локали¬
зацию и механизмы реализации мутации на клеточном
уровне. Менделирующие заболевания нервной системы
могут наследоваться по аутосомно-доминантному, ауто-
сомно-рецессивному и Х-сцепленному типам.Аутосомно-доминантный тип наследования бо¬
лезни имеет место в тех случаях, когда патологический
ген является доминирующим и закономерно приводит к
развитию симптоматики даже в гетерозиготном состоя¬
нии, т.е. на одной из двух гомологичных неполовых хро¬
мосом (аутосом). Обычно при аутосомно-доминантных
заболеваниях в браке больного и здорового членов се¬
мьи распределение аллелей имеет вид Аа х аа (где А -
доминантный мутантный ген, а-рецессивный нормаль¬
ный ген). Данный тип наследования характеризуется
следующими признаками:
40Глава 11) прямая передача болезни от одного из роди¬
телей потомкам («вертикальная» передача болезни), в
том числе передача заболевания детям от больного отца;
нередко может прослеживаться манифестация заболева¬
ния в нескольких поколениях;2) соотношение больных и здоровых лиц у по¬
томков больного индивидуума близко к 50%; соответ¬
ственно, для каждого из детей - потомков больного ро¬
дителя риск унаследовать мутантный ген (т.е. риск воз¬
никновения заболевания) составляет 50%;3) мужчины и женщины поражаются обычно в
равной степени; в редких случаях (например, при торси¬
онной дистонии) может наблюдаться более высокая пе-
нетрантность гена и более тяжелое течение заболевания
у лиц определенного пола - чаще у женщин.Типичный пример родословной, в которой имеет
место наследование аутосомно-доминантного заболева¬
ния, представлен на рис. 8. Среди неврологических за¬
болеваний аутосомно-доминантный тип наследования
характерен для хореи Гентингтона, нейрофиброматоза,
эссенциального тремора, торсионной дистонии, ряда
форм спиноцеребеллярных атаксий и др.Доминантные гены обладают различной пенет-
рантностъю. Под пенетрантностью понимают вероят¬
ность проявления мутантного гена у его носителей. В
том случае, когда заболевают все члены семьи, имею¬
щие мутантный ген, пенетрантность гена составляет
100%. При неполной пенетрантности мутантного гена
отдельные члены семьи, заведомо являющиеся носите¬
лями мутации («облигатные» носители), могут на про¬
тяжении всей жизни оставаться клинически здоровыми,
но при этом они передают мутантный ген и заболевание
своим детям. В таких случаях говорят о «пропуске по¬
коления» в родословной (см. рис. 8, индивидуум Ш-5).
12 3 4 510Рис. 8. Родословная семьи с аутосомно-доминантным заболеваниемОбщие принципы генодиагностики
42Глава 1В целом при отсутствии признаков аутосомно-доминан-
тного заболевания у родителей больного возможны сле¬
дующие объяснения: а) неполная пенетрантность мутан¬
тного гена у одного из родителей, являющегося носите¬
лем мутации; б) ложное отцовство; в) возникновение
мутации de novo в гамете одного из родителей или у боль¬
ного на ранней постзиготической стадии.Еще одним свойством доминантных генов, кото¬
рое следует принимать во внимание при генеалогичес¬
ком анализе, является различная экспрессивность. Экс¬
прессивность гена - это степень тяжести и широта спек¬
тра клинических проявлений болезни у носителей мута¬
ции. Высокая экспрессивность мутантного гена приво¬
дит к появлению развернутой клинической картины бо¬
лезни у лиц, унаследовавших мутантный ген. При низ¬
кой экспрессивности гена заболевание может у отдель¬
ных носителей мутации проявляться в виде различных
«стертых» форм. Так, например, гены аутосомно-доми-
нантной торсионной дистонии характеризуются непол¬
ной экспрессивностью, при этом у носителей патологи¬
ческого гена могут наблюдаться как тяжелые генерали¬
зованные формы заболевания, так и разнообразные
«стертые» варианты (forme fruste), в частности - фокаль¬
ные дистонические синдромы (кривошея, писчий спазм,
блефароспазм), изолированный постуральный тремор
рук и т.д. Правильная диагностика таких «стертых» форм
болезни в родословной исключительно важна для меди¬
ко-генетического консультирования, поскольку лица как
с развернутой клинической картиной, так и имеющие
forme fruste в одинаковой степени являются носителями
мутантного гена и могут передать его в следующее по¬
коление, что приведет к появлению новых случаев бо¬
лезни у потомков.Аутосомно-рецессивньш тип наследования на¬
блюдается при заболеваниях, для манифестации кото¬
Общие принципы генодиагностики 43рых необходимо присутствие мутантного гена в гомози¬
готном состоянии, т.е. на обеих гомологичных хромосо¬
мах, унаследованных от родителей. Чаще всего при ауто-
сомно-рецессивных заболеваниях первичный молекуляр¬
ный дефект заключается в повреждении фермента, а па¬
тологический эффект проявляется при критическом сни¬
жении его активности ниже определенного порогового
значения (5-10%) - как это и имеет место при повреж¬
дении обеих копий гена. Гетерозиготные носители име¬
ют «одинарную дозу» мутации; благодаря второй (нор¬
мальной) копии гена активность белка у них составляет
около 50%, что обычно вполне достаточно для поддер¬
жания соответствующей функции на физиологическом
уровне, поэтому они остаются клинически здоровыми.
В классических случаях аутосомно-рецессивиого насле¬
дования генотип родителей больных имеет вид Аа х Аа
(где а - рецессивный мутантный ген А - доминантный
нормальный ген). Данный тип наследования характери¬
зуется следующими особенностями:1) болезнь проявляется в одном поколении сре¬
ди сибсов (т.е. среди братьев-сестер - детей одной роди¬
тельской пары), родители при этом клинически здоро¬
вы («горизонтальная» передача болезни);2) доля пораженных сибсов среди всех потом¬
ков определенной родительской пары составляет около
25% (возможные вариации этой цифры в разных семьях
обусловлены случайным характером наследования оп¬
ределенной комбинации родительских хромосом); риск
развития заболевания у каждого ребенка соответствую¬
щей родительской пары также составляет 25%;3) у родителей больных лиц часто имеет место
кровнородственный брак (именно в кровнородственном
браке наиболее высока вероятность того, что дети унас¬
ледуют от обоих родителей две мутантные хромосомы,
имеющие общее генетическое происхождение);
4) мужчины и женщины поражаются в равной
степени.Типичный пример родословной с аутосомно-ре-
цессивным заболеванием представлен на рис. 9. Такой
тип наследования характерен для болезни Фридрейха,
гепатолентикулярной дегенерации, спинальной амиот-
рофии Верднига-Гофманаи Кугельберга-Веландер, атак-
сии-телеангиэктазии и ряда других моногенных заболе¬
ваний нервной системы.О-• смОО (_) 11(ь1 2 3 4 5 6 7 8Рис. 9. Родословная семьи с аутосомно-рецессивным
заболеваниемПри проведении генеалогического анализа в се¬
мьях с предполагаемым аутосомно-рецессивным типом
наследования следует принимать во внимание одно важ¬
ное обстоятельство. Как было указано выше, в соответ¬
ствии с менделевскими законами доля сибсов, поражен¬
ных аутосомно-рецессивным заболеванием, должна со¬
ставлять около % от общего числа детей в поколении.
Поскольку для современной структуры семей характер¬
но сравнительно небольшое число детей (1-3 ребенка),
в большинстве случаев аутосомно-рецессивные болезни
проявляются в виде единичных (спорадических) случа¬
ев, и наследственно-семейный характер заболевания
бывает очевидным далеко не всегда. В такой ситуации
Общие принципы генодиагностики 45отсутствие семейного анамнеза не снимает вопрос о ге¬
нетической природе болезни и не исключает 25%-й риск
ш »явления ее повторных случаев при рождении других
детей у данной родительской пары.Еще один источник ошибок в оценке данного типа
наследования проиллюстрирован на рис. 10 на примере
большой семьи с аутосомно-рецессивной мышечной
дистрофией, обследованной нами в одном из горных
пзолятов Северного Кавказа. В данной высокоинбред-
пой семье заболевание наблюдается у 12 родственников
п ? 3 разных поколений, что, на первый взгляд, противо¬
речит модели аутосомно-рецессивного наследования.
()днако ни в одном случае в данной родословной нет11 рямой передачи болезни от родителей детям, а в преде¬
лах каждой конкретной родительской пары характер сег¬
регации подчиняется всем закономерностям, характер¬
ным для аутосомно-рецессивной патологии. Таким об¬
разом, сам по себе факт наличия болезни в нескольких
поколениях обширной родословной не исключает ауто-
еомно-рецессивный тип наследования, и ключевым при-
■ таком здесь является манифестация симптомов у части
потомства (-25%) при клинически здоровых родителях,
являющихся облигатными гетерозиготными носителями
мутации.Правило об отсутствии прямой передачи аутосом¬
но-рецессивной болезни в следующее поколение имеет
редкие исключения: такое бывает возможным в ситуа¬
ции, когда больной вступает в брак либо с другим боль¬
ным с тем же заболеванием (тип брака аа х аа), либо с
гетерозиготным носителем мутации в том же гене
(аа х аА). В первом случае все дети унаследуют 2 копии
мутантного гена и будут больными, во втором случае за¬
болеет половина детей.
IV4^Рис. 10. Родословная большой семьи с аутосомно-рецессивным заболеванием, манифестирующим у ряда род¬
ственников из 3 поколенийГлава 1
Общие принципы генодиагностики 47Пример представлен на рис. 11 (показана родос¬
ловная наблюдавшейся нами семьи, отягощенной болез¬
нью Фридрейха).IVРис. 11. Родословная семьи с псевдодоминантным наследо¬
ванием болезни ФридрейхаВ данной семье больной отец (III-1) вступил в
кровнородственный брак с троюродной сестрой, являю¬
щейся гетерозиготным носителем мутантной хромосо¬
мы, унаследованной от общего предка (носительство
мутации подтверждено при ДНК-исследовании). В ре¬
зультате заболевание манифестировало у отца и 3 его
детей, т.е. в 2 последовательных поколениях. Такой осо¬
бый характер передачи аутосомно-рецессивного заболе¬
вания называется псевододоминантным. В отличие от
истинного аутосомно-доминантного типа передачи бо-
48Глава 1лезни при псевдодоминантном наследовании болезнь
регистрируется обычно лишь в 2 поколениях и не затра¬
гивает серии последовательных поколений и боковых
ветвей родословной. Еще одним признаком псевдодоми-
нантного наследования является то, что оно чаще всего
имеет место в случаях кровнородственных браков, по¬
скольку в соответствующих семьях частота носительства
мутантного рецессивного гена среди родственников зна¬
чительно выше общепопуляционной. Наконец, при псев¬
додоминантном наследовании число пораженных сиб-
сов в каждом поколении больше обычной для аутосом-
но-рецессивного наследования цифры 25%.При локализации мутантного гена в Х-хромосо-
ме имеет место наследование, сцепленное с полом. В
абсолютном большинстве случаев такие гены являются
рецессивными, а тип наследования заболевания - Х-сцеп-
ленным рецессивным. Поскольку X и У-хромосомы не
комплементарны, у мужчин даже рецессивный ген, рас¬
положенный на единственной Х-хромосоме, не имеет
своей пары (состояние гемизиготности) и является ма¬
нифестирующим; напротив, у женщин-гетерозит мута¬
ция на одной из Х-хромосом компенсируется нормаль¬
ным геном, расположенным на второй копии Х-хромо-
сомы. Таким образом, при Х-сцепленном рецессивном
типе наследования заболевание проявляется у мужчин,
унаследовавших от матери мутантную хромосому. Дан¬
ный тип наследования имеет следующие признаки:1) заболевают только мужчины (исключения из
этого правила крайне редки и рассматривают¬
ся ниже);2) заболевание передается клинически здоровы¬
ми женщинами-носительницами половине
сыновей (посредством передачи им мутантной
Х-хромосомы);
Общие принципы генодиагностики 493) отсутствует прямая передача болезни от муж¬
чин их сыновьям (поскольку сыновья всегда
наследуют от отца нормальную У -хромосому);4) все дочери больных-мужчин являются клини¬
чески здоровыми гетерозиготными носитель¬
ницами мутации.Пример родословной с Х-сцепленным рецессив¬
ным заболеванием показан на рис. 12.Рис. 12. Родословная семьи с Х-сцепленным рецессивным
чаболеваниемХ-сцепленное рецессивное наследование свой¬
ственно прогрессирующей мышечной дистрофии Дю-
шенна и Бекера, адренолейкодистрофии, спинально-буль-
барной амиотрофии Кеннеди и некоторым другим на¬
следственным неврологическим заболеваниям. В связи
с тем, что женщины-носительницы мутантного гена
50Глава 1обычно остаются клинически здоровыми (пропуск по¬
колений в родословной) и передают болезнь лишь поло¬
вине сыновей, при Х-сцепленном типе наследования
проследить семейный характер болезни бывает весьма
непросто, а число спорадических случаев намного пре¬
вышает частоту семейных форм. Об этом следует по¬
мнить при обследовании больных и проведении меди¬
ко-генетического консультирования.Иногда у женщин носительство мутантного гена
на одной из Х-хромосом не компенсируется присутстви¬
ем нормально функционирующей копии гена на второй
Х-хромосоме. Это случается, например, при различных
цитогенетических нарушениях - синдроме Шерешевс-
кого-Тернера (генотип ХО, т.е. отсутствие одной Х-хро-
мосомы), транслокации критического участка Х-хромо-
сомы, а также при высокой частоте инактивации нор¬
мальной Х-хромосомы (феномен аномальной лайониза-
ции). Такие женщины являются манифестными носи-
телъцами гена; у них может наблюдаться развернутая
клиническая картина или отдельные симптомы соответ¬
ствующего Х-сцепленного заболевания, что приводит к
серьезным затруднениям в клинической диагностике.
Случаи манифестного носительства у женщин являются
исключительно редкими (не более 1-2% от общего чис¬
ла больных с Х-сцепленными рецессивными заболева¬
ниями) и нуждаются в соответствующем подтверждении
с использованием цитогенетических методов и методов
ДНК-анализа.В тех случаях, когда ген, локализованный на X-
хромосоме, определяет развитие доминантного призна¬
ка, имеет место Х-сцепленный доминантный тип насле¬
дования. Для него характерны следующие особеннос¬
ти:1) все дочери больного отца наследуют заболе¬
вание; передача заболевания от отца сыну не¬
Общие принципы генодиагностики 51возможна (сыновья наследуют от отца здоро¬
вую У-хромосому);2) вероятность рождения больного ребенка лю¬
бого пола от больной матери равна 50%;3) в каждой родословной число больных женщин
в 2 раза больше, чем больных мужчин.Пример родословной с Х-сцепленным доминан¬
тным типом наследования представлен на рис. 13.-Оо4Т и /Т• 18 9і1"!*10 11 12 13 141 іої2 3 4 5 6 7 8 9 10 11 12Рис. 13. Родословная семьи с Х-сцепленным доминантным
чаболеваниемДанный тип наследования является исключитель¬
но редким. В неврологии практически единственным
примером заболевания с Х-сцепленным доминантным
типом наследования является наследственная моторно¬
сенсорная демиелинизирующая невропатия, обусловлен¬
ная мутациями в гене Сх32.Все рассмотренные выше классические менделев-
ские типы наследования свойственны заболеваниям,
обусловленным мутациями ядерной ДНК; их закономер¬
52Глава 1ности определяются механизмом мейотического расхож¬
дения хромосом половых клеток в процессе деления
клеточного ядра. В 80-90-е годы был открыт новый класс
наследственных болезней человека - митохондриальные
цитопатии. Они вызываются мутациями генов в составе
коротких кольцевых молекул ДНК, расположенных в
митохондриях- т.е. в органеллах цитоплазмы, вне кле¬
точного ядра (раздел 1.1). Митохондриальные цитопа¬
тии характеризуются совершенно особым, неменделев-
ским характером передачи заболевания в родословной,
который определяется как митохондриальный (цитоп¬
лазматический, неядерный) тип наследования. Посколь¬
ку набор митохондрий в клетках организма имеет ис¬
ключительно материнское происхождение, митохондри¬
альный тип наследования болезни имеет следующие ос¬
новные признаки:1) заболевание передается от больной матери
всем ее детям;2) мужчины и женщины (сыновья и дочери
больной матери) поражаются в равной сте¬
пени;3) передача болезни по мужской линии невоз¬
можна.На рис. 14 представлена типичная родословная
семьи с митохондриальным (материнским) наследовани¬
ем болезни. По такому типу передаются синдромы
MERRF (миоклонус-эпилепсия с рваными красными
волокнами), MELAS (митохондриальная энцефаломио-
патия с лактат-ацидозом и инсультоподобными эпизо¬
дами) и некоторые другие заболевания нервной систе¬
мы. Более подробно клинико-генеалогические особен¬
ности группы митохондриальных болезней разбирают¬
ся в разделе 3.8.
Общие принципы генодиагностики 5311111АРис. 14. Родословная семьи с заболеванием, наследующим¬
ся по материнскому (митохондриальному) типу
Разной штриховкой обозначены больные с различной степенью тя¬
жести заболевания.Важнейшим этапом обследования больного с по¬
дозрением на наследственное заболевание является сбор
генеалогического анамнеза. Его целью является состав¬
ление родословной, позволяющей проследить передачу
болезни в ряду поколений, установить тип наследова¬
ния болезни и определить круг лиц, принадлежащих к
группе риска и нуждающихся в медико-генетическом
консультировании (в том числе с использованием мето¬
дов ДНК-диагностики).Можно выделить несколько основных этапов, на
которые подразделяется процедура генеалогического
анализа в обследуемой семье.1) Установление наследственной природы болез¬
ни. Предположение о наследственном характере того или
иного заболевания может быть сделано на основании
наличия повторных случаев этого заболевания у род¬
ственников обследуемого больного. В процессе опроса
больного и его родственников нельзя ограничиваться
лишь получением сведений о наличии в семье других
случаев «аналогичного заболевания». Следует помнить
54Глава 1о том, что для наследственных болезней нервной систе¬
мы характерен значительный фенотипический полимор¬
физм, а трактовка членами семьи тех или иных симпто¬
мов, имеющих место у родственников, может быть весь¬
ма субъективной и ошибочной. Поэтому в целях полу¬
чения максимально точной информации необходимо
интересоваться наличием у родственников любых забо¬
леваний, в особенности сопровождающихся какими-либо
неврологическими нарушениями. Это особенно важно
для заболеваний, имеющих мультисистемные и мульти-
органные проявления. Например, миотоническая дист¬
рофия- сравнительно частое наследственное заболева¬
ние с аутосомно-доминантным типом передачи и варьи¬
рующей экспрессивностью мутантного гена- в развер¬
нутых случаях характеризуется миотоническим феноме¬
ном, мышечными атрофиями, кардиомиопатией, катарак¬
той, эндокринными нарушениями и рядом других симп¬
томов; в то же время в некоторых случаях единствен¬
ным проявлением болезни могут быть катаракта либо
нарушение сердечной проводимости. Выявление таких
симптомов у родственников больного миотонической ди¬
строфией позволяет заподозрить семейный характер за¬
болевания и предпринять необходимые меры для под¬
тверждения носительства мутации у лиц, имеющих суб-
клинические признаки болезни.Для получения генеалогической информации
может использоваться анкетирование, при этом решаю¬
щим фактором успеха является адекватный перечень
вопросов анкеты и доступность вопросов для членов
семьи, не имеющих медицинского образования. Очень
важно провести личный осмотр ближайших родствен¬
ников больного, а в необходимых случаях и других чле¬
нов семьи с целью более детальной оценки их клини¬
ческого статуса. При необходимости данные личного
осмотра родственников больного могут дополняться
Общие принципы генодиагностики 55результатами соответствующих лабораторно-инструмен-
глльных методов обследования (ЭЭГ, ЭМГ, рентгено¬
вская и магнитно-резонансная компьютерная томогра¬
фия и др.). При сборе семейного анамнеза следует стре¬
миться использовать и другие источники достоверной
медицинской и генеалогической информации, например
различную медицинскую документацию (выписки из
историй болезни, амбулаторные карты), домовые книги,
архивные данные и т.п.Накопление случаев одного и того же заболева¬
ния в семье может быть не связанным с наследованием
какой-либо мутации, а объясняться действием опреде¬
ленных патогенных факторов, общих для всех членов
семьи (особенности пищевого режима, среды обитания
п т.д.). С этой точки зрения термины «наследственный»11 «семейный» применительно к повторным случаям за-
(юлевания в семье не всегда являются синонимичными11 могут иметь различный патогенетический смысл. Сле¬
дует помнить также о том, что тот или иной клиничес¬
кий синдром (фенотип), напоминающий симптоматику
известного наследственного заболевания, может быть
следствием ряда других болезней ненаследственной при¬
роды. В таком случае говорят о фенокопии наследствен¬
ного заболевания. Например, фенокопиями хореи Ген-
тингтона могут быть ревматическая (малая) хорея, се¬
нильная хорея и другие патологические состояния, что
принципиально меняет прогноз для больного и членов
его семьи и требует проведения тщательного дифферен¬
циально-диагностического поиска. Таким образом, де¬
тальное выяснение особенностей общего и социального
анамнеза, условий жизни, характера трудовой деятель¬
ности и других факторов является важнейшим элемен¬
том обследования больного с подозрением на наслед¬
ственное заболевание.
56Глава 12) Составление родословной. Для четкого гра¬
фического отображения родословной и повышения «ин¬
формационной емкости» представления генеалогических
данных существуют общепринятые правила и унифици¬
рованная система условных обозначений. Строгое сле¬
дование соответствующим рекомендациям при состав¬
лении родословных является абсолютно необходимым
требованием, позволяющим врачам и другим специали¬
стам, работающим в системе медико-социальной помо¬
щи, медицинской статистики, судебной криминалисти¬
ки и т.д., разговаривать «на одном языке» при анализе
различных вопросов наследственной патологии и гене¬
тической идентификации. В 1995 году международной
рабочей группой по стандартизации родословной были
предложены обновленные рекомендации по стандарти¬
зованной номенклатуре родословной человека [Bennett
R. et al., 1995]. Их следует придерживаться в практичес¬
кой деятельности при представлении генеалогической
информации. На рис. 15 приводятся основные символы,
используемые в настоящее время при составлении ро¬
дословных.Составление родословной проводится от пробан¬
да к ближайшим и отдаленным родственникам (пробанд-
лицо, через которое осуществляется регистрация всей
семьи). Лица, принадлежащие к одному поколению, рас¬
полагаются в родословной строго в одном ряду. Каждый
член семьи кодируется индивидуальным индексом (1-3,
II-4, III-12 и т.д.), отражающим номер поколения (римс¬
кая цифра, нумерация сверху вниз) и порядковый номер
в соответствующем поколении (арабская цифра, слева
направо). В некоторых случаях один из представителей
той или иной родительской пары может не отображать¬
ся в родословной, если он клинически здоров и не име¬
ет прямого отношения к передаче мутантного гена.
Общие принципы генодиагностики 57□ ■ 1О • 2О ♦ 3И © 4■ • 50 0 в©Шь-о10111213141й151617Рис. 15. Основные символы, рекомендуемые к использова¬
нию при составлении родословной: 1 - лица мужского пола; 2 -
лица женского пола; 3 - пол неизвестен (а - здоровые, б - больные);
4 - несколько человек (а - число известно, б - число неизвестно); 5
пробанд; 6 - умершие; 7 - брак; 8 - родственный брак; 9 - сибсы
(дети одной родительской пары); 10 — монозиготные близнецы; 11 -
дизиготные близнецы; 12 - облигатный гетерозиготный носитель
мутантного гена (клинически здоров и не заболеет); 13 - носитель
мутантного гена на пресимптоматической стадии (имеет высокий
риск заболеть по достижении соответствующего возраста); 14 - без¬
детный брак; 15 - выкидыш; 16 - медицинский аборт; 17 - усы¬
новление.
58Глава 1Составленная родословная должна дополняться
так называемой легендой - письменным изложением
дополнительных сведений, имеющих значение для об¬
следования изучаемой семьи и генеалогического анали¬
за (национальность, место рождения и адреса включен¬
ных в родословную членов семьи, имеющиеся результа¬
ты клинико-инструментального обследования, другая
касающаяся родственников информация, полученная из
различных медицинских документов, и т.п.).3) Определение типа наследования. После состав¬
ления родословной и обоснования наследственного ха¬
рактера изучаемого заболевания необходимо установить
тип наследования болезни в данной семье. При этом ха¬
рактер сегрегации клинического синдрома сопоставля¬
ется с приведенными выше признаками, свойственны¬
ми всем известным типам наследования. При решении
вопроса о типе наследования болезни важно принимать
во внимание целый ряд обстоятельств, таких как: а)воз-
можность неполной пенетрантности мутантного гена
(«пропуск поколения»); б) вариабельная экспрессивность
мутантного гена (наличие у части родственников «стер¬
тых» форм заболевания); в) близкородственные браки;г) ранняя смерть одного из родителей (который мог не
дожить до возраста манифестации болезни и поэтому
характеризовался родственниками как «здоровый»);д) место рождения родителей и более отдаленных пред¬
ков (происхождение родителей больного и/или их пред¬
ков из одного и того же сравнительно изолированного
населенного пункта может косвенно свидетельствоватьо возможности определенной кровнородственной связи
между ними) и т.д. При наличии нескольких случаев за¬
болевания в семье и несоответствии типа передачи ка¬
кой-либо определенной модели наследования (менделев-
ской или митохондриальной) может быть сделан вывод
Общие принципы генодиагностики 59о том, что данное заболевание является полигенным
(мультифакториальным); это означает, что соответству¬
ющие члены семьи наследуют не один основной мутан-
П1ЫЙ ген, а высокую генетическую предрасположен¬
ность, которая может привести либо не привести к бо-
исзни в зависимости от конкретных модифицирующих
факторов среды и индивидуальных особенностей генома.Следует подчеркнуть, что правильно установлен-
ш.тй тип наследования уже сам по себе имеет важное
диагностическое значение. В качестве примера можно11 редставить типичную ситуацию, при которой в консуль¬
тируемой семье выявлено несколько случаев прогресси¬
рующей мышечной дистрофии с проксимальным распре¬
делением мышечных атрофий и парезов. В случае X-
сцепленного рецессивного типа наследования такого
синдрома следует заподозрить мышечную дистрофию
) [.юшенна/Бекера, что требует поиска повреждений в гене
или белке дистрофине с помощью ДНК-диагностики и11 ммуногистохимического анализа мышечных биоптатов;
с другой стороны, если указанный клинический синд¬
ром характеризуется всеми признаками аутосомно-рецес-
сивной передачи, врач должен заподозрить диагноз ко-
иечностно-иоясной мышечной дистрофии (формы Эрба-
Рота) и исследовать другие гены и/или белки мышечно¬
го волокна. При этом прогноз заболевания и расчеты
г енетического риска в консультируемой семье будут раз¬
личными при разных формах патологии.4) Определение круга лиц в изучаемой семье, от¬
носящихся к группе риска и нуждающихся в проведе¬
нии медико-генетического консультирования (т.е. лиц,
которые могут являться носителями мутантного гена);
расчет конкретной величины генетического риска у кон¬
сультируемых родственников; осуществление комплек¬
са мер, направленных на профилактику новых случаев
60Глава 1заболевания в семье, отягощенной наследственным за¬
болеванием. Детальный анализ этих вопросов в свете
современных возможностей ДНК-диагностики представ¬
лен в главе 5.1.3. Характеристика основных типов
мутаций, вызывающих
наследственные заболевания
нервной системыГеномы различных индивидуумов в рамках одно¬
го биологического вида идентичны с точки зрения об¬
щего набора генов и их тонкой внутренней структуры. В
то же время существует значительное число межинди-
видуальных различий, связанных с теми или иными ва¬
риациями нуклеотидной последовательности ДНК. Та¬
ким образом, любой ген (в более широком смысле —
любой генетический локус) может существовать в виде
различных вариантов - аллелей. Аллели различаются
между собой по составу нуклеотидов в определенных,
ограниченных участках ДНК. Количество аллелей одно¬
го гена может составлять от 2-3 до нескольких десят¬
ков.Любое изменение структуры ДНК, возникающее
спонтанно либо индуцированное путем целенаправлен¬
ного воздействия физическими или химическими фак¬
торами, называется мутацией. Мутации ведут к возник¬
новению новых аллелей соответствующих генов и ле¬
жат в основе генетической изменчивости в живой при¬
роде. Во многих случаях мутация не имеет каких-либо
заметных фенотипических последствий, например при
ее локализации в функционально незначимых областях
молекулы ДНК. Другим примером «молчащей мутации»
является такая замена нуклеотида, при которой новый
Общие принципы генодиагностики 61кодон имеет прежний информационный смысл, т.е. ко¬
дирует ту же самую аминокислоту и поэтому не меняет
структуру белка. Такие нейтральные мутации рассмат¬
риваются как нормальные полиморфизмы, они не эли¬
минируются отбором и могут иметь достаточно высо¬
кую частоту в популяции. Изучение полиморфизмов ДНК
широко используются в генетической идентификации
личности и целом ряде базовых методов молекулярно¬
генетического анализа.В отличие от нейтральных мутаций (полиморфиз¬
мов), патологические мутации приводят к нарушению
механизма транскрипции/трансляции гена либо к син¬
тезу аномального белкового продукта. В литературе об¬
щий термин «мутация» нередко используется примени¬
тельно именно к таким случаям нарушений структуры
гена, которые имеют несомненное патогенетическое зна¬
чение. Аналогичным образом, при дальнейшем изложе¬
нии материала нами под мутациями будут пониматься
любые патогенетически значимые повреждения молеку¬
лы ДНК, приводящие к развитию болезни.Мутации могут быть генными и хромосомными.
Хромосомные мутации затрагивают количественный
состав либо структуру хромосом. Такие мутации выяв¬
ляются при цитогенетическом исследовании в процессе
проведения световой микроспокопии специально окра¬
шенных клеточных хромосомных препаратов. Хромосом¬
ные мутации обычно являются результатом разовых на¬
рушений деления клеточного ядра в мейозе конкретной
половой клетки, поэтому вызванные ими хромосомные
болезни чаще всего представлены спорадическими слу¬
чаями. Генные мутации представляют собой более тон¬
кие изменения нуклеотидного состава генов; заболева¬
ния, обусловленные генными мутациями, передаются в
родословной в соответствии с рассмотренными выше
основными типами наследования. Именно генные мута¬
62Глава 1ции и связанные с ними моногенные болезни нервной
системы являются основным предметом настоящей мо¬
нографии.Генные мутации могут быть классифицированы
на основании характера изменений нуклеотидного со¬
става гена, а также на основании механизма нарушения
функции соответствующего молекулярного продукта.
Основные типы повреждений генов при любых наслед¬
ственных заболеваниях человека в целом идентичны;
однако ряду наследственных заболеваний нервной сис¬
темы свойственны определенные особенности спектра
мутаций в соответствующих генах, знание которых не¬
обходимо при проведении ДНК-диагностики.Генные мутации могут быть подразделены на
точковые и структурные. Точковые мутации {point
mutation) затрагивают один нуклеотид либо 1-2 сосед¬
них нуклеотида, тогда как к структурным мутациям от¬
носятся более протяженные дефекты гена.Среди точковых мутаций чаще всего встречают¬
ся нуклеотидные замены в кодирующей области гена,
т.е. в экзонах. В том случае, если нуклеотидная замена
сопровождается изменением аминокислотного шифра
соответствующего кодона, такая мутация ведет к изме¬
нению состава полипептидной цепи вследствие встраи¬
вания в нее «ошибочной» аминокислоты. Мутации дан¬
ного типа называются лшссенс-мутациями (missense - т.е.
с изменением «смысла» повреждаемого кодона). Тяжесть
фенотипических проявлений болезни в этом случае бу¬
дет определяться функциональной значимостью мутан¬
тного участка белка; наиболее тяжелые последствия име¬
ют обычно миссенс-мутации, затрагивающие активные
центры белковой молекулы. Иногда нуклеотидная заме¬
на приводит к замещению информационно значимого
кодона на стоп-кодон; в результате этого на мутантном
Общие принципы генодиагностики 63участке соответствующей молекулы мРНК происходит
преждевременный обрыв трансляции и образование
«усеченной» (truncated), функционально дефектной мо¬
лекулы белка. Такие нуклеотидные замены обозначают¬
ся термином нонсенс-мутации (;nonsense - «бессмыслен¬
ные», нарушающие информационную роль кодона).Достаточно распространенным типом мутантных
аллелей являются аллели, образующиеся в результате
выпадения (делеции) или вставки (инсерции) нуклеоти¬
дов в кодирующей области гена. Если число нуклеоти¬
дов при делениях и вставках некратно трем (например,
в случае точковой мутации с выпадением одного нукле¬
отида), такая мутация приводит к нарушению нормаль¬
ного отсчета кодирующих триплетов, в результате чего
все последующие кодоны в полинуклеотидной цепи при¬
обретают другой аминокислотный смысл (рис. 16).AGTCACGCTTTACGTATAAAGGSer—His —Ala—Leu—Arg —Ile —Lys —
делеция Снормальная
. нуклеотидная
последовательность
. синтез нормального
белкаAGTCACGCTTTACGTATAAAGGсдвиг рамки считывания
генетического кодаAGTCAGCTTTACGT ATAAAGGSer—Gin—Leu—-Туг—Val—STOP •* синтез "бессмысленного"полипептида и обрыв
трансляции из-за появления
аномального стоп-кодона1’ис. 16. Мутация со сдвигом рамки считывания генетичес-
|ч)го кода
64Глава 1Другими словами, данный тип мутации сопровож¬
дается сдвигом рамки считывания генетического кода
{frameshift)-, мутации со сдвигом рамки ведут к синтезу
«бессмысленного» чужеродного белка, который быстро
деградирует под воздействием внутриклеточных про-
теаз.Если мутация со сдвигом рамки либо нонсенс-
мутация расположены в 5’-области гена (т.е. ближе к его
началу), они сопровождаются отсутствием синтеза с дан¬
ной копии гена сколько-нибудь значимой в функциональ¬
ном отношении полипептидной молекулы и полной
потерей активности белка - так называемые нулевые
мутации (null mutation). Разумеется, такие мутации яв¬
ляются весьма тяжелыми и нередко либо несовместимы
с жизнью, либо характеризуются развитием выражен¬
ных мультисистемных проявлений болезни. В невроло¬
гии ярким примером такого рода является атаксия-теле-
ангиэктазия, обусловленная повреждением обеих копий
гена ATM, кодирующего белок семейства фосфатидили-
нозитол-киназ. Около 90% известных на сегодня мута¬
ций в гене ATM представляют собой нулевые мутации,
приводящие к полной инактивации белка и развитию
множественных симптомов поражения головного моз¬
га, кожи, сосудистой, иммунной, эндокринной, костной
систем, онкологических осложнений [Gilad S. et al., 1996].Точковые и структурные мутации могут проис¬
ходить не в собственно кодирующих участках гена, а на
самом стыке экзонов и интронов - в сайтах сплайсинга
(сплайсинговые мутации). Такие мутации нарушают нор¬
мальные механизмы созревания первичного РНК-транс-
крипта и приводят к неправильному вырезанию интро-
на либо к удалению из молекулы РНК информационно
значимой экзонной последовательности. В результате
этого происходит значительное нарушение структуры
Общие принципы генодиагностики 65белка, ведущее обычно к развитию тяжелых клиничес¬
ких проявлений болезни. В редких случаях мутации мо¬
гут затрагивать различные регуляторные последователь¬
ности (например, в области промотора или интронов),
влияющие на характер экспрессии гена; такие регулятор¬
ные мутации обычно сопровождаются не нарушением
структуры или функции белковой молекулы, а количе¬
ственными изменениями содержания белка в клетке.При некоторых наследственных заболеваниях
нервной системы основным типом структурных мута¬
ций являются протяженные делеции, охватывающие
значительную область гена или даже несколько близле¬
жащих генов. Такие делеции, как правило, характеризу¬
ются тяжелыми биохимическими и фенотипическими
последствиями. Классическими примерами могут слу¬
жить прогрессирующая мышечная дистрофия Дюшен-
па/Бекера и аутосомно-рецессивная проксимальная спи¬
нальная амиотрофия. Более чем в 60% случаев мышеч¬
ной дистрофии Дюшенна/Бекера заболевание обуслов-
нено разнообразными делениями гена дистрофина на
ч | юмосоме Хр21.2, причем тяжесть симптоматики у кон-
кретного больного определяется протяженностью деле¬
нии, ее влиянием на рамку считывания кодонов и лока-
иизацией по отношению к функционально значимым
доменам дистрофина [Koenig М. et al., 1989]. У 98% боль-
н 1.1 х с различными клиническим вариантами аутосомно-
рецессивной проксимальной спинальной амиотрофии
(болезни Верднига-Гофмана и Кугельберга-Веландер)
| и II 1аруживается делеция обеих копий гена SMN на хро¬
мосоме 5ql 3, что приводит к отсутствию у больных нор¬
мального транскрипта данного гена [Lefebvre S. et al.,
1995].При некоторых неврологических заболеваниях
i I руктурная мутация может заключаться в дупликации
66Глава 1гена или его части. Так, болезнь Пелицеуса-Мерцбахера
(одна из форм лейкодистрофий детского возраста) мо¬
жет вызываться дупликацией или точковыми мутация¬
ми гена протеолипидного протеина (хромосома Xq22)
[Sistermans Е. et al., 1998]. Наиболее частой причиной
демиелинизирующей формы наследственной моторно¬
сенсорной невропатии (болезнь Шарко-Мари-Тута типа
1 А) является полная дупликация гена РМР22 на хромо¬
соме 17pl 1.2 [Lupski J. et al., 1991]. Интересно отметить,
что делеция этого же хромосомного участка ведет к раз¬
витию совершенно другой клинической формы невро¬
патии - так называемой наследственной невропатии с
предрасположенностью к параличам от сдавления
[Chance P. et al., 1993].В последние годы был открыт принципиально
новый тип структурных мутаций, характерный для це¬
лого ряда нейродегенеративных заболеваний. Он схема¬
тично представлен на рис. 17.НормаI ... CTG AAG ССС CAG CAG CAG ... CAG CCG CCG ССС ... |... Leu Lys Pro Gin Gin Gin ... Gin Pro Pro Pro...12-37 тандемных
повторовМутацияj... CTG AAG ССС CAG CAG CAG CAG CAG CAG ... CAG CAG CCG CCG CCC...... Leu Lys Pro Gin Gin Gin Gin Gin Gin ... Gin Gin Pro Pro Pro39 - 80 и более
тандемных повторовРис. 17. Экспансия тандемных тринуклеотидных повторов
(на примере спиноцеребеллярной атаксии-1)В рамке показан нуклеотидный состав части гена с областью
тринуклеотидных повторов (экзон 1), под рамкой указана соответ¬
ствующая последовательность аминокислот в составе белка. Тан¬
демным САв-триплетам гена (выделены жирным шрифтом) соот¬
ветствует полиглутаминовый участок белка.
Общие принципы генодиагностики 67Как видно на рисунке, в некоторых участках генов
нуклеотидная последовательность представлена
цепочкой тандемных тринуклеотидных повторов -
например, «цитозин-аденин-гуанин», или (CAG)n. Число
таких повторов в норме варьирует в строго
определенных пределах; другими словами, существует
большое число нормальных аллелей соответствующих
генов, отличающихся между собой количеством
тандемных тринуклеотидных повторов [Willems Р., 1994].
Сущность мутации заключается в патологическом
увеличении числа копий данных повторов,
превышающем определенные пороговые значения,
специфичные для каждой нозологической формы.
Данный тип мутаций обозначается как экспансия
тринуклеотидных повторов [Иллариошкин С.Н. и др.,
1995]. Мутантный удлиненный участок гена является
весьма нестабильным, что нередко приводит к
дальнейшему изменению (чаще - нарастанию) числа
повторов при передаче гена в следующее поколение. В
связи с этим мутации по типу экспансии
тринуклеотидных повторов получили название
динамические мутации [Richards R., Sutherland G., 1992].Происхождение динамических мутаций представ¬
ляет собой двухэтапный процесс (рис. 18). На первом
этапе в результате редких мутационных событий проис¬
ходит образование аллелей с «промежуточным» числом
тринуклеотидных повторов, превышающим общепопу¬
ляционную норму, но не достигающим патологического
порога. Такой аллель представляют собой «премутацию»:
он не приводят к развитию болезни у данного
индивидуума, но характеризуется значительной генети¬
ческой нестабильностью и повышенной вероятностью
перехода в «полную мутацию» в последующих поколе-
11иях, что и приводит к манифестации болезни у потом¬
ков [Richards R., Sutherland G., 1992; Willems P., 1994].
68Глава 1Таким образом, «промежуточные» аллели являются сво¬
еобразным резервуаром, постоянным источником новых
мутаций в популяции.ооооОООоооонормальный аллельо о о о О О О О О о о о опремутацияклиническиздоровыеОООО ФФФФФФ ФФШWWФ ОООО больные
мутантный аллельРис. 18. Двухэтапный механизм происхождения динамичес¬
ких мутацийЗвёздочками обозначены тандемные тринуклеотидные повторы,
кружками-другие нуклеотидныепоследовательности гена. Возник¬
новение аллеля с «промежуточным» числом повторов может быть
связано с одним или несколькими мутационными событиями.К настоящему времени известно уже более десят¬
ка наследственных заболеваний нервной системы, обус¬
ловленных динамическими мутациями [Rosenberg R.,
1996; Brice А., 1998]. Наиболее обширную группу со¬
ставляют заболевания с аутосомно-доминантным типом
наследования (хорея Гентингтона, доминантные атаксии),
обусловленные экспансией CAG-повторов в транслиру¬
емых областях соответствующих генов. Поскольку трип¬
лет CAG кодирует аминокислоту глутамин, такие мута¬
ции приводят к пропорциональному удлинению и изме¬
нению свойств белковых полиглутаминовых участков,
Общие принципы генодиагностики 69в норме играющих важную роль в реализации сложных
межмолекулярных взаймодействий [HousmanD., 1995;
Ross С., 1995]. Результатом мутации является формиро¬
вание нерастворимых амилоидогенных белковых комп¬
лексов, ведущее к гибели специфических популяций ней¬
ронов. Предполагается, что в патогенезе указанной груп¬
пы болезней важное значение имеет также нарушение
взаимодействия мутантных полиглутаминовых цепей с
белками-регуляторами апоптоза, что может приводить к
запуску процессов программируемой гибели нейронов
|HousmanD., 1995; IkedaH. et al., 1996]. Экспансиятри-
нуклеотидных повторов может иметь место и в некоди¬
рующих областях генов - при атаксии Фридрейха, мио-
тонической дистрофии, синдроме ломкой Х-хромосомы.
В этих случаях мутации обусловливают нарушение нор¬
мальных механизмов транскрипции гена и угнетение
белкового синтеза [Willems Р., 1994; Pandolfo М., 1998].При всех заболеваниях, обусловленных динами¬
ческими мутациями, тяжесть клинических проявлений
прямо пропорционально коррелирует с величиной экс¬
пансии тринуклеотидных повторов, т.е. со степенью тя¬
жести генетического дефекта. Динамический характер
мутаций и тенденция к нарастанию числа повторов при
передаче гена являются молекулярной основой феноме¬
на антиципации, свойственного доминантным «тринук-
деотидным» заболеваниям и заключающегося в появле¬
нии все более ранних и тяжелых случаев болезни в каж¬
дом последующем поколении [Brice А., 1998]. Было по¬
казано также, что необычные свойства динамических
мутаций объясняют так называмый эффект отцовской
(материнской) передачи, характерный для некоторых из
н их заболеваний. Так, при хорее Гентингтона наиболее
тяжелый акинетико-ригидный вариант болезни (вариант
1$сстфаля) развивается обычно у детей, унаследовавших
(юлезиь по отцовской линии; это связано с преимуще¬
70Глава 1ственной нестабильностью мутантного аллеля и «дра¬
матическим» нарастанием числа повторов в мужском
гаметогенезе [Telenius H. et al., 1993; Kremer В. et al.,
1995]. Напротив, при миотонической дистрофии увели¬
чение степени экспансии повторов наблюдаются обыч¬
но при передаче гена по женской линии, в связи с чем
случаи тяжелой врожденной миотонической дистрофии
наблюдается исключительно при наследовании болезни
от матери [Mulley J. et al., 1993]. Данный феномен (зави¬
симость фенотипических проявлений заболевания от
пола больного родителя) носит название импринтинг.
Различная степень экспансии повторов лежит в основе
весьма варьирующей экспрессивности «тринуклеотид-
ных» заболеваний - от субклинических и «мягких» форм
до тяжелых развернутых случаев с быстрым, фатальным
течением [Иллариошкин С.Н. и др., 1995; 1999].Некоторые наследственные заболевания нервной
системы обусловлены экспансией более сложных по сво¬
ей структуре повторяющихся участков ДНК, состоящих
из десятков нуклеотидов. В их числе можно назвать ми-
оклонус-эпилепсию Унферрихта-Лундборга, обусловлен¬
ную экспансией 12-нуклеотидного повтора в 5’-области
гена CST6 на хромосоме 21 q22 [Lafreniere R. et al., 1997;
Vitraneva K. et al., 1997], а также семейные формы при-
онных болезней (болезнь Крейтцфельдта-Якоба), кото¬
рые в части случаев могут вызываться увеличением ко¬
пий длинного транслируемого повтора гена прионного
белка на хромосоме 20p [Collinge J., 1997].Для описания различных типов мутаций в том или
ином гене разработана специальная номенклатура
[Beutler E., 1993; Beaudet A., Tsui L., 1993; Antonarakis S.
et al., 1998]. Она предполагает использование унифици¬
рованных символов, позволяющих четко обозначать ло¬
кализацию мутации в гене, ее характер (точковая мута¬
Общие принципы генодиагностики 71ция, делеция, вставка и т.д.), последствия мутации (за¬
мена аминокислоты, появление стоп-кодона) и некото¬
рые другие особенности. Номенклатура мутаций носит
универсальный характер и позволяет исследователям,
работающим в различных областях медицинской гене¬
тики, разговаривать «на одном языке» при сопоставле¬
нии и анализе данных мутационного скрининга. Основ¬
ные элементы действующей номенклатуры генных му¬
таций человека представлены в Приложении 2.В редких случаях развитие наследственного не¬
врологического заболевания может быть связано не с
непосредственными мутациями в гене, а с вторичным
нарушением его функции вследствие так называемого
негативного позиционного эффекта, или эффекта по-
чожения. Примером является лице-лопаточно-плечевая
мышечная дистрофия - аутосомно-доминантное заболе¬
вание, обусловленное делецией вариабельного участка
размером 20-250 кб в субтеломерной части хромосомы
4q35 [van Deutekom J. et al., 1993].r ->гетерохроматинрегуляторные транскрибируемыйэлементыгенJо;=гфс;фVблок транскрипции
генаI'll с. 19. Негативный позиционный эффект при лице-лопа-
гочно-плечевой мышечной дистрофии
72Глава 1Как было показано (рис. 19), мутирующий учас¬
ток молекулы ДНК не содержит каких-либо генов; од¬
нако, делеция этого участка приводит к изменению струк¬
туры хроматина в данной хромосомной области и к опос¬
редованному ингибированию транскрипции близлежа¬
щего гена (FRG1), расположенного проксимальнее уча¬
стка разрыва. Предполагается, что такой дистантный ме¬
ханизм нарушения экспрессии структурно сохранного
гена, связанный с негативным позиционным эффектом
крупной внегенной делеции, и обусловливает появление
неврологической симптоматики у больных лице-лопаточ-
но-плечевой мышечной дистрофией [Fisher J.,
Upadhyaya М., 1997; Kleinjan D., van Heyningen V., 1998].По своему патогенетическому механизму все му¬
тации, вызывающие развитие наследственных заболева¬
ний нервной системы, могут быть подразделены на сле¬
дующие 5 основных групп:1. Мутации, ведущие к потере функции (loss-of-
function). К данной группе относятся любые структур¬
ные изменения генов (точковые мутации, делеции, экс¬
пансия тринуклеотидных повторов в некодирующих об¬
ластях генов и др.), приводящие к ингибированию про¬
цессов транскрипции/трансляции или нарушению нор¬
мальной структуры и функциональных свойств белка.2. Мутации, ведущие к появлению новой функции
(gain-of-function). Данный тип мутаций, не затрагивая
нормальную функцию белка, характеризуется появлени¬
ем у мутантного протеина новых свойств, обладающих
цитотоксичным эффектом и проводящих к постепенной
гибели нейронов. Такой механизм характерен для ауто-
сомно-доминантных болезней, обусловленных экспанси¬
ей CAG-повторов в транслируемой области гена: как
было рассмотрено выше, удлинение полиглутаминовых
участков в составе мутантных белковых молекул сопро-
Общие принципы генодиагностики 73иождается образованием необратимых межмолекулярных
«сшивок» и формированием токсичных внутриклеточ¬
ных полимерных комплексов.У Мутации, обладающие доминантным негативным
»ффектом (dominant negative effect). Доминантный нега¬
тивный эффект проявляется в том случае, когда продукт
мутантного аллеля ингибирует функцию нормальных
белковых молекул. Примером может служить аутосом-
I ю-доминантная дофа-зависимая дистония, обусловлен¬
ная повреждением гена ГТФ-циклогидролазы 1: белко¬
вый комплекс данного фермента имеет мультимерную
структуру и состоит из 10 идентичных субъединиц, по¬
этому встраивание в состав комплекса даже одной му¬
тантной молекулы приводит к образованию функцио¬
нально дефектных гетеромеров и преждевременной дег¬
радации фермента [Hirano М. et al., 1998].4. Мутации, изменяющие «дозу гена» (gene dosage
effect). Нарушение «дозы гена» имеет место при его де-
псциях или дупликациях, что может сопровождаться на¬
рушением нормальной цитоархитектоники соответству¬
ющего молекулярного продукта. Так, например, делеция
или дупликация гена миелинового белка РМР22 приво¬
дит к нарушению внутренней структуры и укладки мие¬
лина периферических нервов [Chance P., Fischbeck К.,1994], что сопровождается развитием различных форм
наследственных демиелинизирующих невропатий (см.
раздел 3.1.3).5. Мутации, обусловливающие количественные изме-
нения первичных молекулярных продуктов гена (мРНК
п белка). Такой механизм наблюдается обычно при ло¬
кализации мутаций в регуляторных областях генов.Знание механизмов, посредством которых на кле¬
точном и биохимическом уровне реализуется патологи¬
ческий эффект различных мутаций, является необходи¬
74Глава 1мым условием для разработки новых эффективных ме¬
тодов лечения наследственных заболеваний человека
(включая комплексное патогенетическое лечение и раз¬
нообразные методы генной терапии).1.4. Методы ДНК-диагностикиИсходным материалом для проведения ДНК-ди-
агностики заболеваний, обусловленных мутациями ядер-
ных генов, могут служить любые клетки организма, со¬
держащие ядро. Обычно для этих целей используются
лейкоциты, выделяемые из 5-20 мл периферической кро¬
ви. В некоторых случаях (например, при митохондри¬
альных энцефаломиопатиях, обусловленных мутациями
митохондриальной ДНК с преимущественой экспресси¬
ей мутации в мышечной и нервной ткани) более адек¬
ватным источником ДНК являются биоптаты мышц. Ге¬
нодиагностика может также проводиться на основе ис¬
следования ДНК, выделяемой из клеток эпителия поло¬
сти рта, кожных фибробластов и т.д. При проведении
пренатальной ДНК-диагностики у плода (обычно на 10-
21 -й неделе беременности) источником ДНК служат био¬
птаты хориона, плаценты, клетки амниотической жид¬
кости (получаемые при амниоцентезе) или лимфоциты
пуповинной крови (кордоцентез).Практически любой анализ структуры фрагмен¬
тов ДНК осуществляется с помощью различных мето¬
дик электрофореза ДНК в агарозном или полиакрила¬
мидном гелях. При этом концентрация и состав геля
определяют размер «пор», разрешающую способность
геля и некоторые другие его характеристики, влияющие
на характер перемещения отрицательно заряженных
молекул ДНК в электрическом поле. В подобранных
стандартных условиях именно подвижность молекул
Общие принципы генодиагностики 75ДНК при электрофорезе является ключевым анализиру¬
емым параметром, зависящим от конкретных характе¬
ристик изучаемого фрагмента ДНК (например, от его
величины или конформации). Полиакриламидные гели
по сравнению с агарозными обладают значительно бо¬
лее высокой разрешающей способностью, но при этом
они более трудоемки с точки зрения приготовления и
технической обработки. Для визуализации молекул ДНК
по результатам проведенного электрофореза может ис¬
пользоваться окраска их различными флюорохромами,
нитратом серебра или бромистым этидием (дающим ха¬
рактерное красноватое свечение при исследовании в про¬
ходящем ультрафиолетовом свете). Более сложный ме¬
тод визуализации ДНК - авторадиография - основан
на введении радиоизотопной метки в состав анализиру¬
емых макромолекул; по окончании электрофореза на гель
с радиоактивно мечеными молекулами ДНК накладыва¬
ется рентгеновская пленка, и после определенной экс¬
позиции положение различных фрагментов ДНК в геле
определяется в виде соответствующих полос на прояв¬
ленной пленке.Все разнообразие процедур, применяемых при
проведении ДНК-диагностики, можно разделить на 2
большие группы - прямые и косвенные методы.1.4.1. Прямая ДНК-диагпостика.Прямая ДНК-диагностика предполагает непос¬
редственное выявление мутации в исследуемом гене. Она
обладает практически абсолютной точностью, требует
для анализа только образец ДНК обследуемого лица и
может проводиться как в семейных, так и в спорадичес¬
ких случаях заболеваний. Для проведения прямой ДНК-
диагностики необходимо точно знать структуру гена (или
76Глава 1конкретного участка гена, содержащего анализируемую
мутацию). Методические подходы, используемые при
прямой ДНК-диагностике того или иного наследствен¬
ного заболевания нервной системы, зависят от характе¬
ра мутаций и молекулярной организации соответствую¬
щего гена.В настоящее время большинство протоколов пря¬
мой ДНК-диагностики базируется на полимеразной цеп¬
ной реакции (ПЦР). Разработка принципов ПЦР стала
поистине революционизирующим этапом в развитии
молекулярной генетики, что нашло свое отражение в
присуждении ее автору К. Муллису Нобелевской премии
за 1993 год в области биологии и медицины. Метод ПЦР
заключается в циклическом синтезе in vitro строго за¬
данных, ограниченных участков ДНК длиной от десят¬
ков до нескольких тысяч п.о. Это позволяет в течение
3-5 часов получить огромное число копий искусствен¬
но синтезированных молекул нужной последовательно¬
сти ДНК (т.е. амплифицировать необходимый участок
ДНК, являющийся предметом молекулярного анализа).
По сути, метод ПЦР как бы «имитирует» на ограничен¬
ном участке гена естественный процесс репликации
ДНК, имеющий место in vivo (см. раздел 1.1): в состав
реакционного раствора с определенной концентрацией
ионов магния и pH входит ДНК-матрица, четыре вида
дезоксирибонуклеотид-трифосфатов, а также термоста¬
бильная ДНК-полимераза, сохраняющая свою актив¬
ность при нагревании раствора до высоких температур.
Реакция инициируется добавляемыми в раствор прай¬
мерами - олигонуклеотидными последовательностями,
фланкирующими «зону интереса» и играющими роль
затравок в процессе комплементарного синтеза ДНК.Сущность метода ПЦР схематично представлена
на рис. 20 и заключается в циклическом чередовании
Денатурация 2-Й ЦИКЛДНК и отжиг
праймеров3-й циклРис. 20. Полимеразная цепная реакция
78Глава 13 основных биохимических этапов: 1) денатурация двой¬
ной спирали ДНК-матрицы при температуре 95°С; 2) гиб¬
ридизация (отжиг) одноцепочечной ДНК-матрицы и
праймеров при температуре 45-60°С, в процессе кото¬
рой праймеры распознают комплементарные им участ¬
ки ДНК-матрицы; 3) полимеризация при температуре 65-
72°С, т.е. синтез комплементарной цепи на ДНК-матри-
це, начинающийся от места гибридизации праймера и
происходящий в направлении 5’-»3\ В последующих
циклах вновь синтезируемые молекулы ДНК становят¬
ся, в свою очередь, матрицей для аналогичного синтеза
новых копий. Поскольку синтез каждой из 2 антипарал-
лельных цепей ДНК начинается от места гибридизации
праймера, эти места и становятся границами синтезиру¬
емого участка. Число указанных циклов в ПЦР состав¬
ляет обычно от 25 до 40, причем в каждом цикле проис¬
ходит удвоение числа копий амплифицируемого участка
ДНК, приводящее к увеличению в геометрической про¬
грессии общего содержания продуктов реакции. Обыч¬
но ПЦР осуществляется в автоматическом режиме на спе¬
циальных программируемых приборах (амплификато-
рах), контролирующих заданные параметры реакции
(температура, длительность отдельных этапов, число
циклов и т.д.).Дальнейший анализ продуктов ПЦР в процессе
прямой ДНК-диагностики предполагает исследование
конкретных особенностей амплифицированного участ¬
ка гена. Так, при заболеваниях, обусловленных экспан¬
сией тринуклеотидных повторов, продукты амплифика¬
ции различаются по своей длине (отражающей различ¬
ное число триплетов в изучаемом участке гена) и, как
следствие - по их скорости движения в геле. Благодаря
этому достигается четкое электрофоретическое разделе¬
ние нормальных и мутантных аллелей и точное опреде¬
Общие принципы генодиагностики 79ление патологически удлиненного фрагмента, т.е ДНК-
диагностика болезни. Таким же сравнительно простым
путем с помощью анализа размеров продуктов ПЦР на
электрофореграмме может проводиться прямая детекция
делеций и вставок в составе амплифицированного учас¬
тка гена. Например, в большинстве случаев аутосомно-
доминантной дофа-независимой дистонии у больных
имеет место одна и та же типичная гетерозиготная деле¬
ния трех нуклеотидов GAG в 5-м экзоне гена DYT1
[Ozelius L. et al., 1997]; таким образом, непосредствен¬
ное выявление при электрофорезе аномально короткого
ПЦР-продукта, отличающегося от нормального фрагмен¬
та на 3 п. о., может служить молекулярным подтвержде¬
нием данного диагноза (рис. 21).1 2 3 4 5 6Рис. 21. Диагностика делеции GAG в гене DYT1 у больных
) и )фа- f геэа в иси мо й дистонией) 1,орожки 2,3,6- больные, дорожки 1,4,5 - контроль. Тонкой ст рел¬
кой обозначен нормальный аллель, жирной стрелкой - мутантный,
более короткий аллель (деления 3 нуклеотидов).
80Глава 1Для обнаружения нуклеотидных замен в анали¬
зируемом участке гена возникает необходимость прове¬
дения секвенирования, т.е. определения точной нуклео¬
тидной последовательности продуктов ПЦР. На практи¬
ке наиболее широкое распространение получило секве-
нирование по классическому методу Сэнгера, техника
которого показана на рис. 22.Исследуемый фрагмент ДНК
праймер+ дезоксирибонуклеотид-трифосфаты
4 ДНК-полимеразаВ каждую из пробирок
добавлен один из 4
ди-дезоксирибонуклеотид-
трифосфатов (терминаторов)Набор образующихся цепей
различной длины, оканчи¬
вающихся на определен¬
ный нуклеотидРис. 22. Принцип секвенирования ДНК по методу Сэнгера
Общие принципы генодиагностики 81Изучаемый фрагмент ДНК (например, продукт ПЦР)
играет роль матрицы, на которой при участии праймера,
дезоксирибонуклеотид-трифосфатов и ДНК-полимера-
зы инициируется направленный комплементарный син¬
тез новых молекул ДНК, осуществляемый одновремен¬
но в 4 параллельных реакциях. Смысл метода в том, что
в каждую из реакций добавляется один из 4 химически
измененных дезоксирибонуклеотид-трифосфатов (терми¬
наторов). В качестве таких терминаторов выступают ди-
дезоксирибонуклеотид-трифосфаты (с1сЮТР, сИАТР,
сЫТТР и сИСТР), которые при встраивании в состав мо¬
лекулы ДНК обрывают дальнейший синтез полинуклео-
тидной цепи. В результате этого в каждой из пробирок
накапливается набор дочерних молекул ДНК различной
длины, оканчивающихся строго на один и тот же нукле¬
отид. После электрофореза продуктов этих реакций в 4
соседних дорожках проводится считывание информации
от более коротких фрагментов к более длинным, при этом
выявляемый пошаговый порядок удлинения цепей и от¬
ражает последовательность нуклеотидов в составе ана¬
лизируемой ДНК-матрицы. Одномоментно может быть
ссквенирован участок ДНК, не превышающий -500 п.о.,
поэтому для исследования более длинных участков гена
они должны быть амплифицированы в виде совокупно¬
сти коротких перекрывающихся фрагментов, каждый из
которых секвенируется отдельно. Обычно для обнару¬
жения мутаций секвенируются отдельные экзоны изу¬
чаемого гена вместе с примыкающими к ним интронны-
ми последовательностями (областями сплайсинга).В некоторых случаях в качестве объекта исследо-
иания удобнее использовать не геномную ДНК, а более
компактную и информационно «насыщенную» кДНК
(гм. раздел 1.1), получаемую после соответствующей
01 »работки образцов тканей, например биопсийного ма¬
82Глава 1териала или клеточных линий лимфоцитов, фиброблас-
тов и т.д. Для этого используется методика обратно-
транскриптазной ПЦР (КТ-РСК): проводится обратная
транскрипция мРНК, и получаемые молекулы кДНК
служат матрицей для полимеразной цепной реакции
(рис. 23).геномная ДНКI' ' ~ 'ТР1 шгзрелая мРНКПЕННШЕтранскрипция,сплайсингобратная транскрипция,
полимеразная цепная реакциякДНКпрямой анализ делеции,
секвенирование и т.д.встраивание в
экспрессионную
клеточную систему,
синтез белкабелок
(электрофорез)Рис. 23. Диагностика мутаций на уровне мРНК и белкаВ последующем кДНК подвергается секвениро-
ванию и другим методам мутационного скрининга, пря¬
мому электрофоретическому исследованию (выявление
делеций, вставок и т.д.) либо встраиванию в экспресси¬
онную систему с целью получения белкового продукта
и его непосредственного анализа. Последний метод осо¬
бенно эффективен для детекции мутаций, ведущих к
синтезу «усеченного» белка (нонсенс-мутации, мутации
Общие принципы генодиагностики 83сплайсинга, крупные делении) - так называемый РТТ-
анализ (.Protein Truncation Test) [Telatar M. et al., 1996].
РТТ-анализ обычно используется при исследовании про¬
тяженных мультиэкзонных генов, таких как ген мышеч¬
ной дистрофии Дюшенна/Бекера, атаксии-телеангиэкта-
зии или нейрофиброматоза 1-го типа.Поскольку точковые мутации могут иметь самую
различную локализацию, для их обнаружения необхо¬
димо проводить тотальное исследование всей кодирую¬
щей области гена. Принимая во внимание тот факт, что
гены многих заболеваний содержат в своем составе де¬
сятки экзонов, полное секвенирование кодирующей об¬
ласти может представлять исключительно трудоемкую
задачу. Для облегчения идентификации мутаций были
предложены различные скрининговые методы анализа,
позволяющие предварительно отбирать предположитель¬
но мутантные фрагменты ДНК, подлежащие секвениро-
ванию. Наиболее распространенным является анализ
конформационного полиморфизма однонитевой ДНК,
или SSCP-анализ (от англ. «Single Strand Conformation
Polymorphism») [Orita M. et al., 1989]. Его принцип по¬
казан на рис. 24. При проведении SSCP-анализа ампли-
фицированные в ПЦР фрагменты ДНК исследуются в
особых неденатурирующих условиях, при которых од-
нонитевые молекулы ДНК сохраняют свою простран¬
ственную организацию в процессе электрофореза. Замена
даже одного нуклеотида приводит к изменению конфор¬
мации исследуемых полииуклеотидных цепей, в резуль¬
тате чего нарушается их электрофоретическая подвиж¬
ность (что может быть выявлено посредством обнару¬
жения на электрофореграмме атипично расположенных
полос).Таким образом, обнаружение при SSCP-анализе
измененного паттерна миграции фрагментов одноцепо-
84Глава 1Рис. 24. Принцип вБСР-анализаА-контроль (2 нормальных аллеля); Б-гетерозиготность по нор¬
мальному аллелю и точковой мутации; 1-исходные двойные цепи
ДНК, подвергающиеся денатурации; 2-разделенные нити ДНК, при¬
нимающие специфическую пространственную конформацию в не¬
денатурирующих условиях; 3-результат электрофореза.Черными и
серыми кружками обозначены нормальные нуклеотиды вкомпле-
ментарных цепяхДНК,черными и серыми треугольниками-мутант-
ные нуклеотиды в тех же сайтах. Короткими стрелками на электро-
фореграмме указаны нормальные фрагменты ДНК, длинными стрел-
ками-мутантные фрагменты ДНК с измененной электрофоретичес¬
кой подвижностью.
Общие принципы генодиагностики 85чечной ДНК позволяет заподозрить наличие мутации в
составе одного из многих изучаемых участков гена, пос¬
ле чего проводится секвенирование данного мутантного
образца для точной характеристики нуклеотидных на¬
рушений. Чувствительность ЗБСР-анализа для детекции
неизвестных мутаций зависит от величины анализируе¬
мого фрагмента, будучи максимальной (80-95%) при
анализе образцов размером от 70 до 200 п.о. Существу¬
ет целый ряд других скрининговых методов поиска не¬
известных точковых мутаций - гетеродуплексный ана¬
лиз, метод денатурирующего градиентного гель-элект¬
рофореза, метод химического расщепления некомпле¬
ментарных сайтов и др. [Оготре М., 1993]. Как и ЗБСР-
анализ, они основаны на регистрации некоторых изме¬
ненных физико-химических свойств амплифицирован-
ных мутантных фрагментов ДНК. Во всех случаях после
предварительного отбора аномальных образцов наличие
мутации подтверждается секвенированием.В конкретных популяциях мутационный поиск
при том или ином наследственном заболевании может
быть существенно упрощен в связи с высокой частотой
так называемых мажорных мутаций, т.е. мутаций, ко¬
торые являются преобладающими и обусловливают зна¬
чительный процент всех случаев заболевания в данной
популяции. Наличие мажорных мутаций может быть
обусловлено либо эффектом основателя (высокая рас¬
пространенность конкретной мутантной хромосомы,
исторически унаследованной от одного предка), либо
частым возникновением одного и того же повреждения
в связи с некоторыми особенностями нуклеотидного
состава данной области гена (так называемые горячие
точки мутаций). Примером такого рода является гепа-
толентикулярная дегенерация. При данном аутосомно-
рецессивном заболевании описано свыше 100 различ¬
86Глава 1ных мутаций в сложно организованном гене медной
АТФ-азы (АТР7В), что значительно затрудняет мутаци¬
онный скрининг. Однако одна и та же точковая мутация
в 14-м экзоне гена, ведущая к замене гистидина на глу¬
тамин, выявляется в славянских популяциях России бо¬
лее чем у 60% больных [1уапоуа-8то1епакауа I. е1 а1.,
1999]. Это позволяет проводить относительно простую
и экономную ДНК-диагностику гепатолентикулярной
дегенерации в большинстве обследуемых семей из дан¬
ной этнической группы, не прибегая к технически слож¬
ному исследованию всей кодирующей области гена.При проведении целенаправленного поиска кон¬
кретных точковых мутаций важная роль принадлежит
использованию рестрикционных эндонуклеаз (рестрик-
таз) - особых клеточных ферментов, выделяемых из
различных штаммов бактерий. Ценным свойством рест¬
рикционных эндонуклеаз является распознавание ими
специфических нуклеотидных последовательностей дли¬
ной от 4 до 10 нуклеотидов, в результате чего фермент
осуществляет рестрикцию, т.е. разрезание этих после¬
довательностей в составе двунитевой молекулы ДНК.
Каждая рестриктаза распознает и разрезает в фиксиро¬
ванном месте строго определенную, специфичную для
себя нуклеотидную последовательность - сайт рестрик¬
ции (сайт узнавания). В настоящее время известно уже
несколько сотен различных рестриктаз и, следователь¬
но, столько же различных вариантов коротких нуклео¬
тидных последовательностей, образующих сайты рест¬
рикции. В тех случаях, когда точковая мутация изменяет
естественный сайт узнавания для определенной рестрик-
тазы, данный фермент не сможет расщепить мутантный
амплифицированный фрагмент ДНК; в некоторых слу¬
чаях, напротив, мутация приводит к появлению нового
сайта узнавания для той или иной рестриктазы, отсут¬
Общие принципы генодиагностики 87ствующего в норме. В обеих описанных ситуациях му¬
тантный и нормальный ПЦР-продукты дадут различные
по длине фрагменты рестрикции, что можно будет легко
обнаружить при электрофорезе (рис. 25).А1 и Iнормальный
аллель г.. . А Є С Т . ,|< >|<—150 п.о. 80 п.о. 230 п.о.1 2 3Рис. 25. Выявление точковой мутации с помощью рестрик¬
ционного анализаД . Амплифицируемый участок гена, содержащий сайт рестрикции
АСтСТ для рестрикционной эндонуклеазы АІиІ. Мутация в—>А
шменяет данную нуклеотидную последовательность, в результате
чого рестрикция ферментом АІиІ блокируется. Б. Электрофорег-
рамма продуктов рестрикции: дорожка 1 - гомозиготность по нор¬
мальному аллелю; дорожка 2- гомозиготность по мутации; 3- ге-
юрозиготное состояние (нормальный аллель+ мутация).мутантныйаллельА1 и IV/\А АСТ
88Глава 1Таким образом, при необходимости быстрой де¬
текции какой-либо определенной точковой мутации за¬
дача сводится к компьютерному поиску соответствую¬
щей рестриктазы, сайт узнавания которой локализован
в месте нарушенной нуклеотидной последовательнос¬
ти; обработка ПЦР-продуктов такой рестриктазой позво¬
лит легко дифференцировать нормальные и мутантные
аллели. Рестрикционный анализ значительно упрощает
обнаружение известных точковых мутаций и в настоя¬
щее время широко используется для прямой ДНК-диаг-
ностики наследственных заболеваний.Существует большое число модификаций метода
ПЦР, разработанных для быстрого сканирования и по¬
иска известных генных мутаций. Кратко упомянем
некоторые из них. 1) Мультиплексная (.мультипрай-
мерная) ПЦР — основана на одновременной ам¬
плификации в одной реакции нескольких экзонов
исследуемого гена, что позволяет проводить экономный
экспресс-скрининг наиболее частых мутаций в гене
[Chamberlain J. et al., 1988]. 2) Аллель-специфическая
амплификация — основана на использовании различных
праймеров, один из которых комплементарен
нормальной, а другой - мутантной последовательности
ДНК; в результате такой реакции в растворе
синтезируются две разновидности ПЦР-продуктов -
нормальные и мутантные, различающиеся по своей
длине [DvorakovaD.,FajkusovaL., 1993]. 3) Метод аллель-
специфических олигонуклеотидов - основан на
гибридизации амплифицированных фрагментов ДНК с
мечеными олигонуклеотидами двух видов,
комплементарными нормальной либо мутантной по¬
следовательности ДНК [Reiss J., 1991]. 4) Метод сайт-
направленной модификации амплифицированной ДНК -
основан на использовании в ПЦР mismatch-праймера,
отличающегося от матричной последовательности на
Общие принципы генодиагностики 891 нуклеотид [НаНаззоэ А. е! а1., 1989]; в результате вклю¬
чения такого праймера в состав мутантного ПЦР-про-
дукта в нем образуется искусственно созданный сайт
рестрикции для одной из рестриктаз, что позволяет
провести ДНК-диагностику с помощью рестрикционного
анализа.Как указывалось выше, для осуществления ПЦР
необходимо точно знать структуру кодирующих участ¬
ков изучаемого гена и примыкающих к ним интронных
областей (без чего невозможен синтез праймеров, ини¬
циирующих реакцию). Некоторые участки гена (напри¬
мер, сверхдлинные области тандемных тринуклеотид-
ных или более сложных повторов) не могут быть эффек¬
тивно амплифицированы в ПЦР. Кроме того, ПЦР-опос-
редованная прямая ДНК-диагностика не всегда легко
применима для анализа больших и сложных по своей
молекулярной организации генов (таких как ген дистро-
фина), выявления протяженных делеций в
гетерозиготном состоянии и в некоторых других случаях.
Поэтому при ряде наследственных заболеваний
используется другой метод детекции изменений
нуклеотидного состава гена - блот-гибридизация. В
общем виде технология блот-гибридизации показана на
рис. 26. Геномная ДНК обрабатывается одной из
рестрикционных эндонуклеаз, после чего образующийся
набор большого числа рестрикционных фрагментов ДНК
различной длины фракционируется в агарозном геле с
разделением их по молекулярной массе. Далее, после
денатурации ДНК, проводится процедура блоттинга -
переноса фрагментов ДНК с геля на нитроцеллюлозный
или нейлоновый фильтр (это осуществляется за счет
действия осмотических сил, вакуума или электрического
поля), в результате чего на фильтре фиксируется точная
реплика ДНК-рестрикта. Денатурированные фрагменты
ДНК гибридизуются с радиоактивно-меченым зондом -
90Глава 1молекулой ДНК длиной несколько сотен нуклеотидов.
После отмывки фильтра и авторадиографии на
рентеновской пленке можно видеть точное положение
и оценить длину фрагментов ДНК, комплементарных
использованному зонду. Таким образом, блот-
гибридизация позволяет идентифицировать
специфические последовательности нуклеотидов в
общем наборе рестрикционных фрагментов геномной
ДНК.Денатурация ДНКНабор рестрикционных
Геномная ДНК фрагментов ДНКРестрикцияэндонуклеазойЭлектрофорез
в агарозном
гелеИзображение на
рентгеновской
пленкеОтмывка
фильтра,графияПеренос ДНК на
нитроцеллюлозный
фильтр (блоттинг)Г ибридизациямеченыйДНК-зондРис. 26. Принцип блот-гибридизации по СаузернуПредставленная технология блот-гибридизации
ДНК носит название блот-гибридизации по методу Сау-
зерна (Саузерн-блоттинг) - по имени английского ис¬
следователя, предложившего данный метод в 1975 г.
Общие принципы генодиагностики 91Аналогичным образом может проводиться гибридиза¬
ция ДНК-зонда с электрофоретически разделенными мо¬
лекулами РНК с целью идентификации специфических
последовательностей тканевых РНК (Нозерн-блоттинг),
а также гибридизация меченых антител со специфичес¬
кими белками (Вестерн-блоттинг, или иммуноблот-
тинг). Модификациями Саузерн-блоттинга являются
методы гибридизации с ДНК-зондом молекул ДНК
(РНК), нанесенных на фильтр без предварительной рес¬
трикции и электрофореза - дот-гибридизация (при на¬
несении на фильтр округлого пятна ДНК) и слот-гибри-
дизация (при вытянутой форме пятна).Метод блот-гибридизации обладает высокой чув¬
ствительностью, однако он весьма трудоемок, предпо¬
лагает использование радиоактивных зондов, предъяв¬
ляет высокие требования к качеству анализируемой ДНК
и требует для проведения больших количеств геномной
ДНК (5-10 микрограмм, что на несколько порядков пре¬
вышает количество ДНК, необходимое для проведения
ПЦР). Тем не менее во многих случаях он играет веду¬
щую роль в прямой ДНК-диагностике, в том числе в
сочетании с другими разнообразными вышеописанны¬
ми методами исследования структуры ДНК. Например,
когда та или иная точковая мутация затрагивает сайт уз¬
навания используемой при бло г-гибридизации рестрик-
тазы, на пленке можно четко зафиксировать отсутствие
соответствующей полосы или появление атипичного
фрагмента ДНК. Удлинение определенного участка изу¬
чаемого гена (например, при болезнях экспансии повто¬
ров) или делеция участка гена также приведут к измене¬
нию размера одного из фрагментов ДНК и появлению
атипичной полосы либо отсутствию полосы на радиоав¬
тографе (рис. 27).
92Глава 112 3 4Рис. 27. ДНК-диагностика мутации в гене дисгрофина с
помощью блот-гибридизации по Саузерну
В дорожке 2 у больного отсутствуют два облигатных фрагмента
ДНК (указаны стрелками), что свидетельствует о протяженной де¬
лении соответствующего участка гена дистрофина. В дорожке 4
(мать больного) указанные стрелками фрагменты визуализируются
в виде полос пониженной интенсивности сигнала (по сравнению с
контролем в дорожках 1 и 3), что свидетельствует о сниженной вдвое
«дозе гена» (гетерозиготное носительство делении).
Общие принципы генодиагностики 93Одним из приложений блот-гибридизации по
Саузерну является диагностика мутаций, характеризую¬
щихся изменением «дозы гена». При наличии делеции
или дупликации гена в гетерозиготном состоянии соот¬
ветствующие полосы на радиоавтографе будут характе¬
ризоваться уменьшением или увеличением интенсив¬
ности и площади сигнала, что можно зафиксировать
визуально либо оценить количественно с помощью ден¬
ситометрии полученного изображения (см. рис. 27).
Определение дозы гена возможно и на основе техноло¬
гий количественной ПЦР [Heid C. et al., 1996], позволя¬
ющей оценивать количественный состав продуктов ам¬
плификации (как правило, для этого применяются авто¬
матические секвенаторы и соответствующее программ¬
ное обеспечение). Использование блот-гибридизации в
ряде случаев позволяет выявить патогенетически значи¬
мые нуклеотидные изменения в составе интронов, кото¬
рые пропускаются при обычных методах скрининга ко¬
дирующей области гена.В последние годы технологии прямой ДНК-диаг-
ностики вышли на качественно новый уровень в связи с
появлением методов так называемого параллельного
молекулярно-генетического анализа, позволяющих про¬
водить одновременный мутационный скрининг большого
числа образцов и значительно ускорить выявление му¬
таций в изучаемых генах [Yershov G. et al., 1996;
McKenzie S. et al., 1998; Syvänen A., 1999]. К ним отно¬
сятся уже упоминавшееся автоматическое секвенирова-
иие (осуществляемое на специальных приборах с флюо¬
ресцентными методами детекции и компьютерным ана¬
лизом нуклеотидных последовательностей), технология
капиллярного электрофореза, метод мультиплексного
аллель-специфического диагностического анализа, прай¬
94Глава 1мер-продлевающее «минисеквенирование», использо¬
вание ДНК-биочипов и целый ряд других новейших
разработок.На последнем из названных методов следует ос¬
тановиться чуть подробнее. ДНК-биочип представля¬
ет миниатюрную гелевую пластину, в ячейки которой
с помощью специальных методов нанесен упорядочен¬
ный набор из десятков и сотен иммобилизованных ко¬
ротких олигонуклеотидов. Данные олигонуклеотиды
специфически гибридизуются с разнообразными флю-
орохромно-мечеными молекулами ДНК, а результат
реакции фиксируется с помощью флюоресцентной
микроскопической техники и анализируется с исполь¬
зованием соответствующего компьютерного программ¬
ного обеспечения. Применение на практике техноло¬
гии ДНК-биочипов может позволить проводить авто¬
матизированный и эффективный анализ большого чис¬
ла известных мутаций и полиморфизмов, осуществ¬
лять быстрое секвенирование крупных фрагментов
ДНК, а также одномоментно анализировать экспрес¬
сию большого числа генов в изучаемой ткани в задан¬
ный интервал времени, что сулит поистине уникаль¬
ные возможности в изучении основ функционирова¬
ния генома [Yershov G. et al., 1996; Brown P.,
Botstein D., 1999]. Многие методы параллельного мо-
лекулярно-генетического анализа находятся пока в
стадии разработки, однако не вызывает сомнений, что
они в самое ближайшие время будут внедрены в прак¬
тику работы широкого круга специализированных ди¬
агностических лабораторий, сделав процедуру мута¬
ционного скрининга при большинстве наследственных
заболеваний рутинной операцией.
Общие принципы генодиагностики 951.4.2. Косвенная ДНК-диагностикаКосвенная (непрямая) ДНК-диагностика исполь¬
зуется при заболеваниях, ген которых достаточно точно
картирован, т.е. локализован в конкретном узком учас¬
тке определенной хромосомы. Важно подчеркнуть, что
косвенная ДНК-диагностика может проводиться даже в
тех случаях, когда какая-либо другая информация о гене
болезни, помимо его хромосомного расположения, от¬
сутствует (т.е. когда неизвестны структура и размер гена,
характер мутаций в нем, кодируемый геном белок и т.д.).Сущность косвенной ДНК-диагностики заключа¬
ется в анализе наследования у больных и здоровых чле¬
нов семьи полиморфных генетических маркеров, распо¬
ложенных в изучаемой хромосомной области и, следо¬
вательно, сцепленных с геном болезни. В качестве таких
маркеров выступают участки ДНК, существующие в виде
нескольких возможных аллельных вариантов и различа¬
ющиеся у разных лиц по тем или иным структурным
особенностям (например, по составу нуклеотидов или
по числу копий динуклеотидных СА-повторов). Благо¬
даря вариабельности состава таких маркерных участков
ДНК обычно бывает возможно дифференцировать ма¬
теринское или отцовское происхождение конкретного
варианта маркера при анализе ДНК обследуемого лица.
Сцепление конкретного маркера с геном болезни озна¬
чает, что данный маркер располагается в непосредствен¬
ной близости от патологического гена, поэтому маркер
и ген болезни после кроссинговера остаются в составе
одного хромосомного сегмента и передаются потомству
как единое целое (рис. 28). Таким образом, благодаря
анализу полиморфных генетических маркеров удается
проследить в ряду поколений наследование каждой из
родительских хромосом.
96Глава 1Г\Г\Г\дА1МВ1Аи иГ\ААА2МВ1чу иРис. 28. Различная степень сцепления исследуемых хромо¬
сомных локусов и их рекомбинация при кроссинговере
М - мутантный аллель, N - нормальный аллель, А и В - генетичес¬
кие маркеры, расположенные в исследуемой хромосомной облас¬
ти. Исследуемый ген тесно сцеплен с маркером В, поэтому после
кроссинговера мутация остается на одной хромосоме с маркерным
аллелем В1, а нормальный ген - с аллелем В2. Маркер А является
более отдаленным, поэтому исходные аллели данного маркера и
исследуемого гена в результате кроссинговера расходятся по раз¬
ным хромосомам.
Общие принципы генодиагностики 97Пример косвенной ДНК-диагностики аутосомно-
доминантного заболевания показан на рис. 29.І■II-21_ пн^ ИІ-2 Д■О"11-4«— 210 п.о.
<— 206 п.о.
I— 202 п.о.
I— 198 п.о.
і— 194 п.о.Рис. 29. Косвенная ДНК-диагноетика аутосомно-доминант-
ного заболеванияЧерным цветом обозначены больные лица, белым - клинически
■здоровые, серым цветом - клинически здоровый родственник, яв¬
ляющийся асимптомным носителем мутантной хромосомы. Иссле¬
дована ДНК родственников из 2 нижних поколений родословной.
В нижней части рисунка показана электрофореграмма с результа¬
тами исследования изучаемого (СА)п-маркера (каждая дорожка геля
соответствует индивиду в родословной, расположенному сверху
от дорожки). Справа от электрофореграммы указаны размеры ал¬
лелей (в парах оснований). Другие объяснения даны в тексте.На первом этапе необходимо определить, какой из алле¬
лей изучаемого маркера сцеплен с мутантным геном в
98Глава 1данной семье у заведомо больных лиц (на рисунке 29
это отцовский аллель размером 210 и.о., поскольку имен¬
но он всегда обнаруживается у больных лиц 11-2, П-З, III-
1 и Ш-2 - дорожки 1, 3, 4 и 6). Дальнейший анализ на¬
следования данного аллеля позволяет проследить пере¬
дачу патологического участка отцовской хромосомы, со¬
держащего мутантный ген. Так, на рисунке видно, что
указанный 210-нуклеотидый аллель передался от боль¬
ного П-З (дорожка 6) его клинически здоровому сыну
Ш-4, входящему в группу риска (дорожка 7); на основа¬
нии этого делается вывод о том, что сын Ш-4 унаследо¬
ван от отца патологическую хромосому и является но¬
сителем мутантного гена (положительный результат кос¬
венной ДНК-диагностики). Напротив, сын Ш-З (дорож¬
ка 5) унаследовал от больного отца П-2 (дорожка 4) дру¬
гой аллель изучаемого маркера размером 206 п.о., рас¬
положенный на «здоровой» хромосоме и сцепленный с
нормальным геном; следовательно, у него результат ДНК-
диагностики отрицательный (носительство мутантного
гена отвергнуто).Как видно из приведенной схемы, косвенная ДНК
диагностика обладает 3 основными недостатками:1) Для ее проведения требуется анализ ДНК не
скольких членов семьи, как правило, из 2-3 поколений:,
таким образом в неполных (неинформативных) семьях
такая диагностика невозможна. Косвенная ДНК-диагно
стика может оказаться невозможной также в том случае,
когда отец и мать имеют одинаковые аллели маркера
что не позволяет дифференцировать генетическое про
исхождение родительских хромосом у детей (в ЭТОЙ СП
туации говорят о неинформативности маркера).2) Для проведения косвенной ДНК-диагност! I
ки у лиц из группы риска необходимо предварительно
диагностировать заболевание хотя бы у одного индивп
Общие принципы генодиагностики 99дуума в родословной; таким образом косвенный подход
неприменим для ДНК-диагностики единичных (спора¬
дических) случаев заболевания.3) При проведении косвенной ДНК-диагности¬
ки всегда существует вероятность ошибки, связанная с
возможной рекомбинацией в мейозе между геном болез¬
ни и исследуемым маркером, в результате чего «патоло¬
гический» маркерный аллель и ген болезни у потомка
разойдутся по разным хромосомам. Величина такой
ошибки прямо пропорциональна расстоянию между мар¬
керным локусом и изучаемым геном: чем ближе они друг
к другу на хромосоме (т.е. чем теснее сцеплены между
собой), тем меньше вероятность их расхождения вслед¬
ствие кроссинговера. Для известных на сегодня генети¬
ческих маркеров, используемых в косвенной ДНК-диаг¬
ностике наследственных заболеваний, вероятность рас¬
хождения с геном болезни в мейозе (т.е вероятность
ошибки) не превышает 1-5%; таким образом, точность
косвенной ДНК-диагностики болезни составляет обыч¬
но^ 95%.На практике для повышения точности и инфор¬
мативности косвенной ДНК-диагностики обычно ис¬
пользуют несколько маркеров, расположенных в изуча¬
емой хромосомной области. Таким образом, для каждой
ич хромосом можно установить и графически реконст¬
руировать последовательную серию аллелей изучаемых:
I епетических маркеров. Такая комбинация аллелей раз-
ми ных локусов на одной хромосоме носит название гап-
т тип.Косвенная ДНК-диагностика не ограничивается
нпц,ко заболеваниями, ген которых картирован, но не
.' I ановлен. В настоящее время косвенная ДНК-диагно-
ми ка широко используется также при заболеваниях, гены
мп орых идентифицированы, но имеют большой размер
и г ножную молекулярную организацию, в связи с чем
100Глава 1непосредственное определение мутаций в них чрезвы¬
чайно затруднено. Если ген заболевания известен, при
проведении косвенной ДНК-диагностики могут исполь¬
зоваться как внешние сцепленные маркеры, так и внут-
ригенные маркеры, непосредственно входящие в состав
кодирующей области или интронов. В качестве приме¬
ров заболеваний с установленными генами, при которых
с успехом применяется косвенная ДНК-диагностика,
можно назвать атаксию-телеангиэктазию, гепатоленти-
кулярную дегенерацию, различные формы аутосомно-
рецессивных прогрессирующих мышечных дистрофий
и др.Косвенный ДНК-анализ может применяться в
качестве дополнительного диагностического метода у
лиц из группы риска при отрицательных результатах
традиционного мутационного скрининга. Так, например,
в семьях, имеющих одного больного ребенка с мышеч¬
ной дистрофией Дюшенна/Бекера, пренатальная ДНК-
диагностика болезни у плода при последующих беремен¬
ностях может включать ряд последовательных молеку¬
лярных тестов [van Essen A. et al., 1997]. На первом эта¬
пе у пробанда проводится скрининг наиболее частых
делений в гене дистрофина с использованием мульти¬
плексной ПЦР, а после обнаружения конкретной деле¬
нии исследуется ее наличие у плода. При отрицатель¬
ном результате этого первого этапа ДНК-анализа далее
может быть проведен Саузерн-блоттинг, позволяющий
выявить ряд делений, дупликаций или точковых мута¬
ций, не выявляемых с помощью мультиплексной ПЦР;
для поиска структурных или точковых мутаций может
использоваться также анализ кДНК дистрофина, полу¬
ченной из образцов тканей пробанда (лимфоциты, мы¬
шечные биоптаты), SSCP-анализ и другие скрининговые
методы исследования кодирующей области гена. Нако¬
нец, при невозможности обнаружения конкретных нук¬
Общие принципы генодиагностики 101леотидных изменений в гене проводится косвенная ДНК-
диагностика с вне- или внутригенными маркерами, по¬
зволяющая оценивать сегрегацию патологического уча¬
стка Х-хромосомы у матери, больного ребенка и плода и
тем самым с высокой вероятностью определять генети¬
ческий статус плода.Данный пример хорошо иллюстрирует важней¬
ший принцип молекулярно-генетической диагностики
наследственных заболеваний - необходимость рацио¬
нального сочетания различных методов, четкую после¬
довательность выбора тех или иных процедур ДНК-ана-
лиза, определяемую особенностями строения гена и ти¬
пичным спектром мутаций, характерным для каждого
заболевания.1.5. Стратегия генетического
картирования и его роль в
идентификации новых генов
наследственных заболеванийВажнейшей задачей молекулярной генетики при¬
менительно к медицине является идентификация генов
наследственных заболеваний человека и выявление кон¬
кретных повреждений в них, приводящих к развитию
фенотипических проявлений болезни. Эта задача может
быть выполнена с помощью нескольких основных под¬
ходов.Первый подход к идентификации генов, оставав-i дайся ведущим приблизительно до начала 90-х годов,I шзируется на имеющейся информации об основном био¬
химическом дефекте (первичном белковом продукте гена), ха¬
рактеризующем изучаемую болезнь [Шишкин С.С., Кали¬
нин В.Н., 1992; Gardner Е. et al., 1991; Collins Е, 1995].
11среход от белкового анализа на уровень ДНК осуще¬
102Глава 1ствлялся через секвенирование очищенного белкового
продукта и получение ДНК-зондов, использование мо¬
ноклональных антител и с помощью некоторых других
методических приемов. Хромосомная локализация гена
в данной схеме поиска является конечным результатом
исследования. Описанный подход, использующий ту или
иную предварительную информацию о функциональном
значении искомого гена, получил название «функцио¬
нальное клонирование» [Collins F., 1995]. Примером ус¬
пешного применения функционального клонирования
является идентификация гена фенилкетонурии. К сожа¬
лению, данный метод может быть применен лишь к весь¬
ма ограниченному кругу заболеваний человека, тогда как
для большинства наследственных болезней первичные
продукты гена или патогномоничные биохимические
маркеры неизвестны.Совершенствование молекулярных технологий
привело к созданию принципиально иной стратегии по¬
иска гена, не требующей каких-либо предварительных
знаний о его функции или первичном биохимическом
продукте. Данная стратегия предполагает идентифика¬
цию гена на основании точного знания его локализации
в определенном хромосомном локусе — «позиционное
клонирование» (менее удачный термин «обратная гене¬
тика») [Collins F., 1992; 1995; Davis М. et al., 1993]. По¬
зиционное клонирование ведет к установлению молеку¬
лярной основы болезни «от гена к белку» и включает
следующие основные этапы: 1) картирование гена бо¬
лезни в определенном участке конкретной хромосомы
(генетическое картирование); 2) составление физичес¬
кой карты изучаемой хромосомной области (физическое
картирование); 3) идентификация экспрессирующихся
последовательностей ДНК в изучаемой области; 4) сек¬
венирование генов-кандидатов и выявление мутаций в
искомом гене у больных лиц; 5) анализ структуры гена.
Общие принципы генодиагностики 103расшифровка последовательности и первичной структу¬
ры его продуктов - мРНК и белка [Leppert М., 1990;
Gardner Е. etal., 1991; Cantor С., Smith С., 1993; Collins F.,1995]. В ряде случаев позиционное клонирование гена
облегчается при обнаружении у больных видимых цито¬
генетических перестроек или определяемых делеций в
критической хромосомной области, позволяющих зна¬
чительно повысить точность картирования мутантного
гена. Выявление таких перестроек способствовало, в
частности, успеху в клонировании генов миодистрофии
Дюшенна/Бекера, нейрофиброматоза 1-го типа, тубе¬
розного склероза, адренолейкодистрофии и других на¬
следственных заболеваний нервной системы.Одним из важных промежуточных результатов
исследовательского проекта «Геном человека» стало со¬
здание все более и более насыщенной транскрипцион¬
ной карты генома, содержащей сведения о тысячах уже
известных генов и экспрессирующихся нуклеотидных
последовательностей. Это способствовало значительно¬
му развитию еще одного подхода к идентификации пер¬
вичного генетического дефекта, при котором после пред¬
варительного картирования мутантного гена проводит¬
ся скрининг подходящих генов-кандидатов, расположен¬
ных в том же хромосомном участке («positional candidate
approach») [Collins F., 1995]. Данный метод предполага¬
ет наличие определенных знаний о патофизиологии изу¬
чаемого заболевания, что дает возможность проводить
рациональный отбор генов-кандидатов для анализа из
большого числа генов, которые могут быть расположе¬
ны в «зоне интереса». Среди неврологических наслед¬
ственных заболеваний, гены которых были идентифи¬
цированы таким образом благодаря анализу подходящих
кандидатов в установленном хромосомном интервале,
можно назвать дофа-зависимую дистонию и фридрейхо-
подобную атаксию с дефицитом витамина Е. По суще¬
104Глава 1ствующим прогнозам, именно анализ «позиционных
кандидатов» станет в ближайшем будущем ведущим ме¬
тодом идентификации генов наследственных болезней,
чему в немалой степени способствует создание и посто¬
янное расширение компьютерных баз данных экспрес¬
сирующихся последовательностей на хромосомах
(«expressed sequence tags») [Collins F., 1995].Таким образом, определение хромосомной лока¬
лизации искомого гена — генетическое картирование -
является первым, ключевым шагом на пути к раскры¬
тию молекулярной основы того или иного наследствен¬
ного заболевания.Существует несколько основных методов, позво¬
ляющих картировать неизвестный ген в конкретном хро¬
мосомном локусе: а) клинико-генеалогический (простей¬
ший и наиболее давний) - основан на анализе наследо¬
вания признаков в больших родословных; примером мо¬
жет служить установление локализации гена на Х-хро-
мосоме в случае передачи болезни по Х-сцепленному
типу; б) цитогенетический - базируется на ассоциации
выявляемых при микроскопии хромосомных перестро¬
ек с определенным клиническим фенотипом; в) метод
гибридизации in situ (в том числе его современная моди¬
фикация - флюоресцентная гибридизация in situ, FISH)
- использует специфическую гибридизацию мРНК и
кДНК искомого гена с денатурированными хромосома¬
ми на метафазных препаратах клетки; г) метод гибрид¬
ных клеток - основан на анализе совместной сегрегации
клеточных признаков и хромосом в клонированных in vitro
гибридных соматических клетках [Фогель Ф., Мотульски А.,
1990; Gardner Е. et al., 1991]. Все эти методы нашли свое
применение в современной молекулярной генетике, од¬
нако они обладают серьезными ограничениями, связан¬
ными как с недостаточной разрешающей способностью,
так и с существованием жестких предусловий, необхо¬
Общие принципы генодиагностики 105димых для проведения исследования (таких как нали¬
чие зондов, доступность селективных систем для отбо¬
ра гибридных клеток и т.п.). Наиболее мощным, про¬
дуктивным и широко используемым в настоящее время
методом картирования генов наследственных болезней
человека является так называемый linkage-анализ - ана¬
лиз сцепления искомого гена с набором точно локализо¬
ванных генетических маркеров [Ott J.,1991; Pulst S.,
1999].Центральное положение linkage-анализа заключа¬
ется в том, что мерой относительного генетического рас¬
стояния между двумя локусами на хромосоме может слу¬
жить частота рекомбинаций между этими локусами в
результате кроссинговера гомологичных хромосом в
мейозе. Чем ближе расположены локусы на хромосоме,
чем больше вероятность того, что они будут наследовать¬
ся как единое целое (группа сцепления); при значитель¬
ной удаленности изучаемых локусов (т.е. слабой степе¬
ни сцепления) они с большей вероятностью разойдутся
после кроссинговера по разным хромосомам. Частота
рекомбинации между локусами 1% принята за единицу
генетического расстояния между ними - 1 сантиморга-
пиду (сМ), что эквивалентно в среднем 1 миллиону и.о.
Следует подчеркнуть, что частота рекомбинаций и, сле¬
довательно, генетическое расстояние, неодинаковы для
мужчин и женщин (больше у женщин), для разных хро¬
мосом, а также для разных участков одной хромосомы
(«горячие точки» рекомбинации) [Cantor С., Smith С.,
1.993; Merette C., Ott J., 1993].Сущность анализа сцепления состоит в сопостав¬
лении наследования патологического признака (болез¬
ни) в родословной с наследованием различных полимор¬
фных генетических маркеров с точно известной хромо-
еомной локализацией. Если все больные или их значи¬
тельная часть в семье имеют, в отличие от здоровых, один
106Глава 11-1 1-2Рис. 30. Принцип анализа генетического сцепления на
примере аутосомно-доминантного заболевания
В данном примере исследованы 4 сцепленных маркера А, В, С и О,
по которым реконструированы гаплотипы. Разные по происхожде¬
нию хромосомы маркированы различными типами штриховки (ис¬
ходная мутантная хромосома обозначена черным цветом). Все боль¬
ные в родословной имеют одну и ту же общую (среднюю) часть
исходной мутантной хромосомы. Например, в нижнем поколении
хромосомы претерпели ряд рекомбинаций, однако у всех больных
сибсов (в том числе у лиц Ш-З и Ш-8) сохраняется один и тот же
мутантный гаплотип по маркерам В и С (гаплотип у ). Напротив,
никто из здоровых сибсов в нижнем поколении не унаследовал от
отца гаплотип у по маркерам В и С (индивидуум Ш-4 унаследо¬
вал хромосому, в которой рекомбинация произошла ниже крити¬
ческого сегмента). Таким образом, сегрегация маркерных аллелей
и анализ гаплотипов свидетельствуют о том, что ген заболевания
расположен в хромосомном сегменте, включающем в себя марке¬
ры В и С. Соответственно, внешними границами участка хромосо¬
мы, в пределах которого расположен мутантный ген, являются мар¬
керы А и О.
Общие принципы генодиагностики 107и тот же аллель исследуемого маркера, это свидетель¬
ствует об отсутствии рекомбинаций между искомым
мутантным геном и данным маркером, т.е. о наличии
сцепления между ними. Пример сцепления между ге¬
ном аутосомно-доминантного заболевания и определен¬
ными генетическими маркерами представлен на рис. 30.Для достоверного доказательства сцепления раз¬
работан специальный математический аппарат [Ott J.,
1991]. Принцип расчета заключается в сопоставлении
вероятностей гипотез о наличии и отсутствии сцепле¬
ния при имеющихся семейных данных и выбранной ча¬
стоте рекомбинаций 0; соотношение этих двух вероят¬
ностей (соотношение правдоподобий) выражает шансы
за и против сцепления. Для удобства используется деся¬
тичный логарифм соотношения правдоподобий - Лод-
балл (от англ. Logarithm of the Odds, или LOD):PoLOD = Logio —-P1/2 , где P - вероятность
полученного распределения семейных данных для сцеп¬
ленных генов с частотой рекомбинаций 9, Р1/2 - вероят¬
ность такого распределения для двух несцепленных сво¬
бодно рекомбинирующих генов (частота рекомбинаций0 = 1/2). Использование логарифмической формы расче¬
та позволяет проводить сложение Лод-баллов, получен¬
ных при анализе отдельных родословных. Для доказате¬
льства генетического сцепления принято значение Лод-
балла +3, которое означает соотношение шансов 1000:1
в пользу наличия генетического сцепления между марке¬
ром и изучаемым признаком. Лод-балл -2 и ниже свиде¬
тельствует о достоверном отсутствии сцепления; значе¬
ния Лод-балла от +3 до - 2 являются, соответственно,
более или менее предположительными с точки зрения
наличия сцепления и нуждаются в дальнейшем подтвер¬
ждении. Частота рекомбинаций 9, для которой был вы¬
108Глава 1явлен максимальный Под-балл, является отражением
наиболее вероятного генетического расстояния между
изучаемыми локусами; ориентировочно считается, что
1% рекомбинаций свидетельствует об очень тесном сце¬
плении, частота рекомбинаций около 5% - о хорошем
сцеплении и частота 10-20% - о некотором умеренном
сцеплении.Расчет ./7с>0-баллов предполагает использование
специального компьютерного программного обеспече¬
ния (программа LIPED, пакет программ LINKAGE и др.)
[Lathrop G., Lalouel J., 1984; Ott J., 1991].Для успеха linkage-анализа необходимо, чтобы
исследуемые семьи были информативны по болезни и
по генетическому маркеру. Первое означает наличие до¬
статочного числа информативных мейозов в родослов¬
ной, позволяющих анализировать расхождение призна¬
ков в данной родословной. С практической точки зре¬
ния это означает наличие большого числа доступных для
анализа больных и здоровых родственников, как прави¬
ло, из нескольких поколений. Информативность по мар¬
керу предполагает его полиморфизм (т.е. существование
большого числа аллелей) и гетерозиготность у ключе¬
вых членов семьи, что позволяет дифференцировать ге¬
нетическое происхождение конкретных аллелей марке¬
ра. До конца 80-х годов основным типом маркеров, ис¬
пользуемых в анализе сцепления, были участки ДНК
хромосом, имеющие в своем составе вариацию в одной
паре оснований и различаемые по наличию или отсут¬
ствию участка рестрикции для соответствующего фер¬
мента, т.е. по длине рестрикционных фрагментов
(«restriction fragment length polymorphism», RFLP)
[Botstein D. et al., 1980]. Новая эра в генетическом кар¬
тировании наступила с открытием класса высокополи¬
морфных маркеров, представляющих собой участки
ДНК, состоящие из вариабельного числа копий тандем¬
Общие принципы генодиагностики 109ных (СА)п-повторов и обладающие чрезвычайно высо¬
кой гетерозиготностью [Ott J., 1991]. Это позволило в
значительной степени разрешить проблему информатив¬
ности используемых маркеров и способствовало суще¬
ственному прогрессу linkage-анализа. По некоторым
оценкам, для скрининга полного гаплоидного генома и
выявления генетического сцепления необходимо иметь
200-300 высокополиморфных маркеров, равномерно рас¬
пределенных по хромосомам [Leppert М., 1990;
Antonarakis S., 1994]. Генетические карты последнего
поколения включают свыше 5000 таких маркеров [Dib С.
et al., 1996], что позволяет считать сегодня задачу уста¬
новления генетического сцепления принципиально воз¬
можной в любых информативных родословных
[Antonarakis S., 1994].Серьезных проблемой, с которой приходится стал¬
киваться при проведении анализа сцепления на серии
семей, является проблема возможной генетической ге¬
терогенности изучаемого клинического синдрома. В слу¬
чае, если изучаемый фенотип может вызываться мута¬
циями в разных генах, механическое сложение получен¬
ных в отдельных семьях положительных (при наличии
сцепления) и отрицательных (при его отсутствии) Лод-
баллов ведет к нивелированию суммарного значения Лод-
балла и ложному выводу о полном отсутствии сцепле¬
ния. Примером может служить аутосомно-доминантная
моторно-сенсорная невропатия I типа, обусловленная
мутациями в разных генах, локализованных на 1-й, 17-й
и других хромосомах [Chance Р., Fischbeck K., 1994].
В этой ситуации особое значение приобретает тщатель¬
ное, детальное обследование больных и семей, направ¬
ляемых для linkage-анализа, с целью отбора максималь¬
но однородных клинических групп. Дополнительным
способом избежать ложно-отрицательного результата
исследования является использование в процессе расче¬
110Глава 1та Лод-баллов специальной программы HOMOG или
аналогичных ей программ, позволяющих оценивать ве¬
роятность генетической гетерогенности при полученном
конкретном наборе семейных данных [Ott J., 1991;
Merette C., Ott J., 1993]. Наиболее действенным подхо¬
дом на первом этапе исследования является анализ сцеп¬
ления в одной большой информативной родословной, что
позволяет заведомо исключить возможность генетичес¬
кой гетерогенности в изучаемой группе больных. Допол¬
нительные сложности при проведении linkage-анализа
связаны с нередко наблюдающейся неполной пенетрант-
ностью и вариабельной экспрессивностью мутантного
гена, наличием фенокопий среди обследуемых членов
семьи, оценкой возраста начала болезни и возможности
доклинического носительства мутации, оценкой распро¬
страненности конкретных аллелей изучаемых маркеров
в популяции и т.д. [Ott J., 1991; Bird Т., 1993; Davis М. et
al., 1993; Pulst S., 1999]. Неверный учет или недооценка
этих факторов могут существенно повлиять на итоговый
результат, поэтому качество подробного клинико-генеа-
логического анализа в изучаемых семьях выступает на
первый план.Разработано много новых методов, представляю¬
щих из себя дальнейшее развитие традиционной страте¬
гии исследования генетического сцепления и существен¬
но повышающих скорость выполнения, методические
возможности и разрешающую способность данного ана¬
лиза в локализации неизвестных генов наследственных
заболеваний человека. Одним из таких методов является
мультилокусный анализ (multipoint linkage analysis), по¬
зволяющий оценивать Лод-6аллы для совокупности сцеп¬
ленных локусов в соответствии с генетической картой
изучаемого хромосомного участка и определять наибо¬
лее вероятную локализацию мутантного гена в пределах
данного участка [Lathrop G. et al., 1984]. В инбредных
Общие принципы генодиагностики 111родословных с аутосомно-рецессивным заболеванием при
наличии предположения об «эффекте основателя» исклю¬
чительно продуктивным зарекомендовал себя метод го¬
мозиготного картирования: он заключается в анализе «го-
мозиготности по происхождению» («homozygosily-by-
descent») и позволяет оценить степень гомозиготности
больных лиц по серии маркеров как результат наследо¬
вания от единого предка общего хромосомного участка,
включающего мутантный ген [LanderE.,BotsteinD., 1988;
Ben Hamida C. et al., 1993; Hillaire D. et al., 1994]. Много¬
обещающим является метод «экономного сканирования
генома», предполагающий преимущественное использо¬
вание маркеров, локализованных в «стратегических» CG-
насыщенных хромосомных областях, богатых экспрес¬
сирующимися последовательностями [Antonarakis S.,
1994]. Предложен также целый ряд других модифика¬
ций классического /ш/ш^е-аиализа [Boehnke М., 1986;
Weeks D., Lange K., 1988; Ott J., 1991; Nelson S. et al.,
1993; Ward P., 1993; Rruglyak L. et al., 1995].Важно подчеркнуть, что анализ сцепления
сохранит свое значение и после идентификации всего
генома человека [Pulst S., 1999]. Например, при изучении
все еще достаточно большой группы наследственных
заболеваний с неустановленными генами первым шагом
на пути к выяснению молекулярного /дефекта может
служить linkage-аутшз и определение хромосомного
локуса болезни, с последующим скринингом подходящих
генов в данной области. Чрезвычайно важной в успехе
генетического картирования является роль клинициста.
Она заключается в адекватном отборе репрезентативных
семей, детальной оценке клинического статуса всех
включенных в исследование членов семьи, точной
диагностике болезни и оценке характера сегрегации
мутантного гена, а также в решении многих других
ключевых вопросов.
ГЛАВА 2ПРОБЛЕМА ГЕНЕТИЧЕСКОЙ
ГЕТЕРОГЕННОСТИ
И КЛАССИФИКАЦИИ
НАСЛЕДСТВЕННЫХ
ЗАБОЛЕВАНИЙ НЕРВНОЙ
СИСТЕМЫС момента первого описания и выделения основ¬
ных групп моногенных наследственных заболеваний не¬
рвной системы проблема их систематизации и номенк¬
латуры оставалась одной из наиболее сложных и запу¬
танных в клинической неврологии и нейрогенетике. Это
обусловлено рядом взаимосвязанных факторов.Во-первых, для наследственных заболеваний не¬
рвной системы характерен исключительно выраженный
меж- и внутрисемейный фенотипический полиморфизм
[Давиденков С.Н., 1934; Калмыкова JI.Г., 1976; Ивано¬
ва-Смоленская И.А. и др., 1998 (a); Harding А., 1984;
1993; Davis М. et al., 1993]. Он проявляется развитием
самых разнообразных, нередко весьма отличающихся
друг от друга неврологических синдромов в рамках од¬
ной нозологической формы, в том числе даже у разных
членов одной семьи. Вариабельная экспрессивность
мутантных генов может приводить к манифестации как
развернутых клинических форм болезни, так и разви¬
тию атипичных ее вариантов, а также «стертых» или
Проблема генетической гетерогенности ... 113«фрустрированных» форм (forme fruste). Для многих
моногенных неврологических заболеваний (таких, напри¬
мер, как миотоническая дистрофия, ряд форм наслед¬
ственных атаксий, торсионной дистонии и др.) полимор¬
физм клинических проявлений и развитие разнообраз¬
ных атипичных и «стертых» форм является скорее пра¬
вилом, чем исключением, что значительно осложняет
систематизацию изучаемых клинических синдромов.Во-вторых, морфологической картине наслед¬
ственных болезней нервной системы также свойствен
значительный полиморфизм, отражающий в первую оче¬
редь вариабельный характер и тяжесть мутаций у конк¬
ретных больных. При ряде заболеваний строгая корре¬
ляция между клиническими и морфологическими при¬
знаками отсутствует. Например, согласно классификации
В. Konigsmark и L. Weiner (1970), несомненной анато¬
мической картине оливопонтоцеребеллярной атрофии
соответствует не менее 5 заметно отличающихся клини¬
ческих синдромов дегенеративных атаксий. Следует от¬
метить, что в неврологии различные генетические фор¬
мы заболеваний могут иметь сходные клинико-морфо¬
логические проявления; с другой стороны, при некото¬
рых наследственных заболеваниях ЦНС (эссенциальныйI ремор, торсионная дистония) на секции какие-либо из¬
менения в веществе мозга отсутствуют. Таким образом,
морфологические критерии (даже в сочетании с особен¬
ностями клинического синдрома) в большинстве случа¬
ев являются недостаточным фундаментом для строгой
классификации наследственных заболеваний нервной
системы.Наконец, до настоящего времени патогенез боль¬
шинства форм наследственных неврологических забо¬
леваний изучен недостаточно, с связи с чем отсутствуют
114Глава 2биохимические и другие рутинные лабораторно-инстру¬
ментальные маркеры, позволяющие провести четкие
нозологические границы среди многообразия сходных
клинических синдромов.В самом общем виде все наследственные заболе¬
вания нервной системы могут быть подразделены на 2
большие группы:1) Заболевания, при которых поражение нервной
системы составляет ведущий симптомокомп-
лекс, «ядро» клинической картины. Именно
данные заболевания, которым посвящены пос¬
ледующие главы настоящей монографии, яв¬
ляются основным предметом изучения кли¬
нической нейрогенетики.2) Заболевания с системными наследственными
дефектами метаболизма, при которых, наря¬
ду с поражением различных органов и тканей,
в качестве одного из проявлений может на¬
блюдаться вовлечение нервной системы. Клас¬
сическими примерами являются различные
«болезни накопления» (гликогенозы, лизосом-
ные болезни), наследственные дефекты обме¬
на аминокислот, пуринов, порфиринов и т.д.
В большинстве случаев данные заболевания
встречаются в педиатрической практике.На протяжении столетия наиболее распространен¬
ной и сохранившей свое значение в нейрогенетике до
настоящего времени была классификация наследствен¬
ных болезней, построенная по топическому признаку и
отражающая уровень поражения и основной клиничес¬
кий синдром заболевания.В современном виде данная классификация мо¬
жет быть представлена следующим образом (таблица 2).
Проблема генетической гетерогенности ... 115Таблица 2Классификация наследственных болезней нервной
системы в соответствии с уровнем поражения■ Наследственные нервно-мышечные болезни:а) прогрессирующие мышечные дистрофии;б) врожденные непрогрессирующие миопатии (структурные
миопатии);в) наследственные болезни мотонейрона (спинальные амиотро-
фии, семейные формы бокового амиотрофического склероза);г) наследственные невропатии (моторно-сенсорные, моторные,
сенсорные, сенсорно-вегетативные);д) наследственные миотонические синдромы;е) наследственные миастенические синдромы;ж) периодический паралич (гипер-, гипо- и нормокалиемические
формы)■ Наследственные болезни центральной нервной системы:а) заболевания с преимущественным вовлечением экстрапира-
мидной системы;б) заболевания с преимущественным вовлечением пирамидной
системы (наследственные спастические параплегии);в) заболевания с преимущественным вовлечением координа-
торных систем (наследственные атаксии);г) заболевания с преимущественным вовлечением когнитивной
сферы (наследственные деменции);д) наследственные формы эпилепсий;е) другие формы наследственных заболеваний центральной
нервной системы (наследственные формы мигрени и других
цереброваскулярных заболеваний, наследственные аномалии
развития мозга и т.д.)■ Наследственные болезни нервной системы с мультиорганны-
ми проявлениями:а) заболевания, характеризующиеся нарушением клеточной
пролиферации (факоматозы);б) наследственные болезни обмена с вовлечением нервной сис¬
темы (митохондриальные болезни, липидозы, гликогенозы идр)
116Глава 2Данная классификация носит в определенной
мере условный характер и не исчерпывает всего много¬
образия возможных комбинаций синдромов, встречаю¬
щихся у больных с наследственной неврологической
патологией. Так, при болезни Фридрейха, традиционно
рассматриваемой в группе наследственных атаксий, в
патологический процесс в одинаковой степени вовлека¬
ются как структуры ЦНС (задние и боковые столбы спин¬
ного мозга), так и периферическая нервная система
(спинномозговые ганглии, чувствительные волокна пе¬
риферических нервов). Хорея Гентингтона с полным
основанием может быть отнесена как к наследственным
экстрапирамидным заболеваниям, так и к наследствен¬
ным деменциям, поскольку для данного заболевания
характерна классическая триада симптомов в виде хоре¬
ического гиперкинеза, прогрессирующей деменции под¬
коркового типа и нарушений в эмоционально-волевой
сфере. При одной из основных форм первично-дегене-
ративных семейных деменций - лобно-височной демен¬
ции - почти в половине случаев отмечается развитие
выраженного синдрома паркинсонизма (так называемая
аутосомно-доминантная лобно-височная деменция с пар¬
кинсонизмом). Можно привести большое количество
таких примеров. Тем не менее представленный подход к
классификации наследственных болезней нервной сис¬
темы остается в целом весьма удобным с практической
точки зрения и в достаточной степени проверен време¬
нем.Важным дополнением приведенной классифика¬
ции является подразделение указанных групп заболева¬
ний на основные типы наследования (аутосомно-доми-
нантный, аутосомно-рецессивный, Х-сцепленный доми¬
нантный и рецессивный, митохондриальный или мате¬
ринский), каждому из которых нередко соответствуют
Проблема генетической гетерогенности ... 117определенные особенности клинической картины, спо¬
собствующие уточнению диагноза.По мере изучения наследственных заболеваний
нервной системы накапливалось все больше данных о
том, что даже в рамках конкретных клинико-морфоло¬
гических форм с определенным типом наследования су¬
ществует более глубокая гетерогенность, результатом
которой является наличие большого числа вариантов
болезни, отличающихся друг от друга наличием или от¬
сутствием различных дополнительных симптомов. Ста¬
ло очевидным, в основе того или иного «единого» фе¬
нотипа может лежать повреждение многих генов и их
белковых продуктов, по-видимому, имеющих отношение
к различным звеньям соответствующего биохимическо¬
го каскада или клеточной функции [Rosenberg R., 1990].
Данное предположение нашло свое блестящее подтвер¬
ждение в последние годы в связи с успехами молекуляр¬
ной генетики и открытием большого числа генов основ¬
ных групп наследственных заболеваний нервной систе¬
мы. Эти открытия представили убедительное обоснова¬
ние положения о генетической гетерогенности наслед¬
ственных болезней нервной системы. Таким образом,
генетическая гетерогенность является одним из ключе¬
вых понятий современной нейрогенетики, имеющим
важнейшее значение в решении целого ряда теоретичес¬
ких и прикладных проблем данной области клиничес¬
кой неврологии.Идентификация генов, разработка разнообразных
методов ДНК-диагностики и установление феномена
генетической гетерогенности наследственных болезней
нервной системы позволили на качественно новом уров¬
не решить проблему построения четкой и упорядочен¬
ной современной номенклатуры этих заболеваний, при¬
ведя к внедрению наиболее совершенного и принципи¬
118Глава 2ально нового геномного подхода к классификации. Ге¬
номная (генетическая) классификация предполагает оп¬
ределение прямой взаимосвязи между конкретной но¬
зологической формой и повреждением определенного
гена, лежащим в основе болезни [Hardy J., Hardy-
Gwinn К., 1998]. Ярким примером может служить груп¬
па аутосомно-доминантных атаксий, систематизация ко¬
торых до недавнего времени была чрезвычайно затруд¬
нена. В последнее десятилетие было показано, что ва¬
риабельность клинико-анатомической картины аутосом¬
но-доминантных атаксий обусловлена генетической ге¬
терогенностью данной группы заболеваний. На сегод¬
няшний день установлено существование, как минимум,
16 самостоятельных генов, локализованных на различ¬
ных хромосомах, мутации которых обусловливают раз¬
витие доминантных атактических синдромов. В резуль¬
тате стала возможной предельно четкая и объективная
молекулярно-генетическая классификация доминантных
атаксий, основанная на прямой (выявление мутации в
конкретном гене) или косвенной ДНК-диагностике (ус¬
тановление сцепления с конкретным хромосомным ло-
кусом). При этом каждому генетическому варианту за¬
болевания присваивается соответствующий условный
индекс, например: спиноцеребеллярная атаксия 1-го
типа (СЦА1, ген расположен на хромосоме 6р22-р23),
спиноцеребеллярная атаксия 2-го типа (СЦА2,, хромо¬
сома 12q24.1), спиноцеребеллярная атаксия 3-го типа
(СЦАЗ или болезнь Мачадо-Джозеф, хромосома 14q32.1)
и т.д. По такому же принципу строится в настоящее вре¬
мя классификация наследственных спастических пара¬
плегий (картированы на хромосомах 15 локусов данных
заболеваний, для 4 из которых идентифицированы му¬
тантные гены), нейрональных цероид-липофусцинозов
(6 хромосомных локусов, в том числе 5 идентифициро¬
Проблема генетической гетерогенности ... 119ванных генов), семейных форм первичного паркинсониз¬
ма (7 локусов, в том числе 3 идентифицированных гена)
и других групп наследственных неврологических забо¬
леваний.В ряде случаев при построении современной клас¬
сификации генетическая индексация дополняется обо¬
значением типа наследования и некоторых других базо¬
вых клинических признаков. Так, в группе аутосомных
форм конечностно-поясной прогрессирующей мышеч¬
ной дистрофии (КПМД) цифровой индекс обозначает тип
наследования (1 - аутосомно-доминантный, 2 - аутосом-
но-рецессивный), а буквенный - конкретный хромосом¬
ный локус. Например, символ КПМД 1В обозначает ауто-
сомно-доминантную форму конечностно-поясной мы¬
шечной дистрофии, ген которой расположен на хромо¬
соме Ц11-ц21, а символ КПМД 2Э - аутосомно-рецес-
сивную форму, обусловленную мутацией гена а-сарког-
ликана на хромосоме \1 ц\2-(\2\.ЪЪ. Аналогичным об¬
разом, группу наследственных моторно-сенсорных не¬
вропатий принято условно подразделять на демиелини-
зирующие (тип I) и аксональные формы (тип II), в рам¬
ках каждой из которых выделяются отдельные генети¬
ческие подтипы, например: НМСН 1А (или СМТ 1А -
от англ. Скагсо1-Мапе-Тоо1И) - обусловлена мутация¬
ми генаРМР22 на хромосоме 17р11.2; НМСН 1В (СМТ
1В) — мутации гена Р0 на хромосоме Ц22-23; НМСН 2В
(СМТ 2В) - аксональная форма, сцепленная с хромосо¬
мой Зц13-22 и т.д. Эти примеры ясно показывают, что
для успеха ДНК-диагностики и практической реализа¬
ции геномного подхода к классификации наследствен¬
ных заболеваний нервной системы чрезвычайно важным
является тесное сотрудничество клинициста-невролога
и врача лаборатории, осуществляющего мутационный
скрининг. Квалифицированный клинико-генеалогичес¬
120Глава 2кий анализ синдрома является краеугольным камнем, на
котором основаны постановка показаний к проведению
ДНК-диагностики, отбор генов, подлежащих исследо¬
ванию, последовательность процедур мутационного
скрининга, а также адекватная трактовка получаемых
результатов (рис. 31).Рис. 31. Общий алгоритм ДНК-диагностики
Проблема генетической гетерогенности ... 121В наиболее полном виде геномная номенклатура
наследственных заболеваний нервной системы представ¬
лена в Приложении 1.Важно подчеркнуть, что вопрос геномной клас¬
сификации наследственных болезней не носит чисто ака¬
демический характер, а имеет ярко выраженную прак¬
тическую направленность. Проведение ДНК-диагности¬
ки у лиц из группы риска, а также пренатальной ДНК-
диагностики с целью профилактики повторных случаев
заболевания в семье, может осуществляться только при
условии идентификации конкретной молекулярной фор¬
мы заболевания у пробанда. В будущем при разработке
эффективных методов лечения основных групп наслед¬
ственных заболеваний нервной системы (особенно с ис¬
пользованием методов генной инженерии) точный ДНК-
диагноз в каждом конкретном случае будет основным
условием для проведения такой специфической терапии.
Весьма показательным примером является болезнь
Фридрейха - аутосомно-рецессивная форма наследствен¬
ных атаксий, обусловленная повреждением гена фратак-
сина на хромосоме 9q 13. После внедрения в практику
прямой ДНК-диагностики было установлено, что целый
ряд редких атактических синдромов, заметно отличаю¬
щихся от «классической» болезни Фридрейха (например,
случаи с поздним началом, сохранностью сухожильных
рефлексов и т.д.), могут быть обусловлены «мягкими»
мутациями в гене фратаксина [Dürr A. et al., 1996; Илла-
риошкин С.Н. и соавт., 1999 (а)]. Это позволило суще¬
ственно расширить нозологические границы болезни
Фридрейха, пересмотреть существовавшие представле¬
ния о распространенности данного заболевания, изме¬
нить подходы к проведению медико-генетического кон¬
сультирования в группе семейных и спорадических атаксий.
122Глава 2Понятие генетической гетерогенности имеет весь¬
ма широкое содержание. Выше были представлены при¬
меры локусной гетерогенности наследственных заболе¬
ваний нервной системы. Локусная гетерогенность свя¬
зана с существованием различных генетических локу-
сов на хромосомах (т.е. различных генов), мутации в
которых могут приводить к развитию сходных синдро¬
мов. Большое значение имеет также другая разновид¬
ность генетической гетерогенности — аллельная гетеро¬
генность, при которой определенное наследственное
заболевание может вызываться различными мутациями
одного и того же гена (иными словами, у больных могут
обнаруживаться различные мутантные аллели одного
гена). Приведем несколько неврологических примеров,
демонстрирующих ключевую роль аллельной гетероген¬
ности в механизмах фенотипического полиморфизма
наследственных болезней нервной системы. Как уже
указывалось в главе 1, гепатолентикулярная дегенерация
может быть обусловлена большим числом (>100) раз¬
личных точковых мутаций гена медной АТФ-азы, обна¬
руживаемых практически в любых областях гена. При
этом одни мутации в данном гене приводят лишь к сни¬
жению функциональной активности фермента различ¬
ной степени, тогда как другие - к его полной инактива¬
ции. В результате тяжесть и характер неврологических
и соматических проявлений мутаций у разных больных
могут варьировать в весьма широких пределах, что и
является одним из факторов выраженного полиморфиз¬
ма клинической картины гепатолентикулярной дегене¬
рации (5 различных клинических форм болезни, по клас¬
сификации Н.В. Коновалова). Более ограниченный ха¬
рактер аллельной гетерогенности характерен для забо¬
леваний, обусловленных «динамическими мутациями»,
поскольку для каждого такого заболевания область му¬
Проблема генетической гетерогенности ... 123тации в гене строго постоянна (см. главу 1). Так, у всех
больных хореей Гентингтона имеет место экспансия три-
нуклеотидных повторов CAG в одном и том же участке
гена IT-15, однако длина патологически удлиненного
тринуклеотидного сегмента может быть различной (от
37 до >120 повторов); при этом у больных с тяжелой
степенью CAG-экспансии развивается ранний акинети-
ко-ригидный вариант Вестфаля, при числе триплетов 45-
55 имеет место классический вариант болезни, а при
минимальной степени генетического дефекта (37-40
триплетов) может наблюдаться позднее начало болезни
с минимально выраженными нарушениями психики.
Таким образом, различные аллели мутантного гена у
больных хореей Гентингтона отличаются лишь числом
тандемных тринуклеотидных повторов, однако и в дан¬
ном случае аллельная гетерогенность сопровождается
вариабельностью клинических проявлений болезни.Наличие аллельной гетерогенности нередко при¬
водит к тому, что у больных с аутосомно-рецессивными
заболеваниями каждая из двух мутантных хромосом со¬
держит разные мутации в анализируемом гене. Такое
состояние называется компаунд-гетерозиготностъю (от
англ. compound - «составной»). Во избежание некото¬
рой терминологической путаницы следует четко подчер¬
кнуть: такие больные являются гомозиготными носи¬
телями мутантного гена в том смысле, что обе копии
соответствующего гена повреждены; в то же время они
являются компаунд-гетерозиготами (компаундами) при¬
менительно к конкретным (разным) мутациям в каждом
из двух аллелей гена. «Истинная» гомозиготность при
аутосомно-рецессивных заболеваниях (т.е. наличие оди¬
наковых мутаций в обоих аллелях гена) обычно имеет
место у больных, родившихся от кровнородственного
брака и получивших от обоих родителей одну и ту же
124Глава 2мутантную хромосому, исторически унаследованную от
общего предка.Еще более сложной является ситуация, когда раз¬
личные мутации в одном гене могут приводить к разви¬
тию совершенно различных клинических фенотипов,
квалифицируемых как самостоятельные нозологические
формы. Ярким примером такой дивергенции является
ген нейрональной а]А-субъединицы потенциал-зависи-
мого кальциевого канала, расположенный на хромосоме
19р 13.1. Различные типы мутаций в данном гене ответ¬
ственны за развитие 3 самостоятельных форм аутосом-
но-доминантных наследственных заболеваний нервной
системы:- экспансия тринуклеотидных САС-повторов в
кодирующей области гена приводит к мани¬
фестации одной из форм доминантных атак¬
сий - спиноцеребеллярной атаксии 6-го типа
(СЦА6); при этом заболевание характеризу¬
ется развитием изолированной медленно про¬
грессирующей атаксии и атрофией верхних
отделов полушарий и червя мозжечка
рЬисЬепко О. е1 а1., 1997];- точковые нонсенс-мутации в данном гене с
преждевременным обрывом трансляции явля¬
ются причиной развития так называемой эпи¬
зодической атаксии 2-го типа (ЭА2); она ха¬
рактеризуется пароксизмальными кратковре¬
менными приступами атаксии, головокруже¬
ния, тошноты и диплопии, которые нередко
купируются ацетазоламидом [ОрЬо1Т Я. е! а1.,
1996; Тоигшег-Ьаззегуе Е., 1999].- наконец, точковые миссенс-мутации в этом же
гене, ведущие к замене аминокислоты в со¬
ставе белка и, предположительно, приобрете¬
Проблема генетической гетерогенности ... 125нию мутантным белком новой функции, яв¬
ляются причиной развития семейной гемип¬
легической мигрени [Carrera P. et al., 1999;
Tournier-Lasserve Е., 1999].Хотя указанные клинические формы имеют не¬
которые общие симптомы (атаксия у больных СЦА6 и
ЭА2, пароксизмальность клинических проявлений при
ЭА2 и семейной гемиплегической мигрени, наличие ат¬
рофии червя мозжечка при всех трех клинических фор¬
мах), данные фенотипы в клиническом смысле, несом¬
ненно, представляют собой самостоятельные заболева¬
ния. Эти заболевания являются аллельными, поскольку
они обусловлены различными мутантными аллелями
единого гена. Такая совокупность аллельных заболева¬
ний, различающихся по своим базовым фенотипическим
проявлениям но связанных с повреждением одного гена,
называется аллельная серия. Манифестация отличающих¬
ся друг от друга заболеваний в рамках каждой аллель¬
ной серии объясняется, наиболее вероятно, разными
функциональными последствиями соответствующих
мутаций (повреждение различных доменов белка, вари¬
абельные механизмы действия мутаций) и рядом других
молекулярных факторов (эффект генов-модификаторов,
особенности сцепленных гаплотипов и т.д.). Существо¬
вание аллельных серий является достаточно распрост¬
раненным феноменом в нейрогенетике, который суще¬
ственно расширяет наши представления о патогенезе
наследственных неврологических заболеваний, функци¬
онировании различных биохимических «цепей» нервной
системы в норме и при патологии, механизмах форми¬
рования конкретных фенотипов. В таблице 3 представ¬
лены некоторые наиболее значимые аллельные серии
наследственных заболеваний нервной системы.
126Глава 2Таблица ЗАллельные серии наследственных заболеваний
нервной системыМутантный ген и
белок (хромосома)Аллельные серии заболеванийа 1 д-субъединица
потенциал¬
зависимого
кальциевого канала
(19р13.1)■ спиноцеребеллярная атаксия 6 типа
(СЦА6);■ эпизодическая атаксия 2 типа;* семейная гемиплегическая мигреньдистрофии (Хр21.2)■ прогрессирующая мышечная дистрофия
Дюшенна;* прогрессирующая мышечная дистрофия
Бекера;• крампи с рецидивирующей миоглобинуриейРМР22 (17р11.2)■ моторно-сенсорная невропатия 1А;" наследственная невропатия спредрасположенностью к параличам от
сдавления;■ болезнь Дежерина-Соттаандрогенный
рецептор
(Хд11.2-д12)" спинально-бульбарная амиотрофия
Кеннеди;* синдром тестикулярной феминизацииГТФ-циклогидрол аз а
1 (14422)■ дофа-зависимая дистония;■ атипичная гиперфенилаланинемиядисферлин (2р13)* конечностно-поясная мышечная дистрофия
типа 2В;■ дистальная миопатия Миошиа-субъединица
мышечного
натриевого канала
(17я13.1—я13.3)■ гиперкалиемический периодический
паралич;■ периодический парамиотонический
паралич;■ врожденная парамиотония Эйленбурга;■ калий-индуцируемая и ацетазол-
чувствительная миотониядигидропиридиновыйрецепторкальциевого канала
С1Ч31-р32)" гипокалиемический периодический
паралич;■ злокачественная гипертермияпротеолипидный
белок (Xq22)■ болезнь Пелицеуса-Мерцбахера;■ Х-сцепленная спастическая параплегиябелок-прекурсор (5-
амилоида (2^21)■ семейная форма болезни Альцгеймера ;■ семейное церебральное кровоизлияние
(голландский тип)
Проблема генетической гетерогенности ... 127Таким образом, с точки зрения современных зна¬
ний, взаимодействие между генотипом и фенотипом но¬
сит весьма сложный, комплексный характер, что нахо¬
дит свое преломление в решении различных вопросов
диагностики и лечения наследственных заболеваний не¬
рвной системы. Указанное взаимодействие в обобщен¬
ном виде представлено на рис. 32.Прогресс в изучении молекулярных основ наслед¬
ственных заболеваний нервной системы позволяет груп¬
пировать их по ряду дополнительных признаков - в за¬
висимости от конкретных целей исследования. Так, весь¬
ма удобным является подразделение на группы болез¬
ней, характеризующихся определенным типом мутаций,
например: заболевания с нестабильными «динамичес¬
кими» мутациями (экспансия тринуклеотидных повто¬
ров); заболевания, обусловленные нарушением дозы гена;
заболевания с протяженными делениями митохондриаль¬
ной ДНК и т.д. Такая классификация весьма важна для
решения целого ряда теоретических и практических воп¬
росов, поскольку в рамках каждой выделенной группы
болезней имеется много общих клинических и патоге¬
нетических характеристик (реализация мутаций на кле¬
точном и биохимическом уровне, закономерности фено¬
типической экспрессии поврежденного гена, методы
ДНК-диагностики, подходы к лечению и профилактике).Более тонкая молекулярно-генетическая субклас¬
сификация предполагает подразделение заболевания на
фенотипы, соответствующие отдельным мутациям в изу¬
чаемом гене. Это имеет основное значение в тех случа¬
ях, когда носительство конкретных мутантных аллелей
гена достоверно ассоциировано с развитием специфи¬
ческих клинических синдромов (что в нейрогенетике
встречается не столь редко). Сущность такой «мутаци¬
онной» классификации можно проиллюстрировать на
примере семейных форм прионных болезней, обуслов-
Рис. 32. Генетическая гетерогенность наследственных заболеваний128 Глава 2
Проблема генетической гетерогенности ... 129ленных мутациями гена прионного белка на хромосоме
20р. Как известно, прионные болезни традиционно под¬
разделяются на болезнь Крейтцфельдта-Якоба, синдром
Герстманна-Штреусслера, фатальную семейную инсом-
нию и куру, однако существование большого числа «про¬
межуточных» клинико-морфологических вариантов су¬
щественно осложняет клиническую дифференциацию
конкретных синдромов данных заболеваний. На сегод¬
няшний день установлено, что отдельные мутации в гене
прионного белка чаще всего приводят к развитию впол¬
не определенных вариантов прионных болезней: напри¬
мер, мутация P102L ассоциирована с синдромом Герст-
манна-Штреусслера, мутация Е200К- с особым фено¬
типом болезни Крейтцфельдта-Якоба, мутация D178N- с фатальной семейной инсомнией [Goldgaber et al., 1989;
Collinge J., 1997; Windl O. et al., 1999]. С другой сторо¬
ны, описан выраженный полиморфизм прионных болез¬
ней, обусловленных одной и той же мутацией, что часто
не позволяет использовать даже у больных внутри еди¬
ной родословной какое-то одно традиционное клиничес¬
кое обозначение [Prusiner S., Hsiao K., 1994]. В связи с
этим высказывается предположение о том, что класси¬
фикация и номенклатура семейных форм прионных бо¬
лезней должны базироваться главным образом на иден¬
тификации конкретной мутации в обследуемой семье, а
не на описательных клинико-морфологических терми¬
нах [Prusiner S., Hsiao К., 1994; Collinge J., 1997; Windle О.
et al., 1999].Изучение специфической роли белковых про¬
дуктов соответствующих мутантных генов знаменует
собой появление фунщионалъного подхода к классифи¬
кации наследственных заболеваний нервной системы.
Данный подход раскрывает патогенетическую сущность
болезни и «точку приложения» патологического процес-
130Глава 2са при конкретных нозологических формах. В зависи¬
мости от функции мутантного белка изучаемые заболе¬
вания могут быть разделены на 2 большие группы:1) Энзимопатии и патогенетически близкие к
ним заболевания с повреждением транспортных белков.
Примерами являются: рецессивная форма дофа-зависи-
мой дистонии (ген тирозин-гидроксилазы), атаксия с де¬
фицитом витамина Е (ген транспортера а-токоферола),
болезнь Фридрейха (ген фратаксина - трансмембранно¬
го переносчика железа) и другие преимущественно ауто-
сомно-рецессивные болезни. Общим для данной груп¬
пы болезней является полное или частичное (ниже кри¬
тического порога) нарушение функциональной активно¬
сти мутантного белка, ведущее к соответствующим био¬
химическим и фенотипическим проявлениям.2) Заболевания, обусловленные повреждением
структурных белков клетки (компонентов клеточных
мембран, рецепторов, нейрофиламентов и т.д.). Эти за¬
болевания являются наиболее многочисленными в не¬
врологии и могут наследоваться в соответствии с раз¬
личными типам менделевской передачи.В рамках последней группы болезней формиру¬
ются все новые и новые подгруппы, объединяющие па¬
тогенетически сходные нозологические формы, что со¬
провождается постоянным обновлением и расширени¬
ем терминологического арсенала нейрогенетики. Так, в
самые последние годы было открыто существование осо¬
бого класса наследственных болезней нервной системы,
объединяемых новым термином каналопатии
(таблица 4). К ним относятся различные пароксизмаль¬
ные нервно-мышечные и центральные синдромы (семей¬
ные миотонии, периодический паралич, эпизодические
атаксии, наследственные формы эпилепсий и др.), обус¬
ловленные мутациями генов белковых субъединиц ион-
Проблема генетической гетерогенности ... 131Таблица 4Молекулярная характеристика группы
каналопатийЗаболеваниеТип вовлекаемого ионного
каналаХромосомная
локализация генаМышечные ионные
каналыМиотонии Томсена и Бекерахлорный канал7я35Врожденная парамиотония
Эйленбурганатриевый канал17я23Калий-индуцируемая /ацетазол-чувствительнаямиотониянатриевый канал17я23Периодический
парамиотонический параличнатриевый канал\lq22Гиперкалиемическая форма
периодического параличанатриевый канал17я23Врожденная миастениянатриевый канал2д, 17рГипокалиемическая форма
периодического параличакальциевый канал\ф2Злокачественная гипертермиякальциевый каналI д32, 19с]13.1Болезнь центрального стержнякальциевый канал113.1Нейрональные ионные
каналыДоброкачественные семейные
неонатальные судорогикалиевый канал8ц24, 20ц 13.3Эпизодическая атаксия
1-го типакалиевый канал12р 13Пароксизмальный хореоатетоз
со спастичностьюкалиевый канал (?)1рГенерализованная эпилепсия с
фебрильными судорогами
плюснатриевый канал2я23—31,
19я13.1Эпизодическая атаксия
2-го типакальциевый канал19ц 13.1Семейная гемиплегическая
мигренькальциевый канал113.1Спиноцеребеллярная атаксия
6-го типакальциевый канал19Ч13.1Лобная эпилепсия с ночными
пароксизмамикальциевый канал20я13.2-13.3Идиопатическая
генерализованная эпилепсиякальциевый канал2Ч22-231 Іаследственная
гиперэксплексияхлорный канал5ч32
132Глава 2ных каналов - натриевых, калиевых, кальциевых, хлор¬
ных [Bulman D., 1997; Jurkat-Rott К. et al., 1999]. Моле¬
кулярный анализ каналопатий исключительно важен для
установления тонких механизмов проницаемости и воз¬
будимости клеточных мембран, процессов секреции и
передачи клеточных сигналов. В качестве других при¬
меров функциональной классификации можно назвать
объединение ряда форм прогрессирующих мышечных
дистрофий в группу саркогликанопатий (заболеваний,
обусловленных мутациями саркогликанов - белков мы¬
шечной сарколеммы) [Bushby К., 1999], выделение в
рамках нейродегенеративных заболеваний особых групп
синуклеинопатий и таупатий (заболеваний, обусловлен¬
ных патологией а-синуклеин-ассоциированных или
тау-ассоциированных белков) [Hardy J., Hardy-Gwinn К.,1998] и т.д. Важно подчеркнуть, что именно функцио¬
нальный подход к систематизации наследственных не¬
врологических заболеваний служит основой для разра¬
ботки соответствующих патогенетических методов ле¬
чения.Таким образом, в настоящее время, на рубеже
столетий, «остов» наиболее совершенной классифика¬
ции наследственных заболеваний нервной системы, от¬
ражающей молекулярные основы патологического про¬
цесса, полностью сформирован. Центральным звеном
этой классификации является идентификация первично¬
го молекулярного дефекта (гена, белка и конкретной
мутации) у обследуемых больных. При этом генетичес¬
кая характеристика изучаемого синдрома может допол¬
няться рядом ключевых клинико- генеалогических при¬
знаков. Такое построение классификации представляет¬
ся наиболее оптимальным: во-первых, оно соответству¬
ет нуждам повседневной практики, обеспечивая точность
диагностики и предусматривая активную роль клиници¬
Проблема генетической гетерогенности ... 133ста в процессе диагностического поиска; во-вторых, оно
ориентировано на фундаментальные исследования, име¬
ющие целью разработку патогенетических и этиотроп-
ных методов лечения наследственных заболеваний не¬
рвной системы.
ГЛАВА 3ДНК-ДИАГНОСТИКА
НАСЛЕДСТВЕННЫХ
МОНОГЕННЫХ БОЛЕЗНЕЙ
НЕРВНОЙ СИСТЕМЫ3.1. НАСЛЕДСТВЕННЫЕ НЕРВНО-
МЫШЕЧНЫЕ БОЛЕЗНИНервно-мышечные болезни занимают первое ме¬
сто по частоте среди всех наследственных моногенных
неврологических заболеваний. По данным A. Emery
(1991), суммарная распространенность всех наследствен¬
ных нервно-мышечных болезней составляет 250-300 х
10~6 (т.е. приблизительно 1 случай на 3500 населения).
Принимая во внимание тот факт, что для основной час¬
ти заболеваний из данной группы характерно неуклон¬
но-прогрессирующее течение и отсутствие эффективных
методов лечения, нервно-мышечные болезни следует
признать одной из наиболее актуальных проблем кли¬
нической неврологии. Профилактика повторных случа¬
ев нервно-мышечных болезней в семьях «высокого рис¬
ка» является на сегодняшний день единственным эффек¬
тивным средством борьбы с этими тяжелыми и нередко
фатальными недугами, при этом центральное место к
системе профилактических мероприятий занимает ДНК
диагностика.3.1.1. Прогрессирующие мышечные дистрофииПрогрессирующие мышечные дистрофии (ПМД)
представляют собой клинически и генетически гетеро¬
ДНК- диагностика наследственных болезней ... 135генную группу наследственных заболеваний, характери¬
зующихся первичным поражением скелетной мускула¬
туры невоспалительного характера. Распространенность
данных заболеваний весьма высока - около 200 случаев
на 1 млн. населения [Emery А., 1991]. Общей характери¬
стикой ПМД является развитие нарастающих мышечных
атрофий и парезов вследствие прогрессирующей деге¬
нерации миоцитов, обусловленной поражением струк¬
турных белков сарколеммы или ключевых ферментов
скелетных мышц. Прогресс в изучении данной группы
заболеваний связан с раскрытием в последние годы
структурных и молекулярных основ, регулирующих фун¬
кционирование мышечного волокна [Иллариошкин С.Н.,
Иванова-Смоленская И.А., 1998; Campbell K., 1995;
Worton R., 1995; Emery A., 1998].В настоящее время клиническая классификация
11МД базируется, главным образом, на характере распре¬
деления мышечных атрофий и парезов (конечностно-
поясные, дистальные, лице-лопаточно-плечевые, оку-
лофарингеальные ПМД), который дополняется типом
наследования болезни (аутосомные и Х-сцепленные фор¬
мы). Более детальная классификация в рамках данных
групп ПМД предполагает идентификацию первичного
молекулярного дефекта, лежащего в основе болезни.*. 1.1.1. Х-сцепленные ПМД Дюшенна и Бекера
(дистрофинопатии)Прогрессирующие мышечные дистрофии Дюшен-
ма и Бекера относятся к самым частым формам мышеч¬
ных дистрофий (соответственно, 1 на 3500 и 1 на 20 000
рожденных мальчиков) и наследуются по Х-сцепленно-
чу рецессивному типу [Emery А., 1991]. Форма Дюшен-
пл начинается в возрасте 3-6 лет со слабости мышц та-
юного пояса и проксимальных отделов ног, нередко со¬
136Глава 3провождающейся псевдогипертрофиями икроножных,
ягодичных, дельтовидных и других мышц, кардиомио-
патией. В дальнейшем характерна быстрая генерализа¬
ция парезов и атрофий, нарастающая обездвиженность,
развитие контрактур и дыхательных нарушений; гибель
больных наступает обычно на 2-3-м десятилетии жизни
[Темин П.А. и др., 1997; Emery A., 1993; Specht L.,
Kunkel L., 1993]. Форма Бекера традиционно рассмат¬
ривается как «мягкий» клинический вариант ПМД Дю-
шенна с более поздним началом болезни (в 12-15 лет),
относительно доброкачественным течением и сохранно¬
стью способности к самостоятельной ходьбе на протя¬
жении 15-20 лет от момента появления первых симпто¬
мов [Темин П.А. и соавт., 1997]. Женщины, являющие¬
ся гетерозиготными носительницами мутантного гена,
как правило, остаются клинически здоровыми, однако
иногда у них могут наблюдаться отдельные субклини-
ческие проявления носительства мутации - умеренно вы¬
раженная мышечная слабость, увеличение объема икро¬
ножных мышц, высокий уровень в крови мышечного
фермента креатинфосфокиназы [Emery A., 1993;
Specht L., Kunkel L., 1993].ПМД Дюшенна и Бекера являются аллельными
заболеваниями и обусловлены мутациями одного гена в
хромосомном локусе Хр21 [Goodfellow Р. et al., 1985;
Monaco A. et al., 1986; Koenig M. et al., 1987]. Данный
ген является самым большим из известных на сегодня
генов человека и имеет весьма сложную молекулярную
организацию: он содержит, как минимум, 5 промоторов,
свыше 80 экзонов, состоит из 24 ООО кб и кодирует бе¬
лок с молекулярной массой 427 килодальтон, получив¬
ший название «дистрофии» [Koenig М. et al., 1987;
Hoffman E. et al., 1992]. В норме в мышечном волокне
дистрофии локализуется на цитоплазматической
ДНК- диагностика наследственных болезней .137поверхности сарколеммы, являясь важной составной
частью цитоскелета и обеспечивая связь между
актиновыми филаментами (т.е. сократительным
аппаратом мышечного волокна) и сарколеммой [Ahn A.,
Kunkel L., 1993; Campbell K., 1995; HoffmanE., Kunkel L.,
1989]. Известна также изоформа дистрофина,
экспрессирующаяся в центральной нервной системе
[Tinsley J. et ah, 1994]; с отсутствием данной изоформы
белка в мозге может быть связана умственная отсталость,
имеющая место у 1/3 больных ПМД Дюшенна.Приблизительно 55-65% всех случаев ПМД Дю-
шенна/Бекера обусловлены делециями гена дистрофина
различной протяженности, 5-10% случаев - дуплика¬
циями части гена, у остальных больных имеют место
точковые мутации [Forrest S. et ah, 1988; Hu X. et ah,
1988; Koenig M. et ah, 1989]. Делении в гене дистрофина
распределяются отнюдь не равномерно по его длине, а
преимущественно группируются вокруг двух областей
гена, образуя так называемые «горячие точки» делеций- в 5’-области гена (экзоны 2-20) и в его дистальной
части в области экзонов 44-53 [Koenig М. et ah, 1987;
Forrest S. et ah, 1988]. Интересно отметить, что прокси¬
мальные делеции гена чаще выявляются при семейных
формах болезни, тогда как дистальные делеции обычно
ассоциированы со спорадическими случаями (т.е. воз¬
никшими как результат новой мутации); при выявлении
проксимальной делеции гена повторный риск заболева¬
ния в семье почти на порядок выше, чем при дисталь¬
ной (соответственно, 30% и 4%) [Passos-Bueno М. et ah,1992].Открытие генетического дефекта при ПМД Дю¬
шенна и Бекера дало возможность с молекулярных по-
чиций объяснить причину различий в клинической кар¬
тине этих форм миопатий. Тяжелая форма Дюшенна раз¬
138Глава 3вивается обычно при наличии мутаций, повреждающих
рамку считывания кодирующей области гена либо нару¬
шающих структуру функционально значимых доменов
дистрофина [Hoffman Е. et al., 1988; Monaco A. et al.,
1988; Koenig M. et al., 1989]. Результатом таких мутаций
является грубое нарушение синтеза или функции дист¬
рофина. Напротив, у больных миопатией Бекера имеют
место, как правило, внутренние делеции или дуплика¬
ции гена, не приводящие к сдвигу рамки считывания
кодонов; вследствие этого синтезируется измененный,
«усеченный» дистрофии, сохраняющий частичную фун¬
кциональную активность [Hoffman Е. et al., 1988;
Monaco A. et al., 1988]. He случайно у больных ПМД
Дюшенна отмечается полное отсутствие дистрофина при
иммуногистохимическом исследовании мышечных био-
птатов с помощью антидистрофиновых антител, тогда
как при миопатии Бекера в препарате выявляются не¬
большие дистрофин-позитивные участки (рис. 33)
[Hoffman Е. et al., 1988]. Указанная теория «рамки счи¬
тывания», объясняющая характер клинической картины
в зависимости от типа мутации, подтверждается в абсо¬
лютном большинстве (92%) всех случаев ПМД Дюшен¬
на и Беккера [Koenig М. et al., 1989]. В зависимости от
характера молекулярного дефекта могут наблюдаться
клинически и иммуногистохимически «промежуточ¬
ные» варианты болезни [Tinsley J. et al., 1994]; описан
также ряд необычных мышечных синдромов, обуслов¬
ленных мутациями гена дистрофина - крампи с рециди¬
вирующей миоглобинурией и сниженной толерантнос¬
тью к физической нагрузке, изолированная кардиомио-
патия, доброкачественная миопатия позднего возраста
[Doriguzzi С. et al., 1997; Palmucci L. et al., 2000]. В связи
с молекулярным единством ПМД Дюшенна, ПМД Беке¬
ра и указанных выше атипичных вариантов миопатий в
ДНК- диагностика наследственных болезней... 139литературе в последнее время принято объединять эти
формы общим термином дистрофинопатии [Griggs R.,
Fischbeck К., 1992].АБВРис. 33. Иммуногистохимический анализ мышечных био-
птатов на дистрофии у больных ПМД Дюшенна/Бекера
А. Контроль. Б. ПМД Бекера (на фоне нарушения нормальной ок¬
раски мышечных волокон на дистрофии выявляются отдельные
дистрофин-позитивные участки). В. ПМД Дюшенна (полное отсут¬
ствие дистрофин-позитивных мышечных волокон).В редких случаях заболевание в своем разверну¬
том виде может манифестировать у женщин, являющихся
I етерозиготными носительницами мутантного гена; это
140Глава 3наблюдается при хромосомных транслокациях и пере¬
стройках, затрагивающих критический хромосомный
сегмент Хр21, при синдроме Тернера (генотип ХО), а
также при несбалансированной инактивации Х-хромо-
сом в раннем эмбриогенезе [Griggs R., Fischbeck K., 1992;
Specht L., Kunkel L., 1993]. Во всех указанных случаях
наличие мутантного гена на одной из Х-хромосом не
компенсируется нормальным геном гомологичной хро¬
мосомы, что и приводит к появлению типичных симп¬
томов болезни у женщин.«Золотым стандартом» при постановке диагноза
дистрофинопатий является исследование мышечной тка¬
ни на дистрофии (иммуногистохимический анализ, им-
муноблоттинг) (рис. 33). Этот метод, однако, весьма до¬
рог, технически сложен и требует проведения биопсии
мышцы. Поэтому в настоящее время широко применя¬
ется неинвазивная диагностика ПМД Дюшенна/Бекера
с использованием различных молекулярно-генетических
подходов на клетках крови. Наиболее простым методом
ДНК-диагностики дистрофинопатий является так назы¬
ваемая мультиплексная (мультипраймерная) ПЦР
[Chamberlain J. et al., 1988]. Она заключается в одновре¬
менной амплификации набора наиболее часто мутирую¬
щих экзонов гена дистрофина; при электрофорезе про¬
дуктов реакции отсутствие одного или нескольких экзо¬
нов будет свидетельствовать о делеции, т.е. служить мо¬
лекулярным подтверждением диагноза [Chamberlain J.,
et al., 1988; Multicenter Study Group, 1992]. Комбиниро¬
ванное использование нескольких мультиплексных ре¬
акций позволяет диагностировать до 98% всех делеций,
имеющих место у больных ПМД Дюшенна/Бекера
[Beggs A. et al., 1990]. На рис. 34 показан пример пря¬
мой ДНК-диагностики мышечной дистрофии Дюшенна
при мультиплексной ПЦР (выявление различных деле¬
ций в гене дистрофина).
ДНК- диагностика наследственных болезней... 1411 2 3 4 5 45 1 9 1 7•< 44Рис. 34. Прямая ДНК-диагиостика мышечной дистрофии
Дюшенна с помощью мультиплексной полимеразной цеп¬
ной реакцииУ каждого из обследуемых лиц одновременно амплифицированы 4
экзона гена дистрофина (экзоны 17,19,44 и 45; стрелки указывают
на соответствующие продукты амплификации). Дорожка 1 - конт¬
роль, дорожки 2-5 - больные мышечной дистрофией Дюшенна с
различными делениями гена дистрофина (дорожки 2 и 5 -делеция
экзона 45, дорожка 3 - делеция экзона 44, дорожка 4 - делеция экзо-
нов 17 и 19).Для диагностики носительства мелких или ред¬
ких делеций в гене дистрофина, а также при наличии
других типов мутаций (вставки и дупликации, точковые
мутации) могут применяться более сложные методы
ДНК-анализа, такие как Саузерн-блоттинг с использо¬
ванием кДНК-зондов (см. рис. 27 на стр. 92), SSCP- и
гетеродуплексный анализ отдельных экзонов гена, раз¬
личные методы исследования кДНК, полученной из лим¬
фоцитов или мышечных биоптатов с помощью обратно-
транскриптазной ПЦР, а также ряд других подходов
[Darras В. et al., 1988; Forrest S. et al., 1988; Roberts R. et
142Глава 3al., 1992; Prior T. et al., 1994; Tubiello G. et al., 1995; van
Essen A., 1997]. Важно отметить, что указанные ДНК-
методы не только значительно повышают процент вы¬
являемое™ мутаций в гене дистрофина у обследуемых
больных, но и позволяют осуществлять диагностику
мутаций у женщин-носительниц (последнее невозмож¬
но при проведении мультиплексной ПЦР, поскольку у
женщин-носительниц делеция на мутантной хромосоме
«маскируется» наличием нормального экзона, амплифи-
цируемого с другой Х-хромосомы).В 25-30% всех случаев ПМД Дюшенна/Бекера у
больного не удается выявить доступными методами
повреждений в гене дистрофина, но при этом имеется
необходимость установления генетического статуса как
лиц из группы риска в обследуемой семье (сибсы мужс¬
кого пола, родственники больного по женской линии),
так и плода у женщин-носительниц (для решения воп¬
роса о сохранении или прерывании беременности). В
таких ситуациях может использоваться косвенная ДНК-
диагностика, позволяющая проследить наследование
мутантной хромосомы в изучаемой семье. Для этой про¬
цедуры необходимо наличие образцов ДНК либо заве¬
домо больного ребенка, либо заведомо здоровых брать¬
ев пробанда. Поскольку для гена дистрофина характер¬
на высокая частота внутригенных рекомбинаций, при
проведении косвенной ДНК-диагностики ПМД Дюшен¬
на/Бекера используются обычно несколько маркеров,
расположенных в проксимальной, дистальной и цент¬
ральной частях гена либо тесно сцепленных с геном и
фланкирующих его с теломерного и центромерного кон¬
ца [Clemens Р. et al., 1991; Feener C. et al., 1991]. На рис.
35 показан результат косвенной ДНК-диагностики ПМД
Дюшенна у сибсов больного ребенка в большой семье.
ДНК- диагностика наследственных болезней ... 143©-и■{Ц1-2Л сЬ© сЬ сЬ сЬ11-1 П-2 Н-З П-4 П-5 П-6 П-7-216П.О.
• 166 п.о.■ 50 п.о.Рис. 35. Косвенная ДНК-диагностика мышечной дистрофии
ДюшеннаИсследован локус рЕКГ 87-15 (рестрикция ферментом Ват Н1).
Мутантный материнский аллель визуализируется после рестрикции
в виде двух фрагментов ДНК длиной 166 и 50 п.о. (жирные стрел¬
ки), нормальный материнский аллель - в виде фрагмента длиной
216 п.о. (тонкая стрелка). Дочери П-З, П-4, П-6 и П-7 унаследовали
от матери мутантный аллель маркера и являются носителями му¬
тантной хромосомы, дочь П-5 унаследовала от матери нормальный
аллель маркера (носительство мутантной хромосомы исключено).
Вероятность ошибки, связанной с возможной рекомбинацией меж¬
ду маркерным локусом и геном дистрофина- около 2%.Серьезной проблемой при проведении ДНК-ди-
агностики в семьях с ПМД Дюшенна/Бекера является
чрезвычайно высокая частота спонтанных мутаций
(10'4 на поколение), что частично может объясняться его
144Глава 3гигантским размером [Tinsley J. et al., 1994]. Предпола¬
гается, что около трети всех случаев ПМД Дюшенна/
Бекера обусловлены мутациями de novo [Darras В. et al,1988]. Возникновение новых мутаций в гене дистрофи-
на может происходить на любых этапах гамето- и онто¬
генеза, в результате чего единая клетка-предшественник
дает начало смешанной популяции нормальных и мутан¬
тных клеток (феномен соматического и гонадного мо-
заицизма). Возможность соматического и гонадного
мозаицизма существенно видоизменяет расчеты генети¬
ческого риска при проведении медико-генетическом кон¬
сультирования у членов семьи, относящихся к группе
риска (братья-сестры пробанда, родственники по женс¬
кой линии). Более подробно этот вопрос будет рассмот¬
рен в главе 5.3.1.1.2. Аутосомные формы конечностно-поясных
ПМДАутосомные формы конечностно-поясных ПМД
(в старой номенклатуре - ПМД Эрба-Рота) являются
относительно редкими, их суммарная распространен¬
ность составляет приблизительно 50 случаев на милли¬
он населения [Emery A., 1991].Клиническая картина конечностно-поясной ПМД
(КПМД) характеризуется началом болезни с мышц та¬
зового пояса и бедер, с последующим вовлечением мус¬
кулатуры плечевого пояса и проксимальных отделов рук
и постепенной генерализацией процесса [Shields R., 1994;
van der Kooi A. et al., 1994]. Диагноз КПМД может быть
поставлен только после исключения многочисленных
гено- и фенокопий, таких как митохондриальные, вос¬
палительные, эндокринные, метаболические миопатии,
дистрофинопатии, спинальные амиотрофии и др.
[Bushby K., 1995]. КПМД может передаваться как по
ДНК- диагностика наследственных болезней ... 145аутосомно-доминантному, так и по аутосомно-рецессив-
ному типу (соответственно, КПМД1 и КПМД2). Значи¬
тельно более частым является аутосомно-рецессивное на¬
следование, при котором заболевание нередко проявля¬
ется в семье как единичный (спорадический) случай.Аутосомно-рецессивные КПМД. Конечностно-
поясной вариант мышечной дистрофии с аутосомно-ре-
цессивным типом наследования характеризуется выра¬
женной генетической гетерогенностью. К настоящему
времени известно сущестование, как минимум, 9 локу-
сов данных заболеваний, причем для 7 форм идентифи¬
цированы гены и их белковые продукты [ИллариошкинС.Н., Иванова-Смоленская И.А., 1998; Weiler Т. et al.,
1998; Bushby K., 1999; Driss A. et al., 2000]. В соответ¬
ствии с современной геномной классификацией, ауто-
сомно-рецессивные мышечные дистрофии (КПМД2) и
соответствующие хромосомные локусы обозначаются
буквенными символами в порядке их открытия -
КПМД2^4, 2В, 2С, 2D и т.д. (см. приложение 1). Знание
первичных молекулярных дефектов при большинстве
форм аутосомно-рецессивных КПМД позволяет подраз¬
делить их на 3 большие группы: саркогликанопатии,
каньпаинопатии и дисферлинопатии.К саркогликанопатиям относятся мышечные ди¬
строфии, обусловленные мутациями в генах особых
с труктурных белков мышечной мембраны - саркоглика-
иов [Tinsley J. etal., 1994; Ozawa E. etal., 1995; Worton R.,
1995]. Саркогликаны являются элементами сложного
оелкового комплекса мышечного волокна, состоящего из
/щстрофина и дистрофин-ассоциированных протеинов
саркогликанов, дистрогликанов, синтрофинов, дист-
робревина, мерозина и др. (рис. 36).Различные компоненты данного белкового комп¬
лекса (обозначаемого как дистрофин-гликопротеиновый
146 Глава З
ДНК- диагностика наследственных болезней ... 147комплекс) образуют единую «ось», обеспечивающую
эластичную связь между филаментами актина, сарколем¬
мой и внеклеточным матриксом. Считается, что такая
структурно-функциональная организация дистрофин-
гликопротеинового комплекса способствует фиксации
миоцита к элементам внеклеточного матрикса и стаби¬
лизирует мышечную мембрану, предохраняя ее от по¬
вреждения в процессе мышечных сокращений
[Campbell K., 1995; Ozawa E. et al., 1995]. Ключевое зна¬
чение в стабилизации всего дистрофин-гликопротеино-
вого комплекса придается дистрофину, поэтому у боль¬
ных ПМД Дюшенна/Бекера имеют место вторичные из¬
менения дистро- и саркогликанов мышечной мембраны
[Tinsley J. et al., 1994]. Первичные саркогликанопатии
связаны непосредственно с патологией этих белков, тог¬
да как уровень и локализация дистрофина в мышечном
волокне могут оставаться нормальными. Известны 4
формы аутосомно-рецессивных КПМД, относящихся к
первичным саркогликанопатиям: 1) КПМД2С, обуслов¬
ленная мутациями гена у-саркогликана на хромосоме
13q 12 [Noguchi S. et al., 1995]; 2) КПМД2Д обусловлен¬
ная мутациями гена а-саркогликана (адгалина) на хро¬
мосоме 17ql2—21.33 [Roberds S. et al, 1994]; 3) КПМД2Е,
вызываемая мутациями гена ß-саркогликана на хромо¬
соме 4q 12 [Lim L. et al., 1995]; 4) КПМД27% связанная с
мутациями гена 5-саркогликана на хромосоме 5q33-34
[Nigro V. et al., 1996]. При всех формах саркогликанопа-
тий отмечается конечиостно-поясной тип распределения
парезов скелетных мышц, тогда как тяжесть клиничес¬
кой картины определяется характером молекулярного
дефекта. При миссенс-мутациях с заменой аминокисло¬
ты заболеванию обычно свойственно позднее начало (на
2-3-м десятилетии жизни) и сравнительно благоприят¬
ное течение [Lim L. et al., 1995; Piccolo F. et al., 1995];
напротив, при дефектах, грубо нарушающих синтез бел¬
148Глава 3ка (делеции со сдвигом рамки, нонсенс-мутации), имеет
место ранний дебют заболевания и быстрое прогресси¬
рование - так называемая «тяжелая детская аутосомно-
рецессивная мышечная дистрофия», или «дюшенно-по-
добная мышечная дистрофия» [Campbell K., 1995; Lim L.
et al., 1995; Piccolo F. et al., 1995]. Клинически диффе¬
ренциальная диагностика саркогликанопатий с ПМД
Дюшенна/Бекера как правило представляет собой весь¬
ма непростую задачу, особенно учитывая тот факт, что
для саркогликанопатий достаточно характерны задерж¬
ка раннего двигательного развития, псевдогипертрофии
икроножных мышц, в некоторых случаях - кардиомио-
патия [Bushby К., 1999].К калъпаинопатии относится форма КПМД2^4,
обусловленная мутациями в гене на хромосоме 15ql5,
кодирующем синтез мышечно-специфичного фермента
кальпаина-3 [Richard I. et al., 1995]. Данный фермент
принадлежит к семейству внелизосомных внутриклеточ¬
ных цистеиновых протеаз; предположительно, его фун¬
кции могут быть связаны с регуляцией процессов диф-
ференцировки мышечной ткани и влиянием на экспрес¬
сию факторов транскрипции [Wang К. et al., 1989]. В
отличие от саркогликанопатий, для КПМД2А характер¬
на тенденция к более позднему началу болезни и более
медленному прогрессированию, нормальное физическое
развитие в раннем детстве, длительно наблюдающееся
преобладание слабости в мышцах-аддукторах бедра по
сравнению с абдукторами, отсутствие мышечных псев¬
догипертрофий и кардиомиопатии [Bushby K., 1999]. Все
эти признаки не могут, однако, служить абсолютно на¬
дежной основой для клинической диагностики данной
формы КПМД.К дисферлинопатиям относится большое число
аллельных вариантов аутосомно-рецессивной ПМД,
ДНК- диагностика наследственных болезней ... 149обусловленных мутациями в гене дисферлина на хромо¬
соме 2р 13 [Bashir R. et al., 1994; 1998; Bejaoui K. et al.,
1995; Passos-Bueno M. et al., 1995; Liu J. et al., 1998;
Weiler T. et al., 1999]. Дисферлин представляет собой но¬
вый белок, ассоциированный с мышечной мембраной,
функции которого окончательно не установлены; выс¬
казывается предположение, что дисферлин может уча¬
ствовать в процессах регуляции слияния миобластов
[Bashir R. et al., 1998; Liu J. et al., 1998]. Для дисферли-
нопатий характерен чрезвычайно широкий фенотипичес¬
кий полиморфизм. Чаще всего они манифестируют в
виде конечностно-поясного варианта мышечной дистро¬
фии (КПМД25), особенностями которого являются нор¬
мальное раннее развитие, начало болезни в конце 2-го
десятилетия жизни, относительно «мягкое» течение, пре¬
обладание слабости в задних группах мышц ног (осо¬
бенно при распространении процесса дистально), чрез¬
вычайно высокий уровень сывороточной креатинфосфо-
киназы (>50-100 раз выше нормы) [Passos-Bueno М. et
al., 1995; Bushby K., 1999]. Аллельными вариантами дис-
ферлинопатий являются также некоторые редкие фор¬
мы дистальных ПМД - миопатия Миоши (характеризу¬
ющаяся слабостью и атрофией дистальных отделов ног,
преимущественно - икроножной мышцы) и тибиальная
дистальная миопатия [Miyoshi K. et al., 1986; Bashir R. et
al., 1998; Liu J. et al., 1998]. Более того, в литературе пред¬
ставлены наблюдения двух уникальных инбредных ро¬
дословных (одна из них описана нами в дагестанском
горном изоляте, вторая - канадскими исследователями
в изоляте индейцев Манитобы), в которых у родствен¬
ников с идентичными мутациями в гене дисферлина
имела место либо КПМД2Б, либо миопатия Миоши
|Illarioshkin S. et al., 1996; Weiler T. et al., 1996; 1999].
Данный пример показывает, что отнесение различных
150Глава 3фенотипических вариантов ПМД к группе дисферлино-
патий только по клиническим данным невозможно и
должно основываться на молекулярных методах иссле¬
дования.Совсем недавно, в 2000 году, был идентифициро¬
ван еще один генетический дефект в группе аутосомно-
рецессивных конечностно-поясных мышечных дистро¬
фий, ответственный за развитие KI\WIJ\2G\ данный мо¬
лекулярный вариант болезни обусловлен мутациями в
гене саркомерного белка телетонина (хромосома 17ql 1—12)
[Moreira Е. et al., 2000].Ведущее значение в дифференциальной диагнос¬
тике аутосомно-рецессивных КПМД имеет определение
типа наследования болезни, а также иммуногистохими-
ческий анализ биоптатов мышц с использованием анти¬
тел к соответствующим белкам. У больных с саркогли-
канопатиями окраска на дистрофии остается нормаль¬
ной; в то же время дефект любого из белков-саркоглика-
нов обычно приводит к дезинтеграции всего саркогли-
канового комплекса и нарушению нормального окраши¬
вания препарата при использовании антисаркогликано-
вых антител (рис. 37) [Ozawa Е. et al., 1995]. Аналогич¬
ным образом другие молекулярные формы аутосомно-
рецессивных мышечных дистрофий диагностируются с
помощью выявления специфического белкового дефек¬
та кальпаина-3 или дисферлина в мышечном препарате
[Anderson L. et al., 1998; Matsuda С. et al., 1999]. После
того как иммуногистохимический анализ позволил оп¬
ределить потенциальный ген-кандидат, может ставить¬
ся вопрос о проведении мутационного скрининга коди¬
рующей области данного гена, обнаружении конкретных
мутаций и проведении прямой ДНК-диагностики у лиц
из группы риска [Beckmann J., Bushby К., 1996; DugganD. et al., 1997; Bushby K., 1999]. Принимая во внимание
ДНК- диагностика наследственных болезней... 151большие размеры генов аутосомно-рецессивных КПМД
и разнообразие описанных мутаций, прямая ДНК-диаг-
носгика этих заболеваний до сегодняшнего времени пред¬
ставляет серьезные трудности и может проводиться лишь
в небольшом числе высокоспециализированных лабора¬
торий.АБРис. 37. Иммуногистохимический анализ мышечных био-
I ітатов на а-саркогликан у больного КГІМД2/)Д. Контроль. Б. Конечностно-поясная мышечная дистрофия типа
20 (нарушение нормальной окраски мышечных волокон на а -сар-
когликан).
152Глава 3uСТСТ GAGCT ТСАТ GT G G Т G G T CA A110 1?0 1ЗЙi/VwWWVwifWWvviW'лу:UCTCTGAGCTTCATGATGTGGT CA A
110 170 1^0Рис.38. Прямая ДНК-диагностика дисферлинопатии в семье
с КПМД2В и миопатией МиошиА. Точковая мутация TG573/574AT в гомозиготном состоянии (фраг¬
мент нуклеотидной последовательности экзона 3 гена дисферлина).
Мутантные нуклеотиды указаны стрелками. Б. Рестрикционный
анализ продуктов амплификации экзона 3 с использованием фер¬
мента Rca I. Мутация TG573/574AT создает новый сайт рестрикции
для Rca I. Нормальный аллель имеет размер 250 п.о., мутантный
аллель разрезается на фрагменты 160 и 90 п.о. Дорожка 1 - конт¬
роль, дорожка 2-гетерозиготный носитель мутации 573-574TG->AT,
дорожка 3 - гомозиготный носитель мутации 573-574TG—>АТ
(больной).В нашей стране впервые прямая ДНК-диагностика от¬
дельных случаев аутосомно-рецессивных КПМД была
ДНК- диагностика наследственных болезней ... 153проведена двумя группами исследователей - нейрогене-
тическим отделением НИИ неврологии РАМН в семье с
КПМД25 и миопатией Миоши (рис. 38) и сотрудниками
Медико-генетического научного центра РАМН и Инсти¬
тута молекулярной генетики РАН в семьях с КПМД2А
[Липатова H.JI. и др., 2000]. В достаточно больших ин¬
формативных семьях предпочтительным подходом яв¬
ляется косвенная ДНК-диагностика болезни (анализ ге¬
нетического сцепления с одним из локусов на хромосо¬
мах 2, 4, 5, 9, 13, 15, 17 и 19) [Beckmann J., Bushby K.,
1996; Weiler T. et al., 1998; Bushby K., 1999].Комбинированное использование методов белко¬
вой и генной диагностики позволило оценить сравни¬
тельную частоту конкретных форм аутосомно-рецессив-
ных мышечных дистрофий: в большинстве изученных
популяций мира не менее 40% больных с КПМД имеют
первичную патологию кальпаина-3, около 10-25% боль¬
ных страдают дисферлинопатиями и различными вари¬
антами саркогликанопатий (из них чаще всего встреча¬
ется КПМД2Д связанная с патологией а-саркогликана),
тогда как все остальные формы аутосомно-рецессивных
КПМД являются более редкими [Bushby K., 1999]. В
отдельных популяциях эти цифры могут существенно
различаться, причем знание о преобладании в той или
иной популяции определенной молекулярной формы
мышечной дистрофии (и даже определенной мутации)
позволяет значительно упростить идентификацию по¬
вреждений в гене и проведение ДНК-диагностики в се¬
мьях соответствующего этнического происхождения.Аутосомно-доминантные КПМД. Семьи с ауто-
сомно-доминантным наследованием являются весьма
редкими среди всех случаев КПМД (не более 10%)
[Bushby K., 1999]. Общими клиническими особеннос¬
тями данных заболеваний являются достаточно выражен-
154Глава Зная вариабельность возраста начала болезни, незначи¬
тельное повышение уровня сывороточной креатинфос-
фокиназы (обычно в 4-6 раз, нередко может быть в нор¬
ме), а также сравнительно медленное прогрессирование.
Важно отметить, что примерно у 8% пациентов с кли¬
ническим диагнозом аутосомно-доминантной КПМД
может выявляться генетический дефект, характерный для
лице-лопаточно-плечевой мышечной дистрофии (см.
раздел 3.1.1.3), поэтому исключение последней формы
ПМД с помощью ДНК-диагностики должно быть пер¬
вым шагом при обследовании таких больных [van der
Kooi A. et al., 1994].На сегодня известны 5 различных генетических
локусов аутосомно-доминантных КПМД - на хромосо¬
мах 5q (форма КПМД7Л), lqll-21 (КПМДІЯ), Зр25
(КПМД і С), 6q22 (КПМД 7Я) и 7q (КПМД /А’) [Speer М.
et al., 1999; Messina D. et al., 1997; Minetti C. et al., 1998;
Hauser M. et al., 2000; Muchir A. et al., 2000]. Первичные
молекулярные дефекты при 3 формах идентифицирова¬
ны. Форма КПМД 1А обусловлена мутациями структур¬
ного саркомерного белка мышечного волокна миотили-
на [Hauser М. et al., 2000]. Форма КПМД/С вызывается
мутациями в гене мышечного белка кавеолина-3, выпол¬
няющего сигнальные регуляторные функции [McNally Е.
et al., 1998; Minetti C. et al., 1998]; предполагается, что
кавеолин-3 может иметь отношение к регуляции процес¬
сов гликолиза в скелетной мышце [McNally E. et al., 1998].
Форма КГІМД /ß является аллельной с аутосомно-доми¬
нантной мышечной дистрофией Эмери-Дрейфуса (см.
раздел 3.1.1.6) и обусловлена мутациями гена ламина А/С,
кодирующего белки ламины - компоненты ядерной мем¬
браны [Muchir A. et al., 2000].До настоящего времени в мире опыт ДНК-диаг-
ностики аутосомно-доминантных КПМД весьма невелик
и ограничивается, главным образом, возможностью кос-
ДНК- диагностика наследственных болезней ... 155пенного ДНК-тестирования в информативных семьях, в
которых подтверждено генетическое сцепление с одним
из указанных локусов. В семьях с КПМД 7/4, КПМД / В и
КПМД/ С в случае обнаружения мутаций в генах миоти-
лина, ламина А/С и кавеолина-3 возможно проведение
прямой ДНК-диагностики у лиц из группы риска.3.1.1.3. Лице-лопаточно-плечевые ПМДЛице-лопаточио-плечевая форма мышечной дис¬
трофии, впервые описанная Ландузи и Дежерином в 1885
году, имеет распространенность 1 случай на 20 ООО и
наследуется по аутосомно-доминантному типу [Lunt Р.,
Нагрег Р., 1991]. Название данной формы ПМД отража¬
ет соответствующий специфичный для нее паттерн рас¬
пределения мышечной слабости и атрофий, хотя на по¬
здней стадии болезни может отмечаться также вовлече¬
ние в патологический процесс мускулатуры дистальных
отделов рук, тазового пояса и ног. Лице-лопаточно-пле-
чевую ПМД отличает широкая вариабельность клини¬
ческих проявлений с точки зрения как возраста начала
болезни, так и ее тяжести. Описаны атипичные вариан¬
ты болезни - лопаточно-перонеальный и лоиаточно-пле-
чевой, характеризующиеся отсутствием или минималь¬
ной степенью вовлечения лицевой мускулатуры
[Finzsimons R., 1999].Основной ген лице-лоиаточно-плсчевой ПМД
картирован в теломерной части длинного плеча 4-й хро¬
мосомы, в области 4q35 [Wijmenga С. et al., 1990;
Sarfarazi M. et al., 1991]. Данный хромосомный локус и
соответствующий молекулярный вариант болезни при¬
нято обозначать «лице-лопаточно-плечевая мышечная
дистрофия, тип 1А» [Fisher J., Upadhyaya М., 1997]. В
2% семей показано отсутствие сцепления с основным
локусом 4q35 (лице-лопаточно-плечевая мышечная ди¬
156Глава 3строфия 1В), что свидетельствует о генетической гете¬
рогенности аутосомно-доминантной лице-лопаточно-
плечевой ПМД [Gilbert J. et al., 1993]. Упомянутые выше
атипичные лопаточно-перонеальные варианты болезни
обусловлены чаще всего повреждениями основного гена
на хромосоме 4q35 [Fisher J., Upadhyaya М., 1997]. В
отдельных наблюдениях, однако, не было выявлено сцеп¬
ления лопаточно-перонеальной формы ПМД с данным
локусом [Tawil R. et al., 1995], а для одного из генов ло-
паточно-перонеального мышечного синдрома показано
сцепление с хромосомой 12q [Wilhelmsen К. etal., 1996].Практически во всех семейных и спорадических
случаях лице-лопаточно-плечевой мышечной дистрофии
1А у больных имеет место делеция участка в субтело-
мерной области хромосомы 4q35, состоящего из боль¬
шого числа тандемных повторов длиной 3,3 кб [van
Deutekom et al., 1993]. Чем большей является протяжен¬
ность делеции, тем тяжелее клинические проявления
заболевания [Fitzsimons R., 1999]. Предполагается, что
данная делеция посредством изменения структуры хро¬
матина в области 4q35 приводит к нарушению экспрес¬
сии близлежащего гена FRG1 или других генов, распо¬
ложенных проксимальнее точки разрыва, что и лежит в
основе развития болезни [Fisher, Upadhyaya, 1997;
Kleinjan, van Heyningen, 1998]. Как указывалось в главе
1 (см. рис. 19 на стр. 71), такой необычный механизм
мутации носит название негативного позиционного эф¬
фекта (эффект положения). Указанная делеция в локусе
4q35 затрагивает сайт распознавания для рестрикцион¬
ной эндонуклеазы ЕсоШ, поэтому прямая ДНК-диагно¬
стика болезни может проводиться с помощью рестрик¬
ционного анализа с данным ферментом и последующей
блот-гибридизации с соответствующим зондом. Диагноз
лице-лопаточно-плечевой ПМД достоверен в случае вы¬
явления атипично короткого рестрикционного ЕсоШ-
ДНК- диагностика наследственных болезней ... 157фрагмента длиной менее 28 кб (в норме >35 кб) [Fisher,
Upadhyaya, 1997].Определенные затруднения при проведении пря¬
мой ДНК-диагностики лице-лопаточно-плечевой мышеч¬
ной дистрофии связаны с тем, что рестрикция геномной
ДНК ферментом ЕсоШ приводит к появлению в рест¬
рикционной смеси фрагментов ДНК гомологичного уча¬
стка хромосомы 10q26, имеющих сходные размеры с
фрагментами хромосомы 4q35 [Tawil R. et al., 1998;
FitzsimonsR., 1999]. Эти затруднения удается преодолеть
либо с помощью электрофореза в пульсирующем поле,
обеспечивающего более тонкое разделение крупных
фрагментов ДНК, либо путем дополнительной обработ¬
ки рестриктазой Bln 1, специфически расщепляющей
«посторонние» 1С^26-генерированные фрагменты ДНК
[DeiddaG. etal., 1996]. Использование таких усовершен¬
ствованных процедур ДНК-анализа позволяет успешно
диагностировать заболевание в 95% случаев лице-лопа-
точно-плечевой ПМД [Galuzzi G. et al., 1999]. В остаю¬
щихся 5% случаев может иметь место: а) «промежуточ¬
ная» длина рестрикционного фрагмента от 28 до 35 кб,
что затрудняет трактовку электрофореграммы; б) крос-
синговер между гомологичными участками хромосом
4q3 5 и 10q26 с транслокацией делетированного lOq-фраг-
мента в субтеломерную область хромосомы 4q, что мо¬
жет приводить к болезни и не диагностироваться опи¬
санными методами ДНК-анализа; 3) форма лице-лопа-
точно-плечевой ПМД 1В, не сцепленная с локусом 4q35
|Tawil R. et al., 1998; Fitzsimons R., 1999]. В настоящее
иремя в мире накоплен большой опыт практической
ДНК-диагностики лице-лопаточно-плечевой ПМД,
включая диагностику асимптомных случаев носитель-
сгва мутации и пренатальную ДНК-диагностику
| Deidda G. etal., 1996; Fisher, Upadhyaya, 1997; Galuzzi G.
e! al., 1999].
158Глава 33.1.1.4. Миотоническая дистрофияМиотоническая дистрофия представляет собой
наиболее частую форму мышечной дистрофии взрослых
(распространенность - 1 на 7500 человек) [Harper Р.,1989]. Заболевание наследуется по аутосомно-доминан-
тному типу и характеризуется мультисистемностью по¬
ражения с широкой вариабельностью клинических про¬
явлений, основными из которых являются миотония,
мышечные атрофии и парезы скелетной мускулатуры
(преимущественно дистальных отделов конечностей и
лица), катаракта, кардиомиопатия, эндокринные нару¬
шения. В семьях с миотонической дистрофией весьма
характерным является феномен антиципации, проявля¬
ющийся манифестацией более тяжелых и все более ран¬
них форм болезни в каждом последующем поколении
[Harper Р., 1989].Ген, ответственный за развитие миотонической
дистрофии (DMPK), локализуется на хромосоме
19ql3.2-13.3 и кодирует синтез белка миотонинпроте-
инкиназы [Brook J. et al., 1992]. Миотоническая дистро¬
фия - яркий представитель заболевания с «динамичес¬
ким» типом мутации: у больных имеет место экспансия
тандемных тринуклеотидных повторов СТО (цитозин-
тимин-гуанин), расположенных в З’-нетранслируемой
области гена [Buxton J. et al., 1992; Fu Y. et al., 1992;
Mahadevan M. et al., 1992]. Нормальные аллели гена
DMPK содержат до 30 копий CTG-триплетов, тогда как
на мутантных хромосомах число этих тринуклеотидных
повторов увеличено в десятки и сотни раз (до 4000 CTG-
повторов). Показана тесная корреляция между числом
CTG-повторов и тяжестью клинического синдрома: боль¬
ные с небольшой степенью экспансии триплетов (40-
160 копий) имеют минимальные клинические проявле¬
ния, например - только катаракту в качестве единствен¬
ДНК- диагностика наследственных болезней ... 159ного симптома [Brook J. et al., 1992; Harley H. et al., 1993].
Напротив, больные с развернутой клинической карти¬
ной имеют большее число копий триплетов, а наиболь¬
шая степень экспансии CTG-повторов отмечена при тя¬
желой врожденной форме миотонической дистрофии
[Lavedan С. et al., 1993]. Причиной антиципации в семь¬
ях с миотонической дистрофией является генетическая
нестабильность мутантного тринуклеотидного сегмен¬
та [Harper P. et al., 1992]. Интересно, что нестабильность
CTG-повтора заметно выше при передаче гена по мате¬
ринской линии [Lavedan С. et al., 1993; Mulley J. et al.,1993], поэтому врожденная форма миотонической дист¬
рофии наблюдается исключительно при рождении де¬
тей от больных матерей. В редких случаях наблюдается
уменьшение (вплоть до нормы) длины мутантного по¬
втора у потомков, что сопровождается развитием более
легкой клинической картины или асимптомным течени¬
ем [Abeliovich D. et al., 1993]. Такие случаи обычно на¬
блюдаются при отцовской передаче гена; в качестве при¬
чины обсуждается возможность селекции против боль¬
ших аллелей в мужском гаметогенезе либо неблагопри¬
ятного влияния больших аллелей на мужскую фертиль¬
ность [Harley Н. et al., 1993; Lavedan С. et al., 1993].Предполагается, что экспансия тринуклеотидных
повторов при миотонической дистрофии может обуслов¬
ливать нарушение транскрипции/трансляции, либо вли¬
ять на стабильность соответствующей мРНК, результа¬
том чего является угнетение синтеза миотонинпротеин-
киназы [Harley Н. et al., 1993.; Willems P., 1994]. Другим
гипотетическим механизмом реализации данной мута¬
ции может быть негативное влияние патологически уд¬
линенного тринуклеотидного сегмента на экспрессию
некоторых близлежащих генов в локусе 19ql3 [Willems P.,
1994; Klesert Т. et al., 1997].
160Глава 3Непосредственное определение мутантного алле-
ля гена миотонической дистрофии путем электрофоре¬
тического разделения ПЦР-продуктов может проводить¬
ся только у больных с небольшой степенью экспансии
CTG-повторов (50-100 триплетов); эффективность ам¬
плификации более длинных и сверхдлинных мутантных
аллелей гена DMPK недостаточна для непосредствен¬
ной визуализации продуктов ПЦР при электрофорезе.
Поэтому чаще всего для прямой ДНК-диагностики мио¬
тонической дистрофии используется блот-гибридизация
рестрицированной геномной ДНК или продуктов амп¬
лификации с (CTG)n- зондами [Harley Н. et al., 1993;
Lavedan С. et al., 1993]. При такой диагностике мутант¬
ный аллель нередко визуализируется как смазанный про¬
тяженный след на радиоавтографе (рис. 39), что являет¬
ся отражением особенностей амплификации сверхдлин¬
ных тринуклеотидных аллелей либо следствием сомати¬
ческого мозаицизма мутантного CTG-повтора в клетках
крови [Lavedan С. et al., 1993]. Нами совместно с Инсти¬
тутом молекулярной генетики РАН проведена прямая
ДНК-диагностика болезни в 30 российских семьях с
миотонической дистрофией: экспансия тринуклеотидных
CTG-повторов в одном из аллелей гена у обследован¬
ных больных выявлена более чем в 90% семей, при этом
величина экспансии повторов составила от 130 до 1500
CTG-копий (рис. 39).Выявление нами нормальной длины тринуклео-
тидного сегмента гена у небольшой части обследован¬
ных больных миотонической дистрофией соответствуе'1
наблюдениям других авторов [Thornton С. et al., 1994] и
может быть обусловлено либо казуистическими случая¬
ми точковых мутаций в гене миотонинпротеинкиназы
[Harley Н. et al., 1993], либо существованием других ло
кусов, ассоциированных с данным клиническим синдромом
ДНК- диагностика наследственных болезней... 161Рис. 39. Прямая ДНК-диагностика миотонической дистро¬
фии с помощью блот-гибридизации по Саузерну
Дорожка 1 - отсутствие мутации (нормальная длина обоих аллелей
I ена миотонинпротеинкиназы), дорожка 2 - больной миотоничес¬
кой дистрофией (экспансия тринуклеотидных CTG-повторов в од¬
ном из аллелей гена). Мутантный аллель обозначен длинной стрел¬
кой, нормальный аллель - короткой стрелкой.Генетическая гетерогенность миотонической ди¬
строфии была подтверждена в 1998 году: второй локус
«классического» дистального фенотипа миотонической
дистрофии был картирован на хромосоме 3q [Ranum L.
et al., 1998]. Более того, в том же участке хромосомы 3q
расположен ген особого и чрезвычайно редкого заболе¬
вания- так называемой проксимальной миотонической
миопатии (PROMM) [Ricker К. et al., 1999]. Предполага¬
йся, что указанные клинические синдромы (второй ва¬
риант дистальной миотонической дистрофии и прокси¬
162Глава 3мальная миотоническая миопатия) могут являться ал¬
лельными заболеваниями, обусловленными мутациями
одного гена [Thornton С., AshizawaT., 1999], однако под¬
тверждение этой гипотезы станет возможным лишь пос¬
ле идентификации данного гена на хромосоме 3q.3.1.1.5. Другие формы ПМДДистальные ПМД (дистальные миопатии). Дис¬
тальные миопатии представляют собой клинически и
генетически гетерогенную группу заболеваний, характе¬
ризующихся избирательным или преимущественным
поражением дистальной мускулатуры нижних и/или вер¬
хних конечностей. В соответствии с типом наследова¬
ния, возрастом начала болезни и локализацией мышеч¬
ных атрофий и парезов дистальные миопатии подразде¬
ляются на несколько самостоятельных клинических
форм [Griggs R., Markesbery W., 1994; Mastaglia F.,
Laing N., 1999]. Современная классификация основных
форм дистальных миопатий представлена на рис. 40.Как видно на этой схеме, наиболее четко тип ди¬
стальной миопатии дифференцируется в начальной ста¬
дии болезни, когда возможно определение паттерна вов¬
лечения скелетной мускулатуры «в чистом виде». С го¬
дами, по мере прогрессирования заболевания при боль¬
шинстве форм в основном сохраняется относительная
селективность поражения соответствующих групп мышц,
однако наблюдается постепенное распространение про¬
цесса на более проксимальные отделы и иногда - про¬
тивоположные конечности и мускулатуру шеи. Напри¬
мер, на поздней стадии «кистевой миопатии» Веландер
может наблюдаться вовлечение мышц ног, а при осталь¬
ных формах дистальных миопатий (за исключением ти-
биальной миопатии Удда) заболевание, начавшись с ди¬
стальной мускулатуры ног, может распространяться на
ДНК- диагностика наследственных болезней ... 163Рис. 40. Современная клинико-генетическая классификация
дистальных миопатийверхние конечности [М^адНа К, Ьаш^К, 1999]. Важ¬
ным дифференциально-диагностическим признаком яв¬
ляется преимущественное вовлечение передней или зад¬
ней группы мышц ног: так, для миопатии Миоши наи¬
более характерным признаком (наряду со значительно
164Глава 3повышенным уровнем сывороточной креатинфосфоки-
назы) является более грубое поражение икроножной
мышцы и связанные с этим затруднения при ходьбе на
носках, тогда как при большинстве других форм дисталь¬
ных миопатий наблюдается преимущественное пораже¬
ние передней (тибиальной) группы мышц с невозмож¬
ностью ходьбы на пятках. У больных дистальными мио-
патиями Нонака и Веландер при биопсии пораженных
мышц, помимо типичных дистрофических изменений,
выявляются кольцевые вакуоли и тубулофиламентозные
включения, что сближает данные формы дистальных ми¬
опатий с редким аутосомно-рецессивным заболеванием
скелетных мышц - наследственным миозитом с включе¬
ниями [Mitrani-Rosenbaum S. et al., 1996].На сегодняшний день известна хромосомная ло¬
кализация 6 генов дистальных миопатий (см. рис. 40),
однако лишь для миопатии Миоши идентифицирован
сам ген и его молекулярный продукт - белок дисферлин
[Liu J. et al., 1998]. Как указывалось в разделе 3.1.1.2,
мутации в гене дисферлина могут приводить к манифес¬
тации не только миопатии Миоши, но и другой формы
аутосомно-рецессивной мышечной дистрофии -
КПМДЖ Принимая во внимание тот факт, что ген дис¬
ферлина включает 55 экзонов и имеет длину кодирую¬
щей области около 6,9 кб, поиск мутаций в данном гене
и прямая ДНК-диагностика миопатии Миоши весьма
трудоемки и на практике редко осуществимы. Для мо¬
лекулярной диагностики миопатии Миоши более удоб¬
ным является стандартный для всех ПМД подход, осно¬
ванный на выявлении характерного белкового дефекта
при иммуногистохимическом исследовании биоптатов
мышц с помощью антител к дисферлину [Matsuda С. et
al., 1999]. Для всех других форм дистальных миопатий
может ставиться вопрос о проведении косвенной ДНК-
диагностики на основе анализа генетического сцепле-
ДНК- диагностика наследственных болезней ... 165ния с локусами 2р13, 2q31-33, 9pl-ql и 14q 11
[Mastaglia F., LaingN., 1999]. Однако установление сцеп¬
ления (т.е. получение диагностически значимого Лод-
балла) осуществимо лишь в очень небольшом числе се¬
мей с несколькими больными родственниками, а пра¬
вильно выбрать необходимый для исследования хромо- ■
сомный локус только на основании клинической карти¬
ны не представляется возможным. Поэтому в целом кос¬
венная ДНК-диагностика дистальных миопатий имеет
пока весьма ограниченное значение. В будущем, после
идентификации генов дистальных миопатий и их белко¬
вых продуктов будет возможной стандартная иммуноги-
стохимическая диагностика болезни и прямое опреде¬
ление мутаций у больных лиц и их родственников из
группы риска.Окулофарингеальная ПМД. Окулофарингеальная
форма ПМД представляет собой миодистрофию позднего
возраста, обычно манифестирующую на б-м десятиле¬
тии жизни. В большинстве случаев тип наследования
болезни аутосомно-доминантный; описаны единичные
семьи с аутосомно-рецессивной передачей окулофарин-
геальной ПМД [Tomé F., Fardeau М., 1994]. Заболевание
клинически характеризуется развитием прогрессирую¬
щей слабости и атрофий проксимальных отделов конеч¬
ностей, расстройствами глотания и фонации, птозом,
нарушением движений глазных яблок и (на поздней ста¬
дии) слабостью лицевой мускулатуры, а патоморфоло-
I ически - миодистрофическими изменениями указанных
групп скелетных мышц и появлением патогномоничных
филаментозных внутриядерных включений в мышечных
волокнах [Tomé F., Fardeau М., 1994; Emery А., 1998].Ген окулофарингеальной ПМД на хромосоме
I4ql 1.2-13 ответственен за синтез ядерного белка
1’АВР2, служащего фактором полиаденилирования мРНК
| lirais В. et al., 1998]. У всех больных окулофарингеаль-
166Глава 3ной ПМД в мутантном гене обнаруживается увеличение
числа копий тринуклеотидных повторов GCG в 1 -м эк-
зоне гена: в норме ген содержит 6 тандемных копий по¬
второв GCG, кодирующих полиаланиновый участок в N-
терминальной области белка, тогда как у больных число
повторов в мутантном гене увеличено до 8-13 копий
[Brais В. et al., 1998; Grewal R. et al., 1999; Mirabella M. et
al., 2000]. Предполагается, что удлинение полиаланино-
вого участка белка РАВР2 приводит к олигомеризации
мутантных белковых молекул, что и лежит в основе об¬
разования внутриядерных филаментозных включений. В
нормальной популяции у 2% лиц на одной из хромосом
обнаруживается «промежуточный» аллель гена, имею¬
щий 7 повторов GCG, наличие которого в гетерозигот¬
ном состоянии не сопровождается каким-либо симпто¬
мами. Однако у гомозиготных носителей аллеля (GCG)
развивается клиника окулофарингеальной ПМД, причем
в таких семьях имеет место аутосомно-рецессивный тип
наследования болезни [Brais et al., 1998]. Гомозиготность
по более длинным GCG-повторам (т.е. двойная доза за¬
ведомо мутантного аллеля) характеризуется развитием
более тяжелой клинической картины и более ранним
дебютом симптомов (в среднем на 18 лет раньше чем у
гетерозиготных носителей мутации) [Blumen S. et al.,1999]. Таким образом, с точки зрения мутационного
механизма окулофарингеальная ПМД является уникаль¬
ным заболеванием с двух точек зрения: во-первых, при
этой форме миодистрофии имеет место наиболее корот¬
кая экспансия повторов по сравнению со всеми другими
«тринуклеотидными» заболеваниями; во вторых, различ¬
ная по тяжести, но одинаковая по характеру мутация в
одном и том же гене может служить причиной развития
как аутосомно-доминантной, так и аутосомно-рецессив-
ной формы болезни.
ДНК- диагностика наследственных болезней ... 167Открытие гена РАВР2 и универсального типа
мутации в нем позволяет проводить сравнительно про¬
стую и достоверную прямую ДНК-диагностику окуло-
фарингеальной мышечной дистрофии, основанную на
амплификации тринуклеотидного участка гена с помо¬
щью ПЦР Подтверждением диагноза служит обнаруже¬
ние у больного на электрофореграмме двух различных
фрагментов ДНК, один из которых имеет нормальный
размер (б повторов GCG), а другой - патологически уд¬
линен и соответствует экспансии GCG-повторов >8 ко¬
пий. Вывление «промежуточного» ПЦР-продукта с 7
повторами GCG является диагностически значимым в
том случае, если оба аллеля имеют такой размер либо
второй аллель является заведомо мутантным (>8 повто¬
ров GCG).ПМД Эмери-Дрейфуса. Мышечная дистрофия
Эмери-Дрейфуса относится к редким формам ПМД и
имеет ряд специфических особенностей клинической
картины: болезнь начинается в детском или юношеском
возрасте, характеризуется относительно медленным про¬
грессированием миодистрофии (или даже стационарным
течением), ранним развитием типичных контрактур в
локтевых, голеностопных и межпозвонковых суставах,
а также поражением проводящей системы сердца
[Emery А., 1998]. Тип наследования в абсолютном боль¬
шинстве случаев - Х-сцепленный рецессивный. Ген за¬
болевания локализован на хромосоме Xq28 и кодирует
белок с неустановленной функцией эмерин [Bione et al.,1994]. Эмерин экспрессируется в клетках мышечной и
других тканей, локализуется наядерной мембране и, воз¬
можно, принимает участие в регуляции экспрессии ге¬
нов [Manilal S. et al., 1996]. При ПМД Эмери-Дрейфуса
описано более 60 различных мутаций в гене эмерина
| Yates J., Wehnert М., 1999]. Идентификации мутации у
конкретного больного предполагает проведение скринин¬
168Глава 3га всей кодирующей области гена, поэтому прямая ДНК-
диагностика болезни на сегодня не является рутинной и
может проводиться лишь в высокоспециализированных
лабораториях. Более доступным молекулярным диагно¬
стическим тестом является обнаружение дефекта белка
эмерина в биоптатах мышц при иммуногистохимичес-
ком анализе и Вестерн-блоттинге [Manilal S. et al., 1996;
Nagano A. et al., 1996].В единичных семьях описаны аутосомно-доми-
натный и аутосомно-рецессивный варианты ПМД Эме¬
ри-Дрейфуса. Оба они обусловлены различными по тя¬
жести мутациями в гене на хромосоме 1 q21.3 (LMNA),
кодирующем белки внутренней поверхности ядерной
мембраны - ламины А/С, тесно связанные с эмерином
[Bonne G. et al., 1999; Di BarlettaM. et al., 2000]. Очевид¬
но, что мутации в данном гене могут иметь различную
пенетрантность и действовать как доминантные (вызы¬
вая болезнь в гетерозиготном состоянии) или как рецес¬
сивные (не проявляясь у гетерозигот и приводя к пато¬
логии мышц только при наличии у больного двух му¬
тантных хромосом). По мнению М. Di Barletts с соавто¬
рами (2000), поиск мутаций в гене LMNA следует про¬
водить у всех больных с ранними контрактурами плече-
перонеальных мышц и/или синдромом «ригидного по¬
звоночника» даже при отсутствии типичного разверну¬
того фенотипа ПМД Эмери-Дрейфуса.Как уже указывалось выше, аутосомные формы
ПМД Эмери-Дрейфуса являются аллельными заболева¬
ниями с КПМД типа 1В.Миопатия Бетлема. Миопатия Бетлема представ¬
ляет собой редкую аутосомно-доминантную форму ПМД,
которая некоторыми авторами относится к группе врож¬
денных миопатий. Заболевание характеризуется мани¬
фестацией симптомов в раннем детском возрасте, доб¬
рокачественным течением с практически нормальной
ДНК- диагностика наследственных болезней ... 169продолжительностью жизни, конечностно-поясным ти¬
пом распределения мышечных атрофий, сохранностью
лицевых мышц. Важнейшим диагностическим призна¬
ком миопатии Бетлема является развитие ранних (нередко
врожденных) контрактур в межфаланговых, локтевых и
голеностопных суставах [МоЫге М. е! а1., 1988]. Данная
форма мышечной дистрофии является генетически ге¬
терогенной. Оба известных гена миопатии Бетлема, ло¬
кализованные на хромосомах 2Ц22.3 и 2q37, кодируют
синтез различных субъединиц (а1/а2 и аЗ) коллагена
VI типа - белка скелетных мышц, обеспечивающего
связь базальной пластинки с гликопротеинами внекле¬
точного матрикса |.ТоЬз1з О. е1 а1., 1996]. К настоящему
времени в мире в нескольких десятках семей получен
опыт прямой ДНК-диагностики миопатии Бетлема на
основе мутационного скрининга генов коллагена VI.3.1.2. Врожденные миопатииК группе врожденных миопатий относится боль¬
шое число наследственных заболеваний скелетных
мышц, проявляющихся в раннем постнатальном перио¬
де генерализованной мышечной слабостью и гипотони¬
ей, сухожильной арефлексией, задержкой моторного раз¬
вития, нормальным или незначительно повышенным
уровнем сывороточной креатинфосфокиназы [П.А. Те¬
мин и соавт., 1998]. Клинически на первом этапе у боль¬
ных обычно диагностируется неспецифический симп-
томокомплекс, определяемый как синдром «вялого ре¬
бенка». В отличие от других возможных этиологичес¬
ких причин данного синдрома (спинальная амиотрофия
15ерднига-Гофмана, атоническая форма детского цереб¬
рального паралича и др.), при врожденных миопатиях
имеет место первично-мышечный характер процесса,
который может быть подтвержден на основании харак¬
170Глава 3терных электромиографических и морфологических дан¬
ных [Goebel H., Lenard Н., 1992]. С патогенетической
точки зрения врожденные миопатии могут быть подраз¬
делены на 2 группы. К первой группе относятся заболе¬
вания, обусловленные повреждением белков сарколем¬
мы мышечного волокна. В этом случае у больных при
микроскопии обнаруживается дистрофический паттерн
поражения мышц и нередко - постепенное прогресси¬
рование процесса, в связи с чем они иногда обозначают¬
ся в литературе как врожденные мышечные дистрофии.
Ко второй группе относятся заболевания, обусловлен¬
ные структурными аномалиями белков цитоскелета мы¬
шечного волокна - так называемые врожденные струк¬
турные миопатии.Типичными примерами врожденных мышечных
дистрофий являются 3 самостоятельные молекулярные
формы.1) «Классическая восточная» форма (мерозин-
негативная) - обусловлена мутациями 6д2-сцепленного
гена, ответственного за синтез белка мерозина (а2-цепи
ламинина) [Helbling-Leclerc A. et al., 1995]. Белок меро-
зин является важным компонентом базальной мембра¬
ны мышечного волокна (см. рис. 36), и его дефект со¬
провождается нарушением структуры сарколеммы и раз¬
витием тяжелой формы врожденной мышечной дистро¬
фии, нередко в сочетании с гиподенсивными очагами в
белом веществе полушарий мозга. Совсем недавно была
описана особая форма врожденной мышечной дистро¬
фии с вторичным дефектом мерозина, характеризующа¬
яся (наряду с поражением проксимальной скелетной
мускулатуры) ранним вовлечение диафрагмальной мыш¬
цы и дыхательной недостаточностью; ген данной фор¬
мы картирован на хромосоме 1 q42 [Brockington М. et al.,
2000].
ДНК- диагностика наследственных болезней ... 1712) Интегрин-негативная форма врожденной мы¬
шечной дистрофии - обусловлена мутациями \2ql3-
сцепленного гена, ответственного за синтез белка интег-
рина ос 7 (рецептора мерозина на мышечной мембране)
[НауаэЫ У. е! а1., 1998]. Данная форма врожденной мы¬
шечной дистрофии характеризуется более благоприят¬
ным течением.3) Врожденная мышечная дистрофия Фукуямы -
обусловлена мутациями 9q31 -сцепленного гена, ответ¬
ственного за синтез белка фукутина (компонента базаль¬
ной мембраны в мозге и мышцах) [КоЬауаэЫ К. е1 а!.,
1998]. Характерной клинической особенностью данно¬
го заболевания является сочетание тяжелой врожденной
мышечной дистрофии с пороками развития мозга (мик-
рополигирия, лиссэнцефалия, гидроцефалия), сердца и
глаз.Все перечисленные заболевания наследуются по
аутосомно-рецессивному типу Как и при прогрессиру¬
ющих мышечных дистрофиях, на практике молекуляр¬
ная диагностика указанных форм врожденных мышеч¬
ных дистрофий осуществляется посредством выявления
специфических белковых дефектов при иммуногистохи-
мическом анализе мышечных биоптатов (исследование
с использованием антител к мерозину, интегрину а7,
фукутину). В конкретных семьях после идентификации
поврежденного белка может быть проведено исследова¬
ние кодирующей области соответствующего гена с це¬
лью выявления мутации и проведения прямой ДНК-ди-
агностики у пробанда и пренатальной ДНК-диагности-
ки у плода; с этой же целью возможно проведение кос¬
венной ДНК-диагностики с соответствующими генети¬
ческими маркерами.Основными представителями врожденных струк¬
турных миопатий являются различные формы немали¬
новых миопатий, болезнь центрального стержня, цент-
172Глава 3ронуклеарная (миотубулярная) миопатия, миофибрил-
лярные (десминовые) миопатии и ряд других редких
синдромов, для большинства их которых характерен ауто-
сомно-доминантный или (реже) аутосомно-рецессивный
тип наследования [Goebel H., Lenard H., 1992]. Общим
для всей группы заболеваний является формирование тех
или иных структурных аномалий мышечного волокна -
таких как мелкие палочкообразные тельца по перифе¬
рии и в центре мышечного волокна, особые централь¬
но-расположенные азурофильные стержни, центральные
скопления ядер, десминовые включения и т.п. Указан¬
ные морфологические феномены связаны с нарушением
нормальной структуры ряда белков цитоскелета мышеч¬
ных волокон - а-актина, а-тропомиозина, миотубуля-
рина, десмина, аВ-кристаллина, тропонина Т1 и др.;
установлены соответствующие гены и мутации в них,
вызывающие дефекты вышеназванных структурных бел¬
ков у больных врожденными миопатиями [Kaplan J.-C.,
Fontaine В., 1998; Vicart P. et al., 1998; Wallgren-
Petersson C., 1998; Engel A., 1999; Nowak K. et al., 1999;
Johnston J. et al., 2000]. Показано также сцепление неко¬
торых форм врожденных миопатий с рядом других хро¬
мосомных локусов (см. приложение 1), однако соответ¬
ствующие гены их белковые продукты до настоящего
времени не установлены.Основным методом диагностики врожденных
структурных миопатий является выявление характерных
морфологических маркеров отдельных форм данных за¬
болеваний при проведении гистологического и гистохи¬
мического исследования биопсий скелетных мышц. В
литературе описаны единичные случаи прямой ДНК-
диагностики врожденных структурных миопатий в се¬
мьях с дентифицированными мутациями соответствую¬
щих генов [Vicart P. et al., 1998; Nowak К. et al., 1999],
причем на практике ДНК-анализ при этих заболеваниях
ДНК- диагностика наследственных болезней ... 173принципиально важен, главным образом, для пренаталь¬
ной диагностики генетического статуса плода и профи¬
лактики повторных случаев заболевания в отягощенной
семье. Косвенная ДНК-диагностика при данных заболе¬
ваниях практически не применяется, поскольку в силу
генетической гетерогенности отдельных морфологичес¬
ких подтипов врожденных структурных миопатий дос¬
товерно решить вопрос о том, какой хромосомный ло-
кус необходимо исследовать, не представляется возможным.3.1.3. Наследственные невропатииНаследственные формы невропатий составляют
до 60-70% всех хронических полиневропатий в различ¬
ных популяциях и встречаются с частотой не менее 1 на
2500 [Emery А., 1991 ]. Данная группа заболеваний весь¬
ма гетерогенна. Клиническая классификация наслед¬
ственных невропатий предполагает подразделение их на
3 основные группы: 1) наследственные моторно-сенсор¬
ные невропатии; 2) наследственные моторные невропа¬
тии; 3) наследственные сенсорные и сенсорно-вегетатив¬
ные невропатии. Данное деление основано на характере
симптомов и типе волокон периферических нервов, пре¬
имущественно вовлекаемых в дегенеративный процесс
[Dyck P. et al., 1993]. Имеется также ряд других редких
форм наследственных невропатий, многие из которых
являются проявлением различных системных генетичес¬
ких нарушений обмена (амилоидные невропатии, пор-
фирийные невропатии, некоторые метаболические лей-
кодистрофии и др.).Наследственные моторно-сенсорные невропатии
(НМСН) являются самыми распространенными в рас¬
сматриваемой группе заболеваний и имеют наибольшее
значение в клинической практике [Dyck P. et al., 1993;
Chance P., Fischbeck К., 1994]. Большинство заболева¬
174Глава 3ний из группы НМСН имеют весьма сходные клиничес¬
кие проявления и на протяжении многих десятилетий
обозначались как «болезнь Шарко-Мари-Тута»
(Charcot-Marie-Tooth) [Harding А., 1995]. Характерным
для них являются развитие хронически прогрессирую¬
щей слабости и атрофии дистальной мускулатуры ног,
угнетение сухожильных рефлексов (в первую очередь
ахилловых), расстройства чувствительности по полинев-
ритическрому типу, деформация стоп («стопы Фридрей-
ха»), расстройства походки по типу «степпажа»; на по¬
здней стадии может присоединяться слабость и атрофия
дистальных отделов рук, деформация кистей [Dyck P. et
al., 1993]. В основе болезни лежат дегенеративные из¬
менения миелиновой оболочки или аксонов двигатель¬
ных и чувствительных волокон периферических нервов
и спинномозговых корешков. Заболеваниям из группы
НМСН свойственен различный (как аутосомный, так и
Х-сцепленный) характер наследования.В соответствии с данными электрофизиологичес-
ких и морфологических методов исследования можно
четко дифференцировать 2 основных типа НМСН -
НМСН I и НМСН II [Dyck P. et al., 1993; Timmerman V. et
al., 1998]. Тип I НМСН характеризуется, по данным
электронейромиографии, значительным снижением ско¬
рости проведения по двигательным (менее 38 м/с) и чув¬
ствительным волокнам периферических нервов, а мор¬
фологически - сегментарной гипертрофической демие-
линизацией нервов с формированием «луковичных го¬
ловок» («onion bulbs»). Таким образом, НМСН I пред¬
ставляет собой демиелинизирующую форму полиневро¬
патии (миелинопатию). Напротив, для НМСН типа II
характерно первичное поражение аксонов периферичес¬
ких нервов, при этом скорости проведения импульса по
периферическим нервам в пределах нормы или умерен¬
ДНК- диагностика наследственных болезней ... 175но снижены, а на биопсии структура миелина остается
сохранной (аксональная форма полиневропатии, или
аксонопатия).Помимо указанных выше двух основных типов
моторно-сенсорных полиневропатий, иногда в литера¬
туре под рубрикой НМСН выделяют также ряд сравни¬
тельно редких синдромов, отличающихся от классичес¬
кого фенотипа Шарко-Мари-Тута теми или иными осо¬
бенностями клинической картины [Dyck P. et al., 1993;
Harding А., 1995; Timmerman V. et al., 1998]:а) НМСН III (синдром Дежерина-Сотта) - харак¬
теризуется дебютом симптомов на протяжении первых
лет жизни, резко выраженной гипертрофией перифери¬
ческих нервов («гипертрофический неврит»), выражен¬
ным снижением скорости проведения импульса по дви¬
гательным волокнам (менее 10 м/с), ранней инвалидиза-
цией и нередко наблюдаемым повышением уровня бел¬
ка в ликворе;б) НМСН IV (болезнь Рефсума) - самостоятель¬
ное заболевание, связанное с нарушением обмена фита-
новой кислоты, при котором двигательная полиневро¬
патия сочетается с атаксией, ихтиозом и другими симп¬
томами;в) редкие формы НМСН, характеризующиеся со¬
четанием преимущественно моторной невропатии с
нижним спастическим парапарезом (НМСН V), атрофи¬
ей зрительных нервов и глухотой (НМСН VI), пигмент¬
ным ретинитом (НМСН VII);г) врожденная гипомиелинизирующая полинев¬
ропатия - характеризуется нарушением формирования
миелиновой оболочки периферических нервов с рожде¬
ния, значительным отставанием ребенка в двигательном
развитии, резким снижением скорости проведения им¬
пульса по периферическим нервам.
176Глава 33.1.3.1. Демиелинизирующие моторно-сенсорные
невропатии (НМСН тип I)Демиелинизирующие формы НМСН (НМСН I)
являются гораздо более распространенными, чем аксо¬
нальные. В абсолютном большинстве случаев заболева¬
ния из группы НМСН I наследуются по аутосомно-до-
минантному типу. В настоящее время идентифицирова¬
ны 4 гена аутосомно-доминантных НМСН I.СМТ 1А. Основной ген, ответственный за разви¬
тие большинства случаев НМСН I, расположен в хро¬
мосомной области 17р11.2 и кодирует синтез структур¬
ного белка периферического миелина РМР22 {Peripheral
Myelin Protein) [Lupski J. et al., 1991; Patel P. et al., 1992;
Timmerman V. et al., 1992; Valentijn L. et al., 1992]. Дан¬
ный хромосомный локус и соответствующая форма мо-
торно-сенсорной невропатии получили обозначение
СМТ типа 1А (аббревиатура от англ. Charcot-Marie-
Tooth). Тип наследования заболевания - аутосомно-до-
минантный. СМТ 1А имеет необычный молекулярный
механизм, который определяется особенностями строе¬
ния критической хромосомной области 17р11.2. Данный
механизм схематично представлен на рис. 41.Как видно на схеме, миелиновый ген РМР22 флан¬
кируется высокогомологичными повторами ДНК, меж¬
ду которыми в мейозе может происходить неравный крос-
синговер как результат спаривания ложно ориентирован¬
ных друг против друга теломерного и центромерного по¬
второв. В конечном счете образуются 2 различные му¬
тантные хромосомы: одна из них содержит тандемную
дупликацию области длиной 1,5 мб, другая - делецию
той же области, причем в обоих случаях область хромо¬
сомной перестройки захватывает ген РМР22 [Chance Р.
et al., 1994]. Таким образом, ген РМР22 является дозо-
чувствительным: гаметы с дупликацией РМР22 дают
начало СМТ 1 А, тогда как делеция этого гена лежит в ос-
ДНК- диагностика наследственных болезней ... 1771,5 мб< »Те„ГоГ'ЙГен РМР 22 Ц~рНЫЙО—Ген РМР22Ген РМР22Дупликация (3 мб) ,Ш—■—Ш—с-Ген РМР22 Ген РМР22ДелецияРис. 41. Механизм неравного кроссинговера с участием гена
РМР22Л, Структура критической хромосомной области 17р 11.2. Б. Нерав¬
ный кроссинговер между двумя хроматидами (гомологичные тело-
мсрный и центромерный повторы разных хроматид спариваются
между собой, приводя к неэквивалентному обмену хромосомными
участками). В. Продукты неравного кроссинговера: показано обра-
юиание двух мутантных хромосом с дупликацией и делецией
1'М1>22-содержащего хромосомного участка.
178Глава 3нове развития особой формы доминантного заболевания
периферических нервов - наследственной невропатии с
предрасположенностью к параличам от сдавления (см.
ниже). Показано, что около 70-80% больных демиели-
низирующими моторно-сенсорными невропатиями име¬
ют указанную дупликацию в локусе 17pll.2 [Wise С. et
al., 1994], причем она часто возникает de novo в резуль¬
тате многократной реализации рассмотренного механиз¬
ма неравного кроссинговера [Chance P., Fischbeck К.,
1994]. В отдельных случаях заболевание обусловлено точ-
ковыми мутациями гена РМР22 [Roa В. et al., 1993; Nelis
Е. et al., 1994], что подтверждает этиологическую роль
данного гена в развитии СМТ 1А. По-видимому, след¬
ствием указанных точковых мутаций является повыше¬
ние уровня экспрессии белка РМР22 - аналогично тому,
что имеет место при дупликации гена. Предполагается,
что изменение уровня экспрессии РМР22 может нару¬
шать баланс между процессами пролиферации, останов¬
ки роста и дифференцировки шванновских клеток, при¬
водя в конечном счете к гипертрофической демиелини-
зации периферических нервов [Gabriel J. et al., 1997].Другие молекулярные формы аутосомно-доминан-
тных НМСН I. В более редких случаях доминантная
моторно-сенсорная невропатия может быть связана с
мутациями генов миелинового белка Р0 на хромосоме
lq22-23 (СМТ типа IB) [Hayasaka К. et al., 1993;
Kulkens T. et al., 1993] и коннексина-32 (Cx32) на хромо¬
соме Xql3.1 (СМТ типа IX) [Bergoffen J. et al., 1993].
Форма СМТ 1В наследуется по аутосомно-доминантно-
му, а СМТ IX - по Х-сцепленному доминантному типу
с ограниченной пенетрантностью у женщин. Белок Р(|
является важнейшим компонентом периферического ми¬
елина, регулирующим укладку слоев миелиной оболоч¬
ки, а коннексин-32 относится к белкам межклеточных
контактов и формирует связи между соседними швап-
ДНК- диагностика наследственных болезней ... 179новскими клетками (коннексиновые каналы) [Chance Р.,
Fischbeck К., 1994]. К настоящему времени в данных ге¬
нах описано большое число точковых мутаций (более
50 в гене Р0 и более 150 в гене Сх32), приводящих к
нарушению структурно-функциональной организации
миелиновой оболочки периферических нервов и разви¬
тию СМТ 1В и СМТ IX [Nelis Е. et al., 1999]. Клиничес¬
кая картина этих форм НМСН практически неотличима
от СМТ 1А. Недавно был идентифицирован еще один
ген аутосомно-доминантной НМСН типа I: он располо¬
жен на хромосоме 10q21.1-22.1 и кодирует белок EGR2
{Early Growth Response), предположительно являющий¬
ся фактором транскрипции и контролирующий проли¬
ферацию и миелинизацию шванновских клеток
| Warner L. et al., 1998]. Мутации гена EGR2 являются
чрезвычайно редкой причиной развития аутосомно-до¬
минантной НМСН I (описано несколько точковых мута¬
ций EGR2 в единичных семьях).Синдром Дежерина-Сотта. врожденная гипоми-
слинизирующая полиневропатия и синдром Русси-Леви.
Интенсивные исследования генов РМР22, Р0 и EGR2 по¬
казали, что данные гены ответственны не только за клас¬
сические аутосомно-доминантные случаи НМСН типа
I, но также за развитие описанных выше тяжелых (не¬
редко врожденных) форм демиелинизирующих невропа-
11 i й — синдрома Дежерина-Сотта и врожденной гииоми-
слинизируютцей полиневропатии [Nelis Е. et al., 1999].
) 1анные заболевания представляют собой экстремальные
нлрианты фенотипического спекгра демиелинизирующих
iii-вропатий: они обусловлены наиболее тяжелыми по
гноим последствиям гетерозиготными точковыми мута¬
циями de novo генов РМР22, Р0 и EGR2 или гомозигот¬
ными мутациями EGR2 и РМР22 (т.е. двойной дозой
мутантного гена), в результате чего грубо нарушаются
механизмы формирования миелиновой оболочки пери¬
180Глава 3ферических нервов [Chance P., Fischbeck К., 1994;
Haites N. et al., 1998; Parman Y. et al., 1999; Warner L. et
al., 1999]. Исходя из вышесказанного, методы ДНК-ди-
агностики в указанных случаях синдрома Дежерина-
Сотта и врожденной гипомиелинизирующей полиневро¬
патии не отличаются от методов ДНК-диагностики, при¬
меняемых для всей группы НМСНI (см. ниже). Совсем
недавно был открыт еще один генетический вариант син¬
дрома Дежерина-Сотта с аутосомно-рецессивным типом
наследования [Boerkoel С. et al., 2001]: он обусловлен
мутациями в гене периаксина - белка цитоскелета, уча¬
ствующего в реализации мембранно-белковых взаимо¬
действий и стабилизации структуры зрелого миелина.На протяжении многих десятилетий в литерату¬
ре описывалось особое заболевание, известное под на¬
званием «синдром Русси-Леви». Для него характерно
развитие типичной моторно-сенсорной невропатии с низ¬
кими скоростями проведения импульса по периферичес¬
ким нервам, а также «полая стопа», постуральный тре¬
мор и легкие дискоординаторные симптомы [Dyck P. et
al., 1993; Harding А., 1995]. Многими авторами синдром
Русси-Леви рассматривался как «стертая форма» болез¬
ни Фридрейха [Harding А., 1995]. Развитие электрофи-
зиологических, морфологических и молекулярно-гене-
тических методов показало, что синдром Русси-Леви не
является самостоятельной нозологической формой, а
представляет собой своеобразный фенотипический ва¬
риант НМСН I: данные электронейромиографии и био¬
псии нервов у больных с синдромом Русси-Леви иден¬
тичны таковым при НМСН I [Dyck P. et al., 1993], а мо¬
лекулярно-генетический анализ выявляет у этих боль¬
ных мутации в генах Ро и РМР22 [Plante-Bordeneuve V.
et al., 1999]. Таким образом, термин «синдром Русси-
Леви» имеет лишь историческое значение и в настоящее
время не применяется.
ДНК- диагностика наследственных болезней ... 181Наследственная невропатия с предрасположенно¬
стью к параличам от сдавления. Наследственная невро¬
патия с предрасположенностью к параличам от сдавле¬
ния (ННППС) представляет собой аутосомно-доминан-
тное заболевание, характеризующееся развитием реци¬
дивирующих демиелинизирующих мононевропатий,
обусловленных повышенной чувствительностью пери¬
ферических нервов к сдавлению [Иллариошкин С.Н. и
др., 1998; Оошёег Я. е1а1., 1995; StбgbauerF. е1а1.,2000].
У больных с молодого возраста отмечаются острые по¬
вторные эпизоды безболевых параличей периферичес¬
ких нервов, проявляющиеся парезами, парестезиями и
расстройствами чувствительности в соответствующих
зонах. Развитию параличей способствуют мелкие трав¬
мы и даже весьма незначительное и кратковременное
сдавление нервов, например - после работы за письмен¬
ным столом (повреждение локтевого нерва) или при
сидении нога на ногу, на коленях, на корточках (паралич
малоберцового нерва). В 10% всех случаев развития па¬
раличей наблюдается полное восстановление в течение
первых 24 часов; более характерным является отсрочен¬
ное восстановление (на протяжении ряда месяцев), в том
числе с сохранением той или иной резидуальной симп¬
томатики [Stбgbauer Е Й а1., 2000]. По мере прогресси¬
рования болезни постепенно могут развиваться симмет¬
ричные или асимметричные амиотрофии в дистальных
отделах конечностей, свисающая стопа, угнетение сухо¬
жильных рефлексов, «пятнистые» или диффузные рас¬
стройства чувствительности, что сближает клиническую
картину ННППС с симптоматикой НМСН I. При элект-
рофизиологическом исследовании у больных ННППС
отмечается снижение скорости проведения по двигатель¬
ным и чувствительным волокнам периферических не¬
рвов, наиболее отчетливое в местах компрессии нервных
стволов, а также удлинение дистальной латентности
182Глава 3[Verhagen W. et al., 1993; Gouider R. et al., 1995]. Био¬
псия нервов выявляет характерные изменения миелина
с формированием колбасовидных утолщений - так на¬
зываемых «томакул» (отсюда синоним заболевания -
«томакулярная невропатия»), а также сегментарную де-
миелинизацию [Verhagen W. et al., 1993; Stôgbauer F. et
al., 2000].Как указывалось выше, молекулярной основой
ННППС является делеция области длиной 1,5 мб (вклю¬
чая ген РМР22) как следствие неравного кроссинговера
хромосомных участков в локусе 17р 11.2 (см. рис. 41)
[Chance P. et al., 1993; 1994]. Таким образом, ННППС и
СМТ 1А обусловлены разнонаправленными нарушени¬
ями дозы гена РМР22 (деления или дупликация) в ре¬
зультате одного и того же мутационного события. Такой
механизм предполагает, что распространенность ННППС
и СМТ 1А в популяции должна быть примерно одина¬
ковой - около 1 на 3000 [Chance P., Fischbeck К., 1994].
Между тем истинная распространенность ННППС в
прошлом явно недооценивалась, что обусловлено отно¬
сительно доброкачественным течением заболевания и
отсутствием выраженных клинических симптомов у
большого числа носителей мутантного гена, нередко не
обращающихся за медицинской помощью [Gouider R. et
al., 1995].В единичных случаях к развитию ННППС могут
приводить также точковые мутации гена РМР22, приво¬
дящие (как и делеция гена) к снижению экспрессии
РМР22 и дезорганизации периферического миелина
[Stngbauer F. et al., 2000].Прямая ДНК-диагностика аутосомно-доминант-
ных НМСН I. Принимая во внимание тот факт, что око¬
ло 80% всех случаев НМСН I обусловлены мутациями
гена РМР22, обычно ДНК-диагностика НМСН I начи¬
нается с исследования именно этого гена и предполага¬
ДНК- диагностика наследственных болезней... 183ет в первую очередь выявление основной мутации - дуп¬
ликации области 1,5 мб в локусе 17р 11.2. Наиболее рас¬
пространенным подходом для этого является исследова¬
ние числа аллелей высокополиморфных (СА)п-маркеров,
входящих в состав дуплицируемого участка [Сискеу С.
ег а1., 1995]. В норме любой маркер имеет по два аллеля
(по одному с каждой хромосомы), тогда как в случае
дупликации на мутантной хромосоме 1,5 мб-участка
общее число аллелей каждого маркера у больного СМТ
1А равно трем. Маркеры амплифицируются в ПЦР, пос¬
ле чего продукты амплификации разделяются с помо¬
щью электрофореза в полиакриламидном геле (рис. 42).1 2 3Рис. 42. ДНК-диагностика СМТ 1А с использованием
полиморфных маркеров 0178122 (верхняя часть геля) и
1)178921 (нижняя часть геля)Дорожки 1 и 3-больные СМТ 1А, дорожка 2-больной с другой
формой наследственной невропатии. У больных СМТ 1А отмечает¬
ся дупликация исследуемой хромосомной области (в дорожке 1 ви¬
зуализируются по 3 аллеля каждого из маркеров, в дорожке 3 визу¬
ализируются 3 аллеля маркера 0178122 и двойная доза нижнего
аллеля маркера 0178921). Отдельные аллели изучаемых маркеров
(>бозначены стрелками.
184Глава 3Дупликация 17р11.2 диагностируется либо на основа¬
нии регистрации трех различных ПЦР-продуктов (если
все аллели имеют различную длину), либо на основании
двойной интенсивности сигнала одной из полос (если
два аллеля являются одинаковыми и «накладываются»
друг на друга на электрофореграмме). Разновидностью
указанного метода анализа полиморфных маркеров из
области 17pl 1.2 является флюоресцентная гибридизация
in situ (FISH) с маркерными зондами, позволяющая ре¬
гистрировать у больных СМТ 1А три пятна от гибриди-
зованного флюоресцирующего маркера [Lupski J. et al.,1991]. Существует также ряд других, более сложных в
техническом плане подходов для прямой регистрации
дупликации критической хромосомной области 17р 11.2:
а) блот-гибридизация по Саузерну с использованием раз¬
личных зондов из дуплицируемой области; б) гель-элек-
трофорез в пульсирующем поле, позволяющий регист¬
рировать изменение подвижности сверхкрупных (дуп¬
лицированных) фрагментов ДНК, полученных после ре¬
стрикции геномной ДНК и гибридизованных с соответ¬
ствующими зондами; в) обнаружение (на основе рест¬
рикции и последующей гибридизации) рекомбинантно¬
го участка в локусе 17pl 1.2, послужившего источником
дупликации [Patel P. et al., 1992; Raeymaekers P. et al.,
1992; Timmerman V. et al., 1997].При отсутствии данных за дупликацию в области
17р 11.2 проводится последовательный поиск точковых
мутаций в генах РМР22, Р0, Сх32 и ERG2. Порядок ис¬
следования генов может варьироваться в зависимости
от относительной частоты встречаемости мутаций в них
в различных популяциях, а также в зависимости от осо¬
бенностей генеалогии конкретной родословной: напри¬
мер, при обследовании семьи с подозрением на Х-сцеп-
ленный доминантный тип наследования НМСН I в пер¬
ДНК- диагностика наследственных болезней... 185вую очередь должен исследоваться ген Сх32. Для поис¬
ка точковых мутаций обычно используются стандартные
процедуры скрининга отдельных экзонов (SSCP-анализ,
гетеродуплексный анализ и др.) с последующим прямым
секвенированием предположительно мутантных образ¬
цов ДНК либо тотальное секвенирование всей кодирую¬
щей области гена [Nelis E. et al., 1999].Рис. 43. ДНК-диагностика наследственной невропатии с
I федрасположенностыо к параличам от сдавления с исполь¬
зованием полиморфного маркера Э178921
У больных отца и сына (дорожки 2 и 3) отмечается «потеряI егерозиготности» и наличие лишь одного маркерного аллеля.
Вследствие делеции изучаемой хромосомной области у сына на¬
блюдается видимое «отсутствие» отцовского аллеля (сын
унаследовал от больного отца мутантную хромосому, не дающуюII родукта амплификации).■12 3Для диагностики делеции в области 17р11.2 у
больных ННППС используются те же методы, что и для
186Глава 3выявления дупликации при СМТ 1А. При этом анализ
высокополиморфных маркеров (СА)п позволяет диагно¬
стировать феномен «потери гетерозиготности» у боль¬
ного родителя и отсутствие родительского аллеля у боль¬
ного ребенка (рис. 43). Диагностика делеции возможна
также на основании прямого обнаружения половинной
дозы гена РМР22 (например, при блот-гибридизации ге-
номной/амплифицированной ДНК или количественном
определении ПЦР-продукта участка гена с использова¬
нием автоматического секвенатора). Нейрогенетическим
отделением НИИ неврологии РАМН проведен молеку¬
лярно-генетический анализ большой серии рецидиви¬
рующих невоспалительных невропатий на основе иссле¬
дования гена РМР22. Полученные нами результаты сви¬
детельствуют о том, что деления РМР22 обусловливает
около 30% случаев рецидивирующих невропатий различ¬
ной локализации и более половины семейных форм дан¬
ных заболеваний (в том числе при семейной невропатии
лицевого нерва). Принимая во внимание сложность диф¬
ференциальной диагностики ННППС с другими форма¬
ми наследственных невропатий, а также несомненно су¬
ществующую недооценку распространенности ННППС,
можно сделать вывод о том, что прямая ДНК-диагнос-
тика является ключевым методом исследования и позво¬
ляет в абсолютном большинстве случаев безошибочно
диагностировать ННППС, не прибегая к необходимос¬
ти проведения такой инвазивной процедуры как биопсия
нерва. ДНК-диагностика должна применяться не толь¬
ко у больных, но и у их клинически здоровых родствен¬
ников из группы риска, поскольку заболевание может
иметь стертое и асимптомное течение. Больным со сво¬
евременно диагностированной ННППС необходимо ре¬
комендовать соответствующую коррекцию стиля жизни,
физических нагрузок и профессиональной ориентации.
ДНК- диагностика наследственных болезней ... 187что имеет решающее значение для профилактики нео¬
братимых и инвалидизирующих осложнений данного
заболевания. ДНК-диагностика ННППС позволяет из¬
бежать необоснованного направления больного к хирур¬
гу, поскольку оперативное лечение, являющееся в ряде
случаев наиболее эффективным и радикальным методом
терапии обычных туннельных компрессионных синдро¬
мов, при ННППС неэффективно и лишено смысла.Аутосомно-рецессивные формы НМСН I. Деми-
елинизирующие моторно-сенсорные невропатии могут
наследоваться и по аутосомно-рецессивному типу
(СМТ 4). Такие заболевания представляют собой исклю¬
чительную редкость и описаны в единичных семьях
[Harding А., 1995]. При подозрении на аутосомно-рецес-
сивную форму НМСН I необходимо тщательное клини¬
ческое и электрофизиологическое обследование родите¬
лей пробанда с целью исключения у них субклинически
протекающей невропатии, т.е. для исключения аутосом-
но-доминатного наследования. На сегодняшний день из¬
вестны 7 самостоятельных генетических вариантов ауто-
сомно-рецессивных демиелинизирующих моторно-сен-
сорных невропатий - они сцеплены с хромосомами
8ql3—21 (СМТ 4А) [Ben Othmane К. et al., 1993 (а)],1 lq23 (СМТ 4В1) [Bolino A. et al., 1996], 11р15 (СМТ
4B2) [Ben Othmane K. et al., 1999], 5q23-33 (СМТ 4C)
| Le Guern E. et al., 1996], 8q24.3 (CMT 4D) [Kalaydjieva L.
et al., 1996], 10q21.1-22.1 (СМТ 4E) [Warner E. et al., 1998]
и 19ql3.1-13.3 (CMT 4F) [Delague V. et al., 2000]. Bee
указанные формы аутосомно-рецессивных демиелини-
шрующих моторно-сенсорных невропатий описаны в
семьях с кровнородственными браками. Общими осо¬
бенностями клинической картины являются более тяже¬
лое течение и частое сочетание с глухотой и атрофией
фительных нервов.
188Глава 3Гены 3 форм аутосомно-рецессивных демиелини-
зирующнх моторно-сенсорных невропатий к настояще¬
му времени идентифицированы. Форма СМТ 4В1 обус¬
ловлена мутациями в гене тирозин-фосфатазы (МТМ112),
имеющей отношение к белку аксонального цитоскелета
миотубулярину [ВоНпо А. е! а1., 2000]; детальные меха¬
низмы повреждения асконов периферических нервов при
СМТ 4В1 не установлены. Форма СМТ 4Е представляет
собой тяжелую врожденную гипомиелинизирующую
полиневропатию, обусловленную гомозиготностью по
мутациям в гене Е0112 [\Уатег Е. е! а1., 1998]; как ука¬
зывалось выше, гетерозиготные мутации в этом же гене
являются причиной развития более мягкой аутосомно-
доминантной формы демиелинизирующей моторно-сен¬
сорной невропатии. Наконец, в основе развития СМТ 40
лежат мутации в гене сигнального белка N01161, регу¬
лирующего процессы роста и дифференцировки шван-
новских клеток и взаимодействие между шванновскими
клетками и аксоном, а также участвующего в поддержа¬
нии целостности структуры зрелого аксона
[Ка1аус1]1еуа Ь. е! а1., 2000]. Интересно отметить, что ген
еще одной формы - СМТ 4Б - расположен в том же ло-
кусе на хромосоме 19ql 3, что и ген, кодирующий белок
цитоскелета периаксин, мутации в котором вызывают
аутосомно-рецессивный вариант синдрома Дежерина-
Сотта (см. выше). Весьма вероятно, таким образом, что
СМТ 4¥ и указанный вариант синдрома Дежерина-Сот-
та являются аллельными заболеваниями.В связи с тем, что четкие фенотипические разли¬
чия между отдельными формами аутосомно-рецессив-
ных демиелинизирующих моторно-сенсорных невропа¬
тий (за исключение СМТ 4Е) отсутствуют, прямая ДНК-
диагностика данных заболеваний возможна только пос¬
ле обнаружения достоверного сцепления с соответству¬
ющими хромосомами. При развитии тяжелого аутосом-
ДНК- диагностика наследственных болезней ... 189но-рецессивного варианта гипомиелинизирующей поли¬
невропатии, проявляющейся с момента рождения ребен¬
ка, можно диагностировать форму СМТ 4Е и предпри¬
нять исследование гена EGR2, которое проводится стан¬
дартными методами мутационного скрининга. В осталь¬
ных случаях при обследовании больших информативных
родословных возможно проведение анализа генетичес¬
кого сцепления с маркерами из соответствующих локу-
сов и в случае подтверждения высокого 77од-балла- про¬
ведение косвенной ДНК-диагностики у членов семьи из
группы риска.3.1.3.2. Аксональные моторно-сенсорные невропатии
(НМСН тип II)НМСН типа II являются относительно редкими
заболеваниями (4-12 случаев на 100 ООО) [De Jonghe P.
et al., 1998]. Большинство заболеваний из группы НМСН
II наследуются по аутосомно-доминантному типу, опи¬
саны также отдельные семьи с аутосомно-рецессивным
и Х-сцепленным рецессивным наследованием
| Harding А., 1995]. По сравнению с НМСН I, для
IIMCH II характерен более поздний возраст начала бо¬
лезни (в среднем на 10 лет позже), меньшее вовлечение
мелких мышц кисти и меньшая степень угнетения сухо¬
жильных рефлексов; при отдельных молекулярных ва¬
риантах НМСН II может выявляться также ряд допол¬
нительных симптомов, не свойственных демиелинизи-
нующим формам НМСН [Dyck P. et al., 1993; Chance P.,
l'ïschbeck К., 1994].Молекулярная генетика аксональных форм мотор¬
но-сенсорных невропатий остается малоизученной. Име-
li н циеся данные свидетельствуют в пользу несомненной
к-нстической гетерогенности НМСН II: на различных
чромосомах картированы как минимум 7 самостоятель¬
ных локусов НМСН II. Аутосомно-доминантные формы
IIMCH II сцеплены с хромосомами 1 рЗ5—36 (локус
190Глава 3СМТ 2А), 3ql3—22 (СМТ 2В), 7р14 (СМТ 2D) и 7qll-
21 (СМТ 2F) [Ben Othmane К. et al., 1993 (b); Kwon J. et
al., 1995; Ionaseseu V. et al., 1996; Ismailov S. et al., 2001].
Характерной особенностью формы СМТ 2В является вы¬
раженный сенсорный компонент и развитие трофичес¬
ких язв нижних конечностей, а СМТ 2D - более отчет¬
ливое и раннее вовлечение рук. Совсем недавно груп¬
пой российских ученых из Медико-генетического науч¬
ного центра РАМН был идентифицирован еще один ге¬
нетический вариант доминантной аксональной мотор-
но-сенсорной невропатии (СМТ 2Е), связанный с мута¬
цией гена одного из белков аксонального цитоскелета -
белка легкого нейрофиламента (ген расположен на хро¬
мосоме 8р21) [Mersiyanova I. et al., 2000]. Известны два
локуса аутосомно-рецессивной формы НМСНII: они кар¬
тированы на хромосомах lq21.2-21.3 [Bouhouche A. et
al., 1999] и 19ql3.3 [Leal A. et al., 2001]. Наконец, X-
сцепленная рецессивная форма НМСН II, ассоциирован¬
ная с глухотой и умственной отсталостью (синдром
Cowchock), связана с хромосомным локусом Xq24-26
[Priest J. et al., 1995]. В отличие от перечисленных дис¬
тальных вариантов НМСН II (фенотип Шарко—Мари-
Тута), Takashima Н. с соавторами описали особую фор¬
му аксональной НМСН с преимущественно проксималь¬
ным типом вовлечения скелетной мускулатуры; ген дан¬
ного аутосомно-доминантного заболевания был карти¬
рован на хромосоме 3ql3.1 [Takashima Н. et al., 1999].
Для всех генетических вариантов аксональных НМС11
при наличии достаточно больших родословных возмож
но исследование высокополиморфных маркеров из ука¬
занных хромосомных локусов с целью анализа генетн
ческого сцепления; при получении достоверных данны х
в пользу сцепления с определенным локусом может про
водиться косвенная ДНК-диагностика у родственники!!
из группы риска. В 8р21 -сцепленных семьях с домина! i
ДНК- диагностика наследственных болезней ... 191тной формой аксональной НМСН (СМТ 2Е) возможна
прямая ДНК-диагностика болезни на основе обнаруже¬
ния мутации в гене легкого нейрофиламента.Описаны казуистические случаи, когда у больных
с фенотипом НМСН II (т.е. при нормальных или субнор¬
мальных скоростях проведения импульса по перифери¬
ческим нервам) при ДНК-анализе выявлялись мутации
в генах Сх32 и Р0- т.е. молекулярные дефекты, обычно'
свойственные демиелинизирующим формам наслед¬
ственных невропатий [De Jonghe P. et al., 1998]. Мута¬
ции в указанных генах могут выявляться также у боль¬
ных, имеющих при электронейромиографическом иссле¬
довании «промежуточные» значения скоростей прове¬
дения импульса (между 30 и 40 м/с). Таким образом,
данные гены должны становиться объектами анализа в
тех редких случаях, когда точное отнесение наследствен¬
ной невропатии к демиелинизирующим или аксональ-11 ым формам на основании клинических, электрофизио-
ногических и гистологических данных затруднено. По
мнению Timmerman V. et al. (19966), мутационный скри¬
нинг гена Сх32 целесообразно проводить при НМСН II,
когда в обследуемой родословной не зарегистрировано
случаев передачи болезни от отца сыну (т.е. когда не ис¬
ключен Х-сцепленный тип наследования, характерный
для СМТ IX).L 1.3.3. Моторные, сенсорные и сенсорно¬
вегетативные невропатииНаследственные моторные невропатии - группа
редких заболеваний периферических нервов, характери¬
зующихся классическим дистальным фенотипом мышеч¬
ных атрофий и парезов, нормальными или слегка сни¬
ми 11ыми скоростями проведения импульса по двигатель-
нмм волокнам и сохранностью проведения по сенсор¬
ным волокнам периферических нервов. При многих фор¬
192Глава 3мах этих заболеваний выявляются признаки поражения
спинальных структур (например, периферического мо¬
тонейрона или пирамидного тракта), поэтому нередко
наследственные моторные невропатии (точнее-нейроно-
патии) обозначаются в литературе как «спинальный ва¬
риант болезни Шарко-Мари-Тута» [De Jonghe P. et al.,
1998]. На основании особенностей клинической карти¬
ны и типов наследования выделяют 7 форм наследствен¬
ных моторных невропатий (нейронопатий), для четырех
из которых установлена локализация мутантных генов.
Локус дистальной формы типа II (возраст начала симп¬
томов между 20 и 40 годами) расположен на хромосоме
12q24 [Timmerman V. et al., 1996a]; локус дистальной
формы типа V (характеризующейся преимущественным
вовлечением рук) расположен на хромосоме 7р
[Christodoulou К. et al., 1995]; локусы редких форм дис¬
тальных моторных нейронопатий VI и VII типов (ослож¬
ненных, соответственно, тяжелым респираторным дис¬
трессом и параличем голосовых связок) , картированы
на хромосомах 11 ql3—21 и 2ql4 [McEntagart m. et al.,
2001]. Известна также ювенильная моторная нейроно-
патия с вовлечением пирамидального тракта (форма
Jerash), ген которой локализован на хромосоме 9р21.1—12 [Christodoulou К. et al., 2000].Наследственные сенсорные и сенсорно-вегетатив¬
ные невропатии представляют собой редкие формы не¬
вропатий, характеризующиеся дистальными нарушени¬
ями чувствительности, трофическими расстройствами и
язвами в пораженных областях (вплоть до мутиляций),
остеомиелитом и деструкцией костной ткани дисталь¬
ных отделов конечностей. В ряде случаев на поздней
стадии могут присоединяться амиотрофии и слабость
дистальной мускулатуры конечностей. В основе данных
заболеваний лежит прогрессирующая дегенерация ней¬
ронов заднекорешковых ганглиев, обычно в том или ином
ДНК- диагностика наследственных болезней ... 193сочетании с поражением систем периферического веге¬
тативного и двигательного нейронов. Выделяют 4 ос¬
новных клинико-генетических варианта наследственных
сенсорных и сенсорно-вегетативных невропатий, пере¬
дающиеся по аутосомно-доминантному и аутосомно-ре-
цессивному типам. Ген аутосомно-доминантной сенсор¬
ной невропатии I типа был картирован на хромосоме 9q22
в семьях различного этнического происхождения
[Nicholson G. et al., 1996; Bejaoui K. et al., 1999]. Ген не¬
вропатии III типа (син.: синдром Рейли-Дея, семейная
вегетативная дисфункция) - аутосомно-рецессивного
заболевания, распространенного почти исключительно
в популяции евреев ашкенази - картирован на хромосо¬
ме 9q31—33 [Blumenfeld A. et al., 1993]. Непосредствен¬
ные генетические и белковые дефекты не установлены.Для охарактеризованных выттте генетических ва¬
риантов наследственных моторных и сенсорно-вегета-
гивных невропатий возможно проведение косвенной
ДНК-диагностики в случае подтверждения в изучаемой
семье сцепления с одним из известных хромосомных
локусов. При наследственных моторных невропатиях
необходимо проводить дифференциальную диагности¬
ку с наиболее частыми формами спинальной амитрофии
| De Jonghe Р. et al., 1998] и с этой целью исключать го¬
мозиготную делецию гена SMN на хромосоме 5q (см.
далее - раздел 3.1.4).3.1.4. Спинальные амиотрофии и другиенаследственные болезни мотонейронаВ современной номенклатуре нейродегенератив-
IH.IX заболеваний в последние годы принято выделять
особую группу болезней, характеризующихся дегенера¬
цией периферического двигательного нейрона - так на¬
чинаемые болезни мотонейрона (motor neuron diseases)
11 cigh Р., Ray-Chaudhuri K., 1994]. К данной категории
194Глава 3относятся, главным образом, боковой амиотрофический
склероз (который в небольшом числе случаев может но¬
сить наследственно-семейный характер) и спинальные
амиотрофий. В настоящем разделе основное внимание
будет посвящено спинальным амиотрофиям - гетероген¬
ной группе наследственных заболеваний нервной систе¬
мы, связанных с поражением мотонейронов передних
рогов спинного мозга и в некоторых случаях - ствола
мозга, в результате чего развиваются денервационные
атрофии и парезы соответствующих мышечных групп.3.1.4.1. Проксимальные спинальные амиотрофии
детского возрастаСреди всех спинальных амиотрофий заболевания
с ранним началом и преимущественным вовлечением
проксимальной мускулатуры являются наиболее распро-
страненными и изученными формами (1:10 ООО)
[Emery А., 1991; International SMA Consortium, 1992]. Тип
наследования в абсолютном большинстве случаев - ауто-
сомно-рецессивный. В соответствии с особенностями
течения проксимальные аутосомно-рецессивные спи¬
нальные амиотрофии подразделяются на три основных
клинических варианта: 1) острая инфантильная форма
Верднига-Гофмана (1-й тип спинальной амиотрофии) -
характеризуется развитием тяжелой генерализованной
мышечной слабости и гипотонии на протяжении пер¬
вых 3 месяцев жизни и наступлением летального исхода
вследствие дыхательных нарушений на 1-2 году жизни
ребенка; 2) «промежуточная» или хроническая инфан¬
тильная форма (2-й тип), при которой ребенок заболева¬
ет в раннем детстве, может научиться самостоятельно
сидеть, однако обычно не способен стоять или ходить
без поддержки; 3) хроническая ювенильная форма, или
болезнь Кугельберга-Веландер (3-й тип) - характеризу¬
ется началом болезни на 1-м или 2-м десятилетии жиз¬
ДНК- диагностика наследственных болезней ... 195ни и более медленным течением на протяжении десяти¬
летий [International SMA Consortium, 1992].Все три формы аутосомно-рецессивной спиналь¬
ной амиотрофии являются аллельными заболеваниями,
обусловленными мутациями одного гена, локализован¬
ного на длинном плече 5-й хромосомы [Gilliam T. et al.,
1990; Melki J. et al., 1990]. Данный ген, идентифициро¬
ванный в 1995 г. и состоящий из 8 экзонов, получил обо¬
значение SMN (аббревиатура от англ. Survival Motor
Neuron - «ген выживаемости мотонейрона»)
[Lefebvre S. et al., 1995]. Ген SMN представлен в облас¬
ти 5ql3 двумя копиями - теломерной (SMNT), располо¬
женной дистально ближе к теломере хромосомы, и цен¬
тромерной (SMNC), расположенной более проксималь¬
но [Lefebvre S. et al., 1995]. Высокая нуклеотидная гомо¬
логия между SMNT и SMNC способствует нарушению
нормальных механизмов кроссинговера и выраженной
генетической нестабильности критического участка хро¬
мосомы 5ql3, результатом чего является образование
гамет, в которых ген SMNT утрачен или замещен его
центромерной копией SMNC [Hahnen E. et al., 1996;
Campbell L. et al., 1997; Lefebvre S. et al., 1998]. В целом
около 98,6% больных проксимальной спинальной ами-
( > I рофией имеют гомозиготную делецию всего гена SMN1'
либо его части [Cobben J. et al., 1995; Lefebvre S. et al.,
1995; Rodrigues N. et al., 1995]; в то же время центро¬
мерные копии гена обычно остаются интактными либо
обнаруживаются в увеличенном числе копий [Taylor J.
et al., 1998; Vitali T. et al., 1999]. В единичных случаях11 роксимальной спинальной амиотрофии показано нали¬
чие патогенетически значимых точковых мутаций в геие
SMNT [BussagliaE. et al., 1995; Lefebvre S. et al., 1995].
lice эти данные свидетельствуют о непосредственном
новлечении теломерной копии гена SMN (SMNT) в па-
Iогенез аутосомно-рецессивной проксимальной спиналь¬
196Глава 3ной амиотрофии. Поскольку для различных по тяжести
вариантов спинальной амиотрофии (1-й, 2-й или 3-й тип)
характерен один и тот же первичный молекулярный де¬
фект - деления гена SMNT, имеются дополнительные
генетические факторы, модифицирующие характер кли¬
нических проявлений болезни. К таким факторам отно¬
сятся протяженность делеции и вовлечение близлежа¬
щего гена белка-ингибитора апоптоза (NAIP), число со¬
хранных копий гена SMNC и др. [Lefebvre S. et al., 1995;
Velasco E. et al., 1996; Vitali T. et al., 1999]. Показано, в
частности, что у больных с наиболее «мягким» феноти¬
пом - болезнью Кугельберга-Веландер - число копий
гена SMNC составляет в среднем 4, тогда как при тяже¬
лой форме (болезнь Верднига-Гофмана) число копий
SMNC обычно не превышает 1-2 [Campbell L. et al., 1997;
Taylor J. et al., 1998; Vitali T. et al., 1999]. В связи с этим
предполагается, что экспрессия гена SMNC имеет функ¬
циональное значение и в определенной степени может
компенсировать (хотя бы частично) отсутствие у боль¬
ных «основного» гена SMNT.Исследование гена SMN открыло возможность
проведения прямой ДНК-диагностики аутосомно-рецес-
сивной спинальной амиотрофии детского возраста. Ос¬
новной технической проблемой при этом является не¬
обходимость дифференцировать теломерную (значимую)
копию SMN от гомологичной центромерной копии. Нук¬
леотидные последовательности теломерной и центромер¬
ной копий гена SMN практически идентичны, за исклю¬
чением 7-го и 8-го экзонов: по каждому из этих экзонов
SMNT отличается от SMNC на один нуклеотид
[Lefebvre S. et al., 1995]. Указанные различия нуклеотид
ного состава SMNT и SMNC позволяют дифференциро
вать эти гены с помощью сравнительно простого мето
да, предложенного van der Steege G. с соавт. в 1995 году.
ДНК- диагностика наследственных болезней... 197Метод основан на использовании рестрикционных
эндонуклеаз, имеющих сайты распознавания в указан¬
ных вариабельных участках 7-го и 8-го экзонов вМЫ.
Данные ферменты расщепляют амплифицированные
экзоны 8М№ на более короткие фрагменты, тогда как
для тех же экзонов БМЫ7 рестрикция невозможна (рис. 44).1 2 3 4 5I*ис. 44. Прямая ДНК-диагностика аутосомно-рецессивной
проксимальной спинальной амиотрофии детского возраста
11 родукты амплификации 8-го экзона гена SMN обработаны рест-
риктазой Dde I. Длинной стрелкой указан продукт амплификации
гена SMNT, короткими стрелками - продукт амплификации гена
SMNC, разрезанный рестриктазой на 2 фрагмента. Дорожка 1 - мар¬
кер, дорожка 2 - больной спинальной амиотрофией Кугельберга-
Неландер (гомозиготная делеция гена SMN'1'), дорожки 3,4- боль¬
шие с другими молекулярными формами спинальных амиотрофий
( I ют гомозиготной делеции гена SMN1), дорожка 5 - контроль.Таким образом, отсутствие ЭМЬТ-специфичных
фрагментов ДНК позволяет установить гомозиготную
198Глава 3делецию гена Б МИ1 и тем самым диагностировать ауто-
сомно-рецессивную проксимальную спинальную амиот-
рофию (формы Верднига-Гофмана, Кугельберга-Велан-
дер или «промежуточный» вариант болезни).Как указывалось выше, у небольшого числа боль¬
ных (<2%) на одной из мутантных хромосом может иметь
место не полная деления гена БММ1, а точковая мутация
в нем, в связи с чем ПЦР-продукт данного аллеля будет
визуализироваться на электрофореграмме. В этих слу¬
чаях для ДНК-диагностики болезни необходимо опре¬
деление дозы гена Б МЫ'1, т.е. доказательство наличия у
больного одной копии БМЫ1 по сравнению с двумя ко¬
пиями данного гена в контрольных образцах. Для ана¬
лиза дозы гена требуется постановка реакции в особых
стандартизированных условиях (количественная ПЦР):
отсутствие одной копии БМЫ1 может быть определено
либо визуально - на основании вдвое меньшей интен¬
сивности окрашивания соответствующего фрагмента
ДНК по отношению к норме, либо с помощью компью¬
терного анализа относительной площади флюоресцен¬
ции пиков ДНК. Для этой же цели может использовать¬
ся денситометрия радиоавтографа, полученного при про¬
ведении блот-гибридизации. С практической точки зре¬
ния, выявление половинной дозы гена БМИ1 (т.е. его
делеции в гетерозиготном состоянии) позволяет, при
наличии соответствующей клинической картины, с вы¬
сокой степенью вероятности склониться к диагнозу ауто-
сомно-рецессивной 5qlЗ-cцeплeннoй спинальной ами-
отрофии. Однако достоверная диагностика в этом слу¬
чае возможна только при условии идентификации точ-
ковой мутации во втором аллеле гена.Наш собственный 4-летний опыт проведения пря¬
мой ДНК-диагностики данных заболеваний в семьях из
европейской и центральной части России показал, что
при анализе строго отобранной серии классических форм
ДНК- диагностика наследственных болезней ... 199спинальной амиотрофии более чем в 90% случаев выяв¬
ляется гомозиготная делеция 7-го и 8-го экзонов гена
SMNT. Это соответствует данным большинства авторов
и позволяет заключить, что в нашей стране данный тип
мутации также является ведущей причиной развития
аутосомно-рецессивной спинальной амиотрофии детс¬
кого возраста. Таким образом, сравнительно простая
ДНК-диагностика спинальной амиотрофии возможна в
абсолютном большинстве случаев данного заболевания.
Следует отметить, однако, что в отдельных популяциях
(особенно характеризующихся высоким уровнем инбри¬
динга и «эффектом основателя») соотношение основных
типов мутаций может иметь существенные особеннос¬
ти. Так, нами совместно с канд. мед. наук Г.Х. Багыевой
в 1998-1999 гг. были обследованы 8 семей с аутосомно-
рецессивной спинальной амиотрофией из Ахалского ве-
лаята Туркменистана: гомозиготная делеция 7-го и 8-го
экзонов гена SMNT была выявлена лишь в 2 семьях, что
может свидетельствовать о накоплении других мутаций
SMNT либо о генетической гетерогенности данного за¬
болевания в туркменской популяции.Диагностика гетерозиготного носительства деле¬
нии гена SMNT у родителей больных спинальной ами¬
отрофией основывается на описанных выше методах
определения дозы гена. В случае выявления у обоих ро¬
дителей носительства делеции в гетерозиготном состоя¬
нии можно сделать вывод о том, что каждая из мутант¬
ных хромосом унаследована пробандом от родителей, а
не возникла de novo. В такой ситуации риск повторного
возникновения заболевания в семье для каждого из сиб-
сов равен 25%, что требует подтверждения с помощью
прямой ДНК-диагностики (в том числе возможно про-
медение пренатальной ДНК-диагностики с целью про¬
филактики рождения больного ребенка).
200Глава 33.1.4.2. Спинально-бульбарная амиотрофия Кеннеди
(болезнь Кеннеди)Спинально-бульбарная амиотрофия Кеннеди -
редкое заболевание, характеризующееся Х-сцепленным
рецессивным типом наследования и проявляющееся у
мужчин в относительно позднем возрасте (обычно после
40 лет). Типичная клиническая картина включает мед¬
ленно прогрессирующую мышечную слабость, амио гро-
фии и фасцикуляции проксимальных отделов конечнос¬
тей, бульбарные симптомы денервационного характера
(дизартрия, дисфагия, фибрилляции языка), а также ха¬
рактерные эндокринные расстройства (гинекомастия,
тестикулярная атрофия) [Kennedy W. et al., 1968]. На
поздней стадии может вовлекаться проксимальная мус¬
кулатура ног.Заболевание обусловлено повреждением гена ан¬
дрогенного рецептора, расположенного в локусе Xql 1.2—12 [La Spada A. et al., 1991]. У всех больных амиотрофи-
ей Кеннеди имеет место экспансия тандемных тринук-
леотидных повторов CAG в 1-м экзоне гена: в норме
число копий CAG-повторов составляет 9-36, тогда как
больные амиотрофией Кеннеди имеют увеличенное чис¬
ло тандемных повторов - от 38 до 72 [La Spada Ä. et al.,
1991; Igarashi S. et al., 1992; Amato A. etal., 1993]. Такой
характер мутации на белковом уровне проявляется па¬
тологическим удлинением соответствующего полиглута-
минового участка белка, что лишь в небольшой степени
влияет на нормальную функцию андрогенного рецепто¬
ра (у больных отмечается лишь умеренное снижение чув¬
ствительности к действию андрогенов). Как и при дру¬
гих «полиглутаминовых» болезнях, поражение ЦНС при
болезни Кеннеди связывается с тем, что мутантный бе¬
лок приобретает новые цитотоксические свойства и спо¬
собствует формированию патологических внутриядер
ныхвключений [Mhatre A. etal., 1993; HousmanD., 1995
ДНК- диагностика наследственных болезней... 201Li М. et al., 1998]. При этом с увеличением числа CAG-
повторов и длины полиглутаминового участка заболева¬
ние характеризуется более тяжелым течением и более
ранним началом [Igarashi S. et al., 1992; La Spada A. et al.,1992]. Интересно отметить, что точковые мутации в дан¬
ном гене, приводящие к инактивации андрогенного ре¬
цептора, сопровождаются развитием совершенно другого
заболевания - так называемого синдрома тестикуляр¬
ной феминизации [Gottlieb В. et al., 1998]. Таким обра¬
зом, различные по своей сущности мутации в гене анд¬
рогенного рецептора, по-разному влияющие на функцию
данного белка, лежат в основе принципиально различ¬
ных форм патологии.1 2 3 4 5Рис 45. Прямая ДНК-диагностика спинально-бульбарной
; I м иотрофии Кеннеди]1,орожка 1 - маркер, дорожки 2,3 - контроль, дорожка 4 - больной
пшнально-бульбарной амиотрофией Кеннеди, дорожка 5 - мать
(юл ьного (гетерозиготная носительница мутации). Длинной стрелкой
указан мутантный аллель (экспансия САО-повторов гена андроген¬
ного рецептора), короткой стрелкой - нормальный аллель.
202Глава 3Прямая ДНК-диагностика болезни Кеннеди яв¬
ляется относительно несложной и основана на ПЦР-ам-
плификации фрагмента 1-го экзона гена, содержащего
тринуклеотидный участок. У больных мужчин мутант¬
ный аллель (продукт единственной Х-хромосомы) чет¬
ко определяется благодаря более медленной электрофо¬
ретической подвижности, что является следствием уве¬
личенного числа тринуклеотидных С AG-повторов (рис.
45, дорожка 4). У женщин-носительниц на электрофо-
реграмме визуализируются нормальный и мутантный
аллели (рис. 45, дорожка 5), что позволяет достоверно
диагностировать наличие мутации в гетерозиготном со¬
стоянии. В отягощенных семьях возможно проведение
ранней пресимптоматической ДНК-диагностики болез¬
ни у лиц мужского пола, а также пренатальной ДНК-
диагностики.3.1.4.3. Семейный боковой амиотрофический склероз
Боковой амиотрофический склероз - тяжелое де¬
генеративное заболевание ЦНС позднего возраста (сред¬
ний возраст начала составляет около 55 лет). Оно харак¬
теризуется прогрессирующей гибелью центральных и
периферических мотонейронов с развитием парезов ске¬
летной и бульбарной мускулатуры, амиотрофий, фасци-
куляций и пирамидной спастичности; летальный исход
(главным образом, вследствие дыхательных нарушений)
наступает в среднем через несколько лет от момента
появления первых симптомов болезни [Leigh P., Ray-
Chaudhuri К., 1994]. Распространенность заболевания
составляет 4-6 случаев на 100 ООО населения. В боль¬
шинстве случаев боковой амиотрофический склероз
представляет собой спорадическое заболевание мульти-
факториальной этиологии. У 5-10% больных, однако
может выявляться положительный семейный анамнез,
ДНК- диагностика наследственных болезней203свидетельствующий о моногенном аутосомно-доминан-
тном или (гораздо реже) аутосомно-рецессивном насле¬
довании заболевания среди родственников [Siddique Т.,
Deng H., 1996]. Клиническая картина семейных и спо¬
радических форм бокового амиотрофического склероза
в большинстве случаев практически идентична (некото¬
рые исключения рассмотрены ниже), однако в целом для
семейных форм заболевания характерен более ранний
дебют симтомов.Приблизительно у 20% больных с аутосомно-
доминантной формой бокового амиотрофического скле¬
роза заболевание обусловлено мутациями гена цитозоль¬
ного фермента Cu/Zn супероксиддисмутазы (SOD1) на
хромосоме 21q22 [Rosen D. et al., 1993]. К настоящему
времени известно свыше 50 мутаций, распределенных
по всем экзонам SOD1 (они в основном представлены
точковыми мутациями, реже вставками и делениями)
[Siddique Т., Deng П., 1996; Rowland L., 1998]. Некото¬
рые из мутаций описаны лишь в отдельных семьях, дру¬
гие являются мажорными в определенных популяциях.
Так, мутация Ala4Val обусловливает половину всех слу¬
чаев данной молекулярной формы семейного бокового
амиотрофического склероза в Северной Америке
[Cudkowicz М. et al., 1997]. Мутации в гене SOD1 обна¬
руживаются также у 2-5% больных со спорадической
формой бокового амиотрофического склероза [Jones С.
et al., 1995; Andersen R et al., 1997]. Предполагается, что
в этих случаях имеет место «ложно-отрицательный се¬
мейный анамнез» (ранняя смерть или поздняя манифес¬
тация болезни у одного из родителей) либо неполная
пенетрантность мутантного гена [Rowland L., 1998].
Интересно отметить, что мутация Asp90Ala в ряде слу¬
чаев приводит к развитию болезни лишь будучи в гомо¬
зиготном состоянии и, таким образом, может являться
204Глава 3причиной аутосомно-рецессивной формы бокового ами¬
отрофического склероза [Andersen P. et al., 1997].В казуистических случаях отдельные мутации
SOD1 могут быть ассоциированы с весьма атипичными
вариантами болезни - например, характеризующимися
длительным течением свыше 20 лет [Aoki М. et al., 1994]
либо отсутствием вовлечения верхнего мотонейрона
[Cudkowicz М. et al., 1997; Rowland L., 1998]. В связи с
этим для семейных случаев обсуждается возможность
пересмотра классических клинических критериев боко¬
вого амиотрофического склероза (так называемых «кри¬
териев Эль-Эскориаль»), По мнению Cudkowicz М. et
al. (1997), для достоверной диагностики семейных слу¬
чаев болезни может быть достаточным наличие следую¬
щих двух признаков: 1) симптоматика поражения пери¬
ферического мотонейрона в трех конечностях; 2) обна¬
ружение мутации в гене SOD1. Таким образом, мутаци¬
онный скрининг SOD 1 может являться ценной диагнос¬
тической процедурой в семейных (особенно атипично
потекающих) случаях бокового амиотрофического скле¬
роза. В семьях с идентифицированной мутацией возмож¬
но проведение прямой ДНК-диагностики у лиц из груп¬
пы риска. В целом, однако, практическое значение ДНК-
диагностики при данном заболевании остается на сегод¬
ня сравнительно ограниченным, поскольку вероятность
выявления мутации невелика: как указано выше, в 80%
аутосомно-доминантных случаев заболевание не связа¬
но с мутациями в гене SOD1.В отличие от классической фатальной формы бо¬
кового амиотрофического склероза с поздним началом
в литературе описаны редкие семьи так называемого юве¬
нильного бокового амиотрофического склероза. Для него
характерно начало болезни чаще всего на 2-м десятиле¬
тии жизни, вариабельное соотношение выраженности
ДНК- диагностика наследственных болезней ... 205поражения центрального и периферического мотоней¬
ронов, а также весьма медленное прогрессирование, в
некоторых случаях даже не влияющее на естественную
продолжительность жизни [Siddique T., Deng Н., 1996].
Ювенильный боковой амиотрофический склероз может
наследоваться по аутосомно-доминантному и аутосом-
но-рецессивному типу. Ген аутосомно-доминантной фор¬
мы картирован на хромосоме 9q34 [Chance P. et al., 1998].
Аутосомно-рецессивная форма является генетически ге¬
терогенной: один локус расположен на хромосоме 2q33
(тунисский вариант болезни), другой (более частый ва¬
риант болезни, обнаруживаемый в Северной Африке и
Европе) - на хромосоме 15ql5.1-22.1 [Hentati A. et al.,
1994а; 1998]. Первичные молекулярные дефекты, ответ¬
ственные за развитие ювенильного бокового амиотро¬
фического склероза, до настоящего времени не установ¬
лены. В информативных семьях возможно проведение
косвенной ДНК-диагностики на основе анализа сцепле¬
ния с генетическими маркерами хромосом 2q, 9q и 15q.3.1.5. Нервно-мышечные каналопатииБольшое число наследственных нервно-мышеч¬
ных заболеваний в соответствии с характером первич¬
ного молекулярного дефекта может быть отнесено к ка-
налопатиям - заболеваниям, обусловленным мутациями
генов ионных каналов [Bulman D., 1997; Jurkat-Rott К. et
al., 1999]. Само представление о каналопатиях в невро¬
логии было в значительной степени связано с изучени¬
ем именно нервно-мышечных заболеваний, поскольку
поперечно-полосатая мышца относится к возбудимым
тканям, нормальное функционирование которых опре¬
деляется в первую очередь взаимодействием трансмем¬
бранных ионных потоков и состоянием ионных каналов.
206Глава 3Патология мышечного хлорного канала, обуслов¬
ленная точковыми мутациями гена хлорного канала
(CLC-1) на хромосоме 7q35, лежит в основе развития 2
аллельных форм наследственной миотонии - аутосом-
но-доминантной врожденной миотонии Томсена и ауто-
сомно-рецессивной генерализованной миотонии Бекера
[Koch М. et al., 1992]. Обе формы характеризуются на¬
рушением механизмов релаксации скелетных мышц пос¬
ле их сокращения (миотоническая реакция) в связи со
снижением проводимости мышечной мембраны для
ионов хлора, в норме обеспечивающих стабилизацию
мембранного потенциала покоя после акта сокращения.
Форма Бекера является более тяжелой и обычно сопро¬
вождается постепенным развитием постоянной мышеч¬
ной слабости даже вне миотонических приступов. С
современных позиций более тяжелое течение миотонии
Бекера может быть объяснено гомозиготностью по му¬
тациям гена хлорного канала (т.е. двойной дозой мутан¬
тного гена).Ряд других аутосомно-доминантных миотоничес¬
ких синдромов - врожденная парамиотония Эйленбур-
га, калий-индуцируемая и ацетазол-чувствительная ми-
отония, периодический парамиотонический паралич -
связаны с мутациями одного и того же гена а4-субъеди-
ницы мышечного натриевого канала (SCN4A) на хромо¬
соме 17ql3 [BulmanD, 1997; Kaplan J., Fontaine В., 1998].
Последняя из упомянутых форм (периодический пара¬
миотонический паралич) с клинической точки зрения
представляет собой как бы «переходный» синдром меж¬
ду миотониями и периодическим параличом. Более того,
одна из классических форм периодического паралича -
гиперкалиемическая форма - также обусловлена мута¬
циями в гене SCN4A [Bulman D., 1997]. Для гиперкали-
емической формы периодического паралича характерны
ДНК- диагностика наследственных болезней ... 207манифестирующие в детском возрасте кратковременные
приступы вялого паралича скелетных мышц (провоци¬
руемые, в частности, холодом или отдыхом после физи¬
ческой нагрузки), которые сопровождаются повышени¬
ем уровня калия в сыворотке крови; у ряда больных в
межприступном периоде выявляются симптомы миото-
нии век, языка и дистальных мышц рук. Таким образом,
повреждения а4-субъединицы мышечного натриевого
канала ответственны за целый спектр аллельных частич¬
но сходных нервно-мышечных заболеваний - от чистых
миотонических синдромов до периодических параличей
с миотоническими феноменами. Патофизиологической
основой данных синдромов является нарушение инак¬
тивации (закрытия) натриевого канала, в результате чего
имеет место патологическая «утечка» натриевого тока в
клетку и генерация потенциала действия; при этом со¬
пряженный калиевый ток движется через мембрану в
противоположном направлении, с накоплением ионов
калия в межклеточном пространстве и последующей
гиперкалиемией. Все это приводит к повторным пролон¬
гированным приступам деполяризации мышечной мем¬
браны (миотонический феномен) и, в наиболее тяжелых
случаях, к ее длительной рефрактерности (мышечные
параличи).Гипокалиемическая форма периодического пара¬
лича отличается от гиперкалиемической формы более
поздним началом, большей тяжестью и длительностью
приступов (от часа до нескольких дней), сниженным
уровнем сывороточного калия в момент приступа, а так¬
же провоцирующей ролью приема углеводной пищи и
физических нагрузок. Тип наследования аутосомно-до-
минантный. Гипокалиемическая форма периодического
иаралича вызывается мутациями гена САСЖЛАЗ, ко¬
дирующего синтез а,-субъединицы дигидроииридин-чув-
208Глава 3ствительного кальциевого канала (хромосома Ц31-32)
[Ви1шап Б., 1997; 1игка1-КоЦ К. ег а1., 1999]. Предпола¬
гается, что мутации приводят к уменьшению кальцие¬
вого тока внутрь миоцита, в результате чего содержание
ионов кальция в саркоплазме не достигает уровня, не¬
обходимого для инициации мышечного сокращения. При
дисфункции кальциевого канала, кроме того, имеет ме¬
сто компенсаторная аккумуляция ионов калия в клетке в
силу инсулин-опосредованной стимуляции натрий-кали-
евого насоса (что сопровождается гипокалиемией в мо¬
мент приступа).Некоторые мутации в указанном гене САСЖЛ АЗ
приводят к развитию самостоятельного аутосомно-до-
минантного нервно-мышечного заболевания - злокаче¬
ственной гипертермии. Она характеризуется приступа¬
ми генерализованной мышечной ригидности, гипертер¬
мии, метаболического ацидоза, рабдомиолиза и миогло¬
бинурии, которые провоцируются применением некото¬
рых анестетиков и деполяризующих мышечных релак¬
сантов [.Гигкап-ЯоЦ К. е! а1., 1999]. Еще один молекуляр¬
ный вариант злокачественной гипертермии может быть
связан с мутациями в гене саркоплазматического риано-
дин-чувствительного рецептора кальциевого канала
(КУШ) на хромо соме 19сх13.1 [.Тигкап-КоЦ К. е! а1., 1999].
Известно также несколько дополнительных хромосом¬
ных локусов аутосомно-доминантной злокачественной
гипертермии (см. приложение 1). Патофизиологической
основой указанных синдромов является активация со¬
ответствующего кальциевого канала, вызывающая пато¬
логический массивный выброс в цитоплазму мышечно¬
го волокна ионов кальция, депонированных в саркоп-
лазматическом ретикулуме; это сопровождается мышеч¬
ным сокращением и выраженным тканевым гипермета¬
болизмом с активизацией гликогенолиза, гиперпродук¬
ДНК- диагностика наследственных болезней ... 209цией лактата, нарушениями окислительного цикла и де¬
струкцией мышечного волокна.Примером патологии натриевых каналов скелет¬
ных мышц, регулируемых активностью никотиновых
ацетилхолиновых рецепторов (н-холинорецепторов), яв¬
ляются врожденные миастенические синдромы. Это ге¬
терогенная группа заболеваний, которые наследуются по
аутосомно-доминантному или, реже, аутосомно-рецес-
сивному типу и характеризуются ранней (нередко с рож¬
дения) манифестацией миастеноподобного феномена
различной степени тяжести и генерализации. Данные
заболевания обусловлены мутациями генов, кодирующих
a-, ß- и s-субъединицы мышечного н-холинорецептора
(хромосомы 2q и 17p) [Engel A. et al., 1996]. Мутации
приводят к нарушению инактивации и «протеканию»
сопряженного с рецептором натриевого канала, что со¬
провождается повреждением концевой пластинки вслед¬
ствие переизбытка катионов в постсинаптической обла¬
сти. Кроме того, нарушается порог безопасности нервно-
мышечной передачи в условиях «блока деполяризации»,
связанного с суммированием пролонгированных потен¬
циалов концевой пластинки в ответ на обычную физио¬
логическую стимуляцию [Engel A. et al., 1996].При указанных формах наследственных миото-
ний, пароксизмальных миоплегий, врожденной миасте¬
нии, злокачественной гипертермии и близких к ним не¬
рвно-мышечных заболеваний принципиально возможна
прямая ДНК-диагиостика с помощью стандартных под¬
ходов мутационного скрининга. На практике, однако,
ДНК-диагностика данных заболеваний пока не получи¬
ла широкого распространения. Это связано с их относи¬
тельной редкостью, сложностью мутационного поиска
п генетически гетерогенных группах, а также доступно¬
стью рутинных и достаточно надежных лабораторно¬
210Глава 3инструментальных тестов (электромиография и др.); в
то же время диагностика конкретной молекулярной фор¬
мы в рамках указанных групп заболеваний пока не име¬
ет существенного клинического значения для выбора
тактики лечения или определения прогноза.3.2. Наследственные заболеванияс преимущественным вовлечением
экстрапирамидной системы3.2.1. Хорея Гентингтона и другие формы
наследственной хореиХорея (болезнь) Гентингтона - тяжелое дегене¬
ративное заболевание головного мозга, распространен¬
ность которого в большинстве популяций мира состав¬
ляет 4-10 случаев на 100 ООО населения. Хорея Гентин¬
гтона наследуется по аутосомно-доминантному типу.
Морфологической основой болезни является атрофия
подкорковых узлов (главным образом полосатого тела и
бледного шара), а также гибель нейронов и атрофия коры
больших полушарий с развитием наружной и внутрен¬
ней гидроцефалии и прогрессирующим снижением об¬
щей массы мозга [Hayden М., 1981].Принято выделять две основных клинических
формы заболевания - гиперкинетическую и акинетико-
ригидную [Иванова-Смоленская И.А. и др., 1998 (а);
Penney J., Young А., 1998]. Гиперкинетическая форма
является наиболее типичной и характеризуется началом
болезни на 4-6 десятилетии жизни; у больных нараста¬
ют размашистые генерализованные хореические гипер-
кинезы и деменция подкоркового типа, развивающиеся
на фоне выраженных эмоционально-волевых нарушений.
Постепенное развитие экстрапирамидной ригидности,
ДНК- диагностика наследственных болезней ... 211кахексии и вегатативно-трофичееких нарушений знаме¬
нует собой переход болезни в так называемую позднюю
акинетико-ригидную форму, представляющую собой
финал заболевания. Самостоятельное значение имеет
первичный ювенильный акинетико-ригидный вариант
болезни Гентингтона (вариант Вестфаля), наблюдаемый
в 5-10% случаев. У таких больных заболевание мани¬
фестирует на 1-2-м десятилетии жизни и характеризу¬
ется особенно неблагоприятным течением, с развитием
мышечной ригидности, контрактур, изменений поведе¬
ния и психики, эпилептических припадков, миоклоний,
пирамидных симптомов, атаксии. Хореические гиперки-
незы могут быть выражены незначительно или вообще
отсутствовать, в связи с чем диагностика данного вари¬
анта болезни весьма затруднена. Ювенильный вариант
Вестфаля наблюдается обычно при передаче мутантно¬
го гена от больного отца, особенно если наследование
болезни по мужской линии происходило в нескольких
поколениях подряд (так называемый «эффект о тцовской
передачи») [Hayden М., 1981; Folstein S., 1989]. Еще од¬
ним характерным для хореи Гентингтона феноменом,
связанным преимущественно с мужской передачей му¬
тантного гена, является антиципация - нарастание тя¬
жести болезни и появление ее в более молодом возрасте
в «нижних» поколениях родословной [Folstein S., 1989;
Penney J., Young A., 1998].Хорея Гентингтона характеризуется неуклонно
прогредиентным течением. Больные обычно погибают
через 15-20 лет от момента появления первых симпто¬
мов вследствие интеркуррентных заболеваний, травм или
суицидальных действий. При ювенильной акинетико-
ригидной форме длительность болезни значительно
меньше. Напротив, в случае манифестации симптомов
после 60-70 лет заболевание прогрессирует медленно,
212Глава 3а выраженная деменция развивается реДко; такой клини¬
ческий вариант с поздним началом и относительно бла¬
гоприятным течением чаще наблюдается при наследо¬
вании мутантного гена от матери [Penney J., Young А.,1998].Принимая во внимание выраженный фенотипи¬
ческий полиморфизм хореи Гентинггона, а также неспе¬
цифический характер изменений, выявляемых при ис¬
пользовании рутинных лабораторно-инструментальных
методов исследования (расширение боковых желудочков
и субарахноидального пространства больших полуша¬
рий при КТ и МРТ, депрессия a-ритма на электроэнце¬
фалограмме), важной задачей является внедрение в прак¬
тику методов ДНК-диагностики данного заболевания.В 1983 году ген хореи Гентингтона был картиро¬
ван на коротком плече 4-й хромосомы (локус 4р 16.3) в
одном из самых первых успешных исследований гене¬
тического сцепления у человека [Gusella J. et al., 1983].
Это послужило основой для разработки косвенной пре-
симптоматической и пренатальной ДНК-диагностики
хореи Гентингтона в информативных семьях [Евграфов О.В.
и др., 1991; Иванова-Смоленская И.А. и др., 1992;
Meissen G. et al., 1988]. Благодаря накопленному в мире
многолетнему опыту такой диагностики и некоторым
клинико-генетическим особенностям хореи Гентингто¬
на (позднее начало, практически полная пенетрантность
мутантного гена, фатальное течение и др.) данное забо¬
левание стало классической «моделью» для изучения
различных аспектов медико-генетического консультиро¬
вания, основанного на ДНК-тестировании [World
Federation of Neurology Research Committee and Research
Group on Huntington’s chorea, 1989; Huggins M. et al.,
1990].Спустя 10 лет после картирования локуса болез¬
ни Гентингтона данный ген был идентифицирован
ДНК- диагностика наследственных болезней ... 213Международной исследовательской группой под руко¬
водством J. Gusella [The Huntington’s Disease Collaborative
Research Group, 1993]. Ген имеет размер 180 кб, состоит
из 67 экзонов и кодирует белок с молекулярной массой
348 кДа и неизвестной функцией, получивший название
«гентингтин». В своем первом экзоне ген содержит тан¬
демную последовательность (CAG)n-nOBTOpOB, число
которых в норме составляет обычно от 6 до 32, а на му¬
тантных хромосомах - от 36 до 180 [Kremer В. et al.,
1994]. Таким образом, сущность мутации при хорее Ген-
тиштона заключается в экспансии внутригенных тандем¬
ных тринуклеотидных повторов. Экспансия повторов
CAG имеет место в транслируемой области гена и при¬
водит к синтезу аномального белка, содержащего про¬
порционально удлиненный полиглутаминовый участок
(каждый триплет CAG соответствует одной аминокис¬
лоте глутамина в полипептидной цепи). Предполагает¬
ся, что в результате происходит формирование патоло¬
гических полиглутамин-опосредованных связей мутан¬
тного гентингтина с молекулами ряда белков ЦНС, что
обусловливает его цитотоксичность и в конечном счете
селективную гибель нейронов, характерную для хореи
Гентингтона [Albin R., Tagle D., 1995; Ross С., 1995].
Такой патогенез болезни подтверждается выявлением в
мозге больных хореей Гентингтона характерных внут¬
риядерных включений, содержащих мутантный гентин¬
гтин и представляющих собой высокомолекулярные ами¬
лоидоподобные агрегаты [Li H. et al., 1995]. Интересно
отметить, что лица, имеющие «двойную дозу» мутант¬
ного гена (гомозиготные носители патологически удли¬
ненного CAG-участка), клинически неотличимы от боль-
ных-гетерозигот по экспансии повторов [Durr A. et al.,1999].Анализ гена гентингтина у представителей основ¬
ных этнических групп мира показал, что 0,75% всех хро¬
214Глава 3мосом характеризуются числом CAG-повторов, «проме¬
жуточным» между нормой и заведомо патологическими
значениями [Kremer В. et al., 1994]. Такие аллели, по-
видимому, представляют собой премутацию и служат
(в случае дальнейшего нарастания числа повторов) посто¬
янным источником полных мутаций de novo в популя¬
ции [Myers R. et al., 1993; Nance М., 1996]. Важно под¬
черкнуть, что аллели с числом CAG-повторов 36-39 яв¬
ляются «зоной неполной пенетрантности»: часть лиц с
указанным числом повторов могут иметь типичные про¬
явления хореи Генгтингтона, тогда как у других носите¬
лей таких аллелей заболевание не проявляется даже в
самом преклонном возрасте - вплоть до 95 лет
[Rubinsztein D. et al., 1996; Brinkman R. et al., 1997;
McNeil S. et al., 1997]. Очевидно, что возможность раз¬
вития заболевания при наличии хромосомы с числом
CAG-повторов 36-39 определяется взаимодействием раз¬
нообразных экзо- и эндогенных факторов, которые мо¬
гут иметь как протективное, так и патологическое зна¬
чение [Nance М., 1996]. В случае наследования хромо¬
сом с числом CAG-повторов >40 пенетрантность гена
становится полной, и болезнь неизбежно проявится при
условии достижения соответствующего возраста.Прямая ДНК-диагностика хореи Гентингтона ос¬
нована на амплификации с помощью ПЦР участка пер¬
вого экзона гена, содержащего тринуклеотидный CAG-
сегмент, с последующим электрофоретическим разделе¬
нием продуктов амплификации. Наличие мутации диаг¬
ностируется на основании выявления аномального уд¬
линенного фрагмента ДНК, содержащего увеличенное
число CAG-повторов. На рис. 46 показана типичная элек-
трофореграмма агарозного геля, позволяющая полуко
личественным экспресс-методом достоверно визуализи
ровать заведомо мутантные аллели.
ДНК- диагностика наследственных болезней... 2151 23456789 10Рис. 46. Прямая ДНК-диагностика хореи Гентингтона
(электрофорез в агарозном геле)Дорожка 1 - маркер, дорожки 2-4,7-10 - больные хореей Гентинг-
тона, дорожки 5,6- контроль. Длинной стрелкой указан мутант¬
ный аллель (экспансия САО-повторов гена гентингтина), короткой
стрелкой - нормальный аллель.пег «В*м утантны <
аллелинорм альны е
аллелиРис. 47. Прямая ДНК-диагностика хореи Гентингтона
( шектрофорез в полиакриламидном геле)Каждому члену семей, указанных над электрофореграммой,
соответствует одна дорожка электрофореза. Дорожки 1,4, 6,9 —
больные хореей Гентингтона, дорожки 2 и 10 - носители мутации
на пресимптоматической стадии (обозначены штриховыми
с имволами), дорожки 3, 5, 7, 8 - здоровые лица, не являющиеся
I к жителями мутации. Стрелкой указан аллель с «промежуточным»
числом тринуклеотидных САО-повторов.
216Глава 3При необходимости определения точного числа
САО-повторов (например, в случаях носительства «про¬
межуточных» аллелей) ДНК-диагностика проводится с
использованием полиакриламидного геля, обладающе¬
го значительно более высокой разрешающей способно¬
стью (рис. 47). Нейрогенетическим отделением НИИ
неврологии РАМН в 1993-1999 г.г. накоплен уникаль¬
ный опыт прямой ДНК-диагностики хореи Гентингтона
в российской популяции. Нами обследовано >200 боль¬
ных и их клинически здоровых родственников из груп¬
пы риска (свыше 90 неродственных семей), что позво¬
лило сформулировать основные принципы медико-гене¬
тического консультирования и провести анализ разно¬
образных клинико-генетических корреляций при данном
заболевании [Иллариошкин С.Н. и др., 1996 (а); Илла-
риошкин С.Н., 1997; Иванова-Смоленская И.А. и др.,
1998 (б); Клюшников С.А., 1998; Клюшников С.А. и др.,2000]. Было показано решающее значение прямого ДНК-
анализа для подтверждения клинического диагноза хо¬
реи Гентингтона, а также для диагностики спорадичес¬
ких и атипичных вариантов заболевания (включая ран¬
ние и поздние акинетико-ригидные формы).Обследование клинически здоровых родственни¬
ков больных хореей Гентингтона позволило нам выде¬
лить группу из 25 носителей заведомо мутантного алле-
ля в возрасте от 18 до 48 лет (пресимптоматическая ДНК-
диагностика). При использовании набора нейропсихо-
логических тестов было установлено, что такие докли¬
нические носители мутантного гена характеризуются
рядом достоверных когнитивных и личностных измене¬
ний - повышением уровня реактивной и личностной
тревожности, снижением объема памяти, нарушением
практического и абстрактного мышления, нарушением
концентрации внимания [Клюшников С.А., 1998]. Это
позволяет сделать вывод о тонких изменениях в психо¬
ДНК- диагностика наследственных болезней ... 217логической сфере у носителей мутации задолго до ма¬
нифестации развернутой картины хореи Гентингтона.
Подтверждением ранних признаков когнитивной дисфун¬
кции у доклинических носителей мутантного гена хо¬
реи Гентингтона могут служить изменения, выявляемые
у них при исследовании когнитивных вызванных потен¬
циалов - в частности, достоверное удлинение латентно¬
сти и снижение амплитуды волны Р300 (рис. 48). Полу¬
ченные нами данные существенно дополняют результа¬
ты ряда зарубежных авторов [Hahn-Barma V. et al., 1998;
Kirkwood S. et al., 1999] и свидетельствуют о существо¬
вании при хорее Гентингтона достаточно продолжитель¬
ной стадии «предболезни». Этот вывод имеет принци¬
пиальное теоретическое и практическое значение и дол¬
жен учитываться при решении морально-этических про¬
блем консультирования таких «асимптомных» лиц, а
также (в будущем) при решении вопроса о назначении
превентивной терапии.У 42 обследованных нами клинически здоровых
лиц из группы риска (дети и братья-сестры больных хо¬
реей Гентингтона) результат прямой ДНК-диагностики
оказался отрицательным (число САО-повторов в обоих
аллелях гена <35). Таким образом, носительство мута¬
ции было отвергнуто, и данные лица были сняты с дина¬
мического наблюдения. В целом в российской популя¬
ции при исследовании свыше 600 хромосом нами ни в
одном случае не было выявлено «пограничных» алле¬
лей из «зоны неполной пенетрантности» (36-39 CAG-
повторов), затрудняющих интерпретацию результатов
ДНК-анализа. Это совпадает с данными других авторов
об исключительной редкости таких аллелей гена ген-
тингтина [Kremer В. et al., 1994; Rubinsztein D. et al., 1996].
15 случае выявления носительства подобного «погранич¬
ного» аллеля сделать однозначное заключение о прогнозе
у консультируемого лица не представляется возможным.
218Глава 3А~\ответы на
)”■ незначимый
стимулЛ>ответы на
значимый
стимулУРис. 48. Когнитивные вызванные потенциалы на пресимп-
томатической стадии хореи ГентингтонаА. Норма (возраст испытуемого 25 лет). Показаны формы кривой в ответ
на предъявление незначимого (слева) и значимого (справа) стимулов [Гнез-
дицкий В.В., 1997]. Б. Когнитивные вызванные потенциалы у 25-летнего
клинически здорового носителя мутантного гена хореи Гентингтона (му¬
тантный аллель содержит 44 копии САО-повторов). Отмечается удлине¬
ние латентности пика Р300 до 450 мс и снижение его амплитуды до 4,9
мкВ (возрастные нормы — соответственно 320 мс и 9,4 мкВ).
ДНК- диагностика наследственных болезней ... 219Такие лица (как и их дети) должны оставаться в группе
«высокого риска» и находиться под постоянным наблю¬
дением врача-нейрогенетика.Исследование гена гентингтина у больных с раз¬
личными клиническими формами хореи Гентингтона
дало нам возможность детально проанализировать вза¬
имоотношения между генотипом и фенотипическими
проявлениями болезни. Была установлена достоверная
обратная корреляция между степенью экспансии САО-
повторов и возрастом, в котором манифестируют пер¬
вые симптомы хореи Гентингтона (коэффициент корре¬
ляции г=—0,79, р<0,001). На рис. 49 показан график экс-
потенциальной зависимости между числом CAG-повто-
ров и возрастом начала хореи Гентингтона, по которому
можно ориентировочно рассчитать вероятный возраст
дебюта заболевания у асимптомных носителей конкрет¬
ного мутантного аллеля. Данная взаимосвязь была наи¬
более четко выражена для длинных аллелей (>52 повто¬
ров), тогда как при небольшой степени экспансии CAG-
I ювторов возраст начала болезни варьировал в достаточ¬
но широких пределах, что следует учитывать при опре¬
делении прогноза у консультируемых лиц из группы рис¬
ка. В соответствии с выявленной закономерностью, все
наблюдавшиеся нами случаи ювенильного акинетико-
ригидного варианта заболевания были связаны с наиболь¬
шей степенью тяжести генетического дефекта - число
(AG-повторов в мутантном аллеле у этих больных со¬
ставило от 59 до 78. Обратная корреляция между степе¬
нью тяжести мутации и возрастом манифестации хореи
I ентингтона, а также взаимосвязь акинетико-ригидного
варианта Вестфаля с наиболее тяжелыми мутациями
показаны также рядом зарубежных авторов на примере
других популяций [Andrew S. et al., 1993; Duyao M. etal.,
1 *>93; Telenius H. et al., 1993; Trottier Y. et al., 1994].
62О -1 * 1 » 1 » 1 ' 1 т 1 1 , 1 1 1 1 Т 1 1 . 1 . 1 » 1 ■ 1 Т , 1 ■ 1 > 1 » 1 » 1 Т 1 . 35 37 39 41 43 45 47 49 51 53 55 57 59 61 63 65 67 69 71 73 75 77 79 81Число САв-повторовРис. 49. Корреляция между числом тринуклеотидных САв-повторов мутантного гена и возрастом манифестации
симптомов хореи ГентингтонаНа диаграмме показан пример прогностического расчета ориентировочного возраста, в котором может ожидаться начало
заболевания у носителя мутантного гена с числом копий СА(3-повторов 45.
ДНК- диагностика наследственных болезней ... 221Минимально выраженная экспансия CAG-повто-
ров нередко обусловливает поздний и относительно доб¬
рокачественный фенотип заболевания с отсутствием вы¬
раженной деменции [Kremer В. et al., 1993; Britton J. et
al., 1995]. Нами впервые была показана достоверная пря¬
мая корреляция между числом CAG-повторов в мутант¬
ном аллеле и темпом прогрессирования неврологичес¬
ких и психических симптомов хореи Гентингтона
[Illarioshkin S. et al., 1994]. Как и при анализе возраста
начала болезни, данная корреляция оказалась более стро¬
гой при длине повтора, превышающей определенное
«пороговое» значение (>50 С AG-триплетов). Таким об¬
разом, у носителей длинных, более «злокачественных»
аллелей число копий CAG-повторов является ведущим
фактором, определяющим дебют и прогноз заболевания.
В то же время, при небольшой степени экспансии CAG-
повторов течение болезни является более вариабельным,
что свидетельствует о влиянии на него у таких больных
различных дополнительных молекулярных и/или экзо¬
генных факторов.Весьма важным является вопрос о характере не¬
стабильности мутантного аллеля в зависимости от пола
больного родителя, передающего патологический ген.
Наши данные по этому вопросу (рис. 50) полностью со-
кпадают с результатами других авторов и свидетельству¬
ют о том, что в большинстве случаев передачи мутации
от больного отца и в трети всех случаев передачи мута-
I щи от больной матери у потомков наблюдается нарас¬
тание числа копий CAG в мутантном гене [Duyao М. et
al., 1993; Telenius Н. et al., 1993; Kremer В. et al., 1995].
11аследование мутации от матери нередко сопровожда¬
ется уменьшением числа повторов или отсутствием из¬
менений в мутантном аллеле [Kremer В. et al., 1995].
Случаи передачи гена хореи Гентингтона
по мужской и женской линииЧисло САв-повторовРис. 50. Нестабильность мутантного аллеля при передаче гена хореи Гентингтона по отцовской и материнской линии
Черные квадратики — отцовская передача гена, белые кружки — материнская передача гена.222 Глава 3
ДНК- диагностика наследственных болезней ... 223Крайняя степень экспансии повторов наследуется почти
исключительно по мужской линии [Telenius H. et al., 1993;
Kremer В. et al., 1995; Ranen N. et al., 1995; Nance M.,
1996]. Таким образом, генетическая нестабильность
мутантного аллеля реализуется преимущественно в муж¬
ском гаметогенезе, что и обусловливает «эффект отцов¬
ской передачи», характерный для хореи Гентингтона.
Нарастание длины повтора при передаче мутантного гена
в ряду поколений закономерно сопровождается более
ранней манифестацией симптомов болезни у потомков
(в наших наблюдениях в среднем на 4-10 лет). Это явля¬
ется молекулярной основой антиципации в семьях, отя¬
гощенных хореей Гентингтона.По сравнению с хореей Гентингтона другие виды
наследственных хореических гиперкинезов являются
значительно более редкими. Генерализованная хорея с
аутосомно-доминантным типом наследования может
быть одним из атипичных фенотипов дентаторубро-пал-
лидолюисовой атрофии (ДРПЛА), обусловленной экс¬
пансией CAG-повторов в гене на хромосоме 12р (фено¬
тип «псевдохореи»). Характеристика ДРПЛА и подхо¬
ды к прямой ДНК-диагностике данного нейродегенера-
тивного заболевания подробно рассматриваются в раз¬
деле 3.3.1. Ген наследственной доброкачественной хо¬
реи - редкого аутосомно-доминантного заболевания, ха¬
рактеризующегося появлением гиперкинезов в детском
возрасте, непрогрессирующим течением и отсутствием
когнитивных нарушений, локализован на длинном пле¬
че 14-й хромосомы. На хромосоме 9q21 картирован ген
хореи-акантоцитоза (нейроакантоцитоза) - аутосомно-
рецессивного заболевания, проявляющегося сочетанием
мультисистемных неврологических проявлений (хорея,
атаксия, деменция, амиотрофии и др.) с характерными
«звездчатыми» изменениями мембран эритроцитов.
224Глава 3В соответствующих информативных семьях возможно
проведение косвенной ДНК-диагностики наследствен¬
ной доброкачественной хореи и хореи-акантоцитоза с ис¬
пользованием полиморфных маркеров, сцепленных с ло-
кусами 14q и 9я13. Молекулярная диагностика (и в пер¬
вую очередь анализ тринуклеотидных САО-повторов в
гене гентингтина) является ключевым исследованием при
дифференцировании наследственных хореических син¬
дромов с различными фенокопиями - ревматической
(малой) хореей, церебральными васкулитами при неко¬
торых специфических инфекциях и системных заболе¬
ваниях соединительной ткани, сенильной хореей и т.д.
В качестве примера приводим следующее клиническое
наблюдение.Больной М.Н., 13 лет, заболел остро: на следую¬
щий день после прививки от дифтерии появились насиль¬
ственные движения в руках, ногах, туловище, изменилась
походка. После стационарного обследования по месту
жительства направлен в НИИ неврологии РАМН с подо¬
зрением на хорею Гентинггтона. Семейный анамнез не
отягощен. При поступлении в неврологическом статусе:
распространенный крупноразмашистый хореический ги¬
перкинез в конечностях, туловище; «танцующая» поход¬
ка; отмечается также ритмичный тремор пальцев вы¬
тянутых рук и тремор покоя в дистальных отделах рук;
со стороны координации, рефлекторной и интеллектуаль-
но-мнестической сферы существенных нарушений не от¬
мечено. На протяжении месяца указанные гиперкинезы
постепенно регрессировали. При компьютерной томогра¬
фии головного мозга выявлено симметричное расширение
субарахноидального пространства больших полушарий, III
и боковых желудочков. Комплексное ультразвуковое иссле¬
дование сосудов мозга (включая дуплексное сканирование):
грубое двустороннее распространенное стенозирующес
ДНК- диагностика наследственных болезней225атеросклеротическое поражение магистральных артерий
головы. Результаты прямой ДНК-диагностики (исследо¬
вание гена гентингтина): выявлены 2 аллеля с нормальным
числом, копий тандемных САС-повторов (менее 30 повто¬
ров). Диагноз хореи Гентингтона исключен. Принимая во
внимание характер течения заболевания, особенности кли¬
нического синдрома и данные лабораторно-инструмен¬
тальных исследований, состояние больного расценено как
постпрививочный инфекционно-аллергический васкулит
сосудов головного мозга на фоне выраженного церебраль¬
ного атеросклероза.Следует подчеркнуть, что в настоящее время в
развитых странах мира в связи с большими успехами в
лечении ревматизма и других системных заболеваний
соединительной ткани хорея Гентингтона (включая ее
атипичные варианты) является основной причиной раз¬
вития генерализованного хореического гиперкинеза.
Поэтому во всех подобных случаях должно предусмат¬
риваться прямое ДНК-тестирование с помощью доступ¬
ных молекулярно-генетических методов.3.2.2. Наследственные дистонииНаследственные дистонии представляют собой
клинически и генетически гетерогенную группу заболе¬
ваний с различными типами наследования мутантного
гена и широкой вариабельностью фенотипических про¬
явлений болезни. В подавляющем большинстве случаев
клинический синдром характеризуется развитием гене¬
рализованной дистонии с вовлечением, в первую оче¬
редь, аксиальной мускулатуры, что принято обозначать
термином «торсионная дистония» [Маркова Е.Д., 1975;
I ‘>89; РаИп 8., 1988]. Торсионная дистония может пере-
ч; жаться по аутосомно-доминантному, аутосомно-ре-
иессивному и Х-сцепленному типам, однако в большин¬
226Глава 3стве случаев наблюдается классический аутосомно-до-
минантный путь наследования [Eldridge R., 1970;
Ashizawa T., Gasser T., 1998]. Существует также группа
редких наследственных дистонических синдромов с бо¬
лее избирательной локализацией и особыми характери¬
стиками гиперкинезов, например пароксизмальность их
возникновения, фокальный характер дистонии и др.'
[Fahn S., 1988; Jankovic J., Fahn S., 1998].3.2.2.1. Торсионная дистонияДетальный анализ клинического полиморфизма
торсионной дистонии позволил Е.Д. Марковой в 1975
году подразделить это заболевание на две основные фор¬
мы: ригидную и гиперкинетическую. Ригидная форма
торсионной дистонии начинается обычно в детском или
юношеском возрасте и характеризуется постоянным по¬
вышением мышечного тонуса по экстрапирамидному
типу, формированием фиксированных патологических
поз, развитием контрактур на поздней стадии болезни
[Ткачев Р.А., Маркова Е.Д. и др., 1972; Маркова Е.Д.,
1975; 1983]. Аналогичная форма торсионной дистонии
была описана японскими исследователями под названи¬
ем «наследственная прогрессирующая дистония»
(«hereditary progressive dystonia») [SegawaM. et al., 1972;
1976]. Segawa М. и соавторы отметили, что для данной
клинической формы дистонии характерным является
наличие дневных флюктуаций в моторике больных -
ухудшение состояния к вечеру и улучшение двигатель¬
ных функций утром или после дневного сна. Заболева¬
ние чаще наблюдается у женщин [Segawa М. et al., 1976;
MarkovaE., Ivanova-Smolenskaya I., 1995]. Гиперкинети-
ческая форма торсионной дистонии, которая обычно на¬
чинается на 1-м десятилетии жизни, характеризуется
развитием вычурных «скручивающих» генерализован¬
ных гиперкинезов конечностей, туловища и шеи, резко
ДНК- диагностика наследственных болезней ... 227усиливающихся при ходьбе и приводящих к тяжелой
инвалидизации больного [Маркова Е.Д., 1975; 1989].
В западной литературе для обозначения данного забо¬
левания чаще используются термины «генерализованная
дистония с ранним началом» («early-onset generelized
dystonia») либо «идиопатическая торсионная дистония»
(«idiopathic torsion dystonia») [ZiberN. etal., 1984; Fahn S.,
1988; Jankovic J., Fahn S., 1998]. Гиперкинетическая
форма торсионной дистонии особенно часто встречает¬
ся в этнической группе евреев ашкенази - около 40-50
случаев на 100 ООО [ZiberN. et al., 1984].В основе фенотипических различий двух данных
форм торсионной дистонии лежат разнонаправленные
изменения метаболизма основных нейротрансмиттеров
в головном мозге: при ригидной форме выявлено сни¬
жение центральной дофаминергической и серотонинер-
гической трансмиссии с одновременным повышением
активности холинергической системы, тогда как для ги-
перкинетической формы характерны обратные соотно¬
шения [Бархатова В.П., 1988; LeWitte Р., 1993]. Это яви¬
лось теоретической предпосылкой для назначения боль¬
ным с ригидной формой торсионной дистонии замести¬
тельной терапии препаратами L-дофа. Такое лечение
было предложено в начале 70-х годов и выявило «дра¬
матический» эффект малых доз препарата, сохраняю¬
щийся на протяжении многих лет при отсутствии серь¬
езных побочных эффектов [Ткачев P.A. и др., 1972; 1974;
Segawa М. et al., 1976]. Результаты лечения дали основа¬
ние Nygaard Т. и соавторам в 1988 году предложить для
обозначения данной клинической формы торсионной
дистонии термин «дофа-зависимая» или «дофа-чувстви-
I ельная дистония» («dopa-responsive dystonia»). Напро¬
тив, при гиперкинетической форме торсионной дисто¬
нии назначение L-дофа не вызывает улучшения состоя¬
ния и даже приводит к усилению гиперкинезов («дофа-
228Глава 3независимая дистония») [Маркова Е.Д., 1975; 1989; Ива-
нова-Смоленская И.А. и др., 1998 (а)].Дофа-зависимая дистония. В 1993 году Т. Nygaard
с соавторами картировали ген аутосомно-доминантной
дофа-зависимой дистонии на длинном плече 14-й хро¬
мосомы [Nygaard Т. et al., 1993]. Вскоре после этого ло¬
кализация мутантного гена на хромосоме 14q была под¬
тверждена японскими исследователями в семьях с «на¬
следственной прогрессирующей дистонией» [Tanaka Н.
et al., 1995], что позволило на молекулярном уровне ус¬
тановить нозологическую идентичность понятий «на¬
следственная прогрессирующая дистония» и «дофа-за-
висимая дистония». Выделенный ген кодирует синтез
одного из ключевых ферментов дофаминового обмена -
ГТФ-циклогидролазу 1 (ГЦГ-1) [IchinoseH. et al., 1994].
В свете современных представлений патогенез аутосом-
но-доминантной дофа-зависимой дистонии схематично
показан на рис. 51. Фермент ГЦГ-1 регулирует превра¬
щение ГТФ в дигидронеоптерин-трифосфат; последний,
в свою очередь, является субстратом для образования
тетрагидробиоптерина (ВН4) - кофактора тирозин-гид-
роксилазы, ответственной за синтез дофамина из тиро¬
зина. При наличии мутаций в гене ГЦГ-1 нарушается
функция данного фермента и весь последующий биохи¬
мический каскад, что приводит к недостаточности син¬
теза дофамина в нейронах черной субстанции [Ozelius L.,
Breakefield X., 1994]. Синтез дофамина может быть эф¬
фективно восстановлен при введении его предшествен¬
ника - L-дофа, что и имеет место у больных дофа-зави-
симой дистонией. Подтверждением патогенетической
значимости выявленных мутаций в гене ГЦГ-1 является
значительное снижение уровня активности этого фер¬
мента (<20% от исходного уровня) у обследованных боль¬
ных дофа-зависимой дистонией [Ichinose Н. et al., 1994;
Hirano М. et al., 1998].
ГТФтирозинтирозин-гидроксилазадофа-
декарбоксилазадофамин 1_-дофаГТФ-циклогидролаза 1дигидронеоптерин-трифосфатб-пирувоил-тетрагидроптерин-синтетаза6-пирувоил-тетра-гидроптеринсепиаптерин-редуктазатетрагидробиоптерин (ВН4)дигидроптеридин-редуктазаквиноид-дигидробиоптерин
Рис. 51. Биохимический путь синтеза дофаминаДНК- диагностика наследственных болезней ... 229
0-Г0оРис. 52. Родословные обследованных семей с дофа-зависимой дистонией
Черные символы — генерализованная форма дистонии, серые символы — фокальные формы, штриховые символы
«стертая форма» (forme fruste).
ДНК- диагностика наследственных болезней ... 231К настоящему времени идентифицировано свы¬
ше 40 различных (преимущественно точковых) мутаций
в кодирующей области гена ГЦГ-1 у больных из различ¬
ных популяций мира [Ichinose Н. et al., 1994; 1995;
Bandmann О. et al., 1996; 1998; Furukawa Y. et al., 1996;
Jarman P. et al., 1997; Hirano M. et al., 1998; Steinberger D.
et al., 1998]. Нами впервые был проведен мутационный
скрининг гена ГЦГ-1 в российских семьях с дофа-зави-
симой дистонией [Маркова Е.Д. и др., 2000; Illarioshkin S.
et al., 1998]. На рис. 52 показаны наиболее информатив¬
ные родословные семей с дофа-зависимой дистонией,
обследованных в нейрогенетическом отделении НИИ
неврологии РАМН. Нами выявлены 5 точковых мутаций
в гене ГЦГ-1 (таблица 5), четыре из которых являются
новыми и не описанными ранее в других популяциях.Таблица 5Идентифицированные мутации в гене ГЦГ-1
у больных дофа-зависимой дистонией
в российских семьяхЭ к з о нНуклеотиднаязаменаБиологический
эффект мутации1ATG -> AAGМ et 1 02Lys1AC G -> AAGThr94Lys2TGT -> TGGCys141T rp4AGT -> ACTS er1 76Th r6С G А-> TG АArg21 6stop
232Глава 3Таким образом, наши данные подтверждают мне¬
ние других авторов об уникальном характере мутаций в
абсолютном большинстве случаев аутосомно-доминан-
тной дофа-зависимой дистонии. В связи с этим прямая
ДНК-диагностика данного заболевания требует прове¬
дения тотального скрининга всей кодирующей области
гена. Наиболее общепринятым подходом является про¬
ведение 88СР-анализа отдельных экзонов гена с после¬
дующим прямым секвенированием образцов с аномаль¬
но подвижными фрагментами ДНК. На рис. 53 показан
пример такой ДНК-диагносгики.в'СТвСтсААвАС/АОСсстввА.т с«в*Ж * - '■ 'Заршй\ • ■—Рис. 53. Идентификация мутации 281 С->А в первом экзоне
гена ГЦГ -1 (прямое секвенирование)Стрелкой обозначена область мутации (нуклеотидная замена С—>А).
ДНК- диагностика наследственных болезней ... 233Учитывая сравнительно небольшой размер коди¬
рующей области гена (6 коротких экзонов), некоторые
исследователи предпочитают проводить непосредствен¬
ное секвенирование всей кодирующей области, не при¬
бегая к SSCP-анализу.Многие авторы отмечают, что выявляемые мута¬
ции в кодирующей области гена ГЦГ-1 обусловливает
лишь половину всех случаев дофа-зависимой дистонии
[Ichinose Н. et al., 1994; Bandmann О. et al., 1996; 1998;
Furukawa Y. et al., 1996]. Данный факт принято объяс¬
нять генетической гетерогенностью данной формы дис¬
тонии либо возможной локализацией повреждений в
некодирующих участках ГЦГ-1 (промоторная зона, инт-
ронные мутации с нарушениями сплайсинга и др.)
[Furukawa Y., Kish S., 1999]. По нашим данным, в рос¬
сийских семьях с несомненным аутосомно-доминантным
наследованием болезни мутации в гене ГЦГ-1 обнару¬
живаются более чем в 80% случаев. Напротив, в спора¬
дических случаях и при возможном аутосомно-рецессив¬
ном наследовании дофа-зависимой формы дистонии
мутации ГЦГ-1 крайне редки. Это позволяет склонить¬
ся в пользу точки зрения о роли других генов в развитии
основной части спорадических и аутосомно-рецессив-
ных случаев дофа-зависимой дистонии (см. далее).5-летний опыт молекулярного анализа гена ГЦГ-I в семьях различного этнического происхождения по¬
казал, что у небольшой части гетерозиготных носителей
мутации заболевание может проявляться не классичес¬
ким генерализованным фенотипом ригидной формы ди¬
стонии, а разнообразными атипичными клиническими
нариантами - такими как фенотипы атетоидного цереб¬
рального паралича и спастической параплегии, паркин¬
сонизм (в том числе паркинсонизм с L-дофа-индуциро-
нлнными дискинезиями), фокальная дистония конечно¬
234Глава 3стей, оромандибулярная дистония [Bandmann О. et al.,
1996; 1998; SteinbergerD. etal., 1998; Steinberger D. etal.,
1999; Tassin J. et al., 2000]. Мы наблюдали больных, у ко¬
торых единственным проявлением носительства мутан¬
тного гена ГЦГ-1 была непостоянная эквиноварусная
поза стоп (главным образом, при ходьбе) либо изолиро¬
ванный статокинетический тремор рук [Markova E. et al.,
1999]. Во всех указанных случаях клиническая симпто¬
матика значительно регрессировала после назначения
малых доз L-дофа. Таким образом благодаря ДНК-тес-
тированию стало понятным, что клинические проявле¬
ния дофа-зависимой дистонии являются чрезвычайно
вариабельными, а диагностические рамки данной фор¬
мы наследственной дистонии - весьма широкими. Это-
позволяет говорить о гораздо более высокой пенетрант-
ности мутантного гена дофа-зависимой дистонии, чем
традиционно цитируемая цифра 35-40% [Nygaard Т. et
al., 1988]. Ранее при анализе пенетрантности явно недо¬
оценивалась роль различных атипичных и «стертых» слу¬
чаев болезни, поскольку без ДНК-диагностики генети¬
ческий статус таких лиц не мог быть определен с досто¬
верностью. В последних работах авторы включали в сег¬
регационный анализ всех выявленных носителей мута¬
ций в гене ГЦГ-1, имеющих даже минимальные клини¬
ческие проявления: в результате значение пенетрантно¬
сти гена оказалось равным 80-100% [Steinberger D., et
al., 1998], что близко и к нашей оценке (>70%).Интересно отметить, что наличие мутаций ГЦГ-1
в гомозиготном состоянии ведет к манифестации со¬
вершенно особого заболевания - атипичной неонаталь¬
ной гиперфенилаланинемии [Blau N. et al., 1995;
Furukawa Y., Kish S., 1999]. Оно обусловлено блоком
активности тирозин-гидроксилазы (см. рис. 51), глубо¬
ким сочетанным дефицитом всех катехоламинов, а так¬
ДНК- диагностика наследственных болезней ... 235же повышением содержания в крови фенилаланина (пос¬
леднее связано с тем, что при выраженной недостаточ¬
ности тетрагидробиоптерина страдает не только тиро-
зин-гидроксилаза, но и фенилаланин-гидроксилаза).
Клинически данный синдром характеризуется неонаталь¬
ной задержкой двигательного развития, гипотонией, эпи¬
лептическими припадками, умственной отсталостью.
У некоторых больных при особой комбинации мутант¬
ных аллелей гена ГЦГ-1 наблюдается «промежуточный»
фенотип болезни с задержкой психомоторного развития
и речи, прогрессирующей дистонией и умеренным по¬
ложительным эффектом при назначении L-дофа
[Furukawa Y. et al., 1998]. Было показано, что при синд¬
роме атипичной гиперфенилаланинемии имеет место
практически нулевая активность ГЦГ-1, при «промежу¬
точной» форме она обычно не превышает 1—2%, у боль¬
ных дофа-зависимой дистонией она составляет 5-20%,
а при значениях активности фермента выше 30-40% ка¬
кие-либо клинические проявления недостаточности ГЦГ-1
отсутствуют [Ozelius L., Breakefield X., 1994; Furukawa Y.
et al., 1998; Furukawa Y., Kish S., 1999]. Таким образом,
заболевания, обусловленные патологией гена ГЦГ-1,
представляют собой особый континуум взаимно пере¬
крывающихся состояний - от «стертых» субклиничес-
ких форм дофа-зависимой дистонии на одном конце спек¬
тра до тяжелой неонатальной гиперфенилаланиемии на
другом; при этом степень тяжести клинического синд¬
рома увеличивается по мере нарастания дозы мутантно¬
го гена и дефицита активности фермента ГЦГ-1.Сравнительно недавно были открыты два новых
самостоятельных аутосомно-рецессивных варианта дофа-
зависимой дистонии, связанных с повреждением других
генов биохимической цепи синтеза дофамина
(см. рис. 51). Один из них обусловлен мутациями в гене
236Глава 36-иирувоил-тетрагидроптерин-синтетазы (6-ПТГС), ка¬
тализирующей вторую реакцию в указанной биохими¬
ческой цепи [Hanihara Т. et al., 1997]. До настоящего вре¬
мени описана лишь одна больная с гомозиготной мута¬
цией в гене 6-ПТГС, имевшая «мягкую» генерализован¬
ную симптоматику дофа-зависимой дистонии с типич¬
ными дневными флюктуациями симптомов [Hanihara Т.
et al., 1997]. Второй вариант аутосомно-рецессивной
дофа-зависимой дистонии обусловлен мутациями в гене
тирозин-гидроксилазы, локализованном на хромосоме1 lpl 1.5 [KnappskogP. etal., 1995; Ludecke В. et al., 1995],
что приводит к нарушению функции данного фермента
и блоку синтеза дофамина. Назначение этим больным
L-дофа также оказывает выраженный положительный
эффект. По сравнению с аутосомно-доминантной дофа-
зависимой дистонией, при дистонии с дефититом тиро-
зин-гидроксилазы более часто наблюдается развитие син¬
дрома паркинсонизма (в том числе в самом дебюте бо¬
лезни) и несколько реже - дневные флюктуации тяжес¬
ти симптомов [Furukawa Y., Kish S., 1999]. Очевидно,
что исследование активности ферментов 6-ПТГС и ти¬
розин-гидроксилазы должно рассматриваться в качестве
важных диагностических биохимических тестов в ауто-
сомно-рецессивных и спорадических случаях дофа-за-
висимой дистонии.Дофа-независимая торсионная дистония. Основ¬
ной ген дофа-независимой дистонии, получивший обо¬
значение DYT1, был картирован в 1989 году в хромо¬
сомном локусе 9q32-34 [Ozelius et al., 1989]. Локализа¬
ция мутантного гена в области 9q34 была подтверждена
в большинстве семей с аутосомно-доминантной дофа-
независимой генерализованной дистонией в различных
этнических группах мира [Kramer P. et al., 1990; 1994].
Анализ серии DYT1-сцепленных высокополиморфных
ДНК- диагностика наследственных болезней ... 237маркеров показал, что в популяции евреев ашкеиази (ха¬
рактеризующейся очень высокой частотой данной фор¬
мы дистонии) у больных отмечается один и тот же ха¬
рактерный гаплотип, свидетельствующий о едином ге¬
нетическом происхождении дофа-независимой дистонии
в данной этнической группе [Bressman S. et al., 1994;
Risch N. et al., 1995].В 1997 ген DYT1 был идентифицирован
[Ozelius L. et al., 1997]. Его продукт - неизвестный ранее
белок торсин А, имеющий определенную гомологию с
семейством белков «теплового шока», играющих роль в
предохранении клеток от термического стресса. Эти бел¬
ки имеют АТФ-связывающую активность и, помимо ре¬
гуляции термотолерантности, могут участвовать в про¬
цессах образования везикул, конформационных измене¬
ниях белков, регуляции клеточных сигналов и функцио¬
нировании митохондрий [Parsell D., Lindquist S., 1993;
Ozelius L. et al., 1997]. Augood S. с соавторами показали,
что торсин А экспрессируется в дофаминергических ней¬
ронах компактной части черной субстанции, а также в
мозжечке и гиппокампе [Augood S. et al., 1998]. В сово¬
купности вышеуказанные данные свидетельствуют о
возможной роли торсина А в процессах высвобождения
нейромедиаторов в нигро-стриарной системе мозга и/или
поддержании энергетического потенциала дофаминер¬
гических нейронов и нейронопротекции.На сегодняшний день единственной мутацией,
выявленной в гене DYT1 у больных дофа-независимой
формой торсионной дистонии, является делеция трех
нуклеотидов GAG в 946-м положении 5-го экзона гена
(в гетерозиготном состоянии). Эта делеция ведет к утра¬
те аминокислоты глутамата в карбоксильной части бел¬
ка. Высокая частота данной мажорной мутации описана
у больных самых различных национальностей в популя¬
238Глава 3циях Европы и Северной Америки [Ozelius L. et al., 1997;
Valente E. et al., 1998; Lebre A. et al., 1999; Kamm C. et al.,
1999]. Если в этнической группе евреев ашкенази пре¬
обладающее носительство делении GAG у больных обус¬
ловлено в основном «эффектом основателя» и наследо¬
ванием общей предковой мутантной хромосомы, то в
других этнических группах данная деления возникает
повторно в результате многократных независимых му¬
тационных событий. Последнее обстоятельство подтвер¬
ждается наличием у этих больных разных гаплотипов
по маркерам 9q34, сцепленным с мутантной хромосо¬
мой [Lebre A. et al., 1999; Kamm C. et al., 1999]. Возник¬
новение делеции GAG de novo было подтверждено в 2
семьях, исследованных нами совместно с американски¬
ми исследователями из Гарвардского Университета
[Klein C. et al., 1998 (а)]. Многократное возникновение
одной и той же мутации у различных больных является
сравнительно редким феноменом в клинической нейро¬
генетике. При дофа-независимой дистонии это связано
с существованием в гене DYT1 тандемного 24-нуклео-
тидного повтора в виде двух не полностью гомологич¬
ных копий, включающих GAG-димер; делеция первого
GAG-элемента данного димера приводит к существен¬
ному повышению степени гомологии соседних повто¬
ров, «провоцируя» возникновение данной мутации
[KleinC. et al., 1998 (а)].Анализ клинико-генетических корреляций пока¬
зал, что универсальная делеция GAG в гене DYT1 обна¬
руживается не только у больных с типичной генерали¬
зованной формой дофа-независимой дистонии с ранним
дебютом. В некоторых семейных случаях данная мута¬
ция была выявлена также у больных с фокальными (пис¬
чий спазм), мультифокальными и сегментарными фор¬
мами дофа-нечувствительной дистонии, а также у боль¬
ДНК- диагностика наследственных болезней239ных с атипичными клиническими проявлениями болез¬
ни (постуральный тремор рук, заикание вследствие дис¬
тонии оральной мускулатуры и т.п.) [Миклина Н.И.,
1999; Gasser Т. et al., 1998 (б); Lebre A. et al., 1999;
Kamm C. et al., 1999; Slominsky P. et al., 1999]. Делеция
GAG при указанных вариантах заболевания обнаружи¬
вается весьма редко, однако данный факт позволяет су¬
щественно расширить спектр клинических проявлений
9q34-cnen ленной формы наследственной дистонии и
должен приниматься во внимание при медико-генети¬
ческом консультировании. В ряде случаев делеция GAG
в изучаемой области гена DYT1 была выявлена также у
больных, не имевших семейного анамнеза [Ozelius L. et
al., 1997; Valente E. et al., 1998; Slominsky P. et al., 1999;
Brassat D. et al., 2000]. Большинство указанных случаев,
по-видимому, объясняются неполной пенетрантносгью
мутантного гена [Brassat D. et al., 2000] либо отсутстви¬
ем достоверной генеалогической информации.В нейрогенетическом отделении НИИ невроло¬
гии РАМН в 1998-2000 гг. обследовано 46 больных с
различными клиническими вариантами дофа-независи-
мой дистонии, включая как семейные, так и споради¬
ческие случаи заболевания [Миклина Н.И., 1999; Мар¬
кова Е.Д. и др., 2000; Slominsky Р. et al., 1999]. Обоб¬
щенный анализ результатов тестирования больных на
носительство делеции GAG в гене DYT1 представлен
на рис. 54. Суммарно данная мажорная делеция выявле¬
на нами у 28 больных из 18 семей (69,2% обследован¬
ных российских семей с дофа-независимой дистонией).
Как видно на рис. 54, большинство случаев с идентифи¬
цированной делецией составляет классическая генера¬
лизованная дофа-нечувствительная дистония с ранним
началом симптомов (55%) и фокальная дистония конеч¬
ностей (36%), а у небольшой части носителей мутации
240Глава 3заболевание манифестировало в виде атипичных «стер¬
тых» форм (9%). В группе больных с отрицательными
результатами ДНК-диагностики генерализованная дис¬
тония наблюдалась лишь у 25% больных, тогда как буль-
шую часть случаев составили фокальные и атипичные
формы заболевания.Делеция GAG в гене Мутация в гене DYT1DYT1 не выявленагенерализованная форма
Я фокальные формы□ атипичные и стертые формы (forme fruste)Рис. 54. Частота делеции GAG в гене DYT1 при различных
клинических вариантах дофа-независимой дистонииТаким образом, согласно нашим данным, основ¬
ным показанием для проведения ДНК-анализа на носи-
тельство делеции GAG в гене DYT1 является клиника
генерализованной или фокальной дистонии, нечувстви¬
тельной к препаратам L-дофа, особенно при манифеста¬
ции симптомов на первом десятилетии жизни. По мне¬
нию ряда авторов, в спорадических случаях генерализо¬
ванной дистонии анализ DYT1-делеции следует прово¬
дить при дебюте болезни до 26 лет, манифестации сим¬
птомов с мускулатуры конечностей и развитии генера¬
лизованной формы дистонии [Brassat D. et al., 2000;
Bressman S. et al., 2000].
ДНК- диагностика наследственных болезней ... 241Обнаружение универсальной делении GAG в гене
DYT1 у значительной части больных дофа-независимой
дистонией дает возможность проведения простой и дос¬
товерной ДНК-диагностики в большинстве семейных и
части спорадических случаев заболевания, а также у кли¬
нически здоровых родственников из группы риска в кон¬
сультируемых отягощенных семьях. На рис. 21 (с. 79)
показана типичная электрофореграмма полиакриламид¬
ного геля, на которой у носителей делеции четко опре¬
деляется более короткий фрагмент амплифицированной
ДНК. Делеция GAG приводит к исчезновению сайта ре¬
стрикции для рестриктазы BseRI, поэтому во многих ла¬
бораториях для прямой ДНК-диагностики делеции ис¬
пользуется обработка продуктов амплификации фермен¬
том ifaeRI с последующим электрофорезом ДНК в ага¬
розном геле (рис. 55). По нашему мнению, второй метод
прямой ДНК-диагностики болезни является предпочти¬
тельным, поскольку он более специфичен и позволяет
абсолютно точно диагностировать именно искомый три-
нуклеотидный GAG-дефект в изучаемой области гена.До настоящего времени не описан ни один боль¬
ной с дофа-независимой дистонией, который бы имел
другие мутации в гене DYT1 (помимо делеции GAG).
Не исключено, что другие мутации в гене DYT1 либо
являются «клинически немыми», либо приводят 14 со¬
вершенно иному фенотипу болезни, либо, наконец, яв¬
ляются эмбрионально летальными и несовместимы с
жизнью [Ozelius L. et al., 1997]. Ответ на этот вопрос,
который по-видимому будет получен в самое ближай¬
шее время, чрезвычайно важен как для дальнейшего со¬
вершенствования генодиагностики дофа-независимой
формы дистонии, так и для более глубокого понимания
патофизиологии дистонического гиперкинеза.
Рис. 55.Прямая ДНК-диагностика дофа-независимой
дистонии с помощью рестрикционного анализа
Продукты амплификации 5-го экзона обработаны рестриктазой Bse
RI. Дорожка 1 ~ маркер, дорожка 2 - контроль (нативный продукт
амплификации без рестрикции, указан длинной стрелкой), дорожки
3,4,7 - больные дофа-независимой дистонией, имеющие делецию
GAG в гене DYT1, дорожки 5 и 6 - больные без делеции GAG.
Короткой жирной стрелкой указан мутантный аллель (более
длинный рестрикционный фрагмент), короткими тонкими стрелками
-продукты рестрикции нормального аллеля.Известна особая «смешанная» форма аутосомно-
доминантной дистонии, детальное описание которой
представлено C.Klein с соавторами [Й1ет C. et al., 1998
(б)]. В семьях отмечается неполная пеНетрантность му¬
тантного гена, раннее начало болезни и отсутствие чув¬
ствительности к препаратам L-дофа, что сближает дан¬
ное заболевание с DYT1 -ассоциированной дистонией.
Однако в отличие от DYT1-дистонии при «смешанной»
наследственной дистонии у большинства больных раз¬
виваются фокальные формы заболевания с преимуще¬
ственным вовлечением краниоцервикальной мускулату¬
ры (шея, лицо, артикуляционная мускулатура), тогда как
генерализация процесса имеет место лишь у небольшо¬
го числа больных.
ДНК- диагностика наследственных болезней ... 243В 1997 г. в результате исследования 2 семей с
данной формой дистонии ген заболевания (локус DYT6)
был локализован на хромосоме 8p21-q22 [Almasy L. et
al., 1997]. Таким образом была подтверждена генетичес¬
кая гетерогенность дофа-нечувствительной формы тор¬
сионной дистонии. В соответствующих информативных
семьях при исключении у больных носительства деле¬
ции GAG в гене DYT1 возможен анализ сцепления с
локусом DYT6 на 8-й хромосоме и (в случае подтверж¬
дения сцепления) - проведение косвенной ДНК-диагно¬
стики в группе риска.3.2.2.2. Пароксизмальная дистонияТермин «пароксизмальная дистония»
(«пароксизмальная дискинезия») объединяет
гетерогенную группу наследственных дистонических
синдромов, характеризующихся различными типами
передачи гена и своеобразными патофизиологическими
характеристиками, основной из которых является
пароксизмальность и обратимость дистонических
гиперкинезов [Bhatia К., 1999]. Одним из вариантов
пароксизмальной дистонии является пароксизмальный
дистонический хореоатетоз (пароксизмальная
некинезигенная дискинезия) [Demirlciran М., Jankovic J.,
1995]. Это заболевание имеет аутосомно-доминантный
тип наследования и проявляется приступами сложных
непроизвольных движений (дистония, хореоатетоз),
возникающих в покое. В типичных случаях приступ
начинается как гемидистония, но постепенно гиперкинез
охватывает все конечности, туловище, шею и речевую
мускулатуру; сознание в момент приступа остается
сохранным. Гиперкинезы длятся от нескольких минут
до нескольких часов, нередко сопровождаются аурой
(нерестезии) и могут быть спровоцированы приемом
алкоголя или кофеина. Частота двигательных
244Глава 3пароксизмов обычно снижается с возрастом. В 1996 году
двумя группами исследователей ген данного заболевания
был картирован на хромосоме 2q33-35 (локус DYT 8)
[Fink J. et al., 1996; Fouad G. et al., 1996]. В семьях с
пароксизмальным дистоническим хореоатетозом
возможно проведение косвенной ДНК-диагностики с
маркерами из критической области 2q33-35.Осложненной формой пароксизмального дисто-
нического хореоатетоза является другой редкий наслед¬
ственный синдром - так называемый пароксизмальный
хореоатетоз со спастичностью [Auburger G. et al., 1996].
Данное заболевание наследуется по аутосомно-доминан-
тному типу и проявляется приступами дистонически-
хореоатетозных гиперкинезов продолжительностью не¬
сколько минут, обычно провоцируемых физической на¬
грузкой; у части больных развивается постоянный (иног¬
да прогрессирующий) нижний спастический парапарез.
Ген пароксизмального хореоатетоза/спастичности лока¬
лизован на хромосоме 1р (локус DYT 9), в тесной бли¬
зости от кластера генов калиевого канала [Auburger G.
et al., 1996]. Предположительно данный синдром обус¬
ловлен мутацией одного из генов указанного кластера,
т.е. он может относиться к растущей группе каналопа-
тий [BulmanD., 1997]. Окончательное решение вопроса
о принадлежности пароксизмального хореоатетоза со
спастичностью к каналопатиям станет возможным пос¬
ле клонирования гена болезни и установления первич¬
ного молекулярного дефекта.Особую разновидность пароксизмальных дисто¬
ний представляет собой пароксизмальный кинезигенный
хореоатетоз [Bhatia K., 1999], отличающийся тем, что
приступы дискинезии у больных тесно связаны с вне
запными движениями конечностей (чаще ног), напри¬
мер при беге или резком вставании с кресла. Провоци
рующими факторами могут служить стресс, действие
ДНК- диагностика наследственных болезней ... 245неожиданных сенсорных стимулов, усталость, холод,
менструация. Длительность двигательных пароксизмов
короткая (от 3 до 40 сек.), нередко приступу предше¬
ствует аура в виде парестезий или мышечного напряже¬
ния, сознание не нарушается. Заболевание наследуется
по аутосомно-доминантному типу. В некоторых случаях
пароксизмальным кинезигенным дискинезиям, манифе¬
стирующим в детском и юношеском возрасте, могут
предшествовать доброкачественные младенческие судо¬
роги [Szepetowski P. et al., 1998]. Ген пароксизмального
кинезигенного хореоатетоза - как изолированного, так
и осложненного младенческими судорогами - картиро¬
ван на 16-й хромосоме в локусе 16р 11.2—q 11.2 (локус
DYT 10) [Tomita Н. et al., 1999; Bennett L. et al., 2000].
Это позволяет осуществлять косвенную ДНК-диагнос-
тику носительства мутантной хромосомы в информатив¬
ных семьях.3.2.2.3. Другие формы наследственной дистонииФокальная дистония с поздним началом. Поздние
фокальные дистонии (спастическая кривошея, писчий
спазм, спастическая дисфония и т.п.) представляют со¬
бой самую распространенную форму идиопатической
дистонии и встречаются в общей популяции с частотой
3,4 на 100 000 населения [Nutt J. et al., 1988]. Абсолют¬
ное большинство случаев идиопатических поздних фо¬
кальных дистоний являются спорадическими, и их эти¬
ология остается не вполне понятной. Есть основания
предполагать определенную роль генетических факто¬
ров в их развитии, поскольку в литературе описаны еди¬
ничные семьи с аутосомно-доминантным типом переда¬
чи различных фокальных форм дистонии [Jankovic J.,
Nutt J., 1988; Waddy H. et al., 1991; Stojanovic M. et al.,
1995]. В одной из таких больших семей, в которой у ряда
246Глава 3родственников из 2 поколений в возрасте от 28 до 70 лет
манифестировали симптомы фокальной дистонии раз¬
личной локализации (кривошея, спастическая дисфония,
оромандибулярная дистония, писчий спазм), В. Leube с
соавторами картировали ген заболевания на хромосоме
18р [Leube В. et al., 1996]. В современной номенклатуре
данный хромосомный локус имеет обозначение DYT7.
В изучаемой семье были выявлены несколько родствен¬
ников, являвшихся носителями мутантного гаплотипа и
не имевших неврологической симптоматики, что свиде¬
тельствует о низкой пенетрантности мутантного гена. В
дальнейшем теми же авторами изучалась ассоциация
маркеров критического участка хромосомы 18р с боль¬
шой группой изолированных случаен фокальной идио-
патической дистонии в ряде европейских популяций
[Leube В. et al., 1997; Leube В., Auburger G., 1998]. Ре¬
зультаты этих исследований оказались противоречивы¬
ми и не позволяют пока с достоверностью судить о роли
локуса DYT7 в этиологии спорадических фокальных
дистоний. В литературе имеется описание больного с
делецией участка хромосомы 18р (включавшего локус
DYT7), у которого наблюдался полиморфный невроло¬
гический синдром в виде дистонии, пирамидной симп¬
томатики и зрительных расстройств [Kakinuma S. et al.,
1994]; этот факт может служить дополнительным под¬
тверждением локализации одного из генов дистонии на
коротком плече 18-й хромосомы. Суммируя вышесказан¬
ное, оптимальным при обследовании семейных случаев
фокальной дистонии является следующий алгоритм
ДНК-тестирования: 1) исключение делеции GAG в гене
DYT1; 2) анализ генетического сцепления с локусом
DYT7 на хромосоме 18р и (в случае подтверждения сцеп¬
ления) - косвенная ДНК-диагностика у родственников
из группы риска.
ДНК- диагностика наследственных болезней ... 247Миоклоническая дистония (миоклонус-дисто-
ния). Миоклоническая дистония представляет собой ред¬
кое заболевание, характеризующееся появлением на пер¬
вом десятилетии жизни миоклонического гиперкинеза
в сочетании с дистонией мускулатуры шеи, рук и лица, а
также уменьшением выраженности симптомов после
приема алкоголя [Jankovic J., Fahn S., 1998]. Заболева¬
ние имеет непрогрессирующее течение. Описаны как
спорадические, так и семейные (аутосомно-доминант-
ные) случаи миоклонической дистонии. В семейных слу¬
чаях у некоторых родственников заболевание может про¬
являться в виде изолированного миоклонического гипер¬
кинеза. В 1999 году C.Klein с соавторами картировали
ген миоклонической дистонии на хромосоме llq23 (ло-
кус DYT 11) [Klein C. et al., 1999]. Исследуя подходящие
гены-кандидаты в данной хромосомной области, они же
показали, что заболевание обусловлено мутацией гена
02-рецептора дофамина и нарушением дофаминергичес-
кой трансмиссии. Интересно отметить, что у некоторых
больных членов семьи отмечался также ряд психиатри¬
ческих и аффективных симптомов (фобии, депрессия,
панические атаки), что, по-видимому, связано с ролью
02-рецепторов в регуляции эмоционально-волевой сфе¬
ры [Klein C. et al., 1999]. Таким образом, учитывая фе¬
нотипический полиморфизм моклонической дистонии,
исследование гена D2-рецептора дофамина с целью вы¬
явления мутации и проведения прямой ДНК-диагности¬
ки может быть рекомендовано в семьях с аутосомно-до-
минантными двигательными нарушениями (изолирован¬
ный эссенциальный миоклонус, миоклоническая дисто¬
пия), особенно в случае сочетания указанных двигатель¬
ных симптомов с аффективными и эмоционально-воле-
ными расстройствами.Не все случаи аутосомно-доминантной миокло-11 ической дистонии связаны с патологией 02-рецепторов.
248Глава 3Второй самостоятельный локус данного заболевания был
недавно картирован на хромосоме 7q21-23 [Nygaard Т.
et al., 1999]. Данная форма, возможно, является наибо¬
лее частым генетическим вариантом миоклонической
дистонии [Klein C. et al., 2000]. Первичный молекуляр¬
ный дефект не установлен.Дистония-паркинсонизм. Данным термином объе¬
диняется гетерогенная группа заболеваний, характери¬
зующихся сочетанием дистонических гиперкинезов и
фиксированных патологических поз с синдромом пар¬
кинсонизма различной степени тяжести, как правило,
не чувствительным к препаратам L-дофа. Отсутствие
эффекта L-дофа принципиально отличает данный синд¬
ром от дофа-зависимой формы торсионной дистонии
(раздел 3.2.2.1) и ювенильного паркинсонизма (см. да¬
лее), при которых паркинсонизм и дистония представ¬
ляют собой ведущие фенотипические проявления болез¬
ни и являются дофа-чувствительными. Наиболее изучен¬
ными (как с клинической, так и с молекулярной точки
зрения) являются 2 самостоятельные формы дистонии-
паркинсонизма.Х-сцепленный синдром дистония-паркинсонизм
(«синдром Lubag») является весьма редким популяци¬
онно-специфичным заболеванием: он наблюдается, глав¬
ным образом, у аборигенов Филиппинских островов
[Ashizawa Т., Gasser Т., 1998]. Тин наследования - X-
сцепленный рецессивный. Заболевание начинается на 2—
4 десятилетии жизни с фокальных дистонических гипер¬
кинезов лица, оромандибулярной мускулатуры, шеи, ту¬
ловища, конечностей, с постепенным развитием генера¬
лизованной формы дистонии. По мере прогрессирова¬
ния болезни постепенно присоединяется синдром пар¬
кинсонизма, практически не реагирующий на примене¬
ние L-дофа-содержащих препаратов. Патологический ген
ДНК- диагностика наследственных болезней ... 249(DYT3) локализован в перицентромерной области
Х-хромосомы, в локусе Xql3.1 [Graeber М. et al., 1992;
Haberhausen G. et al., 1995].Дистония-паркинсонизм с острым началом пред¬
ставляет собой аутосомно-доминантное заболевание,
характеризуклцееся внезапным появлением дизартрии,
дисфагии, дистонических спазмов, брадикинезии и по¬
стуральной неустойчивости [Dobyns W. et al., 1993]. Сим¬
птомы нарастают обычно на протяжении нескольких
часов, дней или недель, без последующего существен¬
ного прогрессирования неврологической симптоматики.
У отдельных больных в течение ряда лет может наблю¬
даться негрубо выраженная постоянная дистония конеч¬
ностей, после чего отмечается характерное острое появ¬
ление других симптомов заболевания. Симптоматика
манифестирует обычно на 2-3-м десятилетии жизни и
может быть спровоцирована интенсивной физической
нагрузкой, лихорадкой, родами или эмоциональным
стрессом. До настоящего времени в литературе описаны
лишь четыре семьи с данным заболеванием, и в трех из
них ген дистонии-паркинсонизма с острым началом был
картирован в хромосомном локусе 19ql3 (локус DYT 12)
[Kramer P. et al., 1999].Как для Х-сцепленной (Lubag), так и для 19q-
сцепленной формы дистонии-паркинсонизма возможно
проведение косвенной ДНК-диагностики в информатив¬
ных родословных. Принципиальным для корректного
выбора генетических маркеров является анализ клини¬
ческой картины (особенно характер манифестации сим¬
птоматики) и типа наследования болезни в каждой об¬
следуемой семье.В литературе имеются описания семей с аутосом-
по-доминантными формами дистонии, в которых дос¬
товерно исключено сцепление болезни с основными оха¬
рактеризованными выше хромосомными локусами
250Глава 3[Ahmad F. et al., 1993; Holmgren G. et al., 1995; Bressman S.
et al., 1996; Bentivoglio A. et al., 1997]. Эти данные под¬
тверждают выраженную генетическую гетерогенность
наследственной дистонии и свидетельствуют о наличии
ряда других, пока не идентифицированных генов, по¬
вреждение которых лежит в основе развития наслед¬
ственных дистонических синдромов.3.2.3. Гепатолентикулярная дегенерацияГепатолентикулярная дегенерация, или болезнь
Вильсона-Коновалова - тяжелое аутосомно-рецессив-
ное наследственное заболевание нервной системы и
внутренних органов, характеризующееся генетически
обусловленным нарушением метаболизма меди и избы¬
точным ее отложением в различных органах и тканях (в
первую очередь - в мозге, печени и почках) [Иванова-
Смоленская И. А. и др., 1998 (a); Scheinberg I., Stemlieb I.,
1984]. Без лечения заболевание имеет неуклонно про¬
грессирующее течение, с неизбежным летальным исхо¬
дом спустя 5-10 лет от момента появления первых сим¬
птомов. Гепатолентикулярная дегенерация отличается
значительным полиморфизмом неврологических и со¬
матических проявлений, что нашло свое отражение в
существовании различных классификаций клинических
форм болезни.В нашей стране общепринятой является подраз¬
деление гепатолентикулярной дегенерации на 5 основ¬
ных форм, предложенное в 1960 году классиком отече¬
ственной неврологии Николаем Васильевичем Конова¬
ловым. В соответствии с классификацией Н.В. Конова¬
лова, выделяются следующие формы болезни:1) брюшная форма (преневрологическая) - ма¬
нифестирует в возрасте от 5 до 17 лет и характеризуется
ДНК- диагностика наследственных болезней ... 251различными вариантами поражения печени, нередко
принимающими злокачественное «галопирующее» течение;2) аритмогиперкинетическая форма - возникает
в раннем возрасте и проявляется полиморфными инва-
лидизирующими экстрапирамидными гиперкинезами
(чаще торсионно-дистонического характера), интеллек¬
туальным снижением и тяжелыми висцеральными рас¬
стройствами;3) дрожательно-ригидная форма - возникает в
возрасте от 15 до 25 лет и проявляется различным соот¬
ношением ригидности, статокинетического тремора,
психических и висцеральных проявлений;4) дрожательная форма — характеризуется по¬
здним началом (20-30 лет), наиболее доброкачествен¬
ным течением и постепенным нарастанием характерно¬
го дрожательного гиперкинеза, длительной сохраннос¬
тью интеллекта;5) экстрапирамидно-корковая форма - представ¬
ляет собой финал перечисленных выше форм и харак¬
теризуется присоединением к типичным двигательным
расстройствам пирамидных парезов и очаговых эпилеп¬
тических пароксизмов. Под нашим наблюдением в ней-
рогенетическом отделении НИИ неврологии РАМН на
протяжении 40-летнего периода находились свыше 550
больных гепатолентикулярной дегенерацией, что позво¬
лило подтвердить обоснованность вышеуказанной сис¬
тематизации клинических проявлений болезни и деталь¬
но изучить особенности течении отдельных ее форм
[Иванова-Смоленская И.А. и др., 1998 (а)].Клиническая диагностика гепатолентикулярной
дегенерации базируется на выявлении таких характер¬
ных изменений медно-белкового обмена, как рогович¬
ное кольцо Кайзера-Флейшера, снижение концентрации
в сыворотке крови медь-содержащего белка церулоп-
лазмина, гиперэкскреция меди с мочой, повышение кон¬
252Глава 3центрации свободной фракции меди и снижение кон¬
центрации связанной фракции меди в сыворотке крови,
повышение содержания меди в биоптате печени.Гепатолентикулярная дегенерация являет собой
редкий пример наследственного заболевания, для кото¬
рого благодаря знанию основного патогенетического
фактора еще до идентификации гена были разработаны
высокоэффективные методы лечения. Современная па¬
тогенетическая терапия гепатолентикулярной дегенера¬
ции основана на использовании медь-элиминирующих
препаратов - главным образом, Э-пеницилламина, три-
ентина и солей цинка [Тимербаева С.Л., 1991; Иванова-
Смоленская И. А. и др., 1998 (а); 8с1гетЬещ1., 81егпНеЬ I.,
1984]. Б-пеницилламин и триентин - хелатные комп¬
лексны, образующие с медью прочные высокоаффин¬
ные соединения, которые легко выводятся из организма
с мочой. Действие ионов цинка основано на конкурент¬
ном ингибировании металлотеининов, что затрудняет
всасывает меди в кишечнике и вытесняет ее из белко¬
вых тканевых депо. В настоящее время своевременно
начатая терапия гепатолентикулярной дегенерации либо
предотвращает появление клинических признаков забо¬
левания (превентивное лечение), либо ведет к их пол¬
ному или частичному регрессу, восстанавливая трудо¬
способность и существенно улучшая состояние больных
[Лекарь П.Г., Макарова В.А., 1984; Иванова-Смоленс¬
кая И.А. и др., 1998 (а)]. В связи с возможностью эф¬
фективного патогенетического лечения ключевым при
гепатолентикулярной дегенерации является вопрос адек¬
ватной и максимально более ранней диагностики болез¬
ни. Между тем до настоящего времени общепринятая
лабораторно-инструментальная диагностика болезни
остается весьма непростой, а в 5% случаев результаты
исследования обмена меди бывают сомнительными, осо¬
ДНК- диагностика наследственных болезней ... 253бенно в начальной стадии болезни [Шайнберг Г., 1996].
Таким образом, весьма актуальным и жизненно необхо¬
димым становится внедрение в практику надежных ме¬
тодов ДНК-диагностики гепатолентикулярной
дегенерации.Ген гепатолентикулярной дегенерации локализо¬
ван на длинном плече 13-й хромосомы (локус 13ql4.3)
[Frydman М. et al., 1985]. Картирование гена позволило
разработать методы косвенной ДНК-диагностики болез¬
ни, в том числе на пресимптоматической и пренаталь¬
ной стадии. В России впервые успешная косвенная ДНК-
диагностика гепатолентикулярной дегенерации была
проведена сотрудниками нейрогенетического отделения
НИИ неврологии РАМН совместно с лабораторией ДНК-
диагностики Медико-генетического научного центра
РАМН [Воронцова Н.И. и др., 1996]. Ген болезни, полу¬
чивший обозначение АТР7В, идентифицирован незави¬
симыми исследовательскими группами из США и Ка¬
нады в 1993 году [Bull Р. et al., 1993; Tanzi R. et al., 1993].
Ген ATP7B содержит 22 экзона и кодирует синтез медь-
транспортирующей АТФ-азы P-типа. Данный белок в
норме обеспечивает встраивание меди в молекулу апо-
церулоплазмина в аппарате Гольджи и экскрецию с жел¬
чью избытка меди в составе церулоплазмина и других
медь-содержащих белков клетками печени [Сох D., 1995;
Chowrimootoo G. et al., 1996]. Результатом нарушения
функции АТР7В у больных гепатолентикулярной деге¬
нерацией является токсическое накопление меди в пе¬
чени, а в последующем и в других тканевых депо.После открытия первичного молекулярного де¬
фекта появилась реальная возможность осуществлять
непосредственный анализ повреждений в гене, ведущих
к развитию гепатолентикулярной дегенерации, сопос¬
тавлять характер мутаций в различных популяциях,
254Глава 3а также внедрить прямые методы ДНК-диагностики дан¬
ного заболевания. К настоящему времени у больных ге-
патолентикулярной дегенерацией из большого числа
популяций мира идентифицировано уже более 100 раз¬
личных мутаций в гене АТР7В, в основном представля¬
ющих собой нуклеотидные замены и короткие делеции/
вставки [BullP. et al., 1993; Tanzi R. et al., 1993; Thomas G.
et al., 1995; Shah A. et al., 1997]. Большинство больных
являются компаунд-гетерозиготами, т.е. имеют различ¬
ные мутации АТР7В на каждой из двух мутантных хро¬
мосом. В разных этнических группах спектр мутаций
гена АТР7В у больных имеет свои особенности. Наибо¬
лее частой мутацией является замена С—>А в 1069-м
кодоне гена (экзон 14), ведущая к замещению амино¬
кислоты гистидин на глютамин. Указанная мутация
Hisl069Gln встречается в большинстве европейских и
северо-американских популяций (в среднем 20-30% всех
мутантных хромосом) [Tanzi R. et al., 1993; Thomas G. et
al., 1995; Shah A. et al., 1997]. Кодон 1069 гена ATP7B
представляет собой «горячую точку» мутаций, посколь¬
ку замена Hisl069Gln ассоциирована с разными гапло-
типами [Tanzi R. et al., 1993] и, следовательно, возника¬
ет неоднократно у различных больных в результате не¬
зависимых повторных мутационных событий на разных
хромосомах. Другие относительно частые кластеры му¬
таций, характерные для определенных популяций мира,
выявляются преимущественно во 2-м, 3-м, 8-м, 15-м,
16-м и 18-м экзонах гена [Bull P. et al., 1993; Thomas G.
et al., 1995; Shah A. et al., 1997].В связи с большим размером гена АТР7В и раз¬
нообразием мутаций в нем прямая ДНК-диагностика
гепатолентикулярной дегенерации представляет собой
непростую задачу. Стандартный подход основан на SSCP-
анализе отдельных экзонов гена с последующим прямым
ДНК- диагностика наследственных болезней ... 255секвенированием образцов, имеющих аномальную элек¬
трофоретическую картину Существенно упрощает пря¬
мую ДНК-диагностику болезни установление характер¬
ного спектра мутаций АТР7В в конкретной изучаемой
популяции и выделение мажорных мутаций, встречаю¬
щихся в данной популяции с высокой частотой. Обнару¬
жение таких мажорных мутаций позволяет проводить
ДНК-диагностику в данном регионе и/или этнической
группе, избегая необходимости исследовать всю коди¬
рующую область гена. Указанный подход был реализо¬
ван нами при проведении молекулярно-генетического
анализа гепатолентикулярной дегенерации в славянских
семьях, проживающих в европейской части России [Ка-
рабанов A.B. и др., 1998; Ivanovä-Smolenskaya I. et al.,
1999]. Нами были обследованы 42 больных гепатолен¬
тикулярной дегенерацией, не связанных кровным род¬
ством и поступивших в нейрогенетическое отделение
НИИ неврологии РАМН в 1992-1998 гг. Мажорная му¬
тация Hisl069Gln (14-й экзон гена) была обнаружена в
обследованной группе почти на половине мутантных
хромосом (рис. 56). Аналогичные результаты о преобла¬
дании Hisl069Gln (73% хромосом) были получены в эт¬
нически близкой польской популяции [Czlonkowska A.,1997]; интересно отметить также, что в недавно прове¬
денном исследовании замена Hisl069Gln выявлена на
43% мутантных хромосом в популяции больных гепато¬
лентикулярной дегенерацией в Башкирии [Карунас A.C.,1998].По нашим данным, суммарно данная мутация вы¬
является в гомозиготном состоянии или в компаунде с
другими мутациями более чем у 60% больных-славян в
европейской части России, что позволяет считать выяв¬
ление данной нуклеотидной замены важнейшим диаг¬
ностическим тестом при обследовании больных в ука-
256Глава 3Рис. 56. Прямая ДНК-диагностика мажорной мутации
№51069С1п в гене АТР7В у больных гепатолентикулярной
дегенерациейА. БЗСР-анализ 14-го экзона гена. Дорожка 2- гомозиготность по
нормальному аллелю, дорожки 1,4,5 - гетерозиготы по нормаль¬
ному аллелю и мутации, дорожки 3,6,7 - гомозиготы по мутантно¬
му аллелю. Длинные стрелки указывают на продукты амплифика¬
ции нормального аллеля, короткая стрелка - на аномальный элект¬
рофоретический фрагмент (мутация). Б. Прямое секвенирование 14-
го экзона. Стрелкой обозначена область мутации (нуклеотидная за
мена С—>А в 1069-м кодоне).
ДНК- диагностика наследственных болезней ... 257занной этнической группе. В настоящее время в нашей
клинике скрининг на мутацию 1Нз1069Ст1п в 14-м экзо-
не гена АТР7В является неотъемлемой частью диагнос¬
тического алгоритма у больных гепатолентикулярной де¬
генерацией.В нашей работе были исследованы также некото¬
рые другие экзоны гена, которые, согласно литератур¬
ным данным, наиболее часто повреждаются у больных
гепатолентикулярной дегенерацией (экзоны 15,16 и 18);
проведенный анализ показал отсутствие мутаций в дан¬
ных областях АТР7В, за исключением делеции с1с13400С
в 15-м экзоне, выявленной нами в нескольких семьях.
Можно заключить, что спектр мутаций у больных сла¬
вянского этнического происхождения в нашей стране
существенно отличается от большинства других изучен¬
ных популяций мира.Большое число повреждений в гене АТР7В и раз¬
нообразие возможных комбинаций мутаций у компаунд-
гетерозигот, несомненно, являются основными причи¬
нами выраженного фенотипического полиморфизма ге¬
патолентикулярной дегенерации. Анализ больших серий
случаев гепатолентикулярной дегенерации, проведенный
различными авторами, позволил установить основные
закономерности корреляций между характером мутаций
в гене АТР7В и особенностями клинической картины
заболевания. Наиболее неблагоприятное течение болез¬
ни с ранним началом и тяжелыми соматическими про¬
явлениями (фульминантный «вильсоновский» гепатит и
др.) обычно характерно для нонсенс-мутаций, приводя¬
щих к преждевременному обрыву трансляции и отсут¬
ствию белкового продукта с мутантной копии гена
[ТЪотаэ О. еХ а1., 1995]. Миссенс-мутации в большин¬
стве случаев характеризуются более мягким течением и
развитием полиморфных неврологических и соматичес¬
ких проявлений гепатолентикулярной дегенерации, что
258Глава 3обусловлено наличием у синтезируемого мутантного
белка частичной функциональной активности
[Thomas G. et al., 1995; Shah A. et al., 1997.]. Следует
отметить, что некоторые миссенс-мутации, затрагиваю¬
щие функционально важные участки белка АТР7В, так¬
же приводят к тяжелым фенотипическим проявлениям
[Shah A. et al., 1997]. В наших исследованиях было уста¬
новлено, что носительство мутации Hisl 069Gln (особен¬
но на обеих мутантных хромосомах) характеризуется
достаточно тяжелым течением болезни и развитием ряда
осложнений в ответ на назначение D-пеницилламина
(таких как выраженная тромбоцитопения) [Ivanova-
Smolenskaya I. et al., 1999]. Наш вывод о неблагоприят¬
ном течении болезни у носителей мутации Iiisl069Gln,
подтверждаемый результатами дискриминантного ана¬
лиза (рис. 57), совпадает с мнением Shah А. с соавтора¬
ми (1997), но противоречит данным других исследова¬
телей [Bull P. et al., 1993; Thomas G. et al., 1995]. По-
видимому, фенотипическая экспрессия данной мутации
определяется совокупностью генетических и иных фак¬
торов, специфичных для каждой конкретной популяции
(например, наличие модифицирующих генов, уровень
инбредности, характер питания, природно-средовые фак¬
торы и т.д.) [Thomas G. et al., 1995; Shah A. et al., 1997].
Указанные наблюдения имеют важное практическое зна¬
чение и демонстрируют необходимость принимать во
внимание характер выявленных у больного мутаций в
гене АТР7В при решении вопросов лечения и определе¬
нии прогноза болезни.Иллюстрацией роли прямой ДНК-диагностики
гепатолентикулярной дегенерации на пресимптоматичес-
кой стадии болезни является следующее наше наблюде¬
ние.Больная М-ц Т., 26 лет, находилась на обследо¬
вании и лечении в нейрогенетическом отделении НИТ1
ДНК- диагностика наследственных болезней ... 259Рис. 57. Результаты дискриминантного анализа у больных
гепатолентикуляриой дегенерациейПоказано расположение наблюдений трех групп больных в много¬
мерном пространстве признаков (основных клинико-биохимичес-
ких показателей) в проекции на плоскость канонических коорди¬
нат. Квадратные символы - гомозиготные носители мутации
1-Пз 1069(Нп; треугольные символы - больные, имеющие мутацию
Н1э 106901п в компаунде с другой мутацией; круглые символы —
носители других мутаций в гене АТР7В. На рисунке видно, что на¬
блюдения разных групп формируют относительно компактные кла¬
стеры, причем гомозиготы по мутации ШзЮ6901п являются наи¬
более изолированными, что свидетельствует об их существенных
отличиях по анализируемым клинико-биохимическим параметрам
от двух других групп.
260Глава 3неврологии РАМН с 31.03 по 30.04.97 г. Больна с 1990
года, когда в возрасте 19 лет впервые появились носо¬
вые кровотечения. Спустя 2 года присоединилось и ста¬
ло нарастать дрожание рук и головы. В декабре 1996
году нейроофтальмологом выявлено кольцо Кайзера-
Флейшера, диагностирована гепатолентикулярная де¬
генерация (дрожательная форма). При поступлении: со¬
стояние удовлетворительное, существенных изменений
со стороны внутренних органов нет. В неврологичес¬
ком статусе: гипомимия; скандированная речь; тремор
головы по типу «нет-нет»; крупноразмашистый «пор¬
хающий» тремор рук с выраженным интенционным
компонентом; со стороны мышечного тонуса, рефлек¬
торной сферы - без особенностей; пошатывание при
ходьбе. В общем анализе крови отмечается снижение
содержания тромбоцитов до 120 ООО. В сыворотке кро¬
ви снижено содержание церулоплазмина - 12 мг% (в
норме 25-45 мг%) и общей меди —4,3 мкмоль/л (в нор¬
ме 12-24 мкмоль/л). При компьютерной томографии
головного мозга патологических изменений плотности
вещества мозга не обнаружено; отмечается слабое
расширение 3-го и боковых желудочков, слабое расши¬
рение охватывающей цистерны, сильвиевых щелей, су-
барахноидального пространства больших полушарий.
Ультразвуковое исследование органов брюшной полос¬
ти: печень не увеличена в размерах, отмечается диф¬
фузная неоднородность ткани печени; эхоструктура
паренхимы селезенки однородная, контуры чет¬
кие, ровные, размеры не увеличены.При молекулярно-генетическом исследовании
была выявлена мутация Hisl 069Gln в гомозиготном со¬
стоянии, что подтвердило ранее поставленный диаг¬
ноз гепатолентикулярной дегенерации.В отделении обследована младшая сестра боль¬
ной. У консультируемой 18-летней девушки в течение
ДНК- диагностика наследственных болезней261последнего года отмечались повторные кровотечения
из носа при отсутствии каких-либо неврологических
симптомов. Кольцо Кайзера-Флейшера отсутствова¬
ло, уровень церулоплазмина в сыворотке крови соста¬
вил 18,5 мг%, что не позволяло с достоверностью су¬
дить о ее клиническом статусе (норма 25—45 мг%).
Данные других лабораторно-инструментальных мето¬
дов обследования (за исключением сниженного количе¬
ства тромбоцитов в крови -108 ООО) в пределах нор¬
мы. При проведении ДНК-диагностики было выявлено,
что консультируемая (как и ее больная сестра) являет¬
ся гомозиготным, носителем мутации 1И$1069С1п в гене
АТР7В. Девушке был поставлен диагноз «доневрологи-
ческая стадия гепатолентикулярной дегенерации» и
начато лечение солями цинка по стандартной схеме.
Больная продолжает находиться под наблюдением, на
фоне постоянного лечения неврологической симптома¬
тики не отмечается.Таким образом, в данном наблюдении иденти¬
фикация мутации в гене АТР7В оказалась решающим
диагностическим тестом при проведении обследования
больной.Полученный нами опыт прямой ДНК-диагности¬
ки заболевания должен лечь в основу системы медико¬
генетического консультирования и профилактики гепа¬
толентикулярной дегенерации в нашей стране.3.2.4. Эссенциальный тремор и семейный
паркинсонизмВ ряду экстрапирамидных заболеваний особое ме¬
сто занимают болезни, ведущим клиническим проявлени¬
ем которых является дрожательный гиперкинез; они пред¬
ставлены, в первую очередь, эссенциальным тремором и
различными формами первичного паркинсонизма.
262Глава 3Их высокая распространенность в популяции, значитель¬
ный удельный вес наследственных факторов в патогене¬
зе и сложности в дифференциальной диагностике опре¬
деляют большое практическое значение ДНК-техноло-
гий для анализа данной группы заболеваний. Достиже¬
ния молекулярной генетики последних лет позволили по-
новому взглянуть на проблемы этиопатогенеза, нозоло¬
гической принадлежности и диагностики наследствен¬
ных форм эссенциального тремора и первичного пар¬
кинсонизма.3.2.4.1. Эссенциальный треморЭссенциальный тремор - наиболее частое экст-
рапирамидное заболевание человека: его распространен¬
ность составляет от 0,4% до 6,7% среди лиц моложе 40
лет и достигает 8-13% на восьмой и девятой декадах
жизни [Louis E., Oilman R., 1996; Brin M., Koller W., 1998].
Заболевание носит относительно доброкачественный
характер и характеризуется медленно прогрессирующим
симметричным статокинетическим тремором рук часто¬
той 8-10 Гц, реже - тремором головы, губ, голоса, туло¬
вища и ног [Иванова-Смоленская И.А., 1979; Lou J.S.,
Jankovic J., 1991].Генетические исследования эссенциального тре¬
мора значительно осложняются существованием боль¬
шого числа фенокопий, а также тем фактом, что данный
тип тремора нередко описывается у членов семей с раз¬
личными формами других наследственных заболеваний- торсионной дистонии, семейных форм болезни Пар¬
кинсона, некоторых наследственных невропатий и др.
[Baxter D., Lai S., 1979; Young R., 1986; Jankovic J., 1989;
Brin M., Koller W., 1998]. Семейные формы составляют,
в среднем, около половины всех случаев эссенциально¬
го тремора и наследуются по аутосомно-доминантному
ДНК- диагностика наследственных болезней ... 263типу с неполной пенетрантностью и вариабельной эксп¬
рессивностью мутантного гена [Иванова-Смоленс-
кая И.А., 1983; Young R., 1986; Lou J.S., Jankovic J., 1991;
Brin M., Koller W., 1998]. В ряде семей наблюдается фе¬
номен антиципации, с появлением в последующих по¬
колениях более ранних и более тяжело протекающих
случаев заболевания. Детальный анализ феноменологии
эссенциального тремора позволяет выделить несколько
клинических вариантов данного заболевания [Иванова-
Смоленская И.А., 1979; 1983], что свидетельствует о его
генетической гетерогенности.Результаты недавних молекулярно-генетических
исследований подтвердили предположение о генетичес¬
кой гетерогенности эссенциального тремора. В настоя¬
щее время показано существование, как минимум, двух
хромосомных локусов, ответственных за развитие «чис¬
тых» форм эссенциального тремора. J. Gulcher с соавто¬
рами (1997) исследовали большую серию исландских
семей с эссенциальным тремором, предположительно
имеющих общие генетические корни, и локализовали ген
на хромосоме 3ql3. Данный хромосомный локус полу¬
чил обозначение ЕТМ1 (от англ. Essential Tremor
Mutated). Средний возраст манифестации болезни в
изученных семьях составил около 26 лет, каких-либо
клинических особенностей болезни в изученной попу¬
ляции отмечено не было [Gulcher J. et al., 1997]. Следует
подчеркнуть, что ЕТМ1-сцепленная форма эссенциаль¬
ного тремора не ограничена лишь территорией Ислан¬
дии. Недавно нами был обследован изолят с эссенци¬
альным тремором на территории северного Таджикис¬
тана, и в нескольких семьях доказано сцепление болез¬
ни с локусом ЕТМ1 на хромосоме 3ql3 [Illarioshkin S. et
al., 2000].
264Глава 3J. Higgins с соавторами на примере 4 обширных
американских семей, в которых наблюдался отчетливый
феномен антиципации, показали существование друго¬
го локуса эссенциального тремора на хромосоме 2р22-
25 [Higgins J. et al., 1997; 1998]. В одной из этих семей
методом детекции экспансии повторов RED (Repeat Ex¬
pansion Detection) было показано, что возможной моле¬
кулярной основой болезни является «динамическая»
мутация (экспансия тринуклеотидных CAG-повторов) в
не установленном пока гене [Higgins J. et al., 1997]. Дан¬
ный хромосомный локус обозначен как ЕТМ2. В неко¬
торых семьях с эссенциальным тремором сцепление
болезни с локусами ЕТМ1 и ЕТМ2 достоверно исклю¬
чено, что доказывает дальнейшую гетерогенность дан¬
ного заболевания [Illarioshkin S. et al., 2000].Отдельно следует остановиться на взаимоотно¬
шении эссенциального тремора и болезни Паркинсона.
Как хорошо известно из практики, дифференциальный
диагноз между этими заболеваниями (особенно при де¬
бюте тремора) нередко бывает весьма непростым и мо¬
жет потребовать определенного промежутка времени -
когда становится очевидным либо присоединение к тре¬
мору явных клинических проявлений паркинсонизма,
либо (в случае эссенциального тремора) сохранение ти¬
пичного моносимптомного характера болезни. В рабо¬
тах И.А. Ивановой-Смоленской (1979, 1983) проводил¬
ся детальный анализ точки зрения некоторых авторов,
считавших эссенциальный тремор «стертой» формой бо¬
лезни Паркинсона. Основанием для такого предположе¬
ния послужило нередко наблюдаемое в пределах одной
семьи наличие случаев болезни Паркинсона и эссенци¬
ального тремора у различных родственников [Young R.,
1986; Jankovic J., 1989]. До самого последнего времени
большинством исследователей возможность этиологи¬
ДНК- диагностика наследственных болезней ... 265ческой взаимосвязи эссенциального тремора и болезни
Паркинсона отвергалась. Новый импульс данной дис¬
куссии был дан М. Раггег с соавторами в 1999 году. Ука¬
занные авторы картировали один из локусов аутосомно-
доминантной болезни Паркинсона на хромосоме 4р14-
16.3 (см. далее) и показали в изучаемой семье наличие
4р-сцепленного «мутантного» гаплотипа у ряда родствен¬
ников, имевших лишь изолированный постуральный
(непаркинсоновский) тремор. В свете этих данных нельзя
исключить, что в основе определенных вариантов бо¬
лезни Паркинсона и эссенциального тремора могут ле¬
жать близкие механизмы генетической предрасположен¬
ности [Зрасеу Б., \¥оос1Т\[., 1999].Опыт косвенной ДНК-диагностики эссенциаль¬
ного тремора в мире практически отсутствует. В связи с
доброкачественным характером болезни, возможностя¬
ми медикаментозной терапии (анаприлин, клоназепам,
гексамидин) и неполной пенетрантностью мутантного
гена эссенциальный тремор является одним из немно¬
гих наследственных неврологических заболеваний, при
котором определение генетического статуса лиц из груп¬
пы риска в семье или пренатальная ДНК-диагностика у
плода не имеют первоочередного медико-социального
значения. Более существенной представляется задача
использования ДНК-анализа для дифференциальной
диагностики эссенциального тремора с многочисленны¬
ми гено- и фенокопиями, но это станет возможным лишь
после открытия генов эссенциального тремора и обна¬
ружения в них соответствующих мутаций.3.2.4.2. Семейные формы первичного паркинсонизмаПонятием «первичный паркинсонизм» объединя¬
ются дегенеративные заболевания головного мозга, ха¬
рактеризующиеся прогрессирующей избирательной ги¬
266Глава 3белью нейронов нигро-стриарной системы и развитием
изолированного синдрома паркинсонизма (тремор покоя,
мышечная ригидность, олиго-брадикинезия). Согласно
современным представлениям, выделяют две основные
нозологические формы первичного паркинсонизма -
болезнь Паркинсона и ювенильный паркинсонизм
[Шток В.Н. и др., 1998]. В развитии первичного паркинсо¬
низма значительную роль играют наследственные фак¬
торы, в связи с чем молекулярно-генетический анализ
стал в последние годы одним из наиболее приоритет¬
ных направлений в исследовании данных заболеваний.Болезнь Паркинсона (семейные формы). Болезнь
Паркинсона представляет собой второе по частоте (пос¬
ле болезни Альцгеймера) нейродегенеративное заболе¬
вание человека, которое встречается во всех популяциях
мира и имеет распространенность 2-4% среди лиц стар¬
ше 65 лет [De Rijk М. et al., 1995]. Заболевание характе¬
ризуется типичным тремором покоя рук и других частей
тела частотой 4-6 Гц, экстрапирамидной мышечной ри¬
гидностью, брадикинезией, постуральной неустойчиво¬
стью, вегетативными нарушениями, постепенно присо¬
единяющимися когнитивными расстройствами подкор¬
кового типа и неуклонно прогрессирующим течением
[Голубев В.Л. и др., 1999; Marsden C.D., 1994]. Морфо¬
логическая картина болезни Паркинсона включает де¬
генерацию и гибель дофаминергических нейронов ком¬
пактной части черной субстанции, а также наличие в
сохранных клетках особых интранейрональных эозино¬
фильных включений - телец Леви [Gibb W., Lees А.,
1988]. Использование L-дофа (предшественник дофами¬
на) и других препаратов, корригирующих характерные
для болезни Паркинсона центральные нейротрансмит-
терные нарушения, позволяет значительно улучшить
двигательную активность и качество жизни больных на
ДНК- диагностика наследственных болезней ... 267протяжении ряда лет. Однако современная терапия не
предотвращает дальнейшего неуклонного прогрессиро¬
вания болезни и появления большого числа тяжелых
осложнений, многие из которых являются следствием
проводимого лечения [Голубев B.JL и др., 1999;
MarsdenC.D., 1994].Большинство случаев болезни Паркинсона явля¬
ются спорадическими. У этих больных заболевание име¬
ет мультифакториальную природу с отчетливой генети¬
ческой предрасположенностью, о чем подробно говорит¬
ся в главе 4. В то же время в литературе неоднократно
описаны семейные формы болезни Паркинсона, когда
заболевание имело место у нескольких родственников
из одного или разных поколений [Spacey S., Wood N.,1999]. По различным оценкам, частота таких семейных
форм составляет в среднем около 10-20% всех случаев
болезни Паркинсона [Marder K. et al., 1996; Duvoisin R.,
1998; Gasser Т., 1998; Elbaz A. et al., 1999]. Особенно
значимо роль наследственности выступает при анализе
редко встречающихся больших семей, в которых болезнь
Паркинсона наблюдается у большого числа родственни¬
ков и наследуется как моногенный признак; в большин¬
стве таких описанных родословных отмечен аутосомно-
доминантный тип наследования с неполной пенетрант-
ностью мутантного гена [Duvoisin R., 1998; Spacey S.,
Wood N., 1999]. Таким образом, в некоторых случаях
развитие болезни Паркинсона определяется поврежде¬
нием одного основного гена. Для этих случаев примени¬
мы все основные подходы к медико-генетическому кон¬
сультированию и ДНК-диагностике, которые использу¬
ются в семьях с другими наследственными моногенны-
ми болезнями, рассматриваемыми в данной главе.В 1997-1999 гг. были открыты 4 генетических ло-
куса, ответственных за развитие аутосомно-доминантных
случаев болезни Паркинсона, причем для двух из них
268Глава 3идентифицированы мутантные гены. Первый из них рас¬
положен на хромосоме 4q21-23 (данный локус обозна¬
чен символом PARK1) и кодирует синтез а-синуклеина- основного белкового компонента телец Леви
[Polymeropoulos М. et al, 1997]. К настоящему времени,
несмотря на проведенное в мире обследование сотен
семей с болезнью Паркинсона, описаны лишь 2 точко-
вых миссенс-мутации в гене а-синуклеина: одна из них
обнаружена в небольшом числе итальяно-греческих се¬
мей, имеющих общее происхождение из одного среди¬
земноморского региона [Polymeropoulos М. et al., 1997;
Athanassiadou A. et al., 1999], а вторая - в семье этничес¬
ких немцев [Krüger R et al., 1998]. Обе мутации предпо¬
ложительно приводят к изменению пространственной
организации а-синуклеина, следствием чего может яв¬
ляться нарушение его процессинга или деградации, при¬
водящее к формированию амилоидоподобных белковых
агрегатов [Spacey S., WoodN., 1999].Нами было проведено прямое секвенирование
гена а-синуклеина в 9 российских семьях с несомнен¬
ным аутосомно-доминантным наследованием болезни
Паркинсона (рис. 58), при этом мутаций в кодирующей
области а-синуклеина выявлено не было. Результаты
исследований различных авторов и наши собственные
данные свидетельствуют о том, что мутации гена а-си-
нуклеина представляют собой весьма редкую причину
семейных форм болезни Паркинсона [Vaughan J. et al.,
1998; Scott W. et al., 1999].Следующий ген аутосомно-доминантной болез¬
ни Паркинсона был выделен в 1998 году. Он локализо¬
ван на хромосоме 4р14 (локус PARK5) и кодирует син¬
тез убиквитин-карбокси-терминальной гидролазы L1
(UCH-L1) [Leroy E. et al., 1998]. Данный белок состав¬
ляет 1-2% от всего белкового состава мозга, обнаружи-
2й 3 5 6 >/Пт4от»■яйой ой4575г5757я57^р*"4/8 Р-г0т*?ЕмГьТТ)0-гаоис. 58. Примеры родословных обследованных семей с аутосомно-доминантной болезнью ПаркинсонаДНК- диагностика наследственных болезней ... 269
270Глава 3вается (как и а-синуклеин) в тельцах Леви и играет важ¬
ную роль в процессах протеолиза полимерного убикви-
тина. Предварительные данные свидетельствуют о том,
что частота мутаций в гене UCH-L1 среди всех семей¬
ных случаев болезни Паркинсона чрезвычайно низка
[Lincoln S. et al., 1999], однако в целом генетика данной
молекулярной формы болезни Паркинсона остается ма¬
лоизученной.Еще один хромосомный локус (PARK3), ассоци¬
ированный с развитием аутосомно-доминантной болез¬
ни Паркинсона, картирован на хромосоме 2р13 [Gasser Т.
et al., 1998а]. Пенетрантность мутантного гена в иссле¬
дованной немецкой семье составила около 40%, что по¬
зволяет обсуждать роль локуса PARK3 в развитии
определенной части спорадических случаев болезни
Паркинсона. Наконец, еще в одной обширной родослов¬
ной М. Farrer с соавторами в 1999 году обнаружили сцеп¬
ление локуса болезни Паркинсона (PARK4) с хромосом¬
ной областью 4р 14-16.3; в данной семье (как отмеча¬
лось в разделе 3.2.4.1) «мутантный» гаплотип был ассо¬
циирован не только с болезнью Паркинсона, но также
со случаями «чистого» эссенциального тремора
[Farrer М. et al., 1999]. Указанное наблюдение может сви¬
детельствовать об определенной этиологической взаи¬
мосвязи эссенциального тремора и, как минимум, од¬
ной из генетических форм болезни Паркинсона. В лите¬
ратуре описаны также семьи с аутосомно-доминантной
болезнью Паркинсона, в которых все вышеперечислен¬
ные генетические локусы были исключены [Gwinn-
HardyK. etal., 2000].Таким образом, уже сейчас вполне очевидно мно¬
гообразие генетических вариантов семейного идиопати-
ческого паркинсонизма. По-видимому, развитие заболе¬
вания может быть обусловлено повреждением болыпо-
ДНК- диагностика наследственных болезней ... 271го числа генов и их белковых продуктов, взаимосвязан¬
ных между собой посредством участия в функциониро¬
вании общих метаболических путей (например, регуля¬
ции процессов протеолиза, обеспечении нейронального
белкового транспорта и т.п.). С этих позиций формиро¬
вание амилоидоподобных белковых комплексов, входя¬
щих в состав телец Леви, может представлять собой уни¬
версальную патологическую реакцию как результат на¬
рушенного на различных этапах процессинга нейрональ¬
ных белков [Spacey S., WoodN., 1999].Генетическая гетерогенность семейных случаев
болезни Паркинсона существенно осложняет проведе¬
ние ДНК-диагностики в отягощенных семьях. Тоталь¬
ный скрининг кодирующей области генов а-синуклеина
и UCH-L1 является с практической точки зрения неце¬
лесообразным ввиду чрезвычайной редкости мутаций в
них при семейном паркинсонизме. То же самое, по-ви¬
димому, будет относиться и к каждому другому конкрет¬
ному гену, когда они станут доступными для прямого
анализа. Одним из путей повысить эффективность ДНК-
тестирования является выделение отдельных подгрупп
больных, в каждой из которых особенности клиники
могли бы помочь правильному выбору генов для мута¬
ционного скрининга. Например, для мутаций в локусе
PARJC1 (ген а-синуклеина) характерно более раннее на¬
чало и быстрое прогрессирование болезни
[Polymeropoulos М. et al., 1997], для РА11К4-ассоцииро-
ванной формы (как уже было отмечено) характерно со¬
четание в семье случаев болезни Паркинсона и эссенци¬
ального тремора [Farrer М. et al., 1999] и т.д. Сейчас мы
находимся лишь на самом начальном этапе анализа кли-
нико-генетических корреляций и определения характер¬
ного фенотипа отдельных генетических форм болезни
Паркинсона. Дальнейшее накопление и обобщение этих
272Глава 3данных на основе широкого международного сотрудни¬
чества позволит выработать более четкий алгоритм для
проведения ДНК-диагностики и медико-генетического
консультирования в семьях, отягощенных наследствен¬
ными случаями болезни Паркинсона.Ювенильный паркинсонизм. Ювенильный пар¬
кинсонизм представляет собой особую наследственную
форму первичного паркинсонизма, детально изученную
лишь в последние 10-15 лет [ЫагаЬауазЫ Н. ег а1., 1986;
ТакаЬазЫ Н. е! а1., 1994; Ыпка\¥а, Тзир Б., 1996; Гаакт J.
е1а1., 1998]. Заболевание распространено повсеместно и
несколько чаще встречается у женщин. Тип наследова¬
ния в большинстве описаний - аутосомно-рецессивный,
в связи с чем многие случаи ювенильного паркинсониз¬
ма в небольших по размеру семьях являются споради¬
ческими. Дебют симптомов чаще всего приходится на
2—4-е десятилетие жизни, реже на более ранний возраст.
Клиническая картина ювенильного паркинсонизма имеет
ряд особенностей по сравнению с классической болез¬
нью Паркинсона. Главными из них являются: частое со¬
четание паркинсонизма с дистонией; наличие флюктуа¬
ций в выраженности симптомов паркинсонизма и дис¬
тонии на протяжении дня (ухудшение состояния к вече¬
ру, улучшение утром или после дневного сна); отсутствие
(даже при многолетнем течении болезни) деменции и
других психических расстройств и, наоборот, частое со¬
четание паркинсонизма с повышением сухожильных
рефлексов и другими пирамидными симптомами; пре¬
красный многолетний терапевтический эффект неболь¬
ших доз Г-дофа (в среднем 200-250 мг Ь-дофа в сутки в
комбинации с карбидофой), который, однако, нередко
осложняется присоединением размашистых дискинезий
и усилением выраженности симптомов дистонии. Тече¬
ние заболевания медленно прогрессирующее (на протя¬
жении десятилетий), прогноз относительно благоприятный.
ДНК- диагностика наследственных болезней ... 273Патологанатомическая картина ювенильного пар¬
кинсонизма характеризуется отсутствием телец Леви в
нейронах структур среднего мозга (это главное отличие
от классической болезни Паркинсона) и гибелью нейро¬
нов с реактивным глиозом в компактной части черной
субстанции и в области locus ceruleus [Takahashi Н. et al.,
1994].Основной ген аутосомно-рецессивного ювениль¬
ного паркинсонизма картирован в хромосомной облас¬
ти 6q25.2-27 (локус PARK2) [Matsumine Н. et al., 1997].
Данный ген был выделен в 1998 году: он содержит 12
экзонов и кодирует новый белок паркин, состоящий из
465 аминокислот [Kitada Т. et al., 1998]. В нейроне пар¬
кин локализован в комплексе Гольджи и цитозоле; им-
муногистохимическим методом показано, что у больных
ювенильным паркинсонизмом паркин отсутствует во
всех отделах головного мозга [Shimura et al., 1999]. Точ¬
ная функция паркина в клетке до настоящего времени
не установлена. У больных ювенильным паркинсониз¬
мом выявлено большое число различных мутаций в гене
паркина, представленных как структурными перестрой¬
ками (делеции, реже дупликации различной протяжен¬
ности), так и точковыми мутациями [Hattori N. et al., 1998;
Lucking С. et al., 1998; 2000; Abbas N. et al., 1999].Специфический характер мутаций в гене парки¬
на определяет некоторые особенности методов мутаци¬
онного скрининга у больных ювенильным паркинсониз¬
мом. На первом этапе (особенно в инбредных семьях, в
которых больные с высокой вероятностью являются но¬
сителями 2 копий идентичной мутантной хромосомы)
целесообразно исключить гомозиготную делецию того
или иного участка гена. Для этой цели проводится амп¬
лификация отдельных экзонов гена (в том числе в муль¬
типлексной ПЦР), после чего на электрофореграмме
гомозиготная делеция диагностируется на основании от¬
274Глава 3сутствия ПЦР-продукта соответствующего экзона или
группы экзонов. На следующем этапе анализа может
быть использован специально разработанный протокол
полуколичественной мультиплексной амплификации,
позволяющий исследовать дозу гена и диагностировать
гетерозиготные делении или дупликации исследуемых
экзонов [Lucking С. et al., 2000]. Наконец, для диагнос¬
тики точковых мутаций в оставшихся случаях проводится
секвенирование кодирующей области гена (в том числе
после предварительного SSCP-анализа) на образцах ге¬
номной ДНК или кДНК. Комбинированное использова¬
ние указанных методов ДНК-диагностики позволяет
выявлять мутации в гене паркина более чем в 70% се¬
мейных и спорадических случаев ювенильного паркин¬
сонизма, причем чаще всего мутации выявляются в груп¬
пе больных с началом болезни до 20 лет [Lucking С. et
al, 1998; 2000; Abbas N. et al., 1999].Под нашим наблюдением на протяжении 25 лет
находится 6 семей с типичным аутосомно-рецессивным
ювенильным паркинсонизмом, а также ряд изолирован¬
ных случаев данного заболевания. Родословные двух из
этих семей (в каждой из которых больны 3 сибсов) по¬
казаны на рис. 59. В 1999 году нами совместно с колле¬
гами из Национального Института Здоровья и Медицин¬
ских Исследований Франции (INSERM, Париж) прове¬
дена первая у российских больных прямая ДНК-диагно-
стика ювенильного паркинсонизма. В представленных
на схеме родословных больные оказались компаунд-ге¬
терозиготами по различным делециям во 2-м, 3-м и 7-м
экзонах гена паркина (рис. 60). Приводим краткие вы¬
писки из историй болезни пациентов в одной из наблю¬
давшихся нами семей (родословная А на рис. 59).Больная Тр. II-3, в возрасте 17 лет впервые отме¬
тила скованность в правой ноге и подволакивание ноги
ДНК- диагностика наследственных болезней ... 275&Рис. 59. Родословные обследованных семей с ювенильным
паркинсонизмомпри ходьбе. Спустя несколько лет скованность распрос¬
транилась на левые конечности, присоединилось дро¬
жание рук. В 23 года впервые обследована в НИИ не¬
врологии РАМН, где после назначения небольших доз
Ь-дофа (до 0,5 г в сутки в составе мадопара-125) был
достигнут значительный терапевтический эффект: пол¬
ностью исчезло дрожание, существенно уменьшилась
выраженность экстрапирамидной ригидности, улучши¬
лась походка. Спустя 6 лет на фоне непрерывного лече¬
ния появились отчетливые дискинезии пика дозы, вы¬
раженность которых постепенно нарастала, что стало
существенно ограничивать самообслуживание. После¬
днее стационарное лечение в НИИ неврологии РАМН
проводилось в возрасте 44 лет. В неврологическом ста¬
276Глава 3тусе на фоне отмены Ь-дофа отмечалась выраженная
олигобрадикинезия, повышение мышечного тонуса по
экстрапирамидному типу в конечностях (больше спра¬
ва), среднеамплитудный ритмичный тремор покоя рук.
Походка скованная, «шаркающая», в резко замедленном
темпе, при ходьбе содружественные движения рук от¬
сутствуют. Сухожильные рефлексы оживлены, без чет¬
кой разницы сторон. Гиперсаливация; речь монотонная,
слабо модулированная. При назначении 1 таблетки на-
кома наблюдалось исчезновение тремора рук и значи¬
тельный регресс акинетико-ригидного синдрома, при
этом на фоне максимального действия препарата в тече¬
ние 30-40 минут появлялись размашистые хореоформ-
ные гиперкинезы конечностей (особенно ног), подерги¬
вания головы и дискинезии мимических мышц. Наряду
с этим периодически, вне четкой связи с временем при¬
ема Ь-дофа, отмечаются кратковременные эпизоды рез¬
ко выраженных генерализованных гиперкинезов в виде
баллистических движений конечностей, «подбрасыва¬
ния» туловища на кровати; в момент гиперкинезов са¬
мостоятельная ходьба невозможна. При КТ патологичес¬
ких изменений в веществе головного мозга не выявле¬
но. После «лекарственных каникул» больной назначена
Ь-дофа (наком) в суточной дозе 0,75 г в 6 приемов в ком¬
бинации с паркопаном и клоиазепамом; на фоне данной
схемы лечения удалось практически полностью купиро¬
вать дискинезии, больная продолжала прием препара¬
тов на протяжении 12 лет со стабильным терапевтичес¬
ким эффектом. В возрасте 56 лет скончалась от острого
нарушения мозгового кровообращения.Больной Тр. П-4, пробанд, в возрасте 23 лет стал
отмечать скованность в ногах во второй половине дня и
после умеренной физической нагрузки; скованность
полностью проходила после сна. На протяжении
нескольких лет скованность в ногах постепенно
ДНК- диагностика наследственных болезней ... 277нарастала и распространилась на руки, больной стал
отмечать дрожание рук, значительно изменилась походка,
появились пропульсии. В возрасте 28 лет были впервые
назначены препараты Ь-дофа (2 капсулы в день мадопара-
250) в сочетании с циклодолом (4 мг в день); спустя
несколько дней от начала лечения скованность
практически исчезла (она отмечалась в ногах лишь в
утренние часы до приема Ь-дофа), нормализовалась
походка. С 29 лет наблюдается в НИИ неврологии, на
протяжении многих лет принимал препараты Ь-дофа в
различных комбинациях с центральными
холинолитиками, без существенных побочных эффектов.
Последнее стационарное обследование в Институте
проводилось в возрасте 54 лет. На фоне отмены
препаратов отмечается выраженное генерализованное
повышение мышечного тонуса по экстрапирамидному
типу, отчетливая брадикинезия, гипомимия, тремор
покоя рук средней интенсивности, уменьшающийся при
целенаправленных движениях. Походка «шаркающая»,
при ходьбе отсутствуют синкинезии рук, отмечаются
редкие пропульсии, постуральная неустойчивость.
Сухожильные рефлексы оживлены, симметричные;
выявляются рефлексы орального автоматизма. На МР-
томограммах головного мозга отмечается слабое
расширение III и боковых желудочков. После назначения
адекватной схемы приема препаратов (Ь-дофа 0,375-0,5
г в день в 2 приема в составе накома, циклодол 2 мг в
день в 2 приема) отмечено значительное уменьшение
выраженности симптомов паркинсонизма. Эффект
сохраняется на протяжении многих лет, больной
проживает отдельно от семьи и полностью обслуживает
себя.Больной Тр. П-7, заболел в возрасте 19 лет, когда
с тала беспокоить скованность и утомляемость ног к
концу дня. Через несколько месяцев присоединилась
АВА1IУХк1(10 1 3 экзон 12э к з о н 2Э К 3 о н 9экзон 3 экзон 123 К 3 о н 23 К 3 о н 9Ь 1 |
л * Л. )1 ;^ )1 1-в о о-400А А ]III- 1 ОО О— 5003 0 н 3 экзон 123 К 30 н 2 экзон 9Рис. 60. Прямая ДНК-диагностика делеций в гене паркнна с помощью анализа дозы гена
А. Больной с делецией двух нуклеотидов А в в положении 202-203 во втором экзоне (мутация выявлена в гетерозигот¬
ном состоянии). Область мутации («расщепление» единого пика на 2 фрагмента) указана стрелкой. Б. Больная с делецией
экзона 3 в гетерозиготном состоянии. Область мутации указана стрелкой: отчетливо видно уменьшение дозы гена
(высота и площадь пика экзона 3 у данной больной вдвое меньше по сравнению с таким же пиком в контроле). В.
Контроль (здоровый индивидуум).278 Глава 3
ДНК- диагностика наследственных болезней ... 279скованность в мышцах рук, появилось дрожание в ко¬
нечностях (больше слева), изменилась походка. С 21 года
наблюдается в НИИ неврологии РАМН; в Институте
впервые были назначены препараты Ь-дофа, которые
больной с отличным эффектом принимал на протяже¬
нии многих лет (первоначально - чистую Ь-дофу в
дозе до 3 г/сут, а в дальнейшем - в составе накома в дозе
0,5 г в день). Последний раз поступил для коррекции
схемы лечения в возрасте 34 лет в связи с усилением
тремора и ухудшением самообслуживания. В невроло¬
гическом статусе на фоне отмены препаратов отмечалась
легкая гипомимия, значительное повышение мышечно¬
го тонуса по экстрапирамидному типу в конечностях
(больше слева), выраженная генерализованная брадики-
незия, мелкоамплитудный тремор покоя рук (8>В). При
ходьбе туловище напряжено, наклонено вперед, походка
«шаркающая», мелкими шажками, ахейрокинез слева.
Сухожильные рефлексы на руках оживлены, на ногах вы¬
сокие, отмечается клонусоид стоп, рефлекс Россолимо с
двух сторон, рефлексы орального автоматизма. Отмеча¬
ются изменения в эмоциональной сфере (эйфоричность,
некоторая расторможенность), а также негрубое интел¬
лектуальное снижение. При КТ головного мозга выяв¬
ляется умеренное расширение ликворосодержащих про¬
странств. Больному был назначен более дробный прием
прежней суточной дозы накома в комбинации с цикло-
долом до 6 мг в день, в результате чего полностью ис¬
чезло дрожание, значительно уменьшилась скованность,
улучшилась ходьба и самообслуживание. В возрасте 42
лет больной скончался от прободной язвы желудка и
желудочного кровотечения.Молекулярно-генетическое исследование было
проведено в 1999 году у пробанда: при анализе гена пар-
кина в экзоне 2 выявлена деления двух нуклеотидов АО
280Глава Jв положении 202-203 (см. рис. 60 А). Диагноз: аутосом-
но-рецессивный ювенильный паркинсонизм.Таким образом, в представленных случаях у 3
больных в семье наблюдались практически все «класси¬
ческие» клинические признаки ювенильного паркинсо¬
низма - раннее начало болезни, хороший терапевтичес¬
кий эффект сравнительно небольших доз L-дофа, сохра¬
нявшийся на протяжении многих лет, отсутствие демен-
ции, наличие пирамидной симптоматики, наличие ран¬
них L-дофа-индуцированных дискинезий (у больнойII -3), а также суточных флюктуаций выраженности сим¬
птомов паркинсонизма (у больного II-4).После того как появилась возможность достовер¬
ной молекулярной диагностики ювенильного паркинсо¬
низма, было показано существование ряда атипичных
случаев данного заболевания. Так, у отдельных больных- носителей мутаций в гене паркина заболевание в раз¬
вернутой стадии было практически неотличимо от клас¬
сической болезни Паркинсона, а первые симптомы мог¬
ли появляться только после 50-летнего возраста [Tassin J.
et al., 1998; Lucking С. et al., 2000]. В других наблюдени¬
ях у больных, имевших мутации в гене паркина, клини¬
ческая картина соответствовала дофа-зависимой форме
торсионной дистонии [Abbas N. et al., 1999; Tassin J. et
al., 2000]. Нередко у остальных больных в описанных
семьях наблюдалась типичная клиника ювенильного
паркинсонизма с ранним началом симптомов (что обыч¬
но является решающим фактором в клинической диаг¬
ностике ювенильного паркинсонизма в обследуемой се¬
мье). С практической точки зрения, отмеченные наблю¬
дения демонстрируют важность тщательного анализа
клинической картины у всех больных родственников, а
также необходимость проведения ДНК-анализа с целью
правильной диагностики ювенильного паркинсонизма и
ДНК- диагностика наследственных болезней ... 281его дифференцирования от ранних случаев болезни Пар¬
кинсона и других сходных заболеваний.Второй локус аутосомно-рецессивного паркинсо¬
низма (PARK6), отличпющегося от «классических» слу¬
чаев ювенильного паркисонизма несколько более по¬
здним началом (от 32 до 48 лет), отсутствием дистонии
в начале болезни и отсутствием дневных флюктуаций,
был совсем недавно картирован на хромосоме 1р35—36
[Valente E. et al., 2001]. Заболевание характеризуется весь¬
ма медленным течением и стабильным многолетним
эффектом леводопа-препаратов, а также частыми лево-
допа-индуцированными дискинезиями. Соответствую¬
щий мутантный ген не установлен.На той же хромосоме
картирован локус еще одной молекулярной формы ауто-
сомно-рецессивного паркинсонизма (PARK7) [van
Duijn C. et al., 2001]. В семьях с аутосомно-рецессив-
ным паркинсонизмом, в которых не было выявлено му¬
таций в гене паркина, целесообразно исследовать сцеп¬
ление с данными новыми локусами на хромосоме 1р и в
случае подтверждения сцепления - проводить косвен¬
ную ДНК-диагностику у братьев-сестер пробанда.В единичных семьях с типичной клинической
картиной ювенильного паркинсонизма описан аутосом-
но-доминантный тип передачи болезни [Dwork A. et al.,
1993; Ishikawa A., MiyatakeT., 1995]. Это свидетельствует
о вовлечении в патогенез ювенильного паркинсонизма
и других генов, белковые продукты которых могут вза¬
имодействовать с паркином в сложных биохимических
реакциях, имеющих отношение к обмену дофамина.
282Глава 33.3. Наследственные заболеванияс преимущественным вовлечением
координаторных систем
(наследственные атаксии)Группа наследственных атаксий является чрезвы¬
чайно гетерогенной. Данные заболевания имеют распро¬
страненность 5-10 на 100 ООО населения [Bobowick А.,
Brody J., 1975] и могут передаваться по аутосомно-до-
минантному, аутосомно-рецессивному и Х-сцепленному
типам, причем в рамках каждого типа наследования вы¬
деляется большое число самостоятельных атактических
синдромов [Harding А., 1984; Conner К., Rosenberg R.,
1993]. Общей клинической характеристикой наслед¬
ственных атаксий является прогрессирующее расстрой¬
ство координации движений, манифестирующее в мо¬
лодом или зрелом возрасте, нередко в сочетании с ря¬
дом других неврологических симптомов [Иванова-Смо¬
ленская И.А. и др., 1998 (a); Harding А., 1984; Conner К.,
Rosenberg R., 1993]. До последнего времени одной из
наиболее сложных проблем нейрогенетики была разра¬
ботка адекватной классификации наследственных атак¬
сий. Это обусловлено их выраженным меж- и внутрисе¬
мейным полиморфизмом, существованием большого
числа атипичных форм и отсутствием четких критериев
прижизненной диагностики различных вариантов дан¬
ных заболеваний [Калмыкова Л.Г., 1976; Иллариош-
кин С.Н. и др., 1999 (б); Harding А., 1984]. Традицион¬
ный клинико-морфологический подход к систематиза¬
ции наследственных атаксий, основанный на специфи¬
ческом паттерне нейродегенеративных изменений в каж¬
дом конкретном случае, оказался непригодным для прак¬
тического невролога [Иллариошкин С.Н. и др., 1999 (б)].
Стремление выделить все описанные в литературе вари¬
анты наследственных атактических синдромов в отдель¬
ДНК- диагностика наследственных болезней ... 283ные нозологические единицы привело к неоправданно¬
му усложнению предлагавшихся классификаций, в ко¬
торых число форм атаксий достигало 60-80 и более [Кал¬
мыкова Л.Г., 1976; Barbeau A. et al., 1984; Harding А.,
1984; Rosenberg R., 1990]. Современный этап развития
учения о наследственных атаксиях характеризуется вне¬
дрением принципиально новой номенклатуры данных
заболеваний, основанной на знании соответствующих
генетических дефектов и использовании методов ДНК-
диагностики.3.3.1. Аутосомно-доминантные спиноцеребел-
лярные атаксии3.3.1.1. Клинико-генетическая характеристика ауто-
сомно-доминантных атаксийГруппа аутосомно-доминантных спиноцеребел-
лярных атаксий включает ряд самостоятельных форм,
сходных по своей клинической картине, основным па¬
томорфологическим изменениям и характеру генетичес¬
кого дефекта. До внедрения молекулярных методов ис¬
следования в практику единой общепринятой классифи¬
кации данных заболеваний не существовало. В настоя¬
щее время все доминантные спииоцеребеллярные атак¬
сии четко подразделяются на отдельные нозологические
единицы с помощью ДНК-диагностики [Иллариош-
кин С.Н. и др., 1996 (б); 1999 (б); Rosenberg R., 1995;
Klockgether T., Dichgans J., 1997; Stevanin G. et al., 2000].
Следует отметить, что даже на фоне современных зна¬
ний о молекулярных основах наследственных заболева¬
ний степень генетической гетерогенности аутосомно-
доминантных атаксий (а это не менее полутора десятков
различных генов!) является весьма впечатляющей. В све¬
те такого многообразия генетических дефектов стано¬
вится вполне понятным выраженный полиморфизм этих
284Глава 3заболеваний, остававшийся загадкой на протяжении
многих десятилетий - начиная с 1891 года, когда Р. Menzel
впервые описал одну из форм дегенеративных атаксий с
аутосомно-доминантным типом наследования. Благода¬
ря созданию геномной классификации аутосомно-доми-
нантных атаксий ряд широко применявшихся ранее тер¬
минов (таких как «церебеллооливарная атрофия Холм¬
са», «семейная оливопонтоцеребеллярная атрофия Мен-
целя», «семейная атаксия Пьера Мари» и т.п.) в настоя¬
щее время вышли из употребления и представляют лишь
исторический интерес [Иллариошкин С.Н. и др.,
1996 (б); Conner K., Rosenberg R., 1993].По состоянию на конец 2001 года, установлено
существование 16 хромосомных локусов аутосомно-до-
минантных спиноцеребеллярных атаксий, в том числе
для 10 из них идентифицированы мутантные гены [Orr Н.
et al., 1993; Kawaguchi Y. et al., 1994; Koide R. et al., 1994;
Ranum L. et al., 1994; Flanigan K. et al., 1996; Sanpei K. et
al., 1996; David G. et al., 1997; Zhuchenko O. et al., 1997;
Holmes S. et al., 1999; Koob M. et al., 1999; Matsuura T. et
al., 1999; Worth R et al., 1999; Zu L. et al., 1999; Herman-
Bert A. et al., 2000, Yamashita I. et al., 2000; Matsuura T. et
al., 2000; Miyoshi Y. et al., 2001; Nakamura K. et al., 2001].
Для обозначения каждого локуса и соответствующего ге¬
нетического варианта атаксии ему присваивается соот¬
ветствующий условный индекс (в порядке открытия).
Так, спиноцеребеллярная атаксия 1-го типа (СЦА1) пред¬
ставляет собой первую форму доминантных атаксий, для
которой была установлена хромосомная локализация
гена (локус 6р22-23). Ген следующей формы - спино-
церебеллярной атаксии 2-го типа (СЦА2) был картиро¬
ван на хромосоме 12q24.1, ген спиноцеребеллярной атак¬
сии 3-го типа (СЦАЗ, болезнь Мачадо-Джозеф) локали¬
зован на хромосоме 14q32.1 и т.д. Самостоятельное ис¬
ДНК- диагностика наследственных болезней285торические название, отражающее характер обнаружи¬
ваемых на секции морфологических изменений в мозге,
имеет особая редкая форма аутосомно-доминантной атак¬
сии - так называемая дентаторубро-паллидолюисова
атрофия (ДРПЛА). Символы СЦА9 и СЦА15 зарезерви¬
рованы для характеристик локусов неустановленных ге¬
нетических форм аутосомно-доминантной атаксий. Свод¬
ные данные по молекулярной генетике аутосомно-доми-
нантных атаксий представлены в таблице 6. Необходи¬
мо добавить, что в ряде семей с аутосомно-доминант-
ными атаксиями достоверно исключено сцепление с из¬
вестными на сегодня и указанными в таблице хромосом¬
ными локусами [Giunti P. et al., 1999; Worth P. et al., 1999],
что свидетельствует о существовании еще одного или
нескольких не установленных пока генов данных забо¬
леваний.Как видно из таблицы, при 9 из 10 форм доми¬
нантных спиноцеребеллярных атаксий с идентифициро¬
ванными генами механизм мутации заключается в экс¬
пансии тринуклеотидных повторов, а еще при одной фор¬
ме (СЦА10) выявлена экспансия пентануклеотидного
повтора. За исключением СЦА8 и СЦА12, при которых
имеет место удлинение нетранслируемых повторов раз¬
личной конфигурации, для остальных 6 форм характер¬
на экспансия тандемных С AG-триплетов в кодирующей
области гена, приводящая к экспансии полиглутамино-
вого участка в составе соответствующего белка. Таким
образом, большинство молекулярных форм доминант¬
ных атаксий с идентифицированными генами относятся
к классу «полиглутаминовых» болезней.К этому же классу болезней относятся хорея Ген-
тингтона и спинально-бульбарная амиотрофия Кеннеди,
рассмотренные в предыдущих разделах главы 3. Как и
при указанных заболеваниях, реализация патологичес¬
286Глава 3кого действия мутации на клеточном уровне при доми¬
нантных атаксиях связана с нарушением полиглутамин-
опосредованных межмолекулярных связей в ткани моз¬
га и формированием внутриядерных включений в деге¬
нерирующих нейронах [Ross С., 1995; Burke J. etal., 1996;
PaulsonH. etal., 1997; Skinner P. etal., 1997; HolmbergM.
et al., 1998].При большинстве рассматриваемых форм ауто-
сомно-доминантных атаксий наблюдается ряд «класси¬
ческих» клинико-генетических характеристик, свой¬
ственных и другим заболеваниям с динамическими му¬
тациями [Иллариошкин С.Н. и др., 1995; Zoghbi Н., 1996;
Klockgether Т., Dichgans J., 1997; Stevanin G. et al., 2000].
К таким характерным для доминантных атаксий особен¬
ностям относятся:а) обратная корреляция между числом трипле¬
тов в мутантном аллеле и возрастом манифестации сим¬
птомов болезни;б) прямая взаимосвязь между степенью экспан¬
сии повторов и тяжестью клинических проявлений (у
больных с большим числом тринуклеотидных повторов
в соответствующем гене наблюдается тенденция к раз¬
витию наиболее «злокачественных» форм заболевания
с быстрым прогрессированием и присоединением ряда
дополнительных симптомов);в) феномен антиципации (утяжеление клиники
в ряду поколений), обусловленный нестабильностью
повтора и нарастанием его длины при передаче мутант¬
ного гена потомкам;г) феномен «отцовской передачи», связанный с
преимущественным удлинением мутантного повтора в
мужском гаметогенезе и проявляющийся манифестаци¬
ей более ранних и тяжелых случаев болезни у потомков
больного отца;
ДНК- диагностика наследственных болезней ... 287д) происхождение мутаций de novo от имеющих¬
ся в популяции редких аллелей с «промежуточным» чис¬
лом тринуклеотидных повторов. Такие аллели сами по
себе не приводят к болезни, но являются генетически
нестабильными и способны переходить в «полную му¬
тацию» при передаче гена потомкам. Прямое доказатель¬
ство существования «промежуточных» аллелей было
представлено, в частности, для СЦА7 [Stevanin G. et al.,
1998; Giunti P.et al., 1999].Механизм генетической нестабильности мутант¬
ного аллеля при аутосомно-доминантных атаксиях име¬
ет особую природу. Показано, что у больных СЦА1 и
СЦА2 мутантный ген содержит непрерывный (CAG)n-
повтор, тогда как нормальные аллели генов прерывают¬
ся отдельными вставками других триплетов [Chung М.
et al., 1993; Sanpei К. et al., 1996]; предполагается, что
эти вставки служат стабилизирующим фактором, утрата
которого с образованием непрерывной тандемной кон¬
фигурации ведет к появлению генетически нестабиль¬
ного мутантного аллеля [Zoghbi Н., 1996]. Учитывая не¬
сомненное клинико-генетическое сходство аутосомно-
доминантных атаксий, а также наличие антиципации при
СЦА4, СЦА5, СЦА11 и СЦА14, многими авторами выс¬
казывалось предположение о том, что данные формы
атаксий с неизвестными пока генами также обусловле¬
ны динамической экспансией тандемных CAG-содержа-
щих или иных тринуклеотидных повторов [Flanigan К.
et al., 1996; Zoghbi Н., 1996; Klockgether Т., Dichgans J.,
1997; Worth P. et al., 1999].Белковые продукты генов аутосомно-доминант¬
ных атаксий получили обозначения «атаксин-1», «атак-
син-2» и т.д. (см. таблицу 6). Их нормальная функция в
клетке не установлена. Исключение составляют формы
СЦА6, СЦА12 и СЦА17, продукты генов которых яв¬
ляются, соответственно, а1Д-субъединицей потенциал-
Молекулярно-генетическая характеристика аутосомно-доминантных атаксий Таблица 6Генетическая форма аутосомно-
доминантной атаксииХромосомныйлокусИдентифицированный
ген и его белковый
продуктХарактер
мутации
в генеЛокализация
области повтора
в генеЧисло понормальныеаллеливторовмутантныеаллелиСпиноцеребеллярная атаксия 1 -го
типа (СЦА1)6р22-23атаксин-1экспансияСАС-повторовэкзон<3739-83Спиноцеребеллярная атаксия 2-го
типа (СЦА2)12Ч24.1атаксин-2экспансияСАО-повторовэкзон<2932-77Спиноцеребеллярная атаксия 3-го
типа (СЦАЗ) / болезнь Мачадо-Джозеф14Ч32.1атаксин-3экспансияСАв-повторовэкзон<4355-86Спиноцеребеллярная атаксия 4-го
типа (СЦА4)1бц22.1??---Спиноцеребеллярная атаксия 5-го
типа (СЦА5)1Ц1377---Спиноцеребеллярная атаксия 6-го
типа (СЦА6)19р 13а1А-субъединица
потенциал-зависимого
кальциевого каналаэкспансияСАв-повторовэкзон<1821-30Спиноцеребеллярная атаксия 7-го
типа (СЦА7)Зр 12-21.1атаксин-7экспансияСАв-повторовэкзон<3537-300Спиноцеребеллярная атаксия 8-го
типа (СЦА8)13ц21обозначения нет
(в стадии изучения)экспансияСТС-повторовЗ’-область гена(не транслируемая)<34107-127Спиноцеребеллярная атаксия 10-го
типа (СЦА10)22д1377---Молекулярно-генетическая характеристика аутосомно-доминантных атаксий Продолжение Таблицы 6Генетическая форма аутосомно-
доминантной атаксииХромосомныйлокусИдентифицированный
ген и его белковый
продуктХарактер
мутации
в генеЛокализация
области повтора
в генеЧисло повторовнормальныеаллели.мутантныеаллелиСпиноцеребеллярная атаксия 11-го
типа (СЦА11)15ч14-21.377--Спиноцеребеллярная атаксия 12-го
типа (СЦА12)5я31-33регуляторная
субъединица белковой
фосфатазы РР2АэкспансияСАв-повторов5’-область гена<2866-78Спиноцеребеллярная атаксия 13-го
типа(СЦА13)^13.3-13.4?9-- .-Спиноцеребеллярная атаксия 14-го
типа (СЦА14)^!3.4ч}(ег?7---Спиноцеребеллярная атаксия 16-го
типа (СЦА 16)8я22,1-24.17?---Спиноцеребеллярная атаксия 17-го
типа (СЦА 17)6q27фактор транскрипции
ТВРэкспансияСАв-повторовэкзон<4247-55Дентаторубро-паллидолюисова
атрофия (ДРПЛА)ИрИ-гегатрофинэкспансияСАв-повторовэкзон<3649-88288 ГлаваЗ ДНК-диагностика наследственных болезней... 289
290Глава 3зависимого кальциевого канала [Zhuchenko О. et al.,
1997], регуляторной субъединицей белковой фосфатазы
РР2А [Holmes S. et al., 1999] и фактором транскрипции
ТВР [Nakamura K. et al., 2001].Как видно из таблицы 6, степень экспансии три-
нуклеотидных повторов наиболее выражена при СЦА7;
эта же форма характеризуется чрезвычайно высокой не¬
стабильностью мутантного аллеля при передаче гена в
следующее поколение. Описаны (при отцовской переда¬
че мутации) крайне тяжелые врожденные случаи СЦА7
с гибелью больных в течение первых нескольких меся¬
цев жизни, обусловленные экстремальной экспансией
CAG-повторов до 250-300 копий [Benton C. et al., 1998;
Johansson J. et al., 1998]. С другой стороны, патологи¬
ческие аллели гена а]А-потенциал-зависимого кальцие¬
вого канала (CACNL1A4), ответственные за развитие
СЦА6, генетически весьма стабильны и характеризуют¬
ся наименьшим среди всех «полиглутаминовых» забо¬
леваний порогом числа тандемных повторов, приводя¬
щим к болезни. Генетические характеристики мутант¬
ных аллелей у больных СЦА1, СЦА2, СЦАЗ, СЦА12,
СЦА17 и ДРПЛА являются в значительной степени сход¬
ными. Интересно отметить, что в описанных редчайших
случаях гомозиготности по мутациям СЦА1 и СЦА6 кли¬
ническая картина болезни не отличалась от обычных
случаев, обусловленных экспансией CAG в одном алле-
ле гена [Goldfarb L. et al., 1996; Matsuyama Z. et al., 1997];
в то же время, у гомозигот по мутации СЦАЗ двойная
доза мутантного гена характеризовалась «аддитивным»
эффектом и приводила к более раннему и более небла¬
гоприятному варианту заболевания [Lang A. et al., 1994;
Kawakami H. et al., 1995].Каждая из генетических форм рассматриваемых
заболеваний, характеризуясь в первую очередь про¬
грессирующей атаксией мозжечкового типа, имеет в то
ДНК- диагностика наследственных болезней ... 291же время определенные особенности неврологического
синдрома, течения и морфологической картины болезни.Спиноцеребеллярная атаксия 1-го типа. Заболе¬
вание начинается обычно на 3-4-м десятилетии жизни.
Первым симптомом чаще всего бывает неловкость при
быстрой ходьбе и беге. Спустя несколько лет развива¬
ются атактическая походка, неустойчивость в позе Ром¬
берга, дискоординация и интенционный тремор в руках,
дизартрия. Типичным проявлением СЦА1 уже на ран¬
ней стадии болезни является сочетание мозжечковых
нарушений с пирамидной симптоматикой (повышением
сухожильных рефлексов, стопными и кистевыми пато¬
логическими знаками, клонусами, спастическим повы¬
шением тонуса в ногах). В ряде случаев может наблю¬
даться тремор головы, нистагм, в далеко зашедшей ста¬
дии - нарушения глотания и фонации, снижение глубо¬
кой чувствительности, тазовые расстройства, деменция,
атрофия зрительного нерва. С течением времени боль¬
ные перестают ходить и обслуживать себя, наступает глу¬
бокая инвалидизация; в большинстве случаев причиной
смерти (обычно спустя 10-15 лет от момента появления
первых симптомов) являются инфекционные осложне¬
ния. КТ/МРТ-картина характеризуется расширением су-
барахноидальных пространств полушарий и червя моз¬
жечка, истончением средней ножки мозжечка и демие-
линизацией поперечных волокон моста, расширением IV
желудочка, большой цистерны, цистерн ствола, в ряде
случаев - атрофическими изменениями больших полу¬
шарий мозга. На секции выявляется дегенерация коры
мозжечка и демиелинизация его белого вещества, деге¬
нерация нижних олив, ядер и поперечных волокон мос¬
та мозга; в процесс могут вовлекаться также проводни¬
ки спинного мозга (в первую очередь - спиноцеребел-
иярные тракты), клетки передних рогов, кора больших
полушарий.
292Глава 3Спиноцеребеллярная атаксия 2-го типа. Невроло¬
гическая картина у больных СЦА2 сходна с симптома¬
тикой, выявляемой при СЦА1. Важными (хотя и не аб¬
солютными) дифференциально-диагностическими при¬
знаками, нередко отмечаемыми у больных СЦА2 даже
на ранней стадии болезни, являются медленные сакка-
ды, угнетение сухожильных рефлексов, статокинетичес¬
кий тремор рук. Нейровизуализационные (КТ, МРТ) и
морфологические изменения вещества мозга при СЦА2
в основном не отличаются от таковых при СЦА1.Спиноцеребеллярная атаксия 3-го типа (болезнь
Мачадо- -Джозеф ). Данная форма характеризуется выра¬
женным полиморфизмом клинических проявлений и
вариабельным возрастом манифестации болезни. Поми¬
мо прогрессирующей мозжечковой атаксии, у больных
может наблюдаться разнообразная экстрапирамидная
симптоматика (в первую очередь - дистония и синдром
паркинсонизма), амиотрофии, моторно-сенсорная невро¬
патия, симптомы поражения пирамидного тракта. У не¬
которых больных с данной формой описаны наружная
офтальмоплегия, крупные фасцикуляции периоральной
мускулатуры, феномен «выпученных глаз» (широко рас¬
крытые глазные щели с фиксированными глазными яб¬
локами); обнаружение этих симптомов существенно об¬
легчает клиническую диагностику. В литературе описа¬
ны единичные случаи, когла болезнь Мачадо-Джозеф у
отдельных членов семьи проявлялась в виде изолиро¬
ванного синдрома паркинсонизма [Giunti P. et al., 1995]
или генерализованной дистонии [Mtinchau A. et al., 1999].
Продолжительность заболевания обычно не превышает
20 лет. При КТ и МРТ у больных выявляется выражен¬
ное расширение IV желудочка, атрофия ствола мозга, на
поздней стадии - расширение субарахноидальных про
странств полушарий и червя мозжечка. На секции отме¬
чается в первую очередь дегенерация зубчатых ядер, вер
ДНК- диагностика наследственных болезней293хней и средней ножек мозжечка, ядер ствола мозга, чер¬
ной субстанции, внутреннего сегмента бледного шара,
передних рогов спинного мозга.Спиноцеребеллярная атаксия 4-го типа. СЦА4
представляет собой редкую форму аутосомно-доминан-
тных атаксий, для которой характерно начало болезни
на 4-м или 5-м десятилетии жизни и сочетание прогрес¬
сирующих координаторных нарушений с сенсорной
невропатией[Т1а1^апК. е1 а1., 1996]. В описанных япон¬
ских семьях СЦА4 характеризовалась развитием изоли¬
рованной мозжечковой атаксии, медленным течением и
атрофией червя мозжечка на МР-томограммах [>^аока
И. е1 а1., 2000]. Морфологическая картина СЦА4 до на¬
стоящего времени неизвестна.Спиноцеребеллярная атаксия 5-го типа. Средний
возраст начала болезни при СЦА5 около 40 лет, в невро¬
логическом статусе у больных отмечается развитие срав¬
нительно изолированной атаксии походки. Динамичес¬
кая атаксия в конечностях и дизартрия являются менее
выраженными, признаки мультисистемного поражения
мозга отсутствуют. Темп прогрессирования СЦА5 мед¬
ленный, больные на протяжении десятилетий сохраня¬
ют способность к самостоятельной ходьбе и самообслу¬
живанию. Заболевание практически не влияет на есте¬
ственную продолжительность жизни. На МР-томограм¬
мах выявляется атрофия червя и полушарий мозжечка
при сохранности ствола мозга. Морфологическая кар¬
тина СЦА5 не описана.Спиноцеребеллярная атаксия 6-го типа. Клини¬
ческая картина СЦА6 в целом аналогична таковой при
СЦА5 (за исключением несколько более поздней мани¬
фестации симптомов) и характеризуется развитием срав-
11ительно изолированной мозжечковой атаксии с весьма
медленным прогрессированием болезни. Интересно от¬
метить, что в данном гене а]Д-потенциал-зависимого
294Глава 3кальциевого канала, помимо экспансии CAG-повторов,
известны и другие мутации - нуклеотидные замены типа
миссенс и нонсенс. Они приводят к развитию двух дру¬
гих аллельных заболеваний - соответственно, эпизоди¬
ческой атаксии 2-го типа (см. далее) и семейной гемип¬
легической мигрени [Ophoff R. et al., 1996; Carrera R et
al., 1999; Tournier-Lasserve E., 1999]. У некоторых боль¬
ных с небольшой экспансией CAG-повторов (20-23 по¬
втора) или нонсенс-мутациями в гене а1Л-потенциал-за-
висимого кальциевого канала может наблюдаться кли¬
ническая картина, «промежуточная» между СЦА6 и эпи¬
зодической атаксией, начало болезни с приступообраз¬
ных эпизодов неустойчивости, головокружения, тошно¬
ты, рвоты и диплопии, с последующим постепенным
развитием постоянной медленно прогрессирующей атак¬
сии [Jodice C. et al., 1997]. При использовании методов
нейровизуализации у больных обнаруживается расши¬
рение субарахноидальных пространств полушарий и чер¬
вя мозжечка. Морфологические изменения при СЦА6
ограничены главным образом атрофией мозжечка (по¬
лушарий, червя) и нижних олив.Спиноцеребеллярная атаксия 7-го типа. Характер¬
ным признаком СЦА7 является сочетание мозжечково¬
пирамидного синдрома с прогрессирующей дегенераци¬
ей сетчатки (вплоть до полной слепоты). Частым явля¬
ется также наличие пирамидной симптоматики. Важно
отметить, что нарушения зрения у больных могут на
несколько лет опережать развитие координаторных рас¬
стройств либо, напротив, присоединяться значительно
позднее [Benton C. et al., 1998; Johansson J. et al., 1998],
что осложняет клиническую диагностику данной фор¬
мы аутосомно-доминантной атаксии. Как уже отмечалось
выше, данная форма атаксии характеризуется выражен¬
ной генетической нестабильностью мутантного аллеля
и антиципацией (главным образом при передаче мутан¬
ДНК- диагностика наследственных болезней295тного гена по мужской линии); описаны семьи с СЦА7,
в которых разница в возрасте начала болезни у лиц из
разных поколений составляет 30 и более лет. При КТ и
МРТ выявляются распространенные атрофические из¬
менения со стороны структур мозжечка и ствола мозга.
На секции в веществе мозга обнаруживаются измене¬
ния, в основном аналогичные таковым у больных СЦА1
и СЦА2.Спиноцеребеллярная атаксия 8-го типа. Данная
недавно открытая форма характеризуется вариабельным
возрастом манифестации симптомов (от 18 до 73 лет) и
развитием сравнительно изолированной мозжечковой
атаксии, иногда в сочетании с пирамидной спастичнос-
тью и снижением вибрационной чувствительности. За¬
болевание отличается медленно прогрессирующим те¬
чением. МР-томография выявляет изолированную атро¬
фию мозжечка. Морфологическая картина СЦА8 не опи¬
сана.Спиноцеребеллярная атаксия 10-го типа. Первые
симптомы появляются обычно на 2-4-м десятилетии
жизни. Заболевание характеризуется развитием изоли¬
рованной мозжечковой атаксии, которая у 2/3 больных
может сочетаться с генерализованными или сложными
парциальными эпилептическими припадками. В редких
случаях припадки могут явиться первым проявлением
болезни. Результаты обследования больных с использо¬
ванием методов нейровизуализации и морфологическая
картина СЦА10 не описаны.Спиноцеребеллярная атаксия 11 -го типа. Клини¬
ческая картина СЦА11 характеризуется симптоматикой
изолированной мозжечковой атаксии с медленным, срав¬
нительно благоприятным течением. Средний возраст, в
котором манифестирует заболевание, составляет около
25 лет. При проведении КТ/МРТ-исследования у боль-
I (ых выявляется атрофия мозжечка. Сведения о морфо¬
296Глава 3логической картине данной формы аутосомно-доминан-
тной атаксии отсутствуют.Спиноцеребеллярная атаксия 12-го типа. Для
СЦА12 характерен вариабельный возраст манифестации
болезни (от 8 до 55 лет, чаще на 4-м десятилетии жиз¬
ни). У больных отмечается прогрессирующая мозжеч¬
ковая атаксия в сочетании с акционным тремором рук
тремором головы, повышением сухожильных рефлексов,
нарушением функции глазодвигательных нервов, а так¬
же (на поздней стадии болезни) деменцией и легкими
паркинсоническими симптомами. При КТ и МРТ выяв¬
ляется атрофия мозжечка и коры больших полушарий.
Морфологическая картина СЦА12 не описана.Спиноцеребеллярная атаксия 13-го типа. Данная
форма имеет существенные отличия от большинства
описанных фенотипов доминантных атаксий. Для СЦА13
характерно начало болезни в детском возрасте и сочета¬
ние координаторных нарушений с умеренно выражен¬
ной умственной отсталостью. В связи с ранним дебю¬
том симптомов говорить об антиципации в обследован¬
ных семьях не представляется возможным. Заболевание
отличается медленным течением, самостоятельная ходь¬
ба становится невозможной к 40 годам. На МР-томог-
раммах выявляется атрофия червя мозжечка, расшире¬
ние IV желудочка, атрофия задних отделов моста и про¬
долговатого мозга. Данных о морфологической картине
СЦА13 в литературе нет.Спиноцеребеллярная атаксия 14-го типа. В един¬
ственной описанной японской семье с данной формой
атаксии средний возраст начала болезни составил 27 лет
(от 12 до 42), а фенотип характеризовался сочетанием
мозжечковой атаксии с аксиальными миоклониями (ми-
оклонии были особенно типичны для больных с более
ранним началом заболевания); у наблюдавшихся боль¬
ных симптоматика прогрессировала весьма медленно.
ДНК- диагностика наследственных болезней ... 297При МР-томографии выявлена атрофия червя и (на бо¬
лее поздней стадии) полушарий мозжечка. В родослов¬
ной отмечен четкий феномен антиципации. Морфоло¬
гическая картина СЦА14 не описана.Спиноцеребеллярная атаксия 16-го типа. При дан¬
ной редкой форме доминантной атаксии первые симп¬
томы могут дебютировать в возрасте от 20 до 66 лет, а в
клинической картине наблюдается развитие сравнитель¬
но изолированного мозжечкового синдрома (иногда в
сочетании с тремором головы) и медленное прогресси¬
рование болезни. При МР-томографии выявляется ат¬
рофия полушарий и червя мозжечка. Морфологическая
картина СЦА16 не описана.Спиноцеребеллярная атаксия 17-го типа. Заболе¬
вание характеризуется возрастом начала болезни от 19
до 48 лет и развитием прогрессирующей мозжечковой
атаксии, которая в ряде случаев может сочетаться с де-
менцией, симптомами паркинсонизма и эпилептически¬
ми припадками. При КТ/МРТ выявляется атрофия моз¬
жечка и больших полушарий мозга; морфологическая
картина неизвестна.Дентаторубро-паллидолюисова атрофия. Данное
заболевание является популяционно-специфичным и
встречается почти исключительно в Японии (в Европе и
Северной Америке описаны лишь единичные случаи
ДРПЛА). Симптоматика ДРПЛА чрезвычайно полимор¬
фна. Заболевание может начинаться в возрасте от 1-го
до 6-го десятилетия жизни и проявляется в виде 2 ос¬
новных клинических вариантов. При небольшой степе¬
ни экспансии С Ав-повторов имеет место более поздний
дебют и развитие хореоатетоза, атаксии, психических
нарушений (данный фенотип иногда обозначается в лите¬
ратуре как «псевдохорея»). У больных с выраженным
генетическим дефектом болезнь проявляется в более
раннем возрасте в виде сочетания атактических рас¬
298Глава 3стройств с синдромом прогрессирующей миоклонус-эпи-
лепсии и деменцией. Продолжительность болезни обыч¬
но не превышает 15-20 лет. Морфологически ДРПЛА
характеризуется дегенеративными изменениями в зуб¬
чатом ядре, наружном сегменте бледного шара и их про¬
екционных зонах в красном и люисовом ядрах, атрофи¬
ей коры больших полушарий. При МР-томографии го¬
ловного мозга у больных выявляется атрофия мозжечка,
ствола мозга (особенно покрышки) и больших полуша¬
рий, а в некоторых случаях - демиелинизация в белом
веществе перивентрикулярной области и семиовально¬
го центра больших полушарий мозга.Следует еще раз подчеркнуть, что неврологичес¬
кая картина большинства форм аутосомно-доминантных
атаксий является весьма полиморфной, а указаннные
выше обобщенные клинические критерии каждой фор¬
мы носят лишь ориентировочный «усредненный» харак¬
тер и далеко не всегда применимы у постели конкретно¬
го больного. На практике в большинстве случаев диф¬
ференцировать отдельные генетические варианты атак¬
сий без обнаружения мутации в конкретном гене не пред¬
ставляется возможным. Таким образом, единственным
достоверным методом специфической диагностики ауто¬
сомно-доминантных атаксий является прямое ДНК-тес-
тирование [^еуапт О. е1 а1., 2000].3.3.1.2. ДНК-диагностика аутосомно-доминантных
атаксийПри всех формах аутосомно-доминантных атак¬
сий с идентифицированными генами (СЦА типа 1, 2, 3,6, 7, 8, 10, 12, 17 и ДРПЛА) возможно проведение пря¬
мой ДНК-диагностики, основанной на амплификации
тринуклеотидного (при СЦА 10 пентануклеотидного)
участка гена. При электрофорезе продуктов амплифика¬
ции мутантный аллель четко определяется как патоло¬
ДНК- диагностика наследственных болезней... 299гически удлиненный фрагмент ДНК, содержащий уве¬
личенное (надпороговое) число тринуклеотидных повто¬
ров. Для большинства форм атаксий различия между
верхней границей нормальных аллелей и нижней грани¬
цей мутантных аллелей составляют как минимум не¬
сколько триплетов (9-12 нуклеотидов и более), в связи
с чем качественное электрофоретическое разделение
нормальных и мутантных аллелей может проводиться
по упрощенному экспресс-протоколу с использованием
агарозного геля (рис. 61).Ч-Рис. 61. Прямая ДНК-диагностика СЦА1 (электрофорез в
агарозном геле)Дорожка 1 - маркер, дорожки 2,3,5,7 - больные СЦА1, дорожки 4
п 8 -носители мутантного гена на пресимптоматической стадии,
дорожка 6-родственник из группы риска, у которого носительство
мутации отвергнуто, дорожка 9 - здоровый контроль. Длинной
стрелкой указан мутантный аллель (экспансия САО-повторов гена
СЦА1), короткой стрелкой - нормальный аллель.При необходимости точного количественного оп¬
ределения числа тринуклеотидных повторов разделение
продуктов амплификации проводится в полиакриламид-I юм геле, обладающем более высокой разрешающей спо¬
собностью (альтернативой является использование осо¬1 23456789
300Глава 3бой высокоразрешающей агарозы). Это требуется, в ча¬
стности, при проведении ДНК-диагностики СЦА6, по¬
скольку при данной форме аутосомно-доминантной атак¬
сии разница между нормальными (<18 САО-повторов)
и мутантными аллелями (>21 САО-повторов) является
весьма небольшой и требует точной количественной
оценки длины амплифицированных фрагментов гена. В
редких случаях при выраженной экспансии тринуклео-
тидных повторов (более 100 копий) ПЦР-диагностика
болезни не представляется возможной; это обусловлено
весьма низкой эффективностью амплификации аллелей,
содержащих сверхдлинные тринуклеотидные тракты.
Такая проблема обычно возникает при анализе гена в
ювенильных и ранне-детских случаях доминантных атак¬
сий (особенно СЦА7), обусловленных крайней степенью
тяжести мутации. Выявление указанных сверхдлинных
аллелей проводится путем блот-гибридизации геномной
ДНК с (САО)п-зондами.Серьезной проблемой при прямой ДНК-диагно¬
стике аутосомно-доминантных атаксий является генети¬
ческая гетерогенность данных заболеваний и необходи¬
мость «перебора» большого числа генов для выявления
мутации в одном из них у конкретного больного. Следо¬
вательно, разработка рационального алгоритма генети¬
ческого скрининга представляет собой важнейшую за¬
дачу при обследовании больных и семей с аутосомно-
доминантными атаксиями. Такой алгоритм может бази¬
роваться на двух основных принципах - анализе клини¬
ческой картины и сравнительной частоте различных
форм атаксий в изучаемой популяции. Те или иные осо¬
бенности клинического синдрома могут позволить за¬
подозрить конкретную генетическую форму атаксии и
начать поиск мутации с анализа соответствующего гена.
Так, наличие у больного с доминантной атаксией экст-
рапирамидной симптоматики позволяет с высокой ве¬
ДНК- диагностика наследственных болезней ... 301роятностью предположить экспансию CAG-повторов в
гене СЦАЗ (болезнь Мачадо-Джозеф), сочетание атак¬
сии с дегенерацией сетчатки требует в первую очередь
исследования гена СЦА7 и т.д. Как указывалось выше,
однако, большинство случаев аутосомно-доминантных
атаксий характеризуются отсутствием каких-либо пато-
гномоничных симптомокомплексов, которые могли бы
служить надежными опорными пунктами для выбора
«нужного» гена. В такой ситуации последовательность
анализа генов определяется частотой отдельных генети¬
ческих форм атаксий. Распределение аутосомно-доми¬
нантных атаксий на генетические формы чрезвычайно
варьирует в различных странах и географических регио¬
нах, однако в целом свыше 70% данных заболеваний в
изученных популяциях мира приходятся на СЦА1, СЦА2,
СЦАЗ и СЦА6. В Северной Америке, Японии, Китае и
ряде европейских стран (Германия, Португалия, Фран¬
ция) преобладающей формой является СЦАЗ/болезнь
Мачадо-Джозеф - от 21% до 50% случаев атаксий
[Maruyama H. et al., 1995; Ranum L. et al., 1995; Schuls L.
et al, 1995; Silveira I. et al., 1996; Tang B. et al., 2000], на
Кубе и в Южной Корее ведущей формой является СЦА2
[Gispert S. et al., 1993; Kyu Jin D. et al., 1999], тогда как в
Италии и Великобритании превалирует СЦА1 - от 26%
до 51% всех семей с аутосомно-доминантными атаксия¬
ми [Giunti Р. et al., 1994; Pareyson D. et al., 1999]. Таким
образом, в каждой конкретной популяции у обследуе¬
мых больных с аутосомно-доминантными атаксиями
ДНК-диагностику следует начинать с анализа тех генов,
мутации в которых являются наиболее частыми в дан¬
ной популяции.Предполагается, что различия в частоте отдель¬
ных форм атаксий в разных популяциях могут быть свя-
чаны с некоторыми молекулярными факторами - напри¬
мер, определенными межпопуляционными различиями
302Глава 3в распределении числа повторов нормальных аллелей,
являющихся источником мутаций de novo; большое зна¬
чение придается также историческим и географическим
особенностям возникновения и распространения отдель¬
ных мутаций. Так например предполагается, что СЦАЗ/
болезнь Мачадо-Джозеф в большинстве семей Север¬
ной Америки, Японии и Австралии обязана своим про¬
исхождением большим оригинальным семьям португаль¬
ских островов Флорес и Сан-Мигель, откуда заболева¬
ние было распространено в эти страны португальскими
мореплавателями в XVII веке [Rosenberg R., 1992; Burt Т.
et al., 1996].Формы атаксий сСЦА1 (40%)Рис. 62. Частота основных форм аутосомно-доминантных
атаксий в российской популяцииНами в 1994-1999 гг. проведен молекулярно-ге¬
нетический скрининг больных с аутосомно-доминант-
ными атаксиями в российской популяции, позволивший
установить частоту основных молекулярных форм этих
заболеваний в нашей стране (рис. 62). В нашей выборке
в 10 из 25 обследованных неродственных семей (40%
семей) заболевание было обусловлено экспансией САв-
повторов в гене СЦА1. В небольшом числе семей с ауто-СЦАЗ (12%)СЦА7 (4%)
СЦА6 (4%)неустановленным
молекулярным
актом (40%)
ДНК- диагностика наследственных болезней... 303сомно-доминантными атаксиями нами были выявлены
мутации в генах СЦА2, СЦАЗ, СЦА6 и СЦА7 (рис. 63).
В 44% семей у больных были выявлены нормальные зна¬
чения числа тринуклеотидных повторов в исследуемых
генах. Это позволяет заключить, что в данных случаях
заболевание обусловлено повреждением других генов
аутосомно-доминантных атаксий (например - СЦА4,
СЦА5, СЦА16 и т.д.), непосредственный мутационный
анализ которых в настоящее время невозможен. Резуль¬
таты наших исследований показывают, что в российс¬
кой популяции прямая ДНК-диагностика аутосомно-до¬
минантных атаксий должна начинаться с анализа (САО)п-
повтора в гене СЦА1 на хромосоме 6р22-23 [Иллари-
ошкин С.Н. и др., 1996 (б); 1999 (б)].12 3 4Рис. 63. Прямая ДНК-диагностика СЦА6
Дорожка 1 - маркер, дорожка 2 - больной СЦА6, дорожка 3 - больной
с другой генетической формой аутосомно-доминантной атаксии,
дорожка 4 - здоровый контроль. Мутантный аллель с экспансией
САС-повторов указан длинной стрелкой, нормальные аллели - ко¬
роткими стрелками. Электрофорез продуктов амплификации
1 троведен с использованием высокоразрешающей агарозы МБ (2,5%).
304Глава 3В обследованных нами семьях с СЦА1 в 6 случа¬
ях оказалось возможным проведение прямой ДНК-ди-
агностики носительства мутантного гена у клинически
здоровых родственников из группы риска - детей боль¬
ных СЦА1. В 4 случаях было диагностировано пресим-
птоматическое носительство мутации; у 2 тестируемых
лиц результат ДНК-диагностики был отрицательным, и
носительство мутантного гена отвергнуто (см. рис. 61)
[Иллариошкин С.Н. и др., 1997]. Лица, являющиеся но¬
сителями мутантного гена, были взяты под динамичес¬
кое наблюдение с целью оказания им помощи в реше¬
нии ряда психологических, медицинских и социальных
проблем.Нами были обследованы свыше 40 больных с раз¬
личными спорадическими формами атрофий мозжечка,
и ни в одном случае у них не было выявлено мутаций в
изучаемых генах. Это совпадает с результатами других
авторов [Kyu Jin D. et al., 1999; Pujana M. et al., 1999] и
свидетельствует о том, что новые мутации в рассматри¬
ваемых генах представляют собой исключительную ред¬
кость. Можно заключить, что при отсутствии у больно¬
го с мозжечковой атаксией семейного анамнеза вероят¬
ность обнаружения мутации в одном из генов аутосом-
но-доминантных атаксий крайне мала, и проведение
ДНК-диагностики у такого больного с практической точ¬
ки зрения нецелесообразно.Как указывалось выше, для 6 форм аутосомно-
доминантных атаксий известна хромосомная локализа¬
ция мутантного гена, однако сам ген и мутации в нем
пока не установлены (см. таблицу 6). В настоящее вре¬
мя оценить частоту данных форм атаксий не представ¬
ляется возможным, поскольку их диагностика требует
подтверждения генетического сцепления с соответству
ющими хромосомными локусами, что выполнимо лишь
ДНК- диагностика наследственных болезней ... 305в редко встречающихся больших информативных родос¬
ловных. При обследовании таких больших семей ДНК-
анализ проводится в 2 этапа: 1) скрининг известных му¬
таций (экспансия тандемных повторов) в генах СЦА-1,2, 3, 6, 7, 8, 10, 12, 17 и ДРПЛА; 2) при исключении
данных мутаций в идентифицированных генах проводит¬
ся анализ сцепления с маркерами остальных локусов
аутосомно-доминантных атаксий - на хромосомах
16q22.1 (СЦА4), llql3 (СЦА5), 15ql4—21.3 (СЦА11),
19ql3.3—13.4 (СЦА13), 19ql3.4-qter (СЦА14) и 8q22.1-
24.1 (СЦА16). При подтверждении сцепления с одним
из локусов в обследуемой семье может проводиться кос¬
венная ДНК-диагностика болезни у лиц из группы риска.3.3.2. Болезнь ФридрейхаБолезнь Фридрейха является наиболее часто
встречающейся формой наследственных атаксий. В ев¬
ропейских популяциях распространенность заболевания
составляет 2-5 случаев на 100 ООО населения, а частота
гетерозиготного носительства мутации - около 1 на 100
человек [Harding A., 1984]. Заболевание наследуется по
аутосомно-рецессивному типу и в значительной части
семей проявляется в виде единичных случаев. Оба пола
заболевают с одинаковой частотой. В типичных случаях
болезнь Фридрейха развивается на первом-втором деся¬
тилетии жизни и проявляется сочетанием характерных
неврологических и экстраневральных симптомов [Ива¬
нова-Смоленская И.А. и др., 1998 (a); Geoffroy G. et al.,
1976; Harding A., 1981; 1984]. Неврологическая картина
характеризуется смешанной (мозжечково-заднестолбо¬
вой) атаксией, дизартрией, сенсорной полиневропатией
с выпадением сухожильных рефлексов, парезами и ами-
306Глава 3отрофиями, стопными пирамидными знаками и наруше¬
нием глубокой чувствительности. К экстраневральным
проявлениям болезни Фридрейха, которые в ряде случа¬
ев могут предшествовать появлению неврологических
расстройств, относятся скелетные деформации (сколи¬оз, «стопа Фридрейха»), кардиомиопатия, сахарный ди¬
абет, гипогонадизм. В последние 20 лет широкое рас¬
пространение получили «классические» критерии диаг¬
ноза болезни Фридрейха, предложенные Geoffroy G. с
соавторами (1976) и Harding А. (1981). Они включают
начало болезни до 20 лет, наличие атаксии, дизартрии,
ранней сухожильной арефлексии, симптома Бабинского
и одного из указанных выше экстраневральных прояв¬
лений.Патоморфологически болезнь Фридрейха харак¬
теризуется гибелью афферентных волокон задних кореш¬
ков спинного мозга и периферических нервов, а также
комбинированной дегенерацией задних и боковых стол¬
бов спинного мозга; на поздней стадии процесса дегене¬
ративные изменения обнаруживаются также в ядрах ство¬
ла мозга, мозжечке, больших полушариях [Harding A.,
1984]. Течение заболевания неуклонно прогрессирую¬
щее, больные обычно погибают через 15-20 лет от мо¬
мента появления первых симптомов.На рубеже 90-х годов ген болезни Фридрейха был
картирован на длинном плече 9-й хромосомы в локусе
9ql3-q21 [Chamberlain S. et al., 1988; Hanauer A. et al.,
1990], что дало возможность проведения косвенной
ДНК-диагностики заболевания в отягощенных семьях.
В нашей стране первый опыт косвенной ДНК-диагнос¬
тики болезни Фридрейха в ряде информативных семей,
основанной на анализе высокополиморфных маркеров
из критической хромосомной области, был получен нами
совместно с лабораторией ДНК-диагностики Медико¬
ДНК- диагностика наследственных болезней ... 307генетического научного центра РАМН [Пугачев В.В. и
др., 1996]. В 1996 году большой международной иссле¬
довательской группой был идентифицирован новый ген
Х25 (другое название данного гена - FRDA), мутации в
котором приводят к возникновению болезни Фридрейха
[Campuzano V. et al., 1996]. Было показано, что развитие
заболевания обусловлено экспансией тринуклеотидных
GAA-повторов, локализованных в 1-м интроне генаХ25.
В норме число копий тринуклеотидных GAA-повторов
в гене Х25 варьирует от 7 до 22, тогда как у больных
болезнью Фридрейха оно составляет от 120 до 1700
[Campuzano V. et al., 1996; Dbrr A. et al., 1996; Filla A. et
al., 1996]. У абсолютного большинства больных экспан¬
сия GAA-повторов обнаруживается на обеих мутантных
хромосомах. В единичных случаях заболевание может
вызываться сочетанием экспансии GAA в одном аллеле
с наличием точковой мутации во втором аллеле гена Х25
[Campuzano V. et al., 1996; Filla A. et al., 1996;
BidichandaniS. et al., 1997], причем хромосомы с точко-
выми мутациями составляют не более 2% от общего
числа всех мутантных хромосом.Ген Х25 кодирует белок, состоящий из 210 ами¬
нокислот и получивший название «фратаксин». В клет¬
ке фратаксин локализуется в митохондриях
[Campuzano V. et al., 1997]. В норме наиболее высокая
экспрессия фратаксина характерна для мышцы сердца,
спинного мозга, скелетной мускулатуры, поджелудочной
железы и печени, тогда как у больных болезнью Фрид¬
рейха уровень мРНК фратаксина в тканях значительно
снижен [Campuzano V. etal., 1996; 1997]. Это свидетель¬
ствует о нарушении экспрессии мутантного гена как цен¬
тральном молекулярном механизме, лежащем в основе
развития болезни. Предполагается, что значительное уве¬
личение числа копий GAA-повторов в 1-м интроне Х25
308Глава 3ведет к нарушению созревания первичного транскрип¬
та, препятствуя вырезанию интрона из молекулы зрелой
мРНК, что приводит к блоку трансляции и отсутствию
нормального фратаксина [Rosenberg R., 1996]. Редкие
случаи точковых мутаций в кодирующей части гена Х25
ведут к синтезу функционально дефектного фратаксина
[Bidichandani S. et al., 1997]. Результаты исследований
последних лет свидетельствуют о ключевой роли фра¬
таксина в регуляции митохондриального транспорта
железа [Babcock М. et al., 1997; KoutnikovaH. etal., 1997].
Нарушение функции фратаксина в результате мутаций в
гене Х25 сопровождается повышением содержания же¬
леза в митохондриях, снижением резистентности к ок-
сидантному стрессу и окислительным повреждением
митохондрий, снижением синтеза АТФ и нарушением
энергетического метаболизма клетки [Wilson R., Roof D.,
1997; DelatyckiM. etal., 1999; Lodi R. etal., 1999; WongA.
et al., 1999]. Важное значение в патогенезе болезни при¬
дается также развивающейся недостаточности Fe-S-за-
висимых субъединиц ферментов дыхательной цепи
митохондрий [Rötig A. et al., 1997]. Таким образом, с со¬
временных позиций болезнь Фридрейха рассматривает¬
ся как особая разновидность митохондриальной болез¬
ни, обусловленная повреждением ядерного гена [Rötig А.
et al., 1997]. Не случайно в клинической картине заболе¬
вания доминируют симптомы поражения наиболее энер¬
гозависимых органов-мишеней (ЦНС, сердце, ске¬
летные мышцы, поджелудочная железа).Прямая ДНК-диагностика болезни Фридрейха
основана на ПЦР-амплификации тринуклеотидного уча¬
стка гена Х25. При электрофорезе продуктов амплифи¬
кации мутантные аллели определяются как фрагменты
ДНК увеличенной длины, нередко в виде смазанного
пятна (последнее обусловлено соматическим мозаициз-
ДНК- диагностика наследственных болезней... 309мом мутантного САА-повтора и особенностями ампли¬
фикации сверхдлинных тринуклеотид-содержащих ал¬
лелей) (рис. 64). В типичных случаях у больных отсут¬
ствуют аллели нормальной длины. Эффективность амп¬
лификации значительно снижается по мере нарастания
числа триплетов, поэтому аллели с наибольшей степе¬
нью экспансии повторов (свыше 1000 копий) на элект-
рофореграмме могут отчетливо не визуализироваться. У
таких больных для прямой ДНК-диагностики применя¬
ется более трудоемкий метод блот-гибридизации [Оигг А.
е!а1., 1996].1 2 3 4 5 6 7Рис. 64. Прямая ДНК-диагностика болезни Фридрейха
Дорожка 1 - маркер, дорожки 2-4 и 6 - больные с болезнью
< 1>ридрейха, дорожка 5 - гетерозиготный носитель мутации, дорожка
7 - контроль. Область мутантных аллелей с различной степенью
экспансии ОАА-повторов обозначена фигурной скобкой,
нормальный аллель указан стрелкой.Некоторые лаборатории предпочитают использо¬
вать блот-гибридизацию для рутинной ДНК-диагносги-
ки болезни Фридрейха. В редких случаях, когда боль¬
310Глава 3ной является компаунд-гетерозиготой по экспансии ОАА-
повторов в одном аллеле и точковой мутации в другом
аллеле гена, на электрофореграмме будут видны 2 про¬
дукта амплификации - увеличенной (экспансия повто¬
ров) и нормальной длины. В такой ситуации сам по себе
факт обнаружения одного патологически удлиненного
аллеля при наличии соответствующей клинической кар¬
тины позволяет с высокой вероятностью склониться в
пользу диагноза болезни Фридрейха, однако окончатель¬
ное подтверждение возможно только после выявления
точковой мутации на второй хромосоме, которое прово¬
дится с помощью стандартных подходов (чаще всего
используется 88СР-анализ и секвенирование кодирую¬
щей области гена).В 1997-2000 гг. нами были обследованы 28 боль¬
ных из 20 семей, поступивших в нейрогенетическое от¬
деление НИИ неврологии РАМН с подозрением на бо¬
лезнь Фридрейха. При проведении прямой ДНК-диаг-
ностики экспансия тринуклеотидных вАА-повторов в
1-м интроне гена Х25 была выявлена у больных из 15
семей, что явилось молекулярным подтверждением ди¬
агноза болезни Фридрейха (см. рис. 64). У 5 больных
(все - изолированные случаи) результат ДНК-диагнос¬
тики оказался отрицательным, и таким образом данный
диагноз был исключен. Число ОАА-повторов у больных
варьировало в достаточно широких пределах - от 100
до 1200 (680 ± 350), тогда как в норме во всех случаях
оно было менее 50 повторов, что вполне соответствует
данным других авторов. Нами не было выявлено каких-
либо различий в распределении повторов среди боль¬
ных различных этнических групп. Распределение мутан¬
тных аллелей гена Х25 по числу тринуклеотидньк ОАА-
повторов, выявленное в нашем исследовании, представ¬
лено на рис. 65. У 19 больных из 14 семей выявлена эк¬
ДНК- диагностика наследственных болезней ... 311спансия GAA-повторов в гомозиготном состоянии. В
одной семье у 4 больных сибсов, наряду с экспансией
GAA-повторов на одной из хромосом, был выявлен так¬
же аллель с нормальной длиной повтора, что позволяет
предполагать у них наличие точковой мутации гена на
второй мутантной хромосоме. Таким образом, в нашей
выборке больных свыше 96% не связанных общим про¬
исхождением мутантных хромосом характеризовались
экспансией GAA-повторов в соответствующем участке
гена Х25.Частота20.00%18.00%160С%- '14.00%-12.00%-}-'Число GAA-пювторовРис. 65. Распределение мутантных аллелей гена Х25 по числу
вАА-повторов в российской популяцииВсе больные с «классическим» фенотипом болез¬
ни Фридрейха, характеризовавшимся началом болезни
до 20 лет, быстрым прогрессированием и сочетанием
312Глава 3типичных неврологических и экстраневральных прояв¬
лений, имели гомозиготную экспансию вАА-повторов.
При этом число повторов у них в одном или (чаще) обо¬
их аллелях гена было значительным и всегда превыша¬
ло 400 копий. У больных из 2 семей, имевших неболь¬
шую степень экспансии вАА-повторов в обоих аллелях
гена (от 200 до 500 копий), клиническая картина была
весьма атипичной: она характеризовалась необычно по¬
здним началом (22-48 лет), относительно медленным
течением (на протяжении многих десятилетий), а также
отсутствием в клинической картине таких «облигатных»
для болезни Фридрейха проявлений как симптом Бабин-
ского, кардиомиопатия, скелетные деформации [Илла-
риошкин С.Н. и др., 1999 (а, б)]. Аналогичный «мяг¬
кий» вариант болезни с необычно медленным прогрес¬
сированием и сохранностью мышцы сердца наблюдал¬
ся нами у больных, являвшихся компаунд-гетерозигота¬
ми по экспансии вАА-повторов и точковой мутации в
гене Х25. Еще у одного больного, имевшего экспансию
вАА-повторов на обеих мутантных хромосомах, клини¬
ческая картина характеризовалась крайне необычным для
болезни Фридрейха синдромом - спастической атакси¬
ей [Иллариошкин С.Н. и др., 2000]. Приводим краткую
выписку из истории болезни этого пациента, демонст¬
рирующую решающую роль прямого ДНК-анализа в
постановке диагноза атипичных случаев болезни Фрид¬
рейха.Больной Ф.А., 30 лет, в возрасте 14 лет впер¬
вые стал отмечать ограничение движений в стопах,
утомляемость в ногах при ходьбе. С 16 лет изменился
почерк, появились жалобы на «перебои» в работе серд¬
ца. До 18 лет заметного прогрессирования болезни не
отмечалось. В 1986 году во время службы в армии на¬
росла слабость в ногах, изменилась походка, в это же
ДНК- диагностика наследственных болезней ... 313время больной стал ощущать скованность в ногах, уси¬
ливающуюся при длительном, беге и ходьбе. В 1988 году
обследован в военном госпитале, где впервые отмече¬
на деформация стоп, повышение тонуса в ногах, атак¬
сия, изменения на ЭКГ. В дальнейшем постепенно на¬
растала выраженность атаксии, развился нижний спа¬
стический парапарез, с 23 лет. стала невозможной са¬
мостоятельная ходьба, ухудшилась речь. Неоднократ¬
но находился на обследовании в неврологических ста¬
ционарах с диагнозами «атипичная форм.а болезни
Фридрейха со спастическим парапарезом», «наслед-
ственная спастическая атаксия», «осложненная фор¬
ма наследственной спастической параплегии». В 1998
году поступил в нейрогенетическое отделение НИИ не¬
врологии РАМН для уточнения диагноза.Общий и семейный анамнез не отягощен. При
поступлении: левосторонний сколиоз грудного отдела
позвоночника, деформация стоп по типу «фридрейхов-
ских». В неврологическом статусе отмечаются сла¬
бость конвергенции и горизонтальный нистагм; дис-
фония; речь растянутая, дизартричная. Снижен темп
движений в дистальных отделах рук; легкая гипотро¬
фия и снижение силы межкостных мышц кистей и
мышц области Иуро1кепаг. Выраженный нижний па¬
рапарез с резким повышением мышечного тонуса по
спастичесому типу в четырехглавой мышце бедра с
двух сторон и приводящих группах мышц. В голенос¬
топных суставах движения практически отсутству¬
ют, контрактуры; в положении лежа возможно огра¬
ниченное сгибание ног в коленных суставах и кратков¬
ременное отрывание пятки от постели. Сухожильные
и периостальные рефлексы с рук оживлены, выявля¬
ются симптомы Якобсона-Ласка, Тремнера, Гофмана
с обеих сторон. Коленный и ахиллов рефлексы высокие,
поликинетичные, с расширением, рефлексогенных зон,
314Глава 3выявляются 2-сторонние симптомы Бабинского, Рос-
солимо, Оппенгейма, отмечается заъцитная реакция в
виде тройного сгибания нижних конечностей в ответ
на укол. Гиперметрия и адиадохокинез в руках, мимопо-
падание при пальценосовой пробе с двух сторон; почерк
неровный, изменен по типу макрографии. Координатор-
ные пробы в ногах проверить не удается из-за спасти¬
ческого парапареза. Отчетливая атаксия туловища в
положении сидя; стоять не может даже с поддерж¬
кой. Болевая и тактильная чувствительность в конеч¬
ностях снижена по дистальному типу (в ногах до уров¬
ня коленного сустава и в руках до уровня верхней тре¬
ти предплечий); суставно-мышечное чувство наруше¬
но в пальцах стоп, вибрационная чувствительность сни¬
жена в дистальных отделах конечностей.Клинические анализы крови и мочи, а также ру¬
тинные биохимические показатели (включая уровень
глюкозы крови) в пределах нормы. На ЭКГ выявлены
диффузные изменения миокарда боковой стенки левого
желудочка (миокардиодистрофия). Анализ данных, по¬
лученных при электронейромиографии, свидетельству¬
ет о поражении сенсорных волокон периферических нер¬
вов (преимущественно на ногах), при сохранном прове¬
дении возбуждения по двигательным волокнам. При
исследовании соматосенсорных вызванных потенциа¬
лов определяется грубое нарушение функции сомато¬
сенсорных периферических путей на участке «средин¬
ный нерв-плечевое сплетение» с обеих сторон. .ДНК-диагностика: при исследовании области
тринуклеотидных ОАА-повторов в гене Х25 выявлено
увеличение длины амплифицированного фрагмента в
обоих аллелях (число САА-повторов составило 600 и 700
копий). Клинический диагноз: болезнь Фридрейха.Взаимоотношению между характером генетичес¬
кого дефекта и особенностями клинической картины
ДНК- диагностика наследственных болезней ... 315при болезни Фридрейха посвящено большое число ис¬
следований. Наши собственные данные и аналогичные
данные других авторов показывают, что с нарастанием
числа GAA-повторов мутантного гена имеет место бо¬
лее ранняя манифестация болезни [Иллариошкин C.H.,
1997; Иллариошкин С.Н. и др., 1999 (а, б); Dürr A. et al.,
1996; Filla A. et al., 1996]. Более того, такие тяжелые
проявления болезни Фридрейха, как кардиомиопатия и
сахарный диабет, развиваются только у тех больных,
число GAA-повторов у которых превышает 800 копий
[Filla A. et al., 1996]. С другой стороны, при небольшой
степени экспансии GAA-повторов могут иметь место
разнообразные атипичные, «доброкачественные» вари¬
анты болезни - такие как болезнь Фридрейха с поздним
началом (на 5-м и даже 6-м десятилетии жизни!), бо¬
лезнь Фридрейха с сохранными сухожильными рефлек¬
сами или со спастичностью (что обусловлено меньшей
выраженностю сенсорной полиневропатии) и др. [Dürr
A. et al., 1996; Ragno М. et al., 1997; Coppola G. et al.,
1999]. Взаимосвязь между числом триплетов и тяжес¬
тью заболевания наиболее четко выражена для мень¬
шего из двух мутантных аллелей, поскольку он является
более «сохранным» с функциональной точки зрения и
непосредственно определяет количество активного фра-
таксина в клетке [Filla A. et al., 1996]. Характер прояв¬
ления точковых мутаций в гене Х25 зависит от влияния
мутации на экспрессию гена: миссенс-мутации, при
которых фратаксии сохраняет определенный уровень
остаточной активности, проявляются «мягким» фено¬
типом болезни; в то же время повреждения, ведущие к
угнетению синтеза белка (нонсенс-мутации, мутации в
сайтах сплайсинга), могут характеризоваться «класси¬
ческим» тяжелым вариантом заболевания с ранним на¬
чалом симптомов [Filla A. et al., 1996; Bidichandani S. et
ul., 1997; Forrest S. et al., 1998; Cossee M. et al., 1999].
316Глава 3Таким образом, в настоящее время стало очевид¬
ным, что истинный спектр клинических проявлений бо¬
лезни Фридрейха гораздо шире, чем это предполагалось
до введения в практику методов прямого ДНК-тестиро-
вания. В целом, по данным ряда авторов, около 30-40%
больных с экспансией GAA-повторов в гене Х25 харак¬
теризуются теми или иными отклонениями от «класси¬
ческих» критериев болезни Фридрейха [Campuzano V.
et al., 1996; Dürr A. et al., 1996; Filla A. et al., 1996], в
связи с чем многие случаи заболевания оставались до
последнего времени не диагностированными. Это позво¬
ляет пересмотреть в сторону увеличения данные об ис¬
тинной распространенности болезни Фридрейха. Мож¬
но заключить, что исследование гена Х25 показано во
всех аутосомно-рецессивных и спорадических случаях
атаксий дегенеративной природы (независимо от возра¬
ста дебюта заболевания), особенно при наличии в кли¬
нической картине хотя бы отдельных симптомов, свой¬
ственных типичному фенотипу болезни Фридрейха (на¬
пример, кардиомиопатии, нарушений глубокой чувстви¬
тельности и т.д.).В настоящее время выявление с помощью ДНК-
диагностики случаев болезни Фридрейха на ранней и
пресимптоматической стадии приобретает особое зна¬
чение в связи с потенциальными возможностями пре¬
вентивной терапии, перспективы которой становятся все
более реальными в свете новых знаний о патогенезе бо¬
лезни. Первые подходы к патогенетической терапии бо¬
лезни Фридрейха уже сейчас оживленно обсуждаются в
литературе (применение десферала и других хелатных
соединений с целью коррекции нарушенного митохонд¬
риального транспорта железа, назначение коэнзима Q и
всего комплекса препаратов, используемых при лечении
митохондриальных болезней, и др.) [Cossee М. et al.,1997]. По-видимому, в ближайшем будущем можно ожи¬
ДНК- диагностика наследственных болезней ... 317дать появления этих и целого ряда других принципиаль¬
но новых методов лечения болезни Фридрейха в арсена¬
ле практических врачей-неврологов.3.3.3. Другие формы наследственных атаксий3.3.3.1. Атаксия, обусловленная изолированным дефи¬
цитом витамина ЕДанное заболевание представляет собой редкую
форму наследственной атаксии, большинство случаев
которой описано в северной Африке и странах Среди¬
земноморского региона. В литературе оно нередко обо¬
значается как «синдром AVED» (от англ. Ataxia with
Vitamin Е Deficiency), а также как «атаксия Фридрейха с
дефицитом витамина Е» вследствие ее значительного
клинико-морфологического сходства с болезнью Фрид¬
рейха [Stumpf D. et al., 1987; DiDonato S., 1995]. Тип
наследования - аутосомно-рецессивный.Клиническая картина AVED практически неотли¬
чима от «классического» варианта болезни Фридрейха:
заболевание начинается чаще всего в возрасте от 4 до 18
лет и характеризуется прогрессирующими нарушения¬
ми координации движений, дизартрией, угнетением су¬
хожильных рефлексов, нарушением суставно-мышечной
и вибрационной чувствительности [Harding A. etal., 1985;
Stumpf D. et al., 1987; Kayden H., 1993]. В отличие от
болезни Фридрейха, при витамин Е-дефицитной атак¬
сии лишь у 19% больных имеет место кардиомиопатия,
значительно реже встречаются также скелетные дефор¬
мации и другие экстраневральные проявления. При ес¬
тественном течении болезни в большинстве случаев уже
на 3-м десятилетии жизни больные прикованы к посте¬
ли и теряют способность к самообслуживанию. На сек¬
ции выявляются изменения, идентичные таковым при
318Глава 3болезни Фридрейха, главным из которых является ком¬
бинированная дегенерация проводников в задних и бо¬
ковых столбах спинного мозга [Harding А., 1984].Даже в начальной стадии заболевания у больных
отмечается резкое снижение концентрации витамина Е
в сыворотке крови. Это обусловлено повреждениями гена
на хромосоме 8q, ответственного за синтез микросомаль-
ного белка - транспортера а-токоферола; данный белок
осуществляет в клетках печени встраивание а-токофе¬
рола (активной молекулярной формы витамина Е) в со¬
став липопротеидов очень низкой плотности [Ouahchi К.
et al., 1995]. В норме а-токоферол в комплексе с липоп-
ротеинами очень низкой плотности представляет собой
главную и наиболее стабильную фракцию витамина Е в
сыворотке крови. Мутации гена приводят к инактива¬
ции соответствующего белка и нарушению печеночной
секреции а-токоферола в кровь. Результатом этого яв¬
ляется глубокий системный дефицит витамина Е в орга¬
низме, несмотря на нормальную абсорбцию пищевого
а-токоферола в тонком кишечнике. Предполагается, что
в основе патогенеза данной формы наследственной атак¬
сии лежит недостаточность антиоксидантной активнос¬
ти, реализуемой витамином Е на уровне клеточных мем¬
бран [Kayden Н., 1993; Ouahchi К. et al., 1995].Прямая ДНК-диагностика синдрома AVED с кли¬
нической точки зрения имеет ограниченное значение,
поскольку диагноз данного заболевания обычно устанав¬
ливается на основании сравнительно простого биохими¬
ческого исследования (содержание витамина Е в сыво¬
ротке крови). Выявление мутаций в гене транспортера
а-токоферола может помочь в дифференциальной диаг¬
ностике синдрома AVED с вторичными витамин Е-де-
фицитными состояниями, характеризующимися сниже¬
нием содержания витамина Е и развитием прогрессиру¬
ДНК- диагностика наследственных болезней ... 319ющей атаксии (синдром Бассена Корн Цвей га, синдром
мальабсорбции и др.) [Kayden H., 1993]. Принимая во
внимание сходство клинической картины синдрома
AVED и болезни Фридрейха, молекулярно-генетическое
обследование таких больных должно всегда предусмат¬
ривать исключение экспансии GAA-повторов в гене Х25
(генетический дефект, характерный для болезни Фрид¬
рейха). Для прямой ДНК-диагностики синдрома AVED
используются стандартные методы мутационного скри¬
нинга кодирующей области гена транспортера а-токо-
ферола [Hentati A. et al., 1996; Cavalier L. et al., 1998].
При установлении диагноза AVED больным назначает¬
ся пожизненная заместительная терапия витамином Е в
суточной дозе от 5-10 мг/кг веса, что способствует нор¬
мализации уровня витамина Е в крови, приводит к оп¬
ределенному уменьшению выраженности неврологичес¬
кой симптоматики и полностью предотвращает дальней¬
шее прогрессирование болезни.3.3.3.2. Эпизодические атаксииЭпизодические (периодические, пароксизмаль¬
ные) атаксии представляют собой гетерогенную группу
аутосомно-доминантных заболеваний, которые характе¬
ризуются повторяющимися приступообразными эпизо¬
дами координаторных нарушений различной длительно¬
сти и интенсивности, нередко в сочетании с другими
симптомами [Gancher S., Nutt J., 1986]. Приступы обыч¬
но начинаются в детском возрасте и могут отмечаться с
частотой от нескольких в день до 1-2 в месяц. В боль¬
шинстве случаев частота приступов атаксии снижается
с течением времени.С клинической и генетической точки зрения мож¬
но выделить несколько основных форм эпизодических
атаксий. Эпизодическая атаксия 1-го типа характеризу¬
320Глава 3ется кратковременными (от нескольких секунд до не¬
скольких часов) острыми эпизодами мозжечковой атак¬
сии, сопровождающимися тошнотой, рвотой, головок¬
ружением, двоением, дизартрией, иногда - зрительны¬
ми нарушениями. Приступы могут быть спровоцирова¬
ны резкими движениями, испугом и другими эмоцио¬
нальными факторами [Van Dyke D. et al., 1975]. В меж-
приступном периоде часто отмечаются миокимии в ли¬
цевой мускулатуре и других группах мышц. Эпизодичес¬
кая атаксия-1 обусловлена разнообразными точковыми
мутациями гена потенциал-зависимого калиевого кана¬
ла (KCNA1), расположенного на хромосоме 12р 13
[Browne D. et al., 1994]. Патофизиологической основой
пароксизмов является уменьшение калиевого тока из
клетки во время потенциала действия и нарушение ка-
лий-зависимой реполяризации мембраны нейрона. По¬
скольку значительная экспрессия гена KCNA1 отмеча¬
ется в корзинчатых клетках мозжечка, предполагается,
что приступы атаксии связаны с нарушением моторного
контроля вследствие дисбаланса между тормозными и
возбуждающими медиаторными путями [Jurkat-Rott К.
et al., 1999]. В некоторых случаях у больных с подтверж¬
денными мутациями в гене KCNA1, помимо типичных
острых эпизодов атаксии, могут иметь место парциаль¬
ные эпилептические припадки [Zuberi S. et al., 1999].При эпизодической атаксии 2-го типа у больных
развиваются сходные с вышеописанными мозжечково¬
вестибулярные пароксизмы, которые, однако, являются
более длительными и могут продолжаться до несколь¬
ких дней. Острые эпизоды атаксии при данной форме
провоцируются эмоциональными и физическими стрес¬
сами, приемом алкоголя и кофе и купируются ацетазо-
ламидом (диакарбом) в суточной дозе 0,5-0,75 г
[Baloh R., Winder А., 1991]. В межприступном периоде
ДНК- диагностика наследственных болезней ... 321у больных эпизодической атаксией-2 выявляются нис¬
тагм и расстройства координации различной степени вы¬
раженности. С течением времени выраженность посто¬
янных мозжечковых нарушений может нарастать, и на
поздней стадии клиническая картина данной формы эпи¬
зодической атаксии начинает все больше напоминать
клинику прогрессирующей аутосомно-доминантной спи-
поцеребеллярной атаксии 6-го типа, описанную в разде¬
ле 3.3.1. Это сходство усиливается частым обнаружени¬
ем у больных эпизодической атаксией-2 атрофии червя
мозжечка при использовании методов нейровизуализа¬
ции (КТ, МРТ). Сходство эпизодической атаксии-2 и
СЦА6 имеет под собой четкую молекулярную основу:
показано, что эти заболевания являются аллельными и
обусловлены различными типами мутаций в гене а -
субъединицы потенциал-зависимого кальциевого кана¬
ла (CACNL1A4) на хромосоме 19р 13.1 [Zhuchenko О. et
al., 1997; Tournier-Lasserve E., 1999]. Большинство слу¬
чаев эпизодической атаксии-2 вызываются точковьтми
мутациями в гене CACNL1 A4, ведущими к обрыву транс¬
ляции [Ophoff R. et al., 1996; Denier C. et al., 1999]. В то
же время, как указывалось в разделе 3.3.1, у больных
СЦА6 имеет место экспансия тринуклеотидных CAG-
иовторов в данном гене [Zhuchenko О. et al., 1997]. По
мнению Zhuchenko О. с соавторами (1997), разные фе¬
нотипы атаксии при повреждении одного и того же гена
обусловлены различным характером нарушения функ¬
ции потенциал-зависимого кальциевого канала у каж¬
дой категории больных. Недавно было показано, одна¬
ко, что эти различия не носят принципиального харак¬
тера: у некоторых больных с фенотипом эпизодической
атаксии-2 заболевание может быть связано с небольшой
степенью экспансии CAG-повторов в гене CACNL1A4
(20-23 копий повторов) [Jodice C. et al., 1997]. Удиви¬
322Глава 3тельно, что еще один тип мутаций в данном гене (точ-
ковые миссенс-мутации с заменой аминокислоты) обус¬
ловливает развитие совершенно особого аутосомно-до-
минантного заболевания - семейной гемиплегической
мигрени [Саггега Р. е! а1., 1999]. В некоторых случаях
точковые мутации в гене САСМЛ А4 могут приводить к
манифестации «смешанных» фенотипов, представляю¬
щих собой комбинацию вышеуказанных неврологичес¬
ких проявлений - например, сочетание эпизодической
ацетазол-чувствительной атаксии с эпизодической гемип¬
легией [.Теи I. е1: а1., 1999] или сочетание приступов аце¬
тазол-чувствительной гемиплегической мигрени с посто¬
янной прогрессирующей атаксией [ ОрЬюй’К. е1а1., 1996;
ВаШэйш 8. е! а1., 1999]. Полиморфизм комбинаций раз¬
личных острых пароксизмов и постоянных прогресси¬
рующих неврологических расстройств при данных ал¬
лельных заболеваниях связан, наиболее вероятно, с мно¬
гообразием функций кальциевых каналов и особеннос¬
тями их распределения в ЦНС [ВаШэйш 8. е1 а1., 1999].Известна еще одна форма аутосомно-доминант-
ной эпизодической атаксии - так называемая «периоди¬
ческая вестибуломозжечковая атаксия», характеризую¬
щаяся приступами диплопии, осциллопсии, мозжечко¬
вой атаксии, тошноты и рвоты. Приступы провоциру¬
ются внезапными изменениями положения головы, мель¬
канием объектов перед глазами больного [Патр К. е1.
а1., 1996]. В межприступном периоде отмечается нару¬
шение плавных движений глазных яблок, спонтанный
нистагм, угнетение оптокинетического нистагма, а так¬
же постепенное появление и нарастание выраженности
постоянных координаторных нарушений. Данная форма
эпизодической атаксии является нозологически самосто¬
ятельной, поскольку в соответствующих семьях исклю¬
чено сцепление с известными локусами эпизодических
атаксий 1-го и 2-го типов [Патр К. й а!., 1996].
ДНК- диагностика наследственных болезней ... 323К настоящему времени в мире накоплен доста¬
точный опыт прямой ДНК-диагностики эпизодических
атаксий 1-го и 2-го типов, осуществляемой на основе
мутационного скрининга генов KCNA1 и CACNL1A4.
Выбор конкретного гена для исследования определяет¬
ся особенностями клинической картины, позволяющи¬
ми достаточно четко дифференцировать между собой
основные формы эпизодической атаксии (длительность
атактических пароксизмов, особенности их возникнове¬
ния, реакция на ацетазоламид). Необходимо также при¬
нимать во внимание любые пароксизмальные невроло¬
гические расстройства у ближайших родственников об¬
следуемого лица (мигрень, эпизоды преходящего геми¬
пареза и т.д.), поскольку полиморфизм клинических про¬
явлений эпизодических атаксий (особенно атаксии 2-го
типа) может носить не только межсемейный, но внутри¬
семейный характер. В большинстве случаев проводится
SSCP-анализ отдельных кодирующих участков гена, пос¬
ле чего наличие мутации в образце с измененными элек¬
трофоретическими характеристиками подтверждается
прямым секвенированием изучаемого фрагмента ДНК
[Denier C. et al., 1999; Jen J. et al., 1999]. Принимая во
внимание простое строение гена KCNA1 (один кодиру¬
ющий экзон, 1448 п.о.), многие исследователи для ДНК-
диагностики эпизодической атаксии-1 предпочитают
сразу проводить прямое секвенирование кодирующей
области гена, амплифицированной в трех последователь¬
ных реакциях ПЦР [Zuberi S. et al., 1999]. При обследо¬
вании больных с предположительным диагнозом эпизо¬
дической атаксии-2 на первом этапе анализа целесооб¬
разно исключить экспансию тринуклеотидных CAG-no-
второв в гене CACNL1A4 в соответствии с протоколом,
используемым для прямой ДНК-диагностики СЦА6.
324Глава 33.3.3.3. Лтаксия-твлеангиэктазияАтаксия-телеангиэктазия (синдром Луи- Бар. син¬
дром Бодера-Седжвика) является относительно частой
формой наследственных атаксий, распространенность
которой составляет около 1 на 100 ООО населения. Тип
наследования - аутосомно-рецессивный.Атаксия-телеангиэктазия характеризуется мульти-
системными и весьма полиморфными клиническими
проявлениями, которые не ограничены лишь поражени¬
ем ЦНС, а вовлекают также кожу, сосудистую, иммун¬
ную, эндокринную и костную системы [Shiloh Y., 1995;
Gatti R., 1998]. Заболевание обычно начинается на 1-2-м
году жизни ребенка и проявляется прогрессиругцими рас¬
стройствами ходьбы и координации движений, к кото¬
рым могут присоединяться другие неврологическике
симптомы (хореоатетоидные гиперкинезы, мышечная
гипотония, угнетение сухожильных рефлексов, наруше¬
ние психического развития и др.). Весьма характерны¬
ми являются изменения мелких сосудов в виде симмет¬
ричных телеангиэктазий, обычно локализованные на
склерах и конъюнктиве глаз, веках, ушных раковинах,
спинке носа, области локтевого сгиба и подколенной
ямки. К важнейшим диагностическим критериям атак-
сии-телеангиэктазии относятся частые инфекционные
заболевания (обусловленные дисплазией тимуса и дефи¬
цитом иммуноглобулинов классов A, G и Е), прогери-
ческие изменения кожи и волос, различные онкологи¬
ческие осложнения, скелетные деформации, эндокрин¬
ные расстройства (гипогонадизм, задержка роста, сахар¬
ный диабет).Продолжительность болезни составляет около 20
лет, больные погибают обычно от инфекционных и он¬
кологических осложнений. Нейропатоморфологические
изменения включают дегенерацию полушарий и червя
ДНК- диагностика наследственных болезней ... 325мозжечка, зубчатых ядер, нижних олив, проводников
спинного мозга [Gatti R., 1998].Основной ген, ответственный за развитие атак-
сии-телеангиэктазии, локализован на хромосоме 11 q22-
23 и получил обозначение ATM (от англ. Ataxia-
Teleangiectasia Mutated) [Savitsky К. et al., 1995]. Белко¬
вый продукт данного гена относится к семейству фос-
фатидилинозитол-киназ - важнейших передатчиков внут¬
риклеточных сигналов, реализующих свои функции по¬
средством фосфорилирования молекул-мишеней различ¬
ных компартаментов клетки. Основной функцией дан¬
ного белка, локализованного в ядре, является контроль
клеточного цикла, в частности приостановка митоза в
ответ на разрыв двойной цепи ДНК с целью предостав¬
ления клетке возможности репарации поврежденной
молекулы ДНК и поддержания стабильности генома
[Hoekstra М., 1997]. Клеточный ответ на повреждение
структуры ДНК осуществляется путем модулирования
белковым продуктом гена ATM активности ряда внут¬
риклеточных субстратов (с-АЫ, р53 и др.), изменения
экспрессии регуляторных генов и (в некоторых тканях)
индукции апоптоза мутантных клеток [Lehmann А.,
Carr А., 1995; Rotman G., Shiloh Y., 1998; Brown К. et al.,
1999]. Предполагается также, что белок ATM принима¬
ет участие в процессах аксонального и синаптического
транспорта [Brown К. et al., 1999]. Таким образом, муль¬
тисистемность клинических проявлений атаксии-теле-
ангиэктазии обусловлена важной общебиологической ро¬
лью белка ATM в функционировании клеток различных
тканей, а также его взаимодействием с большим числом
сигнальных путей клетки.На сегодняшний день у больных атаксией-теле-
апгиэктазией описано свыше 250 мутаций в гене ATM
[Concannon R, Gatti R., 1997]. Большинство из них при¬
326Глава 3водят к раннему обрыву трансляции и практически пол¬
ному отсутствию белка в клетке [Gilad S. et al., 1996;
Lakin N. et al., 1996]. Реже выявляются короткие деле¬
нии, не нарушающие рамку считывая, и «мягкие» мута¬
ции в сайтах сплайсинга, сохраняющие частичную воз¬
можность нормального сплайсинга первичного транс¬
крипта [McConville С. et al., 1996; Gilad S. et al., 1998]. В
случаях указанных «мягких» мутаций заболевание мо¬
жет проявляться в виде атипичных, относительно доб¬
рокачественных и более поздних вариантов (например,
изолированная мозжечковая атаксия без телеангиэкта-
зий и признаков мультисистемной патологии), адекват¬
ное распознавание которых практически невозможно без
использования молекулярных методов диагностики. В
связи с тем, что мутации в гене ATM приводят к полно¬
му или частичному угнетению синтеза нормального бел¬
ка, наиболее универсальным методом молекулярной ди¬
агностики атаксии-телеангиэктазии является Вестерн-
блоттинг образцов тканей больного с антителами к бел¬
ку ATM [Rotman G., Shiloh Y., 1998]. Более трудоемким
является скрининг гена ATM с целью выявления мута¬
ций и прямой ДНК-диагностики болезни. В связи с боль¬
шим размером кодирующей области гена (13 кб, 66 эк-
зонов) такой анализ может проводиться лишь в высоко¬
специализированных и хорошо оснащенных лаборато¬
риях. На практике, с целью проведения в отягощенной
семье пренатального ДНК-анализа плода (такая необхо¬
димость возникает при наличии в семье больного ре¬
бенка с несомненным клиническим диагнозом атаксии-
телеангиэктазии) нередко проводится косвенная ДНК-
диагностика с исследованием маркеров из критической
хромосомной области 1 lq22—23.В последние годы было показано, что женщины,
являющиеся гетерозиготными носительницами мутации
ДНК- диагностика наследственных болезней ... 327и гене ATM, имеют значительно повышенный (в 4-9 раз)
риск заболеть раком молочной железы по сравнению с
общей популяцией [Athma P. et al., 1996; Broeks A. et al.,
.'ООО]. Имеются также данные, свидетельствующие о
иысокой частоте инактивации гена ATM в соматических
клетках больных Т-пролимфоцитарной лейкемией
| Kotman G., Shiloh Y., 1998]. В связи с этим в литературе
обсуждается возможность выполнения геном ATM фун¬
кции супрессора опухолевого роста, связанной, по-ви-
димому, с его участием в интимных механизмах деления
клетки [Rotman G., Shiloh Y., 1998]. Таким образом, иден¬
тификация мутаций в гене ATM имеет значение не толь¬
ко для ДНК-диагностики атаксии-телеангиэктазии, но
также может служить одним из молекулярных тестов,
позволяющих оценивать предрасположенность к неко¬
торым формам онкологических заболеваний.Атаксия-телеангиэктазия является генетически
гетерогенным заболеванием. Приблизительно у 6% боль¬
ных не удается выявить мутаций в гене ATM; в этих слу¬
чаях заболевание обусловлено мутациями нового гена-
репаратора ДНК - MRE11A, расположенного в хромо¬
сомной области 11 q21 (т.е. в непосредственной близос¬
ти от гена ATM) [Stewart G. et al., 1999]. В целом
MRJE11 A-ассоциированные случаи атаксии-телеангиэк-
газии характеризуются более благоприятным течением
по сравнению с классической формой, вызываемой му¬
тациями гена ATM. Принимая во внимание близкое рас¬
положение обоих генов на хромосоме 1 lq, дифференци-
лция двух форм атаксии-телеангиэктазии с помощью
.шализа генетического сцепления является весьма про¬
блематичной [Stewart G. et al., 1999]. У больного с фено¬
типом атаксии-телеангиэктазии при исключении повреж¬
дений ATM может проводиться ДНК-диагностика мута¬
ций в гене MRE11 А.
328Глава 33.3.3.4. Врожденная гипоплазия мозжечкаДанным термином объединяется большая группа
атактических синдромов, характеризующихся нарушени¬
ем нормального отногенетического развития и диффе-
ренцировки как различных частей мозжечка, так и от¬
дельных клеточных слоев его коры [Bruyn G., 1991]. К
мозжечковой дисгенезии может приводить воздействие
разнообразных экзогенных факторов (таких как внутри¬
утробная цитомегаловирусная инфекция, авитаминоз,
применение рентгенотерапии у матери во время бере¬
менности и др.). Не менее чем в половине случаев врож¬
денная гипоплазия мозжечка носит наследственный ха¬
рактер и связана с мутациями различных ядерных генов
[Sanner G., Hagberg В., 1974; Harding А., 1984]. Чаще
всего наблюдается аутосомно-рецессивный тип наследо¬
вания болезни; в отдельных семьях заболевание может
передаваться по аутосомно-доминантному и Х-сцеплен-
ному типам.Клиническая картина врожденной гипоплазии
мозжечка достаточно стереотипна: на фоне общей за¬
держки двигательного развития ребенка уже на первом
году может отмечаться интенционный тремор конечно¬
стей, тремор головы, осцилляции туловища в положе¬
нии сидя, а в последующем с большой задержкой фор¬
мируется неловкая, атактическая походка. К концу пер¬
вого десятилетия жизни состояние ребенка стабилизи¬
руется. В ряде случаев у больных с врожденой
гипоплазией мозжечка может наблюдаться умственная
отсталость, дизартрия, глазодвигательные нарушения, ат¬
рофия зрительных нервов, глухота, пирамидная симпто¬
матика, гидроцефалия и другие симптомы [Harding А.,
1984; Bruyn G., 1991]. Сочетание врожденной атаксии с
умственной отсталостью и другими дополнительными
неврологическими симптомами или аномалиями разви-
ДНК- диагностика наследственных болезней ... 329гия вещества мозга наиболее характерно для аутосомно-
рецессивных форм врожденной гипоплазии мозжечка,
тогда как аутосомно-доминантные и Х-сцепленные ва¬
рианты болезни чаще проявляются сравнительно изо¬
лированной задержкой развития моторики и недостаточ¬
ностью координации. Патоморфологически при боль¬
шинстве форм заболевания отмечается частичная или
полная агенезия червя мозжечка, реже - односторонняя
или тотальная агенезия полушарий мозжечка, гетерото-
пии клеток коры мозжечка, гипоплазия гранулярного
слоя коры, разнообразные сочетанные аномалии (менин-
гомиелоцеле, агенезия мозолистого тела и т.п.)
[Harding А., 1984].Молекулярная генетика наследственных форм
гипоплазии мозжечка изучена недостаточно, что обус¬
ловлено редкостью больших информативных родослов¬
ных, позволяющих проводить анализ генетического сцеп¬
ления и хромосомное картирование мутантного гена. Ген
одной из форм аутосомно-рецессивной младенческой
спиноцеребеллярной атаксии картирован на хромосоме
I0q23-q24.1 [Nikali К. et al., 1995]. Другая аутосомно-
рецессивная форма врожденной непрогрессирующей
атаксии, характеризующаяся гипоплазией гранулярного
слоя коры мозжечка, умственной отсталостью и низко-
рослостью, сцеплена с хромосомой 11 q 14—21
| Megarbane A. et al., 1999]. Сотрудниками нейрогенети-
ческого отделения НИИ неврологии РАМН в 1995-1996
гг. была описана новая форма Х-сцепленной врожден¬
ной гипоплазии мозжечка, выявленная в большой семье
бурятского этнического происхождения: заболевание
п роявлялось непрогрессирующей атаксией, офтальмоп¬
легией, пирамидными симптомами и сохранным интел¬
лектом [Illarioshkin S. etal., 1996]. Ген данного заболева-11 ия картирован нами в хромосомном локусе Хр 11.21 -q24
330Глава 3[Illarioshkin S. et al., 1996], а несколько лет спустя ука¬
занная локализация гена была подтверждена итальянс¬
кими и французскими исследователями в другой семье с
аналогичной формой Х-сцепленной врожденной гипоп¬
лазии мозжечка [Bertini Е. et al., 2000]. Описана также
еще одна форма врожденной спиноцеребеллярной атак¬
сии в сочетании с сидеробластической анемией, насле¬
дующаяся по Х-сцепленному рецессивному типу
[Allikmets R. et al., 1999]; данное заболевание обусловле¬
но повреждением гена АВС7, кодирующего АТФ-связы-
вающий белок внутренней мембраны митохондрий, уча¬
ствующий в обмене железа.Возможности ДНК-диагностики врожденных ги¬
поплазий мозжечка ограничиваются мутационным скри¬
нингом гена АВС7 при сочетании врожденной атаксии с
сидеробластической анемией, а также косвенной ДНК-
диагностикой носительства мутантной хромосомы в се¬
мьях, в которых подтверждено сцепление болезни с
вышеуказанными хромосомными локусами.3.4. Наследственные заболеванияс преимущественным вовлечением
пирамидной системы
(наследственные спастические
параплегии)Дегенеративные заболевания ЦНС с преимуще¬
ственным вовлечением пирамидной системы являются
генетически гетерогенными и могут наследоваться по
аутосомно-доминантному, аутосомно-рецессивному и X-
сцепленному рецессивному типам [Behan W., Maia М.,
1974; Harding А., 1984; Reid Е., 1999]. Их распростра¬
ненность в европейских популяциях составляет около10 на 100 000 [Polo J. et al., 1991]. Ядром клинической
ДНК- диагностика наследственных болезней ... 331картины данных заболеваний является прогрессирующий
нижний спастический парапарез, в некоторых случаях
сочетающийся с рядом дополнительных неврологичес¬
ких симптомов. В большинстве семей имеет место ауто-
сомно-доминантное наследование болезни, соответству¬
ющее оригинальным описаниям A.Strumpell (1880-1904 гг.);
именно для аутосомно-доминантных случаев наслед¬
ственной спастической параплегии правомерно исполь¬
зование широко распространенного термина «болезнь
Штрюмпеля».С клинической точки зрения принято подразде¬
лять наследственную спастическую параплегию на 2
основные группы: а) изолированная («чистая») наслед¬
ственная спастическая параплегия; б) параплегия
«плюс», то есть форма, осложненная развитием призна¬
ков мультисистемного поражения мозга (а иногда и дру¬
гих органов) [Harding А., 1984]. Изолированная форма
спастической параплегии является наиболее частой и в
большинстве случаев наследуется по аутосомно-доми-
нантному типу. Рецессивным типам наследования чаще
соответствуют различные осложненные варианты забо¬
левания.3.4.1. Аутосомно-доминантные спастические
параплегииДля аутосомно-доминантных генетических вари¬
антов наследственной спастической параплегии харак¬
терна широкая вариабельность возраста дебюта клини¬
ческой симптоматики - от первого до седьмого десяти¬
летия жизни. Больные предъявляют жалобы на скован¬
ность и быструю утомляемость ног при ходьбе и беге,
судороги в мышцах ног, после чего постепенно разви-
наются типичные нарушения ходьбы (спастическая по¬
332Глава 3ходка), при многолетнем течении формируются контрак¬
туры и деформации стоп. Характерно преобладание спа-
стичности над парезами. В руках повышение мышечно¬
го тонуса и парезы мышц наблюдаются исключительно
редко и лишь в самой поздней стадии заболевания. При
неврологическом осмотре у больных, помимо симпто¬
мов вовлечения пирамидного тракта (спастичость, рез¬
ко повышенные сухожильные рефлексы, клонусы, сто¬
пные и кистевые патологические знаки) может отмечать¬
ся также легкий интенционный тремор, нистагм, сни¬
жение вибрационной чувствительности, в части случаев
имеют место расстройства мочеиспускания [Harding А.,
1984; Durr A. et al., 1994; Reid Е., 1999]. Появление ука¬
занных дополнительных симптомов (которые обычно
являются негрубыми и выявляются после многолетнего
течения болезни) не противоречит диагнозу «чистой»
спастической параплегии. В отдельных семьях описаны
осложненные формы аутосомно-доминантной спастичес¬
кой параплегии, когда у больных нижний спастический
парапарез сочетается с дегенерацией сетчатки, атрофи¬
ей зрительных нервов, сенсорной невропатией, амиот-
рофиями, экстрапирамидными расстройствами, демен-
цией, эпилептическими припадками, нарушением пиг¬
ментации кожи и др. [Harding А., 1984; Reid Е., 1999].Течение заболевания медленно прогрессирующее,
серьезная инвалидизация наступает через 15-20 лет от
момента появления первых субъективных жалоб. На сек¬
ции выявляется дегенерация пирамидных путей в боко¬
вых столбах спинного мозга (особенно в каудальных от¬
делах), изменения обнаруживаются также в задних стол¬
бах, спиноцеребеллярных и переднем пирамидном трак¬
тах; на поздних стадиях болезни может отмечаться вов¬
лечение пирамидных волокон в стволе мозга и частич¬
ная гибель клеток Беца двигательной зоны коры боль¬
ших полушарий [Behan W., Maia М., 1974].
ДНК- диагностика наследственных болезней ... 333В соответствии с современной номенклатурой,
различные локусы и соответствующие им генетические
варианты наследственных спастических параплегий обо¬
значаются символами SPG1, SPG2 и т.д. (от англ. Spastic
Paraplegia Gene). К настоящему времени известны 8 са¬
мостоятельных генетических вариантов аутосомно-доми¬
нантных спастических параплегий, сцепленных со сле¬
дующими хромосомами: 14ql 1.2-24.3 (SPG3); 2р21-24
(SPG4); 15q 11.1 (SPG6); 8q24 (SPG8); 10q23.3-24.2
(SPG9); 12q 13 (SPG10); 19ql3.1 (SPG12) и 2q24-34
(SPG13) [I-Iazan J. et al., 1993; 1994; Fink J. et al., 1995;
Hedera P et al., 1999; Reid E. et al., 1999; 2000; Seri M. et
al., 1999; Fontaine B. et al., 2000]. В ряде семей локализа¬
ция мутантного гена с известными хромосомными ло-
кусами исключена [Reid Е. et al., 1999], что свидетель¬
ствует о существовании еще одного или нескольких, пока
не установленных генетических вариантов доминантных
параплегий.Анализ клинико-генетических корреляций в се¬
мьях с аутосомно-доминантными спастическими пара¬
плегиями в целом показал отсутствие каких-либо суще¬
ственных фенотипических различий между отдельны¬
ми формами: в абсолютном большинстве семей, незави¬
симо от хромосомного локуса, имеет место клиничес¬
кая картина «чистой» спастической параплегии
| Figlewicz D., Bird Т., 1999]. Специфический осложнен¬
ный фенотип характерен лишь для формы SPG9: у этих
больных нижний спастический парапарез сочетается с
катарактой, гастроэзофагеальным рефлюксом и амиот-
рофиями [Seri М. et al., 1999]. Определенной особенно¬
стью формы SPG3 является более ранний возраст мани¬
фестации симптомов болезни (в среднем в обследован¬
ных семьях заболевание проявлялось в возрасте <10 лет)
| Reid Е., 1999].
334Глава 3Примерно в 40-50% всех обследованных семей с
аутосомно-доминантными спастическими параплегиями
заболевание связано с локусом SPG4 на 2-й хромосоме
[ReidE., 2000]. Таким образом, SPG4 является наиболее
распространенным генетическим вариантом данных забо¬
леваний. Соответствующий ген, получивший название «спа-
стин», был идентифицирован в 1999 году: он состоит из 17
экзонов и кодирует один из белков AAA-семейства (ААА-
белки представляют собой АТФ-азы, обладающие разно¬
образными клеточными функциями) [Hazan. J. et al., 1999].
Предполагается, что белок спастин является типичным
белком-шапероном и имеет отношение к формированию
белковых комплексов, критических для поддержания струк-
турно-функциональной целостности длинных аксонов, со¬
ставляющих кортикоспинальный тракт [Hazan J. et al., 1999].
Мутации в спасшие приводят к повреждению цитоскеле¬
та и гибели наиболее дистальных отделов аксонов цент¬
ральных мотонейронов, что и лежит в основе развития спа¬
стической параплегии. У больных SPG4 выявлено свыше
30 различных точковых мутаций, делеций и вставок, рас¬
пределенных вдоль всей кодирующей части спастина
[Hazan J. et al., 1999; Fonknechten N. et al., 2000]. Обращает
на себя внимание, что до настоящего времени в кодирую¬
щей области данного гена не выявлено ни одного клини¬
чески «немого» нуклеотидного полиморфизма; в связи с
этим высказывается мнение, что любые изменения в нук¬
леотидном составе гена являются патогенетически значи¬
мыми и могут приводить к развитию болезни
[Fonknechten N. et al., 2000]. Рядом авторов показано, что
мутации в локусе SPG4 могут быть связанными с развити¬
ем не только «чистой» спастической параплегии (что на¬
блюдается у большинства больных), но и синдромов, ос¬
ложненных эпилептическими припадками и когнитивны¬
ми нарушениями вплоть до деменции [Heinzlef О. et al.,
1998; Fonknechten N. et al., 2000].
ДНК- диагностика наследственных болезней ... 335Из всех форм аутосомно-доминантных спастичес¬
ких параплегий прямая ДНК-диагностика в настоящее
время возможна лишь для SPG4. Она является весьма
трудоемкой и заключается в тотальном молекулярно-ге¬
нетическом анализе кодирующей области гена спастина
- либо посредством прямого секвенирования всех экзо-
нов гена, либо с предварительным мутационным скри¬
нингом отдельных экзонов с помощью SSCP-анализа и
других стандартных методов скрининга [FonknechtenN.
et al., 2000]. Учитывая высокую частоту формы SPG4,
анализ гена спастина целесообразно проводить во всех
случаях аутосомно-доминантных спастических парапле¬
гий, в которых точное определение генетического локу-
са с помощью анализа сцепления не представляется воз¬
можным. Как указывалось выше, характерной особен¬
ностью доминантных форм наследственной спастичес¬
кой параплегии является весьма длительное субклини-
ческое течение заболевания. Совсем недавно было уста¬
новлено, что почти четвертая часть всех носителей му¬
тации в гене спастина являются на момент обследова¬
ния асимптомными или, во всяком случае, не предъяв¬
ляют никаких субъективных жалоб [Fonknechten N. et
al., 2000]. Это наблюдение подчеркивает значение ДНК-
диагностики в проведении медико-генетического кон¬
сультирования в семьях, отягощенных аутосомно-доми-
нантной наследственной спастической параплегией.В случае сочетания спастичности с катарактой,
гастроэзофагеальным рефлюксом и амиотрофиями с
весьма высокой вероятностью можно диагностировать
в обследуемой семье форму SPG9, что позволяет прово¬
дить у лиц из группы риска косвенную ДНК-диагности-
ку болезни с помощью исследования маркеров из хро¬
мосомной области 10q23.3-24.2 [Seri М. etal., 1999]. Для
остальных форм аутосомно-доминантных спастических
параплегий проведение косвенной ДНК-диагностики у
336Глава 3лиц из группы риска возможно лишь при условии уста¬
новления в обследуемой семье генетического сцепления
с одним из известных вышеуказанных хромосомных ло-
кусов (SPG3, 6, 8, 10, 12 и 13). Идентификация данных
генов позволит в будущем провести детальные клини¬
ко-генетические сопоставления, изучить сравнительную
частоту основных генетических вариантов доминантных
спастических параплегий в различных популяциях и на
этой основе разработать целостную систему прямой
ДНК-диагностики данных заболеваний. Интересно от¬
метить, что в ряде семей с SPG3 и SPG8 наблюдался
феномен антиципации, который весьма характерен для
аутосомно-доминантных заболеваний с «динамически¬
ми» мутациями (экспансией тринуклеотидных повто¬
ров)- таких как миотоническая дистрофия, хорея Ген-
тингтона, доминантные атаксии. Более того, K. Benson с
соавторами (1998) при использовании в семьях с доми-
нантыми параплегиями специального метода RED
(.Repeat Expansion Détection) показали, что у некоторых
из этих больных геном содержит удлиненные CAG/CTG-
тракты, которые, возможно, имеют патогенетическое зна¬
чение. Таким образом, нельзя исключить, что часть мо¬
лекулярных форм аутосомно-доминантных спастических
параплегий, гены которых пока не установлены, может
со временем быть отнесена к растущей группе нейроде-
генеративных заболеваний, обусловленных экспансией
тринуклеотидных повторов.3.4.2. Рецессивные спастические параплегииНа долю рецессивных (аутосомных и Х-сцеплен-
ных) форм приходится около 20-30% всех семей с на¬
следственными спастическими параплегиями, причем в
данной группе большинство составляют аутосомно-ре-
ДНК- диагностика наследственных болезней ... 337цессивные случаи, тогда как семьи с Х-сцепленным ре-
1 (.ессивным наследованием представляют собой исклю¬
чительную редкость [Harding А., 1984]. Особенностью
заболеваний из группы рецессивных спастических па¬
раплегий является весьма частое развитие разнобразных
осложненных форм (параплегия «плюс»), а также не¬
сколько более ранний (по сравнению с доминантными
формами) средний возраст манифестации первых симп¬
томов болезни. Морфологические изменения в спинном
и головном мозге в целом аналогичны таковым у боль¬
ных аутосомно-доминантными спастическими парапле¬
гиями, однако при рецессивных формах существенно
чаще наблюдаются более распространенные изменения,
включающие дегенерацию мозжечка, спинномозговых
I англиев и периферических нервов, атрофию дисков зри¬
тельных нервов, различные врожденные дизгенезии и
т.д. [Harding А., 1984; Reid Е., 1999].На сегодняшний день выделяют 2 самостоятель-
иых генетических варианта Х-сцепленных рецессивных
параплегий и 5 генетических вариантов аутосомно-ре-
нсссивных параплегий.Осложненная форма Х-сцепленной спастической
параплегии, характеризующаяся сочетанием спастично-
сти с умственной отсталостью и отсутствием т. extensor
pollicis longus, сцеплена с хромосомным участком Xq28
(локус SPG1) и обусловлена мутациями в гене молеку-
пы адгезии нейронов LI (Ll-CAM) [Jouet М. et al., 1994].
Iбелковый продукт Ll-CAM - это гликопротеин клеточ¬
ной поверхности. Он является представителем суперсе¬
мейства молекул адгезии класса иммуноглобулина G и
ответственен за созревание, миграцию нейронов и фор¬
мирование их аксональных контактов с другими нейро¬
нами, клетками глии и мышцами в процессе онтогене-
| пческого развития либо после травмы [Sonderegger Р.,
338Глава 3Rathjen F., 1992; Jouet M. et al., 1994]. Такое многообра¬
зие функций свидетельствует о важной роли L1-CAM в
нейрогенезе и предполагает возможность множествен¬
ных сочетанных аномалий развития при повреждении
данного гена. Не случайно, помимо указанной формы
осложненной спастической параплегии, мутации в гене
L1 -САМ могут вызывать также развитие Х-сцепленной
гидроцефалии, агенезии мозолистого тела и ряда других
врожденных пороков развития ЦНС [Jouet М. et al., 1994].Вторая генетическая форма Х-сцепленной рецес¬
сивной спастической параплегии сцеплена с хромосом¬
ным участком Xq22 (локус SPG2) и обусловлена мута¬
циями в гене протеолипидного белка (PLP) [Saugier-
Veber P. et al., 1994]. Интересно отметить, что различ¬
ные точковые мутации в данном гене приводят к мани¬
фестации как «чистой» спастической параплегии, так и
осложненной ее формы (в сочетании с умственной от¬
сталостью) [Cambi R et al., 1996; Reid E., 1999]. Более
того, мутации в гене PLP (чаще всего дупликации) явля¬
ются молекулярной основой еще одного Х-сцепленного
синдрома - болезни Пелицеуса-Мерцбахера, которая от¬
носится к группе лейкодистрофий и характеризуется
врожденной гипотонией, нистагмом, задержкой психи¬
ческого развития, прогрессирующими пирамидными,
мозжечковыми и дистоническими нарушениями [Saugier-
Veber P. et al., 1994]. Продукт гена PLP — протеолипид-
ный белок - играет ведущую роль в созревании олиго-
дендроцитов и компактной укладке миелиновых слоев
[Griffiths I. et al., 1995]. Предполагается, что дупликации
и другие мутации, влияющие на функцию данного белка
в отношении созревания олигодендроцитов, проявляются
тяжелым фенотипом болезни Пелицеуса-Мерцбахера,
тогда как мутации PLP, затрагивающие лишь процессы
компактизации миелина, приводят к манифестации бо¬
ДНК- диагностика наследственных болезней ... 339лее легких аллельных заболеваний - «чистой» и ослож¬
ненной форм спастической параплегии [Reid Е., 1999].Локусы аутосомно-рецессивных форм наслед¬
ственной спастической параплегии картированы на хро¬
мосомах 8ql2—13 (SPG5), 16q24.3 (SPG7), 15ql3—15
(SPG11), 3q27-28 (SPG14) и 14q22-24 (SPG15)
|HentatiA. et al., 1994 (6); De Michele G. et al., 1998;
Martinez Murillo F. et al., 1999; Vazza G. et al., 2000;
Hughes C. et al., 2001]. Ген формы SPG7, получивший
название «параплегии», был недавно идентифицирован:
он кодирует синтез АТФ-зависимой металлопротеазы,
локализованной на внутренней мембране митохондрий
и играющей роль в процессах протеолиза и организа¬
ции/деградации белковых комплексов [Casari G. et al.,1998]. Как и спастин (продукт гена доминантной формы
SPG4), белок параплегии относится к ААА-семейству
клеточных АТФ-аз [Pearce D., 1999]. В случае мутаций вI траплегине у больных с 8РС7-формой заболевания вы¬
является системный дефект окислительного фосфори-
лирования, что может быть связано с нарушением сбор¬
ки субъединиц комплексов дыхательной цепи митохон¬
дрий и приводить к дегенерации длинных аксонов цент¬
ральных мотонейронов [Casari G. et al., 1998; Pearce D.,1999]. Следовательно, 8Р07-форма аутосомно-рецессив-
ной спастической параплегии является ядерно-кодируе-
мым митохондриальным заболеванием. Для митохонд¬
риальных заболеваний характерен полиморфизм клини¬
ческих проявлений, и SPG7 подтверждает данное пра¬
вило, поскольку различные мутации параплегина могут
манифестировать как в виде «чистых» форм спастичес¬
кой параплегии, так и в виде осложненных ее вариантов
(в сочетании с атрофией зрительных нервов, атрофией
м >ры мозжечка или больших полушарий мозга) [Reid Е., 1999].
Такой же клинический полиморфизм характерен и для
340Глава 3формы SPG11, ген которой локализован на 15-й хромо¬
соме, но пока не идентифицирован: часть больных SPG11
имеют изолированную спастичность в ногах, тогда как в
других случаях SPG11 спастический парапарез сочета¬
ется с агенезией мозолистого тела, умственной отстало¬
стью и дизартрией [Martinez Murillo F. et al., 1999]. Для
SPG5 характерна изолированная спастическая парапле¬
гия с ранним началом болезни [Flentati A. et al., 1994 (б)].
Форма SPG14 описана лишь в одной семье, в которой у
всех больных спастичность манифестировала в сравни¬
тельно позднем возрасте (около 30 лет) и сочеталась с
дистальной моторной невропатией и умственной отста¬
лостью [Vazza G. et al., 2000]. Для SPG15 (синдром Кьел-
лина) характерно сочетание спастичности с пигментной
дегенерацией сетчатки, дистальными амиотрофиями
конечностей, дизартрией и интеллектуальным снижени¬
ем [Hughes С. et al., 2001].Таким образом, анализ клинико-генетических
корреляций в семьях с рецессивными и (в меньшей сте¬
пени) доминантными спастическими параплегиями по¬
казал определенную условность общепринятого подраз¬
деления данных заболеваний на «чистые» и осложнен¬
ные формы [Figlewicz D., Bird Т., 1999; Reid Е., 1999].
Более правильным было бы говорить о достаточно об¬
ширном спектре спастических синдромов, клиническая
картина которых может дивергировать или, напротив,
перекрываться как для аллельных, так и для разиоло-
кусных заболеваний. В обобщенном виде современная
клинико-генетическая характеристика наследственных
спастических параплегий представлена в таблице 7.Для полноты картины следует упомянуть также
еще два аутосомно-рецессивных заболевания с установ¬
ленными хромосомными локусами, близких по своей
клинической картине к группе спастических параплегий:
ДНК- диагностика наследственных болезней ... 341а) Синдром Шегрена-Ларссона - своеобразная
форма осложненной спастической параплегии, характе¬
ризующаяся непрогрессирующей спастичностью в соче¬
тании с тяжелой умственной отсталостью и врожден¬
ным ихтиозом. Заболевание обусловлено мутациями гена
жирной альдегид-дегидрогеназы (ЕАЫЭН), расположен¬
ного на хромосоме 17р11.2 [Эе Ьаигепг1 V. е! а1., 1996].Таблица 7Клинико-генетическая характеристика
наследственных спастических параплегийА ут осомно-дом и н ан т н ы е с п а с т и чески епараплегии:2р21 -24:($ Р 6 4 - генсп а сти н );2 я 24-34
8 д 2 4(Б Р О 1 3 );
(Б Р в 8);г|1 0 я 2 3 .3 -2 4 .2 (Э Р в 9); Щ1 2ц 1 31 4 я 1 1 .2-2 4 .
1 5я 1 1 .1
1 9 д 1 3.1(Б Р в 1 0);
3 (Э Р в 3);
(в Р в 6);
(в Р в 1 2)Л у т осомно-оеиессивны е с п ас т и ч е с к и епараплегии:8 ч 1 2 - 1 3(Э Р в 5);1 5я 1 3-1 5(в Р С 1 1');16 Я 2 4.3(Б Р в 7 - ге нпараплегии);;И 3 Я 2 7 -2 8
1 1 4я22-24(Э Р в 1 4) ШЯ
(вРв 1 5)Х-сиепленны ерецессивные спастическиепараплегии:X я 2 2(8 Р в 2 - генР 1-Р );|Хя2 8(Э Р в 1 - ген1. 1 -С А М ) Ш111Примечание: черным фоном выделены осложненные фор¬
мы (параплегия «плюс»), белым фоном - «чистые» формы, серым
фоном - формы, которые могут манифестировать как «чистым»,
I нк и осложненным фенотипомб) Спастическая атаксия Шарлевуа-Саженё, рас-
гматриваемая как «промежуточная» форма между на¬
342Глава 3следственными атаксиями и наследственными спасти¬
ческими параплегиями. Ген заболевания картирован в
локусе 13ql 1 [Richter A. et al., 1999].в) Описаны семейные аутосомно-рецессивные
случаи спастической симметричной формы детского це¬
ребрального паралича (ДЦП), и соответствующий гене¬
тический локус был картирован в области 2q24-25
[McHale D. et al., 1999]. Установленный интервал на хро¬
мосоме 2q частично перекрывается с интервалом, в ко¬
тором ранее был картирован ген аутосомно-доминант-
ной формы спастической параплегии SPG13 (см. выше).
Таким образом, несмотря на различия в клинике и типе
наследования спастической формы ДЦП и SPG13-фор¬
мы спастической параплегии Штрюмпеля, нельзя исклю¬
чить, что эти два спастических синдрома являются ал¬
лельными заболеваниями, обусловленными разными
мутациями одного гена [Fontaine В. et al., 2000]. Этот
вопрос окончательно может быть разрешен только пос¬
ле идентификации молекулярной основы обоих заболе¬
ваний и клонирования соответствующего гена (генов) на
хромосоме 2q.Прямая ДНК-диагностика форм спастической
параплегии SPG1, SPG2 и SPG7 проводится путем стан¬
дартных методов мутационного скрининга генов L1-
САМ, PLP и параплегина (чаще всего с помощью пред¬
варительного SSCP-анализа отдельных экзонов и пос¬
ледующего секвенирования мутантных образцов). Клю¬
чевым условием для правильного выбора гена, подле¬
жащего исследованию, является анализ клинической
картины и типа наследования (см. таблицу 6). Так, ма¬
нифестация болезни исключительно у лиц мужского пола
и отсутствие в родословной прямой передачи болезни
от отца сыну свидетельствует об Х-сцепленном рецес¬
сивном наследовании. В такой ситуации возможными
ДНК- диагностика наследственных болезней ... 343генетическими вариантами болезни являются SPG1 и
SPG2, причем в пользу первой формы (ген L1-CAM)
может свидетельствовать сочетание спастической пара¬
плегии с задержкой умственного развития и аномалией
развития 1-го пальца рук. Важным маркером, позволя¬
ющим заподозрить аутосомно-рецессивную форму SPG7
(ген параплегии), является дефект митохондриального
окислительного фосфорилирования, который диагнос¬
тируется на основании выявления в биоптате мышц ха¬
рактерных гистохимических и структурных нарушений
(наличие «рваных красных волокон», негативная реак¬
ция волокон при окраске на цитохром с-оксидазу, ульт-
раструктурные изменения митоходрий) [Casari G. et al.,1998]. Молекулярная диагностика синдрома Шегрена-
Ларссона обычно проводится на основании измерения
активности жирной альдегид-дегидрогеназы в клетках
крови и фибробластах и окончательно подтверждается
при выялении мутаций в гене FALDH [Rizzo W. et al.,1999]. Для остальных форм аутосомно-рецессивной спа¬
стической параплегии возможно проведение косвенной
ДНК-диагностики у лиц из группы риска в небольшом
числе информативных семей, для которых подтвержде¬
но сцепление с одним из известных хромосомных локу-
сов (SPG5, SPG11, SPG14 и т.д.).3.5. Наследственные заболеванияс преимущественным вовлечением
когнитивной сферы
(наследственные деменции)3.5.1. Семейные формы болезни АльцгеймераБолезнь Альцгеймера представляет собой одно из
наиболее частых нейродегенеративных заболеваний че¬
344Глава 3ловека и ведущую причину деменции в современном
обществе [Rocca W. et al., 1991; Schellenberg G., 1995].
Ее распространенность у лиц старше 65 лет составляет
около 3% и неуклонно повышается в более пожилых
возрастных группах [KatzmanR, 1976]. Заболевание ха¬
рактеризуется прогрессирующим ослабоумливающим
процессом, центральное место в развитии которого за¬
нимают нарушения памяти, являющиеся наиболее ран¬
ним и типичным проявлением болезни. Спустя несколь¬
ко лет от начала болезни закономерно присоединяются
расстройства праксиса, речи, счета, письма, ориентиров¬
ки и узнавания, у больных могут отмечаться острые пси¬
хотические эпизоды, эпилептические припадки, разно¬
образные экстрапирамидные симптомы. В конечном сче¬
те развивается глубокая тотальная деменция с распадом
личности, тотальная афазия, общее физическое истоще¬
ние. Средняя продолжительность заболевания составляет
около 10 лет. На секции выявляется диффузная атрофия
мозга, с большей выраженостью изменений в теменных
и затылочных областях. При микроскопическом иссле¬
довании обнаруживаются два основных гистологичес¬
ких признака болезни Альцгеймера: 1) амилоидные (се¬
нильные) бляшки в паренхиме мозга, главным компо¬
нентом которых являются фибриллярные агрегаты гид¬
рофобного пептида Р-амилоида (А|3), состоящего из 39-
43 аминокислот; 2) нейрофибриллярные клубки, пред¬
ставляющие собой спаянные попарно скрученные фи-
ламенты измененных нейронов, легко выявляемые при
импрегнации серебром [McKee A. et al., 1991]. Харак¬
терно также выпадение нейронов, глиоз, наличие еди¬
ничных телец Леви. Отмеченные изменения наиболее
выражены в коре больших полушарий и гиппокампе.Большинство случаев болезни Альцгеймера име¬
ют мультифакториальную природу и являются спора¬
дическими. В то же время многочисленные популяци
ДНК- диагностика наследственных болезней ... 345онные исследования показали, что 25-40% случаев бо¬
лезни Альцгеймера могут быть семейными, т.е. в семье
пробанда имеется, как минимум, еще один больной с
■ >тим заболеванием [Van Broeckhoven С., 1995]. Важная
роль генетических факторов в развитии болезни Альц¬
геймера подтверждается высокой конкордантностыо по
болезни среди монозиготных близнецов [Breitner J. et al.,1993]. Анализ большого числа семей с болезнью Альц-I еймера позволил установить бимодальное распределе-
пие значений возраста дебюта симптомов, причем ус¬
ловной границей между ранними и поздними семейны¬
ми случаями болезни принято считать возраст 58 лет [Van
Broeckhoven С., 1995]. В ранних случаях семейной бо¬
лезни Альцгеймера заболевание обычно наследуется как
аутосомно-доминантный признак, связанный с повреж¬
дением одного основного гена. Поздние же семейные
случаи болезни Альцгеймера являются гетерогенными;
чаще всего в этих случаях имеется полигенно обуслов¬
ленная предрасположенность к болезни Альцгеймера,II ри которой накопление повторных случаев болезни сре¬
ди родственников связано с действием комплекса гене¬
тических и средовых факторов, общих для членов дан¬
ной семьи. По некоторым оценкам, наследственные мо-
иогенные формы составляют в целом около 5-10% слу¬
чаев болезни Альцгеймера [Van Broeckhoven С., 1995;
Whitehouse P., 1997], и именно им посвящен данный раз-
чел монографии. Механизмы генетической предраспо¬
ложенности и соответствующие методы ДНК-анализа
мри ненаследственных формах болезни Альцгеймера
подробно разбираются в главе 4, посвященной мульти-
(факториальным заболеваниям нервной системы.На сегодняшний день идентифицированы 3 гена,
мутации которых приводят к развитию наследственных
(аутосомно-доминантных) форм болезни Альцгеймера с
ранним началом симптомов. Один из них - ген белка-
346Глава 3предшественника [3-амилоида, локализованный на хро¬
мосоме 21q21 и обозначаемый аббревиатурой АРР (от
англ. Amyloid Precursor Protein) [Kang J. et al., 1987;
St George-Hyslop PH. et al., 1987; Goate A. et al, 1991].
Ген состоит из 19 экзонов, причем аминокислотная пос¬
ледовательность [3-амилоида (Ар) кодируется частью эк¬
зонов 16 и 17; данная аминокислотная последователь¬
ность расположена в карбоксильной части белка АРР
[Kang J. et al., 1987; Yoshikai S. et al., 1990]. В норме бе¬
лок АРР подвергается протеолизу под воздействием а-,
р- и у-секретаз; два последних протеолитических пути
приводят к высвобождению интактных молекул Ар, что
само по себе не сопровождается развитием болезни
[Schellenberg G., 1995; Van Broeckhoven С., 1995]. Все
восемь известных патогенных точковых мутаций АРР
расположены в 16-м и 17-м экзонах гена и ведут к нару¬
шению Р- и у-секретазного процессинга белкового про¬
дукта АРР, результатом чего является гиперсекреция
пептида Ар или преимущественная секреция более длин¬
ных, склонных к быстрой фибриллярной агрегации форм
Ар [Cai X. et al., 1993; Jarrett J. et al., 1993]. В обоих
случаях высвобождаемый пептид Ар приобретает ами¬
лоидогенные свойства - процесс, лежащий в основе фор¬
мирования сенильных бляшек в паренхиме мозга. Инте¬
ресно отметить, что мутация Glu693Gln в 17-м экзоне
гена АРР приводит к развитию другого заболевания с
близким патогенезом - так называемого наследственно¬
го церебрального кровоизлияния голландского типа,
обусловленного изменением стенок церебральных сосу¬
дов вследствие отложения в них амилоидных депозитов
[Levy Е. et al., 1990]. Более того, оба АРР-ассоциирован-
ных заболевания (деменция альцгеймеровского типа и
церебральное кровоизлияние) могут наблюдаться у раз¬
личных родственников в одной и той же родословной -
как это описано в семье с мутацией Gly692Ala в 17-м
ДНК- диагностика наследственных болезней ... 347экзоне гена АРР [Hendriks L. et al., 1992]. В целом, мута¬
ции в гене АРР представляют собой большую редкость:
во всем мире они выявлены лишь в 20 семьях и, по при¬
близительным оценкам, обусловливают не более 5% всех
случаев семейной болезни Альцгеймера с ранним нача¬
лом симптомов [Van Broeckhoven C., 1995; Blacker D.,
Tanzi R., 1998].Два других гена, обусловливающие основную
часть случаев ранне-семейной болезни Альцгеймера и
расположенные на хромосомах 14q24.3 и lq31-42, были
клонированы в 1995 году [Sherrington R. et al., 1995; Levy-
Lahad E. et al., 1995; Rogaev E. et al., 1995]. Эти гены
являются высокогомологичными и кодируют родствен¬
ные мембранные белки - пресенилины (соответствен¬
но, пресенилин-1 и пресенилин-2). В мозге пресенили¬
ны экспрессируются преимущественно в нейронах и
локализованы в эндоплазматическом ретикулуме тел
нейронов и их дендритов [Cook D. et al., 1996; Kovacs D.
et al., 1996]. Предполагается, что одна из функций пре-
сенилинов может быть связана с регуляцией внутрикле¬
точного транспорта мембранных белков, в том числе
белка-предшественника ß-амилоида (АРР) [Hutton М.,
Hardy J., 1997; Sisodia S. et al., 1999]. Мутации в генах
пресенилинов сопровождаются гиперпродукцией амило¬
идогенных форм пептида Aß, формирующих сенильные
бляшки [Scheuner D. et al., 1996; Citron M. et al., 1997].
Этот феномен обусловлен, наиболее вероятно, активи¬
зацией у-секретазного протеолиза АРР в условиях «за¬
держки» данного белка в эндоплазматическом ретику¬
луме [Hutton М., Hardy J., 1997]. Другой возможный
механизм патогенного эффекта мутантных пресенили-
пов может заключаться в индуцировании апоптоза (про¬
граммируемой клеточной гибели) вследствие нарушен¬
ной регуляции кальциевого гомеостаза в эндоплазмати-
' iccKOM ретикулуме и активизации свободнорадикальных
348Глава 3реакций [Mattson М., 1997; Blacker D., Tanzi R., 1998]. В
этом случае выявляемое нарушение процессинга АРР в
клетках, экспрессирующих мутантные пресенилины,
носит вторичный характер по отношению к реализуемо¬
му «апоптотическому каскаду». В целом, чуть более по¬
ловины всех семейных случаев болезни Альцгеймера с
ранним началом обусловлены мутациями в генах пресе-
нилинов; при этом основная часть случаев связана с пре-
сенилином-1 (около 100 описанных семей, >50 точко-
вых миссенс-мутаций), тогда как повреждения гена пре-
сенилина-2 встречаются весьма редко (лишь 3 описан¬
ных мутации) [Blacker D., Tanzi R., 1998; Campion D. et
al., 1999]. Следует подчеркнуть, что 70% всех извест¬
ных мутаций в генах пресенилинов являются уникаль¬
ными (т.е. каждая из них была выявлена лишь в какой-
то одной семье).Большинство мутаций в генах АРР и пресенили¬
нов характеризуются полной пенетрантностью к концу
6-го десятилетия жизни и неизбежно приводят к мани¬
фестации болезни при условии достижения носителем
мутации соответствующего возраста. Анализ клинико¬
генетических корреляций показал отсутствие каких-либо
существенных различий между фенотипами отдельных
молекулярных форм болезни Альцгеймера, за исключе¬
нием возрастных рамок появления первых симптомов
болезни. При повреждении гена АРР заболевание мани¬
фестирует в возрасте 39-67 лет (чаще всего от 40 до 50
лет), несколько более позднее начало болезни наблюда¬
ется у больных с мутациями в гене пресенилина-2 (в
среднем от 50 до 65 лет), тогда как в случае мутаций в
гене пресенилина-1 заболевание носит наиболее агрес¬
сивный и ранний характер (начало болезни от 24 до 56
лет) [Blacker D., Tanzi R., 1998; Campion D. et al., 1999;
Lovestone S., 1999]. Некоторые мутации в гене пресени¬
лина-1 могут в единичных случаях вызывать развитие
ДНК- диагностика наследственных болезней ... 349т ипичного фенотипа болезни Альцгеймера, характери¬
зующегося сочетанием ранней деменции с нижним спа¬
стическим парапарезом [Campion D. et al., 1999].В значительном числе семей с ранней болезнью
Лльцгеймера мутации в генах АРР и пресенилинов были
исключены [Rogaev Е. et al., 1995], что свидетельствует0 дальнейшей генетической гетерогенности ранней фор-
мы заболевания.Прямая ДНК-диагностика болезни Альцгеймера
представляет собой непростую задачу, что связано с ге¬
нетической гетерогенностью, сравнительно большими
размерами изучаемых генов и отсутствием в них мажор-
ных мутаций. Опыт такой диагностики в мире имеется
п ишь в сравнительно небольшом числе хорошо оснащен¬
ных лабораторий, специализирующихся на молекуляр¬
но-генетическом анализе данного заболевания. Прини¬
мая во внимание, что аутосомно-доминантные формы
болезни Альцгеймера характеризуются чаще всего ран¬
ним началом заболевания, с практической точки зрения
мутационный скрининг целесообразно ограничивать
семьями, в которых хотя бы у части родственников сим¬
птомы болезни манифестировали до 60 лет [Campion D.
et al., 1999; Lovestone S., 1999]. Правильному выбору1 сна, подлежащего исследованию, может способствовать
предварительное установление сцепления болезни с од¬
ним из йзвестных локусов на хромосомах 1 q, 14q или
.11 q. Если такой анализ сцепления невозможен ввиду
небольшого размера обследуемых семей, мутационный
анализ генов проводится в следующей последователь¬
ности: пресенилин-1, АРР, пресенилин-2 (в соответствии
с частотой встречаемости мутаций в данных генах)
|( ampion D. et al., 1999]. Поиск мутаций в пресенили-
пах проводится как на геномной ДНК (SSCP-анализ +
прямое секвенирование отдельных экзонов), так и на
к ДНК из лимфоцитов крови, которая амплифицируется
350Глава 3в виде полного набора перекрывающихся фрагментов и
подвергается тотальному секвенированию [Alzheimer’s
Disease Collaborative Group, 1995; CampionD. et al., 1999].
Для ДНК-диагностики мутаций в гене АРР обычно из¬
бирательно секвенируются 16-й и 17-й экзоны гена, ко¬
дирующие структуру основного патогеннного продукта
белка АРР - пептида Aß. При исключении мутаций в
исследованных областях всех указанных генов некото¬
рые авторы рекомендуют также секвенировать экзоны7, 8 и 18 гена АРР, соответствуюхцие функционально
значимым участкам АРР-белка [Campion D. et al., 1999].
По данным разных авторов, изучавших различные по¬
пуляции и выборки больных с ранними аутосомно-до-
минантными случаями болезни Альцгеймера, общая ча¬
стота выявления мутаций в генах АРР и пресенилинов
варьирует от 18% до 100% [Hutton М., Hardy J., 1997;
Cruts М. et al., 1998; Campion D. et al., 1999]. Поскольку
даже в семьях с подтвержденным сцеплением секвени-
рование может не выявлять нарушений нуклеотидного
состава кодирующей части гена (это возможно при ло¬
кализации мутаций в интронах, промоторной области и
др.), в таких семьях доступным методом молекулярной
диагностики может служить косвенная ДНК-диагностика
с использованием маркеров мутантного хромосомного
локуса.3.5.2. Прионные болезниПрионные болезни (трансмиссивные спонгиофор-
мные энцефалопатии) представляют собой редкие фа¬
тальные нейродегенеративные заболевания, поражающие
приблизительно 1 человека на миллион населения в год
[Collinge J., 1997]. Значительное внимание к данной
проблеме в последнее десятилетие обусловлено двумя
ДНК- диагностика наследственных болезней ... 351обстоятельствами: во-первых, уникальной биологией
прионных болезней, вызываемых особым трансмиссив¬
ным агентом - «инфекционным» белком прионом; во-
вторых, тем обстоятельством, что недавно описанная
эпидемия спонгиоформной энцефалопатии крупного
рогатого скота (так называемое «бешенство коров») мо¬
жет нести серьезную угрозу для человеческой популя-I щи при употреблении в пищу тканей зараженных жи¬
вотных [Зуев В.А. и др., 1999; Prusiner S., Hsiao К., 1994;
Collinge J., Rossor М., 1996; Collinge J., 1997].Описан ряд форм прионных болезней животных
(коровы, овцы, козы), а также 4 прионных болезни че¬
ловека - болезнь Крейтцфельдта-Якоба, синдром Гер-
стманна-Штреусслера (Шейнкера), фатальная семейная
ннсомния и куру [Зуев В.А. и др., 1999; Prusiner S. et al.,
1993; Prusiner S., Hsiao K., 1994]. Свыше 80-85% всех
случаев прионных болезней составляет спорадическая
t юлезнь Крейтцфельдта-Якоба. Небольшая часть случаев
болезни Крейтцфельдта-Якоба, а также синдром Герст-
манна-Штреусслера и фатальная семейная инсомнияII редставляют собой моногенные наследственные забо¬
левания, передающиеся по аутосомно-доминантному
типу. Известен также ятрогенный вариант болезни Крей-
I цфельдта-Якоба, который, как и куру, манифестирует
в виде инфекций. Инфекционность ятрогенного вариан¬
та болезни Крейтцфельдта-Якоба обусловлена попада¬
нием в организм или непосредственно в мозг прионного
белка от инфицированного человека (это может проис¬
ходить, например, при введении инфицированного эк¬
стракта трупного гипофиза с целью лечения эндокрин¬
ных заболеваний, при трансплантации инфицированнойI вердой мозговой оболочки, роговицы и т.д.). Инфекци-
он ность куру - заболевания у аборигенов одного из пле-
мсн восточных островов Папуа Новой Гвинеи - связана
352Глава 3с обычаем каннибализма и употребления в пищу мозга
умерших соплеменников.Клиническая картина болезни Крейтцфельдта-
Якоба (основного представителя прионных болезней) ха¬
рактеризуется подостро развивающейся и быстро про¬
грессирующей деменцией в сочетании с миоклониями и
другими экстрапирамидными проявлениями, атаксией,
пирамидными симптомами, супрануклеарным парали¬
чом, эпилептическими припадками, слепотой [Зуев В.А.
и др., 1999; Ргштег Б. е1 а1., 1993]. На ЭЭГ на фоне гру¬
бых общемозговых изменений выявляются типичные пе¬
риодические комплексы типа острая волна-медленная
волна, следующие с частотой 1-2 в секунду; при КТ и
МРТ-исследовании обнаруживаются нарастающие атро¬
фические изменения головного мозга. Заболевание обыч¬
но начинается в возрасте 45-75 лет, причем пик прихо¬
дится на возраст от 60 до 65 лет. Около 70% больных
погибают в течение 6 месяцев от начала болезни, реже
заболевание может длиться на протяжении 2 и более лет.
Описанный недавно новый вариант болезни Крейтцфель-
дта-Якоба, связанный, как предполагается, с употреб¬
лением в пищу инфицированной говядины (следствие
эпидемии спонгиоформной энцефалопатии коров), от¬
личается необычно ранним началом болезни - на тре¬
тьем и даже втором десятилетии жизни [СоШ^е 1,
КоэзогМ., 1996]. Фенотипические проявления других
форм прионных болезней имеют много общего с пред¬
ставленной клиникой болезни Крейтцфельдта-Якоба.
При синдроме Герстманна-Штреусслера наиболее ран¬
ним и доминирующим проявлением болезни является
атаксия, к которой могут присоединяться когнитивые
нарушения, экстрапирамидные симптомы, параличи взо¬
ра и другие симптомы; продолжительность болезни со¬
ставляет от 2 до 10 лет. Для фатальной семейной инсом-
ДНК- диагностика наследственных болезней ... 35311 ии характерны прогрессирующие нарушения сна и цир¬
кадных ритмов, гипертермия, дезориентированность,
галлюцинации, разнообразные экстрапирамидные и пи¬
рамидные симптомы, атаксия. Клиника куру сходна с
проявлениями синдрома Герстманна-Штреусслера (моз¬
жечковая атаксия в сочетании с деменцией, тремором,
миоклониями); в связи с ликвидацией каннибализма дан-
I юе заболевание в настоящее время практически не встре¬
чается.Классическая патологоанатомическая триада, ха¬
рактерная для прионных болезней, включает спонгио-
формную вакуолизацию серого вещества головного моз¬
га, астроцитарный глиоз и утрату нейронов; в ряде слу¬
чаев в мозге обнаруживаются также компактные или
диффузные (мультицентрические) амилоидные бляшки,
состоящие главным образом из протеазо-резистентной
патологической изоформы прионного белка (последний
морфо-гистохимический признак особенно характерен
для синдрома Герстманна-Штреусслера) [Beck Е.,
Daniel Р., 1987; Prusiner S. et al., 1993].Центральным патогенетическим звеном в биоло¬
гии прионных болезней являются конформационные
изменения собственного белка клетки - прионного бел¬
ка РгР (от англ. Prion Protein). Заболевание развивается
it результате трансформации (на посттрансляционной
с тадии) нормального клеточного прионного белка (РгРс)
к его патологическую изоформу, получившую обозначе¬
ние PrPSc (от англ. Scrapie - названия одной из основ¬
ных разновидностей прионных болезней животных)
| Prusiner S., 1982; Collinge J., 1997; Wadsworth J. et al.,
1999]. В основе пространственной организации клеточ¬
ного белка РгРс лежат а-спиральные пептидные моно¬
меры, которые легко расщепляются клеточными проте¬
идами, тогда как вторичная структура PrPSc представле¬
354Глава 3на в основном р-тяжами, организованными в крупные
нерастворимые фибриллярные агрегаты, устойчивые к
действию протеаз [Wadsworth J. et al., 1999]! Патологи¬
ческая изоформа PrPSc играет роль матрицы и взаимо¬
действует с окружающими нормальными молекулами
РгРс, инициируя спонтанную конверсию последних в
prpso j-j результате этого в клетке запускается «цепная»
реакция саморепликации молекул PrPSc, приводящая к
гибели нейронов, высвобождению PrPSc во внеклеточ¬
ное пространство и формированию амилоидных бляшек
[Prusiner S. et al., 1990; Collinge J., 1997; Wadsworth J. et
al., 1999]. Таким образом, весь экспотенциальный про¬
цесс последовательных конформационных изменений
РгРс инициируется появлением в клетке хотя бы одной
«агрессивной» молекулы PrPSc. По современным пред¬
ставлениям, это может происходить тремя путями
[Collinge J., 1997; Wadsworth J. et al., 1999]. Во-первых,
конверсия PrPc в патологическую изоформу PrPSc мо¬
жет провоцироваться изменением первичной аминокис¬
лотной структуры пептидной молекулы в результате на¬
следования мутации в гене, кодирующем синтез прион-
ного белка (см. далее). Такой механизм лежит в основе
наследственных форм прионных болезней. Во-вторых,
молекулы PrPSc могут попадать в клетку в результате
прямой инокуляции инфицированного биологического
материала (через кровь, трансплантацию твердой моз¬
говой оболочки и роговицы, с пищей и т.д.) - механизм,
лежащий в основе трансмиссивных (в том числе ятро-
генных) форм прионных болезней. Наконец, при наибо¬
лее распространенных спорадических формах прионных
болезней спонтанная конверсия РгРс в PrPSc, как пред¬
полагают, может происходить в результате соматической
мутации гена прионного белка в клетке-мишени либо в
результате редких стохастических событий с той или
иной молекулой РгРс, провоцируемых определенными
ДНК- диагностика наследственных болезней ... 355ередовыми факторами (среди таких факторов показана,
в частности, важная роль низких значений pH в клетке).
Приведенная схема удовлетворительно объясняет наи¬
более удивительное свойство прионных болезней - воз¬
можность развития наследственных, инфекционных и
спорадических форм данных заболеваний.Около 10-15% случаев прионных болезней име¬
ют наследственно-семейный характер (семейные формы
болезни Крейтцфельдта-Якоба, а также синдром Герст-
манна-Штреусслера и фатальная семейная инсомния)
[Collinge J., 1997]. Тип наследования аутосомно-доми-
нантный. Развитие семейных форм прионных болезней
связано с наследованием мутаций в гене прионного бел¬
ка (PRNP). Ген PRNP локализован в дистальном участ¬
ке короткого плеча 20-й хромосомы (локус 20pl2-pter),
он имеет протяженность 16 кб и состоит из 2 экзонов,
второй из которых кодирует полную аминокислотную
последовательность прионного белка PrP [Kretzschmar IT
et al., 1986; Robakis N. et al., 1986]. Точная функция при¬
онного белка до настоящего времени не установлена. У
больных семейными формами прионных болезней опи¬
сано свыше 25 мутаций в гене PRNP, которые могут быть
подразделены на 2 класса: 1) точковые мутации в коди¬
рующей области гена (они представлены в основном
миссенс-мутациями, ведущими к аминокислотным за¬
менам в белке РгР, гораздо реже выявляются нонсенс-
мутации с обрывом трансляции); 2) вставки дополни¬
тельных копий октапептид-кодирующего повтора в про¬
ксимальной части гена между кодонами 51 и 91 (в нор¬
мальном гене этих повторов пять, у больных может от¬
мечаться до 9 дополнительных копий повторов)
[Prusiner S., Hsiao К., 1994; Collinge J., 1997; Windl О. et
al., 1999].В целом, семейные случаи болезни Крейтцфель¬
дта-Якоба характеризуются более ранним началом и бо¬
356Глава 3лее продолжительным течением по сравнению со спо¬
радическими случаями заболевания [Brown Р., 1992].
Клиническая манифестация каждой конкретной мутации
имеет свои определенные особенности. Более 70% слу¬
чаев семейной болезни Крейтцфельдта-Якоба связаны
с мутацией G->A в 200-м кодоне, ведущей к замене глу-
тамата на лизин (Glu200Lys), причем в большинстве из¬
вестных популяционных кластеров данной формы бо¬
лезни (Ливия, Чили, Испания, Тунис) мутация Glu200Lys
имеет единое генетическое происхождение [Lee Н. et al.,
1999]. Для носителей мутации Glu200Lys характерен
средний возраст манифестации болезни 50-56 лет и срав¬
нительно редкая выявляемость амилоидных бляшек на
секции. Еще более раннее начало болезни Крейтцфель¬
дта-Якоба (в среднем 46 лет) типично для мутации
Aspl78Asn, причем у больных нередко отмечаются ран¬
ние расстройства памяти и отсутствие периодических
комплексов на ЭЭГ [Brown Р., 1992]. Эта же мутация
выявлена во всех случаях фатальной семейной инсом-
нии [Collinge J., 1997]. Интересно, что молекулярным
различием между фатальной семейной инсомнией и
Aspl78Asn-BapnaHTOM болезни Крейтцфельдта-Якоба яв¬
ляется генотип по полиморфизму в 129-м кодоне гена
PRNP: больные фатальной семейной инсомнией обыч¬
но гомозиготны по метионину в кодоне 129, тогда как
при болезни Крейтцфельдта-Якоба выявляется гомози-
готность по валину в данном кодоне [Prusiner S., Hsiao К.,
1994]. Описан также целый ряд других мутаций PRNP,
выявляемых у больных с болезнью Крейтцфельдта-Яко¬
ба. При синдроме Герстманна-Штреусслера наиболее
частыми мутациями являются Prol02Leu, Alall7Val,
Phel98Ser и Gln217Arg; первая из указанных мутаций
характеризуется началом болезни после 40 лет, медлен¬
ным прогрессированием (до 5 лет и более) и превалиро¬
ДНК- диагностика наследственных болезней ... 357ванием мозжечкового синдрома, мутация Alall7Val -
более ранним дебютом симптомов (38 лет), высокой
частотой псевдобульбарных нарушений и редкостью
атаксии и миоклоний, тогда как у носителей мутаций
Phel98Ser и Gln217Arg на секции, помимо типичных
спонгиоформных изменений и мультицентрических ами¬
лоидных бляшек, выявляются множественные нейрофиб-
риллярные клубки [BrownP., 1992; Prusiner S. et al., 1993].
При мутациях в гене PRNP по типу вставок дополни¬
тельных копий октапептид-кодирующих повторов обыч¬
но наблюдается раннее начало (в среднем около 34 лет),
весьма длительное течение (до 7 лет), редкое развитие
миоклоний и периодических комплексов на ЭЭГ, а так¬
же весьма полиморфная патоморфологическая картина,
что позволяет обозначить данный фенотип как «смешан¬
ный» (болезнь Крейтцфельдта-Якоба/синдром Герстман-
н а-Штр еу сс л ер a) [BrownP, 1992].Как видно из представленного анализа, клинико¬
морфологические проявления мутаций в гене PRNP чрез¬
вычайно вариабельны, что в ряде случаев существенно
затрудняет отнесение конкретного синдрома к традици¬
онно выделяемым вариантам прионных болезней (бо¬
лезнь Крейтцфельдта-Якоба, синдром Герстманна-
Штреусслера или фатальная семейная инсомния). Наи¬
более яркий пример такого полиморфизма описан
Collinge J. с соавторами у 3 родственников, имевших одну
и ту же мутацию в гене PRNP: один из больных имел
классическую клинику болезни Крейтцфельдта-Якоба
и спонгиоформиые изменения в коре мозга, другой род¬
ственник имел атактический вариант болезни (близкий
к синдрому Герстманна-Штреусслера) и множественныеI 'rP-позитивные амилоидные бляшки на аутопсии, тогда
как у третьего родственника из той же семьи наблюда¬
лась изолированная деменция и на секции отсутствова¬
358Глава 3ли спонгиоформные изменения в веществе мозга
[Collinge J. et al., 1992]. В связи с указанными наблюде¬
ниями, рядом ведущих специалистов по данной пробле¬
ме предложена более адекватная номенклатура семей¬
ных форм прионных болезней, основанная на «мутаци¬
онном» принципе [Prusiner S., Hsiao К., 1994; Collinge J.,
1997; Windl О. et al., 1999]. Так, при обнаружении конк¬
ретных мутаций, соответствующий вариант заболевания
предпочтительнее обозначать как «прионная болезнь
[Prol02Leu]» (вместо «атактический вариант синдрома
Герстманна-Штреусслера»), либо «прионная болезнь
[Aspl78Asn, Metl29]» (вместо «фатальная семейная ин-
сомния») и т.д. Такая номенклатура позволяет точно
обозначать этиологию изучаемой формы прионного за¬
болевания и избежать любых возможных нечеткостей и
двусмысленностей в формулировках [Prusiner S.,
Hsiao К., 1994].На практике абсолютно достоверная диагности¬
ка прионных болезней базируется, главным образом, на
морфологическом и иммуногистохимическом секцион¬
ном исследовании. В мире существует опыт прижизнен¬
ной диагностики прионных болезней при проведении
биопсии мозга, однако эта процедура не может быть ре¬
комендована для широкого использования. В последние
годы для диагностики нового варианта болезни Крейтц-
фельдта-Якоба предложено иммуногистохимическое ис¬
следование на PrPSc лимфоретикулярных тканей - на¬
пример, биоптатов миндалин [Collinge J., 1997]. В се¬
мейных случаях прионных болезней важнейшим и дос¬
тупным методом достоверной прижизненной диагнос¬
тики заболевания является выявление патогенных мута¬
ций в гене PRNP (прямая ДНК-диагно стика). Принимая
во внимание небольшой размер кодирующей области
гена (759 п.о.), с технической точки зрения мутацион¬
ДНК- диагностика наследственных болезней ... 359ный анализ PRNP является относительно несложным и
обычно основывается на тотальном секвенировании гена
[Prusiner S., Hsiao К., 1994; Windl О. et al., 1999]. Встав¬
ки и делении октапептидных повторов легко выявляют¬
ся при рутинном электрофорезе продуктов амплифика¬
ции в агарозном геле, позволяющем разделять нормаль¬
ные и мутантные фрагменты ДНК, имеющие разную
длину. В некоторых случаях (особенно при наличии оп¬
ределенных особенностей клинической картины) прямую
ДНК-диагностику целесообразно начинать с исследова¬
ния тех или иных известных мутаций в гене PRNP, кото¬
рое проводится посредством гибридизации ДНК с ал-
лель-специфическими олигонуклеотидами либо с помо¬
щью рестрикционного анализа с соответствующими ре¬
стрикционными эндонуклеазами [Prusiner S., Hsiao К.,
1994]. Результаты интенсивного молекулярного скринин¬
га гена PRNP на больших сериях случаев с возможным
или предполагаемым диагнозом прионного заболевания
показывают, что частота встречаемости наследственных
прионных болезней может быть выше, чем частота на¬
следственных форм болезни Альцгеймера и других на¬
следственных деменций, за исключением болезни Ген-
гингтона [Windl О. et al., 1999]. По мнению S. Prusiner и
К. Hsiao (1994), возможность прионных болезней долж¬
на рассматриваться во всех случаях атипичных нейроде¬
ген еративных заболеваний, в связи с чем необходимо
предусматривать проведение соответствующих молеку-
лярно-генетических и иммуногистохимических исследо¬
ваний.При спорадических и инфекционных формах при-
опных болезней показано существование генетической
предрасположенности, одним из ведущих факторов ко¬
торой является полиморфизм в 129-м кодоне гена PRNP,
определяющий встраивание аминокислоты валина или
360Глава 3метионина в соответствующем положении прионного
белка. Абсолютное большинство больных со споради¬
ческой формой болезни Крейтцфельдта-Якоба и ятро-
генной формой заболевания, связанной с введением че¬
ловеческого гормона роста, являются гомозиготами по
данному полиморфизму [Collinge J. et al., 1991; Palmer М.
et al., 1991]. Предполагается, что повышенная предрас¬
положенность к развитию прионных болезней у гомози¬
гот по 129-му кодону обусловлена абсолютной гомоло¬
гией молекул прионного белка, что облегчает межмоле-
кулярное взаимодействие и, следовательно, конверсию
РгРс в PrPSc [Collinge J., 1997]. Напротив, гетерозигот-
ность валин/метионин в 129-м кодоне имеет определен¬
ный протективный эффект, что подтверждается более
поздним (на 10-20 лет) началом болезни у гетерозигот
по сравнению с гомозиготами при некоторых наслед¬
ственных формах прионных болезней [Baker Н. et al.,
1991]. Клиническое и прогностическое значение опре¬
деления ДНК-полиморфизма в 129-м кодоне гена PRNP
у лиц из группы риска в настоящее время активно об¬
суждается в литературе.3.53. Другие формы наследственных деменций3.5.3.1. Лобно-височная деменция с паркинсонизмомЛобно-височная деменция составляет около 15-
20% всех случаев первично-дегенеративных деменций,
причем почти у половины больных имеется положитель¬
ный семейный анамнез, свидетельствующий об аутосом-
но-доминантном типе наследования [Chow Т. etal., 1999].
Особую форму представляет собой лобно-височная де¬
менция с паркинсонизмом - аутосомно-доминатное за¬
болевание, описываемое еще в литературе под названи¬
ями «паркинсонизм-деменция с паллидо-понто-ниграль-
ной дегенерацией», «паллидо-нигро-люисова дегенера¬
ДНК- диагностика наследственных болезней361ция», «комплекс расторможенность-деменция-паркин-
сонизм-амиотрофии» и т.п. [Wilhelmsen К. et al., 1994;
Bird Т., 1998; Clark L. et al., 1998]. Заболевание начина¬
ется обычно после 45 лет и проявляется прогрессирую¬
щей деменцией лобного типа в сочетании с разнообраз¬
ными двигательными нарушениями — брадикинезией,
гипомимией, мышечной ригидностью, расстройствами
походки, пирамидными симптомами, амиотрофиями и
фасцикуляциями, парезами глазодвигательной мускула¬
туры и др. На секции выявляются дегенеративные изме¬
нения лобных и теменно-височных долей полушарий
мозга, базальных ганглиев, черной субстанции, гиппо¬
кампа, а также своеобразные филаментозные включения
в нейронах и клетках глии. Атрофия лобно-теменно-ви-
сочных долей и расширение боковых желудочков, осо¬
бенно передних рогов, отчетливо визуализируются на
рентгеновских и магнитно-резонансных компьютерных
томограммах.Ген лобно-височной деменции с паркинсонизмом
локализован на хромосоме 17q21 [Wilhelmsen К. et al.,1994]. Продуктом данного гена является клеточный бе¬
лок тау, ассоциированный с микротубулярным аппара¬
том нейронов [Hutton М. et al., 1998; Poorkaj P. et al.,
1998]. К настоящему времени у больных аутосомно-до-
минантной лобно-височной деменцией с паркинсониз¬
мом идентифицировано около 20 мутаций в гене тау
(главным образом, миссенс-мутаций и мутаций в сайтах
сплайсинга) [Clark L. et al., 1998; Hutton M. et al., 1998;
Poorkaj P. et al., 1998; Houlden IT et al., 1999; Rizzu P. et
al., 1999]. Мажорные мутации в гене тау не выявлены.
Предполагается, что мутации приводят к нарушению
взаимодействия тау с микротрубочками цитоскелета ней¬
ронов, что сопровождается гиперфосфорилированием
данного белка, формированием нерастворимых тау-фи-
ламентов и гибелью нейронов [Hong М. et al., 1998].
362Глава 3Прямая ДНК-диагностика лобно-височной демен-
ции с паркинсонизмом проводится на основе секвени-
рования кодирующей области тау, обычно в сочетании с
предварительным SSCP-анализом всех 13 экзонов гена.
При молекулярно-генетическом анализе больших групп
больных с деменциями было показано, что мутации в
гене тау обусловливают примерно 13-17% всех случаев
лобно-височной деменции и около 45% семейных слу¬
чаев данного заболевания [Houlden Н. et al., 1999; Rizzu P.
et al., 1999]. Необходимо сделать определенную скидку
на то, что в указанных работах исследование некодиру¬
ющих областей гена тау не проводилось. Полученные
данные могут свидетельствовать о генетической гетеро¬
генности наследственных форм деменции лобного типа.3.5.3.2. Деменция при семейной церебральной амило¬
идной ангиопатии британского типаДанное заболевание было описано в 70-е годы в
больших семьях британского и датского этнического
происхожения, в связи с чем в литературе оно иногда
обозначается как «семейная деменция британского
типа», «семейная датская деменция» или «офтальмо-ото-
энцефалическая гередопатия». Тип наследования ауто-
сомно-доминантный. Заболевание манифестирует на
пятом десятилетии жизни и проявляется прогрессирую¬
щей деменцией, спастичностью, мозжечковой атаксией,
катарактой, снижением слуха. При морфологическом
исследовании выявляется тяжелая распространенная
амилоидная ангиопатия сосудов мозга, периваскулярные
амилоидные бляшки (преимущественно в области гип¬
покампа) и нейрофибриллярные клубки, что сближает
данное заболевание с болезнью Альцгеймера. Ген забо¬
левания расположен на хромосоме 13ql4 и получил обо¬
значение IMB2B, или BRI (от англ. British) [Vidal R. et
al., 1999]. Продукт гена - интегральный мембранный
ДНК- диагностика наследственных болезней ... 363белок с неустановленной функцией. В британской и дат¬
ской семьях с данной формой деменции у больных вы¬
явлены 2 различных мутации в гене BRI (соответствен¬
но, точковая мутация в стоп-кодоне и дупликация учас¬
тка длиной 10 п.о. со сдвигом рамки), каждая из кото¬
рых ведет к «проскакиванию» нормального сайта для
стоп-кодона и патологическому удлинению мутантного
белка [Vidal R. et al., 1999; 2000]. Синтезируемый в ре¬
зультате белок содержит в своей карбоксильной части
чужеродную последовательность из 34 аминокислотных
остатков, которая в высвобожденном виде представляет
собой амилоидогеиный пептид, становящийся основой
формирования нерастворимых амилоидных фибрилл.Принимая во внимание небольшой размер гена
BRI (всего 267 кодонов), в подходящих семьях и изоли¬
рованных случаях с подозрением на данную редкую фор¬
му наследственной деменции наиболее удобным мето¬
дом прямой ДНК-диагностики болезни является тоталь¬
ное секвенирование кодирующей области гена.3.5.3.3. Семейные деменции, ассоциированные
с хромосомой 3В 1999 году R. Davis с соавторами описали 2 се¬
мьи с новой формой пресенильной деменции, наследо¬
вавшейся по аутосомио-доминантному типу. Заболева¬
ние начиналось на пятом десятилетии жизни и характе¬
ризовалось когнитивным снижением в виде нарушений
концентрации и внимания, нарушения зрительно-про¬
странственной ориентировки, расстройств памяти (вы¬
раженных в меньшей степени, чем при болезни Альц¬
геймера). По мере прогрессирования болезни развива¬
лась глобальная деменция, у некоторых больных отме¬
чались также эпилептические припадки. На секции выяв¬
лялись особые эозинофильные нейрональные включе¬
ния в клетках глубоких слоев коры больших полушарий
364Глава 3и подкорковых ядер (особенно в области черной субстан¬
ции). Анализ данных включений показал, что их основ¬
ным биохимическим субстратом является полимеризо-
ванный нейросерпин - 410-аминокислотный нейрональ¬
ный белок, относящийся к классу ингибиторов серино-
вых протеиназ (серпинов). В обеих семьях у больных
были выявлены точковые миссенс-мутации в гене ней-
росерпина, расположенном в хромосомном локусе 3q26
[Davis R. et al., 1999]. Показано, что мутантный сейро-
серпин приобретает нестабильную патологическую кон¬
формацию, способствующую его фибриллярной поли¬
меризации и фатальному накоплению в клетке сейро-
серпин-позитивных нерастворимых комплексов. Таким
образом, данный тип наследственной деменции можно
отнести к растущему числу «конформационных болез¬
ней» (аналогично прионным болезням, болезни Альц¬
геймера, болезни Паркинсона, множественным систем¬
ным атрофиям и ряду других нейрогенеративных забо¬
леваний). Ведущим проявлением «конформационных
болезней» является прогрессирующая дегенерация спе¬
цифических популяций нейронов, поскольку аккумуля¬
ция белковых комплексов несет основную опасность
именно для длительно живущих и неделящихся (пост-
митотических) клеток, наиболее типичным примером
которых являются нейроны.Сотрудниками французского исследовательского
центра INSERM U289 (рук. лаборатории - проф. A. Brice)
проведено секвенирование гена нейросерпина в 38 се¬
мьях с аутосомно-доминантными деменциями альцгей-
меровского и неальцгеймеровского типа, в которых пред¬
варительно были исключены мутации в генах АРР, пре-
сенилинов и тау. Мутаций в гене нейросерпина у обсле¬
дованных больных выявлено не было, что свидетельству¬
ет об исключительной редкости первичной нейросерпи-
ДНК- диагностика наследственных болезней ... 365нопатии в общей группе наследственных деменций.
Систематический опыт прямой ДНК-диагностики нейросер-
пин-ассоциированной деменции в мире на сегодняшний
день отсутствует.На 3-й хромосоме в локусе 3pl 1.1—ql 1-2 карти¬
рован локус другой самостоятельной формы аутосомно-
доминантного ослабоумливающего заболевания - так
называемой «семейной неспецифической деменции»,
клинически имеющей некоторое сходство с лобно-ви-
сочной деменцией [Brown J. et al., 1995]. Генетическое
сцепление примерно с той же группой маркеров показа¬
но еще в одной аутосомно-доминантной семье, в кото¬
рой у пробаида наблюдалась деменция альцгеймеровс-
кого типа с атипичной нейроморфологической картиной
(отсутствие нейрофибриллярных клубков) [Poduslo S. et
al., 1999]. Дальнейший клинико-генетический анализ
позволит более точно определить нозологические гра¬
ницы и клинический спектр данной формы наследствен¬
ной деменции.3.5.3.4. Болезнь ГентингтонаОдним из наиболее распространенных моноген-
ных нейродегенеративных заболеваний, характеризую¬
щихся развитием деменции, является болезнь Гентинг-
тона. В литературе иногда выделяется особая и крайне
редкая психическая форма болезни Гентингтона, при
которой в клинической картине на протяжении длитель¬
ного времени преобладают нарушения интеллектуаль-
но-мнестических функций при сохранной моторике и
почти полном отсутствии хореических гиперкинезов
[Иванова-Смоленская И.А. и др., 1998 (а)]. Такие боль¬
ные, как правило, становятся пациентами психиатричес¬
ких стационаров. Диагностика психической формы бо¬
лезни Гентингтона весьма непроста и базируется на пря¬
366Глава 3мой ДНК-диагностике соответствующей тринуклеотид-
ной мутации в гене на 4-й хромосоме. Молекулярная
генетика и ДНК-диагностика болезни Гентингтона де¬
тально рассматриваются в разделе 3.2.1.3.6. Наследственные формы эпилепсийЭпилептические припадки различных типов мо¬
гут быть проявлением около 150 наследственных невро¬
логических болезней, в большинстве случаев присоеди¬
няясь на поздней стадии дегенеративного процесса ЦНС.
Самостоятельное значение имеют наследственные забо¬
левания, в структуре которых стойкий эпилептический
синдром является доминирующим. Эти заболевания при¬
нято подразделять на две большие группы: а) идиопати-
ческие наследственные эпилепсии, при которых эпилеп-
тизация нейронов обусловлена специфическими дефек¬
тами генов, контролирующих возбудимость нейрональ¬
ных мембран; б) симптоматические наследственные эпи¬
лепсии, при которых поражение нейронов со стойким
эпилептическим синдромом является частью распрост¬
раненного системного дегенеративного процесса, обус¬
ловленного дефектами структурных белков или фермен¬
тов клетки. Ко второй группе относится в первую оче¬
редь синдром прогрессирующей миоклонус-эпилепсии,
а также ряд наследственных заболеваний обмена.3.6.1. Идиопатические наследственные
эпилепсииНаследственность играет важную роль в разви¬
тии эпилепсии. При большинстве форм эпилепсии име¬
ет место полигенно-детерминируемая предрасположен¬
ность, а возникновение заболевания является результа¬
ДНК- диагностика наследственных болезней ... 367том взаимодействия совокупности ряда генетических и
средовых факторов [Карлов В.А., 1990; SteinleinO., 1999].
В то же время вклад некоторых генов является настоль¬
ко значимым, что мутации в любом из них являются
достаточным условием для манифестации менделирую-
щих (чаще всего аутосомно-доминантных) форм эпилеп¬
сии. Следует добавить, что моногенное наследование
наиболее характерно для генерализованных форм эпи¬
лепсии и в меньшей степени — для эпилепсии с парци¬
альными припадками.3.6.1.1. КаналопатииВсе наследственные формы эпилепсии с установ¬
ленными генами обусловлены повреждением ионных
каналов, имеющих прямое отношение к механизмам
поляризации мембраны нейронов. Это позволяет отнес¬
ти данные заболевания к группе каналопатий.Заболевания, обусловленные повреждением кали¬
евых каналов. В нервной системе имеются разные типы
калиевых каналов, специфичные для определеных групп
нейронов. Одним из наиболее изученных является М-
канал, контролирующий медленную активацию и инак¬
тивацию трансмембранного потока ионов калия, порог
электровозбудимости нейронов и ответ на синаптичес¬
кое высвобождение нейротрансмиттеров [Wang H.-S. et
al., 1998]. М-канал имеет соматодендритическую лока¬
лизацию в полиморфных нейронах коры больших полу¬
шарий и гиппокампа, а также периферических симпати¬
ческих нейронах. Он образован двумя взаимодействую¬
щими субъединицами KCNQ2 и KCNQ3, относящими¬
ся к KQT-семейству потенциал-зависимых калиевых
каналов, причем обе белковые субъединицы ассоцииро¬
ваны с тубулином и протеинкиназой А, формируя слож¬
ный сигнальный комплекс с множественными функциями
368Глава 3[Wang H.-S. et al., 1998; Cooper E. et al., 2000]. Гены
KCNQ2 и KCNQ3 локализованы, соответственно, на
хромосомах 20ql3.3 и 8q24. Мутации в этих генах при¬
водят к развитию одной их форм генерализованной эпи¬
лепсии детского возраста - доброкачественных семей¬
ных неонатальных судорог [Biervert C. et al., 1998;
CharlierC.et al., 1998; Singh N. et al., 1998]. Данное ауто-
сомо-доминантное заболевание характеризуется корот¬
кими тонико-клоническими припадками, моторными ав¬
томатизмами и эпизодами апноэ в первые дни жизни ре¬
бенка (иногда резкие пароксизмальные движения плода
регистрируются внутриутробно на протяжении после¬
дних 2 месяцев беременности), спонтанным выздоров¬
лением спустя несколько недель и дальнейшим нормаль¬
ным психомоторным развитием [Ronen G. et al., 1993].
Риск последующей трансформации доброкачественных
неонатальных судорог в генерализованную эпилепсию
составляет около 16%, в связи с чем KCNQ3-acconHH-
рованная форма неонатальных судорог, возможно, явля¬
ется аллельным заболеванием с генерализованной иди-
опатической эпилепсией, картированной в том же хро¬
мосомном локусе 8q24 [Zara F. et al., 1995].После клонирования генов KCNQ2 и KCNQ3
соответствующие белковые продукты стали предметом
интенсивных функциональных исследований. Экспрес¬
сия мутантного белка KCNQ2 в эксперименте показала
резкое угнетение калиевого тока во время потенциала
действия, что приводит к нарушению калий-зависимой
реполяризации нейрональных мембран [Biervert C. et al.,1998]. Интересно отметить, что повреждения еще одно¬
го родственного гена KQT-семейства калиевых каналов
(ген KCNQ1 на хромосоме 11) ответственны за развитие
наследственной формы аритмии с синкопальными со¬
стояниями - синдрома удлиненного интервала QT, обус¬
ДНК- диагностика наследственных болезней ... 369ловленного нарушением нейрогенной регуляции сердеч¬
ного ритма [Wang Q. et al., 1996].Как указывалось в разделе 3.3.3.2, мутации в гене
принципиально другого типа калиевых каналов (ген
KCNA1 на хромосоме 12р 13) являются причиной раз¬
вития эпизодической атаксии 1-го типа. У некоторых
больных с подтвержденными мутациями в гене KCNA1
острые эпизоды атаксии сочетаются с парциальными
эпилептическими припадками [Zuberi S. et al., 1999];
более того, анализ описанных в литературе семейных
случаев эпизодической атаксии-1 показывает более вы¬
сокую частоту эпилепсии у больных родственников по
сравнению со здоровыми членами семей. Эти наблюде¬
ния подтверждают роль дисфункции нейрональных ка¬
лиевых каналов в генерации эпилептических феноменов
(как и пароксизмов атаксии) и свидетельствуют о том,
что и другие разнообразные гены калиевых каналов яв¬
ляются очевидными кандидатами при поиске генов се¬
мейных форм эпилепсии у человека.Заболевания, обусловленные повреждением на¬
триевых каналов. Нейрональный потеициал-зависимый
натриевый канал представляет собой белковый комплекс,
состоящий из большой трансмембранной а-субъедини-
цы (структурно-функциональной основы канала) и двух
малых (3-субъединиц (модулирующих инактивацию Na+-
канала и уровень экспрессии а-субъединицы) [Wallace R.
et al., 1998]. Повышение проницаемости нейрональных
мембран для ионов натрия является хорошо изученным
и важным фактором эпилептогенеза. Совсем недавно
было показано, что дефекты а- и -субъединиц нейро¬
нального натриевого канала, обусловленные мутациями
соответствующих генов SCN1A и SCN1B на хромосо¬
мах 2q24 и 19ql 3.1, приводят к развитию так называе¬
мой «генерализованной эпилепсии с фебрильными су¬
370Глава Здорогами плюс» [Wallace R. et al., 1998; Escayg A. et al.,
2000 (б)]. Данное заболевание наследуется по аутосом-
но-доминантному типу с неполной пенетрантностью и
характеризуется фебрильными судорогами (наблюдаемы¬
ми не только в типичном возрасте <5 лет, но и позднее),
а также различными видами афебрильных генерализо¬
ванных припадков (тонико-клонических, миоклоничес-
ких, атонических, абсансов) или их сочетанием, что мо¬
жет сопровождаться развитием фенотипа миоклоничес-
ки-астатической эпилепсии [SchefferI.,Berkovic S., 1997].Функциональные исследования мутантного бел¬
ка SCN1B на клеточной системе in vitro продемонстри¬
ровали замедление инактивации (закрывания) нейро¬
нальных натриевых каналов, что, по-видимому, сопро¬
вождается патологической «утечкой» тока ионов Na+ в
клетку, провоцирующей деполяризацию и гипервозбу¬
димость нейрональной мембраны [Wallace R. et al., 1998].
Предполагается, что взаимосвязь припадков с повыше¬
нием температуры тела обусловлена влиянием темпера¬
турного фактора на проницаемость натриевых каналов.Интересно отметить, что, помимо мутаций в ге¬
нах натриевых каналов, фенотип данной формы эпилеп¬
сии может быть связан также с повреждением гена
у2-субъединицы рецептора ГАМК [Baulac S. et al., 2001].Заболевания, обусловленные повреждением каль¬
циевых каналов. Потенциал-зависимые кальциевые ка¬
налы играют ключевую роль в высвобождении нейро¬
трансмиттеров. Нейрональный кальциевый канал явля¬
ется мультимерным белковым комплексом, в состав ко¬
торого входит а! -субъединица, формирующая остов ка¬
нала, и вспомогательные субъединицы р, у и а2й
[Escayg A. et al., 2000 (а)]. Известно, как минимум, семь
различных генов, кодирующих субъединицу аг и 4 го¬
мологичных гена, кодирующих субъединицы Р,-Р4 каль
ДНК- диагностика наследственных болезней ... 371циевого канала. Взаимодействие между и р-субъе¬
диницами регулирует амплитуду кальциевого тока, его
потенциал-зависимость и кинетику активации и инак¬
тивации [Бе \Уаагс1 М., СатрЬеП К., 1995]. В 2000 году
А. Escayg с соавторами, проводя скрининг гена р4-субъе-
диницы кальциевого канала (САСХВ4. хромосома 2q22-
23) в большой выборке семейных случаев эпилепсии, об¬
наружили мутации в данном гене в 2 семьях с аутосом-
но-доминантной идиопатической генерализованной эпи¬
лепсией [ЕБсау§ А. е1 а1., 2000 (а)]. Интересно, что одна
из выявленных мутаций была также обнаружена у боль¬
ного с семейной эпизодической атаксией, близкой по кли¬
ническим характеристикам к эпизодической атаксии
2-го типа (ацетазол-чувствительной). Основной же вари¬
ант эпизодической атаксии-2, как указывалось в разделе3.3.3.2, обусловлен нонсенс-мутациями гена другой
субъединицы кальциевого канала - а 1 д (ген С'АСЖ, 1А4)
|ОрЬю:£Г К е! а1., 1996]. Таким образом, синдромы каль¬
циевых каналов, аналогично описанным выше КСЫА1 -
ассоциированным синдромам калиевых каналов, свиде¬
тельствуют о патогенетической взаимосвязи между эпи¬
лептическими припадками и острыми пароксизмами
атаксии при каналопатиях. Конкретный механизм пато¬
генетического эффекта мутаций в гене САСМВ4 заклю¬
чается в нарушении взаимодействия мутантной Р4-субъе-
диницы с а,А-субъединицей кальциевого канала, что
приводит к сокращению быстрого компонента инакти¬
вации канала и снижению потока ионов кальция в акти¬
визированные нейроны в процессе генерации потенциа¬
лов действия [Escayg А. е! а1., 2000].Нарушение проницаемости мембран для ионов
кальция как основа эпилептогенной гипервозбудимости
нейронов может опосредоваться и другим молекулярным
механизмом - патологией нейрональных никотиновых
572Глава 3рецепторов ацетилхолина (н-холинорецепторов). Нейро¬
нальные н-холинорецепторы относятся к суперсемейству
лиганд-зависимых ионных каналов, контролирующих
быструю синаптическую трансмиссию [Groot
Kormelink Р., Luyten W., 1997]. Различные подтипы ней¬
рональных н-холинорецепторов различаются по соста¬
ву входящих в них а- и ß-субъединиц. Активация нейро¬
нальных н-холинорецепторов сопровождается значитель¬
ным повышением содержания кальция в клетке благо¬
даря прямому пассажу ионов кальция через рецептор¬
ный канал, и именно таким путем ацетилхолинергичес-
кие нейроны регулируют обширный спектр клеточных
событий [Rathouz М. et al., 1996]. Показано, что мута¬
ции в генах а - и ß^-субъединиц нейронального н-холи-
норецептора (соответственно, ген CHRNA4 на хромо¬
соме 20ql3.2-13.3 и ген CHRNB2 на хромосоме 1р21),
лежат в основе развития аутосомно-доминантной лоб¬
ной эпилепсии с ночными пароксизмами [Steinlein О. et
al., 1995; De Fusco M. et al., 2000]. Это единственная пока
форма эпилепсии с парциальными припадками, для ко¬
торой идентифицированы мутантные гены. Еще один
локус аутосомно-доминантной лобной эпилепсии с ноч¬
ными пароксизмами картирован на хромосоме 15q24, в
тесном сцеплении с кластером генов а3-, а5- и ß4-cy6be-
диниц нейрональных н-холинорецепторов [Phillips H. et
al., 1998]. Можно предположить, что патогенез данного
генетического варианта заболевания также связан с на¬
рушением проницаемости нейрональных мембран для
ионов кальция, обусловленным повреждением одного из
указанных генов н-холинорецепторов в области 15q24.
В ряде семей, отягощенных аутосомно-доминантной
лобной эпилепсией с ночными пароксизмами, сцепле¬
ние со всеми указанными хромосомными локусами дос¬
товерно исключено.
ДНК- диагностика наследственных болезней ... 373ДНК-диагноетика эпилептических каналопатий.
К настоящему времени у больных наследственными эпи¬
лепсиями из различных популяций мира выявлено око¬
ло 20 мутаций в генах ионных каналов (Точковые мута¬
ции, делеции и вставки), большинство из которых явля¬
ются уникальными и не повторяются в разных семьях
[81ет1ет О., 1999]. Принимая во внимание сложную
мультиэкзонную организацию генов ионных каналов,
поиск мутаций в них является весьма трудоемким и пред¬
полагает исследование всей кодирующей области гена с
помощью стандартных методов мутационного скринин¬
га, описанных в главе 1. При отборе генов-кандидатов
для исследования необходимо ориентироваться на кли¬
ническую картину заболевания и сравнительную часто¬
ту отдельных генетических форм каналопатий (так, по¬
вреждения гена КСКТС)2 обусловливают свыше 90% всех
случаев доброкачественных семейных неонатальных су¬
дорог с идентифицированными мутациями, тогда ген
КСМ^З вовлекается гораздо реже и должен исследовать¬
ся во вторую очередь). Большое число фенокопий суще¬
ственно осложняет задачу, поэтому на сегодняшний день
иоиск мутаций в генах ионных каналов рекомендуется
ограничить только несомненными семейными случая¬
ми заболеваний. При обнаружении мутации в изучаемой
семье возможно проведение прямой ДНК-диагностики
у клинически здоровых лиц, относящихся к группе риска.Несмотря на технические сложности и сравни¬
тельно небольшой опыт прямой ДНК-диагностики эпи¬
лептических каналопатий, такая диагностика представ¬
ляется исключительно важной с клинической точки зре¬
ния. Дело в том, что механизм действия многих извест¬
ных антиконвульсантов основан на модулировании фун¬
кции нейрональных ионных каналов. Например, карба-
мазепин и фенитоин оказывают свой противосудорож-
374Глава 3ный эффект через потенцирование инактивации натрие¬
вых каналов [McDonald R., Kelly К., 1994]. Более того,
знание молекулярных звеньев патогенеза различных
форм эпилепсии является необходимым для разработки
новых противосудорожных лекарственных препаратов.
Таким образом, идентификация мутации в конкретном
гене в будущем должна стать одной из ключевых пред¬
посылок для проведения специфической, патогенетичес¬
ки обоснованной терапии наследственных форм эпилепсии.3.6.1.2. Другие кандидатные локусыНаследственные формы генерализованных эпи¬
лепсий. Наиболее распространенной формой судорож¬
ного синдрома являются фебрильные судороги - крат¬
ковременные (обычно до 15 минут) тонико-клонические,
тонические или клонические приступы, связанные с ли¬
хорадочным состоянием и наблюдающиеся примерно у
3% детей в возрасте до 5 лет [Никанорова М.Ю. и др.,
1997]. В 2-7% случаев у лиц, имевших в детстве феб¬
рильные судороги, позднее развиваются стойкие формы
эпилепсии с афебрильными припадками. В большинстве
наблюдений фебрильные судороги имеют сложное по-
лигенное наследование, однако описаны отдельные се¬
мьи с аутосомно-доминантной формой данного заболе¬
вания. Гены аутосомно-доминантных фебрильных судо¬
рог картированы на хромосомах 2q23-24, 8ql3—21 и
19р13.3 [Wallace R. et al., 1996; Johnson E. et al., 1998;
Peiffer A. et al., 1999]. В локусе 2q23-24 расположены
несколько весьма подходящих генов-кандидатов (пять
субъединиц натриевого канала и один из генов калие¬
вых каналов), один из которых, по-видимому, и обуслов¬
ливает развитие 2q-cneniieHHoft формы фебрильных су¬
дорог.Еще одной прогностически благоприятной фор¬
мой наследственной эпилепсии раннего детского возра¬
ДНК- диагностика наследственных болезней ... 375ста являются доброкачественные семейные младенчес¬
кие судороги. Они проявляются кластерами генерализо¬
ванных афебрильных припадков различных типов (кло-
нических, тонических или атонических), реже кластера¬
ми парциальных припадков, манифестирующих в воз¬
расте от 3 до 12 месяцев, хорошо поддающихся терапии
обычными антиконвульсантами и прекращающихся в
более позднем возрасте без признаков задержки психо¬
моторного развития [Echenne В. et al., 1994]. «Чистый»
фенотип доброкачественных семейных младенческих
судорог сцеплен с длинным плечом 19-й хромосомы
[Guiponti М. et al., 1997]либо длинным плечом 2-й хро¬
мосомы [Malacarne М. et al., 2001], тогда как фенотип,
осложненный пароксизмальным хореоатетозом - с пе¬
рицентромерным участком 16-й хромосомы
[Szepetowski P. et al., 1998]. Иногда последняя форма так¬
же может манифестировать в виде изолированных мла¬
денческих судорог [Caraballo R.et al., 2001]. Обе ука¬
занные формы четко наследуются по аутосомно-доми-
нантному типу. Сочетание доброкачественных семейных
младенческих судорог с пароксизмальным хореоатето¬
зом позволяет предполагать наличие общих патогенети¬
ческих механизмов развития этих двух клинически раз¬
личных пароксизмальных феноменов.Миоклонические эпилепсии представляют собой
гетерогенную и обширную группу заболеваний, при ко¬
торых центральным клиническим симптомом являются
полиморфные миоклонические припадки. Самой частой
формой из них (до 10% всех идиопатических эпилеп¬
сий) является ювенильная миоклоническая эпилепсия
Янца, характеризующаяся доброкачественным течени¬
ем, началом болезни на втором десятилетии жизни, со¬
четанием проксимальных миоклоний и генерализован¬
ных припадков, манифестирующих чаще в утренние часы
[Карлов В.А., 1990]. Значительная часть случаев явля¬
376Глава 3ются семейными, однако тип наследования не вполне
уточнен: наиболее вероятным считается аутосомно-ре-
цессивный тип наследования либо аутосомно-доминан-
тный с неполной пенетрантностью. Гены предрасполо¬
женности к ювенильной миоклонической эплепсии кар¬
тированы на хромосомах 6р 11—12 и 15ql4 [Liu A. et al.,
1996; Emslie F. et al., 1997], однако не вызывает сомне¬
ний дальнейшая генетическая гетерогенность данного
заболевания. Генетический локус другой формы миок¬
лонической эпилепсии, описанной лишь в японской по¬
пуляции - семейной доброкачественной миоклоничес¬
кой эпилепсии взрослых (тип наследования аутосомно-
доминантный), картирован на хромосоме 8q24
[Mikami М. et al., 1999]. Наконец, известна еще одна ред¬
кая форма миоклонической эпилепсии, манифестирую¬
щая в раннем детском возрасте, наследующаяся по ауто-
сомно-рецессивному типу и проявляющаяся частыми
длительными миоклоническими припадками, тонико-
клоническими припадками и фебрильными судорогами
[Zara F. et al., 2000]. Ген указанной аутосомно-рецессив-
ной младенческой миоклонической эпилепсии картиро¬
ван в хромосомном локусе 16р 13, в тесном сцеплении с
геном потенциал-зависимого хлорного канала (CLCN7)
[Zara F. et al., 2000]. Поскольку хлорные каналы имеют
прямое отношение к реполяризации клеточных мемб¬
ран, данный ген является очевидным кандидатом для
аутосомно-рецессивной младенческой миоклонической
эпилепсии.В 1999 году М. Durner с соавторами картировали
ген аутосомно-рецессивной идиопатической генерализо¬
ванной эпилепсии юношеского возраста, проявляющей¬
ся тонико-клоническими припадками и абсансами без
миоклонического компонента, на хромосоме 8р 11—12
[Durner М. et al.v 1999]. В этом же локусе расположен
ДНК- диагностика наследственных болезней ... 377ген р,-субъединицы никотиновного ацетилхолинового
рецептора. Принимая во внимание установленную роль
никотиновых ацетилхолиновых рецепторов в развитии
наследственных форм эпилепсии (см. выше), данный ген
может рассматриваться как подходящий кандидат для
аутосомно-рецессивной идиопатической генерализован¬
ной эпилепсии юношеского возраста.Детская абсансная эпилепсия составляет от 5 до
15% всех случаев генерализованных эпилепсий у детей.
Данное заболевание чрезвычайно гетерогенно и может
проявляться как изолированными абсансами с возник¬
новением припадков в 2-8 лет и исчезновением симпто¬
матики к пубертатному периоду, так и более стойким
синдромом, при котором абсансы сочетаются с тонико-
клоническими и миоклоническими припадками [Ника-
норова М.Ю., 1997]. Последний вариант болезни может
наследоваться предположительно по аутосомно-рецес-
сивному типу; ген данной формы детской абсансной эпи¬
лепсии картирован в хромосомном локусе 8q24 [Fong G.
ct al., 1998], более теломерно по отношению к KCNQ3-
;ICCOIщированной форме доброкачественных семейных
неонатальных судорог. Данный пример позволяет про¬
иллюстрировать одну важную особенность наследствен¬
ных идиопатических эпилепсий - частое расположение
локусов этих заболеваний в виде протяженных класте¬
ров на определенных хромосомах. Наиболее отчетливый
кластер расположен на длинном плече хромосомы 8: как
было указано при изложении предыдущего материала, в
области 8q локализованы гены, как минимум, четырех
различных форм наследственных эпилепсий. Другие хо¬
рошо изученные кластеры наследственных эпилепсий
расположены на хромосомах 15q и 20q. Можно предпо¬
ложить, что такое достаточно компактное расположение
локусов наследственных эпилепсий отражает кластериза¬
378Глава 3цию на хромосомах гомологичных генов, принадлежа¬
щих к определенным семействам и кодирующих белки с
близкими клеточными функциями.Наследственные формы парциальных эпилепсий.
В ряде семей различного этнического происхождения
описана новая форма наследственной парциальной эпи¬
лепсии -латеральная височная эпилепсия (парциальная
эпилепсия со слуховыми симптомами) [Poza J. et al.,1999]. Заболевание начинается на втором-третьем деся¬
тилетии жизни, проявляется простыми редко возникаю¬
щими парциальными припадками (слуховыми, зритель¬
ными) с вторичной генерализацией, межприступными
височно-затылочными эпилептиформными ЭЭГ-призна¬
ками и доброкачественным течением. Тип наследования
аутосомно-доминантный, пенетрантность мутантного
гена составляет около 80%. Заболевание сцеплено с хро¬
мосомным сегментом 10q23.3-24.1, содержащим ряд
подходящих генов-кандидатов (гены кальциевых и ка¬
лиевых каналов, нейротрансмиттерных рецепторов и др.)
[Ottman R. et al., 1995; Poza J. et al., 1999].Семейная парциальная эпилепсия с вариабельны¬
ми очагами представляет собой редкое заболевание, на¬
чинающееся в возрасте от 5 до 25 лет и характеризую¬
щееся повторными (преимущественно ночными) парци¬
альными припадками с локализацией эпилептических
очагов в лобных, височных и значительно реже затылоч¬
ных отделах мозга [Xiong L. et al., 1999]. Наследование
аутосомно-доминантное с неполной пенетрантностью
мутантного гена, характерен благоприятный ответ на
лечение традиционными антиконвульсантами. Основной
локус данного заболевания картирован в хромосомной
области 22ql 1—12 [Xiong L. et al., 1999]; имеются пред¬
варительные данные о локализации второго генетичес¬
ДНК- диагностика наследственных болезней ... 379кого локуса парциальной эпилепсии с вариабельными
очагами на хромосоме 2 [Scheffer I. et al., 1998].Роландическая эпилепсия (доброкачественная
эпилепсия 'детского возраста с центротемпоральными
спайками) связана с наличием эпилептического очага в
мозговой коре на одной стороне центральной борозды
[Карлов В.А., 1990]. Клиническая картина характеризует¬
ся односторонними лицевыми и фарингооральными при¬
падками и центротемпоральными пиками и высокоамп¬
литудными острыми волнами на ЭЭГ; прогноз заболе¬
вания благоприятный, припадки обычно прекращаются
к 15 годам. В значительной части случаев роландичес¬
кая эпилепсия манифестирует как менделирутощее ауто-
сомно-доминантное заболевание. Мутантный ген лока¬
лизован в хромосомной области 15ql4 [Neubauer В. et
al., 1998]. Совсем недавно была описана аутосомно-ре-
цессивная форма роландической эпилепсии в сочетании
с пароксизмальной дистонией и писчим спазмом, ген
которой картирован на хромосоме 16р 11.2-12 [Guerrini
R. et al., 1999]. В этом же локусе расположен ген добро¬
качественных младенческих судорог с пароксизмальным
хореоатетозом (см. выше), поэтому нельзя исключить
возможности того, что данные заболевания являются
аллельными.Для всех перечисленных форм наследственных
эпилепсий принципиально возможно проведение косвен¬
ной ДНК-диагностики, которая может оказаться весьма
ценной для выявления в семье асимптомных носителей
мутантной хромосомы, нуждающихся в тщательном на¬
блюдении и определенных рекомендациях в отношении
стиля жизни, профессиональной ориентации и т.п. На
практике, однако, такая диагностика практически не
проводится из-за крайней редкости необходимых для
380Глава 3этого информативных семей с несколькими больными
родственниками (это связано не только с небольшим
размером семей, но и с относительно низкой пенетрант-
ностью мутантного гена при большинстве форм наслед¬
ственных идиопатических эпилепсий).3.6.2. Симптоматические наследственные
эпилепсии: синдром прогрессирующей
миоклонус-эпилепсииСиндром прогрессирующей миоклонус-эпилеп¬
сии - гетерогенная группа фатальных дегенеративных
заболеваний ЦНС, характеризующихся миоклоническим
гиперкинезом, эпилептическими припадками и нарас¬
тающими признаками тяжелого мультисистемного по¬
ражения мозга [Вегкхтс Б. е1 а1., 1993]. Большинство
заболеваний, проявляющихся синдромом прогрессиру¬
ющей миоклонус-эпилепсии, носят наследственный мо-
ногенный характер. Систематизация данной группы за¬
болеваний стала возможной благодаря решающему вкла¬
ду молекулярно-генетических методов исследований.3.6.2.1. Миоклонус-эпилепсия Унферрихта-
ЛундборгаМиоклонус-эпилепсия Унферрихта-Лундборга
(болезнь Унферрихта-Лундборга) - относительно ред¬
кое наследственное заболевание нервной системы, пе¬
редающееся по аутосомно-рецессивному типу. Распрос¬
траненность его составляет около 1 случая на 100 ООО
населения, а в некоторых популяциях (Финляндия, се¬
верная Африка) - гораздо выше (1:20 ООО) [ОеШоп Р. е!
а1., 1990; Вегкоую Б. е1 а1., 1993]. Заболевание начинает¬
ся в возрасте 6-15 лет и проявляется сочетанием фото-
и аудио сенситивных миоклоний действия с миоклони-
ДНК- диагностика наследственных болезней ... 381ческими и тонико-клоническими эпилептическими при¬
падками. Типично чередование «благоприятных» и «не¬
благоприятных» дней, заметно различающихся между
собой по степени выраженности и частоте миоклоний и
эпилептических пароксизмов. Дополнительными прояв¬
лениями болезни на ее поздней стадии могут быть атак¬
сия, пирамидные симптомы и изменения личности. Бо¬
лезнь Унферрихта-Лундборга характеризуется медлен¬
но прогрессирующим течением, продолжительность за¬
болевания варьирует от 10 до 30 лет. На секции выявля¬
ется гибель нейронов и глиоз в зубчатом ядре и коре моз¬
жечка, медиальных отделах зрительного бугра, нижних
оливах, спинном мозге.Ген, ответственный за возникновение миоклонус-
эпилепсии Унферрихта-Лундборга (СБТб), локализован
на 21-й хромосоме в локусе 2lq22.3 [ЬеЬез^а А. е1 а1.,
1993]. Молекулярный продукт гена - белок цистатин В,
являющийся ингибитором ряда внутриклеточных лизо-
сомальных протеаз [РеппассЫо Ь. е! а1., 1996]. Около 90%
случаев заболевания обусловлены резким увеличением
числа копий (экспансией) тандемно повторяющегося 12-
нуклеотидного участка в некодирующей 5’-области гена:
в норме имеется 2-3 копии данного повтора, а у боль¬
ных число копий достигает 90, что приводит к патоло¬
гическому удлинению мутантного аллеля на 600-900 п.о.
и подавлению транскрипции гена. У остальных больных
обнаруживаются точковые мутации в пределах кодиру¬
ющей рамки (к настоящему времени описано 5 таких
мутаций) [РеппассЫо Г. е! а1., 1996; Ьайгешёге Я. е! а1.,
1997; Укгапеуа К. е1 а1., 1997]. Конечным результатом
указанных видов мутаций является угнетение экспрес¬
сии и внутриклеточных регуляторных функций циста-
гина В. После установления молекулярной основы ми-
оклонус-эпилепсии Унферрихта-Лундборга было дока¬
382Глава 3зано, что выделявшиеся ранее «балтийский» и «среди¬
земноморский» варианты болезни не имеют самостоя¬
тельного нозологического значения и являются аллель¬
ными синдромами [Ьайгешеге Я. е1 а1., 1997].Возможность сравнительно простой прямой
ДНК-диагностики болезни Унферрихта-Лундборга яв¬
ляется весьма ценной, поскольку ранее данный диагноз
мог быть поставлен только на основании исключения
целого ряда других причин прогрессирующей миокло-
нус-эпилепсии (см. ниже), что требовало проведения
большого числа трудоемких и инвазивных обследований
(биопсия мышцы, кожи, ректальной субмукозной обо¬
лочки, биохимический скрининг ряда болезней накоп¬
ления и т.д.).1 2 3 4 5 6 78 91011Рис. 66. Прямая ДНК-диагностика миоклонус-эпилепсии
Унферрихта -ЛундборгаДорожка 1 -контроль, дорожки 3-5-больные миоклонус-эпилеп-
сией Унферрихта-Лундборга (отсутствие продукта амплификации
вследствие экспансии 12-нуклеотидного участка в 5’-области гена
цисгатина В), дорожки 2,6-11 - больные с другими формами миок¬
лонус-эпилепсии. Стрелкой указан продукт амплификации 5’-обла-
сти гена, имеющий нормальный размер.ДНК-диагностика болезни Унферрихта-Лундбор¬
га заключается в ПЦР-амплификации участка гена, со¬
держащего область тандемных повторов, с последующим
разделением продуктов реакции в агарозном геле. Экс-■Ир!тт
ДНК- диагностика наследственных болезней ... 383пандированная область мутантного аллеля богата Об¬
основаниями и является в связи с этим ПЦР-резистент-
ной, поэтому диагноз подтверждается на основании от¬
сутствия на электрофореграмме продукта амплификации
(рис. 66). Альтернативным методом является блот-гиб-
ридизация обработанной рестриктазами геномной ДНК
с соответствующим зондом, что позволяет непосред¬
ственно визуализировать удлиненный мутантный аллель.
В небольшом числе случаев, обусловленных точковыми
мутациями гена С8Т6, ДНК-диагностика болезни осно¬
вывается на прямом секвенировании кодирующей обла¬
сти гена. Под наблюдением нейрогенетического отделе¬
ния НИИ неврологии РАМН находится большая группа
больных с синдромом прогрессирующей миоклонус-эпи-
лепсии. По нашим данным, не менее чем в четверти слу¬
чаев заболевание обусловлено мутациями в гене СБТб.
В качестве иллюстрации приводим следующее клини¬
ческое наблюдение.Больной Ч.А., 16лет, с 8-летнего возраста стра¬
дает генерализованными судорожными припадками с
утратой сознания, с этого же времени отмечаются
миоклонии рук при целенаправленных движениях, спус¬
тя 2 года присоединились нарушения походки, речи, рас¬
стройства памяти. На фоне противосудорожной те¬
рапии (бензонал, финлепсин, клоназепам, позднее — де-
пакин хроно 900 мг/сутки) припадки повторялись с ча¬
стотой 1 в 2 месяца, отмечена четкая тенденция к уси¬
лению выраженности миоклоний, их генерализации,
нарастанию тяжести атаксии и когнитивных наруше¬
ний. При поступлении в неврологическом статусе: вер¬
тикальный нистагм с ротаторным компонентом, па¬
рез конвергенции; речь дизартричная, эксплозивная,
тремор языка. Распространенные миоклонические ги-
перкинезы оромандибулярной мускулатуры, мышц шеи,
верхних и нижних конечностей, усиливающиеся при
384Глава 3постуральном напряжении, волнении. Среднеамплитуд-
ный статокинетический тремор конечностей, нечет¬
кость и интенционный тремор при выполнении коор-
динаторных проб, шаткость в позе Ромберга и при ходь¬
бе. Отмечается снижение круга интересов, памяти,
ограничение абстрактного мышления, депрессивный
фон настроения. При компьютерной томографии го¬
ловного мозга выявлено расширение субарахноидалъного
пространства червя мозжечка и больших полушарий,
расширение цистерн моста и межполушарной щели.
ЭЭГ-исследование: выраженные диффузные изменения
биоэлектрической активности мозга с признаками вов¬
лечения мезенцефалъных структур, эпилептиформная
активность в лобно-передневисочных отделах левого
полушария, фотопароксизмалъныереакции, нарастание
множественных спайков при гипервентиляции.Больному проведена биопсия кожи, не выявив¬
шая изменений, характерных для нейрональных церо-
ид-липофусцинозов (см. далее). Содержание лактата и
пирувата в крови — в пределах нормы. При ДНК-иссле-
довании выявлена экспансия минисателлитного повто¬
ра в 5 '-области гена СБТб в гомозиготном состоянии.
Клинический диагноз: миоклонус-эпилепсия Унферрих-
та—Лундборга.3.6.2.2. Миоклонус-эпилепсия с рваными краснымиволокнами (синдром МЕЛКР)Данное сравнительно недавно описанное заболе¬
вание с материнским типом наследования относится к
группе митохондриальных энцефаломиопатий и деталь¬
но рассматривается в разделе 3.8.3.6.2.3. Наследственные болезни накопленияНейрональные цероид-липофусцинозы. К нейро¬
ДНК- диагностика наследственных болезней ... 385нальным цероид-липофусцинозам (общее название -
болезнь Баттена) относится группа наследственных бо¬
лезней обмена, характеризующихся накоплением аутоф-
люресцентных липопигментов в нейронах, а также дру¬
гих клетках различных тканей организма (кожа и слизи¬
стые оболочки, костный мозг, печень, селезенка, почки,
лиматические узлы). Нейрональные цероид-липофусци-
нозы являются одними из наиболее частых нейродеге-
неративных заболеваний детского возраста (приблизи¬
тельно 1 на 12 500 новорожденных) и обусловливают до
четверти всех случаев наследственных заболеваний не¬
рвной системы у детей [Rider J., Rider D., 1988;
Boustany R., Kolodny E., 1989]. В соответствии с возрас¬
том появления первых симптомов, клинической карти¬
ной и характером наследования, выделяют несколько
основных типов нейрональных цероид-липофусцинозов-
инфантильный (болезнь Сантавуори), поздний инфан¬
тильный (тип Янского-Бильшовского), ювенильный (бо¬
лезнь Шпильмейера-Фогта), взрослый (болезнь Куфса)
и некоторые другие [Berkovic S. et al., 1993]. Все пере¬
численные варианты нейрональных цероид-липофусци-
нозов наследуются по аутосомно-рецессивному типу, а
болезнь Куфса - по аутосомно-рецессивному и по ауто-
сомно-доминантному типам. В наиболее общем виде кли¬
ническая картина нейрональных цероид-липофусцино-
зов характеризуется миоклониями, полиморфными ре¬
зистентными эпилептическими припадками (тонико-кло-
ническими, атоническими, атипичными абсансами), рег¬
рессом психомоторного развития, спастичностью, пиг¬
ментной дегенерацией сетчатки, атрофией зрительных
нервов. Постепенно нарастает диффузная атрофия коры
больших полушарий, визуализируемая на рентгеновских
и магнитно-резонансных компьютерных томограммах.
Длительность заболевания от появления первых симп¬
томов до фатального исхода составляет от 5 до 15 лет,
386Глава 3при поздних формах течение более медленное. Важней¬
шее место в диагностике нейрональных цероид-липофус-
цинозов имеет ультраструктурное исследование биопта-
тов кожи и ректальной слизистой, которое позволяет
обнаружить характерные интранейрональные лизосо-
мальные включения, имеющие вид гранулярных осмио-
фильных образований, криволинейных профилей или
«отпечатков пальцев».К настоящему времени показано существование,
как минимум, 6 генов, мутации которых могут приво¬
дить к развитию нейрональных цероид-липофусцинозов
[МПсЫэоп Н. е1 а1., 1998; 8ауикоз1<а М. еХ а1., 1998;
ЗеггаЮза I. е1 а1., 1999; ЯаШа Б. е1 а1., 1999]. Пять из этих
генов клонированы и отвечают за синтез различных ли-
зосомальных белков двух видов:1) лизосомальные ферменты - пальмитоил-про-
теинтиоэстераза (ген СЬШ на хромосоме 1р32) и три-
пептидил-пептидаза I (ген СЬЫ2 на хромосоме 11р15);2) структурные трансмембранные лизосомаль¬
ные белки (гены СЬ№ на хромосоме \3q22, СЬ№ на
хромосоме 1бр12 и СЬШ на хромосоме 8р23). Еще один
локус (СЕМ6) картирован на хромосоме 15q21—23.У больных с различными вариантами нейрональ¬
ных цероид-липофусцинозов идентифицировано около
75 разных мутаций в указанных генах, причем большин¬
ство этих мутаций являются уникальными для конкрет¬
ных обследованных семей.Болезнь Лафоры. Болезнь Лафоры представляем
собой редкое заболевание нервной системы с аутосом-
но-рецессивным типом наследования. Первые симпто
мы появляются обычно между 10 и 18 годами жизни,
заболевание проявляется типичной для миоклонус-эпи
лепсий триадой симптомов - миоклониями, тонико-кло
ническими припадками и прогрессирующим снижени
ДНК- диагностика наследственных болезней387ем когнитивных функций вплоть до тяжелой деменции.
Особенностью данного заболевания является частое со¬
четание генерализованных тонико-клонических припад¬
ков с фокальными затылочными пароксизмами (зритель¬
ные галлюцинации, скотомы, преходящая корковая сле¬
пота). В развернутой стадии у больных нередко развива¬
ются также мозжечковая атаксия, атрофия зрительных
нервов, дегенерация сетчатки, расстройства сфинктеров.
При использовании методов нейровизуализации обна¬
руживаются диффузные атрофические изменения полу¬
шарий большого мозга и мозжечка. Прогноз неблагоп¬
риятный, больные обычно погибают через 2-12 лет от
начала заболевания; в редких случаях с поздним нача¬
лом (после 20 лет) длительность заболевания может до¬
стигать нескольких десятилетий [Вегкоую Б. е1 а1., 1993].
Специфическим морфологическим признаком болезни
является наличие в нейронах ЦНС и клетках ряда дру¬
гих тканей особых гранулярно-филаментозных включе¬
ний - телец Лафоры. В этих включениях определяется
накопление разветвленных амилопектин-подобных по¬
лисахаридов. Соответственно, основным методом при¬
жизненной диагностики болезни является обнаружение11 ри световой микроскопии телец Лафоры в клетках про¬
токов потовых желез, для чего проводится биопсия кожи.I ен лафорин, обусловливающий развитие примерно 80%
случаев болезни Лафоры, локализован на хромосоме
Ц24. Он отвечает за синтез белковой тирозин-фосфата-
1Ы, играющей важную роль в контроле метаболизма гли¬
когена [БеггаЮза I. е1 а1., 1999]. Имеются данные о суще¬
ствовании, как минимум, еще одного локуса болезни
Пафоры [Мтазз1ап В. еХ а1., 1999].Сиалидозы. Сиалидозы представляют собой ред¬
кие аутосомно-рецессивные болезни обмена, проявляю¬
щиеся синдромом прогрессирующей миоклонус-эпилеп-
388Глава 3сии, расстройством зрения (симптом «вишневой косточ¬
ки»), периферической и кохлеарной невропатией, лице¬
вым дизморфизмом и другими симптомами. Начало за¬
болевания приходится на детский и подростковый воз¬
раст, его продолжительность при большинстве форм
сиалидозов не превышает нескольких лет. В основе па¬
тогенеза сиалидозов лежит недостаточноть лизосомаль-
ной а-УУ-ацетилнейраминидазы (сиалидазы), ведущая к
накоплению гликопротеидов, олигосахаридов и глико¬
липидов в ряде органов и тканей [Всгко\тс Б. е1 а1., 1993].
Диагностическое значение при сиалидозах имеет уста¬
новление снижения активности нейраминидазы и (при
некоторых формах) (3-галактозидазы в лейкоцитах и фиб-
робластах, определенную роль играет также обнаруже¬
ние повышенной экскреции олигосахаридов с мочой.
Заболевание может быть обусловлено как первичным
дефицитом нейраминидазы, связанным с мутацией его
гена на хромосоме 6р21.3, так и с дефектом белка катеп-
сина А, предотвращающего деградацию стабилизатора
нейраминидазы (соответствующий ген локализован на
хромосоме 2(^13.1) [Ра1теп е1 а1., 1986]. В последнем
случае у больных имеет место сочетанный дефицит ней¬
раминидазы и (3-галактозидазы (галактосиалидоз).Синдром прогрессирующей миоклонус-эпилеп-
сии может иногда наблюдаться при ряде других наслед¬
ственных болезней обмена - СМг-ганглиозидозе (забо¬
левание обусловлено недостаточностью фермента (3-га-
лактозидазы), болезни Гоше (недостаточность (3-глюко-
цереброзидазы), болезни Краббе (форма лейкодистро-
фии, обусловленная недостаточностью галактоцеребро-
зидазы) и др.Методы ДНК-диагностики прогрессирующих
миоклонус-эпилепсий, относящихся к наследственным
болезням накопления, на практике не получили пока
ДНК- диагностика наследственных болезней ... 389широкого распространения. Традиционная диагностика
данных заболеваний базируется на проведении соответ¬
ствующих биохимических и морфологических тестов.3.7. ФакоматозыТермин «факоматозы» (от греч. phakos - пятно)
объединяет группу генетически обусловленных прогрес¬
сирующих заболеваний, представляющих собой дис- и
гиперплазии эктодермальных и мезодермальных тканей
(нервная система, кожа, сетчатка, внутренние органы).
Облигатным проявлением факоматозов, определившим
общее название данных болезней, служат особые пиг¬
ментные пятна и ангиомы на коже и/или сетчатке глаза.
Весьма характерной для факоматозов является также
склонность к развитию разнообразных доброкачествен¬
ных и злокачественных опухолей. В исследованиях пос¬
ледних лет было показано, что молекулярной основой
факоматозов является повреждение генов-супрессоров
опухолевого роста, контролирующих различные стадии
клеточной пролиферации и дифференцировки.3.7.1. НейрофиброматозНейрофиброматоз (болезнь Реклингхаузена) пред¬
ставляет собой наиболее распространенную форму фа¬
коматозов и встречается в популяции с частотой при¬
близительно 1:3500. Заболевание характеризуется раз¬
витием опухолей преимущественно эктодермального
происхождения с поражением нервов, кожи и ЦНС, на¬
личием типичных кожных пигментных пятен типа «кофе
с молоком» (café au lait), аномалиями костного скелета,
а также рядом других клинических проявлений. Тип на¬
следования аутосомно-доминантный с пенетрантностью,
390Глава 3близкой к 100%. В зависимости от распространенности
и локализации новообразований заболевание подразде¬
ляется на периферическую и центральную форму - со¬
ответственно, нейрофиброматоз-1 и нейрофиброматоз-2
[ОШтапп О. еТ а1., 1997]. Обоснованность такого под¬
разделения нейрофиброматоза на самостоятельные фор¬
мы подтверждается и на молекулярном уровне благода¬
ря идентификации соответствующих мутантных генов.3.7.1.1. Нейрофиброматоз 1-го типаНейрофиброматоз 1-го типа («классический»)
проявляется множественными нейрофибромами по ходу
периферических нервов, которые определяются в виде
безболезненных округлых узелков в толще кожи, варьи¬
рующих по своим размерам и локализации (туловище,
конечности, краниоцервикальная область). В некоторых
случаях нейрофиброматоз может носить весьма ограни¬
ченный сегментарный характер (например, медиасти-
нальные нейрофибромы в сочетании с опухолями в со¬
ответствующем кожном сегменте), однако гораздо чаще
он является генерализованным. Сегментарный нейро¬
фиброматоз связывают с соматическими мутациями на
определенной стадии эмбриогенеза, приводящими к вов¬
лечению ограниченного клона клеток и соматическому
мозаицизму. Описан нейрофиброматоз толстого и тон¬
кого кишечника с повторными кровотечениями. Неред¬
ко появление нейрофибром сопровождается гипертро¬
фией пораженных участков тела (слоновость) и внутрен¬
них органов. Типичные для нейрофиброматоза-1 пигмен¬
тные проявления носят характер пятен цвета кофе с мо¬
локом и «веснушчатых гроздьев» на коже, а также узел¬
ков Лиша - патогномоничных белесоватых пятен на ра¬
дужке (гамартом). У 3-15% больных нейрофибромы
имеют склонность к злокачественному перерождению.
ДНК- диагностика наследственных болезней ... 391Несмотря на «периферический» характер нейрофибро-
матоза-1, у части больных может наблюдаться вовлече¬
ние ЦНС с развитием опухолей другой гистологичес¬
кой природы - астроцитом зрительных путей, эпенди-
мом, менингиом. Для нейрофиброматоза-1 характерен
также ряд дополнительных клинических проявлений,
таких как умственная отсталость, эндокринные расстрой¬
ства (феохромоцитома, нарушение роста и полового со¬
зревания), аномалии развития сосудов (коарктация аор¬
ты, сосудистые аневризмы), изменения скелета (сколи¬
оз, псевдоартроз), эпилептические припадки и др.
[McGaughran J. et al., 1999; DeBella К. et al., 2000].Еще до открытия гена нейрофиброматоза-1 было
установлено, что при данном аутосомно-доминантном
заболевании нередкое выявление больных, не имеющих
семейного анамнеза, может быть связано с высокой час¬
тотой возникновения новых мутаций [Huson S. et al.,
1989]. Темп мутирования гена нейрофиброматоза-1 яв¬
ляется одним из наиболее высоких при всех известных
заболеваниях человека (до 6,5х 10“5 гамет на поколение,
или почти 1 на 10 ООО гамет), а примерно 50% случаев
заболевания представляют собой мутации de novo
[Samuelsson В., Akesson Н., 1988; Huson S. et al., 1989;
Clementi M. et al., 1990]. Такая высокая частота спонтан¬
ных мутаций может объясняться очень большими раз¬
мерами гена и/или определенными особенностями его
внутренней структуры. Данное предположение блестя¬
ще подтвердилось в 1990-1995 г., когда ген заболевания
(NF1), локализованный на хромосоме 17ql 1.2, был вы¬
делен и полностью охарактеризован [Wallace М. et al.,
1990; Marchuk D. et al., 1991; Li Y. et al., 1995].Ген NF1 действительно является чрезвычайно
протяженным и сложно организованным: он имеет дли¬
ну около 350 кб, состоит из 60 экзонов и экспрессирует¬
392Глава 3ся, помимо нервной системы, в большом числе различ¬
ных тканей [Li Y. et al., 1995; Shen M. et al., 1996]. Ген
NF1 кодирует белок нейрофибромин, являющийся суп¬
рессором опухолевого роста. Нейрофибромин содержит
в своем составе особый домен, общий для семейства
белков-активаторов ГТФ-азы; посредством этого доме¬
на нейрофибромин в норме взаимодействует с продук¬
том протоонкогена RAS, ингибируя его функцию и та¬
ким образом реализуя свой супрессорный эффект в от¬
ношении клеточной пролиферации [Shen М. et al., 1996;
Gutmann D., 1998]. У больных нейрофиброматозом-1
идентифировано свыше 200 различных мутаций в гене
NF1, нарушающих его регулирующую роль в каскаде
событий онкогенеза. Характер мутаций весьма специ¬
фичен: более 80% из них ведут к синтезу нефункцио¬
нального «усеченного» белка либо к полному отсутствию
транскрипта (нонсенс-мутации, мутации в сайтах сплай¬
синга, делеции и вставки со сдвигом рамки, крупные
делеции, охватывающие весь ген или его значительную
часть) [Heim R. et al., 1994; Abernathy C. et al., 1997;
Osborn M., Upadhyaya M., 1999; Ars E. et al., 2000;
Messiaen L. et al., 2000]. Остальные мутации представ¬
ляют собой внутренние делеции без сдвига рамки и мис-
сенс-мутации, затрагивающие функционально важные
участки нейрофибромина (например, вышеупомянутый
ГТФ-азный домен) [Fahsold R., 2000]. Мутации распре¬
делены в пределах кодирующей области NF 1 достаточ¬
но равномерно, и лишь экзоны 10а-10с, 31 и 37 пред¬
ставляют собой области с относительно высокой часто¬
той повреждения (от 6% до 30% всех выявленных мута¬
ций) [Messiaen L. et al., 1999; 2000]. Описаны также слу¬
чаи нейрофиброматоза-1, обусловленного цитогенетичес¬
кими перестройками, затрагивающими критический хро¬
мосомный сегмент 17q 11.2 [Ledbetter D. et al., 1989;
ДНК- диагностика наследственных болезней ... 393Messiaen L. et al., 2000].Рядом исследователей было показано, что фор¬
мирование и особенно злокачественная трансформация
нейрофибром обусловлены инактивацией второй копии
гена NF1 или повреждением другого гена-супрессора
опухоли (р53) в клеточном клоне в результате сомати¬
ческой мутации [Menon A. et al., 1990; GutmarmD., 1998;
Eisenbarth I. et al., 2000]. По мнению I. Eisenbarth и соав¬
торов (2000), разнообразие дополнительных соматичес¬
ких мутаций в различных опухолях у больных нейро-
фиброматозом-1 является одной из основных причин
фенотипического полиморфизма заболевания. Таким
образом, в развитии нейрофибром может иметь значе¬
ние «двойная инактивация» reHaNFl как следствие двух
самостоятельных мутаций, одна из которых унаследова¬
на, а вторая возникает de novo в определенной опухоле¬
вой клетке на постзиготической стадии. Роль каскада
мутационных событий в процессе злокачественной эво¬
люции опухолей нервной системы более детально ана¬
лизируется в главе 4.Идентификация мутаций в гене NF1 и прямая
ДНК-диагностика нейрофиброматоза 1-го типа значи¬
тельно затрудняются гигантским размером гена, высо¬
кой частотой мутаций de novo, фактическим отсутстви¬
ем мажорных мутаций, а также наличием большого числа
гомологичных локусов. Принимая во внимание тот факт,
что абсолютное большинство мутаций в гене NF1 при¬
водят к синтезу «усеченного» нейрофибромина, мута¬
ционный анализ проводится в первую очередь на РНК/
белковом уровне с помощью РТТ-метода (Protein
Truncation Test): из мРНК лейкоцитов крови путем об-
ратно-транкриптазной ПЦР амплифицируют отдельные
участки кДНК, встраивают их в экспрессионную систе¬
му и анализируют образующийся белковый продукт
394Глава 3[Heim R. et al., 1994; Osbom M., Upadhyaya M., 1999;
Fahsold R., 2000; Messiaen L. et al., 2000]. Данный метод
может дополняться целым рядом других традиционных
технологий мутационного скрининга - таких как SSCP,
гетеродуплексный анализ, градиентный денатурирующий
гель-электрофорез, блот-гибридизация, прямое секвени-
рование отдельных экзонов гена, а также (с учетом ве¬
роятности хромосомных перестроек) флюоресцентная
гибридизация in situ и цитогенетический анализ
[Abernathy C. et al., 1997; Fahsold R., 2000; Messiaen L. et
al., 2000]. Использование различной комбинации пере¬
численных методов позволяет выявлять мутации в гене
NF1 в 47-95% случаев нейрофиброматоза 1-го типа. К
настоящему времени в мире прямая ДНК-диагностика
данного заболевания успешно проведена у нескольких
сотен больных.При наличии в обследуемой семье одного или
нескольких больных с типичной клинической картиной
нейрофиброматоза-1 предсказательное тестирование у
лиц из «группы риска» и пренатальное тестирование
может осуществляться с помощью косвенной ДНК-ди-
агностики (анализ генетических маркеров из области
17ql 1.2). Для нейрофиброматоза-1, с его большим ге¬
ном и обилием редких мутаций, косвенная ДНК-диаг¬
ностика является значительно более простой и эконом¬
ной по сравнению с прямой, поэтому она не потеряла
своего значения и сегодня. Тесное сцепление используе¬
мых маркеров с геном NF1 позволяет проводить косвен¬
ную ДНК-диагностику нейрофиброматоза-1 с точностью
свыше 98% [Ward K. et al., 1990].3.7.1.2. Нейрофиброматоз 2-го типаНейрофиброматоз-2 (центральная форма нейро¬
фиброматоза) характеризуется развитием 2-сторонних
ДНК- диагностика наследственных болезней ... 395иеврином (шванном) VIII пары черепно-мозговых нервов,
реже задних корешков спинного мозга, а также цереб¬
ральных менингиом [Gutmann D. et al., 1997]. Неврино-
ма слуховых нервов может быть единственным проявле¬
нием заболевания. Лишь у небольшого числа больных
выявляются единичные кожные пятна цвета кофе с мо¬
локом и опухоли периферических нервов. К важным
диагностическим признакам нейрофиброматоза-2 отно¬
сятся задняя субкапсулярная катаракта и ретинальные
гамартомы, выявляемые, соответственно, в 67% и 22%
случаев [Ragge N. et al., 1995]. Обычно симптомы забо¬
левания манифестируют после 20 лет. Таким образом,
клиническая картина нейрофиброматоза-2 заметно от¬
личается от таковой у больных нейрофиброматозом-1.
В связи с этим считается, что термин «болезнь Реклин-
гхаузена» к пациентам с нейрофиброматозом-2 непри¬
меним и должен быть зарезервирован только для «клас¬
сических» случаев периферического нейрофиброматоза
1-го типа.Ген нейрофиброматоза-2 (NF2) локализован на
хромосоме 22ql2.2 и был идентифицирован в 1993 году
двумя исследовательскими группами [Rouleau G. et al.,
1993; Trofatter J. et al., 1993]. Продукт данного гена со¬
стоит из 587 аминокислот и имеет значительную гомо¬
логию с белками ERM-семейства, обеспечивающими
связь компонентов цитоскелета с белками клеточной
мембраны. J. Trofatter с соавторами предложили для обо¬
значения кодируемого геном NF2 белка название «мер-
лин» (акроним от названий белков ERM-семейства), тог¬
да как группой G. Rouleau было предложено название
«шванномин». Аналогично нейрофибромину, белок мер-
лин (шванномин) является ингибитором опухолевого
роста. Предполагается, что влияние данного белка на
клеточный рост реализуется через специфические пере¬
396Глава 3стройки цитоскелета клетки и/или нарушение клеточ¬
ной адгезии [Gutmann D. et al., 1999; Stokowski R., Cox D.,2000]. В настоящее время имеется большое число дан¬
ных, свидетельствующих о том, что для формирования
шванном и менингиом необходима «двойная инактива¬
ция» гена NF2 в клетках опухолевого клона [Trofatter J.
et al., 1993; Sainz J. et al., 1994]. В типичных случаях
нейрофиброматоза-2 одна из мутаций является наследу¬
емой, тогда как второй аллель утрачивается на постзи-
готической стадии в результате спонтанной делении уча¬
стка 22ц12.2-локуса, содержащего ген NF2. Показано,
что инактивация нормального аллеля (унаследованного
от здорового родителя) в результате спонтанной деле¬
нии гена в клетках опухолевого клона обнаруживается в
большинстве образцов опухолей, полученных от боль¬
ных нейрофиброматозом-2 [Rouleau G. et al., 1993;
Trofatter J. et al., 1993; Kluwe L. et al., 2000]. Более того,
инактивация гена NF2 в опухолевых клетках выявляет¬
ся также у многих больных со спорадическими форма¬
ми менингиом, шванном, меланом, карцином молочной
железы, злокачественных мезотелиом и некоторых дру¬
гих видов опухолей [Bianchi A. et al., 1994; 1995;
Ruttledge М. et al., 1994; Sainz J. et al., 1994;
Wellenreuther R. et al., 1995]. Это подтверждает универ¬
сальное значение повреждений гена NF2 в механизмах
онкогенеза как при наследственных (нейрофиброматоз-2),
так и при спорадических формах новообразований.У больных нейрофиброматозом-2 идентифици¬
ровано несколько десятков мутаций, расположенных
практически во всех 17 экзонах гена NF2. Примерно
половина из них являются точковыми мутациями (в боль¬
шинстве случаев ведущими к синтезу нефункциональ¬
ного «усеченного» белка), тогда как 1/3 мутаций пред¬
ставляют собой крупные геномные делеции в локусе
ДНК- диагностика наследственных болезней ... 39722ql2.2 [Rouleau G. et al., 1993; Evans D. et al., 1998;
Zucman-Rossi J. et al., 1998; Legoix R et al., 1999]. Мута¬
ции с преждевременным обрывом трансляции сопровож¬
даются развитием более тяжелого и раннего фенотипа
болезни, тогда как относительно доброкачественные
формы нейрофиброматоза-2 характерны для миссенс-
мутаций, меняющих ту или структурную аминокислоту
белка [Parry D. et al., 1996; Ruttledge M. et al., 1996;
Evans D. et al., 1998].При проведении прямой ДНК-диагностики ней¬
рофиброматоза-2 обычно используется SSCP-анализ эк-
зонов гена NF2 с последующим секвенированием му¬
тантных образцов ДНК. Кроме того, учитывая возмож¬
ность крупных делеций с вовлечением reHaNF2, Р. Legoix
с соавторами (1999) предложили исследовать серию внут-
ригенных полиморфных маркеров с целью выявления
феномена потери гетерозиготности (утраты аллеля). Со¬
временный комбинированный подход позволяет при ис¬
следовании клеток крови идентифицировать мутации у
34-84% больных нейрофиброматозом-2, причем выяв-
ляемость мутаций в среднем почти вдвое выше при се¬
мейных формах заболевания по сравнению со споради¬
ческими [Evans D. et al., 1998; Kluwe L., Mautner V.-F.,
1998; Zucman-Rossi J. et al., 1998]. Такая разница обус¬
ловлена наличием у ряда спорадических больных сома¬
тического мозаицизма по мутации, возникающего при
повреждении гена NF2 в на постзиготической стадии
[Evans D. et al., 1998; Kluwe L., Mautner V.-F., 1998]. Вы¬
сокая частота соматического мозаицизма существенно
осложняет медико-генетическое консультирование у
больных нейрофиброматозом-2. Поскольку отсутствие
мутации в лейкоцитарной ДНК не исключает возмож¬
ности ее существования в пораженных тнанях, в спор¬
ных диагностических случаях для повышения чувстви¬
398Глава 3тельности мутационного скрининга может быть реко¬
мендовано дополнительное исследование гена NF2 в
опухолевой ткани - например, в биоптатах кожных опу¬
холевых узелков [Kluwe L. et al., 2000]. В семейных слу¬
чаях заболевания определенное значение для установ¬
ления генетического статуса родственников из группы
риска имеет и косвенная ДНК-диагностика носительства
мутантной хромосомы, информативность которой, по
данным М. Ruttledge с соавторами (1993), является весь¬
ма высокой.3.7.2. Туберозный склерозТуберозный склероз (болезнь Бурневилля-Прин-
гла) представляет собой заболевание, характеризующее¬
ся формированием в различных органах особых диспла-
стических опухолевидных образований - гамартом.
Частота туберозного склероза составляет около 1 на
10 ООО рожденных детей [Osborne J. et al., 1991]. Тип
наследования аутосомно-доминантный с практически
полной пенетрантностью, однако яркой особенностью
генетики туберозного склероза является чрезвычайно вы¬
сокая частота спорадических случаев (60-70%), обуслов¬
ленных новыми мутациями генов болезни [Sampson J. et
al., 1989]. Соответственно, во многих случаях семей¬
ный анамнез у больных, не имеющих потомства, отсут¬
ствует.Клиническая картина туберозного склероза явля¬
ется весьма полиморфной [Roach Е. et al., 1998]. Наибо¬
лее часто имеет место поражение мозга, кожи, ночек и
сердца, реже в процесс вовлекаются легкие, костный
скелет, селезенка, эндокринные железы и другие орга¬
ны. Заболевание проявляется обычно уже в первые годы
жизни резистентными эпилептическими припадками и
разнообразными кожными изменениями. Эпилептичес¬
кий синдром обусловлен локализацией гамартом (неред¬
ДНК- диагностика наследственных болезней399ко кальцифицированных) в коре больших полушарий;
опухолевые узлы могут располагаться также в белом ве¬
ществе, мозжечке, стволе, спинном мозге, выступать в
полость боковых желудочков. Наблюдаются как длитель¬
ные ремиссии, так и учащение припадков с возрастом.
При тяжелом течении туберозного склероза отмечается
отставание в умственном развитии ребенка. В мозге мо¬
гут развиваться также астроцитомы, эпендимомы. Кож¬
ные изменения являются важнейшим диагностическим
признаком болезни и включают множественные розова¬
то-красные папулезные узелки {adenoma sebaceum) в виде
«бабочки» по обе стороны носа, шагреневые утолщения
кожи, депигментированные и «кофейные» пятна на коже
туловища и конечностей, единичные и множественные
фибромы. Для туберозного склероза характерны также
опухоли сетчатки, рабдомиома сердца, ангиомиолипо-
мы, кисты и карциномы почек, опухоли других внутрен¬
них органов, кистозные изменения фалант пальцев кис¬
ти с участками периостального утолщения и другие про¬
явления. Наиболее ценным методом диагностики явля¬
ется рентгеновская компьютерная томография головно¬
го мозга, позволяющая выявлять двусторонние кальци-
фикаты в перивентрикулярной области, базальных ганг¬
лиях, мозжечке.Туберозный склероз является генетически гете¬
рогенным. Типичный фенотип данного заболевания мо¬
жет быть обусловлен мутациями двух самостоятельных
генов - TSC1 на хромосоме 9q34.1-34.2 и TSC2 на хро¬
мосоме 16р 13.3 [European Chromosome 16 Tuberous
Sclerosis Consortium, 1993; van Slegtenhorst M. etal., 1997].
Среди больных и семей с туберозным склерозом отме¬
чено преобладание 2-го типа заболевания (ген TSC2),
сцепленного с 16-й хромосомой.Ген TSC1 содержит 23 экзона и кодирует новый
белок - гамартин, не имеющий существенной гомоло¬
400Глава 3гии с известными ранее белковыми семействами
[van Slegtenhorst М. et al., 1997]. Ген TSC2 состоит из 41
короткого экзона, имеет сравнительно небольшую про¬
тяженность (около 45 кб) и кодирует транскрипт дли¬
ной 5,5 кб; белковый продукт гена TSC2, туберин, ассо¬
циирован с клеточной мембраной [European Chromosome
16 Tuberous Sclerosis Consortium, 1993; Wienecke R. et al.,1995]. Аналогично белку нейрофибромину, туберин со¬
держит домен с ГТФ-азной активностью, что свидетель¬
ствует о его роли в митогенезе, регуляции клеточного
цикла и нейрональной дифференцировке [Wienecke R.
et al., 1995; Jones A. et al., 1999]. В эксперименте показа¬
но, что гамартин и туберин взаимодействуют друг с дру¬
гом и имеют, по данным иммунофлюоресценции, общую
клеточную локализацию, что может свидетельствовать
о реализации этими белками близких клеточных функ¬
ций [Plank Т. et al., 1998]. Центральной из таких функ¬
ций белковых продуктов TSC1 и TSC2 является их фун¬
кция супрессии опухолевого роста, что получило пря¬
мое подтверждение на трансгенных моделях у живот¬
ных [Kobayashi Т. et al., 1997]. Молекулярно-генетичес¬
кий анализ тканей гамартом и других опухолей у боль¬
ных туберозным склерозом 1-го и 2-го типов показал не
только наличие унаследованного мутантного аллеля гена
(TSC1 или TSC2), но и инактивацию второго аллеля
вследствие спонтанной соматической мутации в опухо¬
левом клоне на постзиготической стадии [Carbonara С.
et al., 1996; Henske E. et al., 1996; Sepp T. et al., 1996;
van Slegtenhorst M. et al., 1997; Au K. et al., 1999]. Вто¬
рая, приобретенная мутация обычно характеризуется ран¬
ним обрывом трансляции либо представляет собой круп¬
ную делецию соответствующего хромосомного участка,
проявляющуюся феноменом потери гетерозиготности.
Интересно, что повторные «комплементирующие» му¬
ДНК- диагностика наследственных болезней ... 401тации у одного и того же больного могут быть различ¬
ными и возникать независимо в разных соматических
клетках, давая начало разным опухолям [Carbonara C. et
al., 1996]. Таким образом, как и при нейрофиброматозе,
у больных туберозным склерозом опухолевая трансфор¬
мация клеток предполагает «двойную инактивацию» и
отсутствие белковых продуктов генов-супрессоров опу¬
холи (при нейрофиброматозе это касается генов NF1 и
NF2, а при туберозном склерозе - TSC1 и TSC2).К настоящему времени в различных популяциях
мира обследованы на молекулярно-генетическом уров¬
не сотни больных туберозным склерозом, у которых
идентифицировано свыше 200 патогенетически значи¬
мых мутаций в генах TSC1 и TSC2 [van Bakel I. et al.,
1997; van Slegtenhorst M. et al., 1997; Kwiatkowska J. et
al., 1998; Jones A. et al., 1999; Mayer K. et al., 1999;NiidaY.
et al., 1999]. Большинство выявленных мутаций состав¬
ляют нонсенс-мутации, мутации со сдвигом рамки, му¬
тации в сайтах сплайсинга с укорочением транскрипта и
крупные геномные перестройки (включая делецию це¬
лого гена), гораздо реже встречаются миссенс-мутации,
ведущие к замене аминокислоты в функционально зна¬
чимых доменах (например, ГТФ-азном домене тубери-
на). Примерно у 80% больных выявляются мутации в
гене TSC2 (туберозный склероз 2-го типа), в остальных
случаях заболевание обусловлено мутациями гена TSC1
(туберозный склероз 1-го типа) [Jones A. et al., 1999;
Niida Y. et al., 1999]. Единственным существенным кли¬
ническим различием между двумя генетическими фор¬
мами туберозного склероза является более тяжелое те¬
чение заболевания у больных с мутациями гена TSC2,
что проявляется, в частности, значительно более высо¬
кой частотой умственной отсталости при туберозном
склерозе-2 [Jones A. et al., 1999]. Отчетливых «горячих
402Глава 3точек» в генах туберозного склероза, имеющих особен¬
но высокий темп мутирования, не выявлено. Мажорные
мутации также практически не зарегистрированы, за ис¬
ключением нескольких точковых мутаций как в TSC1,
так и в TSC2, выявленных различными авторами у ряда
неродственных больных [van Slegtenhorst М. et al., 1997;
Kwiatkowska J. et al., 1998; Jones A. et al., 1999].ДНК-диагностика туберозного склероза представ¬
ляет собой непростою задачу. Поиск мутаций следует
начинать с гена TSC2, поскольку вероятность обнару¬
жения мутации в нем заметно выше, чем для гена TSC1.
Используемые при туберозном склерозе методы мута¬
ционного скрининга практически идентичны для обоих
генов. Учитывая доминирующую роль мутаций, приво¬
дящих к укорочению транскрипта и синтезу «усеченно¬
го» белка, часто используемыми являются РТТ-метод
(анализ белковых продуктов экспрессии кДНК), а также
другие подходы, основанные на РНК-технологиях (об-
ратно-транскриптазная амплификация и анализ набора
фрагментов, покрывающих всю длину кДНК) [Wilson Р.
etal., 1996; van Bakel I. et al., 1997; Mayer K. et al., 1999].
Для выявления мутаций в кодирующих областях иссле¬
дуемых генов обычно используется SSCP, гетеродуплек-
сный анализ и прямое секвенирование отдельных экзо-
нов, а для детекции возможных крупных генных пере¬
строек - электрофорез в пульсирующем поле, блот-гиб-
ридизация по Саузерну и «сверхдлинная» ПЦР
[Kwiatkowska J. et al., 1998; Jones A. et al., 1999; Niida Y.
et al., 1999]. В зависимости от используемой комбина¬
ции методов, мутации в генах TSC1 или TSC2 выявля¬
ются (при исследовании ДНК клеток крови) у 59-80%
всех больных туберозным склерозом и почти в 95% се¬
мейных случаев заболевания [Jones A. et al., 1999;
Mayer K. et al., 1999; Niida Y. et al., 1999]. Более низкая
ДНК- диагностика наследственных болезней ... 403выявляемо сть мутаций в спорадических случаях тубе¬
розного склероза обусловлена частым соматическим мо-
заицизмом у этих больных (мутация происходит в эмб¬
риогенезе вне клеточной закладки кроветворного рост¬
ка) [Jones A. et al., 1999]. Поскольку при туберозном скле¬
розе достаточно высока доля спорадических случаев, а
также в связи с проблемой соматического и гонадного
мозаицизма, косвенная ДНК-диагностика болезни в груп¬
пе риска не получила широкого распространения.3.7.3. Синдром Гиппеля-ЛиндауСиндром Гиппеля-Линдау (церебро-ретино-вис-
церальный ангиоматоз) характеризуется сочетанием ге-
мангиобластом мозжечка и спинного мозга с ангиомато-
зом сетчатки, а также опухолями ряда висцеральных ор¬
ганов [Horton W. et al., 1976]. Ориентировочная частота
заболевания составляет 1 на 36 ООО рожденных детей
[Maher Е. et al., 1991]. Тип наследования аутосомно-до-
минантный с неполной пенетрантностью мутантного
гена. Первые симптомы появляются обычно на втором-
четвертом десятилетии жизни. Клиническая картина ге-
мангиобластом ЦНС вышеуказанной локализации харак¬
теризуется гипертензионным синдромом (наиболее гроз¬
ным является развитие окклюзионной гидроцефалии),
атаксией, нистагмом, дизатрией, спастическим парапа¬
резом. Суммарно, около 14% всех случаев гемангиобла-
стом ЦНС обусловлены синдромом Гиппеля-Линдау
[Maddock I. et al., 1996]. Ангиоретикулема сетчатки мо¬
жет на поздней стадии болезни приводить к отслойке
сетчатки, глаукоме и другим серьезным осложнениям,
вплоть до слепоты. К типичным опухолевым поражени¬
ям внутренних органов при синдроме Гиппеля-Линдау
относятся феохромоцитома, рак почек и поджелудочной
железы, часто обнаруживаются также кистозные изме¬
404Глава 3нения в почках, поджелудочной железе, печени и других
органах. У некоторых больных могут выявляться геман-
гиомы лица, надпочечников, легких, печени.Ген заболевания, локализованный на хромосоме
Зр25-26, является эволюционно консервативным, ши¬
роко экспрессируется в различных тканях и включает 3
небольших экзона, кодирующих белковый продукт из 213
аминокислот [Latif F. et al., 1993]. В современной но¬
менклатуре ген синдрома Гиппеля-Линдау и его белко¬
вый продукт принято обозначать символом VHL (от англ.
Von Hippel-Lindau). VHL является классическим супрес¬
сором опухолевого роста: инактивация обоих аллелей
гена является ключевым событием в патогенезе нео¬
плазм, типичных для синдрома Гиппеля-Линдау (ана¬
логично тому, что имеет место при нейрофиброматозе и
туберозном склерозе) [Iliopoulos О. et al., 1995]. В клет¬
ке, дающей начало опухолевому клону, унаследованная
мутация в одном аллеле гена VHL дополняется постзи-
готической инактивацией нормального аллеля, что про¬
исходит чаще всего в результате делеции критического
участка хромосомы Зр25-26 [Crossey P. et al., 1994 (а);
Prowse A. et al., 1997]. Универсальная роль VHL как суп¬
рессора опухолевого роста подтверждается обнаруже¬
нием полной инактивации данного гена в опухолевой
ткани у ряда больных со спорадическими формами фе-
охромоцитомы, рака толстой кишки, рака почки [Latif F.
et al., 1993; Kenck С. et al., 1996; Zhuang Z. et al., 1996;
Gallou C. et al., 1999]; в этих случаях мутации в обоих
аллелях гена происходят в соматических клетках на по-
стзиготической стадии развития.Механизм противоопухолевого действия белка
VHL был предметом интенсивных экспериментальных
исследований. Установлено, что в норме VHL ингиби
рует удлинение растущей цепи мРНК путем связывания
ДНК- диагностика наследственных болезней ... 405с белками-факторами элонгации - элонгином В и С
[Duan D. et ab, 1995]. Таким образом, инактивация VHL
сопровождается нарушением контроля транскрипции в
клетке. Этот контроль в первую очередь касается генов,
экспрессирующихся в условиях гипоксии и кодирующих
ангиогенные пептиды - например, эндотелиальный фак¬
тор роста, гипоксия-индуцируемый фактор роста-1 и др.
[Iliopoulos О. etal., 1996; Maxwell Р. et al., 1999]. Предпо¬
лагается, что именно неадекватно повышенная экспрес¬
сия ангиогенных пептидов в результате инактивации
VHL лежит в основе формирования сосудистых опухо¬
лей, столь характерных для синдрома Гиппеля-Линдау
[Iliopoulos О. et al., 1996; Mukhopadhyay D. et al., 1997].Спектр мутаций, характерных для синдрома Гип¬
пеля-Линдау, весьма широк: он включает крупные де-
леции всего гена или его части, нонсенс-мутации и му¬
тации со сдвигом рамки, ведущие к преждевременному
обрыву трансляции, мутации в сайтах сплайсинга, ко¬
роткие внутренние делеции и вставки без нарушения
рамки считывания, миссенс-мутации в кодирующей об¬
ласти гена [Crossey Р. et al., 1994 (б); Richards F. et al.,
1994; Chen F. et al., 1995; Zbar B. et al., 1996; Pack S. et
al., 1999]. Анализ корреляций генотип-фенотип показал,
что миссенс-мутации гена VHL у больных с синдромом
Гиппеля-Линдау достоверно чаще сопровождаются раз¬
витием феохромоцитомы по сравнению с «инактивиру¬
ющими» типами мутаций (крупными делециями или
мутациями с обрывом трансляции) [Crossley Р. et al., 1994
(б); Chen F. et al., 1995; Zbar B. et al., 1996]. Указанная
взаимосвязь является настолько значимой, что позволя¬
ет с клинико-генетической точки зрения подразделить
синдром Гиппеля-Линдау на 2 типа - без клинических
проявлений феохромоцитомы и с таковыми (соответ¬
ственно, тип 1 и тип 2). В 43% случаев синдрома Гиппе-
406Глава 3ля-Линдау с феохромоцитомой (тип 2) обнаруживаются
нуклеотидные замены в 238-м кодоне - Arg238Gln и
Arg238Trp [Chen F. et al., 1995].Прямая ДНК-диагностика синдрома Гиппеля-
Линдау базируется на SSCP-анализе и тотальном сек-
веквенировании кодирующей области гена VHL, допол¬
няемыми (с учетом частичных и полных делений гена)
блот-гибридизацией по Саузерну и флюоресцентной гиб¬
ридизацией in situ [Crossey P. et al., 1994 (6); Pack S. et
al., 1999; Hes F. et al., 2000]. При синдроме Гиппеля-
Линдау, осложненном феохромоцитомой, в качестве пер¬
вого шага целесообразно провести прицельное исследо¬
вание возможных мутаций в 238-м кодоне гена VHL с
помощью ПЦР-рестрикционного анализа. При выборе
наиболее предпочтительного метода ДНК-анализа и оп¬
ределении прогноза болезни весьма полезным является
обширный каталог мутаций VHL и соответствующих
фенотипов, составленный В. Zbar с соавторами (1996).
Указанная стратегия молекулярного скрининга позволя¬
ет идентифицировать до 80% мутаций в гене VHL у боль¬
ных синдромом Гиппеля-Линдау [Chen F. et al., 1995;
Zbar В. et al., 1996; Pack S. et al., 1999]. У некоторых
больных с синдромом Гиппеля-Линдау описан сомати¬
ческий мозаицизм по мутации, в том числе случаи, ког¬
да в клетках крови обнаруживались 2 популяции ДНК -
нормальная и мутантная [Sgambati М. et al., 2000]. Для
выявления данного феномена, существенно влияющего
на расчеты риска при медико-генетическом консульти¬
ровании, могут применяться специальные методы - на¬
пример, секвенирование отдельных видов клонирован¬
ной геномной ДНК [Sgambati М. et al., 2000]. ДНК-диаг-
ностика имеет большое значение для подтверждения
синдрома Гиппеля-Линдау в атипичных случаях с «не¬
полной» клинической картиной заболевания (изолиро-
ДНК- диагностика наследственных болезней ... 407ванная гемангиобластома мозжечка и т.д.).3.7.4. Лтаксия-телеангиэктазияДанное аутосомно-рецессивное заболевание, ко¬
торое в некоторых ранее использовавшихся классифи¬
кациях не вполне корректно относилось к группе фако-
матозов, рассматривается в разделе 3.3, посвященном
наследственным атаксиям.Генетические механизмы некоторых других забо¬
леваний, традиционно рассмативаемых в группе фако-
матозов (синдром Штурге-Вебера, кожно-оболочечно-
спинальный ангиоматоз Кобба и др.), до настоящего вре¬
мени не установлены.3.8. Митохондриальные
энцефаломиопатии3.8.1. Молекулярные основымитохондриальных болезнейМитохондриальные энцефаломиопатии пред¬
ставляют собой группу заболеваний, относящихся к осо¬
бому классу наследственной патологии человека - ми¬
тохондриальным болезням (митохондриальным цитопа¬
тиям). В самом общем виде митохондриальные болезни
можно определить как заболевания, обусловленные ге¬
нетическими и структурно-биохимическими дефектами
митохондрий и сопровождающиеся нарушением ткане¬
вого дыхания [ВельтищевЮ.Е.ДеминП.А., 1998; Крас¬
нопольская К.Д., Захарова Е.Ю., 1998; Shoffner J.,
Wallace D., 1992; DiMauro S., 1993].Митохондрия - это внутриклеточная органелла,
играющая центральную роль в энергетическом метабо¬
лизме клетки. Функция митохондрий заключается в
408Глава 3ступенчатом высвобождении химической энергии окис¬
ления органических субстратов и ее превращении в энер¬
гию макроэргических фосфатов. Таким образом, в ми¬
тохондриях обе стороны двуединого процесса - аэроб¬
ное окисление и окислительное фосфорилирование -
являются сопряженными. Аэробное окисление крупных
органических молекул реализуется через образование
более простых органических соединений (глюкоза, жир¬
ные кислоты, глицерол, аминокислоты) и их дальней¬
шее окисление до специфических промежуточных про¬
дуктов (ацетил-КоА), вступающих в цикл трикарбоно-
вых кислот Кребса. В цикле Кребса реализуется серия
окислительно-восстановительных реакций, в которых
атомы водорода акцептируются адениновыми и флави-
новыми нуклеотидами (НАД+ и ФАД+). Финальной ста¬
дией является окисление восстановленных форм НАДН
и ФАДН2 в так называемой дыхательной цепи митохон¬
дрий - системе из 5 ферментативных комплексов, пос¬
ледовательно транспортирующих электроны и протоны
на кислород с образованием молекул воды. Энергия, выс¬
вобождающаяся в процессе переноса электронов, накап¬
ливается в виде макроэргических фосфатов (АТФ и др.).
Основные химические реакции происходят на внутрен¬
ней мембране митохондрий, имеющей значительную
площадь благодаря многочисленным складкам (кристам),
инвагинирующим в митохондриальный матрикс. Поми¬
мо участия в энергетическом метаболизме, митохондрии
выполняют также ряд дополнительных важных функ¬
ций - таких как синтез аминокислот, пиримидинов, ли¬
пидов, гема и других метаболитов.Сложность и многообразие митохондриальных
функций находят свое прямое отражение на белковом
уровне: в митохондриях локализовано около 1000 раз¬
личных полипептидов, что составляет почти 10% всего
белкового пула типичной эукариотической клетки. Уни¬
ДНК- диагностика наследственных болезней ... 409кальной особеностью митохондрий является тот факт,
что источником митохондриальных белков являются 2
различных самостоятельных генома клетки - ядерный и
собственно митохондриальный. Абсолютное большин¬
ство белков, принимающих участие в функционирова¬
нии митохондрий, синтезируются в ядре клетки и впос¬
ледствии транспортируются к соответствующему мито¬
хондриальному компартменту. В то же время 13 поли¬
пептидов (все из них являются компонентами дыхатель¬
ной цепи) синтезируются генами, входящими в состав
митохондриальной ДНК.Митохондриальная (внеядерная, цитоплазмати¬
ческая) ДНК представляет собой автономную по отно¬
шению к ядру генетическую систему, организованную в
виде двуцепочечной кольцевой молекулы (рис. 67). Ми¬
тохондриальную ДНК (мтДНК) иногда условно называ¬
ют М-хромосомой. Структура мтДНК полностью рас¬
шифрована: молекула мтДНК состоит всего из 16569 п.о.
и содержит 37 генов. Эти гены кодируют синтез 2 видов
рибосомальной РНК, 22 видов транспортной РНК (не¬
обходимых для синтеза белка в митохондриях), а также
13 белков, входящих в состав I, III, IV и V комплексов
дыхательной цепи митохондрий.Значительная часть белков этих комплексов, а
также все белки II комплекса дыхательной цепи, коди¬
руются ядерной ДНК. Гены мтДНК расположены очень
компактно, не прерываясь интронными вставками и не
образовывая комплексов с белками-гистонами. Отсут¬
ствие «гистонной защиты», а также относительное не¬
совершенство системы репарации и высокая подвержен¬
ность воздействию свободных радикалов, образуемых
при аэробном окислении, способствуют весьма высокой
скорости накопления мутаций в мтДНК в онто- и фило¬
генезе. Повышенный темп мутирования мтДНК обус-
410Глава 3D-петляСиндром Кернса-СейраПрогрессирующая наружная
офтальмоплегия(типичная делеция 5 кб) _тATPase 8MELASMELAS
Болезнь Лея
Неспецифическая
энцефаломиопатияГпухота, вызваннаяприменениемаминогликозидовАтрофия зри¬
тельных нервов
ЛебераMELASПрогрессирующая наружная
офтальмоплегия
Миопатия (в том числе
с кадиомиопатией)Атрофия зри¬
тельных нервов
ЛебераПрогрессирующая
наружная офталь¬
моплегия —*Атрофия зри¬
тельных нервов
Лебера —IНеспецифическаяэнцефаломиопатияПрогрессирующая
наружная офталь¬
моплегияАтрофия зри¬
тельных нервов'
ЛебераАтрофия зри¬
тельных нервов
Лебера (в том
числес дистонией)Атрофия зри¬
тельных нервов
ЛебераНейросенссглухотаMERRFMERRF/MELASNARPБолезнь ЛеяРис. 67. Митохондриальная ДНК человека и основные
мутации при митохондриальных энцефаломиопатиях
Показано подразделение кольцевидной молекулы мтДНК на отдельные
гены. Гены мтДНК указаны вдоль внутренней поверхности кольца:- символы ND 1 - ND 6 и ND 4L обозначают семь субъединиц
комплекса I дыхательной цепи (NADH-ubiquinone oxidoreductase);- символ Cyt b обозначает субъединицу комплекса III (ubiquinoi-
cytochrome с oxidase oxidoreductase);- символы СОХ I - СОХ III обозначают три субъединицы комплекса IV
(cytochrome с oxidase);- символы ATPase 6 и 8 обозначают две субъединицы комплекса V (ATP
synthase);- символы 16S и 12S обозначают гены рибосомальных РНК;- однобуквенные символы обозначают гены транспортных РНК (в соот¬
ветствии с однобуквенными символами 22 транспортируемых амино¬
кислот, указанными в приложении 2).В центре кольца указаны границы типичной делеции протяженностью
5 кб, обусловливающей развитие синдрома Кернса-Сейра и прогресси¬
рующей наружной офтальмоплегии. Наиболее частые точковые мутации,
обусловливающие развитие митохондриальных энцефаломиопатий, обо¬
значены вдоль наружной поверхности кольца.
ДНК- диагностика наследственных болезней ... 411ловливает высокую частоту спорадических случаев ми¬
тохондриальных болезней.Число митохондрий в клетках различных тканей
может составлять от сотен до нескольких тысяч, а каж¬
дая митохондрия содержит от 2 до 10 копий мтДНК.
Общее же количество молекул мтДНК в клетке может
достигать десятков тысяч. Например, в одном кардио-
миоците может содержаться до 50 ООО молекул мтДНК.
Каждая молекула мтДНК реплицируется самостоятель¬
но, деление митохондрий в цитоплазме также представ¬
ляет собой автономный процесс. В результате клеточно¬
го деления (митоз, мейоз) различные молекулы мтДНК
в составе митохондрий в случайном порядке переходят
в цитоплазму дочерних клеток. В обычной ситуации во
всех клетках и тканях организма имеется один и тот же
нормальный вид мтДНК - состояние, обозначаемое как
гомоплазмия. При возникновении мутации в мтДНК и
дальнейшей амлификации мутантного клона возникает
гетероплазмия, т.е. состояние, при котором в клетке (тка¬
ни) существует совокупность двух различных популяций
мтДНК - нормальной и мутантной. Процентное содер¬
жание нормальной и мутантной мтДНК в конкретной
ткани может варьировать в широких пределах (от 0 до
100%), что в значительной степени определяет тяжесть
соответствующих клинических проявлений болезни.
Поскольку репликация митохондрий и мтДНК является
автономной по отношению к клеточному циклу, а их
распределение по дочерним клеткам имеет случайный
характер, в организме имеет место митотическая сег¬
регация мтДНК, проявляющаяся постоянным динами¬
ческим изменением соотношения нормального и мутан¬
тного пулов мтДНК по мере обновления клеточных по¬
пуляций. Следовательно, уровень гетероплазмии явля¬
ется различным как в разных тканях, так и в одной и той
же ткани в различные периоды жизни. В определенных
412Глава 3тканях митотическая сегрегация мтДНК может приво¬
дить к полному замещению нормальных молекул мтДНК
мутантными, с переходом от состояния гетероплазмии к
состоянию гомоплазмии по мутантной мтДНК. Таким
образом, по образному выражению A. Schon и S. DiMauro(1994), по своей сути вся митохондриальная генетика
является популяционной генетикой, основанной на зако¬
номерностях динамики соотношения между нормальной
и мутантной популяциями мтДНК. Данные закономер¬
ности имеют большое значение для понимания клини¬
ческих особенностей и методов молекулярной диагнос¬
тики митохондриальных болезней (см. далее).Тяжесть поражения отдельных тканей и органов
при митохондриальных болезнях определяются не толь¬
ко характером генетического дефекта мтДНК и уровнем
гетероплазмии, но и их потребностью в энергии и зави¬
симостью клеточных функций от эффективности окис¬
лительного фосфорилирования. Наиболее энергозависи¬
мыми являются ЦНС, скелетная и сердечная мускулату¬
ра, глаз, почки, эндокринные железы, поэтому именно
эти ткани наиболее часто и закономерно вовлекаются в
патологический процесс при митохондриальных дефек¬
тах. Для манифестации симптоматики со стороны того
или иного органа или ткани необходимо, чтобы мито¬
хондриальная дисфункция и нарушение окислительно¬
го фосфорилирования в них превысили определенный
критический порог, специфический для каждой ткани.
Например, для ЦНС таким пороговым значением явля¬
ется содержание мутантного вида мтДНК в нейронах
около 60%; если у больных содержание мутантной
мтДНК в ЦНС превышает эту цифру, в клинической кар¬
тине развиваются неврологические симптомы. Для дру¬
гих тканей порог переносимости митохондриальной дис¬
функции выше. Тяжесть митохондриальных болезней с
ДНК- диагностика наследственных болезней ... 413течением времени неуклонно нарастает, что обусловле¬
но возрастным снижением активности процессов окис¬
лительного фосфорилирования и накоплением мутаций
мтДНК в стареющих тканях.Митохондриальные болезни могут наследовать¬
ся по различным типам, что зависит от характера гене¬
тического дефекта при конкретном заболевании. По¬
скольку вся мтДНК в организме имеет материнское про¬
исхождение (из цитоплазмы яйцеклетки), в случае пере¬
дачи митохондриальной мутации потомству в родослов¬
ной имеет место материнский тип наследования - когда
заболевание проявляется у всех детей больной матери.
Если мутация происходит в ядерном гене, кодирующем
синтез митохондриального белка, заболевание переда¬
ется по классическим менделевским законам (чаще все¬
го по аутосомно-доминантному и аутосомно-рецессив-
ному типам). Наконец, при ряде форм митохондриаль¬
ных болезней мутация мтДНК возникает de novo на ран¬
ней стадии онтогенеза, приводя в результате митотичес¬
кой сегрегации к преимущественному накоплению му¬
тантного вида мтДНК в тканях-мишенях; в такой ситуа¬
ции имеет место спорадический случай болезни.Рассматриваемые в настоящей главе митохондри¬
альные энцефаломиопатии представляют собой частную
группу митохондриальных болезней, в клинической кар¬
тине которых превалирует поражение мозга и попереч¬
но-полосатой мускулатуры.3.8.2. Клинико-генетическая характеристика
митохондриальных энцефаломиопатийОбщепринятая классификация митохондриаль¬
ных энцефаломиопатий базируется на сочетании гене¬
тической и биохимической характеристики молекуляр-
414Глава Зного дефекта и особенностей клинической картины за¬
болевания. С генетической точки зрения митохондриаль¬
ные энцефаломиопатии могут быть разделены на три
группы заболеваний [БіМаиго Б., 1993]: а) заболева¬
ния, обусловленные мутациями собственно митохондри¬
альной ДНК; б) заболевания, обусловленные мутациями
ядерной ДНК; в) заболевания, обусловленные наруше¬
нием взаимодействий между митохондриальным и ядер-
ным геномами.Как указывалось выше, для первой группы харак¬
терно материнское наследование или спорадический ха¬
рактер болезни, для второй и третьей групп - все основ¬
ные типы менделевского наследования. Более тонкое
подразделение данных болезней проводится в соответ¬
ствии с конкретным типом мутации (точковые мутации,
делеции, дупликации). В рамках каждой подгруппы мож¬
но дифференцировать ряд наиболее устойчивых клини¬
ческих синдромов, краткая характеристика которых пред¬
ставлена ниже. Однако выделение подобных клиничес¬
ких синдромов при митохондриальных энцефаломиопа-
тиях весьма условно. В значительной части случаев ми¬
тохондриальных энцефаломиопатий симптоматика мо¬
жет носить «смешанный» характер, когда у больных на¬
блюдается причудливое сочетание разнообразных муль-
тисистемных и мультиорганных проявлений (атаксия,
деменция, эпилептические припадки, мышечная сла¬
бость, невропатии, поражение сердечной мышцы, диа¬
бет, задержка роста и др.), затрудняющее отнесение изу¬
чаемого симптомокомплекса к какой-либо конкретной
известной клинической форме. Такая множественность
поражения различных по происхождению и функциям
тканей, кажущаяся «необъяснимость» сочетания симп¬
томов (в основе чего лежит близкий порог чувствитель¬
ности к энергетическому дефициту в органах-мишенях)
являются важнейшими ключами к правильной диагнос¬
ДНК- диагностика наследственных болезней ... 415тике митохондриальной болезни. Следует добавить, что
митохондриальным энцефаломиопатиям свойственен
чрезвычайно выраженный внутрисемейный полимор¬
физм: у различных родственников (чаще у сибсов) одна
и та же мутация может манифестировать как в виде раз¬
вернутого и фатального страдания, так и в виде отдель¬
ных непрогрессирующих симптомов либо может вооб¬
ще не иметь никаких фенотипических проявлений. Та¬
кая крайняя степень внутрисемейного полиморфизма
обычно связана с различным уровнем гетероплазмии и,
соответственно, различным процентным содержанием
мутантной мтДНК у членов семьи.3.8.2.1. Заболевания, обусловленные мутациями ми¬
тохондриальной ДНКС клинической точки зрения данные заболевания
являются наиболее четко очерченными формами мито¬
хондриальных энцефаломиопатий. Их принято подраз¬
делять на синдромы, обусловленные: а) точковыми му¬
тациями мтДНК; б) крупными делениями и дупликаци¬
ями мтДНК.Точковые мутации митохондриальной ДНК. Точ-
ковые мутации мтДНК при митохондриальных энцефа-
ломиопатиях могут затрагивать как структурные гены
белковых субъединиц дыхательной цепи, так и гены
транспортных РНК (тРНК). Хорошо изученными пред¬
ставителями данной подгруппы являются синдромы
MERFF, MELAS, NARP, болезнь Лея, наследственная
атрофия зрительных нервов Лебера [Вельтищев Ю.Е.,
Темин П.А., 1998; Краснопольская К.Д., Захарова Е.Ю.,
1998; DiMauro S., 1993; Schapira A. et al., 1998; Chinnery P.
et al., 1999 (a)].Синдром MERRF (устоявшееся навание-акроним
от англ. Myoclonus Epilepsy with Ragged-Red Fibers) -
миоклонус-эпилепсия с рваными красными волокнами -
416Глава 3в типичных случаях характеризуется началом заболева¬
ния на втором десятилетии жизни и развитием прогрес¬
сирующей миоклонус-эпилепсии в сочетании с мозжеч¬
ковой атаксией, деменцией, миопатией, нейросенсорной
тугоухостью, задержкой физического развития. На сек¬
ции выявляются распространенные дегенеративные из¬
менения в базальных ганглиях, ядрах ствола мозга (вклю¬
чая черную субстанцию), коре и ядрах мозжечка, про¬
водниках спинного мозга. Важнейшим диагностическим
признаком является обнаружение при исследовании
мышечных биоптатов так называемого феномена «рва¬
ных красных волокон» (при окраске методом Гомори-
трихром). «Рваные» красные волокна отражают факт
митохондриальной дисфункции и представляют собой
миофибриллы со своеобразно измененными краями
вследствие пролиферации митохондрий и формирования
митохондриальных агломератов по периферии мышеч¬
ного волокна. При электронно-микроскопическом иссле¬
довании данных агломератов патологические изменения
митохондрий определяются вполне отчетливо (аномалии
формы и размеров, нарушение конфигурации крист,
наличие паркристаллических включений и др.). Другим
важным маркером болезни является выявляемый гисто¬
химически дефицит цитохром с-оксидазы в мышечных
волокнах. Характерно повышение уровня лактата в кро¬
ви (лактат-ацидоз). Указанные лабораторные признаки
не являются специфичными для только для синдрома
МЕШУ7 и могут с различной частотой и выраженнос¬
тью обнаруживаться у больных с другими формами ми¬
тохондриальных энцефаломиопатий. В семьях, отяго¬
щенных МЕЯЯ?, обычно наблюдается материнское на¬
следование болезни. Свыше 95% случаев МЕМУ обус¬
ловлены заменами нуклеотидов мтДНК в положениях
8344 (А-Ю) и 8356 (Т—»С), затрагивающими ген лизи-
новой тРНК (тРНК1у5); это приводит к нарушению ми¬
ДНК- диагностика наследственных болезней ... 417тохондриального синтеза лизин-обогащенных белков
вследствие преждевременного обрыва трансляции
|Shoffner J., Wallace D., 1992; DiMauro S., 1993;
ChomynA., 1998]. Мутации являются гетероплазмичес-
кими, и процентное содержание мутантной мтДНК тес¬
но коррелирует с выраженностью дефектов окислитель¬
ного фосфорилирования и тяжестью клинических про¬
явлений болезни [Shoffner J., Wallace D., 1992].Синдром MELAS (от англ. Mitochondrial
Encephalomyopathy, Lactic Acidosis, and Stroke-like
episodes) — митохондриальная энцефаломиопатия, лак¬
тат-ацидоз и инсультоподобные эпизоды — обычно ма¬
нифестирует в 5-20 лет. Заболевание проявляется в пер¬
вую очередь острыми инсультоподобными эпизодами с
развитием очаговых изменений в затылочной и темен¬
но-височной областях мозга и появлением соответству¬
ющей неврологической симптоматики (парезы, корко¬
вые расстройства зрения, судороги, кома, приступы го¬
ловной боли и рвоты и др.). Появление очагов связыва¬
ют с преходящей дисфункцией окислительного фосфо¬
рилирования в паренхиме мозга, а также структурно¬
метаболическими нарушениями в стенках артериол и
капилляров; характерной особенностью таких «смешан¬
ных» по генезу инфарктов мозга является относительно
быстрое восстановление. При синдроме MELAS могут
наблюдаться также миопатические проявления (повы¬
шенная утомляемость и непереносимость физических
нагрузок), деменция, атаксия, дегенерация сетчатки, ней-
росенсорная глухота, низкорослость, диабет, кардиоми-
опатия и целый ряд других мультиорганных проявлений.
Характерен значительный уровень лактат-ацидоза в кро¬
ви и спинномозговой жидкости, при биопсии скелетных
мышц нередко выявляется феномен «рваных красных
волокон». Синдром MELAS наследуется по материнс¬
кому типу, однако исключительная вариабельность кли¬
418Глава 3нических проявлений может весьма затруднять оценку
семейного анамнеза. У больных с синдромом MELAS
описано, как минимум, 8 точковых мутаций в генах
мтДНК, причем 5 из них локализованы в различных уча¬
стках гена тРНКЬеи(иш) (см. рис. 67) [DiMauro S., 1993;
Servidei S., 1997; Chinnery P. et al., 1999 (а)]. Наиболее
частой мутацией является замена A->G в положении
3243 (около 80% больных), а в целом мутации указанно¬
го гена лейциновой тРНК обнаруживаются почти в 95%
случаев MELAS. В редких случаях у больных MELAS
описаны точковые мутации в генах других тРНК и гене
СОХ Ш-субъединицы IV комплекса дыхательной цепи
(рис. 67). Все мутации обнаруживаются в гетероплазми-
ческом состоянии.Синдром NARP (от англ. Neuropathy, Ataxia,
Retinitis Pigmentosa) - невропатия с атаксией и пигмен¬
тным ретинитом — характеризуется, в соответствии с
названием, развитием прогрессирующей периферичес¬
кой невропатии с мышечной слабостью, мозжечковой
атаксии и пигментной дегенерации сетчатки. Как и при
других митохондриальных энцефаломиопатиях, клини¬
ческая картина может быть весьма вариабельной, с на¬
личием или отсутствием у родственников ряда допол¬
нительных симптомов (задержка психомоторного разви¬
тия, эпилептические припадки, деменция). Исследова¬
ния на лактат-ацидоз и другие маркеры митохондриаль¬
ной дисфункции не всегда информативны. Тип наследо¬
вания болезни материнский. У всех больных с синдро¬
мом NARP обнаруживается гетероплазмическая мутация
T-»G в положении 8993 (ген АТФазы 6 - субъединицы
V комплекса дыхательной цепи) [Harding A. et al., 1992].
Уровень гетероплазмии является решающим для харак¬
тера манифестации данной мутации: при содержании
мутантной мтДНК <78% заболевание может проявлять¬
ся изолированными расстройствами зрения, несколько
ДНК- диагностика наследственных болезней ... 419более высокий уровень мутантной мтДНК сопровожда¬
ется развитием различных вариантов синдрома NARP,
тогда как у больных с уровнем мутантной мтДНК 90% и
более наблюдается драматически иной фенотип с быст¬
рым фатальным исходом - болезнь Лея [Wallace D., 1993].Болезнь Лея (подострая некротизирующая энце-
фаломиелопатия) представляет собой наиболее тяжелую
клиническую манифестацию недостаточности окисли¬
тельного фосфорилирования. Она проявляется в груд¬
ном и раннем детском возрасте в виде дыхательных на¬
рушений, отставания психомоторного развития ребен¬
ка, экстрапирамидных симптомов, атаксии, спастичнос-
ти, атрофии зрительных нервов, а также подострых эпи¬
зодов тяжелой клинико-биохимической декомпенсации
на фоне интеркуррентных инфекций. Весьма характерен
лактат-ацидоз, особенно в период обострения симпто¬
матики. Продолжительность жизни обычно не превы¬
шает 5 лет. Типичными изменениями в веществе мозга
являются билатеральные некрозы в области зрительно¬
го бугра, базальных ганглиев, мозжечка и ствола мозга,
диффузная демиелинизация и глиоз. Болезнь Лея разви¬
вается при носительстве ряда точковых мутаций мтДНК
в состоянии гомоплазмии либо гетероплазмии с высо¬
ким (>90%) содержанием мутантного вида мтДНК. На¬
пример, нуклеотидные замены в генах АТФазы 6, ND1
и тРНКУа|, ассоциированные в гетероплазмическом со¬
стоянии с NARP и MELAS, в случае перехода к гомоп¬
лазмии обычно принимают более тяжелое течение и про¬
являются в виде болезни Лея [Wallace D., 1993; Campos Y.
et al., 1997; Chinnery P. et al., 1999 (a)]. В связи с этим в
одной родословной могут наблюдаться различные фе¬
нотипы матерински-наследуемых митохондриальных
энцефаломиопатий.Наследственная невропатия зрительных нервов
Лебера проявляется острой и безболевой утратой цент¬
420Глава 3рального зрения. Спонтанное и обычно неполное вос¬
становление остроты зрения возможно лишь в редких
случаях болезни. В острой стадии на глазном дне опре¬
деляются изменения в области дисков зрительных не¬
рвов (перипапиллярная телеангиэктатическая микроан-
гиопатия, отечность дисков). В некоторых описанных
родословных зрительная невропатия сочеталась с дис¬
тонией и билатеральным стриарным некрозом (фенотип,
близкий к болезни Лея), периферической полиневропа¬
тией, спастическими парезами, скелетными деформаци¬
ями и другими дополнительными симптомами. Дебют
заболевания обычно приходится на второе и третье де¬
сятилетия жизни, хотя известны более ранние и более
поздние случаи леберовской невропатии. Тип наследо¬
вания материнский. Невропатия Лебера может вызывать¬
ся рядом точковых мутаций в генах комплексов I, III и
IV дыхательной цепи (см. рис. 67). Наиболее частыми
являются замены G3460A в гене ND 1, G11778A в гене
ND 5 и Т14484С в гене ND 6 комплекса I - свыше 95%
случаев заболевания [Shoffner J., Wallace D., 1992;
Servidei S., 1997; Chirmery P. et al., 1999 (а, б)]. Обычно
данные мутации обнаруживаются в гомоплазмическом
состоянии и характеризуются неполной пенетрантнос-
тью: заболевают около 50% мужчин и 10% женщин, в
связи с чем некоторые случаи манифестируют как спо¬
радические [Chinnery P. et al., 1999 (б)]. Причины такой
неполной пенетрантности не вполне ясны. Предполага¬
ется, что для манифестации зрительных расстройств
может быть необходимо одновременное присутствие си-
нергистических «вторичных» мутаций в мтДНК, нали¬
чие которых у пациентов с невропатией Лебера было по¬
казано рядом исследователей [Johns D., Berman J., 1991;
Brown M. et al., 1992].Помимо указанных основных фенотипов, у боль¬
ных с точковыми мутациями мтДНК могут наблюдаться
ДНК- диагностика наследственных болезней ... 421и другие клинические варианты митохондриальной не¬
врологической патологии, например прогрессирующая
наружная офтальмоплегия, миопатия (изолированная
или в сочетании с кардиомиопатией), различные вари¬
анты неспецифической энцефаломиопатии (например,
синдром деменция/хорея) и т.д. [DiMauro S., 1993;
Servidei S., 1997; Chinnery P. et al., 1999 (а)]. Некоторые
из соответствующих мутантных сайтов указаны на рис. 67
(см. также приложение 1).Делеции и дупликации митохондриальной ДНК.
Данные типы мутаций мтДНК обычно возникают de novo
в соматических клетках на ранней стадии эмбриогенеза
и приводят к манифестации спорадических случаев ми¬
тохондриальных болезней. Наследование этих мутаций
невозможно, поскольку, как предполагается, ооциты с
крупными перестройками мтДНК не способны дать на¬
чало развитию эмбриона [DiMauro S., 1993]. Типичным
неврологическим проявлением делеций/дупликаций
мтДНК является прогрессирующая наружная офтальмоп¬
легия, которая может входить в структуру сложных муль-
тисистемных синдромов. Наиболее изученным из них
является синдром Кернса-Сейра. Это заболевание харак¬
теризуется развитием на 1 -2-м десятилетии жизни про¬
грессирующей наружной офтальмоплегии (птоз, огра¬
ничение движений глазных яблок, иногда двоение) в
сочетании с пигментной дегенерацией сетчатки, атрио¬
вентрикулярной блокадой сердца, миопатией, атаксией,
нейросенсорной глухотой, эндокринными расстройства¬
ми. При развернутой клинической картине большинство
больных погибают от патологии сердца через 10-20 лет
от начала заболевания. Нередко в клинике могут наблю¬
даться те или иные редуцированные варианты синдрома
Кернса-Сейра. При лабораторных исследованиях обыч¬
но выявляется повышение уровня белка в спинномозго¬
вой жидкости, лактат-ацидоз, феномен «рваных красных
422Глава 3волокон» в мышечных биоптатах. Около 80% больных с
синдромом Кернса-Сейра имеют те или иные делеции
митохондриального генома протяженностью от 2 до 10,4
кб, причем 30-40% из них имеют идентичную делецию
4977 нуклеотидов, захватывающую сегмент мтДНК от
гена АТФазы 8 до reHaND 5 (рис. 68а) [Moraes C. et al.,
1989; DiMauro S., 1993; Wallace D., 1993]. Описаны бо¬
лее редкие случаи, когда при тех или иных вариантах
синдрома Кернса-Сейра у больных выявлялись дупли¬
кации или точковые мутации мтДНК [DiMauro S., 1993;
Servidei S., 1997]. Все описанные мутации являются ге-
тероплазмическими, и различные уровни гетероплазмии
в разных тканях-мишенях обусловливают значительный
полиморфизм клинических проявлений болезни.Ярким примером указанного полиморфизма явля¬
ется синдром Пирсона — злокачественная младенческая
форма поражения костного мозга с панцитопенией, обус¬
ловленная теми же крупными делециями мтДНК, что и
синдром Кернса-Сейра. Основное молекулярное разли¬
чие между данными заболеваниями заключается в том,
что при синдроме Кернса-Сейра содержание мутатной
мтДНК в мышцах достигает 80% против ~5% в крове¬
творных клетках, тогда как у больных с синдромом Пир¬
сона наблюдаются обратные соотношения [Schon E.,
DiMauro S., 1994]. Более того, с возрастом пропорция
мутантной мтДНК в мышечной ткани повышается, а в
клетках костного мозга, напротив, снижается вследствие
интенсивной митотической сегрегации в сторону нормаль¬
ных стволовых клеток. С клинической точки зрения дра¬
матизм ситуации заключается в том, что больные с синд¬
ромом Пирсона, у которых в детстве ценой множествен¬
ных трансфузий удалось компенсировать фатальный де¬
фект кроветворения, фактически обречены спустя 10-15
лет заболеть другим тяжелейшим и некурабельным ми-
тоходриальным недугом - синдромом Кернса-Сейра.
ДНК- диагностика наследственных болезней ... 423Более доброкачественной формой по сравнению
с синдромом Кернса-Сейра является так называемая про¬
грессирующая наружная офтальмоплегия с рваными
красными волокнами - медленно прогрессирующее за¬
болевание молодого возраста, не влияющее на продол¬
жительность жизни и проявляющееся парезом глазод¬
вигательных мышц, блефароптозом, проксимальной сла¬
бостью скелетной мускулатуры. Иногда единственным
проявлением болезни может быть 2-сторонний блефа-
роптоз. Примерно у 50-70% указанных больных выяв¬
ляются делеции или, гораздо реже, дупликации мтДНК
(в последнем случае иногда наблюдается сочетание гла¬
зодвигательных нарушений с диабетом) [Moraes С. et al.,
1989; DiMauro S., 1993; Wallace D., 1993]. Как отмеча¬
лось выше, у остальных больных с прогрессирующей
наружной офтальмоплегией митохондриальной приро¬
ды заболевание обусловлено разнообразными толковы¬
ми мутациями мтДНК (рис. 67).3.8.2.2. Заболевания, обусловленные мутациями ядер-
ной ДНК и нарушением межгеномных взаи¬
модействийБольшинство митохондриальных белков кодиру¬
ются ядерным геномом, поэтому известно значительное
число митохондриальных болезней, наследуемых в со¬
ответствии с менделевскими типами наследования [Вель-
тищев Ю.Е., Темин П.А., 1998; Краснопольская К.Д.,
Захарова Е.Ю., 1998; DiMauro S., 1993]. Биохимическая
классификация данных болезней предполагает их под¬
разделение на следующие классы:1) Дефекты транспорта митохондриальных
субстратов. К ним относятся синдромы дефицита кар-
нитина и ферментов его метаболизма (карнитин-паль-
митоилтрансферазы, карнитин-ацилкарнитин-транслока-
зы и др.). Примером заболевания с идентифицирован¬
424Глава 3ным генетическим дефектом является аутосомно-рецес-
сивный синдром недостаточности карнитин-пальмито-
илтрансферазы, обусловленный мутациями гена данно¬
го фермента на хромосоме 1р.2) Дефекты утилизации митохондриальных суб¬
стратов. К ним относятся дефекты ферментов р-окисле-
ния жирных кислот (соответствующие мутантные гены
локализованы на хромосомах 1,2 и 12), недостаточность
пируват-карбоксилазы, а также дефекты компонентов
пируват-дегидрогеназного комплекса - пируват-декар-
боксилазы (гены субъединиц а и |3 на хромосомах X и
3), дигидролипоил-трансацетилазы и дигидролипоил-
дегидрогеназы (один из генов на хромосоме 7). Все фор¬
мы наследуются па аутосомно-рецессивному либо X-
сцепленному рецессивному типам.3) Дефекты ферментов цикла Кребса - фумара-
зы, сукцинат-дегидрогеназы и а-кетоглутарат-дегидро-
геназы (гены локализованы на хромосомах 1 и 7). Тип
наследования ферментопатий цикла Кребса аутосомно-
рецессивный.4) Дефект сопряжения окисления и фосфорили-
рования (болезнь Люфта). Этот редчайший синдром опи¬
сан лишь у 2 неродственных больных и обусловлен не¬
способностью митохондрий конвертировать энергию
окисляемых органических субстратов в АТФ, что сопро¬
вождается гипертермией, непереносимостью тепла, про-
фузным потом, полифагией, полидипсией, тахикардией.
Тип наследования и мутантный ген не установлены.5) Дефекты дыхательной цепи. Комплексы 1-У
дыхательной цепи митохондрий содержат приблизитель¬
но 83 белковых субъединицы, причем 69 из них кодиру¬
ются ядерным геномом. В связи с этим известно доволь¬
но большое число клинических синдромов недостаточ¬
ности комплексов 1-У, наследуемых по менделевскому
типу и предположительно обусловленных мутациями
ДНК- диагностика наследственных болезней ... 425генов ядерной ДНК. Однако на сегодняшний день в ли¬
тературе описано лишь несколько случаев выявления
мутаций в ядерных генах, кодирующих белки дыхатель¬
ной цепи и локализованных, в частности, на хромосо¬
мах 3,5,9,11 и 22. Такие мутации идентифицированы в
генах AQDQ, NDUFS8 и NDUFV1 комплекса I, сукци-
натдегидрогеназы комплекса II, а также SURF1 и SC02- вспомогательных белков, участвующих в биогенезе
комплекса IV (сборка субъединиц комплекса, поддержа¬
ние его пространственной организации и т.п.).Клиническая картина митохондриальных болез¬
ней, обусловленных мутациями ядерных генов, чрезвы¬
чайно вариабельна. Первые симптомы появляются чаще
всего в раннем детском возрасте, реже болезнь манифе¬
стирует в более позднем периоде. Характерны следую¬
щие основные фенотипы: а) подострая некротизирую-
щая энцефалопатия (болезнь Лея); б) различные вари¬
анты фатальной детской мультисистемной энцефаломи-
опатии с лактат-ацидозом, нарушениями дыхания и не¬
редким поражением сердечной мышцы; в) прогрессиру¬
ющая миоклоническая эпилепсия (судороги обычно ре¬
зистентны к антиконвульсантной терапии); г) синдром
Кернса-Сейра; д) прогрессирующая детская полиодист-
рофия (синдром Альперса) - комбинированная дегене¬
рация серого вещества головного мозга; е) прогрессиру¬
ющая лейкодистрофия; ж) доброкачественная детская
митохондриальная миопатия, непереносимость физичес¬
кой нагрузки; з) синдром миалгии-миоглобинурии [Вель-
тищев Ю.Е., Темин П.А., 1998; Краснопольская К.Д.,
Захарова Е.Ю., 1998; DiMauro S., 1993; Chinnery P. etal.,
1999 (а)]. При многих формах неврологическая патоло¬
гия сопровождается поражением сердца (кардиомиопа-
тия), печени (печеночная недостаточность, гепатомега-
лия, цирроз), почек (проксимальная тубулопатия), ки¬
шечника (синдром мальабсорбции, потеря массы тела),
426Глава 3метаболическими кризами (гипогликемические и аци-
дотические приступы с рвотой, диареей, расстройства¬
ми сознания), разнообразными аномалиями развития.Особую группу составляют заболевания, при ко¬
торых можно предполагать нарушение взаимодействий
между ядерным и митохондриальным геномами. Одно
из них - синдром множественных делеций мтДНК. Тип
наследования чаще всего аутосомно-доминантный, в
типичных случаях симптомы дебютируют на 2-3-м де¬
сятилетии жизни. В клинической картине наблюдаются
весьма полиморфные мультисистемные проявления: про¬
грессирующая наружная офтальмоплегия, миопатичес-
кий синдром с вовлечением дыхательной мускулатуры,
атаксия, нистагм, невропатии периферических, слуховых
и зрительных нервов, катаракта, гипопаратиреоз, задер¬
жка роста, липоматоз и др. На компьютерных томограм¬
мах выявляются очаги пониженной плотности в базаль¬
ных ганглиях, семиовальном центре и ножках мозга. При
лабораторных исследованиях выявляется лактат-ацидоз
в крови, феномен «рваных красных волокон» и сниже¬
ние активности цитохром с-оксидазы в мышечных био-
птатах. Прогноз неблагоприятный, больные обычно по¬
гибают в течение 15-20 лет вследствие дыхательных
нарушений. Кардинальным признаком данного заболе¬
вания являются сочетанные делеции многих участков
митохондриального генома, грубо нарушающие струк¬
туру мтДНК и функционирование различных митохонд¬
риальных генов (рис. 68Б) [2еу1аш М. etal., 1989]. Пред¬
полагается, что в основе такого типа мутаций лежит по¬
вреждение регуляторных белков, кодируемых ядерными
генами и контролирующих митохондриальный биогенез,
в частности-процессы репликации мтДНК [ШМаиго 8.,
1993; Саггогго Я. е1 а1., 1998]. При исследовании боль¬
ших семей с аутосомно-доминантным синдромом мно¬
ДНК- диагностика наследственных болезней ... 427жественных делеций мтДНК была показана его гетеро¬
генность: три генетических локуса данного синдрома
картированы на хромосомах 10q24(PE01), Зр14.1—21.2
(РЕ02) и 4q35 (РЕОЗ) [Chinnery P. et al., 1999 (а);
Kaukonen J. et al., 2000]. Ген последней формы иденти¬
фицирован: он кодирует фермент аденинового обмена
аденин-нуклеотид-транслоказу 1 (ANT1); нарушение
функции данного фермента в результате мутаций ANT1
сопровождается патологией метаболизма аденинового
нуклеотида и нарушением репликации мтДНК
[Kaukonen J. et al., 2000].12 12 12лтт—ШИ 4шШ>А Б ВРис. 68. Диагностика перестроек и количественных дефектов
мтДНК с помощью блот-гибридизации по Саузерну
Дорожка 1 -здоровый контроль, дорожка 2-больной. А. Делеция
мтДНК у больного с синдромом Кернса-Сэйра. Тонкой стрелкой
обозначена нормальная мтДНК, жирной стрелкой - мутантная уко¬
роченная молекула мтДНК с делецией 4977 п.о. Б. Синдром множе¬
ственных делеций мтДНК. Стрелкой обозначена нормальная моле¬
кула мтДНК, скобкой - мутантные укороченные молекулы с деле¬
ниями различной длины. В. Синдром истощения мтДНК. Тонкой
стрелкой обозначены фрагменты ядерной ДНК, служащие внутрен¬
ним стандартом; жирной стрелкой обозначена мтДНК: у больного
(дорожка 2) по сравнению со здоровым индивидом четко опреде¬
ляется заметно сниженная интенсивность сигнала, свидетельству¬
ющая о количественном дефекте мтДНК.
428Глава 3Известна аутосомно-рецессивная форма синдро¬
ма множественных делеций мтДНК - так называемая
митохондриальная нейрогастроинтестинальная энцефа-
ломиопатия (МП\Ю1Е). Заболевание манифестирует в
более молодом возрасте и проявляется тем же набором
типичных для митохондриальных болезней неврологи¬
ческих симптомов в сочетании с выраженной дисфунк¬
цией желудочно-кишечного тракта (синдром кишечной
псевдообструкции с приступами повторной рвоты, диа¬
реей, потерей массы тела). У больных МЫСЕЕ выявлено
выраженное снижение активности фермента тимидин-
фосфорилазы, обусловленное мутациями соответствую¬
щего гена на хромосоме 22ql3.32-qter [МбЫпо I. е1 а1.,
1999]. Таким образом, в основе болезни лежит генети¬
чески детерминированная патология метаболизма тими-
дина, приводящая к нарушению репликации и/или под¬
держания молекулы мтДНК.Синдром истощения мтДНК представляет собой
состояние, при котором у больных имеется не качествен¬
ный, а количественный дефект мтДНК - т.е. резкое сни¬
жение числа копий молекул мтДНК (рис. 68 В) [МогаеБ С.
е1 а1., 1991; Б1Маиго Б., 1993]. Характерно, что истоще¬
ние мтДНК наблюдается только в строго определенных
тканях (например, только в мышцах, только в печени, в
мышцах и почках и т.д.). Клиническая картина зависит
от вовлечения конкретных тканей и обычно включает в
различных комбинациях миопатию (в том числе врож¬
денную), судорожный синдром, печеночную и почечную
недостаточноть, кардиомиопатию. Характерен лактат-
ацидоз, феномен «рваных красных волокон», обнаружи¬
вается комбинированная недостаточность комплексов
дыхательной цепи, содержащих мтДНК-кодируемые
субъединицы (I, III-V). Заболевание носит врожденный
характер или манифестирует на 1 -2-м году жизни и имеет
ДНК- диагностика наследственных болезней ... 429обычно фатальное течение. В большинстве описанных
случаев синдрома истощения мтДНК зарегистрирован
аутосомно-рецессивный тип наследования. Генетический
дефект не установлен. Предполагается, что заболевание
обусловлено повреждением ядерного гена, контролиру¬
ющего репликацию мтДНК. Известна также относитель¬
но доброкачественная вторичная форма данного синд¬
рома, при которой истощение мтДНК в клетках вызвано
применением анти-ВИЧ препарата зидовудина.3.8.3. ДНК-диагностика митохондриальных
энцефаломиопатий.В силу особенностей генетики митохондриальных
энцефаломиопатий ДНК-диагностика этих заболеваний
имеет ряд принципиальных отличий от традиционных
подходов, применимых для менделирующих заболева¬
ний. Обобщение многолетнего опыта обследования боль¬
ных с митохондриальной патологией в ведущих иссле¬
довательских центрах мира позволило разработать чет¬
кий и последовательный диагностический алгоритм,
обеспечивающий наибольшую эффективность диагнос¬
тического поиска [Краснопольская К.Д., Захарова Е.Ю.,
1998; Shoffner J., Wallace D., 1992; DiMauro S., 1993;
Chinnery P. et al., 1999 (а)]. Данный алгоритм включает
несколько основных этапов (рис. 69).1) На первом этапе проводится детальный кли¬
нико-генеалогический и лабораторно-инструментальный
анализ, целью которого является накопление доказа¬
тельств в пользу обоснованного предположения о мито¬
хондриальной природе изучаемого заболевания. Наибо¬
лее очевидно о митохондриальной болезни может сви¬
детельствовать: а) материнский тип наследования (с уче¬
том всех полиморфных и даже субклинических прояв¬
430Глава 3лений у сибсов - детей больной матери); б) своеобраз¬
ный характер синдрома (наличие мультисистемной и
мультиорганной патологии с вовлечением органов, раз¬
личных по эмбриональному происхождению и функциям);в) прогрессирующее течение, наличие метаболических
кризов (последнее особенно актуально для митохондри¬
альных синдромов раннего детского возраста); г) повы¬
шение уровня лактата в крови и спинномозговой жид¬
кости (в том числе на фоне пищевой и физической на¬
грузки); д) аминоацидурия и органическая ацидурия.
Поиск мультиорганной патологии должен проводиться
целенаправленно, с использованием необходимых пара¬
клинических методов, с целью выявления явной или
скрытой кардиомиопатии, почечной тубулопатии, гепа-
тоцеллюлярной дисфункции, диабета, недостаточности
гормона роста, атрофии ворсин кишечника, изменений
в мазке крови и пунктате костного мозга, миопатии, пе¬
риферической, кохлеарной и зрительной невропатии,
патологии сетчатки, петрификатов и очаговых измене¬
ний в веществе мозга.2) Уже в результате проведенного вышеуказан¬
ного комплексного обследования может быть достаточ¬
но четко идентифицирован один из известных клини¬
ческих синдромов, обычно обусловленных точковыми
мутациями мтДНК (MELAS, MERRF, NARP, атрофия
зрительных нервов Лебера). В таком случае ДНК из кле¬
ток крови может исследоваться на наличие соответству¬
ющих известных мутаций мтДНК. Молекулярный ана¬
лиз облегчается тем, что каждый из указанных синдро¬
мов ассоциирован со сравнительно ограниченным чис¬
лом мутаций мтДНК, часть которых являются мажор¬
ными и имеются у абсолютного большинства больных.
При выявлении искомой мутации в лимфоцитах диаг¬
ноз может считаться окончательно подтвержденным.
КЛИНИЧЕСКОЕОБСЛЕДОВАНИЕанализ клинического синдромаанализ типа наследованиядополнительные методы:- кровь (лактат, глюкоза, КФК );- моча (аминокислоты,
органические кислоты);- спинномозговая жидкость
(лактат, белок);
рентгенография черепа
(петрификаты);- ЭКГ, эхокардиография;- ЭЭГ, электромиография;- рентгеновская и магнитно-
резонансная компьютерная
томография- поиск мультиорганной
патологииНЕТСпецифический синдром
точковых мутаций мтДНК ?
(MELAS, MERRF, NARP и т.д.)ДААнализ лимфоцитов
крови на известные
точковые мутации
мтДНКмутациявыявленаСпецифическийдиагнозбиохимический и• световая ииммунологическииэлектроннаяанализ компонентовмикроскопия;дыхательной цепи• гистохимияМолекулярно¬
генетический анализ;перестройки мтДНК;
известные точковые
мутации мтДНК;
секвенирование мтДНКРис. 69. Современный алгоритм обследования больного с митохондриальной энцефаломиопатиейДНК- диагностика наследственных болезней ... 431
432Глава 3Однако такой наиболее благоприятный «сценарий» на
практике реализуется далеко не всегда.3) На рассматриваемых этапах основная пробле¬
ма заключается в том, что легко доступные клетки кро¬
ви не являются идеальным источником для поиска му¬
таций мтДНК. Клетки кроветворной системы размно¬
жаются весьма быстро, поэтому в силу митотической
сегрегации мутантной мтДНК ее содержание в крови
весьма вариабельно (от 0% до 60-80%). Более того, в
эмбриогенезе кроветворный росток может вообще не
унаследовать мтДНК мутантного вида, тогда как при этом
в пораженных тканях (мозг, мышцы и др.) число копий
мутантной мтДНК будет патогенетически значимым.
Например, пациенты с леберовской атрофией зритель¬
ных нервов и MERRF обычно имеют достаточно высо¬
кое содержание мутантной мтДНКв лимфоцитах (что
облегчает ДНК-диагностику этих болезней), тогда как
при MELAS и особенно синдроме Кернса-Сейра ситуа¬
ция обратная. Таким образом, отсутствие мутаций при
исследовании ДНК лимфоцитов отнюдь не исключает
митохондриальной природы болезни. Гораздо более ин¬
формативным источником являются скелетные мышцы,
поскольку, во-первых, отсутствие клеточных делений в
данной постмитотической ткани способствует «удержа¬
нию» митохондрий с мутантной мтДНК, а, во-вторых,
мышечная ткань является одной из основных «мише¬
ней» патологического процесса при митохондриальных
энцефаломиопатиях. Поэтому следующий шаг при об¬
следовании больного с неидентифицированной мутаци¬
ей предполагает проведение биопсии скелетной мышцы
(обычно четырехглавой или дельтовидной).4) Образцы мышечных биоптатов целесообразно
делить на 3 части - одна для микроскопического иссле¬
дования (гистология, гистохимия и электронная микро-
ДНК- диагностика наследственных болезней ... 433скопил), вторая для энзимологического и иммунологи¬
ческого анализа (изучение характеристик компонентов
дыхательной цепи) и третья - для молекулярно-генети¬
ческого анализа. Как и при исследовании ДНК крови,
анализ мтДНК мышцы начинается с исследования изве¬
стных точковых мутаций или перестроек, специфичес¬
ких для изучаемого синдрома. Поиск известных мута¬
ций на мышечном материале позволяет в большинстве
случаев успешно осуществлять ДНК-диагностику болез¬
ни. Следует добавить, что обычно наряду с мутантной
мтДНК в мышце выявляется нормальный вид мтДНК, и
в такой ситуации весьма ценным является дополнитель¬
ное определение уровня гетероплазмии (анализ дозы
мутантного гена с помощью количественной ПЦР или
блот-гибридизации по Саузерну). Установление процен¬
тного соотношения между нормальной и мутантной
мтДНК позволяет на молекулярном уровне объективи¬
зировать тяжесть поражения и оценить прогноз заболе¬
вания.5) При отсутствии известных мутаций мтДНК в
мышечной ткани следующим этапом молекулярно-гене-
тического анализа является секвенирование всей цепи
мтДНК. Это исследование, однако, является не только
достаточно трудоемким и дорогостоящим, но и связано
с определенными сложностями в интерпретации полу¬
чаемых результатов. Поэтому предварительно необходи¬
мо провести весь комплекс морфогистохимических и
биохимических анализов мышечного биоптата с целью
получения абсолютно достоверных данных, позволяю¬
щих подтвердить первичную митохондриальную пато¬
логию и по возможности установить конкретное звено
митохондраильной дисфункции. Так например, важны¬
ми маркерами могут служить выявляемые цитохром-с-
оксидазно-негативные и «рваные красные» волокна, на¬
434Глава 3рушение окисления пирувата и скорости синтеза АТФ,
дефекты активности отдельных субъединиц комплекса
дыхательной цепи и т.д. В ряде случаев указанные на¬
рушения позволяют весьма точно локализовать биохи¬
мический уровень поражения и предположительно ус¬
тановить ген или группу генов мтДНК, в которых может
иметь место мутация.6) Наконец, проводится секвенирование митохон¬
дриального генома с целью выявления нового варианта
мутации мтДНК. Как уже указывалось, особенностью
мтДНК является высокая частота спонтанных мутаци¬
онных событий, поэтому большую сложность представ¬
ляет обоснование патогенетической значимости той или
иной новой нуклеотидной вариации, выявленной у об¬
следуемого больного. Основными доказательствами того,
что обнаруженная и неизвестная ранее мутация мтДНК
является причиной болезни, считаются следующие при¬
знаки: а) гетероплазмия по данной мутации (гетероплаз-
мия свидетельствует о сравнительно недавнем происхож¬
дении мутации либо о том, что она несовместима с жиз¬
нью в состоянии гомоплазмии); б) более высокое содер¬
жание мутантной мтДНК в пораженных тканях по срав¬
нению с интактными; в) локализация мутации в эволю-
ционно консервативной области мтДНК, особенно если
соответствующая аминокислотная замена может суще¬
ственно нарушить структуру и функцию генного продукта;г) отсутствие данной мутации в нормальной популяции
(группе контроля). Последние 2 критерия применимы и
для мутаций, выявляемых в гомоплазмическом состоя¬
нии [СЫппегу Р. е! а1., 1999 (б)].С технической точки зрения, скрининг на извес¬
тные мутации проводится на основе метода ПЦР: амп-
лифицированный изучаемый участок мтДНК подверга¬
ется обработке подходящей рестрикционной эндонукле¬
ДНК- диагностика наследственных болезней... 435азой либо гибридизации со специфическим олигонукле¬
отидом. Пример такой ДНК-диагностики при синдроме
MELAS на примере мажорной мутации A3243G пока¬
зан на рис. 70.Рисунок 70. Прямая ДНК-диагностика мутации 3243А—>G
при синдроме MELASИсследование проведено на геномной ДНК, продукты амплифика¬
ции обработаны ресгриктазой Ара I. Дорожка 1 - маркер, дорожки
2 и 3 - больные с синдромом М ELAS, имеющие мутацию 3243А—Ю
в гетероплазмическом состоянии (данная мутация создает сайт ре¬
стрикции для фермента Ара I), дорожка 4 - здоровый контроль.
Нормальный фрагмент амплифицированной мтДН К имеет длину 330
п.о. (короткая стрелка), мутантная мтДНК разрезается ферментом
на 2 фрагмента длиной 213 и 117 п.о. (длинные стрелки).Перестройки и количественные дефекты мтДНК
(единичные и множественные делеции, дупликации, ис¬
тощение мтДНК) регистрируются методом блот-гибри-
дизации по Саузерну после обработки мтДНК рестрик¬
ционными эндонуклеазами (рис. 68 A-В). В последние1 23 4
436Глава 3годы в практику все шире входят новые высокоскорост¬
ные методы анализа ДНК, значительно облегчающие мо¬
лекулярную диагностику (автоматическое секвенирова-
ние, использование биологических микрочипов и др.).Идентификация генетического дефекта у пробан¬
да дает возможность проведения ДНК-диагностики у
родственников в группе риска - в первую очередь у бра-
тьев-сестер пробанда и у детей больной матери. В прин¬
ципе, при митохондриальном наследовании все сибсы -
дети одной матери являются носителями определенного
количества мутантной мтДНК, однако ее содержание в
крови и других тканях у сибсов чрезвычайно вариабель¬
но. Поэтому анализ уровня гетероплазмии позволяет с
известной осторожностью оценивать относительный
риск, прогноз возможного заболевания и необходимость
превентивного лечения у клинически здоровых лиц. Что
касается пренатальной ДНК-диагностики, то ее прове¬
дение в традиционном виде при митохондриальных бо¬
лезнях в настоящее время не рекомендуется. Дело в том,
что содержание мутантной мтДНК в ворсинах хориона
не дает однозначного ответа на вопрос о характере пора¬
жения мозга и других тканей плода, а также о дальней¬
шей динамике митотической сегрегации мутантной
мтДНК в тканях на протяжении жизни [Poulton J. et al.,1998]. В литератере описаны отдельные случаи искусст¬
венного прерывания беременности при выявлении в вор¬
синах хориона крайне высокого содержания исследуе¬
мой митохондриальной мутации [Harding A. et al., 1992],
однако этот вопрос до последнего времени остается дис¬
куссионным. Альтернативой может быть преимпланта-
ционная ДНК-диагностика, позволяющая в некоторых
случаях накануне фертилизации in vitro проводить пред¬
варительный отбор яйцеклеток, не содержащих опреде¬
ляемого количества мутантной мтДНК [Poulton J. et al., 1998].
ДНК- диагностика наследственных болезней ... 437Наконец, поистине революционизирующая статья была
опубликована в 1997 году в журнале «Lancet»: авторы
сообщили об успешном рождении здорового ребенка
после процедуры замещения цитоплазмы яйцеклетки,
содержащей мутантные митохондрии, цитоплазмой до¬
норской яйцеклетки и последующего оплодотворения in
vitro [Cohen J. et al., 1997]. Этот метод, однако, пред¬
ставляющий собой, по существу, «митохондриальное
клонирование», связан с серьезными и вряд ли разре¬
шимыми в обозримом будущем этическими проблемами.Диагностика мутаций в ядерных генах при мито¬
хондриальных энцефаломиопатиях, наследующихся по
менделевскому типу, широкого распространения не по¬
лучила и используется лишь для решения специальных:
исследовательских задач в небольшом числе профиль¬
ных лабораторий. На практике более доступным и целе¬
сообразным методом специфической диагностики дан¬
ной подгруппы митохондриальных болезней является
соответствующий биохимический скрининг.
ГЛАВА 4Молекулярно-генетический
анализ наиболее
распространенных
мультифакториальных
заболеваний нервной системыВ предыдущих главах была подробно представ¬
лена методология исследования моногенных мендели-
рующих заболеваний нервной системы, развитие кото¬
рых детерминируется повреждением одного основного
гена. В силу сравнительной простоты «генетической
системы» данных болезней прогресс в их изучении явил¬
ся весьма впечатляющим этапом развития медицинской
науки, а надежные методы ДНК-диагностики стали ес¬
тественным, а в ряде случаев даже рутинным подходом
в арсенале практического врача. В то же время молеку¬
лярно-генетический анализ мультифакториальных болез¬
ней, или болезней предрасположенности, представляет
собой гораздо более сложную задачу, в решении кото¬
рой делаются лишь первые шаги. Это связано с суще¬
ствованием для мультифакториальных болезней большо¬
го числа генов, каждый из которых на разных этапах
вносит свой вклад в формирование клинического фено¬
типа. При этом вероятность реализации имеющейся на¬
следственной предрасположенности к определенному
заболеванию (т.е. вероятность проявления или непрояв-
ления патологического фенотипа) определяется резуль¬
татом взаимодействий совокупности генов и факторов
внешней среды.
Молекулярно-генетический анализ439Существуют различные модели, описывающие
механизм формирования и реализации наследственной
предрасположенности к тому или иному мультифакто-
риальному заболеванию. Согласно одной из них, разви¬
тие заболевания и его тяжесть обусловлены суммарным
аддитивным эффектом мутантных аллелей ряда «малых»
генов, эффекты которых в отдельности незначительны
и лишь при определенной «неблагоприятной» комбина¬
ции могут превысить функциональный порог, приводя к
болезни. С другой стороны, при некоторых формах па¬
тологии нельзя исключить, что на фоне аддитивного
действия нескольких генов решающим фактором мани¬
фестации болезни является влияние основного локуса,
условно патогенный эффект которого «запускается» спе¬
цифическим неблагоприятным средовым фактором [Ли-
льин Е.Т. и др., 1990; Бочков Н.П., 1997]. В последнем
случае указанный основной локус определяет наиболь¬
ший удельный вес наследственной предрасположеннос¬
ти. Накопление в семье повторных случаев заболевания
связано с действием общих факторов риска (как генети¬
ческих, так и средовых), но при этом, ввиду сложного и
многоступенчатого механизма реализации имеющейся
предрасположенности, характер наследования болезни
в семье не подчиняется простым менделевским моде¬
лям. В целом, общая частота мультифакториальных за¬
болеваний в популяции является весьма высокой.К числу наиболее распространенных мультифак¬
ториальных заболеваний, имеющих значительную гене¬
тическую составляющую, относятся гипертоническая
болезнь, атеросклероз и сахарный диабет. В последние
были достигнуты большие успехи в раскрытии патоге¬
нетических механизмов этих широко распространенных
заболеваний человека, связанные с идентификацией ге¬
нов рецепторов липопротеидов и инсулина, ангиотензин-
440Глава 4ковертирующего фермента, факторов свертывания, ре¬
гуляторных пептидов, различных сигнальных молекул и
т.д. [Бочков Н.П., 1997; Пузырев В.П., 2000; Weissman S.,
1995; Thomson G., Esposito M., 1999]. При этом не мень¬
шую роль в развитии указанных болезней играет харак¬
тер питания, эмоциональный стресс и другие социаль-
но-средовые факторы. Среди наиболее распространен¬
ных психоневрологических полигенных заболеваний с
наследственной предрасположенностью можно назвать
шизофрению, маниакально-депрессивный психоз, бо¬
лезнь Паркинсона, болезнь Альцгеймера, рассеянный
склероз, миастению и др. Ниже представлены некото¬
рые наиболее общие подходы к анализу молекулярно¬
генетических механизмов мультифакториальных заболе¬
ваний нервной системы, которые уже сегодня нашли свое
отражение в клинике благодаря появлению специфичес¬
ких ДНК-тестов.4.1. Анализ генетических ассоциацийАнализ генетических ассоциаций (исследование
полиморфизмов в генах-кандидатах) является в настоя¬
щее время ведущим подходом при изучении роли гене¬
тической составляющей в патогенезе мультифакториаль¬
ных заболеваний человека, в том числе заболеваний не¬
рвной системы [Lander E., Schork N., 1994]. В его осно¬
ве лежит тот отмеченный выше факт, что при мульти¬
факториальных болезнях важнейшим фактором наслед¬
ственной предрасположенности является совокупное
действие комплекса генов, определяющих характер клю¬
чевых биохимических и иммунологических процессов в
организме (особенности метаболизма, характер иммун¬
ного ответа, эффективность энергетических реакций и
т.д.). Вычленение из общего «генетического фона» наи¬
Молекулярно-генетический анализ441более значимых генов (т.е. оказывающих наиболее су¬
щественное влияние на вероятность развития того или
иного заболевания) и составляет сущность анализа ас¬
социаций [Lander E., SchorkN., 1994; Thomson G., 1995
(a); Elston R., 1998; Thomson G., Esposito M., 1999].Теоретической предпосылкой анализа ассоциаций
является существование в большинстве генов человека
особых вариабельных участков (полиморфизмов), игра¬
ющих роль генетических маркеров и различающихся у
отдельных индивидуумов по нуклеотидному составу —
например, по нейтральной однонуклеотидной замене или
по числу коротких нуклеотидных повторов. Таким об¬
разом, каждый их этих генов может существовать в виде
достаточно большого числа различных вариантов (алле¬
лей). Анализ генетических ассоциаций заключается в
сравнении частоты маркерных аллелей определенного
гена в группе больных с частотой данных аллелей в об¬
щей популяции. В том случае, если у больных наблюда¬
ется достоверное отклонение распределения маркерных
аллелей по сравнению с контрольной группой, напри¬
мер достоверное преобладание у них определенного ал-
леля, делается заключение о наличии ассоциации между
данным аллелем гена и изучаемым заболеванием. Таким
образом, есть принципиальное различие между рассмот¬
ренным в главе 1 генетическим сцеплением и генети¬
ческой ассоциацией: генетическое сцепление предпола¬
гает взаимосвязь между локусами (маркерным локусом
и локусом болезни), тогда как в случае генетической ас¬
социации речь идет о взаимосвязи заболевания с конк¬
ретным аллелем изучаемого генетического локуса.Возможны 2 основные причины выявленной ге¬
нетической ассоциации. Во-первых, повышенная (пони¬
женная) частота тех или иных аллелей конкретного гена
в группе больных может свидетельствовать о том, что
442Глава 4данные варианты гена и его белкового продукта оказы¬
вают специфическое модулирующее действие на функ¬
ционирование биохимических каскадов, определяющих
механизм развития патологического процесса. В такой
ситуации изучаемый ген имеет прямое этиологическое
значение в реализации наследственной предрасположен¬
ности к заболеванию, а соответствующие аллели гена
могут служить генетическими маркерами и факторами
риска (антириска) по данному заболеванию. Во вторых,
генетическая ассоциация может быть следствием того,
что изучаемый маркер чисто механически находится в
тесном сцеплении с одним из «этиологически значимых»
генов. В этом случае говорят о неравновесном сцепле¬
нии маркерного аллеля и другого, неизвестного гена,
ответственного за возникновение заболевания. Неравно¬
весное сцепление предполагает, что у значительного
большинства больных имеется одна и та же мутантная
хромосома, распространившаяся в популяции от обще¬
го предка, а изучаемый маркерный аллель не играет са¬
мостоятельной патогенетической роли и расположен в
непосредственной близости от «истинного» гена болез¬
ни [Elston R., 1998; Thomson G., Esposito М., 1999]. Не¬
зависимо от того, какая из двух возможных причин ге¬
нетической ассоциации имеет место при конкретном за¬
болевании, выявление такой ассоциации является важ¬
нейшим шагом в уточнении молекулярных механизмов
наследственной предрасположенности к данному забо¬
леванию: в первом случае речь идет непосредственно об
идентификации гена предрасположенности, во втором -
об установлении хромосомной локализации такого гена.При анализе генетических ассоциаций могут ис¬
пользоваться как выборки изолированных случаев забо¬
левания, так и группы семей, в которых имеет место на¬
копление повторных случаев данного заболевания. Клю¬
Молекулярно-генетический анализ443чевым требованием является одинаковый этнический
состав группы больных и контрольной группы, поскольку
частоты конкретных аллелей изучаемых генетических
маркеров могут существенно различаться в разных по¬
пуляциях [Thomson G., 1995 (б); Thomson G., Esposito М.,1999]. Еще одним важным условием при изучении гене¬
тических ассоциаций является знание (хотя бы в общих
чертах) некоторых звеньев патогенеза изучаемого муль-
тифакториального заболевания, поскольку на основании
этого может быть отобран круг соответствующих генов-
кандидатов для проверки теоретической гипотезы об их
возможной роли в формировании предрасположеннос¬
ти к данному заболеванию.В неврологии классическим примером выражен¬
ной генетической ассоциации является значение аполи-
попротеинаЕ как важнейшего эндогенного фактора риска
в развитии поздней формы болезни Альцгеймера - за¬
болевания, которым в мире страдают около 20 млн. че¬
ловек. Аполипопротеин Е (апоЕ) представляет собой
белок с молекулярной массой 34 кДа, кодируемый ге¬
ном на хромосоме 19ql3.2 [Mahley R., 1988]. АпоЕ иг¬
рает ключевую роль в метаболизме липидов (особенно
холестерина), способствуя их перераспределению меж¬
ду клетками различных органов [Mahley R., 1988;
Mahley R. et al., 1996]. В 1993 году W. Strittmatter с соав¬
торами установили, что апоЕ является одним из протеи¬
нов, специфически связывающихся с Р-амилоидом (ос¬
новным компонентом амилоидной бляшки - морфоло¬
гического маркера болезни Альцгеймера); это послужи¬
ло основанием для детального анализа ассоциации апоЕ
и болезни Альцгеймера. Известно, что ген апоЕ имеет 3
основных аллеля (s2, еЗ и s4), отличающихся единич¬
ными нуклеотидными заменами и определяющих суще¬
ствование 3 изоформ белка апоЕ, причем в общей попу¬
444Глава 4ляции аллель s3 наиболее распространен. В серии ис¬
следований, проведенных в 1993-1996 гг., было установ¬
лено, что аллель s4 гена апоЕ встречается достоверно
чаще у больных с поздней формой болезни Альцгейме¬
ра - как семейной (50%), так и спорадической (40%), в
то время как в контрольной группе его частота не пре¬
вышает 14-16% [Strittmatter W. et al., 1993; Saunders A.
et al., 1993; Locke P. et al., 1995; Schellenberg G., 1995;
Strittmatter W., Roses A., 1996]. Более того, риск разви¬
тия на протяжении жизни болезни Альцгеймера в зави¬
симости от генотипа апоЕ является доза-зависимым: у
гомозиготных носителей аллеля s4 он является наивыс¬
шим и составляет около 90% (т.е. 90% лиц с генотипом
s4/s4 заболеют при условии достижения возраста -70-
75 лет), у гетерозиготных носителей в4 он равен 47%,
тогда как лишь 20% лиц, не имеющих аллеля в4, заболе¬
ют болезнью Альцгеймера в пожилом возрасте [Corder Е.
et al., 1993; vanBroeckhovenC., 1995; Blacker D.,Tanzi R.,1998]. Доза «неблагоприятного» аллеля s4 напрямую
коррелирует также с интенсивностью формирования
амилоидных бляшек в мозге больных с болезнью Аль-
цеймера [Schmechel D. et al., 1993]. Интересно отметить,
что средний возраст начала заболевания у гомозиготных
носителей аллеля s4 является достоверно более ранним
(68 лет) по сравнению с гетерозиготными носителями
апоЕ-е4 (75 лет) и лицами с другими генотипами апоЕ
(84 года). Другими словами, каждая копия аллеля s4
«приближает» возраст начала болезни Альцгеймера на
5-9 лет [Corder Е. etal., 1993; Blacker D., Tanzi R., 1998].
С другой стороны, носительство аллеля s2 (особенно
двух его копий) обладает протективным эффектом в от¬
ношении развития на протяжении жизни болезни Альц¬
геймера [Corder Е. et al., 1994]. Подтверждением патоге¬
нетической значимости ассоциаций аллелей апоЕ и бо¬
Молекулярно-генетический анализ445лезни Альцгеймера являются данные о взаимосвязи раз¬
личных генетических вариантов аиоЕ с особенностями
клинической картины заболевания и реакции на прово¬
димое лечение [Lovestone S., 1999]. Указанные законо¬
мерности действия различных вариантов гена апоЕ в
качестве мощных факторов риска и антириска болезни
Альцгеймера справедливы практически для всех иссле¬
дованных популяций мира [Schellenberg G., 1995].Открытие роли гена апоЕ в развитии болезни
Альцгеймера в позднем возрасте поставило вопрос о кли¬
ническом значении обнаружения у конкретных индиви¬
дуумов «критического» аллеля апоЕ-в4. Как известно, в
целом для пожилых людей риск развития болезни Аль¬
цгеймера сам по себе является достаточно высоким и
повышается с каждым прожитым годом. Так, распрост¬
раненность болезни в группе лиц 65-74 года составляет
3%, для следующего десятилетия - 18% и для лиц стар¬
ше 85 лет - около 47% [Katzman R., 1976]; следователь¬
но, в связи с неуклонным постарением населения все
большее число пожилых людей будет попадать в катего¬
рию относительного высокого риска по болезни Альц¬
геймера. Таким образом, изменение степени этого рис¬
ка в зависимости от наличия конкретных аллелей апоЕ
не носит «драматический» характер и, по-видимому, не
является в настоящее время основанием для проведения
широкомасштабных программ генетического тестирова¬
ния пожилого населения, направленных на установле¬
ние вероятности развития у них болезни Альцгеймера
[Seshardi S. et al., 1995; National Institute on Aging/
Alzheimer’s Association Working Group, 1996; Lovestone S., 1999].
Такое тестирование, по-видимому, могло бы основывать¬
ся на комбинированном исследовании гена апоЕ и дру¬
гих полиморфных генов, определенные варианты кото¬
рых привносят дополнительный риск в отношении разви¬
446Глава 4тия болезни Альцгеймера [Roses А., 1997; Sheu К. et al.,
1999; Zubenko G. et al., 1999]. В будущем, при внедре¬
нии в практику высокоэффективных методов лечения
болезни Альцгеймера, прогностическое ДНК-тесгиро-
вание пожилых лиц с выявлением носительства апоЕ-в4
и других «неблагоприятных» аллелей может принять
более активный и целенаправленный характер, связан¬
ный с необходимостью формирования достоверной груп¬
пы «высокого риска» и возможностью проведения та¬
ким клинически здоровым людям превентивной тера¬
пии. Несколько иначе обстоит дело с ДНК-диагности-
кой аллельных вариантов гена апоЕ у пожилых больных,
обратившихся в клинику с нарушениями памяти и дру¬
гими симптомами, позволяющими предполагать у них
раннюю стадию болезни Альцгеймера. Большинством
авторов признается, что при наличии сложностей в кли¬
нической диагностике обнаружение аллеля s4 (и особен¬
но генотипа s4/s4) может, наряду с результатами других
методов исследования, служить важным дополнитель¬
ным диагностическим маркером, косвенно подтвержда¬
ющим диагноз болезни Альцгеймера [Saunders A. et al.,
1996; Lovestone S., 1999]. Окончательное решение это¬
го вопроса требует дальнейшего накопления данных (в
особенности - проспективного наблюдения за тестиро¬
ванными лицами и анализа репрезентативных серий сек¬
ционных случаев), поэтому в настоящее время практи¬
ческая роль ДНК-тестирования гена апоЕ с целью ран¬
ней диагностики болезни Альцгеймера остается дискус¬
сионной [Blacker D., Tanzi R., 1998].Таким образом, в клинической медицине болезнь
Альцгеймера является первым примером распространен¬
ного заболевания, для которого установлен ведущий ге¬
нетический фактор предрасположенности. В связи с
этим болезнь Альцгеймера может рассматриваться как
Молекулярно-генетический анализ447своеобразная модель для разработки методологических
аспектов ДНК-тестирования при мультифакториальных
болезнях человека (включая решение медицинских, со-
циально-правовых и этических вопросов) [Roses А.,1997]. Если при наследственных моногенных болезнях
мутационный анализ касается сравнительно немногочис¬
ленной группы риска в отягощенных семьях, то при бо¬
лезни Альцгеймера результаты ДНК-тестирования, по¬
тенциально несущие благоприятный или неблагоприят¬
ный прогноз, затрагивает десятки миллионов людей.
Именно поэтому, как указывалось выше, внедрение в
практику методов ДНК-анализа апоЕ и других генов
предрасположенности к болезни Альцгеймера не может
форсироваться и должно основываться только на строго
доказанных и многократно подтвержденных положени¬
ях. На сегодняшний день очевидно, что сам по себе ген
апоЕ не является единственным детерминантом «гене¬
тической судьбы» обследуемых лиц, что подтверждает¬
ся следующими хорошо известными фактами: а) не ме¬
нее чем в половине случаев болезнь Альцгеймера разви¬
вается у людей, не иметощих «неблагоприятного» алле-
ля anoE-s4; б) у носителей аллеля s4 риск развития бо¬
лезни Альцгеймера, хотя и значительно превышает об¬
щепопуляционный, но все же не является абсолютным;3) отмеченная выше ассоциация апоЕ-в4 и болезни Аль¬
цгеймера заметно снижается в популяции наиболее по¬
жилых лиц, переживших «критический» возраст (>80
лет) [van Broeckhoven С., 1995; Roses А., 1997; Sheu К. et
al., 1999]. Обобщая сказанное, можно еще раз подчерк¬
нуть, что ген апоЕ является лишь одним из звеньев слож¬
ной «мозаики» взаимодействующих факторов, опреде¬
ляющих возможность развития позднего варианта бо¬
лезни Альцгеймера.
448Глава 4Молекулярные механизмы, объясняющие повы¬
шенную предрасположенность к болезни Альцгеймера
у носителей ал л ел я апоЕ-в4, были предметом интенсив¬
ных исследований последних лет. Основное внимание
при этом уделялось изучению кинетики межмолекуляр-
ных взаимодействий различных изоформ белка апоЕ,
каждой из которых свойственны свои особенности кон¬
формации пептидной молекулы. Имеющиеся в литера¬
туре данные позволяют предполагать, что е4-изоформа
белка характеризуется более выраженным амилоидоген¬
ным потенциалом благодаря высокому сродству к (3-ами¬
лоиду, а также относительно непрочным связыванием с
белком тау (гиперфосфорилирование свободных моле¬
кул которого лежит в основе образования характерных
для болезни Альцгеймера нейрофибриллярных клубков)
[Schmechel D. et al., 1993; Weisgraber К. et al., 1994;
Schelleberg G., 1995; van Broeckhoven C., 1995]. Экспе¬
рименты на трансгенных животных свидетельствуют о
прямом участии апоЕ в формировании фибриллярных
депозитов Р-амилоида [Mattson М., 1997]. Обсуждается
также роль апоЕ как сигнального протеина, регулирую¬
щего механизмы липидного транспорта (в частности,
мобилизации «строительного» холестерина) в процессе
синаптогенеза и регенерации нейрональных мембран
[Mahley R. et al., 1996]. Последняя гипотеза подтверж¬
дается значительным (в несколько сот раз) повышением
уровня экспрессии апоЕ при экспериментальном повреж¬
дении седалищного нерва [Ignatius М. et al., 1986]. Наи¬
более вероятным представляется комплексный характер
участия апоЕ в патогенезе болезни Альцгеймера.Предположение об универсальной роли апоЕ в
механизмах повреждения и регенерации нейронов обус¬
ловило многочисленные исследования, направленные на
анализ генетических ассоциаций между конкретными
аллелями апоЕ и рядом других мультифакториальных
Молекулярно-генетический анализ449заболеваний центральной нервной системы (инсульт,
болезнь Паркинсона, множественные системные атрофии
и др.). В этих исследованиях наиболее определенные ре¬
зультаты были достигнуты при изучении группы нейро-
дегенеративных заболеваний. J. Schneider с соавторами(1995) обнаружили повышенную частоту аллеля s4 у боль¬
ных кортико-базальной дегенерацией, болезнью Пика и
прогрессирующим супрануклеарным параличом. В не¬
скольких недавних исследованиях было показано, что у
больных болезнью Паркинсона ген апоЕ модулирует воз¬
раст начала заболевания, поскольку носительство аллеля
s4 было достоверно ассоциировано с более ранним дебю¬
том симптомов (на 4-7 лет) [Zareparsi S. et al., 1997; Krüger
R. et al., 1999]. В то же время, распределение различных
аллелей апоЕ в группе больных болезнью Паркинсона не
отличается от такового в общей популяции [Rubinsztein
D. et al., 1994; Koller W. et al., 1995]. Наши предваритель¬
ные данные, полученные при изучении спорадической
болезни Паркинсона в российской популяции, также сви¬
детельствуют в пользу более раннего начала заболевания
у носителей «неблагоприятного» аллеля s4, что может
служить подтверждением роли апоЕ в механизмах пред¬
расположенности к данному заболеванию. На рис. 71 по¬
казан пример тестирования гена апоЕ у больных болез¬
нью Паркинсона.При болезни Паркинсона проводился анализ ас¬
социаций с целым рядом других генов-кандидатов, кото¬
рые, предположительно, могут быть связаны с различны¬
ми звеньями патогенеза данного заболевания. В числе
таких кандидатов исследовались гены дофаминовых ре¬
цепторов, переносчиков дофамина, моноаминоксидаз,
I юкоторых ферментов, метаболизирующих разнообразные
1 жзотоксины или участвующих в реакциях окислительного
фосфорилирования, митохондриальные гены и др. (таб-
н ица 8).
450Глава 41 2 3 4 5Рис.71. Анализ полиморфизма в гене апоЕ при болезни
ПаркинсонаЭлектрофореграмма продуктов рестрикции (использована рестрик-
таза Нка I). Дорожки 14-больные с ранней формой болезни Пар¬
кинсона, дорожка 5 - больной с поздней формой болезни Паркин¬
сона. Внизу под каждой дорожкой обозначены генотипы больных.
Стрелкой указан фрагмент длиной 72 п.о., специфичный для аллеля
е4. Три из четырех обследованных больных с ранней формой бо¬
лезни Паркинсона являются носителями аллеля е4 в гомо- или гете¬
розиготном состоянии.Проведенные исследования показали, что в от¬
дельных выборках больных болезнью Паркинсона на¬
блюдается изменение распределения аллельных часто!
генов дофаминовых 02-рецепторов, моноаминоксидаз А-
и В-типов, тирозин-гидроксилазы, Ы-ацетилтрансфера-
зы, 4-гидроксилазы цитохрома Р450 (С \T2D6), Е2-субъе-
диницы а-кетоглутарат-дегидрогеназного комплекса и
некоторых митохондриальных полиморфизмов
[БЬоАЬег I е1а1., 1993; И^исЫ 8. ег а1., 1995; ТЧшзЬаит Я.,
Ро1утегорои1о8 М., 1997; \¥оос!К., 1997; КоЬауаэЫ Т. е!
а1„ 1998;№с1ю1т.е1а1., 1999; Зрасеу 8., \УоосШ., 1999|.
Молекулярно-генетический анализ451Таблица 8
Анализ генетических ассоциаций
при болезни ПаркинсонаИсследованный ген-кандидат (белок)РезультатанализаАполипопротеин Е(апоЕ)+Дофаминовый 02-рецептор (01Ш2)+ -Дофаминовый ОЗ-рецептор (01103)-Дофаминовый 04-рецептор (01Ю4)-Транспортер дофамина (0АТ1)-Моноаминоксидаза А-типа (МАО-А)+ -Моноаминоксидаза В-типа (МАО-В)+ -Тирозин-гидроксилаза+ -Е2-субъединица а-кетоглутарат-дегидрогеназного комплекса+Семейство генов цитохрома Р450 (СУР1А1, СУР206, СУР2Е1)+ -Глутатион-трансферазы М1 и Т1-Ы-ацетилтрансфераза 2 (ЫАТ2)-ЫАОРН-редуктаза-Митохондриальные полиморфизмы
(нуклеотидные положения 3397, 4336, 5460 и др.)+ -Примечание: “+” означает подтвержденную ассоциацию (повышение частоты
определенного аллеля гена у больных болезнью Паркинсона или
модулирующий эффект аллеля на возраст начала заболевания), “+ означает
ассоциацию, выявленную лишь в отдельных выборках обследованных больных,
означает отсутствие ассоциации.Однако указанные генетические ассоциации при болез¬
ни Паркинсона не во всех случаях могут быть подтвер¬
ждены при исследовании других популяций, что может
отражать существование определенных межпопуляцион-
ных различий аллельных частот изучаемых генов, а так¬
же различий удельного веса разнообразных генетичес¬
ких и средовых факторов в патогенезе болезни Паркин¬
сона [Spacey S., Wood N., 1999]. Таким образом, в меха¬
низмах предрасположенности к болезни Паркинсона
нажную роль играет совокупное действие ряда генов,
452Глава 4контролирующих активность дофаминергической транс¬
миссии, процессы детоксикации и клеточной энергети¬
ки. Вклад этих генетических факторов, детерминирую¬
щих определенный «метаболический фон» у близких
родственников, а также сходный характер разнообраз¬
ных экзогенных воздействий у членов одной семьи ле¬
жат в основе нередко наблюдающегося накопления по¬
вторных случаев болезни Паркинсона в обследуемых
семьях; не случайно положительный семейный анамнез
по болезни Паркинсона выявляется у 10,3% больных с
этим заболеванием (против 3,5% в контрольной группе)
[ElbazA. etal., 1999].Прогрессирующий супрануклеарный паралич
(ПСП) представляет собой второй по частоте (после бо¬
лезни Паркинсона) синдром паркинсонизма дегенератив¬
ной природы [Bowel J. et al., 1997]. В абсолютном боль¬
шинстве случаев ПСП является спорадическим заболе¬
ванием, имеющим мультифакториальную природу, по¬
этому в последние годы основное внимание было сфо¬
кусировано на поиске генов предрасположенности к дан¬
ному тяжелому страданию. Характерным нейрогистохи-
мическим критерием ПСП является формирование ли¬
нейных нейрофибриллярных филаментов, образованных
преципитатами гиперфосфорилированной формы кле¬
точного белка тау [Bergeron С. et al., 1998]. В связи с
этим рядом авторов исследовалась генетическая струк¬
тура тау у больных ПСП. Показано, что в группе боль¬
ных данным заболеванием достоверно чаще встречает¬
ся сочетание ряда специфических полиморфных участ¬
ков гена тау, образующих характерный редкий гаплотип,
практически не наблюдающийся у лиц контрольной груп¬
пы [Baker М. et al., 1999; Higgins J. et al., 1999]. Данный
гаплотип сопровождается экспрессией в веществе мозга
(особенно в стволе - первичной «мишени» патологичес¬
Молекулярно-генетический анализ453кого процесса при ПСП) особой изоформы белка тау,
взаимодействие которой с микротубулярным аппаратом
нейронов и клеток глии существенно меняется
[Chambers С. et al., 1999]. Предполагается, что такой ме¬
ханизм способствует образованию нейрофибриллярных
преципитатов, являющихся важнейшим звеном патоге¬
неза ПСП. Таким образом, доказанная ассоциация ПСП
с определенными генетическими вариантами тау, а так¬
же (как указывалось выше) с аллелем в4 гена апоЕ по¬
зволяет говорить об идентификации, как минимум, 2
генов предрасположенности к развитию ПСП. Для уточ¬
нения диагностического и прогностического значения ре¬
зультатов ДНК-тестирования указанных генов в группе
риска (например, у родственников больных ПСП) тре¬
буется дальнейший анализ катамнестических данных на
основе многолетних наблюдений за обследованными
группами клинически здоровых лиц.4.2. Иммуногенетический анализ
мультифакториальных
заболеваний нервной системыИнтенсивные исследования механизмов генети¬
ческой предрасположенности проводились на протяже¬
нии многих лет в группе аутоиммунных заболеваний
нервной системы. Одними из ведущих факторов риска
при данных заболеваниях являются гены главного комп¬
лекса гистосовместимости человека. Данный генетичес¬
кий кластер, содержащий около 200 генов, расположен
на коротком плече 6-й хромосомы [Thorsby Е., 1997].
Белковые продукты главного комплекса гистосовмести¬
мости представляют собой поверхностные клеточные
антигены, наиболее полно представленные в лейкоци¬
тах, которые распознаются иммунной системой хозяина
454Глава 4в качестве «своих» и дифференцируются от поступае-
мых в организм чужеродных антигенных детерминант.
В соответствии с этим семейство генов главного комп¬
лекса гистосовместимости человека получило название
HLA-комплекс (от англ. Human Leukocyte Antigen). Фун¬
кция HLA-комплекса заключается в обеспечении конт¬
роля за специфичностью и корректностью реакций им¬
мунного ответа организма; обсуждается также роль ан¬
тигенов, кодируемых HLA-комплексом, в развитии раз¬
личных клонов иммунокомпетентных клеток в процес¬
се эмбрионального развития [Фогель Ф., Мотульски А.,
1989]. Гены главного комплекса гистосовместимости
тесно сцеплены с рядом других генов иммунного ответа
на 6-й хромосоме и представляют собой важнейший эле¬
мент сложной целостной системы, регулирующих кон¬
такт клеток со средой.Комплекс HLA включает ряд локусов, разделен¬
ных на три класса и обозначаемых символами А, В, С,
DR, DQht-д. [Зотиков Е.А., 1982; Bodmer J. etal., 1990].
Характерной особенностью генов каждого локуса HLA
является их полиморфизм, т.е. существование большого
числа аллельных вариантов, которые могут идентифи¬
цироваться как на белковом уровне (специальными ан¬
тисыворотками), так и с помощью соответствующих
молекулярно-генетических методов. Это делает систему
HLA весьма удобной для изучения генетических ассо¬
циаций. Исследование системы HLA представляет со¬
бой один из разделов иммуногенетики. В широком смыс¬
ле слова иммуногенетика включает в себя также генети¬
ку групп крови и ряда других показателей иммунного
гомеостаза.Наибольшее развитие иммуногенетические иссле¬
дования получили при таком распространенном аутоим¬
мунном заболевании нервной системы, как рассеянный
Молекулярно-генетический анализ455склероз. Данные, полученные при анализе различных
популяций, свидетельствуют о том, что у больных рас¬
сеянным склерозом достоверно повышена частота встре¬
чаемости антигенов HLA-A3, HLA-B7, HLA-DR2, HLA-
DR3, HLA-DQ6 и некоторых других [Завалишин И.А.,
1990; Гусев Е.И. и др., 2001; Olerup О., Hillert J., 1991;
Dyment D. et al., 1997]. При рассеянном склерозе отме¬
чена также ассоциация болезни с определенными гап-
лотипами HLA, включающими специфические аллель¬
ные комбинации локусов главного комплекса гистосов¬
местимости (в указанных комбинациях, в частности, об¬
наруживается преобладание аллеля HLA-B7- возможно,
как следствие неравновесного сцепления с основным
гаплотипом локуса DRB1) [Гусев Е.И. и др., 2001;
Hauser S. et al., 1989; Allen M. et al., 1994]. Имеются дан¬
ные о существовании ряда «протективных» антигенов
(таких как HLA-B17, HLA-B40 и HLA-DR1), выявляе¬
мых у больных рассеянным склерозом достоверно реже,
чем в контроле, а также об определенной взаимосвязи
между конкретными антигенными детерминантами и
особенностями течения заболевания [Завалишин И.А.,
1990; Гусев Е.И. и др., 2001; Tiwari J., Terasaki P., 1985].Анализ ряда других генов-кандидатов, имеющих
важное значение в механизмах реализации иммунного
ответа, позволил установить существование дополни¬
тельных звеньев генетической системы предрасположен¬
ности к рассеянному склерозу. К таким звеньям отно¬
сятся гены особого семейства провоспалительных цито-
кинов - факторов некроза опухоли а и (3 (TNF-а, TNF-P),
для которых показано преобладание определенных «не¬
благоприятных» аллелей в группе больных рассеянным
склерозом [Fugger L. et al., 1990; McDonnell G. et al.,1999]. Поскольку гены фактора некроза опухоли тесно
сцеплены с локусом HLA-DR на 6-й хромосоме, указан¬
456Глава 4ное специфическое распределение аллелей TNF может
быть в какой-то степени обусловлено наличием досто¬
верной ассоциации между аллелями HLA-комплекса и
рассеянным склерозом. Однако обнаружение повышен¬
ного уровня TNF в демиелинизирующих бляшках и спин¬
номозговой жидкости больных рассеянным склерозом
[Selmaj K. et al., 1991; Sharief М., Henteges R., 1992], а
также положительный эффект растворимой формы TNF
на активность процесса у больных рассеянным склеро¬
зом [Dyment D. et al., 1997] свидетельствует о патогене¬
тической значимости фактора некроза опухоли при дан¬
ном заболевании. Еще одним геном предрасположенно¬
сти к рассеянному склерозу является ген ß-цепи рецеп¬
тора Т-лимфоцитов: показана ассоциация «неблагопри¬
ятных» аллелей данного гена с прогредиентной формой
рассеянного склероза и экспериментальным аллергичес¬
ким энцефаломиелитом, особенно при сочетанном на¬
следовании с определенными аллелями комплекса HLA
[Goverman J. et al., 1993; Hockertz M. et al., 1998].Таким образом, в настоящее время не вызывает
сомнений существование наследственной предрасполо¬
женности к рассеянному склерозу. Она определяется
суммарным действием ряда взаимосвязанных генов им¬
мунного ответа, ведущими из которых являются гены
главного комплекса гистосовместимости. Предполагает¬
ся, что наследование специфических аллелей указанных
генов во взаимодействии с рядом природно-средовых
факторов может формировать повышенную либо пони¬
женную восприимчивость иммунной системы к воздей¬
ствию определенного этиологического фактора, «запус¬
кающего» патогенетический каскад при рассеянном скле¬
розе, например - гипотетического вирусного агента [За-
валишин И.А., 1990]. Существование наследственной
предрасположенности к рассеянному склерозу подтвер¬
Молекулярно-генетический анализ457ждается наличием семейных случаев данного заболева¬
ния (до 10%), более высокой конкордантностью по рас¬
сеянному склерозу среди монозиготных близнецов по
сравнению с дизиготными парами, а также более высо¬
кой (в 20-30 раз) заболеваемостью рассеянным склеро¬
зом среди родственников больных по сравнению с об¬
щей популяцией [Завалишин И.А., 1990; Гусев Е.И. и
др., 2001; Dyment D. et al., 1997]. В связи с этим ДНК-
тестирование и выявление конкретных полиморфизмов
в указанных генах имеет прогностическое значение для
определения степени риска заболевания рассеянным
склерозом у родственников больных (в более широком
смысле - для выявления круга лиц, предрасположенных
к рассеянному склерозу). Результаты иммуногенетичес-
кого анализа могут также играть определенную диагно¬
стическую роль, особенно в некоторых сложных клини¬
ческих ситуациях. Так, например, по мнению ряда авто¬
ров, выявление у больных ретробульбарным невритом
аллеля HLA-B7 позволяет, по-видимому, с большей уве¬
ренностью говорить о возможности дебюта рассеянно¬
го склероза и значительном риске развития в дальней¬
шем соответствующего распространенного поражения
центральной нервной системы.Ассоциация с аллелями главного комплекса гис¬
тосовместимости была выявлена еще при некоторых
мультифакториальных заболеваниях нервной системы -
синдроме Гийена-Барре, миастении, нарколепсии и др.,
хотя полученные результаты были не столь убедитель¬
ными, как при рассеянном склерозе [Rees J. et al., 1995;
ThorsbyE., 1997; Thomson G., Esposito М., 1999]. У боль¬
ных синдромом Гийена-Барре, развившимся после ин¬
фекции Campylobacter jejuni (один из возможных этио¬
логических факторов данной формы полинейропатии),
показано достоверное преобладание определенного ал-
458Глава 4леля гена фактора некроза опухоли, в то время как у
Сатру1оЪа&ег-негативных пациентов с синдромом Гий-
ена-Барре такой ассоциации с геном фактора некроза
опухоли выявлено не было [Ма I. е! а1., 1998]. Патогене¬
тическая роль данного гена в развитии синдрома Гийе-
на-Барре подтверждается повышенным содержанием
фактора некроза опухоли в сыворотке больных и корре¬
ляцией его уровня с тяжестью заболевания [8ЬапеГМ. е!
а1., 1993]. 0.8ке1е с соавторами (1999) показали взаимо¬
связь между различными клиническими вариантами
миастении (наличие или отсутствие тимомы, возраст
начала болезни) и наличием определенных аллелей ге¬
нов фактора некроза опухоли а и (3. Можно заключить,
что при изученных аутоиммунных заболеваниях нервной
системы предрасположенность к той или иной болезни
детерминируется деятельностью сложной полигенной
системы, контролирующей ключевые этапы иммунного
ответа организма. Определение ведущих генетических
факторов риска и разработка эффективных методов кор¬
рекции выявляемого иммунного дисбаланса должны
стать в будущем важнейшими элементами стратегии ак¬
тивной профилактики данных тяжелых заболеваний не¬
рвной системы у лиц с повышенной предрасположенностью.4.3. Молекулярное тестирование
при онкологических заболеваниях
нервной системыОдной из областей клинической и фундаменталь¬
ной медицины, претерпевшей наиболее значительную
эволюцию в свете новейших достижений молекулярной
генетики, является онкология. Согласно современным
представлениям, большинство онкологических заболе¬
ваний обусловлены определенным каскадом спонтанных
соматических мутаций в генах, контролирующих различ¬
Молекулярно-генетический анализ459ные фазы клеточного деления и дифференцировки; в ряде
случаев аналогичный механизм опухолевой трансформа¬
ции инициируется хромосомными перестройками, ви¬
русной интеграцией в геном или менделевским насле¬
дованием генной мутации [Фогель Ф., Мотульски А.,
1990; Гарькавцева Р.Ф., Гарькавцев И.В., 1999;
KnudsonA., 1977; VogelF., 1979; Yunis J., 1983;Bishop J.,
1991]. Конечным результатом является нарушение рав¬
новесия в сложной полигенной системе, состоящей из
ингибиторов и активаторов клеточного цикла, результа¬
том чего является приобретение клеткой и соответству¬
ющим клеточным клоном способности к неконтролиру¬
емой пролиферации [Фогель Ф., Мотульски А., 1990;
Гарькавцева Р.Ф., Гарькавцев И.В., 1999; Bishop J., 1991].
Как и при других мультифакториальных заболеваниях,
в возникновении опухоли важная роль принадлежит сре-
довым факторам, определяющим, в частности, конкрет¬
ные особенности мутагенеза в генах-мишенях. Все ука¬
занные механизмы имеют универсальный характер и на¬
прямую применимы к нейроонкологии, что находит свое
отражение во внедрении в практику новых методов ДНК-
анализа при опухолях центральной и периферической
нервной системы. Это будет проиллюстрировано на при¬
мере некоторых наиболее распространенных видов пер¬
вичных опухолей мозга.Наиболее частой разновидностью первичных опу¬
холей мозга как в детском, так и во взрослом возрасте
являются астроцитомы. По степени злокачественности
они традиционно классифицируются на ряд стадий (ас¬
троцитома, анапластическая астроцитома, мультиформ-
ная глиобластома) - в зависимости от гистологических
особенностей, отражающих наличие митозов, некроза,
ядерных и цитоплазматических атипий, эндотелиальной
пролиферации в ткани опухоли [Fetell М., 1995]. Пере¬
ход от одной стадии к другой сопровождается последо¬
460Глава 4вательной серией генетических изменений в опухолевых
клетках [Cavenee W., 1992].Одним из важнейших генов, играющих роль суп¬
рессора опухоли, является ген р53, локализованной на
коротком плече 17-й хромосомы [Bogler О. et al., 1995].
Белковый продукт гена р53 ответственен за реализацию
разнообразных клеточных функций, включая контроль
цикла клеточного деления, репарацию ДНК и стабиль¬
ность генома; он действует как фактор транскрипции
через взаимодействие со специфическими последова¬
тельностями ДНК генов-мишеней. Мутации, вовлекаю¬
щие ген р53, обнаруживаются в опухолевых клетках при¬
близительно в 40% всех астроцитом, особенно у лиц
ранне-взрослого возраста [Hill J. et al., 1999]. Чаще всего
у больных имеет место делеция гена р53 на одной из
гомологичных хромосом в опухолевом клеточном кло¬
не; в результате этого в опухолевых клетках остается
лишь одна копия гена р53, что проявляется феноменом
потери гетерозиготности при исследовании ткани опу¬
холи. Выявление повреждения гена р53 имеет несомнен¬
ное прогностическое значение, поскольку данный ген
опосредует реакцию опухоли в ответ на проведение лу¬
чевой терапии [Haas-Kogan D. et al., 1999].Последующая эволюция астроцитарной опухоли
включает в себя ряд дальнейших генетических измене¬
ний клеток опухолевого клона, схематично представлен¬
ных на рис. 72. Делеции гена RB1 на хромосоме 13q (дан¬
ный ген кодирует белок pRB, подавляющий клеточный
рост), а также протяженного локуса на хромосоме 19q
являются типичными формами мутаций, определяющи¬
ми трансформацию опухоли в анапластическую астро-
цитому [Ritland S. et al., 1995; Hill J. et al., 1999]. Боль¬
шое число генов-супрессоров опухоли (таких как ММАС,
кодирующий пептид с фосфатазной клеточно-регулятор¬
ной активностью) локализовано на 10-й хромосоме
Молекулярно-генетический анализ461[Ichimura К. et al., 1998]. В связи с этим в 70% случаев
глиобластом в клетках опухоли выявляется феномен по¬
тери гетерозиготности по хромосомному локусу 10q23
[Biernat W. et al., 1997]; более того, обнаружение мута¬
ций в гене ММАС может считаться важным диагности¬
ческим маркером, свидетельствующим о нарастании сте¬
пени злокачественности астроцитарной опухоли [Hill J.
et al., 1999]. Еще одним семейством генов, ингибирую¬
щих цикл клеточного деления, являются гены циклин-
зависимых киназ р 14, р 15 и р 16 на хромосоме 9р; не
случайно делеции локуса 9р являются характерным мо¬
лекулярно-генетическим признаком мультиформных гли¬
областом [Ueki К. et al., 1996].Помимо мутаций в генах белков-ингибиторов
клеточной пролиферации, в развитии астроцитарных
опухолей важнейшую роль играет повышение экспрес¬
сии генов, стимулирующих клеточный цикл (такие гены
обозначаются общим термином онкогены). Как прави¬
ло, активизация онкогенов происходит посредством их
амплификации (т.е. увеличения числа копий) в результа¬
те хромосомной нестабильности в клетках опухоли, свой¬
ственной абсолютному большинству злокачественных
новообразований. Классическим примером онкогена
является ген рецептора эпидермального фактора роста
(EGFR), белковый продукт которого является важным
стимулятором роста астроцитов. Амплификация гена
EGFR на хромосоме 7q практически не встречается при
доброкачественных астроцитомах, но выявляется более
чем в трети случаев глиобластом, особенно у пожилых
больных [Hunter S. et al., 1995]. Как показано на рис. 72,
злокачественная трансформация астроцитарных опухо¬
лей ассоциирована также с амплификацией ряда других
онкогенов, локализованных, в частности, на хромосомах
1 q, 9q, 12q и т.д. [Hill J. et al., 1999].
462Глава 4Рис. 72. Генетическая эволюция астроцитарной опухолиВ настоящее время имеется большое число ра¬
бот, свидетельствующих о существовании аналогичных
генетических механизмов, способствующих формирова¬
нию и эволюции других разновидностей первичных опу¬
холей центральной нервной системы (олигодендрогли¬
ом, медуллобластом, менингиом, эпендимом и др.), каж¬
дой из которых свойственен свой спектр мутантных ге¬
нов в клетках опухолевого клона. Чаще всего в клетках
данных опухолей описываются как амплификация ряда
онкогенов, так и различные мутации (в первую очередь
Молекулярно-генетический анализ463- делеции) в хромосомных локусах lp, 6q, 8р, 9q, 11р-
llq, 17р 13, 19ql3, 22ql2.2 и др., приводящие к ингиби¬
рованию активности соответствующих пептидов-регуля¬
торов клеточного цикла [Ritland S. et al., 1995; Raffel C.
et al., 1997; Hill J. et al., 1999]. Один из наиболее извест¬
ных примеров - инактивация гена NF2 в клетках раз¬
личных видов наследственных (нейрофиброматоз-2) и
спорадических опухолей нервной системы - подробно
проанализирован в разделе 3.7.1.2.Таким образом, формирование опухоли представ¬
ляет собой многоэтапный процесс, центральными зве¬
ньями которого являются повышенная экспрессия он¬
когенов и инактивация генов-супрессоров опухоли, про¬
исходящие в соматических клетках-мишенях под воздей¬
ствием разнообразных мутагенных факторов. Несмотря
на известную ограниченность современных знаний о всей
«палитре» генетических повреждений, инициирующих
опухолевую пролиферацию, ДНК-диагностика соответ¬
ствующих мутаций в ряде случаев может способствовать
решению различных вопросов клинической нейроонко¬
логии. Это касается в первую очередь вопросов класси¬
фикации, дифференциальной диагностики и прогноза
опухолей мозга. Так, на сегодня представляется совер¬
шенно очевидным, что каждой гистологической разно¬
видности опухоли соответствуют свой собственный «ге¬
нетический профиль»; более того, дальнейшая деталь¬
ная молекулярная характеристика опухолей мозга при¬
водит к постепенному выделению новых нозологичес¬
ких подгрупп данных заболеваний [Mohapatra G. et al.,1998]. Наконец, накопление знаний о молекулярной ге¬
нетике опухолей мозга является важнейшей предпосыл¬
кой разработки различных подходов к генной терапии.
В силу некоторых особенностей опухолевых процессов
в центральной нервной системе (относительно компак¬
464Глава 4тная локализация совокупности мутантных клеток, от¬
сутствие клеточных делений в нейронах и др.) они явля¬
ются идеальным объектом для использования ряда ген¬
но-инженерных конструкций [Каграй в. е1 а1., 1996].
Детальное рассмотрение вопросов генной терапии на¬
ходится за пределами темы настоящей монографии; ука¬
жем лишь, что на современном этапе развития нейроон¬
кологии и молекулярной медицины различные методы
генной терапии с успехом находят все большее распрос¬
транение не только в эксперименте, но и в отдельных
протоколах клинических испытаний у больных со зло¬
качественными формами глиальных опухолей мозга
[В1бтег и. е! а1., 1996; РаШаПаЬ-ЗЬаукИН., 1999;Риеуо I.
е1а!., 1999].
ГЛАВА 5СОВРЕМЕННЫЕ НАУЧНЫЕ,
ЭТИЧЕСКИЕ
И ОРГАНИЗАЦИОННЫЕ
АСПЕКТЫ МЕДИКО-
ГЕНЕТИЧЕСКОГО
КОНСУЛЬТИРОВАНИЯ
В НЕВРОЛОГИИ *Профилактика наследственной патологии и
уменьшение «генетического груза» в популяции пред¬
ставляет собой одну из важнейших медицинских и со¬
циально-экономических задач, которые стоят перед со¬
временным индустриальным обществом, испытываю¬
щим беспрецедентное давление неблагоприятных
экологических факторов. Эта задача является тем более
актуальной, когда речь идет о наследственных заболева¬
ниях нервной системы, характеризующихся в большин¬
стве случаев тяжелой физической и психической инва-
лидизацией больных. Профилактика наследственных
заболеваний может быть первичной (предупреждение
зачатия или рождения больного индивида) и вторичной
(разнообразные способы коррекции патологического
генотипа) [Бочков Н.П., 1997]. На современном этапе
развития клинической нейрогенетики возможности вто¬*Глава написана с участием канд. мед. наук С.А. Клюшникова.
466Глава 5ричной профилактики наследственной патологии (т.е.
патогенетического лечения нейрогередитарных заболе¬
ваний) достаточно ограниченны, поэтому на первый план
выходит предотвращение появления «генетически боль¬
ного» потомства. Этой цели служит медико-генетичес¬
кое консультирование - особый вид специализирован¬
ной медицинской помощи, направленный на предупреж¬
дение появления повторных случаев наследственных
заболеваний в отягощенных семьях.Медико-генетическое консультирование включа¬
ет несколько основных задач [Козлова С.И. и др., 1996;
Бочков Н.П., 1997; Доклад научной группы ВОЗ, 1997]:а) установление точного диагноза и типа наследова¬
ния заболевания в консультируемой семье;б) расчет генетического риска у консультируемых род¬
ственников, включая (при возможности) точное установ¬
ление их генетического статуса с помощью методов ДНК-
диагностики (прогностическое тестирование);в) определение прогноза потомства и определение
наиболее эффективного способа профилактики новых
случаев заболевания (в том числе с помощью пренаталь¬
ной ДНК-диагностики плода на ранних сроках беремен¬
ности);г) объяснение консультируемым лицам смысла полу¬
ченной и проанализированной информации, решение
возникающих юридических и морально-этических про¬
блем;д) помощь консультируемой семье в решении целого
ряда других вопросов, касающихся планирования жиз¬
ни, репродуктивного поведения и возможности деторож¬
дения, психологической поддержки, социальной адап¬
тации и т.д.Точная диагностика наследственного заболевания
нервной системы и установление типа наследования па¬
Современные аспекты . . . 467тологического фенотипа являются первым необходимым
этапом медико-генетического консультирования обратив¬
шегося за помощью лица (семьи). Этот этап может осу¬
ществляться, в зависимости от конкретных медико-орга-
низационных условий, врачом общей практики, невро¬
логом, врачом-генетиком медико-генетической консуль¬
тации либо (в идеале) квалифицированным
специалистом в области клинической нейрогенетики.
Особенности генеалогического анализа в неврологии и
постановки диагноза наследственного заболевания не¬
рвной системы подробно проанализированы в главе 1
(раздел 1.2) и в соответствующих частных разделах гла¬
вы 3. Хотелось бы еще раз подчеркнуть значение деталь¬
ного обследования всех членов консультируемой семьи,
а также необходимость выявления у родственников лю¬
бых скрытых проявлений носительства мутантного гена,
в том числе с использованием комплекса дополнитель¬
ных лабораторно-инструментальных методов исследова¬
ния.Адекватно проведенный клинико-генеалогичес-
кий анализ позволяет выделить в консультируемой се¬
мье так называемую группу риска, которая и является
основным объектом медико-генетического консультиро¬
вания. Под группой риска понимаются родственники
пробанда, которые в соответствии с типом наследова¬
ния могут являться носителями мутантного гена и име¬
ют высокий риск заболеть данным наследственным за¬
болеванием или передать мутантный ген потомству. На¬
пример, при хорее Гентингтона, характеризующейся
аутосомно-доминантным типом наследования, практи¬
чески полной пенетрантностью и поздним возрастом
начала болезни, к группе риска следует относить детей
пробанда, а также его братьев-сестер: для каждого из
этих лиц при рождении теоретический риск унаследо-
468Глава 5Таблица 9Расчетные значения вероятностей фенотипов
потомства при известных типах брака (для
менделирующих заболеваний)Типнаследо¬ванияГенотипотцаГенотипматериВероятность фенотиповв случае рождения сыновейв случае рождения дочерейбольныездоровыеносителимутацииздоровые(мутациинет)больныездоровыеносителимутацииздоровые(мутациинет)Аугосом-АААА100100ноАААа100100доми-ААаа100100нангныйАаАА100100АаАа3/401/43/401/4Аааа1/201/21/201/2ааАА100100ааАа1/201/21/201/2аааа001001Аутосом-АААА001001но-АААа01/21/201/21/2рецессив-ААаа010010ныиАаАА01/21/201/21/2АаАа1/41/21/41/41/21/4Аааа1/21/201/21/20ааАА010010ааАа1/21/201/21/20аааа010010X-ААА100100сцеп-ААа1/201/2100ленныйАаа001100доми¬аАА100100нантныйаАа1/201/21/201/2ааа001001X-ААА001001сцеп-ААа1/201/201/21/2ленныйАаа100010рецес¬аАА00I010сивныйаАа1/201/21/21/20ааа100100Примечание: вероятности фенотипов в таблице представлены для случаев с
полной пенегрангносгью мутантного гена. А - доминантный аллель (мутантный в
случае доминантных заболеваний), а — рецессивный аллель (мутантный в случае
рецессивных заболеваний).
Современные аспекты . . . 469вать мутацию и заболевание составляет 50%, но при этом
для здоровых лиц риск заболеть постепенно снижается
с каждым прожитым десятилетием (особенно после
прохождения наиболее «опасного» возраста в 40-60 лет).При болезни Фридрейха, наследующейся по аутосом-
но-рецессивному типу и начинающейся обычно на вто¬
ром десятилетии жизни, основную группу риска состав¬
ляют братья и сестры пробанда (теоретический риск
25%), причем степень риска заболевания выше у млад¬
ших сибсов, не достигших «критического» возраста ма¬
нифестации болезни. При Х-сцепленной мышечной ди¬
строфии Дюшенна/Бекера к группе риска по заболева¬
нию относятся лица мужского пола - родственники
пробанда по материнской линии; помимо этого, сестры
пробанда также имеют высокий (50%) риск унаследо¬
вать от матери мутантный ген и, оставаясь клинически
здоровыми, передать заболевание мужчинам в следую¬
щее поколение.Как видно из представленных примеров, выделе¬
ние группы риска неразрывно связано с определением у
данной категории лиц генетического риска - т.е. веро¬
ятности появления у них или их потомства соответству¬
ющего наследственного заболевания. Генетический риск
до 5% оценивается как низкий и не считается противо¬
показанием к деторождению в консультируемой семье.
Риск от 6 до 20% принято считать средним, а свыше 20%- высоким. При среднем и высоком риске рекоменда¬
ции относительно возможности деторождения опреде¬
ляются в основном тяжестью медицинских и социальных
последствий конкретного заболевания, а также возмож¬
ностями и доступностью пренатальной ДНК-диагности¬
ки [Козлова С.И. и др., 1996].До использования методов ДНК-диагностики ге¬
нетический риск при менделирующих заболеваниях
470Глава 5определяется путем вероятностных расчетов, основан¬
ных на закономерностях сегрегации патологического фе¬
нотипа при том или ином типе наследования. Точность
расчета генетического риска зависит от информативно¬
сти родословной и возможности установления геноти¬
пов консультируемых родителей. Если генотипы роди¬
телей (тип брака) известны или могут быть предполо¬
жены с высокой вероятностью, оценка риска
манифестации патологического фенотипа у потомков
сравнительно проста - см. таблицу 9 [Козлова С.И. и
др., 1996]. В необходимых случаях делается поправка на
неполную пенетрантность мутантного гена, коэффици¬
ент инбридинга и некоторые другие факторы, влияющие
на проявление фенотипа. Значительно сложнее расчет
генетического риска при неизвестных генотипах роди¬
телей. Обычно это имеет место при наличии единичных
случаев заболевания в семье, особенно если изучаемый
синдром является генетически гетерогенным: в такой
ситуации оценка сегрегации признаков в родословной
неосуществима, и расчет генетического риска приходится
проводить для различных возможных типов наследова¬
ния. В этих случаях используются специальные матема¬
тические формулы, основанные на теории вероятности.
Данные формулы при расчете общего риска принимают
во внимание априорные факторы (менделевские законо¬
мерности предполагаемого типа наследования и сравни¬
тельную частоту возможных генетических вариантов
болезни), а также фактическое наличие у консультируе¬
мого лица больных или здоровых родственников (что
определяет апостериорную вероятность рассматривае¬
мой гипотезы). При неизвестном типе брака рождение
каждого последующего ребенка (здорового или больного)
существенно дополняет имеющуюся генетическую ин¬
формацию и видоизменяет величину апостериорной ве¬
роятности.
Современные аспекты . . . 471_При мультифакториальных заболеваниях с поли-
генно-детерминируемой наследственной предрасполо¬
женностью суммарный генетический риск зависит от
большого числа факторов и определяется с помощью
специальных таблиц эмпирического риска.Возможность ДНК-диагностики существенно
повышает точность определения генетического риска,
позволяя от вероятностных моделей перейти к однознач¬
ному (дефинитивному) определению генотипов и ожи¬
даемых фенотипов. В практике медико-генетического
консультирования методы ДНК-анализа могут исполь¬
зоваться для пренатальной, преимплантационной и ран¬
ней доклинической диагностики носительства мутации.
Пренатальная ДНК-диагностика предполагает определе¬
ние генотипа плода на ранних сроках беременности (пер-
вый-второй триместр, предпочтительнее на 8-12-й не¬
деле), что дает возможность при обнаружении мутации
сделать медицинский аборт. Образцы ДНК плода полу¬
чают из биоптатов хориона (хорионбиопсия), плаценты
(плацентобиопсия), клеток амниотической жидкости (ам-
ниоцентез) или лимфоцитов пуповинной крови (кордо-
центез). К сожалению, все указанные процедуры, про¬
водящиеся под контролем ультразвука, являются инва¬
зивными и сопряжены с определенным риском
прерывания беременности, оцениваемым в среднем как
1,5-2%. Новым неинвазивным методом в пренатальной
диагностике является анализ ядросодержащих клеток
плода, присутствующих в крови матери [Золотухина Т.В.,
Шилова Н.В., 1999]. Эти клетки (трофобластные клет¬
ки, лимфоциты, гранулоциты, эритробласты плода) по¬
являются в организме матери в результате трансплацен¬
тарного переноса, способствуя развитию толерантности
материнского организма по отношению к плоду; их кон¬
центрация составляет, ориентировочно, 105-10~8. В на¬
стоящее время наиболее серьезной проблемой является
472Глава 5недостаточная эффективность существующих методов
сортировки и детекции плодных клеток, получаемых из
крови матери. Одним из возможных решений данной
проблемы является многократная амплификация гено¬
ма единичных клеток плода с помощью полимеразной
цепной реакции. По-видимому, перспективы более ши¬
рокого использования анализа клеток плода из крови
матери с целью пренатальной ДНК-диагностики связа¬
ны с дальнейшим развитием методов обогащения попу¬
ляции плодных клеток.Если в обследуемой семье мутация изучаемого
гена известна (т.е. выявлена ранее у больного родствен¬
ника или клинически здорового носителя), при прена¬
тальной ДНК-диагностике плода молекулярное тести¬
рование облегчается - например, путем использования
соответствующей рестрикционной эндонуклеазы. В том
случае, когда предварительный генетический материал
больного члена семьи (носителя) недоступен для иссле¬
дования, поиск мутаций у плода осуществляется стан¬
дартными методами мутационного скрининга - в зави¬
симости от генетических особенностей изучаемого за¬
болевания, подробно разбираемых в главе 3. Выявление
конкретных мутаций у плода позволяет с абсолютной
достоверностью устанавливать его генетический статус
и определять прогноз. Несколько иначе обстоит дело с
пренатальной ДНК-диагностикой, осуществляемой кос¬
венными методами. Такая диагностика обычно прово¬
дится в тех случаях, когда изучаемый ген не идентифи¬
цирован (известна лишь его хромосомная локализация)
либо он имеет сложную структуру, что технически зна¬
чительно затрудняет поиск мутаций. Точность косвен¬
ной ДНК-диагностики, позволяющей оценивать насле¬
дование мутантной хромосомы, определяется близостью
патологического гена и используемого маркера (марке¬
ров). Например, если ген болезни и изучаемый маркер
Современные аспекты . . . 473расположены друг от друга на расстоянии 2 сМ, вероят¬
ность рекомбинации между данными локусами (и сле¬
довательно, вероятность ошибки) составляет 2%; соот¬
ветственно, точность диагностики носительства мутан¬
тной или нормальной хромосомы в этом случае
составляет около 98%. Идеальным является использо¬
вание внутригенных маркеров в комбинации с тесно
сцепленными маркерами, фланкирующим и изучаемый
ген с теломерной и центромерной сторон.Весьма перспективным направлением профилак¬
тики наследственной патологии является преимпланта-
ционная ДНК-диагностика. Сущность ее заключается в
исследовании ДНК клеток раннего зародыша, развитие
которого инициируется in vitro путем искусственного
оплодотворения (альтернативой является анализ ДНК
полярных телец овулировавших яйцеклеток). Имплан¬
тация зародыша в матку осуществляется только после
исключения у него носительства мутантного гена. Та¬
ким образом исход соответствующей беременности ста¬
новится, с генетической точки зрения, однозначно бла¬
гоприятным. В неврологии наибольший опыт преимп-
лантационной ДНК-диагностики получен при мышечной
дистрофии Дюшенна/Бекера, спинальной амиотрофии и
некоторых других моногенных заболеваниях [Verlmsky Y.,
Kuliev А., 1993; Fallon L. et al., 1999]. До настоящего вре¬
мени основной проблемой преимплантационной ДНК-
диагностики является недостаточно высокий процент
приживаемости имплантируемых в матку зародышей (не
более 20-30%). Однако с развитием микрохирургичес¬
кой техники и общим прогрессом гинекологии и эндок¬
ринологии можно ожидать новых существенных успе¬
хов в техническом и медико-биологическом обеспечении
преимплантационной диагностики наследственных за¬
болеваний.
474Глава 5Методы ДНК-диагностики носительства мутаций
у клинически здоровых лиц, принадлежащих к т руппе
риска, принципиально не отличаются от аналогичных
подходов, используемых для ДНК-диагностики заболе¬
вания на развернутой стадии. При наличии возможнос¬
ти проведения ДНК-диагностики эта процедура стано¬
вится центральным звеном медико-генетического кон¬
сультирования у лиц из группы риска, трансформируя
определенные теоретические величины рассчитанного
риска либо в сторону многократного повышения (вплоть
до 100% при обнаружении мутации), либо, напротив, в
сторону полного исключения риска наследственного за¬
болевания. Определение генотипа клинически здоровых
консультируемых лиц, обозначаемое термином прогнос¬
тическое (предсказательное) тестирование, представ¬
ляет собой едва ли не первый пример в истории клини¬
ческой медицины, демонстрирующий возможность на¬
учно обоснованного прогноза будущей судьбы
клинически здорового человека за многие годы до ожи¬
даемого дебюта тяжелого (нередко - неизлечимого) за¬
болевания. Поэтому закономерно, что осуществление
прогностического ДНК-тестирования связано с решени¬
ем серьезных правовых и этических вопросов, рассмат¬
риваемых во второй части настоящей главы.Медико-генетическое консультирование у род¬
ственников пробанда существенно осложняется возмож¬
ностью новой мутации у больного со спорадическим
случаем болезни. Совокупность связанных с этой воз¬
можностью проблем наиболее удобно рассмотреть h;i
примере ПМД Дюшенна и Бекера. Как указывалось в
главе 3 (раздел 3.1), не менее трети всех случаев данных
форм мышечной дистрофии обусловлены спонтанными
мутациями de novo в гене дистрофина [Darras В. et al.
1988], в связи с чем вопросы коррекции величины гене
тического риска для новых мутаций являются особенно
Современные аспекты . . . 475актуальными и наиболее изученными именно при ПМД
Дюшенна/Бекера. В случае мутации de novo прогноз для
членов консультируемой семьи с ПМД Дюшенна/Беке¬
ра зависит от того, на каком этапе гамето- или онтогене¬
за данная мутация возникла. Если новая мутация в гене
дистрофина возникает в зрелой яйцеклетке, будет иметь
место истинный спорадический случай болезни; при этом
все клетки больного, имеющие одну и ту же материнс¬
кую хромосому, будут иметь идентичный мутантный ге¬
нотип. Если же мутация возникает после оплодотворе¬
ния (т.е. на стадии эмбриогенеза), то клетки-потомки
мутантного клона будут иметь идентичную мутацию в
гене дистрофина, тогда как другая популяция клеток
организма останется нормальной (рис. 73). Указанное
явление носит название соматического мозаицизма. В
случае соматического мозаицизма тяжесть и распрост¬
раненность симптоматики определяются соотношением
нормального и мутантного клеточных клонов организ¬
ма и тем, какие ткани развиваются из мутантного клона
(чем раньше в эмбриогенезе возникла мутация, тем боль¬
шее число тканей будут вовлечены в патологический
процесс). В обеих рассмотренных ситуациях заболева¬
ние проявляется как спорадический случай, а риск по¬
вторного появления мутантной хромосомы у братьев-
сестер пробанда не отличается от общепопуляционного.В ряде случаев новая мутация может возникнуть
в родительской половой клетке на ранней (митотичес¬
кой) стадии гаметогенеза - на уровне первичных поло¬
вых клеток будущей матери или отца (рис 74). Это дает
начало разделению клеток-потомков на нормальную и
мутантную популяции гамет - феномен гонадного мо-
шицизма. Наличие гонадного мозаицизма видоизменя¬
ет расчеты риска при медико-генетическом консульти¬
ровании. Так, при ПМД Дюшенна/Бекера братья и сест¬
ры больного, оцениваемого как «спорадический» случай
476Глава 5мутантный клон
клетокРис. 73. Соматический мозаицизмМутация, возникшая de novo на одной из хромосом в соматической
клетке, обозначена черным кружком.
Современные аспекты477мутантный клон
половых клетокРис. 74. Гонадный мозаицизмМутация, возникшая de novo на одной из хромосом в первичной
половой клетке, обозначена черным кружком.
478Глава 5(т.е. когда у матери не выявляется гетерозиготное носи-
тельство мутации), имеют генетический риск не менее
5-10% унаследовать ту же мутацию - в силу теорети¬
ческой возможности того, что их мать является гонад¬
ным мозаиком [Bakker E. et al., 1989; van Essen Ä. et al.,
1992].Наконец, мутация может возникнуть у будущей
матери (отца) на ранней стадии их эмбрионального раз¬
вития в такой клетке, которая дает начало развитию и
дифференцировке зародышевых тканей. В этом случае
мать (отец) будут являться соматическими мозаиками,
но при этом все их гаметы будут мутантными. В такой
ситуации дальнейшее наследование мутантной хромо¬
сомы у потомков (и, соответственно, наследование бо¬
лезни у мужчин или наследование носительства у жен¬
щин) будет полностью подчиняться классическому X-
сцепленному рецессивному типу. Родительский
соматический мозаицизм, однако, является еще одним
источником ошибок при медико-генетическом консуль¬
тировании: поскольку обычные процедуры генодиагно¬
стики базируются на исследовании ДНК клеток крови,
данный анализ может показать нормальный генотип ро¬
дителя, тогда как половые клетки обследуемого родите¬
ля являются мутантными.Наличие соматического или гонадного мозаициз-
ма может быть подтверждено молекулярно-генетичес-
кими методами, например - с помощью сравнительного
анализа гаплотипов больных и здоровых лиц в инфор¬
мативных родословных [Lebo R. et al., 1990; Melis M. et
al., 1993]. В тех случаях, когда такое исследование не¬
возможно, соответствующие поправки на мозаицизм при
оценке величины генетического риска делаются на ос¬
нове эмпирических данных. Например, для локуса дис-
трофина в спорадических случаях ПМД Дюшенна/Беке¬
Современные аспекты479ра возможность материнского соматического/гонадного
мозаицизма составляет около 7% [van Essen A. et al.,
1997].Свои особенности имеет медико-генетическое
консультирование в случае новых мутаций при заболе¬
ваниях, обусловленных экспансией тринуклеотидных
повторов. В главе 1 (раздел 1.3, рис. 18) подробно рас¬
сматривался механизм возникновения динамических
мутаций de novo. Источником такой мутации обычно
является имеющийся у здорового родителя аллель с «про¬
межуточным» числом повторов, который в гаметогене¬
зе трансформируется в полную мутацию и ведет к раз¬
витию болезни у ребенка. При таком механизме генети¬
чески нестабильный «промежуточный» родительский
аллель может претерпевать повторные аналогичные
трансформации при передаче гена следующим детям.
Таким образом, при спорадических случаях тринуклео¬
тидных болезней, обусловленных новой мутацией у про¬
банда, для сибсов риск унаследовать заболевание явля¬
ется гораздо более высоким, чем считалось ранее
[Goldberg Y. et al., 1993]. Поэтому в подобной ситуации
(даже при отсутствии явной мутации у родителей!) оп¬
равданной является постановка вопроса о проведении
прямой ДНК-диагностики у всех братьев-сестер пробан¬
да. Следует помнить, что при установлении мутации de
novo возможность наличия в семье наследственного за¬
болевания при видимом отсутствии «наследственности»
нередко с недоверием встречается тестируемыми лица¬
ми, что требует от врача особого такта и аргументиро¬
ванности при проведении медико-генетического консуль¬
тирования.В случае выявления у плода носительства мута¬
ции (мутантной хромосомы) вопрос о продолжении или
прерывании беременности должен решаться строго ин¬
480Глава 5дивидуально, с учетом всей совокупности медицинских
и социальных факторов, главными из которых являются
тяжесть прогнозируемого заболевания, предполагаемый
возраст манифестации симптомов, возможность эффек¬
тивной патогенетической терапии и т.д. При ожидаемом
рождении ребенка с генотипом «высокого риска» в от¬
ношении курабельного заболевания (например, гепато-
лентикулярной дегенерации) оправданной является ре¬
комендация сохранить беременность, поскольку суще¬
ствующие методы превентивной терапии позволяют
полностью предотвратить развитие симптомов этой тя¬
желой болезни (см. главу 3, раздел 3.2.3). Сложнее при¬
нять решение при выявлении у плода мутации, связан¬
ной с высоким риском развития неизлечимого заболева¬
ния. При ряде таких заболеваний (например, торсионной
дистонии) пенетрантность мутантного гена не полная, а
его экспрессивность может быть весьма вариабельной,
в связи с чем носительство мутации само по себе от¬
нюдь не является абсолютным предиктором манифеста¬
ции у будущего ребенка тяжелых проявлений болезни.
Следует помнить также, что многие заболевания (клас¬
сический пример - хорея Гентингтона) проявляются
обычно лишь в позднем возрасте, предоставляя носите¬
лю мутации несколько десятилетий для активной, пол¬
ноценной жизни, а также оставляя реальный шанс до¬
жить до внедрения в практику эффективных методов
лечения болезни. Таким образом, в данном случае пра¬
во будущей матери сохранить плод даже при получении
положительного результата пренатальной ДНК-диагно¬
стики имеет под собой серьезные моральные и меди¬
цинские основания. Прерывание беременности представ¬
ляется наиболее целесоообразным при однозначно вы¬
соком риске развития у будущего ребенка тяжелого
некурабельного заболевания, манифестирующего в ран¬
Современные аспекты . . . 481нем детстве (например, при выявлении у плода мутации
в гене дистрофина и прогнозируемом развитии у ребен¬
ка мышечной дистрофии Дюшенна). В любом случае
окончательное решение по результатам пренатальной
ДНК-диагностики всегда остается за супругами. Врач ни
в коем случае не имеет права оказывать какое-либо пси¬
хологическое давление; его роль заключается в деталь¬
ном информировании родственников о смысле и резуль¬
татах проведенных исследований, величине генетичес¬
кого риска, а также в объяснении (в доходчивой форме)
всех нюансов существующей ситуации и прогноза.Как уже было отмечено выше, практика проведе¬
ния прогностического ДНК-тестирования у клинически
здоровых лиц из группы риска привела к интенсивному
развитию новой области медицинской генетики, связан¬
ной с оценкой этических аспектов указанного вида ме¬
дицинской помощи и обеспечением правовой защиты
консультируемых лиц [Доклад научной группы ВОЗ,
1997]. На этапе зарождения и выработки основных прин¬
ципов генетического консультирования вопросам мо¬
рально-этического порядка не всегда уделялось долж¬
ное внимание. Так, считалось, что врач-генетик обязан
в первую очередь выполнить свой врачебный долг по
отношению к обществу — проводить профилактику рож¬
дения физически неполноценных лиц в семьях, отяго¬
щенных тяжелой наследственной патологией. Первона¬
чально во многих странах практиковался метод полного
информирования лиц из групп риска о том, что их ждет
в будущем, без учета психологического статуса тестиру¬
емых лиц, в категоричной форме рекомендовалось пре¬
рывание беременности и т.д. Описаны случаи дискри¬
минации и произвола со стороны работодателей, требо¬
вавших сведения о генетическом статусе подчиненных
от страховых компаний [ЫаЮ’шег М. ег а!., 1992]. Одна¬
482Глава 5ко по мере совершенствования общественных отноше¬
ний во всех развитых странах основы концепции гене¬
тического консультирования были подвергнуты серьез¬
ному пересмотру. Значительно большее внимание стало
уделяться соблюдению интересов отдельной семьи и
каждого ее члена, права родителей иметь ребенка, права
каждого человека на жизнь. Во многом это связано с
достигнутым общемировым прогрессом в области со¬
блюдения прав человека.Непосредственным поводом к более пристально¬
му вниманию специалистов-генетиков к вопросам эти¬
ческого порядка стало внедрение в практику методов,
пригодных для пресимптоматического ДНК-тестирова-
ния лиц из группы риска при таких тяжелых неизлечи¬
мых заболеваниях, как хорея Гентингтона, доминантные
формы наследственных мозжечковых дегенераций, бо¬
лезнь Фридрейха и т.д. В наиболее концентрированном
виде весь спектр этических вопросов может быть рас¬
смотрен на примере проведения консультирования па¬
циентов и клинически здоровых лиц из семей, отягощен¬
ных хореей Гентингтона. В главе 3 уже отмечалось, что
данное заболевание с экспансией тринуклеотидных САО-
повторов, благодаря специфическим особенностям кли¬
ники, течения и генетики (поздний возраст начала, ан¬
тиципация, эффект «отцовской передачи» и т.д.), стало
своеобразной «моделью» при разработке вопросов ме-
дико-генетического консультирования и прогностичес¬
кого тестирования в современной нейрогенетике, а так¬
же в решении проблем морально-этического и юриди¬
ческого порядка. Важнейшая из существующих
проблем — возможность нанесения тяжелейшей психо¬
эмоциональной травмы доклиническому носителю па¬
тологического гена при сообщении ему о позитивном
результате ДНК-тестирования без должной психологи¬
Современные аспекты . . . 483ческой подготовки. Само по себе носительство гена та¬
кого заболевания как хорея Гентингтона автоматически
означает, что человек обречен заболеть неизлечимым
страданием (пример которого у него уже находится пе¬
ред глазами в лице больного родственника), постепенно
разрушающим личность, приводящим к глубокой инва-
лидизации и неизбежной смерти через 15-20 лет от его
начала. В литературе неоднократно описаны случаи аф¬
фективного поведения и суицидальных попыток в по¬
добных ситуациях [Farrer L., 1986; Lawson К. etal., 1996;
AlmqvistE. etal., 1999; Meiser В., Dunn S., 2000]. Может
ли вообще врач-генетик брать на себя функцию «верши¬
теля человеческих судеб», выносящего в ряде случаев
фактически смертный приговор клинически здоровому
человеку? Сообщать ли тестируемому лицу результаты
ДНК-диагностики, если он этого желает? В какой форме
сообщать ему и его родственникам результаты исследо¬
вания и какой процент врачебной «лжи во имя спасе¬
ния» допустим в данной ситуации? Каков будет после¬
дующий трудовой анамнез лица - носителя патологи¬
ческого мутантного гена, особенно если работа связана
с высокой степенью ответственности? Что делать, если
носитель гена - несовершеннолетний? Как построить
тактику дальнейшего наблюдения за таким «потенциаль¬
ным больным»? Эти проблемы вызвали острейшую дис¬
куссию не только среди генетиков, но и среди специали¬
стов по этике и медицинской деонтологии, социологов,
юристов и законодателей, психологов, непосредственно
членов семей, отягощенных наследственной патологией.Разработка организационно-методических прин¬
ципов проведения медико-генетического консультирова¬
ния в семьях, отягощенных хореей Гентингтона (а также
любой другой тяжелой наследственной неврологической
патологией), стала возможна благодаря напряженной
484Глава 5работе специалистов различных профилей под эгидой
международных профессиональных и непрофессиональ¬
ных организаций по борьбе с хореей Гентингтона. В ча¬
стности, весь контроль за проведением ДНК-тестирова-
ния и последующего наблюдения за больными и лицами
из группы риска был возложен на непрофессиональную
Всемирную Ассоциацию хореи Гентингтона (IHA), ос¬
нованную в 1979 году, в которую вошли, наряду с невро¬
логами, генетиками, психологами и специалистами по
этике, больные и члены их семей. Ассоциация представ¬
ляет собой объединение добровольных национальных
обществ борьбы с хореей Гентингтона из более чем 30
стран мира (в том числе и из России). В настоящее вре¬
мя Всемирная Ассоциация хореи Гентингтона является
весьма влиятельной организацией со значительными
финансовыми возможностями, обеспечивающей
поддержку важнейших научных исследований по данному
заболеванию, а также проведение международных семи¬
наров и научных форумов, посвященных проблеме хо¬
реи Гентингтона. Под ее попечительством организуют¬
ся и проводятся всевозможные съезды и конгрессы вра¬
чей, больных хореей Гентингтона и членов их семей,
спортивные, культурно-массовые мероприятия, во вре¬
мя которых царит доброжелательная непринужденная
атмосфера. Все делается для того, чтобы укрепить в этих
людях веру в возможность противостоять тяжелому не¬
дугу, всячески облегчить их существование. Оказывает¬
ся помощь в организации подобных мероприятий на
национальном и региональном уровне. Вся необходимая
информация в большом объеме имеется в глобальной
сети Интернет на соответствующих сайтах (www. huntington-
assoc, сот, neuro-www. mgh. harvard, edu/forum/
HuntingtonsDiseaseMenu.html, hdfoundation.org и др.).
Современные аспекты485Сложный комплекс этических и правовых про¬
блем был обсужден на совместной конференции IHA и
профессиональной Международной группы по изучению
хореи Гентингтона при Всемирной Федерации Невро¬
логии, состоявшейся в 1985 году во Франции. Решения
и рекомендации по всему спектру вопросов были окон¬
чательно приняты на аналогичной конференции в Кана¬
де (1989) и опубликованы в виде своеобразного свода
этических норм и правил, которым должны следовать
как специалисты, осуществляющие пресимптатическую
ДНК-диагностику, так и лица, проходящие это тестиро¬
вание. Данный протокол был уточнен и расширен в 1994
году, после идентификации патологического гена и раз¬
работки прямой ДНК-диагностики болезни [International
Huntington Association arid World Federation of Neurology
Research Group on Huntington’s Chorea, 1994]. Специально
оговаривается, что тщательное выполнение разработан¬
ных рекомендаций является важнейшим принципом ме-
дико-генетического консультирования и служит интере¬
сам как врача, так и тестируемого лица. Документ реко¬
мендован для стран всего мира, где возможно проведение
подобной работы - с учетом местных особенностей. Учи¬
тывая особую важность и сложность вопроса и большое
практическое значение вышеуказанного протокола для
практики проведения медико-генетического консульти¬
рования в целом, ниже приводятся основные положе¬
ния разработанных рекомендаций (на примере хореи
Гентингтона):1) Каждый индивидуум, желающий пройти тестирова¬
ние, имеет право на получение полной информации о
заболевании, его развитии и последствиях, сущности те¬
стирования, возможных результатах.2) Решение о прохождении тестирования должно быть
добровольным, без принуждения со стороны кого бы то
ни было, даже близких родственников.
486Глава 53) Необходимым условием проведения медико-генети¬
ческого консультирования является информированное
согласие, которое означает, что дееспособный индиви¬
дуум полностью ознакомлен с представленной ему ин¬
формацией, адекватно ее понимает и принимает реше¬
ние об обследовании самостоятельно, без какого-либо
давления извне.4) Тестирование несовершеннолетних детей по просьбе
родителей (либо в случае усыновления) не допускается,
поскольку позитивный ответ может привести к такому
решению вопросов наследства, имущества, образования
и др., которые будут ущемлять права ребенка - недеес¬
пособного лица. Исключение из этого правила возмож¬
но лишь с целью пренатального тестирования при на¬
ступлении беременности у несовершеннолетней девоч¬
ки из группы риска, а также при вступлении
несовершеннолетних лиц из группы риска в брак.5) Каждый индивидуум должен иметь возможность
пройти тестирование независимо от его финансового по¬
ложения.6) Тестирование должно проводиться коллективом спе¬
циалистов разного профиля (как клиницистов, так и
молекулярных генетиков); результаты ДНК-диагности-
ки должны сообщаться клиницистам, как правило, не¬
посредственно перед встречей с тестируемым лицом,
чтобы исключить предвзятость и возможные манипуля¬
ции результатами со стороны отдельных специалистов.7) Информация о результатах тестирования строго кон¬
фиденциальна и не подлежит разглашению без письмен¬
ного согласия тестируемого, за исключением специаль¬
но оговоренных судебных случаев.8) При желании пройти тестирование лицом с 25%-
ным риском (т.е. когда его родители клинически здоро¬
вы и не тестировались, а кто-либо из бабушек и деду¬
шек болен аутосомно-доминантным заболеванием) со¬
Современные аспекты . . . 487беседование и тестирование предлагается также родите¬
лям, имеющим 50%-ный риск. В случае их согласия они
имеют предпочтение при тестировании перед лицом с
25%-ным риском. Отказ их не влияет на даль¬
нейшее прохождение тестирования лиц с 25%-ным риском.9) Перед тестированием проводится комплексное ней-
ропсихологическое и неврологическое обследование,
причем консультируемый может отказаться от этого об¬
следования под расписку, сохраняя за собой право на
тестирование. При выявлении «суицидальной готовно¬
сти» просьба о проведении тестирования отклоняется.10) На обдумывание полученной информации тестиру¬
емое лицо получает не менее 1 месяца. К концу этого
времени он имеет право и возможности: а) отказаться от
дальнейшего тестирования вообще; б) сдать кровь для
выделения ДНК в научных целях; в) сдать кровь для
выделения ДНК с целью хранения; г) продолжить об¬
следование. В случае отказа (на любом этапе) консуль¬
тируемого лица от дальнейшего участия в ДНК-тести-
ровании такое решение не должно служить основанием
для каких-либо дискриминационных мер со стороны
медицинского персонала.11) Пренатальное тестирование проводится в случае,
когда будущий родитель знает о своем повышенном риске
развития заболевания (по данным прямой или косвен¬
ной ДНК-диагностики), а также в случае, когда он име¬
ет априорный 50%-ный риск (так называемое исключа¬
ющее тестирование).12) Результат ДНК-анализа сообщается индивидууму,
проходящему тестирование, немедленно после его по¬
лучения при непосредственном общении с врачом-гене-
тиком; сообщение по почте и телефону не допускается.13) После сообщения результата ДНК-анализа прово¬
дятся последующие встречи с консультируемым незави¬
симо от характера полученного результата - с целью
488Глава 5обеспечения всяческой психологической поддержки ис¬
пытуемому и разрешения всех возникающих вопросов.Таким образом, при проведении генетического
консультирования и скрининга следует придерживаться
нескольких ключевых принципов, таких как: а) добро¬
вольность; б) конфиденциальность; в) информирован¬
ное согласие; г) приоритет интересов консультируемого
лица; д) соблюдение в полном объеме прав консультиру¬
емого лица; е) постоянная психологическая, правовая и
медицинская поддержка на всех этапах медико-генети¬
ческого консультирования [Доклад научной группы ВОЗ, 1997].Согласно разработанным рекомендациям, при
консультирующих учреждениях должны создаваться спе¬
циальные этические комитеты, в состав которых, поми¬
мо специалистов по медико-генетическому консульти¬
рованию, предусматривается вхождение психологов, пси-
хиатров, специалистов по медицинской этике и
деонтологии, праву, юристов, служителей культа, а так¬
же нередко родственников больных. Подобные комите¬
ты осуществляют контроль за соблюдением вышеизло¬
женных этических норм и правил, проводят работу со
здоровыми родственниками обследуемых, настраивая их
на всестороннюю помощь и поддержку, которую они
должны оказывать своим близким. Основное значение
придается максимальному соблюдению прав личности,
в том числе возможности получения достоверной инфор¬
мации и необходимой психологической поддержки; та¬
ким образом, предотвращается ухудшение качества жиз¬
ни при получении испытуемым позитивного результата
ДНК-тестирования.По сообщениям многих авторов [Meissen G. et al.,
1991; Nance M. et al., 1991], нередко испытуемые пред¬
почитали получить позитивный ответ о носительстве
мутантного гена хореи Гентингтона, нежели оставаться
Современные аспекты . . . 489в неведении в группе 50%-ного риска, руководствуясь
при этом тем, что при позитивном ответе можно будет
адекватно спланировать свою дальнейшую трудовую де¬
ятельность и соответствующим образом перестроить
стиль и приоритеты жизни. Из протокола нетрудно за¬
метить, что работа с тестируемым лицом во многом на¬
правлена на формирование у него собственного мнения
о необходимости прохождения тестирования без какого-
либо нажима извне. В случае позитивного результата
тестируемый самостоятельно решает вопрос о целесо¬
образности дальнейшего деторождения, любые катего¬
рические запреты в этом случае недопустимы. Возмож¬
ные варианты выбора для носителей генов наследствен¬
ных болезней представлены в таблице 10.С практической точки зрения примерную схему
проведения медико-генетического консультирования при
хорее Гентингтона и других тяжелых наследственных
заболеваниях согласно рекомендациям наиболее автори¬
тетных международных организаций можно представить
следующим образом. Путем генеалогического анализа
(по родословным) определяется круг лиц из группы рис¬
ка. Им предлагается пройти ДНК-тестирование, причем
на обдумывание дается около 1 месяца. Обычно в сред¬
нем за этот период 35-40% лиц отказываются от после¬
дующего прохождения ДНК-диагностики.Остальных освидетельствует комиссия в составе
генетика, психиатра, психолога, невролога, в отдельных
случаях юриста - на предмет определения дееспособно¬
сти и возможности сообщить откровенно результаты
тестирования. Ряд пациентов соглашается пройти тес¬
тирование «с научной целью», но отказывается узнать
его результаты. После работы подобной комиссии оста¬
ется 25-30% лиц, которым подробно объясняются ре¬
зультаты ДНК-тестирования и дальнейшее поведение в
490Глава 5Таблица 10Возможные варианты выбора для носителей генов
наследственных заболеваний[Доклад научной группы ВОЗ, 1997]Время выявле¬
ниягенетическогорискаВозможные мерыЧастота принятия
этих мерДо выбора
партнераОтказ от супружестваНевысокая(редко)Выбор партнера, который
не является носителем *Очень низкаяВыбор партнера обычнымОчень высокаяпутемПосле выбораРешение не иметь детейВысокая только впартнера и доотношении тяжелыхбеременностиболезней при отсут¬(более часто)ствии возможности
пренатальной диаг¬
ностикиРешение не отказыватьсяВысокая в отноше¬от беременности в надеждении менее тяжелыхна благоприятный исходболезнейРешение обратиться в
службы пренатальной ди¬
агностикиОчень высокаяИскусственное оплодотво¬
рение спермой донора или
другая форма искусствен¬
ного зачатияОчень высокаяРазвод и выбор другогоОчень низкаяпартнера *После рожде¬Принятие ситуации и ле¬Высокаяния больногочениеребенкаПринятие ситуации, но от¬Низкая(чаще всего)каз от лечения**Отказ от ребенкаНизкаяПримечание: * относится только к рецессивным заболеваниям; ** вариан¬
ты выбора, возможные до первой беременности, относятся также к беремен¬
ностям. наступившим после рождения больного ребенка
Современные аспекты . . . 491случае положительного результата. Практикуется, напри¬
мер, такое объяснение: «У Вас обнаружены некоторые
изменения в гене, которые при определенных неблагоп¬
риятных условиях могут привести к развитию заболева¬
ния в более или менее отдаленном будущем. Для профи¬
лактики необходимо делать следующее ... Помимо это¬
го, мы бы рекомендовали Вам задуматься над решением
следующего круга вопросов ...». Если, к примеру, тес¬
тируемому лицу 20 лет, то специально подчеркивается,
что развитие заболевания возможно лишь через 15-20
лет, а к тому времени можно ожидать появления эффек¬
тивных методов патогенетической и/или генной терапии
(что весьма вероятно, учитывая бурный прогресс гене¬
тики) и т.д.Отечественные специалисты, осуществляющие
медико-генетическое консультирование, сталкиваются с
аналогичными проблемами морально-этического поряд¬
ка. Их решение должно проводиться на основе всего
накопленного мирового опыта, с учетом специфики рос¬
сийского менталитета и текущего общественно-эконо¬
мического уклада. В качестве примера приводим наше
наблюдение пренатальной ДНК-диагностики хореи Ген¬
тингтона в одной из консультированных семей.В кабинет медико-генетического консультирова¬
ния при нейрогенетическом отделении НИИ невроло¬
гии РАМН обратилась молодая семейная пара (мужчи¬
на 25 лет, женщина 23 лет) по поводу диагностики воз¬
можного носителъства гена хореи Гентингтона у
плода 12-13 недель. Поводом для обращения послужи¬
ла информация, полученная супругом незадолго до бра¬
косочетания, о наличии в семейном анамнезе хореи Ген¬
тингтона. Сведения были отрывочными: известно, что
якобы болела бабушка по линии отца, скончавшаяся в
возрасте 62 лет. При этом генетический статус отца
492Глава 5известен не был. Со слов сына, отцу 46 лет, он занима¬
ется активной трудовой деятельностью, готовится к
защите докторской диссертации, жалоб на состояние
здоровья не предъявляет, желания пройти генетичес¬
кое консультирование не имеет. После беседы и полу¬
чения информированного согласия на обследование на
первом, этапе было принято решение первоначально ус¬
тановить генетический статус супруга. Из анамнеза
жизни известно, что пациент, в детстве рос и разви¬
вался нормально. Имеет высшее образование, работа¬
ет программистом, на одном из предприятий г. Влади¬
востока. При неврологическом осмотре каких-либо от¬
клонений от нормы не выявлено. Психологическое
тестирование по экспресс-шкале ММБЕ отклонений от
нормы не выявило. Углубленное исследование с исполь¬
зованием батареи психологических тестов выявило лег¬
кое нарушение абстрактного мышления, среднюю сте¬
пень снижения практического мышления и памяти, не¬
резкое снижение внимания и концентрации, высокий
уровень личностной тревожности при нормальном
уровне реактивной тревожности; обследование по
шкале депрессии отклонений профиля от нормальных
показателей не выявило. С помощью исследования ког¬
нитивных вызванных потенциалов Р300 объективизи¬
ровано наличие легких когнитивных нарушений (удли¬
нение латентности и снижение амплитуды пика вол¬
ны Р300).При проведении прямой ДНК-диагностики выяв¬
лено наличие экспансии тринуклеотидных САО-повто-
ров в одном из аллелей гена гентингтина (свыше 40 по¬
второв). После предварительной подготовительной
беседы пациенту сообщены данные ДНК-тестирова-
ния, результаты восприняты адекватно. На следующем
этапе проводилась генодиагностика с использованием
ДНК, выделенной из ворсин хориона: супруге пробанда
Современные аспекты . . . 493проведена биопсия хориона с получением биологичес¬
кого материала в количестве, пригодном для экстракции
ДНК (биопсия проведена в клинике акушерства и гине¬
кологии ММ А им. И.М.Сеченова - кандидат мед. наук
В.И. Ланчинский). Результаты ДНК-диагностики пред¬
ставлены на рисунке 75. У плода (дорожка 4) четко ви¬
зуализируется мутантный аллель гена (показан длинной
стрелкой).1 2 3 4Рис. 75. Пренатальная ДНК-диагносгика хореи Гентингтона
Дорожка 1 - маркер, дорожка 2 - консультируемая женщина, до¬
рожка 3 - муж консультируемой женщины (носитель мутации на
пресимптоматической стадии), дорожка 4 - результат ДНК-анализа
плода (биопсия хориона). Длинной стрелкой указан мутантный ал¬
лель (экспансия САО-повторов гена гентингтина), короткой стрел¬
кой - нормальный аллель.Таким образом, выявлено наличие носительства
мутации у плода 13 недель. Проинформированной о ре¬
зультатах теста женщине была предоставлена возмож¬
ность самой решить вопрос о необходимости сохране¬
ния или прерывания беременности по медицинским по¬
казаниям. Было принято решение прервать
беременность.
494Глава 5Данное наблюдение демонстрирует важные орга¬
низационные аспекты медико-генетического консульти¬
рования, в частности - обязательное предварительное
нейропсихологическое обследование тестируемого муж¬
чины из группы риска (исключение «суицидальной го¬
товности»), Следует отметить, что при отсутствии ин¬
формированного согласия отца будущего ребенка на
ДНК-исследование проведение пренатальной ДНК-ди-
агностики у плода привело бы к установлению носитель-
ства мутантного гена у данного мужчины и, следователь¬
но, к нарушению его прав (в этом случае просьба матери
о проведении пренатального ДНК-тестирования долж¬
на была бы быть отклонена).По данным Е. А1тцу1з1 с соавторами (1999), про¬
водивших анализ результатов катамнестического наблю¬
дения за лицами, получившими положительный резуль¬
тат пресимптоматической ДНК-диагностики хореи Ген-
тингтона, опасения относительно крайне высокого риска
суицидальных действий и тяжелых психических рас¬
стройств у таких людей оказались преувеличенными.
Частота указанных катастрофических последствий в
группе «информированных носителей мутации» соста¬
вила 2%, что практически не отличается от частоты суи¬
цидальных действий у больных хореей Гентингтона.
Дополнительными факторами риска, повышающими
вероятность суицидальных попыток у доклинических
носителей мутации, являются наличие каких-либо пси¬
хиатрических проблем у данных лиц на протяжении 5-
летнего периода, предшествовавшего ДНК-анализу, а
также безработный статус тестируемого лица [А1тду1з1 Е.
й а1., 1999]. Эти данные следует принимать во внимание
при решении вопроса о прогностическом ДНК-тестиро-
вании.
Современные аспекты . . . 495В 1995 году на базе нейрогенетического отделе¬
ния НИИ неврологии РАМН была создана Ассоциация
помощи больным хореей Гентингтона России, интегри¬
рованная в структуру 1НА. На основании полученного
опыта работы Ассоциации нами отработаны собствен¬
ные этические и организационные принципы проведе¬
ния медико-генетического консультирования в семьях
больных, страдающих тяжелой наследственной невро¬
логической патологией; эти принципы базируются на
рекомендациях международных исследовательских групп
и организаций с учетом российского законодательства о
здравоохранении [Клюшников С.А. и др., 2000]. Тести¬
руемые лица предварительно подвергаются всесторон¬
нему клиническому и нейропсихологическому обследо¬
ванию с установлением профиля личности. При необхо¬
димости тестируемые лица осматриваются психиатром.
Строго соблюдается принцип добровольности обследо¬
вания и конфиденциальности полученных результатов.
По нашему мнению, лица с маскированной или явной
депрессией, а также длительное время наблюдавшие тя¬
желых больных в собственных семьях и имеющие пло¬
хой преморбидный психологический фон, наиболее
«опасны» в плане аффективных психических реакций и
неадекватного восприятия результатов прогностическо¬
го ДНК-тестирования. С такими лицами должна прово¬
диться серьезная подготовительная работа. В некоторых
случаях, очевидно, оптимальным выходом является от¬
каз от дальнейшего обследования или перенесение ДНК-
тестирования на более поздний срок. Основанием для
отсрочки проведения прогностического ДНК-тестирова¬
ния может служить также выявление личных и ситуаци¬
онных факторов, которые влияют на осознанность и доб¬
ровольность решения консультируемого лица пройти
496Глава 5обследование (давление третьих лиц, наличие недавней
психотравмирующей ситуации, желание консультируе¬
мого произвести неадекватные страховые действия и др.)
[С)иа1с1 К., 1993]. Во всех случаях необходим постоян¬
ный мониторинг неврологического и психологического
статуса больных и членов их семей с целью максималь¬
ной адаптации больных и выявленных доклинических
носителей мутации к новым условиям жизни. Создан¬
ная Ассоциация помощи больным хореей Гентингтона
занимается также информационно-образовательной де¬
ятельностью (распространение буклетов, брошюр, ма¬
териалов о работе аналогичных зарубежных ассоциаций
и т.д.).Применяемый нами подход является важнейшим
шагом к решению разнообразных проблем прогности¬
ческого ДНК-тестирования и представляет собой пер¬
вый в нашей стране опыт создания комплексной систе¬
мы медико-генетического консультирования (клиника +
ДНК-лаборатория, а также реализация на базе одного
учреждения предварительного психологического тести¬
рования, собственно генодиагностики, постлабораторно-
го наблюдения и деятельности Ассоциации помощи боль¬
ным). В будущем, по мере внедрения в практику эффек¬
тивных методов лечения наследственных заболеваний,
считавшихся некурабельными, принципы медико-гене-
тического консультирования будут подвергаться посте¬
пенному пересмотру. В частности, прогностическое
ДНК-тестирование будет носить более настоятельный и
императивный характер, поскольку максимально раннее
выявление асимптомных носителей мутантного гена ста¬
нет ключевым фактором для инициации превентивного
лечения и предотвращения манифестации болезни.
Современные аспекты . . . 497Закончился XX век. Перевернута еще одна
страница истории человечества, истории науки. С
идентификацией генома человека завершается эпоха
становления структурной геномики, открывшая
исследователям беспрецедентные возможности анализа
молекулярного субстрата наследственности. Следующим,
несравненно более сложным шагом является выяснение
тонких механизмов функционирования генома, включая
временную и тканевую специфичность взаимодействия
генов и их белковых продуктов, регуляцию экспрессии
генов, разработку эффективных подходов к
целенаправленному воздействию на генетический
аппарат клетки. Данное приоритетное направление
исследований, определяемое термином функциональная
геномика, даст в руки врача реальную возможность
коррекции генетических дефектов, позволив перейти от
экспериментальных протоколов генной терапии к ее
широкому использованию в клинической практике. При
этом базовые принципы молекулярной медицины и
молекулярной неврологии, обобщенные в своем
современном понимании в настоящей книге, станут
неотъемлемой частью арсенала врача-невролога. Такова
непреложная логика поступательного развития науки,
таковы требования жизни и общечеловеческого
прогресса.
ПРИЛОЖЕНИЕ 1СВОДНЫЙ КАТАЛОГ ГЕНОВ
НАСЛЕДСТВЕННЫХ
ЗАБОЛЕВАНИЙ
НЕРВНОЙ СИСТЕМЫУсловные обозначения,
использованные в таблицах:Типы наследования:АД - аутосомно-доминантный;АР - аутосомно-рецессивный;ХД - Х-сцепленный доминантный;ХР - Х-сцепленный рецессивный;М - материнское наследование;С - спорадические формы.Типы мутаций:В - вставки;Д - делении;ДП - дупликации;ТМ - толковые мутации;ЭТП - экспансия тринуклеотидных повторов;ЭПП - экспансия пентануклеотидных повторов.
Неустановленные гены и типы мутаций обозначены
значком вопроса (?).OMIM - номер по каталогу V.A.McKusick “Online
Mendelian Inheritance in Man” (электронная версия:
http://www.ncbi.nlm.nih.gov/Omim/searchchromim.html).
Таблица IIЗаболевания нервной системы, обусловленные мутациями в ядерных генахЗаболеваниеТипнаследо¬ванияХромосомнаялокализациягенаОбозначениелокуса(гена)БелковыйпродуктгенаХарактермутацииОМІМНервно-мышечные болезниМышечные дистрофииДистальные формы миопатий:- ВеландерАД2р13\УОМ97604454-ЛэнгаАД14ч 11МРОІ07160500-МиошиАР2р 13ММдисферлинТМ, В254130- Нонака( с кольцевыми вакуолями)АР9р 1—яОМЯУ77601073- тибиалъная (У66)АД2яЗ1-33тмэ■>7600334- Фейта (с ларингеальным
и фарингеальным параличом)АД5Ч31МР027158580Конечностно-поясные формы
мышечных дистрофий:-тип 1ААД5ч22-34ІХЗМША7?159000-тип 1ВАД1Я11 —21УЗМОІВ77159001— тип ІСАДЗр25ГАМБІЄ (САУЗ)кавеолин-3тм,д601253- тип ЮАД6^22ІЛМОШ7?603511-тип 1ЕАДчІЛЗМОІЕ77- тип 2ААР15д 15.1—д 15.3Ь0М02А (САРЮ)кальпаин-3ТМ, Д, В253600Приложение 1 499
Продолжение таблицы 11Заболевания нервной системы, обусловленные мутациями в ядерных генах ЗаболеваниеТипнаследо¬ванияХромосом¬наялокализациягенаОбозначениелокуса(гена)БелковыйпродуктгенаХарактермутацииОМІМ- тип 2САР13я121ЛМЭ2С (БОСО)у-саркогликанТМ,Д253700- тип 20АР17я12—21.331£гМ020 (БОСА)а-саркогликан(адгалин)ТМ,В600119— тип 2ЕАР4ql2Ь0М02Е (БОСВ)(3-саркогликантм,в604286- тип 2РАР5ч33-341ЛМ02Р (ЭОСО)5-саркогликантм601287— тип 2йАР\1ц\1-12иЗМБ2Стелетонинтм601954- тип 2НАР9дЗ1—33ШМ02Н?7254110- тип 21АР19ч13.3ЬСМБ21?7Лице-лопаточно-плечевая
мышечная дистрофияАД4я35РЭНМ01А?Д158900Лопаточно-перонеальная
мышечная дистрофияАД12я13.3—158РМ?7181430Миотоническая дистрофия:— аАД19ч13.3ОМІ (ОМРК)миотонин-протеинкиназаэтп160900-бАДЗчОМ2?7602668Мышечная дистрофия Бетлема:— аАД2Ц22.3СОЬ6А1 /СОЬ6А2а 1 /а2-субъединицы
коллагена VI типатм158810-бАД2р37СОЬбАЗаЗ-субъединица
коллагена VI типатм158810о,С5Продолжение таблицы 11ЗаболеваниеТипнасле¬дова¬нияХромосом¬наялокализациягенаОбозначениелокуса(гена)БелковыйпродуктгенаХарактермутацииОМІММышечная дистрофия Дюшенна/БекераХРХр21.2ОМО (БУБ)дистрофиид, дп.ТМ310200Наследственный миозит
(миопатия) с включениямиАР9р 1 —яНІВМ97601073Окулофарингеальная
ЭТП мышечная дистрофияАД, АР14ц 11.2-13ОРМО (РАВР2)полиаденилин-
ассоциированный белок164300,257950Поздняя мышечная дистрофия
с буллезным эпидермолизомАР8я24-я1егМО-ЕВБплектинтм, д226670Проксимальная миотоническая
миопатияАДЗчРШЭММ99600109Мышечная дистрофия Эмери-Дрейфуса:— аХРХя28ЕОМИэмеринДА ТМ310300-бАДІЯ21.2ЕОМБ2 (ЬМЫА)ламин А/СТМ181350-вАРЦ21.2ЕОМБ2 (УША)ламин А/СТМ604929Врожденные миопатии:Интегрин-негативная формаАР12Ч13ІТОА7интегрин а7тм600536«Классическая» восточная форма
(мерозин-негативная)АР6ч2МОС1А (ЬАМА2)мерозин (а2-цепь
ламинина)ТМ156225Форма с вторичным дефицитом
мерозинаАР1Ч42МБСІВ77604801Форма е контрактурами-
мышц-разгибателей позвоночника
(врожденная миопатия со спинальной
ригидностью)АР1 р35—36КЭМО!?7602771500 Приложение 1 Приложение 1
Продолжение таблицы 11Форма Фукуяма | АР | 9ц31 \ РСМБ | фукутин | ТМ, Д | 253800Врожденные непрогрессирующие
миопатии:- актиновая миопатияАД, АР1442.1АСТА1а-актинТМ102610— болезнь центрального стержняАД19я13.1ОХ> (ЮНІ)рианодиновый
рецептор кальциевого
каналаТМ117000- миотубулярная миопатияХРХ428МТМХ (Св2)миотубуляринТМ310400-миофибриллярная
(десминовая) миопатия (а)АД2я35ОИМ (ОЕЭ)десминТМ601419- миофибриллярная миопатия (б)АД, АР1Ц22.3-23.1СЯУАВаВ-кристаллинТМ123590-миофибриллярная миопатия (в)АД10я22.3МБМ??— немалиновая миопатия (а)АД1 я21 —23ИЕМ1 (ТРМЗ)а-тропомиозин 3ТМ161800— немалиновая миопатия (б)АД, АР1442.1ИЕМ2 (АСТА1)а-актинТМ256030- немалиновая миопатия (в)АР2ц72№М2 (ЛЕВ)небулинТМ161650— немалиновая миопатия (г)АР19ЯІ3.4АИМ (ТШТ1)тропонин Т1ТМ605355Наследственные невропатииГигантоаксональная невропатия
(с утолщенными аксонами)АР16424.1йАЛ??256850Наследственные моторно-сенсорные
невропатии (НМСН):• болезнь Рефсума
(НМСН IV)АР1 Ор 11.2—ріегРАНХ (РНУХ)фитаноил-КоА-гидроксилазаД тм266500* врожденная
гипомиелинизирующая
полиневропатияАД, АР10я21.1-22.1ЕОЯ2белок ЕОЯ2ТМ605253Продолжение таблицы 11ЗаболеваниеТипнасле¬дова¬нияХромосом¬наялокализациягенаОбозначениелокуса(гена)БелковыйпродуктгенаХарактермутацииОМІМ® гипертрофический неврит-
болезнь Дежерина—Сотта
(НМСН III):— тип ЗААД, АР17р 11.2РМР22белок перифериче¬
ского
миелина РМР22ТМ145900— тип ЗВАД1422-23мргбелок перифериче¬
ского миелина Р0ТМ145900- ЕСК-ассоциированный типАД10421.1-22.1ЕОЯ2белок Е0112ТМ145900— периаксин-ассоциированный типАР19ц13.13—13.2РИХпериаксинТМ145900• дистальный фенотип НМСН -
болезнь Шарко-Мари-Тута
(СМТ), в том числе:демиелинизирующие формы (НМСН I):— тип ІААД17р 11.2СМТ1А (РМР22)белок
периферического
миелина РМР22ДП, ТМ118220— тип 1ВАД1422-23смпв (мрг)белок
периферического
миелина Р0ТМ118200-тип IXхдХ4ІЗ.ІСМТ IX (Сх32)коннексин-32Д, тм302800— ЕСК2-ассоциированный типАДЮ421.1-22.1ЕОІ12белок ЕОЯ2тм129010502 Приложение 1 Приложение 1 503
Продолжение таблицы И- тип 4ААР8ql3-q21.1CMT4A??214400- тип 4В1АР1 lq23CMT4B1миотубулярин-ассоциированнаятирозин-фосфатаза(MTMR2)ТМ601382- тип 4В2АР1 lp 15CMT4B2??604563— тип 4САР5q23-33CMT4C??601596- тип 4D (Ьот)АР8q24.3CMT4D (HMSNL)белок NDRG1ТМ601455- тип 4ЕАР10q21.1-22.1CMT4E (EGR2)белок EGR2ТМ605253- тип 4FАР19ql3.1-13.3CMT4F (PRX?)периаксин (?)?602260аксональные формы (НМСНII):- тип 2ААДlp36CMT2A??118210- тип 2ВАД3q13-22CMT2B??600882- тип 2DАД7pl4CMT2D??- тип 2ЕАД8p21CMT2E(NEFL)белок легкого
нейрофиламентаТМ162280- аутосомно-рецессивный тип (а)АРlq21.2—21.3??- аутосомно-рецессивный тип (б)АР19ql3.3?9—Х-сцепленный тип
(синдром Cow chock)ХРXq24-26NAMSD??310490• наследственная невропатия
с предрасположенностью
к параличамот сдавленияАД17pl 1.2HNPP (PMP22)белок периферического
миелина Р22Д,ТМ162500• НМСН с врожденной катарактой
и лицевым дизморфизмомАР18qterCCFDN??604168Продолжение таблицы 11ЗаболеваниеТипнасле¬дованияХромосом¬наялокализациягенаОбозначениелокуса(гена)БелковыйпродуктгенаХарактермутацииOMIMПроксимальный фенотип НМСНАД3ql3.1HMSNP99Наследственные моторныенекпопатии Ґнейронопатиі-О: . .— тип // (спинальный вариант
фенотипа Шарко-Мари-Тута)АД12q24.3HMN2■)9158590-тип VАД7р15HMN5 (SMAD1)9600794-тип VIАР11 q 13-21HMN69604320-тип VIIАД2q 14HMN7?158580-тип 1егазИАР9р21.1-12HMNJ9605726Наследственная
невралгическая амиотрофияАД17q24начА9162100Наследственная сенсорная и
сентпно-легетативная невропатия:- тип IАД9q22.1-q22.3HSN1?7162400- тип III (семейная дизавтономия,
синдром Райли-Дея)АР9q31-q33HSAN3?7223900Периферическая невропатия
с агенезией мозолистого телаАР15qACCPN?7218000Семейная амилоидная невропатия:— транстиретин-ассоциированный
типАД18ql 1.2—12.1PALBтранстиретинТМ176300— Иова-типАД1 lq23-24APOA1аполипопротеин AlТМ107680— финский типАД9q33GSNгелсолинТМ105120504 Приложение 1 Приложение 1
Продолжение таблицы 11Наследственные болезни
мотонейронаБоковой амиотрофический склероз,
«классический» фенотип
(семейная форма)АД21я22.1-22.2АЬБ! (5СЮ1)Си/гп супероксид-
дисмутазаТМ105400Боковой амиотрофический
склероз, ювенильный фенотип:— аАР2Ч33Аьэг?7205100-бАД9ч34АЦ34?7602433-вАР15415.1-22.1АЬБ5?7602099Дистальные спинальные
амиотрофии (прогрессирующие):- с фенотипом болези
Шарко-Мари-ТутаАД12424.3нмга??158590- с преимущественным
вовлечением рукАД7р 15БМАОІ?7600794- тяжелая младенческая форма
с респираторным дистрессомАР1І4ІЗ-21НМШ?7604320- взрослая форма с параличом
голосовых связокАД2414НМШ?7158580- ювенильная форма с пирамидными
симптомами (Іегазії)АР9р21.1-12НМ№??605726Дистальная непрогрессирующая
врожденная спинальная амиотрофия
с контрактурамиАД12Ч23-24ЭМАЬ??600175ЗаболеваниеПроксимальные спинальные
амиотрофии(формы: Верднига-Гоффмана,
«промежуточная»,Кугельберга-Веландер) Спинально-бульбарная
амиотрофия КеннедиДругие нервно-мышечные
заболевания Болезнь Броди (крампи и
псевдомиотония, обусловленная
физической нагрузкой)Вакуолярная нейромиопатия
Врожденная миастения:Типнасле¬дова¬нияАРХРХРАДАДХромосом¬наялокализациягена5я 13Х411.2-1216р 1219р 13АД2ц24-3217р 11-12Обозначениелокуса(гена)БМА (ЭМЫ)БВМА (АЯ)АТР2А1(ЗЕЯСАІ)СНЇШАСНКЫВ!Продолжение таблицы IIБелковыйпродуктгенабелок БММ
(«белок выживаемости
мотонейронов»)Характермутацииандрогенныйрецепторкальциевая АТФ-аза
саркоплазматического
ретикулума
(БЕКСА!)мышечныйацетилхолиновыйрецептор,а-субъединицамышечныйацетилхолиновыйрецептор,Р-субъединицаД, тмэтптмТМтмОМІМ253300,253400,253550313200108730100690100710О,О-41506 Приложение 1 Приложение 1_
Продолжение таблицы 11-вАД, АР17р 13СНКЫЕмышечныйацетилхолиновыйрецептор,8-субъединицаТМ100725-г (младенческая форма)АД17р1егБІМ?7254210Врожденная миотония:-миотония БекераАР7ч35СЬС-1мышечный хлор¬
ный
каналТМ255700-миотония ТомсенаАД7ч35СЬС-1мышечный хлорный
каналТМ160800Врожденная парамиотония
(болезнь Эйленбурга)АД\lq\2.1-13.3ЗСЖАмышечный натриевый
канал, а-субъединицаТМ168300Врожденный фиброз
экстраокулярных мышцАД12сепСБЕОМ??135700Злокачественная гипертермия:-аАД19я 13.1мнэ! (ятал)рианодиновый
рецептор кальциевого
каналаТМ180901-бАД17я 11.2-24мшг?1154275-вАД7р21-22мнэз??154276-гАДЗчІЗЛМН34??600467-дАД1 яЗ1—32МШ5 (САСИЫАЗ)Дигидропиридино-
вый рецептор
кальциевого канала,
а1-субъединицаТМ601887-еАД5рМН36??601888ЗаболеваниеКалий-индуцируемая и ацетазол-
чувствительная миотония,
периодический
парамиотонический параличКардиоскелетная миопатия
с нейтропенией и измененными
митохондриями (синдром Барса)
Метаболические миопатии:— гликогеноз 11 типа (Помпе)— гликогеноз VII типа (Таруи)- гликогеноз IX типа- гликогеноз X типа- гликогеноз XI типа- дефицит
каритин-пальмитоил-
трансферазы - гликогеноз V типа (Мак—Ардля)Тип
наследова
нияАДХРАРАРАРХРАРАРАРХромосом¬наялокализациягена17Ч13.1—13.3Хц2817д2311 я 131сеп~ч32Хд137р12—1311р 15.41р32Обозначениелокуса(гена)ЗСЫ4А04.5ОААРУвМРБКМРвК1РвАММЦЭНАСРТ2Продолжение таблицы 11Белковыйпродуктгенамышечныйнатриевыйканал.а-субъединицабелок в4.5а-глюкозидазамышечнаяфосфорилазамышечная
фосфофруктокиназафосфоглицерат-киназамышечная
фосфоглицерат-мутазалактат-дегидрогеназакарнитин-пальмитоил-трансферазаХарактермутацииТМТМ, Д, ВТМ, Д, ВТМ, ДТМТМТМТМ, ДТМОМІМ168300,168350.302060232300232600232800311800261670150000255110Сэ''О508 Приложение 1 Приложение 1
Продолжение таблицы 11периодический паралич:— гиперкалиемическая формаАД17ч 13.1—13.3ЭСЖАмышечный натриевый
канал,
а-субъединицаТМ170500— гипокалиемическая формаАД1чЗ1—32САСЫЫАЗдигидропиридиновый
рецептор кальциевого
канала,
а1-субъединицаТМ170400Прогрессирующая наружнаяофтальмоплегия(включая осложненные формы)- синдром множественных делений
митохондриальной ДНК:-аАД10ч24РЕ01?7157640-6АДЗр 14.1—21.2РЕ02??-вАД4ч35РЕОЗ (АШ1)аденин-нуклеотид-
транслоказа 1 (А1МТ1)ТМ601227—гАР22д13.32^1егМИШЕтимидин-фосфорилазаТМ550900Синдром Уокера-Варбурга
(болезнь «мышца-глаз-мозг»)АР1р32-34МЕВ (WWS)7?253280ииндром Мебиуса (врожденный
паралич лицевого нерва):— аАДЗя21-22МВБ17?157900-бАД1(^21.3-22.1мвэг?7157900Продолжение таблицы 11ЗаболеваниеТипнасле¬дованияХромосом¬наялокализациягенаОбозначениелокуса(гена)БелковыйпродуктгенаХарактермутацииОМІМЭкстрапирамидные заболеванияАхл-пг.пмнп-пепессивный паркинсонизм-аАР1р35-36РАЯК677605909-бАР1р36РАЛК7??Болезнь Вильсона(гепатолентикулярнаядегенерация)АР13ч14.3ТОГО (АТР7В)Медь-транспортирую-
щая АТФ-аза Р-типа
(АТР7В)ТМ, Д, В227900Болезнь Галлервордена-ШпатцаАР20р 12.3—13нэв77234200Копелнь Паркинсона (семейная форма):-аАД4ц21-23РАМП(БИСА)а-синуклеинТМ601508-6АД4р14РАІІК5(исн-ы)Убиквитин карбокси-
герминальная гидрола-
заЫ (иСН-Ы)ТМ191342-вАД2р13РАЇІКЗ77602404-гАД4р14—16.3РАІЖ4??Гентингтоно-подобный синдром(НиМіпріоп-Ііке')'. — аАД20рньш??603218-бАР4р 15.3Ш1Х>277604802Дистония-паркинсонизм: -синдром ЬиЬах 1 ХР | ХцІЗ.І | ОУТЗ | ? | ? | 314250510 Приложение 1 Приложение 1
Продолжение таблицы 11— дистония-паркинсонизм
с острым началомАД19д13ОУТ12??128235Дофа-зависимая торсионная дистония:— аАД\4q22ОУТ5
(йСН I)ГТФ-циклогидролаза Iтм,д128230-бАР11р11.5ТНтирозин-гидроксил азатм191290-вАР1Ц22.3-23.3б-РТСЭ6-пирувоил-тетрагидробиоптерин-синтетазатм261640Дофа-независимая торсионная дистония:— аАД9ц34ОУТ1торсин Ад128100-бАД8р21-я22ОУТ6?9602629Миоклоническая дистония:-аАДЩ23ОУТ11 (ОРШ)02-рецептор дофаминатм159900-бАД7я21—23?9Наследственная
доброкачесвенная хореяАД14цвен?9118700Наследственный гениоспазмАД9ч13-21ОЭМ!99190100Нейроакантоцитоз(хорея-акантоцитоз)АРСНАС?9200150Нейроферритинопатия (аутосомно-
доминантная болезнь базальных
ганглиев взрослого возраста)АД19ч13.3итьлегкая цепь
ферритина•В134790Пароксизмальные дистонии:- пароксизмальный дистонический
хореоатетоз (пароксизмальная
некинезигенная дискинезия)АД2я33-35ОУТ8??118800о,Продолжение таблиь.оі і дЗаболеваниеТипнасле¬дованияХромосом¬наялокализациягенаОбозначениелокуса(гена)БелковыйпродуктгенаХарактермутацииОМІМ- пароксизмальный хореоатетозАД1рОУТ97?601042- пароксизмальный кинезигенныйАД16р11.2-Ч11.2БУЛО9?128200Фокальная дистонияАД18рОУТ797602124Хопея ("болезнь-) ГентингтонаАД4р16.3НО (ІТ-15)гентингтинЭТП143100'Эг.г.р.ШШЯПЬНЫЙ тпемоп: : -АД3я13ЕТМ1 (РЕТ1)97190300-бАД2р22-25ЕТМ277602134Ювенильный паркинсонизмАР6о25-27РАР.К2паркинД, В, ТМ600116Наследственныеатаксии и папаплепиАтаксия с изолированным
пегЬипитом витамина ЕАР8ЧАУЕО (ТТР1)транспортера-токофероладв277460Дтяігг-иа-трпеянгичктязия' —-аАР11422-23АТ (АТМ)белок семейства
фосфатидилинозитол-
киназтм, д, в208900-бАРЩ21АТШ (МЯЕ11А)белок МЯЕ1 1Атм604391Аутосомно-доминантные
спиноцеребеллярные атаксии:- спиноцеребеллярная атаксия1-го типа (СЦА1)АД6р22-р23ЭСА!атаксин-1ЭТП164400512 Приложение 1 Приложение 1
- спиноцеребеллярная атаксия
2-го типа (СЦА2)АД\2а[24АЭСА2атаксин-2ЭТПшлицы 1 1
183090-спиноцеребеллярная атаксияЗ-го типа
(СЦАЗ, болезнь Мачадо-Джозеф)АД14ч32.1ЭСАЗ (М.ГО)атаксин-3ЭТП109150— спиноцеребеллярная атаксия
4-го типа (СЦА4)АД\6q22A5СА47?600223- спиноцеребеллярная атаксия
5-го типа (СЦА5)АД11сепЭСА5??600224- спиноцеребеллярная атаксия
6-го типа (СЦА6)АД19р 13.18СА6 (САСЫЬ1А4)потенциалзависимый
кальциевый канал,
ам-субъединицаЭТП183086— спиноцеребеллярная атаксия
7-го типа (СЦА7)АДЗр12—р21.1ЪСАЛатаксин-7ЭТП164500- спиноцеребеллярная атаксия
8-го типа (СЦА8)АД13Ч21ЭСА8атаксин-8ЭТП603680- спиноцеребеллярная атаксия
10-го типа (СЦА10)АД22Ч13ЭСАЮ??603516- спиноцеребеллярная атаксия
11-го типа (СЦА11)АД15ч14—21.3БСАП??604432—спиноцеребеллярная атаксия
12-го типа (СЦА12)АД5яЗ1—335СА12 (РР2А)регуляторная
субъединица белковой
фосфатазы РР2АЭТП604326- спиноцеребеллярная атаксия
13-го типа (СЦА13)АД19ч 13.3—13.4БСАІЗ77605259— спиноцеребеллярная атаксия
14-го типа (СЦА14)АД19ч13.4^ег8СА147?605361- спиноцеребеллярная атаксия
16-го типа (СЦА16)АД8ч22.1—24.1БСАІб7?Продолжение таблицы 11ЗаболеваниеТипнасле¬дованияХромосом¬наялокализациягенаОбозначениелокуса(гена)БелковыйпродуктгенаХарактермутацииОМІМ- спиноцеребеллярная атаксия
17-го типа (СЦА 17)АД6ц21БОБ 17фактор транскрипции
ТВР7600075Аутосомно-рецессивная мозжечковаяятякг.ия г. окуппмотопной аптаксией: . .-аАР9р13АОА1??208920-бАР9ц34АОА2?7208920Болезнь (атаксия) ФридрейхаАР9я13БІША (Х25)фратаксинЭТП229300Врожденная гранулярная
гипоплазия мозжечкаАР1ІЧІ4-21СХАІ7?213200Врожденная Х-сцепленная
гипоплазия мозжечкаХРХр11.21-ч24СЬА2??302500Дентаторубро-паллидолюисоваатрофияАД12р12—ріегОЯРЬАатрофинЭТП125370Заднестолбовая атаксия
с пигментным ретинитомАР1яЗ1—32АХРС177176250Младенческаяспиноцеоебелляпная атаксияАР10я23-24.1105СА?7271245Няг.петтгвенные спастические параплегии:• аутосомно-доминантные формы-
бопечнь Штпюмпеля: -аАД14я 11.2—24.3ЭРвЗ??182600-бАД2р21-24ЭР04спастин (АТФ-аза из
семейства ААА белков^ТМ, Д, В182601-вАД15ч-11.1БРвб??600363514 Приложение 1 Приложение 1 515
Продолжение таблицы 11-гАД8q24SPG8?7603563-дАД10q23.3-24.2SPG9?601162-еАД12ql3SPG 10?604187-жАД19q 13.1SPG12?604805-зАД2q24-34SPG 13?605280• аутосомно-рецессивные
формы:-аАР8q12—13SPG5??270800-бАР16q24.3SPG7 (PGN)параплегии(АТФ-зависимаямитохондриальнаяметаллопротеаза)Д, в, тм602783-вАР15q13—15SPG11??604360-гАР3q27-28SPG 14?7605229-дАР14q22-24SPG 15??— г(синдром Шегрена-Ларссона)АР17p 11.2SLS (FALDH)жирная альдегид-
дегидрогеназаТМ270200• Х-сцепленные формы:-аХРXq28SPG1 (MASA)белок Ы-САМ (фак¬
тор адгезии нейронов)тм303350-бХРXq22SPG2 (PLP)протеолипидныйбелоктм312920Сидеробластическая анемия со
спиноцеребеллярной атаксиейХРXql3.1-13.3XLSA/A (ASAT)белок АВС7тм301310Синдром Артса (фатальная
Х-сцепленная атаксия с глухотой
и потерей зрения)ХРXq21.2-24ARTS97301835Продолжение таблицы 11ЗаболеваниеТипнасле¬дованияХромосом¬наялокализациягенаОбозначениелокуса(гена)БелковыйпродуктгенаХарактермутацииОМІМСиндром Жуберта(атаксия с окуломоторной апраксией.
умственной отсталостью и гипоплази¬
ей червя мозжечка)АР9q34.3JBTS197213300Спастическая атаксия
(болезнь Шарлевуа-Сажене)АР13ч11—12SACS97270550Спастическая форма детского
церебрального паралича
(семейные случаи)АР2ч24-25SCP77603513Эпизолические атаксии:- эпизодическая атаксия 1-го типаАД12р 13ЕА1 (KCNA1)потенциал-зависимый
калиевый каналтм160120- эпизодическая атаксия 2-го типаАД19р 13.1ЕА2 (CACNL1A4)потенциал-зависимый
кальциевый канал,
аід-субьединицаТМ108500Наследственные деменцииБолезнь Альцгеймера
(семейные <ктмы):- аАД21 q2АОІ (АРР)предшественник(3-амилоидаТМ104760- 6АД14q24.3АОЗ (РБ1)пресенилин-1ТМ104311- вАД1q31—42АБ4 (РБ2)пресенилин-2ТМ600759516 Приложение 1 Приложение 1 517
Продолжение таблицы 11Болезнь Ярви-Хакола-Насу
(мембранозная липодисгрофия,
поликистозная липомембранозная
остеодисплазия со склерозирующей
лейкоэнцефалопатией)АР19ql3PLO-SL?7221770Лобно-височная деменция
с паркинсонизмомАД17q21FTDP (МАРТ)тауТМ601630Пресенильная нейросерпин-
ассоциированная деменция (семейная
энцефалопатия с нейросерпиновыми
включениями)АД3q26FEN1Bнейросерпинтм604218Прионные болезни
(семейная форма болезни
Крейтцфельдта-Якоба,
синдром Герстманна-Штреусслера,
фатальная семейная инсомния)АД20pl2-pterPRNPприонный белоктм, в123400137440600072Семейная деменция британского/
датского типа (семейная
церебральная амилоидная ангиопатия
британского типа)АД13ql4ITM2B (BRI)интегральный
мембранный белок
(Bri)тм,дп117300176500Семейная неспецифическая
деменцияАД3pl 1.1—ql 1.2DMT177600795Синдром Йенсена (оптико¬
акустическая невральная атрофия
с деменцией)ХРXq22DFN1белок DDPтм311150'-'ї4DПродолжение таблицы IIЗаболеваниеТипнасле¬дованияХромосом¬наялокализациягенаОбозначениелокуса(гена)БелковыйпродуктгенаХарактермутацииOMIMНаследственные эпилепсииДетская абсансная эпилепсия
(семейная форма)АР (?)8q24EC Al??600131Доброкачественные семейные
неонатальные судоооги:-аАД20ql3.3EBN1 (KCNQ2)нейрональный
калиевый канал,
<32-субъединицатм, Д, В121200-бАД8q24EBN2 (KCNQ3)нейрональный
калиевый канал,
Оз-субъединицатм121201Доброкачественные семейные
младенческие судороги:— изолированныеАД19ql2BFIC?7601764— с пароксизмальным хореоатетозомАД16pl2—ql2ICCA??602066Генерапизованная эпилепсия с
гЬебпильными сулооогами плюс:— тип 1АД19q 13.1GEFSP1 (SCNA1B)нейрональный
натриевый канал,
(31-субъединицатм604236- тип 2АД2q24GEFSP2 (SCNA1A)нейрональный натриевый
канал, а-субьединицаТМ604233-тип 3АД5q31.1-33.1GEFSP3 (GABRG2)-субъединица
рецептора ГАМК518 Приложение 1 Приложение 1
Идиопатическая генерализованная
эпилепсия (семейные формы):-аАДЕСІ99600669-бАР8р11—12Еетг?9Латеральная височная эпилепсия
(парциальная эпилепсия со слуховыми
симптомами)АД10423.3-24.1ЕРТ?9600512лооная эпилепсия с ночными
пароксизмами:-аАД20ц 13.2Е№Ы(СН1ША4)нейрональныйн-холино-рецептор,а.|-субъединицатм, В600513-бАД15424Е№Ь299603204-вАД1р21Е№ЬЗ(сшшвг)нейрональныйн-холино-рецептор,р2-субъединицатм605375Младенческая миоклоническая
эпилепсияАР16р13ЕІМ?7605021Пиридоксин-чувствительная(пиридоксин-зависимая)эпилепсияАР5ч31РЭЕ97226100Прогрессирующая эпилепсия с
умственной отсталостью («северная
эпилепсия», «финская
эпилепсия»)АР8р1егЕРМИ77600143Роландическая эпилепсия
(семейная форма)АД15ц 14ЕСТ79117100іПродолжение таблицы 11ЗаболеваниеТипнасле¬дованияХромосом¬наялокализациягенаОбозначениелокуса(гена)БелковыйпродуктгенаХарактермутацииОМІМРоландическая эпилепсия с
пароксизмальной дистониейАР16р11.2—12??Семейная доброкачественная
миоклоническая эпилепсияАД8ч24ВАНУІЕ9?601068Семейная парциальная эпилепсия
с вариабельными очагами:АД22411-12РРЕ\Ф1??604364-бАД2РРЕУР2??Синдром прогрессирующей• болезнь ЛафорыАР6424ЕРМ2А (МЕЬР)лафорин (белковая
тирозин-фосфатаза)Д, ТМ, В254780• миоклонус-эпилепсия
Унферрихта-ЛундборгаАР21422.3ЕРМ1 (СЭТВ)цистатин Вв,тм254800• нейрональные цероид--аАР1р32СЬШ (РРТ)пальмитоил-протеинтиоэстеразаТМ,'В, Д256730-бАР11 р 15сшгтрипептидил-
пептидазаIТМ, В, Д204500520 Приложение 1 Приложение 1 521
-вАР16р 12СШЗтрансмембранный
лизосомальный белоксимзД, ТМ, Вулицы 1 I204200-гАР13ц22СЫЧ5трансмембранный
лизосомальный белок
ШЧ5ТМ,Д256731-дАР15я21—23ОЛЧб99601780-еАР8р23СЦч[8трансмембранный
лизосомальный белок
СЦ48ТМ600143• сиалидозы:-аАР6р21.31ЧЕШа-7\/-ацетилнейр-
аминидаза (сиалидаза)ТМ256550- б (галактосиалидоз)АР20ч13.1ОБЬ (РРЭВ)катепсин Атм, Д, в256540Фебрильные судороги
(семейная форма):-аАД8Ч13-21РЕВ199602476-бАД19р 13.3РЕВ299602477— вАД2Ч23-24РЕВЗ99604403ювенильная миоклоническая эпилепсия (семейная форма):-а?8Ч13—21Е.1М199254770-б?15ч14е.тм2?9604827Продолжение таблицы 11ЗаболеваниеТипнасле¬дованияХромосом¬наялокализациягенаОбозначениелокуса(гена)БелковыйпродуктгенаХарактермутацииОМ1МАцерулоплазм инемияПрочиеАРмоногенные наЗц23-25СРцерулоплазминТМ, В604290Идиопатическая кальцификация
базальных ганглиев
(болезньФара), семейная формаАД14я11.2-21.3шее?9213600Лейкодистрофии:— адренолейкодистрофияХРХц28А1Х) (АМЫ)белок пероксисомной
мембраны (А1ЮР)тм, д, в202370- атаксия детского возраста
с центральнойАРЗч27У\УМ??603896- болезнь АлександераАР11413даШУ1флавопротеин-1МАПН-убихинон-оксидоредуктазытм203450- болезнь КанаванаАР17р 13 —р1егАЭР (АСУ2)аспартоацилаза
(аминоацилаза 2)тм,д271900- болезнь Краббе (глобоидно-
клеточная лейкодистрофия)АР14Ч31ОХ)галактоцере брозидазатм.д
дп, тм245200312080— болезнь Пелицеуса—Мерцбахера
-лейкодистрофия с моторно¬
сенсорной невропатией и синдромом
Ваарденбурга-ХиршпрунгаХРАДXq22
22 я 13БОХЮфактор транскрипции
ЭОХЮтм277580522 Приложение 1 Приложение 1
- метахромотическая
лейкодистрофияАР22q 13.31
—qterMLD (ARSA)арилсульфатазаТМ, Д, В250100Мегалэнцефалическая лейкоэнцефа-
лопатия с субкортикальными кистамиАР22qtelMLC77604004Наследственная гиперэкплексияАД, АР5q32STHE (GLRA1)глициновый рецептор,
al-субъединицаТМ149400Семейная гемиплегическая мигреньАД19p 13.1MPH1 (CACNL1A4)потенциап-зависимый
кальциевый канал,
aiA-субъединицаТМ141500Синдром Мартина-Белл
(синдром ломкой Х-хромосомы):-аХРXq27.3FRAXA (FMR1)белок FMR1ЭТП309550-6ХРXq28FRAXE (FMR2)белок FMR2этп309548Синдром РетгаХД(?)Xq28RTT (MECP2)метил-CpG-
связывающий
белок 2 (МеСР2)ТМ312750Факоматозы:— нейрофиброматоз 1-го типаАД17ql 1.2NF1нейрофиброминТМ, Д, В162200- нейрофиброматоз 2-го типаАД22ql2.2NF2мерлин / шванноминТМ, д101000-туберозный склероз 1-го типаАД9q34.1-34.2TSC1гамартинТМ,Д191100- туберозный склероз 2-го типаАДІбрІЗ.ЗTSC2туберинтм,д191092- синдром Гиппеля-ЛиндауАД3p25-26VHLбелок VHLД, ТМ, В193300Продолжение таблицы 11ЗаболеваниеТипнасле¬дованияХромосом¬наялокализациягенаОбозначениелокуса(гена)БелковыйпродуктгенаХарактермутацииOMIMо,Kj4^IОГОЖКгоЦеребральные кавернозные
мальформации (семейные формы)
-а ЦАДАСИЛ (церебральная аутосомно-
доминантная артериопатия с субкор¬
тикальными инфарктами и лейкоэн-
цефалопатией)АДАДАДАД(NOTCH3)7ql 1.2-217р 13-153q25.2-2719р 13.1ССМ1 (Kritl)ССМ2ССМЗCADASILбелок КпНбелок Notch 3ТМ,ДТМ604214603284603285125310Церебро-артериапьный амилоидоз
(семейное церебральное кровоизлияние):— голландский тип- исландский тип (амилоидоз VI)
Церебро-тендинальный
ксантоматоз АДАДАР21q2120р11.22q33-qterСУАР (АРР)HCHWA (CST3)СТХ (CYP27)прекурсор [3-амилоидацистатин Сстерол-27-гидроксилазаТМТМТМ,В104760105150213700онгожSгоKj
Таблица 12ЗаболеваниеТипнаследованияГени его белковый продуктХарактермутацийАтаксия / лейкодистрофияСгруппа митохондриальных геновдАтаксия / миоклонии / глухотаМтРНКсгринТМГлухота, вызванная применением
аминогликозидовМ125 (рРНК)ТМДвусторонний некроз полосатого теламАТФ-синтетаза, субъединица 6ТМДеменция / хореястрнк тРгттоФанТМИзолированная митохондриальная
миопатиямтрнк лег'1ч11Н тРНК пролинТММиоклонус-эпилепсия с рваными
красными волокнами (MERRF)мтРНК мт"тмMERRF / MELASмтРНК ттн, тРНКсер"“тмМитохондриальная миопатия
/ невропатиясгруппа митохондриальных геновдМитохондриальная миопатия
с кардиомиопатиеймтрнк лега<интмМитохондриальная энцефаломиопатиястРНК треонин, тРНК """""тм,дМитохондриальная энцефаломиопатия,
лактат-ацидоз, ,инсультоподобные эпизоды (MELAS):-амтрнк ватш тРНК яе'п'ин тРНК цист-НИтм-бмцитохром с-оксидаза, субъединица IIIтм— всгруппа митохондриальных геновдOiKjOsПродолжение таблицы 12Наследственная невропатиязрительныхнервов Лебера:— амКАОН-убихинон-оксидоредуктаза,
субъединицы 1-6ТМ-бмубихинол-цитохром с-
оксидоредуктазаТМ-вмцитохром с-оксидаза,
субъединицы I, IIIТМТМ-гНаследственная невропатия
зрительных нервов ЛебераммЫАОН-убихинон-оксидоредуктаза,
субъединицы 4, 6ТМНевропатия, атаксия, пигментный ретинит
(ЫАК.Р)мАТФ-синтетаза, субъединица 6
тРНКТМтмНейросенсорная глухота
Прогрессирующая наружная
офтальмоплегиям-а-б мстРНК аспараги" тРНК изолеицин^
тРНКгруппа митохондриальных геновтмд
Продолжение таблицы 12ЗаболеваниеТипнаследованияГен и его белковый
продуктХарактермутацийСиндром Кернса-СейраСгруппа митохондриальных геновД. дпСиндром (болезнь) Лея(подострая некротизирующая энцефалопатия):— амАТФ-синтетаза, субъединица 6ТМ-бмЫАБН-убихинон-оксидоредуктаза,
субъединица ГТМ-вмтРИКТубулопатия / диабет /
мозжечковая атаксиямгруппа митохондриальных геновДПФатальное врожденное
мультисистемное заболеваниемтРНК п'Ре0ИинТМЭнцефаломиопатия / диабетмтРНК глутаминоваяТМ528 Приложение 1
ПРИЛОЖЕНИЕ 2НОМЕНКЛАТУРА ГЕННЫХ
МУТАЦИЙ ЧЕЛОВЕКАВажнейшим элементом номенклатуры генных
мутаций является общепринятое краткое обозначение
каждой из 20 аминокислот, входящих в состав белка
(таблица 13). Оно может осуществляться либо путем
записи 3 ключевых букв английского названия ами¬
нокислоты, либо условным однобуквенным символом.
Современная номенклатура предполагает преимуще¬
ственное использование однобуквенных обозначений
аминокислот.Для обозначения мутаций используется стан¬
дартизованная нумерация аминокислот в белке и нук¬
леотидов в составе гена. Отсчет аминокислот ведется
с первого положения в полипептидной цепочке, от¬
счет нуклеотидов начинается с первого нуклеотида в
составе первого смыслового кодона и ведется на про¬
тяжении всей кодирующей области (исключая интро-
ны). Нуклеотиды, находящиеся в составе экзонов,
обозначаются заглавными буквами (А, Т, С, в), в со¬
ставе интронов - маленькими (а, 1, с, §).Нуклеотиды, входящие в состав интронов или
других некодирующих областей гена, нумеруются ис¬
ходя из их расположения по отношению к ближайше¬
му экзону: если нуклеотид расположен левее экзона
(т.е против рамки считывания), он записывается со
знаком «-», если он находится правее экзона (вдоль
530Приложение 2Таблица 1:Общепринятая номенклатура аминокислотАминокислотаТрехбуквенныйсимволОднобуквенныйсимволАланинAlaААргининArgRАспарагинAsnNАспарагиноваяAspDкислотаВалинValVГ истидинHisHГлицинG lyGГлутаминGinQГлутаминоваяGluEкислотаИзолейцинIleIЛейцинLeuLЛизинLysКМетионинMetMПролинProPСерииSersТирозинTyrYТреонинThrTТриптофанTrpwФенилаланинPheFЦистеинCysС
Приложение 2531рамки считывания), он записывается со знаком «+».
Например, запись «-3» означает третий нуклеотид
слева от начала первого экзона (т.е. от первого коди¬
рующего нуклеотида); запись «15-1» означает пер¬
вый интронный нуклеотид слева от экзона, начинаю¬
щегося 15-м нуклеотидом; запись «34+3» означает
третий нуклеотид интрона, следующего за экзоном,
оканчивающимся 34-м нуклеотидом (рис. 76).Запись мутаций может осуществляться как
посредством обозначения нуклеотидных изменений
в ДНК, так и посредством обозначения изменений
аминокислотного состава белка (в тех случаях, когда
такое изменение имеет место). В случае первого опи¬
сания в литературе определенной мутации ее обозна¬
чение должно даваться как в нуклеотидной, так и в
аминокислотной системе записи (если это возможно).Относительно несложной является регистра¬
ция точковых мутаций. Для записи нуклеотидной за¬
мены приводятся номер мутировавшего нуклеотида,
обозначение исходного нуклеотида и (через стрелку)- обозначение нового нуклеотида, например: 2360->Т
(замена О на Т в положении 236 экзона); 1174+3 А—>С
(замена А на С в 3-м положении интрона, примыкаю¬
щего справа к 1174-му нуклеотиду экзона) и т.д. Ког¬
да известна полная геномная последовательность,
интронные мутации могут обозначаться символом
«ГУБ» и номером соответствующего интрона. Так,
если в указанном выше примере 1174-й нуклеотид
замыкает 5-й экзон, то предствленная интронная му¬
тация может быть обозначена как 1У85+ЗА—>С (за¬
мена А на С в 3-м положении 5-го интрона).Для записи замены аминокислоты приводятся
последовательно исходный вариант аминокислоты, ее
порядковый номер в составе белка и новый вариант
ЭКЗОН 1ЭКЗОН 2ИНТРОН1ИНТРОН2аддсаАЮССОСТАОАСТТд1даа ... аиадОТССАТТ ...СС Ад1:с дс аа-3-2-11 23456789101112131414+1114+3 15-2 15 11714+215-1 16 18 2032/ 34 34+1 \ 34+3
34+2Рис. 76. Принципы обозначения нуклеотидов в гене
Приложение 2533аминокислоты, возникший в результате мутации, на¬
пример: Y59S (замена тирозина на серин в 59-м кодо¬
не). Эта же мутация может быть обозначена как
Tyr59Ser (однако, как уже было указано, однобуквен¬
ное обозначение аминокислот признается предпочти¬
тельным). Для обозначения стоп-кодона, ведущего к
обрыву трансляции, используется символ «X». Напри¬
мер: R67X (нонсенс-мутация с заменой аргинина на
стоп-сигнал в 67-м кодоне).Делеции принято обозначать символом «del»,
который следует после номера мутантного нуклео¬
тида, например: 877delC (делеция нуклеотида С в
положении 877); 966-968delATT (делеция трех нук¬
леотидов ATT в положении 966-968). Вставки (ин-
серции) обозначаются символом «ins» с указанием
нуклеотидного интервала, в который встроился но¬
вый нуклеотид, например: 911-912insG (вставка G
между нуклеотидами, находящимися в положении 911
и 912). Для обозначения протяженных делеций и вста¬
вок обычно указывается лишь общее число мутант¬
ных нуклеотидов. Например, запись 1634del27 озна¬
чает делецию участка длиной 27 нуклеотидов, начи¬
ная с 1634-го нуклеотида.Вариабельное число простых нуклеотидных
повторов обозначается следующим образом:
1337(GT)8-19 (т.е. динуклеотид GT, начиняющийся с
1337-го положения, может повторяться от 8 до 19 раз).
Исходя из этого, мутация по типу экспансии тринук-
леотидных повторов может быть записана следующим
образом: 23(CAG)56 (т.е. 56 повторов триплета CAG,
начинающегося с 23-го нуклеотида).Точковые мутации, являющиеся полиморфизма¬
ми (т.е. либо не приводящие к изменению аминокис¬
лоты, либо ведущие к замене аминокислоты на функ¬
534 Приложение 2ционально равноценную ей) обозначаются символа¬
ми, разделенными косой чертой, например: 1366C/G
(С либо G в положении 1366). Аналогичным образом
полиморфизмы обозначаются и в аминокислотной
системе записи: 455Leu/Val, 455L/V (лейцин либо ва-
лин в кодоне 455).Если в одном и том же аллеле зарегистрирова¬
ны 2 мутации, они указываются в квадратных скоб¬
ках через точку с запятой, например: [866G—>Т;
1001 А—>С]. При аутосомно-рецессивных заболевани¬
ях для записи 2 мутаций, расположенных в различ¬
ных аллелях одного гена, эти мутации также указы¬
ваются в квадратных скобках и объединаются знаком
«+»: [1997G-*T + 2001А—>G].В некоторых случаях для уточнения
используемой нуклеотидной нумерации (геномная
ДНК или кДНК) перед номером нуклеотида
указывается особый индекс с точкой: для геномной
ДНК используется индекс g. («genomic»), для к ДНК- индекс с. («cDNA»).
ПРИЛОЖЕНИЕ 3Словарь специальных
генетических терминов,
использованных в текстеАвторадиография - метод выявления макромолекул, меченных радиоактивной
меткой и разделенных гель-электрофорезом, по их способности ос¬
тавлять след на рентгеновской пленке.Аллель - один из возможных альтернативных вариантов гена, отличающийся
от других вариантов определенными особенностями нуклеотидного
состава:• доминантный - аллель, наличие одной копии которого доста¬
точно для проявления соответствующего фенотипического при¬
знака;• мутантный - вариант гена, несущий патогенетически значи¬
мую мутацию;• нормальный (дикого типа) - вариант гена, функция которого
не изменена;• «промежуточный» (при заболеваниях с динамическими му¬
тациями) - аллель, представляющий собой «премутацию», в
составе которого число копий тандемных тринуклеотидных
повторов превышает общепопуляционную норму, но не дости¬
гает нижней патогенетически значимой границы экспансии
повторов;• рецессивный - аллель, фенотипически проявляющийся толь¬
ко в гомозиготном состоянии и подавляющийся доминантным
аллелем.Аллельная серия - совокупность самостоятельных заболеваний, различающих¬
ся по своим базовым фенотипическим проявлениям, но связанных с
повреждением одного и того же гена.Аллельная гетерогенность - наличие в рамках единой нозологической фор¬
мы различных генетических вариантов заболевания, вызываемых раз¬
личными мутантными аллелями (различными мутациями) одного
гена.Аллельные заболевания - фенотипически различные заболевания, обуслов¬
ленные повреждением одного и того же гена.Амплификация - увеличение числа копий определенного фрагмента ДНК (на¬
пример, на основе полимеразной цепной реакции).Антикодон - нуклеотидный триплет в составе молекулы тРНК, комплементар¬
но взаимодействующий со специфическим кодоном мРНК в процес¬
се трансляции.Антиципация - появление более ранних и более тяжелых случаев заболевания
в каждом последующем поколении.Аутосома - неполовая хромосома (любая кроме хромосом X и У).
536Приложение 3Блот-гибридизация (блоттинг) - метод идентификации макромолекул, разде¬
ленных гель-электрофорезом и фиксированных на твердом матрик¬
се, путем гибридизации содержимого образцов с мечеными компле¬
ментарными зондами:• Всстерн-блоттинг (иммуноблоттинг) - идентификация иско¬
мого белка путем гибридизации разделенных гель-электрофо¬
резом белковых молекул с меченым антителом;• Нозерн-блоттинг- идентификация искомой последовательно¬
сти РНК путем гибридизации разделенных гель-электрофоре-
зом молекул мРНК с меченым комплементарным ДНК-зондом;• Саузерн-блоттинг - идентификация участка ДНК, содержа¬
щего искомую нуклеотидную последовательность, путем гиб¬
ридизации разделенных гель-электрофорезом фрагментов ДНК
с меченым комплементарным ДНК-зондом.Болезни :• аутосомныс - обусловленные мутацией гена, локализованно¬
го в аутосоме;• доминантные - обусловленные доминантным мутантным ал-
лелем;• менделиругощие - наследующиеся в соответствии с законами
Менделя;• митохондриальные - обусловленные генетическими и струк-
турно-биохимическими дефектами митохондрий;• моногенные - обусловленные мутацией одного главного гена;• мультифакториальные (полигенные, болезни предрасполо¬
женности) - обусловленные совместным действием ряда ге¬
нов и средовых факторов;• наследственные - связанные с повреждениями генов, пере¬
дающимися потомству;• «полиглутаминовые» - обусловленные экспансией трансли¬
руемых тандемных повторов САв, что сопровождается пропор¬
циональным удлинением полиглутаминового участка в соста¬
ве белка;• рецессивные - обусловленные двумя мутантными рецессив¬
ными аллелями;• сцепленные с полом - обусловленные мутацией гена, лока¬
лизованного в половых хромосомах;• хромосомные - обусловленные нарушением количественного
состава или структуры хромосом;• экспансии - обусловленные экспансией (патологическим уве¬
личением числа копий) тандемных тринуклеотидных повторов.Вставка (инсерция) - тип мутации, заключающийся во встраивании в опреде¬
ленный участок ДНК «лишней» нуклеотидной последовательности.Вырожденность - свойство генетического кода, в силу которого каждая из 20
аминокислот может кодироваться несколькими возможными комби¬
нациями из 3 нуклеотидов.Гамета - зрелая половая клетка с гаплоидным набором хромосом.Гаплоидный - содержащий одинарный (однокопийный) набор хромосом.Гаплотип - комбинация конкретных аллелей сцепленных генов (локусов) на
одной хромосоме.
Приложение 3 537Гемизиготность - отсутствие одного из двух аллелей в определенном локусе.Ген - транскрибируемый участок молекулы ДНК, последовательность которо¬
го заключает в себе всю информацию, необходимую для синтеза мо¬
лекулы белка или РНК.Генетическая ассоциация - достоверная взаимосвязь между заболеванием и
определенным аллелем изучаемого гена.Генетическая гетерогенность - наличие в рамках единого фенотипа различ¬
ных молекулярных вариантов заболевания, имеющих разную генети¬
ческую основу.Генетический код - соответствие кодирующих нуклеотидных триплетов (ко¬
донов) в молекуле нуклеиновой кислоты определенным аминокис¬
лотам в составе белка.Генетический маркер - полиморфный участок ДНК строго определенной ло¬
кализации, многообразные аллели которого позволяют дифференци¬
ровать различные по происхождению хромосомы и анализировать их
расхождение в родословной.Генетический риск - вероятность появления определенного наследственного
заболевания у консультируемого лица или у его потомков.Генетическое сцепление - близкое расположение генов (локусов) на хромо¬
соме, позволяющее им наследоваться как единое целое.Генная диагностика (генодиагностика) - см. ДНК-диагностика.Генная инженерия - совокупность методов и технологий (в том числе техно¬
логий получения рекомбинатных молекул ДНК и РНК) по выделе¬
нию генов из организма, осуществлению манипуляций с генами и
введению их в другие организмы.Генная терапия (генотерапия) - совокупность биотехнологических и меди¬
цинских подходов, направленных на внесение изменений в генети¬
ческий аппарат соматических клеток человека в целях лечения.Генокопия - клинический синдром, манифестирующий под маской известно¬
го наследственного заболевания с установленным генетическим де¬
фектом, но обусловленный повреждением другого гена.Геном - а) совокупность всех генов определенного биологического вида; б)
полная генетическая система отдельной клетки или конкретного орга¬
низма.Геномная (генетическая) классификация - классификация наследственных
заболеваний, основанная на взаимосвязи между конкретной нозоло¬
гической формой и повреждением определенного гена, лежащим в
основе болезни.Генотип - набор генов, определяющих развитие фенотипического признака или
ряда признаков организма.Гетерозигота - носитель двух различных аллелей в определенном локусе.Гете роз и готн ость - наличие двух различных аллелей в определенном локусе.Гетероплазмия - наличие в клетках и тканях смешанной популяции нормаль¬
ных и мутантных молекул митохондриальной ДНК.Гетерохроматин - конденсированные участки хромосом, содержащие в основ¬
ном некодирующие высокоповторяющиеся последовательности ДНК.Гибридизация (ренатурация) - взаимодействие комплементарных цепей ДНК
(или ДНК и РНК), приводящее к образованию двуцепочечной моле¬
кулы:• дот (слот)-гибридизация - модификация Саузерн-блотгинга,
при которой проводится гибридизация с ДНК-зондом молекул
538Приложение 3ДНК (РНК), нанесенных на фильтр в виде округлых или вытя¬
нутых пятен;• in situ - реакция гибридизации меченого ДНК- или РНК-зонда
с денатурированной хромосомной ДНК клеток на гистологи¬
ческих препаратах;• FISH (fluorescent in situ hybridization)-модификация гибрид¬
изации in situ с использованием меченных флюорохромами
ДНК-зондов.Гистоны - ядерные белки, образующие комплекс с молекулой ДНК и принима¬
ющие участие в формировании и поддержании структуры хромосом.Гомозигота - носитель двух одинаковых аллелей в определенном локусе.Гомозиготность - наличие двух одинаковых аллелей в определенном локусе.Гомология - сходство в последовательности макромолекул между организма¬
ми одного или разных видов:• гомологичные участки ДНК (гены) - участки ДНК, имею¬
щие сходство в нуклеотидной последовательности;• гомологичные хромосомы (гомологи) - пара хромосом, име¬
ющих одинаковое цитогенетическое строение и набор генов.Гомоплазмия - наличие в клетках и тканях митохондриальной ДНК одного
вида (нормальной или мутантной).«Горячая точка» - участок ДНК с высокой частотой возникновения мутаций
или рекомбинаций.Группа риска - члены консультируемой семьи, которые могут являться носи¬
телями мутантного гена и имеют высокий риск заболеть данным на¬
следственным заболеванием или передать мутантный ген потомству.Дезоксирибонуклеотидтрифосфаты - молекулы-предшественники ДНК, со¬
стоящие из дезоксирибозы, одного из азотистых оснований (А, Т, С
или G) и трех остатков фосфорной кислоты.Дезокси рибонуклеиновая кислота (ДНК) - нитевидная самореплицирующа-
яся молекула, состоящая из отдельных дезоксирибоза-содержащих
нуклеотидов и являющаяся хранителем генетической информации,
закодированной посредством триплетного кода:• геномная - тотальная ДНК, выделенная из ядерных клеток;• кДНК (комплементарная) - одноцепочечная молекула, по¬
лученная путем обратной транскрипции мРНК и содержащая
только кодирующие участки гена;• мтДНК (митохондриальная) - кольцевидная молекула, лока¬
лизованная в митохондрии;• рекомбинантная - состоящая из фрагментов различного про¬
исхождения;• цитоплазматическая - см. мтДНК.Деления - тип мутации, заключающийся в утрате (выпадении) определенной
нуклеотидной последовательности.Денатурация - переход молекулы ДНК из двуцепочечной формы в одноцепо¬
чечную вследствие разрыва водородных связей между комплементар¬
ными основаниями.Диплоидный - содержащий двойной набор хромосом (по 2 копии каждой из
аутосом и 2 половые хромосомы).ДНК-диагностика (генодиагностика) - совокупность методов по выявлению
изменений в структуре генома:• косвенная (непрямая) - основана на анализе наследования в
Приложение 3539ряду поколений мутантной хромосомы с помощью полиморф¬
ных генетических маркеров;• преимплантационная - диагностика мутации на ранней ста¬
дии развития зародыша, предшествующей его имплантации в
стенку матки;• пренатальная - диагностика мутации или мутантной хромо¬
сомы у плода;• пресимптоматическая - диагностика мутации или мутант¬
ной хромосомы у клинически здорового индивида, относяще¬
гося к группе риска;• прямая - основана на непосредственной идентификации му¬
тации в изучаемом гене.ДНК-полимераза - фермент, осуществляющий комплементарный синтез (реп¬
ликацию) ДНК.Домен - участок аминокислотной последовательности белка, связанный с оп¬
ределенной функцией.Дупликация -тип мутации, заключающийся в удвоении той или иной нуклео¬
тидной последовательности.Зигота - оплодотворенная яйцеклетка, образующаяся при слиянии двух гамет.Зонд - меченная радиоизотопом или флюорохромом молекула ДНК (РНК), гиб-
ридизующаяся с изучаемым комплементарным участком геномной
ДНК или РНК.Изолят - относительно изолированная в силу географических и иных причин
популяция, в которой имеет место высокая частота кровнородствен¬
ных браков, приводящая с течением времени к высокой степени ге¬
нетического родства членов популяции.Импринтинг - различный характер экспрессии гена в зависимости от того,
каким родителем он передан.Инбредный (применительно к браку, семье, популяции и т.д.) - характеризую¬
щийся высокой степенью генетического родства.Инбридинг - брак между индивидами, находящимися в близком генетическом
родстве.Инсерция - см. вставка.Интрон - некодирующий участок гена, находящийся между кодирующими об¬
ластями (экзонами) и удаляемый из первичного РНК-транскрипта в
процессе сплайсинга.Информативность (в контексте косвенной ДНК-диагностики и анализа сцеп¬
ления) - возможность дифференцировать родительские хромосомы
и анализировать их расхождение в родословной с помощью полимор¬
фных генетических маркеров.Картирование - определение локализации генетических элементов на хромо¬
соме:• генетическое - определение расположения изучаемого гена
по отношению к другим локусам в определенной хромосом¬
ной области;• физическое - установление точной последовательности сай¬
тов и физического расстояния между ними в изучаемой хромо¬
сомной области.Килобаза (кб) - единица длины молекулы ДНК, равная тысяче пар оснований.Клеточный цикл - последовательность событий в клетке между двумя деле¬
ниями.
540Приложение 3Клон - генетически однородное потомство одной клетки.Клонирование (гена) - встраивание выделенного гена (фрагмента ДНК) в ге¬
ном реципиентной клетки, с последующей многократной реплика¬
цией и получением большого числа копий изучаемого гена в составе
данной клеточной системы:• позиционное (обратная генетика) - технология выделения и
клонирования гена, основанная на знании его хромосомной ло¬
кализации, при отсутствии предварительной информации о
первичном биохимическом продукте данного гена;• функциональное - технология выделения и клонирования
гена, основанная на имеющейся информации о первичном бел¬
ковом продукте данного гена.Кодон - тринуклеотидная последовательность в составе ДНК и мРНК, кодиру¬
ющая определенную аминокислоту или определяющая окончание
трансляции:• стоп (нонсенс) - кодон, являющийся сигналом окончания
трансляции.Компаунд-гетерозигота (компаунд) - больной с аутосомно-рецессивным за¬
болеванием, имеющий два различных мутантных аллеля одного гена.
Компаунд-гетерозиготность - наличие на гомологичных хромосомах двух раз¬
ных мутантных аллелей гена у больного с аутосомно-рецессивным
заболеванием.Комплементарность - свойство нуклеотидов образовывать пары по правилу
А-Т, С-<} за счет формирования водородных связей между ними в
двуцепочечной молекуле ДНК или гибриде ДНК/РНК.
Комплементарный - основанный на взаимодействии нуклеотидов по правилуА-Т, СМ}.Кроссииговер - обмен участками ДНК, происходящий между гомологичными
хромосомами в мейозе:• неравный - обмен неравноценными участками ДНК вслед¬
ствие неправильного спаривания гомологичных хромосом в
мейозе.Лайонизация - случайная инактивация одной из двух Х-хромосом в женской
соматической клетке (механизм, обеспечивающий сбалансирован¬
ность генетического материала у индивидов мужского и женского
пола).Лод-балл - логарифм соотношения правдоподобий, выражающий количествен¬
ную оценку генетического сцепления.Локус - область локализации определенного генетического элемента на хро¬
мосоме.Локусная гетерогенность - наличие в рамках единой нозологической формы
различных генетических вариантов заболевания, вызываемых мута¬
циями самостоятельных генов в разных хромосомных локусах.
Маркер - см. Генетический маркер.Мегабаза (мб) - единица длины молекулы ДНК, равная миллиону пар основа¬
ний.Медико-генетическое консультирование - особый вид специализированной
медицинской помощи, направленный на предупреждение появления
повторных случаев наследственных заболеваний в отягощенных се¬
мьях.Мейоз - особое двухступенчатое деление клетки, приводящее к образованию
из одной диплоидной клетки четырех гамет, имеющих гаплоидный
набор хромосом.
Приложение 3 541Менделевские законы - основные законы наследственности.Митоз - основной способ деления эукариотических клеток, обеспечивающий
образование из одной диплоидной клетки двух генетически идентич¬
ных дочерних клеток с таким же набором хромосом.Мозаицизм - наличие в организме генетически отличающихся популяций кле¬
ток, имеющих разные геномы:• гонадный - наличие у индивида двух генетически различных
популяций половых клеток (нормальной и мутантной), что
обусловлено возникновением мутации в одной из клеток-пред-
шественников на ранней стадии гаметогенеза;• соматический - наличие у индивида в какой-либо соматичес¬
кой ткани, помимо нормальных клеток, популяции клеток с
мутантной ДНК, что обусловлено возникновением мутации в
одной из соматических клеток на постзиготической стадии
развития.Мутация - любое изменение структуры ДНК:• генная - изменение последовательности нуклеотидов в опре¬
деленном участке молекулы ДНК;• динамическая - мутация по типу экспансии тандемных три-
нуклеотидных повторов;• мажорная - встречающаяся с высокой частотой в определен¬
ной популяции;• миссенс - замена нуклеотида, сопровождающаяся изменени¬
ем аминокислотного шифра кодона и ведущая к замене амино¬
кислоты в составе белка;• нейтральная (молчащая) - не сопровождающаяся изменени¬
ем фенотипа;• нонсенс - замена нуклеотида, приводящая к замещению ин¬
формационно значимого кодона на стоп-кодон и сопровожда¬
ющаяся преждевременным обрывом трансляции;• нулевая - приводящая к отсутствию синтеза сколько-нибудь
значимого функционального продукта;• регуляторная - затрагивающая регуляторные последователь¬
ности гена и нарушающая его экспрессию;• со сдвигом рамки - приводящая к нарушению нормального
отсчета кодирующих триплетов;• сплайсинговая - затрагивающая сайт сплайсинга и приводя¬
щая к неправильному вырезанию интрона либо к удалению из
молекулы РНК информационно значимой экзонной последо¬
вательности;• структурная - приводящая к протяженному (мультинуклеотид-
ному) дефекту гена;• точковая (точечная) - затрагивающая один нуклеотид либо
1-2 соседних нуклеотида;• хромосомная - нарушение количественного состава либо
структуры хромосом.Неравновесное сцепление - неслучайное распределение частот аллелей ге¬
нетических маркеров в исследуемой группе лиц по сравнению с об¬
щей популяцией.Нуклеиновые кислоты - особый класс биоорганических макромолекул (ДНК,
РНК), способных к саморепликации и обеспечивающих хранение и
передачу генетической информации.
542Приложение 3Нуклеотид - элементарный мономер, из которого состоят нуклеиновые кислоты;содержит в своем составе остаток сахара (дезоксирибоза, рибоза),
азотистое основание и фосфатную группу.Обратная генетика - см. позиционное клонированиеОбратная транскриптаза - фермент, осуществляющий обратную транскрип¬
цию (перевод мРНК в молекулу кДНК).Олигонуклеотид - короткая однонитевая молекула ДНК или РНК.Онкоген - ген, принимающий участие в позитивном контроле клеточного цикла;повышение экспрессии онкогенов играет важную роль в развитии
опухолей.п.о. - пара оснований (единица длины молекулы ДНК).Олигонуклеотид - короткая однонитевая молекула ДНК или РНК.Пенетрантность - вероятность фенотипического проявления мутантного гена.Полимеразная цепная реакция (ПЦР) - метод циклического синтеза in vitro
огромного числа копий строго определенного участка ДНК длиной
от десятков до нескольких тысяч п.о.:• количественная - реакция, по результатам которой оценива¬
ется количественный выход продуктов амплификации с помо¬
щью соответствующих сканирующих устройств;• мультиплексная (мультипраймерная) - одновременная ам¬
плификация в одной реакции нескольких участков исследуе¬
мого гена;• обратно-транскриптазная - реакция, в которой матрицей слу¬
жат молекулы кДНК, получаемые в результате обратной транс¬
крипции мРНК.Полиморфизм (применительно к элементам генома) - существование несколь¬
ких аллельных вариантов гена или генетического маркера в популяции.Потеря гетерозиготности - наличие лишь одного аллеля в определенном локу¬
се; отсутствие второго аллеля обычно является результатом делеции
соответствующего хромосомного сегмента.Праймер - олигонуклеотид, выполняющий роль «затравки» и инициирующий
синтез полинуклеотидной цепи на ДНК- или РНК-матрице.Пробанд - лицо, через которое осуществляется регистрация консультируемой
семьи.Прогностическое (предсказательное) ДНК-тестирование-проведение ДНК-
диагностики у клинически здорового члена отягощенной семьи из
группы риска, позволяющее установить его генетический статус, риск
развития заболевания, вероятный возраст манифестации и характер
течения болезни.Промотор - регуляторный участок гена, с которым связывается РНК-полимера-
за перед началом транскрипции.Процессинг (РНК) - «созревание» РНК, в процессе которого первичный РНК-
транскрипт претерпевает ряд модификаций (удаление интронов,
полиаденилирование и др.), превращаясь в молекулу зрелой мРНК.Рамка считывания - последовательность нуклеотидов, представляющая со¬
бой ряд последовательно расположенных кодирующих триплетов,
начиная со стартового кодона и заканчивая стоп-кодоном.Рекомбинация - процесс перераспределения генетического материала роди¬
телей при передаче потомству, приводящий к созданию новой ком¬
бинации генов при соединении родительских гамет.Ренатурация - см. Гибридизация
Приложение 3 543Репарация - исправление повреждений в молекуле ДНК и восстановление ее
нативной первичной структуры.Репликация - процесс самовоспроизведения молекул ДНК, происходящий в
результате комплементарного синтеза новой цепи ДНК на существу¬
ющей матричной цепи.Репрезентативная (информативная) семья - семья с наследственным забо¬
леванием, в которой имеется достаточное число больных и здоровых
родственников из разных поколений (достаточное число информа¬
тивных мейозов), что позволяет оценивать расхождение признаков в
изучаемой родословной при анализе генетического сцепления.Рестрикционная эндонуклеаза (рестриктаза)- фермент бактериального про¬
исхождения, распознающий специфическую нуклеотидную последо¬
вательность длиной от 4 до 10 п.о. и разрезающий двунитевую моле¬
кулу ДНК в этом месте.Рестрикционный анализ - метод анализа ДНК с использованием рестрикци¬
онных эндонуклеаз.Рибонуклеиновая кислота (РНК) - нитевидная молекула, состоящая из от¬
дельных рибоза-содержащих нуклеотидов и выполняющая в клетке
многообразные посреднические функции в процессе передачи гене¬
тической информации:• информационная - см. матричная;• матричная (мРНК) - молекула РНК, последовательность ко¬
торой комплементарна экзонам гена и служит матрицей для
синтеза белка;• первичный РНК-транскрипт (преРНК) - точный «слепок»
определенного участка ДНК-матрицы, образующийся в резуль¬
тате транскрипции гена;• рибосомальная (рРНК) - молекула РНК, входящая в состав
рибосом;• транспортная (тРНК) - молекула РНК, отвечающая за транс¬
портировку строго определенной аминокислоты в процессе
трансляции; специфичность связывания тРНК с аминокисло¬
той определяется наличием в составе тРНК тринуклеотидного
антикодона, комплементарного соответствующему аминокис¬
лотному кодону в составе мРНК.РНК-полимераза - фермент, осуществляющий синтез РНК на ДНК-матрице.Сайт - определенное место (позиция) в молекуле ДНК:• рестрикции (узнавания) - специфическая нуклеотидная пос¬
ледовательность длиной 4-10 п.о., распознаваемая рестрикци¬
онной эндонуклеазой;• сплайсинга - специфическая нуклеотидная последователь¬
ность на границе между экзоном и интроном, выполняющая
важную сигнальную роль для правильного протекания сплай¬
синга.Сантиморганида (сМ) - единица генетического расстояния между локусами;1 сМ соответствует частоте рекомбинационных событий между ло¬
кусами 1% и эквивалентна в среднем 1 миллиону п.о.Сегрегация - расхождение среди потомков родительских хромосом, опреде¬
ленных хромосомных сегментов либо аллелей конкретных генов (мар¬
керов), а также расхождение в потомстве тех или иных фенотипичес¬
ких признаков.
544Приложение 3Секвенирование - определение последовательности нуклеотидов в молекуле
ДНК или последовательности аминокислот в молекуле белка.Сибсы - братья и сестры, потомки одной родительской пары.Сплайсинг - процесс удаления интронов из первичного РНК-транскрипта, в
результате которого образуется молекула мРНК, содержащая лишь ко¬
дирующие экзонные последовательности и служащая матрицей для
синтеза белка:• альтернативный - различный характер «вырезания» интро¬
нов из одного и того же первичного РНК-транскрипта, обеспе¬
чивающий возможность синтеза одним геном различных бел¬
ков.Спорадический случай - единичный случай заболевания в обследуемой се¬
мье.Стоп-кодон - нуклеотидный триплет, являющийся сигналом окончания транс¬
ляции.Сцепление - см. Генетическое сцепление.Тандемные повторы - множественные и расположенные друг за другом ко¬
пии определенной последовательности ДНК, число которых у раз¬
личных индивидов является чрезвычайно вариабельным:• динуклеотидные - множественные копии последовательнос¬
ти из 2 нуклеотидов (обычно СА), используемые в качестве ге¬
нетических маркеров;• тринуклеотидные - множественные копии последовательно¬
сти из 3 нуклеотидов, увеличение числа которых (экспансия)
является одним из наиболее распространенных типов мутаций
в нейрогенетике (динамические мутации).Теломера - концевой участок хромосомы.Тип наследования - одна из основных характеристик любого моногенного
заболевания, описывающая закономерности сегрегации патологичес¬
кого фенотипа в родословной:• аутосомно-доминантный - связан с наследованием доминан¬
тного аллеля, расположенного на неполовой хромосоме;• аутосомно-рецессивный - связан с наследованием рецессив¬
ного аллеля, расположенного на неполовой хромосоме;• митохондриальный (материнский, цитоплазматический)-
связан с наследованием от матери всеми потомками мутант¬
ной митохондриальной ДНК;• псевдодоминантный - особый случай наследования аутосом-
но-рецессивного заболевания, при котором болезнь регистри¬
руется в двух и более последовательных поколениях родослов¬
ной в результате браков между больными индивидами либо
между больным индивидом и гетерозиготным носителем му¬
тантного гена;• Х-сцепленный (доминантный и рецессивный) - связан с
наследованием доминантного или рецессивного аллеля, рас¬
положенного на Х-хромосоме.Трансгенные животные (модели) - экспериментальные животные, геномы
которых включают в себя чужеродный генетический материал, ис¬
кусственно введенный с помощью методов генной инженерии.Транскрипция - комплементарный синтез молекулы РНК на одноцепочечной
ДНК-матрице:• обратная - комплементарный синтез молекулы кДНК, при ко¬
тором роль матрицы выполняет мРНК.
Приложение 3545Трансляция - синтез на рибосомах полипептидной цепи, в которой порядок
аминокислот соответствует порядку кодирующих триплетов мРНК-
матрицы.Факторы транскрипции - особые белки, осуществляющие активацию или реп¬
рессию гена при взаимодействии с молекулой ДНК.Фенокопия - клинический синдром, манифестирующий под маской опреде¬
ленного наследственного заболевания, но имеющий иную (негене¬
тическую) природу.Феномен «отцовской (материнской) передачи» - характерен для заболева¬
ний с динамическими мутациями и проявляется развитием более ран¬
них и более тяжелых случаев болезни при передаче мутантного гена
по отцовской (материнской) линии.Фенотип - совокупность наблюдаемых признаков, контролируемых определен¬
ными генами во взаимодействии со средовыми факторами.
Фенотипический полиморфизм - вариабельность фенотипа болезни при по¬
вреждении определенного гена.Фрустрированная форма (forme fruste) - «стертая» форма заболевания, ха¬
рактеризующаяся неполной клинической картиной и развитием лишь
отдельных симптомов из большого числа клинических проявлений,
обычно свойственных данному заболеванию.Хиазма - участок контакта гомологичных хромосом в мейозе, в котором проис¬
ходит межхромосомный обмен генетическим материалом (кроссин-
говер).Хроматиды - хромосомные копии, образующиеся в результате репликации:• сестринские - продукты репликации одной хромосомы;• несестринские - продукты репликации разных хромосом.
Хроматин - комплекс между ДНК и белками в ядрах клеток.Хромосома - гиперспирапизованная молекула ДНК в комплексе с ядерными
белками- гистонами, представляющая собой дискретную единицу
генома в ядре клетки:• аутосома - неполовая хромосома (у человека - 22 пары ауто-
сом и одна пара половых хромосом X и Y);• мутантная - содержащая мутацию в определенном локусе;• М-хромосома - кольцевидная молекула митохондриальной
ДНК;• половая - одна из пары хромосом X и Y, детерминирующих
пол (XY - мужской, XX - женский);• рекомбинантная - образующаяся в результате рекомбинации
(кроссинговера) в мейозе и состоящая из фрагментов хромо¬
сом, имеющих разное генетическое происхождение.Центральная догма (молекулярной биологии) - основной путь передачи ге¬
нетической информации в клетке в направлении ДНК -> РНК -» бе¬
лок.Центромера - центральная перетяжка хромосомы, разделяющая ее на корот¬
кое и длинное плечо.Экзон - кодирующий участок гена, сохраняющийся в молекуле зрелой мРНК.
Экспансия тринуклеотидных повторов - особый тип мутаций (динамичес¬
кие мутации), заключающийся в патологическом увеличении числа
копий внутригенных тандемных последовательностей, состоящих из
3 нуклеотидов.
546Приложение 3Экспрессивность - степень фенотипической выраженности патологического
признака (заболевания) у носителей мутантного гена.Экспрессия (гена) - переход гена в активное состояние, при котором происхо¬
дит реализация записанной в нем генетической информации, приво¬
дящая к синтезу первичных молекулярных продуктов гена - РНК и
белка.Эухроматин - активный хроматин с низкой плотностью упаковки, содержа¬
щий основное число активно транскрибируемых генов.Эффект основателя - высокая частота встречаемости идентичных локусов у
коренных жителей инбредной популяции как результат наследова¬
ния от единого предка общих по происхождению хромосомных учас¬
тков.Эффект положения (негативный позиционный эффект) - нарушение эксп¬
рессии гена в результате изменения его нормального хромосомного
«окружения» (например, при делеции смежного хромосомного учас¬
тка и перемещении гена в область гетерохроматина).
Библиографический указательАйала Ф., КайгерДж. (Ayala F., KigerJ.) Современная генетика (пер. с англ.) М.: Мир,
1987. - в 3-х т.Арефьев В.А., Лисовенко JI.A. Англо-русский толковый словарь генетических терминов.
М.: Изд-во ВНИРО, 1995. - 407 с.Бархатова В.П. Нейротрансмитгеры и экстрапирамидная патология. М.: Медицина, 1988.
- 176 с.Бочков Н.П. Клиническая генетика. М.: Медицина, 1997. - 288 с.Вельттцев Ю.Е., Темин П.А. Митохондриальные болезни. В кн.: Наследственные болезни
нервной системы (под ред. Ю.Е. Вельтищева, П.А. Темина). М.: Медицина, 1998:
346-471.Воронцова Н.И., Полещук В.В., Тимербаева C.JI. и др. Молекулярно-генетический анализ
болезни Вильсона-Коновалова и его роль в ранней диагностике и медико-генетичес¬
ком консультировании. В кн.: Молекулярная генетика наследственных болезней и
медико-генетическое консультирование. М.: МОНИКИ, 1996: 14-19.Гарькавцева Р.Ф., Гарькавцев И.В. Молекулярно-генетические аспекты злокачествен¬
ных новообразований. Вестник РАМН 1999; 2: 38-44.Голубев В.Л., Левин Я.И., Вейн А.М. Болезнь Паркинсона и синдром паркинсонизма. М.:
МЕДпресс, 1999. - 416 с.Горбунова В.Н. Молекулярные основы медицинской генетики. СПб.: Интермедика, 1999.
-212 с.Горбунова В.H., Баранов B.C. Введение в молекулярную диагностику и гемотера¬
пию наследственных заболеваний. СПб.: Специальная литература, 1997.-287 с.Гусев Е.И., Бойко A.H., Судомоина М.А.,Фаворова О.О. Клиническая генетика рассеян¬
ного склероза. М.: Журн.. неврол. и психиатр, им. С.С. Корсакова, 2001; 9: 61-68.Давиденков С.И. Проблема полиморфизма наследственных болезней нервной системы.
Л.: Изд-во ВИЭМ, 1934. - 139 с.Докпад научной группы ВОЗ. Борьба с наследственными болезнями. Женева, 1997. - 133 с.Евграфов О.В., Гроппа С.А., Проскурина Л.А., Тагиев Э.Ш. Использование метода
полимеразной цепной реакции в пресимптоматической диагностике хореи
Гентингтона. В кн.: Наследственные болезни и медико-генетическое консультирова¬
ние. М.: МОНИКИ, 1991: 75-78.Завалиишн И.А. Современные представления об этиологии рассеянного склероза. Журн.
невропатол. и психиатр, им. С.С. Корсакова 1990; 2: 3-8.Золотухина Т.В., Шилова И.В. Клетки плода в крови матери: новый неинвазивный
подход в пренатальной диагностике наследственных болезней. Вестник РАМН 1999;
12: 45-48.Зотиков Е.А Антигенные системы человека и гомеостаз. М.: Наука, 1982. - 236 с.Зуев В.А., Завалиишн И.А., Рошель В.М. Прионные болезни человека и животных. М.:
Медицина, 1999. - 192 с.
548 Библиографический указательИванова-Смоленская И. А. Клинические варианты эссенциального тремора. Журн.
невропатол. и психиатр, им. С.С. Корсакова 1979; 3: 291-298.Иванова-Смоленская И.А. Эссенциальный тремор (клинический полиморфизм, генетика,
патогенез, лечение). В кн.: Наследственные заболевания нервной системы. Саратов,
1983: 31-33.Иванова-Смоленская И.А. Клинические и молекулярно-генетические аспекты изучения
наследственных заболеваний нервной системы. Журн. неврол. и психиатр, им. С.С.
Корсакова 1996; 1: 29-33.Иванова-Смоленская И.А., Мэ/сельская Т.И., Миловидов Ю.К. и др. Моле'кулярно-
генетический анализ болезни Гентингтона (обзор зарубежной литературы). Журн.
неврол. и психиатр, им. С.С. Корсакова 1992; 4: 97-100.Иванова-Смоленская И.А., Маркова Е.Д., Иллариошкин C.H., Никольская H.H. Моно-
генные болезни центральной нервной системы. В кн.: Наследственные болезни
нервной системы (под ред. Ю.Е. Вельтищева, П.А. Темина). М.: Медицина, 1998
(а): 9-104.Иванова-Смоленская И.А., Овчинников И.В., Иллариошкин С.Н. и др. Молекулярно¬
генетическое тестирование в диагностике спорадических случаев хореи Гентингто¬
на. Журн. неврол. и психиатр, им. С.С. Корсакова 1998 (б); 3: 19-22.Иллариошкин С.Н. Наследственные моногенные заболевания нервной системы:
молекулярный анализ и клинико-генетические сопоставления. Автореф. дис. ...
докт. мед. наук. М., 1997.Иллариошкин C.H., Иванова-Смоленская. И.А. Молекулярные основы прогрессирую¬
щих мышечных дистрофий (обзор). Журн. неврол. и психиатр, им. С.С. Корсакова
1998; 10: 55-62.Иллариошкин С.Н., Иванова-Смоленская И.А., Маркова Е.Д. Новый механизм мутации
у человека: экспансия тринуклеотидных повторов (обзор). Генетика 1995; 31: 1478—
1489.Иллариошкин C.H., Иванова-Смоленская И.А., Маркова Е.Д. и др. Анализ экспансии
тринуклеотидных повторов как нового механизма мутации при хорее Гентингтона:
теоретические и прикладные аспекты. Генетика 1996 (а); 32: 103-109.Иллариошкин C.H., Овчинников И.В., Иванова-Смоленская И.А. и др. Молекулярно¬
генетический подход в изучении доминантных спиноцеребеллярных атаксий. Журн.
неврол. психиатр, им. С.С. Корсакова 1996 (б); 1: 37-41.Иллариошкин C.H., Иванова-Смоленская И.А., Лимборская С.А. и др. Пресимптомная
ДНК-диагностика спиноцеребеллярной атаксии 1-го типа. Генетика 1997; 33:
693-698.Иллариошкин C.H., Адарчева Л.С., Евграфов О.В. и др. Наследственная невропатия со
склонностью к параличам от сдавления. Неврол. журнал 1998; 4: 8-12.Иллариошкин C.H., Друзина Е.Б., Багиева Г.Х. и др. Болезнь Фридрейха: истинный
спектр клинических проявлений в свете возможностей ДНК-диагностики. Журн.
неврол. и психиатр, им. С.С. Корсакова 1999 (а); 8: 31-34.Иллариошкин C.H., Иванова-Смоленская И.А., Маркова Е.Д. и др. Наследственные
атаксии: от клинико-морфологического анализа к ДНК-тестированию. В кн.:
Проблемы нейрогенетики, ангионеврологии, нейротравматологии (под ред.
Е.М. Бурцева, Е.Д. Марковой). Иваново, 1999 (б): 48-59.Иллариошкин C.H., Ершова М.В., Клюшников С.А. и др. Спастическая атаксия как
редкий клинический вариант болезни Фридрейха. Неврол. журнал 2000; 5: 40-43.Калмыкова Л. Г. Наследственная гетерогенность болезней нервной системы. М.: Медицина,
1976.- 319 с.Карабанов A.B., Иванова-Смоленская И.А., Овчинников И.В. и др. Значение молекуляр¬
но-генетического анализа в диагностике гепатолентикулярной дегенерации. В сб.:
Актуальные вопросы неврологии, нейрохирургии и медицинской генетики. Уфа,
1998: 102-103.
Библиографический указатель 549Карлов В.А. Эпилепсия. М.: Медицина, 1990. - 336 с.Карунас A.C. Молекулярно-генетическое изучение болезни Вильсона-Коновалова в
Башкортостане. Автореф. дис. ... канд. мед. наук. М., 1998.Киселев Л.Л. Геном человека и биология XXI века. Вестник РАН 2000; 70: 412-424.Клюшников С. А. Диагностика хореи Гентингтона на доклинической стадии и при атипичных
вариантах заболевания (клинические и молекулярно-генетические сопоставления).
Автореф. дис. ... канд. мед. наук. М., 1998.Клюшников С.А., Иванова-Смоленская И.А., Никольская H.H. и др. Этические пробле¬
мы медико-генетического консультирования на примере хореи Генгтингтона.
Российский мед. журнал 2000; 2: 32-36.Козлова С.И., Демикова Н.С., Семанова E., Блинникова O.E. Наследственные синдро¬
мы и медико-генетическое консультирование. М.: Практика, 1996. - 416 с.Коновалов Н.В. Гепато-церебральная дистрофия. М.: Медгиз, I960 - 556 с.Краснопольская К.Д., Захарова Е.Ю. Современные достижения в диагностике и
профилактике митохондриальных болезней. Журн. неврол. и психиатр, им. С.С.
Корсакова 1998; 8: 49-56.Лекарь П.Г., Макарова В.А. Гепатоцеребральная дистрофия. Д. : Медицина 1984. - 208 с.Лильин Е.Т., Богомазов Е.А., Гофман-Кадошников П.Б. Генетика для врачей. М.:
Медицина, 1990,- 256 с.Липатова Н.Л., Крахмалева И.H., Шишкин С.С. и др. Сравнительное изучение особен¬
ностей строения гена кальпаина 3 (CANP-3) и гена дистрофина (DMD) в российских
семьях, отягощеных первичными миодистрофиями. В сб.: Второй (четвертый)
Российский съезд медицинских генетиков (Тез. докл.) Курск, 2000; Часть I:
115-116.Льюин Б. (Lewin В.) Гены (пер. с англ.) М.: Мир, 1987. - 544 с.Маркова Е.Д. Клиника и лечение торсионной дистонии. Клин. мед. 1975; 9: 89-92.Маркова Е.Д. Вопросы клинического полиморфизма при торсионной дистонии.
В кн.: Наследственные заболевания нервной системы. Саратов, 1983: 46-47.Маркова Е.Д. Особеннности клиники, патогенеза и лечения торсионной дистонии в детском
возрасте. Журн. невропатол. и психиатр, им. С.С. Корсакова 1989; 8: 32-35.Маркова Е.Д., Сломинский П.А., Иллариошкин С.Н. и др. Молекулярно-генетический
анализ торсионной дистонии в России. Генетика 2000; 36: 952-958.Миклина Н.И. Молекулярно-генетический анализ торсионной дистонии. Автореф. дис.
... канд. мед. наук. М., 1999.Никанорова М. Ю Абсансные эпилепсии детского возраста. В кн.: Диагностика и лечение
эпилепсий у детей (под ред. П.А. Темина, М.Ю. Никаноровой).
М.: Можайск-Терра, 1997: 254-263.Никанорова М.К)., Темин П.А., Кобринский Б.А. и др. Фебрильные судороги. В кн.:
Диагностика и лечение эпилепсий у детей (под ред. П.А. Темина, М.Ю. Никаноро¬
вой). М. Можайск-Терра, 1997: 140-164.Пугачев В.В.. Иллариошкин C.H., Прокунина Л.В. и др. ДНК-диагностика атаксии
Фридрейха с клинико-генетическим анализом. В кн.: Молекулярная генетика
наследственных болезней и медико-генетическое консультирование.
М.: МОНИКИ, 1996: 22-34.Пузырев В.П Состояние и перспективы геномных исследований в генетической
кардиологии. Вестник РАМН 2000; 7: 28-33.Пузырев В.П.. Степанов В.А. Патологическая анатомия генома человека. Новосибирск:
Паука, 1997 - 224 с.Темин П.А., Белозеров Ю.М., Страхова О.С., Никанорова М.Ю. Клинический и генети¬
ческий полиморфизм и вопросы современной терапии Х-сиепленных
прогрессир\ющнх мышечных дистрофии Дюшенна и Бекера. Вестник практич.
550 Библиографический указательневрол. 1997; 3: 85-101.Темин П.А., Белоусова Е.Д., Сухорукое B.C. Врожденные структурные миопатии и
дистрофии. В кн.: Наследственные болезни нервной системы (под ред. Ю.Е.
Вельтищева, П.А. Темина). М.: Медицина, 1998: 200-237.Тимербаева C.JI. Эффективность длительной патогенетической терапии при гепато-
церебральной дистрофии (клинико-катамнестическое исследование). Автореф. дис.
... канд. мед. наук. М., 1991.Ткачев Р.А., Маркова Е.Д., Бархатова В.П. и др. Результаты наблюдений при лечении L-
ДОПА некоторых наследственных экстрапирамидных заболеваний. Москва:
«Larodopa» Roche 1972: 141-148.Ткачев Р.А., Маркова Е.Д., Бархатова В.П. и др. Лечение препаратами L-ДОПА боль¬
ных торсионной дистонией и наследственной паплидарной дегенерацией Галлервор-
дена-Шпатца. В кн.: Дегенеративные заболевания нервной системы. Уфа, 1974:
196-200.Фогель Ф., Мотульски А. (Vogel F, Motulsky А.) Генетика человека (пер. с англ.). М.:
Мир, 1990. - в 3-х т.Шайнберг Г. (Scheinberg Н.) Болезнь Уилсона. В кн.: Внутренние болезни. М.: Медици¬
на, 1996; Т.8: 198-202.Шишкин С.С., Калинин В.Н. Медицинские аспекты биохимической и молекулярной
генетики. М.: ВИНИТИ, 1992. - 215 с.Шток В.Н., Левин О.С., Федорова Н.В. Экстрапирамидные расстройства.
М.: РМАПО, 1998. - 128 с.Abbas N., Lucking С.В., Ricard S. et а/. A wide variety of mutations in the parkin gene are
responsible for autosomal recessive parkinsonism in Europe. Hum. Mol. Genet.
1999; 8: 567-574.Abeliovich D., Lerer /., Pashut-Lavon I. et al. Negative expansion of the myotonic dystrophy
unstable sequence. Am. J. Hum. Genet. 1993; 52: 1175-1181.Abernathy C.R., Rasmussen S.A., Stalker H.J. et al. NF1 mutation analysis using a combined
heteroduplex/SSCP approach. Hum. Mutat. 1997; 9: 548-554.Ahmad F, Davis M.B., Waddy H.M. et al. Evidence for locus heterogeneity in autosomal
dominant torsion dystonia. Genomics 1993; 15: 9-12.Ahn A.H., Kunkel L.M. The structural and functional diversity of dystrophin. Nat. Genet.
1993;3:283-291.Albin R.L., Tag/e D.A. Genetics and molecular biology of Huntington’s disease. T.I.N.S.
1995;18:11-14.Allen М., Saudberg-Wollheim М., Sjogren K. et al. Association of succeptibility to multiple
sclerosis in Sweden with HLA class II DRB1 and DQB1 alleles. Hum. Immunol. 1994;
39: 41-48.Allikmets R., Raskind W.H., Hutchinson A. et al. Mutation of a putative mitochondrial iron
transporter gene (ABC7) in X-linked sideroblastic anemia and ataxia (XLSA/A). Hum.
Mol. Genet. 1999; 8: 743-749.AlmasyL., Bressman S.B., Kramer P.L. etal. Idiopathic torsion dystonia linked to chromosome
8 in two Mennonite families. Ann. Neurol. 1997; 42: 670-673.Almqvist E.W., Bloch М., Brinkman R. et al. A worldwide assessment of the frequency of
suicide, suicide attempts, or psychiatric hospitalization after predictive testing for
Huntington disease. Am. J. Hum. Genet. 1999; 64: 1293-1304.Alzheimer s Disease Collaborative Group. The structure of the presenilin 1 (SI82) gene and
identification of six novel mutations in early onset AD families. Nat. Genet. 1995; 11:
219-222.Amato A.A., Prior T.W., Barohn R.J. et al. Kennedy’s disease: a clinicopathologic correlation
with mutations in the androgen receptor gene. Neurology 1993; 43: 791-794.Andersen P., Nilsson P., Keranen M. et al. Phenotypic heterogeneity in motor neuron disease
Библиографический указатель 551patients with CuZn SOD mutations in Scandinavia. Brain 1997; 120: 1723-1737.Anderson L.V.B., Davison K., Moss J.A. et ah Characterization of monoclonal antibodies to
calpain 3 and protein expression in muscle from patients with limb-girdle muscular
dystrophy type 2A. Am. J. Pathol. 1998; 153: 1169-1179.Andrew S.E., Goldberg Y.P., Kremer B. et al. The relationship between trinucleotide (CAG)
repeat length and clinical features of Huntington’s disease. Nat. Genet. 1993; 4:398-403.Antonarakis S.E. Genome linkage scanning: systematic or intelligent? Nat. Genet. 1994;
8:211-212.Antonarakis S.E., and the Nomenclature Working Group. Recommendations for a nomenclature
system for human gene mutations. Hum. Mutat. 1998; 11: 1-3.Aoki M., Ogasawara M., Matsubara Y. et ai Familial ALS in Japan associated with H46R
mutation in Cu/Zn SOD gene: possible new subtype of familial ALS. J. Neurol. Sci.
1994; 126: 77-83.Ars E., Serra E., Garcia J. et al. Mutations affecting mRNA splicing are the most common
molecular defects in patients with neurofibromatosis type 1. Hum. Mol. Genet. 2000; 9:
237-247.Ashizawa T., Gasser T. Genetics of movement disorders. In: Jankovic J., Tolosa E. (eds.)
Parkinson’s disease and movement disorders, 3rd ed. Baltimore: Williams & Wilkins,
1998: 909-943.AthanassiadouA., Voutsinas G., Psiouri L. et al. Genetic analysis of families with Parkinson’s
disease that carry the Ala53Thr mutation in the gene encoding a-synuclein. Am. J.
Hum.Genet. 1999; 65: 555-558.Athma P., Rappaport R., Swift M. Molecular genotyping shows that ataxia-teleangiectasia
heterozygotes are predisposed to breast cancer. Cancer Genet. Cytogenet. 1996;
92: 130-134.Au K.-S., Hebert A.A., Roach E.S., Northrup H. Complete inactivation of the TSC2 gene
leads to formation of hamartomas. Am. J. Hum. Genet. 1999; 65: 1790-1795.Auburger G., RatzIaffT., Lunkes A. et al. A gene for autosomal dominant paroxysmal
choreoatetisis / spasticity (CSE) maps to the vicinity of a potassium channel gene
cluster on chromosome lp, probably within 2 cM between D1S443 and D1S197.
Genomics 1996; 31: 90-94.Augood S.J., Penney J.B., Friberg I.K. et al. Expression of the early-onset torsion dystonia
gene (DYT1) in human brain. Ann. Neurol. 1998; 43: 669-673.Babcock M., de Silva D., Oaks R. et ai Regulation of mitochondrial iron accumulation by
Yfhlp, a putative homolog of Frataxin. Science 1997; 276: 1709-1712.Baker H.F., Pouf ter M., Crow T.J. et al. Amino acid polymorphism in human prion protein
and age at death in inherited prion disease. Lancet 1991; 337: 1286.Baker M., Litvan /., Hou/den H. et ai Association of an extended haplotype in the tau gene
with progressive supranuclear palsy. Hum. Mol. Genet. 1999; 8: 711-715.Bakker E., Veenema H., den Dunnen J. T. et al. Germinal mosaicism increases the reccurrence
risk for ‘new’ Duchenne muscular dystrophy mutations. J. Med. Genet.
1989; 26:553-559.Baloh R.W., Winder A. Acetazolamide-responsive vestibulocerebellar syndrome: clinical and
oculographic features. Neurology 1991; 41: 429-433.Bandmann O., Nygaard T.G., Surtees R. et ai Dopa-responsive dystonia in British patients:
new mutations of the GTP-cyclohydrolase I gene and evidence for genetic heterogeneity.
Hum. Mol. Genet. 1996; 5: 403-406.Bandmann O., Va/ente E.M., Holmans P. et ai Dopa-responsive dystonia: a clinical and
molecular genetic study. Ann. Neurol. 1998; 44: 649-656.Barbeau A., Sadibelouis M, Sadibelouis A., Roy M. A clinical classification of hereditary
ataxias. Can. J. Neurol. Sci. 1984; 11: 501-505.Bashir R., Britton S., Strachan T. et al. A gene related to Caenorhabditis elegans
552 Библиографический указательspermatogenesis factor fer-1 is mutated in limb-girdle muscular dystrophy type 2B.
Nat. Genet 1998; 20: 37-42.Bashir R., Strachan T., Keers S. et al. A gene for autosomal recessive limb-girdle muscular
dystrophy maps to chromosome 2p. Hum. Mol. Genet. 1994; 3: 455-457.Battistini S., Stenirri S., Piatti M. et al. A new CACNA1A gene mutation in acetazolamide-
responsive familial hemiplegic migraine and ataxia. Neurology 1999; 53: 38-43.Baulac S., Huberfeld G., Gourfmkel-An I. et al. First genetic evidence of GABA(A) receptor
dysfunction in epilepsy: a mutation in the gamma2-subunit gene. Nat. Genet. 2001;
28: 46-48.Baxter D. W., Lai S. Essential tremor and dystonia syndrome. Adv. Neurol 1979; 24: 373-377.Beaudet A.L. Making genomic medicine a reality. Am. J. Hum. Genet. 1999; 64: 1-13.Beaudet A.L., Tsui L.-C. A suggested nomenclature for designating mutations. Hum. Mutat.
1993; 2: 245-248.Beck E., Daniel P.M. Neuropathology of transmissible spongiform encephalopathies. In:
Prusiner S.B., McKinley M.P. (eds.) Prions: Novel infectious pathogens causing scrapie
and Creutzfeldt-Jakob disease. San Diego: Academic Press, 1987: 331-385.Beckmann J.S., Bushby K.M.D. Advances in the molecular genetics of the limb girdle type of
autosomal recessive progressive muscular dystrophy. Cur. Opin. Neurol.
1996; 9: 389-393.Beggs A.H., Koenig M., Boyce F.M., Kunkel L.M. Detection of 98% of DMD/BMD gene
deletions by polymerase chain reaction. Hum. Genet. 1990; 86: 45-48.Behan W.M.H., MaiaM. StriimpeH’s familial spastic paraplegia: genetics and neuropathology.
J. Neurol. Neurosurg. Psychiatry 1974; 37: 8-20.Bejaoui K., Hirabayashi K., Hentati F. et al. Linkage of Miyoshi myopathy (distal autosomal
recessive muscular dystrophy) locus to chromosome 2p 12—14. Neurology 1995;
45: 768-772.Bejaoui K., McKenna-Yasek D., Hosier B.A. etal. Confirmation of linkage of type 1 hereditary
sensory neuropathy to human chromosome 9q22. Neurology 1999; 52: 510-515.Ben Hamida C., Doerflinger N., Belal S. et al. Localization of Friedreich’s ataxia phenotype
with selective vitamin E deficiency to chromosome 8q by homozygosity mapping. Nat.
Genet. 1993; 5: 195-200.Bennett L., Roach E., Bowcoclc A. A locus for paroxysmal kinesigenic dyskinesia maps to
human chromosome 16. Neurology 2000; 54: 125-130.Bennett R.L., Steinhaus K.A., Uhrich S.B. et al. Recommendations for standardized human
pedigree nomenclature. Am. J. Hum. Genet. 1995; 56: 745-752.Ben Othmane K., Hentati F, Lennon F. et al. Linkage of a locus (CMT4A) for autosomal
recessive Charcot-Marie-Tooth disease to chromosome 8q. Hum. Mol. Genet. 1993 (a);
2: 1625-1628.Ben Othmane K., Middleton L.T., Loprest L.J. et al. Localization of a gene (CMT2A) for
autosomal dominant Charcot-Marie-Tooth disease type 2 to chromosome 1 p and evidence
for genetic heterogeneity. Genomics 1993 (b); 17: 370-375.Ben Othmane K., Johnson E., Menold M. et al. Identification of a new locus for autosomal
recessive Charcot-Marie-Tooth disease with focally folded myelin on chromosome 1 lpl 5.
Genomics 1999; 62: 344-349.Benson K.F, Horwitz M., Wolff J. et al. CAG repeat expansion in autosomal dominant
familial spastic paraparesis: novel expansion in a subset of patients. Hum. Mol. Genet.
1998;7:1779-1786.Bentivoglio A.R., Del Grosso N.. Albanese A. et al. A large Italian family affected by
idiopathic torsion dystonia with complete penetrance not linked to DYT1. J. Neurol.
Neurosurg. Psychiatry 1997; 62: 357-360.Benton C.S., de Silva R., Rutledge S.L. et al. Molecular and clinical studies in SCA-7 define
a broad clinical spectrum and the infantile phenotype. Neurology 1998; 51: 1081-1086,
Библиографический указатель 553Berkovic S.F., Cochius J., Andermann E., Andermann F. Progressive myoclonus epilepsies:
clinical and genetic aspects. Epilepsia 1993; 34 (Suppl. 3): S19-30.Bergeron C., Davis A., Lang A.E. Corticobasal degeneration and progressive supranuclear
palsy presenting with cognitive decline. Brain Pathol. 1998; 8: 355-365.Bergoffen J., Scherer S.S., Wang S. et ai. Connexin mutations in X-linked Charcot-Marie-
Tooth disease. Science 1993; 262: 2039-2042.Bertini E., des Portes V., Zanni G. et al. X-linked congenital ataxia: a clinical and genetic
study. Am. J. Med. Genet. 2000; 92: 53-56.Beutler E. The designation of mutations. Am. J Hum. Genet. 1993; 53: 783-785.Bhatia K.P. The paroxysmal dyskinesias. J. Neurol. 1999; 246: 149-155.Bianchi A.B., Hara T, Ramesh V et al. Mutations in transcript isoforms of the
neurofibromatosis 2 gene in multiple human tumour types. Nat. Genet. 1994; 6: 185-192.Bianchi A.B., Mitsunaga S.-I., Cheng J.Q. et al. High frequency of inactivating mutations in
the neurofibromatosis type 2 gene (NF2) in primary malignant mesotheliomas. Proc.
Natl. Acad. Sci. USA 1995; 92: 10854-10858.Bidichandani S.I., Ashizawa T., Patel P.I. Atypical Friedreich ataxia caused by compound
heterozygosyty for a novel missense mutation and the GAA triplet-repeat expansion.
Am. J. Hum. Genet. 1997; 60: 1251-1256.Biernat W., Tohma Y., Yonekcrwa Y. et ai Alterations of cell cycle regulatory genes in primary
(de novo) and secondary glioblastomas. Acta Neuropathol. 1997; 94: 303-309.Biervert C., Schroeder B.C., Kubisch C. et ai A potassium channel mutation in neonatal
human epilepsy. Science 1998; 279: 403-406.Bione S., Maestrini E., Rivella S. et ai Identification of a novel X-linked gene responsible for
Emery-Dreifuss muscular dystrophy. Nat. Genet. 1994; 8: 323-327.Bird T.D. Are linkage studies boring? Nat. Genet. 1993; 4: 213-214.Bird T.D. Genotype, phenotypes, and frontotemporal dementia. Neurology 1998;
50: 1526-1527.Bishop J.M. Molecular themes in oncogenesis. Cell 1991; 64: 235-248.Blacker D., Tanzi R.E. The genetics of Alzheimer disease: Current status and future prospects.
Arch. Neurol. 1998; 55: 294-296.Blau N., Jchinose H., Nagatsu T. et ai A missense mutation in a patient with guanosine
triphosphate cyclohydrolase I deficiency missed in the newborn screening program. J.
Pediatr. 1995; 126: 401-405.Blömer V., Naldini L., Verma J.M. et ai Applications of gene therapy to the CNS. Hum. Mol.
Genet. 1996; 5: 1397-1404.Blumen S.C., Brais B., Korczyn A.D. et ai Homozygotes for oculopharyngeal muscular
dystrophy have a severe form of the disease. Ann. Neurol. 1999; 46: 115-118.Blumenfeld A., Slaugenhaupt S.A., Axelrod F.B. et ai Localization of the gene for familial
dysautonomia on chromosome 9 and definition of DNA markers for genetic diagnosis.
Nat. Genet. 1993; 4: 160-164.Bobowick A.R., Brody J.A. Epidemiology of neurodegenerative system disorders. In: Vinken
P.J., Bruyn G.W. (eds.) Handbook of clinical neurology, vol. 21. Amsterdam: Elsevier,
1975: 3-42.Bodmer J.C., March S.G.E., Albert E. Nomenclature for factors of the HLA system, 1989.
Immunol. Today 1990; 11: 3-10.Boehnke M. Estimating the power of a proposed linkage study: a practical computer simulation
approach. Am. J. Hum. Genet. 1986; 39: 513-527.Boerkoel C.F., Takashima H., Stankiewicz P. et ai Periaxin mutations cause recessive
Dejerine-Sottas neuropathy. Am. J. Hum. Genet. 2001; 68: 325-333.Bogler O., Huang H.J., Kleihues P., Cavenee W.K. The p53 gene and its role in human brain
tumors. Glia 1995; 15: 308-327.
554 Библиографический указательBolino A., Muglia М., Conforti F.L. et al. Charcot-Marie-Tooth type 4B is caused by
mutations in the gene encoding myotubularin-related protein-2. Nat. Genet. 2000;
25: 17-19.Bolino A., Brancolini V, Bono F. et al. Localization of a gene responsible for autosomal
recessive demyelinating neuropathy with focally folded myelin sheaths to chromosome11 q23 by homozygosity mapping and haplotype sharing. Hum. Mol. Genet. 1996;
5: 1051-1054.Bonne G., di Barletta M.R., Varnous S. et ai Mutations in the gene encoding lamin A/С cause
autosomal dominant Emery-Dreifuss muscular dystrophy. Nat. Genet. 1999; 21:285-288.Botstein D., H'bite R.L., Skolnick M.H., Davis R.W. Construction of a genetic linkage map in
man using restriction fragment length polymorphisms. Am. J. Hum. Genet. 1980; 32:
314-331.Bouhouche A., Benomar A., BiroukN. etal. A locus for an axonal form of autosomal recessive
Charcot-Marie-Tooth disease maps to chromosome 1 q21.2—q21.3. Am. J. Hum. Genet.
1999; 65: 722-727.Boustany R.-M.N., Kolodny E.H. Neurological progress: The neuronal ceroid lipofuscinoses
(a review). Rev. Neurol. 1989; 145: 105-110.Bowel J.H., Maraganore D.M., McDonnell S.K., Rocca W.A. Incidence of progressive
supranuclear palsy and multiple system atrophy in Olmsted County, Minnesota, 1976
to 1990. Neurology 1997; 49: 1284-1288.Brais B., Bouchard J.-R, Xie Y.-G. et al. Short GCG expansions in the PABP2 gene cause
oculopharyngeal muscular dystrophy. Nat. Genet. 1998; 18: 164-167.Breitner J.C.S., Gatz М., Bergem A.l.M. et al. Use of twin cohorts for the research in
Alzheimer’s disease. Neurology 1993; 43: 261-267.Brassat D., Camuzat A., Vidailhet M. et at. Frequency of the DYT1 mutation in primary
torsion dystonia without family history. Arch. Neurol. 2000; 57: 333-335.Bressman S.B., de Leon D., Kramer P.L. et al. Dystonia in Ashkenazi Jews: Clinical
characterization of a founder mutation. Ann. Neurol. 1994; 36: 771-777.Bressman S.B., Sabatti C., Raymond D. et al. The DYT1 phenotype and guidelines for
diagnostic testing. Neurology 2000; 54: 1746-1752.Bressman S.B., Warner T.T., Almasy L. et al. Exclusion of the DYT1 locus in familial
torticollis. Ann. Neurol. 1996; 40: 681-684.Brice A. Unstable mutations and neurodegenerative disorders. J. Neurol. 1998; 245: 505-510.Brin M.F., Koller W. Epidemiology and genetics of essential tremor. Mov. Disord. 1998; 13
(Suppl. 3): 55-63.Brinkman R.R., Mezei M.M., Theilmann J. et al. The likelihood of being affected with
Huntington disease by a particular age, for a specific CAG size. Am. J. Hum. Genet.
1997; 60: 1202-1210.Britton J.W., Uitti R.J., Ahlskog J.E. et al. Hereditary late-onset chorea without significant
dementia: genetic evidence for substantial phenotypic variation in Huntington’s disease.
Neurology 1995; 45: 443-447.Brockington М., Sewry C.A., Herrmann R. et al. Assignment of a form of congenital muscular
dystrophy with secondary merosin deficiency to chromosome lq42. Am. J. Hum.
Genet. 2000; 66: 428-435.BroeksA., UrbanusJ.H.M., FlooreA.N. et al. ATM-heterozygous germline mutations contribute
to breast cancer-susceptibility. Am. J. Hum. Genet. 2000; 66: 494-500.Brook J.D., McCurrach M.E., Harley H.G. et al. Molecular basis of myotonic dystrophy:
expansion of a trinucleotide (CTG) repeat at the 3’ end of a transcript encoding a protein
kinase family member. Cell 1992; 68: 799-808.Brown P. The phenotypic expression of different mutations in transmissible human spongiform
encephalopathy. Rev. Neurol. 1992; 148: 317-327.Brown J., Ashworth A., Gydesen S. etal. Familial nonspecific dementia maps to chromosome
Библиографический указатель 5553. Hum. Mol. Genet. 1995; 4: 1625-1628.Brown K.D., Barlow C., Wynshaw-Boris A. Multiple ATM-dependent pathways: an explanation
for pleiotropy. Am. J. Hum. Genet. 1999; 64: 46-50.Brown M.D., Voljavec A., Lott M.T. et al. Mitochondrial DNA complex I and III mutations
associated with Leber’s hereditary optic neuropathy. Genetics 1992; 130: 163-173.Brown P.O., Botstein D. Exploring the new world of the genome with DNA microarrays. Nat.
Genet. 1999; 21 (Suppl.): 33-37.Browne D.L., Gancher S.T., NuttJ.G. etal. Episodic ataxia-myokymia syndrome is associated
with point mutations in the human potassium channel gene, KCNA1. Nat. Genet. 1994;
8: 136-140.Bruyn G.W. Hereditary congenital cerebellar atrophy. In: de Jong J.M.B.V. (ed.) Handbook of
clinical neurology, vol.60: Hereditary neuropathies and spinocerebellar atrophies.
Amsterdam: Elsevier, 1991: P.285-297.Bull P.C., Thomas G.R. Rommens J.I. et al. The Wilson disease gene is a putative copper
transporting P-type ATPase similar to the Menkes gene. Nat. Genet. 1993; 5: 327-337.Bulman D.E. Phenotype variation and newcomers in ion channel disorders. Hum. Mol. Genet.
1997; 6: 1679-1685.Burke J.R., Enghild J.J., Martin M.E. et al. Huntingtin and DRPLA proteins selectively
interact with the enzyme GAPDH. Nat. Med. 1996; 2: 347-350.Burt T., Currier B., Kilburn C. et al. Machado-Joseph disease in east Arnhem Land, Australia:
chromosome 14q32.1 expanded repeat confirmed in four families. Neurology 1996; 46:
1118-1122.Bushby K.M.D. Diagnostic criteria for the limb-girdle muscular dystrophies: report of the
ENMC Consortium on Limb-Girdle Dystrophies. Neuromusc. Disord. 1995; 5: 71-74.Bushby K.M.D. The limb-girdle muscular dystrophies - multiple genes, multiple mechanisms.
Hum. Mol. Genet. 1999; 8: 1875-1882.Bushby K.M.D. Making sense of the limb-girdle muscular dystrophies. Brain 1999;
122: 1403-1420.Bussaglia E., Clermont O., Tizzano E. et al. A frame-shift deletion in the survival motor
neuron gene in Spanish spinal muscular atrophy patients. Nat. Genet. 1995; 11: 335-337.Buxton J., She/bourne P., Davies J. et al. Detection of an unstable fragment of DNA specific
to individuals with myotonic dystrophy. Nature 1992; 355: 547-548.CaiX., Golde T.E., Younkin S.G. Release of excess amyloid-ß protein from a mutant amyloid-
ß protein precursor. Science 1993; 259: 514-516.Cambi F., Tang X.-M., Cordray M.S. et al. Refined genetic mapping and proteolipid protein
mutation analysis in X-linled pure hereditary spastic paraplegia. Neurology 1996; 46:
1112-1117.Campbell K.P. Three muscular dystrophies: loss of cytoskeleton-extracellular matrix linkage.
Cell 1995; 80: 675-679.Campbell L., Potter A., Ignatius J. et al. Genomic variation and gene conversion in spinal
muscular atrophy: implications for disease process and clinical phenotype. Am. J. Hum.
Genet. 1997; 61: 40-50.Campion D., Dumanchin C., Hannequin D. etal. Early-onset autosomal dominant Alzheimer
disease: prevalence, genetic heterogeneity, and mutation spectrum. Am. J. Hum. Genet.
1999; 65: 664-670.Campos Y, Martin M.A., Rubio J.C. et al. Bilateral striatal necrosis and MELAS associated
with a new T3308C mutation in the mitochondrial ND1 gene. Biochem. Biophys. Res.
Commun. 1997; 238: 323-325.Campuzano V, Montermini L., Lutz Y. et al. Frataxin is reduced in Friedreich ataxia patients
and is associated with mitochondrial membranes. Hum. Mol. Genet. 1997; 6:1771-1780.Campuzano V, Montermini L., Molto M.D. et al. Friedreich’s ataxia: autosomal recessive
556Библиографический указательdisease caused by an intronic GAA triplet repeat expansion. Science 1996; 271: 1423-1427.Cantor C.R., Smith C.L. Consequences of mapping and sequencing the human genome for
neurologic diseases. In: Rosenberg R.N., Prusiner S.B., DiMauro S. et al. (eds.) The
molecular and genetic basis of neurological disease. Boston: Butterworth-Heinemann,
1993: 977-987.Caraballo R., Pave/c S., Lemainque A. et al. Linkage of benign familial infantile convulsions
to chromosome 16p 12— 13q suggests allelism to the infantile convulsions and
choreoathetosis syndrome. Am. J. Hum. Genet. 2001; 68: 788-794. ,Carbonara C., Longa L., Grosso E. et a!. Apparent preferential loss of heterozygosity at
TSC2 over TSC1 chromosomal region in tuberous sclerosis hamartomas. Genes
Chromosomes Cancer 1996; 15: 18-25.Carrera P., Piatti M., Stenirri S. et a!. Genetic heterogeneity in Italian families with familial
hemiplegic migraine. Neurology 1999; 53: 26-33.Carrozzo R., Hirano M, Fromenty B. et al. Multiple mtDNA deletions features in autosomal
dominant and recessive diseases suggest distinct pathogeneses. Neurology 1998;
50: 99-106.Casari G., De Fusco M., Ciarmatori S. et al. Spastic paraplegia and OXPHOS impairment
caused by mutations in paraplegin, a nuclear-encoded mitochondrial metalloprotease.
Cell 1998; 93: 973-983.Cavalier L., Ouahchi K., Kcryden H.J. et al. Ataxia with isolated vitamin E deficiency:
heterogeneity of mutations and phenotypic variability in a large number of families. Am.
J. Hum. Genet. 1998; 62: 301-310.Cavenee W.K. Accumulation of genetic defects during astrocytoma progression. Cancer 1992;
70: 1788-1793.Chamberlain J.C., Gibbs R.A., RanierJ.E. et al. Deletion screening of the Duchenne muscular
dystrophy locus via multiplex DNA amplification. Nucl. Acids Res. 1988;
16: 11141-11156.Chamberlain S., Shaw J., Rowland A. et al. Mapping of mutation causing Friedreich’s ataxia
to human chromosome 9. Nature 1988; 334: 248-250.Chambers C.B., Lee J.M., Troncoso J.C. et al. Overexpression of four-repeat tau mRNA
isoforms in progressive supranuclear palsy but not in Alzheimer’s disease. Ann. Neurol.
1999; 46: 325-332.Chance P.F., Abbas N.. Lensch M. W. et al. Two autosomal dominant neuropathies result from
reciprocal DNA duplication/deletion of a region on chromosome 17. Hum. Mol. Genet.
1994; 3: 223-228.Chance P.F., Alderson M.K., Leppig K.A. et al. DNA deletion associated with hereditary
neuropathy with liability to pressure palsies. Cell 1993; 72: 143-151.Chance P.F., Fischbeclc K.H. Molecular genetics of Charcot-Marie-Tooth disease and related
neuropathies. Hum. Mol. Genet. 1994; 3: 1503-1507.Chance P.F., Rabin B.A., Ryan S.G. et al. Linkage of the gene for an autosomal dominant form
of juvenile amyotrophic lateral sclerosis to chromosome 9q34. Am. J. Hum. Genet.
1998; 62: 633-640.Charlier C., Singh N.A., Ryan S.G. et al. A pore mutation in a novel KQT-like potassium
channel gene in an idiopathic epilepsy family. Nat. Genet. 1998; 18: 53-55.Chen F., Kishida T., Yao M. et al. Germline mutations in the von Hippel-Lindau disease
tumor suppressor gene: correlations with phenotype. Hum. Mutat. 1995; 5: 66-75.Chinneiy P.F., Howell N., Andrews R.M., Turnbull D.M. Clinical mitochondrial genetics. J.
Med. Genet. 1999 (a); 36: 425-436.Chinnery P.F., Howell N.. Andrews R.M., Turnbull D.M. Mitochondrial DNA analysis:
polymorphism and pathogenecity. J. Med. Genet. 1999 (6); 36: 505-510.Chomyn A. the myoclonic epilepsy and ragged-red fiber mutation provides new insights into
human mitochondrial function and genetics. Am. J. Hum. Genet. 1998; 62: 745-751.
Библиографический указатель557Chow T.W., Miller B.L., Hayashi V.N., GeschwindD.H. Inheritance of frontotemporal dementia.
Arch. Neurol. 1999; 56: 817-822.Chowrimootoo G.F.E., Ahmed H.A., Seymour C.A. New insights into the pathogenesis of
copper toxicosis in Wilson’s disease: evidence for copper incorporation and defective
canalicular transport of caeruloplasmin. Biochem. J. 1996; 315: 851-855.Christodoulou K., Kyriakides T., Hristova A.H. et al. Mapping of a distal form of spinal
muscular atrophy with upper limb predominance to chromosome 7p. Hum. Mol. Genet.
1995;4:1629-1632.Christodoulou K., Zamba E., Tsingis M. et al. A novel form of distal hereditary motor
neuronopathy maps to chromosome 9p21.1—pi2. Ann. Neurol. 2000; 48: 877-884.Chung M.-Y., Ranum L.P.W., Duvick L.A. et al. Evidence for a mechanism predisposing to
intergenerational CAG repeat instability in spinocerebellar ataxia type 1. Nat. Genet.
1993; 5: 254-258.Citron M., Westaway D., Xia W. et al. Mutant presenilins of Alzheimer’s disease increase
production of 42-residue amyloid p-protein in both transfected cells and transgenic
mice. Nat. Med. 1997; 3: 67-72.Clark L.N., Poorkaj P., Wszo/ek Z. et al. Pathogenic implications of mutations in tau gene in
pallido-ponto-nigral degeneration and related neurodegenerative disorders linked to
chromosome 17. Proc. Natl. Acad. Sci. USA 1998; 95: 13103-13107.Clemens P.R., Fenwick R.G., Chamberlain J.S. et al. Carrier detection and prenatal diagnosis
in Duchenne and Becker muscular dystrophy families, using dinucleotide repeat
polymorphisms. Am. J. Hum. Genet. 1991; 49: 951-960.dementi M., Barbujani G., TurollaL., Tenconi R. Neurofibromatosis-1: a maximum likelihood
estimation of mutation rate. Hum. Genet. 1990; 84: 116-118.Cobben J.M., van der Steege G., Grootscho/ten P. et al. Deletions of the survival motor
neuron gene in unaffected siblings of patients with spinal muscular atrophy. Am. J.
Hum. Genet. 1995; 57: 805-808.Cohen J., Scott R., Schimmel T. et al. Birth of infant after transfer of anucleate donor oocyte
cytoplasm into recipient eggs. Lancet 1997; 350: 186-187.Collinge J. Human prion disease and bovine spongiform encephalopathy (BSE). Hum. Mol.
Genet. 1997; 6: 1699-1705.Collinge J., Rossor M. A new variant of prion disease. Lancet 1996; 347: 916-917.Collinge J., Brown J., Hardy J. et al. Inherited prion disease with 144 base pair gene
insertion. 2. Clinical and pathological features. Brain 1992; 115: 687-710.Collinge J., Palmer M.S., Diyden A.J. Genetic predisposition to iatrogenic Creutzfeldt-Jakob
disease. Lancet 1991; 337: 1441-1442.Collins F.S. Positional cloning: let’s not call it reverse anymore. Nat. Genet. 1992; 1: 3-6.Collins F.S. Positional cloning moves from perditional to traditional. Nat. Genet. 1995;
9: 347-350.Concannon P., Gatti R.A. Diversity of ATM mutations detected in patients with ataxia-
teleangiectasia. Hum. Mutat. 1997; 10: 100-107.Conner K.E., Rosenberg R.N. The hereditary ataxias. In: Rosenberg R.N.; Prusiner S.B.,
DiMauro S. et al. (eds.) The molecular and genetic basis of neurological disease.
Boston: Butterworth-Heinemann, 1993: 697-736.Cook D.G., Sung J.C., Go/de T.E. et al. Expression and analysis of presenilin 1 in a human
neuronal system: localization in cell bodies and dendrites. Proc. Natl. Acad. Sci. USA
1996; 93: 9223-9228.Coppola G., De Michele G, Cavalcanti F et al. Why do some Friedreich’s ataxia patients
retain tendon reflexes? A clinical, neurophysiological and molecular study. J. Neurol.
1999; 246: 353-357.Cooper E.C., Aldape K.D., Abosch A. et al. Colocalization and coassembly of two human
brain M-type potassium channel subunits that are mutated in epilepsy. Proc. Natl. Acad.
558 Библиографический указательSoi. USA 2000; 97: 4914-4919,Corder E.H., Saunders A.M., Risch N.J. et al. Protective effect of apolipoprotein E type 2
allele for late onset Alzheimer’s disease. Nat. Genet. 1994; 7: 180-184.Corder E.H., Saunders A.M., Strittmatter W.J. et al. Gene dose of apolipoprotein E type 4
allele and the risk of Alzheimer’s disease in late onset families. Science
1993;261: 921-923.Cossée M., Campuzano V, Koutnikova H. et at. Frataxin fracas. Nat. Genet 1997; 15: 337-338.Cossée M., Diirr A., Schmitt M. et a!. Friedreich’s ataxia: point mutations and clinical
presentation of compound heterozygotes. Ann. Neurol. 1999; 45: 200-206.Cox D.W. Genes of the copper pathway. Am. J. Hum. Genet. 1995; 56: 828-834.Crossey P.A., Foster K, Richards F.M. et ai Molecular genetic investigations of the mechanism
of tumourigenesis in von Hippel-Lindau disease: analysis of allele loss in VHL tumours.
Hum. Genet. 1994 (a); 93: 53-58.Crossey P. A., Richards F.M., Foster K. et al. Identification of intragenic mutations in the von
Hippel-Lindau disease tumour suppressor gene and correlation with disease phenotype.
Hum. Mol. Genet. 1994 (6); 3: 1303-1308.Cruts M., Van Duijn C.M., Backhovens H. et ai Estimation of the genetic contribution of
presenil in-1 and -2 mutations in a population-based study of presenile Alzheimer disease.
Hum. Mol. Genet. 1998; 7: 43-51.Cudkowicz M.E., McKenna-Yasek D., Sapp P. et ai Epidemiology of mutations in superoxide
dismutase in ALS. Ann. Neurol. 1997; 39: 128-131.Cudrey C, Chevil/ard C., Le Paslier D. et ai Assignment of microsatellite sequences to the
region duplicated in CMT1A ( 17p 12): a useful tool for diagnosis. J. Med. Genet. 1995;
32: 231-233.Czlonkowska A., Rodo M., GaidaJ. et ai Very high frequency of the Hisl069Gln mutation in
Polish Wilson disease patients. J. Neurol. 1997; 244: 591.Damji K.F.. Allingham R.R.. Pollock S.C. et ai Periodic vestibulocerebellar ataxia, an
autosomal dominant ataxia with defective smooth pursuit, is genetically distinct from
other autosomal dominant ataxias. Arch. Neurol. 1996; 53: 338-344.Darras B.T., Koenig M, Kunkel L.M., Francke U. Direct method for prenatal diagnosis and
carrier detection in Duchenne/Becker muscular dystrophy using the entire dystrophin
cDNA. Am. J. Med. Genet. 1988; 29: 713-726.Dcn>enport W.J., Siegel A.M., Dichgans J. et al. CCM1 gene in families segregating cerebral
cavernous malformations. Neurology. 2001; 56: 540-543.David G., Abbas N.. Stevanin G. et ai Cloning of the SCA7 gene reveals a highly unstable
CAG repeat expansion. Nat. Genet. 1997; 17: 65-70.Davis M.B., Rosenberg R.N., Harding A.E. Molecular genetics and neurologic disease: an
introduction. In: Rosenberg R.N., Prusiner S.B., DiMauro S. et al. (eds.) The molecular
and genetic basis of neurological disease. Boston: Butterworth-Heinemann, 1993: 3-20.Davis R.L., Shrimpton A.E., Holohan P.D. et ai Familial dementia caused by polymerization
of mutant neuroserpin. Nature 1999; 401: 376-379.DeBella À., Szudek J., Friedman J. M. Use of the National Institutes of Health criteria for
diagnosis of neurofibromatosis 1 in children. Pediatrics 2000; 105: 608-614.De Fusco M., Becchetti A., Patrignati A. et ai The nicotinic receptor (32 subunit is mutant
in nocturnal frontal lobe epilepsy. Nat. Genet. 2000; 26: 275-276.Deidda G. Cacurri S., Piazzo N., Felicetti L. Direct detection of 4q35 rearrangements
implicated in facioscapulohumeral muscular dystrophy (FSHD). J. Med. Genet. 1996;
33: 361-365.De Jonghe P., Timmerman V, van Broeckhoven C. 2nd Workshop of the European CMT
Consortium: 53rd ENMC International Workshop on classification and diagnostic
guidelines for Charcot-Marie-Tooth type 2 (CMT2-HMSN II) and distal hereditary
motor neuropathy (distal HMN-spinal CMT). Neuromusc. Disord. 1998; 8: 426-431
Библиографический указатель 559De/ague V., Bareil C., Tuffeiy S. et al. Mapping of a new locus for autosomal recessive
demyelinating Charcot-Marie-Tooth disease to 19q 13.1-13.3 in a large consanguineous
Lebanese family: exclusion of MAG as a candidate gene. Am. J. Hum. Genet. 2000; 67:
236-243.Delatycki M.B., Camakaris J., Brooks H. et al. Direct evidence that mitochondrial iron
accumulation occurs in Friedreich ataxia. Ann. Neurol. 1999; 45: 673-675.De Lauremi V, Rogers C.G., Hamrock D.J. et al. Sjögren-Larsson syndrome is caused by
mutations in the fatty aldehyde dehydrogenase gene. Nat. Genet. 1996; 12: 52-57.Demirkiran M., JankovicJ. Paroxysmal dyskinesias: clinical features and classification. Ann.
Neurol. 1995; 38: 571-579.De Michele G., De Fusco M., Cavalcanti F. et al. A new locus for autosomal recessive
hereditary spastic paraplegia maps to chromosome 16q24.3. Am. J. Hum. Genet. 1998;
63: 135-139.Denier C., DucrosA., Vahedi K. et al. High prevalence of CACNA1A truncations and broader
clinical spectrum in episodic ataxia type 2. Neurology 1999; 52: 1816-1821.De Rijk M.C., Breteler M.M., Graveland G.A. et al. Prevalence of Parkinson’s disease in the
elderly: the Rotterdam study. Neurology 1995; 45: 2143-2146.De Waard M., Campbell K.P. Subunit regulation of the neuronal aIA Ca2+ channel expressed
in Xenopus oocytes. J. Physiol. 1995; 485: 619-634.Dib C., Faure S., Fizames C. et al. A comprehensive genetic map of the human genome based
on 5,264 microsatellites. Nature 1996; 380: P. 152-154.Di Barletta M.R., Ricci E., Galuzzi G. et al. Different mutations in the LMNA gene cause
autosomal dominant and autosomal recessive Emery-Dreifuss muscular dystrophy. Am.
J. Hum. Genet. 2000; 66: 1407-1412.DiDonato S. Can we avoid AVED? Nat. Genet. 1995; 9: 106-107.DiMauroS. Mitochondrial encephalomyopathies. In: Rosenberg R.N., Prusiner S.B., DiMauroS. et al. (eds.) The molecular and genetic basis of neurological disease. Boston:
Butterworth-Heinemann, 1993: 665-694.Dobyns W., Ozelius L., Kramer P.L. et al. Rapid-onset dystonia-parkinsonism. Neurology
1993; 43: 2596-2602.Doriguzzi C., Palmucci L., Mongini T. et al. Systematic use of dystrophin testing in muscle
biopsies: results in 201 cases. Eur. J. Clin. Invest. 1997; 27: 352-358.Driss A., Amouri R., Ben Hamida C. et al. A new locus for autosomal recessive limb-girdle
muscular dystrophy in a large consanguineous Tunisian family maps to chromosome
19ql3.3. Neuromusc. Disord. 2000; 10: 240-246.Duan D.R., Pause A., Burgess W.H. et al. Inhibition of transcription elongation by the VHL
tumor suppressor protein. Science 1995; 269: 1402-1406.Duggan D.J., Gorospe J.R., Fanin M. et al. Mutations in the sarcoglycan genes in patients
with myopathy. New Engl. J. Med. 1997; 336: 618-624.Durner M., Zhou G., Fu D. et al. Evidence for linkage of adolescent-onset idiopathic generalized
epilepsies to chromosome 8 - and genetic geterogeneity. Am. J. Hum. Genet. 1999; 64:
1411-1419.Dürr A., Brice A., Serdaru M. et al. The phenotype of «pure» autosomal dominant spastic
paraplegia. Neurology 1994; 44: 1274-1277.Dürr A., Cossee M., Agid Y et al. Clinical and genetic abnormalities in patients with
Friedreich’s ataxia. New Engl. J. Med. 1996; 335: P. 1169-1175.Dürr A., Hahn-Barma V., Brice A. et al. Homozygosity in Huntington’s disease. J. Med.
Genet. 1999; 36: 172-173.Duvoisin R.C. Role of genetics in the cause of Parkinson’s disease. Mov. Disord. 1998;
13 (Suppl. 1): 7-12.Duyao M., Ambrose C., Myers R. et al. Trinucleotide repeat length instability and age of onset
560 Библиографический указательin Huntington’s disease. Nat. Genet. 1993; 4: 387-392.Dvorakova D., Fajkusova L. Direct analysis of R408W mutation in phenylalanine hydroxylase
gene by allele-specific PCR amplification. Hum. Mol. Genet. 1993; 2: 323.DworkA.J., BalmacedaC., Fazzini E.A. etal. Dominantly inherited, early-onset parkinsonism:
neuropathology of a new form. Neurology 1993; 43: 69-74.Dyck P.J., Chance P., Lebo R., Carney J.A. Hereditary motor and sensory neuropathies. In:
Dyck P.J., Thomas P.K.., Griffin J.W. et al. (eds.) Peripheral neuropathy, 3rd ed.
Philadelphia: W.B. Saunders, 1993: 1094-1136.Dvment D.A., Sadnovich A.D., Ebers G.C. Genetics of multiple sclerosis. Hum. Mol. Genet.
1997;6: 1693-1698.Echenne B., Humbertclaude V, Rivier F et al. Benign infantile epilepsy with autosomal
dominant inheritance. Brain Dev. 1994; 16: 108-111.Eisenbarth /., Beyer K., Krone W., Assum G. Toward a survey of somatic mutation of the NF1
gene in benign neurofibromas of patients with neurofibromatosis type 1. Am. J. Hum.
Genet. 2000; 66: 393-401.Elbaz A.. Grigoletto F, Balderschi M. et al. Familial aggregation of Parkinson’s disease: A
population-based case-control study in Europe. Neurology 1999; 52: 1876-1882.E/dridge R. The torsion dystonias: literature review and genetic and clinical studies. Neurology
1970; 20: 1-78.Elston R.C. Linkage and association. Genet. Epidemiol. 1998; 15: 565-576.Emery A.E.ll. Population frequencies of inherited neuromuscular diseases - a world survey.
Neuromusc. Disord. 1991; 1: 19-29.Emeiy A.E.H. Duchenne muscular dystrophy, 2nd ed. Oxford: Oxford University Press, 1993.Emery A.E.ll. The muscular dystrophies. B. M. J. 1998; 317: 991-995.Emslie F.V., Rees M., Williamson M.P. et al. Genetic mapping of a major susceptibility locus
for juvenile myoclonic epilepsy on chromosome 15q. Hum. Mol. Genet. 1997; 6:
1329-1334.Engel A.G. Myofibrillar myopathy. Ann. Neurol. 1999; 46: 681-683.Engel A. G., Ohno K., Mi/one M. et al. New mutations in acetylcholine receptor subunit genes
reveal heterogeneity in the slow-channel congenital myasthenic syndrome. Hum. Mol.
Genet. 1996; 5: 1217-1227.Escayg A., De Waard M., Lee D.D. et al. Coding and noncoding variation of the human
calcium-channel (34-subunit gene CACNB4 in patients with idiopathic generalized epilepsy
and episodic ataxia. Am. J. Hum. Genet. 2000 (a); 66: 1531-1539.Escayg A., MacDonaldB.T., Meisler M.H. et al. Mutations of SCN1 A, encoding a neuronal
sodium channel, in two families with GEFS+2. Nat. Genet. 2000 (6); 24: 343-345.European Chromosome 16 Tuberous Sclerosis Consortium. Identification and characterization
of the tuberous sclerosis gene on chromosome 16. Cell 1993; 75: 1305-1315.Evans G.A. The Human Genome Project: application in the diagnosis and treatment of
neurologic disease. Arch. Neurol. 1998; 55: 1287-1290.Evans D.G.R., Trueman L., Wallace A. et al. Genotype/phenotype correlations in type 2
neurofibromatosis (NF2): evidence for more severe disease associated with truncating
mutations. J. Med. Genet. 1998; 35: 450-455.Fahn S. Concept and classification of dystonia. Adv. Neurol. 1988; 50: 1-8.Fahsold R., Hoffmeyer S., Mischung C. et al. Minor lesion mutational spectrum of the entire
NF1 gene does not explain its high mutability but points to a functional domain
upstream of the GAP-related domain. Am. J. Hum. Genet. 2000; 66: 790-818.Fallon L., Harton G.L., Sisson M.E. et al. Preimplantation genetic diagnosis for spinal
muscular atrophy type I. Neurology 1999; 53: 1087-1090.Farrer L. Suicide and attempted suicide in Huntington's disease: implications for preclinical
testing of persons at risk. Am. J. Med. Genet. 1986; 24: 305-311.
Библиографический указатель 561Farrer M, Gwinn-Hardy K., Muenter M. et al. A chromosome 4p haplotype segregating with
Parkinson’s disease and postural tremor. Hum. Mol. Genet. 1999; 8: 81-85.Fathallah-Shaykh H. New molecular strategies to cure brain tumors. Arch. Neurol. 1999; 56:
449-453.Feener C.A., Boyce F.M., Kunkel L.M. Rapid detection of CA polymorphisms in cloned
DNA: application to the 5' region of the dystrophin gene. Am. J. Hum. Genet. 1991; 48:
621-627.Fetell M.R. Gliomas. In: Rowland L.P. (ed.) Meritt’s textbook of neurology, 9,h ed. Baltimore:
Williams & Wilkins, 1995: 336-351.Figlewicz D.A., Bird T. «Pure» hereditary spastic paraplegias: the story becomes complicated.
Neurology 1999; 53: 5-7.Fill a A., De Michele G., Cavalcanti F. et al. The relationship between trinucleotide (GAA)
repeat length and clinical features in Friedreich ataxia. Am. J. Hum. Genet. 1996;
59: 554-560.FinkJ.K., Rainier S., Wilkowski J. et al. Paroxysmal dystonic choreoathetosis: tight linkage
to chromosome 2q. Am. J. Hum. Genet. 1996; 59: 140-145.FinkJ.K., Wu C.B., Jones S.M. et al. Autosomal dominant familial spastic paraplegia: tight
linkage to chromosome 15q. Am. J. Hum. Genet. 1995; 56: 188-192.Fisher J., Upadhyaya M. Molecular genetics of facioscapulohumeral muscular dystrophy.
Neuromusc. Disord. 1997; 7: 55-62.Fitzsimons R.B. Facioscapulohumeral muscular dystrophy. Cur. Opin. Neurol. 1999;
12: 501-511.Flanigan K., Gardner K., Alderson K. et al. Autosomal dominant spinocerebellar ataxia with
sensory axonal neuropathy (SCA4): clinical description and genetic localization to
chromosome 16q22.1. Am. J. Hum. Genet. 1996; 59: 392-399.Folstein S.E. Huntington’s disease: a disorder of families. Baltimore: The Johns Hopkins
University Press, 1989.Fong G.C.Y., Shah P.U., Gee M.N. et al. Childhood absence epilepsy with tonic-clonic
seizures and electroencephalogram 3-4-Hz spike and multispike-slow wave complexes:
linkage to chromosome 8q24. Am. J. Hum. Genet. 1998; 63: 1117-1129.Fonknechten N., Mavel D., Byrne P. et al. Spectrum of SPG4 mutations in autosomal
dominant spastic paraplegia. Hum. Mol. Genet. 2000; 9: 637-644.Fontaine B., Davoine C.-S., Diirr A. et al. A new locus for autosomal dominant pure spastic
paraplegia, on chromosome 2q24-q34. Am. J. Hum. Genet. 2000; 66: 702-707.Forrest S.M., Cross G.S., Flint T. et al. Further studies of gene deletions that cause Duchenne
and Becker muscular dystrophies. Genomics 1988; 2: 109-114.Forrest S.M., Knight M, Delatycki M.B. et al. The correlation of clinical phenotype in
Friedreich ataxia with the site of point mutation in theFRDA gene. Neurogenetics 1998;
1: 253-257.Fouad G.T., Servidei S., Durcan S. et al. A gene for familial paroxysmal dyskinesia (FPD1)
maps to chromosome 2q. Am. J. Hum. Genet. 1996; 59: 135-139.Frydman M., Bonne-Tamir B., Farrer L. et al. Assignment of the gene for Wilson disease to
chromosome 13: linkaqe to the esterase D-locus. Proc. Natl. Acad. Sci. USA 1985; 82:
1819-1821.Fu Y.H., Pizziti A., Fenwick R.G. et al. An unstable triplet repeat in a gene related to myotonic
muscular dystrophy. Science 1992; 255: 1256-1258.Fueyo J., Gomez-Manzano C., Yung W.K.A., Kyritsis A.P. Targeting in gene therapy for
gliomas. Arch. Neurol. 1999; 56: 445-448.Fugger L., Morling N.. Sandbej'g-Wolheim M. et al. Tumor necrosis factor alpha gene
polymorphism in multiple sclerosis and optic neuritis. J. Neuroimmunol. 1990; 27: 85-88.Furukawa Y., Kish S.J. Dopa-responsive dystonia: recent advances and remaining issues to be
addressed. Mov. Disord. 1999; 14: 709-715.
562Библиографический указательFurukawa Y, Kish S.J., Bebin E.M. et at. Dystonia with motor delay in compound
heterozygotes for GTP-cyclohydrolase I gene mutations. Ann. Neurol. 1998; 44: 10-16.Furukawa X, Shimadzu A/., Rajput A.H. et al. GTP-cyclohydrolse I gene mutations in
hereditary progressive and dopa-responsive dystonia. Ann. Neurol. 1996; 39: 609-617.Gabriel J.-M., Erne B., Pareyson D. et al. Gene dosage effect in hereditary peripheral
neuropathy: Expression of peripheral myelin protein 22 in Charcot-Marie-Tooth disease
type 1A and hereditary neuropathy with liability to pressure palsies nerve biopsies.
Neurology 1997; 49: 1635-1640.Ga/lou C., Jo/y D., Mejean A. et al. Mutations of the VHL gene in sporadic renal cell
carcinoma: definition of a risk factor for VHL patients to develop an RCC. Hum. MuL
1999; 13: 464-475.Galuzzi G., Deidda G., Cacurri S. et at. Molecular diagnosis of 4q35 rearrangements in
facioscapulohumeral muscular dystrophy (FSHD): application to family studies for a
correct genetic advice and a reliable prenatal diagnosis of the disease. Neuromusc.
Disord. 1999; 9: 190-198.Gancher S.T., Nutt J.G. Autosomal dominant episodic ataxia: a heterogeneous syndrome.
Mov. Disord. 1986; 1: 239-253.Gardner E.J., Simmons M.J., Snustad D.P. Principles of genetics. New York: John Wiley &
Sons, 1991.Gasser T. Genetics of Parkinson’s disease. Ann. Neurol. 1998; 44 (Suppl. 1): S53-S57.Gasser T., Miitter-Myhsok B., Wszolek Z.K. et at. A susceptibility locus for Parkinson’s
disease maps to chromosome 2p 13. Nat. Genet. 1998 (a); 18: 262-265.Gasser T., Windgassen K., Bereznai B. et at. Phenotypic expression of the DYT1 mutation:
a family with writer’s cramp of juvenile onset. Ann. Neurol. 1998 (6); 44: 126-128.Gatti R.A. Ataxia-teleangiectasia. In: Vogelstein B., Kinzler K.W. (eds.) The genetic basis of
human cancer. New York: McGraw-Hill, 1998: 275-300.Genton P., Miche/ucci R., Tassinari C.A., Roger J. The Ramsay Hunt syndrome revisited:
Mediterranean myoclonus versus mitochondrial encephalomyopathy with ragged-red
fibers and Baltic myoclonus. Acta Neurol.Scand. 1990; 81: 8-15.Geoffroy G., Barbeau A., Breton G. et at. Clinical description and roentgenological
evaluation of patients with Friedreich’s ataxia. Can. J. Neurol. Sci. 1976; 3: 279-286.Gibb W.R., Lees A.J. The relevance of the Lewy body to the pathogenesis of idiopathic
Parkinson’s disease. J. Neurol. Neurosurg. Psychiatry 1988; 51: 745-752.Gitad S., Chessa L., Khosravi R. et al. Genotype-phenotype relationships in ataxia-
teleangiectasia (A-T) and A-T variants. Am. J. Hum. Genet. 1998; 62: 551-561.Gitad S., Khosravi R., Shkedy D. etal. Predominance of null mutations in ataxia-teleangiectasia.
Hum. Mol. Genet. 1996; 5: 433-439.Gilbert J.R., Stajich J.M., Watt S. et at. Evidence for heterogeneity in facioscapulohumeral
muscular dystrophy. Am. J. Hum. Genet. 1993; 53: 401-408.Gilliam T.C., Brzustowicz L.M., Castilla L.H. et al. Genetic homogeneity between acute and
chronic forms of spinal muscular atrophy. Nature 1990; 345: 823-825.Gispert S., Twelts R., Orozco G. et al. Chromosomal assignment of the second locus for
autosomal dominant cerebellar ataxia (SCA-2) to chromosome 12q23-24.1. Nat. Genet.
1993; 4: 295-299.Giunti P., Stevanin G., Worth P. et al. Molecular and clinical study of 18 families with ADCA
type II: evidence for genetic heterogeneity and de novo mutation. Am. J. Hum. Genet.
1999; 64: 1594-1603.Giunti P., Sweeney M.G., Harding A. E. Detection of the Machado-Joseph disease/ spinocerebellar
ataxia three trinucleotide repeat expansion in families with autosomal dominant motor
disorders, including the Drew family of Walworth. Brain 1995; 118: 1077-1085.Giunti P., Sweeney M. G., Spadaro M. et al. The trinucleotide repeat expansion on chromosome
6p (SCA1) in autosomal dominant cerebellar ataxias. Brain 1994; 117: 645-649.
Библиографический указатель 563Goate A.M., Chartier-Harlin M.C., Mullan M.C. et al. Segregation of a missense mutation
in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature 1991;
349: 704-706.Goebel H.H., Lenard H.G. Congenital myopathies. Handb. Clin. Neurol. 1992; 62: 331-368.Goldberg Y.P, Andrew S.E., TheilmannJ. et al. Familial predisposition to recurrent mutations
causing Huntington’s disease: genetic risk to sibs of sporadic cases. J. Med. Genet.
1993; 30:987-990.Goldfarb L.G., Vasconcelos O., Platonov FA. et al. Unstable triplet repeat and phenotypic
variability of spinocerebellar ataxia type 1. Ann. Neurol. 1996; 39: 500-506.Goldgaber D., Goldfarb L.G., Brown P. et al. Mutations in familial Creutzfeldt-Jakob disease
and Gerstmann-Sträussler-Scheinker’s syndrome. Exp. Neurol. 1989; 106: 204-206.Goodfellow P.N., Davies K.E., Ropers H.H. Report of the committee on the genetic constitution
of the X and Y chromosomes. Cytogenet. Cell Genet. 1985; 40: 296-352.Gottlieb B., Lehvanslaiko H., Beitel L. et al. The androgen receptor gene mutation database.
Nucl. Acids Res. 1998; 26: 234-238.Gouider R., LeGuern E., Gudenheim M. et al. Clinical, electrophysiologic, and molecular
correlations in 13 families with hereditary neuropathy with liability to pressure palsies
and a chromosome 17p 11.2 deletion. Neurology 1995; 45: 2018-2023.Goverman J., Woods A., Larson L. et al. Transgenic mice that express a myelin basic protein-
specific T cell receptor develop spontaneous autoimmunity. Cell 1993; 72: 551-560.Graeber M.B., Kupke K.G., Müller U. Delineation of the dystonia-parkinsonism syndrome
locus in Xql3. Proc. Natl. Acad. Sci. USA 1992; 89: 8245-8248.Grewal R.P., Karkera J.D., Grewal R.K., Detera-Wadleigh S.D. Mutation analysis of
oculopharyngeal muscular dystrophy in Hispanic American families. Arch. Neurol.
1999; 56: 1378-1381.Griffiths ]., Montague P., Dickinson P. The proteolipid protein gene. Neuropathol. Appl.
Neurobiol. 1995; 21: 85-96.Griggs R.C., Fischbeck K.H. X-linked muscular dystrophies. In: Vinken P.J., Bruyn G.W.,
Klawans H.L. (eds). Handbook of clinical neurology, vol. 62. Amsterdam: Elsevier,
1992: 117-144.Griggs R.C., Markesbery W.R. Distal myopathies. In: Engel A.G., Franzini-Armstrong C.
(eds). Myology, 2nd ed. New York: McGraw-Hill., 1994: 1246-1257.Grompe M. The rapid detection of unknown mutations in nucleic acids. Nat. Genet. 1993;
5: 111-117.Groot Kormelink P., Licyten W. Cloning and sequence of full-length cDNAs encoding the
human neuronal nicotinic acetylcholine receptor (nAChR) subunits beta-3 and beta-4
and expression of seven nAChR subunits in the human neuroblastoma cell line SH-
SY5Y and/or IMR-32. FEBS Lett. 1997; 400: 309-314.Guerrini R., Bonanni P., Nardocci N. et al. Autosomal recessive Rolandic epilepsy with
paroxysmal exercise-induced dystonia and writer’s cramp: delineation of the syndrome
and gene mapping to chromosome 16pl2—11.2. Ann. Neurol. 1999; 45: 344-352.Guiponti M., Rivier F., Vigelano F. et al. Linkage mapping of benign familial infantile
convulsions (BFIC) to chromosome 19p. Hum. Mol. Genet. 1997; 6: 473-477.Gulcher J.R., Jonsson P., Kong A. et al. Mapping of a familial essential tremor gene, FETJ,
to chromosome 3ql3. Nat. Genet. 1997; 17: 84-87.Gusella J.F., Wexler N.S., Conneally P.M. et al. A polymorphic DNA marker genetically
linked to HD. Nature 1983; 306: 234-238.Gutmann D.H. Recent insights into neurofibromatosis type 1. Arch. Neurol. 1998; 55: 778-780.Gutmann D.H., Aylsworth A., Carley J. et al. The diagnostic evaluation and multidisciplinary
management of neurofibromatosis 1 and neurofibromatosis 2. J.A.M.A. 1997; 278: 51-57.Gutmann D.H., Sherman L., Seftor L. et al. Increased expression of the NF2 tumor suppressor
564Библиографический указательgene product, merlin, impairs cell motility, adhesion and spreading. Hum. Mol. Genet.
1999; 8: 267-275.Gwinn-Hardy K.A., Crook R., Lincoln S. el al. A kindred with Parkinson’s disease not
showing genetic linkage to established loci. Neurology 2000; 54: 504-507.Haberhausen G., Schmitt /., Kohler A. et al. Assignment of the dystonia-parkinsonism
syndrome locus, DYT3, to a small region within a 1.8-Mb YAC contig of Xql3.1. Am.
J. Hum. Genet. 1995; 57: 644-650.Hahn-Barma V, Deweer B., Durr A. et al. Are cognitive changes the first symptoms of
Huntington’s disease? A study of gene carriers. J. Neurol. Neurosurg. Psychiatry 1998;
64: 172-177.Hahnen E., SchonlingJ., Rudnik-Schoneborn S. et al. Hybrid survival motor neuron genes in
patients with autosomal recessive spinal muscular dystrophy: new insights into molecular
mechanisms responsible for the disease. Am. J. Hum. Genet. 1996; 59: 1057-1065.Haites N.E., Ne/is E., van Broeckhoven C. 3rd Workshop of the European CMT Consortium:
54lh ENMC International Workshop on genotype/phenotype correlations in Charcot-
Marie-Tooth type 1 and hereditary neuropathy with liability to pressure palsies.
Neuromusc. Disord. 1998; 8: 591-603.Haliassos A., Chomel J.C., Tesson L. etal. Modification of enzymatically amplified DNAfor
the detection of point mutations. Nucl. Acids Res. 1989; 17: 3606-3607.Hanauer A., Cheny M., Fujita R. et al. The Friedreich’s ataxia gene is assigned to chromosome
9q 13—q21 by mapping of tightly linked markers and shows linkage disequilibrium with
D9S15. Am. J. Hum. Genet. 1990; 46: 133-137.Hanihara T., Jnoue K., Kawanishi C. et al. 6-Pyruvoyl-tetrahydropterin synthase deficiency
with generalized dystonia and diurnal fluctuation of symptoms: a clinical and molecular
study. Mov. Disord. 1997; 12: 408-411.Harding A.E. Fredreich’s ataxia: a clinical and genetic study of 90 families with an analysis of
early diagnostic criteria and interfamilial clustering of clinical features. Brain 1981; 104:
589-620.Harding A.E. The hereditary ataxias and related disorders. Edinburgh: Churchill Livingstone,
1984. - 266 p.Harding A.E. The DNA laboratory and neurological practice. J. Neurol. Neurosurg. Psychiatry
1993; 56: P.229-233.Harding A.E. From the syndrome of Charcot, Marie and Tooth to disorders of peripheral
myelin proteins. Brain 1995; 118: 809-818.Harding A.E., Holt I.J., Sweeney M.G. et al. Prenatal diagnosis of mitochondrial DNA8993
T—>G disease. Am. J. Hum. Genet. 1992; 50: 629-633.Harding A.H., Matthews S., Jones S. et al. Spinocerebellar degeneration associated with a
selective defect of vitamin E absorption. New Engl. J. Med. 1985; 313: 32-35.Hardy J., Hardy-Gwinn K. Genetic classification of primary neurodegenerative disease. Science
1998; 282: 1075-1079.Harley H.G., Rundle S.A., McMillan J.C. et al. Size of the unstable CTG repeat sequence in
relation to phenotype and parental transmission in myotonic dystrophy. Am. J. Hum.
Genet. 1993; 52: 1164-1174.Harper P.S. Myotonic dystrophy. Philadelphia: Saunders, 1989.Harper P.S., Harley H.G., Reardon W., Shaw D.J. Anticipation in myotonic dystrophy: new
light on an old problem. Am. J. Hum. Genet. 1992; 51: 10-16.Hattori N., Kitada T., Matsumine H. et al. Molecular genetic analysis oif a novel parkin gene
in Japanese families with autosomal recessive juvenile parkinsonism: evidence for
variable homozygous deletions in the parkin gene in affected individuals. Ann. Neurol.
1998;44:935-941.Hauser M.A.. Horrigan S.K., Salmikangas P. et al. Myotilin is mutated in limb girdle
muscular dystrophy 1A. Hum. Mol. Genet. 2000; 9: 2141-2147.
Библиографический указатель 565Hauser S.L., Fleischnick E., Weiner H. et al. Extended major histocompatibility complex
haplotypes in patients with multiple sclerosis. Neurology 1989; 39: 275-277.Hayasaka K., Himoro M., Sato W. et al. Charcot-Marie-Tooth neuropathy type 1B is associated 4
with mutations of the myelin P0 gene. Nat. Genet. 1993; 5: 31-34.Hayashi Y.K., Chou F.-L., Engvall E. et al. Mutations in the integrin a7 gene cause
congenital myopathy. Nat. Genet. 1998; 19: 94-97.Hayden M.R. Huntington’s chorea. Berlin: Springer-Verlag, 1981.Hazan J., Fonknechten N., Mavel D. et al. Spastin, a novel AAA protein, is altered in the most
frequent form of autosomal dominant spastic paraplegia. Nat. Genet. 1999; 23: 296-303.Hazan J., Fontaine B., Bruyn R.P.M. et al. Linkage of a new locus for autosomal dominant
familial spastic paraplegia to chromosome 2p. Hum. Mol. Genet. 1994; 3: 1569-1573.Hazan J., Lamy C., Melki J. et al. Autosomal dominant familial spastic paraplegia is
genetically heterogeneous and one locus maps to chromosome 14p. Nat. Genet. 1993;5: 163-167.Hedera P., Rainier S., Alvarado D. et al. Novel locus for autosomal dominant hereditary
spastic paraplegia, on chromosome 8q. Am. J. Hum. Genet. 1999; 64: 563-569.Heid C.A., Stevens J., Livak K.J., Williams P.M. Real time quantitative PCR. Genome Res.
1996; 6: 986-994.Heim R.A., Silverman L.M., Färber R.A. et al. Screening for truncated NF1 proteins. Nat.
Genet. 1994; 8: 218-219.Heinzlef O., Paternotte C., Mahieux F. et al. Mapping of a complicated familial spastic
paraplegia to locus SPG4 on chromosome 2p. J. Med. Genet. 1998; 35: 89-93.Helbling-Leclerc A., Zhang X., Topaloglu H. et al. Mutations in the laminin a2-chain gene
(LAMA2) cause merosin-deficient congenital muscular dystrophy. Nat. Genet. 1995; 11:
216-218.Hendriks L., van Duijn C.M., Cras P. et al. Presenile dementia and cerebral haemorrhage
linked to a mutation at codon 692 of the ß-amyloid precursor protein gene. Nat. Genet.
1992;1:218-221.Henske E.P., Scheithauer B.W., Short M. P. et al. Allelic loss is frequent in tuberous sclerosis
kidney lesions bur rare in brain lesions. Am. J. Hum. Genet. 1996; 59: 400-406.Hentati A., Bejaoui K., P ericak-Vance M.A. et al. Linkage of recessive familial amyotrophic
lateral sclerosis to chromosome 2q33-q35. Nat. Genet. 1994 (a); 7: 425-428.Hentati A., Deng H.-X., Hung W.-Y. etal. Human a-tocopherol transfer protein: gene structure
and mutations in familial vitamin E deficiency. Ann. Neurol. 1996; 39: 295-300.Hentati A., Ouahchi K., Pericak-Vance M.A. et al. Linkage of a commoner form of recessive
amyotrophic lateral sclerosis to chromosome 15q 15—q22 markers. Neurogenetics 1998;
2: 55-60.Hentati A., Pericak-Vance M.A., Hung W.-Y. et al. Linkage of «pure» autosomal recessive
familial spastic paraplegia to chromosome 8 markers and evidence of genetic locus
heterogeneity. Hum. Mol. Genet. 1994 (6); 3: 1263-1267.Herman-Bert A., Stevanin G., Netter J.-C. et al. Mapping of spinocerebellar ataxia 13 to
chromosome 19ql3.3—ql3.4 in a family with autosomal dominant cerebellar ataxia and
mental retardation. Am. J. Hum. Genet. 2000; 67: 229-235.Hes F., ZewaldR., Peeters T. et al. Genotype-phenotype correlations in families with deletions
in the von Hippel-Lindau (VHL) gene. Hum. Genet. 2000; 106: 425-431.Higgins J.J., Adler R.L., Loveless J.M. Mutational analysis of the tau gene in progressive
supranuclear palsy. Neurology 1999; 53: 1421-1424.Higgins J.J., Loveless J.M., Jankovic J., Patel P.I. Evidence that a gene for essential tremor
maps to chromosome 2p in four families. Mov. Disord. 1998; 13: 972-977.Higgins J.J., Pho L.T., Nee L.E. A gene (ETM) for essential tremor maps to chromosome
2p22-p25. Mov Disord 1997; 12: 859-864.
566 Библиографический указательHiguchi S., Muramatsu T., Arai H. et al. Polymorphisms of dopamine receptor and transporter
genes and Parkinson’s disease. J. Neural Transm. 1995; 10: 107-113.Hill J.R., Kuriyama N.. Kuriyama H., Israel M.A. Molecular genetics of brain tumors. Arch.
Neurol. 1999; 56: 439-441.Hillaire D., Leclerc A., Favre S. et al. Localization of merosin-negative congenital muscular
dystrophy to chromosome 6q2 by homozygosity mapping. Hum. Mol. Genet.
1994;3:1657-1661.Hirano M., Yanagihara T., Ueno S. Dominant negative effect of GTP cyclohydrolase 1
mutations in dopa-responsive hereditary progressive dystonia. Ann. Neurol. 1998; 44:
365-371.Hockertz M.K., Paty D.W., Beall S.S. Susceptibility to relapsing-progressive multiple sclerosis
is associated with inheritance of genes linked to the variable region of the TcR ß locus:
use of affected family-based controls. Am. J. Hum. Genet. 1998; 62: 373-385.Hoekstra M.F. Responses to DNA damage and regulation of cell cycle checkpoints by the ATM
protein kinase family. Curr. Opin. Genet. Dev. 1997; 7: 170-175.Hoffman E.P., Brown R.H., Kunkel L.M. Dystrophin: the protein product of the Duchenne
muscular dystrophy locus. Biotechnology 1992; 24: 457-466.Hoffman E.P., Fischbeck K.H., Brown R.H. et al. Characterization of dystrophin in muscle-
biopsy specimens from patients with Duchenne’s or Becker’s muscular dystrophy. New
Engl. J. Med. 1988; 318: 1363-1368.Hoffman E.P., Kunkel L.M. Dystrophin abnormalities in Duchenne/Becker muscular dystrophy.
Neuron 1989;2:1019-1029.Holmberg M., Duyckaerts C., Dürr A. et al. Spinocerebellar ataxia type 7 (SCA7): a
neurodegenerative disorder with neuronal intranuclear inclusions. Hum. Mol. Genet.
1998; 7: 913-918.Holmes S.E., O'Hearn E.E., Mclnnis M.G. et al. Expansion of a novel CAG trinucleotide
repeat in the 5’ region of PPP2R2B is associated with SCA12. Nat. Genet.
1999; 23: 391-392.Holmgren G., Ozelius L., Forsgren L. et al. Adult-onset idiopathic torsion dystonia is
excluded from the DYT1 region (9q34) in a Swedish family. J. Neurol. Neurosurg.
Psychiatry 1995; 59: 178-181.Hong M., Zhulcareva V, Vogelsberg-Ragaglia V. etal. Mutation-specific functional impairments
in distinct tau isoforms of hereditary FTDP-17. Science 1998; 282: 1914-1917.Horton W.A., Wong V, Eldridge R. Von Hippel-Lindau disease - clinical and pathological
manifestations in 9 families with 50 affected members. Arch. Intern. Med. 1976;
136: 769-777.Houlden H., Baker M., Adamson J. et al. Frequency of tau mutations in three series of non-
Alzheimer’s degenerative dementia. Ann. Neurol. 1999; 46: 243-248.Housman D. Gain of glutamines, gain of function? Nat. Genet. 1995; 10: 3-4.Hu X., Burghes A.H.M., Ray P.N. et al. Partial gene duplication in Duchenne and Becker
muscular dystrophy. J. Med. Genet. 1988; 25: 369-376.Huggins M., Block M., Karani S. et al. Ethical and legal dilemmas arising during predictive
testing for adult onset disease: the experience of Huntington’s disease. Am. J. Hum.
Genet. 1990; 47: 4-12.Hughes C.A., Byrne P.C., Webb S. et al. SPG 15, a new locus for autosomal recessive
complicated HSP on chromosome 14q. Neurology 2001; 56: 1230-1233.Huson S.M., Compston D.A.S., Clark P., Harper P.S. A genetic study of von Recklinghausen
neurofibromatosis in south east Wales. I. Prevalence, fitness, mutation rate, and effect of
parental transmission on severity. J. Med. Genet. 1989; 26: 704-711.Hutton M., Hardy J. The presenilins and Alzheimers disease. Hum. Mol. Genet. 1997:
6: 1639-1646.Hutton M., Lendon C.L., Rizzu P. et al. Association of missence and 5’-spIice-site mutations
Библиографический указатель 567in tau with the inherited dementia FTDP-17. Nature 1998; 393: 702-705.Ichimura K., Schmidt E.E., Miyakawa A. et al. Distinct patterns of deletion on lOp and lOq
suggest involvement of multiple tumor suppressor genes in the development of astrocytic
gliomas of different malignancy grades. Genes Chromosomes Cancer 1998; 22: 9-15.Ichinose H., Ohye T, Segawa M. et al. GTP cyclohydrolase I gene in hereditary progressive
dysonia with marked diurnal fluctuation. Neurosci. Lett. 1995; 196: 5-8.Ichinose H., Ohye T., Takahashi E. etal. Hereditary progressive dystonia with marked diurnal
fluctuation caused by mutations in the GTP cyclohydrolase I gene. Nat. Genet. 1994; 8:
236-242.Igarashi S., Tanno Y, Onodera 0. et al. Strong correlation between the number of CAG
repeats in androgen receptor genes and the clinical onset of features of spinal and bulbar
muscular atrophy. Neurology 1992; 42: 2300-2302.Ignatius M.J., Gebicke-Harter P.J., Skene J.H.P. et al. Expression of apolipoprotein E during
nerve degeneration and regeneration. Proc. Natl. Acad. Sci. USA 1986; 83: 1125-1129.Ikeda H., Yamaguchi ML, Sugai S. et al. Expanded polyglutamine in the Machado-Joseph
disease protein induces cell death in vitro and in vivo. Nat. Genet. 1996; 13: 196-202.Iliopoulos O., Kibel A., Gray S., Kaefin W.G.Jr. Tumour suppression by the human von
Hippel-Lindau gene product. Nature Med. 1995; 1: 822-826.Iliopoulos O., Levy A. P., Jiang C. et al. Negative regulation of hypoxia-inducible genes by the
von Hippel-Lindau protein. Proc. Nat. Acad. Sci. 1996; 93: 10595-10599.Illarioshkin S.N., Igarashi S., Onodera O. et al. Trinucleotide repeat length and rate of
progression of Huntington’s disease. Ann. Neurol. 1994; 36: 630-635.Illarioshkin S.N., Ivanova-Smolenskaya I.A., Rahmonov R.A. et al. Clinical and genetic study
of familial essential tremor in an isolate of Northern Tajikistan. Mov. Disord. 2000; 15:
1020-1023.Illarioshkin S.N., Ivanova-Smolenskaya I. A., Tanaka H. etal. Clinical and molecular analysis
of a large family with three distinct phenotypes of progressive muscular dystrophy. Brain
1996; 119: 1895-1909.Illarioshkin S.N., Markova E.D., Slominsky PA. et al. Analysis of the GTP cyclohydrolase I
gene in Russian families with dopa-responsive dystonia. Arch. Neurol. 1998; 55: 789-792.Illarioshkin S.N., Tanaka H.t Markova E.D. et al. X-linked non-progressive congenital
cerebellar hypoplasia: clinical description and mapping to chromosome Xq. Ann. Neurol.
1996; 40: 75-83.International Huntington Association and World Federation of Neurology Research Group
on Huntington’s Chorea. Guidelines for the molecular genetics predictive test in
Huntington disease. Neurology 1994; 44: 1533-1536.International SMA Consortium. Meeting report: International SMA Consortium Meeting.
Neuromusc. Disord. 1992; 2: 423-428.Ionasesku V, Sear by C., Sheffield VC. et al. Autosomal dominant Charcot-Marie-Tooth
axonal neuropathy mapped on chromosome 7p (CMT2D). Hum. Mol. Genet. 1996;
5: 1373-1375.Ishikawa A., Miyatake T. A family with hereditary juvenile dystonia-parkinsonism. Mov.
Disord. 1995; 10: 482-488.Ishikawa A., Tsuji S. Clinical analysis of 17 patients in 12 Japanese families with autosomal-
recessive type juvenile parkinsonism. Neurology 1996; 47: 160-166.Ivanova-Smolenskaya LA., Ovchinnikov I. V, Karabanov A. V. et al. The Hisl 069Gln mutation
in the ATP7B gene in Russian patients with Wilson disease [letter]. J. Med. Genet.
1999; 36: 174.Jankovic J. Essential tremor and Parkinson’s disease. Ann. Neurol. 1989; 25: 211-212.Jankovic J., Fahn S. Dystonic disorders. In: Jankovic J., Tolosa E. (eds.) Parkinson’s
disease and movement disorders, 3rd ed. Baltimore: Williams & Wilkins, 1998: 513-551.
568 Библиографический указательJankovic J., Nutt J.G. Blepharospasm and cranial-cervical dystonia (Meige’s syndrome):
familial occurrence. Adv. Neurol. 1988; 49: 117-123.Jarman P.R., Bandmann O., Marsden C.D., Wood N.W. GTP cyclohydrolase I mutations in
patients with dystonia responsive to anticholinergic drugs. J. Neurol. Neurosurg.
Psychiatry 1997; 63: 304-308.JarrettJ.T., Lansbury PT. Seeding ‘one-dimentional crystallization’of amyloid: A pathogenic
mechanism in Alzheimer’s disease and scrapie? Cell 1993; 73: 1055-1.058.Jen J., Yue Q., Nelson S.F et al. A novel nonsense mutation in CACNA1A causes episodic
ataxia and hemiplegia. Neurology 1999; 53: 34-37.Jöbsis G.J., Keizers H., Vreijing J.P. et al. Type VI collagen mutations in Bethlem myopathy,
an autosomal dominant myopathy with contractures. Nat. Genet. 1996; 14: 113-115.Jodice C., Mantuano E., Veneziano L. et al. Episodic ataxia type 2 (EA2) and spinocerebellar
ataxia type 6 (SCA6) due to CAG repeat expansion in the CACNA1A gene on chromosome
19p. Hum. Mol. Genet. 1997; 6: 1973-1978.Johansson J., Forsgren L., Sandgren O. etaJ. Expanded CAG repeats in Swedish spinocerebellar
ataxia type 7 (SCA7) patients: effect of CAG repeat length on the clinical manifestation.
Hum. Mol. Genet. 1998; 7: 171-176.Johns D.R., Berman J. Alternative simultaneous complex I mitochondrial DNA mutations in
Leber’s hereditary optic neuropathy. Biochem. Biophys. Res. Commun. 1991; 174:
1324-1330.Johnson E.W., Dubovsky J., Rich S.S. et al. Evidence for a novel gene for familial febrile
convulsions, FEB2, linked to chromosome 19p in an extended family from the Midwest.
Hum. Mol. Genet. 1998; 7: 63-67.Johnston J.J., Kelley R.I., Crawford T.O. et al. A novel nemaline myopathy in the Amish
caused a mutation in troponin Tl. Am. J. Hum. Genet. 2000; 67: 814-821.Jones A.C., Shyamsundar MM, Thomas M.W. et al. Comprehensive mutation analysis of
TSC1 and TSC2 - and phenotypic correlations in 150 families with tuberous sclerosis.
Am. J. Hum. Genet. 1999; 64: 1305-1315.Jones C.T., Brock D.J.H., Chancellor A.M. et al. Cu/Zn superoxide dismutase (SOD1)
mutations and sporadic ALS. Lancet 1995; 342: 1050-1051.Jouet M.t Rosenthal A., Armstrong G. et al. X-linked stastic paraplegia (SPG1), MASA
syndrome and X-linked hydrocephalus result from mutations in the LI gene. Nat. Genet.
1994; 7: 402-407.Jurkat-Rott K., Lerche H., Mitrovic N.. Lehmann-Horn F. Teaching course: ion channelopathies
in neurology. J. Neurol. 1999; 246: 758-763.Kakinuma S., Sasabe F., Negoro K et al. 18p-syndrome with bilateral pyramidal tract signs,
dystonia of the lower extremities and concentric visual field defects. Clin. Neurol. 1994;
34: 474-478.Kalaydjieva L., Gresham D., Gooding R. et al. N-myc downstream-regulated gene 1 is
mutated in hereditary motor and sensory neuropathy-Lom. Am. J. Hum. Genet. 2000;
67: 47-58.Kalaydjieva L., Hallmayer J., Chandler D. et al. Gene mapping in Gypsies identifies a novel
demyelinating neuropathy on chromosome 8q24. Nat. Genet. 1996; 14: 214-217.Kamm C., Castelon-Konkiewitz E., Naumann M. et al. GAG deletion in the DYT1 gene in
early-onset idiopathic torsion dystonia. Mov. Disord. 1999; 14: 681-683.Kang J., Lemaire H., Unterbeck A. et al. The precursor of Alzheimer’s disease amyloid A4
protein resembles a cell-surface receptor. Nature 1987; 325: 733-736.Kaplan J.-C., Fontaine B. Neuromuscular disorders: gene location. Neuromusc. Disord.
1998; 8: I-VII.Karpati G., Lochmüller H., Nalbantoglu J., Durham H. The principles of gene therapy for the
Библиографический указатель 569nervous system. T.I.N.S. 1996; 19: 49-53.Katzman R. The prevalence and malignancy of Alzheimer’s disease: A major killer. Arch.
Neurol. 1976; 33: 217-218.Kaukonen J., Juselius J. K., Tiranti V. et al. Role of adenine nucleotide translocator 1 in
mtDNA maintenance. Science 2000; 289: 782-785.Kawaguchi Y, Okamoto T., Taniwaki M. et al. CAG expansions in a novel gene for Machado-
Joseph disease at chromosome 14q32.1. Nat. Genet. 1994; 8: 221-228.Kawakami //., Maruyama H., Nakamura S. et al. Unique features of the CAG repeats in
Machado-Joseph disease. Nat. Genet. 1995; 9: 344-345.Kayden H.J. The neurologic syndrome of vitamin E deficiency: a significant cause of ataxia.
Neurology 1993; 43: 2167-2169.Kenck C., Wilhelm M., Bugert P. et al. Mutation of the VHL gene is associated exclusively
with the development of non-papillary renal cell carcinomas. J. Path. 1996; 179: 157-161.Kennedy W.R., Alter M, Sung J.H. Progressive proximal spinal and bulbar muscular atrophy
of late onset: a sex-linked recessive trait. Neurology 1968; 18: 671-680.Kirkwood S.C., Siemers E., Stout J.C. et al. Longitudinal cognitive and motor changes
among presymptomatic Huntington disease gene carriers. Arch. Neurol. 1999;
56: 563-568.Kitada T., Asakawa S., Hattori N. et al. Mutations in the parkin gene cause autosomal
recessive juvenile parkinsonism. Nature 1998; 392: 605-608.Klein C., Brin M.F., de Leon D. et al De novo mutations (GAG deletion) in the DYT1 gene
in two non-Jewish patients with early-onset dystonia. Hum. Mol. Genet. 1998 (a);
7: 1133-1136.Klein C., Brin M.F., Kramer P. et al. Association of a missense change in the D2 dopamine
receptor with myoclonus dystonia. Proc. Natl. Acad. Sci. USA 1999; 96: 5173-5176.Klein C., Pramstaller P.P., Castellan C.C. et al. Clinical and genetic evaluation of a family
with a mixed dystonia phenotype from South Tyrol. Ann. Neurol. 1998 (6); 44: 394-398.Klein C., Schilling K., Saunders-Pullman R.J. et al. A major locus for myoclonus-dystonia
maps to chromosome 7q in eight families. Am. J. Hum. Genet. 2000; 67: 1314-1319.Kleinjan D.-K., van Heyningen V. Position effect in human genetic disease. Hum. Mol. Genet.
1998; 7: 1611-1618.Klesert T.R., Otten A.D., Bird T.D., Tapscott S.J. Trinucleotide repeat expansion at the
myotonic dystrophy locus reduces expression ofDMAHP. Nat. Genet. 1997; 16:402-406.Klockgether T., Dichgans J. Trinucleotide repeats and hereditary ataxias. Nature Med. 1997;
3: 149-150.Kluwe L., Mautner V.-F. Mosaicism in sporadic neurofibromatosis 2 patients. Hum. Mol.
Genet. 1998; 7: 2051-2055.Kluwe L., Friedrich R.E., Hagel C. et al. Mutations and allelic loss of the NF2 gene in
neurofibromatosis 2-associated skin tumors. J. Invest. Derm. 2000; 114: 1017-1021.Knappskog P.M., Flatmark 7., Mallet J. et al. Recessively inherited L-DOPA-responsive
dystonia caused by a point mutation (Q381K) in the tyrosine hydroxylase gene. Hum.
Mol. Genet. 1995; 4: 1209-1212.Knudson A.G. Genetics and etiology of human cancer. Adv. Hum. Genet. 1977; 8: 1-66.Kobayashi K., Nakatori X, Miyake M. et al. An ancient retrotransposal insertion causes
Fukuyama-type congenital muscular dystrophy. Nature 1998; 394: 388-392.Kobayashi T., Matsumine H., Matuda S., Mizuno Y. Association between the gene encoding
the E2 subunit of the a-ketoglutarate dehydrogenase complex and Parkinson’s disease.
Ann. Neurol. 1998; 43: 120-123.Kobayashi T., Mitani H., Talcahashi R.I. et al. Transgenic rescue from embryonic lethality
and renal carcinogenesis in the Eker rat model by introduction of a wild type TSC2
transgene. Proc. Natl. Acad. Sci. USA 1997; 94: 3990-3993.
570Библиографический указательKoch M.C., Steinmeyer K, Lorenz C. et al. The skeletal muscle chloride channel in dominant
and recessive human myotonia. Science 1992; 257: 797-800.Koenig M., Beggs A.H., Moyer H. et al. The molecular basis for Duchenne versus Becker
muscular dystrophy: correlation of severity with type of deletion. Am. J. Hum. Genet.
1989; 45:498-506.Koenig M., Hoffman E.P., Bertelson C.J. et al. Complete cloning of the Duchenne muscular
dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in
normal and affected individuals. Cell 1987; 50: 509-517.Koide R., Ikeuchi T., Onodera O. et al. Unstable expansion of CAG repeat in hereditary
dentatorubral-pallidoluysian atrophy (DRPLA). Nat. Genet. 1994; 6: 9-13.Koller W., Glatt S., Hubble J. et al. Apolipoprotein E genotypes in Parkinson’s disease with
and without dementia. Ann. Neurol. 1995; 37: 242-245.Königs mark B.W., Weiner L. P. The olivopontocerebellar atrophies: a review. Medicine 1970;
49: 227-241.Koob M.D., Moseley M.L., Schut L.J. et al. An untranslated CTG expansion causes a novel
form of spinocerebellar ataxia (SCA8). Nat. Genet. 1999; 21: 379-384.Koutnikova H., Campuzano V, Foury F et al. Studies of human, mouse and yeast homologues
indicate a mitochondrial function for frataxin. Nat. Genet. 1997; 16: 345-351.Kovacs D.M., Fausett H.J., Page K.J. et al. Alzheimer-associated presenilins 1 and 2:
neuronal expression in brain and localization to intracellular membranes in mammalian
cells. Nat. Med. 1996; 2: 224-229.Kramer PL., de Leon D., Ozelius L.J. et al. Dystonia gene in Ashkenazi Jewish population
is located on chromosome 9q32-q34. Ann. Neurol. 1990; 27: 114-120.Kramer P.L., Heiman G.A., Gasser T. et al. The DYT1 gene on 9q34 is responsible for most
cases of early limb-onset idiopathic torsion dystonia (ITD) in non-Jews. Am. J. Hum.
Genet. 1994; 55: 468-475.Kramer P.L., Mineta M., Klein C. et al. Rapid-onset dystonia-parkinsonism: linkage to
chromosome 19q 13. Ann. Neurol. 1999; 46: 176-182.Kremer B., Almqvist E., Theilmann J. et al. Sex-dependent mechanisms for expansions and
contractions of the CAG repeat on affected Huntington disease chromosomes. Am. J.
Hum. Genet. 1995; 57: 343-350.Kremer B., Goldberg P., Andrew S. et al. A worldwide study of the Huntington’s disease
mutation. New Engl. J. Med. 1994; 330: 1401-1406.Kremer B., Squitieri F., Telenius H. et al. Molecular analysis of late onset Huntington’s
disease. J. Med. Genet. 1993; 30: 991-995.Kretzschmar H.A., Stowring L.E., Westaway D. et al. Molecular cloning of a human prion
protein cDNA. DNA 1986; 5: 315-324.Krüger R., Kuhn W., Müller T. et al. Ala30Pro mutation in the gene encoding a-synuclein in
Parkinson’s disease. Nat. Genet. 1998; 18: 106-108.Krüger R., Vieira-Saecker A.M.M., Kuhn W. et al. Increased susceptibility to sporadic
Parkinson’s disease by a certain combined a-synuclein/apolipoprotein E genotype.
Ann. Neurol. 1999; 45: 611-617.Kruglyak L., Daly M.J., Lander E.S. Rapid multipoint linkage analysis of recessive traits in
nuclear families, including homozygosity mapping. Am. J. Hum. Genet. 1995; 56:
519-527.Kulkens T., Bolhius P.A., Wolterman R.A. et al. Deletion of the serine 34 codon from the
major peripheral myelin protein P() gene in Charcot-Marie-Tooth disease type IB Nat.
Genet. 1993; 5: 35-39.Kwiatkowska J., Jozwiak S., Hall F. et al. Comprehensive mutational analysis of the TSC1
gene: observations on frequency of mutation, associated features, and nonpenetrance.
Ann. Hum. Genet. 1998; 62: 277-285.KwonJ.M., Elliot J.L., Yee W.C. et al. Assignment of a second Charcot-Marie-Tooth type II
Библиографический указатель 571locus to chromosome 3q. Am. J. Med. Genet. 1995; 57: 853-858.Kyu Jin D., Ryurl Oh M, Mi Song S. et al. Frequency of spinocerebellar ataxia types 1, 2, 3,6, 7 and dentatorubral pallidoluysian atrophy mutations in Korean patients with
spinocerebellar ataxia. J. Neurol. 1999; 246: 207-210.Lafreniere R.G., Rochefort D.L., Chretien N. et al. Unstable insertion in the 5’ flanking
region of the cystatin B gene is the most common mutation in progressive myoclonus
type 1, EPM1. Nat. Genet. 1997; 15: 298-302.Lakin N.D., Weber R, Stankovic T. et al. Analysis of the ATM protein in wild-type and ataxia-
teleangiectasia cells. Oncogene 1996; 13: 2707-2716.Lander E.S., Botstein D. Homozygosity mapping: a way to map human recessive traits with
the DNA of inbred children. Science 1988; 236: 1567-1570.Lander E.S., SchorkN.J. Genetic dissection of complex traits. Science 1994; 265: 2037-2048.Lang A.E., Rogaeva E.A., Tsuda T. et al. Homozygous inheritance of the Machado-Joseph
disease gene. Ann. Neurol. 1994; 36: 443-447.La Spada A.R., Wilson E.M., Fischbeck A. et al. Meiotic stability and genotype-phenotype
correlation of the expanded trinucleotide repeat sequence in X-linked spinal and bulbar
muscular atrophy. Nat. Genet. 1992; 2: 301-304.La Spada A.R., Wilson E.M., Lubahn D.B. et al. Androgene receptor gene mutations in X-
linked spinal and bulbar muscular atrophy. Nature 1991; 352: 77-79.Lathrop G.M., Lalouel J.M. Easy calculations of lod scores and genetic risks on small
computers. Am. J. Hum. Genet. 1984; 36: 460-465.Lathrop G.M., Lalouel J.M., Julier C., Ott J. Strategies for multi locus linkage analysis in
humans. Proc. Natl. Acad. Sci. USA. 1984; 81: 3443-3446.Latif F., Tory K., Gnarra J. et al. Identification of the von Hippel-Lindau disease tumor
suppressor gene. Science 1993; 260: 1317-1320.Lavedan C., Hofmann-Radvanyi H., Shelbourne P. et al. Myotonic dystrophy: size- and sex-
dependent dynamics of CTG meiotic instability, and somatic mosaicism. Am. J. Hum.
Genet. 1993; 52: 875-883.Lawson K., Wiggins S., Green T. et al. Adverse psychological events occurring in the first year
after predictive testing for Huntington’s disease. The Canadian Collaborative Study
Predictive Testing. J. Med. Genet. 1996; 33: 856-862.Leal A., MoreraB., Del Valle G. etal. A second locus for an axonal form of autosomal recessive
Charcot-Marie-Tooth disease maps to chromosome 19q 13.3. Am. J. Hum. Genet. 2001;
68: 269-274.Lebo R. V, Olney R.K., Golbus M.S. Somatic mosaicism at the Duchenne locus. Am. J. Med.
Genet. 1990; 37: 187-90.Lebre A.-S., Durr A., Jedynak P. et al. DYT1 mutation in French families with idiopathic
torsion dystonia. Brain 1999; 122: 41-45.Ledbetter D.H., Rich D.C., O’Connell P. et al. Precise localization of NF1 to 17q 11.2 by
balanced translocation. Am. J. Hum. Genet. 1989; 44: 20-24.Lee H.S., Sambuughin N., Cemenakova L. et al. Ancestral origins and worldwide distribution
of the PRNP 200K mutation causing familial Creutzfeldt-Jacob disease. Am. J. Hum.
Genet. 1999; 64: 1063-1070.Lefebvre S., Burglen L., Frezal J.. et al. The role of the SMN gene in proximal spinal
muscular dystrophy. Hum. Mol. Genet. 1998; 7: 1531-1536.Lefebvre S., Burglen L., Reboullet S. et al. Identification and characterization of a spinal
muscular atrophy-determining gene. Cell 1995; 80: 155-165.Legoix P., Legrand M.-F., Ollagnon E. et al. Characterisation of 16 polymorphic markers in
the NF2 gene: application to hemizygosity detection. Hum. Mutat. 1999; 13: 290-293.Le Guern E., Guibot A., Kessali M. et al. Homozygosity mapping of an autosomal recessive
form of demyelinating Charcot-Marie-Tooth disease to chromosome 5q23-q33. Hum.
5 72 Библиографический указательMol. Genet. 1996; 5: 1685-1688.Lehesjoki A.-E., Koskiniemi M., Norio R. et al. Localization of the EPM1 gene for progressive
myoclonus epilepsy on chromosome 21: linkage disequilibrium allows high resolution
mapping. Hum. Mol. Genet. 1993; 2: 1229-1234.Lehmann A.R., Carr A.M. The ataxia-teleangiectasia gene: a link between checkpoint controls,
neurodegeneration and cancer. T.I.G. 1995; 11: 375-377.Leigh P.N., Ray-Chaudhuri K. Motor neuron disease. J. Neurol. Neurosurg. Psychiatry 1994;
57: 886-896.Leppert M.F. Gene mapping and other tools for discovery. Epilepsia 1990; 31
(Suppl.3): 11-18.Leroy E., Boyer R., Auburger G. et al. The ubiquitin pathway in Parkinson’s disease. Nature
1998; 395: 451-452.Leube B., Auburger G. Questionable role of adult-onset dystonia among sporadic dystonia
patients. Ann. Neurol. 1998; 44: 984-985.Leube B., Hendgen T., Kessler K.R. et al. Evidence for DYT7 being a common cause of
cervical dystonia (torticollis) in Central Europe. Am. J. Med. Genet. 1997; 74: 529-532.Leube B., Rudnicki D., RatzlaffT. Idiopathic torsion dystonia: Assignment of a gene to
chromosome 18p in a German family with adult onset, autosomal dominant inheritance
and purely focal distribution. Hum. Mol. Genet. 1996; 5: 1673-1678.Levy E., Carman M.D., Fernandez-Madrid J.J. et al. Mutation of the Alzheimer’s disease
amyloid gene in hereditary cerebral hemorrhage, Dutch type. Science 1990;
248: 1124-1126.Levy-Lahad E., Wasco W., Poorkaj P. et al. Candidate gene for the chromosome 1 familial
Alzheimer’s disease locus. Science 1995; 269: 973-977.LeWitte P.A. Neurochemical investigations of dystonia: catecholamine neurotransmitter synthesis.
In: Segawa M. (ed.) Hereditary progressive dystonia with marked diurnal fluctuation.
Carnforth: Parthenon, 1993: 117-123.Li M., Miwa S., Kobayashi Y. et al. Nuclear inclusions of the androgen receptor protein in
spinal and bulbar muscular atrophy. Ann. Neurol. 1998; 44: 249-254.LiX.-J., Li S.-H., Sharp A.H. et al. A huntingtin-associated protein enriched in brain with
implications for pathology. Nature 1995; 378: 398-402.Li Y, O’Connell, P., Huntsman Breidenbach H. et al. Genomic organization of the
neurofibromatosis 1 gene (NF1). Genomics 1995; 25: 9-18.Lim L.E., Duclos F, Broux O. et al. ß-sarcoglycan: characterization and role in limb-girdle
muscular dystrophy linked to 4q 12. Nat. Genet. 1995; 11: 257-265.Lincoln S., Vaughan J., Wood N. et al. Low frequency of pathogenic mutations in the
ubiquitine carboxy-terminal hydrolase gene in familial Parkinson’s disease. Neuroreport
1999; 10: 427-429.Liu A. W., Delgado E.A., Gee M.N. et al. Juvenile myoclonic epilepsy in chromosome 6p 12—
p 11: locus heterogeneity and recombinations. Am. J. Med. Genet. 1996; 63: 438-446.Liu J., Aoki M., Ilia I. et al. Dysferlin, a novel skeletal muscle gene, is mutated in Miyoshi
myopathy and limb girdle muscular dystrophy. Nat. Genet 1998; 20: 31-36.Locke P., Conneally P.M., Tanzi R.E. et al. APOE and Alzheimer disease: examination of
allelic association and effect on age at onset in both early- and late-onset cases. Genet.
Epidemiol. 1995; 12. 83-92.Lodi R., Cooper J.M., Bradley J.L. et al. Deficit of in vitro mitochondrial ATP production in
patients with Friedreich ataxia. Proc. Natl. Acad. Sei. USA 1999; 96: 11492-11495.LouJ.S., JankovicJ. Essential tremor: clinical correlates in 350 patients. Neurology 1991; 41:
234-238.Louis E.D., Ottman R. How familial is familial essential tremor? The genetic epidemiology
of essential tremor. Neurology 1996; 46: 1200-1205.
Библиографический указатель 573Lovestone S. Early diagnosis and the clinical genetics of Alzheimer’s disease. J. Neurol. 1999;
246: 69-72.Liicking C.B., Abbas N.. Durr A., et al. Homozygous deletions in parkin gene in European
and North African families with autosomal recessive juvenile parkinsonism. Lancet
1998; 352: 1355-1356.Lucking C.B., Durr A., Bonifati V. et al. Association between early-onset Parkinson’s disease
and mutations in the parkin gene. New Engl. J. Med. 2000; 342: 1560-1567.Liidecke B, Dworniczak B, Bartholome K. A point mutation in the tyrosine hydroxylase gene
associated with Segawa’s syndrome. Hum. Genet. 1995; 95: 123-125.Lunt P.W., Harper P.S. Genetic counseling in facioscapulohumeral muscular dystrophy.
J. Med. Genet. 1991; 28: 655-664.Lupski J.R., de Oca-Luna R.M., Slaugenhaupt S. et al. DNA duplication associated with
Charcot-Marie-Tooth disease type 1A. Cell 1991; 66: 219-232.MaJ., Nishimura M., Mine H. et al. Genetic contribution of the tumor necrosis factor region
in Guillain-Barre syndrome. Ann. Neurol. 1998; 44: 815-818.Maddock I.R., Moran A., Maher E.R. et al. A genetic register for von Hippel-Lindau disease.
J. Med. Genet. 1996; 33: 120-127.Mahadevan M., Tsilfidis C., Sabourin L. et al. Myotonic dystrophy mutation: an unstable
CTG repeat in the 3’ untranslated region of the gene. Science 1992; 255: 1253-1255.Maher E.R., Iselius L., Yates J.R.W. et al. Von Hippel-Lindau disease: a genetic study.
J. Med. Genet. 1991; 28: 443-447.Mahley R.W. Apolipoprotein E: cholesterol transport protein with expanding role in cell
biology. Science 1988; 240: 622-630.Mahley R.W., Nathan B.P., Pitas R.E. Apolipoprotein E: structure, function, and possible
roles in Alzheimer’s disease. Ann. NY Acad. Sci. 1996; 777: 139-145.Malacarne M., Gennaro E., Madia F. et al. Benign familial infantile convulsions: mapping of
a novel locus on chromosome 2q24 and evidence for genetic heterogeneity. Am. J. Hum.
Genet. 2001; 68: 1521-1526.Manilal S., Nguyen T.M., Sewry C.A., Morris G.E. The Emery-Dreifuss muscular dystrophy
protein, emerin, is a nuclear membrane protein. Hum. Mol. Genet. 1996; 5: 801-808.Marchuk D. A., Saulino A.M., Tavakkol R. etal. cDNA cloning of the type 1 neurofibromatosis
gene: complete sequence of the NF1 gene product. Genomics 1991; 11: 931-940.Marder K., Tang M.X., Mejia H. et al. Risk of Parkinson’s disease among first-degree
relatives: a community-based study. Neurology 1996; 47: 155-160.Markova E.D., Jvanova-Smolens/caya LA. Phenotypic polymorphism of dopa-responsive dystonia
(in Russia). In: Segawa M., Nomura Y. (eds.) Age-related dopamine-dependent disorders.
Basel: Karger, 1995: 36-40.Markova E.D., Slominsky P.A., Illarioshkin S.N. et al. A novel mutation in the GTP
cyclohydrolase I gene associated with a broad range of clinical presentations in a family
with autosomal dominant dopa-responsive dystonia. Eur. J. Neurol. 1999; 6: 605-608.Marsden C.D. Parkinson’s disease. J. Neurol. Neurosurg. Psychiatry 1994; 57: 672-681.Martinez Murillo F., Kobayashi H., Pegoraro E. et al. Genetic localisation of a new locus for
recessive familial spastic paraparesis to 15q 13— 15. Neurology 1999; 53: 50-56.Maruyama H., Nakamura S., Matsuyama Z. et al. Molecular features of the CAG repeats and
clinical manifestations of Machado-Joseph disease. Hum. Mol. Genet. 1995; 4: 807-812.Mastaglia F.L., LaingN.G. Distal myopathy: clinical and molecular classification. J. Neurol.
Neurosurg. Psychiatry 1999; 67: 703-709.Matsuda C., Aoki M., Hayashi Y.K. et al. Dysferlin is a surface membrane-associated protein
that is absent in Miyoshi myopathy. Neurology 1999; 53: 1119-1122.Matsumine H., Saito M., Shimoda-Matsubayashi S. et al. Localization of a gene for an
autosomal recessive form of juvenile Parkinsonism to chromosome 6q25.2-27. Am. J.
Hum. Genet. 1997; 60: 588-596.
574Библиографический указательMatsuura T, Yamagata T., Burgess D.L. et al. Large expansion of the ATTCT pentanucleotide
repeat in spinocerebellar ataxia type 10. Nat. Genet. 2000; 26: 191-194.Matsuyama Z, Kawakami H., Maruyama H. et al. Molecular features of the CAG repeats of
spinocerebellar ataxia 6 (SCA6). Hum. Mol. Genet. 1997; 6: 1283-1287.Mattson M.P. Advances fuel Alzheimer’s conundrum. Nat. Genet. 1997; 17: 254-256.Maxwell P.H., Wiesener M.S., Chang, G.-W. et al. The tumour suppressor protein VHL
targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature
1999; 399: 271-275.Mayer K., Ballhausen W., Rott H.-D. Mutation screening of the entire coding regions of the
TSC1 and the TSC2 gene with the protein truncation test (PTT) identifies frequent
splicing defects. Hum. Mutat. 1999; 14: 401-411.McConville C.M., Stankovic T., Byrd PJ. et al. Mutations associated withvariant phenotypes
in ataxia-teleangiectasia. Am. J. Hum. Genet. 1996; 59: 320-330.McDonald R.L., Kelly K.M. Mechanisms of action of currently prescribed and newly developed
antiepileptic drugs. Epilepsia 1994; 35: S41-S50.McDonnell G. V, Kirk C. W., Middleton D. et al. Genetic association studies of tumor necrosis
factor a and ß and tumor necrosis factor receptor 1 and 2 polymorphisms across the
clinical spectrum of multiple sclerosis. J. Neurol. 1999; 246: 1051-1058.McEntagart M., Norton N., Williams H. et al. Localization of the gene for distal hereditary
motor neuronopathy VII (dHMN-VII) to chromosome 2ql4. Am. J. Hum. Genet. 2001;
68: 1270-1276.McGaughran J. M., Harris D. I., Donnai D. et al. A clinical study of type I neurofibromatosis
in north west England. J. Med. Genet. 1999; 36: 197-203.McHale D.P., Mitchell S., BundeyS. et al. A gene for autosomal recessive symmetrical cerebral
palsy maps to chromosome 2q24-25. Am. J. Hum. Genet. 1999; 64: 526-532.McKee A.C., Kosik KS., Kowall N.W. Neuritic pathology and dementia in Alzheimer’s
disease. Ann. Neurol. 1991; 30: 156-165.McKenzie S.E., Mansfield E., Rappaport E. et al. Parallel molecular genetic analysis. Eur. J.
Hum. Genet. 1998; 6: 417-429.McKusick V.A., Amberger J.S. The morbid anatomy of the human genome: chromosomal
location of mutations causing disease. J. Med. Genet. 1993; 30: 1-26.McNally E.M., de Sa Moreira E., Duggan D. J. etal. Caveolin-3 in muscular dystrophy. Hum.
Mol. Genet. 1998; 7: 871-877.McNeil S.M., Novelletto A., Srinidhi J. et al. Reduced penetrance of the Huntington’s disease
mutation. Hum. Mol. Genet. 1997; 6: 775-779.Vfegarbane A., Delague V, Salem N., Loiselet J. Autosomal recessive congenital cerebellar
hypoplasia and short stature in a large inbred family. Am. J. Med. Genet. 1999;
87: 88-90.Ideiser B., Dunn S. Psychological impact of genetic testing for Huntington’s disease: an
update of the literature. J. Neurol. Neurosurg. Psychiatry 2000; 69: 574-578.Meissen G.J., Mastromauro C.A., Kiely D.K. et al. Understanding the decision to take the
predictive test for Huntington disease. Am. J. Med. Genet. 1991; 39: 404-410.Aeissen G.J., Myers R.H., Mastromauro C.A. et al. Predictive testing for HD with use of a
linked DNA marker. New Engl. J. Med. 1988; 318: 535-542.Aelis M.A., Cau M., Congiu R. et al. Germinal mosaicism in a Duchenne muscular dystrophy
family: implications for genetic counselling. Clin. Genet. 1993; 43: 247-249.Aelki J., Abdelhak S., Sheth P. et al. Gene for proximal spinal muscular atrophies maps to
chromosome 5q. Nature 1990; 344: 767-768.Aenon A.G., Anderson K.M., Riccardi V.M. et al. Chromosome 17p deletions and p53 gene
mutations associated with the formation of malignant neurofibrosarcomas in von
Recklinghausen neurofibromatosis. Proc. Nat. Acad. Sei. 1990; 87: 5435-5439.
Библиографический указатель 5 75Merette C., Ott J. Finding and excluding gene locations by linkage analysis. In: Rosenberg
R.N., Prusiner S.B., DiMauro S. et al. (eds.) The molecular and genetic basis of
neurological disease. Boston: Butterworth-Heinemann, 1993: 25-28.Mersiyanova I. V, Perepelov A. V, Polyakov A. V. et al. A new variant of Charcot-Marie-Tooth
disease type 2 is probably the result of a mutation in the neurofilament-light gene. Am.
J. Hum. Genet. 2000; 67: 37-46.Messiaen L.M., Callens T., Mortier G. et al. Exhaustive mutation analysis of the NF1 gene
allows identification of 95% of mutations and reveals a high frequency of unusual
splicing defects. Hum. Mutat. 2000; 15: 541-555.Messina D.L., Speer M.C., Pericak-Vance M.A., McNally E.M. Linkage of familial dilated
cardiomyopathy with conduction defect and muscular dystrophy to chromosome 6q23.
Am. J. Hum. Genet. 1997; 61: 909-917.Mhatre A.N., Trifiro M.A., Kaufman M. et al. Reduced transcriptional regulatory competence
of the androgen receptor in X-linked spinal and bulbar muscular atrophy. Nat. Genet.
1993; 5: 184-188.Mikami M., Yasuda T., Terao A. et al. Localization of a gene for benign adult familial
myoclonic epilepsy to chromosome 8q23.3-q24.1. Am. J. Hum. Genet. 1999;
65: 745-751.Minassian B.A., Sainz J., Serratosa J.M. et al. Genetic locus heterogeneity in Lafora’s
progressive myoclonus epilepsy. Ann. Neurol. 1999; 45: 262-265.Minetti C., Sotgia F, Bruno C. et al. Mutations in the caveolin-3 gene cause autosomal
dominant limb-girdle muscular dystrophy. Nat. Genet. 1998; 18: 365-368.Mirabel la M., Silvestri G., de Rosa G. et al. GCG genetic expansions in Italian patients with
oculopharyngeal muscular dystrophy. Neurology 2000; 54: 608-614.Mitchison H.M., Hofmann S.L., Becerra C.H.R. et al. Mutations in the palmitoyl-protein
gene (PPT\ CLN1) causing juvenile neuronal ceroid lipofuscinosis with granular
osmiophilic deposits. Hum. Mol. Genet. 1998; 7: 291-297.Mitrani-Rosenbaum S., Argow Z, Blumenfeld A. et al. Hereditary inclusion body myopathy
maps to chromosome 9pl-ql. Hum. Mol. Genet. 1996; 5: 159-163.Miyoshi K, Kawai H., Iwasa M. et al. Autosomal recessive distal muscular dystrophy as a
new type of progressive muscular dystrophy. Brain 1986; 109: 31-54.Miyoshi Y., Yamada T., Tanimura M. et al. A novel autosomal dominant spinocerebellar ataxia
(SCA16) linked to chromosome 8q22.1-24.1. Neurology 2001; 57: 96-100.Mohapatra G., Bollen A.W., Kim D.H. et al. Genetic analysis of glioblastoma multiforme
provides evidence for subgroups within this grade. Genes Chromosomes Cancer 1998;
21: 195-206.Mohire M.D., Tandan R., Fries T.J. et al. Early-onset benign autosomal dominant limb-
girdle myopathy with contractures (Bethlem myopathy). Neurology 1988;
38: 573-580Monaco A.P., Bertelson C.J., Liechti-Gallati S. et al. An explanation for the phenotypic
differences between patients bearing partial deletions of the DMD locus. Genomics
1988; 2: 90-95.Monaco A.P., Neve R.L., Colletti-Feener C. et al. Isolation of candidate cDNAs for portions
of the Duchenne muscular dystrophy gene. Nature 1986; 323: 646-650.Moraes C.T., DiMauro S., Zeviani M. et al. Mitochondrial DNA deletions in progressive
external ophthalmoplegia and Kearns-Sayre syndrome. New Engl. J. Med.
1989; 320: 1293-1299.Moraes C.T., Shanske S., Tritschler H.-J. et al. MtDNA depletion with variable tissue
expression: A novel genetic abnormality in mitochondrial diseases. Am. J. Hum. Genet.
1991; 48: 492-501.Moreira E.S., Wiltshire T.J., Faulkner G. et al. Limb-girdle muscular dystrophy type 2G is
576 Библиографический указательcaused by mutations in the gene encoding the sarcomeric protein telethonin. Nat. Genet.
2000; 24:163-166.Moreira M.C., Barbot C., Tachi N. et al. Homozygosity mapping of Portuguese and Japanese
forms of ataxiaoculomotor apraxia to 9pl3, and evidence for genetic heterogeneity. Am.
J. Hum. Genet. 20001; 68: 501-508.MuchirA., Bonne G., van der Kooi A.J. et al. Identification of mutations in the gene encoding
lamins A/C in autosomal dominant limb girdle muscular dystrophy with atrioventricular
conduction disturbances (LGMD1B). Hum. Mol. Genet. 2000; 9: 1453-1459.Mukhopadhyay D., Knebel mann B., Cohen H. T. et al. The von Hippel-Lindau tumor suppressor
gene product interacts with Spl to repress vascular endothelial growth factor promoter
activity. Molec. Cell. Biol. 1997; 17: 5629-5639.Mulley J.C., Staples A., Donnelly A. et al. Explanation for exclusive maternal inheritance for
congenital form of myotonic dystrophy. Lancet 1993; 341: 236-237.Multicenter Study Group. Diagnosis of Duchenne and Becker muscular dystriphies by
polymerase chain reaction: A multicenter study. J. A. M. A. 1992; 267: 2609-2615.Mimchau A., Dressier D., Bhatia K.P. et al. Machado-Joseph disease presenting as severe
generalised dystonia in a German patient. J. Neurol. 1999; 246: 840-842.Myers R.H., McDonald M.E., Koroshetz W.J. et al. De novo expansion of a (CAG) repeat in
sporadic Huntington’s disease. Nat. Genet. 1993; 5: 168-173.Nagano A., Koga R., Ogawa M. et al. Emerin deficiency at the nuclear membrane in patients
with Emery-Dreifuss muscular dystrophy. Nat. Genet. 1996; 12: 254-259.Nagaoka (J., Takashima M., Ishikawa K. et al. A gene on SC A4 locus causes dominantly
inherited pure cerebellar ataxia. Neurology 2000; 54: 1971-1975.Nakamura K., Jeong S. Y., Uchihara T. et al. SCA17, a novel autosomal dominant cerebellar
ataxia caused by expanded polyglutamine in TATA-binding protein. Hum. Mol. Genet.
2001; 10: 1441-1448.Nance M.A. Huntington’s disease - another chapter rewritten. Am. J. Hum. Genet. 1996; 59:
1-6.Nance M.A., Leroy B.S., Orr H.T. et al. Protocol for genetic testing in Huntington disease:
three years of experience in Minnesota. Am. J. Med. Genet. 1991; 40: 518-522.Narabayashi H., Yokoshi M., Iizuka R., Nagatsu T. Juvenile parkinsonism. In: Vinken P.J.,
Bruyn G.W., Klawans H.L. (eds.) Handbook of clinical neurology, vol. 49. Amsterdam:
Elsevier Science Publishers, 1986: 153-165.National Institute on Aging/Alzheimer's Association Working Group. Apolipoprotein E
genotyping in Alzheimer’s disease. Lancet 1996; 347: 1091-1095.Natowicz M.. AlperJ.K, A/per J.S. Genetic discrimination and the law. Am. J. Hum. Genet.
1992; 50: 465-475.Nelis E., Haites N.E., Van Broeckhoven C. et al. Mutations in the peripheral myelin genes
and associated genes in inherited peripheral neuropathies. Hum. Mutat. 1999; 13: 11-28.Nelis E., Timmerman V, De Jonghe P., Van Broeckhoven C. Identification of a 5’ splice site
mutation in the PMP-22 gene in autosomal dominant Charcot-Marie-Tooth disease type
1. Hum. Mol. Genet. 1994; 3: 515-516.Nelson S.F, McCusker J.H., Sander M.A. et al. Genomic mismatch scanning: a new approach
to genetic linkage mapping. Nat. Genet. 1993; 4: 11-18.Neuhauer B.A., Fiedler B., Himine/ein B. et al. Centrotemporal spikes in families with
rolandic epilepsy: linkage to chromosome 15q 14. Neurology 1998; 51: 1608-1612.Nicho/l D.J.. Bennett P., Hiller L. el al. A study of five candidate genes in Parkinson’s disease
and related neurodegenerative disorders. Neurology 1999; 53: 1415-1421.Nicholson G.A., Dawkins J.L., Blair I.P. et al. The gene for hereditary sensory neuropathy
type I (HSN-I) maps to chromosome 9q22. l-q22.3. Nat. Genet. 1996; 13: 101-104.Nigro V., de Moreira E., Pi/uso G. ct al. Autosomal recessive limb-girdle muscular dystrophy,
Библиографический указатель 577LGMD2F, is caused by a mutation in the delta-sarcoglycan gene. Nat. Genet. 1996; 13:
195-198.Niida Y., Lawrence-Smith N., Banwell A. et al. Analysis of both TSC1 and TSC2 for
germline mutations in 126 unrelated patients with tuberous sclerosis. Hum. Mutat.
1999; 14: 412-422.Nikali K., Suomalainen A., Terwilliger J. et al. Random search for shared chromosomal
regions in four affected individuals: the assignment of a new hereditary ataxia locus. Am.
J. Hum. Genet. 1995; 56: 1088-1095.Nishino /., Spinazzola A., Hirano M. Thymidine phosphorylase gene mutations in MNGIE,
a human mitochondrial disorder. Science 1999; 283: 689-692.Noguchi S., McNally E.M., Ben Othmane K. et al. Mutations in the dystrophin-associated
protein y-sarcoglycan in chromosome 13 muscular dystrophy. Science 1995;
270: 819-822.Nowak K.J., Wattanasirichaigoon D., Goebel H. et al. Mutations in the skeletal muscle a-
actin gene in patients with actin myopathy and nemaline myopathy. Nat. Genet. 1999;
23: 208-212.Nussbaum R.L., Polymeropoulos M.H. Genetics of Parkinson’s disease. Hum. Mol. Genet.
1997; 6: 1687-1691.NuttJ.C., Muenter M.D., MeltonL.J. etal. Epidemiology of dystonia in Rochester, Minnesota.Adv. Neurol. 1988; 50: 361-365.Nygaard T.G., Marsden C.D., Duvoisin R.C. Dopa-responsive dystonia. Adv. Neurol. 1988;
50: 377-384.Nygaard T.G., Raymond D., Chen C. et al. Localization of a gene for myoclonus-dystonia to
chromosome 7q21-q23. Ann. Neurol. 1999; 46: 794-798.Nygaard. T.G., Wilhelmsen K.C., Risch N.J. et al. Linkage mapping of dopa-responsive
dystonia (DRD) to chromosome 14q. Nat. Genet. 1993; 5: 386-391.Olerup O., Hillert J. HLA class II-associated genetic susceptibility in multiple sclerosis: a
critical evaluation. Tissue Antigens 1991; 38: 1-15.Ophoff R.A., Terwindt G.M., Vergouwe M.N. et al. Familial hemiplegic migraine and episodic
ataxia type-2 are caused by mutations in the Ca(2+) channel gene CACNL1A4. Cell
1996; 87: 543-552.Orita M., Suzuki Y, Sekiya I, Hayashi K. Rapid and sensitive detection of point mutations
and DNA polymorphisms using the polymerase chain reaction. Genomics
1989;5:874-879.Orr H.T., Chung M.-Y., Banfi S. et al. Expansion of an unstable trinucleotide CAG repeat in
spinocerebellar ataxia type 1. Nat. Genet. 1993; 4: 221-226.Osborn M.J., Upadhyaya M. Evaluation of the protein truncation test and mutation detection
in the NF1 gene: mutational analysis of 15 known and 40 unknown mutations. Hum.
Genet. 1999; 105: 327-332.Osborne J.P., Fiyer A., Webb D. Epidemiology of tuberous sclerosis. Ann. NY Acad. Sci.
1991; 615: 125-127.Ott J. Analysis of human genetic linkage, 2nd ed. Baltimore: Johns Hopkins
University Press, 1991.Ottman R., Risch N.. Hauser W.A. et al. Localization of a gene for partial epilepsy to
chromosome lOq. Nat. Genet. 1995; 10: 56-60.Ouahchi K., Arita M., Kayden H. et al. Ataxia with isolated vitamin E deficiency is caused by
mutations in the a-tocopherol transfer protein. Nat. Genet. 1995; 9: 141-145.Ozawa E., Yoshida M., Suzuki A. et al. Dystrophin-associated proteins in muscular dystrophy.
Hum. Mol. Genet. 1995; 4: 1711-1716.Ozelius L.J., Breakefield X.O. Co-factor insufficiency in dystonia-parkinsonian syndrome.
Nat. Genet. 1994; 8: 207-208.
5 78 EuönuoepacjnmecKuü yK.a.3ameJibOzelius L.J., Hewett J.W., Page C.E. et al. The early-onset torsion dystonia gene (DYT1)
encodes an ATP-binding protein. Nat. Genet. 1997; 17: 40-48.Ozelius L.J., Kramer P.L., Moskowitz C.B. et al. Human gene for torsion dystonia located on
chromosome 9q32-q34. Neuron 1989; 2: 1427-1434.Pack S.D., Zbar B., Pak E. et. al. Constitutional von Hippel-Lindau (VHL) gene deletions
detected in VHL families by fluorescence in situ hybridization. Cancer Res. 1999; 59:
5560-5564.Palmer M.S., Diyden A.J., Hughes J.T., Collinge J. Homozygous prion protein genotype
predisposes to sporadic Creutzfeldt-Jakob disease. Nature 1991; 352: 340-342.Pa/meri S., Hoogeveen A.T., Verheijen F.W., Galjaard H. Galactosialidosis: molecular
heterogeneity among distinct clinical phenotypes. Am. J. Hum. Genet. 1986;
38: 137-148.Palmucci L., Mongini T., Chiado-Piat L. et al. Dystrophinopathy expressing as either
cardiomyopathy or Becker dystrophy in the same family. Neurology 2000; 54: 529-530.Pandolfo M. Molecular genetics and pathogenesis of Friedreich ataxia. Neuromuscul. Disord.
1998; 8: 409-415.Pareyson D., Gellera C., Castellotti B. et al. Clinical and molecular studies of 73 Italian
families with autosomal dominant cerebellar ataxia type I: SCA1 and SCA2 are the most
common genotypes. J. Neurol. 1999; 246: 389-393.Pc/rman Y.. Plante-Bordeneuve V, Guiochon-Mantel A. et al. Recessive inheritance of a new
point mutation of the PMP22 gene in Dejerine-Sottas disease. Ann. Neurol. 1999; 45:
518-522.Parry D.M., MacCollin M.M., Kaiser-Kupfer M.J. et al. Germ-line mutations in the
neurofibromatosis 2 gene: correlations with disease severity and retinal abnormalities.
Am. J. Hum. Genet. 1996; 59: 529-539.Parse/I D.A., LindquistS. The function of heat-shock proteins in stress tolerance: degradation
and reactivation of damaged proteins. Annu. Rev. Genet. 1993; 27: 437-496.Pcissos-Bueno M.-R., Bakker E., Kneppers A.L. et al. Different mosaicism frequences for
proximal and distal Duchenne muscular dystrophy (DMD) mutations indicate difference
in etiology and recurrent risk. Am. J. Hum. Genet. 1992; 51: 1150-1155.Passos-Bueno M.-R., Bashir R., Moreira E.S. et al. Confirmation of the 2p locus for the mild
autosomal recessive limb-girdle muscular dystrophy gene (LGMD2B) in three families
allows refinement of the candidate region. Genomics 1995; 27: 192-195.Patel PI., Roa B.B., Welcher A.A. et al. The gene for the peripheral myelin protein PMP-22
is a candidate for Charcot-Marie-Tooth disease type 1A. Nat. Genet. 1992; 1: 159-165.Paulson H.L., Perez M.K., Trottier Y et al. Intranuclear inclusions of expanded polyglutamine
protein in spinocerebellar ataxia type 3. Neuron 1997; 19: 333-344.Pearce D.A. Hereditary spastic paraplegia: mitochondrial metalloproteases of yeast. Hum.
Genet. 1999; 104: 443-448.Peiffer A., Thompson J., Charlier C. et al A locus for febrile seizures (FEB3) maps to
chromosome 2q23-24. Ann. Neurol. 1999; 46: 671-678.Pennacchio L.A.. Lehesjoki A.-E., Stone N.E. et al Mutations in the gene encoding cystatin
B in progressive myoclonus epilepsy (EPM1). Science 1996; 271: 1731-1734.Penney J.B., Young A.B. Huntington’s disease. In: Jankovic J., Tolosa E. (eds,) Parkinson's
disease and movement disorders, 3rd edition. Baltimore: Williams&Wilkins, 1998:
341-355.Phillips H.A., Scheffer I.E., Crossland KM. ei al. Autosomal dominant nocturnal frontal¬
lobe epilepsy genetic heterogeneity and evidence for a second locus at 15^24. Am. J.
Hum. Genet. 1998; 63: 1108-1116.Piccolo F., Roberds S.L., Jeanpierre M. et al. Primary adhalinopathy: a common cause of
autosoma! recessive muscular dystrophy of variable severity. Nat. Genet. 1995;
10: 243-245
Библиографический указатель 5 79Plank T.L., Yeung R.S., Henske E.P. Hamartin, the product of the tuberous sclerosis (TSC1)
gene, interacts with tuberin and appears to be localised to cytoplasmic vesicles. Cancer
Res. 1998; 58: 4766-4770.Plante-Bordeneuve V., Guiochon-Mantel A., Lacroix C. et al. The Roussy-Levy family: from
the original description to the gene. Ann. Neurol. 1999; 46: 770-773.Poduslo S.E., Yin X., Hargis J. et al. A familial case of Alzheimer’s disease without tau
pathology may be linked with chromosome 3 markers. Hum. Genet. 1999; 105: 32-37.Polo J.M., Calleja J., Combarros O. et al. Hereditary ataxias and paraplegias in Cantabria,
Spain. An epidemiological and clinical study. Brain 1991; 114: 855-866.Polymeropoulos M.H., Lavedan C., Leroy E. et al. Mutation in the a-synuclein gene identified
in families with Parkinson’s disease. Science 1997; 276: 2045-2047.Poorkaj P., Bird T.D., Wijsman E. et al. Tau is a candidate gene for chromosome 17
frontotemporal dementia. Ann. Neurol. 1998; 43: 815-825.Poulton J., Macaulay V, Marchington D.R. Mitochondrial genetics’98: Is the bottleneck
cracked? Am. J. Hum. Genet. 1998; 62: 752-757.Poza J.J., Saenz A., Martinez-Gil A. et al. Autosomal dominant lateral temporal epilepsy:
clinical and genetic study of a large Basque pedigree linked to chromosome lOq. Ann.
Neurol. 1999; 45: 182-188.Priest J.M., FischbeckK.H., NouriN. el al A locus for axonal motor-sensory neuropathy with
deafness and mental retardation maps to Xq24-26. Genomics 1995; 29: 409-412.Prior T.W., Papp A.C., Snyder P.J. et al. Heteroduplex analysis of the dystrophin gene:
application to point mutation and carrier detection. Am. J. Med. Genet. 1994: 50: 68-73.Prowse A.H., Webster A.R., Richards F.M. et al. Somatic inactivation of the VHL gene in
Von Hippel-Lindau disease tumors. Am. J. Hum. Genet. 1997; 60: 765-771.PrusinerS.B. Novel proteinaceous infectious particles cause scrapie. Science 1982; 216: 136-144.Prusiner S.B., Hsiao K.K. Human prion diseases. Ann. Neurol. 1994; 35: 385-395.Prusiner S.B., Carlson G.A., DeArmond S.J. et al. Transmisssible and genetic prion diseases
of humans and animals. In: Rosenberg R.N., Prusiner S.B., DiMauro S. et al. (eds.)
The molecular and genetic basis of neurological disease. Boston: Butterworth-Heinemann,
1993: 585-609.Prusiner S.B., Scott M, Foster D. et al. Transgenetic studies implicate interactions between
homologous PrP isoforms in scrapie prion replication. Cell 1990; 63: 673-686.Pujana M.A., Corral J., Gratacds M. et al. Spinocerebellar ataxias in Spanish patients:
genetic analysis of familial and sporadic cases. Hum. Genet. 1999; 104: 516-522.Pulst S.M. Genetic linkage analysis. Arch. Neurol. 1999; 56: 667-672.Quaid K.A. Presymptomatic testing for Huntington disease. Am. J. Hum. Genet. 1993;
53: 785-787.Raeymaekers P., Timmerman V., Nelis E. et al. Estimation of the size of the chromosome
17pl 1.2 duplication in Charcot-Marie-Tooth neuropathy type la (CMTla). J. Med.
Genet. 1992; 29: 5-11.Raffel C., Jenkins R.B., Frederick L. et al. Sporadic medulloblastomas contain PTCH
mutations. Cancer Res. 1997; 57: 842-845.Ragge N.K., Baser M.E., Klein J. et al. Ocular abnormalities in neurofibromatosis 2. Am. J.
Ophthal. 1995; 120: 634-641.Ragno M., De Michele G., Cavalcanti F. et al. Broadened Friedreich’s ataxia phenotype after
gene cloning: minimal GAA expansion causes late-onset spastic ataxia. Neurology
1997; 49: 1617-1620.Ranen N.G., Stine O.C., Abbott M.H. et al. Anticipation and instability of IT-15 (CAG)N
repeats in parent-offspring pairs with Huntington disease. Am. J. Hum. Genet. 1995; 57:
593-602.Rem to S., Zhang Y. Ross B. et al. The neuronal ceroid lipofuscinoses in human EPMR and
580Библиографический указательmndmutant mice are associated eith mutations in CLN8. Nat. Genet. 1999; 23:233-236.Ranum L.P., Limdgren J.K., Schut L.J. et al. Spinocerebellar ataxia type I and Machado-
Joseph disease: incidence of CAG expansions among adult-onset ataxia patients from
311 families with dominant, recessive or sporadic ataxia. Am. J. Hum. Genet. 1995; 57:
603-608.Ranum L.P., Rasmussen P.F., Benzow K.A. et al. Genetic mapping of a second myotonic
dystrophy locus. Nat. Genet 1998; 19: 196-198.Ranum L.P., Schut L.J., Lundgren J.K. et al. Spinocerebellar ataxia type 5 in a family
descended from the grandparents of President Lincoln maps to chromosome 11. Nat.
Genet. 1994; 8: 280-284.Rathouz MM, Vijayaraghavan S., Berg D.K. Elevation of intracellular calcium levels in
neurons by nicotinic acetylcholine receptors. Mol. Neurobiol. 1996; 12: 117-131.Rees J.H., Vaughan R.W., Kondeatis E., Hughes R.A.C. HLA-class II alleles in Guillain-
Barre syndrome and Miller Fisher syndrome and their association with preceding
Campylobacter jejuni infection. J. Neuroimmunol. 1995; 62: 53-57.Reid E. The hereditary spastic paraplegias. J. Neurol. 1999; 246: 995-1003.Reid E., Dear love A.M., Osborn O. et al. A locus for autosomal dominant «pure» hereditary
spastic paraplegia maps to chromosome 19q 13. Am. J. Hum. Genet. 2000; 66: 728-732.Reid E., Dear/ove A.M., Rhodes M, Rubinsztein D.C. A new locus for autosomal dominant
«pure» hereditary spastic paraplegia mapping to chromosome 12q 13, and evidence for
further genetic heterogeneity. Am. J. Hum. Genet. 1999; 65: 757-763.Reiss J. Polymerase chain reaction and its potential role in clinical diagnostics and research.
J. Intern. Med. 1991; 230: 391-395.Richard /., Broux O., Allamand V. et al. Mutations in the proteolytic enzyme calpain 3 cause
limb-girdle muscular dystrophy type 2A. Cell 1995; 81: 27-40.Richards F.M., Crossey P.A., Phipps M.E. et al. Detailed mapping of germline deletions of
the von Hippel-Lindau disease tumour suppressor gene. Hum. Molec. Genet 1994;
3: 595-598.Richards R.I., Sutherland G.R. Dynamic mutations: a new class of mutations causing human
disease. Cell 1992; 70: 709-712.Richter A., Rioux J.D., Bouchard J.P. et al. Location score and haplotype analysis for the
locus for autosomal recessive spastic ataxia of Charlevoix-Sageuney, in chromosome
region 13q 11. Am. J. Hum. Genet. 1999; 64: 768-775.Ricker K., Grim T., Kock M.C. et al. Linkage of proximal myotonic myopathy to chromosome
3q. Neurology 1999; 52: 170-171.Rider J.A., Rider D.L. Batten disease: past, present and future. Am. J. Med. Genet. 1988;
5 (SupplJ: 21-26.Risch N.J., de Leon D., Ozelius L.J. et al. Genetic analysis of idiopathic torsion dystonia in
Ashkenazi Jews and their recent descent from a small founder population. Nat. Genet.
1995; 9: 152-159.Ritland S.R., Ganjit / Jenkins R.B. Region-specific loss of heterozygosity on chromosome
19 is related to the morphologic type of human glioma. Genes Chromosomes Cancer
1995; 12: 277-282.Rizzo W.B., Carney G, Lin Z. The molecular basis of Sjogren-Larsson syndrome: mutation
analysis of the fatty aldehyde dehydrogenase gene. Am J. Hum. Genet. 1999;
65: 1547-1560.Rizzu P., van Syvieten J.C., Joosse M. et al. High prevalence of mutations in the microtubule-
associated protein tau in a population study of frontotemporal dementia in the Netherlands.
Am. J. Hum. Genet. 1999; 64 414-421.Roa B.B., Garcia C.A., Suter U. et al. Charcot-Marie-Tooth disease type 1A: Association
with a spontaneous point mutation in the PMP22 gene. New Engl. J. Med.
1993; 329: 96-101.
Библиографический указатель 581Roach E.S., Gomez M.R., Northrup H. Tuberous sclerosis consensus conference: revised
clinical diagnostic criteria. J. Child Neurol. 1998; 13: 624-628.RobalcisN.K., Devine Gage E. A., Jenkins E.C. etal. Localization of a human gene homologous
to the PrP gene on the p arm of chromosome 20 and detection of PrP-related antigens in
normal human brain. Biochem. Biophys. Res. Commun. 1986; 140: 758-765.Roberds S.L., Leturcq F., Allamand. V. et al. Missense mutations in the adhalin gene linked
to autosomal recessive muscular dystrophy. Cell 1994; 78: 625-633.Roberts R.G., Bobrov/ M., Bentley D.R. Point mutations in the dystrophin gene. Proc. Natl.
Acad. Sci. USA 1992; 89: 2331-2335.Rocca W.A., Hofinan A., Brayne C. et al. Frequency and distribution of Alzheimer’s disease
in Europe: A collaborative study of 1980-1990 prevalence findings. Ann. Neurol. 1991;
30: 381-390.Rodrigues N.R., Owen N., Talbot K. et al. Deletions in the survival motor neuron gene on
5ql3 in autosomal recessive spinal muscular atrophy. Hum. Mol. Genet. 1995; 4:631-634.Rogaev E.I., Sherrington R., Rogaeva E.A. et al Familial Alzheimer’s disease in kindreds
with missense mutations in a gene on chromosome 1 related to the Alzheimer’s disease
type 3 gene. Nature 1995; 376: 775-778.Ronen G.M., Rosales T.O., Connolly M. et al Seizure characteristics in chromosome 20
benign familial neonatal convulsions. Neurology 1993; 43: 1355-1360.Rosen D.R., Siddique T., Patterson D. et al Mutations in Cu/Zn superoxide dismutase gene
are associated with familial amyotrophic lateral sclerosis. Nature 1993; 362: 59-62.Rosenberg R.N. Autosomal dominant cerebellar phenotypes: the genotype will settle the
issue. Neurology 1990; 40: 1329-1331.Rosenberg R.N. Machado-Joseph disease: an autosomal dominant system degeneration. Mov.
Disord. 1992; 3: 193-203.Rosenberg R.N. Autosomal dominant cerebellar phenotypes: the genotype has settled the
issue. Neurology 1995; 45: 1-5.Rosenberg R.N. DNA-triplet repeats and neurologic disease. New Engl. J. Med. 1996; 335:
1222-1224.Roses A.D. A model for susceptibility polymorphisms for complex diseases: apolipoprotein E
and Alzheimer disease. Neurogenetics 1997; 1: 3-11.Ross C.A. When more is less: pathogenesis of glutamine repeat neurodegenerative diseases.
Neuron 1995; 15: 493-496.Rotig A., de Lonlay P., Chretien D. et al Aconitase and mitochondrial iron-sulphur protein
deficiency in Friedreich ataxia. Nat. Genet. 1997; 17: 215-217.Rotman G., Shiloh Y. ATM: from gene to function. Hum. Mol. Genet. 1998; 7: 1555-1563.Rouleau G.A., Merel P., Lutchman M. et al Alteration in a new gene encoding a putative
membrane-organizing protein causes neuro-fibromatosis type 2. Nature 1993;
363: 515-521.Rowland L.P. What’s in a name? Amyotrophic lateral sclerosis, motor neuron disease, and
genetic heterogeneity. Ann. Neurol. 1998; 43: 691-694.Rubinsztein D.C., Hanlon C.S., Irving R.M. et al ApoE genotypes in multiple sclerosis,
Parkinson’s disease, schwannomas and late-onset Alzheimer’s disease. Mol. Cell. Probes
1994; 8: 519-525.Rubinsztein D C., Leggo J., Coles R. et al Phenotypic characterization of individuals with
30-40 CAG repeats in the Huntington disease (HD) gene reveals HD cases with 36
repeats and apparently normal elderly individuals with 36-39 repeats. Am. J. Hum.
Genet. 1996; 59: 16-22.Ruttledge M.H., AndermannA.A., Phelan C.M. et al Type of mutation in the neurofibromatosis
type 2 gene (NF2) frequently determines severity of disease. Am. .1. Hum, Genet. 1996;
59: 331-342.
582Библиографический указательRuttledge M.H., Narod S.A., Dumanski J.P. et al. Presymptomatic diagnosis for
neurofibromatosis 2 with chromosome 22 markers. Neurology 1993; 43: 1753-1760.Ruttledge M.H., Sarrazin J., Rangaratnam S. et al. Evidence for the complete inactivation of
the NF2 gene in the majority of sporadic meningiomas. Nat. Genet. 1994; 6: 180-184.SainzJ., Huynh D.P., Figueroa K. et al. Mutations of the neurofibromatosis type 2 gene and
lack of the gene product in vestibular schwannomas. Hum. Mol. Genet. 1994; 3: 885-891.Sampson JR., Scahill S.J., Stephenson J.B.P. et al. Genetic aspects of tuberous sclerosis in
the west of Scotland. J. Med. Genet. 1989; 26: 28-31.Samuelsson B., Akesson H.O. Relative fertility and mutation rate in neurofibromatosis.
Hereditas 1988; 108: 169-171.Sanner G., Hagberg B. 188 cases of non-progressive ataxic syndromes in childhood.
Neuropediatrie 1974; 5: 224-235.Sanpei K., Takano H., Igarashi S. et al. Identification of the spinocerebellar ataxia type 2 gene
using a direct identification of repeat expansion and cloning technique, DIRECT. Nat.
Genet. 1996; 14: 277-284.Sarfarazi M., Wijmenga C., Upadhyaya M. et al. Regional mapping of facioscapulohumeral
muscular-dystrophy gene on 4q35 - combined analysis of an international consortium.
Am. J. Hum. Genet. 1991; 51: 396-403.Saugier-Veber P., Munnich A., Bonneau D. et al. X-linked spastic paraplegia and Pelizaeus-
Merzbacher disease are allelic disorders at the proteolipid protein locus. Nat. Genet.
1994; 6: 257-262.Saunders A.M., Hulette C., Welsh-Bohmer K.A. et al. Specificity, sensitivity, and predictive
value of apolipoprotein-E genotyping for sporadic Alzheimer’s disease. Lancet 1996;
348: 90-93.Saunders A.M., Strittmatter W.J., Schmechel D. et al. Association of apolipoprotein E allele
E4 with late-onset familial and sporadic Alzheimer’s disease. Neurology 1993; 43:
1467-1472.Savitslcy K., Bar-Shira A., Gilad S. et al. A single ataxia teleangiectasia gene with a product
similar to PI-3 kinase. Science 1995; 268: 1749-1753.Savukoski M, Klockars T., Holmberg V et al. CLN5, a novel gene encoding a putative
transmembrane protein mutated in Finnish variant late infantile neuronal ceroid
lipofuscinosis. Nat. Genet. 1998; 19: 286-288.Schapira A.H.V. Inborn and induced defects of mitochondria. Arch. Neurol. 1998;.
55: 1293-1296.Schellenberg G.D. Genetic dissection of Alzheimer disease, a heterogeneous disorder. Proc.
Natl. Acad. Sci. USA 1995; 92: 8552-8559.Scheuner D., Eckman C., Jensen M. et al. Secreted amyloid beta-protein similar to that in the
senile plaques of Alzheimer’s disease in increased in vivo by the presenilin 1 and 2 and
APP mutations linked to familial Alzheimer’s disease. Nat. Med. 1996; 2: 864-870.Schmechel D., Saunders A., Strittmatter W et al. Increased amyloid (3-peptide deposition in
cerebral cortex as a consequence of apolipoprotein E genotype in late-onset Alzheimer
disease. Proc. Natl. Acad. Sci. USA 1993: 90: 9649-9653.Schneider J.A., Gearing M., Robbins R.S. et al. Apolipoprotein E genotype in diverse
neurodegenerative disorders. Ann. Neurol 1995; 38: 131-135.Schols L., Vieira-Saec/cer Schols S. et al. Trinucleotide expansion within the MJD1gene presents clinically as spinocerebellar ataxia and occurs most frequently in German
SCA patients. Hum. Mol. Genet. 1995, 4: 1001-1005.Schon E.A., DiMauro S. Mitochondrial diseases. KargerGazette 1994; 58: 2-4Scott W.K., Yamctoka L.H.. Stajich J.M. et cil. The alpha-synuclein gene is not a major risk
factor in familial Parkinson disease. Neurogenetics 1999; 2: 191-192.Segawa M., Hosaka A., Miyagcnva F. et at. Hereditary progressive dystonia with marked
diurnal fluctuation. Adv Neurol. 1976; 14: 215-233.
Библиографический указатель 583Segawa M., Ohmi K., Itoh S. et al. Childhood basal ganglia disease with remarkable response
to L-dopa, «hereditary basal ganglia disease with marked diurnal fluctuation». Chiryo
(Tokyo) 1972; 24: 667-672.Selmaj K., Raine C.S., Canella B. Identification of lymphotoxin and tumor necrosis factor in
MS lesions. J. Clin. Invest. 1991; 87: 949-954.Sepp T., Green A. J, Yates J.R. W. Loss of heterozygosity in tuberous sclerosis hamartomas. J.
Med. Genet. 1996; 33: 962-964.Seri M., Cusano R., Forabosco R et al. Genetic mapping to 10q23.3-q24.2, in a large Italian
pedigree, of a new syndrome showing bilateral cataracts, gastroesophageal reflux, and
spastic paraparesis with amyotrophy. Am. J. Hum. Genet. 1999; 64: 586-593.Serratosa J.M., Gardiner R.M., Lehesjoki A.E. et al. The molecular genetic bases of the
progressive myoclonus epilepsies. Adv. Neurol. 1999; 79: 383-398.Servidei S. Mitochondrial encephalomyopathies: gene mutation. Neuromusc. Disord. 1997;
7: XIII-XVIII.Seshadri S., Drachman D.A., Lippa C.F. Apolipoprotein E s4 allele and the lifetime risk of
Alzheimer’s disease - what physicians know, and what they should know. Arch. Neurol.
1995; 52: 1074-1079.Sgambati M.T., Stolle C., Choyke P.L. et al. Mosaicism in von Hippel-Lindau disease:
lessons from kindreds with germline mutations identified in offspring with mosaic
parents. Am. J. Hum. Genet. 2000; 66: 84-91.Shah A. B., Chernov I., Zhang H.T. et al. Identification and analysis of mutation in the Wilson
disease gene (ATP7B): population frequencies, genotype-phenotype correlation, and
functional analyses. Am. J. Hum. Genet. 1997; 61: 6317-328.ShariefM.K., Henteges R. Association between tumor necrosis factor and disease progression
in patients with multiple sclerosis. New Engl. J. Med. 1992; 327: 272-273.Sharief M.K., McLean B., Thompson E.J. Elevated serum levels of tumor necrosis factor-a in
Guillain-Barré syndrome. Ann. Neurol. 1993; 33: 591-596.Scheffer I.E., Berlcovic S.F. Generalised epilepsy with febrile seizures plus - a genetic disorder
with heterogeneous clinical phenotypes. Brain 1997; 120: 479-490.Scheffer I.E., Phillips H.A., O'Brien C.E. et al. Familial partial epilepsy with variable foci:
a new partial epilepsy syndrome with suggestion of linkage to chromosome 2. Ann.
Neurol. 1998; 44: 890-899.Scheinberg I.H, Sternlieb I. Wilson’s disease. Philadelphia: W. B. Saunders, 1984.Shen M. H., Harper P. S., Upadhyciya M. Molecular genetics of neurofibromatosis type 1
(NF1). J. Med. Genet. 1996; 33: 2-17.Sherrington R., Rogaev E.I., Liang Y. et al. Cloning of a gene bearing missense mutations in
early-onset familial Alzheimer’s disease. Nature 1995; 375: 754-760.Sheu K.-F.R., Brown A.M., Haroutunian V. et al. Modulation by DLST of the genetic risk of
Alzheimer’s disease in a very elderly population. Ann. Neurol. 1999; 45: 48-53.Shields R.W. Limb girdle syndromes. In: Engel A.G., Franzini-Armstrong C. (eds.) Myology,
2nd ed. New York: McGraw-Hill, 1994: 1258-1274.Shiloh Y. Ataxia-teleangiectasia: closer to unraveling the mistery. Eur. J. Hum. Genet. 1995;
3: 116-138.Shimura H., Hattori N.. Kubo S. et., al. Immunohistochemical and subcellular localization of
Parkin protein: absence of protein in autosomal recessive parkinsonism patients. Ann.
Neurol. 1999; 45: 668-672.Shoffner J.M., Wallace D.C. Mitochondrial genetics: principles and practice. Am. J. Hum.
Genet. 1992: 51: 1179-1186.Shoffner J.M., Brown M.D., Torroni A. et al. Mitochondrial DNA variants observed in
Alzheimer disease and Parkinson disease patients. Genomics 1993; 17: 171-184.Siddique T., Deng H.-X. Genetics of amyotrophic lateral sclerosis. Hum. Mol. Genet. 1996; 5:
584 Библиографический указатель1465-1470.Silveira I., Lopes-Cendes I., Kish S. et al. Frequency of spinocerebellar ataxia type 1,
dentatorubropallidoluysian atrophy, and Machado-Joseph disease mutations in a large
group of spinocerebellar ataxia patients. Neurology 1996; 46: 214-218.Singh N.A., Char/ier C., Stauffer D. et al. A novel potassium channel gene, KCNQ2, is
mutated in an inherited epilepsy of newborns. Nat. Genet. 1998; 18: 25-29.Sisodia S.S., Kim S.H., Thinakaran G. Function and dysfunction of the presenilins. Am. J.
Hum. Genet. 1999; 65: 7-12.Sistermans E.A., de Coo R.F., de Wijs I.J., van Oost B.A. Duplication of the proteolipid
protein gene is the major cause of Pelizaeus-Merzbacher disease. Neurology 1998; 50:
1749-1754.Skeie G.O., Pandey J.P., Aarli J.A., Gilhus N.E. TNFA and TNFB polymorphisms in
myasthenia gravis. Arch. Neurol. 1999; 56: 457-461.Skinner P.J., Koshby B.T., Cummings C.J. et al. Ataxin-1 with an expanded glutamine tract
alters nuclear matrix-associated structures. Nature 1997; 389: 971-974.S/ominsky P.A., Markova E.D., Shadrina M.I. et al. A common 3-bp deletion in the DYT1
gene in Russian families with early-onset torsion dystonia. Hum. Mutat. 1999;
14: 269-272.Sonderegger P., Rathjen F.G. Regulation of axonal growth in the vertebrate nervous system by
interactions between glycoproteins belonging to two subgroups of the immunoglobulin
superfamily. J. Cell Biol. 1992; 119: 1387-1394.Spacey S.D., Wood N.W. The genetics of Parkinson’s disease. Cur. Opin. Neurol. 1999;
12: 427-432.Specht L.A., Kunkel L.M. Duchenne and Becker muscular dystrophies. In: Rosenberg R.N.,
Prusiner S.B., DiMauro S. et al. (eds). The molecular and genetic basis of neurological
disease. Boston: Butterworth, 1993: 613-631.Speer M.C., Vance J. M., Grubber J.M. etal. Identification of a new autosomal dominant limb-
girdle muscular dystrophy locus on chromosome 7. Am. J. Hum. Genet. 1999; 64: 556-
562.St George-Hyslop P. H., Tanzi R.E., Pol insky P.J. et al. The genetic defect causing familial
Alzheimer’s disease maps on chromosome 21. Science 1987; 235: 885-890.Steinberger D., Topka H., Fischer D., Müller U. GCH1 mutation in a patient with adult-onset
oromandibular dystonia. Neurology 1999; 52: 877-879.Steinberger D., Weber Y., Korinthenberg R. et al. High penetrance and pronounced variation
in expressivity of GCH1 mutations in five families with Dopa-responsive dystonia. Ann.
Neurol. 1998; 43: 634-639.Steinlein O.K. Gene defects in idiopathic epilepsy. Rev. Neurol. (Paris) 1999; 155: 450-453.Steinlein O.K., Mulley J.C., Propping P. et al. A missense mutation in the neuronal nicotinic
acetylcholine receptor a4 subunit is associated with autosomal dominant nocturnal
frontal lobe epilepsy. Nat. Genet. 1995; 11: 201-203.Stevanin G., Dürr A., Brice A. Clinical and molecular advances in autosomal dominant
cerebellar ataxias from genotype to phenotype and physiopathology. Eur. J. Hum.
Genet. 2000; 8: 4-18.Stevanin G, GiuntiP, DcmdG. et al. De novo expansion of intermediate alleles in spinocerebellar
ataxia 7. Hum. Mol Genet. 1998; 7: 1809-1813.Stewart G.S., Maser R.S.. Stankovic T. The DNA double-strand break repair gene hMREl 1 is
mutated in individuals with an ataxia-teleangiectasia-like disorder. Cell 1999: 577-587.Stögbauer F., Young P.. Kuhlenbciumer G. et al. Hereditary recurrent focal neuropathies:
Clinical and molecular features. Neurology 2000; 54: 546-551.Stojanovic M., Cvetkovic D., Kostic VS. A genetic study of idiopathic focal dystonias.
J. Neurol. 1995; 242: 508-511.
Библиографический указатель 585Stokowski R.P., Cox D.R. Functional analysis of the neurofibromatosis type 2 protein by
means of disease-causing point mutations. Am. J. Hum. Genet. 2000; 66: 873-891.Strittmatter W.J., Roses A.D. Apolipoprotein E and Alzheimer’s disease. Annu. Rev. Neurosci.
1996; 19: 53-77.Strittmatter W.J., Saunders A.M., Schmechel D. et al. Apolipoprotein E: High-avidity
binding to ß-amyloid and increased frequency of type 4 allele in late-onset familial
Alzheimer disease. Proc. Natl. Acad. Sci. USA 1993; 90: 1977-1981.Stumpf D.A., Sokol R., Bettis D. et al. Friedreich’s disease: variant form with vitamin E
deficiency and normal fat absorption. Neurology 1987; 37: 68-74.Syvänen A.-C. From gels to chips: “Minisequencing” primer extension for analysis of point
mutations and single nucleotide polymorphisms. Hum. Mutat. 1999; 13: 1-10.Szepetowski P., Rochette J., Berquin P. et al. Familial infantile convulsions and paroxysmal
choreoathetosis (ICCA): a new neurological syndrome linked to the pericentromeric
region of human chromosome 16. Am. J. Hum. Genet. 1998; 61: 889-898.Takahashi H., Ohama E., Suzuki S. et al. Familial juvenile parkinsonism: clinical and
pathologic study in a family. Neurology 1994; 44: 437-441.Takashima H., Nalcagawa M., Suehara M. et al. Gene for hereditary motor and sensory
neuropathy (proximal dominant form) mapped to 3q 13.1. Neuromusc. Disord. 1999; 9:
368-371.Tanaka H., Endo K., Tsuji S. et al. The gene for hereditary progressive dystonia with marked
diurnal fluctuation (HPD) maps to chromosome 14q. Ann. Neurol. 1995; 37: 405-408.Tang B., Liu C., Shen L. et al. Frequency of SCA1, SCA2, SCA3/MJD, SCA6, SCA7, and
DRPLA CAG trinucleotide repeat expansion in patients with hereditary spinocerebellar
ataxia from Chinese kindreds. Arch. Neurol. 2000; 57: 540-544.Tanzi R.E, Petrukhin K., Chernov I. et al. The Wilson disease gene is a copper transporting
ATPase with homology to the Menkes disease gene. Nat. Genet. 1993; 5: 344-350.Tassin J., Dürr A., Bonnet A.M. et al. Levodopa-responsive dystonia: GTP cyclohydrolase I
or parkin mutations? Brain 2000; 123: 1112—1121.Tassin J., Dürr A., de Broucker T. et al. Chromosome 6-Iinked autosomal recessive early-
onset parkinsonism: linkage in European and Algerian families, extension of the clinical
spectrum, and evidence of a small homozygous deletion in one family. Am. J. Hum.
Genet. 1998; 63: 88-94.Tawil R., Figlewicz D.A., Griggs R.C. et al. Facioscapulohumeral dystrophy: a distinct
regional myopathy with a novel molecular pathigenesis. Ann. Neurol. 1998; 43:279-282.Tawil R., Myers G.J., Weißenbach B., Griggs R.C. Scapuloperoneal syndromes. Absence of
linkage to the 4q35 FSHD locus. Arch. Neurol. 1995; 52: 1069-1072.Taylor J.E., Thomas N.H., Lewis C.M. et al. Correlation of SMNt and SMNc gene copy
number with age of onset and survival in spinal muscular atrophy. Eur. J. Hum. Genet.
1998; 6: 467-474.Telatar M., Wang Z., Udar N. et al. Ataxia-teleangiectasia: mutations in ATM cDNA detected
by protein-truncation screening. Am. J. Hum. Genet. 1996; 59: 40-44.Telenius H., Kremer H.P.H., Theilmann J. etal. Molecular analysis of juvenile Huntington’s
disease: the major influence on (CAG) repeat length is the sex of the affected parent.
Hum. Mol. Genet. 1993; 2: 1535-1540.The Huntington’s Disease Collaborative Research Group. A novel gene containing a
trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes.
Cell 1993; 72: 971-983.Thomas G.R., Forbes, J.R., Roberts E.A. et al. The Wilson disease gene spectrum of
mutations and their consequences. Nat. Genet. 1995; 9: 210-217.Thomson G. Analysis of complex human genetic traits: an ordered notation method and new
tests for mode of inheritance. Am. J. Hum. Genet. 1995 (a); 57: 474-486.Thomson G. Mapping disease genes: family based association studies. Am. J Hum. Genet.
586Библиографический указатель1995 (b); 57: 487-498.Thomson G., Esposito M.S. The genetics of complex disease. T.I.G. 1999; 15 (millenium
issue): M17-M20.Thornton С.Л., Ashizawa T. Getting a grip on the myotonic dystrophies. Neurology 1999;
52: 12-13.Thornton C.A., Griggs R.C., Moxley R.T. Myonotic dystrophy with no trinucleotide repeat
expansion. Ann. Neurol. 1994; 35: 269-272.Thorsby E. Invited anniversary review: HLA associated diseases. Hum. Immunol. 1997; 53:
1-11.Timmerman V, De Jonghe P., Simo/covic S. et a/. Distal hereditary motor neuropathy type II
(distal HMN II): Mapping of a locus to chromosome 12q24. Hum. Mol. Genet. 1996
(a); 5: 1065-1069.Timmerman V, De Jonghe P., Spoelders P. et al. Linkage and mutation analysis of Charcot-
Marie-Tooth neuropathy type 2 families with chromosomes Ip35-p36 and Xql3.
Neurology 1996 (6); 46: 1311-1318.Timmerman V, Ne/is E., De Jonghe P. et al. Hereditary neuropathy. In: Emery A.E.N. (ed.)
Neuromuscular disorders: Clinical and molecular genetics. New York: John Wiley &
Sons, 1998: 487-511.Timmerman V, Nelis E., Van Hit! W. et al. The peripheral myelin protein gene PMP-22 is
contained within the Charcot-Marie-Tooth disease type 1A duplication. Nat. Genet.
1992; 1: 171-175.Timmerman V, Rautensrauss B., Reiter L.T. et al. Detection of the CMT1A/HNPP
recombination hotspot in unrelated patients of European descent. J. Med. Genet. 1997;
34: 43-49.Tinsley J.M., Blake D.J., Zuellig R.A., Davies K.E. Inceasing complexity of the dystrophin-
associated protein complex. Proc. Natl. Acad. Sci. USA 1994; 91: 8307-8313.Tiwari J., Terasaki P. Multiple sclerosis. In: HLA and disease associations. New York:
Springer-Verlag, 1985: 152-167.Tome F, FardeauM. Oculopharyngeal muscular dystrophy. In: Engel A.G., Franzini-Armstrong
C. (eds.) Myology, 2nd ed. New York: McGraw-Hill., 1994: 1233-1245.Tomita H., Nagamitsu S., Wakui K. et al. Paroxysmal kinesigenic choreoathetosis locus maps
to chromosome 16pl 1.2-q 12.1. Am. J. Hum. Genet. 1999; 65: 1688-1697.Tournier-Lasserve E. CACNA1A mutations: hemiplegic migraine, episodic ataxia type 2, and
the others. Neurology 1999; 53: 3-4.Trofatter J.A., MacCollin M.M., Rutter J.L. et al. A novel moesin-, ezrin-, radixin-like gene
is a candidate for the neurofibromatosis 2 tumor suppressor. Cell 1993; 72: 791-800.Trottier Y, Bianca/ana V, Mandel J.-L. Instability of CAG repeats in Huntington’s disease:
relation to parental transmission and age of onset. J. Med. Genet. 1994; 31: 377-382.Tubiello G., Carrera P., Soriani N. et al. Mutational analysis of muscle and brain specific
promoter regions of dystrophin in DMD/BMD Italian patients by denaturating gradient
gel electrophoresis (DGGE). Mol Cell. Probes 1995; 9: 441-446.Uekt K., Ono Y., Henson J.W. et al. CDKN2/pl6 or RB alterations occur in the majority of
glioblastomas and are inversely correlated. Cancer Res. 1996; 56: 150-153.Valente E.M., Bentivoglio A.R., Dixon P.H et al Localization of a novel locus for autosomal
recessive early-onset parkinsonism, PARK6, on human chromosome 1 p35-p36. Am. J.
Hum. Genet. 2001; 68: 895-900.Valente E.M., Warner T.T., Jarman P.R. et al. The role of DYT1 in primary torsion dystonia
in Europe. Brain 1998; 121: 2335 -2339.Valentijn L.J., Bolhius P.A., Zorn I. et at The peripheral myelin gene PMP-22/GAS-3 is
duplicated in Charcot-Marie-Tooth disease type 1A. Nat. Genet. 1992; 1: 166-170.Van Bake/ /., Sepp Т., Ward S. et al. Mutations in the TSC2 gene: analysis of the complete
coding sequence using the protein truncation test (PTT). Hum. Mol. Genet.
Библиографический указатель 5871997; 6: 1409-1414.Van Broeckhoven C.L. Molecular genetics of Alzheimer disease: identification of genes and
gene mutations. Eur. Neurol. 1995; 35: 8-19.Van der Kooi A.J., de Visser M, Barth P.G. Limb girdle muscular dystrophy: reappraisal of
a rejected entity. Clin. Neurol. Neurosurg. 1994; 96: 209-218.Van der Steege G., Grootscholten P.M., van der Vlies P. et al. PCR-based DNA test to confirm
clinical diagnosis of autosomal recessive spinal muscular atrophy. Lancet 1995; 345:
985-986.Van Deutekom J.C., Wijmenga C., van Tienhoven E.A. et al. FSHD associated DNA
rearrangements are due to deletions of integral copies of a 3.2 kb tendemly repeated unit.
Hum. Mol. Genet. 1993; 2: 2037-2041.Van Duijn C.M., Dekker M.C.J., Bonifati V et al. PARK7, a novel locus for autosomal
recessive early-onset parkinsonism, on chromosome lp36. Am. J. Hum. Genet. 2001;
69: 629-634.Van Dyke D.H., Griggs R.C., Murphy M.J., Goldstain M.N. Hereditary myokymia and
periodic ataxia. J. Neurol. Sci. 1975; 25: 109-118.Van Essen A.J., Abbs S., Baiget M. et al. Parental origin and germline mosaicism of deletions
and duplications of the dystrophin gene: a European study. Hum. Genet. 1992;
88: 249-257.Van Essen A.J., Kneppers A.L.J., van der HoutA.H. et al. The clinical and molecular genetic
approach to Duchenne and Becker muscular dystrophy: an updated protocol. J. Med.
Genet. 1997; 34: 805-812.Van Slegtenhorst M., deHoogt R., Hermans C. et al. Identification of the tuberous sclerosis
gene TSC1 on chromosome 9q34. Science 1997; 277: 805-808.Vaughan J.R., Farrer M.J., Wszolek Z.K. et al. Sequencing of the a-synuclein gene in a large
series of cases of familial Parkinson’s disease fails to reveal any further mutations. Hum.
Mol. Genet. 1998; 7: 751-753.Vazza G., Zortea M., Boaretto F. et al. A new locus for autosomal recessive spastic paraplegia
associated with mental retardation and distal motor neuropathy, SPG14, maps to
chromosome 3q27-q28. Am. J. Hum. Genet. 2000; 67: 504-509.Velasco E., Valero C., Valero A. et al. Molecular analysis of the SMN and NAIP genes in
Spanish spinal muscular atrophy (SMA) families and correlation between numbers of
copies of cBCD541 and SMA phenotype. Hum. Mol. Genet. 1996; 5: 257-263.Verhagen W.I.M., Gabreels-Festen A.A.W., van Wensen M. et al. Hereditary neuropathy with
liability to pressure palsies: a clinical, electroneurophysiological and morphological
study. J. Neurol. Sci. 1993; 116: 176-184.Vertinsky Y., Kuliev A. New technique in assisted reproduction. New York:
Wiley-Liss, 1993.- 155 p.Vicart P., Caron A., Guicheney P. et al. A missense mutation in the aB-crystallin chaperone
gene cause a desmin-related myopathy. Nat. Genet. 1998; 20: 92-95.Vidal R., Frangione B.. Rostagno A. et al. A stop-codon mutation in the BRI gene associated
with familial British dementia. Nature 1999; 399: 776-781.Vidal R., Revesz T., Rostagno A. et al. A decamer duplication in the 3’ region of the BRI gene
originates an amyloid peptide that is associated with dementia in a Danish kindred
Proc. Natl. Acad.' Sci. USA 2000; 97: 4920-4925.Vitali T., Sossi V, Tiziano F. et al. Detection of the survival motor neuron (SMN) genes bv
FISH: further evidence for a role for SMN2 in the modulation of disease severity in SMA
patients. Hum. Mol. Genet. 1999; 8: 2525-2532.Vitraneva K., D'Amato E., MiaoJ. et al. Unstable minisatellite expansion causing recessively
inherited myoclonus epilepsy, EPM1. Nat. Genet. 1997; 15: 393-396.Vogel F. Genetics of retinoblastoma. Hum. Genet. 1979; 52 1-54.
588 Библиографический указательWaddy H.M., Fletcher A., Harding A.E., Marsden C.D. A genetic study of idiopathic focal
dystonia. Ann. Neurol. 1991; 29: 320-324.Wadsworth J.D.F., Jackson G., Hill A.F., Co/linge J. Molecular biology of prion propagation.
Curr. Opin. Neurol. 1999; 9: 338-345.Wallace D.C. Mitochondrial diseases: genotype versus phenotype. T.I.G. 1993; 9: 128-133.Wallace M.R., Marchuk D.A., Andersen L.B. etal. Type 1 neurofibromatosis gene: identification
of a large transcript disrupted in three NF1 patients. Science 1990; 249: 181-186.Wallace R.H., Berkovic S.F., Howell R.A. et al. Suggestion of a major gene for familial febrile
convulsions mapping to 8q 13-21. J. Med. Genet. 1996; 33: 308-312.Wallace R.H., Wang D. W., Singh R. et al. Febrile seizures and generalized epilepsy associated
with a mutation in the Na+-channel pi subunit gene SCN1B. Nat. Genet. 1998;
19: 366-370.Wallgren-Pettersson C. Genetics of the nemaline myopathies and the myotubular myopathies.
Neuromusc. Disord. 1998; 8: 401-404.Wang H.-S., Pan Z, Shi W. et al. KCNQ2 and KCNQ3 potassium channel subunits:
molecular correlates of the M-channel. Science 1998; 282: 1890-1893.WangK.W., Villalobo A., Roufogalis B.D. Calmodulin-binding proteins as calpain substrates.
Biochem. J. 1989; 262: 693-706.Wang O., Curren M.E., Splawski I. Et al. Positional cloning of a novel potassium channel
gene: KVLQT1 mutations cause cardiac arrhythmias. Nat. Genet. 1996; 12: 17-23.Ward P.J. Some developments on the affected-pedigree-member method of linkage analysis.
Am. J. Hum. Genet. 1993; 52: 1200-1215.WardK, O’Connell P., Carey J.C. et al. Diagnosis of neurofibromatosis 1 by using tightly
linked, flanking DNA markers. Am. J. Hum. Genet. 1990; 46: 943-949.Warner L.E., Mancias P., Butler IJ. et al. Mutations in the early growth response 2 (EGR2)
gene are associated with hereditary myelinopathies. Nat. Genet. 1998; 18: 382-384.Warner I.E., Svaren J., Milbrandt J., Lupslci J.R. Functional consequences of mutations in
the early growth response 2 gene (EGR2) correlate with severity of human myelinopathies.
Hum. Mol. Genet. 1999; 8: 1245-1251.Weeks D.E., Lange K. The affected-pedigree-member method of linkage analysis. Am. J. Hum.
Genet. 1988; 42: 315-326.Weiler T., Bashir R., Anderson L.V.B. et al. Identical mutation in patients with limb girdle
muscular dystrophy type 2B or Miyoshi myopathy suggests a role for modifier gene(s).
Hum. Mol. Genet. 1999; 8: 871-877.Weiler T., Greenberg C.R., Nylen E. et al. Limb-girdle muscular dystrophy and Miyoshi
myopathy in an aboriginal Canadian kindred map to LGMD2B and segregate with the
same haplotype. Am. J. Hum. Genet. 1996; 59: 872-878.Weiler T., Greenberg C.R., Zelinski T. et al. A gene for autosomal recessive limb-girdle
muscular dystrophy in Manitoba hutterites maps to chromosome region 9q31-q33:
evidence for another limb-girdle muscular dystrophy locus. Am. J. Hum. Genet. 1998;
63: 140-147.Weisgraber K.H., Roses A.D., Strittmatter W.J. The role of apolipoprotein E in the nervous
system. Cur. Opin. Lipidol. 1994; 5: 110-116.Weissman S.M. Genetic bases for common polygenic diseases. Proc. Natl. Acad. Sci. USA
1995; 92: 8543-8544.Wellenreuther R., Kraus J.A.. Lenartz D. et al. Analysis of the neurofibromatosis 2 gene
reveals molecular variants of meningioma. Am. J. Pathol. 1995; 146: 827-832.Whitehouse P.J. Genesis of Alzheimer’s disease. Neurology 1997; 48 (Suppl. 7): S2-S7.Wienecke R.t Konig.4., DeClue J.E. Identification oftuberin, the tuberous sclerosis-2 product:
tuberin possesses specific RaplGAP activity. J. Biol. Chem. 1995: 270: 16409-16414.Wijmenga C., Frants R.R., Brouwer O.F et al. Location of facioscapulohumeral muscular
Библиографический указатель 589dystrophy on chromosome 4. Lancet 1990; 336: 651-653.Wilhelmsen K.C., Blake D.M., Lynch T. et al. Chromosome 12-linked autosomal dominant
scapulohumeral muscular dystrophy. Ann. Neurol. 1996; 39: 507-520.Wilhelmsen K.C., Lynch T., Pavlou E. et al. Localization of disinhibition-dementia-
parkinsonism-amyotrophy complex to 17q21-22. Am. J. Hum. Genet. 1994;
55: 1159-1165.Willems P.J. Dynamic mutations hit double figures. Nat. Genet. 1994; 8: 213-215.Wilson P.J., Ramesh V, Kristiansen A. et al. Novel mutations detected in the TSC2 gene
from both sporadic and familial TSC patients. Hum. Mol. Genet. 1996; 5: 249-256.Wilson R.B., Roof D.M. Respiratory deficiency due to loss of mitochondrial DNA in yeast
lacking the frataxin homologue. Nat. Genet. 1997; 16: 352-357.Windl O., Giese A., Schulz-Schaeffer W. et al. Molecular genetics of human prion diseases in
Germany. Hum. Genet. 1999; 105: 244-252.Wise C.A., Garcia C.A., Davis S.N. et al. Molecular analyses of unrelated Charcot-Marie-
Tooth (CMT) disease suggest a high frequency of the CMT1A duplication. Am. J. Hum.
Genet. 1994; 54: 727-729.Wong A., Yang J., Cavadini P. et al. The Friedreich’s ataxia mutation confers cellular
sensitivity to oxidant stress which is rescued by chelators of iron and calcium and
inhibitors of apoptosis. Hum. Mol. Genet. 1999; 8: 425-430.Wood N. Genes and parkinsonism. J. Neurol. Neurosurg. Psychiatry 1997; 62: 305-309.World Federation of Neurology : Research Committee and Research Group on Huntington's
chorea. Ethical issues policy statement on Huntington’s disease molecular genetic
predictive test. J. Neurol. Sci. 1989; 94: 327-332.Worth P.E., Giunti P., Gardner-Thorpe C. et al. Autosomal dominant cerebellat ataxia type
III: linkage in a large British family to a 7.6-cM region on chromosome 15q 14-21.3.
Am. J. Hum. Genet. 1999; 65: 420-426.Worton R. Muscular dystrophies: diseases of the dystrophin-glycoprotein complex. Science
1995; 270: 755-756.Xiong L., Labuda M, Li D.-S. et al. Mapping of a gene determining familial partial epilepsy
with variable foci to chromosome 22ql 1-q 12. Am. J. Hum. Genet. 1999; 65: 1698-1710.Yamashita /., Sasaki H., Yabe I. et al. A novel locus for dominant cerebellar ataxia (SCA14)
maps to a 10.2-cM interval flanked by D19S206 and D19S605 on chromosome 19q 13.4—
qter. Ann. Neurol. 2000; 48: 156-163.Yates J.R.W., Wehnert M. The Emery-Dreifuss muscular dystrophy database. Neuromusc.
Disord. 1999; 9: 199 (http://www.path.cam.ac.uk/emd).Yershov G., Barsky V, Belgovskiy A. et al. DNA analysis and diagnostics on oligonucleotide
microchips. Proc. Natl. Acad. Sci. USA 1996; 93: 4913-4918.Yoshikai S., Sasaki H., Doh-ura K. et al. Genomic organization of the human amyloid beta-
protein precursor gene. Gene 1990; 87. 257-263.Young R.R. Essential-familial tremor. In: Vinken P.J., Bruyn G.W., Klawans H.L. (eds.)
Extrapvramidal disorders. Amsterdam: Elsevier, 1986: 565-581.Yunis J.J. The chromosomal basis of human neoplasia. Science 1983; 221: 227-236.Zara F, BianchiA., Avanzini G. etal. Mapping of genes predisposing to idiopathic generalized
epilepsy. Hum. Mol. Genet. 1995; 4: 1201-1207.Zara F., Gennaro E., Stabile M. et al. Mapping of a locus for a familial autosomal recessive
idiopathic myoclonic epilepsy of infancy to chromosome 16p 13. Am. J. Hum. Genet
2000; 66: 1552-1557.Zareparsi S.. Kaye J., Camicioli R. et at. Modulation of the age at onset of Parkinson s
disease by apolipoprotein E genotypes. Ann. Neurol. 1997; 42: 655-658.Zbar B., Kishida T, Chen l\ et al. Germline mutations in the Von Hippel-Lindau disease
(VHI.) gene in families from North America, Europe, and Japan. Hum. Mutat. 1996; 8
590 Библиографический указатель348-357.Zeviani M., Servidei S., Gellera C. et al. An autosomal dominant disorder with multiple
deletions of mitochondrial DNA starting at the D-loop region. Nature 1989; 339:309-311.Zhuang Z., Emmert-Buclc M.R., Roth M.J. et al. Von Hippel-Lindau disease gene deletion
detected in microdissected sporadic human colon carcinoma specimens. Hum. Path.
1996; 27: 152-156.Zhuchenlco O., Bailey J., Bonnen P. et al. Autosomal dominant cerebellar ataxia (SCA6)
associated with small polyglutamine expansions in the a1A-voltage-dependent calcium
channel. Nat. Genet. 1997; 15: 62-69.Ziber N., Korczyn A.D., Kahana E. et al. Inheritance of idiopathic torsion dystonia among
Jews. J. Med. Genet. 1984; 21: 13-20.Zoghbi H.Y. The expanding world of ataxins. Nat. Genet. 1996; 14: 237-238.Zubenko G.S., Hughes H.B., StifflerJ.S. Neurobiological correlates of a putative risk allele for
Alzheimer’s disease on chromosome 12q. Neurology 1999; 52: 725-732.Zuberi S.M., Eunson L.H., Spauschus A. et al. A novel mutation in the human voltage-gated
potassium channel gene (KvJ.J) associates with episodic ataxia type 1 and sometimes
with partial epilepsy. Brain 1999; 122: 817-825.Zucman-Rossi J., Legoix P., Der Sarkissian H. et al. NF2 gene in neurofibromatosis type 2
patients. Hum. Mol. Genet. 1998; 7: 2095-2101.
С.Н. Иллариошкин, И.А. Иванова-Смоленская, Е.Д. МарковаДНК-диагностика и медико-генетическое
консультирование в неврологииРук. научно-информ. отдела, канд. мед. наук A.C. Макарян
Главный редактор, канд. мед. наук Д. Д. Проценко
Ответственный за выпуск Н.В. Лодыгина
Компьютерная верстка Л.С. Притчина
Обложка Д.Ю. РожковИзд. лиц. № 064889 от 24.12.96. Подписано в печать 13.05.2002.
Формат 84x108/32 , Печать офсетная. Бумага офсетная.
Объем 18,5 псч.л. Тираж 1000 экз. Заказ Л» 1204ООО “Медицинское информационное агентство”. 119435 Москва,
М. Трубецкая ул., д.8 (ММА им. И.М. Сеченова), комн. 733.
Тел./факс 245-86-20, 242-91-10. e-mail: miapubl@mail.ru,
hllp://www.medagency.ruМосковская типография № 2
Министерства Российской Федерации
по делам печати, телерадиовещания и средств
массовых коммуникаций (МПТР России).Тел.: 282-24-91. 129085, Москва, пр. Мира, 105.ISBN 5-89481 - 1О0-77 9 5 8 948 1 10 0 9