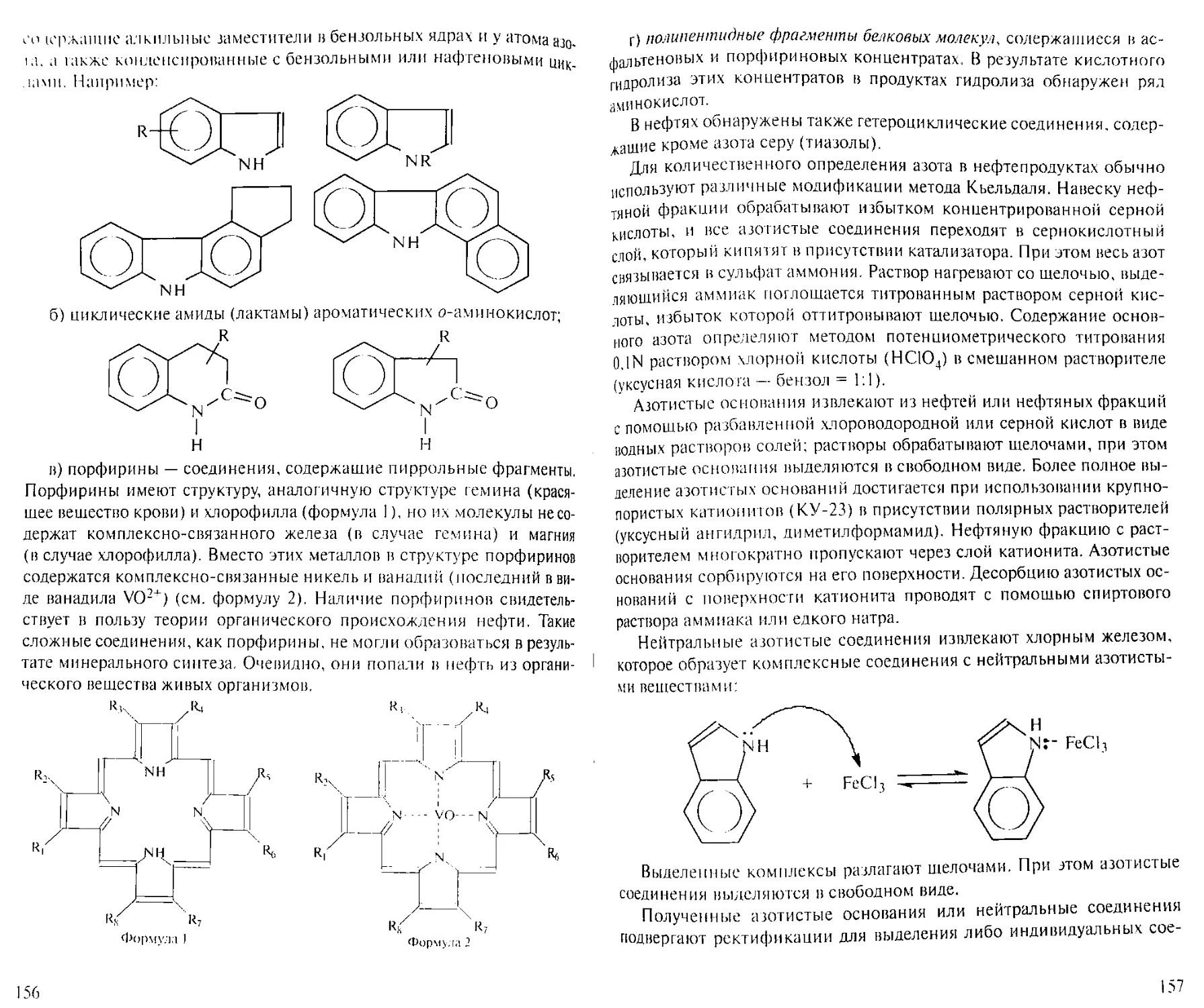
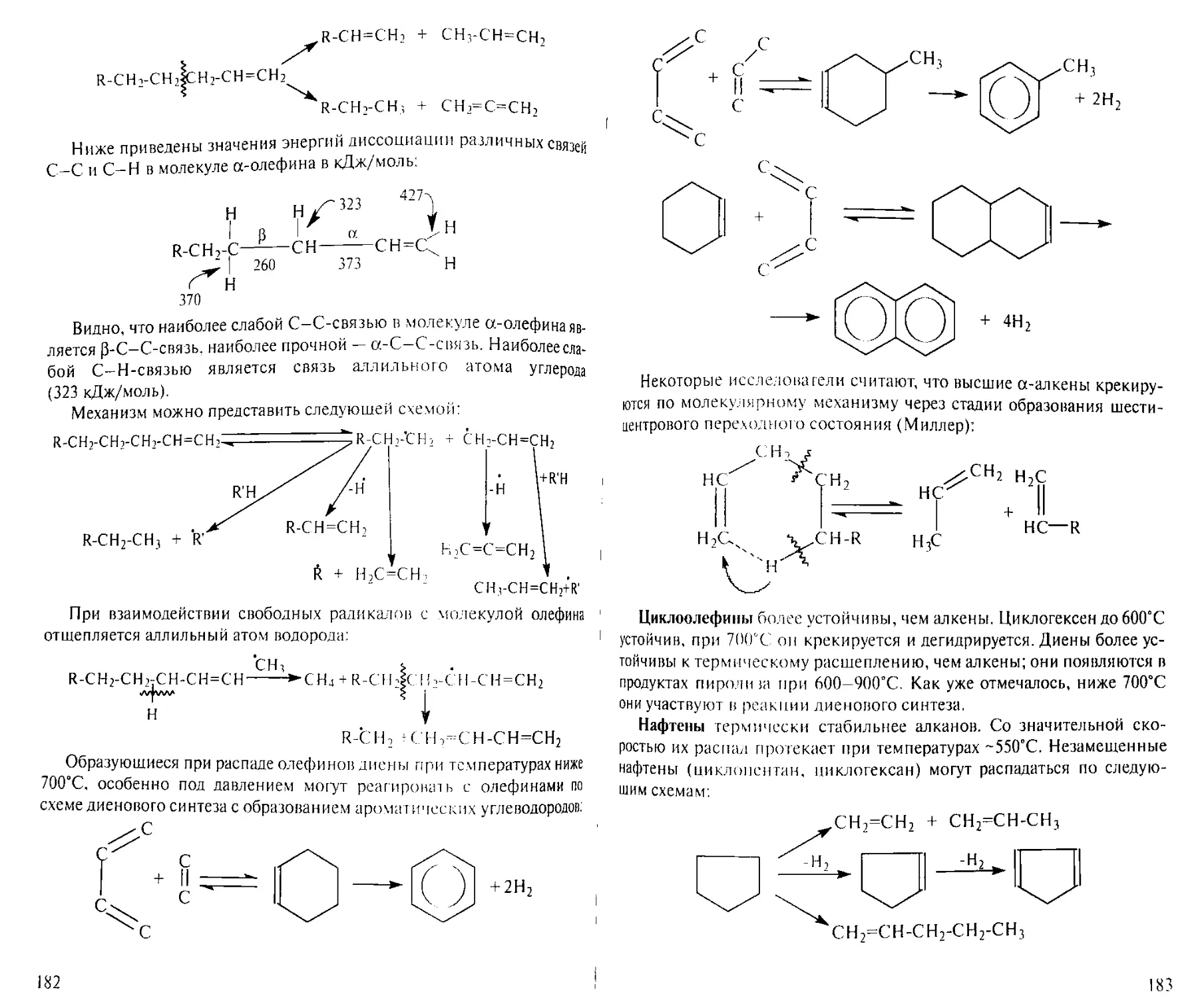
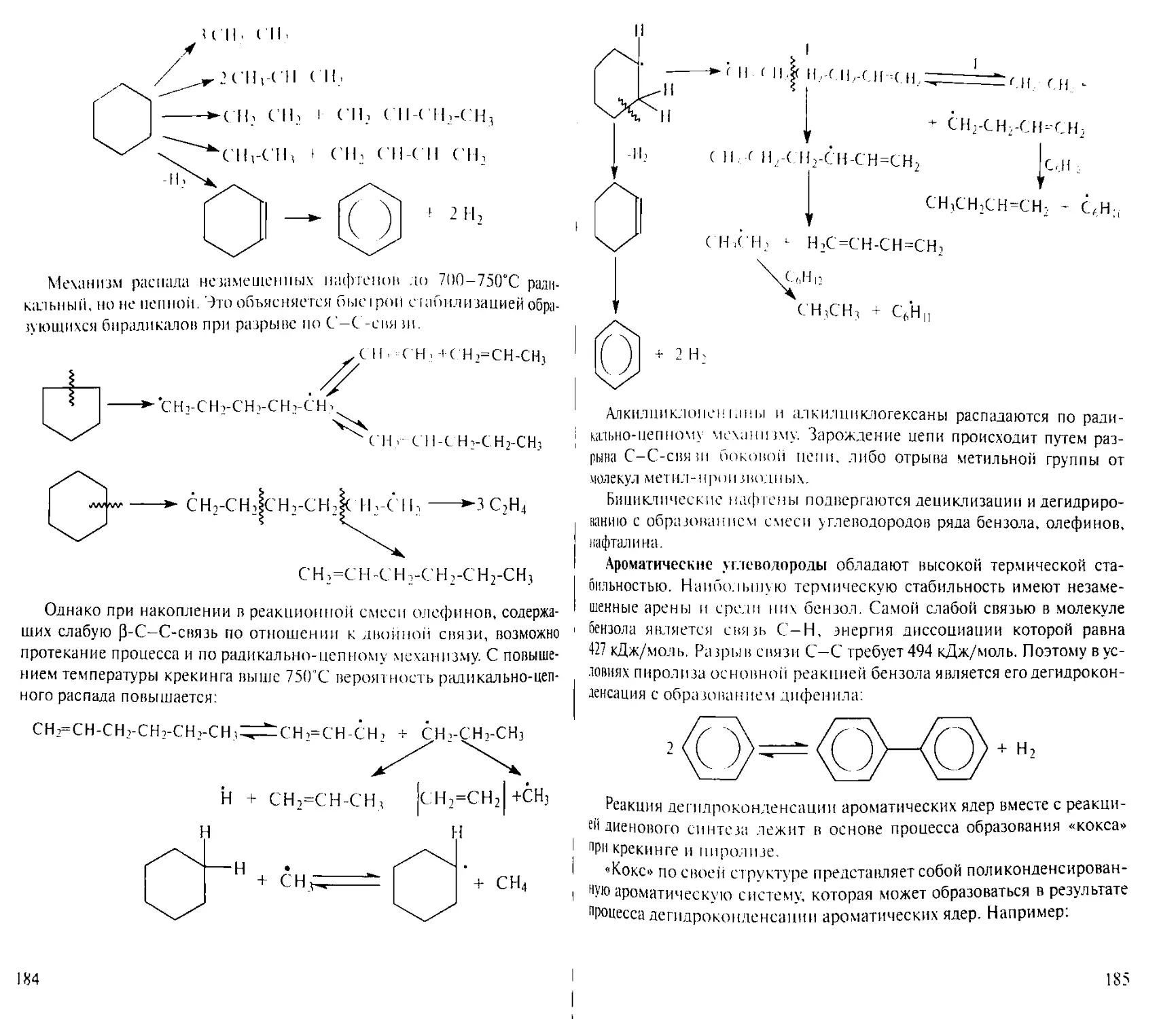
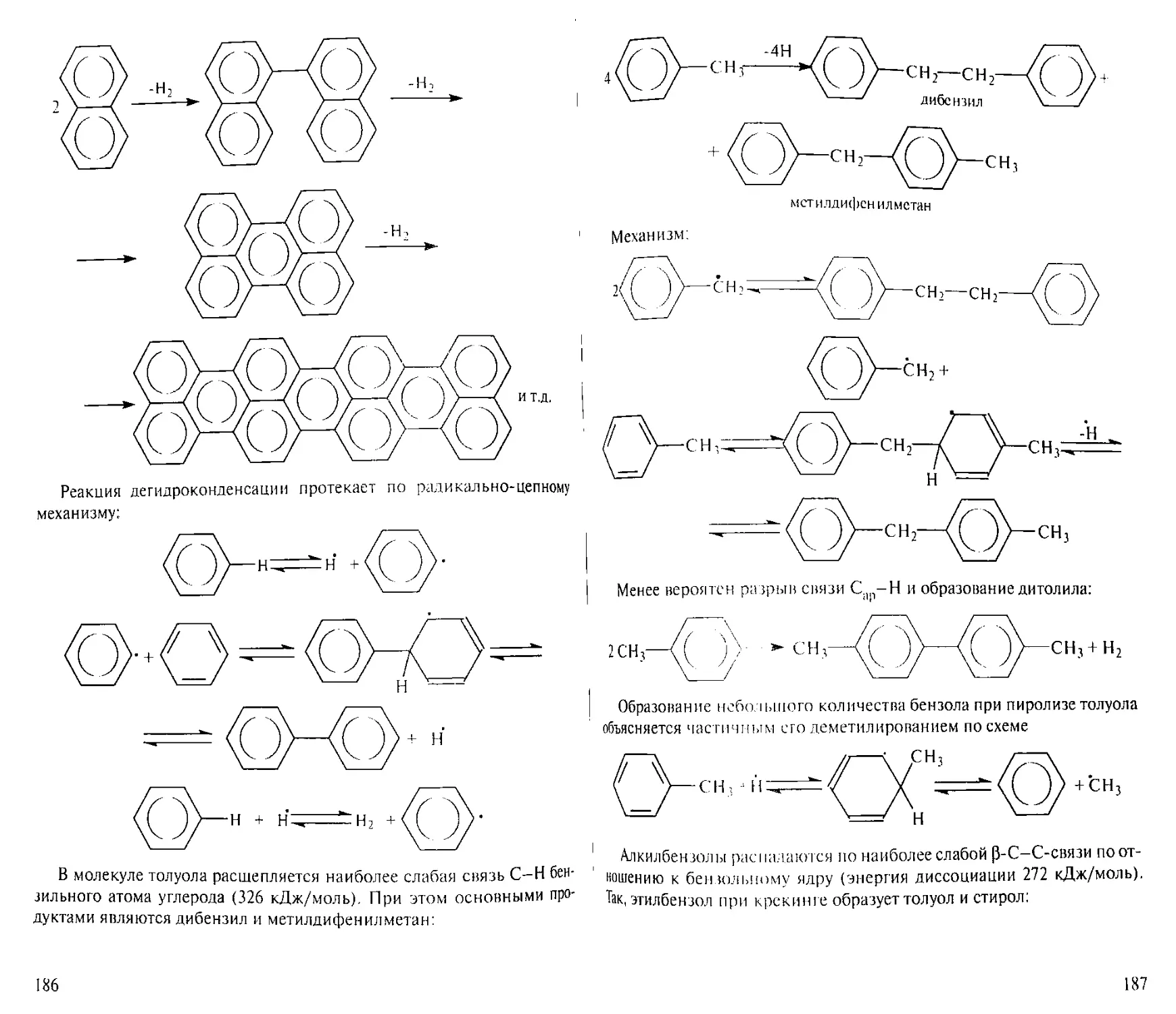
/















































































































Author: Рябов В.Д.
Tags: технология минеральных масел технология нефти и аналогичного сырья химия нефть газ
ISBN: 5-93969-023-8
Year: 2004




Text
фПКчПК
hjoOMbl
ooog/^XX-x4^ .
v
- I fi:4;'.-UC!.rt!. •)ff.". --'' 1
РОССИЙСКИЙ ГОСУДАРСТВЕННЫЙ УНИВЕРСИТЕТ
НЕФТИ И ГАЗА им. И. М. ГУБКИНА
В. Д. Рябов
ХИМИЯ НЕФТИ И ГАЗА
Издание второе, исправленное и дополненное
Утверждено Советом университета
в качестве учебника
Москва
2004
УДК 665.6(075.8)
ПРЕДИСЛОВИЕ
Рецензенты: Л 3. Магарил,
д.т.н., профессор, заведующий кафедрой химии
и технологии переработки нефти Тюменского
государственного нефтегазового университета
В. Н. Перченко,
д.х.н., профессор Института нефтехимического
синтеза им. А. В. Топчиева РАН
В. Д. Рябов
Химия нефти и газа. — М.: Издательство «Техника», ТУМА ГРУПП,
2004. - 288 с.
ISBN 5-93969-023-8
Прицелены современные данные о составе, свойствах, методах анализа углеводородов
и других компонентов нефти и газа.
Рассмотрены химические основы термических и каталитических превращений углево-
дородов и гстероатомных соединений нефти.
Излагаются основные гипотезы неорганического и органического происхождения
нефти,
Книга предназначена в качестве учебника по курсу «Химия нефти и газа» лдя студентов'
хи мн ко-технологических специальностей вузов и факультетов нефтегазового профиля и
может представлять интерес для студентов других специальностей нефтяных вузов, для
специалистов, работающих в области химии и технологии нефти и некоторых других об-
ластях нефтяной и газовой промышленности.
© В. Д Рябов, 2004
© Издательство «Техника», ТУМА ГРУПП, 2004
Химия нефти как наука получила свое развитие is конце XIX века и
по мере накопления знаний о нефти, ее составе и свойствах оконча-
тельно сформировалась в первые десятилетия XX века.
Как учебная дисциплина «Химия нефти» была впервые в мире
включена в учебные планы подготовки инженеров-технологов по пере-
работке нефти С. С. Наметкиным в 1927 г. в Московской горной акаде-
мии, а затем в Московском нефтяном институте им. И. М. Губкина.
С 1930 г. преподавание «Химии нефти» было начато также в Ленин-
градском технологическом институте им. Ленсовета А. Ф. Добрянским.
По мерс развития и дифференциации химии вообще, и химии нефти
в частности, изменялись ее цели, задачи и содержание. В первой поло-
вине XX века химия нефти занималась не только вопросами изучения
химического состава нефтей, свойств углеводородов и других компо-
нентов нефти, но и проблемами переработки нефти: первичной, тер-
мокаталигической, химической: вопросами химической очистки неф-
тепродуктов. В соответствии с этим и составлялись учебные програм-
мы и готовились учебные пособия по химии нефти.
После создания учебных курсов «Технология переработки нефти»
(1933) и «Технология нефтехимического синтеза» (60-е гт.) из програм-
мы курса «Химия нефти» были исключены вопросы технологии пере-
работки нефти и нефтехимического синтеза. В связи с развитием
инструментальных физико-химических методов исследования потеря-
ли значение многие химические методы выделения, количественного
определения и идентификации компонентов нефти, которые раньше
излагались в курсе «Химия нефти».
В настоящее время «Химия нефти и газа» как наука решает следую-
щие задачи:
I. Исследование химического состава нефтей, нефтепродуктов,
газоконденсатов и газов с помощью современных физико-химических
методов анализа.
2. Исследование физико-химических свойств углеводородов и дру-
гих компонентов нефти и их влияния на свойства нефтепродуктов,
установление связи между строением молекул и надмолекулярных
структур компонентов нефти, их способностью к межмолекулярным
взаимодействиям и фазовым переходам и свойствами нефтепродуктов.
3. Исследование химизма и механизма термических и каталитичес-
ких превращений компонентов нефти, в том числе взаимных превраще-
ний углеводородов как высокотемпературных (в процессах переработки
нефти), гак и низкотемпературных, что важно как с аналитической. так
и с геохимической (превращение нефтей в природе) точек зрения.
Рис. 1. Логическая схема и структура курса «Химия нефти и газа»
в связи с другими дисциплинами учебного плана
/I
4. Исследование проблемы происхождения нефти.
В соответствии с этим находится и содержание курса "Химия нефти
и таза» как учебной дисциплины.
На рис. 1 приведена логическая схема и структура курса «Химия
нефти и газа» в связи с другими дисциплинами учебного плана.
Во вводной части курса приводятся краткие сведения о роли нефти
и газа в современном мире, об основных нефтедобывающих странах и
нефтяных месторождениях, предварительные данные о химическом и
фракционном составе нефтей, о химической классификации нефтей.
Эти данные необходимы при изучении главы 1 курса «Физико-хими-
ческие методы исследования состава нефтей и газов». На основании
главы I курса рассматривается материал главы II курса «Химический
состав, свойства и анализ углеводородов и других компонентов нефти
и газа» Из химических свойств компонентов нефти рассматриваются
только те, которые имеют либо аналитическое значение, либо необхо-
димы при и зучении хими зма процессов термической и каталитической
переработки нефтяного сырья.
Методы определения состава различных фракций нефти и нефте-
продуктов обобщены в III главе курса.
В главе IV курса «Гетероатомные соединения нефти» рассматрива-
ются данные о составе, строении, свойствах и анализе кислородных,
сернистых, азотистых, смолисто-асфальтеновых, минеральных компо-
нентов нефти.
Глава V курса рассматривает термические и каталитические превра-
щения углеводородов и других соединений нефти в условиях основных
процессов термокаталнгической переработки нефтяного сырья. В этом
разделе приводятся данные о превращениях углеводородов в условиях
реакций алкилирования и ступенчатой полимеризации и изомеризации
с целью получения высокооктановых компонентов моторных топлив.
В главе VI курса рассматриваются вопросы происхождения нефти и
ее отдельных компонентов.
Логическая схема и структура курса показывают также связь отдель-
ных разделов курса между собой и с другими дисциплинами учебного
плана
Настоящему учебнику предшествовал ряд учебных пособий, подго-
товленных авюром и изданных в РГУ нефти и газа им. И. М. Губкина
в разные годы. Эго конспект лекций по курсу «Химия нефти и газа»
(1976), «Физико-химические методы исследования углеводородов и
других компонентов нефти» (I и II издания 1979 и 1996 гг.), «Химичес-
кий состав, свойства и анализ углеводородов и других соединений неф-
ти» (I и II издания. 1983 и 1997 гг.). «Термические и каталитические
превращения углеводородов и других компонентов нефти» (1982).
ВВОДНАЯ ЧАСТЬ
Нефть является горючим ископаемым наряду с каменным углем,;
бурым углем и сланцами. В отличие от других горючих ископаемых
нефть — жидкость и содержит очень мало минеральных негорючих
примесей, что обусловливает ее высокую теплотворную способность
42000 кДж/кг (10000 ккал/кг). i
Несмотря на высокую теплотворную способность, сырая нефть не
используется в качестве топлива, так как представляет собой богатый
источник углеводородов — ценного сырья для получения нефтепрсы
дуктов (бензина, дизельных топлив, смазочных масел и т. д.) и п роду к-;
тов нефтехимического синтеза.1
Нужно отметить, что другие горючие ископаемые представляют
собой также потенциальные ресурсы углеводородов. Их природные за-;
пасы значительно превосходят запасы нефти. Так, в 1958 г, мировые'
запасы каменного угля были равны 7500 млрд т, тогда как мировые раз-
веданные2 запасы нефти — 33 млрдт. В 1964 г. мировые разведанные за--
пасы нефти в зарубежных странах составляли 40 млрд т, в 1971 г. — |
60 млрдт. В настоящее время мировые разведанные запасы нефти рав-
ны 100 млрдт.3 Следовательно, увеличение разведанных запасов нефти
невелико. В 1964 г. добыча нефти составила 1,4 млрд т. В 1970 г. миро-
вая добыча нефти увеличилась почти вдвое — 2,3 млрд т, в 1971 г. —.
2,5 млрд т. В настоящее время мировая добыча нефти стабилизировав
лась на уровне 2,6 млрд т. 1
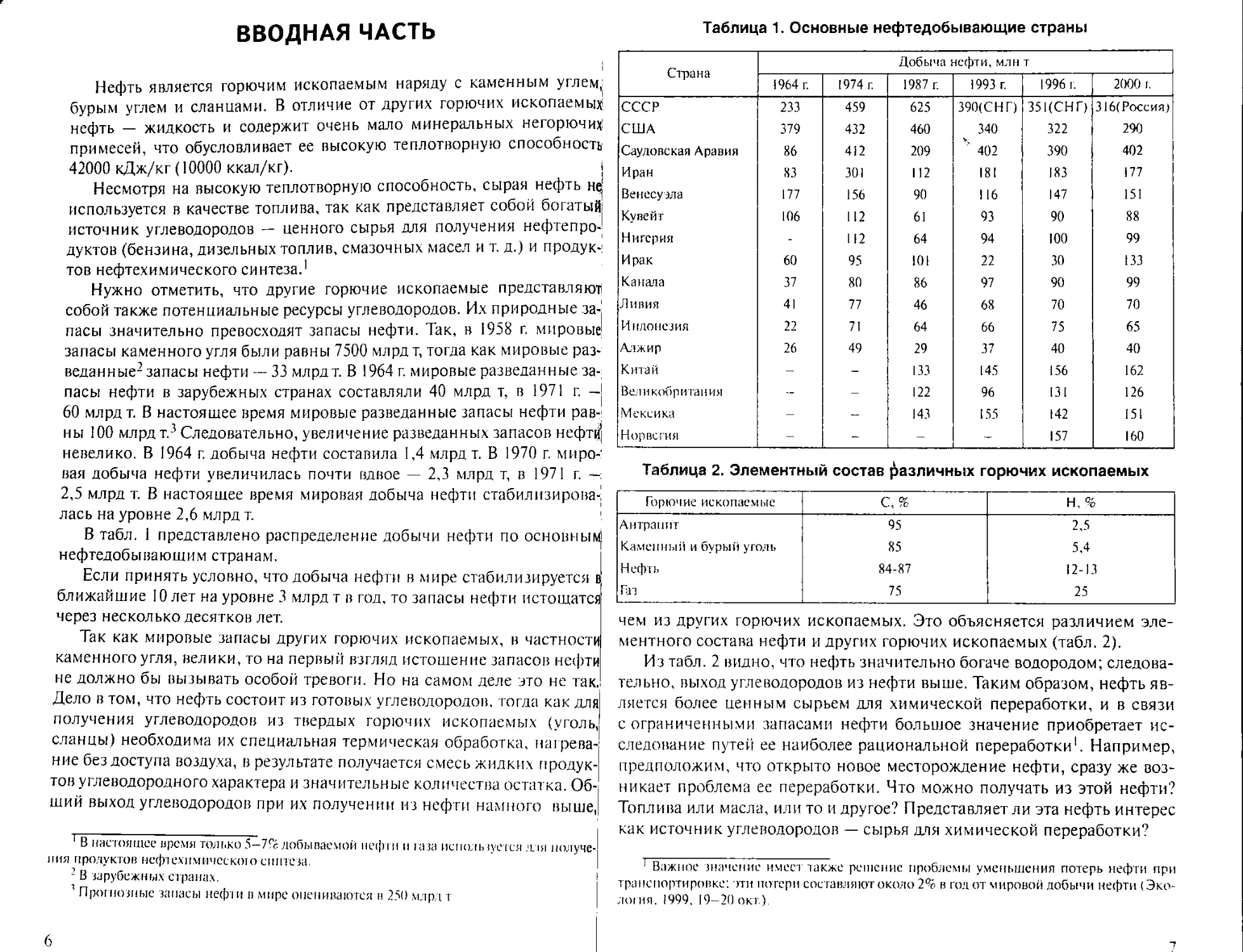
В табл. 1 представлено распределение добычи нефти по основным
нефтедобывающим странам.
Если принять условно, что добыча нефти в мире стабилизируется в
ближайшие 10 лет на уровне 3 млрд т в год, то запасы нефти истощатся
через несколько десятков лет.
Так как мировые запасы других горючих ископаемых, в частности
каменного угля, велики, то на первый взгляд истощение запасов нефти
не должно бы вызывать особой тревоги. Но на самом деле это не так.
Дело в том, что нефть состоит из готовых углеводородов, тогда как для
получения углеводородов из твердых горючих ископаемых (уголь,
сланцы) необходима их специальная термическая обработка, нагрева-
ние без доступа воздуха, в результате получается смесь жидких продук-
тов углеводородного характера и значительные количества остатка. Об-
щий выход углеводородов при их получении из нефти намного выше,
1 В настоящее время только 5-7Гс добываемой нефти и газа используется дчя получе-
ния продуктов нефтехимическою синтеза,
2 В зарубежных странах.
• Прогнозные запасы нефти в мире оно пинаются в 250 млрд г
Таблица 1. Основные нефтедобывающие страны
Страна Добыча нефти, млн т
! 964 г. 1974 г. 1987 г. 1993 г. 1996 г. 2000 I.
СССР 233 459 625 390(СНГ) 35ЦСНГ) 316(Россия)
США 379 432 460 340 322 290
Саудовская Аравия 86 412 209 ' 402 390 402
Иран 83 301 112 181 183 177
Венесуэла 177 156 90 116 147 151
Кувейт 106 112 61 93 90 88
Нигерия - 112 64 94 100 99
Ирак 60 95 101 22 30 133
Канала 37 80 86 97 90 99
Ливия 41 77 46 68 70 70
Индонезия 22 71 64 66 75 65
Алжир 26 49 29 37 40 40
К тай — — 133 145 156 162
Великобритания — 122 96 131 126
Мексика — 143 155 142 151
Норвегия — — — — 157 160
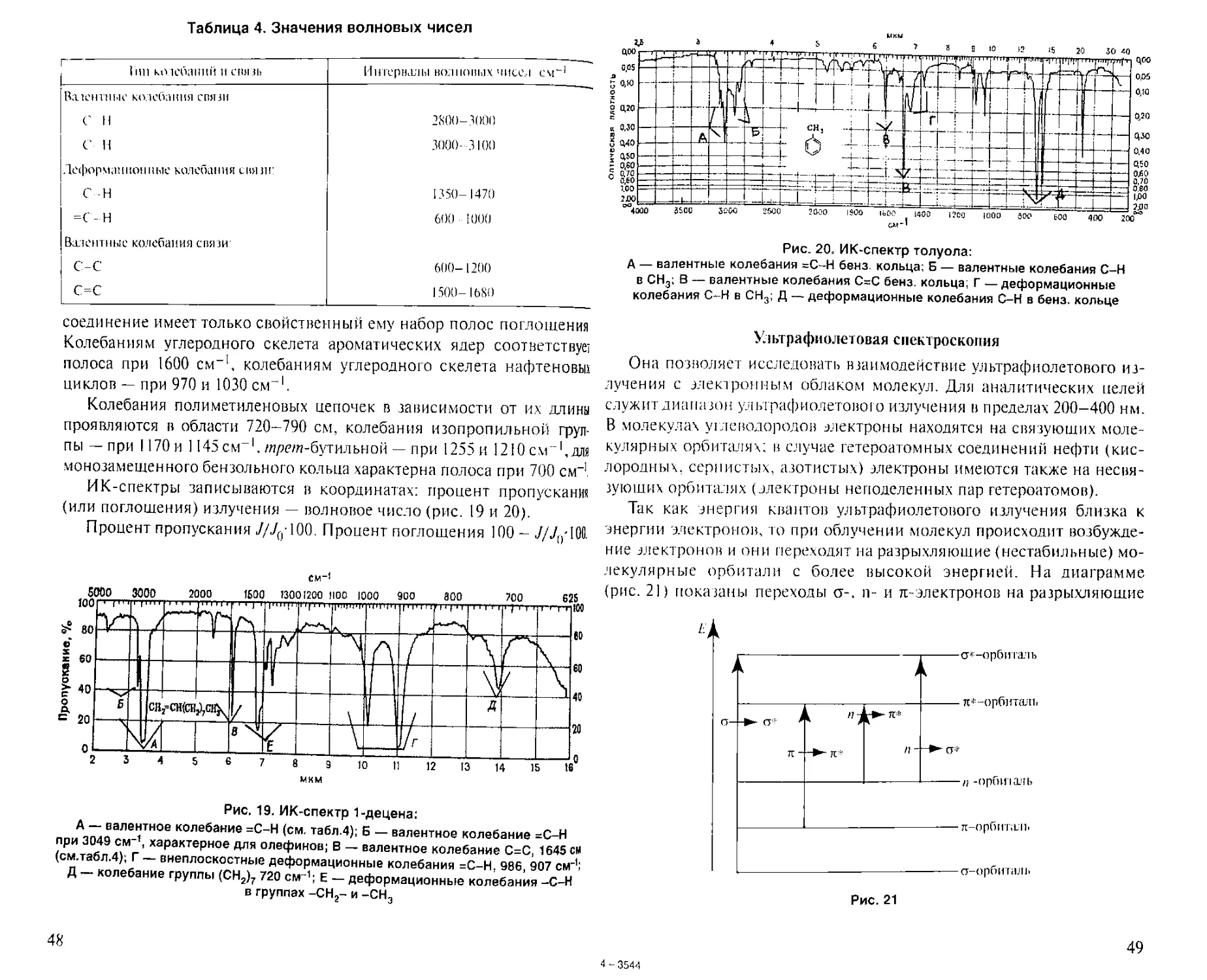
Таблица 2. Элементный состав различных горючих ископаемых
Горючие ископаемые С, % Н, %
Антрапит 95 2,5
Каменный и бурый уголь 85 5,4
Нефть 84-87 12-13
Газ 75 25
чем из других горючих ископаемых. Это объясняется различием эле-
ментного состава нефти и других горючих ископаемых (табл. 2).
Из табл. 2 видно, что нефть значительно богаче водородом; следова-
тельно, выход углеводородов из нефти выше. Таким образом, нефть яв-
ляется более ценным сырьем для химической переработки, и в связи
с ограниченными запасами нефти большое значение приобретает ис-
следование путей ее наиболее рациональной переработки1. Например,
предположим, что открыто новое месторождение нефти, сразу же воз-
никает проблема ее переработки. Что можно получать из этой нефти?
Топлива или масла, или то и другое? Представляет ли эта нефть интерес
как источник углеводородов — сырья для химической переработки?
1 Важное значение имеет также решение проблемы уменьшения потерь нефти при
транспортировке: эти потери составляют около 2% в год от мировой добычи нефти (Эко-
логия. 1999, [9-20 окт).
7
Для того чтобы ответить на эти вопросы, необходимо детальное
исследование химического состава нефти с применением современных
методов физико-химического анализа. Таким образом, изучение,
состава нефтей является одной из важнейших задач химии нефти. ;
В разработке научных основ исследования нефтей выдающаяся!
роль принадлежит трудам российских и советских ученых:
Ф. Ф. Бейльштейна, Д. И. Менделеева, А. А. Курбатова, В. В. Марков-
никова, М. И. Коновалова, Л. В. Гурвича - по изучению химического,
состава кавказских нефтей; С. С. Наметкина, А. В. Топчиева,
Ал. А. Петрова, А. Ф. Добрянского, П. И. Санина — по исследованию
состава ряда нефтей и природных газов нашей страны, А. М. Бутлеро-
ва, В. Н. Ипатьева, Н. Д. Зелинского, С. С. Наметкина, Б. А. Казанско-
го, А. Ф. Платэ — в области каталитических превращений углеводоро-
дов. Существенный вклад в развитие химии углеводородов нефти внес-
ли работы зарубежных исследователей. Это прежде всего работы
немецкого химика К. Шорлеммера по исследованию алканов нефтей,
работы ученых Американского нефтяного института: Вошборна, Рос-
сини, Мейра, а также исследования Уотермана и Флюгера, Ван Неса и
Ван Вестена.
Рассмотрим теперь кратко понятие о нефти как природном объекте.
Нефть представляет собой сложную смесь углеводородов и органичес-
ких соединений серы, азота, кислорода. Это маслянистая жидкость,
обычно темно-бурого цвета, хотя встречаются нефти и светлые. Отно-
сительные плотности нефтей колеблются обычно между 0,8 и 0,9, а мо-
лекулярная масса в пределах 200-300. Нефть обычно залегает в порис-
тых породах — в песках, песчаниках, известняках (коллекторы). Часто
в нефтяном месторождении вода и большие количества газа сопутству-
ют нефти (рис. 2). Вода, имеющая большую плотность, чем нефть, за-
нимает в нефтяном месторождении нижнюю часть, а газ — верхнюю.
Нефть обычно залегает на больших глубинах — от 2 до 5 км, иногда и
на большей глубине, и вследствие этого она находится в довольно
жестких условиях. Температура в пласте может достигать больших зна-
чений — 200°С и более. Давление в нефтяном пласте зависит от глуби-1
Рис. 2
ны залегания и от температуры (уп-i
ругость паров углеводородов); оно!
обычно бывает 10,0-15,0 МПа, но!
может достигать и более высоких;
значений. I
В газовых месторождениях значе-;
ния температуры и давления также!
высоки. В некоторых газовых место-!
рождениях с высокими давлением и
температурой (20,0-40,0 МПа и
100-200ГС), соответствующими надкритической области, в газе может
содержаться значительное количество углеводородов, являющихся
жидкими при нормальных условиях. В процессе добычи газа при его
выходе на поверхность давление снижается и происходит выделение
жидких углеводородов из газовой фазы (так называемая обратная или
ретроградная конденсация). Образующаяся при этом жидкая углеводо-
родная фаза называется конденсатом. Такие месторождения называют-
ся конденсатными. Конденсаты представляют собой смесь алканов,
нафтенов и аренов с концом кипения 300—350°С.
Сравнительно жесткие условия, в которых находятся нефти и газо-
конденсаты, обусловливают практически полное отсутствие таких хи-
мически активных соединений, как алкины, алкадиены, альдегиды,
спирты.
Как уже говорилось, основными элементами, входящими в состав
нефти, являются углерод и водород. Содержание углерода в нефти
об ы ч н о кол ебл етс яви редел ах 8 2 - 8 7 % и водорода 12-13%, На дол ю се -
ры, кислорода и азота приходится обычно 1-5%. Чаще всего встречает-
ся в нефтях сера. Ее содержание в отдельных нефтях может достигать
5-6%. Содержание азота и кислорода обычно не превышает несколь-
ких десятых процента, но может достигать иногда 1,5-2%. Таким обра-
зом, н ефт ь состо и т в ос н о в и о м и з у г лерода и водорода. С л ед о вател ьно,
основными соединениями, входящими в состав нефти, являются угле-
водороды. Кроме углеводородов, нефть содержит другие соединения.
Рассмотрим предварительно в общих чертах химический состав нефти,
ее основные компоненты и фракционный состав.
Краткая характеристика компонентов нефти
Атканы (парафины)
Эти углеводороды составляют основную часть нефти. Обычно со-
держание алканов в нефтях колеблется от 20 до 50%. Некоторые нефти,
так называемые слабопарафинистые или беспарафинистые, содержат
не более \-2% этих углеводородов, другие могут содержать до 80% этих
углеводородов, и они носят название парафинистых нефтей.
Циклоалканы (нафтены, цикланы)
Моной и кл ячеек ие нафтены представлены в нефтях в основном
производными циклопентана и циклогексана. Производные низших
циклов в нефтях не найдены; в небольших количествах в некоторых
нефтях найдены производные высших циклоалканов. Кроме моно-
циклических нафтенов, нефти содержат бициклические, трицикли-
ческие и полициклические углеводороды. Обычно содержание нафте-
нов в различных нефтях составляет 30-50%. Однако в некоторых неф-
8
тях (слабопарафиннстые и беспарафинистые) может быть до 80% наф-
тенов,
Ароматические углеводороды (арены) ;
Этот тип углеводородов слабо представлен в нефтях. Обычно нефти
содержат 15-20% аренов. В некоторых нефтях их содержание может!
достигать 35%. Кроме ароматических углеводородов ряда бензола,!
в нефтях содержатся производные полициклических аренов. Отдель-!
ную группу составляют углеводороды смешанного строения. Молекул
лы таких углеводородов содержат ароматические и нафтеновые кольца
и парафиновые цепи.
Кислородные соединения
Эти соединения представлены в основном фенолами, жирными!
кислотами и нафтеновыми кислотами. Кислоты содержатся главным
образом в средних нефтяных погонах в количестве 1-2%.
Азотистые соединения
Эти вещества представлены в нефтях в основном гетероцикли-!
вескими соединениями.
I
Сернистые соединения ,
В нефтях содержатся меркаптаны, сульфиды, дисульфиды, гетеро-
циклические сернистые соединения (производные тиофана, тиофена).
Смолисто-асфальтеновые вещества
Эти вещества по своей природе представляют собой мн ого кольча-
тые соединения, содержащие нафтеновые, ароматические циклы и ге-
тероциклы с атомами кислорода, азота и серы. Содержание этих соеди-
нений в нефтях может изменяться от нескольких процентов до 10-40%
(в случае смолистых нефтей).
Минеральные вещества
К этим веществам относится вода до (4%) и различные минераль-
ные соли, которые находятся в растворенном в воде состоянии. В неф-
тях также содержатся соли различных металлов и органических кислот,
называемых нефтяными, металлы, входящие в состав некоторых комп-
лексных соединений, а также сера и сероводород.
Кроме перечисленных, в нефтях найдены вещества, которые, как
доказано в настоящее время, образовались из продуктов животного и
растительного происхождения. Эти вещества получили название «био-
логических меток» или «биомаркеров», так как указывают на связь!
нефти с живой природой. К «биологическим меткам» относятся,
например, следующие группы веществ.
Оптически активные вещества
В высококипяших фракциях нефти, имеющих температуру кипения
порядка 450-500°С (300-320°С при 6-8 мм рт, ст.) содержатся вещест-
ва, присутствие которых в этих фракциях вызывает вращение плоскос-
ти поляризации поляризованного луча света. Было установлено, что
такие соединения относятся к полициклическим нафтеновым углево-
дородам (3-5 и более циклов в молекуле). Эти оптически активные со-
единения не могли образоваться путем превращения углеводородов
нефти, так как при синтезе соединений с асимметрическим углерод-
ным атомом всегда образуется рацемическая смесь, не обладающая оп-
тической активностью. Поэтому предполагают, что оптически актив-
ные соединения перешли в нефть из органического вещества вымер-
ших десятки и сотни миллионов лет назад живых организмов. Таким
веществом может быть, например, содержащийся в живых организмах
холестерн н:
Холестерин вращает плоскость луча поляризованного света влево
(против часовой стрелки). Интересно отметить, что продукты превра-
щения холестерина являются правовращающими. Так, из нефтей выде-
лен холестан — углеводород, структура которого соответствует структу-
ре холестерина и который является правовращающим, [а]л =24 :
Оптическая активность органических соединений с точки зрения
термодинамики является маловероятным состоянием, так как это со-
Таблица 3. Зависимость оптической активности нефтей
от их возраста
Нефть третичного периода
Нефть силурийского периода
стояние требует повышенной свободной энергии. Процессы в природе
стремятся к уменьшению свободной энергии. Однако для очень слож-
ных оптически активных соединений процесс образования рацемичес-
кой смеси с минимумом свободной энергии является крайне медлен-
ным процессом (хотя он протекает). Примером служит уменьшение
оптической активности нефтей с увеличением их геологического воз-
раста (табл. 3).
Изопреноидные углеводороды (изопренаны)
Это разветвленные алканы, молекулы которых содержат повторяю-
щееся углеводородное звено, углеродный скелет которого соответству-
ет структуре изопрена:
---СН2-СН —СН2— СН?—
СН,
Установлено, что эти углеводороды могли образоваться из фитола —
непредельного спирта изопреноидной структуры, являющегося составу
ной частью хлорофилла. |
Порфирины
Порфирины являются производными гетероциклического соедине-i
ния пиррола. В виде комплексов с металлами они входят в состав геми-
на — красящего вещества крови и в состав хлорофилла. В нефтях най-
дены как свободные порфирины, так и комплексы порфиринов
с металлами (ванадий, никель). ,
Подробно о «биомаркерах» см. в главе II.
Химическая классификация нефтей
Химическая классификация нефтей строится в зависимости от пре-
обладания в них углеводородов различных рядов. При химической
классификации нефти иногда учитывается содержание гетероатомный
соединений. Предложен ряд методов химической классификации неф-
тей. В 1967 г. А. Э. Конторович с сотрудниками предложили класс и фи-!
кацию, которая строится в соответствии с групповым углеводородным
составом фракции нефти, выкипающей при 250-300°С, т. е. содержа-.
ни ем в этой фракции аренов, нафтенов и алканов1. В зависимости от
преобладания в этой фракции углеводородов одного ряда (выше 50%)
нефти делятся на 3 основных типа:
— метановые — нефти грозненские парафинистые, сураханская, не-
которые румынские, нефти Западной Украины, Татарии, Самотлора,
полуострова Мангышлак:
- нафтеновые — эмбенские, некоторые бакинские (нефть место-
рождения Грязевая Сопка), калифорнийские нефти США;
- ароматические — нефть месторождения Чусовские Городки, май-
копская, нефти Зондских островов.
При содержании во фракции 250—300вС более 25% углеводородов
других рядов нефти относят к смешанному типу: мета но-нафтеновый,
нафтено-мета новый, а ром ат и чес ко-нафте новый и т, д. В этих назван и-
ях первым ставится название углеводородов с меньшим содержанием,
Наиболее распространенными являются нефти метано-нафтеновые
и нафтено-метановые. Ал. А. Петров подразделяет все нефти на 4 типа
(А1, А“, Б". Б1) в зависимости от концентрации алканов, разветвленных
алканов, нафтенов во фракции нефти 200—430"С. Нефти первых двух
типов А1 и А2 характеризуются высокой концентрацией w-алканов и
изопрена нов; нефти типа Б1 и Б2 — высоким содержанием нафтенов.
В нефтях типа А1 содержание w-алканов выше, чем в нефтях типа А2.
Подробно о классификации (химической типизации) нефтей
Ал. А. Петрова см. с. 72.
Кроме химической, имеется технологическая классификация неф-
тей, в соответствии с которой нефти подразделяются на ряд классов в
зависимости от таких характеристик, как содержание серы, содержа-
ние фракций, выкипающих до 350°С, содержание масляных фракций,
парафина и т. д.
Фракционный состав нефтей
Нефть перегонкой (см. гл. I) разделяют на следующие фракции:
1) оензиновую фракцию (от температуры начала кипения нефти до
15(.)Г'С или 180’С или до 200’С);
2) керосиновую фракцию (от конца кипения бензиновой фракции
до -’250°С):
3) керосине-газойлевую фракцию или легкий газойль (от конца ки
пения бензиновой фракции до 35О°С), Остаток от перегонки >35О°(
пазываегея мазутом. Мазут перегоняют в вакууме и получают вакуум
ный газойль (350-550°С ) и остаток выше 550°С — гудрон.
7 Конторович А Э Нефтепром зволя низе толши и условия образования нефти в мезо
зойских отложениях Западно-Сибирской ни змснпости. — Л.: Недра, 1967.
I
12
Глава I
ФИЗИКО-ХИМИЧЕСКИЕ МЕТОДЫ
ИССЛЕДОВАНИЯ НЕФТИ И ГАЗА
§1. Физико-химические методы разделения
компонентов нефти и газа
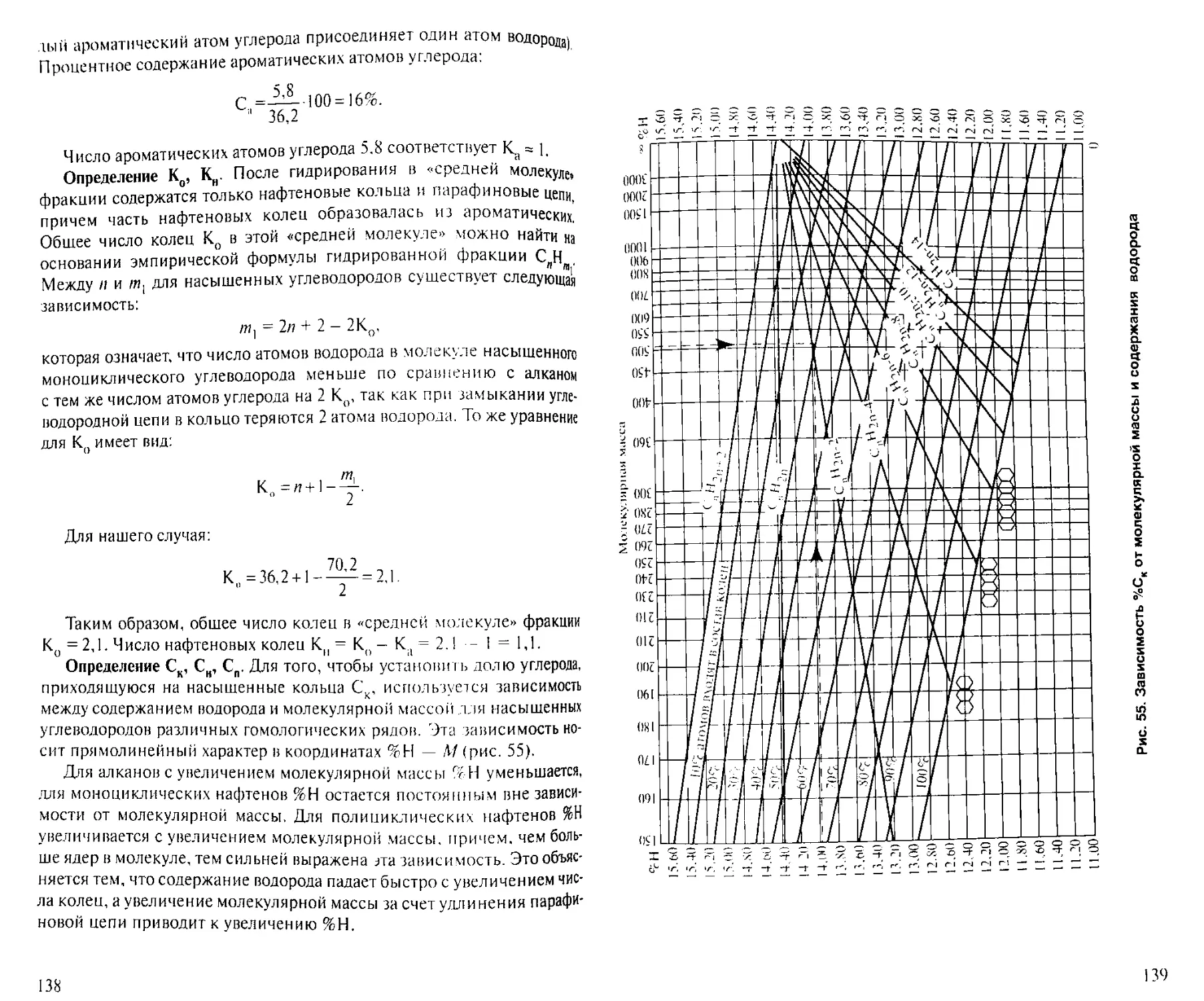
|00-
Исследование химического состава нефти имеет большое значение,
так как оно позволяет научно подойти к решению вопроса о наиболе!
рациональных путях переработки нефти. Рассмотрим кратко общуц
методику исследования химического состава нефти.
В результате работ по исследованию нефтей, проведенных в различ-
ных странах за последние 50 лет, разработана общая методика исследи
вания состава нефти.
Вначале определяют содержание растворенных в нефти газов (ж
ОД-
НО ---У
------------1 1 !-------
100 200 100 мл
Рис. 3. Кривая перегонки
ароматической части
бензиновой фракции
С помощью специальных раство-
рителей можно вытеснить из колонки
раздельно нафтено-алкановую и аро-
матическую части, причем разделение
удается осуществить количественно.
Этот метод разделения неоднократно
проверялся на искусственных смесях.
В книге1, представляющей отчет о ра-
боте 6-го Американского нефтяного
и нститута, при волятся сл еду ющие
данные по исследованию адсорбцион-
ного разделения искусственной смеси
углеводородов. Была приготовлена
искусственная смесь из 17 чистых уг-
леводородов массой 1752 г, в которой
625 г составляли 7 различных арома-
тических углеводородов (выкипаю-
щих до 160СС), а остальную часть 6 ал-
С4) и их состав, затем нефть обезвоживают и обессоливают, определи канов и 4 нафтена. Разделение этой смеси на силикагеле дало 618 г аро-
ют ее основные константы (плотность, температуру застывания, моле- матической части (потери 1.15%) и 1099 г нафтено-парафиновой части
кулярную массу, вязкость при различных температурах, давление на(
сышенных паров, содержание парафина, смол, асфальтенов), эдеме!
нтный состав. Затем проводят перегонку нефти для получения узки!
бензиновых, керосиновых, газойлевых и масляных фракций (интерва»
лы кипения 30-50°С) и остатка1. Перегонка проводится вначале прй
атмосферном давлении до 200’С, а затем в вакууме, чтобы понизит!
температуру перегонки и избежать возможных химических превраще
ним компонентов нефти под действием тепла. Остаток анализируете!
отдельно.
Каждая из полученных фракций подвергается раздельному исследо!
нанию. Например, углеводороды бензиновой фракции разделяют ш
две части — ароматическую и нафтено-алкановую - с помощью адсор
бции на силикагеле (жидкостно-адсорбционная хроматография, см
с. 24) Это разделение возможно потому, что ароматические углеводоро-
ды прочнее адсорбируются на поверхности адсорбента, чем нафтены!
алканы. Если пропускать бензиновую фракцию через стеклянную ко
донку, наполненную мелкоизмельченным силикагелем (SiO7), то аро
матические углеводороды адсорбируются в первую очередь и задержи
ваются в верхней части колонки, а смесь нафтенов и алканов проходи!
в нижнюю часть и по мере накопления вытекает снизу.
(потери 2,34%).
После разделения ароматическая и нафтено-алкановая части под-
вергаются четкой ректификации на специальных ректификационных
коленках с высокой разделительной способностью (100—200 теорети-
ческих тарелок).
В процессе перегонки строят кривую перегонки в координатах тем-
пература кипения — объем дистиллята (рис. 3). Площадки на этой кри-
вой могут соответствовать либо индивидуальным углеводородам, либо
смесям близкокнпяших углеводородов, либо постоянно кипятим азе-
отропным смесям.
После четкой ректификации ароматической и нафтено-парафино-
вой части отдельно изучают узкие фракции ароматических углеводоро-
дов и узкие фракции смесей нафтенов и алканов. Если постоянно ки-
пящая фракция является индивидуальным углеводородом2, то опреде-
ляются константы этого углеводорода и проводится его идентифика-
I г ия ф и зи ко- х и м и чес к и м и метода м и.
Если же какая-либо узкая фракция, отвечающая площадке на кри-
вой, представляет собой смесь двух или нескольких веществ, то для раз-
деления этих веществ можно использовать один из следующих методов:
1) перегонку при пониженном давлении;
1В современных схемах исследования нефтс й выдел я ют также широкую бен з и новуй ПФссини Ф Д.. М.шр Б Д ж . ( грс ш| j А. Д м. У г; i е но л о рол ы г1 er |)т и. ГI е р. с а и i. - 1 о с -
фракцию (начало кипения - 200’С) и ее исследуют .метолом капиллярной г. но-жид [<>" гехп злг1т. 1957
костной хроматографии (с. 133). 2 Эго можно определи п> с по мог шло гаю-жил костной хроматографии (см. с. 28).
14
2) азеотропную и экстрактивную перегонку;
3) кристаллизацию;
4) экстракцию;
5) термическую диффузию;
6) хроматографию.
Некоторые из этих методов часто применяются также для разделе-
ния и анализа нефтяных углеводородов различных широких фракций
нефти. Рассмотрим существо этих методов.
1.1. Разделение углеводородных смесей методами
перегонки, экстракции, кристаллизации,
термической диффузии
Перегонка при пониженном давлении
Пониженное давление применяется для перегонки в двух случаях:
во-первых, как уже говорилось, для перегонки высококипяших фрак-
ций нефти, чтобы избежать возможного разложения углеводородов, и,
во-вторых, для разделения смесей углеводородов, имеющих близкие
температуры кипения при атмосферном давлении и существенно раз-
личающихся при пониженном давлении. Так, смеси нафтенов и слабо-
разветвленных алканов могут быть разделены этим способом, так как
они имеют различные коэффициенты изменения упругости пара с тем-
пературой (рис. 4).
При пониженных давлениях температуры кипения этих углеводо-
родов могут различаться настолько, что возможно их разделение пере-
гонкой.
Для разделения высококипяших углеводородов нефти (температура
кипения выше 350—400°С) следует применять перегонку при глубоком
вакууме с остаточным давлением 0,1-0,001 мм рт. ст.) или молекуляр-
ную перегонку (ост. давл. 0,001 мм рт. ст.).
Рис. 4.1 — нафтен; 2 — алкан
Прибор для молекулярной перегонки состоит из специальной пере-
гонной колбы, в которой поверхность, охлаждающая пары углеводоро-
дов. находится от поверхности перегоняемой жидкости на расстоянии,
меньшем длины свободного пробега молекул, и системы, создающей
вакуум и состоящей из диффузионного и вакуумного насоса. В процес-
се молекулярной перегонки проходит процесс испарения; молекулы
отрываются от поверхности и. достигнув охлаждающей поверхности,
оседают на ней, образуя конденсат.
Однако даже перегонка в глубоком вакууме не может обеспечить
разделения компонентов нефти без их частичного разложения, особен-
но в тех случаях, когда вакуумной перегонке подвергаются тяжелые
нефти или нефтяные остатки типа гудрона.
Эффективным методом разделения высококипяших компонентов
нефти является метод разделения, основанный на ступенчатой
экстракции компонентов тяжелого нефтяною сырья с помощью раст-
ворителя (например, //-пентана), находящегося в сверхкритических ус-
ловиях. При взаимодействии такого растворителя, например, с гудро-
ном, последний частично растворяется в растворителе. Если отделить
раствор и снизить давление, то из раствора выпадает растворившаяся
часть гудрона (это явление называется ретроградной (обратной) кон-
денсацией — см. также с. 9. тле рассматривается это явление в связи
с образованием тазовых конденсатов). Если повышать давление в сис-
теме ступенчато, тем самым повышая растворяющую способность
растворителя, удается постепенно извлекать новые порции компонен-
тов гудрона со все возрастающей молекулярной массой. Такой метод
разделения позволяет' отобрать от гудрона до 90% углеводородов и по-
лучить остаток с температурой кипения выше I()00°С, в то время как
обычной перегонкой удается извлечь из нефти без разложения фрак-
ции с температурой кипения <350°С, что составляет лишь 40-50% тя-
желой нефти.
Основная часть аппарата сверхкритической жидкостной фракцион-
ной экстракции, разработанной китайскими учеными (Ян-Гуан-Хуа и
Вэнг Рен Ан)1, состоит из экстракционной (нижней) части, где проис-
ходит смешение гудрона с пентаном, и ректификационной (верхней)
части, тле происходи г разделение.
Азеотропная и экстрактивная перегонка
Лет кос ть разделения двух компонентов при перегонке определяется
величиной коэффициента относительной летучести:
1 Ки гайский пагсш ZI. 93117577.1.
16
17
где />|( />2 — упругости паров компонентов; у,, у2 — коэффициенты
активности, характеризующие отклонения раствора от идеального.
Если а= 1, то мы имеем нераздельнокипящую смесь. Если добавить
к этой смеси третий компонент, то можно изменить соотношение меж-
ду у и у2 и увеличить а. Третий компонент должен быть подобран так,
чтобы разделяемые вещества обладали различной растворимостью в
нем. Если третий компонент по летучести приближается к разделяемой
смеси, то он образует азеотропную смесь с одним из компонентов. Пе-
регонка в присутствии такого вещества называется азеотропной.
Обычно в качестве третьего компонента при разделении углеводород-
ных смесей методом азеотропной перегонки используют спирты (ме-
тиловый, этиловый), уксусную кислоту, кетоны, простые и сложные
эфиры. С помощью спиртов можно разделять близкокипяшие смеси
аренов и алканов, например смесь бензола (Гкип == 80°С) и 2,2,3-три ме-
тилбутана (Г - 80,9'С): бензол образует азеотропную смесь с этило-
вым спиртом с Ткип = 68,1 °C.
Для разделения смеси бензола (Гкип = 80°С) и циклогексана (7 =
80, ГС) в промышленности применяют метил эти л кетон с 10% воды.
Мет ил этил кетон образует азеотропную смесь с циклогексаном (Гкип
смеси 74°С). Для разделения алканов и нафтенов применяют уксуснукг
кислоту, простые и сложные эфиры.
Если летучесть третьего компонента мала, то перегонка в его при-
сутствии называется экстрактивной. Третий компонент остается в жид-|
кой фазе и удерживает одно из разделяемых веществ, которое лучше1
растворяется в нем. С помощью экстрактивной перегонки можно раз-
делить близкокипяшие смеси аренов и нафтенов. В качестве третьего^
компонента используют фенол или фурфурол. Они преимущественно
растворя ют ароматические углеводороды.
Кристаллизация
Смесь близкокипяших компонентов можно разделить кристаллиза-
цией, которая проводится одним из двух методов.
1. Вымораживают фракцию в чистом виде; один из компонентов
выделяется в виде кристаллов, которые можно отделить центрифуги-
рованием. Процедура повторяется многократно. Таким образом, мож-
но разделить смесь ксилолов, имеющих следующие константы:
Температура Температура
кристаллизации,°C кипения,йС
Ксилол-0 -25 144
Ксилол-Э/ -48 139
Ксилол-77 +13 138,3
2. Если смесь, которую нужно разделить кристаллизацией, обладает
повышенной вязкостью, снижающей скорость кристаллизации, к этой
смеси добавляют подходящий растворитель. Он должен хорошо раство-
рять примеси при температуре кристаллизации углеводорода и быть ле-
тучим (например, бензол, ацетон, диэтиловый эфир, метанол), что
обусловливается необходимостью его последующего удаления. Приме-
ром кристаллизации с растворителем может служить выделение алка-
нов нормального строения из масляных фракций с применением в ка-
честве растворителей смесей толуол — метнлэтилкетон и бензол — аце-
тон. С помощью кристаллизации с растворителями при низких темпе-
ратурах удается разделить изоалканы и нафтены нефтяных фракций.
Для очистки углеводородов от примесей с помощью кристаллиза-
ции эффективным является метод зонной плавки, разработанный
Н. Пфанном. Метод состоит в том, что вещество помешают в трубку,
которая медленно двигается впереди назад через чередующиеся зоны
нагрева и охлаждения. Каждая зона вещества при этом подвергается
многократной перекристаллизации: очищенное вещество концентри-
руется в одной части трубки, а примеси — в другой. Методом зонной
плавки д-нопан ( = 53’С) был очищен от я-гексана1.
К Н11 L 1
Экстракция
Смесь углеводородов можно разделить путем селективного (избира-
тельною) растворения одного из компонентов смеси в подходящем
растворителе. Этот процесс избирательного растворения и называется
экстракцией.
При экстракции растворитель должен образовывать отдельную фа-
зу, т. е. нс должен полностью растворяться в углеводородной смеси.
Массовое отношение между углеводородной смесью и растворителем
должно быть также строго определенным, так как при избытке раство-
рителя разделяемая смесь может полностью раствориться в раствори-
теле. а при избытке разделяемой смеси в ней полностью может раство-
риться сам растворитель. С помощью экстракции можно:
а) отделить арены от алканов и нафтенов;
б) разделить моноцикличсскис, бициклические и трициклические
арены;
в) отделить алканы ог нафтенов.
Для извлечения ароматических углеводородов используют в качест-
ве растворителей ли этилен гл и коль, жидкий сернистый ангидрид, фе-
нол. фурфурол, сульфолан.
Для разделения нафтенов и алканов применяют сложные эфиры.
Так, например, при разделении нафтенов и алканов керосиновых
фракций можно использовать метиловый эфир муравьиной кислоты.
^"RifTcr R Bull Sot ( him. I iancc. I%7 P H'24.
18
19
Термическая диффузия
Принцип разделения углеводородов методом термической диффу,
зии состоит в следующем: если смесь двух веществ поместить между
двумя стенками, одна из которых холодная, другая горячая, то моле-
кулы одного вещества перемешаются к холодной стенке и в силу кон-
векции опускаются вниз, молекулы другого вещества направляются
к горячей стенке и поднимаются вверх. Таким образом, происходит
разделение: один компонент собирается наверху колонки, другой —
внизу. Термической диффузии препятствует обычная диффузия, про-
исходящая за счет разности концентраций. Установлены следующие
закономерности термической диффузии.
I. Из членов гомологического ряда тенденцию двигаться по направ-
лению к холодной стенке имеет компонент с наибольшим числом ато-
мов углерода и наибольшей температурой кипения.
2. В случае смесей с одинаковой температурой кипения по направ-
лению к холодной стенке будет перемешаться компонент с наимень-
шим молекулярным объемом.
3. В смесях вешеств с одинаковыми молекулярными объемами и
температурами кипения будет двигаться к холодной стенке компонент,
молекулы которого имеют наименьшую поверхность.
4. В смесях молекул, имеющих одинаковую молекулярную массу,
молекулярный объем и одинаковую площадь поверхности, компонент
с более высокой температурой кипения будет двигаться к холодной
стенке.
В процессе термодиффузионного разделения происходит образова-
ние концентратов углеводородов. Одна группа углеводородов концент-
рируется в верхней части колонки, другая — в нижней. Однако провес-
ти полное количественное разделение углеводородов невозможно:
в средней части колонки всегда остается значительное количество не-
разделенной смеси. В этом состоит основной недостаток метода терми-
ческой диффузии. Другой недостаток заключается в малой скорости
термодиффузионного разделения: равновесие в колонке устанавлива-
ется в течение нескольких суток. С помощью термической диффузии
можно:
— отделить моноциклические нафтены (перемещаются к горячей
стенке и концентрируются в верхней части колонки) от полицикличес-
ких (перемешаются к холодной стенке и концентрируются в нижней
части колонки):
— выделить из смеси алканов и нафтенов концентрат //-алканов, ко-
торые собираются в верхней части колонки: в средней части колонки
находится смесь изопарафинов и нафтенов, а в нижней концентриру-
ются нафтены;
- получить концентрат изопреноидных углеводородов (до 50%) из
средних фракций нефтей нафтенового основания, в которых содержание
изопреноидных углеводородов измеряется несколькими процентами.
1.2. Хроматографические методы разделения
и анализа углеводородных смесей
Хроматография — это процесс разделения смесей веществ, основан-
ный на их различной сорбционной способности. Сорбцией называется
явление концентрирования вещества в одной из смежных фаз. Можно
дать примеры самого различного сочетания фаз. В случае если смежны-
ми фазами являются газ и твердое тело или жидкость и твердое тело, то
происходит концентрирование вещества на поверхности твердой фазы,
поглощение вещества твердой фазой. Такой процесс называется адсо-
рб ни е й. В сл у ч а е ее л и с м е ж н ы м и с]) а за ми я i зл я ю тс я га з и ж и д к ос т ь и газ
поглощается жидкостью, то такой процесс называется абсорбцией. Ад-
сорбция подразделяется на физическую, которая обусловливается сила-
ми притяжения, и химическую, которая происходи! за счет валентно-
химического воздействия. Существует аналотчнос подразделение про-
цессов абсорбции.
Так как в результате химического взаимодействия может изменить-
ся структура сорбируемых молекул, хроматографическое разделение
углеводородов нужно проводить в условиях, исключающих или сводя-
щих до минимума химическую адсорбцию между молекулами углево-
дородов и сорбентом.
Так, например, аналитическую адсорбционную хроматографию
нужно проводить с использованием инертных, специально обработан-
ных адсорбентов, таких как силикагель, оксид алюминия, активиро-
ванный УГОЛЬ.
Такие адсорбенты, как алюмосиликаты, нельзя использовать при
аналитическом адсорбционном разделении, т. к. в их присутствии воз-
можны химические превращения углеводородов.
Температура, при которой проходит адсорбционное разделение уг-
леводородов, не должна быть повышенной (скорость химической ад-
сорбции возрастает с температурой). Углеводороды обладают различ-
ной адсорбционной способностью в зависимости от их химической
природы и строения. Наибольшей адсорбционной способностью обла-
дают ароматические углеводороды. Значительно ниже адсорбционная
способность нафтенов и алканов. Адсорбционная способность углево-
дородов нефти увеличивается в ряду: алканы < нафтены << мононик-
лические арены < полициклические арены.
Таким образом, хроматография представляет собой физический ме-
тод разделения смесей веществ. В процессе хроматографии разделяе-
мыс вещества распределены между двумя фазами, одна из которых -
неподвижный слой с большой поверхностью, другая подвижна.
В зависимости от характера неподвижной фазы хрома тография под-
разделяется на следующие вилы:
I) адсорбционную хроматографию: неподвижная фаза — твердое
пористое вещество; подвижная — газ или жидкость,
2) распределительную хроматографию: неподвижная фаза — жид-
кость. которая находится на поверхности твердого носителя; подвиж-
ная фаза — жидкость, газ или пар.
Адсорбционная и распределительная хроматографии могут быть'
также подразделены в зависимости от характера подвижной и непод-
вижной фаз. Всего имеется 4 вида хроматографии.
I, Неподвижная фаза —
твердое тело (адсорбционная
хроматография)
1. Неподвижная фаза —
жидкость на твердом носителе
(рас п редел и тел ьн а я
хроматография)
1. Подвижная фаза — жидкость
Жидкостно-адсорбционная
хроматография.
Пример: разделение нефтяных
фракций на ароматическую
и нафтено-парафиновую части
на силикагеле.
2. Подвижная фаза — газ.
Газо-адсорбционная
хроматография. Пример,
разделение газообразных
углеводородов на твердых
адсорбентах (гнперсорбппя),
а также разделение газов на
молекулярных ситах.
1. Подвижная фаза — жидкость.
Жидкостно-распределительная
хроматография. Пример:
хроматография на бумаге,
хроматография в тонком слое
адсорбента. Неподвижной жидкой
фазой служит пленка волы на
поверхности бумаги или оксида
алюминия.
2. Подвижная фаза га j (нар)
Газо-жидкости а я хроматография
Для каждого из 4 видон хроматографии возможны три методики
анализа. Рассмотрим методики анализа для случая ж пл кости о-адсорб-
ционной хроматографии на примере разделения бинарной смеси из ве-
щества сильно адсорбирующегося А (например, смесь ареною и слабо
33
адсорбирующегося В (например, смесь нафтенов и алканов гик на-
зываемая нафтсно-алкановая часть нефтяной фракции;.
Фронтальный анализ
В колонку с адсорбентом заливают смесь вещества А и вещества В.
Вначале из колонки выходит вещество В, затем смесь А В (рис. 5).
Основная масса вещества А остается в колонке и занимает ее верхнюю
часть,
В этом случае можно выделить в чистом виде только слабо адсорби-
рующийся компонент В. Этот метод применяется для отделения про-
дукта от примеси какого-либо вещества, которое сильно адсорбирует-
ся, например для обесцвечивания органических соединений, для отде-
ления смол от нефтепродуктов. Фронтальный метод используется
иногда для выделения нафтено-алкановой части бензинов в неболь-
шом количестве, необходимом для определения ее основных констант.
Однако при разделении нефтяной фракции отделить полностью наф-
тено-алкановую часть от ароматической этой методикой нельзя.
Проявительный анализ
В колонку заливают смесь А -г В. которая пропитывает адсорбент,
а затем добавляют проявитель (растворитель Е). который адсорбирует-
ся слабее, чем компоненты смеси, но при продолжительном добавле-
нии его в колонку он можем сместить вниз (вымыть) слабо адсорбиру-
ющийся компонент В. После добавления проявителя происходит чет-
кое разделение зон А и В; образуются полосы этих компонентов, разде-
ленные полосой проявителя Е (рис. 6). При последующем добавлении
растворителя можно вымыть компонент В полностью. Положительная
особенность этой методики: можно выделить полностью только ком-
понент В. Недостаток: компонент А остается в колонке.
Рис. 5
Рис. 6
Рис. 7
Вытеснительный анализ
В колонку заливается смесь А + В. Она пропитывает адсорбент, а за-
тем пропускают жидкость D, которая сильнее адсорбируется, чем ком-
поненты смеси и вытесняет их из колонки. Происходит следующее.
Вначале из колонки выходит слабоадсорбируюшийся компонент
В (чистый), затем смесь А и В, затем прочно адсорбирующийся компо-
нент А, затем вытеснитель (рис. 7). Недостаток этой методики: зоны:
веществ не разделены зонами растворителя. Поэтому полное разделе-|
ние А и В невозможно. Но можно получить определенное количество'
чистых компонентов А и В.
Для избежания недостатков каждой методики анализа их комбини-
руют. Так, например, если скомбинировать проявительную и вытесни-;
тельную методики анализа, то можно количественно выделить каждый:
компонент. В этом случае порядок выхода из колонки будет следую-
щий: Е, В+Е, Е, А+Е, D (рис. 8). Таким образом, из 4 видов хрома-
тографии и 3 методик анализа можно получить 12 вариантов. Однако
далеко не все эти 12 вариантов находят применение на практике.
Рассмотрим важнейшие из методов хроматографического анализа
в соответствии с характером подвижной фазы.
1.2.1. Жидкостно-адсорбционная хроматография
Подвижная фаза - жидкость. В этом случае хроматография называ-
ется жидкостной. Если в жидкостной хроматографии неподвижной фа-
зой является адсорбент, то имеем случай жидкостно-адсорбционной
хроматографии. Жидкостно-адсорбционная хроматография была впер-
вые разработана в 1903 г, русским ботаником Цветом1 в проявительном
Цвет М. С. Хроматографический адсорбционный анализ. Избранные работы. -
Изд. АН СССР, 1946. - С. 9-39.
24
варианте для разделения отдельных компонентов растительных пиг-
ментов. При этом в колонке получались окрашенные полосы разделен-
ных пигментов (отсюда слово хроматография — цветопись). В химии
нефти жидкостно-адсорбционная хроматография применяется широко
в проявительно-вытеснительном варианте, когда используется комби-
нированная методика анализа: проявительно-вытеснительная. Рас-
смотрим применение этой методики для разделения углеводородов бен-
зиновой фракции. Аналогично с некоторыми модификациями можно
р а здел и ть у гл е водород ы д ру тих н ефтя н ы х ф ра к и и й.
Стеклянная колонка плотно заполняется мелкоизмельченным си-
ликагелем, который предварительно высушивается при 150’С 3—4 часа.
Количество бензина (в мл), необходимое для анализа, определяется по
формуле
Д, = О£У |00
ь
гд eg- м а с с а с и л и к а ге л я, г; а — активность с и л и к а ге л я (мл бен зол а,
поглощенных 1 г силикагеля): b — содержание ароматических углево-
дородов в бензине. определенное методом сульфирования, % об.: к —
коэффициент использования силикагеля (для проявительно-вытесни-
тельного варианта равен О,К).
Рассчитанное количество бензина Л' заливается в колонку. После
пропитывания всего силикагеля осторожно заливают в колонку проя-
витель или смешшошую жидкость (обычно изопентан) в количестве
0.35 мл на 1 г силиюпеля. После поглощения изопентана приливают
вытесняющую жидкость — этиловый спирт (0,3—0.4 мл на I г силика-
геля), а затем после его поглощения — дистиллированную воду. Объем
выходящей по каплям из колонки жидкости (элюата) измеряют граду-
ированным цилиндром: через определенные промежутки времени оп-
ределяют объем и показателъ преломления элюата1.
Из колонки вытекает вначале изопентан 1, затем смесь алканов и
нафтенов с изопентаном 2, чистый изопентан 3, смесь изопентана
с ароматическими углеводородами 4, чистые ароматические углеводо-
роды 5, их смесь с этиловым спиртом 6 и. наконец, этиловый спирт 7.
В процессе отбора фракций строят хроматограмму в координатах объ-
ем элюата — показатель преломления (рис. 9). Полученные фракции
(число их достигает большой величины) объединяют по типам углево-
дородных молекул. Удалив растворители (изопентан отгонкой,
а спирт — растворением в воле), получают отдельно нафте но-алкано-
вую и ароматическую части бензиновой фракции.
ПГаккак угле водороды бен л (новой фракции бесцветны, для их обнаружения (детек-
тирования) на выходе из колонки необходимо определение их констант, например пока-
зателя преломления (детектор-рефрактометр)
25
Рис. 9. Хроматограмма процесса разделения фракции
I
В практике разделения смесей углеводородов методом жидкостью-
адсорбционной хроматографии применяются жидкостные хроматограф
фы высокого давления1. В них скорость хроматографии высока. Из;
хроматографической колонки компоненты смеси выходят вместе с фин на бу
подвижной жидкой фазой (элюентом) и поступают в детектор, В жид-
костной хроматографии высокого давления обычно применяются
ультрафиолетовые детекторы, основанные на различии в поглощении
ультрафиолетового излучения компонентами смеси и элюентом (по-
следний не поглотает ультрафиолетовое излучение), и рефрактомет-
рические детекторы, основанные на различии показателей преломле-
ния компонентов разделяемой смеси и элюента. Сигналы от детектора
поступают к устройству, которое записывает хроматограмму. Если в ко-
лоночной жидкостно-адсорбционной хроматографии, проводимой
при атмосферном давлении в колонке длиной 50 см и диаметром 1—2,5
см, наполненной адсорбентом с размером частиц 100 мкм. эффектив-
ность разделения, проходящего в течение нескольких часов, составля-
ет примерно 50 теоретических тарелок (т. т.), то в высокоэффективной
жидкостной хроматографии высокого давления (длина колонок 10-20
см, диаметр 2-4 мм, размер частиц адсорбента 2-10 мкм, скорость no-j
тока эл юе нта 0,2- 2 мл/мин п од д а вл е н и е м д о 40 • 106 Па) эф фе кти в*;
ность разделения составляет 10000 т. г. Разновидностью жидкостно-ад-1
сорбционной хроматографии является гель-хроматография2. ;
В качестве неподвижной фазы в этом виде хроматографии применя-1
юг слой набухшего геля3 (гидрофобного или гидрофильного). Через
Современное состояние жидкостной хроматографии. Под рел Дж. Киркленда. Пер.
e iiin.i. М . 1974.
Де1срмап Г Ге и.-хромагырафия,— М.: Мир 1970.
В kj'icciии ии рифил ыиях (слей прпмсиягот крахмал. полиакриламид, сульфиро-
toijiju.1l- полимеры: в качеыве IпдрофоГтлы.х — полистрол, каучуки.
колонку, содержащую гель, пропускают смесь разделяемых веществ,
затем элюент. Компоненты смеси распределяются по длине колонки
в зависимосги от размеров их молекул.
Крупные молекулы, которые не могут проникнуть в норы гранул ге-
ля, удерживаются слабо гелем, перемешаются быстро по длине слоя и
оказываются в нижней части колонки. Мелкие молекулы, проникаю-
щие в поры геля, очень медленно смешаются под влиянием элюента и
задерживаются в верхней части колонки.
Компоненты смеси покидают хроматографическую колонку в по-
рядке убывания молекулярных масс, т. е. первым покидает колонку
компонент с наибольшей молекулярной массой Л/, затем компоненты
с молекулярной массой Л/р Л/2, Л/, и т. д. (М > > М2 > и т. д.).
Если в жидкостной хроматографии неподвижной фазой служит
тонкий слой жидкости на поверхности твердого носителя, то в этом
случае имеем жидкостно-распределительную хроматографию.
Этот вид хроматографии был открыт в 1941 г. Мартином и Синжем1
и получил в дальнейшем широкое распространение в виде хроматогра-
маге и тонкослойной хроматографии. И тот и другой вид хро-
матографии имеют ограниченное применение в химии нефти: главным
образом при исследовании гетероагомных соединений (кислородных,
сернистых, азотистых) и олефинов.
В бумажной хроматографии роль подвижной фазы выполняет тон-
кий слой волы, находящийся на поверхности целлюлозы. Бумажная хро-
матография применяется в проявительном варианте. На лист бумаги
(ватман или специальная бумага для хроматографии) наносят микро кап-
лю раствора исследуемой смеси в подходящем растворителе на расстоя-
нии 1—2 см от края (линия старта). Лист бумаги опускают в ванночку
и элюентом и закрепляют вертикально или наклонно так, чтобы нижний
край листа, ближе к которому располагается пятно пробы, касался элю-
ента. Под действием капиллярных сил элюент поднимается вверх, сма-
чивая лист бумаги, и смешает компоненты разделяемой смеси снизу
вверх. Если компоненты разделяемой смеси имеют окраску, то на бума-
ге образуются окрашенные пятна на разных расстояниях от линии стар-
та. Если компоненты анализируемой смеси бесцветны, то пятна прояв-
ляются при обработке бумаги с помощью специальных реагентов.
В отличие от бумажной в тонкослойной хроматографии вместо бу-
маги используют гонкий слой порошка оксида алюминия или оксида
кремния, находящийся на стеклянной пли металлической пластинке.
Для проявления пятен бесцветных веществ в тонкослойной хромато-
графии часто используют пары йода, который, растворяясь в разделен-
ных веществах, окдхгшиваст соответствующие им зоны сорбента.
Marlin Л I Р. Synge R. I. М. . / Biothcni I. 1941 - V. 35. - Р. 1358.
В последнее время разработаны
i 1 инструментальные методы тонкослой-
j Л ной хроматографии. Так, в одном
д II I вариантов вместо пластинки ис пользу.
/1 I \ 1 ется набор тонких стержней из адсор-
/ I I \ I бента. Стержни закрепляются в спецд
I I \ 1 альной вертикальной прямоугольной
I / | рамке. На каждый стержень (в его них
I \ s' \ I ней части на расстоянии 1 2 см от код
I \^/ V ца) наносится по капле раствора акали-
------ гмшЛ зИруемых вешеств (например, тяжелы*
Рис. 10. Хроматограмма образца нефтяных фракций). Нижняя част|
фракции вакуумного газойля: рамки опускается в растворитель -
1—смесь алканов и нафтенов, ЗЛЮСНТ, КОТОрЫЙч поднимаясь по
2 — ароматические углеводороды; ’ смешает компоненты анали
3 — СМОЛЫ С)С)УЛПЛ1 ,
зируемой смеси и разделяет нефтянук
фракцию на насыщенную (верхняя часть стержня), ароматически
(средняя часть стержня) и смолистую части (нижняя часть стержня.
Затем рамку помешают в специальную ячейку, в которой водороднц
пламя пламенно-ионизационного детектора (см. с. 33) выжигает ад-
сорбированные вещества поочередно на каждом стержне, а запись»,
юшее устройство записывает хроматограмму. На рис. I0 представлен,
хроматограмма образца фракции вакуумного газойля одной из кита»
с к их нефтей.
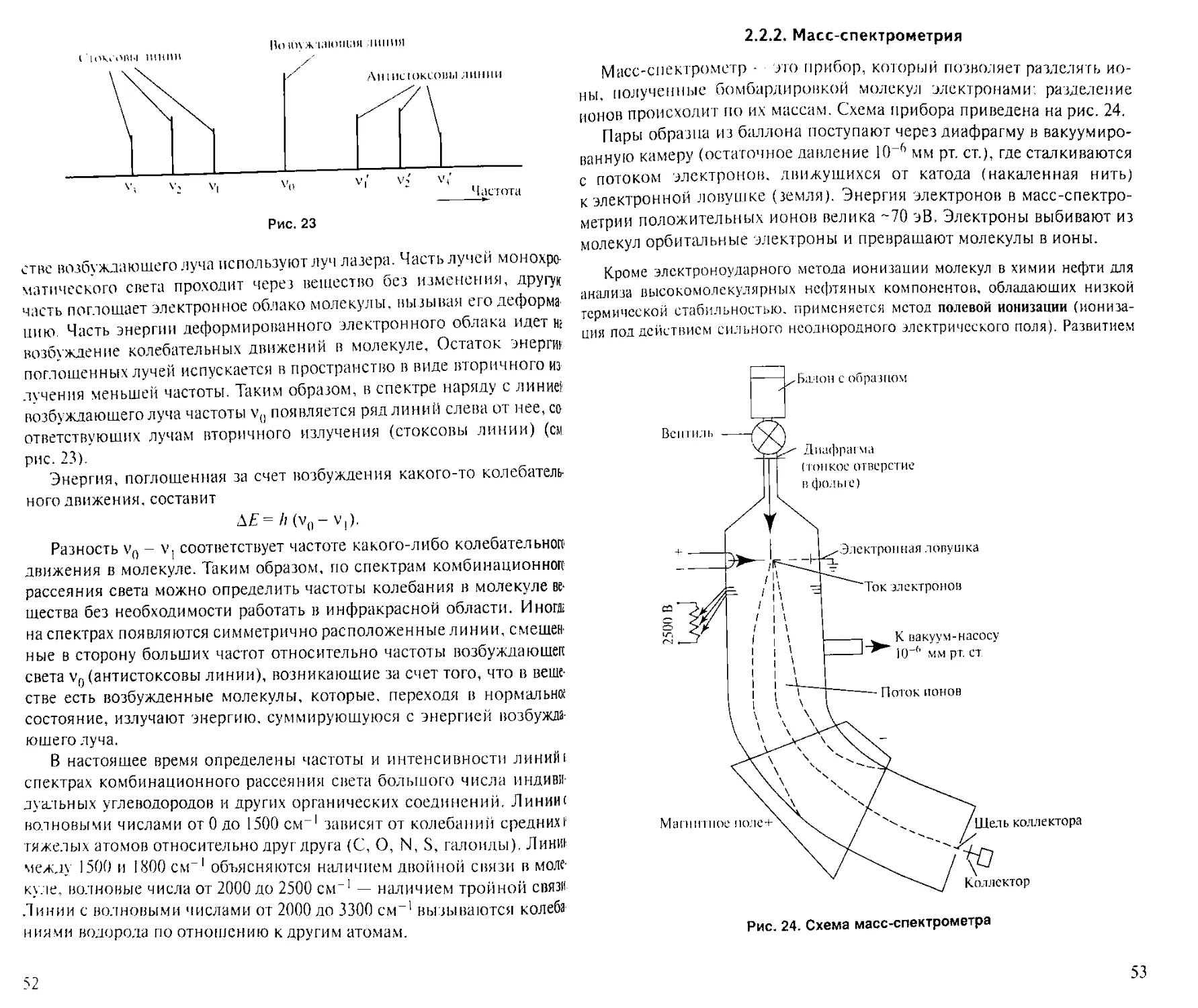
1.2.2. Газовая хроматография
Если подвижная фаза — газ (пар), то в этом случае хроматографии
называется газовой. Газовая хроматография была открыта в 1952 г. Мар-
тином и Джеймсом1. Если в газовой хроматографии неподвижной фа-
зой является жидкость на поверхности твердого носителя, то имеек
случай газо-жидкостной хроматографии, если адсорбент, то газо-ад-
сорбционной хроматографии.
Газовая хроматография осуществляется в проявительном варианте.
Анализ проводится на специальных приборах, хроматографах. Рас-
смотрим принцип работы простейшего хроматографа с детектором по
теплопроводности. На рис. 11 приведена принципиальная схема тако-
го хроматографа в следующих обозначениях: I — ввод образна в испа-
ритель, 2 — ввод газа-носителя (элюента), 3 — счетчик газового поток!
(ротаметр), 4 — детектор (катарометр), 5 — колонка (колонка, детектор
и испаритель термостатированы; температура в испарителе и детекторе
на ~50°С выше, чем в колонке).
1 James А Т, Mart in А. I. Р, // Biochem. J. — 1952 — V. 50 — Р. 679.
Рис. 11. Схема хроматографа с детектором по теплопроводности
Насадочная хроматографическая колонка
Колонка представляет собой металлическую (иногда стеклянную)
трубку длиной до 3 мм и диаметром 2-4 мм, изогнутую в спираль. Та-
кая колонка называется набивной или насадочной. В случае газо-алсорб-
п ион ной хроматографии колонку заполняют порошком адсорбента
(активированный уголь, оксид кремния, оксид алюминия и др.) В слу-
чае газо-жидкосгной хроматографии в колонку засыпают порошок
твердого носителя, на поверхности которого находится тонкий слой
неподвижной жилкой фазы. В качестве твердого носителя используют
обычно огнеупорный кирпич, глины, крупно-пористые стекла.
Перед заполнением колонки носитель пропитывается раствором
м it! ол отучен жидкости в легком растворителе. После отгонки раство-
рителя жидкость остается на поверхности твердого носителя и играет
роль неподвижной жилкой фазы (количество неподвижной жидкой
фазы может составлять от 5 до 40% от массы носителя).
Неподвижная жидкая фаза может быть селективной и нсселектив-
пой. Неселективная фаза разделяет вещества независимо от химичес-
кой природы, только в соответствии с их температурами кипения. В ка-
честве нссслсктивной фазы используют неполярные или малополярные
жидкости (вазелиновое масло, высшие алканы, например сквалан
(Для ра зделения смеси углеводородов различных рядов необхо-
димо использоват ь селективную фазу, так как в такой смеси уитеводо-
ролов могут находиться углеводороды, температуры кипения которых
28
29
очень близки. Селективная фаза должна обладать способностью изби-
рательно удерживать углеводороды различного типа. В качестве селек-
тивных фаз используют полярные вещества, например фталаты (дибу.
тилфталат, диоктилфталат), трикрезилфосфат, полиэтиленгликоль
глицерин. Одной из активных селективных фаз является р,р-дициано-
диэтиловый эфир. Эта фаза в 20 раз сильнее удерживает алкилбензолы
чем алканы, ив 10 раз сильнее, чем нафтены.
Анализируемая смесь поступает в колонку следующим образом. Не-
большая ее проба (0,1 мкл жидкости или 0,5 мл газа) вводится в испари-
тель хроматографа с помощью шприца (место ввода пробы под номером
1 на схеме), подхватывается потоком газа-носителя (элюента) и входит
в колонку. Скорость газа-носителя (мл/мин) постоянна и измеряется
ротаметром (см. схему). Двигаясь в колонке, компоненты смеси распре
деляются по ее длине в зависимости от их растворимости в не подвид,
ной жидкой фазе и их летучести; они образуют в колонке отдельные зд1
ны, отделенные друг от друга зонами газа-носителя, выходят из колон-
ки через различные промежутки времени и поступают в детектор, '
Детектор
Детектор по теплопроводности (рис. 11) основан на измерении раз-
ности теплопроводности газа-носителя и компонентов смеси. В еп}
корпусе имеются два канала, по осям которых натянуты проволочки
(платиновые или вольфрамовые) с одинаковым сопротивлением. Эи
проволочки С, и С, входят в схему мостика Уитстона (рис. 12). по вет-
ням которого идет постоянный ток.
Пока по обоим каналам детектора протекает только газ-носител1
(в качестве газов-носителей используют обычно водород, 1елий, азо!|
аргон _ газы с высокой теплопроводностью), от каждой проволочи
тепло отводится с одной и той же скоростью; проволочки имеют оди-.
наковые температуру и со про'
тивление = Яс2)- Так как в
мостике Уитстона = /?2, тй
в каждой ветви мостика иде(
одинаковый ток и между точка-;
ми А и В нет разности потенциа-
лов. Мостик сбалансирован. Не
как только в канал из колонки
поступает компонент разделяе-
мой смеси (например углеводо-
род, обладающий меньшей теп-
лопроводностью, чем газ-носи-
тель), то тепло от проволочки С
отводится медленнее, она нака-
Рис. 12. Схема мостика Уитстона
ляется сильнее, чем Ср увеличивается ее сопротивление и по ней идет
ток меньшей силы. Тогда между точками А и В появляется разность по-
тенциалов, она усиливается, передается на записывающее устройство и
фиксируется в виде пика. Через некоторое время из колонки выхолит
второй компонент, которому соответствует второй пик и т. л.
Характеристика хроматограммы и хроматографического пика
На рис. 13 представлена хроматограмма трехкомпонентной смеси.
Хроматограмма состоит из следующих частей:
1 — начало хроматограммы, соответствующее времени ввода пробы
в хроматограф;
2 — нулевая линия, которая записывается при прохождении через
детектор чистого газа-носителя;
3 - пики компонентов анализируемой смеси;
4 — пик несорбирующегося компонента (воздух).
Появлению каждого пика на хроматограмме соответствует о преде-
ленное время, называемое временем удерживания (tv j). Это время от мо-
мента ввода пробы в хроматограф до появления максимума пика (т(}, т|?
г-,. т3, соответственно для каждого пика). Чем больше сорбционная
способность или температура кипения компонента (в случае неселек-
тивной фазы), тем больше время удерживания.
Времени удерживания соответствует обз>ем газа-носителя, называе-
мый объемом удерживания ( И );
'у. = ” V
где и — объемная скорость газа, мл/мин. Удерживаемое время и удер-
живаемый объем несорбирующегося компонента обозначают соответ-
ственно тп и И(), Время удерживания и объем удерживания являются
Рис. 13. Хроматограмма
трехкомпонентной смеси
постоянными величинами при
постоянных условиях хрома-
тографирования. В газовой хро-
матографии широко использует-
ся понятие относительного объе-
ма удерживания (И0П|):
У _ У т — т
н - v:i t! — Ч-1
ОГН ~~ tГ/
V,! Cl ' “ ^0
В этой формуле tv,vci и
1\,ит - соответственно время и
объем удерживания стандартно-
го вещества.
О т 11 ос и тел ь 1 {ы й об ье м уд е р-
живания является более надеж-
> I
ной элюнионной .характеристикой компо*
нентов разделяемой смеси, так как в мень-
- илпр чем т и К , зависит от условий
шеи мере, чем 1уд и гу:г
г-тгКнплйяния’ Е зависит от тем-
хроматографирования, отн
пературы колонки и природы неподвижной
фазы
Хроматографический пик может был
широким и узким и зависимости от приро.
лы соответствующего ему компонента, ско-
рости газа-носителя, свойств неподвижной
фазы. .
Пик ограничивается «фронтом» и «ты-
, Нч пис 14 представлен симметричный пик: h - высота,
лом» (рис. 14). На р • Р хроматографического анали-
Ь~ ширина основания. “ Рп||ки. ХСиМметрия бы пае
за встречаются размытые аси «хвостовой», когда размы,
X™' ФроттоваГасимметрия обычно появляется при разделе-
НИИ веществ с низкой упругостью пара: «хвостовая» асимметрия свиде.
тель Z о сорбционной неоднородности поверхности неподвижной
фазы.
Особенности современных газовых хроматографов
В современных газовых громатографах, например «Цвет», ЛХМ-}
8МД, «Хром», «Кристалл 2000М». кроме набивных колонок, имеются
капиллярные хроматографические колонки. Капиллярная колони
представляет собой металлический или стеклянный капилляр, дости-
гающий обычно длины 50 м (иногда до 80 м) с диаметром 0,25-
0,35 мм. Неподвижная жидкая фаза наносится на внутреннюю по-
верхность этого капилляра. Разделительная ..пособность капиллярны!
колонок велика1, она соответствует 50-100 ыс. т. т. С помощью ка-
пиллярных колонок можно разделить любые углеводороды, выкипаю]
шие до 500°C.
В хроматографах наряду с катарометрами используют
чувствительные детекторы, в частности пламенно-ионизационные
(ДИП), действие которых основано на следующем принципе. При
обычных условиях газы не проводят ток, но если под действием пламе-
ни или излучения в газе образуются ионы и электроны, он становится
проводимым. За счет сгорания водорода в детекторе (рис. 15) возника-
ет пламя. Как только в пламя попадает компонент, образуются заря-
женные частицы и между электродами, к которым прилагается напря-
жение -200 В, протекает ток; он усиливается и подается на записываю-
щее устройство.
Рис. 15. Схема ДИП
Кроме пламенно-ионизационных используются другие детекторы,
в частности ионизационные, в которых ионизация осуществляется
с помощью источников радиоактивного излечения - изотопов (v3Ni,
wSr. ’Н.
Программирование температ уры
В газовых хроматографах анализ может проводиться как в изотер-
мическом режиме (температуры колонки и детектора являются посто-
янными в течение всего анализа), так и при переменном температур-
ном режиме с постоянным подъемом температуры. Этот температур-
ный режим необходим тогда, когда анализируемая смесь веществ име-
ет широкий интервал температур кипения (например, фракция нефти).
Если такую смесь анализировать в изотермическом режиме при темпе-
ратуре, близкой к нижнему пределу темпера гуры кипения смеси,то бу-
дут хорошо разделяться только легкокипяшис компоненты смеси; вре-
мя удерживания высококипяших компонентов увеличится, и общая
продолжительность анализа также увеличится. При более высокой
температуре, близкой к концу кипения смеси, будут плохо разделяться
более легкие компоненты. Четкое и быстрое разделение компонентов и по-
лучение хроматограммы с симметричными пиками достигается в том
случае, если в процессе анализа температура повышается по заданному
закону (ступенчато или плавно). Такой метод называют газовой хрома-
тографией с программированием температуры. Скорость подъема тем-
пературы может изменяться в широком интервале (от 0,5 до 30’С/мин).
Адсорбенты в газовой хроматографии
В газо-адсорбционной хроматографии в качестве неподвижной фа-
зы используются адсорбенты. К адсорбенту предъявляются следующие
требования: он должен быть химически инертен к компонентам разде-
1 Разделительная способность i габи in i ы х кол опок I - 3 ты с г. г.
33
ляемон смеси, обладать развитой поверхностью, характеризоваться ли|поверхность такого адсорбента участвует в процессе адсорбции С по-
нейной изотермой адсорбции. Адсорбенты, применяемые в газово|мощью такого адсорбента можно разделить молекулы ^л^даюшие ^аз-
хроматографии, подразделяются. 1личнои адсорбционной способностью, но нельзя разделить смесь углс-
I) неполярные (активированным уголь сажаТ водородов одного и того же ряда, имеющих одинаковую алсорбиион-
2) полярные (оксид алюминия, оксид кремния, алюмо иликаты); ную способность. Хотя их молекулы имеют различные размеры олна-
3) органические полимеры. используются !К0 ЛЮб0Й МОлекуле может соответствовать пора определенного разме-
чает» в практике так что каждая молекула будет задерживаться адсорбентом. Возни-
дифицированные адсорбенты. Модификация адсорбентов необходвкает вопрос, можно ли приготовить адсорбент в котооом лияметпи
ма последующим причинам. Поверхность обычных адсорбентов неод всех пор были бы одинаковы? Ведь если бы удалось приготовить такой
нородна и состоит из участков с сильно различающейся адсорбциомдеорбеит. то можно было бы разделить углеводороды одного типа но
ной способностью. Наличие участков с повышенной активное^ разным и размерами молекул. Одни молекулы т е“ о™,™ ди ото
обусловливает размывание зоны компонента в колонке за счет бо^размера. задерживались бы в порах адсорбента, а более крупные прохо-
прочной адсорбции какой-то части компонент . р ультате ладили бы между гранулами, не задерживаясь, т. е. просеивались бы
компонента на хроматограмме оказывается несимметричным, с хвое Таким веществом может быть только кристачпическое вещество
товой асимметрией. Модификацию таких адсорбентов провод^- определенной кристаллической решеткой, так как только в нем воз-
с целью выравнивания адсорбционной активности различных учас^можно существование объемов одного размера, соответствующих оди-
ков поверхности. В качестве модификаторов используют в небольша паковым по размерам порам. Так как в кристалл как известно нет
количестве (несколько процентов) малолетучие органические пустот. то, чтобы они образовались, нужно удалить из него какие-то хи-
кости (например, вазелиновое масло). Модификаторадсорбируетсянмические составные части вполне определенного размера тогда
наиболее активных участках поверхности£блокирует их), и активное^ кристалле образуются пустоты - поры.
Например, если вода входит в структуру кристаллической решетки,
то. удалив волу можно образовать поры, однако при этом основная
структура кристалла не должна нарушаться. Нельзя, скажем, получить
... . такоП адсорбент из кристаллов CuSO4-5H,O. так как удаление воды
ния углеводородов в процессе хроматографии. Цель модификавд приводит к полному разрушению кристаллической решетки Нужно
в случае таких адсорбентов состоит в подавлении каталитически акт иметь такое вещество, в образовании кристаллической решетки кото
ных центров с помощью химических реагентов. В качестве модификЦого вода не тирада бы основной роли. Такими веществами
торов в этом случае используются щелочи и другие химические реаген некоторые
ты. Для модификации поверхности силикагеля применяют тримета CaO-Al?(\4SiO7-6НЭО
хлорсилан. В процессе модификации происходит замена протону Г
гидроксильных групп поверхности на триметилсилильные группы:
поверхности адсорбента выравнивается. Такая модификация называ
ется физической.
Некоторые адсорбенты (оксид алюминия, алюмосиликаты) содел
жат каталитически активные центры, на которых возможны превращ
являются
минералы из группы цеолитов, например шабазит
г
В 1925 г. Вейнгель и Штейнгоф обнаружили, что обезвоженный ша-
: базит очень легко и быстро сорбирует пары воды, метилового, этилово-
го спирта и пропускает пары ацетона и бензола. В дальнейшем было ус-
тановлено, что диаметр всех пор в шабазите 3,9 . Примерно такой же
д иаметр имени молекулы метиловою, этилового спирта, а диаметр мо-
лекул ацетона и бензола больше, поэтому последние не адсорбируются.
С пособность цеолитов сорбировать молекулы, проникающие в их по-
* . и пропускать через их слой, не задерживая, более крупные молеку-
лы, обус л о в л и васт так называемы й м олеку 'ля рно-ситовой эффект. Этот
Обычный адсорбент содержит огромное число различных пор (mJ ntx’IBBO[,OJI(M<CH обычному си юному эффекту.
ких и крупных), однако не все поры участвуют в процессе адсорбций ‘J L ObIjkl Ус1{1|,омена принципиальная возможность разделе-
Если размеры молекул адсорбируемого вещества больше размеров ма Ж1Я 'f JCBai°IXUUb,x IdX)B иа HC0JKidX’ или молекулярных ситах. Все
ких пор, то молекулы в эти поры не проникают. Следовательно, нек|IW BPCjlcu,BJ4iOi со хш полшилрагы алюмосиликатов, состав ко-
Si—OH +CI-Si(CH3h
В настоящее время в хроматографическом анализе используют
специальные адсорбенты, называемые молекулярными ситами.
Рассмотрим, что собой представляют молекулярные сита и каков'
их значение для разделения и анализа углеводородов, jI
торых и общем пиле следующий:
(Me+, Me++)nO-(AI2O3)x- (SiO2); mH,O
Me+: Na+, КЛ Li Л
Ме++: СаД Ва^, Mg++ и т. д.
Природных цеолитов мало, поэтому цеолиты получают в промыц
ленности синтетическим путем. Для приготовления цеолита с катио
ном Ма+ смешивают с ил и кат натри я, алюминат натрия и гидрат оксид
натрия. Соотношение этих вешеств зависит от того, какого типа цеолд
нужно приготовить. Смесь вводят в кристаллизатор и выдерживай
при 100°С несколько часов. Затем кристаллы промывают водой и де
бавляют глину в качестве связующего агента и формуют. Гранулирован
ный цеолит прокаливают при 650 С. По своей кристаллической стрр
туре цеолит можно рассматривать как соль поликремневой кислоты,
которой часть атомов кремния заменена на атомы алюминия. Порис
тость цеолитов может достигать 50%, поверхность 800-1000 м2/г. Цео
литы подразделяются в зависимости от природы катиона и от тип
кристаллической решетки на следующие марки: !
NaA, СаА. NaX, СаХ, NaY и др.'
Цеолиты обозначают еше так: ЗА, I0X и т. д,: цифра указывает к
размер пор, .
Молекулярные сита обладают рядом интересных свойств.
I) Вследствие малых размеров пор адсорбированная молекула очеи
прочно удерживается (проявляется роль противоположной стенки nti
ры). Этим объясняется высокая адсорбционная способность цеолите
даже при повышенных температурах.
2) Молекулярные сита имеют большое сродство к ненасыщенны
соединениям. Поэтому с их помощью можно отделить этилен от этан!
хотя размеры молекул этих углеводородов одинаковы.
3) Высокая адсорбционная способность молекулярных сит обет
чивает полное поглощение вещества даже при его малой концентрат^
в смеси. Это свойство используется для глубокой осушки газов и дя
очистки их от примесей.
С этой целью применяются молекулярные сита марок ЗА и 4А.
4) С помощью молекулярных сит можно количественно отделит
алканы от изоалканов, аренов и нафтенов. В настоящее время цеолит)
широко используются как катализаторы и в качестве фазы в хромат^
график. Разделение углеводородов с помощью молекулярных сит пр
водят статическим и динамическим методами. По статическому мето-
Д.'1Я неолитов типа А мольное соотношение оксида крем и ил и оксида ал гомики
равно 2. для цеолитов типа X — 2,5-3; Y — 4-6.
ду обезвоженный в вакууме цеолит смешивают с веществом, по дина-
мическому — вещество пропускают через слой цеолита. Для выделения
адсорбированного вещества цеолит нагревают в вакууме при 300-
350°С в течение нескольких часов.
1.2,3. Аналитические задачи в химии нефти,
решаемые с помощью газовой хроматографии
С помощью газовой хроматографии решается ряд аналитических
задач: качественный и количественный анализы, разделение смесей уг-
леводородов и других соединений, выделение в чистом виде отдельных
углеводородов.
Качественный анализ
Задача качественного анализа углеводородных смесей с помошью
газовой хроматографии состоит в определении числа компонентов
смеси и в идентификации каждого компонента. Чтобы определить чис-
ло компонентов, необходимо подобрать условия для разделения всех
компонентов смеси и получить хроматограммы с четкими пиками. Для
идентификации пиков на хроматограмме в хроматограф вводят инди-
видуальные углеводороды (эталоны) при тех же условиях, при которых
проводился анализ смеси. Если время или объем удерживания эталон-
ною углеводорода совпадает с таковыми для какого-то пика анализи-
руемой смеси, то пик идентифицируется.
Количественный анализ
Количественный состав анализируемой смеси определяют, исходя
из того, что содержание компонентов в смеси пропорционально пара-
метрам их пиков (высота, площадь). Имеется несколько методов расче-
та хроматограмм с целью определения количественного состава.
а) Метод внутренней нормализации.
По этому методу процентное содержание какого-либо компонента в
смеси определяют как отношение приведенной площади его пика к
сумме приведенных площадей всех пиков:
Площадь ника определяют как произведение высоты пика на его
ширину на половине высоты пика (рис. 16):
5. = h а.
Приведенная площадь пика компонента S' — это произведение
его плошали Sf на поправочный коэффициент К, учитывающий
чувствительность детектора по отношению к данному компоненту
36
37
________ (В случае детектора по теплопроводное]!
/\ этот коэффициент учитывает различи
/ \ в теплопроводности компонентов разде
А а А_____ляемой смеси):
* Г Д 5/ = S,K= ha К.
/ \ л/2 б) Метод абсолютной калибровки.
/ \ В соответствии с этим методом пр®
f” центное содержание компонента в смес
„ можно найти с помошью калибровочное
Рис. 16
графика в координатах плошадь пика (вц
сота пика) - процентное содержание компонента в смеси. Калибре
вочные графики строятся на основании данных хроматографическое
анализа искусственных смесей, в которых содержание каждого компа
нента известно. По хроматограммам определяют параметры каждое
пика (плошадь или высоту) и строят графики.
в) Метод внутреннего стандарта. ।
Но этому методу в анализируемую смесь вводят стандартное вецд
ство, пик которого на хроматограмме должен четко отделяться от др)
гих пиков. Концентрацию любого компонента смеси рассчитываютп
формуле
С =Д-Я-I00 (%>.
' S’
ТО
где S' — приведенная плошадь пика компонента; 5'ст — приведена
плошадь пика стандартного вешества; R — отношение массы стандар
ного вешества к массе анализируемой смеси.
Выделение веществ в чистом виде
Эта задача решается с помошью препаративной хроматографа
Применяются хроматографические колонки большою диамето
(10-100 мм). Объем жидкой пробы от I до 20 мл; объем пробы га
100-200 мл, Из хроматографической колонки небольшая часть noioj
идет к детектору, а основная часть направляется на разделение. Компа
ненты разделяемой смеси извлекаются из подвижной фазы глубоки
охлаждением с помошью жидкого азота (-196ГС).
Продолжительность разделения и выделения в чистом виде комп^
нентов смеси зависит от размера пробы. Например, разделение смеС|
/-пентана и //-пентана при пробе 20 мл проходит около 2 ч.
§2. Физико-химические методы идентификации
и количественного определения углеводородов
и других компонентов нефти и газа
Ранее мы рассмотрели в основном методы разделения углеводоро-
дов нефтяных фракций, включая выделение индивидуальных углево-
дородов в чистом виде.
После выделения углеводородов в чистом виде следующим этапом
исследования является их идентификация. Идентификация может
быть химической (установление строения углеводорода путем его син-
теза (встречный синтез) и изучение свойств его производных) и физи-
ко-химической, которая основана либо на определении физико-хими-
ческих констант углеводорода — плотности, показателя преломления,
рефракции, либо на исследовании его спектральных характеристик.
Рассмотрим физико-химические методы идентификации углеводо-
родов нефти.
2.1. Физико химические константы углеводородов нефти
и их роль в идентификации компонентов и анализе
углеводородных смесей
Для характеристики индивидуальных углеводородов или нефтяных
фракций необходимо определение их относительной плотности и по-
казателя преломления. Определение этих констант для индивидуаль-
ных углеводородов позволяет идентифицировать их путем сравнения
найденных констант с литературным данными для предполагаемого уг-
леводорода. Совпадение найденных и литературных данных подтверж-
дает предполагаемую структуру yiлеволорола.
Относится иная плотность представляет собой отношение плотнос-
ти углеводорода при 20'С’ к плотности воды при 4'Х’:
Относительные плотности углеводородов возрастают в ряду: алканы
олефины ' нафтены х аромат и чес кис у uic водороды.
В одном гомологическом ряду относительные плотности повыша-
ются с увеличением молекулярной массы Относительную плотность
определяют с точностью до 0,0001 с помошью пикнометров, представ-
ляющих собой стеклянные ампулы объемом обычно от 0,5 до 5 мл,
в которых в шешивают одинаковые объемы исследуемого углеводорода
и волы при 20 С. Вначале находят относительную плотность углеводо-
рода к воде при 20"С Ру//, а затем пересчитывают это значение и нахо-
лят р420 по формуле, учитывающей также величину потери веса пикно-
метра в воздухе:
pj* =0,99703-р^+0,0012.
Показатель преломления характеризует степень преломления луча
света при его прохождении через границу раздела двух сред различной
плотности.
Показатель преломления данного вещества представляет собой,
строго говоря, отношение скорости распространения света в вакууме к
скорости света в веществе:
л, = -.
о
Это — абсолютный показатель преломления. На практике обычно
изучают преломляющую способность среды /?2 относительно воздуха,
для которого/?| = 1,00027:
/к
л = —.
",
Согласно закону Снеллиуса (рис. 17),
sin а. н,
--------------------------— = — = /;.
sin а. /?,
4 I
Если заменить направление света на противоположное, то при не-
котором критическом угле а2 наблюдается явление полного внутрен-
него отражения (рис. 18). Для каждого вещества имеется вполне опре-
деленный угол а2. На измерении этого угла и основано определение
показателя преломления вещества.
Действительно:
sin а, //,
1 4
40
так как
sin а, = sin 90” = 1 и /?,=(.
то
п.
sin ос,
Показатели преломления измеряются с помощью рефрактометров.
Показатель преломления для данного вещества зависит от длины вол-
ны луча света и от температуры. Для большинства углеводородов неф-
ти с увеличением длины волны п уменьшается. Обычно определяют п
для желтой линии в спектре натрия D (I = 5896 ), реже для С и F —
красной и голубой линий в спектре водорода (Хс = 6563 ; Z,F = 4681 ).
Показатель преломления уменьшается для углеводородов в последова-
тельности: ароматические углеводороды > нафтены > олефины > алка-
ны. В одном и том же гомологическом ряду п возрастает с увеличением
плотноеги.
Показатель преломления подчиняется правилу аддитивности для
смесей углеводородов, что позволяет определить содержание того или
иного компонента в бинарной смеси, если известен показатель пре-
ломления зтой смеси /?см и каждого компонента:
//см' 100 = /?] х + /?2( 100 - л).
Кроме простых констант, таких как р и /7, для характеристики угле-
водородов и нефтяных фракций используют сложные константы, явля-
ющиеся функцией простых. Так, широко используются, например,
сложные константы — удельная и молекулярная рефракции. Удельная
рефракция является функцией н и р (формула Лоренца — Лорентца):
Г -
Молекулярной рефракцией называется произведение удельной
рефракции на молекулярную массу:
R = r-М=—----
/г -I- 2
М
Удельная и молекулярная рефракции
молекул сх следующими зависимостями:
связаны с поляризуемостью
4 л N.
г = а,
3 М
41
/? = — /V, a = 2,52J0H ot,
3 J
Таким образом, молекулярная рефракция по Лоренцу - Лорену
является мерой средней поляризуемости молекул, В электромагнитно^
поле видимого света поляризуемость молекул обусловлена смещениел
электронов и равна сумме смешений отдельных электронов. Поэтомуц
молекулярная рефракция носит аддитивный характер. Она являет^
суммой атомных рефракций для насыщенных соединений:
R, н = Ю/?с + 22/?н.
с|0н22 С Н
Атомная рефракция углерода равна 2,418, и атомная рефракция во.1
дорода — 1,100.
Для ненасыщенных соединений к сумме атомных рефракций необ
холимо прибавить инкременты рефракций для кратных связей:
Связь
Инкремент (D-линия)
1,733
2,397
Полученная таким путем (т. е. путем сложения атомных рефракций
и инкрементов) молекулярная рефракция является теоретической ве-
личиной в отличие от экспериментально найденной по формуле Ло-
ренца — Лорентца на основании экспериментального определения л
и р. Совпадение теоретической и найденной молекулярных рефракций
позволяет идентифицировать органическое соединение.
Рассмотрим пример1. Из нефтяной фракции выделен углеводород
с температурой кипения 110°; //D20 = 1,4955 и р420 = 0,866.
Константы углеводорода почти совпадают с константами толуола.
Однако для полной идентификации этого углеводорода необходима
вычислить молекулярную рефракцию и сопоставить ее с теоретической
для толуола:
сн.
и2 + 2 р I.49552 + 2 0.866
Теоретическая молекулярная рефракция:
Rr= 7RC + 8/?н + ЗЯС=С = 7-2,418 + 8-1,100 + 3-1.733 - 30,925,
Совпадение является удовлетворительным, следовательно, угле-
водород идентифицирован. Это толуол.
1 Пример вин из книги В. И Исагуляппа. Г. М. Егоровой. Химия исфги. Руководств
к лабораторным шиятиям — М.; Химия. 1965
Разиину в 0,2-0,5 относят к ошибкам эксперимента.
Атомные рефракции или рефракции связей (инкременты) могут
быть найдены по экспериментальным рефракциям /?>кс химически
чистых соединений. Например, для нахождения атомных рефракций С
и Н поступают следующим образом.
Находят вначале атомную рефракцию группы -СН2~:
/? — /? >кс — D
l\ Ibhlll; АУНиСН?)4СН, “ к
Затем находят
R
и R,
= Л- си,-
Другой сложной константой, применяемой при анализе углеводо
ролов нефти, является интернепт рефракции. Это разность между по
казателем преломления и половиной плотности:
г =
2
Значения ной функции для углеводородов одного и того же ряда
близки. Эго позволяет отнести исследуемый углеводород к определен-
ному гомологическому ряду. Так, алканы, нафтены и ароматические уг-
леводороды, выкипающие до 200°С, имеют следующие значения г.:
Углеводороды
Алканы
Hat}) гены
Арен ы
1,046
1,040
1,063
Ряд аналитических задач решается в химии нефти с помощью дис-
персионных методов, основанных на измерении рефракционной дис-
персии. Дисперсией называют зависимость показателя лучепрелом-
ления оз длины волны. В качестве меры дисперсии может служить
разность показателей преломления для двух длин волн:
Д, ' 4..
Для удобства эту разность умножают на 104. Углеводороды с близки-
ми значениями показателей преломления могут иметь сильно различа-
ющиеся значения дисперсии. Это позволяет иногда провести иденти-
фикацию углеводородов.
42
43
Так1. //t2() для //-ксилола и 1,3-диэтилбензола равны 1,49141 и
1,49146, а значения дисперсии (//рп - /7С20)-104 для этих углеводородов
значительно отличаются: 157,1 и 144,2.
Если (лг - //с)Ч04 разделить на плотность, то получим удельную
дисперсию:
У углеводородов ароматического ряда величина удельной диспер-
сии намного выше, чем у алканов и нафтенов, например у углеводоро-
дов ряда бензола до 200 (170—200). а у нафтенов и алканов — 99. Удель-
ные дисперсии аддитивны, поэтому, зная удельную дисперсию бензи-
новой фракции (8ф ), можно рассчитать содержание ароматических уг-
леводородов в этой фракиии(,г):
бфр100 = 5.,рх + 6„ф(Ю0-л).
Другой сложной константой является относительная дисперсия:
н[; - н(
Wr C.D “ .
Относительная дисперсия может быть применена для идентифика-
ции углеводородов. Зная относительную дисперсию углеводорода
сОрс.п и его можно с помошью специальных таблиц определить,
к какому ряду углеводородов он относится. Относительная дисперсия
cOf.gd1 как указывает Иоффе2, для легких ароматических углеводородов
колеблется в пределах 29-33 ед.
Легкие алканы и нафтены имеют wRC D примерно одинаковую и
равную 17,5. Для тяжелых нафтеновых углеводородов, выделенных из
высококипяших фракций различных нефтей, значение этой константы
18—19. Величина относительной дисперсии сложных углеводородных
смесей связана с содержанием ароматических углеводородов следую-
щим образом:
А= со<0>г.с О -0.015г-17.55).
где I — йодное число; — величина, характерная для ароматичес-
ких углеводородов узкой фракции:
Барковский В. Ф., Горелик С. М., ГорденцепаТ. Б. Физико-химические методы ана-
лиза. — М/ Высшая школа, 1972. — С. I [0.
2 Иоффе Б. В. // ЖОХ. - [946. - [6. - [121.
44
к 100
,<C D (®h c dU -17.55’
17,55 — средняя величина дисперсии для нафтено-парафиновой
части.
Метод относительной дисперсии требует для анализа малых коли-
честв фракции (0,5—1 мл) и является быстрым и удобным. Вместо
(Орс D используется другая константа — дисперсиометрический коэф-
фициент:
величина которого постоянна для деароматизированных бензиновых
фракций и равна 194,4 (кроме бензольной фракции 60-95°С, для кото-
рой эта величина равна 193,9). Дисперсиометрический коэффициент
является аддитивной функцией1, позволяющей определить процентное
содержание ароматических углеводородов л в бензиновых фракциях:
фрак
Г.(
100-C7.X+/)Дк1||аф(100-х).
Л'Т ’ ароматических углеводородов находят с помощью специаль-
ных таблиц в зависимости от температуры кипения фракции.
2.2. Спектральные методы идентификации углеводородов
и других компонентов нефти и газа
2.2.1. Молекулярная спектроскопия
Известно, что при прохождении электромагнитного колебания от
источника излучения через вещество последнее поглощает лучи только
определенной длины волны. В спектре поглощения этого вешества
имеются характерные полосы поглощения, соответствующие частотам
поглощенных лучей.
В чем заключается причина способности органических веществ,
в частности углеводородов, поглощать лучи электромагнитного спектра?
Молекула углеводорода обладает определенным запасом внутрен-
ней энергии. Эта энергия слагается из энергии взаимодействия элект-
ронов с ядрами, энергии колебательного движения атомов, энергии
вращательного движения атомов или групп атомов. Энергия взаимо-
действия электронов с ядрами (энергия электронных переходов)
в Ю—20 раз превышает энергию колебательных движений внутри мо-
лекулы и в 1000 раз энергию вращательных движений.
1 Иоффе Б. В.. Баталин О. ЁГ// Ж ИХ. —1959. — 32 — 2723
45
В зависимости оттого, какие лучи электромагнитного спектра про-
пускать через вещество, могут возбуждаться либо вращательные дви-
жения, либо колебательные, либо электронные переходы, либо все
движения одновременно. Возбуждение того или иного движения в мо-
лекуле происходит тогда, когда его частота совпадает с частотой
электромагнитного колебания. Таким образом, в основе молекулярной
спектроскопии лежит физическое явление резонанса.
Энергия электромагнитных колебаний уменьшается с увеличением
Л. в последовательности: рентгеновские лучи (X. = 0,01- 10 ), ультра-
фиолетовые (10-4000 ), видимый свет (4000-8000 ), инфракрасные
лучи (0,8-300 мкм), микроволны (0,03-100 см), волны радиодиапазо-
на. Энергия радиоволн слишком мала, чтобы возбуждать органические
молекулы.
Микроволны и длинные инфракрасные волны могут возбуждать
только вращательные движения в молекулах. Если частоты колебаний
этих волн совпадают с частотами вращения отдельных частей молекулы,
то происходит резонансное поглощение энергии этих волн, что отразит-
ся в спектре поглощения. Такого рода спектры применяются для тонко-
го структурного анализа органических веществ.
Основополагающими законами молекулярной спектроскопии яв-
ляются законы Бугера, Ламберта и Бера.
Закон Бугера — Ламберта: относительное количество поглощенного
слоем вещества света не зависит от интенсивности падающего света, и
каждый последующий слой среды одинаковой толщины поглощай
одну и ту же долю проходящего через него света.
Закон Бера: поглощение монохроматического света пропорцио-
нально числу молекул поглошаюшего вещества в единице объема раст-
вора.
Оба закона могут быть выражены уравнением Бугера — Ламберта-
Бера:
Z)" 1g у- = е с /,
где ,/(J — интенсивность падающего излучения; J — интенсивность из-
лучения, прошедшего через слой вещества: е — молекулярный коэф-
фициент поглощения: с — концентрация анализируемого вещества,
моль/литр: /-— толщина слоя вещества, см.
Инфракрасная спектроскопия
Обычно инфракрасные спектры органических соединений изуча-
ют в диапазоне 1—25 мкм. При этом линии поглощения в спектре по-
являются за счет колебательных движений в молекулах исследуемого
иетсстнп.
Колебательные движения могут быть линейными (валентными; ю-
формационными и групповыми.
Линейные колебания проходят вдоль связи: при деформационном
колебании меняются углы между валентностями. Ниже приводятся не-
которые типы колебаний С-Н-связи в группе =СНу
вал С И 7 Н ОС С i I м м с гр и ч н ОС
вал с н т н ос ас 11 м м стр и1 [ н ос
—Н——Н—►
дсформаиионнос
симметричное
деформаиионнос
аси ммстричнос
н
Групповые колебания - это колебания скелета органической моле-
кулы. Например, колебания цепочки из метиленовых групп в молекуле
н-алкана, колебания третичной бутильной, изопропильной и других
групп. Энергия и частота линейных колебательных движений выше,
чем деформационных колебаний.
Колебательные движения в молекулах характеризуются частотой,
т. е. числом колебаний в секунду (с_|); в практике инфракрасной
спектроскопии обычно используют вместо частоты волновое число v—
величину, обратную длине волны (см
Если частота какого-либо колебательного движения в молекуле
совпадаете частотой какого-либо луча, то такой луч поглощается веще-
ством (резонансное поглощение) и в спектре появляется полоса погло-
щения.
Каждой функциональной группе, группе атомов и связи в молекуле
исследуемого вещества в спектре соответствуют несколько линий,
отвеч а ю щ и х частот а м п о гл о ще н н ы х л у ч е й. В хим и и нефти зада ча к аче-
ственного анализа углеводородов с помощью ИК-спектров состоит в об-
наружении характеристических частот выделенных из нефти индивиду-
альных соединений.
В табл. 4 приводятся значения волновых чисел валентных и дефор-
мационных колебаний святи С—С и С—Н в молекулах углеводородов.
Групповым колебаниям скелета органических молекул соответству-
ют полосы поглощения в области 700— 1500 см Г Эта часть спектра ор-
ганических молекул очень чувствительна к малейшим изменениям в их
структуре. В этой области, называемой «отпечатком пальцев», каждое
46
47
Таблица 4. Значения волновых чисел
Тип колебаний н спя ль Интервалы волновых чисел см“!
L ——-— Вх 1С1П11 bl С КО.КЧКШНЯ спязи
с н 2800-3000
С VI 3000- экю
Леформанной ные колебания спя яг
он 1350-1470
=С- н 600 1000
Валентные колебания связи
С-С 600-1200
ОС 1500-I6S0
соединение имеет только свойственный ему набор полос поглощения
Колебаниям углеродного скелета ароматических ядер соответствуя
полоса при 1600 см-', колебаниям углеродного скелета нафте но вьц
циклов — при 970 и 1030 см-1.
Колебания полиметиленовых цепочек в зависимости от их длина
проявляются в области 720-790 см, колебания изопропильной груп-
пы — при 1170 и 1145 см-1. mpe/n-бутильной — при 1255 и 1210 см-|,длй
монозамещенного бензольного кольца характерна полоса при 700 см-1.
ИК-спектры записываются в координатах: процент пропусканий
(или поглощения) излучения — волновое число (рис. 19 и 20).
Процент пропускания J/Jo-lOO. Процент поглощения 100 - J/J0'IOO.
Рис, 19. ИК-спектр 1-децена:
А — валентное колебание =С-Н (см. табл.4); Б — валентное колебание =С-Н
при 3049 см"1, характерное для олефинов; В — валентное колебание С=С, 1645 см
(см.табл,4); Г — внеплоскостные деформационные колебания =С-Н, 986, 907 смФ
Д колебание группы (СН2)7 720 см~1; Е — деформационные колебания -С-Н
в группах -СНг- и -СНЭ
48
Оптическая плотность
Рис. 20. ИК-спектр толуола:
А — валентные колебания =С-Н бенз, кольца; Б — валентные колебания С-Н
в СН3; В — валентные колебания С=С бенз, кольца; Г — деформационные
колебания С-Н в СН3; Д — деформационные колебания С-Н в бенз, кольце
Ультрафиолетовая спектроскопия
Она позволяет исследовать взаимодействие ультрафиолетового из-
лучения с электронным облаком молекул. Для аналитических целей
служит диапазон ультрафиолетового излучения в пределах 200-400 нм.
В молекулах углеводородов электроны находятся на связующих моле-
кулярных орбиталях; в случае гетероатомных соединений нефти (кис-
лородных, сернистых, азотистых) электроны имеются также на несвя-
зующих орбиталях (электроны неподеленных пар гетероатомов).
Так как энергия квантов ультрафиолетового излучения близка к
энергии электронов, то при облучении молекул происходит возбужде-
ние электронов и они переходят на разрыхляющие (нестабильные) мо-
лекулярные орбитали с более высокой энергией. На диаграмме
(рис. 21) показаны переходы о-, п- и лээлектронов на разрыхляющие
4
4 - 3544
49
орбитали. Через а то* обозначается переход о-электронов со связущ.
meii на разрыхляющую а*-орбиталь. о-Электроны — это гибридизиро.
ванные электроны, участвующие в образовании простых С—С и С-Н-
связей. Они близко расположены к ядру, прочно с ним связаны, и дм
их перехода на разрыхляющую а-орбиталь необходима большая энер-
гия. Такой энергией обладают ультрафиолетовые лучи с длиной волнв
менее 160 нм (труднодоступная область ультрафиолетового излуче-
ния — вакуумный ультрафиолет). Так как в образовании связей в моле-
кулах ал канон и нафтенов участвуют только о-электроны, то ультрафи-
олетовые спектры для этих углеводородов не характерны.
Через тгтол* обозначается переход теэлектронов двойных связей
непредельных и ароматических углеводородов на разрыхляющие к*,
орбитали; теэлектроны слабее, чем о-электроны, связаны с ядром,г
пя их возбуждения и перехода на разрыхляющие орбитали требуете
меньшая энергия. Так, олефины поглотают ультрафиолетовое излуче-
ние в области 170-180 нм, а диены и ароматические углеводороды-
в области еще более длинных волн (> 200 нм). Таким образом, диено-
вые и ароматические углеводороды дают характерные ультрафиолето-
вые спектры в пределах 200—400 нм.
Энергия квантов ультрафиолетового излучения в 20 раз больик
энергии квантов инфракрасного излучения и приближается к величи-
не энергии, необходимой для разрыва связей в молекулах органически),
соединений. Поэтому ультрафиолетовое излучение сильно возбуждав
молекулы органических соединений, и ультрафиолетовые спектрк
представляют собой довольно грубую картину в отличие от тонкой
структуры инфракрасных спектров.
Ультрафиолетовые спектры записываются в форме зависимости ло-
гарифма молярного коэффициента поглощения 1g е от длины волны!
Молярный коэффициент поглощения е является коэффициента»
в уравнении Бугера — Ламберта — Бера. Кривая УФ-спектра имеет
один или несколько максимумов поглощения, которые характеризуют-
ся соответствующими длинами волн 2.тах. Из рис. 22, на котором при-
веден УФ-спектр нафталина (1) и антрацена (2). видно, что X, , соот-
ветствующие максимумам поглощения антрацена, смещены в длинно-
волновую часть спектра по сравнению с нафталином, Это объясняете
тем, что в антрацене число сопряженных двойных связей больше, че»
в молекуле нафталина. Известно, что с увеличением длины цепи сод
ряжения в молекуле облегчается переход тг-эгс*. Так как для такого пе-
рехода требуется меньшая энергия, поглощается волна большей длины
В габл 5 приведены значения длин волн, соответствующие макси
мхмам поглощения в УФ-спектрах для углеводородов, молекулы кото-
рых содержат различное число сопряженных двойных связей.
Введение алкильных групп в бензольное ядро вызывает' сдвиг обет
Рис. 22
полос поглощения для бензола
в длинноволновую область
спектра. Для смесей алкилбен-
золов максимум поглощения
лежит в области 255-275 нм.
Для алкилнафталинов макси-
мум поглощения находится в
области 275-290 нм; в области
310-330 нм имеются также два
характерных максимума. Для
алкилпроизводных антрацена
и фенантрена характерны ши-
рокие полосы поглощения с
несколькими максиму м а м и,
соответственно в области 310-380 нм и 280-310 нм.
В случае гетероатомных соединений нефти (кислородные, сернис-
тые, азотистые соединения) алифатического ряда УФ-спектры возни-
кают в результате п—мт* перехода неподеленных пар электронов гетеро-
атомов. Полосы поглощения находятся в коротковолновой области
(<200 нм).
Если гетероатом находится рядом с двойной связью или аромати-
ческим ядром, то поглощение происходит в области 200-300 нм (пере-
ход в—>11*).
Спектры комбинационного рассеяний образуются, если вещество об-
лучать монохроматическим светом, причем частота монохроматичес-
кого света должна значительно отличаться от частоты ультрафиолето-
вых лучей, так как они поглощаются электронами. Обычно используют
луч видимого света, например луч, соответствующий синей линии
спектра ртутной лампы (4358 ), В современных спектрографах в каче-
Таблица 5. Значения Хтах для углеводородов с различным числом
сопряженных двойных связей
Углеводород Число сопряженных спя jcii Длина волны, соответствующая максимумам поглощения, им
Этилен 1 162
Бутадиен э 217
Бен ю. । з 203.255
Нафта.1 ин S 228.2S6 314
Ашрацен 7 258, 356, 380
1 Эффект комбинационною рассеяния снега открыт советскими учеными Мандель-
и।гамом и Даплсбср!ом и индийским ученым Раманом в 192JS г.
Рис. 23
ст вс возбуждающего луча используютлуч лазера. Часть лучей монохро-
матического света проходит через вещество без изменения, другук
часть поглощает электронное облако молекулы, вызывая его деформа-
цию. Часть энергии деформированного электронного облака идет не
возбуждение колебательных движений в молекуле, Остаток энерт
поглощенных лучей испускается в пространство в виде вторичного из
лучения меньшей частоты. Таким образом, в спектре наряду с линиег
возбуждающего луча частоты v() появляется ряд линий слева от нее, со
ответствуюших лучам вторичного излучения (стоксовы линии) (см.
рис. 23).
Энергия, поглощенная за счет возбуждения какого-то колебатель-
ного движения, составит
h (v0- vp.
Разность v0 - Vj соответствует частоте какого-либо колебательной
движения в молекуле. Таким образом, по спектрам комбинационное
рассеяния света можно определить частоты колебания в молекуле к*
шества без необходимости работать в инфракрасной области. Иногдз
на спектрах появляются симметрично расположенные линии, смешен*
ные в сторону больших частот относительно частоты возбуждающей
света v0 (антистоксовы линии), возникающие за счет того, что в веще-
стве есть возбужденные молекулы, которые, переходя в нормальное
состояние, излучают энергию, суммирующуюся с энергией возбужда-
ющего луча.
В настоящее время определены частоты и интенсивности линий!
спектрах комбинационного рассеяния света большого числа индиви-
дуальных углеводородов и других органических соединений. Линии с
волновыми числами от 0 до 1500 см-1 зависят от колебаний средних!'
тяжелых атомов относительно друг друга (С, О, N, S, галоиды). Лини!
между 1500 и 1800 см-1 объясняются наличием двойной связи в моле-
куле, волновые числа от 2000 до 2500 см-1 — наличием тройной связи
Линии с волновыми числами от 2000 до 3300 см-! вызываются колеба
ниями водорода по отношению к другим атомам.
52
2.2.2. Масс-спектрометрия
Масс-спектрометр - это прибор, который позволяет раздетигз ио
ны. полученные бомбардировкой молекул электронам.г раде еХ
ионов происходит по их массам. Схема прибора приведена на рис 24
Пары образна из баллона поступают через диафрагму в вакуумиро-
ванную камеру (остаточное давление 10-6 мм от ст i г,^\-т. " Р
с потоком электронов. движущихся
к электронной ловушке (земля). Энергия электронов в масс-спектро
метрии положительных ионов велика -70 эВ. Электроны выбивают ю
молекул ороитальные электроны и превращают молекулы в ионы
Кроме электроноударного метола ионизации молекул в химии нефти для
анализа высокомолекулярных нефтяных компонентов, обладающих низ№й
термической стабильностью, применяется метод волевой ионизации (ионии-
имя пол действием сильного неоднородного электрического поля) Развитием
Балов с образном
Вентиль
(тонкое отверстие
в фольге)
Рис. 24. Схема масс-спектрометра
53
полевой ионтапии является метод полевой десорбции; образец наносится н>
острие вольфрамового эмиттера, помешенного в сильное неоднородное элек]
ричсскос поле. Эмиттер медленно нагревается электрическим током. Иониза
пня молекул происходит вследствие туннелирования наиболее подвижны!
электронов в эмиттер, и образующиеся молекулярные ионы десорбируются;
газовую фазу. Энергия таких молекулярных ионов мала, поэтому они почти н;
подвергаются фрагментации.
Ионы под действием возрастающего электрического поля, прила
женного к сеткам ионной пушки, втягиваются в ионную пушку и уско-
ряются. Диаметр диафрагм сеток увеличивается по .ходу движения ио-
нов, поэтому ионы образуют расходящийся пучок, который попадаг
в магнитное поле. Нейтральные молекулы вы водятся из камеры с по-
мощью вакуумного насоса. Магнитное поле отклоняет ионы от прямо-
линейного движения, и они начинают двигаться по круговым орбита1
под влияниям центростремительной силы магнитного поля:
F= Hzw
где Н — напряженность магнитного поля: < — заряд иона: к — скорост:
иона.
Эта сила уравновешивается центробежной силой:
г
где г— радиус кривизны траектории.
Откуда
_ v т
7
Следовательно, радиус кривизны зависит от отношения массы о
ряду (при постоянных г и Н). Изменяя Н или v (г зависит от прилагав
мого к сеткам ионной пушки потенциала И, можно поочередно фоку
сировать ионы различной массы на коллектор (с помошыо шел1
~0,1 мм). Образующийся при этом ионный ток усиливается и подаете
на записывающее устройство.
На рис. 25 приведен общий вид масс-спектрограммы. Последив
пик соответствует самому тяжелому молекулярному иону, который обя-
зуется при отщеплении от молекулы одного электрона. Остальные пи
ки соответствуют осколочным ионам, образующимся при распаде Mt
пекулярного иона.
Ниже приведена упрощенная схема масс-спектрального распа:
и зо и ро п ил бе и зола:
Молекула изопропилбензола под действием электронного удара
превращается в ан ион-радикал, затем — в катион-ради кал (молекуляр-
ный ион), распад которого приводит к положительным осколочным
ионам различной массы.
С помощью масс-спекзроскопии в химии нефти решается ряд задач:
1) по массе молекулярного иона можно с большой точностью опре-
делить молекулярную массу углеводорода или гетероато мн ого соедине-
ния, выделенного из нефзи. Для этого достаточно иметь очень малые
количества вещества вплоть до 10 12 г;
Рис. 25. Общий вид масс-
спектрограммы
2) по массе молекулярного ио-
на с помощью ЭВМ по специаль-
нои программе можно определить
элементный состав вещества. Дня
этого необходимо иметь приборы
с высокой разрешающей способ-
ностью1 и определить молекуляр * 7
ную массу вещества с точностью
до 10 3-10-4. По масс-спектру ве-
щества можно определить его
Рэ (решающая способное зь определяется степенью ра меления л пух соседних пикон:
7 hccb id массовое число о,-того из ионон; Aw - разность масс соседних
ионов У лучших современных приборов ла величина может быть от 10 до 150 тыс.
54
55
структурную форму iv. Для лого необходимо идентифицировать оско-
лочные ионы по их массам, а затем по фрагментам молекулы воссоз-
дать ее структуру;
?) с помощью масс-спектрометрии низкого разрешения можно оп-
ределить качественный состав углеводородных смесей по массам моле-
кулярных ионов компонентов.
Масс-спектральным анализом можно определить количественный
состав углеводородных смесей.
Дня простых смесей нескольких веществ это можно сделать на ос-
новании интенсивности пиков молекулярных ионов. Пользуясь калиб-
ровочными графиками в координатах интенсивность пика — процент-
ное содержание, составленными для каждого компонента на основа-
нии масс-спектров искусственных смесей компонентов, можно лс
масс-спектрограмме смеси неизвестного состава определить процент-
ное содержание каждого компонента в смеси.
Для получения данных по количественному составу более сложны),
смесей масс-спектрограмму смеси необходимо сравнивать с масс-
спектрограммами индивидуальных углеводородов. С помошью систе-
мы уравнений можно определить количественный состав анализируе-
мой смеси по ее масс-спектру. Предположим, что смесь из п компонен-
тов дает спектрограмму, состоящую из р пиков. Вклады компонент®
смеси в образование этих пиков могут быть различными. Каждый пш
может соответствовать иону, полученному из одного компонента, либо
ионам одинаковой массы, полученным из разных компонентов. Дк
установления того, какова доля участия каждого компонента в образо-
вании пиков, получают отдельные спектрограммы каждого компонен-
та (если известен качественный состав смеси). Эти спектрограмма
сравнивают со спектрограммой смеси и обнаруживают те пики н;
спектрограмме исследуемой смеси, в образовании которых принимаю’
участие все компоненты. Выбирают несколько таких пиков и составля-
ют для них систему уравнений;
= Ai \ + ХЛ.„ + • ^Л,.
= х^ + хЛ + хЛ> +...лД
н.: +x,h, +...Л-Д
• 1 * < 1 ’ • - . ь
= а-А„„ +Х'Л.:, +хЛт> + ---ХЛ. -
где ,7/rtI......Н,и _ высоты пиков, соответствующие массовй'1
числам на спектрограмме смеси; д-р ад, a'v... хп — пар'
циальные давления компонентов смеси, мм рт. ст.; /? \ h-, , ". h ,
I W | 1 Ли J ' I f^2
56
/;„ - высоты пиков на спектрограммах чистых компонентов, прихо-
дящиеся на единицу их давления и соответствующие массовым числам
/лр т2. ту ... т1;.
Решением системы уравнений с помошью ЭВМ находятл^, лд. л3. ...
х В последнее время при исследовании нефтей при меняется метод
хромато-масс-снектрометрии. В этом методе используется газовый хро-
матограф в блоке с масс-спектрометром и ЭВМ. В хроматографе проис-
ходит разделение углеводородов, затем каждый углеводород поступает
в масс-спектрометр: данные его масс-спектра поступают в ЭВМ, кото-
рая расшифровывает спектр и идентифицирует углеводород.
Для исследования гетероатомных соединений нефти большое зна-
чение имеет масс-спектрометрия отрицательных ионов. В отличие от
масс-спектрометрии положительных ионов молекулы анализируемого
вешества подвергаются бомбардировке электронами низкой энергии
(1-3 эВ), которые захватываются молекулами гетероатомных соедине-
ний с образованием анион-радикалов, Отсюда другое название этого
в и да м асе- с пе ктро м етр и и — масс-спектрометрия электронного захва-
та. Углеводороды не поглотают электроны низкой энергии.
Ан ион-ради кал. образовавшийся в результате захвата электрона мо-
лекулой вешества (молекулярный ион) подвергается распаду с образо-
ванием радикалов, анионов и анион-радикалов. В качестве примера
рассмотрим схему масс-снейтрального распада нефтяного сульфида:
Благодаря тому что углеводороды «прозрачны» для электронов низ-
кой энергии, масс-спектрометрическое исследование гетероатомных со-
единений можно проводить, не выделяя их из фракции в чистом виде.
57
2.2.3. Спектроскопия ядерного магнитного резонанса
Кроме массы и заряда, ядро обладает третьей характеристикой -
моментом количества движения, который обусловлен его вращение^
вокруг оси (спином). Поскольку ядро заряжено, его вращение вокру
собственной осп приводит к круговому движению заряда, что фор-
мально аналогично круговому электрическому току (рис. 26),
Этот круговой ток создает магнитное поле, так что вращающееи
ядро подобно крошечному магниту, ось которого совпадает с осью спи-
на. и в результате ядро может характеризоваться магнитным диполь-
ным моментом1. Рассмотрим теперь ядро, магнитный диполь которой
ориентирован под некоторым углом 0 к направлению силовых лини£
постоянного магнитного поля /70 (рис. 27). Это поле обусловливает по-
явление силы, стремящейся расположить ядро-магнит вдоль поля, нс
поскольку ядро вращается и обладает моментом количества движения,
оно сопротивляется этому воздействию, в результате чего наблюдаете
прецессия магнита-ядра: кроме вращения вокруг своей оси, ядро вра-
щается еще вокруг направления постоянного магнитного поля, подоб-
но тому, как прецессирует волчок, если он наклонен по отношению г
силовым линиям гравитационного поля Земли. Угловая скорость этов
прецессии (рад/с) не зависит от угла 0, но зависит от напряженности
постоянного магнитного поля Н[}:
со = ун(г
где у — гиромагнитное отношение, куда входит, в частности, ядерный
магнитным момент.
Рис. 26
'Ядра, имеющие четный заряд
ладают магнитным моментом ЯчпяЛ|°РЯДК°ВЬ,Й Номер^ и четное массовое число, не об-
30. имени „агни^ХХХ /?1’ счетный заряд. но четное массовой
массовое число, имеют магнитный момент / и^сю,цие нечетный заряд и нечетна
ядра изотона углерода и изотопа кремния (В0Д0р0л< фтор’ фосФ°^ так же кая
и четный заряд. Р ‘ $ М1 имеющие нечетное массовое числе
58
Таблица 6. Значения ЯМР для различных ядер
Я : фО Спин Ч(|С 1 (НИ Я М Р (! ПО. !С |4ИЮ1с. М1п
IH 1/2 60
|4\ 1 4.3
19 F? 1/2 36 5
160 0 —
120 0 -
130 1/2 1.3,09
Рассмотрим теперь влияние небольшого магнитного поля //,, пер-
пендикулярного к //0. Оно стремится отклонить диполь (ось спина) в
плоскость XY.. однако действие этого поля незначительно. Если же это
поле начнет вращаться вокруг направления линий магнитного поля Н[}
(т. е. если оно будет переменным), то, когда частота этого поля достиг-
нет частоты прецессии ядра, произойдет поглощение ядром кванта
энергии. Это поглощение называется резонансным.
Тот же самый результат может быть получен, если частота пере-
менного поля остается постоянной, а изменяется напряженность
постоянного магнитного поля. При изменении напряженности пос-
тоянного магнитного поля изменяется частота прецессии ядра, и,
когда она достигает частоты переменного магнитного поля, происхо-
дит резонанс. На практике обычно реализуется этот способ. Таким об-
разом, задача анализа обычно состоит в том, чтобы определить напря-
женность постоянного магнитного поля, при которой наступает резо-
нанс ядер исследуемого образна в переменном поле определенной
частоты V. В этом случае частота v равна частоте ЯМР. В табл. 6 при-
ведены значения ЯМР для ядер различных атомов. Из табл. 6 видно,
что ядра кислорода и углерода, спин которых равен нулю, не являют-
А у/ \ / Рис. 28 ся магнитными, поэтому они неспо- собны к Я М Р. Что же происходит с ядром в мо- мент резонанса? Поглотив квант энер- гии. ядро меняет направление своего спина по отношению к направлению постоянного магнитного поля //0 на вполне определенный угол (рис. 28). Таким образом, ЯМР связан с перео- риентацией спинов ядер. Для получения спектров ядерного магнитного резонанса вещество в ко-
59
Рис. 29
л и мест ве 0,2 мг в запаянной стеклянной ампуле помешается в катушку
(рис. 29, (7), находящуюся в постоянном магнитном поле, которая явля-
ется частью высокочастотного контура (рис. 29, а). Контур настраива-
ют на определенную частоту переменного магнитного поля, затем уве-
личивают напряженность постоянного магнитного поля. При опре-
деленном значении /70 наступает резонанс. Образен поглотает энер-
гию: при этом уменьшается сопротивление контура и уменьшается
напряжение на клеммах. Это изменение напряжения посредством уси-
лителя фиксируется записывающим устройством в виде сигнала. На
рис, 30 приведена часть спектрограммы воды. Сигнал (пик) протонов
воды получается при напряженности поля 1180 Гс при частоте пере-
менного поля 5 МГц.
В практике анализа углеводородов нефти применяют метод протон-
ного магнитного резонанса (ПМР) и ядерного магнитного резонанса
изотопа углерода |3С(ЯМР ,3С).
Протонный магнитный резонанс
Если в молекуле исследуемого вещества все протоны одинаковы
(например, в молекулах воды, метана), то все эти протоны будут резо-
нировать при одной и той же напряженности внешнего поля, поэтому
спектр этих веществ (при условии их абсолютной чистоты) будет состо-
ять из одного пика (рис. 30).
Спектр метилового спирта включает два сигнала (пика) соответ-
ственно числу протонов различного типа (рис. 31): л-метилбензилхло-
рид — три пика (рис. 32), причем площади, ограниченные этими пика-
ми. находятся в отношении 4:2:3, что пропорционально числу протонов
различного типа.
То, что протоны, занимающие различное положение в молекуле ор-
ганического соединения, резонируют при различных напряженностях
внешнего магнитного поля, обусловливается различием электронной
60
h [Lioi нос।и вокруг протонов. Прогоны
I-цо защищены от действия внешнего маг-
нитного поля окружающими электрона-
ми. Под действием внешнего магнитно-
го поля электроны образуют свое маг-
-------II____ нитное поле, направленное противопо-
ложно внешнему. Чем больше электрон -
_____________г—-—т_ пая плотность вокруг протона, тем силь-
ной НЮО 1500 Н. нее будет ослабляться действие внеш не-
Рис. 30____________г0 магнитного поля, и для достижения
резонанса протона нужно усилить пос-
леднее. Плотность электронного облака вокруг протона зависит от его
расположения в молекуле и от влияния других атомов. Так, в молекуле
метилового спирта
Н
н—► С------►()-<—н
н
сильнее всего индуктивному /-эффекту атома кислорода подвержен
протон группы ОН, затем протоны группы СН3. Следовательно, мини-
мальная электронная плотность и минимальное экранирование наб-
людаются у прогона группы ОН, затем группы СН3. Поэтому протон
группы ОН будет резонировать при меньшей напряженности магнит-
ного поля, чем протоны СН3 (см. рис. 31).
В случае протонов ароматических ядер наблюдается особый эф-
фект — эффект к-электронного дезэкранирования. Если молекула
бензола находится в магнитном поле, то л-электроны бензольного яд-
ра вращаются вокруг силовых линий магнитного поля, образуя круго-
Рис. 31
61
A
вой ток. Возникает магнитное поле, силовые линии которого внутри
ядра направлены против внешнего поля (экранирование), а вне ядра-
по полю (дезэкранирование). При этом магнитное поле около прото-
нов бензольного ядра усиливается (рис, 33), что облегчает их резонанс.
Аналогичный эффект наблюдается также в случае протонов, связанных
углеродными атомами двойных связей олефинов и диенов. Этот эф-
фект приводит к тому, что протоны ароматических углеводородов и
протоны при двойных связях резонируют в значительно более слабом
поле, чем протоны насыщенных углеводородов.
Таким образом, резонанс протонов может дать очень пенную ин-
формацию о строении молекулы вешества. о взаимном влиянии атомов
в молекуле. Эта информация определяется из величин химического
сдвига 5 (выражается в миллионных долях — мд.):
5=tf" tf|0".
где Н ~ напряженность магнитного поля, при которой происходит ре-
зонанс протонов данного типа; Нп — напряженность поля, при кото-
рой резонируют протоны эталонного вещества.
В качестве эталонного вешества обычно используют тсграметилси-
лан (ТМС) (CH3)4Si. Протоны в молекуле ТМС рстонируют в более
сильном поле, чем протоны многих других органических соединений.
Кроме шкалы 6, для характеристики химического сдвига принимается
шкала т. Две шкалы связаны между собой отношением
т = 10-6.
На рис. 34 приведен ПМР-спектртолуола с использованием шкал$
Рис. 34. ПМР-спектр толуола
и т, из которого очевидно значительное различие в химических сдвигах
протонов метильной группы (6 ~ 2) и протонов ядра — (8 ~ 7).
Если в молекуле углеводорода у соседних атомов углерода имеются
протоны различного типа (неэквивалентные протоны), то сигналы
этих протонов расщепляются, проявляется тонкая структура спектров.
Это дает ценную информацию о строении углеводородов. Одной из
причин растепления сш налов является спин-спиновое взаимодей-
ствие неэквивалентных протонов. При наложении внешнего магнит-
но го п ол я п р ото н ы //^ н ол о в и н ы м ол е ку: i б у дут и м ет ь с п и и ы, направ-
ленные по полю:
При этом увеличивается эффективное поле, действующее на прото-
ны Н(1 этих же молекул, и они начинают резонировать при несколько
меньшей напряженности внешнего магнитного поля. Протоны //рдру-
гой половины молекул будут иметь спины, направленные против поля:
эффективное поле, действующее на протоны //(Х этой части молекул,
будет ослабляться, и резонанс протонов Ни наступит при большей на-
пряженности внешнего магнитного поля. Аналогичные рассуждения
могут быть применены к протону //(/ Таким образом, каждый протон
даст в спектре дуплет (рис. 35).
Если взаимодействует между собой большее число неэквивалент-
ных протонов, чем в приведенном выше примере, то это приводит к бо-
лее сильному расщеплению пиков.
Так, в случае группы (Т Ц-Cl I 2 (в молекуле этилбромида) будет наб-
людаться картина, представленная на рис, 36. В общем случаен экви-
валентных протонов будут расщеплять пик группы соседних неэквива-
63
Рис, 35
(II, иг
Рис. 36
зснтных им протонов на (л + 1) пиков. Расстояние между линиями, нг
которые расщепляется пик протонов данного чипа. называется кон-
стантой взаимодействия J и измеряется в герцах. Измерение в герцам
возможно потому, что частота прецессии ядра прямо пропорциональна
напряженности постоянного магнитного поля. Значения констант J
дают дополнительную информацию о строении молекулы. В справоч-
никах наряду со значениями химсдвигов приводятся данные о конс-
тантах взаимодействия между неэквивалентными протонами.
В табл. 7 приведены значения химических сдвигов для протонов
различного типа.
Н а ос но ва н и и спектров П М Р не фтя н ы х ф ра к ц и й м ож н о н а йт и со-
отношение между протонами различного типа в усредненной молекуле
фракции, что позволяет составить представление о химическом соста-
ве фракции, о соотношении в ней различных структурных групп.
Ядерный магнитный резонанс изотопа углерода 13С
Кроме протонного магнитного резонанса, для химии нефти боль-
шое значение имеет ядерный магнитный резонанс стабильного изото-
па углерода ,3С.
Этот изотоп содержится в нефти в количестве 1,1%. Частота ЯМР
13С значительно отличается от частоты ЯМР протона, что позволяй
получать четкие спектры ЯМР 13С. По этим спектрам можно судитьо
Таблица 7, Значения химических сдвигов для протонов различного
типа в углеводородах (шкала 8)
Тип протона Химический сдвиг, м л. (эталон Si(CI 1д 5 = 0)
R-CH, 0,9
R-CH.-R' R.CH 1,3 2 0
r2c=ch2 CJ-p-H 5,0 7,3
а С-Н нафталина 7 8
Р С? Н нафталина 7.5 - - - —
64
34 32 30 28 2(i 24 22 20 IS 16 14 12 10 8 6 4 2 0 мд
Рис. 37. Спектр ЯМР 13С 2-метипбутана
числе типов атомов углерода, занимающих различное положение
и молекуле углеводорода (оно равно числу пиков на спектрограмме).
Например, у циклогексана — I пик. //-гексана — 3 пика, метилцикло-
пентана — 4 пика. Химические слюни щю атомов углерода, занимаю-
щих различные положения в молекулах углеводородов, существенно
различаются. По згому спектры ЯМР 13С позволяют провести четкую
идентификацию углеводородов различных рядов, в то время как
спектры ПМР в случае насыщенных углеводородов несут мало инфор-
мации из-за слабого различия в химических сдвигах протонов различ-
ного типа. На рис. 37 и ЗУ приведены спектры ЯМР 13С 2-мегилбутана
и 3-метил гептана и для сравнения на рис. 39 — спектр ПМР 3-метил-
гептана
Рис. 38. Спектр ЯМР '3С 3-метилгептана
65
5 - 3544
Рис. 39. Спектр ПМР 3-метилгептана
2.2.4. Спектроскопия электронного
парамагнитного резонанса (ЭПР)
Этот вид спектроскопии применяется для исследования систем, со-
держащих неспаренные электроны. В химии нефти - это смолисто-ас-
фальтеновые вещества и комплексные соединения металлов перемен-
ной валентности с гетероатомными соединениями нефти (порфирины,
смолы, асфальтены).
Явление ЭПРоткрыто в 1944 г. советским физиком Е. К. Завойским.
В чем оно заключается?
Электрон, как и ядро атома, имеет заряд и характеризуется момен-
том количества движения, обусловленным вращением вокруг своей
оси, то есть спином. Следовательно, электрон представляет собой маг-
нитный диполь, т. е. является крошечным магнитом. При отсутствии
внешнего магнитного поля все магнитные моменты электронов распо-
лагаются хаотично и имеют одинаковую энергию Е(}. Поэтому суммар-
ный магнитный момент будет равным нулю.
Если образец вещества, содержащего неспаренные электроны, ока-
жется в постоянном магнитном поле, то ось магнитного диполя элект-
рона может ориентироваться двояко: по направлению силовых линий
магнитного поля (нижний уровень энергии) и против направления си-
ловых линий магнитного поля (верхний уровень энергии).
Разность между энергиями этих состояний электрона во внешнем
магнитном поле определяется уравнением
AE = gp//,
где# - константа, равная 2,0023; [3 - магнетон Бора, равный для сво-
oojHoro электрона 0,9273-10^ эрг/эрстед; Н- напряженность маг-
нитного поля
66
Отношение числи эликiронов. находящихся на верхнем /роли-;
jiiepritu nf, к числу электронов. находящихся на нижнем уровне эн;р-
гни определяется законом распределения Ьольпмана
К!
где К- постоянная Ьольпмана; 7- абсолютная температура.
Если на свободный электрон, находящийся на нижнем уровне энер-
гии, будет действовать переменное магнитное поле частотой v, сило-
вые линии которою перпендикулярны силовым линиям постоянного
магнитного поля, го при условии
hv -= g р/А
где h — постоянная Планка, электрон поглотает энергию магнитно-
го поля, равную А/Г. и переходит на более высокий уровень энергии и
ось магнитного диполя оказывается направленной против поля. Од-
новременно с такой же вероятностью будут проходить переходы
электронов с верхнего уровня на нижний с выделением энергии. Но
так как л, больше /р и переходы с нижнего уровня на верхний будут
преобладать, то в резулыате будет происходить поглощение образцом
энергии переменного магнитного поля, что фиксируется прибором
в виде пика.
Для снятия спектра образен помешают в резонатор микроволново-
го генератора X = 3 см. создающего переменное магнитное поле и на-
ходящегося в постоянном внешнем магнитном поле, и увеличивают
постепенно напряженность последнего, пока не произойдет поглоще-
ние образцом энергии резонатора, те. пока не наступит резонанс.
В современных приборах ЭПР записывается не сама кривая погло-
щения (как в спектрах ЯМР), а ее первая и вторая производные. На
рис, 40 представлена кривая поглощения ЭПР, первая и вторая произ-
водные от этой кривой.
С помощью спектров ЭПР можно не только обнаружить неспарен-
ные электроны в образце, но и определить их количество, т. к. оно про-
порционально плошали под кривой поглощения. С этой целью ис-
пользуют эталонные образцы с известным числом парамагнитных
центров в единице массы. Сравнение интенсивности сигналов эталона
и исследуемого вещества позволяет определить число парамагнитных
Центров [з последнем.
Кроме того, с помощью ЭПР можно получить информацию о яд-
рах, соседних с неспаренным электроном, если спины ядер не равны
нулю. Это объясняется тем. что магнитные поля ядер влияют на на-
пряженность поля, действующего на электрон (уменьшают или увели-
чивают его).
67
5’
Рис. 40. Сигнал ЭПР: а — кривая поглощения; в — первая производная
от кривой поглощения; с — вторая производная от кривой поглощения
Если вблизи электрона находятся несколько эквивалентных ядеря.
то резонансный сигнал распадается на /Улиний:
/V = 2nJ+ 1,
где J — спин ядра.
Например, в случае сигнала метильного радикала (вблизи неспарен-
ного электрона находятся 3 протона со спином 1/2) число линий будет.
N = 2’3' 1/2 + 1=4.
Относительная интенсивность линий при и эквивалентных прото-
нах совпадает с коэффициентами в биноме Ньютона (г/ + b)”, Есд1
/} = 3 (метил), то соотношение будет равно 1:3;3:1.
Нефтяные смолы и асфальтены и нефтяные порфирины м о гут со-
держать в своем составе комплексно связанные металлы с переменно!
валентностью, чаше всего ванадий, спин ядра которого равен 7/2. Есдо
неспаренный электрон будет находиться вблизи ядра ванадия, то егс
сигнал расщепится на 8 линий:
/V= 2trJ + 1 = 2-17/2 + 1-8.
Таким образом, изучение спектров ЭПР нефтяных смол, асфальте
нов и других высокомолекулярных соединений нефти позволяет оп-
68
рсделигь в них число парамагнит ных центров (неспаренных электро-
нов) и определить качественно и количест пенно содержащиеся в них
металлы.
2.2.5. Атомно-абсорбционная спектроскопия
Атомно-абсорбционная спектроскопия широко применяется для
количественного определения металлов в нефтях и нефтепродуктах,
Принцип метола заключается в следующем При пропускании элект-
ромагнитного излучения (в ультрафиолетовом диапазоне) через среду,
содержащую свободные атомы какого-либо металла, происходит воз-
буждение атомов и переход их из низкого в более высокое энергетичес-
кое состояние; при этом происходит поглощение (абсорбция) опреде-
ленных лучей, что отражается на спектре в виде линий поглощения,
В качестве источника излучения в атомно-абсорбционной спектро-
скопии используют специальные лампы с полым катодом. Лампа пред-
ставляет собой стеклянный или кварцевый баллон, в котором имеются
два электрода: катод чашкообразной формы из определенного металла
и анол (из любого материала). Лампа заполнена инертным газом с низ-
ким давлением. Под напряжением 100-200 В вначале происходит ра-
зогрев лампы, а затем тлеюшпй разряд, под действием которого обра-
зуются положительные ионы из инертного газа, которые, бомбардируя
катод, выбивают из пего атомы металла, происходит как бы распыле-
ние катода. Атомы металла поглотают энергию, возбуждаются, а за-
тем, переходя в исходное состояние, испускают характеристическое
излучение, состоящее из нескольких дискретных линий. Так. для ни-
келя в пределах 4 нм наблюдается 4 основных линии, из которых глав-
ной является линия при 232 им. Излучение этой волны и проходит
через атомизированный образен (остальные лучи отсекаются монох-
роматором).
Чтобы атомизировагь исследуемый образец (какое-либо соедине-
ние металла), его необходимо подвергнуть действию высокой темпера-
туры за счет тепла высокотемпературного пламени или за счет электри-
ческого нагрева. В качестве пламенного источника высокой температу-
ры используют чаше всего ацетиленовое пламя (2200°С), которое обра-
зуется в виде гонкой полосы в специальной горелке, в смесительную
камеру которой поступают под давлением ацетилен, воздух и впрыски-
вается форсункой образен В современных приборах чаше используют
непламенные источники высокой температуры, в частности графито-
вую трубку (так называемую графитовую кювету), нагреваемую током
большой силы (до 500 А) при низком напряжении (диаметртрубки нес-
колько миллиметров, и длина — около сантиметра). Образец помещают
в графитовую кювету и нагревают по специальной программе: вначале
69
его нагревают в течение минуты примерно до ЗОО'С для испарен^
растворителя, затем еше примерно в течение минуты до 170(ГС для его.
рання органических веществ и, наконец, до 2500-300047 .для разлом
ния неорганического соединения на атомы. Программа подбираем-
ой ытным путем для каждого металла, В процессе анализа измеряю-
высоту пика на ленте самописца, появляющегося при температур
в кювете 2500-3000’С,
По высоте пика с помощью градуировочного графика определяю
содержание металла в образце (в мкг/мл или в миллионных долях). Дк
построения калибровочного графика для данного металла готовят™
называемые стандартные растворы. Это ряд растворов какой-нибу^
соли этого металла в подходящем растворителе с различи oil известно;
концентрацией металла. Каждый раствор анализируют, измеряют вы
соту соответствующего ему пика и строят калибровочный г рафик в ко-
ординатах высота пика - содержание металла.
70
Глава II
УГЛЕВОДОРОДЫ НЕФТИ
И ПРОДУКТОВ ЕЕ ПЕРЕРАБОТКИ
§1. Алканы (метановые углеводороды)
1.1. Содержание в нефтях
Все нефти содержат большее или меньшее количество алканов.
Обычно их содержание в нефтях колеблется от 20 до 50%. В парафи-
нистых нефтях содержание алканов достигает 60% и более; в нефтях
малопарафинистых их содержание может упасть до 1-2%. Если рас-
смотреть распределение алканов по фракциям нефти, то наблюдается
следующая обшая для всех нефтей закономерность: содержание алка-
нов падает с увеличением температуры кипения фракции. В парафино-
нафтеновых нефтях алканы находятся в низкокипяших фракциях (до
300’С). В парафинистых нефтях их содержание может быть значитель-
ным даже в высококипяших фракциях.
В табл. 8 приводятся данные по содержанию алканов в некоторых
грозненских нефтях.
Алканы нефтей подразделяются на алканы с прямой цепью (н-алка-
ны) и с разветвленной цепью (/-алканы). Во многих нефтях /-алканы яв-
ляются слаборазветвленными, молекулы которых содержат I или 2 ме-
тила в главной цепи. В нефтях некоторых типов содержатся заметные
количества сильно разветвленных алканов с регулярным расположени-
ем метилов в главной цепи. Это изопреноидные углеводороды или
изопренаны (см. с. 83). Их количество может достигать 3—4% на нефть.
Таблица 8. Содержание алканов в нефтяных фракциях (%)
7"К1|[] фракции, С Н^фтепо-парафин истая нефть Парафинистая нефть
60-95 50 62
95 122 43 57
122 150 42 61
150-200 24 57
200-250 1 1 46
250 -300 0 29
300 350 0 .32
330 400 0 24
71
В некоторых нефтях л-алканы или изопренаны отсутствуют и со-
держится очень небольшое количество (4-10%) сильно разветвлен^-,
алканов.
В зависимости от состава и соотношения алканов различного ти^
Ал. А. Петров предложил химическую классификацию (типизацию
нефтей. В соответствии с этой классификацией неф! и подразделяю^
на 4 группы: А'. А2, Б2, Б1. Для определения типа нефти проводят^
хроматографический анализ с применением капиллярных колонок эф.
фективностью 25-30 тыс. теор. тарелок с неполярной НЖФ в режим;
программирования температуры от 110 до 32СГС со скоростью подъем
температуры ГС/мин. Хроматограммы нефтей разного типа сущесг
венно отличаются и могут рассматриваться как «отпечатки пальцев,
нефтей. Так, на хроматограмме нефти типа А1 четко видны пики я-ц
канов и изопренанов.
Хроматограммы нефти типа Б1 представляют собой лишь кривук
нафтенового фона с полным отсутствием пиков. Нефти А2 и Б2 занима-
ют промежуточное положение. Для дополнительной характеристик
нефтей методом масс-спектрометрии исследуется сослав фракии?
200-430°С нефти (% содержания алканов, нафтенов и аренов). Эт
фракция была выбрана потому что ее состав наиболее правильно отра-
жает средний состав нефти для различных нефтей, т. к. эта фракция са-
мая неизменяемая часть нефтей («тело нефти»).
Нефти тип a /l7 являются нефтями парафинового или нафтено-па-
рафинового основания. Содержание алканов в этих нефтях обычно
30—40%, иногда выше, причем л-алканы составляю!', как правило.
25-30% от суммы алканов; отношение л-алканы/изопренаны »|
К нефтям типа А1 относятся нефти Татарии (Ромашкино), Западной
Сибири, США и Казахстана (Узень). Нефти типа А1 содержат главным
образом слабо разветвленные /-алканы. Так, из 16 изомеров октана
в нефти месторождения Понка (США) 49,4%- составляет л-октан,
38,6— метилгептаны, 11% - дизамешенные изомеры и только 1%-
тризамешенн ые.
Нефти тина А22 нефти парафино-нафтенового и нафтено-парз-
финового основания. Содержание алканов обычно 15-25%, причеч
л-ал каны составляют от суммы алканов 12-15%. т. с, в 2 ра за ниже, w
Рис.. 41, Хроматограмма нефти типа
А1 (большие пики — н-алканы,
малые — изопренаны)
Рис. 42. Хроматограмма нефти
типа Б1
72
В нефтях А1. Отношение н-алканы/изопренаны < 1, К этим нефтям от-
носятся некоторые нефти Азербайджана и Прикаспия.
Нефти типа Ь2 — главным образом нефти нафтенового основания.
Они, как правило, не содержат нормальных алканов, а содержание изо-
алканов обычно 8-20%, причем изопренаны составляют часто 16-30%
от суммы изоалканов. К этим нефтям относятся некоторые нефти Гру-
зии и Северного Кавказа.
Нефти типа Б1. Это нафтеновые, нафтено-ароматические нефти.
В этих нефтях нет я-ал капов и изопренанов, а содержание сильно раз-
ветвленных изоалканов составляет 8-9%.
Это нефти месторождений Грязевая Сопка. Нафталан (Азербайд-
жан), Русское (Западная Сибирь). Например, в нефти месторождения
Грязевая Сопка октаны имеют следующий состав.%: н-октан — 7; ме-
тилгептаны — 37,7: диметил гексаны — 35: триметилпентаны — 20,3.
1.2. Физические свойства алканов
Шесть алканов — газы при обычных условиях (метан, этан, пропан,
бутан, изобутан, неопентан). Температуры кипения этих углеводоро-
дов. °C:
Метан
Этан
Пропан
Бутан
И зобута н
Неопентан
-161,6
-88,5
—42,2
0,5
12,2
9.45
Начиная с изопентана ( ТКН]] = 28'С) и пентана (ТКИ|| = 36°С) метано-
вые углеводороды — жидкости. Для углеводородов нормального строе-
ния увеличение на -СН?- группу повышает температуру кипения
в среднем на ЗО'С. затем по мере увеличения молекулярной массы эта
величина уменьшается. Гели сравнить температуры кипения алканов
нормального и изостроения, то ра зветвленные углеводороды имеют бо-
лее низкие температуры кипения, чем углеводороды с прямой цепью.
Начиная с С[Г1-С|7, алканы с прямой цепью — твердые вещества.
Температура плавления, "С:
Гексадекан С!ЬН34 18,1
Гептадекан С]7Н22.0
Температура плавления алканов с прямой цепью повышается с уве-
личением числа углеродных атомов в молекуле. При переходе от угле-
водорода с нечетным числом атомов углерода к углеводороду с четным
числом атомов углерода увеличение температур плавления больше, чем
при переходе от четного числа атомов углерода к нечетному (рис. 43).
73
Рис. 43
В нефтях содержится несколько больше алканов с нечетным число»
атомов углерода, что объясняется образованием этих углеводородов ю
жирных кислот животного происхождения путем их декарбоксиляци»
(животные жиры являются производными жирных кислот с четны»
числом углеродных атомов):
С15Н31СООН->С|5Н32 + СО2
С17Н35СООН -> С|7Н3~ + со2
Температуры плавления изоалканов зависят в большой степени с:
их строения, причем изоалканы несимметричного строения имею?
меньшую температуру плавления, чем соответствующие н-алканк.
Изоалканы симметричного строения иногда имеют высокую темпера-
туру плавления. Температуры плавления некоторыых алканов, °C:
я-Гексакозан, С26 +57
5-бутилдоказан,С26 +21
9-бутилдоказан, C2f) +1,3
11 -бутилдоказан, С26 О
н- Октан -56
2,2,3,3-тетраметилбутан +106,6
Молекулы высших алканов с прямой цепью в кристаллическом со-
стоянии располагаются в пространстве таким образом, что все атоми
углерода лежат в одной плоскости:
В расплавленном состоянии некоторая доля молекул принимай
в пространстве форму спирали с диаметром 5 : часть молекул сохрэ
инет плоскую структуру, являющуюся более энср1етически выгодной.
1, к. в лом случае молекулы находятся в заторможенной конформации.
Имеется ряд эмпирических зависимостей, связывающих между со-
бой простые констан ты метановых углеводородов. Зависимость темпе-
ратуры плавления от числа атомов углерода выражается формулой Пат -
терсона и Кинса.
где с — число атомов углерода: у — коэффициент, равный нулю для уг-
леводорода с четным числом атомов углерода и единице — с нечетным
числом.
По этой формуле можно найти, что для углеводорода с числом ато-
мов углерода, равным оо. 7f| [ = 137,8”С.
Показатели преломления и плотности у ал капов меньше, чем у дру-
гих углеводородов: эти константы увеличиваются с увеличением моле-
кулярной массы. Между ними установлена довольно точная эмпири-
ческая «зависимость:
-0,52167р.;п +1,03104.
Спектральные характеристики алканов
В ИК-спектрах для валентных колебаний С—Н характерно погло-
щение в области 2850- 3000 см1. Деформационные колебания С— Н-
связи находятся в области меньших частот (1350—1470 см-1)1.
В УФ области доступных г ши волн алканы не поглощают.
Спектры протонного магнитного резонанса позволяют четко опре-
делить на основании интегральной кривой резонансного поглощения,
является ли алкан разветвленным или нормального строения. Однако
полная расшифровка и их спектров является трудной, так как для про-
тонов различного типа в молекулах алканов разница в химических
сдвигах невелика.
Ядерная магнитная спектроскопия изотопа |3С дает возможность
четко определить строение алканов, например изопентана (рис. 44).
1.3. Химические свойства алканов
Алканы, будучи насыщенными, способны только к реакциям заме-
щения. Однако их реакционная способность мала, особенно в ионных
П1ри 1200 п 1250 см 1 паблюлаюгея полосы iioi.loineniisi. характернзуюшнс коле-
бания скелета iреiбугилиioit [руины, а при 1145 п 1170 ем"1 — колебания скелета то-
проннльпой группы. Д )я прямых упас i ков углеротного сколе га ( -СН->~) при п > Дхарак-
Н’рна полоса при 720 см '. обусловленная маяпшковыхп! колебаниями -С Н,- [руины
ЧЦД * wUij । j 1
36 32
т м с
28 24 20 16 12 a
Рис. 44, Спектр ЯМР 13С изопентана
о 5
реакциях, При обычных условиях эти углеводороды не реагируют с та-
кими электрофильными реагентами, как серная и азогная кислоты.
Более активны эти углеводороды в реакциях радикального замещения
(хлорирование, нитрование по Коновалову, сульфохлорирование,
сул ьфоок и сл е н ие):
ChH|4 + HNO3------► CfiHj^NOo + HiO
(10-13%)
С6Н |4 + SO? + Cl2 CftH|3SO2Cl + HCI
C6H|4 +SO2 + l/2O2-^-*" c6h13so2oh
Работами Ола и сотрудников показано, что в присутствии очень
сильных кислот (HOSO3F, HBF4, HSbF6 и др., называемых сверхкисло-
тами) алканы подвергаются различным реакциям, протекающим по
ионным механизмам.
В курсе химии нефти рассматриваются лишь определенные хими-
ческие свойства алканов, которые имеют либо аналитическое значе-
ние, либо значение для изучения деструктивной переработки нефти.
Комплексообразование
Газообразные алканы образуют твердые комплексы с водой. Эл
комплексы относятся к так называемым соединениям включения или
клатратным соединениям. Комплексы углеводородных газов с водой
образуются при пониженной температуре (-(ГС). Иногда в газопрово-
дах они могут быть причиной закупорки, В присутствии молекул га^
вода («хозяин») кристаллизуется с образованием клеток, в которых зак-
лючены молекулы алкана («гость»). Образование клатратов газообраз-
ных алканов с водой лежит в основе обессоливания морской воды. Так.
76
,(NHP<O * * • (Nh2)2CO .
.(НЖО*
CO- -
(NH2)2 • , ;
• <OC(NHib. . .ocOW2
** - •
’OCCNFb)?. . . CCKNbbh----------------
Рис. 45. Спиралевидные гексагональные каналы комплекса
пропан при давлении 0,4 МПа и температуре 2СС образует в морской
воде кристаллическое соединение с водой C3HS17H2O.
Алканы нормального строения, начиная с гептана, образуют при
комнатной температуре -соединения включения с мочевиной
H?N—СО— NH2. В лих соединениях молекулы мочевины («хозяин»)1
соединяются между собой с помощью водородных связей и образуют
спиралевидные гексагональные каналы, в которых находятся молеку-
лы алкана («гость») (рис. 45).
Шаг этой спирали 3,7 , а диаметр гексагонального канала 4,9 .
Диаметр эффективною поперечного сечения молекулы алкана нор-
мального строения 3,8-4,2 . Поэтому молекулы нормальных алканов
умещаются в этом канале в отличие от молекул изоалканов, эффектив-
ный диаметр молекул которых значительно больше. Благодаря этому
комплексообразованием с мочевиной можно отделить нормальные ал-
каны от разветвленных алканов. Однако слаборазветвленные алканы,
моле ку л ы котор ы х и м с ю г у ч а сто к прямой цепи из 10 ато м о в у гл ерод а,
также образуют устойчивые комплексы с мочевиной.
Тиомочевина H2N-C(S)-NH? образует соединения включения
с изопарафинами. Диаметр гексагонального канала, образованного
молекулами тиомочевины в соединении включения, равен 7 , в этот
канал могут быть легко включены молекулы даже сильноразветвлен-
ных алканов. Молекулы углеводородов в соединениях включения мо-
чевины и тиомочевины удерживаются с помощью сил Ван-дер-Вальса.
Возможно также наличие слабых водородных связей.
Изомеризация алканов
Алканы подвергаются изомеризации в присутствии кислотных
капп и заторов. Легко изомеризуются алканы, содержащие третичный
углеродный атом, труднее — изоалканы с четвертичным атомом угле-
рода. Углеводороды с прямой цепью занимают промежуточное поло-
жение. Например, для гексанов скорость изомеризации падает в ряду:
2-мстилпентан > //-гексан > 2,2-диметилбутан.
Зермины «гость» и «хшяин» приняты и химии клатратных соединений.
77
С повышением температуры относительная устойчивость разветв.
ленных алканов падает по сравнению с //-алканами. Этим и объясняет
с я малое содержание в нефтях сильно разветвленных алканов.
С высокой скоростью протекает изомеризация алканов в присут-
ствии бромида алюминия, Этот катализатор растворим в углеводородах,
и поэтому реакция в его присутствии проходит в гомоген них условиях.
//-Гептан в присутствии нескольких процентов AlBr, в течение
10-20 ч при комнатной температуре превращается в сложную смесь
всех своих изомеров. Эта смесь может быть разделена с помощью пре-
паративной газо-жидкостной хроматографии, и изомеры гептана могут
быть получены в чистом виде. Это очень важно для получения модель-
ных углеводородов для газо-жидкостной хроматографии, т. к. синтез
индивидуальных изоалканов обычными методами (например, по реак-
ции Вюрца) требует значительно большего времени.
Реакция изомеризации индивидуальных алканов в промышленнос-
ти имеет ограниченное применение. Это в основном реакции изомери-
зации w-бутана в /-бутан и н-пентана в /-пентан.
Чаше проводят изомеризацию фракций прямой перегонки нефти
с целью получения высокооктановых компонентов бензина.
Реакция изомеризации играет большую роль в современных про-
цессах каталитической переработки нефти — риформинге. гидрокре-
кинге, каталитическом крекинге.
Термическое разложение алканов
Под действием тепла при повышенных температурах алканы разла-
гаются. При этом проходят две основные реакции: дегидрирование и
расщепление по связи С—С, С повышением молекулярной массы алка-
нов реакция с расщеплением связи С-С начинает преобладать. Газооб-
разные алканы заметно расщепляются при 800—900°С\ Высшие углево-
дороды — при 500-60СГС. При этом из молекулы алкана образуется
алкан и а-алкен меньшей молекулярной массы:
R-CH2-Ch|ch2-CH2-CH3----► r„ch=CH2 + CH-CHrCH3
Дегидроциклизация алканов
Алканы с числом атомов С 6 и более превращаются в ароматически
углеводороды в присутствии оксидов некоторых металлов:
В процессе этой реакции происходит дегидрирование алкана с пос-
п еду ю шей циклизацией продуктов дегидрирования. В присутствии
платинового катализатора наряду с ароматическими углеводородами
в этой реакции образуются также циклопентаны:
2 СН3-СН2-СН2-СН2-СН2-СН3
1.4. Газообразные алканы
Газообразные алканы составляют основную часть природных и по-
путных газов, газов газоконденсатных месторождений, а также газов
переработки нефти.
Природные газы — это такие газы, которые образуют в земной коре
самостоятельные залежи.
Попутными называются газы, которые сопутствуют нефти и выде-
ляются при ее добыче.
Природные газы состоят в основном из метана (90-99% об.) и содер-
жат не более 100 г/м3 паров бензина. Такие газы называются «сухими».
Попутные газы содержат кроме метана (30-70%) значительные ко-
личества газообразных алканов и паров бензина — более 100 г/м3. Газы
газоконденсатных месторождений состоят в основном из метана и со-
держат более 100 г/м3 паров жидких углеводородов, т, е. являются
«жирными».
Кроме углеводородов они могут содержать:
Г Сероводород — обычно до 3%. Сероводород содержится как в га-
зах, сопровождающих сернистые нефти, так и в природных газах. На-
пример, газ оренбургского месторождения содержит 1,3—4,5% серово-
дорода, Природный газ месторождения Ляк во Франции содержит 15%
сероводорода. Газ астраханского месторождения содержит 16-20% се-
роводорода.
2. Углекислый газ (обычно от 0,1 до 7%), иногда до 40% и более.
3. Азот — до 30%, аргон и гелий (содержание которого в некоторых
американских газах достигает 7%). В российских газах содержание азо-
та и гелия может составлять до 0.02—0,05%, В табл. 9 приведен состав
некоторых природных и попутных газов (% об,).
СНгСНгСН2-СН2<нг^
Сг?О5
Методы анализа углеводородных газов
Перед тем как провести полный анализ природного или попутного
газа или какого-нибудь газа нефтепереработки, содержащего непре-
дельные углеводороды, проводят предварительный анализ, т. е. опреде-
ляют основные компоненты газа, наличие и количество неорганичес-
78
79
Таблица 9. Химический состав некоторых природных
и попутных газов
Место- рождение сн4 C;Hfl СЛНК бдН [() CSH|2 СО, H,S N2 Приме, чанне
С lanpono.ib 98, 8 0,3 0.2 0.1 — 0.2 — 0,4 Природ, ный
Л як 69.1 2.8 0.8 0,6 0.9 9,7 15,3 — Природ- ный
Ромашкин» 40,0 19.5 18.0 7.5 4.9 од — 10 Попутный
Волгоград 96.3 1.2 0.5 0.1 — 0.1 — 1.8 Попутный
’Сухой-
Уренгой 98,5 0.1 — — 0,21 — 1,1 природ-
(Тюм. обл ) ный
’Cy.xoib
Оренбург 81,5 3.1-5.4 1-2.1 0.47-2.8 0.4-2.8 1.0-3,2 1,9-4,5 2.4-7.4 природ- ный Газокон-
Астрахань 47-54 2.0-5,5 0 9-1.7 04-0.9 0,3-1 6 18-21 20-26 -2,0 денсатное место* рождение
ких составных частей: СО,, О2, Н2, H,S. непредельных у тле водородов г
алканов. Это позволяет определить, к какой категории относится дан
ный газ и выбрать метод его полного анализа.
Для предварительного анализа удобны приборы, основанные на аб-
сорбции, то есть на поглощении основных частей газа различными хи-
мическими поглотителями, Примером таких аппаратов может служите
аппарат ВТИ-21.
Чтобы определить химический состав какого-либо газа на аппара-
тах такого типа, образец газа определенного объема последовательнс
пропускают через абсорберы, содержащие различные химические пог-
лотители, и измеряют объем газа после поглощения в каждом абсорбе-
ре. Изменение объема газа позволяет вычислить процентный состав
каждого компонента (% об).
Для определения СО, в качестве поглотителя используют водный
раствор гидроксида калия или натрия:
СО2 + 2 КОН-> К2СО3 + Н,О.
Дня определения кислорода — либо щелочной раствор пирогаллол
(1,2,3-триоксибензол);
2C6H3(ONa)3+ 1/2 О,-> (NaO)1Cf)H,-C6H,(ONa)3 - Н2О,
либо раствор гидросульфита натрия:
Na,S2O4 + 2NaOH + О, Na,SO4 + Na,SO, + Н?О.
1 ВТ И — Всесою зный геплотехнический hiicth тут
Для определения содержания олефинов газ пропускают через
раствор брома в воде (бромную воду), олефины реагируют с бромом,
превра ша я с ь в д ибро м и л ы:
R С Н = С Н 2 + Во-----► R С Н—С Н}
I I
В г В г
Содержа н и е вод о р ода. у г л с р ода 1г с у м м ы ал канон в газе о п ред ел я ю т
путем их полного окисления. Вначале определяют водород и оксид уг-
лерода, пропуская газ через трубку, наполненную СиО, нагретым до
280сС, Происходит окисление:
Нт + Си() —> Си т Н,О.
СО + СиО -ж Си + СО,.
Водород окисляется в воду, пары которой конденсируются. При
окислении оксида углерода образуется такой же объем углекислого га-
за. Поэтому уменьшение объема газа происходит только за счет окис-
ления водорода, что позволяет определить его содержание. После по-
глощения углекислого газа раствором шел очи рассчитывают процент-
ное содержание СО. Для определения СО можно использовать также
его поглощение суспензией Cu,SO4h р-нафтола в концентрированной
серной кислоте:
Cu,SO4 + 2СО -э Cu,SO4'2CO.
Алканы определяют, пропуская газ через слой оксида меди при
850-950еС. Происходит их полное окисление. Например:
СН4 + 4СиО -ж СО, -I- 2Н,0 + 4Сн.
Углекислый газ. образующийся при лом, поглотают раствором
шелочи. По разности обьемов газа определяют содержание алканов.
Если в газе содержится азот, го он остается в остатке после анализа всех
остальных компонент о в.
Наиболее быстрым и универсальным метолом полного анализа га-
зов является метол газовой хроматографии. Анализ проводят на газо-
вых хроматографах. В качестве неподвижной фазы используют твердые
адсорбенты, иногда модифицированные добавлением небольшого
количества (5-10%) жидкой фазы (вазелиновое масло). Вазелиновое
масло закрывает наиболее активные участки поверхности адсорбента,
в результате чего активность различных участков поверхности вырав-
нивается, Это способствует образованию симметричных пиков на хро-
матограмме. В ре пульта ге модификации поверхность адсорбента
покрывается тонким слоем масла, через который проявляются силы
адсорбции. Поэтому, хотя вазелиновое масло является неполярным ве-
ществом. в случае полярных абсорбентов (SiO2. А1,ОД такая фаза явля-
йся селективной и опа сильнее удерживает ненасыщенные угле вод о -
3 - 3544
80
81
ролы. Ес.Iи газы содержат неорганические компоненты, то необходимо
применение молекулярных сит, так как обычные сорбешы плохо pay
деля ют неорганические газы.
Для выбора методики иодного хроматографическою анализа газа
необходимы данные предварительного анализа. Рассмотрим один из
вариантов хроматографического анализа газа. Предположим, что при
предварительном анализе какого-либо газа установлено, что он состо-
ит из водорода, кислорода, азота, оксида углерода, алканов и олефц.
нов, Если ввести пробу этого газа в хроматограф с насадочной колон-
кой, заполненной адсорбентом, модифицированным вазелиновые
маслом, с применением в качестве газа-носителя водорода и проводить
анализ при комнатной температуре, то хроматограмма будет иметь вид
изображенный на рис. 46.
По хроматограмме1 подсчитывают процентное содержание каждого
углеводорода, содержание суммы С2 и суммы СН4, О2, СО, N2. Из хро-
матограммы видно, что с помощью обычного адсорбента не удается
разделить СН4, О2, N2 и СО, а также смесь этана и этилена. Для их раз-
деления газ анализируютс применением цеолита NaX и водорода в ка-
честве газа-носителя (аргон и гелий не используются, так как они име-
ют теплопроводности, близкие к теплопроводности оксида углерода)
Получают хроматограмму, изображенную на рис, 47.
На основании этой хроматограммы и предыдущей можно подсчи-
тать процентное содержание О2, СН4, N2, СО, С2Н4 и С2Н6. Хотя при-
менение молекулярного сита позволяет разделить неуглеводородные
компоненты, а также метан, этан и этилен, полностью анализ газа на
этом адсорбенте провести нельзя, так как он прочно удерживает алке-
ны. Содержание водорода определяют, применяя цеолит NaX и аргон
(\Н4. Для отделения СО2 от смеси С2НДС,Н4 в качестве лснотвижной фазы исПОПР
,’укл >фир три лилентликоля и //-масляной кислоты, нанесенной в количестве
1кхщель сферохром-1 (Страшилина Л. А // Нефтехимия, — 1982 Т. 22. — С. 69).
н качестве газа-носигсля (гелии применять нс п
водность близка к ченлопроводности воаопо-т » п * С'° Ш,;И)Нрс-
грамму с пиком водорода, остальные пики не и °Дучают хромата-
пика подсчитывают пропентнос содержани • 1)ИКСИруюг вь/соте
либровочную кривую "высота пика % в°лорода, применяя ка-
зультате анализа искусственных смесей^ НОДОрола*>' полученную в ре-
1.5. Жидкие алканы нефтей
Жидкие алканы содержатся в бензиновых и керосиновых фракциях
нефтей. Подробно исследованы алканы лишь бензинов, выкипающих
до 150- 180°С (сюда относятся углеводороды состава С5-С|0). Фрак-
ции, выкипающие выше, исследованы хуже. Это объясняется тем, что
с повышением температуры кипения резко возрастает число изомеров.
Так, у октана — 18, у нонана — 35 изомеров. Число изомеров примерно
удваивается при переходе от одного гомолога к следующему. У додека-
на С[2Нэй (температура кипения 212.2°С) 335 изомеров. В нефтях най-
дены все изомеры пентана, гексана, гептана. Из 18 изомерных октанов
выделено 17. Из 35 изомерных нонанов в нефтях найдено 34 изомера.
В нефтях найдены и выделены все алканы нормального строения,
вплоть до С36Н74 (гексатриаконтан).
На XV Мировом конгрессе по биомаркерам сообщалось о выделе-
нии из нефти алканов состава С40-С)(Ю. В 60-х годах в средних фракци-
ях американских и некоторых советских нефтей были идентифициро-
ваны разветвленные алканы изопреноидного типа1, называемые изо-
пренанамн, в молекулах которых метильные группы находятся в глав-
ной цепи в положениях 2, 6, 10, 14, т. е. молекулы изопренанов состоят
из соединенных между собой углеводородных фрагментов, имеющих
такое же строение углеродного скелета, как и молекула изопрена. Наи-
более распространенными в нефтях углеводородами этого типа явля-
ются фитан (С2ОН42) 2.6,10,14-тетраметилгексадекан и пристан
(CflJH4()) 2,6,10.14-тетрамегилпентадекан:
СН3 СН3 CH, СНз
rK|trf=316°C
СНз СНз СНз СНз
rD^R,Whiiclu.4Ki Е/ТТёЫк-d I си - 196!. — V 21 - Р. 768. Красавченко М. И.
11 лр//Нефтехимия |969 T9 С 65).
пристан,
Г =299°С
1 кип 4
83
6’
Рис. 48. Предполагаемая схема образования фитана и пристана из фитола1
Всего 8 нефтях обнаружено более 20 углеводородов яого типа со-
става (С()-С\5). Концентрация изопреноидных алканов в нефтях тиш
А достигает 3—4% (иногда и выше) Нефти типа Б обычно не содержа?
утих углеводородов, либо содержат их в очень небольшом количестве
Предполагается, что источником образования изопренанов в неф
тях является непредельный алифатический спирт фитол, который вхо-
дит в состав хлорофилла растений. Предполагаемая схема образование
фитана и пристана из фитола приведена на рис. 48.
В процессе превращения фитола в земной коре в результате отщеп-
ления гидроксильной группы (дегидратации) и насыщения водорода
кратно)) связи (перераспределение водорода) образовался фитат, от-
щепление одного атома углерода и гидроксильной группы (вследствие
окисления) с последующим насыщением остатка молекулы водороде*
приводило к образованию пристане! Изопреноидные углеводород
с числом атомов углерода менее !9 также, по-видимому. образовали^
в результате деструкции фитола. Что же касается углеводородов этот
тина с числом углеродных атомов более 20 (например, таких каклико-
пан С4ЛНК2 и сквалан С3()Н()2 и продукты их распада), го они могли об-
разоваться из других веществ природного происхождения, таких,
например, как ликопин (С4(,Н56) и оквален (CV)H-(J).
Изопренаны могут иметь правильное (регулярное) строение (фи
тан, пристан) и нерегулярное строение (сквалан, ликопин):
сквалан
2,6.К). 15.19 23-нсксамс!птгетрако зап
1 В формуле фитола ыеяочками обозначены асиммсiрические ашмы углсрогшЫ!
раненые центры) пол Nr.’7 nil. Клиньями обозначены могильные [рейны. nanparoiW-
от ещашшы в сторону чи ште.ш.
84
Недавно и нефтях обнаружены так называемые Т-образные изопрс
папы;
Для изопренилов характерна оптическая активность, так как в их
молекулах имеются асимметричные атомы углерода (хиральные цент-
ры) Так. в молекуле пристана имеются 2 таких атома углерода (обозна-
чены звездочкой). Число оптических изомеров равно 22 = 4.
горкаЛ 1>Иыс и юмсры
Интересно отметить, что в составе хлорофилла фитол имеет только
конфигурацию 7R. I IR (ем. рис. 48). В процессе превращения фитола
л природных условиях образуется равновесная смесь оптических изо-
меров пристана (6R,IOS: 6SJ0S’ 6R.I0R 2; I: 1).
Жидкие алканы как компоненты топлив
Жидкие алканы входят в сое гав то пл и в для карбюраторных двигате-
лей (бензины авиационный, автомобильный), ,тля дизельных двигате-
лей (дизельные топлива, га зойли) и реакт ивных. В каждом топливе они
выполняют определенную функцию и должны обладать определенны-
ми свойствами. Гак, алканы, содержащиеся в бензинах, должны иметь
высокую детонационную стойкость; алканы дизельных топлив должны
85
Ofvinnb епоеоошчn.10 |егм> i«Kll.iiiMen>ii1,01. алканы рсактивщ,
,,;1|W ,S co способное uno к ле. кому ..оепаамепеник, долж,,^
.алан. HiiiKUMH температурами ............ниш. I ассмтрим,
обусловлены эги требования.
' В карбюраторных двигателях зажигание рабочей смеси (или паи
бензина с воздухом) происходит е помошью искры, которая возник
в момент достижения определенной степени сжатия рабочей сма,
(типенпе 10-1.2 МПа, температура может достигать 300-400‘С.
В 'соответствии с теорией А. Н. Баха, в этих условиях происходит оИ[
дение углеводородов с образованием гидропероксидов еще до того, и
искра воспламенит рабочую смесь.
RH + 02 -> R-O-O-H.
Легко подвергаются такому окислению алканы нормального строе
ния. и трудно окисляются разветвленные алканы. 1ак, //-гексан окис
ляется более, чем в 100 раз быстрее, чем 2.2-диметилбу ганчтообъяс
няется особенностями строения этих углеводородов Сопоставим ф0^
мулы w-гексана и 2,2-диметилбутана (неогексан):
С Н з-С Н 2-С Н 2-С Н 2-С Н 2-С Н з
!! ;( -( ( 11 .-( Нч
//-гексан
с н з
2,2-лимс гплбу ган
иcore кеа и
Молекула //-гексана содержит 4 метиленовые [руины, атомы вода
рода которых более подвижны, чем атомы водорода мезильных групп.
Молекула неогексана содержит только одну группу СНЭ, которая
экранирована трудноокисляюшимися метильными группами, что сни-
жает вероятность окисления атомов водорода метиленовой группы
Кроме того, если отрыв атома водорода от экранированной группы
СН? и происходит, то реакционная способность образующегося ради-
кала понижена. Поэтому, если в бензине имеется новы i пен ное содер-
жание //-алканов, концентрация гидроперокентов в рабочей смесг
может быть значительной(и они могут подвергаться взрывному paw
жен и ю erne до возникновения искры2. После ввода искры и воспламе-
нения топлива образование и разложение гидронсроксндов могут про-
должи гься перед фронтом пламени, поэтому горение топлива буле1
Паушкпл Я. М.. Апельсин С. В. Вишнякова I, II Гемго.нн ия неф i ихпмнческок
сиигеза Ч. I М. Химия, 1973,-С. 217
- Перекисная шорня A. H. Баха хорошо описывает процесс лшлкофл шого (jih>
icMtrcpaiypHoin) окисления угли ио'городов. В случае иарофапюго (высокотемпсрапГ
ною) окисления предложен иной механизм пронесен (им. Mai арил 1 I’. Корзун Н. Вт
лр.'/Нсфн-и ни. - 2001 - № 5. • С 7).
неравномерным и можс г завершиться мг новен игл м воспламенением
рабочей смеси (лето на пней). Кел и скорое и, нормального безлитонапи-
онного горения 2О--ЗО м/с, то скорость детонационного [прения 1.5
2 км/с. Удар такой взрывной волны вызывает стук в двигателе и приво-
дит к быстрому сю износу.
Детонационная сгойкосгь углеводородов характеризуется октано-
вым числом. Алканы нормального строения обладают низким октано-
вым числом, разветвленные алканы имеют высокое октановое число,
поэтому их присутствие желательно в карбюраторных топливах. За эта-
лон хорошего карбюраторною топлива принят изооктан (октановое
число 100). Октановое число //-гептана принято равным нулю. Октано-
вые числа некоторых изоалканов:
Изооктан (2,2.4-гримстил пентан) 100
Триптан (2,2,3-тримстилбутан) 106
Неогексан (2,2-димстилбутан) 92
Если [з карбюраторном двигателе воспламенение горючей смеси яв-
ляется принудительным, то в дизельном двигателе горючая смесь топ-
лива и воздуха самовоспламеняется: в сжатый нагретый воздух (давле-
ние 4 МПа. температура 550-60(ГС) впрыскивается топливо. Чем лег-
че самовоспламеняется топливо, тем легче запуск двигателя и равно-
мернее нарастание давления при его сгорании.
Наиболее желательными компонентами дизельных топлив являют-
ся алканы с прямой цепью, так как они легче воспламеняются. За эта-
лон хорошего дизельного топлива принят //-цетан С|6Н34, цетановое
число которого принято равным 100. Меньшим цетановым числом об-
ладают изоалканы и нафтены, еще меньшим — ароматические углево-
дороды. Цетановое число а-метил нафталина принято равным нулю.
В реактивных топливах алканы с прямой цепью должны отсутство-
вать, так как их кристаллизация при пониженных температурах может
явиться причиной закупорки топливных фильтров двигателя. Слабо-
разветвленные алканы желательные компоненты этих топлив.
1.6. Твердые алканы
Твердые алканы находятся во фракциях нефти, кипящих выше
ЗОСГС. Гептадекан имеет температуру кипения ЗОЗ'С и температуру
плавления 2ГС. Твердые алканы присутствуют во всех нефтях, в пара-
финистых нефтях их содержание может быть равно 7—20% и более.
В нефти полуострова Мангышлак содержание твердых алканов дости-
гает 26-30%. В высших фракциях парафинистых нефтей содержание
твердых алканов может достигать 45-55%. Эти углеводороды выделяют
из высших фракций нефтей кристаллизацией в присутствии раствори-
телей (депарафинизация). После очистки гача — сырого продукта де-
86
парафннизапии получаю! парафин — твердое ислое, полуцрозрачя&;
не шесто с температурой плавления 40—60' С и плотностью 0,86—0,94
(’рслнчя молекулярная масса парафина - 500, что соответствует угле-
во юродам с V) 40 атомами углерода в ней и. В расплавленном состоц.
нии плотность парафина равна 0,77-0.79. При запинании парафу
хмсныпает свои объем на 15—17%. Парафин представляет собой сло^.
н\ю смесь алканов нормального строения и твердых изоалканов
206')с примесью нафтенов с длинными боковыми пенями. Твердые^
капы входя г также is состав нерезв на. Церезин — твердое воскообразное
вещество с температурой плавления 60-85Т и молекулярной массой
до 700, что соответствует углеводородам, содержащим 40-50 атомов уг-
лерода в пени. Церезины обладают мелкокристаллической структурой
Они состоят, в основном, из слаборазветвленных изоалканов, но со-
держат также алканы нормального строения, нафтены и даже аромати-
ческие углеводороды с боковыми да к ил иным и пенями. Церезин полу
чают депарафинизацией остаточных рафинатов (после деасфальтиза-
ции гудрона). Церезин можно получить также из горючею минерал
озокерита. Озокерит — твердая пористая порода, пропитанная смесью
твердых углеводородов и смол. Органическая часть озокерита отделя-
ется от минеральной плавлением, и после отгонки легких фракций и
удаления смол адсорбционной очисткой пол уч а гот церезин. В отличие
от парафина, который химически инертен, церезин легко реагируете
серной, азотной н хлорсульфоновон кислотами. Парафин и церезин
находят применение в бумажной, спичечной, электротехнической
парфюмерной промышленности. Они применяются гакже при произ-
водстве консистентных смазок и являются пенным сырьем /из я .хими-
ческой переработки.
1.7. Анализ алканов нефтяных фракций
Количественное определение алканов
В бензиновых фракциях содержание алканов чаше всего определя-
ют методом анилиновых точек. Анилиновая точка (АТ) -- это темпера-
тура взаимного растворения равных объемов анилина и фракции, Оп-
ределяют анилиновую точку фракции, затем из фракции удаляют аро-
матические углеводороды (либо сульфированием, либо методом жид-
кое т н о - ад с о р б и и о и н о й х ромато гра ф и и) и определяют анилине вую
точку дсаро.матизирован ной фракции. На основании л тих двух анили-
новых точек рассчитывают процентное содержание ароматических уг-
леводородов и нафтенов. По разности определяют’ содержание алканов
гсумхга //-алканов и /-алканов).
Раздельное определение //-алканов и /-алканов основано па избира-
тельной адсорбции /г-алканов цеолитом СаА (5А). В поры неолита (5Ai
88
проникают только //-алканы. Анализ провопят пропуская пары фрак-
ции и токе водорода через слой неолита при температуре на 15~20'С
ньппетемпературы кипения фракции. Зная увеличение массы неолита
и величину навески, рассчитывают содержание //-алканов’.
В керосин0'™зой.зевых фракциях i оч н ое о предел е н и е су м ма рн о го со-
кржания алканов (//-алканы и изоалканы) представляет собой труд-
ную задачу, не решенную до настоящею времени, Определение содер-
жания нормальных алканов в ji их фракциях основано на их комплек-
сообразовании с карбамилом. Количество кароамида (в молях на моль
смеси алканов фракии и). необходи мое для образован и я комплекса,
рассчитывают по формуле
/?? = 0,683 н + ] 5 j t
гле // - число атомов углерода, соответствующее средней молекуляр-
ной массе алканов фракции.
Нафтено-парафиповую часть фракции перемешивают при комнат-
ной температуре с раствором карбамила в метаноле (например, (70 г
нафтсио-парафиновой части, 100 i карбамила, 70 г метанола) в течение
30-60 мин. Комплекс фильтруют на вакуум-фильтре, промывают пен-
таном, изопентаном или пстро.чейным эфиром.
При обработке комплекса горячей водой происходит его разложе-
ние, карбамил растворяется в воде, и выделяются нормальные алканы,
которые экстрагируют эфиром и после высушивания раствора и отгон-
ки эфира пол уч а ют в чистом виде. Зная их массу и массу взятой для
анализа фракции, определяют процентное содержание //-алканов.
Во фрикциях выше 300'С и в сырой нефти содержание //-алканов
можно определить, выделяя их метолом низкотемпературной кристал-
лизации (при -20..-50:’С) с применением специальных растворителей
(ацетон, метил этил кет он. бензол, толуол и др.). Так, количественное
определение твердых алканов (парафин) в нефтях основано на их крис-
таллизации из обессоленной нефти в десятикратном избытке раство-
рителя — смесь а негой а и бен зола в соотношении 1:2 (метод
ГрозНИИ).
Выделение алканов
Выделение //-алканов из бензиновых и средних фракций может
быть осуществлено с помощью неолита СаА (5А), который, как уже от-
мечалось, избирательно поглотает //-алканы. Адсорбцию можно про-
водить как в паровой, так и в жидкой фазах при атмосферном или слег-
ка повышенном давлен пи, Адсорбция в паровой фазе протекает со зна-
чительно большей скоростью, чем в жилкой, причем чем вышетемпе-
1 Кпигконск! 1й ! FT, IpvTiicином 1: В // Химия и технология топлив и масел. --
1%2. - №3. - С 61
89
paiApit, гем выше скорость адсорбции. Цеолит, используемый для в
имения //-алканов, должен быть предварительно обработан щелочью
для подавления каталитически активных центров. Без такой обработке
в процессе разделения возможны химические превращения //-алканов
(в частности, изомеризация) особенно при парафазном разделении
когда температура может достигать 370’0. Десорбцию проводят, нагре
вая неолит в вакууме, либо пропуская пары легких алканов состав
С-.
Выделение //-алканов из масляных фракций с применением неоли
та СаА также возможно при температуре 270-30()°С. Масляная фрак.
ння должна быть предварительно очищена от смолист ых веществ к
ароматических углеводородов, так как последние могу г блокирован
поры цеолита. Выделение //-алканов из средних фракций и масляньи
фракций проводят также метолом комплексообразования с карбами-
дом. Из масляных фракций и нефтей твердые w-алканы выделяют так
же методом низкотемпературной кристалл и за ин и в присутствии раст-
ворителей.
Выделение изоалканов в чистом виде представляет собой сложную
задачу.
Из фракций можно последовательно удалить арома i ичсскис углево-
дороды,/т-алканы, шестичленные нафтены (дегидрирование и превра-
щение их в ароматические углеводороды). Остается труд неразделимая
смесь из никлопентановых углеводородов и изоалканон. Разделение
этой смеси можно провести с помощью активированного угля1 или це-
олита 13 X в п а ро во и фазе2. Поел ед н и й м сто д применим дл я у гл е водо-
роде в состава до С)().
Идентификация алканов
Алканы нормального строения и изопренаны фракций нефти могут
быть идентифицированы с помощью газовой хроматографии. Каждый
пик, соответствующий //-алкану или изопарафину, может быть иденти-
фицирован с помощью эталонных углеводородов метолом «меток»
Изоалканы, выделенные из нефти в чистом виде, могут быть иденти-
фицированы физико-химическими и химическими мс юлами анализа
Классические примеры химической идентификации можно найти
в работах В. В. Марковникова по исследованию кавказских нефтей*.
Так. из фракции 49-5ГС бакинской нефти Марковников выделил хи-
мическим путем алкан общей формулы констан ; ы которогобы-
v ^Лр.Л М Ушакова ИЛ,.. Гее, И С„ Сап,,,, П. И //Нсф.ек,,,,,,,,. - 1966,-
-Вгаппоок J.V. Luke L. A.. Anaht Chem - I9W. - v4l) - р а Ик
И,.,. АНiTT-TTr'"* ,И'""/ По-' Л « " I В Ьыком -Н-
90
Г]И близки к константам 2,2-димегилбутана (7'Kmi -- 49,7X5). Этот угле-
[1ОДОродбыл идентифицирован следующим образом.
Вначале было проведено нитрование углеводорода (по метолу Ко-
новалова):
СНт
I 11 (Гс I
Н,С—С— СН2-СН3 + HONO2----► Н3С—С-СН-СНз + Н3О
L HjC 'N(>2
Затем было проведено восстановление. При восстановлении нитро-
соединений жирного ряда (вторичных и первичных) наряду с аминами
часто получаются альдегиды и кетоны. Причина этого заключается
и следующем:
314\
—* CH-NH? + 2Н2О
77~*' cMn—он
-| >( J s' 5
Н 2О
При восстановлении нитрососдинения Марковников получил
пинаколин
идентификация которою (получение семнкарбазона) позволила уста-
новить, что исследуемый углеводород является 2,2-диметилбутаном.
В настоящее время для идентификации углеводородов нефти широко
используются физико-химические методы исследования. Ниже приво-
дится пример физико-химической идентификации изоалкана, выде-
ленного из американской нефти1. Этот углеводород содержался во
фракции нефт, имевшей! температуру кипения около 156иС, его пока-
затель преломления др2'1 - 1,4077 был близок к показателю преломле-
ния 2,4-ди мстил окта на Масс-спектральным анализом была установ-
лена молекулярная масса этого утлеволорода (по массе молекулярного
иона), соответствующая углеводороду состава С|()Н22' По данным
ПМР-сиекгра углеводород содержал 4 метильные группы. Кроме 2,4-
лиметилоктана ( Л 155.9 С, н 25 = 1,4069), полученным данным со-
КИН I 7
1 MairB, I . Bonen / // J. ( hem f jig Data 1967. -- V 12 - P. 432.
91
ответствовали также 2.2-диметилоктан (Гк]|(| 156,9 С. 1,4060)
4.4-лиметилоктан (7КШ| = 157,5°С, Др25 - 1,4122).
2,4-дим стил октан
2,2-диметилоктан
4,4-димстилоктан
Необходимо было доказать, какая из этих структур соответствуй
структуре выделенного углеводорода,
ИК-спектр показал, что в углеводороде нет третичной бутильной
группы (отсутствие полосы поглощения при 1250 см-1), В спектреЯМр
|3С углеводорода отсутствовал сигнал, соответствующий трем эквива-
лентным метильным группам. Следовательно, отпала структура 2,2-ди-
метилоктана. С другой стороны, в ИК-спектре углеводорода были по-
лосы, соответствующие изопропильной группе (1145 и 1170 см-1) и бу-
тильной группе (720 см-1), что позволило установить для углеводорода
структуру 2,4-д и метилокта на. Дополнительно эта структура была подт-
верждена масс-спектральным анализом (по массам осколочных ионов)
§2. Циклоалканы (нафтены) нефтей
Циклоалканы составляют основную массу углеводородов нефти,
Обычно нефти содержат40-70% циклоалканов. Содержание этих угле-
водородов в некоторых нафтеновых нефтях может достигать иногда
80%. Распределение циклоалканов по фракциям нефти примерно рав-
номерно. Несмотря на то что исследования химического состава наф-
тенов продолжаются уже в течение более ста лет, эти углеводороды,
особенно высших фракций нефти, являются наименее изученными уг-
леводородами нефти, Это объясняется сложностью их состава, обус-
ловленной разнообразной их изомерией.
2.1. Номенклатура и изомерия циклоалканов
Для циклоалканов характерна структурная, геометрическая и кон-
формационная изомерия.
92
Структурные изомеры циклоалканов отличаются
тожением и структурой алкильных заместителей я Р‘'МИЧНЬ14 Р»спо-
атомов углерода в циклах. Например, некоторы * ПК*е числом
С|Г1Н2о:
СН,
|-мстил-2-и.ю-
пропилииклогсксан
изомер с вици-
нальным располо-
жением алкилов
1 - мсти л-2-про лил-
ии клогсксан
изомер с вици-
нальным располо-
жением алкилов
сн2сн2сн3
1-метил-З-пропил-
ци клогсксан
1-метил- 1-пропил-
ииклогсксан
изомер с геминальным
(гем) расположением
алкилов
Н3С^
н 2С н 2с н 2с н3
1-метил-3-бутил-
циклогскса и
Пространственная (геометрическая) изомерия циклоалканов обус-
ловливается как различным расположением алкильных заместителей
по отношению к плоскости, в которой лежит цикл (цис-транс-изоме-
рия монониклических углеводородов), так и различным взаимным рас-
положением циклов и алкильных заместителей в молекулах полицикли-
ческих углеводородов (цис-, транс-изомеры, эндо-, экзо-изомеры)1:
транс- 1,2-димстил-
ииклопентан
цис-1.2-лимстил-
пиклопентан
^Эндо-няуз реннин, жю-паружвыи (грен)
93
цм1‘-бииикло(313.0)-октан, /и/ю//('-бипнкло(3,3,1))ок тан,
i u i c-п c i i тал а н тра ни- r i сн тала н
энло-2-мстилбицикло(3,31())октан
экзо-2-мстилбицикло(3,3,0)октан
Конформационная изомерия цикяоа.гканов обусловлена различным
расположением атомов в пространстве, причем конформеры с различ-
ным расположением углеродных атомов в пространстве могут взаимно
превращаться друг в друга в результате вращения вокруг простых угле-
род-углеродных связей.
Так. у циклогексана имеются три конформера:
конформация «кресла»
конформаиия «ванны»
«т вист» - ко нформ а ц и я
94
Наиболее устойчива koi i форм а и и я "кресла-. в которой вы: с ос с.:
СН находятся в заторможенной конформации, наименее
[руппы 2 )Опмаиия «ванны-/, так как в последней имеются .две
тОЙЧИВя кип 4 1 f...
оненные энергетически невыгодные СН^-грунпы:
у монозамешенных циклогексанов заместители находятся в эквато-
риальном положении. Например, в случае метилниклогексана:
Аксиальная форма (а)
(5%)
Экваториальная форма (с)
(95%)
Аксиальная форма менее устойчива, так как метильная группа вза-
имодействует с атомами водорода в положениях 3,5.
Все монозамешенные циклогексаны находятся преимущественно в
экваториальной форме. У двух/вмещенных 1,2-производных цикло-
гексана транс-изомер может находиться в двух формах: е.е и а,а. Фор-
ма е,е более устойчива. У полизамещенных циклогексанов конформа-
ционная изомерия еще более сложная. В связи с этим разнообразием
форм изомерии число возможных изомеров циклоалканов резко воз-
растает с увеличением молекулярной массы.
2.2. Циклоалканы, найденные в нефтях
Первые исследования никлоалкапов нефти принадлежат русским
ученым1. Впервые Ф. Ф. Бенлыитейн и А. А. Курбатов высказали пред-
положение, что основная масса yiлеводородов кавказских нефтей со-
стоит из гексагидрогенещрованных гомологов бензола.
В. В. Марковников и В, Н. Оглоблин2 выделили из кавказской! неф-
ти и изучили свойства углеводородов состава C-Hu. C\H]h.
ЦНц., С10Н2(). Они назвали их нафтенами (от греческого слова «наф-
уМарконинкин В. В. И пЗр.гру in. М. Ии ,АН ССС Р. 1955 - С 4Э 522
'Марковников В. В., Омоб.иш В Н. Исследование кавкаккои нефш : ЖРФ\О.
6(1883). - С. 237- 26S. .ЧП 354
та* — нефть) и высказали точку зрения, что нафтены состоят методу
из гекса гидробензолов (т. е. производных циклогексана), но содер^
\ глеводороды с меньшим и большим (чем шесть) числом атомов урч/
рола » писал что «нет никаких данных чтобы считан
в В Марковников писал, i |П0Меров. полооно гомолог»,
нафтены состоящими из одног V изомернь,х производи
бензола. Может быть, мы имеем
различных ядер...»1- г в. в. Марковников и М. И, Ко-
Действительно, позже- _ метилциклопентана в кавказской
„овалов сообщили о присутств
нефти. . .
В работах Н, Д. Зелинского2 показано, что нафтены легких фракций
нефти в значительной степени состоят из алкилпроизводных цикло-
пентана.
В настоящее время установлено, что циклоалканы нефтей состоя?
из моно-, ди-, три- и полициклических углеводородов, содержат^
циклогексановые и циклопентановые кольца, причем углеводороды
с шестичленными кольцами преобладают3. Производные низших цик-
лоалканов (циклопропана и циклобутана) не найдены в нефтях. В сиди
низкой термодинамической устойчивости их присутствие в нефтях ма-
ловероятно. Более вероятно присутствие углеводородов, содержащих
в циклах более шести атомов углерода. В научной литературе есть от-
дельные сообщения об обнаружении в нефтях циклогептана и его го-
мологов.
Моноцикл ические нафтены содержатся в основном во фракциях до
ЗОО’С. Бициклические углеводороды появляются в средних бензино-
вых фракциях (130-150°C) и сохраняются в высококипяших фракциях.
Трициклические нафтены находятся во фракциях выше 200'С4.
Моноциклические нафтены содержатся в основном в бензиновых и
керосино-газойлевых фракциях. Это алкил производные и и ют о пентана
и циклогексана (моно-, ди- и триал килпроизводные). В бензиновых
фракциях преобладают метилпроизводные: в значительно меньшей
степени представлены углеводороды, содержащие этильные группы,
очень мало содержание углеводородов с пропильными и бутильными
группами.
В бензиновых фракциях найден ииклопентан (до 0,5%), циклогек-
сан (до 7%), метил циклопентан (до 5%), метил циклогексан (до
1 Марковников В В. Итбр.труды — М.. Итл АН СССР. 1955.
- Зелинский Н Д Собр трудов. Т 2 — М Изд. АН СС СР. 1955 С. 504.
1 Куклинский А Я., Пушкина Р. А // Нефтехимия — 19X0 № I
4 Высоким содержанием нафтенов характеризуется нафталанская нефть (Азерб
респ.). Фракция згой нефти н к.-450’С содержит 38.1% моношпсшческих нафтенов.
39% би- и три циклических нафтенов и всего 2,9% изопарафинов, при полном отсутствии
„-алканов (Г. Г. Ашумов.Р. М. Ачасва // Азерб хим. жури. - 1976. - № I — С 88)
96
(]НОгда до 20%). В общем виде моноциклические нафтены,
10' "'с' п бензиновых фракциях, можно представить следующими
найденные
структу'рами:
R
где R -СН5. -Ср-Ц, редко -С>Н7. -С4Н9.
7/кис-изомеры производных цикзюпентана и циклогексана ™ «
шаютнад цис-изомерами. гексана прсоб-
Геминальные замещенные ],]-диметшши!/™^
циклогексан содержатся, как ,,РХ™ „X™ ” и~ил-
(обычнодо 10% от суммы других изомеров) И зтг ИХ количествах
.одних аиклопентана и индекс Тчи еТьно ПР°И3’
опт по сравнению с другими изомера^о ХХя 1 1?Х Т’ИЧ"
тшаклические нафтены представлены
СН3
R
Бициклические нафтены появляются уже во фракции 130-1506С и со-
аержатся «основном в средних фракциях нефти. В нефтях найдены1:
транс-декалин, или
биш1кло(4,4,0)лскан
ф/с-пснталан, или
бини кло(3,3,0)октан
^Жирные точки в формулах обояычакн атомы волорола, расположенные нал плос
костью страницы
7-3544
97
транс- гидри нда н,
бииикло(4,3,0)нонан
дициклогексил
В 1963 г. появилось сообщение, что из калифорнийской нефтц
(США) были выделены методом термической диффузии и идентцф^
цированы так называемые мостиковые углеводороды1.
бииикло(3,2,1)
октан
норборнащ бииикло-
(2,2,1)гептан
бицикло(2,2,2)-
октан
бицикло(3,3,1)нонан
а также их алкильные производные. Эти углеводороды были найдены н
в советских нефтях2.
Трициклические нафтены содержатся в средних фракциях нефти
Первым трициклическим нафтеном, выделенным из нефти был ада-
мантан, относящийся к ряду три циклодека на:
В 1933 г. чехословацкий химик С, Ланда выделил этот углеводород
из нефти месторождения Годонин. Молекула углеводорода симметрич-
^Lindcnian L. Р. Lc Tourneau R. L. // 6 World Petroleum Congress Section 5, paper H
(1963)
2 Солодков В. К., Воробьева Н С., Мнхновская А. А.. Петров Ал А. // Нефтей
мня - 1967.-Т. 7.-С. 511
98
на. вписывается в шар и термОд$
аярЖтка угяеподородд пкая туески сга6и
йнмамантаном (от греческого е; Ki"< У алмаза по Крмст^>1чес-
лантана: Гл, = 269”С. 7 = . Л°«а '<ала1Чант>1 он бЫл наз.
тщафип). плотность 1Х)7'"тич^\С(найленам КонсПнты
С. Ланды адамантан был ИаЦден 25 л„
Долгое время е„„тез ад, «нефПх 0CJIe «садедопания
он был синтезирован в 1941 г- П ‘ Не^Яав<спся Со
порошим выходом изомеризаХХ^ 8 * 1957 < «-ходом
'КГМ,И-’Род111(1,вд^ ^ею
tH тад не на (])
Метиладамантаны были впервые обнаружены в нефтях С. Ландой
111966 г. Позднее в американских нефтях и нефтях Бакинскою и Ро-
машки некого месторождений были также найдены (во фракциях
Х)0-250°С) гомологи адаман гана — метил- и зтиладамантан.
В 1967 г. предложен метол синтеза гомологов адамантана изомери-
зацией пергидроароматинеских углеводородов (Шнайдер):
В 1966 г. Ланда с сотрудниками выделил нт нефти тетраиикло-
(ШЛ2Л05’10)додскан:
1 ScliJcycr Р V |< //.) Alli ( I1CIII Sot. I0S7 v 70 Р 3292.
который можно рассматривать как производное адамантана, у кото^.
го положения 2 и 5 связаны этановым мостиком.
В высших фракциях нефти содержатся полициклические нафтену
мотекулы которых представляют собой системы конденсированных^,
5 циклов с короткими боковыми цепями (стераны и тритерпаны).
В высших фракциях нигерийской нефти и некоторых нефтей СССР
обнаружены стераны - холестан. эргостан и стигмастан’:
холестан: R= -Н;
эргостан: -СНу
стигмастан: R=-CH7CH1
и тритерпаны, например гопан:
Стераны и тритерпаны являются оптически активными и относятся
к так называемым «биологическим меткам», свидетельствующим
о связи нефти с живой природой (см. с. 10).
2.3. Физические свойства нафтенов
Нафтены имеют более высокие температуры кипения и плавления!'
показатели преломления, чем соответствующие по числу атомов угле-
рода алканы (табл. 10).
Это объясняется тем, что вследствие более правильной, более жест-
кой структуры молекулы нафтенов плотнее упаковываются в жидко'1
или твердом состоянии, что увеличивает силы меж молекул яркого вза-
В формулах в в иле клиньев изображены связи, направленные оз с Гранины, гтупктн
ром - за страницу
100
Таблица 10. Сравнение
Упеволород 1 'Аиг,- с Г <
Пешан 1 36 Г 130
Цикдопопан 49 -94
рксан 69 - 95
Фимогексан j 8’1 6.3
имолеиствия и обусловливает ботее
плавления, чем у алканов.
алканов и нафтенов
ТУ ’ "л 2^3?!
1,3577 0,6264
1,4064 0,7454
Г 3750 0,6594
1,4264 0,7781
температуры кипения и
Введение метильной группы в молекулу циклопентана и™
гексана резко нарушает симметрию молекуп что п ииЯпо-
нию температуры плавления метил пи клопе м™ „РИ,ЮДИТ к Уменьше-
но сравнению с циклопентаном и циклогексаном ?.1етилци,и10Гексана
^нк-joitKCdHOM. Например Т ас-
Циклогексан , , _ ше v-
.. +6,5
Метилциклогексан
-126,3
Нафтены имеют значительно более высокие о™^
соответствующие алканы с прямой цепью z______ вые числа, чем
Пентан
Циклопентан
Гексан
Циклогексан
(моторный метод):
62
87
77
Метилииклогсксан
ИК-спектры циклов
72
алканов похожи на ИК-спектры алканов. Цикло-
шаны не поглощают ультрафиолетового излучения в области длин
волн выше 2000 А, поэтому УФ-спектры не характерны для этих углево-
дородов. Спектры ПМР нафтенов с алкильными группами представля-
ют собой сложную картину вследствие спин-спинового расщепления.
Нарве, 49 приводятся спектры ПМР для циклогексана (а) и метил-
цнклогексана (б).
* f
Спектры ПМР циклогексана (а) и метилциклогексана (б)
101
иеские свойства нафтенов
2Д Химич м напоминают свойств
еские свойства нафтенов во много.
Химические мм
алканов. р комплексов с тиомочевинои
Образование ко. чения (клатратные соеднве.
нпйвуют соединения в
Нафтены образуют
ния) с тиомочевинои. rC-NH2
S
“ «большей степени зависит от яр#.
Стабильность этих соединении - ов Поэтому с помощью ж
С апркул углеводородов и ихР ого строения и едина».
Хин моноииклические нафте»
вой Молекулярной массы^а т^* болес прочные комп.«
от полициклических (нос,
с тиомочевинои). ация нафтенов
Д «г., легко дегидрируются над Pt, РФ Nil*
циклогексан и его гомологи. обр,а10В.жием ароматичен
TSO-320'С, либо над Сг,О3 Р заи11И циклогексана и его тома®
углеводород01’- «Я^Хи ДОС была открыта в 19П г. Н.Д.1
гов нал катализаторами Pt.
линеким .
нафтены с шестичленным никлом Это геи
(например, U-Диметилциклогексан) мос™^,ещеннь,с Циклогексаны
ролы ряда адамантана. Дегидрирова’ние "° ,ь,е нафтены, углеводе
с температурой кипения более ЗОО’С проволХ« '"'еСКИХ наФтеиов
егзии порошкообразного платинированного 1"^°" фазе " пРИсут-
шшеЗООС. ОГ0 Угля при температурах
/-«-замешенные циклогексаны и мостика,,
вращаться в ароматические углеводороды I? НафТены Могуг 1>Р^
чемусловия реакции Зелинского. Однако п ,! *естких Условиях
расщепление С-С-связей: р том происходит также и
На реакции Зелинского основано определение циклогексановых уг-
леводородов в бензиновых и керосиновых фракциях нефти. Деарома-
тизированную нефтяную фракцию пропускают над Pt при 300°С, при
этом циклогексановые углеводороды превращаются в ароматические.
В катализате определяют процентное содержание образовавшихся
ароматических углеводородов, которое равно процентному содержа-
нию циклогексановых углеводородов в деаромат изирован ной фракции.
Циклизация алкилпроизволных нафтенов
Над платиной при ЗОО'С алкилзамешенные нафтены с алкильной
прямой цепью, содержащей три и более атомов углерода, циклизуются
с образованием бициклических углеводородов:
Дегидрогенизация шестичленных нафтенов является основнойре
акцией, протекающей при риформинге бензиновых фракций (о
гл. IV). В условиях реакции Зелинского ииклопентан и его гомологи-
дегидрируются2. Не подвергаются дегидрогенизации также некоторг
^СН3-СН1-СН2-СН1 „
н,с—с 1-1 Г —Д_
н! Jh^7
сн2
бутилпиклопентан
' к*.шискии Н Д. Собр трудов — М Изя АН СССР 1955 - С. 46ft.
Ииклолиидш и его гомологи дегидрируются на л оксилом хрома при 450’С.
02
103
4-мстил пенталин
л и дан
В 1971 г. Ал. А. Петровым была показана возможность циклизации
I . 1,3.3-тетра метил циклогекса на над платиной:
Изомеризация нафтенов
В присутствии кислот Льюиса (например: AlClv А1Вг3) нафтены
подвергаются изомеризации (сжатие и расширение циклов, структур-
ная изомеризация боковых цепей).
Равновесную структурную изомеризацию проводят в присутствии
бромистого алюминия при комнатной температуре в течение 2-3 дней
до полного достижения термодинамического равновесия. При этом
происходит накопление в смеси наиболее термодинамически устойчи-
вых углеводородов. В частности, бициклические нафтены состава ClfJ
превращаются в основном в шронс-декалин, трициклические углеводо-
роды — в производные адамантана. В результате изомеризации проис-
ходит упрощение состава нафтеновой части исследуемой фракции неф-
ти. что, в свою очередь, упрощает хроматограмму и позволяет на ее ос-
новании определить соотношение между нафтеновыми углеводородами
различных молекулярных масс. На рис. 50 приведены1 хроматограммы
смеси нафтенов фракции 150-190°С одной из бакинских нефтей.
Равновесная структурная изомеризация позволяет приготовить
практически все возможные изомеры любого нафтенового углеводоро-
да. Так. изомеризацией гидриндана в присутствии AlBr^ в течение од-
ного дня с последующим разделением смеси с помощью препаратив-
ного хроматографа было получено 16 индивидуальных б и ни кланов
состава С9Нi6. На синтез этих углеводородов обычным путем приш-
лось бь! потратить несколько лет.
1 ( о.юлков В. К.. Михпонская А А.. Смирнов Б А Пиров Ам А -7 Нефтехимия. -
11)Ы 9 - С 49I ......
104
о
Время
Рис. 50. Хроматограммы смеси нафтенов фракции 150-190сС
до изомеризации (а) и после изомеризации (б)
Ce.ieKMiiMiax изомеризация с)наякияциклоцентана заключается в се-
тектиином расширении никла только за счет о-метиленовой группы
хзкила (метил практически нс участвует в расширении цикла):
СЩ 30—40%
Реакцию проводя! при комнатной температуре и в течение неболь-
шого промежутка времени (в отличие от равновесной изомеризации),
чтобы препятс I нова гь дал внешней изомеризации образовавшихся цик-
логексановых углеводородов. Последние затем дегидрированием прев-
ращают в ароматические углеводороды. На основании строения арома-
тических углеводородов судят о строении исходного диалкилцикло-
пентана.
Изомеризацию и дегидрирование можно совместить в одну стадию,
применяя бифункциональный катализатор, например, катализатор
платформинга, содержащий как изомеризующие (кислотные), так и
дети др и ру ю ш и е цен тр ы.
2.5. Анализ нафтенов
Количественное определение нафтенов
Методом количественного определения нафтенов в бензиновых
фракциях является метод анилиновых точек (АТ). АТ зависит от хими-
ческого состава фракции и, st частности, от содержания нафтенов. Зная
АТ нафтено-парафиновой части фракции после удаления из исходной
Фракции ароматических углеводородов, с помошью специальных таб-
тиц можно определить процентное содержание нафтенов. Подробно
мог метод будет рассмотрен далее.
Раздельное количественное определение циклогексановых и цик-
ЗДентановых углеводородов основано на реакции Зелинского. Пред-
105
положим, что нафте но-параф II новая часть бензиновой фракции содер-
ж и т 5 0 % и а ф те н о в (о п редел е н о м ето до м АТ). Н е об х oj i и м о о п редел ид
сколько из этих 50% приходится на циклогексановые я сколько на цнк-
лопснтановые углеводороды. Для этого нафтено-алкановую часть
фракции подвергают дегидрированию над платиновым катализаторов
при 300-321ГС и получают катализат, содержащий, предположим, 20§
ароматических углеводородов^. Следовательно, процентное содержа-
ние циклогексановых углеводородов в нафтено-ал каповой части фрак-
ции также -20 % и содержание циклопентановых углеводородов ~3(Ж
Точное количественное определение нафтенов в керосино-газой-
левых фракциях представляет собой трудную задачу, не решенную до
настоящего времени.
Достаточно точно можно определить только содержание нафтенов,
молекулы которых содержат шестизвенные циклы. С этой целью наф-
тено-алкановую часть подвергают либо парофазной, либо жидкофаз-
ной дегидрогенизации (в зависимости от температуры кипения узкой
фракции) над платиновым катализатором. При этом ал кил циклогекса-
ны превращаются в алкилбензолы; ал кил гидри нданы — в алкилинда-
ны; алкилдекалины — в алкилнафталины:
алкилде калин
ал к ил нафталин
Выделение нафтенов и их идентификация
Выделение нафтенов даже из легких фракций нефти в чистом
или в концентрированном состоянии представляет собой трудную^'
дачу, Из нафтено-алкановой части нефтяной фракции легко можновк
делить алканы с прямой цепью (цеолит 5А для бензиновых фракции,
комплексообразование с мочевиной для керосино-газойлевых фР^
ций), а оставшуюся смесь изоалканов и нафтенов полностью раздел^
очень трудно вследствие близости физико-химических свойств этихуг
леводородов. В настоящее время эта задача может быть решена части*
п-п-иГиг™с де! курированием циклогексановых углеводородов над плат»*
' ВИЯХ Може1 идти иик;1изация углеводородов. Для предотвращения никли
ими в катали затор необходимо ввести небольшое коли
чсство железа.
106
нос помоиiwo i срмичсской диффузии или путем адсорбпионно/о раз-
деления на молекулярных ситах 13Х или на активированном угле.
В процессе iсрмичсской диффузии алканы (в первую очередь //-ал-
каны) перемешаются к горячей стенке термодиффузионной колонки1,
венду конвекции поднимаются вверх и концентрируются в верхней
части колонки: нафтены движутся к холодной стенке и собираются в
нижней части колонки. Ал. А. Петрову и сотрудникам с помощью тер-
мической диффузии из фракции 125-150°С одной из бакинских неф-
тей удалось выделить концентрат бициклических нафтенов (С8-С9).
Дня это io нафтено-ал каповая часть этой фракции была подвергнута
термической диффузии в колонке высотой 1,5 м. Зазор между холод-
ной и горячими стенками — 0,3 мм. Температура горячей стенки
+70йС, холодной <5 С. Время опыта составляло 103 ч. В результате би-
циклические нафтены сконцентрировались в нижней части, а моно-
ииклические нафтены и алканы — в верхней части колонки. Анализом
бициклических нафтенов с помощью капиллярной хроматографии
было идентифицировано 18 индивидуальных углеводородов. На
рис, 51 приводятся хроматограммы нафтено-алкановой части и би-
циклического концентрата2,
Для разделения изоалканов и нафтенов средних фракций нефтей
возможно использование активированного угля3. Нафтено-алкановую
часть фракции (без //-алканов) подвергают жидкостно-адсорбционной
хроматографии на активированном угле марки БАУ. Изооктаном элю-
ируют полиад кил замещенные нафтены, //-гексаном элюируют изоал-
каны. Последними из колонки выходят наиболее прочно абсорбирую-
щиеся нафтены с дл и н н ой б о к о во й цепью.
Хорошее разделение изоалканов и нафтенов бензиновых фракций
достигается с помощью обработанного щелочью молекулярного сита
Рис. 51. Хроматограммы нафтено-алкановой части фракции 125-150 С (а)
и бициклических нафтенов (б)
УсГгермичеекои чиффузии см. с. 20.
“Силочков В. К., Воробьев Н. С.. Михновская А. А., Петров Ал. А.// Нефтехимия. -
1967. -. №7 _ f । ।
1 Р(меНбср[ Л. М . Ушакова И. Б... Гёнсх И. С.. Саиии П. И. // Нефтехимия. - 1%6. --
Г 6- - С 659.
107
их в паровой фазе1. Разделение обусловливается тем, что изоалаН1
сорбируются сильнее нафтенов на этом адсорбенте.
Для выделения нафтенов наряду с физико-химическими метода»
используют и химические. Так, с помошью реакции Зелинского ц||(,
тогексановые углеводороды нафтено-алкановон части фракции moj.
но превратить в ароматические. После удаления последних остаевд
смесь алканов и алкилциклопентанов, которые можно разделит,
с помошью термической диффузии. Полным гидрированием вы»
ленных ароматических углеводородов получают циклогексановыеуь
леводороды.
д и я выделения полициклических нафтенов, в том числе и адамац.
тана, используют комплексообразование с тиомочевиной2. Нефтяную
фракцию обрабатывают тиомочевиной и метанолом (1:1) при комич-
ной температуре в течение нескольких часов. Тиомочевина берете?
в небольшом количестве (10% от массы фракции). Аддукт тиомочеви-
ны и полициклических нафтенов отфильтровывают, промывают н-пен-
таном, разлагают водой. При этом выделяются полициклические наф
тены, которые после тщательной сушки снова обрабатывают тиомоче-
виной и метанолом в тех же соотношениях и получают концентрат ада-
мантана. Выделение адамантана из этого концентрата проводят с по-
мощью препаративной газовой хроматографии.
Для выделения адамантана и алкиладамантанов используют также
их низкую реакционную способность по сравнению с другими три
циклическими углеводородами. Так, из нафтено-алкановои части
фракции 200-250’С балаханской нефти термической диффузией би;
выделен концентрат трициклических нафтенов3, который пропустили
через слой катализатора Pt/Al2O3 при 420°С. В этих условиях все три-
циклические нафтены (кроме алкиладамантанов), а также бицикли-
ческие нафтены подверглись либо реакции дегидрирования, либо кре-
кингу. После удаления из катализата образовавшихся ароматически
углеводородов и продуктов крекинга была получена смесь алкилада-
мантанов.
Идентификация нафтенов. Эффективным методом идентификации
нафтенов в смеси является метод газо-жидкостной хроматограф^1
с применением эталонных углеводородов (метод меток). Если выделе-
ны индивидуальные нафтены, то для их идентификации использую?
спектральные методы (инфракрасная спектроскопия, масс-спектро-
скопия, спектроскопия ЯМР |3С). ПМР-спектры несут мало информ
пии о структуре молекулы нафтена из-за малого различия в химичес-
^Bnmnock J. V, Luke L, A. //1 Chrom - 1969. - V 39
-БагрийЕ И , Амосова Е. И.. Санин П. И //Нефтехимия. - !966Т. 6, - №5'
I. бю.
3 Санин П. И, // Успехи химии, - 1976. - №8. — С. 1377.
кнх сдвигах протонов различного типа, пики которых перекрываются
исчедствие спи н-с л и нового взаимодействия.
Спектры ЯМР 1 ’С позволяют более четко интерпретировать струк-
туру нафтенов.
' Для идентификации нафтенов, выделенных в чистом вине можно
использовать химические методы, в частности дегидрирование и се-
лективную изомеризацию. Если индивидуальный нафтен, выделенный
из фракции, в условиях реакции Зелинского превращается в аромати-
ческий у|леводород, с л слова гсльно. молекула этого v гл с водорода со-
держит шестизвенный незамещенный геминально цикл. Построению
образовавшегося ароматического углеводорода можно судить о строе-
нии молекулы нафтена. Например, если в результате дегидрирования
нафтена неизвестного строения был получен л/е/пя-ксилол
то нафтен имел структуру 1,3-ли метил циклогексана
Если дегидрирование нафтена в условиях реакции Зелинского не
проходит, то что возможно по следующим причинам:
1) у гл е водород я вз i яется п ро и з вол н ы м ц и кл о пе н тан а;
2) углеводород является геминально-замешенным циклогексаном
или мостиковым нафтеном.
Если углеводород является производным циклопентана, то он не
будет претерпевать заметных превращений при пропускании его над
катализатором Pt/C при температурах 300—350’С1. В этих условиях ге-
^алыю-замещенный циклогексан и мостиковый нафтен могут пре-
вратиться в ароматические углеводороды в результате расщепления
С’-С-связи и дегидрирования. Например:
Д|я плен i пфикапии и рои j водно! о никло пентана можно использовать селек гивную
jniiiiK’) с последукнппм тсгилрироианисм в ароматический углеводород
108
109
§ 3. Ароматические углеводороды нефти (арены)
и углеводороды смешанного строения
Арены представлены в нефти монониклическими и полицикличес-
кими. Обычно нефти содержат 15-20% аренов. В ароматических (смо-
листых) нефтях их содержание доходит до 35%. В зависимости от рас-
пределения ароматических углеводородов по фракциям нефти можно
подразделить на три группы:
I) нефти, ароматические углеводороды которых (в основном, поли-
циклические) концентрируются в высших фракциях. Это тяжелые
смолистые нефти с плотностью > 0,9;
2) нефти, ароматические углеводороды которых концентрируются
в основном в средних фракциях. Плотность таких нефтей 0,85-ОД
нефти этих двух классов относятся главным образом к нафтеновыми
нафтено-ароматическим;
3) нефти, ароматические углеводороды которых сконцентрированы
в легких фракциях (до 300’С). Это парафинистые нефти.
Во фракциях до 200°С (бензиновые фракции) содержатся только го-
мологи бензола. В нефтях найдены все гомологи бензола, включая Q
Монозамешенные гомологи бензола, содержащие 4 и более атомов уг-
лерода в боковой цепи, встречаются редко. Наиболее распространен-
ными являются толуол, этилбензол, ксилолы (.и-ксилол преобладает
как более термодинамически устойчивый), затем триметилбензоМ
далее идут кумол, пропилбензол, метилэтилбензолы.
Во фракциях 200—350°С преобладают алкилбензолы. главным обра-
зом ди- и три замешенные, молекулы которых содержат метильньк
группы и алкильную группу состава С7-Ск. Кроме гомологов бензол
в зтих фракциях содержатся гомологи нафталина (моно-, би-, три*"
ПО
тетраметилнафталины), Найденм „
ЛИН встречается редко Во фХйЯА '°Л°™ Л"фе"илу- Н“г1т‘~
бензола и гомологов нафтена соХаТся лХ ,0Ma10Г0),
лоролы. в молекулах которых изол~нЛ Р К“НЫ ~ угле,!°-
заны с углеводородным мостиком, например: Р°Матические ялРа сня~
В высших фракциях содержатся в небольшом количестве также го-
мологи полициклических углеводородов с конденсированными коль-
ttrilf П « Z » ' ’ ‘ - • •
нами, таких как:
I ( антрацен
фенантрен
мирен
хризен
перилен
ОСНОВНОЙ .ЖС ЧЯСТЬ ЭТИХ углеводородов KOHllCll 1 рИрУСДСЯ В гудрощ;
Широко представлены в высших фракциях нефicii углеводороды сме-
шанного строения, молекулы которых содержа! наряду с ароматичес-
кими кольцами нафтеновые кольца и алкильные боковые цепи. Более
подробно мы рассмотрим эти углеводороды специально,
3.1. Физические свойства ароматических
углеводородов
Ароматические углеводороды имеют более высокие температуры
кипения, чем соответствующие им по числу атомов углерода нафтены
(табл. 11). Это объясняется более плотной упаковкой молекул аромати-
ческих углеводородов (плоское кольцо), а также более сильным физи-
ко-химическим взаимодействием между молекулами — наличием
я-электронов (исключение составляют бензол и циклогексан, имею-
щие близкие свойства).
Алкилбензолы симметричного строения имеют более высокие тем-
пературы плавления. Например, и-ксилол ( + !3.2°С) и о-ксилол
(-25’С). С увеличением числа углеводородных атомов в боковой цепи
константы алкилбензола приближаются к константам алканов. Арома-
тические углеводороды избирательно растворяются в определенные
растворителях, таких как гликоли, метанол, жидкий S02, анилин.
Молекулы аренов в нефтях ассоциированы как между собой, таки
с молекулами алканов и нафтенов. Образование ассоциатов подтверж-
дается отклонением от аддитивности физико-химических свойств би-
нарных смесей аренов, а также смесей аренов с нафтенами и алканами1.
3.2. Химические свойства ароматических углеводородов
Образование комплексов с пикриновой кислотой (пикратов)
Полициклические арены (в частности, нафталин, антрацен и их го-
мологи) легко образуют пикраты. Бензол и его гомологи не образуют
стабильных комплексов и могут служить растворителями при комплек-
сообразовании, Пикраты получают по следующей методике.
Таблица 11
Углеводород т *с КИШ п 20 ""1 Рд __
Мет ил циклогексан I01 — 126 0.7692
Толуол I Ю.8 -95,5 ОА670
' Шимонаев Г С. // ЖФХ. ~ |973 - Т 17. - Выи. К). - С 3002.
Бронштейн Л а.. Шсхтер Ю. Н , Школьников Б. М., Сидоренко Н Н. //Химия"
технология топлив и масел. — I977 — 112 - С. 24.
I I2
К нефтяной фракции или расп вору арена добавляют раствор ггикри-
новой кислоты (в алею нс. хлороформе, спирте). Смесь поло (рева ют;
при охлаждении выпадаю! кристаллы пикратов— молекулярных сое-
динений пикриновой кислю ы с аренами. Образование комплекса про-
исходит за счет донорно-акцепторного взаимодействия арена (донор
^электронов) с пикриновой кислотой (акцептор л-электронов).
В комплексе молекулы углеводорода и пикриновой кислоты распола-
гаются друг над другом в параллельных плоскостях:
Пикраты ароматических углеводородов представляют собой твер-
дые кристаллические вещества желтого цвета, имеющие четкие темпе-
ратуры плавления. Каждому пол и ни юли чес кому углеводороду соответ-
ствует пикрат с определенной температурой плавления. По температу-
ре плавления пикрата можно идентифицировать полициклический
ароматический углеводород.
Комплексообразование с пикриновой кислотой используется также
как метод выделения полициклических аренов. Пикраты легко разла-
гаются горячей водой. Пикриновая кислота растворяется в воде, а по-
лициклические арены выделяются в свободном виде.
Полициклические арены образуют комплексы также и с другими
ароматическими полинитросоединениями, в частности с 1,3,5-три-
нитробензолом.
Скорость комплексообразования зависит от структуры ароматичес-
кого углеводорода и структуры гюлинитросоединения. Наиболее проч-
ные комплексы образуют молекулы ароматических углеводородов,
которых все связи между атомами располагаются в одной плоскости.
Если какие-то элементы структуры молекулы выходят из плоскости,
комплексообразование затрудняется. То же самое следует сказать и
° пЭДи нитросоединениях. Так, молекула 1,3,5-тринитробензола имеет
плоское строение, а в молекуле пикриновой кислоты, вследствие
113
-3544
пространственных затруднений между гидроксильной группой и нит-
рогрхппами, последние не находятся в плоскости бен зольного ядра.
По иомх пикриновая кислота вступает в комплексообразование
с меньшей скоростью, чем 1,3,5-трш нитробензол. Однако комплексо-
образование с 1,3,5-тринитробензолом не используется в аналитичес-
ких нетях, так как из комплекса трудно выделить углеводороды (три-
нитробензол не растворяется в воде).
Сульфирование ароматических углеводородов
Все ароматические углеводороды нефтей легко сульфируются
концентрированной серной кислотой:
H+Hdso2OH
SO2OH +н2о
Так как сульфирование является обратимой реакцией, то для пре-
дотвращения десульфирования необходимо связывать выделяющуюся
воду. С этой целью к концентрированной серной кислоте добавляют
пентоксид фосфора (смесь Каттвинкеля). Сульфирование нефтяных
фракций применяют для извлечения ароматических углеводородов.
Сульфирование проводят при небольших температурах (обычно при
комнатной), чтобы исключить изомеризацию углеводородов и окисли-
тельную дегидрогенизацию шестизвенных нафтенов; применяют из-
быток серной кислоты. Если ароматический углеводород содержит ко-
роткие алкильные цепи, то образующаяся сульфокислота растворяется
в избытке серной кислоты и переходит в сернокислотный слой.
Если алкильная группа большая, то образующаяся сульфокислота
будет плохо растворяться в серной кислоте и частично оставаться в уг-
леводородной фазе. Поэтому извлечь полностью ароматические угле-
водороды с длинными алкильными цепями (например, из керосино-
газойлевой фракции) сульфированием по методике сульфирования
бензиновых фракций нельзя.
Полученные при сульфировании сульфокислоты можно превратить
в углеводороды, нагревая их с водой, с соляной кислотой (или
обработкой водяным паром):
+ Н 2SO4
Однако эта реакция протекает не со всеми углеводородами гладка
полому в настоящее время она не применяется при анализе.
1 14
Гилриронаиие ароматических углеводородов
Bchkvi ii его io.mo.tohj j jijpiipyiojoi в присутствии Pt и Pd при КОМ
Патной температуре и лавлении 0,3 0,5 МПа:
R
В случае менее акт иных никелевых катализаторов требуются более
высокие температуры (150 250 С) и давление до 12 МПа. Следует от-
метить, что бензол i парируется легче алкилбензолов.
Нафталин iидрирустся значительно легче бензола. Гидрирование
протекает ступенчато' вначале быстро образуется тетралин, который
затем медленно гидрируется с образованием декалина:
Конденсация полициклических ароматических углеводородов
с малеиновым ангидридом
Антрацен и ею гомологи при нагревании с малеиновым ангидридом
образуют аддукты (реакция Дильса -- Альдера):
Базируясь на згой реакции, можно удалить из нефтяной фракции
антраценовые углеводороды.
Фенантрен и его гомологи реагируют с малеиновым ангидридом
под влиянием ультрафиолетового облучения:
115
Полученные аддукты легко разрушаются фотохимически с образо-
ванием фенантреновых углеводородов. Этот метод позволяет выделить
эти углеводороды из нефтяных фракций1,
Пербромирование ароматических углеводородов
В присутствии бромистого алюминия ароматические углеводороды
полностью бромируются в ядро (Г. Г. Густавсон):
Вг В г
Пербромиды имеют четкие температуры плавления. По температу-
ре плавления пербромида можно идентифицировать анализируемый
углеводород.
Конденсация с формальдегидом (формолитовая реакция)
В присутствии концентрированной серной кислоты ароматические
углеводороды конденсируются с формальдегидом с образованием смо-
лообразных веществ бурого цвета:
R R R
Эта очень чувствительная реакция, позволяющая обнаружить да*г
следы ароматических углеводородов в нефтяной фракции, применяет-
ся для контроля полноты удаления ароматических углеводородов hj
нефтяной фракции адсорбционным методом. Опыт проводится следу
юшим образом: на часовое стекло наливают немного концентрирован-
1 Леквейшвйли Э. Г. и др. // Нефтехимия. - 1979 - N? 5. - С 35.
116
ной серной кислоты, добавляют несколько капель формалина и 2-3
капли исследуемой фракции. Появление бурой или желтой окраски
указывает на присутствие ароматических углеводородов.
3.3. Анализ ароматических углеводородов
Количественное определение
I. Методом количественного определения ароматических углеводо-
родов во фракциях (до 200 С) является метод анилиновых точек. Зная
анилиновую точку исходной фракции и анилиновую точку этой фрак-
ции после удаления ароматических углеводородов, можно рассчитать
содержание ароматических углеводородов.
2. Быстрым методом определения объемного содержания аромати-
ческих углеводородов в бензиновых фракциях является метод сульфиро-
вания. Определенный объем бензиновой фракции энергично встряхива-
ют в сульфаторе с и збытком раствора фосфорного ангидрида в концент-
рированной серн о й кислоте; ароматические углеводороды сульфируют-
ся и в виде сульфокислот переходят в серную кислоту. Затем измеряют
объем фракции. По уменьшению объема фракции можно рассчитать
объемное процентное содержание ароматических углеводородов.
3. Довольно точно (ошибка 2-3%) можно определить содержание
ароматических углеводородов в бензиновой фракции с помощью жид-
костно-адсорбционной хроматографии.
Бензиновую фракцию разделяют на ароматическую и нафтено-ал-
кановую части, используя проявительно-вытеснительный метод жид-
костно-адсорбционной хроматографии. Зная массу фракции, взятой
.ия разделения, и массу выделенных ароматических углеводородов,
можно рассчитать содержание последних.
На основе жидкостно-адсорбционной хроматографии разработаны
быстрые методы количественного определения ароматических углево-
дородов в бензиновых и керосине-газойлевых фракциях с применени-
ем флюоресцирующих индикаторов. Образец фракции, содержащий
флюоресцирующий индикатор, обладающий такой же адсорбционной
способностью, как и ароматические углеводороды, вводят в хромато-
графическую колонку, затем добавляют спирт.
После того как фракция полностью пропитает адсорбент, колонку
освещают ультрафиолетовым светом. Измеряют длину флюоресцирую-
щей зоны ароматических углеводородов и. отнеся ее к длине части
колонки, заполненной адсорбентом, рассчитывают процентное содер-
жание ароматических yi ле водородов (установлено, что длина зоны аре-
нов пропорциональна их содержанию, см. также §5).
В случае керосино-газойлевых фракций, содержащих полицикли-
ческие ароматические углеводороды, флюоресцирующие вультрафио-
117
летовом свете, нет необходимости в применении специальных индика-
торов
Разработан’ метод, позволяющий определять в этих фракциях как
суммарное содержание ароматических углеводородов, так и отдельно
содержание гомологов бензола и полициклических аренов. Для опре-
деления суммарного содержания ароматических углеводородов в тон-
кую стеклянную колонку (длина — 50 см, диаметр — 2,4-2,8 см), со-
держащую силикагель, вводят 0,4 мл фракции, затем вытеснитель -
спирт. Когда фракция пропитает весь адсорбент, колонку освещают
ультрафиолетом. Измеряют флюоресцирующую зону и рассчитывают
общее содержание аренов, отнеся длину флюоресцирующей зоны к
длине части колонки, заполненной адсорбентом. Для определения по-
лициклических аренов в качестве адсорбента используют оксид алюми-
ния, на котором происходит разделение алкилбензолов и полицикли-
ческих аренов. В этом случае флюоресцирующая зона содержит только
полициклические арены. Отнеся длину этой зоны к длине зоны аренов
(при адсорбции на силикагеле), определяют процентное содержание
полициклических аренов и процентное содержание ал кил бензолов.
4. Точным методом количественного определения ароматических
углеводородов во фракциях до 200°С является метод, основанный на
определении дисперсиометрического коэффициента:
= /?г пс 1Qi,
FC лс-1,04
где /?с и fiF — показатели преломления фракции соответственно для
красной и голубой линий спектра водорода.
Зная Z)FC(J) фракции, />ЕСн нафтено-алкановой части, /)ГС1 аромати-
ческих углеводородов, можно по правилу аддитивности рассчитатьX-
процентное содержание ароматических углеводородов:
100 Ю„=ЛсД + Лсн(Ю0-Л'),
^ЕСн = *94,4 (постоянная величина для всех фракций, выкипаюшихдо
200 С, кроме бензольной фракции, для которой эта величина равна
193,3). Z?FCa берут из таблиц для различных узких фракций.
5. Для количественного определения ароматических углеводородов
используются также методы, основанные на ультрафиолетовой и ин-
фракрасной спектроскопии. Методы основаны на том. что интенсив-
ности полос в спектрах поглощения ароматических углеводородов про-
порциональны их концентрации во фракции. Так. например, предло-
жены методы определения ароматических углеводородов в бензиновых
и керосино-газойлевых фракциях на основании интенсивности поло-
МаС^Р-'|981В-№ П°СТИИКОва Н' Т tl Х”М|1Я 11 '«пология топлив к
118
сы поглощения при 1600 см J (валентные колебания связи О-С в аро-
матическом кольне) в ИК-спектрах1 и интенсивностей полос при
198 нм (для бензольных углеводородов) и 225 нм (для нафталиновых
углеводородов) в УФ-спектрах2.
Выделение ароматических углеводородов
Выделение ароматических углеводородов легких и средних фракций
проводят с помощью жидкостно-адсорбционной хроматографии (проя-
вительно-вытеснительный вариант) с применением в качестве элюента
(проявителя) изопентана и в качестве вытеснителя — этилового спирта3.
Этим же методом можно выделить также ароматические углеводороды
масляных фракций нефти и разделить эти углеводороды на моноцикли-
ческие, бициклические и полициклические. Для этого в хроматографи-
ческую колонку с силикагелем добавляют исследуемую масляную фрак-
цию в виде раствора в легком алкане, например в изопентане. После
того как адсорбент пропитается, в колонку заливают вначале изопен-
тан, затем бензол и спирто-бензольную смесь (1:1). Отбирают пробы
элюата, выходящего из колонки, и определяют их показатели прелом-
ления. Вначале из колонки выходит смесь нафтенов и алканов, затем
моноииклические арены (и от 1,5 до 1,53), затем бициклические (п от
1,53 до 1,55), затем полициклические арены (д от 1,55 и выше).
Для выделения полициклических ароматических углеводородов ис-
пользуют также и химические методы.
Так, многократной обработкой пикриновой кислотой можно выде-
лить из средних и высших фракций нефти значительную долю поли-
циклических аренов в виде пикратов. При разложении пикратов водой
выделяются полициклические арены.
Фенантреновые углеводороды можно выделить из нефтяных фрак-
ций в виде аддуктов с малеиновым ангидридом и после гидролиза
ангидридных групп в аддуктах с последующим фотохимическим разло-
жением образовавшихся ди карбоновых кислот получить эти углеводо-
роды в чистом виде.
Если из нефтяной фракции необходимо только удалить ароматичес-
кие углеводороды (без их выделения в чистом виде), то с этой целью
проводят сульфирование фракции избытком концентрированной сер-
ной кислоты или смесью Кагтвинкеля (раствор пентоксида фосфора
веерной кислоте). В случае бензиновых фракций все арены переходят
в слой кислоты в виде сульфокислот.
гКовачсва К., Димов Н.. Панкона М. // Химия и технология тошшв и масел. - 1979. -
№2. - С. 57. Бакулин Р А. и др. // Нефтепереработка и нефтехимия. - 1979. - №5. - С. 32.
'Чередниченко В. П., Фалеев В. С Расширение и уточнение программы исследо-
вания нефтей. Грозный. 1976. - С . 80.
5 Рябов В. Д. Физико-химические методы исследования углеводородов и других ком-
понентов нефти и газа — М : ГАНГ, 1996. — С. 28
119
В случае средних и высших фракций нефти часть сульфокислот
остается в углеводородном слое, и для их удаления необходимо промы-
вать углеводородный слой водным раствором щелочи. Промывка мо-
жет привести к образованию стойких эмульсий. Поз тому лучше перед
сульфированием к фракции добавлять л-гексан или //-гептан, чтобы
уменьшить растворимость арен сульфо кислот.
Идентификация ароматических углеводородов
Для идентификации ароматических углеводородов используют как
физико-химические методы (УФ-, ИК-спектроскопия, масс-спектро-
метрия, ЯМР-спектроскопия, газовая хроматография), так и химичес-
кие методы.
Для химической идентификации ароматических углеводородов
можно использовать получение их кристаллических производных
(пикраты в случае полициклических углеводородов, пербромиды -
в случае алкилбензолов).
Классические примеры применения химических методов для иден-
тификации и выделения ароматических углеводородов нефти находим
в трудах В. В. Марковникова1. Сульфированием различных фракций
бакинской нефти были получены сульфокислоты, которые переводили
в соли. Соли очищали кристаллизацией. Очищенные соли превращали
в хлорангидриды сульфокислот действием пятихлористого фосфора:
последние превращали в амилы действием спиртового раствора амми-
ака. Из очищенных солей сульфокислот получали исходные аромати-
ческие углеводороды, нагревая соли с соляной кислотой в запаянных
ампулах.
3.4. Углеводороды смешанного строения
Это полициклические углеводороды, молекулы которых содержат
ароматические, нафтеновые кольца и алкильные группы. Иногда их
называют гибридными углеводорода ми. Простейшие гибридные угле-
водороды находятся в керосиновых фракциях. Например, тетралини
его гомологи:
Особенно много гибридных углеводородов в масляных фракциях-
О строении этих углеводородов известно мало. Предполагают, что их
1 Марковников В. В. И збр. грул ы. - М . И чл. А Н С С СР. 1955 - - С 368.
120
молекулы содержит бензольные, нафтил и новые ядра, конденсирован-
ные с циклошднановыми и циклогексановыми кольцами. Например’
С. Р. Сергиенко1 предполагает для гибридных углеводородов масля-
ных фракций следующие структуры:
Полициклические ароматические и гибридные углеводороды с ко-
роткими боковыми цепями ухудшают вязкостные свойства масел и
уменьшают их стабильность против окисления, поэтому они являются
нежелательными компонентами нефтяных смазочных масел.
§4. Ненасыщенные углеводороды нефти
и продуктов ее переработки
Ранее существовала точка зрения, что алкены либо не содержатся
и нефтях, либо содержатся в незначительных количествах.
В конце 8()-х годов прошлого века было показано, что в ряде нефтей
Восточной Сибири, Татарии и других районов СССР содержание оле-
финов может доходить до 15—20% от массы нефти. Установлено, что
олефины ряда неф гей имеют высокую молекулярную массу и являют-
ся. по-видимому, продуктами радиолиза алканов под действием естест-
венного радиоактивного излучения в условиях залежи. Известно, что
при радиолизе алканов происходит распад связей С-Н.
Ненасыщенные углеводороды (олефины и диолефины) содержатся
в продуктах термической и термокаталитической переработки нефтя-
ных фракций (в (азах и жидких продуктах термического и каталитичес-
кого крекинга, пиролиза, коксования и т. д,).
^Сергиенко С Р Высокомолекулярные соединения нефти — М.: Гостов гсхизда к
1959.
121
4.1. Олефины (алкены)
Га зооб ра з н ы е эти л е н о в ы е у гл е водород ы (эт и л е н, п ро п и л е н, бутил е-
ны) находятся в газах термической и каталитической переработки неф.
тп. Газы термического крекинга под давлением (крекинг жидкофаз-
ный. 470-520’С, давление 20-50 атм) содержат 20—25% об. олефиновн
газы пиролиза (800-900’С, давление около I атм) содержат40- 50% об.
олефинов. При сравнении этих данных видно, что с уменьшением дав-
лен ня процесса содержание олефинов в газах увеличивается.
В табл. 12 представлены данные по химическому составу газов раз-
личных процессов термической переработки нефти и каталитического
крекинга.
Жидкие продукты процессов термической и каталитической пере-
работки нефти содержат также значительные количества олефинов.
Так, бензины термического крекинга содержат 30—35% олефинов, бен-
зины каталитического крекинга — до 10% олефинов. Углеводороды
с двойной связью, содержащиеся в нефтепродуктах, можно подразде-
лить на следующие группы:
1) олефины нормального и изостроения;
2) циклоолефины (циклогексен, циклопентен и их гомологи);
3) ароматические углеводороды с двойной связью в боковой цепи
(стирол, инден, их гомологи).
Общее содержание непредельных углеводородов в различных
фракциях бензина термического креки нга уменьшается с увеличением
температуры кипения фракций; уменьшается также и содержание
алкенов, в то время как содержание циклоолефинов и с тиролов увели-
чивается.
Таблица 12. Состав газов термических процессов
и каталитического крекинга нефтяного сырья, % об.
Ком попет Термический крекинг Коксование Пирочи 5 К а пиитический крекинг
H-, 0.4 I-2 -l.fi 1-1.5
сн4 I6 20 20 30 В) -20 8-12
CHf, 19- 20 15-20 Ь И) 8-10
25-28 5-K.I 1 2 10-15
9-Ю I0-I5 1 2 20-25
5- 7 3- 5 8-12
С ?Н4 2-3 I0-I5 20 30 2-3
С'Л, 9 - К) 20-25 12 15 10-15
9 И) IO I5 1 2 15-20
I 5 .3 -5 --
I22
Таблица 13
Уисволори 1 Лип ’С Р?’
Пешая Vi -120 0.626
1-ПС1ПСН 30 - 165 0.64
Таблица 14
Уисводорп J Лп.г С V'c р4-°
2Д-Дцмети.!-2 буи-н 7 3..2 7 S 4 0.70SS
I - Гексен 63.5 -140 0.6740
Алкены — С । -Сд газы. С 5-С |7 — жидкости. Остальные — твер-
дые вешества о.-Алкепы нормального строения имеют более низкие
температуры кипения и плавления (табл. 13), чем соответствующие ал-
каны нормальною строения, но более высокие плотности и показате-
ли преломления:
Разветвленные алкены симметричного строения имеют значитель-
но более высокие температуры кипения, температуры плавления и
плотности (табл. I4), чем остальные изомеры
Обычно щ/г-июмеры алкенов имеют более высокие температуры
кипения, чем транс-\\ то меры Олефины имеют более высокие октано-
вые числа, чем cnoiвстствуюпшс алканы с прямой пенью (l-гептен и
3-гептен имеют октановые числа, соответственно равные 65 и 95).
Адсорбционная способность олефинов ниже, чем адсорбционная
способност ь аромат ических углеводородов, но выше, чем насыщенных
углеводородов. На лом различии основано количественное определе-
ние олефинов в нефтепродуктах метолом адсорбционной хроматогра-
фии в присутствии ф.тjoopeciтирующих индикаторов (§5).
4. 1.1. Химические свойства алкенов
Рассмотрим хим ичсские свойства олефинов, имеющие специальное
значение для химии нефт и.
Присоединение водорода
Водород присоединяется к олефинам при комнатной температуре
пприсутствии мелкой тмельченпой плагины или палладия; в присут-
ствии никеля при INO'C:
р— ( | [— (: 11, f н J К—С Н 3—С Н з
Нужно отмстить. что в )гих условиях ароматические углеводороды
не подвергаются гидрированию, полому путем гидрирования можно
определить содержание олефинов в креки нг-бен зинах. При определе-
нии содержания олефинов в нефтяных продуктах методом гидрирова-
123
НИМ OlipC.lC.lMfOl водородное число, то есть число миллилитров водоро-
ла. приведенных к нормальным условиям (760 мм pi. сь, О С), погло-
щепных 1 г вещества.
В настоящее время метод водородных чисел не используется вана-
литических целях.
Галоидирование
Аналитическое значение имеет реакция присоединения галоидов к
олефинам. При комнатной температуре реакция присоединения селек-
тивно проходит лишь в случае йода: бром и хлор не только присоеди-
няются. но и замещают атомы водорода. Чтобы исключить реакциюза-
мешения, бромирование нужно проводить в темноте, при пониженных
температурах:
R—СН=СН2 + Вг2 R— CH—(j?H2
Вг В г
В этом случае реакция бромирования может служить для отделения
непредельных углеводородов от остальных углеводородов и их выделе-
ния в виде либромидов перегонкой (ГК(1Г| дибромидов намного выше
ГКИ|] углеводородов).
На реакциях присоединения брома и йода основано количествен-
ное определение олефинов в нефтяных продуктах методами бромныхи
йодных чисел. Бромное или йодное число — это количество граммов
соответственно брома или йода, поглощенное 100 г исследуемого про-
дукта. Метод йодных чисел (метод Маргошеса1) состоит в следующем.
К навеске анализируемого продукта (0.1-0,3 г) добавляют избыток
титрованного раствора йода. Часть йода реагирует с олефинами, содер-
жащимися в навеске: избыток йода определяется титрованием тио-
сульфатом натрия в присутствии крахмала как индикатора.
2Na3S2O3 + J2 2NaJ + Na2S4O6.
Разность между количеством взятого йода и количеством йода, не
вступившего в реакцию и определенного титрованием, равняется ко-
личеству йода, вступившего в реакцию (д). Зная величину навески (Я
можно легко рассчитать йодное число:
ич J00 °
ь '
и на его основе — процентное содержание олефинов:
Могол Марюшеса молот давать заниженные результаты вследствие неполноты
присоединения йода к олефинам с пониженной реакционной способностью-
124
где И.Ч.теор — теоретическое йодное число, рассчитанное из предполо-
жения, что исследуемое вещество целиком состоит из олефинов:
Отсюда
где М— молекулярная масса анализируемого продукта; 254 — молеку-
лярная масса йода.
Метод Кауфмана состоит в том, что на навеску действуют титрован-
ным раствором брома в метаноле в темноте. Затем в колбу вносят и збы-
ток 10%-го раствора йодида калия; при этом происходит взаимодей-
ствие непрореагировавшего брома с йодидом калия с образованием эк-
вимолекулярного количества йода, который оттитровывают тиосуль-
фатом натрия. В случае метода Кауфмана возможны завышенные ре-
зультаты вследствие реакции замещения.
Присоединение серной кислоты
Серная кислота различной концентрации может абсорбировать раз-
личные газообразные олефины с образованием алкилсульфатов:
98%-ная H2SO4 поглотает С2Н4; 80%-ная — С3Н6; 75—77%-ная — бу-
тен-1; 55-58%-ная — изобутилен.
Для удаления олефинов из крекинг-бензинов применяют 80%-ную
серную кислоту:
R—СН=СНч + HOSChOH
/ **
R—СН—СН3
о—SO2OH
Озонирование алкенов
Озон количественно с большой скоростью присоединяется к алке-
нам при комнатной температуре. При этом ароматические углеводоро-
ды, находящиеся вместе с алкенами в нефтепродукте, практически не
реагирует с озоном:
о—о
Оз / .
Н5с— СН=СН — СНЬ-СН1--------►Н3с— НСХ /СН — СН2 СНз
о
Разрушение озонида под действием молекулярного водорода при-
водит к образованнию карбонильных соединений — альдегидов или
125
кетонов, по составу которых можно судить о положении двойной cBi).
зи в молекуле алкена:
Hi
CH—CH.-CH?-^
.-CHj + HjO
На реакции озонирования алкенов основано количественное опре-
деление алкенов в нефтепродуктах на приборе АДС (анализатор двой-
ных связей).
Присоединение ацетата ртути
Ацетат ртути присоединяется к олефинам при комнатной темпера-
туре в присутствии метилового спирта:
R—сн—сн—R'-Hg
дсжон
RCH---(j?HR’ + СН3СООН
Н3СО HgOC—сн3
о
Продукт реакции, представляющий собой белый осадок, может
быть легко разрушен действием разбавленного раствора хлороводород- '
ной кислоты, при этом олефины выделяются в чистом виде. Этот метод
позволяет отделить олефины от смеси других углеводородов и выде-
лить их в чистом виде. Например, для выделения олефинов из бензина
каталитического крекинга определенное количество бензина обраба-
тывают ацетатом ртути в метаноле; от реакционной смеси отгоняют во-
дяным паром остальные углеводороды: остаток разлагают разбавлен-
ной хлороводородной кислотой и выделяют олефины
4. 1.2. Количественное определение, выделение
и идентификация олефинов
Количественное определение олефинов в нефтяных продуктах про-
водится методом бромных и йодных чисел, методом жидкостно-ад-
сорбционной хроматографии в присутствии флюоресцирующих инди-
каторов (см. 5.1), а также озонометрическим метолом.
Отделение олефинов от нефтепродуктов можно проводить с по-
мощью 80%-ной серной кислоты; для выделения их в чистом виде
используют реакцию с ацетатом от,.
разработаны методы анадиза 8 "Поящее время
хроматографическим путем. В газовыйСОЖР*^иих олефины
гор. содержащий адсорбент с нанесХ ТОГра* '''««тируется
серной кислотой. В хроматограф вв^Хя ? 7 "Юность 80%.
ролукта. один из них поступает непосг>е„? °бразца Фр«ции нефтей
гой вначале проходит через реактор ссерТоГ^ “ *Р°^ограф X
мато-рамм позволяет определить на них ПХ 'M°TOiL X
новым углеводородах,, и идентифиц И’С00Тве^Вующие
олефинов, наРялу со спектральными“ неХ" "Х' нде”™Ф™Х
методы, ак, для установления положен,,, ° ИСП0ЛЬЗУЮт химические
мефина применяют озонирование. Двойно« св«и „ молекуле
4.2. Диолефины нефтяных продуктов (диены)
В продуктах парофазного крекинга и пиролиза может содержаться
от 5 до 15% диолефинов. Это — бутадиен, пиперилен, циклопентадиен.
Все эти диолефины являются диолефинами с конъюгированными свя-
зями.
Диолефины с конъюгированными связями вступают в реакцию яи
енового синтеза (реакция Дильса - Альдера). Реакционная способ-
ность диенов записи, от их строения. Из двух возможных компланар-
пых систем диенов (1) и (2); ’й
только диены с цисоидным расположением двойных связей (2) вступа-
ют в реакцию. Это бутадиен в цисоидной конформации:
Циклопентадисн реагирует при комнатной температуре, так как для
него возможно только цисоидное расположение двойных связей. Бута-
диен - при температурах выше ЮОТ. Цис- пи пери лен не вступает в ре-
акцию диенового синтеза. Для него характерна только трансоидная
конформация, ид кик цисоидная конформация тусюйчива Hj-jaQf.
1 л. 1 к и ва н и ч мсжлх могильной группой и томом водорода
Поэтому в реакцию вступает дгронс-пиперилсн в цисоидной кон-
формации.
Диеновые углеводороды образуют комплексные соединения с ам-
миачным раствором ацетата одновалентной меди при температурах
0-1 (ГС. При нагревании эти комплексные соединения разрушаются
с выделением диенов. Отделение диенов и их количественное опреде-
ление в нефтепродуктах основано на реакции диенового синтеза. Об-
разец нефтепродукта нагревают с малеиновым ангидридом в течение
нескольких часов, затем отделяют от смеси не вступивший в реакцию
малеиновый ан гидр ид действием водного раствора щелочи. После про-
мывки и сушки образца отгоняют углеводороды и получают в остатке
кристаллы аддукта, по массе которых можно рассчитать процентное
содержание диенов в нефтепродукте.
§ 5. Определение состава нефтяных фракций
и нефтяных продуктов
Определение химического состава нефтяных фракций и других
нефтяных продуктов имеет большое значение, так как их эксплуатаци-
онные свойства определяются химическим составом.
Как следует из предыдущих глав, определение индивидуального хи-
мического состава даже бензиновых фракций нефти представляет со-
I2X
бой донолыю сложную ишачу и г ребует значительного времени, поэто-
му такой аналн j проводится в особых случаях. Обычно используют бо-
лее бышрые мсюлы анализа, которые позволяют определить групповой
или структурно-групповой состав нефтяных фракций.
Групповой состав низших и средних фракний нефти показывает со-
держание в л их фракциях учлеводоролов различных групп (содержа-
ние аренов, нафюной и алканов).
Детализированный групповой состав, кроме содержания углеводоро-
дов различных трупп, показывает содержание подгрупп углеводородов
вход я ш и х восстав каждой г р у п п ы. Структурно-групповой состав пыс-
ших масляных ({.тракций нефти дает представление о соотношении раз-
личных структурных трупп (ароматических ядер, нафтеновых ядер, ал-
кановых пенен) и т ак называсмой *средней молекуле» данной масляной
фракции, то есть такой гипотетической молекуле, элементный состав
которой одинаков с элементным составом масляной фракции при равен-
стве молекулярных масс.
Для характеристики масляных фракций обычно используют струк-
турно-групповой состав. Точно определить групповой состав масляных
фракций, как ото делается для бензиновых фракций, нельзя, так как
масляные фракции содержат много углеводородов смешанного строе-
ния, которые нельзя отнести ни к алканам, ни к аренам, ни к нафтено-
вым углеводородам.
5.1. Определение группового состава и детализированного
группового состава бензиновых фракций
Определение группового состава основано на различии физико-хи-
мических свойств углеводородов различных рядов, в частности, на раз-
личии простых и комбинированных констант. Так как эти константы
являются аддитивными величинами, можно рассчитать групповой сос-
тав, зная константы углеводородов каждой группы и константы иссле-
дуемой фракции. Наиболее простым методом определения группового
состава бензиновых фракций нефти является метод анилиновых точек.
Анилиновая точка (АТ) — это температура, при которой происходит
взаимное растворение смеси равных объемов анилина и углеводорода.
Чем легче углеводород растворяется в анилине, тем ниже его анилино-
вая точка. Анилиновая точка увеличивается при переходе от углеводо-
родов ароматического ряда (~2(ГС) к нафтенам (~60°С) и от нафтенов
калканам (70-80'С). Олефины и циклоолефины имеют более низкие
АТ, чем соответствующие насыщенные углеводороды.
Одним из методов определения анилиновой точки является метод
равных объемов. По этому методу берут равные объемы анилина и уг-
леводородов (но 2 мл) и при перемешивании нагревают до полного
растворения. За анилиновую точку принимается температура начала
129
помутнения раствора при его охлаждении. Анилин должен бытьсвеже-
перегнанным и высушенным, его чистота проверяется путем определе-
ния анилиновой точки н-гептана (70,ГС).
Для анализа бензина методом анилиновых точек его разгоняют
с помошью ректификационной колонки на следующие фракции:
60-95°С — бензольная;
95 -122 С — толуольная;
122-150°С — содержит этилбензол, ксилолы;
150-175’С — ароматические углеводороды CQ:
175-200°С — ароматические углеводороды С,().
Каждую фракцию исследуют отдельно. Определяют анилиновую
точку каждой фракции; затем извлекают из каждой фракции аромати-
ческие углеводороды (сульфированием или адсороциси) и определяют
АТ деароматизированных фракций. Содержание ароматических угле-
водородов рассчитывают по формуле1
где К — анилиновый коэффициент, равный массовому процентному
содержанию ароматических углеводородов, при котором разностьАТ
равна ГС; г, — анилиновая точка исходной фра к и ин. С; /у —анилино-
вая точка фракции после удаления ароматических у иеводородов, °C.
Вел ичи н ы К на й де и ы дт я л юбо й и з на зван н ы х ф р з к 11 и й п утем при-
готовления искусственных смесей углеводородов, выкипающих в пре-
делах кипения этих фракций:
К=Т/(/2-/1).
Коэффициент К для каждой фракции находят в таблицах в зависи-
мости от объемного процентного содержания ароматических углеводо-
родов, которое можно определить сульфированием После определе-
ния содержания ароматических углеводородов на основании значения
с помощью специальных таблиц находят содержание нафтенов вде-
ароматизированных фракциях.
По разности находят процентное содержание алканов.
Пример2. Анилиновые точки фракции 60 -95 С бен шла до и после
удаления ароматических углеводородов:
= 48Т; f2 = 59Х.
Приближенная объемная концентрация ароматических углеводоро-
дов в этой фракции (метод сульфирования) — 12Г. Тогда К по таблице
равно 1,15.
’В настоящее время имеются другие, боде с быстрые и юн11ыс мезолы оп ределения
ароматических углеводородов (с 154).
- Иса1улянц В. И., Егорова Г. М. Химия нефти. Руконокпю к лабораторным заняти-
ям - М. Химия, 1965. — С. 179
130
Следовательно. А = 1.15(59 - 48) - 12.65% мае. По лрхтой таблице
11Я (, = М С находим, что концентрация нафтенов в деароматизиро-
rauHPii фракции Ы)-9р С равна 36%. мае. Тогда в исходной фракции со-
держание нафтенов равно:
31,45%
Содержание алкинов:
/ ’ 100 - 12,65 - 31,45 = 55,9.
При определении Тта.шифрованного группового состава бензиновых
фракций помимо проиентого содержания аренов (% Ар.), нафтенов
(Ф Н) и алканов (Ф А) находят процентное содержание циклопентано-
вых (% Н( _) и циклогексановых углеводородов, алканов нор-
мального строения (% //-А) и z/30-алканов (% /-А).
Раздельное определение циклогексановых и циклопентановых уг-
леводородов основано как уже говорилось, на реакции Н. Д. Зелинс-
кого. Раздельное определение алканов с прямой цепью и изоалканов
проводится с помощью молекулярного сита 5А, избирательно погло-
щающею алканы _ прямой пенью. На рис. 52 приведена схема опреде-
ления летали л ip; 'вл и hoi о i рун нового состава бензинов прямой гонки.
После опрели ici।ни i ру iiiioboiо состава бензиновую фракцию разделя-
ют на аромаiипсскхю и нифгено-алкановую части с помощью жилко-
стно-адсорбшн mi ion хрома 101 рафии. Одну часть смеси нафтенов и ал-
канов npoi । \ сж л к > । нал Pt/( ’ при 300"С; любым методом в катализате
определяю! соц-ржание ароматичсских углеводородов, которое чис-
ленно равно содержанию шест и звенных нафтенов в нафтено-алкано-
вой части бен шна. Н ipyioii части смеси нафтенов и алканов с по-
мощью нсолн юн ип)Ч'Л.сляк)т содержание алканов с прямой цепью,
Групповой сослан нафтено-ал каповой части (содержание нафтенов,
//-алканов, и юллканов.) можно определить методом газо-адсорбпион-
ной хрома mi рафии применением молекулярною сита 13Х1.
Для опрслс. К‘Н ИЯ 1р типового состава крек ин г-бензинов или жидких
продуктов пиролп ш анализ проводят последующей схеме:
I. Определен ис и отделение лиолсфинон от всей массы анализируе-
мого бен шна i homi )пн.ю малеиновою аш идрила.
2 Опрею ichhc наире тельных углеводородов в бензине после удале-
ния диенов (метол бромных или йодных чисел).
3. Отдслсниг п вторичное определение непредельных углеводоро-
дов с помои нал S6 пропен гной серной кислоты и получение смеси аре-
нов. нафтенов и алканов.
Fl’Ти ифов Р, И.. Негрона В. И. // Химия и технология топлив и
•кд .1. 1970. >
131
Дегилрогепи запил
Р(/С ЗИИ'С
Рис. 52. Схема определения детализированного группового
состава бензинов прямой гонки
> н
4. Дальнейший анализ проводится по схеме для бензинов прямом
гонки.
Групповой состав олефинов крекинг-бензинов или жидких продук-
тов пиролиза можно определить следующим обра зом:
1. После отделения диеновых углеводородов от креки нг-бензина
или от фракции смолы пиролиза выделяют олефины с помощью уксус-
нокислой ртути.
2. Гидрируют выделенные олефины над Ni при 150 С. При этом ал-
кены превращаются в алканы, циклоолефины — в нафтены, а алкенил-
бензолы — в алкилбензолы.
3. Определяют групповой состав продукта гидрирования методом
анилиновых точек.
Находят процентное содержание алканов, равное процентному со-
держанию алкенов, процентное содержание нафтенов, равное проиент*
132
Рис, 53. Хроматограмма
и ;иканы
ному содержанию циклоолефинов, и про-
центное содержание алкилбензолов, равное
процентному содержанию алкенилбензолов.
Быстрым методом определения группово-
го состава бензинов крекинга и риформинга
(определение олефинов, ароматических угле-
водородов, суммы нафтенов и алканов) явля-
ется метод жидкостно-адсорбционной хрома-
тографии в присутствии флюоресцирующих
веществ.
К исследуемому бензину добавляется в не-
большом количестве смесь флюоресцирую-
щих веществ (полициклические соединения,
выделяемые из нефтяных смол, вместе с жиро-
вым красителем судан III). Полученный раст-
вор заливают в стеклянную колонку с силика-
гелем. после его впитывания заливают вытес-
нитель (дгпловый спирт). По высоте колонки происходит распределение
углеводородов вмеси? с соответствующими красителями (рис. 53). Кра-
ситель судан концентрируется на границе раздела спирт: ароматические
углеводороды. Колонку ос веша ют ультрафиолетовым светом; каждая зо-
на в ультрафиолетовом свете имеет определенную окраску, кроме зоны
нафтенов и ал канон, которая не окрашена. Измеряют /тину каждой
зоны и рассчитывают объемное содержание каждой группы углеводоро-
дов, допуская. чю длина каждой зоны пропорциональная объему углево-
дородов. Групповой и одновременно индивидуальный состав бензино-
вых фракций успешно определяется методом газо-жидкостной хрома-
тографии с применением капиллярных колонок. Таким образом, можно
определить групповой и индивидуальный состав легких бензиновых
фракций с температурой кипения до 150- 180°С. С этой целью образец
бензина анали ntpvioi на хроматографе с применением капиллярной ко-
лонки и получаю! довольно сложную хроматограмму. Каждый пик мож-
но идентифицировать с помощью модельных углеводородов, затем мож-
но определит!) концентрацию каждого углеводорода и, следова1елызо,
рассчитать [рунповой и детализированный групповой составы,
5.2. Определение детализированного группового состава
керосино-газойлевых фракций
Групповой состав керосин о-газойле вых фракций нефти (200—
35О°С) может быть определен методом анилиновых точек, но прибли-
зительно. так как анилиновые коэффициенты для керосино-газойле-
вых фракций являются весьма приближенными величинами, вслед-
ствие сложности химического состава этих фракций.
133
bn ice точно можно определить труп новой состав кероенно-газойле-
вых фракций комбинированным методом, основанным па применении
ж и дкостно-адсорбнионной хроматографии, ком i итсксообразовання,
четкой ректификации и спектрального анализа. Комбинированный ме-
тод анализа керосино-газойлевых фракций позволяет определить дета-
лизированный групповой состав кероси но-газойле вых фракций. На
рис. 54 приводится схема этого анализа.
Вначале широкую фракцию разделяют на узкие 50 -с фракции пере-
гонкой в вакууме. Каждую фракцию разделяют с помощью жидкостно-
адсорбционной хроматографии (ЖАХ) на силикат еле на две части: аре-
ны (Ар) и смесь алканов и нафтенов (LH, А). Арены разделяют методом
ЖАХ на А12О3 с получением фракции аренов с одним бензольным яд-
ром (Ар,) (алкилбензолы, алкилинданы. алкил тетрад ины) и фракции
полициклических аренов (Ар2) (алкилнафталины, алкилдифенилы, ал-
кил антрацены и т. д.)- Каждая из этих фракций может оыть подвергну-
та хромато-масс-снейтральному анализу. Из смеси алканов и нафтенов
комплексо-образован нем с карбамидом можно выделить
w-алканы (я-А) и определить их количество и состав методом ГЖХ. Ос-
тавшуюся после выделения д-алканов смесь /-алканов и нафтенов под-
вергают парофазной или жидкофазной дегидрогенизации на катализа-
торе Pt/C (в зависимости от температуры кипения фракции). Образо-
вавшийся катализат, состоящий из аренов, производных никлопентана
и изоалканов, анализируют: определяют содержание аренов (Ар), кото-
рое приблизительно равно процентному содержанию нафтенов с шес-
тизвенным циклом, и далее разделяют катализат по той же схеме, чтои
для 50°-х фракций исходной широкой фракции. Арены (Ар) разделяю!
на производные бензола (Б), которые образовались из нафтенов с од-
ним шестичленным циклом, и на производные полициклических аре-
нов (П), которые образовались из нафтенов с несколькими шестичлен-
ными циклами.
5.3. Определение структурно-группового состава
масляных фракций
Как уже говорилось, точное определение групповою состава воз-
можно только в случае бензиновых фракций: приблизительные ре-
зультаты можно получить для керосино-газойлевых фракций. Что же
касается высших фракций, то определение их групп о во го состава не-
возможно вследствие наличия большого количеенш углеводородов
смешанного строения. Поэтому химический состав фракций чаше
всего характеризуют их структурно-групповым составом1.
Дополни i ел ьно химический сослан маслю пах фракций можел бьнь охаракк-
рилован содержанием д-алканов и углеводородов с ароматическими кольцами (моно-
циклические. Сил циклические и полициклические).
134
Рис. 54. Схема определения детализированного группового состава КГФ
135
показывает соотношение различна,
.чпукзерно-тпупповои " в вредней молекулы» фрак®,
Хныч групп, входа"*"' "Ханне атомов углерода, составлю
струкгм ., _ процентное содер* тветственно процентное»
Н8ХОаЯТ ^веские ядра: " %С" X нафтеновые кольца и Мщ.
ишхарома!^ углероаа. составив ’
жание угперода- входяшеговсос-
держание атомов >_ Hffloe содержание
новые да""йаходаТ также К„ - °®шее ых колеи в ..средней молекул,
тав колеи. _ _ число нафтен молекулы» масляное,
тических ко-' улярНуЮ формулу *ср ения элементного соста-
фракипи. • дят „а основании Р этой фОрмуте6ы.
ФраКмолекудаРН*>й массы фракиии.б^'няписать структурную формулу
“'^числами и можно было бы H.Q бу жгко „
«среднем mo --во..состав.
- -nvMTii когда it и т в «средней
”’Т'
»«™ ™!К”"
(2 Н ? Эта молек\ляРн‘,н ч
лер^ё11ИЛте’гра'ПИНа‘
1 \ /I >
S
<-—-• ”sz”^”₽:s"ss”' *=
.............................
Так, К, = 1 "
К.. = 0,66
'Н->
С lb
ж
о
20
%С1, =100-50 = 50
/лам» высших фракций отвечают формулы
- лт-nuv*
%са =-—100 = 30
%Ck. =30+20=50
X.
Охтнако «средним молекулам» —
с дробными значениями н и т и. следовательно, нельзя написать струк-
турную формулу «средней молекулы» фракции и рассчитать по ней
структурно- групповой состав.
136
Поэтому для определения структурно
высших фракций нефти имеется ряд метта^м °ð С0СТава сре™их и
вой для всех них является так называемой Метолологической осно-
группового анализа. «прямой метол» структурно-
Прямой метод
Нахождение структурно.грулл
да основано на определении содержания воды л Помошью этого мето-
массы исследуемой фракции до и 110С1е y?'’fOf)a " молекулярной
рим конкретный пример. ' 0 ее г11"Рарования, Рассмот-
Предположим, что спетияи
фракции равна 500. а элементный ападгТ™’' Масса (Л/) како«-либо
С-*’®, Н - 13%. Определим м;еХн™Т(СЛеДУЮЩИе Ре3ул^
ду фракции Cz/Hw. р ую Эмпирическую) форму-
Вначале находим простейшую формулу фракции „
соотношение между углеродом „ водородоу, ,Г“ейТ
„ CH,7S М. = 13.804
Для нахождения эмпирической формулы находим разделив м ,.,а
Следовательно, эмпирическая формула С;|Н/(, 7S или С\б.2Н64 4- Если
провести полное гидрирование фракции, то все ароматические ядра
превратятся в нафтеновые. и число атомов водорода в «средней молеку-
ле» фракции увеличится на число атомов углерода, находившихся в аро-
матических ядрах, так как при полном гидрировании каждый углерод-
ный атом ароматического ядра присоединяет один атом водорода.
Предположим, ч то после гидрирования фракция имела следующий
элементный состав: С — 86%. Н — 14%, М = 505,7. Произведя расчет,
найдем, что эмпирическая формула после гидрирования (Cf/H,)() выра-
жается формулой: С 2Н7() 2- Таким образом имеем следующие исход-
ные данные ;ыя расчета:
до гидрирования: Л/ — 500; С16 2Н()4
после гидрирования: М = 505,7; С76 2Н7() 2.
Необходимо определить основные показатели структурно-группо-
вого состава,
Определение С , Ка. Увеличение числа атомов водорода в «средней
молекуле» фракции после гидрирования: 70,2 - 64,4 = 5,8. Следова-
тельно, число ароматических атомов углерода в «средней молекуле»
фракции до гидрирования равно 5,8 (так как при гидрировании каж~
137
ibiii ароматический атом углерода присоединяет один атом водороду
Процентное содержание ароматических атомов углерода:
с =Д1.100 = 16%.
" 36,2
Число ароматических атомов углерода 5.8 соответствует Ка *= 1.
Определение Ко, Кд. После гидрирования в «средней молекуле»
фракции содержатся только нафтеновые кольца и парафиновые цепи,
причем часть нафтеновых колеи образовалась из ароматических.
Обшее число колец Ко в этой «средней молекуле» можно найти на
основании эмпирической формулы гидрированной фракции
Между п и для насыщенных углеводородов существует следующая
зависимость:
т} - 2/7 + 2 - 2К0,
которая означает, что число атомов водорода в молекуле насыщенного
моноциклического углеводорода меньше по сравнению с алканом
с тем же числом атомов углерода на 2 Ко, так как при замыкании угле*
водородной цепи в кольцо теряются 2 атома водорода. То же уравнение
для К() имеет вид:
Для нашего случая:
Таким образом, обшее число колец в «средней молекуле» фракции
Ко = 2,1. Число нафтеновых колеи Кн = Ко - Ка = 2.1 - I = 1,1.
Определение Ск, Сн, Сп, Для того, чтобы установить долю углерода,
приходящуюся на насыщенные кольца Ск, используется зависимость
между содержанием водорода и молекулярной массой для насыщенных
углеводородов различных гомологических рядов. Эта зависимость но-
сит прямолинейный характер в координатах %Н — Л/ (рис. 55).
Для алканов с увеличением молекулярной массы % И уменьшается,
для моноциклических нафтенов %Н остается постоянным вне зависи-
мости от молекулярной массы. Для полициклических нафтенов %Н
увеличивается с увеличением молекулярной массы, причем, чем боль-
ше ядер в молекуле, тем сильней выражена зта зависимость. Это объяс-
няется тем, что содержание водорода падает быстро с увеличением чис-
ла колец, а увеличение молекулярной массы за счет удлинения парафи-
новой цепи приводит к увеличению %Н.
138
Молекулярная масса
Рис. 55. Зависимость %СК от молекулярной массы и содержания водорода
139
График (рис. 55) составлен из предположения, что все полиции
веские углеводороды нефти являются ката-конденсированными
нейного
или ангулярного
строения. Хотя в нефтях содержатся полициклические углеводороды и
другого строения, ката-конденсированные углеводороды условно выб-
раны потому, что они занимают промежуточное положение среди всех
полициклических углеводородов по числу атомов углерода, приходя-
щихся на 1 кольцо:
П с ри- конде нс ирова н ное Н с кон де i ic и роиа н нос строе ние
строение (4 атома С на I кольцо) (6 атомов С на I кольцо)
Ката-конденсированнос строение
(5 атомов С на 1 колыю)
На графике для нашего случая (Н = 14%; М = 505,7) находим: Ск =
29%, Следовательно, Сн = Ск- Са = 29 - 16 = 13% и С„ =100 — 29 = 7^-
Для определения структурно-группового состава прямым методом
необходимо определение элементного состава и проведение гидриро-
вания фракции, что делает этот метод трудоемким. Поэтому разрабо-
тан ряд упрошенных методов определения структурно-группового сос-
тава. Наибольшее распространение имеет метод Тадема или метод
п-р-М,
140
Метод /ьр-Д/i
По этому методу необходимо точно
НИИ <дл>1 вязких фракций р 70> На °Х’елить "о“ Р4М и Мфрак_
.тают структурно-групповой состав с n„J И этих даиных вычис-
ти номограмм. ' ОЩ|>Ю специальных формул
Тадема установил линейную зависимость
мп углерода, составляющих различима У солеРжанием ато-
CJ и константами фракции: стРУктурные группы (Са, С„,
%С —-----г b' Др + с' Дн,
(!)
а также зависимость между числом колец и теми же константами:
К = а' + Ь'МДр + с'МДн, (2)
где Л/ - средняя молекулярная масса; Др - разность между плот-
ностью фракции и плотностью гипотетического алкана в жидком сос-
тоянии с бесконечно большим числом углеродных атомов; Дн — соот-
ветствующая разность для показателя преломления; й, 6, с, а', Ь\ с' —
числовые константы.
Для гипотетического алкана выбраны следующие константы:
п/ = 1,4750; р420 = 0,8510.
В соответствии с формулами (1) и (2) составлены номограммы, с по-
мощью которых можно легко определить структурно-групповой состав
фракции.
Метод д-р-Л/ был исследован на большом количестве различных
высших (фракций и показал близкую сходимость с результатами анали-
за прямым методом.
Прямой метол, а также метол /?-р-Л/ применимы, если во фракции
содержится до 2% S, до 0,5% N и до 0,5% О, Если содержание серы вы-
ше, то в формулы вводят поправку на серу.
Метод //-р-Л7 нельзя применять для высокоароматизированных
фракций, для которых %С;(/%СИ > 1,5 и число ароматических колен
больше 50% от общего числа колец.
В этом случае применяются, кроме прямого метода, модификации
метола л-р-Л/, в частности метод Хезельвуда, В этом методе, кроме л, р
и Д/, необходимо определение так называемого интерцепта плотности:
( нН
Др= p;"-v -О.Н35.
П’анес известен как метол n-d-М.
141
уделения структурно- группой о cocm,
В последнее основанные на нспользо^
получили Рае”^( I гЬопмуле ее «средней мо-
** По cnSw ПМР « ЯМР ' JJ’ctpn ее состава. С помошьюспад-
с„н„, й “рО11еНТНОе С0Д~
атомов углерода ( л> Чй
%С>-М0(),
11 J„
где — интегральная интенсивность сигналов ароматических атомов
углерода в области 120-150 м. д.; Jo — суммарная интегральная интен-
сивность сигналов в спектре.
Количество ароматических атомов углерода в «средней молекуле»
фракции находят по формуле
Са = Ч-
На основании спектра ПМР находят долю ароматических протонов-
где JH, — интегральная интенсивность ароматических протонов в об-
I -- нп Е J pcui ОПЦЛ ri Г I I VI ic-ri |>1 IVO I и ы 1*1 VI J м 1 W IX 11 A I J рм 1 ипио D UU’
ласти i,4-8,3 м. д.; JH — интегральная интенсивность протонов на-
сыщенных структур в области 0,5-4 м. д.
Умножая долю ароматических протонов на число атомов водорода
в «средней молекуле», получают число «ароматических» атомов водо-
рода:
Н = m-h .
d И
Число атомов углерода в насыщенных структурах получают, вычи-
тая из общего числа атомов углерода в «средней молекуле» (/г) число
ароматических атомов углерода (Са):
С.. = л - С...
I it.lL t.l
Число атомов водорода в насыщенных структурах получают, вычи-
тая из общего числа атомов водорода в «средней молекуле» (/и) число
«ароматических» атомов водорода (Н ):
Число алкильных заместителей ароматическо! о ядра (N) в «средней
молекуле» находят по формуле:
1 (лоролникнв В. Д. ЯМР-спсктроскония как метол исследования химическогосос-
гана яеф|си. В кв. Инструментальные методы нсслслования нефти. — Новосибирск
Наука, 19X7 - С 49-67.
142
/V = /л
где доля протонов, находящихся у сх-углеродных атомов боковых
цепей; х отношение водорода к углероду в насыщенных структурах:
Н„«
нас
Число узловых атомов углерода (С;) находят по формуле
Число ароматических колен в «средней молекуле» находят по фор-
муле
Ка
Обшее количество колец в «средней молекуле» (Ко) находят на ос-
новании зависимости между т и /г
т - 2/? + 2 - 2К, - н------
100
Откуда
гу , т
/ф = п 4-1 -т-------------
2V 100 J
Количество нафтеновых колец получают, вычитая из общего числа
колец (/ф) число ароматических колец:
к„ = К„ - ка.
Степень замещения атомов водорода ароматических ядер (о) нахо-
дят по формуле
Н
а
0,5-1,05;
1,05-2;
На основании спектров ПМР можно рассчитать соотношение про-
тонов различного типа в «средней молекуле» фракции по интенсивнос-
ти соответствующих сигналов (шкала 5), м. д.:
протоны СН.
протоны СН,- и СП-групп
протоны а-углсродных атомов
боковых цепей аренов
протоны ароматических ядер
143
Глава III
ГЕТЕРОАТОМНЫЕ СОЕДИНЕНИЯ
И МИНЕРАЛЬНЫЕ ВЕЩЕСТВА НЕФТИ
§ I. Кислородные соединения нефти
Содержание кислорода в нефтях невелико (О, I -2%). В нефтях нахо-
дятся следующие кислородные соединения: нефтяные кислоты и
фенолы. Значительное количество кислорода нефти приходится на
смолы — вещества, которые, кроме С, Н и О, содержат N и S.
Нефтяные кислоты находятся в средних фракциях нефтей, выкипа-
ющих выше 250°С, в количестве нескольких процентов и представляют
собой смесь органических кислот, в которой преобладают алифатичес-
кие и нафтеновые кислоты.
Нафтеновые кислоты — кислоты обшей формулы С/гН2/||СООН,
являются производными нафтеновых углеводородов — циклопентана
и циклогексана, откуда и происходит их название. Установлено, что
нафтеновые кислоты в зависимости от типа могут быть, в основном,
либо производными циклолентана, либо циклогексана, причем кар-
боксильная группа, как правило, удалена от ядра на 1 -5 атомов углеро-
да. Обшии вид формулы, отвечающей структуре моноциклических
нафтеновых кислот:
где п = 1-5.
Алифатические (жирные) кислоты представлены в нефтях как кис-
лотами линейного строения, так и изостроения. в гом числе изопрено-
идного строения.
Ароматические кислоты нефтей являются производными бензола и
полициклических аренов.
Следует отметить, что состав нефтяных кисло! соответствует типу
нефти. Так в нефтях типа А преобладают алифатические кислоты;
в нефтях типа Б — нафтеновые.
В высших фракциях нефти могут находиться кислоты, являющиеся
п рои з вод н ы м и у гл е водороло в с м е ш а н н о го строе н и я.
Сырые нефтяные кислоты, выделенные из нефти, представляют со-
бой темные маслянистые жидкости с неприятным запахом. Они слабо
144
растворимы в воде. Температура застывания нефтяных кислот зависит
от соотношения алифатических и нафтеновых кислот и может быть
очень низкой - около -80’С (в случае, если преобладают в смеси наф-
теновые кислоты).
Химические свойства нефтяных кислот. Нефтяные кислоты могут
быть превращены в сложные эфиры, амиды, галоидан гидриды:
ROH, Н(+)
-(СН2)/;СООН
(CH2)„COOR
(CH2)„CONH2
^—-^(CH^COCl
Соли щелочных металлов этих кислот обладают хорошими моющи-
ми свойствами (мылонафт). Соли меди и марганца растворимы в угле-
водородах, лают ярко окрашенные растворы и применяются в качестве
катализаторов жидкофазного окисления углеводородов. Водный раст-
вор калиевых солен нафтеновых кислот (40%) используется в качестве
ускорителя роста растений.
Рассмотрим выделение, количественное определение и идентифи-
кацию нефтяных кислот. Нефтяные кислоты выделяют из средних
фракций нефти (250-4()0°С). Для этого фракцию обрабатывают 10%-м
водным раствором карбоната натрия. Натриевые соли кислот перехо-
дят в водный раствор, из которого можно выделить свободные нефтя-
ные кислоты, подкисляя раствор минеральной кислотой.
Количественное определение кислот в нефтяных фракциях осущес-
твляется метолом кислотных чисел. Кислотное число — это число мил-
лиграммов КОН, необходимое .для нейтрализации 1 г вещества. Кис-
лотное число определяют титрованием навески нефтепродукта спирто-
вым раствором гидроксила калия потенциометрическим титрованием
или титрованием в присутствии индикаторов.
Изучение состава нефтяных кислот и их идентификацию можно
провод ить дну мя 11 утя ми.
1. Смесь кисло! превращают в метиловые эфиры, а эфиры — в угле-
водороды и о схеме (Н. Д. Зел и н с к и й):
С Н;,ОН. НО
- Н ?О
HsH 4|Н|
RnrC-rOCHj----------
* Na+CjHsOH
СН2—ОН + СН,ОН;
145
’0'3544
PJ7 HsH 21H1
R—CH7OH-------R—СНЛ-J ► R—CH5 + HJ
” ? Zn + HC1
Строение полученных углеводородов точно соответствует строению
исходных нефтяных кислот. Состав этой углеводородной смеси, по
которому судят о составе кислот, может быть установлен обычными ме-
тодами исследования нефтяных фракций. Например, наличие проит
водных циклогексана в этой смеси и, следовательно, наличие цикло-
гексан-карбоновых кислот в нефтяных кислотах можно определить
с помошью реакции каталитического дегидрирования (образование
ароматических углеводородов). Именно таким путем проводил иссле-
дование нафтеновых кислот Н. Д. Зелинский. При изучении кислот,
выделенных из бакинских нефтей, Н. Д. Зелинский показал, что при
дегидрировании углеводородов, полученных из этих кислот, образуют-
ся следы ароматических углеводородов, на основании чего был сделан
вывод, что кислоты бакинских нефтей содержит очень мало шести-
звенных циклов.
2. Проводится ректификация кислот или их метловых эфиров
с получением узких фракций, а затем в каждой фракции идентифици-
руются кислоты химическими или физико-химическими методами.
Для доказательства строения циклической части нафте новой кис-
лоты ее переводят в углеводород по вышеприведенной схеме. Получен-
ный углеводород подвергают дегидрированию в ус товиях реакции Зе-
линского (Pt/C. 300°С). Если образуется ароматический углеводород,
это доказывает то, что молекула нафтеновой кислоты содержит шес-
тичленный цикл; иногда проводят дегидрирование метиловых эфиров
^з т ^з доказано приссзствие в бакинской
нефти циклогексанкарбоновой кислоты (А. Е. Чичипабин):
Для установления строения боковой цепи нафтеновых кислот (оп-
ределение числа атомов углерода между карбоксил ьной группой и ник-
лом) можно использовать ряд методов, которые в настоящее ярем»
имеют методологическое значение. Рассмотрим только один из ник-
метод постепенного расщепления.
146
Метод постепенного расщепления
По этому метол проводят постепенное расщепление боковой цепи
нафтеновой кислоты: в итоге получается кислота, имеющая на один
атом углерода меньше, чем исходная.
Вначале исследуемая кислота подвергается этерификации, а затем
превращается посте.тонательно в спирт, четвертичную аммониевую
соль, олефин и новую кислоту':
СН.ОН гц ।
R-CHrCH;-COOH -------Q*R-CH,-CHrCOOCH( —1 1 »
н Na+C,H5OH
—► r-( нн ,-с 11 ,-он-н- > r-ch,-ch,5chX <ch'>»n
О д
—► R-( l b < 11, С I HN(CH,),Br— i
4
<+) e
—►K-CH -( ti=CH?+ HNfCHOjBr
R-CH2~COOH
+ CO2 + H2O
Если в pc a luiiic лих превращений образуется неизвестная кисло-
та. которую не упился идентифицировать, полученную кислоту под-
вергают тем же превращениям, что и исходную, и так до получения
продукта, который ноллается идентификации.
Кроме кислсн нефти содержат фенолы (особенно много фенолов в
смолистых нефiя\). И j нефтей выделены крезолы (о-, ксилено-
лы. о-этилфснол. ли лилфенолы, [3-нафтол и другие фенолы. Фенолы
можно выдели и» следующим образом: при обработке нефтяной фрак-
ции водным рас 1 вором едкого натра ('10%) в водный раствор перехо-
дят феноляты натрия вместе с солями нефтяных кислот. Из водного
раствора фенолы и кислоты могут быть выделены действием минераль-
ной кислоты. Рм cic.icHMc нефтяных кислот и фенолов можно провести
с помошью Волжин раствора соды, в котором фенолы нс растворяются.
§ 2. Сернистые соединения
Содержание серы в нефтях месторождений России изменяется
Обычно в предсчах 0.05 3%. Сернистыми (более 0,6% серы) являются
нефти Башкортостана и Татарстана, а также Западной Сибири. В ба-
лансе нефтедобычи нашей страны сернистые нефти занимают ведущее
147
10'
Mrcio и составляют около NOfr- Основная масса серы содержится
фракциях топлив и масса (аоОО'с). fl(1
('ера находи юя в нефтях в виде простою вещества, ссроводоро
в opiaiimiccKiix соединениях и смолистых веществах.
(с’/’о кил простое вещество содержится в нефтях в Р^сгворенно
состоянии. При нагревании нефти (в процессе перегонки) сера частиц^
но реагирует е у пае водорода ми (легче с ароматическими):
2RH I 2S -» R-S- R + H2S.
Сероводород содержится в некоторых нефтях в растворенном состо
янищ часто появление его во фракциях нефти является следстииец
термического разрушения сернистых соединеный.
Сера и сероводород вызывают коррозию металлов, кроме того се
роводород очень токсичен.
Основная масса серы входит в состав органических сернистых сое-
динений и в состав смолисто-асфальтеновых веществ. В нефтях найде-
ны меркаптаны R-SH, сульфиды R-S-R, дисульфиды R-S-S-R
производные тиофена, тиофана и гианиклогексана В вастоящее время
насчитывается свыше 200 различных сернистых соединений, найден-
ных и идентифицированных в нефтях.
Тиолы (меркаптаны), содержащие от I до 9 атомов углерода (в общей
сложности более 40), выделены из бензиновых фракций нефтей (вое- '
новном, алифатические). Следует отметить, что содержание меркапта-
новой серы в нефтях составляет 0,1-15% от общего содержания серы
Меркаптаны в бензиновых фракциях нефтей преобладают над другими
сернистыми соединениями. С повышением температуры кипения
фракций их содержание быстро уменьшается.
По своим химическим свойствам меркаптаны напоминаютспирты,
но атом водорода в группе SH более подвижен, поэтому меркаптаны
реагируют легко с основаниями и даже с оксидами металлов, властнос-
ти с оксидом ртути:
2R-.SH + HgO -> (RS)iHg -г Н?О.
Отсюда их название — меркаптаны (тегсшшш captans — связываю-
ший ртуть).
Тиолы извлекают из нефтяных фракций дейеншем водных раство-
ров гидроксида натрия и моноланоламина: в аналитических не.ш
возможно использование солей некоторых металлов (нитрат серебра,
плюмбит натрия).
Меркаптаны, содержащиеся в бензинах, окислением воздухом
в присутствии катализаторов (Си2С12) превращаются в дисульфид
(облагораж ива н ие бштзи _____
! -- J ' г'л II
Окисление меркаптанов азотной КИСЛ()1(). п
лотам: ои 'фиволиг к су.тьфокис-
R-SH R-SO2-OH
При термическом разложении меркаптан-
Под действием водорода при повышенных разрыяает« связь C-S.
присутствии катализаторов происходит ати„пНИ?Х и ««пературах в
5 mrc ШеПДение H2S (гидроочистка,:
R—CH’-CHj—SH-j—CHj__CH>__
I ’ С1Г СНг-------R + H,S
R—CH=CH2 + H2s
R—CH,-CH,
Сульфиды (пштфиры) наиболее pacnrw™,
>,средних фракциях нефти, где они составляют 50-йог бензиновы* и
нистых соединении. Сульфиды нефтей попп->„„ °т Суммы сер’
ческие (диалкн.1сул1,фнды) н алициклические' соле10™ ал11фати’
виикле (тнанпкланыу Последние nneofiv. ' одержашне атом серы
нефти. Из бен зиновых фраки, ? “ срвдни* Фракциях
во более 5(3 ) ZZc " Z"°2 ИДен™*^ирова-
нейтральные вен,ее, „а Однако в ±ЛКИЛСМЬфИДЫ ~
проявляют основные свойства, благодаря тому чтоатоХ КИСЛ°Т °Н"
свободные электронные пары: ’ серы содержит
R S R
* »
На этом свойстве сульфидов основан промышленный способ иг
экстракции из нефтяных фракций; способ их
R S R (- н ()SOj ц—-----f R— s— [q QSO3 Н
Н
Судьфидьт легко ОКИСТЯЮТСЯ ГН О ЫЮГЧ У ет
сульфоксидов „сульфонов 2 2’ ’ HN0,) С 06paJ0'>aH„eM
О
В аналитических целях для удаления сульфидов из фракций нефти
149
148
используют их способность образовывать комплексы с различнымиак
ней торам 11 эл ектронов:
BF> Hg(NO3)2. А1С13, Hg(OOCCH3)2. TiCh, SnCI2, AgNO3
R
/S: AICI3, /S^AgNCh
R R
Эти комплексы можно разложить водным раствором аммиака и вы-
делить сульфиды.
Термическое разложение сульфидов приводит к образованию серо-
водорода и углеводородов:
СНз-СН?-5-СН,-СН, H.S + 2СН-)=СН-).
Дисульфиды R-S-.S-R' находятся в нефтях в небольшом количестве
во фракциях до 300°С. На них приходится 1-15% всей серы. Восста-
новление дисульфидов водородом в момент выделения (Zn + уксусная
кислота) приводит к образованию меркаптанов:
Н Н
R-sIS-R
2[Н]
—R-SH -ь R’-SH
Тиацикланы. Это соединения, молекулы которых содержат пяти-и
шестичленные никлы с атомом серы в цикле, причем пятичленные ти-
аникланы (тиофаны) преобладают нал шестичлснпыми (тианиклогек-
саны). Обычно тиапикланы содержат алкильные заместители и кон-
денсированные нафтеновые и ароматические кольпа. Например:
Н2С-СН—R
/ \
Н2С сн2
" S
FFG .СН—R?
ди ал к ил г и а п и кл о гс кс а н
алкилбен пи о», рал
алкилтиофан
алкилтиалснталан
Тиофены — это гетероциклические сернистые соединения, произ-
водные тиофена:
150
Так же как и в случае тиофанов, молекулы тиофенов, найденных
в нефтях, содержат алкильные группы и конденсированные нафтено-
вые и ароматические кольца:
ал к ил бен п иофен
циклогекса нобензтиофен
в нефтях найдено более 20 гетероциклических сернистых соедине-
ний — это, в основном, ал к ил производные тиофана, а также в неболь-
шом количестве ал кил производные тиофена и тиациклогексана.
Электронодонорные свойства серы в тиофанах ниже, чем в алифа-
тических сульфидах. Еще в меньшей мере эти свойства проявляются
в алкилтиофенах, что объясняется сопряжением свободных электрон-
ных пар атома серы с л-электронами двойных связей. Поэтому тиофе-
ны и тиофан ы труднее образуют комплексы с акцепторами электронов,
чем диалкил сульф иды. Также трудно образуют комплексы арил- и диа-
рнлеульфиды. благодаря этому можно с помощью таких акцепторов
электронов как азотнокислая ртуть или азотнокислое серебро отделить
алифатические сульфиды от ароматических и гетероциклических сер-
нистых соединений!, так как комплексы образуются только с алифати-
ческими сульфидами.
В керосиновых и масляных фракциях нефти содержатся сернистые
соединения полициклического строения. Выделение этих соединений
в чистом виде представляет собой сложную задачу. Их выделение воз-
можно в концентрированном виде вместе с полициклическими арома-
тическими углеводородам и. Исследование таких концентратов позво-
лило предположи гь. что сернистые соединения высших фракций нефти
представляют собой производные бензтиофена, бензтиофана, дибенз-
тиофена и других гетероциклических сернистых соединений, содержа-
щих 3-5 ароматических и нафтеновых колец.
В 1987 г. в запалjiо-сибирских нефтях методом масс-спектрометрии
отрицательных ионов обнаружены сульфоксиды и сульфоны'. О масс-
спектрометрии отрицательных ионов см. гл= 1 §2.
Присутствие сернистых соединений в нефтепродуктах и в сырье не-
которых процессов переработки нефти крайне нежелательно. Актив-
ные сернистые соединения (S, H?S, HSR) вызывают коррозию метал-
лов. Сернистые соединения, находящиеся в топливах, при сгорании
образуют диоксид серы, вызывающий коррозию двигателей. Даже нич-
тожные их примеси в сырье для платформинга вызывают отравление
платинового катализатора. Удаление серы из нефтяных продуктов про-
ППмаковВ С.. Ляпина К К. и пр. // ДАН СССР. - 1987. - Т 296. - № 3. - С. 619.
151
водится с помошью гидроочистки, которая состоит в том, что нефтя-
ной продукт подвергается действию водорода при 300—450°С и 1,7-
7 МПа над катализаторами, состоящими из сульфидов и оксидов ме-
таллов переменной валентности. При этом сера, входящая всоставсер-
нисты.х соединений, превращается в сероводород, который удаляется
с газами:
Н3с—СН R
/ \ 41 ИД- HjC-CHi-CH-CH, + H2S
По способности к гидродесульфированию сернистые соединения
можно расположить в следующий ряд:
дисульфиды > тиолы > сульфиды > тиофаны > тиофены
Количественное определение серы в нефтяных фракциях проводят
путем сожжения определенной навески вешества. При сгорании про-
дукта сера превращается в диоксид серы, который улавливается с по-
мощью титрованного раствора Na2CO; (0,05N или 0.1 N). После сож-
жения титруют раствор соды с помошью 0.05N или 0JN растворов
НС1.
Параллельно ставят холостой опыт сожжения продукта, не содержа-
щего серы. Расчет ведут по формуле:
где И() — объем НСI в холостом опыте, — объем HCI ;ьтя основного
опыта; /V— нормальность раствора НС I; 16 — грамм-эквивалент серы;
Р — навеска, г.
Для легких фракций хорошие результаты ласт анализ путем сожже-
ния в лампе со специальным фитилем, Однако, лаже в случае легких
фракций, анализ осложняется тем, что происходит неполное сгорание
нефтепродукта, фитиль покрывается углистыми отложениями, В нас-
тоящее время широко применяется наиболее удачный вариант лампо-
вого метода, который называегся «пиролитическим ламповым мето-
дом», По этому методу навеску анализируемого продукта подвергают
сильному нагреву в кварцевой ампуле (рис. 56), пары вещества и про-
дуктов разложения поступают в пламя лиоксановой горелки1, где про-
исходит их полное сгорание. Твердые продукты пиролиза, оставшиеся
в ампуле, окислякн кислородом, который вводят в ампулм с помошью
капилляра. Продукты полного окисления навески (СО7. Н7О, 502)под
'В модификации лого метола (прибор Н П. Волынского. ИЯ.ХС им. А В. Топчиева)
вместо диоксаноиой горелки используют тазовую микрогорелку.
152
Рис., 56. Принципиальная схема прибора для пиролитического
лампового определения серы
действием водоструйного насоса из зоны сгорания током воздуха увле-
каются в абсорбер с титрованным раствором соды, где происходит их
поглощение. В холостом опыте проводят сжигание диоксана в течение
того же времени, что н во время основного опыта. В холостом опыте
учитывается влияние углекислого газа, образовавшегося, главным об-
разом. при сгорании диоксана, на титр раствора соды.
Одним из эффскзивных физико-химических методов количествен-
ного определения серы является метод рентгено-флюоресцентного
анализа. Этот метол основан на зависимости наблюдаемой интенсив-
ности спектра флюоресценции серы (под действием рентгеновских лу-
чей) от содержания серы в анализируемом образце1.
Для количественною определения отдельных групп сернистых сое-
динений применяют, наряду с пиролитическим ламповым методом и
химическими методами, физико-химические методы анализа.
I. Сероводород определяют по разности двух количественных опре-
делений серы ло и после его удаления из нефтепродукта с помощью
хлористого кадмия:
H.S +CdCl2->CdS + 2HCI.
2. Свободная сера может быть определена по разности количествен-
ных определений ;ю и после ее удаления из нефтепродукта с помошью
металлической ртути:
Hg з S —> HgS.
41 В Борзунов
в нефlenpo.iyx гач В
бирск- Наука. 19X7
Г М Великова. О выборе оптимального метола определения серы
кп Инструментальные методы исследования нефти - Поноси-
сь I
153
3, Содержание меркаптановой серы можно определить по разности
двух определении серы до и после удаления меркаптанов плюмбитом
натрия:
СНЮН
R-S-H + Na2PbO2 ------►(R-SbPb + 2NaOH
Другим способом определения меркаптанов является амперметра
веское титрование меркаптанов водным раствором AgNO3. Меркапта-
ны могут быть определены также потенциометр и ясским титрованием
азотнокислым аммиакатом серебра Ag(NH ЬтЬОк
RS- + Ag^--► R-S-Ag
4. Сульфиды определяют потенциометрическим титрованием навес-
ки нефтепродукта раствором KJO3 в 90%-ной уксусной кислоте, При
этом происходит окисление сульфидов в сульфоксиды:
2 R-S-R+ KJO3 + 2НС1 —► 2 R-S-R - KG I L JC1 + Н2О
I
О
Предварительно из нефтепродукта необходимо удалить S и Н2$, :
Меркаптаны можно не удалять, но при этом в результатах определения
делают поправку, так как меркаптаны окисляются в дисульфиды. Ди-
сульфиды в этих условиях дальше не окисляются.
5. Дисульфиды определяют после всех компонентов активной серы
(RSH. S, H^S) и после удаления S и H0S. Вначале проводят восстанов-
ление дисульфидов, действуя на навеску активным водородом:
Zn + CHKOOH
R-S-f-S-R' + 2[Н] ----► RSH - R'SH
Затем определяют образовавшиеся меркаптаны и рассчитывают со-
держание дисульфидной серы;
и сульф И Л "" ‘S “
где и S2 — содержание меркаптановой серы до и после восстанов-
лен ия,
6. Остаточную серу определи ют по разности между обшим содержа-
нием серы в нефтепродукте и суммарным содержанием серы всех ос-
тальных групп сернистых соединений. В остаточную серу входит сера
малоактивных ароматических сульфидов, тиофенов и тиофанов, а так-
же с ул Ьф О КС И Д Ы И Сул ьф о н ы.
1 С d
§ 3. Азотистые соединения
С одержи н 11 с и зоти в н ефтях епгт^ »
(Обычно ло 0.3 >. но в отдельных случае мОже“™ ™'о I Т д"™
При перегонке эти „е.цества X “ НеФЖ
тых соединении, которые таких, путем 110падаЮт,, нефтяные фра^ии’
Интересно отметить что. видимо, этим и объясняете поп и X
держания азота в нефтяных фракциях по мере увеличения их 'Хера-
туры кипения. А ин истые соединения нефтей подразделяются на две
основные , руины; азотистые основания и .нейтральные» (слабооенов-
ные) соединения 4мп1/с1иые основания. находящиеся в низших и сред-
них фракциях нефтей, являются, в основном, алкильными или uhiLo-
алкильными производными пиридина и хинолина:
пиридин хинолин
В высших фракциях идентифицированы бензохннолины и бензоак-
ридины, моле куя 1,1 которых содержат несколько конденсированных
бензольных иди нафтеновых циклов. Например:
.м-беню.тано.шн 1,2-бсн юакрилин
Содержание азотистых основании в нефтях может составлять
20-40%. от обшею количества азотистых соединений.
Нейтральные (слабоосновныс) азотистые соединения, иденшфипи-
рованные в нефтях. пол разделяются на:
а) производные индола и карбазола
карбазол
155
l'ii юржаnine алкильные заместители в бензольных ядрах и у атома азо
la. а также конденсированные с бензольными или нафтеновыми инк
. тми. Например:
б) циклические амиды (лактамы) ароматических о-аминокислот;
R R
Н Н
в) порфирины — соединения, содержащие пиррольные фрагменты.
Порфирины имеют структуру, аналогичную структуре гемина (крася-
щее вещество крови) и хлорофилла (формула 1), но их молекулы не со-
держат комплексно-связанного железа (в случае гемина) и магния
(в случае хлорофилла). Вместо этих металлов в структуре порфиринов
содержатся комплексно-связанные никель и ванадий (последний в ви-
де ванадила VO2+) (см. формулу 2), Наличие порфиринов свидетель-
ствует в пользу теории органического происхождения нефти. Такие
сложные соединения, как порфирины, не могли образоваться в резуль-
тате минерального синтеза. Очевидно, они попали в нефть из органи-
ческого вешества живых организмов.
156
г) полапентидные фрагменты белковых молекул, содержащиеся в ас-
фальтеновых и порфириновых концентратах, В результате кислотного
гидролиза этих концентратов в продуктах гидролиза обнаружен ряд
аминокислот.
В нефтях обнаружены также гетероциклические соединения, содер-
жащие кроме азота серу (тиазолы).
Для количественного определения азота в нефтепродуктах обычно
используют различные модификации метода Кьельдаля. Навеску неф-
тяной фракции обрабатывают избытком концентрированной серной
кислоты, и все азотистые соединения переходят в сернокислотный
слой, который кипятят в присутствии катализатора. При этом весь азот
связывается в сульфат аммония. Раствор нагревают со шелочыо, выде-
ляющийся аммиак поглощается титрованным раствором серной кис-
лоты, избыток которой оттитровывают щелочью, Содержание основ-
ного азота определяют методом потенциометрического титрования
0.1N раствором хлорной кислоты (НС1О4) в смешанном растворителе
(уксусная кислота — бензол = 1:1).
Азотистые основания извлекают из нефтей или нефтяных фракций
с помошью разбавленной хлороводородной или серной кислот в виде
иодных растворов солей: растворы обрабатывают щелочами, при этом
азотистые основания выделяются в свободном виде. Более полное вы-
деление азотистых оснований достигается при использовании крупно-
пористых катионитов (КУ-23) в присутствии полярных растворителей
(уксусный ангидрид, ди метил формам ид). Нефтяную фракцию с раст-
ворителем многократно пропускают через слой катионита. Азотистые
основания сорбируются на его поверхности. Десорбцию азотистых ос-
нований с поверхности катионита проводят с помошью спиртового
раствора аммиака или едкого натра.
Нейтральные азотистые соединения извлекают хлорным железом,
которое образует комплексные соединения с нейтральными азотисты-
ми веществами:
Выделенные комплексы разлагают щелочами. При этом азотистые
соединения выделяются в свободном виде.
Полученные азотистые основания или нейтральные соединения
подвергают ректификации для выделения либо индивидуальных сое
157
;кпении. пин) копнен гразов. Далее следует идентификация азотистых
от hi нении фи шко-химическими методами (масс-спектрометрия
Ilk , >Ф-ci 1скгроскопия). В последние 10-15 лез для идентификации
а Д) 1 нс i ы х сое шнений них смесях успешно испод ь метод хрома-
ю-масс спектрометрии Иногда используют химические методы иден-
। ификаиии ( пои целью получают кристаллические производные -
пикраты, хлористоводородные соли, хлорплатинагы. .фиг идентифика-
ции а югистых соединений нейтрального характера используют восста-
понленпе алюмо гидридом лития (1лА1НД. При лом амиды и нитрилы
превращаются в амины. Гетероциклические соединения не подверга-
ются изменениям.
§4. Смолисто-асфальтеновые вещества (САВ)
После отгонки легких, средних и масляных фракций нефти в остат-
ке получают гудрон — сложную смесь тяжелых углеводородов и САВ
высокой молекулярной массы. Количество этих веществ в высокосмо-
листых нефтях может достигать 40%. В высококонцентрированном ви-
де САВ находятся в природе в виде природных битумов. Для выделения
САВ навеску нефти разбавляют 20-крат ным кол идее том легкого алка-
на (пентан, изопентан, гексан). При стоянии из раствора осаждается
нерастворимая часть — асфальтены, которые отделяют фильтрованием
и взвешивают. Для выделения и определения асфальтенов в твердых
нефтепродуктах (битумы, гудроны) навеску растворяю! в бензоле, аза-
тем раствор разбавляют избытком изопентана и фильтруют. Фильтрат
(раствор остальных смолистых веществ п углеводородов) пропускают
через слой адсорбента (силикагеля), на котором адсорбируется остав-
шиеся нефтяные смолы вместе с углеводородами (маслами). Масла,
которые адсорбируются значительно слабее чем смолы, легко вымыва-
ются с поверхности силикагеля пентаном, изопентаном или петролей-
ным зфиром Нефтяные смолы вытесняют с поверхности адсорбента
спирто-бел зольной смесью. Их получают в чистом в иле отгонкой раст-
ворителя. Таким образом, можно разделить и количественно опреде-
лить асфальтены, масла и смолы Нефтяные смолы состоят из нейт-
ральных смол и асфальтогеновых кислот. Асфалыогеноныс кислоты от-
личаются от нейтральных смол не только своими кислотными свой-
ствами. но и лучшей растворимостью в органических растворителях
< например, в спирте), На этом основано их отделение от нейтральных
с мол
Центральные смолы — густые вязкие вещества бурого цвета. Их
ш 'ы и, 1J и молекулярная масса в пределах 600 - 700. Что представ-
ило! собой эти вещества по химической природе? Основными струк-
турными единицами нейтральных смол являются конденсированные
кольчатые системы, связанные между собой алифатическими пеночка-
ми и состоящие из ароматических (преимущественно) нафтеновых и
гетероциклических колене алкильными боковыми цепями:
Нейтральные смолы растворимы во многих растворителях, в том
числе и в алканах.
Нейтральные смолы пол влиянием нагрева, освещения, под
действием кислот легко подвергаются химическим превращениям, уп-
лотняются и превращаются в асфальтены.
Асфальтены твердые высокоплавкие хрупкие вещества черного
цвета, не растворимые в алканах, но растворимые в ароматических уг-
леводородах и ipyinx растворителях. Молекулярная масса равна
2000-3000. И ногда выше — до 6000. Если растворы асфальтенов нагре-
вать или освещать, то они подвергаются конденсации и превращаются
в карбены и карбо иды, нерастворимые продукты еше большей молеку-
лярной массы. Молекулы асфальтенов можно рассматривать как про-
дукты конденсации нескольких молекул нейтральных смол.
Между молекулами САВ существуют межмолекулярные взаимодей-
ствия (ММВ), 1з результате образования водородных связей, слабых до-
норно-акпеп горных связей, л-л- и р-л-сопряжения, диполь-диполь-
ного взаимодействия и сил Ван-дер-Ваальса. Наиболее сильно эти вза-
имодействия проявляются в асфальтенах. Вследствие ММВ асфальте-
ны обычно находятся в нефти в форме коллоидных частиц-полиассо-
циатов (мипелл), образующих «ядро», сольватированное полярными
молекулами нефтяных смол, (сольватный слой). Такая частица называ-
ется сложной структурной единицей (ССЕ) нефтяной дисперсной сис-
темы (нефть, тяжелые фракции нефти, нефтяные остатки) и находится
вереде углеводородов (дисперсионная среда), а совокупность ССЕ на-
зывается дисперсной фсиой.
Будучи увеличены электронным микроскопом, ССЕ в сильно
разбавленных растворах нефтяных дисперсных систем представляют
собой округлые частицы овальной формы, размером от 20—30 до 106 ,
чаще всего 6-300 нм.
Рентгеноструктурным анализом установлено, что большинство ас-
фальтенов. выделенных из нефти, имеют' слоистое строение и состоят
из 5—6 почти плоских «пластин», образующих «пачки». Каждая «плас-
159
тина» представляет собой систему из конденсированных ароматически
колец (3-6). нафтеновых и гетероциклических колец., обрамленную а.ъ
кильными боковыми цепочками, короткими и длинными, нормально-
го или изостроения. Диаметр пластины (ее ароматической части) 9-15
, расстояние между пластинами 3.5—3,7 . Общая толщина «пачки*
16-20 , Пластины связаны в «пачке» углеводородными или гетероа-
томными цепочками, возможно также л-тг-взаимодейезвие между аро-
магическими фрагментами соседних пластин. Ароматические поликон-
денс крова иные фрагменты пластин, содержат, по-видимому; неболь-
шое число ароматических колец. На это указывает относительно не-
большая величина Л, — 260 нм, соответствующая максимуму в Уф.
спектре многих асфальтенов. Причем с увеличен нем молекулярной
массы асфальтенов не наблюдается батохромнын сдвиг максимума при
X = 260 нм. что свидетельствует о том. что с увеличением молекулярной
массы асфальтенов не происходит увеличение пол и конденсированных
ароматических фрагментов.
Методом ЭПР установлено, что асфальтены содержат от 10|8до IO]|J
на грамм парамагнитных центров (стабильных радикалов), и это число
увеличивается с увеличением степени ароматичности асфальтенов.
Другая точка зрения на природу асфальтенов высказана в работе1,
согласно которой пачечная структура асфальтенов под вергается сомне-
нию и высказывается предположение, что наряду с пачечной кристал-
лической структурой (доля которой невелика) основная масса асфаль-
тенов представлена углеродными структурами, имеющими линейно-
полимерную форму.
Легкость химических превращений асфальтенов можно объяснить,
в частности, наличием парамагнитных центров.
Л сфад ьтогеновые кисдоты — это те м н ы е вязкие вещества, ра створи -
мые в щелочах, в спирте. Их молекулы содержат несколько гидрок-
сильных и карбоксильных групп. В остальном их структура близка
структуре нейтральных смол.
Смолистые вещества, содержащиеся в нефтяных продуктах (нап-
ример, в маслах), ухудшают их свойства, повышают склонность масел
к окислению и осадкообразованию. Пот тому тля получения товарные
масел необходимо удаление этих вещест в и з масляной фракций, что
достигается различными методами очистки масел с помощью селек-
тивных растворителей или адсорбентов. Остатки от перегонки (мазут,
гудрон), а также крек ин г-остатки служат сырьем для получения иску-
сстве иных битумов. Битумы находят широкое применение в про-
мышленности (строительная промышленность. дорожное строитель-
ство, производство бетонов, асфальтов, для приготовления лаков).
Г, Андреева Л, И Фунламеп 1а.и>ныс аспекты химии нс(|ли Приролзсма1
и асфальтенов — Новосибирск. Наука. 1995 192 с
160
Качество би ту мои, предназначенных, в частности, для дорожного
строит ел ьс гва, записи! oi содержания в них различных смолистых ве-
ществ. Так, асфальтены придают битумам твердость, повышают их
температуру размягчения. а нейтральные смолы и аофальтогсновые
кислоты обеспечивают эластичность битумов и повышают их проч-
ность.
§5. Минеральные компоненты нефти
К минеральным компонентам нефти относятся вода, соли, раство-
ренные в ней (соли шелочных и шелочпоземельных металлов: NaCl,
СаС12. MgCl2, Na,SO4 и т. д.), сероводород и его соли. Кроме того, в
нефти содержатся различные металлы в составе солей органических
кислот (нафтеновых, жирных, асфальтогеновых), а также в составе
комплексных соединении. При полном сгорании образца нефти или
остатка от перегонки нефти все металлы и некоторые неметаллы в ви-
де оксидов и солей переходят в золу. При сгорании часть некоторых
цементов (В, Си.. V. Ni и др.) в виде летучих соединений теряется. По-
лому озоление необходимо проводить в специальных герметических
сосудах. Содержание золы в нефти очень мало и составляет сотые или
десятые доли процента. Спектральным анализ элементов, входящих в
состав золы, позволил расположить эти элементы в порядке уменьше-
ния их содержания в следующий ряд:
S, О. N. Р, V. К, Ni, Si, Са, Fe. Mg, Na, Al, Mn,
Ph, Ag, Au, Cu, U, Sn, As,
Всего в нефтях найдено более 50 элементов, 15-20% от массы золы
составляют щелочные и щелочноземельные металлы (в нефти содер-
жатся в виде солей нефтяных кислот, фенолов, входят в состав асфаль-
тенов). Элементы подгруппы мели (Си, Ag) концентрируются, в основ-
ном, в остатке (в асфальтенах и смолах). Металлы подгруппы цинка
(Zn, Cd. Hg) содержатся, главным образом, в высококипяших фракци-
ях и остатках в виде металл о порфиринов и солей нефтяных кислот.
Элементы III группы (В, Al. Са, Jn. Т1) присутствуют в нефтях в виде
комплексов с фенолами и кислотами. Такие элементы IV группы, как
кремний, свинец, олово и элементы V группы — фосфор, мышьяк,
сурьма, висмут, обнаружены во фракциях нефти в виде элементоорга-
нических соединений. Из элементов V подгруппы в нефтях найден
только ванадий в виде ванадия-порфиринов (летучие соединения) и
в составе комплексов с асфальтенами и смолами (с участием атомов N,
S. О), В нефтях содержатся галогены Cl, Br, J; фтор не обнаружен. Из
элементов VIII группы найдены Fe, Со, Ni в виде комплексных соеди-
И -3544
161
пенни с асфальтенами и смолами, из радиоактивных элементов —
и радий (в виде комплексов с порфиринами)1
Интересно отметить, что ванадий и никель, являясь микроэлемен-
тами в земной коре, по содержанию в нефтях занимают первое место
среди металлов. В золе некоторых нефтей содержание никеля может
доходить до 36. а ванадия — до 60%. Причем ванадий содержится пре-
имущественно в золе сернистых и смолистых нефтей. Для определения
микроэлементов нефтей используют обычно атомно-абсорбционную
спектроскопию, эмиссионную спектроскопию и нейтронный актива-
ционный ан tin из.
В случае атомно-абсорбционной спектроскопии (.ДАС) исследуется
спектр поглощения специально подготовленного образна нефтепродук-
та, который подвергают высокотемпературному воздействию, напри-
мер вводят в пламя (пламенная ААС) или в специальную камеру с высо-
кой температурой — так называемый атомизатор (беспламенная ААС).
Через пары образца пропускают соответствующий монохроматический
свет, который поглотают только атомы анализируемого элемента. По
интенсивности полосы поглощения с помощью калибровочного графи-
ка рассчитывают процентное содержание элемента (см, гл. 1 §2).
Эмиссионная спектроскопия (ЭС) основана на исследовании
спектров испускания атомов образца, введенных в зону высоких темпе-
ратур (например, вольтова дуга). Этот вид спектроскопии применяют
для определения элементов, имеющих низкие энергии возбуждения
(Li, Na, К, Sr, Ва, В, Al, R S и др.). Элементы, имеющие высокую энер-
гию возбуждения (Be, Mg, Со, Pt, Zn, Cd, Ag. Au, Hg, Pb и др.), анали-
зируют методом атомно-абсорбционной спектроскопии2.
Принцип нейтронного активационного анализа заключается в сле-
дующем3. Образец облучают нейтронами с так называемыми тепловы-
ми скоростями. Нейтрон захватывается ядром атома, при этом образу-
ется радиоактивный изотоп того же элемента, По периоду полураспада
изотопа можно определить элемент. По интенсивности у-излучения
при распаде изотопа определяют его количество.
В-И Ц Неф, схимпя. >979. - 19. - №5.
разделение и измерение Ки 2 уСте|,с 'Ха,1сс Лж Хифп.е Т. Химическ®
ко-химические ~ С •' ’’•'бое В. Д. Фи»
ГАНГ. 1996. — С. XX У -1С,»оюролов и других компонентов нефти. - М-
Они, I. В. Инс 1 рументальные методы антпнч м г с 711
41,1 dHcLHUd. — М . Госагомизаат, (963 — С/п
162
Глава IV
ТЕРМИЧЕСКИЕ И КАТАЛИТИЧЕСКИЕ
ПРЕВРАЩЕНИЯ УГЛЕВОДОРОДОВ
И ДРУГИХ КОМПОНЕНТОВ НЕФТИ И ГАЗА
§1. Некоторые аспекты физической химии
углеводородов
В этой главе бу-лу [ рассмотрены зависимости некоторых характе-
ристик связей в мол деводородов (в частности, энергии диссо-
циации) от электронно!! структуры связей, типы разрыва связей с об-
разованием нестабильных углеводородных частиц (ионов и радика-
лов), а также термодинамические аспекты термических превращений
углеводородов.
1.1. Основные характеристики связей
в молекулах углеводородов.
Типы разрыва связей
В молекулах углеводородов связи между атомами С и Н осуществля-
ются с помощью а и к-электронов. В молекулах алканов углерод нахо-
дится в первом валена ном состоянии (з/Д-гибридизация), т, е. в образо-
вании каждой вален тности атома углерода участвуют орбитали одного
з-электрона и трех р- электронов.
В этиленовых углеводородах атомы углерода, связанные двойной
связью, находя гея во в тором валентном состоянии (зр2-гибридизация).
Только два р и один з-валентные электроны каждого атома углерода
при двойной связи участвуют в образовании трех гибридных о-связей,
а один р-электрон ос тается свободным, и он участвует в образовании л-
связи.
В ацетилене атом углерода находится в третьем валентном состоя-
нии (.^-гибридизация), т. с. только один р- и один 5-электрон участву-
ют в гибридизации, в образовании о-связей. два р-электрона участвуют
^образовании двух тг-связей.
Доля з-электро! того облака в орбитале при з/Д-гибридизации равна
при .sp2-гибрид и ищи и — 33%, а при sp- гибридизации — 50%.
Так как 5-электроны находятся ближе к ядру, чем р-электроны, уве-
личение доли з-электронного облака в гибридизованной орбитале ато-
углерода повышает способность последнего притягивать электро-
е. повышает его электроотрицательность:
и-
163
Электроотрицательность
Углеводород % s-облака атома умерода (эВ)
Этан 25
Этилен 33,3 2,62
Ацетилен 50.0 2.75
Вследствие различной электроотрицательности атомов углерода
различаются характеристики связей в молекулах углеводородов.
Основные характеристики связей — это длина связи (измеряется
в нм1 или ), дипольный момент связи (измеряется в дебаях2), энергия
диссоциации связи (выражается в кДж/моль). В табл. 15 приводятся
значения этих характеристик для этана, этилена и ацетилена.
Характеристики связей зависят не только от валентного состояния
атома углерода, но и от взаимного влияния атомов в молекуле углево-
дорода (индукционный эффект, а также эффект сопряжения в случае
не нас ы шен н ых у гл е вол ородов).
Так, например, в молекулах изопентана характеристики связей
С-С, так же как и С-Н, различны (различны длины, дипольные мо-
менты, энергии диссоциации связей), хотя различия эти и малы:
Сц
Электроотрицательность атомов углерода падает в ряду: Cj > С2>
С33. а энергии диссоциации связей С-Н возрастают' С-.-Н < С2-Н<
С-Н,
В молекуле бутадиена имеется сопряжение между л -электронами
двойных связей. Это приводит к укорочению простой свя зи между атома-
ми 2 и 3 и некоторому удлинению двойных связей по сравнению со стан-
дартными значениями:
Таблица 15. Характеристики связей в молекулах этана,
этилена и ацетилена
Углеводород Энергия лис связи С-Нт кДж/моль Длина сияш. им Дипольный момент С-Н, дебаи
С-Н С - С
Этан 4Ю 0,1094 0.1540 0.307
Этилен 435 0.1084 0.(332 0,629
Ацетилен 473 0,1064 0.1201 1.05
Тнм = !0-ум
2Дебай равен IO~IS зл статич ед. чем
’С], С,. С, — соответственно первичный, вторичный и третичный атомы углерода.
I64
2
0.1337 нм 0,1483 нм
Ьс (этан) = 0.154 нм
(этилен) = 0,1332 нм
Связь между атомами 2 и 3 характеризуется определенной крат-
ностью, т. е. является частично двойной связью,
В молекулах бензола кратность связи С-С равна 1,5, поэтому эта
связь является очень прочной и ее полный разрыв не происходит даже
при температурах пиролиза 700-900°С.
Связи в молекулах углеводородов могут разрываться гомолитически
п гетерол итически.
Гомолитический разрыв связей приводит к образованию свободных
радикалов. Связь ра зрывается так, что у каждого осколка молекулы ос-
тается по одному электрону:
CH,.|-CH,Z^=±CHj + Гн,
сн,-си4н^=^сн,-сн2 + н
Гомолитический разрыв происходит чаще всего при термических
пре лра ше н и я х у । л е вол t )pi ж о j з.
Гетерол итичсск и й разрыв приводит к образованию ионов. Связь
разрывается так, что пара электронов связи педиком переходит к одно-
му из атомов, образующих связь:
> ______- ©
С!1:-|н^-------(СН4) + СН3
карбанион карбкатион
> _______©
( Н ( Н -----------(СНй-СН-, + енг
карбкатион гидридиои
СИ. СН*-|н "(СНз-СН))- + Н®
карбанион протон
Углеводородные радикалы, ионы (карбкатионы, карбанионы), атом
водорода (Н ). i идридион (Н:) и протон (Н *) являются промежуточ-
ными частицами в процессах термокаталитических превращений угле-
водородов, Они обладают высокой реакционной способностью. Гете-
ролитический разрыв связей происходит только при каталитических
превращениях углеводородов. Кроме полного разрыва связей может
происходить частичный их разрыв (отрыв одного электрона или силь-
ная поляризация свя зи) под действием активного центра катализатора
(акцептора электронов — А):
165
Гомолитический разрыв связи требует меньше 'энергии, чем гетеро-
литический, так как в последнем случае необходимо затратить энергию
на преодоление электростатического взаимодействия ионов. Еще мень-
шая энергия требуется для отрыва одного электрона (олноэлектронный
перенос). Впервые концепция одноэлектронного переноса при гетеро-
литических реакциях была высказана советским ученым О, Ю. Охло-
быстиным1. В настоящее время установлена значительная роль одноэ-
лектронного переноса в каталитических превращениях углеводородов3.
Важной характеристикой связи является энергия, необходимая
для гомолитического ее разрыва, называемая энергией диссоциации
связи.
Следует отличать понятие энергии связи (/:’) от понятия энергии
диссоциации связи (D ) дл я сложных м ол е ку л (' б о л с е ч е м д ву хатом-
ных).
Так, например, энергия разрыва связи НО-Н в молекуле Н2О,т.е.
тепловой эффект реакции Н?О = НО’ + Н’, составляет 495 кДж/моль,
в то время как средняя энергия О-Н в молекуле Н2О. равная половине
теплоты ее атомизации, т. е. половине теплового эффекта реакции
Н,О= Н + Н + Q.
составляет 460 кДж/моль.
Другой пример. Энергия разрыва одной из связей С—Н в молекуле
СН4 (СН4 = СН3 + Н) равна 428 кДж/моль, средняя же энергия связей
С-Н в СН4 равна 415 кДж/моль. Несовпадение mcpi ни разрыва связи
С—Н и ее средней энергии объясняется тем, что при диссоциации свя-
зи С—Н молекулы метана происходит изменение геометрической кон-
фигурации системы и валентного состояния атома упорола. Молекула
метана — тетраэдр, угол между валентностями составляет 109°28\ Ме-
тил — плоская частица. Угол между валентностями равен 120°.
Чтобы перейти от тетраэдрической молекулы СН4 гибридиза-
ция атома углерода) к плоской структуре мстила (т/г3-гибридизация
Билсвич К. А., Охлобыстин О. Ю. // Успехи химии - 196S - Г 37 - С. 21Ы>
Охлобыстин O.IO. Перенос электронов в иронических реакциях Ростов-на Дону
Изл. Ростов, ун-та. 1974.
" Виншепкая М. В., Романовский Б. В // Жури, физ хим. 1997. - Т 67. - № 5. '
166
Рис. 57
атома углерода), необходимо затра-
тить энергию, поэтому знергия дис-
социации связи С-Н больше сред-
ней энергии этой связи1.
Энергия диссоциации связи рав-
на энергии активации распада мо-
НМ реакция соединения радикалов идет с нулевой ' °6раТ"
шш. Поэтому для экспериментального определения , “ ргиеи акти|,а‘
аши связи С-С необходимо знать пвнеиТ Р™ диссоии-
распада молекулы по этой связи от температурь? К°НСТанТЫ СК0Р°сти
-D
К~А-екг,
где Л — частота колебаний разрываемой связи, 1013Сч.
Тоже уравнение в логарифмической форме имеет вид:
D
in К = In К{}
RT'
Построив эту зависимость графически (рис 57), определяют тангенс
угла наклона прямой и вычисляют D.
Как и другие ,\арак1еристики связей (длина, дипольный момент и
т. д.), энергия диссоциации связи зависит от ее положения в молекуле
углеводорода. В табл. 16 приведены значения энергий диссоциации не-
которых связей С С и С-Н.
Из приведенных данных видно, что энергия диссоциации связи
С-С изменяется при изменении длины углеводородной цепи и поло-
жения связи в молекуле. Энергия диссоциации связи С-Н изменяется
। d зависимости от характера углеродного атома — первичный, вторич-
ный, третичный. При замене атома водорода в этане на фенильный ра-
дикал энергия диссоциации связи С-С уменьшается по сравнению с
незамещенным этаном. Вообще легкость гомолитического разрыва
связи С-С и, следовательно, уменьшение энергии диссоциации соот-
ветствующей связи находится в прямой зависимости от устойчивости
образующихся при лом свободных радикалов. Так, энергия диссоциа-
нии связи C.^^^-C, шф гексафен ил эта на всего -42 кДж/моль.
Энергия гетсроли 1 ического разрыва связей значительно выше, чем
Политического. Значения энергий гетеролитического разрыва связей
'ПоиелукПпИе<'|7^^е1«>л<>Р™1а от - М8 кДж/ммк Виер-
|«СН,--Н = 440 кДж/M.™. < И
вам случае происходи i переход и з .уг - '«сняется высокой электроогрииа
Повышение значения Г) во втор°м случае о
атома углсрола в Ум нллентиом состоянии.
167
Таблица 16. Энергия диссоциации связей С-С и С-Н1
У Г.1СНО Юрод И СПЯ Jh Энергия разрыва, кДж/моль
< 16 СП. 352
СН,=СН, 503
iСНЕсн 964
сд.-сн. 337
333
(СН^СН-СН, 314
(СНР2СН-СН(СН.)2 281
с(1н5-сн3 377
с6щсн2-сщ 268
С(,Н5С(СН2)2-СН3 251
сьц-н 428
СЙН--Н 410
Я-С4Н(ГН 394
(СН3)2СНСН2-Н 390
(С нрд-н 377
С6Н,СН2-Н 348
с()н5-н 427
могут быть вычислены на основании данных но потенциалам иониза-
ции радикалов:
R -
R' + е
и значений энергий гомолитического разрыва связей. Для простых мо-
лекул углеводородов они могут быть найдены экспериментально (ме-
тод электронного удара).
Гринсфельдером2 вычислены значения гетерол яти чес кого разрыва
центральной связи С-С в молекуле гексана. Это значение равно
1089 кДж/моль. Для различных связей С-Н значение энергии гетеро-
литического разрыва изменяется в пределах 670— 1689 кДж/моль.
1.2. Термическая стабильность углеводородов
Термическая стабильность углеводородов различных рядов опре*
деляется энергией Гиббса образования углеводородов из простых ве-
ществ и зависимостью этой энергии от температуры:
Приведены усредненные данные, вол у не иные ран ичиги ми методами (пиролиз,
ллектроиный улар, фотоионизация и др.).
2 Гринсфельдср Б. С. В кц; Химия углеводородов нефти Т 2 — М.: Гостоптехиздаь
1958 -С. 114.
AG""''1’
= A+BT.
Коэффициент А и В можно легко найти, если известна энергия
Гиббса при двух температурах. Например, для метана:
С + 2Н, <=ДСН, Д(7°298к =—50867 кДж/моль,
к =-12799 кДж/моль;
50867 — А + /?*298,
АСжк = -12799 = Л + 700.
Откуда А = -79019, В - 94,6
Зная А и В. получаем уравнение зависимости энергии Гиббса обра-
зования метана от температуры:
=-79019 + 94,6 Г
Аналогичные уравнения можно найти для любого углеводорода,
(если есть справочные данные) и с помощью этих уравнений можно
построить прямые, выражающие зависимость энергии Гиббса от тем-
пературы (рис. 58).
Из рис. 58 видно, что с повышением температуры энергия Гиббса
образования углеводородов различных рядов изменяется различным
образом. Быстрее повышается энергия Гиббса алканов и циклоалканов.
Энергия Гиббса для аренов и алкенов увеличивается медленнее. В слу-
чае же ацетилена энергия Гиббса уменьшается с увеличением темпера-
туры. Различный характер изменения энергий Гиббса образования уг-
леводородов с температурой приводит к уменьшению различий в энер-
гии 700 900 1100 1300 1500 Г, К
Рис. 58, Зависимость энергии Гиббса образования углеводородов
от температуры: 1 — СН4; 2,5 — С2Н6, С3Н8; 6, 8,11 С„Н2л+2,
3 —С2Н4; 7, 9, 10 — арены; 4 — С2Н2
169
И1я\ Гиббса с повышением температуры. Вследствие этого меняется
cool ношение гермолипам и чес к их устойчивостей углеводородов. До
температуры 500°С алканы являются наиболее устойчивыми. Так, до
900 К этан более устойчив, чем бензол, после 900 К осн юл более устой-
чив. чем этан. Зная уравнение зависимости энергии Гиобса от темпера-
туры, можно найти температуру, выше которой теоретически возмож-
но разложение углеводородов. Например. ,ия метана
Л(7ГР - -79019 + 94,674
Если Дб = 0, то Т= 835 К,
Аналогично, зная уравнение зависимости энергии Гиббса реакции
крекинга какого-либо углеводорода от температуры, можно найти тем-
пературу, при которой возможен крекинг этого углеводорода в данном
направлении. Предположим, что необходимо найти температуру, при
которой возможен крекинг пентана с образованием метана и бутена
СНз-СГЬ-СНу-СН-СН3 - СИ," С Н. = С Н —СН2-СН,
В справочнике находим значения энергии Гиббса образования из
простых вешеств для всех трех углеводородов при двух различных тем-
пературах:
Г. к ЛС’г .кДж/мол, реакции, кДж/мол ь
сн4 Бутен-1 (+11 .
298 50X06 L 72139 *212 -29485
700 12779 177697 f 01/42/. -28743
Составляем уравнение зависимости G? реакции от температуры:
ДС = Я+77 298.
ДС° = А+В 7(H)
Откуда находим: А = 72621; В = 144,8.
Следовательно, уравнение зависимости энернш Гиббса реакции
крекинга от температуры имеет вид
АС-/1 = 72621 - 144.8 7'.
При Л С = 0 находим температуру (510 К), выше которой возможно
протекание реакции крекинга пентана с образованисм метана и бутена.
Следует отметить. что если реакция термодинамически возможна
в данных условиях, то это не означает, что она реально осуществима.
5^ ю реакцию можно осуществить, если
в данных условиях она протекает со значительной скоростью. Напри-
170
мер, реакция распада молекул алканов с числом атомов углерода п > 7
с образо ва н и е м у гл е рода и вод о р ода
С/(Н 2п+2^ + + 2)Н
термодинамически возможна при 25’С, однако реально она не осуще-
ствима при лой гсмпсратуре. т. к. скорость ее бесконечно мала.
§2. Химизм и механизм термических превращений
углеводородов и других компонентов нефти и газа
Основными процессами термической переработки нефтяного
сырья являются термический крекинг, коксование и пиролиз.
Термический крекинг
Термический крекинг мазута под давлением (470-520", давление 2-
5 МПа) с получением бензина является устаревшим процессом. В на-
стоящее время установки крекинга мазута под давлением не строятся.
В последнее время развиваются процессы термического крекинга
тяжелых очищенных дистиллятов нефти, главная цель которых — по-
лучение не бензина, а крекинг-остатка с низким содержанием асфаль-
тенов и металлов, пригодного для получения игольчатого кокса1.
Важным термическим процессом является также процесс легкого
крекинга гудрона (висбрекинг — в переводе с англ, «снижение вязкос-
ти») с получением небольших количеств газа, бензина и дизельной
фракции (суммарно -20) и котельного топлива (-80%). Этот процесс
протекает при температуре 450-480'С, давлении 0,2 МПа и малом вре-
мени контакта.
Коксование
Процессы коксования нефтяных остатков служат для получения
пектродного кокса (выход 10- 40% в зависимости от сырья и типа про-
цесса). Кроме кокса образуются газ (выход 10-20%) и дистилляты кок-
сования (50 70?/)
Условия процесса коксования: температура 450-550°С (иногда и
выше) и небольшие давления, близкие к атмосферному.
Пиролиз
Процесс пиролиза служит для получения газообразных олефинов,
главным обра юм л илена-. Сырье для пиролиза — бензин прямой пе-
гИ।ольча[ый кош. мже высокой степени чистого и механической прочности -
применяется для и иокшлепия ыектролов при произволе гве стали и алюминия.
"О составе niton термической и каталитической переработки нефтяною сырья см
гаПл. 12.
S71
регонки, керос и но-газойлевые фракции, а также природные и поир.
ные газы. Пиролиз протекает при небольшом давлении -о.| МПаипрц
температуре 700-900’С
Рассмотрим химизм и механизм превращения у iеводородов раз-
личных рядов в условиях вышеназванных процессов
11 Термически* превращения углеводородов
Алкалы расщепляются как по связям С -I тлк и -е связям С-Н
Так как энергия диссоииаиии связи С С меньше чем с ня зей C-H.fc.
лее вероятен риспад С-С-свяэей При распаде по( i -связи (ремтм
крекинга» из молекулы алкана в простом случае V'dj н*'!сн алкен им-
кан меньшей молекулярной массы
CHrCH; CH* HrCH Z== < Н. ( н (Н ( Н.-СН,
зибо
СН^Н^ЭМГН-СН, ^—( Н ( Н Н < Н~СН?
При распаде по С Н-спи зим промах* »л и: : н :?.>> инне алкшш
( Н, СН. С Н, (Н. ( Н ( Н Н
Для ни ни их алканов < пан пропан) реакция юм лиронания яые-
етс* преобладающей Это объясняется тем чтпим- н их низших »•
камов число С - Н сяя зей значительно прсныгнз * лс »<> ( (-сваей
Для высших алканов преобладает реакция хрт* юо t
Наиболее термически устойчив метан Ко uu *р установщик
термическая деструкция метана термо зин зм>* < .* ♦« -и мм о а на яря
температурах выше 560 ( |Я.и к» Одна*»» ли ноельной «»•
рос тью метан рас паллете я только при ОМ ( и и При гем першу*
рах более 1400’С метан распадается п<>лнгхгьн- <i ч; герол и водород
Однако еези время пребывания метана в к»не ,н ем пера туры Л*
ло, можно получить, кроме углерода и во л про и. силен и ацетилов
I до 10<^ об ) На этом основано промышленноле ер^и шодство аветя*
лена нз метана
Этан менее устойчив, чем метан ^аме^чач инфекция настутяР
при температуре ботее Ж. При Х00 (. зтан раииеп ляется со WW-
тсльной скоростью Основной реакцией при ком является peэкlЯlЯЖ
гидрирования
СНгСН,^-- СН2Ч Н, * Н2
172
в меньшей степени происходит распад по сняли С С Пропан рас-
лелтяется .кччг. чем метан и этан. Заметная деструкция начинается
выше 450 I и проходит подиум направлениям
lН,CHvCH
СЛНЪ + Н;
С?Н4 Т сн«
Ьх।jh ншнммс: имeiно растепляться при температурах более
4WC и по 1нер;4цчсм тем же прекращениям, что и пропан, т с. лет мл-
лиронаник' и р.кшеи тению по сия пт С С
Начинам < iuth.um. рас me и темне по снят С- С становится нреоб-
лаюшим Нычит л ik.ihh растептякпея только по С ( снм»чм,
причем ib' мс|ч- х нс чтения числа aiомов уIлерода н молекуле потряс-
чкч скоро*." kpcknuid По шнным М Д Тияичеена. относительная
^мтрокчь крек ю'|ич« иоротон (г) различной молекулярной массы
К Я с h ’ \ k'llii’H I JU I И 110 И
И крч 11 ♦ i d g . * _ -i ( ч - - < > - in * 10 - .V 1 " ( , 46 IX»
Такие р< *нн х !»<• 1иченне скорости крекинга объясняется уменьше-
нием жернти i И .. v ОЦИДНИИ I. НН тей (' ( с ростом числа атомов у 1леро-
м (колебаге (ьии> шиженин \г леродных атомов цепи) и увеличением
числа с ня в* и ( ( < м-t юи терт ней диссоциации
Место ра фына н мо iex \ тс а ткана определяется энергиями диссоци-
ации святей и тямт ।емперятурой и давлением При умеренных тем-
пературах 40” хпо ( рлфын происходит ближе к середине молекулы,
т е по наибо п г ( мбим ( ня »ям Так. в молекуле октана энергии днссо-
аяаиии различны* г ня и»и имеют следующие жачения:
((<<((( С
us i;; <|4 ип А |4 322 335 кДж/моль
Прслпочтии'он'к'к разрыва ближе к середине молекулы пол-
тхерждает(.я н-рмм шнамическими расчетами Например, для
«акции крекинга 1сэама
кДж/моль
( Н < Л,(1 4 С.Н1; 35 4
< „,ц..^= ( н6 • < ,н,6 3’1
171
С повышением температуры место разрыва становится неопределен-
ным. Уменьшение давления способствует разрыву ближе к концу моле-
кулы, так как при этом образуются продукты с большей упругостью
пара, что находится всоответствии с принципом Ле-Шагелье: при повы-
шении давления место растепления смешается к центру молекулы,так
как при этом образуются продукты с меньшей упру гостью пара.
Изоалканы крекируются несколько легче, чем алканы нормального
строения, хотя некоторые изоалканы симметричного строения терми-
чески стабильны.
В настоящее время установлено, что термический крекинг боль-
шинства углеводородов протекает по радикально-не и ному механизму
Существование свободных радикалов было дока за и о работами Гомбер-
га (1900) и Панета (1929). Так, для радикала СН ; был найден полупери-
од жизни, равный 0,006 с.
Радикально-цепной механизм крекинга алканов был впервые выдви-
нут американским химиком Райсом (в ЗО-х гг. XX сю юзия). В соответ-
ствии с этим механизмом крекинг проходит чере з слса\ юшие этапы:
а) зарождение цепи (образование свободных рал и канон). При терми-
ческом распаде всех алканов, начиная с этана, за рождение пепи проис-
ходит в результате разрыва свя зи С-С При ном t юра зуются свобод-
ные радикалы:
снгсн4сн^ -- снгсн; < н-;
В случае алканов с большим числом атомов у t ле рода при не очень
высоких температурах крекинга (350-4504). как уже отмечалось, раз-
рыв углеводородной цепи происходит посерел и не. т. е. по наиболее
слабым связям С—С. При более высоких темпера п рах могут разры-
ваться и другие С-С-связи. Значительно менее вероятен при зарожде-
нии пепи разрыв С-Н-связей углеводородов, и он возможен только
в случае низших алканов (этан, пропан) при соударении их молекулсо
стенкой реактора при высоких температурах. Гак как атомы водорода
являются внешними атомами в молекулах, го при сох дарении со стен-
кой реактора может произойти отрыв атома водорода:
СНгСн|н СНгСН; Н':
б) продолжение цепи (реакции свободных радикалов). Свободные
радикалы обладают высокой реакционной способностью, так как
держат неспаренный электрон. Поэтому они стремятся слабил изире-
ваться и подвергаются различным превращениям.
Распад радикалов. Радикалы распадаются по 0-евязям (С—С или
менее вероятно С—Н), находящимся в [5-пол ожени и по отношению к
атому углерода, несущему холостой электрон:
н
•
CH,-CH-С l-l-CH,
СН,-СН=СН2
i CHrtH-CH-CH,
\
' H
CHrCH^CH-CH,
Относительно тоичивыми к распаду являются низшие алифати-
ческие ради ка. । и. < ’ Н ;. С Н ;С Н /
Такие радикалы, как CH.-CH/. CH,-CH‘-CHV ’(’(СН,), мот
распадаться io.ii.h с p i зрыном [3-С Н-связсй. Другие радикалы распа-
даются с разрывам юк [i-( С. шк и ji-C-Н-связей. причем разрыв
р-С-С-свя зеи ч р(. ] i к ас । легче. 'Энергия диссоциации для различных
р-спязей С ( в pa шка ia\ и (меняется в пределах 42—84 кДж/моль.
Изомери ищи» р^оккилов. Первичные радикалы могут изомеризо-
ваться в более стамильныс вторичные. Изомеризация протекает в ре-
зультате внутримо в ку шрной миграции атома водорода из положения
4 или 5 в положение i через циклическое переходное состояние (1,4- и
1.5-изомеризация) 11 а пример. 1.5-изомеризация:
Н2СХ CH-R
сн2
( II,.( H2-CH2-CH2-CHR
1.2- и 1,3-и зомери шнии мало вероятны, вследствие низкой ста-
бильности соот вс тс 1 ну инн их переходных состояний.
Следует от мет и п». чю изомеризация радикалов протекает зна-
чительно медлен ее. чем их распад по (i-связи.
Реакции радика ina ( моцекуцами углеводородов (реакции передачи
иепи). Эти реакции являются основными, приводящими к развитию
иепи. Низшие свободные радикалы (атомарный водород, CHV
при столкновении с молекулами алканов отрывают от них ато-
мь> водорода
I7S
R-H + H* — И’ + R’
R-н + CH3^=^= CH4 + R'
Другие радикалы (состава C-j, C4 и выше) также могут вступать в эти
реакции при повышенных давлениях (2-5 МПа) и при температурах до
600°С. Однако в условиях пиролиза они значительно легче распадают-
ся, чем вступают в эти реакции. При взаимодействии радикала с моле-
кулой углеводорода, в частности алкана, возможен отрыв атомов водо-
рода от первичных, вторичных и третичных атомов углерода. Вероят-
ность отрыва первичного, вторичного и третичного атомов водорода
зависит как от легкости разрыва связи С-Н, так и от количества соот-
ветствующих атомов водорода в молекуле углеводорода (т. е. от вероят-
ности соударения радикала с атомами водорода различного типа):
г
-С„ср-Н + R=^ -С11ср + RH
>Clrr-H + >С,и + RH
>стр-н + R^±->crp + RH
Легкость разрыва связей С-Н при соударении с радикалом характе-
ризуется константами скоростей соответствующих реакций:
где К"тр, Кт, — константы скорости образования соответственно
третичного, вторичного и первичного радикалов: о , o[tT, дтр - пред-
экспоненииальные множители;
Япер ~ °вт
£1|ер, ЕаГ £тр — соответствующие энергии активации реакции отрыва
атомов водорода под действием радикала.
Энергии активации вышеназванных реакций можно вычислить по
уравнению Семенова — Полян и для экзотермических реакций, зная
значения их тепловых эффектов (Q):
£= 11,5 - 0,25 Q.
Вычислив значения £пер, £гп, Е^, можно найти отношения конс-
тант скоростей реакции отрыва атома водорода от первичного, вторич-
ного и третичного атомов углерода и найти относительные скорости
отрыва соответствующих атомов водорода:
^пер Ат^гр = ^пер: ^вг Кр'
Райс считал, что отношение этих скоростей при 600°С равно И2.Ю’
176
т, е. третичные атомы водорода отщепляются в 10 раз быстрее, а вто-
ричные — в 2 раза, чем первичные1.
Рассмотрим расчет относительных скоростей отрыва первичных,
вторичных и третичных атомов водорода при 600°С (873 К)
Известно, что тепловые эффекты соответствующих реакций отрыва
атомов водорода составляют:
Спср = 2! кДж/моль: е|п. = 42 кДж/моль: QTp = 57кДж/моль.
Рассчитанные по формуле Семенова Поляни соответствующие
значения энергий активации:
^пср 43 кДж/моль, 38 кДж/моль, £тр — 33 кДж/моль.
Находим отношения констант скоростей реакции, которые равны
отношению скоростей отрыва атомов водорода:
к
Al,.-,,’ I
кг
я/
TW00-W(1
-JQ 14.11X73
= е
JWWKW0
~Ю 191 |Ji71
Видно, что отношение скорости отрыва вторичных и первичных
помов водорода то же, ч то и предложенное Райсом (~2). Однако отно-
шение скоростей от рыва третичного и первичного атомов водорода от-
личается (4 вместо 10). Это объясняется тем, что Рамс рассчитывал зна-
чения констант скоростей отрыва атомов водорода на основе энергий
диссоциации соответствующих связей, что неточно.
Зная относительные скорости отрыва различных атомов водорода,
можно рассчитать относительные вероятности их отрыва и, следова-
тельно, относительные вероятности образования соответствующих
радикалов:
= и 7/
пер нер пер1
jy = / п
ВТ ВТ г ВГ
И/ = и 7?
г гр тр птр’
гяе И'пср’ ^)Г, относительные вероятности образования соответ-
ственно первичного, вторичного и третичного радикалов; ^]ер, Ивт,
относительные скорости образования соответствующих радика-
Я011: Ппср’ "иг ’* соответственно число первичных, вторичных и тре-
тичных атомов водорода в молекуле.
1 Температура сильно влияет на соот ношение скоростей отрыва различных атомов
'дюрола. При небольших температурах (300-400 К) относительная активность атомов
‘шороза сильно ра ншчас гея. Например, по отношению к СН^ при 455 К относи [ельные
^Biuiiocm первичных, вторичных и третичных атомов водорода относятся как 1.7.50.
||П[11,||испис‘.м темпера гуры различия в относительных активностях сглаживаются.
177
Зная относительные вероятности Wzncp, И^., можно рассчитат
состав продуктов термического крекинга алканов. Расчетные данные
имеют хорошую сходимость с экспериментальными для простейшие
алканов (см. ниже);
в) Обрыв цепи. Обрыв цепи происходит при столкновении радика
лов. когда их концентрация в системе становится значительной:
СН3 + СН3^=^ СН3-СН3
сн3-сн2-сн2 + сн3
сн4 + снгсн=сн2
На основе радикально-цепного механизма Райса можно рассчитать
состав продуктов крекинга низших алканов.
Рассмотрим конкретный пример термического крекинга бутана при
температуре 500-600°С. В этих условиях бутан подвергается преимуще-
ственно расщеплению по связям С-С:
сн3-сн2-сн2-сн3^=^
сн4+ сн3-сн=сн2
CH3-CHrCH2-CH3z^zz^ с2Н4 + с2н6
Зарождение цепи проходит в результате разрыва связи С-С:
СН3-СН|СНГСН3~
2 СН3-СН2
либо
сн|сн2-сн2-сн3^-*~ сн3 + сн2-сн2-сн3
Столкновение свободных радикалов с молекулами бутана приводит
к развитию цепи. При этом возможен отрыв атомов водорода от пер-
вичного или вторичного атомов углерода с образованием бутила или
вторичного бутила:
I направление
сн,-сн2-сн2-сн, + сн3^-— сн4 + сн,-сн2сн2сн2
бутил
|[ направление
СН,-СН2-СН,-СН, + CH3^Z СН4 + СН3-СН2-СН-СН3
нт. бутил
Бутил распадается с образованием этилена и этана:
СН3-СН2|СН2-СН2^=^
г СНГСН2
этилен
178
и далее:
СНГСН2 + С4ню^1=±
СНГСН3 + *С4Н()
этан ~
Вторичный бу I ил распадается с образованием пропилена и метана
СН3-СН-СН,|СН,^=:|СН3-СН=СН2 + СН,
[I далее
си;, - с4н10^=±: |сн4|+ с4н9
Соотношение между продуктами распада бутила и вторичного бути-
ла определяется соотношением вероятностей их образования:
И/6иц.-| = %) =1'6 = 6,
= С™ А, =2'4 = 8
~б’
Следовательно, и з каждых 14 молекул бутана 8 молекул будут прев-
ращаться во вторичный бутил и затем в пропилен и метан, а 6 молекул
будут превращаться в бутил и затем в этилен и этан:
8С4Н1О = 8С3Н6 + 8СН4,
6 С4Н1() = 6 С2н4 + 6 с2н6.
Суммируя эти уравнения, получаем:
14 С4Н ,0 - 8 С\Н6 + 8 СН4 + 6 С2Н4 + 6 С2Н6.
Расчет мольного содержания каждого продукта реакции дает следу-
ющие значения: C\Hf, 28%. СН4 - 28%, С2Н4- 21%, С2Нб - 21%.
Результаты аналога продуктов крекинга близки к вышеприведен-
ным данным.
Радикальный механизм термического расщепления углеводородов
(распад радикалов по |%связи С-С) объясняет преобладание этилена
в газообразных продуктах пиролиза:
R-СНэ-СН .-С Н.-С Н2 - — R-CH2-CH2 + сн2=сн3
R + СН2=СН2
1?
179
,V,счетных значении. что объясни,.
О ,нако »««« ли',е"1’ Т«пени изомеризации первичных рад.
ея X' капнем » - Р— с образованием пр».
калов во вторичные - ____
пилена: --^.сНз-СНуСН-СН, ’
R-CHrCHrCHrCHi- сн.
______R-eH2 + CHr=cH сн’
рнню с алканами термически более стабильны.
Алкены по сравнен!но с ен) „ условиях термического кре-
Низшие ^е*ины‘Хгаются следующим превращениям.
книга и пиролиза под» Р
Дегидрирование;
700-900 С н
СНгсн=сн2^ СН2=С=СН2 . Н2
]000°С
СН2=СН2^=^: CFECH + Н2
Механизм дегидрирования включает расщепление связи С-Н и
последующее отщепление атома водорода от радикала:
н|сн=сн2^=±: сн?=сн + *н
* I
СН2=СН ± сн=сн + н
В случае пропилена разрывается наиболее слабая С— Н-связь ал-
лильного атома углерода:
н|сн2-сн=сн,WH + СНгСН-СН2
алл ил
Образующийся аллил стабилизируется в результате разрыва р-С-Н-
связи:
±:СН2=С=СН? + Н
аллен
Полимеризация, При атмосферном давлении полимеризация олефи-
нов термодинамически возможна до 50(У'С:
2 CH2=CH>Z<—— С Н ?=С Н -С Н ?С Н :
Под давлением полимеризация протекает при более высоких темпе- ,
ратурах. Полимеризация проходит через стадии образования бирали- ।
кала, который взаимодействует затем с молекулой олефина: 1
С Н 2= С Н 2 t Н д-‘С Н з
180
СНз-СН?
сНгСНгСН3-СН? V
-и ^З-июмерищция
~Н3—CHj
Дегидроконденсация.’
2
Механизм этой реакции вмючает разрыв связи С-Н с образован.,
ем винила с последующим его присоединением к этилену:
Н2С=Сн|н^=^н + сн=сн2
снгсн + н2с=сн2=сн2=сн-сн2-сн2^н2с=сн-сн=сн2
Пропилен в сталии развития цепи может расщепляться с образова
нием этилена
СН3-СН=СН2
’Н2-СН,
Бутены и пентены кроме вышеприведенных реакций подвергаются
крекингу с разрывом наиболее слабой fl-связи С-С или В-связи С-Н
по отношению к двойной связи;
.СН2=СН-СН=СН? + Н7
С >7()()°сЛ
,'ЛЛТЛЛ £ / <5оо“с
Н 2С=С. Н -С Н |С F(v—► пол и меризаиия
Н2ООСН2 + СН4
В стадии зарождения цепи происходит распад по [З-С-С-связи:
н2с=сн-сн2|сн5^—Г-Н2С=СН-СН2 + сн3
Затем
H2C=CH-tH2^=^H2C=C=CH2 + н
Н£СН-СН2-СНз " ’СН3— ^сн4 + Н2С=СН-СН-СН3
Н/>СН-СН-СНз.^-..ж Н2С>СН-СН-СН2 + н
Для молекул высших алкенов при 400—450аС преобладающим явля-
ется распад по Р-С-С-связи с образованием олефинов меньшей моле-
кулярной массы, либо алкана и диена:
181
^xR-CH=CH2 + СНз-СН=СН2
R-CH2-CH2|cH2-CH=CH2
>KR-CH2-CH;, + сн2=с=сн1
Ниже приведены значения энергий диссоциации различных связей
С-С и С-Н в молекуле а-олефина в кДж/моль:
370
Видно, что наиболее слабой С-С-связью в молекуле а-олефинаяв-
ляется р-С-С-связь, наиболее прочной - а-С-С-связь. Наиболее сла-
бой С-Н-связью является связь аллильного атома углерода
(323 кДж/моль).
Механизм можно представить следующей схемой:
- СН2-СН=СН2
R-CH2-CH3 + к
R-CH7-CH?-CFb-CH=CH?
!-• — « "
R-CH=CH
При взаимодействии свободных радикалов с
CH i-CH=CH?+R'
*
молекулой олефина
отщепляется аллильный атом водорода:
r-ch2-ch27ch-ch=ch
Cl-ь, >
----^СН4 + R-CH2|CH2“CH-CH=CH2
У
R ( II: 1 CIC-СН-СН=СН2
Образующиеся при распаде олефинов диены при температурах ниже
700 С, особенно под давлением могут реагировать с олефинами по
схеме диенового синтеза с образованием ароматических углеводородов:
н
182
Некоторые исследователи считают, что высшие а-алкены крекиру-
ются по молекулярному механизму через стадии образования шести -
центрового переходного состояния (Миллер):
^сн2 Н2С
нс^
+
НС—R
Н3С
Циклоолефины более устойчивы, чем алкены. Циклогексен до 600“С
устойчив, при 70(ГС он крекируется и дегидрируется. Диены более ус-
тойчивы к термическому расщеплению, чем алкены; они появляются в
продуктах пиролит при 600-900°С. Как уже отмечалось, ниже 700°С
они участвуют в реакции диенового синтеза.
Нафтены термически стабильнее алканов. Со значительной ско-
ростью их распад протекает при температурах ~550°С. Незамещенные
нафтены (циклопснтан, циклогексан) могут распадаться по следую-
щим схемам:
СН2=СН2 + СН2=СН-СН3
СН2=СН-СН2-СН2-СН3
183
Механизм распада незамещенных нафтенов до 700—750°С ради-
кадьный, но не пенной. Это объясняется owcipoii стабилизацией обра-
зующихся бирадикалов при разрыве по С-С -спя ш.
IC ( Н К Н2=СН-СН1
----► С Н 2- С Н г С Н 2- С Н 2- с н,
^СН < СН-С Н2-СН2-СН3
СН2=СН-СН2-СН2-СН2-СН3
Однако при накоплении в реакционной смеси олефинов, содержа-
щих слабую 0-С-С-связь по отношении к двойной связи, возможно
протекание процесса и по радикально-цепному механизму. С повыше-
нием температуры крекинга выше 750 С вероятность радикально-цеп-
ного распада повышается:
СН2=СН-СН?-С1Ъ-СН.-СН
н + сн2=сн-сн3
184
II
II
II
CH.CH -
снгс н2-аь-сн-,
( II.( ikiuhch-ch-. ffH .
f
СН?СН2СН=СН2 - GH:i
C HX H? H2C=CH-CH=CH?
\cflHn
( H:CH< + C6H|J
2 H>
Алкилииклоисн ыны и ал кил циклогексаны распадаются по ради-
кально-цепному механизму. Зарождение цепи происходит путем раз-
рыва С-С-связи боковой цепи, либо отрыва метильной группы от
молекул мет ил-и рои звони ых.
Бициклические нафтены подвергаются дециклизации и дегидриро-
ванию с образованием смеси углеводородов ряда бензола, олефинов,
нафтали на.
Ароматические углеводороды обладают высокой термической ста-
бильностью. Наибольшую термическую стабильность имеют незаме-
щенные арены и среди них бензол. Самой слабой связью в молекуле
бензола является связь С-Н, энергия диссоциации которой равна
427 кДж/моль. Разрыв связи С—С требует494 кДж/моль. Поэтому в ус-
ловиях пиролиза основной реакцией бензола является его дегидро кон-
денсация с образованием дифенила:
Реакция де гидро конденсации ароматических ядер вместе с реакци-
61 диенового синтеза лежит в основе процесса образования «кокса»
при крекинге и пиролизе.
«Кокс» по своей структуре представляет собой пол и конденсирован-
ную ароматическую систему, которая может образоваться в результате
процесса деглдроконленсании ароматических ядер. Например.
185
Реакция дегидроконденсации протекает по радикально-цепному
механизму:
В молекуле толуола расщепляется наиболее слабая связь С—Н бен-
зильного атома углерода (326 кДж/моль). При этом основными про-
дуктами являются дибензил и метилдифен ил метан:
186
мстилдифснилметан
Механизм:
Менее вероятен разрыв связи С -Н и образование дитолила;
Образование небольшого количества бензола при пиролизе толуола
объясняется частичным его деметилированием по схеме
Алкилбензолы распадаются по наиболее слабой р-С-С-связи поот-
ношению к бензольному ядру (энергия диссоциации 272 кДж/моль),
Так, этилбензол при крекинге образует толуол и стирол:
187
2.2. Термические превращения высокомолекулярных
компонентов нефти в жидкой фазе
В сырье термического крекинга (висбрекинга) и коксования (мазут
гудрон) содержится значительное количество высокомолекулярных
соединений нефти.' углеводородов, смол и асфальтенов. Так, в состав
гудрона могут входить алканы С2П-С4П. полициклические ароматичес-
кие и нафтено-ароматические углеводороды, молекулы которых содер-
жат несколько колен и боковые алкильные пени, смолы, молекулы
которых содержат 3-6 ароматических и нафтеновых колец или гетеро-
циклов с алкильными боковыми пенями, а также асфальтены, молеку-
лы которых могут содержать до 20 и более колен, боковые алкильные
цепи и углеводородные мостики. Суммарное содержание асфальтеноп
и смол в гудроне может достигать 50-60%.
В состав асфальтенов входят такие металлокомплексные соедине-
ния, в которых атом металла с переменной валентностью координиро-
ван с гетероатомами (S, N, О) молекул асфальтенов и смол.
Соединения, входящие в состав нефтяных остатков, обладают не-
высокой термической стабильностью. Это объясняется тем, что в моле-
кулах этих соединений содержатся слабые С--С-связи в алкильных
боковых цепях и углеводородных мостиках.
Низкая термическая стабильность асфальтенов объясняется также
тем, что их молекулы содержат значительное число парамагнитных
центров (1()’М019 на грамм).
Нефтяные остатки при обычных условиях представляют собой
структурированные коллоидные системы, состоящие из дисперсион-
ной среды (углеводороды) и дисперсной фазы (ассоциированные моле-
кулы смол и асфальтенов).
188
в процессе термического крекинга и коксования значительная
часть сырья находится в жилкой фазе. Расщепление молекул компо-
нентов сырья в жилкой фазе имеет свои особенности. Во-первых, го-
молитический разрыв связи уысводорода в жидкой фазе не приводит
к быстрому образованию двух разобщенных радикалов, как это проис-
\од.цт в газовой фа зе. Эл о объясняется тем, что расщепляющаяся моле-
кула сольватирована, । е. находится в тесном окружении других моле-
кул, как бы 13 «клст ке». Xl-Ля ioio чтобы вырваться из этого окружения,
образовавшийся радикал должен получить дополнительную энергию
(энергия активации лиффузии). Величина этой энергии зависит от ха-
рактера сольватация, т. с. межмолекулярного взаимодействия между
радикалом и окружающими молекулами.
Если сольватация является неспецифической (слабое межмолеку-
лярное взаимодействие, обусловленное силами Ван-дер-Ваальса), то
энергия активами и диффузии невелика. Примером может служить рас-
пад молекулы, окруженной неполярными молекулами алканов.
В случае если молекула углеводорода окружена полярными молеку-
лами, например, молекулами ароматических углеводородов или смо-
листых веществ, то энергия активации диффузии будет значительно
больше, вследствие более сильного межмолекулярного взаимодей-
ствия окружающих молекул (диполь-дипольное, донорно-акцептор-
ное) и сильного их взаимодействия с образовавшимся радикалом.
В этом случае радикал будет легче вступать в химическое взаимодей-
ствие с окружающими молекулами (присоединение к кратным свя-
зям), чем диффундировал ь из клетки,так как энергия активации хими-
ческого вза и моле метни я
R* ж а -э RA' -э RA + Н‘
будет значительно меньше энергии активации диффузии. Поэтому при
жидкофазном крекинге ул ле водородной смеси с высоким содержанием
ароматических углеводородов выход газа и легких фракций будет не-
большим, так как ароматические углеводороды являются «ловушками»
тля радикалов.
Вместе с тем ароматические углеводороды, особенно полицикли-
ческие. будут полверл аться реакции дегидрогенизаиионной конденса-
ции с образованием асфальтенов и кокса по схеме
аро м ат и чес к и е у л л с водороды —> с м ол ы —> асфал ьте н ы ->
карбены —> карбонды —> кокс.
Высокое содержание в сырл?е ароматических углеводородов, я вл я
кшлихся хорошими растворителями для асфальтенов, также благопри
ятсплует выходу кокса, так как при этом пороговая концентрация
^Фальтенов в растворе, при которой начинается новообразование,
будет высокой.
189
Еще более сложная картина наблюдается в том случае, если пропс-
холит распад молекул смол или асфальтенов. Смолы и асфальтены на-
ходятся в углеводородных растворах при повышенных концентрации
в ассоциированном состоянии. Надмолекулярная структура асфальте-
нов представляет собой несколько ассоциированных молекул асфаль-
тенов (пакеты из 2—5 молекул). Смолы также ассоциированы.
При термической деструкции асфальтенов и смол происходит раз-
рыв наиболее слабых С-С-связей боковых цепей и углеводородных
мостиков. Образовавшиеся радикалы малоподвижны вследствие зна-
чительных размеров, к тому же они находятся в составе надмолекуляр-
ной структуры, связаны с остальной ее частью силами межмолекуляр-
ного взаимодействия. Поэтому они могут только реагировать либо
с соседними молекулами внутри ассоциата (взаимодействие с л-элект-
ронами ароматических ядер с образованием прочных связей с аромати-
ческими атомами углерода), либо с соседними ассоциатами асфальте-
нов или смол. В процессе термического распада асфальтенов исчезают
слабые связи, появляются прочные. Происходит сшивка молекул
асфальтенов в ассоциате и соединение ассоциатор, друг с другом. В ре-
зультате этого образуются более крупные част ицы, обладающие худ-
шей растворимостью в углеводородах.
Если углеводородная дисперсионная среда, окружающая асфальте-
ны, содержит мало ароматических углеводородов, то продукты конден-
сации асфальтенов выделяются из раствора в виде капель и в дальней-
шем происходит превращение этих капель в кокс
Если углеводородная дисперсионная среда вы сокоароматизирована
и хорошо растворяет асфальтены, то по мере их конденсации не проис-
ходит их выделения из раствора, а образуется студень-трехмерная
структурированная система, внутри которой находятся молекулы угле-
водородной среды (масла). Кокс образуется в результате дальнейшей
сшивки этой структуры, при этом дисперсионная среда вытесняется.
Таким образом, в конечном итоге высокохюлсктлярные соединения
нефти (ВМС) (полициклические ароматические уч л с водороды, смолы
и асфальтены) в результате термической деструкции превращаются
в низкомолекулярные соединения (НМС) и карбоилы (кокс):
I смолы
ВМС у асфальтены
I углеводороды
190
Механизм конденсации асфальтенов в кокс является радикально-
цепным.
в стадии зарождения пени происходит распад молекулы асфальтена
„о слабой С—С-связи. например, боковой цепи с образованием
асфальтенового (А. ) и алифатического (R') радикалов:
А'. + R*
В стадии развития пени эти радикалы взаимодействуют с другими
моле кул а ми ас ф ал ь г е н о в:
а; + А-ч А'А*
А + R' А. + RH
А'А АА” + Н
А’А + А А'АА
Длина цепи составляет около 120-150 звеньев.
Обрыв цепи происходит тогда, когда в результате поликонденсации
образуется высокомолекулярный малоактивный радикал, в котором
свободная валентность делокализована. Этот радикал не может про-
должать реа к ц и о 11 н у ю цепь.
Обрыв цепи может произойти также в результате рекомбинации ра-
дикалов
А’,' ж ' А' --► А"А'
Однако этот путь обрыва цепи маловероятен, вследствие малой под-
вижности и больших размеров асфальтеновых радикалов.
§3. Химизм и механизм каталитических превращений
углеводородов и других компонентов нефти и газа
В этом разделе будут рассмотрены химизм и механизм преврашений
углеводородов в процессах каталитического крекинга, риформинга,
гидрокрекинга, в реакциях алкилирования, ступенчатой полимериза-
ции, изомеризаци и.
Вышеназванные превращения углеводородов протекают обычно в
Условиях гетерогенного катализа в присутствии твердых или жидких
(№) катализаторов. Твердые катализаторы, применяемые в нефтепе-
реработке, могут быть подразделены на:
1) металлические катализаторы, например: Pt, РФ Ni,
191
2) полх'проводники, например.' ZnO, Cr^O^ WS2, MoO,:
3) изоляторы1, например, алюмосиликатные катализаторы, Д^Ор
Гетероген но-каталитическая реакция нал твердыми пористыми ка-
тализаторами протекает через следующие сзади и.
1) диффузию реагентов к поверхности катализатора (внешняя и
внутрен няя);
2) активированную адсорбцию:
3) химическое превращение;
4) десорбцию продуктов реакции;
5) диффузию продуктов реакции в объем.
Скорость всего процесса определяется наиболее медленной стади-
ей. Если такой стадиен является диффузия, то процесс протекает итак
называемой диффузионной области, скорость его мало зависит от тем-
пературы и процесс описывается уравнениями скорости диффузии.
Если реакция тормозится диффузией, то необходимо применять круп-
нопористые катализаторы, либо применять сильно измельченный,
например пылевидный, катализатор, что позволит увеличить доступ-
ную поверхность катализатора.
Если наиболее медленной стадией является химическая реакция,то
процесс находится в кинетической области. Скорость процесса будет
зависеть от температуры, так как повышение температуры увеличивает
скорость химической реакции в 2-3 раза при увеличении температуры
на 10’С. Однако повышение температуры надо проводить до опреде-
ленного предела, иначе реакция перейдет в диффузионную область. ।
Каждая стадия гетерогенно-каталитического процесса представляет
собой очень сложное явление. Стадию химического превращения или
механизм химических превращений на катализаторах мы рассмотрим
позже. Сейчас же рассмотрим механизм активированной адсорбции
молекул на поверхности катализатора.
При активированной адсорбции молекула удерживается на поверх-
ности валентно-химическими силами. Продолжительность пребыва-
ния молекулы на поверхности катализатора выражается уравнением
с
т = тоеЛ7.
где Q — теплота адсорбции, кДж/моль: — минимальное время жизни
молекулы на поверхности (при 0 = 0, Т= 10n- 1016 с.
За время пребывания на поверхности молекула перемешается с од-
ного адсорбционного центра на другой. Поэтому существует другое
понятие — время жизни молекулы на адсорбционном центре:
В |ки?,7елнее время н ряде работ показано. что в онречеленных условиях оксид мю
МИНИН ЯИЛЯСТСЯ полуIфОНОДНИком.
192
це Q' - величина энергетического барьера ,ъи перехода с одного
центра на другой.
Приняв, что т'() - т(), найдем отношение
т' RT '
Если Q = 41.9 кДж/моль, О' = 20,9 кДж/моль. а Г = 300 К то т/т' =
1000.
Таким образом, за время жизни на поверхности катализатора моле-
кула совершает oi ромное количество скачков.
3.1. Каталитический крекинг
Создание процесса каталитического крекинга было обусловлено
необходимое идо смягчить условия крекинга нефтяных продуктов (по-
низить темпера гуру и давление), повысить выход бензина и улучшить
его качество. Наиболее активным катализатором крекинга углеводоро-
дов является хлорил алюминия. Впервые крекинг в присутствии Al Вг}
и AICI3 — был проведен в России Густавсеном. Под действием хлорида
алюминия крекинг, например, парафина начинается при 100°С, а при
200°С протекает с высокой скоростью. Недостатки процесса крекинга в
присутствии этого катализатора состоят в повышенном расходе хлори-
да алюминия и невозможности его регенерации, а также в том, что при
его разложении под действием влаги воздуха выделяется хлорид водо-
рода, сильно корродирующий аппаратуру.
В настоящее время широко используются в качестве катализаторов
крекинга алюмосиликатные катализаторы (аморфные и кристалличес-
кие), которые менее активны, чем хлорид алюминия, но лишены всех
его недостатков. Эти катал и заторы обладают высокой механической
прочностью, высокой химической и термической стабильностью, не
вступают в необратимое химическое взаимодействие с сырьем, легко
реактивируются выжигом углистых отложений (кокса). Крекинг в при-
сутствии алюмосиликатных катализаторов проходит в паровой фазе
при 400—50(ГС и давлении, близком к атмосферному. Сырьем служат
вакуумные газойли с концом кипения 500°С и выше, к которым иног-
да добавляют дсасфалътизаты нефтяных остатков (около 20%).
В настоящее время в нашей стране разрабатываются, а за рубежом
действуют установки каталитического крекинга мазута.
Алюмосиликатные катализаторы представляют собой гранулиро-
ванные вещества, обладающие высокой удельной поверхностью (400—
193
^3- 3544
ЦНЮ M2/1 ). По своей химической природе алюмосиликатные катализа-
торы являются слабыми кислотами. Так, их можно титровать бутила-
мином в присутствии индикатора л-ди метиламиноазобензола.
Кристаллическая решетка аморфных алюмосиликатов может рас-
сматриваться как трехмерная кристаллическая решетка поликремне-
вой кислоты, в которой часть атомов кремния заменена на атомы алю-
миния:
В промышленности синтез аморфного алюмосиликатного катали-
затора (ААК) проводят следующим способом.
При взаимодействии водных растворов силиката натрия и сульфата
алюминия образуется силикат алюминия, который в среде минераль-
ного масла под действием поверхностного натяжения превращается
в гранулы геля; гранулы после промывки и сушки прокаливают при
600-800°С.
В лабораторных условиях смешивают в необходимых пропорциях
гель гидрата оксида алюминия и гель кремневой кислоты. Из получен-
ной смеси гелей формуют гранулы, которые промывают, высушивают
и прокаливают. При прокаливании происходит химическое взаимодей-
ствие кремневой кислоты и гидрата оксида алюминия и образуется
алюмосиликат.
Химический состав алюмосиликатного катализатора иногда рас-
сматривают как смесь оксида алюминия и оксида кремния, находя-
щихся в химическом соединении, и записывают химическую формулу
алюмосиликатного катализатора следующим образом: «А^Оз'^^г
хН2О.
Необходимо отметить, что ни оксид алюминия, ни оксид кремния,
взятые в отдельности, не являются катализаторами крекинга. Их меха
нические смеси также неактивны. Однако, если добавить к гелю кре^
невой кислоты даже небольшое количество геля гидрата оксида злю
миния, при прокаливании такой смеси получается активный катали®
194
тор крекинга углеводородов. Зависимость активности ААК от содержа-
ния химически связанного оксида алюминия изучалась Г. М. Панчен-
ковым. Им найдено, что максимум активности катализатора соответ-
ствует 3 0 % о кси ла ал ю м и н и я.
В настоящее время в нефтеперерабатывающей промышленности
широко применяются кристаллические алюмосиликатные неолитсо-
держащие катализаторы крекинга. Эти катализаторы содержат до 20%
цеолитов на аморфной алюмосиликатной матрице. Цеолитсодержа-
шие катализаторы крекинга значительно активнее аморфных (выход
бензина выше на 30-50%).
Наиболее активны катализаторы, содержащие цеолиты Y, В их при-
сутствии процесс превращения углеводородов идет глубоко. Состав
продуктов крекинга с применением ААК и цеолитов существенно раз-
личается. Так, бензин1, полученный при крекинге на ААК, содержит
30% ароматических углеводородов. 42% нафтенов, 12% олефинов, 12%
алканов: а бензин, полученный из того же сырья на цеолите, содержит
50% ароматических углеводородов, 22% нафтенов, 5% олефинов и 25%
алканов. Однако, цеолиты, несмотря на их высокую активность, сами
не применяются обычно в качестве катали заторов крекинга, так как
вследствие малых размеров пор при крекинге не используется пол-
ностью внутренняя поверхность гранул цеолита (крекинг протекает в
диффузионной области). Это можно иллюстрировать следующими
данными: средний диаметр пор ААК равен 30—50 , а цеолитов 3-10 ;
средний эффективный диаметр молекул полициклических углеводоро-
дов в сырье каталитического крекинга может достигать 13-15 и бо-
лее. Поэтому применяют цеолитсодержашие катализаторы.
Продукты крекинга (бензин и газ) с применением аморфного и цео-
литсодержашшо алюмосиликатных катализаторов также различаются по
составу, хотя и в меньшей мере, чем с использованием чистых цеолитов.
В табл. 17 приведен состав бензинов каталитического крекинга на
аморфном (А) и цеолитсодержащем (В) алюмосиликатных катализа-
торах2.
В газе крекинга на цеолитсодержащем катализаторе больше изобу-
тана и меньше бутенов, чем в газе крекинга на аморфном алюмосили-
катном катализаторе.
Алюмосиликатные катализаторы содержат воду, связанную хими-
чески и адсорбированную физически. Физически адсорбированная во-
ла понижает активность катализатора, поэтому ее необходимо удалить
прокад и ва н и е м п р и 500 -600“С.
Эс. Тарами Достижения н области изучения каталитическою акшвирования yi.ie-
вояородов в гетерогенном катализе Доклад на IX Нефтяном конгрессе в Японии, 1976 I.
-В И. Суханов Каталитические процессы в нефтепереработке. - М.; Химия, 1979,
С 39.
195
Таблица 17. Состав продуктов крекинга
Катал изагор Сое гаи, ‘ ’
Нафтены Ароматические у тле вол о ролы Ояефияы
А 43.2 20,7 2ХД S’J '
В 53.3 13 9 29.3 3,5
Химически связанная вода играет, по-видимому, большую роль
в механизме каталитического действия, так как полное удаление воды
из катализатора (прокаливание при ЮОО’С) приводит к потере его ак-
тивности.
В соответствии с современными представлениями на поверхности
алюмосиликатных катализаторов имеется 2 типа каталитических цент-
ров: протонные и апротонные. Ниже представлены предполагаемые
структуры этих центров:
протонные центры
апротонные центры
катМиз осуществлТется^одТадными]10М(>С111И1<аТНЫХ каталииторо“
воды, хемисорбированной m протонами. 9то либо протоны
алюминия (структура h к оРд,п<апиоино ненасыщенным атомом
атомом алюминия (структур ™Дроксильнш Win, сорбированных
всех этих структурах imm С ' ИЛИ своболнМ'Х (структура 111). Во
сильной поляризации связи o^?6₽eTaeT "«лнижность вследствие
снойств координационно
На поверхнос! и цеолитов поливалентных металлов также находятся
как апротонные активные центры (координационно ненасыщенные
атомы алюминия), так и протонные активные центры. Дополнительно
к протонным центрам, структура которых указана выше на рисунке,
в цеолитах поливалентных металлов имеются протонные центры,
обусловленные протонизацией молекул воды, сорбированных катио-
нами металла:
Мс" Н2О-^----Мс+"ОН’ ....Н®
В апротонных центрах носителями каталитической активности яв-
ляются координационно ненасыщенные атомы алюминия с координа-
ционным числом 5,4 или 3. Вероятность существования атомов алюми-
ния с координат!ионным числом 3 мала (структура V), вследствие очень
высокой активности таких атомов. Наиболее вероятно наличие атома
алюминия с координационным числом 4 в алюмокислородном тетра-
эдре АЮ4 (структура IV).
Механизм взаимодействия реагирующих молекул углеводородов
с активными центрами катализатора можно представить следующим
образом.
Протоны, входящие в состав протонных кислотных центров, взаи-
модействуют с молекулами углеводородов, превращая их в карбокати-
оны, которые подвергаются дальнейшим превращениям. В соответ-
ствии с теорией катализа полиэдрами1, выдвинутой И, М. Колеснико-
вым и Г. М. Панченковым, координационно ненасыщенный атом
алюминия в апротонном центре является акцептором электронов.
В процессе хемосорбции, предшествующей химической реакции,
электроны реакционноспособных центров молекул углеводорода пе-
реходят на вакантные орбитали координационно-ненасыщенных ато-
мов алюминия. Образующиеся при этом катион-радикалы углеводо-
родов подвергаются дальнейшим превращениям.
Наличие в алюмосиликатных катализаторах протонных и апротон-
ных кислотных нептров доказано с помощью ИК-спектроскопии.
Найдено, что при адсорбции аммиака или органических оснований на
поверхности алюмосиликатного катализатора существуют соединения,
которые можно рассматривать как продукты взаимодействия основа-
ния с апротонным (а) и протонными (б) центрами.
NH3
а
n*la»>>e>iKO» Г М . Комют И М. // ЖФХ. - 1970. - № 4. - С. 900; Кохесни-
хон И. М.//ЖФХ - 1975. -№ 9. - С. 2175.
Рассмотрим химизм и механизм каталитического крекинга углево-
дородов.
Алканы
Как и при термическом крекинге, при каталитическом крекинге
происходит распад молекул алкана с ооразованием в простейшем случае
молекулы алкана и молекулы алкена меньшей молекулярной массы:
R-CHrCH2-CHrCH3^=^R-CH5 - СН2-СН-СН3
Протекает также дегидрирование с образованием олефинов. Одна-
ко. по сравнению с термическим крекингом каталитический крекинг
имеет ряд особенностей:
1, Скорость каталитического крекинга алканов в 40—60 раз больше
скорости их термического крекинга при од иен г и той же температуре
(500°С).
2. Жидкие продукты каталитического крекинга алканов имеют пре-
имущественно изостроение. Следовательно, в отличие от термическо-
го крекинга при каталитическом крекинге прогскасг интенсивно изо-
меризация.
3. Газообразные продукты каталитического крекинга состоят из уг-
леводородов состава С3 и С4 (пропилен, пропан, изобутилен, изобу-
тан, бутены), в то время как в газообразных продуктах термического
крекинга преобладают углеводороды состава С',. С, (метан, этан и
этилен).
4. Скорость каталитического крекинга чистых алканов резко воз-
растает в присутствии следов олефинов
5. Изоалканы крекируются с большей скоростью. чем алканы с пря-
мой цепью.
Все эти особенности химизма каталитического крекинга, а также
кислотный характер катализаторов крекинга привели к гипотезе об
ионном механизме каталитического крекинга углеводородов через ста-
дию образования карбокатионов. Эта гиггозе за была выдвинута амери-
канскими учеными Хансфордом, Гринсфельлсром и Томасом. Карбо-
катион — это положительно заряженный углеводородный ион, кото-
рый можно рассматривать как продукт отщепления от молекулы алка-
на протона и пары электронов:
Н
R-C-R. + н
н н
Как видно из этой схемы, для образования карбокатиона из молеку-
лы алкана необходимо гетерол и ти чес кое растепление связи С-Н. ко-
торое может проходить как под действием апротонного, так и протон-
ного кислотного центра:
R
\ i © ©
R—С|’-Н + H^=^R3c + Н2
R
Карбокатионы могут быть первичными, вторичными и третичными:
первичный
снгсн-снгсн
вторичный
©
СНг(<СНт
СН3
третичный
Третичные карбокагионы более устойчивы, чем вторичные и пер-
вичные, так как положительный заряд на третичном атоме углерода
частично компенсирован смешением электронов от трех алкильных
групп.
Образование карбокатиона из олефинов на поверхности алюмоси-
ликатного кашли iaropa происходит при взаимодействии протона ката-
литического центра с лэ электронами двойной связи:
R-НС —СЬЬ
©
Н г- -1
© : ________
11^------R.HC==CH)^---R-СН-СНз
© \
L н-1
_ -^R-СН-СНз
©
Стабильные карбокагионы были идентифицированы на поверхнос-
ти алюмосиликат hoi о катализатора с помошью УФ-спектров, Так, при
адсорбции дифенил этилена на алюмосиликате был идентифицирован
дифенил мети лкарбока! ион
©
н _ ©
(Сf,Н 5)=С Н 2 (С6Н фС-С Нт
Образовавшийся карбокатион либо взаимодействуете молекулами
Других угле вол оролов, либо подвергается растеплению. Это приводит
к продолжению реакционной цепи.
Рассмотрим сталии цепного механизма каталитического крекинга
углеводородов.
^ebbA.N. in «Actesdn dctixicmu emigres internalional Catalyse*. Fechnip, Paris. 1961. -
P. IM
А. Зарождение цепи (образование карбокатионов). В случае катали-
тического крекинга чистых алканов следы алкена, необходимого для
образования карбокатиона, могут возникнуть в результате чисто тер-
мического крекинга алкана:
R-CH^CH2-CH2-CH-,^=^R-CH-, СН2=СН-СН;.
Далее алкен адсорбируется на кислотном протонном центре и прев-
ращается в карбокатион:
©_____ ©
СН,=СН-СНз ~ Н^=^СН:-СН-СН.
Как указывалось выше, образование карбокатиона возможно также
непосредственно из молекулы алкана путем отщепления гидрид-иона
под действием протонного центра:
или апротонного центра катализатора:
Al © Z
R--H'^ R - HtAI^
Возможность такого пути образования карбокашона подтверждает-
ся тем, что спектры изобутилена и изобутана, адсорбированных на од-
ном и том же катализаторе, идентичны1.
Б. Развитие цепи. Карбокатионы могут пол вериться различным
превращениям, что приводит к развитию цепного процесса каталити-
ческого крекинга. Все превращения карбокатионов обусловлены их
нестабильностью, стремлением стабили зоваться.
Изомеризация. Первичные карбокатионы стремятся превратиться во
вторичные за счет гидридного переноса (2.1-сдвиг гидрид-иона):
-СН2-СН3—
Вторичные — в третичные за счет алкильного переноса. При ал киль-
ном переносе происходит переход алкиланиона (метил, зтил), причем
разрывается связь в ^-положении по отношению к атому углерода с по-
ложительным зарядом:
СН3 СНз
1
-СНГСН-СН2^ — СНз-С-СЖ
перенос Н ®
Жермен Дж. Каталитические превращения угле во торило в — М.: Мир 1972. — С 57-
снгсн
1
н?с—сн2
• © _____
снгсн-сн2^-----
Другие алкильные группы (пропил, бутил и т, д.) в условиях катали-
тического крекинга легче отщепляются в виде карбокатионов, чем
участвуют в алкильном переносе,
Гидридный 2,1-сдвиг протекает, по-видимому, по схеме
d
CH3-CH-CH2-CH3
Алкильный перенос1 проходит через стадию образования некласси-
ческого трехпен 1 ponoi о карбокатиона (л-комплекса):
Расщепление карбокатионов. Карбокатионы стремятся стабилизиро-
ваться путем разрыва P-связи С—С с образованием алкена и нового
карбокатиона:
|Р ®-----------*RCH?+ СН7=СН2
R-С И i 1 ’С Н 2-СН 2*-К 2
/ . V к ди 7-связи) обусловливается следую-
Разрыв р-свя ш С С (и ис а- J енН0Г0 атома углерода проис-
uw. под действием положительно з р аиИЯ)уа- и р-С-С-свя-
ходит сильное смешение злектронов j значительно удале-
ц-связь С-С поляризуется слабее, так как
на от положительно пряженного атома у
а
ЙСТВИСМ КИСЛМ Л ЬЮ'
' B UO »ИЛ«МОМУ. м».У
й‘-аи.|и очень сильных нртонны* кисло!
'.'Частвовап, и алкиланиины С v С д и ДР'
R-CH,
to
Эго облегчает гстеролитический разрыв p-связи. Разрыв а- и у-свя-
icii энергетически невыгоден, так как приводи! к ооразованию двух
нестабильных частиц.
Так как в процессе каталитического крекинга вторичные и третич-
ные карбокатионы преобладают вследствие изомеризации, то при их
расщеплении образуются углеводороды С3 и С4, которые в основном и
составляют газы каталитического крекинга (в газах термического кре-
кинга преобладают углеводороды С, и С2):
R-CHrCHjcHrCH-CH?-»—R-CH2-CH2 - С H2-CH-CH3
© ©
НГС-СН3^=^К-СН2 + СН?-С-СН;
СНз СНз
Причем легкость разрыва p-связи увеличивается в ряду:
с2-С, < С2-С2 < С2-С3 < С<-С4.
где С।, С2, С3. С4 — соответственно первичный, вторичный, третичный
и четвертичный атомы углерода.
Расщепление карбокатиона по p-связи протекает тем легче, чем
больше молекулярная масса образующегося при распаде нового иона.
Расщепление карбокатионов может проходить также по р-связн
С-Н по отношению к заряженному атому углерода'
© ________ ©
СНз-СН-СН-СНз^—^СНз-СН=СН-СН-; + н
В за имодействие карбокатионов с углеводародам и. Стал к и ваяс ь с мо-
лекулой алкана, карбокатион отрывает от нес гидр ид-ион:
©
R + СН3-СН-СН;^----r:h Ч- сн;сн-сн3
* (• ] ©
\ /Н
В эту реакцию вступают главным образом низшие карбокатионы
СН3 ,С2Н5 ,С3Н7 , Карбокатионы большей молекулярной массы мо-
гут также отрывать гидрид-ионы от молекул углеводородов, однако они
легче расщепляются, чем вступают в эту реакцию.
Сталкиваясь с молекулами олефинов, низшие карбокатионы при-
соединяются к двойной связи так же. как и протон:
СНз + СН2 — CH-CH2-R~ СНз-СН?-СН-СН2-К ,
202
либо отшешяю, оирвд-ион с обра ю)1анисм стаби1ьнт
карбокатионов: льных длш^ьных
— R :Н
CIC-CH-CH-R
+ CH2=CH-CH-R
аллильный
карбокатион
Аллильный карбокатион обладает пониженной реакционной спо-
собностью вследствие гою, что положительный заряд делокализован
между тремя атомами углерода:
HC~СН—СН—R
Ч-------!
В, Обрыв цени:
© е______
Н А ------Н А
© 6____
СН-гСНН-( Н; + А^-Д^НА + СНГСН=СН-СН3
Алкены
Алкены крекируются значительно легче, чем алканы (при терми-
ческом крекише труднее). Это объясняется, по-видимому, большей
способностью алкенов хемосорбироваться на активных центрах ката-
лизатора (наличие лабильных л-электронов).
Скорость каталитического крекинга алкенов на 2-3 порядка выше
скорости их терм и веского крекинга.
При каталитическом крекинге алкены подвергаются следующим
химическим превращениям: крекингу, изомеризации, перераспределе-
нию водорода, циклизации.
Ра с с м огр и м химизм и меха н и з м эт и х п ре вра щен и и.
При крекинге происходит растепление по связи С-С в р-положе-
нии по отношению к двойной связи. При этом из молекулы алкена об-
разуются в простейшем случае две молекулы алкенов:
R-CH2-CH2-(. Н;| НИ H-CH2^^R-CH2-CH=CH2 + СНз-СН=СН2
Реально крекшп протекае т сложнее.
Механизм; .молекула алкена адсорбируется на активном центре
тали затора, присоединяет протон и превращается в карбокатион, ко-
7°рый затем раса цепляется по р-связи:
©____
И-СН2-СНгСН2-СН2-СН=СН2 + ---
203
s (+) _______
к — С H - с Н - с Н 4 • -с Н 2" с I I—с Н з
(+)
I с н - с Н= с Н 2 + R-С Н 2-С н 2с Hi R-с Н 2-С Н-СН2
®+ сн2=сн-сщ
Так как первичные карбокатионы изомеризуются во вторичные,
а затем происходит их распад, в газообразных продуктах каталитичес-
кого крекинга содержатся значительные количеств пропилена.
Изомеризация алкенов. В условиях каталитического крекинга алке-
ны могут подвергаться изомеризации с перемещением двойной связи
и скелетной изомеризации.
а) перемещение двойной связи:
СН^СН-СНгСНт^^СН.-СН-СН-CHs
бугсн-l бутен-2
Механизм:
; Н 7= С Н-С Н 7-С Н1 + Н < — С Н i-C н -с и - с И; -- С Hi- С Н=С Н -CH i
в) скелетная изомеризация:
ch2=ch-ch2-r^
Механизм:
С+)
СН2 = СН—СН2—R + н
СНз---СН
алкил.
перенос
204
Перераспределение водорода. Впервые перераспределение водорода в
процессе превращения алкенов в присутствии кислотных катализато-
ров и аб л юд ал С. С. Наметкин1, П од де й ст ви ем катал и зато pa (Н , S О 4)
алкены полимеризовались. Часть полимеров теряла водород и превра-
щалась в смолистые вещества, другая часть за счет этого подвергалась
гидрированию и превращалась в смесь алканов. С. С. Наметкин назвал
яу реакцию реакцией легидрогидрополимеризации2:
4 C4HS -> 2 С8Н |() -э CSH14 + С8Н ]К
диен алкан
X
трисн —> смолы
Впоследствии было установлено, что эта реакция протекает по ион-
ному механизму (ионное дегидрирование-гидрирование)3,
В условиях каталитического крекинга (повышенные температуры,
алюмосиликат ный катализатор) процесс происходит глубже.
Олефин, адсорбированный на поверхности, постепенно теряет во-
дород, образующиеся более ненасыщенные углеводороды полимеризу-
ются: полимеры, постепенно обедняясь водородом, превращаются
в кокс, а водород насыщает другие молекулы олефинов, превращая их
калканы. Поэтому длительный контакт олефина и алюмосиликатного
катализатора приводит к образованию кокса и алкана:
2 с=С -С -С - С-С -э С-С-С-С-С-С + С=С-С=С-С-С
полимеры (смолы)
кокс
Механизм превращения олефина в этом процессе можно предста-
вит ь следу ю 1 не й схемой:
1) Н2С=СН-С Н2-СН2-СН2-СНА + Н^—Н,С-СН-(СН2)гСНя
гексен-1
©
2) IhCtil СН-(СН?)2-СНз + СН3-СН-(СН2)з-СНз-<—
н_______________________________i
- П,С СН-СН-(СН2)2-СН3 + СНДСН2).ГСЩ
гексан
^Наметкин . Абаку минская Л. Н, // ЖОХ. — 1932. Т. 2 С
2НамсткннС С ТЫиралныс труды -- М Изл-во АН ССС, 1949. —
1 Курсанон Д Н . Ларисе 3 Н. Калиикин М. И.. Лойм Н М. Ионное гидриро-
«ие. - Химия J979
205
© — н
3) н?С=СН-СН-СН-СН2-СНз^=^Н2С=СН-СН = СН-СН3-СНз
Н 1,3-гексадиен
В дальнейшем гексадиен может превратиться в гексатриен и далее
в продукты уплотнения.
Циклизация. Алкены в условиях каталитического крекинга образуют
циклоалканы, циклоалкены и ароматические углеводороды:
-----1—снз
Циклизация, по-видимому, протекает в результате ионного дегид-
рирования олефина через стадию образования диена и триена:
@
СНз-СН3-СНгСН2=СН21^СНз-СН2-СН2-СН-СН-С Н2 ~Н
СНз-СН2-СН=СН-СН=СН2
CHi
смолы
С гЦ
►сн3-сн-сн=сн-сн=сн
Нафтены
Каталитический крекинг нафтенов протекает в -1000 раз быстрее,
км гсрмичсскип. Нафтены подвергаются расшеплению по С—С-свя-
ЗЯМ (расщепляются кольцо или боковая цепь), а также реакциям изо
Меризации и дегидрогенизации.
Рассмотрим химизм и механизм этих реакций.
Расщепление (упрощенная схема):
2 СН3-СН=СН
2
СН3-СН2-СН2-СН2-СН=СН2
Механизм: в первой стадии образуется циклогексильный карбока-
тион
который затем расщепляется по p-связи. Образующийся при этом не-
насыщенный первичный карбокатион изомеризуется во вторичный,
который расщепляется:
СН2-СН2-СН2-СН2-СН=СН2
©
-Н3С-СН=СН2+СН2-СН=СН2
и далее:
СН2
/СН2 + Н3ССН=СН2
сн2
Образование гексена менее вероятно и возможно в результате изо
Аризации ненасыщенного первичного карбокатиона по схеме
н;с-снгснгснгсн=сн2^снгснгсн:-сн-сн=сн:
х X н.с
~
сн?-сн2-сн2-сн-сн=сн2 ©снч
Н2С
н2с—сн2
=±СНГСН2-СН2-СН2-СН=СН2 СН\ /СН2
н2с—сн2
Дегидрирование:
Механизм: пол действием протонного кисло! ного центра катализа-
тора происходит образование карбокатиона и затем растепление
P-связи С-Н:
Н2С^ СН
СН2
В дальнейшем от аллильного атома углерода молекулы циклогексе-
на может отшепиться гидрил-ион:
RH,
208
затем протон:
в бензол.
"о!,НалОГ...СЧСме инклогексадиен превращается
Изомеры юиии. В условиях каталитического кшС
«ки возможна реакция изомеризации ииклогекХт » РМОД"И1Ш"-
ЙВ в ииклопентаноиые (сужение кольца) «•новых углеводоро-
Например
Обратная реакция (расширение никла) термодинамически невоз-
можна при температурах каталитического крекинга (500°С)
Механизм сжашя никла можно объяснить следующей схемой:
“3544
209
rnllB гнлрнл-чопа от молекулы циклогексана
BH.w.ne происходи' 1 * ' К1П.ИО1П, 0(Л1, протона катализатора),
но I действием какою jii > пбокатионе обра 1уется протонирован-
Ыем н ииклогекси^ - £ которое разрывается „о
„ос ииклоиропановое колыю (Д
связи СН,-СН. аш1И „зомеризании -icpe) стадию протони-
Ароматические углеводороды
Голоядерные ароматические углеводороды (бен юл. нафталин) в ус-
ловиях каталитического крекинга практически не подвергаются прев-
ращениям; крекинг толуола в этих условиях про.х<пнт незначительно.
Высшие алкилбензолы крекируются легко, причем скорость их прев-
ращения выше скорости термического крекинга В отличие от терми-
ческого крекинга расщепляется не р-, и ot-связь углерод — углерод бо-
ковой цепи алкилбензола;
СНГ-СНт-СГЬ-И
Hl- —I
СН2СН-СН2-И
Это находит объяснение с позиции гипотезы об образовании
л-комплекса бензольного ядра с активным центром катализатора. Со-
гласно гипотезе об образовании л-комплекса. в процессе крекинга об-
разуется я-комплекс между активным центром катали затора и я-элект-
ронам и бензольного ядра, что приводит к поляризации о.-с вяз и С-С и
облегчает ее гетеролитический разрыв:
( H?-CH-CHrR
Кроме отщепления боковой цепи происходит перемещение боко-
вых цепей по кольцу (изомеризация), которая находит объяснение
с точки зрения карбений-ионной теории.
Например, изомеризация .м-ксилола в /?-ксилол:
1 Петров Ат. А. Химия вафтснон. — М Наука, 1971 С 162
Таким образом, выше мы рассмотрели возможные схемы механиз-
мов превращения углеводородов с помощью карбений-ионной теории.
Качественная сторона этой теории получила общее признание. Она
объясняет многие особенности химизма каталитического крекинга уг-
леводородов. Слабым местом этой теории является количественная
сторона, так как на ее основе не удается предсказать ход процесса ката-
литического крекинга индивидуальных углеводородов и рассчитать
состав продуктов крекинга. Следует также отметить, что существова-
ние карбокатионов на поверхности алюмосиликатных катализаторов
при температурах крекинга не доказано экспериментально. Возможно,
что промежуточными частицами при каталитическом крекинге углево-
дородов являются не карбокатионы (о-комплексы), для образования
которых необходим полный гетеролитический разрыв связей, а по-
верхностные комплексные соединения углеводородов с активными
центрами катализатора. Такими соединениями могут быть я-комплек-
сы, для образования которых требуется меньшая энергия, чем для об-
разования а-комп. тексов:
R-CH=CH-R’
R-CHrCH-R'
аибо катион-радикалы. Катион-радикалы могут образоваться врезуль-
татеодноэлектронного переноса от молекулы углеводорода к активно-
му Центру катали затора:
2Н
Отрыв одного электрона энергетически более выгоден, чем отрыв
двух электронов от атома углерода1 (гетеролитический полный разрыв
связи).
Карбений-ионный механизм каталитического крекинга углеводоро-
дов полностью отрицается в работах Г М. Панченкова и И. М. Колес-
никова2, которые считают, что носителем каталитической активности
алюмосиликатных катализаторов являются не протоны, а координаци-
онно-ненасыщенные атомы алюминия алюмокислородных тетраэдров
и крекинг ал кил бен зол о в протекает через стадию образования катион-
радикалов за счет перехода к-электрона на вакантную 3d-орбиталь ато-
ма алюминия. В соответствии с представлениями об одноэлектронном
переносе и теорией полиэдров И. М. Колесникова, механизм каталити-
ческого крекинга алкилбензолов с участием апротонного центра ката-
лизатора можно представить следующими схемами:
vhmui^ Д '^геРГН' Теория возмущений молекулярных орбиталей в органической
химии. — М.: Мир, 1977. — С. 420.
Г М; ^ченков, Колее”иков// ЖФХ - 1974 - хй 4 - С 900 И. М. Колес
ников//ЖФХ.-1975,-№9.-С 2175: И. М. Колесников//ЖПХ - I960.-Ml"
719
СНз-СН-СНз^^Н + сн2=сн-сн3
CH
В табл. 18 приводится сравнение химизма превращений углеводоро-
дов при термическом крекинге (пиролизе) и каталитическом крекинге.
Устойчивость углеводородов к химическим превращениям возрас-
тает при термическом и каталитическом крекинге в следующих рядах:
Термический крекинг:
парафины < олефины < нафтены < алкиларены <
г< тл оя де р н ы е а ро м а т и ч ес к и е у гл е водороды.
Катал ити чес к и й креки н г;
олефины < алкиларены < нафтены < парафины <
голоядерные ароматические углеводороды.
3.2. Превращения углеводородов и других компонентов
нефти и газа в гидрогенизационных процессах переработки
Процессы переработки углеводородов нефти под давлением водоро-
да (гидрокрекинг, гидроочистка и риформинг) в последнее время при-
обретают все возрастающее значение. Это объясняется рядом причин.
I- В связи с ограниченными запасами нефти перед нефтеперераба-
тывающей промышленностью стоит задача углубленной переработки
нефти с получением максимального количества топлив, смазочных ма-
Сел и сырья для нефтехимической промышленности.
В настоящее время глубина переработки нефти в нашей стране со-
ставляет -65%. Достичь более глубокой переработки нефти невозможно
без применения водорода. Это объясняется тем, что содержание водо-
Р°ла в нефтях невысоко (12-13%) вследствие значительных количеств
смолистых веществ и полициклических ароматических углеводородов,
тогда как содержание водорода в светлых нефтепродуктах выше (напри-
МеР, в бензинах 15-16%). Поэтому для повышения выхода светлых неф-
Таблица 18. Сравнение химизма превращений углеводородов
при термическом и каталитическом крекинге
Углеводороды Реакции
Термический крскиш (пиролиз) Кш д [и гинее кий крекинг
Алканы [. Растепление по связям С-С (в [азах преобладают углеводороды С}, С ,) 2. Дегидрирование низших ал капов 1 Растепление по связям С-С (в । a sax преобладают \ Lie водороды С;. Сд 2.;Ici si.ipnpo[?ание '. (4 тдерн запоя
Олефины. 1. Растепление по связям С-С 2. Дегидрирование 3. Полимеризация (под давлением) 4. Реакция с диеновыми углеводородами, тионовый синтез(иол давлением) 1 Расnieii.пн।не по связям С -С 2 . Дегп л pi [ронял не .' И >омсри зания 4 11ерсрасцрелеление fio три ла (1 илродегидро- [io нннерп шипя) 5. 11пыи дитя
Диолефины Диеновый синтез (пол давлением) I изр», пропояимеризания, при во on идя к образованию
Нафтены 1. Растепление посвязям С-С 2. Дегидрирование 1 Pde ItlCTl. [СНИС посвязям С-С 2 I ci и пнфогшние 3. Г1 дглсри гашш
Ароматические 1. Конденсация с поразова 1 Р;!С!пенлепне алкилбен-
у i леводороды нием мио|и?шершэ1х Я). юн гю </.-связям С-С
углеводородов 2. Распгеплепие алкил- бензолов по [3-связям С С 2 И iU\!rpn кшия
компонентов насышение водородом и растепление тяжелых
7 В ' ЧТ0Д0СТИгае™ в процессе гидрокрекинга.
пользование волопоа™ИТИЧгСК°ГО ри*°РМИ||га и । и.трокрекинга ис-
без реактивации ла способствУет Длительной работе катализаторов
рует ненасышеннм К КаК водороа под повышенным давлением гидри-
на поверхности
•> «кокс». Водород подавляет та кже р^иии^Г"1"' " "ревраШСНИЮ
магических углеводородов, привод™М(ZЮ "Лрж,’"Д"са1Р1и ар0'
отравлению катализаторов. ₽ к коксооора топанию и
нистые и другие ге™оэтом3ннеИ1еЛЬНЫ,( коли'1сс7"ач содержатся сер-
странесернистые и высокосернистые”« Т’ “ т' ме,а;,;,ь1- В нашей
рабатываемых нефтей. Пои It Фи’ состав-:,ЯЮ1 от пере-
Р работке таких нефтей (перегонка ат-
214
«осферная, вакуумная, каталитический крекинг) в нефтепродукты по-
падает значительная доля гетероатомных соединений, ухудшающих их
качество. Для гидрирования гетероатомных соединений, а также поли-
циклических и ненасыщенных углеводородов нефтепродуктов в про-
мышленности широко применяются процессы гидроочистки. В этих
процессах в присуici вин катализаторов под давлением водорода про-
исходит насыщение двойных связей и гидрогенолиз связей С-S, С— N,
С-0 с удалением гстероатомов в виде сероводорода, аммиака и водьц
а также удаление тяжелых металлов (Ni, V и др.). Гидроочистке подвер-
гаются также мазуты (коюльное топливо) и сырье для процессов ката-
литического крекинга и риформинга.
В процессе гидрокрекинга наряду с насыщением водородом и рас-
щеплением полициклических аренов и смол также происходит удале-
ние серы, азота и кислорода, т. е. гидроочистка.
3.2.1. Гидрокрекинг
Гидрокрекинг во шик на базе процессов деструктивной гидрогени-
зации каменного угля, угольных и сланцевых смол. Эти процессы по-
лучили развитие в 30 -40-е годы XX века с целью получения искус-
ственных моторных топлив. Затем интерес к процессам получения топ-
лив на базе каменного угля ослаб, так как такие топлива не могли кон-
курировать с более дешевыми нефтяными.
Процессы гидрокрекинга нефтяного сырья начали развиваться
в 50-60-х голах XX века. Значение их возросло особенно в последнее
время в связи с проблемой углубленной переработки нефти и повыше-
нием требований к качеству нефтепродуктов.
Сырьем процесса гидрокрекинга обычно являются тяжелые нефтя-
ные дистилляты. нефтяные остатки (мазут, гудрон),тяжелые и высоко-
сернистые нефти.
Гидрокрекинг очень гибкий процесс. В зависимости от сырья и
условий в результате гидрокрекинга можно получать различные нефте-
продукты: высокооктановый бензин, реактивное или дизельное топли-
во,смазочные масла, котельное топливо.
Гидрокрекинг может протекать в одну стадию или в две стадии.
Водностадийном процессе наряду с гидрированием и крекингом
компонентов сырья происходит гидроочистка от азотистых, сернистых
соединений и металлов.
В двухступенчатом процессе стадия гидроочистки и частичного 1ид-
рирования угле водородов отделена от стадии гидрокрекинга.
Одностадийный гидрокрекинг гудрона, тяжелых смолистых нефтей
проводят при 400- 500rС и давлении водорода от 7,0 до 20 МПа. При
яом из сырья удаляется до 90-98% серы и металлов. В качестве ката-
215
шшоров используют бифункциональные оксидные катализаторы
содержащие как крекирующие, так и гидрирующие центры.
Продуктами гидрокрекинга этого типа в зависимости от степени
превращения сырья являются: газ (3-5%), бензиновая фракция
70-175°С (12-22%); фракция 170-370’С (30—60%), газойлевая фрак-
ция 370-540’С (20-10%) и остаток >540’С (40-5%), который служит
котельным топливом,
В большинстве процессов гидрокрекинга в качестве сырья исполь-
зуют широкие тяжелые фракции (например, вакуумный газойль); в ка-
честве компонентов сырья могут быть использованы тяжелые фракции
продуктов вторичных процессов.
Процессы этого типа проводят при 370-4 50’С и давлении 10-
24 МПа (процессы высокого давления) или 7,0-10,0 МПа (процессы
среднего давления). Они могут быть одноступенчатыми или двухсту-
пенчатыми. Продуктами этих процессов являются газ, бензин, газойли
(легкие или тяжелые) и очищенные от гетероатомов и металлов остат-
ки, выкипающие выше 380°С (мазуты), которые могут служить либо
котельным топливом, либо сырьем для каталитического крекинга.
В случаях двухступенчатого гидрокрекинга тяжелых нефтяных
фракций в первой ступени в присутствии оксидных катализаторов при
350-400’С и давлении 10-15 МПа происходит насыщение сырья водо-
родом в результате разрыва к-связей С-С, связей C-S, C-N, С-0;
происходит также разрыв наиболее слабых о-связей С—С. Ниже при-
водятся усредненные значения энергий этих связей.
Связь Е, кДж/моль
я С-С (олефины) 167
C-S 272
C-N 335
С-0 377
Энергия диссоциации наиболее слабых о-С-С-связей составляет
294 кДж/моль. Если сопоставить приведенные значения энергий свя-
зей, то видно, что наиболее легко должны расщепляться и гидриро-
ваться я-С-С-связи, затем С-S, слабые о-С—С-связи, затем связи
С—N и С—О. Однако в присутствии катализаторов легче разрываться и
гидрироваться будут связи C -S, C-N, С-О, а затем уже о-связи С-С,
так как разрыв связей С—S, С—N, С—О облегчается в результате элект-
ро нодо норн ой способности атомов S, N, О и их более прочной хемо-
сорбции на каталитически активных центрах.
При разрыве связей С—S, С—N и С—О и насыщении осколков водо-
родом происходит удаление гетероатомов в виде H2S, NH3 и Н2О. Наи-
более легко происходит удаление из сырья серы и труднее — азота и
кислорода. Таким образом, происходят гидроочистка и частично кре-
кинг. Ниже приводятся схемы реакций, протекающих в этом процессе.
216
1. Гидрирование алифатических
двойных связей;
R-CH-CH-R~±:R-CHrCH2-R
2, Частичное гидрирование полициклических ароматических
углеводородов.
3, Гидрогенолиз гетероатомных соединений:
217
4, Гидрогенолиз алканов и алкильных боковых цепей. Происходит
в небольшой степени, разрываются наиболее слабые связи:
R-CH2icH2-R-t^J^R СН3 + CH3R
Во II ступени гидрокрекинга тяжелого дестиллятного сырья, каки
в одноступенчатом, применяют активные бифункциональные катализа-
торы: оксиды и сульфиды вольфрама, молибдена, никеля, а также метал-
лы — на кислотном носителе (оксид алюминия, аморфные алюмосили-
каты, цеолиты). Они содержат каталитически активные центры двух
типов: 1) гидрирующие, 2) крекирующие и изомеризующие, В случае ка-
тализаторов Pt, Ni на кислотных носителях такими центрами являются:
— атомы металлов, которые выполняют функцию гидрирования.
Это — гидрирующие активные центры;
— кислотные центры носителя (протонные и апротонные), на кото-
рых протекает расщепление и изомеризация углеводородов.
Гидрирующие центры, находящиеся рядом с крекирующими, пре-
пятствуют деактивации последних, так как ускоряют гидрирование ад-
сорбированных ненасыщенных углеводородов, что препятствует их уп-
лотнению и превращению в кокс. В случае оксидных катализаторов,
таких как Со2О3-МоО3-А12О3 или WO3-A13O3, МоО3-А13О3, гидрирую-
щую функцию выполняют оксиды Со, Mo, W. Крекинг и изомеризация
проходят на кислотных центрах оксида алюминия.
Сульфиды W, Mo, Ni содержат как гидрирующие, так и кислотные
активные центры.
Природа активных центров оксидов и сульфидов металлов может
быть объяснена исходя из электронных представлений в катализе1.
1 Вол ькен штейн Ф. Ф. Электронная теория катализа па полупроводниках. — М.‘
Физматиздат, I960 — С. 187
218
Свободная <оаа -
зона провод и м ост и
Запре11[ с 1111 ая !°11 а
Оксиды и сульфиды металлов
(W, Mo, Ni, Со и др.) являются по-
лупроводниками. В полупроводни-
ках переход электронов из валент-
ной зоны в зону проводимости тре-
бует преодоления энергетического
барьера, так называемой энергии
а кт и ва ц и и эл е ктро провод н ост и
(к) (рис. 59). Это объясняется тем.
В ал е и т п а я и> н а
Рис. 59
JOB металле атомы - нейтральные частицы
мены. В оксидах или сульфидах находятся ион КТроны °бобщес-
?Ува электронов от ионов требуется энергия П МетаЛпов’ и Для от-
крой? оксидов-изоляторов) начинают п °ЭТ°Му оксиды метал-
нагревании*
Полупроводники подразделяются на л-полуппл
к) и/’-полупроводник и (дырочные) R по™™ ВОЛНИ|<И (электрон-
всегда имеются llpm Л
ясостава(избыток металла или металлоида) Hann ИометРическо-
в имеется примесь пинка (1(„нк
рои электронов). При нагревании про—ион ПРИМесЬ10’ дон°-
илутем отщепления электронов; Р°ИСХОЛИТ ^изация атомов цин-
Zn —> Zir Ч- ё~,
Электрон может переходить на ион цинка:
Zn2-’ + ё-—» Zn+
нот иона Zn+ к иону пинка Zn2\ то есть электрон блуждает по крис-
таллической решетке оксида цинка. Причем соответственно появля-
ются «блуждающие» ионы пинка.
Оксид цинка с избыточными атомами цинка является типичным
электронным полупроводником («-полупроводником). Если же в ок-
сиде металла в избытке находится металлоид, который является акцеп-
тором электронов, то в решетке оксида или сульфида появляются
•дырки» за счет перехода электронов от ионов металла к металлоиду.
Этот переход ускоряется с повышением температуры. Например, в
оксиде никеля (II) присутствует избыточный кислород. Он захватыва-
дэлектроны у Ni2'*' и превращает его в Nli3+.
Ni3+захватывает, в свою очередь, электрону соседнего иона Ni2+, то
гсть происходит перемещение дырок (положительных зарядов); возни-
кает дырочная проводимость или р-проводимость. Оксид никеля (II)
является р-пол у проводи и ком.
Следует отметить, что ряд промышленных катализаторов гидрокре-
кинга являются «-полупроводниками. Это Fe2O3, WO3, МоО3. На по-
верхности таких катализаторов могут находиться свободные электро-
219
ны, При вMHiMoneiicTiMUi с которыми молекулы углеводородов, водоро-
да или других компонентов реакционной смеси подвергаются гомоли-
тическому растеплению с образованием свободных радикалов, атомов
водорода.'Таким образом, механизм гидрокрекинга над окисными ка-
тализаторами (п-полупроводниками) — радикальный:
AWA*
блуждающий
электрон
Образовавшийся радикал или атомарный водород продолжает цеп-
ной процесс крекинга.
Промышленные сульфидные катализаторы являются ^-полупро-
водниками. Они содержат примесь серы. Так, формула промышленно-
го катализатора — сульфида вольфрама WS2 2. а нс WS,. Избыточная
сера в сульфидах обусловливает образование «дырок»., под влиянием
которых происходит гетеролитический разрыв связей в углеводородах
с образованием карбокатионов. Молекулы водорода пол действием
«дырок» могут также подвергаться гетеролит и чес кому растеплению с
образованием протонов:
©
R
Карбокатионы и протоны продолжают цепной процесс превраще-
ния углеводородов, Таким образом, на сульфидных катализаторах про-
текают ионные реакции превращения углеводородов (крекинг, изоме-
ризация). Вместе с тем на поверхности сульфидных катализаторов
имеются также неспаренные электроны, которые обусловливают про-
текание радикальных реакций, в частности реакции гидрирования.
Следовательно, сульфидные катализаторы являются бифункцио-
нальными.
Во второй ступени двухступенчатого гидрокрекинга тяжелых неф-
220
ТЯНЫХ фракции, а ткже в процессе одноступенчатого гидрокрекинга
протекает гидрогенизация сернистых, азотистых, кислородных соеди-
нений, которые не подверглись гидрогенизации в первой ступени (или
содержатся в сырье одноступенчатого гидрокрекинга). Схемы превра-
щений этих соединений приведены выше.
Углеводороды сырья подвергаются глубоким превращениям. Рас-
смотрим основные превращения углеводородов в процессе гидрокре-
кинга.
Алканы в условиях гидрокрекинга над бифункциональным катали-
затором подвергаются превращениям потрём направлениям!
Крекинг с участием кислотных центров с насыщением продуктов
растепления водородом:
> н®
r-ch,-ch|ch2-r---------►RCH=CH2 + CHrR
R-CH2-CH3
Гидрогенолиз на гидрирующих активных центрах:
R-CH7-CH)|cHvR“^R-CHtCH^ + CHrR
< ПЭ "
Изомеризация на кислотных центрах:
© *[нз
R С Н 2-СН,-С Н2-R-H » R-СН-СНг-R
Крекинг и изомеризация протекают с участием обоих центров ката-
лизатора (гидрируюших-дегидрируюших и изомеризующих), и зарож-
дение их происходит на центрах гидрирования-дегидрирования.
Хотя в условиях высокого давления водорода равновесие гидриро-
вание-дегидрирование для алканов смещено в сторону гидрирования,
небольшое количество олефинов образуется:
R-С Н 2-С Н R-С Н =С Н 2 + Н2
Образовавшийся олефин сорбируется на кислотных центрах ката-
лизатора и превращается в карбокатион, который инициирует реакции
изомеризации или крекинга.
Гидрогенолиз алканов протекает, по-видимому, по радикальному
механизму. Квантовомеханические расчеты показывают, что электрон-
ная плотность у атомов «С» в алканах падает в ряду:
С > С > С > С
Ш1ерн '“'втор трет четверг
221
Поэтому, если катализатор обладает электроноакцепторными свой-
ствами (например, Ni/Al3O3), то разрываются связи между атомами уг-
лерода с повышенной электронной плотностью, если электронодонор-
ными (Pt/SiO?), то разрываются связи между атомами углерода с пони-
женной электронной плотностью1.
Соотношение между реакциями гидрогенолиза, крекинга и изоме-
ризации определяется соотношением между кислотной и гидрирую-
шей-дегидрируюшей функциями катализатора, а также давлением
водорода и температурой.
Повышение давления водорода тормозит протекание ионных про-
цессов крекинга и изомеризации алканов. Это объясняется следующи-
ми причинами. Во-первых, повышение давления водорода, препят-
ствуя дегидрированию алканов, понижает концентрацию олефинов,
а следовательно, снижает скорость изомеризации и крекинга, так как
олефины являются промежуточными продуктами этих реакций. Во-
вторых, с повышением давления водорода повышается концентрация
на поверхности катализатора поляризованных молекул водорода, акти-
вированных под действием активных электроноакцепторных центров
катализатора, напримералюмокислородных полиэдров (Д1О4).
При взаимодействии карбокатионов с этими молекулами водорода
они преврашаются в молекулы углеводородов:
© © ©
R + Н - Н“
I
©
RH + Н
Вместе с тем необходимо отметить, что повышение давления водо-
рода способствует гидрогенолизу алканов на гидрирующих активных
центрах.
В отдельных случаях гидрогенолиз н-алканон является желательным
(процессы гидроген и за цион ной депарафинизации дизельных топлив и
масел). Катализаторами этих процессов являются Pt на неолите или це-
олит с высоким содержанием оксида кремния. Поры этих катализато-
ров имеют малые размеры, и в них проникают преимущественно //-ал-
каны, которые и подвергаются гидрогенолизу. Установлены2 относи-
тельные скорости гидрогенолиза алканов состава С6, С7: //-гептан —
1,00; 2-метил гексан — 0,52; 2, З-д и метил пентан — 0,09; л-гексан — 0,71;
2-метилпентан — 0,38; 2,2-диметил-бутан — 0,09.
1 I cvnskii 1. I. // J. Caial. - 1979. - V 58. - № 1. - Р 144
Weis/ Р. В. Ij Kinetics and Catalysis in Petroleum Processing. Preprint of the Tenth World
Petroleum Congress Review Papers — № 5. — p.5.
222
Нафтены в условиях гидпомо-
ра щен иям: Р крекинга подвергаются следующим прев-
L Крекинг длинных боковых цепей нз
щением водородом продуктов крекинга ЛОТНЫХ литрах с насы-
дит расщепление колец. В случае катал’ В Меньшей степе»и происхо-
гидрируюшие центры (металлы - Pt Рн* кг?Р0В’ Имеющих активные
колец: ’ ’ пРотекает гидрогенолиз
СН3
^СН3<Н2-СН-СНГСН2-СН3
2. Изомеризация. Моноциклические насЬтем., г 11IIL,
п'енпнов- П°ЛВергаЮТСЯ ИЗОМеРизацни с образован
Бициклические нафтены — производные декалина и гидриндана —
подвергаются изомеризации с образованием производных пентала на:
R
либо деструктивной изомеризации в ал кил циклопентаны:
Ароматические умеводороды в условиях гидрокрекинга претерпева-
ют следующие превращения:
Гидрирование с последующим расщеплением циклогексановых ко-
лец. Гидрированию подвергаются., в основном, полициклические аро-
матические углеводороды. В первую очередь гидрируются незамещен-
ные ароматические кольца; конечными продуктами являются алкил-
бензолы, дальнейшее гидрирование которых нежелательно, так как это
приводит к снижению октанового числа бензина.
223
224
Реакциям гидрирования полициклических
повышенное давление водорода кмгл Х aptH0B способствуют
центров катализатора и бо “скопи . ™°СТЬ г^Р»РУюшкх
лических аренов (ПА) к гидрирующим Ь^“^узии молекУЛ полицик-
ииатора. Учитывая значительные отшр реКирующим центрам ката-
алюмосил и каты (как компоненты катали?Ы M0J1CKyjl ПА- аморфные
ПОРЫ. предпочтительнее по^сравХни^с^пеп^011’ Ш,,р0К,1е
Так как в силу этой причины скопп " (УЗКИС ™РЫ)’
веских аренов на катализаторах содержаших^'Т0^41^ полицикли“
шои. то значительную роль начнут Ифать другие реакции деп|дрош?к'
лизания алкиларенов и реакция диенового синтеза, что может прX'
™ к повьпиению концентрации ПА и ухудшению свойств продуХ
гллрокрекиш а. приду к юн
Дегидроциклизация алкиларенов
Хотя повышенное давление и более низкие температуры гндрокре-
M1HI.I (по к равнению с риформингом) не способствуют легидроцикли-
зации. зга реакция может протекать в значительной степени. Так1 ал-
^"-"'"•-"-рои-водные ПА в условиях гидрокрекинга при
.88 С и к.. МПа В значительной степени подвергаются дегидроцикли-
ЗИНИИ, ОЧСВИДПО. по схеме
Реакция диенового синтеза
Этой реакции способствует повышенное давление в процессефш-
рокрекинза. Полициклические ароматические углеводороды могу,
выступать как и роли диенофила, гак и в роли «диена
‘Energy ягк! Fuels. - 1989. № 3. Р 611.
225
’5-3544
Крекинг(деалкилирование)
Алкиларены расщепляются на кислотных лен грз\ • а i ал и затора по
«-связям:
Механизм деалкилирования аналогичен мсл-лич згой реакции
при каталитическом крекинге (см. VI). На ; h.iphp лих металличес-
ких центрах возможен гидрогенолиз длинных пы. линей с образо-
ванием алкилбензолов с короткими боковыми целями
Изомеризация
В условиях гидрокрекинга на кислотных пен । р-н :нисходит изоме-
ризация боковых цепей алкилбензолов и их перемещение по кольну.
3.2.2. Гидроочистка
Процессы гидроочистки нефтепродуктов по очи ш развитие в свя-
зи с увеличением доли сернистых нефтей в ш?Ф г сг:с реработ ке и повы-
шением требований к качеству топлив, масел, л оме сырья для ката-
литических процессов (каталитического крекинга, риформинга). Нель
процессов гидроочистки — удаление из нефтепродуктов гетероатомов
(О, N, S) и металлов, а также гидрирование непредельных и частично
а ро ма тич ес ки х у г ле водородов.
В качестве катализаторов применяют вещества. устойчивые по от-
ношению к отравляющему действию серы. Это оксиды и сульфиды ме-
таллов переменной валентности — Ni. Со, Mo, W. Металлические ката-
226
>И»торы(\'|. P,.pd). нес
ность. не использ, ют Пр„ г,ЦроочХыс^» ка^птическ, ю акт,,в-
клея кататнмторы гидроочистки и ' Так как они быстро птпч»
мидму гидроочистки различных неЛ™ Прежде чем перейти L Д.
и катализаторы. „Ри которых npo^n*«W»". рассмотрим условия
ние олефинов и ароматических тгле°ХОппЛеСТРУКТИВНОС '«рнровд
дажны оьпь онределяюшими при вьХТ*' Как эт» «ЗД я
рмусуслониямн падения гетероатомов Нс ВИЙ гидР°»<<истк„ Н|.
аве олефинов протекает легко в присутствий“?УКТИ8ное "Цирова-
торов < Pi. fd. используемых в ваде м^лко ЛИЧеСКИХ ката"'™-
твбо на носителях с развитой поверхности?,ИСПерсны* порошков,
силикагель, пины,, в присутствии наиболее акт И"Ировднны« Уголь,
й и Pd. а также Г ю, полное гидрирование одД НЫ* Катал«»™М
хает при компа тпоп температуре и атмосфХХ™ ниТ”0” ПР°те'
К< H=CH2^R.CHrCH2
20 С, атм. данл.
В случае мсыл.шчсского никеля в мелкодисперсном состоянии
(никель Ренея) ;.ля iилриронания двойной связи олефинов требуется
более высокая i м г1 ерат ура (-1 Х0"С) и атмосферное давление водорода.
Легче гидрируючя олефины с концевой двойной связью, труднее -
олефины с пен ipuльным расположением двойной связи, особенно ес-
ли двойная спя <ь ,4.раниронанэ объёмистыми углеводородными груп-
пами. Цис-м юмеры олефинов гидрируются легче, чем транс-изомеры.
Полное i парирование диенов до алканов протекает примерно в тех
же условиях, чини илрирование олефинов, однако при этом требуется
больше времени ! илрирование протекает ступенчато:
лисп -к олефин -> алкан.
В промышленности и в лабораторных условиях иногда ставится за-
йма селектившл<> парирования диеновых и ацетиленовых углеводо-
родов в их смеси с (ефинами. Для этой цели используют комплексы
платины и пл ! и < ра (личными лигандами, а также некоторые суль-
фиды (напрпмс-ц PdS. NiyS}). Так, удается селективно гидрировать
«присутствии m г I । ^комплексных катализаторов аллен и метилаце-
тклен до прони т-Нг! н пропан-пропиленовой фракции газов пиролиза1.
Циклопенталисн селективно гидрируется я циклопентен над
MS/AI/L при а I мосферном давлении и 75-85’С1. В случае оксидных
'Беренб;|н>м А (
I !-м1н Л И . Кире ли кий В. В. и лр, // Нефтепереработка и
•• 12 С 26 V)
227
катализаторов (оксиды Ni, Со. Cr, Mo, W) для гидрирования олефинов
и диенов требуются жесткие условия.
В нефтеперерабатывающей промышленности полное гидрирование
олефинов до алканов проводят с целью подготовки сырья для катали-
тического риформинга, если сырьем служат бензины термического или
каталитического крекинга. При давлении 2—3 МПа и температуре
350-400°С в присутствии катализатора на основе оксидов Со, Mo, AI
(алюмокобальтомолибденовый катализатор) происходит практически
полное гидрирование олефинов в алканы. Гидрирование диеновых уг-
леводородов в олефины, необходимое для стабилизации бензинов тер-
мического крекинга, пиролиза, а также гидрирование наиболее актив-
ных и поэтому малостабильных олефинов бензинов каталитического
крекинга протекает над оксидными катализаторами в более мягких ус-
ловиях: температура 300—350°С, давление 0,5-2 МПа.
Ароматические углеводороды гидрируются труднее, чем олефины и
диены. Наиболее трудно гидрируется бензол, что объясняется равно-
мерным распределением д-электро иной плотности в молекуле. Для
гидрирования бензола в присутствии Pt и Pd при комнатной темпера-
туре требуется повышенное давление водорода 0,3-0.5МПа:
В случае никелевых катализаторов, гидрирование бензола и алкил-
бензолов проводят при 150-250°С и давлении до 12 МПа.
Следует отметить, что сернистые соединения, даже в виде примеси,
в ароматических углеводородах подавляют активность металлических
катализаторов. Поэтому в промышленных условиях гидрирование аро-
матических углеводородов нефтепродуктов проводят на оксидах и
сульфидах. В присутствии оксидов Ni. Со. Mo, W гидрирование бензо-
ла и его гомологов протекает в весьма жестких условиях: 350-450’С и
давление водорода 10-25 МПа. Следует отметить, что. как правило, ал-
кил бензолы гидрируются труднее бензола, вследствие пространствен-
ных затруднений при адсорбции на поверхности катализатора.
Нафталин гидрируется легче бензола (Ni2O3, 1,2- 1,5МПа, 180—
200°С1 2 3). Это объясняется неравномерным распределением я-электрон-
ной плотности в его молекуле. Скорость гидрирования нафталина до
1 Мухина Т Н , Кричко А А,, Черных С. Г] и лр. // I ЬЬтехимия - 1979. - Т. 19. -
№. 5— С 663.
- Селективное гидрирование нафталина в тетралин лоеппаится также при 260 С.
3 МПа над PdS/A 1,0, (Мухина Т Н. Крич ко А А , Черных С II и др // Нефтехимия. -
1979 -Т. 19. — № 5. — С. 663).
228
тетралина в несколько раз выше скорости гидрирования бензола в оди
наковых условиях:
Однако дальнейшее гидрирование тетралина в декалин протекает
значительно труднее, чем гидрирование бензола:
При гидрировании моноалкил нафталинов значительно легче гид-
рируется незаметен ное ароматическое кольцо:
R 2()0(’Ct -ЮМПа
— W.,
оксиды С г, Си
Соотношение углеводородов I и II в продуктах реакции равно при-
мерно 2:1.
При гидрировании антрацена легче гидрируется среднее кольцо:
150(1С, ЮМ Па
9.10-лигидроантраисн
I
тетрагидроантрацен
П
9,10-дигидроантрацен (1) преобладает в продуктах реакции. После-
дующее гидрирование соединений 1 и II протекает значительно труд
нее. Фенантрен селективно гидрируется в 9,10-дигидрофенантрен
229
в присутствии Pd при комнатной температуре и неоольшом давлении
водорода1:
В настоящее время установлено, что гидрирование непредельных и
ароматических углеводородов проходит через с i алию образования
л-комплекса с активными центрами капы и затора в результате перехо-
да я-электронов на вакантные ^/-орбитали металла или его ионов.
Ниже приведена схема механизма ступенчатой гидрогенизации
бензола над Pt-катализатором:
На стадии образования циклогексена возможна десорбция послед-
него. В продуктах гидрирования бензола в ряде случаев обнаружены
небольшие количества циклогексена
Таковы в общих чертах условия и закономерное ! и нилеструктивно-
го гидрирования непредельных и ароматических углеводородов.
Рассмотрим теперь некоторые вопросы химизма и механизма прев-
ращения гетероатомных соединений в условиях процессов гидроочист-
ки. Особенности превращения гетероатомных соединений в условиях
1 J. Org Clicjn. — J980. — V 45 - XT 14. - P. 2797.
230
гидроочистки, по сравнению с гид
что катализаторы гидроочистки облай к обуело|,ли»аются тем,
слабыми кислотными свойствами чем °бЫЧН° значительно более
В процессе гидроочистки происходит „я,тализаторы гидрокрекинга,
с насыщением осколков воДородом. ПриХ'^ C~S' C~N' С~°
ходит разрыва простых С-С-связей maur пРак™чески не пропс-
иолиз связи eLlm|-s. При этом меркаптанr,W|,orc’
на алканы, сероводород: сульфиды распадаются
RSH + H2->RH + H S,
RSR' + 2H2-*RH + R'H + H s.
Связь С —S ппочнгсС с _ 2
кие сульфиды труднее подвергаются И арОматичес'
Тиофан и его производные подвергают™ У'
с такой же скоростью, что н алифатические сульфвдьг:°Л'”У "Р"Мерно
► CHrj2H-CHrCH3 + H2S
сн3
Скорость гидрогенолиза тиофена в несколько раз меньше скорости
гидрогенолиза i иофана. Это объясняется повышенной прочностью
связи С-S вследствие сопряжения неподеленных электронных пар
атома серы и я- ыектронов двойных связей. Установлено, что на оксид-
ных катализа юрах гидроочистки в обычных условиях гидрогенолиз ти-
офена протекает но схеме
2Н2---► Н2ОСН-СН=СН2 + H2S
н2
СНГСН2-СН=СН2---► С4НЮ
Образование тиофана не наблюдается:
I Ц + 2Н2 -/-*[
Дибензтиофсн превращается труднее тиофена с образованием ди-
фенила и никли с кс ил бен зола:
231
Дисульфиды четко конверт a jo гея гндрогсиоли iy и лают в конечном
итоге алканы и сероводород:
R-S-S-R 2 R-SH 2 RU I 2 II,$
А чп со ежится » нефтепродуктах в основном в составе гетерот-
-К ,х сХненин - производных пиррола и пиридина. Гидрогено-
"и св" и C-N протекает труднее, чем связи С -S. поэтому в процессе
1ИЗ связи u I ртся значительно труднее, чем сера.
TTXSпс^гидрогенолизу, по'-видимому. производные
пиррола:
НЛ’-СН-СНз
Труднее — производные пиридина, так как сопряженная электрон-
ная система в молекуле пиридина значительно более устойчива, чем
в молекуле пиррола:
*- СН3-
H-CH2-CH3-CH2-NH
Нз
2---
-NH
3
R-СДНц
R
Если ядро пиридина конденсировано с бензольным ядром (напри-
мер, в молекуле хинолина или изохинолина), то вначале гидрируется
кольцо, содержащее азот, а затем бензольное:
232
Это подтверждается тем, что в бензине гидрокрекинга 80-90% азо-
тистых оснований приходится на долю алкиланилинов1. Кислородные
соединения гидрируются по схемам, аналогичным приведенным
ранее.
Рассмотрим кратко химизм основных промышленных процессов
гидроочистки.
Гидроочистки сырья для платформинга.
Если сырьем для платформинга является прямогонный бензин, то
целью гидроочистки является максимальное удаление серы, азота, а так-
же микрокод к ч ест в мышьяка и свинца, так как соединения всех этих
элементов являются ядами для катализаторов платформинга, причем
мышьяк и свинси вызывают необратимое отравление катализаторов.
Гидроочистку проводят обычно в присутствии алюмо-кобальт-мо-
либденового катализатора (А12О,-СоО-МоО3) при 380-420СС и давле-
нии водорода 2.5—4 МПа.
Если к сырью платформинга добавляются бензины вторичного про-
исхождения. то при гидроочистке происходит также полное гидриро-
вание олефинов-
Селективная гидроочистка (облагораживание) бензинов термичес-
кого и каталитического крекинга проводится с целью удаления серы,
азота, а также гидрирования диенов и некоторых олефинов (алкенил-
бензолов). которые снижают стабильность бензинов к окислению.
Катализатор — алюмо-кобальт-молибденовый, температура 340—
360’С, давление 1-2 МПа. В случае более активного сульфидного ни-
кель-вольфрамового катализатора — температура 300 С, давление
0,5 МПа.
Кроме гидрогенолиза гетероатомных соединений, гидрирования
диенов и части олефинов, происходит также изомеризация олефинов.
Гидроочистку керосиновых фракций проводят с целью получения ре-
активного топлива. Применяют обычно алюмо-кобальт-молибдено-
ПВайков А. Я., Ьелыювский“В Г. // Химия и технология топлив и масел- - 1960. -
№ 5. • С. 34.
233
Bbiii катализатор, температура 300—40СГС, давление э-6 МПа, При
этом происходит гидрогенолиз сернистых, азотистых, кислородных
соединений и смолистых веществ и повышается термоокислительная
стабильность топлив. В случае, если нужно понизить содержание аро-
матических углеводородов (алкилнафталинов). применяют более ак-
тивные сульфидные катализаторы, тогда происходит гидрирование
ароматических колен.
Гидроочистку газойлей проводят для получения дизельных топлив.
Если используют газойлевую фракцию прямой переi инки, то условия и
химизм гидроочистки аналогичны условиям и химизму гидроочистки
керосиновых фракций. Если используют газойли каталитического кре-
кинга и коксования, необходимы более жесткие условия и более актив-
ные катализаторы, так как для получения стабильною дизельного топ-
лива с высоким цетановым числом необходимо гидрирование олефи-
нов и основной части ароматических углеводородов: температура до
400сС, давление до 10 МПа.
Гидроочистка сернистых вакуумных газойлей и другого тяжелого
сырья для процесса каталитического крекинга приводит к гидрирова-
нию полициклических ароматических углеводородов, металлоорга-
нических, смолистых соединений и способствует более длительной и
эффективной работе катализаторов крекинга, повышению выходам
качества бензина. Процесс гидроочистки тяжелого сырья для катали-
тического крекинга проводят при температуре 350-4002С. давлении
5- 6 МПа, катализаторы: алюмо-кобальт- или алюмо-никель-молиб-
деновые.
Гидроочистка (облагораживание) является шключительным этапом
производства высококачественных смазочных масел: температура
250-375’С, давление водорода: 2-5 МПа. катали шор — алюмо-ко-
бальт-молибденовый. В этих сравнительно мягких \ ел о вия.х проходит
в основном гидрогенолиз кислородных, сернистых и азотистых соеди-
нений и разрушение смолистых веществ. В результате улучшается цвет,
стабильность масел к окислению, снижается кислю ное число, содер-
жание азота, серы и кислорода. Гидрирование ароматических углеводо-
родов не проходит.
3.2.3. Каталитический риформинг
Каталитический риформинг — процесс превращения низкооктано-
вых бензинов с целью повышения их октанового числа или получения
арома 1 ических углеводородов для химической переработки
Для получения высокооктанового бензина в качес тве сырья приме-
няют фракцию 80—180 С. В результате каталитического риформинга
получают бензин с октановым числом 80-85. Для получения аромати-
234
чес к их углеводородов используют узкие фракции: 60 85° С — для полу-
чения бензола: 8>-110’С — толуола; 110- 140°С — ксилолов.
Процессы каталитического риформинга подразделяются:
1) процессы с ка гализаторами на основе оксидов металлов. Это про-
цессы гидроформинга. Катализаторами гидроформинга являются ок-
сиды молибдена, хрома, кобальта. Температура 480-540°С, давление
водорода 1- 2 МПа, в настоящее время не применяются;
2) процессы с применением катализаторов, содержащих платину.
Это процессы платформинга. Катализаторы платформинга состоят из
платины (обычно 0.3-0,6% мае.), нанесенной на у-оксид алюминия,
который активируют НО или HF с целью повышения его кислотной
активности (содержание С1 или F 0,5—1%). Кроме оксида алюминия,
в качестве носи гелей применяют аморфные алюмосиликаты и цеоли-
ты. Темпера гура процессов платформинга 470- 540 С, давление водо-
рода 2,5- 5 М Па.
В последнее время получили распространение более эффективные
платино-рениевыс катализаторы, с применением которых удается сни-
зить давление водорода до 1-1,5 МПа, повысить выходи качество бен-
зина. Рении препятствует рекристаллизации (укрупнению частиц) пла-
тины на поверхности носителя, благодаря чему высокое число плати-
новых акт ивных петров сохраняется длительное время.
Гидроформиш был одним из первых процессов каталитического
риформинга В настоящее время он потерял свое значение вследствие
малой активности оксидных катализаторов по сравнению с платино-
выми и уступил место платформингу. Катализаторы платформинга
(особенно плагнно-рениевыс) работают непрерывно, без реактивации
длительное время (1-2 гола). Это объясняется тем, что в присутствии
платины пол давлением водорода происходит гидрирование ненасы-
щенных у|лсно;1ородов, что препятствует их уплотнению и образова-
нию кокса, снижающего активность катализатора. Реактивацию ката-
лизатора проводят, пропуская через него воздух, разбавленный дымо-
выми газами (содержание кислорода до 2%) при 300-450*С и 1-2 МПа.
Проводят также реактивацию обработкой водородом. Общая продол-
жительность работ ы катализатора - несколько лет. Значительное вли-
яние на длительность работы катализаторов нлатформигиа оказываю!
азотистые и сернистые соединения, а также примеси в сырье мышьяка
и свинна. Азотистые соединения подавляют активность кислотных
центров. Сернистые соединения вызывают обратимое отравление пла-
тины (активность восстанавливается после реактивации), а мышьяк и
свинец необратимо отравляют платину, Поэтому в сырье платфор-
минга должно быть нс более 0,002% серы, 0,510“4% азота и полностью
должны отсутствовать мышьяк и свинец. Это достигается блаюдаря
гидроочистке сырья в реакторе гидроочистки, содержащем алюмо-ко-
235
р,। 11,1 мошбленовый кикипнагор или алюмо iihkcju.-молибденовый
к л 1.1 in лиор.
Химизм и механизм основных реакций,
протекающих при платформинге
Катали шторы платформинга являются бифункпиональными, так
как содержат два типа активных центров: кисло! ныс активные центры
носителя (в дальнейшем будем условно обозначать Н + ) и металличес-
кие активные центры (атомы платины)1.
В процессе платформинга на этих активных центрах углеводороды
нефтяной фракции подвергаются глубоким превращениям. Об этом
свидетельствуют данные группового состава сырья (прямогонного бен-
зина) и продукта его превращения при платформинге (табл. 19).
Из табл. 19 видно, что в процессе платформинга происходит в ос-
новном превратен не ал капов и нафтенов, в результате чего повышает-
ся содержание ароматических углеводородов и изоалканов, что соотве-
тственно приводит к повышению октанового числа платформата.
Алканы подвергаются в процессе платформинга дегидроциклизации
на платиновых активных центрах с образованием ароматических угле-
водородов и алкил циклопентанов:
Таблица 19
Углеводороды Содержание, мае.
в сырье и продукте платформинга
Ароматические 10 50
Нафтеновые 40 4
И юпарафины 15 28
/г-Ал канн 35 18
! Показано также (Бурсиан Н Р, Коган С. Б.. Давыдова 3. П. // Кинетика и катз-
1из — 1968. — Т IX — С. 661), что на поверхности гипомоплатиновых катализаторов
нахолягся катионы плагины;
236
На кислотных
мери талия;
тел трах (возможно на ионах платины; проходит
и '/> -
R - С Н r С Н .-С Н1 -R- (j Н - С Н
СНч
и на платиновых центрах — гидрогенолиз’
Pt
---►RCH, + R’H
Нафтены. Шсетизвеиные нафтены подвергаются дегидрированию
на платиновых центрах с образованием ароматических углеводородов:
Апкаацакаонентаны по реакции дегидроизомеризации превращают-
ся в ароматические углеводороды:
Частично проходит гидрогенолиз колец нафтенов:
237
Однако изомеризация может проходить и с участием платиновых
активных центров. Например, изомеризация этилбензола в ксилолы
по-видимому, протекает по схеме
Рассмотрим более подробно химизм и мехами зм основных реакций,
которые приводят к образованию в процессе платформинга аромати-
ческих углеводородов.
Дегидрирование шестичленных нафтенов
Это основная реакция, за счет которой образуются ароматические
углеводороды при платформинге. Повышенное давление водорода сни-
жает глубину дегидрирования шестизвенных нафтенов, при яом увели-
чивается степень гидрогенолиза и изомеризации пик .апекса новых уг-
леводородов в циклопентановые. особенно в случае высокой кислотной
активности носителя. С этой точки зрения процесс платформинга необ-
ходимо проводить при пониженном давлении водорода, но достаточ-
ном для предотвращения коксообразонания на катализаторе.
В соответствии с современными представлениями, циклогексано-
вые углеводороды дегидрируются на плагиновых активных центрах по
последовательной схеме:
238
jt-KDMH к кс гс-комплскс
Скоросп. lei н гриронания шестизвенных нафтенов зависит от н\
строения В некоторых случаях дегидрирование невозможно (хч измене-
ния у i лево. мро.инм о скелет молекулы. Примером может служить прев-
ращение s см мешенных циклогексанов в условиях платформ и i mi.
В прис\и.!вии оифунктшонального капни затора платформинга,
например Pi А! •()., возможно превращение 1.1-ди метил циклогексана
по схеме
Вначале происходит дегидрирование 1,1 -ли метил циклогексана на
платиновых а и г hbhi.iv центрах: образовавшийся ли метил циклогексен
на кислотном пси iре превращается в карбокатион, который подверга-
ется алкильномх переносу с последующим дегидрированием.
Мостиковые инк.тотексаны при платформинге превращаются варе-
ны также с предварительным расщеплением связей С -С.
239
Бициклические нафтены, содержащие циклогексановое кольцо,
также легко дегидрируются в условиях дегидрирования мононикличес-
ких шестизвенных нафтенов:
Следует отметить, что циклопентан и его производные не дегидри-
руются над платиной в условиях, когда легко проходит дегидрирование
циклогексановых углеводородов. Это объясняется тем, что активные
центры платинового катализатора быстро деактивируются в начальный
момент процесса в результате прочной адсорбции продуктов дегидри-
рования, в частности циклопентадиена:
В условиях платформинга образованию циклопентал иеновых угле-
водородов препятствуют, с одной стороны, повышенное давление во-
дорода, с другой — быстрая изомеризация алкилииклопентенов:
В отличие от платиновых катализаторов, при риформинге на
оксидных катализаторах дегидрирование пиклопентановых углеводо-
родов с образованием циклопентадиенов проходит легко (если давле-
ние водорода невелико).
240
Изомеризация ал кил циклопентанов в циклогексановые углеводороды
с последующим дегидрированием последних
(реакция дегндроизомеризации)
Для реакции изомеризации метилциклопентана в циклогексан
имеем
ДС= - 3600 + Ю Г
При температурах платформинга (-500йС) Д6 > 0: следовательно,
реакция гермодинамически маловероятна. Равновесие сметено влево:
равновесный сое тав при 500 С 5% циклогексана и 95% мети л цикло-
пентана.
Однако вследствие тою, то циклогексан быстро и количественно
дегидрируется в бензол и таким образом выводится из зоны реакции,
происходит смешение равновесия реакции изомеризации в сторону
циклогексана. Изомеризация протекает с участием как платиновых,
так и кислотных центров катализатора:
Дегидроциклизация алканов
Реакция дегидроциклизации была открыта в 1936 г. почти одновре-
менно в трех лабораториях нашей страны. Б. Л, Молдавский и Г. Д, Ка-
му шер открыли, что алканы, начиная с гексана, при 450-470 С в при-
сутствии оксида хрома превращаются в ароматические углеводороды
с тем же числом атомов углерода1.
Гексан превращался в бензол с выходом 17%, гептан — в толуол
с выходом 26%. Продукты реакций содержали также до 10% олефинов.
В. М Каржсв, Г. М ( с вс рьян она и А. Н. Сизова провели циклизацию
Утканы с<»сга.«Г-4.(\Температурах выше 450*С над окиелами хромамолиб-
тена и jpyi их mcjjj.t.joh rci и.трируются с обра иншпием алкенов и диенов. яих
мкилбензолы с коро । кими боковыми пенями (С,. С р дегидрируются в ст и
гомолог и
'6-3544
241
алканов при 500-550°С в присутствии медно-хромового катализатора и
получили аналогичные результаты.
Б. А. Казанский и А. Ф. Платэ установили, что ароматизация алка-
нов протекает над катализатором Pt/C при 305-310'С. Однако выходы
ароматических углеводородов были значительно ниже, чем в опытах
Молдавского. Например, в случае дегидроциклизации гексана выход
бензола составил всего ~2%. Таким образом, в отличие от оксида хрома
платиновый катализатор осушествлял также С--циклизацию1.
Так, при дегидроциклизации гептана в катализа те содержалось
около 10% гомологов цикл о пентана. Таким образом, в присутствии
платины циклизация алканов протекает по двум направлениям. На-
пример:
СНД
- 4Н?
ЗСН3СН2СН2СН2СН2СН2СнД
сн2сн
С н.
+ 2Н2
Последующие работы в области дегидропиклизапии позволили ус-
тановить другие закономерности зтой реакции:
— при дегидроциклизации алканов образуются изомерные аромати-
ческие углеводороды, что объясняется различными вариантами замы-
кания молекулы в цикл:
При дегидроциклизации алканов C|(J и выше. наряду с алкилбензо-
лами, образуются нафталин и алкилнафталины:
1 Реакция С5-дсп1дроииклиза1 иги а.тканов протекаеi 1 олько нл Pl; 11 а лруг их м еталлах
(Pd. Os. 1г), активных в реакции дегидрирования. Cs-.iei ил рои и кд и яшию осуществить
не удается (В. К. Скварснко // Успехи химии. - 197f Вып 12 - С. 2164; Ю. В. Фо-
мичев Н. В. Госпнская. Б. А Казанский//ДАН СССР. |%Х [Mi. - 3X3).
242
разветвление прямой цепи алканов повышает скорость реакции
Скорость де i идроциклизации возрастает, например, в ряду
— алкилбен золы и ал кил циклопентаны, имеющие алкильную груп-
пу с числом у । л сродных атомов четыре и более, также подвергаются де-
гидроциклизации:
СН3
сн3
- изоалканы, имеющие в прямой цепи пять атомов углерода, в при-
сутствии Сг/)-(-А1?О3 также подвергаются ароматизации. В этом случае
ароматизации должна предшествовать изомеризация, проходящая на
кислотных центрах оксида алюминия:
СН3
Как было отмечено ранее, при дегидроциклизации алканов над ок-
сидными катализаторами в продуктах реакции было найдено значи-
1R’
243
тельное количество олефинов. Это позволило предположить, что оле-
фины являются промежуточными продуктами при дегидроциклизации
над этими катализаторами.
Для механизма дегидроциклизации была предложена следующая
схема:
1, С-С-С-С-С-С ОС-С-С-С-С
Однако работами Б. А. Казанского и его учеников показано, что при
дегидроциклизации над оксидом хрома молекулы олефинов не подвер-
гаются сами циклизации: происходит их дальнейшее дегидрирование и
превращение в диены и триены. которые затем циклизуются1:
В присутствии катализатора Pt/C механизм дегидроциклизации
иной. Установлено, что С5-циклизаиия протекает пузем непосред-
ственного замыкания молекулы алкана в никл с образованием алкил-
циклопентана:
С() дезидроциклизация алканов на плагине проходил' через стадию
образования олефинов (диенов и трисиов) Прямое замыкание в ше-
Выхолы зояуолаиз 1-ieinena. 1.3-ieiiraanen;j и iciiiaipuciki при jci и,| рои мкл и займи
относятся как 1:5:150 (М. Ц. Розешаря. Е. С. Мортиков. Б. Л Казанский //ДАН
С ССР. - 1966. - Г 166 - С 619.
244
стюкнный ЦИКЛ не происходит2. Таким образом. С,- и С,-дегидро-
циклизация ал канон над платиновым катализатором протекает по раз-
ным механизмам, очевидно, на различных активных центрах платины.
В процессе платформинга все вышеприведенные реакции циклиза-
ции и ароматизации проходят в присутствии Pt/AI?O? или Pt/алюмоси-
ликат, которые являются оифункииональными катализаторами.
И если на кт ал и заторе Pt/C, как было показано выше^ выходы аро-
магических \ i.jcводородов малы из-за преимущественного протекания
С^-циклмзани11. ю ни катализаторах платформинга дегидроциклиза-
ция протекае! с оольшнми выходами ароматических углеводородов.
Это объясняется тем. что образовавшиеся при С5-циклизации алкил-
циклопен 1аны изомеризуются на кислотных центрах в циклогексано-
вые углеводороды. которые дегидрируются в ароматические;
Термодинамически возможный выход ароматических углеводоро-
дов при дегидроциклизации алканов уменьшается с повышением дав-
ления водорода и снижением температуры. Поэтому в обычных усло-
виях платформинга на алюмоплатиновых катализаторах при давлении
водорода 3-4 МПа дегидроциклизация алканов проходйт в небольшой
степени. С применением полиметаллических катализаторов (напри-
мер, Pt-Re-ка гал и затора) при пониженном давлении водорода (1-
1.5 МПа) ароматизация алканов протекает в значительно большей сте-
пени. Повышение температуры до 500-520°С (жесткий режим плат-
форминга) также способствует ароматизации алканов, однако при этом
для снижения скорости гидрокрекинга необходимо применять катали-
заторы с пониженной кислотной функцией носителя.
3.3. Превращения углеводородов в реакциях
полимеризации, алкилирования и изомеризации
(синтез высокооктановых компонентов топлив)
Топлива для современных карбюраторных двигателей должны об-
ладать высокими октановыми числами (90-93). Получение таких топ-
лив возможно путем компаундирования, т. е. добавления к базовым
бензинам высокооктановых углеводородов или их смесей. В табл. 2
приводятся октановые числа некоторых высокооктановых углеводо-
родов.
Исспулянп Г. В . Роэсшарл М И. // Нефтехимия. 1978. N.2
245
Таблица 20. Октановые числа углеводородов
У|леиодород Октановое число
Моторный метил Иссле.тователнекий метод
1. Букш 92 94
2. Изобутан 9S 101
з. Неогексан (2.2-диметилбутан) 93 92
4. 2.3-лимсгилбутан 94 102
5. Триптан (2.2.3-триметилбутан) 102 106
6. Изооктан (2,2,4-три метил пента и) 101) 100
7. Циклопентан 87 101
К. Бензол1 107 113
9. Толуол 101
10 Этилбензол 97 103
‘Бензол и его гомолош облазают высокой токсичностью, особенно опасен бензол.
В США и Западной Европе содержание бензола в гол птах нс должно превышать 0,5%
Индивидуальные высокооктановые изоалканы (наиболее желатель-
ные компоненты карбюраторных топлив) получают либо полимериза-
цией олефинов с последующим гидрированием полимеров, либо алки-
лированием изобутана олефинами. Необходимый для процесса алки-
лирования изобутан получают изомеризацией w-бутана. Изомериза-
цией пентана и гексана получают изопентан и изогексаны, которые
используют в качестве добавок, повышающих октановые числа голов-
ных фракций бензинов. В высокооктановых автомобильных бензинах
должно содержаться 20-30% мае. изоалканов, для получения которых
часто используют изомеризацию легких фракций нефти. Ароматичес-
кие углеводороды ряда бензола получают в процессе платформинга,
либо алкилированием бензола олефинами. В России содержание аро-
матических углеводородов в высокооктановых бензинах — 50—55%.
В США и странах Западной Европы содержание ароматических угле-
водородов в автомобильных бензинах не превышает 30%. Наиболее
желательными компонентами дизельных и реактивных топлив явля-
ются высшие слаборазветвленные алканы, которые легко воспламеня-
ются и имеют низкие температуры кристаллизации. Эти углеводороды
можно получать изомеризацией соответствующих алканов нормаль-
ного строения.
Большинство реакций алкилирования, полимеризации и изомери-
зации углеводородов, лежащих в основе процессов синтеза компонен-
тов топлив, проходят в присутствии кислотных катализаторов в жид-
кой или паровой фазе.
246
Рассмотрим некоторые особенности кислотного катализа в жилкой
фазе.
Катализаторами реакций алкилирования, полимеризации и изоме-
ризации являются сильные кислоты; протонные (например, серная
фосфорная, фтористоводородная), называемые кислотами Бренстеда^
и апротонные (например, хлорид алюминия, фторид бора), называе-
мые кислотами Лыоиса2.
Следует отметить, что кислоты Льюиса, взаимодействуя со многи-
ми водоролсодержашими полярными веществами, образуют комплек-
сы, являющиеся донорами подвижных протонов, как и кислоты
Бренстеда:
AICI, + НС1^=^Н + [А1С!4]’
ВН + Н2О^—H ^BFxOHI
При взаимодействий кислот Лыоиса (например, BF3, SbF5, TaF$,
AsF5) с сильными кислотами Бренстеда (например, HF, HSO3F) обра-
зуются очень сильные кислоты, называемые «сверхкислотами»3. Это
HF-BF3, Hi -SbFs. HOSO2F-SbF5 и т. д, К сверхкислотам относят так-
же безводные прогонные кислоты, более сильные, чем 100%-я серная
кислота. Это. например, FSO2OH, CISO2OH, НСЮ4, CF3SO3H.
Для сравнения силы кислот, то есть их способности отдавать протон
основанию (воле) в водных разбавленных растворах, определяют кон-
центрацию ионов Н3(У и кислотность водных растворов (по шкале
pH):
pH = -log[H3Ob],
Сравнением значений pH для разбавленных водных растворов кис-
лот одной и (ой же концентрации, можно оценить их относительную
силу.
В случае концентрированных кислот (малое содержание воды) меж-
ду кислотностью и концентрацией ионов Н3О+ нет связи и шкала pH
теряет смысл
Сверхкислоты могут существовать только в безводной среде (под
влиянием волы они разлагаются), поатомудля измерения их силы шка-
ла pH также нс применима.
Кислотность с верх кислот определяют, используя в качестве акцеп-
тора протонов слабое органическое основание (обозначим буквой В),
ГП<) и ме н и л a i с мн о у чс п<) юБ ре н стела (В го listed), который и 1923 г. дал определение
кислоты, как иешесим. стpcMsinteiося отдать протон.
~ По имени американского химика Льюиса (Lewis), который в 1923 т. выдвинул кон
Иепиию кислогы как акцептора элсктронон, а основания как донора электронов.
1 Буренин И I’. Орлов Д. С.. Шакун А Н. Катализ на сверхкислотах. Тематический
обзор - М : 11НИИТ Знифгсхим. 1979.
247
)f< lr;( ,(VCt W ПР1М iB ... H i' ’ " ‘ 1“’ ,Г lrt
hl.;(l, ь, ..1. Bl I К OHIO HUHI.III. р.ПИМНПЧ ПС
n II \ "ни X
Чем vhimu'V мо JM mu чих иг us tci смспнп. Hiipauo, io ecih чем
m .Kt ммшенцмннч HI Г и .»» ik »ihch i и‘11 i 11 I • I B|. icm выше си-
м кМЫ k0lllinil|UUHKI Bl I и В в p.B । в* II IMCpHII. С ПО-
MOIHMO \ ИЛ риф пи к" I ОНИ11 U ПС к 1 pt К К» 1 (II111 I I Л t Ч I h ИМ 1111 Л ) [ ИХ ЛИИНЫХ
iWVMHIMIUnn ф\ НКШ1Ю к и С lot IB Ч I II I л ММ С I Л. \Л|1ЛК I Г|Н! 1УЮ1ЦуК) силу
KIU. ioin I’.K'KI HCAVI lit) формуле
1 Bl I
',K- l"' III I
। u* М11И‘ konciaiH.i основноеfи c i.idoio npi.iinriciм>iо основания
В, которое используют в качеепк пнликлюрд
Для 100%-и серной кислшы /(, 12; лля винт сильной фтор-
ссльфоновой кислоты //п 15; л ли сверх мк лты f-SOjH-SFj
Н:л - -14, то есть на кислота в 10 миллионов р.и ио лее сильная, чем
100%-я H,SO4.
В настоящее время установлено. ч го реакции нолимсри заиии. алки-
лирования, изомеризации в жилкой фа »с в ирису к твиии кислотных
катализаторов проходят по ионному мехами imv мере з ci алию образова-
ния карбокатионов (карбений-ионон).
Карбокатионы образуются путем присоединения протонов катали-
затора к молекулам алкенов:
R-HC=CH; а Н-Д ^=^[RCH-l Нф\
либо путем отрыва катализатором гидрид-и он а от молекулы углеводо-
рода;
/ \ Н "А
R(hJ-------►R*....Д - н;
При зтом карбокатион находится в ионной паре с противоионом
(анион катализатора). В процессах алкилирования, полимеризации и
и .томеризации углеводородов катализатор обычно не растворен в уг-
кводородном слое, полому имеются две фазы — углеводородная фа-
и слои кд tilth затора. В зависимости от соотношения углеводород"
нои и неорганической части ионная пара может растворяться либо
в о зной из них фаз. либо концентрироваться на границе раздела фаз.
( ..мннетствснно реакция может протекать либо в слое катализатора.
24*
Illlll » H \ I I' - 111 I 11 J л I J HI P.1 < It,г ,5Hb<) ИЛ ' JMH И ,1'" ?,.! ; , Z-y ,. . ! )
jICCi np< 111> .1-'I li л irh'j Нфи.-тпи фа;*' !<) !'/,П’,.Г .
щи. Ill *. pv.n.i инны и ш>ни<»И парс мера и/шины
амин »п I la-I iiUHiihiiuui. нфГнжа! иона невсдика. р^г ;/? - гхл' < др-
ена.к>и iHjfjouhh) В у 1лгводороднми слои можс?
ионная пара и к Ширин карбонлион содержит больше дне. > row-i
у I.терпла । 12 Is-». чю н практике встречается редко (адкилирс«ймит
аромат nevi их \ i icводородов высшими олефинами). Чаше всего при
алкилировании и подимеританин используют олефины состава
С\ ( по > г । <му ионные пары, образованные с участием >ти» олефи-
нов, .in ин рас ।ворям)гея в слое катализатора (кислоты». Благодаря
высокой ли । в-к 1ричсскои проницаемости катализатора ионы в ион-
ной парс рлюбшены та счет сольватации молекулами кислоты и
сушес! ну к о и юлированно друг от друга. Карбокатион, изолирован-
ный «н Hpoi 11ВЫЮН.1. облддас! высокой реакционной способностью и
н таи модеме । их с। с большой скоростью с другими молекулами углево-
дородов на [рднинс раздела фаз. Полому в условиях катализа в жид-
кои фа ic pcai i1 и и проходят чаше всею на поверхности раздела фаз и
.и я у вс точения >пш поверхности необходимо очень сильное переме-
шивании
В присутстии свсрхкис.тот легко происходит протонирование не
только ненасыщенных углеводородов, но и алканов:
(,Н. - Н'^^СЛН '
Обри ювавшиися карбокатион отшепляет молекулу водорода и
превращается в карбениевый ион:
Н2 - СНз-СН-СН2-СН3
3.3Л. Ступенчатая полимеризация олефинов
Ступенчатая полимеризация олефинов приводит к получению низ-
комолекулярных полимеров (смеси димеров, тримеров и т. дЛ. В каче*
стве катализаторов в промышленности обычно при меня ют фосфорную
кислоту и пеолиты. В лабораторных условиях можно испатьзовать сер-
ную кислоту. Полимеризация изобутилена в присутствии 0%-й серной
кислоты при 20-35еС приводит к образованию димеров.
249
CH; CH;
При более высоких температурах и концентрациях катализатора по-
лимеризация осложняется перераспределением водорода (протекает
реакция гидродегидрополимеризации, приводящая к частичному об-
разованию изоалканов и смол).
Гидрированием смеси изооктиленов можно получить изооктан.
Ступечатую полимеризацию олефинов в промышленности в присут-
ствии фосфорнокислого катализатора Н;POyAI-,0SiO2 проводят при
температурах 120-200’С и давлении 2-6 МПа (в зависимости от оле-
фина).
Полимеризацией фракции олефинов С4-С5 получают полимербен-
зин (Гк = 60-200°С), который имеет высокое октановое число 100 (по
исследовательскому методу).
Рассмотрим механизм ступенчатой полимеризации олефинов на
примере полимеризации изобутилена.
Протон катализатора присоединяется к молекуле олефина и образу-
ется карбокатион:
© ©
Н2О^-СН3 + н---------►!!;(' С СН; ,
СН3 СН;
который присоединяется к другой молекуле олефина.
►Н;С-С-СН2-С-СН3
СН; СН3
Образовавшийся /ирет-октильный карбокатион может присоеди-
ниться к новой молекуле изобутилена и дать карбокатион три мера, но
может стабилизироваться. Стабилизация возможна путем отщепления
протона от соседних с заряженным атомом углерода первичного либо
вторичного атомов углерода.
Отщепление протона /ире/и-октильного карбокатиона от первично-
го атома углерода проходит в четыре раза быстрее, чем от вторичного.
Этим определяется соотношение изооктиленов (I и II) в продуктах ре-
акции
250
Н3С-С-СН=С-СН3 (20%)
СН3 СН3 п
Такое соотношение изомеров может быть объяснено меньшей тер-
модинамической устойчивостью изомера с внутренней двойной
связью, вследствие пространственного затруднения, возникающего
между третичной бутильной группой и метильной группой при двой-
ной связи в изомере И:
3.3.2. Алкилирование изоалканов алкенами
Синтез изооктана
Изооктан получают алкилированием изобутана изобутиленом или
бутеном в присутствии 98-99%-й H2SO4 при 5-15°С либо в присут-
ствии HF при 25-35°С. Давление - до 0,6 МПа1.
Алкилирование изобутана протекает по схеме
Из
СНч СН3
Кажущееся протекание реакции против правила Марковникова на
основе ее приведенной формальной схемы легко объясняется ионным
механизмом:
1 Активными катализаторами алкилирования изобутана олефинами являются также
цеолиты f Е. С. Мортиков // Химия и технология топлив и масел. - 1974. - №7. - С. 13;
Долинский С Э.. Плахогиик В А.. Мортиков Е. ( Нефтепереработка и нефтехимия //
1997. _. №| _ с 63.
251
©
Основной побочной реакцией при алкилировании изобутана изобу-
тиленом является реакция полимеризации изобутилена, приводящая
к образованию смеси полимеров, подвергающихся затем сложной по-
лимеризации (дегидрогидрополимеризации) с образованием смолооб-
разных веществ и смеси алканов.
Для того чтобы свести до минимума полимеризацию, применяют не
менее, чем трехкратный мольный избыток изобутана в реакционной
смеси. Кроме 2,2,4-триметилпентана продукты алкилирования изобу-
тана изобутиленом содержат другие изомеры октана (2,2,3-, 2.3,4- и
2,3,3-триметилпентаны), а также изоалканы состава С\-С7.
Последние образуются в результате распада некоторых сложных
карбокатионов и последующего ионного гидрирования образующихся
олефинов:
СН3
©Is ©
НзС-С-СН|сН-СНз^=^СНз-С=СН-СНз © СН3-СН-СН3
сн3 СНз СНз\дЦ
CH3-CH-CH2-CH3
СНз
смесью бутенов6 вТотоп °быЧНо ПР°ВО^Т алкилиронание изобутана
и бутен-2 Алкилиповянм И НарЯДу С изобУтиленом содержатся бутен-1
гидридных перемещений » п ‘ ' V) в Результате алкильных и
катионе (I): ИИ в первично образующемся октильном карбо-
252
Синтез триптана, неогексана и 2,3-диметилбутана
Триптан наряду с другими изомерами гептана образуется при ката-
литическом алкилировании изобутана пропиленом. В обычных усло-
виях алкилирования изобутана бутенами в присутствии серной кисло-
ты алкилирование пропиленом идете малой скоростью. Полому, ал-
килирование изобутана пропиленом проводят с применением HF при
больших избытках изобутана при 10°С и небольшом давлении,
Основными продуктами реакции являются 2,2- и 2,3-
димет илпентаны. Это объясняется следующей схемой:
Н
I / (+) (+)
н,с-с=сн2 -----------сн?-сн-сн2
— Н^С~(|Х+) ъ снгсн2-сн3
сщ
253
гидридный
перенос
(СНЖС:Н
/-СдН ю
2,3-диметилпентан
Н3С сн3
2,2-диметил пентан
алкильный
перенос
Образование небольшого количества (4-6%) триптана объясняется
следующей схемой:
СН3СН3 СН3СН3
Триптан можно получить термическим алкилированием изобутана
пропиленом и в аналогичных условиях — неогексан алкилированием
изобутана этиленом:
сн3
, ' 35 МПа
н3с—СН + Н2С=СН-СН3'^
1 ’ 500° С
СН3
СНз
:н3с-с-€н-снз
СН3СН3
254
с н;!
I 30 МПа
Н3С-~СН + Н.С=СН,^=
I 500°С
С Н ч.
Механизм термического алкилирования — радикальный:
СН3
2) н3с—С* + Н3С=СН2^
СН3
СН3
сн3
сн3
Каталитическое алкилирование изобутана этиленом протекает
труднее, чем изобутиленом и пропиленом. В качестве катализатора ис-
пользуют хлорид алюминия и цеолиты. Основным продуктом реакции
является 2,3-диметилбутан:
СН3
н3с—СН + Н2С=СН2^
сн.
<^н3
Н3С-СН-СН-СН3
сн3
Это можно объяснить следующей схемой механизма реакции:
255
н сщ
3.3.3. Изомеризация алканов
Изомеризацию н-ал к а н о в п ро вод ят в п ро м ы ш л е н ност и с цел ыо п о-
лучения низкомолекулярных изоалканов (изобутан, изопентан, изо-
мерные гексаны), а также низкозастывающих слабораз ветвленных
высших алканов.
Низкомолекулярные изоалканы добавляют к автомобильным бен-
зинам1 для повышения октановых чисел головных фракций, выкипаю-
щих до 70’С. Для получения таких высокооктановых добавок проводят
изомеризацию низкокипяших фракций прямогонных бензинов.
Изобутан и изопентан служат также исходными продуктами для по-
лучения непредельных углеводородов — изобутилена и изопрена. Изо-
бутан является также сырьем процессов алкилирования. В качестве ка-
тализаторов изомеризации в промышленности используют хлорид
алюминия и бифункциональные катализаторы (платина на цеолите
или оксиде алюминия).
Термодинамически более глубокому протеканию изомеризации
с образованием сильноразветвленных изоалканов способствуют пони-
женные температуры, поэтому наибольшее значение имеют процессы
низкотемпературной изомеризации с применением высокоактивных
катализаторов. В лабораторных условиях для получения эталонных
изоалканов изомеризацию w-алканов проводят в присутствии хлорида
или бромида алюминия; последний удобен тем, что растворим в угле-
водородах.
В присутствии AICI-, в качестве промоторов применяют хлорид во-
дорода и небольшие количества (доли процента) олефинов. Без хлори-
да водорода реакция практически не иле г. Эго свидетельствует о реша-
ющей роли в катализе подвижного протона сильной кислоты HAICI4.'
НС) + А1СЪ^~~ Н'|А1С14|
В отсутствие олефинов реакция протекаете очень малой скоростью.
Роль олефина заключается в образовании карбокатиона, который про-
должав цепной процесс. Таким образом, механизм изомеризации ал-
BdJOjJWM компонентом смесей лля получения автомобильного бен лит в России
слхжиг бензин кагалнтичсского риформинга. шикокишпиие фракции которого имеют
низкие октановые числа.
256
капов в ирису тел вин хлорила алюминия и промоторов (олефина и хло
рила водорода) можно представить следующей схемой:
Н’А1С14^=^Н ' + А1СЦ-
©
С Н2=СН-СНгС1ф 4- Н^^СНз-СН-СН.-СЖ
СН?-СНГС1-Ь-СЖ + СНз-^СН;.^
СНз
:СНгСН-СН3 + CHrCH-CH2-CHj
CHi
Про гон катали затора присоединяется к молекуле олефина с образо-
ванием вторичного карбокатиона, который изомеризуется в результате
алкильного и гидридного переноса в третичный. Третичный карбока-
тион отщепляет гидрид-ион от молекулы бутана, и образуется вторич-
ный карбокатион, то есть продолжается процесс изомеризации бутана.
В последнее время в качестве катализаторов изомеризации предло-
жены сверхкислоты — комплексы протонных и апротонных кислот,
например: HF-BF^; HF-SbF5; HSO^F-SbF^.
В присутствии этих катализаторов в атмосфере водорода изомериза-
ция алканов протекает быстро — в течение нескольких часов при невы-
соких температурах (20-50°С), наиболее благоприятных для образова-
ния сильно разветвленных изоалканов с высоким октановым числом.
Согласно представлениям, развиваемым Ола!, в присутствии сверх-
кислот карбокатионы образуются непосредственно из молекул алка-
нов. Вначале происходят их протонирование поо-связи и образование
карбон и евого иона, в котором постулируется существование 3-центро-
вой и 2-:злектронной связи:
карбон новый
ион
Карбон левый ион затем распадается с выделением молекулы водо-
рода и образованием карбен и евого иона:
1 Olah (ТА // .1. Am. Chcm "Sot. - 1972 - V 94 — Р. 808.
17-3544
257
В последнее время1 развиваются представления о катион-радикаль
ном механизме изомеризации алканов в присутствии цеолитов. Возмо
жен следующий механизм изомеризации и-иентана;
П 1
Хлорид алюминия и другие кислоты Лыоиса., а также сверхкислоты,
будучи активными катализаторами изомеризации, имеют существен-
ный недостаток: они загрязняют реакционную смесь, требуется ее
нейтрализация, промывка, что приводит к образованию сточных вод.
Этого недостатка лишены твердые гранулированные катализаторы
на основе оксида алюминия, алюмосиликатов, цеолитов.
В присутствии бифункциональных катализаторов (Pt на твердом
носителе кислотного типа) изомеризацию алканов (бутан, пентан или
соответствующие фракции бензинов прямой iонки> проводят в усло-
виях. близких к условиям платформинга (350- 450 С. давление водоро-
да 3—4 МПа), Роль водорода заключается в пола (злении реакции кре-
кинга.
Непрореагировавшие н-алканы отделяют от и зоалканов с помошью
молекулярных сит и направляют на рециркуляцию.
В последнее время разработаны процессы низкотемпературной изо-
меризации в присутствии более акт ивных алюмоплатиновых катализа-
торов (Ю0-200°С, давление водорода 2,0-7.0 МПа).
1 Випшецкай М. В,. Ромамояский Б. В.//Ж фи > химии 1993 №9. - С. 923.
258
Изомеризацию высших алканов (сырье - парафины, гачи, петро-
латумы. фракции прямой юнки высокопарафинистых нефтей) прово-
дят на бифункциональных катализаторах (Pt, Pd, Re на кислотных но-
сителях) при температурах 350—47(ГС под давлением водорода 2-
7 МПа. В промышленности этот процесс получил название гидроизо-
мери за пи и.
В этом процессе кроме изомеризации алканов происходит гидриро-
вание аренов и гидроочистка (удаление азотистых, сернистых и смо-
листых веществ).
Гидрон зомсризапней получают либо компоненты высококачествен-
ных смазочных масел с высоким индексом вязкости и низкой темпера-
турой замерзания, либо компоненты реактивных и дизельных топлив.
Гидроизомеризацией керосино-газойлевых фракций парафинистых
нефтей получают низкозастываюшие дизельные топлива. Так, гидрои-
зомеризацией дизельного топлива Мангышлакской нефти*
(Г^ ( = -4’С) над катализатором Pt на цеолитсодержащем алюмосили-
кате при давлении водорода 3 МПа и 340-3804С получают дизельное
топливо с TjacT = -35°С.
Изомеризация алканов на бифункциональных катализаторах проте-
кает, по-видимому, с участием как дегидрирующих, так и кислотных
центров2.
На платиновых активных центрах происходит дегидрирование моле-
кулы алкана. Образующийся олефин, взаимодействуя с кислотным
центром носителя, образует вторичный карбокатион, который! изомери-
зуется в т ретичный. Последний присоединяет гидрид-ион от другой мо-
лекулы алкана, либо стабилизируется путем выброса протона с образова-
нием изоалкена, который гидрируется на платиновом центре в изоалкан.
Побочной реакцией при изомеризации алканов на кислотных и би-
функциональных катализаторах является реакция крекинга, которая
протекает в результате распада карбокатионов Соотношение между
изомеризацией и крекингом изменяется в пользу крекинга при увели-
чении молекулярной массы алкана. Так. если изомеризация бутана
практически протекает без побочных реакций, то. начиная с пентана,
образуются продукты крекинга и их выход увеличивается с увеличени-
ем молекулярной массы алкана.
Начиная с гексана, изомеризация протекает неселективно. Для по-
давления реакции крекинга изомеризацию проводят при возможно
низких температурах и повышенных давлениях водорода.
1 Дырпн В~ТЛквин1ср м’ С и яр. // 11еф|ехимия IWO 20. №2. -С. 237.
3 И юмсримиия гткже моле! протекать юлько па плагинонык активных астрах
НО. М Жоров И и)мсри;ания уыешиоролов // Химия и гехиология М.' Химия.
1'ЖЗ. ( [7ХЭ
!?
254
Глава V
ПРОИСХОЖДЕНИЕ НЕФТИ
И ЕЕ КОМПОНЕНТОВ.
ПРЕВРАЩЕНИЕ НЕФТИ В ПРИРОДЕ
Вопрос о происхождении нефти с давних пор и по настоящее время
является предметом исследования ученых. Был выдвину! ряд гипотез,
объяснявших происхождение нефти, однако многие из предложенных
гипотез не были подтверждены расчетами и экспериментами. Между
тем, правильное решение вопроса о происхождении нефти могло бы
облегчить поиски месторождений, объяснить некоторые особенности
нефтей, показало бы пути синтеза нефти и j природною сырья.
Среди гипотез происхождения нефти есть гипотезы чисто умозри-
тельного плана, почти не подтвержденные ни научными фактами, ни
расчетами. К таким гипотезам относится, в частности, космическая,
согласно которой нефть является продуктом превращения углеводоро-
дов, попавших на землю из космоса (В. Д. Соколов, 1892).
Основная часть гипотез имеет под собой и теоретическую, и экспе-
риментальную базы. Их можно разбить на две группы — гипотезы
неорганического происхождения нефти и гипотеты органического
происхождения нефти. Из неорганических i иного з наибольшее рас-
пространение получила гипотеза Д И. Менделеева.
5.1. Гипотезы минерального происхождения нефти
Д. И. Менделеева и других ученых
Эта гипотеза была выдвинута Менделеевым в )879 г. Выдвигая эту
гипотезу, Д. И, Менделеев опирался на работы французских химиков
(в частности. Бертло) по синтезу карбидов металлов и по изучению их
свойств. Известно, что карбиды металлов реагируют с водой с образо-
ванием углеводородов:
СаС2 + 2 Н -0--►Са(ОН) НС ~ СИ
АЙС, + |2Н,0----►ЗСЩ 4AI(OHh
Менделеев допускал, что в земной коре находятся карбиды метал-
лов, которые могут взаимодействовать с водой, проникающей по тре-
щинам, с образованием углеводородов. Эти углеводороды при повы-
шенной температуре, давлении и под каталитическим действием пород
могли, по мнению Менделеева, подвергнуться различным реакциям и
образовал ь углеводородные смеси. Менделеев обратил внимание на то,
260
что нефть залегает часто недалеко от горных массивов, и объяснил это
в свете своей гипотезы тем, что вблизи горных хребтов в земной коре
могут существовать трещины, по которым вода проникает на глубину.
Гипотеза Д, И. Менделеева в свое время являлась наиболее аргументи-
рованной. В настоящее время существуют очень серьезные возражения
против этой гипотезы:
I. На глубине в 40-50 км вследствие пластичности горных пород не
может быть 1 решин. по которым вола могла бы проникнуть в недра.
2. Нефть встречается не только в складчатых областях Земли; гораз-
до чаше встречаются платформенные месторождения (Менделеев об
этом не знал).
3. Непонятно, каким образом вода могла попадать из области низ-
ких давлений (на поверхности) в области высоких давлений в недрах.
4. Гипотеза Менделеева не может объяснить оптическую активность
нефти и наличие в нефти таких сложных соединений, как порфирины,
изопреноидные углеводороды и другие «биологические метки».
5. Гипотеза Менделеева не может объяснить, почему нефть приуро-
чена к осадочным породам.
В 1950 г. профессор Н. А, Кудрявцев предложил магматическую гипо-
тезу образования нефти, согласно которой на большой глубине в мантии
земли при высоких температурах и давлениях взаимодействие углерода и
водорода приводи т к образованию радикалов СН. СП? и CHV Эти ради-
калы перемешаются в мантии пол действием перепада давления в зоны
глубинных разломов в земной коре и далее по этим разломам в области,
находящиеся ближе к поверхности, где вследствие снижения темпера-
туры они взаимодействуют друге другом, а также с оксидами углерода и
водородом, что приводит к образованию углеводородов.
На грандиозные масштабы выделения глубинных газов (СО2, СО,
Нт, СН4) обратил внимание В. И, Вернадский, образно назвав этот
провесе «дыханием земли».
Близких взглядов на неорганическое происхождение нефти при-
держиваются А. И. Кравцов и ). Ь. Искалюк, которые считают, что
нефти образовались при взаимодействии оксидов углерода, водорода
и метана, вылсляюшихся из мантии земли по глубинным разломам в
земной коре1.
Пол действием высоких температур, лавлений и под каталитичес-
ким действием оке илов металлов (Г с. Ni и лр.). содержащихся в i орных
породах, ироисхолит взаимодействие оке илов углерода с водородом
с обраюванием углеводородов, которые понадают в естественные ло-
вушки — будущие месторождения.
’ Вепмье И А. Кр.няюва 'Жизнь из нефти или нефть из житии» (Химия и жизнь,
1976. № 12. - (. 5 I) приводятся данные о выделении огромных количеств СО3, СО. Н2
и СН4 Г1ри и свержении нулканон на Куриэьских ociponax
261
Вместе с гем в настоящее время большинство ученых в области гео-
химии и химии нефти являются сторонниками органического проис-
хождения нефти из липидного (жирового) вещества планктонных ор-
ганизмов.
5.2. Гипотеза органического происхождения нефти
из органического вещества, рассеянного
в осадочных породах
Основоположником современной органической теории происхож-
дения нефти является выдающийся русский ученый И. М. Губкин.
Обобщив работы отечественных и зарубежных исследователей в облас-
ти органического происхождения нефти, и в первую очередь труды
В. И. Вернадского, который в 1927 г. высказал мысль о том, что исход-
ным веществом нефтей являются организмы. И. М. Губкин разработал
научно обоснованную теорию происхождения нефти, которую изло-
жил в своей монографии «Учение о нефти».
Исходным веществом при образовании нефти и газа И. М. Губкин
считал сапропель, представляющий смесь органических остатков расти-
тельного и животного происхождения в смеси с минеральными отло-
жениями.
Большие скопления органического вещества вмесге с минеральны-
ми осадочными отложениями могли образоваться в результате массо-
вого отмирания планктона. Планктон — эго совокупность организмов,
населяющих водное пространство континентальных и морских водое-
мов, имеющих ограниченную подвижность. Планктон подразделяется
на фитопланктон (водоросли, в том числе микроскопические, бакте-
рии) и зоопланктон (ракообразные, простейшие). Планктонные орга-
низмы размножаются с большой скоростью, особенно в благоприят-
ных условиях: в лагунах, озерах, устьях рек, где хорошо прогревается
вода и много питательных вешеств. Исключительно быстро могут
размножаться диатомовые морские водоросли. Подсчитано, что за 8
дней размножения в благоприятных условиях масса яих водорослей
может достигнуть массы Земли.
Миллионы лет назад, когда климат на Земле Сиял значительно теп-
лее, имелись весьма благоприятные условия для размножения планк-
тона. В результате усиленного размножения образовывались огромные
скопления этих организмов. Периодически происходило, по-видимо-
му, их массовое вымирание из-за недостатка кислорода и питательных
веществ. Мертвые организмы оседали на дно, где смешивались с илом
и песком, приносимым реками. Органическое вещество планктонных
организмов состоит в основном из липидов (жироподобные вещества)
и содержи г также углеводы и белки. Липиды представляют собой сме-
262
си: а) масел — сложных эфиров глицерина и алифатических кислот
счетным числом атомов углерода, состава Cf4-C36, причем углеводо-
родные цепи молекул могут быть насыщенными и ненасыщенными,
прямыми или разветвленными; б) восков — сложных эфиров карбоно-
вых кислот (аналогичных тем, которые входят в состав масел) и выс-
ших алифатических спиртов или алициклических спиртов (стеролов).
В образовании липидного вещества могут также участвовать оксикис-
лоты, в составе его могут находиться свободные карбоновые кислоты,
стеролы, алифатические терпены и углеводороды с прямой и разветв-
ленной цепью. Количество липидов в водорослях может достигать
SO-20 %, в животных организмах и бактериях 30-50%. Каковы же эта-
пы превращения липидов в углеводороды нефти? Попадая в минераль-
ные осадки, органическое вещество мертвых организмов подвергалось
уплотнению и образовывало вместе с глинистыми осадками твердое уг-
ле подобное вещество, называемое керогеном. Этот этап превращения
органического вещества называется диагенезом1. Вместе с уплотнением
органического вещества (поликонденсацией) происходили гидролити-
ческие и восстановительные процессы: гидролиз жиров, восстановле-
ние кислородных соединений, декарбоксиляиия кислот, насыщение
двойных связей, отщепление аминогрупп от молекул аминокислот и
т. д. В этих процессах могли принимать активное участие ферменты (би-
окатализаторы) бактерий. Часть органическою вещества превращалась
бактериями в газ (метан). По мере увеличения количества минеральных
осадков происходило постепенное погружение керогена, повышение
температуры и давления и на глубине 1-3 км начиналась решающая фа-
за генезиса нефти — образование углеводородов из органического ве-
щества и их превращения. Эта стадия получила название катагенез2.
В згой стадии при температуре 130-150°С происходила перестройка
ор1анических молекул, разрыв наиболее слабых связей С-С алкильных
фрагментов молекул, связей атомов углерода с гетероатомами (N, S, О),
а также разрыв донорно-акцепторных связей органических молекул с
неорганической составляющей керогена (глинистыми минералами).
Таким образом, из керогена постепенно образовывались углеводороды.
Образующиеся углеводороды под действием капиллярных сил пере-
мешались в слое осадочных порол и скапливались, образуя месторож-
дения. В процессе перемещения углеводородов под действием повы-
шен noii температуры, давления и каталитического влияния горных по-
род (особенно глинистых) продолжались их химические превращения,
что приветило к образованию сложной смеси углеводородов, какой и
является нефть.
1 01 греческих слов а/л/ - завершение, генез - возникновение.
- От 1РСЧССКИХ слог, ката - приставка, означающая движение вниз, усиление или
мвершсиие процесса, и генез
263
Нсоохолимо подчеркнуть роль глин в процессе образования углево-
юро юв. 1лпны составляют 50% всей массы осадочных пород.
Раоотами А. В. Фроста и Б. Брукса показано, что при 150—250°С под
icHciвием глин плут реакции превращения кислородных соединений;
при атом образуется сложная смесь углеводородов различных рядов.
Рассмотрим возможные пути образования углеводородов.
Алканы образовались в результате декарбоксиляции жирных кислот:
R-COOH -> СО.+ RH
и восстановления высших спиртов (см. состав липидов):
R-OH + 2 |Н] -у RH + Н2О.
Слаборазветвленные изоалканы 2-метил- и 3-метилалканы попали
в нефть непосредственно из «материнского» вещества нефти, где они
содержатся, Происхождение изопренанов и других терпеновых соеди-
нений рассмотрено ранее (см. гл. II).
В настоящее время установлено, что в нефтях содержатся алканы
состава С4()-С1()(). Такие алканы не могли образоваться в результате
декарбоксиляпии жирных кислот, так как кислоты с таким большим
числом углеродных атомов не найдены в природе. Их образование воз-
можно в результате радикального присоединения кислот к а-алкенам
(продукты термического расщепления длинных алкильных цепей мо-
лекул асфальтенов):
▼ । -СО,
R-СН-СНэ + R'-CH-COOH------►R-CH,CH,-CH-COOH------►R-(CH2h-R’
| I ' ' I
I---------- H R’
Циклоалканы (нафтены). Основным исходным веществом при об-
разовании нафтенов легких и средних фракций нефти явились, по-ви-
димому, жирные кислоты. А. И. Богомоловым установлено, что при
нагревании высших алифатических карбоновых кислот с алюмосили-
катным катализатором при температурах 200—250%' образуются моно-
циклические и бициклические нафтены.
Температуры в нефтяных пластах ниже (150-170°С), а в стадии ка-
тагенеза, когда могли проходить подобные превращения жирных кис-
лот под воздействием природных алюмосиликатов (глин) температуры
были еще ниже. Однако длительность протекания этих процессов мог-
ла компенсировать их низкую скорость. Возможный механизм таких
превращений:
О
264
о
Полициклические нафтены высших фракций нефтей являются
продукыми воссfановления (ионного гидрирования*) в стадии катаге-
неза стероидных спиртов, стеринов (холестерина, стиг.мастерина, эр-
гостерина и др.), широко представленных в животном мире:
Ароматические углеводороды. В органическом веществе --предшест-
веннике нефти, содержание ароматических структур крайне мало. По-
этому образование аренов связано со вторичными процессами преоб-
разования органического вещества, главным образом алифатических
кислот в стадии катоюнеза, очевидно, по схеме
кислота алкил циклогексан -> алкилбензол
А. И. Богомоловым показано, что нагревание алифатических
кислот с алюмосиликатами при 2()(ГС приводит к смеси углеводородов,
в которой содержание аренов может достигать 40%. Превращение
шестичленных нафтенов в арены проходит, вероятно, в результате
ТО ионном титрировании -~пегидрировании см гл IV §3.
2Точками в формулах обозначены метильные группы.
265
ионного дегидрирования и окислительного дегидрирования с участием
к и с л ородн ы х соед и нений:
Из этой схемы видно, что при взаимодействие кеюна и алкилцик-
логексана при участии протонных центров алюмосиликата образуются
арен и циклоалкан. Полициклические арены высших фракций нефти
являются продуктами ионного (или окисли ic.tbhoi oi ici курирования
и крекинга в стадии катагенеза сгеринов и лр^'их i 11 i: ю*»11 и а х алицик-
лических соединений. Например:
Рассмотрим кратко происхождение других компонентов нефти.
Кислородные соединения - кислоты, сложные эфиры могли образо-
ваться из липидного вещества планктона, состоя шею. как уже отмеча-
лось, из сложных эфиров глицерина и высших карбоновых насыщен-
ных и ненасыщенных кислот, эфиров кислот с высшими и алицикли-
ческими спиртами.
Предшественником изопреноидных кислот являются, ио всей веро-
ятности, фитол и продукты его превращения, (см. гл II §1).
Алкилфсно.ты и нафтолы образовались, вероятно, в результате
превращения таких природных продуктов, как лигнин, прелставляю-
266
ший собой смесь сложных фенольных соединений, и витамины. На-
пример- в результате превращений витамина К (содержит нафтохино-
новый фрагмент), витаминов Е или токоферолов (содержат алкилфе-
нольный фрагмент), производных флавона, кумарина и других при-
родных соединений, широко представленных в растительном мире.
Ниже приведены примеры соединений этого типа:
Витамин К,
пт К - ненасыщенный
радикал
Токоферолы
(отд 11 ч а юте я строе наем
радикала R)
Производное кумарина —
умпеллиферон
Найденные в высших нефтяных фракциях полициклические фено-
лы являются, по-вилимому, продуктами превращения полицикличес-
ких природных соединений (стероидных спиртов, например, холесте-
рина) в процессе катагенеза:
267
268
Сернистые соединения. Содержание серы в нефтях значительно вы-
ше, чем в органических соединениях — предшественниках нефти. По-
этому основная масса сернистых соединений нефти имеет вторичное
происхождение. В настоящее время установлено, что алифатические
сернистые соединения и некоторые гетероциклические (тиофаны) яв-
ляются продуктами осернения углеводородов нефти при их взаимодей-
ствии с серой и сероводородом. Установлено, что сероводород образу-
ется в результате жизнедеятельности анаэробных бактерий, которые
восстанавливают сульфаты, содержащиеся в горных породах место-
рождений, используя их кислород:
Na.SO, + 4 Н, -> Na?S + 4 Н?О.
Z 4 Z Z Z
Сульфиды подвергаются гидролизу с образованием сероводорода:
Na.S + 2 Н7О о 2 NaOH + H?S.
Сероводород, являясь сильным восстановителем, взаимодействует
с оксидом железа с образованием серы:
H2S + Fe2O3 -> S + 2 FeO + H2O.
В стадии диагенеза сероводород может присоединяться к олефинам,
содержащимся в исходном органическом веществе с образованием ти-
олов, сульфидов и дисульфидов:
^-7—-►R-CH-S-CH-R
А I I П
СН3 СН3
^^►R-CH-S-S-CH-R
101 | | in
СНз СНз
2R-CH=CHr^*2 R-CH-SH
1 I
СН3
269
Превращение тиолов (I) в дисульфиды (HI) происходит в результате
взаимодействия тиолов с оксидами металлов переменной валентности,
например с оксидами железа:
2 R-SH + Fe2O3 -> R-S-S-R + Н2О 4- FeO.
Известно, что углеводороды (легче ароматические, труднее насы-
щенные) реагируют с серой с образованием тиолов, сульфидов при
температуре 150--170°С:
2 RH + 2S^ R-S-R- H.S,
R-S-R -> R'CH=CH2 + R'"'SH.
Возможно также образование тиофанов:
Н<—СН?
*4“
R-CH2-CH2-CH2-CH2-R + 2S---► / \ + H2S
Тиофены образуются, возможно, в результате дегидрирования тио-
фанов под действием серы:
Н 2С—С н- R НС-----С- R
/ \ + 2S------- II \\ - 2 HjS
Н2С ,СН2 нс. сн
Азотистые соединения нефтей имеют биологическое происхожде-
ние, т. е. попали в нефть из органических остатков растительных и жи-
вотных организмов и бактерий, содержащих белки, хлорофиллы, геми-
ны, алкалоиды (в случае высших растений).
Белки, содержащие 15-19% азота, в стадии диагенеза подвергались
под действием бактерий распаду с образованием аминокислот; основ-
ная часть аминокислот в стадии катагенеза разлагалась и превраща-
лась в азотсодержащие соединения, в том числе гетероциклические,
например:
h2n-ch-ch
соон снгсн2- Н С + С°2 + NH3 н
270
Хлорофиллы и гемины распадались с образованием порфиринов.
Алкалоиды, многие из которых содержат азотсодержащие гетероцикли-
ческие фрагменты (фрагменты пиррола, изохинолина) в процессе пре-
образовании органического вещества в стадии катагенеза превращались
в производные пиррола, пиридина, хинолина, карбазола (см. гл. III §3).
Смолисго-асфальтеновые вещества. Образование нефтяных смол
{нейтральные смолы и асфальтеновые кислоты) относится к стадии диа-
генеза. В результате реакций конденсации (в том числе и окислитель-
ной с участием бактерий) молекул исходного органического вещества
образовались более сложные молекулы. В стадии катагенеза наряду
с последующей сшивкой молекул с участием серы проходили реакции,
приводящие к отщеплению алкильных групп (при каталитическом воз-
действии глинистых пород), реакции окислительного дегидрирования
циклогексановых колец (в том числе и участием серы) с образованием
ароматических ядер.
Асфальтены можно рассматривать как остаток керогена, нс превра-
тившегося в нефть. В стадии катагенеза асфальтены могли образовать-
ся из смол в результате их термической или окислительной конденса-
ции. Асфальтены являются наименее изменяемой частью нефтей
в процессе их превращения в природе, так как их углеводородная часть
не подвергается биологическому разложению. Как показано работами
Ал. А. Петрова и сотрудниками1 углеводородный состав продуктов пи-
ролиза асфальтенов нефти типа Б' и нефти типа А! одного и того же
месторождения идентичен.
5.3. Превращение нефти в земной коре
В процессе нефтсобразования по мере увеличения глубины залегания
вмещающих порол вследствие повышения температуры и давления в
нефти проходили процессы деструкции сложных молекул, удаления гете-
роатомов; нефть обогащалась алканами, и облегчался ее фракционный
состав. Такая нефть по составу может быть охарактеризована как нефть
типа А1 (по классификации Ал. А. Петрова). Глубина залегания нефти
этого типа -2000 м, а температура в пласте в среднем 90~100’С. В этих ус-
ловиях существование бактерий мало вероятно и, следовательно, бакте-
риальное окисление (биодеградация) алканов не проходило.
Вследствие геотектонических смещений в земной коре, отложения,
вметающие нефть типа А1, могли оказаться ближе к поверхности и на
глубине 1200 м при средней температуре 40 С происходило развитие
бактерий, которые поглощали //-алканы, изоалканы, и нефть превра-
щалась в зависимости от длительности биологического воздействия
в нефть типа А2, а затем в нефти типа Б2 и Б1.
^ Петров А ) Л У| леволоролы нсф | и М/ Наука. 19X4. — С. 247.
271
5.4. Превращение нефти в окружающей среде.
Экологические аспекты
В процессе транспортирования нефти по трубопроводам, железно-
дорожным транспортом в цистернах, морским путем в танкерах случа-
ются аварии, в результате которых происходит разлив нефти на поверх-
ности суши и воды, что является экологической катастрофой, так как
компоненты нефти, в большинстве своем ядовитые вещества, отравля-
ют растительные и животные организмы, которые либо погибают, либо
замедляют свой рост и развитие.
Нефть, попавшая в окружающую среду после аварийных разливов,
подвергается сложным физико-химическим превращениям, таким как
испарение, растворение в воде, эмульгирование в воде, окисление,
конденсация под действием солнечной радиации наиболее химически
активных веществ (смолы, асфальтены). включая окислительную кон-
денсацию (с участием кислорода воздуха); нефть подвергается превра-
щениям под действием бактерий (биодеградация). Остановимся на
особенностях этих превращений. Рассмотрим вначале испаряемость
углеводородов нефти.
При одной и той же молекулярной массе максимальной испаряе-
мостью обл ада ют ал ка н ы. П р и ч е м с ред и ал ка н о в л и гч е вс е го и с паря ют-
ся изоалканы. Труднее испаряются нафтены, еще труднее ароматичес-
кие углеводороды.. Это объясняется различным межмолекулярным
взаимодействием углеводородов различных рядов. Наиболее сильное
межмолекулярное взаимодействие — у ароматических углеводородов.
Молекулы ароматических углеводородов имеют плоское строение в
циклической части, поэтому они плотно упаковываются в жидком и
кристаллическом состоянии и для того, чтобы молекула оторвалась от
поверхности и перешла в газовый объем, необходимо затратить энер-
гию на преодоление этого межмолекулярного взаимодействия. Мень-
ше межмолекулярное взаимодействие у нафтенов. Еще меньше межмо-
лекулярное взаимодействие у алканов и изоалканов, Изоалканы имеют
несимметричное строение и плотно не упаковываются в жидком состо-
янии, поэтому молекулы легко отрываются от поверхности и переходят
в объем. Парафинистые нефти типа А| будут легче испарятся, чем наф-
тено-ароматические нефти типа Б,. Так как скорость испарения опре-
деляется диффузией паров в газовый объем и при повышенной кон-
центрации паров над поверхностью пленки нефти диффузия будет за-
трудняться, то движение воздуха над пленкой разлитой нефти будет
способствовать уменьшению концентрации паров над пленкой и их
испарению. Низкокипяшие летучие углеводороды, которые уносятся
ветром, могут быть идентифицированы обонянием человека, будучи
даже в весьма низкой концентрации. Также легко идентифицировать
272
углеводороды нефти, растворенные или эмульгированные По запаху
можно обнаружишь следы углеводородов бензина при концентрации
0,005 млн в холодной воде и почувствовать резкий запах при конце-
нтрации 0,01 млн Следы тяжелых фракций нефти можно обнару-
жить в воде при более высокой концентрации 1- 25 млн-1. Допусти-
мый предел содержания нефти в питьевой воде в разных странах от 0,1
до 1 млн-1. Он определяется появлением неприятного вкуса воды. Ры-
ба в водоемах, содержащих 0,01 млн-1 углеводородов в воде, быстро
приобретает неприятный запах и вкус. Пары легкокипящих углеводо-
родов при испарении с поверхности нефтяной пленки увлекают с со-
бой более тяжелые углеводороды (с температурой кипения до 200°С) и
в ветряную погоду разносятся на большие расстояния, так что вблизи
разлива нефти через 2-3 дня при температуре меньше 20°С концент-
рация паров углеводородов в воздухе будет близка к предельно допус-
тимой или слабо превышать ее. При этом легкие нефти типа А могут
терять до 30% своей массы.
Важным свойством компонентов нефти является их растворимость
в воде. Все углеводороды слабо растворяются в воде, но их раствори-
мость отличается друг от друга. Растворимость углеводородов повыша-
ется в ряду алканы < нафтены < арены (табл. 21). Причем с увеличени-
ем длины углеводородной цепи на два атома углерода растворимость
понижается на порядок. В качестве примера слабой растворимости вы-
сококипяших углеводородов можно привести следующие расчетные
данные: на каждые 250 км2 в Ла-Манше можно растворить 1 т додекана
С12Н?6, а для растворения 1 тонны октадекана С1КН^, потребовалась бы
вся поверхность этого пролива (длина 500 км, ширина от 35 до 180 км).
Эмульгирование нефтей в воде зависит от поверхностного натяже-
ния нефти, ее плотности и вязкости.
Поверхностное натяжение нефти зависит от температуры и состава
нефти. С повышением температуры поверхностное натяжение умень-
шается.
Так, поверхностное .натяжение бензола при 20°С равно 28,8 дин/см,
а при 80°С — 20,3 дин/см.
При одинаковом числе атомов углерода в молекуле поверхностное
натяжение на границе углеводород - воздух увеличивается с увеличе-
нием плотности в ряду алканы < нафтены < арены.
У различных нефтепродуктов поверхностное натяжение на границе
раздела углеводород - воздух увеличивается также с увеличением
ПЛОТНОСТИ'.
бензин керосин смазочное масло.
Эти закономерности находятся в соответствии с уравнением
Маклеода (1923).
273
18-3544
Таблица 21. Растворимость углеводородов нефти в воде
X i iciui.iopo i формула т , ч кип* J1 HJJHOL’ГЪ р4'() Растворимость а воде
Пентан CSH|2 36.2 0,626 0.036% ((70сС)
Гексан 69,0 0,660 0,014% (150°С)
Гептан с?н l(1 98,0 0,684 0,0052% (150’С)
Нонан 151.0 0,718 10 млн-1
Гсксадекан 1(Дз4 287,5 0 774 0,001 млн~*
Нафтены Циклопентан CsH|0 49,3 0,751 Незначительная
Циклогексан с(,н|2 80,7 0,779 Незначительная
Арены Бензол сбн(. 80.1 0,879 0,082%
Толуол 110.6 0,366 0,047%
Нафталин ад 217.9 1,145 0.002%
Нефтепродукты Бензин — н.к - 180 0 72-0.75 0,004%
Керосин — 180-300 0,80-0 82 0,003%
Масло — >300 0.83-0.86 0,002%
о = с (D - б/)4.
где с — константа; D — плотность жидкости•_ d — плотность паров над
жидкостью.
В случае поверхностного натяжения нефть — вода наблюдается дру-
гая закономерность: с повышением плотности поверхностное натяже-
ние уменьшается. Вследствие этого нефти и нефтепродукты с высокой
плотностью будут легче эмульгироваться в воде. Так, тяжелые нефти
типа Б будут легче эмульгироваться, 1. е. образовывать эмульсии типа
нефть в воде (прямые эмульсии), чем нефти типа А. При эффективном
эмульгировании вводе 1 мл нефти может давать 15-1012 капель с обшей
поверхностью 12 м2.
Однако прямая эмульсия лаже тяжелых нефтей, полученная меха-
ническим эмульгированием, быстро расслаивается.
Более стабильными являются так называемые обратные эмульсии
типа вода в нефти. Особенно, если нефть тяжелая (типа Б) и содержит
много асфальтенов, играющих роль эмульгаторов. Такая эмульсия мо-
жет содержать 50—80% воды и не расслаиваться в течение нескольких
месяцев, образуя на поверхности воды слон толщиной 1 мм (мусс).
После испарения части углеводородов плотность «мусса» приближает-
274
ся к плотности волы, и при волнении пленка разбивается на куски, ко-
торые, захвснив из виды минеральные частицы. могут затонуть
Сточки зрения повеления нефти в окружающей среле большое зна-
чение имеет такая характеристика нефти, как вязкость. Обычно для
нефти измеряют кинематическую вязкость. Она измеряется в стоксах.
Наибольшей вязкостью из углеводородов обладают нафтены, так как
они имеют строение часто несимметричное, в пространстве они зани-
мают такое положение, которое препятствует движению слоев жидкос-
ти друг по отношению к другу. Наименьшую вязкость имеют алканы.
Ароматические углеводороды занимают промежуточное положение.
Тяжелые нефти типа Б имеют большую вязкость, чем нефти типа А.
Нефть пониженной вязкости (легкая нефть типа А) может расте-
каться на водяной поверхности теоретически до образования мономо-
лекулярного слоя, но практически это не происходит, так как вслед-
ствие быстрого испарения легких компонентов нефти и их растворения
в воле вязкость гг поверхностное натяжение нефти увеличиваются и
скорость распространения нефтяной пленки снижается. Следователь-
но, растекание нефти по водной поверхности — это самотормозяший-
ся процесс. Если пленка очень тонкая (0,002 мм), то она практически
не задерживает проникновение кислорода в волу и не препятствует тем
жизненным процессам, которые протекают в воде. Но если слои более
толстые (примерно от нескольких миллиметров до 10 мм), то проник-
новение кислорода воздуха задерживается на 5-20%. Однако это не
влияет существенно на жизнедеятельность организмов. Темно окра-
шенные пленки нефти поглотают свет на 80-90%, и тогда процесс фо-
тосинтеза в воде затрудняется. Вследствие этого замедляется выделе-
ние кислорода растениями и концентрация кислорода значительно
падает, что может вызвать угнетение жизнедеятельности организмов и
даже при больших скоплениях их гибель. В летнее время такая толстая
темная пленка приводит к нагреву воды и уменьшению содержания
кислорода, что также губительно сказывается на жизнедеятельности
организмов.
Теперь рассмотрим, какие химические превращения происходят с
компонентами нефти при попадании ее в атмосферу и воду. Под
действием солнечного ультрафиолетового излучения, с длиной волны
примерно 300-350 нанометров, происходит окисление углеводородов в
атмосфере. Легче всего окисляются алканы нормального строения:
изоалканы н нафтены также легко окисляются, а ароматические угле-
водороды ряда бензола, особенно с короткими боковыми цепями,
окисляются мслленно. Но если ароматический углеводород имеет
длинную алкильную цепочку, тогда окисление протекает интенсивно,
особенно у соседнего с бензольным кольцом углеродного атома. Про
лукты окисления — спирты, альдегиды, кегоны. кислоты, как более тя
жсгыс пнем падают па поверхность воды или суши и подвергаются
дальнейшему окислению как кислородом воздуха, так и биологическо-
му окислению с участием бактерий и водных растений. Те углеводоро-
ды. которые оказываются в водной среде, также легко подвергаются
окислению, особенно если они интенсивно эмульгированы и поверх-
ность контакта их с водой будет велика. Они подвергаются как хими-
ческому, так и биологическому окислению с участием бактерий и вод-
ных растений. Следует отметить, что почти все углеводороды подверга-
ются биологическому разложению под действием бактерий (биодегра-
дации). Легче всего подвергаются биодеградации алканы нормального
строения, затем изоалканы и нафтены, труднее подвергаются этому
процессу ароматические углеводороды, особенно те, которые не имеют
длинных боковых цепей. Однако полициклические ароматические уг-
леводороды, такие как бензпирен, пирен, легко подвергаются биодег-
радации. Это объясняется тем, что, кроме строительного материала для
клетки, которым является углерод, эти углеводороды несут в себе по-
вышенный запас энергии. При благоприятных условиях все аромати-
ческие углеводороды, даже бензол, могут разлагаться отдельными ви-
дами бактерий, грибов и микроскопических водорослей. Все арены об-
ладают избыточной энергией, которая необходима бакзериям, чтобы
осуществить синтез белка. Например, бензпирен — очень опасное кан-
церогенное вещество, которое вызывает образование опухолей у жи-
вотных, довольно легко биологически разлагается. Если смешать 3 г
бензпирена с 1 л ила сточных вод, то происходит быстрое разложение
бензпирена (за час разлагается примерно 80% вещества). В отличие от
углеводородов, эмульгированных или растворенных в воде, окисление
которых протекает очень легко вследствие высокой поверхности соп-
рикосновения с кислородом, в пленке нефти окисление происходит
медленнее, так как необходима диффузия кислорода через пленку. Есть
данные, что в поверхностном слое разлитой нефти может окисляться
ежедневно 2 т на км2. Окисление в этом слое ускоряется металлами.
В нефти много металлов, которые находятся в виде комплексных сое-
динений (в особенности металлы переменной валентности) с такими
веществами, как смолы и асфальтены. Металлы ускоряют окисление.
Сернистые соединения нефтей являются ингибиторами окисления.
Они замедляют окисление. В поверхностном слое лучше будет окис-
ляться легкая нефть, нафтеновая или нафтено-парафинистая нефть, не
содержащая или содержащая мало ванадия и никеля. В отличие от би-
ологического химическое окисление происходит гораздо медленнее.
Очистка сточных вод от бензпирена химическим окислением не очень
эффективный процесс. Известно, что за 5 часов окисляется 30% бенз-
пирена, через 5 суток — 50%, и затем окисление прекращается, потому
что продукты окисления ингибируют процесс окисления. Гетероатом-
276
Бензпирен
С ООН
Н20 з С02
/СООН 101
|О1 О=с __
о—с.
^соон
нс^
С ООН
Рис. 60. Биологическое
окисление бензпирена, бензола и фенолов
277
ные соединения нефти (кислородные, сернистые, азотистые) окисля-
ются по-разному. Нефтяные кислоты практически не окисляются. Фе-
нолы окисляются легко либо биологически, либо кислородом воздуха,
на чем и основано обезвреживание фенольных сточных вод. Азотистые
и сернистые соединения окисляются медленно. Быстро превращаются
под действием кислорода и солнечного света смолисто-асфальтеновые
вещества, полициклические ароматические углеводороды. Эти компо-
ненты под действием кислорода воздуха и солнечного света уплотня-
ются и превращаются в более тяжелые углеподобные вещества, кото-
рые оседают на дно водоема. На рис. 60 показано, как проходит биоло-
гическое окисление бензпирена, бензола и фенола. Под действием
ферментов микроорганизмов происходит атака кислорода по двойным
связям одного из бензольных колен бензпирена. Образуются две гид-
роксильные группы и атом кислорода в виде мостика. В дальнейшем
под действием кислой среды происходит распад этого мостика, образу-
ются три гидроксильные группы и положительно заряженный атом
углерода. В такой частице имеются гидрофильная и гидрофобная час-
ти. Такая молекула, являясь поверхностно-активным веществом, легко
проникает в клетку и блокирует наиболее активные (электронодонор-
ные) участки ДНК, тем самым нарушая синтез белка и вызывая обра-
зование опухолей у животных. Таким образом, не сам бензпирен опа-
сен как таковой, а продукты его биологического окисления, так как
они легко проникают через клеточные мембраны и блокируют ДНК.
На рис. 60 представлена схема биологического окисления бензола.
Окисление идет по-разному, втом числе с образованием фенола. Бензол
очень трудно окисляется. Фенол легко окисляется. Имеются способы
окисления фенольных сточных вод пропусканием воздуха через слой
сточной воды. Происходит окисление фенолов в хиноидную структуру
и ее распад до кислоты и в конечном итоге до СО2 и воды.
Фенол сам по себе хотя и ядовит, но опасность для организма пред-
ставляет не столько фенол, сколько продукты его окисления, которые
образуют очень прочные комплексы с биокатализаторами (фермента-
ми) и нарушают процесс биохимического синтеза в орг анизме. В рабо-
те1 исследован состав нефти Усинского месторождения, подвергшейся
превращениям в окружающей среде, после аварийных разливов в 1990
и 1994 гг. и показано, что химический и фракционный составы нефтей
претерпели глубокие изменения в результате испарения, окисления
биодеградации углеводородов, втом числе и полициклических аренов.
Вместе с тем в результате экстракции из растительных остатков в неф-
ти по сравнению с нативной повысилось содержание некоторых н-ал-
канов и высших изопренанов.
Рябой В. Д., Горладзе Г. Н., Котенев В. Н. и др. // Наука и технология углеводо-
родов, — 2000 — №4 — С. 44,
278
ЛИТЕРАТУРА
1. Бурейан Н. Л Технология изомеризации парафиновых углеводо-
родов. - Л.: Химия. 1985-
2. Бранд Дж., Эгминтон Г. Применение спектроскопии в органичес-
кой химии (пер. с англ.). — М.: Мир. 1967.
3. Веденеев В. И., ГурвичЛ. В., Кондратьев В. Н. и др. Энергии разры-
ва химических связей. Потенциалы ионизации и сродства к электро-
ну. - М.: Изд-во АН СССР, 1962.
4. Вигдергауз М. С. Газовая хроматография как метод исследования
нефти. - М.: Наука, 1973.
5. lypeee А. А., Фукс И. Г,, Лашхи В. Л. Химмотология. — М.: Химия,
1986.
6. Добрянский А. Ф. Химия нефти. — Гостоптехнздат, 1961.
7. Жермен Дж. Гетерогенный катализ. — М.: ИЛ, 1961.
8. Жермен Дж. Каталитические превращения углеводородов. — М.:
Мир, 1972.
9. Инструментальные методы исследования нефти. — Новосибирск;
Наука, 1987.
10. Иоффе Б. В. Рефрактометрические методы химии. — М.; ГХИ,
I960.
11. Иоффе Б. В., Костиков Р. Р., Разин В. В. Физические методы оп-
ределения строения органических соединений (под рея. Ь. В. Иоф-
фе) -- М.; Высшая школа, 1984.
12. Исагулянц В. И., Егорова Г. М. Химия нефти. Руководство к лабо-
раторным занятиям. — М.: Химия. 1965.
13. Калении И. В. Химия гидрогенизанионных процессов в перера-
ботке топлив. — М.: Химия. 1970.
14. Камьянов В. Ф., Аксенов В. С., Титов В. И. Гстероатомныс компо-
ненты нефти -- Новосибирск: Наука, 1982.
15. Kenneth Е. Peters, /. Michael Moldowan. Hie Bio marker Guide. -
New Jersy, 1993
16. Капщина Л. А., Куплетския H. Б. Применение инфракрасной,
ультрафиолетовой и ЯМР-спекгроскопии в органической химии. --
М.; Высшая школа, 1971.
17. Кирсо У. ).. Стом Л- М.. Белых Л. И.. Ирха И. И. Превращение
токсичных и канцерогенных веществ в гидросфере, 1аллинн: Валгуе,
1988.
18, Леви Г.. Нельсон /'. Руководство к ялерному магнитному резонан-
су углерода для химиков-органиков (пер. с англ.). М,: Мир, 1975.
19. Магарил Р. 3. Теоретические основы химических процессов пе-
реработки нефти: Учебн. и особ, для вузов. — Л.: Химия. 1985.
279
20. Наметкин С. ('. Химин нефти. — М.: изд. АН СССР, 1955,
21. Нельсон-Смит /I. Нефть и экология моря. — М.: Прогресс,
19" - 302 с.
22. Нефедов Б. К., Радченко £ Д., Алиев Р. Р. Катализаторы пронес-
сов углубленной переработки нефти. — М.: Химия, 1992,
23. Панченков Г, М., Лебедев В. П. Химическая кинетика и ката-
лиз. — М.: Химия, 1974.
24. Петров Ал, А. Химия нафтенов. — М.: Наука, 1971.
25. Петров Ал. А. Химия алканов. — М.: Наука, 1974.
26. Петров Ал. А. Углеводороды нефти. — М,: Наука, 1984.
27. Поконова Ю. В, Химия высокомолекулярных соединений неф-
ти. — М,: Изд-воЛГУ, 1980.
28. Полякова А. А. Молекулярный масс-спектральный анализ орга-
нических соединений. — М.: Химия, 1983.
29. Рябов В, Д. Термические и каталитические превращения углево-
дородов и других соединений нефти: Учебн. пособ. — М.: МИНХи ГП,
1982.
30. Рябов В. Д. Физико-химические методы исследования углево-
дородов и других компонентов нефти: Учебн. пособ. — Мл ГАНГ,
1996.
31. Рябов В. Д. Химический состав, свойства и анализ углеводоро-
дов и других компонентов нефти и газа: Учебн. пособ. — М.: ГАНГ,
1997.
32. Сафиева Р. 3. Физикохимия нефти. — М.: Химия, 1998.
33. Сергеенко С Р, Таимова Б. А., Талалаев В. И. Высокомолекуляр-
ные неуглеводородные соединения нефти. — Мд Наука, 1979.
34, Соколов В. А., Бестужев М. А., Тихом олова Т. В. Химический со-
став нефтей и природных газов в связи с их происхождением. — М.:
Недра, 1972.
35. Суханов В. П. Каталитические процессы в нефтепереработке. —
3-изд. пер, и доп. — М.: Химия, 1979.
36, Сюняев Р. 3., Сафиева Р. 3. Коллоидные структуры асфальте-
нов. — М.; Нефть и газ, 1994.
37. Сюняев 3. И., Сафиева Р. 3., Сюняев Р. 3. Нефтяные дисперсные
системы. — М.: Химия, 1990.
38. Томас Ч. Промышленные каталитические процессы и эффектив-
ные катализаторы. — М.: Мир, 1973.
39. Унгер Ф, Г, Андреева Л. И. Фундаментальные аспекты химии
нефти, — Новосибирск: Наука, Сибирская издательская фирма РАН,
1995.
40. Богомолов А. И., Гайле А. А,, Громова В. В, и др. Химия нефти и га-
за: Учебн. пособ. для вузов / Под ред. В. А. Проскурякова, А. Е. Драб-
кина. — 3-е изд. доп. и испр. — СПб: Химия, 1995.
280
41. Поконова Ю. В., Гайле А. А., Спиркин В, Г. и др. Химия нефти /
Подред. 3. И. Сюняена. — Л.: Химия, 1984.
42. Юинг Г. В. Инструментальные методы химического анализа. —
М.: Госатомиздат, 1963.
43. Заикин В. Г., Варламов А. В., Мыкая А. И., Простаков Н. С. Осно-
вы масс-спектрометрии органических соединений. — М.: МАИК «На-
ука/Интер-периодика», 2001.
281
СОДЕРЖАНИЕ
ПРЕДИСЛОВИЕ ................ ........... ................ •. 3
ВВОДНАЯ ЧАСТЬ.......................... ...............6
Краткая характеристика компонентов нефти ......9
Химическая классификация нефтей ................12
Фракционный состав нефтей ................. 13
Глава 1. ФИЗИКО-ХИМИЧЕСКИЕ МЕТОДЫ
ИССЛЕДОВАНИЯ НЕФТИ И ГАЗА ........................... 14
§ 1. Физико-химические методы разделения
компонентов нефти и газа..............................14
1.1. Разделение углеводородных смесей методами перегонки,
экстракции, кристаллизации, термической диффузии ... 16
Перегонка при пониженном давлении .............16
Азеотропная и экстрактивная перегонка .........17
Кристаллизация............................... 18
Экстракция................................. 19
Термическая диффузия........................ 20
1.2. Хроматографические методы разделения и анализа
углеводородных смесей ............................21
Фронта льный анализ...................... .....23
Прояви тельный анализ..........................23
Вытесн ительный анализ....................... 24
1.2.1. Жидкостно-адсорбционная хроматография.....24
1.2.2. Газовая хроматография.................... 28
Насадочная хроматографическая колонка .........29
Детектор .................................. 30
Характеристика хроматограммы
и хроматографического пика.....................31
Особенности современных газовых хроматографов ..... 32
Программирование температуры ................ 33
Адсорбенты в газовой хроматографии.............33
1.2.3. Аналитические задачи в химии нефти,
решаемые с помошыо газовой хроматографии..........37
Качественный анализ......................... 37
Количественный анализ..........................37
282
Выделение веществ в чистом виде................38
§ 2. Физико-химические методы идентификации
и количественного определения углеводородов
и других компонентов нефти и газа..................39
2.1. Физико-химические константы углеводородов нефти
и их роль в идентификации компонентов и анализе
у гл е водород н ых с месей ........................39
2.2. Спектральные методы идентификации углеводородов
и других компонентов нефти и газа..................45
2.2.1. Молекулярная спектроскопия ..................45
Инфракрасная спектроскопия ....................46
Ультрафиолетовая спектроскопия ...................49
2.2.2. Масс-спектрометрия ........................ 53
2.2.3. Спектроскопия ядерного магнитного резонанса .58
Протонный магнитный резонанс......................60
Ядерный магнитный резонанс изотопа углерода 13С .... 64
2.2.4. Спектроскопия электронного парамагнитного
резонанса (ЭПР) ............................. 66
2.2.5. Атомно-абсорбционная спектроскопия...... 69
Глава И. УГЛЕВОДОРОДЫ НЕФТИ
И ПРОДУКТОВ ЕЕ ПЕРЕРАБОТКИ ................................71
§ I, Алканы (метановые углеводороды) .................. 71
1,1. Содержание в нефтях ......................... 71
1,2. Физические свойства алканов......................73
Спектральные характеристики алканов ..............75
1.3. Химические свойства алканов ................... 75
Комплексообразование .............................76
Изомеризация алканов..............................77
Термическое разложение алканов..................78
Дегидроциклизация алканов ........................78
1.4. Газообразные алканы .............................79
Методы анализа углеводородных газов..............-79
1.5. Жидкие алканы нефтей ............................S3
Жидкие алканы как компоненты топлив ..............85
283
1Xv Твердые алканы . .................................87
1.7. Анализ алканов нефтяных фракций..................88
Количественное определение алканов ..............88
Выделение алканов ...............................89
Идентификация алканов ...................... 90
§ 2. Циклоалканы (нафтены) нефтей...................... 92
2.1. Номенклатура и изомерия циклоалканов........... 92
2.2. Циклоалканы, найденные в нефтях................ 95
2.3. Физические свойства нафтенов .................. 100
2.4. Химические свойства нафтенов....................102
Образование комплексов с тиомочевинои ........ 102
Дегидрогенизация нафтенов..................... 102
Циклизация алкилпроизводных нафтенов.......... 103
Изомеризация нафтенов...........................104
2.5. Анализ нафтенов............................ 105
Количественное определение нафтенов.............105
Выделение нафтенов и их идентификация...........106
§ 3. Ароматические углеводороды нефти (арены)
и углеводороды смешанного строения ...................110
3.1. Физические свойства ароматических
углеводородов .................................... 112
3.2. Химические свойства ароматических
углеводородов ................................... 112
Образование комплексов с пикриновой кислотой
(пикратов) .................................. 112
Сульфирование ароматических углеводородов ......114
Гидрирование ароматических углеводородов........115
Конденсация полициклических ароматических
углеводородов с малеиновым ангидридом ..........115
Пербромирование ароматических углеводородов.....116
Конденсация с формальдегидом
(формолитовая реакция) ..................... 116
3.3. Анализ ароматических углеводородов..............Н?
Количественное определение ......................И?
Выделение ароматических углеводородов...........119
284
Идентификация ароматических углеводородов.....120
3.4. Углеводороды смешанного строения............. 120
§4. Ненасыщенные углеводороды нефти и продуктов
ее переработки................................. j2j
4.1. Олефины (алкены) ...........................122
4.1.1. Химические свойства алкенов ........... 123
Присоединение водорода.......................123
Галоидирование .............................. 124
Присоединение серной кислоты .................125
Озонирование алкенов ........................125
Присоединение ацетата ртути................ 126
4.1.2. Количественное определение,
выделение и идентификация олефинов .............126
4.2. Диолефины нефтяных продуктов (диены)........127
§ 5. Определение состава нефтяных фракций
и нефтяных продуктов ............................ 128
5,1. Определение группового состава и детализированного
группового состава бензиновых фракций ......... 129
5.2. Определение детализированного группового
состава керосино-газойлевых фракций .............133
5.3. Определение структурно-группового состава
масляных фракций............................... 134
Прямой метод............................. 137
Метод н-р-Л/! ........................... 141
Глава III. ГЕТЕРОАТОМНЫЕ СОЕДИНЕНИЯ
И МИНЕРАЛЬНЫЕ ВЕЩЕСТВА НЕФТИ .........................144
§ 1. Кислородные соединения нефти ..................144
Метод постепенного растепления............. 147
§ 2. Сернистые соединения ........................ 147
§ 3. Азотистые соединения......................... 1^5
§4. Смолието-асфальтеновые вещества (САВ)..........158
§ 5. Минеральные компоненты нефти...................161
285
Глава IV. ТЕРМИЧЕСКИЕ И КАТАЛИТИЧЕСКИЕ
ПРЕВРАЩЕНИЯ УГЛЕВОДОРОДОВ
И ДРУГИХ КОМПОНЕНТОВ НЕФТИ И ГАЗА .......................163
§ I. Некоторые аспекты физической химии углеводородов...163
ЕЕ Основные характеристики связей
в молекулах углеводородов. Типы разрыва связей..163
1.2. Термическая стабильность углеводородов ........168
§ 2. Химизм и механизм термических превращений
углеводородов и других компонентов нефти и газа.......171
2.Е Термические превращения углеводородов ..........172
2.2 . Термические превращения высокомолекулярных
компонентов нефти в жидкой фазе........... ......... 188
§ 3. Химизм и механизм каталитических превращений
углеводородов и других компонентов нефти и газа.......191
З .Е Каталитический крекинг........................193
Алканы..........................................198
Алкены........................................ 203
Нафтены ........................................206
Ароматические углеводороды .....................210
3 .2. Превращения углеводородов и других компонентов
нефти и газа в гидрогенизапионных
процессах переработки ..........................213
3.2.Е Гидрокрекинг .,................................. 215
Дегидроциклизация алкиларенов................. 225
Реакция диенового синтеза .................... 225
Крекинг (деалкилирование) .................... 226
Изомеризация....................................226
3.2.2. Гидроочистка .......,......................226
3.2.3. Каталитический риформинг...................234
Химизм и механизм основных реакций,
протекающих при платформинге....................236
Дегидрирование шестичленных нафтенов ...........238
Изомеризация ал кил циклопентанов
в циклогексановые углеводороды
с последующим дегидрированием последних
(реакция дегидроизомеризации) ................... 241
286
Дегидроциклизация алканов ....,............241
3 .3. Превращения углеводородов в реакциях
полимеризации, алкилирования и изомеризации
(синтез высокооктановых компонентов топлив) ...245
3.3.1. Ступенчатая полимеризация олефинов ... 249
3.3.2. Алкилирование изоалканов алкенами......251
Синтез изооктана ........................ 251
Синтез триптана, неогексана и 2,3-диметилбутана.253
3.3.3. Изомеризация алканов................. 256
Глава V. П РОИСХОЖДЕНИЕ НЕФТИ
И ЕЕ КОМПОНЕНТОВ.
ПРЕВРАЩЕНИЕ НЕФТИ В ПРИРОДЕ...................... 260
5.1. Гипотезы минерального происхождения нефти
Д. И. Менделеева и других ученых ......... 260
5,2. Гипотеза органического происхождения нефти
из органического вешества, рассеянного
в осадочных породах......................... 262
5.3. Превращение нефти в земной коре ..........271
5.4. Превращение нефти в окружающей среде.
Экологические аспекты..........................272
ЛИТЕРАТУРА ...................................... 279
i
д
I
287
Учебное издание
Владимир Дмитриевич Рябов
ХИМИЯ НЕФТИ И ГАЗА
Все права защищены, Никакая часть данного издания
не может быть воспроизведена в любой форме
или любыми средствами, электронными,
механическими, включая фотографирование,
ксерокопирование или иные средства копирования
или сохранения информации,
без письменного разрешения издательства.
Редактор Ю. Н, Кузьмичева
Оформление и верстка В. В. Земсков
Формат 60x90 ’/и,- Печать офсетная.
Тираж 1000 экз. Заказ № 3544
Отпечатано в полном соответствии
с качеством предоставленных диапозитивов
в ОАО «Можайский полиграфический комбинат»,
143200, г. Можайск, ул. Мира, 93,