















































































































































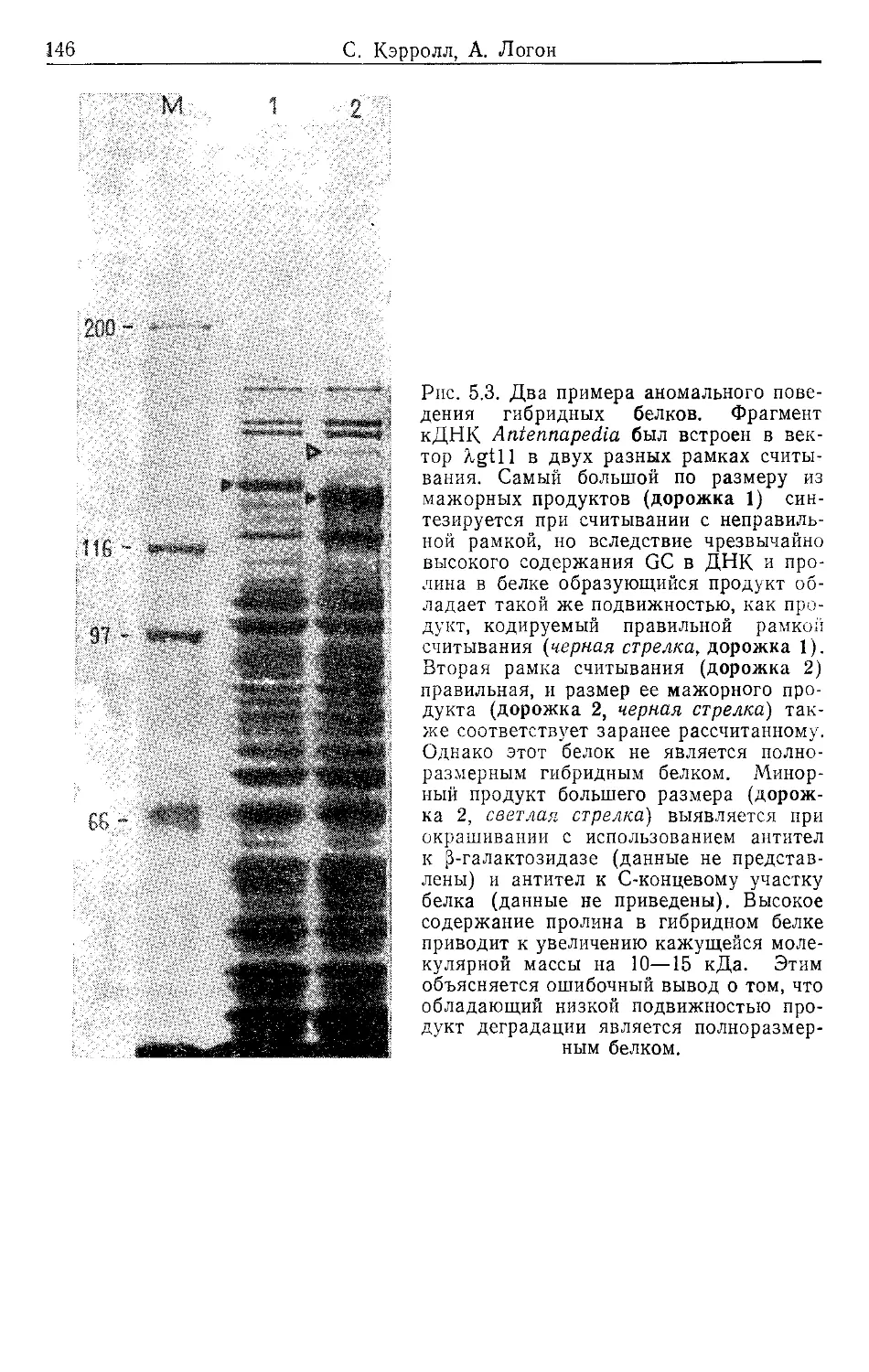



























































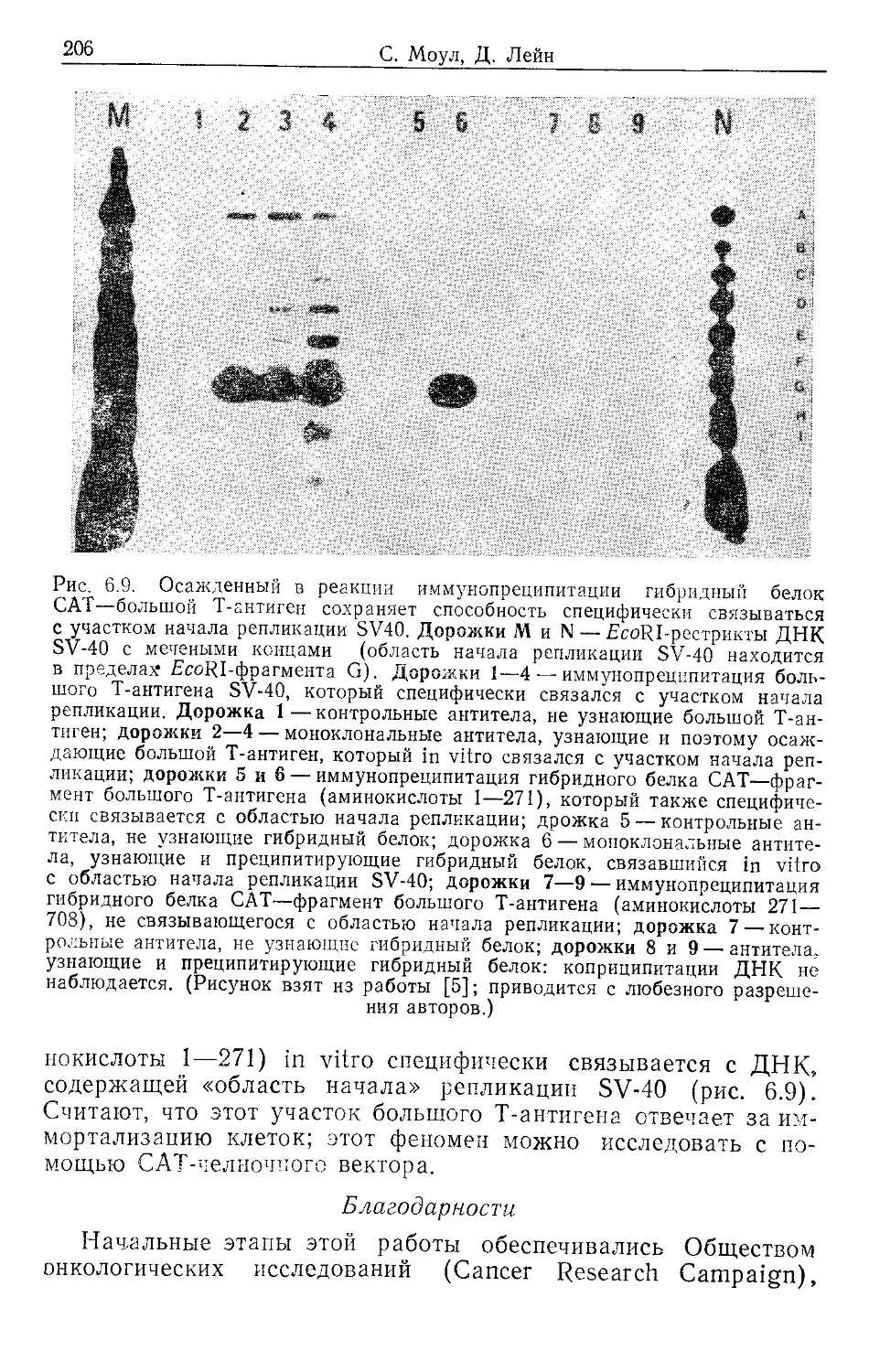





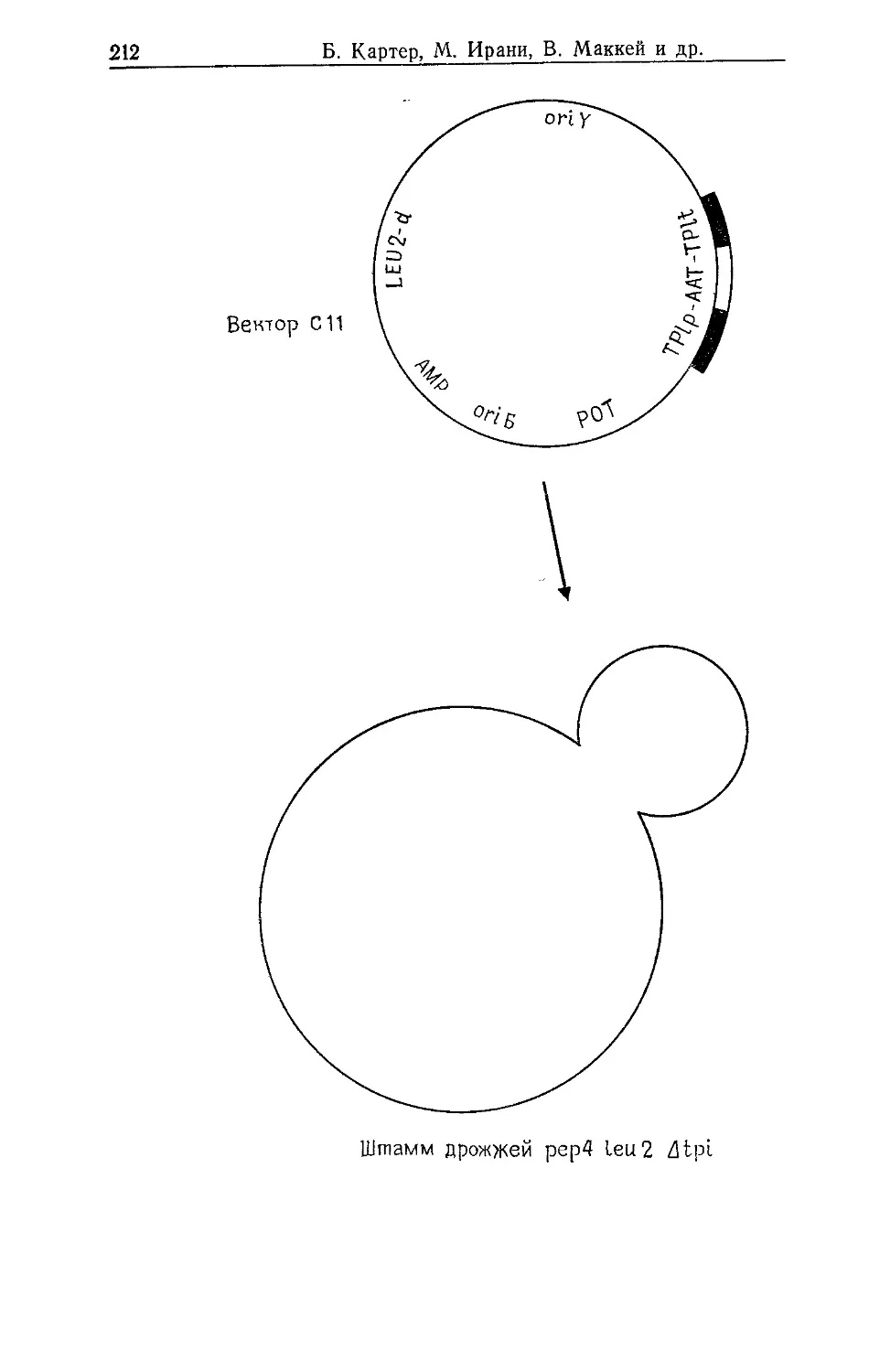









































































































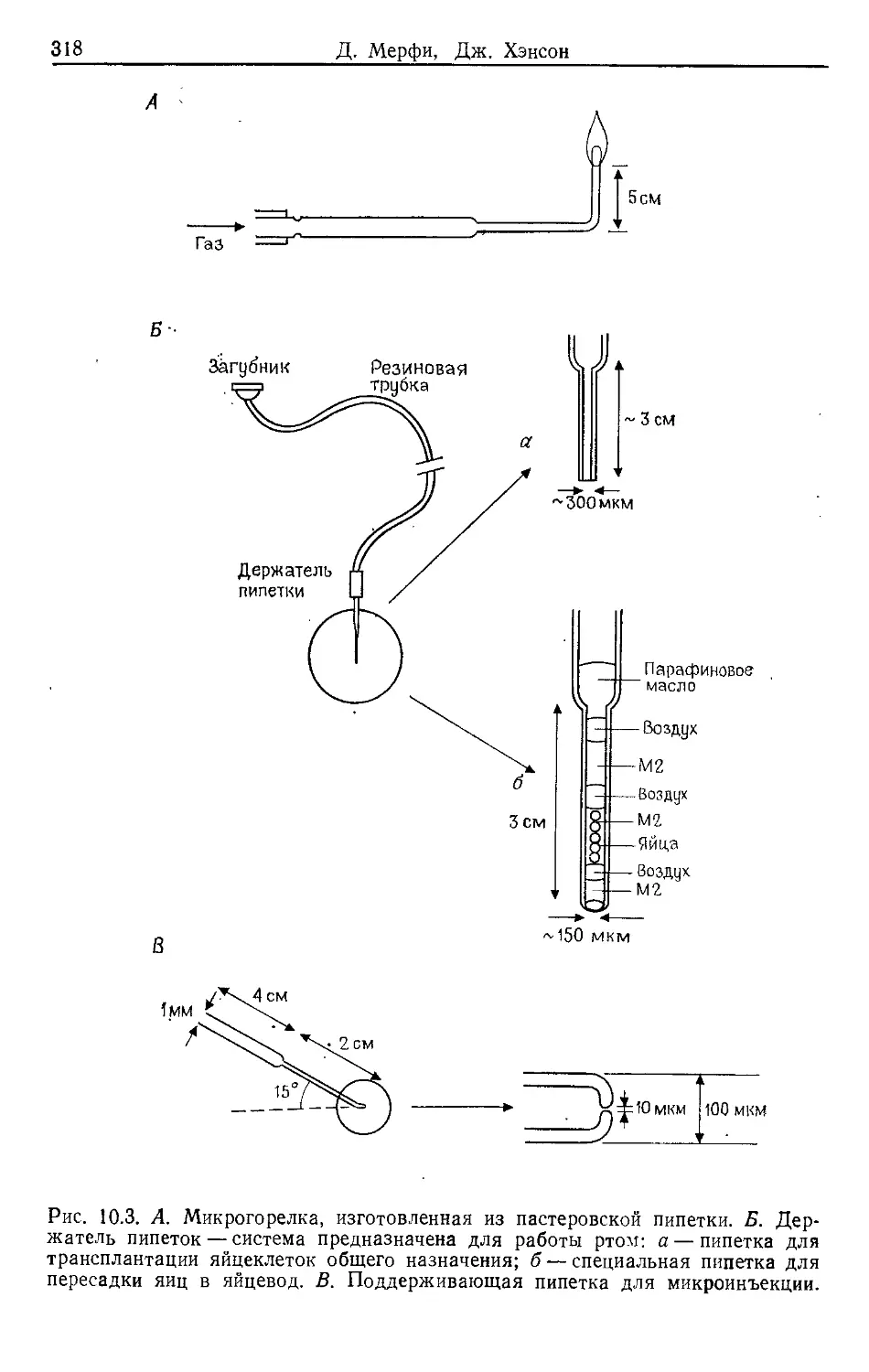















































Author: Гловер Д.
Tags: общая генетика общая цитогенетика иммуногенетика эволюционное учение видообразование филогенез генетика молекулярная биология биотехнологии
ISBN: 5-03-001270-2
Year: 1989




DNA cloning
Volume III
A PRACTICAL APPROACH
Edited by D. M. Glover
Cancer Research Campaign, Eukaryotic
Molecular Genetics Research Group,
Department of Biochemistry, Imperial
College of Science and Technology,
London SW7 2AZ, UK
IRL Press
Oxford. Washington DC
b 9 2 2 2 /,
Новое в клонировании ДНК.
Методы
Под редакцией
Д. ГЛОВЕРА
Перевод с английского
канд. биол. наук П. Л. ИВАНОВА,
канд. биол. наук М. И. ОВАДИС,
канд. биол. наук Т. Ю. ПЕРЕСЛЕНИ
под редакцией
канд. биол. наук П. Л. ИВАНОВА
Москва «Мир» 1989
ББК 28.04
Н74
УДК 575 + 577.2
Новое в клонировании ДНК. Методы: Пер. с англ./Под
Н74 ред. Д. Гловера. — М.: Мир, 1989. — 368 с., ил.
ISBN 5-03-001270-2
Книга авторитетного коллектива авторов из Англии, США и ФРГ — третий
том практического руководства по молекулярной биологии и генной инженерии.
Описана техника использования различных векторов, новые эукариотические экс-
прессирующие системы, способы выделения некоторых продуктов экспрессии.
Два первых тома руководства выпущены издательством «Мир» в 1988 г. (в одной
книге).
Для молекулярных биологов, генетиков, биотехнологов.
и
1903010000—445
--------------95—89
041(01)—89
ББК 28.04
Редакция литературы по биологии
НАУЧНАЯ •
им. И. И.
КПИСИуГо
ISBN 5-03-001270-2 (русск.)
ISBN 1-85221-049-4 (англ.)
© 1987 IRL Press Limited
© перевод на русский язык. Ива-
нов П. Л., Овадис М. И., Пересле-
ни Т. Ю„ 1989
ОТ РЕДАКТОРА ПЕРЕВОДА
В минувшем году вышел в свет в русском переводе сборник
«Клонирование ДНК» (М., Мир, 1988), посвященный молеку-
лярной и физико-химической биологии.
Вышедшая в оригинале в двухтомном варианте книга «Кло-
нирование ДНК» привлекла внимание исследователей не как
вспомогательный материал, а как самостоятельный научный
труд, включающий методические разработки самого высокого
уровня.
Перевод третьего тома этого издания мы предлагаем совет-
скому читателю под названием «Новое в клонировании ДНК».
Настоящая книга представляет собой сборник методических
разработок, охватывающий широкий спектр современных под-
ходов к молекулярному клонированию и экспрессии генетиче-
ского материала. Сборник включает десять самостоятельных
глав, написанных авторами, работающими в ведущих медико-
биологических центрах США, Великобритании и ФРГ.
Поскольку в своем предисловии к английскому изданию
Д. Гловер раскрывает замысел книги, а также ее структуру
и логику, нет необходимости в разверн'утбм:.комментарии. Тем
не менее я хочу отметить следующее. Сейчас- уже можно утвер-
ждать, что методическая литература, во всяком случае в обла-
сти молекулярной биологии и биотехнологии, оформилась в са-
мостоятельный жанр научной литературы. Этот жанр, хотя
и имеет свои традиции, весьма неоднороден, и прежде всего
это касается формы изложения и подачи материала. На одном
полюсе — получившие широкое распространение методические
разработки ряда зарубежных фирм, представляющие собой уз-
коцелевые технологические описания-методики (то, что назы-
вают «know-how»), которые, естественно, ориентированы на
использование продукции собственной фирмы, но при этом
несут и много независимой информации, полезной и нужной в ла-
бораторной практике. На другом же полюсе — энциклопедиче-
ские издания, охватывающие большое число методов и предла-
5
6
От редактора перевода
тающие читателю развернутые теоретические экскурсы по каж-
дому освещаемому вопросу. Лабораторное руководство «Новое
в клонировании ДНК» относится именно к этой последней кате-
гории, составляя, на наш взгляд, гармоничный тандем с уже
переведенным первым сборником данной книги. Хочется на-
деяться, что новая книга будет по достоинству оценена иссле-
дователями, работающими в области молекулярной биологии
и генетики.
Книгу переводили: П. Л. Иванов (предисловие и гл. 7—10),
М. И. Овадис (гл. 3 и 4) и Т. Ю. Переслени (гл. 1, 2, 5 и 6).
П. Иванов
ПРЕДИСЛОВИЕ
Мы выпускаем в свет третий том нашей серии, посвященной
молекулярному клонированию и генноипженерному манипули-
рованию с ДНК — факт, который, безусловно, служит еще одним
свидетельством того, что данные методические подходы за-
нимают теперь центральное место в экспериментальной моле-
кулярной биологии. Серия была запланирована как продолже-
ние и дополнение уже существующих методических пособий по
молекулярной биологии и генной инженерии, в частности попу-
лярной книги Т. Маниатиса, Э. Фрича и Дж. Сэмбрука (Т. Ма-
niatis, Е. Fritsch, J. Sambrook. Molecular cloning, Cold Spring
Harbor Laboratory Press, New York, 1982)*.
Книга этих авторов — лабораторное руководство, охваты-
вающее практически все основные методические приемы техно-
логии рекомбинантных ДНК, и неудивительно, что на него
ссылаются в своих работах большинство авторов как данного
тома, так и двух первых томов.
Первый том нашей серии посвящен уже разработанным или
разрабатываемым в настоящее время системам молекулярного
клонирования в Е. coli. Второй том2 включает описание других
систем хозяин/вектор, используемых наряду с системами на
основе Е. coll для клонирования и экспрессии генов в прокарио-
тических и эукариотических клетках. Содержание третьего тома
весьма разнообразно; представленные здесь методические раз-
работки охватывают широкий спектр современных подходов к
молекулярному клонированию и экспрессии генетического ма-
териала. Как и при знакомстве с любыми узкопрофессиональ-
ными изданиями, рассчитанными на достаточно подготовленных
специалистов, некоторым нашим читателям наверняка придет-
ся поначалу обратиться к книгам более общего характера.
Я по-прежнему рекомендую: Watson, Tooze, Kurtz «Recombi-
nant DNA: A Short Course» (Scientific American Books, New
York, 1983)3; Old, Primrose «Principles of Gene Manipulation»
7
8
Предисловие
(Blackwell, Oxford, 1985), а также свою книгу «Gene Cloning:
The Mechanics of DNA Manipulation» (Chapman and Hall, 1984).
Первая глава данного тома касается использования плаз-
мидных векторов, имеющих в своем составе высокоспецифич-
ные промоторы РНК-полимераз некоторых бактериофагов. Та-
кие векторные молекулы позволяют получать радиоактивно
меченные зонды, которые благодаря своим уникальным свойст-
вам все шире применяются сейчас в исследованиях структуры
и функций нуклеиновых кислот. Подобные системы на основе
космидных векторов рассматриваются во второй главе. Эти век-
торы разработаны для того, чтобы сделать менее трудоемкими
«прогулки» по хромосомам высших организмов. Фаговые про-
моторы здесь расположены таким образом, что синтезируемый
радиоактивно меченный зонд соответствует концевой области
клонированной ДНК, а это облегчает поиск клонов, содержа-
щих перекрывающиеся сегменты ДНК.
Альтернативные методы скрининга космидных библиотек,
описанные в гл. 3, предполагают селекцию космидных клонов
с использованием феномена гомологичной рекомбинации in vi-
vo. Остальные главы книги посвящены вопросам, связанным
с экспрессией клонированных генов. Для многих белков мле-
копитающих удалось осуществить высокопродуктивную внутри-
клеточную экспрессию в Е. coll. Однако гетерологические бел-
ки, локализующиеся в цитоплазме, часто образуют трудно
растворимые агрегаты, что значительно осложняет получение
нативного продукта. В гл. 4 описаны эффективные способы
выделения активных растворимых продуктов из нерастворимых
белков цитоплазмы Е. coli. Вероятность деградации специфиче-
скими бактериальными протеиназами многих эукариотических
белков, синтезируемых в Е. coli, может быть существенно сни-
жена, если их экспрессировать в виде гибридных белков. Такие
составные белки, в которых бактериальный компонент обычно
представлен [3-галактозидазой, можно использовать в качестве
иммуногенов для получения антисыворотки и моноклональных
антител к клонированному эукариотическому белковому доме-
ну. Эти вопросы рассматриваются в двух главах — одна посвя-
щена получению поликлональной антисыворотки, а другая —
методам гибридной технологии. В последующих главах книги
описаны современные эукариотические экспрессирующие систе-
мы: в гл. 7 — дрожжевая, далее в трех главах — системы на
основе культивируемых клеток млекопитающих и трансгенные
животные. В частности, описана система экспрессии с исполь-
зованием векторов, которые несут гены, обеспечивающие воз-
можность их индуцибельной амплификации; это позволяет
снимать токсическое действие антибиотиков, введенных в куль-
туральную среду. Клонированные в таком векторе гены также
Предисловие
9
амплифицируются, что приводит к высокому выходу экспресси-
руемых продуктов.
Ретровирусные векторы, описанные в предпоследней главе,
находят сейчас все более широкое применение в практике моле-
кулярно-биологических исследований. Мы постарались не по-
гружаться в узкоспециальные проблемы, связанные с ретрови-
русными векторами, а рассмотреть принципиальные вопросы,
касающиеся конструирования и использования подобных
систем.
Заключительная глава книги посвящена одному из основных
на сегодняшний день экспериментальных подходов, позволяю-
щих вводить новые гены в целый многоклеточный организм
(в нашем случае это мышь), — микроинъекции ДНК в оплодо-
творенную яйцеклетку. Тем не менее этот подход — лишь один
из нескольких возможных путей решения данной задачи и, ве-
роятно,. альтернативные экспериментальные приемы найдут от-
ражение в других лабораторных руководствах.
Итак, очевидно — и это явственно прослеживается в структу-
ре данной серии,— что технология рекомбинантных ДНК вно-
сит свой вклад практически во все области биологического
поиска. Однако книги способны отразить лишь текущее состоя-
ние дел, и поэтому мы не претендуем на исчерпывающий охват
того методического арсенала, который продолжает непрерывно
совершенствоваться и расширяться. Вместе с тем мне хочется
надеяться, что данная книга сможет высветить принципиальные
направления в развитии экспериментальной базы молекуляр-
ной биологии. Свидетельством ее успеха будет ее место на ра-
бочем столе молекулярного биолога. Я надеюсь, что так слу-
чится и что описанные нами методы окажутся полезными иссле-
дователям, экспериментирующим в самых разных областях
молекулярной биологии.
В заключение — и это наиболее значимая часть мною ска-
занного— я хотел бы поблагодарить всех авторов, вложивших
свой труд в подготовку данной книги.
Дэвид М. Гловер
1 Имеется перевод: Маниатис Т„ Фрич Э., Сэмбрук Дж. Методы генетиче-
ской инженерии. Молекулярное клонирование. — М.: Мир, 1984.
2 В русском переводе два первых тома вышли объединенными: Клониро-
вание ДНК (под ред. Д. Гловера). — М.: Мир, 1988.
3 Имеется перевод: Уотсон Дж., Туз Дж., Курц Д. Рекомбинантные
ДНК. — М.: Мир, 1986.
СПИСОК АВТОРОВ
С. R. Bebbington
Celltech Ltd, 244—250 Bath Road, Slough SL1 4DY, UK
А. M. C. Brown
Department of Microbiology and Immunology, HSE 407 Uni-
versity of California Medical Center, San Francisco, CA 94143,
USA
S. B. Carroll
Laboratory of Molecular Biology, University of Wisconsin —
Madison, 1525 Linden Drive, Madisson, WI 53706, USA
B. L. A. Carter
ZymoGenetics Inc., 2121 North 35th Street, Seattle, WA 98103,
USA
J. Hanson
National Institute of Medical Research, The Ridgeway, Mill
Hill, London NW7 1AA, UK
С. C. G. Hentschel
Celltech Ltd, 244—250 Bath Road, Slough SL1 4DY, UK
M. Irani
ZymoGenetics Inc., 2121 North 35th Street, Seattle, WA 98103,
USA
I. Jackson
MRC Clinical and Population Cytogenetics Unit, Western Ge-
neral Hospital, Crewe Road, Edinburgh, UK
D. P. Lane
Imperial Cancer Research Fund, Clare Hall Laboratories,
Blanche Lane, South Mimms, Potter’s Bar, Herts EN6 3LD,
UK
A. Laughon
Laboratory of Genetics, University of Wisconsin — Madison,
445 Henry Mall, Madison, WI 53706, USA
H. Lehrach
European Molecular Biology Laboratory, Postfach 10.2209, 69
Heidelberg, FRG
Список авторов
11
Р. F. R. Little
Department of Biochemistry, Imperial College, London SW7
2AZ, UK
V. L. MacKay
ZymoGenetics Inc., 2121 North 35th Street, Seattle, WA98T.3,
USA
F. A. O. Marston.
Celltech Ltd, 244—250 Bath Road, Slough SL1 4DY, UK
5. E. Mole
Imperial Cancer Research Fund, Clare Hall Laboratories,
Blanche Lane, South Mimms, Potter’s Bar, Herts EN6 3LD,
UK
D. Murphy
National Institute of Medical Research, The Ridgeway, Mill
Hill, London NW7 1AA, UK
A. Poustka
European Molecular Biology Laboratory, Postfach 10.2209, 60
Heidelberg, FRG
R. L. Seale
ZymoGenetics Inc., 2121 North 35th Street, Seattle, WA 98103,
USA
M. R. D. Scott
Department of Microbiology and Immunology, HSE 407 Uni-
versity of California Medical Center, San Francisco, CA 94143,
USA
A. V. Sledziewsky
ZymoGenetics Inc., 2121 North 35th Street, Seattle, WA 98103,
USA
R. A. Smith
ZymoGenetisc Inc., 2121 North 35th Street, Seattle, WA 98103,
USA
ГЛАВА 1
ПРИМЕНЕНИЕ ПЛАЗМИД, СОДЕРЖАЩИХ
ПРОМОТОРЫ, СПЕЦИФИЧЕСКИЕ ДЛЯ ФАГОВЫХ
РНК-ПОЛИМЕРАЗ
Питер Ф. Р. Литтл и йэн Дж. Джексон
(Peter F. R. Little, Ian I. Jackson)
1.1. Краткое содержание главы
В этой главе рассматриваются методы синтеза и использо-
вания в экспериментальных целях РНК, полученной in vitro
с помощью РНК-полимераз фагов SP6, Т7 и ТЗ. Основное вни-
мание мы уделили использованию радиоактивных РНК-зондов
для рутинного анализа нуклеиновых кислот и не пытались из-
ложить, кроме как в самых общих чертах, более специальные
аспекты их применения, например для гибридизации in situ,
в качестве смысловой мРНК или трансляционных матриц. По
таким вопросам мы постарались дать ссылки на ключевые ра-
боты, которые позволят хорошо разобраться в технических воз-
можностях этих методов.
1.2. РНК-попимеразы бактериофагов
1.2.1. Основные положения
Уже давно известно, что многие бактериофаги Е. coli коди-
руют РНК-полимеразы, способные взаимодействовать лишь со
специфическими промоторами фаговой ДНК- Фаги Т7 и ТЗ —
первые тому примеры [1], а относительно недавно полимераза
с подобными свойствами была обнаружена и у фага SP6 Sal-
monella typhhnurium [2].
Фаговые РНК-полимеразы во многих отношениях отлича-
ются от полимераз клетки-хозяина. Они обычно имеют неболь-
шую молекулярную массу (90—100 000 дальтон), представляют
собой мапомеры и предъявляют весьма ограниченные, но тем
не менее высокоспецифические требования к промотору. В про-
тивоположность этому РНК-полимераза Е. coli представляет
собой большую гетеромультимерную молекулу, способную ини-
циировать синтез РНК с большого числа премоторных и подоб-
ных промоторным последовательностей [3]. На практике такие
различия дают возможность использовать фаговые РНК-поли-
12
Плазмиды, несущие промоторы для фаговых РНК-полчмераз 13
меразы для инициации синтеза РНК in vitro и получения спе-
цифических одноцепочечных молекул РНК. С помощью РНК-
полимеразы Е. coll это сделать нелегко.
Первым ферментом, широко использовавшимся для синтеза
РНК-зондов, стала РНК-полимераза фага SP6 [4]. Это в ос-
новном было обусловлено исключительно высокой стабильно-
стью фермента и возможностью получать его в больших коли-
чествах с помощью простых методов — в отличие от ферментов
фагов Т7 и ТЗ. Однако генетическая организация фагов Т7 и
ТЗ исследована достаточно полно, что дало возможность полу-
чить их ферменты путем экспрессии клонированных генов,
и сейчас они легко доступны. Принцип использования фаговых
РНК-полимераз идентичен для всех трех ферментов (рис. 1.1).
Обычно полимеразы применяются для транскрипции линейных
матриц, но существует и другой подход, основанный на таком
природном свойстве этих ферментов, как преждевременная тер-
минация цепи в присутствии низкой концентрации нуклеозид-
трифосфатов. Достоинство этого подхода состоит в том, что он
не требует линейной матрицы, однако получаемые при этом
молекулы-зонды имеют разную длину, что делает их непригод-
ными для работы при использовании приводимых здесь ме-
тодик.
1.2.2. В чем преимущество РНК-зондов?
РНК-зонды обладают всеми преимуществами одноцепочеч-
ных ДНК-зондов: это отсутствие конкурентной гибридизации
молекул зонда друг с другом, высокая удельная радиоактив-
ность по сравнению с продуктом ник-трансляции и не представ-
ляющее трудностей определение длины молекул. Дополнитель-
ное преимущество этих зондов по сравнению с одноцепочечной
ДНК — большая простота их получения. Транскрипты можно
отделить от матрицы путем ее ДНКазного расщепления и фе-
нольной экстракции, тогда как получение одноцепочечных
ДНК-зондов требует более трудоемкой процедуры выделения
их из геля. В то же время РНК-зонды в большей степени под-
вержены деградации, чем соответствующие им ДНК-зонды, и
об этом нельзя забывать. Самая распространенная проблема,
встающая при использовании РНК-зондов, — неоднозначность
интерпретации в случае получения отрицательного результата.
1.2.2.1. Уникальные свойства РНК-зондов
РНК-зонды могут быть использованы вместо ДНК-зондов
во всех случаях, причем обычно с минимальными модификация-
ми методики. Большая стабильность РНК-РНК-дуплексов мо-
14
П. Литтл, И. Джексон
г кДНК Г
d Sp» г «ДНК г h , '
________£,-» г __________________р h h
г 5|~» г t г, h
У РНК
Рис. 1.1. Принципы использования вектороз, содержащих последовательности
фаговых полимераз, а. Нерекомбинантный вектор. Стрелка у буквы S обозна-
чает транскрипционный промотор SP6, с которого начинается считывание по-
лилинкера с множественными сайтами ктонироваппя; сайты расщепления для
двух рестриктаз обозначены г в h. б. Фрагмент кДНК (или любой другой
ДНК) с соответствующими липкими концами встроен в сайт г вектора, в. Ре-
комбинантная молекула расщепляется по сайту А; образуется фрагмент, вклю-
чающий промотор, кДНК. и сайт терминации h на конце, г. Фрагмент матрицы
с сайтом А, в котором будет происходить терминация «сквозной» РНК-полиме-
разной реакции, д. При инкубации матрицы с соответствующей РНК-полиме-
разой и иуклеотидтрифосфатамп (NTP) образуются РНК-транскрипты, колли-
неарные участку ДНК от промотора S до сайта терминации А. Транскрипты
можно отделить от матрицы воздействием ДНКазы.
жет повысить вероятность артефактов при гибридизации (не-
специфическая гибридизация зонда с рРНК в «нозерн»-анализе
может оказаться серьезной проблемой).
Протекторный эффект при использовании РНК-зондов
в опытах по защите от воздействия РНКазы равноценен наблю-
даемому эффекту для одноцепочечпых ДНК-зондов [10]; РНК-
зонды пригодны также при использовании нуклеазы S1 или
нуклеазы из проростков золотистой фасоли. Пои гибридизации
в растворе РНК-зонды будут защищать от нуклеазного расщеп-
Плазмиды, несущие промоторы для фаговых РНК-полимераз 15
ления только высокогомологичные им последовательности; де-
градация же некоторых внутренних участков, где гомология
прерывается, будет приводить к образованию фрагментов ха-
рактерной длины. Это позволяет избирательно изучать интере-
сующие последовательности в присутствии других, родственных
последовательностей. Так был исследован специфический
транскрипт одной из последовательностей «гомео-бокса» в при-
сутствии других последовательностей этого семейства [11],
были выявлены различия у транскриптов гена коллагена ти-
па II у человека и его гомолога у мыши [12], а также была
обнаружена al-антитрипсиновая мРНК человека на фоне про-
дуктов гомологичного гена мыши [G. Kesley, R. Lovell-Badge,
личное сообщение].
Чувствительность метода обнаружения РНК в эксперимен-
тах по защите от РНКазы очень высока. В работе Мелтона
и др. [5] в простой модельной системе было выявлено 0,5 пкг
глобиновой мРНК- Это эквивалентно одному транскрипту из
106 в стандартном анализе 40 мкг суммарной РНК. Цинн и
др. [13] сообщают о такой же чувствительности метода при
исследовании транскриптов гена p-интерферона в препаратах
суммарной клеточной РНК. Подобная чувствительность, позво-
ляющая обнаруживать один транскрипт из миллиона (это со-
ставляет менее одного транскрипта на клетку), приближается
к пределу чувствительности «Нозерн»-блот-гибридизации. О’Хэр
и др. [14] с помощью «Нозерн-гибридизации показали, что
содержание транскрипта гена white в клетках Drosophila почти
в 10 000 раз меньше, чем содержание транскрипта активного
гена. Однако для такого анализа потребовалось 2 мкг или
более poly-(А)+-РНК (что эквивалентно примерно 100—200мкг
суммарной РНК) по сравнению с 20—40 мкг суммарной РНК,
необходимыми для анализа, основанного на протекторном
эффекте. Применение РНК-зондов для гибридизации в растворе
позволяет обнаруживать специфические мРНК, проводить
их количественную оценку при наличии небольшого количе-
ства материала и быстро проанализировать большое число
проб.
Одно важное преимущество метода с использованием про-
текторного эффекта в РНКазной реакции по сравнению с лю-
бым другим методом состоит в том, что он позволяет точно оп-
ределить в препарате содержание РНК, гомологичной зонду.
Описание метода и пример его использования приведены
в разд. 1.4.3. Обратите внимание на то, что метод дает абсолют-
ные цифры (количество молекул на клетку), а не относитель-
ную величину, полученную путем сравнения с концентрацией
некой стандартной мРНК, например мРНК актина.
16
П. Литтл, И. Джексон
RVIId7 SP6 250 М Н Bg Р В R
pSP62 SP« 42 М Н Р S X В Sm Sa R
pSP64 SP6 5 М Н Р S X В Sm Sa
pSPfcs SP6 ' 9М R Sa Sm В X S PH
pSP64dt SP6 2М S X В Sm Sa R
pGEM3 SP6 7м H Sp P S X В Sm К Sa R 8МТ7
pT7-l Т7 6 м R Sa Sm В X S P H
BLUESCRIBE i ТЗ 9 М H Sp P S X В Sm К Sa R 5МТ7
BLUESCRIPT ТЗ 17 М К A Xh S С H R5 R P Sm, .,. ,B Se Xb N E Sa2 Bx Sa 1IMT7
Рис. 1.2. Векторы каждого вектора для клонирования приведены только содержат промоторы SP6, сайты, используемые для Т7 и ТЗ. Для клонирования,
а расстояние между сайтом инициации транскрипции и началом полилинкера
дано в числе нуклеотидов (н). Структура RVlld7 детально описывается в ра-
боте [19], pSP62 — в работе [4], pSP64 и pSP65 — в работе [5]. В работе [6]
приведены детальные данные о строении еще восьми плазмид, четыре из кото-
рых содержат промоторы SP6 и Т7 и много добавочных других сайтов клони-
рования. Структура pSP64dl приводится в работе [7]. Транскрипты, получен-
ные на этой плазмиде, имеют очень короткую лидерную последовательность.
Плазмида pGEM3 была сконструирована и производится фирмой Promega
Biotech (Madison, USA), pT71 была сконструирована и производится фирмой
United States Biochemical Corp. (Cleveland, USA). Плазмиды bluescribe и blue-
script сконструированы и производятся фирмой Stratagene (San Diego, USA).
Последние плазмиды имеют то преимущество, что им свойственна «-компле-
ментация гена lacZ, т. е. цветная реакция на вставку, подобно плазмидам серии
pUC. Bluescript может быть получена также и в виде одноцепочечной ДНК,
так как она содержит область начала репликации М13. Сокращения, исполь-
зуемые при обозначении сайтов рестрикции: А — Apall, В — BamHI; Bg —
BgLII; Вх — BsfXl; C — Clal; E — Eaglll; H — Hind\H; K — Kpnl; N — Noll;
P — PsH; R — EcoRl; S — Sall; Sa — Sad; Sa2 — Sadi; Sm — Smal; Se—
Spel; Sp — Sphl; X — Xmal; Xb — Xbal; Xh — Xhol. [На рисунке ошибка:
вместо буквы «м» следует читать букву «н». — Ред.]
1.2.3. Выбор векторов
В настоящее время известно множество векторов, содержа-
щих промоторы для РНК-полимераз фагов SP6, Т7 и ТЗ в ком-
бинации с различными последовательностями. Данные о наибо-
лее распространенных плазмидиых векторах приведены на
рис. 1.2. Выбор вектора — это в известной мере дело вкуса ис-
следователя, хотя при этом могут учитываться и некоторые
другие факторы.
1.2.3.1. Выбор клонируемого сайта
Этот выбор зависит от природы клонируемой последователь-
ности ДНК, однако следует с осторожностью относиться к ре-
стриктазпым сайтам, расположенным поблизости от сайта ини-
Плазмиды, несущие промоторы для фаговых РНК-полимераз 17
циации синтеза РНК, учитывая, что в некоторых случаях РНК-
полимеразе для работы нужен небольшой отрезок избыточной
«лидерной» РНК. С другой стороны, если зонд будет использо-
ваться в экспериментах по защите от действия РНКазы
(разд. 1.4.3), то нежелательно, чтобы меченый транскрипт-зонд
оказался защищенным по всей длине, поскольку защищенный
фрагмент тогда будет неотличим по размеру от продуктов не-
полного РНКазного расщепления. Если известны нуклеотидная
последовательность матрицы и расположение интронов или
5’- или З’-фланкирующих участков, предпочтение следует отда-
вать ферментам, расщепляющим молекулу ДНК именно в этих
участках. Но и в таких случаях могут возникать трудности,
если существуют альтернативные сайты инициации или терми-
нации или в исследуемом препарате наблюдается высокое со-
держание РНК, не подвергшейся сплайсингу.
Если же «рабочая» последовательность (вставка) для пред-
лагаемого зонда будет защищаться по всей длине, следует
иметь в виду следующее: введение клонируемой последователь-
ности в сайт полилинкера, наиболее удаленный от промотора
(например, сайт HindVIA в pSP65 или Есо R1 в pSP64, или под-
ходящий сайт, выбранный в зависимости от используемого про-
мотора в плазмидах pGEM), приведет к тому, что до 55 пар
оснований векторной последовательности сохранится у 5’-кон-
ца зонда. Эта последовательность не будет гибридизоваться
с мРНК, и в случае, если вставка будет защищена мРНК по
всей длине, при обработке РНКазой образуется фрагмент, легко
отличимый по размеру от нерасщепленного зонда. Это позволя-
ет четко различать неспецифически защищенные зонды от зон-
дов, защита которых обеспечена гибридизацией со специфиче-
ской мРНК. Такую возможность предоставляет и сайт Pvu II,
расположенный в плазмидах pSP64 и pSP65 на расстоянии
180 п. н. по направлению транскрипции полилинкера. В pGEM-
векторах данный сайт расположен также за полилинкером отно-
сительно направления транскрипции полимеразой SP6. Это
весьма удобно, поскольку позволяет получить зонд в котором
содержится до 180 пар нуклеотидов векторной последователь-
ности, дающих возможность убедиться в полноте расщепления
(имейте в виду, однако, что при воздействии РпиП образуются
тупые концы; см. разд. 1.2.3.2).
В заключение отметим, что нуклеотидные последователь-
ности большинства векторов известны, и если возникает необ-
ходимость использовать зонды для гибридизации с другими
плазмидами, следует убедиться в том, что последовательность
полимера не будет вступать в перекрестную гибридизацию
с другими плазмидами.
2—1186
181
П, Литтл, Й, Джексон
1.2.3.2. Выбор сайта терминации транскрипта
Для ограничения длины матричного фрагмента можно ис-
пользовать любую рестриктазу. Необходимо соблюдать лишь
одно условие: выбранный фермент не должен вносить разрыв
между премоторным сайтом инициации транскрипции и встро-
енной ДНК. Не имеет значения, образуется ли под действием
рестриктазы один фрагмент или несколько — транскрибировать-
ся будет только фрагмент, содержащий промотор.
В ряде случаев может происходить неправильная инициация
транскрипции на выступающих З’-концах и реже на тупых кон-
цах [8]. В результате транскрипция идет в обратном направле-
нии, и тогда образуется продукт, комплементарный нужному
нам зонду. В некоторых случаях это не осложняет работы, по-
скольку доля «обратных транскриптов» по отношению к тран-
скриптам, начинающимся с промотора, очень невелика, и они
не будут мешать, если полученный зонд используется для гиб-
ридизации на фильтрах или in situ. Но если зонд должен быть
использован в эксперименте по защите от РНКазы, могут на-
блюдаться ложные сигналы в результате гибридизации двух
цепей и их взаимного протектирования. При этом детектиру-
ются специфические устойчивые к РНКазе фрагменты и совсем
не выявляется тестируемая РНК. Количество обратных транс-
криптов может составлять величину того же порядка, что и ис-
следуемая мРНК, а это сильно затруднит ее выявление.
Если известна подробная рестрикционная карта клонирован-
ной последовательности, то набор линейных матриц, получен-
ных разрезанием этой последовательности по разным сайтам,
позволяет получить ряд последовательно удлиняющихся зондов.
Их можно использовать для построения транскрипционной кар-
ты клонирования последовательности.
1.2.4. Выбор РНК-полимеразы
Выбор РНК-полимеразы может быть продиктован несколь-
кими соображениями — в том числе выбором вектора. Однако
следует иметь в виду следующее. РНК-полимеразы фагов Т7
и ТЗ обычно производят путем экспрессии клонированных ге-
нов. Поэтому они значительно дешевле РНК-полимеразы SP6
и их можно использовать в больших количествах. Вместе с тем
РНК-полимеразы фагов Т7 и 73 обладают перекрестной специ-
фичностью, особенно при низких концентрациях солей, при ко-
торых полимераза фага Т7 способна инициировать транскрип-
цию на ТЗ-промоторе и наоборот. Вследствие этого векторы,
содержащие оба промотора, могут обеспечивать синтез само-
комплементарной РНК- Это может и не мешать; например, для
Плазмиды, несущие промоторы для фаговых РНК-пол им ер аз 19’
матриц, обычно используемых при синтезе РНК-транскриптов,
терминированных в заданной точке, такое встречное иницииро-
вание транскрипции может приводить к образованию транс-
криптов, имеющих ничтожно малую длину. В наших опытах,
если судить по включению внесенной метки, полимераза SP6
оказывается всегда менее эффективной, чем полимераза Т7.
Возможно, это объясняется лимитирующим количеством радио-
активных нуклеозидтрифосфатов (NTP), используемых в реак-
циях транскрипции. При низкой концентрации нуклеотидов,
полимеразы Т7 и ТЗ прекращают транскрипцию в пределах
первых 15 оснований, считая от сайта инициации. Ферменты
сходят с матрицы, происходит реинициация и возобновляется
синтез неполноценных коротких транскриптов. В обычно ис-
пользуемых векторах на основе фагов Т7 и ТЗ при транскрип-
ции этого начального участка не требуется включения UTP.
Поэтому они эффективно транскрибируются даже при низких
концентрациях этого нуклеотида ([9] и W. F. McAllister личное
сообщение). До сих пор неизвестно, обладает ли аналогичными
свойствами полимераза SP6. Как бы то ни было при использо-
вании промотора фага SP6 транскрипция первых 15 оснований
требует присутствия всех четырех нуклеотидов, и это, по всей
вероятности, уменьшает эффективность транскрипции матрицы
полимеразой SP6 в присутствии низких концентраций UTP.
Если же транскрипция первых 15 оснований прошла успешно,
то вероятность терминации и реипициации сильно уменьшается.
1.3. Методы
При работе с препаратами РНК требуется большая акку-
ратность, чтобы избежать загрязнения растворов и реакцион-
ных смесей РНКазой. Для этого следует соблюдать все необхо-
димые стандартные меры предосторожности, и в том числе все
процедуры выполнять в перчатках. Очень полезно обрабаты-
вать растворы диэтилпирокарбонатом (ДЭПКом): добавьте
в них ДЭПК до концентрации 0,1% (объемная концентрация),
оставьте на 1 ч, а затем автоклавируйте, чтобы разрушить
ДЭПК.
Необходимо помнить, что трис-буферы нельзя эффективно
обработать ДЭПКом, и их необходимо автоклавировать. Чувст-
вительные к нагреванию реактивы (такие, как нуклеотиды)
следует готовить на стерильной, обработанной ДЭПКом воде,
и работать с ними надо в стерильных условиях. Для эффектив-
ного подавления активности РНКазы используют ингибитор из
плаценты (РНКазин), при этом полностью сохраняется актив-
ность РНК-полимеразы. Необходимо помнить о том, что широ-
ко используемый ингибитор РНКазы ванадил-нуклеотидный
2*
20
П. Литтл, И. Джексон
комплекс (ВНК) резко подавляет активность РНК-полимеразы
в результате конкурентного ингибирования и поэтому его при-
менения следует избегать.
1.3.1. Получение ДНК-матрицы
Плазмидпые ДНК, используемые в реакциях транскрипции,
в идеале следует выделять центрифугированием в градиенте
плотности CsCI с бромидом этидия (CsCl/EtBr). При необхо-
димости более быстрого получения матрицы используют метод
быстрого щелочного лизиса в присутствии додецилсульфата
натрия (ДСН, CDS) [15], после чего осаждая полиэтиленгЛи-
колем [16] или спермидином [17], получают ДНК, которая
может служить подходящей матрицей в реакциях с полимера-
зами SP6 и Т7. С помощью таких методов нам никогда не уда-
валось получить столь же хорошие матрицы, как при очистке
материала в градиенте плотности хлористого цезия, и во всех
случаях, когда это возможно, мы рекомендуем использовать
более сложный метод очистки в градиенте CsCl/EtBr.
Выбор точной последовательности операций по очистке ДНК
в градиенте CsCl/EtBr — вопрос, решаемый в каждом конкрет-
ном случае. Однако важно, чтобы конечный препарат ДНК не
содержал примесей EtBr, так как они могут резко подавлять
синтез РНК. Наиболее подходящий способ выполнения этого
требования — воспользоваться методикой, приведенной в
табл. 2.10, начиная со стадии 6 и далее. Детальное описание
модифицированной методики щелочного лизиса в присутствии
ДСН также можно найти в табл. 2.9 и 2.10.
Крайне важно, чтобы матричная ДНК была полностью ли-
неаризована, поскольку сверхспирализованные ДНК — эффек-
тивные матрицы для всех фаговых РНК-полимераз и на них
синтезируются очень длинные транскрипты. Это снижает выход
транскриптов требуемого размера и, кроме того, может суще-
ственно снизить специфичность зондов, получаемых в реакциях,
содержащих смеси молекул.
После обработки рестриктазой мы выделяем чистую ДНК
путем депротеинизации фенолом, хлороформом, осаждаем ее
этанолом или изопропанолом, промываем 7О°/о-ным этанолом
и, наконец, ресуспендируем ДНК в растворе, содержащем 10 мМ
трис-НС1 (pH 7,4) и 1 мМ ЭДТА (ТЭ). Эти общие процедуры,
ссылки на которые несколько раз встречаются в этой и следую-
щей главе, приведены в табл. 1.1. Препараты многих рестрик-
таз загрязнены примесью РНКазы и поэтому использовать их
надо с большой осторожностью. В реакции транскрипции ко-
нечная концентрация ДНК должна составлять примерно 20—
40 мкг/мл, поэтому для удобства ее следует растворять в ТЭ
до конечной концентрации, превышающей 200 мкг/мл.
Плазмиды, несущие промоторы для фаговых РНК-полимераз 21
Таблица 1.1. Общие методы очистки от белков и концентрирования
нуклеиновых кислот
1. Экстракция фенолом. Добавьте к раствору ДНК или РНК равный объем
фенола, насыщенного ТЭ1. Перемешивайте, переворачивая колбу, до полу-
чения гомогенной смеси. Разделите фазы центрифугированием в микро-
центрифуге в течение 5 мин в настольной центрифуге или в центрифуге
Sorvall с ротором НВ4 при 2000 об/мин в течение 10 мин. Осторожно от-
берите верхнюю (водную) фазу пипеткой или микропипеткой и перенесите
ее в чистую пробирку.
2. Экстракция смесью фенол—хлороформ. К водной фазе, полученной на ста-
дии 1, добавьте равный объем смеси (соотношение объемов фенол : хлоро-
форм=1 : 1). Перемешайте смесь и разделите фазы, как описано в п. 1.
3. Экстракция хлороформом. Водную фазу, полученную на стадии 2, экстра-
гируйте равным объемом хлороформа. Разделите фазы, как описано в п. 1.
4. Осаждение этанолом. К водной фазе, полученной на стадии 3, добавьте
1/10 объема ЗМ ацетата натрия (pH 5,4) и два объема этанола. Охлади-
те смесь до —70 °C или поставьте в сухой лед. Осадите нуклеиновую кис-
лоту центрифугированием в микроцентрифуге в течение 10 мин млн в цент-
рифуге Sorvall с ротором НВ4 при 8000 об/мин в течение 10 .мин. Осто-
рожно слейте надосадочную жидкость. При необходимости промойте оса-
док 70%-ным этанолом при комнатной температуре и повторно осадите
центрифугированием, как описано выше. Удалите избыток этанола с по-
мощью фильтровальной бумаги и дайте осадку высохнуть на воздухе.
Ресуспендируйте его в нужном объеме ТЭ.
5. Осаждение изопропанолом. Этот метод может с успехом заменить осажде-
ние этанолом, когда необходимо свести к .минимуму объем осаждаемого
материала. Добавьте от 0,6 до 1 объема изопропан-2-ола к водной фазе,
полученной на стадии 3. Затем обработайте ее точно так же, как при
осаждении этанолом на стадии 4 (включая промывание 70%-ным этано-
лом)
') ТЭ — 10 мМ трис-НС1 (pH 7.5) и 1 иМ ЭДТА.
1.3.2. Реакция транскрипции
Метод получения высокоспецифичных полноразмерных
РН1<-зондов приведен в табл. 1.2, а метод определения включе-
ния нуклеозидтрифосфатов в РНК — в табл. 1.3.
Реакция транскрипции, катализируемая полимеразами SP6
и Т7, чувствительна к концентрации рибонуклеозидтрифосфа-
тов. При низких концентрациях полимеразная реакция имеет
тенденцию к преждевременной терминации и образованию не-
полных транскриптов. Полноразмерные транскрипты обычно
образуются, когда концентрация нуклеотидов превышает
250 мкМ. В принципе при получении радиоактивных зондов
стремятся к получению максимальной удельной радиоактивно-
сти путем увеличения удельной радиоактивности нуклеозидтри-
фосфата-предшественника. В то же время очевидно, что при
работе с нуклеозидтрифосфатами в концентрации 250 мкм не-
реально использовать меченые предшественники с высокой
22
П. Литтл, И. Джексон
Таблица 1.2. Синтез РНК-зондов
А. Реактивы
ЮХбуфер для транскрипции (10ХТВ):
400 мМ трис-НС1 (pH 7,5)
60 мМ MgCl2
10 мМ спермидин
10 X рибонуклеозидтрифосфаты (10 X rNTP)
5 мМ АТР, СТР и GTP (каждого),
250 мкМ UTP,
РНКазин, ингибитор РНКазы из плаценты,
РНК-полимераза фагов SP6 или Т7.
ДНКаза, свободная от РНКазы
Б. Методика
1.
2.
3.
4.
5.
6.
о мкл
5 мкл
1,5 мкл ( ~ 80 ед.)
5 мкл
26,5 мкл
1 мкл (~0,1—1 мкг)
5 мкл (50 .мкК’и).
Смешайте при комнатной температуре, добавляя ДНК только после раз-
бавления ЮхТВ:
10ХТВ
ЮмМ дитиотрейтол
РНКазин
lOXrNTP
Н2О
ДНК-матрица
зар.иТР
Отберите две аликвоты по 0,5 мкл для осаждения ТХУ (см. табл. 1.3).
Затем добавьте 1 мкл РНК-полимеразы (10—20 ед.)
Инкубируйте смесь при 37 °C 1 ч.
Добавьте 1 мкл (1 ед.) ДНКазы, свободной от РНКазы1’.
Добавьте 5 мкл 10% ДСН и 1 мкл 0,5 М ЭДТА.
Отберите две аликвоты по 0,5 мкл для осаждения ТХУ (см. табл. 1.3).
Экстрагируйте смесь равным объемом смеси фенол—хлороформ (в соот-
ношении объемов 1:1), как описано .в табл. 1.1. Отберите водную фазу,
повторно экстрагируйте органическую фазу и объедините обе водные фа-
зы. На этой стадии можно также удалить невключившиеся нуклеотиды,
пропуская смесь через колонку с сефадексом G-50 (medium), уравновешен-
ную раствором, содержащим 40 мМ трис-НС1 (pH 7,5), 5 мМ ЭДТА и
0,5% ДСН. После прохождения всего объема смеси через колонку собе-
рите материал, связавшийся с сефадексом.
Осадите РНК этанолом, как описано в п. 4 табл. 1.1. предварительно до-
бавив 5 мкг дрожжевой тРНК в качестве носителя.
') Данная стадия не обязательная. Подробнее это описано в тексте.
удельной радиоактивностью; вместо этого в смесь вносят 20—
100 мкКи меченого нуклеозидтрифосфата, а концентрацию того
же немеченого нуклеотида уменьшают. Создаваемые при этом
условия оказываются не оптимальными для синтеза полнораз-
мерных транскриптов. Несколько меньшая длина зонда не яв-
ляется препятствием для его использования при гибридизации
на фильтрах или in situ, но очень затрудняет интерпретацию
результатов экспериментов по защите от действия РНКазы. На
практике обычно используют некую среднюю концентрацию
нуклеозидтрифосфатов, чтобы можно было получить максималь-
Плазмиды, несущие промоторы для фаговых РНК-полимераз
23
Таблица 1.3. Определение процента включившихся в РНК
нуклеозидтрифосфатов с помощью осаждения ТХУ
А. Реактивы
Бумага ватман 540
Смесь 5% трихлоруксусной кислоты и 1% пирофосфата натрия (раствор
ТХУ)
Б. Методика
1. Нанесите 0,5 мкл реакционной смеси на небольшой (размером примерно
5X5 мм) -кусочек бумаги ватман 540. Такие фильтры можно пометить
карандашом или шариковой ручкой, что дает возможность проводить
одновременно несколько определений.
2. Измерьте радиоактивность фильтра, используя эффект Черенкова, опре-
делив тем самым общую радиоактивность пробы. (При использовании
35S или 3Н перед определением радиоактивности в сцинтилляторе фильт-
ры следует высушить, и, кроме того, для промывки ТХУ рекомендуется
ставить две параллели.)
3. Промойте фильтр примерно 20 мл раствора ТХУ, встряхивая его в подхо-
дящей пробирке с завинчивающейся крышкой, или большим объемом
ТХУ в конической колбе. Вылейте раствор ТХУ и замените его свежим
раствором. Повторите отмывку 5 раз или -более до тех пор, пока радио-
активная метка не перестанет смываться с фильтра. (Если одновременно
отмываются много фильтров, объем ТХУ следует увеличить.)
4. Высушите фильтр и снова определите его радиоактивность, используя
эффект Черенкова, чтобы получить значение радиоактивности кислотоне-
растворимой фракции. * 5
но возможную удельную радиоактивность продукта и в то же
время свести к минимуму преждевременную терминацию
транскрипции.
Транскрипция с участием полимеразы SP6, по-видимому,
наиболее чувствительна к низким концентрациям АТР и UTR
[5] и гораздо менее чувствительна к концентрациям СТР или
GTP. Концентрация GTP наименее важна для синтеза полно-
размерного продукта, но G — первая «буква» транскрипта,
и уменьшение его концентрации приводит к уменьшению выхо-
да продукта за счет снижения эффективности инициации. По-
этому идеальным меченым рибонуклеозидтрифосфатом пред-
ставляется СТР. Однако по иронии судьбы во многих лаборато-
риях, включая и нашу, используют меченый UTP, который под-
ходит для этого менее всего. Такой выбор обусловлен тем, что
UTP — первый радиоактивный предшественник РНК из выпус-
каемых фирмой Amersham International, который был апроби-
рован в реакции с 5Р6-полимеразой.
По методике, приведенной в табл. 1.2, UTP используется
в концентрации 25 мкМ, при которой более 90% транскриптов
являются полноразмерными. Количество вносимой в реакцию
метки может быть увеличено; при увеличении количества высо-
комеченого UTP вдвое удельная радиоактивность продукта поч-
24
П. Литтл, И. Джексон
ти удваивается. Условия реакции, приведенные в этой таблице,
можно изменить — уменьшить ее количественные параметры
и тем самым повысить в реакционной смеси относительное со-
держание радиоактивного UTP. В случае использования
РНК-зонда для гибридизации на фильтрах или in situ, когда
требуется очень высокая удельная радиоактивность зонда,
а его размеры не столь существенны, можно вообще обойтись
без холодного UTP и тем самым уменьшить объем реакцион-
ной смеси. 100 мкКи UTP с удельной радиоактивностью
400 Ки/ммоль при объеме реакционной смеси 20 мкл соответ-
ствуют концентрации 12,5 мкМ, которая позволяет получать
зонды подходящей длины, с максимальной удельной радиоак-
тивностью.
При гель-электрофорезе меченых транскриптов было обнару-
жено, что сами матрицы различаются по способности давать
полноразмерные транскрипты. Даже в условиях такой низкой
концентрации нуклеозидтрифосфатов, как 4 мкМ, при исполь-
зовании в качестве матрицы ДНК гена йргКмыши синтезиру-
ется значительное количество полноразмерной РНК-зонда, тогда
как на ДНК «гомео-бокса» при концентрации нуклеозидтрифос-
фатов 15 мкМ полноразмерных транскриптов почти не синтези-
руется (И. Дж. Джексон, неопубликованные данные).
При гель-электрофорезе транскриптов, образовавшихся в ус-
ловиях преждевременной терминации, как правило, не обнару-
живается дискретных больших фрагментов. Между тем некото-
рые последовательности, по-видимому, содержат внутренние
слабые сайты терминации; в результате в геле обнаруживаются
полосы, соответствующие достаточно большим, но неполнораз-
мерным транскриптам. Количество таких полос уменьшается
по мере возрастания количества полноразмерных транскриптов
при использовании более высоких концентраций нуклеотидов,
но даже при самых высоких концентрациях они никогда не
исчезают полностью. Именно эти дискретные продукты, полу-
чающиеся в результате преждевременной терминации, служат
основным источником трудностей при анализе результатов
опытов по защите от действия РНКазы. Следовательно, требу-
ется проводить анализ меченого зонда с помощью гель-электро-
фореза до его использования, особенно в случае экспериментов
по защите от РНКазы.
Необходимо обратить внимание на следующие вопросы, ка-
сающиеся метода, приведенного в табл. 1.2.
1. ДНК-матрицу (примерно 0,1 — 1 мкг на реакцию) нужно
вносить в реакционную смесь после разбавления буфера, по-
скольку 10-кратнын буфер содержит спермидин в концентрации
10 мМ. В концентрации, превышающей 4 мМ спермидин может
приводить к выпадению ДНК в осадок [7].
,Плазмиды, несущие промоторы для фаговых РНК-полимераз 25
2. По-видимому, присутствие плацентарного РНКазина
существенно для максимального выхода полноразмерных транс-
криптов.
3. Останавливают реакцию и удаляют матрицу обработкой
ДНКазой. Неясно, необходим ли этот этап, если учесть, что
матрица не подвергается денатурации ни на одной из стадий
получения зонда. Кроме того, существует опасность разрушения
зонда примесями РНКазы. Присутствие ДНК-матрицы при гиб-
ридизации в растворе может мешать гибридизации лишь при
температурах, близких к температуре плавления (Тпл) ДНК,
когда ее цепи могут на отдельных участках начать расходить-
ся и вступать во взаимодействие с РНК-зондом, защищая его
от воздействия РНКазы и тем самым искажая результаты
опыта.
4. По окончании реакции о величине включения нуклеоти-
дов в РНК. можно судить по количеству кислотонерастворимого
материала, осажденного трихлоруксусной кислотой (ТХУ).
В табл. 1.3 приведен метод быстрого осаждения ТХУ.
Для каждого заданного времени реакции обычно ставят по
две параллельных пробы. Фоновые значения радиоактивности
ТХУ-нерастворимой фракции определяют до добавления фер-
мента. Включение метки в РНК (в кислотонерастворимую
фракцию) к концу реакции обычно составляет 30—90%. Обра-
тите внимание на то, что при использовании этого метода ана-
лиза не требуется знать точного объема реакционной смеси,
поскольку общая радиоактивность и радиоактивность включив-
шейся в РНК метки измеряются на одном и том же фильтре.
Однако для определения суммарного количества радиоактивной
метки, включившейся в зонд, необходимо знать точный объем
реакционной смеси.
5. Для удаления невключившихся в РНК нуклеотидов
необязательно пропускать реакционную смесь через сефадекс
G-50. При осаждении РНК этанолом обычно происходит доста-
точно полное удаление основной части нуклеотидов, особенно
при высоком их включении.
1.3.3. Получение немеченой РНК или РНК
с низкой удельной радиоактивностью и использование
других изотопов и альтернативных меток
Метод получения большого количества немеченых или обла-
дающих низкой удельной радиоактивностью РНК-транскриптов
лишь немногим отличается от приведенного в табл. 1.2. Все
четыре нуклеотида должны присутствовать в концентрации
500 мкМ, а меченый нуклеотид добавляют в количестве, необ-
ходимом для получения требуемой удельной радиоактивности.
26
П, Литтл, И, Джексон
Условия инкубации те же, что и указанные в таблице, и через
час после начала инкубации можно добавить дополнительное
количество полимеразы. Используя эту методику, на ДНК-мат-
рице можно получить выход РНК с молярным соотношением
1 : 10. Обратите внимание на данные о том, что как матрица,
так и фермент часто могут служить компонентами, лимитирую-
щими протекание реакции [5]; следовательно, для достижения
наиболее высокого выхода продукта реакции желательно повы-
сить концентрацию матрицы.
Сообщалось об использовании меченных 3SS а-нуклеозидтри-
фосфатов (NTP) и биотинилированных нуклеотидов [6]. И те,
и другие меченые нуклеотиды включаются в РНК менее эффек-
тивно, чем обычные NTP; однако и в том, и в другом случае
выход меченой РНК достаточен для работы. Детальные пропи-
си методик не опубликованы, но обычно их можно получить
у авторов.
1.3.4. Методические трудности
Методические трудности обычно связаны с тем, что получа-
ют нулевое или низкое (<5%) включение метки в ТХУ-нера-
створимую фракцию или не происходит образования полнораз-
мерных транскриптов. Поэтому следует обратить особое внима-
ние на следующие моменты.
1.З.4.1. Нуклеозидтрифосфаты (NTPj
Рибонуклеозидтрифосфаты при хранении распадаются. Во-
обще следует с осторожностью относиться к реактивам, кото-
рые хранились более одного-двух месяцев. Для получения опти-
мальной удельной радиоактивности и высокого суммарного
включения метки радиоактивные нуклеотиды нужно использо-
вать в течение одной или самое большее двух недель. При при-
готовлении компонентов реакционной смеси следует помнить
о необходимости нейтрализации растворов.
1.3.4.2. ДНК-матрица
ДНК-матрица не должна содержать примесей солей и бро-
мида этидия (EtBr). Всегда используйте в качестве контроля
препарат ДНК, заведомо способный к эффективной транскрип-
ции, чтобы контролировать возможные изменения в протекании
реакции, связанные с использованием новых растворов; это по-
может выявить недостаточную степень чистоты препарата ДНК-
Если тщательно следовать методикам, приведенным в табл. 1.1
и 2.10, то осложнений не возникнет. Необходимо убедиться, что
Плазмиды, несущие промоторы для фаговых РНК - полимераз
27
удален весь фенол. Для этого полученные препараты линейных
матриц следует промыть 70%-ным этанолом при комнатной
температуре. Наконец, избегайте внесения ДНК в 10х буфер
для транскрипции с высокой концентрацией спермидина — это
может вызвать выпадение ДНК в осадок.
1.3.4.3. Возможные неудачи при синтезе
полноразмерных транскриптов
Причиной низкого включения метки и образования коротких
транскриптов часто могут служить факторы, перечисленные
выше в разд. 1.3.4.1 и 1.3.4.2, так же как и загрязнение буфер-
ных растворов РНКазой. Если все указанные факторы исклю-
чены, то тогда, возможно, матрица содержит один или несколь-
ко терминаторов транскрипции. Такое препятствие в свою оче-
редь можно преодолеть, увеличив концентрацию нуклеотидов.
Если же и это не помогает или если это нежелательно из-за
необходимости получить определенную удельную радиоактив-
ность, возможно, целесообразнее переключиться на получение
другого, более короткого транскрипта. В работе Крига и Мел-
тона [6] показано также, что в одном случае снижение темпе-
ратуры транскрипции с 40 до 30 °C привело к увеличению выхо-
да полноразмерных транскриптов. По-видимому, можно исполь-
зовать такой подход. Если ни одно из перечисленных изменений
методики не дает желаемого результата, не остается другого
выхода, кроме как выделять полноразмерные транскрипты из
суммарного материала, фракционированного в агарозном или
акриламидном геле.
1.4. Использование РНК-зондов
В этом разделе рассматриваются основные методы исполь-
зования РНК-зондов при исследовании нуклеиновых кислот
в обычной лабораторной практике. В раздел включены деталь-
ные описания методов, в которых РНК используются вместо
ДНК-зондов при «Саузерн-» и «нозерн-» гибридизации и в опы-
тах по защите от действия РНКазы.
Следует снова подчеркнуть, что необходимо принимать все
меры предосторожности для удаления возможных примесей
РНКазы из растворов и с посуды. Соответствующие методы
приводятся в разд. 1.3; кроме того, рекомендуется после завер-
шения синтеза РНК использовать растворы, содержащие 10 мМ
ванадил-рибонуклеотидные комплексы (ВНК). После приобре-
тения достаточного опыта работы с РНК некоторые меры
предосторожности можно опустить, но в начале работы их не-
обходимо соблюдать.
28
П, Литтл, И. Джексон
Таблица 1.4. Условия гибридизации по Саузерну
1. Для переноса по Саузерну и иммобилизации ДНК на фильтрах исполь-
зуются стандартные условия (см., например, работу (28]); соблюдайте
особую осторожность, чтобы не касаться фильтров руками.
2. Для удаления избытка солей промойте фильтр в растворе 5XSSC в тече-
ние нескольких минут.
3. Проведите предварительную гибридизацию фильтра в растворе, содержа-
щем:
0,1% (весовая концентрация) фикола 400,
0,1% (весовая концентрация) БСА (бычий сывороточный альбумин),
0,1% (весовая концентрация) поливинилпиролидона.
50 мМ Na-фосфатный буфер (pH6,5),
0,1% (весовая концентрация)
250 мг/мл обработанной ультразвуком и денатурированной ДНК
спермы лосося
10 мкг/мл PolyC
10 мкг/мл PolyA
50% (объемная концентрация) деионизованного формамида,
10 ,мМ ванадилнуклеотидиый комплекс.
Инкубируйте смесь при 50 °C не менее 1 ч. Инкубация смеси в течение
более длительного срока не имеет очевидного преимущества. На фильтр
площадью 15X20 см нанесите 10—20 мл раствора.
4. Проведите реакцию гибридизации на фильтре размером 15x20 см в 10 мл
раствора, использовавшегося для предварительной гибридизации, в кото-
рый дополнительно внесено 106 имп/мин/мл 32Р-меченной РНК-зонда. Ин-
кубируйте при 50°C 17 ч (в течение ночи) или более длительное время.
5. После гибридизации промойте фильтр при 65 °C 3 раза по 30 мин 500 мл
раствора, содержащего 2XSSC, 0,1% (весовая концентрация) ДСН,
а затем один раз в растворе 0.1XSSC, 0,1% ДСН. Условия последней
жесткой отмывки могут быть изменены в зависимости от требований,
предъявляемых к специфичности зонда. Иногда целесообразно изменить
температуру или концентрацию солей.
6. По нашим наблюдениям нет абсолютной необходимости в обработке
фильтра РНКазой после отмывки, хотя это и может привести к снижению
фоновой радиоактивности. Такую обработку наиболее удобно проводить
при 37 °C в растворе 2XSSC, содержащем 10—100 мкг/мл РНКазы А,
предварительно подвергнутой тепловой обработке.
1.4.1. Гибридизация по Саузерну
При использовании РНК-зондов не требуется вносить ка-
кие-либо изменения в условия фракционирования в геле и пе-
реноса на нитрицеллюлозные или найлоновые мембранные
фильтры.
Предложено множество различных вариантов гибридизаци-
онных смесей; в табл. 1.4 мы подробно приводим простой ме-
тод, при котором используются те же растворы, что и при обыч-
ной гибридизации с ДНК-зондами. Строго говоря, основная
часть компонентов этой гибридизационной смеси не является
принципиально необходимой для протекания реакции гибриди-
зации. И если мы пользуемся именно этой методикой, то лишь
исключительно для удобства.
Плазмиды, несущие промоторы для фаговых РНК-полимераз 29
Таблица 1.5. «Нозерн»-гибридизация
1. Используйте стандартную методику гель-электрофореза РНК [28]. Удоб-
нее всего работать с содержащими формальдегид агарозными гелями„
а для переноса РНК и анализа ее на фильтрах можно использовать мето-
ды, описанные в работе [28]. Мы получали хорошие результаты с найло-
новыми фильтрами Hybond (Amersham International), следуя инструкциям-
фирмы-производителя.
2. Проведите предварительную гибридизацию фильтров в точности так, как
это описано в табл. 1.4.
3. Проведите гибридизацию на фильтрах в точности так, как это описано-
в табл. 1.4. Однако в данном случае оптимальная температура гибриди-
зации составляет 55—60 °C в зависимости от свойств используемого зонда
(содержание GC-nap, гомология с исследуемым материалом и длина).
Время гибридизации — 17 ч.
4. Промойте фильтры в трех сменах по 500 мл раствора 2XSSC, 0,1% ДСН
и затем в 0.1XSSC, 0,1% ДСН при 65 °C; время каждой отмывки — 20—
30 мин. Условия последней отмывки можно изменить, повышая или по-
нижая температуру, с тем чтобы добиться наибольшей контрастности по-
лос, которые по интенсивности включения метки близки к фоновым зна-
чениям.
Использование РНК-зондов в блот-гибридизации по Саузер-
ну, по-видимому, увеличивает чувствительность метода — глав-
ным образом из-за отсутствия конкурентной гибридизации зон-
да «на себя», препятствующей гибридизации зонд—мишень
(такая же чувствительность может быть достигнута и при ис-
пользовании кДНК или других одноцепочечных ДНК-зондов;
но такие зонды, как правило, сложнее получить).
Другую методику выявления ДНК на «Саузерн-фильтрах»
разработали Черч и Гилберт [18]: предложенный ими подход
получил название геномного секвенирования. По сообщению
авторов, метод позволяет детектировать 3 фг ДНК при 10-днев-
ной экспозиции, что несомненно свидетельствует о достижении
самой высокой на сегодня чувствительности метода. Подробные
методики приводятся в отдельной работе [18]. В общих чертах
этот метод состоит в использовании ДНК, «пришитой» с по-
мощью ультрафиолетового облучения к найлоновым мембран-
ным фильтрам, которую гибридизуют с высокорадиоактивной
32Р-меченной РНК. в растворе, содержащем 1% (весовая кон-
центрация) БСА, 1 мМ ЭДТА, 0,5 М NaHPO4 (pH 7,2) и 7%
(весовая концентрация) додецилсульфата натрия (ДСН). Не
ясно, какой именно фактор в данном случае обусловливает
столь высокую чувствительность метода.
1.4.2. «Нозерн»-гибридизация
Метод «Нозерн»-гибридизации по существу идентичен гибри-
дизации по Саузерну и детально описан в табл. 1.5.
30
П, Литтл, И. Джексон
В случае «Нозерн»-гибридизации РНК-зонды оказались го-
раздо более чувствительными, чем ДНК-зонды. Мелтон и др.
[5] сообщали о достигнутом ими 10-кратном повышении чувст-
вительности метода при использовании РНК-зондов; как выяс-
нилось, это общее правило. РНК-РНК-гибриды более стабильны
в растворах формамида, чем аналогичные ДНК-ДНК-гибриды,
что и порождает проблему неспецифичности гибридизации.
В частности, связывание некоторых зондов, обладающих спо-
собностью к перекрестной гибридизации с рибосомной РНК,
стало серьезной проблемой, и для выявления специфических
гибридов приходится варьировать степень жесткости отмывки.
В общем случае лучше с осторожностью относиться к полосам
РНК, по размеру совпадающим с 28- или 18S-PHK, до тех пор,
пока не будут получены еще и другие доказательства того, что
они появились в результате специфической гибридизации. Для
удаления неспецифических гибридов некоторые исследователи
предлагают проводить отмывку в присутствии РНКазы. В этом
случае фильтры нельзя использовать повторно.
1.4.3. Защита от РНКазы
Защита от воздействия РНКазы, по всей вероятности, наи-
более чувствительный и простой метод выявления и анализа
специфических РНК [13]. Этот метод позволяет обнаружить
даже столь малое количество РНК, как 0,1 пг [13]. Его пре-
имущества по сравнению с использованием нуклеазы S1 [10]
сводятся к простоте получения зонда, высокой чувствительности
анализа и большей эффективности ферментативной обработки
на конечных стадиях (обработка РНКазой вместо нуклеазы
S1). По-видимому, на этой стадии гибрид менее чувствителен
к «избыточной» обработке РНКазой, чем к обработке нуклеа-
зой S1.
1.4.3.1. Методика
В табл. 1.6 приведена методика эксперимента по защите от
действия РНКазы.
Следует обратить внимание на следующие моменты:
1. Для определения уровня фоновой гибридизации и провер-
ки полноты ферментативного расщепления следует ставить
контрольные пробы, содержащие только зонд (без исследуемой
РНК) или зонд и дрожжевую тРНК.
2. Показано, что самые разные зонды эффективны в гибри-
дизации при 45 °C в 80%-ном растворе формамида. Однако,
если содержание A-I-U или G + C сильно отличается от обычно-
го или область гомологии между зондом и исследуемой РНК
очень коротка, необходимо изменить температуру реакции.
Плазмиды, несущие промоторы для фаговых РНК-полимераз 31
Таблица 1.6. Определение устойчивости к обработке РНКазой
А. Реактивы
Перекристаллизованный, деионизованный формамид1*
Буфер для гибридизации2*:
80% формамид
40 мМ Pipes (pH 6,7)
400 мМ NaCl
1 мМ ЭДТА
Раствор РНКазы3*:
40 мкг/мл РНКазы А
2 мкг/мл РНКазы Т1
10 мМ трис (pH 7,5)
5 мМ ЭДТА
300 мМ NaCl
Б. Методика
1. Осадите пробы РНК, как описано в табл. 1.1. Для реакции достаточно
40 мкг суммарной РНК.
2. Растворите осадок РНК в 30 мкл буфера для гибридизации.
3. Растворите радиоактивный зонд в буфере для гибридизации и добавьте
1 мкл к каждой реакционной смеси. (Вносите в каждую пробу по 105—
106 имп/мин РНК.)
4. Инкубируйте смеси при 45 °C в течение ночи4*.
5. Добавьте 300 мкл раствора РНКазы. Инкубируйте при 30 °C 1 ч.
6. Добавьте 30 мкл 10% ДСН и 50 мкг протеиназы К. Инкубируйте при
37 °C 10 мин.
7. Экстрагируйте пробы смесью фенол—хлороформ, как описано в табл. 1.1,
а затем до осаждения этанолом добавьте к водной фазе 5 мкг носителя —
дрожжевой тРНК.
>) Формамид перекристаллизовывают путем замораживания и деионизуют его, встря-
хивая с бифункциональной ионообменной смолой фирмы Biorad в течение 30 мии.
2) Формамид хранят отдельно в замороженном состоянии и добавляют в буфер не-
посредственно перед использованием.
3) Раствор готовят непосредственно перед использованием: нуклеазную активность
в отношении двухцепочечных участков ингибируют, прогревая фермент на кипящей во-
дяной бане в течение 5 мин.
4) На этой стадии может оказаться полезным повысить на 10 мин температуру ре-
акции до 80 °C перед инкубацией в течение ночи. Если это сделано, важно удалить мат-
рицу, использовавшуюся для получения зонда, обработкой ДНКазой.
3. Очень важно контролировать температуру при обработке
РНКазой. Специфичность расщепления практически нечувстви-
тельна к избытку фермента; в то же время соотношение специ-
фического и фонового гибридизационных сигналов может силь-
но варьировать в зависимости от физических условий реакции.
При повышении температуры фон обычно уменьшается, но сле-
дует учитывать, что в этих условиях дуплексы poly (U: А), по
всей видимости, чувствительны к воздействию РНКазы, и по-
этому при гель-электрофорезе могут обнаруживаться ложные
фрагменты (Robb Krumlauf, личное сообщение).
4. После обработки РНКазой защищенные фрагменты раз-
деляют с помощью электрофореза в полиакриламидных гелях
в присутствии мочевины и визуализируют путем радиоавтогра-
32
П, Литтл, И. Джексон
Таблица 1.7. Электрофорез в полиакриламидном геле
в присутствии мочевины
А. Реактивы
1, Трис-боратный электродный буфер (ТБЭ) готовят в виде исходного
20Xраствора. На 1 л раствора вносят:
216 г сухого триса,
110 г борной кислоты,
20,4 г ЭДТА, Na-соль.
2. Для приготовления геля берут 57 г акриламида (особо чистого; для
электрофореза), 3 г био акрил а ми да, 460 г мочевины и растворяют при-
мерно в 900 мл воды. Для более быстрого растворения можно подо-
греть раствор. После растворения перечисленных компонентов добавьте
примерно 20 г бифункциональной ионообменной смолы фирмы Biorad
и для деионизации встряхивайте около 30 мин. Не увеличивайте слиш-
ком сильно время деионизации. Профильтруйте через нитроцеллюлоз-
ный фильтр с диаметром пор 0,45 мкм. Добавьте 50 мл 20XТБЭ и
доведите объем до 1 л,
3. Буфер для нанесения — 7 М мочевина, 1ХТБЭ, 0,1% ксилолцианол,
0,1% бромфеноловый синий.
В. Методика
1. Подготовьте стекла и соберите их в блок для заливки геля. Лучше
всего использовать очень тонкие (0,3 мм) спейсеры и одно стекло си-
ликопизировать для более легкого отделения геля после электрофореза.
2. Дегазируйте смесь для приготовления геля в вакуум-эксикаторе в те-
чение .нескольких минут. Раствора объемом примерно 75 мл с избытком
хватит на приготовление большого (шириной 30 см) «секвенирующего»
геля. Раствор должен иметь комнатную температуру, поскольку при
более низкой температуре гель может заполимеризоваться недоста-
точно быстро, чтобы сформировались кармашки.
3. Добавьте 650 мкл свежеприготовленного 10%-ного персульфата аммо-
ния и 100 мкл М,Ы,М',М'-тетраметилэтилендиамина (ТЕМЭД). Залейте
гель, вставьте гребенку (гребенку типа «акульи зубы» вставляют пло-
ской стороной к гелю). Гель должен полимеризоваться по меньшей
мере в течение 1 ч.
4. Поместите гель в прибор для электрофореза. Проведите префорез в те-
чение 30 мин или Дольше в условиях электрофореза (см. пункт 6).
5. Добавьте к каждой пробе по 5 мкл буфера для нанесения. Для рас-
творения быстро перемешайте пробы. Поместите пробы на .кипящую
водяную баню 'на 5 мин.
6. Нанесите пробы на гель. Проводите электрофорез при мощности
40 Вт в случае геля шириной 30 см или 25—30 Вт при ширине геля
20 см. Во время префореза и электрофореза напряжение возрастет при-
мерно с 950 до 1200 В.
7. Время электрофореза зависит от предполагаемого размера защищенных
от РНКазы фрагментов. В качестве грубого маркера для определения
времени электрофореза используют краситель бромфеноловый синий,
который в 6%-ном ПААГ движется как фрагмент размером -примерно
40 оснований, и ксилолцианол, соответствующий фрагменту примерно
в 120 оснований.
8. После окончания электрофореза осторожно разъедините стекла. Зафик-
сируйте гель на стекле, поместив его на 15 мин, в раствор, содержа-
щий 10%-ный метанол и 10%-ную уксусную кислоту, затем в течение
Плазмиды, несущие промоторы для фаговых РНК-полимераз 33
П родолжение
15 мии подсушивайте его на воздухе, заверните в пленку Clingfilm и по-
ставьте на радиоавтографию. Как вариант гель можно перенести на бу-
магу, плотно прижав к нему сухой лист ватман 3 ММ и затем сняв со
стекла бумагу вместе с гелем. Завернутый в пленку Clingfilm гель на
бумаге можно высушить в вакуумной сушке с подогревом и поста-
вить на радиоавтографию в стандартной кассете. Высушенные гели
дают при радиоавтографии более четкие и более интенсивные полосы.
фии. Эти гели идентичны используемым при секвенировании
ДНК, а методика проведения электрофореза приведена в
табл. 1.7. Параллельно следует ставить электрофорез интакт-
ного зонда, взятого в разных разведениях. Обычно берут раз-
ведение 1:1000 или 1:10 000 относительно общей радиоактив-
ности гибридизационной смеси, но конкретная степень разбавле-
ния должна выбираться в соответствии с предполагаемым ко-
личеством РНК, обеспечивающей защитный эффект.
Маркеры при электрофорезе могут быть получены примене-
нием матриц, на которых синтезируются транскрипты известной
длины. Обычно матрицы можно транскрибировать в одной ре-
акции, чтобы получить смесь фрагментов нужной длины для
электрофореза.
1.4.3.2. Определение содержания РНК в опытах по защите
от действия РНКазы
Использование РНК-зондов в экспериментах по защите от
действия РНКазы позволяет точно рассчитать содержание
в клетке защищающей РНК. Пример такого расчета приведен
в табл. 1.8.
Удельная радиоактивность меченых нуклеозидтрифосфатов
при синтезе зонда известна. Отсюда можно вычислить и удель-
ную радиоактивность самого зонда. (Она равняется молярной
удельной радиоактивности меченого нуклеотида, деленной на
число соответствующих оснований в последовательности зонда.)
Если нуклеотидная последовательность данного зонда неизвест-
на, надо приблизительно оценить количество содержащихся в
нем меченых оснований, исходя из того, что в среднем содер-
жание каждого нуклеотида составляет 25%. Зная общую ра-
диоактивность зонда, внесенного в гибридизационную смесь,
можно определить число его молей.
После гель-электрофореза и радиоавтографии интенсивности
полос, соответствующих защищенному от действия РНКазы
фрагменту (фрагментам) и серийным разведениям зонда, срав-
ниваются (это желательно делать с помощью денситометриче-
ского сканирования автографов, экспонированных в режиме
3-1186
34 П. Литтл, И. Джексон
Таблица 1.8. Пример расчета содержания мРНК в клетке в экспериментах
по защите мРНК от действия РНКазы
1. Условия синтеза зонда: концентрация СТР равна 25 мкМ. Радиоактив-
ность 1 мкл реакционной смеси при ее определении с использованием,
эффекта Черенкова составляет до промывки ТХУ 5-Ю5 имп./мин. Таким
образом, 25 пмолей СТР дают 5-10® имп./мин,
2. Зонд имеет в длину 600 нуклеотидов, и в нем содержится 150 остат-
ков С. Следовательно, 5-Ю6 имп./мин соответствуют 25/150 пмоль зонда.
В гибридизационную смесь вносят 10б имп./мин зонда (радиоактивность
кислотонерастворимой фракции), что соответствует 25/150 10®/5- 10б пмоль,
или 0,033 лмоль.
3. Сканирование радиоавтографа показывает, что радиоактивность защищен-
ного от действия РНКазы фрагмента (длиной 200 нуклеотидов) составля-
ет 20% радиоактивности зонда в разведении 1/1000. Поскольку защищен-
ный фрагмент короче зоида, эквимолярное по отношению к зонду количе-
ство защищенной РНК будет иметь суммарную радиоактивность, равную
200/600 радиоактивности зонда. Отсюда содержание защищенного фраг-
мента составляет 0,033X600/200X0,2X1/1000 пмоль в реакционной смеси,
или 1,98хЮ~17 * * * * * моль.
4. Согласно Авогадро, 1 моль вещества содержит 6,02-1023 молекул. Таким
образом, в процессе реакции защищенными от действия РНКазы оказы-
ваются 1,98-6,02-Ю6 молекул исследуемой мРНК, или 12-10® молекул.
5. В каждой гибридизации участвует 40 мкг суммарной РНК, которая по-
лучена из 8-Ю6 клеток. Следовательно, на 1 клетку приходится 12/8 мо-
лекул, или 1,5 молекулы исследуемой РНК.
линейной характеристики рентгеновской пленки). С учетом раз-
личий в размерах зонда и защищенного от действия РНКазы
фрагмента, а также степени разбавления можно подсчитать, ка-
кая часть меченого зонда оказалась защищенной от действия
РНКазы. Таким образом определяют число молей защищенно-
го зонда. Поскольку зонд находится в большом избытке по
сравнению с каждой из индивидуальных мРНК, можно считать,
что все соответствующие ему индивидуальные мРНК сгибриди-
зуются полностью. Отсюда можно вычислить, какое число мо-
лей исследуемой мРНК присутствовало в реакции гибридиза-
ции, и, исходя из числа Авогадро, подсчитать число молекул
мРНК- Если ткань, из которой выделяют исследуемую РНК,
гомогенна, то можно определить содержание РНК в одной
клетке. В противном случае эту величину оценивают приблизи-
тельно (в среднем суммарное содержание РНК в клетке млеко-
питающего составляет 5 пг). Так подсчитывают число молекул
исследуемой мРНК, приходящихся на одну клетку.
Если требуется определить представленность данной мРНК,
необходимо знать ее размер, чтобы, исходя из числа молей
последовательности, защищенной от РНКазы, можно было оп-
ределить количество мРНК, участвующей в реакции гибридиза-
ции. Если в реакционной смеси присутствует суммарная РНК,
Плазмиды, несущие промоторы для фаговых РНК-полимераз 35
установив содержание в ней poly (А)+РНК, можно вычислить
общее количество мРНК, участвующей в реакции; остается оп-
ределить, какую часть этого количества составляют мРНК, за-
щищенные от действия РНКазы.
1.5. Специальные методы
В этом разделе не будет приведено подробное описание ме-
тодик. В нем вкратце рассматриваются специальные методы,
которые могут оказаться полезными. Подробное описание мето-
дов, как правило, не трудно найти в оригинальных работах,
ссылки на которые мы приводим.
1.5.1. Гибридизация in situ
Радиоактивные молекулы РНК все шире используются
в качестве зондов в гибридизации in situ для выявления специ-
фических мРНК и определения их локализации в срезах тка-
ней. Эти методы впервые были описаны сотрудниками лабора-
тории Анджерера, и для детального ознакомления с ними мы
отсылаем читателей к работам [19, 20].
В реакции транскрипции могут использоваться радиоактив-
ные нуклеотиды, меченные 32Р, 35S или 3Н. Изотоп 32Р дает
такой радиоактивный сигнал, который не удается локализовать
на уровне отдельной клетки, но который легко можно обнару-
жить с помощью радиоавтографии всего за несколько дней.
Следует иметь в виду, что излучение 32Р невозможно достаточ-
но эффективно регистрировать с помощью стандартной эмуль-
сии; гораздо лучше это делать, экспонируя меченый препарат
с рентгеновской пленкой (Д. Дэвидсон, личное сообщение;
Р. Купмен, личное сообщение). Излучение 35S имеет гораздо
меньшую энергию, чем излучение 32Р; поэтому оно обеспечива-
ет лучшее разрешение, и, кроме того, меченные этим изотопом
нуклеотиды имеют более высокую удельную радиоактивность,
чем нуклеотиды, меченные тритием. Рибонуклеозидтрифосфаты,
меченные 35S, довольно эффективно включаются в РНК под
действием полимераз фагов SP6 или Т7.
Меченные тритием зонды дают самое высокое разрешение
на тканевых срезах, залитых эмульсией. Удельная радиоактив-
ность 3Н-меченных нуклеозидтрифосфатов не превышает 40—
60 Ки/ммоль, и поэтому их следует вносить в реакционную
смесь в концентрации 100 мкМ или выше, не добавляя холод-
ных трифосфатов.
Важным контролем, необходимым при гибридизации in situ,
служит гибридизация с зондом, который заведомо не будет
взаимодействовать ни с одной из РНК препарата. Это исклю-
3
36 П. Литтл, И. Джексон
чает вероятность получения ошибочного положительного ре-
зультата, обусловленного неспецифическим связыванием радио-
активного зонда клетками определенного типа, как это иногда
наблюдается (Р. Хаффнер, личное сообщение). Обычно удобно
в качестве контроля использовать РНК-транскрипт, комплемен-
тарный зонду («обращенный» зонд), т. е. молекулу, полученную
в результате транскрипции матрицы в обратном направлении.
Использование векторов, промоторы которых обеспечивают
транскрипцию и в прямом, и в обратном направлении, таких,
как плазмиды pGEM, позволяет синтезировать обе цепи.
1.5.2. Биологическая активность РНК,
синтезированной in vitro
1.5.2.1. Кэпирование и трансляция
На 5’-конце мРНК, выделяемой из эукариотических клеток,
обычно содержится так называемый «кэп» — структура, необхо-
димая для обеспечения стабильности мРНК и ее трансляции.
Такая структура может быть ферментативно присоединена
к 5’-концу синтезированной in vitro РНК, но это дорогой спо-
соб. Опубликованы работы, в которых показано, что фрагмент,
аналогичный кэпу, можно ввести в реакцию транскрипции и он
присоединится к 5’-концу синтезированной РНК [6, 21]. Ис-
пользуемая при этом методика совпадает с описанной
в разд. 1.3.3 с той лишь разницей, что аналог кэпа
G(5’)ppp(5’)G, mG (5’) ppp (5’) Gm или mG(5’)ppp(5’)G вклю-
чается в состав РНК при концентрации 500 мкМ, a GTP — при
концентрации 50 мкМ. Более подробное описание методики
можно найти в другой работе [6].
Показано, что РНК, как содержащая, так и не содержащая
кэп, может транслироваться in vitro в лизатах ретикулоцитов
и in vivo при микроинъекции в ооциты Xenopus [22, 23].
1.5.2.2. Другие функции РНК
РНК, синтезированные in vitro, были успешно использованы
для изучения сплайсинга in vivo и in vitro, поскольку с их по-
мощью можно получить большие количества псевдопервичных
транскриптов с высокой удельной радиоактивностью, что позво-
ляет обнаруживать даже низкий уровень ферментативной ак-
тивности [4]. Для получения таких субстратов не требуется
использования каких-то особых методик.
Транскрипты, представляющие собой цепи, комплементар-
ные мРНК — «антисмысловые» транскрипты, — также были
Плазмиды, несущие промоторы для фаговых РНК-полимераз 37
использованы во многих экспериментах, например для блокиро-
вания трансляции мРНК, инъецированных в ооциты Xenopus
[24], или для блокирования специфической трансляции продук-
та гена krilppell в эмбрионах Drosophila [25].
1.5.3. Специальные методы картирования;
«прогулка по геному»
Были сконструированы специальные векторы, в которых со-
держались промоторы для РНК-полимераз фагов SP6, Т7 или
ТЗ, расположенные в непосредственной близости от сайтов кло-
нирования распространенных фаговых и космидных векторов.
Наличие таких промоторов делает возможным быстрый синтез
радиоактивных РНК-зондов, специфичных по отношению к кон-
цам клонированного фрагмента ДНК; полученные зонды пред-
ставляют собой идеальный инструмент для «прогулки по гено-
му». Это общий метод выделения фрагментов ДНК, располага-
ющихся в геноме по соседству с уже клонированным в составе
соответствующего вектора участком ДНК. Постановка таких
экспериментов с использованием космид описана в работе [26]
и гл. 2 этой книги.
1.5.4. Секвенирование РНК-транскриптов
РНК можно секвенировать с помощью метода, идея которо-
го идентична идее дидезокси-терминации Сэнгера.
Были опубликованы подробные описания методик с исполь-
зованием РНК-полимераз фагов Т7, SP6 [27] и фага ТЗ [9].
Суть метода состоит в использовании З’-дезоксианалогов рибо-
нуклеозидтрифосфатов в низких концентрациях, чтобы синтези-
руемые молекулы РНК статистически терминировались при
включении модифицированных оснований. Затем, используя
стандартные гели, определяют нуклеотидную последователь-
ность РНК, причем и принципиально, и методически такое сек-
венирование идентично методу Сэнгера.
Возможности этих методов еще не до конца раскрыты. В ча-
стности, использование радиоактивных З’-дезоксинуклеотидов
существенно увеличивает точность секвенирования, но пока еще
не все из них имеются в продаже. Секвенирование РНК облег-
чается при использовании систем с промоторами, работающими
в двух направлениях (например, плазмид pGEM, векторов
phagescript и bluescript). Это уменьшает количество генноин-
женерных манипуляций, необходимых для определения нуклео-
тидной последовательности обеих цепей.
38
П. Литтл, И. Джексон
1.6. Заключение
В настоящее время все более широкое применение находит
использование РНК в качестве специфических гибридизацион-
ных зондов, а также в качестве субстратов и реагентов при
анализе широкого круга биологических процессов. Чрезвычай-
но высокая специфичность кодируемых фагами полимераз и
небольшие размеры их промоторов (во многих случаях это все-
го 15 пар оснований) открывают путь новым методическим под-
ходам, которые не следует игнорировать. Возможность исполь-
зования готовых коммерческих наборов или отдельных компо-
нентов для синтеза РНК in vitro с помощью РНК-полимераз
фагов SP6, Т7 и ТЗ значительно облегчает решение экспери-
ментальных задач, в которых требуется синтезировать меченые
зонды, а некоторые уникальные функции и специфические свой-
ства РНК в ряде случаев делают описанные системы просто не-
заменимыми.
Благодарности
Работа П. Ф. Р. Литтла обеспечивалась грантом, предостав-
ленным совместно Советом по медицинским исследованиям и
Ассоциацией по изучению рака. И. Дж. Джексон — сотрудник
Листеровского института профилактической медицины; его ра-
бота над книгой финансировалась Советом по медицинским ис-
следованиям.
Литература
1. Chamberlin М., Kingston R., Gilman М., Wiggs J., de Vera A. (1983). In:
Methods in Enzymology, Wu R., Grosman L., Moldave K. (eds), Academic
Press, New York, Vol. 101, p. 540.
2. Butler E. T„ Chamberlin M. J. (1982). J. Biol. Chem., 257, 5772.
3. Losich R., Chamberlin M. (eds.) (1976). RNA Polymerase, Cold Spring Har-
bor Laboratory, Cold Spring Harbor, NY.
4. Green M. R., Maniatis T., Melton D. A. (1983). Cell, 32, 681.
5. Melton D. A., Krieg P. A., Rebagliati M. R., Maniatis T., Zinn K-, Green M. R.
(1984). Nucleic Acids Res., 12, 7035.
6. Krieg P. A., Melton D. A. (1987). Methods in Enzymology, in press.
7. Nam H. G., Loechel S., Fried H. M. (1986). Gene, 46, 57.
8. Schenborn E. T„ Mierendorf R. C. (1985). Nucleic Acids Res., 13, 6223.
9. Klement J. F., Ling M.-L„ McAllister It". F. (1987). Gene Analysis Techniques,
in press.
10. Berk A. J., Sharp P. A. (1977). Cell, 12, 721.
11. Jackson 1. J., Schofield P„ Hogan B. (1985). Nature, 317, 745.
12. Lovell-Badge R. H., Bygrave A. E., Bradley A., Robertson E., Evans M. J.,
Cheah K. S. E. (1985). Cold Spring Harbor Symp. Quant. Biol., 50, 707.
13. Zinn K-, di Maio D., Maniatis T. (1984). Cell, 34, 865.
14. O'Hare K., Levis R„ Rubin G. M. (1983). Proc. Natl. Acad. Sci. USA,
80, 6917.
Плазмиды, несущие промоторы для фаговых РНК-полимераз 39
Birnboim Н, С., Doly J. (1979). Nucleic Acids Res., 7, 1513.
Lis J. T. (1980). In: Methods in Enzymology, Grossman L., Moldave K.
(eds.), Academic Press, New York, Vol. 65, p. 347.
Gosule L. C„ Schellman J. A. (1978). J. Mol. Biol., 121, 311.
Church G. M., Gilbert W. (1984). Proc. Nat. Acad. Sci. USA, 81, 1991.
Lynn D. A., Angerer L. M„ Bruskin A. M., Klein W. H., Angerer R. C.
(1983). Proc. Natl. Acad. Sci. USA, 80, 2656.
Сох К. H„ DeLeon D. V., Angerer L. M.t Angerer R. C. (1984). Dev. Bicl.,
101, 485.
Konarska M. M., Padgett R. A., Sharp P. A. (1984). Cell, 38, 731.
Krieg P. A., Melton D. A. (1984). Nucleic Acids Res., 12, 7057.
Drummond D. R., Armstrong J., Colman A. (1985). Nucleic Acids Res., 13,
7375.
Melton D. A. (1985). Proc. Natl. Acad. Sci. USA, 82, 144.
Rosenberg U. B., Preiss A., Seifert E„ Jackie H„ Knipple D. C. (1985). Nature,
313 703
Cross S' H„ Little P. F. R. (1986). Gene, 49, 9.
Axelrod U. O„ Kramer F. R. (1985). Biochemistry, 24, 5716.
Maniatis T., Fritsch E. F., Sambrook J. (1982). Molecular Cloning — A Labo-
ratory Manual, Cold Spring Harbor Laboratory, NY. [Имеется перевод:
Маниатис T., Фрич Э., Сэмбрук Дж. Методы генетической инженерии. Мо-
лекулярное клонирование. — М_: Мир, 1984.]
ГЛАВА 2
ВЫБОР И ИСПОЛЬЗОВАНИЕ КОСМИДНЫХ ВЕКТОРОВ
Питер Ф. Р. Литтл
(Peter F. Р. Little)
2.1. Введение
2.1.1. Краткое содержание главы
Идеальный вектор для клонирования ДНК высших эукариот
должен обладать как можно большей емкостью, чтобы можно
было обойтись меньшим количеством циклов скрининга библио-
теки для выделения клонированных перекрывающихся фрагмен-
тов крупного гена. В нынешнем поколении векторов наиболь-
шей емкостью обладают космидные векторы, но используют их
пока реже, чем векторы, полученные на основе фага L. Объяс-
няется это частично непредсказуемостью поведения космидных
векторов, а также существенно большей трудоемкостью мето-
дов получения космидных библиотек по сравнению с эквива-
лентными фаговыми библиотеками. В задачу данной главы вхо-
дит описание ряда разработанных в нашей лаборатории мето-
дик, а также характерных случаев и наиболее распространен-
ных ошибок, мешающих созданию полноценной библиотеки.
Мы должны подчеркнуть, что техническая сложность проводи-
мых экспериментов затрудняет детальный анализ всех аспектов
космидного клонирования. Составленный нами свод методик
должен рассматриваться лишь как руководство и допускает
возможные отклонения. Однако следует иметь в виду, что не
имеющие большого опыта исследователи не должны серьезно
от них отклоняться до тех пор, пока не приобретут сами доста-
точного навыка, чтобы позволить себе импровизировать.
2.1.2. Космидные векторы
Первые космидные векторы разработали Коллинз и Хон [1]
для решения технических проблем, связанных с введением
больших фрагментов ДНК в Esherichia coli. Большинство мето-
дик получения компетентных клеток Е. coli обеспечивает пере-
нос молекул ДНК размером до 10—15 т. п. н., а попытки ис-
пользовать ДНК большей длины приводят к резкому уменьше-
нию эффективности трансформации. Решающее значение для
успешного внедрения больших плазмид в бактериальные клет-
40
Выбор и использование космидных векторов
41
ки имеет, по-видимому, перенос ДНК через мембрану или, воз-
можно, процессы, происходящие сразу после проникновения
ДНК в периферию цитоплазмы.
Для преодоления этих трудностей Коллинз и Хон [1] ввели
cos-последовательности фага X в подходящий (4—5 т. п.н.)
плазмидный вектор для клонирования. Это позволило использо-
вать метод упаковки фага in vitro [2], который полностью иск-
лючает наблюдающиеся при трансфекции сложности, связан-
ные с размерами ДНК. Спаренная соз-последовательность и
примерно 200 п. н. с каждой стороны составляют функциональ-
ный блок, требующийся для упаковки ДНК в частицы фага X.
Для успешной упаковки не требуется каких-либо специфиче-
ских сегментов ДНК. Нужно только, чтобы две соз-последова-
тельности находились в составе одной молекулы ДНК, будучи
разделены участком в 38—52 т. п. н.; таковы предельные разме-
ры молекул ДНК, пригодных для упаковки данного фага. Ре-
комбинантные космидные молекулы состоят из вставочного
фрагмента ДНК длиною 30—45 т. п. н., например ДНК человека
и крсмидного вектора, соединенных между собой в результате
реакции лигирования, так что формируются длинные конкатеме-
ры космида — человеческая ДНК. — космида. Этим обеспечива-
ется специфическое распределение cos-сайтов, необходимое для
успешной упаковки такой ДНК in vitro в реакции, которая
в сущности аналогична процессам, протекающим in vivo. Эф-
фективность упаковки in vitro достигает 109 бляшек (стериль-
ных пятен) на 1 мкг ДНК фага X. Это означает, что не более
5% молекул образуют функциональные фаговые частицы. Од-
нако эффективность трансдукции космидной ДНК гораздо ни-
же— не более 106 колониеобразующих единиц (к. о. е.) в расче-
те на 1 мкг ДНК, а обычно еще меньше.
2.1.3. Выбор векторов
Основным критерием выбора именно космидного вектора
должна быть необходимость клонирования очень больших фраг-
ментов ДНК; в противном случае всегда легче и быстрей скон-
струировать фаговую библиотеку.
Существует много различных космидных векторов (см. Ма-
ниатис и др. [3], Пауэле и др. [4]). Наиболее важное значение
из их свойств имеют лекарственная устойчивость, характер реп-
ликона и наличие подходящих сайтов рестрикции. Рассмотрим
их по порядку.
2.1.3.1. Лекарственная устойчивость
По довольно сложным и пока еще недостаточно изученным
причинам присутствие космид часто приводит к замедлению
клеточного роста, нередко сопровождающемуся недостаточной
42
П. Литтл
устойчивостью к широко используемым антибиотикам, таким,
как ампициллин, что может уменьшать селективное давление
на космиду. Кроме того, на начальной, медленной стадии роста
космидосодержащих клеток может наблюдаться также образо-
вание сателлитных колоний. Большинство космидных векторов
содержит детерминанту устойчивости к ампициллину, но при
прочих равных условиях эта детерминанта не является опти-
мальной. Устойчивость к тетрациклину не имеет указанных не-
достатков, однако этот антибиотик нестабилен. В некоторых
векторах используется устойчивость к стабильным антибиоти-
кам—канамицину (или неомицину), детерминируемая двумя
различными генами. Их главный недостаток — большое время,
требующееся для максимальной экспрессии (разд. 2.3.5.3).
Некоторые космиды несут кроме прокариотических маркеров
лекарственной устойчивости гены,, обеспечивающие селекцию
эукариотических клеток. Наиболее удобные из них — это гены
тимидинкиназы и устойчивости к антибиотику G418 [4]; но в
принципе можно использовать и некоторые другие. Наличие
селектированного маркера, облегчающего введение ДНК в эу-
кариотические клетки, может служить решающим критерием
при выборе вектора. Описаны подходы, предполагающие «спа-
сение» космидной ДНК в подобных экспериментах, что часто
требует определенных комбинаций детерминант устойчиво-
сти [5].
2.1.3.2. Репликон
Большинство космид, пригодных для использования в
Е. coli, — производные pBR322, основанные на репликоне рМВ1.
Система контроля этого репликона не идеальна для использо-
вания в космидных векторах (см. обсуждение у Литтла и Крос-
са [6]) и может вызвать перегрузку библиотек маленькими
космидами в результате res Д-независимых делений. Наличие
промоторов в ДНК-вставке сопряжено с дополнительными про-
блемами, связанными с нарушением регуляции [6—8]. Исполь-
зование области начала репликации фага % снимает многие,
но не все трудности [7]. В двух космидах имеется область на-
чала репликации R6K. Неизвестно, как она соотносится с реп-
ликонами рМВ1 или X. Проблема выбора репликона подверга-
лась тщательному анализу в ряде масштабных экспериментов
по клонированию; предварительные данные опубликовали Кол-
сон и др. [9].
2.1.3.3. Сайты рестрикции
Первичным условием при выборе векторов должно быть на-
личие подходящих для клонирования сайтов (обсуждавшихся
в разд. 2.1.4.1). Кроме того, существенное значение имеет пра-
Выбор и использование космидных векторов
43
ви^тьная локализация этих сайтов: она должна обеспечивать
возможность конструирования «плеч» вектора с помощью про-
цедуры, описанной ИшТоровицем и Берком [10], наличие сай-
тов,'фланкирующих сайт (ы) клонирования (что позволяет рас-
щеплять клонируемую ДНК вне векторной части), и, наконец,
присутствие между парами cos-последовательностей сайтов, об-
легчающих образование космидных «плеч» [11].
2.1.4. Стратегия клонирования
2.1.4.1. Выбор ферментов для клонирования
Чаще всего стратегия клонирования основана на использо-
вании частичных гидролизатов Sau3M и введении полученных
фрагментов в сайт ВапгЪП. Это вполне удовлетворительная сис-
тема, и к настоящему времени создано множество подходящих
векторов с сайтами ВатШ. Однако анализ полученных таким
путем клонов, например для идентификации космид с перекры-
вающимися вставками, затруднен, поскольку слияние Sau3AI-
и BamHI-сайтов приводит в большинстве случаев к элиминации
сайта BamHl и в результате концы вставленной ДНК не могут
быть проанализированы. По этой причине трудно анализиро-
вать концы, перекрывающиеся в небольшой степени (т. е. такие
ДНК, у которых гомологичны только концы вставок). Клони-
рование частичных гидролизатов ДНК, полученных с помощью
ферментов рестрикции, узнающих шестинуклеотидные последо-
вательности, в том же самом сайте позволяет анализировать
все клонированные фрагменты. Репрезентативность библиотеки,
полученной таким путем, теоретически очень высока [12], а на
практике представленность клонов в библиотеках, полученных
на основе частичных гидролизатов £coRI и Sau3M, очень близ-
ка, если не идентична [9]. Преимущество более легкого анализа
клонов существенно для экспериментов, связанных с картиро-
ванием или «прогулкой» по геному.
2.1.4.2. Процедура клонирования
Существует два основных метода получения фрагментов
эукариотической ДНК подходящих размеров: 1) физическое
фракционирование частичного гидролизата и отбор фракции
нужного размера (разд. 2.2.1.3) и 2) дефосфорилирование сум-
марного частичного гидролизата с последующим использовани-
ем естественного отбора по размеру за счет упаковки in vitro —
отбираются фрагменты размером 35—45 т. п.н. (разд. 2.2.1.4).
Первый метод более расточителен в плане исходного материа-
ла: требуется приблизительно 300 мкг ДНК, чтобы получить
44
П. Литтл
фракционированные фрагменты в количестве, достаточном Для
конструирования хорошей библиотеки (~106 колоний на 1 мкг
эукариотической ДНК). Фосфатазный метод более экономичен
в отношении исходной ДНК, но значительно менее эффективен
(никогда не удается получить более 5/104 клонов в расчете на
1 мкг, а в нашей практике обычно и того меньше).
2.2. Методы
2.2.1. Получение ДНК для клонирования
2.2.1.1. Выделение ДНК из клеток или тканей
При конструировании космидной библиотеки, вероятно, наи-
более важное значение имеет качество используемой ДНК-
Лишь около трети фрагментов ДНК длиной 40—50 т. п. н., по-
лученных из молекул с исходным размером 100 т.п.н., содер-
жат сайты рестрикции на обоих концах. Разорванные молекулы
конкурируют в реакции лигирования, что делает невозможным
создание хорошей библиотеки. При выделении ДНК необходи-
мо соблюдать следующее условие: на каждой из стадий выде-
ления (частичный гидролиз, градиентное центрифугирование и
т. д.) рекомендуется проверять ДНК. в 0,2%-ном агарозном геле.
Инструкции по применению этих гелей приведены в разд. 2.3.4.
Для получения высокомолекулярной ДНК мы используем ме-
тод, детали которого описаны в табл. 2.1. Метод этот пригоден
при работе со свежими и с замороженными клетками и тканями.
2.2.1.2. Частичный рестриктазный гидролизат
Мы получали пробные частичные гидролизаты, используя
двукратные серийные разведения фермента. Подходящий гидро-
лизат определяли по методу Сида и др. [13]. Обычно мы ис-
пользуем два-три варианта условий частичного гидролиза, что-
бы получить различную кинетику реакции. Метод, описанный
здесь, очень сходен с методом, описанным Маниатисом и др.[3].
1. Используйте ДНК, очищенную, как описано в разд. 2.2.1.1,
и разводите ее только непосредственно перед каждым исполь-
зованием. Отберите 180 мкл и добавьте 20 мкл соответствующе-
го 10-кратного рабочего буферного раствора рестриктирующей
эндонуклеазы (рестриктазы) и тщательно перемешайте.
2. Отберите аликвоту в 40 мкл и распределите остаток —по
20 мкл в 8 других пробирок. Пометьте их номерами 1—9.
3. Добавьте избыток рестриктазы к пробирке № 1, содержа-
щей аликвоту 40 мкл. Мы обычно используем 2 мкл рестрикци-
онной эндонуклеазы в концентрации 10—20 ед./мкл (за 1 ед.
Выбор и использование космидных векторов 45
Таблица 2.1. Получение высокомолекулярной эукариотической ДНК
Растворы
1. Рвсуспендируйте или гомогенизируйте приблизительно 10* 4 s * * *—109 клеток (или
0,о—1 г ткани) в 5 мл 1XSSC1. Нужно быть уверенным, что ресуспенди-
ро^аиие полное.
2. Добавьте 5 мл 100 мМ трис-НС1-буфера (pH 7,5), содержащего 100 мМ
NaCl, 10 мМ ЭДТА, 1%-ный саркозил. Тщательно перемешивайте 2—3 мин,
чтобы лизировать клетки. Добавьте протеиназу К до концентрации
100 мкг/мл. Инкубируйте при 55 °C в течение 2 ч.
3. Один раз экстрагируйте фенолом, добавив равный объем фенола, насы-
щенного ТЭ. Смешайте мягким переворачиванием закрытую пробирку и
разделите фазы центрифугированием (табл. 1.1). Отберите водную фазу
в чистую пробирку и проведите дальнейшую экстракцию равным объемом
смеси фенол-хлороформ (1 : 1). Наконец экстрагируйте один раз равным
объемом чистого хлороформа. Если использовать слишком много клеток,
то первый фенольный экстракт будет очень плотным, почти твердым.
Если это так, разведите ДНК лизирующим раствором, указанным в п. 2.
Первый фенольный экстракт часто неоднороден, но интерфаза должна
быть, насколько это возможно, отделена от фенольной фазы. Последую2
щие фенол-хлороформный и хлороформный экстракты должны быть более
прозрачными.
4. Диализуйте конечную водную фазу против 4 л ТЭ2, содержащего 100 мМ
NaCl, при 4 °C в течение 17 ч (ночь) или дольше.
5. Повторите диализ против 4 л ТЭ при 4 °C 24 ч.
в. Теперь ДНК готова к использованию. Концентрацию нужно определить
электрофорезом в геле с использованием в качестве маркера ДНК фага Л
в известной концентрации, так как препарат содержит РНК, которая
будет приводить к неточным результатам при спектроскопии при 260 нм.
Если все было сделано правильно, то концентрация ДНК будет 100—
300 мкг/мл.
1 1XSSC=O,15 М NaCl, 0,015 М цитрат натрия.
3 ТЭМ0 мМ трис-НС1 (pH 7,5), 1 мМ ЭДТА.
принято количество фермента, расщепляющего полностью 1 мкг
ДНК фага X в течение 1 ч). Тщательно перемешайте, чистым
носиком отберите 20 мкл и добавьте в пробирку № 2. Переме-
шайте и повторите эту процедуру с последующими пробирками.
В пробирку № 9 ничего не добавляйте.
4. Инкубируйте при 37 °C 30 мин, затем перенесите пробирки
в лед.
5. Проверьте длину молекул ДНК в 1%-ном агарозном геле
(аликвота 5 мкл). Вы должны увидеть полную или почти пол-
ную рестрикцию в пробирке № 1 и постепенное уменьшение
полноты гидролиза в направлении к пробирке № 8. Если это
наблюдается, проанализируйте 10 мкл реакционной смеси в
0,2%-ном агарозном геле (разд. 2.3.4). Подберите такие усло-
вия рестрикции, чтобы ДНК размером более 50 т. п. н. и менее
40 т. п. н. при фотографировании 0,2%-ного геля не были видны.
Тогда нужный гидролизат будет не этот — в котором основная
часть ДНК находится в этом интервале, — а следующий за ним.
46 П. Литтл
Обсуждение этого вопроса приводится в статье Сида и др. [JB].
Для препаративной рестрикции используйте найденные точные
условия частичного гидролиза и — для гарантии — близкие-ва-
рианты (±50% фермента). (
6. Важно воспроизвести препаративную реакцию точна при
найденных условиях. Из-за больших объемов, вероятно, понадо-
бится заранее нагреть раствор ДНК до нужной температуры.
Рассчитайте объем фермента, требующийся для реакции. Не
меняйте концентрации буфера, фермента или ДНК. Постарай-
тесь получить как можно больше расщепленной ДНК: потре-
буется 200—300 мкг для очистки в градиенте концентрации са-
харозы (разд. 2.2.3).
7. Проверьте ДНК электрофорезом в 1%-ном геле — вы
должны увидеть едва заметную размытую зону свечения ниже
зоны исключенных размеров. Если это так, отберите маленькую
аликвоту из каждой пробы для анализа в 0,2%-ном агароз-
ном геле.
8. Объедините частично гидролизованную ДНК и экстраги-
руйте один раз фенолом, один раз хлороформом и осадите эта-
нолом, как описано в табл. 1.1. Ресуспендируйте ДНК в буфере
ТЭ до концентрации 300 мкг/мл.
2.2.1.3. Очистка фракции 30—50 т. п. н. частичного гидролизата
в градиенте концентрации сахарозы
1. Мы использовали для этой цели сахарозные градиенты.
Приготовьте 40- и 10%-ные (вес/объем) растворы сахарозы в
буфере, содержащем 1 М NaCl, 20 мМ трис-НС1 (pH 7,1),
20 мМ ЭДТА, 0,3% (по объему) саркозила.
Мы пользовались ротором Beckman 28/27.1 и 38 мл-градиен-
тами. Градиенты наслаивайте смесителем за 1—1,5 ч до цент-
рифугирования и убедитесь, что остается место для образца
ДНК; если это необходимо, отберите 1 или 2 мл. Для формиро-
вания градиента можно также наслоить накануне в пробирку
равные объемы 40, 35, 30, 25, 20, 15 и 10% сахарозы и оставить
на ночь. Аккуратно наслоите в каждую пробирку 1мл раствора
ДНК, но не более 300 мкг на градиент. Мы обычно используем
3 градиента и одну пробирку для уравновешивания. Центрифу-
гируйте при 26 000 об/мин 16 ч при 10 °C.
2. Раскапайте градиент, проколов дно пробирки иглой, при-
соединенной к шлангу насоса, и соберите фракции по 0,7—1 мл.
ДНК нужного размера должна быть приблизительно в 15—25-й
фракциях. 15 мкл из каждой фракции проверьте электрофоре-
зом в 0,2%-ном геле. Не забудьте добавить в лунку с маркера-
ми раствор сахарозы в высокосолевом буфере, использованном
в градиенте, так чтобы его концентрация в пробе составляла
Выбор и использование космидных векторов 47
около 30%. Это существенно, так как высокая концентрация
сожг очень влияет на подвижность ДНК-
S. Осадите ДНК из фракции, предположительно содержащей
фрагменты нужного размера добавлением двух объемов этано-
ла. Осаждение сахарозы можно предотвратить добавлением до-
полнительных 2 мл 70%-ного этанола. Тщательно перемешайте
и оставьте при —20 °C по крайней мере на ночь или, если это
возможно, дольше. Осадите ДНК центрифугированием, как опи-
сано в табл. 1.1. Тщательно промойте осадок дважды 70%-ным
этанолом и ресуспендируйте приблизительно в 50 мкл буфера
ТЭ. Около 1 мкл нанесите на 0,2%-ный гель, чтобы идентифи-
цировать нужные фракции (50—40 т. п. н.). Соберите эти фрак-
ции и доведите до 0,3 М NaCl. Осадите ДНК двумя объемами
этанола. Соберите осадок центрифугированием и ресуспенди-
руйте ДНК в буфере ТЭ до конечной концентрации около
1—2 мкг/мкл.
2.2.1.4. Обработка фосфатазой
1. Получите частичные рестрикционные гидролизаты, как
описано в разд. 2.2.1.2, ресуспендируйте ДНК в концентрации
200 мкг/мл после осаждения этанолом в 10 мМ трис-НС1
(pH 7,5) и 10 мМ ЭДТА.
2. Добавьте избыток фосфатазы кишечника теленка (ФКТ) —
примерно 0,5 ед./мкг ДНК (это количество может варьировать
в зависимости от условий). Инкубируйте 20 мин при 37°C, за-
тем добавьте NaCl до 200 мМ.
3. Проведите фенольную экстракцию, осадите ДНК этано-
лом и промойте 70%-ным этанолом, как описано в табл. 1.1.
4. Ресуспендируйте ДНК в ТЭ-буфере до концентрации
1 мкг/мкл.
Чтобы в ФКТ-реакции избежать активации ДНКазы двух-
валентными катионами, необходимо поддерживать высокую
концентрацию ЭДТА.
2.2.2. Приготовление вектора
Выбор ферментов, используемых для двойного расщепления
космидного вектора при приготовлении «плеч» по методу Иш-
Горовица и Берка [10], определяется характеристиками данной
космиды. На рис. 2.1 приведена схема общего метода для лю-
бого космидного вектора. Методика, детально описанная в
табл. 2.2, касается семейства космид Лориста [7, 8], где фермент
X — это SstI, фермент Y — BsiEII, а сайт клонирования
ЯшсПП.
Альтернативная методика состоит в обработке фосфатазой
вектора, расщепленного в сайте клонирования. Результат зави-
48
П. Литтл
cos
Ж—М--
С COS'
К----------ЛИ-----у
I
с с
I
! I Лигаза
I ♦
I
[ Вставка 40 т.п.н.
Упаковка
4
Рис. 2.1. Конструирование рекомбинантов по методу Иш-Горовица и Бер-
ка [10]. с — сайт клонирования, х и у — другие уникальные рестрикционные
сайты. Космиды расщепляют один препарат по сайту х, другой по сайту у
и обрабатывают фосфатазой. Затем каждый препарат разрезают по сайту с.
Нужные «плечи»—те, которые несут cos-сайты в правильной (показано
маленькой стрелкой) ориентации. Желательно, хотя и необязательно, индиви-
дуально выделять необходимые фрагменты до лигирования. Методика по-
строена так, чтобы исключить образование мультимеров космид, которые могли
бы эффективно вовлекаться в упаковку.
сит от качества препарата ФК.Т; фермент используют в мини-
мальном количестве в, буфере, описанном в разд. 2.2.1.4. В ре-
зультате реакции образуются желаемые нелигируемые концы
векторной ДНК-
Наш опыт показывает, что ФКТ не только удаляет концевые
фосфаты, но часто также повреждает липкие концы. Клониро-
вание материала, обработанного фосфатазой, всегда менее эф-
фективно, чем аналогичные эксперименты с «плечами»; послед-
ний метод, хотя и более трудоемкий, более надежен в работе.
Выбор и использование космидных векторов 49
Таблица 2.2. Приготовление «плеч» космидного вектора
1. Проведите полную рестрикцию двух препаратов «лористВ», одного SstI,
а щругого BstEII. Пользуйтесь инструкциями фирмы-изготовителя при
рестрикции. Отберите небольшую аликвоту для контроля на этапе 3.
2. Обработайте каждый гидролизат ФКТ («Molecular bioiogy grade»,
Boehringer), Добавьте избыток ФКТ прямо в рестрикционную смесь и
продолжайте инкубацию еще 30 ,мин при 37 °C. (Мы использовали 56 ед.
ФКТ для дефосфорилированпя 150 мкг в объеме около 500 мкл.) Экстра-
гируйте фенолом и затем хлороформом, как указано в табл. 1.1. Осадите
ДНК изопропанолом, промойте 70%-ным этанолом и ресуспендируйте
в ТЭ (150 мкг ДНК в 300 мкл ТЭ).
3. Отберите аликвоты обработанных фосфатазой «плеч» и убедитесь, что они
не способны лигироваться в течение ночи при использовании стандарт-
ных буферов и условий для лигирования (разд. 2.2.3). В качестве контро-
ля вы можете использовать аликвоту, полученную на этапе 1.
4. Если лигирования нет, обработайте оба образца ffindlll. Нужно быть
уверенным в хорошем качестве фермента и брать минимальное его ко-
личество для полной рестрикции. Проверьте полноту реакции электрофоре-
зом аликвоты в 1%-ном агарозном геле.
5. Если реакция на этапе 4 прошла полностью, проведите лигирование в али-
квоте каждого препарата отдельно. Проверьте полноту лигирования элек-
трофорезом реакционной смеси в 1%-ном агарозном геле. Вы должны
увидеть, что фрагмент размером около 10 т. п. н., являющийся большим
фрагментом в каждой рестриктазной реакции, замыкается сам на себя.
Не должно быть материала, расположенного выше. Мы обнаружили, что
лигирующие буфер и фермент могут повлиять на подвижность фрагмен-
тов, так что поставьте соответствующие контроли.
6. Если все препараты лигированы правильно, экстрагируйте фенолом всю
реакционную смесь, полученную на этапе 4. Осадите ДНК из водной
фазы изопропанолом, как в табл. 1.1, промойте 70%-ным этанолом и
растворите в небольшом объеме ТЭ. Выход каждого «плеча» должен со-
ставить около 1 мкг/мкл.
2.2.3. Реакция лигирования
Лигирование следует проводить в буфере, рекомендованном
фирмой-изготовителем. Используйте только нейтрализованный
АТР, поскольку при работе с не-нейтрализованным АТР pH
реакционной смеси может значительно отличаться от опти-
мального.
1. Приготовьте следующую реакционную смесь:
ДНК -вставка
«Плечо» 1
«Плечо» 2
Ю-кратный лигазный буфер
Вода
5 ед. ДНК-лигазы фага Т4
2. Инкубируйте при 22 °C 2 ч или при 15 °C 17 ч.
3. После этого проверьте аликвоту электрофорезом в 0,2 %-
ном агарозном геле. Лигирование вектора должно быть очевид-
3 мкг
1,5 мкг
1,5 мкг
2 мкл
20 мкл (конечный объем)
4—1186
.50
П. Литтл
пым, а лигирование вставши — едва заметным, так как исполь-
зован 10-кратный молярный избыток «плеч».
4. Если лигирование вектора имеет место, переходите к ре-
акции упаковки. Если лигирования не наблюдается, то, вероят-
но, в реакционную смесь цопали сахароза и(или) саркозил из
градиента. Чтобы поправить дело, вновь осадите ДНК двумя
объемами этанола, отцентрифугируйте и промойте осадок ДНК
70%-ным этанолом. Дайте осадку высохнуть на воздухе, вновь
растворите его в буфере для лигазной реакции и повторите ли-
гирование.
2.2-4. Упаковка
Коммерческие экстракты для упаковки высокоэффективны,
но дороги. Тем не менее они наиболее удобны, и мы обычно
успешно используем экстракты Amersham и Stratagene.
1. Упакуйте 4 мкл лигированной смеси точно так, как ука-
зано изготовителем. Не питайтесь брать в реакцию больший
объем лигазной смеси.
2. По истечении необходимого времени добавьте в реакцион-
ную смесь 1 мл буфера для разведения фага [10 мМ трис-НС1
(pH 7,4), 10 мМ MgSO4, 0,01% (вес/объем) желатина].
3. Определите титр фата в 1 и 10 мкл реакционной смеси,
как описано в разд. 2.2.5.3.
2.2.5. 0Ь1сев библиотеки
2.2.}-1- Выбор клеток
Основное условие — клетки должны быть дефектными по
способности к рекомбинации. Обычно это бывает обусловлено
мутацией гена гесА, приводящей к дефекту по рекомбинации
и к чувствительности к ультрафиолету. Мутации гесА часто
могут ревертировать, и поэтому важно использовать аллель
гесА, ревертирующую с низкой частотой (например, гесА-деле-
цию). Мы получаем хорошие результаты со штаммами ED8767,
являющимися гесАБб supE supE hsdS~met~ [14], и 1046, по-види-
мому, идентичным FD8767- Клетки должны быть проверены на
присутствие аллеля гесА до их чувствительности к ультрафио-
лету (методика описана Маниатисом и др. [3]).
Другие гдс-мутации нужно использовать с осторожностью.
Генотип recBCsbcB, который, как сообщалось, стабилизирует
фаги X, содержащие повторы ДНК «голова к голове» [15], не
поддерживает роста плазмид с репликоном рМВ1, и мы столк-
нулись с серьезными затруднениями при размножении космид,
Выбор и использование космидных векторов
51
। ______Таблица 2.3, Адсорбция и высев упакованных космид___________
1. Добавьте по 10 мл свежей насыщенной культуры Е. colt ED8767 в расче-
те на каждый миллилитр упаковывающей смеси. Инкубируйте при 37 °C
30'мин.
2. Разведите в 25 мл подогретой L-среды (не содержащей антибиотиков) и
инкубируйте 1 ч при 37 °C.
3. Осадите клетки на низкой скорости (5 мин, 2000 об/мин) в препаративной
центрифуге типа Sorvall RC5 и суспендируйте в 1,2 мл L-среды.
4. Высевайте на соответствующие чашки не более чем по 500 мкл этой
смеси.
полученных на основе pBR322 и л в клетках recBCsbcBrecA,.
которые, по имеющимся данным, поддерживают рост
pBR322 [16].
2.2.5.2. Подготовка клеток
Мы растим культуру ED8767 до насыщения (ночь) при 37 °C
и эффективной аэрации в L-среде, содержащей 10 мМ MgSO4
и 0,1% (вес/объем) мальтозы. Добавление последней, по-види-
мому, не столь существенно. Имеется несколько более сложных
методик подготовки клеток, которые, может быть, несколько-
более эффективны, но мы не видели необходимости их исполь-
зовать.
2.2.5.3. Адсорбция упакованных космид на Е. coli
Упаковывающие экстракты могут убивать бактерии, несущие
космиды. Если объемное соотношение разведенной упаковываю-
щей смеси и стационарной культуры больше, чем 1:4—1:2,
титр космид резко снижается. Мы обнаружили также, что ка-
кой-то компонент упаковывающей смеси способен подавлять
рост бактерий на чашках, даже если он присутствует,в большем
разведении, чем 1:4. Мы разработали методику (табл. 2.3),.
которая, по-видимому, позволяет решить эту проблему и обес-
печивает достаточно времени для того, чтобы даже медленно
экспрессирующийся ген неомицин-фосфотрансферазы (КпУ)
экспрессировался полностью. Эта методика может быть исполь-
зована как для аналитических, так и для препаративных целей,
но не следует высевать на чашки более 500 мкл конечной сус-
пензии клеток.
2.2.5.4. Эффективность процедуры клонирования
Эффективность получения космид сильно варьирует, и мож-
но ожидать конечного выхода 104—106 колоний в расчете на
1 мкг эукариотической ДНК- В среднем мы получали величину
4*
52
П. Литтл
5-Ю4—5-Ю5, однако обычны и более низкие значения. В случае
генома человека, согласно статистическим расчетам Кларка и
Карбона [17], требуется 345 000 космид для 99%-ной представ-
ленности генов в библиотеке (принимая емкость вектора
40 т. п. н. и размер генома 3 109 т. п. н.).
2.2.6. Рассев библиотеки
2.2.6.1. Высев на агар для амплификации библиотеки
Обычно на чашку диаметром 14 см можно высеять 400—
'500 мкл суспензии клеток, полученных, как описано в
разд. 2.2.5.3. В стандартном опыте (50 000 космид из реакции
упаковки) хорошим выходом можно считать около 17 000 коло-
ний на чашку. Применяйте чашки с толстым слоем L-arapa
(100 мл на чашку), в который добавлен соответствующий ан-
тибиотик. Если возможно, используйте чашки, разлитые один-
два дня назад, тогда они не будут слишком влажными. После
высева, если нужно, подсушите чашки в стерильном боксе и
инкубируйте, перевернув вниз крышкой, в течение 24 ч.
Теперь с чашек может быть сделан смыв. Добавьте 2 мл
L-среды с 15% глицерола (стерильный глицерол следует доба-
вить к L-бульону непосредственно перед использованием),
сделайте смывы с каждой чашки и объедините их, тщательно
смешайте и храните при —70 °C или в жидком азоте. Титр бак-
терий должен составлять примерно (1—2)-109/мл; таким обра-
зом вы получите многократно размноженную библиотеку.
Мы успешно проводили скрининг амплифицированных биб-
лиотек, однако при этом наблюдались различия в росте коло-
ний: встречались как большие, так и очень мелкие. Космиды с
.Х-репликоном могут быть использованы для получения ампли-
фицированных библиотек, но нельзя предусмотреть заранее
вероятное нарушение представительства тех или иных специфи-
ческих клонов.
2.2.6.2. Высев непосредственно на фильтры
Все фильтры, которые мы использовали (как найлоновые,
так и нитроцеллюлозные), оказались умеренно токсичными для
бактерий. Промывание фильтра водой и стерилизация помога-
ют, но не снимают эту проблему полностью. Лучшие результа-
ты были получены с космидами, адсорбцию которых на Е. coli
провели в точности так, как это описано в разд. 2.2.5.3, и затем
высевали. Можно ожидать снижение титра на 20%, но оно мо-
жет достигать и 100%. Некоторые штаммы E.coli очень плохо
растут на пайлоновых фильтрах, и поэтому необходимо прове-
Выбор и использование космидных векторов 53
рить эффективность высева на данных мембранах по сравнению
с агаром. Штамм ED8767 вполне для этого подходит.
Если библиотека высевается прямо на фильтр, то в этом
случае для приготовления оптимальных реплик важно следо-
вать методике, описанной в разд. 2.2.6.3. Особенно существенно
время, необходимое для роста колоний, поскольку мы обнару-
жили, что бактерии на фильтрах имеют тенденцию расти мед-
леннее, чем на агаре.
2.2.6.3. Получение реплик для скрининга библиотеки
Мы работаем в основном с 1,2мкм-найлоновыми фильтрами
фирмы Pall/Biodyne и используем методы Ханаана и Меселсо-
на [18], предусматривающие получение матричного фильтра и
реплики. Мы обнаружили, что нельзя смешивать фильтры раз-
ных типов при получении реплик (например, Biodyne плохо
реплицируется на нитроцеллюлозе). Поэтому используйте на
всех'этапах только фильтры одного, типа.
Инструкции, приведенные в табл. 2.4, пригодны для получе-
ния амплификационной библиотеки; в случае первичных биб-
лиотек они нуждаются в некоторых изменениях.
2.2.7. Скрининг фильтров с космидными клонами
Фильтры можно скринировать различными методами. Мы
предпочитаем тот, который позволяет использовать как РНК-,
так и ДНК-зонды без заметных изменений в методике
(табл. 2.5).
Зонды должны иметь высокую удельную радиоактивность
(^108 имп./мин/мкг) и могут быть мечены любым подходящим
методом.
Наилучшие результаты скрининга мы получали с РНК-зон-
дами, приготовленными с помощью РНК-полимераз фагов SP6
или Т7 (см. гл. 1 и разд. 2.3.3, табл. 2.11), однако следует пред-
принять меры против деградации зонда. Мы обычно используем
реакцию с SP6- или Т7-полимеразами с достаточным количест-
вом 32P-UTP и ДНК-матрицы для получения 30—80% включе-
ния в РНК- Реакцию останавливают однократной экстракцией
хлороформом, РНК осаждают изопропанолом, а осадок ресус-
пендируют в небольшом объеме буфера ТЭ, содержащего вана-
дил-нуклеотидный комплекс (ВНК) в концентрации 10 мМ. При
этом удалять невключенные нуклеотиды не требуется. Не дена-
турируйте РНК-зонд до использования.
После гибридизации фильтры высушивают на воздухе (если
планируется повторно гибридизовать фильтры с другой пробой,
не высушивайте их полностью — см. инструкцию Pall/Biodyne)
54
П. Литтл
Таблица 2.4. Приготовление фильтров-реплик для гибридизации
1. Приготовьте разведения амплифицированного штока. Вам потребуется око-
ло 50 мл суспензии плотностью около 2-Ю4 кл/мл. Оттитруйте суспензию
на покрытых фильтрами чашках с L-агаром1, содержащим 30 мкг/мл ка-
намицина. (Учтите, что при высеве на фильтры титр может оказаться
примерно на 20% меньшим из-за сниженной выживаемости бактерий.)
2. Поместите фильтр, подписанный с одной стороны карандашом, на чашку
со свежим F-агаром2, содержащим антибиотик, и дайте ему увлажниться.
Не используйте слишком влажные чашки — сначала подсушите их до
удаления явных следов влаги. Нанесите 0,5 мл суспензии клеток на
фильтр, отступив от края фильтра на 0,5—1см (примите меры к сохране-
нию стерильности). Дайте чашкам постоять и убедитесь, что жидкость
впиталась в фильтр. После высыхания инкубируйте перевернутые чашки
12 ч при 37 °C. Наносите столько бактерий, чтобы получить 20 000 колоний
на чашку, а всего 3-105 (или больше, если это необходимо).
3. Через 12 ч колонии должны быть маленькими (приблизительно 0,5 мм).
Этот размер важен, и поэтому уточните время инкубации, если это необ-
ходимо. Приготовьте реплику следующим образом:
а) поместите нужное число фильтров, подписанных карандашом, на чаш-
ки с L-агаром, содержащим антибиотик, чтобы увлажнить их;
б) на ровную поверхность положите подложку из какой-либо фильтро-
вальной бумаги (ватман 3 ММ) и на нее поместите исходный фильтр
колониями кверху;
в) аккуратно уложите второй фильтр поверх исходного — его нельзя сдви-
гать, даже если он лег неточно;
г) положите второй лист бумаги 3 ММ поверх фильтров и сильно приж-
мите стеклянной чашкой, равномерно распределяя тяжесть;
д) снимите чашку и бумагу с бактериальных фильтров и осторожно на-
колите фильтры в нескольких местах для маркировки. Сделайте два
прокола рядом, а три других асимметрично;
е) разделите оба фильтра одним резким движением и поместите фильтры
колониями кверху на две чашки.
Исходный фильтр можно заморозить для повторного использования (по
методу Ханаана и Меселсона [18]) или хранить при 4 °C до месяца. Реп-
лику нужно подрастить 3—5 ч при 37 °C. Должны получиться колонии
диаметром около 1 мм или меньше. Они могут быть использованы для
скрининга.
4. Колонии лизируют и обрабатывают точно так, как описано в инструкциях
Pall/Biodyne. Лизис проводят, помещая фильтры в 0,5 М NaOH, 1,5 М
NaCl на 5 мин и затем переносят в 3 М Na-ацетат (pH 5,5) на 5 мин.
Фильтры высушивают на воздухе и прогревают при 80 °C 1 ч. Храните
их при 4 °C.
5. Промойте фильтры в большом объеме 5XSSC3 до предварительной гиб-
ридизации. Обратите внимание на ориентацию фильтров, так как реплика
является зеркальным отражением исходного фильтра.
1 L-arap: 10 г бактотриптоиа Difco, 5 г дрожжевого экстракта Difco, 5 г NaCl и 15 г
агара в 1 л.
2 F-агар: L-arap+5% глицерола.
3 SSC — см. примечание к табл. 2.1.
и экспонируют при —70 °C с пленкой Kodak XAR-5. Экспозиция
в течение ночи обычно дает достаточно интенсивный гибридиза-
ционный сигнал с РНК-зондом и менее интенсивный с меченой
ДНК> но это зависит от специфики космид.
Выбор и использование космидных векторов
55
Таблица 2.5. Гибридизация фильтров
1. Промойте фильтры избытком 5XSSC при комнатной температуре 5 мин.
2. Для предварительной гибридизации используйте следующий раствор:
бХденхарт [0,1% (весовой) фиколла 400, 0,1% (весовой) БСА
0,1% (весовой) поливинилпиролидона]
5XSSC1
50 мМ Na-фосфвтный буфер (pH 6,5)
0,1%-ный ДСН
250 мкг/мл ДНК спцрмы лосося, предварительно обработанной ультра-
звуком и денатурированной
10 мкг/мл ро!у(С)
10 мкг/мл poly (А)
50%-ный формамид (деионизованный)
При ДНК-гибридизации предварительно инкубируйте при 42 °C 4 ч или
ночь. При РНК-гибридизации — 1 ч. В обоих случаях требуется 3—5 мл
раствора в расчете на один фильтр.
3. Гибридизация проводится в 2,5—3 мл свежего буфера на фильтр. Буфер
того же состава, что и в п. 2, но дополнительно содержит 10s имп./мин
на 1 мл соответствующей 32Р-меченной РНК- или ДНК-зонда. Для
РНК-зонда к смеси добавляют 10 мМ ВНК и гибридизуют 17 ч при 50 °C.
ДНК-гибридизацию проводят 2 дня при 42 °C. На стадии предварительной
гибридизации мы обычно вводили в качестве конкурирующей ДНК дена-
турированный космидный вектор в .концентрации 0,5—1 мкг/мл.
4. После гибридизации фильтры промывают одинаково независимо от того,
РНК или ДНК использовались в качестве зонда. Первые три раза — 1 л
2XSSC, 0,1% ДСН при комнатной температуре; по 20 мин. Конечная
промывка — 30 мин при 65 °C в 0,1 SSC, 0,1% ДСН. Условия последней,
более жесткой промывки, могут быть изменены. Эта промывка нужна
только при использовании высокогомологичных проб.
1 См. примечания к табл. 2.1.
Колонии, дающие положительную реакцию, затем нужно
отобрать и провести повторный скрининг не менее трех раз для
того, чтобы однозначно идентифицировать отдельные колонии.
Не пытайтесь отобрать индивидуальную колонию уже в первом
цикле отбора. Фильтры сморщиваются при —70 °C, что очень
затрудняет точный отбор колоний. Отберите нужную зону, рас-
сейте клетки и повторно скринируйте их. Очень легко ошибить-
ся при отборе, поэтому не исключено, что операцию придется
проверить. При использовании высококопийных векторов на ос-
нове фага К можно экспонировать фильтры при комнатной тем-
пературе с тем, чтобы избежать их сморщивания.
Процедура, альтернативная рассеву колоний, состоит в от-
боре всей зоны, дающей положительный ответ, и ресуспендиро-
вании клеток в 1 мл L-среды с антибиотиком. Взятые из него
аликвоты (обычно от 1 до 100 мкл) можно сразу нанести на
свежие фильтры и скринировать их (разд. 2.2.6).
56
П. Литтл
2.2.8. Хранение космидных библиотек
2.2.8.1. Хранение бактерий
Амплифицированные библиотеки достаточно стабильны при
хранении как при —70 °C, так и в жидком азоте в L-среде с
15% глицерола. Аликвоты амплифицированной библиотеки
(разд. 2.2.6.1) хранят в 1-мл флакончиках и оттаивают по мере
надобности. Мы не замечали заметного уменьшения титра за
два-три цикла оттаивания, но обычно флакончики после этого
выбрасывают.
2.2.8.2. Хранение упакованных космид
Космиды могут храниться как упакованные фаговые части-
цы в течение по крайней мере 6 мес при 4 °C. Не стоит хранить
упаковочную смесь, поскольку мы наблюдали постепенное
уменьшение титра при хранении даже в течение одной недели.
Космиды Лориста могут быть упакованы, а также переупакова-
ны в Е. coll с использованием суперинфекции или путем высева
на специальный штамм, упаковывающий in vivo.
2.2.8.3. Упаковка космидной библиотеки
с использованием суперинфекции
Это хороший метод для «оздоровления» библиотеки, которая
может быть загрязненной или содержать много маленьких де-
леционных производных. Он приводит к дальнейшей амплифи-
кации библиотеки, которая, однако, почти наверняка будет
непредставительной. Методика приведена в табл. 2.6.
Полученный лизат содержит упакованные космиды и супер-
инфицирующие фаги, например SepB или L47-1, причем титры
фагов будут сильно зависеть от скорости роста и характера ин-
фекционного цикла. Часто получается около 106—107 космид
или фагов в 1 мл. При высеве упакованных космид суперинфи-
цирующий фаг может осложнить работу в результате лизиса
клеток. Чтобы преодолеть это, лучше всего использовать штамм
ED8767, который лизогенизирован фагом с той же областью
иммунности, что и L47-1 (йши434) или SepB (imm21). Эти клет-
ки по определению иммунны к суперинфекции. Нужно отметить,
что космиды-производные фага X сообщают клеткам иммунитет
к фагам, несущим детерминанту Zimm1, и в этом случае для
суперинфицирования можно использовать только imm2'- или
1’тт434-С7~-фаги. Не рекомендуется применять суперинфицирую-
щие фаги Хшг, так как они способны расти на всех лизогенных
бактериях.
Выбор и использование космидных век торов
57
Таблица 2.6. Упаковка космид путем суперинфекции жидкой культуры
1. Вырастите 100 мл космидной библиотеки в L-среде в присутствии анти-
биотика и 10 мМ MgSO4. Внесите достаточно бактерий, чтобы получить
исходно Лбоо около 0,08. Выращивайте клетки при энергичной аэрации
до Лбоо=О,4. Это может произойти быстро (2 ч) или гораздо медленнее
(>4 ч).
2. Инфицируйте Е. coll с множественностью не менее 1 любым фагом к, не
способным к лизогенизации, т. е. С1~ (не используйте kvir). Мы брали
разные векторные фаги — L47.1 [19] или чаще SepB [3].
3. Следите за клеточным ростом. Значение Исоо должно подняться примерно
до 1,0, а затем упасть до 0,5 вследствие лизиса не позднее чем за 4 ч.
Когда лизис станет заметным, добавьте 1 мл хлороформа, встряхивайте
1—2 мин. Осадите клеточные остатки центрифугированием при 4000 об/мин
в лабораторной центрифуге типа Sorvall RC5. Соберите надосадочную
жидкость.
4. Оттитруйте надосадочную жидкость, содержащую свежеупакованную кос-
мидную библиотеку.
Упакованные таким образом библиотеки могут долгое время
храниться при 4 °C, и с ними можно работать, используя те же
методы, что и в случае обычных космидных или фаговых биб-
лиотек.
2.2.8.4. Упаковка индивидуальных космид
с использованием суперинфекции
Этот метод используют для хранения, для трансформации
космидами специальных линий клеток и для «спасения» космид,
начавших терять стабильность (разд. 2.4.7.1). Методика описа-
на в табл. 2.7. Для хранения упакованных космид просто от-
центрифугируйте суспензию верхнего слоя агара в фаговом бу-
фере, полученную, как указано в п. 3 табл. 2.7. Скорость цент-
рифугирования должна быть достаточной, чтобы осадить
суспендированный агар. Надосадочную жидкость отберите и
храните над каплей хлороформа.
2.2.8.S. Переупаковка путем высева первичной библиотеки
на хелперный штамм
Подробные сведения о специальных линиях упаковывающих
клеток описаны в другой работе [20].
Получены клеточные линии, лизогенизировапные дефектны-
ми Мтт434-фагами, со следующими генотипическими характе-
ристиками и биологическими свойствами: 1) CIts, т. е. стабиль-
ны только при 32 °C и индуцируют развитие фага при 42 °C;
2) Sami, т. е. дефектны по лизису (что позволяет получить вы-
сокий выход фаговых частиц) и содержат делению Ь2, которая
резко снижает эффективность вырезания из хромосомы, и, на-
58
П. Литтл
Таблица 2.7. Упаковка космид путем суперинфекции культур на чашках
1. Внесите 200 мкл насыщенной (ночной) культуры космидосодержащих
бактерий вместе с 2,5 мл расплавленного при 55° верхнего агара на чаш-
ку диаметром 9 см с L-агаром и антибиотиком.
2. Дайте застыть верхнему агару, затем капните 5—40 мкл суспензии фага
в буфере для разведения1, содержащих 10* * * 4—105 * * В б. о. е. соответствующего
супериифицирующего фага (разд. 2.2.8.3), на центр чашки. Дайте высох-
нуть и инкубируйте при 37 °C в течение ночи.
3. Там, где фаги лизировали клетки, на следующее утро должна быть видна
прозрачная зона. Отберите эту зону концом пастеровской пипетки и пе-
ренесите в 1 мл буфера для разведения фага (разд. 2.2.4). Добавьте
3 капли хлороформа и мягко перемешайте. Оставьте при 4 °C на 2—5 ч
и затем оттитруйте надосадочную фракцию — следите, чтобы при этом не
захватить хлороформ. Используйте в качестве реципиента любые клетки.
Титры могут сильно варьировать. Поэтому проще вносить 1, 10 и 100 мкл
фага с 200 мкл насыщенной культуры прямо в верхний агар.
1 Буфер для разведения фагов: 10 мМ трис-НС1 (pH 7,4), 10 мМ MgSO«, 0,01%
(вес/объем) желатина.
конец, 3) red3, т. е. дефектны по системе фаговой рекомбинации.
Если космиду (предварительно упакованную in vitro) ввести в
эти клетки, клетки будут расти в присутствии соответствующего
антибиотика при 32 °C. Если же перенести клетки на 42 °C, ре-
прессор (продукт гена CI) инактивируется, дефектный профаг
получит сигнал исключиться из хромосомы и, хотя ему это не
удастся, в больших количествах будут синтезироваться компо-
ненты фаговых головок и отростков, в которые может упако-
ваться резидентная космида. Клетки затем лизируют хлорофор-
мом и получают высокий выход космид и незначительное коли-
чество частиц хелперного фага (109 * * * космид на 1 мл по сравне-
нию с 104 фага). Этот процесс служит примером упаковки
in vivo.
В принципе можно получать библиотеки непосредственно
в таких клеточных линиях, но дело в том, что эффективность
высева космид на температурочувствительных штаммах всегда
ниже. Для упаковки in vivo используют два штамма — ВНВ3175
и DK22. Оба они лизогенны по одному и тому же фагу 7.3169
CIi3 Ь2red.3 Sam7). ВНВ3175 представляет собой
HL202, лизогенный по фагу 7.3169 [20], a DR22 — производное
DK.1, лизогенное по фагу Х3169. (Д. Карнит, личное сообщение.)
Оба штамма — гесД-мутанты.
Методика упаковки in vivo приведена в табл. 2.8.
Эта техника пригодна для получения больших количеств
упакованных космид (фосмид). Х-ДНК внутри фаговой частицы
линейна и содержит одноцепочечные липкие концы (cos). Такую
же структуру имеют и упакованные космиды; космиды с не
замкнутыми cos-сайтами мы называем фосмидами. Упаковка
Выбор и использование космидных векторов 59
Таблица 2.8. Приготовление фосмид с помощью упаковывающих in vivo
клеточных линий
I. Введите соответствующую упакованную космиду в клетки Е. coli
ВНВ3175 или DK.22. Не допускайте нагревания клеток выше 32 °C на лю-
бой стадии работы. Высейте их на L-arap, содержащий антибиотики, и
растите при 32 °C в течение ночи.
2. Отберите устойчивые к антибиотику колонии, рассейте их в течение ночи
в двух чашках и инкубируйте одну при 32 °C, другую при 42 °C. Сначала
засевайте ту чашку, которая будет инкубироваться при 42 °C. Растите их
ночь.
3. Отберите колонии, которые совсем не растут при 42 °C. Не пытайтесь ра-
ботать с колониями, обнаруживающими признаки роста при непермиссив-
ной температуре. Если колония достаточно большая, введите ее в L-cpe-
ду, содержащую антибиотик, и растите до Леоо=О,3 при 32 °C.
4. Быстро прогрейте культуру до 45 °C и держите при этой температуре
14 мин. Измерьте 46оо; этот показатель должен повыситься по сравне-
нию с этапом 3.
5. Растите при 37 °C с перемешиванием по крайней мере в течение 3 ч, но
не более 17 ч. Измеряйте Л6оо на ранних этапах — эта величина должна
повыситься до 0,6—0,7. Превышение этого уровня будет означать, что.
в культуре растут нелизогенные клетки.
6. После этого лизируйте клетки добавлением хлороформа (2 мл/л). При
лизисе должны быть видны хлопья. Отцентрифугируйте клеточные остат-
ки при 4000 об/мин в препаративной центрифуге и оттитруйте осветлен-
ный лизат. В данном случае хелперный фаг мало жизнеспособен и по-
этому для титрования космид не нужно использовать Л-лизоген. Для тит-
рования примените методику, описанную в разд. 2.2.5.3.
in vivo по описанной методике более эффективна в случае кос-
мид, дающих высокий выход; нам удавалось получить хороший
результат с космидами — производными фага X и некоторыми
производными pBR322, но в последнем случае только при ус-
ловии, что они относились к высокопродуктивным космидным
клонам. Фосмиды удобно использовать для быстрого рестрик-
ционного картирования, методика которого вкратце изложена
в разд. 3.2.2, с применением техники так называемого cos-кар-
тирования [6, 21].
Фосмиды эффективны также для микроинъекции в клетки
и в опытах по трансфекции.
2.2.8.6. Хранение ДНК
Космидная ДНК может быть упакована in vitro подобно
ДНК фага X. Однако эффективность упаковки космид сущест-
венно ниже, так как препарат содержит в основном мономерные
кольца с одним cos-сайтом, которые не могут служить субстра-
том в упаковочной реакции. Эффективность упаковки, как пра-
вило, колеблется между 103 и 106 в расчете на 1 мкг ДНК-
Однако есть смысл прибегнуть к этому методу, если не удалось
использовать бактериальный и фосмидный подходы.
60
П. Литтл
2.2.9. Получение космидной ДНК
Существует много способов выделения плазмидной ДНК из
бактериальных клеток. Приводимая нами методика проста и
вполне воспроизводима, но ее почти всегда можно заменить на
другую. Мы описываем процедуру наработки космид Лориста —
производных фага X. Для других космидных векторов при не-
обходимости можно ввести небольшие модификации.
2.2.9.1. Размножение бактерий
Размножайте клетки в L-среде с 30 мкг/мл канамицин-суль-
фата. Никогда не выращивайте космидсодержащие клетки без
антибиотика и всегда — с хорошей аэрацией. Для получения-
мини-препаратов мы выращиваем 5 мл в 50-миллилитровой про-
бирке, для препаративного выделения — 500—1000 мл в колбе
на 2 л с энергичной аэрацией. Засев произведите введением-
одной колонии или небольшого количества стационарной куль-
туры и инкубируйте в течение ночи (17 ч) при 37 °C. Некоторые
космиды обусловливают плохой рост клеток; в таких случаях
культура не достигает стационарной фазы и ее нужно культи-
вировать дольше. Для космид, не являющихся производными
фага X, мы не рекомендуем проводить амплификацию в присут-
ствии хлорамфеникола. Это может повысить выход, но может
также привести к преимущественной репликации делеционных
производных исходных рекомбинантов.
2.2.9.2. Мини-препараты
Метод, приведенный в табл. 2.9, взят из литературы [10] и
незначительно модифицирован.
Для рестрикционного анализа мини-препарата ДНК обраба-
тывайте 5 мкл ДНК в 10 мкл реакционной смеси. Добавьте
1 мкг РНКазы (прокипяченной) в начале рестрикции — не уда-
ляйте РНК раньше, так как она стабилизирует неочищенные
препараты. При аккуратной работе эта методика позволяет по-
лучить ДНК, которая служит хорошим субстратом для рестрик-
ции. Если вы обнаружили, что это не так, экстрагируйте препа-
рат фенолом, осадите этанолом нуклеиновую кислоту, промойте
осадок 70%-ным этанолом (табл. 1.1), растворите его и повто-
рите процедуру рестрикции снова. Этот этап не всегда необхо-
дим. Чаще всего неудачи бывают связаны с неполным удалени-
ем осадка или плохой промывкой от солей.
Выбор и использование космидных векторов 61;
Таблица 2.9. Получение мини-препаратов космидной ДНК
1. Отцентрифугируйте 1,5 мл насыщенной культуры в микроцентрифуге-
1 мин при комнатной температуре.
2. Слейте культуральную среду и вытрите ватным тампоном ее остатки.
К осадку добавьте 200 мкл холодного раствора I1. Тщательно ресуспенди-
руйте клетки. Добавьте несколько крупинок лизоцима (храните лизоцим
в морозильной камере над осушителем — он портится довольно быстро).
Перемешайте и оставьте при комнатной температуре на 5 мин.
3. Добавьте 400 мкл свежеприготовленного раствора II* 2 комнатной темпера-
туры. Перемешайте, 2—3 раза перевернув пробирку. Резко не встряхи-
вайте, но убедитесь, что раствор однороден. Перенесите в лед на 5—
10 мин.
4. Добавьте 200 мкл холодного раствора III3. Смешайте тщательно, но не
резко (не используйте Wortex) и оставьте на льду на 10 мин.
5. Открутите в микроцентрифуге при комнатной температуре 10 мин.
6. Удалите липкий и студнеобразный осадок зубочисткой. Осадок часто раз-
мазывается по стенкам пробирки и имеет неприглядный вид — не беспо-
койтесь, это нормально.
7. Добавьте 480 мкл изопропанола при комнатной температуре и хорошо пе-
ремешайте. Заморозьте в сухом льду и перенесите в микроцентрифугу,
не давая растаять. Открутите 10 мин при комнатной температуре. Не ос-'
тавляйте пробирку в сухом льду слишком долго — лишь на время, необ-
ходимое для замораживания.
8. Слейте надосадочную жидкость — на дне пробирки должен быть виден
осадок. Добавьте 1 мл 70%-ного этанола при комнатной температуре.
Разбейте осадок резким встряхиванием пробирки, открутите 5 мин в мик-
роцентрифуге и слейте. Для высушивания осадка оставьте перевернутую-
вверх дном пробирку на 10 мин. Вытрите ватным тампоном избыток эта-
нола. Ресуспендируйте осадок в 50 мкл ТЭ и оставьте при 55 °C по крайней
мере иа 10 мин для растворения.
1 Раствор I: 25% (вес/объем) глюкозы; 50 мМ трис-НС1 (pH 8,0), 10 м.М ЭДТА
(pH 8,0). Храните и используйте холодным.
2 Раствор II: 0,2 М NaOH, 1% (вес/объем) додецилсульфата натрия. Готовьте рас-
твор каждый раз заново и используйте растворы NaOH, хранящиеся в пластиковой,
а не в стеклянной посуде.
3 Раствор III: ЗМ К-ацетат, доведенный уксусной кислотой до pH 4,8. Храните
и используйте холодным.
2.2.9.3. Препаративное выделение
из культур объемом 500—1000 мл
Метод приведен в табл. 2.10.
Эффективность этой процедуры, однако, всегда разочаровы-
вает— она гораздо ниже, чем можно предположить, работая с
мини-препаратами. Вместе с тем 50 мл культуры дают по мень-
шей мере 200—300 мкг космиды. Выход нерекомбинантных
космидных векторов — производных фага X также низкий: мы
редко получали более 200 мкг из 1 л. Это обусловлено меха-
низмами контроля репликации этих производных фага X.
2.3. Специальные методы
Настоящий раздел охватывает широкий спектр методов, ко-
торые могут пригодиться работающим в данном направлении,.
62
П. Литтл
Однако детальное описание всех аспектов этих методов здесь
не приводится.
2.3.1. Клонирование с помощью рекомбинации
Этот метод впервые разработали Лерах и др. [20] и Линден-
майер и др. [22]. Подробные методики можно найти в указан-
ных работах, а также в гл. 3 данной книги.
Клонирование с помощью рекомбинации позволяет с наи-
меньшими усилиями скринировать большое множество космид.
В основе метода лежит тот факт, что фаг X в общем случае не
является трансдуцирующим фагом, т. е. упаковывается только
ДНК, расположенная между двумя cos-последовательностями,
разделенными достаточным расстоянием.
В основе метода лежит использование двух векторов —• од-
ного космидного, а другого плазмидного, не имеющих областей
взаимной гомологии и различающихся по маркерам лекарствен-
ной устойчивости, например, Ктт и Арт. Оба вектора способны
временно поддерживаться в гес+-окружении в штаммах для
упаковки in vivo (разд. 2.2.8.5), причем клетки содержат обе
эписомы.
Индукция профага приводит к упаковке космиды Kmr, но не
плазмиды Арт. Однако если между фрагментами ДНК, клони-
рованными в космиде и в плазмиде, имеется гомология, то меж-
ду гомологичными последовательностями может с частотой 10-3
произойти рекомбинация, в результате которой оба вектора
объединяются в одну крупную кольцевую космиду. Только в
этом случае упаковка обеспечит возможность трансдукции обо-
их маркеров Ктг и Арт. Поэтому для клонирования какого-либо
гена достаточно лишь вставить небольшой фрагмент его ДНК
в плазмиду-зонд, содержащуюся в линии хелперных клеток,
суперинфицировать их космидной библиотекой (упакованной,
как указано в разд. 2.2.8.5), индуцировать профаг в хелперных
клетках и высеять их на чашки с Кт и Ар. Колонии с двойной
лекарственной устойчивостью должны в принципе содержать
ген, клонированный в космиде, в которую интегрировала плаз-
мида-зонд.
Этот эффективный метод требует точного соблюдения мето-
дик и тщательного подбора штамма. Он должен использоваться
при необходимости многократно выделять один и тот же ген
из различных библиотек или точно и специфично объединить
между собой последовательности ДНК. Этот метод значительно
упрощает процесс выделения гена, однако может приводить к
разрывам ДНК в точке рекомбинации. Существуют методики
для отбора обратных рекомбинационных событий, ведущих к
вырезанию интегрированой плазмиды-зонда.
Выбор и использование космидных векторов
63
олигону-
клеотид
г\
I Частичный
1 гидролиз
R
______________________R
'R .
____ —R
Рис. 2.2. Cos-картирование. Радиоактивные олигонуклеотиды, комплементар-
ные левому или правому липкому концу, гибридизируют с соответствующим
сайтом. Затем проводят частичный гидролиз рестриктазой R, разделение фраг-
ментов электрофорезом в агарозном геле и радиоавтографию, позволяющую
идентифицировать все меченые фрагменты, полученные в результате непол-
ного гидролиза. Молекулярную массу истинных рестрикционных фрагментов
легко вычислить путем вычитания меньших величин из больших.
2.3.2. Cos-картирование
Cos-картирование разработали Раквиц и др. [21] для кар-
тирования клонов фага X. Эта процедура используется также
и в случае космидных клонов [6]. Основная проблема, возника-
ющая при анализе космидных клонов путем рестрикционного
картирования, связана с их размером: у них слишком много
сайтов для наиболее широко используемых ферментов, чтобы
осуществить быстрое картирование. Cos-картирование обходит
эту трудность, используя частичный рестриктазный гидролиз
для идентификации сайтов и определения порядка их распре-
деления. Схематично этот процесс показан на рис. 2.2. Левый
или правый липкие концы метят, подвергая их отжигу с мечен-
ным 32Р 12-членным фрагментом ДНК, комплементарным лево-
му или правому липкому концу фага X. Затем ДНК подвергают
частичному рестриктазному гидролизу, а радиоактивные фраг-
менты путем определения стартового участка обнаруживают
радиоавтографией после фракционирования их по размеру в-
агарозном геле. При этом выявляются все фрагменты с меченым
концом.
'•64
П. Литтл
Таблица 2.10. Препаративное выделение космидной ДНК
1. Осадите центрифугированием клетки из 500 мл культуры и добавьте
к осадку 20 мл холодного -раствора I1’. Добавьте кристаллический лизо-
цим (на кончике шпателя) и тщательно перемешайте. Оставьте на 10 мин
при комнатной температуре.
2. Добавьте 60 мл раствора II1’ и перемешайте пипеткой или чем-нибудь по-
добным. Не встряхивайте резко. Оставьте на льду на 10 мин.
3. Добавьте 45 мл холодного раствора III1’. Перемешайте, как на этапе 2.
Оставьте на 30 мин на льду.
4. Центрифугируйте 15 мин при 5000 об/мин в роторе GSA на центрифуге
Sorvall RC5 (или в аналогичных условиях). Слейте надосадочную жид-
кость через тонкую ткань, чтобы удалить крупные плавающие хлопья
клеточных остатков. Соберите надоса-дочную жидкость и добавьте 0,6 объ-
ема пропан-2-ола. Оставьте на льду на 30 мин. Выпавший тяжелый оса-
док открутите при низких скоростях. Удалите остатки надосадочной
жидкости и ресуспендируйте осадок в ТЭ. Объем ТЭ зависит от размера
ультрацентрифужной пробирки, которую вы потом будете использовать.
Мы пользуемся вертикальным ротором VTi 65 с 5-миллилитровыми про-
бирками и поэтому ресуспендируем осадок в 5 мл ТЭ, разливая затем
пробу в две пробирки.
5. Принимая объем ДНК за у мл, добавьте 0,1 у мл раствора бромистого
этидия (10 мг/мл) и 1,1у г CsCl любой квалификации. Оставьте в темно-
те на 30 мин -при комнатной температуре. Должен образоваться тяжелый
преципитат белков и прочих клеточных остатков. Открутите препарат на
средней скорости 5 мин. Это позволит разделить преципитат на флоти-
рующие хлопья (если это белок) и осадок. Тщательно отберите прозрач-
ный раствор, стараясь не захватить клеточных остатков. Центрифугируйте
соответствующее время в ультрацентрифуге при 20 °C — мы используем
ротор VTi 75 фирмы Beckman, 17 час при 50 000 об/мин.
6. Соберите полосу (нижнюю), соответствующую суперспирализованнот!
плазмиде в минимальном объеме (х мл) и перенесите в подходящую
50-миллилитровую полипропиленовую пробирку (Falcon 2070 или анало-
гичную).
7. Наполните пробирку водо-насыщенным н-бутанолом, закройте и встряхни-
те. Дайте фазам разделиться и отбросьте верхнюю (органическую). Обыч-
но это составляет около 45 мл.
8. Повторите этап 7. Раствор CsCl/ДНК увеличится в объеме в результате
разбавления водой из насыщенного н-бутанола. Удалите все следы орга-
нической фазы.
9. Добавьте х мл дистиллированной воды, чтобы получить у мл раствора
ДНК/CsCl.
10. Добавьте у мл изопропанола, заморозьте на сухом льду, дайте оттаять
и центрифугируйте со средней скоростью. Тщательно промойте осадок при
комнатной температуре 70 %-ным этанолом. Высушите на воздухе и ресус-
пендпруйте ДНК в 10 мМ трис (pH 7,5), 1 мМ ЭДТА (см. табл. 1.1).
Для растворения ДНК инкубируйте 10 мин при 55 °C и используйте по
мере необходимости.
1 Растворы I, II и III описаны в примечании к табл. 2.9.
Данный метод может быть также распространен на анализ
космид, упакованные; in vivo в хелперных клетках, т. е. фосмид,
описанных в разд. 2.2.8.5. Детальное описание этой методики
можно найти у Литтла и Кросса [6]. Проблема выделения фос-
Выбор и использование космидных векторов 65
мид из космид, не являющихся производными фага К, обсуж-
дается в разд. 2.2.8.5.
Первоначально указанная методика представляется несколь-
ко сложной, однако, будучи освоена в повседневной практике,
она оказывается удобной и очень быстрой. Основная трудность
связана с переносом индивидуальных космид из клеток Е. coli
ED8767 в штамм ВНВ3175. Это можно сделать с помощью ме-
тодики, детально описанной в разд. 2.2.8.4, но штамм Е. coli
ВНВ3175 иногда ведет себя непредсказуемо — он часто ревер-
тирует, утрачивая температурную чувствительность, либо спон-
танно индуцируется. В наших опытах более стабильным был
штамм DK22.
2.3,3. «Прогулка по геному»
Недавно сконструированы векторы, содержащие промоторы
SP6, Т7 или ТЗ, непосредственно примыкающие к сайтам для
клонирования ([8] и неопубликованные результаты). Это по-
зволяет быстро и легко приготовить меченные 32Р РНК-зонды,
специфичные в отношении концов клонированной ДНК. и по-
этому очень подходящие для использования в качестве зондсв
для идентификации новых космид, вставки в которых перекры-
ваются со вставкой в исходной космиде. Этот процесс выделения
перекрывающихся клонов известен как «прогулка» по геному,
причем использование промоторных систем SP6/T7/T3 может
существенно уменьшить время и труд, затраченный на проведе-
ние такой «прогулки», поскольку при этом отпадает необходи-
мость в идентификации концов клонированных фрагментов с
помощью рестрикционного картирования и физического выде-
ления этих фрагментов для приготовления новых зондов.
Реакции транскрипции наиболее эффективно проводят на
ДНК-матрицах, очищенных в CsCl (разд. 2.2.9.3). Важно тща-
тельно очистить ДНК с помощью следующей процедуры, удоб-
ной в работе и не требующей много усилий:
1. Выделите плазмиду из градиента в объеме х мл
(~ 0,5—1 мл).
2. Экстрагируйте дважды 50 мл водонасыщенного н-бута-
нола; бромид этидия будет удаляться бутанолом, а раствор
ДНК увеличится в объеме.
3. Добавьте х мл с тем, чтобы получить конечный объем у.
4. Добавьте у мл изопропанола, хорошо перемешайте, замо-
розьте в сухом льду и отцентрифугируйте, чтобы осадить ДНК-
5. Промойте осадок 70%-ным этанолом при комнатной тем-
пературе.
6. Соберите осадок центрифугированием, высушите его на
воздухе и ресуспендируйте в подходящем объеме ТЭ (табл. 1.1).
5—1186
F6
П. Литтл
Таблица 2.11. Транскрипция космид in vitro
1. Мы использовали 20 мкг рекомбинантной космиды для одной реакции
транскрипции (это эквивалентно 2 мкг маленького вектора семейства
pUC). Если нужно расщепить матрицу для получения терминирующих
сайтов, рестриктируйте ее соответствующим ферментом и затем обрабо-
тайте ДНК один раз фенолом и один раз хлороформом. Осадите ДНК
изопропанолом, промойте 70%-ным этанолом, высушите и ресуспендируй-
те в ТЭ. Если нужна неочищенная ДНК, то ее можно использовать
сразу.
2. Транскрипционная реакция должна проводиться при комнатной темпера-
туре, чтобы избежать осаждения нуклеиновых кислот спермидином.
На 100 мкл смеси используйте следующие компоненты и концентрации:
20 мкг ДНК
40 мМ трис-НС1 (pH 7,5)1
6 мМ MgCl2u
2 мМ спермидин-НС1 (Sigma)1
100 мкг/мл БСА1) (Pentex, фракция V, или другой высокой чистоты)
10 мМ дитиотреитола (ДТТ)
100 ед. РНКазина
400 мкМ АТР
400 мкМ СТР
400 мкМ GTP
50 mkKh32P-UTP (800 Ки/ммоль, Amersham, 5Р6-чистоты); если
нужно, то холодный UTP.
40 ед. полимеразы SP6 или Т7 (Biolabs, Boehringer или др. фирмы).
3. Инкубируйте 60 мин при 40 °C. Отберите аликвоты до добавления фермен-
та и через 60 мин.
4. Остановите реакцию добавлением равного объема хлороформа, встряхни-
те и разделите фазы центрифугированием. Осадите нуклеиновые кислоты
из водной фазы изопропанолом. Осадок после центрифугирования ресус-
пендируйте в ТЭ, содержащем 10 мМ канадилнуклеотидов. Если в этом
нет необходимости, не денатурируйте образец, не пытайтесь удалить мат-
рицы и используйте РНК как можно скорее.
*) Эти реагенты можно хранить в виде 5-кратного буфера состава: 200 мМ трис-HCI
(pH 7,5); 30 мМ MgCl2; Ю мМ спермидин-НС1, 500 мкг/мл БСА. Данный буфер может
быть стерилизован фильтрованием и храниться при 4 °C. 7 * * * * * * * * * * *
7. Реакцию транскрипции проведите, как описано в
табл. 2.11.
Важно проверить транскрипционную смесь, поскольку мы
получаем в некоторых случаях низкую эффективность включе-
ния, что обычно связано с деградацией зондов, которые, естест-
венно, не годятся для гибридизации. Проверьте включение
осаждением ТХУ на фильтрах DE81 либо ватман 540 (описание
методики приведено в гл. 1). При использовании РНК-полиме-
разы фага Т7 включение в отсутствие холодного UTP достигает
90%, а с полимеразой фага SP6 — 30—60% (в нашем случае
БРб-полимераза была всегда менее эффективной, чем 77-фер-
мент) .
Выбор и использование космидных векторов
67
Можно делать РНК-транскрипты только на концах клониро-
ванной ДНК — либо расщепляя матрицу рестриктазами, либо
используя пониженные концентрации UTP. При концентрации
UTP 0,5 мкМ спонтанно образуются транскрипты длиной в
среднем 800—1000 п.н., причем для этого даже не требуется
наличия терминирующего сайта расщепления [8].
РНК-зонды очень эффективны для скринирования библио-
тек— они дают более сильный сигнал, чем меченые двухцепо-
чечные ДНК, хотя, несомненно, более чувствительны к деграда-
ции. Условия гибридизации детально описаны в разд. 2.2.7.
2.3.4. 0,2%-ные агарозные гели
Они могут быть приготовлены с использованием любого
стандартного аппарата для электрофореза в агарозном геле.
Гели можно заливать на стеклянных пластинах, поскольку их
довольно трудно переносить к УФ-трансиллюминатору.
1. Используйте, например, поликарбонатную крышку, кото-
рая по размерам соответствовала бы гелю, но была бы на 2 см
со всех сторон больше.
2. Придавите крышку чем-нибудь тяжелым к стеклянной
пластине и налейте 1%-ную агарозу слоем высотой 0,5 см во-
круг крышки. Она будет выполнять роль более твердой рамки
для содержащегося внутри 0,2%-ного геля. Покрывать 1%-ной
агарозой дно не нужно.
3. Когда гель застынет, осторожно удалите крышку.
4. Поставьте гребенку, отступя на 2 см от агарозной рамки
и воткнув зубья в обрамляющую 1%-ную агарозу — не волнуй-
тесь, что вы ее слегка нарушите. Затем заполните центральную
полость 0,2 %-ной агарозой и оставьте застывать.
5. Очень осторожно удалите гребенку — лунки будут выгля-
деть спаявшимися, но откроются под буфером.
6. Очень бережно залейте гель буфером — не наливайте
буфер на поверхность 0,2 %-ной агарозы, так как струя может
повредить гель.
Гели можно приготовить, добавляя в них и в используемый
для разделения буфер бромистый этидий (0,5 мкг/мл), что лег-
че, чем пытаться окрасить гель по окончании электрофореза.
Однако это несколько уменьшает разрешающую способность
геля.
Гели очень чувствительны к перегрузке, и нужно провести
несколько пробных электрофорезов, чтобы установить нужный
объем и количество наносимой ДНК- Мы используем гребенки
с зубьями размером 0,75x0,15 см, погружаемые в гель толщи-
ной 0,5 см. Это дает возможность без труда вносить в лунки
5*
68
П. Литтл
20—25 мкл и фракционировать свыше 100 нг ДНК, избежав
серьезной перегрузки.
Гели должны бы разгоняться быстро, чтобы скорее выявить
примерные размеры анализируемых фрагментов ДНК, но, к со-
жалению, нужны низкие градиенты напряжения (для оптималь-
ного разделения обычно 1 В/см). Для разделения используется
тот же буфер, что и для стандартных агарозных гелей.
Наиболее удобным маркером является ДНК фага %, фрак-
ционируемая в геле в виде либо мультимеров, либо рестриктаз-
ных фрагментов длиною 25—30 т. п. н. При электрофорезе в
гелях при условиях, описанных в данном разделе, не удается
достичь достаточно хорошего разрешения Х-мультимиров, одна-
ко все же можно достичь хорошего разрешения форм размером
48 и 96 т. п. н.
ДНК, используемая для получения библиотеки, должна об-
ладать меньшей электрофоретической подвижностью, чем %-ди-
меры размером 95 т. п. н., и не должна содержать более легкий
материал. Нужно, однако, быть осторожным, поскольку в пере-
гружаемом геле ДНК может двигаться с большей или меньшей
подвижностью, чем ожидается.
Анализ сахарозных градиентов можно провести путем элек-
трофореза аликвот, взятых непосредственно из градиента. Не-
обходимо при этом, чтобы маркеры содержали в той же кон-
центрации сахарозу, соли и ЭДТА. Иначе нельзя будет сравнить
их подвижность с ДНК из градиента.
Очень высокого разрешения можно добиться с помощью
OFAGE или электрофореза в переменном электрическом поле
[23, 24]. Мы обнаружили, что при высоком содержании соли в
градиентах использование инверсии электрического поля мало-
эффективно. Мы не применяли в своей работе OFAGE.
2.4. Проблемы
Цель этого раздела — описать некоторые из наиболее общих
проблем, с которыми мы столкнулись при попытках сконструи-
ровать библиотеки, и предложить пути их решения.
2.4.1. Исходная ДНК
Необходимо, чтобы исходная ДНК была очень высокого ка-
чества. В 0,2%-ном геле не должна обнаруживаться ДНК раз-
мером менее 100 т. п.н. В противном случае попытки сконструи-
ровать библиотеку оказываются неэффективными. Обработка
РНКазой и переосаждение уменьшают молекулярную массу
ДНК, поэтому мы никогда их не используем. Метод, представ-
ленный в разд. 2.2.1.1, работает хорошо и очень прост. Не ис-
.Выбор и использование космидных векторов 69
пользуйте старые культуры клеток или ткани, находившиеся
при комнатной температуре, так как они часто слегка дегради-
рованы.
2.4.2. Сахарозные градиенты
ДНК из сахарозных градиентов кажется иногда деградиро-
ванной. Такой вывод можно сделать, если высокомолекулярные
фракции дают заметные полосы в зоне ниже 25 т.н.п. Сходные
проблемы касаются солевых градиентов. Для надежности все
растворы автоклавируйте (для сахарозы достаточна обработка
при ПО °C). Нельзя добиться успеха при создании библиотеки,
если ДНК подверглась деградации. Иногда деградация ДНК
происходит даже в процессе электрофореза в 0,2 %-ном геле в
течение ночи. Это трудно контролировать — убедитесь, что
аппарат для геля чист и растворы свежие и автоклавированные.
2.4.3. Неудачное лигирование или упаковка
Это обычно бывает связано с избытком соли, детергента или
сахарозы. Такой вывод можно сделать, если вектор не лигиру-
ется сам на себя. Дефекты упаковки трудно идентифициро-
вать— проверьте тщательно экстракты с ДНК фага X и убеди-
тесь, что буферы для разведения и т. д. не загрязнены
детергентом, использованном для мытья посуды. Если экстрак-
ты работают, если происходит лигирование вектора, но колоний
при высеве не образуется, хотя высеваемые клетки в порядке,
то дефектна, по-видимому, клонируемая ДНК- Переосадите ее
и повторите лигирование. Если и это не помогает, значит, по-
вреждены липкие концы ДНК и нужно получить свежий пре-
парат ДНК.
2.4.4. Высеваемые клетки
Всегда проверяйте способность высеваемых клеток поддер-
живать рост фага К — делайте это обычно путем пробного вы-
сева фага на этих клетках. Не так часто, но случается, что
клетки приобретают устойчивость к фагу X, и тогда в них не
будут размножаться космиды и другие упакованные молекулы.
Всегда проверяйте высеваемые клетки на загрязнение путем
посева их (без добавления экстракта) на L-arap, содержащий
подходящий антибиотик. Таким же образом проверьте все рас-
творы (SM, MgSO4 и т. д.). Особенно опасайтесь загрязнений
медленно растущими формами, например дрожжами и псевдо-
монадами), которые ведут себя очень похоже на медленно рас-
тущие клетки Е. coli, несущие космиду. Для идентификации
70
П. Литтл
загрязнения следует оставить чашки после анализа на несколько
дней при комнатной температуре. Наиболее вероятный путь по-
явления подобных загрязнений — серийное пассирование высе-
ваемых клеток (использование одной культуры как исходной
для следующей). Всегда возвращайтесь к штоку. E.coli
ED8767 — строгий гесА и ревертирует редко, но все же стоит
его регулярно проверять на УФ-чувствительность.
2.4.5. Высев
Тщательно следуйте методике, описанной в разд. 2.2.6.
В случае некоторых клеточных линий высев на фильтры непри-
емлем. Поэтому необходимо проверять способность клеток рас-
ти на фильтрах. Для этого следует высевать их параллельно
на L-arap, содержащий антибиотик (с тем, чтобы убедиться,
что используемые бактерии содержат рекомбинантную кос-
миду).
Переход к препаративным реакциям упаковки-лигирования
не всегда сводится к простому увеличению объема: десятикрат-
ное увеличение объема экстракта не во всех случаях обеспечи-
вает десятикратное увеличение выхода. Будьте очень аккурат-
ны, увеличивая масштабы — лучше всего увеличить все пара-
метры точно в соответствии с условиями, диктуемыми реакцией
в малом объеме.
Большинство мембран снижает выживаемость бактерий, рас-
тущих на их поверхности. Этот эффект можно свести к миниму-
му путем предварительной промывки фильтра и автоклавирова-
ния, как это описано в разд. 2.2.6.2. Всегда, однако, титруйте
библиотеку на фильтрах, а не экстраполируйте результаты, по-
лученные при высеве непосредственно на L-arap с антибиоти-
ком — это может оказаться выдачей желаемого за действи-
тельное.
2.4.6, Дифференциальный рост космид
Для амплифицированных библиотек не характерна случай-
ная представленность клонов; одни клоны могут существовать
в недостатке, другие — в избытке, но в определенных обстоя-
тельствах это может-и не иметь значения. Помимо дифферен-
циального роста (разд. 2.4.7) это может объясняться другими
причинами, а некоторые космиды всегда обусловливают обра-
зование мелких колоний и медленный рост клеток.
Выбор и использование космидных векторов 71
2.4.7. Проблемы, связанные с вектором
2.4.7.1. Нестабильные последовательности
Космиды довольно часто нестабильны и спонтанно пере-
страиваются или становятся меньше (это может происходи: >
в 1—5°/о случаев). Чем это объясняется, установить трудно,
и можно допустить любую из нескольких причин: наличие ин-
вертированных дупликаций, области гомологии в эукариотиче-
ской ДНК с сильными прокариотическими промоторами, опера-
торными участками и сайтами связывания репрессоров [7]. Мы
идентифицировали космиды, обусловливающие замедление рос-
та клеток-хозяев; при выращивании культур, по-видимому, будет
происходить обогащение клональной библиотеки делеционными
производными космид, не влияющими на рост клеток. На прак-
тике мало что можно сделать с нестабильными космидами, за
исключением попыток выделить и хранить полноразмерные мо-
лекулы из культуры, обогащенной делеционными вариантами.
Для этого можно использовать упаковку in vivo и суперинфек-
цию на чашках (разд. 2.2.8.3—2.2.8.6). Препараты, полученные
из фаголизатов, будут обогащены полномерными космидами,
поскольку только такие космиды укладываются в рамки огра-
ничений по размеру, характерных для реакции упаковки. Одна-
ко при этом могут быть упакованы и мультимеры делеционных
производных, так что этот метод может оказаться неэффек-
тивным.
Чтобы уменьшить вероятность делеций, важно заложить
космиды на хранение как можно раньше после их получения
(т. е. хранить часть той же бактериальной суспензии, которую
использовали для получения исходных мини-препаратов) и как
можно меньше растить космидиые штоки. Рекомендуем также
хранить упакованные космиды.
2.4.7.2. Неслучайная представленность клонов
Даже в неамплифицированных библиотеках всегда найдутся
клоны либо в небольшом количестве, либо в избытке. Чем это
объясняется, точно не установлено [7], но не исключено, что это
отражает существование «неклонируемых» последовательностей.
Снижение показателя представленности клона даже в два раза
повышает для него вероятность вовсе не встретиться в статисти-
чески достаточной (по размерам) библиотеке.
Один из источников этой вариабельности, а именно транс-
крипционное блокирование области начала репликации из-за
наличия гомологов промоторных зон клонируемой ДНК, может
быть частично элиминировано в результате использования сай-
72
П. Литтл
тов для клонирования, защищенных терминаторами РНК-ноли-
меразы. Одно такое семейство космид, основанное на Х-репли-
коне, уже получено [7].
2.5. Заключение
Клонирование в космидах технически труднее, чем клониро-
вание в фаге X; его следует избегать, за исключением тех слу-
чаев, когда оно диктуется интересами постановки эксперимента
или решения каких-либо биологических проблем. Методики,
детально описанные в этой главе, в большинстве случаев на-
дежны в работе. Однако не следует рассматривать их как нечто
обязательное. Описаны и другие методы и, конечно, возможны
различные модификации.
Благодарности
Я приношу благодарности Салли Кросс за ее помощь в про-
ведении экспериментов, описанных в этой главе, а также Тоби
Гибсону, Алану Кулсону, Джону Салстону, Гансу Лераху, Но-
рин Марри и многим другим за их поддержку и обсуждения.
Эта работа субсидирована Medical Research Council и Cancer
Research Campaign.
Литература
1. Collins Hohn B. (1978). Proc. Natl. Acad. Sci. USA, 75, 4242.
2. Becker A„ Gold M. (1975). Proc. Natl. Acad. Sci. USA, 72, 581.
3. Maniatis T„ Fritsch E. F., Sambrook J. (1982). Molecular Cloning. A Labo-
ratory Manual, Cold Spring Harbor Laboratory Press, Cold Spring Harbor,
NY. [Имеется перевод: Маниатис T., Фрич Э., Сэмбрук Дж., Методы гене-
тической инженерии. Молекулярное клонирование. — М.: Мир, 1984.]
4. Pouwels Р. Н., Enger-Valk В. Е., Brammar W. J. (1986). Cloning Vectors.
A Laboratory Manual, Elsevier, Amsterdam.
5. Brady G., Funk A., Mattern J., Schutz G., Brown R. (1985). EMBO J., 41,
2583.
6. Little P. F. R„ Cross S. H. (1985). Proc. Natl. Acad. Sci. USA, 82, 3159.
7, Gibson T. J., Coulson A. R., Sulston J. E„ Little P. F. R. (1987). Gene,
in press.
8. Cross S. H„ Little P. F. R. (1986). Gene, 49, 9.
9. Coulson A„ Sulston J., Brenner S., Karn J. (1986). Proc. Natl Acad. Sci
USA, 83, 7821.
10. Ish-Horowicz D„ Burke J. F. (1981). Nucleic Acids Res., 9, 2989.
11. Bates P. F„ Swift R. A. (1983). Gene, 26, 137.
12. Seed B. (1982). Biopolymers, 21, 1793.
13. Seed B., Parker R. C., Davidson N. (1982). Gene, 19, 201.
14. Murray N. E„ Brammar IF. J., Murray R. (1977). Mol. Gen Genet., 150, 53
15. Leach D. R. F., Stahl F. W. (1983), Nature, 305, 448.
16. Vapnek D„ Alton N. R„ Bassell C. L„ Kushner S. R. (1976). Proc. Natl Acad
Sci. USA, 73, 3492.
Выбор и использование космидных векторов
73
17. Clarke L„ Carbon J. (1976). Cell, 9, 91.
18. Hanahan D., Meselson №.. (1983). In: Methods in Enzymology, Wu R., Gros-
sman L. and Moldave K. (eds.), Academic Press, NY, Vol. 100, p. 333.
19. Loenen W. A. №., Brammar W. J. (1980). Gene, 10, 249.
20. Poustka A., Rackwitz H. R., Frischauf A.-M., Hohn B., Lehrach H. (1984).
Proc. Natl. Acad. Sci. USA, 81, 4129.
21. Rackwitz H. R., Zehetner G., Mutialdo H., Delius H., Chai J. H„ Frischauf A.,
Lehrach H. (1985). Gene, 40, 259.
22. Lindenmaier W., Dittmar K. E. J.t Hauser H., Necker A., Sebald W. (19851.
Gene, 39, 33.
23. Carle G. F., Frank M., Olson M. V. (1986). Science 232, 65.
24. Schwartz D. C„ Cantor C. R. (1984). Cell, 37, 67.
ГЛАВА 3
ГЕНЕТИЧЕСКИЕ ПОДХОДЫ К КЛОНИРОВАНИЮ,
МОДИФИКАЦИИ И ХАРАКТЕРИСТИКЕ
ИНДИВИДУАЛЬНЫХ КОСМИДНЫХ БИБЛИОТЕК
КЛОНОВ
Анна-Мария Постка и Ханс Лерах
(Annemarie Poustka, Hans Lehrach)
3.1. Введение
Настоящая глава посвящена генетическим подходам к вы-
делению, генноинженерному манипулированию и анализу инди-
видуальных космидных клонов или библиотек клонов. В ней
представлены методики упаковки космидных клонов в лямбо-
доидные фаговые частицы и отбора нужных космид (или плаз-
мид) из полных библиотек путем гомологичной рекомбинации.
Рассмотрены также способы модификации отобранных клонов
или групп клонов с помощью вставок или замещений определен-
ных последовательностей.
Космидные векторы — плазмиды, несущие сод-последователь-
ности, распознаваемые компонентами системы упаковки фага Z
[1], представляют собой удобный инструмент для клонирования
и анализа больших фрагментов геномов, картирования хромо-
сом и клонирования генов, размер которых превышает 20 т. п.н.
Хотя в настоящее время разработаны приемы, позволяющие
конструировать космидные клоны и оперировать с космидными
библиотеками, все же неизбежно возникают трудности при по-
лучении и анализе больших библиотек, необходимых для кло-
нирования генома млекопитающих; в частности, вызывает за-
труднение введение специфических модификаций во вставки в
тех случаях, когда их нельзя прямо «увязать» с характеристи-
ками рестрикционной карты. Эти трудности могут быть весьма
существенными. Во многих случаях для их преодоления исполь-
зуются методы генетики бактерий, позволяющие упростить или
облегчить решение этих задач.
3.2. Работа с космидными клонами,
полученными путем упаковки in vivo
Идея создания и применения космидных векторов основана
на возможности упаковки in vitro последовательностей ДНК,
содержащих cos-сайты, в частицы фага X. При этом получается
инфекционный фаг, который затем с высокой эффективностью
74
Космидные клоны и клоны библиотек
75
вводит свою ДНК в бактерию-хозяина. Те же реакции, приво-
дящие к эффективной и избирательной упаковке космид в час-
тицы фага Л, можно провести и in vivo [2—5]. В этом случае
упаковку обеспечивает система, запускающаяся в бактериаль-
ной клетке либо при индукции профага в ответ на тепловую
инактивацию термочувствительного репрессорного белка, либо
при индуцировании клетки хелперным фагом. При этом способе
существенно возрастают эффективность процессов переноса
генетической информации между разными клетками и взаимо-
превращения различных (кольцевых и линейных с выступаю-
щими концами) форм ДНК (рис. 3.1).
В наших опытах высокий титр космидных лизатов получал-
ся и при суперинфекции, и при индукции профага. Суперинфек-
ция обладает тем явным преимуществом, что снимает необходи-
мость в специальном штамме для упаковки in vivo. При это:.',
однако, часто получаются сильно варьирующие титры, а из-за
загрязнения образующихся лизатов частицами хелперного фага
суперинфекцию нельзя использовать для получения препаратов
чистой линеаризованной космидной ДНК- Высокая концентра-
ция упаковывающего фага в лизатах к тому же заставляет
использовать иммунные к нему рецпписнтные штаммы (напри-
мер, ВНВ3169 или ВНВ3175) [5], чтобы избежать лизиса бак-
терий-реципиентов. Индукция резидентного профага дает,
напротив, более стабильные титры упакованных космид. Введе*.
76
А, Постка, X. Лерах
ние соответствующих мутаций в a/f-сайт профага позволяет
резко уменьшить загрязнение препаратов ДНК хелперным фа-
гом (или дефектными частицами) или в случае хелперных cos~
фагов полностью его исключить. '
Упаковку космид in vivo можно применять при многих видах
работ с космидными клонами и библиотеками. Основное досто-
инство данного подхода при амплификации библиотек — прос-
тота работы с упакованными космидами. Как и библиотеки,
полученные на основе Z-векторов, космиды хорошо хранятся
при 4 °C в виде фаговых суспензий, что позволяет легко оттит-
ровать космидный препарат. При комнатной температуре пре-
парат стабилен и хорошо выдерживает транспортировку.
Амплификация перед упаковкой in vivo, подобно любой
амплификации библиотеки, чревата уменьшением представлен-
ности библиотеки и может привести к повторному выделению
идентичных клонов. Как и в других случаях, этот риск может
быть снижен раздельной амплификацией и упаковкой незави-
симых фракций космидной библиотеки.
Другое преимущество использования упаковки in vivo осно-
вывается на действии кодируемого %-геномом фермента терми-
назы, который как при упаковке, так и в отдельной реакции
in vitro разрезает исходно кольцевую космидную молекулу, пе-
реводя ее в линейную форму. ДНК, выделенная из фаговых
частиц, образовавшихся в процессе упаковки in vivo, избира-
тельно, разрезается терминазой по cos-сайту, так что образу-
ются одноцепочечные концы длиной 12 нуклеотидов. Такая
линейная ДНК нужна, например, для рестрикционного анализа
космидных клонов методом частичного рестриктазного гидроли-
за [6, 7], для анализа клонов с использованием ЕМ-гетеродук-
плексов [3], для перевода космид перед введением в эукарио-
тические клетки в линейную и (или) конкатемерную формы [4].
Кроме того, выступающие концевые одноцепочечные участки
используются, например, для избирательного мечения одного
или другого конца комплементарными олигонуклеотидами.
В других случаях одноцепочечные концы могут пригодиться для
выделения фрагментов при помощи гибридизации.
Третье достоинство упаковки in vivo — возможность исполь-
зования ее для избирательного и высокоэффективного переноса
космид между различными штаммами-хозяевами. Проводя по-
следовательные циклы упаковки и повторного инфицирования,
космиды или космидные библиотеки можно легко (и избиратель-
но) переносить между штаммами-хозяевами с различным гено-
типом; при этом космиды будут постоянно или временно под-
вергаться воздействию других генетических элементов (плазмид,
транспозонов). Поэтому упаковка in vivo представляет собой
важный этап многих подходов к генетическим манипуляциям с
Космидные клоны и клоны библиотек
77
космидными клонами. Это особенно существенно для описанных
ниже методов рекомбинации космид. В этом случае упаковка
in vivo обеспечивает две независимые важные функции — пере-
нос'^осмид между различными штаммами-хозяевами и селекцию
космидных клонов, рекомбинировавших с плазмидой-зондом.
3.3. Отбор космид с помощью гомологичной рекомбинации
3.3.1. Гомологичная рекомбинация в клетках Е. coll
может применяться в качестве метода,
альтернативного гибридизации колоний
Идентификация специфических космидных клонов может
проводиться либо (в редких случаях) с применением селектив-
ной системы, специфичной для клонируемой последовательности,
либо, как правило, путем отбора клонов, несущих последова-
тельности, гомологичные специфическому ДНК-зонду. Эта про-
цедура обычно проводится методом гибридизации при высокой
плотности колоний [8].
Генетические способы отбора требуют значительно меньших
затрат труда, чем методы скрининга, что позволило разработать
новые подходы, позволяющие проводить генетическую селекцию
клонов, содержащих последовательности, гомологичные любой
желаемой последовательности-зонду. Это дало возможность
расширить применение этого метода, распространив его на за-
дачи, обычно решаемые с помощью более трудоемких (и часто
менее специфичных) методов скрининга. В общих чертах в ос-
нове всех этих подходов лежит следующее обстоятельство: хотя
в случае большинства специфических эукариотических последо-
вательностей нельзя обеспечить генетическую селекцию в клет-
ках Е. coll, можно сконструировать пары векторов, позволяющие
селектировать продукты гомологичной рекомбинации, обуслов-
ленной гомологией последовательностей между клонами. Такая
селекция основана на том факте, что процессы, подобные реп-
ликации ДНК, транскрипции, мобилизации плазмид или упаков-
ке ДНК в фаговые частицы, могут происходить только по cis-
схеме в отношении соответствующего регуляторного элемента
(области начала репликации, промотора, сигнала мобилизации
или упаковки). Чтобы исключить рекомбинацию между вектор-
ными последовательностями, нужно подбирать векторные пары,
не обладающие взаимной гомологией (или в случае рекомбина-
ции фагов без взаимной гомологии в предела?; той области, ко-
торая определяется маркерами, используемыми для селекции).
Соблюдение этого главного принципа лежит в основе и скри-
нинга фаговых клонов с использованием рекомбинации между
78 А. Постка, X, Лерах
фагами [9] или между фагом и плазмидой [10], и скрпниш-а
космидных (или плазмидных) библиотек с использованием ре-
комбинации космида-плазмида [4, 11]. Ниже мы остановимся
на последнем варианте, предназначенном для отбора специфи-
ческих клонов из репрезентативных библиотек, которые относи-
тельно полно представляют геном млекопитающих. j
/
3.3.2. Принципы космидно-плазмидной рекомбинации
Главное условие для проведения экспериментов по космид-
но-плазмидной рекомбинации — это отсутствие гомологии в по-
следовательностях обоих векторов, так как это исключит воз-
можность их гомологичной рекомбинации между ними. Кроме
того, необходимо ввести космидную библиотеку, исходно скон-
струированную и амплифицированную в гесА~-клетке, в полно-
ценный по рекомбинации штамм, несущий селективную плазми-
ду (плазмиды). Наконец, необходимо обеспечить возможность
селекции рекомбинантных космид после их повторного переноса
в гесА~-хозяина с тем, чтобы убедиться в стабильности клонов
при их дальнейшем размножении.
Принцип метода схематически показан па рис. 3.2. Космид-
ные библиотеки, сконструированные в гесД~-клетке, упаковы-
вают в Х-частицы, чтобы получить лизат с высоким титром
трансдуцирующих частиц. Аликвоты упакованной библиотеки
используют затем для заражения гес+-клеток, в которые пред-
варительно введена последовательность (или последовательно-
сти) -зонд, клонированная в плазмиде, не имеющей гомологии
с космидным вектором. Через 30 мин — 2 ч, в течение которых
должна произойти рекомбинация, космиды вновь упаковывают
в фаговые частицы. Упакованные космиды используют для за-
ражения гесА“-хозяина, проводя селекцию по обоим маркерам
лекарственной устойчивости — плазмидному и космидному. По-
скольку плазмида не несет соз-последовательностей, она будет
перенесена в реципиентные клетки только в случае рекомбина-
ции с гомологичной последовательностью в космидном клоне.
Для того чтобы восстановить исходную космиду, можно вновь
разделить два клопа в результате второго рекомбинационного
события и после переноса в нового хозяина идентифицировать
ревертантов на индикаторных чашках. Как будет сказано ниже,
точное восстановление, однако, не всегда возможно (или жела-
тельно) .
3.3.3. Отбор рекомбинантов
Технически легкий и селективный перенос космидных кло-
нов (обусловленный использованием упаковки in vivo) удобен
для отбора рекомбинантов, поскольку только последовательно-
Космидные клоны и клоны библиотек
79
Упакованная космидная
’библиотека (KmR)
Инфицирование
гее Рё- клетки,
несущей плазмиду
/l/js
I
Рекомбинация
I
Упаковка in vivo
I
Инфицирование
rec Рё- клетки,
селекция на
Арки
Переупаковка
in vivo
Селекция Кгпк
Рис. 3.2. Принципиальная схема рекомбинации между космидой и плазмидой.
80 А, Постка, X. Лерах j
i
сти, находящиеся в cts-положении по отношению к соз-сайтэм,
будут упаковываться в фаговые частицы. Рекомбинант, образо-
вавшийся за один (или любое нечетное число) рекомбинацион-
ный акт, содержит селектируемую плазмиду, которая интегри-
рована в космиду. Поэтому он способен после упаковки пере-
носить лекарственную устойчивость плазмиды и космиды в
реципиентную клетку. Такие клетки и, следовательно, i/лоны
рекомбинантных молекул можно легко отобрать. Для этого по-
пуляцию клеток, зараженных всем множеством упакованных
космид, среди которых есть небольшое число рекомбинантов*,
высевают на чашку с двумя антибиотиками и производят отбор
по обоим маркерам лекарственной устойчивости.
Данная система сочетает, с одной стороны, высокую избира-
тельность cis-этапа, которая обеспечивает селективную упаков-
ку последовательностей, несущих cos-сайты, с другой — очень
сильный, но не обладающий cis-специфичностью отбор по генам
устойчивости к антибиотикам. Такое сочетание дает возмож-
ность получить очень низкий фон. (Однако, как будет указано
ниже, иногда по непонятной причине все же наблюдается сла-
бый фон.)
Можно рассматривать и другие подходы к селекции, допол-
няющие или заменяющие селекцию при упаковке. Такие подхо-
ды могут пригодиться в особых случаях — например, когда
отбираемые рекомбинантные молекулы слишком велики, чтобы
быть упакованными в Х-частицы, или когда проводят селекцию
на высокую частоту рекомбинационных событий. При таких
способах селекции может, в частности, использоваться контро-
лируемая внешними условиями репликация одного из Двух
репликонов (такую возможность дают, например, космидные
производные с температурочувствительной областью начала
репликации R6K) или специфическая мобилизация плазмидных
или космидных последовательностей.
3.3.4. Векторы
Хотя исторически сложилось так, что многие плазмидные
(или космидные) векторы были сконструированы на основе не-
большого числа очень похожих репликонов (ColEl, РМВ1 и
рА15) [12], используются и специальные векторы с другими
областями начала репликации. Получению таких векторов, осу-
ществленному лишь недавно, отчасти способствовало осознание
их потенциальной ценности для систем космидной рекомбина-
ции. Для этой цели мы сконструировали семейство космидных
(pcos2EMBL, рсозЗЕМВЕ, pros5EMBL, pcos6EMBL) и плаз-
мидных (pslEMBL) векторов. В них использованы 0- и у-реп-
ликоны природной многокопийной плазмиды R6K [13] (из них
Космидные клоны и клоны библиотек
81
взяты области начала репликации) и ген устойчивости к кана-
мицину из транспозона Тп903 как селективный маркер ([4];
Эрик и др., в печати; Крэг, неопубликованные данные).
ци в одном из перечисленных выше векторов нет последо-
вательностей, гомологичных pUC8 [14] или сходным плазмидам.
Поэтому рекомбинация между клонами в космидных библиоте-
ках, сконструированных на основе этих векторов, и клонами на
основе pUC или сходных плазмид будет зависеть только от го-
мологии в последовательностях-вставках. Библиотеки, созданные
в pcos3, pcos4, pcos5 и рсозб, могут быть скринированы также
с помощью последовательностей, клонированных в pBR322 [15]
и родственных плазмидах, несущих такой же ген устойчивости
к тетрациклину. Этот ген имеется, однако, в pcos2EMBL, так
что библиотеки, сконструированные на ее основе, не могут быть
скринированы с использованием рекомбинантных плазмид —
производных pBR322. Напротив, у плазмиды pslEMBL [5] нет
последовательностей, гомологичных многим распространенным
космидным векторам — производным рМВ1 (например,
pcos4EMBL [Пустка, Брэди, Шуц и Лерах, неопубликованные
данные]), и поэтому она может быть использована в рекомби-
национных экспериментах с космидными библиотеками, полу-
ченными на основе этих векторов. pslEMBL — плазмида раз-
мером 4,2 т. п. н., несущая ген устойчивости к канамицину, об-
ласть начала репликации R6K, /acZa-комплементирующую
последовательность и полилинкер из М13шр9[16].
Недавно начали применять другие космидные или плазмид-
ные векторы на базе репликонов, отличных от семейства ColEl,
которые также используют в рекомбинационных экспериментах
в сочетании с подходящими векторами. В качестве примеров
можно назвать селективные плазмиды, несущие область начала
репликации R1 [11], космиды на основе pSClOl [17] и космид-
ные векторы, использующие область начала репликации фа-
га X [18].
3.3.5. Штаммы
Основными штаммами для селекции клонов с помощью го-
мологичной рекомбинации являются упаковывающие in vivo
гесА~- и гес+-штаммы, используемые для этапов амплификации
и рекомбинации. Штаммами, исходно сконструированными и
использованными нами [5] и применявшимися позднее в сход-
ной системе [И], были: 1) ВНВ3175, полученный из HL202
(5моО-производного DH1 [endAl, hsdR17 (rk~ mk+), supE44,
thi-1, recAl, gyrA96, relAl] [19]) путем лизогенизации X3159
(kimm434, cits, b2, red3 Sam7), и 2) BHB3169, который был
аналогично получен из W3110 rk~ mk+ [10]. Фаг Х3169, исполь-
6—1186
®2 А, Постка, X, Лерах Д
зованный для лизогепизации, несет температурочувствите/ш-
ный (imm.434) ген репрессора CI, обусловливающий индукйию
при изменении температуры. К генетическим характеристикам
профага, ответственными за реакцию упаковки in vivo, Отно-
сятся I) мутация Sam7, которая, блокируя бактериальный ли-
зис, приводит к повышению эффективности упаковки; 2) деле-
ния Ь2, введенная для уменьшения частоты исключения профа-
га, и 3) мутация в гене red, предотвращающая red-зависимую
рекомбинацию.
Одна из трудностей, с которой сопряжено использование
ВНВ3175, состоит в крайне низкой эффективности высева пер-
вичных библиотек, содержащих ДНК млекопитающих. Объяс-
няется это, по-видимому, индукцией профага из-за гесЛ-зави-
симого расщепления репрессора [20], обусловленного некото-
рыми особенностями ДНК млекопитающих. Наоборот, эффек-
тивность высева упакованных космидных клонов, предваритель-
но амплифицированных в клетках Е. coll, идентична обнаружи-
ваемой для штамма DH1. Исходя из анализа данной модели,
был создан упаковывающий штамм, несущий профаг ind~Cl857
(Крэй, неопубликованные данные), в котором эффективность
высева первичных библиотек оказалась такой же, что и для
библиотек, предварительно амплифицированных а Е. coli.
Этот штамм, однако, обладает сниженной эффективностью упа-
ковки. Описаны также и другие штаммы (разд., 3.4.2 и гл. 2).
3.3.6. Генетическая селекция клонов
Как сказано выше, приведенные здесь методики использу-
ются для отбора уникальных космидных (или плазмидных)
клонов из больших библиотек с применением генетической се-
лекции, а не скрининга библиотек путем гибридизации коло-
ний. Этот подход получил широкое распространение; использо-
вание в качестве селекционных зондов последовательностей,
клонированных в pUC8 и pUC9, позволяет выделить многие
космидные клоны из библиотек мыши и человека (см., напри-
мер, [5, 6, 22]).
С тем, чтобы продемонстрировать универсальность рассмат-
риваемого генетического подхода, амплифицированные библио-
теки были скринированы и с помощью гибридизации и гене-
тическим способом. Полученные клоны оказались в этих слу-
чаях одинаковыми. Как уже упоминалось, генетическому под-
ходу присущ определенный недостаток: это необходимость ам-
плификации, которая может привести к уменьшению полноты
библиотеки и выделению идентичных клонов. Такой проблемы
не возникает в случае гибридизационного скрининга первич-
ных библиотек. Вероятность нежелательных последовательное-
Космидныеклоны и клоны библиотек 83
тей\ можно значительно уменьшить путем амплификации (или
упаковки in vivo) независимых фракций первичной библио-
теки.
Необходимо рассмотреть и ряд других потенциальных труд-
ностей. Одна из них заключается в том, что на космидные-
клоны неизбежно воздействует рекомбинационная система клет-
ки-хозяина, что может привести к перестройкам в космидных
последовательностях. В наших опытах такие перестройки были
достаточно редкими, возможно, из-за короткого времени пре-
бывания космид в гес+-условиях, а также благодаря отбору
полноразмерных клонов, что предопределяется самой системой
селекции. Стабильность космид в процессах рекомбинации и
реверсии к исходной структуре была испытана, в частности, во
многих последовательных циклах рекомбинации — реверсии,
и никаких свидетельств перестроек обнаружено не было [5].
Теоретически селекции присуще ограничение предельных
размеров, устанавливаемых для рекомбинантов упаковываю-
щей системой Z [23]. Однако эти трудности легко обойти, если
использовать маленькие (2,9—5 т. п. н.) плазмиды. Средний
размер космидного клона в наших библиотеках составлял 45
т. п. н., и существующий запас размера (до предельного) впол-
не достаточен, чтобы покрыть прирост размера образующихся
продуктов рекомбинации. Кроме того, можно ожидать, что ог-
раничения по размеру, накладываемые одиночным актом упа-
ковки, менее строгие, чем те, которые накладываются повтор-
ными упаковками, необходимыми для образования фаговых
бляшек [24].
Использование методики генетической рекомбинации осуще-
ствляется и в том случае, если селекционная плазмида и кос-
мидный клон отличаются друг от друга по последовательнос-
тям — например, когда наблюдается полиморфизм клонирован-
ного участка или когда клонированы разные копии одного се-
мейства генов или когда в космидпоп ДНК присутствуют
интронные последовательности, которых нет в составе се-
лекционной плазмиды. Поскольку кроссинговеры, ведущие к
интеграции и разделению двух клонов, будут, как правило,,
происходить в различных местах в пределах области гомолог?;:?,
а-временно возникающие ошибки спаривания при их репарации
могут создавать дополнительные непредсказуемые изменения,
то последовательности, обнаруженные в ревертантной космпде
(или селекционной плазмиде), могут отличаться от исходных.
Однако возможные изменения ограничены областью гомологии
последовательностей и, стало быть, проблема легко решается
повторным выделением той же космиды с использованием дру-
гой гомологичной последовательности. Эта трудность не возни-
кает в случае «прогулки по хромосоме», при которой каждая’
6*
84
А, Постка, X. Лерах
последовательность, которая будет использоваться на следую-
щем этапе, уже содержится в немодифицированном виде в пре-
дыдущем клоне. Помимо гомологичной рекомбинации в клетке
функционируют и другие рекомбинационные механизмы, что
может приводить к образованию фона случайных клонов, рас-
познаваемых селекционной системой. Эти возникающие с низ-
кой частотой фоновые события особенно заметны (и ведут к
ошибкам), если в библиотеке вообще не содержится клонов с
последовательностями, гомологичными искомым. Часто такие
фоновые клоны, образовавшиеся в результате негомологичной
рекомбинации, могут быть идентифицированы на стадии ре-
версии, так как в> этом случае наблюдается очень низкая час-
тота возникновения ревертантов. В силу сказанного характе-
ристика ревертантов должна включать как меру общей пред-
осторожности идентификацию оставшихся последовательностей
селекционного плазмидного вектора.
Основное достоинство генетического подхода — быстрота и
эффективность метода; достаточно большое число эксперимен-
тов можно проводить параллельно. Поэтому не возникает
нужды в повторном скрининге, что делает этот метод особенно
удобным там, где требуется повторное выделение многих кос-
мид (например, при «прогулке по хромосоме»). Другое пре-
имущество генетического метода — очень высокая чувствитель-
ность гомологичной рекомбинации к ошибочному спариванию
последовательностей [25, 26]. Данное обстоятельство может
быть особенно удобным, например, при выделении космид, со-
держащих конкретные копии тех или иных семейств генов, или
в экспериментах по «прогулке по хромосоме». Такие экспери-
менты упрощаются при работе с некоторыми из наших новых
векторов (Эрих и др., в печати), содержащих А/оЛ-сайты, флан-
кирующие инсерционный сайт, что позволяет провести быстрое
и избирательное клонирование концевых фрагментов вставки
в производных pUC18 или pEMBL18, содержащих АЫ1-сайты
в полилинкере.
Представляется возможность не только отбирать специфи-
ческие космидные клоны из космидных библиотек, но и исполь-
зовать специфические космиды для выделения клонов из слож-
ных плазмидных библиотек. Этот подход можно использовать
и для выделения клонов кДНК, содержащих последовательнос-
ти, кодируемые вставочным фрагментом данного космидного
клона.
3.3.7. Модификация клонов
Второе важное применение гомологичной рекомбинации —
использование изменений, происходящих при рекомбинации
и (или) реверсии для внесения специфических или даже слу-
Космидные клоны и клоны библиотек
85
Рис. 3.3. Направленная интеграция последовательностей путем гомологичной
рекомбинации.
чайных изменений в структуру космиды. Эти изменения могут
быть внесены в любое место во вставке или в векторе (безот-
носительно к существованию уникальных рестрикционных сай-
тов) одновременно с выделением клонов методом гомологич-
ной рекомбинации.
Известно два основных пути: желаемая структура может
быть образована в один этап, например, в результате рекомби-
нации в пределах короткого района гомологии — при введении
последовательностей, содержащихся в селекционной плазмиде,
в специфический участок космидного клона (рис. 3.3). Приме-
ром может служить подстановка энхансерных или премотор-
ных последовательностей в участок, предшествующий гену, вве-
дение селективных маркеров или индикаторных генов (устой-
чивости к неомицину, хлорамфеникол-ацетилтрансферазы, ^-га-
лактозидазы) под контроль промотора, который несет космида,
или создание случайных составных сегментов, образованных
гомологичными генами, принадлежащими космиде и плазмиде.
При необходимости может быть использован перенос в клетки-
хозяева, непермиссивные для репликации одного из этих ком-
понентов, что позволяет элиминировать независимо реплициру-
ющиеся плазмиды, возникающие в результате вторичных ре-
комбинационных событий. Сходным образом селекционные
плазмиды с последовательностями, гомологичными космидному
вектору, используют для введения специфических последова-
тельностей (например, маркеров, селектируемых в эукариоти-
86
А. Постка, X. Лерах
ческих клетках, или элементов, обеспечивающих опосредован-
ные транспозонами делеции) во все или отдельные клоны кос-
мидной библиотеки.
Может также быть использован и альтернативный двухсту-
пенчатый процесс, приводящий к обмену последовательностями
между плазмидой и космидой. При использовании подходящей
системы селекции или переноса космида или плазмида может
быть получена раздельно. Поэтапно, например, можно ввести
мутации, индуцированные в коротком районе гена, клонирован-
ного в упаковывающейся плазмиде, обратно в космиду. Наобо-
рот, можно перенести последовательности из космиды в плаз-
миду для дальнейшего анализа. Как показано на рис. 3.4, та-
кой процесс особенно хорошо контролируется в двухэтапном
варианте метода, когда исходная маркерная последователь-
ность (например, /яс-оператор) вводится в результате двойной
рекомбинации в тот локус космиды, который должен быть
мутагенизирован. Обмен этой последовательности на последо-
вательности, несущие желательную мутацию, легко обнаружить
с помощью теста на присутствие или отсутствие /яс-оператора
(см. методику получения реверсий).
Модификация космидных клонов путем гомологичной реком-
бинации — исключительно эффективный и высокоизбиратель-
ный процесс. В большинстве случаев, когда работают с интакт-
ными космидами, подходящие сайты рестрикции либо трудно
идентифицировать, либо их вовсе нет. И преимущество генети-
ческих подходов обычно состоит в том, что они требуют значи-
тельно меньших затрат труда.
3.4. Методики
3.4.1. Конструирование космидных библиотек
Космидные библиотеки можно построить с помощью любой
из обычных методик (гл. 2). Мы используем разработанный
нами вариант [5] и сходную с ним методику Бэйтса и Свифта
[27]. В наших векторах два cos-сайта разделены тупоконечным
сайтом для клонирования. Эта методика, представляющая со-
бой модификацию способа космидного клонирования, предло-
женного Иш-Горовицем и Берком [28], предполагает частичный
гидролиз рестриктазами ЗашЗА или Mbol для случайной фраг-
ментации ДНК с последующим дефосфорилированием получен-
ных фрагментов (вместо разделения по размерам), чтобы из-
бежать образования клонов, содержащих составные (лигиро-
ванные) вставочные последовательности. «Плечи» космидного
вектора получают обработкой PvuII; расщепленные РгшП-сай-
Космидные клоны и клоны библиотек
87
Рис. 3.4. Замещение последовательностей путем гомологичной рекомбинации.
Таблица 3.1. Среды и методики высева космидных библиотек
А. Реактивы
1. L-среда
10 г бактотриптона (Difco), 5 г дрожжевого экстракта (Difco), 10 г
NaCl, Н2О до 1 л, доведите pH до 7,2 и автоклавируйте.
2. L-arap
L-среда, содержащая 15 г агара на 1 л.
3. Ампициллин, исходный раствор
30 мг/мл ампициллина растворите в 50%-ном этаноле и храните при.
—20 °C.
4. Канамицин, исходный раствор
30 мг/мл канамицина растворите в воде и храните при —20 °C.
5. L-агар с антибиотиками
Расплавьте L-arap и дайте ему остыть до 50 °C. Добавьте 1 мл исход-
ного раствора антибиотика на 1 л L-arapa.
6. X-Gal (5-бромо-4-хлоро-3-индолил-бета-П-галактозид)
Растворите 20 мг/мл X-Gal в N-N-диметилформамиде. Добавляйте
40 мкл на чашку.
7. Буфер для разведения фага К
10 мМ трис-НС1 (pH 7,6), 10 мМ MgSO4, 1мМ ЭДТА.
8. 10X.HMFM
Растворите 6,3 г К2НРО4, 0,45 г Na-цитрата, 0,09 г MgSO4. 7Н2О,
0,9 г (NH4)2SO4, 11,8 г КН2РО4, 44 г глицерола в воде до конечного
объема 100 мл.
Б. Высев космидных библиотек
I Подготовка клеток DH1
1. Засейте единичной колонией L-среду.
2. Растите ночь при 37 °C с хорошей аэрацией.
3. Осадите клетки центрифугированием 10 мин при 4000 об/мин в роторе
Sorvall НВ4 или GSA.
4. Ресуспендируйте осадок в 0,5 объема исходной культуры 10 мМ
MgSO4. Полученные таким способом клетки жизнеспособны в течение
2—4 нед.
II Подготовка клеток ВНВ3175
1. Пересейте несколько независимых колоний зубочисткой на 2 чашки
с L-агаром и оставьте на ночь: одну чашку — при 30 °C, другую — при
42 °C, чтобы проверить клетки на термочувствительность.
2. Посейте отобранную температурочувствительную колонию на чашку
с L-агаром и растите ночь при 30 °C.
3. Смойте клетки с половины чашки в 500 мл L-среды и растите при
30 °C до OD6oo=0,31).
4. Осадите клетки центрифугированием 10 мин при 4000 об/мин в роторе
Sorvall НВ4 или GSA.
5. Ресуспендируйте осадок в 25 мл 10 мМ MgSO4.
Ill Высев
1. Для обычной (10 см) чашки возьмите 0,1 мл суспензии клеток до Ю4
упакованных космид, смешайте и инкубируйте 15 мин при подходящей
температуре (30 °C для температурочувствительных лизогенов, 37 °C
для других штаммов).
2. Разведите в 10 раз L-средой, подрастите 1 ч при нужной температуре
и сконцентрируйте клетки снова, как описано выше.
3. Высейте на селективную среду или на нитроцеллюлозный или иайлоно-
вый фильтр, помещенный на чашку с L-средой на 1 ч, до переноса на
селективные чашки2). Увеличьте все количественные параметры в 10 раз
для чашек (22X22 см).
*) Высеваемые клетки, содержащие профаг, по-видимому, более стабильны при хра-
нении. если приготовлены из экспоненциально растущей культуры.
2) Если применяется селекция на устойчивость к ампициллину, то нет нужды
в предварительной экспрессии в течение 1 ч.
Космидные клоны и клоны библиотек
89
ты дефосфорилируют для того, чтобы исключить возможность
формирования олигомеров вектора, и расщепляют BamHI-сайт.
Избирательное лигирование липких концов (благодаря фосфа-
тазной обработке) в сочетании с отбором по размеру, прису-
щим упаковывающей системе фага А., обеспечивает образование
лишь конструкций с правильной структурой. Параллельно с
использованием аналогичных этапов можно конструировать
космидные библиотеки и библиотеки на основе А-векторов за-
мещения. Обычный выход составляет приблизительно 105 кос-
мидных клонов и 106 А-клонов в расчете на I мкг ДНК- Кос-
мидные библиотеки амплифицируют, высевая колонии при вы-
сокой плотности (105 клонов на чашку размером 22x22 см).
После выращивания в течение ночи колонии смывают в среде
LB + HMFM (табл. 3.1), разделяют на аликвоты и хранят при
—70 °C. Используемые среды и реактивы, методики высева кле-
ток, титрования и высева космидных библиотек описаны в
табл. 3.1. Титрование упаковывающего фага требует supp-
клетку (например, NM538 [29]).
3.4.2. Упаковка in vivo
В табл. 3.2 приведены методики упаковки in vivo и ампли-
фикации космидных библиотек с использованием суперинфек-
ции. Аликвоты амплифицированной библиотеки переносят в
жидкую культуру, подращивают до ранней логарифмической
фазы (приблизительно 0,3 OD6oo) и заражают фагом А3169 с
высокой множественностью, чтобы обеспечить упаковку in vivo.
Необходима хорошая аэрация. Продуктивность упаковки повы-
шается в течение по крайней мере 3—5 ч. После этого бактерии
собирают центрифугированием, концентрируют в 10 раз и ли-
зируют хлороформом, что приводит к высвобождению космид.
Титрование должно проводиться на клетках-хозяевах, иммун-
ных к использованному хелперному фагу (imtn434 для 7,3169),
чтобы исключить его летальное действие на реципиентные клет-
ки. Другая возможность состоит в том, что предварительно оп-
ределяют титр хелперного фага, а титрование проводят при
достаточно низкой множественности заражения и небольшом
числе клеток, чтобы уменьшить взаимодействие с упаковываю-
щим фагом.
В табл. 3.3 приведена методика аналогичной процедуры для
библиотек, содержащихся в упаковывающей in vivo клетке-хо-
зяине (например, ВНВ3175). Этот этап может быть использован
для вторичной амплификации космидной библиотеки после
амплификации путем суперинфецирования первичной 'библиоте-
ки. Равным образом эта методика приложима для амплифика-
90
А. Постка, X. Лерах
Таблица 3.2. Упаковка in vivo с использованием суперинфекции
1. Разведите аликвоту библиотеки, полученной в штамме Е. coll DH1 до
OD6oo=0,05 в L-среде с антибиотиками.
2. Вырастите культуру при хорошей аэрации до ODeoo=0,3 и суперинфици-
руйте red-S/^aroM1» с множественностью заражения около 50.
3. Инкубируйте 30 мин при 42 °C, затем перенесите на 37 °C и встряхивайте
3—4 ч2)_
4. Соберите клетки центрифугированием в роторе GSA Sorvall 10 мин
5000 об/мин. Ресуспендируйте в 0,05 исходного объема культуры в бу-
фере для разведения №> и добавьте хлороформ до 5—10%. Энергично
перемешайте при комнатной температуре и удалите клеточные остатки
центрифугированием.
5. Оттитруйте космиды в надосадочной фракции, высевая их на клетки
штамма ВНВ3175, как описано в табл. 3.1. Ожидаемые титры для кос-
мид— 108—109/мл, для хелперного фага—10” частица/мл.
’) Мы обычно используем фаг, полученный тепловой индукцией клеток шп
ВНВ3169.
2) Титр повышается вплоть до 4 ч, по снижается за ночь.
3) См. табл. 3.1.
ции и упаковки библиотек, сконструированных непосредствен-
но в соответствующих упаковывающих in vivo клетках-хозяе-
вах.
Методика, аналогичная описанной в табл. 3.3, применима
также в случае реакций упаковки, проводимых для получения
больших количеств линеаризованной космидной ДНК, например,
для электронно-микроскопического анализа. В этом случае до-
статочно использовать в качестве инокулята пробу из жидкой
культуры, выращенной из проверенной температурочувствитель-
ной колонии. После получения лизата используют обычную ме-
тодику приготовления ДНК фага Л, in vivo.
Реакции упаковки в небольшом объеме применяют для пере-
носа космидных клонов между различными хозяевами. Их про-
водят как с использованием суперинфицирования, так и индук-
ции профага.
3.4.3. Рекомбинация космид
Для отбора уникальных клонов из сложной библиотеки (на-
пример, генов млекопитающих) применяется методика, изложен-
ная в табл. 3.4. Космиды могут быть получены упаковкой биб-
лиотеки с использованием суперинфекции (табл. 3.2) или путем
индукции лизогена (табл. 3.3).
В тех случаях, когда отбор плазмид производится по устой-
чивости к ампициллину, мы считаем необходимым высевать
Космидные клоны и клоны библиотек
91
Таблица 3.3. Амплификация и упаковка in vivo путем индукции лизогена
1. Сделайте смыв клеток ВНВ3175 с чашки в L-среде или (для хранения
при —70 °C) в L-среде + среда Хогнесса для замораживания1». Засейте
клетки в 400 мл L-среды, содержащей антибиотики, в 2-литровой колбе
Эрленмейера до ODeoo=0,05.
2. Вырастите культуру при 30 °C до OD60o=0,30.
3. Перенесите колбу в водяную баню с температурой 45 °C на 25 мин.
Необходимо, чтобы культура прогрелась до 45 °C. Перенесите колбу в ка-
чалку и инкубируйте в течение 4 ч при 37°С2).
4. Соберите клетки центрифугированием, как описано в табл. 3.1, и ресус-
пендируйте осадок в 0,05 исходного объема культуры в буфере для раз-
ведения фага L.
5. Лизируйте клетки и оттитруйте космиды, как указано в табл. 3.2. Ожи-
даемый титр космид: 108—109 на 1 мл3>.
') Среда Хогнесса для замораживания: 3 г NaCl, 12,6 г К2НРО«, 0,9 г Na-цитрата.
0,09 г MgSO4, 1,8 г (NHJ2SO4, 3,6 г КН2РО4, 88 г (70 мл) глицерола на 1 л.
2) Титр повышается вплоть до 4 ч инкубации, но падает при инкубации в течение
ночи.
3) Титры могут значительно варьировать, сильнее, чем при упаковке с использовани-
ем суперинфекции. В таких случаях часто лучше повторить упаковку, чем пытаться
работать с лизатом со слишком низким титром. В гес4--клетках концентрация инфек-
ционного упаковывающего фага достаточно низкая, в то время как загрязнение препа-
ратов ДНК — продуктами абортивной упаковки — могут быть весьма велики. Фоновые
количества хелперного фага выше в гес+-клетках. Библиотеки могут храниться при 4 °C
значительное время (в буфере для разведения фага % или в Ь-среде+10 мМ MgSCM-
Для очень длительного хранения может оказаться рациональной концентрация космид
в ступенчатом градиенте плотности CsCl (оттитруйте фракции, чтобы выявить косми-
ды) и хранение аликвот в CsCI. Перед работой нужно отдиализовать их против неболь-
шого объема буфера для разведения фага К.
клетки, инфицированные рекомбинантными космидами, на нит-
роцеллюлозные фильтры, которые помещают на поверхность
чашки, содержащей ампициллин (табл. 3.5). Высев на фильтры
уменьшает расщепление антибиотика в агаре ампициллиназой,
содержащейся в лизате. Аналогичные предосторожности следует
применить и для работы с некоторыми другими маркерами ус-
тойчивости к антибиотикам.
Рекомбинационные эксперименты в аналитическом варианте,
используемые, например, для интеграции или замены последова-
тельностей в индивидуальных космидах (разд. 3.4), могут быть
проведены по такой же методике с соответствующим уменьше-
нием объемов (см. табл. 3.6 или 3.7). Упаковку in vivo можно
осуществить путем индукции профага или с использованием су-
перинфекции. Мы обычно проверяем и очищаем рекомбинанты
переупаковкой в аналогичном варианте (табл. 3.6), чтобы ис-
ключить варьирующий и труднообъяснимый фон колоний с двой-
ной резистентностью, содержащих нерекомбинировавшие кос-
мидные и плазмидные последовательности.
Способ удаления селекционной плазмиды описан в табл. 3.7.
Реверсии можно получить в упаковывающем гес+-штамме с по-
мощью метода, аналогичного методу, описанному в табл. 3.6.
92
А. Постка, X. Лерах
Таблица 3.4. Рекомбинация
1. Отберите две изолированные колонии штамма ВНВ3169, несущего плаз-
миду-зонд, засейте каждую на половину чашки с L-агаром, содержащим
ампициллин. Растите в течение ночи при 30 °C. Контрольную чашку ин-
кубируйте при 42 °C. Прогрейте в течение ночи при 30 °C 100 мл L-cpe-
ды, содержащей 30 мкг/мл ампициллина.
2. Смойте термочувствительные клетки ВНВ3169, содержащие селекционную
плазмиду, с чашки, инкубировавшейся при 30 °C—каждую половину
чашки в 100 мл прогретой L-среды с ампициллином.
3. Растите клетки с аэрацией при 30 °C до OD6ao = 0,30.
4. Добавьте MgSO4 до концентрации 5 мМ, затем лизат, содержащий при-
близительно 109 космид. Дайте колбе постоять 15 мин при 30 °C. Пере-
несите колбу в качалку на 30 °C на 90 мин до добавления 15 мкг/мл ка-
намицина.
5. Проведите индукцию при 42°C в водяной бане с покачиванием в течение
25 мин.
6. Перенесите на 37 °C и энергично встряхивайте 3 ч.
7. Осадите клетки центрифугированием в роторе GSA Sorvall при
6000 об/мин 10 мин.
8. Слейте надосадочную фракцию как можно тщательнее. Отберите алик-
воту для титрования.
9. Ресуспендируйте осевшие клетки в 10 мл буфера для разведения фага X
(табл. 3.1).
10. Добавьте 1 мл хлороформа и энергично встряхивайте время от времени
в течение 30 мин или дольше при комнатной температуре.
11. Осветлите лизат центрифугированием при условиях, указанных выше.
Слейте надосадочную фракцию.
12. Оттитруйте лизат на клетках DH1 (табл. 3.1) на чашках с канамицином.
Ожидаемый титр: 10s—1091).
*) Для контроля высейте отдельно клетки, а также продукт пробной незаконной ре-
комбинации, проведенной с участием интактного селекционного плазмидного вектора
(например, pUC8). В качестве позитивного контроля может быть использована любая
плазмида, обладающая гомологией с космидным вектором (ожидаемая частота реком-
бинации составляет 10-2—10-3).
Ревертанты идентифицируют, определяя наличие или отсутствие
/ас-операторной последовательности, свойственной селекционной
плазмиде pUC; эта последовательность в /ас+-хозяине будет
конкурентно связывать /ас-репрессор, и, следователь-
но, способствовать индукции хромосомного гена lac.
Клетки, содержащие плазмиду pUC (как компонент реком-
бинантной конструкции), будут голубыми, в то время как про-
дукты реверсии теряют селекционную плазмиду, и соответству-
ющие колонии будут белыми.
Благодарности
Мы благодарим Анну-Марию Фришауф за ее вклад в раз-
работку описанных методик.
Космидные клоны и клоны библиотек
93=<
Таблица 3.5. Селективный высев рекомбинантных космид
1. За день до опыта посейте две ночные культуры штамма ВНВ3175 и рас-
тите их при 30 °C. Это делается для того, чтобы иметь на следующий
день по крайней мере одну культуру температурочувствительпых клеток.
Проверьте температурочувствительность высевом клеток на 30 и 42 °C.
2. Возьмите аликвоту лизата, содержащую 108—109 космид (но не ботее
2 мл), дайте испариться хлороформу, оставив лизат в открытом флаконе
на 10 мин при 37 °C. К остывшему лизату добавьте 2 мл суспензии кле-
ток ВНВ3175.
3. Оставьте на 15 мин при 37 °C. Добавьте 15 мл L-среды и встряхивайте
в течение 60 мин при 30 °C.
4. Отцентрифугируйте в роторе НВ4 при 4000 об/мин 5 мин. Удалите тща-
тельно надосадочную фракцию и ресуспендируйте осадок в 1,5 мл L-среды.
5. На чашку (24X24 см) с L-агаром, содержащим 15 мкг/мл канамицина и
30 мкг/мл ампициллина, наложите нитроцеллюлозный фильтр фирмы
Schleicher Schuell, высейте суспензию и инкубируйте 24—36 ч. при 30 °C0.
’) Ожидаемая частота рекомбинантов, содержащих уникальную последовательность^
в космидной библиотеке генома млекопитающих, варьирует от 2-I0-6 до Ю“3 в зависи-
мости от типа последовательности, ее длины и степени гомологии с зондом. Колонии,
которые растут группами, ближе к краю и в складках фильтра, часто бывают фоновыми.
Хорошие колонии через 24—36 ч почти прозрачны, неплотные, круглые и имеют корич-
неватый цвет.
Таблица 3.6. Переупаковка потенциальных рекомбинантов1 >
1. Отберите до 24 колоний, ресуспендируйте каждую в 200 мкл L-среды
с 15 мкг/мл канамицина, 30 мкг/мл ампициллина.
2. Подрастите их с аэрацией 2—3 ч при 30 °C, перенесите на 15 мин в 42 °C.
Уберите на 37 °C и покачивайте 2 ч.
3. Добавьте каплю хлороформа, встряхните, оставьте при комнатной тем-
пературе на 15 мин. Осветлите лизат центрифугированием и соберите на-
досадочную фракцию.
4. Распределите 150 мкл суспензии клеток DH1 (табл. 3.1) по чашке с L-ага-
ром, на которую помещен фильтр фирмы Schleicher и Schuell. Капните-
5 мкл каждой надосадочной фракции на засеянную чашку.
5. Инкубируйте 2 ч при 37 °C.
6. Перенесите фильтр на чашку с L-агаром, содержащим ампициллин и ка-
яамицин.
7. Инкубируйте при 37 °C в течение ночи.
') Этот этап нужен, чтобы избавиться от ложных клонов, возникших в результате
трансформации плазмидой-зондом или ведущих свое происхождение от клеток, пережив-
ших обработку хлороформом. На этой стадии происходит также очистка клонов и эли-
минация смешанных колоний, возникших при заражении клетки более чем одной кос-
мидой. В качестве альтернативы может быть использован метод упаковки in vivo с ис-
пользованием суперинфекции (табл. 3.2).
Таблица 3.7. Отбор ревертантных космид
1. Отберите маленькие колонии, выросшие после переупаковки, в 200 мкл
L-среды, содержащей 15 мкг/мл канамицина и 5 мМ MgSO4.
2. Подрастите 2 ч при 37 °C. Добавьте 20 мкл гей+-фага 3171 с титром
около 1011 ед/мл.
3. Качайте 2 ч при 37° и добавьте каплю хлороформа.
4. Осветлите центрифугированием и соберите надосадочную фракцию.
94
А. Постка, X. Лерах
Продолжение
5. Добавьте 5 мкл падосадочной фракции к 150 мкл суспензии клеток штам-
ма DHi (табл. 3.1). Высейте на чашки с L-агаром, в котором растворе-
ны 40 мкл 2% X-Gal в диметилформамиде, содержащем 30 мкг/мл кана-
млцина.
6. Инкубируйте ночь при 37 °C. Белые колонии содержат клоны-ревертанты,
появляющиеся с частотой от 0,1—90% среди фоновых голубых колоний,
несущих рекомбинантные космиды.
Литература
1. Hohn В., Collins J. (1980). Gene, 11, 291.
2. Fukamaki Y., Shimada К.., Takagi Y. (1976). Proc. Natl. Acad. Sci. USA, 73,
3238.
3. V ollenweider H. J., Fiandt Al., Rosenvold E. C., Szybalski W. (1980). Gene,
9, 171.
4. Lindenmaier W., Hauser H., Greiser de Wilke I., Schuetz G. (1982). Nucleic
Acids Res., 10, 1243.
5. Poustka A., Rackwitz H.-R., Frischauf A.-M., Hohn B., Lehrach H. (1984).
Proc. Natl. Acad. Sci. USA, 81, 4129.
6. Rackwitz H. R„ Zehetner G., Murialdo H., Delius H., Poustka A., Fri-
schauf A.-M., Lehrach H. (1985). Gene, 40, 259.
7. Zehetner G., Frischauf A.-M., Lehrach H. (1987). In: Nucleic Acid and Pro-
tein Sequence Analysis — A Practical Approach, Bishop M. J. and Raw-
lings C. J. (eds.), IRL Press, Oxford, UK, p. 147.
8. Hanahan D., Meselson Al. (1980). Gene, 10, 63.
9. Caroil D., Aijoka R. S. (1980). Gene, 10, 273.
.10. Seed B. (1983). Nucleic Acids Res., 11, 2427.
11. Lindenmaier W„ Dittmar K. E. J., Hauser H., Necker A., Sebald W. (1985).
Gene, 39, 33.
12. Pouwels P. H., Eng'er-Valk В. E., Brammar W. J. (1986). Cloning Vectors,
A Laboratory Alanual,Elsevier, Amsterdam.
13. Kolter R„ Helinski D. R. (1978). Plasmid, 1, 571.
14. Messing J., Vieira J. (1982). Gene, 19, 259.
15. Bolivar F„ Rodriguez R. L., Greene P. J., Betlach M. C., Heynecker H. L„
Boyer H. W. (1977). Gene, 2, 95.
16. Yamisch-Perron C., Vieira J., Messing J. (1985). Gene, 33, 103.
17. Brady G., Jantzen H. M., Bernard H. U., Brown R. Hashimoto-Goto H
(1984). Gene, 27, 223.
18. Little P. F. R„ Cross S. H. (1985). Proc. Natl. Acad. Sci USA, 82 3159
19. Hanahan D. (1983). J. Mol. Biol., 166, 577.
20. Ossanna N„ Peterson K. R„ Mount D. W. (1986). Trends Genet., 2, 55.
21. Cami B„ Rourilsky P. (1978). Nucleic Acids Res., 5, 2381.
22. Hofker M. H., vanOmmen G. J. B., Bakker E„ Burmeister M., Pearson P. L.
(1986). Hum. Genet., 74, 270.
23. Feiss M„ FisherR. A., Crayton M. A., Egner C. (1977). Virology, 77 281.
24. Struck D. l\., Durica D. S„ Young R. (1986). Gene, 47, 221.
25. Shen P., Huang H. V. (1986). Genetics, 112, 441.
26. Watt V. M., Hngles C. J., Urdea M. S., Rutter W. J. (1985) Proc Natl Acad
Sci. USA, 82, 4768.
27. Bates P. F„ Swift R. A. (1983). Gene, 26, 137.
28. Ish-Horovitz D„ Burke J. F. (1981). Nucleic Acids Res., 9, 2989.
29. Frischauf A.-M., Lehrach H„ Poustka A., Murray N. (1983). J. Mol. Biol., 170,
ГЛАВА 4
ВЫДЕЛЕНИЕ ЭУКАРИОТИЧЕСКИХ ПОЛИПЕПТИДОВ,
ПРОДУЦИРУЕМЫХ В КЛЕТКАХ Е. coll
Файона А. О. Марстон
(Fiona А. О. Marston)
4.1. Введение
С разработкой в 70-х годах методов работы с ДНК in vitro
наметились два возможных направления развития этого подхо-
да. Первое — получение труднодоступных природных белков,,
а второе — конструирование новых белков путем мутагенеза
in vitro. В наши дни появилась возможность экспрессировать
клонированные гены в различных прокариотических и эукарио-
тических клетках-хозяевах. В частности, в клетках Е. coli может
быть осуществлен эффективный и контролируемый синтез реком-
бинантных полипептидов.
Гибридные гены можно конструировать таким образом, что
детерминируемые ими чужеродные белки будут локализоваться
в цитоплазме клеток Е. coli либо, если лидерную последователь-
ность встроить перед кодирующей, будут секретироваться нару-
жу через клеточную мембрану. Как правило, когда рекомби-
нантные полипептиды остаются в клетке, они накапливаются в
больших количествах (до 25% от суммарного белка клетки),
чем когда они секретируются клеткой (менее 1 % суммарного
белка клетки). Однако многие полипептиды, экспрессируемые в
цитоплазму, образуют малорастворимые агрегаты [1J, и в этом
случае для работы с ними требуются специальные методы их
перевода в растворимое состояние.
В этой главе основное внимание сосредоточено на методах
выделения активных растворимых белковых продуктов из нера-
створимых белков, продуцируемых в цитоплазме Е. coli. Учиты-
вая эмпирический характер методик растворения и выделения
белков, подобранных для каждого индивидуального белка, в дан-
ной главе приводятся описания конкретных примеров таких ме-
тодов. Рассматриваются также методы, используемые для очист-
ки эукариотических полипептидов, экспрессируемых в виде рас-
творимых продуктов в цитоплазму или секретируемых клеткой.
В одной главе невозможно дать описание всех опубликованных
методов, более подробный обзор литературы приведен в отдель-
ной работе [1].
95.
96
Ф, Марстон
Рис. 4.1. рСТ70 — плазмида Е. coll, способная экспрессировать мет-прохимо-
зин под контролем /гр-промотора [4] (рисунок приводится с разрешения ав-
торов).
4.1.1. Векторы, используемые для экспрессии
Высокий уровень накопления «чужеродного продукта» в клет-
ках-хозяевах часто обусловливается инициацией транскрипции
сильным промотором [2] и наличием в каждой клетке большого
числа копий гетерологичного гена [3]. Однако чрезвычайно важ-
но, чтобы экспрессия плазмидных генов была контролируемой,
так как плазмиды сохраняются в клетках-хозяевах на протяже-
нии многих поколений [4]. Это существенно, поскольку синтез
рекомбинантных продуктов — метаболическая нагрузка на клет-
ки [5]; клетки, утратившие плазмиды, при размножении вскоре
опережают в росте клетки, содержащие плазмиды. Строгий
контроль экспрессии позволяет сначала получить большую био-
массу клеток, не экспрессирующих гетерологичный ген, с после-
дующим появлением небольшого числа поколений со включенной
экспрессией этого гена.
В первых экспрессирующих плазмидах для запуска транс-
крипции использовали сильные метаболические <trp- или 1ас-
промоторы (рис. 4.1), а рекомбинантный ген встраивали за уча-
стком промотор-оператор. Однако при использовании систем ме-
таболической регуляции, контролирующих работу этих оперонов,
было обнаружено, что экспрессия плазмидных генов была ско-
рее конституитивной, чем контролируемой [6], и в результате
при выращивании клеток плазмиды утрачивались. Экспрессия
контролируется лучше при использовании плазмид с терморегу-
лируемой системой транскрипции [4], содержащих такие промо-
торы, как PR и PL, полученные из фага X. Контроль работы та-
ких промоторов может осуществляться температурочувствитель-
ным продуктом гена Х-репрессора с 1а$7, денатурирующим при
температуре выше 37 °C. Плазмида, содержащая Рк-промотор
фага X, изображена на рис. 4.2. Другая особенность этого век-
Экспрессия эукариотических полипептидов в Е. coli
97
Рис. 4.2. Плазмида pMG168 Е. coli— вектор с двумя точками начала реплика-
ции, способный экспрессировать большие количества мет-прохимозина при тем-
пературах выше 37°C [4] (рисунок приводится с разрешения авторов).
тора состоит в том, что он содержит две точки начала реплика-
ции; в результате благодаря возможности контролировать число
копий плазмиды в клетке можно повысить эффективность на-
копления продукта [7]. При начале репликации с одной из то-
чек образуется низкое число копий плазмиды, при начале репли-
кации с другой точки, находящейся под контролем %-промотора
PR — большое число копий. При температуре 34 °C или ниже
репликация плазмид осуществляется с «низкокопийной» точки
начала репликации. Процесс репликации, начинающейся с дру-
гой точки, находится под контролем с/857-репрессора. Таким об-
разом можно вырастить большую биомассу клеток, затем под-
нять температуру культивирования до 38—42°C: происходят де-
натурация репрессора с /857 и как следствие — увеличение числа
копий плазмиды и включение экспрессии встроенного чужерод-
ного гена [8J. При использовании таких экспрессирующих векто-
ров могут быть наработаны большие количества рекомбинант-
ных белков. Обычно на их долю приходится 20—25% суммарно-
го белка клетки [4]. Удавалось осуществлять успешную нара-
ботку таких белков в самом различном количестве — от объема
лабораторной колбы до объема ферментера (100—200 л). Здесь
имеется широкое поле деятельности для будущих исследовате-
лей, особенно в области физиологии микроорганизмов и раз-
работки термоиндуцибельных систем [4].
7—1186
98
Ф. Марстон
4.1.2. Методы экспрессии
Имеются два общих подхода к осуществлению внутриклеточ-
ной экспрессии клонированных генов. Соответствующий ген мо-
жет быть клонирован в одной рамке считывания с синтетически-
ми или бактериальными кодирующими последовательностями и
экспрессироваться с образованием гибридного белка. Другой
способ — непосредственная экспрессия встроенного гена. По-
требность в экспрессии эукариотических полипептидов в составе
гибридных белков возникла, когда было обнаружено, что уро-
вень экспрессии эукариотических белков в клетках Е. coli огра-
ничивается по той причине, что они распознаются клеткой как
чужеродные и разрушаются [9]. Это особенно наглядно прояви-
лось в случае полипептидов небольшого размера. При сшивании
эукариотического гена с бактериальным синтезировались гибрид-
ные продукты, которые накапливались в клетке в значительно
больших количествах [Ю, 11]. Однако, если требуется получить
эукариотический полипептид в чистом виде, возникает необхо-
димость в методе, позволяющем правильно расщепить гибрид-
ный белок. С другой стороны, непосредственная экспрессия од-
ного эукариотического гена дает возможность получить нужный
белковый продукт. Однако первичные продукты трансляции не-
сут на своем N-конце остаток метионина. В клетках Е. coli име-
ются ферменты, осуществляющие при необходимости эффектив-
ное отщепление метионина от природных белков; однако в слу-
чае рекомбинантных белков эти ферменты работают не столь
эффективно [1]. Таким образом, белки, полученные в результате
прямой экспрессии эукариотического гена, могут содержать не-
свойственный природному белку N-концевой метионин.
Белки, имеющие в своем составе сигнальную последователь-
ность, способны секретироваться через цитоплазматическую
мембрану Е. coli в периплазматическое пространство или по
направлению к наружной мембране клетки и далее во внекле-
точную среду. Экспрессия чужеродных белков с последующей
их секрецией имеет некоторые преимущества перед экспрессией
белков, остающихся внутри клетки. Если сигнальная последова-
тельность подвергается правильному процессингу, концевая ами-
нокислота рекомбинантного белка будет идентична его природ-
ному аналогу. Секреция в периплазму может также предотвра-
тить деградацию полипептида. Самое большое преимущество
систем экспрессии такого типа состоит в том, что в процессе
секреции формируются дисульфидные связи и образуются имею-
щие правильную конформацию активные продукты.
Экспрессия эукариотических полипептидов в Е. coli 99
4.2. Методы лизиса клеток
Клеточная стенка Е. coll имеет сложную структуру и состоит
из трех четко различимых слоев: наружного, липополисахарид-
ного (ЛПС) слоя, к которому прилегает двуслойная мембрана
(наружпяя клеточная мембрана), и пептидогликанового слоя,
тесно связанного с наружной клеточной мембраной. Цитоплаз-
матическая мембрана отделена от пептидогликанового слоя
периплазматическим пространством.
Для выделения нерастворимых или растворимых белков, на-
ходящихся в цитоплазме, и клеточная стенка Е. coll, и цито-
плазматическая мембрана должны быть разрушены. При выде-
лении белков, секретируемых в периплазму, требуется большая
избирательность; в этом случае используются методы, при кото-
рых целиком или частично удаляется клеточная стенка, но не
разрушается цитоплазматическая мембрана.
4.2.1. Полный лизис клеток
Клетки Е. coll могут быть лизированы ферментами, такими,
как лизоцим, в сочетании с детергентами [12]. Можно исполь-
зовать также и методы механического разрушения клеток (гид-
родинамические), например, с помощью Френч-пресса или гомо-
генизатора Мэнтон-Голина. Третья возможность — разрушение
ультразвуком, но этот метод применяют только в случае неболь-
ших объемов растворов.
В табл. 4.1 приведен метод эффективного лизиса клеток ли-
зоцимом и дезоксихолевой кислотой (ДОХ). Клетки Е. coli,
•способные продуцировать рекомбинантные белки, переходящие
в цитоплазме в нерастворимое состояние, могут приобрести ус-
тойчивость к лизису. Такое явление наблюдалось при выработ-
ке клетками Е. coli активатора тканевого плазминогена [13].
При этом для достижения эффективного лизиса пришлось уве-
личить соотношение объемов лизирующего буфера и клеточной
массы с 3 : 1 до 9 : 1.
Во многих работах описано использование обработки уль-
тразвуком для лизиса клеток Е. coli, продуцирующих рекомби-
нантные белки. В этом случае при использовании малых объе-
мов типичная методика выглядит следующим образом:
1. Суспендируйте 1 г влажных клеток в 6 мл лизирующего
буфера (см. примечания к табл. 4.1).
2. Добавьте лизоцим до конечной концентрации 1 мг/мл.
3. Инкубируйте суспензию при 25 °C 15 мин.
4. Поместите суспензию в лед и охладите до температуры
ниже 4 °C.
7
100
Ф. Марстон
Таблица 4.1. Лизис 10 г (вес сырого осадка) клеток Е. coli
с использованием лизоцима и дезоксихолата (упрощенный вариант методики,
приведенной в работе [13])
1. Суспендируйте 10 г (вес сырого осадка) клеток Е. coli в 30 мл лизирую-
щего буфера1) с помощью гомогенизатора, поддерживая температуру
ниже 10 °C.
2. Добавьте 80 мкл раствора 50 мМ ФМСФ (PMSF2’), а затем 0,8 мл рас-
твора лизоцима3).
3. Инкубируйте суспензию во льду в течение 20 мин, периодически встряхи-
вая.
4. Добавьте при перемешивании 40 мг дезоксихолевой кислоты.
5. Перенесите суспензию в водяную баню, нагретую до 37 °C, и перемешай-
те стеклянной палочкой.
6. Когда суспензия станет вязкой и ее будет трудно перемешивать, добавьте
при перемешивании 200 мкл дезоксирибонуклеазы4).
7. Инкубируйте суспензию при комнатной температуре 30 мин или до поте-
ри ею вязкости.
*) Состав лизирующего буфера: 50 мМ трис-НС1 (pH 8,0), 1 мМ ЭДТА, 100 мМ NaCl.
50 мМ раствор
2) Исходный раствор ФМСФ (феиилметилсульфоиилфторида):
в 100%-ном метаноле. Готовят непосредственно перед использованием.
3) Исходный раствор лизоцима: 10 мг/мл в лизирующем буфере.
—20 °C.
4) Исходный раствор дезоксирибонуклеазы: 1 мг/мл в лизирующем
непосредственно перед использованием.
Хранится при
буфере. Готовят
5. Проведите обработку ультразвуком (например, на ульт-
развуковом дезинтеграторе Model W225R, Heat System, Ultra-
sonics Inc.) два раза по 15 с.
Стадии 1 н 3 можно опустить, но тогда время обработки
ультразвуком следует увеличить: три'раза по 20 с.
Чтобы определить, насколько эффективно прошел клеточный
лизис, в световом микроскопе подсчитывают количество интакт-
ных клеток до и после процедуры лизиса. Можно также опре-
делять и изменение количества растворимого белка.
4.2.2. Получение сферопластов
Для выделения содержимого периплазмы клетки Е. coli об-
рабатывают лизоцимом, затем подвергают легкому осмотическо-
мушоку[15] (табл. 4.2). В описанном методе используется вы-
сокая концентрация трис-HCl для дестабилизации наружной
клеточной мембраны, позволяющей лизоциму проникнуть через
слой пептидогликанов. ЭДТА способствует этому процессу,
a Mg2+ — препятствует.
4.3. Нерастворимые белки
Нерастворимые рекомбинантные белки обычно накапливают-
ся в клетках Е. coli в виде дискретных включений. Эти включе-
ния можно увидеть при использовании фазово-контрастного
микроскопа (рис. 4.3).
Экспрессия эукариотических полипептидов в Е. coli
101
Таблица 4.2. Получение сферопластов Е. coli
1. Суспендируйте 1 г (вес сырого осадка) клеток в 25 мл 200 мМ трис-НС1
(pH 8,0).
2. Разбавьте суспензию равным объемом раствора, содержащего 200 мМ
трис-НС1 (pH 8,0) и 1 М сахаразу.
3. Добавьте 0,5% объема 100 мМ ЭДТА (pH 7,6).
4. Добавьте лизоцим до конечной концентрации 60 мкг/мл.
5. Разбавьте суспензию в два раза водой и инкубируйте при 23°C.
6. Отберите небольшую часть суспензии, разбавьте в 50 раз водой и измерь-
те поглощение при 450 нм. Образование сферопластов завершается, когда
оптическая плотность за 10 с падает на 80—85%; обычно это происходит
менее чем за 30 мин.
7. Для стабилизации сферопластов добавьте Mg2+ до конечной концентрации
20 мМ.
Они выглядят как большие, преломляющие свет агрегаты,
часто расположенные на полюсах клетки. О накоплении нераст-
воримого белка в процессе наращивания клеточной массы мож-
но судить по появлению таких включений, а также с помощью
ДСН-электрофореза в полиакриламидном геле (ПААГ).
4.3.1, Выделение белковых включений
4.3.1.1. Центрифугирование
При использовании описанного выше метода лизиса клеток
Е. coli из них высвобождаются белковые включения, которые,
как правило, остаются в нерастворимой форме. Поскольку ре-
комбинантный белок находится в виде дискретных включений,
его выделение из клеточного лизата само по себе может слу-
жить стадией очистки. Включения эти обладают достаточно вы-
сокой плотностью, и поэтому их можно выделять центрифугиро-
ванием. Средний размер и плотность включений могут варьиро-
вать в зависимости от природы экспрессируемого белка [16].
Это подтверждается опубликованными данными о выделении
включений при использовании разных скоростей центрифугиро-
вания (величина центробежного ускорения): от 500 до 12 000 g
[1]). Обычно можно эмпирически определить скорость центри-
фугирования и время, необходимые для осаждения включений,
такие, чтобы другие специфические белки оставались в надоса-
дочной фракции. Типичный метод выбора оптимальных условий
разделения выглядит следующим образом.
1. Центрифугируйте клеточный лизат при скоростях, обеспе-
чивающих величину центробежного ускорения в интервале от
500 до 12 000 g в течение 10 мин.
2. Отберите надосадочные фракции.
3. Разведите надосадочные фракции и осадки в ДСН-буфере
для нанесения на ПААГ до концентрации белка 5 мг/мл.
102
Ф. Марстон
Рис. 4.3. Фазово-контрастная микрофотография клеток Е. coli НВ101, проду-
цирующих прохимозин.
4. Проведите электрофорез надосадочных фракций и осадков
в ПААГ [17], нанося на гель 50—150 мкг белка.
Удовлетворительная степень относительной чистоты и выход
экспрессируемого белка свидетельствуют об оптимальных усло-
виях функционирования; в противном случае полученные ре-
зультаты могут служить основанием для проведения эксперимен-
та, в котором стадии 1-—4 повторяются с использованием других
условий центрифугирования.
4.3.1.2. Методы отмывки включений
Как показано на рис. 4.4, после выделения включения могут
быть загрязнены различными примесями. Дальнейшую очистку
можно осуществить путем отмывки включений растворами, со-
любилизирующими примеси, но не рекомбинантный белок. Так,
например, два основных примесных компонента, загрязняющих
включения прохимозина, по всей видимости, — белки наружной
Экспрессия эукариотических полипептидов в Е. coli
103
Рис. 4.4. Электронная микрофотография включений прохимозина, выделенных
из Е. coli. Можно увидеть примеси материала, связанного с этими включениями
(фотография предоставлена и воспроизводится с разрешения Р. Сэгрю, Р. Нью-
сэма и отдела электронной микроскопии Кентского университета (Кентербери,
Великобритания).
мембраны клеток Е. coli [18]. Это предположение основывалось
на том, что они в значительной степени солюбилизируются в
растворе тритона Х-100 и ЭДТА (в отсутствие ионов Mg2+). Ме-
тод отмывки включений прохимозина этими реагентами [18]
приведен в табл. 4.3.
Как описано в работе Шонера и др. [14], для отмывки вклю-
чений бычьего гормона роста (6ГР) использовалась мочевина.
Поскольку для отмывки растворимых белков от включений
обычно применяются денатурирующие агенты, важно использо-
вать такую концентрацию мочевины, которая обеспечивает ми-
нимальную степень растворения. В методе очистки 6ГР
Таблица 4.3. Отмывка белковых включений тритоном Х-100 и ЭДТА
1. Лизируйте клетки лизоцимом и дезоксихолатом (табл. 4.1).
2. Центрифугируйте клеточный лизат при 12 000g 5 мин при 4 °C.
3. Ресуспендируйте осадок в 9 объемах лизирующего буфера1’, содержащего
0,5% (объемная концентрация) тритона Х-100 и 10 мМ ЭДТА.
4. Инкубируйте суспензию 5 мин при комнатной температуре.
5. Центрифугируйте суспензию при 12 000g 5 мин при 4 °C.
6. Проанализируйте осадок и надосадочную фракцию методом ДСН-электро-
фореза в ПААГ [17].
*) Состав лизирующего буфера приведен в примечании к табл. 4.1.
104
Ф. Марстон
Таблица 4.4. Отмывка с использованием мочевины
1. Суспендируйте 1 г (вес влажного осадка) замороженного осадка клеток
в 5 мл 0,1 М трис-НС1 (pH 7,3).
2. Лизируйте клетки лизоцимом, а потом обработайте ультразвуком (см.
разд. 4.2.1).
3. Центрифугируйте лизат при 1000g 5 мин при 4 °C.
4. Центрифугируйте надосадочную фракцию при 27 000 g 15 мин при 4 °C.
5. Суспендируйте осадок в 1 мл воды, разделите суспензию на аликвоты
по 100 мкл и поместите их в конические микроцентрифужные пробирки.
6. Центрифугируйте суспензию при 27 000g 15 мин при 4 °C.
7. Ресуспендируйте каждый осадок в 100 мкл 0,1 М трис-НСЬбуфере
(pH 8,5), содержащего мочевину нужной концентрации (например, 0,5 М,
1,0 М, 2,0 М, 5,0 М).
8. Центрифугируйте суспензию! при 27 000g 15 мин при 4 °C
9. Ресуспендируйте каждый осадок в 100 мкл воды.
10. Проанализируйте аликвоты по 10 мкл, отобранные из проб надосадоч-
ных фракций и ресуспендированных осадков, методом ДСН-электрофоре-
за в ПААГ [17].
(табл. 4.4) при последовательной отмывке 0,5, 1,0 и 2,0 М моче-
виной растворяется основная часть белковых примесей, но не
растворяется 6ГР. В 5,0 М мочевине растворяется примерно
10% 6ГР, тогда как 5,0 М мочевина, содержащая 1% тритона
Х-100, частично растворяет 6ГР, но не растворяет загрязняющие
белковые примеси.
Белковые примеси могут оказывать влияние на эффектив-
ность растворения, расщепления и восстановления исходной кон-
формации рекомбинантных белков. В качестве исходного мате-
риала для таких экспериментов часто отдают предпочтение вы-
деленным и отмытым белковым включениям.
4.3.2. Подходы к расщеплению гибридных белков
Как описано в разд. 4.1, гибридные белки состоят из бакте-
риального или синтетического полипептида, связанного с эукари-
отическим полипептидом. Обычно бывает нужно выделить толь-
ко эукариотический белок. Для этого используется следующий
подход. Конструируют гибридный ген, в продукте которого име-
ется точка расщепления, расположенная между участком цепи,
кодируемым бактериальной или искусственно синтезированной
последовательностью, и участком, кодируемым последователь-
ностью эукариотического гена. Было описано множество подхо-
дов, решающих проблему расщепления, которые грубо можно
разделить на два типа: химические и ферментативные (табл.
4.5). Выбор конкретного подхода определяется свойствами чу-
жеродного белка, идеальная ситуация — это возможность ис-
пользования сайта расщепления, отсутствующего в последова-
тельности чужеродного белка.
Экспрессия эукариотических полипептидов в Е. coli__________105
Таблица 4.5. Возможные варианты расщепления гибридных белков
Узнаваемая последовательность Расщепляющий агент Источник данных
-Asp-Pro- Кислый pH [19]
-Met- Бромистый циан [1Ц
-Arg- или -Lys- -Arg- -Lys- -(Asp)4-Lys- 1 -Ile-Glu-Gly-Arg-x- Трипсин [Ю]
Пептидаза из клостридий [20]
Эндопротеиназа лиз-С [21]
Энтерокиназа [22]
Фактор Ха [23]
-Pro-x-Gly-Pro-y- Коллагеназа [24]
| указывает расщепляемую пептидную связь.
4.З.2.1. Химическое расщепление
Один из примеров аминокислотной последовательности, под-
дающейся химическому расщеплению, — дипептид, содержащий
кислотолабилъную аспартат-пролиновую связь. Дипептид ис-
пользовался для сшивки двух гибридных белков, в состав кото-
рых входит ТгрЕ-бГР и TrpLE-бГР [19J. Белок TrpLE-acnapa-
гин-пролин-бГР составлял до 5% суммарного белка клеток Е.
coli; он агрегировал с образованием включений. Методика выде-
ления и расщепления этого белка приведена в табл. 4.6. Дан-
ный белок расщепляется раствором 70 %-ной муравьиной кис-
Таблица 4.6. Кислотное расщепление гибридных белков TrpLE-бГР
1. Получите клеточный лизат, используя обработку ультразвуком.
2. Центрифугируйте лизат при 12 100 g 10 мин при 4 °C.
3. Ресуспендируйте осадок в 70 %-ной муравьиной кислоте и 6 М гуанцдин-
хлориде до получения концентрации белка 0,85—1 мг/мл.
4. Инкубируйте в течение максимум 72 ч при 37 °C.
5. Диализуйте пробы расщепленных белков против 1000-кратного объема
раствора 50 мМ трис-НС1 (pH 7,6), 0,1 мМ ЭДТА и 1 мМ ФМСФ
при 4 °C.
6. Проанализируйте диализованные пробы с помощью ДСН-электрофореза
в ПААГ [17].
106
Ф. Марстон
Таблица 4.7. Расщепление бромистым цианом гибридных белков:
(J-галактозидаза-А-цепь инсулина и p-галактозидаза—В-цепь инсулина
1. Ресуспендируйте включения, выделенные из 24 г влажного осадка клеток
в 40 мл раствора, содержащего 6 М гуанидинхлорид и 1% (объемная
концентрация) 2-меркаптоэтанола (2-МЭ).
2. Центрифугируйте суспензию при 21 000g 1 ч.
3. Диализуйте надосадочную фракцию в течение ночи против 20 л воды.
4. Растворите образовавшийся преципитат в 25 мл 70%-ной (объемная кон-
центрация) муравьиной кислоты.
5. Добавьте 1,3 г бромистого циана.
6. Инкубируйте смесь в течение ночи при комнатной температуре.
7. Удалите муравьиную кислоту и избыток бромистого циана на роторном
испарителе.
лоты (объемная концентрация) и 6 М гуанидин-HCl; это жест-
кие условия, которые, по-видимому, приводят к развертыванию
молекул агрегировавших гибридных белков, делая связь аспа-
рагин —- пролин доступной для воздействия растворителя. Де-
натурирующие агенты удаляют с помощью диализа, заменяют
их на подходящий буфер и создают условия, позволяющие мо-
лекулам 6ГР снова принять нативную конформацию. Одна из
проблем, возникающих при таком подходе, состоит в том, что
на N-конце рекомбинантного белка остается остаток пролина,
и, следовательно, такой белок может отличаться от природно-
го. В приведенном примере обнаружилось, что активность свя-
зывания с рецептором 6ГР, полученного из гибридного белка,
по сравнению с активностью интактного природного 6ГР была
гораздо ниже. Это могло быть обусловлено несколькими воз-
можными причинами: дополнительным остатком .пролина, мо-
дификацией аминокислотных остатков при низком значении pH
или принятием 6ГР неправильной конформации после денату-
рации и кислотного расщепления [19].
В случае полипептидов, не содержащих метионина, для об-
разования составного белка обычно используют метиониновую
сшивку, которую в будущем можно расщепить бромистым циа-
ном. Одним из примеров такого подхода служит присоединение
^-галактозидазы к А- и В-цепям инсулина [11]; соответствующая
методика приведена в табл. 4.7. Гены гибридных белков, содер-
жащих А- и В-цепи, клонированы и экспрессируются по отдель-
ности. Перед расщеплением бромистым цианом агрегировавшие
белки инкубировали в 6 М гуанидинхлориде и 1%-ном (объем-
ная концентрация) 2-меркаптоэтаноле. Такая обработка должна
приводить к развертыванию молекул гибридных белков и ос-
лаблению всех имеющихся внутримолекулярных или межмоле-
кулярных дисульфидных связей. Опять же такая предваритель-
ная обработка может оказаться необходимой, чтобы связь ме-
тионин-Х стала доступной для расщепления.
Экспрессия эукариотических полипептидов в Е. coli 107
... cys-asn-arg-arg-asn-ser-met~phe
TOG AAC AGG ОАО ААТ ТОТ ATG ТТТ
Рис. 4.5. Структура линкерного участка, соединяющего проинсулиновые доме-
ны в гибридном белке, состоящем из ^-галактозидазы и множественных тан-
демных копий проинсулина [25].
В случае многих гибридных белков гетерологичный полипеп-
тид составляет только небольшую часть гибридной молекулы.
Для увеличения выхода гетерологичного белка были получены
такие гибридные белки, которые представляли собой продукт
экспрессии множественных копий эукариотического гена, при-
соединенных к одной-единственной последовательности бактери-
ального гена. Примером может служить соединение тандемно-
организованных проинсулиновых генов с N-концевым фрагмен-
том р-галактозидазы [25]. Аминокислотная последовательность,
связывающая домены проинсулина, изображена на рис. 4.5. Для
расщепления гибридного белка использовался бромистый циан.
Однако при использовании этого метода из одной молеку-
лы гибридного белка образуется только одна молекула чистого
проинсулина, расположенная на С-конце. Все другие мономер-
ные молекулы проинсулина имеют на своем С-конце дополни-
тельную последовательность, состоящую из пяти аминокислот
(аргинин-аргинин-аспарагин-серин-гомосерин). Поэтому для по-
лучения чистого проинсулина требуется дальнейшее расщепление
трипсином и карбоксипептидазой В. При использовании этой
системы экспрессии такое же количество гетерологичного белка
получали и в случае негибридной системы, состоящей только из
тандемноорганизованных проинсулиновых генов.
4.3.2.2. Ферментативное расщепление
Из методик, приведенных в табл. 4.6—4.8, очевидно, что Для
солюбилизации гибридных белков для последующего их рас-
щепления необходимы жесткие условия, такие, как присутствие
6 М гуанидинхлорида или 8 М. мочевины. В том случае, когда
последовательность, связывающая бактериальный и эукариоти-
ческий полипептиды, должна быть расщеплена с помощью фер-
мента, необходимы такие условия, при которых гибридный бе-
лок находится в растворенном состоянии, а фермент вместе с
тем сохраняет свою активность. В клетки Е. coli был введен
рекомбинантный ген, и осуществлена экспрессия гибридного бел-
ка хлорамфеникол-ацетилтрансфераза (САТ)-лиз-арг-кальцито-
нин; цель, которая при этом преследовалась, состояла в расщеп-
лении этого белка клостридиальной пептидазой (клострипаи-
ном), которая разрывает связь между аргинином и аминокисло-
108
Ф. Марстон
Таблица 4.8. Расщепление гибридных белков САТ-кальцитонии
клостридиальной пептидазой клострипаином
1. Суспендируйте 10 г влажного осадка клеток в 30 мл лизирующего бу-
фера1’ и лизируйте клетки, следуя методике, описанной в табл. 4.1.
2. Центрифугируйте клеточный лизат при 12 000g 30 мин при 4 °C.
3. Ресуспендируйте осадок в 9 объемах лизирующего буфера, содержащего
0,5% (объемная концентрация) тритона Х-100 и 10 мМ ЭДТА.
4. Инкубируйте суспензию 5 мин при комнатной температуре.
5. Центрифугируйте суспензию при 12 000g 30 мин при 4 °C.
6. Ресуспендируйте осадок до конечной концентрации 40 мг/мл в 100 мМ
трис-НС! (pH 7,8), содержащем 8 М мочевину и 0,14 М 2-меркапто-
этанол.
7. Инкубируйте в течение 10 мин при 37 °C.
8. Разбавьте суспензию равным объемом воды.
9. Добавьте 1 весовую часть жлострипаина к 40 весовым частям гибридного
белка.
10. Инкубируйте в течение 15 мин при 37 °C.
И. Добавьте трифторуксусную кислоту до конечной концентрации 5% (объ-
емная концентрация).
12. Инкубируйте 20 мин при 4 °C.
•13. Удалите кислотонерастворимые белки Е. coli центрифугированием при
15 000 в течение 10 мин при 4 °C.
*) Состав лизирующего буфера приведен в примечании к табл. 4.1.
той, связанной с карбоксильной группой аргининов остатка.
Синтезированный в клетках Е. coli гибридный белок находится
в нерастворимом состоянии, но его можно расщепить, поскольку
клострипаин стабилен даже в 6 М мочевине. Белковые включе-
ния выделяют и сначала инкубируют в 8 М мочевине и 2-мер-
каптоэтаноле для развертывания молекулы гибридного белка
(табл. 4.8). Затем концентрацию мочевины снижают в два раза,
разбавляя перед добавлением клострипаина. После осаждения
кислотой (для удаления некоторых белков Е. coli и пептидазы)
кальцитонин очищают методом обратнофазовой жидкостной хро-
матографии под высоким давлением (ЖХВД; reverse phase
h. р. 1. с.) [20]. На этой стадии кальцитонин представляет собой
смесь восстановленной и окисленной форм. Ресуспендирование
кальцитонина в буфере при pH 8,5 в присутствии ЭДТА способ-
ствует тиол-дисульфидной перегруппировке и образованию 1,7-
дисульфидной связи. Окисленная форма кальцитонина оконча-
тельно очищается с помощью второго цикла обратнофазовой
жхвд.
Как правило, гибридные белки конструируются таким обра-
зом, чтобы они содержали уникальные точки расщепления. Так,
например, в кальцитонине нет остатков аргинина, и, следователь-
но, можно осуществить расщепление, катализируемое клостри-
паином, не рискуя расщепить заодно и эукариотический поли-
пептид.
Экспрессия эукариотических полипептидов в Е. coli 109
1 5 10
Туг - Gly - Gly - Phe - Met - Thr - Ser - Glu - Lys - Ser -
15 20
. Gin - Thr - Pro - Leu - Vai - Thr - Leu - Phe - Lys - Asn -
25 30
Ala - lie - lie - Lys - Asn - Ala - His - Lys - Lys - Gly
Рис. 4.6. Аминокислотная последовательность p-эндорфина [10].
Гибридный белок [3-галактозидаза-р-эндорфин, экспрессируе-
мый в клетках Е. coli, напротив, содержит внутренние точки
расщепления в последовательности [3-эндорфина [10] (рис. 4.6).
Этот белок конструировался так, чтобы его можно было расще-
пить трипсином, специфически расщепляющим те пептидные свя-
зи, которые образованы карбоксильной группой лизина или ар-
гинина с соседней аминокислотой. В данном гибридном белке
остаток аргинина располагался непосредственно перед N-концом
эукариотического полипептида; в то же время, хотя в составе
p-эндорфина нет остатков аргинина (рис. 4.6), в нем имеется
пять остатков лизина. Для того чтобы не происходило расщеп-
ления самого [3-эндорфина, эти остатки лизина обратимо бло-
кируются in vitro. Нерастворимый гибридный белок выделяли
из клеточных лизатов центрифугированием (табл. 4.9; данные из
работы [10]) и обрабатывали ангидридом цитраконовой кисло-
ты при pH 9,0. В этих условиях остатки лизина модифицируются
цитракониловыми группами. После расщепления трипсином для
ингибирования протеиназ добавляют ФМСФ, после чего доводят
pH до 3,0 для удаления цитракониловых остатков. Гетерологич-
ные полипептиды, выделенные из гибридных белков, лиофилизи-
руют, перерастворяют и очищают с помощью стеклянного порош-
ка. Опиатная активность p-эндорфина, полученного таким мето-
дом, была продемонстрирована в опытах in vitro на культуре
клеток; кроме того, было показано, что полученные полипептиды
специфически связываются с рецепторами [3-эндорфина. Это бы-
ли первые опубликованные данные о биологически активном
эукариотическом полипептиде, синтезированном в клетках Е. co-
ll [10].
Приведенные в тексте примеры ферментативного расщепле-
ния предполагают использование протеиназ, узнающих специ-
фические аминокислотные остатки. Известны протеиназы, узна-
ющие более или менее протяженные аминокислотные последо-
вательности (табл. 4.5). Само собой разумеется, что чем длин-
нее узнаваемый сайт расщепления, тем с меньшей вероятностью
данная последовательность присутствует в гетерологичном бел-
ке. Одним из примеров тому может служить тетрапептидная по-
по
Ф. Марстон
Таблица 4.9. Расщепление трипсином гибридного белка
[3-галактозидаза—[3-эндорфин
1. Приготовьте клеточный лизат с помощью обработки ультразвуком
(разд. 4.2.1) и выделите белковые включения центрифугированием при
21 000 в течение 30 мин.
2. Суспендируйте белковый осадок, полученный из 6 г влажного осадка
клеток, в 10 мл раствора, содержащего 6 М гуанидинхлорида и 1%
2-меркаптоэтанола.
3. Центрифугируйте при 36 000g 60 мин.
4. Добавьте 30 мкл ангидрида цитраконовой кислоты (Fisher), три раза
по 10 мкл с интервалом в 15 мин. В течение этого времени поддержи-
вайте pH между 9 и 11, добавляя 2 м NaOH.
5. Диализуйте раствор в течение ночи против раствора 50 мМ бикарбоната
аммония.
6. Добавьте трипсин (Worthington) до конечной концентрации 0,5 мг/мл
и инкубируйте 12 ч при 37 °C.
7. Добавьте ФМСФ до конечной концентрации 1 мМ и продолжайте инку-
бировать еще 60 мин.
8. Добавьте муравьиную кислоту до конечной концентрации 1% (объемная
концентрация), затем полностью лиофильно высушите раствор.
9. Растворите высушенный [3-эндорфин в буфере трис-НС1 pH 7,6 до полу-
чения концентрации белка 0,5—1 .мкг/мкл.
10. В пластмассовой центрифужной пробирке на 15 мл добавьте 0,5 мл ло-
шадиной сыворотки (Grand Island Biological) к 0,5 мл Р-эндорфина,
а затем добавьте 50 мг стеклянного порошка1’ (140 меш, Corning Glass
Works).
11. Перемешивайте суспензию в течение 30 с, затем центрифугируйте 5 мин
при 2000g.
12. Удалите надосадочную фракцию и добавьте к осадку 3 мл воды.
13. Перемешайте суспензию и центрифугируйте ее, как описано в п. 11.
14. Удалите надосадочную жидкость и ресуспендируйте осадок в 1 мл
50%-ного (объемная концентрация) ацетона в 0,25—5,0 М НС1.
15. Перемешивайте в течение 30 с, затем центрифугируйте, как описано
в п. 11.
16. Перенесите надосадочную фракцию в чистую пробирку и полностью вы-
сушите ее от ацетона тонкой струей азота, инкубируя пробирку в во-
дяной бане с температурой 45 °C.
17. Высушенные пробы растворяют в трис-НС! (pH 7,6).
’) Перед использованием стеклянный порошок один раз промывают водой, прока-
ливают в течение 24 ч при 120 °C и до использования хранят в вакуумном эксикаторе.
следовательность, узнаваемая фактором свертывания крови
Ха. Нагаи и др. [23] встраивали такую последовательность на
границе между последовательностями XcII и р-глобина. Экспрес-
сируемый гибридный белок нерастворим, и поэтому его выделяли
в виде включений, промывали, а затем растворяли в мочевине
(табл. 4.10). Примеси других белков, которые в дальнейшем
могут помешать формированию нативной конформации р-цепи
глобина, удаляли путем ионообменной хроматографии и гель-
фильтрации. Обе стадии очистки проводили в присутствии дена-
турирующих агентов. Для удаления денатурирующих агентов
Экспрессия эукариотических полипептидов в Е. coli 111
Таблица 4.10. Расщепление гибридного белка IcII—р-глобин фактором
свертывания крови Ха
1. Суспендируйте 100 г влажного осадка клеток Е. coli в 8 мл раствора,
содержащего 50 мМ трис-НС1 (pH 8,0), 25% сахарозы (весовая концент-
рация), 1 мМ ЭДТА.
2. Добавьте 200 мг лизоцима с последующим добавлением MgCl2, МпС12 и
дезоксирибонуклеазы I до конечных концентраций соответственно 10 мМ,
1 мМ и 10 мкг/мл.
3. Инкубируйте смесь 30 мин, затем добавьте 200 мл раствора, содержа-
щего 0,2 М NaCl, 1% (весовая концентрация) дезоксихолевой кислоты.
1,6% (объемная концентрация) Nonidet Р-40, 20 мМ трис-НС1 (pH 7,5)
2,0 мМ ЭДТА.
4. Для выделения включений центрифугируйте лизат при 5000g 10 мин.
.5. Суспендируйте осадок в растворе тритона Х-100 и ЭДТА, как описано
в п. 3 табл. 4.3, но используйте 1 мМ ЭДТА.
6. Проведите отмывку и центрифугирование включений, как описано
в и. 4 и 5 табл. 4.3.
7. Повторяйте п. 5 и 6 этой таблицы до получения плотного осадка.
8. Растворите осадок в буфере, содержащем мочевину”.
"9. Уравновесьте колонку с СМ-сефарозой (CAl-Sepharose, Pharmacia) раз-
мером 4X10 см буфером, содержащим мочевину, и нанесите на псе гиб-
ридный белок, растворенный в буфере с мочевиной.
10. Промывайте колонку буфером с мочевиной до тех пор, пока поглощение
элюируемого материала при 280 нм не станет равным нулю.
11. Элюируйте гибридный белок 1 л линейного градиента 0—0,2 М NaCl
в буфере с мочевиной.
12. Уравновесьте колонку с сефакрилом S-200 (Sephacryl S-200, Pharmacia)
размером 5X60 см раствором, содержащим 5 М гуанидинхлорид, 50 мМ
трис-НС1 (pH 8,0) 1 мМ ЭДТА, 1 мМ ДТТ.
13. Нанесите гибридный белок, элюированный с колонки с СМ-сефарозой,
на колонку с сефакрилом S-200 и элюируйте его уравновешивающим бу-
фером, состав которого указан в п. 12.
14. Проанализируйте фракции, собранные при гель-фильтрации методом
ДСН-электрофореза в ПААГ [17] и отберите фракции, содержащие гиб-
ридный белок (примерно 160 мг).
15. Тщательно диализуйте очищенный гибридный белок против раствора, со-
держащего 50 мМ трис-НС1 (pH 8,0), 100 мМ NaCl.
16. Добавьте 5 мг активированного2’ фактора свертывания крови Ха к диа-
лизованному гибридному белку.
17. Тщательно диализуйте расщепленный гибридный белок против воды и
лиофильно высушите.
Состав буфера, содержащего мочевину: 8 М мочевина, 25 мМ трис-ацетат
4рН 5,0), 1 мМ ЭДТА, I мМ ДТТ.
2) Фактор свертывания крови Ха активировали ядом гадюки Русселя, иммобилизо-
•ваиным иа сефарозе 6В (Sepharose 6В).
очищенный гибридный белок до расщепления фактором Ха диа-
лизовали [23].
Имеется также ряд эукариотических полипептидов, экспрес-
сируемых в различных формах, у которых сайты ферментатив-
ного расщепления находятся на границе между повторяющими-
ся последовательностями. Пентапептид энкефалин экспрессиро-
вался как продукт конкатеннированного гена, содержащий 11
112
Ф. Марстон
энкефалиновых последовательностей, каждая из которых отде-
лена от предыдущей двумя остатками аргинина [26J. Эти гены
были присоединены к участку гена малого t-антигена вируса
SU-40 и экспрессировались с образованием гибридных белков.
Метод получения мономеров включает инкубацию с трипсином
для их расщепления и последующее удаление остатков аргини-
на карбоксипептидазой В. Для выделения и расщепления пепти-
дов можно использовать следующую методику.
1. Прокипятите интактные клетки в течение 5 мин в 60 мМ
трис-НС1 (pH 6,8), 2% (весовая концентрация) ДСН, 5% 2-мер-
каптоэтанола, 3 М мочевины.
2. Проведите разделение белков методом ДСН-электрофоре-
за в 12,5% ПААГ [17].
3. Элюируйте белки с ПААГ; соответствующий срез геля из-
мельчите и встряхивайте его в течение 16 ч при 37 °C в раство-
ре 50 мМ трис-НС1 (pH 8,0), 1 мМ СаС12 и 0,1% (весовая кон-
центрация) ДСН.
4. Осветлите элюат фильтрацией раствора и инкубируйте его
с трипсином (Worthington, TPCK-treated) при конечной концен-
трации 10 мкг/мл в течение 16 ч при 37°С.
5. Прокипятите гидролизат в течение 15 мин, охладите и за-
тем проинкубируйте с карбоксипептидазой В (Serva) при конеч-
ной концентрации 0,1 мкг/мл в течение 60 мин при 37 °C.
6. Прокипятите гидролизаты еще раз в течение 15 мин.
Пептидные гормоны, получаемые по этому методу, облада-
ют опиоидной активностью; это было показано in vitro по их
способности угнетать перистальтику подвздошной кишки морс-
кой свинки.
4.3.3. Гибридные белки, рассчитанные на специальные
процедуры выделения
Гибридные белки можно конструировать таким образом, что
их выделение в чистом виде упрощается. В этом случае бакте-
риальные или синтетические нуклеотидные последовательности,
присоединенные к эукариотическому гену, кодируют полипепти-
ды, которые избирательно присоединяются к определенным ре-
агентам, используемым для хроматографии. В трех примерах,
приведенных в табл. 4.11, гибридные белки сконструированы с
расчетом на их выделение с помощью аффинной хроматографии.
Как схематично изображено на рис. 4.7, для осуществления это-
го процесса нужно использовать следующую методику.
1. Лизируйте клетки и выделите белковые включения.
2. Солюбилизируйте включения в денатурирующем растворе.
3. Нанесите солюбилизированные белки на аффинную колон-
Экспрессия эукариотических полипептидов в Е. coli
113
Таблица 4.11. Некоторые гибридные белки, рассчитанные на специальные
методы выделения
Гибридный белок Лиганд илн субстрат, используемые для очистки Источник данных
р-Г алактозидаза—X га-Аминофенил-13-Б-тиогалактозид [27]
CAT—X Хлорамфеникол [20]
Б ело к-А—X IgG (28]
X—полиаргинин Катионообменник [29]
ку и отмойте белки Е. coli уравновешивающим буфером, содер-
жащим денатурирующие агенты.
4. Проведите специфическую элюцию гибридного белка.
Гибридный белок САТ-кальцитонин [20] (табл. 4.11) был
сконструирован так, чтобы его можно было очистить субстрат-
аффинной хроматографией. Когда обнаружилось, что гибрид-
ный белок находится в нерастворимом состоянии, стало очевид-
ным, что этот подход неприемлем. Другие гибридные белки,
приведенные в табл. 4.11, были также сконструированы с расче-
том на очистку с помощью аффинной хроматографии. Для вы-
деления составного белка р-галактозидаза-Х использовали
и-аминофенил-^-П-тиогалактозид (субстратный аналог р-галак-
тозидазы [27]) и белок А, специфически связывающийся с фраг-
ментом Fc иммуноглобулина G [28]. Эти гибридные белки, син-
тезируемые в клетках Е. coli, растворимы; метод их очистки
описан в разд. 4.4.
Подход, использованный Сассенфельдом и Бруэром [29],.
отличается тем, что гибридный белок был сконструирован так,
чтобы его можно было очистить методом катионообменной хро-
матографии. Урогастрон человека (фактор роста эпидермиса)
был получен в форме гибрида с С-концевым полиаргининовым
фрагментом. Аргинин — основная аминокислота; поэтому при
кислотном значении pH полиаргинин положительно заряжен и
прочно связывается с катионообменннком. Белки, наоборот, яв-
ляются кислыми и поэтому с ним не связываются. Гибридный
белок урогастрон—полиаргинин нерастворим, но при использо-
вании подхода Сассенфельда и Бруэра [29] не проводят выде-
ления нерастворимых включений (табл. 4.12). Вместо этого
клетки разрушают обработкой ультразвуком при pH 9,5 в буфе-
ре, содержащем 5 М мочевину, и гибридный белок оказывается
во фракции растворимых белков. Проводят две стадии катионо-
обменной хроматографии: одну—до ферментативного расщепле-
ния гибридного белка (табл. 4.12, этапы 6—8) и другую после
расщепления (табл. 4.12, этапы 13—15). Во время первой ста-
дии катионообменной хроматографии гибридный белок связыва-
8-1186
114
Ф. Марстон
Лизат клеток
Е. coli содержит
гибридные белки
Нанесение на колонку
Отмывка белков
Е со И
Рис. 4.7. Схема, иллюстрирующая принцип очистки гибридных белков.
Экспрессия эукариотических полипептидов в Е. coli 115
Таблица 4.12. Очистка и расщепление гибридного белка
урогастрон—полиаргинин
1. Культивируйте клетки Е. coli в ферментере на 7 л до OD6oo=2,2.
2. Осадите клетки из объема 800 мл центрифугированием при 4000 в тече-
ние 20 мин при 20 °C.
3. Ресуспендируйте клетки в 20 мл трис-буфера (pH 9,5), содержащего мо-
чевину’), и лизируйте их ультразвуком (разд. 4.2.1).
4. Центрифугируйте клеточные лизаты при 16 000g в течение 60 мин при
20 °C.
5. Отбросьте надосадочную фракцию и доведите pH до 5,5 НС1.
6. Уравновесьте колонку с SP-сефадексом (SP-Sephadex, Pharmacia) раз-
мером 1X2 см трис-буфером (pH 5,5), содержащим мочевину2’.
7. Нанесите надосадочную фракцию на колонку с SP-сефадексом, а затем
промойте колонку 10 мл трис-буфером (pH 5,5), содержащим мочевину.
8. Элюируйте гибридный белок 45-мл линейного градиента 0—300 мМ NaCl
в трис-буфере (pH 5,5), содержащем мочевину. Соберите фракции объ-
емом 1,5 мл.
9. Соберите фракции, содержащие гибридный белок, и доведите pH раство-
ра до 8,1 1 М NaOH.
10. Добавьте 100 мкл карбоксипептидазы В, иммобилизованной на сефаро-
зе3’, и инкубируйте 2 ч при 22 °C, осторожно переворачивая пробирку
со смесью.
11. Отделите комплекс карбоксипептидаза В—сефароза на воронке со стек-
лянным фильтром.
12. Диализуйте расщепленный гибридный белок в течение ночи при 4 °C
против 8 л 40 мМ трис-ацетатного буфера (pH 5,5).
13. Добавьте мочевину до конечной концентрации 5 М и нанесите образец
иа колонку с SP-сефадексо.м (1X1 см), предварительно уравновешенную
трис-буфером (pH 5,5), содержащим мочевину.
14. Промойте колонку 5 мл уравновешивающего буфера и элюируйте урога-
строн 30 мл линейного градиента 0—125 мМ в буфере с pH 5,5.
15. Соберите фракции объемом 1 мл и определите в них активность уро-
гастрона [31].
0 Состав трис-буфера (pH 9,5), содержащего мочевину: 5 М мочевина, 40 мМ трис-
ацетатный буфер, доведенный NaOH до pH 9,5.
2) Состав трис-буфера (pH 5,5), содержащего мочевину: 5 М мочевина, 40 мМ трис-
ацетатный буфер (pH 5,5).
з) 20 мг карбоксипептидазы В (B-DFP Type I, Sigma, UK), суспендированной в
10 мл 0,1 М Na2HCO3 (pH 8,3), добавляли к 10 мл CNBr-сефарозы и проводили реакцию
в течение 16 ч при 4 °C.
ется катионообменником, кислые белки Е. coli смываются с ко-
лонки, а затем гибридный белок элюируют солевым раствором.
Полиаргининовые остатки расщепляются карбоксипептидазой
В, и во время второй стадии катионообменной хроматографии
урогастрон (р1=5,8±0,3, [31]) проходит через колонку без
задержки, тогда как все положительно заряженные белки или
пептиды связываются катионообменником. Такой метод обеспе-
чивает выход 39% урогастрона, чистота которого превышает
95%. Два решающих момента при использовании этого подхо-
да — это доступность полиаргинина и эффективность получения
белка с «правильным» С-концом при отщеплении карбоксипеп-
тидазой аргининового «хвоста».
8'
116
Ф. Марстон
Клетки
Ферментативное
или механическое
разрушение
Клеточный лизат
Центрифугирование
Выделенные (отмытые)
включения
Инкубация с
солюбилизирующим^
агентами
Солюбилизированный
белок
“Белки, экспрес-
сируемые в
аутентичной
форме
Химическое или
ферментативное
расщепление
Отделенный
полипептид
Замена
буферного
раствора
Ренату рированныи
активный белок
Гибридные
белки
Рис. 4.8. Основные этапы выделения активных, растворимых эукариотических
белков из нерастворимых включений клеток.
4.3.4. Растворение белков, синтезируемых клетками Е. coli
в чистом виде и в виде гибридных белков
Отделение растворимых, активных продуктов из нераствори-
мого исходного материала, продуцируемого в клетках Е. coli, —
процесс, состоящий по меньшей мере из двух стадий (рис. 4.8).
Белковые включения выделяют, растворяют, а потом создают
Экспрессия эукариотических полипептидов в Е. coli
117
Таблица 4.13. Реагенты, обеспечивающие выход эукариотических
полипептидов из нерастворимых включений в раствор
Реагент
Растворяемый эукариотический полипептид’)
Гуанидин-НС1 (5—8 М) А- и В-цепи инсулина Гормон роста крупного рогатого скота
Мочевина (6—8 М) Урокиназа Прохимозин
ДСН у-Интерферон Гормон роста лосося ^-Интерферон
Щелочной pH (>9,0) Интерлейкин-2 Прохимозин
Ацетонитрил/пропанол Гормон роста цыпленка regA-белок фага Т4
’) Более подробное описание дается в другой работе [1].
условия для формирования правильной конформации (ренату-
рации). При необходимости гибридные белки перед ренатураци-
ей расщепляют (разд. 4.3.2). Имеется ряд агентов, используе-
мых для перехода в раствор полипептидов, входящих в состав
нерастворимых включений (табл. 4.13).
Такие денатурирующие агенты, как мочевина и гуанидинхло-
рид, нарушают ионные взаимодействия. Нативные белки, подвер-
гающиеся воздействию высоких концентраций этих растворите-
лей, оказываются полностью развернутыми, но ковалентные
связи, в том числе дисульфидные мостики, остаются интактны-
ми. При экстремальных значениях pH нарушаются ионные взаи-
модействия между полипептидными цепями, а детергенты и ор-
ганические растворители разрушают гидрофобные связи между
боковыми цепями полипептидов. Эффективность солюбилизации
белков каждым из таких агентов может варьировать. Это про-
исходит вследствие того, что взаимодействия между белками в
нерастворимых включениях неодинаковы, поскольку зависят от
того, какие именно белки входят в их состав [1J.
В табл. 4.14 приводится схема, которая может служить от-
правной точкой для изучения процесса солюбилизации. При ис-
пользовании реакционных смесей в таких объемах, как 1—2 мл,
можно исследовать влияние на солюбилизацию большого числа
агентов. Поскольку при этом ставится задача получить активный
растворимый белок, после того, как будет определена эффектив-
ность его солюбилизации данным агентом, проводится дальней-
шая оптимизация процесса растворения с учетом последующего
свертывания молекулы белка и восстановления нативной кон-
формации (ренатурации).
118
Ф. Марстон
Таблица 4.14. Определение эффективности перехода эукариотических
полипептидов в раствор из нерастворимых включений
1. Выделите белковые включения и, если это необходимо, отмойте их от
примесей других белков (разд. 4.3.1).
2. Суспендируйте включения в растворяющих агентах так, чтобы конечная
концентрация белка не превышала 5 мг/мл. При работе с небольшими
объемами включения суспендируют путем перемешивания, а большие объ-
емы суспендируют с помощью гомогенизатора, стараясь свести к мини-
муму пенообразование.
3. Инкубируйте суспензии в течение разных периодов времени при комнат-
ной температуре. Рекомендуемое время инкубации составляет 1—16 ч.
Большие объемы можно инкубировать при их осторожном перемешива-
нии.
4. Для осаждения нерастворимого материала центрифугируйте суспензии.
Как правило, подходящий режим центрифугирования 12 000g в течение
10—30 мин.
5. Ресуспендируйте осадки и разбавьте надосадочные фракции буфером для
нанесения1). Проанализируйте белковый состав методом ДСН-электрофо-
реза в ПААГ [17].
>) Буфер для нанесения (2Х): 20%-ный глицерол (объемная концентрация), 10%-ный
2-меркаптоэтанол (объемная концентрация), 6% ДСН (весовая концентрация), 0,125 М
трнс-НС1 pH 6,8.
Полипептиды в растворе сохраняют способность к агрегации,
поскольку у них имеются внутримолекулярные ковалентные свя-
зи, такие, как дисульфидные мостики. Если необходимо пол-
ностью разрушить эти связи, к солюбилизирующему агенту до-
бавляют восстановители тиоловых групп — дитиотрейтол (ДТТ)
или 2-меркаптоэтанол (2-МЭ). Другой метод, который может
быть использован для разрушения внутримолекулярных дисуль-
фидных связей, — получение производных в форме S-сульфона-
тов. Такой подход был использован в случае А- и В-цепей ин-
сулина [11] (табл. 4.15). Есть ли необходимость в полном раз-
рушении таких связей, можно определить, только исследуя про-
цесс ренатурации белка и восстановления его активности. Не
всегда необходимо полностью разрушать дисульфидные связи
между отдельными цепями молекулы; при ренатурации белко-
вой молекулы можно создать условия, обеспечивающие тиол-
дисульфидный обмен с образованием мономерных молекул, со-
единенных дисульфидными связями (разд. 4.3.5).
Когда подобраны подходящие солюбилизирующие агенты,
исследуют условия солюбилизации. На конечный выход активно-
го белка после его ренатурации могут влиять многие парамет-
ры реакции растворения:
1) рн,
2) температура инкубации,
3) время воздействия растворителя,
4) ионный состав растворителя,
Экспрессия эукариотических полипептидов в Е. coli
119
Таблица 4.15. Получение S-сульфонированных производных
А- и В-цепей инсулина
1. Выделите А- и В-цепи инсулина из гибридных белков с р-галактозидазой,
взяв для начала 24 г влажного осадка клеток (см. табл. 4.7).
2. После обработки CNBr и высушивания в роторном испарителе суспен-
дируйте остаток в 50 мл 8 М гуанидинхлорида.
3. Для получения S-сульфонатов добавьте 1 г тетратионата натрия и 2 г
сульфита натрия.
4. Доведите pH до 9,0 NaOH.
5. Инкубируйте смесь в течение 24 ч при комнатной температуре при пе-
ремешивании.
6. Доведите значение pH до 5,0 уксусной кислотой.
7. Дважды диализуйте смесь против 3 л воды.
5) концентрация растворяющего агента,
6) концентрация суммарного белка,
7) соотношение растворитель/белок,
8) наличие или отсутствие тиоловых реагентов,
9) модификация тиоловых групп, например, при образовании
S-сульфонатов.
Условия, используемые для растворения полипептидов, вхо-
дящих в состав нерастворимых включений, довольно жесткие
(табл. 4.13) и могут приводить к образованию производных
аминокислотных остатков. Цианат-ионы в растворах мочевины
способны модифицировать остатки лизина и привести к сшивке
полипептидных цепей. Во избежание этого растворы мочевины
готовят непосредственно перед использованием, а в качестве
дополнительной меры предосторожности их можно деионизовать
с помощью бифункциональной ионообменной смолы. Учитывая
свойства растворителей, следует свести к минимуму время рас-
творения полипептидов. Это особенно важно при использовании
значений рН>9. В таких условиях может произойти потеря
амидных групп аспарина и глутамина. При щелочном значении
pH возможны также специфические модификации аминокислот-
ных последовательностей.
4.3.5. Ренатурация
После солюбилизации эукариотических полипептидов осу-
ществляется их ренатурация в буфере, не содержащем денату-
рирующих агентов или других солюбилизаторов. Чтобы достичь
этого, можно использовать диализ или разбавление. Путем раз-
бавления можно контролировать одновременно изменения кон-
центрации растворителя и гетерологичного полипептида. Что
касается растворения, то здесь имеется ряд параметров, влияю-
щих на выход получаемого активного белка:
120
Ф. Марстои
1. Скорость перехода от условий солюбилизации к условиям
ренатурация.
2. Степень чистоты полипептида.
3. Концентрация полипептида.
4. pH.
5. Ионный состав растворителя.
6. Наличие или отсутствие тиоловых реагентов.
Существуют методики, при которых ренатурация белка про-
текает в две стадии; считают, что на первой стадии белки при-
нимают конформацию, близкую к нативной, а на второй — про-
цесс ренатурации завершается полностью. Примером этого слу-
жит процесс перевода прохимозина из 8 М мочевины в
буферный раствор при pH 10,7; после инкубации значение pH
доводится до 8,0 [18]. Другой подход — перевод растворенных
белков в раствор с низкой концентрацией мочевины, где проис-
ходит частичная ренатурация [30].
Важное значение, по-видимому, имеет степень чистоты выде-
ленного полипептида, поскольку примеси могут препятствовать
процессу ренатурации. В этом плане целесообразно выделить
нерастворимые включения, так как в данном случае возможна
очистка полипептидов в присутствии некоторых растворителей
до их ренатурации. Ионообменную хроматографию можно про-
водить в присутствии мочевины, даже при её концентрации 8М.
Гуанидинхлорид несет электрический заряд и мешает процессу
ионообменной хроматографии, но не препятствует гель-фильтра-
ции, Методы ЖХВД и высокоскоростной жидкостной хромато-
графии (ЖХВС, f. р. 1. с) могут быть использованы для очистки
полипептидов в присутствии ряда растворяющих агентов, приве-
денных в табл. 4.13; в частности, ЖХВД можно использовать
для систем, содержащих органические растворители. Другие ме-
тоды, использовавшиеся для очистки полипептидов перед их
ренатурацией, — это высокоскоростное центрифугирование [30]
и экстракция органическими растворителями [31].
В процессе ренатурации белка одним из определяющих па-
раметров является его концентрация, поскольку необходимо,
чтобы внутримолекулярные взаимодействия преобладали над
межмолекулярными. Это особенно существенно в том случае,
когда нативный активный белок содержит внутримолекулярные
дисульфидные связи. Когда дисульфидные связи в; нераствори-
мых белковых включениях оказываются разрушенными при ис-
пользовании тиоловых реагентов или образовании производ-
ных — S-сульфонатов, восстановление правильных дисульфид-
ных связей достигается применением смесей восстановленных и
окисленных тиоловых реагентов, таких, как глутатион. Должны
быть выбраны оптимальные концентрации и соотношение вое-
Экспрессия эукариотических полипептидов в Е. coll 121
Таблица 4.16. Методы контроля ренатурации эукариотических
полипептидов, выделенных из клеток Е. coll
1. ДСН-электрофорез в ПААГ в присутствии и в отсутствие 2-меркаптоэта-
нола с последующей окраской гелей.
2. ДСН-электрофорез в ПААГ в присутствии и в отсутствие 2-меркаптоэта-
нола с последующим вестерн-блот-анализом и иммунологическим ана-
лизом.
3. Аналитическая хроматография (ЖХВД и ЖХВС).
4. Определение биологической активности.
5. Иммунологическое тестирование, например, методами радиоиммунологиче-
ского (РИА, RIA) и твердофазного иммуноферментного анализа (ИФА,
ELISA).
6. Связывание с рецепторами — для пептидных гормонов.
становленного и окисленного реагентов; при pH ренатурацион-
ного буфера, равном 8,0 или большем, тиоло-дисульфидный об-
мен происходит быстро.
При высоких значениях pH обмен тиоловых и дисульфидных
групп может происходить и в отсутствие экзогенных тиоловых
реагентов; источником свободных тиоловых групп может служить
сам белок [18].
Разрабатывая методику ренатурации белка, следует оценить
эффективность этого процесса для подбора оптимальных усло-
вий. Имеется ряд способов анализа ренатурировавших полипеп-
тидов; некоторые из них перечислены в табл. 4.16. ДСН-элект-
рофорез в ПААГ и аналитическая ЖХВД и ЖХВС могут быть
использованы для определения степени агрегации конечного
продукта. С помощью ДСН-электрофореза в ПААГ в присутст-
вии и в отсутствие 2-меркаптоэтанола возможно выявление не-
правильных, межмолекулярных дисульфидных связей. Если в
исследуемых белках все еще остаются примеси других белков,
интерпретировать результаты, полученные при окраске гелей,
может быть нелегко. Эта проблема преодолевается при исполь-
зовании метода вестерн-блот-анализа, предполагающего перенос
фракционированных белков на нитроцеллюлозный фильтр и ис-
пользование специфической антисыворотки для анализа получен-
ного фильтра-реплики. Лучший способ определения эффектив-
ности ренатурации — анализ конечной стадии выхода активного
белка. Это можно сделать путем определения его биологической
активности, что осуществимо для многих ферментов. Однако в
ряде случаев для пептидных гормонов (и метаболических моду-
ляторов) может возникнуть необходимость в определении их
биологической активности in vivo, и препятствием для проведе-
ния таких опытов может стать их высокая стоимость или не-
доступность соответствующего объекта. В таких случаях для
выявления биологически активной конформации применяют им-
мунологические методы или методы с использованием специфи-
ческих рецепторов.
122
Ф. Марстон
Рис. 4.9. Очистка расщепленного клострипаином CAT-кальцитонина с помощью
обратнофазовой хроматографии ЖХВД. В и О указывают положение восста-
новленной (В) и окисленной (О) форм кальцитонина.
4.3.6. Примеры растворенных и ренатурированных
эукариотических полипептидов, выделенных из клеток Е. coll
Полипептиды небольших размеров, синтезируемые в клетках
Е. coli в виде нерастворимых гибридных белков, также прихо-
дится переводить в растворимую форму, но тщательное опреде-
ление условий их ренатурации не обязательно. Типичный при-
мер— [1-эндорфин [10]; этот полипептид, по-видимому, спонтан-
но ренатурирует либо в процессе его выделения из гибридного
белка (табл. 4.9), либо в ходе очистки при экстракции стеклян-
ным порошком. Кальцитонин человека — полипептид, который
состоит из 32 аминокислот и включает два остатка цистеина,
образующие в биологически активной молекуле дисульфидную
связь. При разделении гибридного белка (табл. 4.8) отделенный
от бактериального полипептида кальцитонин находится в двух
формах: восстановленной и окисленной, которые можно обна-
Экспрессия эукариотических полипептидов в Е. coli 123
Таблица 4.17. Анализ кальцитонина человека, полученного в результате
расщепления пептидазой из клостридий гибридного белка CAT—кальцитонин
i. Выделите гибридный белок CAT-кальцитонин из клеток Е. coli, раство-
рите его и проведите расщепление (см. табл. 4.8).
2. Уравновесьте колонку для ЖХВД Synchropak RP-P размером 1,0x25 см
раствором, содержащим 30% (объемная концентрация) ацетонитрила,
0,1% (объемная концентрация) трифторуксусной кислоты (ТФУ).
3. Нанесите надосадочную фракцию, полученную на стадии 13 табл. 4.8, на
колонку для проведения обратнофазовой хроматографии при скорости
протекания 2 мл/мин. Измеряйте поглощение при 225 нм.
4. Через 20 мин проведите элюцию кальцитонина линейным градиентом 30—
45% (объемная концентрация) ацетонитрила в 0,1% (объемная концент-
рация) ТФУ (рис. 4.8).
5. Соберите фракции R и О и лиофильно высушите их.
•6. Перерастворите полипептид в конечной концентрации 0,5 мг/мл в 50 мМ
трис-HCl (pH 8,5), 2 мМ ЭДТА.
7. Контролируйте окисление кальцитонина с помощью аналитической обрат-
нофазовой ЖХВД, как описано в п. 2 и 3 данной таблицы.
8. Когда переход в окисленную форму кильцитонина завершится, снова про-
ведите очистку окисленного кальцитонина обратнофазовой ЖХВД, как
описано в п. 2 и 3 данной таблицы.
ружить методом аналитической ЖХВД (рис. 4.9). Были подоб-
раны условия, при которых весь кальцитонин находится в окис-
ленной форме [20] (табл. 4.17).
Получение ^-глобина из гибридного белка, синтезированного
в клетках Е. coli, — пример процесса очистки полипептида, на-
ходящегося в денатурированном состоянии [23].
Исходный гибридный белок вначале был очищен с помощью
ионообменной хроматографии в присутствии 8 М мочевины,
а затем — с помощью гель-фильтрации в присутствии 5 М гу-
анидинхлорида (табл. 4.10). Несколько других примеров ис-
пользования такого подхода можно найти в другой работе [30].
Методика солюбилизации и двухстадийной ренатурации про-
химозина теленка [18] приведена в табл. 4.18. Важное значе-
ние имеет время инкубации в мочевине, так как оно влияет па
общий выход белка. Считается, что это время необходимо для
проникновения мочевины и нарушения белковых взаимодейст-
вий в нерастворимых включениях. Выход активного фермента
также зависит от концентрации белка в растворе мочевины и
щелочи. В случае мочевины это может отражать изменение эф-
фективности дезагрегации нерастворимых включений при увели-
чении соотношения денатурирующий агент : белок. Существует
предположение, что при щелочном pH между молекулами про-
химозина происходит взаимный тиол-дисульфидный обмен. По-
этому оптимальна такая концентрация белка, при которой мо-
номеры образуются предпочтительнее, чем агрегаты. Поскольку
образование «правильных» дисульфидных связей может начать-
ся при pH 10,7, во время инкубации при pH 8,0, по-видимому
124
Ф. Марстон
Таблица 4.18. Растворение и ренатурации прохимозина теленка,
синтезированного в клетках Е. coli
1. Суспендируйте 100 г влажного осадка клеток Е. coli в 300 мл лизирую-
щего буфера1’ и лизируйте клетки, используя лизоцим и дезоаксихолат
(см. табл. 4.1).
2. Выделите белковые включения и промойте их тритоном Х-100 и ЭДТА
(см. табл. 4.3).
3. Суспендируйте осадки в 900 мл раствора, содержащего 8 М мочевину
(деионизованную), 50 мМ трис-НС1 (pH 8,0), 1 мМ ЭДТА, 50 мМ NaCl.
4. Инкубируйте суспензию 1 ч при комнатной температуре.
5. Медленно при помешивании добавьте суспензию к 9 объемам раствора
50 мМ КН2РО4( pH 10,7), 1 мМ ЭДТА, 50 мМ NaCl и поддерживайте
значение pH равным 10,7 путем добавления КОН.
6. Инкубируйте раствор 15—30 мин при комнатной температуре.
7. Доведите pH раствора до 8,0 НС1.
8. Инкубируйте раствор 30 мин при комнатной температуре.
9. Разбавьте раствор равным объемом 20 мМ трис-НС1 (pH 8,0), 1 мМ
ЭДТА и сконцентрируйте его в 4 раза на приборе фирмы «Amicon»
фильтры Н1Р10-8.
10. Добавьте 300 мл суспензии DE-52, предварительно уравновешенной буфе-
ром для ионообменной хроматографии2’, перемешайте и инкубируйте 1 ч
при 4 °C.
И. Соберите DE-52 на воронку со стеклянным фильтром с помощью слабо-
го вакуумного насоса.
12. Ресуспендируйте DE-52 в 3 объемах буфера для ионообменной хромато-
графии, содержащего 350 мМ NaCl, инкубируйте ее 30—60 мин при 4 °C.
13. Соберите DE-52 на воронку со стеклянным фильтром с помощью слабого
вакуумного насоса, так чтобы по возможности свести к минимуму цено-
образование. Прохимозин находится в фильтрате.
’) Состав лизирующего буфера приведен в табл. 4.1.
2) Состав буфера для ионообменной хроматографии: 20 мМ трис-НС1 (pH 8,0), 1 мМ
ЭДТА, 50 мМ NaCl.
происходит полная ренатурация белка с образованием стабиль-
ной, нативной конформации. В результате очистки ренатуриро-
вавшего прохимозина с помощью обработки ионообменной смо-
лой (табл. 4.18) и колоночной ионообменной хроматографии [18]
выход прохимозина составлял 22%, а степень чистоты превыша-
ла 90% (табл. 4.19).
Аполипопротеин Е — эукариотический полипептид, синтези-
руемый в клетках Е. coli, где он находится в нерастворимом
состоянии, но его очищают с помощью аффинной хроматографии
в присутствии денатурирующих агентов [32] (табл. 4.20). Для
этого используют хроматографию на гепарин-сефарозе с 2 М
мочевиной. В этих условиях эукариотический белок, чтобы уз-
навать лиганд, должен частично принять нативную конформа-
цию. Далее используются еще две стадии очистки, одна из кото-
рых— это гель-фильтрация на сефакриле S-300 в присутствии
6 М гуанидинхлорида. Затем после диализа, при котором белок
ренатурирует, осуществляется конечная стадия очистки с исполь-
Экспрессия эукариотических полипептидов в Е. coli 125
Таблица 4.19, Выход и степень очистки прохимозина, выделяемого
из клеток Е. coil [18]
Стадия очистки уОбъем, : мл -Химозин, мг/мл Белок, мг/мл Степень чистоты Выход, %
Клеточный лизат 400 1,4(3,37) 17,5 0,08 100
Экстракт, полученный при обработке моче- виной и щелочью 5000 0,06 0,15 0,40 53,5
Обработка DE-52 . 515 0,32 0,53 0,60 29,4
Колоночная хромато- графия на DE-52 Собранный пик про- химозина 67 1,85 1,86 0,995 22,2
зованием препаративного изоэлектрофокусирования в геле Im-
mobiline.
Все полипептиды, для которых мы выше описали методики
растворения и ренатурации, либо имеют небольшие размеры,,
либо содержат не более шести остатков цистеина, образующих
максимум три дисульфидные связи. Однако в клетках Е. coli
экспрессировались белки, имеющие сложную структуру, а имен-
но урокиназа [33]' и тканевой активатор плазминогена (тАП,
tPA) [13]. Методики растворения этих белков приводятся в
табл. 4.21. и 4.22 соответственно. Оба белка растворяли в отсут-
ствие экзогенных тиоловых реагентов, но восстановление натив-
ной конформации происходило в присутствии восстановленной и
окисленной форм глутатиона. В обоих случаях для ренатурации
необходимы определенная концентрация белка и определенное
соотношение и концентрация тиоловых реагентов. Суммарные
величины выхода урокиназы не приводятся, но для тАП они
очень низкие — менее 0,1% от суммарного клеточного белка.
Это может объясняться тем, что тАП имеет сложную мультидо-
менную структуру и содержит 10 или более дисульфидных свя-
зей, что и затрудняет получение хорошего выхода белка с на-
тивной конформацией. Причиной низкого выхода может служить
также отсутствие гликолизирования белка, продуцируемого
клетками Е. coli.
В клетках Е. coli была осуществлена также экспрессия бел-
ков, содержащих большое число субъединиц, и осуществлено их
выделение из нерастворимых комплексов. Для получения му-
тантных гемоглобинов осуществляли синтез в клетках Е. coll
^-глобина в виде гибридного белка ^-галактозидаза — ^-глобин
и выделяли этот белок методом, описанным в разд. 4.3.2.2 [23]
(табл. 4.10). После расщепления гибридного белка, диализа и
лиофилизации конформацию гемоглобина восстанавливают, ис-
пользуя следующую методику [23].
126
Ф. Марстон
Таблица 4.20. Растворение и ренатурация аполипопротеина Е человека,
синтезированного в клетках Е. coli
1. Смешайте 33 г влажного осадка клеток и 22 г оксида алюминия
(Buehler, Evanston, IL) и измельчите в мелкий порошок пестиком в ох-
лажденной до 4 °C ступке.
2. Обработайте растертые клетки 300 мл раствора 6 М мочевины в 0,1 М
аммоний-бикарбонатном буфере1*.
3. Центрифугируйте клеточный экстракт при 25 000 об/мин в течение
50 мин при 4 °C (ротор SW28, Beckman).
4. Повторно экстрагируйте осадок 200 мл. раствора 6 М мочевины в 0,1 М
бикарбонатном буфере и центрифугируйте в режиме стадии 3.
5. Соберите надосадочные фракции, полученные на стадиях 3 и 4, и диа-
лизуйте их против трех смен раствора 2 М мочевины в 25 мМ аммоний-
бикарбонатном буфере1 2*.
6. Уравновесьте 200 мл суспензии гепарин-сефарозы раствором 2 М моче-
вины в 25 мМ аммоний-бикарбонатном буфере.
7. Добавьте диализованную надосадочную фракцию к гепарин-сефарозе и
инкубируйте на качалке в течение ночи при 4 °C.
8. Заполните гепарин-сефарозой стеклянную колонку (4X3,5 см).
9. Промойте колонку, пропустив через нее с помощью насоса 300 мл рас-
твора 2 М мочевины в 25 мМ аммоний-бикарбонатном буфере при ско-
рости протекания 25 мл/ч.
10. Элюируйте связавшийся материал 50 мл раствора 1,0 М бикарбоната
аммония в 2 М мочевине.
11. Диализуйте элюат против раствора 5 мМ бикарбоната аммония и лио-
фильно высушите его.
12. Растворите высушенный белок в буфере для гель-фильтрации3 4*, содержа-
щем 6 М гуанидинхлорид.
13. Уравновесьте колонку с сефакрилом S-300 (2,5X30 см) буфером для
гель-фильтрации, содержащем 4 М гуанидинхлорид.
14. Нанесите растворенный белок на колонку с сефакрилом S-300 и элюи-
руйте его буфером для гель-фильтрации, содержащем 4 М гуанидин-
хлорид.
15. Тщательно диализуйте элюированный аполипопротеин Е против 5 мМ би-
карбоната аммония и лиофильно высушите его.
) Состав 0,1 М. аммоний-бикарбонатного буфера: 0,1 М NH4HCO3 (pH 7,8), 2 мМ
ФМСФ, 0,1% (объемная концентрация) Trasylol (Mobay, New York, NY).
2) Состав 25 мМ аммоннй-бикарбонатного буфера: 25 мМ NH4HCO3 (pH 7,4), 2 мМ
ФМСФ, 2 мМ ЭДТА, 0,1% (объемная концентрация) Trasylol, 0,1% (объемная концент-
рация) 2-меркаптоэтанола.
s) Буфер для гель-фильтрации: 0,1 М трнс-НС1 (pH 7,4), 1 мМ ЭДТА, 1% (объем-
ная концентрация) 2-меркаптоэтанола.
1. Растворите [3-глобин до достижения конечной концентрации
5 мг/мл в растворе, содержащем 8 М мочевину, 50 мМ трис-
НС1, pH 8, 0, 1 мМ ДТТ.
2. Разбавьте 0-глобин до концентрации белка 0,3 мг/мл.
3. Смешайте ^-глобин (1 М) с избытком (1,2 М) цианогема
и а-глобина.
4. Проведите восстановление ренатурировавшего гемоглоби-
на дитионатом натрия в атмосфере СО в присутствии каталазы
в концентрации 1 мкг/мл.
Экспрессия эукариотических полипептидов в Е. coli
127
Таблица 4.21. Растворение и ренатурация урокиназы,
синтезированной в клетках Е. coli
1. Суспендируйте 100 г влажного осадка клеток в 900 мл раствора, содер-
жащего 0,05 М трис-НС! (pH 8,0), 0,005% (объемная концентрация)
твин-80.
2. Разрушьте клетки, сделав однократный пассаж в гомогенизаторе Ман-
тон-Голина при давлении 5000 фунт/квадратный дюйм.
3. Центрифугируйте лизат при 4700g в течение 45 мин при 4 °C.
4. Ресуспендируйте осадок в растворе, содержащем 5 М гуанидинхлорид,.
0,005 М трис-НС! (pH 8,0), 0,05% (объемная концентрация) твин-80, и
инкубируйте при перемешивании в течение ночи при 4 °C.
5. Центрифугируйте суспензию при 4700g в течение 45 мин при 4 °C.
6. Отберите надосадочную фракцию и разбавьте ее до поглощения при
280 нм, равного 1,0. Состав буфера при этом должен быть следующим:
1,0 М гуанидинхлорид, 0,05 М трис-НС! (pH 8,0), 0,005% (объемная
концентрация) твин-80, 2,0 мМ восстановленный глутатион и 0,02 мМ
окисленный глутатион.
7. Инкубируйте раствор в течение 18—36 ч при 4 °C.
8. Диализуйте раствор против 0,05 М трис-НС1 (pH 8,0), 0,005% (объемная
концентрация) твин-70, 0,05 М NaCl и осветлите его центрифугирова-
нием.
9. Уравновесьте колонку размером 5X5 см с DE-52 (ватман) раствором,
содержащим 0,05 М трис-НС! (pH 8,0), 0,005% (объемная концентрация)
твин-80, 0,05 М NaCl.
10. Нанесите надосадочную фракцию, полученную на стадии 8, на колонку
с DE-52 и соберите раствор, не связавшийся с колонкой.
11. Уравновесьте колонку с бензамидин-сефарозой (2,5X10 см) 0,05 М
трис-НС1 (pH 8,0).
12. Нанесите образец, полученный на стадии 10, и промывайте колонку рас-
твором, содержащим 0,05 М трис-НС! (pH 8,0), 1 М NaCl до достиже-
ния постоянного поглощения при 280 нм.
13. Элюируйте урокиназу с колонки с бензамидин-сефарозой раствором
0,05 М трис-НС! (pH 8,0), содержащим 1 М аргинин.
Два примера белков, состоящих из нескольких субъединиц^
образование которых удалось осуществить в клетках Е. coli, —
это инсулин [11] и IgG (антитела к раково-эмбриональному
антигену, СЕА; [34]). В обоих случаях гены, кодирующие раз-
ные субъединицы, клонировали и экспрессировали независимо
друг от друга. А- и В-цепи инсулина синтезировали в виде гиб-
ридных белков и были выделены, очищены и S-сульфонированы,
как описано в табл. 4.7 и 4.15. S-сульфонированные производные
очищали ионообменной хроматографией и обратнофазовой
ЖХВД перед восстановлением исходной конформации белков.
Цепи A (SSO3_) и В (SSO3~) инсулина формировали натив-
ную молекулу при pH 9,6—10,6; при этом выход активного в
радиоиммунных реакциях инсулина составлял 10—15% [11].
Процесс получения нативного белка анти-CEA-IgG приведен
в табл. 4.23. Используемый подход состоит в следующем: полу-
чают полностью восстановленные формы тяжелых и легких це-
пей в неочищенных экстрактах и солюбилизируют их [34]. За-
128
Ф. Марстон
Таблица 4,22. Растворение и ренатурация тПА, синтезированного
в клетках Е. coll
1. Суспендируйте 1 г влажного осадка клеток в 9 мл лизирующего буфера1'
и лизируйте клетки лизоцимом и дезоксихолатом (см. табл. 4.1).
2. Выделите белковые включения центрифугированием при 12 000g в течение
5 мин при 4 °C.
3. Ресуспендируйте включения в 9 объемах буфера, содержащего мочевину2',
с помощью гомогенизатора.
4. Инкубируйте суспензию 1 ч при 30 °C.
5. Медленно добавьте суспензию при 30 °C к 9 объемам раствора, содержа-
щего 20 мМ трис-НС1 (pH 8,5), 0,01% (объемная концентрация) твин-80,
0,25 М NaCl, 1 мМ восстановленный глутатион, 0,1 мМ окисленный глута-
тион.
6. Инкубируйте в течение еще 2 ч при 30 °C.
7. Доведите pH раствора до 7,4.
'8. Для удаления примесей денатурирующих агентов тщательно диализуйте
раствор при 4 °C против следующей смеси: 20 мМ трис-НС1 (pH 8,5),
0,01 % (объемная концентрация) твин-80, 0,25 М NaCl, 1 мМ восстанов-
ленного глутатиона, 0,1 мМ окисленного глутатиона.
!) Состав лизирующего буфера: 50 мМ трнс-НС1 (pH 7,5), 50 мМ 1 мМ ЭДТА.
2) Состав буфера: 8 М мочевина (деионизованная), растворенная в 20 мМ трис-НС1
(pH 8,5), 0,01% твин-80, 0,25 М NaCl.
тем для постепенного возврата к нативной конформации прово-
дят диализ в буфере, в котором происходит обмен тиоловых и
дисульфидных групп и создаются мягкие условия протекания
восстановительных процессов. Самый высокий выход активного
анти-CEA-IgG из экстрактов Е. coll, определяемый иммунофер-
ментным методом, составлял 3—5% от ожидаемого максималь-
ного выхода.
4.4. Растворимые белки
4.4.1. Внутриклеточная экспрессия
Известны примеры эукариотических полипептидов, продуци-
руемых в клетках Е. coli в виде растворимых гибридных или
обычных белков [1]. В некоторых случаях в растворенном со-
стоянии находится только часть полипептида; остальной белок
входит в состав нерастворимых включений. Многие процедуры
очистки, описанные для таких белков, предусматривают стадию
аффинной хроматографии; это либо иммунологические методы
очистки, либо аффинная хроматография с использованием суб-
страта [1].
Как упоминалось ранее (разд. 4.3.3), для облегчения очист-
ки, а также для оптимизации условий экспрессии пре-52-белка
вируса гепатита В он продуцируется в виде гибридного белка с
Экспрессия эукариотических полипептидов в Е. coli 129
Таблица 4.23. Реконструкция полных молекул анти-СЕА IgG из тяжелых
f-цепей и легких х-цепей, синтезированных по отдельности в клетках Е. coli
Данная методика подходит как для клеток, содержащих тяжелые “f-цепи, так
и для клеток, содержащих легкие х-цепи,
1. Суспендируйте 1 г замороженной клеточной массы в 9 мл лизирующего
буфера1*.
2. Добавьте 2-меркаптоэтанол до конечной концентрации 0,1 М.
3. Инкубируйте суспензию в течение 1 ч при 37 °C.
4. Центрифугируйте суспензию при 15 000 об/мин (ротор Sorvall SS-34)
в течение 30 мин.
Для реконструкции IgG необходимо следующее:
5. Смешайте надосадочные фракции, содержащие тяжелые f-цепи и легкие
х-цепи, полученные на стадии 4, и разбавьте их раствором, содержащим
8 М гуанидинхлорид, 50 мМ трис-НС1 (pH 8,0), 1 мМ ЭДТА, так, чтобы
предполагаемая концентрация IgG составляла 25 ,мкг/мл.
6. Диализуйте раствор в течение 1—2 ч при 4 °C против буфера для диали-
за* Е. 2*, содержащего 20 объемов деионизованной 8 М мочевины.
7. Перенесите диализный мешочек (мешочки) в 20 объемов того же буфе-
ра и добавьте насыщенный N2 буфер для диализа примерно на 15 ч;
при этом конечная концентрация мочевины должна достигнуть 1 М.
8. Добавьте в каждый диализный мешочек сывороточный альбумин до кон-
центрации 0,5 мг/мл.
9. Перенесите диализные мешочки в раствор Na-фосфатного буфера, содер-
жащего 0,1 мМ ФМСФ (раствор не дегазируется).
10. Диализуйте в течение 2 ч при 4 °C.
') Состав лизирующего буфера: 7,6 М гуанидинхлорид, 50 мМ трис-НС1 (pH 8,0),
1 мМ ЭДТА.
2) Состав буфера для диализа: 50 мМ Na-глициновый буфер (pH 10,8), 10 мМ гли-
цин-этиловый эфир, 1 мМ ЭДТА, 1 мМ восстановленный глутатион, 0,1 мМ окисленный
глутатион.
^-галактозидазой (пре-52-^-гал) [27]. Гибридный белок состав-
лял до 30% суммарного белка клетки. Выделение белка из на-
досадочной фракции после лизиса клеток осуществляли мето-
дом аффинной хроматографии на колонке с иммобилизованным
аналогом субстрата (табл. 4.11). Гибридный белок элюируется
с колонки 0,1 М. боратом натрия, pH 10,0. При использовании
такой одноступенчатой методики практически без потерь дости-
гается 75-кратная очистка гибридного белка со степенью чисто-
ты более 90%.
Лимфотоксип человека [35] и белок р24 gag ретровируса,
вызывающего СПИД (синдром приобретенного иммунодефици-
та) [36], были синтезированы в клетках Е. coll в виде негибрид-
ных растворимых белков. В обоих случаях использовалась од-
ностадийная иммунологическая очистка, при которой гетероло-
гичный белок выделяли из суммарного растворимого белка
Е. coli. Методика очистки лимфотоксина выглядит следующим
образом.
1. Уравновесьте колонку с сефарозой (объемом 20 мл), конъ-
югированной с моноклональными антителами, буфером, имею-
9—1186
130
Ф. Марстон
щим состав: 0,05 М трис-НС1 (pH 7,0), 0,15 М NaCl 2 мМ ЭДТА
(TBS).
2. Уравновесьте колонку раствором, содержащим 0,1 М ук-
сусную кислоту (pH 4,5), 150 мМ NaCl.
3. Снова уравновесьте колонку буфером TBS.
4. Добавьте к лизату клеток Е. coli сульфат аммония до ко-
нечной концентрации 40% (весовая концентрация) и соберите
осадок центрифугированием.
5. Растворите осадок в небольшом объеме раствора, содер-
жащего 0,1 М трис-НС1 (pH 7,4), 5 мМ ЭДТА, и нанесите на
колонку (скорость протекания один объем колонки в час).
6. Промывайте колонку буфером TBS, содержащим 0,05%
(объемная концентрация) твин-20, до достижения постоянной
величины поглощения при 280 нм.
7. Элюируйте лимфотоксин раствором, содержащим 0,1 М ук-
сусную кислоту (pH 4,5), 150 мМ NaCl.
8. Сразу же доведите pH элюата до 7,8 добавлением 0,1 объ-
ема 1 М трис-НС1 (pH 8,5).
Белок p24gag вируса СПИДа элюируют с колонки с имму-
носорбентом 2 М тиоцианатом калия (KSCN) [36], который
удаляют путем диализа против Na-фосфатного буфера.
Недавно были опубликованы данные об экспрессии Cu/Zn-
супероксид-дисмутазы человека (СОД) в виде негибридного
белка в клетках Е. coli [37]. Белок находился в растворимом
состоянии, обладал ферментативной активностью и составлял
примерно 13% суммарного клеточного белка. Выделение СОД
из фракции растворимых белков Е. coli включает несколько ста-
дий:
1. Добавление СОД до конечной концентрации 2% (весовая
концентрация) (для избирательного осаждения бактериальной
СОД).
2. Добавление КС1 к надосадочной фракции до конечной кон-
центрации 0,3 М для удаления следовых количеств ДСН.
3. Осаждение примесных белков 60%-ным (весовая концент-
рация) раствором сульфата аммония.
4. Диализ против 20%-ного раствора фосфата калия.
5. Ультрафильтрация (концентрирование) и гель-фильтрация
на фрактогеле 55 (Fractogel 55).
6. Ионообменная хроматография на DE-52 (элюция возраста-
ющими концентрациями КС1).
Степень очистки СОД превышала 90%, но выход составлял
только 10%. Однако это было эквивалентно выходу 2 г СОД
из 50 л суспензии клеток Е. coli, поглощение которой при
600 нм составляло 120 ед. На этом примере отчетливо видно,.
Экспрессия эукариотических полипептидов в Е. coli
131
Таблица 4.24. Системы, используемые для секреции гетерологичных белков
в клетках
Белок—носитель сигнальной
последовательности
Белки, секретируемые клетками Е. coll
Источник
данных
^-Лактамаза Проинсулин [38
Легкая цепь IgG [39
а-Фибриноген А [40
отрА FMDV VP1 [41
Гормон роста человека [42
ompF (3-Эндорфин [43
phoA Фактор роста эпидермиса [44
Гормон роста человека Гормон роста человека [45
Урокиназа Урокиназа [46
какое количество рекомбинантного белка может быть выделено
из клеток Е. coli, когда уровень экспрессии высок, а белок раст-
ворим, причем даже если выход белка после очистки находится
на среднем уровне.
4.4.2. Секреция
Клетки Е. coll в естественных условиях не секретируют боль-
ших количеств белка. Однако они продуктивно секретировали
ряд эукариотических полипептидов в периплазматическое прост-
ранство и несколько полипептидов в культуральную среду
(табл. 4.24). Уровни экспрессии невысоки (<1% от суммарного
белка клеток Е. coli) по сравнению с таковыми при внутрикле-
точной экспрессии, однако здесь требуется меньшая степень
очистки, чем в случае цитоплазматических продуктов.
Метод очистки ^-эндорфина, секретируемого в среду клетка-
ми Е. coli [43], приведен в табл. 4.25. Для подтверждения того
факта, что p-эндорфин действительно секретировался в культу-
ральную среду, а не проникал туда из периплазматического
пространства, определяли активность периплазматических фер-
ментов-маркеров ^-лактамазы [47] (табл. 4.26) и щелочной фос-
фатазы [48] (табл. 4.27). После очистки рекомбинантных поли-
пептидов, секретируемых в среду, гель-фильтрацией и обратно-
фазовой ЖХВД определяли их аминокислотный состав в целом
и N-концевые аминокислотные последовательности. Результаты
определения N-концевых аминокислотных последовательностей
выявили гетерогенность N-концов, но во всех полипептидах от-
сутствовали сигнальные последовательности. Гетерогенность,
по-видимому, обусловливается действием протеиназ [42].
Была осуществлена секреция из клеток Е. coli гибридного
белка: белок А-инсулиноподобный фактор роста 1 (ИФР-1)
*32
Ф. Марстон
Таблица 4.25. Выделение ^-эндорфина, секретируемого клетками Е. coli
в культуральную среду
1. Лиофильно высушите 1 г культуральной среды досуха.
2. Растворите осадок в 50 мл воды и обессольте его пропусканием через
колонку (2,6x95 см) с сефадексом G-10, уравновешенную 0,1 М уксус-
ной кислотой.
3. Лиофильно высушите фракцию, не связавшуюся с G-10.
4. Растворите осадок в 4 мл воды и нанесите раствор на колонку (1,2Х
Х140см) с сефадексом G-25 (fine), уравновешенную 0,1 М уксусной кис-
лотой.
5. Промывайте колонку при скорости протекания раствора 20 мл/ч и собе-
рите фракции объемом по 2 мл.
6. Уравновесьте колонку для обратнофазовой хроматографии (RP-300,
4,6X250 мм, Brown Lee) 0,1% ТФУ (объемная концентрация).
7. Проанализируйте пробы, собранные с сефадекса G-25, с помощью обратно-
фазовой хроматографии ЖХВД, проводя элюцию линейным градиентом
0,1% ТФУ (объемная концентрация)/50% (объемная концентрация) ацето-
нитрилом/0,075% (объемная концентрация) ТФУ в течение 50 мин при
скорости протекания 1 мл/мин.
Таблица 4.26. Определение активности ^-лактамазы
1. Приготовьте 19 мМ раствор нитроцефина в диметилсульфоксиде.
2. Разбавьте нитроцефин в 100 раз 0,1 М фосфатным буфером (pH 7,0).
3. Поместите в спектрофотометр кювету с 1 мл раствора нитроцефина и
скомпенсируйте величину поглощения при 30 °C (установка нуля).
4. Добавьте 0,1 мг соответствующим образом разбавленного исследуемого
раствора.
5. Определите изменение поглощения при 495 нм.
6. Рассчитайте активность fJ-лактамазы, исходя из значения емМ495"м= 1,406
(в кювете с длиной оптического пути 1 см).
Таблица 4.27. Определение активности щелочной фосфатазы
1. Прогрейте 2-амино-2-метил-1-пропанол (2А2М1П) при 35 °C до полного
перехода в жидкое состояние.
2. Поместите 86 г 2А2М1П в стакан объемом 1 л, добавьте 500 мл воды
и перемешайте.
3. Добавьте примерно 120 мл 1 М НС! и перемешайте.
4. Взвесьте 264 мг MgSO4-7H2O и растворите в 200 мл воды в отдельном
стакане.
5. Влейте раствор соли магния в раствор 2А2М1П и перемешайте,
6. Нагрейте раствор до 30 °C и, поддерживая эту температуру, доведите pH
до 10,5 добавлением 1 М. НС1.
7. Доведите объем раствора водой до 1 л.
8. Смешайте в кювете 1,4 мл раствора 2A2Min/Mg2+ и 50 мкл соответству-
ющим образом разбавленного исследуемого раствора.
9. Поместите в спектрофотометр кювету и скомпенсируйте поглощение при
температуре 30 °C.
10. Добавьте к раствору 50 мкл 0,48 М 4-нитрофенплфосфата п переме-
шайте.
11. Определите изменение поглощения при 405 нм.
12. Рассчитайте активность щелочной фосфатазы, исходя из значения
8mm495hm= 1,881 (в кювете с длиной оптического пути 1 см).
Экспрессия эукариотических полипептидов в Е. coli 133
Таблица 4.28. Определение активности алкоголь-дегидрогеназы
1. Внесите в кювету для спектрофотометра 950 мкл 0,1 М глицина (pH 10,0),
2,5 мМ NAD+.
2. Добавьте 50 мкл 4 М этанола, перемешайте и уравновесьте поглощение
раствора в спектрофотометре при 25 °C.
3. Добавьте 0,5 мл исследуемого раствора, соответствующим образом раз-
бавленного.
4. Измерьте изменение поглощения при 340 нм.
5. Рассчитайте активность алкоголь-дегидрогеназы, исходя из значения
8мм495нм=6,22.
[28]. Этот белок был сконструирован так, чтобы он секретиро-
вался клеткой и его можно было выделить с помощью аффинных
методов (табл. 4.11). Сигнальная последовательность белка А
из Staphylococcus aureus в клетках Е. coli распознается и раз-
рушается, а комплекс белок А-ИФР секретируется в культураль-
ную среду. Очистку комплекса производят с помощью сефаро-
зы, конъюгированной с IgG, расщепляют элюируемый гибрид-
ный белок гидросиламином при pH 9,0 и получают ИФР.
Когда оценивают уровень секреции в периплазматическое
пространство, эффективность образования сферопластов опре-
деляют по периплазматическим маркерным ферментам. Кроме
того, контролируется степень лизиса клеток с помощью опреде-
ления активности такого цитоплазматического фермента, как
алкоголь-дегидрогеназа [49] (табл. 4.28).
Максимальный уровень экспрессии, достигаемый в примерах,
приведенных в табл. 4.23, составляет около 0,02% [42], что эк-
вивалентно выходу 2 мг белка/л культуральной среды. Таким
образом, несмотря на то что секреция белков из клеток Е. coli
позволяет обойти трудности, связанные с работой с нераствори-
мыми белковыми включениями, и облегчает очистку белков, для
повышения выхода белка требуется дальнейшее усовершенство-
вание методики. Отметим, что уровень секреции инсулина, со-
ставляющий 1 % суммарного клеточного белка, считается доста-
точным для его промышленного получения [51].
4.5. Заключительные замечания
В этой главе приводится большое количество методик полу-
чения активных растворимых белков из нерастворимых агрега-
тов, присутствующих в клетках Е. coli. Уникальность структуры
каждого белка, по всей видимости, требует наличия специфи-
ческого метода его солюбилизации и последующей ренатура-
ции, которые должны быть разработаны эмпирически.
Кроме одного или двух исключений, приведенные методы вы-
деления работают в случае аналитических количеств материа-
ла, когда используется не более 50 г влажного осадка клеток.
134
Ф, Марстон
В случае препаративного выделения белка при осуществлении
процессов растворения и ренатурации возникает ряд проблем.
Реакции, для протекания которых требуются высокие кон-
центрации мочевины или гуанидина, являются дорогостоящими;
кроме того, в результате образуются большие количества отра-
ботанного денатурирующего агента, который необходимо уда-
лять. Основная проблема, однако, заключается в том, что вслед-
ствие сложности многих реакций трудно осуществить их адекват-
ное масштабирование. Это касается, например, реакции обрати-
мого цитраконилирования остатков лизина и ренатурации таких
мультимерных белков, как IgG.
В данной главе мы не рассматривали процесса удаления не-
белковых примесей из препаратов рекомбинантных полипепти-
дов. Если полипептиды должны вводиться животным или в ко-
нечном итоге предназначены для лечения человека, то необходи-
мо удаление липополисахаридов. Недавно было опубликовано
два обзора, посвященных этому вопросу [51, 52].
Если эукариотический белок синтезируется в клетках Е. coli,
идентичны ли структура и конформация этого белка соответст-
вующим характеристикам природной молекулы? В некоторых
случаях ответ на этот вопрос будет отрицательным. При пря-
мой экспрессии могут образовываться молекулы с отсутствую-
щими в природных молекулах N-концевыми остатками метиони-
на. В клетках также не осуществляются определенные посттран-
сляционные модификации, протекающие в эукариотических
клетках, такие, как ацетилирование, аминирование и гликолизи-
рование. Однако ацетилирование и аминирование успешно проте-
кали на очищенных рекомбинантных полипептидах in vitro [1J.
Такие аналитические методы, как исследование кругового
дихроизма и рентгеноструктурный анализ, могут быть использо-
ваны для определения конформации ренатурированных и раст-
воримых рекомбинантных белков. Немногочисленные имеющиеся
к настоящему времени данные обнадеживают. Например, спект-
ры, полученные при исследовании кругового дихроизма ренату-
рировавшего прохимозина [53] и секретируемого клетками
Е. coll гормона роста человека [42], существенно не отличаются
от спектров природных белков. Был также получен в кристалли-
ческом виде ^-глобин, продуцируемый клетками Е. coli и вклю-
чившийся в состав гемоглобина. Исследование дифракции рент-
геновских лучей с разрешением 2,8 А выявило весьма небольшие
различия между природным и мутантным рекомбинантным бел-
ками; эти различия объясняют изменениями, возникающими при
генноинженерных манипуляциях [23]. Хотя и существуют про-
блемы, связанные с очисткой эукариотических полипептидов,
выделяемых из клеток Е. coli, такая система используется для
получения ряда белков, используемых в медицине [1].
4.6. Приложение
Комбинации хозяин—вектор, используемые в системах экспрессии
Экспрессируемый полипептид Тил экспрессии Штамм Плазмида Источ- ник данных
Белок р24 gag вируса Прямая 294 р24 DE [36]
СПИДа р24 DE Д Hind D III [32]
Аполипопротеин Е Прямая сбОО pVT 170 pVT 194
[20]
Кальцитонин-gly че- В виде гиб- E103S pMG
ловека ридного бел-
ка
₽-Э.ндорфин То же RR1 p₽-gaZ p₽-gaZ-end НО]
р-Эндорфин Секреция KRI N99 pNMIOl [43]
Энкефалин В виде гиб- CSR603 ридного бел- PRE31 [26]
ка
Фактор роста эпидер- Секреция С600 PTA1529 [44]
миса (урогастрон) а-Цепь фибриногена А YK537 » JM105 pl 16.9 [40]
р-Глобин В виде гиб- Q413 pLc IIFx-P-гло- [23]
ридного бел- бин (nic~)
ка
Гормон роста бычий Прямая RV308 pCZlOl [14]
Гормон роста бычий В виде гиб- НВ101 pTEBGH [19]
ридного бел- pSBBGH
ка
Гормон роста чело- Секреция RV308 pOmpA-hGH2 [42]
века Гормон роста чело- » 294 pPreHGH 207-2 [45]
века В виде гиб- JM83 pAPH-1
Белок npe-S2 вируса pWS3 [27]
гепатита В ридного бел-
ка
Тяжелая цепь IgG Прямая W3110 pyCEA trp 207-7 [34]
Легкая цепь IgG » W3I10 pKCEAtrp 207-1 [34]
Легкая цепь IgG Секреция НВ101 pRI 12 [39]
A-цепь инсулина В виде гиб- 294 ридного бел- pIAl Hi]
ка
В-цепь инсулина То же D1210 pIBl [П[
Препроурокиназа Прямая ММ294 pULB1135 [46]
Прохимозин » НВ101 pCT 70 [181
Проинсулин В виде гиб- JM103 pTac/PI [25]
ридного бел- plac 239/PI
Проинсулин Секреция НВ101 P287.47 [38]
Тканевой активатор PR13 P241.1947 P241.CB15 P234.CB16
Прямая E103S pMG 178 [13]
плазминогена
135
136
Ф. Марстон
Благодарности
Хочу выразить благодарность Тиму Харрису и Питеру Лоу
за плодотворное обсуждение рукописи и критические замеча-
ния. Я особенно благодарна Маргарет Тернер за подготовку ма-
шинописного текста. Хочу также поблагодарить всех, кто дал
разрешение привести в этой главе свои данные.
Литература
1. Marston F. А. О. (1986). Biochem. J., 240, 1.
2. Harris Т. J. R. (1983). In: Genetic Engineering, Williamson R. (ed.), Acade-
mic Press, London, UK, Vol. 4, p. 127.
3. O’Farrell P. H., Polisky B., Gelfand D. H. (1978). J. Bacteriol., 134, 645.
4. Caulcott C. A., Rhodes P. M. (1986). Trends Biotechnol., 4, 142.
5. Carrier M. J., Nugent M. E., Tacon IV. C., Primrose S. B. (1983). Trends
Biotechnol., 1, 109.
6. Caulcott C. A., Lilley G., Wright E. M., Robinson M. K., Yarranton G. T.
(1985). J. Gen. Microbiol., 131, 3355.
7. Yarranton G. T„ Wright E„ Robinson M. K., Humphreys G. O. (1984). Gene,
28, 1293.
8. Wright E., Humphreys G. 0., Yarranton G. T. (1986). Gene, in press.
9. Wetzel R., Goeddel D. V. (1983). In: Peptides, Gross E. and Meienhofer J.
(eds.), Academic Press Inc., New York, Vol. 5, p. 1.
10. Shine J., Fettes I., Lan N. C. Y., Roberts J. L., Baxter J. D. (1980). Nature,
285, 456.
11. Goeddel D. V., Kleid D. G„ Bolivar F., Heyneker H. L., Yansura D. G„
Crea R., Hirose T., Kraszewski A., Itakura K., Riggs A. D. (1979). Proc Natl.
Acad. Sci. USA, 76, 106.
12. Burgess R. R„ Jendrisak J. J. (1975). Biochemistry, 14, 4634.
13. Harris T. J. R., Patel T., Marston F. A. O., Little S„ Emtage J. S., Opdenak-
ker G., Volckaert G., Rombauts IF., Billiau A„ De Somer P. (1986) Mol.
Biol. Med., 3, 279.
14. Schoner R. G„ Ellis L. F„ Schoner В. E. (1985). BIO/TECHNOL, 3, 151.
15. Witholt B., Boukhout M., Brock M., Kingma J., van Heerikhuizen H., de
Leif L. (1976). Analyt. Biochem., 74, 160.
16. Taylor G., Hoare M., Gray D. R., Marston F. A. O. (1986). BIO/TECHNOL,
17. Laemmli U. R. (1970). Nature, 227, 680.
18. Marston F. A. O., Lowe P. A„ Doel M. T., Schoemaker J. M., White S., AngalS.
(1984). BIO/TECHNOL, 2, 800.
19. Szoka P. R„ Schreiber A. B., Chan H., Murthy J. (1986). DNA, 5, 11.
20. Bennett A„ Rhind S. R„ Lowe P. A„ Hentschel С. C. G. (1984) U’l< Patent
GB2140810A. ' "
21. Allen G., Paynter C. A., Winther M. D. (1985). J. Cell Sci Suppl 3, 29
22. Belagaje R. M., Mayne N. G„ Van Frank R. M., Rutter W. J. (1984) DNA
3, 120.
23. Nagai R„ Perutz M. F., Poyart C. (1985). Proc. Natl Acad. Sci USA 82
7252.
24. Lee J. M., Ullrich A. (1984). European Patent Application No. 0128733
25. Shen S.-H. (1984). Proc. Natl. Acad. Sci. USA, 81, 4627.
26. Hostomsky Z., Smrt J., Paces V. (1985). Gene 39, 269.
27. Offensperger W., Wahl S., Neurath A. R., Price P., Strick N., Rent S. В. H.
Christman J. R., Acs G. (1985). Proc. Natl. Acad. Sci. USA, 82, 7540.
Экспрессия эукариотических полипептидов в Е, coli 137
28. Uhlen М., Nilsson. В. (1986). Abstracts of the Engineering Foundation 3rd
Conferencee on Recovery of Bioproducts, Uppsala, Sweden, p. 20.
29. Sassenfeld H. M., Brewer S. J. (1984). BIO/TECHNOL, 2, 76.
30. Builder S. E„ Ogez J. R. (1985). US Patent No. 4511502.
31. Konrad M. W„ Lin L. S. (1984). US Patent No. 4450103.
32. Vogel T., Wcisgraer K. H„ Zeevi M. I., Ben-Artzi H„ Levanon A. Z„
Rail S. C., Jr., Innerarity T. L., Hui D. Y„ Taylor J. M., Kanner D„ Yavin Z.,
Amit B„ Aviv H„ Gorecki M., Mahley R. IF. (1985). Proc. Natl. Acad. Sci,
USA, 82, 8696.
33. .Winkler M. E., Blaber M, Bennett G. L„ Holmes W., Vehar G. A. (1985).
BIO/TECHNOL, 3, 990.
34. Cabilly S., Riggs A. D„ Pande H., Shirely J. E., Holmes IF. E., Rey M.,
Perry L. J., Wetzel R-, Heyneker H. L. (1984). Proc. Natl. Acad. Sci. USA,
81, 3273.
35. Gray P. W., Aggarwal В. B., Benton С. V., Bringman T. S., Henzel W. S.,
Jarnett J. A., Leung D. W., Moffat B„ Ng P„ Svederesky L. P., Palladi-
no M. A., Medwin G. E. (1984). Nature, 312, 721.
36. Dowbenko D. J., Bell J. R., Benton С. V., Groopman J. E., Nguyon H., Vetter-
lein D., Capen D. J., Laskey L. A. (1985). Proc. Natl. Acad. Sci. USA, 82,
7748.
37. Hartman J. R., Gelles T., Yavin Z., Bartfield D., Kanner D., Aviv H.,
Gorecki M. (1986). Proc. Natl. Acad. Sci. USA, 83, 7142.
38. Talmadge K, Gilbert W. (1982). Proc. Natl. Acad. Sci. USA, 79, 1830.
39. Zemel-Dreasen O., Zamir A. (1984). Gene, 27, 315.
40. Lord S. T. (1985). DNA, 4, 33.
41. Henning U., Cole S. T., Bremer E., Hindernach I., Schaller H. (1983). Eur. J.
biochem., 136, 233.
42. Hsiung H. M„ Mayne N. A., Becker G. W. (1986). BIO/TECHNOL, 4,
991.
43. Nagahari K, KanayaS., Monakata K, AoyagiY., Mizushima S. (1985). EMBO
J., 4, 3589.
44. Oka T., Sakamoto S., Miyoshi K.-L, Fuwa T., Yoda K, Yamasaki M., Tamu-
ra G., Miyake T. (1985). Proc. Natl. Acad. Sci. USA, 82, 7212.
45. Gray G. L., Baldridge J. S., McKeown К S., Heyneker H. L., Chang C. N.
(1985). Gene, 39, 247.
46. Jacobs P„ Cravador A., Loriau R., Brockly F., Colau B., Churchana P., Van
Eisen A., Herzog A., Bollen A. (1985). DNA, 4, 139.
47. Bush K., Sykes R. B. (1984). In: Methods in Enzymatic Analysis IV, Berg-
meyer H. U. (ed.), Verlag Chemie, GmBH, p. 280.
48. Bretaudiere J.-P., Spillman T. (1984). In: Methods in Enzymatic Analysis IV,
Bergmeyer H. U. (ed.), Verlag Chemie, GmbH, p. 75.
49. Wagner F. IF., Burger A. R., Vallee B. L. (1983). Biochemistry, 22, 1857.
50. Emerick A. IF., Bertolani B. L., Ben-Bassat A., White T. J., Konrad M. IF.
(1984). BIO/TECHNOL, 2, 165.
51. Sofer G. <1984). BIO/TECHNOL, 2, 1035.
52. Sharma S. K. (1986). Biotechnol. Appl. Biochem., 8, 5.
53. Sugrue R., Lowe P. A., Marston F. A. O., Pain R., Freedman R. B. (1987),
In preparation.
ГЛАВА 5
ПОЛУЧЕНИЕ И ОЧИСТКА ПОЛИКЛОНАЛЬНЫХ
АНТИТЕЛ К ГЕТЕРОЛОГИЧНЫМ ФРАГМЕНТАМ
ГИБРИДНЫХ БЕЛКОВ, СОДЕРЖАЩИХ
^-ГАЛАКТОЗИДАЗУ
Син Б. Кэрролл и Аллен Логон
(Sean В. Carroll, Allen Laughon)
5.1. Введение. Векторы для экспрессии гибридных бепков,
содержащих р-гапактозидазу
Возможность экспрессии клонированных эукариотических
генов в клетках Е. coli способствовала углубленному изучению
множества белков, представляющих интерес для фундаменталь-
ных научных исследований и медицины. В тех случаях, когда
нативный негибридный белок экспрессируется недостаточно эф-
фективно, часто экспрессия белков или их фрагментов в виде
гибридов с полипептидами Е. coli, такими, как ^-галактозидаза,
оказывалась более успешной. К тому же гибридные белки мож-
но легко очищать с помощью хроматографических методов, раз-
работанных для ^-галактозидазы. Эукариотические белки, экс-
прессируемые в составе гибридных продуктов, были с успехом
использованы при изучении иммунологически важных участков
поверхностных антигенов [1], функций рекомбинантных поли-
пептидов [2], при получении иммунологических зондов, необхо-
димых для исследования ранее не изученных антигенов [3—6J,
для экспрессии вариантных форм белковых субъединиц и для
выделения и исследования клонов ДНК из экспрессирующихся
библиотек генов [8—10]. Технология работы с экспрессирующи-
ми векторами достигла столь высокого уровня развития, что ста-
ло возможным осуществлять в клетках Е. coli достаточно эф-
фективную экспрессию практически любой кодирующей
последовательности с образованием гибридного продукта, кото-
рый можно выделить с помощью разнообразных биохимических
методов и использовать его либо в различных функциональных
исследованиях либо в качестве иммуногена. Синтез чужеродного
полипептида в виде гибридного белка с ^-галактозидазой, по
всей вероятности, значительно увеличивает стабильность этого
полипептида в клетках Е. coli. По-видимому, стабильность бел-
ка, а не сила промотора — наиболее важный фактор для успеш-
ной экспрессии рекомбинантных белков в бактериях.
В этой главе описываются две широко распространенные
векторные системы, используемые для экспрессии клонирован-
ных генов с образованием гибридных белков, включающих
138
Поликлональные антитела к фрагментам гибридных белков 139
Левый EcoRT » . - - ' Правый
конец ь!асг а 857 S100 КО*!Ч
।------------------------и---------------------------------1
I
3' GAC сет A^S GCG 5'
Gin Phe Glu Ala
Рис. 5.1. Структура Xgtll. EcoRI вносит разрыв в кодон, кодирующий глута-
мин,— 17-й кодон, начиная от З'-конца кодирующего участка lacZ [8].
галактозидазу Е. coli, и приводятся методы выделения гибрид-
ных полипептидов и получения и очистки поликлональных анти-
тел, специфичных к эукариотическому компоненту такого
гибридного белка. Рассматриваемые векторные системы-—это
система Xgtll, разработанная Янгом и Дэвисом [8], и система,
основанная на использовании плазмид pUR, разработанная Рю-
тером и Мюллер-Хиллом [11]. Система с фаговым вектором бо-
лее предпочтительна для поиска и выделения генов с исполь-
зованием антител в качестве зондов для скрининга библиотек
кДНК. Плазмидная система оказалась более простой и эффек-
тивной для экспрессии охарактеризованных клонированных
фрагментов ДНК. Методы, приведенные в этой главе, представ-
ляют наибольший интерес для исследователей, которые, выделив
Xgtl 1-клоны с помощью антител, должны очистить экспресси-
руемый гибридный белок и работать с ним, а также для иссле-
дователей, намеренных экспрессировать ДНК, клонированную
в других векторах.
5.1.1. Xgtll
Экспрессирующий вектор Agtll был детально описан в пре-
дыдущем томе этой серии [12]. Структура векторной системы в
общих чертах представлена на рис. 5.1. В составе фага имеется
ген lacZ, содержащий единственный сайт для рестриктазы
£coRI. Встраивание последовательностей гетерологичной ДНК
в сайт EcoRl приводит к утрате активности ^-галактозидазы.
Для тестирования активности фермента используют хромоген-
ный субстрат 5-бром-4-хлор-3-индолилф-Н-галактозид (X-gal):
при наличии активного фермента образуются голубые бляшки,
при отсутствии — бесцветные. Когда последовательность, коди-
рующая гетерологичный белок, находится в одной открытой
рамке считывания с lacZ, возможен синтез гибридного белка,
включающего гетерологичный чужеродный белок и ^-галактози-
дазу. Кроме того, фаг кодирует температурочувствительный бе-
лок-репрессор (с1857) и в результате амбер-мутации (S100)
теряет способность к лизису клеток, так что при индукции, выз-
ванной повышением температуры с 32 до 42 °C, в клетке накап-
140
С, Кэрролл, А, Логон
ливаются большие количества кодируемых фагом продуктов,
в том числе гибридные белки, включая ^-галактозидазу. Фаг
может размножаться и лизировать клетки-хозяева, содержащие
супрессор амбер-мутации RY1090, или выступать в качестве ли-
зогенизирующего агента (штамм RY1089).
В задачу авторов не входит описание методов конструирова-
ния рекомбинантов Xgtll, кодирующих гибридные белки, по-
скольку в одной из книг данной серии уже имеется соответству-
ющая глава [12]. Мы рассмотрим лишь методы скрининга и
очистки детерминируемых вектором гибридных белков Xgtll,
которые можно использовать как для выявления нужных кло-
нов, так и для получения антисыворотки к гибридным белкам.
5.1.2. Плазмидные векторы семейства pUR
Во многих случаях вследствие меньшего размера плазмид
и их более простых рестрикционных карт легче получить нуж-
ные рекомбинантные конструкции в плазмидных векторах, а не
в фагах. Рютер и Мюллер-Хилл [И] сконструировали ряд плаз-
мид pUR290, pUR291 и pUR292 для получения в клетках Е. co-
li гетерологичных белков, синтезируемых в виде гибридных бел-
ков с ^-галактозидазой (рис. 5.2). У З’-конца кодирующей об-
ласти lacZ встроены четыре уникальных сайта рестрикции
(рис. 5.2'5) (в пределах вектора помимо CZaI-сайта на З'-конце
гена lacZ имеется второй сайт Clal). Во всех указанных векто-
рах положение встроенных рестрикционных сайтов относительно
McZ-кодирующего участка таково, что любая гетерологичная
последовательность может быть введена в них в той же рамке
считывания, что и lacZ.
В отличие от Xgtll гибридные белки с ^-галактозидазой, ко-
дируемые плазмидами pUR, обычно сохраняют ферментативную
активность. Примерно 18 С-концевых аминокислотных остатков
^-галактозидазы, отсутствующие в гибридных белках, синтези-
руемых в Xgll, необходимы для проявления ее ферментатив-
ной активности. По нашим данным, гетерологичные фрагменты
гибридных белков, включающих ^-галактозидазу, также более
стабильны в клетках Е. coli, когда соответствующие гены экс-
прессируются в pUR-векторах, а не в Xgtll. Возможно, это сви-
детельствует о влиянии конформации ^-галактозидазы на кон-
формацию гетерологичного белкового фрагмента в составе бел-
кового гибрида.
Плазмиды pUR содержат участок плазмиды pBR322, вклю-
чающий область начала репликации и ^-лактамазный ген, детер-
минирующий устойчивость к ампициллину. Экспрессия гена
lacZ подавляется тогда, когда эти плазмиды находятся в клет-
ках-хозяевах 71-18 (\.(lac]pro)F'lacl4 lacZ АМ15 pro+ supE),
Поликлональные антитела к фрагментам гибридных белков
141
EcoRI
V
CIO Phe Cln Leu Ser Ala Gly Arg Tyr His Tyr Gin Leu Vai Trp Cys Gin Lys
CAA TTC CAC CTC ACC CCC OCT CCC ТЛС CAT TAC CAC TTC GTC TCC TCT CAA AAA. TAA LacZ
BamHI__Sall——"Psd Hindi!! Clal
V V V V
TGC TGT CAA AAA GGG GAT CCG TCG ACC TCC AGC CAA OCT TAT CGA TCA p'TC’90
TCG TCT C GG GCA TCC CTC GAC CTC CAG CCA AGC TTA TCG АТС pUR291
TGC TCT CA 6 GGG АТС CCT CGA CCT GCA CCC AAG CTT АТС GAT pUR292
Рис. 5.2. Структура плазмид pUR290, pUR291 и pUR292. А. Расположение
сайтов для клонирования на З'-конце кодирующей области lacZ. Б. Сайты для
клонирования в векторах pUR во всех трех рамках считывания. Выше приве-
дена последовательность lacZ дикого типа, начиная с сайта i’coRI до конца
кодирующего участка [11].
где благодаря наличию аллеля lad4 происходит повышен-
ное образование /ас-репрессора. Синтез ^-галактозидазы инду-
цируется изопропил-[3-И-тиогалактопиранозидом (ИПТГ).
5.2. Конструирование и клонирование гибридных белков,
содержащих ^-галактозидазу
Имеются три основных побудительных мотива к использова-
нию гибридных белков, содержащих ^-галактозидазу. В зависи-
мости от характера конкретной задачи они сводятся к следую-
щему.
1. Клонирована последовательность ДНК, причем кодируе-
мый продукт не охарактеризован. Для изучения продукта дан-
ного гена исследователю нужно получить антитела.
2. Имеются антитела к белку (белкам), с помощью кото-
рых путем скрининга библиотеки экспрессирующихся клонов
(например, клонированных в Xgtll) выделен предположительно
соответствующий клон ДНК. Задача сводится к проверке этого
142 С. Кэрролл, А. Логон
клона: необходимо показать, что антитела, полученные с по-
мощью данного клона, действительно взаимодействуют со специ-
фическим антигеном.
3. В распоряжении исследователя имеется охарактеризован-
ная кДНК, но ему требуются антитела, поскольку 1) нужный
белок содержится в клетке в небольшом количестве или его труд-
но очистить, 2) либо необходимо получить антисыворотку, специ-
фичную к определенному участку или субъединице данного бел-
ка. Последний подход обладает значительно большей избира-
тельностью, чем скрининг группы моноклональных антител,
и гораздо проще, чем выделение пептидных фрагментов путем
ферментативного или химического расщепления.
Независимо от того, как был получен клон ДНК, сначала
его секвенируют (определяют нуклеотидную последователь-
ность), определив таким образом положение всех открытых ра-
мок считывания и удобных сайтов рестрикции. Установив нукле-
отидную последовательность ДНК, можно предсказать молеку-
лярную массу гибридного белка и оценить степень деградации
выделенного продукта. Далее, при необходимости можно повто-
рить генноинженерные эксперименты или перейти к выделению
гибридных белков и получению специфических антител.
Стыковку генов можно осуществить и не зная нуклеотидной
последовательности фрагмента чужеродной ДНК- Чужеродную
ДНК «сшивают» с геном lacZ в каждой из трех возможных от-
крытых рамок считывания, а синтез гибридных белков, превы-
шающих по размеру р-галактозидазу, детектируют с помощью
ДСН-электрофореза в ПААГ. При таком подходе может возник-
нуть ряд проблем. Обычно если неизвестна полная нуклеотидная
последовательность ДНК, то неизвестны и границы его кодирую-
щего участка. Поэтому необходимо правильно выбрать сайт ре-
стрикции, соответствующий месту соединения белков в гибрид-
ном продукте. Для решения этой проблемы гены сшивают по
нескольким сайтам рестрикции, так чтобы хоть одна из получае-
мых конструкций включала как можно большую часть кодирую-
щей области. Другая проблема, с которой мы столкнулись, — это
наличие двух перекрывающихся открытых рамок считывания в
одном гене. В случае гена Antennapedia плодовой мушки (Dro-
sophila) определение нуклеотидной последовательности ДНК
сыграло решающую роль при выяснении того, какая из двух от-
крытых рамок считывания кодирует белок in vivo [13]. Нако-
нец, in vivo в клетках Е. coli может происходить протеолиз гиб-
ридных белков, и во многих случаях получаемый гибридный бе-
лок имеет неполную длину, что приводит к неадекватным оцен-
кам генноинженерных конструкций, полученных без знания нук-
леотидной последовательности клонированной ДНК.
Поликлональные антитела к фрагментам гибридных белков 143
5.2.1. Клонирование гибридных генов в векторе Xgtl 1
Метод клонирования кДНК в векторе Xgtll подробно опи-
сали в первом томе этой серии Хьюинх, и Янг и Дэвис [12].
Бактериальные штаммы RY1088, RY1089 и RY1090, необходи-
мые для работы с Xgtll, можно получить из Американской кол-
лекции тканевых культур (American Type Culture Collection,
12301 Parklawn Drive, Rockville, MD 20852, USA).
8-, 10- или 12-членные AcoR 1-линкеры имеются в продаже
(New England Biolabs); они используются для лигирования
концов рестрикционных фрагментов таким образом, чтобы ко-
дирующий участок встраивался с сохранением рамки в EcoRl-
сайт Mgll. По нашим данным, в качестве промежуточного эта-
па при клонировании гибридных генов в Xgtll удобно встраи-
вать сшитые с линкерами фрагменты ДНК в EcoRl-сайт подхо-
дящей плазмиды, например pUC18 или рЕМВЫ8.
5.2.2. Клонирование гибридных генов в плазмидах pUR
Встройте чужеродную ДНК в сайты BamHl, Sall, Pstl,
Hindllt или Clal плазмид pUR290, pUR291 или pUR292, исполь-
зуя природные сайты рестрикции или реконструируя подходя-
щие рестрикционные сайты с помощью синтетических ДНК-лин-
керов. Если это возможно, вводите чужеродную последователь-
ность ДНК в нужной ориентации, используя два различных лип-
ких конца (например, BamHl и //indlll).
5.2.2.1. Подготовка векторной ДНК
Процент клонов, содержащих вставки гетерологичной ДНК,
резко возрастает, если концы векторной ДНК перед лигирова-
нием обработать щелочной фосфатазой из кишечника теленка
(ФКТ, CIP). Удаление 5’-концевых фосфатов векторной ДНК
препятствует внутримолекулярному лигированию. Поскольку
РНК в качестве субстрата для щелочной фосфатазы является
активным конкурентом ДНК, важно, чтобы препарат ДНК плаз-
миды pUR был свободен от РНК-
1. Выделите ДНК стандартным методом и дважды прове-
дите ее очистку равновесным центрифугированием в 50% (ве-
совая концентрация) CsCl [14].
2. Проведите расщепление 1—5 мкг ДНК подходящей ре-
стриктазой при концентрации ДНК 100 мкг/мл.
3. После полного расщепления добавьте 10 ед. ФКТ (Boe-
hringer) и инкубируйте в течение 15 мин при 37 °C.
4. Выделите линейную ДНК плазмиды pUR с помощью
электрофореза в агарозном геле (0,8%-ная легкоплавкая ага-
роза) [14].
144
С. Кэрролл, А. Логон
5.2.2.2. Приготовление гетерологичного фрагмента ДНК
Проведите расщепление 1—5 мкг гетерологичной ДНК под-
ходящей рестриктазой и выделите нужный фрагмент методом
электрофореза в агарозном геле. Если необходимо использовать
ДНК-линкеры, после расщепления рестриктазой затупите кон-
цы ДНК. Тупые концы формируют путем достраивания укоро-
ченных З’-концов фрагментом Кленова ДНК-полимеразы I или
удаления выступающих З’-концов под действием 3’-5’-эндонук-
леазной активности ДНК-полимеразы Т4 [14]. После пришива-
ния линкеров к фрагменту с тупыми концами [14] надо полу-
чить липкие концы путем расщепления линкерной ДНК с по-
мощью подходящей рестриктазы.
5.2.2.3. Лигирование ДНК плазмид pUR с гетерологичной ДНК
и трансформация
Лигируйте приблизительно 0,1 мкг очищенной, обработан-
ной фосфатазой ДНК плазмиды pUR с эквимолярным количе-
ством очищенной встраиваемой ДНК в объеме реакционной
смеси 10 мкл. Используйте полученную после лигирования
смесь для трансформации подходящего для этой цели штамма
71-18 E.coli [15], высевая клетки на чашки с 1XYT (8 г трип-
тона, 5 г дрожжевого экстракта, 5 г NaCl, 15 г агара на 1 л),
содержащие 50 мкг/мл ампициллина.
5.2.3. Скрининг клонов Xgt11
5.2.З.1. Скрининг фаговых клонов на наличие вставок
После лигирования и проведения упаковки in vitro высейте
фаг на чашки с клетками RY 1090 в верхнем агаре, содержа-
щем X-gal и ИПТГ [12]. Отберите бесцветные бляшки (отсут-
ствие окраски указывает на наличие вставки). Выделите фаго-
вую ДНК [12] и для выявления вставочного фрагмента расще-
пите ее EcoRl, а для определения ее ориентации — рестрикта-
зой, вносящей разрыв внутрь вставки.
5.2.3.2. Скрининг клонов Igtl 1 на продуцирование
гибридных белков
Осуществите упаковку фага, который будет использоваться
для получения гибридного белка, и для получения лизогенной
культуры высейте его на чашки со штаммом RY1089 при 32 °C
[12]. Лизогенные бактерии размножаются при 32 °C, но не при
42 °C — из-за наличия температурочувствительного репрессора.
Лизогенные бактерии культивируют и затем анализируют их
Поликлональные антитела к фрагментам гибридных белков 145-
Таблица 5.1. Скрининг лизогенов Xgtll, продуцирующих гибридные белки
1. Оставьте на ночь лизогенные культуры в NZCYM1’ при 30 °C.
2. Внесите ночную культуру в 2 мл NZCYM при разбавлении I : 100 и куль-
тивируйте до достижения плотности 4-10s клеток/мл при 30 °C.
3. Добавьте ИПТГ до концентрации 1 мМ и поместите культуру на 15 мин
в водяную баню с температурой 45 °C.
4. Инкубируйте индуцированные культуры в течение 2 ч при 37 °C и осади-
те клетки центрифугированием в микроцентрифуге в пробирках объемом
1,5 мл в течение 1 мин.
5. Лизируйте осажденные клетки в 200 мкл электрофоретического буфера
для нанесения образцов21 и прогрейте их при 100 °C в течение 5 мин.
6. Быстро встряхните смесь и осветлите лизат центрифугированием в микро-
центрифуге в течение 5 мин.
7. Фракционируйте 25 мкл каждую надосадочную фракцию путем электро-
фореза в 7,5% ПААГ, содержащем ДСН.
!) Состав NZCYM: в 1 л содержится 10 г NZ-амнна, 5 г NaCl» 5 г дрожжевого экст-
ракта, 1 г казаминовых кислот, 1 г MgSO4; pH доводят до 7,5 добавлением 2 М NaOH.
2) Состав буфера для нанесения образцов: 80 мМ трис-НС1 (pH 6,9), 2% ДСН, 0,1 М
дитиотрейтол, 0,004%-ный бромфеноловый синий.
белки с помощью ДСН-электрофореза в ПААГ, как это описа-
но в табл. 5.1. На рис. 5.3 приведен пример ДСН-электрофоре-
за в ПААГ двух препаратов суммарного белка, содержащих
гибридные продукты, экспрессируемые рекомбинантными XgtlL
5.2.4. Скрининг клонов, полученных в плазмидах pUR
5.2. 4.1. Скрининг плазмидных клонов на наличие вставок
Отберите устойчивые к ампициллину трансформанты сте-
рильными зубочистками в 1 мл аликвоты среды 2XYT, содер-
жащей 50 мкг/мл ампициллина, и выращивайте в течение ночи
при 37 °C. Выделите из проб плазмидную ДНК с помощью бы-
строго метода с использованием кипячения, описанного в книге
Маниатиса и др. [14]. Обработайте ДНК рестриктазами, узна-
ющими сайты в пределах встроенной ДНК и в прилегающих
плазмидных векторах для выявления клонированной чужерод-
ной ДНК и определения ее ориентации.
5.2. Д.2. Скрининг плазмидных клонов на продуцирование
гибридных белков
Разбавьте ночные культуры клонов, предположительно про-
дуцирующих гибридные белки, средой 2XYT, содержащей
50 мкг/мл ампициллина, в соотношении 1 : 100 и выращивайте
при 37 °C в течение 2 ч. Осадите клетки, лизируйте их и фрак-
ционируйте лизат путем электрофореза, как описано в
разд. 5.2.3.2 и табл. 5.1. Пример электрофоретического разделе-
10—1186
146
С. Кэрролл, А. Л огон
Рис. 5.3. Два примера аномального пове-
дения гибридных белков. Фрагмент
кДНК Antennapedia был встроен в век-
тор Xgtll в двух разных рамках считы-
вания. Самый большой по размеру из
мажорных продуктов (дорожка 1) син-
тезируется при считывании с неправиль-
ной рамкой, но вследствие чрезвычайно
высокого содержания GC в ДНК и про-
лина в белке образующийся продукт об-
ладает такой же подвижностью, как про-
дукт, кодируемый правильной рамкой
считывания (черная стрелка, дорожка 1).
Вторая рамка считывания (дорожка 2)
правильная, и размер ее мажорного про-
дукта (дорожка 2, черная стрелка) так-
же соответствует заранее рассчитанному.
Однако этот белок не является полно-
размерным гибридным белком. Минор-
ный продукт большего размера (дорож-
ка 2, светлая стрелка) выявляется при
окрашивании с использованием антител
к ^-галактозидазе (данные не представ-
лены) и антител к С-концевому участку
белка (данные не приведены). Высокое
содержание пролина в гибридном белке
приводит к увеличению кажущейся моле-
кулярной массы на 10—15 кДа. Этим
объясняется ошибочный вывод о том, что
обладающий низкой подвижностью про-
дукт деградации является полноразмер-
ным белком.
Поликлональные антитела к фрагментам гибридных белков 147
ния белкового экстракта, содержащего гибридный продукт, экс-
прессированный в pUR-векторе, приведен на рис. 5.4.
5.2.5. Соотношение размера гетерологичного фрагмента
и выхода синтезируемого гибридного белка
Размер гетерологичного белкового фрагмента — важный па-
раметр при клонировании гибридных белков, содержащих ^-га-
лактозидазу. Стабильность (а следовательно, и выход) гибрид-
ных белков, как правило, снижается при увеличении размера
чужеродного фрагмента. Во многих случаях желательно, чтобы
в состав гибридных белков входили короткие фрагменты. Для
получения специфических антител вполне достаточными оказы-
ваются фрагменты гетерологичной ДНК, кодирующей антиген
длиной всего 200 п.н. [16]. Вместе с тем использование корот-
ких фрагментов может привести к тому, что при небольшой от-
носительной массе чужеродного полипептида (по сравнению
с массой ^-галактозидазы) титр специфичных к чужеродному
белку антител у иммунизированных животных окажется низ-
ким. Аффинная очистка антител в этом случае также менее
эффективна вследствие низкой емкости аффинных колонок,
в которых связанные с носителем гибридные белки несут корот-
кие гетерологичные фрагменты. Один из путей решения этой
проблемы состоит в увеличении молярного соотношения гетеро-
логичный фрагмент: ^-галактозидаза в гибридном белке. Это
достигается пришиванием к гену lacZ множественных тандем-
ных копий гетерологичного фрагмента ДНК. На рис. 5.5 приве-
дены сравнительные результаты клонирования и экспрессии
одной из пяти копий кодирующего фрагмента гена ftz Droso-
phila в pUR291. Выход гибридного белка, кодируемого одной
копией этого гена, примерно в десять раз превышает выход
белка, кодируемого пятью тандемными копиями.
• 5.2.6. Характеристика экспрессируемых гибридных продуктов
При интерпретации картины экспрессии гибридных белков
в клетках Е. coli возникает ряд проблем. Во-первых, если в век-
тор был встроен фрагмент ДНК, нуклеотидная последователь-
ность которого не известна, можно наблюдать появление экс-
прессированного гибридного белка, достаточно длинного, но тем
не менее с неправильной рамкой считывания. Пример такого
рода приведен на рис. 5.3, где встраивание кДНК в две разные
рамки считывания в одном рестрикционном сайте приводило
к появлению в клетке двух основных белковых продуктов поч-
ти одинакового размера. Молекулярная масса одного из таких
гибридных белков составляла около 135 кДа, но синтезировал-
10*
148
С, Кэрролл, А. Логон
ся он при трансляции с неправильной рамкой считывания
(рис. 5.3, дорожка 1). Это подчеркивает то, что до конструиро-
вания гибридных генов желательно иметь информацию об их
нуклеотидных последовательностях.
Во-вторых, гибридный белок может подвергаться деградации
и in vivo, что будет приводить к накапливанию в качестве ос-
новного продукта неполноразмерного белка. На рис. 5.3 (до-
рожка 2) приведен пример, когда получаемый белковый про-
дукт примерно на 10 кДа легче полноразмерного продукта. При
секвенировании соответствующей необычной кДНК было обна-
ружено, что существует третий тип аномального поведения
продукта. Хотя основной синтезируемый в этом случае гибрид-
ный белок не является полным, в геле он обладает подвижно-
стью полноразмерного продукта. Это, по-видимому, объясняет-
ся необычайно высоким содержанием в белке пролина, что при-
Н И
Анти-
gal
МНИ
hy
МН И
и
В аак ►
*
I»
Поликлональные антитела к фрагментам гибридных белков
149
водит к завышению электрофоретической оценки его молекуляр-
ной массы.
Чтобы снизить вероятность получения ошибочных резуль-
татов, мы советуем исследовать лизат, содержащий гибридные
белки, с помощью вестерн-блот-анализа с использованием ан-
тител к р-галактозидазе (рис. 5.3) — это позволяет детектиро-
вать все минорные продукты. Кроме того, все получаемые ре-
зультаты необходимо сопоставить с анализом нуклеотидной
последовательности клонированный ДНК. Для идентификации
продуктов клонированных генов можно также использовать вто-
рые антитела, специфичные к антигенным детерминантам, ко-
торыми предположительно должен обладать гибридный белок.
Поскольку уровень экспрессии гибридных белков высок, мы
рекомендуем использовать иммуноферментный метод выявления
антигенов с помощью вестерн-блот-анализа (табл. 5.2) ввиду
надежности и быстроты этого метода.
5.3. Экспрессия гибридных генов
и очистка синтезируемых продуктов
В зависимости от используемого вектора, уровня экспрессии
гибридного белка и его растворимости для выделения чистого
интактного белка, который’ будет использоваться в иммунологи-
Рис. 5.4. Индукция гибридного белка ^-галактозидаза — продукт гена hairy.
Представлены группы, включающие по три дорожки. Электрофорез вели
в 7,5% ПААГ в присутствии ДСН. М —маркеры, Н — неиндуцированные, И —
индуцированные действием ИПТГ клетки 71-18, содержащие плазмиду с гиб-
ридным геном, включающим ген р-галактозидазы и ген hairy Drosophila. Левая
группа из трех дорожек — окраска кумасси; центральная — вестерн-блот, окра-
шенный с помощью аффинноочищенных кроличьих антител к р-галактозидазе
(anti-p-gaf); правая группа — вестерн-блот, окрашенный с помощью аффинно-
очищенных кроличьих антител к белку Hairy (anti-hy). Для окрашивания
обоих вестерн-блотов вторым антителом служит конъюгат щелочной фосфа-
тазы с анти-кроличьими IgG козы (табл. 5.1). Гибридная плазмида — это
pUR290, в которую встроена кДНК — открытая рамка считывания размером
1011 п. н., соответствующая транскриптам гена hairy. Стрелка слева указывает
местоположение р-галактозидазы (мол. масса 116 000), окрашиваемой антите-
лами к Р-галактозидазе, но не антителами анти-Hairy. Нижняя стрелка справа
указывает местоположение гибридного белка р-галактозидаза-hairy с мол. мас-
сой 160 к Да в экстрактах индуцированных клеток, окрашиваемых антителами
и к р-галактозидазе, и к белку Hairy. Гибридный белок обнаруживается
и в пенпдуцированных клетках, что указывает на неполную репрессию его син-
теза продуктом гена lad4. Верхние стрелки справа указывают на слабую поло-
су индуцированного белка, окрашиваемого антителами и к р-галактозидазе,
и к Hairy. Полоса соответствует белку, мол. масса которого на 5000 Да пре-
вышает массу белка 160кДа, указывая на то, что основной продукт по край-
ней мере на 5000 Да меньше полноразмерного гибридного белка. Экстракты
готовили, как описано в разд. 5.2.4.2. Аффинную очистку антител проводили,
как описано в разд. 5.4.2. Клон кДНК hairy, используемый для конструирова-
ния гибридного гена, был любезно предоставлен С. Рашлоу.
150
С, Кэрролл, А. Логон
мономер
Поликлональные антитела к фрагментам гибридных белков
151
ческих исследованиях, применяются различные методы. В неко-
торых случаях легко получить достаточное количество нативно-
го гибридного белка для биохимического исследования его гете-
рологичного фрагмента. В этом разделе мы приведем детальные
описания методов очистки гибридных белков с самыми различ-
ными уровнями экспрессии. Методы позволяют получать 1 —
10 мг гибридного белка за один цикл очистки. Этого количества
достаточно для гипериммунизации животных среднего размера
(например, кроликов) гибридными белками, содержащими ге-
терологичные фрагменты любой величины. Хотя, как правило,
для получения высокоактивной антисыворотки к нативному
белку лучше всего подходят клоны с полноразмерной кДНК, нам
удавалось получать антисыворотку к гибридным белкам, содер-
жащим маленькие гетерологичные фрагменты (<10 кДа). Ан-
тисыворотка к небольшим вставкам может быть особенно по-
лезна в качестве зондов при функциональных или топологиче-
ских исследованиях (см. разд. 5.2.5).
5.3.1. Индукция синтеза гибридных белков
Максимальный уровень экспрессии гибридных белков в оп-
ределенной степени зависит от используемых условий индук-
ции. Для двух описанных здесь векторных систем применяется
прямая индукция, результаты которой, по нашим наблюдени-
ям, мало различаются для этих двух систем.
5.3.1 Л. Индукция штаммов, лизогенных по Igtll
Для получения больших количеств гибридных белков, коди-
руемых последовательностями, клонированными в фаге Xgtll,
необходимо «излечить» лизогенный штамм от плазмиды
—
Рис. 5.5. Соотношение размера и стабильности гибридного белка. Два 7,5°/о-ных
ДСН-полиакриламидных геля с нанесенными одинаковыми образцами: один
окрашен кумасси, а с другого сделан перенос на нитроцеллюлозный фильтр
с помощью вестерн-блот-анализа, после чего он окрашен аффинноочищенными
антителами к чужеродному фрагменту гибридного белка. На гель наносили
экстракты из индуцированных ИПТГ клеток 71-18, содержащих плазмиды
pUR291 со встроенными последовательностями ftz. Дорожки, обозначенные
«мономер», содержат продукт экспрессии плазмид, несущих одну копию нук-
леотидной последовательности /Zz-гомео-бокса (268 п.н.), состыкованного с
геном lacZ. Дорожки, обозначенные «пента-мер», содержат продукт экспрес-
сии плазмид, несущих пять тандемных копий ftz (268 п.н.), состыкованных
в одной рамке, считываются с lacZ. Стрелки указывают местоположение гиб-
ридных белков соответственно моно-мерных и пента-мерных относительно ftz.
Экстракты готовили, как описано в разд. 5.2.4.2, а вестерн-блот-анализ прово-
дили, как описано в табл. 5.2. Выход гибридных белков из экстрактов, полу-
ченных при обработке клеток ДСН, составлял примерно 50 мкг/мл в случае
1-мерного и 5 мкг/мл в случае 5-мерного ftz.
152 С, Кэрролл, А. Л огон
Таблица 5.2. Обнаружение антител к индивидуальным антигенам
с помощью вестерн-блот-анализа
1. Получите лизат клеток, продуцирующих гибридные белки, как описано
в разд. 5.2 и табл. 5.1. Нанесите его на 7,5%-ный ПААГ для проведения
ДСН-электрофореза. Нанесите на соседние дорожки маркерные высоко-
молекулярные белки, включая и р-галактозидазу.
2. После проведения электрофореза перенесите белки на нитроцеллюлозные
фильтры с помощью прибора для электро-блот-анализа (например, Hoefer
Thransphor ТЕ 41), следуя прилагаемой к прибору инструкции.
3. После переноса проведите окрашивание фильтров в течение 3 мин кра-
сителем PonceauS, разбавленным 1:10 дистиллированной водой; промойте
фильтры дистиллятом для отмывки красителя. По интенсивности окраски
можно определить эффективность переноса и расположение дорожек с об-
разцами. Для обозначения границ между дорожками сделайте разметку
карандашом.
4. Инактивируйте свободные участки поверхности нитроцеллюлозного фильт-
ра (для снижения фона), инкубируя фильтр в буфере PBS1), содержащем
3% (весовая концентрация) БСА, в течение ночи при 4 °C или 2 ч при
37 °C.
5. После инкубации в растворе БСА полоски фильтров инкубируют в тече-
ние 2—3 ч со специфическими антителами, разбавленными PBS, содержа-
щим 5% (объемная концентрация) сыворотки неиммунизированной козы.
Очищенные аффинными методами антитела активны при концентрации
0,5—5,0 мкг/мл, а вся сыворотка — при минимальном разбавлении 1 : 50.
После инкубации тщательно промойте фильтры по два раза большими объ-
емами буферов PBS, ВВ5-свин1 2> и PBS. Общее время отмывания должно
составлять 60 мин — по 10 мин на каждую отмывку. Интенсивная отмыв-
ка необходима для уменьшения фонового окрашивания; в некоторых слу-
чаях отмывку можно проводить в течение более короткого времени.
6. Добавьте второе антитело — конъюгаты козьих антикроличьих IgG со
щелочной фосфатазой, разбавленные до концентрации 1 мкг/мл (обычно
1 : 1000) в буфере PBS, содержащем 5% сыворотки неиммунизированной
козы; инкубируйте 2—3 ч при комнатной температуре. После инкубации
тщательно промойте фильтры растворами PBS, BBS-твин и TBS3’ в тече-
ние 60 мин, промывая фильтры каждым буфером по меньшей мере по
два раза.
7. После последней промывки буфером TBS погрузите фильтры в свежепри-
готовленный буфер, содержащий субстрат для щелочной фосфатазы4’.
После развития окраски, промойте их три раза дистиллированной водой.
Дайте фильтру постоять в воде в течение 10 мин и затем высушите его
на фильтровальной бумаге.
1) Состав фосфатного буфера PBS: 10 мМ калий-фосфатного буфера и 150 м.М NaCl
(pH 7,2).
2) BBS-твин — боратный буфер, содержащий твии 20. Состав: 0,1 М. борная кислота,
0,025 М борат натрия, 1 М NaCl 0,1% (объемная концентрация) твин 20 (pH 8,3).
3) TBS: трис-буфер, содержащий 50 мМ трис-НС1 (pH 7,5), 150 мМ NaCl.
4) Состав буфера, содержащего субстрат для щелочной фосфатазы: 2 мл красителя
Nitro Blue Tetrazolium (Sigma) в концентрации 1 мг/мл в 50 мМ Na2CO3 (pH 9,5), 2 мл
0,5 М На2СОз (pH 9,5), 15,5 мл дистиллированной Н2О, 0,1 мл 1 М MgCl2 и 0,2 мл бром-
хлорнндолнлфосфата (Sigma), растворенного в концентрации 5 мг/мл в диметилсульф-
оксиде.
Поликлональные антитела к фрагментам гибридных белков
153
рМС9 (laclq), определяющей устойчивость к ампициллину. Как
правило, снятие /ас-репрессии довольно быстро приводит к по-
явлению мутантных форм гибридного белка. Однако для до-
стижения значительного (в 3—4 раза) увеличения выхода бел-
ка допускается пассирование «извлеченного» лизогенного штам-
ма в течение непродолжительного времени.
Поддерживайте штоки лизогенной культуры, содержащей
рМС9, чтобы избежать потери клона в результате отрицатель-
ной селекции. Проведите несколько пассажей лизогенной куль-
туры при температуре 32 °C в NZCYM в отсутствие антибиоти-
ка (состав NZCYM приведен в сноске к табл. 5.1). Протести-
руйте культуру на потерю устойчивости к антибиотику, высе-
вая лизогенный штамм на чашки с NZCYM, содержащие и не
содержащие ампициллин. «Излеченная» лизогенная культура
в отсутствие продукта lac!q будет продуцировать больше белка,
чем «неизлеченная» культура даже в условиях полной индук-
ции ИПТГ.
Выращивайте лизогенную культуру в течение ночи при 32 °C.
Разбавьте стационарную культуру 1 : 100 и выращивайте ее
в течение 2 ч при 32 °C в условиях хорошей аэрации. Из одного
литра культуры обычно получают 2—20 мг гибридного белка.
Индуцируйте культуру путем быстрого повышения температуры
до 42 °C, поместив ее в водяную баню, предварительно нагре-
тую до 44 °C. Следите за температурой с помощью термометра
в течение периода индукции — около 15 мин. Снова перенесите
культуру на 37 °C и продолжайте культивирование ее в тече-
ние 2—3 ч.
5.3.1.2. Индукция клонов, содержащих плазмиду pUR
с гибридными генами
Вырастите ночную культуру в среде 2XYT, содержащей
50 мкг/мл ампициллина. Разбавьте ночную культуру 1 : 100
средой 2XYT, содержащей 50 мкг/мл ампициллина, и выращи-
вайте ее при 37 °C в течение 2 ч. В 0,5 л культуры обычно со-
держится 5—50 мг гибридного белка. Добавьте ИПТГ до кон-
центрации 0,5 мМ и продолжайте выращивать культуру при
37 °C в течение 2 ч
5.3.2. Осаждение клеток и выделение гибридного белка
Охладите индуцированные клетки в ледяной бане и для
уменьшения протеолиза добавьте фенилметилсульфонилфторид
(ФМСФ, PMSF) до концентрации 1 мМ. Центрифугируйте
клетки при 5000 g в течение 5 мин. Ресуспендируйте клеточные
осадки в одном из следующих буферов для экстракции.
154-
С. Кэрролл, А. Логон
1. Для препаративного электрофореза (см. разд. 5.3.3.1) ре-
суспендируйте клетки при энергичном встряхивании в 1/12
объема буфера для нанесения, содержащего 1,5% ДСН. Про-
грейте экстракт при 100 °C в течение 5 мин.
2. Для выделения белков с помощью иммуноаффинной хро-
матографии (разд. 5.3.3.2) или гель-фильтрации (разд. 5.3.3.3)
ресуспендируйте клетки при 4 °C в 1/50 объема 50 мМ натрий-
фосфатного буфера (pH 7,0), содержащего 10 мМ 2-меркапто-
этанол, 10 мМ ЭДТА и 1 мМ ФМСФ. Добавьте лизоцим до
концентрации 0,2 мг/мл и инкубируйте смесь в ледяной бане
в течение 30 мин. Заморозьте экстракт при —70 °C и оставьте
на 30 мин. Быстро разморозьте экстракт в водяной бане при
37 °C так, чтобы его температура не поднималась выше 4 °C,
и тщательно обработайте ультразвуком 5X20 с, в промежутках
охлаждая пробу так, чтобы поддерживать температуру 4 °C.
Добавьте при тщательном перемешивании 5 М NaCl до кон-
центрации 0,5 М и затем для осветления экстракта центрифуги-
руйте его при 13 000 g в течение 10 мин. Все дальнейшие про-
цедуры проводите, поддерживая температуру экстракта равной
4 °C.
3. Для хроматографии на АФТГ-агарозе (разд. 5.3.3.4) ре-
суспендируйте клетки в ледяной бане в 1/50 объема раствора,
содержащего 0,2 М трис-НС1 (pH 7,6), 10 мМ MgOAc, 5% гли-
церола, 10 мМ 2-меркаптоэтанол, 0,25 М NaCl и 1 мМ ФМСФ.
Инкубируйте клетки с лизоцимом (концентрация 0,2 мг/мл)
в течение 30 мин в ледяной бане, быстро заморозьте при
—70 °C и оставьте на 30 мин. Обработайте экстракт ультра-
звуком и осветлите его, как описано в п. 2, но без добавления.
4. Некоторые белки плохо экстрагируются с помощью мето-
дов, приведенных в пп. 2 и 3. Гибридные белки могут остаться
в клеточных обломках (дебрисе), осаждаемых центрифугирова-
нием после обработки ультразвуком. Нам удавалось солюби-
лизировать некоторые из наиболее труднорастворимых гибрид-
ных белков обработкой клеточного дебриса мочевиной. Экстра-
гируйте дебрис, суспендируя его в буфере, содержащем
мочевину (8 М мочевина, 0,5 М трис-НС1 (pH 7,9), 0,5 М NaCl,
1 мМ ЭДТА, 30 мМ 2-меркаптоэтанол, 1 мМ ФМСФ), из расче-
та 20 мл буфера на 1 л исходной клеточной культуры. Энергич-
но перемешайте суспензию встряхиванием и центрифугируйте ее
при 13 000 g в течение 10 мин. Диализуйте растворенный экст-
ракт против 25—50 объемов раствора, содержащего 50 мМ
трис-НС1 (pH 7,9), 0,5 М NaCl, 10% глицерола, сначала при
комнатной температуре в течение 2—3 ч, затем в течение ночи
при 4 °C. Центрифугируйте экстракт при 13 000 g 10 мин. Такой
экстракт, полученный при обработке мочевиной, может быть
обогащен гибридным белком, который затем очищают с по-
Поликлональные антитела к фрагментам гибридных белков
155
мощью одного из методов, приведенных в разд. 5.3,3. Обогаще-
ние экстракта гибридным белком при обработке мочевиной по-
казано на рис. 5.6.
5.3.3. Выделение гибридного белка — выбор метода
Мы пришли к выводу, что для самых разнообразных имму-
нологических исследований необходимо получать максимально
возможное количество нативного белка. Это легче всего дости-
гается с помощью классических хроматографических методов,
детально описанных в разд. 5.3.3.3, которые наиболее успешно
работают при использовании экспрессирующих систем на осно-
ве плазмид pUR. Другие методы применяют, когда возникают
сложности с выходом, растворимостью или стабильностью гиб-
ридного белка. Например, к выделению гибридных белков мето-
дом ДСН-электрофореза в ПААГ следует прибегать в послед-
нюю очередь, а не использовать его сразу из-за его простоты,
поскольку денатурированные ДСН белки меняют свои иммуно-
генные свойства и с их помощью не всегда удается получить
антисыворотку к нативному белку. Если антисыворотка в ос-
новном будет использоваться в вестерн-блот-анализе, то в этом
случае денатурированные под действием ДСН иммуногены мо-
гут оказаться пригодными. Иммуноаффинные методы очистки
можно эффективно использовать в ограниченном масштабе,
поскольку, к сожалению, элюируемые белки, как правило, не-
обратимо денатурированы реагентами, используемыми для раз-
рушения комплексов антиген—антитело. С помощью хромато-
графии на АФТГ-агарозе можно получить чистый растворимый
белок, но метод не очень эффективен и дает очень низкий вы-
ход белков, содержащихся в клетке в небольшом количестве
или обладающих низкой стабильностью. Выбор того или иного
метода поможет сделать анализ результатов прикидочного вы-
деления и очистки.
5.З.З.1. Препаративный ДСН-электрофорез
в ПААГ гибридных белков
Препаративный электрофорез в акриламидном геле может
быть использован на любой стадии очистки гибридного белка
для его выделения в качестве неповрежденного, но денатуриро-
ванного иммуногена. ДСН-лизаты клеток, полученные, какопи-
сано выше в разд. 5.3.2, после осветления центрифугированием
могут быть непосредственно нанесены на гель. Белок примерно
из 15 мл клеток, лизированных в 1,25 мл буфера для нанесе-
ния, содержащего 1,5% ДСН, можно исследовать, используя
одну пластину геля с площадью поперечного сечения 2,0 см2.
156
С, Кэрролл, А. Логон
Поликлональные антитела к фрагментам гибридных белков 157
При электрофорезе частично очищенных и обогащенных белко-
вых экстрактов можно выделить интактный гибридный белок
из расчета не менее 1 мг белка на одну пластину геля.
Для выявления области геля, содержащей гибридный бе-
лок, после электрофореза вымочите гель в охлажденном 0,1 М
КС1. Холодный раствор, содержащий ионы К+, преципитируют
ДСН в дорожках геля, в результате чего появляются соответ-
ствующие фракционированным белкам белые полосы. Такую
полосу, соответствующую гибридному белку, можно вырезать
из геля острой бритвой. Это устраняет необходимость фиксации
и окраски кумасси полосы геля с исследуемым белком или па-
раллельных дорожек геля.
5.3.3.2. Выделение гибридных белков иммуноаффинными методами
Экстракты, выделенные, как описано в разд. 5.3.2 (п. 2, 3
или 4), могут быть непосредственно нанесены на аффинные
колонки, содержащие антитела к ^-галактозидазе без какого-
либо предварительного фракционирования (например, удале-
ния нуклеиновых кислот). Поэтому гибридные белки, характе-
ризующиеся низкой стабильностью или низким содержанием
в клетке, могут быть легко выделены из разбавленных экстрак-
тов без деградации, возможной при более длительных методах
выделения.
1. Клоны с иммобилизованными антителами к ^-галактози-
дазе можно приготовить, как это описано в разд. 5.4, или ис-
пользовать коммерческие препараты. Возможность повторного
использования колонки зависит от свойств антител (монокло-
нальных или поликлональных) и жесткости элюирующих рас-
творов. Колонки с поликлональными антителами (полученные,
как здесь описано) могут быть использованы более 20 раз без
значительного уменьшения их связывающей способности. Ко-
лонка, содержащая 5 мг аффинноочищенных поликлональных
антител к р-галактозидазе, обычно связывает по меньшей мере
1 мг гибридного белка.
Рис. 5.6. Выделение и очистка гибридного белка, содержащего 3-галактозида-
зу. Сравнение выхода и степени чистоты гибридного белка ^-галактозидаза—ftz,
выделенного из штамма, лизогенного по Zgtl 1, тремя различными методами:
1) 8 М мочевиной, 1,5% ДСН, 50 мМ натрий-фосфатным буфером (pH 7).
Каждый экстракт в объеме, эквивалентном 20 мкл исходной культуры, нано-
сили на 7,5%-ный ПААГ, содержащий ДСН; гель окрашивали кумасси. Экс-
тракт, полученный при обработке мочевиной, обогащен полноразмерным гиб-
ридным белком с мол. массой 175 кДа (черная стрелка, FTZ). Белок, получен-
ный из натрий-фосфатного экстракта, далее очищали с помощью хроматографии
на АФТГ-агарозе (правая дорожка). Материал, очищенный иа АФТГ-агарозе.
состоял в основном из полноразмерного белка и одного основного (85 кДа)
пика продукта деградации ^-галактозидазы (185, светлая стрелка). Рисунок
взят из работы Кэрролла и Скотта [6].
158
С. Кэрролл, А, Логон
2. Нанесите на колонку экстракт, содержащий гибридный
белок, при 4 °C для уменьшения протеолиза. Промойте колон-
ку буфером PBS (его состав указан в сноске к табл. 5.2)
в объеме, равном объему колонки, при 4 °C. Все последующие
стадии проводите при комнатной температуре. Промывайте
колонку буфером BBS-твин (состав указан в сноске к табл. 5.2)
до тех пор, пока в элюирующем растворе не останется белка.
Уравновесьте колонку буфером PBS. Проведите элюцию раство-
ром, содержащим 4 М гуанидин-НС1, 10 мМ трис-НС1 (pH 8,0),
соберите пик и диализуйте его против нескольких смен буфера
PBS. Основная часть белка выпадает в осадок и может быть
собрана с помощью центрифугирования. Для проведения имму-
низации ресуспендируйте преципитат в небольшом объеме PBS
и далее следуйте методике, приведенной в разд. 5.4. Сразу же
после использования снова уравновесьте колонку PBS. При ра-
боте возможно использование и других элюирующих растворов,
но более слабые элюенты не смогут разрушить прочные связи
комплексов антиген—антитело, и в результате емкость колонки
уменьшится, так как большое число участков аффинного связы-
вания окажется занятым.
5.3.3.3. Хроматография гибридных белков методом гель-фильтрации
При использовании большинства хроматографических мето-
дов желательно удалить из белковых экстрактов нуклеиновые
кислоты. После этого гибридные белки могут быть очищены
с помощью ряда хроматографических методов [17]. Самый про-
стой метод, позволяющий отделить гибридные белки больших
размеров от меньших по размеру полипептидов E.coli, — это
гель-фильтрация. Нуклеиновые кислоты удаляют, а белок кон-
центрируют осаждением стрептомицин-сульфатом и сульфатом
аммония (табл. 5.3). Некоторые гибридные белки могут быть
осаждены и очищены при использовании низких концентраций
(25%) сульфата аммония. Для других требуются более высокие
его концентрации. Это можно определить в прикидочных опы-
тах с использованием разных концентраций (NH^SCh. Сефа-
крил S-300 (Pharmacia) хорошо подходит для очистки гибрид-
ных белков, содержащих ^-галактозидазу. ^-Галактозидаза —
тетрамерный белок (молекулярная масса мономера составляет
116 кДа). Поэтому гибридные белки элюируются как молекулы,
молекулярная масса которых превышает 460 кДа. На колонке
размером 2,5x50 см можно хроматографировать 5 мл экстрак-
та, полученного из 1 л культуры клеток Е. coli. На рис. 5.7
представлены типичный профиль элюции гибридного белка с ко-
лонки S-300 и анализ фракций элюированного пика методом
ДСН-электрофореза в ПААГ.
Поликлональные антитела к фрагментам гибридных белков
15»
Таблица 5.3. Гель-фильтрация гибридных белков
1. Добавьте к экстракту, полученному, как описано в разд. 5.3.2 (п. 2),
0,1 объема 30%-ного (весовая концентрация) стрептомиции-сульфата в
буфере для выделения.
2. Перемешивайте смесь в течение 15 мин при 4 °C, а затем отцентрифуги-
руйте ее при 13 000 g в течение 10 мин.
3. Добавьте к надосадочной фракции насыщенный раствор (NHJzSCU
(pH 8,0) до концентрации 25—40% в зависимости от того, какой гибрид-
ный белок вы выделяете. (NH4)2SO4 добавляйте по каплям при осторож-
ном перемешивании. Продолжайте перемешивание еще в течение 30 мин
при 4 °C.
4. Центрифугируйте смесь при 13 000g в течение 30 мин. Отберите надоса-
дочную фракцию и отбросьте ее. Из оставшейся суспензии осадите суль-
фат аммония центрифугированием при 4000g в течение 1 мин. Осторожно
удалите оставшийся раствор. Растворите осадок в буфере для нанесения
образцов из расчета 5 мл буфера на 1 л исходной культуры..
5. Центрифугируйте этот раствор при 13 000g перед нанесением его на ко-
лонку размером 2,5X50 см с сефакрилом S-ЗОО, уравновешенную буфером
для нанесения образцов1’.
6. Установите скорость протекания 1 мл/мин. Собранный по поглощению
при 280 нм пик гибридного белка можно дополнительно проверить на на-
личие ^-галактозидазной активности с помощью цветной реакции. Мы об-
наружили, что гибридный белок содержится в первом основном элюируе-
мом пике поглощения при 280 нм.
7. Гибридный белок выходит в объеме примерно 20 мл и может быть скон-
центрирован осаждением (NHJzSCU.
') Состав буфера для нанесения образцов: 20 мМ трнс-НС1 (pH 7,5), 150 мМ. NaCl,
20 мМ 2-меркаптоэтаиол и 1 мМ ФМСФ. Для увеличения растворимости некоторых бел-
ков можно добавить тритон Х-100 до 0,1% (объемная концентрация).
5.3.3.4. Аффинная хроматография
на аминофенилтиогалактопиранозиде (АФТГ, APTG)
Стиэре [18] первым разработал, а Джермино и др. [2]
использовали для хроматографии гибридных белков метод вы-
деления р-галактозидазных производных полипептидов на ос-
нове их сродства (способности к связыванию) к лактозному
аналогу АФТГ (табл. 5.4). На практике с помощью этого мето-
да можно получать исключительно чистый материал, но с не-
высоким выходом. Поэтому метод неэффективен в случае очи-
стки белков, находящихся в клетке в небольших количествах
[19]. На рис. 5.6 в одном из примеров продемонстрировано
качество препарата гибридного белка, очищенного на АФТГ-
агарозе.
5.3.3.5. Заключение по методам очистки гибридных белков
Особенности разных методов очистки гибридных белков
суммированы в табл. 5.5. В зависимости от исходной концен-
трации, стабильности гибридного белка и цели эксперимента
* I I
ндгвл
Маркерные
белки "pfc„rs
(NH^SOv
'-осадок д|Д||
РастЮрйЯИ®
Фракадща|Я
И»® ::£ЙЙЕИ|В
ч Кэрролл, А, Логон
f®3
®>
ЧЛе
Поликлональные антитела к фрагментам гибридных белков 161
Таблица 5.4. Аффинная хроматография на АФТГ-агарозе
1. Проведите осаждение экстракта, полученного, как описано в разд. 5.3.2
(п. 3), стрептомицин-сульфатом и сульфатом аммония (см. разд. 5.3.3.3
и табл. 5.3, стадии 1—4).
2. Ресуспендируйте (НН4)25О4-осадок в растворе, содержащем: 10 мМ
трис-НС1 (pH 7,7), 0,25 М NaCl, 10 мМ MgCl2, 1 мМ ЭДТА, 10 мМ 2-мер-
капгоэтанол, 1 мМ ФМСФ и 0,1% тритона Х-100 (объемная концентра-
ция) из расчета 5 мл раствора на I л исходной культуры.
3. Диализуйте экстракт против 100 объемов того же буфера, сменив его
один раз через 3 ч.
4. Центрифугируйте экстракт для удаления осадка и нанесите на колонку,
заполненную 10 мл АФТГ-агарозы (Sigma).
5. Промойте колонку 3—5 объемами упомянутого выше буфера с тритоном
Х-100, затем 2—3 объемами того же буфера без тритона Х-100 до тех
пор, пока с колонки совсем не перестанет сходить белок.
6. Промойте колонку 0,2 М раствором бората натрия, pH 10,0.
7. Осадите элюированный белок сульфатом аммония, добавленным до ко-
нечной концентрации 50%. Ресуспендируйте осадок в подходящем буфере.
Иммунизацию лучше всего проводить в PBS.
может использоваться каждый из четырех приведенных в таб-
лице методов. Все эти хроматографические методы могут при-
меняться и для очистки больших количеств белка.
5.4. Получение и очистка поликлональных антител
При использовании методов, приведенных в разд. 5.3, мож-
но получать гибридные белки в разном виде и различных ко-
личествах. По нашим данным, для максимального расширения
области применения этих белков лучше всего при получении
антисыворотки использовать антигены, близкие по своим свой-
ствам к нативным, и инъецировать их в умеренных количест-
Рис. 5.7. Очистка гибридного белка, содержащего ^-галактозидазу. А. Профиль
элюции гибридного белка |3-галактозидаза-На1гу, хроматографированного на
сефакриле S300, как описано в разд. 5.3.3.3. Материал пика собирали по
поглощению при 280 нм. На колонку с S300 размером 2,5X50 см наносили
5 мл экстракта, полученного из 500 мл исходной культуры клеток, индуциро-
ванных ИПТГ. После пропускания примерно 40 мл (свободный объем колон-
ки) раствора собирали фракции по 2 мл. Пик гибридного белка сходит
в 9-18 фракциях. Б. Окрашенный кумасси 7,5% ДСН-ПААГ после электрофо-
реза клеточных экстрактов и фракций, соответствующих пику, полученному
на S300. Стрелки слева указывают местоположение гибридного белка р-галак-
тозидаза-Hairy {верхняя стрелка) п Р-галактозидазы (нижняя стрелка).
На дорожки геля наносили: маркерные белки; лизат — 20 мкл из 10 мл гру-
бого лизата (разд. 5.3.2, п. 2); (НН4)28О4-осадок, 20 мкл из 5 мл ресуспенди-
рованного осадка, полученного при осаждении 25% (NH4)2SO4 (разд. 5.3.3.3
и табл. 5.3); растворимая фракция — 20 мкл из 5 мл растворимой фракции
ресуспендированного осадка, полученного при осаждении 25% (NH4)2SO4
(разд. 5.3.3.3 и табл. 5.3); 9—18 — пробы по 20 мкл из фракций объемом
2 мл, собранных при элюции белка с S300.
11—1186
162
С. Кэрролл, А. Логон
Таблица 5.5. Сравнение методов очистки гибридных белков
Метод Степень чистоты „ Степень натив- __ антигена Выход ного белка Примечания
Препара- тивный ДСН-элект- рофорез в ПААГ Самое высокое со- 0,1—1 мг Низкая, анти- Легкий, быст- отношение интакт- белка на ген денатури- рый метод ный гибридный бе- гель рован лок: р-галактози- даза; продукты деградации не вы- деляются; неболь- шое загрязнение белками Е. coli
Иммуноаф- финная очистка Выделяются все С одной Низкая, но вы- Легкий, быст- белки, содержа- колонки ше, чем при об- рый метод щие ^-галактози- сходит работке ДСН; дазу; примеси бел- 1—2 мг антиген в ос- ков Е. coli отсутст- белка новном пред- вуют ставляет со- бой нераство- римый преци- питат
Гель-фильт- рация Некоторое загряз- С одной Растворимый, Более дли- нение белками колонки нативный белок тельный ме- Е. coli сходит тод, иногда 1—10 мг сложно избе- белка жать протео- лиза
Хроматогра- фия на АФТГ-ага- розе Выделяются в ос- С одной В основном Более дли- новном содержа- колонки растворимый тельный ме- щие ^-галактози- сходит белок (степень тод; низкий дазу продукты 1—5 мг денатурации за- выход бел- болыпих размеров; белка висит от pH) ков, содержа- небольшое загряз- щихся в клет- нение белками ках в малых Е. coli количествах; иногда трудно избежать про- теолиза
вах. Нельзя однозначно сказать, что лучше, использование мо-
ноклональных или поликлональных антител. И те и другие
антитела имеют свои преимущества для определенного круга
задач. Мы рекомендуем сперва получать только поликлональ-
ные антитела или поликлональные антитела вместе с монокло-
нальными в основном потому, что в первую очередь исследова-
тель должен удостовериться в том, что он получает материал,
специфически реагирующий с антигеном при постановке имму-
нологических реакций. Любой гибридный белок, способный при
его введении в организм вызывать активное образование поли-
Поликлональные антитела к фрагментам гибридных белков
163
клональных антител, может успешно использоваться при имму-
низации для получения гибридом. Ниже приводятся схемы им-
мунизации кроликов различными антигенами.
5.4.1. Схемы иммунизации
1. При использовании денатурированных ДСН антигенов,
элюируемых из геля, необходимо, чтобы гель содержал макси-
мально возможное количество белка. Это количество колеб-
лется от 0,1 до 1 мг/мл в зависимости от стабильности гибрид-
ного белка и того, насколько он был очищен до электрофореза.
Животные плохо переносят акриламид, и поэтому антиген сле-
дует вводить в минимальном объеме. Измельчите срез геля,
пропустив его несколько раз через шприц. Смешайте пять объ-
емов полного адъюванта Фрейнда (Gibco) с четырьмя объема-
ми геля с помощью двух шприцов, соединенных переходником
из нержавеющей стали. (Такой переходник можно сделать, спа-
яв вблизи оснований две стальные иглы № 18 для инъекций.)
Полностью эмульгируйте антиген так, чтобы суспензия имела
стабильную консистенцию, и сделайте инъекцию зафиксирован-
ному или находящемуся под анестезией кролику (весом 2,2—
2,7 кг) внутримышечно в несколько участков тела. Через 14
и 21 день повторите инъекции раствором антигена в неполном
адъюванте Фрейнда. На 28-й день возьмите у животного кровь
из ушной вены (~(20 мл, что не представляет опасности для
жизни животного). Кровь можно брать еженедельно до тех
пор, пока животное остается здоровым, а дополнительные вве-
дения антигена можно проводить примерно каждые шесть —
восемь недель.
2. При использовании антигенов, полученных с помощью
хроматографических методов и поэтому, по крайней мере час-
тично, растворимых, смешайте 0,2—2,0 мг интактного гибрид-
ного белка, растворенного в 1 мл физиологического раствора
или PBS с 1,25 мл полного адъюванта Фрейнда и получите го-
могенную эмульсию, как описано выше. Сделайте зафиксиро-
ванному кролику подкожные инъекции в несколько мест.
Повторите инъекции 0,2—1,0 мг антигена в неполном адъюван-
те. Фрейнда на 14-й и 21-й день и с 28-го дня начинайте брать
кровь. Для определения титра антител берите кровь еженедель-
но и повторите введение 0,2—1,0 мг антигена, когда титр анти-
тел начнет уменьшаться. После получения достаточного коли-
чества антисыворотки, пока титр антител остается достаточно
высоким, возьмите под анестезией кровь из сердца кролика.
11*
164
С. Кэрролл, А. Логон
ь. Антитела к
pi1™ ^-галактозидазе
tx__ Антитела к гетеро-
логичному фрагменту
Меспецифические
ig
не связываются с
колонкой
Рис. 5.8. Схема получения антител, специфичных по отношению к гетероло-
гичному фрагменту гибридного белка. Кроликов иммунизируют гибридным
белком, выделенным с помощью того или иного подходящего метода (навер-
ху). Для удаления основной массы антител к ^-галактозидазе антисыворотку
сначала пропускают через колонку с сефарозой с иммобилизованной р-галак-
тозидазой. Несвязавшийся на колонке материал наносят на другую колонку,
содержащую ковалентно связанный гибридный белок: иммобилизованные на
сефаразе антитела к ^-галактозидазе сшиты диметилпимелимидатом-2НС1 с
^-галактозидазным фрагментом гибридного белка. Связавшиеся антитела
элюируют и после заключительного контрольного этапа, т. е. пропускания
через колонку с иммобилизованной ^-галактозидазой (не показано), анализи-
руют их специфичность по отношению к чужеродному фрагменту гибридного
белка.
Аффинная колон-
ка с гибридным
белком (иммо-
билизованные на
сефарозе молеку-
лы анти-р-галак-
тозидазы, сшитые
с помощью ДМП с
гибридным
белком)
Элюция антител,
специфичных к гетероло-
гичному фрагменту гибрид-
ного белка
Поликлональные антитела к фрагментам гибридных белков 165
5.4.2. Получение антисыворотки и работа с ней
Во многих случаях необходимо или желательно получить
очищенные с помощью аффинных методов антитела, специфи-
ческие по отношению к определенному антигену. Ниже приво-
дятся детальные методы фракционирования антисыворотки
к гибридным белкам, позволяющие получать антитела, специ-
фичные по отношению к гетерологичному фрагменту белка.
Общая схема выделения специфических антител приведена на
рис. 5.8.
5.4.2.1. Выделение антисыворотки из крови кролика
Соберите кровь в центрифужную пробирку объемом 50 мл.
С помощью деревянного шпателя удалите со стенок пробирки
образовавшиеся сгустки и поместите палочку в сгусток, образо-
вавшийся в центре пробирки. Поместите кровь в холодильник
(температура 4 °C) на ночь. На следующий день удалите сгус-
ток, прикрепившийся к шпателю, и центрифугируйте оставшую-
ся жидкость при 13 000 g в течение 10 мин. Полученная сыво-
ротка должна быть желтого цвета. В ней либо совсем не долж-
но быть следов гемолиза, либо они должны быть сведены к ми-
нимуму. Добавьте к сыворотке 20% (весовая концентрация)
NaNs (высокотоксичное соединение!) до конечной концентра-
ции 0,02% и храните до использования в замороженном состоя-
нии при температуре —20 °C.
5.4.2.2. Выделение анти-3-галактозидазной фракции
антисыворотки
Для определения количества специфичных к гибридному
белку антител, присутствующих в сыворотке, и для получения
препарата антител, не загрязненного антителами к ^-галактози-
дазе, антисыворотку к гибридному белку сначала наносят на
аффинную колонку, содержащую иммобилизованную ^-галакто-
зидазу. Для подготовки такой колонки используют 5—7 мг очи-
щенной ^-галактозидазы E.coli (Sigma) диализованной против
PBS. Метод подробно приводится в табл. 5.6.
Антитела выделяют, как описано в табл. 5.7. Поскольку
фракция антител, специфичных к гетерологичному фрагменту
гибридного белка не связывается с колонкой, ее собирают без
значительных потерь, промывая колонку PBS. Элюируемая
фракция антител, специфичных к ^-галактозидазе, служит хо-
рошим контролем в опытах, где используются антитела к гиб-
ридным белкам, и может применяться для приготовления аф-
финных колонок для выделения гибридных белков (см. ниже).
166
С. Кэрролл, А, Логон
Таблица 5.6. Присоединение белков к CNBr-активированной сефарозе 4В
1. Все операции следует проводить в вытяжном шкафу: бромистый циан
может оказывать летальное воздействие на организм человека. Со всей
посуды и инструментов, использовавшихся для работы с CNBr, необходи-
мо удалить его следы. Для этого их нужно промыть разбавленным рас-
твором NaOH или замочить в этом растворе и оставить на ночь в вытяж-
ном шкафу, чтобы удалить летучее вещество.
2. Промойте сефарозу 4В (Pharmacia) пятью объемами охлажденной дистил-
лированной воды на воронке с грубым стеклянным фильтром. Для связы-
вания 10 мг белка или 1—2 мл белкового раствора приготовьте как ми-
нимум 1—2 мл сефарозы 4В.
3. Суспендируйте промытую сефарозу равным объемом 2,5 М калий-фосфат-
ного буфера (pH 12,2) 1> в химическом стакане при осторожном переме-
шивании и поместите в ледяную баню.
4. В отдельном сосуде и в максимально возможно закрытом вытяжном шка-
фу растворите 1 г CNBr и 1 мл ацетонитрила; это количество необходимо
для активации 10 мл геля.
5. Добавляйте в течение 2 мин по каплям раствор CNBr к гелю при осто-
рожном перемешивании. Продолжайте перемешивание еще в течение
8 мин.
6. Поместите активированную сефарозу на воронку со стеклянным фильт-
ром и тщательно промойте ее десятью объемами холодной дистиллирован-
ной воды, а затем десятью объемами холодного PBS2).
7. Возьмите из вытяжного шкафа воронку и добавьте к раствору белка ак-
тивированную сефарозу; осторожно встряхивайте в течение ночи. Раствор
белка не должен содержать триса или свободных аминогрупп, и его сле-
дует предварительно диализовать против PBS.
8. На следующий день отфильтруйте смесь с помощью воронки со стеклян-
ным фильтром и определите в фильтрате концентрацию не связавшегося с
сефарозой белка. Суспендируйте сефарозу 4В в равном объеме раствора,
содержащего 1 М этаноламин, 10 мМ трис-НС1 (pH 8,5), и оставьте на
2 ч при 4 °C для того, чтобы заблокировать оставшиеся свободными уча-
стки связывания. В конце промойте и уравновесьте связавшуюся с белком
сефарозу 4В в растворе PBS с 0,02% NaN3 и храните при 4 °C. Приготов-
ленная таким образом сефароза сохраняет стабильность более года и оста-
ется активной после многих циклов работы на ней.
]) Состав 2,5 М калий-фосфатиого буфера (pH 12,2): 353,4 г К3РО4 и 145,4 г К2НРО4
на 1 л воды.
2) Состав PBS приводится в сноске к табл. 5.2.
5.4.2.3. Выделение антител, специфичных по отношению
к гетерологичному фрагменту гибридного белка
Метод, описанный в этом разделе, — самый надежный ме-
тод выделения антител к гетерологичному фрагменту гибридно-
го белка. По нашим данным, он пригоден даже в случае наи-
более «трудных» в этом отношении гибридных белков. При
использовании этого метода можно получить 1—2 мг антител
к гетерологичному фрагменту гибридного белка за один цикл
работы. Метод позволяет преодолеть проблемы растворимости
и низкого выхода путем связывания гибридного белка непо-
средственно из грубого экстракта смолой, содержащей специ-
Поликлональные антитела к фрагментам гибридных белков 167
Таблица 5.7. Выделение антител с помощью аффинной хроматографии
1. Заполните колонку смолой с иммобилизованным антигеном до такой вы-
соты, чтобы скорость протекания составляла не более 1 мл/мин.
2. Для заполнения колонки смолу суспендируйте в PBS; промойте колонку
для удаления нековалентно связавшегося антигена. Для предотвращения
возможного загрязнения элюируемых антител свободным антигеном прово-
дите элюцию буфером с высокой ионной силой (например, содержащим
4 М гуанидин-HCl или 0,5 М уксусную кислоту, или 1 М глицин-НС1,
pH 2,8). Определяйте поглощение сходящего с колонки материала при
280 нм, используя регистрирующий спектрофотометр. Когда пик сойдет
полностью, снова уравновесьте колонку, промыв ее 3—5 объемами PBS.
3. На колонку, содержащую 5 мг ^-галактозидазы или 5 мл смолы, сшитой
с гибридным белком (табл. 5.6), нанесите 10—20 мл антисыворотки. Про-
водите элюцию PBS и промывайте колонку буфером BBS-твин* 1* до тех
пор, пока с нее не перестанет сходить белок. Уравновесьте колонку PBS.
Для обеспечения наиболее полной элюции элюируйте связавшиеся анти-
тела 4 М гуанидинхлоридом. Диализуйте собранный пик против 100 объ-
емов PBS1* как минимум три раза, диализуя раствор против каждой сме-
ны буфера не менее трех часов. Снова уравновесьте колонку 3—5 объ-
емами PBS.
4. Определите выход антител, измерив содержание белка в диализованном
растворе с помощью биуретовой реакции или измерив поглощение при
280 нм. Поглощение при 280 нм, равное 1,4, соответствует концентрации
IgG I мг/мл. Храните IgG в 0,02%-ном растворе NaN3 при —20 °C, разлив
их на аликвоты. Избегайте повторного замораживания.
*) Состав этих буферов приводится в сноске к табл. 5.2.
фический антиген, которую можно активировать для образова-
ния ковалентной сшивки с гибридным белком [6,20] (табл. 5.8).
1. Приготовьте колонку, содержащую иммобилизованные
антитела к ^-галактозидазе, смешав 5—10 мг антител к ^-га-
лактозидазе (аффинноочищенных, как описано в табл. 5.7)
с 5 мл сефарозы 4В, активированной бромистым цианом, как
описано в табл. 5.6.
2. Нанесите на колонку экстракт, содержащий гибридный
белок, как описано в табл. 5.8. Такой экстракт (см. разд. 5.3)
может быть получен в любом буфере, и нет необходимости уда-
лять из него нуклеиновые кислоты или разные белковые при-
меси. Если интересующий вас гибридный белок растворен в бу-
фере для экстракции, не содержащем денатурирующих агентов
(например, в 0,1 М калий-фосфатном буфере, pH 7,0), его мож-
но сразу наносить на колонку.
3. Промойте колонку и проведите реакцию образования сши-
вок с белком (табл. 5.8).
4. Промойте колонку элюирующим буфером и снова урав-
новесьте ее PBS, как описано в табл. 5.7. Нанесите на колон-
ку несвязавшуюся фракцию антисыворотки с колонки, содержа-
щей иммобилизованную ^-галактозидазу, промойте ее и элюи-
руйте фракцию, содержащую антитела к гибридному белку, как
168
С, Кэрролл, А, Логон
Таблица 5.8. Подготовка аффинной колонки с иммобилизованными
гибридными белками
1. Заполните колонку объемом 5 мл сефарозой 4В, к которой «пришито»
5—10 мг аффинноочищенных антител к ^-галактозидазе, как описано в
табл. 5.6. -
2. Уравновесьте колонку PBS’> при 4 °C. Нанесите на нее при 4 °C обогащен-
ный экстракт индуцированной лизогенной культуры или клона, несущего
плазмиду pL’R, содержащий не менее 3—5 мг интактного гибридного бел-
ка. Промойте колонку одним объемом PBS при 4 °C, затем, уже при
комнатной температуре, промойте ее тремя объемами BBS-твин1’, двумя
объемами 0,1 М бората натрия (pH 8,2) и двумя объемами 0,2 М триэта-
ноламина (pH 8,2) для удаления несвязавшегося белка и подготовки для
сшивания связавшегося гибридного белка с иммобилизованными антитела-
ми к ^-галактозидазе.
3. Для пришивания белка удалите смолу из колонки, суспендируя ее в де-
сяти объемах 0,2 М триэтаноламина либо 50 мМ диметилпимелимидата-2
НС1 (Pierce); pH раствора должен составлять 8,2. Перемешайте смесь,
осторожно покачивая ее в течение 45 мин при комнатной температуре.
4. После осуществления сшивки для осаждения гранул центрифугируйте
смолу в мягком режиме в течение нескольких секунд в настольной цент-
рифуге. Удалите раствор и промойте гранулы еще раз десятью объемами
20 мМ триэтаноламина (pH 8,2). Ресуспендируйте гранулы в двух объ-
емах PBS и снова заполните колонку смолой в PBS.
5. Перед проведением аффинного выделения антител к гибридному белку
предварительно промойте колонку, как описано в табл. 5.7.
6. Колонку следует хранить при 4 °C в PBS1', содержащем 0,02% NaN3.
Смола остается стабильной! более года и сохраняет способность к связы-
ванию антител после многих циклов работы.
’) Состав этих буферов приведен в сноске к табл. 5.2.
это описано в табл. 5.7. Диализуйте раствор с антителами про-
тив нескольких смен PBS и определите содержание в нем
белка.
5. Регистрируя оптическую плотность белкового материа-
ла с помощью соединенного с колонкой высокочувствительного
спектрофотометра, нанесите еще раз фракцию диализованных
антител, полученных на стадии 4, на аффинную колонку с им-
мобилизованной ^-галактозидазой, уравновешенную PBS для
удаления возможных примесей антител к ^-галактозидазе. Те-
перь фракция, сходящая с колонки, должна содержать только
антитела к чужеродному фрагменту гибридного белка.
5.4.2.Д. Определение специфичности антител методом
вестерн-блот-анализа
Для определения специфичности антител к чужеродному
фрагменту гибридного белка на каждой стадии их очистки важ-
но отличать антитела к ^-галактозидазному компоненту гибрид-
ного белка от антител к гетерологичному его фрагменту. Са-
мый простой способ для этого — определение активности анти-
Поликлональные антитела к фрагментам гибридных белков
169
тел с помощью вестерн-блот-анализа [21], когда на соседние
дорожки геля наносят гибридный белок и р-галактозидазу и
можно сравнивать эффективность взаимодействия с ними ан-
тител.
1. Приготовьте лизат индуцированных и неиндуцировапных
клеток, содержащих экспрессирующий вектор, как описано
в разд. 5.2.
2. После ДСН-электрофореза проведите блот-анализ образ-
цов, как описано в табл. 5.2.
3. Инкубируйте фильтр с разными фракциями антител, как
описано в табл. 5.2.
4. Препарат антител, очищенных стандартным способом, как:
описано в этом разделе, не будет связываться с ^-галактозида-
зой или антигенами Е. coli, а будет взаимодействовать со всеми
формами гибридного белка, содержащими антигенные детерми-
нанты гетерологичного фрагмента (рис. 5.4, крайняя правая
дорожка).
Благодарности
Эксперименты с гибридными белками проводились в лабо-
ратории доктора Мэтью Скотта. Мы благодарны ему за под-
держку. Для проведения некоторых иммунологических иссле-
дований мы использовали методики, разработанные в лаборато-
рии доктора Б. Дэвида Столлара (Tufts University, Boston, МА),
которые были освоены С. Б. Кэрроллом, стажировавшимся там.
Мы благодарны доктору Джону Тэмкуну за критические заме-
чания, сделанные при прочтении этой главы и за плодотворное
обсуждение некоторых аспектов работы с гибридными белками.
Мы благодарны Кэти Инуайте за подготовку рукописи. Эта
работа обеспечивалась грантом HD18163, выделенным Нацио-
нальными институтами здоровья, и грантом NP6502, выделен-
ным Метью П. Скотту Американским онкологическим общест-
вом (ASC). Работа А. Логона обеспечивалась дотацией, предо-
ставляемой Американским онкологическим обществом; работа
С. Б. Кэрролла обеспечивалась дотацией, предоставляемой Ра-
ковым фондом Дамон Раньон-Уолтер Винчелл DRG-659, и до-
тацией GM-09756, предоставляемой Национальными института-
ми здоровья.
Литература
1. Collins Ж Е. et al. (1986). Nature, 323, 259.
2. Germino J., Gray J. G., Charboneau H., Vanaman T„ Bastia D. (1983).
Proc. Natl. Acad. Sci. USA, 80. 6848
3. White R. A. H„ Wilcox M. (1984). Cell, 39 163
4. Beachy P. A., Helfand S. L„ Hogness D.’s. (1985). Nature, 313, 545.
170
С. Кэрролл, А. Логон
5. DiNardo S., Kuner J. М., Theis J., O’Farrell P. H. (1985). Cell, 43, 59.
6. Carroll S. B„ Scott M. P. (1985). Cell, 43, 47.
7. Paul J. I., Schwarzbauer J. E., Tamkun J. IF., Hynes R. O. (1986). J. Biol.
Chem, 261, 12258.
8. Young R. A., Davis R. W. (1983). Proc. Natl. Acad. Sci. USA, 80, 1194.
9. Young R. A., Davis R. IF. (1983). Science, 222, 778.
10. Schwarzbauer J. E., Tamkun J. IF, Lemischka I. R., Hynes R. 0. (1983). Cell,
35, 421.
11. Rather U„ Muller-Hill B. (1983). EMBO J., 2, 1791.
12. Huynh T. V., Young R. A., Davis R. IF. (1984). In: DNA Cloning. A Practi-
cal Approach, Glover D. (ed.), IRL Press, Oxford, Vol. 1, p. 49. [Имеется
перевод: Клонирование ДНК. Методы. Книга I. Под ред. Д. Гловера.—
М.: Мир, 1988.]
13. Laughon A., Boulet А. М„ Bermingham J. R., Layman R. A., Scott М. Р.
(1986). Mol. Cell. Biol., 6, 4676.
14. Maniatis T., Fritsch E. F., Sambrook J. (1982). In: Molecular Cloning, A La-
boratory Manual, Cold Spring Harbor Laboratory Press, Cold Spring Harbor,
NY. [Маниатис T, Фрич Э, Сэмбрук Дж. Методы генетической инженерии.
Молекулярное клонирование. — М.: Мир, 1984.]
15. Morrison D. А. (1979). In: Methods in Enzymology, Wu R. (ed.), Academic
Press, NY, Vol. 68, p. 326.
16. Tamkun J. IF., DeSimone D. IF., Fonda D., Patel R. S., Buck C„ Hor-
withz A. F„ Hynes R. 0. (1986). Cell, 46, 271.
17. Fowler A. V., Zabin I. (1983). J. Biol. Chem., 258, 1454.
18. Steers E„ Cuatrecasas P., Pollard H. P. (1971). J. Biol. Chem., 246, 196.
19. Struck D. K., Maratea D„ Young R. (1985). J. Mol. Appl. Gen., 3, 18.
20. Schneider C„ Newman R. A., Sutherland D. R. Asser V., Greaves M. F.
(1982). J. Biol. Chem, 257, 10766.
21. Towbin H„ Staehelin T„ Gordon J. (1979). Proc. Natl. Acad. Sci. USA, 76,
4350.
ГЛАВА 6
ПОЛУЧЕНИЕ МОНОКЛОНАЛЬНЫХ АНТИТЕЛ К
ГИБРИДНЫМ БЕЛКАМ; ПРОДУЦИРУЕМЫМ
В КЛЕТКАХ Е. COLI
Сара Е. Моул и Дэвид П. Лейн
(Saral Е. Mole, David Р. Lane)
6.1. Введение
Получение моноклональных антител к полипептидному про-
дукту генетически охарактеризованной открытой рамки считы-
вания (ОРС) открывает широкие возможности для иммунохи-
мического и функционального анализа такого продукта. Этот
метод сложнее, чем описанный в предыдущей главе метод
получения поликлональных антител к продукту ОРС, и должен
рассматриваться скорее как дополнительный, а не альтернатив-
ный подход. Общая схема получения и использования моно-
клональных антител приведена на рис. 6.1. Мы настоятельно
рекомендуем каждому исследователю получать и поликлональ-
ные, и моноклональные антитела. Основные преимущества мо-
ноклональных антител, а именно возможность получения их
в неограниченном масштабе и высокая специфичность, очень
существенны в том случае, когда возникает необходимость
в углубленном исследовании продукта ОРС, а не просто в его
выявлении. Наличие моноклональных антител облегчает коли-
чественный анализ продукта ОРС и часто дает возможность
быстро получать его в биологически активной форме. Специ-
фичность моноклональных антител также может быть использо-
вана для тонкого картирования их участков связывания (эпито-
пы) на молекуле полипептида — продукта ОРС; эта информа-
ция дополняется данными о способности антител усиливать или
подавлять биологическую активность продукта ОРС.
Для наработки моноклональных антител надо индуцировать
сильный иммунный ответ. Для этого необходимы достаточно
большие количества иммуногена. Выделение же больших коли
честв нативного белка не всегда представляется возможным.
Однако клонирование гена в подходящем бактериальном экс-
прессирующем векторе способно обеспечить повышенное обра-
зование кодируемого белка, делает возможным использование
очищенного белка в качестве иммуногена. Такая система позво-
ляет клонировать определенные фрагменты меньшего размера,
входящие в состав гена известного белка, или, наконец, откры-
171
172
С. Моул, Д. Лейн
Идентифи-
кация
. ОРС
Клониро-
вание
в pUR
Очистка
белка
Инъециро-
вание
кролика
Инъециро-
вание
мыши
Использование сыво-
ротки для выявления
полноразмерных
продуктов ОРС в
клетках или ткани
Скрининг на
наличие анти-
тел к белки ,
кодируемому
^^СлйяИйе'У$$$$^
идентификация и'-
клонирование
Скрининг гибрмдомных
клеток на образование
антител к белку,
кодируемому
ОРС
Иммунопреци-
питация
и блоттинг
; содержащих
антитела к белкам^
^кодируемым ОРС;'
и выделение
Иммуно-
аффинная
очистка
Иммуноцитохимия
Картирование
эпитопа
Иммунологический
анализ
Клонирование
вставки pUR
в рЗЕМСаЦ.
Экспрессия в
бактериальных и
эукариотических
клетках
Функциональный
анализ ОРС
Рис. 6.1. Схема, демонстрирующая этапы получения моноклональных антител
к гибридным белкам, продуцируемым в клетках Е. coli, и области их возмож-
ного применения.
тые рамки считывания, кодирующие неизвестный белок, с целью
получения моноклональных антител к определенным участкам
молекулы такого продукта, а также тонкого картирования
эпитопов, распознаваемых уже известными антителами.
Бактериальные экспрессирующие векторы, особенно удобные
для проведения такой работы, включают семейство плазмид
pUR [1], несущих у З’-конца гена lacZ набор дискретных сай-
Моноклональные антитела к гибридным белкам из Е. coli 173
тов рестрикции (см. гл. 5 и рис. 6.2). Такие сайты имеются
в каждой из трех возможных р&мок считывания, что позволяет
экспрессироваться любому рестрикционному фрагменту, содер-
жащему открытую рамку считывания, в составе гибридного
белка, включающего ^-галактозидазу. Это дает определенные-
преимущества, поскольку всего несколько бактериальных бел-
ков превышают по размеру ^-галактозидазу, а экспрессию гена
lacZ можно индуцировать изопропил — {TD-тиогалактозидазой
(ИПТГ), что обеспечивает обильный синтез больших количеств
гибридного белка, облегчая тем самым его идентификацию и
выделение с помощью ДСН-электрофореза в полиакриламид-
ном геле.
Трудности, с которыми можно столкнуться при наработке
моноклональных антител, связаны с получением достаточного
количества иммуногена и последующим скринингом сыворотки
для выявления нужной иммунологической активности. Исполь-
зование гиперэкспрессии гибридных белков может облегчить
преодоление этих трудностей, поскольку дает возможность вы-
делять большие количества белкового продукта с помощью
электрофореза или хроматографии. При скрининге сыворотки
и полученных затем гибридных клонов можно применять метод
радиоиммунного анализа (РИА, RTA) или метод твердофазно-
го иммуноферментного анализа (ИФА, ELISA).
Использование гибридных белков для получения монокло-
нальных антител имеет особое преимущество: участки молекулы
нативного белка, обладающие сильной или слабой иммуноген-
ностью, могут изменить свои иммуногенные свойства при экс-
прессии в составе большого гибридного белка. Это особенно
важно при получении антител к иммунологически неактивным
(«молчащим») участкам молекулы белка. В случае большого
Т-аптигена вируса SV-40 получено много разных моноклональ-
ных антител, узнающих антигенные детерминанты на N- и
С-концах нативного белка, но всего несколько видов антител,
узнающих структуры, расположенные в центре молекулы. Мы
успешно использовали этот подход для получения антител
к такому иммунологически неактивному участку большого Т-ан-
тигена.
6.2. Клонирование и экспрессия фрагментов гена
Большая часть методов, приводимых в’этом разделе, деталь-
но описана в другой работе [2]. Новые или сильно отличающие-
ся методы, используемые нами, приводятся в табл. 6.1—6.3.
Выбор в качестве экспрессирующих векторов плазмид pUR
имеет определенное преимущество, связанное с наличием у них
сайтов для различных рестриктаз, которые могут использовать-
174 С, Моул, Д. Лейн
Таблица 6.1. Скрининг колоний
1. Нанесите колонии на размеченный нитроцеллюлозный фильтр, наложен-
ный на чашку с L-агаром, содержащим 100 мкг/мл ампициллина (Sigma
Chemical Company) и 500 мкМ ИПТГ, (IPTG, Sigma Chemical Company),
а также на чашку — дубликат исходной чашки. Если это возможно, по-
ставьте негативный и позитивный контроли. Выращивайте колонии в тече-
ние ночи при 37 °C.
2. Лизируйте бактерии, инкубируя нитроцеллюлозный фильтр в атмосфере,
насыщенной парами хлороформа, в течение 30 мин.
3. Проведите насыщение фильтра, инкубируя его в течение 30 мин при ком-
натной температуре в 25%-ном растворе сухих белков молока, разбавлен-
ном PBS1’ в соотношении 1:2.
4. Промойте фильтр в PBS и инкубируйте его с нужными антителами2’,
разбавленными, если это необходимо, PBS, содержащим 1 % бычьего сыво-
роточного альбумина (БСА; BSA, Sigma Chemical Company) и 5%-ную
сыворотку эмбриона коровы (СЭК), в течение по меньшей мере 1 ч при ком-
натной температуре.
5. Промойте фильтр PBS, а затем 2 раза по 15 мин PBS, содержащим 1%
NP-40 (Sigma Chemical Company) и еще 2 раза по 15 мин PBS.
6. Инкубируйте фильтр в растворе, содержащем предварительно адсорбиро-
ванные антитела3’ второго порядка, связанные с пероксидазой хрена, не
менее 1 ч при комнатной температуре.
7. Промойте фильтр PBS, затем 2 раза по 15 мин PBS, содержащим 1%
NP-40, и 2 раза по 15 мин PBS.
8. Непосредственно перед употреблением разбавьте 50 мг 4-хлор-1-нафтола
(Aldrich), растворенного в 1 мл этанола в смеси PBS в соотношении
1 : 100, и профильтруйте раствор. Добавьте 30%-ный пероксид водорода
при разбавлении 1 : 5000 и используйте его для окраски фильтра. Если
колонии дают положительную реакцию, промойте фильтр PBS и высуши-
те. Храните фильтр в темноте.
*) Состав PBS: 150 мМ NaCl, ЗмМ. КС1, 8мМ NajHPO-i, 1,5 мМ КН2РО4.
2) Для скрининга могут использоваться моноклональные антитела или поликлональ-
ная антисыворотка,
3) Желательно провести предварительную адсорбцию антител второго порядка экст-
рактом лизированных клеток Е. coli (табл. 6.2). Антитела второго порядка должны об-
ладать специфичностью по отношению к животному, из крови которого выделяли анти-
тела первого порядка, использовавшиеся при скрининге, например антимышиные имму-
ноглобулины кролика, конъюгированные с пероксидазой хрена (DAKO), козьи анти-
тела к иммуноглобулинам кролика и т. д.
ся для клонирования фрагментов генов. Для того чтобы иметь
возможность контролировать индуцибельную экспрессию гена
lacZ, обусловленную активацией плазмидного гена, при исполь-
зовании в качестве векторов плазмид pUR необходимо наличие
бактериальных штаммов, не продуцирующих собственную
^-галактозидазу. Для этих целей успешно использовались два
штамма lacZ: ВМН 71-18 [3j; (lac pro) Athi supEF'
lac IQZ_AM15pro+ и F'llrecA [4]; (lac pro)Athi rifA str A rec A
F'lac IQIZ proF Основное различие между ними заключается
в гесЛ-фенотипе второго штамма, который позволяет свести
к минимуму возможность реорганизации рекомбинантных кон-
струкций. Однако, по нашим данным, гибридный белок более
активно экспрессируется при использовании в качестве штам-
Моноклональные антителу к гибридным белкам из E.coli 175
Таблица 6.2. Предварительная адсорбция на экстракте антител второго
порядка, конъюгированных с пероксидазой хрена.
1. Вырастите 2 мл ночной культуры нужного штамма Е. соЦ (например,
штамма ВМН 71-18, содержащего плазмиду* 1 pUR290) в условиях индуци-
рованной ИПТГ экспрессии lacZ.
2. Перенесите 1 мл культуры в пробирку для микроцентрифуги объемом
\1,5 мл и центрифугируйте в течение 30 с.
3. Суспендируйте осадок в 50 мкл 5%-ного ДСН и прокипятите в течение
5 мин для лизиса клеток.
4. Охладите суспензию и разбавьте ее в соотношении 1 :50 PBS, содержа-
щем 1% БСА, 5% СЭК. Храните при —20 °.
5. Добавьте к экстракту лизированных клеток разбавленные в 100 раз ан-
титела второго порядка, конъюгированные с пероксидазой хрена. Иинку-
бируйте в течение ночи при 4 °C.
6. Для осаждения образовавшихся иммунных комплексов смесь можно от-
центрифугировать, а надосадочную фракцию непосредственно исполь-
зовать.
ма-хозяина ВМН 71-18. Поэтому во избежание потери экспрес-
сируемой конструкции из-за возможных перестроек желательно
как можно скорее получить препарат рекомбинированной плаз-
миды и хранить ее в виде очищенной ДНК.
6.2.1. Конструирование вектора,
экспрессирующего гибридный белок
Клонирование ОРС предполагает следующие операции:
1. Встройте фрагмент гена или ОРС в рестрикционный сайт
в подходящей рамке считывания (если таковая известна) pUR.
2. Проведите трансформацию компетентных (lacZ) клеток
Е. coll.
3. Проведите скрининг полученных колоний для выявления
трансформантов. Это можно осуществить двумя способами:
а) Проведите скрининг на наличие трансформантов, ис-
пользуя в качестве зонда тот же радиоактивно мечен-
ный фрагмент ДНК, который был использован для
клонирования. В соответствующих условиях встроен-
ный фрагмент будет гибридизоваться с радиоактив-
ным зондом, и затем с помощью радиоавтографии
можно будет выявить колонии, дающие положитель-
ный ответ. Этот метод позволяет обнаружить наличие
одной или большего числа копий вставки, ориентиро-
ванных любым образом.
б) Проведите скрининг на наличие антигенных детерми-
нант, кодируемых клонируемым фрагментом. Этот
метод позволяет обнаруживать клоны, содержащие
вставку в рамке считывания и в правильной ориента-
ции, так что гибридный белок может эффективно
176
С. Моул, Д/ Лени
Таблица 6.3. Подготовка проб для ДСН-электрофореза
в полиакриламидном геле
1. Вырастите 2 мл ночной культуры клона, дающего положительную реак-
цию* на наличие гибридного белка, в L-среде, содержащей 100 мкг/мл ам-
пициллина и 500 мкМ. ИПТГ. Или, если имеется опасность деградации
белка, добавьте ИПТГ, когда культура находится в середине логарифми-
ческой фазы роста (OD6oo=0,5), и выращивайте ее в течение 1—2 ч.
2. Охладите культуру во льду и перенесите 1,5 мл в микроцентрифужную
пробирку объемом 1,5 мл.
3. Центрифугируйте смесь в течение 30 с и отберите над осадочную фракцию.
4. Промойте осадок PBS и ресуспендируйте в 300 мкл 1Хбуфера для нане-
сения. '
5. Прокипятите в течение 5 мин перед нанесением проб объемом 5—25 мкл
на гель.
экспрессироваться. Методика с использованием моно-
клональных антител или поликлональной антисыворот-
ки к полипептиду, кодируемому клонированным фраг-
гентом гена, приведена в табл. 6.1.
4. Исследуйте бактериальные экстракты, полученные из по-
ложительных клонов, методом ДСН-электрофореза в ПААГ
(предпочтительно 7,5—8,5%) на наличие в них гибридного бел-
ка. Экспрессия может быть индуцирована с помощью ИПТГ
(табл. 6.3).
5. Определите количество вставок, присутствующих в кло-
нах. Это можно осуществить с помощью рестриктазы EcoRI,
поскольку при расщеплении EeoRI-сайтов (фланкирующих по-
лилинкер со вставкой) образуется фрагмент («EcoRI-кассета»),
по размеру превышающий клонированный фрагмент на 100 ос-
нований при наличии одной копии вставки и примерно в два
раза больший при наличии двойной вставки. Если EcoRI-сайты
имеются внутри клонированного фрагмента, то для определе-
ния числа вставок требуется провести частичный гидролиз
EcoRI.
6.2.1.1. Пример
Селекцию клонов, полученных при клонировании Hindlll D-
и Hind III A-фрагментов вируса SV-40 на экспрессию lacZ, про-
водили методом ДСН-электрофореза в ПААГ. Это показано на
рис. 6.2.
Наличие большого числа копий экспрессирующего вектора
в сочетании с индуцибельной экспрессией гена lacZ обеспечива-
ет синтез большого количества гибридного белка. С учетом
большой молекулярной массы данных белков это делает не-
сложным их выявление при гель-электрофорезе. Было установ-
лено, что в 1 мл индуцированной под действием ИПТГ ночной
Моноклональные антитела к'гибридным белкам из Е. coli 177
Рис. 6.2. Окрашенные кумасси полиакриламидные гели после ДСН-электро-
фореза, демонстрирующие наличие экспрессии гибридного белка, содержаще-
го ^-галактозидазу. А — бактериальный штамм-хозяин ВМН 71-18. Дорож-
ка М — маркеры молекулярной массы (Мг); дорожка 1—неиндуцированный
штамм-хозяин; дорожка 2 — индуцированный штамм-хозяин; дорожка 3 —
неиндуцированный штамм-хозяин, содержащий плазмиду pUR292; дорож-
ка 4—индуцированный штамм-хозяин, содержащий pUR292; дорожка 5 —
индуцированный штамм-хозяин, содержащий pUR292 со встроенным Zfindlll D-
фрагментом SV-40 (гибридный белок включает ^-галактозидазу и фрагмент
большого Т-антигена; аминокислоты 271—448; дорожка 6—индуцированный
штамм-хозяин, содержащий плазмиду pUR290 со встроенным Hindlll D-фраг-
ментом SV-40. Б — бактериальный штамм-хозяин F’llrecA. М — маркеры мо-
лекулярной массы (Мг); дорожка 1—неиндуцированный штамм-хозяин; до-
рожка 2 — индуцированный штамм-хозяин; дорожка 3 — неиндуцированный
штамм-хозяин, содержащий плазмиду pUR290; дорожка 4 — индуцированный
штамм-хозяин, содержащий pUR29O; дорожка 5 — индуцированный штамм-
хозяин, содержащий плазмиду pUR290 со встроенным ffindlll А-фрагментом
SV-40 (гибридный белок включает ^-галактозидазу и фрагмент большого
Т-антигена; аминокислоты 448—708); Дорожка 6 — индуцированный штамм-
хозяин, содержащий плазмиду pUR291 со встроенным ffindlll А-фрагментом
SV-40 (гибридный белок Р-галактозидаза-VPl); дорожка 7 — штамм-хозяин,
содержащий плазмиду pUR292 с встроенным ffindlll А-фрагментом SV40. (Ри-
сунок взят из работы [9]; приводится с любезного разрешения авторов.)
12—1186
178
С. Моул, Д. Лейн
культуры, где экспрессировался встроенный Z/indlll D-фраг-
мент SV-40, содержалось примерно 40 мкг гибридного
белка.
//шсИП D-фрагмент SV-40 кодирует последовательность
аминокислот 271—447 большого Т-антигена и в плазмиде
pUR292 находится в одной рамке считывания с lacZ
(figal. T.HD). ЯогсИН A-фрагмент в одной ориентации кодирует
последовательность аминокислот 447—708 большого Т-антигена;
в этой ориентации он клонирован в pUR290 в одной рамке
с lacZ (pga/.T.HA). В противоположной ориентации этот фраг-
мент кодирует последовательность аминокислот 69—362 VP1
и клонирован в одной рамке с lacZ в плазмиде pUR292
(flga/.VPl.HA).
Из результатов ДСН-электрофореза в ПААГ можно видеть,
что гибридные белки fJGal.T.HA и {3Gal.VPl.HA, по-видимому,
частично деградировали, в то время как гибридный белок
pGal.T.HD, судя по всему, совершенно стабилен. При индукции
ИПТГ в течение меньшего периода времени не наблюдалось
изменений в степени деградированности pGal.T.HA. Из этого
следует, что некоторые гибридные белки, кодируемые клониро-
ванными фрагментами и продуцируемые в бактериальной клет-
ке-хозяине, непосредственно после синтеза подвергаются про-
теолизу.
Вестерн-блот-анализ гибридного белка pGal.T.HA, при кото-
ром в качестве зонда использовались моноклональные антитела
к большому Т-антигену, показал, что гибридный белок подвер-
гался деградации по всей длине. Например, антитело РАЬ423
(рис. 6.3), узнающее эпитоп, расположенный в С-концевом
участке большого Т-антигена, связывается только с полосой на
электрофореграмме, соответствующей белку с самой большой
молекулярной массой, т. е. полноразмерным гибридным белком.
Антитело РАЬ204, узнающее эпитоп, занимающий участок меж-
ду аминокислотами 453 и 469 [5], взаимодействует с разными
полосами, которые соответствуют как полноразмерным белко-
вым продуктам, так и полипептидам с меньшей молекулярной
массой, чем (3-галактозидаза.
Гибридные белки различаются по своей растворимости так
же, как и по стабильности. Было исследовано шесть гибридных
белков, кодируемых клонированными в pUR-векторах рекомби-
нантными генами, содержащими разные фрагменты большого
Т-антигена вируса SV-40. Два из этих шести белков в основном
обнаруживались в надосадочной фракции экстракта лизирован-
ных клеток, три — преимущественно в нерастворимой фракции
экстракта и один — в одинаковой мере присутствовал в обеих
фракциях.
1 2 3 4 f 2 3
< 'сО-к /'.у
Рис, 6.3. Вестерн-блот-анализ гибридных белков, включающих ^-галактозидазу
и фрагмент большого Т-антигена. Дорожка 1—штамм ВМН 71-18 Е. coli,
содержащий pUR292; дорожка 2 — штамм F’llrecA Е. coli, содержащий гиб-
ридный белок р-галактозидаза—фрагмент большого Т-антигена (аминокисло-
ты 271—448); дорожка 3 — штамм F’llrecA Е. coli, содержащий гибридный
белок ^-галактозидаза—фрагмент большого Т-антигена (аминокислоты 448—
708); дорожка 4 — экстракт клеток SVA31E7, содержащий большой Т-анти-
ген. А—при блоттинге использовались антитела РАЬ204, связывающиеся с
участком большого Т-антигена между аминокислотами 453 и 469. Б — при
блоттинге использовались антитела РАЬ423, связывающиеся с С-конпевой об-
ластью большого Т-антигена. Для визуализации связывания антител с ан-
тигеном использовалась пероксидазная система иммунодетекции (описанная
в табл. 6.8). (Рисунок взят из работы [9]; приводится с любезного разре-
шения авторов.)
12*
180
С, Моул, Д. Лейн
6.3. Получение моноклональных антител к гибридным белкам
Линии клеток, продуцирующих моноклональные антитела,
обычно получают от грызунов, например мышей и крыс, даю-
щих хороший иммунный ответ на иммуноген. Готовят суспензию
клеток селезенки этих животных и вызывают их слияние с мие-
ломными клетками. Полученные в результате слияния клетки
обладают свойствами и тех, и других родительских клеток, со-
четая в себе способность к неограниченному размножению, свой-
ственную миеломным родительским клеткам, со способностью
продуцировать специфические антитела, присущей В-лимфоци-
там селезенки. Выделение клона клеток, продуцирующих одно
антитело, позволяет нарабатывать эти моноклональные антите-
ла в больших количествах.
Клонирование фрагментов генома, содержащих ОРС, в экс-
прессирующих векторах позволяет нарабатывать для иммуниза-
ции большие количества чистого белка. Это делает получение
моноклональных антител к белкам, кодируемым последователь-
ностями ОРС, стратегически простой, хотя и в некоторой степе-
ни трудоемкой задачей. Основные требования при подобной ра-
боте— наличие хорошей культуры клеток и использование вы-
сокочувствительного метода анализа, позволяющего отличать
антитела к продуктам ОРС от антител, узнающих векторные
фрагменты гибридного белка. Отлаживание такого метода—•
основное «узкое место» рассматриваемого подхода. После полу-
чения специфических моноклональных антител к продукту, ко-
дируемому встроенным фрагментом, их можно использовать для
выявления, количественного анализа и очистки белкового про-
дукта ОРС.
6.3.1. Получение антигена
Выделение гибридных белков, содержащих ^-галактозида-
зу, было детально описано в предыдущей главе. Для иммуниза-
ции мышей требуются относительно небольшие количества ан-
тигена, поэтому его можно наработать любым методом. Хотя
белковый материал, полученный в результате препаративного
ДСН-электрофореза в ПААГ, не всегда обладает такими имму-
ногенными свойствами, которые присущи большинству нативных
препаратов, он имеет то преимущество, что вызываемый им им-
мунный ответ обусловлен устойчивыми к денатурации эпитопа-
ми. Используемый нами метод выделения нерастворимых гиб-
ридных белков приведен в табл. 6.4. Моноклональные антитела
к таким эпитопам часто удается с успехом использовать в по-
следующем детальном иммунохимическом анализе белковых
антигенов. Наличие абсолютно чистого иммуногена не требует-
Моноклональные антитела к гибридным белкам из Е. coli 181
Таблица 6.4. Выделение гибридных белков с помощью ДСН-электрофореза
в ПААТ
А. Получение экстракта
Этот метод предназначен для выделения нерастворимых белков, находя-
щихся в осадочной фракции клеточного лизата.
1. Вырастите 150 мл ночной культуры, продуцирующей гибридный белок,
в L-среде, содержащей 100 мкг/мл ампициллина и 500 мкМ ИПТГ.
2. Осадите клетки центрифугированием при 2500 об/мин в течение 5 мин
при 4 °C в роторе GS-3 Sorvall и ресуспендируйте осадок клеток в 10 мл
раствора, содержащего 10 мМ трис-НС1 (pH 8,0), 2 мМ ЭДТА, 50 мМ
NaCl и 0,25 мг/мл лизоцима.
3. Инкубируйте смесь при комнатной температуре в течение 15 мин, затем
добавьте тритон Х-100 до конечной концентрации 0,2% (объемная кон-
центрация), MgCl2 до концентрации 10 мМ и ДНКазу до концентрации
1 мкг/мл. Инкубируйте в течение 30 мин при комнатной температуре.
4. Центрифугируйте смесь при 5000 об/мин в течение 5 мин при 4 °C в рото-
ре SS-34 Sorvall и суспендируйте осадок обломков клеток, содержащий
гибридный белок, в 30 мл электрофоретического буфера для нанесения.
Храните при —20 °C.
В. Препаративный гель-электрофорез
1. Проведите электрофорез в ПААГ в присутствии ДСН, используя гель с
одной большой препаративной дорожкой.
2. Отрежьте узкую вертикальную полоску геля и окрасьте ее кумасси.
3. Из оставшегося геля вырежьте область, соответствующую гибридному
белку.
4. Проведите электроэлюцию белка из геля в буфер для ДСН-электрофореза.
5. Разлейте раствор на аликвоты и храните при —20 °C.
6. Концентрацию белка можно определить путем сравнения с результатами
электрофореза известных количеств чистой р-галактозидазы в ПААГ в
присутствии ДСН.
ся, поскольку будут отбираться моноклональные антитела толь-
ко к эпитопам, свойственным продуктам клонированной встав-
ки. Выделение растворимых гибридных белков детально рас-
сматривается в предыдущей главе.
6.3.2. Иммунизация животных
Цель первоначальной иммунизации — получение иммунного
ответа, выражающегося в активном синтезе антител к белку,
кодируемому вставкой, и использование поликлональной анти-
сыворотки для проведения скрининга на наличие антител, спе-
цифических к этому белку. Как правило, для иммунизации ис-
пользуют 3—6-мес самок мышей BALB/c. При двух первых
инъекциях антиген вводят в полном адъюванте Фрейнда, а при
последующих инъекциях — в неполном, за исключением послед-
ней инъекции перед взятием селезенки, которую делают без
адъюванта (см. ниже). Все инъекции могут быть внутрибрю-
шинными. Мышей следует иммунизировать группами по 4—
182
С. Моул, Д. Лейн
Таблица 6.5. Схема иммунизации
День1)
Количество
инъекцируе-
мого анти-
гена, мкг
Адъювант2)
Исследование крови
1 — — Предварительное иссле-
3 50 Полный адъювант Фрейнда дование крови
24 50 Полный адъювант Фрейнда —
34 — — Исследование крови
35 50 Неполный адъювант Фрейнда —
42 — — Исследование крови
43 50 Неполный адъювант Фрейнда —
50 50 Неполный адъювант Фрейнда
57 — — Исследование крови
’) При необходимости иммунизацию можно продолжить.
2) При всех инъекциях вводят 100 мкл белка в PBS (концентрация белка
500 мкг/мл) и 100 мкл адъюванта. Смешайте адъювант с раствором белка с помощью
двух шприцов, соединенных переходником. Вводите антиген внутрибрюшинно.
6 животных, чтобы обеспечить наличие резервных доноров
В-лимфоцитов на случай, если слияние пройдет неудачно. Удоб-
ная схема иммунизации приведена в табл. 6.5. Тогда как нали-
чие ответа на введение ^-галактозидазы обычно можно выявить
после двух инъекций, антитела к белку, кодируемому вставкой,
могут появиться существенно позже. Поэтому, прежде чем де-
лать вывод о том, что иммуноген не вызывает иммунного отве-
та, имеет смысл провести как минимум семь инъекций.
6.3.3. Выявление антител к белку,
кодируемому клонированным фрагментом ДНК
Очень важно установить, имеются ли в сыворотке иммуни-
зированной мыши антитела к белкам, кодируемым вставочными
последовательностями. Для этого используют ряд подходов в
зависимости от того, насколько легко получить полноразмер-
ный нативный продукт ОРС. Если известно, что ОРС экспрес-
сируется в культивируемых клетках определенной линии или
что ее продукт в достаточном количестве присутствует в опре-
деленной ткани, тогда можно определить активность сыворотки
в отношении данных клеток или тканей с помощью иммуноци-
тохимических методов, иммунопреципитации или вестерн-блот-
анализа. По нашим данным, при работе с ОРС млекопитаю-
Моноклональные антитела к гибридным белкам из Е. coli
183
щих и вирусов во всех трех перечисленных методах можно
использовать малоочищенную сыворотку, содержащую поликло-
нальные антитела, не сталкиваясь с проблемами возникновения
значительного фона. Контролями служат сыворотка неиммуни-
зированной мыши и антиф-галактозидазная сыворотка иммуни-
зированного животного. Если об экспрессии ОРС ничего не .из-
вестно, тогда следует отдать предпочтение другой стратегии,
основанной на информации о нуклеотидной последовательности
ОРС. Один из подходов, детально описанный в предыдущей
главе, состоит в следующем; избирательно удаляют антитела
к fj-галактозидазе из сыворотки с помощью иммуноадсорбции
па колонке с иммобилизованной fj-галактозидазой, а затем
с помощью вестерн-блот-анализа определяют способность не-
связавшейся фракции взаимодействовать с гибридным белком,
но не с ^-галактозидазой. Другой подход, описанный в разд. 6.5,
состоит в субклонировании ОРС в другом векторе в составе
другого гибридного белка. Взаимодействие антисыворотки
с продуктом нового рекомбинантного гена в экспериментах по
прямому скринингу колоний или при вестерп-блот-анализе под-
твердит наличие антител, специфичных по отношению к про-
дукту клонированного фрагмента. Однако следует иметь в ви-
ду, что если использовать методику, описанную в разд. 6.5,
16 С-концевых аминокислот ^-галактозидазы переносятся на
CAT-вектор в составе ЕсоШ-«кассеты». Следовательно, необхо-
димо ставить тесты с той же самой контрольной сывороткой
и, кроме того, любую сыворотку следует проверять на способ-
ность взаимодействовать с самим новым вектором (как содер-
жащим, так и несодержащим pUR-полилинкер). Условие, абсо-
лютно необходимое для перехода к следующей стадии рабо-
ты, — получение достоверного специфического иммунного отве-
та на продукт клонированного фрагмента, по крайней мере
с помощью одного из перечисленных методов.
6.3.3Л. Примеры
На рис. 6.4 показана иммунопреципитация экстрактов инфи-
цированных вирусом SV-40 клеток CV-1 моноклональными ан-
тителами РАЬ204 (А) и мышиной антисывороткой к гибридно-
му белку (рис. 6.4, Б). И те, и другие антитела дают специфи-
ческую иммунопреципитацию с большим Т-антигеном вируса
SV-40. Это говорит о наличии иммунного ответа на инъекцию
иммуногена — гибридного белка, включающего фрагмент боль-
шого Т-антигена.
Гидридный белок содержит в своем составе фрагмент ами-
нокислотной последовательности большого Т-антигена (амино-
кислоты 271—448), не узнаваемый ни одним из имеющихся
184
С. Моул, Д. Лейн
А Б
ПИ ЛИ
Рис. 6.4. Иммунопреципитация меченных
355-метионином экстрактов клеток CV-1,
инфицированных и псевдоинфицирован-
ных вирусом SV-40. Дорожки п — псев-
доинфицированные клетки CV-1, дорож-
ки и — инфицированные клетки CV-1.
А — иммунопреципитация моноклональ-
ными антителами к большому Т-антиге-
ну; 5 — экстракты после иммунопреци-
питации сывороткой мыши, иммунизиро-
ванной гибридным белком ^-галактози-
даза — большой Т-антиген (аминокисло-
ты 271—448), не взаимодействующим ни
с одним из известных моноклональных
антител к большому Т-антигену. Новые
гибридомные клоны, продуцирующие мо-
ноклональные антитела, были получены
в результате слияния с использованием
спленоцитов такой мыши. (Рисунок
взят из работы [9]; приводится с лю-
безного разрешения авторов.)
Моноклональные антитела к гибридным белкам из Е. coli
185
моноклональных антител к большому Т-антигену. Таким обра-
зом, с помощью данного метода были получены антитела к ра-
нее иммунологически неактивной области белка.
6.3.4. Скрининг гибридом
После удачного слияния клеток необходимо провести бы-
стрый и эффективный скрининг гибридом. В большинстве слу-
чаев для слияния требуются три этапа скрининга, и поскольку
для каждого эксперимента используется свыше 500—1000 лу-
нок, это означает, что для выявления наличия специфических
антител нужно проанализировать до 3000 проб. Кроме того,
кинетика роста гибридных клеток такова, что результаты скри-
нинга должны быть известны в течение первых 48 ч после от-
бора проб. Единственный реальный способ обработать такое
количество проб — использование метода, который позволяет
провести скрининг, дающий визуальные результаты, на план-
шетах с лунками маленького объема. Скрининг не обязательно
дает абсолютно однозначные результаты и допускается выяв-
ление некоторого количества ложных положительных клонов.
Затем его можно повторить и получить более определенный от-
вет, работая только с относительно небольшим количеством
проб, давших положительный ответ при первом скрининге.
Один из наиболее удобных методов скрининга — твердофазный
иммупофермептный анализ (ИФА, ELISA), при котором анти-
ген наносится на дно лунок пластикового планшета; он связы-
вает антитела, продуцируемые гибридомой, а наличие этого
комплекса обнаруживается с помощью антител к иммуноглобу-
линам, конъюгированных с определенным ферментом. Описание
иммуноферментного анализа (ИФА) приведено в табл. 6.6.
Другой способ скрининга удивительно универсален. Как по-
казал наш опыт — это прямое иммуноцитохимическое окраши-
вание фиксированных клеток (табл. 6.7). Данный метод очень
удобен, поскольку под микроскопом приходится исследовать
содеожимое только тех лунок, в которых после добавления суб-
страта развилась окраска, а характер распределения субстрата
может дать большую информацию о специфичности антител.
Поскольку планшеты могут храниться, с их помощью удобно
исследовать взаимодействие антител, получаемых в результате
одного слияния, с разными клеточными линиями. Возможности
метода позволяют, кроме того, исследовать инфицированные
вирусом или стабильно трансформированные клеточные линии.
После первого скрининга стоит провести серию дополнитель-
ных скринингов, которые позволят точно установить, обладают
ли полученные антитела необходимой специфичностью. На эти
опыты не накладывается таких ограничений, как на предвари-
186
С. Моул, Д. Лейн
Таблица 6.6. Метод твердофазного иммуноферментного теста
(ИФА, ELISA) на анализ к белковым антигенам
Необходимое оборудование
1. Пластиковые планшеты (Falcon 3912)
2. Многоканальная автоматическая пипетка (8-канальная, объемом на 5—
50 мкл, Flow 8 channel 5—50 мкл).
3. Спектрофотометр, регистрирующий результаты ИФА в планшете (ELISA
plate reader) (его наличие не является абсолютно необходимым).
Реактивы
1. Раствор белка-антигена с концентрацией 0,1-—1 мкМ. в PBS.
2. Раствор для инактивации не связавших антиген поверхностей планшета:
3% БСА в PBS.
3. Раствор для отмывки: 0,1% NP-40 в PBS.
4. Антиглобулины (кроличьи антитела к мышиным иммуноглобулинам),
конъюгированные с пероксидазой хрена (DAKO), разбавленной 1 : 1000
PBS, содержащим 10% СЭК.
5. Раствор субстрата: растворите 3'3'5'5'-тетраметилбензидин (Miles Chemi-
cals) в диметилсульфоксиде (Sigma Chemical Company) в концентрации
1 мг/мл. Разбавьте раствором PBS 1/100 и профильтруйте через бумагу
ватман № 1. В конце добавьте Н2О2 до конечной концентрации 0,01 %.
6. Раствор для остановки реакции: 1 М H2SO4.
Методика
1. Подготовьте планшет, добавив в каждую лунку по 50 мкл раствора белка
и инкубируя с ним в течение ночи при 4 °C. Слейте раствор белка.
Планшеты можно хранить при —20 °C в течение неограниченно долгого
времени.
2. Проведите насыщение лунок, внеся в каждую по 50 мкл раствора для
инактивации пластика и инкубируя в течение 1 ч при комнатной темпе-
ратуре. Слейте этот раствор, промойте планшеты PBS и добавьте в каж-
дую лунку аликвоты по 20—100 мкл надосадочной фракции из лунок
планшета с гибридомами с помощью многоканальной пипетки. (При пе-
реходе от одного ряда лунок к другому промывайте наконечники 70%-ным
этанолом, а при переходе на другой планшет смените их.) Инкубируйте в
течение 3 ч.
3. Промойте планшеты 3 раза буфером для отмывки и добавьте в каждую
лунку по 50 мкл конъюгата фермента с антителом второго порядка. Ин-
кубируйте в течение 1 ч, затем промойте планшеты 3 раза буфером для
отмывки и один раз PBS.
4. Добавьте по 50 мкл на лунку свежеприготовленного раствора субстрата и
инкубируйте в течение необходимого времени до развития окраски. Оста-
новите развитие окраски добавлением 50 мкл раствора для остановки
реакции.
5. Определите степень развития окраски в лунках с помощью спектрофото-
метра (ELISA plate reader). Результаты удобнее всего количественно оце-
нивать с помощью калибровочной кривой, где величины оптической плот-
ности располагаются в диапазоне значений 1—10.
тельный скрининг, и здесь можно использовать более сложные
методы, пригодные только для небольшого числа проб. В этом
случае годятся и вестерн-блот-анализ, и иммунопреципитация
(см. табл. 6.8 и 6.9). Если последовательность, кодируемая
ОРС, доступна только как часть экспрессируемого данным век-
тором составного белка, включающего ^-галактозидазу, тогда
Моноклональные антитела к гибридным белкам из Е. coli
187
Таблица 6.7. Окрашивание клеток
1. Для скрининга мышиной сыворотки высейте клетки на чашки диаметром
90 мм; нижнюю поверхность чашек разделите фломастером на квадраты
размером 10ХЮ мм. Для скрининга гибридом внесите клетки в планшет
так, чтобы в каждой лунке в объеме 100 мкл находилось 104 клеток.
Оставьте клетки осаждаться на ночь при 30 °C.
2. Промойте буфером PBS и фиксируйте клетки в течение 2 мин смесью
50% ацетона — 50% метанола. Промойте тем же раствором для фиксации
и дайте им высохнуть.
3. Инкубируйте с надосадочной фракцией моноклональных антител или с
взятой в разных разбавлениях мышиной сывороткой в течение 3 ч во
влажной камере (при работе с планшетами в каждую лунку вносите по
50 мкл надосадочной фракции или разбавленной сыворотки, при работе с
90 мм-чашками капните по 2 мкл раствора каждого антитела на клеточ-
ный монослой на размеченные участки с определенной площадью).
4. Промойте пять раз PBS. Инкубируйте в течение 3 ч со связанными с
пероксидазой хрена кроличьими антимышиными иммуноглобулинами, раз-
бавленными 1 : 100 PBS, содержащим 1% БСА, 10% СЭК.
5. Промойте пять раз PBS и окрасьте клетки следующим образом: возьмите
насыщенный раствор о-дианизидина (Sigma Chemical Company) в этано-
ли и разбавьте его PBS в соотношении 1 : 100. Профильтруйте такой све-
жеразбавленный раствор и добавьте пероксид водорода (30%) в разведе-
нии 1 :5000. Добавьте этот раствор к фиксированным клеткам и оставьте
их в темноте в течение 1—24 ч. Промойте препараты водой и для хране-
ния добавьте азид натрия до конечной концентрации 0,2%.
скрининг может включать предварительный анализ методом
ИФА на наличие антител как к ^-галактозидазе, так и к гиб-
ридному белку, а затем проведение вестерн-блот-анализа двух
белков с использованием гибридом, продуцирующих антитела
только к гибридному белку, но не к ^-галактозидазе (кон-
троль). Однако мы рекомендуем, если возможно, перенести
вставку в другой вектор (как описано в разд. 6.5), поскольку
это позволит выявить специфические кодируемые вставкой ан-
титела даже в присутствии антител к ^-галактозидазе.
6.3.5. Слияние
Оборудование и реактивы, необходимые для слияния, пере-
числены в табл. 6.10.
После получения у иммунизированной мыши четкого специ-
фического иммунного ответа на встроенный фрагмент можно
начать подготовку к проведению слияния. Несмотря на нали-
чие многочисленных эмпирических данных о решающих факто-
рах, от которых зависит успех слияния, оно все-таки не всегда
проходит со стабильно положительным результатом, и стоит
провести несколько повторных слияний. Наибольших усилий
в подобных экспериментах требуют клонирование, выделение
антител и их характеристика; поэтому рекомендуется исследо-
вать полученные при слиянии клоны лишь в тех случаях, ког-
да есть много здоровых растущих клонов (100 и более).
188 С. Моул, Д. Лейн
Таблица 6.8. Вестерн-блот-анализ
1. Проведите разделение белковых экстрактов с помощью ДСН-электрофореза в
ПААГ. При необходимости проведите электрофорез окрашенных маркеров
молекулярной массы (BRL).
2. Проведите перенос белков с геля на нитроцеллюлозный фильтр (Schleicher
and Schnell) в камере для электроблоттинга [15].
3. Инкубируйте фильтр в буфере для насыщения (3% БСА в PBS) в тече-
ние 1 ч.
4. Промойте фильтр PBS и при необходимости нарежьте его на полоски оп-
ределенного размера. Инкубируйте каждую полоску в растворе антите-
ла1’, используемого для скрининга, в течение 3—18 ч.
5. Промойте фильтр PBS, затем 2 раза по 15 мин PBS, содержащим 1%
NP-40, и 2 раза по 15 мин PBS.
6. Инкубируйте фильтр в растворе предварительно адсорбированных антител
второго порядка2’, конъюгированных с пероксидазой хрена, в течение
3—18 ч.
7. Промойте фильтр PBS, затем 2 раза по 15 мин PBS, содержащим 1%
NP-40, и 2 раза по 15 мин PBS.
8. Разбавьте непосредственно перед постановкой реакции раствор 50 мг/мл
4-хлор-1-нафтола (Aldrich) в этаноле PBS в соотношении 1 : 100 и про-
фильтруйте. Для развития на фильтре окраски добавьте пероксид водоро-
да (30%), разбавленный 1 : 5000. Фильтры, где развилась заметная окрас-
ка, промойте PBS и высушите. Храните в темноте.
') Для скрининга можно использовать моноклональные антитела или поликлональ-
ную сыворотку.
2) Желательно предварительно истощить антитело второго порядка экстрактом ли-
зированных клеток Е. coli (табл. 6.2). Антитела второго порядка должны быть специ-
фичными по отношению к животному, служившему хозяином при получении антител, ис-
пользовавшихся при скрининге, например кроличий антимышиный иммуноглобулин, свя-
занный с пероксидазой хрена (DAKO) или антикроличьи иммуноглобулины козы, и т. д.
6.3.5.1. Последняя инъекция антигена
Перед слиянием донорной мыши следует последний раз вве-
сти антиген. Цель такой «подхлестывающей» инъекции — изби-
рательно стимулировать пролиферацию ответственных за син-
тез специфических антител В-лимфоцитов и локализовать их
в определенном органе, в данном случае в селезенке. Эту инъ-
екцию не проводят, когда в сыворотке еще сохраняется доволь-
но высокая концентрация антител к вводимому иммуногену.
Поэтому иммунизированному животному не следует делать
инъекций антигена по крайней мере в течение месяца перед
«подхлестывающей» инъекцией. Если гибридный белок раство-
рим, последняя инъекция может быть внутривенной (введение
в хвостовую вену), поскольку этот путь введения антигена осо-
бенно эффективен. Инъецируют от 10 до 100 мкг белка в объ-
еме 0,5 мл физиологического раствора или PBS без адъюванта.
Чтобы антиген был стерильным, перед инъекцией пропустите
его раствор через фильтр с диаметром пор 0,22 мкм. Если гиб-
ридный белок труднорастворим, не фильтруйте антиген и не
делайте внутривенной инъекции, а вместо этого внутрибрюшин-
Моноклональные антитела к гибридным белкам из Е. coli
189
Таблица 6.9. Иммунопреципитация1)
А. Радиоактивное мечение in vivo клеток, инфицированных вирусом SV-4O
1. Предварительно инкубируйте монослой клеток (CV-1), инфицированных
вирусом SV-40 (степень слияния 90%). в безметпониновоп среде — моди-
фицированной Дульбекко среде Игла (DMEM, Flow) в течение 30 мин
при 37 °C.
2. Пометьте клетки, инкубируя их с 358-метионином (Amersham, 50 мкКи на
90мм-чашку Петри) в безметиониновой среде DMEM при 37 °C в течение
необходимого времени.
Б. Получение клеточных экстрактов
1. Промойте клетки трис-буфером с солями2’. Затем еще добавьте тот же
буфер, но содержащий 10 мМ ЭДТА, и инкубируйте 10—15 мин при 37 °C.
2. Осадите клетки центрифугированием при 1000 g в течение 5 мин и прове-
дите лизис, инкубируя их в растворе NET8,o3), содержащем 1% NP-40,
в течение 30 мин при 4 °C. Для осаждения клеточных обломков центрифу-
гируйте суспензию при 15 000 g при 4 °C в течение 30 мин или в микро-
центрифуге при 4 °C в течение 5 мин.
3. Удалите из надосадочной фракции неспецифически реагирующий материал,
инкубируя его с сывороткой неиммунизированного кролика (10 мкл сыво-
ротки на 107 клеток) в течение 1 ч, а затем — с клетками Staphylococcus
aureus Cowan I4’ (50 мкл на 107 клеток) в течение 15 мин в ледяной ба-
не. Осадите иммунные комплексы центрифугированием в течение 1 мин в
микроцентрифуге и инкубируйте надосадочную фракцию с клетками Sta-
phylococcus aureus Cowan 1, еще 15 мин в ледяной бане и центрифугируйте,
как описано выше. Теперь надосадочная фракция готова к использованию
в реакции иммунопреципитации. В.
В. Иммунопреципитация
1. Каждую реакцию иммунопреципитации проводите с таким объемом над-
осадочной фракции, который эквивалентен примерно ЗХЮ6 клеток. Инку-
бируйте клеточный экстракт с 100 мкл надосадочной фракции антител пли
с 10 мкл мышиной антисыворотки в течение ночи при 4 °C. Клеточные
экстракты, инкубировавшиеся с моноклональными антителами, далее ин-
кубируйте с 5 мкг антимышиных иммуноглобулинов (DAKO) и 5 мкл
нормальной сыворотки кролика в течение 2 ч при комнатной температуре.
2. Инкубируйте все клеточные экстракты с 50 мкл суспензии S. aureus Co-
wan 1 в течение 15 мин при 4 °C. Центрифугируйте 1 мин в микроцентри-
фуге.
3. Промойте осадки клеток, последовательно ресуспендируя их в следующих
буферных растворах и переосаждая центрифугированием:
NETso, содержащий 1% NP-40;
1% NP-40, 0,1% ДСН, 500 мМ LiCl, 50 мМ трис-НС1 (pH 8,0);
1% NP-40, 600 мМ NaCl, 50 мМ трис-НС1 (pH 8,0);
1% NP-40, 0,1% ДСН, 100 мМ NaCl, 50 мМ трис-НС! (pH 8,0);
0,05% NP-40, 1% БСА, 150 мМ NaCl, 5 мМ ЭДТА, 50 мМ трис-НС1
(pH 8,0);
NETs.o
4. Суспендируйте каждый осадок в буфере NETs.o, перенесите в* чистую про-
бирку и центрифугируйте, как описано выше. Ресуспендируйте каждый
осадок в 50 мкл ДСН-содержащего буфера5’ для нанесения на ПААГ, про-
кипятите в течение 2 мин и отцентрифугируйте. Перенесите надосадочную
фракцию в чистую пробирку, прокипятите в течение 5 мин и проведите
электрофорез в ПААГ.
190
С. Моул, Д. Лейн
Продолжение
5. Окрасьте и отмойте гель и инкубируйте его в течение 1 ч в 3 объемах
раствора EN3HANCE (New England Nuclear). Промывайте водой в тече-
ние 1 ч, высушите и проведите радиоавтографию.
’) Здесь приведена методика, используемая нами при анализе антисыворотки, по-
лученной к белкам вируса SV-40. Ее несомненно можно применять и при исследеванин
других вирусных или клеточных белков.
2) Состав этого раствора: 137 мМ. NaCl, 5 мМ КС1, 0,7 мМ NajHPCh, 25 мМ трис-НС1
(pH 7,4).
з) Состав NETg0: 150 мМ NaCl, 5 мМ ЭДТА (pH 8,0), 50 мМ трис-НС1 (pH 8,0).
4) Подготовьте клетки S. aureus Cowan 1, дважды промыв их 10 объемами NETg 0,
содержащего 1% NP-40, а затем ресуспендируйте их в исходном объеме буфера.
5) Состав для нанесения на ДНС-содержащий ПААГ: 62,5 мМ трис-НС1 (pH 6,8),
2% ДСН, 100 м.Ч ДТТ, 10;% глицерола, 0,05% бромфенолового синего.
по введите непрофильтрованный антиген за шесть, пять и че-
тыре дня до инъекции. Слияние надо проводить через 72—96 ч
после последней инъекции.
6.3.5.2. Подготовка миеломных клеток
Родительские клетки гибридомы, используемые при слия-
нии, должны быть в очень хорошем состоянии, поэтому их сле-
дует получить из центра, где имеется коллекция культур клеток
(например, из Американской национальной коллекции культур
клеток), или из лаборатории, где хорошо налажено получение
гибридом. Клетки лучше не хранить в виде культуры в течение
продолжительного времени, а работать со свежеразмороженной
порцией, хранившейся до того в жидком азоте. Клетки должны
оттаять по крайней мере за 10 дней до слияния, так чтобы их
можно было пересеять как минимум дважды до использования
в среде DMEM, содержащей 10%-ную сыворотку эмбриона
коровы (СЭК). Суспендируйте клетки в соотношении 1 :3 в све-
жей среде (DMEM, содержащей 20% СЭК) вечером накануне
слияния. Подготовьте клетки к слиянию, дважды отмыв их
в DMEM, не содержащей сыворотки, и, наконец, ресуспендируя
в 5 мл DMEM. На одно слияние вам потребуется 107 клеток
(это примерно две чашки Петри диаметром 90 мм). Хорошо
удается слияние линий миеломных клеток SP20/AG14 и NS-1
с клетками, образующими почти слитный монослой.
6.3.5.3. Взятие клеток селезенок донора
1. Забейте мышь, переломив ей шейные позвонки.
2. Протрите шерсть этанолом, в ламинарном боксе возьмите
селезенку с помощью стерильных инструментов и поместите ее
в стерильную чашку Петри диаметром 90 мм с 3—4 мл среды
DMEM, содержащей 10% СЭК.
Моноклональные антитела к гибридным белкам из Е. coli
191
Таблица 6.10. Оборудование и реактивы, необходимые для слияния клеток
А. Оборудование
Низкоскоростная центрифуга, ламинарный бокс, термостат для культиви-
рования в атмосфере СОг, термостат на 37 °C, пластиковые планшеты на
96 лунок для культивирования клеток, набор хирургических инструментов,
многоканальная автоматическая пипетка и стерильные наконечники к ней,
пластиковые кругло донные центрифужные пробирки, пластиковая посуда
и пипетки, обычно используемые при работе с культурами клеток.
Б. Реактивы
Сыворотка плода коровы. Она должна производиться фирмой, постоянно
поставляющей проверенные партии сыворотки, используемой для роста
гибридом, например Hybritech.
Раствор полиэтиленгликоля (ПЭГ). ПЭГ получают в виде стерильного
50%-ного раствора (весовая концентрация) в 75 мМ Hepes (фирма
Boehringer Mannheim ФРГ, каталоговый номер 783641).
Селективная среда. Мы предпочитаем работать со средой гипоксантин-аза-
серин (ГА), а не с более часто используемой средой гипоксантин/амино-
птерин/тимидин (ГАТ), поскольку в среде ГА происходит более энергич-
ный рост клеток. ГА готовят в виде раствора 100-кратной концентрации,
растворяя 136 мг гипоксантина и 10 мг азасерина в 100 мл стерильной
дистиллированной воды. Затем раствор стерилизуют фильтрацией и хра-
нят в виде небольших аликвот при —20 °C.
Среда для культивирования клеток. Модифицированная Дульбекко среда
Игла (DMEM), содержащая 2 мМ L-глутамин и 100 мкг/мл гентамицина.
Диметилсульфоксид (ДМСО DMSO).
3. С помощью двух игл № 19 и шприцов на 1 мл очень тща-
тельно гомогенизируйте селезенку.
4. Добавьте к гомогенату 15 мл DMEM, не содержащей
сыворотки, и перенестите клетки в центрифужную пробирку.
Дайте суспензии отстояться в течение 5 мин, затем осторожно
отберите и перенесите не содержащую твердых частиц суспен-
зию в чистую центрифужную пробирку.
5. Промойте клетки один раз DMEM и ресуспендируйте их
в 5 мл DMEM.
6.3.5.4. Слияние
1. Смешайте клетки двух типов в круглодонной, центрифуж-
ной пробирке и осадите их вместе центрифугированием.
2. Удалите надосадочную фракцию, используя водоструйный
насос. Среду следует удалить самым тщательным образом, да-
же если при этом потеряется небольшое количество клеток.
Затем добавьте 300 мкл раствора ПЭГ (33%-ный раствор
в DMEM, подогретый до 37 °C).
3. Ресуспендируйте осадок, энергично встряхивая пробирку,
и центрифугируйте при 1200 g в течение 5 мин.
4. Аккуратно отберите избыток ПЭГ, добавьте 5 мл DMEM
и очень осторожно ресуспендируйте осадок пипеткой с широ-
ким носиком.
192
С. Моул, Д. Лейн
5. Перенесите суспензию клеток в 45 мл DMEM, содержа-
щей 20% СЭК и ГА, и с помощью многоканальной пипетки вне-
сите клетки в 5—10 планшетов на 96 лунок.
6. После слияния поместите планшеты с клетками в СО2-
инкубатор и постарайтесь не трогать их в течение 10 дней.
Только один раз, на 3-й день, проверьте клетки, чтобы убедить-
ся в отсутствии «зароста» и зафиксировать массовую гибель
клеток в процессе селекции.
7. На 10-й день осторожно исследуйте содержимое планше-
тов: выставьте их на свет и осмотрите с нижней стороны. На
положительный рост гибридных клеток указывают пожелтение
культуральной среды и наличие небольших белых пятен на дне
некоторых лунок. Под микроскопом в 10—30% лунок должны
обнаруживаться большие сферические колонии, характеризую-
щиеся сильным двойным лучепреломлением (рис. 6.5).
8. На этой стадии для проведения скрининга на наличие
нужных антител отберите пробы по 50 мкл или из всех лунок,
или только из тех лунок, в которых колонии различимы без
микроскопа.
9. Повторите скрининг на 13-й и 17-й дни.
Клетки из всех лунок, оказавшиеся при скрининге положи-
тельными, надо субкультивпровать. Для этого переносите содер-
жимое этих лупок в 24-луночный планшет и добавьте в каж-
дую лунку по 0,5 мл свежей среды. Клетки из такого планшета
могут служить источником для клонирования клеток данной
гибридомной линии; кроме того, их можно рассеять на чашке
Петри диаметром 90 мм и заморозить. Если слияние оказалось
очень удачным, можно сразу не справиться с размножением
и клонированием слишком большого количества клеток. В та-
кой ситуации лучше выбрать только пять или десять положи-
тельных колоний для немедленного их размножения, а осталь-
ные заморозить до проведения дальнейших исследований. После
размножения колоний в планшете с 24 лунками скрининг
следует повторить и, кроме того, провести и более жесткий
скрининг другим способом. Если в результате слияния во мно-
гих лунках наблюдается рост клеток и обнаруживаются анти-
тела к ^-галактозидазе, но не обнаруживается антител к белко-
вому продукту встроенного фрагмента, тогда единственно, что
остается делать, — это повторить слияние в тех же условиях,
в которых проводилось предыдущее. Если, однако, в результате
слияния вообще образуется небольшое число клонов гибридных
клеток, тогда стоит попробовать внести изменения в методику
слияния. К ним относятся использование так называемого «пи-
тающего» слоя для роста клеток и внесение в среду добавок.
Моноклональные антитела к гибридным белкам из Е. coli
193
Рис. 6.5. Рост клеток гибридомы. А. Небольшая колония клеток з лупке. Ta-
in с колонии без мЕкроскопа пс видны и должны появляться через 7—9 дней
после слияния. Б. Большая колония, 17-й день роста. Колония начинает за-
полнять лунку, среда приобретает желтый цвет. Если лунка содержит антитела
нужной специфичности, клетки необходимо рассеять на следующий день или
через день. (Фотография любезно поедсставлена доктором Э. Б. Лейном,
ICRE, Clare Hail.)
13—11 36
194
С. Моул, Д. Лейн
1. Питающий слой. В случае удачно прошедших слияний
этот слой не требуется, но если клетки растут плохо, здесь он
может помочь. Для него мы используем линию диплоидных
фибробластов, внесенных в планшеты с 96 лунками за 2 дня до
слияния в количестве 104 клеток на лунку.
2. Добавки, вносимые в среду. Если партия сыворотки не
отвечает оптимальным требованиям, то может оказаться полез-
ным добавление в среду смеси растворимых компонентов. При-
готовьте среду DMEM, как обычно, добавив в нее гентамицин и
L-глутамин, а затем добавьте бычий инсулин (0,2 ед/мл), пиру-
ват (0,45 мМ) и оксалоацетат (1 мМ).
6.3.6. Замораживание гибридомных клеточных линий
Родительские миеломные клетки и сами гибридомные клет-
ки могут храниться в жидком азоте в течение длительного вре-
мени (в течение 10 лет определенно). Процедура заморажива-
ния проста, но ее следует опробовать на родительских клетках
до слияния. При замораживании используют специальную
смесь. Поскольку смеси, пригодные для клеток других типов,
в случае гибридом не работают, остановимся на следующем
методе.
1. Замораживать следует хорошо растущие клетки, поэтому
надо создать оптимальные условия: накануне замораживания
выращивать клетки в течение ночи, суспензировав их в свежей
среде в соотношении 1 :3.
2. Осадите клетки центрифугированием, отберите надосадоч-
ную фракцию и осторожно ресуспендируйте их в охлажденной
во льду смеси для замораживания (94% СЭК, 6% диметилсуль-
фоксид) в соотношении 0,3 мл смеси на 5хЮ6 клеток.
3. Разделите клетки на аликвоты и внесите их в стерильные
флаконы, где они будут заморожены (по 0,2—0,3 мл на фла-
кон), упакуйте флаконы в большой полиуретановый контейнер
с ватой и поместите в холодильник на —70°C на 24 ч. Затем
флаконы надо перенести в сосуд с жидким азотом.
4. Убедитесь в том, что при замораживании хорошо сохра-
няется жизнеспособность клеток. Для этого через несколько
дней разморозьте контрольный флакон; кроме того, всегда
следует помещать флаконы с замороженными клетками данной
гибридомы по крайней мере в два разных сосуда с жидким
азотом.
6.3.7. Клонирование клеток гибридом
Для того чтобы полученная гибридома не была загрязнена
примесями других гибридом и была генетически стабильной
линией клеток, она должна представлять собой клон, получен-
Моноклональные антитела к гибридным белкам из Е. coli
195
Таблица 6.11. Клонирование в мягком агаре
А. Реактивы
'Раствор А: 3%-ный раствор легкоплавкой агарозы (Sea prep 15145, FMC
•Corporation) в дистиллированной в стеклянном дистилляторе воде. Раствор
стерилизуют автоклавированием и хранят при 37 °C.
Раствор Б: DMEM двукратной концентрации стерилизуют фильтрованием,
а затем добавляют 20% СЭК, 2 мМ L-глутамин и 100 мкг/мл гентамицина.
Б. Методика
1. Подготовьте флаконы для клонирования (Falcon, 2054; по две на каждую
концентрацию клеток), содержащие 105, 104, 103 и 102 живых клеток в
150 мл среды DMEM. Чтобы суспензия клеток не содержала комков, пи-
петпруйте ее и держите сосуды с клетками при комнатной температуре.
2. Смешайте равные объемы растворов А и Б. Добавьте по 2 мл с.месп в
каждый из флаконов и перемешайте путем пипетирования. Инкубируйте
флаконы с к. дткамп при 4 °C 45 мин, а затем поместите их в термостат в
атмосферу СО2.
3. Когда через 10—21 день во флаконах с самым низким разбавлением ста-
нут разли шмы колонии, соберите их с помощью стерильной пастеровской
пипетки.
4. Внесите отобранные колонии, выросшие в агарозе, в 1 мл свежей среды
(DMEM, содержащей 20% СЭК, L-глутамин и гентамицин) и поместите
суспензию в 24-луночный планшет для культур клеток.
5. Через 2 дня содержимое планшета можно тестировать на наличие ан-
тител.
6. Клетки, выращиваемые в таких условиях, растут исключительно быстро,
так что замораживать их можно всего через несколько дней после скри-
нинга.
7. Все отобранные колонии должны быть положительными, и клонирование
следует повторять до тех пор, пока это не будет достигнуто.
ный из одной клетки. Существует целый ряд методов клониро-
вания, но, на наш взгляд, наиболее подходящий из них — кло-
нирование в мягкой агарозе. Соответствующая методика описа-
на в табл. 6.11.
6.3.8 Загрязнение
При наличии практических навыков и тщательной работе
при выращивании клеток в планшетах содержимое лунок не
должно подвергаться бактериальному заражению. Совершенно
необходимо не переполнять лунки, поэтому никогда не вносите
в лунку более 100 мкл среды. В случае если зароет обнаружен
в лупке планшета, содержащего важные для работы гибрид-
ные клоны, осторожно внесите в лунку 100 мкл 70%-ного эта-
нола, затем отберите из нее все жидкое содержимое и оставьте
в сухом месте. При загрязнении лунки с клетками, секретирую-
щими антитела, ее содержимое можно спасти пассированием
клеток in vivo. Введите содержимое лунки мыши BALB/c, ко-
торой за 2—10 дней до этого было внутрибрюшинно введено
200 мкл полного адъюванта Фрейнда. При удачном стечении
13!
196
С. Моул, Д. Лейн
обстоятельств через 10—14 дней у мыши в результате развития
асцита наблюдается вздутие живота. С помощью шприца с иг-
лой большого диаметра отсосите некоторое количество асцитной
жидкости. Внесите 0,1 мл этой жидкости в 2 мл культуральной
среды (DMEM с 20% СЭК) и исследуйте суспензию под мик-
роскопом. Обычно гибридомные клетки легко различимы и их
сразу же можно клонировать (см. разд. 6.3.7). Оставшуюся
асцитную жидкость можно исследовать на предмет наличия
специфических антител; это исследование нужно проводить,
используя большое количество ее разведений, поскольку асцит-
ная жидкость может по содержанию антител быть в 1000 раз
более концентрированной, чем культуральная среда, содержа-
щая секретируемые клетками антитела.
6.4. Применение моноклональных антител
Моноклональные антитела с успехом применяются во многих
иммунологических методах; некоторые примеры их использова-
ния детально рассматриваются в этом разделе. Моноклональ-
ные антитела легко очищаются с помощью хроматографии на
сефарозе, связанной с белком A (Protein A Sepharose), и их
легко пометить либо непосредственно путем иодирования, либо
непрямым способом с использованием антиглобулиновых реаген-
тов. Непрямое мечение антител, связанных с биотином, с ис-
пользованием стрептавидиновых или авидиновых конъюгатов
особенно удобно, если в опыте используется не одно, а несколь-
ко моноклональных антител, и мы здесь приводим простой ме-
тод их биотинилирования.
Гомогенность и специфичность центров связывания монокло-
нальных антител делают их особенно удобными для использо-
вания в иммуноаффинной хроматографии. В табл. 6.15 мы при-
водим высокоэффективный метод получения иммуносорбентов
для колонок и их применения, а также различные варианты
условий элюции, которые позволили нам очень эффективно
с сохранением их биохимической активности очистить макромо-
лекулы, обладающие высокой чувствительностью к воздействию
оазличпых агентов и находящиеся в низкой концентрации.
Моноклональные антитела могут эффективно использоваться
в иммунопреципитации и в иммуноцитохимических методах.
На ранних этапах наших исследований при использовании и то-
го, и другого метода мы сталкивались с трудностями из-за по-
вышенного фонового сигнала, который отсутствовал при исполь-
зовании поликлональных антител. Нам удалось преодолеть эти
затруднения; в табл. 6.7 и 6.9 приводятся соответствующие ме-
тодики, которые, как мы обнаружили, во многих случаях хо-
рошо работают.
Моноклональные антитела к гибридным белкам из Е. coli 197
• Мы также приводим методику эффективной постановки ве-
стерн-блот-анализа с использованием моноклональных антител
(табл. 6.8). Исходя из нашего опыта, моноклональные антите-
ла четко делятся на две группы: антитела, работающие в таком
анализе, и не работающие антитела. Работающие в блот-анали-
зе антитела воспроизводимо эффективны, но никакими измене-
ниями условий ренатурации белков нам ни разу не удалось
сделать так, чтобы антитела, не работающие в блот-анализе,
вдруг заработали. Интересно, что такие «неработающие» анти-
тела оказываются достаточно эффективными при скрининге ко-
лоний (табл. 6.1), позволяя картировать их некоторые эпитопы.
Наличие разных моноклональных антител к одному и тому
же продукту лежит в основе количественного иммунологическо-
го анализа белка методом «сэндвича», когда одно антитело
используется в качестве твердой фазы для связывания присут-
ствующего в растворе антигена, а другое антитело использует-
ся в качестве меченого зонда для обнаружения иммобилизован-
ного материала. Хотя мы работали как с зондами, меченными
I125, так и с зондами, меченными ферментативно активными
конъюгатами (в последнем случае обычно к антителам при-
соединяли биотин и использовали конъюгат фермента с авиди-
ном), здесь мы приводим описание только первой методики,
поскольку в нашей практике она оказалась более удобной. Этот
метод пригоден также и для выяснения следующего вопроса
о том, конкурируют ли между собой антитела за взаимодейст-
вие со стерически тесно ассоциированными участками, а также
для изучения белок-белковых взаимодействий.
Все преимущества использования моноклональных антител
можно оценить, когда появляется возможность рассматривать
данные об их антигенных детерминантах (эпитопах) в сочета-
нии с оценками функционального эффекта их взаимодействия
с продуктом ОРС. Этот аспект рассматривается в .разд. 6.5.
6.4.1. Очистка моноклональных антител
6.4.1.1. Подклассы IgGZa, IgGZb, IgG3
1. Вырастите гибридомы, если возможно, в центрифужных
стаканах, используя для экономии 5% СЭК- Не используйте
взрослую сыворотку, так как присутствующие в ней иммуно-
глобулины загрязнят ваш препарат.
2. Создайте такие условия для роста клеток, чтобы была
достигнута самая высокая, насколько это возможно, плотность,
добавляя в среду глюкозу (4,5 г/л), когда концентрация кле-
ток достигнет 5Х105 на 1 мл. Продолжайте культивировать
198
С. Моул, Д. Лейн
клетки, пока среда сильно не закислится и жизнеспособность
клеток не упадет ниже 50%.
3. Осадите клетки и клеточные обломки центрифугирова-
нием при 2000g в течение 15 мин и пропускайте надосадочную
фракцию, содержащую антитела, через колонку с белком А,
иммобилизованном на сефарозе (1 л надосадочной фракции че-
рез колонку объемом 5 мл), в течение ночи при скорости про-
текания 60 мл/ч.
4. Промойте колонку 10 объемами PBS и затем проводите
элюцию цитратным буфером, pH 3,0.
5. Соберите элюированные антитела в пробирки, содержа-
щие 1 М трис-НС1 (pH 8,5), чтобы немедленно прошла их
нейтрализация.
Метод очень эффективно работает для всех антител класса
IgG мыши, кроме подкласса IgGl. Эти последние антитела об-
ладают очень низкой аффинностью по отношению к белку А.
Поэтому для получения хорошего их выхода метод следует мо-
дифицировать. Очистка иммуноглобулинов других классов
здесь не рассматривается, поскольку подавляющее большинст-
во гибридом, получаемых в результате гипериммунизации и
слияния, проводимых в соответствии с описанными методика-
ми, продуцирует антитела класса IgG. Мы не советуем вам по-
лучать антитела из асцитной жидкости (разд. 6.3.8), посколь-
ку такие же количества антител, причем более чистых, могут
быть получены при использовании методов, описание которых
приводится выше.
6.4.1.2. Подкласс IgGl
1. Доведите pH гибридомной надосадочной фракции до 8,5
добавлением 0,1 объема 1 М трис-НС1 (pH 8,5). Затем медлен-
но и при перемешивании добавьте сухой сульфат аммония до
50%-ной концентрации при 4 °C и оставьте на ночь при переме-
шивании.
2. Осадите преципитат центрифугированием при 10 000 g
в течение 30 мин и ресуспендируйте его в объеме, равном 1/50
исходного объема в буфере с высокой ионной силой (0,1 М
трис-HCl, рН8,5, ЗМ NaCl), а затем диализуйте против этого
же буфера в течение ночи.
3. Снова осветлите раствор центрифугированием и наносите
на колонку с белком А, предварительно уравновешенную буфе-
ром с высокой ионной силой.
4. Промойте коленку 10 объемами буфера высокой ионной
силы, а затем проведите элюцию так, как это описано для дру-
гих подклассов IgG в разд. 6.4.1.1.
Моноклональные антитела к гибридным белкам из Е. coli
199
Таблица 6.12. Иодирование моноклональных антител
А. Подготовка пробирок, покрытых иодогеном
1. Растворите иодоген (фирма Pierce Chemical Company, 28 600) в хлорофор-
ме в концентрации 0,5 мкг/мл.
2. Разделите раствор на аликвоты по 100 мкл и перенесите их в стеклянные
пробирки (Gallenkamp TES 100 020G 2X6 мм, натриевое стекло). Если при
использовании таких объемов скорость иодирования окажется слишком
высокой, разделите иодоген на аликвоты по 20 мкл.
3. Для испарения хлороформа сушите пробирки в вытяжном шкафу в тече-
ние ночи.
4. Храните в вакуумном эксикаторе при комнатной температуре. Препараты
сохраняются неопределенно долгое время.
Б. Реакция иодирования
1. Внесите в каждую пробирку с иодогеном по 50 мкл раствора антител;
антитела растворяют в 0,5 М. Na-фосфатном буфере, pH 7,5, в концентра-
ции 1 мг/мл).
2. Добавьте в пробирки по 500 мкКп меченного I125 иодида натрия (Amer-
sham; по ~5 мкл исходного раствора) и инкубируйте в течение 2—10 мин.
3. Перенесите содержимое каждой пробирки в 150 мкл PBS, содержащего
1% БСА и 0,01% азида натрия.
4. Фракционируйте раствор на колонке с сефадексом G-50 (объем 5 мл),
используя тот же буфер. Определите "[-радиоактивность фракций и собе-
рите первый пик.
5. Храните материал при 4 °C или при —20 °C.
6.4.2. Твердофазный радиоиммуноанализ
Если получены два антитела к стерически различным уча-
сткам белкового продукта ОРС, их можно использовать для
специфической детекции этого белка с помощью радиоиммуно-
анализа. При этом методе одно из антител (связывающий
агент) иммобилизуется на пластике, из которого изготовлены
лунки планшета. Второе антитело (антитело второго порядка)
иодируется до достижения высокой удельной радиоактивности
(100 мкКи/мкг) с помощью специального метода (табл. 6.12).
Метод твердофазного радиоиммуноанализа подробно приводит-
ся в табл. 6.13.
Этот метод не требует наличия очищенного антигена; при
его использовании получается очень низкий фоновый сигнал,
и он характеризуется хорошей воспроизводимостью (рис. 6.6).
Мы также находим этот метод чрезвычайно ценным при изуче-
нии белок-белковых взаимодействий с использованием пар мо-
ноклональных антител к разным субъединицам мультимерных
белковых комплексов.
6.4.3. Биотинилирование
Очищенные моноклональные антитела легко модифицируют-
ся путем ковалентного связывания с биотиновыми группами.
Этот простой метод может использоваться вместо иодирования
200
С. Моул, Д. Лейн
Таблица 6.13. Твердофазный радиоиммуноанализ
А. Реактивы
Раствор антител для иммобилизации на пластике (20 мкг/мл) в PBS
Иодированное антитело (106 имп./мин/мл) в PBS, содержащем 3% БСА и
0,1% NP-40
Раствор для инактивации пластика — 5°/о БСА в PBS
Раствор для отмывки — PBS, содержащий 0,1% NP-40
Б. Оборудование
Счетчик у-радиоактивности, - планшеты, многоканальные автоматические пи-
петки
В. Методика
1. Иммобилизуйте антитела на пластике, добавляя 50 мкл раствора антител
в каждую лунку и инкубируя планшеты во влажной камере при комнат-
ной температуре в течение ночи.
2. Удалите раствор из планшетов (встряхиванием) и добавьте в каждую лун-
ку по 50 мкл раствора для инактивации свободных поверхностей пластика.
3. Через один час промойте лунки двумя сменами раствора для отмывки и
добавьте в них раствор антигена (чувствительность метода такова, что
позволяет выявлять большинство белковых антигенов в концентрации от
0,1 до 10 пМ.). Инкубируйте 2 ч при комнатной температуре.
4. Отмойте не связавшийся антиген четырьмя сменами раствора для отмыв-
ки, а затем один раз чистым PBS.
5. Добавьте по 50 мкл иодированных антител (50 000 имп./мин) на каждую
лунку п инкубируйте в течение еще двух часов при комнатной температуре.
6. Удалите меченые антитела, промойте планшет, как это делалось ранее,
и высушите его в вытяжкам шкафу или с помощью фена. Отдельные луп-
ки можно вырезать ножницами (или раскаленной проволокой) и опреде-
лить их радиоактивность в у-счетчикс.
антител или их связывания с флуорохромом и позволяет выяв-
лять модифицированное антитело с помощью коммерческих ме-
ченых препаратов стрептавидина или авидина. Подробная мето-
дика биотинилирования моноклональных антител приведена
в табл. 6.14.
6.4.4. Картирование эпитопов
Для того чтобы максимально эффективно использовать мо-
ноклональные антитела в функциональном анализе, необходимо,
чтобы сайты связывания были картированы как можно точнее.
Довольно простой подход к решению этой проблемы состоит
в блот-гибридизации колоний с серией перекрывающихся гиб-
ридных белков, что позволяет идентифицировать искомые по-
следовательности, состоящие примерно из 50 аминокислот. Ме-
тодика блот-гибридизации колоний приводится в табл. 6.1. Сле-
дующий шаг — это синтез перекрывающихся пептидных после-
довательностей для идентификации участка с точностью до
нескольких аминокислот. Мы успешно использовали этот под-
ход при картировании РАЬ204, ингибирующего АТРазу [6],
Моноклональные антитела к гибридным белкам из Е. coli
201
log 10 концентрации Т-антигена
Рис. 6.6. «Сэндвич»-радиоиммунотест на большой 'Г-антиген вируса SV-40 с
использованием двух моноклональных антител к нему, взаимодействующих с
разными эпитопами. По оси ординат отложена радиоактивность (имп/мин)
иодированных антител, связавшихся с аффинноиммобилизованным антигеном,
а по оси абсцисс — концентрация антигена. Исследование проводили в двух
параллельных пробах, и с целью демонстрации стабильности метода на рисун-
ке представлены кривые, полученные для двух параллелей.
геликазу [7] и репликативные функции Т-антигена [8]. Снача-
ла, используя данные по связыванию с гибридным белком ^-га-
лактозидаза— большой Т-антиген и данные по взаимодействию
антитела с делеционными мутантами, мы картировали эту
антигенную детерминанту, определив, что она занимает область
длиной в 64 аминокислотных остатка, расположенную между
остатками 448—509 большого Т-антигена вируса SV-40 [9].
РАЬ204 обнаруживает перекрестную реакцию с эволюционно
консервативным белком хозяина р68, содержание которого из-
меняется в процессе роста клеток [10]. Недавно путем скри-
нинга библиотеки кДНК гипатомы человека в векторе Xgtll
[12] с помощью РАЬ204 был получен набор клонов кДНК, ко-
дирующих р68 [11].
Был секвенирован один из клонов с наиболее короткой по-
следовательностью ДНК, кодирующей эпитоп РАЬ204 в белке
р68. Его аминокислотная последовательность была сопоставле-
на с последовательностью длиной 63 аминокислоты внутри
большого Т-антигена, включающей эпитоп. Методом твердофаз-
ного иммуноферментного анализа (ИФА) изучали взаимодей-
ствие серии из четырех синтетических полипептидов, соответст-
вующих этому участку, с РАЬ204. В результате было установлено,
что эпитоп представляет собой участок длиной 17 амино-
202
С. Моул, Д. Лейн
Таблица 6.14. Биотинилирование моноклональных антител
1. Добавьте раствор эфира биотин-сукцинимида (фирма Miles Ltd., концент-
рация 10 мг/мл) в ДМСО к раствору антител (концентрация 1—3 мг/мл)
в свободном от аминокислот буфере, таком как боратный или Na-бикар-
бонатный при pH 8,8. На 1 мг белка вносят 25—250 мкг эфира. Хорошо
перемешайте и инкубируйте 4 ч при комнатной температуре.
2. Добавьте к смеси NH4C1 из расчета 20 мкл 1 М NH4C1 на 1 мг белка или
на 250 мкг эфира.
3. Для удаления эфира диализуйте смесь или пропустите ее через колонку
с сефадексом G-50.
4. Храните при 4 °C PBS, содержащем 1% СЭК и азид.
кислот, находящийся между аминокислотами 453 и 469 большо-
го Т-антигена [5]. Обращает на себя внимание расположение
этого участка в непосредственной близости от ранее обнаружен-
ного в последовательности Т-антигена универсального (consen-
sus) участка связывания нуклеотидов [73]. Это указывает на
то, что участок, обладающий АТРазной активностью, был иден-
тифицирован правильно.
6.5. Другие применения: челночное перемещение
EcoRI-икассет» с клонированным фрагментом
Особое преимущество при использовании экспрессирующих
плазмид семейств pUR или фагов Xgtll в качестве векторов для
экспрессии гибридных белков дает наличие двух EcoRI-сайтов,
фланкирующих сайт клонирования у З'-конца гена lacZ. Воз-
можности использования таких систем могут быть сильно расши-
рены переносом £соР1-«кассет», содержащих встроенный фраг-
мент, в другой экспрессирующий вектор — например, в вектор
pSEMCatRl (рис. 6.7), сконструированный в нашей лаборато-
рии [5]. Он представляет собой производное pSV2-catr [14],
описанной во втором томе данного издания. Это бактериально-
эукариотический челночный вектор, который можно наработать,
размножая его в бактериальных клетках-хозяевах в условиях
ампициллиновой селекции. Вектор также содержит ген, коди-
рующий хлорамфеникол-ацетилтрансферазу (CAT), который
может экспрессироваться как в бактериальных клетках, так и в
клетках млекопитающих вследствие наличия в нем тандема
промоторов, промотора ранних генов вируса SV-40 и бактери-
ального промотора. У З'-конца гена CAT расположены сайт
полиаденилирования и интрон малого Т-антигена вируса SV-40.
Плазмида pSEMCatRl содержит уникальный EcoRI-сайт
внутри гена CAT, сохраняющий одну рамку считывания с
З'-концом гена lacZ. Таким образом перенос £со/?1-«кассеты»
из плазмиды pUR, в которой содержится участок, кодирующий
гибридный белок, в этот EcoRI-сайт делает возможной экспрес-
Моноклональные антитела к гибридным белкам из Е. coli 203
Таблица 6.15. Связывание моноклональных антител с белком А
А. Реактивы
Сефароза, конъюгированная с белком A (Protein А-Sepharose, Pharmacia)
Диметилпипелимидатдигидрохлорид (ДПД, DPD, Sigma)
0,1 М боратный буфер, pH 9,0 (борная кислота — тетраборат натрия)
0,1 М боратный буфер, pH 8,0
0,02 М этаноламин, pH 8,0 (НС1)
0,1 М цитрат, pH 3,0 (лимонная кислота/цитрат натрия)
Б. Подготовка колонки
1. Пропустите 100 мл гибридомной надосадочной фракции через колонку с
гранулами белок А — сефароза (Protein A Sepharose) в течение ночи при
4 °C. С белок А — сефарозой связываются антитела подклассов IgG2a,
IgG2b или IgG3, но не связываются IgGl.
2. Тщательно промойте колонку 0,1 М боратным буфером, pH 9,0, и ресупен-
дируите гранулы сефарозы в том же буфере при комнатной температуре.
Оставьте аликвоту для проверки степени связывания иммуноглобулина
с гранулами сефарозы (с 1 мл сефарозы должно связываться 0,5—2 мг
иммуноглобулинов).
3. Добавьте для образования сшивок 50 мг ДПД (его следует хранить в ва-
куумном эксикаторе при температуре —20 °C и добавлять к гранулам не-
посредственно после взвешивания). Тщательно смешайте компоненты и
оставьте па 1 ч при комнатной температуре, периодически перемешивая.
4. Осадите гранулы сефарозы центрифугированием, промойте один раз 0,1 М
боратным буфером (pH 8,0) и инкубируйте 10 мин в этаноламине. Тща-
тельно отмойте сефарозу и храните ее в 0,1 М боратном буфере (pH 8,0).
В. Работа с колонкой
1. Промойте колонку буфером, используемым для элюции антигена (таким,
как 0,1 М цитрат; pH 3,0). Убедитесь в том, что с колонки не сходит
белок.
2. Уравновесьте колонку буфером для нанесения антигена (в случае Т-ан-
тигена SV-40 мы используем 20 мМ трис-НС1, pH 8,0, 120 мМ NaCl, 1 мМ
ЭДТА, 0,1% NP-40).
3. Нанесите антиген, пропуская экстракт через очень маленькую (объемом
1 мл) колонку и затем промойте ее как минимум 100 мл буфера.
4. Проведите элюцию, многократно пропуская буфер в направлении, проти-
воположном направлению, в котором антиген наносили. Соберите фракции
по 500 мкл непосредственно в буфер для нейтрализации (200 мкл 1 мМ
трис-НС1; pH 8,8).
5. После завершения элюции снова уравновесьте колонку.
сию клонированного фрагмента гена с образованием включаю-
щего CAT гибридного белка как в бактериальных клетках, так
и в клетках млекопитающих. Это, например, позволяет прово-
дить в клетках млекопитающих эксперименты по локализации
и исследованию функциональной активности продуктов гена,
экспрессирующегося с образованием гибридного белка с CAT.
Кроме того, это дает возможность сравнивать активности гиб-
ридного белка, содержащего CAT, в бактериальных клетках и
клетках млекопитающих, поскольку и в тех, и в других клетках
может работать одна и та же рекомбинантная конструкция.
Для проведения многих таких экспериментов требуются моно-
клональные антитела, обладающие специфичностью по отноше-
204
С, Моул, Д. Лейн
Amp
on
pSEMCatR
5,2 т.п.н.
Интрон малого
t- антигена
и сайт поли-
+Ц аденилиро
:::: вания вируса
J: SV40
Промотор CAT
Промотор об-
ласти ранних
генов вируса
SV-40
EcoRl
CAT
Рис. 6.7. Челночный прокариотически-эукариотический вектор pSEMCats1. Если
смотреть против часовой стрелки, этот вектор включает ген, кодирующий
устойчивость к ампициллину, участок начала репликации pBR322, промотор
области ранних генов SV-40, содержащий ТАТА-бокс Голдберга — Хогнесса,
промоторную область гена CAT (хлорамфеникол-ацетилтрансферазы), содер-
жащую сайт начала транскрипции (этот ген кодирует устойчивость к анти-
биотику хлорамфениколу), интрон малого t-антигена и сайт полиаденилирова-
иия области ранних генов вируса SV-40.
нию к исследуемому гибридному белку. Так, клонируя гены,
кодирующие гибридный белок, в плазмидах pUR, можно полу-
чить новые моноклональные антитела, специфичные по отноше-
нию к продукту ОРС. Перенос этой ОРС в плазмиду
pSEMCatRl, затем позволит in vivo осуществлять экспрессию
гена и проводить эксперименты по локализации и определению
функциональных особенностей гибридного белка, содержащего
CAT, с использованием новых полученных моноклональных ан-
тител.
6.5.1. Примеры
Мы трансфецировали встроенный в плазмиду pSEMCatRl
фрагмент ДНК, кодирующий N-концевой участок (аминокисло-
ты 1-271) большого Т-антигена вируса SV-40 в составе САТ-
гибридного белка, в клетки CV-1; экспрессируемый белок лока-
лизуется в ядре этих клеток (рис. 6.8). При использовании
в реакции иммунопреципитации разных моноклональных анти-
тел, связывающихся с эпитопами, локализованными в разных
участках этого функционального домена, было показано, что
гибридный белок CAT—большой Т-антиген вируса SV-40 (ами-
Рис. 6.8. Специфическая локализация гибридных белков CAT-SV-40 после трансфекции клеток NIH3T3 рекомбинант-
ными конструкциями на основе челночных векторов. А. Реакция гибридного белка CAT-фрагмент большого Т-антигена
(аминокислоты 1—271) с моноклональными антителами, узнающими N-концевой участок Т-антигена, демонстрирует,
что гибридный белок локализуется в ядре. Б. Реакция гибридного белка CAT-фрагмент малого t-антигена (ами-
нокислоты 1—174) с моноклональными антителами, узнающими уникальный участок последовательности t-антиге-
на, демонстрирует, что гибридный белок локализуется в цитоплазме. Для выявления связывания антител с гибрид-
ными белками использовалась пероксидазная система иммунодетекции (как описано в табл. 6.7).
206
С. Моул, Д. Лейн
Рис. 6.9. Осажденный в реакции иммунопреципитации гибридный белок
CAT—большой Т-антиген сохраняет способность специфически связываться
с участком начала репликации SV40. Дорожки М и N — fcoRI-рестрикты ДНК
SV-40 с мечеными концами (область начала репликации SV-40 находится
в пределах EcoRI-фрагмента G). Дорожки 1—4 — иммунопреципитация боль-
шого Т-антигена SV-40, который специфически связался с участком начала
репликации. Дорожка 1—контрольные антитела, не узнающие большой Т-ан-
тиген; дорожки 2—4 — моноклональные антитела, узнающие и поэтому осаж-
дающие большой Т-антиген, который in vitro связался с участком начала реп-
ликации; дорожки 5 и 6 — иммунопреципитация гибридного белка CAT—фраг-
мент большого Т-антигена (аминокислоты 1—271), который также специфиче-
ски связывается с областью начала репликации; дрожка 5 — контрольные ан-
титела, не узнающие гибридный белок; дорожка 6 — моноклональные антите-
ла, узнающие и преципитирующие гибридный белок, связавшийся in vitro
с областью начала репликации SV-40; дорожки 7—9 — иммунопреципитация
гибридного белка CAT—фрагмент большого Т-антигена (аминокислоты 271—
708), не связывающегося с областью начала репликации; дорожка 7 — конт-
рольные антитела, не узнающие гибридный белок; дорожки 8 и 9 — антитела,
узнающие и преципитирующие гибридный белок: коприципитации ДНК не
наблюдается. (Рисунок взят нз работы [5]; приводится с любезного разреше-
ния авторов.)
нокислоты 1—271) in vitro специфически связывается с ДНК,
содержащей «область начала» репликации SV-40 (рис. 6.9).
Считают, что этот участок большого Т-антигена отвечает за им-
мортализацию клеток; этот феномен можно исследовать с по-
мощью САТ-челночиого вектора.
Благодарности
Начальные этапы этой работы обеспечивались Обществом
онкологических исследований (Cancer Research Campaign),
Моноклональные антитела к гибридным белкам из Е. coli
207
а последующие этапы — Королевским фондом по онкологиче-
ским исследованиям (Imperial Cancer Research Fund).
Литература
1. Riither U„ Muller-Hill В. (1983). EMBO J., 2, 1791.
2. Maniatis T., Fritsch E. F., Sambrook J. (eds.) (1982). Molecular Cloning:
A Laboratory Manual, Cold Spring Harbor Laboratory Press, Cold Spring
Harbor, N. Y. [Имеется перевод: Маниатис T., Фрич Э., Сэмбрук Дж. Ме-
тоды генетической инженерии. Молекулярное клонирование. — М.: Мир,
1984.]
3. Messing J., Groneneborn В„ Milller-Hill В., Hof Schneider Р. Н. (1977). Proc.
Natl. Acad. Sci. USA, 74, 3642.
4. Riither U., Koenen M., Otto K., Muller-Hill B. (1981). Nucleic Acids Res., 9,
4087.
5. Mole S. E„ Gannon J. V., Ford M. J., Lane D. P. (1987). Phil. Trans. R. Soc.,
Series B, 317, in press.
6. Clark R., Lane D. P., Tjian R. (1981). J. Biol. Chein., 56, 11854.
7. Stahl H„ Droge P., Knippers R. (1986). EMBO J., 5, 1939.
8. Smale S. T„ Tjian R. (1986). Mol. Cell. Biol., 6, 4077.
9. Mole S. E„ Lane D. P. (1985). J. Virol., 54, 703.
10. Lane D. P„ Hoeffer IF. A. (1980). Nature, 288, 167.
II. Ford M. J., Lane D. P., in preparation.
12. de Wet !. R., Fukushima H., Dewji N. N., Wilcox E., O’Brien J. S., Helin-
ski D. R. (1984). DNA, 3, 437.
13. Seif I. (1984). Virology, 138, 347.
14. Gorman С. M., Moffat L. F„ Howard В. H. (1982). Mol. Cell. Biol., 2,
1044.
15. Burnette IV N. (1982). Anal. Biochem., 112, 195.
ГЛАВА 7
ЭКСПРЕССИЯ И СЕКРЕЦИЯ ПРОДУКТОВ
ГЕТЕРОЛОГИЧНЫХ ГЕНОВ В ДРОЖЖАХ
Брюс Л. А. Картер, Меир Ирани, Вивиан Л. Маккей,
Рои Л. Сил, Анджей В. Следжевски и Роберт А. Смит
(Bruce L. A. Carter, Meher Irani, Vivian L. MacKay,
Ron L. Searle, Andrzej V. Sledziewski, Robert A. Smith)
7.1. Введение
Дрожжи Saccharomyces cerevisiae — удобный объект для
синтеза гетерологичных белков, используемых в фундаменталь-
ных и прикладных исследованиях. Первый рекомбинантный про-
дукт, полученный в дрожжевой системе — вакцина против виру-
са гепатита,—уже завоевал рынок. Вторым таким продуктом,
по всей видимости, будет продуцируемый дрожжами человече-
ский инсулин. Соответствующий технологический процесс, раз-
работанный фирмой Zymogenetics для предполагаемой фирмы-
производителя Novo Industri, включает в себя секрецию дрож-
жевыми клетками в культуральную среду модифицированного
проинсулина и его эффективное превращение in vitro в инсу-
лин.
Экспрессия гетерологичных полипептидов также может
служить удобной тест-системой для исследований в области
физиологии и биохимии дрожжей. Так, например, ген р-галак-
тозидазы Е. coli использовали в дрожжах для изучения таких
вопросов, как 1) ядерные системы адресованного транспорта;
2) функционирование промоторов; 3) механизмы секреции.
Кроме того, экспрессия гетерологичных генов позволяет по-
лучать новую информацию о природе и активности чужеродных
полипептидов. Так, экспрессия в дрожжах области p28sis виру-
са обезьяньей саркомы позволила установить, что биологическая
активность, ассоциированная с фактором роста тромбоцитов
(ФРТ), присуща именно этому фактору, а не цитокинам, кото-
рые могут присутствовать в препаратах ФРТ, выделенных из
животных источников [4].
Дрожжи давно уже входят в число излюбленных объектов
биологов-экспериментаторов; с открытием и введением в иссле-
довательскую практику методов трансформации дрожжевых
клеток экзогенной ДНК [5, 6] интерес к дрожжам пробудился
с новой силой. В значительной степени это объясняется тем,
что дрожжи — организмы эукариотические, что делает их таки-
ми же удобными объектами для биохимических и генетических
208
Продукты гетерологичных генов в дрожжах 209'
исследований (и классических, и молекулярных), как и клетки
Е. coli. Это обстоятельство дает возможность развивать мульти-
дисциплинарные подходы к фундаментальным проблемам био-
логии эукариот, например к изучению митоза. За последние пять
лет наблюдался также значительный интерес к возможному
использованию дрожжевых систем для продуцирования белко-
вых продуктов, имеющих, например, терапевтическую ценность.
Подобный интерес связан прежде всего с осознанием того фак-
та, что Е. coli не может считаться хорошим хозяином для про-
дуцирования большинства белков млекопитающих. Проблемы
здесь в первую очередь качественного, а не количественного
характера. Для выделения интересующего белка из клеток
E.coli часто не удается обойтись без цикла денатурации—рена-
турации. В результате полученный препарат часто теряет зна-
чительную часть биологической активности, свойственной аутен-
тичному белку.
Не следует думать, что дрожжи могут стать панацеей для
решения всех проблем, связанных с экспрессией генов, однако
уже есть примеры (в частности, упоминавшаяся нами вакцина
против вируса гепатита В), когда использование дрожжевых
систем помогло решить такие вопросы, которые не удавалось
обойти в рамках ограничений, присущих Е. со/г-системам.
Помимо цитоплазматической экспрессии дрожжевые систе-
мы позволяют секретировать гетерологичные белки в культу-
ральную среду. Существует несколько причин, по которым се-
креция белкового продукта оказывается предпочтительной. Мно-
гие из человеческих белков, представляющих терапевтический
интерес, секретируются клетками одних типов, тогда как дейст-
вие их направлено на другие клетки. Эти белки часто бывают
богаты цистеином, а множественные дисульфидные связи как
раз служат основным препятствием для успешной ренатурации
полипептидов, продуцируемых Е. coli.
Одно из преимуществ секреции состоит в следующем: в нор-
ме лишь очень немногие из собственных дрожжевых белков
секретируются в среду, и при достаточно высоком уровне гете-
рологичной экспрессии очистка соответствующего продукта не
составляет большого труда. Секреция дрожжами белков, в нор-
ме секретируемых клетками млекопитающих, обычно сопряже-
на с правильным формированием дисульфидных связей и как
следствие с сохранением этими белками биологической актив-
ности. Мы показали, что ФРТ, активная форма которого требует
образования многих дисульфидных связей и димеризации, эф-
фективно синтезируется, собирается и секретируется дрожжевы-
ми клетками, причем его биологическая активность не отлича-
ется от активности препаратов, выделенных из человеческих
тромбоцитов [4].
14—1186
210 Б. Картер, М. Ирани, В. Маккей и др.
Преимуществом секреции является также возможность гли-
козилирования гетерологичных белков, поскольку аппарат гли-
козилирования представляет собой часть аппарата секреции.
Внутренний углеводный кор собирается после транслокации
продукта в эндоплазматический ретикулум, а наружная цепь
формируется в аппарате Гольджи. Следует отметить, что гли-
козилирование наружных цепей в дрожжевых клетках происхо-
дит иначе, чем в клетках млекопитающих. Быстрая секреция
кроме всего прочего позволяет снизить токсическое действие
некоторых гетерологичных белков, проявляющееся при их экс-
прессии в цитоплазму.
Эффективность экспрессии белков человека в дрожжевых
системах зависит от природы самого продукта; известны ус-
пешные опыты, есть здесь и не очень рекламируемые неудачи
(как, например, ^-интерферон, на долю которого приходится
менее 0,1% всех белков, образующихся в дрожжевой клетке).
Вдобавок существуют белки — например, многие из тех, что
участвуют в процессах свертывания крови, — посттрансляцион-
ная модификация которых происходит по такому пути, который
не может реализоваться в клетках дрожжей (в данном случае
это гамма-карбоксилирование остатков глутаминовой кисло-
ты) [7].
7.1.1. Методы, включенные в данную главу
В этой главе мы обсудим методические подходы к экспрес-
сии и секреции гетерологичных белковых продуктов в дрожжах.
Многие манипуляции с ДНК (в частности, конструирование ре-
комбинантных плазмид) проводились с использованием Е. coli.
Поскольку эти методы достаточно полно описаны в отдельных
руководствах [8] и [9], мы не будем на них подробно останав-
ливаться, а сконцентрируем внимание на тех деталях, которые
связаны с особенностями дрожжевых систем. Отметим, что
«дрожжевая» методология подробно изложена в 11-м и 12-м то-
мах книги Methods in Cell Biology [10], целиком посвященной
экспериментальным подходам, разработанным для дрожжевых
клеток. Специальное руководство по генетике дрожжей [11].
опубликованное издательством Cold Spring Harbor, также со-
держит много полезных сведений по методологии биохимичес-
ких и молекулярногенетических исследований на дрожжах.
Различные экспериментальные процедуры, связанные с клони-
рованием генов в дрожжах, приведены в обзоре [12].
7.1.2. Экспрессирующие векторы
Известно много разных типов плазмид, которые можно ис-
пользовать для экспрессии гетерологичных генов в дрожжевых
клетках. Существующие между ними различия подробно описа-
Продукты гетерологичных генов в дрожжах
211
ны в другой работе [12]. На рис. 7.1 схематически показана
экспрессирующая плазмида типа YEp: эти плазмиды способны
к эписомной репликации на дрожжах, что и отражает приве-
денное сокращение (Yeast Episomal plasmid).
Большая часть манипуляций, необходимых для конструиро-
вания плазмид и их размножения, осуществляется в E.coli.
Поэтому такие плазмиды обязательно должны нести область на-
чала репликации, наличие которой необходимо для их реплика-
ции в клетках Е. coli. Чаще всего с этой целью в состав вектора
вводят соответствующие сегменты бактериальных плазмид, на-
пример pBR322. Кроме этого необходим маркер для селекции
трансформантов в E.coli: в случае плазмиды, показанной на
рис. 7.1, эту функцию выполняет ген устойчивости к ампи-
циллину.
Селекция трансформантов в дрожжах имеет принципиаль-
ное значение. В качестве селективного маркера часто исполь-
зуют ген leucine 2 дикого типа, который вводят в состав век-
торной плазмиды. Если маркированный таким образом вектор
трансформирует дрожжевые клетки-хозяева, несущие, дефект-
ную копию гена leucine 2, трансформанты (и только они) по-
лучают возможность расти на среде, обедненной лейцином.
Конечно, можно использовать и другие маркеры ауксотрофности.
Плазмиды типа YEp способны реплицироваться в дрожжевых
клетках благодаря присутствию в их составе области начала
репликации эндогенной дрожжевой плазмиды — так называемо-
го 2 мкм-кольца. Достижение высокой копийности при размно-
жении плазмид, содержащих только область начала репликации
2 мкм-кольца, требует присутствия в клетке-хозяине нативной
2 мкм-плазмиды, обеспечивающей две необходимые для этого
функции по транс-схеме. Гибридные плазмиды, несущие полный
2 мкм-плазмидный геном, достигают высокой копийности даже
в штаммах, в которых отсутствуют эндогенные 2 мкм-плазмиды
(С1'г°-штаммы). Гране-действующие белки, кодируемые генами
repl и гер2, могут «поставляться» нативными 2 мкм-плазмидами
или гибридными плазмидами, которые используются для экс-
прессии. Различные аспекты конструирования и применения
векторов на базе 2 мкм-плазмид для наработки клонированных
генов в дрожжевых клетках обсуждаются в работе Броха [13].
На первый взгляд может показаться, что высокая копий-
ность плазмиды благоприятствует максимальному уровню экс-
прессии. Однако это не всегда так. Дело в том, что некоторые
белки токсичны для дрожжевой клетки. В результате происхо-
дит селекция клеток с пониженным уровнем экспрессии таких
белков, а это обычно бывает обусловлено снижением копийности
плазмиды вплоть до полной ее утраты. В подобных случаях
интеграция плазмиды в какую-нибудь дрожжевую хромосому
14*
212
Б. Картер, М. Ирани, В. Маккей и др.
Штамм дрожжей рер4 leu 2 Zltpl
Z’-AAT-TP^-
Продукты гетерологичных генов в дрожжах
213
при низкой копийности обеспечивает стабильность системы и
достаточно высокий уровень экспрессии [14].
Дрожжевые интегрирующиеся векторы (Yip) не содержат
последовательностей ДНК, способных обеспечить функцию на-
чала репликации в дрожжевых клетках, и, следовательно,
трансформантов можно получить только в том случае, если
произошла рекомбинация и плазмида интегрировалась в дрож-
жевой геном [6].
Интеграция — результат рекомбинации между гомологичны-
ми последовательностями плазмидной ДНК и хромосомы клет-
ки-хозяина. Интегрированная копия плазмидного вектора ока-
зывается фланкированной прямыми повторами: дупликации
подвергается участок взаимной гомологии плазмидной и хромо-
сомной ДНК- Рис. 7.2 иллюстрирует рекомбинационные события
в области гена leucine 2. У многих трансформантов наблюдается
множественная интеграция плазмид в виде тандемно повторяю-
щихся копий (рис. 7.2). Часть интегративных рекомбинационных
событий происходит как двойной кроссинговер. Эта схема может
привести к замещению участка хромосомной ДНК на гомоло-
гичный участок плазмидной ДНК без интеграции векторной
части рекомбинантной плазмиды. Интеграция может произойти
в любом месте генома при условии, что в этом месте находится
последовательность, гомологичная участку плазмидной ДНК-
Единственное исключение составляет геномный сайт плазмидно-
го селективного маркера.
Векторы типа Yip обладают значительно более низкой транс-
формирующей способностью, чем векторы YEp (примерно на
2—3 порядка), однако некоторого повышения эффективности
трансформации клеток этими векторами можно добиться, если
Рис. 7.1. Дрожжевой экспрессирующий вектор. Вектор несет в себе: 1). дрож-
жевую область начала репликации (ori У), взятую из дрожжевой 2мкм-плаз-
миды, 2) ген 1еи2 для селекции трансформированных дрожжевых клеток,
а также 3) последовательности, взятые из pBR322: ori В, обеспечивающую
репликацию вектора в Е. coli, и АМР, обеспечивающую устойчивость к ампи-
циллину (для селекции трансформированных клеток Е. coli). В данном век-
торе использованы промотор и терминатор гена триозофосфат-изомеразы.
В идеальном экспрессирующем векторе эти регуляторные элементы должны
быть разделены сайтом для клонирования. В данном же векторе между про-
мотором и терминатором введена последовательность альфа-1-антктрипсина.
Этот вектор представляет собой сложную рекомбинантную конструкцию и
обеспечивает неселективный рост трансформированных клеток в сложной сре-
де. Селекцию обеспечивают промотор и ген триозофосфат-изомеразы (tri)
Schizosaccharomyces pombe при использовании клеток-хозяев с делецией в об-
ласти гена tri (bdri): только трансформированные клетки могут утилизировать
глюкозу, поскольку у клеток-хозяев отсутствует один из ключевых ферментов
гликолиза. Обозначенный на рисунке штамм-хозяин имеет к тому же дефект-
ный ген leucine 2, поэтому трансформантов можно отбирать на среде, не со-
держащей лейцина. Наконец, данный штамм несет мутацию рер4. Смысл этой
детали объясняется в тексте.
214
Б. Картер, М. Ираии, В. Маккей и др.
или
Leir
Leu+
Рис. 7.2. Возможные варианты интеграции плазмид. Результатом одиночного
рекомбинационного события является единичная интегрированная копня век-
торной последовательности, ограниченная прямыми гомологичными повтора-
ми (а), илн же множественные тандемно организованные копии вектора (б).
Двойной кроссннговер приводит к замещению хромосомной ДНК гомологич-
ными последовательностями рекомбинантной плазмиды, не сопровождающе-
муся интеграцией векторных последовательностей (в).
Продукты гетерологичных генов в дрожжах 215
в их участок, гомоличный дрожжевому геному, ввести двухце-
почечный разрыв.
Какого бы типа векторные плазмиды ни использовались,
для эффективного инициациирования транскрипции необходимо,
чтобы в их составе присутствовал дрожжевой промотор. Чаще
всего используют гликолитические промоторы. Поскольку фер-
менты гликолиза, несмотря на то что они кодируются уникаль-
ными генами, составляют от 1 до 5% суммарного клеточного
белка, можно было предположить (и зто оказалось верно), что
промоторы соответствующих генов относятся к разряду сильных
промоторов. Другое необходимое условие успешной экспрес-
сии — это эффективное терминирование транскрипции. Дрожже-
вые клетки обычно узнают терминаторные последовательности
млекопитающих, однако с точки зрения оптимизации системы
имеет смысл ввести в вектор дрожжевой терминатор (рис. 7.1).
Промотор и терминатор разделены уникальным сайтом рестрик-
ции, что позволяет реализовать стандартный путь создания
рекомбинантной конструкции с интересующим структурным
геном.
Плазмидные векторы, подобные тому, что изображен иа
рис. 7.1, призваны обеспечивать экспрессию гетерологичного
белка в цитоплазму. Экспрессирующие векторы этого типа раз-
работаны в ряде коммерческих и исследовательских лаборато-
рий; по крайней мере некоторые из этих лабораторий предо-
ставляют их для научных изысканий [15, 16].
Сейчас все больший интерес вызывает развитие управляемых
экспрессирующих систем. Дело в том, что промоторы гликоли-
тических генов относятся к конститутивному типу. Конститутив-
ная же экспрессия любого гена, клонированного в мультикопий-
ной плазмиде, приводит к заметному метаболическому сдвигу в
трансформированной клетке. В зависимости от природы гетеро-
логичного продукта этот сдвиг может осложняться токсическими
эффектами (различными как по механизму действия, так и по
степени тяжести), что, по-видимому, определяется также и сте-
пенью выражения данного гена. Низкий уровень трансформации
и медленный рост трансформантов — явления, гораздо более
распространенные, чем это может показаться при чтении лите-
ратуры.
Контролируемые промоторы изменяют ситуацию к лучшему,
поскольку позволяют при выключенной экспрессии получить хо-
роший выход биомассы. В идеале включение экспрессии должно
происходить в тот момент, когда ферментер заполнен сформи-
ровавшейся культурой в ответ на простой запускающий
стимул — изменение условий культивирования. Управляемые
системы экспрессии генов подробно обсуждаются в работе
[17].
216 Б. Картер, М. Ирани, В. Маккей и др.
7.1.3. Векторы для экспрессии/секреции
Для эффективной секреции гетерологичных полипептидов
необходимо, чтобы аминоконцевая последовательность зрелого
белка была состыкована с сигнальной (лидерной) последова-
тельностью дрожжевого секретируемого белка, например ин-
вертазы или кислой фосфатазы. Альтернативный и более совер-
шенный метод заключается в элиминации сигнального пептида
из состава экспрессируемого белка и замене его лидерной по-
следовательностью альфа-фактора, взятой из молекулы дрож-
жевого предшественника альфа-фактора. Альфа-фактор — это
половой фактор, секретируемый дрожжевыми клетками альфа-
типа спаривания. Клетки a-типа продуцируют и секретируют
a-фактор. Факторы спаривания функционируют, вызывая рецип-
рокную остановку в Gi-периоде клеток противоположного типа
спаривания перед конъюгацией [18]. Ген MFa-1 клонирован и
секвенирован [19]. Лидерная последовательность белкового про-
дукта этого гена имеет сложное строение: она содержит участок
из 20 аминокислот с типичным для классической сигнальной
последовательности аминокислотным составом и далее относи-
тельно гидрофобную цепь из 69 аминокислотных остатков, в ко-
торой находятся участки гликозилирования. Гликозилирование
и процессинг пре-про-альфа-фактора изучены довольно деталь-
но [20—22].
Нуклеотидная и соответствующая аминокислотная последо-
вательности лидерной зоны альфа-фактора и непосредственно
примыкающая к ней б'-промоторная последовательность гена
показаны на рис. 7.3. Основные подходы к использованию ли-
дерной последовательности альфа-фактора дрожжей для осуще-
ствления секреции гетерологичных генов описали Брейк и др.
[23]. В структурном гене MFa-1 локализован ЯтсПП-сайт,
который в сайте процессинга молекулы-предшественника захва-
тывает первый из двух повторов, соответствующих GIu-AIa. Это
открывает традиционный, относительно несложный путь присое-
динения зрелых гетерологичных полипептидов к сигнальному
пептиду альфа-фактора. Однако использование природного
//tndIII-сайта может привести к внеклеточной секреции продук-
та, гетерогенного по N-концу: большая часть молекул будет
нести один или два дополнительных N-концевых дипептида Glu-
Ala. Обусловлено это недостаточностью фермента дипептидил-
аминопептидазы — продукта гена stcl3. Фермент этот элимини-
рует повторы Glu-Ala после того, как под действием продукта
гена kex2 произойдет первичное эндопептидазное расщепление
полипептидной цепи предшественника за димером Lys-Arg.
Брейк и др. [23] предложили два способа решения этой про-
блемы. Первый подход заключался в присоединении нуклеотид-
Продукты гетерологичных генов в дрожжах
217
-70 -60 -50 С -40 -30 -20 -10
ACGATTCAAGAATAGTTCAAACAAGAAGATTACAAACTATCAATTTCATACACAATATAAACGACCAAA
А
1 10 20 PstI 30 40 50
AGA ATG AGA TTT CCT TCA ATT TTT ACT GCA GTT TTA TTC GCA GCA TCC TCC GCA
met arg phe pro ser ile phe thr ala val leu phe ala ala ser ser ala
60 Hine II 70 80 90 100
TTA GCT OCT CCA CTC AAC ACT АСА АСА GAA CAT GAA ACG GCA CAA ATT CCG GCT
leu ala ala pro val ASN THR THR thr glu asp glu thr ala gin ile pro ala
110 120 130 140 150
GAA GCT GTC АТС GGT TAG TCA GAT TTA GAA GGG GAT TTC GAT GTT GCT GTT TTG
glu ala val lie gly tyr ser asp leu glu gly asp phe asp val ala val leu
160 170 180 190 200 210
CCA TTT TCC AAC AGC АСА AAT AAC GGG TTA TTG TCT ATA AAT ACT ACT ATT GCC
j>ro phe ser ASN SER THR asn asn gly leu leu phe ile ASN THR Ж ile ala
220 230 AGC 250 Hind III
AGC ATT GCT GCT AAA. GAA GAA GGG GTA TCT TTG GAT AAA AGA GAG GCT GAA GCT
S$er ile ala ala lys glu glu gly val ser leu asp lys arg glu ala glu ala
4*4
Рис. 7.3. Лидерная последовательность альфа-фактора н прилежащая б'-про-
моторная последовательность. Введение направленной танковой нутации
(обозначенной значком А) приводит к появлению внутри промотора //indlll-
сайта, который используется для замещения лидерной последовательности
альфа-фактора желаемой гетерологичной последовательностью. В качестве
альтернативного варианта промотор можно использовать для цитоплазматиче-
ской экспрессии. Направленный мутагенез по сайту, обозначенному ААА
позволяет получать составные белки, в которых отсутствуют повторы Glu-Ala.
Для этой цели можно также изменять последовательности, непосредственно
предшествующие природному /п/гсПП-сайту.
нон последовательности, кодирующей гетерологичный продукт,
к естественному //mdl II-сайту через синтетический ДНК-линкер,
кодирующий второй участок эндопептидазного расщеплении
Lys-Arg непосредственно перед началом гетерологичного белка.
Реализация этого подхода с фактором роста эпидермиса чело-
века позволила получить 80%-ный выход продукта, процесс!:ко-
ванного желаемым образом по новому участку Lys-Arg. 10%
всех молекул имели N-концевой «довесок» состава Glu-Ala-Glu-
Ala-Ser-Leu-Asp-Lys-Arg, поскольку расщепление предшествен-
ника произошло в первичном, природном сайте процессинга.
Второй экспериментальный подход предполагал создание с
помощью направленного мутагенеза рестрикционного сайта
в лидерной последовательности так, чтобы он находился на до-
статочном удалении от места, соответствующего стыку лидерной
и структурной частей секретируемой белковой молекулы (бли-
же к началу гена), чтобы элиминировать повторы Glu-Ala, но в
218
Б. Картер, М. Ирани, В. Маккей и др.
Рис. 7.4. Вектор для экспрессии pJP31 (Пито, неопубликованные данные). Сег-
мент, не имеющий на рисунке специального обозначения, представляет боль-
шой EcoRI/BamHI-фрагмент pJDB 207, из которого удален йтсПП-сайт, при-
сутствующий в 2 мкм-плазмиде. Для этого использовали ДНК-полимеразу I
и ДНК-лигазу. В — Bglll-, Вт — ВатШ- Е —EcoRI; Н —tfindlll; Р — Pstl.
то же время и достаточно близко к первичному сайту расщеп-
ления, чтобы легко можно было его реконструировать с по-
мощью синтетического линкера, не кодирующего дипептиды
Glu-Ala. Процесс этот проиллюстрирован рис. 7.3 (Пиго, не-
опубликованные данные). Замена с помощью направленного
мутагенеза тринуклеотида ТОТ кодоном AGC позволяет сохра-
нить па своем месте в сигнальном пептиде серин и одновремен-
но получить в нуклеотидной последовательности лидера новый
ЯшсПП-сайт. Такой путь был успешно реализован для клони-
рования генов инсулина человека [24], интерлейкинов [25] и
фактора роста тромбоцитов [4].
На рис. 7.4 схематически изображен один из основных век-
торов для экспрессии/секреции в дрожжах гетерологичных бел-
ков pJP 31. В своем составе он содержит промотор альфа-фак-
тора (сильный промотор), лидерную последовательность альфа-
фактора, терминатор adhl и уникальный НшсПП-сайт для
клонирования структурной части интересующего гена.
Конструирование рекомбинантных молекул на базе подоб-
ных векторов, т. е. введение в них гетерологичных структурных
генов, осуществляется с помощью стандартных молекулярноге-
нетических процедур [8, 9]. Аммерер [26] опубликовал интерес-
ную методическую статью, посвященную экспрессии в дрожжах
генов с использованием промотора add. Эффективность лигиро-
Продукты гетерологичных генов в дрожжах
219
вания можно контролировать, анализируя индивидуальные
трансформированные бактериальные клоны. Трансформация
бактерий осуществляется стандартным образом [8, 9]. Ориента-
цию вставок определяют с помощью рестрикционного анализа
плазмидных препаратов. Плазмидные ДНК для трансформации
дрожжей очищают центрифугированием в градиенте плотности
CsCl и хранят при 4 °C.
7.2. Штаммы дрожжей, пригодные для экспрессии,
и их получение
По вполне понятным причинам высокопродуктивные дрож-
жевые штаммы, экспрессирующие гетерологичные белки, далеко
не всегда удается получить из коммерческих лабораторий, ко-
торые в основном занимаются их разработкой. И все-таки бла-
годаря университетским лабораториям и некоторым коммерче-
ским компаниям исследователь всегда может добыть несколько
подходящих штаммов. Кроме того, сконструировать подходящий
штамм-хозяин можно самостоятельно — это вполне реальная
задача.
Приемлемый для большинства целей штамм-хозяин Х4003-5В
можно заказать в коллекции дрожжевых культур при Кали-
форнийском университете (Yeast Genetic Stock Center, Donner
Laboratories, University of California, Berkeley, California, USA).
Этот штамм позволяет проводить эксперименты по экспрес-
сии и секреции чужеродных белков в масштабах качалочной
колбы. Его генотип: mat-a-leu-2-adel-his4-met2-ura3-trp5-gali.
Поскольку штамм Х4003-5В — это /ем2_-мутант, его используют
в паре с плазмидами, маркированными геном 1еи2 дикого типа.
Следует, однако, иметь в виду, что штамм-хозяин, несущий
единственную точковую мутацию в гене, который выполняет
роль селективного маркера для отбора трансформантов, являет
собой недостаточно надежную систему, так как существует опас-
ность реверсии маркера к дикому типу. Это превращается в
проблему, если удается получить лишь небольшое число транс-
формантов: можно ожидать, что ревертанты составляют значи-
тельную часть всех клеток, выросших на среде, не содержащей
лейцина. Такая ситуация, естественно, не может считаться удов-
летворительной. Избежать подобных неприятностей помогают
штаммы типа LL20 (mat-a-his3-ll,15,leu2-3,112), сконструиро-
ванного Лесли Ло из Корнеллского университета, или D234-3B
(mat-a-his3-ll,15-leu2-3-tcml-trpl-ura3), созданного П. Брауном
из Института рака Dana-Farber. Эти штаммы несут двойные
мутации в двух маркерах и пригодны для работы в паре с 1еи2-
или ИдЗ-плазмидами. Двойные точковые мутации снижают ве-
роятность появления ревертантов до 10-10. Несколько подобных
220 Б. Картер, М. Ирани, В. Маккей и др.
Таблица 7.1. Идентификация рер4~- и рер4+-гаплоидиых сегрегацию,
полученных в результате скрещиваний
1. Вырастите колонии на ташках с YEPD-агаром.
2. Смешайте в стеклянной пробирке 3 мл 0,6%-ного расплавленного агара
(охлажденного до 50 °C) с 2 мл раствора N-ацетил D.L-фенилаланин-р-
пафтплового эфира в диметилформамиде (3 мг/мл).
3. Кзлейге смесь поверх к-лтоиний и дайте ей застыть.
4. Аккуратно налейте на поверхность 4 мл раствора, содержащего 5 мг/мл
T'cst Garnet В. С. в 0,1 М трис-НС1 (pH 7,3). Этот краситель производит-
ся фирмой Sigma; обращение с ним требует большой осторожности, по-
. тольку он обладает канцерогенными свойствами.
5. Когда колонии начнут розоветь, удалите раствор красителя и аккуратно
промойте поверхность чашки дистиллированной водой.
6. рер4~ иолсинп приобретают окраску медленнее, чем колонии дикого типа,
и вдобавок не становятся интенсивно красными — они имеют желтоватый
оттенок.
штаммов, обеспечивающих приемлемую эффективность транс-
формации, можно заказать в коллекции дрожжевых культур.
Выбор штамма-хозяина диктуется типом векторной плазми-
ды, которую предполагается использовать для трансформации:
хозяин должен нести мутацию в гене, копия которого в плазми-
де выполняет роль селективного маркера. Так, если доминант-
ным селективным маркером служит плазмидный ген cupl
(обеспечивающий устойчивость к ионам меди), перед исследо-
вателем открывается широкий выбор штаммов-хозяев (гаплоид-
ных, диплоидных или даже не охарактеризованных пивных
дрожжей), поскольку большинство из них чувствительно к ио-
нам меди, а это единственное необходимое условие их пригод-
ности для работы с данным вектором.
В ряде случаев представляется целесообразным введение в
штамм-хозяин рер4-мутаций. Наиболее часто используют мута-
цию рер4-3. Несущие эту мутацию клетки-хозяева не обнаружи-
вают собственно повышенной экспрессии клонированного гена;
они снижают протеолитическую деградацию экспрессируемых
в цитоплазму белков в бесклеточных экстрактах и уменьшают
деградацию секретируемых полипептидных продуктов, которая
может происходить вследствие лизиса клеток. Рер4-мутации
вводят в штамм-хозяин путем стандартных генетических скре-
щиваний. Разделение тетрад аскоспор описано в разд. 7.2.2.3,
а метод идентификации рер4+- и рер4~-колоний приведен в
табл. 7.1.
7.2.1. Хранение дрожжевых культур
Некоторое время дрожжевые культуры можно хранить при
4 °C. На длительное хранение штаммы рекомендуется заклады-
вать в глицероле при —80 °C. Это целесообразно делать с куль-
Продукты гетерологичных генов в дрожжах
221
Таблица 7.2. Хранение дрожжевых культур
1. Налейте в небольшой флакон 2 мл 30%-ного глицерола и неплотно за-
кройте пробкой.
2. Автоклавируйте в течение 20 мин, охладите и плотно закройте.
3. Храните при комнатной температуре.
4. Нарастите 3 мл ночной дрожжевой культуры в среде YEPD’> (или дру-
гой подходящей селективной среде).
5. Перенесите 2 мл культуры во флакон, закройте крышкой и перемешайте.
6. Храните при —80 °C. Культура остается жизнеспособной в течение 5 лет.
7. Пользуйтесь замороженными культурами следующим образом:
а) дайте образцу оттаять на льду
б) микробиологической петлей перенесите некоторое количество материа-
ла в подходящую среду и инкубируйте при 30 °C (25 °C для темпера-
турочувствитель.чых штаммов).
в) снова заморозьте флакон при —80 °C.
) Среда YEPD содержит 1% дрожжевого экстракта, 2% бактопептоиа и 2%
глюкозы.
турами трансформированных дрожжевых клеток, хотя жела-
тельно все-таки ретрансформировать клетки хранящимися пре-
паратами плазмид. Приготовление глицероловых музеев
дрожжевых штаммов описано в табл. 7.2.
7.2.2. Конструирование штаммов
Конструирование штаммов часто пугает новичков в «дрож-
жевой науке» благодаря распространенному мнению о том, что
физическое отделение гаплоидных спор с помощью микромани-
пуляций— занятие весьма трудоемкое и требует известного мас-
терства. В действительности дело обстоит вовсе не так серьезно,
хотя определенные технические навыки безусловно необходимы.
Состав сред для конструирования штаммов приведен в табл. 7.3.
7 .2.2.1. Получение диплоидов
Если две гаплоидные дрожжевые клетки несут индивидуаль-
ные гены, которые планируется совместить в одной гаплоидной
клетке, необходимо провести скрещивание этих штаммов, по-
лучить диплоидные клетки, а затем после мейоза и споруляции
выделить и анализировать гаплоидные споры.
Обычно работают с двумя штаммами противоположных ти-
пов спаривания, имеющими комплементарные маркеры ауксо-
трофности. Небольшие количества каждой культуры (предвари-
тельно выращенной на агаровых чашках) смешивают на чашке
с YEPD-агаром и инкубируют при 30 °C в течение ночи.
Существует и другой способ: на чашку наносят оба штамма
перекрещивающимися штрихами. Клетки переносят с помощью
222
Б. Картер, М. Ирани, В. Маккей и др.
Таблица 7.3. Среды для конструирования штаммов
YEPD-агар
1 % дрожжевого экстракта
2% бактопептона
2% глюкозы
2% агара
Синтетическая полная среда')
0,67% дрожжевого азотистого компонента без аминокислот (Difco)
2% глюкозы
2% агара
по 10 мкг/мл аденина, метионина
по 20 мкг/мл аргинина, гистидина, лейцина и триптофана
10 мкг/мл урацила и лизина
по 50 мкг/мл тирозина, треонина2’ и лейцина
по 60 мкг/мл фенилаланина и изолейцина
Предварительная споруляционная среда
0,8% дрожжевого экстракта
0,3% бактопептона
10% глюкозы
2% агара
Среда для споруляции
1 % ацетата калия
0,1% дрожжевого экстракта
0,05% глюкозы
2% агара
') Лейцин, равно как и любую другую аминокислоту, можно не добавлять в среду,
чтобы получить так называемые чашки с пропуском, т. е. содержащие селективную сре-
ду для соответствующих ауксотрофных мутантов.
2) Стерилизуют фильтрованием и добавляют после автоклавирования среды.
реплик на среду, не содержащую аминокислот, по которым эти
гаплоидные штаммы ауксотрофны. Поскольку на такой среде
могут расти только диплоидные клетки, рост дрожжевой куль-
туры будет наблюдаться только в местах пересечения штрихов.
7.2.2.2. Получение гаплоидных спор
1. Пересейте клетки на селективную среду, чтобы отделить
диплоидов от гаплоидных клеток, и инкубируйте при 30 °C в
течение двух суток.
2. С помощью стерильной петли перенесите колонии на агар
для предварительной споруляции и инкубируйте еще в течение
двух дней при 30 °C.
3. Перенесите клетки на среду для споруляции и инкуби-
руйте при 30 °C в течение четырех или более суток. Проверьте,
происходит ли споруляция, перенеся немного дрожжевой куль-
туральной массы в каплю воды на предметном стекле. В спору-
лирующей культуре должны быть аски, содержащие по четыре
споры и называемые тетрадами. Некоторые аски, как может по-
Продукты гетерологичных генов в дрожжах 223
казаться на первый взгляд, несут только три споры. Однако
перемещение фокальной плоскости микроскопа неизбежно вы-
явит четвертую спору.
4. Отберите небольшое количество культуры и суспендируй-
те в 40-кратном разведении глузулазы, стерилизованной фильт-
рованием (Endo Labs Garden City, NY). Оставьте на качалке
при 30 °C на 1 ч. Вместо глузулазы можно использовать так
называемый улиточный фермент — желудочный сок улитки
Helix pomatia (L’Industri Biologique Francaise, Genevilliers,
France). Оба ферментных препарата разрушают оболочку аска,
но не диссоциируют споры. Продолжительность ферментативной
обработки, необходимой для разрушения асков, может быть
различной в зависимости от штамма дрожжей.
7.2.1.З. Препарирование асков и разделение тетрад аскоспор
Препарирование асков производят: в США — в переверну-
том положении, помещая агаровую пластину так, что аски ока-
зываются на ее нижней поверхности; в Европе — как правило,
ставя чашку с агаром естественным образом. Европейский ме-
тод предполагает введение иглы микроманипулятора между объ-
ективом и поверхностью агара, что диктует выбор объектива
с большим рабочим расстоянием (обычно это объектив 20х).
Желательно, чтобы общее увеличение составляло 150—300 X-
Препарирование асков на перевернутом агаре предъявляет по-
вышенные требования к микробиологической чистоте агара;
впрочем, загрязнения — это не самая серьезная из проблем,
с которыми приходится сталкиваться европейским микробио-
логам.
Применяется несколько типов микроманипуляторов — см.
обзор Шермана [27]. Наибольшее распространение имеют мик-
романипуляторы английской фирмы Singer Instrument Company
Ltd., американской фирмы Lawrence Precision Machine и фран-
цузской de Fonbrune Series A (Beaudion, Paris, France)
(рис. 7.5).
Иглу для микроманипулятора изготавливают с помощью
специального аппарата, однако ее можно сделать и вручную.
1) Нагрейте 2 мм стеклянную палочку на малом пламени
газовой горелки и вытяните ее, чтобы получить тонкий кончик.
2) Для препарирования на перевернутом агаре согните кон-
чик под прямым углом (для «прямого» препарирования этого
делать не следует — изгиб иглы сделает ее контур под микро-
скопом нечетким). Расстояние от точки изгиба до конца иглы
составляет несколько миллиметров и может варьировать в за-
висимости от высоты препарационной камеры (см. ниже).
224
Б. Картер, М. Ирани, В. Маккей и др.
Рис. 7.5. Микроманипулятор de Fonbrune Series А. Справа расположена руч-
ка-регулятор, слева — пневматический исполнительный механизм. Можно раз-
глядеть микроиглу, выступающую из микроманипулятора; ее конец уходит
в препарацнонную камеру, закрепленную на столике микроскопа.
3) Отломите кончик между предметным стеклом и краем
покровного стекла; он должен иметь в диаметре 10—100 мкм.
Шерман [27] описал простую препарацнонную камеру. Она
состоит из металлической рамки, имеющей в плане форму бук-
вы П. Ее внешние размеры — 3X1,5 дюйма1, глубина 0,375 дюй-
ма. Дно камеры образует стеклянная пластина (3x1,5 дюйма),
приклеенная к рамке эпоксидным клеем. Камера может быть
сдетана и целиком из стекла.
1) Пока аски обрабатываются глузулазой, залейте чашку
препарационпым агаром (10 мл на 1 чашку). Подсушите чашку
при 37 °C приблизительно 30 мин. Манипулировать с аскамт; а
спорами легче, если агар не очень влажный, но и не слишком
сухой.
2) Стеклянную пластину размером 3X1,5 дюйма погрузите
в этанол, обожгите в пламени горелки и поместите в пустую
стерильную чашку Петри.
1 1 дюйм равен 2,54 см. — Прим. ред.
Продукты гетерологичных генов в дрожжах 225
3) Стерильным скальпелем вырежьте прямоугольную плас-
тинку препарационного агара (2x5 см) и поместите на приго-
товленное стекло в чашку Петри, стараясь, чтобы между агаром
и поверхностью стекла не оказалось пузырьков воздуха.
4) Обработанную глузулазой дрожжевую массу нанесите
петлей по краю вдоль длинной полосы агаровой пластинки.
5) Поместите стекло с агаром на препарационную камеру
(агаром вниз). Для фиксации его в таком положении можно
использовать вазелин. Препарационную иглу вводят в камеру
через открытую стенку. Постарайтесь не обломить хрупкий кон-
чик иглы.
6) Сфокусируйте микроскоп на поверхности агара и пере-
мещайте столик, пока в поле зрения не появится дрожжевая
суспензия. Выберите кластер из четырех спор — это один аск.
7) Подцепите споры микроиглой. Для этого манипулируйте
иглой таким образом, чтобы она касалась агара в непосредст-
венной близости от аска. Поверхностное натяжение способству-
ет отбору спор: аск начинает двигаться по направлению к игле.
8) Опустите иглу так, чтобы она отделилась от поверхно-
сти агара; споры исчезнут из поля зрения. Если часть аска
осталась на агаре — одна или несколько спор, — не подбирайте
их и не берите в работу отобранные споры, чтобы исключить
опасность отбора по ошибке вместо спор клеток. Удалите споры
с иглы, касаясь ею поверхности агара.
9) После того как целый аск подцеплен микроиглой, пере-
местите камеру так, чтобы в поле зрения оказался ее противо-
положный край, и перенесите все четыре споры на агар. От-
метьте координаты столика. Снова подцепите иглой три из
четырех спор, передвиньте камеру на Змм обратно (приближая
тот край, где нанесена культура) и перенесите эти три споры
на агар. Снова подцепите иглой две из них, передвиньте камеру
еще на 3 мм в ту же сторону, перенесите споры на агар и по-
вторите этот цикл еще раз, чтобы все четыре споры из одного
аска оказались отделены друг от друга.
10) Передвиньте камеру на 3 мм вдоль той стороны, где
нанесена суспензия, выберите новый аск и повторите всю про-
цедуру препарирования. Так следует обработать примерно
15 асков. Регулярное расположение спор имеет важное значе-
ние, так как позволяет идентифицировать и исключить из
анализа посторонние колонии, выросшие не там, где поло-
жено.
11) Снимите камеру со столика (не обломите иглу!).
12) С помощью стерильного шпателя перенесите агаровую
пластину (спорами вверх) на YEPD-чашку.
13) Инкубируйте при 30 °C, пока колонии не вырастут на-
столько, чтобы их можно было пересеять петлей (2—3 дня).
15-1186
226 Б. Картер, М. Ир ани, В. Маккей и др.
14) Пересейте колонии на чашку — каждую тетраду от-
дельно.
15) Проанализируйте генотип каждого клона методом реп-
лик на соответствующую селективную среду.
7.3. Трансформация дрожжей
Интактные дрожжевые клетки можно трансформировать эк-
зогенной ДНК подобно тому, как это делают с клетками бак-
терий [28]. Для того чтобы повысить эффективность дрожжевой
трансформации, следует удалить дрожжевую клеточную стенку
и работать со сферопластами [5, 6]. С этой целью дрожжевые
клетки инкубируют в осмотически стабилизированном растворе,
содержащем ферменты, которые разрушают клеточную стенку.
Дрожжелитической активностью обладают такие «экзотические»
препараты, как экстракт желудка виноградной улитки или фер-
ментные комплексы, используемые в производстве зубной пас-
ты. Сферопласты инкубируют с ДНК в присутствии ионов каль-
ция и полиэтиленгликоля (ПЭГ). ПЭГ вызывает агрегацию
протопластов, провоцируя локальные мембранные спайки и за-
хват ДНК. Поскольку дрожжевые сферопласты не способны к
регенерации клеточной стенки в жидкой среде, трансформиро-
ванные клетки смешивают с плавленым агаром и агаризован-
ную культуру выливают на чашку; погруженные в агар клетки
регенерируют клеточную стенку. Состав среды для регенерации
должен быть подобран так, чтобы расти могли только трансфор-
манты. Например, если клетки-хозяева дефектны по какому-
нибудь гену аминокислотного биосинтетического пути, а транс-
формирующая плазмида несет копию дикого типа эквивалент-
ного гена, трансформантов удобно отбирать на среде, не содер-
жащей соответствующей аминокислоты. Описанная в табл. 7.4
методика трансформации предполагает использование в качест-
ве маркера гена leucine 2. Аналогичная процедура описана
Р. Ротстейном в гл. 3 первого тома данной серии. Небольшие
вариации в методиках отражают конкретный практический опыт
разных лабораторий. Эти методы в принципе равноценны, во
всяком случае трудно найти преимущество одного из них над
другими. В табл. 7.4 описан метод, использованный Zymogene-
tics.
Бактериальные трансформанты, например трансформирован-
ные клетки Е. coll, растут на поверхности агара, и их можно
анализировать методом реплик. Трансформированные же клет-
ки дрожжей растут в толще агара и, следовательно, требуют
индивидуального подхода: выделения каждого клона и его по-
следующего анализа. Часто для этой цели используют стериль-
ные зубочистки — как более дешевую замену петле разового
Продукты гетерологичных генов в дрожжах 227
Таблица 7.4. Трансформация дрожжей: метод с использованием сферопластов
А. Реагенты
1. YEPD: 1% дрожжевого экстракта, 2% пептона, 2% глюкозы (декстрозы).
2. SED1’: 1 М сорбитол2', 25 мМ Ыа2-ЭДТА (pH 8,0), 6,7 мг/мл дитиотрей-
тола.
3. SCE: 1 М сорбитол2’, 0,1 М цитрат натрия, 10 мМ. ЭДТА. Довести pH
раствора до 5,8, добавляя НС1.
4. 1 лузулаза: грубый препарат пищеварительных ферментов улитки Helix
pomatia (Endo Labs, Garden City, NY).
5. Лизирующий буфер: 3% (по весу) саркозил, 0,5 М. трис-НС1 (pH 9),
0,2 М ЭДТА.
6. CaS: 1 М сорбитол, 10 мМ СаС12, 10 мМ трис-НС1 (pH 7,5).
7. ДНК тимуса теленка: 1 мг/мл в стерильной дистиллированной воде.
8. PEG1’: 20% (по весу) PEG 4000, 10 мМ СаС12, 10 мМ трис (pH 7,5).
'9. SOS1’: 1 М сорбитол, 33% (по объему) YEPD, 6,7 мМ СаС12, 14 мкг/мл
лейцина.
10. 1 М сорбитол2’.
11. Верхний агар «—Leu»: 1 М сорбитол, 2,5% агара, 2% глюкозы, 0,67%
дрожжевого азотистого компонента (Difco), не содержащего аминокислот,
по 10 мкг/мл аденина и метионина, по 20 мкг/мл гистидина, аргинина и
триптофана, по 40 мкг/мл урацила и лизина, по 50 мкг/мл тирозина и
треонина, по 60 мкг/мл фенилаланина и изолейцина.
12. Чашки «—leu, +sorb»: то же, что в п. 11, за исключением того, что вме-
сто 2,5%-ного агара используют 2%-ный агар.
13. Агар «—leu»: то же, что в п. 12, с той лишь разницей, что он не содер-
жит сорбитола.
Б. Методика
1-й день: Во второй половине дня или вечером инокулируйте 5 мл YEPD
штаммом, который предстоит трансформировать. Инкубируйте на рота-
ционной качалке при 30 °C.
2-й день:
1. Начните с разведения свежей ночной культуры в 50 раз средой YEPD
(конечный объем 20 мл для трансформации 1—2 плазмидами, 50 мл для
трансформации 3—5 плазмидами, 100 мл для трансформации 6—10 плаз-
мидами). Инкубируйте при 30 °C на качалке при 200—250 об/мин в тече-
ние трех генераций (4,5—6 ч).
2. Когда плотность культуры достигнет (2—4)-107 клеток/мл (OD6M=1,5—
3), осадите клетки центрифугированием и ресуспендируйте в SED
1/10 объема (2 мл) при 30 °C в течение 10 мин.
3. Осадите клетки центрифугированием и ресуспендируйте в 2 мл SCE+
4-20 мкл глузулазы при 30 °C в течение не менее 20 мнн. Проверьте эф-
фективность обработки: отберите 5-мкл аликвоту суспензии клеток, раз-
ведите ее в 45 мкл лизирующего буфера и под фазово-контрастным микро-
скопом определите долю лизировавшихся клеток. 90% — оптимальная ве-
личина, 50—60%—тоже достаточно хорошо, а 100% — вероятна «пере-
держка». Необходимо лишь ослабить толстую и прочную стенку, чтобы
открыть доступ ДНК к мембране и обегчить ее проникновение внутрь
клетки. Слишком жесткое воздействие на клеточную стенку снижает эф-
фективность регенерации.
4. Осадите сферопласты центрифугированием в пробирках с округлым дном
и ресуспендируйте в 2 мл CaS. Снова осадите клетки, слегка подсушите
осадок и ресуспендируйте в 0,2 мл CaS.
5. Разделите суспензию на две части по 0,1 мл. В одну пробирку внесите
0,75—1 мкг плазмидной ДНК; другая пробирка'—контроль («—ДНК»).
Добавление 10 мкг ДНК-носителя (например, ДНК тимуса теленка) по-
15
228
Б. Картер, М. Ирани, В. Маккей и др.
Продолжение
вышает эффективность трансформации, но не является абсолютно необ-
ходимым. Выдержите при комнатной температуре в течение 15 мин.
6. Добавьте 1 мл (10 объемов) раствора ПЭГ, перемешайте и оставьте на
15 мин при комнатной температуре.
7. Осадите клетки центрифугированием и ресуспендируйте в SOS. Инкуби-
руйте при 30 °C в течение 20 мин.
8. Добавьте 6 мл верхнего агара «—Leu top» (расплавленного и охлажден-
ного до 45 °C), быстро перемешайте и немедленно вылейте на чашку
«—Leu + Sorb». Нужно стремиться к тому, чтобы время воздействия на
сферопласты повышенной температуры (45 °C) было минимально корот-
ким. После того как верхний агар застынет, инкубируйте чашки при 30 °C.
Чтобы определить эффективность трансформации или эффективность реге-
нерации, приготовьте серийные разведения суспензии, полученной на ста-
дии 7, в 1 М сорбитоле и высейте в верхнем агаре « + 1еи» на чашки
«+leu + sorb» (20 мкг/мл лейцина).
9. Трансформанты становятся видимыми обычно через 2—3 дня (иногда
значительно позднее). После этого их можно перенести на чашки «—Leu».
’) Стерилизовать фильтрованием; приготавливать каждый раз заново или хранить на
холоду или замороженным непродолжительное время. Все другие растворы можно авто-
клавировать и хранить неопределенно долго. YEPD следует автоклавировать 25 мин; все
остальные растворы не более 15 мин.
2) Используйте свежеприготовленный раствор I <4 сорбитола или храните его замо-
роженным (в холодильнике (+4 °C) раствор не сохраняется).
использования. Трансформантов можно очистить методом от-
дельных колоний на среде, обеспечивающей сохранение плазми-
ды (например, не содержащей лейцин). В экспериментах, на-
правленных на изучение экспрессии клонированных генов,
следует проверить уровень экспрессии гетерологичного белка у
нескольких трансформантов, так как эта величина может силь-
но варьировать.
Период времени, предшествующий появлению трансформан-
тов на чашке, может составлять от трех до одиннадцати суток.
Длительность этого периода зависит от природы экспрессируе-
мого продукта. Как правило, наблюдается отрицательная кор-
реляция между скоростью роста трансформантов и достигаемым
уровнем экспрессии клонированных -генов. Некоторые белки
оказывают на дрожжевую клетку токсическое действие и силь-
но замедляют ее рост. Такие белки удается экспрессировать
в дрожжах только в следовых количествах. Причины до конца
не выяснены. Некоторые аспекты этой проблемы рассматрива-
ются в другой работе [17]. В отдельных случаях токсический
эффект оказывается столь существенным, что получаются мало-
жизнеспособные трансформанты. Здесь может выручить исполь-
зование для трансформации интегрирующегося вектора. При-
мечательно, что в ряде случаев применение низкокопийной
экспрессирующей системы позволяет получить более высокий
уровень экспрессии, чем мультикопийные векторы [14].
Продукты гетерологичных генов в дрожжах
229
Таблица 7.5. Трансформация дрожжей: метод с использованием
ацетата лития
1-й день:
Во второй половине дня или вечером инокулируйте 5 мл YEPD штам-
мом, который предполагается использовать для трансформации. Инку-
бируйте при 30 °C на ротационной качалке.
2-й день:
1. Разведите 1 мл этой свежей ночной культуры в 80 мл YEPD и инкуби-
руйте при 30 °C на качалке при 200—250 об/мин, пока значение OD60o не
достигнет 0,5—1,0 (4,5—6 ч).
2. Осадите клетки центрифугированием в двух стаканах при 8000 об/мин в
течение 5 мин при комнатной температуре в роторе Sorval НВ4 (или эк-
вивалентном).
3. Промойте осадок дистиллированной водой.
4. Ресуспендируйте клетки в 100 мМ ацетата лития и инкубируйте в тече-
ние 30—60 мин при 30 °C.
5. Осадите клетки центрифугированием и ресуспендируйте в 0,4 мл 100 мМ
ацетата лития.
6. Поместите 50 мкл суспензии клеток в пробирку для микроцентрифуги и
добавьте туда же >1 мкг ДНК. Инкубируйте при 30 °C в течение
30 мин.
7. Аккуратно встряхните пробирку и добавьте 0,6 мл раствора, содержаще-
го 10 мМ трис-НС1 (pH 7,5) и 40% ПЭГ-4000.
8. Инкубируйте при 30 °C в течение 1 ч, после чего перенесите на 5 мин
на 42 °C.
9. Осадите клетки центрифугированием в микроцентрифуге в течение 10 с.
10. Поомойте осадок дистиллированной водой и ресуспендируйте клетки в
0,1 мл дистиллированной воды.
11. Высейте на чашки, обеспечивающие селекцию трансформантов (напри-
мер, «—leu»). Трансформанты появятся через 2—3 дня инкубирования
при 30 °C; этот срок может значительно увеличиваться в зависимости от
природы экспрессируемого структурного гена.
Не рекомендуются множественные последовательные пасса-
жи колоний на чашке, так как это способствует отбору клеток
с пониженным уровнем экспрессии (которые быстрее растут).
Предпочтительнее ретрансформация охарактеризованными
плазмидными препаратами, хотя, как уже упоминалось, транс-
форманты можно довольно долго хранить при —80 °C (табл. 7.2).
Для трансформации дрожжей пригоден и другой метод,
связанный с использованием ионов лития (табл. 7.5). Анало-
гичная методика также описана Р. Ротстейном в гл. 3 первого
тома этой серии. Процедура, представленная в табл. 7.5, соот-
ветствует разработке Zymogenetics и приводится для того, что-
бы дать представление о возможных вариантах принципиально
одного и того же метода.
Метод трансформации сферопластов используется наиболее
широко и обеспечивает относительно высокий уровень транс-
формации. Это обстоятельство может иметь большое значение
при скрининге библиотеки, клонированной в дрожжах. Однако
для отбора нескольких трансформантов при субклонировании
230 Б. Картер, М. Ирани, В. Маккей и др.
Таблица 7.6. Прямой перенос плазмид из Е. coli в клетки дрожжей
1. Нарастите 10 мл ночной культуры трансформированных клеток Е. coli
при 37 °C в среде с необходимым для селекции антибиотиком.
2. Осадите бактериальные клетки центрифугированием, ресуспендируйте оса-
док в 0,5 мл 1 М сорбитола в присутствии 20 мМ ЭДТА (pH 8,0) и до-
бавьте 50 мкл раствора лизоцима (10 мг/мл).
3. Поместите на лед на 10 мин и центрифугируйте в течение 5 мин в микро-
центрифуге.
4. Ресуспендируйте осадок в 0,1 CaS (табл. 7.4) и используйте эту суспен-
зию вместо раствора ДНК в методике трансформации, описанной в
табл. 7.4.
конкретной плазмиды это не так важно. Преимущества метода
с использованием ацетата лития сводятся к следующему:
1) приготовление культуры клеток-хозяев для трансформации
не превосходит по трудоемкости приготовление компетентных
бактерий; 2) не возникает проблемы слияния клеток в процессе
трансформации; 3) клетки высевают не в мягком агаре, а не-
посредственно на поверхность твердого агара. В то же время
для метода литиевой трансформации характерна высокая час-
тота появления петит-клеток (клетки с дефектным дыханием).
В этом случае отбирают более крупные колонии нормальных
клеток.
Хотя у нас нет собственного опыта по прямому переносу
генетического материала плазмид из Е. coli в дрожжевые клет-
ки, такой метод существует. Соответствующая процедура опи-
сана в табл. 7.6.
7.4. Анализ трансформантов
Анализу предполагаемых трансформантов обычно придается
большое значение, и мы не остались в стороне от этой пробле-
мы и разработали несколько соответствующих методик. Не нуж-
но объяснить, насколько важно определить, являются ли коло-
нии, отобранные как трансформанты, на самом деле трансфор-
мантами или же это ревертанты. Анализ трансформантов
предполагает гибридизацию с колониями или выделение и ана-
лиз плазмидной ДНК- Кроме того, для оценки экспрессии чу-
жеродного гена в дрожжевой системе весьма важно определить
копийность плазмиды как фактор, оказывающий существенное
влияние на уровень экспрессии.
Селекция колоний трансформантов по их способности расти
на среде, не содержащей лейцина, не исключает возможности
того, что отобранные клетки окажутся не трансформантами,
а ревертантами по гену leucine 2. Проверить генетический ста-
тус таких клеток можно довольно просто, если в работе исполь-
зуются экспрессирующие плазмиды, поскольку не составляет
Продукты гетерологичных генов в дрожжах 231
труда оценить стабильность фенотипа Leu+ путем рассева в не-
селективных условиях.
Предполагаемые трансформанты суспендируют в воде и
рассевают на чашках с YEPD-агаром (100—200 колоний на
чашку). Когда колонии сформируются, их реплицируют на чаш-
ки с агаром, не содержащем лейцина (0,67%-ного дрожжевого
азотистого компонента Difco без аминокислот, 2% глюкозы,
необходимые пищевые компоненты, за исключением лейцина,
2% агара). Сравнивая исходные чашки и реплики, идентифици-
руют колонии, неспособные расти на среде без лейцина из-за
утраты плазмиды.
Гибридизация с колониями позволяет анализировать дрож-
жевые штаммы на содержание специфических нуклеотидных по-
следовательностей. Приготовление дрожжевых образцов не-
сколько отличается от процедуры, описанной для Е. coir, однако
после того, как нитроцеллюлозный фильтр-реплика с ДНК под-
готовлен, дальнейшие операции аналогичны тем, которые ис-
пользуются для бактерий. Соответствующая методика приведена
в табл. 7.7. Другой вариант этого метода описан Р. Ротстейном
в гл. 3 первого тома [29] данной серии.
Препараты плазмидной ДНК, выделенные из дрожжей, мож-
но использовать для рестрикционного анализа, а также для
трансформации клеток Е. coll с целью амплификации плазмиды.
Эти процедуры описаны в табл. 7.8.
Таблица 7.7. Гибридизация с дрожжевыми колониями
1. На поверхность YEPD-агара в чашке Петри поместите нитроцеллюлозный
фильтр и высейте на него клетки петлей илн методом реплик.
2. Инкубируйте в течение ночи при 30 °C.
3. Снимите фильтр и дайте ему подсохнуть.
4. Ниже приведены растворы, в которых последовательно обрабатывают
фильтр. Для этого фильтр накладывают (колониями вверх) на листы ват-
мана ЗММ, пропитанные необходимым раствором:
1) 1 М сорбитол, 20 мМ ЭДТА, 50 мМ дитиотрейтол.
2) 1 М сорбитол, 20 мМ ЭДТА, 1 мг/мл зимолиазы 60 000, 37 °C, 2—3 ч.
Инкубацию следует проводить в закрытом сосуде, чтобы предотвратить
выпаривание жидкости. Процесс образования сферопластов можно
контролировать, как описано в методике трансформации дрожжей. Ука-
занное время является ориентировочно необходимым для формирова-
ния сферопластов.
3) 0,5 М NaOH — 7 мин при комнатной температуре.
4) 0,5 М трис-НС1 (pH 7,0), 10XSSC1’ — 4 мин.
5) 0,5 М трис-НС1 (pH 7,5), 10XSSC — 4 мин.
6) 5XSSC — 2 раза по 2 мин.
5. Высушите фильтр на бумаге в течение 30 мин.
6. Прогрейте фильтр в вакууме при 80 °C в течение 1—2 ч.
О 1XSSC: 0,15 М NaCl —0,015 М цитрат натрия.
232
Б. Картер, М. Ирани, В. Маккей и др.
Таблица 7.8. Быстрый метод выделения плазмид из клеток дрожжей
1. Нарастите 10 мл дрожжевой стационарной культуры (ночной) в синтети-
ческой среде, не содержащей лейцина. 5 мл культуры, выращенной в
жидкой среде YEPD, можно считать эквивалентной заменой для всех
плазмид, кроме наиболее нестабильных.
2. Осадите клетки центрифугированием, ресуспендируйте осадок в 1 мл дис-
тиллированной воды и перенесите в 1,5 мл пробирку для микроцентри-
фуги.
3. Осадите клетки центрифугированием в течение приблизительно 30 с в мик-
роцентрифуге. Ресуспендируйте осадок в 0,6 мл раствора: 1 М. сорбитол,
0,1 М ацетат натрия (pH 5,8), 10 м?Л ЭДТА, 0,1 М 2-меркаптоэтанол,
0,1 мг/мл зимолиазы 60 000 (Miles).
4. Инкубируйте при 37 °C в течение 30 мин, периодически контролируя про-
цесс образования сферопластов под микроскопом.
5. Добавьте 0,2 мл 50 Мм трис-НС1 (pH 8,0), 50 мМ ЭДТА.
б. Добавьте 2 мл днэтилпирокарбоната и инкубируйте при 65 °C в течение
00 мин.
7. Перенесите на лед, добавьте 0,1 мл 5 М ацетата калия и оставьте па 1 ч.
8. Можно отдохнуть н перекусить.
9. Центрифугируйте в течение 5 мин на микроцентрифуге. Удалите надоса-
дочную жидкость, добавьте 2 мл 95%-ного этанола и поместите на
—70 °C на 30—60 мин.
10. Осадите нуклеиновые кислоты центрифугированием и ресуспендируйте
осадок в растворе: 10 мМ трис-НС1 (pH 8,0), — 1 мМ ЭДТА, 0,2 М
NaCl.
И. Повторно осадите нуклеиновые кислоты этанолом, как описано выше,
и ресуспендируйте осадок в растворе 10 мА! трис-НС1 (pH 8,0) и 1 мМ
ЭДТА.
Определить количество копий плазмиды в клетках транс-
формированного штамма можно разными методами, но все эти
методы предполагают выделение суммарной дрожжевой ДНК
с последующим определением соотношения плазмидной и ге-
номной ДНК. Относительно простой способ оценить число копий
высококопийной плазмиды — сравнение интенсивности окраши-
вания бромидом этидия рестрикционных фрагментов, соответст-
вующих плазмидной ДНК и ДНК рибосомных повторов в сум-
марной геномной ДНК- Если суммарную ДНК из трансформи-
рованных дрожжей обработать рестриктазой, расщепляющей
плазмиду и выщепляющей рибосомный повтор, соответствующие
рестрикционные фрагменты можно визуализировать, окрашивая
бромидом этидия агарозный гель после электрофореза. В УФ-
свете эти полосы отчетливо высвечиваются над фоновым свече-
нием всех остальных окрашенных бромидом этидия гетероген-
ных по длине рестрикционных фрагментов. В ДНК гаплоидной
дрожжевой клетки содержится приблизительно 140 копий рибо-
сомных последовательностей, содержание же плазмидной ДНК
может быть оценено количественно при помощи денситометриче-
ского сканирования фотонегативов, полученных при съемке геля.
Интенсивность окрашивания нормализуют относительно длины
Продукты гетерологичных генов в дрожжах 233
фрагментов. Поэтому целесообразно использовать такие рест-
риктазы, которые выщепляют фрагменты сходной длины как из
плазмиды, так и из повторяющихся последовательностей рибо-
сомной ДНК. Рестрикционная карта рибосомного повтора при-
ведена в работе Белла и др.
(Площадь плаз.мидного пика) X (Длина фрагмента рДНК)
Число копий = —---------------------------------------------
(Площадь пика рДНК) X (Длина плазмидного фрагмента) X 140
Альтернативный способ определения числа копий плазми-
ды — это анализ по Саузерну. Суммарную ДНК расщепляют
рестриктазой и рестрикционные фрагменты фракционируют в
геле. Затем ДНК. переносят на нитроцеллюлозный фильтр и
гибридизуют с зондом, представленным (обычно в единственном
числе) как в геноме хозяина, так и в плазмндной ДНК- Коли-
чество копий плазмиды определяют путем денситометрии радио-
автографов. Здесь также следует учитывать относительную
длину фрагментов и степень насыщения фрагментов меченой
пробой.
7.5. Выращивание клеток для осуществления экспрессии
и секреции продуктов клонированных генов
Клетки можно выращивать как на средах с определенным
составом, так и на комплексных средах. Если экспрессирующая
плазмида несет копию гена leucine 2, а дрожжевая клетка-хо-
зяин дефектна по этому гену, трансформированные клетки
обычно выращивают на среде с определенным составом, содер-
жащей все необходимые питательные компоненты, за исключе-
нием лейцина. Рост клеток на этой среде возможен лишь в
в том случае, если они сохраняют плазмиду.
При небольших объемах культуральной жидкости (менее
100 мл) дрожжевые клетки часто выращивают на комплексных
средах — обычно это 1%-ный дрожжевой экстракт, содержащий
2% бактопептона (Difco) и 2% какого-либо источника углеро-
да. Источником углерода может служить глюкоза (если в экс-
прессирующей плазмиде используется гликолитический промо-
тор) или же 1%-ный этанол в сочетании с 3%-ным глицеролом
(если используется промотор генов, кодирующих компоненты
дыхательной цепи, например промотор гена изо-1-цитохрома с).
Рекомендуется забуферить среду при pH 7,0, а также добавить
в нее бычий сывороточный альбумин (БСА) до концентрации
0,01%, способный в некоторых случаях предохранять от разру-
шения секретируемый рекомбинантный белок.
Трансформированные дрожжевые клетки, выращиваемые в
виде колоний на твердом агаре, можно вносить непосредственно
в жидкую среду. Дрожжи хорошо растут в широком интервале
234 Б. Картер, М, Ирани, В. Маккей и др.
температур вплоть до 37 °C. Тем не менее оптимальными усло-
виями следует считать 25—30 °C (если, конечно, не используют-
ся температурочувствительные мутанты, требующие снижения
температуры).
Рост клеток контролируют по изменению оптической плот-
ности при 600 нм или по увеличению количества клеток, опре-
деляемому с помощью счетчика Coulter (Coulter Electronics,
Hialeh, FL). При выращивании дрожжей при 25 °C в сложной
среде количество клеток удваивается через 2,5 ч, а в среде
с определенным составом — через 3 ч. Дрожжевые клетки, транс-
формированные плазмидами, экспрессирующими определенные
белки млекопитающих, растут значительно медленнее. Этот эф-
фект сильно зависит от природы белка: дрожжи способны,
например, экспрессировать альфа-интерферон без заметного
снижения скорости роста, тогда как даже близкородственные
белки, такие как бета-интерферон, обусловливают по меньшей
мере двукратное увеличение времени генерации. К сожалению,
заранее нельзя определить, окажется ли токсичным для дрож-
жевой клетки каждый данный ген, экспрессию которого плани-
руется осуществить.
Чтобы ответить на этот вопрос, через определенные проме-
жутки времени пипеткой отбирают образцы культуры.
7.6. Приготовление образцов для анализа
Чтобы отделить клетки от культуральной среды, культуру
центрифугируют или фильтруют. Если исследуют небольшие
образцы, достаточно двухминутного кручения на микроцентри-
фуге; более крупные образцы (>100 мл) центрифугируют при
8000 об/мин в течение 5 мин (например, в роторе Sorvall НВ4).
С равным успехом можно использовать и другой метод — филь-
трацию через мембраны типа «Millipore» (диаметр пор
0,45 мкм).
Оценка эффективности экспрессии клонированного гена и
анализ ее закономерностей в ряде случаев требует концентри-
рования белкового продукта в среде. Этой цели отвечает про-
цедура ультрафильтрации, разработанная фирмой Amicon.
Дрожжевые клетки разрушают для высвобождения экспрес-
сируемых в цитоплазме белков. Существует несколько способов
разрушения клеток. Мы приводим относительно простой метод,
пригодный для небольших объемов культуры (<100 мл):
1. Отделите клетки от культуральной среды центрифугиро-
ванием или фильтрацией через мембрану Amicon.
2. Промойте клетки подходящим буфером, например фосфат-
но-солевым буфером, содержащим 0,01% БСА, или же буфером
состава: 50 мМ КН2РО4, 2 мМ ЭДТА (pH 8,0) и 1 мМ ФМСФ
Продукты гетерологичных генов в дрожжах 235
(ингибитор протеиназ). Добавьте 2-меркаптоэтанол до концент-
рации 2 мМ.
3. Ресуспендируйте клетки в буфере из расчета 1 мл буфера
на 20 мл исходной культуры.
4. Перенесите небольшие порции суспензии (0,5 мл) в мик-
роцентрифужпые пробирки (на 1,5 мл). Заполните пробирки,
предварительно промытыми кислотой, стеклянными шариками
диаметром 40 мкм (BDH) так, чтобы они едва покрывались
жидкостью.
5. Встряхивайте пробирки на вибромиксере на максималь-
ной скорости. Старайтесь держать пробирку так, чтобы внутри
образовывались интенсивные завихрения. Это требует некоторо-
го навыка и тренировки.
6. Разные штаммы дрожжей и даже один и тот же штамм,
но экспрессирующий различные рекомбинантные белки, требуют
разной продолжительности обработки для полного разрушения
клеток. Если требуется более 5 мин — это уже проблема. Встря-
хивать следует с перерывами не долее 1 мин за один раз, охлаж-
дая пробирку в промежутках между встряхиваниями на льду.
7. Эффективность обработки следует контролировать с по-
мощью светового микроскопа, отбирая кончиком микропипетки
на предметное стекло небольшое количество жидкости. Исполь-
зуйте увеличение 400X или больше. В ряде случаев возможна
недооценка результатов обработки, поскольку дрожжевые клет-
ки могут внешне сохранять морфологическую целостность, не-
смотря па то что клеточная стенка разрушена и содержимое
выходит наружу. Такие клетки легко отличить от интактных под
фазово-контрастным микроскопом. В конце концов встряхивание
со стеклянными шариками приводит к полной деструкции кле-
точной стенки, и в световом микроскопе можно наблюдать лишь
обломки клеток.
8. Отберите жидкость из пробирок при помощи микропи-
петки на 1 мл, опустив копчик до самого дна и не отрывая его
от пробирки. Можно использовать пастеровскую пипетку.
9. Центрифугируйте пробы на микроцентрифуге в течение
5 мин при 4 °C, чтобы осадить клеточные обломки.
10. Отберите надосадочную жидкость и повторите центрифу-
гирование. Отберите надосадочную жидкость и далее обрабаты-
вайте как бесклеточный экстракт.
Для более крупных образцов вместо микроцентрифужных
пробирок можно использовать круглодонные пробирки. Размер
пробирок подбирается в соответствии с объемом образца. Если
жидкость выплескивается через край при встряхивании на виб-
ромиксере, пробирка мала. Если завихрения в пробирке не об-
разуются, возьмите пробирку большего объема, желательно бо-
лее широкую.
236 Б. Картер, М, Ирани, В. Маккей и др.
7.7. Анализ дрожжевой экспрессии
Первичный анализ экспрессируемых в дрожжевой системе
чужеродных белков проводят с помощью электрофореза в геле.
Если эффективность экспрессии белка достаточно высока, со-
ответствующую полосу в геле можно визуализировать кумасси
голубым или путем окрашивания серебром. При низком уровне
экспрессии придется использовать вестерн-блот-анализ с анти-
телами к данному белку. Далее необходимо провести биологи-
ческий анализ для того, чтобы подтвердить наличие у экспрес-
сируемого дрожжевой клеткой продукта ожидаемой биологи-
ческой активности. Нередко белки, продуцируемые в цитоплаз-
му, не обладают соответствующей активностью. Секреция экс-
прессируемого продукта в среду помогает преодолеть эти труд-
ности.
Анализ рекомбинантных продуктов включает такие методы,
как гель-электрофорез с последующим окрашиванием кумасси,
вестерн-блот-анализ, иммуноферментный анализ и другие широ-
ко используемые процедуры, не требующие каких-либо специ-
альных модификаций для работы с дрожжами; поэтому мы их
здесь не описываем.
В отличие от Е. coli дрожжи способны гликозилировать бел-
ки. Поэтому важно определить степень гликозилирования гете-
рологичного продукта. Гликопротеины можно визуализировать
в геле [30] или после переноса на нитроцеллюлозу [31].
7.8. Очистка рекомбинантных белков,
продуцируемых дрожжами
Рекомбинантные белки очищают так же, как и любые другие
белки в бесклеточном экстракте. Методы очистки дрожжевых
белков не отличаются от тех, которые используются для Е. coli
(гл. 4). Работая с дрожжевыми системами, как правило, не при-
ходится беспокоиться по поводу ренатурации денатурированно-
го белкового продукта. В частности, мы не знаем ни одного
примера секретируемого дрожжами белка, который не обладал
бы ожидаемой биологической активностью. Для аналитических
целей очистка секретируемых белков с помощью иммуноаффин-
ной хроматографии представляется предпочтительной, по край-
ней мере по мнению первого автора данной главы.
Благодарности
Брюс Л. А. Картер выражает благодарность А. Р. Гуди за
то, что тот обучил его «безмагниевой» процедуре лигирования.
Продукты гетерологичных генов в дрожжах___________________237
\
\
\ Литература
1. Яр'/ М. Л'.. Hereford L„ Herskowitz I. (1984). Cell, 36, 1057.
2. Giiarente L„ Yocum R. R„ Gifford P. (1982). Proc. Natl. Acad. Sci. USA, 73,
7410.
3. E/ftr S. D., Schauer L., Hansen IF., Esmon P„ Schekman R. (1984). Mol. Cell.
Biol ., 4, 2347.
4. Kelly J. D„ Raines E. IF., Ross R„ Murray M. J. (1985). EMBO, 4, 3399.
5. Beggs J. D. (1978). Nature, 275, 104.
6. Hinnen A. J., Hicks J., Fink G. (1978). Proc. Natl. Acad. Sci. USA, 75,
1929.
7. Hagen F. S„ Gray C. L„ O’Hara P., Grant F. J., Saari G. C„ Woodbury R. G.,
Hart C. E„ Jnsley M., Kisiel IF., Karachi K-, Davie E. W. (1986). Proc. Natl.
Acad. Sci. USA, 83, 2412.
8. Maniatis T., Fritsch E. F., Sambrook J. (1982). Molecular Cloning, Cold
Spring Harbor Laboratiry, Cold Spring Harbor, New York. [Имеется пере-
вод: Маниатис T., Фрич Э., Сэмбрук Дж. Методы генетической инженерии.
Молекулярное клонирование. — М.: Мир, 1984.]
9. Davis R. W„ Botstein D., Roth J. R. (1980). Advanced Bacterial Genetics,
Cold Spring Harbor Laboratory, Cold Spring Harbor, New York.
10. Prescott D. N. (ed.) (1975). Methods in Cell Biology, Volumes 11 and 12,
Academic Press, New York.
11. Sherman F., Fink G. R., Hicks J. B. (1982). Methods in Yeast Genetics, Cold
Spring Harbor Laboratory, Cold Spring Harbor, New York.
12. IFn R., Grossman L., Moldave К (eds.) (1983). Methods in Enzymology,
Volume 101, Academic Press, New York.
13. Broach J. R. (1983). In: Methods in Enzymology, Wu R., Grossman L. and
Moldave I\. (eds.), Academic Press, New York, Vol. 101, p. 307.
14. Smith R. A., Duncan M., Moir D. J. (1985). Science, 229, 1219.
15. Bitter G. A„ Egan К. M. (1984). Gene, 32, 263.
16. Schaber M. D., DeChiara T. M., Kramer R. A. (1986). In: Methods in Enzy-
moiogy, Pestka S. (ed.), Academic Press, New York, Vol. 119, p. 416.
17. Piggott J. R., Watson M. M., Doel S. D„ Goodey A. R., Carter B. L. A.
(1987). In: Yeast Biotechnology, Spencer F. (ed.), Academic Press, New
York.
18. Hartwell L. H. (1973). Exp. Cell Res., 76, 111,
19. Kurjan J., Herskowitz I. (1983). Cell, 30, 933.
20. Julius D., Blair L„ Brake A„ Sprague G., Thorner J. (1983), 32, 839.
21. Julius D., Schekman R., Thorner J. (1984). Cell, 36, 309.
22. Julius D., Brake A., Blair L., Kunlsawa R., Thorner J. (1984). Cell 37
1075.
23. Brake A. J., Merryweather J. P„ Cuit D. G., Herbelein J. A. (1984). Proc Natl
Acad. Sci. USA, 81, 4642.
24. Thim L., Hansen M. T., Norris K., Hoegh I., Boel E., Forstrom J., Anime-
rer G., Fill N. P. (1986). Proc. Natl. Acad. Sci. USA, 83, 6766.
25. Shaw A. R. E., Bleakley R. C., Merryweather J. P., Barr P. J. (1985). Cell
Immunol., 91, 193.
26. Ammerer G. (1983). In: Methods in Enzymology, Wu R., Grossman L. and
Moldave K. (eds.), Academic Press, New York, Vol. 101, p. 192.
27. Sherman F. (1975). In: Methods in Cell Biology, Prescott D. M. (ed.), Aca-
demic Press, New York, vol. 11, p. 189.
28. Jto H., Fukuda Y., Murata K.. Kimura A. (1983). J. Bacteriol., 153, 163.
29. DNA Cloning. A Practical Approach, Glover D. (ed.), (1984). IRL Press,
Oxford, Vol. 1. [Имеется перевод: Клонирование ДНК. Методы. Книга I.
Под ред. Д. Гловера. — М.: Мир, 1988.]
ГЛАВА 8 !
ИСПОЛЬЗОВАНИЕ АМПЛИФИЦИРУЮЩИХСЯ/
ВЕКТОРОВ ДЛЯ ЭКСПРЕССИИ КЛОНИРОВАННЫХ
ГЕНОВ В КЛЕТКАХ МЛЕКОПИТАЮЩИХ j
Кристофер Р. Беббингтон и Кристофер К- Г- Хенчель
(Christopher R. Bebbington, Christopher С. G. Hentschel)
8.1. Введение
8.1.1. Стратегия экспрессии клонированных генов
в клетках млекопитающих
Изучение экспрессии клонированных генов во многом осно-
вано на способности таких генов нормально функционировать
после введения их в культивируемые клетки млекопитающих.
Эта способность помимо всего прочего позволяет получать ред-
кие обычно белки в больших количествах, а также выделять
белковые продукты новых генноинженерных конструкций. Полу-
чение таких белков в клетках млекопитающих имеет то преиму-
щество, что обеспечивает правильную сборку вторичной струк-
туры, нормальную модификацию и полную сохранность функцио-
нальной активности экспрессируемых белков — свойства, выгодно
отличающие их от продуктов экспрессии тех же генов в бакте-
риальных системах.
Во многих случаях для изучения экспрессии генов млекопи-
тающих наиболее приемлема так называемая временная экс-
прессия, происходящая в части клеток в течение нескольких
часов после введения ДНК. Если же требуются большие коли-
чества продукта, то приходится выделять клоны, клетки которых
сохраняют вектор и в ходе пролиферации. Подобное стабильное
наследование вектора достигается двумя путями: использовани-
ем вирусного репликона, например вируса папилломы крупного
рогатого скота (BPV) (см. гл. 8 тома II этой серии [34]), или
в результате интеграции вектора в ДНК клетки-хозяина. Из-
вестно, что любая чужеродная ДНК с низкой частотой способна
встраиваться в неспецифические участки хозяйской хромосомы;
получившиеся клоны можно выявить, если использовать подхо-
дящий селективный маркер. В гл. 6 тома II данного издания
описаны различные селектируемые векторы, а также способы
их введения в культуру клеток. В этой главе мы коснемся ме-
тодов, позволяющих получить высокоэффективную экспрессию
интегрирующихся векторов в стабильно трансфицированных ли-
ниях клеток.
238
\Амплифицирующнеся векторы 239
Гены, экспрессируемые в составе эукариотических векторов,
представляют собой либо геномные копии с аутентичными про-
моторными элементами и сигналами процессинга РНК, либо
последовательности ДНК, состыкованные с гетерологичным про-
мотором и сигналом полиаденилирования. К числу наиболее эф-
фективных транскриптонов относятся рекомбинантные конструк-
ции, содержащие регуляторные сигналы вирусов; такие сигналы
функционируют в клетках многих типов. С другой стороны, даже
высокоэффективные транскрнптопы сами по себе редко обеспе-
чивают максимальный уровень экспрессии; более высоких ре-
зультатов позволяет добиться увеличение числа копий вектора.
Использование с этой целью векторов BPV-типа удобно в
том отношении, что довольно быстро устанавливается множество
копий вируса (до нескольких сотен на клетку). Этим объясня-
ется успешное применение вектора на основе BPV для высоко-
эффективной экспрессии многих белков [1]. Недостатки вирусных
векторов связаны с ограниченным набором клеточных линий,
в которых возможна репликация вектора, а уровень экспрессии
и поддержание эписомного состояния конструкции в целом за-
висят кроме всего прочего и от конкретных последовательностей
ДНК, клонированных в векторе. Альтернативный способ увели-
чения числа копий, описанный в данной главе, предусматривает
селекцию клеток на амплификацию последовательностей вектора
после его интеграции в ДНК клетки-хозяина; такой подход не
имеет принципиальных ограничений на использование того или
иного типа клеток. При этом подходе можно выбрать клетки с
любыми нужными свойствами, например способностью опреде-
ленным образом модифицировать белковый продукт или узна-
вать конкретную регуляторную последовательность ДНК.
В основе обсуждаемой методологии лежит феномен ампли-
фикации генов, рассматриваемый в следующем разделе.
8.1.2. Амплификация генов
Если культивируемые клетки обработать определенными кон-
центрациями некоторых токсических веществ, можно получить
клоны, обладающие повышенной по сравнению с исходной куль-
турой устойчивостью к этим токсинам. Появление таких клонов
часто связано с повышенным образованием именно того необхо-
димого клетке фермента, против которого направлено действие
токсина. Повышенное образование фермента, как правило, бы-
вает следствием повышенного содержания мРНК, а это в свою
очередь обычно отражает увеличение числа копий данного
структурного гена в каждой клетке клона.
Первым геном, искусственно амплифицированным путем по-
добной селекции, был ген дигидрофолят-редуктазы (dhfr); се-
240
К. Беббингтон, К. Хенче.ть
Таблица 8.1. Клонированные гены, способные к амплификации пр;
экспрессии в клетках млекопитающих /
Белок/признак Ген Фенотип Селективный агент ) Источ- / ник I данных
Аденозин-дезаминаза ada ADA Дезоксикофор:>1пш:н («к®) } X (фосфонацетил)-Б-ас- / партат 14]
Аспартат-карбамоил- трансфераза cad CAD 01
Дигидрсдролят-редукта- за dhfr DHFR Mi т;; рсхсат (МТКф М1Х) [61
Г дут амин- синтетаза gs GS Метнонинсульфоксимиц (MCKc, MSX) [71
Мет аллотионеин-1 mtl MT-i Тяжелые металлы, на- пример Cd2+ [81
Тимндин-киназа (де- фектная) tk tk — [91
Множественная лекар- ственная устойчивость hdf Адрнамниин (и др.) [10]
лекцию клонов мышиных клеток и клеток хомяка вели на устой-
чивость к метотрексату — специфическому ингибитору этого фер-
мента (см. обзор [2]). В дальнейшем клонированный ген dhfr
вводили в клетки, лишенные этого фермента, и он также успеш-
но амплифицировался в условиях селекции на устойчивость к
метотрексату. С тех пор описано по крайней мере 12 генов, каж-
дый из которых амплифицировался в ответ на действие соот-
ветствующего ингибитора [3]. Для семи из них удалось клони-
ровать полную кодирующую последовательность (табл. 8.1).
Во всех известных на сегодняшний день случаях не только
геномные копии, но и кДНК любого из этих генов в составе экс-
прессирующего вектора способны к амплификации в трансфици-
рованных культивируемых клетках (см., например, [6—9,11,12]).
Следовательно, вряд ли существует некая специфическая после-
довательность ДНК, ответственная за амплификацию. И хотя
механизм этого явления изучен недостаточно полно, можно все-
таки предположить, что в ходе селекции происходит лишь фик-
сирование определенных случайных событий амплификации,
возникающих независимо с определенной частотой во всех про-
лиферирующих клеточных популяциях. Одна из гипотез связы-
вает эти события с ошибками репликации ДНК [2]. С этой гипо-
тезой согласуется наблюдение о том, что амплифицирующиеся
участки хромосомной ДНК во всех случаях значительно превы-
шают по размеру собственно кодирующую последовательность
фермента (часто амплифицируются фрагменты длиной более
1000 т. п. н.). Точно так же при селекции на амплификацию кло-
нированных генов увеличивается число копий и других последо-
вательностей вектора — они тоже вовлекаются в амплификацию.
____\___________Амплифицирующиеся векторы______ 241
> 8.1.3. Ко-амплификация и ее использование
R
Возможность вести селекцию на амплификацию гена очень
удобнй в целом ряде случаев. Облегчается клонирование генов,
способных к амплификации, а также анализ их структуры в со-
ставе хроматина. Однако в данной главе мы ограничимся вопро-
сами, касающимися использования феномена амплификации для
осуществления избыточной экспрессии белковых продуктов.
Идея заключается в том, что ко-амплификация всех последова-
тельностей рекомбинантной конструкции при селекции на ампли-
фикацию генов-маркеров резко усиливает экспрессию любого
клонированного в ее составе гена, причем повышение продукции
интересующего белка часто оказывается пропорциональным уве-
личению числа копий гена — по крайней мере до тех пор, пока
не будет достигнут максимально возможный уровень экспрессии.
В экспериментах такого рода часто используется ген дигидро-
фолят-редуктазы в комбинации с мутантными клетками СНО,
у которых нет эндогенного фермента.
Селекция на амплификацию данного гена в составе реком-
бинантной конструкции позволила достичь максимально высоко-
го уровня экспрессии для целого ряда секретируемых белков [1].
В принципе любой из генов, перечисленных в табл. 8.1, можно
использовать в качестве маркера амплификации вектора.
В разд. 8.1.4 описано применение некоторых из этих маркеров
при работе с различными клеточными линиями.
8.1.4. Общая характеристика амплифицирующих векторов
Конструкции применяемых векторов отличаются большим
разнообразием и более подробно обсуждаются в разд. 8.3; одна-
ко все они обязательно содержат следующие элементы:
1. Последовательности бактериальных плазмид, включающие
область начала репликации для размножения в Е. coli и селек-
тивный бактериальный маркер.
2. Селективный маркерный ген, функционирующий в клетках
животных.
3. Амплифицируемый маркерный ген.
4. «Кассету» экспрессии, содержащую участок для клониро-
вания интересующего нас гена.
Лучше всего, если селективный маркер является одновремен-
но и маркером амплификации, поскольку при трансфекции ДНК
часто подвергается перестройкам и мутациям, из-за которых не
все клоны, экспрессирующие селективный маркер, будут „ экс-
прессировать и маркер амплификации. Амплифицируемый ген
может служить основой для селекции клонов в том случае, если
1G—1186
242
К. Беббингтон, К- Хенчель
клетки-реципиенты не производят собственного такого же фер-
мента; такая ситуация описана выше, когда для трансфекции
использовали мутантные клетки СНО, дефектные по i белку
DHFR. В этом случае введенный клонированный ген dhff изме-
няет фенотип клеток на DHFR+ (при этом они уже не нуждают-
ся в экзогенных нуклеозидах и глицине).
Можно также использовать амплифицируемый ген как «до-
минантный» селективный маркер при трансфекции клеток, спо-
собных синтезировать соответствующий эндогенный фермент.
В этих случаях подбирают такую концентрацию токсического
вещества, которая убивает родительские клетки,но не детальна
для трансфицированных клеток из-за повышенного уровня экс-
прессии фермента. Не все доминантные селективные маркеры
можно амплифицировать путем селекции: так, бактериальные
маркеры пео и gpt (см. гл. 6 книги II данной серии [34]) не уда-
ется использовать для селекции на амплификацию, так как для
них нет ингибиторов, способных стехиометрически связываться
с этими ферментами. Отметим, что все гены животных, перечис-
ленные в табл. 8.1, относятся, по крайней мере потенциально,
к доминантным амплифицируемым маркерам.
Перечислим характеристики идеального амплифицируемого
маркера:
1. Размер гена не создает препятствий для конструирования
вектора.
2. Ген функционирует как доминантный селективный маркер
в клетках многих типов.
3. Устойчивость клеток к токсическому фактору обусловлена
только амплификацией введенного маркерного гена.
4. Амплификация достигается при использовании разумных
количеств токсического агента (в смысле его растворимости,
а также стоимости).
5. Высокий уровень экспрессии фермента не вредит клетке.
6. Применяемые дозы токсического агента не вызывают по-
бочных эффектов.
Наибольшее распространение в настоящее время получили
векторы с геном dhfr, хотя они и далеки от идеала. Изначально
<//г[г-векторы были сконструированы для с7г/7~-клеток, и в этой
системе они оказались весьма эффективны: число копий на клет-
ку достигало 2000 [12]. При использовании же этих векторов в
клетках с активным геном dhfr нужно обеспечить очень высокую
резистентность к метатрексату — чтобы иметь возможность при-
менить такие дозы этого агента, которые достаточны не только
для элиминации родительских клеток, но и для подавления роста
клонов с амплифицированным эндогенным dhfr. Сконструиро-
ванные для таких целей векторы содержат либо очень сильный
Амплифицирующиеся векторы
243
Рис. 8.1. Амплифицируемый вектор pSV2.d/i/r, предназначенный для исполь-
зования в качестве селективного маркера в клетках СНО, дефектных по
d/ifr [6]. Плазмида имеет в длину 5 т. п. н. и состоит из: 1) фрагмента
PvuII-Hindin SV-40 (323 п. н.), в составе которого находится область начала
репликации, причем фрагмент ориентирован таким образом, что ранний про-
мотор SV-40 регулирует транскрипцию гена dhfr- 2) кДНК гена dhfr размером
735 п. н.; 3) интрона SV-40 (положения 4099—4710 на карте SV-40); 4) фраг-
мента BcZI-fcoRI SV-40 (положения позиции 2770—1782), в котором нахо-
дится сигнал полиаденилирования из области ранних генов SV-40, и 5) фраг-
мента РаиП-fcoRI pBR322 (2295 п. н.), в котором находится область начала
репликации и ген ^-лактамазы, определяющий устойчивость конструкции
к ампициллину.
промотор для эффективной экспрессии dhfr [13], либо кДНК-
копию гена dhfr с такой мутацией в кодирующей последователь-
ности, при которой связывание фермента с метатрексатом сла-
бее, чем в норме [14]. Применение таких векторов ограничено,
так как для селекции необходимы настолько высокие концент-
рации метатрексата, что лимитирующим фактором становится
растворимость этого агента. Следует, однако, подчеркнуть, что
один из векторов на основе dhfr, сконструированный для исполь-
зования в dhfr-клетках (pSV2.^/zfr; рис. 8.1), был успешно ис-
пользован в качестве доминантного маркера в фибробластах
мыши [15].
16!
244
К- Беббингтон, К- Хенчель
Еще один недостаток системы ко-амплификации на основе
гена dhfr связан с тем, что устойчивость к метотрексату зависит
не только от амплификации этого гена [13, 16].
В качестве доминантных селективных маркеров для ряда
клеточных линий успешно используются гены аденилат-дезами-
назы (ada) и глутамин-синтетазы (gs); векторы, несущие эти
гены, хорошо амплифицируются — получено до 500 копий на
клетку в случае использования ada [4] и до нескольких тысяч
копий в случае использования gs [17]. Возможность применения
одного из этих маркеров в каждом конкретном случае зависит
как от уровня эндогенного фермента в клетках разных типов,
так и от частоты амплификации собственного гена клетки. Сле-
дует отметить, что преимущество gs связано с большей доступ-
ностью и дешевизной соответствующего ингибитора — метионин-
сульфоксимина (МСКс), хотя успех селекции связан еще и со
способностью клеток поглощать глутаминовую кислоту из среды
(разд. 8.2.3). Некоторые клеточные линии оказываются дефект-
ными по этому этапу транспорта. С другой стороны, векторы на
основе ada требуют для селекции клонов таких реагентов, кото-
рых в продаже нет, а устойчивость к дезоксиформицину может
быть обусловлена повышенным содержанием аденозин-дезами-
назы [4, 18]. Последнюю проблему удалось решить, используя
специальный метод селекции (разд. 8.2.4).
Ген металлотионеина представляет собой еще один амплифи-
цируемый маркер, работа с которым подробно освещена в дан-
ной главе. Особенно хорошо изучен один из двух генов мыши
m-mtl; этот ген невелик, и его геномную копию удобно клони-
ровать в плазмидных векторах [8]. Попытки использовать дан-
ный ген в качестве доминантного селективного маркера окончи-
лись неудачей (кроме векторов на основе BPV [19]). Поэтому
для первичной селекции требуется дополнительный маркерный
ген. Эти недостатки компенсируются в ряде случаев простым и
недорогим методом селекции клеток на амплификацию мыши-
ного гена m-mtl, хотя число копий на клетку в этой системе не
превышает 100.
Два других гена, способных выполнять функцию амплифи-
цируемых маркеров — cad и tk, — в этой главе описаны лишь в
общих чертах. Геномную ДНК гена cad, способную экспресси-
роваться в космидном векторе, можно использовать как доми-
нантный селективный маркер в клетках СНО [5], однако значи-
тельный размер самого гена сильно затрудняет какие-либо ра-
боты с таким вектором. Описан интересный вариант этой
системы, в котором использовали аспартат-карбамоилтрансфера-
зу. бактерий для комплементирования линии клеток СНО, де-
фектной по cad [20]. Этот ген также способен к амплификации
при селекции в присутствии N-(фосфонацетил)-Г-аспартата и по
Амплифицирующиеся векторы
245
этим признакам подходит в качестве селективного амплифици-
руемого маркера; однако скорее всего область его применения
ограничена этой мутантной линией клеток.
Дефектный ген tk также способен к амплификации [9], одна-
ко использовать его можно лишь в клетках ТК_, причем число
копий весьма ограниченно; поэтому в настоящей главе рассмат-
ривать его мы не будем.
8.1.5. Методы, описанные в данной главе
В разд. 8.2 приведены методики первичной селекции и после-
дующей амплификации генов в векторах на основе dhfr, ada, gs
и m-mtl. В разд. 8.3 обсуждаются факторы, которые следует
учитывать при выборе стратегии амплификации. В разд. 8.4
описаны методы анализа амплификации генов.
8.2. Методики селекции на амплификацию генов
8.2.1. Использование в качестве маркера гена
дигидрофолят-редуктазы
8.2.1.1. Селекция в d/ф—-клетках СНО
Ген dhfr — наиболее распространенный селективный маркер
для систем ко-амплификации. Как правило, в работе использу-
ется ц7г/г_-вариант линии СНО-К1, обозначаемый DUKX-B11
[21]: в этих клетках введенный при трансфекции транскриптон
dhfr выполняет роль селективного маркера. Клетки DUKX-B11
для роста нуждаются не только в нуклеозидах и глицине, но и
в пролине (родительская линия СНО-К1 является рго~-мутан-
том). Целый ряд разных векторов, содержащих ген dhfr, спосо-
бен изменить фенотип клетки на DHFR+, что позволяет им расти
на среде без нуклеозидов и глицина. В этих клетках, например,
успешно применяли векторы, несущие кДНК гена dhfr под конт-
ролем промоторов SV-40 [6] и LTR-MMTV [22], а также главного
промотора поздних генов аденовируса [23]; мини-гены dhfr в
составе этих конструкций содержали 5'-фланкирующие последо-
вательности dhfr [12]. В зависимости от использованного метода
удавалось получить трансформантов с частотой от 2-Ю-5 до
10-3 на 1—10 мкг ДНК. В то же время с учетом крайне низкой
частоты реверсий (<10-7) любой из этих векторов может вы-
ступать в качестве селективного маркера для данной линии
клеток.
Два удобных б/Л/г-вектора представлены на рис. 8.1 и 8.2.
Высокую частоту трансфекции клеток DUKX-B11 обеспечивают
оба вектора (табл. 8.3). Варианты сред для селекции dhfr+-Kno-
нов приведены в табл. 8.2.
246
К- Беббингтон, К. Хенчель
Рис. 8.2. pSVM.rfft/r (6,45 т. п. н.)—альтернативный селектируемый вектор
для клеток, дефектных по гену dhfr [22]. Для регуляции экспрессии кДНК
гена dhfr в векторе pSV2,d/ifr введен промотор в составе длинного б'-концевого
повтора вируса опухоли молочных желез мышей (MMTV-LTR).
8.2.1.2. Методика трансфекции DHFR_-KneTOK СНО
1. Накануне эксперимента высейте в неселективную среду
(табл. 8.2) примерно 106 клеток на чашку Петри диаметром 9 см.
2. Для трансфекции используйте 10 мкг ДНК плазмиды;
трансфекцию проводите СаРО4-методом, описанным в гл. 6 кни-
ги II из этой серии [34]. После завершения трансфекции инку-
бируйте клетки в неселективной среде, чтобы нормализовать
клеточные процессы. На этом этапе эксперимента можно точно
установить число трансфицированных клеток, если подсчитать
число клеток на параллельной чашке (методы подсчета прикреп-
ленных клеток изложены в гл. 6). В качестве контроля исполь-
зуйте клетки, обработанные СаРО4 без плазмидной ДНК-
3. Через 24—48 ч замените среду на селективную (табл. 8.2).
Еще раз смените среду через 3—4 дня, когда станет заметной
значительная гибель клеток (мертвые клетки теряют способность
удерживаться на субстрате и всплывают).
Амплифицирующиеся векторы
247
Таблица 8.2. Среды для селекции по dhfr
Приведены две альтернативные разработки, одна [1] из которых предпола-
гает использование модифицированной Дульбекко среды Игла (DMEM),
а другая [2] — среды MEM-Alpha.
А. Концентрированные растворы
Для неселективной среды:
1. Питательная среда HamF12 (Flow: 12—4321*) или
2. Среда MEM-Alpha с нуклеозидами (Gibco: 041—25711').
Для селективной среды:
1. Среда Игла в модификации Дульбекко (Gibco: 041—18851') или
2. Среда MEM-Alpha без нуклеозидов (Gibco: 041—25611').
200 мМ L-глутамин (Gibco). Хранить при —20 °C в аликвотах по 5 мл.
Заменимые аминокислоты (Gibco), только для ДМЕМ.
Пенициллин-стрептомицин (P/S; Gibco: 043—5070'>). Хранить при —20°C
в аликвотах по 5 мл.
Сыворотка эмбриона коровы (СЭК, FCS): инактивировать прогреванием при
56 °C. Хранить при —20 °C.
Диализованная СЭК (Gibco: 063—6300‘>).
Инактивировать и хранить, как указано выше.
Б. Приготовление сред
1. а. Неселективная среда
HamF12 500 мл
СПК 50 мл
L-глутамин 5 мл
P/S 5 мл
б. Селективная среда
ДМЕМ 500 мл
диализованная СЭК 50 мл
P/S 5 мл
или
2. а. Неселективная среда
MEM-Alpha с нуклеозидами 500 мл
СПК 50 мл
P/S 5 мл
б. Селективная среда
MEM-Alpha без нуклеозидов 500 мл
Диализованная СЭК 50 мл
P/S 5 мл
Ч Номер по каталогу фирмы приведен в качестве примера. Продукция других фирм
может оказаться не хуже, а возможно, и предпочтительней.
4. Каждые 3—4 дня меняйте среду. Иногда через 7—8 дней
на чашке появляется множество мелких колоний, возникающих
в результате временной экспрессии введенного гена dhfr. Поэто-
му для подсчета стабильно трансформированных колоний сле-
дует подождать 10—12 дней.
5. Эффективность трансфекции при использовании векторов,
представленных на рис. 8.1 и 8.2, обычно составляет (1—2) X
X 10-4 трансфицированных клеток на 10 мкг ДНК. Индивидуаль-
248
К. Беббингтон, К- Хенчель
Таблица 8.3. Примеры dfifr-зависимой ко-амплификации
pSVM.dtfr pSV2.;:7ifr
Число трансформантов на чашку
Число трансформантов, секретирующих ткане-
вой активатор плазминогена (тАП)
Максимальный уровень секреции (ед./106 кле-
ток/день)
Максимальная концентрация МТХ, приводящая к
появлению колоний (нМ)
Максимальный уровень секреции после 1 цикла
амплификации (ед./106 клеток/день)
~ 100
20
150
20
1300
-100
50
100
500
1500
Ген тканевого активатора плазминогена (tPA) клонировали в pSV2.rfftfr (рис. 8.1)
и pSVM.dftfr (рис. 8.2). 5 мкг каждой плазмиды использовали для трансфекции около
10s клеток DUKX-B11.
ные колонии используют для получения линий клеток, в которых
анализируют экспрессию интересующего гена.
8.2.1.3. Амплификация в присутствии метотрексата
Концентрация метотрексата, требующаяся для амплификации
гена dhfr, зависит от эффективности экспрессии этого гена, что
отчасти определяется строением выбранного вектора. В клетках
СНО, например, ранний промотор SV-40, входящий в состав
pSV2.dhfr, работает лучше, чем LTR-MMTV из pSVM.d/zfr. По-
этому клоны, трансфицированные pSV2.dh.fr, в среднем более
устойчивы к метотрексату, чем клопы, несущие PSVM.d/ifr. Есте-
ственно, при этом нужно учитывать индивидуальную вариабель-
ность клонов по этому признаку, а это значит, что амплифика-
цию каждого клона следует проводить в широком диапазоне
концентраций метотраксата по следующей методике:
1. Приготовьте концентрированный стерильный раствор ме-
тотрексата в воде (100 мкМ). Этот раствор можно хранить при
—20 °C.
2. Приготовьте несколько чашек Петри, содержащие в селек-
тивной среде отобранные для амплификации колонии (примерно
106 клеток на чашку), и добавьте метотрексат до конечной кон-
центрации 10, 20, 50, 100, 200 и 500 нМ.
3. Через 10 дней отберите устойчивые колонии. Те из них,
которые обладают максимальной резистентностью, можно либо
анализировать по отдельности, либо объединить для проведения
второго цикла селекции при концентрации метотрексата 500 нМ
и выше.
На первом этапе можно получить клоны с десятикратной
амплификацией гена; оценку производят по числу копий или по
Амплифицирующиеся векторы 249
уровню экспрессии сцепленного гена (о методах анализа см.
разд. 8.4). В дальнейшем число копий гена нарастает примерно
пропорционально устойчивости клона к метотрексату; описаны
клоны, содержащие до 2000 копий гена на клетку (после трех
и более циклов амплификации). У высокорезистентных клонов
экспрессия гена, сцепленного с dhfr, может достигать 3- 10s мо-
лекул белка на клетку в день [1].
8.2.2. Использование металлотионеиновых генов
Металлотионеины представляют собой группы небольших
богатых цистеином белков, которые, будучи способны связывать
различные ионы тяжелых металлов, играют важную роль в их
детоксикации. Один из двух генов металлотионеина мыши
(m-mtl) клонирован; показано, что этот ген амплифицируется
при селекции в присутствии ионов Cd2+ [8]. По сравнению с
dhfr ген мыши m-tntl обладает двумя важными особенностями:
во-первых, связывание тяжелых металлов с металлотионеином
само по себе для клетки не опасно; напротив, он предотвращает
другие нежелательные взаимодействия. Поэтому прямых фено-
типических проявлений избыточной экспрессии металлотионеина
не наблюдается. Во-вторых, тяжелые металлы стимулируют
активность промотора m-mtl, так что транскрипция этого гена
возрастает под воздействием Cd2+. По-видимому, оба этих об-
стоятельства определяют не столь высокое, как в случае с dhfr,
число копий гена даже после нескольких циклов селекции на
амплификацию щ-mtl. Кроме того, в клетках СНО-К1 ген
m-mtl вообще не удается использовать как селективный маркер
(наши неопубликованные данные). В силу этих причин в состав
вектора приходится включать другой доминантный селективный
маркер — пео или gpt. Это в свою очередь приводит к тому, что
мышиный ген mtl оказывается активным не у всех трансфектан-
тов, п во многих клонах рекомбинантная конструкция не ампли-
фицируется.
Удобный вектор, содержащий мышиный ген mtl, показан на
рис. 8.3. Ниже приведена методика селекции на амплификацию
векторов с генами пео и mtl в клонах, полученных на основе
клеток СНО-К1.
1. Трансфицируйте клетки методом ко-преципитации СаРО4,
как описано в гл. 6 книги II данной серии [34], используя любую
подходящую среду (например, Ham F12, табл. 8.2).
2. Через 24 ч добавьте агент для селекции гена пео, G418,
до конечной концентрации 0,8 мг/мл.
3. Через 3—4 дня замените селективную среду.
4. Через 7—10 дней должны появиться колонии, устойчивые
к G418. Их можно объединить и использовать на следующем
250
К. Беббингтон, К,, Хенчель
Рис. 8.3. рА Tm-mt (6,6 т. п. н., П. Стефенс, неопубликованные данные) пред-
ставляет собой рАТ153, в которой клонирован фрагмент jEcoRI-Z/mdlll раз-
мером 3,3 т. п. н., содержащий полный ген mil [8].
этапе, а можно выращивать отдельно в виде клеточных линий.
5. В присутствии Cd2+проведите селекцию устойчивых к G418
линий клеток. CdCl2 можно хранить при комнатной температуре
в виде концентрированного стерильного раствора в воде (10 мМ).
Обычно для селекции используют концентрации от 3 до 5 мкМ.
Для оценки частоты амплификации эндогенного гена mt следу-
ет обработать нетрансфицированные клетки СНО-К1 теми же
концентрациями CdCl2 и при той же плотности клеточной попу-
ляции. Частота такой амплификации может быть значительной
при использовании Cd2+ в концентрации 3 мкМ и ниже.
6. Клоны, резистентные к CdCl2, можно сразу использовать
для анализа амплификации генов (методы — см. разд. 8.4),
а можно провести еще один цикл селекции при концентрациях
Cd2+ от 5 до 50 мкМ..
Увеличение числа копий гена, как правило, меньше, чем в
случае d/ifr-зависимой ко-амплификации. Примеры ожидаемых
результатов приведены в, табл. 8.4.
Амплифицирующиеся векторы
251
Клон 1 л Клон 1 Клон 2 Клон 2 20 мкЛ1 20 мкЛ!
Таблица 8.4. Примеры т-т//-зависимой ко-амплификации1 >
Число копий вектора на клет- 1-3 3 30 20
ку Секреция тАП (ед./106 кле- тск/день) 240 260 2400 1800
’) кДНК, кодирующую активатор плазминогена тканевого типа (тАП) под контролем
промотора m-mtl, клонировали в векторе с генами пео и m-mtl. ДНК вводили в клетки
СНО-К1 и два клоиа отобрали для амплификации. На первом этапе отобраны клоны,
устойчивые к Сс1С12 в концентрации 5 мкМ. Эти клетки подвергли второму циклу ампли-
фикации в присутствия 20 мкМ СбСЧ, и устойчивые клоны анализировали на образова-
ние тАП (М. Бендиг и К- Р. Веллингтон, неопубликованные данные).
8.2.3. Амплификация с использованием гена
глутамин-синтетазы
В последнее время мы разработали систему амплификации
вектора, основанную на использовании гена глутамин-синтетазы
(gs) [17]. Глутамин — ключевой продукт многих катаболических
и биосинтетических реакций клетки, необходимое количество
которого должно либо поставляться в клетку извне — из куль-
туральной среды, либо синтезироваться внутриклеточно из глу-
тамата и аммиака в реакции, катализируемой глутамин-синте-
тазой. Хотя известно несколько клеточных линий, нуждающихся
в экзогенном глутамине (поскольку синтез глутамин-синтетазы
у них нарушен), большинство культивируемых клеток, в том
числе и клетки СНО-КД, могут обходиться без глутамина, если
в среде есть глутамат. В этих условиях, естественно, активность
глутамин-синтетазы является жизненно важной для клетки и
угнетение этого фермента специфическим ингибитором метио-
нинсульфоксимином (MCKc, MSX) приводит к ее гибели.
Функционально полноценный клонированный ген gs вводят
в клетки в составе одного из двух экспрессирующих векторов!,
изображенных на рис. 8.4 и 8.5. Поскольку экспрессия любого
из этих векторов в клетках СНО-К1 обеспечивает клеткам устой-
чивость к МСКс, gs-систему можно использовать в качестве
доминантного селективного маркера.
Для селекции клеток СНО-К1, экспрессирующих глутамин-
синтетазу, применяют среду GMEM-S, описанную в табл. 8.5.
Глутамин, который входит в состав обычных сред в относитель-
но высокой концентрации (2 мМ), заменяют на глутамат, содер-
жание которого также превышает норму (500 мкМ вместо
100 мкМ в большинстве сред). Добавление нуклеозидов и повы-
шение концентрации аспарагина снижают метаболические по-
252
К- Беббингтон, К. Хенчель
Рис. 8.4. pSV2.gs (5,5 т. п. н.) содержит кДНК размером 1.2 т. п. н., клониро-
ванную вместо последовательности dft.fr в конструкции pSV2.dhfr [17] (см.
рис. 8.1).
требности в глутамине, хотя и не снимают их полностью; можно
использовать и такие источники углерода, как пируват натрия
и глюкозу.
В такой среде клетки СНО-КД растут хорошо, и токсическое
действие МСКс проявляется лишь при концентрациях выше
3 мкМ. В некоторых случаях удобно использовать среду GMEM-
met (Уилсон, неопубликованные данные; см. табл. 8.5). Для этой
среды характерно высокое содержание метионина. Поскольку
МСКс частично разрушается (обезвреживание) ферментами ка-
таболизма метионина, увеличение концентрации этой аминокис-
лоты приводит (в результате насыщения системы ее утилизации)
к повышению токсического действия МСКс. Клетки СНО-К1 в
этой системе растут хуже, чем в GMEM-S; однако для клеток
с высокой активностью глутамин-синтетазы эта среда более
удобна, поскольку позволяет снизить концентрацию МСКс в
ходе селекции. Селекция на активность глутамин-синтетазы
описана в следующем разделе.
Амплифицирующиеся векторы
253-
Рис. 8.5. pSVL.gs.l (8,5 т. п.н.) содержит фрагмент PyuII-ftindlH SV-40 раз-
мером 323 п. н., в котором расположена область качала репликации. Этот
фрагмент клонирован в WindIII-сайте рСТ54 [29], так что поздний промотор
SV-40 регулирует транскрипцию мини-гена gs размером 4,75 т. п. н., клониро-
ванного между £coRI и BamRI-сайтами рСТ54. Мини-ген gs содержит £coR[
фрагмент кДНК размером 0,94 т. п. н„ а также геномный фрагмент, в котором
расположен единственный интрон gs, З'-конец кодирующей последовательности
и фрагмент 1,5 т. п.н. З'-фланкирующей последовательности [17].
8.2.З.1. Методика селекции векторов,
экспрессирующих гпутамин-синтетазу
1. Рассейте приблизительно 106 клеток в среде GMEM-S
(табл. 8.5) на чашку Петри диаметром 9 см и растите в течение
ночи. (Внимание! Предварительно следует пассировать клетки
в этой среде, чтобы убедиться, что она пригодна для данной
линии клеток.)
2. Трансфицируйте клетки и оставьте их на ночь в неселек-
тивной среде, как описано в разд. 8.2.1.2. По крайней мере три
контрольные чашки обработайте СаРО4 без ДНК-
3. На следующий день добавьте к трансфицированным и
контрольным клеткам МСКс до конечной концентрации 15, 20
и 25 мкМ (приблизительные величины).
4. Еще через 2—3 дня замените среду свежей, содержащей-
МСКс.
254
К. Беббингтон, К. Хенчель
Таблица 8.5. Среды для селекции на активность глутамин-синтетазы
А. Концентрированные растворы
1. Дважды дистиллированная вода, автоклавированная порциями по 400 мл.
2. lOXGlasgow МЕМ без глутамина (GIBCO 042—2541). Храните при 4 °C.
3. 7,5%-ный раствор бикарбоната натрия (GIBCO). Храните при 4 °C.
4. ЮОХзаменимые аминокислоты (ЗАК). Храните при 4 °C.
5. 100Хглутамат+ аспарагин (Г+А): смешайте 600 мг глутаминовой кис-
лоты и 600 мг аспарагина. Доведите до 100 мл дистиллированной водой
и стерилизуйте фильтрованием через 2 мкм-фильтр (Nalgene). Храните
при 4 °C.
6. ЮОХпируват натрия (GIBCO).
7. бОХнуклеозиды: 35 мг аденозина, 35 мг гуанозина, 35 мг цитидина, 35 мг
уридина, 12 мг тимидина доведите до 100 мл водой, стерилизуйте фильт-
рованием и храните при —20 °C в аликвотах по 10 мл.
8. ЮОХметионин: 600 мг метионина растворите в 100 мл воды, стерилизуй-
те фильтрованием и храните при 4 °C.
9. Диализованная СЭК (GIBCO). Инактивируйте прогреванием при 56°C в
течение 30 мин и храните при —20°C.
10. Пенициллин-стрептомицин, 5000 ед./мл. (P/S, GIBCO).
11. 100 мМ МСКс (Sigma): приготовьте раствор 18 мг/мл в культуральной
среде. Стерилизуйте фильтрованием и храните при —20 °C.
Б. Приготовление сред
Смешайте следующие реагенты в указанном порядке в стерильных условиях:
a. GMEM-S
1. Вода 400 мл
2. 10XGMEM 50 мл
3. Бикарбонат натрия 18,5 мл
4. ЗАК ' 5 мл
5. Г+А 5 мл
6. Пируват натрия 5 мл
7. Нуклеозиды 10 мл
8. Диализованная СЭК 50 мл
9. Пенициллин-стрептомицинб мл
GMEM-S содержит заменимые аминокислоты, аланин, аспартат, глицин,
пролин и серин (100 мкМ), глутамат и аспарагин (500 мкМ), аденозин,
гуанозин, цитидин и уридин (30 мкМ) и тимидин (10 мкМ).
б. GMEM-met
1. Вода 410 мл
2. 10XGMEM 50 мл
3. Бикарбонат натрия 18,5 мл
4. ЗАК 5 мл
5. Пируват натрия 5 мл
6. Метионин 5 мл
7. Диализованная СЭК 50 мл
8. Пенициллин-стрептомицинб мл
В среде GMEM-met концентрация глутамата и аспарагина снижена до
<00 мкМ, нуклеозиды отсутствуют, а концентрация метионина увеличена
до 500 мкМ.
5. Через 10—14 дней после трансфекции подсчитайте ко-
лонии.
Подсчет колоний на контрольных чашках очень важен, так
как МСКс способен действовать в качестве селективного агента
ц для нетрансфицированных клеток, у которых амплифициро-
Амплифицирующиеся векторы
255'
Таблица 8.6. Трансфекция клеток СНО-К1 с использованием ^s-векторов1*
Вектор _ Число колоний на 10е клеток при разных концентрациях iMCKc
15 мкМ. 20 мкМ \25 мкМ .30 мкМ 100 мкМ
pSVLGS.l 13,6 9,2 5,6 2,4 0,24
pSV2.G-S 26,4 18 12 12 1,4
Контроль 0,47 0,24 0 0 0
J) Для трансфекции использовали плазмид. примерно 4-Ю6 клеток и 10 мкг каждой из
вался эндогенный ген. Успешное применение pSV2.gs и
pSVLgs.l (рис. 8.4 и 8.5) в качестве доминантных селективных
маркеров основано на том, что экспрессия гена gs в составе-
вектора дает более высокий уровень активности фермента, чем
тот, который достигается после одного цикла амплификации
эндогенного гена. Результаты одного из таких экспериментов
приведены в табл. 8.6. При селекции в присутствии 15 и 20 мкМ
МСКс эндогенная амплификация обусловливает едва заметный
фон резистентных колоний, а частота трансфекций по крайней
мере в 20 раз выше и достигает более чем 10'5 для обоих векто-
ров. Если же селекцию вести при более высоких концентрациях
МСКс, то фон, связанный с эндогенной амплификацией, исчеза-
ет, но и частота трансфекции тоже снижается.
8.2.3.2. Амплификация ^s-векторов
Клеточные линии, для получения которых использовали'
gs-векторы, можно повторно селектировать на устойчивость
к МСКс, пользуясь той же стратегией, что и в случае d/i/r-век-
торов (разд. 8.2.1.3). Независимо от использованного вектора
клеточные линии, полученные при трансфекции клеток СНО-К1,
уже после первого цикла селекции могут образовывать колонии,-
устойчивые к МСКс в концентрации до 500 мкМ, а после трех
циклов — до 10 мМ. Вместе с тем векторы могут существенно
различаться по предельному числу их копий в клетке. У высоко-
резистентных колоний, полученных с применением вектора-
pSV2.gs, максимальное число копий не превышало 10 на клетку,,
в то в,ремя как при использовании pSVLgs.l выделены клоны,,
содержащие до нескольких тысяч копий вектора, в которых при.
этом не обнаружена амплификация эндогенных генов [17].
Еще раз подчеркнем, что для таких высокоустойчивых к
МСКс клеточных линий можно в 10 раз снизить количество
МСКс в селективной среде, если использовать среду GMEM-met
(табл. 8.5).
256
К. Беббингтон, К. Хенчель
8.2.4. Аденозин-дезаминазная система
Для амплификации вектора можно использовать еще один
ген — ada [4]. Существует дефектная по аденозин-дезаминазе
линия СНО, однако векторы, экспрессирующие ген ada, могут
функционировать как доминантные селективные маркеры в
клетках многих типов.
Аденозин-дезаминаза — один из ферментов, участвующих
в метаболизме пуриннуклеозидов. Она катализирует превраще-
ние- различных адениннуклеозидов в соответствующие инозино-
вые аналоги. Хотя этот фермент есть практически во всех клет-
ках и тканях, уровень его активности варьирует в широких
пределах; для культивируемых клеток наличие такой активности
в целом необязательно. Существуют, однако, два способа сде-
лать аденозин-дезаминазу жизненно необходимой для клетки —
при этом условии специфический ингибитор данного фермента
дезоксикоформицин (дКФ) можно использовать в качестве
селективного агента [4]. Первый способ связан с введением
в среду токсического аналога аденозина, например 9-О-ксило-
гуанозиладенина (КсилА). Для превращения этого токсина в
нетоксический инозиновый аналог требуется аденозин-дезамина-
за. Второй способ заключается в увеличении концентрации са-
мого аденозина в среде. Поскольку повышенное содержание
этого нуклеозида токсично для клетки, активная аденозин-деза-
миназа необходима для эффективного снижения внутриклеточ-
ной концентрации аденозина. Соответствующая методика на-
зывается «11 ААи»-селекцией; приготовление культуральных
сред для обеих методик описано в табл. 8.7 [18, 25].
Описано два вектора, экспрессирующих аденозин-дезаминазу
(рис. 8.6 и 8.7). По имеющимся данным, оба вектора дают
сходную частоту трансфекции для клеток СНО DUKX-B11 —
до 10~4 [4].
Количество дезоксикоформицина, необходимое для селекции
трансфектантов, определяется не только конкретной линией кле-
ток, но и составом используемой среды. Так, например, среда
«КсилА» требует меньшего количества этого аналога для дости-
жения токсического эффекта, чем среда «11AAU». Поскольку
разные клетки по-разному реагируют на среду «11AAU» [4],
для первичной селекции лучше использовать «КсилА». С другой
стороны, по имеющимся данным, последующая амплификация
в среде «КсилА» затруднена; возможно, это связано с высокой
частотой появления мутантов, дефектных по аденозинкиназе
[4]. В результате таких мутаций клетки приобретают способ-
ность расти в данной среде и без аденозин-дезаминазы [18];
иными словами, в ней развивается альтернативный механизм
устойчивости к дезоксикоформицину. В силу этих причин пред-
Амплнфицирующнеся векторы
257
Таблица 8.7. Селекция по аденозин-дезаминазе
Описанные методы разработаны для клеток хориокарциномы человека [18] и
фибробластов мыши [25]. Для dhfr~-клеток СНО используют некоторые
другие компоненты [4].
А. Среда для селекции в присутствии КсилА [18].
DMEM (табл. 8.2)
Сыворотка лошади 15%‘>
КсилА2 > 4 мкМ
дДфЗ)
Б. Среда для «1 lAAU-селекции» [25].
DMEM
Сыворотка лошади 15% >’
Аденозин (Sigma) 1,1 мМ
Аланозин3) 0,05 мМ
Уридин (Sigma) 1 мМ
Дезоксикоформиции2*
!) Сыворотка лошади используется в данном случае вместо СЭК, в которой есть
аденозин-дезаминаза; при хранении среды эта ее собственная ферментативная актив-
ность приводит к снижению концентрации КсилА или аденозина. Можно применять диа-
лизованную СЭК, но ее следует добавлять непосредственно перед использованием среды.
Сыворотки обрабатывают, как указано в табл. 8.2.
Дезоксикоформиции используют для угнетения эндогенной активности адеиозин-
дезаминазы. Детали приведены в тексте.
Дезоксикоформиции, аланозии и КсилА можно приобрести в отделе терапии опу-
холей Национального института рака, США [18, 25].
ложена методика, составленная на основе разработок [4, 18
и 251, где среда «КсилА» используется для первичной селекции,
а среда «1IAAU» — для амплификации.
8.2.4.1. Методика сепекции ADA -кпеток
I. Трансфицируйте клетки и инкубируйте их в течение 24—
48 ч, как описано в разд. 8.2.1.2, но в среде «КсилА» (табл. 8.7).
Не забудьте предварительно проверить, хорошо ли растут дан-
ные клетки в этой среде.
2. Добавьте в среду дезоксикоформиции. Оптимальную кон-
центрацию следует определять для каждой конкретной линии
клеток, но обычно она составляет от 3 до 30 нМ.
3. После выделения колоний, устойчивых к дезоксикоформи-
цину, замените среду на «I1AAU» для амплификации. В этой
среде клетки могут выдержать десятикратное увеличение кон-
центрации дезоксикоформицина, поэтому для тестирования
используйте более высокие концентрации.
4. Дополнительные циклы амплификации можно проводить
со всей устойчивой к этому ингибитору популяцией клеток или
с отдельными клонами.
Согласно имеющимся данным, с помощью описанной методи-
ки можно получать клоны, устойчивые к 100 мкМ дезоксикофор-
мицину и содержащие примерно 500 копий вектора на клетку
17—1186
258
К. Беббингтон, К. Хенчель
Рис. 8.6. pSV2.ada [28] содержит фрагмент 1,2 т. п.н. кДНК ada, клонирован-
ный в векторе семейства pSV2. Остальные последовательности те же, что на
рис. 8.1. Размер плазмиды примерно 5,5 т. п.н., а ее использование в качестве
доминантного селективного маркера описано в другой работе [4].
[4 ]. Так же, как и в случае амплификации, зависимой от глу-
тамин-синтетазы, здесь не зарегистрировано увеличения числа
копий эндогенного гена.
8.3. Разработка стратегии амплификации
Появление целого ряда различных амплифицируемых векто-
ров, описанных в разд. 8.2, позволило расширить ассортимент
клеток, используемых для амплификации генов, и не ограничи-
ваться исходными с7г/т~-клетками СНО DUKX-B11. В связи
с этим перед исследователем встала проблема выбора клеток и
векторной системы. В качестве общего правила можно рекомен-
довать в первую очередь рассматривать й/ф-^-систему, посколь-
ку селекция в с7г/г~-клетках СНО обеспечивает высокую часто-
ту трансфекции, а процесс амплификации вектора наиболее
полно изучен именно для этих клеток.
С другой стороны, выбор линий клеток может быть ограни-
чен в силу каких-то особых обстоятельств, обсуждение которых
выходит за рамки данной главы. В некоторых случаях, напри-
Амплифицирующиеся векторы
259
мер, главным условием является пост-трансляционная модифи-
кация экспрессируемого продукта, которую может обеспечить
лишь данная линия клеток, или же определяющую роль играет
ее способность правильно отвечать на конкретный привнесен-
ный регуляторный сигнал. В такой ситуации можно использо-
вать и какую-нибудь из альтернативных систем амплификации.
Несмотря на то что каждая из них (dhfr, gs и ada) применя-
лась в качестве доминантного селективного маркера для опре-
деленной линии клеток, описанные в данной главе векторы
с генами gs и ada можно, по-видимому, использовать в разных
клетках. Дополнительным соображением при выборе системы
Рис. 8.7. p9ada 5—29 (9,2 т. п. н.) — альтернативный вектор, экспрессирую-
щий аденозин-дезаминазу, который можно использовать в качестве доминант-
ного селективного маркера в клетках многих типов [4]. Транскрипцию ada
регулирует главный поздний промотор аденовируса (MLP) и энхансер SV-40,
расположенный в составе фрагмента Avail размером 682 п. н., который содер-
жит участок начала репликации (SVORI); этот фрагмент введен в X/ioI-сайт
MLP. Высокий уровень трансляции обеспечивает взаимодействием продуктов
генов VA (VA-РНК) и трехчастной лидерной последовательностью аденови-
руса (х). В составе конструкции присутствует также З'-участок сплайсинга
генов иммуноглобулинов (:). кДНК dhfr повышает стабильность мРНК [4],
а последовательность (ВсН-Р«Н)-фрагмента SV-40 размером 900 п. и. обес-
печивает сигнал полиаденилирования polyA SV-40. Векторная часть pBR322
соответствует производной этой плазмиды, в которой отсутствует фрагмент
размером около 1,2 т. п. н.; этот участок конструкции содержит ген устойчи-
вости к тетрациклину и бактериальную область начала репликации.
17
260
К. Беббингтон, К. Хенчель
является, конечно, доступность необходимых реагентов. В ка-
честве иллюстрации можно привести малораспространенные
реактивы для т/тсистемы.
Ниже мы рассмотрим детально лишь факторы, имеющие,
по-видимому, важное значение для выбора стратегии экспрессии
гена. Это стабильность амплифицированных последовательно-
стей в отсутствие селекции, структура вектора и частота ампли-
фикации гена.
8.3.1. Стабильность амплифицированного состояния гена
Иногда высокоамплифицированные наборы векторных моле-
кул удается поддерживать в клетках при неселективных услови-
ях культивирования в течение нескольких недель; в других же
случаях амплифицированные последовательности утрачиваются
как только снимается селекция. Существует корреляция между
«стабильностью» или «нестабильностью» амплифицированных
последовательностей и физическим состоянием вектора в геноме
(см. обзор [2]): в стабильных линиях амплифицированные пос-
ледовательности сохраняются в виде копий, интегрированных
в одну или несколько хромосом клетки-хозяина (это можно
продемонстрировать с помощью гибридизации in situ, описанной
в разд. 8.4). Поскольку амплифицированный участок хромосомы
имеет, как правило, большие размеры, его можно выявить при
окрашивании хромосом, так как при этом образуются легко
различимые гомогенно окрашенные районы. С другой стороны,
нестабильные амплифицированные последовательности часто
ассоциированы с небольшими ацентрическими хромосомными
фрагментами, которые называют double minute (DM). Из-за
отсутствия центромер при митозе происходит неравномерное
распределение DM в дочерние клетки. При этом клетки с мень-
шим числом таких фрагментов растут лучше, так что клеточная
популяция постепенно утрачивает DM, а с ними и амплицифи-
цированные последовательности.
Причины появления в одних случаях гомогенно окрашенных
районов, а в других — DM пока неясны. По-видимому, конструк-
ция вектора здесь не играет роли, а определяющее значение
имеют природа клеток-хозяев и характеристика сайта первич-
ней интеграции. Так, например, две наиболее часто применяе-
мые линии клеток хомяка СНО и ВНК-21 обеспечивают преиму-
щественно стабильную амплификацию гена, а у мышиных
фибробластов и некоторых линий опухолевых клеток человека
преобладает DM-форма [2]. Таким образом, если во главу угла
ставить стабильность амплифицированных последовательностей
в отсутствие селекции, то предпочтение следует отдать клеткам
Амплифицирующиеся векторы
261
хомяка; если же культивирование клеток в присутствии селек-
тивного агента не является препятствием, то выбор клеточных
линий становится намного шире.
8.3.2. Строение вектора
Молекулы векторной ДНК, введенные в клетку с помощью
трансфекции, часто соединяются друг с другом, образуя высо-
комолекулярные формы, так что в хромосому клетки-хозяина
в одно и то же место внедряется сразу целый набор плазмид.
Поэтому амплифицируемый маркер и неселектируемый ген не-
обязательно должны находиться в составе одного вектора, а
могут присутствовать в разных плазмидах, которые вводят в
клетку путем ко-трансфекции. С другой стороны, надо учитывать
и возможность интеграции в хромосому лишь «одной копии век-
тора. Поэтому в общем случае лучше использовать один вектор,
в котором есть все нужные гены. Для введения интересующего
гена у многих вектороз, описанных в разд. 8.2, после амплифи-
цируемого гена располагается участок узнавания рестриктазы
BcmHI, в который можно встроить полный транскриптон (рис.
8.1, 8.2, 8.4—8.6).
Определенное влияние на уровень экспрессии гена могут
оказывать его ориентация и положение в составе вектора. Как
правило, лучшие результаты достигаются при одинаковой ори-
ентации обоих генов вектора, когда их транскрипция происходит
в одном направлении; проверить влияние различной ориентации
гена можно в экспериментах по временной экспрессии (см. гл. 6
книга II данной серии [34]), если, конечно, существует доста-
точно чувствительный способ анализа экспрессии интересующего
гена.
В случае когда создание единого вектора в силу тех или
иных причин затруднительно, можно применить селекцию на
интеграцию двух векторов в один участок хромосомы — подход,
описанный Кауфманом и др. [26]. При этом используется век-
тор, содержащий транскриптон dhfr без энхансера, который
обнаруживает низкую эффективность трансфекции в dhfr~-KAeT-
ках СНО (ЗЛО-5). Если же трансфекцию проводить совместно
с вектором, в котором имеется энхансер SV-40, то частота
трансформации возрастает в 20 раз. Этот эффект связан с ин-
теграцией двух векторов в близком соседстве друг с другом, так
что энхансер усиливает экспрессию гена dhfr и тем самым уве-
личивает кажущуюся частоту трансфекции.
8.3.3. Частота амплификации генов
Частота выявления колоний, устойчивых к селективному
агенту, зависит от нескольких факторов, в том числе от типа
клеток, положения интересующей последовательности в геноме,
262
К. Беббингтон, К. Хенчель
концентрации селективного агента (поскольку степень ампли-
фикации, а значит, и степень устойчивости изменяются по мере
увеличения числа копий гена), а также от плотности клеточной
популяции (см. обзор [3]). При этом частота амплификации
отдельного локуса обычно лежит в интервале 101—10~е; иными
словами, амплификацию гена можно, как правило, обнаружить
без особых усилий и не прибегая к специальным приемам для
увеличения ее частоты. В принципе такие приемы существуют,
и связаны они с вмешательством в процессы синтеза ДНК
или с активацией процессов репарации. Нужно учитывать при
этом, что некоторые агенты, на первый взгляд стимулирующие
амплификацию, на самом деле могут просто усиливать процессы
образования колоний в популяциях клеток, растущих при низких
плотностях (подробнее об этом см. работу [3]).
8.4. Контроль амплификации генов
Оценить эффективность амплификации генов можно либо
с помощью гибридизационных методов, позволяющих детектиро-
вать увеличение количества копий специфических последователь-
ностей ДНК или мРНК, либо с помощью количественного ана-
лиза экспрессии белка. Наиболее информативно сравнение дан-
ных по всем трем контрольным параметрам (количества
ДНК, РНК и белка), так как изменения этих параметров не
всегда коррелируют друг с другом. Кроме того, полезную ин-
формацию может дать локализация амплифицированных после-
довательностей на метафазных хромосомах с помощью гибриди-
зации in situ например для оценки стабильности экспрессирую-
щих клонов при культивировании в неселективных условиях.
8.4.1. Определение числа копий гена
Количество копий специфической последовательности ДНК
можно оценить с помощью блот-гибридизации по Саузерну.
Приготовление ДНК из культивируемых клеток описано в гл. 6
книги II данной серии [34], а техника переноса на нитроцеллю-
лозный фильтр и гибридизации с клонированным зондом хоро-
шо изложены в работе [27], и в данной главе мы не будем воз-
вращаться к ним. Общая постановка эксперимента показана на
рис. 8.8.
ДНК выделяют из трансфицированных клеток на разных
стадиях селекции на амплификацию, обрабатывают подходящим
ферментом рестрикции, после чего строго определенные коли-
чества подготовленной таким образом ДНК фракционируют
электрофорезом и переносят на нитроцеллюлозный фильтр.
В этом же геле параллельно разделяют точно известные коли-
Амплифицирующиеся векторы
263
Рис. 8.8. Блот-гибридизационный анализ ДНК из трансфицированных pSVLGS.l
клеток СНО-К1: тест на амплификацию вектора. Дорожки 1—7: ДНК плазми-
ды pSVLgs.l (см. рис. 8.5) после гидролиза ВдтНЛ—стандарт для оценки
количества ДНК. На дорожку 1 нанесено 1,25 нг ДНК, на остальные — дву-
кратные разведения. На дорожки 8—13 наносили по 2,5 мкг геномной ДНК
трансфицированных клонов. Показаны результаты для трех клонов: клона 1
(дорожка 8), селекцию которого вели в присутствии 20 мкМ метионинсуль-
фоксимина; клона 2 (дорожка 9), устойчивого к 20 мкМ МСКс; клона 3 (до-
рожка 10), устойчивого к 30 мкМ метионинсульфоксимина. На дорожке 11
представлена ДНК варианта клона 2, устойчивого к метионинсульфоксимину
в концентрации 250 мкМ. На дорожке 13 — варианта клона 3, устойчивого
к 500 мкМ метионинсульфоксимина. Гибридизацию вели с пробой, специфич-
ной для ДНК вектора. Очевидно, что материал на дорожках 11 —13 содержит
больше копий вектора, чем исходные клоны (дорожки 8—10), что свидетель-
ствует об их эффективной амплификации.
чества плазмиды, содержащей интересующую последователь-
ность (количества измеряют по поглощению в УФ-свете при
длине волны 260 нм). После гибридизации путем визуального
сравнения или с помощью сканирующей денситометрии полос на
радиоавтографе можно оценить количество клонированной пос-
ледовательности в расчете на 1 мкг клеточной ДНК- Если при
этом принять, что общее количество ДНК в диплоидной клетке
млекопитающих примерно равно 6 пкг, то можно произвести
расчет числа копий гена в клетке.
8.4.2. Оценка уровня транскрипции
Уровень РНК в разных клетках можно сравнивать, пользу-
ясь методом «нозерн»-блот-анализа [27]. Общий план экспери-
мента аналогичен тому, который рассматривался в предыдущем
разделе. Стандартный препарат РНК-копий анализируемых пос-
ледовательностей можно приготовить и количественно оценить
с помощью системы транскрипции in vitro SP6 (см. гл. 1 этого
тома).
264
К- Беббингтон, К- Хенчель
Таблица 8,8. Приготовление клеточных экстрактов для определения
активности ферментов
1. Центрифугируйте клетки при 1500 об/мин 5—10 мин в настольной цент-
рифуге,
2. Трижды промойте клетки фосфатно-солевым буфером (PBS, Flow), сус-
пендируя их в PBS с помощью пастеровской пипетки.
3. Ресуспендируйте клетки в 0,25 М сахарозе, 10 мМ трис-НС1 (pH 7,6) и
1 мМ ДТТ при конечной концентрации примерно 5-Ю7 клеток/мл.
4. Обработайте клетки ультразвуком (например, 3 имп. по 15 с каждый).
5. Центрифугируйте в ультрацентрифуге 45 мин при 100 000 g и 4 °C или в
микроцентрифуге в течение 10 мин.
6. Соберите надосадочную жидкость (старайтесь не захватить липидный слой
с поверхности) и поместите на лед или храните при —20 °C, если анализ
проводится позже.
7. Определите общую концентрацию белка, пользуясь, например, методом
Лоури [33],
8.4.3. Оценка эффективности экспрессии белка
Если существует подходящий способ анализа экспрс^опп
белкового продукта гена, клонированного в амплифицирующг м-
ся секторе, то его можно использовать для контроля амплифика-
ции на разных ее этапах. Кроме того, нередко можно следить за
экспрессией амплифицируемого маркерного гена, пользуясь
быстрыми и простыми способами определения дигидрофолят-ре-
дуктазы, глутамин-синтетазы и аденозин-дезаминазы. Это осо-
бенно удобно в тех случаях, когда активность соответствующего
эндогенного гена в используемой линии клеток не охарактери-
зована.
Метод приготовления клеточных экстрактов, в которых
можно определять активность всех трех ферментов, описан в
табл. 8.8: он представляет собой модификацию методов, пред-
ложенных ранее ,[30, 31 и 32]. В табл. 8.9 описан метод анализа
дигидрофолят-редуктазы, основанный на регистрации снижения
Таблица 8.9. Определение дигидрофолят-редуктазы
1. Приготовьте клеточные экстракты и определите концентрацию белка, как
указано в табл. 8.8.
2. Приготовьте следующие реагенты: 100 мМ калий-фосфатный буфер
(pH 7,9); 5 мМ дигидрофолят; 1 мМ NADPH.
3. Приготовьте реакционную смесь в кювете с длиной оптического пути 1 см
и объемом 1 мл: 0,8 мл калий-фосфатного буфера, 10 мкл дигидрофолята,
0,1 мл NADPH, различные объемы клеточного экстракта (до 0,1 мл) и
воды до 1 мл, В качестве контроля используйте ту же смесь, но без кле-
точного экстракта.
4. Инкубируйте кюветы при 37 °C и измеряйте поглощение при 340 нм с ин-
тервалами в 1 мин.
5. Активность фермента можно выразить например так: количество наномо-
лей восстановленного дигидрофолята/мг белка/мин, считая изменение мо-
лярного поглощения равным 12 200 на М-см.
Амплифицирующиеся векторы
265
Таблица 8.10. Определение глутамин-синтетазы
А. Приготовление ионообменных колонок Dowex.
1. Добавьте 0,5 М NaOH к некоторому количеству Dowex-1-CI (8%-ная
сшивка, 200—400 меш).
2. Удалите (отсосав их) мелкие частицы и несколько раз промойте дистил-
лированной водой.
3. Промойте 0,5 М НС1 до тех пор, пока pH не станет ниже 2.
4. Несколько раз промойте дистиллированной водой.
5. Доведите pH до 7, добавляя сухой имидазол.
6. Приготовьте стандартные колонки (например, по 0,8 мл) в наконечниках
для автоматических пипеток на 1 мл (например, голубые наконечники к
пипетке Р 1000 Gilson Pipetman). Конец наконечника следует отрезать,
а в наконечник поместить вату, чтобы гранулы Dowex не проваливались.
7. Промойте колонки дистиллированной водой (несколько миллилитров).
Б. Определение ферментативной активности
1. Необходимые реагенты: 0,5 М трицин натрия (pH 7,6); 100 мМ. АТР
(pH 7,0); 80 мМ NH4C1; 400 мМ MgCl2; 100 мМ 14С-глутамата
(4 мкКи/ммоль).
2. Приготовьте клеточные экстракты, как указано в табл. 8.8.
3. Приготовьте 50 мкл реакционной смеси: 5 мкл трицина натрия; 2,5 мкл
NH4CI; 2,5 мкл MgCl2; определенное количество экстракта (до 25 мкл),
воды до 45 мкл, 5 мкл 14С-глутамата и 7,5 мкл АТР.
4. Инкубируйте при 37 °C разное время, например 5, 10, 15, 20 и 30 мнн.
5. Остановите реакцию, добавив 200 мкл ледяной дистиллированной воды,
и поместите пробирку на лед.
6. Нанесите весь объем на колонку Dowex и удалите первый элюат.
7. Элюируйте глутамин двумя порциями дистиллированной воды по 0,5 мл
каждая; элюат соберите. Глутамат при этом остается на колонке.
8. Добавьте к элюату 10 мл сцинтиллятора (например, Biofluor) и просчи-
тайте в сцинтилляционном счетчике.
9. Просчитайте контрольную реакционную смесь (без клеточного экстракта)
п подсчитайте процент метки, включившейся в глутамин.
10. Подсчитайте количество наномолей прореагировавшего глутамата/мин,
исходя из удельной радиоактивности !4С-глутамата и процента конверти-
рованной метки.
оптического поглощения раствора при длине волны 340 нм в
процессе превращения NADPH и дигидрофолята в NADP+ и
тетрагидрофолят [30]. Радиоизотопный тест на глутамин-синте-
тазу описан в табл. 8.10. При использовании этого метода прос-
леживается превращение 14С-глутамата в глутамин с последую-
щим разделением этих аминокислот с помощью ионообменной
хроматографии.([31] и Уилсон, неопубликованные данные).
Метод спектрофотометрического определения активности, пред-
ложенный Агарвалом и Парксом [321, приведен в табл. 8.11.
Здесь регистрируется снижение оптического поглощения при
265 нм, обусловленное превращением аденозина в инозин.
8.4.4. Гибридизация in situ с метафазными хромосомами
Пред л нгй о мыс методики можно ксиол о для по л уч ОНИ ’
препаратов мстафазных хромосом из культуры прикрепленных
к субстрату клеток и для последующей гибридизации in situ.
266
К- Беббингтон, К. Хенчель
Таблица 8.11. Определение аденозин-дезаминазы
1. Приготовьте клеточный экстракт и определите общую концентрацию бел-
ка, как описано в табл. 8.8.
2. Приготовьте следующие реактивы: 50 мМ калий-фосфатный буфер (pH 7,4);
10 мМ аденозин.
3. Приготовьте реакционную смесь в кювете с длиной оптического пути 1 см
и объемом 1 мл: 0,9 мл калий-фосфатного буфера; 10 мкл аденозина
:,0,1 мкмоль); разные количества клеточного экстракта (до 100 мкл) и во-
ды до 1 мл. Приготовьте ту же смесь, но без экстракта в качестве конт-
роля.
4. Инкубируйте кюветы при 30 °C и измеряйте поглощение при 265 нм с ин-
тервалом в 1 мин.
5. Подсчитайте активность фермента: одна единица активности катализиру-
ет дезаминирование 1 мкМ аденозина в минуту. (А=8,6 в мин/мл при
данных условиях).
Сначала клетки обрабатывают колцемидом, чтобы заблокиро-
вать образование веретена; это приводит к накоплению мета-
фазных клеток в культуре. Такие клетки имеют более округлую
форму и слабее прикреплены к пластику, чем другие клетки по-
пуляции, поэтому при встряхивании флакона они легко отделя-
ются от субстрата. Метафазные клетки затем обрабатывают
гипотоническим раствором (что приводит к их набуханию),
после чего фиксируют в смеси метанол — уксусная кислота.
Когда суспензию таких клеток по каплям наносят на предметное
стекло, клетки лопаются и хромосомы расправляются на стекле
по мере высыхания раствора. Реактивы для этой процедуры
приведены в табл. 8.12, а сам метод описан в табл. 8.13. На сле-
дующем этапе хромосомы обрабатывают РНКазой, чтобы ис-
ключить гибридизацию зонда с хромосомной РНК, и затем
уксусным ангидридом, который ацетилирует хромосомные белки
и тем самым еще более уменьшает неспецифическое связывание
зонда. Хромосомную ДНК денатурируют нагреванием и гибри-
дизуют с зондом, предварительно меченным 1251-СТР. Избыток
меченого зонда удаляют серией промывок в низкосолевом бу-
ферном растворе и покрывают препарат фотоэмульсией. После
необходимой экспозиции выявляют участки образования зерен
серебра, соответствующие сайтам амплификации гена. Необхо-
димые реагенты приведены в табл. 8.14, а сама процедура
описана в табл. 8.15.
Благодарности
Мы хотим поблагодарить Ричарда Уилсона, Пола Стефанса
и Мэри Бендиг за то, что они предоставили нам свои неопубли-
кованные данные. Мы также признательны Ричарду Уилсону и
нашим коллегам из Celltech за сделанные замечания и Джил
Алкок за перепечатку рукописи.
Амплифицирующиеся векторы
267
\Таблица 8.12. Реагенты для приготовления препаратов расправленных
метафазных хромосом
1. PBS. Возьмите таблетки (например, фирмы Flow) и приготовьте раствор
согласно инструкции фирмы-изготовителя. Автоклавируйте и храните
при 4 °C.
2. Колцемид (10 мкг/мл). Растворите 5 мг колцемида в 5 мл воды. Разве-
дите в 100 раз PBS и стерилизуйте фильтрованием через фильтр 0,2 мкм.
Храните при 4 °C.
3. Актиномицин D (15 мкг/мл). Растворите актиномицин D в этаноле до
концентрации 5 мг/мл. Разбавьте в 1000 раз водой.
4. 0,5%-ный раствор хлорида калия. Растворите 0,5 г КС1 в 100 мл воды
и автоклавируйте.
5. Фиксирующий раствор: метанол : уксусная кислота=3 : 1.
6. 45%-ная уксусная кислота, водный раствор.
7. Предметные стекла для микроскопии. Обработайте их следующим образом:
а) на ночь погрузите в 1%-ный «decon»,
б) ополосните в дистиллированной воде,
в) 1 ч выдержите в разбавленной НС1,
г) ополосните в дистиллированной воде,
д) храните погруженными в этанол,
е) перед использованием высушите на воздухе.
Таблица 8.13. Приготовление расплавленных метафазных хромосом
В этой методике используются реактивы, перечисленные в табл. 8.12.
1. Удалите среду с клеток.
2. Добавьте 20 мл инкубационной среды, содержащей 0,2 мл колцемида и
0,5 мл актиномицина D.
3. Инкубируйте клетки при 37 °C в течение примерно 75 мин. (Оптимальное
время надо определять эмпирически для каждого типа клеток; при бо-
лее длительных сроках инкубации актиномицин D следует добавлять
только па последнем часу инкубации.)
4. Сильно встряхните флакон для того, чтобы митотические клетки отдели-
лись от субстрата.
5. Перенесите культуральную среду с митотическими клетками в центри-
фужную пробирку.
6. Центрифугируйте в течение 8 мин при 1200 об/мин в настольной цент-
рифуге для осаждения клеток.
7. Удалите всю надосадочную жидкость, за исключением последних 0,5 мл,
и ресуспендируйте клетки, осторожно встряхивая пробирку.
8. Добавьте по каплям 10 мл 0,5% КС1, осторожно перемешивая суспен-
зию, и инкубируйте 12,5 мин при комнатной температуре.
9. Центрифугируйте клетки при 1200 об/мин в течение 8 мин.
10. Удалите всю надосадочную жидкость, за исключением последних 0,5 чл.
п ресуспендируйте осадок.
11. Очень осторожно, по каплям и с мягким перемешиванием добавляйте хо-
лодный фиксирующий раствор, пока объем не достигнет 5 мл. Сначала
добавляйте раствор очень медленно; после того, как 1 мл фиксирующего
i детвора внесен, можно работать чуть быстрее. И фиксирующий раствор,
и клетки все время надо поддерживать при температуре 0—4 °C.
12. 5 мнп инкубируйте при 4 °C.
13. Центрифугируйте клетки при 1200 об/мин в течение 8 мин.
14. Дважды повторите этапы 10—13.
15. Удалите всю надосадочную жидкость, за исключением последних 0,5 мл,
и ресуспендируйте клетки в 1 мл фиксирующего раствора. Храните при
4 °C.
268
К- Беббингтон, К- Хенчель
Продолжение
16. Фиксированные образцы следует использовать для приготовления препа-
ратов в тот же день.
17. Выньте стекла из этанола и высушите их на воздухе.
18. Капните суспензию фиксированных клеток на стекло и дайте капле рас-
течься. Иногда для растекания лучше сначала капнуть на стекло 45%-ной
уксусной кислотой и затем—препарат.
19. Дайте капле высохнуть и рассмотрите хромосомы в фазово-контрастном
микроскопе. Препараты готовы теперь для гибридизации in situ; хранить
их следует при комнатной температуре, предохраняя от пыли. На этом
этапе можно выявить хромосомы типа double minute.
Таблица 8.14. Растворы для гибридизации in situ
А. Реагенты, которые можно приготовить заранее
1. 20XSSC: растворите в воде 175,3 г хлорида натрия и 88,2 г цитрата нат-
рия. Доведите pH до 7,0, добавляя NaOH или НС1; конечный объем 1 л.
2. РНКаза 100 мкг/мл в 2XSSC: кипятите 10 мин, затем медленно остуди-
те. Разлейте на порции и храните при —20 °C.
3. Этанол.
4. Формамид: деионизируйте, пропуская через ионобменную смолу (BioRad
AG501-X8) до тех пор, пока pH не станет нейтральным. Разлейте на пор-
ции и храните при —20 °C.
5. 0,25 М ЭДТА. Растворите 93 г ЭДТА в воде. Доведите pH до 8,0, до-
бавляя NaOH; конечный объем 1 л. Автоклавируйте.
6. 0,1 М триэтаноламин (pH 8,0). Смешайте: 13,3 мл триэтаноламина и
800 мл воды. Доведите pH до 8,0, добавляя НС1; конечный объем 1 л.
7. Уксусный ангидрид.
8. ЮОХраствор Денхардта. Растворите в воде: 21 г фиколла, 21 г поли-
винилпирролидона, 21 г бычьего сывороточного альбумина (Pentax Frac-
tion V) и доведите объем до 100 мл. Храните в небольших аликвотах
при —20 °C.
9. 20XSSPb. Растворите в 800 мл воды: 174 г NaCl, 27,6 г NaH2PO4-H2O,
7,4 г ЭДТА. Доведите pH до 7,4, добавляя NaOH;. конечный объем 1 л.
10. 50%-ный декетрансульфат. Растворите 500 г декстрансульфата в воде.
Доведите до 1 л и автоклавируйте.
11. ДНК спермы лосося — 10 мг/мл. Кипятите 10 мин и охладите на льду.
Хранить при —20 °C.
12. 1261-СТР (Amersham или другой поставщик). 1500 Ки/ммоль или более
высокая удельная радиоактивность.
13. Ядерная эмульсия Ilford L4 (или равноценная).
14. Проявитель D19 (Kodak или другой поставщик).
15. 1%-кая ледяная уксусная кислота.
16. Фотографический фиксаж (Kodak Kodafix или равноценный).
17. Краска Гимза (BDH-Gurr).
18. Буфер pH 6,8 (поступает в продажу в виде таблеток; BDH или другой
поставщик; приготовить по инструкциям фирмы).
19. Клей резиновый (Dunlop или другой поставщик).
Б. Растворы, которые следует готовить в день эксперимента
1. 2XSSC” (разбавьте концентрированный 20Храствор в 10 раз).
2. Этанол в таких концентрациях: 10% этанол — разведите 100 мл этанола
') SSC: 0,3 М NaCl, 0,03 М цитрат натрия.
Амплифицирующиеся векторы
269
Продолжение
вбдой до 1 л; 50% этанол — разведите 500 мл этанола водой до 1 л;
75% этанол—разведите 750 мл этанола водой до 1 л; 95% этанол — раз-
ведите 950 мл этанола водой до 1 л.
3. Дейатурирующий раствор (70%-ный формамид; 1 мМ ЭДТА; 2XSSC.
На (500 мл: формамида — 350 мл; ЭДТА — 0,2 мл; 20XSSC— 50 мл; во-
ды — 100 мл.
4. Уксусный ангидрид (0,25%-ный раствор уксусного ангидрида в 0,1 М
триэтаноламине, pH 8,0). На 500 мл: уксусного ангидрида—1,25 мл;
0,1 М триэтаноламина—499 мл.
5. Гибридизационный раствор: (50%-ный формамид; 5Храствор Денхардта;
5XSSPE; 10%-ный декстрансульфат; 200 мкг/мл ДНК спермы лосося).
На 1 мл: формамида — 500 мкл; ЮОХраствора Денхардта — 50 мкл;. 20Х
XSSPE — 250 мкл; 50% декстрансульфата — 200 мкл; ДНК спермы лосо-
ся 10 мг/мл — 20 мкл.
Таблица 8.15. Гибридизация in situ
1. Проведите «ник-трансляцию» 50—100 нг пробы по стандартной методике
[27], используя в качестве метки 40 мкКи 1251-СТР.
2. Лиофилизируйте ДНК и растворите ее в 300 мкл гибридизационного
раствора (табл. 8.14).
3. Нанесите на каждое предметное стекло с препаратом хромосом по 20 мкл
РНКазы, накройте покровным стеклом и инкубируйте во влажной каме-
ре в течение 1,5 ч при 37 °C.
4. Аккуратно промойте предметные стекла, окуная их в ванночки с 2XSSC.
Используйте 4 смены 2XSSC. Предметные стекла держите вертикально,
чтобы покровные стекла сами отделились.
5. Дегидратируйте препараты, помещая их на 1 мин в 10, 50, 75, 95 и
100%-ный этанол. Высушите на воздухе и храните в эксикаторе.
6. Денатурируйте хромосомную ДНК в денатурирующем растворе в тече-
ние 4 мин при 65 °C.
7. Промойте дегидратируйте и храните, как на этапах 4 и 5.
8. На 15 мин поместите стекла в раствор 0,25%-кого уксусного ангидрида
при комнатной температуре.
9. Промойте, дегидратируйте и храните, как на этапах 4 и 5.
10. Денатурируйте пробу в буфере для гибридизации в кипящей воде 3 мин.
Сразу же поместите в лед.
И. Нанесите 30 мкл гибридизационной смеси с пробой на стекло и закройте
каплю покровным стеклом. Убедитесь, что под стеклом нет пузырьков
воздуха и жидкость выступает за края покровного стекла. Для этого
следует осторожно опускать покровное стекло пинцетом.
12. Заклейте резиновым клеем.
13. На ночь оставьте во влажной камере при 45 °C.
14. Осторожно удалите покровное стекло с предметного стекла, погрузив
препарат в 5XSSC. Для этого держите стекло пинцетом так, чтобы по-
кровное стекло было обращено вниз, и вторым пинцетом удаляйте клей.
Покровное стекло должно соскользнуть под собственным весом.
15. Инкубируйте стекла в 2XSSC при комнатной температуре 1—2 ч.
16. Промойте 1 ч в 0.5XSSC при 60 °C.
17. Промойте 1 ч в O.IXSSC при 65 °C.
18. Дегидратируйте и храните в эксикаторе.
19. Для нанесения эмульсии нагрейте ее до 55 °C в полной темноте в фото-
комнате.
20. Разбавьте эмульсию равным объемом дистиллированной воды.
270
К- Беббингтон, К- Хенчель
Продолжение
21. Окунайте препараты в эмульсию по одному. (Сначала рекомендуем Оку-
нуть пустое стекло, чтобы убедиться в отсутствии пузырьков и в гомо-
генности эмульсии.) Вытаскивайте стекло осторожно, прижимая один его
край к краю сосуда.
22. На 30 мин оставьте стекла в вертикальном положении в темноте так,
чтобы эмульсия застыла.
23. Экспонируйте препараты в течение 1—10 дней в светонепроницаемой ко-
робочке с осушителем при 4 °C.
24. Перед проявлением дайте препаратам нагреться до комнатной темпера-
туры в течение 1 ч.
25. Проявляйте 5 мин при 20 °C в проявителе D19 в специальной камере для
стекол.
26. Ополосните в стоп-ванне (1%-ная уксусная кислота).
27. Фиксируйте 5 мин в Kodafix (разбавленном 1:4).
28. Промойте в течение 30 мин в проточной воде.
29. Окрасьте в течение 10—20 мин в красителе Гимза, разбавленном в Юра»
буфером pH 6,8. (Разбавленная краска не хранится.)
30. Высушите на воздухе и анализируйте препараты под микроскопом.
Литература
1. Bebbington С. R., Hentschel С. С. G. (1985). Trends Biotechnol., 3, 314.
2. Stark G. R., Wahl G. M. (1984). Annu. Rev. Biochem., 53, 447.
3. Stark G. R. (1986). Cancer Surveys, 5, 1.
4. Kaufman R. J., Murtha P., Ingolia D. E., Yeung C.-Y., Kellems R. E. (1986).
Proc. Natl. Acad. Sci. USA, 83, 3136.
5. de Saint Vincent B. R., Delbruck S., Eckhart W., Meirloth J., Vitto L.,
Wahl G. (1981). Cell, 27, 267.
6. Subramani S„ Mulligam R., Berg P. (1981). Mol. Cell. Biol., 1, 854.
7. Hayward В. E., Hussain A., Wilcon R. H., Woodcock V., McIntosh B., Har-
ris T. J. R. (1986). Nucleic Acids Res., 14, 999.
8. Hamer D. H., Walling M. J. (1982). J. Mol. Appl. Genet., 1, 273.
9. Roberts J. M., Axel R. (1982). In: Gene Amplification, Schimke R. (ed.), Cold
Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
10. Gros P., Heriah Y. B., Croop J. M., Housman D. E. (1986). Nature, 323,
728.
11. Milbrandt J. D., Azizkahn J. C., Hamlin J. L. (1983). Mol. Cell. Biol., 3,
1274.
12. Crouse G. F., McEwan R. N., Pearson M. L. (1983). Mol. Cell. Biol., 3,
257
13. Murray M. J., Kaufman R. J., Latt S. A., Weinberg R. A. (1983). Mol. Cell.
Biol., 3, 32.
14. O’Hare K-, Benoist D., Breathnach R. (1981). Proc. Natl. Acad. Sci. USA,
78, 1527.
15. Deschatrette I., Fougere-Deschatrette C., Corcos L., Schimke R. T. (1985).
Proc. Natl. Acad. Sci. USA, 82, 765.
16. Simonsen C., Levinson A. D. (1983). Proc. Natl. Acad. Sci. USA, 80, 2495.
17. Bebbington C. R., Hentschel С. C. G. (1987) submitted.
18. Yeung C.-Y., Riser M. E., Kellems R. E., Siciliano M. J. (1983). J. Biol. Chem.,
258, 8330.
19. Pavlakis G. N., Hamer D. H. (1983). Recent Progress in Hormone Research,
39 353
20. Ruis J.'C., Wahl G. M. (1986). Mol. Cell. Biol., 6, 3050.
\Амплифицирующиеся векторы 271
2К Urlaub G.. Chasin L. А. (1980). Proc. Natl. Acad. Sci. USA, 77, 4216.
221 Lee F., Mulligan R„ Berg P., Ringold G. (1981). Nature, 294, 228.
23. \Kaufman R. J., Sharp P. A. (1982). Mol. Cell. Biol., 2, 1304.
24. Zanders P. G„ Wilson R. H. (1984). EMBO J., 3, 65.
25. \eung C.-Y., Ingolia D E., Bobonis C., Dunbar B. S., Riser M. E., Sici-
liano M. J., Kellems R. E. (1983). J. Biol. Chem., 258, 8338.
26. Kaufman R. J., Wasley L. C., Spiliotes A. J., Gossels S. D., Latt S. A., Lar-
se\ G. R., Ray R. M. (1985). Mol. Cell. Biol., 5, 1750.
27. Maniatis T., Fritsch E. F., Sambrook J., eds. (1982). Molecular Cloning, Cold
Spring Harbor Laboratiry Press, Cold Spring Harbor, NY. [Имеется перевод:
МаЬиатис T.. Фрич Э., Сэмбрук Дж. Методы генетической инженерии. Мо-
лекулярное клонирование. — М.: Мир, 1984.]
28. Orkln S. Н., Goff S. С., Kelley И7. Лг., Daddona Р. Е. (1985). Mol. Cell. Biol.,
5, 762.
29. Emtage J. S., Angal S., Doel M. T., Harris T. J. R., Jenkins B., Lilley G..
Lowe P. A. (1983). Proc. Natl. Acad. Sci. USA, 80, 3671.
30. Flintoff Ж F., Davidson S. V., Siminovitch L. (1976). Somat. Cell Genet.,
3, 245.
31. Tiemeier D. E., Milman G. (1972). J. Biol. Chem., 247, 2272.
32. Agarwal R. P., Parks R. E. (1978). In: Methods Enzymol., Academic Press,
NY, Vol. 51, p. 502, 51, p. 502.
33. Lowry О. H., Rosenbrough N. J., Farr A. L., Randall R. J. (1951). J. Biol.
Chem., 193, 265.
34. DNA Cloning. A practical Approach, Glover D. (ed.), (1984). IRL Press,
Oxford, Vol. 2. [Имеется перевод: Клонирование ДНК. Методы. Книга II.
Под ред. Д. Гловера. — М.: Мир, 1.988.]
ГЛАВА 9
РЕТРОВИРУСНЫЕ ВЕКТОРЫ
Энтони М. К. Браун и Майкл Р. Д. Скотт
(Anthony М. С. Brown, Michael R. D. Scott)
9.1. Введение
9. 1.1. Почему именно ретровирусы?
Использование ретровирусов в качестве векторных молекул за
последние годы обрело статус наиболее перспективного и уни-
версального метода введения и экспрессии клонированных ге-
нов в эукариотических клетках. Многие свойства ретровирусов
делают их более удобными для таких исследований, чем методы
прямого генетического переноса или использование потенциаль-
ных вирусных векторных систем. Во-первых, относительно не-
большой геном ретровирусов позволяет довольно просто с ним
манипулировать и вводить в его состав чужеродные гены таким
образом, чтобы любой возникающий при этом дефект вирусной
репликации мог бы комплементироваться по транс-схеме. Во-
вторых, вирусы можно легко нарастить в культуре до высокого
титра. Эффективность инфицирования пермиссивных клеток
чрезвычайно высока, что в сочетании с высоким титром вирус-
ных препаратов позволяет получать в культуре почти 100%-ное
заражение клеток-мишеней. После инфицирования единственная
копия ретровирусной ДНК оказывается стабильно интегрирован-
ной в геном клетки-хозяина строго определенным образом, так
что практически все инфицированные клетки способны экспрес-
сировать гены, привнесенные вирусом. Кроме того, ретровирусы
содержат мощные транскрипционные энхансеры, которые суще-
ственно повышают уровень экспрессии клонированных генов
в клетках различных типов.
Хотя применение ретровирусных векторов требует, чтобы
экспрессируемый ген был введен в соответствующий вектор в
строго определенной ориентации (это условие в общем случае
ограничивает их использование лишь для хорошо охарактеризо-
ванных последоательностей) после того, как правильная реком-
бинантная конструкция получена, дальнейшие операции по
наращиванию вируса и инфицированию клеток-мишеней техни-
чески оказываются достаточно простыми.
272
Ретровирусные векторы
273-
9. 1.2. Применение ретровирусных векторов
Наибольшее применение ретровирусные векторы нашли как.
экспрессирующие векторы общего назначения для эксперимен-
тов с культурами эукариотических клеток. Это, однако, не
уменйщает их роли в решении более специализированных задач
молекулярной биологии, генетики, биологии развития, и, воз-
можно, медицины. Некоторые из этих аспектов рассмотрены
ниже.
1. Ретровирусные векторы наряду с тем, что они являются,
эффективным инструментом для осуществления экспрессии ге-
нов в культивируемых клетках, могут быть использованы для
прямого инфицирования клеток in vivo, например путем внутри-
брюшинной инъекции новорожденным мышам [1].
2. С помощью ретровирусной инфекции гемопоэтических
стволовых клеток в культурах костного мозга и их последую-
щей трансплантации облученным мышам можно заменить всю
гемопоэтическую ткань животного клетками, содержащими
ретровирусный вектор [2]. Можно надеяться, что эксперименты
такого рода приблизят возможность лечения ряда наследствен-
ных дефектов методами генотерапии [3].
3. Ретровирусы позволяют вводить ДНК в клетки зароды-
шевого пути путем предимплантационного инфицирования эмб-
рионов или плюрипотентных эмбриональных стволовых клеток.
Последующая трансплантация этого материала позволяет с вы-
сокой эффективностью получать трансгенных животных [4, 5].
4. Инфицирование плюрипотентных стволовых клеток ре-
тровирусными векторами дает возможность получать легко
идентифицируемые маркеры для исследования рядов клеточных
поколений, формирующихся, например, в процессе дифферен-
цировки тканей [6, 7] или в ходе эмбрионального развития [8].
5. Поскольку ретровирусная инфекция обычно приводит
к интеграции единственной копии вирусного генома в ДНК
клетки-хозяина, ретровирусные векторы, несущие чужеродные
гены, могут выполнять роль уникальных генетических маркеров
индивидуальных хромосом в экспериментах по гибридизации
соматических клеток, хромосомному переносу генов, а также в
исследованиях рекомбинационных процессов [9].
6. Ретровирусные векторы можно использовать в качестве
инсерцпонных (включающихся в хромосому) мутагенов как для
культивируемых клеток, так и для трансгенных животных. Вве-
дение в состав вектора маркерного гена, который бы обеспечи-
вал возможность селекции в Е. coli, может значительно облег-
чить дальнейшие манипуляции с последовательностями мутант-
ного локуса [10].
18—11186
274 Э. Браун, М. Скотт
7. Поскольку геномная РНК ретровирусов претерпевает
сплайсинг, интронные последовательности, введенные в состав
ретровирусного вектора, могут точно выщепляться из вирусного
генома. Следовательно, путем клонирования провирусной ДНК
после инфицирования с помощью ретровирусных векторов мож-
но получать полноразмерные кДНК-копии генов среднего разме-
ра [И, 12].
Цель настоящей главы — очертить рамки практического
использования ретровирусов в качестве экспрессирующих векто-
ров общего назначения и описать методы размножения виру-
сов, тестирования и инфицирования, которые являются общими
для большинства процедур, описанных выше. Всех деталей
специализированных экспериментов мы касаться не будем.
Большинство ретровирусных векторов, описанных на сегодняш-
ний день, ведет свое происхожедние либо от вирусов птиц, либо
от вирусов лейкоза мышей (ВЛМ, MLV). В данной главе мы
сконцентрируем внимание на векторных системах типа MLV
и экспериментальных процедурах, связанных с использованием
этих векторов.
Следующий раздел касается некоторых фундаментальных
аспектов биологии ретровирусов, на которых базируется их при-
менение в качестве векторных молекул. Разд. 9.3 дает представ-
ление о различных конструкциях ретровирусных векторов.
В разд. 9.4—9.6 описаны методы размножения вирусов, инфици-
рования клеток и тестирования вирусных препаратов. Заключи-
тельный, разд. 9.7 касается некоторых практических вопросов,
которые следует принимать во внимание при работе с данными
векторами.
9.2. Биология ретровирусов
9.2.1. Жизненный цикл ретровирусов
Ретровирусы представляют семейство РНК-содержащих
вирусов, репликативный цикл которых отличается тем, что вирус-
ный РНК-геном транскрибируется в ДНК-форму; эту функцию
обеспечивает обратная транскриптаза—фермент, кодируемый
вирусным геномом. Жизненный цикл типичного ретровируса,
такого, например, как вирус лейкоза мышей (MLV), показан
в упрощенном виде на рис. 9.1. Вирусные частицы, каждая из
которых содержит две копии одноцепочечного РНК-генома,
образованы центральным нуклеопротеиновым кором, окружен-
ным мембраной, которая несет на своей наружной поверхности
специфическую оболочку из гликопротеинов. Инфицирование
клеток-мишеней инициируется взаимодействием этих гликопро-
теинов со специфическими рецепторами на клеточной поверхно-
сти. По большей части эти рецепторы пока недостаточно хорошо
охарактеризованы. После проникновения в клетку вирусный
геном переводится в форму линейной двуцепочечной ДНК; этот-
процесс катализируется ферментом обратной транскриптазой,
молекулы которой образуются в инфицированных частицах. Это
сложный многоступенчатый процесс, включающий в себя сайт-
специфическое связывание праймеров для обратной транскрип-
ции и серийное снятие с матриц новообразованных цепей ДНК
[13]. Далее линейная ДНК проникает в ядро клетки-хозяина
и принимает кольцевую форму, необходимую, по всей видимости,
для интеграции в клеточный геном. Одна инфекционная вирус-
ная частица дает.начало лишь одной интегрированной копии
вирусной ДНК, и эта копия, как правило, оказывается довольно
стабильной. Если не считать вероятного предпочтения транс-
крипционно активным участкам, интеграция ретровируса не
обнаруживает выраженной специфичности по отношению к ка-
ким-либо определенным участкам клеточной ДНК и в большин-
стве случаев может рассматриваться как событие случайное.
Интегрированная линейная ДНК-копия ретровирусного гено-
ма называется провирусом и имеет характерную структурную'
18’
276
Э. Браун, М. Скотт
особенность — наличие на обоих концах длинных концевых пов-
торов (LTR— long termine repeats, см. разд. 2.2). Именно в
такой структуре ретровирусной ДНК заключена возможность
манипулирования с ней in vitro для создания ретровирусных
векторных систем. Провирус дикого типа имеет три структурных
гена: gag, pol и env, кодирующих соответственно белки кора,
обратную транскриптазу и белки оболочки. 5'LTR несет промо-
тор, с которого осуществляется траскрипция генов интегрирован-
ного провируса, a 3'LTR — сигнал полиаденилирования тран-
скриптов. В случае MLV и многих других ретровирусов
синтезируются два разных РНК-транскрипта приблизительно в
равных количествах: полноразмерный «геномный» транскрипт
и «усеченная» субгеномная мРНК, которая претерпевает сплай-
синг и служит матрицей для трансляции продукта гена env.
Полноразмерные транскрипты обеспечивают трансляцию про-
дуктов генов gag и pol; кроме того, определенная их часть вы-
полняет роль РНК-геномов и упаковывается в вирусные части-
цы. Новообразованные вирусы, включающие в свой состав
молекулы обратной транскриптазы, высвобождаются из клетки
в результате выпячивания и отпочковывапия плазматической
мембраны клеточной поверхности (более детальное описание
.ретровирусной репликации можно найти в работе [13]).
9.2.2. Структура ретровирусного генома и провируса
Все ретровирусы имеют сходную генетическую организацию,
.которую мы рассмотрим на примере MLV. Структуры провируса
MLV, вирусной геномной РНК и сплайсированной субгеномной
.РНК показаны на рис. 9.2. Ниже приводятся основные особен-
ности строения провируса [14]:
1. Наличие на каждом конце провирусной ДНК LTR, кото-
рые содержат последовательности, обеспечивающие инициацию
и терминацию транскрипции, обратную транскрипцию вирусной
РНК и интеграцию в геном клетки-хозяина. Собственно LTR
построены из трех частей: это б'-концевые последовательности
вирусной РНК (U5), З'-концевые последовательности этой РНК
(U3) и короткие повторяющиеся последовательности (R), лока-
лизующиеся у обоих концов вирусной РНК.
2. Присутствие коротких последовательностей (обозначен-
ных РВ(—) и РВ( + ); рис. 9.2), необходимых для инициации
синтеза «плюс»- и «минус»-, частей вирусной ДНК в процессе
обратной транскрипции.
3. «Упаковочная» последовательность Т, обеспечивающая
эффективную упаковку вирусной РНК в вирусные частицы.
4. Транс-действующие вирусные структурные гены gag, pol
.и env (разд. 9.2.1).
Ретровирусные векторы
277
gag pol (env)
R.IJ5 V; — .^ф.АААА
Геномная РНК
5- + 3'
епу
К,05, t W/ГГ^ТаЗ U-iRhAAAA Субгеномная РНК
П ’Г
। gag pol [ env ____
£uT]f[|u£r^-E^^--------------Провирусная ДНК
PB(-J ' РВ(+)
I LTP—\ Sn Sa I LTR--1
Рис. 9.2. Структурная организация проретровируса и вирусных транскриптов
(масштаб не соблюден). Провирусный геном ограничен двумя LTR, которые
фланкируют центральную область, включающую три структурных гена gag,
pol, env. Участки связывания праймера для обратной транскрипции вирусной
РНК—РВ( —) и РВ( + ) соседствуют с LTR. Каждый LTR включает 3'-(U3)-
и 5'-(U5)-концевые участки теномной РНК, соединенные короткой последова-
тельностью (R), которая присутствует у обоих концов геномной РНК. После-
довательность U3 содержит промоторные и энхансерные элементы. Сайт ини-
циации транскрипции локализуется в 5'LTR; терминация РНК-транскриптов
происходит в сайте полиаденилирования внутри 3'LTR. Полноразмерный ге-
номный транскрипт выполняет двоякую функцию: вирусного РНК-генома и
трансляционной матрицы для синтеза продуктов генов gag и pol. Продукт
гена env транслируется на матрице субгеномной РНК, формирующейся в ре-
зультате сплайсинга первичного транскрипта; происходит исключение участка
первичного транскрипта, расположенного между донорным (SD) и акцептор-
ным (Sa) сайтами сплайсинга. Примечательно, что только полноразмерная
геномная РНК содержит сигнальную последовательность, обеспечивающую
упаковку в вирусные частицы. Подробная карта и полная нуклеотидная после-
довательность вируса Mo-MLV приведены в работе [42].
5. Вирусные донорные и акцепторные последовательности
сплайсинга (Sd и Sa), необходимые для образования субгеном-
ной e/w-PHK.
9.2.3. Рецепторы, тропизм и иммунность
Мы рассмотрим лишь некоторые аспекты жизненного цикла
ретровирусов, представляющие интерес в данном контексте —
а именно те, которые имеют отношение к специфичности круга
хозяев ретровирусных векторов. Круг хозяев ретровируса дико-
го тг.'ча можно ограничить как на внеклеточной, так и на внут-
риклеточной стадии его жизненного цикла [15]. При этом осо-
бого внимания заслуживает внеклеточная стадия, включающая
взаимодействие белков вирусной оболочки с поверхностными
клеточными рецепторами.
Оболочечные белки вирусов семейства MLV можно подраз-
делить на несколько классов, взяв за основу природу рецепторов
278
Э. Браун, М. Скотт
клеточной поверхности, с которыми они взаимодействуют. Круг
хозяев, или «тропизм», вируса ограничивается теми клетками,
которые экспрессируют рецепторы, соответствующие данному
классу белков оболочки. Так, например, «экотропный» мыши-
ный ретровирус MLV-Молони инфицирует только мышиные и
крысиные клетки, а «амфотропный» MLV имеет гораздо более
широкий круг хозяев, поскольку белки его оболочки способны
взаимодействовать с рецепторами, присутствующими на клетках
многих видов — от птиц до человека. Большинство векторов,
полученных на основе MLV, представляют собой производные
MLV-Молони. Однако их круг хозяев может легко расшириться,
выйдя за свойственные этому вирусу естественные пределы в
результате псевдотипирования [15], т. е. в результате упаковки
векторного генома в вирусные частицы, оболочка которых обра-
зована белками амфотропного вируса.
Помимо описанного видового тропизма круг хозяев ретрови-
руса в пределах данного вида может ограничиваться также
тканевой специфичностью экспрессии соответствующих клеточ-
ных рецепторов. В целом большинство фибробластов, лимфоид-
ные клетки и многие другие клетки чувствительны к инфициро-
ванию MLV, хотя систематического исследования клеток разных
типов на чувствительность к MLV не проводилось.
Отметим, что, даже если клетка-мишень экспрессирует под-
ходящие для данного ретровируса поверхностные рецепторы,
после заражения эти рецепторы блокируются благодаря клеточ-
ной экспрессии белков вирусной оболочки [15]. Следовательно,
клетка, продуктивно инфицированная репликационно-компе-
тентным ретровирусом, часто оказывается иммунной к суперин-
фицированию вирусом с тем же тропизмом.
9.3. Выбор вектора
9.3.1. Принципы конструирования векторов
Ретровирусы можно рассматривать как природные векторы
для переноса и экспрессии чужеродных генов. Ретровирусы
с высокой степенью онкогенности быстро и весьма эффективно
вызывают опухоли у млекопитающих и, как правило, способны
трансформировать соответствующие клетки в культуре. За од-
ним лишь исключением (разд. 9.3.3), эти вирусы дефектны по
репликации, поскольку онкогенные последовательности клеточ-
ного происхождения заместили у них те вирусные последова-
тельности, которые кодируют транс-действующие функции [16].
Вирусные продукты, необходимые для репликации и интеграл
ции таких дефектных вирусов, обеспечиваются по транс-схеме не-
Ретровирусные векторы
279
трансформирующими репликационно-компетентными вируса-
ми-помощниками (так называемыми хелперами). Логично
в конструировании ретровирусных векторов идти по пу-
ти, намеченном природой, и ориентироваться на дефектные по
репликации векторы, требующие тра«с-комплементации вирус-
ных функций. Эти функции могут обеспечиваться либо хелпер-
ным вирусом, либо хелперными клетками (называемыми также
«упаковывающими» клетками), которые содержат мутантные
провирусы, не способные к упаковке в зрелые вирионы [17].
К цис-действующим вирусным последовательностям, необ-
ходимым вектору для эффективной репликации, относятся оба
LTR, область инициации обратной транскрипции (прилежащая
к LTR) и сигнальная последовательность для упаковки, распо-
ложенная вблизи от 5'LTR [14] (рис. 9.2). Все остальные пос-
ледовательности могут быть заменены, поскольку их утраченные
функции легко комплементируются по транс-схеме. Структура
провируса как бы заранее подготовлена к конструированию на
его основе вектора, так как цис-действующие последовательно-
сти, необходимые для репликации, кластерированы у концов
провирусного генома и отделены от транс-действующих последо-
вательностей, расположенных в центре. Такая организация поз-
воляет легко вводить в геном чужеродные последовательности,
замещая ими вирусные структурные гены.
В разд. 9.3.2 описаны принципиально различные типы векто-
ров, дефектных по репликации. Альтернативная стратегия, пред-
полагающая клонирование в репликационно-компетентных век-
торах, кратко рассматривается в разд. 9.3.3.
9.3.2. Векторы, дефектные по репликации
9.3.2.1. Векторы для одиночных генов
В случае простейшего ретровирусного вектора транс-дейст-
вующие вирусные структурные гены удаляют и на их место
встраивают один или несколько рестрикционных сайтов для
клонирования. Известно несколько векторов этого типа (см.,
например, [18, 19]). В качестве примера вектора, рассчитанного
на клонирование одиночных генов, можно назвать вектор на
основе вируса лейкоза мышей Молони (Mo-MLV), обозначаемый
рМХ1112 (рис. 9.3, а). В этой конструкции полилинкер содержа-
щий множественные рестрикционные сайты для клонирования,
введен на место удаленных вирусных генов gag, pol и епи.
Клонированный в данном векторе чужеродный ген эффективно
экспрессируется благодаря промотору в составе вирусного
5'-LTR, а наличие цис-действующих вирусных последовательно-
стей позволяет этой системе формировать инфекционные части-
280
Э. Браун, М. Скотт
Xh.So, Ba, Sa,H,C|
ig ApR
d Г~ Г]--Ц | |-------------------- pMX1112
Xh Bg
|Sc, Ba,Sa,H C|
ё
neo
ApR
pMX1122
pMX 1 122 neo
|Xh,Sc,E,Ba,sa, H,C[
г
pMX 262
i-------1
1 т.п.н.
Рис. 9.3. Ретровирусные векторы семейства MLV: (a), pMX 1112; (б),
рМХ 1122; (б), рМХ 1122 пео; (г), рМХ 262. Все векторы изображены-линеари-
зованными, в едином масштабе. Указаны уникальные рестрикционные сайты,
используемые для получения рекомбинантных конструкций, сигнал упаков-
ки (Ч*1) и сайты сплайсинга (SD и Sa) в векторе рМХ 1122. При конструиро-
вании вектора рМХ 1122 пео Bg/I-Sa/I-фрагмент (длиной 1,1 т. п.н.), содержа-
щий ген пео, встроили в BamHI-Sa/I-участок рМХ 1122. Отметим, что иници-
ирующий кодон (ATG) гена gag отсутствует в рМХ 1122 и РМХ 1112, но включен
в состав рМХ 262 вместе с открытой рамкой считывания gag (939 п.н.). Каж-
дая векторная плазмида включает 2,6 т. п. н.-фрагмент (волнистая линия), со-
держащий область начала репликации pBR322 и ген устойчивости к ампицил-
лину (ApR). Эти плазмидные последовательности не присутствуют в вирусных
геномах — производных векторной ДНК- Рестрикционные сайты: Xh; Xhol;
Sc, SacII; E, EcoRI; Ba, BomHI; Sa, Sall; H, /findlll; C, Clal; Bg, Bg/II.
цы в присутствии подходящих хелперных компонентов. Подоб-
ные векторы по сравнению с векторами других типов имеют
важные преимущества — простоту, и, судя по нашему опыту,
позволяют получать вирусные препараты со стабильно высоким
титром и устойчивой экспрессией клонированного гена. Более
сложные векторные системы могут оказаться более универсаль-
ными, однако часто их использование наталкивается на труд-
ности, связанные с нестабильностью или низким уровнем
экспрессии.
Ретровирусные векторы
281
Основной недостаток векторов для одиночных генов — зто
отсутствие подходящей селективной системы, которая бы поз-
воляла отбирать рекомбинантные вирусы и осуществлять их
избирательную экспрессию. Иногда этот недостаток удается
компенсировать высоким титром препаратов (поскольку прак-
тически все клетки в чашке могут оказаться инфицированными),
но в целом применение векторов такого типа ограничено. Они
используются главным образом для клонирования генов, имею-
щих ярко выраженное фенотипическое проявление.
9.3.2.2. Векторы для спаренных генов
Гены, для которых нельзя обеспечить селекцию и которые
не сообщают культивируемым клеткам каких-либо примечатель-
ных фенотипических черт, заставляют серьезно задуматься, как
идентифицировать инфицированные клетки, несущие рекомби-
нантный провирус. Решением этой проблемы может быть второй
ген, вводимый в ту же рекомбинантную конструкцию в качестве
селективного маркера. Для этой цели был опробован целый ряд
генов, обеспечивающих возможность селекции, — в том числе
гены, кодирующие неомицин-фосфотрансферазу (пео), тимидин-
киназу (tk), гуанозил-фосфорибозилтрансферазу (gpt), гигро-
мицин-фосфотрансферазу В (hgr) и дигидрофолят-редуктазу
(dhfr) [14, 20, 21].
Векторы, несущие ген dhfr, имеют то преимущество, что
провирусные последовательности можно подвергнуть ампли-
фикации путем метотрексатной селекции; это позволяет повы-
сить уровень экспрессии и титр вирусных препаратов [21]. Тем
не менее наибольшее распространение в качестве доминантных
маркеров получили гены, обеспечивающие селективную лекар-
ственную устойчивость — такие, как пео или hgr. Это объясня-
ется тем, что они хорошо сочетаются со многими клеточными
типами и не требуют специальной среды для селекции. Во всех
известных конструкциях векторов для спаренных генов ген, рас-
положенный ближе к 5'-LTR, экспрессируется в составе геном-
ного вирусного РНК-транскрипта. Для обеспечения экспрессии
более удаленного гена применяют два разных методических
приема: это либо экспрессия в составе отдельной субгеномной
РНК, образующейся в результате сплайсинга вирусной РНК,
либо использование внутреннего промотора, введенного в состав
вектора.
1. Сплайсинг-векторы. Наиболее «естественным» подходом
в конструировании векторов для спаренных генов может счи-
таться имитация механизма, свойственного родительским ретро-
вирусам, а именно использование вирусных донорных и акцеп-
282
Э. Браун, М. Скотт
торных сайтов сплайсинга для образования самостоятельного
субгеномного РНК-транскрипта, аналогичного вирусной env-
РНК (рис. 9.2). Эта РНК становится матрицей для трансляции
дистального гена. В то же время проксимальный ген вводится
в соответствующей рамке считывания на место генов gag и pol
и транслируется в составе несплайсированной РНК- Схема тако-
го вектора (рМХ1122) показана на рис. 9.3, б. Он несет Xhol-
и Bg/II-сайты для клонирования в области «gag», а в области
«env» — множественные сайты для клонирования SacII, BamHI,
Sall, Hindlll и Clal, которые расположены непосредственно за
акцепторным сигналом сплайсинга MLV. В качестве селектив-
ного маркера для этого вектора был выбран ген неомицин-
фосфотрансферазы (пео), обеспечивающий устойчивость к ан-
тибиотику G418. Маркерный ген ввели в область «env» и назва-
ли рекомбинантную конструкцию рМХ1122 пео (рис. 9.3, в).
Теперь оказалось возможным ввести второй ген в область «gag»
и осуществить его коэкспрессию с геном пео. Пример успешного
применения этого вектора описали Браун и др. [12].
Сепко и др. [22] разработали набор сплайсинг-векторов,
аналогичных рМХ1122, в которых ген пео вводится в область
«gag» или «env». Один из них — pZIP-NeoSV(X) 1, несущий ген
пео в области «env» и BamHI-сайт для клонирования в области
«gag», широко используется во многих лабораториях.
Одна из общих неприятностей, связанная с использованием
сплайсинг-векторов подобного типа, обусловлена тем, что эф-
фективность вирусного сплайсинга может резко изменяться, что'
приводит к снижению уровня экспрессии одного из генов клони-
рованной пары. Эта проблема обсуждается ниже в разд. 9.7.
2. Векторы для спаренных генов с внутренним промотором.
Описанные выше сплайсинг-векторы предполагают экспрессию
обоих клонированных генов под контролем промотора, локали-
зованного в пределах 5Z-LTR. Альтернативный подход преду-
сматривает использование дополнительного промотора, введен-
ного в вектор таким образом, чтобы обеспечить экспрессию
дистального (относительно 5'-LTR) гена, тогда как проксималь-
ный ген по-прежнему экспрессируется при участии вирусного'
LTR; иными словами, каждый ген имеет свой собственный неза-
висимый промотор. Как и в случае сплайсинг-векторов один из
клонированных генов здесь также выполняет функцию селектив-
ного маркера (например, пео).
Дополнительные внутренние промоторы вводили в ретрови-
русные векторы в обеих ориентациях относительно направления
транскрипции с вирусного LTR. В тех случаях, когда внутренний
промотор имел встречную ориентацию, продуцирование вируса
несколько ухудшалось, что, по-видимому, было обусловлено
неблагоприятным транскрипционным взаимодействием противо-
Ретровирусные векторы
283
положно направленных промоторов и, возможно, эффектом «ан-
тисмысловой» РНК. Однако во многих случаях неблагоприятные
последствия были сглажены [14]. Небольшое преимущество
именно такой ориентации внутреннего промотора заключается
в том, что ДНК-вставка способна сохранить собственный сиг-
нал полиаденилирования без риска ухудшить образование
вируса. В то же время, если клонированный фрагмент ДНК
транскрибируется в той же ориентации, что и вирусная РНК,
наличие в нем сигнала полиаденилирования может сильно сни-
зить образование вируса из-за преждевременного полиаденили-
рования вирусного геномного транскрипта [14]. Конечно, это
препятствие легко преодолимо просто путем удаления сигнала
полиаденилирования из клонируемой ДНК.
Векторы с внутренним промотором лишены тех недостатков
сплайсинг-векторов, которые связаны с вероятностью неадекват-
ного сплайсинга (разд. 9.7). Однако ретровирусы дикого типа
не содержат внутренних промоторов, поэтому данные векторы
могут давать неудовлетворительную экспрессию клонированных
генов вследствие неблагоприятного взаимодействия промоторов
(даже однонаправленных) [23, 24]. Как бы то ни было на прак-
тике векторы с внутренним промотором хорошо себя зарекомен-
довали во многих случаях и приобретают все большую попу-
лярность.
Конструированием векторов этого типа занимаются многие
лаборатории [7, 23, 25, 26]. Один из примеров — вектор pMV-7,
описанный Киршмейером и др. [26], схематически изображен
на рис. 9.4. pMV-7 относится к семейству векторов на основе
вируса саркомы Молони (Mo-MSV)—близкородственного виру-
са лейкоза Молони (Mo-MLV); РНК Mo-MSV эффективно упа-
ковывается с помощью MLV-хелперов или MLV-упаковывающих
линий клеток. Внутренний промотор для вектора pMV-7 взят из
гена tk вируса простого герпеса и находится в той же транскрип-
ционной ориентации, что и вирусные LTR [26].
9.3.2.3. Векторы для экспрессии составных генов
Многие природные онкогенные ретровирусы экспрессируют гиб-
ридные белки. Происходит это в результате слияния N-концевой
части вирусного гена gag или env с онкогенными последователь-
ностями, захваченными вирусом из ДНК клетки-хозяина. Такая
структура способна обеспечить очень эффективную экспрессию
гибридных белков, поскольку сигналы, влияющие на их тран-
сляцию, относятся к категории весьма активных генетических
элементов, которые в норме обеспечивают масштабное произ-
водство продуктов вирусных генов в инфицированной клетке.
Кроме того, на эффективность синтеза таких белков может
284
Э. Браун, М. Скотт
EcoRI Hindi II
ApR
pMV-7
1т,п.н
Рис. 9.4. Линейная карта вектора pMV-7, содержащего внутренний промо-
тор [26]. pMV-7 — производное Mo-MSV, содержит уникальные рестрикцион-
ные сайты £coRI и //indlll, которые используются для клонирования генов,
функцию селективного маркера выполняет ген пео под контролем /.4-промото-
ра (Ptk) вируса герпеса простого. Волнистой линией обозначены ген устойчи-
вости к ампициллину (zlpR) и другие плазмидные последовательности, которых
нет в вирусных геномах — производных векторной ДНК.
оказывать влияние специфическая субклеточная локализация
сигналов в gag- или епа-последовательностях.
В тех случаях, когда определяющим фактором исследования
является оптимальная экспрессия продукта интересующего ге-
на, а возможные ограничения в субклеточной локализации
приемлемы или даже желаемы, целесообразно использовать
ретровирусный вектор, приспособленный для состыковки генов.
Например, вектор рМХ262 (рис. 9.3, г). Он несет множественные
сайты для клонирования за X’/ioI-сайтом гена gag Mo-MLV и
позволяет получать рекомбинантные конструкции гена gag, эк-
спрессирующие гибридные белки.
Эти белки могут быть адресованы внутренней поверхности
плазматической мембраны благодаря миристинилированию
аминоконцевой части их ^^-последовательности. Основной не-
достаток описанного подхода, свойственный любой системе, в
которой осуществляется экспрессия составного белка, — вероят-
ная лабильность и(или) низкая биологическая активность эк-
спрессируемого продукта.
9.3.2.4. Челночные векторы
Как отмечается в разд. 9.6.3, в ряде случаев желательно
иметь возможность быстро выделить провирусные последова-
тельности из клеток млекопитающих путем молекулярного кло-
нирования,— например, чтобы проанализировать структуру
трансдуцированных последовательностей, если ретровирусный
вектор использовался для удаления интронов из геномной ДНК-
вставки [19]. Для решения этой задачи могут оказаться весьма
полезными «челночные» векторные системы, облегчающие пере-
нос рекомбинантов между животными и бактериальными клет-
ками. Такие векторы содержат эукариотическую область нача-
Ретровирусные векторы
285
ла репликации — либо SV-40 [22], либо вирус полиомы ,[28],—
которая обеспечивает их высококопийную внехромосомную реп-
ликацию в подходящих клетках. В составе провируса присутст-
вует также область начала репликации плазмиды pBR322,
благодаря чему кольцевую реплицирующуюся ДНК легко
можно получить в препаративных количествах путем трансфор-
мации Е. coli. Селекция трансформантов не составляет труда,
поскольку находящийся в векторе ген пео обеспечивает устой-
чивость животных клеток к G418 и бактерий к канамицину или
неомицину (разд. 9.6.3). Примером распространенного ретрови-
русного челночного вектора может служить сплайсинг-вектор
для спаренных генов pZIP Neo SV(X) 1 [22] (разд. 9.3.2.2).
9.3.2.5. Самоинактивирующиеся векторы
Самоинактивирующиеся векторы, или векторы-«самоубийцы»,.
разработаны для того, чтобы исключить влияние вирусных про-
моторов и энхансеров на экспрессию гена, поставленного под
контроль внутреннего промотора [29]. Эти векторы сконструи-
рованы таким образом, что в процессе обратной транскрипции
и интеграции те участки вирусного генома, которые содержат
промоторные и энхансерные элементы, подвергаются делецни.
Провирусные LTR в инфицированных клетках становятся
транскрипционно неактивными. Это способствует нормальной
экспрессии генов, транскрибируемых с внутреннего промотора
и исключает возможность нежелательных последствий включе-
ния (инсерции) провируса, в частности транскрипционной акти-
вации соседних генов [13]. Несмотря на то что титр вирусных
препаратов, полученных на основе подобных векторов, был
достаточно низок (104 к. о. е./мл [29]), самоинактивирующиеся
векторы можно рассматривать как наиболее перспективные
кандидаты для применения в генотерапии человека [3].
9.3.3. Репликационно-компетентные векторы
Пока еще не создано репликационно-компетентного мышин-
ного ретровирусного вектора общего назначения. Объясняется
это как отсутствием подходящих сайтов для введения чужерод-
ных генов в MLV (которые бы не нарушали целостность функ-
ционально важных вирусных последовательностей), так и огра-
ничениями, накладываемыми на размер вирусного генома [4].
Тем не менее одной из удачных попыток можно считать введе-
ние коротких последовательностей в состав вирусных LTR.
Таким путем была получена репликационно-компетентная ре-
комбинантная конструкция на основе MLV, несущая ген супрес-
сорной тРНК Е. соли (SupF) [30], которая оказалась неплохим;
-286
Э. Браун, М, Скотт
гегенетическим маркером, а также удобным инструментом,
облегчающим клонирование прилежащих последовательностей
после инсерции провируса. Однако остается открытым вопрос о
том, могут ли быть введены в состав вирусных LTR и эффек-
тивно экспрессироваться последовательности, кодирующие
белки.
Более универсальный репликационно-компетентный вектор
создан для работы с клетками птиц. Он является производным
вируса саркомы Рауса (RSV), который единственный из высо-
коонкогенных ретровирусов компетентен по репликации. Его
организация имеет ряд необычных особенностей. Так, онкоген
src экспрессируется в составе собственного субгеномного РНК-
транскрипта, который образуется в результате дополнительного
сплайсинга вирусной РНК, а вирусные гены экспрессируются
обычным образом [16]. Векторы, сконструированные на основе
RSV, вместо последовательностей, кодирующих src, несут сайт
для введения фрагментов гетерологичной ДНК [14, 31]. Такая
система обеспечивает чрезвычайно высокую эффективность вве-
дения и экспрессии чужеродных последовательностей и позво-
ляет получать вирусные препараты с очень высоким титром.
К сожалению, узкий круг хозяев этих векторов, ограниченный
лишь клетками птиц, служит помехой для их широкого исполь-
зования. Кроме того, в плане широкого практического примене-
ния (например, для целей генотерапии [3]) использование реп-
ликационно-компетентных векторов, по-видимому, нежелатель-
но, так как эти системы способны промотировать постоянное
наращивание вирусной продукции.
9.3.4. Приготовление вставочных фрагментов ДНК
Самое простое правило, которым следует руководствовать-
ся при подготовке последовательностей для клонирования в рет-
ровирусных векторах, — уменьшение их длины до того миниму-
ма, который только необходим для экспрессии белка. Это
позволит избежать непредсказуемых трудностей сопряженных
с экспрессией или лабильностью системы. В любом случае
вставка должна быть достаточно мала, чтобы суммарный раз-
мер геномной РНК, предназначенной для упаковки, не превы-
шал 10—11 т. п. н. [14]. Идеальным можно считать фрагмент
ДНК, включающий полную открытую рамку считывания с 5'- к
З'-фланкирующими последовательностями, которые были бы
как можно более короткими. Особое внимание следует уделить
элиминации последовательностей, влияющих на транскрипцию
или процессинг РНК, — таких, как промоторы или сигналы
полиаденилирования. Добавочные кодоны AUG «выше» пред-
полагаемой точки начала трансляции также нежелательны, так
Ретровирусные векторы
287
как они могут отрицательно влиять на эффективность трансля-
ции.
Учитывая возможные осложнния, связанные со сплайсингом,
кДНК-вставки (если они доступны) предпочтительны по срав-
нению с последовательностями, содержащими интроны. Хотя
ретровирусные векторы обладают способностью точно удалять
интроны из геномных последовательностей и таким образом
генерировать кДНК-копии клонированных генов, этот процесс,
не всегда достаточно эффективен [14].
9.4. Получение препаратов рекомбинантных вирусов
9.4.1. Получение препаратов,
не содержащих хелперных компонентов
Чтобы получить вирусный препарат на основе дефектного по-
репликации рекомбинантного ретровирусного вектора, плазмид-
ную ДНК, содержащую провирусную форму рекомбинанта,
необходимо ввести в клетки, способные упаковать ретровирусную-
РНК в вирусные частицы. Для этой цели подходят, например,
линии фибробластов, продуктивно инфицированные соответст-
вующим хелперным вирусом — получается вирусный препарат,
представляющий смесь рекомбинантного и хелперного геномов
(разд. 9.4.2). Альтернативный вариант — использование ретро-
вирусных упаковывающих клеточных линий для получения ви-
русных препаратов, свободных от хелперных компонентов.
Клетки «упаковывающих» линий содержат провирусную ДНК
хелперного вируса с делетированным сигналом упаковки. По-
этому собственные вирусные РНК-транскрипты не могут упако-
вываться в вирусные частицы. Однако этот дефект является
только г{«с-действующим, так что хелперный провирус произво-
дит все необходимые вирусные белки, способные обеспечить,
упаковку соответствующих РНК-геномов в вирионы по транс-
схеме. Таким образом, когда рекомбинантный провирус вводит-
ся в упаковывающие клетки, его транскрипты формируют мажор-
ный класс способных к упаковке молекул и клетки продуцируют
вирусные частицы, содержащие почти исключительно рекомби-
нантные РНК-геномы. Такие вирусные препараты считаются
свободными от хелперных элементов, хотя на практике они
иногда содержат в небольшом количестве репликационно-ком-
петентные хелперы [32].
Несмотря на то что свободные от хелперов вирусные препа-
раты дефектны по репликации, они обладают высокой инфекци-
онностью. Благодаря вирусным белкам, заключенным внутри
вириона, эти вирусы эффективно заражают пермессивные
.288
Э. Браун, М. Скотт
клетки-мишени, проходят в них цикл обратной транскрипции и
б форме провирусной ДНК интегрируются в реципиентный ге-
ном [17]. Начиная с этого момента, транскрипция вирусных
генов происходит под контролем вирусных и внутренних промо-
торов, если последние введены в состав вектора. Однако в от-
сутствие продуктов соответствующих генов хелперного вируса
зрелые вирусные частицы больше не образуются.
Описано несколько различных «упаковывающих» клеточных
линий для получения «чистых» препаратов ретровирусных век-
торов семейства MLV [17, 19, 21, 32]. Из них наиболее широкое
применение нашли, пожалуй, линия ф2 [17], упаковывающая
вирусную РНК в экотропные частицы (т. е. такие, которые
способны инфицировать только клетки мыши и крысы), и линия
РА 317 [32], позволяющая получать амфотропные препараты
с широким кругом возможных хозяев, практически не содержа-
щие хелперных вирусов. Известны также упаковывающие клет-
ки для векторов на основе ретровирусов птиц [33].
9.4.1.1. Трансфекция векторной ДНК в упаковывающие клетки
Поскольку все описанные на сегодняшний день упаковываю-
щие MLV-клетки ведут свое начало либо от линий NIH3T3 либо
от линии BALB/сЗТЗ, рекомбинантную ретровирусную ДНК
в них можно легко ввести путем трансфекции, используя стан-
дартный метод кальций-фосфатной преципитации [34]. Непро-
должительная, так называемая временная (transient), тран-
сфекция может оказаться достаточной, чтобы получить вирусный
препарат с низким титром (обычно 10—103/мл). Однако для
достижения более высоких концентраций вируса необходимы
стабильно трансформированные клеточные линии, экспресси-
рующие доминантный селективный маркер. Такие линии позво-
ляют получать препараты вируса с титром 105—106 инфекцион-
ных ед./мл. Титр вирусных препаратов, полученных путем
временной трансфекции, можно повысить, если использовать
гибридный ретровирусный вектор, содержащий полную область
ранних генов вируса полиомы: такая конструкция претерпевает
в упаковывающих клетках мультикопийную внехромосомную
репликацию [35].
Процедура временной трансфекции приведена в табл. 9.1.
Детальное описание кальций-фосфатного метода трансфекции
можно найти в гл. 15 тома II данной серии [34]. Титр вируса,
полученного таким путем, зависит от всех параметров, в норме
оказывающих влияние на эффективность трансфекции (чистота
используемой ДНК, собственная восприимчивость клеток к этой
ДНК и т. п.), а также от свойств конкретной векторной систе-
мы. Некоторые ретровирусные -конструкции воспроизводимо
Ретровирусные векторы
289
Таблица 9.1. Получение вирусных препаратов путем временной
трансфекции ДНК в упаковывающие клетки
1. Высейте упаковывающие клетки при плотности 4-Ю5 на бсм-чашку за
день до эксперимента.
2. Растворите 10—20 мкг векторной ДНК в 0,5 мл стерильного раствора
250 мМ. СаС12. По каплям, постоянно перемешивая, прилейте этот раствор
к 0,5 мл стерильного 2XHBS1’. Оставьте при комнаткой температуре на
15 мин, чтобы сформировался осадок.
3. Нанесите на чашку 0,5 мл СаРО4-ДНК-преципитата, добавляя его по
каплям к среде культивирования.
4. Оставьте на 8—12 ч, затем замените среду.
5. По прошествии 24—72 ч приступайте к сбору вируса (разд. 9.4.3). Опти-
мальное время находят эмпирически, отбирая пробы с интервалом в 12—
24 ч и определяя титр каждого препарата (разд. 9.6.1).
') 2XHBS: 1 л содержит 16 г NaCl, 0,74 г КС1, 0,39 г Na2HPO4-7H2O, 1 г декстрозы,
5 г HEPES. pH доводят до 7,05, добавляя NaOH.
дают более низкий титр, чем другие, и при использовании их
для временной трансфекции могут дать вирусные препараты с
очень и очень низким титром.
Для получения стабильно трансформированных клеточных
линий также используется стандартная процедура кальций-
фосфатной трансфекции и затем селекция на доминантный мар-
кер, который либо введен в состав самого ретровирусного век-
тора, либо находится в составе отдельной вспомогательной
плазмиды, участвующей в котрансфекции. В качестве маркера
обычно используют бактериальный ген пео, обеспечивающий
устойчивость клеток млекопитающих к аналогу неомицина G418.
Процедура трансфекции с последующей селекцией на устойчи-
вость к G418 описана в табл. 9.2.
9.4.1.2. Инфицирование клеток упаковывающих линий
Более сложный технически, но часто дающий более удовлет-
ворительные результаты метод получения «чистых» вирусных
препаратов (т. е. не содержащих хелперных вирусов) преду-
сматривает последовательное использование двух разных упа-
ковывающих линий так, что вирус, продуцируемый первой кле-
точной линией (после трансфекции соответствующей ДНК)
инфицирует затем вторую линию клеток [36] (рис. 9.5). Необ-
ходимое условие при этом: наличие в векторе подходящего
маркера для осуществления селекции инфицированных клеток.
Чтобы «обмануть» свойственный упаковывающим клеткам им-
мунитет к инфицированию вируса той же тропности, вирус «пе-
ресаживают» из амфотропной упаковывающей линии на эко-
тропную или наоборот. Если такое решение вопроса неприемле-
мо, скажем по соображениям возможной биологической опас-
19— 1186
290
Э. Браун, М. Скотт
Таблица 9.2. Получение стабильно трансформированных клеток,
устойчивых к G418
1. Трансфекция осуществляется так же, как описано в табл. 9.1., но следует
взять только 0,5 мкг векторной ДНК (в расчете на одну чашку), смешав
ее предварительно с 10 мкг ДНК-носителя (например, ДНК спермы лосо-
ся). Если вектор не содержит маркерного гена пео, включите в преципитат
0,1 мкг ДНК пео-содержаш,ей плазмиды.
2. Через 24—48 ч после трансфекции, перед тем как клетки достигнут стадии
слияния, перенесите их с каждой бсм-чашки на две Юсм-чашки, содержа-
щие в среде G418 (Geneticin, Gibco). Подходящая концентрация G418
подбирается эмпирически для каждой линии клеток, но для большинства
производных NIH3T3 эта величина составляет приблизительно 400 мкг/мл.
G418 останавливает клеточный рост в течение 24 ч и приводит к гибели
клеток приблизительно через неделю.
3. Когда гибель клеток становится очевидной, среду заменяют: концентра-
цию G418 снижают до 200 мкг/мл и оставляют чашки, пока не станут ви-
димыми колонии, устойчивые к G418.
ности работы с конкретными амфотропными рекомбинантными
вирусами, можно использовать упаковывающую линию ф2:
устойчивость этих клеток к экотропному вирусу можно снизить
путем обработки сублетальными дозами туникамицина [36].
Описанная двухстадийная процедура по сравнению с обыч-
ной трансфекцией вирусной ДНК в упаковывающие клетки
имеет тройное преимущество:
1. Провирусная ДНК, попавшая в клетку в результате
ретровирусной инфекции, как правило, экспрессируется с гораз-
до более высокой эффективностью, чем идентичная ДНК, вве-
денная путем трансфекции [37]. Причины этого явления до
конца не выяснены, однако благодаря более активной продук-
ции вирусной РНК титр препаратов, полученных из инфициро-
ванных упаковывающих клеток, часто значительно превышает
(более чем в 100 раз) титр препаратов, полученных на основе
трансфицированной ДНК.
2. Если в качестве источника вируса используются клетки-
потомки индивидуальных инфицированных колоний, можно
ожидать, что полученный вирусный препарат обнаружит боль-
шее генетическое единообразие, чем тот, который получен из
трансфицированных клеток. Это связано с тем, что трансфици-
рованные клетки часто несут множественные интегрированные
копии введенной ДНК, причем некоторые из них могут подвер-
гаться различного рода перестройкам и мутировать, но все они
вносят свой вклад в состав вирусного препарата. Клетки же
из инфицированных колоний содержат только индивидуальные
копии провирусной ДНК, интегрированные в клеточный геном
определенным образом.
3. Данный метод позволяет изучать структуру провирусной
ДНК (из которой происходят все вирусные геномы) с помощью
Ретровирусные векторы_______ 291
блот-гибридизационного анализа ДНК из отдельных колоний
инфицированных упаковывающих клеток. Это может иметь
большое значение в тех случаях, когда ДНК, клонированная
в составе ретровирусного вектора, изначально содержала
интроны и желательно определить — перед тем, как использо-
вать вирусный препарат в дальнейших экспериментах, — удале-
ны ли эти интроны из вирусного генома (на этапе сплайсинга)
(рис. 5).
9.4.1.3. Трансфекция упаковывающей ДНК
Альтернативой введению ретровирусной векторной ДНК
в упаковывающие клетки для получения вирусных препаратов,
свободных от хелперных компонентов, служит использование
любых других клеток, несущих рекомбинантную провирусную
ДНК, в которые вводится путем трансфекции хелперный геном
MLV с делегированным собственным упаковывающим сигналом.
Эта процедура с высокой эффективностью превращает исходные
клетки в упаковывающие. Метод особенно удобен в тех случаях,
когда требуется получить рекомбинантный вирус из клеток, в
которых уже содержится провирусная ДНК.
9.4.2. Получение препаратов
репликационно-компетентных вирусов
9.4.2.1. Приготовление препаратов хелперного вируса
Препараты хелперных вирусов получают просто путем
трансфекции в соответствующие клетки клонированной ДНК
репликационно-компетектиого вируса в провирусной форме.
Затем производят несколько пассажей вируса, чтобы добиться
полного инфицирования всех клеток в культуре. Подходящим
провирусным клоном для получения хелперного Mo-MLV можно
считать pZAP [38]. В качестве клеток-хозяев для получения
препаратов с высоким титром хорошо зарекомендовали себя
NIH3T3; впрочем, большинство линий быстроделящихся мыши-
ных фибробластов дают хорошие результаты. Чтобы исключить
возможность рекомбинации с эндогенными мышиными ретрови-
русами, можно использовать и крысиные клетки Rat-1 или их
производные. Методика получения препаратов MLV-хелпера
приведена в табл. 9.3.
9.Д.2.2. Приготовление смешанных препаратов хелперного
и рекомбинантного вирусов
Здесь возможны два методических подхода. Один из них
предполагает использование препарата хелперного вируса для
инфицирования клеточной линии, которая уже несет в себе
19!
292
Э. Браун, М. Скотт
дефектный по репликации рекомбинантный провирус (введен-
ный в клетки либо путем трансфекции, либо с помощью обычно-
го заражения), так что в результате происходит «спасение»
рекомбинанта и «разбавление» им препарата хелперного вируса
(разд. 9.5 — методика инфицирования). Другой метод преду-
сматривает ко-трансфекцию хелперного и рекомбинантного про-
вирусов в подходящие клетки-хозяева, что в дальнейшем приво-
дит к инфицированию вирусом всей культуры. Этот подход
обычно обеспечивает более высокий титр рекомбинантного виру-
са в препарате как в абсолютном выражении, так и относитель-
но содержания хелперного вируса. Ниже приведена соответст-
вующая экспериментальная процедура.
1. Проведите трансфекцию клеток NIH3T3, как описано в
табл. 9.2, используя смесь рекомбинантной провирусной ДНК
и клонированной хелперной провирусной ДНК (молярное
соотношение 10: 1).
3.
Ретровирусные векторы
293
2. После трансфекции пассируйте клетки в течение по мень-
шей мере 10 дней, чтобы вирус распространился в культуре.
3. Если рекомбинантный провирус несет селективный маркер,
например пео, рассейте клетки на чашки с селективной средой
и ожидайте появления устойчивых колоний.
4. Объедините колонии и культивируйте их в массе. Собери-
те вирус, как описано в разд. 9.4.3.
9.4.3. Сбор и хранение вируса;
оптимизация условий для повышения титра
Ретровирусные препараты получают из культур упаковываю-
щих клеток или продуктивно инфицированных клеток, просто
собирая среду культивирования, в которой росли клетки. Фло-
тирующие клетки и клеточные обломки удаляют центрифугиро-
ванием при 5000 g в течение 5 мин.
Инфицированные клетки высвобождают вирусные частицы
наиболее активно в экспоненциальной фазе роста. Следователь-
но, максимальное содержание вируса в среде достигается на
Рис. 9.5. Использование ретровирусного вектора для удаления интронов из
клонированного гена и продуцирование вирусов с последовательным исполь-
зованием двух упаковывающих клеточных линий. (Воспроизводится из рабо-
ты [12] по разрешению фирмы-праводержателя Cell Press.) А. Получение ви-
русных препаратов (разд. 5.4.1.2). Ретровирусный вектор рМХ 1122 inf-1.пео
(разд. 9.3.2.2), содержащий мышиный онкоген inf-1 в ^а^-позиции и ген пео
в епо-позиции, ввели в амфотропную упаковывающую линию РА12(21)
СаРО4-трансфекцией; отбирали колонии, устойчивые к G418. Препараты амфо-
тропного вируса (свободные от хелперного вируса), полученные из отобран-
ных С418к-колоний, использовали для инфицирования ф2 и снова отбирали
колонии, устойчивые к G418. Индивидуальные инфицированные колонии вы-
ращивали в виде отдельных линий, анализировали с помощью блот-гибридиза-
ции и использовали в качестве источников вирусных препаратов, свободных
от хелперных вирусов, для дальнейшей работы [12]. Четыре экзона гена int-1
обозначены прямоугольниками (кодирующие области зачернены); ген пео
обозначен крапчатым прямоугольником. Показана предполагаемая структура
провирусных ДНК, интегрировавшихся в геном клетки-хозяина в результате
трансфекции или инфекции (в последнем случае — с утратой интронов int-1).
Указаны также предполагаемые размеры LTR-LTR ХЬаЦХ) -фрагментов.
Б. Блот-гибридизационный анализ провирусной ДНК в трансфицированных и
инфицированных клетках, демонстрирующий элиминацию интронов int-1 из
ретровирусного генома. Суммарную клеточную ДНК расщепляли Xbal, фрак-
ционировали электрофорезом в агарозном геле, переносили на нитроцеллюлоз-
ный фильтр и гибридизовали с меченной 32Р кДНК int-1. Дорожки 1 и 3: ДНК
нетрансфицированных клеток РА 12 и неинфицированных клеток ф2 (выявляет-
ся эндогенный геномный int-1 -фрагмент); дорожка 2: ДНК из клеток РА 12,
трансфицированных рМХ 1122 int-1.пео\ дорожка 4: ДНК из отдельной коло-
нии клеток ф2, инфицированных вирусом, полученным из трансфицированных
клеток РА 12. Стрелками отмечены провирусные фрагменты длиной 6,3 и
4,7 т. п. н., соответствующие изображенным на панели А. (4,7 т. п. н.-фрагмент,
детектируемый в дорожке 2, вероятно, образовался в результате инфицирова-
ния клеток РА 12 вирусом, который они сами продуцируют, поскольку эта ли-
ния не обладает выраженной устойчивостью к суперинфекции [36].)
294
Э. Браун, М. Скотт
Таблица 9.3. Приготовление препаратов хелперного Mo-MLV
1. Приготовьте СаРО4-прецппитат, содержащий 2 мкг/мл ДНК плазмиды
pZAP и 20 мкг/мл ДНК-носителя (ДНК спермы лосося) (см. табл. 9.1).
2. Нанесите 0,5 мл суспензии по каплям к среде культивирования в 5см-чаш-
ке, содержащей 2-Ю5 клеток NIH3T3. Инкубируйте чашку в течение
8-12 ч.
3. Осушите чашку и налейте свежую среду.
4. Когда сформируется клеточный монослой, разделите его и пересейте на
10 новых чашек.
5. После слияния клеток вновь разделите монослой и рассейте на 25 ча-
шек. Далее продолжайте пассирование таким образом в течение 2 пед.
6. Соберите вирус, как описано в разд. 9.4.3.
7. Заморозьте клетки и храните их как источник хелперного вируса.
стадии, непосредственно предшествующей формированию слит-
ного монослоя (так как далее рост клеток останавливается).
Когда монослой сформировался на 80—90%, клетки помещают
в свежую среду и инкубируют еще 12—24 ч, после чего собирают
вирус. Следующий сбор вируса еще через 12—24 ч также позво-
ляет получить препарат с высоким титром. Чем меньше объем
среды, используемой для выделения вируса, тем более высокий
титр имеет полученный препарат. Вирусные препараты можно
затем концентрировать путем центрифугирования при 30 000 g
в течение 12—16 ч при 4 °C, после чего ресуспендировать осадок
в небольшом объеме среды, содержащей 10% сыворотки. Сле-
дует, однако, помнить, что, скажем, 100-кратное концентрирова-
ние препарата всегда приводит к менее чем 100-кратному уве-
личению его «функционального» титра, поскольку определенная
доля вирусных частиц теряет биологическую активность в
результате центрифугирования.
Для достижения максимально высокого титра вирусного
препарата рекомендуется работать с клональными линиями,
ведущими свое начало от отдельных колоний клеток-продуцен-
тов. Это требование приобретает особую важность в тех случа-
ях, когда вирус получают путем трансфекции провирусной
ДНК: индивидуальные клональные линии, выделенные из исход-
ной популяции клеток, часто различаются по уровню вирусной
продукции в 10—100 раз. Если после трансфекции осуществля-
ется селекция с использованием необходимых маркеров, инди-
видуальные колонии отбирают с помощью специальных цилинд-
риков для клонирования, выращивают раздельно в многолуноч-
ком планшете и для каждой определяют титр вируса в культу-
ральной среде, как описано в разд. 9.6.1. Наиболее продуктив-
ные клеточные линии сохраняют и используют в дальнейшей
работе.
Препараты ретровирусов хранят замороженными при —70°C
в среде, содержащей 10% сыворотки. Циклы замораживания —
Ретровирусные векторы
295
оттаивания приводят к снижению титра; поэтому целесообраз-
но важные препараты хранить в виде расфасованных порций.
Определив титр в аликвоте после оттаивания, можно более точ-
но судить о качестве остального препарата.
9.5. Инфицирование клеток-мишеней
9. 5.1. Инфицирование с помощью вирусных препаратов
Краткость и небольшой объем данного раздела могут послу-
жить свидетельством той легкости, с которой ретровирусные
векторы обеспечивают введение клонированных последователь-
ностей в клетки-мишени, при условии, что в руках эксперимен-
татора есть функциональный вирусный препарат. Для фибро-
бластов и клеток многих других типов, экспрессирующих
соответствующие поверхностные рецепторы, эффективность
инфекции приближается к 100%. Ниже приводится рекомендуе-
мая методика.
1. Высейте клетки при плотности 4-Ю5 на бсм-чашку за один
день до инфицирования.
2. Приготовьте концентрированный раствор полибрена
(polybrene Aldrich) в бидистилляте (8 мг/мл), стерилизуйте
фильтрованием и храните при 4 °C.
3. Удалите из чашки культуральную среду и залейте 2 мл
свежей среды, содержащей необходимое количество вируса и
8 мкг/мл полибрена. (Полибрен представляет собой поликати-
он, способствующий связыванию вируса с клеточной поверхно-
стью.)
4. Поместите чашку в инкубатор на 2 ч.
5. Удалите из чашки среду, содержащую полибрен, и налей-
те 5 мл свежей среды. Оставьте клетки при 37°C до рассева по
меньшей мере на 24 ч (или дольше — соответственно продолжи-
тельности одного клеточного цикла).
6. Если вирус несет селективный маркер, клетки рассевают
на подходящую селективную среду.
9. 5.2. Инфицирование путем совместного культивирования
Альтернативный способ осуществления ретровирусной инфек-
ции состоит в совместном культивировании клеток-мишеней с
клетками, активно продуцирующими соответствующий вирус.
Этот метод предпочтителен в тех случаях когда эффективность
инфекции клеток-мишеней низка или когда требуется множест-
венное заражение каждой клетки. Чтобы предотвратить после-
дующее загрязнение культуры клеток-мишеней донорными
296
Э. Браун, М. Скотт
Таблица 9.4. Дот-блот-анализ вирусной РНК
1. Осветлите вирус-содержащую среду путем центрифугирования при
10 000 g в течение 10 мин при 4 °C.
2. Осадите вирусные частицы центрифугированием при 200 000 g в течение
1 ч при 4 °C. Хорошо подсушите осадок.
3. К каждой пробирке добавьте по 10 мкл дрожжевой тРНК (5 мг/мл) в
качестве носителя и интенсивно ресуспендируйте осадок в 0,4 мл буфера
STE1’, содержащего 5 мкг/мл протеиназы К и 0,2% ДСН (SDS) (пред-
варительно выдержанного в течение 15 мин при 37°C).
4. Перенесите каждый • образец в 1,5 мл центрифужную пробирку с защел-
кивающейся крышкой и инкубируйте при 37 °C в течение 30 мин. Экстра-
гируйте 1 раз равным объемом смеси фенол — хлороформ (1:1). Разде-
лите фазы центрифугированием и вновь экстрагируйте водную фазу хло-
роформом. Затем к водной фазе добавьте 0,1 мл объема ЗМ ацетата
натрия и 2 объема этанола. Охладите до —70 °C.
5. Осадите РНК центрифугированием в микроцентрифуге. Удалите жидкость
и подсушите осадки на воздухе. Каждый из них растворите в 20 мкл ТЭ2)
и сделайте серии пятикратных разведений.
6. Увлажните небольшой кусок нитроцеллюлозного или найлонового фильт-
ра, погрузите его в 20XSSC* 1 2 3 4 **, быстро промакните и поместите на лист
фильтровальной бумаги.
7. Инкубируйте образцы РНК при 65 °C в течение 5 мин; нанесите на
фильтр аликвоты по 5 мкл (для этой цели можно использовать аппарат
для дот-блот-гибридизации «minifold» фирмы Schleicher and Schnell).
8. Высушите фильтр под лампой-термоизлучателем и затем прогрейте в ва-
куумной камере в течение 1 ч.
9. Проведите стандартную гибридизацию с «ник-транслированным» меченым
зондом [39].
10. Для учета неспецифического связывания зонда к контрольным образцам
РНК перед этапом 7 добавляют РНКазу А до концентрации 10 мкг/мл.
’) STE: раствор 100 мМ NaCl, 10 мМ трис-НС1, (pH 7,5), 1 мМ ЭДТА.
2) ТЭ: 10 мМ трис-HCl (pH 7,5), 1 иМ ЭДТА.
3) SSC: 0,3 М NaCl, 0,03 М цитрат натрия.
клетками, последние предварительно экспонируют с митомици-
ном С: после этого клетки сохраняют жизнеспособность лишь
в течение нескольких дней и в течение этого срока продуцируют
вирус. Типичная процедура совместного культивирования описа-
на ниже.
1. Рассейте приблизительно 106 вирус-продуцирующих
«донорных» клеток на 10 см-чашке.
2. Обработайте клетки летальной дозой митомицина С, до-
бавив его в среду культивирования до концентрации 10 мкг/мл
и выдержав 1 ч. Затем промойте чашку PBS и замените среду.
3. Высейте на ту же чашку 106 клеток-мишеней, одновремен-
но добавив в среду полибрен до концентрации 8 мкг/мл (если
клетки обнаруживают непереносимость полибрена, экспозицию
с ним можно сократить).
4. Инкубируйте в течение 2—3 дней до формирования слит-
ного монослоя, затем рассейте инфицированные клетки-мишени
на новые чашки в свежей среде.
Ретровирусные векторы
297
9.6. Контроль вирусных препаратов
9.6.1. Титрование вируса
9.6.1.1. Дот-блот-аиализ вирусной РНК
Титр вируса в культуральной среде инфицированных клеток
можно определить с помощью целого ряда методик, в основе
которых лежат физические,. биохимические или биологические
методы анализа. Наибольшее распространение получил способ,
заключающийся в количественном определении вирусной геном-
ной РНК в культуральной среде с помощью гибридизации с
меченным 32Р ДНК-зондом. Эту стандартную процедуру дот-
блот-гибридизационного анализа проводят после концентриро-
вания препарата вируса, как описано в табл. 9.4. Количествен-
ное определение достигается путем сравнения интенсивности
полученного гибридизационного сигнала с интенсивностью
контрольных сигналов гибридизации точно титрованного вируса
(разд. 9.6.1.3) или известных количеств РНК, или ДНК, нане-
сенных на фильтр. Используя разные ДНК-зонды, можно оце-
нить концентрацию индивидуальых компонентов в вирусном
препарате и непосредственно определить, содержит ли вирус
интересующие нас последовательности.
9.6.1.2. Тест иа активность обратной транскриптазы
Поскольку ретровирусные частицы содержат РНК-зави-симую-
ДНК-полимеразу, определение активности этого фермента мо-
жет служить удобным критерием титра вирусного препарата.
Однако этот метод неприменим к препаратам, не содержащим
хелперных элементов, поскольку нетрансфицированные упаковы-
вающие клетки спонтанно продуцируют заметные количества
частиц, несущих обратную транскриптазу [17]. Для препаратов,
в которых присутствует хелперный вирус, тест на активность
обратной транскриптазы представляет собой наиболее простой
способ качественого анализа большого числа образцов. Количе-
ственное титрование требует приготовления серии стандартов
для построения калибровочной кривой.
Методика определения обратной транскриптазы приведена
в табл. 9.5. Вирусную полимеразу высвобождают мягким лизи-
сом вирионов детергентом, а затем тестируют на poly (С)-матрице
с oligo(dG)-затравкой. Включение метки (меченного 3Н dGTP)
в новосинтезированный полинуклеотид служит мерой фермен-
тативной активности препарата; эта величина в свою очередь
отражает титр вируса. Культуральную среду с ожидаемым вы-
соким титром вируса (<103/мл) можно анализировать непосред-
298
Э. Браун, М. Скотт
Таблица 9.5. Определение обратной транскриптазы
Понадобятся следующие растворы:
1 М трис-НС1 (pH 8,1)
1 М MgCl2
1 мМ ДТТ
10% NP40
10 мг/мл poly(C) (Boehringer Mannheim)
1 мг/мл oligo(dG) (Collaborative Research)
1 мКи/мл 3H-dGTP' (Amersham, 37 Кл/ммоль).
1. Приготовьте 2X реакционную смесь, содержащую 100 мМ трис-НС1
(pH 8.1), 2мМ MgCl2, 20 мМ ДТТ, 0,1% NP40.
2. К 250 мкл 2Х реакционной смеси (этого количества хватит на 10 анали-
зов) на льду добавьте 25 мкг poly(C), 6 мкг oligo(dG) и 10 мкКи
3H-dGTP‘>.
3. Добавьте 25 мкл последней смеси к 25 мкл содержащей вирус среды или
концентрированной вирусной суспензии и инкубируйте при 37°C в тече-
ние 2—4 ч.
4. Нанесите весь объем реакционной смеси на лист ДЭАЭ-бумаги (W hat-
man, DE-81) размером 2X2 см и подсушите на воздухе.
5. Промойте бумагу в 0,5 м Na2HPO4 при комнатной температуре в течение
30 мин, пять раз меняя раствор.
6. Промойте дважды водой, и затем дважды—95%-ным этанолом. Высуши-
те под лампой-термоизлучателем.
7. Поместите бумагу в сцинтиллятор и просчитайте радиоактивность на
сцинтилляционном счетчике.
’) В другом варианте методики, удобном для скрининга большого числа образцов,
предполагается использование 32P-dGTP вместо субстрата, меченного 3Н. Реакционные
пробы наносят в ряд на полоску ДЭАЭ-бумаги и после промывки бумагу экспонируют
с рентгеновской пленкой. В каждой реакции используется 0,1—0,5 мкКи 32P-dGTP; неме-
ченый dGTP добавляют до концентрации 10 мк.М. Подробнее см. работу Коффа
и др. [40].
ственно. В случае более низкого титра следует предварительно
сконцентрировать препарат путем осаждения вируса, как описа-
но в табл. 9.4 (пп. 1 и 2), и затем тщательно ресуспендировать
осадок на льду в 100 мкл буфера для разведения вируса (50 мМ
трис-НС1, pH 8,1, 10 мМ дитиотрейтола, 10 мМ MgCl2,
100 мкг/мл бычьего сывороточного альбумина).
9.6.1.3. Титрование по частоте трансдукции маркера
Оба описанных выше метода определения титра вируса стра-
дают одним общим недостатком — необходимостью строить ка-
либровочные кривые на основе точных стандартов. Кроме того,
этими методами нельзя различить дефектные вирусы и полно-
ценные инфекционные частицы. Очевидно, что наиболее
информативными следует считать такие методы титрования ви-
руса, которые основаны на определении количества инфекцион-
ных частиц с помощью биологических тестов. В случае реплика-
ционно-компетентного MLV этой цели отвечает анализ бляшек
с использованием ХС-клеток [15]. Однако обычно нет нужды
Ретровирусные векторы
299
применять этот метод в повседневной практике. Препараты
рекомбинантных вирусов, несущих селективные маркеры (на-
пример, ген пео), можно точно титровать, определяя частоту
трансдукции маркера после инфицирования восприимчивых
клеток-мишеней серийными разведениями препарата. При этом
.подсчитывают количество пео-устойчивых колоний, возникших
в результате инфекции, принимая во внимание соотношение, с
которым инфицированные клетки рассевали на селективную
среду. Титр препарата определяют как количество колониеобра-
зующих единиц (к. о. е.) в 1 мл вирусной суспензии. Точно так
же определяют титр вирусов, несущих гены, которые детерми-
нируют любой примечательный (но обязательно селективный)
признак, например трансформирующие онкогены.
9.6.2. Анализ структуры и экспрессии
рекомбинантного вируса
Принимая во внимание непредсказуемое поведение некото-
рых новых ретровирусных конструкций (разд. 9.7), как правило,
имеет смысл проверять структуру и экспрессию рекомбинантных
ретровирусов с помощью анализа их ДНК и РНК, а также пу-
тем прямой детекции экспрессируемых белковых продуктов
(если, конечно, это возможно). Для векторов, не экспрессирую-
щих примечательный фенотип, такого рода молекулярный
анализ особенно важен.
Наилучший способ определения структуры вирусной геном-
ной РНК—анализ провирусной ДНК-формы генома, присутст-
вующей в инфицированных клетках, например, с помощью
блот-гибридизации клеточной ДНК [39]. На рис. 9.5 показан
вариант такого анализа, демонстрирующий удаление интронов
из гена, клонированного в ретровирусном векторе. Как правило,
клеточную ДНК расщепляют рестриктирующими ферментами,
которые узнают специфические сайты в пределах каждого из
вирусных LTR, но не затрагивают внутренние последовательно-
сти; так, в случае векторов семейства Mo-MLV для этой цели
часто используют рестриктазы Xbal и Sad. Следует подчерк-
нуть, что в тех случаях, когда инфицированные клетки имеют
мышиное происхождение, гибридизационная проба для блот-
анализа не должна содержать MLV-последовательностей. В про-
тивном случае родственные MLV эндогенные провирусы ослож-
нят интерпретацию картины гибридизации. При необходимости
проводят детальное структурное картирование или секвениро-
вание ДНК рекомбинантного провируса, используя клонирован-
ную провирусную ДНК-
Экспрессию вирусных РНК-транскриптов изучают с помощью
стандартной процедуры «нозерн»-блок-анализа. При этом рабо-
300
Э. Браун, М. Скотт
тают как с poly(A)-содержащей, так и с суммарной РНК, вы-
деленной из инфицированных клеток (39].
9.6.3. Выделение провирусной ДНК путем клонирования
Для решения целого ряда задач желательно иметь способ,
позволяющий легко выделять рекомбинантный провирус из
инфицированных клеток путем молекулярного клонирования.
В частности, это относится к использованию ретровирусных
векторов для удаления интронов, так как клонирование прови-
руса дает возможность после инфекциононго цикла получать
кДНК-копии интересующего нас гена. Кроме всего прочего
клонирование провируса наилучшим образом позволяет иссле-
довать и подтвердить генетическую структуру любой ретрови-
русной конструкции после того, как она претерпела цикл вирус-
ной репликации. Клонирование провирусов включает обычные
генноинженерные манипуляции, в том числе создание геномной
библиотеки и ее скрининг гибридизационными методами. Одна-
ко заметим, что это непростая задача ввиду того, что обычно
лишь одна копия искомого провируса присутствует в клеточном
геноме.
Для клонирования цео-содержащих провирусов предложены
две упрощенные методики; каждая из них предполагает селек-
цию искомого клона в клетках Е. coli на устойчивость к канами-
цину. Одна из методик представляет собой модифицированную
процедуру «спасения» маркера и предусматривает прямое кло-
нирование провируса в плазмидном векторе. Другая методика,
гораздо более эффективная, требует применения специальных
ретровирусных «челночных» векторов, описанных в разд. 9.3.2.4.
9.6.З.1. Прямое клонирование провируса в ппазмидиом векторе
Эта процедура описана здесь лишь в общих чертах, но при-
менима ко всем ретровирусным векторам, несущим ген пео.
Геномную ДНК выделяют из инфицированных клеток, расщеп-
ляют рестриктазой, для которой имеются сайты узнавания
только внутри провирусных LTR (например, ХЬа\), и затем
фракционируют путем препаративного гель-электрофореза, что-
бы получить материал, обогащенный интересующим провирус-
ным ДНК-фрагментом. Этот фрагмент лигируют с плазмидным
вектором, обработанным ХЬа\ или «на себя», если провирус со-
держит область начала репликации pBR 322, после чего вводят
полученную конструкцию в клетки Е. coli путем трансформа-
ции. Затем бактериальную культуру разделяют на аликвоты,
разводят в 20 раз и проводят селекцию в среде, содержащей
канамицин (7,5—50 мкг/мл).
Ретровирусные векторы
301
Аликвоты наращивают до стадии стационарной культуры;
каждая из них содержит независимые клоны провирусной ДНК,
имеющей в своем составе ген пео. Описанная процедура клони-
рования требует использования компетентных бактерий, обеспе-
чивающих высокую эффективность трансформации (по меньшей
мере 2-Ю7 мкг), в количествах, позволяющих получить 2-Ю7
трансформантов. Однако высокоэффективная канамициновая
селекция в жидкой среде делает отбор клонов относительно
несложным. Подробно этот метод описали Браун и др. [12].
9.6.3.2. Метод слияния инфицированных клеток с cos-клетками
Так называемые ретровирусные челночные векторы (один из
прототипов — pZIP-Neo SV(X) 1 -[22]) разработаны специально
для того, чтобы облегчить клонирование соответствующих про-
вирусных ДНК. Как описано в разд. 9.3.2.4, «челночные» свой-
ства pZIP-Neo SV(X) 1 обусловлены сочетанием в единой конст-
рукции трех элементов: области начала репликации SV-40,
области начала репликации pBR322 и гена пео.
Для вектора pZIP-Neo SV(X) 1 описана эффективная проце-
дура получения плазмидных клонов этого провируса из ин-
фицированных клеток, которая применима и для других анало-
гичных челночных векторов [22]. Инфицированные клетки сли-
вают с cos-клетками (линия клеток обезьяны, экспрессирующая
Т-антиген SV-40). В присутствии Т-антигена происходит много-
кратная инициация репликации ДНК в области начала репли-
кации SV-40, локализованной в интегрированном провирусе, а
эксцизиционная рекомбинация приводит к высвобождению мно-
жественных копий кольцевых молекул ДНК, содержащих про-
вирусные последовательности. Эти кольцевые плазмиды очища-
ют по методу Херта, трансформируют полученной ДНК
компетентные клетки Е. coli и отбирают клоны, устойчивые к
канамицину. Подробная экспериментальная процедура приведе-
на в табл. 9.6.
Обычно эффективность данного метода составляет 103 кана-
мицин-устойчивых бактериальных колоний на 107 клеток млеко-
питающих. Показано, что большая часть клонов имеет ожидае-
мую структуру (кольцевая провирусная ДНК с единственным
LTR), хотя приблизительно 20% клонов обнаруживает различ-
ные перестройки ДНК вследствие рекомбинационных событий
[22].
9.7. Потенциальные проблемы
Несмотря на то что использование ретровирусов в качестве
векторов основано на обширном знании биологии природных
ретровирусов, в вопросах, связанных с репликацией и экспресси-
302
Э. Браун, М. Скотт
Таблица 9.6. Клонирование провирусной ДНК с использованием
слияния инфицированных клеток с cos-клетками
и феномене «спасения» маркера [22j
1. Смешайте равные количества вектор-содержащих клеток и cos-клеток,
рассеяв по '/г слитного монослоя каждой культуры на свежую 10см-
чашку.
2. На следующий день расплавьте небольшое количество полиэтнленгликоля
1000 (Baker), охладите до 40 °C, разведите средой, не содержащей сы-
воротки (1 : 1), и стерилизуйте фильтрованием.
3. Промойте клетки три раза средой, не содержащей сыворотки, и хорошо
подсушите.
4. Проведите слияние клеток: аккуратно наслоите на чашку 2 мл 50%-ного
раствора ПЭГ в среде и оставьте при комнатной температуре на 1 мин
(оптимальное время инкубации варьирует в зависимости от конкретной
партии ПЭГ; однако слишком длительная обработка может оказаться
гибельной для клеток).
5. Мягко, но быстро промойте клетки 10 мл среды, не содержащей сыво-
ротки. Оставьте клетки в этой среде.
6. Инкубируйте при 37 °C в течение 1—3 дней, заменяя среду каждый день.
7. Промойте клетки 2 раза буфеном PBS. Лизируйте клетки по методу Хер-
та в 2 мл 10 мМ раствора, содержащего трис-НС1 (pH 7,4), 10 мМ
ЭДТА и 2% ДСН. Перенесите в пробирку типа «Oak Ridge» и добавьте
5 М NaCl до конечной концентрации 1,25 М.
8. Инкубируйте при 4 °C в течение 6 ч, затем центрифугируйте при 38 000 g
в течение 45 мин.
9. Экстрагируйте один раз смесью фекол/хлороформ и один раз хлорофор-
мом, как описано в п. 4 табл. 9.4, затем осадите нуклеиновые кислоты
из последней водной фазы двумя объемами изопропанола. Выдержите
при —20 °C по меньшей мере 2 ч и осадите нуклеиновые кислоты цент-
рифугированием. Ресуспендируйте осадок в буфере ТЭ1’, добавьте ацетат
натрия до 0,3 М и снова осадите нуклеиновую кислоту двумя объемами
изопропанола.
10. Ресуспендируйте осадок в 200 мкл буфера ТЭ, содержащего 100 мМ
NaCl. Добавьте РНКазу А (лрокипяченую) до 1 мкг/мл и инкубируйте
при 37 °C в течение 30 мин. Охладите до комнатной температуры.
11. Добавьте спермин до концентрации 5 мМ и оставьте при комнатной тем-
пературе на 10 мин. Центрифугируйте в течение 10 мин в микроцентри-
фуге и подсушите осадок. Промойте мягко, но интенсивно в 0,5 мл сме-
си, содержащей 75%-ный этанол, 0,3 М ацетат натрия, 10 мМ ацетат
магния. Снова центрифугируйте в течение 10 мин и подсушите осадок.
12. Ресуспендируйте ДНК в 50 мкл ТЭ, содержащего 100 мМ, и трансфор-
мируйте Е. coli НВ101. Проведите селекцию трансформантов в присутст-
вии 50 мкг/мл канамицинсульфата.
1) ТЭ: 10 мМ трис-НС! (pH 7,5), I мМ ЭДТА.
ей, остается много неясного. В ряде случаев пробелы в наших
знаниях наглядно иллюстрируются тем, что ретровирусные век-
торные конструкции ведут себя иначе, чем мы можем предполо-
жить. Некоторые из возможных затруднений в работе, связан-
ных с присутствием в последовательностях-вставках промоторов,
сигналов полиаденилирования или преждевременных старт-
сигналов трансляции, уже упоминались в разд. 9.3.2.2. и 9.3.4.
Другие, менее поддающиеся прогнозированию проблемы, сопря-
Ретровирусные векторы
303
женные с использованием ретровирусных векторов, обсуждаются
в данном разделе. Это не должно обескураживать читателя,
поскольку примеры успешного применения ретровирусных векто-
ров широко освещаются в литературе. Тем не менее эксперимен-
татор должен иметь представление о возможных трудностях
в работе с ретровирусными векторами и помнить о том, что один
и тот же вектор может обнаруживать совершенно разные свой-
ства в зависимости от конкретной клонированной в нем последо-
вательности.
9.7.1. Неэквивалентная экспрессия спаренных генов
Одна из проблем, с которой часто приходится сталкиваться
при использовании векторов для спаренных генов, заключается
в том, что не удается перенести и экспрессировать одновременно
оба маркера. Это особенно типично для сплайсинг-векторов
(разд. 9.3.2.2.), способных обеспечить эффективную экспрессию
обоих клонированных генов только в том случае, если эффектив-
ность сплайсинга вирусной РНК близка к 50%. К сожалению,
мы пока мало что знаем о закономерностях сплайсинга ретро-
вирусной РНК. Существует наблюдение, что последовательности,
введенные в проксимальный (относительно промотора) сайт для
клонирования, могут влиять на формирование субгеномной РНК,
в составе которой экспрессируется ген, клонированный в ди-
стальном сайте [20, 25]. В частности, это было показано для
вектора рМХ 1122пео (рис. 9.3, в) с протоонкогеном с-тус,
введенным в область «gag». Когда провели селекцию на экспрес-
сию этой конструкцией с-тус (по его способности «коопериро-
ваться» с EJ-ras при ко-трансфекции первичных фибробластов),
полученные трансформированные клетки экспрессировали бе-
лок Мус, но в них не удалось детектировать субгеномную пео
RNA (М. Скотт, Г. Вармус, неопубликованные данные). Кроме
того, при трансфекции рМХ 1122 тус-neo ДНК в клетки NIH3T3
оказалось, что экспрессия дистального гена пео сильно ингиби-
руется. Это следовало из очень низкой эффективности транс-
формации клеток в С418к-фенотип в сравнении с результатами,
достигаемыми при трансфекции родительской векторной ДНК
рМХ 1122 пео (табл. 9.7). В этом случае также не удалось об-
наружить субгеномной neo-РНК (М. Скотт и Г. Вармус, неопуб-
ликованные данные). Точно так же введение с-тус «выше» v-ras
в рМх1122 ингибировало сплайсинг вирусной РНК- В то же
время при введении в «gag»-caftT рМХ1122 пео онкогена int-1
наблюдалась эффективная экспрессия субгеномной пео-РНК
[12], что указывает на специфичность ингибирования сплайсин-
га вирусной РНК, определяемую природой вставки.
.304
Э. Браун, М. Скотт
Таблица 9.7. Трансфекционная и трансдукционная активность
ретровирусных векторов, демонстрирующая ингибирование
экспрессии гена пео (разд. 9.7.1 и 9.7.2)
Вектор Эффективность трансфек- ции’) (селекция на фенотип G418R), колоиий/мкг Эффективность трансдук- ции2) (селекция на фенотип G418R), к. о. е./мл
рМХ1122пео 1,3 103 2,510&
рМХ1122тус. пео 70 3,0105
Ч Эффективность трансфекции определяли, трансфицируя клетки NIH3T3 с после-
дующей селекцией на фенотип G418R; количественно выражается числом О418К-колоний,
•полученных в расчете на 1 мкг векторной ДНК.
2) Эффективность трансдукции определяли в параллельных экспериментах по ко-
тр.ансфекции клеток iNIH3T3 вейторной ДНК репликационно-к'омпетецтного хелпера
pZAP. Трансдуцируютий вирус, продуцируемый С418К-колониями, полученными в ре-
зультате трансфекции, титровали, инициируя клетки NIM3T3. Количественная оценка вы-
ражена числом С418К-колониеобразующих единиц в 1 мл культуральной жидкости.
Векторы для спаренных генов с внутреннем Промотором не
эксплуатируют вирусную систему сплайсинга. Однако нередко
и они не могут обеспечить эквивалентную экспрессию обоих ге-
нов. Кроме тех случаев, когда LTR или внутренний промотор
оказываются неспособными эффективно функционировать в
инфицируемых клетках конкретного типа, экспрессия любого
маркера может подавляться также вследствие неблагоприятно-
го взаимодействия двух промоторов. Эмерман и Темин [23],
работая с вектором, содержащим внутренний промотор, обнару-
жили, что селекция, направленная на экспрессию 5'-гена, вызы-
вает эпигенетическую супрессию З'-промотора и наоборот. Хотя
этот эффект, зависящий от специфического сочетания используе-
мых промоторов, по-видимому, наиболее ярко проявляется в век-
торах, основанных на ретровирусах птиц, он также свойствен
и векторам, принадлежащим к семейству M.LV [241.
9.7.2. Генетическая нестабильность
Ретровирусные векторы имеют тот серьезный недостаток, что
все они генетически нестабильны. Это может проявляться либо
в форме массированных делеций, которые легко обнаруживают-
ся с помощью блот-гибридизационного анализа, либо в виде точ-
ковых мутаций, которые труднее детектировать и потому они
более коварны. По всей видимости, РНК-полимераза и обратная
транскриптаза, реплицирующие вирусный геном, не обладают
сколько-нибудь заметной корректирующей активностью и часто-
та спонтанных мутаций у ретровирусов необычайно высока
[14, 20]. По имеющимся оценкам, частота мутирования ретро-
вирусных векторов достигает 0,5% в расчете на каждый цикл
репликации '[20].
Ретровирусные векторы
305
Одной из возможных причин делеций в ретровирусных гено-
мах служит аномальный сплайсинг вирусной РНК. Хотя в MLV-
векторах сигнал для упаковки расположен «ниже» вирусного
донорного сайта сплайсинга (относительно направления тран-
скрипции), так что молекулы, претерпевшие сплайсинг с участи-
ем именно этого сайта, неспособны упаковываться в вирусные
частицы, криптические сайты сплайсинга, присутствующие во
вставке, способны вызывать нежелательные последствия. Деле-
нии и перестройки могут происходить также в результате реком-
бинационных событий с участием эндогенных мышиных
ретровирусных последовательностей, рекомбинации в процессе
трансфекции или же ошибок при обратной- транскрипции и
интеграции [14]. Кроме всего прочего в некоторых случаях де-
лении могут неумышленно отбираться при селекции на экспрес-
сию определенного маркерного гена. Примером этого может
служить эксперимент (табл. 9.7), демонстрирующий особенности
сплайсинг-вектора для спаренных генов рМХ 1122 тус.чгео: в
присутствии последовательности с-тус ингибируется сплайсинг
вирусной РНК (разд. 9.7.1). Несмотря на низкую эффективность
трансфекции, обнаруживаемую, этим вектором при селекции на
устойчивость к G418, трансфицированные клетки активно проду-
цировали пео-трансдуцирующий вирус (табл. 9.7). Однако пос-
ледующий анализ показал, что эти вирусные частицы несут
делецию с-тус Скотт, Г. Вармус, неопубликованные дан-
ные). Следовательно, селекция на эффективную экспрессию гена
г.ео привела к утрате последовательностей с-тус, ответственных
за ингибирование образования субгеномной neo-мРНК- Высокая
частота делеций, обусловливающая повышенный уровень экс-
прессии селективного маркера, также свойственна и векторам
с внутренним промотором, однако пока такие данные получены
только для векторов на основе ретровирусов птиц [41].
Благодарности
Пользуясь случаем, мы хотели бы поблагодарить многих
наших коллег за ценные советы и дискуссии, и в особенности
Гарольда Вармуса: именно в его лаборатории мы провели боль-
шинство своих собственных экспериментов с ретровирусными
векторами. Мы также благодарны Конни Сепко за предоставле-
ние методики клонирования провируса, а также Р. Киршмейеру
и Б. Вейнстейну за разрешение описать вектор pM.V7. А. Браун
работает по специальной стипендии Американского общества по
изучению лейкемии.
20—1186
306
Э. Браун, М. Скотт
Литература
1. Bright man В. К., Pattengale Р. К-, Fan Н. (1986). J. Virol., 60, 68.
2. Williams D. A., Lemischka I. R„ Nathan D. G„ Mulligan R. C. (1984). Natu-
re, 310, 476.
3. Anderson W. F. (1984). Science, 226, 401. ,—
4. Jahner D., Haase K., Mulligan R. C., Jaenisch R. (1985) ./Proa) Natl. Acad.
Sci. USA, 82, 6927. /
5. Robertson E., Bradley A., Kuehn M., Evans M. (1986). Nature/323, 445.
6. Dick J. E., Magli M. C„ Huszar D., Philips R. A., Bernstein A, (1985). Cell,
42, 71. /
7. Price J., Turner D„ Cepko C. (1987). Proc. Natl. AcaxUSci. USA, 84, 156.
8. Soriano P., Jaenisch R. (1986). Cell, 46, 19.
9. Weis J. H„ Nelson D. L., Przyborski M. J., Chaplin D. D., Mulligan R. C.,
Housman D. E., Seidman J. G. (1984). Proc. Natl. Acad. Sci. USA, 81,
4879
10. King W., Patel M. D., Label L. I., Goff S. P„ Nguyen-Huu M. C. (1985).
Science, 228, 554.
11. Shimotohno K., Temin H. (1982). Nature, 299, 265.
12. Brown A. M. C., Wildin R. S., Prendergast T. J., Varmus H. E. (1986). Cell,
46, 1001.
13. Varmus H. E. (1982). Science, 216, 812.
14. Coffin J. (1984). In: RNA Tumor Viruses, Weiss R., Teich N., Varmus H. and
Coffin J. (eds.), Cold Spring Harbor Laboratory, Cold Spring Harbor, New
York, Vol. 2, p. 36.
15. H7eiss R. (1982). In: RNA Tumor Viruses, Weiss R., Teich N., Varmus H.
and Coffin J. (eds.), Cold Spring Harbor Laboratory, Cold Spring Harbor,
New York, Vol. 1, p. 209.
16. Bishop J. M., Varmus H, E. (1982). In: RNA Tumor Viruses, Weiss R.,
Teich N., Varmus H. and Coffin J. (eds.), Cold Spring Harbor Laboratory,
Cold Spring Harbor, New York, Vol. 1, p. 999.
17. Mann R., Mulligan R. C„ Baltimore D. (1983). Cell, 33, 153.
18. Kreigler M„ Perez C. F„ Hardy C., Botchan M. (1984). Cell, 38, 483.
19. Sorge J., Wright D„ Erdman V. D„ Cutting A. E. (1984). Mol. Cell. Biol., 4.
1730.
20. Dougherty J. P„ Temin H. M. (1986). Mol. Cell. Biol., 7, 4387.
21. Miller A. D., Law M. F., Verma I. M. (1985). Mol. Cell. Biol., 5, 431.
22. Cepko C. L„ Roberts В. E„ Mulligan R. C. (1984). Cell, 37, 1053.
23. Emerman M„ Temin H. M. (1984a). Cell, 39, 459.
24. Emerman M., Temin H. M. (1986). Nucleic Acids Res., 14, 9381.
25. E„ Eglitis M. A., Kantoff P. W„ Anderson W. F. (1986). Biotechniques,
26. Kirschmeier P. T., HouseyG.M., Johnson M. D„ Perkins A. S., Weinstein I. B.,
submitted for publication.
27. Dickson C„ Eisenman R„ Fan H. (1984). In: RNA Tumor Viruses, Weiss R.,
Teich N., Varmus H. and Coffin J. (eds.), Cold Spring Harbor Laboratory,
Cold Spring Harbor, New York, Vol. 2, p. 135.
28. Berger S. A„ Bernstein A. (1985). Mol. Cell. Biol., 5, 305.
29. Yu S. F„ von Ruden T., Kantoff P. W„ Garber C„ Sieberg M., Ruther U,
Anderson W. F., Wagner E„ Gilboa E. (1986). Proc. Natl. Acad. Sci USA,
83, 2566.
30. Reik W„ Weiber H„ Jaenisch R. (1985). Proc. Natl. Acad. Sci. USA 82
1141. ’ ’
31. Hughes S., Kosik E. (1984). Virology, 136, 89.
32. Miller A. D„ Baltimore C. (1986). Mol. Cell. Biol., 6, 2895.
33. Watanabe S„ Temin H. M. (1983). Mol. Cell. Biol., 3, 2241.
Ретровирусные векторы
307
34. Gorman С. (1985). In: DNA Cloning — A Practical Approach, Glover D. M.
(eaj, IRL Press, Oxford, Vol. II, p. 143. [Имеется перевод: Клонирование
ДНК. Методы. Книга II. Под ред. Д. Гловера. — М.: Мир, 1972].
35. Korman А. /., Franz J. D„ Strominger J. L., Mulligan R. C. (1987). Proc.
Natl. Acad. Sci. USA, in press.
36. Miller A. D., Trauber D. R., Baltimore C. (1986). Somatic cell Mol. Genet.,
12, 175.
37. Hwang L. S„ Gilboa E. (1984). J. Virol., 50, 417.
38. Hoffmann J. W., Steffen D., Gusella J., Tabin C., Bird S., Lowing D., Wein-
berg R. A. (1982). J. Virol.. 44, 144.
39. Maniatis T., Fritsch E. F., Sambrook J. (1982). Molecular Cloning, Cold
Spring Harbor Laboratory, Cold Spring Harbor, New York. [Имеется пере-
вод: Маниатис T., Фрич Э., Сэмбрук Дж. Методы генетической инженерии.
Молекулярное клонирование. — М.: Мир, 1984.]
40. Goff S., Traktman Р„ Baltimore D. (1981). J. Virol., 38, 239.
41. Emerman M., Temin H. M. (1984b). J. Virol., 50, 42.
42. Appendix В in RNA Tumor Viruses (1985). Weiss R., Teich N., Varmus H.
and Coffin J. (eds.), Cold Spring Harbor Laboratory. Cold Spring Harbor,
New York, Vol. 2, p. 766.
20*
ГЛАВА 10
ПОЛУЧЕНИЕ ТРАНСГЕННЫХ МЫШЕИ ПУТЕМ
МИКРОИНЪЕКЦИИ КЛОНИРОВАННОЙ ДНК
В ОПЛОДОТВОРЕННЫЕ ЯЙЦЕКЛЕТКИ
Дэвид Мерфи и Дженнифер Хэнсон
(David Murphy, Jennifer Hanson)
10.1. Введение
10.1.1. Трансгенные животные
За последнее десятилетие удалось осуществить молекуляр-
ное клонирование и характеризовать структуру множества ге-
нов млекопитающих. Функциональное содержание и механизмы
регуляции этих генов исследуются теперь в экспериментах по
переносу генетического материала. Рекомбинантные конструкции
на основе последовательностей дикого типа или их мутантных
производных вводят путем трансфекции в культивируемые клет-
ки [1] для того, чтобы идентифицировать г{ис-действующие ре-
гуляторные элементы и изучить физиологические последствия
экспрессии генных продуктов. Однако,, даже если для интере-
сующего гена и существует подходящая культивируемая ткане-
вая система, возможности исследования генной экспрессии в
таких экспериментах in vitro ограничены. В конце концов функ-
ции генов и закономерности их экспрессии следует изучать, ис-
ходя из сложности целого организма. Был разработан целый
ряд методик, позволяющих вводить интересующие нас последо-
вательности ДНК в клетки зародышевого пути мышей и других
млекопитающих. Включившись в геном данного организма, та-
кие чужеродные последовательности, называемые трансгенами,
устойчиво наследуются в ряду поколений. Весьма важное значе-
ние имеет тот факт, что трансгены часто экспрессируются и вы-
зывают изменения в системе тканевой специфичности, физиоло-
гических реакциях, а иногда во всей программе развития орга-
низма. Следовательно, открывается путь к изучению функцио-
нальной роли и регуляции экспрессии интересующих нас клони-
рованных генов на уровне целого организма — в данном случае
это так называемый трансгенный организм.
10.1.2. Использование клеток зародышевого пути
Для получения трансгенных мышей существуют три основ-
ных методических подхода; все три метода предполагают вме-
шательство экспериментатора в предимплантационную фазу раз-
вития организма (рис. 10.1). Перечислим эти методы:
308
Получение трансгенных мышей
309
МЫШЬ
Рис. 10.1. Возможные пути «интервенции» в зародышевый путь. Такое вмеша-
тельство в предимплантационную стадию развития (стадия от оплодотворения
до имплантации) позволяет вводить в организм млекопитающих новые гены
(разд. 10.1.2).
1. Инфицирование предимплантированных эмбрионов реком-
бинантными ретровирусами.
2. Манипулирование с эмбриональными стволовыми клетками
(ES- или ЕК-клетки).
3. Микроинъекции ДНК в оплодотворенное одноклеточное
яйцо.
10.1.2.1. Ретровирусы
Инфицирование предимплантированных эмбрионов рекомби-
нантными ретровирусами — относительно несложная процедура,
не требующая дорогостоящего оборудования. Тем не менее лю-
бой интересующий ген необходимо сначала переклонировать в
вирусный вектор и эту рекомбинантную конструкцию ввести в
соответствующие клетки, чтобы получить клеточную линию, про-
дуцирующую инфекционные рекомбинантные вирусные частицы.
Здесь, однако, следует иметь в виду, что размер вставки, кото-
рую способен акцептировать вирусный вектор, ограничен (около
10 т. п. н.); кроме того, интересующая ДНК может содержать
310
Д. Мерфи, Дж. Хэнсон
сигналы сплайсинга или терминирующие сигналы, которые во
многих случаях оказывают неблагоприятное влияние на жиз-
ненно важные вирусные функции. Эмбрионы инфицируют сле-
дующим образом: восьмиклеточную морулу освобождают от
яйцевой оболочки и помещают в культуральную чашку с фибро-
бластами, продуцирующими рекомбинантный вирус. После ин-
фицирования эмбрионов, достигших стадии бластоцисты, вводят
в матку псевдобеременной «суррогатной» матери; часть эмбрио-
нов продолжает нормально развиваться и в положенный срок
превращается в трансгенных детенышей. За интеграцию ретро-
вируса ответственны прецизионные механизмы, которые обеспе-
чивают включение в геном (инсерцию) одиночной интактной
копии провирусной ДНК, содержащей интересующий нас ген и
фланкированной длинными концевыми повторами ретровируса
(LTR). Гарантированное включение в геном только одной копии
провируса представляется существенным преимуществом данно-
го метода в сравнении с другими известными подходами. Одна-
ко следует подчеркнуть, что, хотя в результате одного акта ин-
теграции в геном клетки действительно вводится только одна
копия вирусного генома, нельзя исключить возможность множе-
ственной интеграции в одной клетке; тогда в каждой клетке
эмбриона будут обнаруживаться множественные инсерции. В ре-
зультате получится организм, мозаичный по числу вирусных
инсерций в клетках. Поэтому для выведения чистых линий, при-
годных для исследования экспрессии введенных генов, понадо-
бится широкомасштабный аутбридинг. Известно несколько удач-
ных попыток экспрессировать трансгены в организме мыши в
составе ретровирусных векторов [2, 3]. Однако влияние сильных
вирусных контролирующих элементов, содержащихся в LTR, на
характер экспрессии трансгенов изучено еще недостаточно.
10.1.2.2. Эмбриональные стволовые клетки
ES- и ЕК-клетки представляют собой плюрипотентные эмб-
риональные стволовые клетки, предшественники которых были
взяты из бластоцисты [4,5]. ES-клетки можно культивировать
in vitro и после необходимых манипуляций вернуть в живой ор-
ганизм, инъецируя их в бластоцисту. Клетки колонизируют эмб-
рион и участвуют в его нормальном развитии, давая начало
клеткам всех типов соматических тканей и нередко клеткам за-
родышевого пути [6]. Прежде чем инъецировать в бластоцисту,
в ES-клетки вводят клонированные гены — либо путем транс-
фекции [7], либо инфицируя их рекомбинантными ретровируса-
ми [8]. Практическое достоинство этой методической схемы со-
стоит в том, что она дает возможность проводить селекцию или
скрининг трансформированных ES-клеток на определенный же-
Получение трансгенных мышей 311
лаемый параметр. Это может быть число копий трансгена, его
хромосомная локализация или характер экспрессии. Все первич-
ные трансгенные животные — мозаичные организмы, состоящие
из трансген-содержащих и несодержащих клеток; поэтому для
получения чистых трансгенных линий необходима селекционная
работа. Между тем довольно часто ES-клетки оказываются не-
способными колонизировать зародышевый путь, что, по всей ве-
роятности, объясняется накоплением хромосомных нарушений в
процессе культивирования и селекции in vitro.
Проблему мозаичности первичных трансгенных животных
легко преодолеть, если использовать трансформированные и
прошедшие селекцию ES-клетки в качестве доноров ядер. Про-
нуклеус оплодотворенной яйцеклетки можно удалить, чтобы
затем в эту яйцеклетку трансплантировать донорное яйцо, кото-
рое обычно берут из другого оплодотворенного яйца [9]. Ядра
ES-клеток, несущие трансгены, способны колонизировать эну-
клеированные яйца, которые затем продолжат свое нормальное
развитие. В результате может получиться животное, в каждой
клетке которого будет содержаться трансген.
В будущем, вероятно, появится возможность адресовать му-
тации конкретным генам в мышином организме путем введения
в ES-клетки соответствующим образом измененных in vitro кон-
струкций в качестве субстрата для гомологичной рекомбинации
[Ю, 11].
10.1.2.3. Микроинъекции
Этот подход остается пока наиболее популярным у исследо-
вателей, занятых получением трансгенных мышей, несмотря на
то что он требует высокой квалификации и дорогостоящего обо-
рудования. Это неудивительно — быстрота и надежность данного
метода с лихвой окупают все его недостатки.
I. Краткая характеристика метода. Процесс получения транс-
генных мышей с помощью микроинъекций схематически пока-
зан на рис. 10.2. Сущность каждого этапа экспериментальной
схемы изложена ниже со ссылками на соответствующие разделы
данной главы, где приводится более детальная информация.
1. Донорных мышей-самок с экспериментальной суперовуля-
цией (разд. 10.2—10.4) скрещивают с самцами-производителями.
2. Через 12 ч после спаривания (ПК — посткоитум) вскрыва-
ют яйцеводы донорных самок, выделяют оплодотворенные одно-
клеточные яйца (разд. 10.2, 10.3 и 10.6) и помещают их в куль-
туру (разд. 10.2, 10.3 и 10.5).
3. Далее в оплодотворенные яйца инъецируют очищенную
клонированную ДНК (разд. 10.4.1).
312
Д. Мерфи, Дж. Хэнсон
Гиперовуляция X •*—
О Стерильным
(после ваз-
эктомии)
Псевдобеременная
реципиентная
,, самка
Коллекция преимплантированных эмбрионов
Оплодотворенное
1-клеточное яйцо
Мышь-кормилица
Пересадка
в яйцевод
Рождение естест-
Кесарево сечение
♦ и вскармливание
фрагмента ДНК
венным путем
Выделение ДНК из
тканей хвоста
Анализ выделенной ДНК
Идентификация трансгенных мышей
Скрещивание Анализ экспрессии генов
Рис. 10.2. Схема получения трансгенных мышей путем микроинъекции клони-
рованной ДНК в оплодотворенные одноклеточные яйца.
4. Яйца, пережившие инъекцию, возвращают в естественную
среду, пересаживая их в яйцевод псевдобеременной (12 ч ПК)
реципиентной самки. Псевдобеременных самок получают, спа-
ривая половозрелых самок с самцами, подвергшимися вазекто-
мии (разд. 10.2.3.3).
5. Некоторая часть трансплантированных яиц продолжает
развиваться положенное время, и затем рождаются детены-
ши — естественным образом или с помощью кесарева сечения
(разд. 10.2.3.8).
Получение трансгенных мышей 313
6. Трансгенных животных в помете идентифицируют с по-
мощью гибридизационного анализа высокомолекулярной геном-
ной ДНК, выделенной из тканей хвоста (разд. 10.4.2).
1. Затем трансгенных животных скрещивают, чтобы получить
чистую линию, и анализируют экспрессию трансгена
(разд. 10.5).
II. Структурная организация экзогенной ДНК, введенной в
клетку путем микроинъекции и внедрившейся в геном. Точный
механизм, обеспечивающий интеграцию инъецированной ДНК
в хромосомы клетки-хозяина, неизвестен, однако некоторое
представление о характере процесса можно составить на основе
анализа структурной организации интегрированной ДНК в ге-
номе трансгенных мышей [12]. Примерно у 70% трансгенных
мышей во всех соматических клетках ц клетках зародышевого
пути имеется экзогенная ДНК; что свидетельствует о том, что
интеграция обычно происходит до первого цикла’ репликации
ДНК. Остальные 30% трансгенных животных обнаруживают ту
или иную степень мозаичности, что, вероятно, является следст-
вием интеграции ДНК на более поздних стадиях — уже после
первого цикла репликации. Количество копий трансгена в гено-
ме сильно варьирует и может достигать нескольких тысяч. В то
же время в клетках первичных трансгенных животных обычно
имеется только один участок интеграции; множественные копии
трансгена, как правило, представляют серию тандемных повто-
ров внутри единственного локуса. Участок интеграции в геноме
клетки-хозяина, по всей видимости, строго не детерминирован и
определяется случайным образом. Интеграционные события на-
блюдались во многих разных аутосомах [13], в Х-хромосоме
[14] и У-хромосоме (Д. Мэрфи, Дж. Хэнсон и В. Хоган, не-
опубликованные данные).
III. Эффективность микроинъекционного метода. Эффектив-
ность процедуры получения трансгенных мышей путем микро-
инъекций сильно варьирует от эксперимента к эксперименту.
В оптимальных условиях 60—80% яиц переживает инъекцию.
Из них 10—30% имплантируется в организм реципиентной сам-
ки, продолжает нормально развиваться и дает начало потомст-
ву. 10—30% новорожденных мышей оказываются трансгенными.
В какой-то степени успех эксперимента зависит от превходящих
'условий, например от сноровки экспериментатора. Однако опре-
деляющее значение здесь имеют качество инъецируемой ДНК
(разд. 10.4.1) и выбор линии мышей (разд. 10.2.2.1); поэтому
эти два параметра следует строго контролировать.
Нижеследующие разделы посвящены детальному описанию
методик получения, разведения и анализа трансгенных мышей
314
Д. Мерфи, Дж. Хэнсон
и включают рекомендации по уходу за животными, а также
методики микроинъекции, манипулирования с ДНК и анализа
экспрессии генов в трансгенных животных.
10.2. Обращение с животными и их содержание
Данный раздел касается непосредственной работы с живы-
ми животными, и его следует рассматривать только как сбор-
ник рекомендаций для новичков. Всем манипуляциям с живот-
ными должен обязательно обучать квалифицированный и опыт-
ный сотрудник. Две из описанных методик — вазэктомия и пе-
ресадка яйцеклетки в яйцевод — в сущности являются хирурги-
ческими операциями, проводимыми на живых мышах; поэтому
обучение здесь должно проводиться под тщательным контролем
и руководством опытного наставника. Новичкам следует некото-
рое время поработать с трупным материалом для приобретения
необходимых технических навыков и лишь затем переходить к
живым анестезированным объектам. Если животное в ходе опе-
рации обнаруживает любые признаки страдания, эксперимента-
тор обязан его немедленно умертвить переламыванием шейных
позвонков (разд. 10.2.3.1).
10.2.1. Содержание животных
Получение и содержание трансгенных мышей подразумева-
ет, что исследователь должен обеспечить нормальное существо-
вание сотен или даже тысяч особей. Очевидно, что за такую ра-
боту можно браться только при условии наличия всей необхо-
димой технологической базы. Основные требования здесь тако-
вы:
1. Достаточные площади для клеточного содержания живот-
ных.
2. Оборудование для мойки и, желательно, стерилизации
клеток.
3. Регулярная замена подстилки.
4, Вентиляция.
5. Снабжение водой и кормом.
6. Контроль параметров внешней среды.
7. Ветеринарная служба.
В Великобритании условия содержания лабораторных живот-
ных и работы с ними строго регламентированы специальным за-
конодательным актом от 1986 г. Учреждения, в которых разре-
шено проводить эксперименты на животных, обязаны иметь не-
обходимый сертификат стандартного образца. Все процедурные
Вопросы, касающиеся работы с животными, находятся под стро-
Получение трансгенных мышей.315
гим контролем правительственных органов. Для работы с жи-
вотными каждый исследователь должен получить допуск, доку-
ментированный в форме персональной лицензии. Кроме того,
получившие такой допуск лица обязаны в своей работе строго
следовать условиям так называемой проектной лицензии, кото-
рая выдается на конкретную научную программу. Ученые, рабо-
тающие в других странах, должны придерживаться соответст-
вующих национальных правил. И наконец, помимо всех офици-
альных регламентаций экспериментатор должен помнить, что
обеспечение нормальной жизнедеятельности подопытных живот?
ных — его наиважнейшая обязанность.
10.2.2. Требования, предъявляемые к животным
и оборудованию
10.2.2.1. Лабораторные животные
Для экспериментов по получению трансгенных мышей требу-
ется относительно большая популяция лабораторных животных.
Экспериментальная схема (рис. 10.2) предполагает использо-
вание различных линий на разных этапах работы. Рассмотрим
каждый из этих этапов в отдельности.
I. Животные для получения оплодотворенных яйцеклеток.
Оплодотворенные яйцеклетки выделяют из организма самки-
донора после ее спаривания с самцом-производителем. Выбор
линии мышей для этой стадии работы имеет принципиальное
значение. Бринстер и др. [15] сравнивали эффективность всей
процедуры получения трансгенных мышей, отбирая в качестве
исходного материала оплодотворенные яйцеклетки инбредных
мышей С57В1/6 и гибридные яйцеклетки C57B1/6XCBA/J. Бы-
ло показано, что многие контрольные параметры различаются
у мышей разных линий. В частности, это касается формирова-
ния яйцеклеток в организме донорной самки и жизнеспособности
яиц, перенесших инъекцию. В целом работа с гибридными яйце-
клетками оказалась в восемь раз эффективнее, чем аналогичные
манипуляции с инбредными яйцеклетками. Инбредные зиготы
следует использовать только в тех случаях, когда предъявляют-
ся специальные требования к генетической конструкции живот?
ных.
Микроинъекции в Б2-зиготы, полученные от скрещиваний
CBA/JXC57B1/6 или 10 гибридных самцов F1 с самками в сос?
тоянии суперовуляции, дали отличные результаты. Для подобной
работы необходимы следующие животные.
1. По меньшей мере 20 гибридных самцов-производителей
F1, которые содержатся индивидуально. Половозрелые самцы
316
Д. Мерфи, Дж. Хэнсон
считаются хорошими производителями в возрасте от восьми ме-
сяцев до года. К самцам индивидуально подсаживают самок че-
рез день. Поведение животных постоянно контролируется
(разд. 10.2.3.4); в случае низкой активности самца его заме-
няют.
2. Группа неполовозрелых (12—14 г) гибридных самок F1,
которые подвергаются суперовуляции, спаривается с гибридными
самцами F1 и используется затем в качестве доноров оплодот-
воренных яйцеклеток. Количество самок диктуется масштаба-
ми планируемого эксперимента. Десять самок дают по меньшей
мере 250 яиц, пригодных для микроинъекций.
3. Большая группа мышей линий CBA/J и С57В1/6 для вос-
производства гибридных донорных самок F1 (межлинейное скре-
щивание). Альтернативный вариант — часто более простой и де-
шевый— заказывать молодых гибридных самок F1 в питомнике.
II. Животные-реципиенты для трансплантации яйцеклеток.
Псевдобеременные реципиентные самки (12 ч ПК) выполняют
роль «суррогатной» матери, вынашивая трансплантированные
оплодотворенные яйцеклетки, в которые была инъецирована
экзогенная ДНК.
Чтобы получить псевдобеременных мышей, самок в стадии
эструса спаривают с самцами, подвергшимися вазэктомии (под-
робно операция вазэктомии описана в разд. 10.2.3.3). Самцы мо-
гут быть любой линии, однако наилучшие результаты дают сам-
цы линии Parkes благодаря своей высокой половой активности.
Оперированные самцы содержатся в индивидуальных клетках.
20—30 таких самцов спаривают через день с самками, достиг-
шими нужной стадии эстрального цикла (разд. 10.2.3.4. и
табл. 10.2); такая группа вполне обеспечивает получение пяти
псевдобеременных самок в день. Самки могут быть любой ли-
нии с хорошими материнскими характеристиками. Они должны
быть половозрелыми и весить не менее 19 г.
111. Мыши-кормилицы. Желательно иметь группу резервных
животных, способных при необходимости обеспечить вскармли-
вание детенышей, родившихся путем кесарева сечения
(разд. 10.2.3.8). Это необязательное требование, оно относится
к разряду рекомендаций
10.2.2.2. Необходимое оборудование
В табл. 10.1- представлено оборудование, требующееся для
проведения хирургических операций и препарационных про-
цедур, описанных в данной главе. Необходимые химические
реагенты упоминаются в тексте соответствующих разделов.
Получение трансгенных мышей
317
Таблица 10.1. Хирургическое1» и секционное оборудование и материалы
1. СО2-инкубатор для культур тканей (37°C, 5% СО2).
2. Весы для взвешивания животных.
3. 70%-ный этанол в пластиковой спринцовке.
4. Бумажная ткань.
5. Секционные ножницы (большие и малые).
6. Две пары заостренных часовых пинцетов.
7. Хирургическая шелковая нить (№ 5).
8. Изогнутые хирургические иглы (размер 10, трехгранные).
9. Спиртовая горелка.
10. Осветитель с волоконным световодом (например, Schott KL 1500).
11. Секционный стереомикроскоп (например, Nikon SMZ-10TD).
12. 9мм-скобки и аппликатор (Clay Adams2»).
13. Пинцет с тонкими тупыми губками.
14. Артериальный зажим (I’/s")-
15. Стерильные шприцы на 1 мл разового пользования.
16. Стерильные разовые иглы 0,1X16 мм.
17. 9см-чашки Петри.
18. 35мм-чашки для культур клеток.
19. Капилляры из твердого стекла с внешним диаметром 1,5 мм (BDH, но-
мер по каталогу 32124).
’) Хирургический и секционный инструментарий иам поставляет фирма Holborn
Surgical and Veterinary Instruments Ltd. (Великобритания).
2) Можно заказать у фирмы Arnold R. Horwell Ltd. (Великобритания).
Таблица 10.2. Идентификация стадий астрального цикла у самок мышей
Стадия астрального цикла
Состояние влагалища
А. Диэструс
Б. Проэструс
В. Эструс (овуляция)
Г. Метаэструс
Слабое раскрытие, ткани синюшные и влажные
Зияющее раскрытие, ткани розово-красные и
влажные. Складки на дорсальной и вентральной
поверхности
Зияющее раскрытие, ткани розовые и влажные.
Выраженные складки
Ткани бледные и сухие. Белесый налет
10.2.2.3. Пипетки для трансплантации яйцеклеток
Следует заранее приготовить микропипетки для транспланта-
ции яйцеклеток двух типов: общего назначения и специальные,
(рис. 10.3,5). Все пипетки изготавливают из прочных стеклян-
ных капилляров BDH. Рабочая система состоит из пипетки,
держателя, резиновой трубки и загубника. Методика изготовле-
ния пипеток описана в табл. 10.3.
318
Д. Мерфи, Дж. Хэнсон
Парафиновое
масло
Воздух
---М2
~ 150 мкм
100 мкм
Рис. 10.3. А. Микрогорелка, изготовленная из пастеровской пипетки. Б. Дер-
жатель пипеток — система предназначена для работы ртом: а — пипетка для
трансплантации яйцеклеток общего назначения; б — специальная пипетка для
пересадки яиц в яйцевод. В. Поддерживающая пипетка для микроинъекции.
Получение трансгенных мышей
319
Таблица 10.3. Изготовление трансплантационных пипеток
А. Пипетки общего назначения (рис. 10.3 Б, 1) .
1. Сделайте из пастеровской стеклянной пипетки микрогорелку, как показано
на рис. 10.3, А. Подключите газ; высота пламени должна быть около 1 см.
2. В пламени микрогорелки размягчите среднюю часть стеклянного капилля-
ра BDH. Выньте капилляр из пламени и резко потяните за концы. Тяни-
те до тех пор, пока две половинки не оторвутся друг от друга. Если пи-
петки получились слишком длинными (более 5 см) или слишком узкими
(менее 200 мкм), отчеркните лишнее алмазным резцом и аккуратно обло-
майте кончик.
3. Внутренний диаметр пипетки должен равняться приблизительно 300 мкм,
и кончик должен быть ровным без зазубрин по краям.
Б. Специальные трансплантационные пипетки
1. Эта стадия аналогична описанной выше для пипеток общего назначения,
за тем исключением, что внутренний диаметр пипетки должен составлять
около 150 мкм.
2. Отполируйте кончик на огне, осторожно касаясь им пламени микрогорел-
ки. Как только пламя приобретает желтую «натриевую» окраску, выни-
майте пипетку из огня. Проверьте чтобы отверстие пипетки имело доста-
точный размер для захвата яйцеклетки (>120 мкм).
10.2.3. Манипуляции с животными
10.2.3.1. Умерщвление мышей
Рекомендуемый способ умерщвления мышей — переламыва-
ние шейных позвонков. Эту операцию можно проделывать очень
быстро, причиняя животному минимум страданий. Возьмите
мышь за хвост и посадите ее на крышку клетки. Не( отпуская
хвоста, подождите пока животное, устремившись вперед, вытя-
нется так, что задние лапки почти зависнут в воздухе, а перед-
ние прочно вцепятся в прутики клетки. Крепко сожмите шею
мыши у основания черепа большим и указательным пальцами
или прижмите ее в этом месте шпателем и одновременно резко
потяните за хвост: шейные позвонки разрушаются в зоне при-
жима.
10.2.3.2. Анестезирование мышей
1. Приготовьте «100»%-ный анестезирующий раствор
(Avertin stock, смешав 10 г трибромэтилового спирта (Fluka,
номер по каталогу 90710) и 10 мл третичного амилового спир-
та (BDH, номер по каталогу 27214), Храните раствор при 4 °C
в защищенном от света месте.
2. Для получения рабочего раствора разбавьте концентри-
рованный раствор стерильной водой до 2,5%. Храните при
4 °C в темном месте.
320
Д. Мерфи, Дж. Хэнсон
Рис. 10.4. Показано, как держать мышь (Б) и как вводить в брюшинную по-
лость анестетик (А).
3. Рабочий раствор используют из расчета 15—17 мкл на
1 г живого веса. Точную дозу определяют всякий раз, когда
готовят 1ОО°/о-ный раствор, так как эффективность его действия
варьирует для разных партий исходных веществ.
4. Анестетик вводят внутрибрюшинно. Животное взвешива-
ют и определяют требуемую дозу. Набирают 2,5%-ный раствор
в стерильный шприц на 1 мл разового пользования, снабжен-
ный иглой 0,5X16 мм (следите за тем, чтобы в шприц не по-
пал воздух). Одной рукой берут мышь так, как показано на
рис. 10.4, Б. Другой рукой вводят иглу со шприцем в брюшную
полость (рис. 10.4,А). Старайтесь не затронуть желчный пу-
зырь и диафрагму. Вводят раствор и через короткое время вы-
таскивают иглу. О случайном подкожном введении анестетика
свидетельствует образовавшийся пузырь.
5. Полная анестезия длится 30—60 мин. Этого времени
вполне достаточно для выполнения хирургической операции.
После операции животное помещают в спокойное теплое место.
10.2.3.3. Вазэктомия самцов
Половозрелых самцов стерилизуют вазэктомией и спарива-
ют с половозрелыми самками, чтобы получить псевдоберемен-
ных мышей. Пригодны любые линии животных с высокой по-
ловой активностью (например, Parkes).
1. Операцию вазэктомии проводят на молодых, здоровых
половозрелых (оптимально — двухмесячных) самцах.
2. Животное анестезируют, как описано в разд. 10.2.3.2,
и кладут его на спину на крышку от 9 см-чашки Петри.
3. Протирают нижнюю часть брюшка 70%-ным спиртом и
выщипывают пинцетом шерсть в области разреза; разрез в
1,5 см планируется на уровне линии, соединяющей верхние
части бедер (рис. 10.5, А и Б).
Получение трансгенных мышей 321
4. Пинцетом приподнимают кожу на нижней части брюшка
и надрезают ее большими секционными ножницами (10.5, Л).
Чтобы предотвратить кровотечение, края разреза растягивают
(обратной стороной ножничных лезвий.
! 5. Разрезают брюшную стенку (рис. 10.5, 5) и так же рас-
тягивают разрез, чтобы остановить кровотечение.
6. У одного края разреза на брюшную стенку накладывают
один шов, оставляя запас шелковой нити (рис. 10.5, В).
7. С одной стороны вытягивают из разреза наружу жировой
сальник и вместе с ним семенник, эпидидимис и семявынося-
щий проток (рис. 10.5, Г).
8. Находят семявыносящий проток, расположенный под се-
менником, и освобождают его от поддерживающих связок
(рис. 10.5, Д).
9. Пинцетом загибают проток в виде петли. Накаляют до-
красна большой пинцет с тупыми губками и сжимают раска-
ленными концами эту петлю. Петля разрушается, и концы про-
тока прижигаются (рис. 10.5, Е).
10. Разделяют концы семявыносящего протока (рис. 10.5,
Ж).
11. Внутренние органы вкладывают обратно в брюшную по-
лость с помощью тупого пинцета (рис. 10.5,3).
12. Проделывают все аналогичные операции (пп. 7—11) со
вторым семявыносящим протоком.
13. Зашивают брюшную стенку, накладывая не менее двух
швов (рис. 10.5, Я).
14. На кожу накладывают скобки (рис. 10.5, Д').
15. Оперированных самцов содержат в индивидуальных
клетках. Через несколько недель после операции их можно
спаривать для получения псевдобеременных самок.
10.2.3.4. Скрещивание мышей
Для большинства целей, например для поддержания и вос-
производства популяций нормальных и трансгенных мышей,
животные должны спариваться и размножаться относительно
свободно, с минимальным посторонним вмешательством. Разве-
дение мышей контролируют только путем регламентированной
подсадки самок и самцов. Однако в ряде случаев необходимо
строго контролировать время овуляции, копуляции и оплодот-
ворения. Это делается для решения двух задач:
1. Получение оплодотворенных гибридных яйцеклеток F2
для микроинъекций.
2. Получение псевдобеременных гибридных самок F1, вы-
полняющих роль суррогатной матери для трансплантированных
эмбрионов.
21—1186
322
Д. Мерфи, Дж. Хэнсон
Получение трансгенных мышей
323-
Рис, 10.5. Операция вазэктомии на самце мыши (разд. 10.2.3.3.). Буквами,
обозначены: Т — testis; Ер—эпидидимис, V — vas deferens.
Здесь спаривание контролируют, либо используя внешние-
условия (естественное спаривание), либо применяя гонадотро-
пины, которые вызывают у самок гиперовуляцию. Естественное
спаривание половозрелых, но молодых (от 6 нед до 4 мес) гиб-
ридных самок F1 и гибридных самцов-производителей F1 в
принципе обеспечивает получение оплодотворенных яиц для
микроинъекций. Тем не менее предпочтение следует отдать спа-
риванию самцов F1 и неполовозрелых самок в состоянии су-
перовуляции. Суперовуляция позволяет резко увеличить коли-
чество оплодотворенных яйцеклеток в расчете на одну самку
по сравнению с естественным спариванием (в первом случае
около 30, во втором 10). Следовательно, гораздо меньшее чис-
ло особей может обеспечить необходимое количество яиц для
микроинъекций. Псевдобеременных реципиентных самок полу-
чают путем естественного спаривания половозрелых F1 гибрид-
ных самок и стерилизованных вазэктомией (или каким-либо
другим способом) самцов.
21*
:324
Д. Мерфи, Дж. Хэнсон
Ниже приводится режим контролируемого естественного
спаривания мышей.
1. Самцы-производители, стерилизованные самцы и полово-
зрелые самки содержатся при постоянном световом цикле: тем-
ное время с 17.00 до 06.00, светлое время с 06.00 до 17.00. Та-
кой режим освещенности обеспечивает овуляцию у самок каж-
дые 4 дня через 5 ч после наступления темноты, т. е. примерно
в полночь. Самцы копулируют с овулирующими самками в се-
редине темнового периода (примерно в 00.30), и оплодотворе-
ние происходит через 0,5—2 ч после копуляции. Оплодотворен-
ные яйца выделяют примерно в полдень на следующий день;
они остаются пригодными для микроинъекций до полуночи.
Псевдобеременные реципиентные мыши пригодны для транс-
плантации донорных оплодотворенных яйцеклеток в течение
всего дня после их спаривания с бесплодными самцами.
2. За день до того, как понадобятся нужные самки, в пери-
од между 16.00 и 18.00 подсаживают одну самку в клетку к
самцу (производителю или стерильному). Если имеется доста-
точно большая популяция самок, их можно отбирать наугад,
зная, что в среднем каждая четвертая находится в данный мо-
мент в фазе экструса и, следовательно, готова к спариванию.
'Однако предпочтительно отбирать самок целенаправленно, оп-
ределяя их эстральный статус по состоянию влагалища
(табл. 10.2). От 50 до 80% самок, относимых к фазе эструса
на основании внешнего осмотра, беременеют.
3. Утром исследуют самок на наличие копуляционной проб-
ки, которая служит свидетельством успешного спаривания. Это
белая масса, состоящая из коагулированного белка и закрыва-
ющая вход во влагалище. В большинстве случаев пробка легко
различима, но иногда она расположена достаточно глубоко,
поэтому мышей следует осматривать тщательно, используя
гладкий затупленный зонд. К полудню копуляционная пробка
обычно растворяется.
Процедура получения и спаривания самок, подвергшихся
суперовуляции, приведена в табл. 10.4
10.2.3.5. Культивирование оплодотворенных мышиных яйцеклеток
Эксперименты по получению трансгенных мышей предпола-
гают необходимость культивирования оплодотворенных мыши-
ных яйцеклеток вне их естественной среды в течение 3—36 ч.
Оплодотворенные яйцеклетки выделяют через 12 ч после ко-
пуляции и затем в какой-то момент в течение следующих 12 ч
инъецируют в них чужеродную ДНК. После этого яйцеклетки
должны вернуться в свою естественную среду, которую в дан-
ном случае представляет яйцевод псевдобеременной суррогат-
Получение трансгенных мышей
325
'.Таблица 10.4. Получение и спаривание подвергшихся гиперовуляции самок
1. Приготовьте концентрированные растворы гонадотропинов: фолликуло-
стимулирующего гормона (ФСГ, FSH) и хорионического гонадотропина
человека ХГТ, hCG)!>.
2. Примерно в 12.00 введите 5—10 неполовозрелым гибридным самкам F1
(12,5—14 г; 4—5 нед) до 5 м. ед. ФСГ (т. е. ио 100 мкл концентрирован-
ного 50 мед./мл раствора). Животное удерживают одной рукой, как по-
казано на рис. 10.4, Б, и инъецируют гормон внутрибрюшинно (рис.
10.4, Л), стараясь не задеть диафрагму н желчный пузырь.
3. Через 46—48 ч каждой мыши инъецируйте внутрибрюшинно 5 м. ед. ХГТ
(100 мкл раствора 50 мед/мл) н немедленно подсадите к самцу произ-
водителю.
4. На следующее утро проверьте наличие копуляционной пробки (разд.
(10.2.3.4). Обычно беременеет 80—100% самок.
’) FSH (торговое название Folligon) н hCG (торговое название Chorulon) поставля-
ет Interved Laboratories Ltd. (Великобритания). Оба гормона растворяют в стерильной
воде или 0,9% NaCl, раствор разливают на 1 мл-алнквоты и хранят при —70 °C.
ной матери. Трансплантацию яйцеклеток (разд. 10.2.3.7) мож-
но провести либо вскоре после инъекции (на одноклеточной
стадии), либо после ночного культивирования (на двухклеточ-
ной стадии) (рис. 10.10, В). В любом случае яйцеклетки транс-
плантируют псевдобеременным самкам 12 ч ПК.
Яйцеклетки на одно- и двухклеточных стадиях поддержива-
ют в М16-микрокапельной культуре в СО2-инкубаторе для
культивирования тканей (37 °C, 5% СО2). Микрокапельная
культура приготавливается так: маленькие аликвоты среды
М16 (20—40 мкл) наносят в определенном порядке в культу-
ральную 35мм-чашку и покрывают слоем жидкого парафиново-
го масла, чтобы предотвратить выпаривание. Все манипуляции
с яйцеклетками вне инкубатора — их выделение или микро-
инъекции — проводят в среде М2, содержащей HEPES-буфер.
Приготовление сред М2 и М16 подробно описано в табл. 10.5.
В состав обеих сред входит бычий сывороточный альбумин
(БСА), который снижает клейкость яйцеклеток, обеспечивает
их микропотребности и связывает токсические вещества. Одна-
ко некоторые партии БСА оказываются непригодными для
культивирования яйцеклеток; поэтому каждый раз нёЪбходи-
мо проверять новые препараты на их способность поддержи-
вать жизнеспособность яйцеклеток в течение нескольких деле-
ний и только после этого использовать их для культивирова-
ния яйцеклеток, прошедших микроинъекцию.
10.2.3.6. Выделение оплодотворенных одноклеточных яйцеклеток
1. Не позднее, чем за 1 ч до намеченной процедуры, подго-
товьте две культуральные 35 мм-чашки со средой М16 и две
такие же чашки с М16-микрокапельными культурами. Помес-
Таблица 10.5. Приготовление культуральных сред М2 и М16
Все среды приготавливают в стерильных разовых пластиковых флаконах, ис-
пользуя стерильные разовые пипетки. Вымытая стеклянная посуда может
оказаться загрязненной детергентами, токсичными для яйцеклеток.
А. Концентрированные растворы компонентов
1. ЮХА на 100 мл 5,534 г NaCl
0,356 г КС1
0,162 г КН2РО4
0,293 г MgSO4X7H2O
2,61 г лактата натрия
1,0 г глюкозы
0,06 г пенициллина
0,05 г стрептомицина
Взвесьте нужное количество каждого компонента (кроме лактата натрия)
в мерной колбе на 100 мл. Лактат натрия взвесьте в стаканчике и перенеси-
те в мерную колбу. Обмойте стаканчик бидистиллятом, слейте в ту же кол-
бу н доведите объем до 100 мл.
2. 10ХВ на 100 мл 2,101 г NAHCO3
0,01 г фенолового красного
3. ЮОхС на 10 мл 0,036 пирувата натрия
4. 100XD на 10 мл 0,252 г СаС12Х2Н2О
Взвесьте указанные вещества в мерных колбах на 10 и 100 мл и доведите
объем до 100 мл бидистнллированной водой
5. ЮХЕ на 100 мл 5,958 г HEPES
0,252 фенолового красного
Взвесьте компоненты в стаканчике и растворите в 50 мл бидистиллята.
Доведите pH до 7,4, добавив 0,2 М NaOH, перенесите раствор в 100 мл
мерную колбу и доведите объем до 100 мл, обмывая стаканчик н перенося
смыв в колбу. Профильтруйте все концентрированные растворы через
мембранные фильтры Millipore (0,45 мкм) в стерильные пластиковые про-
бирки и храните при —20 °C. Рабочие растворы можно хранить при 4 °C:
A, D и Е — в течение 3 мес, В и С — в течение только 2 нед.
Б. П риготовление сред М2 и М16 из концентрированных растворов
Концентрированный раствор М2 М16
ЮХА 10 мл 10 мл
ЮХВ 1,6 мл 10 мл
юохс 1,0 мл 1,0 мл
100XD 1,0 мл 1,0 мл
ЮХЕ 8,4 мл —
Бнднстиллят 78 мл 78 мл
Бычий сывороточный 400 мг 400 мг
альбумин (БСА) Общий объем 100 мл 100 мл
1. Отмерьте в колбе нужное количество бидистиллята.
2. Внесите указанное количество концентрированных растворов в воду, вся-
кий раз тщательно промывая пипетку (несколько раз набирая и выпуская
жидкость из колбы).
3. Добавьте БСА и аккуратно перемешайте до полного растворения.
4. Пропустите через фильтр Millipore 0,45 мкм, используя большой стериль-
ный разовый шприц. Разлейте среды в стерильные флаконы.
5. Храните при 4 °C. Каждую неделю следует готовить свежую среду.
326
\ Получение трансгенных мышей 327
-----------------------------—22---------------------------
тите их для уравновешивания в СО2-инкубатор (37 °C, 5%
СО2). Подготовьте также четыре культуральные 35-мм чашки
со средой М2 и оставьте при комнатной температуре.
2. Забейте донорную самку с копуляционной пробкой, как
описано в разд. 10.2.3.1.
3. Положите животное на спину и обмойте брюшко 70%-
ным этанолом из спринцовки.
4. Разрежьте кожу в нижней части живота, вскройте брюш-
ную полость и подцепите одно плечо репродуктивного тракта,
как показано на рис. 10.6, А Найдите свернутый яйцевод, кото-
рый находится между яичником и маткой.
5. Захватив матку тонким пинцетом, вытяните репродуктив-
ный тракт наружу. При помощи тонких ножниц проткните и
отделите от яйцевода мембрану (мезометрий), соединяющую
репродуктивный тракт с брюшной стенкой.
6. Отрежьте яйцевод от яичника (рис. 10.6, Б).
7. Осторожно поддерживая яйцевод заостренным часовым
пинцетом, отрежьте яйцевод от матки (рис. 10.6, В).
8. Положите яйцевод в одну из приготовленных М2-ча-
шек.
9. Отпрепарируйте яйцевод из другого рога репродуктивно-
го тракта и поместите в ту же М2-чашку.
10. Осмотрите яйцеводы в секционном стереомикроскопе при
10—20Хувеличении. Найдите набухшую ампулу, содержащую
кумулусную массу (оплодотворенные яйцеклетки, окруженные
кумулусными клетками; рис. 10,6, Г).
11. Используя два заостроенных пинцета, проткните ампу-
лу, чтобы эта масса вытекла наружу (рис. 10,6, Д). В некото-
рых случаях яйцеклетки из ампулы приходится вытаскивать
при помощи пинцета. Пустой яйцевод выньте из культуральной
чашки. Те же операции проделайте с другим яйцеводом.
12. С помощью микропипетки для переноса яйцеклеток об-
щего назначения (разд 10.2.2.3) соберите кумулусную массу
и все индивидуальные яйцеклетки и диспергируйте в свежей
чашке со средой М2.
13. Смешайте кумулусную массу с 50 мкл раствора гиалу-
ронидазы (Sigma, номер по каталогу Н3884) в среде М2. Об-
работка гиалуронидазой в течение нескольких минут позволяет
отделить яйца от кумулусных клеток. Пипетирование яйцекле-
ток вверх-вниз ускоряет процесс.
14. Соберите яйцеклетки и промойте дважды в свежей сре-
де М2, чтобы удалить следы фермента. При отмывках поста-
райтесь удалить все кумулусные клетки.
15. Промойте яйцеклетки дважды в приготовленных
М16-чашках и затем перенесите их в микрокапельную куль-
туру.
Рис. 10.6. Выделение оплодотворенных яйцеклеток (разд. 10.2.3.6). Буквами
обозначены: Г — грудина; К — кишечник; Я — яичник; Яв — яйцевод; М —
матка; Як — яйцеклетки; Ар — ампула.
Получение трансгенных мышей 329
16. Инкубируйте при 37 °C в атмосфере 5%' СО2 вплоть до
микроинъекции.
10.2.3.7. Трансплантация яйцеклеток
1. Анестезируйте псевдобеременную реципиентную мышь
(12 ч пк), как описано в разд. 10.2.3.2.
2. Положите животное на живот на крышку от 9 см-чашки
Петри. Обмойте спину 70%-ным этанолом.
3. Выщипайте тонким пинцетом шерсть в области намечае-
мого разреза: поперечный надрез делают на расстоянии 1 см
от позвоночника на уровне последнего ребра (рис. 10.7, А).
4. Большими острыми ножницами сделайте проникающий
разрез (в 1 см) стенки тела. Края разреза растяните, чтобы
остановить кровотечение.
5. Найдите в разрезе яичник (он имеет оранжевую окраску).
Тонкими острыми ножницами сделайте дополнительный 3—
5 мм-разрез на расстоянии нескольких миллиметров от яични-
ка (рис. 10.7, А). Растяните его края, чтобы уменьшить крово-
течение.
6. Наложите один шов на один из краев разреза и оставьте
запас шелковой нити (рис. 10.7, Б).
7. Вытяните наружу сальник, используя тонкий пинцет с
тупыми губками. Яичник, яйцевод и матка прикреплены к саль-
нику и вытягиваются вместе с ним (рис. 10.7, В).
8. Прикрепите к сальнику артериальный зажим так, чтобы
не повредить яичник. При помощи этого зажима репродуктив-
ный тракт закрепляется на спине самки таким образом, чтобы
витки яичника оказались сверху (рис. 10.7, Г и 10.8, А).
9. Перенесите животное вместе с крышкой, на которой оно
лежит, на столик секционного стереомикроскопа, снабженного
осветителем с волоконным световодом. Осмотрите яйцевод при
увеличении 10—20Х (рис. 10.8, А).
10. Входное отверстие яйцевода — воронка — располагается
в полости под яичником и прикрыто витками яйцевода. Акку-
ратно сместите витки яйцевода вниз, чтобы освободить полость.
Иногда воронку можно разглядеть за прозрачной мембраной,
покрывающей яичник, яйцевод и полость. Выберите участок
мембраны (желательно над воронкой), свободной от капилля-
ров, и проткните ее в этом месте пинцетом. Теперь можно
очень осторожно вытянуть воронку из полости, пользуясь ма-
леньким заостренным пинцетом (рис. 10.8, А).
11. Приготовьте яйцеклетки для трансплантации. Отберите
не более 30 яйцеклеток, переживших микроинъекцию, из мик-
рокапельной культуры и промойте их в среде 512. Введите
яйцеклетки в специальную микропипетку для трансплантации
Рис. 10.7. Первый этап операции по пересадке оплодотворенных яйцеклеток
в яйцевод псевдобеременной самки: подготовка репродуктивного тракта.
Рис. 10.8. Микрохирургическое введение материала в яйцевод (разд. 10.2.3.7).
332 Д, Мерфи, Дж. Хэнсон
яйцеклеток (разд. 10.2.2.3) и рис. 10.3, Б, б). Сначала заполните
носик пипетки до изгиба жидким парафиновым маслом: вязкое
масло облегчает микроманипулирование с яйцеклетками. Затем
захватите маленький пузырек воздуха, потом — каплю среды
М2, потом — еще пузырек воздуха. После этого захватывают
яйцеклетки в капле М2. Яйцеклетки следует засасывать в пи-
петку в минимальном объеме среды так, чтобы они выстроились
в ряд. Затем захватывают еще пузырек воздуха и каплю М2.
Рис. 10.3.5, б иллюстрирует сказанное.
12. Вернемся к мыши. Тампоном из бумажной ткани удали-
те подтеки биологических жидкостей и кровяные сгустки во-
круг яйцевода. Рис. 10.8, А—Г представляет фотографии раз-
ных стадий демонстрационной операции, проводившейся на
трупном материале; поэтому ткани выглядят неестественно су-
хими и безжизненными. На рис. 10.8, Д представлена фотогра-
фия настоящей операции на живой мыши. Обратите внимание,
что яйцевод покрыт слоем жидкости и крови, что затрудняет
работу.
13. При помощи тонкого пинцета захватите воронку яйцево-
да и вытяните ее наружу из-под витков яйцевода через разрыв
мембраны, так чтобы отверстие яйцевода оказалось доступным
для специальной трансплантационной микропипетки (рис. 10.8,
А).
14. Введите кончик пипетки в отверстие воронки. Это отвер-
стие нельзя разглядеть, поэтому локализуют его «на ощупь»,
осторожно тыкая пипеткой; при попадании в отверстие яйцево-
да ткань «поддается» и кончик микропипетки погружается в
воронку яйцевода (рис. 10.8,5).
15. Выдуйте содержимое пипетки. Этот процесс контролиру-
ют по появлению пузырьков воздуха в ампуле. После появле-
ния третьего пузырька, можно быть уверенным, что яйцеклетки
попали в ампулу. На фотографиях 10.8, Б, В и Г акцент на
переносе содержимого пипетки в ампулу сделан с помощью
специального красителя.
16. Выньте пипетку из яйцевода. Снимите артериальный за-
жим и, захватив пинцетом с тупыми губками сальник, введите
обратно в полость тела весь репродуктивный тракт. Зашейте
разрез одним или двумя швами; на кожу наложите скобки.
17. Если имеется достаточный запас «уколотых» яйцеклеток,
повторяют операцию на другой половине тела, используя для
трансплантации второе плечо репродуктивного тракта.
10.2.3.8. Кесарево сечение и вскармливание
Довольно часто лишь небольшое число «уколотых» яйцекле-
ток продолжает нормально развиваться в организме суррогат-
ной матери. В этих условиях эмбрионы получают избыточное
Получение трансгенных мышей
333;
Таблица 10.6. Операция кесарева сечения
1. Забейте мышь, как описано в разд. 10.2.3.1.
2. Обмойте брюшко 70% ным этанолом, разрежьте н раздвиньте кожу в ниж-
ней его части, вскройте брюшную полость и отделите матку с эмбрионами.
3. Вскройте матку, выньте эмбрионов, аккуратно освободив нх от материн-
ских тканей. Отрежьте пуповину.
4. Оботрите хлопчатобумажной тканью нос н рот каждого детеныша н ак-
куратно сдавите грудную клетку пинцетом, чтобы стимулировать дыхание.
5. Положите детенышей на влажную тканевую подстилку в теплое место.
Таблица 10.7. Вскармливание
1. Отнимите ненадолго мышь-кормнлнцу от собственных детенышей.
2. Быстро перемешайте приемышей и естественный приплод.
3. Попытайтесь заставить мышь помочиться на детенышей. Мочеиспускание
можно обычно вызвать, взяв мышь так, как показано на рнс. 10.4, Б.
4. Верните самку-кормнлицу в клетку н оставьте в покое на 1—2 ч.
5. Удалите часть естественного приплода, чтобы общее количество детенышей
не превышало 12. Одна мышь не может выкормить большее потомство.
питание и вырастают существенно более крупными, чем эмбри-
оны нормального помета. Если эмбрионы оказываются слиш-
ком большими, чтобы пройти родовой канал, беременность про-
должается больше положенных 19—20 дней; на 21—22-й день
появляется опасность того, что детеныши погибнут в матке.
Единственный выход из сложившейся ситуации — кесарево се-
чение и вскармливание детенышей самкой-кормилицей. Вскарм-
ливание необходимо также в случае неожиданной гибели мате-
ри нормально родившихся детенышей. Мыши-кормилицы долж-
ны иметь собственных детенышей примерно того же возраста^
что и приемыши, и принадлежать к линии с хорошими мате-
ринскими характеристиками. Самки линии Parkes отличные
кандидаты на роль кормилиц не только благодаря своему за-
ботливому характеру, но еще и потому, что их собственные бе-
лые детеныши легко отличимы от черных или «агути»
СВА/С57В1 гибридных приемышей. Операции кесарева сечения
и вскармливания описаны соответственно в табл 10.6 и 10.7.
10.3. Микроинъекции
10.3.1. Оборудование для микроинъекций
В табл. 10.8 перечислены оборудование и инструменты, не-
обходимые для проведения микроинъекций в яйцеклетки мыши.
На рис. 10.9 изображен аппарат для микроинъекций Nikon —
Leitz. Ряд технических вопросов, касающихся оснащения экс-
периментов обсуждается ниже.
334
Д. Мерфи, Дж. Хэнсон
Таблица 10.8. Оборудование для микроинъекций
1. Инвертированный микроскоп (например, Nicon-Diaphot; см. разд. 10.3.1.1).
2. Правый микроманипулятор Leitz.
3. Левый микроманипулятор Leitz.
4. Два держателя инструмента Leitz.
5. Инструментальные трубки Leitz.
6. Стеклянные капилляры Leitz с внешним диаметром 1 мм (номер по
каталогу 520119).
7. Опорная плита. .
8. Микрокузница (например, De Fondbrune1’)
9. Аппарат для вытягивания микропипеток (например, Korf Model 720* 1 2) 3 4 5 6 7)
10. Fluorinert electronic liquid (3M company, FC77)
11. Легкое парафиновое масло.
12, Капилляры для инъекционных пипеток (Clark Electromedical Instr., номер
по каталогу GC 100TF— 152))
13. Микрометрический шприц Agla MS013>
14. 5см-иглы для шприца № 26.
15. Стеклянные пластины с лункой.
16. 50мл-стеклянный шприц со стеклянным поршнем.
17. Соединительные шланги Tygon, внутренний диаметр 3/32», наружный
диаметр 5/32».
18. Стойка с зажимом.
19. Алмазный резак.
20. Шприцы на 1 мл разового пользования.
*) Поставляет Microinstruments (Oxford) Ltd. (Англия).
2) Поставляет Clark Electromedical Instruments (Англия).
3) Поставляет Wellcome Reagents Ltd. (Англия).
10.3.1.1. Микроскоп
Инвертированный микроскоп, используемый в данных экс-
периментах, должен иметь следующие характеристики.
1. Выпрямляющую изображение оптику.
2. Фиксированный столик (т. е. фокусировка достигается
только перемещением объектива).
3. Конденсор с большим рабочим расстоянием.
4. 10 X окуляры.
5. 4Х объектив для работы при небольшом увеличении.
6. 40X объектив для микроинъекции.
7. Подходящую оптическую систему.
Наилучшей оптической системой для визуализации внутрен-
них структур одноклеточного яйца является система дифферен-
циального контраста Номарского (DIC). Однако эта оптика
•стоит очень дорого, и к тому же она требует, чтобы инъекцион-
ная камера обязательно была стеклянной. Система модуляци-
онного контраста Хоффмана не позволяет получить столь же
высокое разрешение, как DIC-оптика, но гораздо дешевле и
совместима с пластиковыми инъекционными камерами. Если
.7
Получение трансгенных мышей
335-
Рис. 10.9. Общий вид микроинъекционного комплекса Nikon — Leitz. 1 — мик-
рометрический шприц Agla, соединенный пластиковой трубкой, заполненной
жидким парафиновым маслом с поддерживающей микропипеткой; 2 — левый
микроманипулятор Leitz, управляющий поддерживающей микропипеткой; 3 —
инвертированный микроскоп Nicon Diaphot; 4 — правый микроманипулятор-
Leitz, управляющий инъекционной микропипеткой; 5 — видеомонитор (в комп-
лект не входит); 6 — опорная плита; 7— стеклянный шприц на 50 мл, соеди-
ненный заполненной воздухом пластиковой трубкой с инъекционной микро-
пипеткой; 8 — стеклянная пластина с лункой для инъекционной камеры; 9 —
правая инструментальная трубка Leitz с закрепленной в ней инъекционной
пипеткой (левая инструментальная трубка не видна за окулярами микроско-
па); 10 — фотокамера (в комплект не входит).
системы Номарского и Хоффмана недоступны, можно исполь-
зовать оптику для микроскопии в светлом поле, которая в дан-
ном случае предпочтительна по сравнению с фазово-контраст-
ной микроскопией.
10.3.1.2. Микроманипуляторы
Микроинъекция предполагает микроманипулирование с оп-
лодотворенными яйцеклетками при помощи двух микропипеток:
поддерживающей пипетки, которая удерживает яйцеклетку при
укалывании, и инъекционной пипетки, которая прокалывает
яйцеклетку и вводит в нее раствор ДНК. Движениями обеих
пипеток управляют при помощи микроманипуляторов. Наи-
336
Д. Мерфн, Дж. Хэнсон
большее распространение получили микроманипуляторы фирмы
Leitz.
Микроманипулятор Leitz прост в обращении и снабжен
удобной рычажной системой контроля малых горизонтальных
перемещений в двух плоскостях. Два микроманипулятора мон-
тируются по обе стороны микроскопа (рис. 10.9). Левый управ-
ляет поддерживающей пипеткой, а правый — инъекционной
пипеткой. Микроманипуляторы и микроскоп устанавливают на
опорной плите так, чтобы эти три элемента имели фиксирован-
ное расположение относительно друг друга. Опорная плита
призвана также уменьшать вибрации. Описание опорной пли-
ты, адаптированной к системам Nicon и Leitz, приведено в от-
дельной книге [16].
10.3.1.3. Инъекционная камера
Стеклянные инъекционные камеры совместимы с инвертиро-
ванными микроскопами любой оптической системы. Силикони-
зируйте стеклянную пластину с лункой, погрузив ее в 3%-ный
раствор дихлорметилсилана в хлороформе. Промойте стекло
большим количеством воды и затем еще раз вымойте с каким-
нибудь детергентом. Обмойте стекло этанолом и оботрите насу-
хо хлопчатой тканью. Нанесите в центр лунки плоскую каплю
среды М2 (в поперечнике ее размер не должен превышать
1 см). Сверху наслоите легкое парафиновое масло, чтобы пред-
отвратить высыхание. Установите инъекционную камеру на
столике микроскопа и, используя объектив 4Х, сфокусируйте
оптику на дне капли.
10.3.1.4. Изготовление поддерживающих микропипеток
Поддерживающая пипетка изображена на рис. 10.3, В. Та-
кая пипетка служит обычно на протяжении всей серии экспери-
ментов. Изредка ее заменяют на новую, так как постепенно
она загрязняется и ее просвет суживается. Заранее можно под-
готовить большое количество поддерживающих пипеток и хра-
нить их в чашке Петри.
1. Нагрейте капилляр Leitz (твердое стекло, внешний диа-
метр 1 мм) в пламени микрогорелки. Чтобы стекло равномерно
размягчилось, капилляр следует медленно вращать. Затем ка-
пилляр растяните за концы, одновременно вынув его из пла-
мени. Постарайтесь получить прямолинейно вытянутые капил-
ляры.
2. Алмазным резцом отчеркните оттянутый конец на рас-
стоянии примерно 2 см от начала сужения и отломите лишнее
стекло. Осмотрите капилляр в секционном микроскопе. Кончик
Получение трансгенных мышей 337
должен быть совершенно ровным без зазубрин, и сам капил-
ляр должен быть абсолютно прямым. Внешний диаметр оття-
нутого конца должен укладываться в интервале 80—150 мкм.
Капилляры, не удовлетворяющие перечисленным требованиям,
выбраковываются.
3. Поместите оттянутый капилляр в микрокузницу (Defond-
brune). Сфокусируйте объектив 4Х на кончике капилляра и
подведите к нему почти вплотную, но без касания филамент
микрокузницы. Нагрейте филамент и следите за постепенным
оплавлением кончика капилляра. Когда его внутренний диа-
метр достигнет 10—15 мкм, отключите нагрев.
4. Аккуратно переведите капилляр в горизонтальное поло-
жение. Подведите филамент снизу на уровне 1—2 мм от конца
капилляра (филамент подводят почти вплотную, но без каса-
ния). Включите нагрев филамента. Когда кончик капилляра
изогнется и отклонится от горизонтали на 15°, отключите на-
грев. Контролировать этот процесс можно невооруженным гла-
зом или под небольшим увеличением. Осторожно выньте изго-
товленную микропипетку из аппарата.
5. Микропипетка готова для установки в микроманипуля-
тор. Соедините инструментальную трубку Leitz с микрометри-
ческим шприцом Agla отрезком шланга Tygon длиной 1 м. За-
полните систему легким парафиновым маслом; удостоверьтесь
в отсутствии пузырьков воздуха.
6. Заполните микропипетку жидкостью Fluorinert electronic
liquid при помощи разового шприца на 1 мл с 5 см-иглой
№ 26. Вставьте пипетку в заполненную маслом инструменталь-
ную трубку и затяните стопорное кольцо. Аккуратно закрепите
инструментальную трубку в левом микроманипуляторе. Шприц
Agla отрегулируйте так, чтобы жидкость перестала вытекать
из пипетки, но в то же время в нее не подсасывался воздух.
7. При помощи микроманипулятора установите микропипет-
ку над инъекционной камерой так, чтобы ее кончик располагал-
ся горизонтально. При использовании объектива 4Х в поле
зрения микроскопа должна появиться тень микропипетки. Опус-
тите пипетку в инъекционную камеру. Удостоверьтесь, что в
пределах поля зрения пипетка свободно перемещается в гори-
зонтальной плоскости и не задевает дно инъекционной камеры.
8. Отрегулируйте шприц Agla так, чтобы мениск между
Fluorinert electronic liquid и средой М2 располагался поблизос-
ти от конца микропипетки.
10.3.1.5. Подготовка инъекционной микропипетки
Инъекционную пипетку изготавливают из тонкостенной стек-
лянной трубки с внешним диаметром 1 мм. Предпочтительны
капилляры с внутренним рубчиком, поскольку изготовленные
22—1186
338
Д. Мерфи, Дж. Хэнсон
из них инъекционные пипетки легко заполняются раствором
ДНК с тыльного конца за счет капиллярных сил. Пипетки вы-
тягивают на специальном аппарате. Подходящую температуру
нагрева и растягивающее усилие подбирают опытным путем.
Хорошей считается пипетка, выходное отверстие которой име-
ет диаметр 1 мкм. Меньшее отверстие быстро закупорится,
а более широкий кончик сильно травмирует яйцо. Инъекцион-
ные пипетки готовят по мере надобности.
Работают с микроинъекционной пипеткой с помощью пра-
вого микроманипулятора. Инструментальную трубку Leitz со-
единяют отрезком шланга Tygon длиной приблизительно 1 м
с 50 мм-шприцем со стеклянным поршнем (поршень можно сма-
зать топким слоем парафинового масла). Система заполнена
воздухом. В инъекционную пипетку вводят раствор ДНК с
тыльной стороны, используя капиллярный эффект. Пипетка
считается заполненной, когда жидкость покажется на рабочем
конце. Следите за тем, чтобы в раствор ДНК не попали загряз-
нения, например тальк с перчаток или ферменты с незащищен-
ных рук. Установите пипетку в инструментальную трубку и
закрепите последнюю в держателе микроманипулятора. Ис-
пользуя микромапипулятор, установите инъекционную пипетку
так, чтобы ее кончик располагался над инъекционной камерой.
Затем осторожно опустите кончик пипетки в каплю среды М2,
контролируя перемещение пипетки под микроскопом (исполь-
зуйте объектив 4Х). Удостоверьтесь, что пипетка может сво-
бодно перемещаться в вертикальной плоскости.
10.3.2. Микроинъекция в оплодотворенные одноклеточные
яйцеклетки мыши
1) Смонтируйте и подготовьте приборы для микроинъекции,
как описано в разд. 10.3.1. При помощи трансплантационной
пипетки общего назначения отберите примерно 20 оплодотво-
ренных яйцеклеток из М16-микрокапелыгой культуры (поддер-
живаемой при 37°C). Промойте дважды яйцеклетки водой,
введите их в минимальном объеме жидкости в пипетку и за-
тем выдуйте в инъекционную камеру. Контролируйте поведение
яйцеклеток в инъекционной камере визуально, используя объ-
ектив 4Х. Постарайтесь собрать яйцеклетки группой под инъ-
екционной и поддерживающей пипетками. Следите за тем, что-
бы вместе с яйцами в инъекционную камеру не попали пузырь-
ки воздуха — это может полностью ее нарушить: не только
яйцеклетки потеряются, по и всю инъекционную камеру при-
дется готовить заново.
2) Перемещая обе пипетки в вертикальном направлении,
установите их в одной плоскости с яйцеклетками так, чтобы
Получение трансгенных мышей 339
было удобно ими манипулировать. Подведите кончик поддер-
живающей пипетки вплотную к одному из яиц и, создав не-
большое разрежение с помощью шприца Agla, зафиксируйте
его на конце пипетки.
3) Переместите яйцо в центр поля зрения. Подключите
объектив 40XDIC и сфокусируйте его на яйцеклетке. Переме-
щая фокальную плоскость вверх-вниз, локализуйте больший из
двух пронуклеусов (обычно это мужской пронуклеус). Манипу-
лируя шприцем Agla, поддерживающей пипеткой и инъекцион-
ной пипеткой, поверните яйцеклетки так, чтобы большой про-
нуклеус оказался по центру пипетки, но ближе к наружной
стороне яйцеклетки (рис. 10.10,А). Немного повысьте разре-
жение с помощью микрометрического шприца, чтобы яйцеклет-
ка плотнее удерживалась микропипеткой. При этом оболочка
яйцеклетки может деформироваться, но не допускайте, чтобы
она сама вытягивалась в пипетку.
4) Сфокусируйте объектив на пронуклеусе. Подведите кон-
чик инъекционной пипетки к оболочке яйцеклетки. Перемещая
при помощи микроманипулятора инъекционную пипетку в
вертикальной плоскости, установите ее на одном уровне (т. е. в
той же фокальной плоскости) с пронуклеусом.
5) Уколите яйцеклетку, оболочка легко прокалывается инъ-
екционной иглой. Когда игла окажется внутри пронуклеуса,
нажмите на поршень инъекционного шприца. Далее ожидается
одно из трех возможных последствий: 1) Пронуклеус начинает
набухать. Это свидетельствует об успешной инъекции; продол-
жайте двигать поршень, пока пронуклеус не увеличится в
объеме примерно вдвое (рис. 10.10,5), затем быстро выньте
инъекционную пипетку. 2) Небольшой прозрачный пузырек по-
является у конца инъекционной пипетки и оболочка яйцеклет-
ки набухает. Следовательно, осталась непроколотой мембрана
яйцеклетки, которая очень эластична. Чтобы проткнуть мем-
брану, продолжайте перемещать иглу внутрь яйцеклетки до
его противоположной стороны, затем подвиньте ее назад так,
чтобы кончик оказался снова внутри пронуклеуса, и продол-
жайте вдвигать поршень шприца. 3) Ничего не происходит.
Это, вероятно, следствие закупорки иглы. Замените иглу и, ес-
ли это необходимо, раствор ДНК.
6) Иногда после укола видно, как в перивителлиповое про-
странство начинают вытекать цитоплазматические гранулы.
Это симптом лизиса яйцеклетки. Если такой эффект повторя-
ется с другим яйцом, замените иглу. Инъекционную иглу-пи-
петку следует заменять также в случае ее загрязнения или за-
купорки.
7) После инъекции подключите объектив 4Х и отделите
«уколотые» яйцеклетки от интактных. Уколотые яйцеклетки це-
22*
Рис. 10.10. А. Микроинъекционная камера под большим увеличением (Х400,
система дифференциального интерференционного контраста — DIC). Слева
видна поддерживающая микропипетка, «присосавшаяся» к яйцевой оболочке.
Инъекционная пипетка просматривается справа. В центральной части яйца хо-
рошо виден большой мужской пронуклеус с многочисленными ядрышками.
Б. Микроинъекция и как следствие — набухание мужского пронуклеуса.
В. Двухклеточный зародыш (DIC, Х400).
Получение трансгенных мышей
341
лесообразно разделить на две группы — пережившие инъекцию
и погибшие.
8) Яйцеклетки следует держать в среде М2 не более
15 мин. Опытный экспериментатор способен за это время уко-
лоть от 15 до 40 яиц. Как только все яйцеклетки данной пар-
тии уколоты, жизнеспособные эмбрионы изымаются из камеры,
их дважды промывают в среде М16 и снова помещают в мик-
рокапельную культуру Ml6, где они поддерживаются вплоть
до трансплантации в яйцевод псевдобеременной самки
(разд. 10.2.3.7). Погибшие яйцеклетки удаляют. Яйцеклетки
можно трансплантировать псевдобеременной реципиентной
самки либо в тот же день, либо после ночного культивирова-
ния — к этому времени эмбрионы оказываются уже двухкле-
точными (рис. 10.10, В).
10.4. Манипулирование с ДНК
10.4.1. ДНК для микроинъекции
Одно из преимуществ описываемой процедуры получения
трансгенных мышей по сравнению с другими методами состоит
в том, что любую клонированную ДНК (в лямбоидном, плаз-
мидном или космидном векторе) можно ввести в зиготу, где
она интегрируется в геном и оказывается включенной в форми-
рование зародышевого пути. Дополнительные манипуляции,
такие, как переклонирование в специальные векторы и получе-
ние вирусных препаратов, здесь не требуются. Следует, одна-
ко, подчеркнуть, что эффективность микроинъекции в большой
степени зависит от качества и свойств вводимой ДНК [15].
Ниже мы рассмотрим этот вопрос подробнее.
10.4.1.1. Физические параметры ДНК
Путем микроинъекции в эмбрионы мыши можно вводить
клонированную ДНК любого размера, вплоть до лямбда-клонов
длиной 50 т. п. н. [17]. Линейная ДНК интегрируется в пять
раз эффективнее, чем сверхспирализованная [15], причем не
наблюдается сколько-нибудь заметного предпочтения в отно-
шении концов, образованных разными рестриктазами [15, 18].
10.4.1.2. Раствор ДНК
Инъецируемая ДНК должна быть растворена в буфере сле-
дующего состава: 10 мМ трис-HCli (pH 7,4); 0,1—0,25 мМ
ЭДТА. Более высокие концентрации ЭДТА, а также MgCl2 да-
342
Д. Мерфи, Дж. Хэнсон
же в малых дозах оказывают на клетку токсическое действие.
Концентрация вводимой ДНК и число копий трансгена в гено-
ме клетки не коррелируют друг с другом, причем очень высо-
кие концентрации ДНК токсичны для яйцеклетки [15]. По су-
ществующим оценкам, при микроинъекции в пронуклеус вво-
дится 1—2 ил раствора ДНК. Концентрация вводимой ДНК
обычно составляет от 1 до 5 мкг/мл. Следовательно, в яйце-
клетку вводится приблизительно 500 копий клонированного
фрагмента ДНК среднего размера. Некоторые исследователи
готовят ряд разведений раствора ДНК (например, 1; 2,5;
5 мкг/мл) и используют их в одной серии уколов, заменяя
инъекционные пипетки.
10.4.1.3. Прокариотические ДНК
С помощью микроинъекции в яйцеклетку можно вводить
последовательности ДНК как эукариотического, так и прока-
тиотического происхождения; и те и другие в равной мере ока-
зывают влияние на зародышевый путь. Однако присутствие по
соседству (в составе одного инъецируемого фрагмента ДНК)
прокариотических векторных последовательностей может силь-
но ингибировать экспрессию некоторых эукариотических транс-
генов, например глобина [19—21], альфа-фетопротеина [22] и
актина [23]. Чада и др. [19] сообщают, что тканеспецифичную
экспрессию экзогенного ^-глобинового гена удалось осущест-
вить только в отсутствие плазмидных последовательностей.
Аналогично Таунесу и др. [21] удалось повысить уровень экс-
прессии человеческого ^-глобинового трансгена в 1000 раз пу-
тем элиминации плазмидных последовательностей. В некото-
рых же вариантах экспериментов бактериальные кодирующие
последовательности (например, ген хлораменификол-ацетил-
трансферазы, CAT) специально вводят в состав гибридных кон-
струкций в качестве индикаторных элементов, позволяющих
оценить эффективность работы эукариотического промотора.
В отличие от некоторых векторных последовательностей ген
CAT не ингибирует прилежащие эукариотические регуляторные
элементы. Была осуществлена экспрессия этого гена под кон-
тролем аА-кристаллинового промотора [24] и длинного конце-
вого повтора вируса саркомы Рауса [25]. Не исключено, что
ингибирующий эффект присущ лишь вполне определенным про-
кариотическим последовательностям, в частности некоторым
элементам, входящим в состав широко используемых векторов
для молекулярного клонирования па основе фага лямбда и
pBR322. Вследствие этого сейчас принято удалять из реком-
бинантных конструкций, содержащих предназначенные для
микроинъекций клонированные эукариотические гены, все век-
Получение трансгенных мышей 343
торные последовательности с тем, чтобы максимально повы-
сить надежность экспериментов с трансгенами, а также улуч-
шить их количественный и качественный выход.
10.4.1.4. Приготовление и очистка препаратов ДНК
для микроинъекции
ДНК для микроинъекций можно выделять из космидных
или плазмидных клонов любым стандартным методом, напри-
мер, используя обработку бактериальной массы лизоцимом и
тритоном Х-100 или же лизируя бактерии щелочью и далее
отделяя сверхспирализованные молекулы в градиенте плотнос-
ти CsCl в присутствии бромида этидия [26]. Процедура полу-
чения высококачественной плазмидной ДНК, не требующая
ультрацептрифугирования [27], приведена в табл. 10.9.
Клонированные фрагменты ДНК отделяют от вектора с по-
мощью рестриктазной обработки и последующего препаратив-
ного электрофореза в агарозном геле. Препараты ДНК, пред-
назначенные для микроинъекции, не должны содержать каких-
либо вредных примесей, токсичных для яйцеклетки, а также
твердых частиц, которые могут закупорить просвет микроиглы.
Адекватный метод очистки ДНК описан в табл. 10.10.
10.4.2. Идентификация трансгенных мышей
Трансгенных животных идентифицируют с помощью гиб-
ридизационного анализа высокомолекулярной геномной ДНК,
выделенной из тканей хвоста. Процедура выделения ДНК опи-
сана в табл. 10.11.
Метод анализа выделенной ДНК выбирают, исходя из при-
роды трансгена. Если трансген имеет гомологию с каким-либо
участком генома мыши, используют блот-гибридизацию [26,
28[. Если же гомология с мышиной ДНК отсутствует или она
незначительна, можно ограничиться дот-блот- или слот-блот-
анализом [29].
10.4.2.1. Блот-гибридизационный метод
Чужеродный ген, интегрированный в мышиный геном, мож-
но обнаружить и отличить от эндогенного гомолога по нестан-
дартной картине рестриктазного расщепления геномной ДНК.
Даже в том случае, когда трансген идентичен эндогенному ге-
ну, в геноме он окружен другими нуклеотидными последова-
тельностями и с другим расположением сайтов рестрикции и
потому будет выявляться в составе других рестриктазных фраг-
ментов. Если же трансген происходит из другого биологическо-
344
Д. Мерфи, Дж. Хэнсон
Таблица 10.9. Выделение плазмидной ДНК
1. Внесите отдельную бактериальную колонию в 5 мл среды и инкубируйте
в течение ночи. Полученную ночную культуру разведите до 100 мл све-
жей средой и оставьте при интенсивном встряхивании при 37 °C на ночь.
2. Соберите бактериальную массу центрифугированием, например при
7000 об/мин в течение 5 мин в роторе Sorvall SS34 пли НВ4. Удалите по
возможности всю надосадочную жидкость.
3. Ресуспендируйте осадок в 5 мл раствора, содержащего 50 мМ глюкозу,
25 мМ трис-НС1 (pH 8,0) и 10 мМ ЭДТА. Затем добавьте 10 мл свеже-
приготовленного раствора: 0,2 М NaOH, 0,1% ДСН; перемешайте и по-
местите на лед на 5 мин.
4. Добавьте 5 мл охлажденного на льду ацетата калия (pH 8) % тщатель-
но перемешайте и оставьте на льду на 5 мни. Центрифугируйте при
8000 об/мин в течение 5 мин.
5. Перенесите жидкость в чистую центрифужную пробирку и смешайте с
0,6 объема изопропанола (12 мл). Центрифугируйте при 8000 об/мин в
течение 5 мин.
6. Удалите надосадочную жидкость и ресуспендируйте осадок в 2 мл ТЭ2>.
Добавьте 2 мл охлажденного на льду 5 М LiCl, перемешайте i: центри-
фугируйте при 1000 об/мин в течение 5 мин.
7. Перенесите жидкость в чистую пробирку и добавьте 8 мл этанола. Ин-
кубируйте при —70 °C в течение 30 мин, затем центрифугируйте при
10000 об/мин в течение 10 мин.
8. Удалите надосадочную жидкость и ресуспендируйте осадок в 0,5 ТЭ, со-
держащего 40 мкг/мл РНКазы А3>. Инкубируйте при 37 °C в течение
15 мин.
9. Добавьте 0,5 мл 2,5 М NaCl, содержащего 20% PEG 6000, и оставьте на
ночь при 4 °C. Центрифугируйте при 10 000 об/мин в течение 5 мин, осу-
шите осадок и ресуспендируйте его в 0,5 мл ТЭ.
10. Добавьте 0,5 мл хлороформа, встряхните на вибромиксере при
10 000 об/мин в течение 1 мин, затем перенесите водную фазу (верхний
слой) в чистую пробирку. Повторите экстракцию хлороформом.
11. К водной фазе добавьте 0,5 мл фенола4), встряхните и центрифугируйте
при 10 000 об/мин в течение 2 мин. Перенесите водную фазу (верхний
слой) в пробирку с 50 мкл 3 М ацетата натрия. Добавьте 550 мкл изо-
пропанола и перемешайте.
12. Выдержите при —70 °C по меньшей мере в течение 15 мин, затем цент-
рифугируйте при 10 000 об/мин в течение 5 мин. Добавьте к осадку 1 мл
холодного 70%-ного этанола, встряхните на вибромиксере и снова цент-
рифугируйте в течение 2 мин при 10 000 об/мин.
13. Подсушите осадок плазмидной ДНК в вакууме и ресуспендируйте его
в 0,5 мл ТЭ.
|) 5 м ацетат калия (pH 8,0) готовят, смешивая равные объемы ЗМ ацетата калия
и 2 М уксусной кислоты.
2) ТЭ: Ю мМ. трис-НС1 (pH 7,5), 1 мМ ЭДТА.
8) РНКазу А (Sigma, номер по каталогу R5125) растворяют в ТЭ и инкубируют на
кипящей водной бане в течение 10 мии, чтобы инактивировать возможные примеси
4) Фенол (перегнанный, квалификации «для нуклеиновых кислот» Gibco-BRL, номер
по каталогу 5509UA) перед использованием уравновешивают ТЭ.
го вида, он может отличаться от гомологичного мышиного гена
еще и по расположению внутренних сайтов рестрикции.
На рис. 10.11 показаны результаты одного из экспериментов
по рестриктазно-гибридизационному анализу ДНК. От сурро-
гатной матери, которой были трансплантированы оплодотворен-
Таблица 10.10. Очистка ДНК для микроинъекции с использованием
стеклянных микрогранул
А. Приготовление стеклянных микрогранул
1. Смешайте 250 мл стеклянной пудры флинтглас1’ с 500 мл стерильной
воды* 2’. Дайте отстояться в течение 1 ч. Удалите осевшее стекло и затем
отцентрифугируйте тонкую стеклянную взвесь.
2. Ресуспендируйте этот осадок в 200 мл воды. Добавьте 200 мл концентри-
рованной азотной кислоты и доведите до кипения. Эту операциию выпол-
няйте в вытяжном шкафу.
3. Дайте суспензии охладиться, затем отцентрифугируйте стеклянные микро-
гранулы. Отмойте кислоту, многократно ресуспендируя осадок в воде и
снова центрифугируя, до тех пор, пока промывная вода не приобретет
нейтральный pH.
4. Храните микрогранулы при комнатной температуре в виде 50%-ной взве-
си в стерильной воде.
Б. Выделение из агарозного геля и очистка фрагментов ДНК,
1. Обработайте ДНК подходящей рестриктазой в рекомендуемых условиях.
2. Фракционируйте гидролизованную ДНК в агарозном геле в fivcbepe
ТАЭ3’, содержащем 50 мкг/мл этидиумбромида. Визуализируйте ДНК в
длинноволновом УФ-свете, вырежьте интересующий фрагмент в мини-
мальном объеме агарозного геля и поместите его в предварительно взве-
шенную 1,5мл-пробирку эппендорф.
3. Определите объем и вес геля. Добавьте 2 объема 6 М Nal4’. Инкубируй-
те при 37 °C, время от времени встряхивая в вибромиксере, пока гель
полностью не растворится.
4. «Оживите» взвесь стеклянных микрогранул, встряхнув ее на миксере.
Добавьте ее к солюбилизированному гелю из расчета 1 мкг взвеси на
2 мкг присутствующей в растворе ДНК. Охладите на льду в течение 1 ч,
время от времени перемешивая.
5. Отцентрифугируйте микрогранулы, чтобы получить неплотный осадок, кру-
тите на малой скорости непродолжительное время (1—2 с в микроцент-
рифуге).
6. Удалите надосадочную жидкость и суспендируйте стеклянный осадок в
6М Na! (0,5 первоначального объема геля). Снова осадите микрограну
лы центрифугированием (1—2 с в микроцентрифуге).
7. Суспендируйте осадок в спиртовом промывном растворе5’ (0,5 объема ге-
ля) и центрифугируйте. Повторите этот этап еще раз.
8. Удалите по возможности всю надосадочную жидкость, ио не давайте
осадку высохнуть. Немедленно добавьте не менее 50 мкл раствора, со-
держащего 10 мМ трис-НС1 (pH 7,4) и 0,2 мМ ЭДТА. Элюируйте ДНК,
инкубируя пробу при 37 °C в течение 15 мин.
9. Отцентрифугируйте стекло и перенесите раствор, содержащий ДНК, в чи-
стую пробирку6’.
10. Чтобы удалить загрязняющие примеси, пропустите раствор ДНК через
колонку с сефадексом G-50, уравновешенную 10 мМ трис-НС1-буфером
(pH 7,4), содержащим 0,2 мЕ ЭДТА, и профильтруйте через фильтр AVI-
Проге 0,45 мкм7’.
11. Определите концентрацию ДНК на спектрофотометре или путем срав-
нительной колориметрии в присутствии бромида этидия.
’) Стеклянную пудру флинтглас поставляют стекольные компании (например, Eagle
Ceramics Inc., MD 20852, USA).
2) Соблюдайте стерильность, так как бактерии неплохо размножаются во взвеси
стеклянных микрогранул.
3) 20ХТАЭ-буфер: 0,8 М трис-НС1 (pH 8,0), 0,4 М ацетат натрия, 0,02 М ЭДТА.
4) 6М Nal готовят, растворяя 90,8 Nal и 0,5 г Йа2$Оз в воде — конечный объем
100 мл. Раствор фильтруют через фильтр Nalgene 0,45 мкм и добавляют к фильтру не-
сколько кристаллов Na2SO3 (Na2SO3 полностью не растворяется). Хранят при 4 °C в за-
щищенном от света месте.
5) Спиртовой промывной раствор: 50%-ный этанол, 0,1 М. NaCl, 10 мМ трис-НС1
(pH 7,5) и 1 мМ ЭДТА. Храните при —20 °C.
в) На этой и последующих стадиях используйте пробирки и наконечники для мик-
ропипеток, предварительно промытые в профильтрованной воде.
7) Millipore, тип HV, номер по каталогу SJHV004NS.
346
Д. Мерфи, Дж. Хэнсон
Таблица 10.11. Выделение геномной ДНК мыши из тканей хвоста
1. Удерживая мышь, как показано на рис. 10.4, Б, острыми ножницами бы-
стро отрежьте кончик хвоста (приблизительно 1 см) и поместите образец
в 700 мкл раствора, содержащего 50 мМ трис-НС1 (pH 8,0), 100 мМ
ЭДТА, 100 мМ NaCl, 1% ДСН, 100 мкг/мл протеиназы К1’. Прежде чем
отпустить животное, пометьте его (ухо или лапку), чтобы впоследствии
можно было его узнать.
2. Измельчите ткань ножницами и инкубируйте при 50 °C в течение ночи,
желательно при перемешивании.
3. Наутро добавьте 5 мкл РНКазы А2* (1 мг/мл) и инкубируйте при 37 °C
не менее 1 ч.
4. Добавьте 700 мкл фенола, насыщенного ТЭ. Осторожно перемешивайте
до гомогенного состояния (на это уйдет примерно 20 мин).
5. Разделите фазы центрифугированием при 10 000 об/мин в течение 10 мин
в микроцентрифуге. Перенесите вязкую водную фазу (верхний слой)
вместе с интерфазой в чистую пробирку.
6. Добавьте 700 мкл смеси фенол — хлороформ3* и аккуратно перемешайте.
Центрифугируйте при 10 000 об/мин в микроцентрифуге и перенесите
верхнюю водную фазу в чистую пробирку. На этот раз интерфазу не бе-
рите.
7. Добавьте 800—1000 мкл 100%-ного этанола и аккуратно перемешайте.
При этом формируется хорошо заметный белый волокнистый оса-
док. Осадите его центрифугированием (2 мин) в микроцентрифуге.
8. Удалите надосадочную жидкость и промойте осадок 1 мл 70%-ного эта-
нола. Снова осадите ДНК в микроцентрифуге (1 мин). Удалите жид-
кость и подсушите осадок в вакууме.
9. Добавьте к осадку ДНК 250 мкл ТЭ и инкубируйте при 37 °C в течение
нескольких часов, чтобы ДНК полностью растворилась.
10. Определите концентрацию ДНК на спектрофотометре.
’) Sigma, номер по каталогу Р-0390.
2) Sigma, номер по каталогу R-5I25. РНКазу растворяют в ТЭ и инкубируют на ки-
пящей водяной бане в течение 10 мин для того, чтобы инактивировать возможные при-
меси ДНКазы.
3) Смесь равных объемов фенола и хлороформа уравновешивают ТЭ и добавляют
к ней 1/50 объема изоамилового спирта.
ные яйцеклетки с введенным в них фрагментом гена c-fos чело-
века, получили одиннадцать детенышей (Д. М. Дж. X.,
Р. Трейсман, Б. Хоган, неопубликованные данные). Блот-гибри-
дизационный анализ ДНК, выделенной из тканей хвоста и за-
тем гидролизованной PstI, показал, что фрагмент экзогенной
ДНК, отличающийся по расположению внутренних Ps/1-сайтов
от эндогенного гена, присутствует только у мышат 1,4,7 и И.
Гибридизационные полосы, обусловленные эндогенными по-
следовательностями c-fos, могут служить контролем на коли-
чество и качество выделенной ДНК-
10.4.2.2. Слот(и дот]-бпот-анапиз
Слот (и дот)-блот-анализ относится к экспресс-методам, т. е.
он относительно прост и требует немного времени. Однако этот
вид гибридизационного анализа применим лишь в случае
Получение трансгенных мышей
347
Рис. 10.11. Блот-гибридизационный анализ мышиной высокомолекулярной
ДНК, выделенной из тканей хвоста. ДНК выделяли из одиннадцати мышей,
которые развились из оплодотворенных яйцеклеток, переживших инъекцию
человеческой ДНК (фрагмент гена c-fos). Выделенную ДНК расщепляли
рестриктазой Psfl, фракционировали электрофорезом в 0,8%-ном агарозном
геле, переносили на нитроцеллюлозный фильтр и гибридизовали с c-fos-зон-
дом. Стрелками указаны детектируемые эндогенные мышиные (М) и экзо-
генные человеческие (Ч) фрагменты c-fos.
трансгенов, не имеющих участков, гомологичных ДНК организ-
ма-хозяина (например, это гибридный ген с вирусной или про-
кариотической кодирующей частью). Процедура анализа опи-
сана в табл. 10.12, а результат одного из таких экспериментов
показан на рис. 10.12. Слот- или дот-блот-анализ следует про-
водить параллельно на двух одинаковых фильтрах, один из
которых гибридизуют с трансгенным зондом, а другой — с
эндогенной последовательностью. Количество ДНК в препара-
те трудно оценить точно из-за присутствия в нем РНК. Поэто-
му необходимо провести гибридизацию с эндогенным мыши-
ным зондом, для того чтобы исключить ложные «негативы» и
в какой-то степени облегчить количественное сравнение гиб-
ридизационных сигналов.
10.4.2.3. Идентификация гомозиготных трансгенных мышей
Мыши, гомозиготные по трансгену, получаются в результа-
те скрещивания гетерозиготных родителей. Гомозиготные, ге-
терозиготные (по трансгену) и не содержащие трансгена по-
томки появляются у таких родителей в соотношении 1:2:1.
Гомозиготные особи выявляются благодаря двойному количе-
ству в их геномах трансгенной ДНК по сравнению с гетерози-
готными родителями, братьями или сестрами. Далее их гомо-
348
Д. Мерфи, Дж, Хэнсон
Таблица 10.12. Слот (или дот)-блот-гнбридизация геномной ДНК
1. В аликвоту раствора, содержащего 10 мкг ДНК, добавьте NaOH до кон-
центрации 0,3 М.
2. Инкубируйте на кипящей водяной бане в течение 10 мин. Охладите на
льду.
3. Выдержите препарат при комнатной температуре в течение 5 мин, за-
тем добавьте ацетат аммония до 2 М.
4. Предварительно вырезанный лист нитроцеллюлозы1) вымочите в 20XSSC2’
в течение по меньшей мере 5 мин и соберите аппарат для слот3’- или
дот-блоттинга4) согласно инструкции фирмы-изготовителя.
5. В ячейки аппарата нанести подготовленные препараты ДНК.
6. Выньте нитроцеллюлозный фильтр и подсушите его на воздухе при ком-
натной температуре.
7. Прогрейте фильтр в вакууме при 80 °C в течение 2 ч.
8. Поместите фильтр на предгнбридизацию в течение ночи при 42 °C в рас-
творе, содержащем 50% деионизованного формамида5’, 5XSSC, 5Храс-
твор Денхардта6’, 0,1% ДСН, 50 мМ натрий-фосфатный буфер (pH 7,5),
200 мкг/мл фрагментированной ДНК спермы лосося7’. Фильтр поместите
в пластиковый пакет и залейте небольшим количеством раствора. Пакет
необходимо герметично запаять.
9. Добавьте к раствору меченый зонд7’ так, чтобы его концентрация не пре-
вышала 25 нг/мл. Инкубируйте при 42 °C в течение ночи, желательно при
медленном покачивании.
10. Отмойте фильтр при 65 °C два раза по 30 мин в растворе, содержащем
2XSSC и 0,1% ДСН, и еще дважды в растворе 0,1 SSC — 0,1% ДСН
(желательно при покачивании).
11. Экспонируйте фильтр с рентгеновской пленкой.
l) Schleicher and Schull, 0,45 мкм, ВА85.
2) 20XSSC: 3 М. NaCl — 0,3 М цитрат натрия,
s) Например, Schleicher and Shull, Minifold II Slot-Blotter (номер по каталогу
SRC 072/0).
4) Например, Schleicher and Schtill Аномер по каталогу SRC-96).
5) Формамид смешивают со смолой Dowex XG8, инкубируют при помешивании в те-
чение 1 ч и затем фильтруют через бумагу ватман 1М.М. Формамид Fluka 47670 не тре-
бует деионизации.
•) 5ОХ раствор Денхардта: 1% Фиколла, 1% БСА (Франция V) и 1% поливинилпнр-
родидона.
7) Раствор ДНК спермы лосося готовят, растворяя сухое вещество (Sigma, номер по
каталогу D-1626) в воде до концентрации 10 мг/мл, и обрабатывают ультразвуком или
пропускают вязкий раствор через шприцевую иглу № 18. Некоторые экспериментаторы
дополнительно экстрагируют раствор смесью фенол — хлороформ, осаждают ДНК эта-
нолом и снова растворяют в воде. Затем ДНК следует денатурировать на кипящей
водяной баие в течение 10 мин.
8) Двуцепочечную ДНК-пробу надо предварительно денатурировать.
зиготность подтверждают генетическими методами. Для иден-
тификации гомозиготных животных можно использовать либо
обычный блот-гибридизационный анализ, либо слот (или дот)-
анализ: выбор метода зависит от наличия или отсутствия го-
мологии между трансгеном и эндогенным мышиным геном (см.
выше). В любом случае принципиальное значение имеет коли-
чественная оценка гибридизационного сигнала. Такую возмож-
ность дает сравнение результатов параллельных гибридизаций
с использованием в качестве зонда трансгена и эндогенной
ДНК мыши. На рис. 10.12 приведен пример слот-блот-анализа
Получение трансгенных мышей
349
Ж’*;
’ eSJHIiliW дои
уусй»<<' йй’’^И5 йй
Вййййййй:;^йьйй\3*й1йкй8^и1ййй:1йййййй-ййй1
ВЙййДШ:-йШййй^:::::::ЙМ.^№>'№~^
ШШШШкШ '•$ illiili
И|1|ЙД:ЙДШЙйВД8Ж
||fi|||S|®^
Рис. 10.12. Слот-блот-анализ геномной ДНК, выделенной из тканей хвоста
(разд. 10.4.2.3).
ДНК мышей, несущих в качестве трансгена фрагмент ДНК ви-
руса SV-40, который не обнаруживает гомологии с мышиной
ДНК- Исследовали потомков (А, В и С) гетерозиготных по
трансгену родителей (их гетерозиготность считается генетичес-
ки установленной). Два одинаковых фильтра с ДНК из тканей
хвоста гибридизовали один — с радиоактивно меченной ДНК
SV-40, чтобы выявить трансген, а другой — с радиоактивно ме-
ченной мышиной последовательностью, в данном случае с ге-
ном c-fos. Одинаковая интенсивность гибридизационного сигна-
ла с зондом c-fos для всех трех образцов указывает на то, что
количество ДНК на фильтре в каждом случае одинаково. Тог-
да гибридизация с зондом SV-40 позволяет сделать вывод, что
мышь А имеет в геноме больше трансгенных последовательнос-
тей, чем особи В и С. Количественное денситометрическое ска-
нирование или просчитывание радиоактивных пятен на сцин-
тилляционном счетчике показало, что в геномной ДНК А со-
держится в два раза больше вирусной ДНК, чем в ДНК В и
С, и, следовательно, особь А можно считать потенциально го-
мозиготной по трансгену. Этот вывод подтверждается резуль-
татами генетического анализа. Мышь А (это самец) скрестили
с нормальной самкой F1 и ДНК родившихся детенышей иссле-
довали с помощью слот-анализа. Поскольку все 10 мышей ока-
зались гетерозиготными по трансгену, их отец (мышь А) го-
мозиготен.
350
Д. Мерфи, Дж. Хэнсон
10.5. Разведение трансгенных мышей
и анализ экспрессии экзогенных генов
10.5.1. Получение трансгенных линий
После того как первичные трансгенные животные иденти-
фицированы (разд. 10.4.2), необходимо заняться их скрещива-
нием для получения трансгенных линий. Разумеется, такие ли-
нии должны быть получены прежде, чем животное-основатель
будет умерщвлено для детального исследования. Лучше, если
животное-основатель самец. Самца можно быстро скрестить с
несколькими самками, тогда как самка-основатель линии долж-
на выносить и выкормить хотя бы один помет,, прежде чем ее
можно будет забить. Желательно провести гибридизационный
анализ и идентифицировать трансгенных животных первого
поколения еще при жизни родителей.
В большинстве случаев 50% потомков, полученных от скре-
щивания животных-основателей с животными дикого типа, ока-
зываются трансгенными, причем как родитель-основатель, так
и потомки первого поколения гетерозиготны по трансгену. Од-
нако необходимо иметь в виду, что некоторые трансгенные жи-
вотные-основатели представляют собой мозаичные организмы,
и, следовательно, трансгенными окажется меньшая часть по-
томства первого поколения. Всех детенышей следует анализи-
ровать, как описано в разд. 10.4.2.3, пока не будет выведена
гомозиготная линия. В ряде случаев гомозиготные линии полу-
чить не удается, поскольку от 5 до 15% трансгенных инсерций
в гомозиготном состоянии детальны.
10.5.2. Экспрессия генов в трансгенных мышах
За последние несколько лет накоплено очень много экспе-
риментальных данных по экспрессии экзогенных ДНК в транс-
генных мышах и обобщить всю эту информацию в рамках дан-
ной главы невозможно. Поэтому мы отсылаем заинтересованно-
го читателя к обзору Палмитера и Бринстера [12]. Здесь же
мы сформулируем лишь некоторые общие принципы.
Примерно половина всех линий трансгенных мышей не экс-
прессирует трансген. По-видимому, это связано либо с присут-
ствием в трансгене ингибиторных последовательностей (напри-
мер, некоторых последовательностей прокариотического проис-
хождения; см. разд. 10.4.1), либо с особенностями конкретного
сайта интеграции трансгена: экзогенная ДНК может интегри-
роваться в транскрипционно неактивные области хромосом,
Если же экзогенная ДНК экспрессируется в трансгенном орга-
низме, этот процесс в большинстве случаев контролируется
Получение трансгенных мышей 35[
присутствующими в ней регуляторными элементами. Тем не
менее прилежащие последовательности клеточной ДНК способ-
ны оказывать влияние на экспрессию трансгена. В ряде случа-
ев наблюдаются неожиданные отклонения в характере экспрес-
сии чужеродных последовательностей в трансгенных организ-
мах, которые, вероятно, обусловлены эффектом положения
трансгена в геноме клетки-хозяина, например взаимодействием
трансгена с эндогенным эхансером.
Размер и локализация последовательностей, необходимых
для обеспечения правильной экспрессии, зависят от природы
гена. Некоторые гены, например инсулина [30], эластазы [31,
32] и аА-кристаллина [24, 25], «обходятся» несколькими сотня-
ми нуклеотидных пар, предшествующих началу гена. Для дру-
гих генов, например AFP, требуется 5'-регуляторная зона раз-
мером 10 т. п. и [22]. Некоторые гены, например глобиновые
гены человека [33], нуждаются не только в б'-регуляторных
зонах, предшествующих началу гена, но также и в последова-
тельностях, расположенных за точкой инициации транскрип-
ции.
10.5.3. Применение трансгенной технологии
Трансгенная технология таит в себе большие возможности,
в первую очередь в плане фундаментальных научных исследо-
ваний. Эксперименты с трансгенными мышами призваны обес-
печить прогресс в биологии млекопитающих — в частности бла-
годаря развитию следующих направлений научного поиска.
1. Анализ г{цс-действующих контролирующих элементов,
ответственных за тканевую специфичность: за временную и фи-
зиологическую регуляцию экспрессии генов.
2. Анализ физиологических последствий (на уровне целого
организма) различного рода отклонений в экспрессии нормаль-
ных генов или мутировавших генов, в том числе онкогенов.
3. Получение новых мутантов. Рецессивные мутанты часто
возникают в популяции трансгенных мышей вследствие внедре-
ния трансгена в последовательность того или иного функцио-
нального гена. Мутировавший ген можно выделить путем кло-
нирования, используя трансген в качестве гибридизационной
пробы [34]. В перспективе можно будет осуществлять направ-
ленный мутагенез, вводя в мышей экспрессирующиеся «анти-
смысловые» конструкции, адресованные конкретным генам.
Подобные эксперименты уже приводятся на растениях [35].
Помимо фундаментальных исследований трансгенная техно-
логия открывает широкие перспективы для развития целого ря-
да прикладных областей, в частности биотехнологии и сельско-
352
Д. Мерфи, Дж. Хэнсон
го хозяйства. Сейчас уже получены трансгенные свиньи, овцы
и кролики [36]. Однако, по-видимому, пройдет еще немало вре-
мени, прежде чем трансгенные животные займут подобающее
им место в системе сельскохозяйственного производства.
Благодарности
Мы хотели бы выразить благодарность работникам Графи-
ческого бюро NIMR за помощь в подготовке иллюстраций. Ра-
бота Дэвида Мерфи и Дженнифер Хэнсон финансировалась
Советом по медицинским исследованиям (MRC). Мы выражаем
благодарность Б. Хоган за большую консультативную помощь
и за то, что она дала нам возможность реализовать поисковые
проекты у себя в лаборатории.
Литература
1. Gorman С. (1985). High efficiency gene transfer into mammalian cells. In:
DNA Cloning — A Practical Approach, Glover D. M. (ed.), IRL Press, Oxford,
Voi. II, p. 143—190. [Имеется перевод: Клонирование ДНК. Методы. Кни-
га II. Под ред. Д. Гловера. — М.: Мир, 1972.]
2. Jaliner D., Haase К-, Mulligan R., Jaenisch R. (1985). Proc. Natl. Acad. Sci.
USA, 82, 6927.
3. van der Putten H., Botteri F. M., Miller A. D., Rosenfeld M. G., Fan H.,.
Evans R. M., Verma I. M. (1985). Proc. Natl. Acad. Sci. USA, 82, 6148.
4. Evans M. Kaufman M. H. (1981). Nature, 292, 154.
5. Martin G. R. (1981). Proc, Natl, Acad. Sci. USA, 78, 7634.
6. Bradley A., Evans M. H„ Kaufman M. H„ Robertson E. (1984). Nature 309,
255.
7. Lovell-Badge R. H.. Bygrave A. E., Bradley A., Robertson E., Evans M. J.,
Cheah K. S. E. (1985). Cold Spring Harbor Symp. Quant. Biol. 50, 707.
8. Robertson E., Bradley A., Kuehn Mi., Evans M. (1986). Nature, 323 445.
9. McGrath J., Solter D. (1983). Science, 220, 1300.
10. Smithies O., Gregg R. G., Boggs S. S., Koralewski M. A., Kucherlapati R. S.
(1985). Nature, 317, 230.
11. Thomas K. R-, Folger K. R., Capecchi M. R. (1986). Cell, 44, 4189.
12. Palmiter R. D., Brinster R. E. (1987). Annu. Rev. Genet., in press.
13. Lacy E„ Roberts S„ Evans E. P., Burtenshaw M. D., Constantini F. (1983)
Cell, 34, 343.
14. Krumlauf R„ Chapman V. M„ Hammer R. E., Brinster R. L„ Tilgham S. M.
(1985). Nature, 319, 224.
15. Brinster R. L., Chen H. Y., Trumbauer M. E„ Yagle M. K, Palmiter R. D.
(1985). Proc. Natl. Acad. Sci. USA, 82, 4438.
16. Hogan B„ Constantini F„ Lacy E. (1986). Manipulating the Mouse Embryo —
A Laboratory Manual, Cold Spring Harbor, New York.
17. Constantini F„ Lacy E. (1981). Nature, 294, 92.
18. Grosschedl R„ Weaver D., Baltomore D., Constantini F. (1984). Cell, 38,
647.
19. Chada K, Magram J., Raphael K, Radice G., Lacy E„ Constantini F (1985)
Nature, 314, 377.
20. Magram J., Chada K-, Constantini F. (1985). Nature, 315, 338.
21. Townes T. M., Lingrel J. B., Chen H. Y., Brinster R. L„ Palmiter R. D. (19854
EMBO J., 4, 1715.
Получение трансгенных мышей
355
22. Krumlauf R., Hammer R. Е., Tilghman S. М., Brinster R. L. (1985). Cold
Spring Harbor Symp. Quant. Biol., 50, 371.
23. Shani M. (1986). Mol. Cell Biol., 6, 2642.
24. Overbeek P. A., Chepelinsky A., KJiillan J. S., Piatgorsky J., Westphal M.
(1985). Proc. Natl. Acad. Sci. USA, 82, 7815.
25. Overbeek P. A„ Lai S.-Р., van Quill K. R., Westphal H. (1986). Science, 231,
1574.
26. Maniatis T., Fritsch E. F„ Sambrook J. (1982). Molecular Cloning: A Labora-
tory Manual, Cold Spring Harbor, New York. [Имеется перевод: Ma-
ниатис Т., Фрич Э., Сэмбрук Дж. Методы генетической инженерии. Молеку-
лярное клонирование. — М.: Мир, 1984.]
27. Treissman R. (1985). Cell, 43, 889.
28. Southern Е. (1974). J. Mol. Biol, 98, 503.
29. Kafatos F. C., Jones W. C., Efstratiados A. (1979). Nucleic Acids Res, 7,.
1541.
30. Hanahan D. (1985). Nature, 315, 115.
31. Or nit z D. M., Palmiter R. D„ Hammer R. E„ Brinster R. L., Swift G., MacDo-
nald R. J. (1985). Nature, 313, 600.
32. Ornitz D. M. Palmiter R. D., Messing A., Hammer R. E., Pinkert C. A.,
Brinster R. L. (1985). Cold Spring Harbor Symp, Quant. Biol, 50, 399.
33. Kollias G., Wrighton N., Hurst J., Grosveld E. (1986). Cell, 46, 89.
34. Woychik R. P„ Stewart T. A., Davis L. G., D’Eustachhio P., Leder P. (1985).
Nature, 318, 36.
35. Ecker J. A., Davis R. W. (1986). Proc. Natl. Acad. Sci. USA, 83, 5372.
36. Hammer R. E., Pur sei V. G., Rexroad С. E., Wall R. J. Bolt D. J.7.
Ebert R. M., Palmiter R. D„ Brinster R. L. (1985). Nature, 315, 680.
СПИСОК ИСПОЛЬЗОВАННЫХ СОКРАЩЕНИЙ
АФП — альфа-протеин
АФТГ — аминофенилтиогалактопиранозид
б. о. е. — бляшкообразующая единица
БСА — бычий сывороточный альбумин
ВПК — ванадил-нуклеоитдный комплекс
ГАТ — гипоксантин/аминоптерин/тимидин
ДМСО — диметилсульфоксид
ДПД — диметилпимелидатдигидрохлорид
ДСН — додецилсульфат натрия (SDS)
ДТТ — дитиотрейтол
ДЭПК — диэтилпирокарбонат
ЖХВД — жидкостная хроматография под высоким давле-
нием
ЖХВС — высокоскоростная жидкостная хроматография
ИПТГ — изопропил-р-П-тиогалактозид
ИПФ — инсулиноподобный фактор
ИФА— иммуноферментный анализ (ELISA)
МЭ— меркаптоэтанол
ЛПС — липополисахариды
ОРС — открытая рамка считывания
тПА (тАП) — активатор плазиногена тканевого типа
ПААГ — параакриламидный гель
ПК — пост коитум
ПЭГ — полиэтиленгликоль (PEG)
РИА — радиоиммунологический анализ (RIA)
РНКазин— ингибитор рибонуклеазы
CAT — хлорамфеникол — ацетилтрансфераза
СЭК (СПК) — сыворотка эмбриона (плода) коровы (FCS)
СПИД — синдром приобретенного иммунодефицита
ТХУ — трихлоруксусная кислота
ТЭ— ЮмМ трис-HCl и 1 мМ ЭДТА
ФКТ — фосфатаза кишечника теленка
ФЛГСФ — фенилметилсульфонилфторид
ФРТ — фактор роста тромбоцитов
ФСГ— фолликулостимулирующий гормон
ХГТ — хорионический гонадотропин
ЭДТА — этилендиаминотетраацетат
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
L-arap 54
F-агар 54
Агарозные гели 67
Адрнозин-дезаминаза 266
Аденозин-дезаминазная система 256—
257
Алкоголь-дегидрогеназа 133
Альфа-фактор 216—217
Амплификация векторов, разработка
стратегии 258—262
— генов 239—240, 261—266
— космидных библиотек см. Библио-
теки космидные, амплификация
Амплифицнрованное состояние гена
260
Ампнлифнцирующие векторы 241—
245
Анестезированное состояние мышей
319
Антиген, получение 180
Антитела к белкам, кодируемым
вставочными ДНК 182
— к гибридным белкам, получение
171—207
— моноклональные 196—207
----биотинилование 199, 202
----иодирование 199
----к гибридным белкам, 180—
196
Аполипопротеин Е человека из Е. Co-
li 126
Афинная хроматография гибридных
белков 159
----на АФТГ-агарозе 161
Белки гибридные см. Гибридные бел-
ки
— нерастворимые 100
— растворимые, внутриклеточная
экспрессия 128
— рекомбинантные, очистка 236
— синтезируемые в Е. coli 116
Белок А, связывание с моноклональ-
ными антителами 203
— CAT—большой антиген 206
бета-галактозидаза, гибридный бе-
лок 138—169
Библиотеки космидные 74—94
---амплификация 52, 75, 91
---высев 50—52, 69, 88
---клонирование, модификация н
характеристика 74—94
--- хранение 56
Биотилирование антител 199, 202
Блот-гибридизациониый анализ 263,
343, 346—349
Вазэктомия самцов 320—323
Вектор (ы) амплифицирующиеся
238—270
— гетерологичных генов 211—219
— гибридных белков 175
— используемые для экспрессии 96
— космидные 47, 80
---выбор 40—72
--- маркеры лекарственной устойчи-
вости 41
--- приготовление плеч 47
---репликоны 42
— ретровируса, выбор 279
—1—генетическая нестабильность 304
---дефект по репликации 279
——применение 273
---репликационно-комплементар-
ные 285
---самоинактивирующиеся 285
— строение 261
— p9arfa 259
— pATm-mt 250
— рМХ262 280, 284
— pSEMCat 204
— pSV2. ada 258
23
355
356
Предметный указатель
— pSV2. dhfr 243
— pSV2 gs 252
— pVSL gs 253
— pSVM dhfr 246
Векторы-«самоубийцы» 285
Вектор—хозяин, используемые комби-
нации 136
Вестерн-блот-аналнз 152, 179, 188
Вирус (ы) лейкоза мышей (MLV) 274
— рекомбинантные, получение препа-
ратов 287—295
— саркомы Молони (Mo-MSV) 283
— саркомы Рауса (RSV) 286
— SV-4O, инфицирование н псевдоин-
фицирование клеток CV-1 184
Гель-фильтрация гибридных белков
158
Ген krtippell 37
Геномное секвенирование 29
Гены в трансгенных мышах, экспрес-
сия 350
— гетерологичные, продукты 208—
236
— спаренные, неэквивалентная экс-
прессия 303
— экзогенные, анализ экспрессии 350
Гибридизация на фильтрах 54—55
— in situ 35
Гибридные белки, выделение 113—
115, 155, 162
--клонирование в Xgtll и pUR
143
---нерастворимые, выделение 181
---очистка, схема 114—115
---получение антител к ним 171—
207
---распределение в клетках 178
---р асщепленне 104— 112
---с ^-галактозидазой 138—169
---вестерн-блот-анализ 179
— гены, экспрессия 149
Гибридизация in situ 268—270
Гибридомные клеточные линии, замо-
раживание 194
Гибридомы, клонирование клеток
193—195
— слияние клеток 187—194
— скрининг 185—187
Гиперовуляция 325
Глутамин-синтетаза 265
— экспрессирующие векторы 253
Голдберга — Хогнесса ТАТА-бокс 204
Гомозиготные трансгенные мыши,
идентификация 347
Дигидрофолят-редуктаза, определе-
ние 264
ДМЕМ 190
ДНК, выделение крупномасштабное
61
—'геномная, слот-блот-анализ 346,
349
— для инъекции, очистка 345
— манипулирование с ней 341
— мини-препараты 60—61
— частичный гидролизат, очистка в
сахарозном градиенте 46
— приготовление вставочных фраг-
ментов 286
— частичное расщепление для кло-
нирования космид 43—47
Дот-блот-анализ 296—297, 346
Дрожжи, анализ экспрессии гетеро-
логичных белков 236
— в изучении гетерологичных бел-
ков, преимущества 208
— получение нужных штаммов 219—
226
— трансформация 226—233
— рер4 leu2 Atpi 212
ДСН-электрофорез гибридных бел-
ков 155, 176—177
Животные, содержание и манипуля-
ции с ними 314, 319
ЖХВД (жидкостная хроматогра-
фия при высоком давлении) 108
ЖХВС (высокоскоростная жидкост-
ная хроматография) 120
Иммунизация животных 181—182
Иммуноаффинный метод выделения
белков 157
Иммуноглобулины подкласса G
(IgG) 197
Иммуноокрашивание клеток 185, 187
Иммунопреципитация, метод 189
— экстрактов клеток, инфицирован-
ных вирусом 184
Иммуноферментный анализ (ИФА,
ELISA) 184—486
Инъекционная камера 336
Иш-Горовица и Берка метод 43, 48,
86
Кальцитонин—ТАТ, расщепление
108
«кассета» EcoRI 183
— челночные перемещения 202
Предметный указатель
357
-cos-картирование космид 62
Кесарево сечение 332—333
— селезенки 190
— слияние 187—194
Клетки зародышевого пути 308
— ЕК и ES 309—311
— cos 301—302
Клетки-мишени, инфицирование
295—296
Клонирование и фосфатазные реак-
ции 47, 143
— клеток в мягком агаре 194—195
— космид 69
— с помощью рекомбинации 62
— стратегия 43
— фрагментов 173—179
Клонированная ДНК, получение
трансгенных животных 308—352
Клонируемые сайты 16
Клоны, полученные в pUP, скрининг
145 -
Клострипаин 107, 122
Ко-амплификация 241, 248
Космидная ДНК, получение мини-
препаратов 60—61
Космидные библиотеки см. Библио-
теки космидные
— векторы см. Векторы космидные
— клоны, генетическая селекция 82
---модификация 83
— рекомбинанты, посев 88
--- упаковка и реупаковка 89
---cos-картирование 63
Космиды, дифференциальный рост 70
— клонирование 43, 48, 86
— конструирование библиотек 86
— Лориста 47
— превращения 75
— рекомбинация 77—86, 90, 92
— рекомбинантные, высев, переупа-
ковка, отбор 93
•--транскрипция in vitro 66
— упаковка 50, 56—58, 74—77, 89.
91
— pcos (2, 3, 5, 6) EMBL 80
Кумулусная масса 327
«Кэп» 36
Кэпирование и трансляция 36
P-Лактамаза, определение активности
132
Лигирование, получение космидных
библиотек 49, 69
Лизис клеток 99—100
Л изогены, индукция 91
— Xgtl 1, скрининг 144—145
Лориста космиды 47, 60
Лямбда gtll, скрининг клонов 144—
145
Металлотионеины, гены 249
Метотрексат, значение для амплифи-
кации генов 248
Миеломные клетки, слияние 190
Микрогорелка (из пастеровской пи-
петки) 318
Микрогранулы, приготовление 345
Микроинъекционная камера 340
Микроинъекция в оплодотворенные
яйцеклетки 338
— при получении трансгенных мы-
шей 311, 333—335
Микроманипуляторы 335
Микролппеткн инъекционные 336—
338
Микроскоп инвертированный 334
Молони вирус 278, 283
Моноклональные антитела см. Анти-
тела моноклональные
Найлоновые фильтры для скрещива-
ния космидных библиотек 52
Нозерн-анализ, РНК-зонды 29
Номарского система 334—335
Окрашивание клеток 183
Пероксидаза хрена 175
Петит-клетки 230
Пипетки для трансплантации яйце-
клеток 317, 319
Плазмидная ДНК 20, 344
Плазмиды, варианты интеграции 214
— векторы с SP6-, Т7- или ТЗ-промо-
торами 16
—'выделение из дрожжевых клеток
232
— семейства dUR 140
Плазмида рАТтМТ 250
— рСТ70 96
— pGEME 16
— pJP31 218
— pMG168 97
- pSV2.dhfr 243
— pSV2.GS 252
— pSVLGS 253
— pSVM.dhfr 246
— 'pT7-l 16
— YEP 211
358
Предметный указатель
Плазминогена активатор (тПа) из
Е. coli 128
Поликлональные антитела к гибрид-
ным белкам 138—169
Полимераза SPS 12—38
---реакция транскрипции 21, 66
Полимераза ТЗ 12—38
— Т7, реакция транскрипции 21—25,
66
Полипептиды, экспрессия в Е. coli
95—144
— эукариотические из Е. coli 95—144
— эукариотические из Е. coli, раст-
воримые и ренатурированные 122
Провирусная ДНК, выделение 300
«Прогулка по геному» 37, 65
Промоторы лямбда (X) 96
— lac 96
— SP6 12—38
— Т7 12—38
— trp 96
Проретровирус, структура 277
Прохимозин 102—103
— примесный компонент 102
— теленка из Е. coll 124—125
ПЭГ 191
Радиоиммунные тесты 199—201
Радиоиммуноанализ 198—200
Растворы для гибридизации 268
Рекомбинантный вирус, анализ струк-
туры и экспрессии 299
Ренатурация белков 119—121
Ретровирусные векторы 272—305
Ретровирус(ы), биология 274—278
— контроль препаратов 297—301
— получение трансгенных животных
309
—' рспликационно-компетентные, по-
лучение 291
— сбор и хранение 293—294
--хелперный и рекомбинантный, по-
лучение смешанных препаратов
291'
Рецепторы ретровируса 277
Рибонуклеаза плацентарная, ингиби-
тор '19, 25
РНК, предосторожности при работе
19
— содержание в клетке 34
мРНК, защита от РНКазы 14, 30—
35
— копирование 36
— синтезированная in vitro, функ-
ции 36
РНКазин 19, 25
РНК-зонды для «прогулки по гено-
му» 37
— полноразмерные 21
— преимущества и использование 13,
27
—'протекторный эффект 14, 30—35
— скрининг космидных библиотек 53
РНК-полимеразы 12—38
— выбор фермента 18
— SP6 и Т7 21-25
CAT-кальцитонин расщепленный,
очистка 122
Саузерн-гибридизация с РНК-зонда-
ми 28
Сахарозные градиенты 46, 69
Секвенирование SP6- и Т7-транскрип-
тов 37
Селекция на амплификацию генов,
методики 245—258
Скрещивание мышей 21
Слияние клеток 187—194
— получение гибридом 187—194
Слот-блот-анализ 346—'349
Сплайсинг-векторы 281
Среда Хогнесса 91
Среды для высева космидных биб-
лиотек 86
— для конструирования штаммов
221, 222
— для селекции 247, 254
— YEPD 221
Стволовые клетки эмбриональные
(ES и ЕК) 310
Суперинфекция 90
Сферопласты 100—101, 227
«Сэндвич», радиоиммунотест 201
ТАТА-бокс 204
ТАЭ-буфер 345
Трансгенная техология 351
Трансгенные животные, получение
308—352
— мыши, идентификация 343—3 9
----схема получения 312
Трансгенный организм 308
Транскриптаза обратная 297—298
Транскрипция вектора, оценка vdob-
ня 263
— in vitro 66
----тест на включение нуклеотидов
23
Трансфекция векторной ДНК в клет-
ки 288—291
— клеток СНО-К1 255
Предметный указатель
359
DHFR^-клетки СНО 246
Трансформанты, анализ 230
Трансформация дрожжей 226—233
Трансформированные клетки, устой-
чивые к G418 290
ТЭ, состав 21, 296, 302, 344
Упаковка, реакция in vitro 50
Упаковывающие клетки 288—291
Урокиназа из Е. coli 127
Фактор роста тромбоцитов 208
Фосмиды 58—59
Фосфатаза щелочная, определение
активности 132
7
Хогнеса среда 91
Хоффмана система 334—335
Хроматография, белокА—сефароза
196, 203
Хромосомы метафазные 265, 267
Челночные векторы 204—'205, 284,
301
— перемещения £соР1-«кассет» 202
Экспрессия белка, оценка эффектив-
ности 264
— клонированных генов 98
— неэквивалентная 303
P-Эндорфин, аминокислотная после-
довательность 109
— экспрессия в E.coll 132
Эпидидимис 323
Эпитопы, картирование 200—202
Эстральный цикл у мышей, стадии
317
Яйцеклетки оплодотворенные, куль-
тивирование и выделение 324—329
— трансплантация 317, 314—322
ОГЛАВЛЕНИЕ
От редактора перевода................................................. 5
Предисловие......................................................... 7
Список авторов ....................................................... 10
Глава 1. Применение плазмид, содержащих промоторы, специфические
для фаговых РНК-полимераз. Питер Ф. Р. Литтл и Йэн Дж
Джексон (Peter F. R. Little, lan J. Jackson) ....
1.1. Краткое содержание главы................................
1.2. РНК-полимеразы бактериофагов............................
1.2.1. Основные положения..................................
1.2.2. В чем преимущество РНК-зондов?......................
1.2.2.1. Уникальные свойства РНК-зондов..................
1.2.3. Выбор векторов......................................
1.2.3.1. Выбор клонируемого сайта........................
1.2.3.2. Выбор сайта терминации транскрипта..............
1.2.4. Выбор РНК-полимеразы................................
1.3. Методы..................................................
1.3.1. Получение ДНК-матрицы...............................
1.3.2. Реакция транскрипции................................
1.3.3. Получение немеченой РНК или РНК с низкой удельной ра-
диоактивностью и использование других изотопов и альтер-
нативных меток.............................................
1.3.4 Методические трудности...............................
1.3.4.1. Нуклеозидтрифосфаты (NTP) . .................
1.3.4.2. ДНК-матрица.....................................
1.3.4.3. Возможные неудачи при синтезе полноразмерных транс-
криптов .................................................
1.4. Использование РНК-зондов................................
1.4.1. Гибридизация по Саузерну............................
1.4.2. «Нозернх-гибридизация...............................
1.4.3. Защита от РНКазы....................................
1.4.3.1. Методика........................................
1.4.3.2. Определение содержания РНК в опытах по защите от
действия РНКазы..........................................
1.5. Специальные методы......................................
1.5.1. Гибридизация in situ................................
1.5.2. Биологическая активность РНК, синтезированной in vitro
1.5.2.1. Кэпирование и трансляция........................
1.5.2.2. Другие функции РНК..............................
1.5.3. Специальные методы картирования; «прогулка по геному»
12
12
12
12
13
13
16
16
18
18
19
20
21
25
26
26
26
27
27
28
29
30
30
33
36
36
36
37
Оглавление
361
1.5.4. Секвенирование РНК-транскриптов.........................37
1.6. Заключение..................................................38
Литература................................................38
Глава 2. Выбор и использование космидных векторов. Питер Ф. Р. Литтл
(Peter F. R. Little).......................................... 40
2.1. Введение.....................................................40
2.1.1. Краткое содержание главы.................................40
2.1.2. Космидные векторы........................................40
2.1.3. Выбор векторов.......................................41
2.1.3.1. Лекарственная устойчивость.......................41
2.1.3.2. Репликон.............................................42
2.1.3.3. Сайты рестрикции.....................................42
2.1.4. Стратегия клонирования...................................43
2.1.4.1. Выбор ферментов для клонирования.................43
2.1.4.2. Процедура клонирования...............................43
2.2. Методы.......................................................44
2.2.1. Получение ДНК для клонирования.......................44
2.2.1.1. Выделение ДНК из клеток или тканей...................44
2.2.1.2. Частичный рестриктазный гидролизат...................44
2.2.1.3. Очистка фракции 30—50 т. п. н. частичного гидролизата
в градиенте концентрации сахарозы..............................46
2.2.1.4. Обработка фосфатазой.................................47
2.2.2. Приготовление вектора....................................47
2.2.3. Реакция лигирования.....................................49.
2.2.4. Упаковка.................................................50
2.2.5. Высев библиотеки.........................................50
2.2.5.1. Выбор клеток.....................................50
2.2.5.2. Подготовка клеток................................
2.2.5.3. Адсорбция упакованных космид на Е. coli . . . .
2.2.5.4. Эффективность процедуры клонирования .
2.2.6. Рассев библиотеки....................................
2.2.6.1. Высев на агар для амплификации библиотеки
2.2.6.2. Высев непосредственно на фильтры.................
2.2.6.3. Получение реплик для скрининга библиотеки
2.2.7. Скрининг фильтров с космидными клонами . . . .
2.2.8. Хранение космидных библиотек.........................
2.2.8.1. Хранение бактерий................................
2.2.8.2. Хранение упакованных космид......................
2.2.8.3. Упаковка космидной библиотеки с использованием су-
перинфекции ..............................................
2.2.8.4. Упаковка индивидуальных космид с использованием су-
перинфекции ..............................................
2.2.8.5. Переупаковка путем высева первичной библиотеки на
хелперный штамм...........................................
2.2.8.6. Хранение ДНК.....................................
2.2.9. Получение космидной ДНК..............................
2.2.9.1. Размножение бактерий ... ..............
2.2.9.2. Мини-препараты...................................
2.2.9.3. Препаративное выделение из культур объемом 500—
1000 мл......................'........................61
2.3. Специальные методы.........................................61
2.3.1. Клонирование с помощью рекомбинации....................62
2.3.2. Cos-картирование.......................................63
2.3.3. «Прогулка по геному»...................................65
2.3.4. 0,2%-ные агарозные гели................................67
362
Оглавление
2.4. Проблемы...................................................68
2.4.1. Исходная ДНК............................................68
2.4.2. Сахарозные градиенты....................................69
2.4.3. Неудачное лигирование или упаковка..................... 69
2.4.4. Высеваемые клетки.......................................69
2.4.5. Высев...................................................70
2.4.6. Дифференциальный рост космид............................70
2.4.7. Проблемы, связанные с вектором..........................71
2.4.7.1. Нестабильные последовательности.....................71
2.4.7.2. Неслучайная представленность клонов.................71
2.5. Заключение.................................................72
Литература...............................................72
Глава 3. Генетические подходы к клонированию, модификации и харак-
теристике индивидуальных космидных библиотек клонов. Анна-
Мария Постка и Ханс Лерах (Annemarie Poustka, Hans Leh-
rach) ...............................................................74
3.1. Введение...................................................74
3.2. Работа с космидными клонами, полученными путем упаковки
in vivo.........................................................74
3.3. Отбор космид с помощью гомологичной рекомбинации . . 77
3.3.1. Гомологичная рекомбинация в клетках Е. coli может при-
меняться в качестве метода, альтернативного гибридизации
колоний.......................................................77
3.3.2. Принципы космидно-плазмидной рекомбинации ... 78
3.3.3. Отбор рекомбинантов.....................................78
3.3.4. Векторы.................................................80
3.3.5. Штаммы..................................................81
3.3.6. Генетическая селекция клонов............................82
3.3.7. Модификация клонов......................................84
3.4. Методики...................................................86
3.4.1. Конструирование космидных библиотек.....................86
3.4.2. Упаковка in vivo........................................89
3.4.3. Рекомбинация космид.....................................90
Литература...............................................94
Глава 4. Выделение эукариотических полипептидов, продуцируемых в
клетках Е. coli. Файона А. О. Марстон (Fiona А. О. Marston) 95
4.1. Введение...................................................95
4.1.1. Векторы, используемые для экспрессии....................96
4.1.2. Методы экспрессии.......................................98
4.2. Методы лизиса клеток.......................................99
4.2.1. Полный лизис клеток.....................................99
4.2.2. Получение сферопластов..................................100
4.3. Нерастворимые белки........................................100
4.3.1. Выделение белковых включений...........................101
4.3.1.1. Центрифугирование..................................101
4.3.1.2. Методы отмывки включений...........................102
4.3.2. Подходы к расщеплению гибридных белков ... 104
4.3.2.1. Химическое расщепление..............................Ю5
4.3.2.2. Ферментативное расщепление..........................Ю7
4.3.3. Гибридные белки, рассчитанные на специальные процедуры
выделения....................................................112
4.3.4. Растворение белков, синтезируемых клетками Е. coli в чи-
стом виде и в виде гибридных белков..................110
4.3.5. Ренатурация............................................119
Оглавление
363
4.3.6. Примеры растворенных н ренатурированных эукариотиче-
ских полипептидов, выделенных из клеток Е. coli . . . 122
4.4. Растворимые белки.........................................128
4.4.1. Внутриклеточная экспрессия............................128
4.4.2. Секреция..............................................131
4.5. Заключительные замечания..................................133
4.6. Приложение................................................135
Литература.............................................136
Глава 5. Получение и очистка поликлональных антител к гетерологич-
ным фрагментам гибридных белков, содержащих (3-галактози-
дазу. Син Б. Кэрролл и Аллен Логон (Sean В. Carroll, Allen
Laughori).............................................................138
5.1. Введение. Векторы для экспрессии гибридных белков, содержа-
щих ^-галактозидазу.............................................138
5.1.1. /.gt 11................................................139
5.1.2. Плазмидные векторы семейства pUR.......................140
5.2. Конструирование и клонирование гибридных белков, содержа-
щих ^-галактозидазу.............................................141
5.2.1. Клонирование гибридных генов в векторе Xgtll . . . 143
5.2.2. Клонирование гибридных генов в плазмидах pUR . . 143
5.2.2.1. Подготовка векторной ДНК...........................143
5.2.2.2. Приготовление гетерологичного фрагмента ДНК • . 144
5.2.2.3. Лигирование ДНК плазмид pUR с гетерологичной ДНК
и трансформация..............................................144
5.2.3. Скрининг клонов %gt 11.................................144
5.2.3.1. Скрининг фаговых клонов на наличие вставок . . 144
5.2.3.2. Скрининг к.тоноов %gt 11 на продуцирование гибридных
белков.......................................................144
5.2.4. Скрининг клонов, полученных в плазмидах pUR . . . 145
5.2.4.1. Скрининг плазмидных клонов на наличие вставок 145
5.2.4.2. Скрининг плазмидных клонов на продуцирование гиб-
ридных белков................................................145
5.2.5. Соотношение размера гетерологичного фрагмента и выхода
синтезируемого гибридного белка ...................... 147
5.2.6. Характеристика экспрессируемых гибридных продуктов 147
5.3. Экспрессия гибридных генов н очистка синтезируемых продук-
тов ........................................................149
5.3.1. Индукция синтеза гибридных белков......................131
5.3.1.1. Индукция штаммов, лизогенных по ?.gt 11 . . 131
5.3.1.2. Индукция клонов, содержащих плазмиду pUR с гиб-
ридными генами..........................................
5.3.2. Осаждение клеток и выделение гибридного белка
5.3.3. Выделение гибридного белка — выбор метода . . . .
5.3.3.1. Препаративный ДСН-электрофорез в ПААГ гибридных
белков .................................................
5.3.3.2. Выделение гибридных белков иммуноаффинными мето-
дами ..............
5.3.3.3. Хроматография гибридных белков методом гель-филь-
трации .................................................
5.3.3.4. Аффинная хроматография на аминофенилтиогалактопи-
ранозиде (АФТГ, APTG)...................................
5.3.3.5. Заключение по методам очистки гибридных белков
5.4. Получение и очистка поликлональных антител.............
5.4.1. Схемы иммунизации..................................
5.4.2. Получение антисыворотки и работа с ней.............
364
Оглавление
5.4.2.1. Выделение антисыворотки из крови кролика . . . 165
5.4.2.2. Выделение анти-^-галактозидазной фракции антисыво-
ротки ......................................................165
5.4.2.3. Выделение антител, специфичных по отношению к ге-
терологичному фрагменту гибридного белка . . . 166
5.4.2.4. Определение специфичности антител методом вестерн-
блот-анализа................................................168
Литература..............................................169
Глава 6. Получение моноклональных антител к гибридным белкам, про-
дуцируемым в клетках Е. coll. Сара Е. Моул, Дэвид П. Лейн
(Sara Е. Mole, David Р. Lane).................................171
6.1. Введение..................................................171
6.2. Клонирование и экспрессия фрагментов гена.............173
6.2.1. Конструирование вектора, экспрессирующего гибридный
белок.........................................................175
6.2.1.1. Пример.............................................176
6.3. Получение моноклональных антител к гибридным белкам 180
6.3.1. Получение антигена.....................................180
6.3.2. Иммунизация животных...................................181
6.3.3. Выявление антител к белку, кодируемому клонированным
фрагментом ДНК...............................................182
6.3.3.1. Примеры............................................183
6.3.4. Скрининг гибридом......................................185
6.3.5. Слияние................................................187
6.3.5.1. Последняя инъекция антигена........................188
6.3.5.2. Подготовка миеломных клеток........................190
6.3.5.3. Взятие клеток селезенки донора.....................190
6.3.5.4. Слияние............................................191
6.3.6. Замораживание гибридомных клеточных линий . . . 194
6.3.7. Клонирование клеток гибридом...........................194
6.3.8. Загрязнение.......................................... 195
6.4. Применение моноклональных антител.........................196
6.4.1. Очистка моноклональных антител.........................197
6.4.1.1. Подклассы IgG2a, IgG2b, IgG3.......................197
6.4.1.2. Подкласс IgGl......................................198
6.4.2. Твердофазный радиоиммуноанализ.........................199
6.4.3. Биотинилирование.......................................199
6.4.4. Картирование эпитопов..................................200
6.5. Другие применения: челночное перемещение 5соР1-«кассет»
с клонированным фрагментом......................................202
6.5.1. Примеры................................................204
Литература..............................................207
Глава 7. Экспрессия и секреция продуктов гетерологичных генов в дрож-
жах. Брюс Л. А. Картер, Мейл Ирани, Вивиан Л. Маккей,
Рои Л. Сил, Анджей В. Следжевски и Роберт А. Смит
(Bruce L. A. Carter, Meher Irani, Vivian L. MacKay, Ron L.
Searle, Andrzej V. Sledzievski, Robert A. Smith) .... 208
7.1. Введение..................................................208
7.1.1. Методы, включенные в данную главу......................210
7.1.2. Экспрессирующие векторы................................210
7.1.3. Векторы для экспрессии/секреции........................216
7.2. Штаммы дрожжей, пригодные для экспрессии и их получение 219
7.2.1. Хранение дрожжевых культур......................... . 220
7.2.2. Конструирование штаммов................................221
Оглавление
365
7.2.2.1. Получение диплоидов.............................
7.2.2.2. Получение гаплоидных спор.......................
7.2.2.3. Препарирование асков и разделение тетрад аскоспор
7.3. Трансформация дрожжей..................................
7.4. Анализ трансформантов..................................
7.5. Выращивание клеток для осуществления экспрессии и секреции
продуктов клонированных генов ...............................
7.6. Приготовление образцов для анализа.....................
7.7. Анализ дрожжевой экспрессии............................
7.8. Очистка рекомбинантных белков, продуцируемых дрожжами
Литература..................................................
Глава 8. Использование амплифицирующихся векторов для экспрессии
клонированных генов в клетках млекопитающих. Кристофер
Р. Беббингтон и Кристофер К- Г. Хенчель (Christopher R. Beb-
bington, Christopher C. G. Hentschel)..........................
8.1. Введение...............................................
8.1.1. Стратегия экспрессии клонированных генов в клетках мле-
копитающих ...............................................
8.1.2. Амплификация генов................................
8.1.3. Ко-амплификация и ее использование................
8.1.4. Общая характеристика амплифицирующих векторов
8.1.5. Методы, описанные в данной главе..................
8.2. Методики селекции на амплификацию генов................
8.2.1. Использование в качестве маркера гена дигидрофолят-ре-
дуктазы...................................................
8.2.1.1. Селекция в dhfr~-клетках СНО...................
8.2.1.2. Методика трансфекции DHFR-'Клеток СНО
8.2.1.3. Амплификация в присутствии метотрексата
8.2.2. Использование металлотионеиновых генов............
8.2.3. Амплификация с использованием гена глутамин-синтетазы
8.2.3.1. Методика селекции векторов, экспрессирующих глута-
мин-синтетазу...........................................
8.2.3.2. Амплификация gs-векторов.......................
8.2.4. Аденознн-дезаминазная система.....................
8.2.4.1. Методика селекции АПА+-клеток..................
8.3. Разработка стратегии амплификации......................
8.3.1. Стабильность амплифицироваиного состояния гена
8.3.2. Строение вектора..................................
8.3.3. Частота амплификации генов........................
8.4. Контроль амплификации генов............................
8.4.1. Определение числа копий гена......................
8.4.2. Оценка уровня транскрипции........................
8.4.3. Оценка эффективности экспрессии белка.............
8.4.4. Гибридизация in situ с метафазными хромосомами
Литература................................................
221
222
223
226
230
233
234
236
236
237
238
238
238
239
241
241
245
245
245
245
246
248
249
251
253
255
256
257
258
260
261
261
262
262
263
264
265
270
Глава 9. Ретровирусные векторы. Энтони М. К. Браун и Майкл
Р. Д. Скотт (Anthony М. С. Brown, Michael R. D. Scott) . . 272
9.1. Введение.....................................................272
9.1.1. Почему именно ретровирусы?...............................272
9.1.2. Применение ретровирусных векторов........................273
9.2. Биология ретровирусов........................................274
9.2.1. Жизненный цикл ретровирусов..............................274
9.2.2. Структура ретровирусного генома и провируса . . . 276.
9.2.3. Рецепторы, тропизм и иммунность..........................277
366
Оглавление
9.3. Выбор вектора....................................................278
9.3.1. Принципы конструирования векторов.............................278
9.3.2. Векторы, дефектные по репликации..............................279
9.3.2.1. Векторы для одиночных генов...............................279
9.3.2.2. Векторы для спаренных генов...............................281
9.3.2.3. Векторы для экспрессии составных генов .... 283
9.3.2.4. Челночные векторы.........................................284
9.3.2.5. Самоинактивирующиеся векторы..............................285
9.3.3. Репликационно-компетентные векторы............................285
9.3.4. Приготовление вставочных фрагментов ДНК .... 286
9.4. Получение препаратов рекомбинантных вирусов .... 287
9.4.1. Получение препаратов, не содержащих хелперных компо-
нентов .....................................................287
9.4.1.1. Трансфекция векторной ДНК в упаковывающие клетки 288
9.4.1.2. Инфицирование клеток упаковывающих линий . . 289
9.4.1.3. Трансфекция упаковывающей ДНК...............................291
9.4.2. Получение препаратов репликациопно-компетеитных виру-
сов ........................................................291
9.4.2.1. Приготовление препаратов хелперного вируса . . . 291
9.4.2.2. Приготовление смешанных препаратов хелперного и ре-
комбинантного вирусов.................................291
9.4.3. Сбор и хранение вируса; оптимизация условий для повыше-
ния титра................................................293
9.5. Инфицирование клеток-мпшеней..........................295
9.5.1. Инфицирование с помощью вирусных препаратов . . . 295
9.5.2. Инфицирование путем совместного культивирования . . 295
9.6. Контроль вирусных препаратов..........................297
9.6.1. Титрование вируса..................................297
9.6.1.1. Дот-блот-анализ вирусной РНК...................297
9.6.1.2. Тест на активность обратной транскриптазы . . . 297
9.6.1.3. Титрование по частоте трансдукции маркера . . . 298
9.6.2. Анализ структуры и экспрессии рекомбинантного вируса 299
9.6.3. Выделение провирусной ДНК путем клонирования . . 300
9.6.3.1. Прямое клонирование провируса в плазмидном векторе 300
9.6.3.2. Метод слияния инфицированных клеток с cos-клетками 301
9.7. Потенциальные проблемы................................301
9.7.1. Неэквивалентная экспрессия спаренных генов . , . . 303
9.7.2. Генетическая нестабильность........................304
Литература...........................................305
Глава 10. Получение трансгенных мышей путем микроинъекции клони-
рованной ДНК в оплодотворенные яйцеклетки. Дэвид Мерфи
и Дженнифер Хэнсон (David Murphy, Jennifer Hanson) 308
10.1. Введение.........................................308
10.1.1. Трансгенные животные..........................308
10.1.2. Использование клеток зародышевого пути .... 308
10.1.2.1. Ретровирусы...............................309
10.1.2.2. Эмбриональные стволовые клетки............310
10.1.2.3. Микроинъекции.............................311
10.2. Обращение с животными н их содержание..........314
10.2.1. Содержание животных...........................314
10.2.2. Требования, предъявляемые к животным и оборудованию 315
10.2.2.1. Лабораторные животные.....................315
10.2.2.2. Необходимое оборудование..................316
10.2.2.3. Пипетки для трансплантации яйцеклеток .... 317
10.2.3. Манипуляции с животными.......................319
Оглавление 367
10.2.3.1. Умерщвление мышей................................319
10.2.3.3. Анестезирование мышей............................319
10.2.3.3. Вазэктомия самцов................................320
10.2.3.4. Скрещивание мышей................................321
10.2.3.5. Культивирование оплодотворенных мышиных яйцекле-
ток ......................................................324
10.2.3.6. Выделение оплодотворенных одноклеточных яйцекле-
ток ......................................................325
10.2.3.7. Трансплантация яйцеклеток........................329
10.2.3.8. Кесарево сечение и вскармливание.................332
10.3. Микроинъекции............................................333
10.3.1. Оборудование для микроинъекций.........................333
10.3.1.1. Микроскоп........................................334
10.3.1.2. Микроманипуляторы................................335
10.3.1.3. Инъекционная камера..............................336
10.3.1.4. Изготовление поддерживающих микропипеток . . 336
10.3.1.5. Подготовка инъекционной микропипетки .... 337
10.3.2. Микроинъекция в оплодотворенные одноклеточные яйце-
клетки мыши.................................................338
10.4. Манипулирование с ДНК................................... 341
10.4.1. ДНК для микроинъекции..................................341
10.4.1.1. Физические параметры ДНК.........................341
10.4.1.2. Раствор ДНК......................................341
10.4.1.3. Прокариотические ДНК.............................342
10.4.1.4. Приготовление и очистка препаратов ДНК для микро-
инъекцип .................................................343
10.4.2. Идентификация трансгенных мышей........................343
10.4.2.1. Блот-гибриднзациониый анализ.....................343
10.4.2.2. Слот (и дот)-блот-анализ.........................346
10.4.2.3. Идентификация гомозиготных трансгенных мышей 347
10.5. Разведение трансгенных мышей н анализ экспрессии экзоген-
ных генов.....................................................350
10.5.1. Получение трансгенных линий............................350
10.5.2. Экспрессия генов в трансгенных мышах...................350
10.5.3. Применение трансгенной технологии......................351
Литература.............................................352
Список использованных сокращений...............................354
Предметный указатель...........................................355
Уважаемый читатель!
Ваши замечания о содержании книги, ее
оформлении, качестве перевода и другие просим
присылать по адресу:
129820, Москва
1-й Рижский пер., д. 2,
издательство «Мир»
НАУЧНОЕ ИЗДАНИЕ
Кристофер Р. Беббингтон, Энтони М. К. Браун,
Син. Б. Кэрролл и др.
НОВОЕ В КЛОНИРОВАНИИ ДНК. МЕТОДЫ
Под ред. Дэвида М. Гловера
Заведующий редакцией чл.-корр. АН СССР Т. М. Турпаев. Зам. зав. ре-
дакцией М. Д. Гроздова. Научн. редактор Л. Г. Тер-Саркисян. Мл. редак-
тор М. С. Карнюшина. Художник А. С. Захаров. Художественные редакто-
ры А. Я. Мусин, Л. М. Аленичева. Технический редактор Т. А. Мироши-
на. Корректор В. С. Соколов
ИБ JMe 7073
Сдано в набор 23VI.89. Подписано к печати 5Х.89. Формат 60X90!/ie.
Бумага типографская № 1. Печать высокая. Гарнитура Литературная.
Объем 11,5 бум. л. Усл. печ. л. 23,0. Усл. кр.-отт. 23,0. Уч. изд. л. 25,17.
Изд. № 4/6534. Тираж 6100 экз. Зак. 1186. Цена 2 р. 80 к.
Издательство «Мир» В/О «Совэкспорткнига» Государственного комитета
СССР по делам издательств, полиграфии и книжной торговли. 129820
ГСП, Москва, 1-й Рижский пер., 2.
Московская типография № 11 Госкомпечати СССР.
113105, Москва, Нагатинская ул., д. 1.