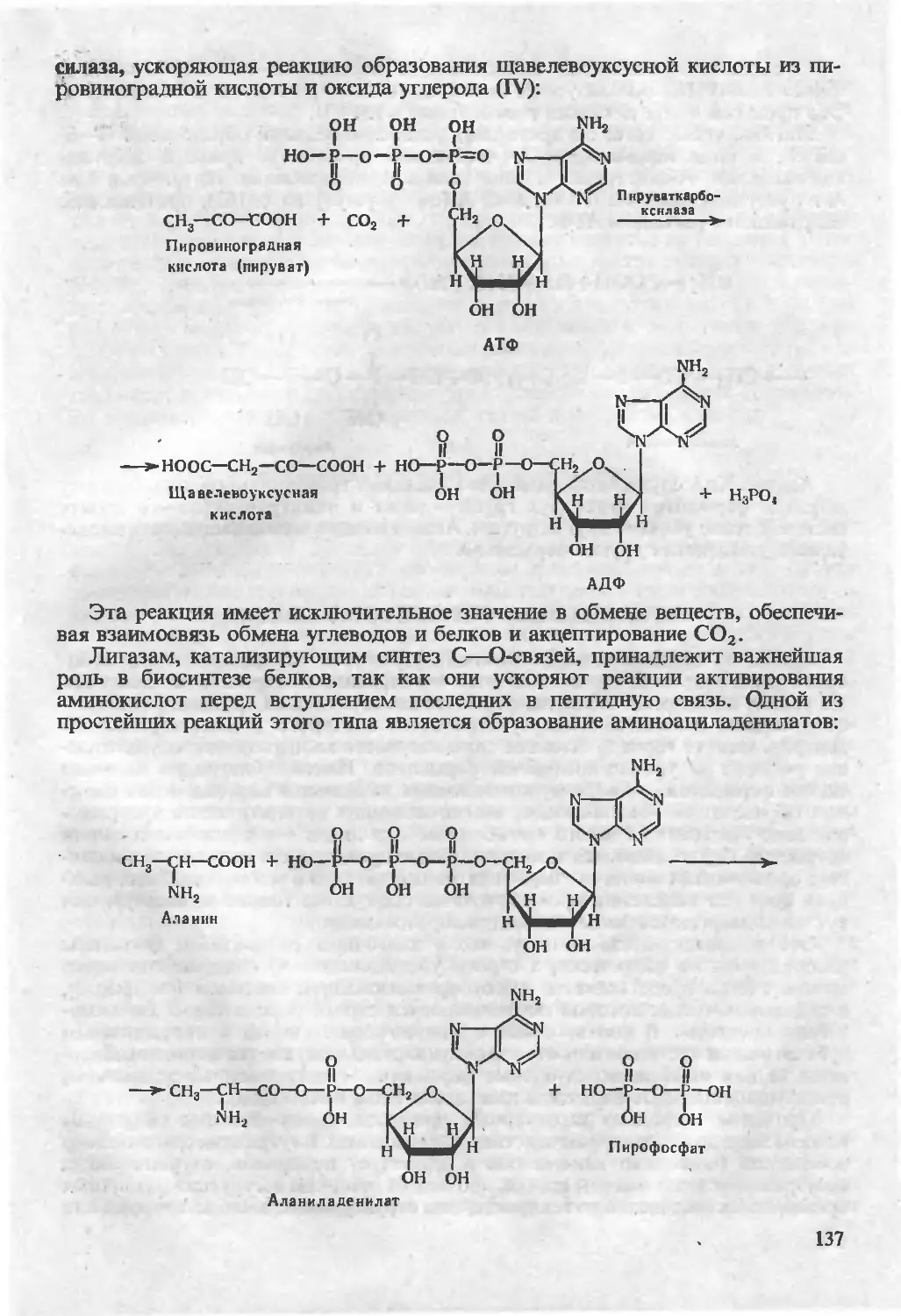
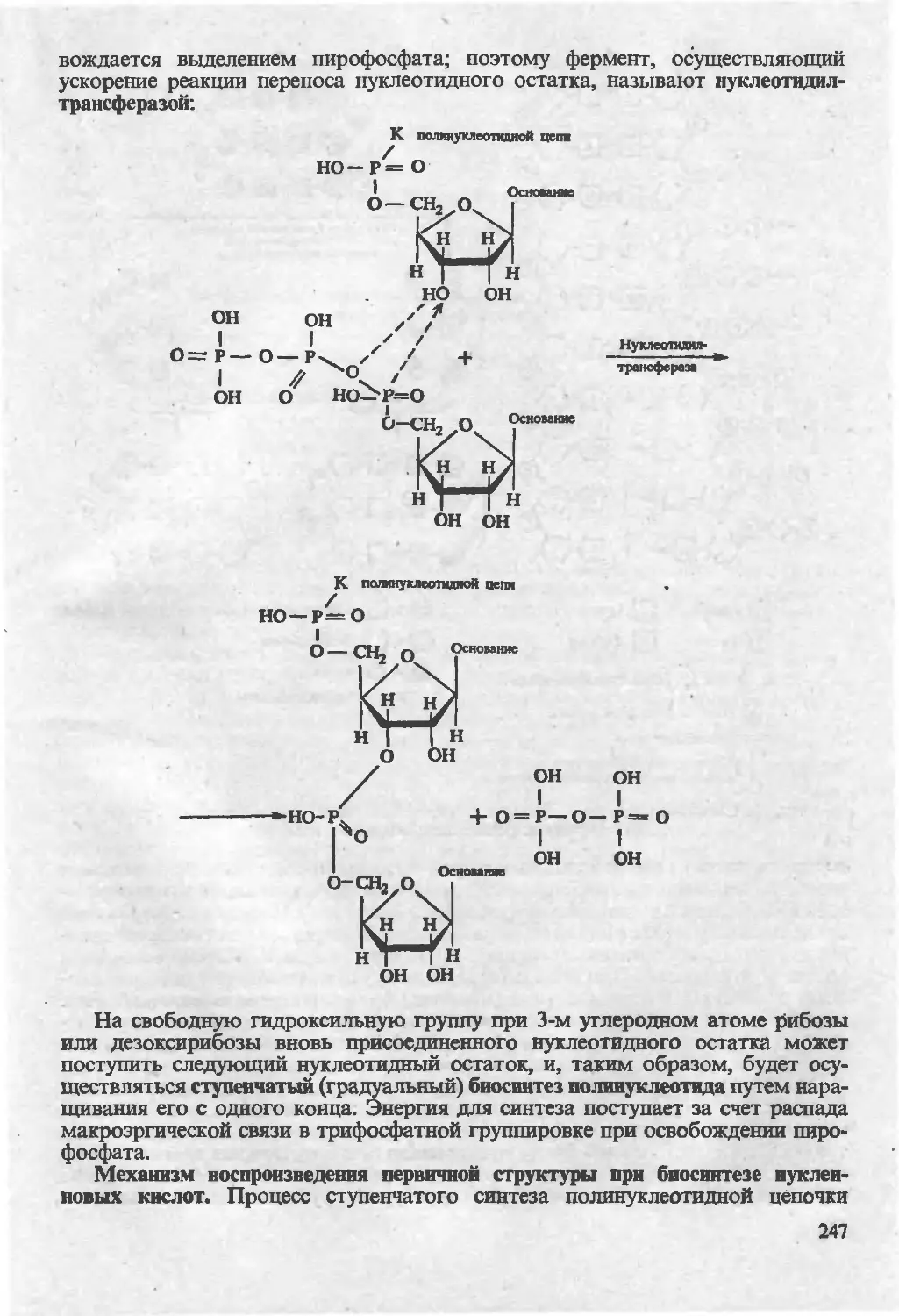
/



































































































































































































































































































































































































































































































































Author: Филиппович Ю.Б.
Tags: общая биохимия биохимия учебник для вузов
ISBN: 5-89218-046-8
Year: 1999




Text
Ю. Б. ФИЛИППОВИЧ
осно
Ю. Б. ФИЛИППОВИЧ
основы
БИОХИМИИ
Издание четвертое, переработанное
идополненное
Рекомендовано Управлением Учебного книгоиздания,
библиотек и медиатек Министерства образования
Российской Федерации
в качестве учебника д ля студентов
высших учебных заведений,
обучающихся по направлению
и специальности ’’Химия” и "Биология”
/
«АГАР»
Москва
«ФЛИНТА»
Москва
1999
№
Санкт-Петербург
ББК 28.072 (28.072)
Ф53
Рецензент: главный научный сотрудник Института физико-химической биологии им.
А. Н. Белозерского Московского государственного университета им. М. В. Ломоносова
д-р биол. наук, СоросовсКИй проф. Й. К. Наградой
Филиппович Ю. Б.
Основы биохимии: Учеб, для хим. и биол. спец. пед. ун-тов и ин-тов. — 4-е изд.,
перераб. и доп. — М.: изд-во ’’Агар”, 1999.—512 с.: ил.
ISBN 5-89218-046-8
Описаны строение и метаболизм белков, нуклеиновых кислот, углеводов, липидов,
минеральных веществ, биологически активных соединений, процессы биологического
окисления и регуляции обмена веществ. Издание (1-е—1969 г., 2-е—1985 г., 3-е—1993 г.)
отличается существенным обновлением материала, усилением проблемного характера
изложения всех глав, включением элементов молекулярной биологии и биотехнологии.
ISBN 5-89218-046-8
© Ю. Б. Филиппович, 1999
©’’Агар”, 1999
ПРЕДИСЛОВИЕ
Исключительная динамичность развития биохимии обусловила необходи-
мость переработки третьего издания учебника, вышедшего в свет несколько
лет тому назад. Это выразилось не только в приведении учебного материала
в соответствие с современным состоянием биохимических знаний, но и в из-
менении подходов к изложению и соотношению отдельных разделов в нем,
вплоть до введения элементов молекулярной биологии и биотехнологии,
наиболее ярко раскрывающих практический потенциал современной биологи-
ческой химии.
При отборе материала для четвертого издания учебника учитывалось, как
и ранее, значение определенных разделов биохимии для формирования отчет-
ливых представлений по общей биохимии, а также то, что развитие самой
биохимии в отдельных ее частях идет неравномерно: за последнее время
произошли огромные сдвиги в изучении строения и обмена некоторых групп
органических соединений. Поэтому в книге уделено много внимания строению
белков, нуклеиновых кислот и ферментов, рассмотрены особенности белковых
тел как носителей жизни, обращено внимание на принцип комплементарности
в строении нуклеиновых кислот и его значение в матричном биосинтезе
природных полимеров, изложены современные представления о биологиче-
ском окислений, регуляции обмена веществ и взаимосвязи обмена соединений
различных классов. Там, где это уместно, освещены вопросы использования
достижений биохимии в развитии новых направлений в биологических науках
(химическая систематика, молекулярные основы наследственности, изменчи-
вости и эволюции и др.), медицине (наследственные болезни, биохимическая
диагностика, стратегия химиотерапии, взаимодействие вирусов и клеток
и т. п.), сельском хозяйстве (биохимическая паспортизация генетического фон-
да, экологическая биохимия, клеточная инженерия и др.) и промышленном
производстве (инженерная энзимология, техническая биохимия, фармацевти-
ческая химия, микробиологический синтез и т. п.).
Перечисленные материалы, в том числе и те из них, которые касаются
философских, социальных и морально-этических аспектов современной биохи-
мии (проблема сущности жизни, перспективы развития генетической инжене-
рии, соотношение физической, химической и биологической форм движения
материи и др.), обеспечивают необходимый уровень подготовки будущих
учителей для преподавания в средней школе не только курса органической
химии и курса общей биологии, но и для успешного решения всего комплекса
задач по реализации основных направлений реформы общеобразовательной
и профессиональной школы, равно как и перестройке высшего педагогиче-
ского образования в нашей стране. Решительная направленность последней на
развитие самостоятельной работы студентов обеспечивается широким привле-
чением дополнительной литературы, рекомендуемый перечень которой чита-
тель найдет в конце учебника. Кроме того, одновременно с выходом учебника
3
в свет издательство «Прометей» опубликует методические разработки «Свод-
ные вопросы для подготовки студентов к безмашинному самоконтролю по
курсу биохимии», в которых отражен многолетний опыт преподавания биохи-
мии в МПГУ им. В. И. Ленина. Он складывается из широкого использования
модульного принципа изучения курса, рейтинговой системы оценки знаний
и проведения экзамена в форме студенческих докладов, рекомендуемые темы
которых и литература к ним помещены в этом же методическом руководстве.
Учитывая, что при изложении основ такой стремительно развивающейся
науки, как биохимия, неизбежны'как различные толкования тех или иных ее
проблем, так и отдельные неточности, автором будут с признательностью
приняты советы, пожелания и замечания по содержанию данной книги.
Автор выражает искреннюю признательность д-ру биол. наук, Соросовс-
кому проф. Н. К. Наградовой за тщательное рецензирование рукописи и цен-
ные советы по ее содержанию и канд. биол. наук О. П. Курановой—за по-
мощь в подготовке библиографии и рисунков.
Автор
ВВЕДЕНИЕ
Биологическая химия—наука о качественном составе, количественном со-
держании и преобразованиях в жизненных процессах соединений, образующих
живую материю.
Как самостоятельная научная дисциплина, биохимия оформилась во
второй половине XIX в., когда в ряде университетов были созданы кафедры
биохимии, написаны учебники, начали издаваться научные журналы, и курс
биохимии стал обязательной составной частью учебных планов при подготов-
ке биологов и медиков. Причинами для выделения биохимии в отдельную
науку были значительные успехи, достигнутые органической химией в изуче-
нии многочисленных природных соединений и физиологией в области ис-
следования процессов, протекающих в животных и растительных организмах
(поэтому на заре возникновения биохимию называли физиологической
химией). Кроме того, развитие биохимии теснейшим образом связано
с потребностями практики—медицины, сельского хозяйства и промыш-
ленности.
Особенно бурный процесс развития биохимии характерен для последних
десятилетий. Этому способствовало в первую очередь прогрессирующее
применение в биохимических исследованиях новых физико-химических ме-
тодов. Исключительную роль в расширении возможностей научного поиска
в биохимии сыграло внедрение в практику биохимических работ рентге-
ноструктурного анализа, электронной микроскопии, газовой, жидкостной,
гелевой и капиллярной хроматографии, метода меченых атомов, инфра-
красной и ультрафиолетовой спектрофотометрии, флуоресцентного и по-
лярографического анализа, электрофореза, метода молекулярных сит, масс-
спектрометрии, разделения веществ в гравитационном поле ультрацентри-
фугированием, методов дисперсии магнитооптического вращения, магнитного
кругового дихроизма, электронного парамагнитного резонанса, ядерного
магнитного резонанса и др.
Введение новых методических приемов каждый раз поднимало биохими-
ческую науку на более высокую ступень познания закономерностей жизнеде-
ятельности организмов, открывало новые уровни исследования живого. Свое-
образной чертой последнего периода в развитии биохимии является широкое
применение скоростных методов анализа в соединении с автоматическим
контролем. Это в значительной мере облегчает и ускоряет выполнение намеча-
емых научных программ. В настоящее время полностью автоматизированы:
количественное определение ряда соединений—аминокислот в белковых гид-
ролизатах, моно- и дисахаридов в биологических жидкостях (кровь, моча
и др.); выяснение первичной структуры пептидов, белков и нуклеиновых
кислот; проведение исследований по кинетике ферментативного катализа
и при серийных определениях энзиматической активности; элементарный ана-
лиз природных соединений; синтез пептидов, олигонуклеотидов и белков;
5
процедуры хроматографического и гельфильтрационного фракционирования
природных соединений; денситометрирование хроматограмм, электрофоре-
грамм и авторадиограмм с выходом на компьютерные системы; проведение
работ по установлению уровня включения радиоактивных предшественников
в те или иные соединения метаболического фонда. В нашей стране созданы
жидкостные и газовые хроматографы, секвенаторы, масс-спектрометры
и рентгеновские дифрактометры высокого класса, используются ЭВМ для
управления автоматизированными системами анализа и обработки получа-
емой информации.
Разделы современной биохимии. Современная биологическая химия охваты-
вает большую область человеческого знания. В связи с огромным объемом
фактического материала и разнообразием теоретических обобщений биохимия
распадается на ряд отделов, каждый из которых имеет самостоятельное
значение. В зависимости от подхода к изучению живой материи биохимию
делят на статическую, динамическую и функциональную. Статическая биохи-
мия занимается исследованием химического состава организмов. При этом
в понятие химического состава включают как качественный состав (и строе-
ние) соединений, так и количественное их содержание в тех или иных биологи-
ческих объектах. Динамическая биохимия изучает превращение химических
соединений и взаимосвязанных с ними превращений энергии в процессе жиз-
недеятельности органических форм. Функциональная биохимия выясняет связи
между строением химических соединений и процессами их видоизменения,
с одной стороны, и функцией субклеточных частиц специализированных кле-
ток, тканей или органов, включающих в свой состав упомянутые вещества,— с
другой.
Деление это в значительной мере условно. Практически в ходе биохимиче-
ских исследований все три раздела тесно переплетаются друг с другом: ведь
в живом организме состав и строение веществ неотделимы от их преобразова-
ний, равно как и от функций тех структур, органов и тканей, в которых эти
вещества находятся.
В зависимости от объекта или направления исследований современная
биохимия распадается на несколько самостоятельных разделов.
Общая биохимия рассматривает закономерности строения, содержания
и преобразования в процессе жизнедеятельности организмов химических со-
единений, общих для живой материи в целом.
Биоорганическая химия выясняет физико-химические основы функциониро-
вания важнейших систем живой клетки, используя идеи, методы и приемы
химии, включая структурный и стереохимический анализ, частичный и полный
синтез природных соединений и их аналогов, разработку препаративных
и технологических методов получения природных веществ и их химической
модификации в непосредственной связи с биологической функцией этих
соединений.
Биоиеоргаиическая химия исследует структуру и функциональную актив-
ность комплексов неорганических ионов с органическими молекулами (лиган-
дами), их участие в процессах жизнедеятельности вплоть до изучения возмож-
ности использовать координационные соединения в качестве моделей биоло-
гических систем.
Биохимия животных изучает состав животных организмов и превращения
в них веществ и энергии.
Биохимия растений исследует состав растительных организмов и превраще-
ний в них веществ и энергии.
Биохимия микроорганизмов выясняет те же вопросы, что и два предыдущих
раздела биохимии, но объектом исследования являются микроорганизмы.
б
Медицинская биохимия исследует
состав и превращение веществ и энер-
гий в организме человека в норме и па-
тологии.
Ветеринарная биохимия изучает те
же вопросы у животных.
Техническая биохимия выясняет со-
став важнейших пищевых продуктов,
изучает превращения, происходящие
при их производстве и хранении, а так-
же разрабатывает способы применения
биохимических процессов в промыш-
ленности.
Сравнительная биохимия сопостав-
ляет состав и пути видоизменения ве-
ществ у организмов различных систе-
матических групп, в том числе в эволю-
ционном аспекте (эволюционная био-
химия).
Радиационная биохимия изучает из-
менение состава и обмена веществ
в организме при действии на него ио-
низирующих излучений и разрабатыва-
ет методы биохимической защиты от
радиации.
Квантовая биохимия сопоставляет
свойства, функции и пути превращения
в организме соединений, имеющих био-
Рис. 1. Соотношение современной биохимии
и сопредельных разделов фундаментальной
биологии:
заштрихованная часть параллелепипеда—разделы, относя-
щиеся к биохимии
логическое значение,ч: их электронными
характеристиками, полученными при помощи квантово-химических расчетов.
Космическая биохимия занимается исследованием биохимических проблем,
связанных с освоением человечеством космического пространства.
Фундаментальное значение биохимии для ряда сопредельных наук насто-
лько возросло, что в настоящее время не так-то просто очертить ее границы.
Оригинальную попытку сделать это предприняла недавно редколлегия журна-
ла «Биохимия» (1989, т. 54, вып. 1), исходя из уровня сложности объекта
исследования, его биологической функции и методологического подхода, что
наглядно представлено на рис. 1. С этой точки зрения современную биохимию
следовало бы определить как науку, раскрывающую закономерности жизнеде-
ятельности на уровне молекул, субклеточных частиц, клеток, организма, био-
логических сообществ и биосферы при посредстве физических, химических
и биологических методов исследования.
Значение биохимии в биологии, медицине, сельском хозяйстве и промыш-
ленности. Из приведенного далеко не полного перечня основных разделов
современной биохимии ясно ее огромное теоретическое и практическое зна-
чение'. Биохимия приобретает все большее основополагающее значение
в биологии, так как проникновение в самую глубокую сущность жизненных
явлений, управление жизнедеятельностью организма человека, животных,
растений и микроорганизмов будет достигнуто только тогда, когда биохи-
мическая наука позволит расшифровать в достаточной мере набор, строение
и свойства химических соединений, из которых слагается все живое, и выяс-
нить закономерности их превращений в процессе существования живой ма-
терии.
7
Вместе с тем уже на данном этапе своего развития биохимия является
фундаментом для решения многих вопросов в биологии, медицине, животно-
водстве, растениеводстве, промышленности микробиологического синтеза, пи-
щевой промышленности и т. д.
В недрах биохимии, на стыке биологии, химии, физики, математики и ки-
бернетики, зародилась наука об особенностях строения и свойств молекул,
обеспечивающих существование биологической формы движения материи,—
молекулярная биология. Шаги этой молодой науки столь стремительны, что
порой превосходят оображение: заложены основы для понимания механизма
биологического катализа и, следовательно, управления процессами жизнеде-
ятельности, выявлены кардинальные закономерности специфического биосин-
теза макромолекул, все больший размах приобретают работы по генетической
инженерии. В результате исследования процессов преобразования нуклеино-
вых кислот под влиянием физических факторов и химических агентов найдены
принципиально новые подходы к пониманию явлений изменчивости и наслед-
ственности в природе.
Успехи в изучении структуры белков и нуклеиновых кислот заложили
прочные основы для развития биохимической систематики, молекулярной
эволюции и биохимической генетики, благодаря чему описательный характер
биол гических наук все более изменяется в направлении познания сущности
биологических явлений. Возникли такие новые области биологических
наук, как химическая филогения, экологическая биохимия, химическая зоо-
логия, фитохимическая экология. Изучение роста, развития и дифферен-
цировки растительных и животных форм уже невозможно без знания
молекулярных основ этих процессов. Даже такие, казалось бы, совершенно
далекие от биохимии области биологии, как популяционный анализ, ав-
торегуляция численности особей и другие, получают объяснение на мо-
лекулярном уровне.
Настала эра химической биологии.
В медицине успехи биохимии определяют стратегию создания и примене-
ния лекарственных веществ, являются источником новых методов для
диагностики заболеваний и основой для поразительных открытий, каса-
ющихся выяснения причин тех или иных патологических процессов в ор-
ганизме. В частности, оказалось, что многие болезни, передающиеся по
наследству, возникают в результате нарушения отдельных звеньев обмена
белков, углеводов, липидов, нуклеиновых кислот, гормонов и т. п. Это
послужило причиной для выделения в отдельную ветвь медицины учения
о молекулярных основах патологии и, в частности, об энзимопатиях, т. е.
нарушениях функций ферментов, приводящих к развитию заболевания.
Глубокое проникновение в биохимию вирусов, выяснение строения и условий
саморепродукции многих из них, расшифровка механизма взаимодействия
вирусного и клеточного геномов, а также взаимообусловленности метаболи-
зма вирусных частиц и субклеточных структур клетки-хозяина позволили
в ряде случаев создать эффективные методы борьбы с вирусными заболева-
ниями.
Открытие в течение последнего десятилетия нового класса биологически
активных пептидов—эндорфинов и энкефалинов, в новом свете представило
проблему психотерапии и управления поведенческими реакциями живот ых
и человека, сделало крайне актуальным вопрос о социальных последствиях
развития биохимии.
Наконец, выполнение государственной научно-технической программы по
выяснению полной первичной структуры ДНК генома человека, представлен-
ной 3 миллиардами нуклеотидных остатков, открывает неисчерпаемые и пока
8
трудно предсказуемые перспективы поистине революционных преобразований
в устранении наследственных болезней.
Применение многочисленных и разнообразных химических препаратов
в животноводстве и растениеводстве, базирующееся на данных биохимии
и физиологии, способствует подъему продуктивности указанных отраслей
сельского. хозяйства и производительности труда. Особенно важно полное
удовлетворени > потребностей сельского хозяйства в микроэлементах, витами-
нах, белковых добавках (в виде кормовых дрожжей и белково-витаминных
концентратов), синтетических аминокислотах (треонин, триптофан, лизин)
и кормовых антибиотиках, а также производство высокоэффективных и эколо-
гически безопасных средств защиты растений, в том числе инсектицидов 3-го
и 4-го поколений, созданных в результате изучения регуляторов роста насеко-
мых—гормонов (экдизона и ювенильных гормонов) и антигормонов.
Наряду с отмеченным выше практическим значением для сельского
хозяйства биохимия все более становится его теоретической основой.
Это выражается в разработке методов раннего прогнозирования про-
дуктивности сельскохозяйственных животных по биохимическим тестам
(структурное состояние ДНК, характеристика степени полиморфизма белков
и ферментов и др.), биохимической паспортизации генетического фонда
с целью отбора пар для скрещивания при выведении новых высокопро-
дуктивных сортов растений и пород животных, использовании данных
о множественных формах ферментов у родителей для получения высо-
когетерозисного потомства и для контроля завершенности селекционного
процесса. В результате проведения фундаментальных биохимических ис-
следований непрерывно обогащаются представления о регуляции роста
и развития растений и животных путем целенаправленного изменения
их генотипов, вырисовываются перспективы создания при посредстве ге-
нетической и клеточной инженерии форм, обладающих уникальными, заранее
предусмотренными качествами. Одним из наиболее ярких примеров фун-
даментальных исследований такого рода являются работы по переносу
в клетки высших растений генов, обеспечивающих фиксацию молекулярного
азота.
Всестороннее изучение биохимии микроорганизмов и открывшиеся при
этом широкие перспективы их практического использования привели
к со данию промышленности микробиологического синтеза вместо разрознен-
ных бродильных производств. Ее продукцию составляют кормовой белок (а
в будущем—пищевой белок), минокислоты все без исключения антибиоти-
ки (кормовые и медицинские), многие витамины и практически все гормоны
и ферменты. В ближайшие годы расширится ассортимент и возрастет объем
производства сложнейших химических соединений, получаемых путем
микробиологического синтеза. Залогом этого служат большой размах
и высокие темпы биохимических исследований жизнедеятельности микроор-
ганизмов, равно как и использование методов генетической и клеточной
инженерии для создания микроорганизмов—суперпродуцентов перечислен-
ных веществ. Именно так получены штаммы-суперпродуценты Bacillus
subtilis, Esche ichia coli и Brevibacterium flavum, накапливающие в 1л
культуральной среды до 10 г триптофана, 20 г треонина и 80 г лизина
соответственно.
Во многих отраслях промышленности широко используют достижения
технической биохимии. Это относится прежде всего к пищевой промыш-
ленности; хлебопечение, виноделие, сыроварение, консервирование продуктов,
производство чая, растительных и животных жиров и масел, переработка
молока, мяса и т. п. непрерывно совершенствуются в результате введения
9
новых технологических схем, основанных на все более углубляющихся пред-
ставлениях о биохимических процессах, которые осуществляются при перехо-
де сырья в готовый продукт. В кожевенной, текстильной, крахмалопаточной
и мясной промышленности нашли применение разнообразные ферментные
препараты.
Можно привести еще много примеров, иллюстрирующих огромное значе-
ние биохимической науки в области теории и практики. Применение хими-
ческих превращений, характерных для природных процессов, все более стано-
вится той силой, которая преобразует химическую промышленность. К их
числу относятся биологический катализ; матричный принцип биосинтеза, ме-
хано-химические явления, акцептирование энергии света при фотосинтезе,
хранение и передача информации в биологических системах, энергетика био-
логического окисления.
Вполне закономерно поэтому, что сформировалась перспективная научно-
техническая отрасль—биотехнология, разрабатывающая научные основы
производственных процессов, в которых используются принципы химических
превращений, присущих биологическим объектам. Она включает техническую
биохимию, микробиологию, генетическую инженерию, использование культур
животных и растительных клеток, а также иммобилизованных ферментов
(инженерная энзимология). Намечено выйти при посредстве биотехнологиче-
ских схем на промышленное производство инсулина, гормона роста, интерфе-
рона, простагландинов, сахарных сиропов из целлюлозы и крахмала, рас-
тительных белков в качестве заменителей животных белков и др., а также
резко увеличить производство ферментов, незаменимых аминокислот, пита-
тельных добавок к кормовым смесям и т. п. В последующих главах учебника,
при рассмотрении отдельных разделов биохимии, будут приведены соответст-
вующие конкретные материалы.
История развития отечественной биохимии. Российские ученые внесли
большой вклад в развитие биохимии. Основоположником отечественной
биохимии по праву считают А. Я. Данилевского (1839—1923), который
возглавил в Казанском университете первую в "России кафедру биохимии
и создал первую русскую школу биохимиков. А. Я. Данилевский сделал ряд
крупных открытий. Им разработан оригинальный метод очистки ферментов
путем их адсорбции с последующей элюцией. Он впервые высказал идею об
обратимости действия биологических катализаторов—ферментов и, исходя
из этого, осуществил синтез белковоподобных веществ—пластеинов. Занима-
ясь изучением белков, А. Я. Данилевский предположил, что составляющие их
структурные единицы соединены друг с другом —СО—NH-связями, которые
были названы впоследствии пептидными. По современным представлениям,
белковая молекула построена из остатков аминокислот, соединенных пептид-
ными связями.
Большие заслуги в развитии отечественной биохимии принадлежат
М. В. Ненцкому (1847—1901). В 1891 г. он создал первую в России биохими-
ческую лабораторию при институте экспериментальной медицины в Петер-
бурге. М. В. Ненцким совместно с рядом сотрудников (Л. Мархлевский,
С. Салазкин, В. 1улевич и др.) был выполнен ряд биохимических исследова-
ний. К их числу относятся работы по изучению химического состава хлоро-
филла и гемина, выяснению механизма биосинтеза мочевины и ряда вопросов
обмена белков.
Из числа выдающихся открытий в области биохимии, сделанных русскими
учеными и их школами, следует назвать открытие витаминов (Н. И. Лунин,
.1880) и проферментов (И. П. Павлов и Н. П. Шеповальников, 1899), разработ-
ку хроматографического метода разделения пигментов и других близких по
10
строению природных веществ (М. С. Цвет, 1903), исследование процесса фо-
тосинтеза (К. А. Тимирязев), изучение закономерностей обмена азотистых
соединений в растениях (Д. Н. Прянишников) и др.
В 1921 г. А. Н. Бах организовал в Москве Научно-исследовательский био-
химический институт Народного комиссариата здравоохранения, а в 1935 г.
он же возглавил переведенный из Ленинграда в Москву Институт биохимии
Академии наук СССР, названный впоследствии его именем. А. Н. Бах из-
вестен как выдающийся биохимик, заложивший основы теории дыхания
и высказавший гипотезу об участии перекисей в окислении органических
соединений.
Огромный вклад в развитие биохимии внесли такие крупнейшие ученые,
как Н. Н. Иванов (автор 8-томного труда «Биохимия культурных растений»);
А. Р. Кизель, известный своими работами в области обмена белков, а также
как автор практического руководства по биохимии растений; Н. Я. Демьянов,
разработавший методы анализа растительного сырья; Я. О. Парнас, плодо-
творно работавший в области медицинской биохимии и предложивший ряд
оригинальных биохимических методов анализа; С. Я. Капланский, изучавший
патологию обмена аминокислот; Н. М. Сисакян, посвятивший многие свои
труды философским вопросам биохимии, выяснению механизма биосинтеза
белков в растениях и изучению биохимии субклеточных структур; Б. Н. Сте-
паненко, внесший большой вклад в изучение химии и биохимии углеводов;
В. А. Букин, разработавший ряд крупных проблем в учении о витаминах;
А. Н. Белозерский, автор классических трудов по биохимии нуклеиновых кис-
лот; И. В. Березин, основавший школу инженерной энзимологии; Ю. А. Ов-
чинников, заложивший основы работ по ионной проницаемости биологиче-
ских мембран; В. С. Ильин, высказавший принципиально новые идеи в регу-
ляции обмена веществ, и многие другие биохимики, внесшие большой вклад
в развитие биохимической науки.
Важнейшие биохимические центры. В настоящее время в нашей стране
насчитывается несколько крупных биохимических центров. Прежде всего сле-
дует отметить Институт биохимии имени А. Н. Баха РАН, который в течение
нескольких десятилетий возглавлял академик А. И. Опарин (1894—1980)—
автор теории происхождения жизни на Земле, ряда работ по технической
биохимии и многих экспериментальных исследований по биохимии систем,
моделирующих простейшие формы жизни. В институте биохимии им.
А. Н. Баха широко проводятся работы по выяснению строения и функций
фотосинтетического аппарата у растений и исследованию азотистого обмена
у растений (ими в течение нескольких десятилетий руководили академик
А. А. К рас невский и чл.-корр. РАН В. Л. Кретович), изучению закономер-
ностей самосборки биологических структур (Б. Ф. Поглазов), инженерной эн-
зимологии (Б. И. Курганов), выделению и изучению свойств белковых ин-
гибиторов ферментов (В. В. Мосолов), биоэнергетике, структуре мембранного
аппарата клеток и ряду других проблем.
Другим крупным биохимическим центром является Институт медицинской
и биологической химии Академии медицинских наук. Здесь академик
В. Н. Орехович впервые открыл проколлаген, что положило начало большой
серии работ по предшественникам белков в мировой науке. В 1937 г. здесь же
академиком А. Е. Браунштейном впервые была открыта (совместно
с М. Г. Крицман) реакция переаминирования аминокислот с кетокислотами,
ознаменовавшая новую главу в биохимии обмена белков и изучении пиридок-
салевого катализа.
Фундаментальные работы по биохимии проводятся на биологическом
факультете Московского государственного университета им. М. В. Ломоносова,
11
где кафедру биохимии животных возглавлял академик С. Е. Северин—ученый
разносторонних познаний и широкой эрудиции, а кафедрой молекулярной
биологии заведует академик А. С. Спирин, осуществивший приоритетные ис-
следования в области биосинтеза белков. Здесь же, в Институте физико-
химической биологии имени А. Н. Белозерского, возглавляемом академиком
В. П. Скулачевым, интенсивно изучается ряд перспективных проблем биохимии
и молекулярной биологии.
В 1959 г. был основан Институт молекулярной биологии АН СССР, нося-
щий имя академика В. А. Энгельгардта, открывшего (совместно с М. Н. Лю-
бимовой) ферментативные свойства белка мышц, изучившего новые пути
распада углеводов и впервые высказавшего идею об окислительном фос-
форилировании. В. А. Энгельгардт был пионером в изучении биологических
явлений на молекулярном уровне, и его работы по механохимии мышц,
по существу, открыли эру молекулярной биологии. Основные проблемы
этой новой науки находятся сейчас в центре внимания коллектива упо-
мянутого института, добившегося существенных успехов в расшифровке
структуры и мех низма действия ферментов (А. Е. Браунштейн), строения
и функций транспортных рибонуклеиновых кислот (А. А. Баев), регуляции
активности генома, нуклеосомной организации хроматина и ряде других
нап явлений.
Крупный биохимический центр представлен Институтом молекулярной
генетики РАН, где ведутся работы по изучению физики биополимеров, биосин-
теза белков и нуклеиновых кислот и другие исследования. Здесь интенсивно
исследуют те особенности структуры и свойств ДНК, которые могут играть
роль в выполнении ею биологических функций, изучают процесс биосинтеза
РНК на ДНК в качестве матрицы и регуляцию этого биосинтеза, разрабатыва-
ют вопросы биохимической генетики.
Многие фундаментальные биохимические проблемы решают в Институте
биоорганической химии им. М. М. Шемякина и Ю. А. Овчинникова РАН, где
осуществляются исследования первичной структуры пептидов, белков и нукле
иновых кислот, троения и проницаемости мембран, ферментативного катали-
за, белково-нуклеинового взаимодействия и ряд других.
Центр работ по биохимии нуклеиновых кислот растений сложился в 70-е
годы в отделе биохимии и цитохимии ВИР им. Н. И. Вавилова, где под
руководством академика В. Г. Конарева проводятся работы по изучению
строения и функций хроматина ядра, структурного состояния ДНК, видовой
специфичности белков и др.
Механизм биосинтеза белков во всех его аспектах исследуют в Институте
белка РАН (биологический центр, г. Пущино-на-Оке Московской обл.), биохи-
мические механизмы мутагенеза—в Сибирском отделении РАН (г. Новоси-
бирск), применение ферментов для лечения некоторых видов лейкозов—на
кафедре биохимии Университета дружбы народов, использование ферментов
для диагностики—в Институте энзимологии Академии медицинских наук,
механизм свертывания белков крови и структуру и функции кардиоактивных
пептидов—в Кардиоцентре (г. Москва); проблемы биохимии насекомых—на
кафедре органической и биологической химии МПГУ им. В. И. Ленина и т. д.
Таким образом, десятки больших и малых научных коллективов решают
многие насущные вопросы биохимической науки. Их усилия объединяет Рос-
сийское биохимическое общество.
Новые направления развития отечественной биохимии. Значительную роль
в развитии биохимической науки в нашей стране сыграли научные программы
и организационные мероприятия, а также решение ряда материально-техниче-
ских вопросов, позволившие не только обеспечить существенное продвижение
12
вперед в фундаментальной и прикладной биохимии, но и создать современ-
ные, не уступающие зарубежным, а в ряде случаев и превосходящие их
технические средства для проведения тончайших биохимических исследований.
Этому в немалой степени содействовало создание ряда новых научных ор-
ганизаций, среди которых следует отметить Институт биологии гена РАН
и проблемную научно-исследовательскую лабораторию диагностики вирусов
растений при МГУ им. Ломоносова. Все они уже внесли существенный вклад
в развитие биохимии и тесно соприкасающихся с нею наук. Не меньшее
значение имеет организация в этот же период Научно-производственного
центра медицинской биотехнологии Минздрава и инженерного центра «Био-
инженерия» при Межотраслевом научно-техническом комплексе «Биотехноло-
гия», деятельность которых направлена на внедрение достижений биохимии
и неразрывно связанной с нею молекулярной биологии в медицину и сельское
'хозяйство.
Участие российских ученых в развитии мировой биохимии. Это предопре-
делило выход отечественной биохимии на передовые рубежи в мировой
биохимической науке, где Всесоюзное биохимическое общество занимало
прочные позиции в Международном биохимическом союзе, основанном 6 ян-
варя 1955 г. Признанием большого вклада русских ученых в развитие био-
химии явилось избрание президентом Международного биохимического со-
юза на период с 1976 по 1979 г. академика А. А. Баева, являвшегося до
своей кончины (1995 г.) вице-президентом этого высшего форума биохимиков
всего мира, а президентом Федерации Европейских Биохимических Обществ
(ФЕБО) на период 1984—1986 гг.—академика Ю. А. Овчинникова. Состоя-
лось 6 Всесоюзных биохимических съездов (последний в 1991 г. в г. Санкт-
Петербурге), 9 симпозиумов по структуре и функции клеточного ядра, 9—по
биохимии углеводов, 6—по биохимии циклических нуклеотидов, 10 объ-
единенных симпозиумов биохимических обществ СССР и Франции, 18—
СССР и 1ермании и 6—СССР и Италии. Со дня основания Международного
био имического союза проведено 16 международных биохимических конгрес-
сов (последний—в 1994 г. в г. Стокгольме, Швеция), а с момента начала
работы ФЕБО—23 конференции ФЕБО (последняя—в 1995 г. в г. Базеле,
Швейцария). На всех перечисленных как внутрисоюзных, так и междуна-
родных форумах были широко представлены исследования наших ученых
по всем разделам биохимии.
Важнейшая периодика по биохимии. С 1936 г. в нашей стране выходит
журнал «Биохимия», издается ряд журналов биохимического профиля—«Мо-
лекулярная биология», «Прикладная биохимия и микробиология» и др. На-
чиная с 1950 г. публикуется ежегодник «Успехи биологической химии» (к
1996 г. вышло в свет 36 томов), с 1966 г.—серия монографий под общим
названием «Биологическая химия», где представлены обзоры по важнейшим
направлениям современной биохимии (в 1991 г. опубликован 39-й том),
а с 1972 г.—серия монографий под названием «Молекулярная биология»
(в 1991 г. издан 29-й том).
Методы биохимии. Как и всякая наука, биохимия располагает специ-
фическими методами научного исследования. Общая их черта состоит в том,
что при изучении обмена веществ исследуемое химическое соединение или
набор определенных соединений вводят в системы, обладающие свойствами
живого, и изучают их превращения. В качестве упомянутых систем ис-
пользуют либо целые организмы, либо п реживающие органы, тканевые
срезы, тканевые и клеточные культуры, тканевые кашицы, экстракты и го-
могенаты, а также выделенные из клеточного содержимого специфические
субклеточные структуры. Для выяснения судьбы добавленных к той или
13
иной системе соединений биохимия использует разнообразные химические
методы анализа и различные физико-химические методы, перечисленные
выше. Вместе с тем для изучения структуры и функций биополимеров,
особенно в сравнительно биохимическом аспекте, все более внедряются
иммунохимические и радиоиммунологические методы, метод адресованных
реагентов, метод ДНК-ДНК-, ДНК-белок- и ДНК-РНК-гибридизации, ме-
тод кинетики реассоциации нуклеиновых кислот, метод нейтронного рассея-
ния, специфические методы исследования кинетики действия ферментов
и многие другие. Некоторые из названных методических подходов будут
рассмотрены ниже в соответствующих главах учебника.
ГЛАВА I
ХИМИЧЕСКИЙ СОСТАВ ОРГАНИЗМОВ
Общий химический состав. По современным данным, биомасса единовре-
менно живущих на Земле организмов (а их насчитывается около 2 млн.
видов) составляет 1,8х 1012 — 2,4 х 1012 т в пересчете на сухое вещество,
причем ежегодно ими продуцируется около 10“ т сухого в< щества. В ор-
ганизмах, соста, ляющих биомассу Земли, обнаружено свыше 60 химических
элементов. Среди них условно выделяют группу элементов, встречающихся
в составе любого организма, независимо от видовой принадлежности
и уровня организации последне о. К их числу относят С, N, Н, О, S, Р, Na, К,
Са, Mg, Zn, Fe, Мп, Си, Со, Мо, В, V, I и CL Первым шести элементам
приписывают исклю ительную роль в биоси темах, так как из них построены
важнейшие соединения, составляющие основу живой материи,— белки,
нуклеиновые кислоты, углеводы, липиды и др.; последующие десять
называют «металлами жизни»—они крайне важны для поддержания
структуры и функциональной активности биополимеров; бор и ванадий
весьма существенны для растительных и животных объектов соответственно,
а хлор образует наиболее распространенный анион. Остальные элементы,
обнаруженные в биомассе, встречаются в живой природе не столь сис-
тематически, а биологическое значение их во многих случаях еще не
выяснено.
По количественному содержанию в живом веществе элементы делят на
три категории: макроэлементы, концентрация которых превышает 0,001% (О,
С, Н, Са, N, Р, S, Mg, Na, Cl, Fe), микроэлементы, доля которых составля т
от 0,001 до 0,000001% (Мп, Zn, Си, В, Мо, Со и многие другие) и ультрамик-
роэлементы, содержание которых не превышает 0,000001% (Hg, Au, U, Ra
и др.).
Из макроэлементов в наибольшем количестве в биомассе содержатся О, С,
Н, N и Са. Из них только О и Са широко пред тавлены в земной коре. Многие
элементы, содержащ еся в литосфере в значительном количестве (Si, Al, Fe
и др.), в органическом мире встречаются сравнительно в невысоких концент-
рациях. Аналогичная картина свойственна, по данным академика А. П. Вино-
градова, количественным соотношениям элементов в гидросфере и живых
существах, ее насел ющих, хотя качественный состав первой и второй почти
совпадает. Таким образом, прямой зависимости между расп остранением
химических элементов в неорганической и органической природе нет, однако
это не означает, что между первой и второй отсутствует какая-либо связь.
Наоборот, установлено, что между организмом и средой сущее вует тонкая
взаимозависимость. Так, например, те элементы, которые легко образуют
растворимые и газообразные соединения, составляют основную массу биосфе-
ры (С, N, Р, S), хотя в земной коре их содержание относительно невелико.
Элементы, которые не дают водорастворимых соединений, широко распро-
странены в неорганической природе, а в составе организмов встречаю ся
15
в ничтожных количеств х (Si, Fe, Al). Таким образом, доступность элементов
для биосферы играет решающую роль в построении живого вещества.
Отмечена определенная зависимость между биологической ролью элемен-
тов и их местом в периодической системе Менделеева. Органический мир
построен главным образом из легких элементов. В подавляющем большинст-
ве случаев при переходе от легких к тяжелым элементам в пределах одной
и той же подгруппы возрастает токсичность элементов и параллельно этому
падает содержание их в биомассе (Zn, Cd, Hg). Элементы некоторых подгрупп
взаимозаменяют друг друга в биологических объектах (Са, Sr, Ва). Функцио-
нальное значение элементов ряда подгрупп своеобразно; например, элементы
восьмой подгруппы (Fe, Со, Ni) являются преимущественно компонентами
биоактивных соединений. В последнее время активно обсуждается вопрос
о биологическом значении Se, F, Si, Sn, As, Cr, Pb, W и других элементов.
Полагают, что Н, О, С, N и Р, составляющие вместе более 99% живого
вещества, играют выдающуюся роль в явлениях жизни благодаря наличию
у них комплекса особых качеств. Первое из них состоит в способности
образовывать кратные связи. Вследствие этого С, например, превосходит Si
в отношении числа и разнообразия возможных соединений, обладающих
уникальными свойствами. Второе качество заключается в том, что атомы
упомянутых элементов, отличаясь малыми размерами, образуют относительно
плотные молекулы с минимальными межа омными расстояниями. Такие моле-
кулы более устойчивы к действию тех или иных химических агентов. И нако-
нец, третье качество присуще в основном Р и S и лишь в небольшой мере N.
Оно сводится к возникновению на базе указанных элементов некоторых
специфических соединений, при расщеплении которых выделяется повышенное
количество энергии, используемой для процессов жизнедеятельности.
Многочисленные макро- и микроэлементы, образующие живую материю,
присутствуют в последней в виде разнообразных химических соединений.
Примерно 75% биомассы составляет вода, хотя ее содержание в организмах
различных видов сильно колеблется (от 40—60% у древесных растении до
99% у медузы). Вода играет огромную роль в создании условий для жизнеде-
ятельности. Она образует ту среду, в которой протекают физико-химические
процессы, обеспечивающие постоянное возобновление живого вещества, а так-
же участвует в реакциях гидролиза.
Вторым по количественному содержанию в биологических объектах, но,
несомненно, первым и главным то значению классом соединений являются белки.
В среднем можно принять, что в сухом веществе организмов содержится 40—50%
белка. Растительному миру свойственно отклонение от этой средней величины
в сторону понижения, а животному—повышения. Микроорганизмы обычно
богаче белком (некоторые вирусы являются почти чистыми белками). Таким
образом, в среднем можно принять, что 10% биомассы на Земле представлено
белком, т. е. его количество измеряется величинами порядка (0,9—1,2) х 1012 т.
В биохимии давно уже утвердилось положение о выдающейся роли белка
в осуществлении жизненных функций. Обладая рядом специфических качеств,
которые подробно будут рассмотрены ниже, белковые тела являются прин-
ципиальной составной частью живых систем. Как выяснено в последние годы,
очень важную роль в существлении жизненных процессов играют нукле-
иновые кислоты (передают информацию о специфическом воспроизведении
структуры важнейших биополимеров), высшие углеводы (обеспечивают меж-
клеточные контакты и др.), некоторые виды липидов (участвуют в образова-
нии мембранного аппарата клеток).
Остальные 50% сухого вещества организмов представлены соединениями
других классов. Это—нуклеиновые кислоты (их доля в сухом веществе до-
16
вольно стабильна и равна нескольким процентам), углеводы и липиды (их
содержание в организмах сильно варьирует, причем в растительном мире
преобладают углеводы, а в животном—липидй) и минеральные вещества
(составляют в среднем около 10% от сухого вещества биомассы).
Кроме белков, нуклеиновых кислот, углеводов, липидов и минеральных
веществ в составе организмов найдены в незначительных количествах углево-
дороды, спирты, альдегиды, кетоны, карбоновые кислоты, кетокислоты, ами-
нокислоты, эфиры, амины и разнообразные другие соединения. У некоторых
видов животных, растений и микроорганизмов такие вещества накапливаются
в значительных количествах и могут служить систематическим признаком
(например, некоторые аминокислоты). Многие из упомянутых соединений
обладают мощным физиологическим действием и выполняют роль ускори-
телей или замедлителей жизненных процессов. Их иногда объединяют под
названием биологически активных соединений, хотя химически они очень раз-
нообразны. Это—витамины, гормоны, ростовые вещества, биостимуляторы,
коэнзимы, антибиотики, фитонциды и т. п. Сюда же относятся вещества,
возникающие в качестве промежуточных продуктов при тех или иных хими-
ческих реакциях в организме. Эти соединения называются метаболитами.
Среди соединений, входящих в состав организмов, принято выделять пласти-
ческие и энергетические вещества. Пластические вещества служат строительным
материалом при формировании внутриклеточных структур, клеток и тканей. Это
главным образом белки, нуклеиновые кислоты, некоторые виды липидов
и высокомолекулярных, углеводов. Энергетические вещества выполняют роль
пос тавщиков энергии для процессов жизнедеятельности, распадаясь при этом до
СО2 и воды. К ним относятся низкомолекулярные и некоторые высокомолекуляр-
ные (гликоген, крахмал) углеводы и отдельные группы липидов (в основном жиры).
Приведенная классифика-
ция носит весьма условный
характер. Так, например, мно-
гие биоактивные соединения
несут в организме пластичес-
кую функцию (некоторые фер-
менты); вместе с тем в опреде-
ленных условиях пластические
соединения могут использо-
ваться как субстрат для окис-
ления, т. е. играют энергети-
ескую роль. Часто трудно
провести границу между мета-
болитами и биоактивными со-
единениями, так как послед-
ние возникают в процессе хи-
мических превращений пер-
вых. Ни к одной из этих кате-
горий нельзя отнести соедине-
ния, вырабатываемые для осу-
ществления специфи еских
функций (яды, пигменты, аро-
матические вещества, алкало-
иды и т. п.). Суммарные дан-
ные о химических соединени-
ях в составе биомассы Земли
представлены на рис. 2.
17
При оценке химического состава организмов следует иметь в виду, что,
видим , не все элементы, присутс вующие в биоло ических объектах, необ-
ходимы для осуществления процессов жизнедеятельности. Изучение по реб-
ности животных, ра тений и микроорганизмов в пределенных элементах
показало, что всем без исключения организмам абсолютно необходимы С, Н,
N, О, Р и S. Все живы существа нуждаются в Mg, Na, К, Са, Fe, Zn, Мп, Си,
Со и Мо. Велика роль таких элементов, как Cd, Se, Li, В, Cl, Вг, I и V. В то же
время значение Al, As, Si, Сг, F, Rb и W для жизнеде тельности органических
форм выяснено еще недостаточно. С новых позиций рассматривают биологи-
ческую роль лантанид в и ряда других элементов, о суждается проблема
антагонизма и синергизма в действии микроэлементов.
Химический состав клетки. Перейдем теперь от данных, характеризующих
химический состав живого вещества в целом, к рассмотрению содержания
важнейших химических соединений в мельчайшей структурной единице живых
opi анизмов—клетке. Примером может служить ростейшая жи ая система—
бак ериальная клетка (табл. 1).
Таблица 1
Примерный ’ mi ки состав кл< i кишечно! палочки
Компонент Содержание в клетке, % Средняя молеку- лярная масса, дальтои1 Среднее число молекул в клетке Число видов молекул
Вода 70 18 4 1O10 1
Н органический ионы 1 40 2,5 108 20
Углеводы и их предшественники 3 150 2-108 200
Аминокислоты и их предшественники 0,4 120 3-107 100
Нуклеотиды и их предшеез шники 0,4 300 1,2 107 200
Липиды и их п дшественники 2 750 2,5 -107 50
Другие низкомолекулярны, зешества 0,2 150 1,5-107 250
Белки 15 4-10* 10® 3000
ДНК 1 2,5 -109 1 1
РНК б — —
В том числе:
16SpPHK 5 -105 3 10* 1
23SpPHK 1-10* 3-10* 1
тРНК 2,5-10* 4 10s 60
мРНК 1-10® 10э 1000
1 Согласие Леждуе системе единиц СИ молекулярная масса (М) измеряется в атомных единицах
массы (а.е.м.); 1 а.е.м.«1,66057 х I0-2’ кг. В биохимии молекулярную массу макромолекул принято выражать
в дальтонах; 1 дальтон=1 а.е.м. В дальнейшем в некоторых понятных случаях обозначение «М» не указывается.
Из данных табл. 1 видно, что при ограниченном числе молекул ДНК
и рибосомальных РНК клетка содержит несколько тысяч различных белков, около
тысячи информационных РНК и сотни разнообразных низкомолекулярных
соединений, относящихся к тем или иным классам органических веществ (крайняя
правая графа таблицы). Число молекул высокомолекулярных соединений
в бактериальной клетке сравнительно невелико и измеряется в основном
десятками и сотнями тысяч, а низкомолекулярных—десятками миллионов, тогда
как самая большая молекула—ДНК, с молекулярной массой в несколько
миллиардов, присутствует в бактериальной клетке в единственном числе.
Указанные соотношения в общем характерны для клеток любых организмов, хотя
18
Рец птор-
наязона
Вакуоль
> Ядро
Хромосомы
Митохондрия
Лизосома
Плазматическая
мембрана
Ядерная
оболочка
Ядрышко
свободныеридосом.
Цитозоль
Вазальные
/77 льца
Шероховатый
эндоплазмати-
ческий ретику-
лум
Рис. 3. Строение клетки
Гладким
эндоплазмати-
ческий ретину
лум
Комплекс Гольджи
в клетках более высокоорганизованных форм число макромолекул измеряется
сотнями миллионов и даже миллиардами, а общее число молекул достигает
1013—1015. Считают, что 1 мкм3 протоплазмы содержит около 40 млрд,
молекул.
При помощи обычной и сканирующей электронной микроскопии получены
детальные данные о внутреннем строении клеток: обнаружена тонкая структу-
ра, представленная субклеточными образованиями, каждому из которых при-
суща определенная функция или ряд функций (рис. 3 и рис. на форзаце
учебника).
Еще более элементарно организованная живая система, являющаяся, види-
мо, нижним пределом жизни (если не считать таковым вирусы и вироиды),
представлена микоплазмами насчитывающими несколько десятков видов
и более 100 представителей. Эти мельчайшие тельца, обладающие всеми
свойствами живого, способные расти и размножаться на искусственных пита-
тельных средах, в десятки и даже сотни раз меньше упомянутой выше бакте-
риальной клетки. Имея размеры (0,15—0,30) х (1,0—1,25) мкм, они крайне
полиморфны, так как ограничены от внешней среды тончайшей (7,5 нм)
двухслойной гибкой мембраной. В них содержится 4% ДНК ярко выражен-
ного ДТ-типа в виде единственной биспиральной кольцевой структуры с мо-
ле! улярной массой от нескольких сотен миллионов до миллиарда даль он
(600000—1.700.000 нуклеотидных пар); 8% РНК (в том числе все три вида
рибосомальных РНК слабо выраженного АУ-типа и полный набор трансп )рт-
ных РНК); до пятисот индивидуальных белков (М=9000 - 200000), среди
которых тестирован до 40 ферментов; липиды, углеводы, липополисахариды
и другие вещества. По сравнению с бактериальной клеткой их структура
19
Рис. 4. Жизненный цикл и ультраструк-
тура микоплазмы:
1—мембрана; 2—рибосомоподобные образования; 3—
фибриллярный ДНК-содержащий материал; 4—недиф-
ференцированная терминальная зона; 5—пузырек
предельно проста (рис. 4), а молеку-
лярный состав предопределен набо-
ром только тех соединений, которые
абсолютно необходимы для обеспече-
ния фундаментальных, элементарных
актов жизнедеятельности.
Многочисленные и разнообразные
биополимеры, входящие в состав жи-
вого вещества (см. табл. 1), в значи-
тельной мере реально существуют
в виде биокомплексов, т. е. соедине-
ний нуклеиновых кислот и белков, по-
лисахаридов и белков, липидов и бел-
ков, полисахаридов и липидов, раз-
личных белков друг с другом и т. п.
Благодаря этому возникают новые
свойства и качества, не присущие био-
полимерам в свободном состоянии.
Поэтому изучению структуры и функ-
циональной активности биокомплек-
сов в современной биохимии уделяют
большое внимание.
Бысшей ступенью надмолекулярной организации биополимеров в клетке
являются субклеточные частицы (см. рис. 3 и 4). Сочетание белков с липидами
дает начало мембранам эндоплазматической сети, митохондрий, лизосом
и т. п. Соединение белков с полисахаридами характерно для клеточных стенок.
Рибонуклеиновые кислоты, взаимодействуя с белками, образуют рибонуклео-
протеиновые частицы, в том числе рибосомы. Комплексирование ДНК с бел-
ками и небольшим количеством РНК приводит к образованию хроматина,
а на его основе—хромосомного и, в конечном счете, ядерного аппарата
клетки.
В настоящее время биохимики уделяют особое внимание исследованию
функциональной деятельности субклеточных структур: ядра, митохондрий,
пластид, рибосом, лизосом, гиалоплазмы (основное вещество) и др. Раз-
работаны специальные методы препаративного разделения субклеточных
единиц при помощи ультрацентрифугирования, т. е. центрифугирования
при очень быстром (несколько десятков и даже сотен тысяч оборотов
в минуту) вращении ротора центрифуги. Развивающиеся при этом цент-
робежные силы характеризуются фактором разделения (см. с. 36), т. е.
отношением ускорения центробежной силы к ускорению силы тяжести,
обозначаемой буквой g. Значения факторов разделения, при которых можно
добиться осаждения из гомогената тех или иных субклеточных частиц,
приведены на рис. 5.
Разделения субклеточных частиц можно добиться также путем пропуска-
ния гомогенатов через колонку с гелем сефарозы. Частицы разного размера
фракционируются здесь по принципу молекулярного сита (см. с. 30): сначала
из колонки выходят ядра, затем митохондрии и лизосомы, вслед за ними—
обломки эндоплазматической сети клетки (микросомы) и, наконец, свободные
рибосомы.
Отдельные фракции субклеточных частиц используют для приготовления
так называемых бесклеточных систем, позволяющих выявить функциональ-
ную активность тех или иных структурных элементов клеточного содержимо-
го. Работы с бесклеточными системами позволили впервые проникнуть в сущ-
20
Центрифугирование при различны» значения» фактора
разделения
15мин,
2000g
25мин,
15000g
Зч,
100000g
Мито-
хондрии
и лизо-
-сомы
Гиалоплазма
Прочие суВкле-
почные частицы
Гомогенат
Улыпрацентрифугиробание 8 градиенте
плотности сахарозы
ЗОмин,
юоооод,.
линеиныи
градиент
Зч,
100000g.
ступенча-
тый гра-
, диент
Оч,
200000g..
линеиныи
градиент
Рис. 5. Упрощенная схема диф-
ференциального центрифугиро-
вания гомогената клеток печени
крысы (по Ж.-К. Ролан, А. Село-
ши и Д. Селоши, 1978)
Гомогенизацию ткани и последующее цент-
рифугирование ведут при О С. Цифры с ле-
вой стороны пробирок указывают моляр-
ные концентрации растворов сахарозы,
обеспечивающих в данной зоне пробирки
плотность раствора, при которой дальней-
шее оседание определенных субклеточных
частиц не происходит
Мито--^
хондрии
Лизосо-
мы
Плазматичес-
кие мемо раны
ES Аппарат Гольджи
SES Микросомы
11ч,
20000g
Ядрышки
Ри Весомы Мембраны
энооплазмати-
ческой сети
ность окислительно-восстановительных процессов в клетке, раскрыть законо-
мерности биосинтеза в ней нуклеиновых кислот и белков и сделать ряд других
важных открытий.
Оказалось, что в ядрах, где сосредоточена почти вся клеточная ДНК,
идет как ее биосинтез, так и новообразование всех видов РНК. В митохонд-
риях интенсивно протекают процессы биологического окисления, сопряжен-
ного с образованием важнейшего макроэргического соединения—аденозинт-
рифосфорной кислоты (АТФ), вследствие чего их считают энергетическими
центрами клетки. Функция лизосом сводится к осуществлению процессов
деструкции биополимеров при участии разнообразных гидролитических
ферментов, которыми они очень богаты. Рибосомы, представляющие по
современным данным механохимические машины молекулярных размеров,
обеспечивают биосинтез всех клеточных белков. Мембраны эндоплазматиче-
ского ретикулума делят клетку на ряд .отсеков (компартменты), обеспечивая
компартментализацию (обособленность) ряда химических процессов в ней,
избирательный перенос веществ из одной части клетки в другую, равно как
и протекание ряда химических реакций при участии ферментов, встроенных
в мембраны эндоплазматической сети. Центриоли имеют отношение к та-
кому важнейшему процессу, как перемещение хромосом в клетке при ее
делении.
Закономерное сочетание деятельности субклеточных частиц лежит в основе
жизнедеятельности клетки, регуляции обмена веществ в ней, быстрой
перестройки клетки на новые стационарные режимы функционирования,
21
обеспечивает экономное расходование вещества и существенное увеличение
скорости многоступенчатых биохимических превращений. Именно благодаря
этому в природе осуществляются непрерывное обновление и саморепродукция
живого вещества, непрерывный и пока еще во многом таинственный процесс
жизни.
Рассмотрению матер альных основ его на уровне молекул посвящены
последующие главь.
ГЛАВА П
БЕЛКИ
Этот раздел является наиболее важным в курсе биологической химии, так
как белковые тела играют выдающуюся роль и в построении живой материи,
и в осуществлении процессов жизнедеятельности.
Почему именно белки являются важнейшим субстратом жизни? Потому, что
они обладают рядом особенностей, которые несвойственны никаким другим
органическим соединениям.
1. Гигантские молекулы белков отличаются неисчерпаемым разнообразием
структуры при строгой ее специфичности у данного, конкретного белка.
2. Белкам присуща способность к внутримолекулярным взаимодействиям, что
обеспечивает динамичность структуры их молекул, изменчивость и пластичность
их формы, обратимость переходов из глобулярного состояния в фибриллярное.
3. Обладая многоликими по химическим свойствам радикалами аминокис-
лотных остатков в составе полипептидных цепей, белковые молекулы в целом
и их отдельные части способны вступать в разнообразные химические и физичес-
кие взаимодействия как друг с другом, так и с нуклеиновыми кислотами;
полисахаридами, липидами и т. п., образуя надмолекулярные комплексы, со-
ставляющие основу субклеточных структур.
4. Важнейшей особенностью механизма возникновения указанных комплек-
сов является безошибочное «узнавание» их ингредиентами друг друга и проте-
кание самого процесса ассоциации по принципу самосборки.
5. Молекулы белков закономерно изменяют свою структуру под влиянием
внешнего воздействия и восстанавливают исходное состояние при его снятии,
причем это явление может сопрягаться с акцептированием и преобразованием
энергии.
6. Многие белки обладают уникальной способностью каталитически уско-
рять химические реакции, протекающие в живом веществе.
7. Белкам присущи регуляторные, защитные, токсические, транспортные,
сократительные, структурные, рецепторные, модуляторные, морфогенные, за-
пасные и многие другие функции.
Даже этот далеко не полный перечень особенностей белковых тел, отлича-
ющих их от других природных биополимеров, свидетельствует об их важней-
шей роли в обеспечении атрибутов жизни.
Только детально изучив строение белков и их свойства, можно понять как
перечисленные особенности белков, так и их функции; поэтому обычно курс
биохимии открывается разделом «Белки».
ЭЛЕМЕНТАРНЫЙ СОСТАВ БЕЛКОВ
Белки, или протеины (от греч. протос—первый, главный),—высокомолеку-
лярные органические вещества, характеризующиеся строго определенным эле-
ментарным составом и распадающиеся до аминокислот при гидролизе.
23
Они содержат (в %): углерода—50—55, водорода—6,5—7,3, азота—15—
18, кислорода—21—24, серы—до 2,4 и золы—до 0,5. Особенно характерный
показатель—процентное содержание азота. В большинстве случаев оно состав-
ляет 16%, поэтому по содержанию белкового азота часто вычисляют содержа-
ние белка в кормах и продуктах питания. Для этого величину, выражающую
процентное содержание белкового азота в препарате, умножают на фактор
пересчета, равный 6,25, который выводят путем деления: 100:16=6,25.
ВЫДЕЛЕНИЕ БЕЛКОВ
Хотя около двух десятков белков (инсулин, рибонуклеаза, лизоцим, цитохром
с, соматотропный гормон, 0-липотропин, ацилпереносящий белок, а-бунгароток-
син, кобротоксин, кардиотоксин, ингибитор гастрина, ингибитор трипсина,
аполипопротеин, ферредоксин др.) удалось синтезировать и, таким образом,
сделать первый шаг на очень трудном пути их искусственного создания, синтезы
эти очень сложны, трудоемки, длительны и дороги. Поэтому единственно
реальным методом получения белков служит выделение их из природных
источников. Но и это нелегкая задача, так как белки обладают особой чувствитель-
ностью к действию большинства химических реагентов (кислот, щелочей, органи-
ческих растворителей и др.) и разрушаются от обычных процедур, применяемых
при очистке веществ (нагревание, перегонка, возгонка, экстракция и т.п.).
Возможен также автолиз (самопереваривание) выделяемых белков. Белок очень
легко теряет свои природные, присущие ему в естественном состоянии нативные
свойства (растворимость, биологическую активность и т. п.), и переходит в денату-
рированное состояние. Чтобы избежать денатурации белка в процессе его выделе-
ния, все операции проводят в мягких условиях: при низкой (не выше + 5° С)
температуре, избегая действия резких химических реагентов.
Впервые белок (клейковина) был выделен Я. Беккари из пшеничной муки
в 1728 г. Эту дату принято считать годом зарождения химии белка. В 1762 г.
началась работа по изучению белка молока—казеина (А. Халлер), затем
с 1789 г.—белков крови (А. Фуркруа). За два с половиной столетия из природ-
ных источников получены сотни различных белков и изучены их свойства.
Для успешного выделения белка из биологического объекта необходимо
тончайшее измельчение тканей вплоть до разрушения клеточных стенок. Для
этого используют (рис. 6) специальные валковые (Л) или шаровые (Б) мель-
ницы, в которых исходный материал многократно продавливается между тесно
сближенными валками или расплющивается непрерывно сталкивающимися
шарами. С этой же целью широко применяют гомогенизаторы (В и Г), в кото-
рых материал либо измельчается острыми ножами, вращающимися с огромной
скоростью (12000 об/мин и более), либо растирается между пришлифованными
стенками стеклянных пробирки и пестика, либо в замороженном состоянии
продавливается через фильеры специального пресса (Д').
Хорошие результаты дает метод разрушения клеточных оболочек путем
попеременного замораживания и оттаивания ткани. Роль «рабочего инструмен-
та» выполняют здесь кристаллики льда, разрывающие стенки клеток и освобож-
дающие клеточное содержимое. Применяют также метод «азотной бомбы»,
заключающийся в насыщении суспендированных клеток газообразным азотом
под высоким давлением, которое затем резко сбрасывают,—- азот, проникший
внутрь клеток, выделяется в виде газа и «взрывает» их.
Классификация методов разрушения (дезинтеграции) биологического материа-
ла, отражающая разнообразие подходов, используемых с этой целью при
выделении белков и других соединений из природных объектов, приведена в табл. 2.
24
Рис. 6. Устройство приборов для измельчения биологического
материала при выделении белков;
А—валковая мельница (at и а2—валки, вращающиеся навстречу друг другу; прямая
стрелка указывает направление поступления измельчаемого материала); Б—шаровая
мельница —корпус; б2—шары; стрелка указывает направление вращения корпуса);
В—ручной гомогенизатор (et—пестик; в2—корпус); Г—механический гомогениза-
тор (г,—нож; г2—камера для измельчения материала; г3—корпус с электродвига-
телем и пусковым устройством; г4—крышка); Д—рабочая камера прибора для из-
мельчения пресс-методом (ду—пространство, заполненное биологическим материа-
лом; д2—плунжеры для продавливания материала; д3—стенки камеры; —
перегородка с тончайшими отверстиями)
Когда достигнуто тонкое измельчение материала, переходят к следующему
этапу—извлечению белков. Белки извлекают чаще всего солевыми растворами,
взятыми в концентрации 8—10%. Подавляющая часть белков хорошо раство-
рима в таких солевых растворах. Различные соли обладают разным раство-
ряющим действием по отношению к белку. Ниже приведен ряд катионов
и анионов, расположенных в порядке убывания их растворяющего действия:
Li+>K+>Na+
Р2ог >В40Г >РОГ >CNS“ >НСО^ >1“ >сг
Классификация дезинтегрирующих воздействий по их природе
(по Б. А. Фихте и Г. А. Юревичу, 1988)
Таблица 2
Физические Химические Энзиматические Биологические
механические немеханические
Баллистические; экструзионные; ультразвуковые; газодекомпрес- сорные; гидро- ударные; электро- гидроударные; комбинирован- ные Осмотический, тепловой или хо- лодовый шок; за- мораживание — оттаивание; замо- раживание — вы- сушивание; деги- дратация—ре- гидратация; мед- ленная газовая декомпрессия; фазовые перехо- ды при высоких давлениях Действие щело- чей, кислот, солей, детерген- тов, антибиоти- ков, хелатных агентов, органи- ческих раствори- телей Действие бакте- риологических, дрожжелитичес- ких, миколитичес- ких ферментов и иммобилизован- ных литических ферментов Действие фагов, бактериоцинов и других киллеров (убийц), внутри- клеточных пара- зитов, плазмидо- подобных факто- ров. Ингибирова- ние синтеза кле- точной оболочки. Автолиз
25
Таким образом, максимальным растворяющим действием должен обла-
дать пирофосфат лития, минимальным —хлорид натрия.
Так как на растворение белков сильное влияние оказывает pH среды, боль-
шинство солей применяют в виде буферных смесей (фосфатных, ацетатных,
боратных, цитратных и т. п.). В последнее время широко используют буфер-
ные смеси, составленные с применением органических соединений—трис-
(оксиметил)-аминометана (HOCH2)3CNH2 и его соли с НС1 (трис-буфер);
диэтилбарбитуровой кислоты и ее натриевой соли (веронал-мединаловый бу-
фер); 1Ч,Ь1-бис-(2-оксиэтил)-глицина (HOCH2CH2)2NCH2COOH (бициновый
буфер); N-[трис (оксиметил)-метил ]-глицина (HOCH2)3CNHCH2COOH (три-
циновый буфер) и т. п.
Хорошие результаты дает извлечение белков спирто-солевыми смесями.
Металлопротеины, образующиеся при этом, обладают разной растворимо-
стью в спирте, что позволяет извлекать индивидуальные белки при различной
концентрации спирта. Весьма широко применяют для экстракции белков
глицерин, предохраняющий их от денатурации.
Извлечению белков из биологического материала способствует его
обработка детергентами: додецилсульфатом натрия, дезоксихолатом нат-
рия, тритоном X-100, алкил гликозидами и триалкиламмониопропансульфо-
натами:
Деэоксихолат натрия
сн3
н3с—с—сн2—
СН3 СН3 Тритон Х-100
{ п-(Третюктал)-феинловый эфир полиэтленглихоля]
(О-СН2— CH2—), _ 1(j-OH
сн2он сн о
н/I----°ч I + II
/н \-O-(CH2)7-CH3 CH3-(CH2)u-N-(CH2)3-S-O
H^pLJfi сн3 А
ОН Долецнл-лиметил-
Окил-р, D-глюкопиранозид .ммониопропансульфокэт (ТМ -12)
Перечисленные детергенты ослабляют гидрофобные белково-липидные
и белок-белковые взаимодействия, результатом чего являются деструкция
биологических мембран и высвобождение из них структурных и функциональ-
ных белковых компонентов.
Сказанное выше о выделении белков в основном относится к животным
тканям Выделение белков из растительного материала неизмеримо труднее:
здесь сложнее разрушить стенку клеток, более вероятна денатурация белков за
счет их взаимодействия с дуб шьными веществами и т. п. Поэтому при выделе-
нии белков из вегетативных органов растений используют специфические
приемы: обработку тканей водно-эфирной смесью, резко повышающей прони-
цаемость оболочки растительной клетки (метод Чибнелла), экстракцию бел-
ков смесью фенола, уксусной кислоты и воды (метод Синджа) и др.
26
Рис. 7. Разностные диаграммы высали-
вания белков:
А—белки сыворотки крови человека; Б—бе пси ге-
молимфы шелкопряда. Каждый сюлбик на диа-
грамме соответствует тому количеству белка, кото-
рое выпало в осадок при изменении концентрации
Na2SO4 на 0.005 (в единицах идейности раствора).
Стрелки указывают на концентрации Na2SO4. явля-
ющиеся пограничными для оi деления фракций друг
от друга
После экстракции смеси белков из биологического материала проводят
разделение полученной смеси на индивидуальные белки, поскольку в состав
растительных и животных тканей входит множество различных белков, экс-
трагирующихся в той или иной пропорции в процессе выделения. Фракциони-
рование белков ведут разными способами: солями, органическими раство-
рителями, электрофоретически, хроматографически, методом молекулярных
сит и т. п.
Метод фракционирования белков солевыми растворами основан на том, что
каждый индивидуальный белок разделяемой смеси осаждается из нее при
определенной концентрации той или иной соли, в то время как другие белки
при данной концентрации соли остаются в растворе. Процесс осаждения
белка из раствора под действием соли называется высаливанием. При
дальнейшем насыщении солью выпадает следующий индивидуальный белок
и, таким образом, последовательно наращивая содержание соли в реакцион-
ной среде, можно один за другим выделить относительно чистые индивиду-
альные белки. Чтобы определить те границы концентрации соли, в которых
происходит осаждение определенного белка, строят разностные диаграммы
(рис. 7).
Из органических растворителей для фракционирования белков широко при-
меняют метиловый и этиловый спирты (во избежание денатурации белка
процесс ведут при температуре около 5° С). В этих случаях также предвари-
тельно строят разностные диаграммы. На рис. 8 приведены данные о концен-
трациях этилового спирта при осаждении разных фракций белков сыворотки
крови человека. Этот метод нашел широкое применение при выработке заме-
нителей крови, так как позволяет извлекать из нее необходимые ингредиенты
и длительно сохранять их. При фракционировании белков иногда сочетают
высаливание и осаждение спиртом, применяя спиртосолевые растворы.
Метод осаждения ионами тяжелых металлов находит ограниченное приме-
нение при фракционировании белков и осуществляется в большинстве случаев
в сочетании с другими методами фракционирования (высаливание, осаждение
органическими растворителями). Метод основан на взаимодействии ионов Hg,
Zn, Са, Ba, Pb, Fe, Си, U и других тяжелых металлов с реакционноспособными
аминокислотными радикалами белковой молекулы (Hg2 +—с сульфгидриль-
ными группами, Zn2 + —с имидазольными, Са2+ и Ва2+—с радикалами
фосфосерина и т. п.).
27
Рис. 8. Диаграмма фракционирования белков плазмы
крови человека этиловым спиртом
Метод электрофореза основан на способности различных белков переме-
щаться под Действием электрического поля с неодинаковой скоростью (а иногда
и в противоположных управлениях) в растворе, на влажной фильтровальной
бумаге или в другой твердой опорной среде. Напряжение при этом устанавлива-
ют от нескольких сотен до нескольких тысяч вольт, силу тока—в несколько
десятков миллиампер. Скорость передвижения белковых молекул определенно-
го вида к аноду или катоду является функцией электрического заряда, молеку-
лярной массы и формы молекул, ионной силы, pH и состава буферного
раствора, а также приложенных потенциалов. Сочетание перечисленных факто-
ров всегда специфично для каждого индивид ального белка, и естественно, что
разные белки обладают различной электрофоретической подвижностью.
Наибольшее распространение получил электрофорез в твердых поддер-
живающих средах: целлюлозе, ацетилцеллюлозе, геле агар-агара, крахмаль-
ном и полиакриламидном гелях. На электрофореграммах белки выявляют
с помощью красителей: амидового черного (амидошварц 10В), бромфеноло-
вого синего, кумасси бриллиантового голубого и др.:
Однако в 100—200 раз чувствительнее реакция обнаружения белков посред-
ством окрашивания их в гелях аммиачными комплексами серебра.
Особенно перспективен метод электрофореза в твердых средах, приготов-
ленных из сополимера акриламида и метиленбисакриламида, имеющего трех-
мерную структуру:
28
В зависимости от соотношения в реакционной среде акриламида и N,
N-метиленбисакриламидд трехмерная структура в процессе сополимеризации
получается мелко-, средне- или крупноячеистой. При электрофорезе белковые
молекулы с разной степенью легкости проходят через указанные ячейки, что
и обеспечивает фракционирование белков. Так, при фракционировании белков
сыворотки крови человека посредством электрофореза на бумаге наблюдают
6 фракций, в крахмальном геле—10, а в полиакриламидном геле—16 фрак-
ций (рис. 9).
Еще большая дробность фракционирования белковых смесей вплоть до
выделения индивидуальных белков обеспечивается при проведении электрофо-
реза в поддерживающих средах с градиентом pH. Поскольку в этом случае
положение белка на том или ином уровне в колонке или тонком слое носителя
определяется значением его изоэлектрической точки, метод получил название
изоэлектрического фокусирования (синонимы—электрофокусирование, изота-
хофорез). Градиент pH создается при посредстве специально синтезированных
для этой цели О. Вестербергом полиаминополикарбоновых кислот—амфоли-
нов, состав которых выражается следующей формулой:
—CHS-N—(СНг)х—N—сн,—
(СНг)х R
I
N
где х=2—3, a R—Н или—(СН2)Х—СООН. Молекулярная масса амфолинов
колеблется от 300 до 1000, причем в зависимости от соотношения в их
молекулах третичных атомов азота и карбоксильных групп в водных раство-
рах амфолинов обеспечивается диапазон pH от 3 до 10.
Хроматографический метод разделения белковых смесей заключается в про-
пускании подлежащей фракционированию смеси белков через колонку, за-
Рис. 9. Разрешающая способность твердых поддер-
живающих сред при фракционировании белков сы-
воротки крови человека методом электрофореза:
(Л—целлюлоза; Б, В—крахмальный и полиакриламидный гели). На
электрофореграммах Л, Б. В—левая мощная белковая фракция—
альбумин; правая—комплекс у-глобулиноа, между ними—фракции
остальных глобулинов. Во всех случаях исследован один и тот же
препарат, нанесенный на зону старта в количестве 100, 80 и 5 мкл
соответственно
- •> IM
Б фаЯ|ОПМЖ
° ием
Рис. 10. Механизм ионообменной хро-
матографии белков
Механизм ионообменной хроматографии сводится
к вытеснению противоионов, связанных с анионны-
ми и катионными центрами ионообменника (Г) ио-
ногенными группировками белковых молекул (II)
и связыванием последних с ионообменником за счет
электростатического взаимодействия. В зависимо-
сти от вида белка и особенно от соотношения в его
составе радикалов главным образом дикарбоновых
аминокислот и диаминокислот суммарный эффект
этих связей варьирует, вследствие чего разные белки
С неодинаковой скоростью вытесняются с иоиооб-
менника ионами проявителя (III и IV) и выходят из
колонки раздельно. Затем ионообменник регепиру-
ют (F)
полненную адсорбентом. В качестве адсорбента применяют производные
целлюлозы и сефадекса (см. с. 325), несущие ионообменные группировки
(аминоэтильные, сульфоэтильные, карбоксиметильные и др.), гель фосфата
кальция, силикагель и другие носители. В тех случаях, когда носители из-
бирательно связывают строго определенные белки, фракционирование бел-
ковых смесей идет очень эффективно. Такой вид хроматографии называют
хроматографией сродства или аффинной хроматографией. За последние 5 лет
широкое распространение получила металлохелат аффинная хроматография
белков, основанная на их способности обратимо сорбироваться на иммо-
билизованных ионах переходных металлов, используя свободные коорди-
национные связи последних.
Одной из ее модификаций является хроматография, основанная на гид-
рофобном взаимодействии носителя (фенил- или октилсефароза) с соответст-
вующими неполярными аминокислотными радикалами белковых молекул.
Применение эффективных сорбентов (различные производные силикагеля)
и высокого давления при элюировании с них белков привело к возникновению
ряда вариантов жидкостной хроматографии высокого разрешения.
Механизм ионообменной хроматографии белков представлен на рис. 10.
Для элюции адсорбированных белков с колонки используют солевые рас-
творы, имеющие различные концентраци и значения pH. Если изменение
концентрации солевого раствора осуществляется в процессе снятия белков
с одной и той же колонки, такую элюцию называют градиентной. Элюат
собирают небольшими порциями (по несколько миллилитров) в отдельные
пробирки при помощи специальной установки—коллектора (сборщика) фрак-
ций. Таких проб в процессе фракционирования белков на колонке хроматогра-
фическим методом получают обычно несколько сотен. В результате проведе-
ния цветной реакции на белок или путем измерения поглощения каждого
раствора в ультрафиолетовой области спектра устанавливают содержание
белка в каждой пробе. По этим данным строят кривую, из рассмотрения
которой становится ясным, сколько фракций белка содержится в элюате
и в какой серии пробирок находится каждая фракция. Одноименные пробы
объединяют и из полученного раствора выделяют чистый белок.
Фракционирование белков методом молекулярных сит основано на раз-
личной скорости перемещения белковых молекул (в зависимости от молеку-
лярной массы) через колонку, запол енную специальным материалом—сефа-
дексом1. Сефадекс получают путем обработки эпихлергидрином полисахарида
1 Его название образовано из начальных слогов трех слов: separation (разделение), pharmacia
(название шведской фирмы, выпускающей реактивы) и dextran (декстран). Применяют также'
30
декстрана, в результате чего между молекулами последнего возникает то или
иное число поперечных связей:
В цепь
К 3-му углеродному атому
остатка глюкозы соседней цепи
декстрана
Аналогично этому обработка другого полисахарида—агарозы 2,3-ди-
бром-пропанолом в резко щелочных условиях приводит к возникновению
сетчатого полимера сефарозы, дающего гели, пригодные для фракционирова-
ния белков с молекулярными массами в сотни тысяч дальтон.
В зависимости от числа поперечных связей в сефадексе и размера белковых
молекул осуществляется та или иная степень проникновения последних внутрь
гранул сефадекса, заполняющих колонку. Большие белковые молекулы, не
способные пройти через сито из ячеек в гранулу, быстро выносятся из колонки
с током элюента; мелкие молекулы белка, проникшие внутрь зерна сефадекса,
удерживаются некоторое время последним и выходят из колонки с последу-
ющими порциями элюата. Таким образом, колонка с сефадексом как бы
просеивает через себя молекулы белка по их размерам, выпуская, как это ни
парадоксально, первыми самые крупные молекулы (рис. 11). Так как гранулы
сефадекса сильно разбухают в растворителе и образуют гель, этот метод
называют также методом гельфильтрации.
Интересен предложенный П. А. Альбертсоном метод фракционирования
белков с помощью двухфазных систем, составленных из водных растворов
полимеров. При этом способе фракционирование осуществляется в мягких
условиях (физиологические значения ионной силы и pH среды), а водные
растворы полимеров оказывают стабилизирующее действие на разделяемые
белки. Метод позволяет работать с любыми количествами вещества,
аналогичные сефадексу материалы—молселект (сефадекс фирмы «Реанал», Венгрия), акрилекс
и биогель Р (на базе полиакриламида), сефакрил (получают из аллилдекстрана и N, N'-метилен-
бисакриламида), сефарозу (получают из агарозы) и др. Перечисленные материалы нашли широкое
применение не только для фракционирования белков, но и для разделения нуклеиновых кислот,
высших углеводов и других биополимеров (см. также с. 325).
31
Рис. 11. Механизм разделения веществ по молеку-
лярной массе на колонке с сефадексом:
А—колонка в начале работы: светлые кружки—зерна сефадекса:
темные точки—молекулы белков разной молекулярной массы, толь-
ко что внесенные в верхнюю часть колонки; Б—по мере продвижения
проявителя по колонке (сверху вниз) осуществляется перенос молекул
белков, однако только меньшие молекулы белков проникают внутрь
зерен сефадекса; В—мелкие молекулы белков резко отстают от круп-
ных его молекул, задерживаясь в зернах сефадекса. Первыми из
колонки выйдут большие молекулы
используя простейшую аппаратуру. Распределение белков между фазами опре-
деляет я различиями в физико-химических свойствах фракционируемых белков
и полимеров, входящих в состав верхней и нижней фаз: на основе комплекса
свойств тех и других происходят взаимодействия (образование ионных и водо-
родных связей, гидрофобные взаимодействия и др.), приводящие к накоплению
определенного белка или группы белков в одной из полимерных фаз.
ОЧИСТКА БЕЛКОВ
Белки, выделенные описанными способами, всегда содержат некоторое
количество низкомолекулярных примесей, особенно ионов солей. Для полного
освобождения от этих примесей белки подвергают дальнейшей очистке путем
диализа, электродиализа, ультрафильтрации и перекристаллизации, гельфиль-
трации и другими способами.
Очистка белков методом диализа состоит в длительном, в течение несколь-
ких суток, пропускании воды через сосуд, в который погружен диализацион-
ный мешочек. Его готовят из материалов, хорошо проницаемых для низ-
комолекулярных соединений и ионов, но не пропускающих крупных молекул
белка. Материалом для полупроницаемых мембран служит целлофан, размер
пор которого можно изменять в широких пределах обработкой раствором
ZnCl2. Внутрь диализационного мешочка (камеры) помещают раствор белка.
Камеру, как правило, снабжают капиллярной трубкой. В нее в первые часы
диализа устремляется часть белкового раствора, объем которого возрастает
вследствие поступления воды внутрь камеры (рис. 12./).
Даже после длительной очистки белка путем диализа в нем остается
некоторое количество ионов, сорбированных, белковыми частицами. Для
полного удаления загрязняющих белок ионов используют метод электро-
диализа: возле полупроницаемых мембран диализационной камеры поме-
Рис. 12. Установки для очистки бел-
ка путем диализа (I) и электроди-
ализа (II):
1—трубка для подведения воды; 2—корпус
диализатора; 3—диализационный' мешочек;
4—трубка для отвода воды и регулирования ее
уровня; 5—капиллярная трубка; А—диализа-
ционная камера; Б—корпус электродиализато-
ра; а и Oj—полупроницаемые мембраны;
б и —электроды; стрелками указано направ-
ление движения воды
32
щают электроды, на которые подают напряжение, в результате чего остатки ионов
поступают в омывающую мембраны воду и выносятся из аппарата (рис. 12.77).
Полупроницаемые мембраны высокой прочности из нецеллюлозных мате-
риалов с калибре ванными порами, через которые проходят низкомолекуляр-
ные вещест а. то не проникают высокомолекулярные белки, используют для
очистки белков методом ультрафильтрации. Белковый раствор продавливают
через такую мембрану сжатым газом или центробежной силой. Белок, оста-
ющийся в процессе ультрафильтрации на мембране, не только очищается от
низкомолекулярных соединений и ионов, но и концентрируется. Если подоб-
рать такой размер пор в мембране, что через них будут проходить не очень
крупные белки, а большие белковые молекулы будут задерживаться, то уль-
трафильтрация может служить для фракционирования белков.
Долгое время считали, что белковые вещества существуют только в аморф-
ном состоянии. Однако в 1906 г. наш соотечественник А. Д. Розенфельд впер-
вые закристаллизовал оксидазу, выделенную из редьки, а в 1926 г. американ-
ский ученый Д. Самнер получил другой кристаллический белок—фермент
уреазу (от лат. urea—мочевина). Затем в течение последующих лет были
получены в кристаллическом состоянии десятки белков.
В настоящее время доказано, что любой белок может существовать в кристал-
лической форме. В основе различных приемов кристаллизации белка лежит
принцип очень медленного приближения к той критической точке, в которой белок
из растворенного состояния переходит в осадок. В этом случае белковые молекулы
успевают закономерным путем объединиться в надмолекулярные агрегаты и дают
кристалл ческие структуры. Практически такое медленное осаждение белков
достигается путем введения соли через полупроницаемую мембрану или при
медленном испарении воды из содержащего соль белкового раствора вплоть до
точки высаливания. В последнее время получила распростране н кристаллизация
белков с использованием орга-
нических соединений: 2-метил-
2,4-пентадиола и полиэтилен-
гликоля; с их помощью удалось
закристаллизовать ряд белков,
не поддававшихся этому тради-
ционными методами.
Несколько видов белковых
кристаллов приведено на
рис. 13. Кристаллические бел-
ки,, особенно полученные в ре-
зультате перекристаллизации,
отличаются высокой степенью
чистоты.
Хорошие результаты дает
очистка белков от низкомо-
лекулярных примесей при по-
мощи гельфильтрации. Зерна се-
фадекса достаточно долго удер-
живают низкомолекулярные ве-
щества, и белковая фракция
успевает за это время выйти из
колонки в чистом виде. Фильт-
рацию через гель сефадекса ши-
роко используют для обессоли-
вания белковых растворов.
Рис. 13. Виды белковых кристаллов: химотрипсина (Л),
пепсина (Б) белка дрожжей (В), рибонуклеазы (Г)
2—3502
33
ОЦЕНКА ГОМОГЕННОСТИ БЕЛКА
1 23456789 10
взято белка.
Зля растворения, г
Рис. 14. Оценка гомогенности белка
по независимости растворимости
от количества твердой фазы:
сплошной линией показан ход кривой в случае
гомогенного белка, пунктиром—при наличии
примеси
Вопрос о том, является ли полученный бел-
ковый препарат индивидуальным белком или
смесью белков, имеет важнейшее значение. Так
как белки >ыделяют из биологических объектов,
содержащих тысячи и деся ки тысяч индивиду-
альных белков (экспериментально в клетках
эукариот доказано наличие более 1200 отдель-
ных белков, но теоретически их число прибли-
жается к 10000), всегда можно ожидать, что
в составе изолированного белка есть примесь
других белков; это может привести к непра-
вильным заключениям и выводам о свойствах
исследуемого белка. Поэтому в белковой хи-
мии большое внимание уделяют оценке гомоген-
ности, т. е. однородности белков.
Для этого предложено почти полтора десятка
методов. Белок признают гомогенным, если увеличение его количества в твер-
дой фазе не сопровождается возрастанием содержания белка в растворе
(рис. 14). Наличие одной белковой полосы при электрофорезе в полиакрила-
мидном геле, одного пика на выходной кривой (либо одной белковой зоны на
лас шке) при хроматографировании или гельфиль рации, резкой границы
пограничного слоя белок—растворитель при ультрацентрифугирс вании бел-
ка являются наиболее надежными показателями гомогенности белкового пре-
парата. В большинстве случаев гомогенны кристаллические белки. Если кри-
вая распределения белковых частиц по высоте при использовании метода
свободной диффузии подчиняется теоретической кривой распределения Гаусса,
белок гомогенен. Достижение в процессе очистки белка стабильного содержа-
ния свободных HS-групп или содержания радиоактивной метки (при рабо е
с меченым белком), равно как и постоянного уровня биологической (фермен-
тативной, гормональной и т. п.) активности, свидетельствует в пользу гомо-
генности белка. Выявление единственной N-концевой и единственной С-кон-
цевой аминокислоты при анализе белкового препарата также является призна-
ком гомогенности выделенного белка.
Работами последних лет доказано, что каждый индивидуальный белок
состоит из молекул только одного вида, строго одинаковой молекулярной массы,
состава и строения. Поэтому столь важно получить гомогенный препарат белка.
МОЛЕКУЛЯРНАЯ МАССА БЕЛКОВ
Молекулярную массу белка измеряют различными физическими методами:
гравиметрическим, вискозометрическим, осмометрическим, ультрафильтраци-
онным, гельфильтрационным, электрофоретическим, оптическим. Полученную
величину принято называть физическим значением молекулярной массы белка;
она определяется с точностью в несколько сотен, а иногда и тысяч дальтон.
У многих белков в настоящее время выяснена первичная структура, что дает
возможность рассчитать молекулярную массу с точностью до сотых долей
дальтона, т. е. получить химическое значение молекулярной массы белка.
Обычно оперируют значениями физических молекулярных масс белков, варьиру-
ющих для всего спектра природных белков в очень широких пределах (табл. 3).
34
Молекулярная масса, степень асимметрии молекул
и изоэлектрические точки некоторых белков
Таблица 3
Белок Молекулярная масса Степень асимметряи молекулы Изоэлектрическая точка
Миоглобин кашалота 17600 3,0 7,0
Пепсин 35000 4,0 1,1
Альбумин яичный 46000 4,4 4,6
Кмоглобин лошади 68000 * 4,3 6,6
у-Пюбулин человека 160000 6,0 , 7,3
Каталаза 250000 5,8 6,7
Фибриноген человека 450000 17,5 5,4
Уреаза 483000 4,3 4,9
Тиреоглобулин свиньи 630000 9,2 4,5
Антипневмококковый сывороточный глобулин лошади 920000 20,1 4,4
Кмоцианин улитки 6600000 4,8 4,7
Определение молекулярной массы белка гравиметрическим методом ве-
дут в аналитических ультрацентрифугах (рис. 15). Принцип количес венно-
го изучения размеров взвешенных частиц в центробежном поле А. В. Думан-
ский выдвинул еще в 1913 г.,
и он же пытался определить
размеры частиц по скорости
оседания в обычных лабора-
торных центрифугах. Однако
первая ультрацентрифуга с оп-
тической приставкой, позволяю-
щей наблюдать и фотографиро-
вать процесс седиментации (осе-
дания частиц, была построена
шведе им физико-химиком
Т. Сведбергом десятью годами
позже. При вращении ее ротора
развивалось центробежное уско-
рение, превышающее ускорение
силы тяжести всего в 150 раз.
В современ ых типовых
ультрацентрифугах это пре-
вышение достигает 300000,
Рис. 15. Устройство ультрацентрифуги:
1 и 2—верхняя и нижняя стальные плиты; 3—
стенка стального цилиндра; 4—стальная игла с под-
вешенным ротором (П); 5—привод ротора; б—
термопара; 7—монтажная плита; 8—электромаг-
нитный затвор; ©—металлический патрубок, соеди-
няющий вакуумную камеру с фотоаппаратом; 10,
13—верхнее и нижнее кварцевые окна; 12—сталь-
ной хвостовик ротора в направляющей втулке; 14—
гнездо ротора для ячейки, в которой наблюдают
оседание белковых частиц
35
а в специализированных—900000—1200000. Данную величину называют
относительным центробежным ускорением или фактором разделения: <2>p=co2r/g,
где ш—угловая скорость ротора, рад/с; г-—расстояние от центра ротора до
середины ячейки с раствором белка, см; g—ускорение силы тяжести, см/с2.
Таким образом, например, запись 100000 g означает, что центробежное уско-
рение в месте расположения ячейки в роторе ультрацентрифуги превышает
ускорение силы тяжести в 100000 раз. Величина достигаемого в ультрацент-
рифуге фактора разделения—одна из важнейших ее характеристик.
Исследуемый раствор белка помещают в небольшую прозрачную для
световых лучей ячейку, которую вставляют в специальное гнездо ротора.
Молекулы белка в этой ячейке под действием центробежной силы, развива-
емой при вращении ротора, постепенно оседают на дно. Фотоприставка
к ультрацентрифуге позволяет делать снимки содержимого ячейки через опре-
деленные промежутки времени и определять таким образом скорость оседа-
ния белковых частиц. Определение молекулярной массы белка методом уль-
трацентрифугирования ведут двумя способами: по скорости седиментации
белковых молекул и по седиментационному равновесию.
В первом случае измеряют скорость перемещения в ячейке ультрацент-
рифуги границы растворитель—белок, относя ее к величине развиваемого
ентробежного ускорения, ,т. е. находят константу седиментации: s=v/oi2r, где
v—скорость перемещения границы растворитель—белок, см/с, ш2г—центро-
бежное ускорение, см/с2. Размерность s выражают в секундах. Величина
константы седиментации, равная 10“13 с, принята за единицу и названа све-
дбергом (S, или Св). Вводя значение экспериментально найденной константы
седиментации в формулу, рассчитывают молекулярную массу белка:
/>(1-р/ст)’
где R—газовая постоянная; Т—температура (по шкале Кельвина); D—коэффи-
циент диффузии; р—плотность растворителя; ст—плотность белковых частиц.
Во втором случае измеряют концентрацию белка Cj и с2 в двух точках ячейки
на расстоянии jq и х2 от центра ротора в тот момент, когда после определенного
срока работы ультрацентрифуги в ячейке установится седиментационное равно-
весие, т. е. равенство между числом оседающих и диффундирующих в обратном
направлении молекул белка. Расчет молекулярной массы белка ведут по
формуле, вводя в нее экспериментально найденные значения с2, jq и х2.
2КТ1пс2/С1
(1-р/ст)со2(х|-х?)’
где ш—угловая скорость, а остальные обозначения те же, что в предыдущей
формуле.
Из остальных перечисленных выше физических методов определения моле-
кулярной массы белков широко применяют еще два: гельфильтрационный
и электрофоретический.
Первый состоит либо в определении объема элюента, необходимого для
выноса белка из колонки с гелем сефадекса (Ие), и соотнесении его со свобод-
ным объемом колонки (Ко), либо в установлении длины пробега белка в тон-
ком слое сефадекса на пластинке. И тот и другой показатели связаны зависи-
мостью со значением молекулярной массы белка. Пользуясь калибровочными
графиками, построенными по VJ Vo или по длине пробега маркерных (т. е.
с известной молекулярной массой) белков, находят молекулярную массу
исследуемого белка.
36
Второй метод состоит в измерении пути пройденного белком при электро-
форезе в полиакриламидном геле; здесь тоже существует зависимость между
молекулярной массой белка и длиной пробега. Еще более отчетливо она
выявляется при сопоставлении торможения пробега белка при переходе от
электрофореза его в геле с меньшим содержанием акриламида к электрофоре-
зу в геле с большим содержанием акриламида. В этом случае также строят
калибровочные графики по маркерным белкам и по ним находят молекуляр-
ную массу неизвестного белка.
Другие физические методы определения молекулярных масс белков: по
светорассеянию, вязкости и осмотическому давлению белковых растворов, по
данным рентгеноструктурного анализа и электронной микроскопии—ис-
пользуют крайне редко.
Некоторое распространение имеет химический метод. Сущность его заклю-
чается в количественном определении в составе белка элемента или аминокисло-
ты, содержащихся в нем в наименьшем количестве. Затем проводят расчет
минимальной молекулярной массы, исходя из того, что в молекуле белка не
может быть менее одного атома элемента или одного аминокислотного
остатка. Однако об истинной молекулярной массе белка этот метод не всегда
дает правильное представление. Например, гемоглобин (белок крови человека
и животных) содержит 0,34% железа. Так как в гемоглобине не может содер-
жаться менее одного атома Fe, то минимальная молекулярная масса гемоглоби-
на рассчиты ае ся по пропорции: 0,34 части Fe соответствует 100 частям белка,
56 частей Fe (один атом) соответствуют х частям белка, отсюда х=(56-100)/
0,34 = 16 500. Истинная молекулярная масса гемоглобина равна 66 000—68 000,
т. е. учетверенной минимальной молекулярной массе. Это естественно, так как
молекула гемоглобина содержит. 4 атома Fe. Таким образом, химический метод
определения молекулярной массы белков в определенной мере условен.
ФОРМА БЕЛКОВЫХ МОЛЕКУЛ
О форме белковых молекул судят на основании математической обработки
данных, получаемых при ультрацентрифугировании белковых растворов. Для
этой же цели широко используют метод двойного лучепреломления в потоке.
Он основан на изменении оптической характеристики раствора белка, находя-
щегося в движении, по срав ению с раствором, находящимся в покое: в пер-
вом происходит ориентация вытянутых белковых частиц по направлению
движ ния раствора, и это сопровождается феноменом двойного лучепреломле-
ния. Кроме того, форма белковых частиц может быть установлена при непо-
сред твенном наблюдении в электронном микроскопе. Исчерпывающие дан-
ные о форме белковых молекул и топографии их поверхности дает рентгеност-
руктурный анализ.
С помощью этих методов выяснено, что белковые частицы бывают и ша-
рообразными, и сильно удлиненными—до нитевидных образований. В боль-
шинстве случаев они имеют вытянутую форму и построены асимметрично.
Степень асимметрии выражают отношением длинной оси частицы b к ее
короткой оси а. Данные о степени асимметрии (bid) молекул некоторых
белков приведены в табл. 2, а их форма показана на рис. 16.
Белки, у которых степень асимметрии равна 1 (частицы шарообразны),
довольно редки. Чаще всего эта величина бывает в пределах от 3 до 6 (эллип-
совидные или палочкообразные молекулы). В некоторых случаях степень
асимметрии достигает 200 и более (нитевидные белковые частицы). В общем,
37
Рибонуклеаза
М’ОЩГ.&фнм
Пепсин Гемоглобин р-Лактоглобыин
М-35000:3]>1Унм Н-бтчрЗрнм м^0000:бр^рнм
Альбумин flp-Глобулин * dtt~ Глобулин .
СыВороточный сывороточный сывороточный
М=69000;Зб*^нм М-ЯОШЛМЧфнм М-300000;3^9рнм
Фибриноген 61=950ООО^р^Онм
Лр Глобулин б-Глобулин
сывороточный р.-Липопротеин сывороточный
М=2ШШ); 50*300нм СыВороточный М-300&Ю; 99*2(5 нм
ГМЗООООВ;/&*/&нм ’
Коллаген
М-350ООО; 1р*300,0нм
Рис. 16. Форма молекул некоторых белков
Под названием каждого белка указаны его молекулярная масса в дальтонах
и размеры молекулы в нанометрах
длина белковых молекул средней молекулярной массы лежит в пределах
нескольких десятков, а толщина—нескольких нанометров.
Более поздние работы, в которых была детально раскрыта полная струк-
тура некоторых белков, показали, что белковые молекулы асимметричны во
всех трех измерениях. Так, молекула миоглобина—одного из белков мышеч-
ной ткани (М = 17 600)—имеет размеры 2,5 х 3,5 х 4,5 нм (см. с. 71 и 72). Мно-
гим белкам, особенно ферментам, присуща многолопастная структура с на-
личием в их молекулах глубоких расщелин, выпячиваний и впадин, на дне
которых располагаются функционально активные центры.
АМИНОКИСЛОТНЫЙ СОСТАВ БЕЛКОВ
Одним из наиболее распространенных методов исследования химического
состава белковых тел является гидролиз. Белок на ревают с растворами
кислот или щелочей при температуре 100—105° С примерно в течение суток.
Чаще всего используют 20%-ный раствор НС1, обеспечивающий глубокий
гидролиз белка с минимальным разрушением аминокислот, из которых он
построен. В последнее время для ускорения реакции гидролиза белков ис-
пользуют иммобилизованные (закрепленные на носителях) протеоли ические
ферменты и ионообменные смолы, что обес ечивает полное соответствие
содержания аминокислот в гидрол зате соотношению их в белке.
Впервые А. Браконно (1820), используя кислотный гидролиз, выделил из
белка (желатины) аминокислоту—глицин, а Н. Любавин (1871) установил,
что при ферментативном гидролизе белки распадаются до аминокислот.
В последующее время было доказано, что аминокислоты являются почти
единственными продуктами гидролиза белков. Первая аминокислота была
получена из сока спаржи в 1806 г. С тех пор из растений, животных и микроор-
ганизмов выделено несколько сотен различных аминокислот; в составе белков
их обна ужено около 30, остальные существуют в свободном виде.
38
В настоящее время фракционирова-
ние аминокислот белковых гидролиза-
тов ведут методом ионообменной хро-
матографии. В колонках с ионообмен-
ными смолами удается разделить
набор белковых аминокислот на со-
ставляющие в течение 1,5—2 ч. Поток
проявителя из колонки направляют
в смеситель, куда поступает раствор
нингидрина, реагируя с которым каж-
дая из аминокислот образует сине-фио-
летовый Руэмана. Подробно эта реак-
ция описана в практикуме по общей
биохимии1. Краска подается далее
в фотоэлектроколориметр, связанный
с самописцем, на ленте которого плот-
ность окраски записывается в виде пи-
ков; высота и ширина их соответству-
ют содержанию аминокислоты в гид-
ролизате белка. Будучи скомпонованы
в единый агрегат, перечисленные эле-
менты и дополнительные устройства
к ним образуют прибор, получивший
название автоматического анализатора
аминокислот (рис. 17). Этот прибор ра-
ботает автоматически по заданной
Рис. 17. Принципиальная схема определения
аминокислот в гидролизате с помощью авто-
матического анализатора:
1, 2, 3—сосуды с раствором нингидрина, буферным раство-
ром для малой колонки, буферным раствором для большой
колонки соответственно, 4, 5, 6—иасосы для подачи соот-
ветстауюших растворов; 7. 8—малая и большая хромато-
графические колонки; 9—смеситель; 10—реакционная баня;
/Z—фотоэлектроколориметр; 12—самописец
программе и определяет содержание аминокислот в гидролизате белка (вклю-
чая и выполнение необходимых расчетов) за несколько часов почти без
участия экспериментатора. Если анализатор оборудован компьютером, то на
это уходит не более 2 ч.
Найденные в белках аминокислоты принято делить на две категории:
постоянно встречающиеся и иногда встречающиеся в белках. Постоянно
встречающихся в белках аминокислот насчитывается 18. Их названия, фор-
мулы и сокращенные обозначения приведены в табл. 4.
Кроме 18 аминокислот в состав белков постоянно входят еще два амида:
амид аспарагиновой кислоты—аспарагин и амид глутаминовой кислоты—
глутамин (сокращенно асн и глн, либо N и Q):
C-CHj-CH-COOH
H,N‘ '
NH,
Аспарагин
С-СН,-СН,-СН-СООН
HjN// NHt
Глутамин
К группе иногда встречающихся в составе белков аминокислот при-
надлежат оксипролин (оксипирролидин-а-карбоновая кислота); оксилизин (а,
8-диамино 8-оксикапроновая кислота); орнитин (а, 8-диаминовалериановая
кислота); 3,5-дииодтирозин; а-аминоизомасляная кислота; N-метил-, N,N-
диметил- и N,N,N-триметиллизин; N-метил , К,К'-диметил- и К,К'-диме-
тиларгинин; у-карбоксиглутаминовая кислота (гла); Р-карбоксиаспарагиновая
1 Здесь и далее даны ссылки на «Практикум по общей биохимии» Ю. Б. Филипповича,
Т. А. Егоровой и Г. А. Севастояновой. 2-е изд., М., 1982.
39
кислота (аса)-, З-К-мстилгистидин; Ы,ТЧ-диметилпролин; метиловые эфиры асп
и глу и некоторые другие аминокислоты. Их структуры легко вывести исходя
из формул, приведенных в табл. 4; строение орнитина, а-аминоизомасляной
и у-карбоксиглутаминовой кислот таково:
H,N-(СН,)з- СН-СООН
МН*
Орнитин
НаСк хоон
с
Н3с/ \чна
«- Аминомзомасл яма я
кислота
Н00С\т ₽ -
СН-СН,—СН—СООН
ноос/
%-Карбокснглутлммновая кислота
Таблица 4
Аминокислоты, постоянно ветре ающиеся в составе белков
Аминокислота Формула Сокращен- ное обозна- чение Когда, кем и в каком белке обнаружена
Глицин (аминоуксусная кислота) СН2—СООН NH2 гли, G 1820 г., А. Бракон- но, в желатине
Аланин (а-аминопропио- новая кислота) СНЭ—СН—СООН NH2 ала, А 1888 г., Т. Вейль, в фиброине шелка
Валин (а-аминоизовале- риановая кислота) снзХ ^СН—СН—СООН CH3Z 1 3 NH2 вал, V 1879 г., П. Шют- ценберже, в альбу- мине
Лейцин (а-аминоизокапро- новая кислота) СН3Ч ;сн—сн2—сн—соон сн/ I nh2 лей, L 1820 г., А. Бра- конно, в белках шерсти и мышц
Изолейцин (а-амино-Р-метил- валериановая кис- лота) 'сн.,—сн,—сн—сн—соон 1 1 снэ nh2 иле, I 1904 г., Ф. Эрлих, в фибрине крови
Аспарагиновая (аминоянтарная) кислота ноос—сн2—сн—соон nh2 асп, D 1868 г., Г. Ритт- хаузен, в расти- тельном белке
Глутаминовая (а-аминоглутаро- вая) кислота ноос—сн,—сн,—сн—соон 1 nh2 глу, Е 1866 г., Г. Ритт- хаузен, в расти- тельном белке
Серин (а-амино-Р-окси- пропионовая кис- лота) но—сн2—сн—соон nh2 сер, S 1863 г., Э. Крамер, в серицине шелка
Треонин (а-амино-Р-окси- масляная кислота) сн,—сн—сн—соон 1 1 он nh2 тре, Т 1921 г., Д. Зелин- ский и В. Садиков, в кератине гусино- го пера
Цистеин (а-амино-Р-тиол- пропионовая кис- лота) HS—сн2—сн— соон nh2 i/uc, С 1901 г., Г. Эмбден, в яичном белке
40
Продолжение табл. 4
Аминокислота
Формула
Сокращен-
ное обозна-
чение
Когда, кем и в каком
белке обнаружена
Метионин
(а-амино-у-метил-
тиомасляная кис-
лота)
Аргинин
(а-аминр-8-гуани-
динвалериановая
кислота)
Лизин
(а, е-диаминока-
проновая кислота)
Гистидин
(а-амино-Р-имида-
золилпропионовая
кислота)
Пролин
(пирролидин-а-
карбоновая кисло-
та)
Фенилаланин
(а-амино-'Р-фенил-
пропионовая кис-
лота)
Тирозин
(а-амино-р-пара-
оксифенил-про-
пионовая кислота)
Триптофан
(а-амино-Р-индо-
лилпропионовая
кислота)
СН3—S—СН2—СН2—СН—СООН
nh2
H,N— С—NH—(СН,).—СН—СООН
II 3 I
NH NH2
h2n—СН2—СН2—СН2—СН2—СН—СООН
nh2
N—С—СН,—СН—СООН
II II I
НС СН NHt
V
н
HtC-CH,
Н4С сн-соон
V/
I
н .
/гк-си
СН ^C-CHj-CH-COOH
сн-сн
// ч
но-с с—снх-сн-соон.
CH=CH NHS
СН
НС С----C-CHt-CH-COOH
J II II I
НС С CH NHj
I
Н
мет, М.
1922 г„ Ю. Мёл-
лер, в казеине
арг, R 1895 г., С. Хедин,
в кератине рога
лиз, К
гис, Н
про, Р
фен, F
тир. Y
три, W
1889 г., Э. Дрек-
сель, в казеине
1896 г., А. Коссель
и С. Хедин, в бел-
ке спермы осетра
и казеине
1901 г., Э. Фишер,
в казеине
1881 г., Э. Шульце
и Ю. Барбиери, в
растительном бел-
ке
1849 г., Ф. Бопп,
в казеине
1901 г., Ф. Гопкинс
и Д. Коль, в казеи-
не
Важной особенностью белковых аминокислот является их оптическая ак-
тивность. За исключением глицина, все они построены асимметрично и,
следовательно, будучи растворены в воде или соляной кислоте, способны
вращать плоскость поляризации света. Значение удельного вращения в боль-
шинстве случаев составляет от 20 до 30° влево или вправо и лишь иногда
выражается большими или меньшими величинами. Из 17 оптически деятель
ных белковых аминокислот 7 характеризуются в водных растворах правым
(+) и 10 левым (—) вращением, но все они относятся к L-ряду.
Лишь в составе гликопротеинов клеточных стенок бактерий и в антибиоти-
ках обнаружены D-a-аминокислоты: фен, глу, ала, лей, вал, про и др.
Тонкая структура аминокислот изучена методом рентгеноструктурного
анализа. При его посредстве удалось построить объемные модели аминокислот;
41
Неполярные
алифатические
Неполярные
ароматические
Гидроксил-
содержащие
Дикарбоновые и их амиды
Основные
Глицин Фенилаланин Серии
"Ч
Н. ХСН /Ж
Пролин
н2с—СН
н/ СН
NH s
о
СН
О
Триптофан
Н\М /ОН
V 8
Н -
Иминокислота
N
i
Н
С
U
о
N
I
Н
N
I
Н
N
I
Н
Глутаминовая
кислота
СОО-
<?Н2
сн2
ср ZOH
с
II
о
снон
СН z<
N Хс
। а
Н О
Тирозин
Аспарагиновая
кислота
СОО”
сн2
СН ^он
''с
It
о
NH2 о
9
СН
н^сн
n"
I
н
N
Н
Лизин
Цистеин
Нч,
N
I
Н
SH
I
сн2
СН он
с
II
о
Аспарагин
Н
Глутамин
NH2 о
С*
сн2
сн2
СН он
X Z
с
Серосодержащие
Метионин
СН,
£ }
£н2
<?Н2
СН ZOH
хс
II
о
NHJ
сн2
сн2
СН;
СИ
СН
Аргинин
NH< J
NH
сн
СН
СН
Н ZCH О"
N XCZ
ft 6
Гистидин
HC = N
HN СН
чс*
сн2
нхzch он
N XCZ
Н &
Рис. 18. Объемные модели аминокислот. Приведенные формулы аминокислот в какой-то мере
имитируют пространственные модели
некоторые из них показаны на рис. 18.
Степень детализации представлений
о пространственной структуре амино-
кислот иллюстрирует рис. 19, на кото-
ром приведены данные о межатомных
расстояниях и валентных углах между
атомами в к исталлической модифика-
ции глицина, полученные методом рент-
геноструктурного анализа. Аналогичные
данные этим же методом получены для
всех постоянно встречающихся i составе
белков аминокислот. На них основаны
современные представления о тонком
строении полипептидно" цепи в высших
структурах белковых молекул.
Рентгеноструктурный анализ кри-
сталлов аминокислот показал, что ве-
Рис. 19. Строение молекулы глицина в струк-
туре а-глицина
Эта модификация глицина возникает в процессе его кристал-
лизации из воды при медленном испарении последней; рас-
стояния между атомами даны в нанометрах
дущая роль в возникновении структуры кристаллов аминокислот и ее поддер-
жании принадлежит разветвленной системе водородных связей, возникающих
между молекулами аминокислоты, закономерно расположенными в структуре
кристалла (рис. 20). Свойство аминокислот образовывать водородные связи
сохраняется и тогда, когда аминокислота является составной частью полипеп-
тидной цепи. Вследствие этого возникновение а-спиральной конформации
(см. ниже) полипептидной цепи можно рассматривать как процесс внутренней
кристаллизации полипептида.
Если расценить 18 постоянно встречающихся в составе белков а-аминокис-
лот с точки зрения их совокупных химическ х свойств, то поража т крайнее
разнообразие взаимодействий, возможных между их радикалами.
Радикалом аминокислоты принято называть группировку атомов в ее
молекуле, связанную с а-углеродным атомом и не пр нимающую участия
в формировании полип птидной цепи. Химическая природа радикалов
(табл. 4) позволяет осуществлять реакции солеобразования (по Ь1Н2- и СООН-
группам), окисления и восстановления (по HS- и SS-группировкам), алкилиро-
вания, ацилирования и этерификации (по NH2-, ОН-, НО — и'СООН-
группам), амидирования (по СООН-группам , нитрования и галогенирования
(по ароматическим ядрам), дезаминирования i осредством азотистой кислоты
(по КН2-группам), фосфорилирования и сульфатирования (по ОН-группам),
сочетания с диазосоединениями (по ароматическим и гетере циклическим
Рис. 20. Водородные связи в структуре а-глицина
Каждая водйродная связь показана пунктирной линией, атом кислорода в верхней
группе молекул глицина экранирован атомом азота. Расстояния между атомами даны
в нанометрах
43
ядрам) и т. п. Все эти химические процессы являются основой для химической
изменчивости белковых препаратов, при обработке их соответствующими
реагентам . Некоторые из указанных реакций протекают в живых организмах
(солеобразование, окисление, восстановление, ацилирование, этерификация,
амидирование, фосфорилирование).
Физические свойства радикалов аминокислот также весьма разнообразны.
Это касается прежде всего длины радикалов и их объема (табл. 5).
Таблица 5
Характеристика радикалов аминокислот
Аминокислота Длина, ям Объем, нм3 Аминокислота Длина, нм Объем, нм3
Глицин 0,15 0,0051 Треонин 0,40 0,0631
Аланин 0,28 0,0322 Цистеин 0,43 0,0579
Валин 0,40 0,0863 Метионин 0,69 0,1121
Лейцин 0,53 0,1134 Аргинин 0,88 0,1257
Изолейцин 0,53 0,1134 Лизин 0,77 0,1210
Аспаргиновая кислота 0,50 0,0584 Гистидин 0,65 0,0890
Глутаминовая кислота 0,63 0,0855 Тирозин 0,77 0,1388
Серин 0,38 0,0360 Фенилаланин 0,69 0,1366
Триптофан 0,81 0,1755
От длины, объема и взаиморасположения радикалов аминокислот, составля-
ющих белковую молекулу, зависят объем, форма и рельеф поверхности белко-
вой частицы. Радикалы гли, ала, вал, лей, иле, фен и три неполярны, а остальных
аминокислот—полярны в той или иной мере. Это определяет степень раствори-
мости белков в различных растворителях. Таким образом, разнообразие радика-
лов аминокислот по химической природе и физическим свойствам тесно связано
с полифункциональностью и специфическими особенностями белковых тел.
Именно эти свойства выделяют белки из ряда других 1риродных биополимеров
и наряду с другими особыми их качествами (биокаталитическая активность,
образование сл жных комплексов с другими биополимерами, способность
образовывать надмолекулярные структуры, дена урация и ренатурация, дина-
мические переходы между глобулярным и фибриллярным состоянием, неисчер-
паемое разнообразие и вместе с тем высокая специфичность структуры молекул
и т. п.) обеспечивают им роль материальной основы жизненных процессов.
Если по качественному составу все разнообразие структурных элементов
белковой молекулы укладывается в основном в 18 перечисленных выше а-
аминокислот, то общее количество аминокислотных остатков в белковой
молекуле изменяется в широких пределах. Принимая среднюю молекулярную
массу аминокислотного остатка равной 115, легко подсчитать коэффициент
поликонденсации аминокислот при образовании белковой молекулы. Так, для
белка с М= 17000 он будет равен (17000:115) примерно 148, для белка
с М=44000—примерно 380 и т. п.
Таким образом, одни и те же 18 аминокислот и два амида—аспарагин
и глутамин—многократно повторяются в белковой молекуле, причем каждая
в разной пропорции (табл. 6).
Длительное время считали, что максимальная длина полипептидной цепи
ограничена примерно ты ячью аминокислотных остатков, как, например,
у ДНК-полимераз и аминоацил-тРНК-синтетаз, обладающих молекулярной
массой около 100 тыс. Да. Однако в течение последнего десятилетия ситуация
резко изменилась: была расшифрована первичная структура Р- и Р'-субъединиц
ДНК-зависимой и РНК-полимеразы из 1342 и 1407 (Ю. А. Овчинников и др.,
44
... 1981), фактора VIII свертывания крови из 2332 (Г. Вехар и др., 1984) и субъединицы
тиреоглобулина из 2750 (М. Люк, 1985) аминокислотных остатков в единой
полипептидной цепи. Но и это не предел: недавно выяснена (М. Koenig et al., 1988)
последовательность 3685 аминокисл тных остатков в дистрофине—палочковид-
ном мембр ином белке цитоскелета мышечных клеток человека. Таким образ м,
некоторые белки оказались поистине гигантскими полипептидами. Вместе с тем
длина непрерывной полипептидной цепи у подавляющего большинства белков
колеблется от нес ольких десятков до нескольких сотен аминокислотных звеньев,
даже если их М намного превышает 100 тыс. Да: они составлены из полипептид-
ных цепей (субъединиц) сравнительно небольших размеров.
В настоящее время детально изучены качественный состав и количест-
венное содержание аминокислот у многих сотен белков. Сопоставление этих
данных позволило установить некоторые закономерности. Как правило, такие
аминокислоты, как лей, лиз, асп и глу, содержатся в белках в значительных
(10—15%) количествах. Наоборот, доля три, цис и гис редко превышает
1,5—2%. Содержание остальных аминокислот колеблется обычно между
приведенными выше крайними величинами. Иле в белках почти всегда ме-
ньше, чем лей; в таких же соотношениях находится содержание в белках
гис и арг, тре и сер, а также асп и глу.
Таблица 6
Аминокислотный состав некоторых белков (А—процентное содержание
аминокислоты, Б—число аминокислотных остатков в молекуле)
Аминокислота Миоглобин человека Пепсин Альбумин яичный 1емоглобин человека
А Б А Б А Б А Б
Аланин 5,7 12 4,5 18 6,7 35 9,0 72
Глйцин 6,3 15 8,1 38 3,1 19 4,2 40
Валин 5,3 7 7,1 21 7,1 28 10,3 62
Лейцин 12,2 17 10,4 27 9,2 32 14,0 72
Изолейцин 5,0 8 10,0 28 7,0 25 0 0
Пролин 4,0 5 4,9 15 3,6 14 4,8 28
Фенилаланин 6,2 7 6,7 14 7,7 21 7,3 30
Тирозин 2,4 2 9,4 18 3,7 9 2,9 12
Триптофан 3,6 2 3,5 6 1,2 3 1,9 6
Серин 4,6 7 13,2 44 8,2 36 4,4 32
Треонин 2,9 4 9,5 28 4,0 16 5,2 32
Цистин 0 0 — — 0,5 2 0 0
Цистеин 0 0 1,5 4 . 1,4 5 1,0 6
Метионин 2,5 3 2,1 5 5,2 16 1,2 6
Аргинин 2,7 2 1,0 2 5,7 15 3,3 12
Гистидин 8,2 9 0,5 1 2,4 7 8,8 38
Лизин 16,1 20 0,4 1 6,3 20 9,6 44
Аспарагиновая кислота 9,2 8 16,6 44 9,3 32 9,6 30
Глутаминовая кислота 17,3 14 11,3 27 16,5 52 6,6 24
Аспарагин — 3 — — — — — 20
Глутамин — 7 — — — — — 8
Итого: 115,3 153 120,7 341 108,8 387 105,0 574
Примечание. Цистив, отмеченный в таблице, возникает в белке при взаимодействии двух остатков цистеина
с образованием дисульфидного мостика (см. ниже) и не рассматривается как самостоятельная белковая
аминокислота.
45
Пр веденные выше закономерности касаются белков с полным набором
аминокислот. Необходимо иметь в виду, что некоторые белки характеризу-
ются ссвершенно специфическим аминокислотным составом. Так, например,
сальмин —протамин из молок семги, на 85,2% состоит из арг, на 9,1%—из
сер и в небольших количест ах содержит ала, гли, вал, иле и про. Фиброин
шелка тутового шелкопряда содержит (в %) 29,7 ала, 43,6 гли, 12,8 тир, 16,2
сер, тогда как процентное содержание 11 других ами окисЛот незначительно.
Свойства того или иного белка в значительной мере определяются набо-
ром и соотношением в нем аминокислот. Некоторые из таких зависимостей
известны. Так, изоэлектрическая точка белка (см. табл. 3), т. е. pH среды, при
котором отсутствует перенос белка в электрическом поле, зависит от соот-
ношения катионных и анионных групп. Сравнительная оценка из< электриче-
ских точек 300 белков показала, что их распределение подчиняется почти
симметричной одновершинной кривой с пиком при pH 4,5. Это свидетельст-
вует о том, что у большинства природных белков дикарбоновые аминокисло-
ты реобладают над диаминокислотами, сообщая белкам суммарный от-
рицательный заряд. Высокое содержание про, гли и асп в белке способствует
его хорошей растворимости в спирте; большое число полярных групп в моле-
куле белка приводит к появлению эластичности и т. п.
ПЕПТИДЫ
Одним из важных химических свойств «-аминокислот, зависящим от одно-
временного присутствия в молекуле аминной и карбоксильной групп, является
их способность в определенных условиях образовывать пептиды. Схема этого
процесса, протекающего по типу реакции поликонденсации, такова:
H2N—CH—COOH+H2N—CH—СООН-» H2N—CH—СО—NH—CH—СООН+Н2О
сн3 СН2 I сн3 сн2 1
SH 1 SH
Аланин Цистеин АланилПистеин (дипептид)
В действительности в организме процесс идет много сложнее. При изуче-
нии обмена белков эта реакция будет рассмотрена детально. Указанная реак-
ция образования пептидов из аминокислот имеет большое значение для
понимания химического строения белковых тел.
В результате реакции поликонденсации аминокислот можно получить со-
единения, составленные из многих аминокислотных остатков с очень высо-
кими молекулярными массами. Такие соединения называют полипептидами,
а —СО—NH-группировки в них—пептидными группами иЛи пептидными
связями. Пептиды могут быть получены также при неполном гидролизе
белков.
Так как аминокислоты в составе пептидов находятся в форме ацилов, то
в названии пептида им придается характерное для ацилов окончание -ил.
Название концевой аминокислоты со свободной карбоксильной группой оста-
вляют без изменений. Наименование пептида начинают с аминокислоты,
сохранившей свободную «-аминогруппу.
Термин пептиды сейчас утратил свое первоначальное значение, так как
когда-то под пептидами понимали конечные продукты переваривания белков,
46
СНз
С Нз—С—О—С О—N Н—С Н—i
СНз R
БОК-аминокислота
СНз
Нз
соон + ci—сн2-
Хлорметилированный
полимер
_fl-C0_N„_C„_CO-O-CH1-Q-^^
(!н
Спз
сн2
R
БОК-аминоацилполимер
Снятие зашиты С H2N-rpynnw
аминокислоты
'2
СН3—С + СО;
сн3
Изобутилен
СНз
сн3—i—О—СО—NH
h2n—сн—со—о—сн2
Аминоацйлполимер
R’
R
Присоединение БОК-аминОкислоты
по освободившейся аминогруппе
СНз
БОК-аминокислота
Рис. 21. Схема твердофазного синтеза пептидов и белков
т. е. по существу—аминокислоты. Поэтому логично было продукты перева-
ривани , составленные из двух аминокислотных оста ков (пептидов), назы-
вать дипептидами, а из многих—полипептидами. Гораздо точнее пептиды
именовать гетерополиаминокислотами, т. е. соединениями, составленными из
того или иного числа различных аминокислот. Однако термин пептиды проч-
но вошел в химию белков, но в новом значении.
В лабораторных условиях пептиды могут быть получены разнообразными
методами, общей чертой которых является обязательная защита в одной из
реагирующих аминокислот аминной, а в другой—карбоксильной группы,
с тем чтобы они могли вступить в реакцию конденсации только лишь по
оставшейся свободной (или активированной в результате присоединения хими-
ческого реагента) карбоксильной (СООН-) или аминной (NH2-) группе. Наи-
большую известность приобрел метод твердофазного синтеза пептидов, пред-
ложенный Р. Меррифилдом (рис. 21). СООН-группу исходной аминокислоты
здесь защищают присоединением к полимеру, a NH2-rpynny присоединяемой
47
аминокислоты—урет-изобутилоксикарбонильным (БОК) радикалом. Метод
поддается автоматизации, и на его основе создан автоматический синтезатор,
при посредстве которого синтезируют не только пептиды, но и белки.
Из природных источников выделено несколько сотен индивидуальных
пептидов и во многих случаях детально изучены их строение, свойства и био-
логическая активность. Приведем несколько примеров.
Глутатион (у-глутамилцистеинилглицин, у-глу-цис-гли)—один из наиболее
широко распространенных внутриклеточных пептидов, принимающий участие
в окислительно-восстановительных процессах в клетках и переносе аминокис-
лот через биологические мембраны:
НООС—СН—СН2—СН2—СО—NH—СН—СО—NH—СН2—СООН
I I '
NH2 СН2 SH Остаток глицина
к__________у_____________/ к_у___)
Остаток глутаминовой Остаток цистеина
кислоты
Глутатион открыт Ф. Гопкинсом в 1921 г. Он представляет собой кристал-
лический порошок с t„n = 190—192° С. Из раствора в 0,5 н. H2S04 выпадает
в виде нерастворимого меркаптида меди при добавлении Си2О.
Приведенная выше формула соответствует так называемому восстановлен-
ному глутатиону (HS-глутатиону). В клетке наряду с восстановленной формой
глутатиона всегда присутствует кисленная форма (SS-глутатион), переходя-
щая в восстановленную при посредстве фермента-глутатионредуктязы:
НООС—СН2—NH
I
со
I -2Н
2 СН—CH2SH
+ 2H
NH2 NH
I I
CH—(CH2)2— co
I
COOH
HS-DiyramoH (восстановленная, сульфгидрильная форма)
НООС—CH2—NH NH—СН2—СООН
I I
СО со
I I
СН—СН2— S— S— СН2— СН
I I
NH2 NH NH NH2
II II
CH— (CH2)2—CO CO—(CH2)2—CH
I I
COOH COOH
SS-Глутатион (окисленная, дисульфидная форма)
Офтальмовая кислота (у-глутамил-а-аминобутирилглицин)—антагонист
глутатиона, столь же широко распространена в природе, как и сам глутатион:
4S
ноос—сн—(сн2)2—co—nh—сн—co—nh—сн2—соон
Jtur pt. Остаток'глицина
Остаток глутаминовой Q.pj
кислоты '»_____^2_______I
Остаток а-амино-
масляной кислоты
Офтальмовая кислота
Присутствуя в клетках в ничтожных количествах, составляющих от 0,1 до
0,001 концентрации глутатиона, офтальмовая кислота действует как ингиби-
тор в процессах, идущих с участием глутатиона.
Карнозин (Р-аланилгистидин; $-ала-гис)—пептид, содержащийся в мышцах
животных:
H,N—СН,—СН,—СО-NH—СН—СООН
> » I I
Остаток р-смнии»
Остаток гистидина
. Кариоми
Он препятствует накоплению и устраняет продукты перекисного окисления
липидов, участвует в поддержании буферной емкости мышечного сока, уско-
ряет процесс рас ада углеводов в мышцах и в виде фосфата вовлекается
в энергетический обмен в мышце. Впервые выделил карнозин из мышечной
ткани и выяснил его строение В. С. 1улевич.
Роль пептидов в процессах жизнедеятельности крайне многообразна. Мно-
гие из них служат гормонами (см. гл. XII), некоторые представлены сильней-
шими ядами (яды змей, жаб, улиток, пауков, насекомых, высших грибов,
микробов), мощными антибиотиками, рилизинг-факторами (способствуют
синтезу и высвобождению гормонов), регуляторами клеточного деления, пере-
носчиками молекул и ионов через биологические мембраны, регуляторами
психической деятельности. Значительное число природных пептидов синтези-
ровано; кроме того, искусственным путем получены сотни их аналогов, неко-
торые из которых обладают более сильным биологическим действием, нежели
их натуральные предшественники. И те и другие находят широкое прак-
тическое применение. На рис. 22 приведено строение некоторых из них.
СТРУКТУРА БЕЛКОВОЙ МОЛЕКУЛЫ
Среди большого числа разнообразных гипотез лишь одна выдержала
испытание временем—это нолипептидная теория строения белковой молекулы,
впервые предложенная Э. Фишером (1902) на базе выдвинутых А. Я. Данилев-
ским идей о роли —СО—NH-связей в строении белка. Согласно этой теории
белковые молекулы представляют собой гигантские полипептиды, по-
строенные из нескольких десятков, а иногда и сотен остатков постоянно
встречающихся в составе белков аминокислот (см. табл. 6).
49
гли—асн
/ 4 5 \б 7 8 9
иле з иис—про—лей—гли (NH,)
42 /
тир—Цис
Окситоцин
глн—асн.
/ 4 s \б 7 8 9
фен 3 цис—про—арг—тли (N Н2)
42 /
тир—Цис
Вазопрессин
диокси-
ала—»-три
| ал
окси- —-цис /
про XD-Tpe
Фаллоидин
/
D-фен
L
леи
орн
'*-вал'—*
\
лей
♦
D-фен
/
про
Грамицидине
Пироглу—тис—jipo (NH2)
Тиролиберин
1 2 3 4 5
Тир—гли—гли—фен—мет
Мет-энкефалин
—фен-
асн 5 6 /фен
лиз 4 8 три
1 2 I I
Ала—гли—цис 3 9 лиз
\ /
цис 12 ц 10 тре
сер
_,вал_.
фен ^про
/ \
фен про
I I
про ала
V /
про фен
’ч»-фен-^'
Соматостатин
Антаманид
Пироглу-пироглутаминовая
кислота.
NH
Рис. 22. Биологически активные пептиды
Расшифровку сокращений см. В Т&бл. 4. Цифры около названий аминокислотных остатков обозначают ях местоположение
в молекуле, считая от N-копца к С-концу пептида. Стрелки начинаются от аминокислот, участвующих в образовании пептидной
связи СООН-группами. Соседние остатки цистеина в циклах связаны дисульфидными мостиками. Ijjynna NH2, заключенная
в скобки, обозначает амидную группу. Подразумевается, что аминокислотные остатки соединены пептидными связями. Окситоцин
и вазопрессин—гормоны задней доли гипофиза. Поступая в кровь, первый из них стимулирует сокращение мышечных волокон,
расположенных вокруг альвеол молочных желез и мышц матки, второй—действует главным образом на гладкие мышцы
кровеносных сосудов н участвует в регуляции водного баланса организма на уровне почек. Фаллоидин—~ ядовитое начало
мухомора, вызывающее в ничтожных концентрациях гибель организма вследствие выхода ферментов и К+ из клеток; связь между
остатками цистеина и триптофана в молекуле фаллоидина образована за счет взаимодействия сульфгидрильной группы и пир-
рольного кольца их радикалов. 1рамицидин-^антибиотик, действующий на многие грамположительныв бактерии (пневмококки,
стрептококки, стафилококки и др.), изменяет проницаемость биологических мембран для низкомолекулярных соединений и вызы-
вает гибель клеток. Ткролиберин—гормон, синтезирующийся в гипоталамусе и обеспечивающий высвобождение (отсюда его
другое название—рилизинг-фактор), а возможно, и усиление биосинтеза в гипофизе другого гормона—тиротропина, который,
в свою очередь, контролирует деятельность щитовидной железы в образование в ней гормона—тироксина; указанная иерархия
в регуляции биосинтеза гормонов является ярким примером участия пептидов в регуляции обмена веществ. Мет-энкефалин-—
пеитапетид, возникающий в нервной ткани и изменяющий (ослабляющий) болевые ощущения. Он связывается с рецепторами, на
которые воздействуют наркотики, например морфин, н представляет собой эндогенное вещество психотропного действия.
Соматостатин—пептид, тормозящий освобождение из передней доли гипофиза синтезируемого там гормона роста—сомато-
тропного гормона. Антаманид—циклический декапептнд, образующий с Na+, Са 2 + и некоторыми другими катионами комплексы,
которые являются прообразами структур, ответственных за перенос ионов через биологические мембраим
50
О том, что белковые молекулы построены именно таким образом, свиде-
тельствуют следующие факты:
1. В нативных белках удается обнаружить очень небольшое число свобод-
ных NH2. и COOH-групп, так как концевые аминокислоты составляют нич-
тожную долю огромной полипептидной цепи белка.
2. При гидролизе белков идет постепенное высвобождение NH2. и СООН-групп
в строгом соотношении 1:1,т. е. происходит распад пептид ных—СО—NH-связей.
3. При взаимодействии биурета H2N—СО—NH—СО—NH2 с растворами
щелочи и медного купороса развивается сине-фиолетовое окрашивание (реак-
ция на наличие —СО—NH-связей); все белки дают яркую биуретовую реак-
цию, т. е. в их молекулах действительно есть большое число пептидных связей.
4. Полипептидная природа ряда белков доказана химическим синтезом.
5. При рентгеноструктурном анализе строения белков удается наблюдать
непрерывную полипептидную цепь с характерным для исследуемого белка
расположением в ней аминокислотных остатков.
6. Методы, используемые для определения чередования аминокислотных
остатков в белках (см. ниже), однозначно свидетельствуют о последователь-
ном (в ряде случаев—выборочном, селективном) расщеплении пептидных
связей в составе именно полипептидных цепей.
Характерной особенностью строения полипептидной цепи является то, что
атомы С и N в ее хребте, составленном из монотонно повторяющих-
ся—СО—СН—NH-ф >агментов, располагаются приблизительно в одной плос-
кости, в то время как атомы и радикалы >CHR-группировок направлены
к этой плоскости под углом 109°28'. При этом в соседних аминокислотных
остатках расположение атомов Н и радикалов противоположно. Сказанное
ясно из рассмотрения рис. 23, Л и структуры фрагмента полипептидной цепи
молекулы инсулина (см. с. 54).
Другая особенность строения полипептидной цепи заключается в особом
характере —СО—NH-связи. Расстояние между атомами С и N в пептидной
связи 0,1325 нм, т. е. меньше нормального расстояния между а-углеродным
атомом и атомом N той же цепи, равного 0,146 нм. Вместе с тем оно
превышает расстояние между атомами С и N, соединенными двойной связью
(0,127 нм). Таким образом, связь С и N в —СО—NH группировке может
рассматриваться как промежуточная между простой и двойной вследствие
сопряжения л-электронов карбонильной группы со свободными электро ами
атома азота. Это определенным образом сказывается на свойствах полипеп-
тидов и белков: по месту пептидных связей легко осуществляется таутомерная
перегруппировка, приводящая к образованию енольной формы пептидной
связи, отличающейся повышенной реакционной способностью:
H2N—СН—СО—NH—СН—СООН ?±H2N—СН—С=N—СН—СООН
R' R" R' ОН • R"
Третья своеобразная черта строения пептидной связи состоит в том, что все
входящие в ее состав атомы располагаютс в одной плоскости (рис. 23, В):
такая конфигурация называется планарной (плоской) и поддерживается дело-
кализацией элек ронной плотности двойной связи карбонильной группы на
Связь между углеродом и азотом (рис. 23, Б).
Наконец, четвертое свойство полипептидной цепи заключается в том, что
ее остов, построенный из —NH—СН—СО-фрагментов, окружен разнообраз-
ными по своей химической природе боковыми цепями, что можно видеть при
рассмотрении структуры фрагмента молекулы инсулина (см. с. 5ф.
51
Боковые цепи функционально многолики. Они представлены (см. табл. 4
и рис. 18) жирными (ала, вал, лей, иле}, ароматическими (фен) и гетероцикличе-
скими (про, три, гис) радикалами. Многие из них несут свободные аминные
(лиз, арг), карбоксильны (асп и глу), гидроксильные и фенольные (сер, тре,
тир), тиольные (цис), амидные (асн, глн) и другие функциональные группы.
Взаимодействуя с окружающими молекулами растворителя (вода), свободные
NH2. и СООН-группы ионизируются, образуя катионные и анионные центры
молекулы белка. В зависимости от их соотношения белковая молекула полу-
чает суммарный положительный или отрицательный заряд, а также характе-
ризуется тем или иным значением pH среды при достижении изоэлектрической
точки белка. Число и соотношение катионных, анионных и иных групп в неко-
торых белках приведены в табл. 7.
Резкое преобладание в молекуле пепсина карбоксильных групп над амин-
ными требует высокой концентрации водородных ионов для перевода его
в изоэлектрическое состояние, в то время как почти равное число тех или
других в молекуле миоглобина сопровождается изоэлектрической точкой,
близкой к pH нейтральной среды (см. табл. 3).
Строение радикалов оказывает большое влияние на многие другие свойст-
ва белков и особенно на пространственную конфигурацию полипептидной
цепи: образующиеся солевые, эфирные, дисульфидные и другие мостики за-
крепляют относительное расположение участков цепи при формировании
третичной структуры белка (см. ниже). Радикалы также в основном определя-
ют круг химических реакций, свойственных белковым телам, и оказывают
наряду с другими факторами существенное влияние на функциональную ак-
тивность белков.
Долгое время представления о структуре белковой молекулы ограничива-
лись признанием полипептидной цепи как ее основы без какой-либо детализа-
ции закономерностей чередования аминокислотных остатков или конфигура-
ции цепи. Лишь разработка новых методов определения последовательности
расположения остатков аминокислот в полипептидной цепи и усовершенство-
вание рентгеноструктурного анализа белковых кристаллов позволили продви-
нуться вперед в понимании как первой (закономерности чередования амино-
кислот), так и второй (закономерности в конфигурации полипептидной цепи)
проблем.
В настоящее время в достаточной мере разработан вопрос о первичной,
вторичной, третичной и четвертичной структурах белковой молекулы. Пред-
ставление о четырех уровнях структуры белковой молекулы впервые выдвинуто
52
Рис. 23. Стандартные значения межатомных расстояний (в нанометрах) и ва-
лентных углов в полипептидной цепи (Л) и делокализация ^л ктронной плот-
ности по пептидному звену (£), приводящая к стабилизации его плоской
конфигурации (В и Г)
53
Фрагмент полипептидной цепи молекулы инсулина (1 - 9-й аминокислотные
остатки цепи Б)
Отток
фежпдакмп (1)
NH2
со
NH
Х_/от
сн=сн
НС-СН
НЭС
НзС
Отток
аалкв» (2)
Остаток
•стропов 0)
О.
HjN
Отток
HK.1WH1 (5)
Остаток
цистеина (7)
СО
I
NH
.о
н—с—сн,—
I
СО
I
NH
C-CHj-CHj—С -н
н.с.
н3с
СО
Отток
глупит (4)
NH
н —с— сн2— с=сн
со
NH
СИ—СН2-С~Н
Отток
жИопк («)
СО
I
NH
Н—С—СН2—S
со
NH
н — с—н
Отток Пттои (Я)
СО
NH
Отток
серккж (9)
Н—С — СН/)Н
со
Линдерстром-Лангом, исходя из методических соображений. Естественно, что
все перечисленные уровни структуры сосуществуют в белковой молекуле и их
уникальное сочетание в каждом конкретном случае определяет общее строение
белковой частицы.
Первичная структура белка. Под первичной структурой белка понимают
последовательность в расположении аминокислотных остатков в одной или
нескольких полипептидных цепях, составляющих молекулу белка. Зная пер-
вичную структуру белка, можно написать его полную химическую формулу.
54
Схема 1. Последовательность операций при установлении
первичной структуры белка
Белок
Раскрытие дисульфидных
мостиков окислением белка
надмуравьиной кислотой
Денатурированный белок
Селективный гидролиз в присутствии
ферментов или избирательная деструкция
при помощи химических методов
Смесь пептидов
Фракциониров ни s методом
высоковольтного электрофореза
Основные Нейтральные Кислые
пептиды пептиды пептиды
I___________________L__________________I
Субфракционирование методами
эл ктрофоре и хроматографии
Индивидуальны пептиды
Определение последовательности
аминокислотных остатков
в каждом пептиде
Первичная структура двух полных наборов
индивидуальных пептидов
Воссоздание первичной
структуры белка
Первичная структура белка
Учитывая, что в составе белковой молекулы содержится как минимум несколь-
ко десятков аминокислотных остатков, опред ление место] сложения каждого
из них составляет весьма сложную задачу. Ее решение может быть дости нуто
в результате осуществления ряда операций, приведенных на схеме 1.
Раскрытие дисульфидных связей в молекуле белка (схема 1) необходимо,
с одной стороны, для обеспечения последующего этапа фрагментирования
пол (пептидной цепи белка и, с другой—для превращения всех остатков цис
в белке в остатки цистеиновой кислоты, не разрушающейся при дальнейшем
анализе:
В цепь—NH—СН—СО—в цепь । в цепь—NH—СН—СО—в цепь
сн2 о 1 сн2
1 HCOOH; Н2О2 1
S — . SO3H
(NH4)2MoO4
1 SO3H
сн2 1 1 сн2
В цепь—NH—СН—СО—в цепь 1 в цепь—NH—СН—СО—в цепь
55
Селективный гидролиз денатурированного белка ведут протеолитическими
ферментами, чаще всего либо трипсином (по остаткам арг и лиз), либо
химотрипсином (по остаткам три, фен и тир)-.
H(NH—СН—С0)х—NH—СН—СО—(NH—СН—СО),—NH-
R' (СН2)4
nh2
R"
Н20
Трипсин
i—сн—со—(nh—сн—со),—он
(СН2)3 1'"
NH—С—NH,
II
NH
H(NH—*рН—CO)£-NH—сн—<
R’
+ H(NH—СН—CO)j—NH—СН—с; + H(NH—СН—СОЪ—он
•он I I 'ОН I
(£н2)„ R" (|н2)3 R"
NH2 NH—С—NH2
NH
Можно деструктировать белок и химическими агентами, например, бром-
цианом—по остаткам мет, 2,4-динитрофторбензолом—по остаткам цис,
N-бромсукцинимидом—по остаткам тир или три и т. п.:
H(NH—СН—CO)jj—NH—СН—СО—(NH—СН—CO)j—ОН
R'
CHj—CHj-S—сн3
90%-муравьиная кислота,
комнатная температура
HBr+CHjSCN
R'
H(NH—СН—CO)—NH
CO + H(NH—CH—CO)—OH
✓°
<fH2
R'
В результате получают два-три различных набора пептидов изучаемого
белка, что обеспечивает воссоздание его первичной структуры на заверша-
ющем этапе работы.
Фракционирование пептидов является многоступенчатым, длительным,
трудоемким, но необходимым элементом схемы определения первичной
структуры белка. Для этой цели используют преимущественно высоковольт-
ный (до 10000 В) электрофорез на бумаге, особенно на заключительном этапе
фракционирования, и хромато рафию различных видов, в том числе ионооб-
менную и распределительную.
Определение последовательности аминокислотных остатков в индивидуаль-
ных пептидах— наиболее ответственная процедура при установлении первичной
структуры белков. В настоящее время эту работу ведут преимущественно либо
фенилизотиоцианатным методом Эдмана, либо масс-спектрометрическим.
Фенилизотиоцианатный метод Эдмана (1950) реализуется в специально со-
зданном для этой цели приборе, получившем название секвенатор (от англ.
sequence—последовательность). Принципиальная схема устройства твердо-
фазного секвенатора й принцип его работы показаны на рис. 24. В современ-
ных моделях секвенатора удается определить чередование аминокислотных
остатков не только в коротких пептидах, но и непосредственно в полипептид-
ных цепях белка, проходя до 70 аминокислотных звеньев в его молекуле.
56
Рис. 24. Схема устройства секвенатора (пояснения в тексте)
Метод Эдмана сводится к обработке фенилизотиоцианатом белка или
пептида (рис. 25), присоединенного через С-концевую аминокислоту к инерт-
ному носителю (полистиролу или пористому стеклу) в колонке секвенато-
Рис. 25. Химические реакции, протекающие при секвенировании пептидов и белков (поясне-
ния в тексте)
57
Рис. 26. Принципиальная схема уст-
ройства масс-спектрометра:
I—инжекторное устройство для ввода образца; 2—
ионизационная камера; 3 в 5—система фильтров;
4—электромагнит; 6—детектор; 7—привод само-
писца; 8—лента самописца
ра (реакция сочетания). После промывки колонки растворителями (метанол,
дихлорэтан) образовавшийся фенилтиокарбамилпептид подвергают воз-
действию безводной трифторуксусной кислоты, в результате чего высво-
бождается аниливотиазолинон и в его составе N-концевая аминокислота
(реакция отщепления), а укороченный на один аминокислотный остаток
пептид или белок остается связанным с носителем. Анилинотиазолинон
поступает в коллектор фракций и, далее, после превращения в водной
среде в фенилтиогидантоин (реакция конверсии)—на хроматографическое
определение отщепившейся N-концевой аминокислоты. Будучи промыт
растворителями, секвенатор готов к следующему циклу работы в авто-
матическом режиме.
Масс-спектрометрический метод определения первичной структуры пепти-
дов, приложимый пока лишь к сравнительно коротким, примерно, 10-членным
пептидам, состоит в фрагментации аминокислотного типа эфиров ацилпеп-
тидов с последующим разделением полученных при этом положительно заря-
женных фрагментов. Фрагментацию осуществляют воздействием электронно-
го удара, а разделение фрагментов—в масс-ст ектрометре (рис. 26). В резуль-
тате получают масс-спектр фрагментов пептида (рис. 27), расшиф овывая
который, делают заключение о первичной структуре пептида. Хотя расшиф-
лей ала
~ ала ала ОМе .
339
^/Х0,5
Рис. 27. Масс-спектр метилового эфира N-деканоиллейцилаланил-
аланилаланина
На рисунке приведены массовые числа, соответствующие аминокислотному типу фрагмен-
тации пептида; как ясно нз названия исследованного соединение, аминогруппа N-концевой
аминокислоты—лейцина ацилирована и несет деканоильную
группировку, а карбоксильная группа С-концевой аминокислоты—аланина, этерифициро-
вана и несет метильную группу; именно такое производное пептида фрагментируется по
аминокислотному типу, т. е. преимущественно по пептидным связям с образованием
фрагментов, характеризующихся соответствующими массовыми числами
58
ровка масс-спектра фрагментов пептида весьма сложна, однако, исходя из
знания аминокислотного состава пептида и используя электронно-вычисли-
тельные машины для просчета различных вариантов сочетания аминокислот-
ных остатков в пептиде, находят тот вариант структуры, расчетные данные
которого совпадают с экспериментальными.
Заключительный этап работы при выяснении порядка следования амино-
кислотных остатков в белковой молекуле—воссоздание первичной структуры
белка на основании данных о последовательности их расположения в пеп-
тидах, полученных при селективном гидролизе. Так как избирательное
расщепление пептидных связей при исследовании первичной структуры
белков ведут не менее чем двумя агентами, обеспечивающими разрыв
пептидных связей в разных точках поли ептидной цепи, то первичные
структуры того и другого набора полученных пептидов неизбежно перекрыва-
ются. Это открывает возможность воссоздания первичной структуры белка
(рис. 28). Если при посредстве секвенатора удалось выяснить чередование
аминокислотных звеньев в фрагментах белковой молекулы значительного
размера, то это существенно упрощает процесс воссоздания ее первичной
структуры.
Другие методы определения последовательности расположения аминокис-
лотных остатков в белковой молекуле основаны главным образом на ис-
пользовании ферментов, способных ускорять реакции последовательного от-
щепления аминокислот либо с N-, либо с С-конца полипептидной цепи. Однако
они не получили еще столь широкого распространения и аппаратурного
оформления, как рассмотренные выше. В последние годы ведущее место
в выяснении первичной структуры белков занял метод, который условно
можно назвать генетическим: он основан на выведении последовательности
аминокислот в белковой молекуле исходя из структуры гена, кодирующего
биосинтез этого белка. Именно так была расш фрована структура самых
длинных полипептидных цепей—фактора VIII свертывания крови (2332 ами-
нокислотных остатка) и субъединицы тиреоглобулина (2750 аминокислотных
остатков). Нельзя не упомянуть также только что появившиеся сообщения об
определении первичной структуры белков методом лазерной фотодиссоциации;
учитывая его высочайшую чувствительность (для анализа достаточно 5 нмоль
белка при молекулярной массе 50 кДа), он, по видимому, имеет большое
будущее.
Первым белком, первичную структуру которого удалось расшифровать
благодаря работам Ф. Сангера и сотр. (1951 —1953), был инсулин быка (за это
в 1958 г. Ф. Сангеру была присуждена Нобелевская премия). Инсулин—бе-
лок-гормон, регулирующий углеводный обмен. Он синтезируется в поджелу-
дочной железе в виде предш ственника, структура которого показана на
рис. 29. При нарушении биосинтеза инсулина у человека раз: геа тся сахарное
мочеизнурение, или диабет.
Субъединица инсулина (М=6000) состоит из 51 аминокислотного остатка,
собранных в две полипептидные цепи (Л и Б), которые соединены двумя
дисульфидными мостиками.
Согласно справочнику Л. Крофта на август 1975 г. была выяснена пер-
вичная структ ра 932 соединений пептидной природы, из числа которых не
менее 800—белки. Учитывая, что в последующее время расшифровывали
от нескольких десятков до нескольких сотен первичных структур ежегодно
(в 1976 г.—76; в 1977 г.—380), список белков с полностью известным чере-
дованием аминокислот в их молекулах к январю 1985 г. достиг 2657. В ряде
случаев определена первичная структура одного и того же белка, но выде-
ленного из разных организмов. Так, первичная структура инсулина, миогло-
59
Ю
Глн—вал—лей—тре—асп—вал—глн—вал—ала—лей—вал—лиз— сер—
т-1, , ,
20
сер—фен—глу—глу—фен—асн—ала—асн—иле—про—лиз—асн—тре—
___________________т-2 _________________ж я Т-з
С-ХУШ-1
30
гис—арг—фен—фен—тре—лей—вал—лей—глу—иле—ала—про—
____________________________Т-4___________________
С-9-2 C-XI-5
40 50
гли—ала—ли^—асп—лей—фен—сер—фен—лей—лиз—гли—сер—
____________ .___________Т-5___________- -_______
60
сер—глу—вал—про—глн—асн—асн—про—асп—лей—глн—ала—гис—
_____________________ Т-6___________________________
____________________________________________________ С-Х-2_ж
70
ала—гли—лиз—вал—фен—лиз—лей—тре—тир—глу—ала— ала —
_________т Т-7 в Т-8 _
С-ХХ1-1 ж С—XXII—4 ,_C-1V-4
Тл-ХХХШ-6
80
иле—глн—лей—глу—вал—асн—гли—ала—вал—ала—сер—асп—ала—
т-8__________________________________________________
д ________________________с-1_______________________
90 100
тре—лей—лиз— сер —лей—гли—сер — вал — гис — вал — сер — лиз—
________. ______________т-9_____________________
ж п с-ххп-5
ПО
гли—вал—вал—асп—ала—гис—фен—про—вал—вал—лиз—глу—ала—
____________________т-ю____________________ж «
C-XVI-5
120
иле—лей—лиз—тре—иле—лиз—глу—вал—вал—гли—асп—лиз—три—
Т-п г т т-12 ________Т-13__________
ж с ххп-з ~~~
130
сер—глу—глу—лей—асн—тре—ала—три—тре—иле—ала—тир—
__________________________Т-14______________________
и____________С—УШ—4___________ _________С—VII—4____
140 150
асп—глу—лей—ала—иле—иле—иле—лиз—лиз—глу—мет—лиз—асп—
____________________________ _Т—15_ Т-16
д, „ Тл-ХХ^Ч я -I ТЛ-ХХУ1-2 Г|
CNBr
153
ала—ала
Т-17 Рис. 28. Воссоздание первичной группы леггемоглобина по
данным о первичной структуре пептидов, полученных при
его селективном гидролизе трипсином (Т), химотрипсином
(С) и термолизином (Тл)—микробной протеиназой, специфичной к оста-
ткам гидрофобных аминокислот, а также при его расщеплении бромциа-
ном CNBr
Леггемоглобин—кислородсвязывающий гемолротенн бобовых растений, играющий важную роль
в фиксации молекулярного азота клубеньковыми бактериями. Т—1, Т—2 и т. д.—триптические
пептиды; С—1, С—IV и т. д.—химотриптические; Тл—XXX, Тл—XXXI и т. д.—термолизиновые;
CNBr—цианобромидные пептиды. Перекрывающиеся зоны стрелок отмечают перекрытия пеп-
тидов из разных гидролизатов, а на стыках стрелок расположены точки селективного распада
полипептидной цепи. Первичная структура фрагментов, содержащих аминокислотные остатки с 1-го
по 24-й и с 29-го по 58-й, определена с помощью секвенатора
60
Рис. 29. Первичная структура проинсулина быка
С-лептид отщепляется от проинсулина в процессе превращения его в инсулин под воздействием
специфического фермента, ускоряющего гидролиз пептидных связей по остаткам аргинина
в позициях 3 [ и 60
бина и гемоглобина изучена у 20 видов, цитохрома с—у 60, лизоцима—у 10
.и т. д. Ниже приведены первичные структуры перечисленных белков, характер-
ные для человека:
61
Инсулин из поджелудочной железы человека, цепь А (21 аминокислотный остаток):
Гли-им-вал-глу-глн-цис-цис-тре-сер - им-цис-сер-лей- гпир-глн-лей-
глу-асн-тир-цис-асн;
цепь В (30 аминокислотных остатков) -
Фен - вал - асн - глн - гис - лей - цис - гли - сер - гис-лей-вал-глу-ала-мй-тир-
лей-вал-цис-гли- глу-арг-гли-фен-фен-тир-тре-про-лиз-тре.
Цитохром с из сердца человека (104 аминокислотных остатка):
СН3СО-гли-асп-вал-глу-лиз-гли-лиз-лиз-им-фек-им-мет-лиз-цис-
(сер-глн)-цис-гис-тре-вал-глу-лиз-гли-гли-лиз-гис-лиз-тре-гм-про-
асн-лей-гис-гм-лей-фен-гли-арг-лиз-тре-гли-глн-ала-про-гм-тир-
сер-тир-тре-ала-ала-асн-лиз-асн-лиз-гли-им-иле-три-гли-глу-асп-
тре-мй-мет-глу-тир-мй-глу-асн-про-лиз-лиз-тир-им-про-гли-тре-
лиз-мет-им-фен-вал-гли-им-лиз-лиз-лиз - глу-глу-арг-ала-асп-лей-
им - ала - тир - лей - лиз - лиз - ала-тре-асн-глу.
Миоглобин человека (153 аминокислотных остатка):
Гли-мй-сер-асп-гли-глу-три-глн-мй-вал-лей-асн-вал-три-гли-лиз-
вал-глу-ала-асп-им-про-гм-гис-гли-глн-глу-вал-мй-им-арг-лей-
фен-лиз-гли-гис-про-глу-тре-мй-глу-лиз-фен-асп-лиз-фен-лиз-гис-
лей-лиз-сер-глу-асп-глу-мет-лиз-ала-сер-глу-асп-лей-лиз-лиз-гис-
гли-ала-тре-вал-мй-тре-ала-мй-гли-гли-им-мй-лиз-лиз-лиз-гли-
гис-гис-глу-ала-глу-им-лиз-про-мй-ала-глн-сер-гис-ала-тре-лиз-
гис-лиз-вал-про-им-лиз-тир-мй-глу-фен-им-сер-глу - цис-им-им-
ели-вал-мй-гли-сер-лиз-гис-про-гли-ат-фен-гли-ала-асп-ала-глн-
гли-ала-мет-асн-лиз-ала-мй-глу-лей-фен-арг-лиз-асп-мет-ала-сер-
асн-тир-лиз-глу-мй-гм-фен-глн-гм.
а -Цепь гемоглобина человека (141 аминокислотный остаток):
Вал-лей-сер-про-ала-асп-лиз-тре-асн-вал-лиз-ала-ала-три-гли-лиз-
вал-гли -ала -гис -ала -гли -глу -тир -гли -ала -глу -ала -лей -глу -арг- мет-
фен - лей - сер - фен - про - тре - тре-лиз-тре-тир-фен-про-гис-фен-асп-лей-
сер-гис-гли-сер-ала-глн-вал-лиз-гли-гис-гли-лиз-лиз-вал-ала-асп-
ала-лей-тре-асн-ала-вал-ала-гис-вал-асп-асп-мет-про-асн-ала-лей-
сер-ала-лей-сер-асп-мй-гис-ала-гис-лиз-мй-арг-вал-асп-про-вал-
асн-фен-лиз-мй-мй-сер-гис-цис-лей-лей-вал-тре-мй-ала-ала-гис-
лей-про-ала-глу-фен-тре-про-ала-вал-гис-ала-сер-мй-асп-лиз-фен-
лей- ала -сер -вал -сер -тре -вал-мй- тре -сер-лиз -тир -арг.
(3 - Цепь гемоглобина человека (146 аминокислотных остатков):
Вал - гис - лей - тре - про - глу - глу - лиз - сер - ала - вал - тре - ала - лей - три - гли -
лиз - вал - асп - вал - асп - глу - вал - гли - гли - глу - ала - мй - гли - арг - мй - мй-
вал -вал -тир -про -три -тре -глн -арг -фен -фен -глу -сер -фен -гли -асп -лей-
сер -тре -про -асп-ала -вал -мет -гли- асн-про-лиз -вал -лиз -ала -гис -гли-
лиз-лиз-вал-лей-гли-ала-фен-сер-асп-гли-лей-ала-гис-лей-аен-асн-
мй-лиз-гли-тре-фен-ала-тре-мй-сер-глу-мй-гис-цис-асп-лиз-лей-
гис-вал-асп-про-глу-асн-фен-арг-лей-лей-гли-асн-вал-мй-вал-цис-
вал-мй-ала-гис-гис-фен-гли-лиз-глу-фен-тре-про-про-вал-глн-ала-
ала-тир-глн-лиз-вал-вал-ала-гли-вал-ала-асп-ала-мй-ала-гис-лиз-
тир-гис.
Лизоцим молока человека (130 аминокислотных остатков):
Лиз - вал - фен - глу - арг - цис - глу-лей-ала-арг-тре-лей-лиз-арг-мй-гли -
мет-асп-гли - тир-арг-гли-им-сер-мй-ала-асн-три-мет-цис-лей-ана-
лиз-три-глу-сер-гли-тир-асн-тре-арг-ала-тре-асн-тир-асн-ала-гли-
асп-арг-сер-тре-асп-тир-гли-им-фен-гли-им-асн-сер-арг-тир-три-
цис-асн-осп-гли-лиз-тре-про-гли-ала-вал-асн-ала-цис-гис-лей-сер-
цис-сер-ала-лей-мй-глн-асп-асн-им-ала-асп-ала-вал-ала-цис-ала-
лиз - арг - вал-вал-арг-асп-про-глн-гли-им-арг-ала-три-вал-ала-три-
арг-асн-арг- цис -глн -асн -арг -асп -вал-арг -глн -тир -вал -глн -гли -цис -
гли-вал.
62
В Институте биоорганической химии им. М. М. Шемякина выполнен ана-
лиз первичной структуры Р- и Р'-субъединиц ДНК-зависимой РНК-полимера-
зы—белков, имеющих очень длинные полипептидные цейи, 1342 и 1407
аминокислотных остатков соответственно; аспартатамино-трансферазы из
сердца свиньи—412; леггемоглобина из клубеньков желтого люпина—153;
LIV (лей, иле, вал)—связывающего белка из кишечной палочки—344; нейро-
токсинов из яда среднеазиатской кобры—72; белков L10 и L25 из рибосом
кишечной палочки—164 и 94 соответственно; а-субъединицы ДНК-зависимой
РНК-полим зразы—329; инсектотоксина из яда среднеазиатского скорпиона—
62; зрительного родопсина—348; фактора элонгации G—701; у-субъединицы
ГТФ-связывающего белка и у-субъединицы цикло -ГМФ-фосфодиэстеразы из
сетчатки крупного рогатого скота—69 и 87 соответственно; О39-белка из
мозжечка быка—341; а- и Р-субъединиц Na+, К+-АТФазы из почек свиньи—
1016 и 302 аминокислотных остатков соответственно.
В нашей же стране расш фрованы первичные структуры пепсина свиньи—
313 остатков (В. М. Степанов и др.); стурина—27 (А. Б. Силаев); актиноксан-
тина—107 (П. Решетов); лактогенного гормона —198 (Н. А. Юдаев); полиэд-
ренного белка вируса ядерного полиэдроза—244 (С. Б. Серебряный и др.);
нейротоксина из яда среднеазиатского скорпиона—65 (Е. Тришин и др.);
холестерин—гидроксилирующего цитохрома Р-450 из митохондрий коры над-
почечников быка—481 (В. А. Чащин и др.); а- и Р-субъединиц лютеинизиру-
ющего гормона из гипофиза кита—96 и 118 соответств нно (В. С. Карасев
и др.); кальмодулина из мозга человека—148 (Ю. Б. Алахов и др.)- гуанилспе-
цифичной РНКазы из нескольких видов аспергилла—102 (С. В. Шляпников
и др.); рестриктазы Есо RV из кишечной палочки—241 (А. А. Баев и сотр.);
нейротоксина III из актинии—48 (Т. А. Зыкова и др.); нейраминидазы, струк-
турного белка нуклеопротеина и гемагглютинина вируса гриппа—470, 498
и 566 соответственно (А. Б. Беклемишев и др.); В4 полипептида глицинина из
сои—251 (Е. С. Захарова и др.); гепаторедоксина из митохондрий печени
быка—117 (В. Л. Чащин и др.); асп ргиллопепсина А-320 (В. И. Остромыс-
ловская и др.); фосфорибозилами оимидазол-сукцино-карбоксамидсинтетазы
пекарских дрожжей—306 (Н. А. Мясников и др.); белка VP1 вируса ящура —
254 (А. М. Онищенко и др.); онкобелка р53 мыши—390 (П. М. Чумаков и др.);
пролактина из гипофиза человека—204 (Н. П. Мертвецов и др.); инсек оток-
сина из яда погребного паука—35 (Н. Ж. Сагдиев и др.); белка PV2 вируса
гриппа—759 (Н. А. Петров и др.); А- и В-субъединиц токсина шигеллы—315
и 89 соответственно (А. А. Баев и сотр.); а-субъединицы Na+, К+-АТФазы из
мозга человека (III форма)—1013 (О. И. Макаревич и др.) и Р субъединицы
фосфодиэстеразы цГМФ—852 (В. М. Липкин и др.) аминокислотных остатка.
Каждый из белков, первичные структуры которых приведены выше (за
исключением гемоглобина) представлен одной полипептидной цепью боль-
шей или меньшей длины. Чередование аминокислотных остатков в полипеп-
тидной цепи индивидуального белка неповторимо и специфично. В некоторых
случаях молекулы белков построены из двух или более полипептидных цепей,
соединенных друг с другом ковалентными связями. Примером такого рода
может служить молекула инсулина (см. рис. 29). В большинстве же своем
молекулы белков состоят из нескольких полипептидных цепей, удерживаемых
силами слабых взаимодействий.
При рассмотрении первичной структуры белковых тел возникает прин-
ципиально важный вопрос: осуществляются ли в белках все потенциально
возможные комбинации из аминокислотных остатков, их составляющих, или
существуют некоторые сочетания аминокислотных остатков, характерные
для многих, а может быть, и для всех белковых тел? Ведь белки и пептиды
63
за счет перестановки аминокислотных остатков в полипептидной цепи мо-
гут давать огромное число изомеров:
Число аминокислот- Возможное Число аминокислот- Возможное
ных остатков в число изомеров ных остатков в число изомеров
молекуле молекуле
2 2 7 5040
3 6 8 40320
4 24 9 362780
5 120 10 3362780
6 .1 720 20 ~2-1018
Понятно, что при увеличении числа аминокислотных остатков в белковой
молекуле до нескольких сотен количество возможных изомеров может достигнуть
астрономических величин. Однако анализ чередования аминокислот в белках,
проведенный в последние годы, привел к открытию ряда закономерностей,
позволяющих утверждать, что в белках реализуются далеко не все возможные
первичные структуры. Прежде всего, в различных белках, а часто и в одном и том
же, встречаются идентичные (тождественные) пептидные группировки. Особая роль
в структурном подобии белков принадлежит тождественным трипептидным
группировкам, но в некоторых случаях и более обширные фрагменты совпадают
по порядку чередования аминокислотных остатков. Так, в 52 рибосомных белках
тождественные трипептидные блоки повторяются 657 раз, тетрапептидные—86,
пентапептидные—И, гексапептидные—3, гептапептидные—ни одного раза.
Вместе с тем в полипептидных цепях есть аналогичные пептидные группировки,
отличающиеся друг от друга взаимозаменяемыми аминокислотными остатками,
т. е. такими остатками аминокислот, которые близки по строению или биогенети
чески, например: гли—сер, гли—ала, лей—иле, лей—вал, глу—асп и др.
Установлено, что в ряде случаев первичные структуры различных белков
включают 50% и более тождественных пептидных ф агментов.
Кроме того, значительные совпадения первичной структуры характерны
для белков, выполняющих сходные биологические функции. Это показано,
например, для семейства ферментов, ускоряющих реакции дегидрирования
разнообразных субстрактов, а также для многих гидролаз, у которых последо-
вательность аминокислотных остатков вблизи активного центра крайне близка
и стандартна. Особенно высоким структурным подобием отличаются белки,
выполняющие у разных видов одну и ту же функцию, что наглядно выявляется
при сопоставлении первичных структур инсулина (табл. 8), а также цитохро-
мов, гистонов, гипофизарных гормонов и других белков. У цитохромов,
выделенных из синезеленых, красных и бурых водорослей, первичные структу-
ры совпадают на 48—67%. Таким образом, видовая специфичность первичной
структуры гомологичных белков сводится к ограниченному числу аминокис-
лотных замен в полипептидной цепи в строго определенных положениях.
Богатейший материал этого рода дали анализы первичной структуры
аномальных гемоглобинов человека, где замена всего лишь одной аминокис-
лоты в а-, Р", у- или б-цепи сопровождается возникновением каждый раз
нового типа аномального гемоглобина. Таких гемоглобинов известно сейчас
более двухсот. Они являются источником заболеваний человека, относящихся
к категории молекулярных болезней. Так, например, при замене в Р-цепи
гемоглобина человека шестого остатка глу на остаток вал возникает аномаль-
ный гемоглобин S. Это приводит к образованию эритроцитов серповидной
формы с меньшим временем жизни, результатом чего является тяжелое
наследственное заболевание—серповидная анемия.
64
Таблица 8
Различия в первичных структурах А- и В-цепсй инсулинов разного происхождения
Источник инсулина Номера аминокислотных остатков
Цепь А Цепь В
4 8 9 10 1 2 3 27 29 30
Кашалот, кит ф нвал, глу тре сер иле фен вал асн тре лиз ала
свинья (принят за эталон
сравнения)
Человек тре
Бык, собака ала вал
Коза, овца ала гли вал
Слон ала гли вал тре
Кит сейвал ала тре
Лошадь гли
Кролик ала вал сер
Крыса асп ала вал лиз лиз сер
мет
Мышь асп лиз лиз сер
мет
Утка глу асн про ала ала сер тре
Цыпленок гис асн тре ала ала сер
Примечание. В остальных позициях цепей А И В инсулина разного происхождения чередование аминокис-
лотных остатков идентично; инсулин трески резко отличается по первичной структуре от инсулинов, перечислен-
ных в таблице видов.
Вторичная структура белка. Строго линейная полипепгидная цепь присуща
крайне ограниченному числу белков. Одним из таких белков является фиброин
шелка—белок, выделяемый гусеницами шелкопряда. В силу особых условий
формирования шелкового волокна в мощном мускульном прессе гусеницы
нитевидные молекулы фиброина, почти лишенные обрамляющих главную
полипептидную цепь радикалов, ориентируются вдоль шелкоотделительного
протока и плотно упаковываются по ходу шелкового волокна, приобретающе-
го на некоторых участках псевдокристаллическое строение. Рентгеноструктур-
ный анализ дал возможность выявить в первую очередь именно линейный
характер полипептидных цепей в фиброине шелка и, таким образом, в ЗО-е
годы нашего столетия возникло представление о белковой молекуле как
полностью вытянутой полипеп идной цепи с периодом идентичности
в 0,71 нм (подобной той, что изображена на рис. 23).
Однако позднее также рентге графически было найдено, что даже в фиб-
риллярных белках, не говоря уже о глобулярных, очень редко удается обнару-
жить полностью растянутые полипептидные цепи. Рентгеновские снимки посто-
янно указывали на наличие в белках цепей, каким-то образом сложенных или
скрученных, для которых повторяемость расположения структурных элементов
составляла 0,54 нм. На основании анализа рентгенограмм растянутых и нор-
мальных волокнистых белков, таких, как кератин волоса, В. Астбери и Ф. Белл
(1941) впервые предложили модель свертывания полипептидной цепи. Она
вполне объясняла наблюдаемый в опытах период идентичности в 0,54 нм и его
изменение при растяжении белкового волокна. Эта модель была уже построена
с учетом того, что валентные углы и межатомные расстояния, характерные для
пептидной связи и ее ближайшего окружения, не должны нарушаться.
Линия на моделирование как средство решения проблемы структуры бел-
ковой молекулы была продолжена Л. Полингом и Р. Кори (1949—1951). Они
i—3502
65
выдвинули критерии условий построения модели, отражающей возможное кон-
формационное состояние полипептидной цепи в белковой молекуле.
Исходя из выдвинутых критериев, Л. Полингом и Р. Кори были построены
спирали а- и б-типа, а Б. Л оу и Р. Бейбуттом—л-спираль.
Так как только х; рактеристика а-спиральной конформации цепи совпадала
с величинами, наблюдаемыми при рентгеноструктурном анализе белковых
кри таллов, то именно она и была признана Л. Полингом и Р. Кори в качестве
одного из структурных элементов, реально существующих в белковых моле-
кулах (рис. 30). Сейчас представление о существовании а-конформации поли-
пептидной цепи в белках является общепринятым.
Как видно из рисунка, а-спираль характеризуется предельно плотной упа-
ковкой скрученной полипептидной цепи, так что все пространство внутри
мыслимого цилиндра, в пределах которого идет закручивание, заполнено. На
каждый виток правозакрученной а-спирали приходится 3,6 аминокислотных
остатков, радикалы которых направлены всегда наружу и немного назад
(вверх на модели), т. е. отклонены в сторону начала полипептидной цепи.
Правый ход спирали легко установить, если смотреть в торец спирали со
стороны N-концевой аминокислоты полипептидной цепи (начало молекулы):
на модели ясно видно, что закручивание полипептидной цепи идет по часовой
стрелке. Шаг спирали (расстояние между витками) составляет 0,54 нм, а угол
подъема витка равен 26°. Период идентичности, т. е. длина отрезка спирали,
полностью повторяющегося по ходу ее, составляет 2,7 нм (18 аминокислотных
остатков).
Важнейшую роль в формировании и поддержании а-спиральной конфигу-
рации полипептидной цепи играют водородные связи, возникающие между
—СО- и —NH-группами хребта полипептидной цепи, расположенными на
соседних витках спирали (рис. 30). И хотя энергия этих связей невелика,
большое количество их приводит к значительному энергетическому эффекту,
в результате чего а-спиральна конфигурация достаточно устойчива и жестка.
Предполагают, что л-электроны СО- и NH-группировок полипептидной цепи
могут вступать во взаимодействие через водородные связи, осуществляющие
контакт соседних витков а-спирали. В результате на спирализованных участ-
ках белковой молекулы возникают зоны сопряжения электронов. Они могут
служить для передачи энергии возбуждения электронов, что имеет огромное
значение для осуществления химических реакций и трансформации одного
вида энергии в другой. Таким образом, под вторичной структурой белковой
молекулы подразумевают ту или иную конфигурацию характерную для од-
ной или нескольких полипептидных цепей, входящих в состав Молекулы.
Не следует представлять дело так, что в любом белке полипептидная цепь
полностью спирализована. Такие случаи очень редки. Видимо, для каждого
белка характерна та или иная степень спирализации полипептидной цепи:
Белок
Парамиозин
Миоглобин
Темоглобин
Альбумин сывороточный свиньи
Альбумин куриного яйца
Лизоцим куриного яйца
Вирус табачной мозаики (субъединица)
Пепсин
Рибонуклеаза
Химотрипсиноген
Доля спиральной
конфигурации. %
100
75
75
50
45
35
30
28
17
11
66
о
Рис. 30. Модели и схема а-спирали
На модели ot-спирали (слева) зачернеты участки спирали, обращенные к наблюдателю; пунктиром обозначены водородные связи между СО- и NH-группами, расположенными на соседних
участках спирали; атомы водорода показаны в виде маленьких кружочков. На схеме а-спирали (рядом) опущены все радикалы и показан ход хребта полипептидной цепи соответственно
таковому в модели. Правее даны изображения а-спирали и я-спирали (крайняя справа) с учетом планарности пептидных связей
Таким образом, в белковых молекулах спирализованные участки полипеп-
тидной цепи закономерно чередуются с линейными.
В природных белках существуют лишь право акрученные а-спиральные
конформации полипептидных цепей, что сопряжено с наличием в белковых
телах аминокислот только L-ряда (за исключением особых случаев).
Наряду с а-спиральной структурой в белковых молекулах наблюдается
также P-структура. Под последней подразумевают слоистые структуры,
образуемые при сочетании участков полипептидной цепи, находящихся в Р-
конформации, т. е. в виде линейно постр енных пептидных фрагментов.
Линейная конформация этих фрагментов удерживается благодаря воз-
никновению водородных связей между параллельно идущими и сбли-
женными на расстояние 0,272 нм (соответственно длине водородной связи
между СО- и NH-группами) участками полипептидной цепи (рис. 31).
Подобные структуры в массовом количестве представлены в волокнистых
белках, например в фиброине шелка. Однако и в глобулярных белках
P-структуры присутствуют систематически и часто превалируют над а-
структурами.
Возникновение а- и P-структур в белковой молекуле является следствием
того, что аминокислоты и в составе полипептидных цепей сохраняют прису-
щую им способность к образованию водородных связей. Таким образом,
крайне важное свойство аминокислот—соединяться друг с другом водород-
ными связями в процессе образования кристаллических препаратов—реализу-
ется в виде а-спиральной конформации или p-структуры в белковой молекуле.
Следовательно, возникновение указанных структур допустимо рассматривать
как процесс кристаллизации участков полипептидной цепи в пределах одной
и той же белковой молекулы (рис. 32).
Способность к образованию водородных связей, являющихся движущей
силой при возникновении а- и p-структур в белковой молекуле, присуща
разным аминокислотам в неодинаковой степени. Среди них вычленяют группу
спиралеобразующих аминокислот, куда входят ала, глу, глн, лей, лиз, мет
и гис. Если остатки перечисленных аминокислот сконцентрированы в какой-то
части полипептидной цепи или превалируют в ее составе, то а-спирализация
осуществляется очень гладко. Наоборот, такие аминокислоты, как вал, иле,
тре, тир и фен, способствуют образованию P-слоев полипептидной цепи. 1ли,
сер, асп, асн и про имеют отношение к преимущественному возникновению
неупорядоченных фрагментов в ее составе. Рис. 33 иллюстрирует один из
вариантов сосуществования а- и P-структур в одном и том же белке.
Третичная структура белка Сведения о чередовании аминокислотных оста-
тков в полипептидной цепи (первичная структура) и наличие в белковой
молекуле специализированных, слоистых и неупорядоченных ее фрагментов
(вторичная структура) еще не дают полного представления ни об объеме, ни
о форме, ни тем более о взаимном расположении участков полипептидной
цепи по отношению друг к другу. Эти особенности строения белка выявляют
при изучении его третичной структуры, под которой понимают общее рас-
положение в пространстве составляющих молекулу одной или нескольких поли-
пептидных цепей, соединенных ковалентными связями. Эту структуру ранее
обозначали как архитектонику белковых молекул.
Решение этой сложнейшей в химии белка проблемы связано с усовершенст-
вованием рентгеноструктурного анализа, выразившимся в увеличении раз-
решающей способности метода до 0,14 нм. Поскольку межатомные расстоя-
ния в молекулах органических соединений составляют 0,1—0,2 нм, столь
высокая разрешающая способность дает возможность точно установить рас-
положение каждого атома полипептидной цепи в пространстве, т. е. построить
68
Рис. 31. P-Структура белков:
А—возникновение 0-структуры; пунктиром обозначены водородные связи между СО- и NH-группами
латерально расположенных полипептидных цепей; Б—плоский слой, образованный антипараллельно рас-
положенными молекулами полиглицина; I—фрагмент объемной модели отдельной молекулы полиглицина;
II—слой из объемных моделей молекул полиглицина; В—0-слой, составленный из двух фрагментов
полипептидной цепи, построенных с учетом планарности пептидных связей
исчерпывающую модель белковой молекулы. По разрешающей способности
рентгеноструктурный анализ превосходит только метод ядерного магнитного
резонанса (0,03—0,09 нм), все более часто применяемый для изучения третич-
ной структуры белков. Кроме того, для этой же цели используют электронную
микроскопию, круговой дихроизм, дисперсию оптического вращения и ней-
тронную кристаллографию, не говоря уже о массовом предсказании третич-
ной структуры белков по их первичной структуре при помощи ЭВМ.
69
Рис. 32. Модели свертывания полипептидной цепи в а-спираль с образованием системы
водородных связей между витками спирали:
А—в виде бумажной ленты с выписанной на ней структурой полипептидной цепи; водородные связи, являющиеся
источником взаимодействия, приводящим к спиралнзации, показаны пунктиром; Б—в виде хребта полипептидной цепи,
составленного из объемно изображенных NH, СО- н CHR-rpynn; атомы водорода н кислорода, участвующие в образова-
нии водородных саяэей, помечены знаками «+» и «—» соответственно. Модель в виде бумажной ленты может быть
использована в курсе химии средней школы и на кружковых занятиях со школьниками по органической химии
Первая модель молекулы белка—миоглобина (рис. 33), отражающая его тре-
тичную структуру, была создана Дж. Кендрю с сотр. (1957). На рис. 34 приведены
схематические изображения третичной структуры молекул миоглобина, рибо-
нуклеазы, лизоцима и химотрипсиногена. Несмотря на большие трудности, за
истекшие три с половиной десятилетия удалось установить третичную структу-
ру почти трехсот белков, причем более трех десятков из них—в СССР. В нашей
стране, в частности, выяснена структура пепсина, леггемоглобина, аспартатами-
нотрансферазы, глицеральдегид-3-фосфатдегидрогеназы, пластоцианина, фи-
тогемагглютинина, у-кристаллина, пирофосфатазы, ряда рибосомальных бел-
ков, актиноксантина, лектина, лейцинаминоп птидазы, а- и Р-интерферона
лейцинспецифичного белка, родопсина, тубулина, гидрогеназы, метанмоноок-
сигеназы и др. Ниже некоторые из них будут охарак еризованы более подробно.
Пола ают, что третичная структура белковой молекулы определяется ее
первичной структурой, так как решающая роль в j оддержании характерного
для третичной структуры расположения полипептидной цепи в пространстве
70
Рис. 33. а- и P-Структуры во фрагм нте молекулы цитохрома bs и а-спиральные участки в моле-
куле миоглобина:
А—фрагменты полипептидной цепи цитохрома (остатки 21—25, 28—34, 50—54 и 73—79), образующие p-слой, показаны
толстыми линиями, сам слой отмечен значками ₽; четыре a-структуры (остатки 34—39, 40—50, 54—63,64—73) изображены в аиде
цилиндров; между попарно объединенными а-спиралями располагается простетичсская группа цитохрома Ь5, содержащая атом
железа; Ь—-восемь а-спиральных участков в молекуле миоглобина (обозначены буквами латинского алфавита) резко преобладают
над изгибами полипептидной цепи (обозначены сочетаниями АВ, ВС и т. д.) по протяженности и пересекаются под различными
углами; черный диск в верхней левой части молекулы—группа гема с атомом железа в центре
71
70
В
Рис. 34. Третичные структуры миоглобина (Л), лизоцима (Б), рибонук-
леазы (В) и химотрипсиногена (Г)
Во всех случаях показаны конфигурация н расположение в пространстве хребта полипептидной
цепи. Радикалы аминокислотных остатков нигде не обозначены, кроме трех радикалов в моле-
куле химотрипсиногена. А—заштрихованный диск—гем; пунктир—место прикрепления его
к хребту полипептидной цепи посредством радикала гистидина; хорошо заметны а-спирали,
составляющие 75% полипептидной цепи миоглобина; цифрами указаны номера аминокислот-
ных остатков в полипептидной цепи (см. также рис. 33, б); Б—символами—8—S-обозначены
дисульфидные мостики, образуемые при окислении радикалов цистеина; заметно, что «-спи-
ральная конфигурация присуща не более чем 1/3 полипептидной цепи; в верхней части молекулы
видно углубление (щель), предназначенное для размещения субстрата, расщепляемого лизоци-
мом; В—а-спиральных конфигураций почти не видно; остальные условные обозначения, как
в А и Б; Г —ясно выраженных а-спиралей не наблюдается;—S—S-связи обозначены точками,
здесь же цифрами указаны порядковые номера соответствующих остатков цистеина; в центре
молекулы выделены штриховкой радикалы гистидина (пятиугольники) и радикал серина (корот-
кий двурогий отрезок), образующие активный центр химотрипсина (после активирования
химотрипсиногена), ответственный за гидролиз пептидных связей
принадлежит взаимодействию радикалов аминокислот друг с другом. Воз-
можные типы связей между радикалами представлены на рис. 35. Особое
значение в поддержании третичной структуры белка придают дисульфидным
мостикам: именно они в ряде белков (см. рис. 29, 34 и 35) прочно фиксируют
расположение участков полипептидной цепи (или цепей) по отношению друг
к другу. Таким образом, местоположение в молекуле белка остатков цистеина
(и других аминокислот) предопределяет характер межрадикальных связей и,
следовательно, третичную структуру. Конечно и здесь, при замыкании дисуль-
фидных мостиков, в частности, реализуются далеко не все возможности их
возникновения, резко отличающиеся от расчетных (по Т. Крейтону, при пяти
SS-связях в белковой молекуле число их комбинаций достигает 945, при
10—654729075, а при 25—превышает 5,8• 1О30).
72
Рнс. 35. Типы связей между радикалами аминокислотных остатков в белковой
молекуле:
а—электростатическое взаимодействие; б—водородные связи; в—взаимодействие неполярных боко-
вых цепей, вызванное вталкиванием лиофобных радикалов в «сухую зону» молекулами растворителя
(так называемая «жирная капля»); г—дисульфидные связи. Двойная изогнутая линия обозначает хребет
полипептидной цепи
Кроме ковалентных связей третичная структура белковой молекулы
поддерживается силами слабых взаимодействий (рис. 35). Рассмотрение пол-
ных химических структур некоторых белков показало, что в их третичной
структуре отчетливо выявляются зоны, где сконцентрированы гидрофобные
радикалы аминокислот и полипеПтидная цепь фактически обматывается
вокруг гидрофобного ядра. Более того, в ряде случаев в белковой молекуле
обособляются два и даже три гидрофобных ядра, в связи с чем возникает двух-
или трехъядерная структура. Такой тип строения молекулы характерен Для
многих белков, обладающих каталитической функцией (рибонуклеаза, лизо-
цим и др.).
Данные о полной химической структуре молекул ряда белков явились
отправным пунктом для создания учения о доменном принципе строения
белковой молекулы. Под доменом понимают обособленную область молекулы
белка, обладающую в определенной степени структурной и функциональной
автономией. У ряда ферментов, например, обособлены кофер ентсвязываю
щие домены. С учением о доменном принципе строения белков связано
постепенно внедряющееся в белковую химию развитие представлений об
элементах однотипности, блочности, стандартности третичной структуры бел-
ков, об ограниченности набора пространственных упаковок полипептидных
цепей, реально существующих в природных белках.
Сейчас домены считают фундаментальными элементами структуры белко-
вых молекул, и соотношение и характер компоновки а-спиралей и p-слоев, как
полагают, дает для понимания эволюции белковых молекул и филогенетиче-
ских связей больше, чем сопоставление первичных структур. Причина этого
лежит в том, что в процессе эволюции происходило слияние доменов, их
удвоение, возникновение псевдосимметричных доменов из повторяющихся
субдоменов; некоторые из этих событий связаны с дупликацией генов и други-
ми изменениями генетического аппарата.
Есть много оснований полагать, что третичная структура белковой моле-
кулы возникает совершенно автоматически. Движущей силой, свертывающей
73
полипептидную цепь белка в строго определенное трехмерное образование,
является взаимодействие амин кислотных радикалов с молекулами окружа-
ющего растворителя. При этом лиофобные радикалы вталкиваются внутрь
белковой молекулы, образуя там сухие зоны («жирная капля»), а лиофиль-
ные—ориентируются в сторону растворителя. В некоторый момент достига-
ется энергетически выгодная конформация молекулы в целом, и белковая
молекула стабилизируется.
Самоорганизация полипептидной цепи определенного белка в только ему
присущую уникальную пространственную структуру, т. е. процесс возникнове-
ния третичной структуры, проходит в несколько этапов (рис. 36).
На конформацию возникшей глобулы оказывают сильное влияние такие
факторы, как pH среды, ионная сила раствора, а также взаимодействие
белковых молекул с другими веществами, что лежит в основе регу ищии
обмена веществ, в частности аллостерической регуляции активности фер-
ментов.
Разработка представлений о самоорганиз ции белковых глобул сопровож-
далась не только введением понятия о доменах, о чем уже было сказано выше,
но и новым подходом к характеристике уровней структуры белковых тел:
к ним добавился упомянутый уже доменный уровень и надвторичная структу-
ра. Под последней подра умевают закономерности возникновения в процессе
свертывания полипептид ой цепи элементарных структур, представленных
P-слоями (Р, Р'-структура), сочетанием а-спиральных участков (а, а'-структура)
или тех и других одновременно (рис. 37). Преобладаю-
щей среди надвторичных структур оказалась топология
греческого ключа и греческого орнамента.
Существенно, что фрагменты полипептидной цепи,
соотв тствующие доменам и даже субдоменам, способны
независимо поддерживать близкую к нативной структу-
1 ру. Так, цианбромидный фрагмент (121 —316-й аминокис-
лотные остатки) и его субдомен (205—316-й остатки)
термолизина (протеолитического фермента из термо-
фильной бактерии) самопроизвольно образуют стойко
удерживаемую нативную структуру. Весь же процесс
формирования третичной структуры таких, например,
п белков, как угольная ангидраза, а- и Р-лактоглобулин,
фосфо глицераткиназа и лактамаза, занимает всего лишь
0,2 с. Вместе с тем выявлены факт ры, лимитирующие
скорость складывания полипептидов в процессе возник-
новения третичной структуры; к их числу относится цис-
транс-изомеризация связи X—про (где X—любая ами-
ш нокислота), ускоряемая пептидилпролил цис-транс-изо-
Рис. 36. Структурные превращения полипептидной цепи при форми-
ровании белковой глобулы
На 1 этапе происходят локальные взаимодействия иа различных участках всей полипептидной
цепи, в результате которых возникают флуктуирующие а-спирали и p-слои. Образование
а-спиралей инициируется с той точки полипептида, где сосредоточены остатки дикарбоиовых
аминокислот, и терминируется в зоне остатков диаминокислот, тогда как центральная часть
возникающих а-спиралей занята аминокислотными остатками с гидрофобными радикалами.
На этом этапе самоорганизации белковой молекулы возникает максимально возможное количе-
IV ство а-спиральных конформаций полипептида, охватывающее большую часть полипептидной
цепи. Поэтому иллюстрируемая гипотеза самоорганизации белковых молекул получила назва-
ние гипотезы избыточных спиралей. На II этапе осуществляется направленное сближение
зародышевых структур, «схлопывание» а-спиралей и возникновение одного или нескольких
гидрофобных ядер за счет контактов гидрофобных радикалов аминокислот а-спнралей. В этот
момент возникает глобулярная структура. Ш этап сводится к компактизации зародышевых
структур н преобразованию промежуточной, высокоспиральной глобулы в нативную глобулу.
На IV этапе возникает окончательная, характерная для данного белка третичная структура
молекулы
74
Рис. 37. Надвторичные структуры белка
меразой. Рассмотренный механизм самоорганизации белковых глобул полу-
чил изящное подтверждение в работах (начиная с 1988 года) по синтезу
искусственных белков. Их создано к настоящему времени более двух десятков.
Исходя из представлений о приуроченности а-спиралей, p-слоев и участков
неорганизованной полипептидной цепи к определенным аминокислотным по-
следовательностям в синтетически полученных полипеп идах, удалось сконст-
руировать белковые молекулы, где автоматически и самопроизвольно воз-
никали элементы вторичной и надвторичной структуры, неизбежно занима-
ющие заранее рассчитанные позиции при формировании пространственной
структуры молекулы такого искусственного белка. Так созданы методом
белковой инженерии синтетические белки получившие условные названия
«Феликс» (от англ, four helices, т. е. из 4-х а-спиралей), «альбебетин» (построен
из а-спиралей и p-слоев в соотношении 1:2, т. е. аль: бебе) и т. п. Более того,
они обладали заранее запрограммированными свойствами и биологической
активностью.
Вместе с тем в течение последнего десятилетия взгляды на саморегуляцию
процесса свертывания полипептидных цепей белков при формировании их
третичной структуры in vivo существенно изменились Оказалось, что приве-
денные выше преобразования (см. предыдущую стр. и рис. 36) осуществля-
ются не сами по себе, а под лиянием особых, пр дназначенных именно для
этой цели специфических белков, названных шаперонами (так в Англии назы-
вали пожилую даму, удерживающую от непродуманных контактов молодую
девушку, впервые выходящую в свет под ее руко одством). Будучи в большин-
стве своем трубчатыми олигомерами, составленными из 10—90 кДа субъеди-
ниц, объединенных в налож иные друг на друга семичленные кольца, они
вовлекают внутрь олигомера пока еще неорганизованную полипе тидную
цепь и контролируют ход образования ею вторичных и надвторичных струк-
тур и их взаимную пространственную укладку (см. рис. 36), обеспечивающую
возникновение третичной, присущей нативной (функционально значимой) гло-
буле данного белка.
Однако и эта концепция, по-видимому,-—не последнее слово в далеко
неоднозначно решаемой проблеме складывания (фолдинга) полипептидных
цепей. Исходя из экспериментальных и теоретических подходов, разработан-
ных в процессе исследований кода белкового синтеза (см. ниже, гл. VII)
высказано предполож :ние (Л. Б. Меклер и Р. Г. Идлис; Г. И. Чипенс) о су-
ществовании общего стереохимического генетического кода, позволяющего
понять, как трехмерные молекулы белков сформировались из линейных поли-
пептидных цепей. Суть вопроса сводится к наличию кода взаимодействия
75
аминокислотных остатков друг с другом, что обеспечивает реализацию ин-
формации, уже заложенной в первичной структуре белка (и, естественно,
в мРНК и ДНК) в виде элементов вторичной и надвторичной структур и,
в конечном счете, в виде уникальной третичной структуры данного белка.
Четвертичная структура белка. Ранее уже было отмечено, что крупные
молекулы белков состоят, как правило, из субъединиц со ср внительно небо-
льшой молекулярной массой. Такие молекулы называют эпимолекулами
(сверхмолекулы) или мультимерами, а составляющие их элементы—субъеди-
ницами или протомерами.
Структура, характеризующаяся наличием в белковой эпимолекуле опреде-
ленного числа полипептидных цепей (субъединиц), занимающих строго фикси-
рованное пространственное положение, вследствие чего белок обладает той
или иной биологической активностью, называется четвертичной.
От четвертичной структуры следует отличать олигомерное и агрегированное
состояние белка. Структура, характеризующаяся существованием в составе
белковой частицы нескольких полипептидных цепей, число которых изменяет-
ся в определенной пропорции, называется олигомерной. Крайне важно, что,
--'<'МОТря на относительное постоянство числа полипептидных связей в олиго-
лире белка и их упорядоченное расположение, у олигомера не возникает
биологической активности. Так, например, сывороточный альбумин быка
существует в виде мономера (М = 68000), димера (М = 136 000), тримера
(М = 204 000) и тетрамера, причем мономеры, объединяясь в олигомерные
структуры, располагаются в составе ди-, три- и тетрамера упорядоченно.
Однако это не сопровождается возникновением каких-либо новых качеств по
сравн нию с теми, которыми обладает мономер данного белка.
Под агрегированным состоянием белка подразумевают такую структуру
белковых частиц, которая представлена неопределенным и изменяющимся
в широких пределах числом полипептидных цепей. При этом агрегация моно-
меров тоже не приводит к формированию каких-либо особых свойств у белка,
находящегося в таком структурном состоянии. Так, цитохром с (и другие
цитохромы) обладают резко выраженной способностью к агрегации молекул
друг с другом, но этому явлению не сопутствует изменение свойств фермента.
Сейчас выяснена четвертичная структура нескольких сотен белков.
В 1965 г. информация о четвертичной структуре ограничивалась примерно
20 белками; в 1970 г. появилась первая солидная сводка, включавшая 108 бел-
ков, а в 1976 г. Д. Дарнелл и И. Клотц опубликовали таблицу, содержавшую
перечень свыше 500 белков и данные о молекулярной массе мультимеров,
протомеров и количестве последних в эпимолекуле.
Оказалось, что число субъединиц в эпимолекулах колеблется в очень широких
пределах: )Т 2 до 162. Наиболее часто в составе молекул-мультимеров насчиты-
вается 2 или 4 протомера, гораздо реже—6, 8, 10, 12 или 24 и в редчайших
случаях—их нечетное количество. Четвер ичной структурой обладают в основ-
ном белки с молекулярной массой выше 50000—60000, а белки с меньшими
молекулярными массами существуют, как правило, в виде мономеров. Критиче-
ским пределом молекулярной массы белковой молекулы, сверх которого белок
в большинстве случаев обладает четвертичной структурой, считают 100 000. Что
касается молекулярных масс субъединиц, то они принимают самые разнообраз-
ные значения, от нескольких тысяч (например, 6000 у инсулина) до 330000 (у
каждой из двух субъединиц тиреоглобулина—белка щитовидной железы,
ответственного за биосинтез гормона тироксина—см. гл. XII).
Классическим примером белка, имеющего четвертичную структуру, явля-
ется гемоглобин (рис. 38). Молекула гемоглобина (М = 68000) построена из
четырех субъединиц с М = 17000 каждая. Первичная, вторичная и третичная
76
Рис. 38. Четвертичная структура белковых молекул:
А—модели субъединиц гемоглобина типа а (слева) и р (справа). Блоки, из которых составлены модели,
характеризуют распределение электронных плотностей в разных частях молекулы; черная (слева) и белая (справа)
линии указывают расположение хребта полипептидной цепи; Б—трехмерная модель молекулы гемоглобина;
субъединицы типа а (светлые) и р (темные) расположены по углам почти правильного тетраэдра, темные
диски—группы гема; В—модель молекулы вируса табачной мозаики: снаружи видны белковые субъединицы,
темная спираль—нуклеиновая кислота; Г—расположение субъединиц в молекуле вируса табачной мозаики
(разрез)
структуры субъединиц молекулы гемоглобина полностью выяснены. Они
оказались попарно идентичными и были названы с бъединицами типа аир.
Субъединица типа а представлена полипептидной цепью из 141 аминокис-
лотного остатка, Р—из 146. Третичная структура их сходна. Четыре субъеди-
ницы (две типа а и две типа Р) соединяются в единую молекулу гемогло-
бина, располагаясь в углах почти правильного тетраэдра (рис. 38, Б). Та-
ким образом, возникает почти шаровидная молекула с парам трами
0,50 х 0,55 х 0,64 нм.
Принципиал ый интерес для будущего учителя химии и биологии пред-
ставляет вопрос о том, как взаимосвязаны структура гемоглобина с его
функцией—способностью связывать, переносить и легко отдавать кислород.
Это явление детально изучается в средней школе. Непосредственно молекула
кислорода присоединяется к Fe2+, закрепленному в центре молекулы гема
(рис. 39), который, в свою очередь, удерживается в гидрофобном кармане
каждой из субъединиц, будучи присоединен координационными связями
к имидазольным радикалам гистидина, расположенным в дистальной и про-
ксимальной частях полипептидной цепи, образующей а- или Р-протомер гемо-
глобина. Присоединение кислорода к Fe2* идет без изменения валентности
последнего на одну из его свободных координационных связей; при этом
радиус атома Fe2+ уменьшается и он вместе с О2 перемещается в плоскость
порфиринового кольца. Здесь он удерживается до тех пор, пока молекула
гемоглобина не будет перенесена в ткань с более низким содержанием О2, где
и происходит обратный процесс отдачи кислорода. И связывание О2, и его
высвобождение сопровождается конформационными изменениями структуры
а- и Р-субъединиц гемоглобина и их взаимного расположения в мультимере.
77
Проксимальный
фрагмент
Рис. 39. Структура активного центра и механизм
связывания кислорода субъединице гемоглоби-
на (пояснения в тексте)
На рис. 38 приведена также схе-
ма строения сложного белка (нукле-
протеина)—вируса табачной мозаи-
ки Его гигантская молекула
(М » 40 000 000) содержит небольшое
количество (около 6%) РНК, осталь-
ное приходится на белок. Белковая
часть склад геается из большого
числа (2130) субъединиц с М = 17 500
каждая. Молекула вируса табачной
мозаики представляет собой полую
палочку длиной около 300 нм и тол-
щиной примерно 17 нм, с отверсти-
ем в центре диаметром в 4 нм. Каж-
дая субъединица имеет размеры
2x7 нм. Субъединицы расположены
по спирали, каждый виток которой
образован примерно 16 субъедини-
цами. Молекула нуклеиновой кисло-
ты, следуя спиральному располо-
жению субъединиц, проходит между
их рядами. Вдоль молекулы рас-
полагается свыше ста витков из бел-
ковых субъединиц.
Самое поразительное явление, на-
блюдающееся при изучении четвер-
тичной структуры белковых молекул, состоит в том, что объединение протоме-
ров в молекулу мультимера осуществляется самопроизвольно. Предполагают,
что в молекуле каждого протомера есть сп< пифические уча тки, взаимодей-
ствующие с таковыми в других протомерах. При соединении протомеров
в мультимер возникают ионные связи. В их формировании принимают участие
ионы металлов и иногда низкомолекулярные органические соединения. Однако
наибольший вклад в поддержание целостности структуры мультимеров вносят
силы слабых взаимодействий, а именно—гидрофобные взаимодействия и водо-
родные связи; суммарный эффек тех и других достаточно велик, чтобы
обеспечить стабилизацию четвертичной структуры белков. Как вне организма,
так, видимо, и в клетках мультимеры способны обра имо диссоциировать на
протомеры.
Принципиально важно, что малейшее изменение третичной структуры
протомеров делает невозможным соединение их в молекулы мультимера, что
резко сказывается на биологической активности белка. Поскольку третичная
структура белка задается его первичной структурой, а также зависит от ряда
других факторов (pH среды, концентрация солей и т. п.), то даже незначитель-
ное изменение первичной структуры белка или стандартных условий в клетке
приводит к изменению функциональной активности белков. Указан ые явле-
ния лежат в основе регулярных процессов в организме.
СВОЙСТВА БЕЛКОВ
Различают химические, физические и биологические свойства белков.
Химические свойства белков отличаются исключительным разнообразием.
Обладая аминокислотными радикалами различной химической природы, бел-
78
ковые тела способны давать широкий круг реакций. При рассмотрении
свойств аминокислот эти реакции уже были перечислены. Все они характерны
и для белков.
Особо важную роль в обеспечении определенных структурных особен-
ностей белковых молекул и ряда их биологических свойств имеют реакции
между радикалами аминокислотных остатков в пределах одной и той же
белковой молекулы. При этом выявляется отчетливая зависимость третичной
структуры белков от ряда внешних условий: pH среды, концентрации солей
в растворе, окислительно-восстано штельного режима клетки и др. Типы этих
реакций отмечены на рис. 35. Весьма характерна для белков реакция гид-
ролиза пептидных связей. Располагая значительным числом основных и кис-
лых групп, белки проявляют амфотерные качества.
Некоторые физические свойства белков (молекулярная масса, двойное луче-
преломление, подвижность в электрическом поле) рассмотрены выше. Кроме
них, для белков характерны оптические свойства, заключающиеся в способ-
ности вращать плоскость поляр зации света (оптическая ак ивность белков),
рассеив ть световые лучи ввиду значительных размеров белковых частиц
и поглощать ультрафиолетовые лучи. Перечисленные оптические свойства
белков используют при их коли ественном определении, измерении молеку-
лярной массы и т. п.
Одним из характерных физических свойств белков является их способность
адсорбировать на поверхности молекул , (а иногда и захватывать внутрь моле-
кулы) низкомолекулярные органические соединения и ионы. С этим свойством
белков св зана их транспортная функция в организме: некоторые белки явля-
ются хорошими переносчиками продуктов обмена.
К числу биологических свойств белков относят в первую очередь их биока-
талитическую активность. Благодаря особому строению молекулы или нали-
чию активной группы, соединенной с белком, многие белки обладают способ-
ностью кат литически ускорять ход химических реакций. Это свойство белков
играет огромную роль в осуществлении процессов жизнедеятельности. Оно
будет детально рассмотрено в главе о ферментах. Другое не менее важное
биологическое свойство белков—их гормональная активность, т. е. способ-
ность воздействовать на целые группы реакций в организме. Ряду белков
присущи токсические свойства, патогенная активность, защитные и рецептор-
ные функции, триггерное поведение, ответственность за явления клеточной
адгезии и, как следствие этого, морфогенеза и т. п.
Огромна пластическая роль белков: в сочетании с другими макромолекула-
ми они дают начало смешанным биополимерам—нуклеопротеинам, липопро-
теинам и гликопротеинам, которые, в свою очередь, обеспечивают возникнове-
ние субклеточных структур и надклегочных образований в организме. Харак-
терно, что информация о том, как будет построена та или иная субклеточная
структура, содержится именно в белковых молекулах. Сочетание своеобразных
химических, физических и биологических свойств белков обеспечивает именно
этому классу органических соединений центральную роль в явлениях жизни.
Еще одно своеобразное качество белковых тел—денатурация. Белки, об-
ладающие всеми характерными природными свойствами, называются натив-
ными. Часто под влиянием очень мягкой обработки, например легкого встря-
хивания, и тем более при грубых физических или химических воздействиях
белки быстро теряют нативность и переходят в денатурированное состояние.
Изменение уникальной структуры нативного белка, сопровождающееся поте-
рей характерных для него свойств: растворимости, биологической активности,
электрофоретической подвижности и т. п., называется денатурацией. Денату-
рация, как правило, затрагивает третичную и частично вторичную структуры
79
белковой молекулы и не сопровождается какими-либо изменениями первичной
структуры. Поэтому при денатурации белка нарушаются главным образом
дисульфидные мостики, солевые и водородные связи, а также гидрофобные
взаимодействия в белковой молекуле. При определенных условиях денатури-
рованный белок можно частично или полностью вернуть к исходному состоя-
нию. Такой белок называют ренатурированным. Современ ая фундаменталь-
ная биология уделяет огромное внимание нарушению нативной конформации
белков, связывая с нею важнейшие свойства клеток и, в частности, денатураци-
онный механизм повреждения последних.
НОМЕНКЛАТУРА И КЛАССИФИКАЦИЯ БЕЛКОВ
Несмотря на то что первичная, вторичная и четвертичная структуры белков
изучены в значительной степени и прогресс в этой области продолж ется, до
сих пор не создано ни строгой номенклатуры, ни научной классификации
белков. Названия белкам дают по случайным признакам, чаще всего принимая
во нимание источник выделения белка (например, наименование авидин—
белок яйца—происходит от лат. avis—птица; казеин—белок молока—от
лат. caseus—сыр;фазеолин—главный запасной белок фасоли—Phaseolus vul-
garis и т. п.) или учитывая растворимость белка в тех или иных агентах, форму
молекулы, аминокислотный состав и т. п.
Столь же несовершенна и классификация белков. В зависимости от поло-
женного в основу классификации признака среди белков выделяю г те или иные
узкие или широкие группы. Так, характеризуя белки по степени сложности,
среди них выделяют две большие группы: простые и сложные белки. К про-
стым белкам, или протеинам, относят белки, дающие при гидролизе только
аминокислоты. Сложными белками называют вещества, состоящие из протеи-
на (простого белка) и добавочной группы небелковой природы. Поэтому ранее
было принято называть сложные белки протеидами, т. е. подобными протеи-
нам. Однако сейчас от этого термина отказались, и в зависимости от химиче-
ской природы добавочной группы эти белки называют хромопротеинами,
липопротеинами, гликопротеинами, нуклеопротеинами, металлопротеинами
и т. п. Простые белки часто обозначают как однокомпонентные, а сложные—
как двухкомпонентные.
По форме частиц белки делят на фибриллярные (волокнистые) и глобуляр-
ные (корпускулярные). Фибриллярные белки характеризуются очень высоким
отношением b/а (несколько десятков единиц), их молекулы нитевидны и обыч-
но собраны в пучки, которые образуют далее волокна. К числу фибриллярных
белков принадлежат фиброин шелка, кератин волоса, коллаген кожи и др.
Белки, имеющие невысокое отношение b/а (в пределах нескольких единиц) и,
следовательно, палочкообразную форму молекулы, называют корпускуляр-
ными (корпускула—частица) или глобулярными. Подавляющее число природ-
ных белков относится к корпускулярному типу.
По отношению к некоторым условно выбранным растворителям среди
белков различают протеиноиды, альбумины, глобулины и проламины.
К протеиноидам относят белки, не растворяющиеся в обычных раствори-
телях белков: воде, солевых и водно-спиртовых смесях. Данное качество
присуще почти всем фибриллярным белкам. Однако в специфических агентах
протеиноиды хорошо растворяются: так, например, фиброин шелка полно-
стью переходит в раствор при обработке дихлоруксусной кислотой, безводной
плавиковой кислотой, концентрированным раствором роданида лития или
бромида калия и т. п.
80
К альбуминам причисляют белки, отлично растворяющиеся в воде и креп-
ких солевых растворах; в последнем случае принимают, что для альбуминов
характерна растворимость в водном растворе (NH4)2SO4, где к нцентрация
сульфата превышает 50% от насыщения. При переходе к очень концент-
рированным растворам (NH4)2SO4, вплоть до полностью насыщенных, аль-
бумины высаливаются.
К глобулинам принадлежат белки, не растворимые в воде, но растворимые
в солевых растворах умеренных концентраций. Характерным признаком глобу-
линов считают их полное осаждение при полунасыщении раствора (NH4)2SO4.
Проламины представляют группу белков, растворимых в 60—80%-ном
водном растворе этилового спирта.
По аминокислотному составу некоторые белки о личаются своеобразием,
и это тоже дает основание делить их на группы. Так, белки, содержащие
в составе молекулы 80—90% арг и ог аниченный набор (6—8) других амино-
кислот, относят к группе протаминов ( ростейшие белки). Они широко пред-
ставлены в молоках рыб. Типичным представителем их является, например,
сальмин из молок семги, содержащий (в %) 85,2 арг, 9,1 сер, 5,8 про, 3,1 вал,
3,0 гли, 1,6 иле, 1,1 ала и лишенный всех других аминокислот, обычно встреча-
ющихся в белках. Другая группа белков со своеобразным аминокислотным
составом—гистоны. Эти белки отличаются высоким содержанием основных
аминокислот: арг, лиз и гис (не менее 30%) и в значительных количествах
содержатся в ядрах клеток. Спирторастворимые белки—проламины—также
имеют характерный аминокислотный состав: в них много глу (20—50%) и про
(10—15%), в связи с чем они и получили свое название. Проламины выделены
только из растительных объектов.
Пр веденная классификация крайне несовершенна. В ее основу положены
случайные признаки, зачастую приводящие к противоречиям ’ и путанице.
Например, деление белков на простые и сложные с развитием аналитических
методов и уточнением состава белковых тел все более затрудняется, так как
тонкий анализ во многих случаях позволяет обнаружить в составе типичных
простых белков незначительные, но стабильные примеси соединений, не явля-
ющихся аминокислотами (металлы, аминосахара и др.). Яичный альбумин,
например, долгое время считали типичным простым белком, но недавно в нем
найдено около 2% маннозы. К глобулинам, как было указано выше, относят
белки, не растворимые в воде и высаливающиеся при 50%-ном насыщении
раствора (NH4)2SO4. Однако существует большая группа белков, раствори-
мых в воде, как альбумины, но высаливающихся, как глобулины (их называют
псевдоглобулинами). Число таких примеров можно было бы умножить. Поэ-
тому предприняты попытки, основываясь на успехах химии и биохимии
белков, дать им научно обоснованную классификацию.
Первая попытка состоит в классификации белков в соответствии с особен-
ностями их вторичной и третичной структуры. Согласно этой классификации
среди глобулярных белков выделяют 4 класса: а, Р, а+Р и а/p. К классу
а-белков относятся глобулярные белки, содержащие только а-спиральные
конформации в количестве не менее 60% от составляющей их полипептидной
цепи; к классу р-белков—содержащие только P-структуры в виде, как прави-
ло, не менее двух антипараллельных цепей; к классу а+Р-белков—содержа-
щие те и другие структуры в предел х одной и той же полипептидной цепи,
причем один домен собран из a-структур, а другой—из P-структур; к классу
а/Р-белков—содержащие многочисленные а- и P-структуры, либо чередующие-
ся вдоль полипептидной цепи, либо расположенные так, что один или несколь-
ко p-слоев окружены несколькими а-спиралями каждый. Домены у а/Р-белков
составлены, как правило, из а- и P-структур (рис. 40).
81
Рибонуклеаза Триозофосфатизотраза
(а+£-6елок) (а/fi-белок)
Миоглобин Рубредоксин
(а-белок) (fi-белок)
Миоеемэритрин
(а-белок)
Эрабутоксин
(fi-белок)
ФлаВобоксин
(a/fi-белок)
Рис. 40. Классификация глобулярных белков в соответствии с предста-
вительством и расположением в их молекулах а- и Р-структур:
А—расположение а- и p-структур (обозначены кружками и прямоугольниками соответственно) в а-,
Р-, а+р- и а/р-белках; стрелками указан ход полипептидной цепи от N-конца к С-концу молекулы;
миоглобин—белок мышц, ответственный за связывание кислорода; рубредоксин—железо протеин,
принимающий участие в окислительно-восстановительных процессах; рибонуклеаза—фермент,
ускоряющий реакцию гидролиза рибонуклеиновых кислот; триоаофосфатизомераза—фермент, ка-
тализирующий превращение фосфоглицеринового альдегида в фосфо ди оксиацетон; Б—строение
трех представителей а-, р- и а/р-белков, где a-структура представлена в виде спиралей, а р-
струхтура—в виде стрелок; миогемэритрин—желеэопротеин, связывающий, подобно миоглобину,
кислород (два атома железа показаны заштрихованными кружками, расположенными между двой-
ными а-сплралями); эрабутоксин—нейротоксин белковой природы, выделенный из яда морской
змеи (пунктиром показаны 4 дисульфидных мостика в его молекуле, представленной полипептидной
цепью из 61 аминокислотного остатки); флаводоксин—белок, выполняющий роль переносчика
втомов водорода (в головной части рисунка показана в виде трех конденсированных шестичленных
колец группировка, ответственная за передачу атомов водорода)
Большинство оцененных с этой точки зрения глобулярных белков от-
носите к а/р-классу, которому по численности лишь немного ступает
р-класс; a-класс и а+Р-класс глобулярных белков менее распростри ены,
чем два первых. Следует иметь в виду, что есть крайне немногочисленные
глобулярные белки, полностью лишенные какой-либо канонической вто-
ричной структуры и не относящиеся ни к одному из отмеченных выше
классов.
Вторая попытка сводится к классиф кации белков в соответств и с выпол-
няемыми ими функциями. По этой классификации среди белков выделяют
следующие группы: 1) каталитически активные белки; 2) белки гормоны;
3) регуляторные белки; 4) защитные белки; 5) токсические белки; 6) транс-
портные белки; 7) структурные белки; 8) сократительные белки; 9) рецептор-
ные белки; 10) белки-ингибиторы ферментов; 11) белки вирусных оболочек;
12) белки с иными функциями.
Конечно, деление белков на поименованные выше группы представляе
классификацию еще далекую от совершенства, и в случае б тфункциональных
белков приходится отдавать предпочтение одной, естественно, главной функ-
ции. Но в каждой из перечисленных уже удается подметить некоторые черты
общности структуры, свойств и функциональной акти ности включенных в ее
состав конкретных белков. Это позволяет более глубоко понять соотношение
82
структуры и функции в белковых молекулах, открывает возможности для
обобщения материалов, касающихся взаимодействия белков с соединениями
других классов (нуклеиновыми кислотами, липидами и др.), дает основание
для обсуждения закономерностей эволюции белковых тел, структурных и ге-
нетических основ их видовой специфичности и других насущных для химии
белков и биологии в целом проблем.
ХАРАКТЕРИСТИКА ОТДЕЛЬНЫХ ГРУПП БЕЛКОВ
Каталитически активные белки. Массовая расшифровка первичной и чет-
вертичной структур каталитически активных белков (ферментов) и исчерпы-
вающие сведения о третичной структуре некоторых из них позволили прийти
к обобщениям, касающимся специфики строения белков, относящихся к этой
группе, и связи последней с каталитической функцией этих белков. Не вдаваясь
в детали (см. гл. III), отметим, что специфика строения ферментов сводится
к следующему.
а-Спиральная и Р-складчатая структуры, представленные в том или ином
количестве в их молекулах, тесно прилегая друг к другу и чередуясь, упаковыва-
ются в блоки, обладающие функциональной активностью. Даже неупорядочен-
ные фрагменты полипептидной цепи частично, но своеобразно, структурированы,
образуя, например, ю-петли и другие квазиструктурные элементы. Радикалы
аминокислот, несущие заряды, направлены, как правило, к поверхности глобулы,
и только те из них, которые имеют функциональное значение и ассоциированы
с другими полярными радикалами, ориентированы внутрь глобулы. В противо-
положность этому на поверхности глобулы обнаруживается ограниченное число
гидрофобных радикалов аминокислот; большинство из них обращено внутрь
молекулы, где они объединяются в одно или несколько гидрофобных ядер.
У многих белков-ферментов указанные ядра сходны и служат, если их
несколько, центрами для формирования двух- или многоядерной белковой
глобулы. Так построены многие ферменты-мономеры (см. рис. 34, структура
рибонуклеазы и лизоцима). На границе двух (или более) оформленных или
отчетливо не выраженных частей молекулы фермента располагается активный
центр, локализованный в щели или впадине. Тлубина, форма и размеры
последней соответствуют пространственной структуре субстрата, чем обес-
печивается специфичность действия каталитически активных белков.
Структура самого активного центра организована столь тонко, что делает
возможным строго координированное в пространстве и во времени осуществ-
ление каталитического акта. Большинство ферментов располагает аллостери-
ческими центрами, которые служат для контакта с аллостерическими регулято-
рами их активности. Большая часть каталитически активных белков обладает
четвертичной структурой, которая в значительной мере предопределяет спо-
собность ферментов образовывать изоферменты; с изучением последних свя-
зана новая и увлекательная область ферментологии.
Перечисленные основные особенности строения ферментов показывают,
что между структурой их молекул и способностью ускорять протекание соот-
ветствующих химических реакций существует теснейшая взаимосвязь, харак-
терная именно для этой обширной группы белковых тел.
Белки-гормоны. Характерной особенностью этой группы белков является
способность воздействовать на фундаментальные механизмы регуляции обмена
веществ: проницаемость клеточных мембран и биосинтез вторичных посред-
ников.
83
Изучены структура и биологическая активность нескольких десятков бел-
ковых гормонов (см. гл. XII). Молекулярные массы подавляющего’их числа
лежат между 20 000 и 30 000 Да. Самой важной особенностью белковых гор-
монов является наличие в составе их полипептидных цепей относительно
небольших фрагментов (до нескольких десятков аминокислотных остатков),
которые являются носителями гормональной активности, тогда как остальная
часть полипептидной цепи несет какие-то иные функции, в частности видоспе-
цифические. В составе белковых гормонов выявлены якорные площадки,
обеспечивающие их соединение с рецептором гормона. Их вторичная структу-
ра дополняет или продолжает таковую рецептора гормона, в результате чего
возникает комплементарно завершенный комплекс, необходимый и достаточ-
ный для формирования биологического сигнала.
Регуляторные белки. Исследование группы регуляторных белков осуществ-
ляется в последние годы необычайно интенсивно, так как их функциональная
активность связана с репрессией и дерепрессией генома и регуляцией таких
важнейших процессов, как рост, развитие и морфогенез растений и животных.
Одной из наиболее изученных подгрупп указанных белков являются гисто-
ны (см. с. 81), локализованные в хроматине клеточных ядер, где они ассоци-
ированы с ДНК. Гистоны, входящие в состав хроматина, не отличаются
большим разнообразием и отнесены к пяти видам, которым различные
авторы в разное время присвоили те или иные названия или индексы
(табл. 9).
Таблица9
Классификация, номенклатура и физико-химическая характеристика гистонов
Номенклатура !• Число аминокислот- ных остатков
историческая по Иван и др. по Джонсу и Батлеру по Рассмус- сену и др. современная предполагае- мая по IUPAC » U 3 и в о я 5 Молекулярная масса
Лизиновые Богатые ала, очень богатые лиз /1 I Н1 КАР 20 21000 215
Умеренно лизиновые Богатые лей Богатые сер /2п f2b 11*1 11*2 Н2я H2Z> LAK KAS 1,2 2,5 14500 13800 129 125
Аргининовые Богатые глу и арг Богатые гли и арг fl f2al III IV НЗ Н4 ARE GRK 0,7 0,8 15300 11300 135 102
Примечание. Трехбуквенная номенклатура гистонов основана на перечислении в их составе трех преоблада-
ющих аминокислот (в порядке убывания), где А—аланин, G—глицин, Е—глутаминовая кислота, К—лизин,
L—лейцин, Р—пролин, R—аргинин, S—серин. Однако, несмотря на наглядность, она пока не получила
распространения.
• Первичные структуры гистонов из различных биологических объектов
(эритроциты цыпленка, семенники карпа, зобная железа свиньи, теленка и бы-
ка, семена гороха и многие другие) выяснены и внутри каждого вида гистонов
оказались весьма сходными, более того—консервативными, за исключением,
пожалуй, гистона Н1, варьирующего как по молекулярной массе, так и по
последовательности аминокислотных остатков.
Что касается вторичной и третичной структур гистонов, то они характери-
зуются присутствием коротких а-спиральных участков и доминированием
неупорядоченной полипептидной цепи, образующей на участках, богатых
84
основными аминокислотами, растянутую ‘спираль с 2,5 аминокислотными
остатками на виток и шагом в 0,8 нм.
Соединяясь с ДНК ионными связями и силами слабых взаимодействий,
гистоны стабилизируют ее структуру. Естественно, что дестабилизация ДНК,
необходимая для проявления ее матричной активности, осуществима лишь
при ослаблении (в силу тех или иных причин) связей между ДНК и гистоном,
чем и определяется регуляторная роль гистонов в функционировании енома.
Согласно современным данным, первый уровень структуры хроматина ре-
ализуется в виде нуклеосом (см. гл. VI).
Другая подгруппа регуляторных белков, также локализованная в хромати-
не ядра клетки,—иегистоновые белки. Они изучены в гораздо меньшей степе-
ни, чем гистоновые. Эти белки крайне гетерогенны, так как при их фракцио-
нировании методами электрофореза и изоэлек рофокусирования в полиакри-
ламидном геле обнаружено около 500 полипептидов с молекулярными
массами от 5000 до 200000 Да. Некоторая часть негистоновых белков активно
участвует в дерепрессии генома, препятствуя образованию суперспирализован-
ной ДНК на тех ее участках, где они присоединились в S-периоде цикла
деления клетки.
Негистоновые белки группы высокой подвижности (HMG-белки), изучен-
ные в последние годы достаточно подробно, способствуют присоединению
гистонов к ДНК, формированию нуклеосом и взаимодействию с хроматином
гормон-рецепторных комплексов.
Перечень регуляторных белков, равно как и данные о соотношении их
структуры и функций, непрерывно расширяются. К категории регуляторных
белков относят: более десятка белковых факторов, участвующих в репликации
ДНК (см. гл. VI); белки, ковалентно связанные с ДНК и РНК вирусов и фагов
и инициирующие репликацию нуклеиновых кислот у них; более 50 ядерных
белковых факторов, усиливающих или ослабляющих транскрипционные процес-
сы (см. гл. VI), механизм узнавания которыми специфических последовательно-
стей нуклеотидных остатков в молекуле ДНК при посредстве определенных
олигопептидных фрагментов в составе белковых транскрипционных факторов все
более проясняется; более двух десятков фак оров инициации, элонгации и терми-
нации, контролирующих этапы сборки полип птидных цепей при биосинтезе
белков (см. гл. VII); белки теплового шока (а в более широком понимании—
стрессовые белки), возникающие при тепловом и иных стрессовых воздействиях,
защищающие клетки от повреждения и восстанавливающие метаболизм в них
после снятия физиологического стресса; G-белки (некоторые из них получены
в гомогенном виде и охарактеризованы), регулирующие биосинтез циклических
аденозин- и гуанозинмонофосфатов—вторичных посредников при передаче
гормональных и иных сигналов (см. гл. XIII); онкобелки, противоборство
которых с антионкобелками привод ит к злокачественному перерождению клеток;
кейлоны и антикейл >ны, имеющие отношение к регуляции пролиферации клеток.
Несомненно, что изучение структуры и функций перечисленных выше белков
открывает перспективу познания наиболее глубоких основ регуляции процессов
жизнедеятельности, в том числе на уровне генома; современные представления
о типах взаимодействия регуляторных белков с ДНК показаны на рис. 41.
Защитные белки. К группе защитных белков принадлежат антитела—
вещества белковой природы, вырабатываемые живот ым организмом в ответ на
введение антигенов. Взаимодействуя с госледними, они инактивируют их,
защищая таким образом организм от воздействия чужеродных соединений,
вирусов, бактерий, клеток и тканей. Для обозначения белков, синтезирующих-
ся в организме в ответ на антигенное воздействие, предложен термин—
иммуноглобулины (сокращенно 1g). Так как они впервые были обнаружены
85
Рис. 41. Варианты связывания регуляторных белков с ДНК
В левой части рисунка перекрещенные а-спирали регуляторного белка взаимодействуют с симметрично
расположенными центрами связывания в двойной спирали ДНК, локализованные в большом желобе ДНК.
Узнающая а-спираль представлена тёмным цилиндром, а расположенная над ней другая а-спираль (белый
цилиндр) помогает распознать связывающий центр. В средней части рисунка—рр'а-надвторичиая структура
с атомом Zn в центре (связан с двумя остатками цистеина (С) p-слоя и двумя остатками гистидина (Н)
в а-спирали), обеспечивающая электростатический тип связи с ДНК за счет диполя в pp'ot-структуре
(полярные области заштрихованы). В правой части рисунка—димер регуляторного белка, скрепленный
гидрофобной застежкой богатых лейцином а-спиралей; пунктиром показана ось вращательной симметрии;
заштрихованными прямоугольниками—высокоосновные фрагменты, прямо взаимодействующие с ДНК,
стрелками—парные полуцентры распознавания сайта связывания в ДНК
в медленно движущейся при электрофорезе белков сыворотке крови фра ции
у-глобулинрв, их называют также у-иммуноглобулинами.
Согласно современной классификации, существует 5 видов иммуноглобу-
линов (IgG, IgM, IgA, IgD и IgE), структурные элементы молекул которых
представлены легкими и тяжелыми полипептидными цепями (рис. 42). Их
молекулярные массы лежат в пределах 150000—950 000 Да. Тяжелые цепи
различных иммуноглобулинов обозначают строчными буквами греческого
алфавита соответственно прописным буквам латинского алфавита, которые
служат для указания вид: иммуноглобулина, т. е. для IgG—у (гамма); IgM—ц
(мю), IgA—а (альфа), IgD—5 (дельта) и IgE—е (эпсилон). Тяжелые полипеп-
тидные цепи (Н-цепи, от англ, heavy—тяжелый) у разных видов иммуно-
глобулинов отличаются друг от друга. Наоборот, легкие полипептидные цепи
(L-цепи, от англ, light—легкий) у разных иммуноглобулинов только 2 типов:
х (каппа) и X (ламбда). В свою очередь у некоторых иммуноглобулинов
о мечено существование подвидов, например IgGi, IgG2, IgG3 и IgG4, в небо-
льшой мере отличающихся друг от друга первичной структурой полипептид-
ных цепей. Основную массу иммуноглобулинов сыворотки крови человека
составляет IgG (75—85% от их суммы); IgA содержится в ней в количестве 7— 15%,
IgM—5—10%, IgD—0,3% и IgE—0,003% (тоже от суммы). IgA локализован,
кроме того, в секретах (слеза, слюна, желчь, кишечный сок и т. п.) и лимфе.
Первичная структура тяжелых и легких цепей многих иммуноглобулинов
выяснена: в тяжелых цепях зафиксировано от 439 до 450 (у-цепи) и от 568 до 576
(p-цепи) аминокислотных остатков, а в легких—от 208 до 220. Удивительной
особенностью является то, что как в легких, так и в тяжелых цепях иммуногло-
булинов выявлены вариабельные зоны, расположенные преимущественно в пер-
86
Тяжелая цепь
(446 аминокислотных
остатков)
СООН соон
Ин и KL—вариабельные домены
CL и Сн— онстантные домены
Рис. 42. Структура иммуноглобулина G, человек
Две легкие и две тяжелые полипептидные цепи попарно связаны дисульфидными (—S—S—Нвязями; в каждой из цепей есть
внутренние дисульфидные мостики, отграничивающие разнообразные домены; Сн., Сн», Сн’—константные домены тяжелой цепи
вой половине цепи, т. е. со стороны аминогруппы. В отличие от этого первичные
структуры легких и тяжелых цепей у различных иммуноглобулинов близки во
второй их половине, т. е. со сторонь С-концевой аминокислоты. Вариабельная
часть молекулы ответственна за взаимодействие иммуноглобулинов с разнооб-
разными антигенами, тогда как часть, отличающаяся относ тельным постоян-
ством первичной структуры, выполняет общие для всех иммуноглобулинов
функции (связывание комплемента, фиксация на мембранах).
Высшие параметры структуры у различных иммуноглобулинов еще более
однотипны и получили название иммуноглобулиновой унаковки, характер ду-
ющейся стандартным расположением доменов по отношению друг к другу
и общими закономерностями их пространственной ориентации.
Определенную роль в функционировании иммуноглобулинов играют углево-
ды, входящие в их состав в количестве от 2 до 12%. Они присоединяются
87
к легким и тяжелйм цепям иммуноглобулинов в основном по радикалам
аспарагина и в некоторых случаях—треонина и представлены олигосахаридами,
в состав которых входят N-ацетилглюкозамин, манноза, галактоза, фукоза
и сиаловая кислота в соотношении, в большинстве случаев близком к 4:3:2:1:1.
Таким образом, в строении иммуноглобулинов удивительно тонко сочета-
ется взаимосвязь структуры и функции и необыкновенно наглядно выступает
специфика молекулярной организации белков, характеризующихся определен-
ной, в данном случае защитной, биологической актив! остью.
Кроме иммуноглобулинов защитные функции выполняют белки системы
свертывания крови, интерфероны, интерлейкины, белки-антифризы, гаптог-
лобины, антигены тканевой совместимости, лизоцимы, антивирусные белки
растений и антибактериальные белки насекомых.
Токсические белки. Группа токсических белков в последние годы изучается
весьма интенсивно ввиду ее большого практического значения.
С точки зрения первичной структуры наиболее полная информация полу-
чена о токсических белках змей: она расшифрована у нескольких десятков
токсинов, полученных из ядов лесной, африканской, азиатской, египетской,
королевской, мозамбикской, индийской и других видов кобры, африканской
зеленой и черной древесных змей, морской змеи и др.
Крайне любопытно, что токсины ядов змей, характеризующиеся молекуляр-
ными массами от 6700 до 7000 Да, составлены в большинстве случаев из
60 аминокислотных остатков; примерно такую же величину имеют токсические
полипептиды скорпиона, пчелы и осы; близки к ним (45 аминокислотных остатков)
токсины из пшеничной муки (пуротонин А, летальный для пивных дрожжей),
морских актиний и омелы белой (вискотоксины). В подавляющем большинстве
они являются нейротоксинами, так как, взаимодействуя с холинэнергическими
белками, блокируют передачу нервных импульсов. Их нейротоксическое действие
зависит от третичной структуры, задаваемой в основном многочисленными (4—5)
дисульфидными мостиками (см. структуру эрабутоксина на рис. 40).
Почти столь же детально изучены высокомолекулярные белковые токсины
микроорганизмов и растений. Структура и механизм действия нескольких десятков
из них выяснены. В одних случаях они представлены мультимерами (дифтерийный
и холерный токсины, токсин шигеллы и др.), построенными из одной субъединицы
типа А (20,0; 28,0 и 32,0 кДа соответственно) и пяти субъединиц типа В (25,0; 12,0
и 7,7 кДа соответственно). Контактируя с клеточной поверхностью субъединицами
типа В, они вводят убъединицу типа А внутрь клетки, где она блокирует биосинтез
белков на рибосомах. В других—двухкомпонентны (растительные токсины—
рицин, абрин, модецин, лектин и др.), причем субъединица В связывается
с рецепторами клетки и обеспечивает перенос субъединицы А внутрь ее, что
приводит к инактивации 60S субчастицы рибосомы и прекращению синтеза белка.
В третьих—одноцепочечны (энтеротоксин из стафилококка, гемолизин из
кишечной палочки, стрептолизин и др.) и их крупные полипептидные цепи (от 34 до
ПОкДа) встраиваются в клеточную мембрану, образуют в ней поры, что
сопровождается лизисом клетки. Здесь, как и в случае нейротоксинов, прослежива-
ется тесная связь структуры и функции токсических белков.
Транспортные белки. Представителем транспортных белков является сыво-
роточный альбумин. Он выделен в высокоочищенном состоянии, его получение
налажено препаративно. Основной функцией этого белка является перенос
различных веществ, особенно жирных кислот. Кроме высших жирных кислот,
сродство к которым у сывороточного альбумина весьма высоко, он переносит
разнообразные анионы и катионы. Сывороточный альбумин связывает до
50% кальция, находящегося в крови, служит переносчиком ионов меди из
кишечника в печень, является носителем стероидных гормонов. Молекулярная
88
масса сывороточного альбумина человека ра на 65 000, изоэлектрическая точ-
ка (р/)—4,7 (при ионной силе 0,15). Он способен образовывать димеры
с константой седиментации 6,7S. Первичная структура сывороточного аль-
бумина человека, состоящего из 585 аминокислотных остатков, выяснена.
Из других белков сыворотки крови человека транспортными функциями
обладают церулоплазмин (М = 160000, р/=4,4)—переносит ионы меди из
печени в клеточные органеллы; трансферрин (М = 90000, р/=5,9)—переносит
ионы трехвалентного железа; Р-липопротеин (М = 3200 000, р/=5,4)—перено-
сит. липиды, жирорастворимые витамины и гормоны; та же функция свойст-
венна другим липопротеинам сыворотки крови. Липопротеины очень низкой
плотности у человека ежесуточно переносят 25—50 г эндогенных триглицерит
дов, а другие их виды—холестерол, Р-каротин, фосфолипиды, углеводороды
и ациклические спирты. Широко известными транспортными белками, перено-
сящими кислород, являются гемоглобины позвоночных и ряда низших живот-
ных, гемоцианины моллюсков, ракообразных, паукообразных и мечехвостов,
гемэритрины (коричневые дыхательные белки) кольчатых червей, хлорокруо-
рины (зеленые дыхательные белки) многощетинковых червей, гемованадины
(содержащие ванадий) морских животных (оболочников).
Транспортные белки характерны не только для биологических жидкостей.
В последние годы внимание исследователей особенно привлекли белки, встро-
енные в наружные и внутренние мембраны клеток и обеспечивающие перенос
через них разнообразных низко- и высокомолекулярных веществ. Эти белки
получили название порины, так как они образуют в мембранах поры, через
которые идет транспорт. Некоторые из этих белков (порин I из наружной
мемб эаны кишечной палочки—молекулярная масса 37205 Да, 340 аминокис-
лотных остатков; порин из митохондрий печени крысы—димер из двух
идентичных полипептидов и др.) выделены и охарактеризованы. Сюда же
относятся белки—транслоказы, механизм переноса которыми веществ через
мембраны ясен из рис. 43.
Рис. 43. Перенос веществ через мембраны прн посредстве белков:
А—вращающийся белок-переносчик; Б—подвижный белок-переносчик; В—пора, образован-
ная из белкоа-переносчиков
89
Структурные белки. Многочисленные и разнообразные белки несут в био-
логических объек ах структурные функции.
Эту роль выполняют прежде всего белки, являющиеся компонентами
различных биологических мембран, исключая, конечно, те из них, что облада-
ют иной функциональной активностью (мембранносвязанные ферменты, по-
ровые белки, белки переносчики, рецепторные белки и т. п.).
Данные о молекулярных массах структурных белков мембран про-
тиворечивы. У структурных белков митохондрий сердца и печени быка
они составляют от 20000 до 60000, а у бактерий—от 10000 до 160000 Да.
Структурные белки мембран отли аются резко выраженной способностью
к агрегации. При pH 12 они существуют в виде мономеров, но при
понижении значения pH олигомеризуются с образованием фибриллярных
структур и микрокристаллов. Более того, они способны соединяться
в стехиометрических соотношениях с другими белками (например, ми-
оглобином) и особенно с ферментами, характеризующимися мембранной
локализацией (цитохромами сх и Ь, цитохромоксидазой, малатдегидро-
геназой и др.), причем каталит ческая активность последних при этом
заметно изменяется. Поэтому полагают, что роль структурных белков
мембран не ограничивается только лишь закреплением ферментов в мем-
бране.
Свойства структурных белков мембран в определенн й мере предопределя-
ются их амин кислотным составом. В них содержатся в высокой пропорции
аминокислоты, обладающие гидрофобными радикалами (глицин и аланин—
10—15%, валин, лейцин и изолейцин—в среднем 5—6% каждый, а в ряде
случаев—до 9—13%), и сравни ельно невелико содержание основных и кис-
лых аминокислот. Но самое любопытное состоит в том, что гидр фобные
аминокислоты образуют в полипептидной цепи структурных белков мембран
локальные зоны (сегменты |, включающие 20 и более остатков только гид-
рофобных аминокислот и занимающие в общем до 20% от длины всей
полипептидной цепи. Это способствует возникновению гидрофобных центров
в молекулах структурных белков мембран, в том числе центров, локализован-
ных на поверхности глобулы. Указанное обстоятельство в известной мере
объясняет высокую способность этих белков к агрегации, а также то, что
субъединицы в олигомерах связаны силами слабых взаимодействий.
Из аминокислотного состава структурных белков мембран вытекает еще
одно существен ое следствие. Содержание в их составе аминокислот, препят-
ствующих спирализации, таково, что доля а спирали )й структуры может
составить в среднем 40% полипептидной цепи. Действительно, эксперимен-
тальные данные, полученные методом инфракрасной спектрофотометрии, дис-
персии оптического вращения и кругового дихроизма, убеждают в том, что
значительная часть полипептидной цепочки структурных белков мембран
находится в а-с иралы й форме (от 30 до 50%).
Наконец, еще одно специфическое свойство структурных белков мембран
состоит в том, что все они независимо от источника выделения очень охотно
связывают фосфолипиды при нейтральных значениях pH. Так как последние
сохраняют при этом свой заряд, связывание структурных белков мембран
и фосфолипидов идет за счет сил слабых взаимодействий, т. е. при посредстве
гидрофобных центров белков и углеводородных радикалов фосфолипидов.
Кроме мембранных белков структурные функции несут белки межклеточ-
ного матрикса (коллаген, ретикулин), кристаллины, а также белки ядерного
матрикса и цитоплазматического скелета. Число последних, частично или
полностью охарактеризованных, достигло сейчас уже нескольких десятков
и это одна из самых острых проблем современной белковой химии.
90
Сократительные белки. Сократительные белки локализованы как в мышеч-
ных клетках животных, так и в немышешых клетках примитивных и высоко-
организованных живых существ. К их числу относятся миксомиозин (нитевид-
ный белок с диаметром молекулы 7,0 нм, выделенный из плазмодия гриба
физарума); белки микротрубочек (диаметр субъединицы в трубочке 4,5—
7,0 нм), обеспечивающих движение протоплазмы в растительных и животных
клетках, миозино- и актомиозиноподобные белки фибриллярного аппарата
амебы, ответственные за пер текание ее протоплазмы; белки трубчатых фиб-
рилл, участвующих в движении хромосом в процессе деления клетки; белки
центральных и периферических фибрилл жгутиков и ресничек простейших,
а также жгутиков сперматозоидов’ актин и миозин мышечных волокон.
Кроме ко трактильных свойств, подавляющее большинство перечислен-
ных выше сократительных белков обладает аденозинтрифосфатазной актив-
ностью, т. е. соединяет в себе два качества—способность совершать ме-
ханическую работу и ускорять химическую реакцию. Это свойство сокра-
тительных белков было открыто в 1939 г. В. А. Энгельгардтом
и М. Н. Любимовой, которые в октябрьском номере журнала «Nature» (т. 144,
с. 688) опубликовали результаты своих опытов в статье под названием «Ми-
озин и аденозинтрифосфатаза». Таким образом, эти белки характеризуются
механохимическими свойствами. Общей особенностью, присущей сократи-
тельным белкам, является также то, что их функция зависит от действия
ряда доп лнительных белков—активаторов и регуляторов их активности
и от присутствия низкомолекулярных соединений (Mg ,Са2+, АДФ). Так,
например, деятель ость актомиозинового комплекса мышц регулируется тро-
помиозином и трот нином.
Конкретные сведения о сократительных белках, выделенных из тех или
иных источников, отличаются различной степенью полноты и достоверности,
а в. ряде случаев противоречивы. Наиболее изучены миозин и актин мышц.
Миозин представляет собой волокнистый белок с молекулярной массой
500000 Да, а актин—глобулярный белок с молекулярной массой от 46000 до
58 000 Да (рис. 44). Первичная структура фрагмента цепи миозина протяжен-
ностью до 200 аминокислотных остатков, что составляет примерно десятую
часть его полипептидной цепи, выяснена. Удалось расшифровать также пер
вичную структуру актина из мышц кролика—белка, состоящего из 374 ами-
нокислотных остатков. Актин обладает сильно выраженной способностью
к агрегации, протекающей с образованием надмолекулярных структур в виде
супер пирализованных длинных двойных нитей.
О других сократительных белках свед ния менее полны. Миксомиозин
имеет молекулярную массу около 6 000 000 Да и размеры молекул 7 х 400—
500 нм. Сократительный белок из ресничек простейших ближе к миозину
мышц по молекулярной массе (400000), аминокислотному составу и свойст-
вам. Однако контрактильный белок из фибрилл митотического веретена гло-
булярен (диаметр глобулы от 15 до 20 нм, М = 880 000); для него характерна
субъединичная структура. Тубулин микротрубочек представлен димером с мо-
лекулярной массой 110000 Да.
Выяснение структуры и особенно механизма действия сократительных
белков представляет огромный интерес и еще очень далеко от завершения.
Рецепторные белки. Выделение рецепторных белков в особую группу свя-
зано с интенсивным исследованием механизмов передачи информации в био-
логических системах.
Мишенью действия агентов, несущих сигнальные функции, являются ре-
цепторные белки, локализованные в мембранном аппарате клетки. Одним
из примеров может служить рецептор ацетилхолина—медиатора передачи
91
I п ш
Рис. 44. Схема строения сократительных белков мышц:
Z—структура молекулы миозина, составленной из двух полипептидных цепей, включающих около 1800 ами-
нокислотных остатков каждая. Большая часть молекулы представлена суперспиралью (горизонтальная часть
рисунка), образованной двумя почти полностью спирализованными полипептидными цепями; меньшая—
двумя фрагментами полипептидных цепей в глобулярном состоянии, где содержание а-спиралей достигает
30% (головка, ответственная за связывание актина и АТФазную активность). Хвостовая часть (легкий
меромиозин) отчленяется от головной части (тяжелый меромиозин) в результате протеолитического расщеп-
ления трипсином. Глобулярная часть (головка) отщепляется при протеолизе папаином; II—суперспираль,
составленная из глобул актина. Каждый шар на рисунке соответствует молекуле актина (М—46000);
III—точка прикрепления головной части молекул миозина (показаны стрелками) к суперспирали актина
в процессе мышечного сокращения
нервного импульса (см. гл. III). Он представлен олигомерным белком
(М = 285 000), составленным из пяти субъединиц (а2Руб), образующих ионный
канал (рис. 45). В отсутствие ацетилхолина этот канал закрыт, но после
рецепции секретируемого ацетилхолина он открывается на короткое время (до
момента разрушения медиатора ацетилхолинэстеразой) и пропускает Na+, что
сопровождается изменением степени поляризации клеточной мембраны и пе-
редачей сигнала через синапс нервной клетки.
Рецепция того или иного вида энергии внешней среды органами чувств, как
показали работы последнего времени, имеет единый механизм, складыва-
ющийся из двух этапов: 1) восприятия энергии стимула рецепторными белка-
ми; 2) преобразования энергии стимула особыми белковыми молекулами
в специфическую информацию и передача ее в центральную нервную систему.
Сведения о рецепторных белках и механизме их взаимодействия с сиг-
налами внешней среды весьма скудны. Представителем фоторецепторных
белков является опсин, сущ ствующий в виде соединения с ретиналем (родо-
псин) и изменяющий свою конформацию, что связано с преобразованием
светового сигнала в нервные импульсы в процессе зрительного акта (см.
гл. IV, рис. 60). Из вкусовых рецепторных белков изучен сладкочувствитель-
ный белок (М = 150 000). Его аминокислотный состав характеризуется высоким
содержанием дикарбоновых аминокислот и их амидов, лизина, лейцина, вали-
на и пролина. Он способен связывать моносахариды и дисахариды. Обонятель-
92
Рис. 45. Структура ацетилхолинового рецептора
В верхнем левом углу—олигомерная форма рецептора; показана пространственная структура а-субъединицы (М =40000),
входящей в состав пентамера; окружность в центре пентамера—ионный канал диаметром 0,65 нм. В нижней части рисунка—
развернутая структура а-субъединицы рецептора, встроенной в плазматическую мембрану клетки; спиральные участки полипеп-
тидной цепи а-субъединицы обогащены гидрофобными аминокислотными радикалами, взаимодействующими с остатками высших
жирных кислот фосфолипидной мембраны; с N-конца полипептидные цепи субъединиц гликозилированы и углеводная компонента
в целом составляет примерно 20% массы рецептора; С192 и С193—остатки цистеина, участвующие в рецепции ацетилхолина
ный белок выделен из половых сенсилл самцов дубового шелкопряда; он
взаимодействует с половым феромоном самок этого насекомого. В восприя-
тии звуковых колебаний и их преобразовании в нервные импульсы придают
большое значение холинорецепторным белкам, которые, как показано недав-
но, ответственны за проницаемость клеточной мембраны.
Большие перспективы открываются при изучении рецепторных белков
насекомых, особенно тех, которые осуществляют рецепцию аттрактантов
и репеллентов.
Белки—ингибиторы ферментов. Вещества белковой природы составляют
самую многочисленную группу ингибиторов ферментов, причем наиболее
изучены из ее состава белки, ингибирующие активность протеаз. Белковые
ингибиторы образуют с протеолитическими ферментами стойкие при физио-
логических условиях комплексы, в составе которых фермент полностью или
частично теряет свою активность. Так как константы диссоциации этих ком-
плексов лежат в пределах 10“12—10“9 моль, они действительно отличаются
высокой прочностью, а ингибиторы, входящие в их состав,— большой мощ-
ностью действия.
Выделено и изучено несколько десятков белковых ингибиторов, подав-
ляющих активность трипсина, химотрипсина, карбоксипептидазы, калликре-
ина, эластазы, плазмина и других протеолитических ферментов. Многие из
них получены в гомогенном кристаллическом состоянии. Молекулярные мас-
сы ингибиторов белковой природы колеблются от нескольких тысяч до не-
скольких сотен тысяч, но в основном они представлены белками с моле-
кулярными массами около 6000 Да. Многие из белков—ингибиторов фер-
ментов—являются гликопротеинами. Первичная структура нескольких
93
десятков ингибиторов расшифрована; среди них—ингибиторы трипсина I и II
из поджелудочной железы свиньи и быка, соевых бобов, арахиса и фасоли,
ингибиторы протеиназ из яда гадюки, лимских бобов и ананаса, ингибитор
химотрипсина из картофеля, инактиватор калликреина (тразилол) из легких
быка, ингибитор субтилизина из стрептомицета и др.
Белки вирусных оболочек. Из многочисленных вирусов выделены и изучены
разнообразные белки. Их молекулярные массы колеблются от нескольких
тысяч до нескольких десятков тысяч дальтон. Наряду с основной функцией
(защита нуклеиновой кислоты) некоторые белки вирусных оболочек необ-
ходимы для созревания вирусных частиц, обладают ферментативной актив-
ностью (нейраминидаза, лизоцим и обратная транскриптаза в составе ряда
вирусов) и др.
Первичная структура белковых субъедини многих вирусов выяснена. К их
числу относятся субъединицы трех штаммов вируса табачной мозаики
(159 аминокислотных остатков), субъединицы бактериофагов/? и/2 (129 оста-
тков), бактериофага (131 остаток), бактериофага ZI (50 остатков), субъеди-
ницы вируса желтой мозаики турнепса (190 остатков) и вируса ядерного
полиедроза тутового шелкопряда (244 остатка), су ъединицы бактериофага fd
(50 остатков), а также ряд белков вируса гриппа (гемагг потинин—566
и PV2—759 аминокислотных остатков).
Удивительной особенностью вирусных белков является их способность
к агрегации, вследствие чего даже в отсутствие вирусных нуклеиновых кислот
они способны к самосборке в соответствующие, харак ерные для данного
вируса морфологические структуры (тени вирусов и фагов). Кроме того, их
структура такова, что концевые аминокислоты, как правило, маскированы
в глубине молекулы и труднодоступны для определения.
Белки с иными функциями. Несомненно, что и далее из крайне многочислен-
ных конкретных представителей класса белков могут вычленяться новые
группы с ясно выраженной функциональной активностью и связанной с нею
спецификой структуры и свойств. Так, например, уже сейчас обособляются
группа гемоглобинов, группа фибриллярных белков, группа рибосомальных
белков и др. Это лишь подтверждает высказанное выше мнение о том, что
классификация белков пережив ет сейчас период становления.
ГЛАВА Ш
ФЕРМЕНТЫ
ОБЩЕЕ ПОНЯТИЕ О ФЕРМЕНТАХ
Важнейшим свойством ряда белков, как уже отмечено, является их катали-
тическая активность, теснейшим образом связанная с общими особенностями
их структуры.
Каталитически активные белки называют ферментами (от лат. fermentum—
закваска) или энзимами (отгреч. ен—внутри, зим—закваска). Как вытекает
из происхождения названий этих веществ, первые сведения )б их существова-
нии были получены при изучении процессов брожени .
Роль ферментов в жизнедеятельности животных растений и микроорганиз-
мов колоссальна. Благодаря каталитической функции разнообразные фермен-
ты обеспечивают быстрое протекание в организме или вне его огромного
числа химических реакций. Складываясь в единый ансамбль саморегулиру-
емых биохимических процессов, эти ре кции преобразования веществ состав-
ляют материальную и эне гетическую основу непрерывного самообновл ния
белковых тел, т. е. самой сущности жизненных явлений. Поэтому ферменты
«есть возбудители всех хи ических превращений» (И.П. Павлов), трансдук-
торы (т. е. датчики) в регуляции обмена вещест .
В настоящее время в биологических объектах обнаружено несколько тысяч
индивидуальных ферментов, а несколько сотен из них выделено и изучено.
Подсчитано, что живая клетка может содержать до 1000 различных фермен-
тов, каждый из которых ускоряет ту или иную химическую реакцию.
Биологические катализаторы (ферменты) по раду признаков резко отличаются
от неорганических катализаторов, хотя те и другие лишь ускоряют достижение
равновесия в хими1 еских про ессах, которые протекают сами по себе, но с очень
малыми скоростями. Как и катализаторы еорганической природы, биокатализа-
торы не вызывают каких-либо химических реакций, а лишь ускоряют существую-
щие. Первое различие состоит в том, что по сравнению с катализаторами
неорганической природы ферменты «работают» в очень мягких условиях (низкая
температура, нормальное давление, невысокие значения pH среды и т. п.) и очень
интенсивно. Так, например, гидролитический распад белка до аминокислот
в присутствии неорганических катализаторов (крепких кисло или щелочей)
осуществляется при температуре 100° С и выше за несколько десятков часов. Этот
же процесс при каталит ческом участии специфи еских ферментов протекает за
десятки минут при температуре 30—40° С. Для гидролиза крахмала, как
указывал еще Й. Берцелиус (1836), при нагревании в растворе кислоты нужно
несколько часов, а при участии соответствующ о фермента этот процесс идет при
комнатной температуре всего несколько минут. Ионы Fe каталитически ускоряют
разложение Н2О2 на Н2О и О2. Однако атомы того же Fe, но в составе фермента
каталазы действуют в 10 млрд, раз нергичнее, и всего 1 мг Fe в ферменте
способен заменить в этой реакции 10 т н органического Fe. Таким образом,
95
исключительно высокая каталитическая активность, проявляемая в условиях
нормальной температуры и давления, отличает биокатализаторы от неор-
ганических катализаторов.
Второе различие аключается в том, что ферменты обладают необыкновен-
но высокой специфичностью действия, чего не наблюдается у катализаторов
неорганической природы. Каждый фермент каталитически ускоряет, как пра-
вило, одну-единственную химическую реакцию или в крайнем случае группу
реакций одного типа.
Наконец, ряд различий между биокатализаторами и неорганическими ка-
тализаторами связан с белковой природой ферментов. Сюда относятся термо-
лабильность, зависимость активности от pH среды и наличия активаторов или
ингибиторов и др.
Самая существенная разница между ферментами и обычными катализатора-
ми вскрыта лишь в последние годы. Она состоит в том, что благодаря
уникальной структуре каждого фермента процесс ферментативного катализа
предстае перед нами как серия элементарных превращений вещества, строжай-
шим образом организованных в пространстве и времени. Кооперативность
и жесткая запрограммированность этапов действия—вот что отличает меха-
низм биокатализа от действия катализаторов иной природы, хотя это не
исключает некоторой степени вариабельности как структуры самого фермента,
так и строения промежуточных продуктов в процессе ферментативного катализа.
В природе под каталитическим воздействием ферментов осуществляются
процессы гидролиза, фосфоролиза, переноса различных групп (метильные
радикалы, остатки фосфорной кислоты и т. д.), окисления и восстановления,
расщепления и синтеза, изомеризации и т. п. Практически все химические
преобразования в живом веществе протекают с помощью ферментов. Естествен-
но поэтому, что каталитическая функция ферментов лежит в основе жизнедея-
тельности любого организма. При посредстве ферментов реализуется влияние
как внутренних, генетических, так и внешних, природных факторов на развитие
организма. Благодаря контакту ферментов с лекарственными веществами
и антибиотиками достигается такое изменение ферментативных процессов,
которое способствует излечению от болезней, в то же время изменение фермента-
тивной активности под влиянием микробных токсинов и иных ядов ведет к гибели
организма. Стимуляция роста животных и растений разнообразными препарата-
ми, применяемыми в сельском хозяйстве, в болыпинс ве случаев основана на их
воздействии на процесс биосинтеза или активность тех или иных ферментов.
Тончайшие различия строения ряда ферментов определяют видовые особенности
организмов, а в нарушении биосинтеза некоторых из них заложена причина
возникновения наследственных и других заболеваний. Все это свидетельствует об
огромном значении ферментов для биологии, сельского хозяйства и медицины.
Будучи выделены из организма, ферменты не утрачивают способности
осуществлять каталитическую функцию. На этом основано их практическое
применение в химической, пищевой, легкой и фармацевтической промыш-
ленности. Особое значение для химического производства имеет специфич-
ность ферментов: ведь до 80% затрат в производстве многих химических
вещее в приходится на отделение примесей, возникших в результате побочных
реакций. Проведение синтеза при посредстве высокоспецифичного фермента,
ускоряющего только ту реакцию, которая ведет к образованию нужного
продукта, упрощает технологический процесс. Кроме того, ферменты позволя-
ют осуществлять ряд процессов, выполнение которых обычными методами
органического синтеза остается пока нерешенной проблемой. Так обстоит
дело, например, при получении лекарственных препаратов путем фермента-
тивной трансформации стероидов.
96
МЕТОДЫ ВЫДЕЛЕНИЯ И ОЧИСТКИ ФЕРМЕНТОВ
Долгое время вполне, обоснованно считали, что все ферменты—тела бел-
ковой природы. Однако в начале 80-х годов была неожиданно открыта способ-
ность низкомолекулярных рибонуклеиновых кислот ускорять реакцию превра-
щения предшественников РНК в функционально значимый продукт, т. е.
возникло представление о полирибонуклеотидной природе некоторых фермен-
тов, названных рибозимами (см. гл. VI).
Хотя уже осуществлен лабораторный синтез ряда ферментов—рибонукле-
азы, лизоцима, ферредоксина и цитохрома с, трудно ожидать, что синтетиче-
ское получение ферментов получит широкое распространение в ближайшие
десятилетия ввиду его сложности и дороговизны. Поэтому единственный
реальный в настоящее время способ получения ферментов—это выделение их
из биологических объектов.
Выделяют ферменты так же, как и другие белки, хотя есть приемы,
применяемые преимущественно для ферментов. Из них можно отметить экс-
тракцию глицерином, в котором сохраняются нативные свойства ферментов,
а также метод ацетоновых порошков, состоящий в осаждении и быстром
обезвоживании при температуре не выше —10° С тканей или вытяжек из них,
содержащих ферменты. К их числу относится также получение ферментов
путем адсорбции с последующей элюцией (снятием) с адсорбента. Этот метод
был введен в химию ферментов А. Я. Данилевским и дал мощный толчок
развитию ферментологии. Сейчас адсорбционный метод выделения и очистки
ферментов разработан детально. Наряду с ним широко применяют метод
ионообменной хроматографии, метод молекулярных сит, электрофорез и осо-
бенно изоэлектрофокусирование. Одна из остроумнейших модификаций ад-
сорбционного метода—аффинная хроматография, где адсорбентом служит
вещество, с которым фермент взаимодействует избирательно. В результате
лишь один этот фермент задерживается на колонке, а все сопутствующие ему
выходят с током проявителя. Изменяя характер проявителя, исследуемый
фермент элюируют с колонки. Этим методом достигают очистки фермента
в несколько тысяч раз, применяя всего лишь одноэтапную (аффинная сорб-
ция—элюция) схему выделения.
Для успешного выделения ферментов из клеточного содержимого необ-
ходимо Очень тонкое измельчение исходного материала, вплоть до разруше-
ния субклеточных структур: лизосом, митохондрий, ядер и др., которые несут
в своем составе многие индивидуальные ферменты. Особое внимание при
выделении ферментов уделяют проведению всех операций в условиях, ис-
ключающих денатурацию белка, так как она всегда связана с потерей фермен-
тативной активности. Этому способствует проведение операций в присутствии
защитных добавок, в частности HS-содержащих соединений (цистеина, глута-
тиона, меркаптоэтанола, цистеамина, дитиотреитола и др.):
HS—СН2—СН2—NH2 HS—СН2—СН(ОН)—СН(ОН)—СН2—SH
Цистеамин ' Дитиотреитол
Очень важно поддерживать на всех этапах выделения ферментов низкую
температуру, так как некоторые из них даже при —80° С теряют активность.
Для оценки гомогенности ферментного препарата прибегают к обычным
методам белковой химии. Переломным моментом в усовершенствовании
методов получения высокоочищенных, гомогенных препаратов ферментов
было открытие способности их кристаллизоваться, осуществленное впервые
в 1906 г. А. Д. Розенфельдом (им была получена в виде кристаллов оксидаза
из корней редьки) и приобретшее с 1926 г. широкую известность после работы
97
4—3502
Д. Самнера по получению кристаллической уреазы из бобов канавалии. Нере-
дко о степени чистоты ферментного препарата судят по его биологической,
активности; если активность при дальн йшей очистке не возрастает, препарат
можно считать гомогенным. Из 2003 включенных в список ферментов более
1500 выделено и в той или иной мере очищено, третья часть их закрис алли-
зована, у нескольких сотен выяснена первичная, а у нескольких десятков—
третичная структура.
СТРОЕНИЕ ФЕРМЕНТОВ
По строению ферменты могут быть однокомпонентными, простыми бел-
ками, и двухкомпонентными, сложными белками. Во втором случае в составе
фермента обнаруживается добавочная группа небелк вой природы.
В разное время возникли различные наименования белковой части и доба-
вочной группы в двухкомпонентных ферментах. Все они до сих пор употребля-
ются в литературе, например:
Фермент в целом Белковая часть Добавочная группа
Симплекс Ферон (носитель) Агон (активная группа)
Холофермент Апофермент Кофермент
Добавочную группу, прочно связанную, не отдели мую от белковой части,
называют простетической группой; в отличие от этого добавочную группу,
легко отделяющуюся от апофермента и способную к самостоятельному су-
ществованию, обычно именуют коферментом.
Химическая природа важнейших коферментов была выяснена в 30-е годы
нашего столетия благодаря трудам О. Варбурга, Р. Куна, П. Каррера и др.
Оказалось, что роль коферментов в двухкомпонентных ферментах играют
большинство витаминов (Е, К, Q, Вх, В2, Вб, В12, С, Н и др.) или соединений,
построенных с участием витаминов (коэнзим А, НАД+ и т. п.). Формулы
упомянутых коферментов приведены в гл. IV. Кроме того, функцию кофер-
ментов выполняют такие соединения, как HS-глутатион, многочисленная
группа нуклеотидов и их пр изводных, фосфорные эфиры некоторых моноса-
харидов и ряд других веществ.
Характерной особенностью двухкомпонентных ферментов является то, что
ни белковая часть, ни добавочная группа в отдельности не о >ладают заметной
каталитической активностью. Только их комплекс проявляет ферментативные
свойства. При этом белок резко повышает каталитическую активность доба-
вочной группы, присущую ей в свободном состоянии в очень малой степени;
добавочная же группа стабилизирует белковую часть и делает ее менее уязви-
мой к денатурирующим агентам Таким образом, хотя непосредственным
исполнителем каталити еской функции является простетическая группа, об-
разующая каталитический центр, ее действие немыслимо без участия полипеп-
тидных фрагментов белковой части фермента. Более того, в апоферменте есть
участок, характеризующийс специфической структурой, избирательно свя-
зывающий кофермент. Это так называемый кофермент связывающий домен;
его структура у различных апоферментов, соединяющихся с одним и тем же
коферментом, очень сходна. Ткковы, например, пространственные структуры
нуклеотидсвязывающих доменов ряда дегидрогеназ (см. рис. 53, с. 119).
Иначе обстоит дело у однокомпонентных ферментов, не имеющих доба-
вочной группы, которая могла бы входить в непосредственный контакт с пре-
образуемым соединением. Эту функцию выполняет часть белковой молекулы,
называемая каталитическим центром. Предполагают, что каталитический
центр однокомпонентного фермента представляет собой уникальное сочета-
ние нескольких аминокислотных остатков, располагающихся в определенной
части белковой молекулы. Сказанное иллюстрирует рис. 34, Г, на котором
приведена третичная структура химотрипсиногена—предшественника одно-
компонентного фермента: аминокислотные радикалы остатка сер и двух оста-
тков гис, расположенные в разных точках полипептидной цепи, сблизились
здесь на расстояние в несколько десятых долей нанометра, предобразовав
каталитический центр фермента. На этом же рисунке (Б и В) приведена
третичная структура молекул еще двух ферментов—лизоцима и рибонуклеа-
зы; у них ясно просматривается двухъядерная, двухлопастная конструкция
молекул с углублением (щелью) на границе двух лопастей, в котором распола-
гается каталитический центр.
Чаще всего в каталитических цен рах однокомпонентных ферментов встре-
чаются остатки сер, гис, три, арг, цис, асп, глу и тир. Радикалы перечисленных
аминокислот выполняют здесь ту же функцию, что и кофермент в составе
двухкомпонентного фермента.
Аминокислотные остатки, образующие каталитический центр одноком-
понентного фермента, расположены в различных точках единой полипептид-
ной цепи (см. рис. 34). Поэтому каталитический центр возникает в тот момент,
когда белковая молекула приобретает присущую ей третичную структуру.
Следовательно, изменение третичной структуры фермента под влиянием тех
или иных факторов может привести к деформации каталитического центра
и изменению ферментативной активности
Кроме каталитического центра, образованного сочетанием аминокислот-
ных радикалов или присоединением кофермента, у ферментов различают еще
два центра: субстратный и аллостерический (см. ниже, рис. 51).
Под субстра ным центром понимают участок молекулы фермента, ответ-
ственный за присоединение вещества (субстрата), подвергающегося фермента-
тивному превращению. Часто этот участок называют «якорной площадкой»
фермента, где, как судно на якорь, становится субстрат. Во многих случаях
прикрепление субстрата к ферменту идет за счет взаимодействия с е-амино-
группой радикала лиз, располож иного в субстратном центре. Эту же роль
может выполнять СООН-группа глу, а также HS-группа цис. Однако работы
последних лет показали, что гораздо большее значение здесь имеют силы
гидрофобных взаимодействий и водородные связи, возникающие между ради-
калами аминокислотных остатков субстратного центра фермента и соответст-
вующими группировками в молекуле субстрата.
Понятие о ка алитическом и субстратном центре не следует абсолютизиро-
вать. В реальных ферментах субстратный центр может совпадать (или пере-
крываться) с каталитическим центром. Более того, каталитический центр
может окончательно сформироваться в момент присоединения субстрата. Поэ-
тому часто говорят об активном центре фермента, представляющем сочетание
первого и второго. Активный центр у ф рмент 1 располагается на дне щели
при двухъядерной структуре, например у лизоцима и рибонуклеазы, или на
дне глубокой впадины, как у химотрипсиногена (см. рис. 34).
Аллостерический центр представляет участок молекулы фермента, в ре-
зультате присоединения к которому определенного низкомолекулярного (а
иногда—и высокомолекулярного) вещества изменяется третичная структура
белковой молекулы. Как следствие этого изменяется конфигурация активного
центра, сопровождающаяся либо увеличением, либо снижением каталитиче-
ской активности фермента. Это явление лежит в основе так называемой
аллостерической регуляции каталитической активности ферментов.
99
Рис. 46. Строение, некоторых ферментов-мультимеров:
Л—молекула глутаматдегидрогеназы, составленная из б субъединиц (М = 56000), расположенных по
граням правильного тетраэдра; молекулярная масса фермента 336000; Б—модель молекулы РНК-
полимеразы из пяти субъединиц; две типа а (М—по 40000), две типа 0 (0—М = 150000 и 0'—М = 155000)
и о-фактор (М=90 000); В—схема строения половины молекулы каталазы; каждая субъединица пред-
ставлена дважды ‘ изогнутой палочковидной частицей; Г—полиферментный комплекс, ускоряющий
реакцию окислительного декарбоксилирования (см. с. 101, 160, 352—354); Д—аспартат—карбамцлт-
рансфераза, составленная из 6 каталитических (М = 33 500, обозначены Ci—С6) и 6 регуляторных
(М = 17 000, обозначены Rj—R*; R3 и Re не видны) субъединиц
Значения молекулярных масс ферментов колеблются в широких пределах:
от нескольких тысяч до нескольких миллионов. В природе насчитывается
несколько десятков ферментов, обладающих сравнительно небольшими моле-
кулами (до 50 тыс.). Строение некоторых из них показано на рис. 34. Однако
большинство ферментов представлено белками более высокой молекулярной
массы, построенными из субъединиц (рис. 46). Так, каталаза (М=252 000)
содержит в молекуле шесть протомеров с М=42000 каждый. Молекула
фермента, ускоряющего реакцию синтеза рибонуклеиновых кислот (РНК-по-
лимераза, М=475 000), состоит из 5 неравных субъединиц. Полная молекула
глутаматдегидрогеназы, ускоряющей процесс окисления глутаминовой кисло-
ты (М=336 000), построена из 6 субъединиц с М = 56000.
Способы компоновки протомеров в мультимеры разнообразны. Некото-
рые из них показаны на рис. 46. Крайне важно, что построенный из субъеди-
ниц фермент проявляет максимальную каталитическую активность именно
в виде мультимера: диссоциация на протомеры резко снижает активность
фермента. Не все ферменты-мультимеры построены исключительно из ката-
литически активных протомеров. Наряду с каталитическими в их составе
отмечены регуляторные субъединицы, как, например, у аспартат-карбамил-
трансферазы (рис. 46).
Среди ферментов-мультимеров безусловно преобладают димеры и тетраме-
ры (их несколько сотен), в меньшей мере распространены гексамеры и октамеры
(несколько десятков) и необыкновенно редко встречаются тримеры и пентамеры.
Молекулы ферментов-мультимеров в ряде случаев составлены из субъединиц
двух типов, обозначаемых условно как субъединицы типа А и В. Они сходны друг
с другом, но отличаются по некоторым деталям первичной и третичной
структур. В зависимости от соотношения протомеров типа А и В в мулы имере
последний может существовать в виде нескольких изомеров, которые называют
изозимами. Так, при четырех суб единицах возможны 5 изозимов:
I II III IV V
АААА АААВ ААВВ АВВВ ВВВВ
Это явление хорошо изучено у фермента, ускоряющего в мышцах превра-
щение молочной кислоты в пировиноградную и обратно:
СН3—СН —СООН Окисление > сн3—с—соон
Восстановление q
Молочная кислота Пировиноградная
кислота
100
Так как окисление молочной кислоты (acidum lacticum) сопровождается
отнятием атомов Н, этот фермент называют лактатдегидрогеназой. Молеку-
ла лактатдегидрогеназы (М = 140 000) составлена из четырех субъединиц
(М = 35000), которые условно обозначают Н и М (от англ, heart—сердце
и muscle—мышцы), так как из сердца и скелетных мышц выделены Ivl V ти-
пы лактатде идрогеназы. Следовательно, изозимы лактатдегидрогеназы
таковы:
I II III IV, V
НННН НН нм нимм нм мм мммм
Они отличаются друг от друга по степени активности, некоторым
физическим свойствам (например, молекулярной массе, электрофоретиче-
ской подвижности), локализации в органах и тканях и т. п. В зависимости
от возраста, физиологического состояния и других причин в организме
устанавливается то или иное соотношение изозимов, которому соответст-
вует определенный уровень активности фермента в целом. Изменение соот-
ношения изозимов во всем организме или в отдельных тканях и органах
представляет, таким образом, один из способов регуляции действия фер-
ментов.
В настоящее время интерес к изозимам резко повысился. Оказалось, что
кроме генетт ески детерминированных изозимов существует большая группа
ферментов, обладающая множественными формами, возникающими в резуль-
тате их посттрансля!ционной модификации (см. гл. VII). Множественные
формы ферментов и изозимы в частности используются сейчас для диагности-
ки болезней в медицине, прогнозирования продуктивности животных, подбора
родительских пар при скрещивании для обеспечения максимального гетерози-
са в потомстве и т. п.
Значение пространственной органи ации ферментов особенно ярко вы-
является при изучении строения так называемых мультиэнзимов, т. е.
ферментов, обладающих способностью ускорять одновременно несколько
химических реакций и осуществлять сложные превращения субстрата.
Примером может служить мультиэнзим, ускоряющий реакцию окисли-
тельного декарбоксилирования пировиноградной кислоты (см. гл. VIII).
Этот многоферментный комплекс с М=4 500000 состоит из трех видов
ферментов. Первый из них (Е^ ускоряет реакцию декарбоксилирования
пировиноградной кислоты. В состав комплекса входит 12 димерных
молекул этого фермента (М= 192000), изображенных на рис. 46, Г в виде
крупных белых шаров, расположенных попарно по периметру рисунка.
Второй и третий ферменты, катализирующие окислительно-восстанови-
тельные процессы при окислении пировиноградной кислоты, сосредоточены
внутри мультиэнзимного комплекса. Один из них представлен шестью
димерными молекулами (М = 112 000), другой (£2)—24 протомерами
(Л/=70000) (см. рис. 46, заштрихованные крупные и мелкие шары со-
ответственно).
В тех случаях, когда мультиэнзимный комплекс обслуживает единый,
многоступенчатый процесс биохимических превращений, его н зывают мета-
болоном (от слова метаболизм—обмен веществ). Таковы метаболоны глико-
лиза (см. гл. VIII), биосинтеза ряда аминокислот (см. гл. VII), цикла дикарбо-
новых и трикарбоновых кислот (см. гл. VIII) и др.
В результате слаженного во времени и пространстве действия всех
трех видов входящих в его состав ферментов мультиэнзим с огромной
скоростью осуществляет превращение пировиноградной кислоты. Именно
101
в кооперативном характере каталитического процесса и кроется главное от-
личие биокатализаторов от катализаторов неорганической природы, именно
поэтому интенсивность биокатализа в десятки, сотни и тысячи раз превос-
ходит мощность действия неорганических катализаторов.
Сравнительно недавно выявлена еще одна своеобразная черта в строении
ферментов: некоторые из них являются полифункциональными, т. е. обладают
несколькими энзиматическими активностями, но всего лишь одной полипеп-
тидной цепью. Дело в том, что эта единая цепь при формировании третичной
структуры образует несколько функционально и стерически обособленных
глобулярных участков—доменов, каждый из которых характеризуется своей
каталитической активностью. В последующих главах будут приведены соот-
ветствующие примеры.
При изучении мультиэнзимных комплексов и полифункциональных фер-
ментов удалось понять наиболее важную особенность ферментативного
катализа, а именно—эстафетную передачу промежуточных продуктов реак-
ции от одного компонента каталитической системы к другому без их
высвобождения.
МЕХАНИЗМ ДЕЙСТВИЯ ФЕРМЕНТОВ
Механизм действия однокомпонентных и двухкомпонентных ферментов
однотипен, так как активные центры в их молекулах функционально сходны
между собой.
Ведущую роль в механизме ферментативного катализа играет образо-
вание фермент-субстратных комплексов, на существование которых впервые
указал Д. Браун (1902). На первой фазе ферментативного катализа между
субстратом (или субстратами) и ферментом возникает соединение, в кото-
ром реагенты связаны друг с другом ионной, ковалентной или иного типа
связью. Затем (вторая фаза) суб трат под действием присоединенного к не-
му фермента претерпевает изменение, делающее его более доступным для
соответствующей химической реакции. На третьей фазе происходит сама
химическая реакция и, наконец, образовавшиеся продукты реакции на чет-
вертой фазе освобождаются из фермент-продуктного комплекса. Если обо-
значить фермент Е, субстрат S, активированный субстрат S' и продукт
реакции Р, то указанная последовательность процессов выразится нижесле-
дующей схемой:
/ п III IV
Е+ S&ES&ES 'i^EP^E+P.
Эта схема была первоначально разработана В. 1енри (1903), затем
Л. Михаэлисом и М. Ментен (1913) и подтверждена прямым выделением ES-,
ES' и ЕР-комплексов.
Одним из примеров ферментативного катализа, осуществляемого в со-
ответствии с приведенной схемой, может служить реакция гидролиза аце-
тилхолина. Это соединение служит медиатором (посредником) при передаче
нервных импульсов: в ответ на выделение окончанием нервного волокна
ацетилхолина следует ответная реакция возбуждения нервной клетки. Что-
бы этот процесс протекал непрерывно, после каждого акта передачи нерв-
ного импульса вызвавшая возбуждение порция ацетилхолина (1—2 мкг)
должна быть полностью разрушена. Это достигается посредством реакции
гидролиза ацетилхолина при участии фермента ацетилхолинэстеразы. Гид-
102
Рис. 47. Механизм действия ацетилхолинэстеразы:
Л—активный центр фермента; Б—фермент-су бстратпый комплекс; В—подготовка к преобразованию
(активирование) субстрата; Г—комплекс продуктов реакции с ферм₽нтом (пояснения в тексте)
ролиз осуществляется с огромной скоростью: 1—2 мкг ацетилхолина за
0,1—0,2 мс:
СН3 - СО - О - СН2 - СН2 - N(CH3)3+н2о
Ацспииолп
СНз - СООН+НО - СН2 - СН2 - N(CH3)3
Уксусная Холин
кислота
Ацетилхолинэстераза—однокомпон нтный фермент. В ее активном центре
сосредоточены по меньшей мере 4 аминокислотных радикала—глу, сер, гис
и тир, обеспечивающие последовательное осуществление перечисленных выше
этапов ферментативного катализа.
Сначала между ферментом (ацетилхолинэстераза) и субстратом (аце-
тилхолин) возникает фермент-субстратный комплекс. Он образуется за
счет электростатического взаимодействия между отрицательно заряженной
103
ионизированной СООН-группой радикала глу и положительно заряженным
атомом N молекулы ацетилхолина (рис. 47, /, Б). После образования фер
мент-субстратного комплекса вступают в действие остальные аминокислот-
ные радикалы активного центра ацетилхол нэстеразы. В первую очередь
замыкается связь между углеродом поляризованной СО-группы ацетильного
радикала холина и кислородом ОН-группы остатка сер. Затем возникает
водородная связь между кислородом сложноэфирной связи в молекуле ацетил-
холина и ОН-группой радикала тир (рис. 47, П, В).
Расположение молекулы ацетилхолина и радикалов сер и тир в активном
центре фермента таково, что образование упомянутых связей ослабляет связь
между СО-группой и атомом кислорода сложноэфирной связи в молекуле
ацетилхолина (эффект «дыбы»). В результате для ее разрыва требуется
гораздо меньше энергии, т. е. энергетический барьер оказывается сниженным
вследствие активации молекулы ацетилхолина (^S'-комплекс). Поэтому под
влиянием радикала гис, оттягивающего на себя протон от ОН-группы сер,
упро няется сложноэфирная связь между радикалом сер и ацетильной
группой с одновременным разрывом сложноэфирной связи в молекуле
ацетилхолина и переходом протона от радикала тир к остатку холина
(рис. 47, III, Г). Последний высвобождается из активного центра (рис. 47, IV),
а его место занимает молекула воды. Она ббразует связь с карбонильным
кислородом ацетильной группы и кислородом тир (на рис. 47 этот этап не
показан), после чего протон от остатка гис возвращается к кислороду
ОН-группы сер, а протон воды—к радикалу тир. Одновременно выделяется
второй продукт реакции—уксусная кислота и регенерируется свободный
активный центр ацетилхолинэстеразы (рис. 47, IV, А ), готовый к новому акту
катализа.
В процессе образования фермент-субстратного комплекса и на дальнейших
фазах ферментативного катализа происходят неоднократные изменения тре-
тичной структуры фермента, приводящие к последовательному сближению
с субстратом и ориентации в пространстве тех активных групп, которые
взаимодействуют друг с другом на различных этапах преобразования субстра-
та. Изменение третичной структуры белка невозможно без участия всей или
почти всей полипептидной цепи, образующей белковую молекулу. Следова-
тельно, в каталитическом акте принимает участие по существу вся или почти
вся молекула фермента.
Отдельные этапы взаимодействия фермента и субстрата при фермента-
тивном катализе все более проясняются. В частности, установлено, что за
стадией адсорбции субстрата в активном центре фермента наступает «уз-
навание» субстратным центром фермента той части молекулы субстрата,
которая непосредственно не подвергается химическому преобразованию. За
счет возникающих при этом многоточечных контактов, реализующихся
в виде сил слабого взаимодействия (гидрофобные, водородные и др.), связь
субстрата с ферментом упрочняется. Одновременно с этим в активном
центре фермента «стабилизируется» та часть субстрата, которая в дальней-
шем участвует в химической реакции,—она фиксируется в напряженной
конфигурации, близкой к переходному состоянию субстрата при превраще-
нии его в продукт. В результате реагирующий фрагмент молекулы субстрата
и каталитические группы фермента образуют продуктивный комплекс, где
уже частично осуществлены электронно-конформационные переходы, необ-
ходимые для протекания собственно химической стадии ферментативного
процесса. Это приводит к понижению энергии активации, необходимой для
осуществления химическ й реакции, благодаря энтропийному эффекту вслед-
ствие иммобилизации, закрепления, жесткой ориентации субстрата в актив-
104
ном центре фермента. Таким образом, каждое звено в многостадийной
химической реакции, ускоряемой ферментом, создает почти оптимальные
условия для прохождения ее следующего этапа. Как следствие, химическая
реакция при ферментативном катализе идет в десятки, сотни тысяч раз
быстрее.
Рассмотрение тонкого механизма ферментативного катализа позволяет
понять специфику действия ферментов, отличающую их от катализаторов
неорганического происхождения. Уникаль ая структура и взаимодействие ка-
талитического, субстратного и аллостерического центров фермента обеспечи-
вает кооперативное осуществление многостадийных процессов. Именно упо-
рядоченность реакций в пространстве и времени, их кооперативный характер
отличают действие биокатализаторов, обеспечивая высокую специфичность
и скорость процесса в целом.
КИНЕТИКА ФЕРМЕНТАТИВНЫХ РЕАКЦИЙ
Под ферментативной кинетикой в широком смысле понимают зави-
симость скорости реакции, ускоряемой ферментом, от химической природы
реагирующих- веществ (субстраты, фермент) и условий их взаимодействия
(концентрация, температура, pH среды, наличие активаторов или инги-
биторов и т. п.). Однако зависимость скорости ферментативного процесса
от температуры, pH среды и влияния ингибиторов и активаторов в зна-
чи ельной мере связана с изменением свойств фермента как белкового
тела. Поэтому здесь будет освещен только вопрос о закономерностях,
определяемых природой и концентрацией реагирующих веществ и их из-
менением.
Как показано выше, первой фазой биокаталитического процесса является
образование фермент-субстратного комплекса:
*+1
E+S<==*ES.
*-1
Эта равновесная система может быть охарактеризована соответствующей
константой равновесия. Величину, обратную ей, называют константой дис-
социации фермент-субстратного комплекса или субстратной константой
и обозначают К5:
^=[£][S]/[£S].
Она зависит от природы субстрата и фермента и отражает степень их
сродства. Так, для сахаразы (фермент, ускоряющий реакцию гидролиза са-
харозы) А'5=0,0167М, т. е. концентрация фермент-субстратного комплекса
превышает концентрацию свободных фермента и субстрата примерно
в 60 раз. Чем ниже значение К„ тем выше, следовательно, сродство фермента
к субстрату.
Так как концентрация фермент-субстратного комплекса изменяется вслед-
ствие перехода последнего в продукт реакции с регенерацией свободного
фермента:
1 + 2
ES---->Е+Р,
то значение Ks находят из соотношения констант скоростей прямой (&+1)
и обратной (А'-1) реакций, т. е.
k-i / k+i.
105
К указанному выводу можно прийти, приравнивая скорости прямой и обрат-
ной реакций, что характерно для равновесного состояния системы:
и1=&+1 DtltAl; У2=^-1[^]- Если V! = v2, то тогда *+1[Е] [$]=*_, [£$],
т. е. [£]И/[^]=^-1/к+1=^-
Таким эбразом, субстратная константа, или константа диссоциации, фер-
мент-субстра гного комплекса (jQ характеризует биокаталитический про-
цесс с точки зрения сродства фермента и субстрата и соотношения кон-
стант скорости реакций распада и становления фермент субстратного ком-
плекса.
Так как одновр менно с диссоциацией фермент убстратного комплекса на
исходные вещества происходят превращения субстрата в продукт и распад
комплекса фермент—продукт на составляющие его компоненты:
E+S ?±ES—>Е+Р
для полной характеристики ферментативного процесса введено понятие о кон-
станте Михаэлиса (Кт), которая представляет отношение констант скоростей
всех трех реакций, осуществляющихся в процессе ферментативного катализа:
Кт=(к-1+к+2)/k+i. Кт всегда несколько выше по числовому значению, чем
Ks. Так, Ks для комплекса сахаразы и сахарозы равно 0,0167М, а Кт составляет
0.0280М.
Скорость химической реакции, ускоряемой ферментом (как и скорость
обычной химической реакции), изм< ряют количеством молей субстрата, пре-
вращаемых в единицу времени. Однако принципиально важно, чтобы при этом
поддерживались стандартные условия для проявления активности фермента:
температура 25° С, оптимальное значение pH и, что особенно существенно,
полное насыщение фермента субстратом. Скорость ферментативной реакции,
измеренной при соблюдении перечисленных условий, обозначают V и называ-
ют максимальной скоростью ферментативной реакции. Она определяется кон-
центрацией фермента и константой скорости распада комплекса фермент-про-
дукт: V=k+2[E]-
Скорость ферментативной реакции, наблюдаемую при отсутствии пол-
ного насыщения фермента субстратом, обозначают v. Она в каждый дан-
Рис. 48. Зависимость скорости фермента-
тивной реакции от концентрации субстрата
при постоянной концентрации фермента
По достижении состояния насыщения фермента субстра-
том скорость ферментативной реакции достигает мак-
симального значения V и кривая идет параллельно оси
абсцисс
ный момент пропорциональна концент-
рации фермент-субстратного комплек-
са: t?=fc+2[KS]. Так как =
=*+1/Л_1[£][5], a k+1fk-х = 1/^ т. е.
[£$]-[£][$]/*„ то i>=fc+1 [£][$]/*,.
Таким образом, наблюдаемая скорость
ферментативной реакции зависит от концен-
трации фермента и субстрата и их сродства.
Численно Кт равна той концентрации суб-
страта (в молях на литр), при которой
v составляет 1/2 И (рис. 48).
Концентрацию фермента и субстрата
выражают обычно в микромолях на литр.
Однако ввиду того, что точная молеку-
лярная масса большинства ферментов
и число каталитических (активных) цент-
ров в их молекулах неизвестны применя-
ют условный способ выражения абсолют-
ного количества фермента. За единицу лю-
106
бого фермента (обозначаемую на русск. и нем. Е, а на англ., франц., итал.
и исп. U) принимают то его количество, которое в стандартных условиях
катализирует превращение 1 мкмоль субстрата в 1 мин. Эта величина
(1 мкмоль/мин) является также единицей активности фермента. Указанная стан-
дартная система обозначения количества фермента была введена в 1961 г.
Комиссией по ферментам Международного биохимического союза и прочно
вошла в ферментологию. Ею пользуются и поныне. Как отмечено выше,
согласно этой системе скорость ферментативной реакции выражали числом
микромолей субстрата, превращаемых в 1 мин.
В 1972 г., в связи с переходом на выражение скорости биокаталитических
превращений в молях субстрата, преобразуемого в течение 1 с, было пред-
ложено вместо старой, безымянной единицы ферментативной активности (Е
или U) ввести новую единицу—катал (символ—кат), обозначающую количе-
ство фермента, способное в течение 1 с обеспечить превращение 1 моля суб-
страта (в стандартных условиях). Так как единица ферментативной активности
в 1 кат, соответствующая скорости реакции 1 моль/с, мало реальна (превра-
щения идут с гораздо меньшей скоростью), каталитическую активность в но-
вой системе единиц выражают в микрокаталах (мккат), нанокаталах (нкат)
и пикокаталах (пкат), чему отвечают скорости реакций в микромолях, нано-
молях и пикомолях в секунду соответственно. Старая единица (Е или U) равна
16,67 нкат. В течение ближайших лет предполагают осуществить переход на
выражение ферментативной активности в каталах.
Концентрацию фермента в растворе в указанных единицах (Е) выражают
числом их в 1 мл раствора. В случае сухого препарата фермента приводят
данные о количестве Е на 1 мг. Если в препарате фермента определено
содержание белка, то высчитывают удельную активность ферментного препа-
рата, выражаемую числом Е на 1 мг белка. При наличии данных о числе
Е в 1 мг чистого фермента говорят об удельной активности фермента. Если
известна молекулярная масса фермента, то легко рассчитать число Е на
1 мкмоль его, т. е. молекулярную активность фермента. Следовательно, речь
идет уже не о выражении концентрации фермента, а о выражении активности
фермента либо числом микромолей субстрата, превращенных в 1 мин 1 мг
фермента (удельная активность фермента), либо числом молекул субстрата,
превращенных в 1 мин одной молекулой фермента (молекулярная актив-
ность).
Если известно, сколько активных центров находится в молекуле фермента,
активность его характеризуют числом молекул субстрата, превращенных при
посредстве одного активного центра. Так, молекулярная активность каталазы
равна 5 млн., тогда как активность ее каталитического центра (их в молекуле
каталазы 4) составляет всего 1 млн. 250 тыс. молекул Н2О2.
СВОЙСТВА ФЕРМЕНТОВ
Будучи белками, ферменты обладают всеми их свойствами. Вместе с тем
биокатализаторы характеризуются рядом специфических качеств, тоже выте-
кающих из их белковой природы. Эти качества отличают ферменты от ката-
лизаторов обычного типа. Сюда относятся термолабильность ферментов,
зависимость их действия от значения pH среды, специфичность и, наконец,
подверженность влиянию активаторов и ингибиторов.
Термолабильность ферментов объясняется тем, что температура, с одной
стороны, воздействует на белковую часть фермента, приводя при слишком
высоких значениях к денатурации белка и снижению каталитической функции,
107
Рис. 49. Влияние температуры на
активность фермента (пояснение
в тексте)
Рис. 50. Влияние pH среды на ак-
тивность фермента
а с другой стороны, оказывает влияние на скорость реакции образования
фермент-субстратного комплекса и на все последующие этапы преобразования
субстрата, что ведет к усилению катализа.
Зависимость каталитической активности фермента от температуры вы-
ражается типичной кривой (рис. 49). По характеру кривой видно, что до
некоторого значения температуры (в среднем до 50° С) ка алитическая ак-
тивность растет, причем на каждые 10° С примерно в 2 раза повышается
скорость преобразования субстрата. В то же время постепенно возрастает
количество инактивированного фермента за счет денатурации его белковой
части. При температуре выше 50° С денатурация ферментного белка резко
усиливается и, хотя скорость реакций преобразования субстрата продолжает
расти, актив ость фермента, выражающаяся количеством превращенного суб-
страта, падает.
Детальные исследования роста активности ферментов с повышением тем-
пературы, проведенные в последнее время, показали более сложный характер
этой зависимости, чем указано выше: во многих случаях она не отвечает
правилу удвоения активности на каждые 10° С в основном из-за постепенно
нарастающих конформационных изменений в молекуле фермента.
Температура, при которой каталитическая активность фермента максима-
льна, называется его температурным оптимумом.
Температурный оптимум для различных ферментов неодинаков. В общем
для ферментов животного происхождения он лежит между 40 и 50° С, а расти-
тельного—между 50 и 60° С. Однако есть ферменты с более высоким темпера-
турным оптимумом, например у папаина (фермент растительного происхож-
дения, ускоряющий гидролиз белка) оптимум находится при 80° С. В то же
время у каталазы (фермент, ускоряющий распад Н2О2 до Н2О и О2) оптималь-
ная температура действия заходится между 0 и 10° С, а при более высоких
температурах происходит энергичное окисление фермента и его инактивация.
Зависимость активности фермента от значения pH среды была установлена
свыше 50 лет назад. Для каждого фермента существует оптимальное значение
pH среды, при котором он проявляет максимальную активность. Большин-
ство ферментов имеет максимальную активность в зоне pH поблизости от
нейтральной точки. В резко кислой или резко щелочной среде хорошо работа-
ют лишь некоторые ферменты (табл. 10).
108
Таблица 10
Оптимальные значения pH среды для некоторых ферментов
Фермент Характер катализируемой реакции pH
Пепсин Гидролиз белка 1,5—2,5
Липаза (из семян клещевины) Гидролиз жиров 4,7—5,0
Уреаза » мочевины 7,0
Трипсин » белка 7,8
Аргиназа » аргинина 9,5—9,9
Переход к большей или меньшей (по сравнению с оптимальной) концент-
рации водородных ионов сопровождается более или менее равномерным
падением активности фермента. На рис. 50 представлена кривая, выражающая
типичную зависимость каталитического действия фермента от pH среды.
Влияние концентрации водородных ионов на каталитическую активность
ферментов состоит в воздействии ее на активный центр. При разных значениях
pH в реакционной среде активный центр может быть слабее или сильнее
ионизирован, больше или меньше экранирован соседними с ним фрагментами
полипептидной цепи белковой части фермента и т. п. Кроме того, pH среды
влияет на степень ионизации субстрата, фермент-субстратного комплекса
и продуктов реакции, оказывает большое влияние на состояние фермента,
определяя соотношение в нем катионных и анионных центров, что сказывается
на третичной структуре белковой молекулы. Последнее обстоятельство заслу-
живает особого внимания, так как определенная третичная структура белка-
фермента необходима для образования фермент-субстратного комплекса.
Специфичность—одно из наиболее выдающихся качеств ферментов. Это
свойство их было открыто еще в прошлом столетии, когда было сделано
наблюдение, что очень близкие по структуре вещества—пространственные
изомеры (а- и Р-метилглюкозиды) расщепляются по эфирной связи двумя
совершенно разными ферментами:
в-Метилглюкозид
(гидролизуется по эфирной
связи ферментом из солода)
Р -Мети лгл ю козид
(гидролизуется по эфирной
связи ферментом из семян
горького миндаля)
Таким образом, ферменты могут различать химические оединения, от-
личающиеся друг от друга очень незначительными деталями строения, та-
кими, например, как пространственное расположение метоксильного радикала
и атома водорода при 1-м углеродном атоме молекулы метилглюкозида.
По образному выражению, нередко употребляемому в биохимической
литературе, фермент подходит к субстрату, как ключ к замку. Это знаменитое
правило было сформулировано Э. Фишером в 1894 г. исходя из того, что
специфичность действия фермента предопределяется строгим соответствием
геометрической структуры субстрата и активного центра фермента.
В 50-е годы нашего столетия это статическое представление было заменено
гипотезой Д. Кошланда об индуцированном соответствии субстрата и фермента.
109
Сущность ее сводится к тому, что пространственное соответствие структуры
субстрата и активного центра фермента создается в момент их взаимодействия
друг с другом, что может быть выражено формулой «перчатка—рука». При
этом в субстрате уже деформируются некоторые валентные связи и он, таким
образом, подготавливается к дальнейшему каталитическому видоизменению,
а в молекуле фермента происходят конформаци энные перестройки. Гипотеза
Кошланда, основанная на допущении гибкости активного центра фермента,
удовлетворительно объясняла активирование и ингибирование действия фер-
ментов и регуляцию их активности при воздействии различных факторов.
В частности, конформационные перестройки в ферменте в процессе изменения
его активности Д. Кошланд сравнивал с колебаниями паутины, когда в нее
попала добыча (субстрат), подчеркивая этим крайнюю лабильность структуры
фермента в процессе каталитического акта.
В настоящее время гипотеза Кошланда постепенно вытесняется гипотезой
топохимического соответствия Сохраняя основные полож ния гипотезы взаимо-
индуцированной настройки субстрата и фермента, она фиксирует внимание на
том, что специфичность действия ферментов объясняется в первую очередь
узнаванием той части субстрата, которая не изменяется при катализе. Между
этой частью субстрата и субстратным центром фермента возникают многочис-
ленные точечные гидрофобные взаимодействия и водородные связи.
Несомнен о, что специфичность ферментов объясняется в первую очередь
совпадением пространственных конфигураций субстра а и субстратного центра
фермента. Видимо, только тогда, когда совпадение это достаточно полно,
может образоваться фермент-субстратный комплекс и, следовательно, начаться
процесс ферментативного катализа. Выяснение третичной структуры некоторых
белков-ферментов полностью оправдало такую точку зрения. ТЬк, в молекуле
лизоцима (фермент, ускоряющий гидролиз гликозидных связей в полисахаридах
клеточной стенки бактерий) обнаружена выемка (щель) (см. рис. 34) для присое-
динения субстрата. В эту выемку плотно входит участок молекулы субстрата
протяженностью ровно в шесть звеньев:
N-ацетил- N-ацетил-
глюкоммина му^аминовой
кислоты
Остальная часть молекулы субстрата, состоящая из нескольких сотен остатков
аминосахаров или их производных, остается свободной. Не только общая форма
щели в молекуле лизоцима соответствует параметрам входящего в нее участка
субстрата, но и расположение отдельных аминокислотных радикалов в субстратном
центре этого фермента совершенно точно согласуется с размещением определенных
атомов и атомных групп в молекуле субстрата. Именно за счет указанных
радикалов и групп замыкаются водородные связи между лизоцимом и полисахари-
дом в роцессе стан< вления фермент-субстра гного комплекса (табл. 11).
ПО
Таблица 11
Многоточечное взаимодействие фрагмента молекулы полисахарида
клеточной стенки бактерии с лизоцимом
Буквенные обозначеник полисахаридных звеньев Число связей и слабых взаимодействий Энергия ассоциации. жДж/моль
водородных Ван-дер-Ваальса
А < 1 7 7,5—9,6
В 1 11 11,7—16,3
С 4 30 19,6—23,8
D 2 35 12,1—25,0
Е 3 45 3,8—16,7
F 4 13 6,3—21,0
Одновременно против радикалов каталитического центра (остатки глу
и асп, занимающие в полипептидной цепи лизоцима 35-е и 52-е положения
соответственно) устанавливается гликозидная связь, расположенная между
кольцами D и Е субстрата, и наступает каталитический акт:
Приведенная схема еще раз иллюстрирует механизм ферментативного ка-
тализа.
Детальное изучение специфичности ферментов показало, что пределы ее
у разных ферментов разл чны. Одни ферменты обладают абсолютной специ-
фичностью, т. е. каталитически уск ряют одну-еди ственную реакцию. Приме-
ром такого фермента может служить уреаза. Другие ферменты осуществляют
катализ реакций определенного типа независимо от того, какие конкретные
вещества в них взаимодействуют или распадаются. Основным признаком для
ферментов этого типа является характер разрушаемой или создаваемой связи.
Такие ферменты характеризуются, следовательно, групповой специфичностью.
Некоторые ферменты отличаются стереохимической специфичностью, т. е. дей-
ствуют только на один из пространственных изомеров Примером могут
служи уже упоминавшиеся выше ферменты, расщепляющие а- и Р-метилг-
люкозиды.
Влияние на ферменты активаторов и ингибиторов впервые было обнаружено
при изучении активаторов (стимулин) и ингибиторов (антиферменты)
А. Я. Данилевским с сотр. еще в прошлом столетии. К числу агентов, повыша-
ющих активность ферментов, относя ся ионы многих металлов и некоторые
анионы. Особенно часто активаторами ферментов бывают Mg2+, Мп2+,
Zn2+, К+ и Со2+, а из анионов—С1“. В одних случаях ионы металлов входят
111
аухкугс
Vn ш гЧЦ
Кгюйк комплекс не образуется
^cyBcmpani?® оллост^еский стиму-
41 конкурентный. h аллостерический инги-
Ч) изостерический •' Витор
ингиоитор
Рис. 51. Активирование и ингибирова-
ние действия фермента:
а—аллостерический центр фермента; к—каталити-
ческий центр фермента; с—субстратный центр фер-
мента; I—взаимодействие фермента с субстратом;
полного совпадения конфигураций субстратного
центра фермента и субстрата нет, вследствие чего
нет тесного контакта между ферментом и субстра-
том; активность фермента далека от максимума;
II—в результате взаимодействия фермента с алло-
стерическим стимулятором каталитический центр
сближается с субстратным, причем последний при-
обретает форму, полностью совпадающую с тако-
вой субстрата; вследствие этого образуется полно-
ценный фермент-субстратный комплекс; активность
фермента максимальна; /Л—взаимодействие фер-
мента с аллостерическим ингибитором сопровожда-
ется деформацией субстратного центра, к которому
становится невозможным присоединение субстрата;
фермент не проявляет активности; IV—к субстрат-
ному центру фермента присоединяется конкурент-
ный ингиоитор; активность фермента подавлена,
так как образование фермент-субстратного комплек-
са невозможно
в состав простетической группы фермента, в других—облегчают образование
фермент-субстратного комплекса, в третьих—способствуют присоединению
кофермента к апоферменту, в четвертых—обеспечивают становление чет-
вертичной структуры фермента или же действуют иными путями. Как
выяснено в последнее время, мощное действие на ферменты оказывают
аллостерические активаторы. Они присоединяются по аллостерическому
центру фермента и изменяют третичную структуру белковой молекулы.
В результате этого субстратный и каталитический центры фермента при-
обретают наиболее выгодную для осуществления своих функций конфи-
гурацию (рис. 51, II).
Ингибиторы тормозят действие ферментов. Механизм ингибирующего
действия разнообразен, но в большинстве сводится к двум типам торможения:
необратимому и обратимому. При необратимом торможении ингибитор,
обладающий структурным сходством (изостерией) с субстратом, соединяется
с ферментом, подменяя собой субстрат. Наиболее ярким примером такого
типа торможения является угнетение действия холинэстеразы при помощи
диизопропилфторфосфата, который структурно весьма близок к ацетилхолину
и, являясь псевдосубстратом, легко присоединяется вместо него к ферменту:
НаСч [ /СН,
СН—О—P-О—сн
н^с/ Vh,
Дижзопропмлфторфосфгг
Диизопропилфторфосфат блокирует активный центр холинэстеразы, фос-
форилируя радикал серина (см. рис. 47). Так как образовавший) я диизо-
пропилфосфосерин гораздо прочнее ацетилсерина и распадается очень медлен-
но, активный центр фермента надолго выводится из строя. Это и аналогичные
ему соединения образуют большую группу так называемых нервных ядов, ибо,
приостанавливая гидролиз ацетилхолина, они резко нарушают деятельность
нервной системы. Среди них широко известны многочисленные нсектициды
и боевые отравляющие вещества.
При необратимом ингибировании фермента ингибитор может не обладать
структурным сходством с субстратом, т. е. может модифицировать фермент
вне его активного центра, тем не менее резко изменяя каталитическую актив-
112
ность последнего вследствие нарушения конформации как фермента в целом,
так и его активного центра. Это происходит, например, при алкилировании
сульфгидрильных групп фермента:
Е—SH+ICH2CONH2—Е—S—CH2CONH2+HI
Фермент Иодацетамид (алки- Алкилированный по
лирующий агент) HS-групппам фермент
Недавно обнаружены необратимые инактиваторы ферментов, возникаю-
щие из инертных соединений после их взаимодействия с каталитическим
центром фермента, например:
R— ChsC—СН2—СО—R—R—СН=С=СН—СО—R'
Инертное соединение Необратимый ингибитор
1
Присоединяясь к активному центру фермента, такой необратимый ингибитор,
возникший в процессе ферментативного катализа, соединяется с активным
центром фермента и полностью его блокирует. Поэтому эти ингибиторы
образно называют ингибиторами типа «самоубийц».
Обратимое ингибирование действия ферментов может быть конкурентным
и неконкурентным. Классическим примером конкурентного ингибирования
ферментативной активности является торможение действия дегидрогеназы
янтарной кислоты дикарбоновыми кислотами (малоновая, глутаровая), близ-
кими по структуре к янтарной кислоте: между ними идет конкуренция за
связывание в активном центре фермента.
При неконкурентном торможении ингибитор взаимодействует с апофермен-
том или простетической группой, вследствие чего фермент теряет активность.
Одним из вариантов такого торможения может служить блокирование фер-
ментов тяжелыми металлами (ртуть, мышьяк, свинец и др., которые присое-
диняются к сульфгидрильным группам полипептидной цепи), солями синиль-
ной кислоты, оксидом углерода (II) и др. (присоединяются к железосодер-
жащим простетическим группам и т. п.). В качестве другого варианта
неконкурентного торможения действия фермента следует отметить аллостери-
ческое ингибирование (см. рис. 51, IV).
Специфическую группу ингибиторов ферментов, которой уделяют огром-
ное внимание в последние годы, составляют ингибиторы белковой природы.
Они блокируют действие фермента за счет белок-белковых взаимодействий,
в результате чего закрывается активный центр фермента. Особенно энергично
белковые ингибиторы регулируют деятельность протеиназ клетки, что сильно
сказывается на регуляции обмена веществ в целом, поскольку под воздействи-
ем ингибиторов протеиназ тормозится переход проферментов в ферменты,
отщепление сигнальных пептидов и других пептидных фрагментов при созре-
вании белков, блокируются реакции протеолиза, ведущие к образованию
биологически активных пептидов (гормоны пептидной природы, рилизинг-
факторы и т. п.—см. гл. XII). Так, например, ингибитор белковой природы—
oti-антитрипсин (гликопротеин с М = 52000, получаемый методом генетиче-
ской инженерии из пекарских дрожжей), нашел применение для лечения эм-
физемы (частичного распада) легких, так как он блокирует действие эластазы,
выделяемой в норме нейтрофилами легочной ткани (рис. 52). От 2 до 4 грам-
мов о^-антитрипсина достаточно для заместительной терапии при эмфиземе
легких, развивающейся в результате курения.
Учение об активаторах и ингибиторах ферментов в настоящее время слива-
ется с более широким и общим вопросом о регуляции действия ферментов
113
Рис. 52. а-Антитрипсин и его роль в поддер-
жании целостности легочной ткани
В норме в легких деструктирующее действие эластазы, выде-
ляемой нейтрофилами, блокируется а j-антитрипсином. при-
носимым током крови в составе ец-гл обул ина. Курение вы-
зывает увеличение числа и активности нейтрофилов и, как
следствие этого, усиление действия эластазы и падение эла-
стичности легочной ткани (отмечено пунктиром прн ее изоб-
ражении на рисунке). К тому же продукты сгорания табака
понижают ингибирующие свойства ац-антитрипсина, кото-
рый следовало бы называть антиэластазой, так как ои ин-
гибирует эластазу сильнее, чем^трипсин
в целом. В частности, установлено, что конечный, а иногда и промежуточный
продукт многостадийного процесса биосинтеза может служить аллостериче-
ским ингибитором одной из первых его реакций (см., например, ретроин-
гибирование биосинтеза пиримидиновых нуклеотидов—гл. XIII).
НОМЕНКЛАТУРА ФЕРМЕНТОВ
Ферментология очень долго не распола ала строго научной номенклатурой
ферментов. Наименования ферментам давали по случайным признакам (три-
виальная номенклатура), по названию субстрата (рациональная), по химичес-
кому составу фермента и, наконец, по типу катализируемой реакции и харак-
теру субстрата.
Примерами тривиальной номенклатуры могут служить названия таких фер-
ментов, как пепсин (от греч. пепсис—пищеварение), трипсин (от греч. три-
псис—разжижаю) и папаин (от названия дынного дерева Carica papaja, из сока
которого он выделен). По действию все эти ферменты являются протеолитиче-
скими, т. е. ускоряют гидролиз протеинов (белков). Характерное название
было дано группе окрашенных внутриклеточных ферментов, ускоряющих
окислительно-восстановительные реакции в клетке,—цитохромы (от лат.
citos—клетка и chroma—цвет).
Наибольшее распространение получила рациональная номенклатура, со-
гласно которой название фермента составляется из названия субстрата и ха-
рактерного окончания -аза. Она была предложена более столетия тому назад,
в 1883 г., Э. Дюкло—учеником Л. Пастера. Так, фермент, ускоряющий реак-
цию гидролиза крахмала, получил название амилаза (от греч. амилон—крах-
мал) гидроли а жиров—липаза (от греч. липос—жир), белков (протеинов)—
протеаза, мочевины—уреаза (от греч. уреа—мочевина) и т. п.
Когда методами аналитической химии были достигнуты известные успехи
в расшифровке химической природы простетических групп, возникла новая
номенклатура ферментов Их стали именовать по названию простетической
группы, например геминфермент (простетическая группа —гем), пиридоксаль-
фермент (простетическая группа—пиридоксаль) и т. п.
Затем в названии фермента стали указывать как на характер субстрата, так
и на тип катали ируемой реакции. Например, фермент, отнимающий водород
от молекулы янтарной кислоты, называют сукцинатдегидрогеназой, подчер-
кивая этим одновременно и химическую природу субстрата, и отнятие атомов
водорода в процессе ферментативного действия:
114
НООС—СН2—СН2—СООН—НООС—CH=CH—COOH
Янтарная кислота Дегидрирование
(acidum succinicum)
В 1961 г. Международная комиссия по номенклатуре ферментов пред-
ставила V Международному биохимическому конгрессу проект номе-
нклатуры, построенный на строго научных принципах1. Проект был
утвержден конгрессом, и новая номенклатура прочно вошла в фермен-
тологию. Согласно этой (Московской) номенклатуре название фермента
составляют из химического названия субстрата и названия той реакции,
которая осуществляется ферментом. Если химическая реакция, ускоряемая
ферментом, сопровождается переносом группировки атомов от субстрата
к акцептору, название фермента включает также химическое наименование
акцептора.
Например, пиридоксальфермент, катализирующий реакцию переаминиро-
вания между L-аланином и а-кетоглутаровой кислотой, называется L-аланин:
2-оксоглутарат аминотрансфераза: —
СН3 I СООН 1 \ (СН2)2 сн3 1 со + СООН 1 (СН2)2
н— с—nh2 1 СООН + 1 со 1 СООН 1 СООН 1 н—с—nh2 1 СООН
L-аланин (субстрат) 2-оксоглута- ровая кислота (а-кетоглута- ровая) Пировино- градная кислота L-глутаминовая кислота
В этом названии отмечены сразу три особенности: 1) субстратом является
L-аланин; 2) акцептором служит 2-оксоглутаровая кислота; 3) от субстрата
к акцептору передается аминогруппа.
Названия ферментов по научной номенклатуре неизмеримо выигрывают
в точности, но становятся в ряде случаев гораздо сложнее старых, тривиаль-
ных. Так уреаза (тривиальное название), ускоряющая реакцию гидролиза
мочевины на оксид углерода (IV) и аммиак, по научной номенклатуре имену-
ется карбамид—амидогидролазой:
H2N—СО—NH2 + Н2О—2NH3+СО2
В этом названии дано точное химическое наименование субстрата и указано,
что фермент катализирует реакцию гидролиза амидогруппы. Трегалаза, уско-
ряющая реакцию гидролиза трегалозы, называется трегалоза-1-глюкогид-
ролазой.
В связи со значительным усложнением научных названий в новой номенк-
латуре допускается сохранение наряду с новыми старых тривиальных, рабочих
названий ферментов. Международной комиссией был составлен детальный
список всех известных в то время ферментов, существенно дополненный
в 1972 г. при пересмотре как классификации, так и номенклатуры некоторых
ферментов, где рядом с новым научным названием каждого фермента приве-
дено старое, а также указан химизм катализируемой ферментом реакции
1 Конгресс проходил в Москве 10—16 августа 1961 г.
115
и в некоторых случаях природа фермента. Таким образом, исключается воз-
можность путаницы в наименовании фермента. В 1964 г. список включал 874
фермента; в последующее время он был существенно дополнен и возрос до
1770 ферментов в 1972 г. и до 2003 ферментов в 1979 г.
Каждому ферменту в указанном списке присвоен индивидуальный номер
(шифр). Например, шифр уреазы выражается цифрами 3.5.1.5. Это означает,
что уреаза относится к 3-му классу (первая цифра) ферментов, все представи-
тели которого катализируют реакции гидролиза. Вторая цифра (5) говорит
о том, что уреаза принадлежит к 5-му подклассу этого класса, куда зачислены
все ферменты, ускоряющие гидролиз С—N-связей, не являющихся пептид-
ными. Третья цифра шифра (1) указывает на принадлежность уреазы к подпод-
классу 5-го подкласса, члены которого ускоряют гидролиз линейных амидов,
а последняя цифра (5)—порядковый номер уреазы в этом подпод-
классе.
Упоминавшаяся ранее лактатдегидрогенеза имеет шифр 1.1.1.27, т. е.
относится к 1-му классу ферментов (оксидоредуктазы), к 1-му подклассу
(оксидоредуктазы, действующие на СН—ОН-группировки в качестве до-
норов атомов водорода), к 1-му подподклассу (акцептором атомов
водорода служит никотинамидадениндинуклеотид—см. о нем ниже в этой
главе) и занимает 27-е место в перечне ферментов упомянутого под-
подкласса. Таким образом, шифр абсолютно точно указывает место
фермента в общем списке. В настоящее время принято в научных
публикациях при первом упоминании фермента указывать в скобках
его шифр.
В дальнейшем при рассмотрении классификации и отдельных представи-
телей ферментов мы будем пользоваться как научными (систематическими),
так и тривиальными (рабочими) названиями.
КЛАССИФИКАЦИЯ ФЕРМЕНТОВ И ХАРАКТЕРИСТИКА
НЕКОТОРЫХ ГРУПП
По первой в истории изучения ферментов классификации их делили на две
группы: гидролазы, ускоряющие гидролитические реакции, и десмолазы,
ускоряющие реакции негидролитического распада. Затем была сделана по-
пытка разбить ферменты на классы по числу субстратов, участвующих
в реакции. В соответствии с этим ферменты классифицировали на три
группы. 1. Катализирующие превращения двух субстратов одновременно
в обоих направлениях: A+B&C+D. 2. Ускоряющие превращения двух суб-
стратов в прямой реакции и одного в обратной: Л+В?±С. 3. Обеспечиваю-
щие каталитическое видоизменение одного субстрата как в прямой, так
и в обратной реакции: А&В.
Одновременно развивалось направление, где в основу классификации фер-
ментов был положен тип реакции, подвергающейся каталитическому воздей-
ствию. Наряду с ферментами, ускоряющими реакции гидролиза (гидролазы),
были изучены ферменты, участвующие в реакциях переноса атомов и атомных
групп (феразы), в изомеризации (изомеразы), расщеплении (лиазы), различных
синтезах (синтетазы) и т. д. Это направление в классификации ферментов
оказалось наиболее плодотворным, так как объединяло ферменты в группы не
по надуманным, формальным признакам, а по типу важнейших биохимиче-
ских процессов, лежащих в основе жизнедеятельности любого организма. По
этому принципу все ферменты делят на 6 классов.
116
1. Оксидоредуктазы—ускоряют реакции окисления—восстановления.
2. Трансферазы—ускоряют реакции переноса функциональных групп и моле-
кулярных остатков. 3. ГЪдролазы—ускоряют реакции гидролитического рас-
пада. 4. Лиазы—ускоряют негидролитическое отщепление от субстратов
определенных групп атомов с образованием двойной связи (или присоединя-
ют группы атомов по двойной связи). 5. Изомеразы—ускоряют простран-
ственные или структурные перестройки в пределах одной молекулы. 6.
Лигазы—ускоряют реакции синтеза, сопряженные с распадом богатых энер-
гией связей. Эти классы и положены в основу новой научной классификации
ферментов.
1. Оксидоредуктазы. К классу оксидоредуктаз относят ферменты, катали-
зирующие реакции окисления—восстановления. Общая схема их может быть
представлена следующим образом:
___ Оксидоредуктаза
Субстрат+акцептор . . 1 Субстрат + Акцептор восста-
окисленный новленный
Окисление протекает как процесс отнятия атомов Н (электронов) от суб-
страта, а восстановление—как присоединение атомов Н (электронов) к акцеп-
тору. Если обозначить акцептор буквой А, а субстрат—В, то уравнение
реакции окисления—восстановления при участии оксидоредуктаз примет та-
кой вид:
Оксидоредуктаза
Jot12 I А ~~ —
Характерной особенностью деятельности оксидоредуктаз в живой клетке
является их способность образовывать системы (так называемые цепи окисли-
тельно-восстановительных ферментов), в которых осуществляется многосту-
пенчатый перенос атомов водорода или электронов от первичного субстрата
к конечному акцептору, которым является, как правило, кислород, так что
в результате образуется вода.
Те оксидоредуктазы, которые переносят атомы Н или электроны непосред-
ственно на кислородные атомы, носят название аэробных дегидрогеназ или
оксидаз. В отличие от них оксидоредуктазы, переносящие атомы Н и электро-
ны от одного компонента окислительной цепи ферментов к другому без
передачи их на кислородные атомы называют анаэробными дегидрогеназами
или редуктазами.
Если фермент катализирует реакцию отнятия Н непосредственно от
окисляемого вещества (первичного субстрата), то его называют первичной
дегидрогеназой. Если фермент ускоряет снятие водородных атомов со вто-
ричного субстрата, который получил атомы Н при посредстве первичной
дегидрогеназы (вторичным субстратом может быть кофермент самой пер-
вичной оксидоредуктазы), его называют вторичной дегидрогеназой (см.
гл. X).
Другая особенность оксидоредуктаз состоит в том, что, будучи двухком-
понентными ферментами с весьма ограниченным набором активных групп
(коферментов), они способны ускорять большое число самых разнообразных
окислительно-восстановительных реакций. Это достигается за счет того, что
один и тот же кофермент способен соединяться со многими апоферментами,
образуя каждый раз оксидоредуктазу, специфичную по отношению к тому или
иному субстрату или акцептору.
Еще одна, пожалуй, главная особенность оксидоредуктаз заключается
в том, что они ускоряют протекание химических процессов, связанных
117
с высвобождением энергии. Последняя используется как для обеспечения
синтетических процессов в организме, так и для других нужд.
В природных объектах обнаружено около пятисот индив дуальных ок-
сидоредуктаз. Наиболее распространены оксидоредуктазы, содержащие в ка-
честве активной группы никотинамидадениндинуклеотид, или НАД+ (о строе-
нии нуклеотидов см. гл. VI):
Окисленная форма НАД (НАД*)
Восстиюилеяия форма НДЦ (НАДН)
Более половины известных в настоящее время оксидоредуктаз содержат
НАД* в качестве кофермента. Соединяясь с тем или иным специфическим
белком и образуя таким образом двухкомпонентный фермент, который
сокращенно называют пиридинпротеином, НАД* резко усиливает свою
способность восстанавливаться по ядру никотинамида. В результате пиридин-
протеины способны отнимать от субстратов (спирты, альдегиды, дикарбо-
новые и кетокислоты, амины и др.) атомы Н в виде идрид-ионов (Н~)
И протонов (Н*), окисляя, таким образом, указанные соединения. Все пири-
динпротеины являются анаэробными дегидрогеназами, т.е. не передают
снятые с субстрата атомы водорода на кислород, а посылают их на ближай-
ший в окислительной цепи другой фермент.
Рассмотрим строение и механизм действия одного из пиридинпротеинов—
алкогольдегидрогеназы из печени животных. Это белок с М = 73000, состоя-
щий из двух субъединиц, каждая из которых несет молекулу НАД* и атом Zn.
В процессе отнятия атомов Н от спир а образуется тройной апофермент-
кофермент-субстратный комплекс, удерживаемый Zn2*. Строение этого ком-
плекса и механизм реакции окисления спирта в альдегид представлены на
рис. 53. Непосредственно к никотинамидадениндинуклеотиду от молекулы
спирта переходит один атом водорода в виде гидридного иона (Н~), т.е.
атома водорода, несущего дополнительный электрон. Второй атом водорода,
отнима мы! от молекулы спирта, наоборот, теряет электрон, превращаясь
в протон (Н+), и поступает в реакционную среду. Поэтому уравнение реакции
окисления спирта при участии НАД* записывают так:
R— СН2ОН + НАД*Ч^ R— С< + НАДН + Н
ХН
118
НАД* удерживается на поверхности белковой молекулы связями, возникающими между положительно заряженным атомом азота
пиридинового цикла и отрицательно заряженным атомом серы (из HS-группы), а также между атомами азота пуринового цикла
и атомом серы через посредство Zn2+. Молекула спирта присоединяется к активному центру фермента за счет координационной
связи между атомом О и Zn3+. Каталитическую функцию в переносе атомов водорода от молекулы спирта к НАД* выполняет
имидазольный радикал гистидина. К пиридиновому ядру НАД* присоединяется атом водорода, ранее находившийся в связи
с атомом углерода, несущим спиртовую группу. Атом водорода спиртовой группы протонируется (/). Присоединение НАД*
к апоферменту происходит по нуклеотидсвязывающему домену (Я), составленному из п-спиралеЙ и 0-слоев. Структура нуклеотпд-
связывающего домена близка у всех НАД-зависимых дегидрогеназ; к нижней части домена (0-тяжи А, В и С) присоединяется
фрагмент АМФ, а к верхней (0-тяжи D, Е и F)—фрагмент никотинамидрибозофосфата молекулы НАД*
В любом случае НАД+ получает два электрона за счет присоединения гид-
ридного иона (Н“).
Кроме НАД+ пи идинферменты содержат в качестве коф рмента никоти-
намидадениндинуклеотидфосфат (НАДФ+). Этот кофермент является произ-
водным НАД+, у которого водород ОН-группы 2-го углеродного атома
рибозы аденозина замещен на остаток фосфорной кислоты.
НАДФ+, соединяясь со специфическими белками, образует б лыпую группу
пиридинпротеинов, характеризующуюся своим набором субстратов. Механизм
окисления при участии НАДФ+ в качестве кофермента аналогичен таковому
при посредстве НАД+. Более того, НАДН и НАДФ+, равно как НАДФН
и НАД+, при каталитическом участии специального фермента трансгидроге-
назы—способны обмениваться атомами водорода и электронами:
НАД(Ф)+-
НАДФН+НАД* 7.....- » НАДФ+ + НАДН
трансгидрогеназа
Партнером восстановленных форм пиридинпротеинов в оксидоредукта -
ной цепи, как правило, служат флавопро еины (ФП). Таким флавопротеином,
например, является фермент, несущий в качестве активной группы фосфорили-
рованный витамин В2. Окисленная форма этого флавопротеина (М = 52000)
окрашена. Каждая молекула фермента несет молекулу рибофлавинфосфата
(или флавинмононуклеотида, ФМН), способного принимать и отдавать два
атома Н по атомам N изоаллоксазинового кольца:
119
^он
СН2О— Р = о
Н—ОН Ч°Н
н—<j:—он
н—с—он
^он
<^Н2О— Р = О
Н-^-ОН ОН
н—с—он
н—с—он
о но
Окрашенная форма
(окнсленная)
Бесцветная форма
(восстановленная)
Другим коферментом в флавопротеинах является флавинадениндинуклео-
тид (ФАД):
Флавиналеннндннуклеотнл (ФАД)
ФМН и ФАД, соединяясь с различными апоферментами, дают начало
приблизительно тридцати флавопротеинам, отличающимся различной специ-
фичностью по отношению к субстратам.
Основная функция флавопро еинов—перенос электронов (атомов Н) от
восстановленных пиридинпротеинов к другим компонентам окислительно-
восстановительной цепи, т.е. ФП в большинстве случаев являются вторич-
ными дегидрогеназами. Однако некоторые флавопротеины, особенно с ФАД
в качестве кофермента, могут непосредственно снима ъ атом Н с субстрата.
Коферментами оксидоредуктаз являются также хиноны. Так, соединяясь
с белком, убихиноны образуют убихинонпротенн, являющийся важной состав-
ной частью ансамблей оксидоредуктаз, при посредстве которых осуществля-
ется перенос атомов Н и электронов.
Убихиноны являются производными бензохинона и обладают боковой
цепью, составленной из большого числа изопреноидных остатков:
Оспкж жмещшнсго
беиэояиовя
120
Число изопреноидных фрагментов в боковой цепи (п) колеблется от 6 до 10.
Установлено, что убихиноны принимают участие в окислительно-восстано-
вительных процессах в организме, осуществляя передачу атомов Н:
В растениях эту функцию выполняет похожее на убихинон соединение—
пластохинон:
Пластохинон
Наиболее сложный, но и самый распространенный вариант окислительно-
восстановительного процесса в клетке состоит в окислении атомов Н, снятых
с субстрата, при посредстве цитохромной системы.
Цитохромную систему образуют несколько оксидоредуктаз, имеющих в ка-
честве простатических групп железопорфирины (рис. 54). Еще в 1915 г., двадца-
тью годами ранее О. Варбурга, на возможную роль железосодержащих белков
в биологическом окислении обратили внимание А. Я. Данилевский
и Б. П. Соловцов. Соединяясь с белками различного строения, железопорфирины
4 типов (A, B,ChD) дают начало семейству хромопротеинов, объединяемых под
общим названием—цитохромы. Сейчас известно несколько десятков цитохро-
мов, и список их непрерывно пополняется. Каждый индивидуальный цитохром
обозначают строчной латинской буквой а, Ь, с и d с соответствующим порядко-
вым индексом, например bt, b2, Ь3 и т.д., а класс цитохрома—прописной
латинской буквой А, В, С или D. Принадлежность цитохрома к определенному
классу определяется строением простатической группы (железопорфирина),
а окончательная индивидуальность—строением аг офермента (белка). В послед •
нее время предпочитают наряду с порядковым номером цитохрома указывать
характерную длину волны, при которой отмечается поглощение в видимой части
спектра (например, цитохром Ь6 или 65б3, найденный в хлоропластах, и т. п.).
Первичная структура ряда цитохромов выяснена: оказалось, что их видо-
вая специфичность связана с небольшими различиями в чередовании амино-
кислот. Эти данные принципиально важны для понимания природы видовой
и иной специфичности ферментов: видимо, она определяется различиями
121
nh-co-ch!
COOH
Рис. 54. Строение цитохрома с из сердечной мышцы лошади
Простеппесхая группа цитохрома с представлена жедезопорфирииом (с атомом Fe2+ центре). Связь его с белком осуществля-
ется за счет взаимодействия винильных радикалов гема с HS-группами 14-го и 17-го остатков цистеина полипептидной цепи.
Аминогруппа N-концевого глицина ацетилирована. На объемной модели цитохрома с цифрами указаны номера аминокислотных
остатков, каждый из которых также представлен пространственной структурой
прежде всего в первичной структуре апоферментов. Цитохромы blt b2,
Ь3тх т. д., содержащие одну и ту же простетическую группу, отличаются друг
от друга именно по этому признаку.
На рис. 54 приведена структура цитохрома с из сердца лошади. Молеку-
лярная масса цитохрома с невелика—порядка 13 000. При увеличении в 1 млн.
300 тыс. раз (электронная микроскопия) видно, что полипептидная цепь цито-
122
хрома с свернута в а-спираль длиной в 12—15 нм, которая, в свою очередь,
закручена в спираль второго порядка с диаметром в 4—5 нм, так что группа
гема оказывается изолированной внутри обвивающей ее полипептидной цепи.
Кольцеообразные молекулы легко образуют более крупные агрегаты. Строе-
ние остальных цитохромов изучено менее детально. Однако известно, что
молекулярные массы некоторых из них более высоки. Способность агрегиро-
вать друг с другом, по-видимому, одно из свойств, присущих молекулам
цитохромов.
Именно поэтому цитохромы образуют цитохромную систему, представ-
ляющую собой упорядоченное сочетание в едином комплексе различных
цитохромов, например Ь, с и а.
Цитохромная система способна принимать электроны, снятые с атомов
Н восстановленного убихинона (УХ). Она передает электроны далее по цепи
цитохромов и, наконец, на кислородный атом; последний, соединяясь с иони-
зированными атомами Н, образует молекулу Н2О.
В цепи цитохромов каждый из индивидуальных цитохромов занимает
строго определенное место. Простейший вариант цитохромной системы при-
веден на следующей схеме:
Как видно из схемы, передача электронов в цитохромной цепи осуществ-
ляется за счет изменения валентности атома Fe порфиринового ядра. Из всех
цитохромов только цитохром а, а3 передает электроны на кислород. Поэтому
именно он завершает цепь цитохромов и носит название цитохромоксидазы.
Кроме атомов Fe (в составе гема) цитохром а, а3 содержит также атомы Си,
с которыми связывают его окислительные свойства (см. гл. X, рис. 132).
Таковы характерные черты действия некоторых важнейших оксидоредуктаз
и окислительно-восстановительных систем, обеспечивающих превращение ря-
да веществ в клетке.
2. Трансферазы. В этот класс входят ферменты, ускоряющие реакции пере-
носа функциональных групп и молекулярных остатков от одного соединения
к другому. Это один из наиболее обширных классов: он насчитывает около 500
индивидуальных ферментов. В зависимости от характера переносимых груп-
пировок различают фосфотрансферазы, аминотрансферазы, гликозилтрансфе-
разы, ацилтрансферазы, трансферазы, переносящие одноуглеродные остатки
(метилтрансферазы, формилтрансферазы), и др.
Фосфотрансферазы. Сюда относятся ферменты, ускоряющие реакцию пере-
носа остатка фосфорной кислоты. Эта реакция имеет исключительно важное
значение для жизнедеятельности организма, обеспечивая превращение ряда
органических соединений в фосфорные эфиры, обладающие повышенной хими-
ческой активностью и более легко вступающие в последующие реакции. Перенос
фосфатных групп идет на спиртовые, карбоксильные, азотсодержащие, фосфор-
содержащие и другие группы тех или иных органических соединений. В соот-
ветствии с этим среди фосфортрансфераз различают несколько подподклассов.
123
Донором фосфатных остатков является в больший тве случаев аденозинт-
рифосфорная кислота (АТФ), но возможны и другие их источники. К фосфо-
трансферазам относится, например, гексокиназа—фермент, ускоряющий пере-
нос остатка фосфорной кислоты от молекулы АТФ к глюкозе (с этой реакции
обычно начинается преобразование глюкозы):
Гексокиназа распространена повсеместно. Особенно хорошо изучена ексо-
киназа дрожжей. Ее молекула (М = 96000) составлена из 4 субъединиц. Муль-
тимер устойчив при pH 5. При снижении или повышении pH раствора моле-
кула гексокиназы распадается на 4 протомера (М=24 000), лишенных фосфо-
трансферазной активности. В соответствии с двойственной природой
субъединиц в мультимерах обнаружено 5 изозимов гексокиназы.
Преобразование многих других моносахаридов тоже начинается с их фос-
форилирования при посредстве фосфотрансфераз: так обстоит дело в случае Р*
D-фруктозы, P-D-рибозы (см. с. 338) и ряда других сахаров.
Особое внимание в последнее время уделяют изучению фосфотрансфераз,
обеспечив ющих перенос остатка фосфата с АТФ на белки, -протеинкиназам.
Они переносят фосфат на радикалы сер, тре, тир, лиз и гис ряда белков,
в результате чего резко изменяется биологическая активность последних. Это,
в свою очередь, сказывается на интенсивности протекания химических фоцес-
сов в организме, т. е. на регуляции обмена веществ (см. гл. XIII).
Аминотрансферазы. Эти ферменты ускоряют реакцию переаминирования
аминокислот с кетокислотами и очень важны для обеспечения биосинтеза
аминокислот. Аминотрансферазы двухкомпонентны: простетической группой
их во всех случаях является пиридоксальфосфат, ковалентно присоединенный
к апоферменту через свою альдегидную группу (см. рис. 55, Б) и ионной
связью—через остаток фосфорной кислоты: п
Пирвдоксиафермеит
124
Механизм реакции переаминир вания сейчас хорошо выяснен, а сама реакция
открыта еще в 1937 г. А. Е. Браунштейном и М. Г. Крицман.
Для удобства пирцдоксальфермент обозначим при помощи следующей
О , _
структуры: ^с-Фч>ме»т. сохранив только функционально значимую альде-
гидную группу его кофермента.
На. первой стадии ферментативного катализа простетическая группа
фермента (для простоты принято, что она свободна, а не соединена альдимин-
ной связью с апоферментом через е-аминогруппу радикала лиз) взаимодей-
ствует с аминокислотой, подвергающейся переаминированию. Реакция идет
по аминогруппе аминокислоты и альдегидной группе остатка пиридоксаль-
фосфата:
СООН
I
сн—ын2
сн2
I
соон
соон
О. I
СН — N = CH—фермент + Н2О
С фермент | *
Н' СЦг
I
соон
Аспцмпаюмк жклоп
Фермеит-субстрвтный комплекс
На второй ступени катализа идет преобразование субстрата, выражающее-
ся в данном случае в таутомерной перегруппировке:
соон соон
СН-^-К=?Н-—фермент <. У*. C=N—СН2—фермент
сн2 сн2
соон соон
Эта перегруппировка осуществляется при участии имидазолсодержащих
радикалов остатков гис, входящих в состав каталитического центра фермента:
125
В результате последующего гидро иза освобожд ются кетокислота и фер
мент в виде пиридоксаминофе мента:
СООН СООН
С=N— СН2—фермент+НОН СО+H2N— СН2—фермент
I I
СН2 СН Пиридоксаминофермент
СООН СООН
Щавелевоуксусна кислота
Далее между пиридоксаминоферментом и другой кетокислотой вновь воз-
никает фермент-субстратный комплекс:
СООН СООН
I I
(СН2)2 (СН2)2
I I
С=О+H2N—СН2—фермент т± С=N— СН2—фермент+ Н2О
I I
СООН СООН
а-Кстоглутаровая кислота
Субстрат в нем снова подвергается преобразованию за счет таутомерного
Превращения:
СООН СООН I
(СИЛ <j?==N-j-CH2-фермент СООН (СН2)2 ♦ СН—N—СН-фермент iooH
Полученное соединение гидролизируется, и возникает новая аминокислота:
соон соон
(СН,),
I I
СН—N=CH— фермент + Н.О СН—NH, +
<!оон СООН
Глутаминовая Пиридоксаль*
кислота фермент
Следовательно, в результате серии реакций, включающих в себя попе-
ременное образование фермент-субстратных комплексов, аспарагиновая
кислота переходит в щавелевоуксусную, а а-кетоглутаровая—в глутаминовую.
Это выражается следующим суммарным уравнением:
СООН соон соон соон
chnh2 I _ ч пиридоксаль- (СНгЪ фермент со (СН2)2
1 + 1 —► + 1
со * СНд CHNH
I (аспартат- 1 1
соон ГППИ алшнотранс- LAJUri фераза) соон соон
126
- Центральную роль в пиридоксалевом катализе играет смещение электрон-
ной плотности в фермент-субстратном комплексе:
В результате у а-углеродного атома аминокислотного остатка ослабляют-
ся связи с заместителями (азотом, СООН-группой и др.), вследствие чего легко
осуществляется разрыв соответствующих связей.
Аспартатаминотрансфераза имеет молекулярную массу, равную 93000,
и состоит из двух идентичных субъединиц (М=46500), каждая из которых
соединена с молекулой пиридоксальфосфата. При разбавлении растворов
аспартатаминотрансферазы ее димеры распадаются на каталитически актив-
ные мономеры. Благодаря исследованиям главным образом советских ученых
(А. Е. Браунштейна с еотр., Ю. А. Овчинникова с сотр. и Б. К. Вайнштейна
с сотр.) выяснены первичная и третичная структуры этого фермента, строение
и детальный механизм функционирования его активного центра. Субъединица
цитозольного изофермента аспарта аминотрансферазы из сердца свиньи (дру-
гой изоф рмент локализован в митихондриях) представлена полипептидной
цепью из 412 аминокислотных остатков. Значительная часть ее находится
в а-спиральной конформации, а в обособленном участке глобулы расположен
коферментсвязывающий домен, где локализован активный центр (рис. 55).
Характерно, что коферментсвязывающий домен пиридоксальферментов очень
похож на нуклеотидсвязывающий домен НАД+- и НАДФ+-зависимых дегид-
рогеназ (см. рис. 53, II), что объясняется, видимо, рисутствием пиридинового
цикла в составе того и другого кофермента.
Поскольку аспартатаминотрансфераза состоит из двух субъединиц и несет,
следовательно, два остатка пиридоксальфосфата, в реакции переаминирова-
ния субъединицы работают согласованно, со сдвигом по фазе в использовании
энергии, необходимой для осуществления химических преобразований; вслед-
ствие этого димерная структура фермента дает существенный выигрыш в осу-
ществлении каталитического процесса.
пу лизина
Рис. 55. Один из возмож-
ных вариантов третичной
структуры аспартатамино-
трансферазы (Л) и строе-
ние ее активного центра
(В)
На рис. Б видно, что пиридоксаль-
фосфат соединен альдиминной
связью с Е-аминогруппой остатка
лизина в апоферменте; именно по
этой альдиминной связи присоеди-
няется аминогруппа аминокисло-
ты, вытесняя оттуда е-аминогруп-
127
Ешкозилтрансферазы. Эти ферменты ускоряют реакции переноса гликозиль-
ных остатков из молекул фосфорных эфиров или других соединений к молекулам
моносахаридов, полисахаридов или иных веществ, обеспечивая главным обра-
зом реакции синтеза и распада олиго- и полисахаридов в животном и раститель-
ном мире. Ниже приведено уравнение реакции распада сахарозы при участии
сахароза: ортофосфат-а-глюкозилтрансферазы, или сахарозофосфорилазы:
Аналогично этому действуют крахмалфосфорилаза, гликогенфосфорилаза
и другие гликозилтрансферазы. В случае переноса гликозильных остатков на
Н3РО4 этот процесс называют фосфоролизом, так как он формально аналоги-
чен гидролизу, но вместо элементов воды по месту разрыва кислородного
мостика присоединяются водород и фосфатная группа фосфорной кислоты
(подробнее о гликогенфосфорилазе и механизме ее действия см. гл. VIII).
В последнее время выяснено, что перенос гликозильных остатков особенно
легко осуществляется ферментами данной группы в тех случаях, когда суб-
стратом служит нуклеозиддифосфатмоносахарид. Эта реакция представляет,
видимо, основной путь природного синтеза олиго- и полисахаридов и будет
детально рассмотрена в гл. VIII. Нуклеозцддифосфатсахара являются кофер-
ментами гликозилтрансфераз.
Ацилтраясферазы. Эти ферменты ускоряют перенос ацилов (остатков карбо-
новых кислот) на аминокислоты, амины, спирты и другие соединения. Уни-
версальным источником ацильных групп во всех этих реакциях является
ацил-коэнзим А, который с полным основанием можно рассматривать как
активную группу ацилтрансфераз.
Чаще всего переносу в биологических объектах подвергается ацил уксусной
кислоты—ацетил |СН3—С/ I.
\ Х>/
Коэнзим А (см. формулу на с. 163), соединяясь с ацетильным остатком, кото-
рый занимает место водорода в его HS-группе, образует ацетил-коэнзим А.
Последний служит кофактором в соответствующей реакции переноса. Одним
из примеров реакции трансацилирования является синтез ацетилхолина:
4- Холивацеплтршсферая
СН3-С + НО—СН2—СН2—N (СН3)3 ---------------------►
КоА Ходни
Ацетил-коэнзим А
------► СН3-С +
^О—СН,—СН,— N(CH3)S + HSKoA
Ацетилхслнн Каэизнм А
128
Важное значение среди трансфераз имеют ферменты, ускоряющие перенос
одноуглеродных фрагментов (метильных, ок иметильны , формильных
и т. и.), а также нуклеотидилтрансферазы, катализирующие перенос нуклео-
тидных остатков в процессе синтеза нуклеиновых кислот. Механизм их дей-
ствия будет описан ниже.
3. Гидролазы. К классу гидролаз относят ферменты, ускоряющие реакции
расщепления (а иногда и синтеза) ор анических соединений при участии воды:
R'R"+HOH?±R'H+R"OH. В зависимости от характера субстрата, подвергающе-
гося гидролизу, гидролазы делят на ряд подклассов, среди которых наиболее
важны следующие: 1) эст разы, ускоряющие реакции гидр лиза сложных эфиров;
2) гликозидазы, ускоряющие реакции гидролиза гликозидов, в том числе углеводов;
3) пептид-гидролазы, ускоряющие реакции гидролиза (а в особых случаях
и синтеза) белков, пептидов и других со динений, содержащих пептидные связи;
4) гидролазы, действующие на С—N-связи, отличающиеся от пептидных (например,
амидазы и т. п.). Всего в составе гидролаз насчитывают почти 500 ферментов.
Эстеразы. Эти ферменты катализи уют реакции гидролиза сложных эфи-
ров спиртов с органическими и неорганическими кислотами. Важнейшими
подподклассами эстераз являются гидролазы эфиров карбоновых кислот и фос-
фатазы. В качестве представ теля первого подподкласса рассмотрим липазу.
Липаза ускоряет гидролиз внешних, т. е. ос-сложноэфирных, связей в моле-
кулах триацилглицеринов (жиров):
сн2—о—со—с13н31
I
сн—о—со—с13нзх
сн2—о—со—с13н31
Трипальмитиа
сн2—ОН
Липаза |
4-2НОН~----» СН—О—СО—СХЗНЗХ+2СХЗН31СООН
СН2—ОН Пальмитиновая
кислота
р-Пальмитил-
глицерин
Механизм действия ряда эстераз детально изучен. Один из примеров
рассмотрен в этой главе (см. раздел о механизме действия ферментов). Харак-
теристика липаз дана в гл. IX.
Фосфатазы катализируют гидролиз фосфорных эфиров. Особенно широко
распространены фосфатазы, действующие на сложные эфиры фосфорной кис-
лоты и углеводов, например глюкозо-1-фосфатаза:
Н2О
Г люкозо-1- фосфат
D-Глюкозо-!-
фосфат-
фосфогкдролаза
Глюкоза
Действие фосфатаз проявляется в широком спектре pH от 3 до 9. Большин
ство из них обладает широкой субстратной специфичностью. Особенно важны
для регуляции процессов жизнедеятельности протеинфосфатазы, обеспечиваю
щие отщепление фосфата от фосфорилированных белков, вследствие чего
изменяется их биологическая, в частности ферментативная, активность.
ГЬикозидазы Эти ферменты ускоряют реакцию гидролиза гликозидов.
В зависимости от того, на какой пространственный изомер (а или 0) действует
5 —3502 129
фермент, его относят к а- или Р-гликозидазам. Таким образом, гликозидазы
обладают ярко выраженной пространственной специфичностью. Кроме глико-
зидов, содержащих в качестве агликонов остатки одноатомных спиртов, суб-
стратами, на которые распространяется действие тех или иных гликозидаз,
являются олиго- и полисахариды. Из действующих на олигосахариды глико-
зидаз упомянем мальтазу (а-гликозидаза) и сахаразу (Р-гликозидаза). Они
ускоряют соответственно гидролиз мальтозы и сахарозы:
+ н2о
н
Глюкоза Фруктоза
(a-D-глюкопираиом) (0- D-фруктофурвном)
н2он
н2он
н
Мальтоза (а-D-глюко пиранозидо-
1,4-D-глюкопираноза)
а-Глюкозидаза
(мальтаза)
Г люком
(a-D -глюкопираном)
Из гликозидаз, действующих на полисахариды, наиболее известны амилазы.
В природе существует несколько видов амилаз, ускоряющих реакции гидролиза
гликозидных связей в молекуле крахмала с образованием глюкозы, мальтозы или
олигосахаридов. Их характеристика и механизм действия рассмотрены в гл. VIII.
Гидролиз других природных полигликозидов: целлюлозы, инулина, ксила-
на и т. п.—также ускоряется соответствующими гликозидазами. Некоторые
гликозидазы катализируют также реакции переноса гликозильных остатков,
т. е. являются трансгликозидазами.
Пептцд-гидролазы. Ферменты этого подкласса ускоряют гидролиз пептид-
ных связей в белках и пептидах, а при определенных условиях также и об-
разование пептидных связей, хотя этот путь синтеза белка не является физио-
логическим. Химизм процесса гидролиза белков и пептидов при участии
пептидгидролаз можно выразить следующей схемой:
H2N—СН—СО—
NH—СН—СО
L I
—NH—СН—СООН+(п+ 1)Н2О
Пептид-гидролаза
~ * H2N—СН—COOH+nH3N—СН—COOH + H2N—СН—СООН
I I I
R' R R"
Среди пептид-гидролаз различают протеиназы или пептндил-пептидогид-
ролазы, катализирующие гидролиз небольшого числа внутренних пептидных
связей в белковой молекуле, в результате чего последняя распадается до
пептидов. Они являются, следовательно, эндопептидазами. В отличие от этого
пептид-гидролазы, называемые пептидазами, обеспечивают отщепление от
пептидной цепи свободных аминокислот, будучи экзопептидазами.
Протеиназы в зависимости от механизма их действия на внутренние пеп-
тидные связи в белковой молекуле делят на 4 подподкласса: 1) сериновые
протеиназы, несущие в активном центре радикалы сер и гис, обеспечивающие
130
Осуществление каталитического акта; представителями их являются химотри-
псин и трипсин, выделяемые поджелудочной железой, субтилизин, продуциру-
емый бактериями, и др.; 2) тиоловые (цистеиновые) протеиназы, имеющие
в активном центре остаток t/wc; к их числу принадлежат папаин из латекса
дынного дерева Carica papaya, фицин из латекса фикуса, бр< мелаин из сока
ствола ананаса, катепсин В—внутриклеточный фермент позвоночных и др.;
3) кислые (карбоксильные) протеиназы, имеющие оптимум pH ниже 5 и содер-
жащие радикалы дикарбоновых аминокислот в активном центре; сюда отно-
сятся пепсин, выделяемый слизистой желудка, катепсин D, характеризующий-
ся внутриклеточной локализацией, и ряд кислых протеиназ, продуцируемых
разнообразными микроорганизмами; 4) металлопротеиназы, каталитич ское
действие которых зависит от присутствия ионов металлов (Са2+, Zn2+) в ак-
тивном центре; примерами их могут служить коллагеназа и ряд протеиназ
микробного происхождения (термолизин, компонент проназы и др.).
Пепсин, трипсин и химотрипсин выделяются железистыми клетками в виде
неактивных проферментов—зимогенов: пепсиногена, трипсиногена и химо-
трипсиногена, так как их активные центры блокированы фрагментами полипеп-
тидной цепи, после гидролитического отщепления которых фермент приоб-
ретает активность. Это явление впервые было открыто в лаборатории
И. П. Павлова.
Очень важной особенностью протеиназ является выборочный (селективный)
характер их действия на пептидные связи в белковой молекуле. Так, пепсин
избирательно ускоряет гидролиз пептидных связей, образованных фен и лей-,
трипсин—арг и лиз-, химотрипсин—ароматическими аминокислотами; папа-
ин—арг, лиз и фен и т. д. В результате индивидуальный белок под действием
определенной пептидил-пептидогидролазы расщепляется всегда на строго
ограниченное число пептидов. Это находит прак ическое использование при
определении первичной структуры белков и имеет огромное значение для
регуляции обмена веществ, так как многие продукты селективного гидролиза
белков обладают высочайшей биологической активностью: именно этим
путем из проферментов во никают ферменты, из предшественников гормо-
нов—гормоны и рилизинг-факторы и т. п. Причина избирательного действия
пептидп птидогидролаз заключается в том, что радикал аминокислоты, по
соседству с которой гидролизуется пептидная связь, служит для образования
фермент-субстратного комплекса.
Пептид гидролазы, катализирующие гидролиз пептидов до свободных
аминокислот, могут отщеплять последние от пептида, начиная либо с амино-
кислоты, обладающей свободной ЫН2-грушюй, либо с аминокислоты, име-
ющей свободную СООН-группу. В первом случае их называют аминопеп-
тидазами (а-аминоацилпептид-гидролазы), во втором—карбоксипептидазами
(пептидиламиноацидо-гидролазы).
Схема, поясняющая действие амино- и карбоксипептидаз, а также некото-
рых эндопептидаз, привед на на рис. 56.
Некоторые амино- и карбоксипептидазы обладают специфичностью дей-
ствия, т. е. отщепляют строго определенные N- или С-концевые аминокисло-
ты. Третий подподкласс пептидаз представлен дипептид-гидролазами, или
дипептидазами. Их известно около десяти. Они завершают гидролиз белка.
Недавно из состава пептидаз вычленено еще два подподкласса: дипептидил-
пептидгидролазы, отщепляющие от N-конца полипептида дипептид, и пеп-
тидилдипептид-гидролазы, отщепляющие дипептид с С-конца.
Амидазы. Эти ферменты ускоряют гидролиз амидов кислот. Из них важ-
ную роль в биохимических про ессах в организме играют уреаза, аспарагина-
за и глутаминаза.
131
Лейцин-
амино-
псптидаза
Пепсия Трипсин
Химотрипсин Карбоксипептцдаза А
СН3СН3
СН
СН2
H2N—СН—С
2 «
Лей °
NH2
СН2
СН2
СНз
ОН
ОН
СН2
СНз
NH-CH — С
1
О
,, сн2 ,, сн2 сн, , ,, сн,
| Z * | Z I Z ¥|г
fiNH-CH-cfiNH-CH-C-NH-CH-C0NH-CH2-C-NH-CH2-cfiNH-CH-COOH
В В II II I
Фен
Лей
Тир
И
О
Лиз
Apr
Сер
I
Тир ° Гли ° Гли ° Фен и другие
- нейтральные
аминокислоты
О
Рис. 56. Точки приложения действия протеолитических ферментов на пептидные связи в белковой
молекуле
Уреаза была одним из первых белков-ферментов, полученным в кристал-
лическом состоянии (Д. Самнер, 1926). Это однокомпонентный фермент
(М=480 000); молекула его глобулярна и состоит из 8 равных субъединиц.
Уреаза ускоряет гидролиз мочевины до NH3 и СО2.
Аспарагиназа и глутаминаза ускоряю' гидролиз амидов дикарбоновых амино-
кислот—аспарагиновой и глутаминовой, например:
Аспарагиназ*
НООС—СН-СНа-С + Н»О---------------
1 V (L-ас па par мн-а мм-
XNH* дргцдролаэ*)
Аспарагин
-------- ноос-сн-сн2-соон + NHS
NHt f
Аспарагиновая кислот*
К гидролазам, действующим на С—N-связи, отличающиеся от пептидных,
кроме амидаз относятся ферменты, катализирующие гидролиз С—N-связей
в линейных амидинах. К их числу принадлежит аргиназа. При посредстве
аргиназы аминокислота аргинин гидролизуется на орнитин и мочевину:
Аргиназ*
Н N—С—NH—(СН»)3—СН—СООН + Н2О ------------
II I
NH NHt
Apr шин
------ H2N—С—NH2 + H2N—(CHJ3—СН—соон
и I
О NH,
Мочевина Оригоя
Так как в процессе гидролиза аргинина отщепляется мочевина, то системати-
ческое название аргиназы—L-аргинин-уреогидролаза. Эта реакция широко пред-
ставлена в природе, являясь заключитель ой стадией биосинтеза мочевины—
одного из конечных продуктов распада азотсодержащих веществ. Для аргиназы,
как и для уреазы, характерна абсолютная специфичность действия.
4. Лиазы. К классу лиаз относятся ферменты, ускоряющие негидролитичес-
кие реакции распада органических соединений по связям С—С; С—N; С—О
и т. д. При этом замыкаются двойные связи и выделяются такие простейшие
132
продукты, как СО2, Н2О, NH3 и т. п. Некоторые из этих реакций обратимы,
и соответствующие ферменты в подходящих условиях катализируют реакции не
только распада, но и синтеза. Таким образом, название этого класса ферментов
не всегда соответствует содержанию тех процессов, которые ими ускоряются.
Одной из важнейших групп ферментов этого класса являются углерод-
углерод-лиазы (С—С-лиазы). Среди них особое значение имеют карбоксили-
азы (декарбоксилазы) и альдегид-лиазы.
В природе широко распространены декарбоксилазы кетокислот и амино-
кислот, катализирующие реакции по следующим схемам:
сн3—со-соон
Пировиноградная
кислота
НООС—сн2—со-соон
Щавелевоуксусная кислота
ПируватдекарбоксиММ
(карбокси-лиаза 2-оксо-
пропионовой кислоты)
Оксалоацетатдскарбоксмлаза
-------------------------» сн3—со—соон + со»
(оксалоацапт-карбоксн-
лиаза)
СНг-(СН2)3-СН-С00Н
NH, NHa
Л нами
Лизиндекарбокснлаза
H2N—(СН2)а—NH, + СО,
(L- лизин-карбокси-
лиаза)
Кадаверин
Эти ферменты двухкомпоненты, простетическими группами их во многих
случаях являются фосфорные эфиры водорастворимых витаминов: тиамина
(Вг)—в карбокси-лиазах кетокислот и пиридоксаля (В6)—в карбокси-лиазах
аминокислот.
Механизм реакции декарбоксилирования кетокислот при участии декарбокси-
лаз с тиаминпирофосфатом в качестве кофермента будет рассмотрен в гл. IV.
Механизм реакции декарбоксилирования аминокислот с помощью карбокси-
лиазы с пиридоксальфосфатом в качестве кофермента очень близок к тому,
который действует при переаминировании аминокислот. Здесь тоже возникает
шиффово основание, в котором электронная плотность у а-углеродного атома
аминокислоты резко ослаблена. Вследствие этого сильно ослабляется связь этого
углеродного атома с карбоксильной группой, и последняя легко отщепляется.
Характерным представителем альдегид-лиаз является альдолаза, катализирую-
щая обратимую реакцию расщепления фруктозо-1,6-дифосфата до фосфотриоз:
?н
о=р—о—сн,
I <
/он
. сн,он
Альдолаза I 1
он
дн
H'ijl
он
,, (фруктозо-1,6- I
СН,—О—Р=О дифосфат-
ItA' -Vi'. I триозофосфатлиаза)
НО ОН
он
он
Фруктофуранозо-1,6- дифосфат
Фосфоднокси-
ацетон
;нон ун
:н2—о—р=о
З-Фосфоглицери-
новый альдегид
Эта реакция занимает центральное место в преобразовании углеводов.
Аналогично альдолазе действуют другие альдегид-лиазы.
Другую важную группу лиаз составляют углерод—кислород лиазы (гидро-
лиазы), ускоряющие реакции гидратирования и дегидратирования органиче-
ских соединений. В качестве представителя гидро-лиаз приведем фумарат-
гвдратазу:
1
133
Дегидратирование НООС— С-— Н
НООС—СН (ОН)— СН2— СООН —> II + Н2О
Н— С— СООН
Яблочная кислота 1мдратирование
Фумаровая кислота
Реакции гидратирования и дегидратирования постоянно идут при распаде
и синтезе углеводов и высших жирных кислот, поэтому гидратазы играют
большую роль в жизнедеятельности организмов.
Примером углерод—азот лиаз может служить аспартат—аммиак-лиаза,
ускоряющая реакцию прямого дезаминирования аспарагиновой кислоты:
соон
| Аспартат-аммиах-лиаза
chnh2 , ноос—с—н
I 4 II
СН2 Н—С—COONH4
| Фумарат аммония
СООН
Аспарагиновая
кислота
Этот фермент характерен для бактерий и ряда растений.
Некоторые лиазы ускоряют реакции не только распада, но и синтеза.
Например, из дрожжей выделена L-серин-гидро-лиаза, отщепляющая от серина
воду и присоединяющая сероводород, в результате чего синтезируется амино-
кислота —цистеин:
НО - СН2 - СН - СООН + H2S----------- HS - сн2 - сн - соон + Н2О
I I
nh2 nh2 а
Чтобы отличить такие лиазы от ферментов класса лигаз (которые ускоря-
ют реакции только синтеза и именуются в связи с этим синте азами), их
называют также синтазами.
5. Изомеразы. Ферменты, относящиеся к этому немногочисленному (около
90 индивидуальных ферментов) классу, ускоряют геометрические или струк-
турные изменения в пределах одной молекулы. Эти изменения могут состоять
во внутримолекулярном переносе водорода, фосфатных и ацильных групп,
в изменении пространственного расположения атомных группировок, в пере-
мещении двойных связей и т. п.
Важнейшими изомеразами являются триозофосфатизомераза, фосфогли-
церат-фосфомутаза, альдозомутаротаза и изопентенил-пирофосфатизомераза.
Триозофосфатизомераза ускоряет перенос атомов Н в процессе превращения
3-фосфоглицеринового альдегида в фосфодиоксиацетон и обратно:
Триозофосфатизомераза
снон
он
I
СН2—О—Р=О
он
СН2ОН
С=о ОН
сн2—о—Р=о
он
З-Фосфоглицериновый
альдегид
Фосфодиоксиацетон
134
Превращение, вероятно, идет через общую эндиольную форму.
Фосфоглицерат-фосфомутаза обеспечивает достаточную скорость превраще-
ния 2-фосфоглицериновой кислоты в 3-фосфоглицериновую кислоту и обратно:
СН2ОН он 1 1 ОН Фосфоглицерат-фосфомутаза |
СН—О—Р=О 1 1 t 1 СН2—О—Р—О 1 1
1 1 соон он 2-Фосфоглицерат 1 сноп он 1 соон
З-Фосфоглицерат
Оба процесса имеют громадное • значение в органичес ом мире, так как
представляют важнейшие стадии распада и синтеза углеводов.
Мутаротаза является пред тавителем стереоизомераз, она ускоряет реак-
цию превращения a-D-глюкопиранозы в p-D-глюкопиранозу:
Альдозо-
мутаротазд
a-D -Глюкопираноза
(альдозо-1-
эпимераза )
p-D-Глюкопираиоза
Ферменты этого типа (стереоизомеразы) обеспечивают, в частности, вза-
имопревращения многих пространственных изомеров моносахаридов, и этот
путь является иногда еди ственным для синтеза некоторых из них в природе.
К стереоизомеразам относятся также цис-трансизомеразы, например ретинол-
изомераза, переводящая транс-ретинол в цис-ретинол (см. гл. IV).
Изопентенилпирофосфат-изомераза катализирует реакцию перестройки изо-
пентенилпирофосфата в диметилаллилпирофосфат, что связано с перемещени-
ем двойной связи из 3-го во 2-е положение:
1 СН3 О О
V I II II I
Н2С—С CHj—СН4—О—Р—О—Р—OH + HS—фермент
I Лн ОН /х (изопеитенилпиро-
__________________/ фосфат-изомераза)
Изопеитенилпирофосфат
СН, О О
I II II
—*н3с—су2—СН2—О—Р—О—Р-ОН —*
S—фермент Ан ОН
О—Р—О—Р—ОН + HS—фермент
ОН ОН
Диметилаллилпирофосфат
Изопентенилпирофосфат-изомераза содержит свободны сульфгидрильные
группы, вероятно, в виде радикалов цис в белковой молекуле. Именно благо-
даря им обеспечивается указанная выше реакция, имеющая огромное значение
для синтеза полиизопреноидов и стеролов. •
135
6. Лигазы (синтетазы). Характерные черты действия ферментов этого клас-
са выявлены совсем недавно в связи со значительными успехами в изучении
механизма синтеза жиров, белков и углеводов. Оказалось, что старые пред-
ставления об образовании этих соединений, согласно которым они возникают
при обращении реакций гидролиза, не соответствуют действительности. Пути
их синтеза принципиально иные.
Главная их особенность—сопряженность синтеза с распадом веществ, спо-
собных поставлять энергию для осуществления биосинтетического процесса.
Одним из таких природных соединений является АТФ. При отрыве от ее
молекулы в присутствии лигаз одного или двух концевых остатков фосфорной
кислоты выделяется большое количество энергии, используемой для активи-
рования реагирующих веществ. Лигазы же -каталитически ускоряют синтез
органических соединений из активированных за счет распада АТФ исходных
продуктов. Таким образом, к лигазам относятся ферменты, катализирующие
соединение друг с другом двух молекул, сопряженное с гидролизом пирофос-
фатной связи в молекуле АТФ или иного нуклеозидтрифосфата.
Механизм действия лигаз изучен еще недостаточно, но, несомненно, он
весьма сложен. В ряде случаев доказано, что одно из участвующих в основной
реакции веществ сначала дает промежуточное соединение с фрагментом рас-
падающейся молекулы АТФ, а вслед за этим указанный промежуточный
продукт взаимодействует со вторым партнером основной химической реакции
с образованием конечного продукта.
В качестве примера действия лигазы можно привести синтез пантотеновой
кислоты из а, у-диокси-0, Р-диметилмасляной кислоты и Р-аланина:
АМФ
Пантотеиатсинтетаза, как следует из приведенного уравнения, относится
к группе лигаз, ускоряющих реакции синтеза С—N-связей. Эта группа лигаз
насчитывает около 40 представителей. В настоящее время изучено более 75
различных лигаз, обеспечивающих важнейшие синтетические процессы в жи-
вотной и растительной клетках.
Кроме ускорения реакций синтеза С—N-связей, в частности пептидных,
лигазы катализируют образование связей С—С, С—О и С—S. К группе лигаз,
образующих С—t-связи, относятся карбоксилазы. Они обеспечивают карбок-
силирование ряда соединений, в результате чего происходит удлинение угле-
родн!<х цепей. Одной из важнейших карбоксилаз является* пируваткарбок-
136
силаза, ускоряющая реакцию образования щавелевоуксусной кислоты из пи-
ровиноградной кислоты и оксида углерода (IV):
Пнруваткарбо-
кснлаза
АДФ
Эта реакция имеет исключительное значение в обмене веществ, обеспечи-
вая взаимосвязь обмена углеводов и белков и акцептирование СО2.
Лигазам, катализирующим синтез С—О-связей, принадлежит важнейшая
роль в биосинтезе белков, так как они ускоряют реакции активирования
аминокислот перед вступлением последних в пептидную связь. Одной из
простейших реакций этого типа является образование аминоациладенилатов:
137
С ами оациладенилатов аминок слоты передаются на тРНК, образуя
аминоацил-тРНК, используемые непосредственно при синтезе полипептидов.
Эти процессы будут детально рассмотрены в гл. VII.
Лигазы, кроме того, осуществляют ускорение реакций образования С—S-
связей, являясь ацил-коэизим А-синтетазами. В качестве примера действия
ацил-коэнзим А-синтетазы можно привести образование ацетил-коэнзима
А из уксусной кислоты и коэнзима А (см. формулу на с. 163), протекающее
сопряженно с распадом АТФ:
Ацетил-КоА-синтетаза
СНз—COOH + HS—КоА+АТФ-------------------------►
(ацетат-КоА-лигаза)
О о
--->СН3—СО—S—КоА+АМФ+НО—Р—О—Р—ОН
I I
ОН он
Ацетал-жоэнзим А Пирофосфат
Ацетил-КоА служит коферментом в реакциях трансацилир вания, поэтому
действие ферментов этих двух групп—лигаз и ацилтрансфераз—в живых
системах тесно увязано друг с другом. Аналогичная взаимозависимость харак-
терна и для многих других ферментов.
ЛОКАЛИЗАЦИЯ ФЕРМЕНТОВ В КЛЕТКЕ
Одним из принципиальных отличий ферментов от катализаторов неби-
ологического происхождения является кооперативный характер их действия.
На уровне одиночной молекулы фермента кооперативный принцип реализует-
ся в тонком взаимодействии субстратного, активного и аллостерического
центров. Однако гораздо большее значение имеет кооперативное осуществле-
ние реакций на уровне ансамблей ферментов. Именно благодаря наличию
систем ферментов—в виде мультиэизимных комплексов или еще более слож-
ных образований—метаболонов, обеспечивающих каталитические превраще-
ния всех участников единого метаболического цикла—в клетках с большой
скоростью осуществляются многостадийные процессы как распада, так и син-
теза органических молекул. Ферментативный катализ в многостадийных реак-
циях идет без выделения промежуточных продуктов: только возникнув, они
тут же подвергаются дальнейшим преобразованиям.
Это возможно лишь потому, что в клеточном содержимом ферменты
распределены не хаотически, а строго упорядоченно. С современной точки
зрения клетка представляется высокоорганизованной системой (см. рис. 3),
в отдельных частях которой осуществляются строго определенные биохими-
ческие процессы. В соответствии с приуроченностью их к определенным
субклеточным частицам или отсекам (компартментам) клетки в них локализо-
ваны те или иные индивидуальные ферменты, мультиэнзимные комплексы,
полифункциональные ферменты или сложнейшие метаболоны.
Приведем несколько характерных примеров. Разнообразные гидролазы
и лиазы сосредоточены преимущественно в лизосомах. Внутри этих сравнительно
небольших (несколько нанометров в диаметре) пузырьков, отграниченных
мембраной от гиалоплазмы клетки, протекают процессы деструкции различных
органических соединений до тех простейших структурных единиц, из которых они
138
построены. Сложные ансамбли окислительно-восстановительных ферментов,
такие, например, как цитохромная система, находятся в митохондриях. В этих же
субклеточных частицах локализован набор ферментов цикла дикарбоновых
и трикарбоновых кислот (см. гл. VIII). Фермент! активирования аминокислот
распределены в гиалоплазме, но они же есть и в ядре. В гиалоплазме присутству-
ют многочисленные метаболоны гликолиза, структурно объединенные с таковы-
ми пентозофосфатного цикла, что обеспечивает взаимопереключение дихотоми-
ческого и апотомического путей распада углеводов (см. гл. VIII). В то же время
' ферменты, ускоряющие перенос аминокислотных остатков на растущий конец
полипептидной цепи и катализирующие некоторые другие реакции в процессе
биосинтеза белка, сосредоточены в рибосомальном аппарате клетки. Нуклеоти-
дилтрансферазы, ускоряющие реакцию переноса нуклеотидных остатков при
новообразовании нуклеиновых кислот, локализованы в основном в ядерном
аппарате клетки. Таким образом, системы ферментов, сосредоточенные в тех или
иных структурах, участвуют в осуществлении отдельных циклов реакций. Будучи
тонко координированы друг с другом, эти отдельные циклы реакций обеспечива-
ют жизнедеятельность клеток, органов, тканей и oj ганизма в целом.
ПРИМЕНЕНИЕ ФЕРМЕНТОВ
Ферменты способны осуществлять свою каталитическую функцию и вне
клетки, вне организма. Хотя, находясь с составе соответствующих структур,
ферменты об адают огромной мощностью действия, тем не менее, будучи
выделены из биологических объектов, они сохраняют свою каталитическую
активность. Естественно, что ряд ферментных препаратов используют в практике.
Практическое применение ферментов в народном хозяйстве нашей страны
расширяется с каждым годом. В хлебопекарной промышленности применяют
ферментные препараты (содержат амилазу и протеиназу), выделяемые из
грибов, относящихся к роду Aspergillus. Будучи добавлены в количестве 20—25 г
на 1 т муки, они улучшают качество и аромат хлеба, ускоряют созревание теста
на 30%, сокращают расход сахара на производство высших сортов булочных
изделий вдвое, увеличивают пористость мякиша и объем хлеба на 20%.
В пивоварении и спиртовой промышленности применяют амилазы—фермен-
ты, ускоряющие реакцию осахаривания крахмала. Их производство приняло
у нас промышленный характер. Экон мический эффект применения амилаз
весьма значителен. Так, в пивоварении использование ферментных препаратов
позволяет экономить 165 г ячменя при производстве каждо о декалитра пива.
Применение амилазы при производстве спирта дает возможность полностью
отказаться от зернового солода и одновременно увеличить выход спирта из
сырья на 1,5% при снижении себестоимости декалитра спирта. Широкие
перспективы сулит использование ферментов в виноделии. Пектш олитические
ферменты повышают выход соков из плодов и ягод на 15—20%, виноматериа-
лов—на 5—7%; они также необходимы для осветления фруктовых соков.
Каталаза и глюкозооксидаза длительно сохраняют букет розовых и белых вин.
В кожевенном н меховом производстве для ускорения снятия волоса со шкур
и размягчения кожевенного сырья применяют препараты протеиназ (протелин
и протофрадин), являющихся внеклеточными протеиназами стрептомицетов.
При этом время на осуществление необходимых процессов сокращается в не-
сколько раз сортность и качество шерсти и кож повышается, а условия труда
в этой отрасли производства резко улучшаются. В текстильной промышлен
ностн процесс расшлихтовки тканей ферментными препаратами грибного
и бактериального происхождения ускоряется в 7—10 раз; эти же препараты
139
служат для удаления серицина при размотке коконов тутового шелкопряда
в производстве натурального шелка. В кулинарии применение пептидгидролаз
(протелин и проназа) для обработки мяса перед его приготовлением резко
улучшает качество мясных блюд. В мясной промышленности протеолитические
ферменты применяют для уск рения созревания мяса и повышения выхода мяса
1-го сорта с 15 до 40%. В молочной промышленности использование протеаз
ускоряет созревание сыров вдвое и снижает их себестоимость на 10%. Подсчеты
п казывают, что если полностью обеспечить кожевенную, пищевую и текстиль-
ную промышленность ферментными препаратами (их потребуется несколько
тысяч тонн в год), то за счет этого можно получить звачительную экономию.
Ферменты прим няют и в бытовой химии. Протеазы растительного проис-
хождения, выдерживающие нагревание до 90° С без заметной потери актив-
ности, являются компонентами стиральных порошков и моющих средств.
В стиральные порошки вводят также а-амилазу, глюкозооксидазу и липок-
сигеназу, а уратоксидаза в составе моющих средств способствует удалению
винных и жирных пятен с одежды. Глюкозооксидазу, каталазу и некоторые
другие ферменты добавляют в зубную пасту—они обеспечивают их антимик-
робные свойства и предохранение зубов от кариеса.
Крупномасштабным является производство глюкозы из отходов целлюло-
зы при посредстве целлолитического комплекса ферментов и из крахмала при
помощи глюкоамилазы. Обработка дигестазой (комплекс протеолитических
ферментов, выделяемый из печени крабов) повышает на 3.0% выход товарного
продукта при обработке икры, извлеченной из рыб.
Ферменты находят большое применение в медицине. Пепсин, трипсин,
химотрипсин, липазу и амилазу в виде ферментных препаратов и их смесей
(бетацид, абомин, фестал, панзинорм и др.) применяют для лечения заболева-
ний желудочно-кишечного тракта. Гиалуронидазу (у нас выпуск ют два ее
препарата, выделяемых из семенников быка,—лидазу и ронидазу), деполимери-
' зующую гиалуроновую кислоту и способствующую проникновению лекарствен-
ных средств в пораженную ткань—для лечения забо еваний суставов, отеков,
ран, кровопод теков и т. п. Протеолитические ферменты—плазмин (фибриноли-
зин) и активирующие его стрептокиназу и урокиназу—для растворения тром-
бов в кровеносных сосудах. Дезоксирибонуклеазу из поджелудочной железы или
стрептококка (стрептодорназа)—для лечения заболеваний верхних дыхатель-
ных путей и роговицы глаза, а также для удаления гноя из ран. Аспарагиназу,
беспечивающую дезамидирование аспарагина, незаменимого для роста ряда
опухолей,—при лечении некоторых видов рака. Лизоцим —для лечения конъюн-
ктивитов, цитохром—для устранения явлений кислородного голодания при
заболеваниях сердца, коллагеназу—для рассасывания рубцовых образований,
эластазу—для задержки развития атеросклероза, Р-галактозидазу—для снятия
явления епереносимости молочных продуктов из-за недостаточности этого
фермента в пищеварительном тракте ряда людей, галактокиназу—для выведе-
ния галактозы из тканей при ее патологическом накоплении в них, L-фенилал -
нин аммиак-лиазу—для понижения содержания в крови фенилаланина при
нарушениях его обмена, лизоамидазу—для лечения заболеваний, вызванных
патогенными микроорганизмами (стафилококки, стрептококки и др.).
Специфическую область применения ферментов в медицине составляет
энзимодиагностика: заболевание того или иного органа у человека может быть
тестировано по уровню содержания фермента или соотношению его множест
венных форм (в том числе изозимов) в крови или реже в моче. Так, пактатдеги-
дрогеназа (ЛДГ), аспартатаминотрансфераза (АсАТ), креатинкиназа, изоцит-
ратдегидр геназа и фруктозо-1,6-дифосфат-альдолаза служат для диагноза
инфаркта миокарда; ЛДГ, АсАТ и аланинаминотрансфераза—заболевания
140
1955 I960 1955 1970
Рис. 57. Развитие работ по ин-
женерной энзимологии:
светлыми кружками и тонкой цветной ли-
нией обозначен ход кривой, отражающей
ежегодные публикации по изучению иммо-
билизованных ферментов, темными н тол-
стой цветной линией—число получаемых
ежегодно патентов
печени, у-глутамилтрансфераза—отторжения ор-
ганов при их пересадке, щелочная фосфатаза—
заболевания желтухой, кислая фосфатаза—нару-
шения функции предстательной железы и т. д.
Особенно широко в последние годы процессы
ферментации применяют в химической промыш-
ленности. Использование ферментов для произ-
водства тех или иных химических продуктов ста-
ло массовым явлением. Для этого в большинстве
случаев применяют иммобилизованные ферменты,
т. е. ферменты, закрепленные на носителе, но
сохранившие каталитическую активность, что по-
зволяет использовать их повторно или непре-
рывно.
Еще в 1916 г. Дж. Нельсон и Е. Триффин пока-
зали, что ферменты в водонерастворимой форме
сохраняют каталитическую активность, однако
первая попытка получить иммобилизованные фер-
менты с целью их практического применения была
предпринята только в 1953 г. Н. Трубхофером
и Л. Шлейтом, закрепившими на диазотирован-
ном полиаминополистироле амилазу, пепсин, рибонуклеазу и карбоксипептида-
зу. В течение последующих 12 лет исследования в этой области были един чны-
ми и только начиная с 1965 г. приобрели широкий, а с 1971 г., после проведения
первой конфе енции по инженерной энзимологии, массовый характер (рис. 57).
Для промышленных целей иммобилизуют ферменты в основном микробно-
го происхождения ввиду их доступности, дешевизны (они в 100 раз дешевле,
чем ферменты животного и растительного происхождения), независимости
массового производства от сезона вегетации растении или сроков выращива-
ния животных, короткого периода накопления бактериальной массы для выде-
ления ферментов. Важно, что иммобилизация, как правило, сопровождается
повышением в тысячи и десятки тысяч раз стабильности ферментов, что
создает условия для их использования в качестве гетерогенных катализаторов.
Кроме того, механическое изменение матрицы, на которой закреплен фермент,
открывает возможность варьировать его активность и понять принцип работы
природных механохимических систем.
Сейчас все более отчетливо вырисовывается огромное значение иммобили-
зации'ферментов для осуществления процессов жизнедеятельности, ибо значи-
тельная часть их в клетке иммобилизована сепаратно или в составе ансамблей
в липидном матриксе биологических мембран (например аденилатциклаза,
см. гл. XII, ансамбли оксидоредуктаз, см. гл. X, и многие другие ферменты).
Для иммобилизации ферментов используют захват их полиакриламидным
гелем и иными полимерами при полимеризации составляющих их мономеров,
а также захват гелями, возникающими при желатинизации природных по-
лимеров, например полисахаридов морских водорослей; присо динение фер-
ментов к целлюлозе, сефарозе, сефадексу, крахмалу, декстрану, агарозе
и другим полисахаридам, активированным цианбромидом и другими аген-
тами; привязку ферментов к стеклянным бусинкам через диазосоединение;
ковалентное связывание фермента с азидными и гидразидными группами
col олимера акриламида и акрилгидразина (энзакрила) и других носителей;
соединение фермента с гидратиро занными о сидами металлов, производными
поливинилового спирта, альдег дными и диазогруппами модифицированных
фенольных полимеров, силикагелями и многими другими материалами.
141
ВоВонерастВоримый
носитель
А Б В Г
Рис. 58. Способы перевода фермента в иммобилизованное состояние:
А—присоединение к носителю; образование перекрестных связей между ферментом и носителем;
В—включение в решетчатую структуру полимерного носителя; Г—микрокапсулирование
Представляет интерес иммобилизация ферментов на мембранах аппаратов
для ультрафильтрации. Соответствующие способы перевода ферментов в им*
мобилизованное состояние показаны на рис. 58.
Почти из 2000 известных в настоящее время ферментов иммобилизовано
и используется для целей инженерной энзимологии примерно десятая часть
(преимущественно оксидоредуктазы, гидролазы и трансферазы). Оптималь-
ным методом иммобилизации счи ают включение ферментов в полимерные
гели, массовым способом является адсорбционное и ковалентное присоедине-
ние ферментов к носителям, более или менее распространено включение
в мембраны и микрокапсулирование единичными остаются другие приемы.
Для осуществления химических процессов с помощью иммобилизованных
ферментов применяют колоночные, трубчатые, пластинчатые, двухфазные
и танкерные реакторы разного объема и производительности. Вероятно, первым
реактором, где иммобилизованный фермент использовали в промышлен-
ном масштабе, был реактор для разделения рацемических смесей D- и L-амино-
кислот.
Он был введен в эксплуа ацию в Японии (1969) на аминоацилазе, им-
мобилизованной на ДЕАЕ-сефадексе:
Амииоацилаза
2R—СН—СООН+Н2О------------------► R—СН—COOH+R—СН—COOH+R'OH
I (гидролиз | I
NH—СО—R' Т"мХкис;ют NH—СО—R'
L-ряда)
Ацил D, L - 1мино“ислоты L-амииокислота Ацил D-аминокислоты
t_______________________________________________1
Рацемизация
Одновременно или даже немного раньше началось промышленное получе-
ние инвертного сахара (смесь глюкозы и фруктозы, возникающая в результате
гидролиза сахарозы) с помощью иммобилизованной Р фруктофуранозидазы
(сахараза или инвертаза), получившее сейчас широкое распространение. Им-
мобилизованная инвер аза очень устойчива и за десять лет непрерывной
работы одного из реакторов ее активность упала всего на 10%.
Не меньшее промышленное значение имеет и другой процесс, приводящий
к получению эквимолярной смеси глюкозы и фруктозы: превращение ’люкозы
в фруктозу с помощью иммобилизованной глюкозоизомеразы.
Соответствующие установки работают в США (начиная с 1972 г.), ФРГ,
Дании и Голландии. В нашей стране изомеризацию глюкозы в фруктозу ведут
142
в трубчатом реакторе с глюкозоизомеразой из Actinomyces olivocinereus, им-
мобилизованной на силохроме; за месяц непрерывной работы реактора при
50%-ном уровне превращения глюкозы в фруктозу теряется не более 14%
активности фермента.
В 1974 г. в Японии начат промышленный синтез L аспарагиновой кислоты
при посредстве аспартат-аммиак-лиазы, иммобилизованной на фенолформаль-
дегидной смоле. Однако при высокой степени (99%) превращения фумарата
аммония в L-аспарагиновую кислоту фермент менее устойчив, чем в. составе
иммобилизованных клеток—время его полужизни равно 18 суткам. В этой же
стране функционируют промышленные установки по синтезу L-триптофана из
индола и серина при помощи включенной в волокна триптофансинтазы, а также
по синтезу L-тирозина при посредстве иммобилизованной тирозинфенол-лиазы.
Упомянем еще одно крупномасштабное производство на базе иммобили-
зованной пенициллинамидазы, имеющее огромное значение для фармацевти-
ческой промышленности, так как оно обеспечивает исходным продуктом
синтез аналогов природного пенициллина, отличающихся большей терапевти-
ческой ценностью. Это—синтез 6-аминопенициллановой кислоты:
Бензилпенициллин
NH—СН
S СН3
С + н2о
|чсн3
С— N—СН—СООН
Феналухсускжя Пеинциллаиовая кислота
кислота
Пеншдилин-
амидиа
-----——>
о
Присоединяя по ее аминогруппе иные, чем бензил, радикалы, получают
пенициллины, отличающиеся специфическими качествами.
Сказанным далеко не исчерпывается список химических производств, бази-
рующихся на применении достижений инженерной энзимологии, имеющей
огромное будущее.
Наряду с использованием иммобилизованных ферментов продолжает раз-
виваться переработка химического сырья при : осредстве микроор анизмов,
которые в этом случае являются как бы живыми ферментсоде жащими лабо-
раториями. Наиболее древним химическим производством такого рода явля-
ется получение спирта путем брожения, когда примерно полтора десятка
ферментов, содержащихся в дрожжевых клетках, с огромной скоростью пре-
вращают глюкозу в этиловый спирт. Характерно, что в настоящее время
осуществляется интенсивный переход от класси еских схем бродильной про-
мышленности на новую технологию с использованием иммобилизованных
клеток различных микроорганизмов. Так, этанол получают из глюкозы с по-
мощью иммобилизованных в полиакриламидном геле клеток Saccharomyces
ccrcvisiae.
143
Первый в мире промышленный 1000-литровый реактор проточного типа
по синтезу L-аспарагиновой кислоты из фумарата аммония был запущен
в Японии (1973); в нем использованы иммобилизованные в полиакриламид
ном геле клетки кишечной палочки (Esherichia coli, штамм АТСС № 11303),
содержащие аспартат-аммиак-лиазу:
Аспартат-аммиак-лиаза
НООС—сн=сн—coonh4-------------------------> НООС—сн2—сн—соон
MgCl (1мМ) |
1 nh2
Фумарат аммония Аспарагиновая кислота
Он давал 1915 кг L-аспарагиновой кислоты/сут при 95 %-ном уровне пре-
вращения в нее введенного фумарата аммония. При подкислении элюата до
pH 2,8 и охлаждении до 15° С аспарагиновая кислота выкристаллизовывалась
в виде препарата 100%-ной чистоты. Иммобилизованные клетки кишечной
палочки сохраняют активность аспартат-аммиак-лиазы на 80% в течение 120
дней и на 50%—в течение 600 дней работы реактора, тогда как интактные
клетки—всего в течение 10 дней и на уровне 25% от исходной. Отсюда ясен
выигрыш, приносимый иммобилизацией. Тодом позже на хими1 еском факуль-
тете МГУ им. М. В. Ломоносова была опробована установка с иммобилизо-
ванными клетками кишечной палочки (штамм 85), обладающими аспартат-
аммиак-лиазной активностью с производительностью несколько килограммов
L-аспарагиновой кислоты особой чистоты. Такая установка в научно-исследо-
вательском институте аминокислот (г. Ереван) масштабируется до промыш-
ленного уровня.
Другая аминокислота, производство которой налажено в индустриальном
масштабе,—L-изолейцин. Ее синтезируют из треонина и глюкозы при посред-
стве иммобилизованных клеток Serratia marcescens с выходом до 4 г/л элюата
с колонки реактора. Аналогичным образом получают еще одну незаменимую
аминокислоту—L-лизин:
НООС—СН—(СН2)3—СН—СООН - м,сгоЬа2пит , СН2—(СН2)3—СН—СООН+СО2
nh2 nh2 nh2 nh2
Диаминопимелиновая
кислота Лизни
При посредстве иммобилизованных клеток Corynobacteriutn glutamicutn
производят L-глутаминовую кислоту из глюкозы; Esherichia coli—L-трипто-
фан из индола; Streptococcus faecalis—L-орнитин из L-аргинина. Разработаны
также методы синтеза L-аланина, L-фенилалани а, L-метионина и L-треонина
тоже с помощью иммобилизованных микроорганизмов. Таким образом, нара-
ботка аминокислот L-ряда для питания человека и выращивания сельскохо-
зяйственных животных осуществляется в настоящее время в основном в реак-
торах с иммобилиз ванными клетками.
С помощью иммобилизованных в полиакриламидный гель клеток
Brevibacterium ammoniagens начиная с 1974 г. в Японии в промышленном
масштабе производят яблочную кислоту из фумаровой. У нас в стране от-
работан регламент получения пропионовой, уксусной и пировиноградной
кислот из глюкозы, лактозы или лактата натрия в проточной системе с клет-
ками пропионовокислых бактерий (Propionibacterium shermanii), иммобилизо-
ванными в полиакриламидный гель.
144
Опробованы на лабораторных установках, а частично и в крупномасштаб-
ных вариантах методы синтеза с помощью иммобилизованных клеток различ-
ных микроорганизмов АТФ, НАДФ, глюкозо-6-фосфата, глутатиона, глюко-
новой и 2-кетоглюконовой кислоты, фруктозы (изомеризацией глюкозы), сме-
си глюкозы и фруктозы (гидролизом сахарозы), коэнзима А и ацетил-КоА.
Недавно японским химикам удалось этим же путем перевести пропилен
в оксид пропилена прямым окислением кислородом—это крупное достижение
внедрено в практику.
Особо следует упомянуть использование иммобилизованных клеток в фар-
мацевтической промышленности для синтеза стероидных гормонов и их про-
изводных, синтеза пенициллинов пролонгированного действия и ряда других
лекарственных препаратов. В последнее время развернулись перспективные
работы по иммобилизации методами инженерной энзимологии клеток, кото-
рым при помощи приемов генетической инженерии придана способность
продуцировать важнейшие лекарственные средства—инсулин, интерферон,
a j-антитрипсин. К 2000 г. намечено резко увеличит производство лекарствен-
ных средств методами генетической инженерии.
Еще одна область применения ферментов—аналитическая химия. Она
базируется на изготовлении электродов, покрытых иммобилизованными фер-
ментами. Так, если на платиновый электрод нанести иммобилизованную
глюкозооксидазу, то концентрацию глюкозы можно определить, регистрируя
амперометрически количество выделяющегося на электроде пероксида во-
дорода:
н н ОН н о
1111 Глюкозо-
НОСН2—С—С—С—С—С + О4 + нао------------
| | | | \ оксидаз*
ОН ОН Н ОН Н
Глюкоза
Н Н ОН н о
\ \ \ \ //
—► носн,—с—с—с—с—с + Н2О,
1111 \
он он н он он
Глюкоиовая кислота
Определение высокоспецифично (на электроде реакция идет только с глю-
козой) и осуществляется в течение 1 мин. На базе такого ферментного элек-
трода создан автоматический анализатор глюкозы. При помощи ферментных
электродов определяют сахарозу, мочевину, этиловый спирт, анализируют
загрязнение среды остаточными пестицидами и т. п.
Не исключено, что использование ферментов позволит найти новые источ-
ники для добывания энергии. Опыты в этом направлении уже сделаны и наме-
тились два пути решения этой насущной для человека проблемы.
Первый состоит в прямом преобразовании химической энергии в электри-
ческую в топливных элементах, где используются такие ферменты, как глюко-
зооксидаза, уреаза, гидрогеназа, формиат- и алкогольдегидрогеназа (субстра-
ты—глюкоза, мочевина, водород, муравьиная кислота и этанол соответствен-
но). В случае алкогольдегидрогеназы в качестве фермента и этанола в качестве
субстрата схема получения электрического тока такова:
Алкоголь-
дегидрогеназа
с2н5он+над+------> СН3СНО+НАДН+Н+
145
н
Ф еназинметосульфат
окисленный
(ФМС)
Феназинметосульфат
восстановленный
(ФМС-Hj)
На 1юверхнос1И
ФМС • Н 2------------------► ФМС+2Н + + 2е
платинового электрода
2Н2
Рис. 59. Принципиальная схема акцеп-
тирования солнечной энергии для раз-
ложения воды и получения газообраз-
ного топлива
Второй путь сводится к использованию в качестве источника энергии
продуктов ферментативной реакции, например водорода, выделяющегося при
улавливании энергии солнца (рис. 59). Расчеты показывают, что квадрат раз-
мером 200x200 [км], где размещены акцепторы солнечной энергии—им-
мобилизованные хлоропласты, при КПД, равном 25%, обеспечи РФ энер-
гией и исключит загрязнение внешней среды.
Во многих лабораториях мира ученые настойчиво разрабатывают одну из
самых сложных, но вместе с тем практически наиболее важных проблем—
проблему механизма ферментативного катализа. Когда она будет полностью
решена, современная нам химическая промышл нность уступит место совер-
шенно новому химическому производству, основанному на принципе фермен-
тативного катализа,—производству, где с огромными скоростями, 100%-ными
выходами, избирательно, без побочных продуктов, в мягких условиях (низкая
температура и давление и т.п.) будет осуществляться превращение одних
веществ в другие. Более того, полная расшифровка ферментативных механиз-
мов таких процессов, как фотосинтез, биоси-
нтез белков, фиксация молекулярного азота
и т. п., и воспроизведение их на этой основе
в лаборатории и промышленности могут
в корне изменить способы добывания пище-
вого и непищевого сырья и оказать огромное
влияние на образ жизни человечества.
Круг химических процессов, ускоряемых
ферментами, необычайно широк, а число их
огромно. В природе осуществляется множе-
ство ферментативных превращений, еще не-
доступных воспроизведению в лаборатор-
ных условиях. Все это свидетельствует об
огромном значении ферментов как истинных
двигателей жизненных процессов. Изучая
ферменты, мы невольно были вынуждены
наряду с описанием их строения и свойств
уделить большое внимание ускоряемым ими
химическим превращениям. Этим мы сдела-
ли первый шаг к изучению ряда конкретных
разделов динамической биохимии, изложен-
ных в последующих главах учебника.
ГЛАВА IV
КОФЕРМЕНТЫ, ВИТАМИНЫ И НЕКОТОРЫЕ ДРУГИЕ
БИОАКТИВНЫЕ СОЕДИНЕНИЯ
Из числа известных в настоящее время ферментов примерно 40%, прояв-
ляют свои каталитические функции в присутствии добавочных соединений
небелковой природы—коферментов, или коэнзимов. К числу таких фермен-
тов относятся большинство оксидоредуктаз и трансфераз, все лигазы, значи-
тельная часть лиаз и некоторые изомеразы. Лишь гидролазы не нуждаются
в коферментах и осуществляют каталический процесс исключительно при
посредстве активных центров, образованных аминокислотными радикалами
полипептидной цепи фермента. В подавляющем большинстве случаев кофер-
менты регенерируются в неизменном виде по завершении каталитического акта,
и это отличает их от субстратов ферментативных реакций. Однако в много-
стадийных химических процессах, ускоряемых ферментами, на определенном
этапе кофермент может выступать как субстрат и приходит в исходное состоя-
ние лишь по завершении всей цепи реакций или после химических преобразова-
ний, ведущих к восстановлению уровня его нормального содержания в клетке.
Весьма существенно, что коферментами часто служат витамины. Поэтому
обе эти группы биологически активных соединений рассматривают обычно
совместно.
КОФЕРМЕНТЫ
Химическая природа коферментов крайне разнообразна. Среди них встре-
чаются органические вещества, относящиеся к алифатическому и ароматичес-
кому ряду, а также гетероциклические соединения, как одно-, так и многоядер-
ные. Коферментам нельзя дать единую физико-химическую характеристику,
поскольку эта сборная группа соединений, объединенная лишь одним призна-
ком—способностью соединяться с белками—апоферментами с образовани-
ем каталитически активных холоферментов.
Термин «кофермент» впервые появился в 1897 г., когда Т. Бертран, изучая
свойства лактазы (^-галактозидаза, отщепляющая концевые нередуцирующие
остатки в В-галактозидах), обозначил этим термином активатор этого фермен-
та—Мп . Следующей важной вехой в развитии учения о коферменте было
исследование английских биохимиков А. Гардена и В. Юнга, которые в 1906 г.
доказали наличие в ферментном препарате дрожжей—зимазе термостабиль-
ного отделяемого ультрафильтрацией фактора—козимазы.
Из энзимов, функционирующих с коферментами в качестве обязатель-
ных партнеров каталитических реакций, 675, т. е. почти 85%, используют
для этого соединения нуклеотидной природы: НАД* (~165 ферментов),
НАДФ + (~ 155), и НАД* и НАДФ + (~50), коэнзим А (~80) и АТФ (~ 225).
Они составляют самую многочисленную группу так называемых нуклеотид-
ных коферментов. Не менее распространенным коферментом является пи-
ридоксалъфосфат, роль которого в реакциях переаминирования рассмотрена
ранее (см. с. 127), а функция в процессах декарбоксилирования аминокислот
будет освещена позже (см. с. 267). У ряда ферментов коферментами являются
ФМН и ФАД, а также соединения хиноидной природы. Химическая природа
147
и роль перечисленных коферментов в каталитических процессах описаны
в гл. III.
В качестве коферментов выступают и иные органические соединения. Так,
в окислительно-восстановительных реакциях коферментами служат лъпосвая
кислота, глутатион и железопорфирины, в реакциях переноса гликозильных
остатков и их производных—нуклеозиддифосфатсахара, в реакциях переноса
азотистых оснований при биосинтезе фосфолипидов—цитидиндифосфатхо-
лин и т. п. Механизмы их участия в ферментативных процессах рассмотрены
в последующих главах. Кроме того, функцию коферментов выполняют многие
витамины.
ВИТАМИНЫ
Общее понятие о витаминах и их классификация. Витамины представляют
сборную в химическом отношении группу органических соединений, поэтому
с точки зрения химического строения им нельзя дать общего определения.
Физические свойства веществ, относящихся к витаминам, столь же разнооб-
разны, как и их химическая природа. Физиологическое действие витаминов на
животных, растительн е ткани и микроорганизмы тоже весьма различно,
и отдельные витамины в этом отношении совершенно не похожи друг на
друга.
Витамины объединены в отдельную группу природных органических со-
единений по признаку абсолютной необходимости их для гетеротрофного
организма в качестве дополнительной к белкам, жирам, углеводам и мине-
ральным веществам составной части пищи.
В большинстве случаев витамины не синтезируются гетеротрофными ор-
ганизмами и их недостаток сопровождается возникновением патологических
явлений.
В количественном отношении потребность в витаминах ничтожна: так,
человек в среднем должен потреблять ежеднев о около 600 г (в пересчете на
сухое вещество) основных питательных веществ и только 0,1—0,2 г дополни-
тельных факторов питания—витаминов.
Отсюда ясно, что витамины в организме выполняют каталитические функ-
ции. Во многих случаях, как будет показано ниже, витамины являются состав-
ными частями ферментов и необходимы для их функционирования.
В последнее время изучается роль витаминов для авто рофных организ-
мов, в части сти растений, где витамины синтезируются. Оказалось, что для
жизнедеятельности растений они тоже абсолютно необходимы и выполняют
здесь также главным образом каталитические функции. Следовательно, на
первый план в характеристике витаминов как особой группы соединений
выступает их способность в ничтожных концентрациях обеспечивать осущест-
вление ферментативных процессов.
Таким образом, витамины могут быть охарактеризованы как группа ор-
гани еских веществ, обладающих разнообразным строением и физико-химиче-
скими свойствами, абсолютно необходимых для нормальной жизнедеятель-
ности любого организма и выполняющих в нем непосредственно или в составе
более сложных соединений каталитические и регулярные функции.
Витамины были открыты в 1880 г. нашим соотечественником Н. И. Луни-
ным. Они привлекли внимание именно как дополнительные факторы питания
животных. В своей диссертации «О значении неорганических солей для пита-
ние животных», защищенной 18 сентября 1880 г. в Дерптском (ныне Тартуском)
университете, Н. И. Лунин пришел к выводу, что кроме белков, жиров,
148
сахаров, солей и воды животные (мыши) нуждаются также в других, неизвест-
ных еще веществах. По его мнению, обнаружить эти вещества и изучить их
значение в питании было бы исследованием, представляющим большой
интерес. В дальнейшем работы Н. И. Лунина были подтверждены и развиты
другими учеными. В 1912 г. польский исследователь К. Функ предложил
называть эти неизвестные вещества витаминами, т. е. аминами жизни (от лат.
vita—жизнь), так как одно из них, выделенное и изученное им, содержало
аминогруппу. Термин этот стал затем применяться ко всем обязательным
дополнительным пищевым факторам. И хотя многие из них не содержат
аминогрупп и вообще азота, название «витамины» до сих пор прочно
удерживается в биологии и медицине.
Благодаря усилиям многих биохимиков и физиологов за более чем столет-
нюю историю витаминологии выделено около трех десятков витаминов,
изучены их состав и строение, физиологическое действие и в подавляющем
большинстве случаев осуществлен химический синтез соответствующих препа-
ратов. Среди исследований советского периода выделяются работы
В. Н. Букина, А. В. Палладина, Л. А. Черкес, М. Н. Шатерникова, А. В. Тру-
фанова, В. В. Ефремова, К. М. Леутского, Б. А. Кудряшова, М. И. Смирнова,
Ю. М. Островского, Р. В. Чаговца и др.
При изучении витаминов сначала каждому из них давали название по
имени того заболевания, которое развивалось при отсутствии витамина в пи-
ще. При этом к названию соответствующей болезни добавлялась приставка
анти, так как введение витамина в диету приводило к быстрому излечению
(табл. 12). Заболевания же, развивающиеся при полном отсутствии витами-
нов в пище, стали обозначать как авитаминозы, при недостаточном их по-
ступлении—как гиповитаминозы, при избыточном—как гипервитаминозы.
Позже, по предложению Мак-Коллума (1913), отдельные витамины по мере
их выделения условились обозначать буквами латинского алфавита: А, В,
С и т. д. Наконец, после того как была исследована химическая природа
ряда витаминов, стали вводить химические их названия. В настоящее время
используют все три вида номенклатуры витаминов. Наметилась тенденция
перехода к химическим наименованиям, которые биохимическая секция Меж-
дународного союза по чистой и прикладной химии с 1956 г. признала между-
народными. Важнейшие витамины и их названия перечислены в табл. 12;
некоторые из них (Q, F, В15, U) иногда относят к витаминоподобным ве-
ществам.
Важнейшие витамины и их номенклатура
Таблица 12
Номенклатура Суточная потребность человека, мт
буквенная химическая (официальная международная) физиологическая (по отношению к человеку)
Жирорастворимые
А Ретинол Антиксероф*гальмический 2,5
D Кальцифе ол Антирахитический 0,0025
Е Токотриенол Антистерильный (токоферол) 15,0
К Филлохинон Антигеморрагический 0,25
Q Убихинон — —
F Комплекс ненасыщенных жирных кислот (линолевая, линоленовая и рахидоновая кислоты) 1000
149
Продолжение табл. 12
Номенклатура Суточная потребность человека, мг
буквенная химическая (официальная международная) физиологическая (по отношению к человеку)
Водорастворимые
В1 Ътамин Антиневритный 2,0
В1 Рибофлавин Витамин роста 2,0
Вз Пантотеновая кислота Антидерматитный фактор 12
РР(Вз) Никотиновая кислота и никоти- Антипеллагрический 25
намид
В« Пиридоксин Антидерматитный 2,0
В12 Цианкобаламин Антианемический 0,003
B1S Глюконодиметиламиноацетат Антианоксический 2,0
Вс Птероилглутаминовая кислота Антианемический 0,2
Вт Кврнитин — —
С Аскорбиновая кислота Антискорбутный 75
н Биотин Антисеборрейный 0,15
р Рутин, биофлавоноид Капилляроукрепляющий витамин 50
и S-метилметионин Противоязвенный —
Особенно велико значение витаминов для развивающегос) детского ор-
ганизма, о чем дает представление табл. 13.
Таблица 13
Суточная потребность (мг) в некоторых витаминах для детей в подростков
(по М. И. Смирнову, 1974)
Возраст, лет в. В, в. с рр
7—10 1.4 1,9 1,7 60 15
11—13 1,7 2,3 2,0 72 19
14—17
ЮНОШИ 1,9 2,5 2,2 79 21
девушки 1,7 2,2 1,9 69 18
По растворимости в воде и жировых растворителях витамины делят на две
группы: водо- и жирорастворимые.
Жирорастворимым и некоторым водорастворимым витам нам свойст-
венна витамерия. Явление это состоит в том, что физиологическим
действием, характерным для того или иного витамина, обладает не одно,
а несколько сходных по химическому строению соединений, называемых
витамерами.
По физиологическому д йствию на организм человека витамины принято
делить на следующие группы (табл. 14).
Аналогичное влияние оказывают витамины на процессы жизнедеятель-
ности у животных. Отсутствие или недостаток витаминов в корме приводит
к нарушению нормального развития, замедлению роста, снижению продук-
тивности и другим нежелательным последствиям. Особенно дефицитны
в подавляющем большинстве кормов витамины D, А и Ви. Введение их
150
в рацион питания животных позволяет резко повысить продуктивность
животноводства.
Таблица 14
1]>упповая характеристика некоторых витаминов
(по П. И. Шилову n Т. Н. Яковлеву, 1974)
Труппа витаминов (по лечебно-про- филактическому эффекту) Краткая клинико-физнологическая характеристика Название основных витаминов
z Повышающие общую ре- активность организма Регулируют функциональное состояние центральной нервной системы, обмен ве- ществ и трофику тканей В„ В2, РР, А, С
Антигеморрагические1 Обеспечивают нормальную проницаемость и устойчивость кровеносных сосудов, повы- шают свертываемость крови С, Р, К
Антианемические Нормализуют и стимулируют кроветворе- ние С
Антиинфекционные Повышают устойчивость организма к ин- фекции: стимулируют выработку антител, усиливают защитные свойства эпителия С, А
Регулирующие зрение Усиливают остроту зрения, расширяют поле цветного зрения А, В2, С
Геморрагия (от грен, гейма—кровь в рагг—прорыв)—кровотечение, кра излияш выход крови из сосудов.
Витамин А (ретинол). Изучение этого витамина начато в 1909 г., а синтез
осуществлен в 1933 г. Витамин А имеет несколько витамеров, из которых
наиболее распространенным счи ают витамин Ai (его много в печени
морских рыб):
Ретинол (витамин А])
Витамин Аг отличается от Ai добавочной двойной связью между 3-м и 4-м
углеродными атомами шестичленного цикла (содержится в печени пресновод-
ных рыб). Обе формы (Ai и Аг) существуют в виде ряда геометрических
изомеров, но лишь некоторые из них физиологически активны. Таким образом,
витамин А состоит из смеси циклических ненасыщенных спиртов характерного
химического строения с большим числом сопряженных двойных связей.
Это кристаллическ е тела лимонно-желтого цвета с темпера урой плавле-
ния от 59 до 64° С (в зависимости от вида геометрического изомера), хорошо
растворимые в жирах и жирорастворителях: бензине, серном эфире, хлорофор-
ме, ацетоне и др.
Витамины группы А легко окисляются как в лабораторных условиях
(посредством МпОг), так и в организме. Окисляясь в организме при участии
биокатализатора, ретинол (спирт) превращается в ретиналь (альдегид), тоже
обладающий активностью витамина А:
151
Однако при отсутствии Ог ретинол устойчив даже при 100° С. В тканях
животных организмов, например в печени, витамин А часто находится
в форме сложных эфиров с пальмитиновой кислотой. В таком виде он более
устойчив и, следовательно, может запасаться впрок, высвобождаясь по мере
надобности. К другим тканям и органам ретинол транспортируется, соединя-
ясь с ретинолсвязывающим белком крови, впервые выделенным в 1968 г.
(М=21000, составлен из 181 аминокислотного остатка, первичная структура
выяснена в 1974 г.).
При отсутствии в пище витамина А в организме животного и человека
развивается ряд специфических патологиче ких изменений (А-авитаминоз):
' ослабление зрения (сумеречная, или «куриная», слепота), поражени эпители-
альных тканей (сухость, слущивание эпителия), в том числе роговицы глаза
(сухость ее и воспаление называются ксерофтальмией, отсюда и название
витамина А—антиксерофтальмический). Кроме того, при А-авитаминозе
наблюдается торможение роста, падение в массе и общее истощение ор-
ганизма.
Сухость кожи и слизистых оболочек, способствующая проникновению
в организм болезнетворных микробов, ведет к возникновению дерматитов,
бронхитов и катаров дыхательных путей. Витамин А, предохраняющий от
этих инфекционных заболеваний, относят поэтому к группе антиинфекцион-
ных витаминов.
У растений только при достаточном содержании предшественников вита-
мина А (каротиноиды) происходят нормальное прорастание пыльцы и опло-
дотворение.
Механизм участия витамина А в поддержании нормального состояния
эпителиальных тканей неизвестен. Роль его в поддержании остроты зрения
выяснена: окисленная форма витамина А (ретиналь) в виде цис-изомера
является простетической группой белка—опсина, образуя родопсин—основ-
ное светочувствительное вещество сетчатки (ретины) глаза (отсюда и название
ретинол). Родопсин открыт более столетия тому назад (18/6) Ф. Боллом.
Опсин имеет М = 38850, содержит два олигосахаридных фрагмента, соеди-
ненных с полипептидной цепью из 348 аминокислотных остатков, чередование
которых выяснено. Он вмонтирован в мембрану диска наружного сегмента
фоторецепторной клетки типа палочки, пронизывая мембрану компактно
расположенными семью а-спиралями, к одной из которых пр соединен имин-
ной связью, возникающей при взаимодействии альдегидной группы с Е-ЫНг-
группой лиз29б, 11-цис-ретиналь (рис. 60). Под действием кванта света проис-
ходит превращение цис-ретиналя в транс-форму чувствительность этой реак-
ции—единичный фотон):
152
Рис. 60. Структура фоторецепторного белка—родопсина и его расположение
в мембране диска фоторецепторной клетки (пояснение в' тексте):
в левом верхнем углу рисунка—фрагмент наружного сегмента фоторецепторной клетки, состоящего
примерно из двух тысяч дисков; квадратом обозначено местоположение родопсина в мембране
Это, в свою очередь, возбуждает активность родопсина, вследствие чего
несколько сотен молекул трансдуцина ( ~ 500)—белка (М = 85 000), относящего-
ся к G-семейству (см. с. 457) и являющегося вторым членом биохимического
каскада усиления светового сигнала (это происходит в течение 1 мс), распадает-
ся на а-субъединицу (М = 39000), у которой одновременно гуанозиндифосфат
заменяется на гуанозинтрифосфат (см. с. 458), и димер из р- и у-субъединиц
(М = 35000 и 8000 соответственно). Комплекс а-субъединицы трансдуцина
с гуанозинтрифосфатом взаимодействует с третьим членом каскада усиления—
фосфодиэстеразой циклического гуанозинмонофосфата (состоит из четырех
субъединиц: а—88 кДа, Р—84 кДа и двух у, по И кДа каждая), которая
в присутствии белка, ее активирующего (он является интегральным белком
ретинальной мембраны), превращает в течение секунды несколько сотен моле-
кул цГМФ в линейные. Гидролиз циклического гуанозинмонофосфата сопровож-
дается закрытием натриевых каналов плазматической мембраны фоторецептор-
ной клетки, ее гиперполяризацией и возникновением электрического импульса,
поступающего в синапс внутреннего сегмента и передаваемого в центральную
нервную систему. Проходящий одновременно процесс фосфорилирования С-
концевой части молекулы родопсина (см. рис. 60) снимает его дальнейшее
воздействие на распад трансдуцина, т. е. фоторецепторный цикл завершается.
Квантово-химические расчеты строения ретиналя позволяют более глубоко
понять его возможную роль в зрительном акте. Система из сопряженных
153
двойных связей в молекуле ретиналя создает условия для возникновения
геометрических изомеров в первую очередь (как это следует из значений
порядка связей—см. с. 191) по двойным связям между 9—10-м и 11—12-м
атомами углерода (они здесь минимальны по сравнению с таковыми при
7—8-м и 13—14-м атомами):
Если при взаимодействии цис-ретиналя (в составе родопсина) с квантами света
происходит возбуждение электронов, что можно рассматривать как началь-
ную фазу возникновения электрического импульса, то, видимо, цис-транс-
переход может служить своеобразным блокирующим механизмом, обеспечи-
вающим односторонний ход процесса утилизации световой энергии..
Источниками витамина А для человека являются рыбий жир, печень рыб
и домашних животных, желток яйца, сливочное масло, зеленые части растений
и красномякотные овощи (морковь, перец, томаты и др.). В двух последних
витамин А содержится в виде провитамина, которым является Р-каротин.
Молекула Р-каротина распадается в кишечной стенке человека и животных
с образованием двух молекул витамина Ai (см. с. 418).
Применение витамина А в животноводстве приносит ощутимый эффект.
Когда жарким летом пастбища выгорают и содержание витамина А, точнее
провитамина А—каротина в травах резко падает, у каракульских овец воз-
никает А-авитаминоз, снижающий их плодовитость. Подкормка витамином
А обеспечивает возрастание приплода на 5—7 ягнят на каждые 100 овцематок,
т. е. примерно на 3 млн. ягнят в южных районах нашей страны. Кроме того,
добавление витамина А или каротина в корм молодняку (цыплята, телята,
поросята) обеспечивает их лучшую выживаемость и более быстрый рост,
а включение его в рационы при откорме крупного рогатого скота повышает
прирост живой массы на 12—15%.
Витамин D (кальциферол). Изучение этого витамина начато в 1916 г.;
в 1931 г. он был получен синтетическим путем.
Как и витамин А, витамин D существует в виде нескольких витамеров.
Наиболее распространены витамины D2 и Оз; их можно рассматривать как
производные стеролов (см. гл. IX):
Витамин D3 (эргокальциферол)
Витамин Р3 (холекальциферол)
Провитаминами D2 иОз являются соответственно эргостерол и холесте-
рол, которые переходят в активную форму в результате размыкания связи
между 9-м и 10-м углеродными атомами кольца В под действием солнечной
радиации (холестерол предварительно дегидрируется и переходит в 7-дегид-
рохолестерол, являющийся непосредственно провитамином). Следовательно,
при наличии соответствующих провитаминов (например, 7-дегидрохолестерол
154
I
Околощитовидные
железы
Рис. 61. Превращения витамина D3, их регуляция и воздействие диокси-
производных на фосфорно- кальциевый обмен
у человека) витамин D3 может синтезироваться в организме, и его поступление
с пищей не обязательно.
Витамины D2 и D3— бесцветные кристаллы, плавящиеся при температуре
115—116° С, не растворимые в воде, но хорошо растворяющиеся в жирах
и растворителях жиров (хлороформ, бензол, серный и уксусно-этиловый эфир,
ацетон, спирт). Оба они малостабильны и быстро разрушаются под действием
окислителей (распад идет по двойной связи между 7-м и 8-м углеродными
атомами кольца В) и минеральных кислот.
При отсутствии в рационе витамина D у детей развивается широко извест-
ное заболевание—рахит. Причина его состоит в расстройстве фосфорно-каль-
циевого обмена и нарушении нормального отложения фосфата кальция в кост-
ной ткани. Предполагают, что при D-авитаминозе нарушается всасывание Са
и Р в желудочно-кишечном тракте и образование фосфорных эфиров ряда
органических соединений; вероятно, оба эти процесса взаимосвязаны. В по-
следнее время показано, что всасывание, перенос Са и кальцификация костей
регулируются не непосредственно витамином D3, а его гормонально-актив-
ным метаболитом, сод ржащим оксигруппы в 1-м и 25-м положениях. Именно
он, связываясь с ядерными рец пторами, обеспечивает биосинтез информаци-
онной РНК для наработки Са-связывающих белков и гормонов (кальцитонин
и паратгормон), регулирующих обмен кальция (рис. 61).
Источником витамина D для человека являются рыбий жир, сливочное
масло, желток яйца, печень животных, молоко.
Особенно важен витамин D для кур-несушек и дойных коров. Подсчитано,
что в скорлупу куриного яйца переходит г/ю часть всего Са, с держащ гося
в теле курицы, а с каждым литром молока из организма коровы выносится
более 1 г Са. Даже малейшее нарушение всасывания Са в кишечнике животных
при D-авитаминозе пагубно сказывается на их состоянии и продуктивности.
155
Поэтому витамин D находит широкое применение в животноводстве для
повышения продуктивности птицы и крупного рогатого скота, обеспечивая
рост привесов на 12—15%.
Витамин Е (токоферол). Первые сведения о существовании витамина, регу-
лирующего процесс размножения, появились в 1922 г. Однако только в 1936 г.
из масла пшеничных зародышей и хлопкового масла были выделены три
производных бензопирана, которые оказались витамерами витамина Е: а-, ₽-
и у-токоферолами (от греч. токос—потомство, феро—несу). В 1938 г. а-
токоферол был синтезирован:
Остаток бензопираиа
Остаток гексадекана
p-Токоферол отличается от а-токоферола тем, что лишен метильной группы
в положении 7, а у-токоферол—в положении 5. В последующее время были
выделены еще четыре токоферола, отличающиеся числом и расположением
метильных групп в бензольном ядре.
Токоферолы—бесцветные маслянистые жидкости, хорошо растворимые
в растительных маслах, спирте, серном и петролейном эфире. Химически они
весьма устойчивы; выдерживают нагревание до 100° С с концентрированной
НС1 и 170° С на воздухе; разрушаются под воздействием ультрафиолетового
излучения; оптически активны.
Витамин Е может окисляться до а-токоферилхинона, структура которого
очень близка к структуре витаминов К и Q (см. ниже):
Близость химического строения витаминов Е, К и Q обусловливает сходст-
во механизмов их действия в организме.
Долгое время считали, что значение витамина Е исчерпывается лишь его
влиянием на процесс размножения, так как при отсутствии или недостатке
витамина Е у человека и животных нарушается эмбриогенез (развитие плода
в организме матери) и наблюдаются дегенеративные изменения репродуктив-
ных органов. У растений витамины Е способствуют прорастанию пыльцы.
Однако более глубокое изучение Е-авитаминоза показало ошибочность такого
представления. Е-авитаминоз выражается в нарушении нормального функцио-
нирования и структуры многих тканей: развиваются мышечная дистрофия,
дегенерация спинного мозга и паралич конечностей, жировое перерождение
и т. п., т. е. общее заболевание организма.
Механизм действия витамина Е в организме двоякий. С одной стороны,
витамин Е—важнейший внутриклеточный агент, предохраняющий от окисле-
156
ния жиры и другие легко окисляемые соединения, это один. из самых
сильных природных антиоксидантов, прежде всего липидов. Реагируя с перо-
ксидными радикалами липидов и сами при этом окисляясь, токоферолы
обрывают цепи окисления. С другой стороны, витамин Е функционирует как
структурный компонент биологических мембран, образуя своим углеводо-
родным радикалом молекулярные комплексы с ненасыщенными высшими
жирными кислотами фосфолипидов и стабилизируя (защищая от окисления)
мембраны. Так как это обеспечивает нормальное протекание биохимических
процессов, то понятны те множественные нарушения функций, которые
наблюдаются при Е-авитаминозе. Недавно высказана еще одна точка зрения
на механизм действия витамина Е—возможное участие в регуляции биосин-
теза некоторых ферментов на уровне транскрипции в генетическом аппарате
клетки их матричных РНК. Кроме того, есть данные о том, что витамин
Е контролирует обмен и функции убихинона и имеет, таким образом,
отношение к сопряже ию окисления с фосфорилированием АДФ, т. е. к био-
энергетике организма.
Источником витамин Е для человека являются растительные масла (под-
солнечное, кукурузное, хлопковое, соевое, конопляное и др.), салат, капуста
и зерновые продукты. Потребность в этом витамине ничтожна, так что
Е-авитаминозы и гиповитаминозы—явление очень редкое, тем более, что
витамин Е откладывается во многих тканях (главным образом в жировой).
Запасы его обеспечивают восполнение убыли даже при полном отсутствии
ви амина в пище в течение нескольких месяцев.
Витамин К (филлохинон). Первые наблюдения, указывающие на существо-
вание особо о витамина, регулирующего процесс свертывал я крови, были
сделаны в 1929 г. Дальнейшие работы привели к открытию двух природных
витаминов Ki и Кг, которые оказались производными нафтохинона. Витамин
Ki был синтетически получен в 1939 г.:
Витамин Кг отличается строением боковой цепи, содержащей от 30 до 45
углеродных атомов и несущей соответственно от 6 до 9 двойных связей. Он
специфичен для бактерий и также получен синтетически (Кг<35)). Его общая
формула такова (я принимает значения от 5 до 8):
Кроме витаминов К^и К2, многие производные нафтохинона обладают
аналогичным физиологическим действием. Из них широкое практическое
применение нашел синтезированный в 1942 г. А. В. Палладиным препарат
157
«викасол».(бисульфитное соединение метилнафтохинона, растворимое в воде).
Он является производив м витамина К3 (метилбензохинон):
Викзсод Витамин
Ви амин Kt —желтоватая маслянистая жидкость с температурой кипения
115—145° С, не растворимая в воде. Очень неустойчив при нагревании в ще-
лочной среде и при облучении. Витамин К2(35>—желтые кристаллы с темпе-
ратурой плавления 54° С, еще более неустойчив, чем ви амин Kt. Витамин
К3—желтый кристаллический порошок с температурой плавления 106° С, не
растворим в воде, но растворим в спирте и эфире. Викасол— бесцветный,
мелкокристаллически" порошок.
Витамин К способствует синтезу компонентов, участвующих в свертыва-
нии крови, и положительно влияет на состояние эндотелиальной оболочки
кровеносных сосудов. При недостатке его в пище могут возникать самопроиз-
вольные кровотечения (носовые кровотечения, кровавая рвота, внутренние
кровоизлияния и т. п.). Полагают, что витамин К принимает участие в син-
тезе протромбина и ряда других белковых факторов, необходимых для
свертывания. Протромбин переходит в тромбин, a i оследний вызывает
превращ ни< фибриногена в фибрин, т. е. непосредственно обеспечивает
коагуляцию крови. Витамин Кь следовательно, стоит у самых истоков этой
сложной системы.
Основное назначение витамина К у растений и микробов состоит в перено-
се электронов при осуществлении процесса фотосинтеза. В последнее время
показано, что посттрансляционная модификация белков путем превращения
глутамильных радикалов в у-карбоксиглутамильные (см. с. 301) осуществля-
ется витамин К зависимой карбоксилазой, локализированной в мембране
эндоплазматической сети. Роль витамина К при этом сводится к отнятию
атома водорода от у-углеродного атома радикала глутаминовой кислоты.
Источниками витамина К для человека являются томаты, капуста, тыква,
зеленые части растений, печень животных. Кроме того, витамин К синтезиру-
ется микрс бами, нормально обитающими в кишечнике. Кишечная микрофло-
ра—постоянный поставщик витамина К для человека и животных.
Витамин Q (убихинон). Эта группа жирорастворимых витаминов открыта
совсем недавно. Она очень близка по строению и, вероятно, по функциям
к витаминам Е и К, что явилось формальным основанием для зачисления
убихинонов в разряд витаминов. В 1955 г. убихинон был впервые выделен из
жира животных.
Витамины Q распространены повсеместно. Они найдены в микроорганиз-
мах, растениях, теле человека и животных, в пищевых веществах. Поэтому
чрезвычайно трудно установить их незаменимость в пище и доказать, что они
не синтез руются самим животным организмом. Тем не менее в опытах
с пищевой недостаточностью на обезьянах, крысах, кроликах, цыплятах, ин-
дюках и хомяках показана витаминная активность убихинонов. Полагают, что
если полиизопреноидная боковая цепь витамина Q может легко синтезиро-
ваться в животном организме, то циклическая хиноидная часть, видимо, в нем
158
не создается. Химинес ая структура и механизм действия убихинона рассмот-
рены на с. 121, а его участие в функционировании дыхательной цепи—в гл. X.
Источником витамина Q являются растительные и животные ткани, в ко-
торых протекают интенсивные окислительно-восстановительные процессы.
Так, например, высокой концентрацией убихинона (и = 10) отличаются сер-
дечная мышца, печень и бурая жировая ткань животных, впадающих в зим-
нюю спячку. Убихинон (и = 10) применяют в ерапии сердечно-сосудистых
заболеваний.
Витамин F (комплекс ненасыщенных жирных кислот). В этот комплекс
входят линолевая, линоленовая, арахидоновая и, возможно, некоторые другие
ненасыщенные высшие кислоты. Биологически наиболее активны арахидоно-
вая и линолевая кислоты; линоленовая кислота усилз вает действие линолевой
кислоты. В 1928 г. Гоген и Гантер предложили считать эти три кислоты
витамином. Линолевая и линоленовая кислоты получены синтетически.
Принадлежность высших ненасыщенных жирных кислот к витаминам при-
знается не всеми, так как неизвестна их каталитическая функция в организме
и отсутствуют явные признаки авитаминозов у человека. Однако при исключе-
нии линолевой, линоленовой и арахидоновой кислот из корма крыс и собак
наблюдались яркие симптомы F-авитаминоза: сухость и шелушение кожи,
выпадение шерсти, омер вение кончика хвоста, задержка роста и падение в весе.
Витамин F участвует в регуляции обмена липидов. Особенно важно, что
непредельные высшие жирные кислоты способствуют выведению из организ-
ма животных и человека холестерола, а это препятствует развитию атероск-
лероза. Отмечено также положительное действие витамина на состояние
кожного и волосяного покровов.
Механизм действия витамина F неизвестен. В специальном исследовании
с применением ряда синтетических непредельных кислот выявлено, что биоло-
гическая активность ненасыщенных жирных кислот связана с нали ием двой-
ных связей между 6—7-м и 9—10-м углеродными атомами.
В последние годы прояснился биохимически" эффект действия арахидоно-
вой кислоты: она оказалась предшественником нового типа гормонов—про-
стагландинов:
(5,8,11,14-эйкозатетраеновая)
кислота
Из арахидоновой кислоты и других полиеновых кислот синтезируется около
20 различных простагландинов, оказывающих мощное влияние на обмен
веществ и физиологические функции у человека и животных. В частности, ряд
простагландинов влияет на деятельность гладких мышц сосудов матки и дру-
гих органов и тканей, в связи с чем их используют для лечения гиперп нической
болезни, облегчения родов, прерывания беременности и т. п. (см. с. 463).
Витамин Вг (тиамин). Витамин Bt занимает особое место в истории учения
о витаминах—это первый кристаллический витамин, полученный в лаборато-
рии. Впервые его исследовал польский ученый К. Функ в 1912 г., а уже
в 1913 г. витамиг В! был получен в виде кристаллов, в 1936 г.—синтезирован.
Строение витамина Вх таково:
159
ИС?ЛС—СИ2—СИа°н
Тиазоловый
цикл
Пиримидиновый
цикл
сг
Так как витамин Bi наряду с аминогруппой содержит в молекуле атом S,
ему было дано химическое название—тиамин (от греч. гпион—сера).
В приведенной выше форме, т. е. в виде соли четырехзамещенного ам-
монийного основания (тиаминхлорид), витамин Вх существует в кислой среде.
Для ейтральной и щелочной среды характерна иная структура—с разомкну-
тым тиазоловым кольцом; при этом в молекуле тиамина появляются свобод-
ные альдегидная и сульфгидриль ая группы. Строение молекулы тиамина
подтверждено синтезом.
Тиамин представляет собой мелкие есцветные кристаллы горького вкуса,
хорошо растворимые в воде. Растворы его в кислой среде устойчивы
и выдержи ают нагревание до высоких температур. В нейтральной и особен-
но в щелочной среде тиамин быстро разрушается. При окис ении он
переходит в тиохром—соединение, обладающее ярко-синей флуоресценцией
в ультрафиолетовом свете, благодаря чему, его легко определить количест-
венно.
При Bi-авитаминозе развивается заболевание, получившее название
полиневрита (болезнь «бери-бери»). Оно состоит в прогрессирующей де-
генерации нервных ок< нчаний и проводящих пучков, следствием чего
являются потеря кожной чувствительности, нарушение нормальной мо-
торики желудочно-кише ного тракта, сердечные боли и т. п.; в конце
концов наступает паралич и смерть. Кроме человека, заболеванию под-
вержены птицы, кролики, собаки, крысы, морские свинки и многие другие
животные.
Механизм действия витамина Вг детально исследован. При посредстве
тиамин-пирофосфокиназы (олигомер из субъединиц с молекулярными мас-
сами 27 000 и 30000 Да и максимальной активностью у тетрамера), перенося-
щей остаток пирофосфата с АТФ на тиамин, он превращается в тиамин-
пирофосфат:
н
Тиаминпирофосфэт
Недавно обнаружено, что тиаминпирофосфат может фосфорилироваться
далее при участии тиаминдифосфаткиназы; возникающей тиаминтрифосфат
считают запасной формой тиаминпирофосфата, хотя есть уже данные о его
абсолютной незаменимости для функционирования нервной ткани. Тиамин-
пирофосфат является коферментом декарбоксилаз кетокислот. При декарбок-
силировании, в частности, пировиноградной кислоты сначала образуется про-
межуточное соединение, распадающееся далее с выделением СО2:
160
Образование промежуточного соединения пировиноградной и других а-
кетокислот с тиаминпирофосфатом объясняется своеобразием его электрон-
ной структуры: второй углеродный атом тиазола обладает повышенной элек-
тронной плотностью вследствие д иссоциации от него протона; поэтому к нему
легко присоединяется а-углеродный атом кетокислоты, характеризующийся
дефицитом электронной плотности.
Оксиэтилтиаминпирофосфат, в свою очередь, распадается с высвобождени-
ем тиаминпирофосфата и продукта деструкции пировиноградной кислоты
либо в виде ацетальдегида который далее превращается в этиловый спирт,
либо в виде ацетил-КоА (см. с. 352). Самое главное состоит в том, что при
этом устраняется сама пировиноградная кислота, возникающая в больших
количествах при распаде углеводов (и частично аминокислот). Она является
сильным ядом для нервной системы, действие которого приводит к тяжелым
последствиям, отмеченным выше при рассмотрении Bi-авитаминоза.
Тиаминпирофосфат катализирует также реа ции переноса двууглеродных
фрагментов, будучи коферментом соответствующих ферментов. Нарушение
этих процессов при недостатке витамина Bt сказывается на состоянии ор-
ганизма и тоже проявляется как Bi-авитаминоз. В последнее время при-
влекают внимание некоферментные функции фосфорных производных ви-
тамина В>: участие их в реакциях фосфорилирования, регуляции обмена
фосфолипидов и др.
Источником витамина Bi для человека являются главным образом хлеб
и крупы в тех случаях, когда зерно в процессе технологической обработки не
теряет зародышей и оболочек, которые в основном содержат тиамин (ржаная
мука, неполированный рис и т. д.). Очень много витамина Вх в пекарских
и пивных дрожжах.
Витамин В2 (рибофлавин) Растворы этого витамина ярко-желтого цвета,
характеризующиеся жieлтo-зeлeнoй флуоресценцией, были получены еще в про-
шлом столетии, но лишь в 1932 г. препарат был выделен в концентрирован-
ном виде, а затем получен в виде оранжевых кристаллов в форме игл,
собранных в друзы (Гпл = 282° С), или пластинок (?пл = 290° С). В настоящее
время витамин В2 синтезирован.
6—3502
161
Основу молекулы рибофлавина составляет изоаллоксазин, в котором’соче-
таются бензольный, пиразиновый и пиримидиновый циклы. Производное
изоаллоксазина, метилированное в положениях 6 и 7, а в положении 9 име-
ющее остаток пятиатомного спирта—рибита, и есть витамин В2. Его хими-
ческое название «рибофлавин» отражает нали ие в молекуле остатка рибита
и желтый цвет окисленной формы препарата. По строению это 6, 7-диметил-9-
рибитилизоаллоксазин:
Витамин В2
Рибофлавин химически неустойчив, легко разрушается при кипячении и на
свету. Под действием света он распадается на рибит и 6,7-диметилизоаллокса-
зин, или люмихром. Особенно важна способность рибофлавина легко окис-
ляться и восст навливаться, что лежит в основе биологического действия этого
витамина. Наибольшей способностью присоединять атомы Н обладают ато-
мы N, находящиеся в 1-м и 10-м положениях в молекуле изоаллоксазина; у них
максимальны индексы свободных валентностей (1,470 и 1,035 соответственно),
Xi растеризующие реакционные возможности в данной точке молекулы.
В2-авитаминоз у чело ека выражается в остановке роста, выпадении волос,
поражении слизис ых оболочек (особенно, в уголках рта), быстрой утомля-
емости зрения, понижении работоспособности, нарушении нормального син-
теза гемоглобина; патологические изме ения возникают и в нервной системе.
Механизм действия витамина В2 изучен. В виде фосфорного эфира (по
концевой гидроксильной группе рибита) или в виде еще более сложных
соединений (в частности, с нуклеотидами) рибофлавин является коферментом
оксидоредуктаз (см. с. 119). Их известно около 30. Они осуществляют ряд
важных реакций в органи ме—окисление L и D-аминокислот, альдегидов,
моноаминов, пуриновых оснований (ксантиноксидаза), углеводов (глюкозоокси-
даза) и др. В их активных центрах часто присутствуют ионы Fe, Мо, Со;
некоторые флавопротеины окисляют субстраты молекулярным кислородом,
т. е. являются оксидазами, но среди них есть первичные и вторичные дегидро-
геназы.
Исто [ником витамина В2 для человека явл ются молоко и зеленые овощи;
много витамина В2 в печени и почках животных, »пивных и пекарских дрожжах.
Витамин В3 (пантотеновая кислота). Этот витамин, впервые обнаруженный
в 1933 г., через несколько лет (1939) был получен в кристаллическом состоя-
нии. В 1940 г. удалось расшифровать его химическую струк уру и осуществить
синтез. Оказалось, что это D(+)-(a, у-диокси-Р, Р-диметилбутирил)-Р-алаиин:
СН3
НО—Н2С—i—СН—СО—NH—СН2—СН2—СООН
^н3Ьн
'---------’----------------------Y---------
Остаток а. у-диокси-р, Остаток р-аланина
р-лиметилмасляной кислоты
162
Пантотеновая кислота содержится во всех животных, растительных и мик-
робных об ектах (греч. пантотен—повсюду). Это вязкая, светло-желтая,
маслян стая жидкость, смешивающаяся с водой и уксусной кислотой. Биоло-
гической активностью обладает только правовращающий (+) оптический
изомер. Пантотеновая кислота малоустойчива, легко окисляется и гидролизу-
ется в присутствии кислот и щелочей по месту пептидной (—СО—NH—)-
связи.
При недостатке пантотеновой кислоты в организме человека и животных
развиваются разнообразные патологические явления: поражение кожных
покровов и слизистых оболочек внутрен их органов, дегенеративные измене-
ния ряда органов и тканей (особенно страдают железы внутренней се реции),
потеря волосяного покрова, депигментация волос и т. д. Наиболее ярким
симптомом В3-авитаминоза у человека является онемение пальцев ног,
сопровождающееся покалыванием; затем возникает жгучая боль в пальцах
и подошвах, распространяющаяся до голени («жжение ног»). Все это объяс-
няется тем, что пантотеновая кислота входит в состав исключительно
важного органического соединения—коэнзима А, который зан мает ключе-
вые позиции в синтезе и расщеплении жирных кислот и обеспечивает
осуществление реак ий, необходимых для взаимопревращения углеводов
Коэнзим A I. . , ।____________I
Остаток 3-фосфоале иозии-
5'-дифосфата
Богатым источником пантотеновой кислоты являются дрожжи, печень,
яичный желток, зеленые части растений; в небольших же количествах она
содержится во всех пищевых продуктах. Кроме того, пантотеновая кислота
синтезируется микрофлорой кишечника.
Витамин РР (никотиновая кислота и никотинамид). Никотиновая кислота
и ее амид (никотинамид, ниацин) известны очень давно:
нс ->сн
НС -СН
ЬГ
Никотином! кислота
( fi - пиридиихарбономя)
Амил никотиновой кислоты
(никотинамид)
Однако только в 1937 г. было показано, что эти вещества являются вита-
минами, так как они предохраняют от заболевания пеллагрой и излечивают
163
уже возникшее заболевание: pellagra (итал.) означает «жесткая или шершавая
кожа». Начальные стадии заболевания пеллагрой выражаются в воспалении
слизистых оболочек желудочно-кишечного тракта, а последующие—в воспа-
лении кожи (дерматитах) на участках тела, подверженных освещению солн-
цем. Поэтому он назван витамином РР: по начальным буквам preventive
pellagra (итал.), что означает «предотвращающий пеллагру».
Никотиновая кислота—белое, кристаллическое вещество (tnjI=235,5° С)
слабокислого вкуса, хорошо растворимое (особенно при нагревании) в воде;
она весьма устойчива и при действии обычных химических и физических
агентов не разрушается. Аналогично ведет себя амид никотиновой кислоты,
представляющий бесцветные иглы с ?пл= 131—132° С.
Собственно антипеллагрическим действием обладает только амид никоти-
новой кислоты, а сама она является провитамином. Механизм действия
никоти амида выяснен. Он входит в состав важнейшего кофермента дегид-
рогеназ—никот амидадениндинукл отида (см. с. 118) и его производного—
никотинамидадениндинуклеотидфосфата.
Некоторое количество никотиновой кислоты синтезируется в организме
животных и человека из аминокислоты—триптофана. Этот синтез протекает
при участии витамина В6. Таким образом, РР-авитаминоз развивается
при неполноценном белковом питании (мало триптофана) и недостатке
витамина В6. Поэтому пеллагру в настоящее время расценивают не
как чисто РР-авитаминоз, а как полиавитаминоз, т. е. заболевание, вы-
званное отсутствием ряда витаминов и зависящее от количества триптофана
в диете.
Никотиновая кислота и ее амид широко распространены в ра тигельных
и животных объектах. Источником витамина РР для человека служит
пшеничный хлеб, печень и почки животных, картофель и многие дру ие
продукты.
Витамин В6 (пиридоксин). Витамин В6 сейчас рассматривают как сочетание
трех индивидуальных веществ: пиридоксола, пиридоксаля и пиридоксамина.
Каждое из них обладает свойствами витамина, ибо в организме способно
перейти в пиридоксальфосфат, который собственно и участвует в химических
реакциях, связанных с деятельностью данного витамина:
Пирндоксол
Пщждокслльфосфат
164
Из трех названных выше веществ, образующих пиридоксиновый комплекс,
наиболее изучен пиридоксол, открытый в 1934 г. и синтезированный в 1939 г.
Он представляет собой бесцветные кристаллы (/пл = 160° С), горькие на вкус,
хорошо растворимые в воде и спирте. Растворы пиридоксола устойчивы
к нагреванию с кислотами и щелочами, но быстро теряют активность под
действием света.
Пиридоксальфосфат является коферментом в реакциях декарбоксилирова-
ния ряда аминокислот, а также реакциях пер аминирования аминокислот
с кетокислотами. Механизм реакции переаминирования и участия в нем
пиридоксальфосфата детально рассмотрен в гл. III (см. с. 124—127).
Отсутствие в пище пиридоксина сопровождается резким нарушением
обмена белков, так как реакции переаминирования аминокислот с кетокис-
лотами обеспечивают фонд свободных аминокислот, необходимых для
биосинтеза белковых тел. Основным симптомом В6-авитаминоза является
нарушение кроветворения и развитие различного рода дерматитов, которые
не поддаются лечению никотиновой кислотой. У молодых животных наступа-
ет остановка роста. В последнее время обнаружено, что В6-авитаминоз
сопровождается нарушением обмена липидов, что ведет к развитию атероск-
лероза.
Источником витамина В6 для человека являются говядина, рыба, горох,
яичный желток и зеленые части растений. Так как витамин В6 очень широко
распространен в продуктах питания, то в обычных условиях В6-авитаминоз
у человека не наблюдается.
Витамин В12 (цианкобаламин). Впервые в кристаллическом состоянии вита-
мин В12 получен в 1948 г. Его химическая структура отличается большой
сложностью; она была расшифрована в 1953 г., а затем в результате десяти-
летних усилий (1961—1971) выдающегося органика Р. Б. Вудворда был осу-
ществлен синтез витамина В12.
Молекула витамина В12 состоит (рис. 62) из так называемой планарной
руппы, которая содержит восстановленные пиррольные кольца с атомом Со
в центре, и расположенных перпендик; лярно к ней двух нуклеозидных групп,
имеющих диметилбензимидазол и аденин в качестве азотистых оснований
и a-D-рибофуранозу в качестве углевода.
Планарная группа витамина В12 является хромофором, вследствие чего
игольчатые кристаллы цианкобаламина окрашены в рубиново-красный цвет,
а его водные растворы—в светло-сиреневый. Кристаллы витамина В12 темне-
ют при 210—220° С и плавятся при f~300° С. Витамин В12 хорошо растворим
в воде, спиртах, низших органических кислотах жирного ряда и фенолах, но не
растворяется в бензоле, серном эфире, хлороформе и ацетоне. На свету он
теряет активность, но в темноте может храниться долго и представляет собой
очень устойчивое вещество. Витамин В12 оптически активен. Установлено, что
в природе существует ряд соединений, обладающих активностью витамина
В12. Строение нескольких из них расшифровано: в псевдовитамине В12 вместо
бензимидазола содержится аденин, в факторе А—2-метиладенин, в факторе
В отсутствует нуклеотидная часть и т. д.
При недостатке витамина В12 в пище человека и животных наступает
нарушение нормального кроветворения в костном мозгу, приводящее к забо-
леванию— анемии; поэтому витамин В12 называют антианемическим.
Механизм действия витамина В12 сводится к тому, что некоторые
его формы являются коферментами и, соединяясь с различными апофе-
рментами, обеспечивают возникновение семейства кобамидных ферментов,
ускоряющих важнейшие реакции азотистого, углеводного, нуклеинового
и липидного обмена. Так, при посредстве метилкобаламина в качестве
165
сн2он
кофермента ускоряются реакции переноса метильных групп и осуществляется,
например, биосинтез метионина. В этой реакции витамин В12 действует в паре
с витамином Вс. Аденозилкобаламин входит в состав ферментов, обеспечива-
ющих внутримолекулярный перенос атомов Н и различных х мических групп
(гидроксильные, аминные, карбонилтиоэфирные и др.), в соответствии со
схемой:
Примеры таких ферментативных процессов будут рассмотрены ниже.
Депо витамина В12 у человека находится в печени, где он накапливается
в количестве нескольких миллиграммов. В переносе витамина В12 через ки-
шечную стенку принима т участие соединение белковой природы, специфиче-
ски связывающее витамин,—так называемый внутренний фактор. Поэтому
нарушение синтеза этого фактора приводит к В12 авитаминозу даже при
наличии достаточного количества оследнего в пище- Часть витамина Bi2 по-
166
ступает в организм человека и животных в результате деятельности микро-
бов—симбионтов кишечного тракта. Ра тения не содержат витамина В12,
поэтому его источниками для человека служат мясо, молоко, яйца, но син-
тезируется он только микроорганизмами.
Витамин В12 находит очень широкое практическое применение в жи-
вотноводстве. Так, добавление его к корму свиней и птицы увеличивает
привесы на 15%. У птицы возрастает также яйценоскость. В нашей стране
разработан дешевый способ производства витамина В12 путем микроби-
ологического синтеза (метановое брожение на отходах ацетонобуталового
производства). Налажено также производство кристаллического витамина
В12 для медицинских целей. Часть кристаллического витамина В12 экс-
портируется.
Витамин В15 (пангамовая кислота). В 1950 г. Т. Томияма обнаружил в экс-
тракте печени быка соединение, которое назвал витамином В15. В 1951 г. было
найдено (Е. Кребс с сотр.) аналогичное вещество в водной вытяжке из ядер
косточек абрикосов и на вано пангамовой кислотой. Позже указанное соеди-
нение было выделено в кристаллическом состоянии из проростков риса,
пивных дрожжей, печени и других источников. Оказалось, что этот витамин
очень широко распространен в природе и всегда представлен в семенах
растений, откуда и произошло его название (от греч. пан—всё и гами—семя).
Химический состав и строение пангамовой кислоты выясн ны и подтверждены
синтезом:
НаС Н Н ОН Н
\ Illi
N—СН2—СО—О—СН2—С—с—с—с—соон
НэС^ он <Lh н он
Пангамомя (N, N-диметилглниил-б-глюконоаая) кислота
Пангамовая кислота—гигроскопичный, кристаллический белый порошок,
хорошо растворимый в воде, но не растворимый в эфире, хлороформе
и бензоле.
Пангамовая кислота оказывает положительное влияние на переносимость
кислородного голодания (аноксия) и может быть охарактеризована как анти-
аноксический витамин. Кроме того, она предохраняет от жирового перерожде-
ния печени. Однако до сих пор неизвестно, синтезируется пангамовая кислота
в организме или она должна обязательно поступать извне.
Механизм действия пангамовой кислоты состоит в каталитическом ускоре-
нии реакций переноса метильных групп. В частности, она обеспечивает нор-
мальный ход биосинтеза холина, метионина, креатина и креатинфосфата. Так
как последний активно разрушается при функциональной перегрузке сердца,
пактам; т кальция применяют для лечения пр дынфарктного и послеинфаркт-
ного состояний.
Витамин Вс (птероилглутаминовая кислота). Первые сведения о существова-
нии витамина Вс были получены в 1940 г. в опытах на цыплятах (отсюда
индекс с—от англ, chicken—цыпленок). К 1945 г. была установлена иденти -
ность витамина Вс с фолиевой кислотой, выделенной из листьев шпината
и полученной син етически. Этот витамин более известен под названием
фолиевой кислоты, так как он содержится в значительных количествах в ли-
стьях (от пат. folium—лист). Однако фолиевых кислот было выделено несколь-
ко, и сейчас каждому представителю этой группы витаминов дают точное
название в соответствии с его химической структурой. Структура одной из
фолиевых кислот, птероилмоноглутаминовой, такова:
167
Остаток птеридина Остаток параами- Остаток глута-
нобензойной кис- миновой кис-
Остаток птероевой кислоты
Птероилмоиоглутаминовая кислота
Остальные фолиевые кислоты отличаются от птероилмоноглутаминовой
кислоты наличием большего или меньшего числа (от 3 до 6) остатков глута-
миновой кислоты, Присоединенных к концевому остатку глутаминовой кисло-
ты в виде у-глутамилпептида.
Фолиевая кислота представляет собой игольчатые кристаллы желтого
цвета, содержащие два моля кристаллизационной воды на один моль кислоты.
Они стабильны на воздухе, не могут быть охарактеризованы по температуре
плавления, так как разлагаются при 250° С. Ограниченно растворимы в воде
(25 мг/л), ледяной уксусной кислоте и спиртах, не растворимы в эфире, ацето-
не, хлороформе. При длительном освещении фолиевая кислота разрушается.
В экспериментах установлено, что если в пище животных (например,
цыплят) недостает фолиевой кислоты, у них задерживается рост и нарушается
кроветворение. Очень чувствительны к недостатку витамина Вс молочнокис-
лые бактерии, для которых он является незаменимым ростовым фактором.
Человек редко страдает от Вс-авитаминоза, так как фолиевая кислота син-
тезируется микрофлорой желудочно-кишечного тракта и всегда поступает
в организм в достаточном количестве, но в случае развития этого авитаминоза
у человека он может быть охарактеризован как анемия; вместе с тем развива-
ются множественные нарушения деятельности органов пищеварения.
Фолиевая кислота, будучи коферментом ряда ферментов, переносит одно-
углеродные фрагменты при биосинтезе многих соединений: метильную группу
при биосинтезе метионина и тимина, оксиметильную (—СН2ОН)—при био-
синтезе серина, формильную
—при новообразовании пуриновых оснований и т. п.
Так как указанные соединения играют ведущую роль в обмене белков
и нуклеиновых кислот (недостаток метионина и серина лимитирует биосинтез
белковых тел, отсутствие тимина и соответствующего ему нуклеотида—
биосинтез ДНК, Дефицит пуриновых оснований—новообразование ДН1£
и всех видов РНК), вполне понятны те нарушения жизнедеятельности, которые
наблюдаются при Вс-авитаминозе.
Фолиевая кислота переносит перечисленные выше фрагменты, находясь
в восстановленном состоянии, в виде 5,6,7,8-тетрагидрофолиевой кислоты.
Присоединение фрагментов идет по атому N, находящемуся в 5-м положении,
при участии трифункционального фермента—формил-метенил-метилентетра-
гидрофолат-синтетазы (М фермента из разных источников от 150000 до
225000). Примером может служить перенос формильной группировки при
168
биосинтезе формилметионил-тРНК (с ее присоединения к рибосоме начинает-
ся биосинтез белка —см. с. 290) в соответствии с уравнением
№ -формил-5,6,7,8" тетрагидрофолиевая Метионил-тРНК
кислота (И5-4)ормил-ТГФК)
При переносе метильного радикала ТГФК взаимодействует с витамином В12.
Источниками фолиевых кислот для человека являются многие продукты,
в том числе шпинат, цветная капуста, печень животных, хлеб. Особенно
высоко содержание фолиевой кислоты в пивных и пекарских дрожжах.
Витамин Вт (карнитин). В 1948 г. был открыт (Г. Френкель и сотр.) особый
витамин необходимый для нормального развития и осуществления линьки
у насе омых. Характерное действие этого витамина было выявлено на мучном
хрущаке (Tenebrio molitor), поэтому в наименовании витамина использован
оответствующий индекс.
По химической природе витамин Вт оказался р-окси-у-триметиламиномас-
ляной кислотой:
СН3
I у ₽ «
Н3С—N+—СН2—СН—СН2—СООН
I I
сн3 он
При его отсутствии в пище у ряда насекомых приостанавливается рост
и наблюдается гибель во время линьки. Хотя точных исследований механизма
действия карнитина нет, полагают, что он участвует у насекомых в переносе
метильных групп. Такая его функция не исключена и у позвоночных, у которых
он принимает также активное участие в переносе ацильных радикалов через
клеточные мембраны, т. е. в конечном счете в реакциях окисления и синтеза
высших жирных кислот.
Холин. Это соединение выделено из желчи более столетия тому назад.
Однако только в последнее время ему приписывают функцию витами а:
СН3
I
Н3С—N +—СН2—СН2ОН
I
сн3
сг
Холинхлорид
Холинхлорид—бесцветные гигроскопические кристаллы, хорошо раство-
римые в воде и спиртах.
169
Хотя холин и синтезируется в организме животных, тем не менее в опреде
ленных условиях создается его дефицит и развиваются симптомы х линавита-
миноза, которые выражаются в жировом перерождении печени, кровоизлия-
ниях в почках и других органах, сокращении объема синтеза протромбина,
изменении условно-рефлекторной деятельности.
Механизм действия холина хорошо выяснен. С одной стороны, Он является
партнером соответствующих акцепторов в реакциях переноса метильных
групп при биосинтезе ряда важнейших соединений: метионина, пуриновых
и пиримидиновых оснований и т. п.; с другой стороны, холин входит в качест-
ве составной части в активную группу биока ализатора, ускоряющего синтез
фосфолипидов (см. с. 409). Кроме того, он представляет компонент ацетил-
холина, участвующего в проведении нервного импульса.
Много холина содержится в желтке куриного яйца, печени и почках живот-
ных, капусте, рыбных продуктах.
Витамин С (аскорбиновая кислота). Первые сведения о существовании
особого органического вещества, наличие которого в пище предохраняет от
цинги (скорбута), относятся к 1885 г., когда В. В. Пашутин отверг распростра-
ненное в то время мнение, что цинга является инфекционным заболеванием,
и выдвинул идею об авитаминозе как ее причине. В 1920 г. антицинготный
фактор получил название в тамина С, двумя годами позже он был получен
в чистом виде, а в 1927 г. окончательно расшифрована его природа и дано
химическое название— аскорбиновая кислота. В 1932 г. был осущ ствлен син-
тез витамина С.
Аскорбиновую кислоту можно рассматривать как производное углевода
L-гулозы; поэтому его на: ывают также у-лактоном 2,3-дегйдро-Ь-гулоновой
кислоты:
но—с—н
I
но—с—н
I
н—с—он
но—с—н
(Lh2OH
L-Гулоза
О
|\)Н
но—с—н
I
но—с—н
I
н—с—он
I
но—с—н
I
СНа0Н
L-Тулоном я кислота
О
г
|\)Н
но—с
и
но—с
I
н-с—он
I
но—с-н
I
сн,он
2.3- Дегидро- L-
гулоиомй кислота
НО—С-Н
сн,он
7-Лактон 2,3-aenupo-L-ry-
лоноаор кислоты, или L-
аскорбниоаая кислота
Кислотные свойства аскорбин вой кислоты, не имеющей, как видно из
формулы, свободной карбоксильной группы, зависят от диссоциации водоро-
да гидро (сильной руппы, расположенной у 3-го углеродного атома.
Витамин С—бесцветные кристаллы (гпл. = 192° С), кислые на вкус, хорошо
растворимые в воде и спирте, но не растворимые в бензоле, хлороформе,
эфире и других растворителях жиров. В бескислородной среде кристаллы
аскорбиновой кислоты можно хранить годами, но в присутствии кислорода
или в растворе, особенно щелочном, витамин С быстро разрушается. Разруше-
нию способствуют ионы Fe и Си.
Аскорбиновая кислота легко отдает два атома Н, переходя при этом
в дегидроаскорбиновую кислоту, и наоборот. Это важнейшее свойство лежит
в основе меха изма действия аскорбиновой кислоты в ор анизме: она
является участником окислительно-восстановительных систем и обеспечива-
170
ет, следовательно, нормальное протекание жизненно важных процессов
в тканях.
Окисление L-аскорбиновой кислоты в L-дегидроаскорбиновую кислоту
сопровождается потерей двух протонов и двух электронов:
Аскорбиновая
кислота
Монодегидро-
аскорбиновая
кислота
Анион дегидро-
аскорбино зон
кислоты
L-Дегидро-
аскорбиновая
кислота
Промежуточные продукты, особенно свободный радикал монодегид-
роаскорбиновой кислоты, исключительно реакционноспособны и взаимо-
действуют со многими другими коферментами оксидоредуктаз: глута-
тионом, НАДН, ФАД, цитохромами и т. п. Непосредственно процесс
окисления аскорбиновой кислоты ускоряется аскорбатоксидазой (см.
с. 414).
Аскорбиновая кислота очень широко распространена в природе, присут-
ствуя буквально во всех тканях и органах живогных, растений, а также
в микроорганизмах. Чаще всего она находится в окисленной форме, а в расте-
ниях— в виде связанной аскорбиновой кислоты, так называемого аскорбигена,
возникающего при ее взаимодействии с 3-оксйметилиндолом. Аскорбиген
отличается несколько более слабым физиологическим действием, но более
устойчив к тем или иным физико-хими еским воздействиям.
При недостаточном поступлении витамина С с пищей у человека
обезьян и морских свинок развивается специфичес ое заболевание—цинга.
Болезнь выражается в повышении проницаемости и хрупкости кровеносных
сосудов, вследствие чего возникают спонтанные кровоизлияния и ха-
рактерные изменения кос ей и зубов: зубы быстро разрушаются, рас-
шатываются и выпадают. В основе этих явлений лежат нарушения
синтеза склеивающего межклеточного белка— коллагена вследствие осла-
бления его посттрансляционной модификации (см. гл. VII), выражающейся
в торможении окисления радикалов пролина и лизина в радикалы ок-
сипролина и оксилизина соответственно. В результате синтезируется
нефибриллярный коллаген. Это и вызывает патологические изменения
сосудистых стенок и опорных тканей. Биосинтез аскорбиновой кислоты
не происходит также у некоторых птиц (семейство воробьиных) и у ряда
видов летучих мышей.
При оценке механизма действия аскорбиновой кислоты в настоящее
время большое значение придают возможному ее участию в предохр нении
от окисления активных HS-групп белков, в том числе белков, обладающих
биокаталитической активностью. Эту функцию выполняет Восстановленная
форма аскорбиновой кислоты. Естественно, что нарушение этого процесса
серьезным образом сказывается на состоянии организма. Недавно показано
также, что аскорбиновая кислота является активной группой фермента,
171
ускоряющего гидролиз некоторых тиогликозидов, например синигрина, содер-
жащегося в семенах горчицы и хрена:
/S-QHnO,
сн4=сн-сна-с' + н2о
XN-O-SO3K
Сииигрии
Т иоглико-
Эндаза
--------► СеН|2О, +
Глюкоза
+ CHs=CH-CHI-N=C=S + KHSQ»
Изородановый эфир
аллилового спирта
Аскорбиновая кислота выполняет также роль парного донора в некоторых
монооксигеназных реакциях (см. гл. X).
Источником витамина С для человека служат самые разнообразные про-
дукты растительного происхождения. Особенно много его содержат плоды
шиповника, черная смородина, облепиха, рябина, красный перец, лимоны,
капуста.
Витамин Р (рутин). В 1936 г. из кожуры лимона было выделено (А. Сцент-
Дьердьи и сотр.) вещество, улучшающее состояние капилляров, и названо
витамином Р (от лат. регтпео—проникать). Витамин Р, который в последнее
время условно обозначают как рутин, представляет семейство веществ, близ-
ких по химической структуре. В основе всех их лежит скелет флавона:
В настоящее время известно свыше десятка соединений, обладающих Р-
витаминным действием. Их называют биофлавоноидами. Они отличаются
различной степенью гидроксилирования бензольных колец, входящих в состав
флавонового ядра, а также различными гликозидными группировками, при-
соединяющимися по 3-му углеродному атому пиранового цикла. В качестве
примера приведем структурную формулу рутина, содержащего в своем соста-
ве остаток дисахарида—рутинозы (С12Н21О9):
Химически чистые препараты витаминов P-группы представляют собой
кристаллические вещества желтой или оранжевой окраски, труднораствори-
мые в воде.
При отсутствии витамина Р в пище у животных и человека повышается
проницаемость капилляров, что сопровождается внезапными кровоизлияни-
ями после сдавливания ткани, болью в конечностях, общей слабостью
и быстрой утомляемостью.
172
Предполагают что витамины группы Р участвуют в окислительно-восста-
новительных реакциях, обеспечивая таким образом нормальный ход процес-
сов биологического окисления в организме. Действие витаминов Р и С взаимо-
связано: каждый из них в присутствии другого обладает гораздо более высо-
ким терапевтическим эффект м, чем в одиночку. Видимо, эти витамины
функционируют в окислительно-восстановительных процессах вместе, образуя
парное звено в соответствующей системе.
Источником витамина Р для человека являются те же продукты, в кото-
рых много витамина С, например черная смородина и лимоны. Кроме того,
витамин Р содержится в значительных количествах в бруснике, чернике,
клюкве, сливе, вишне, винограде и других фруктах, а также в гречихе
и перце.
Витамин Н (биотин). Витаминные свойства биотина стали известны еще
в 20-е годы, но только в 1936 г. удалось выделить из 250 кг яичного желтка
1,1 мг кристаллического препарата, названного биотином. В 1942 г. была
установлена его структура, а годом позже осуществлен химический синтез:
HNr г‘ 7NH
I А 3
СН2 сн2
Биотин
Тетероциклическая часть молекулы состоит из имидазольного (J) и тиофе-
нового (В) циклов, а боковая цепь представлена остатком валериановой
кислоты.
Бесцветные, игольчатые кристаллы биотина (/пл. = 220° С) хорошо раство-
ряются в воде, ограниченно—в спиртах и трудно—в серном эфире. Биотин
устойчив к действию молекулярного кислорода и H2SO4, но разрушается под
влиянием Н2О2, бромной воды, НС1, HNO3 и щелочей.
Необходимость биотина для нормальной жизнедеятельности отражена
в самом его названии (от греч. биос—жизнь). При недостатке этого витамина
у человека наступает ряд патологических изменений: воспаление кожных
покровов, выпадение волос, усиленное выделение жира сальными железами
кожи (себоррея). Защита от возникновения себорреи (от лат. sebum—сало
и rheo—теку) и послужило основанием назвать биотин антисеборрейным вита-
мином.
Механизм действия биотина, вероятно, многообразен. Пола ают, что глав-
ная его роль состоит в том, что в качестве кофермента он входит в состав
ферментов, ускоряющих реакции карбоксилирования.
Так, громадное значение для нормального хода биохимических процессов
имеет карбоксилирование ацетил-коэнзима А, открывающее цикл реакций био-
синтеза высших жирных кислот (см. с. 397). Биотин участвует не только
в фиксации СО2, но и осуществляет реакции транскарбоксилирования, т.е.
передачу карбоксильной группы от одного соединения к другому:
R'—СООН + R"H?±R'H+R"—СООН
Использование биотина как кофермента в реакциях карбоксилирова-
ния и транскарбоксилирования важно для синтеза пуриновых оснований,
173
превращения пировиноградной кислоты в щавел воуксусную, а за-
тем—в аспарагиновую кислоту и для осуществления ряда других реак-
ций.
Источником витамина Н для человека являются печень и почки крупного
рогатого скота, куриные яйца, молоко, томаты, соя, морковь, картофель,
горох, овсяная крупа. Возможно также поступление биотина в организм за
счет микробов-симбионтов; так, жвачные животные полностью обеспечивают-
ся биотином в результате деятельности симбионтов.
Витамин U. В 1942 г. Г. Чиней на основании опытов с цыплятами,
у которых вызвали язву скармливанием алкалоида цинкофена, высказал
предположение, что ускоренное заживление язвы при добавлении в корм
свежей зелени, некипяченого молока и сырой печени объясняется при-
сутствием неизвестного фактора, который он назвал витамином U (от
лат. ulcus—язва). В 1954 г. Р. Макрори и сотр. идентифицирова и при-
родный витамин U, выделенный ими из сока капусты в виде кри-
сталлического бромида, с полученным синтети веским бромидом метил-
метионинсульфония. Стало ясно, что витамин U является S-метилме-
тионином:
НЭС соо-
H3CZ NH.
В СССР В. Н. Букин и сотр. синтезировали в 1969 г. хлорид метилмети-
онинсульфония, нашедший широкое применение в качестве противоязвенного
сред тва. Он представляет собой белый кристаллический порошок сладковато-
солоноватого вкуса со слабым запахом капусты, хорошо растворимый в воде
и водно-спиртовых смесях, но не растворимый в этаноле, глицерине и серном
эфире.
Механизм лечебного действия хлорида метилметионинсульфо'Ния объясня-
ется тем, что он является макроэргическим соединением (см. гл. V) и актив-
ным поставщиком метильных групп для химических процессов, связанных
с регенерацией слизистых оболочек органов пищеварения.
Источниками витамина U для человека являются спаржа, капуста, томаты,
сельдерей и зеленый чай.
ДРУГИЕ БИОАКТИВНЫЕ СОЕДИНЕНИЯ
Кроме коферментов и витаминов на процессы жизне; ятельност оказыва-
ют влияние многие другие биологически активные соединения. Среди них
огромная роль принадлежит гормонам (см. гл. XIII), антивитаминам, анти-
биотикам, телергонам, гербицидам, дефолиа там, ростовым веществам и др.
Их изучение позволяет не только более глубоко понять биохимические меха-
низмы живой природы, но и обеспечивает разработку новых подходов к регу-
ляции роста и развития организма, лечению болезней, борьбе с насекомыми •
вредителями и т. п.
Антивитамины представляют соединения, конкурирующие с витаминами
в соответствующих биохимических процессах или выключающие витамины из
процессов обмена веществ путем, их разрушения или связывания.
Примером антивитаминов первого типа, конкурирующих, могут служить
структурные аналоги витамина РР:
174
Соединяясь с аденил >вой кислотой, они способны образовывать псевдоко-
ферменты, имитирующие НАД+ и блокирующие деятельность НАД+-зависи-
мых оксидоредуктаз.
Примером антивитаминов второго типа, выключающих, является ави-
дин—яичный белок, образующий с витамином Н нерастворимый биологиче-
ски неактивный комплекс, препятствующий использованию этого витамина
в обмене веществ.
Для многих витаминов существуют природные или полученные синтетиче-
ским путем антивитамины. Поскольку возбудители инфекционных заболева-
ний—бактерии и вирусы, а также клетки опухолей обладают повышенной
чувствительностью к отсутствию ряда витаминов, антивитамины используют
как терапевтические средства.
Антибиотики—вещества, образуемые микроорганизмами или получаемые
из других природных источников, обладающие антибактериальным, антиви-
русным и противоопухолевым действием. Они вмешиваются в обмен белков,
нуклеиновых кислот и в эне гетические процессы пораженных организмов
и клеток, избирательно зоздейс вуя на опред ленные молекулярные механиз-
мы. Так, в биосинтезе белка (о поименованных ниже этапах б осинтеза белка
см. гл. VII) пуромицин высвобождает недостроенные полипептиды, тетрацик-
лины подавляют присоединение аминоацил-тРНК к рибосоме, хлорамфеникол
(левомицетин)—пептидилтрансферазную р акцию в ней, эритромицин блоки-
рует перемещение рибосомы по информационной РНК, стрептомицин искажа-
ет считывание кода белкового син еза В биосинтезе нуклеин вых кислот
(терминологию см. в гл. VI) противораковые и антибактериальные антибиоти-
ки (актиномицины, митомицин, нов бицин, рифамицин и др.) подавляют
процессы репликации и транскрипции. На энергетические процессы в клетке
воздействуют антимицин (подавляет перенос электронов в цитохромной сис-
теме), олигомицин (подавляет сопряжение окисления с фосфорилированием)
и другие антибиотики. Биосинтез гликопротеинов клеточных стенок бактерий
приостанавливается под действием пенициллинов и D-циклосерина; проница-
емость клеточных мембран нарушается грамицидинами, нистатином и многи-
ми другими антибиотиками.
Химическая природа антибиотиков разнообразна и сложна, среди
них есть пептиды, полиеновые соединения, олициклические вещества
и т.п. Рассмотрение ее выходит за пределы учебника. Однако принцип
действия антибиотиков может быть иллюстрирован на одном из простых
примеров—механизме торможения биосинтеза гликопротеинов клеточной
стенки бактерий D-циклосерином. В состав пептида, необходимого для
биосинтеза пептидогликана клеточной стенки бактерий, входит D аланин.
Его образование из L-аланина, ускоряемое аланин-рацем зой, а также
синтез О-аланил-В-аланина, катализируемый D-аланил-Ь аланин-синтетазой,
175
блокируется конкурентным ингибитором этих двух ферментов—D-циклосе-
рином, структурно сходным с D-аланином:
D-Циклосерин
D-Аланин
В результате циклосерин полностью тормозит размножение ряда бактерий,
выключая процесс сборки гликопротеинов их клеточных стенок. В целом
антибиотики являются наиболее мощными химиотерапевтическими препара-
тами против бактериальных и вирусных инфекций, а также для остановки
опухолевого роста.
Телергоны—вещества, образуемые железами внешней секреции животных,
оказывающие специфическое воздей твие на другие организмы (от греч.
теле—вдали, эрго—действие). Среди них представлены соединения, при-
влекающие особей противоположного пола, а также стимулирующие про-
цессы размножения и предопределяющие половое поведение (половые фе-
ромоны), вещества тревоги и испуга, следовые вещества, вещества ублажения
(псевдоферомоны) и др. Часто в количестве лишь нескольких молекул они
воздействуют на рецепторные клетки, вызывая далеко еще неясные по мо-
лекулярным механизмам, но совершенно однозначные по физиологической
природе реакции.
Так, из пахучих желез 500000 самок тутового шелкопряда было выделено
12 мг кристаллического вещества, после омыления которого был получен
бомбикол (от Bombyx mori—тутовый шелкопряд), в ничтожной концентрации
(всего 1000 молекул/см3 воздуха) привлекающий самцов этого насекомого
и возбуждающий у них соответствующее половое поведение (однообразное
и длительное трепетание крыльев):
Бомбикол (гексадекадиен-10, 12-ол-1)
Один из продуктов желез пчелиной матки—транс-децен-2-он-9-овая
кислота:
в незначительной концентрации тормозит развитие яичников у рабочих пчел,
сохраняя со тветствующую структуру пчелиной семьи.
Реакция тревоги у солдат муравьев-листорезов возникает под действием
выделяемого их железами цитраля:
176
НХ /СН» /
;с=СН-(СН1),-С^ с
НзС7 сн/ 'н
В ответ на запах этого феромона тревоги они кромсают на части все живое,
что попадает в их мощные челюсти.
Как следует из приведенных примеров, телергоны могут действовать на
организм двумя путями—через посредство хеморецепторных клеток или про-
никая в соответствующие ткани и органы. Интерес к этим веществам непре-
рывно возрастает, и зарождается новая наука—телергонология, изучающая
биологически активные вещества наряду с витаминологией, энзимологией
и эндокринологией. Постепенно вырисовываются весьма впечатляющие прак-
тические аспекты изучения телергонов: половые аттрактанты уже широко
используют для борьбы с насекомыми-вредителями, не губя при этом, в силу
видовой специфичности привлекающих веществ, полезную энтомофауну,
а применение телергонов одомашненных млекопитающих перспективно для
увеличения их плодовитости и регуляции численности.
К сожалению, во многих случаях сведения о биологическом действии тех
или иных соединений либо ограничены, либо отсутствуют. Особую опасность
представляют вещества, воздей твующие на генетический аппарат организ-
ма—мутагены и супермутагены. Их обнаружение и разработка методов защи-
ты—одна из самых насущных задач современной экологической биохимии.
Сказанным далеко не исчерпываются сведения о биологически активных
веществах. С руктура и воздействие некоторых из них на обмен веществ
и регуляцию роста и развитие организма (биогенные амины, рилизинг-факто-
ры, опиоидные пептиды и др.) будут освещены в последующих главах, а ин-
формация о гербицидах, дефолиантах, ростовых веществах, нейротрансмит-
терах и т.п. дается в курсах физиологии растений и физиологии человека
и животных.
ГЛАВА V
ОБЩЕЕ ПОНЯТИЕ ОБ ОБМЕНЕ ВЕЩЕСТВ
И ЭНЕРГИИ В ОРГАНИЗМЕ
Характеристика структуры, свойств и функциональной активности фермен-
тов, коферментов, витаминов и ряда других биологически активных соедине-
ний, приведенная в предыдущих главах, неизбежно сопровождалась рассмот-
рением тех биох мических процессов, в которых они участвуют. Этим неволь-
но был сделан первый шаг к зучению некоторых аспектов обмена веществ.
В последующих главах это важнейшее свойство живого будет освещено во всех
его многообразных проявлениях. Здесь же целесообразно сосредоточить вни-
мание на общих закономерностях.
Общие представления об обмене веществ. Обмен веществ и энергии состав-
ляет сущность жизнедеятельности любого организма. Для самого явления
жизни характерен постоянный обмен веществ с окружающей внешней приро-
дой. Живое остается живым до тех пор, пока оно способно строить самое себя
из веществ окружающей среды; живая материя поддерживает собственное
существование путем постоянного и непрерывно протекающего с той или иной
скоростью поглощения химических соединений из внешнего по отношению
к ней мира, преобразования их в конституционные элементы своего тела или
более простые соединения и, наконец, выведения во внешнее пространство
продуктов распада как собственного тела, так и преобразованных в процессе
жизнедеятельности веществ. Этот непрерывный, самосовершающийся и само-
регулируемый круговорот веществ, протекающий в процессе существования
живой материи и сопровождающийся ее постоянным самообновлением, назы-
вается обменом веществ.
Иначе говоря, обмен веществ есть закономерный, самосовершающийся
процесс превращения материи в живых телах.
Преобразование химических соединений, взаимодействие их друг с другом,
разрушение одних и построение других происходит и в неживой материи.
Однако при этом неживая система любого типа не самообновляется, а лишь
видоизменяется. Жизнь, живая материя возникли тогда, когда среди неживых
систем с характерными для них процессами неупорядоченного изменения
веществ в природе появились системы, способные поддерживать собственное
существование путем упорядоченного преобразования и использования факто-
ров окружающей среды. Поэтому только та форма материи, те системы,
которые обладают обменом веществ, получили название живой материи.
Явление жизни необычайно сложно и многогранно. «Как часть планетного
земного живого вещества мы инстинктивно и бессознательно ярко чувствуем
загадку жизни—своего существования и существования жизни»,—писал ака-
демик В. И. Вернадский
В связи со значительными успехами квантовой биохимии, молекулярной
биологии, кибернетики, термодинамики и генетики в последнее время пред-
1 Вернадский В. И. Химическое строение биосферы Земли и ее окружения. М., 1987.
178
приня о много попыток углубить и конкретизировать п ниманис сущности
жизни. С точки зрения квантовой биохимии жизнь определяют как форму
динамической еализации квантовых характеристик атомов. В молекулярной
биологии приходят к обобщению, согласно которому жизнь предел вляется
как макромолекулярная иерархическая организация, характеризующаяся реп-
ликацией, метаболической цикличностью и отточенной регуляцией энергетиче-
ских процессов. При этом большая роль в явлениях жизни отводится нукле-
иновым кислотам. В кибернетике особое внимание уделяют процессам выра-
ботки и передачи информации, характери уя жизн как высокоустойчивое
состояние вещества, использующее для выработки со: раняющих реакций ин-
формацию, кодируемую состоянием отдел ных молекул. С точки зрения
термодинамики наиболее существенным свойством живых организмов являет-
ся устойчивая термодинамическая неравновесность и способность подд ржи-
вать ее на определенном уровне. Если неживая система стремится прийти
в равновесие с окружающей средой, то живая, напротив, старается уйти от
этого равновесия, целенаправленно поддерживая свое неравновесное по от-
ношению к окружающей среде состояние. Исходя из этого, жизнь определяют
как способ существования динамически самосохраняющейся формы мате-
рии—живой природы, сущность которого сводится к грандиозному процессу
циклической трансформации органического вещества, основанному на взаимо
действии синтеза и распад живой протоплазмы, т. е. биотическом круговоро-
те. В этом же аспекте жизнь определяют как потенциально способную к само-
воспроизведению открытую систему сопряженных органических реакций, ка-
тализируемых последовательно и почти изотермично сложными
и специфическими органическими катализаторами, которые сами вырабатыва-
ются этой системой.
Для современного генетика жизнь на Земле вырисовывается как интеграль-
ное существование ДНК, РНК и белков в форме индивидуализированных
личных и видовых целостных структурно-биохимических саморегулирующих-
ся открытых систем со свойствами воспроизведения исторически развива-
ющихся форм генетической информации. При оценке сущности жизни с пози-
ций эволюционного учения на первый план выступают происхождение, ор-
ганизаци и взаимодействие определенных надмолекулярных и клеточных
структур, закономерности возникновения их совокупности в виде самостоя-
тельной биологической единицы. По мнению физико-химиков, жизнь есть
динамическое равновесие мно офазных систем. Все перечисленные подходы
к более глубокому познанию сущности жизни безусловно заслуживают внима-
ния и каждый из них раскрывает новые стороны этого сложнейшего явления,
но каждый из них в отдельности односторонен.
По мере изучения жизни наши представления о роли отдельных, конкрет-
ных химических соединений, равно как и о значении тех или иных процессов
в ее осуществл нии, будут неизбежно изменяться. Кроме того, выход челове-
чества в Космос делает вероятным знакомство с неземными формами жизни,
ве цественная и функциональная характеристики которых могут оказаться
в каждом случае уникальными. Поэ ому определение жизни как особой,
качеств нно отличной биологической формы движения материи, возникающей
на определенном этапе ее развития, является, видимо, наиболее фундамен-
тальным и удачным.
Биологическая форма движения материи характер язуется существованием
саморегулирующихся и самовоспроизводящихся систем, обменивающихся
с окружающей средой веществом и энергией. Поскольку закономерное преоб-
разование веществ лежит в основе как процесса саморегуляции, так и процесса
самовоспр! изведения и условием их осуществления является непрерывное
179
поступление одних веществ в систему и выведение других из нее, обмен
веществ является важнейшим элементом жизни.
Та часть общего процесса обмена веществ, которая выражается в поглоще-
нии, накоплении, усвоении организмом веществ окружающей среды и созда-
нии, синтезе за их счет структурных единиц своего тела, называется анаболиз-
мом или ассимиляцией. Та часть общего процесса обмена веществ, которая
состоит в разрушении веществ, составляющих организм, в распаде элементов
живого тела и выведении продуктов этого распада из организма, называется
катаболизмом или диссимиляцией. Следовательно, обмен веществ есть диалек-
тическое единство противоположных процессов питания и выделения, усвоения
и разрушения, синтеза и распада, иными словами—диалектическое единство
процессов ассимиляции и диссимиляции.
Обмен веществ представляет собой сочетание многих разнообразных
и противоположных процессов. Одни из них представляют процессы физиоло-
гические (питание, выделение и т. п.), другие—физические (сорбция, перенос
и т. п.), третьи—химические (распад, синтез и т. п.). Та часть процессов
обмена веществ в организме, которая заключается в осуществлении хими-
ческих реакций, ведущих к преобразованию индивидуальных химических со-
единений при их распаде и синтезе в процессе жизнедеятельности организма,
называется промежуточным обменом веществ или метаболизмом. Именно про-
межуточный обмен веществ изучает динамическая биохимия.
Масштабы обмена веществ в живой природе колоссальны. Биомасса Земли,
составляющая, по подсчетам ученых, от 1,8 • 1012 до 2,4-1012 т (в пересчете на
сухое вещество), непрерывно обновляет свой состав, поглощая и выделяя
огромные количества химических веществ. По подсчетам А. А. Ничипорови-
ча, растения Земли за год усваивают из атмосферы около 650 млрд, т СО2
и выделяют в атмосферу около 350 млрд, т О2. За этот же срок растения
извлекают из почвы около 5 млрд, т N, около 1 млрд, т Р и 10—15 млрд, т
других минеральных элементов, образуя около 380 млрд, т биомассы (в
расчете на сухое вещество). Только свободно живущие в почве азотфиксиру-
ющие микроорганизмы и клубеньковые бактерии ежегодно связывают около
100 млн. т молекулярного N из воздуха. За 2000 лет весь О2 атмосферы Земли
проходит через живое вещество. Выделение кислорода в результате фотосин-
теза, протекающего в растениях, водорослях и фотосинтезирующих бактери-
ях, составляет от 2 тыс. до 5 тыс. т в одну секунду. Ежедневно на Земле
разрушается до СО2 и Н2О около 1 млрд, т органических соединений. По
данным В. А. Ковды, полное обновление биомассы суши происходит в тече-
ние 200 лет. По мнению В. И. Вернадского, все вещество биосферы в течение
краткого в геологических масштабах периода может пройти через живые ор-
ганизмы. Таким образом, «...жизнь—живое вещество—поистине является
одной из самых могущественных геохимических сил нашей планеты, а вызыва-
емая ею биогенная миграция атомов представляет форму организованности
первостепенного значения в строении биосферы»1.
Если перейти от рассмотрения обмена веществ в масштабе живого вещест-
ва всей планеты к оценке его роли в мельчайшей единице живого—клетке, то
здесь еще более ярко выступает значение обменных процессов в явлениях
жизни. Клетка, имеющая согласно современным представлениям (см. рис. 3)
сложнейшую внутреннюю организацию, является средоточением нескольких
тысяч различны? веществ. В простейшей бактериальной клетке (см. табл. 1)
находится около 300 млн. молекул органических соединений примерно
1 Вернадский В. И. Химическое строение биосферы Земли и ее окружения. М., 1987.
180
5000 наименований и громадное число молекул воды и неорганических солей.
Они не только закономерно расположены в клеточном объеме, но и находятся
в постоянном физическом и химическом движении. Первое осуществляется
благодаря наличию в клетке большого количества протоков, соединяющих
различные ее части друг с другом. Второе состоит в непрерывном преоб-
разовании химических веществ посредством многочисленных реакций распада
и синтеза белков, нуклеиновых кислот, углеводов, липидов и т. п. Все эти
реакции ускоряются сотнями ферментов и осуществляются согласованно во
времени и пространстве, а самое главное—они совершаются с огромными
скоростями и саморегулируются.
Обмен веществ осуществляется при условии и в результате постоянного
взаимодействия живой и неживой материи, организма и среды. Естественно
поэтому, что ход обмена веществ в организме, а часто и сам характер этого
обмена находятся в тесной зависимости, в единстве с условиями внешней
среды. Присущие организмам молекулярные механизмы видоизменения, пре-
образования, воспроизводства и разрушения специфических органических со-
единений (нуклеиновые кислоты, белки, липиды, углеводы и др.) действуют
лишь в определенных, ограниченных интервалах температуры, давления, ра-
диации. Они осуществляются лишь при условии постоянного притока веществ,
пригодных для преобразования, и оттока веществ, которые уже не могут
служить исходным материалом для построения тела данного организма.
Более того, поддержание на известном уровне специфической активности
молекулярных механизмов, преобразующих вещества клетки, зависит от не-
прерывного воспроизведения составных частей самих этих механизмов в еди-
ном процессе обмена веществ. В этом смысле любой организм представляет
неизбежно самонастраивающуюся, саморегулирующуюся систему, закономерно
(и адекватно) изменяющуюся при изменении условий среды, с которыми ор-
ганизм взаимодействует. Таким образом, тип обмена веществ складывается
в процессе жизнедеятельности организма как единство внутренних (консер-
вативных) и внешних (изменчивых) факторов. В этом и состоит диалектиче-
ское единство организма и среды.
Закономерности обмена энергии. Обмен веществ невозможен без сопутству-
ющего ему обмена энергии. Каждое органическое соединение, входящее в со-
став живой материи, обладает определенным запасом потенциальной энергии, за
счет которой может быть совершена работа. ЭТУ энергию принято называть
свободной энергией. Уровни свободной энергии индивидуальных исходных
веществ и продуктов реакции, как правило, различны, вследствие чего в про-
цессе преобразования веществ происходит перераспределение свободной энер-
гии между компонентами реакционной смеси, т. е. протекает обмен энергией
между веществами.
Понятие о макроэргических соединениях. Главными материаль-
ными носителями свободной энергии в органических веществах являются
химические связи между атомами, поэтому при преобразовании химических
связей в молекуле уровень свободной энергии соединения изменяется. Если
изменение уровня свободной энергии соединения при возникновении или
распаде химической связи составляет около 12,5 кДж/моль преобразуемого
вещества, то такая связь по своему энергетическому уровню считается нор-
мальной. Именно такую размерность имеет изменение уровня свободной
энергии при преобразовании большинства связей в органических соединениях.
Однако при новообразовании и распаде некоторых связей уровень свободной
энергии в молекулах ряда органических соединений изменяется в гораздо
большей степени и составляет 25—50 кДж/моль и более. Такие соединения
называются макроэргическими соединениями, а связи, при преобразовании
181
которых наступают столь крупные изменения в энергетическом балансе веще-
ства,—макроэргическими связями. Последние в отличие от обычных связей
обозначают значком Ниже приведены некоторые органические вещее ва
первого (с нормальными связями) и второго (с макроэргическими связями)
типов. В каждом случае в скобках указаны величины уменьшения свободной
энергии (отмечено знаком минус перед цифрами в скобках) в исходных вещест-
вах по сравнению с тако ыми в продуктах их гидролиза (при pH 7, Z=25° С,
кДж/моль):
СН2ОН
снон ОН СН2ОН он
СН2—О— Р=0+Н2О-»СНОН +но—р=о
I I I
он СН2ОН он
Глицерофосфат (—10,0)
Сахаром (—27,7)
Уридиидифосфвтглюкоэа (—28,4)
182
Ацетнлкоэиаям А (-32,9)
ОН он
I I
НООС—СН2—N— С —NH~ Р =О+Н2О->НООС—СН2—N — С —NH2+HO—Р =О
I II I I II I
СН3 NH ОН СН3 NH ОН
Креатинфосфат (—40,1)
СН3—С—О~Р=О + Н2О —► СНз—СООН + НО—р=о
Ан Ан
Аиетилфосфат (—43,7)
ОН ОН
I I
СНз—О—Р=О СН,—О—Р=О ОН
Il I I I
СНОН ОН + Н2О —► СНОН ОН + но—Р=О
1^° 9Н I
сг^ | сГ он
\)~Р=О хон
I
он
1,3-дифосфо лииерииовая кислота (—49,1)
183
о
H,N—С ОН
\~Р=0
I
ОН
о
+ Н,0 — H,N-C^
''''он
ОН
сн, (
11 j
С—О-Р
Карбамилфосфат (—51,5)
)Н СН, он
II II
=0 + Н,0 -+ с-он + НО-Р=О
соон Ан СООН он
Фосфоено лпнровино- т
граднвя кислота СНч
(-58,6) |
С=0
I
соон
Нетрудно видеть, что макроэргические связи представлены преимущест-
венно сложноэфирными, в том числе и тиоэфирными, ангидридными и фосфо-
амидными связями. Однако наиболее интересно, что почти все известные
соеди ения с макроэргическими Связями содержат атомы Р и S, по месту
которых в молекуле эти связи локализованы. Особая роль атомов S и Р в об-
разовании макроэргических связей объясняется следующими причинами:
1) химические связи, свойственные элементам III периода, слабее, чем связи,
характерные для элементов II периода; 2) S и Р образуют более четырех
ковалентных связей; 3) только S и Р в III периоде сохраняют способность
к замыканию кратных связей.
Именно та энергия, которая высвобождается при разрыве макроэргических
связей, поглощается при синтезе органических соединений с более высоким
уровнем свободной энергии, чем исходные. В то же время запасы макроэр-
гических веществ в организме постоянно пополняются путем аккумулирова-
ния энергии, выделяющейся при понижении энергетического уровня распада-
ющихся соединений.
Таким образом, макроэргические вещества выполняют функцию и доно-
ров, и акцепторов энергии в обмене веществ; они служат как аккумуляторами,
так и проводниками энергии в биохимических процессах. Кроме того, им
свойственна роль трансформаторов энергии, так как они способны преоб-
разовывать стационарную форму энергии химической связи в мобильную, т. е.
в энергию возбужденного состояния молекулы.
Последний вид энергии и служит непосредственным источником реакцион-
ной способности молекул; преобразуясь снова в стационарную форму энергии
химической связи, он энергетически обеспечивает видоизменение веществ, их
преобразование, т. е. их обмен в организме.
Все сказанное еще раз подчеркивает тесную взаимозависимость обмена
веществ и обмена энергии.
Природа макроэргических связей. Какие же закономерности
в строении молекул макроэргических соединений обеспечивают их особую
роль в обмене веществ и энергии? Чтобы ответить на этот вопрос, эассмотрим
структуру молекулы аденозинтрифосфорной кислоты (АТФ)—наиболее важ-
ного макроэргического соединения в организме. Впервые внимание на ее роль
в энергетическом обеспечении химических процессов обратил Ф. Липманн
(1939—1941), а механизм распада под действием миозина и переход энергии
макроэргической связи в механическую энергию сокращения мышцы исследо-
вали В. А. Энгельгардт и М. Н. Любимова (1939).
184
Энергия химической связи между двумя концевыми фосфатными остатка-
ми в молекуле макроэргического соединения (АТФ) и энергия связи между
остатком фосфорной кислоты и глюкозы в молекуле немакроэргического
соединения (глюкозо-6-фосфата) примерно одинакова. Работу, выражающу-
юся одной и той же величиной, в обоих случаях необходимо затратить для
отрыва фосфорного остатка и от того, и от другого соединения. Однако при
изучении реакций гидролиза АТФ и глюкозо-6-фосфата выявлена существен-
ная разница в энергетическом балансе этих одноименных реакций: в случае
АТФ изменение свободной энергии составляет 32,5—34,7 кДж/моль, в случае
глюкозо-6-фосфата—всего 13,1 кДж/моль.
Отсюда следует, что понятия «энергия связи» и «макроэргическая связь»
совершенно различны. Первое сводится к характеристике энергетического
уровня химической связи с точки зрения физической химии, т. е. как величины
энергии, необходимой для разрыва связи между атомами. Второе состоит
в учете энергетического эффекта в результате преобразования связи посред-
ством химической реакции.
Здесь-то и выявляется характерная разница между обычной и макроэргиче-
ской связью; последнюю нельзя рассматривать как химическую связь, об-
ладающую повышенным по сравнению с обычной запасом энергии, или как
связь непрочную, которая только и «ждет» случая, чтобы разрушиться взры-
воподобно, с выделением энергии. Ее основная характеристика состоит в том,
что при гидролизе или преобразовании иным путем такой связи выделяет-
ся гораздо больше энергии, чем при гидролизе или преобразовании обычной
связи.
Исследование структуры той части молекулы АТФ, где сосредоточены
макроэргические связи, показало, что строение ее своеобразно. В частности,
распределение зарядов у атомов Р и О в трифосфатной группировке АТФ
таково:
Как видно из приведенной выше формулы аденозинтрифосфорной кисло-
ты, кислородно-фосфорный остов трифосфатной группировки составлен поло-
жительно заряженными атомами О и Р. Электростатическое отталкивание
положительных зарядов в остове трифосфатной части АТФ имеет, видимо,
существенное значение для выделения повышенного количества энергии при
гидролитическом отрыве фосфатных групп. Аналогично, с точки зрения кван-
тово-механических представлений, построены и остальные макроэргические
соединения: для них характерно накопление положительных зарядов у атомов,
непосредственно связанных макроэргической связью, и отрицательных—у
окружающих эти связи атомов:
185
—0,559 0,817
О О
ГС JI+0,365 о+°^80 j,+0.365 Q
0—0.817
Аци-л фосфаты
—0,817
NH-M« о-0.«3
^-0,823
Гуанидинфосфаты
О-0.400
„ „—0,052 J] +0.217 е+0.168 р
Hjv. ' V* *'*’ О
Тноэфнры
Существенно также, что л-электроны атомов О, N и S, связывающих
группировки атомов в макроэргических соединениях, не в состоянии удовле-
творить потребность в электронах этих группировок.
В качестве иллюстраци приведем струк уру ацетилфосфата:
с?
Н3С—C^-g~P=*=O
О’
Как следствие этого, неустойчивая система сопряжения электронов в мак-
роэргических соединениях при гидролизе и других преобрази ваниях легко
и необратимо нарушается.
Роль макроэргических соединений в трансформации энергии. Рассмотрим теперь,
как можно представить механизм превращения стабильной энергии химической
связи в подвижную энергию возбуждения молекулы и обратного процесса—
преобразования мобильной энергии возбуждения в стационарную энергию
химической связи. Именно в этом заключается одна из наиболее существенных
функций макроэргических соединений. Молекула АТФ осуществляет указанную
трансформацию энергии, будучи связана со специфическим белком. Закрепление
АТФ на белковой молекуле сопровождается сближением трифосфатной части
молекулы с пуриновой ее частью. Выделяющаяся при распаде макроэргической
связи между остатками фосфорной кислоты энергия передается на пуриновую
часть молекулы. Она трансформируется при этом в мобильную энергию
возбуждения электронов системы сопряженных двойных связей пуринового цикла,
откуда поступает далее к месту химической реакции, переводя в возбужденное
состояние электроны преобразуемого органического соединения. Это обеспечива-
ет последнему повышенную реакционную способность, создавая, в частности,
потенциальную возможность для обратного преобразования подвижной энергии
возбуждения в стационарную энергию новых химических связей (рис. 63).
Энергия, которая выделяется при распаде макроэргических соединений и за
счет которой может быть совершена та или иная работа, используется не
только для химического синтеза. Она может служить в организме для тепло-
образования, св чения, накопления электричества, выполнения механической
работы и т. п. При этом химическая энергия преобразуется в тепловую,
лучистую, электр ческую, механическую и пр. Принципиально важно то, что
преобразование хими еской энергии в другие ее виды протекает в организме
при обязательном участии соединений с макроэргическими с язями, в част-
ности АТФ. В молекуле АТФ, как было показано выше, происходит трансфор-
мация стабильной энергии макроэргических межфосфа ных химических связей
186
Рис. 63. Трансформация энергии в молекуле АТФ (пояснения
в тексте)
в подвижную энергию возбуждения электронов пуриновой части молекулы;
это и есть, вероятно, первый этап преобразования энергии в организме.
Именно поэтому АТФ занимает центральное место в энергетическом обмене
живой материи.
Рассмотрение механизма трансформации и передачи энергии при распаде
АТФ и аналогичных ей макр эргических соединений позволяет понять одно из
элементарных фундаментальных свойств живой материи. Оно состоит в том,
что в системах, обладающих свойствами живого, энергия, необходимая для
осуществления химической реакции, будучи высвобождена в одной точке,
может быть передана в другую точку, где она непосредственно используется.
Это значит, что в живой природе нет необходимости в непосредственном
контакте путем соударения (что является характерным свойством реакций
в неживой природе) молекулы, поставляющей энергию, с молекулой, нужда-
ющейся в энергии. Это принципиально отличает ход химического процесса
в живых объек ах от такового в неживых.
В живой природе идет непрерывный процесс запасания энергии в виде
химической энергии связей органических веществ. Он состоит главным об-
разом в преобразовании лучистой энергии в химическую (фотосинтез зеленых
растений) и в небольшой мере—в переходе химической энергии из неор-
ганических соединений в органи еские (хемосинтез микроорганизмов).
Как видно из рис. 64, АТФ играет выдающуюся роль как при запасании,
так и при расходовании энергии, т. е. является ключевым веществом в энер-
гетическом обмене организма. Известие много реакций, при посредстве ко-
торых АТФ возникает из других макроэргических соединений, и наоборот,
есть много процессов, приводящих к синтезу макроэргических соединений
при участии АТФ. Такие, например, макроэргические соединения, как кре-
атинфосфат, фосфоенолпировиноградная кислота и 1,3-дифосфоглицериновая
кислота, при взаимодействии с АДФ образуют АТФ с выделением креатина,
187
Рис. 64. Превращения энергии в живой природе (пояснение в тексте)
пировиноградной кислоты и 3-фосфог пицериновой кислоты. Эти и подобные
им соединения принято обозначать как АТФ-генерирующие вещества. Пере-
численные реакции обратимы, и при изв стных условиях равновесие может
быть смещено в сторону распада АТФ.
Общие закономерности трансформации энергии в живых
системах. Обмен энергии в процессе жизнедеятельности не исчерпывается
превращением химической энергии в другие виды ее и наоборот (рис. 64); он
носит более широкий характер. Так, в палочках и колбочках сетчатки глаза
световая энергия превращается в электрическую; в специфических структурах
внутреннего уха звуковая и гидродинамическая энергия переходит в электри-
ческую и т. п.
Трансформация одного вида энергии в другой осуществляется в организ-
мах в морфологически разнообразных элементах—хлоропластах, мышцах
(российские кардиологи недавно впервые выявили новые пути передачи энер-
гии от места ее образования в митохондриях к точке потребления в миофиб-
риллах сердечной мышцы I, рецепторных аппаратах тканей и органов, сетчатке
глаза, люминесцентных органах и т. п.
Однако всем этим разнообразным элементам свойственны некоторые об-
щие черты строения. Они отличаются наличием двухслойных мембран с высо-
ким содержанием липопротеинов в них и присутствием структурного белка,
связывающего в упорядоченные образования достаточно унифицированные
элементарные частицы. Последние включают в свой состав молекулы опреде-
ленного строения, которые, собственно, и осуществляют процесс трансфор-
мации энергии. При этом энергия одного вида поглощается молекулой-преоб-
разователем и превращается в энергию другого вида. Простейшим примером
механизма внутримолекулярного превращения энергии молекулой-преобразо-
вателем служит переход стационарной энергии химических связей трифосфат-
ной группировки молекулы АТФ в подвижную энергию возбужд ния электро-
нов ее пуриновой части (см. рис. 63). Более сложным примером являются
конформационные изменения белковых молекул в процессе преобразования
одного вида энергии в другой (например, мышечное сокращение).
Обмен веществ и энергии представляет единый, неразрывный процесс, где
видоизменение вещества всегда сопровождается выделением или поглощением
свободной энергии и где выделившаяся или поглотившаяся в том или ином
количестве энергия обеспечивает распад или синтез химических связей, т. е. по
существу видоизменение самих веществ. Таким образом, закономерности об-
мена веществ и энергии в живой природе являются частным случаем общего
закона сохранения материи и энергии.
ГЛАВА VI
НУКЛЕИНОВЫЕ КИСЛОТЫ И ИХ ОБМЕН
СТРОЕНИЕ И СВОЙСТВА НУКЛЕИНОВЫХ КИСЛОТ
Химия нуклеиновых кислот в последнее время развивается необычайно
быстрыми темпами, что связано с коренным пересмотром взглядов на биоло-
гическую роль этих соединений. Представление о нуклеиновых кислотах как
об инертных структурных элементах ядра и цитоплазмы оставлено навсегда,
так как нуклеиновым кислотам принадлежит очень важная роль в обеспечении
специфического синтеза биополимеров в организме человека, животных, рас-
тений и микробов.
К нуклеиновым кислотам относят высокомолекулярные соединения, харак-
теризующиеся определенным элементарным составом и распадающиеся при
гидролизе на пуриновые и пиримидиновые основания, пентозу и фосфорную
кислоту. Особенно характерно для нуклеиновых кислот содержание Р (8—
10%) и N (15—16%). Нуклеиновые кислоты содержат также С, Н и О.
Нуклеиновые кислоты были впервые выделены Ф. Мишером более столе-
тия тому назад (1869) из ядер клеток гноя в виде соединения с белком—
нуклеира (от лат. nucleus—ядро), а сам термин предложен А. Косселем
в 1889 г. К концу прошлого века они были получены Р. Альтманом (1899)
в свободном от белка состоянии из животных тканей и дрожжей, а в 1936 г.
А. Н. Белозерским с сотр.—из растительного материала.
Выделение нуклеиновых кислот. Большая часть нуклеиновых кислот в расти-
тельных, животных и бактериальных клетках находится в соединении с белками.
Поэтому, кроме разрушения клеточных оболочек путем гомогенизации биологиче-
ского материала, для выделения нуклеиновых кислот необходимо разорвать связь
между нуклеиновой кислотой и белком. Это достигается обработкой материала
крепким раствором соли, например 10%-ным раствором NaCl. Одновременно
осуществляется извлечение нуклеиновых кислот. После удаления твердого остатка
нуклеиновые кислоты осаждают из охлажденного до 0° С раствора этанолом или
раствором трихлоруксусной кислоты. Осадок нуклеиновых кислот отделяют
центрифугированием, тщательно промывают и высушивают.
Кроме того, применяют фенольный метод выделения нуклеиновых кислот,
при помощи которого можно получать их нативные препараты. Для этого
измельченную в гомогенизаторе при охлаждении ткань заливают водонасы-
щенным раствором фенола, энергично встряхивают смесь в течение 1 ч и цент-
рифугируют ее. При этом содержимое центрифужной пробирки разделяется на
четыре четко отграни енных друг от друга разных по консистенции и цвету
слоя. В верхнем, водном, слое и подстилающем его вязком слое белого цвета
сосредоточена основная масса нуклеиновых кислот. В третьем, желеобразном,
прозрачном, желтоватом слое находится фенол с растворенными в нем белка-
ми. Четвертый, самый нижний, слой коричневого цвета содержит остатки
ткани, денатурированные белки и ничтожную примесь нуклеиновых кислот.
189
Так как в процессе выделения нуклеиновых кислот этим методом удается
избавиться от значительной части белков (протеинов), указанную процедуру
называют фенольной депротеинизацией.
Освободить препараты нуклеиновых кислот от белка можно также путем
обработки их солевого экстракта двойным объемом хлороформа, содержащего
немного изоамилового спирта; после тщательного перемешивания в течение
15 мин до получения стойкой эмульсии смесь центрифугируют и отделяют
верхнюю водную фазу, содержащую нуклеиновые кислоты (денатурирован-
ный белок остается на границе водного и хлороформного слоев); нуклеиновые
кислоты из водной фазы осаждают двойным объемом охлажденного этанола.
Полученные так суммарные препараты нуклеиновых кислот фракциониру-
ют далее, используя более тонкие методы, такие, как хроматография, в том
числе аффинная, фильтрация через гели агарозы и сефарозы, распределение
в двухфазных полимерных системах, ультрацентрифугирование, электрофорез
и др. Большинство этих методов рассмотрено в гл. II и при работе с нукле-
иновыми кислотами отличается лишь в деталях. В итоге получают препараты
индивидуальных нуклеиновых кислот.
Выделение из биологических объектов давно уже не является единственным
методом получения нуклеиновых кислот. Наряду с ним широкое распростра-
нение получил их химический синтез. Разработка химических и инженерных
подходов позволила создать автоматические синтезаторы нуклеиновых кис-
лот, отличающиеся высокой производительностью и надежностью. Так, одна
из марок синтезаторов фирмы ЛКБ (Швеция) обеспечивает автоматическую
сборку молекул нуклеиновых кислот длиной до 160 звеньев при десятиминут-
ной длительности каждого цикла..
Особенно важен химический синтез нуклеиновых кислот для получения генов, их
фрагментов, регуляторных участков нуклеиновых кислот и т. п. Первый синтез гена
аланиновой транспортной рибонуклеиновой кислоты был осуществлен в 1972 г.
Г. Корана. Сейчас число синтезированных генов достигло нескольких десятков.
В СССР синтезированы ген человеческого интерферона а2(М. Н. Колосов и др.,
1982), ген валиновой транспортной РНК (М. Н. Колосов и др., 1983), ген
кальцитонина человека (А. А. Баев и др., 1985), ряд промоторов и др.
Химический состав. При нагревании нуклеиновых кислот с хлорной кисло-
той они распадаются на структурные единицы, из которых построены их
громадные молекулы. Другие кислоты, как, например, НС1, вызывают очень
резкую деструкцию нуклеиновых кислот с выделением NH3, свидетельст-
вующим о разрушении входящих в их состав структурных элементов.
Среди структурных элементов нуклеиновых кислот найдены пиримидино-
вые основания, пуриновые основания, углеводы и фосфорная кислота.
Пиримидиновые основания являются производными гетероциклического со-
единения —пиримидина:
Структурная форму-
ла пиримидина
Межатомные расстоя-
ния, нм, и величины
валентных углов в
молекуле пиримидина
Порядки связей в
молекуле пирими-
дина
190
Приведенная здесь структурная формула в виде шестичленного цикла
с чередующимися двойными связями является, как и в случае бензола, лишь
данью истории. В действительности, как можно заключить из сопоставления
межатомных расстояний, в молекуле пиримидина нет ни типичных двойных,
ни типичных простых связей, а имеет место взаимодействие л-электронов всех
составляющих цикл атомов. Мерой взаимодействия л-электронов служит так
называемый порядок связи, характеризующий силу сопряжения л-электронов
двух соседних атомов. В случае типичной двойной связи силу взаимодействия
л-электронов, т. е. порядок связи, принимают за единицу. В результате дело-
кализации л-электронов в молекулах с сопряженными двойными связями, как,
например, в рассматриваемой молекуле пиримидина, порядки связей прини-
мают дробные значения. Чем больше значение порядка связи, тем сильнее
выражена ее способность к реакциям присоединения.
В составе нуклеиновых кислот найдены следующие производные пирими-
дина (пиримидиновые основания): цитозин, урацил, тимин, 5-метилцитозин и 5-
оксиметилцитозин:
Уращи
(2, 4-даоксипи-
римядин)
TtaxiM 5-метилцитоат 5-оксиметщпозин
(5-меплуращ<л)
Цитозин, урацил и тимин содержатся в нуклеиновых кислотах в значитель-
ных количествах, а 5-метилцитозин и 5-оксиметилцитозин—в ничтожных
и далеко не всегда. Поэтому они называются минорными (экзотическими)
основаниями. По аналогии с редкими аминокислотами в составе белков их
можно было бы назвать иногда встречающимися в составе нуклеиновых
кислот основаниями. В последние годы список минорных оснований пирими-
динового ряда, обнаруженных в составе нуклеиновых кислот, пополнился
(табл. 15).
Пуриновые основания нуклеиновых кислот являются производными бицик-
лического гетероцикла—пурина:
Межатомные расстоя-
ния в молекуле пури-
на, нм
Порядки связей в мо-
лекуле пурина
пиримидинового цикла,
расстановка в формуле
Пиримидине- Имидазоль-
ный цикл ный цикл
Структурная фор-
мула пурина
Здесь, как и в случае
простых и двойных связей условна. Как межатомные расстояния, так и поряд-
ки связей в молекуле пурина указывают на высокую степень сопряжения
л-электронов атомов С и N, составляющих пуриновый цикл.
191
В гидролизатах нуклеиновых кислот всегда обнаруж ваются два производ-
ных пурина—аденин и гуанин:
Аленин
(6-аминопурин)
Гуанин
(2-амино-6-охсипур«ш)
Кроме того, в составе нуклеиновых кислот найдено большое число минор-
ных пуриновых оснований—метилированных производных аденина и гуанина
(табл. 15):
1 -Метиладеннн
2-Мети ла денин
Nj-Диметиладенин
1-Мети лгу анин
7-Метил гуанин
Nj-Днметнлгуанин
Важной особенностью оксипроизводных пиримидина и пурина является
возможность их таутомерных (лактам-лактимных) превращений, например:
Енольная Кетоиная Енольная Кетонная
(лактимная) (лактамная) (лактимная) (лактамная)
форма форма форма форма
Урацил Гуанин
Благодаря этому, в частности, пиримидиновые основания по 1-му атому
N лактамной формы вступают во- взаимодействие с углеводами, найденными
в составе нуклеиновых кислот.
Углеводная составляющая нуклеиновых кислот представлена двумя весьма
сходными моносахаридами правого ряда: рибозой и дезоксирибозой. В свобод-
ном состоянии эти моносахариды существуют во всех возможных таутомер-
ных формах, возникающих за счет. кольчато-цепной таутомерии. В составе
192
нуклеиновых кислот оба моносахарида находятся в p-D-рибофуранозной
форме:
p-D-Рибоза
p-D-2-Дезоксирибоза
По сравнению с P-D-рибозой второй моносахарид (P-D-2-дезоксирибоза)
является соединением, восстановленным по 2-му углеродному атому. Так как
в процессе восстановления происходит отнятие гидроксильной группы, то
полученное производное называют дезоксирибозой, причем цифра 2 указывает
номер углеродного атома рибозы, у которого гидроксильная группа заменена
на атом Н.
Недавно выяснено, что рибоза и дезоксирибоза не является единственными
углеводами, входящими в состав нуклеиновых кислот: в ряде фаговых ДНК
и ДНК некоторых видов раковых клеток найдена глюкоза.
Изучение продуктов гидролиза нуклеиновых кислот привело к важному
выводу: состав продуктов гидролиза нуклеиновых кислот, выделенных из
разных источников, неодинаков. Впервые это было обнаружено при сравнении
состава нуклеиновых кислот, выделенных из зобной железы (тимус) теленка
(тимонуклеиновая кислота) и дрожжей (дрожжевая нуклеиновая кислота).
В дальнейшем было показано, что им соответствует два типа нуклеиновых
кислот, отличающихся по составу, строению и функциям. В соответствии
с характером углеводной компоненты одна из них была названа дезоксирибонук-
леиновой кислотой (ДНК), другая—рибонуклеиновой кислотой (РНК). Между
ДНК и РНК существуют и иные черты сходства и различия по составу (табл. 15).
Таблица 15
Состав нуклеиновых кислот
Химическое соединение ДНК РНК
Пуриновые основания Аденин Гуанин Аденин Гуанин
Пиримидиновые основания Цитозин Ткмин Цитозин Урацил
Углеводы Дезоксирибоза Глюкоза (иногда) Рибоза
Неорганическое вещество Фосфорная кислота Фосфорная кислота
Минорные основания
Пуриновые М6-Метиладенин 1-Метилгуанин З-Метилгуанин 7-Метилгуанин К2-Метилгуанин К2-Диметилгуанин Ne-Метиладенин М6-Диметиладенин 1-Метиладенин 2-Метиладенин 2-Метилтио-Ы6-изопентенила- денин 1Ч2-Метилгуанин М2-Диметилгуанин 1-Метилгуанин 7-Метилгуанин
7—3502
193
Продолжение табл. 15
Химическое соединение ДНК РНК
Пиримидиновые 5-Метилцитозин 5-Оксиметилцитозин Оксиметилурацил Урацил 5-Метилцитозин 5-Оксиметилцитозин К4-Метилцитозин З-Метилцитозин З-Метилурацил Тимин 5-Метиламиноэтил-2-тиоура- цил Дигидроурацил
Как видно из данных табл. 15, ДНК и РНК отличаются также и по
качественному составу пиримидиновых оснований: для первой характер ю
наличие тимина, для второй—урацила. Особенно разительны различия в ми-
норных пуриновых и пиримидиновых основаниях ДНК и РНК: последняя
содержит гораздо более богатый (более 50) их набор.
Наметилась тенденция связывать наличие и распределение в ДНК и РНК
минорных метилированных оснований с рядом важнейших функций нукле-
. иновых кислот: взаимодействием их с белками, в том числе с рядом фермен-
тов, кодированием и передачей информации о биосинтезе макромолекул,
участием в механизме памяти и старения о{ ганизма, регуляцией биосинтеза
нуклеиновых кислот и др.
Молекулярная масса, содержание и локализация в клетке ДНК и РНК; виды
ДНК и РНК. Молекулярную массу ДНК определяют в основном гидродина-
мическим и электронно-микроскопическим методами, хотя это можно делать,
измеряя светорассеяние растворов ДНК и некоторыми другими способами.
В основе гидродинамического метода лежит линейная зависимость константы
седиментации ДНК, определяемой при ультрацентрифугировании растворов
ДНК, от ее молекулярной массы, которую можно установить по калибровочной
кривой или рассчитать по формуле: 0,4451gM = l,819+lg(s°2o.w—2,7), где s°2o ш—
константа седиментации, приведенная путем экстраполирования к бесконечно-
му разведению (s°), стандартной температуре (20° С) и вязкости воды (со).
Электронно-микроскопический метод определения молекулярной массы
ДНК основан на измерении длины вытянутых молекул ДНК. Известно, что на
0,1 нм протяженности ее молекулы приходится масса, равная 197 Да. Ум-
ножая эту величину на экспериментально найденную длину молекулы ДНК,
получают значение ее молекулярной массы.
Выделить нативную ДНК из клеток эукариот необыкновенно трудно, так
как еще не создано соответствующих методов, позволяющих избежать раз-
рыва выделяемых молекул ДНК. Поэтому надежные цифры получены только
для ДНК вирусов и фагов (табл. 16); из них проще извлечь ДНК, осторожно
сняв белковую оболочку.
Как видно из табл. 16, молекулярные массы вирусных и фаговых ДНК
измеряются десятками и сотнями миллионов дальтон. Можно полагать, что
молекулярная масса ДНК эукариот значительно выше. Об этом свидетельст-
вует молекулярная масса ДНК, выделенной с необходимыми предосторож-
ностями из самой большой хромосомы плодовой мушки—дрозофилы. Вся
ДНК хромосомы представлена одной молекулой с М=40 • 109.
Содержание ДНК в клетках организма определенного вида отличается
необыкновенным постоянством, тогда как межвидовые различия по этому
показателю достаточно велики. Количество ДНК в клетке измеряется пико-
граммами (10-12 г), колеблется от 0,01 пг у кишечной палочки до нескольких
пикограммов в гаплоидных клетках высших организмов.
194
Таблица 16
Молекулярные массы вирусных и фаговых ДНК
Объект Молекулярная масса, млн. далы он
Гидродинамический метод Электронно-микроскопический метод
Бактериофаг fd 1,9 —
Вирус полиомы — 3,2
Аденовирус 21 24
Бактериофаг Т7 23—28 22—28
» Т5 66 67
» Т2 123 105—119
» Т4 111^131 116—152
В зависимости от места локализации ДНК в клетке различают ядерную,
митохондриальную, хлоропластную, центриольную и эписомальную ДНК. Ядер-
ная ДНК у эукариот резко превалирует над ДНК других субклеточных струк-
тур. Так, в митохондриях обнаружено от 0,5 • 10-16 до 5 • 10-16 г ДНК, в хло-
ропластах— от 10-1° до 150 • 10-16 г, а в центриолях—2 • 10-16 г, что состав-
ляет несколько процентов от ядерной ДНК. В таком же соотношении
находится содержание ДНК в бактериальной хромосоме и эписомах — внехро-
мосомных, самостоятельно реплицирующихся детерминантах наследствен-
ности у микроорганизмов, обеспеч вающих перенос генетической информа-
ции, например, об устойчивости к антибиотикам (иначе их называют R-
факторами, т. е. факторами резистентности). Обсуждается вопрос о существо-
вании экстрахромосомной ДНК, транспортируемой, или коммуникационной,
ДНК, цитоплазматической мембранной ДНК, мелкодисперсной сверхскрученной
ДНК. По функциональному назначению различают рибосомальную ДНК
(рДНК) и сателлитную ДНК (стДНК).
Кроме внутриклеточной ДНК, существует также ДНК, входящая в состав
вирусов и фагов. Количество ее в вирусах и фаговых частицах значительно
ниже, чем в клетках бактерий (тысячные доли пикограмма и менее).
Молекулярные массы РНК определяют теми же методами, что и ДНК, но,
кроме того, используют электрофорез в полиакриламидном геле, так как
пробег РНК в геле обратно пропорционален их молекулярным массам. Что
касается содержания и локализации РНК в клетках, то оно не отличается ни
однообразием, ни стабильностью: в клетках, где идет интенсивный биосинтез
белков, содержание РНК в несколько раз превышает таковое ДНК (например,
в печени крысы РНК в 4 раза больше, чем ДНК), но там, где синтез белка мал,
соотношение ДНК и РНК может быть обратным (например, в легких крысы
РНК в 2 раза меньше, чем ДНК).
По функциональному значению и молекулярным массам, равно как и по
локализации в клеточном содержимом, РНК делят на следующие виды.
1. Транспортные РНК (тРНК) отличаются сравнительно невысокими значе-
ниями молекулярных масс (17000—35000), локализованы в гиалоплазме клет-
ки, ядерном соке, бесструктурной части хлоропластов и митохондрий. Они
осуществляют кодирование аминокислот и перенос их в рибосомальный ап-
парат клетки в процессе биосинтеза белков.
2. Рибосомальные РНК (рРНК) характеризуются в основном большими
молекулярными массами (550000—700000 у РНК 30—40S субчастиц рибосом,
1,1-106—1,7-106 у РНК 50—60S субчастиц рибосом, но 40000 у 5S РНК
и ~ 50 000 у 5,8S РНК из рибосом); локализованы в рибосомах, являясь их
структурной основой и выполняя в них разнообразные функции.
195
3. Информационные, или матричные, РНК (мРНК) обладают молекуляр-
ными массами, варьирующими в широких пределах (от 300000 до 4-10й).
Возникая в форме высокомолекулярных предшественников в ядре клетки или
на ДНК других субклеточных частиц, мРНК (в виде рибонуклеопротеинов)
перемещаются к рибосомам; в составе последних они выполняют матричную
функцию в процессе сборки полипептидных цепей.
4. Вирусные РНК отличаются разнообразными и высокими молекуляр-
ными массами, лежащими в основном в пределах нескольких миллионов
дальтон. Они являются составными частями вирусных и фаговых рибонуклео-
протеинов, несут всю информацию, необходимую для размножения вируса
в клетках хозяина.
В современной литературе обсуждается вопрос о целесообразности выде-
ления в отдельные категории еще нескольких видов РНК: ядерной, хромо-
сомной, митохондриальных, низкомолекулярных регуляторных, антисмыс-
ловых.
Строение структурных элементов нуклеиновых кислот. При осторожной
обработке РНК водным раствором щелочи (например, 1 н. NaOH или КОН
в течение 18 ч при комнатной температуре) они распадаются на структурные
единицы, каждая из которых состоит из пуринового или пиримидинового
основания, рибозы и остатка фосфорной кислоты. Еще лучше этот процесс
распада осуществляется при действии специфических биокатализаторов, на-
пример рибонуклеазы. ДНК, в противоположность РНК, устойчивы к дей-
ствию разбавленных растворов щелочей при обычной температуре. Поэтому
гидролиз ДНК до структурных единиц, каждая из которых составлена из
пуринового или пиримидинового основания, дезоксирибозы и фосфорной
кислоты, удается осуществить только в присутствии специального катализато-
ра биологического происхождения—дезоксирибонуклеазы. Кислоты, как ми-
неральные (НС1, НСЮ4 и др.), так и органические (НСООН и др.) для такого
рода гидролиза неприемлемы, так как они деструктируют и РНК, и ДНК до
свободных пуриновых и пиримидиновых оснований, углевода и фосфорной
кислоты. Из гидролизатов нуклеиновых кислот составляющие их структурные
единицы были впервые выделены в 1908 г. П. Левиным и Дж. Манделем,
которые назвали их нуклеотидами. При гидролизе РНК образуются рибонукле-
отиды, при гидролизе ДНК—дезоксирибонуклеотиды.
Пуриновые или пиримидиновые основания, рибоза или дезоксирибоза
и фосфорная кислота связаны в молекулах нуклеотидов совершенно однотип-
но. Химическое строение нукле тид >в и их полные и сокращенные названия
таковы:
Рибонуклеотиды
Цитидин-
монофосфат
(ЦМФ)
Г уаиозни-
монофосфат
(ГМФ)
196
Дезокснцитндин-
монофосфат
(дЦМФ)
Дезоксирибонуклеотиды
Дезокситнмидин-
моиофосфат
(дТМФ)
Дезоксиадеиознн-
монофосфат
(дАМФ)
Дезокснгуанознн--
монофосфат
(ДГМФ)
Названия индивидуальных рибо- и дезоксирибонуклеотидов принимаются
по характерному пуриновому или пиримидиновому основанию, а наличие
дезоксирибозы отмечается приставкой «дезокси». Так как остаток фосфорной
кислоты в природных нуклеотидах присоединен к остатку рибозы (или дезок-
сирибозы) по гидроксильной группе при 3-м или 5-м углеродном атомах,
существует два типа нуклеотидов:
I
он
Аденозии-З’-фосфат
Аденозин-б'-фосфат
Чтобы указать местонахождение остатка фосфорной кислоты в молекуле
нуклеотида, нумеруют углеводные атомы в остатке рибозы. Во избежание
путаницы с соответствующей нумерацией атомов в пуриновых или пиримиди-
новых основаниях эти цифры отмечают знаком «'».
Конфигурация молекулы одного из нуклеотидов представлена на рис. 65.
Как можно видеть из рисунка, проекционная формула молекулы цитидин-3'-
фосфата (как и других нуклеотидов), выписанная с учетом истинной кон-
фигурации молекулы этого соединения, значительно отличается от общепри-
нятой проекционной формулы. Это необходимо учитывать при рассмотрении
строения нуклеиновых кислот.
При отщеплении от нуклеотида остатка фосфорной кислоты получается
еще более простое соединение—нуклеозид. Этот термин был впервые пред-
ложен П. Левиным и В. Джекобсом в 1909 г. для обозначения углеводных
производных пуринов, выделенных из РНК. Впоследствии он был распро-
странен на соединения углеводов с пиримидинами. Наименования нуклео-
зидов складываются из названий пуриновых или пиримидиновых оснований
и соответствующих окончаний. В приведенных выше структурных формулах
нуклеотидов легко найти части, соответствующие нуклеозидным остаткам,
а в названиях нуклеотидов—отличить ту часть, которой обозначен остаток
197
• атом углерода
© атрм азота
О атом кислорода
атом фосфора
Рис. 65. Проекционная формула мо-
лекулы цитидин-3-фосфата:
расстояния между атомами даны и нанометрах;'
атомы водорода не обозначены
нуклеозида. Следовательно, нуклеотиды явля-
ются фосфорными эфирами нуклеозидов. Все
сказанное в полной мере относится к дезок-
синуклеотидам и дезоксинуклеозидам.
Так как нуклеозидфосфаты представляют
довольно сильные кислоты, их часто называют
цитидиловой, уридиловой, адениловой и гуа-
ниловой кислотами.
Характерные для каждого вида нуклеино-
вых кислот мононуклеотиды, объединяясь
в количестве нескольких сотен, а иногда и ты-
сяч в единую молекулу, образуют громадные
полинуклеотидные цепи (см. ниже).
Таким образом, по своему химическому
строению нуклеиновые кислоты являются по
лирибонуклеотидами (РНК) и полидезоксирибо-
нуклеотидами (ДНК). Соединение нукле тид-
ных остатков в молекулах РНК и ДНК осу-
ществляется одним и тем же путем:
сложноэфирными мостиками, образуемыми
между парами нуклеотидов остатками фосфор-
ной кислоты. Последние связаны всегда с 3-м углеродным атомом рибозы
(или дезоксирибозы) одного нуклеотидного остатка и с 5-м углеродным
198
атомом рибозы (или дезоксирибозы) другого, как это видно из приведенных
выше изображений фрагментов молекул ДНК и РНК. Если мысленно за-
вершить структуру изображенных выше фрагментов ДНК и РНК концевыми
нуклеотидами, то при 5'-углеродном атоме остатка дезоксирибозы в случае
ДНК (или рибозы в случае РНК) окажется остаток фосфорной кислоты,
а на противоположном конце цепи у З'-углеродного атома остатка дезо-
ксирибозы (или рибозы)—гидроксильная группа. Эти нуклеотидные остатки
образуют 5'- и З'-концы молекул ДНК и РНК. Первый, т. е. 5'-концевой,
нуклеотид принято считать началом молекулы нуклеиновой кислоты, а вто-
рой, т. е. З'-концевой, нуклеотид—ее концом.
Для сочетания нуклеотидных остатков в полинуклеотидные цепи характер-
но то, что гидроксил у 2-го углеродного атома рибозы никогда не вовлекается
в образование эфирного мостика с остатком фосфорной кислоты при синтезе
нуклеиновых кислот. Следовательно, и в ДНК, и в РНК существуют связи
через остаток фосфорной кислоты только от 3-го к 5-му углеродному атому
углевода. Никаких разветвлений цепи ни в ДНК, ни в РНК не обнаружено.
В зависимости от молекулярной массы нуклеиновой кислоты коэффициент
поликонденсации, т. е. число уклеотидных звеньев, связанных в единую поли-
нуклеотидную цепь, варьирует в широких пределах. Если принять среднюю
молекулярную массу нуклеотидного остатка равной 330 то для молекул ДНК
коэффициент поликонденсации выразится десятками и сотнями тысяч звеньев,
а для РНК—всего лишь десятками, сотнями и в некоторых случаях—тысяча-
ми звеньев.
Нуклеотидный состав ДНК и РНК. В табл. 17 и 18 приведены отдельные
примеры соотношения пуриновых и пиримидиновых оснований в ДНК и РНК
некоторых организмов.
Таблица 17
Нуклеотидный состав ДНК
Источник нуклеиновой кислоты Молярные соотношения оснований, % Г+А Ц+т г+ц А+Т
Г А Ц т
Животные Мышь 21,9 29,7 22,8 25,6 1,07 0,81
Осьминог 17,6 33,2 17,6 31,6 1,03 0,54
Растения Пшеница 23,8 25,6 24,6 26.0 0,98 0,94
Лук 18,4 31,8 18,2 31,3 1,02 0,58
Микроорганизмы Туберкулёзная палочка 34,2 16,5 33,0 16.0 1,03 2,08
Стрептококк 16,6 33,4 17,0 33,0 1,00 0,51
Таблица 18
Нуклеотидный состав суммарной РНК
Источник нуклеиновой кислоты Молярные соотношения оснований, % Ц+У г+ц А+У
Г А Ц У
Животные Крыса 32,8 18,7 29,6 18,9 1,06 1,66
Комар 23,5 30,6 19,7 26,1 1,18 0,76
199
Продолжение табл. 18
Источник нуклеиновой кислоты Молярные соотношения оснований* % Г+А Ц+У г+ц А+У
Г А Ц У
Растения Пшеница 30,8 25,2 25,4 18,6 1,27 1,28
Сосна 27,6 25,3 23,4 23,7 1,12 1,04
Микроорганизмы Туберкулезная палочка 33,0 22,6 26,1 18,3 1,25 1,45
Стафилококк 28,7 26,9 22,4 22,0 1,25 1,05
Буквами в них обозначены основания: Г—гуанин, А—аденин, Ц—цито-
зин, Т—тимин, У—урацил, а цифры выражены в молярных процентах (мол.
%), т. е. в процентах от суммы всех найденных в нуклеиновой кислоте
азотистых оснований, содержание каждого из которых вычислено в препарате
нуклеиновой кислоты в молях или его долях.
Анализ таких данных позволил Е. Чаргаффу сформулировать ряд правил
(правила Чаргаффа):
1. У ДНК молярная сумма Г и А (пуриновые основания) равна молярной
сумме Ц и Т (пиримидиновые основания). Эта закономерность не свойственна
РНК, где отношение пуриновых и пиримидиновых оснований изменяется
в широких пределах.
2. В молекулах ДНК число остатков А всегда равно числу остатков Т.
В таком же отношении находятся Г и Ц. В молекулах РНК этого нет, хотя во
многих случаях молярные соотношения оснований близки (здесь имеется
в виду, что Т и У в молекулах ДНК и РНК равноценны).
3. Отношение суммы молярных концентраций Г и Ц к сумме молярных
г+ц
концентраций А и Т у ДНК и А и У у РНК -------------—- у обоих видов
А+Т (или У)
нуклеиновых кислот сильно варьирует. Особенно широки границы измен-
чивости этого показателя в ДНК.
Две первые закономерности в строении ДНК и РНК связывают с особен-
ностями их вторичной структуры. Подобно тому как в стабилизации спираль-
ной формы белковых молекул решающая роль принадлежит водородным
связям между —СО- и —NH-группами, так и в создании вторичной структуры
полинуклеотидной цепи исключительное значение имеют водородные связи,
возникающие между парами оснований: А и Т (в РНК—А и У) и Г и Ц (в
обоих видах нуклеиновых кислот). Основания, образующие пары, в которых
они сочетаются водородными связями, называются комплементарными.
Характер водородных связей между комплементарными основаниями ясен
из рис. 66. С одной стороны, водородные связи замыкаются между амино-
группами и кетогруппами, занимающими определенное положение в молеку-
лах пуриновых и пиримидиновых оснований (6-аминогруппа у А и 4-кетогруп-
па у Т или У, а также 2-амино- и 6-кетогруппы у Г и 2-кето- и 4-аминогруппы
у Ц). В этом и состоит комплементарность, т. е. дополнительность в химиче-
ском строении пуриновых и пиримидиновых оснований: там, где у пуринового
основания расположена аминогруппа, у пиримидинового находится кетогруп-
па, и наоборот. С другой стороны, водородные связи образуются за счет
взаимодействия по атомам N и NH-группам, занимающим в пуриновых
и пиримидиновых циклах 1-е и 3-е положения соответственно, где они опять
200
Рис. 66. Водородные связи между парами оснований в нуклеиновых кислотах:
А—между аденином и тимином; Б—между гуанином и цитозином; В—между парами комплементарных оснований в биспираль-
ном фрагменте молекулы ДНК; стрелками указан антилараллельный ход спиралей в виде заштрихованных лент
201
дополняют друг друга. Именно поэтому в ДНК число молей А равно числу
молей Т, а число молей Г равно таковому Ц. Естественно, что суммарное
содержание пуринов и пиримидинов одинаково.
Третья закономерность в соотношении пуриновых и пиримидиновых ос-
нований связана со специфичностью ДНК и РНК. Поэтому отношение
г+Ц жж ж
А+у (---— называется коэффициентом специфичности нуклеиновых кислот. •
Установлено, что ДНК обладает ясно выраженной видовой специфичностью.
Особенно резкие видовые различия характерны для ДНК,. выделенных из
г+ц
микроорганизмов. У РНК видовая специфичность отношением----выражена
слабее. В табл. 17 и 18 приведены значения коэффициентов специфичности для
ряда тотальных ДНК и РНК. Несмотря на небольшое число произвольно
выбранных объектов, эти таблицы наглядно иллюстрируют сформулирован-
ные выше закономерности. В общем, коэффициент специфичности у ДНК
варьирует от 0,45 до 2,57 у микроорганизмов, от 0,58 до 0,94 у высших
растений и от 0,54. до 0,81 у животных. У тотальной РНК коэффициент
специфичности, как правило, выше единицы.
Долгое время для определения нуклеотидного состава ДНК и РНК ис-
пользовали разделение нуклеотидов, нуклеозидов или свободных азотистых
оснований, полученных в результате деструкции исследуемых нуклеиновых
кислот, при помощи хроматографических методов с последующим спектрофо-
тометрическим анализом природы и содержания каждого из оснований. Одна-
ко в последнее время классические методы анализа нуклеотидного состава
нуклеиновых кислот заменены физическими методами. Так, содержание ГЦ-
пар в составе ДНК сейчас находят по температуре плавления ДНК и по ее
плавучей плотности. В первом случае содержание ГЦ-пар связано с темпера-
турой плавления (гпл) ДНК следующей зависимостью: ГЦ (мол.
%) = (?пл—69,3)-2,44. Эта зависимость справедлива, если ДНК растворена
в стандартном солевом растворе (0,15 моль NaCl и 0,015 моль цитрата натрия
в 1 л, pH 7,0). Во втором случае содержание ГЦ-пар прямо пропорционально
значению плавучей плотности ДНК, пределяемой при ее ультрацентрифуги-
ровании в градиенте плотности CsCl.
Физические методы ускорили процесс накопления фактического материала
по нуклеотидному составу ДНК разных биологических видов и позволили
добавить к правилам Чаргаффа новые закономерности, выявленные
А. Н. Белозерским и его учениками.
Оказалось, что вариабельность нуклео идного. состава ДНК очень высока
у эволюционно, древних таксонов и сравнительно мала у молодых, т. е. на
основании нуклеотидного состава можно судить об эволюционном возрасте
таксона. Кроме того, в природе распространены два типа ДНК: у хордовых
и беспозвоночных животных, высших растений, дрожжевидных организмов,
синезеленых водорослей, ряда бактерий и вирусов преимущественно АТ-тип
ДНК, а у недрожжевидных грибов, актиномицетов, водорослей и ряда бакте-
рий и вирусов в основном ГЦ-тип. Наконец, распределение метилированных
оснований у представителей животного и растительного царства тоже не
случайно. Так, 5-МЦ в значительном количестве присутствует в ДНК высших
растений (2—10 мол. %) и ряда животных (до 3 мол. %), тогда как в ДНК
бактерий его очень мало (менее 0,5 мол. %). N6-MA, напротив, найден у бак-
терий и водорослей, обнаруживается в малых количествах у растений и отсут-
ствует у многих животных. Все это заложило основы для дальнейшей раз-
работки проблем химической таксономии.
202
Выявленные закономерности в соотношении пуриновых и пиримидино-
вых оснований в молекулах ДНК и РНК свидетельствовали о том, что
чередование нуклеотидных остатков в нуклеиновых кислотах носит специфи-
ческий характер. Однако указанные данные не дали и не могли дать представ-
лений об истинной последовательности расположения структурных элемен-
тов, составляющих молекулы нуклеиновых кислот, т. е. об их первичной
структуре.
Первичная структура ДНК. Даже в простейшем случае бактериофага fd
(см. табл. 16), ДНК которого представлена одноцепочечным полидезоксири-
бонуклеотидом с М = 1,9 10 б. Да, в ней должно содержаться примерно 5760
нуклеотидных остатков (1 900000:330, где 330—средняя молекулярная мас-
са нуклеотидного звена), выстроенных в строго определенной последователь-
ности.
Определить первичную структуру такого огромного полидезоксирибону-
клеотида крайне трудно, так как он состоит всего из четырех видов ну-
клеотидных остатков, содержащих в качестве азотистых оснований А, Г Ц и Т,
сравнительно редко метилированных. В случае ДНК большей молекулярной
массы положение еще сложнее. Тем не менее благодаря разработке в период
между 1975 и 1977 гг. двух весьма перспективных методов определения
первичной структуры ДНК удалось достичь решающих успехов в этом от-
ношении.
Первый из них, предложенный Ф. Сангером и Р. Коулсоном, состоит в по-
лучении полного набора убывающих по числу нуклеотидных остатков копий
одноцепочечной ДНК при помощи ДНК-полимеразной реакции (см. с. 249),
разделении их электрофорезом в полиакриламидном геле (по числу наблюда-
емых полос устанавливают число нуклеотидных остатков в ДНК или ее
фрагменте) и выявлении положения дезоксиадениловых, дезоксигуаниловых,
дезоксицитидиловых и дезокситимидиловых остатков на З'-конце каждого из
фрагментов при помощи особых приемов.
Второй, разработанный А. Максимом и В. Гильбертом, сводится к химиче-
ской модификации пуриновых и пиримидиновых оснований в ДНК или ее
фрагментах, избирательном расщеплении фосфодиэфирных связей по месту
модифицированных оснований, разделении продуктов расщепления электро-
форезом в полиакриламидном геле и прямому считыванию структуры фраг-
мента ДНК с полученных электрофореграмм (число полос на электрофоре-
грамме продуктов расщепления фрагмента ДНК с модифицированным азотис-
тым основанием равно числу нуклеотидных остатков, содержащих этот пурин
или пиримидин, а их положение на электрофореграмме указывает место этого
нуклеотидного остатка в анализируемом фрагменте ДНК).
В результате применения указанных методов в их оригинальном или
модифицированном виде определение первичной структуры ДНК в последние
несколько лет приняло массовый характер (табл. 19).
В табл. 19 приведена лишь небольшая часть известных в настоящее вр^мя
первичных структур ДНК. На 1.1.1985 г. были полу ены данные о первичной
структуре 4175 соединений полинуклеотидной природы, составленных из
3 млн. н. о. Всего за 2,5 года, к августу 1987 г., число расшифрованных
первичных структур нуклеиновых кислот возросло до 14 020 при
14855147 н. о. в их составе. На апрель 1993 г. Gen Bank содержал инфор-
мацию о последовательности нуклеотидных остатков в 111911 полинукле-
отидах, собранных из 129968 355 н. о., в том числе о расположении
2 353 635 н. о. в геноме кишечной палочки (50% генома). Все это позволило
поставить в настоящее время фантастическую задачу—определить полную
первичную структуру генома человека, включающего 3 млрд, нуклеотидных
203
пар (н. п.). По расчетам эта программа обойдется в 3 млрд, долларов и займет
несколько лет. Она уже осуществляется у нас (правительственная научно-
исследовательская программа «Геном человека»), в США и Японии. Цель
ее—не только выявить локализацию генов (они составляют 5—10% генома),
но и понять механизмы регуляции их деятельности, а также наметить пути
устранения дефектов в них, приводящих к наследственным болезням. При уже
достигнутой скорости секвенирования ДНК до 280 000 н. п. в сутки и планиру-
емой до 1 млн. н. п. в сутки (имеется в виду использование флуоресцентно-
меченной ДНК и суперкомпьютерной техники, а также применение октадезок-
синуклеотидов заданного строения) решение этой задачи приобретает реаль-
ные очертания, тем более что стоимость чтения одного нуклеотида уже упала
с 1 доллара до нескольких центов. Реальные результаты уже налицо: если
к 1975 г. была выяснена первичная структура всего 25 генов человека, то
к 1985 г. их число составило 900, а к 1992 г. достигло 3618. На четвертой
научной конференции, посвященной оценке хода работ по программе «Геном
человека» (Черноголовка, 9—11 марта 1994 г.) был дан анализ вклада россий-
ских ученых в ее выполнение.
Таблица 19
Число нуклеотидных пар (н.п.) в ДНК некоторых геномов
и генов с полностью установленной первичной структурой
Теном Число н.п. Тен Число н.п.
Грам-положительной бактерии Bacillus subtilis 4214810 Эмбрионального глобина человека 11376
Спирохеты Borrelia burgdorferi, штамм В31 910725 Яичного альбумина 7564
Вируса Эпштейна-Барра 172282 Гормона роста 2600
Хлоропласта табака 155844 Р-субъединицы ДНК-полимеразы сальмонеллы1 1986
Хлоропласта печеночного мха 121024 ДНК-лигазы фага Т41 1469
Аденовируса 35937 Инсулина человека 1430
Митохондрий человека и быка 16659 Токсина шигеллы1 1380
Вируса мозаики капусты 8031 Глицинина сои1 944
Бакгерифага М13 6407 Р-субъединицы Na+, К+ - АТФазы1 912
Вируса 0Х-174 5386 у-субъединицы фосфодиэстеразы цГМФ1 779
Обезьяньего вируса SV-40 5243 Лейкоцитарного интерферона человека1 520
1 Первичные структуры , расшифрованные российскими учеными.
Примечание', только что опубликованы данные о первичной структуре ДНК генома кишечной палочки
(4 700 000 н.п.) и всех 16-ти хромосом пекарских дрожжей (12 000 000 н. п.).
204
Ясно, что первичная структура большинства ДНК представлена двухцепо-
чечными дезоксиполирибонуклеотидами с уникальным чередованием десятков
и сотен тысяч составляющих их дезоксирибонуклеотидных остатков. В качест-
ве примера ниже приведена структура гена человеческого лейкоцитарного
интерферона:
АТЦТГТТЦТЦТГТГЦТГТГАТЦТГЦЦТЦАГАЦЦЦАЦАГЦЛТГГГТААТАГГАГГГЦЦТТГА
ТАЦТцйтГГЦАЦАААТГГГААГААТЦТЦТЦЦТТТЦТЦЦТГЦЦТГААГГАЦАГАЦАТГАЦТ
ттггаттццццАаггаггагтттгатггцааццагттццагааггцтцаагццатцтцтг
ТЦЦТЦЦАТЦАГАТГАТЦЦАГЦАГАЦЦТТЦААЦТЦТТЦАГЦАЦАААГГАЦТЦАТЦТГЦТА
цттгггаацагагццтццтагааааагптццацтгаацттааццагцагцтгаатгацП
ТГГААГЦЦТГЦГТГАТАЦАГГАГГТТГГГГТГГААГАГАЦТЦЦЦЦТГАТГААТГТГГАЦТЦЦ
АТЦ^ТГГЦТГТГААГАААТАЦТТЦЦАААГААТЦАЦТЦТТТАТЦТГАЦАГАГААГАААТАЦА
ГЦЦЦТТГТГЦ^ТГГГАГГТТГТЦАГАГЦАГАААТЦАТГАГАТЦЦТТЦТЦТГТАТЦАААААТ
ТТТТЦААГАААГАПААГГАГГААГГААТАААЦТЦ
Каждый из 520 дезоксирибонуклеотидных остатков, входящих в состав
гена (показана одна его цепь), обозначен прописными буквами (А—дезокси-
адениловая, Г—дезоксигуанйловая, Т—дезокситимидиловая, Ц—дезоксици-
тидиловая, И—5-метил-дезоксицитидиловая кислота). Именно на этой цепи
синтезируется мРНК (см. с. 220), являющаяся матрицей для биосинтеза в лей-
коцитах человека белка-интерферона, оказывающего мощное воздействие на
метаболические процессы и обладающего поэтому огромным терапевтиче-
ским эффектом. Будучи встроен в ДНК кишечной палочки, ген интерферона
обеспечивает биосинтез интерферона в бактериальной культуре, что открывает
возможность для практического получения последнего в фармацевтической про-
мышленности. Это осуществлено, в частности, в Институте биоорганической
химии им. М. М. Шемякина и Ю. А. Овчинникова РАН, где создан соответст-
вующий штамм кишечной палочки, продуцирующий интерферон.
Вторичная структура ДНК. Молекулы природной ДНК в подавляющем
большинстве случаев (лишь ДНК некоторых фагов одноцепочечны) составле-
ны парами (см. табл. 19) взаимозакрученных полидезоксирибонуклеотидных
цепей, каждой из которых свойственно специфическое, но противоположное
чередование нуклеотидных остатков (пунктиром показаны водородные связи
между комплементарными основаниями):
В цепь—АМЛг-А—А—А—U—U—Ц—Г—Т—Т—Т—Ц—ЛЛМг-в цепь
I I I I I I । * I । ।
I I । । I I I । I । ।
В цепь—ЛМА—Т—Т—Т—Г—Г—Г—Ц—А—А—А—Г-Wft-B цепь
Фотография объемной модели молекул ДНК, построенной по этому типу,
представлена на рис. 67, А. Здесь же приведена схема расположения полинук-
леотидных цепей и характерные параметры их (рис. 67, Б), а также схема
расположения комплементарных оснований, связывающих водородными свя-
зями полидезоксирибонуклеотидные цепи друг с другом (рис. 67, В). Впервые
указанная модель молекулы ДНК была предложена Дж. Уотсоном и Ф. Криком
в 1953 г. на основании рентгеноструктурного анализа строения ДНК. Данные,
полученные в течение последующих 45 лет, полностью ее подтвердили, а ее
авторы стали Нобелевскими лауреатами.
На рис. 67 представлены модели так называемой В-формы ДНК. Дело
в том, что в зависимости от ряда условий ДНК существует в виде
205
Рис. 67. Строение молекулы ДНК:
А—модель молекулы ДНК: темные и светлые цепочки атомов, выделяющиеся на рисунке,—обращенная наружу пентозофосфат-
ная цепь. Внутрь модели, перпендикулярно fee длинной оси, направлены пуриновые и пиримидиновые основания, соединяющиеся
водородными связями; атомы, принадлежащие основаниям (заштриховано косо), плотно заполняют центральную часть модели;
Б—пространственное расположение полидезоксирибонуклеотидных цепей в молекуле ДНК; поперечные линии на схеме обознача-
ют плоскости, в которых располагаются пары оснований, связывающие цепи друг с другом; 0,34 нм—расстояние между
остатками соседних дезоксирибонуклеотидов; 1 нм—радиус молекулы; 3,4 нм—шаг двойной спирали; В—расположение ком-
плементарных пуриновых и пиримидиновых оснований в молекуле ДНК; молекулы дезоксирибозы представлены белыми
пятиугольниками, обведенными двойной линией; фосфатные группы—изгибом двойной линии, связывающей остатки дезок-
сирибозы; от каждого остатка углевода отходят основания в виде покрытых точечной штриховкой шестиугольников и пятиуголь-
ников; короткие двойные линии, соединяющие заштрихованные пуриновые и пиримидиновые основания, представляют водород-
ные связи Все спирали—правозакрученные
разнообразных упорядоченных волокнисто-кристаллических структур. Их по-
лучено более десяти, четыре из них: А-, В-, С- и Т-формы ДНК—изучены
методом рентге] оструктурного анализа. При соблюдении общего плана
строения молекулы ДНК в виде биспирального полидезоксириб нуклеотида
они отличаются по ряду параметров, присущих каждой из названных конфор-
мационных модификаций ДНК (табл. 20).
Наиболее сильное изменение конформации происходит при превращении
A-формы ДНК в В-форму. В частности, при этом резко смещается простран-
ственная стр ктура p-D-2-дезоксирибозы в ее составе, вследствие чего угол
наклона плоскости оснований к оси спирали изменяется более чем на 20°
(рис. 68).
В примечании к табл. 20 приведены условия получения кристаллических
волокон ДНК. Однако и в клетке та или иная степень обводненности ее
компартментов или мембран, как и различия в ионной силе окружающей
среды, создает условия для сущее вования ДНК в различных конформациях,
между которыми осу ествляются взаимные переходы. В биологическом
смысле В-форма наиболее адекватна для репликационных процессов А-
форма—для процесса транскрипции, С-форма—для упаковки ДНК в составе
надмолекулярных структур хроматина и некоторых вирусов. Таким образом,
вторичная структура молекул ДНК, видимо, связана с осуществлением инфор-
мационных процессов в живой природе, а именно: A-форма ДНК—с переда-
206
Таблица 20
Характеристика некоторых конформационных состояний ДНК
Показатель А-форма В-форма С-форма Т-форма
Число пар нуклеотидных остатков на виток 11 10 9,3 8,0
Угол наклона плоскостей оснований к оси спирали, ° 20 -2 -6 -6
Угол поворота оснований вокруг оси спира- ли, ° 32,7 36 38,6 45,0
Расстояние комплементарных пар от оси спирали, нм 0,425 0,063 0,213 0,143
Расстояние между нуклеотидными остатка- ми по высоте спирали, нм 0,256 0,338 0,332 0,304
Угол между плоскостями ком лементарных оснований, ° 8 5 . 5 —
Примечание. A-форма получена из водно-солевых растворов, содержащих ионы щелочных металлов, кроме
Li+; рентгеноструктурный анализ—при относительной влажности 75%. В-форма получена из водно-солевых
растворов, содержащих Li+; рентгеноструктурный анализ—при относительной влажности 66%. С-форма
получена в тех же условиях, что В-форма, но в растворах с иным соотношением катионов; рентгеноструктурный
анализ—при относительной влажности.менее 66%. Т-форма (D-форма) выделена из фага Т2; содержит остатки
гликозилированного оксиметилцитозина.
чей информации от ДНК к РНК, В-форма—с умножением количества инфор-
мации и С-форма—с хранением информации.
В последние годы появились данные о возможности существования двух
принципиально новых форм ДНК: Z-формы и SBS-формы. Z-форма ДНК,
открытая в 1979 г. в лаборатории А. Рича, представлена левозакрученными
полцдезоксирибонуклеотидными цепями (рис. 69) в составе биспиральной моле-
кулы, причем фосфатные группы в ней располагаются зигзагообразно (от-
сюда—Z-форма). Диаметр молекулы ДНК в Z-форме равен 1,8 нм (против
2,0 в В-форме ДНК), число оснований в витке—12, расстояние между витка-
ми—0,34 нм, наклон оснований к оси спирали—7°; в Z-форме ДНК лишь
один желоб (вместо двух в А- и В-формах ДНК, см. рис. 67 и 68). Предполага-
ют, что в природной ДНК могут чередоваться
правые (А-, В- и С-формы) и левые (Z-форма)
участки, причем последние могут быть «горячими
точками», где реализуется участие ДНК в ряде
метаболических процессов.
SBS-форма ДНК характеризуется отсутствием
взаимозакручиваиия полидезоксирибонуклеотид-
ных цепей в биспиральную молекулу; они распола-
гаются при тех же параметрах молекулы ДНК бок а
о бок (англ, side by side, отсюда—SBS-форма).
Такая форма ДНК обеспечивает необыкновенно
легкое распаривание и расхождение цепей ДНК,
что крайне существенно при биосинтезе ДНК.
Рис. 68. Полиморфизм вторичной структуры ДНК:
I—Л-форма; II—В-форма; а—вид сбоку; б—вид сверху. В Л-форме ДНК ком-
плементарно связанные азотистые основания нуклеотидных пар отстоят от оси
спирали иа 0,425 нм, а в В-форме—очень близки к осн спирали (см. табл. 20),
вследствие чего в первом случае внутри молекулы возникает полость, а во втором
осуществляется почти полное заполнение внутренней части бнепирального дезок-
сиполнрибонукле отид а азотистыми основаниями
б
207
z-днк
Рис. 69. Сопоставление В- и Z-форм ДНК:
тёмными шарами обозначены межнуклеотидные фосфаты, а соединяющими
их толстыми линиями — ход пентозофосфатноЙ цепи. Отчетливо прослежива-
ются Z-образные изломы в левозакрученных пентозофосфатных цепях Z-ДНК
и отсутствие таких изломов в правозакрученных цепях В-ДНК. Ясно выступа-
ют различия в числе и глубине большого и малого желобов в той и другой
форме ДНК
Рис. 70. Образование четырехцепочечной
шпильки из полинуклеотида с олигопурин-оли-
гопиримидиновой последовательностью:
четвертичная спираль возникает за счет ком-
плементарных взаимодействий в ней полипури-
иовых (тёмные) и полипиримидиновых (белые)
участков в составе двойных спиралей (нижнм
часть рисунка)
Недавно получена информация о существовании фрагментов молекул ДНК
в виде тройных и четверных спиралей. Первая из них получила название
Н-ДНК, так как образуется при pH 4,0, стабилизуясь Н+; при этом тройной
комплекс возникает на пурин-пиримидиновых блоках путем присоединения
к ним полипиримидиновых нитей. Биологический смысл формирования Н-
ДНК пока не ясен. Что касается четверных спиралей ДНК (рис. 70), то их
можно расценивать как один из вариантов перехода к третичной структуре
дезоксирибонуклеиновых кислот, в первичной структуре которых ярко пред-
ставлены олигопиримидиновые и олигопуриновые последовательн сти.
Два вида сил удерживают две полидезоксирибонуклеотидные цепи в би-
спиральной молекуле ДНК. Во-первых, это водородные связи между ком-
плементарными азотистыми основаниями, обращенными внутрь двойной спи-
рали ДНК (см. рис. 66). Образуя водородные связи, основания находятся
в плоскости, перпендикулярной продольной оси В-формы ДНК, так что эти
связи действуют в поперечном направлении. Во-вторых, это силы гидрофоб-
ных взаимодействий между азотистыми основаниями, собранным в «стопку»
вдоль молекулы ДНК; при такой упаковке оснований в водной среде возника-
ют силы, препятствующие контактам неполярных (гидрофобных) оснований
с молекулами воды, вследствие чего основания сближаются, а стопкообразная
упаковка упрочняется вследствие межплоскостных взаимодействий их друг
208
с другом. Их называют поэтому стэкинг-взаимодействиями (т. е. взаимодействи-
ями, направленными вдоль стопки оснований и, в итоге, вдоль молекулы ДНК).
В последнее время силам стэкинг-взаимодействия придают более сущест-
венное, чем .ранее, значение в поддержании вторичной структуры ДНК, а во-
дородным связям между комплементарными парами оснований приписывают
в большей мере направляющую роль во взаимной ориентации оснований
в процессе стэкинг-взаимодействия. Действительно, вклад гидрофобных вза-
имодействий в поддержание вторичной структуры биспиральных полинукле-
отидов возрастает при замене У на Т, т. е. за счет добавочно вводимого
гидрофобного метильного радикала.
В связи с этим в ином свете представляется и устойчивость биспиральных
структур в молекулах ДНК, равно как и механизмов ее нарушения при воздействии
физических и химических агентов в тех или иных условиях. В частности, молекулы
воды и ионы металлов связываются в основном с пентозофосфатным остовом
ДНК, причем катионы располагаются преимущественно в малом желобе двойной
спирали В-формы ДНК. Стабилизирующее действие молекул воды направлено
непосредственно на усиление стэкинг-взаимодействия, что приводит к стабилиза-
ции водородных связей между основаниями. Вместе с тем при ослаблении
стэкинг-взаимодействия молекулы воды конкурентно взаимодействуют с протон-
донорными и протон-акцепторными центрами оснований, способствуя дестабили-
зации и инициируя дальнейший распад двойной спирали. АТ-пары оснований
гидратируются в 2 раза сильнее, чем ГЦ-пары, причем нативность сохраняется при
содержании воды не менее 0,6 г на 1 г ДНК. Все это подчеркивает динамичность
вторичной структуры ДНК и возможность конформационных и иных переходов в йен
под воздействием агентов окружающей среды.
Биспиральные структуры в молекулах ДНК возникают не только при
взаимодействии двух комплементарных полидезоксирибонуклеотидных цепей,
но и в пределах одной и той же цепи. Это происходит в тех случаях, когда
в комплементарных цепях ДНК присутствуют палиндромы—«обратно
$'-гцтцгтатгт1тгтгтГ|г|ааттгт[гЙгГц,1гГг1аГт|л|,ацаатт|тГцлцацл,1Ггаллцлгцт—— ГЦТЦГТАТГТ1’
3'-ЦГАГЦЛГАЦА|АЦЛЦЛц|ц|ГТЛАЦЛ|ЦГПЦ1Г|ц11^Т1А1т|тГТТАа|а| ГТГТГТЩЦТТТГТЦГА -—ЦГАГЦАТАЦАд
г-.ц
- А
ц
аггааацагцт
,ТЦЦТТТГТ11ГА
Ц--Г
А--Т
ц-г
п
Рис. 71. Палиндромы ДНК кишечной палочки (зона лактозного оперона) в линейном (I)
и шпилечном (II) состоянии:
в линейной структуре жирной точкой отмечено место начала формирования шпильки: пунктирными прямоугольниками—
зоны палиндромов; стрелками различной толшины и формы—одноименные палиндромы и их направление
209
бегущие» последовательности нуклеотидных звеньев (от греч. палин—обра-
тно, дроме—бегу). Палиндромные структуры характерны для тех участков
молекулы ДНК, где расположены зоны «узнавания» структур ДНК ферментами
и регуляторными белками. Так, благодаря наличию палиндромов, спирализу-
ются сами на себя, образуя шпильки, цепи ДНК кишечной палочки в зоне
лактозного оперона—структуры, где сосредоточена информация о биосинтезе
Р-галактозидазы, обеспечивающей распад лактозы при наличии ее в культу-
ральной среде (рис. 71). Высказывают предположение, что палиндромы явля-
ются источником четверных спиралей ДНК, которые могут служить для
формирования элементов третичной структуры ДНК.
Третичная структура ДНК. Молекулы, ДНК существуют в виде линейных
и кольцевых форм (рис. 72). В линейной форме находится, видимо, большин-
ство природных ДНК, но ДНК ряда вирусов и фагов, а также ДНК хлороплас-
тов, митохондрий, центриолей и бактериальнь х плазмид обладают кольцево"
структурой. Третичная структура и линейных и кольцевых форм ДНК характе-
ризуется спи ализацией и суперспирализаци й. Между кольцевыми и линейными
формами ДНК, равно как и между ее обычным и суперспирализованным
состоянием, предполагается существование динамических переходов. В ДНК
ряда вирусов (например, вируса полиомы) и митохондриальной ДНК такие
превращения детально изучены (рис. 72).
Сложнее обстоит дело с ДНК из хромосом эукариот. Применяемые мето-
ды выделения сопровождаются в большей или меньшей мере ее деградацией и,
самое главное, отделением от дезоксирибонуклеопро еина в виде которого
она существует в ядерном аппарате клеток, его белковой части, абсолютно
необходимой для поддержания третичной структуры ДНК. Поэтому пробле-
ма третичной структуры ядерной ДНК может быть решена только путем
изучения структуры хроматина ядра и хромосом.
ДНК в хроматине и хромосомах также находится в суперспирализованном
состоянии, причем здесь реализуется несколько уровней суперспирцлизации.
Первый уровень сверхскрученного состояния ДНК в хроматине поддерживает-
ся гистонами, оккупирующими отрезки ДНК протяженностью около 200 н. п.
и образующими элементарную единицу структуры хроматина—нуклеосому
Рис. 72. Третичная структура молекулы ДНК:
а—линейная; 6—кольцевая; в—сверхскрученная кольцевая; г—структура ком-
пактного клубка
210
Рис. 73. Строение нуклеосомы
Основу нуклеосомы составляет белковое ядро (бел-
ковый кор), вокруг которого навита биспиральная
ДНК (1,8 левых сверхвитка, 146+2 н. п., шаг—
2,75 нм); тетрамер гистонов (Н3)2 • (Н4)2 определяет
укладку центрального витка суперспирали ДНК
в желобе белкового кора, а два гетеродимера
(Н2а-Н2в) участвуют в упаковке остальной части
суперспирали ДНК по ~0,5 витка каждый; эти ги-
стоны называют коровыми гистонами; гистон Н1
стабилизирует области входа и выхода ДНК в нук-
леосому, взаимодействуя с линкерной ДНК (от 10
до 80 и. п.), соединяющей нуклеосомы друге другом
(рис. 73). Цепочка нуклеосом образует, в свою очередь, спираль второго
и последующих порядков, вплоть до конденсации в хромосому (рис. 74, Л).
При этом (рис. 74, Б) на один оборот спирали в нуклеосоме приходится уже
80 н. п. (уплотнение ДНК в 6—7 раз), в солениде (6 нуклеосом на виток)—
1200 н. п. (уплотнение в 40 раз), в каждой петле хроматина—60000 н. п.
(уплотнение в 680 раз) и в хромосоме—1,1 *10® н. п. (уплотнение в 1,2-104).
Существенно, что переход ДНК в суперспиральное состояние и обратно
осуществляется при посредстве особой группы ферментов—топоизомераз,
т. е. ферментов, изменяющих пространственную структуру.
Свойства ДНК. ДНК—вещества белого цвета, волокнистого строения,
плохо растворимые в воде в свободном состоянии, но хорошо растворимые
в виде солей щелочных металлов. Они также хорошо растворяются в крепких
солевых растворах.
Так как молекулы ДНК резко асимметричны, их растворы обладают
высокой вязкостью и двойным лучепреломлением. Имея большой отрицатель-
ный заряд, молекулы ДНК подвижны в электрическом поле. Все ДНК оптиче-
ски активны.
При агревании растворов ДНК в интервале температур от 80 до 90° С
происходит «плавление» нуклеиновых кислот, сопровождающееся изменением
вязкости раствора и возрастанием поглощения в ультрафиолетовой части
спектра (при 260 нм). Последнее получило наименование гиперхромного эффекта.
В химическом отношении ДНК довольно инертны, благодаря чему их
долгое время считали индифферентными структурными элементами клеточ-
ного содержимого. Однако постепенно накопились данные о ряде химических
реакций, свойственных нуклеиновым кислотам: они прочно связывают много-
валентные ионы металлов причем Си2+ и Ме4+ дают с ДНК нерастворимые
комплексы. В первую очередь поливалентные катионы вступают в реакцию
с N и О гуанина. Возможно, ионы металлов принимают участие в поддержа-
нии третичной структуры нуклеиновых кислот.
Нуклеиновые кислоты легко вступают во взаимодействие с полиаминами,
например со спермидином [H2N—(СНг)з—NH—(СНг)4—NH2] и спермином
[H2N—(СНг)з—NH—(СНг)4—NH—(CH2)3—NH2], которые участвуют в под-
держании третичной структуры нуклеиновых кислот. Одной из важных реак-
ций ДНК является алкилирование аминогрупп А, Ц и Г. Не меньшее значение
имеет дезаминирование перечисленных азотистых оснований: в первую оче-
редь дезаминируются Г и Ц. И алкилирование, и дезаминирование ДНК лежат
в основе работ по химическому мутагенезу, т. е. по изменению наследст-
венности при помощи химических средств.
211
2 нм 11 нм 30 нм 300 нм 500 нм
Биспираль
ДНК
Нуклео- Суперспираль Диффузный Кластеризованный
сомный нуклеосом хроматин хроматин
тяж (соленоид)
Хромосома
Рис. 74. Уровни суперспирализации ДНК в хроматине:
Л—динамика суперспирализации ДНК—белкового комплекса:
Б степень уплотнения ДНК в процессе суперспирализации (см. текст на с. 211).
212
Структура и функции транспортных РНК. Транспортные РНК были впервые
выделены из так называемой «растворимой» части клетки, т. е. из надосадоч-
ной жидкости клеточного гомогената. Главной функцией этого вида рибонук-
леиновых кислот оказалась способность акцептировать аминокислоты и пере-
носить их в белоксинтезирующий аппарат клетки—рибосому. В связи с этим
их называют транспортными рибонуклеиновыми кислотами (тРНК).
Фракция растворимых РНК, составляющая 1% от сухого вещества клетки
или 10% от суммарной клеточной РНК, очень сложна по составу. Она
включает несколько десятков индивидуальных тРНК, каждая из которых
получена в гомогенном состоянии.
Так как каждая из индивидуальных тРНК способна переносить единствен-
ную аминокислоту в процессе белкового синтеза, конкретные тРНК называют
по имени той протеиногенной аминокислоты, которую она акцептирует (на-
пример, глициновая тРНК, аланиновая тРНК, лизиновая тРНК и т. д., или
сокращенно тРНКгли, тРНКала, тРНКл“ и т. п.). Если одна и та же аминокис-
лота акцептируется несколькими индивидуальными тРНК, то последние назы-
вают изоакцепторными и нумеруют (например, тРНКаал, тРНК^” и т. п.). Так,
обнаружено 4 изоакцепторных тРНКлей, 3 тРНКпро, по 2 у тРНКсер, тРНК"",
тРНКала, тРНК™3, тРНКтир, тРНКгли и тРНКтре.
Химический состав тРНК оказался своеобразным лишь в одном отноше-
нии: по сравнению с другими видами РНК они богаты минорными нуклеотид-
ными остатками. Именно в составе тРНК найдены минорные азотистые ос-
нования, перечисленные в табл. 15. Более того, тРНК содержат нуклеозиды
и нуклеотиды своеобразного строения, как, например, псевдоуридин и неогу-
аниловая кислота:
Псевдоуридин
Список минорных компонентов тРНК непрерывно расширяется и в насто-
ящее время насчитывает около 50. Минорные нуклеотидные остатки составля-
ют примерно 10% всех нуклеотидных звеньев тРНК, причем значительная
часть их (4—5%) представлена псевдоуридиловой кислотой. Предполагают,
что минорные нуклеотидные остатки защищают тРНК от атаки рибонукле-
азами, что имеет существенное значение, так как тРНК функционируют в рас-
творимой части клетки. Кроме того, есть мнение, что некоторые из них
принимают участие, в кодировании аминокислот и важны для «узнавания»
ферментом (аминоацил-тРНК-синтетазой) той тРНК, которая взаимодейству-
ет с определенной аминокислотой в процессе активирования последней. Что
касается соотношения обычных азотистых оснований в составе тРНК, то оно
характеризуется отчетливо выраженным преобладанием суммы Г и Ц над
таковой А и У. Таким образом, тРНК относится к ГЦ-типу РНК.
Как отмечено выше, молекулярные массы тРНК лежат в пределах 17000—
35 000. В большинстве случаев они сосредоточены в более узком диапазоне, от
22000 до 27000, т. е. тРНК содержит от 70 до 80 н. о.
Благодаря тому, что в процессе работы по исследованию первичной струк-
туры тРНК Р. Холли с сотр. удачно применили рибонуклеазу, отличающуюся
213
специфичностью к гуаниловым нуклеотидным остаткам, к деструкции дрож-
жевой тРНКала (при 0° С, pH 7, в присутствии Mg2+), им удалось получить
несколько крупных блоков, содержавших от 10 до 39 н. о., и далее расшифро-
вать первичную структуру каждого из них. В результате была впервые (1965)
установлена полная первичная структура аланиновой тРНК из пекарских
дрожжей (рис. 75). За последующие 5 лет была расшифрована первичная
структура 16 тРНК, за следующие пять—47, еще за пять—115 тРНК и ряда
предшественников тРНК, причем первичные структуры тРНКаал и тРНК|ал из
дрожжей выяснены в нашей стране академиком А. А. Баевым с сотр. в 1967
и 1971 гг. соответственно. Эти данные охватывают тРНК, выделенны в ос-
новном из бактерий и дрожжей, но в ряде случаев касаются тРНК, полученных
из высших организмов (млекопитающих, растений, насекомых и др.). Всего на
апрель 1993 г. выяснена последовательность нуклеотидных остатков в 2011
тРНК, изолированных из представителей животного и растительного царства,
а также из микроорганизмов.
Массовые данные, полученные в результате исследования первичной струк-
туры тРНК, позволили выявить ее некоторые общие закономерности. В 75%
случаев молекулы тРНК от срываются остатком гуанозин-3', 5'-дифосфата и во
всех случаях завершаются ЦЦА-триплетом, причем остаток концевого адено-
зина лужит для связывания (акцептирования) аминокислоты:
Ц—Ц—в цепь
H2N—СН—СО-О ОН
R
Аминоацил-тРНК
В первичной структуре изученных тРНК представлены гомологичные бло-
ки, крайне близкие по чередованию нуклеотидных остатков. Особенно ярко
это выявляется в дигидроуридиловон и псевдоуридиловой петлях (рис. 75). Так,
в псевдоуридиловой петле у всех без исключения исследованных тРНК присут-
ствует тетрануклеотидный фрагмент—ГТ'РЦ—; дигидроуридиловая петля
является сосредоточием остатков не только дигидроуридиловой кислоты, но
и АГ-последовательностей, а также минорных оснований.
Закономерно распределены в первичной структуре тРНК также минорные
нуклеотидные остатки. Это объясняют тем, что только некоторые позиции
в молекулах тРНК доступны действию метилирующих ферментов, с помощью
которых осуществляется модифицирование обычных оснований в метилиро-
ванные производные на уровне уже полностью сформированной молекулы.
Вторичная структура тРНК ясна из рассмотрения рис. 75. Ее характерной
особенностью является спирализация полинуклеотидной цепочки самой на
себя в строго фиксированных комплементарных зонах.
Таких зон насчитывают 4 или 5 в зависимости от наличия или отсутствия
добавочной петли, которая всегда располагается, если она есть, между анти-
кодоновым отростком и псевдоуридиловой петлей. В результате ограниченной
биспирализации возникает однотипная для всех тРНК вторичная структура
в виде клеверного листа (см. рис. 75).
214
(2Н) У
(2Н)
3'
А (ОН)
/гц
5' А
(р)Г —Ц
Г-Ц
цгцг
гцгц
м:
ц—Г
г—ц
У У
АГГЦЦ А
: : :: : ш г
УЦЦГГ ц
ц тт
У(2Н)
ц—г
ц—г
Ц—г
ф
" и
и г ц
Б
ц
У
ц
Ц
• - основания, связанные водородными связями о - неспареиные основания
Пур-пуриновое основание Пир-пиримидиновое основание
Рис. 75. Структура тРНК:
А—первичная н вторичная структуры аланиновой тРНК; Б—обобщенная структура тРНК в виде клеверното листа;
Ч*—остаток псавдоуридилово кислоты; м*Г—остаток Nj-метилгуаниловой кислоты; м2Г—остаток N2-
имеп лгуаниловой кислоты; м*И-остаток И2-метилинозиновой кислоты; И—остаток инозиновой кислоты,
У(2Н)—остаток дигидроуридиловой кислоты; 1—дигидроуридиловая петля; 11—антикодоновая петля; 111—псев-
доуридиловая петля; IV—акцептирующий конец; V—добавочная петля; •—основания, связанные водородными
связями; О—неспаренные основания; Пур—пуриновое основание; Пир—пиримидиновое основание
В период наивысшего расцвета модели тРНК типа клеверного листа каж-
дой ее отграниченной части приписывали строго определенное функциональ-
ное значение. Особенно большую роль в выяснении функций тех или иных
полинуклеотидных последовательностей в молекуле тРНК сыграл метод раз-
резанных молекул, предложенный в лаборатории акад. А. А. Баева и образно
названный акад. В. А. Энгельгардтом методом хирургии молекул. Сущность
его сводится к расщеплению в молекуле тРНК ферментативным или химиче-
ским путем ограниченного числа межнуклеотидных связей, разделении полу-
ченных фрагментов и реассоциации из них молекул тРНК, дефици ных по
одному из фрагментов. Реассоциация молекул тРНК из того или иного набора
ее фрагментов идет при участии сил слабых взаимодействий и осуществляется
по принципу самосборки. Испытание биологической активности таких моле-
кул показало, что представления об однозначной роли дигидроуридиловой,
псевдоуридиловой и других петель и частей молекулы тРНК не вполне спра-
ведливы. Например, за связывание аминоацил-тРНК-синтетазы отвечает не
только дигидроуридиловая петля, но и другие участки молекулы тРНК.
Решение проблемы функциональной активности как фрагментов, так и це-
лой молекулы тРНК, безусловно, связано с прогрессом в изучении ее третич-
ной структуры. На основании расшифровки рентгенограмм, выполненных при
разрешении от 0,6 до 0,25 нм, построены модели молекул ряда тРНК, в част-
ности тРНКфен дрожжей, кишечной палочки и термофильных бактерий,
тРНКасп, тРНКтрн, формилметиониновой тРНК кишечной палочки и многих
других тРНК. Наиболее детально отработана А. Ричем третичная структура
дрожжевой тРНКфен (рис. 76).
215
АнтикодоноОал
петля
Рис. 76. Третичная структура дрожжевой фенилаланиновой тРНК:
Л—Схематическое изображение. Арабскими цифрами указаны номера нуклеотидных остатков. Молекула имеет Г-образную форму, причем антикодовая петля
и акцептирующий конец находятся, как и в плоскостной модели типа клеверного листа, на противоположных концах молекулы. Дигидроуридиловая
и псевдоуридиловая петли вследствие их взаимодействия друг с другом и образования водородных связей между комплементарными азотистыми основаниями
сближены и находятся на сравнительно небольшом расстоянии от продольной оси молекулы. Биспиральные структуры остаются прекрасно выраженными
в антикодоновой и акцептирующих частях молекулы. В функциональные взаимодействия тРНК с ферментом, рибосомой и субстратами вовлекаются
нуклеотидные остатки, расположенные в различных частях нуклеотидной цепи, но сближенные в процессе формирования пространственной структуры молекулы.
В какой-то мере это напоминает механизм возникновения активных центров в молекулах белков. Б—пространственная модель
Рис. 77. Предполагаемая вторичная структура 16S рРНК кишечной палочки, выведенная на
основании максимального спаривания комплементарных оснований.
!—/Г—домены, существенные для понимания закономерностей становления ее третичной структуры (их границы указаны
сплошными и пунктирными линиями)
217
Для тРНК характерны неканонические функции, которым в последнее время
уделяют все большее внимание: сигнальная в геноме вирусов; нематричного
(внерибосомального) переноса аминокислотных остатков на растущие при
биосинтезе пептидогликанов клеточной стенки бактерий или существующие
полипептиды при участии аминоацил-тРНК-протеинтрансфераз; затравочная
для обеспечения действия обратной транскриптазы (см. ниже) и др.
Структура и функции рибосомальных РНК. На долю рибосомальных РНК
(рРНК) приходится основная масса клеточной РНК (80—85% от тотальной
РНК клетки). У всех организмов найдено три вида рРНК, различающихся по
молекулярным массам и локализации в рибосомах (см. гл. VII), обязательной
составной частью которых они являются. Две рРНК высокомолекулярны,
а третья сравнит льно низкополимерна. Кроме того, в рибосомах эукариот
присутствует еще одна низкомолекулярная рРНК.
В зависимости от класса рибосом (70S или 80S) константы седиментации
и молекулярные массы двух (большой и малой) высокополимерных рРНК
несколько различаются. Малая молекула высокополимерной рРНК (констан-
та седиментации 16,0—18,0S, Л/=0,55 • 106—0,79 • 106) локализована в 30—
40S, а большая (константа седиментации 23—29S, М= 1,07 • 106 —1,6 10б)—в
50—60S субчастицах рибосом.
Во всех без исключения рибосомах присутствует низкополимерная 5S
рРНК с молекулярной массой 40 тыс. Да. Она локализована в 50—60S субча-
стицах рибосом. Низкополимерная рРНК с константой седиментации 5,8S
(Л/~50000) характерна только для эукариотических рибосом.
Нуклеотидный состав высокополимерных рРНК варьирует в довольно
широких пределах и по мере усложнения организма все более смещается
в сторону преобладания ГЦ-пар. РНК, выделенные из рибосом митохондрий,
отличаются резким преобладанием АУ-пар, что является одним из аргумен-
тов в пользу выделения митохондриальных РНК в особую группу. Высокомо-
лекулярные рРНК содержат в 2—5 раз меньше минорных оснований, чем
тРНК, и список их гораздо короче.
Нуклеотидный состав 5S рРНК своеобразен—в нем совершенно не пред-
ставлены метилированные основания и лишь в некоторых случаях (например,
у дрожжевых грибков) обнаружена псевдоуридиловая кислота (около
1,3 мол. %). Соотношение главных нуклеотидов у 5S рРНК характеризуется
отчетливым ГЦ-типом.
В последние годы наметились большие успехи в познании первичной
и вторичной структур рРНК. Прежде всего это касается 5S рРНК. Их первич-
ная структура выяснена уже более чем в 500 случаях. Во всех исследованных
до сих пор 5S рРНК, за единичными исключениями, найдено ровно 120 н. о.,
чередование которых в определенной, но небольшой мере варьирует у 5S
рРНК из разных источников, что используется в химической таксономии.
Ниже представлена первичная структура 5S рРНК кишечной палочки:
(р)УГЦЦУГГЦГГЦЦГУАГЦГЦГГУГГУЦЦЦАЦЦУГАЦЦЦЦАУГЦЦГААЦУЦАГААГУГ
10 70 80 90 100 110
АААЦГЦЦГУАГЦГЦЦГАУГГУАГУГУГГГГУЦУЦЦЦЦАУГЦГАГАГУАГГГААЦУГГЦАГ
120
ГЦАУ(он)
Расшифрована также первичная структура нескольких десятков 5,8S рРНК
и их предшественников. В подавляющем числе они содержат 158 н. о.
218
Большие успехи достигнуты в установлении первичной структуры 16;—18S
рРНК. Впервые в 1974 г. Ж. Эбель (Франция) сообщил данные о первичной
структуре 16S рРНК кишечной палочки. Вслед за этим была выяснена первич-
ная структура более ста 16—18S рРНК. Число нуклеотидных звеньев в 16S
рРНК кишечной палочки (рис. 77)—1542, в 18S рРНК печени крысы —1874.
Завершены аналогичные исследования 23S рРНК кишечной палочки, 25S
рРНК пекарских дрожжей и ряда других. 23S рРНК кишечной палочки вклю-
чает 2904 н. о., 28S рРНК печени крысы—4718 н. о. Всего на апрель 1993 г.
выяснено расположение рибонуклеотидных звеньев у 1850 16—18S рРНК (у
100—из археобактерий, у 1400—из эубактерий и хлоропластов, у 350—из
эукариот), а также у 23—28S рРНК, выделенных из 150 представителей про-
и эукариот.
Вторичная структура рРНК характеризуется спирализацией самой на себя
полирибонуклеотидной цепи как у низко-, так и у высокополимерных рРНК.
Спирализация идет за счет взаимодействия комплементарных (Г—Ц
и А—У) оснований, в результате чего в молекулах рРНК возникает то или
иное количество биспиральных участков. Оно достаточно существенно в мо-
лекулах 5S и 5,8S рРНК и несколько менее выражено в молекулах 16—18S
рРНК. Вторичную структуру 5S рРНК иллюстрирует рис. 78, А.
219
Рис. 79. Доказательство сущест-
вования быстрометящейся РНК
(мРНК):
сплошная черная ливня — поглощение при
260 им в пробах после разделения 23S, 16S
и 4S РНК ультрацентрифугированием
в градиенте плотности сахарозы; пунктир-
ная линия — включение 14С-урацила
в РНК. Ход пунктирной кривой показыва-
ет, что максимальное включение предшест-
венника осуществляется в РНК с коэффици-
ентом седиментации около 8S, которая
и представляет собой мРНК
Егм Имп/мин
Дно Верх
пробирки пробирки
Плоскостные биспирализованные и линейные участки молекул 5S и 16S
рРНК, в свою очередь, укладываются в более компактные структуры высшего
порядка, т. е. образуют пространственную, третичную структуру этих моле-
кул. Она детально охарактеризована у 5S рРНК (рис. 78, Ь) и пока еще далека
от окончательного выяснения у 16—18S рРНК. Тем не менее и у последних нет
уже сомнения в том, что осуществляется мощная компактизация постоянных
и вариабельных доменов.
Функциональная роль всех видов рРНК постепенно проясняется: 16—18S
и 23—29S рРНК являются структурной основой для формирования рибонуклео-
протеинового тяжа, который, складываясь в пространстве, дает начало 30—
40S и 50—60S субчастицам рибосомы. Однако этим, конечно, не исчерпывав ся
роль высокополимерных рибосомальных РНК: они могут взаимодей :твовать
с мРНК и аминоацил-тРНК; их участки, видимо, распознаются белковыми
факторами (см. гл. VII), принимающими участие в сборке полипептидной цепи
в рибосоме; не исключено непосредственное взаимодействие друг с другом или
контакт через посредство специфических белков 16—18S рРНК и 23—29S
рРНК, локализованных в разных субчастицах, в процессе образования 70—80S
рибосом при ассоциации субчастиц или в транслирующей рибосоме. Что
касается 5S и 5,8S рРНК, то их функции в должной мере пока еще не изучены.
Определенно установлено лишь, что 5S рРНК в рибосомах бактерий связана
с тремя белками—L5, L18 и L-25 (о рибосомных белках см. ниже, гл. VII),
у археобактерий—с одним-двумя, у эукариот—только с одним (L8); эти
комплексы рассматривают как третью субчастицу рибосомы, в составе кото-
рой 5S рРНК выполняет роль посредника между пептидилтрансферазным
центром и EF—G—связывающими доменами (об этом тоже см. гл. VII).
Структура и функции информ ционных РНК. Существование информацион-
ных рибонуклеиновых кислот (иРНК), или РНК-посредников, в передаче
информации от ДНК в белоксинтезирующий аппарат клетки (мессенджер-
РНК, от англ, messenger—посыльный, курьер, мРНК) было предсказано
А. Н. Белозерским и А. С. Спириным в 1958 г„ исходя из наличия корреляции
между нуклеотидным составом ДНК и РНК.
Двумя годами позже образование мРНК у бактерий было доказано Ф. 1ро
и сотр. в прямых опытах с включением импульсной метки в различные виды
РНК (рис. 79). Это позволило в новом свете истолковать более ранние экспери-
менты Е. Волкина и Л. Астрахана (1956), наблюдавших появление быстроме-
тящейся фракции ДНК-подобной РНК у бактерий после заражения их фагом.
Прямое выделение мРНК было осуществлено в 1962 г.— Е. Баутц
и Б. Холл для этого применили остроумный метод сорбции мРНК, возника-
ющей при заражении кишечной палочки фагом Т4, на ДНК, выделенной из
этого фага и ковалентно связанной с фосфоцеллюлозой. В дальнейшем метод
220
аффинной хроматографии стал ведущим при выделении и очистке мРНК,
особенно после того, как в составе большинства мРНК эукариот был об-
наружен полиадениловый фрагмент значительной протяженности, а в качестве
носителя в колонках применена сефароза, ковалентно связанная с полиуриди-
ловой кислотой цианбромидным методом. Взаимодействие полиадениловых
последовательностей мРНК с полиуридиловыми фрагментами носителя в си-
лу комплементарности первых и вторых приводит к связыванию на такой
колонке только мРНК.
Благодаря применению разнообразных методов и приемов многие мРНК
получены в высокоочищенном состоянии. К их числу принадлежат мРНК,
обеспечивающие матричный биосинтез субъединиц гемоглобина, яичного аль-
бумина, легких и тяжелых цепей иммуноглобулинов, гистонов, кристаллинов
(белки хрусталика глаза), миозина, фиброина шелка, авидина, протаминов,
а-казеина и др. Суммарное содержание мРНК в клетках составляет 2—3% от
тотальной клеточной РНК.
Молекулярные массы мРНК варьируют в широких пределах: от несколь-
ких сотен тысяч до нескольких миллионов дальтон. Так, молекулярная масса
моноцистронной глобиновой мРНК, служащей матрицей для биосинтеза субъ-
единиц гемоглобина, составляет 150000 (константа ее седиментации равна 9S).
Молекулярные массы многих моноцистронных, т. е. кодирующих биосинтез
одного белка, мРНК близки к этой величине. Однако у полицистронных
мРНК, кодирующих биосинтез не одного, а нескольких функционально вза-
имосвязанных белков, молекулярные массы гораздо выше и достигают не-
скольких миллионов дальтон. Примером может служить полицистронная
мРНК гистидинового оперона, выделенная из салмонеллы. Ее молекулярная
масса равна 4 • 106. Впрочем, некоторые моноцистронные мРНК могут об-
ладать молекулярной массой такого же порядка, что характерно, например,
для мРНК фиброина шелка (М = 5 • 106).
Нуклеотидный состав мРНК крайне разнообразен. Для бактериальных
мРНК характерно ДНК-подобие нуклеотидного состава, тогда как у мРНК
эукариот этого не наблюдается. В ряде случаев содерж шие нуклеотидов
в мРНК крайне специфично. Так бывает, в частности, при кодировании ими
биосинтеза резко асимметричных по аминокислотному составу белков. Ярким
примером может служить фиброиновая мРНК. В ее составе содержится
40 мол. % гуанина и 19 мол. % цитозина, что вполне соответствует наличию
в фиброине шелка 42% глицина.
Первичная структура ряда мРНК выяснена. Так, в составе глобиновой
мРНК человека установлено чередование 576 н. о. (не считая полиаденилового
фрагмента), овальбумина цыпленка—1859, липопротеин внешней мембраны
киш чной палочки—332; известны первичные структуры глобиновых мРНК
кролика и африканской шпорцевой лягушки, ряда вителлогениновых (кодиру-
ют биосинтез запасного белка яиц) мРНК, нескольких десятков мРНК, коди-
рующих биосинтез белков хориона яиц насекомых, и т. п.
Строение мРНК специфично: в их составе есть информативные, т. е. работаю-
щие как матрицы в процессе биосинтеза белка, зоны и неинформативные участки.
Из неинформативных участков изучены уже упоминавшиеся ранее полиадени-
ловые фрагменты, находящиеся на З'-конце молекулы мРНК. Длина п лиадени-
ловых фрагментов у разных мРНК колеблется от 50 до 400 н. о. Эти фрагменты
отсутствуют в молекулах гистоновых мРНК. Неподалеку от полиаденилового
фрагмента в молекулах мРНК располагаются небольшие повторяющиеся
последовательности длиной примерно по 30 н. о., тоже не несущие кодирующей
функции, но, возможно, являющиеся акцепторными участками при взаимодей-
ствии мРНК с рибосомой или отдельными белковыми факторами. Кроме того,
221
на 5'-конце мРНК присутствует нуклеотидная последовательность, азотистые
основания в которой метилированы, а один из нуклеозидных остатков—7-
метилгуанозин, присоединен через трифосфатную группировку. Эта часть
молекулы мРНК также неинформативна и называется шапочка или кэп (от
англ, cap—кепка, шапка):
Отмеченное своеобразие
бщенной структуры:
в строении РНК выражается в виде такой обо-
51
КЭП Неинформативный
фрагмент
(30—100 нуклеотид-
ных остатков)
И нформативны й
(транслируемый)
фрагмент
77^----------------------1------13‘
Неинформативный Поли А
фрагмент (50—400
(100—1000 нуклеотидных нуклео-
• остатков) тидных
остатков)
Полагают, что полиадениловая часть молекулы мРНК участвует в про-
цессе созревания мРНК (см. ниже), предопределяет время жизни мРНК,
способствует переносу мРНК из ядра в цитоплазму и принимает участие
в трансляции (см. гл. VII) мРНК. Кэп нужен для защиты мРНК от эк-
зонуклеаз, для связывания белковых факторов при взаимодействии с рРНК
в рибосоме, он играет сигнальную роль при присоединении мРНК к рибосоме
и участвует в трансляции.
Вопрос о вторичной структуре мРНК находится в стадии разработки. Как
и у других видов РНК, полинуклеотидная цепь в молекуле мРНК спирализована
сама на себя. Данные о возможных спариваниях в молекуле мРНК комплемен-
тарных оснований (А—У и Г—Ц), заложенные в компьютер, послужили
основанием для разработки модели глобиновой мРНК кролика (рис. 80). Что
касается третичной структуры мРНК, то о ней пока ничего не известно и можно
лишь констатировать, что она упакована менее компактно, чем рРНК.
На заре изучения мРНК, когда исследовали преимущественно бактериаль-
ные мРНК, характерным для этого вида РНК считали быструю обменива-
222
Рис. 80. Koi i ютерная юдель вторичной
структуры Р-глобиновой мРНК кролика.
Информативный фрагмент (см. рис.) включает 441 н. о.;
неинформативная зона со стороны кэпа—56 н. о. (от
кэпа до инициирующего кодона, с которого начинается
сборка полипептидной цепи р-глобнна), со стороны по-
лиаденилового фрагмента, ве считая его,—94 н. о. (от
терминирующего кодона, при посредстве которого за-
вершается сборка полипептидной цепи р-глобина до
начала полиадениловой части мРНК). Примерно 40%
комплементарных нуклеотидов в составе р-глобиновой
мРНК кролика, как и в других мРНК. спарено, что
показано точками; в этих зонах мРНК снирализована
сама на себя
-----Неинформативный фрагмент
емость, т. е. короткое время жизни, исчисляемое секундами или минутами,
ДНК-подобие нуклеотидного состава, а также значения молекулярных масс,
которым соответствуют константы седиментации в промежуточной зоне меж-
ду константами седиментации транспортных (4S) и малой рибосомальной
(16S) РНК. Исследования мРНК эукариот решительно изменили эти критерии
отнесения РНК к классу информационных. Дело в том, что мРНК эукариот
оказались достаточно долгоживущими молекулами (от нескольких часов до
нескольких дней и даже недель), их нуклеотидный состав был далек от
соотношения нукле< тидных остатков в валовой ДНК, а коэффициенты седи-
ментации варьировали от 9S до 65S. Поэтому в настоящее время перечислен-
ные критерии отпали ввиду их неадекватности свойствам всех мРНК и остался
единственный критерий, сводящийся к следующему: информационной следует
считать ту РНК, которая в последовательности нуклеотидных остатков в мо-
лекуле несет информацию, обеспечивающую синтез специфического белка
непосредственно на ней самой. Таким образом, матричные свойства данной
РНК оказались единственным признаком, который имеет всеобщее значение.
ОБМЕН НУКЛЕИНОВЫХ КИСЛОТ
Изучение обмена нуклеиновых кислот представляется принципиально важ-
ным в системе подготовки учителей на химико-биологических и биолого-
химических факультетах пединститутов, так как круг вопросов, которые при
этом рассматриваются, далеко выходит за рамки биохимии нуклеиновых
кислот и позволяет дать современную трактовку проблемам наследствен-
ности, изменчивости, естественного и искусственного мутагенеза, систематики
и эволюции.
Изучение обмена нуклеиновых кислот имеет огромное значение и в другом
аспекте. Оно сключительно важно для глубокого понимания процессов жиз-
недеятельности организмов. Исследование молекулярных механизмов биосин-
теза пуриновых и пиримидиновых оснований позволило открыть ряд важней-
ших закономерностей в регуляции новообразования этих соединений и сфор-
мулировать некоторые общие принципы регуляции обмена веществ.
Раскрытие механизма специфического биосинтеза огромных молекул полинук-
леотидов, при осуществлении которого с изумительной точностью обеспечи-
вается порядок чередования мононуклеотидных звеньев, их составляющих,
привело к признанию ведущей роли в этом процессе взаимодействия ком-
плементарных пуриновых и пиримидиновых оснований. Это, в свою очередь,
223
дало возможность впервые понять интимный механизм обеспечения специфи-
ческого воспроизведения первичной структуры макромолекул при их биосин-
тезе. Данные о регуляции новообразования нуклеиновых кислот привели
к фундаментальным открытиям, позволяющим сделать первые шаги к объяс-
нению закономерностей не только воспроизведения специфических макромо-
лекул, но также и морфогенеза.
Пути распада нуклеиновых кислот. Распад нуклеиновых кислот в организме
идет достаточно энергично. Так, период полужизни молекул ДНК в тканях
мыши доставляет от 1 до 5 суток; период полужизни большинства мРНК
у эукариот—несколько суток, а у прокариот—всего несколько секунд.
Нуклеиновые кислоты (РНК и ДНК) распадаются в организме при по-
средстве особых ферментов—нуклеаз. Они ускоряют реакцию разрыва меж-
нуклеотидных фосфодиэфирных связей в молекулах нуклеиновых кислот
и принадлежат, следовательно, к более широкой категории ферментов—
фосфодиэстераз.
Нуклеазы, действующие на внутренние межнуклеотидные связи в молеку-
лах ДНК и РНК, называются эндонуклеазами. При их участии осуществляется
деполимеризация нуклеиновых кислот в основном до олигонуклеотидов. Нук-
леазы, ускоряющие реакции последовательного отщепления нуклеотидов от
РНК, ДНК или их фрагментов, начиная с конца полинуклеотидной цепи,
называют экзонуклеазами. Они обеспечивают распад нуклеиновых кислот до
свободных нуклеотидов.
В зависимости от специфичности действия среди нуклеаз различают рибо-
нуклеазы и дезоксирибонуклеазы. Первые ускоряют реакции распада как внут-
ренних, так и внешних (концевых) межнуклеотидных связей в молекулах РНК.
Вторые выполняют такую же функцию по отношению к ДНК. Вместе с тем
существует большая группа неспецифических эндо- и экзонуклеаз, действую-
щих одновременно и на РНК, и на ДНК.
По характеру действия на фосфодиэфирные связи в молекулах нукле-
иновых кислот нуклеазы делят на две категории. Одни из них ускоряют
реакцию гидролиза сложноэфирной связи межнуклеозидного фосфата с 3'-
углеродным атомом остатка рибозы или дезоксирибозы, а другие—с 5'-
углеродным атомом. Поэтому в названиях указанных ферментов всегда под-
черкивается, гидролиз какой из связей ускоряет данная нуклеаза. Однако
ведущим признаком является локализация фосфата в составе олиго- или
мононуклеотидов, возникающих в результате гидролиза нуклеиновой кисло-
ты. Поэтому нуклеазы, расщепляющие связь Р—5'С, называются З'-нукле-
азами, а расщепляющие связь Р—З'С именуют 5'-нуклеазамн:
224
В жнейшими группами нуклеаз являются следующие.
Дезоксирибонуклеазы I (дезоксирибонуклеинат-У-олигонуклеотидгидрола-
зы) ускоряют гидролиз фосфодиэфирных связей в ДНК между остатком
фосфата и З'-углеродным атомом остатка дезоксирибозы:
ДНК + (и — 1) Н2О~*и олиго дезоксирибонуклеотидов.
Представителями ДНКаз I являются панкреатические ДНКазы
(М~31000). Первичная структура панкреатической ДНКазы быка расшиф-
рована: она представлена одной полипептидной цепью из 257 аминокислотных
остатков. Оптимум pH лежит между 6,8 и 8,0. Фермент активируется ионами
Мп2+ и Mg2+ и ингибируется анионами, связывающими указанные катионы,
а также олигонуклеотидами. Механизм действия панкреатических ДНКаз на
ДНК таков, что вначале осуществляются в основном разрывы фосфодиэфир-
ных связей в одной из цепей ДНК. Парные разрывы очень редки, поэтому
деполимеризация идет не сразу. Конечный продукт переваривания ДНК пан-
креатической ДНКазой содержит следы дезоксирибонуклеозид-5'-фосфатов,
немного динуклеотидов и большое количество олигодезоксирибонуклеотид-S'-
фосфатов, составленных в среднем из 4 мономерных звеньев. Панкреатическая
ДНКаза быка существует в виде четырех множественных форм.
Дезоксирибонуклеазы II (дезоксирибонуклеинат-З'-олигонуклеотидгидро-
лазы) в качестве конечного продукта действия образуют олигодезоксирибону-
клеотид-З'-фосфаты со средней длиной молекул в 6 звеньев и небольшие
количества дезоксирибонуклеозид-З'-фосфатов. Молекулярная масса ДНКазы
II селезенки 38 000, оптимум pH 4,5—5,5. Ее молекула состоит из 343 амино-
кислотных остатков и содержит углеводную компоненту, структурной едини-
цей которой является глюкозамин. Фермент активируется ионами Mg2+,
ингибируется ионами SOj-, РО|“, AsO^-, АУ-кополимером, тРНК. Деполи-
меризация ДНК осуществляется путем парных разрывов фосфодиэфирных
связей в молекуле ДНК, в результате чего быстро накапливаются ее большие
фрагменты; особенно хорошо атакуются связи, составленные из парных соче-
таний ГГ и АЦ. Предполагают, что ДНКазы II являются димерами.
Кроме ДНКазы I и II существует большая группа эндонуклеаз, атакующих
как дву-, так и одноцепочечную ДНК. Так, например, у кишечной палочки их
7, среди них—4 низкомолекулярные (12—33 тыс. Да) и 3 высокомолекуляр-
ные (68—114 тыс. Да).
Экзодезоксирибонуклеазы ускоряют реакцию гидролиза молекул ДНК
с образованием дезоксирибонуклеозид-5'-фосфатов. Из кишечной палочки вы-
делено и очищено в разной степени 8 экзодезоксирибонуклеаз (I—VIII), от-
личающихся друг от друга по определенным показателям. В частности, эк-
зодезоксирибонуклеаза III (М = 30000) обладает способностью отщеплять 3'-
фосфат, если он есть, от концевого нуклеотида фрагментов молекулы ДНК.
Рестриктазы—ДНКазы бактериального происхождения, расщепляющие
чужеродные (фаговую) ДНК в строго определенных зонах, которые «узнают-
ся» ферментом. Эти зоны имеют палицдромную структуру. Ниже приведены
два примера, где стрелками указаны точки гидролиза фосфодиэфирных связей:
1
5'—ГААТТЦ—3'
3'—ЦТТААГ—5'
1
1
5'—НЦЦАГГН —3'
3'—НГГТЦЦН—5'
рестриктаза кишечной палочки I (ЕсоД/);
М = 116 000, тетрамер (29 000 х 4)
рестриктаза кишечной палочки II (ЕсоДЯ);
М = 80 000, димер (40000 х 2)
8—3502
225
В зависимости от взаимного располож ния места узнавания и точки рас-
щепления ДНК рестриктазами, их относят к I типу (сайт узнавания далеко,
на расстоянии сотен н. о. от точки расщепления), II типу (расщепление внутри
сайта узнавания) или III типу (расщепление вблизи сайта узнавания). К 1993 го-
ду выделено и охарак еризовано 2393 рестриктазы I типа, 188 рестриктаз
II типа и только 4 рестриктазы III типа. Всего следовательно изучено 2585 ре-
стриктаз.
Так как рестр тктазы расщепляют ДНК на ограниченное число фрагментов,
они нашли применение для определения первичной структуры ДНК. Однако
еще более важная область их практического использования — генетическая
инженерия, так как именно благодаря действию рестриктаз из ДНК выщепля-
ются фрагменты, которые далее встраиваются в бактериальную ДНК, стано-
вятся интегральной частью бактериального генома и приносят бактерии но-
вые, не свойственные ей ранее биохимические признаки, такие, например, как
способность синтезировать интерфе он, инсулин и другие белки, находящие
широкое применение в медицине (рис. 81). В принципе, такие процессы воз
можны и в геноме высших организмов. Легкое встраивание рестрикционных
фрагментов ДНК в реци иентную ДНК (т. е. ДНК, включающую эти фрагме-
нты в свой состав) объясняется тем что при действии рестриктаз в точке
разрыва молекулы ДНК случаются «липкие», комплементарные концы. Ввиду
огромной теоретической ценности и большого практического значения этих
работ в нашей стране начиная с 1975 г. осуществляются комплексные ис-
следования по проекту «Рестриктаза», который планируют на последующих
этапах работы превратить в более широкий проект «Нуклеаза» и привлечь
к его разработке различные научно-исследовательские институты. В 1980 г.
большая группа советских ученых была удостоена 1осударственной премии за
разработку регламентов получения 30 рестриктаз и внедрение их в практику
работ по генетической инженерии.
Рибонуклеазы I (рибонуклеинат-З'-пиримидино-олигонуклеотидгидрола-
зы)—эндонукл азы, ускоряющие ре кцию гидролиза РНК по пиримидино-
вым нуклеот дным остаткам. Ранее их относили к рибонуклеат-нуклеотидо-2'-
трансферазам циклизующим, но, поскольку 2', З'-циклофосфаты в процессе их
действия образуются как промежуточные продукты, гидролиз которых до
З'-фосфатов ускоряется тем же ферментом, в настоящее время рибонуклеазы
(РНКазы) I включены в класс гидролаз.
Представителями РНКаз I являются панкреатические РНКазы, выделенные
из многих объектов. Установлена первичная структура панкреатической РНКа-
зы быка, свиньи, овцы, жирафа, марала, косули, северного оленя, лани,
шиншиллы, мыши, крысы, морской свинки, одногорбого верблюда и нутрии. Во
всех случаях, за исключением двух последних, фермент состоит из 124 аминокис-
лотных остатков. Выяснена третичная структура некоторых панкреатических
РНКаз. Панкреатические РНКазы могут быть однокомпонентными (риб нукле-
аза А) и дьухкомпонентными (рибонуклеаза В, содержащая углеводный остаток
с М = 1350). Механизм действия их на РНК детально исследован и является ярким
дополнением к материалам гл. III, касающимся механизма действия ферментов.
Полипептидная цепь РНКазы, замкнутая четырьмя дисульфидными мо-
стиками (рис. 82,А), принимает вследствие этого пространственную конфи-
гурацию (рис. 82, Б), в определенной мере предопределяющую третичную
структуру (рис. 82, Б) этого фермента. Понять механизм действия панкре-
атической РНКазы помогли данные о структуре ее активного центра
(рис. 82, Г). В нем связываются участки молекулы РНК, содержащие в своем
составе пиримидиновые (цитидиловые и уридиловые) нуклеотидные остатки,
причем парные сочетания ЦА, ЦГ, ЦЦ и ЦУ атакуются в пропорции
226
Расщепление рестриктазой
Включение фрагмента
чужеродной ДНК в
плазмвдную ДНК
Проникновение плазмиды
в бактериальную
клетку-хозяина
Рис. 81. Принципиальная схема проведения работ по гене-
тической инженерии
В качестве фрагментов чужеродных ДНК используют гены, ответственные за
биосинтез биологически значимых елков, их экспрессии может быть ис-
пользована дли биосинтеза этих белков после интеграции плазмцдвой ДНК
в бактериальный геном или дли клонировании генов и их последующей
экспрессии в иных, чем бактериальные, белоксивтезирующих системах
227
Ю ч 20
ала—ала —лиз—фен—глу—арг-гли 4 гис ч-мет-асп-сер—сер-тре-сер—ала — ала
ала—тре
(HN) Лиз-глу
* 1
70 80
— асн-глн -цис -тир-глн-сер-*тир-сер-тре-мет — сер
тре
сер
сер
вал
асп
ала
лей
50 сер
гли
।— асн—лиз-цис-ала
иле
сер
вал
60
глн —ала — вал —цис —сер—глн -лиз-асн
124
(НООС)вал -сер
ала
тре
асн
асп
тир
цис
цис
иле—вал— ала-цис—глу-гл и —асн-про -тир— вал-про-вал
110
иле
гис
асп
4гис-4фен
Ч-.Л20У
арг
асн
90
глу
сер
тре
| 100
лиз —глн — ала - асн -тре -тре-лиз-тир-цис -ала-асн-про-тир-лиз -сер-гли
глу М се
гис —вал—фен +тре т-асн—вал—продлиз-J-цис — арг—асп-лиз—тре—лей —асн—арг—- I
40
Рис. 82. Структура бычьей панкреатической рибонуклеазы и ее активного центра:
А—первичная структура; черными прямоугольниками обозначены—S—S-связи, арабскими цифрами—номера аминокислотных
остатков, пунктиром обведены аминокислотные остатки, входящие в состав активного центра; Б—расположение в пространстве
полипептидной цепи; В—трехмерная модель молекулы при разрешении в 0,5 нм; зачернена впадина, на дне которой располагается
активный центр; Г—структура активного центра, в котором радикалы гис, лиз, фен, сер и тре (их порядковые номера
глн
I
мет
мет
лиз
сер
30
3000:500:240:27. В активном центре фермента остаток пиримидинового нукле-
отида РНК размещается таким образом, что пиримидиновое основание закреп-
ляется в субстратном центре за счет водородных связей с радикалами тре
и сер, занимающими 45-е и 123-е положения в полипептидной цепи и за счет
гидрофобного взаимодействия с радикалом фен (120-й аминокислотный оста-
ток). Вследствие этого остаток фосфата, расположенный между З'-углеродным
атомом рибозы пиримидинового нуклеотида и 5'-углеродным атомом сосед-
него в цепи РНК нуклеотида, фиксируется между 12-м и 119-м остатками гис
каталитического центра. Фиксации межнуклеозидного фосфата в этом положе-
нии способствует также радикал лиз, занимающий в молекуле РНКазы 41-е
положение, но расположенный в ее активном центре на расстоянии 0,5 нм от
вышеупомянутых остатков гис. Вслед за этим наступает непосредственно
каталитический акт, осуществляемый в результате согласованной передачи
протонов и сводящийся к разрыву межнуклеотидной фосфатной связи, об-
разованию 2', З'-циклофосфата рибозы пиримидинового нуклеотида и по-
228
Рис. 82. Продолжение
в полипептидной цепи указаны цифровыми индексами) сближены на расстояние в несколько десятых долей нанометра; Д—
пространственная структура активного центра
следующего гидролиза 2', З'-циклофосфатной связи с восстановлением ис-
ходной структуры активного центра (см. схему реакции).
Существенно, что именно остатки гис, находящиеся в 12-м и 119-м положе-
ниях в полипептидной цепи и сближенные в каталитическом центре фермента,
обеспечивают перенос протонов.
ООО
Эта функция имидазольных радикалов гистидина воспроизводится при
рассмотрении механизма действия многих других ферментов, особенно гидролаз
(см. раздел о гидролизе пептидной связи—с. 263, гликозидной связи—с. 331).
1уанилрибонуклеазы (рибонуклеинат-З'-гуанило-олигонуклеотидгидрола-
зы) ускоряют гидролиз связей по 5'-углеродному атому рибозы остатка гуани-
ловой кислоты и межнуклеотидного фосфата в молекуле РНК, образуя гуано-
зин-З'-фосфат и олигонуклеотиды с остатком гуанозин-З'-фосфата в качестве
концевого нуклеотида. Первичная структура гуанилрибонуклеазы, выделенной
из плесневого гриба аспергилла (Тх-PHКаза), расшифрована (М = 11 000; 104
аминокислотных остатка).
Фермент нашел широкое применение для деструкции РНК при определе-
нии их первичной структуры, и именно при посредстве Tt -РНКазы Р. Холли
с сотр. (1965) впервые получили крупные фрагменты тРНКала (см. с. 213).
Охарактеризовано еще 10 эндорибонуклеаз, выделенных из бактерий, мик-
роскопических грибов, растений и животных.
Как и в случае ДНКаз, существует большая группа (более десяти) экзорибо-
нуклеаз, ускоряющих реакции отщепления рибонуклеотидов по концевым
остаткам РНК, и олигорибонуклеотидов, возникающих при селективном гидро-
лизе РНК под действием эндорибонуклеаз. Таким образом, в результате
деятельности разнообразных нуклеаз нуклеиновые кислоты при распаде дают
сложную смесь индивидуальных рибо- и дезоксирибонуклеозид-3' и 5'-фосфатов.
Кроме перечисленных ферментов в деструкции нуклеиновых кислот прини-
мают участие еще некоторые энзимы, не являющиеся гидролазами фосфоди-
эфирных межнуклеотидных связей, например полинуклеотидфосфорилаза
и урацил-ДНК-гликозидаза.
Полинуклеотидфосфорилаза (полинуклеотид: ортофосфат-нуклеотидил-
трансфераза) в отличие от всех ранее рассмотренных ферментов, участвующих
в деструкции нуклеиновых кислот, является нуклеотидилтрансферазой, т. е.
переносит нуклеотидные остатки с З'-конца РНК на неорганический фосфат
РНК+НДФ
(п нуклеотидных
остатков)
Очоа (1955) и выделен из
многих источников. Он сосредоточен главным образом в микросомальной
и рибосомальной фракциях клеточного содержимого. По грубой оценке его
молекулярная масса близка к 230 000. Скорость реакции фосфоролиза зависит
от конформации и нуклеотидного состава РНК: двухцепочечные и метилиро-
ванные участки устойчивы к действию фермента. Предполагают, что in vivo
полинуклеотидфосфорилаза обеспечивает деградацию клеточных РНК, осо-
бенно мРНК, до нуклеозиддифосфатов, регулирует концентрацию неорганиче-
ского фосфата в клетке и поставляет необходимое количество НДФ для
превращения их в дезоксиНДФ.
Фермент обладает замечательной особенностью: из нуклеозиддифосфатов
и их смесей in vitro он обеспечивает синтез полирибонуклеотидов с соотноше-
нием в их составе мономерных звеньев в той же пропорции, как в исходном
растворе. Поэтому полинуклеотидфосфорилазу широко применяли для син-
теза полирибонуклеотидов того или иного состава, что сыграло выдающуюся
роль в расшифровке кода белкового синтеза.
Урацил-ДНК-гликозидаза ускоряет реакцию отщепления остатка У от по-
врежденной ДНК, где произошло дезаминирование остатка Ц. На возникшем
230
с образованием нуклеозиддифосфатов (НДФ):
РНК+Н3РО,
(л+1 нуклеотидных
остатков)
Фермент открыт М.
Mg3
Полинуклеотид-
фосфорилаза
-Монаго и
с.
апиримидиновом участке одной из цепей ДНК фосфодиэфирная связь гидроли-
зуется с элиминированием дезоксирибозы, З'-фосфат отщепляется при участии
экзодезоксирибонуклеазы III и вместо отсутствующего нуклеотидного остатка
встраивается новый, в данном случае остаток цитидиловой кислоты, при
посредстве ДНК-полимеразной и ДНК-лигазной реакции (см. с. 251 и рис. 84).
ДНК-гликозидазы представляют новую группу ферментов, участвующих
в обмене ДНК. При их посредстве удаляются и иные модифицированные
пуриновые и пиримидиновые основания, после чего в серии последующих
реакций восстанавливается исходная структура ДНК, т. е. эта группа фермен-
тов имеет существенное значение в репарации (восстановлении структуры) ДНК.
Это происходит, в частности, при замене метилированных пуриновых и пирими-
диновых оснований, так как наряду с урацил-ДНК-гликозидазой изучена
3-метиладенин-ДНК-гликозидаза. Всего открыто уже 8 ДНК-гликозйдаз.
Обмен нуклеозидфосфатов. Дезоксирибонуклеозидфосфаты и рибонукле-
озидфосфаты, представляющие собой конечные продукты ферментативной
деструкции нуклеиновых кислот, распадаются далее до еще более простых
соединений. Первая ступень этого распада состоит в отщеплении остатка
фосфорной кислоты:
(нуклеозид)
Гуанозин-3 -фосфат
(нуклеотид)
На второй ступени распада осуществляется перенос остатка рибозы от
нуклеозида на фосфорную кислоту. Эта реакция ускоряется специфическими
для каждого вида нуклеозидов рибозилтрансферазами. Примером может слу-
жить фосфоролиз уридина:
Урнд инфосфера лаза
(урнднн-ортофосфат-
рнбозшправ сфер аза)
Рибозо-1- Урацил
фосфат
Уридин осфорилаза имеет М = 165 к Да, содержит 6 субъединиц по
27,5 кДа, каждая из которых составлена из 253 аминокислотных остатков,
в том числе из семи остатков гистидина; два из них (8-ой и 122-ой) входят
в активный центр фермента.
231
Следовательно, в результате распада нуклеозидфосфатов выделяются
в свободном состоянии рибозо-1-фосфат и все виды пуриновых и пиримиди-
новых оснований, участвующих в построении нуклеиновых кислот. Приведен-
ная схема распада нуклеозидов не является единственной. Возможны и другие
пути распада нуклеозидов. Один из них состоит в гидролизе нуклеозидов,
например:
Аденозин
Аденнн
Рибоза
В свою очередь, и углевод и азотистые основания видоизменяются далее.
Рибоза и рибозо-1-фосфат включаются в реакции обмена, характерные для
углеводов. Эти реакции будут рассмотрены ниже. Пуриновые и пиримидино-
вые основания претерпевают дальнейший распад и превращаются в те или
иные простейшие азотсодержащие продукты, которые далее либо выводятся
из организма, либо откладываются в нем.
Распад пуриновых и пиримидиновых оснований. Первая фаза распада пури-
новых и пиримидиновых оснований заключается в дезаминировании тех из
них, которые обладают аминогруппами. Этот процесс осуществляется при
посредстве специфических аминогидролаз. В результате аденин превращается
в гипоксантин:
Аденин-амино -
гидролаза
Аденин
ын3
Гуанин переходит в ксантин:
Цитозин преобразуется в урацил:
232
Циплия
+ н2о
+ NH3
Уради
Дезаминирование идет не только на уровне свободных пуриновых и пири-
мидиновых оснований, но и на уровне нуклеозидов и нуклеотидов, причем во
втором случае с большей интенсивностью, так как соответствующие нукле-
озид- и нуклеотид-аминогидролазы более активны, чем пурин- или пиримидин-
аминогидролазы. Так, аденозин и аденозинфосфат более энергично превраща-
ются в инозин и инозинфосфат, чем аденин в гипоксантин:
При дальнейшем распаде дезаминированных нуклеозидов и нуклеотидов из их
состава освобождаются гипоксантин, ксантин или урацил.
Дальн йшая судьба дезаминированных пуриновых и пиримидиновых ос-
нований различна. Пшоксантин и ксантин окисляются в мочевую кислоту:
Реакция окисления гипоксантина в ксантин, а последнего—в мочевую
кислоту ускоряется ксантиноксидазой—оксидоредуктазой с широким спект-
ром действия, представляющей собой молибденсодержащий флавопротеин.
Фермент из разных источников обладает молекулярной массой от 280 тыс.
до 360 тыс. Да и при действии диссоциирующих агентов распадается на две
идентичные субъединицы, каждая из которых содержит одну молекулу ФАД,
один атом Мо и 4 атома негеминового железа, связанных с лабильными
атомами серы в кластеры типа Fe2S2. Активированные гипоксантин и ксантин
восстанавливают молибден, последний быстро восстанавливает Р282-кластер
и, наконец, флавин восстанавливает молекулярный кислород.
У ряда животных (человекообразные обезьяны, птицы, рептилии, тутовый
шелкопряд) и человека конечным продуктом распада пуриновых оснований
является мочевая кислота, которая и выводится из организма. Однако
233
у большинства животных и растений есть ферменты и ферментные системы,
способные ускорять реакции дальнейшего распада мочевой кислоты. От назва-
ния мочевой кислоты acidum uricum и по характеру действия, выражающемуся
в расщеплении (лизисе) ее, эти ферменты получили наименование ферментов
уриколиза. В одних случаях (млекопитающие, насекомые) уриколиз сводится
к окислению мочевой кислоты в аллантоин; в других (костистые рыбы)—
процесс более сложен: аллантоин превращается в аллантоиновую кислоту,
а последняя (амфибии, большинство растений) распадается на мочевину и гли-
оксиловую кислоту:
о
кислота
В отличие от гипоксантина и ксантина дезаминированные пиримидиновые
основания подвергаются восстановлению. Так, урацил переходит в дигидроура-
цил; донором атомов Н в этой реакции служит НАДН. В свою очередь,
ди идроурацил претерпевает гидролиз и превращается в N-карбамил-р-аланин,
который далее гидролизуется до Р-аланина и карбаминовой кислоты. Послед-
234
няя либо используется для синтеза мочевины, либо распадается до СО2 и NH3.
Все перечисленные реакции ускоряются соответствующими ферментами:
Карбаминовая кислота и Р-аланин являются конечными продуктами
распада двух пиримидиновых оснований—У и Ц. В случае Т, распадающе-
гося по такой же схеме, вместо Р-аланина образуется Р-аминоизомасляная
кислота. .
Итак, огромные, сложные молекулы полидезоксирибонуклеотидов и по-
лирибонуклеотидов в процессе распада в организмах животных и растений
превращаются в очень простые соединения—главным образом фосфорную
кислоту, СО2 и NH3. У организмов, расположенных на нижних ступенях
эволюционной лестницы, представлен всегда полный набор ферментов,
обеспечивающих распад нуклеиновых кислот именно до этих простейших
продуктов. При переходе к более высокоорганизованным формам ряд
ферментов, участвующих в превращении пуриновых и пиримидиновых ос-
нований, выпадает и конечными продуктами обмена нуклеиновых кислот
у некоторых групп организмов являются более сложные соединения, чем
NH3 и СО2; это мочевина, аллантоиновая кислота, аллантоин и мочевая
кислота.
Механизм биосинтеза нуклеозидфосфатов. Для обеспе ения биосинтеза нук-
леиновых кислот организм должен располагать полным набором дезоксири-
бо- и рибонуклеозидтрифосфатов. Поэтому в любой клетке любого организма
независимо от положения его на эволюционной лестнице беспрепятственно
осуществляется процесс новообразования всех видов нуклеозидтрифосфатов,
нуклеозиддифосфатов и нуклеозидмонофосфатов.
Из трех основных частей нуклеотида—азотистого основания, пентозы
и фосфор той кислоты—последняя в норме всегда присутствует в клетках,
а вторая неминуемо возникает в процессе распада углеводов. Таким образом,
только первая составная часть нукл отида—пуриновое или пиримидиновое
основание, должна создаваться специфическим путем.
Пути возникновения пуриновых и пиримидиновых оснований различны.
Но есть некоторые черты сходства в механизмах их синтеза. К их числу
относятся: 1) широкое использование гли, асн и глн в качестве источников
азота гетероциклических колец; 2) включение в состав пуриновых и пирими-
диновых циклов атомов углерода из СО2 и формиата; 3) построение пурино-
вого основания и завершение синтеза пиримидинового основания на рибозо-
5-фосфате, в результате чего конечными продуктами биосинтеза являются
сразу нуклеозид-5'-фосфаты, а не свободные А, Г, У, Ц и Т; 4) ферментатив-
ный характер всех реакций, осуществляющихся в процессе новообразования
нуклеотидов; 5) возникновение на определенном этапе биосинтеза предшест-
венников, из которых потом формируются уже индивидуальные нуклеозид-
5'-фосфаты.
Рассмотрим сначала механизм биосинтеза пиримидиновых основании. Под-
готовительной реакцией, открывающей этот синтез, является реакция об-
разования карбамилфосфата из NH3 и СО2 при участии АТФ:
ОН
Карбаматкиназа \
мн3+со2+атф-------->АДФ+Н2Ы—С—О—Р=о
II /
о он
Карбамилфосфат
235
Далее при участии специфического фермента остаток карбаминовой
кислоты (карбамил) переносится на аминогруппу аспарагиновой кислоты
с образованием карбамиласпарагиновой кислоты. Эту реакцию рассматри-
вают как первую специфическую реакцию в синтезе пиримидиновых нукле-
отидов:
ЫН2 ------х fOOH
X ' V 2 Аспартат-карбамил-
° \ О-Р=О+ H^N—СН—СООН------------трансфераза >
Карбамил- Аспарагиновая
фосфат кислота
NH2 СОСИ
£ <^Н2 + Н3РО4
0^ ^NH—СН—СООН
Карбамил аспарагиновая
кислота
При сближении NH2- и СООН-групп в молекуле карбамиласпараги-
новой кислоты между ними осуществляется взаимодействие с выделе-
нием молекул воды. Эта реакция катализируется ферментом из класса
гидролаз—дигидрооротазой, названной так от дигидрооротовой кисло-
ты, гидролиз которой она ускоряет вследствие обратимости данной ре-
акции:
Карбамиласпараги-
новая кислота
Дигидрооротовая
кислота
Дигидрооротовая кислота ферментативно окисляется. Снятие двух атомов
Н осуществляется первичной дегидрогеназой либо с НАД+ или НАДФ+, либо
с ФАД в качестве кофермента:
HN
_с.
сн2
сн—соон
НАД+ НАДН+Н+
Оротатредуктаза
Оротовая кислота
В молекуле оротовой кислоты, как видно из ее формулы, уже пред-
образована структура одного из пиримидиновых оснований, а именно урацила.
Достаточно осуществить реакцию декарбоксилирования, и оротовая кислота
превратится в урацил. Однако этот процесс происходит лишь после того, как
оротовая кислота 'соединится с рибозой, образуя нуклеозид, где агликоном
является остаток оротовой кислоты. Нуклеозид такого строения называется
236
оротидином (по аналогии с цитидином и уридином). Так как реакция идет
непосредственно между оротовой кислотой и 5-фосфорибозилпирофосфатом,
то в ее результате возникает оротидин-5'-фосфат. Процесс ускоряется соот-
ветствующей трансгликозидазой. Уравнения реакций, приводящих к синтезу
оротидин-5'-фосфата, таковы:
5-Фосфорибозил-1-пирофосфат
Оротовая кислота
Оротат-фосфорибозил-
трансфераза
Последнее преобразование состоит в декарбоксилировании оротидин-5'-
фосфата:
В результате возникает один из пиримидиновых нуклеотидов—ури-
дин-5'-фосфат. Уридин-5'-фосфат занимает центральное место в биосин-
тезе пиримидиновых нуклеотидов, так как далее может превращаться в
другие пиримидиновые нуклеотиды в соответствии со следующей схе-
мой:
237
уМф Фосфорилирование
Фосфорилирование _
УДФ ------- ------------- УТФ
(Уридин-5-монофосфат) (Уридин-5'-дифосфат) (Уридин-5'-трифосфат)
rpceTBHoaWy? I Аминирование
д Дефосфорилироваияе Дефосфорилироваиие f
дУДФ ЦМФ * . — ЦДФ ** — и. ЦТФ
(Дезоксиуридин-
5-дифосфат)
X
!
(Цнтидин-5'-монофосфат)
(Цитидин-5'-дифосфат) (Цитидин-5' трифосфат)
I Восстановление
Дефосфорилирование Фосфорилирование
дЦМФ ----------—----- дЦДФ ------------------------------ дЦТФ
(Дезоксицитидин- (Дезоксицитидин- (Дезоксицитидии-
5'-монофосфат) 5-дифосфат) 5’-трифосфат)
(Дезоксиуридин-
5-монофосфат)
(Дезокситимидин- (Дазокситимидин-
5'-монофосфат) 5-дифосфат)
(Дезокситимидии-
5'-трифосфат)
Схема 2. Пути превращений пиримидиновых нуклеотидов
Эти превращения пиримидиновых нуклеотидов осуществляются путем ре-
акций восстановления, аминирования и метилирования моно-, ди- и трифос-
форных эфиров нуклеозидов. Последние образуются при взаимодействии
нуклеозидмонофосфатов с АТФ, запасы которой в клетках непрерывно попол-
няются за счет реакции окислительного фосфорилирования. Например:
УМФ+АТФ
Нуклеозидмонофосфаткиназа
(уридилаткиназа)
УДФ+АДФ
Нуклеозид-
УДФ+АТФ ------------------------• УТФ+АДФ
дифосфат-
киназа
Восстановление протекает по гидроксильной группе при 2-м углеродном
атоме рибозы, благодаря чему остаток рибозы переходит в остаток дезок-
сирибозы. Эта реакция свойственна нуклеозиддифосфатам:
с двойной связью между
2’ и з’-углероднымн ато-
мами остатка рибозы
Донором атомов Н для восстановления рибозы при превращении рибонук-
леозиддифосфата в дезоксирибонуклеозиддифосфат служит специальный бе-
лок—тиоредоксин. Будучи составлен из 108 аминокислотных остатков, тиоре-
238
доксин содержит в 32-м и 35-м положениях остатки цис и располагает,
следовательно, двумя HS-группами. Именно они и поставляют атомы Н,
образуя дисульфидный мостик. Окисленный тиоредоксин немедленно перево-
дится в восстановленную форму, получая атомы Н от НАДН при посредстве
фермента тиоредоксинредуктазы.
Кроме обеспечения атомами Н реакции восстановления остатка рибо-
зы, тиоредоксин в восстановленном состоянии способен соединяться с дву-
мя Другими каталитически активными белками Бх и Б2. Последние при
этом активируются и непосредственно ускоряют процесс восстановления
остатка рибозы. Они же подвержены сильному влиянию других алло-
стерических регуляторов активности, в частности АТФ, ГТФ, ТТФ, дАТФ,
дГТФ и др.
Таким образом, превращение рибонуклеозиддифосфатов в дезоксирибо-
нуклеозиддифосфаты идет в соответствии со следующей схемой:
белки-ферменты
Рибонуклеозид- Б,(2«87000)и Б»(2«43000) Дезоксирибо-
дифосфаты "Х. нуклеозид-
Л \ дифосфаты
Тиоредоксин (SH)3 Тиоредоксин (SS)
Т норедоксннредуктаза
НАД+ НАДН+Н+
Что касается реакций аминирования (переход от УТФ к ЦТФ) и метилиро-
вания (переход от дУМФ к дТМФ), то в первом случае источником амино-
группы у бактерий служит NH3, а у млекопитающих—глн, причем введение
аминогруппы осуществляется сопряженно с распадом АТФ; во втором случае
источником метильной группы является №-метилтетрагидрофолиевая кисло-
та, а реакция переноса ее ускоряется тимидилат-синтазой (димер; каждая
полипептидная цепь—316 аминокислотных остатков; первичная и третичная
структуры расшифрованы).
В результате всех этих реакций обеспечивается создание в организме фонда
свободных пиримидиновых нуклеозидтрифосфатов (УТФ, ЦТФ, дЦТФ,
дТТФ), необходимых для синтеза ДНК и РНК.
Важной особенностью биосинтеза пиримидиновых нукл отидов является
саморегуляция этого процесса. Установлено, что такие конечные продукты
биосинтеза пиримидиновых нуклеотидов, как ЦТФ и дЦТФ, ингибируют
деятельность аспартат-карбамилтрансферазы—фермента, ускоряющего пер-
вую реакцию в цепи тех взаимодействий, которые приводят к формированию
пиримидинового цикла. Выявлено, что понижение активности фермента вызы-
вается присоединением ЦТФ по аллостерическому центру фермента. Таким
образом, накопление в клетке избыточного количества ЦТФ и дЦТФ немед-
ленно сказывается на активности аспартат-карбамилтрансферазы и биосинтез
пиримидиновых нуклеотидов замедляется. Антагонистом ЦТФ в ингибирова-
нии деятельности этого фермента является АТФ, активирующая фермент.
Следовательно, торможение или стимулирование биосинтеза пиримидиновых
нуклеотидов зависит от соотношения в клетках организма АТФ и ЦТФ, т. е. от
239
энергетического баланса клетки, от уровня в ней обмена веществ, в частности
от уровня реакций окислительного фосфорилирования, при посредстве кото-
рых высвобождающаяся в процессе окисления органических веществ энергия
запасается в макроэргических связях АТФ.
Двусторонний контроль деятельности первого в цепи реакций биосинтеза
фермента представляет очень четкий механизм регуляции обмена веществ
и используется во многих системах биосинтеза в организме. Именно на
примере биосинтеза пиримидиновых нуклеотидов он исследован наиболее
детально.
В заключение отметим, что одно из производных пиримидиновых нукле-
отидов, а именно 3'-азидо-2', З'-дидезокситимидин, подавляет развитие ретро-
вирусов, в том числе вызывающих синдром приобретенного иммунодефицита
(СПИД):
Предложены и другие производные нуклеотидов для борьбы с этим гроз-
ным заболеванием.
Перейдем теперь к рассмотрению механизма биосинтеза пуриновых основа-
ний. Формирование пуринового кольца сразу идет на рибозо-5-фосфате. Поэ-
тому первой реакцией является взаимодействие глутамина с 5-фосфорибозил-
1-пирофосфатом при каталитическом воздействии гликозилтрансферазы, уско-
ряющей перенос остатка 5-фосфорибозы на амидогруппу глутамина. Видимо,
одновременно протекает гидролиз возникающего 5-фосфорибозилглутамина,
вследствие чего выделяется 5-фосфорибозиламин:
5-Фосфорнбозил-1- пирофосфат
Амидофосфо-
рибозилтрзнс-
фераэа
NHa
Глутаминовая кислота
5-Фосфорибозиламин
Пирофосфат
240
В присутствии АТФ при участии специфической лигазы (аминосинтетаза)
к 5-фосфорибозиламину присоединяется глицин, причем возникает пептидная
связь:
5-Фосфорибозиламин
I CH2NH2 + АТФ
\но<
Фосфорибозил-
глицннамид-
сивтетаза
Глицин
+ АДФ + Н3РО4
5 -Фосфор и бозилгл и ци н а м и д
Молекула 5-фосфорибозилглицинамида удлиняется на один углеродный
атом при посредстве фосфорибозилглицинамид-формилтрансферазы. Ее ко-
ферментом служит тетрагидрофолиевая кислота, присоединяющая форми-
льную группировку по атому азота, занимающему 5-е положение в молекуле
кофермента.
С N-формилтетрагидрофолиевой кислоты осуществляется перенос форми-
льного остатка на Н2Ы-группу 5-фосфорибозилглицинамида. Получившийся
в результате этой реакции 5-фосфорибозилформилглицинамцд взаимодействует
с глутамином в присутствии сопряженно распадающейся АТФ и соответству-
ющей лигазы (см. два первых уравнения на схеме 3 (с. 242)).
Продуктом реакции является производное, где карбонильный кисло-
род пептидной связи замещен на иминогруппу. Затем в результате ряда
преобразований возникшего при этом соединения замыкается имидазоль-
ный цикл. Данный процесс также идет сопряженно с распадом АТФ и
ускоряется специфическим ферментом, который, будучи выделен и очи-
щен, характеризуется относительно невысокой устойчивостью к денату-
рации.
Весьма существенно, что реакция, в результате которой образуется имида-
зольная часть будущего пуринового остатка, практически необратима в от-
личие от подавляющего большинства остальных стадий рассматриваемого
процесса.
Вслед за этим на имидазольном цикле путем ряда ферментативных реак-
ций отмеченного выше типа из аспарагиновой кислоты, СО2 и формиата
строится пиримидиновое кольцо, т.е. в конце концов создается пуриновый
нуклеотид. Схема, включающая главные этапы этого синтеза, такова:
241
JHj-NH2 У CHj-NH
л « QH сС
\ Фосфорибозмглииии- f '-z \
О=Р—O-CH2 NH «мид-фории*травс- О™?-О—СН2 NH
ОН ‘ н н ' - фе?^------► ОН * '
Н
Н Н
но он
н
но
5-Ф осфорибозил-
глицииамид
АТФ
?н
'Г
он
। ^cHjNfi ; nh2
ний: ноос-сн-(сн2)5-с^
X—Hjl 2 N
12
н н
'КАДФ+
Н3РО4
он
0=Р-0-СН
I
он
н
он
h2n-c.
2
H
н
но он
он h2n-
О=Р—0“СН2^.о
он
н
н
(-С'н)
он
О=Р-О-СН.
+ атф+н2о
-«о
НООС-СН-(СН2)5<ХО1+АДФ + Н3РО4
NH2 (н<й
СО2
соон
сн
сн
Аоон
NH-C
<рн н
о=р-о-сн2
. он '
н
(JOOH соон
у<?ч <4
CH-NH-C^ АТФ+HC-jNH^
СООН с--N соон/
<рн 1;2'
0=Р-О-СН2
Ан
С---N
II II
h9n-c сн
NZ
H
н
он
HjNl-C^
N
н2о
н
но он
АДФ+Н3РО4
HN С-----N
н< 1 Л
<j>H % N
О=Р-О-СН2
он
но он
н
С--N
\
С—N
О
Инознн-б'-фосфат (ИМФ)
Схема 3. Механизм биосинтеза пуриновых нуклеотидов
Если подытожить, из каких соединений строится пуриновый цикл, то
окажется, что он возникает из очень простых веществ:
242 «
|_
। Из оксида |
Из формиата
Из приведенной схемы ясно, что 1-й атом азота пуринового цикла ведет
свое происхождение от аспарагиновой кислоты, 3-й и 9-й—от глутамина,
а 7-й—от г ицина. Что касается происхождения атомов углерода пуринового
кольца, то видно, что источниками их явились формиат (2-й и 8-й атомы
углерода), глицин (4-й и 5-й) и СО2 (6-й атом). Химические уравнения,
приведенные на схеме 3, показывают детали включения тех или иных атомов
N и С из состава перечисленных соединений в пуриновую часть нуклеотида
в процессе его биосинтеза.
Пиримидиновое кольцо пиримидин >вых нуклеотидов синтезируется в ор-
ганизме из аналогичных соединений: NH3, СО2 и аспарагиновой кислоты.
Таким образом, исходные вещества для биосинтеза пуриновых и пиримидиновых
оснований в организме исключительно доступны, всегда присутствуют в ием, так
как аммиак, формильная группа и оксид углерода (IV) образуются в процессе
деструкции разнообразных органических соединений или поступают в ор-
ганизм извне, а глутаминовая и аспарагиновая кислоты и их амиды представ-
ляют первичные в большом объеме синтезируемы* аминокислоты, что имеет
большое значение для обеспечения беспрепятственного синтеза этих важней-
ших для организма соединений.
Как следует из схемы 3, пуриновые нуклеотиды в результате последова-
тельных реакций наращивания пуринового цикла на рибозо-5-фосфате воз-
никают в виде инозин-5'-фосфата. Последний способен кисляться в ксан-
тозин-5'-фосфат. В результате аминиро ания первого синтезируется аденозин-
s' фосфат, а второго—гуанозин-5' фосфат. И тот и другой процессы ускоря-
ются специфическими ферментами (см. уравнение реакции на с. 244)..
В свою очередь, пуриновые нуклеозидмонофосфаты превращаются далее
в нуклебзидтрифосфаты. При этом гуанозин-5'-фосфат переходит в гуанозин-
5'-трифосфат по двойной обменной реакции с АТФ:
ГМФ+АТФ ------------------------------------------> ГДФ + АДФ
Нухлеозид-монофосфатгиназа
ГДФ + АТФ ----------------------------------------> ГТФ+АДФ
Нужлеозиддифосфапсиназа
При взаимодействии аденозин-5'-фосфата с АТФ возникает аденозиндифос-
форная кислота; эта реакция открыта А. В. Котельниковой в начале 60-х годов
нашего века:
Аденилатхиназа
АМФ+АТФ
2АДФ
243
Ксантозин-51- фосфат
Глутамин +
Фумаровая
кислота +
ГДФ + Н3РО4
Аспарагиновая
^/кислота + ГТФ
Аденилосукцинат-
синтетаза;
Дденилосукцинат-
Аденозин-5’- фосфат
Ксантозин - 5-фос-
<рат: L-глутамин
Глутамииоваи
кислота + ____
АМФ + Н4РаО7 । 'амидолигаза (обра-
Гуанозин- 5*-фосфат
Она служит субстратом при окислительном фосфорилировании, в результате
которого запасы АТФ в организме непрерывно пополняются, что обеспечивает
достаточное ее количество для превращения всех остальных нуклеозйдмоно-
фосфатов в нуклеозидтрифосфаты.
Синтез дезоксиаденозин-5'-трифосфата (дАТФ) и дезоксигуанозин-5'-три-
фосфата (дГТФ) осуществляется посредством реакции восстановления рибозы
по гидроксильной группе при втором углеродном атоме. В случае дГТФ
реакция восстановления идет на уровне ГДФ с последующим превращением
дГДФ в дГТФ:
Восстановление
ГДФ
дГДФ;
дГДФ 4-АТФ
Дезоксигуанилаткиназа
АДФ+дГТФ
Механизм реакции восстановления остатка рибозы в остаток дезоксирибо-
зы в случае пуриновых рибонуклеозиддифосфатов идентичен рассмотренному
ранее для пиримидиновых нуклеотидов. Сам же процесс восстановления сти-
мулируется дГТФ и дТТФ, но ингибируется дАТФ.
Как и в случае пиримидиновых нуклеотидов, конечные продукты биосин-
теза пуриновых нуклеотидов (ИМФ, АМФ, АДФ, АТФ, ГМФ, ГДФ и ГТФ)
угнетают действие амидофосфорибозилтрансферазы—фермента, ускоряющего
1-ю реакцию в цепи процессов, приводящих к новообразованию пуринового
244
кольца. Таким образом, осуществляется саморегуляция образования пурино-
вых нуклеотидов. Кроме того, при помощи специфического перекрестного
участия АТФ и ГТФ в реакциях, ведущих к превращению ИМФ в ГМФ и АМФ
соответственно, последние синтезируются в организме всегда в строго опреде-
ленном соотношении:
АМФ
АТФ
-----------------ГТФ
Активирование |
-----ИМФ--------- ГМФ
Активирование
Схема эта весьма показательна: она дает представление о принципах
саморегуляции, используемых в организме, когда обеспечивается синтез ряда
веществ в определенной пропорции по отношению друг к другу. Обеспечение
строго определенного соотношения нуклеозидтрифосфатов в организме имеет
исключительное значение, так как из них образуются нуклеиновые кислоты.
Сходные регуляторные процессы отмечены также в синтезе пиримидиновых
нуклеотидов.
Существует еще один путь синтеза пуриновых и пиримидиновых нукле-
отидов в организмах—из свободных пуриновых и пиримидиновых основа-
ний и 5-фосфорибозил-1-пирофосфата. Он не является, конечно, способом
синтеза данных нуклеотидов заново, так как при этом используются гото-
вые пуриновые и пиримидиновые циклы, освободившиеся при распаде
нуклеиновых кислот; он лишь сберегает пуриновые и пиримидиновые ос-
нования от их распада до соответствующих конечных продуктов. Эта
реакция ускоряется специфическими ферментами—фосфорибозилтрансфе-
разами:
Аденин
Аденин-фосфорибозил-
траисфераза
(j)H ОН
НО—Р—О—Р—ОН
Аденозин -51- фосфат
О О
Пирофосфат
Этот путь, в частности, ярко представлен в злокачественных опухолях.
5?45
БИОСИНТЕЗ ДНК И РНК
Выше было показано, что в живой природе осуществляются реакции,
обеспечивающие беспрепятственный синтез дезоксирибо- и рибонуклеозид-5'-
трифосфатов всех видов: дАТФ, дГТФ, дЦТФ и дТТФ, а также АТФ, ГТФ,
ЦТФ и УТФ. Характерно, что новообразование этих соединений регулируется
таким образом, что они возникают зависимо друг от друга в строго опреде-
ленной пропорции. Следовательно, в организме всегда обеспечено существова-
ние всех их одновременно в необходимых концентрациях. -Именно из нукле-
озидтрифосфатов и осуществляется биосинтез нуклеиновых кислот.
Первой характерной чертой специфического биосинтеза нуклеиновых кис-
лот является то, что он протекает только при наличии всех четырех видов
дезоксирибонуклеозидтрифосфатов (дАТФ, дГТФ, дЦТФ и дТТФ) в случае
синтеза ДНК или же в присутствии всех четырех видов рибонуклеозидтрифос-
фатов (АТФ, ГТФ, ЦТФ и УТФ) в случае синтеза РНК. Вторая—состоит в том,
что биосинтез идет при каталитическом воздействии ферментов—ДНК- или
РНК-полимераз. Третья весьма своеобразная черта—необходимость для его
осуществ ения затравки в виде уже готового п линуклеотида, который играет
роль матрицы. Последнее имеет принципиальное значение, так как именно
благодаря этому обеспечивается специфический биосинтез нуклеиновых кислот
со строго заданной последовательностью нуклеотидных остатков в молекуле.
Общая схема биосинтеза дезоксирибонуклеиновых кислот может быть
представлена в следующем виде:
идАТФ
+
лдГГФ
+
лдЦТФ
лдТГФ
Mg1*; ДНК-пшшмерма
(ДНК-иуклеотадил-
транефераза);
ДНК-матрица
дАМФ
дГМФ + 4лН4р2О7
дТГМФ Пирофосфат
ДТМФ
Она была впервые предложена А. Корнбергом (1958) на основании опытов,
проведенных с ДНК-полимеразой, выделенной из кишечной палочки. В том же
году С. Шпигельман тоже из кишечной палочки получил РНК-полимеразу (см. рис.
46). Позже она была изолирована из тканей млекопитающих и других животных.
РНК-полимераза in vitro обеспечивает синтез РНК по аналогичной схеме:
«АТФ ’ АМФ ’
+
лГ1Ф Mg^*; РНК-полимераза ГМФ +• 471Н4Р2О7
лЦТФ (РНК-нужлеотиднлтранс- । фераза); ДНК-затраша ЦМФ Пирофосфат
лУТФ УМФ п
Химизм реакции образования нуклеиновых кислот. Механизм происходящей
при биосинтезе нуклеиновых кислот химической реакции заключается в пере-
носе остатка нуклеозидмоиофосфата от нуклеозидтрифосфата на концевой
нуклеотидный остаток растущей в процессе синтеза полинуклеотидной цепи.
Перенос идет на место атома Н гидроксильной группы, стоящей при 3-м
углеродном атоме рибозы или дезоксирибозы концевого нуклеотида, и сопро-
246
вождается выделением пирофосфата; поэтому фермент, осуществляющий
ускорение реакции переноса нуклеотидного остатка, называют нуклеотидил-
трансферазой:
К полянуклеотидной цели
НО—Р== о
ОН
I
О= Р —
I
ОН
Нуклеотдил-
трансферап
К полинуклестидной цепи
На свободную гидроксильную группу при 3-м углеродном атоме рибозы
или дезоксирибозы вновь присоединенного нуклеотидного остатка может
поступить следующий нуклеотидный остаток, и, таким образом, будет осу-
ществляться ступенчатый (градуальный) биосинтез полинуклеотида путем нара-
щивания его с одного конца. Энергия для синтеза поступает за счет распада
макроэргической связи в трифосфатной группировке при освобождении пиро-
фосфата.
Механизм оспроизведения первичной структуры при биосинтезе нуклеи-
новых кислот. Процесс ступенчатого синтеза полинуклеотидной цепочки
247
Азотистые основания
|д~| Аденин □ Гуанин
[Т| Тимин |н| Цитозин
или Остатки дезоксирибозы
@ Остаток фосфорной кислоты
=~ Водородные связи
Направление наращивания
полидезоксирибоиуклеотидной цепи
и Q Фосфат
=-- Водородные связи
Рис. 83. Схема матри юго биосинтеза нуклеиновых кислот (Л) и удвоения молекул
ДНК (£) при их репликации (пояснение в тексте)
осуществляется на матрице, вдоль которой располагаются один за другим те или
иные дезоксирибо- или рибонуклеозидтрифосфаты, вступающие затем в реак-
цию конденсации с выделением пирофосфата (рис. 83, А). Порядок расположения
нуклеозидтрифосфатов вдоль полинуклеотидной матрицы задается чередовани-
ем нуклеотидных звеньев, присущим ДНК или иногда РНК, выполняющим
матричную функцию. При этом к определенному пуриновому или пиримидино-
вому основанию полинуклеотида (матрица) присоединяется комплементарное
пуриновое или пиримидиновое основание соответствующего нуклеоз дтрифос-
фата. В результате вновь синтезируемая полинуклеотидная цепь в целом будет
комплементарна полинуклеотидной цепи матрицы (рис. 83, А).
Так как формир вание нового полинуклеотида идет на полинуклеотидной
матрице при непрерывном замыка ии водородных связей между комплемен-
тарными пуриновыми и пиримидиновыми основаниями матрицы и нуклеозид-
трифосфатов, то условием функционирования этого механизма является одно-
цепочечная структура матрицы. Поэтому в случае биосинтеза молекул ДНК,
харак еризующихся б спиральной структурой, существенным моментом
248
в биосинтезе следует признать расхождение биспирального полидезоксирибо-
нуклеотида на одиночные полинуклеотидные цепи, на которых, собственно,
и осуществляется сборка комплементарных им полинуклеотидов. В итоге из
одной биспиральной молекулы ДНК образуются две биспиральные молекулы
ДНК, совершенно идентичные друг другу и исходной молекуле ДНК
(рис. 83, Б). Как качественный состав, так и количественное содержание нукле-
отидных остатков в матричной и вновь синтезированной на ней нуклеиновой
кислоте совпадают (табл. 21).
Таблица 21
Молярные соотношения оснований в составе ДНК, синтезированной
в присутствии разных матриц
Происхождение ДНК Характер образца Аденин Тимин Гуанин Цитозин А+Т г+ц А+Г т+ц
Кишечная палочка Затравка 1,00 0,97 0,98 1,05 0,97 0,98
Продукт 1,04 1,00 0,97 0,98 1,02 1,01
Зобная железа Затравка 1,14 1,05 0,90 0,85 1,25 1,05
Продукт 1,19 1,19 0,81 0,83 1,46 0,99
Бактериофаг Т2 Затравка 1,31 1,32 0,67 0,70 1,92 0,98
Продукт 1,33 1,29 0,69 0,70 1,90 1,01
Это дает основание рассматривать изложенный выше механизм биосинтеза
нуклеиновых кислот как гомологическую репликацию, т. е. бесконечное повто-
рение процесса удвоения числа молекул путем прямого, непосредственного
копирования их структуры.
• Описанный выше (см. рис. 83, Б) механизм удвоения молекул ДНК был
доказан в опытах, где исходную ДНК родительских клеток метили тритием
или 15N. В дочерних клетках после 1-го деления метку обнаруживали во всех
молекулах ДНК, но в меньшем количестве, так как она содержалась здесь
лишь в одной из полидезоксирибонуклеотидных спиралей ДНК. Такой способ
удвоения молекул ДНК называется полуконсервативным. После 2-го деления
половина молекул ДНК во вновь образованных «внучатых» клетках оказыва-
ется немеченой, так как гомологическая репликация в процессе 2-го деления
идет как на меченых, так и на немеченых одноцепочечных полидезоксирибону-
клеотидах, возникающих при расхождении биспиральных молекул ДНК, ме-
ченных только по одной из цепей.
Ферменты биосинтеза ДНК. В биосинтезе ДНК участвуют не только ДНК-
полимеразы, но и РНК-полимераза, эндонуклеаза, ДНК-лигаза и другие фер-
менты.
Впервые ДНК-полимераза была выделена из кишечной палочки А. Корн-
бергом и сотр. (1958). Поскольку в поел дующие годы из этой же бактерии
были выделены еще две полимеразы, она получила название полимеразы I.
В клетке кишечной палочки содержится около 400 молекул ДНК-полимера-
зы I—белка (М = 109 000), содержащего один атом Zn и представленного
одной полипептидной цепью. ДНК-полимераза I хорошо связывается с одно-
цепочечной ДНК или с зонами разрыва фосфодиэфирных связей в биспираль-
ной ДНК. Она обладает ясно выраженной ДНК-полимеразной активностью,
т. е. способна ускорять реакцию переноса дезоксирибонуклеотцдильных оста-
тков с дезоксирибонуклеозид-5'-трифосфатов на растущий конец полидезок-
сирибонуклеотида—3'-гидроксильную группу дезоксирибозы его концевого
нуклеозида.
249
Кроме того, ей присущ экзодезоксирибонуклеазная активность в отноше-
нии одноцепочечной ДНК в направлении 3'-»5', т. е. тоже со стороны кон-
цевого дезоксирибонуклеозида, а также в направлении 5'-»3' со стороны
начального дезоксирибонукле тида молекулы. В 1-м случае достигается уда-
ление с наращиваемого в процессе синтеза ДНК 3'-конца молекулы неправиль-
но (ошибочно) присоединенных нуклеотидных остатков, во 2-м—разрушение
прайм ра, необходимого для син еза фрагментов Оказаки (см. ниже). Послед-
нее очень важно. Так, у мутантов кишечной палочки, утративших эту функцию
ДНК-полимеразы I, накапливаются фрагменты Оказаки и биосинтез ДНК
приостанавливается. Сейчас полагают, что ДНК-полимераза I имеет большее
отношение к «созреванию» реплицирующейся ДНК, нежели непосредственно
к полимеразным процессам в репликационной вилке. Последняя функция
более присуща ДНК-полимеразе III.
ДНК-полимераза II присутствует в клетке кишечной палочки в количестве
около 40 молекул (М=90000). Она представлена одной полипептидной цепью.
ДНК-полимераза II плохо соединяется с одноцепочечными ДНК, но отлично
оккупирует точки разрыва в одной из цепей биспираль ой ДНК. Обладает
лишь 10%-ной ДНК-полимеразной активностью по сравнению с ДНК-поли-
меразой I.
Считают, что основной функцией ДНК-полимеразы II является достраива-
ние поврежденных участков в молекуле ДНК, т. е. репарация ДНК. Предпола-
гают, что ДНК-полимераза I in vivo несет эту же функциональную нагрузку.
Главной полимеразой кишечной палочки является ДНК-полимераза III.
Это—сложный, термолабильный белок (М = 300 000), состоящий из субъеди-
ниц а (140000), р (37000), у (52000), 8 (32000), е (25000) и 0 (10000);
полимеразную реакцию осуществляет каталитический кор из а-, в- и 0-
субъединиц, в котором главную роль играет а-субъединица; р-, у- i 8-субъеди-
ницы являются регуляторными и усиливают действие каталитического ядра
ДНК-полимеразы III. В клетке кишечной палочки всего 20 молекул этого
белка. Именно ДНК-п лимераза III ответственна у кишечной палочки за
процесс репликации ДНК. Она работает в ее клетке в комплексе с белковыми
факторами, тоже принимающими участие в репликации ДНК, полностью
обеспечивая главную ступень ее б осинтеза—элонгацию (продолжение) сбор-
ки дезоксиполирибонуклеотидной цепи.
Не менее сложен вопрос о ДНК-полимеразах эукариот. Их номенклатура
оказалась крайне запутанной, поэтому на международной конференции по
ДНК-полимеразам эукариот (США, 1975) она была унифицирована; услови-
лись называть их ДНК-полимеразы-а, -р и -у.
ДНК-полимераза-а впервые обнаружена у млекопитающих
(М = 150000 — 200000). Фермент, полученный из тимуса теленка, содержит
каталитическую (М = 118 000) и регуляторную (М = 54 000 — 64000) субъеди-
ницы. Это—кислый белок (р! 5,5), сильно ингибируемый агентами, блокиру-
ющими HS-группы. Ее полимеразная активность максимальна при pH 7,2
в присутствии биспиральной ДНК, но не АТ-кополимера. Хотя она и об-
на уживается в цитоплазме, ее истинная локализация—в ядре, где сосредото-
чено ~ 25 000 ее молекул.
ДНК-полимераза-p (М=45000) характеризуется устойчивостью к действию
агентов, блокирующих HS-группы, хорошо выраж иной способностью уско-
рять при pH 8,4 реакцию копирования и ДНК, и АТ-кополимера, является
основным белком (р! 9,2). В клетке содержится в количестве ~8000 молекул,
локализованных в ядре и участвующих в репарации ДНК.
ДНК-полимераза-у—кислый белок (pl 5,8) с М = 180000, тетрамер из иден-
тичных субъединиц; ее полимеразная активность, зависит от целостности
250
HS-групп. При pH 8,5 энергично ускоряет репликацию АТ-кополимера и гора-
здо менее активна с природной ДНК. В клетке присутствует в количестве
~ 1000 молекул, локализована в митохондриях, где обеспечивает репликацию
митохондриальной ДНК.
Ни одна из ДНК-полимераз эукариот, в отличие от таковых прокариот,
не обладает нуклеазной активностью. Их биологические функции выяснены
пока недостаточно. Механизм действия ДНК-полимераз представлен на
рис. 83.
Кроме ДНК-полимераз, обладающих у прокариот к тому же нуклеазной
активностью, в биосинтезе ДНК принимает участие ДНК-лигаза. Она открыта
в 1967 г. одновременно в пяти лабораториях. Это белок с М = 96000. В клетке
кишечной палочки, например, насчитывается около 2000 молекул ДНК-лига-
зы. Ее функция состоит в обеспечении каталитического ускорения реакции
образования фосфодиэфирной связи между 3'- и 5'-концами цепей ДНК,
сближенных на расстояние одного нуклеотидного звена и закрепленных на
комплементарной им, но непрерывной другой цепи ДНК. Такая ситуация
возникает во многих случаях: при устранении разрывов в одной из цепей ДНК,
вызванных облучением, действием нуклеаз и т.п.; при соединении новооб-
разованного при репарации или в процессе нормальной репликации фрагмента
с предшествующими или вновь созданными при этом фрагментами ДНК; при
ковалентном замыкании линейной молекулы ДНК в кольцевую структуру
и т. п. Механизм ДНК-ли азной реакции представлен на рис. 84. ДНК-лигаза
нашла применение в работах по синтезу генов и их фрагментов.
Белковые факторы, необходимые для биосинтеза ДНК. Наряду с перечис-
ленными выше ферментами в биосинтезе ДНК принимают участие белковые
факторы, каждый из которых выполняет собственную функцию.
ДНК-связывающий белок впервые выделен В. Альбертсом и Л. Фреем
(1970) из лизатов кишечной палочки, зараженной фагом Т4. Вначале он был
назван белком 32—по номеру гена в хромосоме фага Т4, ответственного за
биосинтез этого белка. Белок характеризуется молекулярной массой 35000
(собственный ДНК-связывающий белок кишечной палочки имеет М=22000),
высокой степенью асимметрии молекулы (Z>/a=4), преобладанием дикарбоно-
вых аминокислот над диаминокислотами. Белок представлен одной полипеп-
тидной цепью и обладает ярко выраженной способностью связываться с одно-
цепочечной ДНК или дефектными участками биспиральной ДНК, резко осла-
бляя взаимодействие полидезоксирибонуклеотидных цепей в ее молекуле, что
выражается в понижении температуры плавления ДНК на 30—40°.
ДНК-связывающий белок активирует ДНК-полимеразы II и III. Сейчас он
выделен и из клеток эукариот. В противоположность ДНК-связывающему
белку существует и выделен белок, биосинтез которого у кишечной палочки
кодируется геном № 5 фаговой хромосомы. Этот белок полностью блокирует
матричную активность ДНК, т. е. является антагонистом ДНК-связывающего
белка.
ДНК-раскручивающий белок описан Дж. Янгом (1971) и назван белком го.
Предполагают, что он обладает нуклеазной активностью и, присоединяясь
к ДНК, разрывает фосфодиэфирную связь одной из ее цепей, вследствие чего
обеспечивается свободное вращение вокруг фосфодиэфирной связи другой
цепи и раскручивание суперспиральной молекулы ДНК. Его антагонист—
ДНК-закручивающий белок вызывает суперен рализацию ДНК, ему присущи
ДНК-зависимая АТФазная активность и механохимические свойства.
При изучении биосинтеза ДНК у кишечной палочки и ее мутантов тести-
ровано еще несколько белков, участвующих в репликации ДНК (рис. 85),
и функции некоторых из них уже выяснены. Среди них особый интерес
251
-ДНК-лигаза;
Дезоксирибоза (вертикальная жирная черта)
с гидроксильными группами (косые тонкие
5' черточки при 3' и 5'-углеродных атомах)
А- Аденин
Г- Гуанин
Ц- Цитозин
Г- Тимин
Рис. 84. Механизм ДНК-лигазной реакции
Реакция идет в три стадии: I—ДНК-лигаза взаимодействует с АТФ (или НАД+) с образованием ферментадепилатного производ-
ного по e-NH2-группе остатка лизина полипептидной цепи фермента и выделением пирофосфата (или иикотинамидрибозофос-
фата); II—остаток адениловой кислоты с крайне активной в химическом отношении аденилатлигазы перебрасывается на
свободную фосфатную группу 5-углеродного атома рибозы концевого нуклеотида; ДНК-лигаза при этом высвобождается;
III—между сближенными активированной аденилатом 5'-фосфатной группой и З'-гидроксильной группой фрагментов цепи ДНК
происходит спонтанное взаимодействие с образованием фосфодиэфирной связи с выделением АМФ
252
представляет хеликаза—фермент, расплетающий двойную спираль ДНК. При
молекулярной массе около 75 000 Да его г олипептидная .цепь из 650 амино-
кислотных остатков (первичная структура выяснена) организована в виде двух
крупных доменов из 286 и 364 аминокислот (в каждом по 2 субдомена)
и пространственно организована так, что может охватывать биспиральную
молекулу ДНК, расплетая ее за счет энергии р спадающейся АТФ.
Этапы биосинтеза ДНК. Исходя из приведенных выше данных о ферментах
и белковых факторах, принимающих участие в биосинтезе ДНК, постулирова-
на схема этого процесса. Она базируется на представлении о существовании
репликативного комплекса ферментов и белковых факторов, необходимых для
осуществления биосинтеза ДНК, локализованных в репликативной вилке, т. е.
той зоне ДНК, где происходит распаривание биспирального полидезоксири-
бонуклеотида и сборка дочерних, новообразуемых цепей ДНК (рис. 85); В со-
ответствии с этой схемой биосинтез ДНК распадается на 3 этапа: инициацию,
элонгацию и терминацию. Следует подчеркнуть, что многие вопросы, каса-
ющиеся механизма биосинтеза ДНК, еще не до конца выяснены и рассмат-
риваемая модель является в определенной мере условной.
Инициация биосинтеза ДНК, понимаемая в узком смысле как начало био-
синтеза дочерних полидезоксирибонуклеотидных цепей на материнских, сво-
дится к созданию на материнской цепи ДНК затравочного олигонуклеотида
со свободной гидроксильной группой на З'-конце этого олигонуклеотида. Без
него не работает ни одна из ДНК-полимераз. Этот короткий (несколько
десятков звеньев) олигорибонуклеотид, заканчивающийся пиримидиновым
нуклеозидом, является праймером, т. е. предшественником будущей цепи
ДНК, и синтезируется при участии фермента типа РНК-полимеразы, получи-
вшего название праймазы (М = 60000, 100 молекул в клетке кишечной палоч-
ки). Только после его создания в синтез включается ДНК-полимераза III, при
посредстве которой синтезируется далее на материнской цепи ДНК дочерняя
цепь ДНК. Впоследствии ковалентно присоединенный к этой новообразован-
ной цепи ДНК фрагмент РНК (праймер) «вырезается» при помощи нуклеаз
и одноцепочечная брешь застраивается олигодезоксинуклеотидным фрагмен-
том тоже при посредстве ДНК-полимеразной (репаразной) реакции (рис. 85).
Инициация биосинтеза ДНК, понимаемая в более широком смысле как
комплекс процессов, приводящих к старту в биосинтезе ДНК, сводится к воз-
никновению репликативного комплекса ферментов и белковых факторов
и формированию репликативной вилки (рис. 85). Она включает присоединение
к биспиральной ДНК (к одноцепочечному ее фрагменту в момент распарива-
ния биспиральной структуры в результате флуктуаций) ДНК-связывающего
белка, ДНК-раскручивающего белка, ДНК-полимеразного комплекса, ДНК-
зависимой РНК-полимеразы (праймазы), а также комплекса белковых факто-
ров, называемого праймосомой и обеспечивающего продвижение репликатив-
ной вилки (рис. 85), ее пространственную организацию и функционирование.
Элонгация биосинтеза ДНК складывается по меньшей мере из двух процес-
сов: репликации участков материнских цепей ДНК и соединения друг с другом
синтезированных при репликации фрагментов новообразуемых цепей ДНК.
Первый осуществляется при посредстве ДНК-полимеразной реакции (см.
рис. 83), идущей со скоростью (в н. о./с) в среднем около 1000 у бактерий,
100—у животных и 10—у растений. Своеобразие его состоит в том, что
репликация идет не непрерывно, а фрагментарно: за один раз у прокариот
синтезируется полидезоксирибонуклеотид с коэффициентом седиментации 8—
10S, т.е. молекулярной массой в несколько сотен тысяч дальтон (примерно из
1000 н. о.). У эукариот фрагменты Оказаки короче, около 200 н. о., что сопо-
ставимо с длиной нуклеосомной ДНК, в чем, по-видимому, есть определенный
253
254
смысл. Эти фрагменты получили название фрагментов Оказаки в честь японского
биохимика, впервые в 1967 г. на VII Международном биохимическом конгрессе
в Токио предложившего схему биосинтеза ДНК, в которой были преодолены
трудности, связанные с антипараллельностью цепей ДНК в ее биспиральной
молекуле. Дело в том, что ДНК-полимер зы способны наращивать полинуклео-
тидную цепь в направлении только 5'->3', т. е. путем переноса нуклеотид ого
остатка с дезоксирибонуклеозидтрифосфата на гидроксильную группу З'-угле-
родного атома ра тущей полидезоксирибонуклеотидной цепи. Но так как цепи
материнской ДНК антипараллельны, т. е. направление чередования нукле твд-
ных звеньев в них противоположно (см. рис. 85), то один и тот же фермент, т. е.
ДНК-полимераза, не может обеспечить сборку дочерних цепей одноврем нно
в направлениях 5'-»3' и 3'->5' в соответствии с перемещением репликационной
вилки при расплетании биспиральной молекулы ДНК. Т. Оказаки предположил,
что ДНК-полимераза ведет синтез ДНК челночным способом: сначала передви-
гаясь вперед в направлении 5'->3' по одной цепи, а затем в том же направлении
5'-»3', но передвигаясь в обратную сторону по другой цепи. Те фрагменты
Оказаки, у которых олигорибонуклеотидные праймеры заменены при участии
ДНК-пол меразы I на дезоксирйбонуклеотидные фрагменты, при посредст <
ДНК-лигазы объединяются вместе (см. рис. 84 и 85) и дают начало дочерней цепи
ДНК. Есть и другой взгляд на механизм преодоления антипаралл льности цепей
ДНК; суть его ясна из рассмотрения рис. 85, Б и легенды к нему.
Вопрос, син езируются ли на одной из цепей родительской ДНК фрагм нты
Оказаки, т. е. вдет ли на ней прерывистый биосинтез цепи дочерней ДНК, а на
другой—непрерывный биосинтез депи дочерней ДНК, остается дискуссион-
ным. Видимо, как правило, происходит прерывистый синтез на обеих цепях
родительской ДНК, а сочетание прерывистого и непрерывного является ис-
ключением, вполне доказанным для репликации некоторых фаговых ДНК.
Решение проблемы одно- или двунаправленной репликац и однозначно: от
точки инициации репликационные вилки распространяются по биспиральной
молекуле ДНК в двух противоположных направлениях.
По поводу терминации биосинтеза ДНК мнения разноречивы. Предполага-
ют, что прекращение репликации ДНК программируется особой нуклеотид-
ной последовательностью, в том числе в виде специальных палиндромов
(«кроличьих ушей») на конце хромосомы. Само собой разумеется, что репли-
кация прекращается, когда встречаются две репликационные вилки при удвое-
нии как кольцевых, так и линейных ДНК. Наконец, возможно достраивание
ведомой цепи за счет праймирования З'-конца материнской цепи с участием
специфического белка, находящ ;гося на ее конце.
Точность репликации ДНК весьма велика—одна ошибка на 1010 нукле-
отидилтрансферазных р акций. Но даже если ошибка допущена, она может
быть исправлена в ходе репарационных процессов.
Биосинтез ДНК на РНК в качестве матрицы. Кроме рассмотр иного выше
механизма биосинтеза ДНК на полидезоксирибонуклеотидной матрице в по-
следние годы открыта система биосинтеза ДНК на РНК при посредстве
Рис. 85. Схема биосинтеза фаговой ДНК в клетке кишечной палочки:
А—плоскостная модель, согласно которой синтезу затравочного олигорибонуклеотида (праймера) под действием ДНК-завнси-
мой РНК-полимсразы (праймазы) способствует белковый комплекс, называемый праймосомой (и—25 кДа, л’—75 1Д», я*—
11 кДа, /—80 кДа, dnaB—300 кДа, dnaC—29 кДа), движущийся в направлении,, противоположном элонгации, и создающий
Новые центры синтеза праймера- Раскручивание биспиральной ДНК па одноцепочечные полндезоксирибоиуклеотвды осуществля-
ется хеликазой (от англ, helix—спираль), а релаксация суперспиральной ДНК—гиразой (от англ, gyration—кругловрашательнов
движение). Б—пространственная модель, демонстрирующая преодоление аятипараллельиости цепей ДНК за счет возникновения
петля ДНК, где чередование фосфоридиэфирных связей на ее восходящем отрезке изменяется на обратное и не препятствует
ДНК-полимеразе вести синтез фрагмента Оказаки на ведомой цепи родительской ДНК в том же направлении, что и на ведущей
цепи. Остальные пояснения в тексте
255
фермента, названного обратной транскриптазой или ревергазой (его называют
также РНК-зависимой ДНК-полимеразой). Этот фермент был обнаружен
в 1970 г. независимо друг от друга Д. Балтимором в составе вируса Раушера
лейкемии мышей и Г. Теминым и С. Мизутани в составе вируса саркомы Рауса.
Ревертаза активируется Мп2+ сильнее, чем Mg2+, и содержит Zn2 . Синтезиро-
ванная ДНК имеет небольшую молекулярную массу и характеризуется конста-
нтами седиментации от 2S до 7S. Как и обычные ДНК-зависимые ДНК-
полимеразы, ревертаза нуждается в праймере, роль которого может играть
тРНК, в чем проявляется регулирующая роль последней в обмене веществ. Ее
молекулярная масса колеблется от 70 до 180 кДа в зависимости от объекта, из
которого она выделена; фермент, как правило, состоит из двух субъединиц.
Значение открытия ревертазы состоит прежде всего в том, что выявлен
дополнительный механизм биосинтеза ДНК, который, как предполагают,
может использоваться для амплификации генов. Исследование ревертазной
реакции крайне существенно для изучения перерождения нормальной клетки
в раковую. Ревертаза нашла применение в молекулярной биологии для син-
теза генов и фрагментов генов и, как следствие этого, в генетической ин-
женерии. Новой областью ее использования является массовая расшифровка
через кДНК первичной структуры РНК и белков.
Учитывая исключительную важность работ по исследованию обратной
транскрипции, в нашей стране начиная с 1972 г. осуществляют проект «Ревер-
таза», в разработке которого принимали участие академии наук СССР и стран
СЭВ. В результате проведенных исследований удалось создать универсальную
систему обратной транскрипции, позволяющую вести синтез ДНК с любой
заданной точки в молекуле РНК при помощи синтетического праймера (ок-
тадезоксирибонуклеотид заданного строения), комплементарного межгенной
зоне одной из фаговых РНК. Кроме того, путем ревертазной реакции получена
ДНК на глобиновой мРНК голубя и гигантских ядерных РНК. Действием
ревертазы in vivo в ДНК введена онкогенная информация, успешно изучается
структура РНК-содержащих опухолеродных вирусов, синтезированы потенци-
альные ингибиторы ревертазы.
Продолжаются интенсивные работы по изучению свойств ревертаз, выде-
ленных из различных опухолеродных вирусов (в том числе человека), синтезу
ингибиторов ревертаз, не действующих на клеточные ДНК-полимеразы, полу-
чению ДНК-транскриптов для работ по генетической инженерии, исследова-
нию интеграции ДНК, синтезированной на вирусной РНК, в геном клетки.
За этот цикл работ группе советских исследователей и ученых стран СЭВ
в 1979 г. присуждена 1осударственная премия СССР. Начиная с 1980 г. проект
«Ревертаза» преобразован в проект «Ревертаза-онкоген».
Биосинтез РНК. Биосинтез РНК осуществляется на ДНК в качестве матри-
цы. С точки зрения передачи информации в живых системах он характеризует-
ся как процесс транскрипции, т.е. переписывания информации, содержащейся
в последовательности нуклеотидных остатков в ДНК-матрице, в последователь-
ность нуклеотидных звеньев в молекуле новообразуемой РНК. Та часть молеку-
лы ДНК, которая копируется в процессе биосинтеза РНК на ней, носит
название транскриптона. Последний содержит информативную и неинформа-
тивную зоны. Транскрибирование информативной зоны приводит к образова-
нию той части новообразуемой РНК, которая впоследствии дает начало РНК
с определенной функциональной активностью. При копировании неинформа-
тивной зоны продуцируется та часть насцентной РНК, которая впоследствии,
в процессе созревания вновь синтезированной РНК, разрушается. Соотноше-
ние информативной и неинформативной частей в транскриптонах прокариот
и эукариот резко различно и составляет в среднем 9:1 и 1:9 соответственно.
256
Неинформативная зона транскриптона содержит регуляторные последова-
тельности, с которыми взаимодействуют многочисленные (их открыто не-
сколько десятков) регуляторные белковые факторы, ускоряющие или замедля-
ющие процесс транскрипции. Они контактируют с определ иными последова-
тельностями нуклеотидных остатков в ДНК присущими им ДНК-
связывающими доменами. Так, например, транскрипционный фактор ША из
ооцитов шпорцевой лягушки содержит РР'ос—надвторичную структуру
с атомом Zn в ее составе (см. рис. 41), так называемый цинковый палец,
обеспечивающий прикрепление к ДНК.
У эукариот основная масса РНК синтезируется в ядре клетки, где локали-
зация биосинтеза различных видов РНК также различается (рис. 86).
Биосинтез РНК осуществляется при посредстве РНК-полимераз, которые
в основном являются ДНК-зависимыми и лишь в некоторых случаях РНК-
зависимыми ферментами. Механизм их действия во многом совпадает с тако-
Рис. 86. Локализация биосинтеза
различных видов РНК в ядре
клетки
Все виды РНК синтезируются на ядерной ДНК
в качестве матрицы. В ядрышке сосредоточена
рДНК, являющаяся матрицей в биосинтезе
предшественников рибосомальных нуклеино-
вых кислот (45S РНК), при созревании которых
возникают 18S и 28S рРНК (короткими стрелка-
ми подчеркивается многоступенчатость процес-
са превращения предшественников в функцио-
нальные РНК). рДНК в определенные периоды
клеточного цикла многократно реплицируется,
что обеспечивает наработку большего количест-
ва рДНК (на рисунке это отмечено трехкра-
тным повторением зоны рДНК в ядрышке). Все
новообразованные РНК выходят в виде нуклео-
протеинов через поры ядерной мембраны,
в цитоплазму, где рРНК используется для об-
разования 40S и 60S субчастиц рибосом,
а мРНК, 5S рРНК в тРНК—при превращении
неактивных рибосом в активные, транслирую-
щие рибосомы
рибосома
вым у ДНК-полимераз: они ускоряют биосинтез РНК из АТФ, ГТФ, ЦТФ
и УТФ тоже путем нуклеотидилтрансферазной реакции. Обеспечение уникаль-
ной первичной структуры новообразуемой РНК происходит за счет спарива-
ния соответствующего рибонуклеозидтрифосфата с комплементарным ему
нуклеотидным остатком матричного полинуклеотида в точке роста молекулы
РНК. Но есть существенные различия: РНК-полимеразы не нуждаются в прай-
мере, обладают способностью избирательно взаимодействовать с зоной про-
мотора ДНК (точка, с которой начинается биосинтез РНК), являются слож-
ными молекулами, построенными из нескольких субъединиц, и др.
Наиболее изучены РНК-полимеразы прокариот, в частности кишечной
палочки. РНК-полимераза кишечной палочки, представленная белком
с М=487 000, состоит из пяти субъединиц: двух а, одной р, одной р' и одной
о. Форма субъединиц, их молекулярные массы и вероятный вариант ком-
поновки показаны на рис. 46.
В целом же у прокариот (изучено несколько десятков видов) молекулярные
массы РНК-полимераз колеблются в довольно широких пределах (а: 36—45
кДа, р: 86—160 кДа, р': 96—175 кДа и с-фактор: 44—107 кДа). Прокари-
отические РНК-полимеразы синтезируют все виды РНК—рибосомальные,
матричные, транспортные и низкомолекулярные. В отличие от них эукари-
отические РНК-полимеразы типа I (М=473 кДа, 6 субъединиц) транскрибиру-
ют гены рРНК; типа II (М = 882 кДа, 10 ±2 субъединицы)—гены, кодирующие
9—3502
257
белки; типа III—гены малых стабильных РНК (тРНК, 5S рРНК) (М ~653 к Да,
9 субъ диниц). Каждая из субъедин ц РНК-полимераз выполняет свою функ-
цию: одни из них узнаю- нуклео идную последовательность в зоне промото-
ра, обогащенную тимидиловыми и адениловыми дезоксирибо-
нуклеотидами (ТАТААТ—блок Прибнова у прокариот, ТАТА—блок Гольдбер-
га—Хогнесса у эукариот), другие обеспечивают нуклеотидилтра с еразную
реакцию при наращива ии цепи РНК, третьи—взаимодействуют с многочис-
ленными транскрипционными белковыми факторами (их насчитывают уже
несколько десятков), регулирующими биосинтез соответствующих РНК путем
контакта с энхансерными (усиливающими) и сайленсерными (ослабляющими)
полинуклеотидными зонами ДНК или путем аткивирования протеинкиназ,
фосфорилирующих как белковый кор РНК-полимераз, так и регуляторные
белки. Транскрипционный процесс завершается либо факторнезависимым спо-
собом (в составе синтезируемой РНК поблизости от ее конца появляются две
короткие последовательности, дающие шпильку, что выводит РНК из транс-
крипционной вилки), либо при посредстве белкового фактора р (М=46094 Да,
первичная структура выяснена, существует в виде гексамера в количестве 1000
молекул на клетку), который присоединяется к транскрипционному центру,
распаривает ДНК—РНКовый комплекс и высвобождает РНК. Для РНК-
полимеразы I, осуществляющей транскрипцию рРНК, сигналом для ее завер-
шения служит присоединение белкового фактора (М=130 кДа) к 18-звенному
терминатору в составе копируемого гена рДНК млекопитающих.
РНК-полимераза преимущественно связывается с тяжелой цепью ДНК и об-
ладает способностью расплетать биспиральную структуру ДНК на ее ограничен-
ном протяжении. Переме ение РНК-полимеразы вдоль цепи РНК происходит
под воздействием присоединяемого в процессе синтеза РНК нукле тида. Биосин-
тез РНК регулируется при посредстве механизма, показанного на рис. 87.
Синтезированные РНК представляют собой РНК-предшественники функ-
ционально-активных рибонуклеиновых кислот, так как наряду с информатив-
ными зонами содержат неинформативные участки. Впервые это явление было
открыто в начале 60-х годов Г. П. Георгиевым и О. П. Самариной, а сами эти
гигантские транскрипты названы ими дРНК (т.е. ДНК-подобными РНК).
В даль ейшем происходит процесс их видоизменения, сопровождающийся
разрушением неинформативных зон. Он носит название процессинга или созре-
вания РНК. При процессинге РНК метилируется (с помощью РНК-метилаз),
в результате чего она обогащается минорными основаниями (особенно в слу-
чае тРНК); в случае мРНК к ней присоединяются кэп и полиаденилатный
фрагмент. Процессинг идет при участии разнообразных ферментов. Именно
при изучении процессинга предшественников РНК было открыто удивитель-
ное явление: способность некоторых низкомолекулярных РНК осуществлять
каталитическую функцию.
Наиболее отчетливо это выявилось при работе с РНКовой компонентой
рибонуклеазы Р—фермента, катализирующего процессинг 5'-концов предшест-
венников тРНК: будучи отделена от белковой, части, РНК оказалась способной
одна ускорять реакцию распада фосфодиэ ирной связи по гуаниловому остатку
при переходе предшественника в зрелую тРНК (рис. 88, А), вследствие чего
получила название рибозима. М ханизм каталитического акта пока не ясен, но
зафиксировано образование рибозим-субстратного комплекса и полное подчи-
нение процесса ферментативной кинетике. Возможен и аутосплайсинг предшест-
венников (рис. 88, Б), где тоже решающую роль играют гуанозиновые остатки.
В последние годы предприняты попытки по замене рибонуклеотидов в составе
рибозимов на дезоксирибонуклеотиды, что привело к созданию химерных
рибозимов (нуклеоз имов) с повышенной стабильностью и активностью, а также
258
по синтезу рибозимов и нукле-
озимов с меньшим числом
нуклеотидных звеньев в их
составе (минизимов). Все это
позволило создать концеп-
цию, согласно которой имен-
но рибонуклеиновые кислоты
занимали центральное место
в химических процессах, свя-
занных с происхождением
жизни на Земле, лишь потом
к ним добавились ДНК и бел-
ки, и постепенно возникла бо-
лее прогрессивная система
хранения и передачи инфор-
мации при новообразовании
биополимеров, обеспечиваю-
щая передачу из поколения
в поколение структурных осо-
бенностей и функций биологи-
ческих макромолекул.
РНК-
Белок- Белок- оелок-
актибатор репрессор ^^фактор)
Рис. 87. Регуляция биосинтеза РНК
Биосинтез РНК начинается с зоны молекулы ДНК, называемой промото-
ром и «узнаваемой» о-фактором; между промотором и информативной
последовательностью нуклеотидных остатков (цистрон) в ДНК распола-
гается зона оператора. Если оиа свободна, т. е. не занята белком-репрес-
сором (его структура детально изучена), РНК-полимеразная реакция осу-
ществляется беспрепятственно. Сначала транскрибируется зона операто-
ра, затем зона цистрона, содержащего информацию о последовательности
аминокислотных остатков в одном или нескольких метаболически связан-
ных белках. Если промотор блокирован белком-репрессором, синтез РНК
не происходит; зона связывания (энхансер) белка-активатора (тоже полно-
стью охарактеризован) может располагаться ие только вблизи, но н в от-
далении от зоны промотора
Выяснилось далее, что аналогичной способностью обладают многие малые
ядерные РНК, существующие в виде комплексов с белками. Все они содержат
много остатков уридиловой кислоты и получили поэтому шифр Ul, U2 и т. д.
РНК. Их изучено более десятка, и большинство из них принимают участие
в каталитическом ускорении созревания мРНК и рРНК, обеспечивая выщепление
неинформативных фрагментов из их предшественников и соединение (сплайсирова-
ние) биологически значимых последовательностей с образованием зрелых молекул.
Они же, по-видимому, ведут альтернативный сплайсинг пред шественников мРНК,
при котором из одной пре-мРНК образуется несколько функционально значимых
мРНК за счет разного расположения информативных зон в их составе. Внутриядер-
ные комплексы низкомолекулярных ядерных РНК с белковыми факторами,
необходимыми для их сборки и функционирования, получили название сплайсосом;
именно они осуществляют специфический сплайсинг при созревании пре-мРНК
и пре-рРНК. Сейчас на первый план при оценке функций малых ядерных РНК
выступает их ведущая роль в регуляции обмена веществ в клетке: открыт новый
класс их, представители которого комплементарны коротким диспергированным
повторам в ДНК, вследствие чего они могут контролировать репликацию ДНК
и ее транскрипцию в качестве транс-регуляторных элементов, взаимодействующих
с энхансерами и сайленсерами промоторной зоны.
Осуществлены подсчеты скорости синтеза РНК. Так, в случае биосинтеза
в клетках печени крысы мРНК для сывороточного альбумина она равна
90—100 н. о./с. Исходя из того, что в печеночной клетке содержится около
40000 молекул этой мРНК, а время ее жизни составляет 3 ч, при указанной
скорости биосинтеза достаточно 3—4 активных генов для того, чтобы полно-
стью обеспечить наработку ее необходимого количества.
Изучение закономерностей биосинтеза нуклеиновых кислот привело к от-
крытию важнейшего механизма воспроизведения специфичности при их ново-
образовании. Механизм этот сводится к взаимодействию комплемент рных
оснований полинуклеотидной матрицы (на которой идет специфический син-
тез) и нуклеозидтрифосфатов, из которых указанный синтез осуществляется.
Таким образом, принцип комплементарности оказался ведущим не только
в строении нуклеиновых кислот, но и в их биосинтезе. Как будет показано
259
A
"рГр A —^-Уон
15-ти членный
олигонуклеотид
Циклический
олигонуклеотид
Рис. 88. Процессинг пре-РНК:
А—пре-тРНК при участии рибозима; Б—пре-рРНК: путем аутосплайсинта (сплошная линия—функцио-
нальная зона; волнистая—неинформативный фрагмент; рГм-гуанозии). Остальные пояснения в тексте
ниже, этот принцип имеет огромное значение при специфическом воспроиз-
ведении первичной структуры белковых молекул. Взаимодействие комплемен-
тарных структур, обеспечивающее воспроизведение специфического строения
макромолекул при их биосинтезе,— один из важнейших законов, сформулиро-
ванных в биохимической науке за последние годы. Вместе с тем матричный,
комплементарный механизм биосинтеза макромолекул, с полным правом, мож-
но отнести к элементарным, фундаментальным свойствам живой материи.
Матричный принцип биосинтеза—это специфика химизма живого. Так, моле-
кулярная биология и биохимия пришли к крупнейшему обобщению, которое
конкретно характеризует специфику живого, отличие живого от неживого на
молекулярном уровне.
ГЛАВА VII
ОБМЕН БЕЛКОВ
ЗНАЧЕНИЕ БЕЛКОВОГО ОБМЕНА
Белковый обмен—стержневой процесс среди многообразных превращений
веществ, свойственных живой материи. С точки зрения материалистической
диалектики само явление жизни в определенной степени представляет собой
«способ существования белковых тел», которые непрерывно самообновляют-
ся, непрерывно строят себя из веществ окружающей среды. Поэтому в живой
природе весь ход обмена веществ подчинен главной цели—воспроизведению
белковых тел.
Все другие виды обмена—углеводный, липидный, нуклеиновый, минераль-
ный и пр.— обслуживают обмен белков, специфический биосинтез белка. Одни
группы процессов, как, например, углеводный обмен, являются в основном
источником углеродных цепей в биосинтезе аминокислот—исходных соедине-
ний для новообразования белков. Другие, как, например, обмен жиров, глав-
ным образом поставляют вещества, при окислении которых в макроэргических
связях АТФ запасается энергия, необходимая для образования пептидных
связей. Третьи (обмен нуклеиновых кислот) обеспечивают хранение и передачу
информации о расположении аминокислотных остатков во вновь синтезируе-
мых белковых молекулах, обслуживая специфическое воспроизведение уни-
кальной структуры протеинов. Четвертые (минеральный обмен) способствуют
становлению или распаду ферментных систем, при посредстве которых идет
синтез белка, или созданию и разрушению субклеточных частиц и структур, на
которых этот синтез осуществляется. Таким образом, многочисленные, разно-
образные и часто очень сложные процессы превращения веществ и трансформа-
ции энергии в живом веществе обслуживают главным образом обмен белковых
тел. Последний, в свою очередь, так регулирует упомянутые превращения, что
создает оптимальные условия для своего собственного осуществления.
Важнейший вопрос при изучении белкового обмена—выяснение механизма
специфического воспроизведения первичной структуры белковых веществ в про-
цессе их биосинтеза. Как было отмечено ранее, первичная структура белка
предопределяет характер третичной структуры белковых молекул, с которой
связана та или иная функциональная их активность. Именно эта сторона
белкового обмена имеет исключительное значение для жизнедеятельности
организмов; именно она создает специфику обмена веществ у организмов
разной степени сложности или уровня развития, именно со специфичностью
белковых тел связана в первую очередь видовая специфичность организмов.
Таким образом, специфическое воспроизведение белковых тел в природе пред-
ставляет основу, фундамент всего процесса обмена веществ, характерного для
того или иного растительного или животного вида.
Проникновение в тончайшие детали белкового обмена, особенно выяс-
нение закономерностей новообразования специфических белков (ферменты,
261
гормоны белковой и пептидной природы и т. п.), дает в руки исследователя
ключ к разгадке многих тайн природы, позволяет наметить новые пути
управления обменом веществ, а следовательно, найти способы управления
развитием организмов, их наследственностью, патологическими процессами
в них и т. п. Все это свидетельствует о том, что нет в биохимии и биологии
в целом более важной и более сложной проблемы, чем проблема обмена белков.
РАСПАД БЕЛКОВ И АМИНОКИСЛОТ
Пути распада белков. Главный, но возможно не единственный путь распада
белков в организме—гидролиз. Гидролитический распад белков протекает
в любой клетке организма в основном в специальных субклеточных элемен-
тах—лизосомах, где сосредоточены гидролитические ферменты и где осуще-
ствляется деструкция высокомолекулярных веществ до низкомолекулярных
метаболитов. Вместе с тем определенная часть ферментов, ускоряющих рас-
пад белков, есть в цитозоле клетки а некоторые из них секретируются,
обеспечивая внеклеточное переваривание белков. В ряде органов и тканей
(пищеварительная система животных, запасающие органы растений и т. п.)
гидролиз белков осуществляется с огромной интенсивностью и в большом
масштабе. Так, в печени крысы ежедневно распадается около 40% белков,
а время полужизни белков важнейших субклеточных структур (ядро, рибосо-
мы, митохондрии) и цитозоля составляет около 5 суток, хотя есть и более
короткоживущие (сутки и менее) и более длительно сущест ующие (до двух-
трех месяцев) белки и ферменты.
В последние годы выяснено, что время полужизни белка в клетке де ер-
минировано природой его N-концевой аминокислоты. Если она легко соединя-
ется с небольшим (М = 8500 Да, 74 аминокислотных остатка, первичная струк-
тура установлена) белком—убиквитином по АТФ-зависимой реакции, то та-
кой убиквитинированный белок атакуется протеиназами и разрушается.
Наиболее подвержены убиквитинированию (перечислены в порядке убывания)
арг, лиз, асп, асн, три, лей, фен, гис, глу, тир, глн, иле. N-концевые аминокис-
лоты, менее подверженные реакции с убиквитином (мет, сер, ала, тре, вал, гли,
цис), относят к ст билизирующим гидролитический распад белков. Подсчи-
тано, например, что время полужизни цитоплазматических белков, имеющих
в качестве N-концевой аминокислоты арг, составляет 2 мин, асп, лиз, лей
и фен—3 мин, про—7 мин, глн и тир—10 мин, глу и иле—30 мин, гли, ала,
сер, вал, тре и мет—20 ч.
Гидролиз белков может быть частичным (до пептидов) и полным (до
аминокислот). При частичном (неполном) гидр лизе в белковой молекуле
распа аются лишь некоторые пептидные связи, как правило, по соседству со
строго определенными аминокислотными радикалами. Этот процесс ускоря-
ется специфическими ферментами—протеиназами (пептидил-п птидгидрола-
зами). В свою очередь, пептиды гидролизуются до аминокислот, что происхо-
дит при участии ряда пе тидаз. И химизм процесса гидролиза белков, и соот-
ветствующие ферменты, его ускоряющие, охарактеризованы ранее (см. гл. ГГГ).
Таким образом, в результате деятельности разнообразных пептидгидролаз
протеиназы и ептидазы) из белков в процессе их гидролиза сначала образу-
ются сложные смеси различных пептидов, а затем смесь свободных белковых
аминокислот. Последние являются конечным продуктом гидролиза белков.
Механизм действия пептидгидролаз в ряде случаев изучен детально. 3tq
касается, например, механизма действия химотрипсина, упрощенная схема
которого представлена на рис. 89.
262
Роль протеиназ в организме не сводится лишь к фрагментированию белко-
вых молекул до пептидов для обеспечения дальнейшего гидролиза последних
до свободных аминокислот. В последнее время все большее значение ридают
именно способности протеиназ селективно расщеплять полипептидные цепи,
в результате чего из белковых предшестве ников возникают функционально
активные белки и многие биологически активные пептиды, в том числе гормо-
ны, рилизинг-факторы, психотропные пептиды и т. п. Это имеет огромное
значение для регуляции обмена веществ. Протеолиз выступав как особая
форма биологического контроля, однонаправленно обеспечивающего иници-
ацию определенного физиологического процесса.
В последние годы привлекли внимание протеиназы, действие которых
активируется Са2 + . Их называют калыгаинами (М = 110 к Да, 2 субъединицы:
каталитическая—80 кДа и регуляторная—30 кДа). Они расщепляют белки по
границам их доменов, связывая минерал ный обмен с регуляцией метаболиз-
ма. Их действие ингибируется калыюстатином.
Активный транспорт аминокислот через биологические мембраны.. Свобод-
ные аминокислоты, возникающие в результате гидролитического распада
белков, используются в основном для ресинтеза белковых тел и лишь некото-
рая их часть подвергается дальней
шей деструкции. Кроме того, содер-
жание свободных аминокислот
в клетке постоянно пополняется за
счет их синтеза de novo, охва-
тывающего весь спектр протеино-
генных аминокислот у аутотрофов
и заменимых аминокислот у гетеро-
трофов. Естественно, что существу-
ют системы транспорта аминокис-
лот через мембраны, обеспечиваю-
щие их перенос как через внешнюю
клеточную мембрану, так и через
систему внутриклеточных мембран,
В цепь
•Н—СНг
О
NH^**
HR
57
Гис'
ер-ОН
В цепь
nh2
CHR
Рис. 89. Упрощенная схема гидролиза пеп-
тидной связи в активном центре химотрип-
сина
Активный центр фермента содержит остатки серина (195-е
положение в полипептидной цепи) и гистидина (57-е положе-
ние в той же полипептидной цепи). Фрагменты единой поли-
пептидной цепи молекулы фермента условно показаны слева
и справа изогнутой линией, в их составе отмечены остатки
сер и гис С их функциональными группами. В активном
центре фермента точно против остатков сер и гис размещает-
ся пептидная связь гидролизуемого белка, образованная
остатком фен (или тир, три, лей), благодаря контакту ради-
кала фен (иля тир, три, лей) с адсорбционным (субстратным)
центром фермента, который иа рисунке ве показан. Вслед-
ствие того, что электронная пара кислорода гидроксильной
группы радикала сер акцецгируется карбонильной группой
пептидной связи, создаются условия для миграции протона
от гидроксила сер к имидазольному радикалу остатка гис
и для ацилирования радикала сер с одновременным раз-
рывом пептидной связи. Одновременно протон от имида-
зольного ядра радикала гис перемещается к NH-rpynne дес-
труктируемой пептидной связи (I). После ацилирования ра-
дикала сер (II) в активный центр фермента входит молекула
воды, инициирующая распад ацильного производного фер-
мента (III) и восстановление исходного состояния активного
центра (IV), готового принять новую молекулу субстрата
или атаковать пептидную связь, образованную остатками
фен, тир, три или лей в том же субстрате
СН—СН2
95 I
ер—О—СО
Н2О
.В цепь у—.
СООН
ер—ОН
В цепь
В цепь
57
Гис
< 195 ’ I
Ш ^Сер—О—СО
Н—СН
НОН
Гис
f В цепь
/
263
Рис. 90. у-1лутамилтрансферазный цикл
у-Етутамилтрансфераза встроена в клеточную мембрану и осуществляет транслокацию аминокислот из
внеклеточного пространства за счет реакции транспептцдирования остатка у-глутаминовой кислоты
с глутатиона или другого у-глутамилпептида на транспортируемую аминокислоту и переноса возник-
шего переносчика, а именно у-гл у тамил аминокислотного производного, во внутриклеточное (или внут-
римембранное) пространство. Здесь благодаря действию у-глутамилциклотрансферазы переносчик рас-
падается на свободную аминокислоту, которая таким образом оказывается перенесенной через мемб-
рану, и пироглутаминовую кислоту, образование которой практически нацело сдвигает реакцию распада
дипептида—переносчика вправо. В результате ряда ферментативных процессов (правая часть рисунка)
происходит ресинтез глутатиона (или другого у-глутамилпептида, если он участвует в переносе амино-
кислот), и цикл может повториться снова
что обеспечивает их участие в обменных процессах, развертывающихся в ком-
партментах клетки.
Проблему активного переноса аминокислот через биологические мемб-
раны интенсивно разрабатывали многие исследователи. А. Майстер (1973)
предложил гипотезу переноса аминокислот через мембраны при посредстве у-
глутамилтрансферазного цикла, сущность которой ясна из рассмотрения
рис. 90. Согласно этой гипотезе, центральную роль в данном процессе играет
фермент у-глутамилтрансфераза. Естественно, что транслокация аминокислот
через биологические мембраны осуществляется также белками-переносчиками
264
(см. рис. 43). Такие системы переноса изучены для гистидина, лейцина, изолей-
цина и валина.
Превращения аминокислот. Соотношения аминокислот в распадающихся
белках и новообразуемых за их счет протеинах, как правило, различны.
Поэтому известная доля свободных аминокислот, возникших при гидролизе
белков и пептидов, обязательно должна быть преобразована либо в другие
аминокислоты, либо в более простые соединения, выводимые из организма.
Все это осуществляется в результате процессов, которые можно объединить
общим названием—превращения аминокислот.
Известны три типа реакций аминокислот в организме: по а-аминогруппе,
карбоксильной группе и радикалу аминокислоты.
Реакции по а-аминогруппе однотипны у всех аминокислот, это в основ-
ном реакции дезаминирования и переаминирования. Столь же однообразен
набор химических процессов по карбоксильной группе аминокислот: это
главным образом декарбоксилирование и образование аминоациладенила-
тов. В отличие от первых двух типов превращений аминокислот преоб-
разования в радикалах аминокислот исключительно разнообразны, многочи-
сленны и, как правило, уникальны для каждой отдельной аминокислоты.
Наконец, есть тип превращений аминокислот, который состоит в образова-
нии пептидной связи между а-аминогруппой одной аминокислоты и карбок-
сильной группой другой. Он осуществляется сложным путем и приводит
к синтезу пептидов и белков. Здесь рассматриваются лишь первые три типа
превращений аминокислот, а синтез из них пептидов и белков—ниже, в этой
же главе.
Реакции по аминогруппе. Наиболее распространенной и важной реакцией
аминокислот по а-аминогруппе является дезаминирование. Оно может идти
четырьмя путями:
1. R—СН-СООН+1/2О2----------->R—СО—COOH+NH3
Кетокислота (окисли-
алгт тельное дезами-
п 2 вирование)
2. R—СН-СООН+2Н-------------------------R-CH2-COOH+NH3
| Предельная кислота
хттт (восстановительное
** 2 дезаминирование)
3. R-CH-COOH+H2O------------->R—СН-COOH+NH3
nh2 он
Оксикнслота (гидро-
литическое деза-
минирование)
н
4. R-C-CH-COOH------------------»R—СН = СН-COOH + NH3
j Непредельная кислота
т_т vu (внутримолекулярное
п л П2 дезаминирование)
Все перечисленные реакции действительно осуществляются в организмах
и каждая из них ускоряется специфическим ферментом; однако распростране-
ние их в природе совершенно различно: очень широко распространена 1-я
реакция, а остальные три встречаются крайне редко, лишь у отдельных групп
организмов.
265
Так как преобладающим является окислительное дезаминирование, рассмот-
рим его подробнее. Процесс этот осуществляется в две стадии. Сначала
аминокислота окисляется в иминокислоту при участии специфической дегид-
рогеназы с НАД+ или НАДФ+ в качестве кофермента и акцептора водорода.
Затем иминокислота спонтанно гидролизуется на кетокислоту и аммиак:
соон
I
сн2
сн2
chnh2
iooH
НАД(Ф)+ НАД(Ф)Н + Н+
Г лутаматдегидрогеназа
СООН
iH
сн2
C=NH
I
СООН
соон
Lrli
k
u
I
COOH
Глутаминовая
кислота
Иминогл гтаровая
кислота
а-Кетоглутаровая
кислота
Обе реакции обратимы, и таким путем из а-кетоглутаровой кислоты
и аммиака в организме образуется глутаминовая кислота.
В некоторых случаях дегидрогеназы аминокислот представлены флаво-
протеинами. Так, из грены шелкопряда выделена дегидрогеназа «-аминокис-
лот, отличающаяся очень высокой активностью и несущая флавиновую груп-
пировку в составе кофермента.
Полная эпимолекула глутаматдегидрогеназы (М = 336 000) составлена из
6 субъединиц с М = 56000 (см. рис. 46). Каждый из протомеров имеет центры
связывания субстрата, кофермента и эффекторов (АДФ и ГДФ активируют,
а АТФ, ГТФ и пиридоксальфосфат ингибируют фермент). Первичная структу-
ра субъединиц ряда глутаматдегидрогеназ, выделенных из разных объектов,
отличается высокой степенью гомологии.
Дегидрогеназы L-аминокислот (в отличие от дегидрогеназ D-амино-
кислот) в тканях животных и растений представлены слабо, и только де-
гидрогеназа L-глутаминовой кислоты проявляет себя исключительно ярко.
Поэтому допускают, что большинство L-аминокислот дезаминируется в
организме путем переаминирования с а-кетоглутаровой кислотой по урав-
нению:
СООН СООН 1
1 сн2 1 R 1 ^аминокислота: R 1 сн2 1
сн2 + 1 с=о CHv-NH2 1 соон оксоглута- раттранс- аминаза ±с=о + 1 СООН сн2 1 сн—nh2
1 соон а-Кетоглутаровая (2-оксоглутаровая Амшюпсиюта Кетокислота соон 1лутамиИовая кислота
кислота
Вслед за этим глутаминовая кислота претерпевает окислительное дезами-
нирование, а выделяющаяся при этом а-кетоглутаровая кислота снова вовле-
кается в реакцию переаминирования с «-аминокислотами.
Выделены специфические трансаминазы, ускоряющие реакцию переамини-
рования между почти всеми белковыми аминокислотами и а-кетоглутаровой
266
кислотой. Механизм реакции переаминирования детально рассмотрен
в гл. III. Реакция переаминирования между L-аминокислотами и а-кетоглута-
рбвой кислотой обратима, поэтому при определенных условиях она служит
для синтеза L-аминокислот из кетокислот и глутаминовой кислоты. Таким
образом, реакцию переаминирования нельзя сводить только к дезаминирова-
нию аминокислот; ее роль в организме гораздо шире.
1лавным продуктом дезаминирования аминокислот являются а-кетокис-
лоты. Лишь в некоторых, особых и мало распространенных случаях в качестве
конечных продуктов дезаминирования аминокислот о мечены предельные или
непредельные жирные кислоты, а также оксикислоты.
Дезаминирование некоторых аминокислот идет своеобразно. Так, серосо-
держащие аминокислоты (цистеин и метионин) дезаминируются путем отщеп-
ления аммиака и сероводорода или метилмеркаптана (CH3SH) соответствен-
но; оксиаминокислоты (серин и треонин)—путем отщепления аммиака и во-
ды; гетероциклические аминокислоты—путем дегидрирования по кольцу
(пролин) с дальнейшим преобразованием продукта дегидрирования и т. д.
Однако и в этих случаях конечными продуктами дезаминирования остаются
кетокислоты и непредельные кислоты.
Реакции по карбоксильной группе. Превращения аминокислот по СООН-
группам сводятся в основном к декарбоксилированию и образованию амино -
ациладенилатов. Декарбоксилирование аминокислот осуществляется сравни-
тельно легко в тканях животных и растений, но особенно широко оно пред-
ставлено у микроорганизмов. Во всех случаях процесс идет по одной и той же
схеме:
Декарбоксилаза
R - СН - СООН--------------> R - СН2 - NH2+СО2
nh2
Простатической группой декарбоксилаз L-аминокислот служит пиридок-
сальфосфат, комплекс которого с различными специфическими белками дает
начало всем многообразным и высокоспецифичным декарбоксилазам L-ами-
нокислот. Выделены и изучены декарбоксилазы аспарагиновой и глутамино-
вой кислот, валина, лизина, аргинина, гистидина, тирозина, триптофана и ряда
других аминокислот. Механизм действия их ясен из схемы, приведенной на
с. 127, и уравнений реакций на с. 133.
В подавляющем большинстве случаев продуктами декарбоксилирования
аминокислот являются амины. Так как они образуются в качестве продуктов
жизнедеятельности и обладают высокой физиологической активностью, их
называют биогенными аминами. Приведем некоторые примеры.
При декарбоксилировании гистидина возникает гистамин:
Гнетидиндекар-
N~ —СН2—СН—СООН боксилаэа <
U У J,„ (L-ГИСТИДИН-
а карбокси—лиаза;
I М = 110 ООО, димер)
СН2—CH2NH2
Гистидин
Гистамин
Он вызывает усиление деятельности желез внутренней секреции и снижает
кровяное давление.
При декарбоксилировании тирозина и триптофана образуются соответст-
венно тирамин и триптамин:
267
сн2—сн2—nh2
СН2—СН2—NH2
Тирании
Триптамин
Последний легко переходит в 5-окситриптамин (серотонин)—соединение, обла-
дающее многогранным физиологическим действием, имеющее, в частности,
отношение к возникновению болевых ощущений при воспалительных процессах.
Декарбоксилирование лизина и аргинина сопровождается образованием
кадаверина и агматина:
H2N-(CH2)s-NH2 H2N—С—(СН2)4—NH2
Кадаверин ||
NH
Агматин
В последнее время важное значение придают тетраметилендиамину (пут-
ресцин), возникающему при декарбоксилировании аминокислоты—орнитина:
H2N-(CH2)3 -СН-СООН------------------->H2N-(CH2)4-NH2+СО2
I декарбоксилаза л
I Путресцин
nh2
Орнитин
Тетраметилендиамин служит в организме исходным соединением для синтеза
спермидина и спермина (см. с. 211). Оба эти вещества—полиамины, обес-
печивающие наряду с диаминами определенные структурные особенности
и функциональную активность рибосом.
Не только амины и диамины являются продуктами декарбоксилирования
аминокислот. При декарбоксилировании глутаминовой кислоты образуется
у-аминомасляная кислота:
НООС - СН - (СН2)2 - СООН h2n - сн 2 - (сн2)2 - соон+со2
Ан2
Глутаминовая кислота у-Аминомасляная кислота
Она накапливается в мозговой ткани и представляет собой нейрогуморальный
ингибитор. Аналогично этому из аспарагиновой кислоты получается 0-аланин:
Аспартат-
НООС - сн - сн2 - соон---------------------------- h2n - сн2 - сн2 - соон + со2
| декарбоксилаза
р-Алании
NH2
Аспарагине вая
кислота
Он принимает участие в синтезе пантотеновой кислоты (см. с. 162).
Второй важной реакцией аминокислот по карбоксильной группе является
образование ими аминоациладенилатов. Эта реакция была отмечена ранее при
рассмотрении ферментов, относящихся к классу лигаз (см. с. 137), и будет
подробно освещена ниже, в этой главе.
268
Превращения аминокислот, связанные с реакциями по радикалу. Напомним
прежде всего, что радикалом аминокислоты принято называть ту часть ее
молекулы, которая не принимает участия в формировании хребта полипептид-
ной цепи. По своей химической природе радикалы аминокислот исключитель-
но разнообразны, что служит материальной основой для многообразия при-
сущих им химических реакций. Естественно, что многие из этих реакций
осуществляются в процессе обмена аминокислот.
Важнейшим типом превращений аминокислот, протекающих с видоизмене-
нием радикалов, является переход одних аминокислот в другие. Благодаря
этому в организме значительно усиливаются возможности для синтеза амино-
кислот. Приведем некоторые примеры.
При окислении фенилаланина образуется тирозин:
h2n—сн—соон
Фенилаланин
Феиилалаиин-4-
гидроксилаза
f НАДФН+Н+ НАДФ
ОН
H2N—СН—СООН
Тирозин
Твдролиз аргинина приводит к образованию аминокислоты—орнитина:
Н2О
H,N—с—NH—(СН2),—СН—СООН —HjN— (СН2)3— СН—СООН + H2N—С—NH,
у । Аргиназа । ||
NH NH2 NHj О
Аргинин Орнитин Мочевина
Из последнего, в свою очередь, возникает либо глутаминовая кислота, либо
пролин:
nh2
(*Н2
сн2
<}н2
^Н—NH,
СООН
Ориитин
269
Окислительно-восстановительные процессы легко протекают по серосодер-
жащим радикалам цистеина и цистина, благодаря чему эти две аминокислоты
переходят друг в друга; химизм этой реакции аналогичен окислению глутати-
она (см. с. 48), а ускоряется она цистеинредуктазой.
При исчерпывающем окислении тиоловой группы цистеина последний
переходит в цистеиновую кислоту, которая, декарбоксилируясь, дает начало
таурину:
ОН
I
SH o=s=o он
_ _ । Декарбоксн- ।
| Окисление | лированис jj
сн2-------- сн2 ------------► o=s=o+co2
I I I
сн—nh2 сн—nh2 сн2
I I I
соон соон сн2—nh2
Цистеин Цистеиновая кислота Таурин
Последний, соединяясь с желчными кислотами, например холевой кисло-
той (см. с. 380), принимает участие во всасывании жиров.
Большое распространение имеет реакция диметилирования метионина. Она
осуществляется при каталитическ »м воздействии метилтрансферазы. Мети-
онин—универсальный поставщик метильных групп в реакциях трансме-
тилирования. При этом он переходит сначала в «активный метионин», со-
единяясь с АТФ:
АТФ:
VL-метионии-
S-аденозил-
трансфераза
S-Аденозилметионии
(„активный метионин")
ОН ОН ОН
НО—Р—О—Р —О—Р-ОН
ООО
Трифосфат
Н4Р2О7 + Н3РО4
Пирофосфат Фосфорная
кислота
270
Реакция ускоряется специфическим ферментом АТФ: L-метионин—S-адено-
зил трансферазой (М = 100 000, оптимум pH 9,5). Далее метильная группа от
S-аденозилметионина передается соединению, которое подвергается метилиро-
ванию. В качестве примера приведем уравнение реакции метилирования глицина:
S-Аденозилметиоиии
—*-НООС—СН2—NH—СН3
N-Метилглиции
(Саркозин)
(СН2)2
HjNcJh
СООН
S-Адеиозилгомоцистеин
Интересная реакция осуществляется по радикалу треонина. Она состоит
в отщеплении радикала в виде ацетальдегида. Эта реакция ускоряется специ-
фическим ферментом—треонинальдолазой, открытым А. Е. Браунштейном
и Г. Я. Виленкиной (1951). Простетической группой его является пиридоксаль-
фосфа . В качестве второго продукта расщепления треонина идентифицирован
глицин:
СИз сн,
СН—ОН Треоятильдшпм | zO СН,—NH,
I —:-----------и с< + ।
ГН МН (L-rpeottim: СООН
Сп—NH, щеплмсгМ'Ляиа) “
СООН
. Треониов Ацеталь- Глицга
легки
Кроме реакций, приводящих к синтезу одних аминокислот из других, по
радикалам аминокислот известно много других превращений (окисление, ме-
тилирование и т. п.). Часто эти реакции сочетаются с процессами декарбок-
силирования и дезаминирования аминокислот. В результате (особенно из
циклических аминокислот) возникают разнообразные вещества, многие из
которых обладают сильным физиологическим действием. Так, например, из
тирозина образуется гормон адреналин (см. гл. ХП). Триптофан служит источ-
ником образования никотиновой кислоты (витамин РР) и индолилуксусной
кислоты (ростовое вещество); цистеин—меркаптуровых кислот (обезврежива-
ние ароматических соединений); аргинин—аргининфосфата и других гуанидин-
фосфатов (макроэргические соединения).
Таким образом, в процессе превращений аминокислот возникает серия
соединений, принимающих участие в регуляции обмена веществ в организме.
271
Это обстоятельство еще раз подчеркивает ведущую роль белкового обмена
в общем обмене веществ организма.
Конечные продукты распада аминокислот. Как было отмечено выше, в ре-
зультате распада аминокислот возникают СО2, NH3, амины, кетокислоты
и в ряде случаев еще достаточно сложные вещества, относящиеся к тем или
иным классам органических соединений. Все они, за исключением СО2 и NH3,
подвергаются в конце концов дальнейшей деструкции. Амины путем окис-
лительного дезаминирования превращаются в карбоновые кислоты:
Моноамино- /7
R—СНа 4- Н2О + О2 --------------- R—Сч 4- NH3 + Н2О2
I оксидаза >р{
NH,
+ Н2О + НАД+
Альдегид-
Легидрогеназа
R—Ск + НАДН + Н+
ОН
Аналогично идет реакция окислительного дезаминирования диаминов при
посредстве диаминоксидазы.
Кетокислоты и карбоновые кислоты, возникающие в результате распада ами-
нокислот, постепенно окисляются, образуя СО2 и Н2О (см. с. 356). Точно так же
в СО2, NH3 и Н2О превращаются все остальные органические вещества, являющи-
еся продуктами распада аминокислот в организме. Таким образом, конечными
продуктами распада аминокислот являются Н2О, СО2 и NH3. Вода поступает
в общий метаболический фонд, оксид углерода (IV) беспрепятственно выводится
из организма и лишь судьба аммиака нуждается в специальном рассмотрении.
Только, у некоторых обитателей гидросферы (медицинская пиявка, крабы,
речной рак, беззубка, каракатица и др.) NH3 непосредственно или в виде солей
аммония выводится в окружающую среду. У подавляющего большинства
растительных и животных видов аммиак, уже в небольших концентрациях
оказывающий вредное влияние на жизнедеятельность организмов, переводит-
ся в безвредные для биологических форм азотистые соединения. К их числу
относятся аспарагин, глутамин и мочевина. У многих животных, особенно
позвоночных, последняя служит для выведения обезвреженного аммиака.
Полагают, что аспарагиновая и глутаминовая кислоты осуществляют первич-
ное связывание NH3 в момент его образования в клетке. Взаимодействие аммиака
с этими кислотами, в результате чего возникают амиды—аспарагин и глута-
мин, ускоряется специфическими ферментами. Оба фермента—аспарагинсинте-
таза и глутаминсинтетаза—принадлежат к классу лигаз, в частности к подклассу
лигаз, ускоряющих реакции образования С—N-связей, и среди них—к подпод-
классу кислотоаммиачных лигаз (амидосинтетазы). Так как необходимым усло-
вием деятельности лигаз является сопряженный с реакцией синтеза процесс
распада АТФ, то уравнение реакции биосинтеза аспарагина имеет такой вид:
Z
Snh,
АДФ + Н3РО, + СН,
(L-аспартат: I
аммиак-лигаза)
СООН
Аспарагин
I ОН
I Аспарагиисинтетаза
СН» + АТФ 4- NH3
СН—NH,
icon
Аспарагтюыя
кислота
272
Аналогично идет реакция биосинтеза глутамина при участии глутаминсин-
тетазы (Ь-глутамат:аммиак-лигаза).
Реакции образования аспарагина и глутамина особенно широко представ-
лены в растительном царстве. Однако и у животных эти амиды возникают не
столь редко: синтез аспарагина доказан в жировом теле насекомых, а синтез
глутамина—в мышцах, мозгу, печени и почках млекопитающих, а также
гемолимфе насекомых. В тканях млекопитающих синтез аспарагина идет из
глутамина, амидная группа которого переносится на у-карбоксильную группу
аспарагиновой кислоты при участии глутаминзависимой аспарагинсинтетазы
сопряженно с распадом АТФ на АМФ и пирофосфат.
Амидирование аспарагиновой и глутаминовой кислот может происходить
и в том случае, если они находятся в связанном состоянии, например, в составе
белковой молекулы. Как известно, радикалы аминокислот, входящих в поли-
пептидную цепь белка, свободны и по ним легко осуществляются те или иные
химические реакции. Одной из таких реакций является амидирование белков:
HI-NH-CH—СО—]—[—NH-CH-CO—NH-CH—СО—NH—(j:H-CO—1—[-NH—(JH—СО—]j—он
К' СНа К" (СН2)2 R'"
1/° I/O
%н \>н
Белок
2л NH3 + 2л АТФ
2л АДФ + 2л НзРО,-*^
Н (—N Н-СН—СО—)-
R'
[-NH—СН—СО—1—ОН
R"'
Амидированный белок
Следовательно, не только свободные аспарагиновая и глутаминовая кисло-
ты, но и белки организма могут быть акцепторами NH3. Тем самым обеспечи-
вается немедленное связывание аммиака в любой точке, где он возникает
в результате обмена веществ. Вместе с тем амидирование белков представляет
процесс посттрансляционной (см. с.301) модификации белков, в результате
которой полностью завершается их синтез. Различная степень посттрансляцион-
ного амидирования является одним из источников микрогетерогенности белка.
Мочевина—основной конечный продукт белкового обмена у многих жи-
вотных (дождевой червь, слизень, акула, лягушка, черепаха и все млекопита-
ющие). Ее биосинтез у высших животных происходит в печени, на что впервые
в конце прошлого столетия обратили внимание М. В. Ненцкий и И. П. Пав-
лов. В печени найдены все необходимые для этого ферменты. У животных, не
способных синтезировать мочевину (рептилии, птицы), печень не обладает
соответствующим ферментативным аппаратом. Новообразование мочевины
идет также в растениях. Путь ее возникновения у животных и растений
одинаков и состоит в следующем. Из NH3, СО2 и АТФ при каталитическом
воздействии фосфотрансферазы (карбаматкиназа) синтезируется карбамилфос-
форная кислота. Механизм этой реакции рассмотрен ранее (см. с. 235). При
участии другой трансферазы (орнитин-карбамилтрансфераза) карбаминовая
273
группир вка переносится от карбамилфосфата на 5-аминогруппу орнитина,
который всегда присутствует в организме, так как легко во никает при гид-
ролизе аргинина. В результате этой реакции синтезируется цитруллин:
. о NH.
h,n-c^ 9” +(4нл
О-Р=°
°н {юон
Карбмпифоефят Орите
NH*
---------------- А»О + НэРО*
Оршсг ирб«- I
ммлтрмсферм» ИЦ
(Ьл
СН-NH»
Аюн
Цкгруллая
в карбаминовую
Далее в действие вступают еще два фермента, обеспечивающие введение
H2N—С—NH—) группировку цитруллина еще одного атома
° / \
азота и превращение ее в гуанидиновую группировку Ih2n—С—NH—), т. е.
NH
переход цитруллина в аргинин.
Донором аминогруппы в этом превращении служит аспарагиновая кисло-
та, а промежуточным соединением на пути от цитруллина к аргинину —
аргинин-янтарная кислота:
NH2
с=о
I
NH +
(<fH2)3
CHNH2
СООН
Цитруллин
СООН Аргииииосукпишт-
| сннтетаза
H2N-CH + АТФ ~ *
|н2
СООН
Аспараги-
NH, ' СООН\
йЯ/
NH '* <~Н^ + АМФ + Н,Р2О7
[<jH2)e СООН Пирофосфат
CHNH2
СООН
Аргинин
кислота
Аргинин-янтарная
кислота
(аргининосукцниат)
Заключительной реакцией в биосинтезе мочевины является гидролиз ар-
гинина и образование орнитина и мочевины. Получающийся при этом ор-
нитин вновь вступает во взаимодействие с карбамилфосфатом, и все перечис-
ленные выше р акции повторяются снова. Поэтому совокупность указанных
реакций, приводящих к образованию мочевины в качестве одного из звеньев,
включающих высвобождение и вовлечение снова в процесс орнитина, получи-
ла название орнитинового цикла (рис. 91).
274
Рис. 91. Орнитиновый цикл (пояснение в тексте)
Фумаровая кислота, как мы увидим ниже, легко превращается в щавелево-
уксусную кислоту, которая путем переаминирования либо аминирования пере-
ходит снова в аспарагиновую кислоту. Следовательно, с одной стороны,
акцептируется молекула NH3, а с другой—возобновляются запасы аспараги-
новой кислоты, участвующей в функционировании орнитинового цикла. Са-
мое важное состоит в том, что в результате деятельности орнитинового цикла
из каждых двух молекул NH3 и одной молекулы СО2, освобожденных в ре-
зультате распада аминокислот (или других соединений), строится одна моле-
кула мочевины.
Новообразование аминокислот. Выше уже было рассмотрено новообразова-
ние аминокислот в природе путем их переаминирования с кетокислотами, т. е.
превращение одних аминокислот в другие. Однако в обоих случаях исходным
продуктом служат уже готовые аминокислоты, которые лишь тем или иным
способом видоизменяются, т. е. получаются путем вторичного синтеза из
предсуществующих аминокислот. Значит, оба эти процесса не решают пробле-
му первичного синтеза аминокислот в организме. Он осуществляется восста-
новительным аминированием кетокислот и прямым аминированием непре-
дельных кислот.
Прямое аминирование непредельных кислот представляет довольно редкую
реакцию и свойственно в основном бактериям и растениям. Хорошо изучено
прямое аминирование фумаровой кислоты; оно ускоряется специфическим
ферментом—аспартат-аммиак-лиазой и идет в соответствии со следующим
уравнением:
соон соон
сн аАмми«эт’ hc-nh2
|| +NH3. I
СН лиаза СН2
соон соон
Фумаровая Аспарагиновая
кислота кислота
Реакция обратима и в известных условиях служит для дезаминирования
аспарагиновой кислоты. Иммобилизованная аспартат-аммиак-лиаза нашла
(см. с. 144) применение для промышленного получения L-аспарагиновой кис-
лоты. Значительные количества аспарагиновой кислоты синтезируются также
путем переаминирования щавелевоуксусной кислоты с глутаминовой кис-
лотой.
275
Восстановительное аминирование представляет главный путь новообразова-
ния аминокислот. Эта реакция есть обращение окислительного дезаминирова-
ния аминокислот. Ее уравнение приведено на с. 265. Если его прочесть справа
налево, то это и будет уравнение реакции восстановительного аминирования
а-кетоглутаровой кислоты.
Другой кетокислотой, подвергающейся активно восстановительному ами-
нированию, является пировиноградная кислота:
СНз. СНз
i = О+NH3 + НАДН + Н + <— <^Н—NH2 + НАД + + Н2О
। дегидрогеназа |
СООН соон
В принципе, возможно восстановительное аминирование любой кетокис-
лоты. Однако активность всех природных дегидрогеназ аминокислот, за ис-
ключением глутамат- и аланиндегидрогеназы, ничтожна, поэтому синтез всех
остальных протеиногенных аминокислот путем восстановительного аминиро-
вания практического значения не имеет. Только аланин и глутаминовая кисло-
та возникают таким способом из пировиноградной и а-кетоглутаровой кис-
лот, являющихся нормальными промежуточными продуктами распада угле-
водов и жирных кислот.
Следовательно, к первичной аспарагиновой кислоте, синтезируемой путем
прямого аминирования, добавляются еще две первичные аминокислоты —
аланин и глутаминовая кислота, образующиеся в результате восстановитель-
ного аминирования. Остальные аминокислоты образуются в результате реак-
ций переаминирования перечисленных аминокислот с соответствующими ке-
токислотами, возникающими в процессе обмена веществ, а также путем
превращения одних аминокислот в другие (рис. 92). Поэтому аланин, ас-
парагиновую и глутаминовую кислоты называют первичными аминокислота-
ми, а все остальные—вторичными.
Растительные и животные организмы резко отличаются друг от друга по
способности синтезировать аминокислоты. В растениях осуществляется бес-
препятственный синтез самых разнообразных аминокислот. Здесь создаются
не только все 18 аминокислот, постоянно встречающихся в белках, но и огром-
ное число так называемых «экзотических» аминокислот. Некоторые из них
иногда находят в составе белков. В растениях сейчас обнаружено несколько
сотен аминокислот, и список их возрастает с каждым годом. Часто та или
иная аминокислота присутствует в растениях строго определенного вида и ее
наличие может служить надежным таксономическим признаком.
В отличие от этого животные синтезируют далеко не все аминокислоты. Из
18 постоянно встречающихся в белках аминокислот в животном организме
синтезируется в среднем только половина их, а остальные—не синтезируются.
Первые (синтезируемые) называются заменимыми аминокислотами, вторые
(несинтезир; емые)—незаменимыми. Между различными видами животных
есть некоторые отличия в наборе заменимых и незаменимых аминокислот, но
в большинстве случаев к незаменимым аминокислотам относятся валин,
лейцин, изолейцин, треонин, метионин, лизин, фенилаланин и триптофан.
Любопытно, что у заменимых аминокислот, по подсчетам Ю. А. Жданова
(1968), в большинстве случаев степень окисления атомов углерода отрицатель-
на, а у незаменимых—всегда положительна. Это свидетельствует о том, что
заменимые аминокислоты эволюционно более молоды (возникли в окисли-
тельной атмосфере планеты) по сравнению с незаменимыми.
276
Рис. 92. Взаимосвязь реакций, лежащих в основе биосинтеза первичных и вто-
ричных аминокислот:
2, 2, 3—первичные аминокислоты: глутаминовая кислота, аланин и аспарагиновая кислота; а—глута-
матдегвдрогеназа» б—аланиндегидрогеназа, в—аспартаза
Если в корме животных недостаточно содержание одной или нескольких
незаменимых аминокислот, то нормальное развитие животного нарушается,
так как биосинтез белка у него идет на низком уровне. Как правило, рас-
тительные белки содержат мало лизина, метионина и триптофана. Дефицит
этих аминокислот в корме сельскохозяйственных животных встречается наи-
более часто. Рационы их неполноценны также по количеству треонина. Введе-
ние в рационы недостающих незаменимых аминокислот позволяет нормализо-
вать рост организма, увеличивает привес на каждую израсходованную кормо-
вую единицу, улучшает использование белков основной диеты, резко
повышает эффективность животноводства. Так, введение в рацион 0,2—0,5%
лизина повышает продуктивность свиноводства и птицеводства на 10—13%
и сокращает расход кормового белка на 25%.
Вполне понятно, что в описанных ситуациях речь идет о незаменимых
аминокислотах L-ряда, поскольку они необходимы для синтеза белка. Однако
L-аминокислоты очень трудно создать путем химического синтеза, при кото-
ром получаются рацематы аминокислот, нуждающиеся в разделении на оп-
тические антиподы. Поэтому основная масса аминокислот для нужд животно-
водства производится путем микробиологического синтеза, т. е. использова-
ния определенных микроорганизмов—продуцентов аминокислот, которые
выделяют те или иные L-аминокислоты прямо в культуральную жидкость
в количестве нескольких граммов на 1 л. В частности, в Институте биохимии
им. А. Н. Баха под руководством чл.-кор. В. Н. Букина разработан экономич-
ный микробиологический метод получения L-лизина, а во Всесоюзном научно-
исследовательском институте генетики и селекции промышленных микроор-
ганизмов его сотрудниками под руководством чл.-кор. В. Г. Дебабова мето-
дами генетической инженерии создан штамм кишечной палочки, выделяющий
в культуральную среду до 30 г L-треонина. Ряд аминокислот получают также
при помощи иммобилизованных бактериальных клеток и ферментов
(см. гл. III). Лишь метионин синтезируют заводским путем в виде рацемата,
который столь же хорошо используется организмом, как и L-метионин.
277
В нашей стране путем микробиологического синтеза готовят лизин на ряде
биохимических заводов с конечной целью довести его производство до 32 тыс.
т в год.
Проблема незаменимых аминокислот актуальна и в питании человека,
которому необходимо ежедневно получать с пищей 1 г L-триптофана, 2—3 г
L-треонина, по 2—4 г L-лейцина, L-метионина и L-фенилаланина, 3—4 г
L-изолейцинаиЗ—5 г L-лизина. По мнению акад. А. Н. Несмеянова, высказанно-
му в докладе на IX Менделеевском съезде по общей и прикладной химии четверть
века тому назад, индустриальный синтез восьми незаменимых аминокислот,
способных заменить белок в питании человека, представляется вполне реальным
и экономически оправданным. За прошедшие годы промышленное производство
как незаменимых, так и заменимых L-аминокислот продвинулось далеко вперед
и синтез некоторых из них занимает все более прочное место в ряду мероприятий,
направленных на повышение полноценности белкового питания человека.
БИОСИНТЕЗ БЕЛКОВ
Проблема биосинтеза белка—одна из двух наиболее важных и острых
проблем современного естествознания: если в неживой природе принципиаль-
но новые пути получения энергии будут найдены благодаря успехам физики
элементарных частиц, то в живой природе решение кардинального вопроса
управления самой жизнью может быть получено в результате познания химии
и биологии белковых тел.
В изучении строения и биосинтеза белка, как в фокусе, скрещиваются пути
решения важн йших вопросов биологической науки: выяснение законов наслед-
ственности и изменчивости управление ростом и развитием организмов,
выявление причин возникновения и разработка методов лечения многих болез-
ней и т. п. Вполне закономерно поэтому, что XX в. физики называют веком
атома, а биохимики—веком белка.
Биосинтез белков в op ai лзме осуществляется весьма интенсивн >. Средняя
скорость сборки полипептидных цепей в клетках бактерий составляет 16—17
аминокислотных остатков в 1 с, дрожжей—7—10, млекопитающих—5—7.
Время синтеза (в с) молекулы глобина в ретикулоцитах кролика pai но 20,
овальбумина в яйцеводах курицы—80; суммарных белков в печени крысы—
80. За 1 мин в ретикулоците кролика синтезир; ется 5 • 104 молекул глобина,
в клетке яйцеводов курицы—6 • 105 молекул овальбумина, в ги антской клетке
заднего отдела шелкоотделительной железы тутового шелкопряда—38-1011
молеку и фиброина шелка.
История развития представлеиий о механизме биосинтеза белков. Путь,
пройденный при разработке одной из центральных проблем современной
биохимии—механизма биосинтеза белков, крайне поучителен и противор чив.
Первой по времени была гипотеза обращения протеолиза. Она восходит
к концу прошлого столетия. В 1886 г. А. Я. Данилевский наблюдал образова-
ние белковоподобных в щест при действии ферментов желудочного сока на
концентрированный раствор пептонов, возникших в результате расщепления
белка пепсином. Позже был расшир н круг ферментов, способных к обраще-
нию протеолиза (трипсин, пепсин, папаин, катепсины), и белков, с неполными
гидролизатами которых такое обращение удавалось осуществ ть (альбуми-
ны, глобулины, фибрин, казеин и др.). Продукты, возникающие в результате
обращения реакции гидролиза белков, назвали пластеинами.
Хотя сейчас ясно, что реакции пластеинообразования не имеют отношения
к природному биосинтезу белков, они вплоть до сегодняшнего дня привлеь а-
278
ют внимание исследователей, так как нашли практическое применение при
переработке непищевых белков в пищевые и синтезе пептидов, в частности
аспартама (метиловый эфир Ь-а-аспартил-Ь-фен лаланина), используемого
как заменитель сахарозы (он в 100 раз слаще нее) в кондитерской промыш-
ленности (коммерческое название—сластилин):
H2N-CH— СО- NH-CH-CO—ОСН3
Аспартам
Определенный вклад в развитие современных представлений о механизме
биосинтеза белков внесли исследования биосинтеза квазипептидных связей (т. е.
не истинных пептидных связей, но близких к ним), осуществленные в 50-е годы
в ряде лабораторий, в том числе у нас в лаборатории А. Е. Браунштейна.
Будучи проведены на таких со динениях, как лутамин, ацетанилид, глутатион
и гиппуровая кислота, эти исследования доказали ферментативный характер
реакций их образования, необходимость энергообеспечения за счет окисли-
тельных процессов и, что самое важное, участие в биосинтезе квазипептидных
связей АТФ.
Столь же существенными оказались результаты разработки гипотезы
транспептидирования в качестве возможного варианта механизма биосинтеза
пептидных связей. В нашей стране исследования в этом направлении интенсив-
но проводились в 50-е годы В. Н. Ореховичем с сотр. При изучении реакций
транспептидирования впервые было показано, что перенос аминоациальных
или пептидильных группировок на аминогруппу аминокислот может проис-
ходить не только с амидной или пептидной связи, но и со сложно фирной
связи. В дальнейшем оказалось, что именно этот механизм лежит в основе
реакции транспептидирования в рибо оме.
Гипотеза подстановки аминокислот, возникшая в тот же период в связи
с внедрением в биохимию метода радиоактивных индикаторов, не оказала
сколько-нибудь существенного влияния на развитие пр дставлений о механиз-
ме биосинтеза белков.
Переломным моментом в развитии подходов к выяснению механизма
биосинтеза белковых тел было установление сцепленности его с био интезом
РНК (Ж. Браше, 1941; Т. Касперсон, 1941). Оно привело к созданию матричной
схемы биосинтеза белков, являющейся фундаментом современны представле-
ний в этой области. Матричный механизм биосинтеза полимеров, обеспечи-
вающий безошибочное воспроизведение их первичной структуры, пр дставпя-
ет одну из наиболее специфических черт живого. Он является превосходным
примером тех принципиально новых закономерностей которые сопровожда-
ют возникновение и развитие биологической формы движ ния материи: мало-
эффективный механизм обычного химического синтеза, основанного на беспо-
рядочном столкновении молекул, заменен здесь направленным, специфиче-
ским синтезом на шаблоне; скорость его в миллиарды раз превышает таковую
в неупорядоченных системах.
Принципиальное значение в разработке вопроса о механизме биосинтеза
белков имело выявление локализации его в рибосомальном аппарате клетки
и создание бесклеточных систем, где единственной структурой, на которой
протекал биосинтез белка, были рибосомы (см. с. 280). Выяснение их строения
и функции принесло неоценимую информацию об этапах матричного биосин-
теза белка и тончайших молекулярных механизмах, которые ему сопутствую .
279
Аминокислоты
РиБонуклеозидтрифосфаты
ATPiMgv
рН5-/^ментн^ние_ и
wmuBupola-
___ кодиро-
вание
ДНК-матрица
РНК-полимераза
(Транскрипция
информации.
Информационная РНК
----------------ь
, г уцВурсглицс
Аминоацил -тРНК
Трансляция информации
Диосинтез пептидных
связей (транспептиди-
рооание)
АктивнаТГрибосома
70-80 S
Белок
g50-60'Si
iyHuacmutfi
Рис. 93. Общая схема матричного биосинтеза белковых тел
(пояснение в тексте)
Как будет показано ниже, наряду с матричным механизмом в природе
существует мультиэнзимный путь биосинтеза белков и пептидов, где специфи-
ческое чередование аминокислот обеспечивается расположением каталитиче-
ски активных белков в мультиэнзимном комплексе.
Матричный механизм биосинтеза белков. Общая схема матричного биосин-
теза белковых тел представлена на рис. 93. Она складывается из трех подгото-
вительных процессов—переноса вещества, энергии и информации в рибосому,
и главного центрального процесса—сборки полипептидных цепей в рибосоме.
Один из элементов указанной схемы (правая верхняя часть рисунка)—транс-
крипция (переписывание) информации о порядке расположения аминокислот-
ных остатков в молекуле синтезируемого белка—рассмотрен ранее. Известно,
что информация об этом закодирована в генетическом аппарате клетки после-
довательностью дезоксирибонуклеотидных остатков в молекуле ДНК. Будучи
преобразована (транскрибирована) в последовательность рибонуклеотидных
остатков в информативной части молекулы мРНК, синтезированной на ДНК
в качестве матрицы, эта информация о первичной структуре белка поступает
в рибо ому. Здесь она переводится (транслируется) с полинуклеотидной после-
довательности в аминокислотную последовательность новообразуемого в ри-
босомальном аппарате белка. Два других процесса—перенос вещества
(18 протеиногенных аминокислот и двух амидов) и .перенос энергии, необ-
ходимой для синтеза пептидных связей (левая верхняя часть рисунка), равно
как и наиболее сложный процесс—сборка полипептидной цепи в активной,
транслирующей рибосоме (центральная часть рисунка), нуждаются в деталь-
ной характеристике. Она дана ниже.
Активирование аминокислот и перенос их в рибосому. Син-
тез пептидной связи из свободных аминокислот протекает с поглощением
280
энергии в количестве около 12 кДж/моль. В связи с этим давно уже была
высказана идея о сопряженности биосинтеза белка с окислительными процес-
сами (А. В. Благовещенский, М. П. Юргенсон, 1937) или с распадом соедине-
ний, содержащих макроэргические связи (Ф. Липман, 1941). Развитие послед-
него направления привело к обнаружению ферментативного процесса активиро-
вания аминокислот. В 1955 г. М. Хогленд впервые предложил общепринятую
сейчас двухэтапную схему этой реакции.
Первый ее этап состоит во взаимодействии аминокислоты с АТФ, в резуль-
тате чего возникает аминоациладенилат и выделяется пирофосфат. Гидролити-
ческий распад последнего при участии пирофосфатазы обеспечивает необ-
ратимость реакции образования аминоациладенилата. Химизм реакции этого
этапа активирования аминокислот рассмотрен ранее (гл. III) при характерис-
тике лигаз, катализирующих синтез С—О-связей (см. с. 137). Второй этап
сводится к переносу аминокислоты с образовавшегося аминоациладенилата на
концевой аденозин акцептирующего стебля тРНК:
Как видно из структурной формулы аминоациладенилата, он представляет
ангидрид аминокислоты и остатка фосфорной кислоты аденозин-5'-фосфата,
причем донором кислорода для ангидридной связи является ОН-группа карбок-
сила аминокислоты. Будучи ангидридами, свободные аминоациладенилаты
с исключительной легкостью вступают в реакцию с аминокислотами даже при
очень небольшой концентрации (10-3 моль), при почти нейтральной реакции
среды (pH 7,4). Реакция осуществляется без участия ферментов и сопровождается
образованием пептидных связей. Столь же энергично они ацилируют свободные,
доступные аминогруппы белковой молекулы. В то же время в срязанн >м
с ферментом состоянии аминоациладенилаты крайне инертны, и об их использо-
вании для синтеза белка прямо из состава комплекса не может быть речи.
Специфичность реакции активирования аминокислот по вышеуказанной
схеме близка к абсолютной. Ее оценивают как сверхспецифичность. Активиру-
ются изомеры аминокислот только природного L-ряда; D-изомеры аминокис-
лот в реакцию не вступают. Однако некоторые гомологи L-аминокислот
активируются наряду с белковыми L-аминокислотами.
281
Каталитическое ускорение активирования каждой про еиногенной амино-
кислоты осуществляется собственным, специфичным только для данной амино-
кислоты ферментом. Эти ферменты обнаружены у представителей различных
классов животных, ра тений (в том числе водорослей) и микроорганизмов,
т. е. они распространены повсеместно. Это указывает на их важнейшую роль
в биосинтезе белков.
Активирующие ферменты изолируют при низкой температуре из так назы-
ваемой рН5-фракции надосадочной жидкости. Последнюю i олучают после
ультрацентрифугирования разведенного гомогената клеток при 105 000 g в те-
чение 1 ч, в результате чего все структурные элементы клеточного содержимо-
го осаждаются. При подкислении надосадочной жидкости СН3СООН (НС1
денатурирует ферменты) до pH 5,3 из нее ыпадает осадок, содержащий смесь
активирующих аминокислоты ферментов. Поэтому первоначально их назы-
вали рН5-ферментами. В соответствии с современной номенкла урой их
называют аминоацил-тРНК-синтетазами (АРСазами).
Из работ последних лет следует, что аминоацил-тРНК-синтетазы в клетках
существуют в виде высокомолекулярных комплексов—кодосом. При констан-
те седиментации 26—29S и М~ 1,4 мегаДа эти комплексы включают несколь-
ко АРСаз (специфичных, например, к ала, гли, глу, лей и сер или к лиз, мет,
арг, лей и три) и ферменты, модифицирующие АРСазы и регулирующие их
активность—протеинкиназы, метилтрансферазы, АДФ-рибозилтрансферазы,
фосфопротеинфосфатазы и др.
Ферменты, активирующие аминокислоты, исключительно лабильны, они
легко денатурируются с потерей активности. Даже разрушение клеток ультра-
звуком при выделении энзиматического препарата и непродолжительное хра-
нение ведут к их инактивации. Для нормальной деятельности активирующих
аминокислоты ферментов необходимо рисутствие в реакционной среде
Mg2+, так как связывание АТФ идет по имидазольному радикалу гистидина
активного центра фермента через ион магния.
Молекулярные массы болыпинств АРСаз (их выделено в высокоочищен-
ном состоянии более 50) близки к 100000, хотя в отдельных случаях достигают
200000 и более (глицил- и фенилаланил-тРНК-синтетазы). Молекулы у боль-
шей части АРСаз являются димерами или тетрамерами (иногда—тримерами)
и лишь у единичных АРСаз (например, у аланил-, аргинил-, аспартил-, валил-
и изолейцил-тРНК-синтетаз) представлены одной полипептидной цепью,
включающей примерно 1000 аминокислотных остатков. Характерно, что
у АРСаз с димерной структурой субъединицы работают сопряженно: высво-
бождение синтезированной аминоацил-тРНК с одной из них происходит в тот
момент, когда к другой вслед за АТФ и аминокислотой присоединяется тРНК.
Следовательно, каждая субъединица в молекуле АРСазы обладает тремя
структурно обособ енными центрами связывания перечисленных субстратов,
причем посадка каждого предшествующего субстрата облегчает акцептирова-
ние последующего. Поэтому в процессе аминоацилирования тРНК легко
возникают тройные фермент-субстратные комплексы. Наименее прочно с АР-
Сазами связывается АТФ (Х,= 1СГ4 моль), на порядок лучше—аминокислоты
(Х,=10-5 моль) и наиболее мощно—тРНК (Кя= 10-® моль). АРСазы*—са-
мые «медленные» ферменты; их молекулярная активность составляет от не-
скольких десятков до нескольких сотен каталитических актов в 1 мин.
Исходя из изучения различными методами активных центров АРСаз и ис-
следования кинетики ускоряемых ими реакций активирс вания аминокислот,
начал проясняться механизм их действия. Первой к АРСазе присоеди яется
АТФ, аденилирующая остаток гистидина в активном центре фермента с выде-
лением пирофосфата:
282
он он
I I
НО—Р—О—Р—он
Пирофосфат
С аденилированной АРСазой взаимодействует активируемая при ее по-
средстве аминокислота, в результате чего возникает аминоациладенилат. По-
следний, обладая выдающейся способностью аминоацилировать любые ради-
калы, содержащие подвижный атом водорода, передает аминоацильную груп-
пу на радикал гистидина активного центра АРСазы, в результате чего
образуется аминоацилированный фермент:
Т pi~co—сн—NH,
N=CHZ £
После вязывания ферментом соответствующей тРНК аминоацильная
группа передается на ОН-группу остатка аденозина ее акцептирующего конца
(уравн ние реакции см. с. 281). У некоторых АРСаз роль остатка гистидина
в осуществлении приведенных выше этапов активирования аминокислоты
может играть радикал глутаминовой кислоты.
При образовании фермент-субстратного комплекса из АРСазы и тРНК
и та и другая претерпевают конформационные перестройки, облегчающие
реакцию аминоацилирования. Однако самым существенным и интригующим
моментом взаим действия АРСазы и тРНК является их способность безоши-
бочно узнавать друг друга в соответствии с абсолютной специфичностью к той
или иной аминокислоте. По-видимому, она базируется на многоточечном
взаимодействии фермента и тРНК, причем существенная роль в узнавании
принадлежит антикодону тРНК.
Долгое время считали, что аминоацильная группа всегда присоединяется
по ОН-группе З'-углеродного атома рибозы концевого аденозина молекулы
тРНК. Оказалось, что иногда эту же функцию может выполнять гидроксил
2'-углеродного атома остатка рибозы. Это наблюдается при активировании
фенилаланина, лейцина и изолейцина при участии соответствующих АРСаз.
В противоположность этому серил- и треонил-тРНК-синтетазы обеспечивают
присоединение аминоацильных групп только по 3'-углеродному атому
283
рибозы, а тирозин- и цистеин-тРНК-лигазы—по 2'- и по 3'-углеродным
атомам. Установлено также, что аминоацильная группа в аминоацил-тРНК
способна перемещаться от 2'- к 3'-углеродному атому рибозы и наоборот,
вплоть до установления равновесия:
Полагают, что аминоацилирование тРНК либо в 2'-, либо в 3'-положении
остатка рибозы концевого аденозина’ предупреждает от ошибок при активиро-
вании стереохимически близких аминокислот (например, валина и треонина).
Впрочем, если даже ошибка произошла, т. е. вместо специфической для дан-
ной тРНК аминокислоты к ней присоединилась какая-то иная, АРСаза может
исправить ошибку сама, так как обладает способностью гидролизовать чуж-
дые ей неспецифические аминоациладенилаты.
Таким образом в процессе активирования неуклонно обеспечивается строго
избирательное присоединение каждой аминокислоты к специфичной для нее
Время инкудации, мин
Рис. 94. Доказательство
включения 14С-лейцина
в белок (7) из состава 14С-
лейцил-тРНК (2) поясне-
ние в тексте)
тРНК и создание набора аминоацил-тРНК, непосре-
дственно поставляющих энергетически обогащенные
аминокислотные остатки в рибосому.
То, что это действительно так, т. е. что аминоацил-
тРНК являются единственным источником аминокис-
лотных остатков при биосинтезе белка, доказано
в опытах с аминоацил-тРНК, содержащими 14С-ами-
нокислоты: по мере инкубации в белоксинтезирующей
системе содержание меченой аминокислоты в препара-
те аминоацил-тРНК падает, но в такой же пропорции
зеркально возрастает в синтезируемом белке (рис. 94).
Активирование аминокислот сопровождается их
кодированием (шифрование): после присоединения
к соответствующей тРНК аминокислота получает код,
или шифр, в виде строго специфичного только для
данной аминокислоты чередования трех нуклеотидных
остатков в антикодоновой петле тРНК (см. с. 216).
Этот триплет оснований называется антикодоном. Ему
соответствует комплементарный кодон в составе
284
мРНК. Взаимодействие кодонов мРНК с антикодонами аминоацил-тРНК
предопределяет порядок чередования аминокислотых остатков в синтезируе-
мом по матричной схеме белке. Это принципиально важное положение доказа-
но экспериментально и известно как «опыт Шапвиля» (по имени одного из
ученых, участвовавших в его проведении). В этом опыте цистеинил-тРНК путем
восстановления по аминоацильному остатку была превращена в аланил-тРНК:
H2N—СН—СО~О—тРНК4®' --стаго-Х H2N—СН—СО-О—tPHK4HC+H2S
ление
CH2SH сн3
Синтезированная так аланил-тРНК цис содержит остаток аланина, присое-
диненный к тРНК, несущей антикодон цистеина. Когда ее ввели в белоксин-
тезирующую систему, в полипептидной цепи образующегося белка позиции
цистеина оказались занятыми остатками аланина. Это однозначно указывает
на то, что именно тРНК кодирует вступление соединенного с ней аминоациль-
ного остатка в заданное положение в новообразуемой белковой молекуле.
Поэтому аминоацил-тРНК-синтетазы одно время называли кодазами или
шифразами; однако комиссия по номенклатуре ферментов не рекомендовала
использовать эти термины, и их сейчас не употребляют.
Матричный механизм сборки полипептидной цепи. Биосин-
тез белков во всех структурных элементах клетки (ядро, митохондрии, хлоро-
пласты, эндоплазматический ретикулум и др.) идет на рибосомах. Поэтому
именно на пути исследования структуры и свойств рибосом, а также механиз-
ма их взаимодействия с исходными для биосинтеза белков соединениями
достигнуты наиболее впечатляющие результаты в выявлении закономерно-
стей новообразования белковых тел.
Строение и свойства рибосом. Рибосомы—мельчайшие тельца
рибонуклеопротеиновой природы. Они содержатся в основном в цитоплазме
растительных, животных и бактериальных клеток в количестве нескольких
десятков тысяч в каждой.
Рибосомы выделяют путем дифференциального центрифугирования кле-
точного содержимого, получаемого после гомогенизации клеток. Известно,
что этим путем удается разделить клеточное содержимое на ряд фракций (см.
гл. I). Рибосомы связаны главным образом с мембранами эндоплазматиче-
ской сети. Поэтому при фракционировании содержимого клетки они осажда-
ются вместе с обломками липопротеиновой мембраны—микросомами.
Для выделения рибосом микросомы обрабатывают дезоксихолатом—ре-
агентом, растворяющим липиды и, следовательно, разрушающим микросо-
мы; затем рибосомы осаждают в изотонических буферных растворах центри-
фугированием при 100000 g в течение 1 ч. При разрушении других субклеточ-
ных частиц (ядра, пластиды, митохондрии) из них также могут быть выделены
рибосомы. В настоящее время выделение рибосом осуществлено из многочис-
ленных объектов—представителей животного и растительного мира, а также
микроорганизмов. Форма и структура рибосом, выделенных из разных источ-
ников, сходна, но по значению константы седиментации их делят на два
класса: 70S и 80S. Первые найдены у всех прокариот, а также содержатся
в ядрах, митохондриях и хлоропластах эукариот; вторые локализованы в ци-
топлазме только эукариот.
70S и 80S рибосомы стабильны при концентрации Mg2+, близкой
к 0,001 М. При повышении содержания Mg2+ до 0,01 М рибосомы дают
димеры или слипаются в более крупные агрегаты, а при понижении до
285
0,0001 М диссоциируют на субчастицы: 70(80)S?^30(40)S+50(60)S. Этот про-
цесс диссоциации—ассоциации рибосом и их субчастиц—контролируется
также пол аминами (см. с. 211) и изменением конц нтрации других двухва-
лентных катионов.
Будучи исключительно рибонуклеопротеинами (их называют также РНП-.
частицами), рибосомы составлены из почти равных количеств РНК и белка,
варьирующих в зависимости от класса рибосом (табл. 22) с определенно
выраженной тенденцией к некоторому преобладанию в них белка.
Таблица 22
Состав рибосом
Класс рибосом 70S 80S
Субчастицы рибосом 30S 50S 40S 60S
РНК 16S 23S и 5S 18S 28S, 5S и 5,8S
Число белков 21 34 31 41
Молекулярные массы белков, тыс. дальтон 12—65 9—31 10—44 10—54
Соотношение РНК/белок, % 37/63 36/64 54/46 41/59
Кроме белков и нуклеиновых кислот, в составе рибосом в ничтожных
оличествах обнаружены другие вещества: Mg2+ и Со2+, ди- и полиамины,
ряд других катионов (Fe3+, Zn2+, Al3+, Ва2+, Sr2+, Ni2+, Сг2+, NH4),
а также латентная рибонуклеаза.
Рибосомы в нативном состоянии, т. е. в живых клетках, сильно гидратиро-
ваны: так, на 1 г сухого вещества рибосом из ретикулоцитов приходится 2,7 г
гидрота ионной воды. Это свидетельствует об исключительно высокой «по-
ристости» нативных рибосом.
Белок и РНК в рибосомных частицах соединены очень непрочными
связями: уже при инкубации с 0,5—1 М растворами солей при низкой темпера-
туре происходит отделение белка от нуклеиновой кислоты. Аналогичная
диссоциация происходит, например, при pH 12 в условиях электрофореза.
Связи белков с нуклеиновыми кислотами в рибосомах оценивают в основном как
электростатические. Высокое содержание в суммарных рибосомальных белках
основных аминокислот (12% лизина, 11 % аргинина и 3% гистидина), заряжен-
ных положительно, и большое число межнуклеотидных фосфатов в рибосо-
мальных РНК, зар женных отрицательно, обеспечивают условия для возник-
новения достаточного количества ионных связей между первыми и вторыми.
Структура и функция рибосомальных высокомолекулярных (16— 18 и 23—28S)
и низкомолекулярных (5S и 5,8S) РНК рассмотрена ранее (см. гл. VI). Что касается
структуры и функции рибосомальных белков, то об этом в последние годы
получена существе ная информация. Как видно из табл. 22, и малые (30—40S),
и большие (50—60S) субчастицы рибосом содержат в своем составе строго
определенное число белков, крайне гетерогенных по молекулярным массам. Белки,
локализованные в малых (30—40S) субчастицах рибосом, обозначают буквой S (от
англ, smoll—малый), в больших (50—60S)—L (от франц, large—большой).
Благодаря применению современ ых методов белковой химии все белки из
рибосом кишечной палочки получены в высокоочищенном гомогенном со-
стоянии, у всех у них определены первичные структуры, у многих из них—
вторичные и третичные структуры. Как правило, молекулы рибосомальных
белков асимметричны, характеризуются наличием значительного количества
286
Рис. 95. Третичные структуры белков S6, S8, S15, S16, S17 и L7 (А) и атомт
ная модель белка L7 (Б):
спиральные участки представлены цилиндрами; р-структуряые—стрелками. В белке S6 N-коицевой
фрагмент 105—135 не обладает третичной структурой. В атомной модели рибосомального белка L7
цифрами обозначены номера N- и С-аминокислотньгх остатков а-спиральных фрагментов полипеп-
ой цепи, а овалом между а-спиралями 51—59 и 93—101 — полость, в которую входят а-спираль
4—11 при образовании димера
51-59
16-М
93-101
93-101
•Л
Б
а-спиралей, глобулярны и ком-
пактны (рис. 95). Некоторые из
них, будучи обогащены гидро-
фобными аминокислотами на
тех или иных участках полипе-
птидной цепи, легко образуют
димеры и даже олигомеры,
а также, взаимодействуя друг
с другом,—ассоциаты в субча-
стицах рибосомы.
В течение последнего деся-
тилетия достигнуты большие
успехи в выяснении простран-
ственного строения рибосом
в целом и локализации в их
субчастицах отдельных струк-
турных элементов (см.
табл. 22). В этом отношении
следует особо подчеркнуть за-
слуги советской школы биохи-
миков и, в частности, сотруд-
ников Института белка в Био-
логическом центре РАН
(г. Пущино-на-Оке Москов-
ской области)—здесь под ру-
Рис. 96. Пространственная структура 70S рибосомы
кишечной палочки (модель):
ЗОБч^бчастица—спереди; ее размеры: длила— ~23 им, ширина—12 ши;
508-субчастица—сзади; ее размеры во всех направлениях около 20—
23 нм; боковые выступы (палец—справа и боковая долл—слева) содер-
жат белки L7/L12 и L1 соответственно. Субчастицы ассоциированы «го-
ловка к головке» и боковой выступ к боковой доле («платформа к L-
ребру»). 308-субчастица закрывает только часть SOS-субчастицы, оставляя
свободным район, примыкающий к L7/L 12-стержню, где, вероятно, раз-
мещаются функционально важные центры рибосомы. По-видимому, при
ассоциации 30S и 50S субчастиц в области впадины между головкой
и телом 308-субчастицы и основанием центрального протуберанца SOS-
субчастицы возникает отверстие (зазор), предназначенное, как полагают,
для размещения молекулы мРНК
ководством акад. А. С. Спирина выполнены фундаментальные исследования
рибосомального аппарата клетки. Топология рибосом представлена на
рис. 96. Сейчас ясно, что основу пространственного строения рибосом задает
третичная структура 16—18S и 23—28S рНК, предопределяющая форму
и конструкцию 30—40S и 50—60S субчастиц рибосомы соответственно. Это
особенно ярко выступает при анализе данных, полученных при выяснении
строения 30S субчастицы рибосомы кишечной палочки А. С. Спириным
и сотр. Оказалось, что морфологическая модель 30S субчастицы предопреде-
ляется 16S рРНК, находящейся в компактной форме, с которой взаимодей-
ствуют и располагаются на ней в определенных позициях белки S1—S21,
найденные в этой субчастице (рис. 97). Аналогично построена 50S субчастица
рибосомы кишечной палочки.
Принципиальное значение для понимания того, как в рибосоме идет синтез
белка, имеют данные о локализации в ней центров, где осуществляются
важнейшие этапы биосинтеза полипептидной цепи: связывание аминоацил-
тРНК, связывание белковых факторов, необходимых для функционирования
рибосомы, считывание информации о порядке расположения аминокислотных
остатков в новообразуемом белке, синтез пептидной связи, удаление тРНК,
высвобождающейся после образования пептидной связи, перенос пептидил-
тРНК из аминоацильного центра в пептидильный центр с одновременным
продвижением мРНК на один триплет нуклеотидных остатков, гидролиз
сложноэфирной связи между полипептидом и тРНК в момент завершения
синтеза белка в рибосоме и др. Поэтому рассмотрение современных представ-
лений о структуре рибосомы логично завершить данными о функциональной
модели рибосомы (Г. Виттман и др., 1974), где показаны ее функциональные
центры (рис. 98).
Механизм биосинтеза белка в рибосоме. Схема, иллюстрирую-
щая механизм биосинтеза белка в рибосоме, представлена на рис. 99. Она
288
30S
50S
30 S 1BSPHK
Рис. 97. Пространственная структура 308-субч стицы рибосомы кишеч-
ной палочки:
А—морфологическая модель; Б—расположение белков и РНК в соответствии с моделью
(белки, находящиеся на заднем плане, заштрихованы)
П-нентр А-центр
мРНК
аа аминокислотный остаток
ПТаза Пептидилтрансфераза
•••••• П-центр (пептидильный центр)
оооооо
A-центр (аминоацильный центр)
Пептидилтрансферазный центр
ГТФазный центр
EF—Тц -связывающий центр
EF—G -связывающий центр
Зона считывания информации
Рис. 98. Функциональная модель рибосомы 70S кишечной палочки
10—3502
289
суммирует представления о матричном биосинтезе белка, сложившиеся на
основании его изучения у бактерий, в частности у кишечной палочки. Из схемы
видно, что биосинтез белка складывается из трех этапов: инициации, элон-
гации и терминации.
Инициация биосинтеза белка у бактерий происходит при участии трех
белковых факторов инициации—IF-1, IF-2 и IF-3 (Initiation Factors 1, 2 и 3).
IF-3 представлен белком с М=21 000—23500. Он вызывает конформационные
изменения в 30S субчастице рибосомы, способствующие связыванию ею IF-1,
IF-2, ГТФ, мРНК и формилметионил-тРНК. Присоединение последней обес-
печивает поступление в рибосомальный аппарат клетки первой, N-концевой
аминокислоты, а именно—формилметионина, которым открывается полипеп-
тидная цепь любого белка, синтезируемого у бактерий. Впоследствии N-
концевой остаток формилметионина в результате посттрансляционных из-
менений белка может отщепляться. тРНК, переносящая формилметионин
в процессе белкового синтеза у бактерий, отличается от тРНК, осуществля-
ющей перенос метионина в процессе сборки полипептидной цепи, т. е. сущест-
вуют тРНКфмет и тРНКмет:
Метионил-тРНК Формилметионил-тРНК
Именно IF-3 присоединяется первым к 30S субчастице рибосомы, открывая
этап инициации белкового синтеза и изымая 30S субчастицу рибосомы из пула
свободных 30S и 50S субчастиц рибосом, возникающих в процессе диссоци-
ации рибосом 70S. Он же способствует созданию на субчастице 30S мРНК-
связывающего центра.
IF-1 и IF-2 представляют белки с молекулярными массами 8900—9400
и 90000—118000 соответственно. IF-1 стимулирует процесс связывания фак-
тора IF-2 с 30S субчастицей рибосомы и способствует присоединению к ней
мРНК. IF-2 играет центральную роль в связывании формилметиониновой
тРНК с субчастицей 30S и в гидролизе гуанозинтрифосфата. Последний
необходим для осуществления инициации биосинтеза белка, хотя полностью
его функция в этом процессе еще не выяснена.
Присо динение IF-1, IF-2, формилметионил-тРНК и ГТФ к 30S субчастице
осуществляется в виде комплекса (см. рис. 99). Как только оно произойдет, к ней же
присоединяется мРНК причем в результате взаимодействия антикодона формил-
метиониновой тРНК с кодоном мРНК (им является триплет АУГ) предопределя-
ется такое расположение мРНК на рибосоме, которое обеспечивает далее
считывание содержащейся в последовательности ее нуклеотидных остатков
информации о ервичной структуре синтезируемого белка. Таким образом, именно
формилметиониновая тРНК помогает мРНК найти на 30S субчастице ту позицию,
которая необходима для трансляции информации о последовательности амино-
кислотных остатков в белке, и именно в этом заключается ее значение в инициации
белкового синтеза у бактерий. У эукариот эту функцию часто выполняет тРНКм",
но опять специфичной структуры, которая отличается от структуры тРНКм",
переносящей остатки метионина в процессе элонгации белкового синтеза.
Вместе с тем сейчас показано, что гораздо большее значение для стабилизации
необходимого расположения мРНК в 30S субчастице имеет присутствующая в ней
290
Рис. 99. Схема биосинтеза белка у кишечной палочки (пояснение в тексте):
три главных этапа матричного биосинтеза белка—инициация, элонгация и терминация—выделены рамками;
белковые факторы инициации и элонгации указаны условными значками, приведенными непосредственно на
схеме; часть условных обозначений дана отдельно в верхнем правом углу рисунка
последовательность Шайна—Дальгарно (короткая—ААГГА или длинная—
УААГГАГГ), взаимодействующая с комплементарным фрагментом на З'-конце
16S рРНК и находящаяся на расстоянии от 5 до 13 н. о. от стартового АУГ кодона.
Пр соединение мРНК к комплексу, состоящему из 30S суб астицы, факто-
ров инициации и ГТФ сопровождается высвобождением фактора инициации
IF-3, выполнившего присущую ему функцию (см. рис. 99). Комплекс, включа-
ющий теперь в свой состав еще мРНК, жадно притягивает 50S субчастицу
и соединяется с ней, образуя 70S рибосому, причем IF-1 в этот момент
покидает рибосому, так как он тоже выполнил свою роль: стабилизацию
в рибосоме мРНК. Сохранившийся еще в возникшей 70S рибосоме IF-2,
связанный с ГТФ, обеспечивает ускорение реакции распада ГТФ на ГДФ
и неорганический фосфа г и высвобожда тся из рибосомы вместе с продуктами
291
гидролиза ГТФ. Выделяющаяся при гидролизе ГТФ энергия, видимо, необ-
ходима для осуществления конформационных перестроек в рибосоме 70S,
в результате чего формилметионил-тРНК стабилизируется в пептидильном
центре рибосомы (см. 1-ю фазу элонгации на рис. 99). Такая рибосома способ-
на теперь вести сборку полипептидной цепи белка заданной структуры, вслед-
ствие чего ее называют транслирующей (активной) рибосомой. В транслиру-
ющей рибосоме идет, следовательно, процесс элонгации белкового синтеза.
В клетках эукариот инициация рибосомального белкового синтеза идет
тоже при участии эукариотических факторов инициации (elF—eukaryotic
Initiation Factors). Их насчитывается девять: eIF-1, eIF-2, eIF-3, eIF-4A, eIF-4B,
eIF-4C, eIF-4D, eIF-5 и eIF-6. Все они, за исключением eIF-2 и eIF-3, представ-
лены одиночными полипептидами с М от 15000 (elF-Г) до 160000 (eIF-5).
Фактор eIF-2, по-видимому, является димером (М = 86000; а-субъединица—
38000; Р-субъединица—48000); eIF-З состоит из нескольких субъединиц при
суммарной молекулярной массе более 500000.
Из девяти перечисленных белковых факторов абсолютно необходимы для
инициации биосинтеза белка у эукариот eIF-2, eIF-З и eIF-5, а остальные факторы
усиливают функции этих трех. Фактор eIF-2 взаимодействует своей Р-субъедини-
цей с метионил-тРНК, а а-субъединицей—с ГТФ, образуя комплекс с субчасти-
цей 40S. Его деятельность контролируется фосфилированием а-субъединицы при
участии специфической протеинкиназы. Фактор eIF-З тоже присоединяется
к субчастице 40S, что сопровождается возникновением на ней мРНК-связыва-
ющего центра и стабилизацией связи с нею комплекса eIF-2 • метионил-
тРНК • ГТФ. После присоединения мРНК к субчастице 40S по мРНК-связыва-
ющему центру она, при участии фактора eIF-5, энергично притягивает 60S
субчастицу, происходит выброс всех присоединенных ранее, в том числе и eIF-5,
белковых факторов, гидролиз ГТФ и закрепление метионил-тРНК в пептидиль-
ном центре возникшей активной, способной осуществлять трансляцию рибосо-
мы 80S. Фактор elF-б обеспечивает диссоциацию 80S рибосомы на субчастицы
40S и 60S, высвобождая 40S субчастицу для нового цикла инициации.
Элонгация биосинтеза белка в бактериальной клетке обслуживается тремя
белковыми факторами элонгации: EF-TU, EF-TS и EF-G (элонгационные факто-
ры трансляции трех типов—и, s и G). У млекопитающих два фактора элонга-
ции: TF-1 и TF2 (трансляционные факторы первый и второй). EF-TU (М=47 000)
и EF-TS (М = 35ООО) бактерий соответствует TF-1 (М= 186000) млекопитаю-
щих, a EF-G бактерий—TF-2 (М = 70000) млекопитающих. EF-G кишечной
палочки имеет молекулярную массу 77321,45 и представлен полипептидом (701
аминокислотный остаток), первичная структура которого выяснена.
Процесс элонгации начинается со связывания аминоацил-тРНК, содержащей
аминокислотный остаток, который должен быть вторым с N-конца молекулы
синтезируемого в рибосоме белка. У бактерий эта аминоацил-тРНК образует
комплекс с EF-TU и ГТФ, в виде которого она присоединяется к аминоацильному
центру транслирующей рибосомы в соответствии с кодом белкового синтеза
(см. ниже), т. е. благодаря взаимодействию комплементарных триплетов анти-
кодона тРНК и кодона мРНК, локализованного против аминоацильного центра
рибосомы (см. рис. 98 и 99). У млекопитающих это происходит при участии TF-1.
По современным данным, не только кодон—антикодоновое взаимодейст-
вие предопределяет отбор соответствующих аминоацил-тРНК для сборки
полипептидной цепи. В этом участвует вся молекула тРНК, так как посттран-
сляционная модификация сильно изменяет ее способность акцептироваться
рибосомой. В процесс декодирования вовлекаются и белки рибосомы: S4, S9,
S13, L2 и L7. Два последних, будучи локализованы в пептидильном центре
рибосомы, образуют с двумя молекулами тРНК тетрамерный комплекс.
292
И аминоацил-тРНК, и ГТФ связываются с EF-TU за счет свободных
HS-групп остатков цистеина его молекулы, причем первичная структура цент-
ра связывания ГТФ (и ГДФ соответственно) на протяжении 42 аминокислот-
ных остатков расшифрована. Активность EF-TU регулируется фосфорилирова-
нием и взаимодействием с pplpp *.
Благодаря расщеплению ГТФ на ГДФ и неорганический фосфат амино-
ацил-тРНК сближается с формилметионил-тРНК, локализованной в пепти-
дильном центре рибосомы, a EF-TU в комплексе с ГДФ и неорганический
фосфат выносятся из рибосомы. EF-TU • ГДФ-комплекс при взаимодействии
с EF-TS и ГТФ преобразуется в EF-T„ • ГТФ-комплекс, способный соединяться
со следующей молекулой аминоацил-тРНК (см. рис. 99).
В пептидильном центре между формилметионил-тРНК и аминоацил-тРНК
происходит реакция, благодаря которой остаток формилметионина перено-
сится на свободную NH2-rpynny аминокислотного остатка, являющегося со-
ставной частью аминоацил-тРНК. В результате возникает дипептидил-тРНК,
т. е. замыкается первая пептидная связь в будущей молекуле белка, а также
образуется деацилированная тРНКфмет (см1 рис. 99). Этот процесс получил
название реакции транспептидирования. Он ускоряется соответствующим фер-
ментом, причем транспептидазная активность присуща рибосомным белкам
L16, L11 и L6 и, вероятно, фрагменту 23S рРНК (см. рис. 98).
Пептидил-тРНК на следующей фазе элонгации переносится на место
тРНКфмет в пептидильном центре рибосомы, а последняя удаляется из него,
перемещаясь в £-центр рибосомы, из которого она затем вытесняется очеред-
ной аминоацил-тРНК и выносится из рибосомы.
Эта ступень элонгации называется транслокацией и происходит при участии
EF-G у бактерий и TF-2 у эукариот, а также сопровождается непременным
гидролизом еще одной молекулы ГТФ (см. рис. 99). В результате транслокации
дипептидил-тРНК занимает место в пептидильном центре рибосомы, тогда как
ее аминоацильный центр полностью освобождается и готов еперь принять
новую аминоацил-тРНК вкупе с EF-TU или TF-1 и ГТФ. Особенно важно, что
при транслокации пептидил-тРНК перемещается в пептидильный центр рибосо-
мы вместе с молекулой мРНК, с которой она связана благодаря антикодон-
кодоновому взаимодействию (см. рис. 98). Это перемещение идет точно на один
триплет нуклеотидных остатков, т. е. напротив аминоацил ного центра ока-
зывается следующий по порядку кодон молекулы мРНК, предопределяющий,
какая аминоацил-тРНК вступит в аминоацильный центр рибосомы и явит-
ся источником очередного аминокислотного остатка в новообразуемом
белке.
Терминация белкового синтеза в рибосоме осуществляется тоже при уча-
стии трех белковых факторов: RF-1, RF-2 и RF-3 у бактерий и ед нственного
белкового фактора—R—у высших организмов (от англ, recognize—узна-
вать). В клетке кишечной палочки насчитывается примерно по 500 молекул
этих белков. Белковые факторы RF-1 и RF-2 имеют молекулярную массу по
45000 и способны распознавать в молекуле мРНК терминирующие сборку
полипептидной цепи кодоны: фактор RF-I—У\Г и У\А, а фактор RF-2—УХА
и УГА. Фактор RF-3 (его называют также S-протеин) стимулирует действие
факторов RF-1 и RF-2.
Как только в аминоацильном центре рибосомы после очередной трансло-
кации терминирующий кодон молекулы мРНК займет соответствующее ме-
сто, к нему присоединяется один из факторов терминации RF-1 или RF-2.
1 РРГРР—3 '-пирофосфо-гуанозин-5 '-пирофосфат.
293
Этим блокируется возможность присоединения молекулы аминоацил-тРНК,
тем более, что терминирующим кодонам не соответствует ни один из антико-
донов в тРНК. Присоедине ие к мРНК фактора RF-1 или RF-2 возбуждает
пептидилэстеразную активность рибосомальных белков, в частности белков LI 1
и L16, локализованных в 50S субчастице (см. рис. 98), вследствие чего гид-
ролизуется сложноэфирная связь между новообразованным п< липептидом
и тРНК. В результате синтезированный в рибосоме белок отделяется от нее.
Одновременно освобождаются тРНК и мРНК, а рибосома 70S распадается на
субчастицы 30S и 50S, поступающие в общий фонд рибосом и их субчастиц,
откуда они черпаются для нового цикла биосинтеза белковой молекулы (см.
рис. 99). В терминации биосинтеза белка по матричной схеме принимает
участие молекула ГТФ. У бактерий она служит аллостерическим регулятором
активности белковых факторов терминации, а у животных распадается на
ГДФ и неорганический фосфат.
Долгое время считали, что у эукариот—единственный распознающий
терминирующие кодоны фактор (eRF). Он был охарактеризован (50 кДа)
и секвенирован. Однако недавно описана группа родственных ему белков,
азванных eRFl. Эти белки выделены из человека, лягушки и дрожжей,
секвенированы (428, 437 и 437 аминокислотных остатков соответственно),
взаимодействуют с тремя терминирующими кодонами и ГТФ-независимы,
что делает контроль терминации биосинтеза белков более надежным:
Мультиэнзимный механизм биосинтеза пептидов. Разработка мультиэнзим-
ного пути биосинтеза пептидов осуществлена Ф. Липманом и сотр. (1968).
В настоящее время доказано, что по меньшей мере 5 антибиотиков пептидной
природы: грамицидин, тироцидин, бацитрацин, циклоспорин и микобацил-
лин—синтезируются без всякого участия нуклеиновых кислот, в том числе
и тРНК, но при этом обеспечивается безошибочная сборка полипептидных
цепей, характеризующихся определенной первичной структурой.
Биосинтез грамицидина S осуществляется при посредстве мультиэнзимной
системы, состоящей из двух белковых фракций с молекулярными массами 280 000
и 100 000. Они выделены из белкового экстракта Bacillus brevis, из которого при
посредстве гельфильтрации через сефадекс G-200 удалось получить две взаимо-
дополняющие друг друга упомянутые выше белковые фракции. Будучи соедине-
ны вместе, в присутствии аминокислот, АТФ и Mg2 + они обеспечивают синтез in
vitro циклического декапептида—грамицидина S (рис. 100).
Легкая белковая фракция (М = 100 000) рацемизирует и активирует D-фенилала-
нин, а тяжелая (М = 280 000)—активирует 4 остальные L-аминокислоты. Активи-
рование аминокислот указанными ферментами осуществляется путем образования
на 1-м этапе аминоациладенилатов перечисленных аминокислот (по реакции между
свободными аминокислотами и АТФ с выделением пирофосфата), а на 2-м—путем
переноса аминоацильных групп с аминоациладенилатов на HS-группы остатков
цистеина, содержащихся в полипептидной цепи самого фермента, с образованием
тиоэфиров аминокислот. В таком состоянии легкая фракция, содержащая активиро-
ванный по СООН-группе D-фенилаланин будучи соединена с тяжелой фр кцией,
содержащей активированные по СООН-группам L-пролин, L-валин, L-орнитин
и L-лейцин, инициирует последова ельный биосинтез пептидных связей между
перечисленными аминокислотами в том порядке, в каком они располагаются
в молекуле грамицидина S. Непосредственно перенос остатка D-фенилаланина
с его тиоэфирной связи (на легкой белковой фракции) на NH2-rpynny остатка
L-пролина, соединенного тиоэфирной связью с субъединицей тяжелой белковой
фракции, осуществляется аминоацилпроводящим белком (АЛБ), присутствующим
в мультиэнзимном комплексе. Это белок сравнительно небольшой молекулярной
массы (около 20 000) с пантотеином в качестве простетической группы:
294
Г- со ОН сн3
I I I I
АПБ СН—О— Р — О—СН2— С—СН—СО—NH— СН2— СН2— СО—NH—СН2—СН2—SH
II II I I
*- NH О СН3ОН
Остаток D-фенилаланина сначала переносится на HS-группу пантотеина,
а затем с нее—на NH2-группу L-пролина. Так возникает первая пептидная
связь в будущей молекуле грамицидина. Далее остаток дипептида D-фен-Ь-
про поступает с тиоэфирной связи (с субъединицей тяжелой белковой фракции)
на HS-группу пантотеина АПБ, а с нее—на NHi-группу остатка валина.
Пе тиди трансферазная реакция повторяется до тех пор, пока не будет синте-
зирован пентапептид D-фен—-про—‘вал—>орн—-лей (см. рис. 100). Так как одно-
временно на расположенном рядом идентичном мультиэнзимном комплексе
синтезируется еще один такой же пентапептид, то они соединяются по типу
«голова к хвосту» и образуют молекулу циклического декапептида—грамици-
дина S (см. рис. 100).
В соответствии с рассмотренным выше механизмом идет биосинтез друго-
го антибиотика пептидной природы—тироцидина:
D- фен—‘про—‘фен—‘D-фен—‘асн
лей— орн—вал-------тир—глн
Здесь мультиэнзимная система осуществляет полную сборку циклического
декапептида, т. е. последовательно синтезируется 10 пептидных связей в соот-
ветствии с первичной структурой пептида. Этот мультиэнзимный комплекс
устроен сложнее, так как состоит из трех белковых фракций: 1) М = 100000—
Рис. 100. Мультиэнзимный механизм биосинтеза половины молекулы грамицидина
(пояснение в тексте)
295
рацемизует (и активирует) D-фенилаланин; 2) М = 230 000—активирует L-
пролин; 3) М=460 000—активирует D-фенилаланин, аспарагин, глутамин,
тирозин, валин, орнитин и лейцин, а также рацемизует фенилаланин.
Недавно доказано, что при посредстве мультиэнзимного комплекса идет
биосинтез микобациллина—циклического 13-членного пептида, бацитрацина
А и циклоспорина А (оба— 11-членные циклические пептиды).
Ф. Липманом предпринята попытка связать матричную и нематричную
схемы биосинтеза белков в единую концепцию. Хотя в последней выяснены не
все детали, несомненно, что в эволюционном аспекте мультиэнзимный путь
сборки пол пептидов заданного строения предшествовал рибосомальному
пути их биосинтеза, и сейчас у микробов сосуществуют оба пути. Весьма
показательна аналогия в деятельности мультиэнзимных систем пептидного
биосинтеза и функционирования синтетазы высших жирных кислот, что указы-
вает на широкое использование в природе принципа мультиэнзимного биосин-
теза достаточно сложных соединений.
Кодирование биосинтеза белка. Выяснение вопроса о том, какой именно
триплет нуклеотидов в составе мРНК кодирует вступление в белок определен-
ной аминокислоты, представляет одну из самых увлекательных страниц со-
временной биохимии и молекулярной биологии.
Как перевести четырехбуквенный (по числу оснований, входящих в мРНК,
т. е. А-аденин, Г-гуанин, Ц-цитозин и У-урацил) код в двадцатибуквенный (по
числу аминокислот, составляющих белковую молекулу)?
Если каждой комбинации нуклеотидов в мРНК приписать способность
кодировать положение одной аминокислоты в белке, то дуплет 1ый код вряд
ли возможен (число пар нуклеотидов менее числа постоянно встречающихся
в белке аминокислот), квадруплетный нереален (число сочетаний слишком
сильно превышает число аминокислот), в то время как триплетный код
наиболее удовлетворяет численному соотношению возможных кодонов и бел-
ковых аминокислот. Указанные расчеты основаны на том, что при соединении
4 нуклеотидов попарно можно получить 16 комбинаций, по три—64, по
четыре—256 комбинаций и т. д.:
Дуплетный код
(4*=16)
Триплетный код
(43=64)
Квадруплетный код
(44=256)
АД УУ ГГ ЦЦ ААА УУУ ггг ЦЦЦ АААА УУУУ ПТГ ЦЦЦЦ
АУ УА ГА ЦА ААУ УУА ГГА ЦЦА АААУ УУУА ПТА ЦЦЦА
АГ УГ ГУ ЦУ УАА АУУ АГГ АЦЦ и т.д.
АЦ УЦГЦ ЦГ АУА УАУ ГАГ ЦАЦ
ААГ УУГ ГГУ ЦЦУ
ГАА ГУУ УГГ УЦЦ
АГА УГУ ГУГ ЦУЦ
ААЦ УУЦ ГГЦ ЦЦГ
ЦАА ЦУУ ЦГГ гцц
АЦА УЦУ ГЦГ ЦГЦ
АУГ У АГ ГАУ ЦАУ
АГУ УТА ГУА ЦУА
АГЦ УАЦ ГАЦ ЦАТ
АЦГ УЦА ГЦА ЦГА
АУЦ УГЦ ГУЦ ЦУГ
АЦУ УЦГ ГЦУ ЦГУ
Посредством ряда остроумных опытов триплетная природа кода белково-
го синтеза доказана экспериментально. Сначала был установлен качественный
состав нуклеотидных остатков, которые входят в состав одного или несколь-
ких кодонов, обеспечивающих вступление в состав белка той или иной амино-
кислоты. Решающую роль здесь сыграло наблюдение М. Ниренберга (1961),
296
который, используя в качестве мРНК полиуридиловую кислоту, впервые
показал, что вступление в полипептидную цепь ф нилаланина кодируется
УУУ-триплетом. Полиуридиловая кислота, поли (У), вводилась им в белок-
синтезирующую бесклеточную систему, составленную из промытых (лишен-
ных мРНК) рибосом, полного набора тРНК, аминоацил-тРНК-синтетаз и ами-
нокислот (некоторые из последних мечены 14С или 15N), АТФ и генерирующих
ее соединений (фосфоенолпируват, ацетилфосфат и подобные им вещества),
ГТФ, Mg2+ и Мп . В этой системе на поли (У) в качестве матрицы шел
синтез пептидов, составленных только из остатков фенилаланина. Благодаря
применению в этой же системе почти полного набора синтетических гомо-
и гетерополирибонуклеотидных матриц, полученных с помощью полинукле-
отидфосфорилазы, в лаборатории С. Очоа в течение года была завершена
работа по выявлению качественного состава кодонов для всех аминокислот.
Самую большую трудность представляло выяснение последовательности
нуклеотидов в кодонах, определяющих положение аминокислоты в белковой
молекуле: ведь при известном качественном составе кодона оставалось
неясным, какое же чередование нуклеотидных остатков в нем (например,
АЦУ, ЦАУ, УАЦ, АУЦ, ЦУА или УЦА) действительно кодирует данную
аминокислоту при биосинтезе белка в рибосоме. Но и эта трудность была
преодолена после того, как было обнаружено, что синтетические трину-
клеотиды, подобно мРНК, могут специфически связывать аминоацил-тРНК
с рибосомами, а в лаборатории Г. Корана были синтезированы поли-
рибонуклеотиды заданного строения (с известной последовательностью ну-
клеотидных остатков) и применены для экспериментального выявления
в бесклеточных белоксинтезирующих системах первичной структуры ко-
донов для каждой аминокислоты.
В результате уже к концу 1965 г. были получены полные данные о коде
белкового синтеза в рибосомальном аппарате клетки (табл. 23). Как видно из
таблицы, из 64 триплетов 61 кодирует последовательность вхождения амино-
кислот в полипептидную цепь в процессе ее биосинтеза в рибосоме. Три
триплета (УАА, УАГ и УГА) не участвуют в кодировании. Однако и они играют
важную роль в биосинтезе белка. Именно эти триплеты распознаются белко-
выми факторами терминации в тот момент, когда они окажутся (по мере
продвижения мРНК через рибосому) в ее аминоацильном центре. А это, как
известно, приводит к завершению синтеза белковой молекулы.
Таблица 23
Код белкового синтеза
Первая буква кодона Вторая буква кодона Третья буква кодона Первая буква кодона Вторая буква кодона Третья буква кодона
У ц А г У ц А г
фен сер. тир цис У иле тре асн сер У
фен сер тир цис и иле тре асн сер и
У лей сер А А иле тре лиз арг А
лей сер три Г мет тре лиз арг Г
лей про гис арг У вал ала асп гли У
лей про гис арг ц вал ала асп гли Ц
Ц лей про глн арг А Г вал ала глу гли А
лей про глн арг Г вал ала глу гли Г
297
Детальное изучение свойств кода белкового синтеза показало, что он
является триплетным, непрерывным, неперекрывающимся, вырожденным
и универсальным.
Триплетность кода доказана в результате проведения ряда хеленаправлен-
ных экс ериментов. Среди них—сопоставление числа мутаций в геноме фага
Т4 с появлением мутантов и их возвратом к исходному типу при обработке
бактериофага химическим мутагеном—акридиновым оранжевым (Ф. Крик);
выяснение минимальной длины фрагмента олигоуридиловой кислоты, способ-
ного связать фенилаланил-тРНК (М. Ниренберг); синтез олигорибонуклеоти-
дов заданного строения и изучение их кодирующих свойств при использовании
в качестве мРНК в белоксинтезирующей бесклеточной системе (Г. Корана);
анализ распределения аминокислотных замен в гомологичных белках (Г. Вит-
тман).
Казалось бы, не осталось уже сомнений в том, что взаимодействие трипле-
тов нуклеотидов (кодонов) в мРНК с триплетами нуклеотидов (антикодона-
ми) в тРНК однозначно определяет связывание соответствующей аминоацил-
тРНК с рибосомой. Однако в последнее время накопились факты, д называю-
щие, что существенное значение для кодирования имеют лишь два из трех
нуклеотидных остатков в кодоне (У. Лагерквист, 1978). Так, в табл. 23, где
сведены данные о коде белкового синтеза, легко заметить, что существуют
семейства кодонов, отличающиеся только третьей буквой—именно она рас-
познается в ряде случаев неоднозначно. Сначала для объяснения этого явления
использовали представление о неполном соответствии правилу комплемен-
тарности сочетания азотистых оснований в кодоне и антикодоне (wobble-
гипотеза, т. е. гипотеза, допускающая «болтанку», неточную подгонку основа-
ний). Сейчас все более придерживаются гипотезы два из трех. Таким образом,
код белкового синтеза, по существу, является квазидуплетным (условно, мни-
модупдетным).
Непрерывность кода белкового синтеза состоит в том, что все входящие
в его состав кодоны располагаются в мРНК, кодирующей биосинтез опреде-
ленного белка, в строгом порядке один возле другого, не будучи разделен-
ными иными моно- или олигонуклеотидными вставками.
Неперекрывающийся характер кода белкового синтеза заключается в том,
что ни один из нуклеотидов одного кодона не является составной частью
другого (соседнего) кодона.
Вырожденность кода сводится к его множественности для тех или иных
аминокислот. Из табл. 23 видно, что вступление в полипептидную цепь как
лейцина, так и аргинина обеспечивается шестью различными кодонами; по-
давляющего большинства других аминокислот—четырьмя или двумя кодона-
ми и лишь в одиночных случаях (метионин и триптофан)—одним кодоном.
Естественно, что вследствие вырожденности кода существуют соответствую-
щие наборы тРНК, взаимодействующих с комплектом кодонов для данной
аминокислоты.
Наконец, универсальность кода белкового синтеза определяется тем, что он
един для всего живого на земле: от простейшего фага или бактерии до венца
творения—человека и неизменен уже более 3 млрд. лет. Однако код белково-
го синтеза в митохондриях существенно отличается от такового прокариот
и эукариот и его происхождение остается загадкой. К тому же у реснитчатых
и микоплазм он тоже отличается от канонического: терминирующие кодоны
у них выполняют кодирующую функцию.
К перечисленным пяти свойствам кода следует добавить еще помехоустой-
чивость, наличие в нем знаков пунктуации (инициирующие и терминирующие
триплеты), серийносвязанность (см. серии кодонов в табл. 23), симметричность
298
(поворот на 180° не нарушает расположения сильных и слабых оснований),
сцепленность состава и расположения оснований в кодоне с полярностью
и размерами аминокислот и, наконец, однозначность (каждый кодон соответст-
вует единственной аминокислоте).
Всегда ли кодирование осуществляется с абсолютной точностью или воз-
можно ложное кодирование и включение аминокислоты в полипептидную
цепь не в соответствии со структурой кодона? Считают, что определенный
уровень ложного кодирования запрограммирован эволюционно; и в тех случа-
ях, когда вследствие мутации изменен один из кодонов в мРНК, он может
транслироваться как неизменённый («ложь во спасение»). Это помогает клеткам,
в том числе и бактериальным, выжить при неблагоприятных мутациях. Нали-
чие ложного кодирования показано и в нефизиологических условиях, в опытах
по синтезу пептидов в бесклеточных белоксинтезирующих системах. Так, на
поли (У) наряду с массированным включением в полипептидную цепь фенила-
ланина отмечено наличие в возникающих на этой матрице пептидах лейцина
и изолейцина, а также в уменьшающихся количествах серина, тирозина и ва-
лина. Ясно, что эти аминокислоты вступают в пептидную фракцию вследствие
ложного кодирования, причиной которого является квазидуплетность (два из
трех) кода белкового синтеза и возможность неполного соответствия кодона
и антикодона в процессе трансляции (wofeWe-гипотеза!). Ложное кодирование
усиливается при увеличении ко центрации в среде Mg , путресцина, сперми-
дина, этанола, а также при понижении pH и температуры инкубации. В физио-
логических условиях его уровень составляет 10-4—10“3 ошибок на кодон
и в определенной мере увеличивает жизненную гибкость и потенции клетки
(синтез вариантов белков и т. п.). Вопросы ложного кодирования в процессе
белкового синтеза успешно изучаются в Институте белка в Пущино под
руководством акад. А. С. Спирина.
Проблема кодирования биосинтеза белков в последние годы обсуждается
еще в одном принципиальном аспекте. Исходя из работ, в которых удалось
доказать способность некоторых белков без участия нуклеиновых кислот
обеспечивать собственное мультиплицирование, накопление и реализацию
присущего им патологического (инфекционного) процесса, В. А. Кордюмом
выдвинута концепция о существовании новой формы биологической инфо-
рмации—пространственной структуры белка, способной к самовоспроизве-
дению.
ПЕРЕНОС НОВООБРАЗОВАННЫХ БЕЛКОВ
ЧЕРЕЗ МЕМБРАНЫ
Значительная часть синтезируемых клеткой белков в зависимости от их
функционального назначения либо переносится через мембраны, либо встраи-
вается в них. Недавно удалось расшифровать молекулярные механизмы,
обеспечивающие перемещение столь больших молекул, какими являются бел-
ки. Оказалось, что проникновение белков через биологические мембраны,
равно как и их встраивание в мембраны, осуществляется с помощью сигналь-
ных пептидов. Такое предположение впервые было высказано в начале 70-х
годов Г. Блобелем и Д. Сабатини и стало известно в дальнейшем как сигналь-
ная гипотеза Блобеля. В настоящее время эта гипотеза подтверждена экспери-
ментально.
Сигнальные пептиды представлены фрагментами полипептидных цепей -
белков в их N-концевой части протяженностью 15—30 аминокислотных
299
остатков. Например, у предшественников альбумина, лизоцима и липопроте-
ина структура сигнальных пептидов такова:
Мет-лиз - три - вал - тре - фен - лей - лей - лей - лей - фен - иле - сер - гли - сер - ала - фен - сер -...
Мет - аре - сер - лей - лей - иле - лей - вал - лей - цис - фен - лей - про - лей - ала - ала - ала - лей - гли -...
Мет - лиз - ала - тре - лиз - лей - вал - лей - гли - ала - вал - иле - лей - гли - сер - тре - лей - лей - ала - гли -...
Легко заметить, что центральная часть сигнальных пептидов содержит
остатки гидрофобных аминокислот, а концевые последовательности обога-
щены гидрофильными аминокислотными радикалами. Такая структура
обеспечивает, при наличии не менее 7 гидрофобных радикалов подряд,
беспрепятственное внедрение сигнальных пептидов в липопротеиновую ме-
мбрану в зоне рецептора сигнального пептида и последующий перенос
белка через нее или закрепление в ней. Установлено, что перенос белков
через биологические мембраны может осуществляться либо одновременно
с трансляцией белка в рибосоме (котрансляционно), ибо по завершении
трансляции и отделения белка от рибосомы (посттрансляционно), но в лю-
бом случае ведущая роль принадлежит сигнальному пептиду (рис. 101).
Последний отщепляется по завершении переноса при участии сигналопеп-
тидазы.
Сигнальная гипотеза Блобеля позволила по-иному подойти к проблеме
активного транспорта макромолекул. Однако последняя не сводится только
к сигнальной гипотезе: получены данные о существовании специальных бел-
ков—поринов, обеспечивающих перенос макромолекул. Расширились пред-
ставления о мембранных белках, распознающих сигнальные пептиды, а неко-
торые из них (М = 34 и 54 кДа) выделены и охарактеризованы; изучена группа
белков (М = 68—72 кДа), заякоривающих новообразованные белки в мемб-
ране; обнаружены рибонуклеопротеиновые частицы, составленные из 7,8 РНК
и нескольких (до 6) пептидов, тоже узнающие сигнальные последовательности
в белках.
ПОСТТРАНСЛЯЦИОННАЯ МОДИФИКАЦИЯ БЕЛКОВ
Не следует думать, что все белки, образовавшиеся в результате рибосо-
мального синтеза, обладают полностью завершенной структурой. Во мно-
гих случаях они синтезируются в виде предшественников и лишь после
протеолитического отщепления пептидного фрагмента приобретают закон-
ченную форму. Примерами такого рода посттрансляционной модификации
белков может служить отщепление сигнальных пептидов по завершении
переноса белков через биологические мембраны (см. рис. 101), фрагмен-
тирование белковых предшественников при образовании из них функциона-
льно активных белков, например трипсина из трипсиногена, инсулина из
проинсулина, или биологически активных пептидов, например гормонов
и рилизинг-факторов. Аналогичный характер носит посттрансляционная
модификация белков, сводящаяся к протеолитическому отщеплению N-
концевого формилметионина или метионина, с которых, как показано
выше, начинается сборка полипептидных цепей в процессе рибосомального
синтеза белков.
Вместе с тем широко представлена посттрансляционная модификация
белков по аминокислотным радикалам. К их числу относятся: гидрокси-
лирование радикала пролина при переходе проколлагена в коллаген; ацети-
лирование N-концевой аминокислоты в белковой молекуле, как, например,
300
£
Рис. 101. Транслокация
белка через липопротеи-
новую мембрану:
А—контрансляционный перенос
белка; Б—посттрансляционный пе-
ренос белка. Сигнальный пептид,
взаимодействуя с рецептором мемб-
раны, обеспечивает поступление но-
вообразуемого на рибосоме белка
в липопротеиновую мембрану.
В случае котрансляциоиного пере-
носа на мембране при посредстве
специфического рецептора закрепля-
ется также рибосома, а сама транс-
локация белка осуществляется за
счет возникновения конформера по-
липептидной цепи. При посттранс-
ляционном переносе в транслокации
участвуют белковые факторы, при-
мыкающие к рецепторам сигналь-
ного пептида. И в том и в другом
случае сигнальный пептид отщепля-
ется от функциональной части белка
при посредстве пептидазы сигналь-
ного пептида; судьба его пока не
выяснена
Сигнальный
пептид
Кодоны сигнального
пептида
Рецептор сигналь-
ного пептида
Рецептор 50S субчастицы
рибосомы
[fry ^МемВоана
-•О -пептидаза сигнального пептида
Б
при биосинтезе яичного альбумина; метилирование радикалов лизина и ар-
гинина при посредстве 8-аденозил-Ь-метионин: протеин N-метил- или О-
метилтрансферазы, особенно широко представленное при посстрансляцион-
ной модификации гистонов, негистоновых ядерных белков и рибосомальных
белков; присоединение олигосахаридных фрагментов к радикалам аспараги-
на, серина и треонина при биосинтезе гликопротеинов; амидирование радика-
лов аспарагиновой и глутаминовой кислоты, что связывают в настоящее
время с изменчивостью белков в онтогенезе и, в частности, при старении;
карбоксилирование радикалов глутаминовой кислоты по у-углеродному ато-
му, в результате чего в белке возникают остатки у-карбоксиглутаминовой
кислоты, необходимые, в частности, для связывания Са2+; аденилирование
и уридилирование радикалов тирозина. К посттрансляционным процессам,
имеющим важнейшее значение для регуляции метаболической активности
генома, относится фосфорилирование гистонов и негистоновых белков хрома-
тина. Один из примеров котрансляционной модификации белков приведен на
рис. 102.
РЕГУЛЯЦИЯ БЕЛКОВОГО СИНТЕЗА
Исходя из матричной гипотезы биосинтеза белка, Ф. Жакоб и Ж. Моно
(1961) впервые предложили весьма многообещающую схему регуляции
белкового синтеза. Следуя концепции один ген—одна мРНК—один белок,
Ф. Жакоб и Ж. Моно связали узловой пункт регуляции белкового синтеза
с ДНК, входящий в состав генетического аппарата клетки. По их мнению,
в генетическом аппарате клетки существуют сообщества структурных
генов, так называемых оперонов, каждый из которых ответствен за вза-
имосвязанный синтез ряда специфических белков. Деятельность опсрона
301
в качестве поставщика мРНК контролируется геном-оператором, который
либо разрешает, либо запрещает запуск гомологической репликации серии
мРНК на ДНК-матрице. В свою очередь, функция гена-оператора кон-
тролируется пр странственно изолированным от него геном-регулятором,
который продуцирует мРНК, необходимую для синтеза белка-репрессора.
Именно белок-репрессор, будучи присоединен к гену-оператору, блокирует его
функцию. Более того, сам белок-репрессор подвержен действию аллостериче-
ских эффекторов, которые, соединяясь с ним, так изменяют его третичную
структуру, что либо стимулируют, либо ингибируют возникновение комплекса
между репрессором и геном-оператором. В качестве аллостерических эффек-
тов часто выступают субстраты (индуцированный синтез ферментов). Накап-
ливаются данные об участии в контроле биосинтеза мРНК гормонов и ряда
других соединений.
Сказанное иллюстрирует схема 4. В нее включены также информосомы, на
уровне которых тоже осуществляется регуляция биосинтеза белков:
о п Е р о н
Информосома I Информосома II
иРНК I
Белок - репрессор
Аллостернчес- Аллостеричес-
кий ингибитор кий стнмуля-
репрессора тор репрессо-
ра
иРНК II
белок I
Полисома II
Специфический
белок II
Схема 4. Регуляция биосинтеза белков
Данная схема отражает только часть тех факторов, которые принимают
участие в регуляции биосинтеза белков. Эта регуляция осуществляется также
на уровне метаболитов при активировании и переносе аминокислот; на уровне
макромолекул при биосинтезе ДНК, различных видов РНК и рибосом; на
уровне субклеточных структур (формирование полисом, роль белково-липид
ных мембран и т. п.), клетки (ядерноцитоплазменные взаимоотношения и др.),
302
5'
Рис. 102. Котрансляционное гликозилирование белка
Олигосахарид, связанный с липидом (долихолпирофосфатом—см. гл. IX), при участии гликоэилтраисферазы переносится
на остаток аспарагина синтезируемого в рибосоме белка, в результате чего возникает гликопротеин
органа и организма (гормональная регуляция) и, наконец, на уровне среды
(например, зависимость точности считывания кода белкового синтеза от
температуры).
БИОСИНТЕЗ БЕЛКОВ И НЕЗАМЕНИМЫХ АМИНОКИСЛОТ
ДЛЯ ПРАКТИЧЕСКИХ ЦЕЛЕЙ
Можно без преувеличения сказать, что в настоящее время человечество
вступило в эру индустриального производства белка—наиболее дефицитного
пищевого продукта на Земле. 11 июля 1987 г. в г. Загребе родился пятимилли-
ардный житель Земли—Матей Гашпар, и, по подсчетам специалистов из
ООН, к 2000 г. население Земли возрастет до 6,1 млрд. Однако уже сегодня
10—15% жителей Земли голодает, а 40% получает неполноценную и недоста-
точную по белку пищу.
Индустриальное производство белков осуществляется сейчас гремя способа-
ми: производство кормовых дрожжей, приготовление белково-витаминных
концентратов и выделение белков из непищевого сырья растительного проис-
хождения.
Производство кормовых дрожжей, содержащих более 50% белка и богатых
витаминами и микроэлементами, организуют на отходах деревообрабатыва-
ющей, целлюлозно-бумажной, сахароваренной и спиртовой промышленности
(опилки, барда и т. п.), а также на отходах переработки растительного сырья
(солома, кукурузные початки и др.).
Белково-витаминный концентрат производят из нефтяного сырья. Впервые
такое производство в небольших масштабах было развернуто во Франции,
причем его продукцию использовали не только в животноводстве, но и в пи-
303
щевой промышленности. В нашей стране вошли в строй Светлоярский, Кстов-
ский, Томский, Новополоцкий, Киришинский и Кременчугский заводы по
производству белково-витаминного концентрата. Однако в процессе их эксплу-
атации возникли экологические проблемы, решение которых наталкивается как
на технические трудности, так и на эмоциональное противостояние населения.
На промышленную основу за рубежом поставлено выделение белков из
непищевого растительного сырья: листьев древесных растений, несъедобных
бобов и семян и т. п. Из такого белка готовят искусственное мясо, искусствен-
ные колбасы и другие пищевые суррогаты, получающие все большее распро-
странение в качестве дешевых заменителей натуральных продуктов. У нас
разработан метод выделения пищевого белка из хлопковых семян. Отличным
источником кормового белка является одноклеточная водоросль хлорелла,
производство которой развивается во всех странах мира, в том числе в нашей
стране, Болгарии, Японии, ФРГ и др.
Применение кормового белка и белково-витаминных концентратов в жи-
вотноводстве исключительно эффектней : улучшается использование белков
основной диеты, возрастают привесы и увеличивается скорость роста молод-
няка, снижается расход кормов на единицу продукции, резко повышается
эффективность производства. Поэтому перед молодой отраслью — промыш-
ленным производством белков—открываются большие перспективы. Созда-
ются мощные, предприятия по микробиологическому синтезу белка на базе
этилового спирта и природного газа с производительностью 300—500 тыс.
т продукции в год. Разрабатывается также регламент промышленного выра-
щивания водородных бактерий, биомасса которых исключительно богата
белком—содержание его достигает 70%.
Проблема обеспечения человечества белком решается и в более глобаль-
ном масштабе. У нас она впервые была рассмотрена в этом аспекте на
IX Менделеевском съезде по общей и прикладной химии (Киев, 1965). Акад.
А. Н. Несмеянов в своем докладе на съезде отметил, что процесс производ-
ства пищи путем выращивания растений и животных, по существу, мало
изменился со времени первобытного скотоводо-земледельческого обще-
ства. Однако еще столетие тому назад Д. И. Менделеев писал: «Как химик,
я убежден в возможности получения питательных веществ из сочетания
элементов воздуха, воды и земли, помимо обычной культуры, т. е. на
особых фабриках и заводах, но надобность в этом еще очень далека от
современности...»
По мнению А. Н. Несмеянова, огромные успехи синтетической химии,
в принципе способной осуществить синтез любого органического вещества,
создали реальную основу для постановки уже сейчас вопроса об индустри-
альном производстве пищи. Из шести составных частей пищи, необходимых
человеку (вода, белки, углеводы, жиры, минеральные соли и витамины),
первая и две последние есть в природе или могут быть легко произведены
в необходимом количестве. Из трех других—углеводы и жиры, являющие-
ся в основном поставщиками энергии, взаимозаменимы и легко переходят
друг в друга. Их синтетическое получение или производство в неограничен-
ных количествах из натурального непищевого сырья не представляет сейчас
трудности.
Таким образом, проблема сводится к индустриальному получению восьми
незаменимых аминокислот, которые вполне могут заменить белок в питании.
Уже сейчас существует достаточное число методов, химических и микроби-
ологических, которые могут послужить основой технического получения ука-
занных аминокислот в достаточных количествах и по ценам, более низким,
чем продукты, содержащие белки.
304
К этой же проблеме, но в иной форме обратился акад. Н. Н. Семенов
в своем докладе на юбилейной сессии, посвященной 250-летию Академии наук
СССР (декабрь 1978 г.). Он поставил вопрос о необходимости моделирования
процесса фотосинтеза, идущего с КПД, равным всего 1,5%, и повышения
последнего до 30%, что создаст предпосылки для получения энергии на
искусственных энергетических полях, а пищи (при посредстве этой энергии)—
на заводах.
Еще один важнейший аспект получения белков для практических целей был
обозначен акад. А. С. Спириным в докладе на юбилейной сессии Академии
наук СССР (март 1987 г.). Он сводится к преодолению клеточного уровня
биосинтеза белков и переходу к масштабированному их синтезу в бесклеточ-
ных системах трансляции непрерывного действия, работающих в проточном
режиме. Это откроет возможность получать биологически значимые белки
(интерферон, инсулин, о^-антитрипсин) и пептиды медицинского назначения,
позволит конструировать и производить белки с любыми заданными свойст-
вами, поднимет на новый уровень изучение закономерностей химической
коэволюции белков и нуклеиновых кислот. Решающую роль здесь играет
наработка необходимых количеств соответствующих мРНК в системах, содер-
жащих РНК-зависимую РНК-полимеразу типа репликазы фага Qp. Уже созда-
на и опробована на РНК-4 вируса мозаики костра, РНК фага MS2 и мРНК
кальцитонина установка для твердофазной трансляции типа реактора непре-
рывного действия. Указанные работы по внеклеточному синтезу белка ведутся
в рамках Государственной научно-технической программы «Новейшие методы
биоинженерии». Уже сегодня в лабораторных условиях на небольших биореак-
торах этим методом можно получать достаточное для дальнейших исследова-
ний количество пептидных гормонов, антигенов для диагностических целей,
белковых токсинов и антитоксинов, антивирусных защитных белков, некото-
рых ферментов. Революция в молекулярной биологии и биотехнологии про-
должается.
ГЛАВА VIII
У ГЛ ЕВОДЫ И ИХ ОБМЕН
СТРОЕНИЕ УГЛЕВОДОВ
Общая характеристика углеводов. К классу углеводов относят органические
соединения, содержащие альдегидную или кетонную группу и несколько спир-
товых гидроксилов. Их элементарный состав выражается общей формулой
СтН2пОп. Из этого правила есть немногочисленные исключения, и хотя указан-
ное определение не является абсолютно точным, оно позволяет наиболее просто
характеризовать эту группу разнообразных органических соединений в целом.
Ни название класса, ни общая формула этих соединений не дают ясного
представления об их химическом характере и строении. Более того, термин
«углеводы», предложенный впервые К. Шмидтом, профессором Юрьевского
университета (1844), наводит на мысль, что их следует рассматривать как
«гидраты» углерода. На самом деле это было бы совершенно неправильно.
Поэтому сравнительно давно (1927) для обозначения этого класса соединений
был предложен другой термин—«глюциды», который, к сожалению, с боль-
шим трудом пробивает себе дорогу в химической номенклатуре.
К углеводам относятся соединения, обладающие разнообразными и часто
совершенно различными свойствами. Среди них есть вещества низкомолекуляр-
ные и высокомолекулярные, кристаллические и аморфные, растворимые в воде
и не растворимые в ней, гидролизуемые и негидролизуемые, способные очень
легко окисляться и сравнительно устойчивые к действию окислителей и т. д. Это
многообразие качеств находится в тесной связи с химической природой углево-
дов, со строением их молекул; оно предопределяет то или иное участие углеводов
в процессах жизнедеятельности и в построении тканей животных и растений.
Во всех без исключения организмах углеводы служат материалом, при
окислении которого выделяется энергия, необходимая для осуществления
химических реакций. Такие углеводы рассматривают как резервные. Наряду
с этим промежуточные продукты окисления углеводов используются для
синтеза многих других органических соединений.
Перечисленные функции углеводов (структурная, энергетическая и метабо-
лическая) рассматривают как канонические. Однако в последнее время выясне-
но, что углеводам присущи многие другие, нестандартные, неканонические
функции. Многие углеводы и углеводсодержащие биополимеры обладают
уникальным строением и специфичностью. Так, групповые вещества крови,
являющиеся гликопротеинами, где 80% молекулы представлены углеводами,
именно за счет ассимметрических центров, стереоизомеров, таутомеров и кон-
формеров последних приобретают поразительную специфичность взаимодейст-
вия. Олигосахаридные фрагменты гликопротеинов и гликолипидов клеточных
стенок выдвинуты как антенны за пределы клеточных оболочек и служат
локаторами, выполняющими рецепторные функции. В частности, при их по-
средстве с клетками связываются белковые токсины (например, холерный,
ботулический, столбнячный, дифтерийный, шигатоксины и др.), бактерии (на-
пример, кишечная палочка с олигосахаридами, составленными из остатков
306
маннозы), вирусы (например, вирус гриппа) и т. п. Структуры олигосахаридных
фрагментов иммуноглобулинов высоковоспроизводимы и умеренно консерва-
тивны, что обеспечивает специфические углеводно-белковые взаимодействия
между доменами этих удивительно тонко организованных защитных белков.
Более 250 ферментов обладают олигосахаридными фрагментами, которые
избирательно взаимодействуют с многочисленными лектинами—белками, даю-
щими конъюгаты с углеводами. Таким образом, наряду с нуклеиновыми
кислотами и белками углеводы с современной точки зрения являются информа-
ционными молекулами, т. е. кодовыми словами в молекулярном языке жизни.
Благодаря этому все более ясно начинают вырисовываться контуры нового
направления в биохимии углеводов—гликобиология и гликотехнология.
В зависимости от состава, строения и свойств, в частности от поведения
при нагревании с разбавленными водными растворами кислот (т. е. в зависи-
мости от отношения к гидролизу), углеводы делят на две группы: простые
и сложные. Простые углеводы не подвергаются гидролизу. Сложные углеводы
при гидролизе распадаются с образованием простых углеводов.
Простые углеводы. Подавляющее большинство простых углеводов имеет
состав СПН2„ОП. Ввиду того что простые углеводы не гидролизуются, их
называют моносахаридами. Все простые углеводы—кристаллические тела,
хорошо растворимые в воде и имеющие, как правило, сладкий вкус. По
химическим свойствам они бифункциональны и могут быть охарактеризованы
как полиок иальдегиды или полиоксикетоны. Это значит, что в их молекулах
содержится альдегидная или кетонная группа и несколько спиртовых (окси-)
групп. В зависимости от числа кислородных атомов в молекуле моносахариды
делят на несколько групп, названи которых образуют из греческих числитель-
ных с добавлением окончания-оза, характерного для углеводов. Ряд простых
углеводов в целом называют поэтому рядом моноз. Сейчас известно более 200
природных моноз. В зависимости от по ожения в молекуле карбонильной
группы моносахариды существуют в виде изомеров: альдоз и кетоз.
Так как в молекулах моносахаридов присутствуют асимметрические атомы
углерода и, следовательно, молекулы в целом построены асимметрично, цанной
группе соединений свойственна оптическая, или зеркальная, изомерия. Как
и у других оптически деятельных соединений, зеркально построенные формы
моносахаридов носят название оптических антиподов. Эквимолекулярные смеси
или соединения последних друг с другом называются рацематами. Стереоизоме-
ры моносахаридов, отличающиеся простра ственным расположением водоро-
да и ОН-группы у соседнего с альдегидной группой углеродного атома,
являются эпимерами. Природные сахара относятся в основном к правому ряду,
что, видимо, связано с особенностями их первичного биосинтеза в растениях.
Из альдоз особенно широко распространены в природе D-рибоза, D-
глюкоза, D-манноза и D-галактоза: с/> с/> ь» т\и н-с—ОН 1 п 1 с/° гн но-с-н ^\н Н-С—ОН
н-с-он но—с—н 1 но—с—н 1 НО—С—н
н—с—он 1 н-с-он 1 н с—он н С—он НО—с н
1 н—с—он 1 н-с—он 1 н-с-он 1
снгон сн2он СН2ОН СН2ОН
D (—)-Рибоза D (4-)-Глюкоза D (4-) -Манноза D (4-)-Галактоза
307
Из кетоз наиболее известными являются D-рибулоза и D-фруктоза; из
гептоз целесообразно отметить D-седогептулозу:
СН2ОН с=о СН2ОН С=О СН2ОН
н—с—он но—с—н но—с—н
н—с—он н—с—он н—с—он
1 СН2ОН II—С— он н—с—он
* СН2ОН н—с—он
СН2ОН
D-Рибулоза D-Фруктоза D-Седогептулоза
Все указанные кетозы активно участвуют в превращениях углеводов в ор-
ганизме.
Характерной особенностью моносахаридов является их ярко выраженная
способность к таутомерным превращениям. Различают два вида таутомерии
моносахаридов: кето-енольную и кольчато-цепную.
Кето-енольная таутомерия моносахаридов состоит в переходе формы
с карбонильным кислородом в альдегидной или кетонной группе в енольную
форму (с ОН-группой при углеродном атоме, связанном двойной связью).
Благодаря кето-енольной таутомерии эпимерные моносахариды могут пре-
вращаться друг в друга.
Кольчато-цепная таутомерия моносахаридов заключается в существовании
кольчатых (циклических) форм и цепной (т. е. с открытой углеродной цепью)
формы моносахарида, находящихся в динамическом равновесии. Мысль о су-
ществовании циклических форм моносахаридов была впервые (1870) высказа-
на профессором Московского высшего технологического училища А. А. Кол-
ли. Замыкание цикла осуществляется при сближении СО-группы моносахари-
да с гидроксилом углеродного атома, удаленного от нее на 3—4 звена. По
карбонильному кислороду проходит реакция присоединения атома водорода
упомянутой спиртовой группы, в результате чего образуется новый гидроксил,
получивший наз ание гликозидного или полуацетального. Одновременно
с этим замыкается кислородный мостик, дающий начало пяти- или шестичлен-
ному гетероциклу (типа фурана или пирана):
Открытая (альдегидная, или оксо-)
форма глюкозы
Открытая форма глюкозы
со сближенными кето-
группой и спиртовым
радикалом 5-го угле-
родного атома
Кольчатая (окис-
ная) форма глю-
козы
308
Углеродные атомы шести- или пятичленного гетероцикла в циклических
формулах моносахарида, а иногда и все остальные боковые группы опускают,
и тогда они при написании приобретают следующий вид:
Глюкофураноза
Глюкопираноза
Глюкопираноза
Жирными линиями изображают те связи между углеродными атомами
цикла, которые направлены к наблюдателю, тонкими—те, что находятся за
плоскостью бумаги; это облегчает пространственное восприятие циклической
формулы. Атомы водорода и ОН-группы представляются расположенными
соответственно над и под плоскостью гетероцикла.
Так как в момент замыкания цикла углеродный атом бывшей СО-группы
становится асимметрическим, появление циклической формы моносахарида
сопровождается возникновением двух новых оптических изомеров. Тот из них,
у которого гликозидный гидроксил направлен в ту же сторону, что и спирто-
вой гидроксил при предпоследнем углеродном атоме моносахарида (опреде-
ляющем принадлежность его к D- или L-ряду), называют a-формой. Проти-
воположная по пространственному расположению гликозидного радикала
форма называется P-формой. Ниже приведены соответствующие примеры
а и p-форм, называемых также анамерами (от греч. ана—вверх, так как
гликозидные радикалы располагаются обычно при 1-м, верхнем углеродном
атоме):
a-D-Рибофура-
ноза
ноза
f-D-Рибофура-
p-D-Глюкопи-
раноза
Кетозы, как и альдозы, образуют кольчатые формы. В качестве примера
рассмотрим кольчато-цепную таутомерию фруктозы. Как и другие моносаха-
риды, фруктоза образует 4 циклические формы, находящиеся в динамичес ом
равновесии с открытой формой (см. с. 310).
Обычно циклические формы моносахаридов в растворе резко преобладают
над открытой цепной формой; кроме того, как правило, одна из циклических
форм моносахарида присутствует в равновесной смеси всех форм в большем
количестве, чем другие. Так, среди 4 циклических форм D-глюкозы резко
преобладает в растворе P-D-глюкопираноза (64%); содержание же альдегид-
ной формы глюкозы в равновесной смеси составляет всего 0,024%. Концент-
рация а и p-глюкофураноз в смеси также ничтожна и все остальное количество
глюкозы представлено a-анамером. В целом пиранозные формы резко преоб-
ладают над фуранозными формами.
309
Кольчато-цепная таутомерия моносахаридов является причиной любопыт-
ного свойства простых углеводов. В кристаллическом состоянии моносахари-
ды находятся только в цикли еской форме. В зависимости от условий кристал-
лизуется либо а-, либо P-форма. Так, при кристаллизации из воды глюкоза
получается в a-форме, а из пиридина—в P-форме. После растворения a-D-
глюкозы в воде вначале наблюдается характерное для нее значение удельного
вращения, равное +112,2°. Однако при стоянии раствора эта величина посте-
пенно снижается и наконец достигает устойчивого значения + 52,5°. Аналогич-
но этому исходное удельное вращение раствора P-D-глюкозы (+17,5°) при
стоянии также изменяется, повышаясь до + 52,5°. Так как в течение некоторого
времени после растворения кристаллических препаратов глюкозы в растворе
удается наблюдать много значений удельного вращения, пока не установится
его равновесное значение, это явление получило название мультиротации или
мутаротации (от лат. multum—много, rotatio—круговращение). Оно связано
с тем, что в растворе устанавливается равновесие между всеми возможными
кольчатыми и цепной модификациями глюкозы, каждая из которых обладает
своим удельным вращением, а их смесь—средним значением удельного вра-
щения (+52,5°).
Конформации углеводов крайне разнообразны. Известно, что шестичлен-
ные, алициклические соединения (циклогексан) существуют в геометрически
различных формах, которые принимает молекула, без нарушения длины вален-
тных связей и углов между ними. Указанные формы называются конформаци-
онными изомерами. Для моносахаридов, которым присуща в основном пира-
нозная структура, конформационная изомерия также характерна. Однако если
для циклогексана известно всего две конформации—типа кресла и типа лодки:
Кресло (сокращенно Лодка (сокращенно
С-от Chair-кресло) В-от Boat-лодка)
то для моносахаридов в пиранозной форме известно 8 конформаций—2 типа
кресла и 6 типа лодки ввиду присутствия в шестичленном цикле гетеро-
атома —кислорода:
310
Из перечисленных 8 конформаций пиранозного цикла наиболее устойчивы
креслообразные. В свою очередь, из них С1-изомер более предпочтителен, так
как подавляющее число заместителей в нем ориентировано в экваториальном,
совпадающем с плоскостью цикла, направлении. Именно в виде С1-конфор-
мации существует большинство моносахаридов, например:
С-D-Глюкоза
(36% в равновесной смеси)
p-D-Глюкоэа
(64% в равновесной смеси)
Естественно, что Р-анамер D-глюкозы преобладает в равновесной смеси
над а-анамером, так как гликозидный гидроксил в конформации С1 у первого
расположен экваториально:
р- D- Глюкозе
a-D- Галактоза
Так обстоит дело не только у анамеров глюкозы, но и у анамеров ряда
других моносахаридов.
Кольчато-цепная таутомерия моносахаридов является свойством, которое
зависит от одновременного присутствия в их молекулах СО-группы и спир-
товых радикалов. Поведение гликозидного гидроксила, возникающего при
образовании циклической формы моносахарида, своеобразно: он гораздо
активнее вступает в химические реакции, чем остальные гидроксильные
группы. Производные циклических моносахаридов, полученные в результате
311
замещения атома Н гликозидного гидроксила на какой-либо радикал, получи-
ли название гликозидов, а сам этот радикал именуют агликоном. Примером
могут служить а- и Р-метилгликозиды (см. с. 109).
В качестве агликона в природных гликозидах, отличающихся, как правило,
сильным физиологическим де“ствием, встречаются многие, часто весьма слож-
ные радикалы. Так, например, в растениях много гликозидов со стероидными
агликонами; они являются сильными ядами и защищают растения от вирусов
и патогенов. Высоким содержанием гликозидов с разнообразными агликонами
отличаются морские беспозвоночные. Многие из этих гликозидов применяют
в медицине. Сложные углеводы являются тоже типичными гликозидами.
Моносахариды дают все типичные реакции по альдегидной группе и по
спиртовым радикалам. Так, по альдегидной группе для них характерны реакции
окисления и восстановления, замещения карбонильного кислорода, поликон-
денсации (осмоления) и др., по спиртовым—образование простых и сложных
эфиров и другие взаимодействия, хорошо известные из курса органической
химии. В биохимии же особое значение придают окислительно-восстанови-
тельным реакциям моносахаридов и реакциям образования их фосфорных
эфиров.
Так, при окислении или восстановлении, например, глюкозы, протекающем
очень легко, получается соответственно кислота или спирт:
г°
сн»он
н-с—он
I
но-с—н
н—с—он
н-с-он
CHSOH
+ [Н]
н-с-он
Н-С-ОН
Н—С—он
сн.он
+ Ю]
он
он
+ГО]
НО-С—он---> но—с—он
н-
н—с—ОН
D-СорЙИТ
D-Глюкоза
н—i—он
D-Глюконовая
кислота
Н-С-ОН
к°
он
D-Сахзрная
кислота
Из фосфорных эфиров особо важное значение имеют глюкозо-1-фосфат,
глюкозо-6-фосфат, фруктозо-1,6-дифосфат, рибулозо-5-фосфат и др.:
Рнбулоэо-5-
фосфат
Эти соединения пр нимают самое активное участие в биохимических про-
цессах, протекающих в живых объектах. Фосфорилирование (т. е. введение
в молекулу моносахарида остатка фосфорной кислоты)—это первый, подго-
товительный этап как к распаду простых углеводов, так и к биосинтезу из них
более сложных углеводов. По мнению А. Е. Чичибабина (1932), фосф рилиро-
312
ванне моносахаридов способствует их переходу из циклической формы в от-
крытую цепную форму.
Важнейшими представителями моноз являются рибоза, глюкоза, манноза,
галактоза и фруктоза. D-Рибоза входит в состав нуклеиновых кислот в качест-
ве структурного элемента нуклеотидных остатков, где она находится в P-D-
фуранозной форме. Кристаллическая рибоза плавится при 87° С, удельное
вращение в водном растворе равно 23,7°. При восстановлении рибозы получа-
ется пятиатомный спирт—рибит, принимающий участие в построении многих
биактивных соединений:
p-D-Рибофураиоз*
н Н н
-+2Н» нон2с—с—с—с—снаон
он он он
D-Рибит
D-Ппокоза -один из наиболее распространенных природных сахаров.
Впервые кристаллический сахар из сиропа, полученного после обработки
крахмала кислотой, выделил К. Киргоф (1811); четверть века спустя Жан
Батист Андре Дюма назвал его глюкозой. В организмах она содержится
в свободном состоянии или в связанной форме, являясь в последнем случае
основой таких важнейших природных соедш ений, как тростниковый (свекло-
вичный) сахар, крахмал, клетчатка и др. Образует кристаллы с /ПЛ = 146°С
у а-глюкопиранозы и 148—150° С—у Р-глюкопиранозы. При нагревании
в пиридине a-форма (удельное вращение +112,2°) превращается в 0-форму
(удельное вращение +17,5°). Поэтому из водных и спиртоводных растворов
кристаллизуется a-D-глюкопираноза, а из растворов в пиридине - P-D-глюко-
пираноза. При восстановлении глюкозы образуется D-сорбит, а при окисле
нии—D-глюконовая и далее сахарная кислота.
D-Галактоза входит в состав ряда сложных углеводов, в том числе молоч-
ного сахара. Кр сталлизуется в виде моногидрата. Безводные кристаллы
плавятся при 164° С; удельное вращение (конечное значение) +81°.
D-Манноза образует природные сложные углеводы— маннаны, часто явля-
ющиеся углеводными компонен ами гликопротеинов и слизей, а также оболо
чек растительных клеток. Маннопираноза плавится при 132° С, а ее а- и 13-
формы имеют разное значение угла вращения (+30 и —17° соответственно);
конечный угол вращения, устанавливающийся в результате мутаротации,
равен +14,5°.
D-Фруктоза встречается как в свободном (например, в составе меда), так
и в связанном (например, в составе тростникового сахара и некоторых высо-
комолекулярных природных углеводов—фруктозанов) виде. Она в 2,5 раза
слаще глюкозы и в 1,7 раза—тростникового сахара. Безводная фруктоза
плавится при 102—104° С. Значение удельного угла вращения в равновесном
(после достижения постоянного значения при мутаротации) состоянии равно
—92°, благодаря чему фруктозу называют левулозой.
В течение последнего времени накопились данные о существовании новой
группы моносахаридов, которая Н. К. Кочетковым и сотр. (1975) названа
гликолактиловыми кислотами. Моносахариды этой группы составлены из
остатков обычных моносахаридов и молочной кислоты, связанных между собой
простой эфирной связью (от лат. acidum lacticum—молочная кислота, происхо-
дит вторая половина названия этой новой группы моносахаридов). Важнейшим
представителем гликолактиловых кислот является мурамовая кислота:
313
СН2ОН
н>----<Хн
ко н>
но/Чи Ибн
/ Н NH,
ноос—сн—сн3
Она представлена кристаллами с /пл= 150 —155° С; удельное вращение (равно-
весное) ее растворов составляет 103—123° в зависимости от значения pH.
N-ацетилмурамовая кислота входит в состав пептидогликанов клеточных сте-
нок бактерий; полисахаридные фрагменты пептидогликанов атакуются лизо-
цимом (см. с. ПО—111).
Сложные углеводы. К категории сложных относят углеводы, молекулы
которых распадаются при гидролизе с образованием простых углеводов. Состав
сложных углеводов выражается общей формулой СтН2„Ов, где т>п. Среди
сложных углеводов выделяют две группы:
1. Олигосахариды—сахароподобные сложные углеводы, характеризующи-
еся сравнительно невысокой (несколько сотен) молекулярной массой, хорошей
растворимостью в воде, легкой кристаллизацией и, как правило, сладким
вкусом. Молекулы олигосахаридов составлены из небольшого числа (от греч.
олигос—малый, немногочисленный) остатков простых углеводов.
2. Полисахариды—высокомолекулярные сложные углеводы, образован-
ные из многих сотен остатков простых углеводов. Их молекулярная масса
составляет обычно сотни тысяч. Они не дают ясно оформленных кристаллов,
и лишь некоторые из них обладают псевдокристаллическим строением.
Полисахариды либо не растворимы в воде, либо дают растворы, напомина-
ющие по свойс вам коллоидные, что можно объяснить большой молекуляр-
ной массой растворенных частиц. Для полисахаридов сладкий вкус не харак-
терен.
Олигосахариды. В зависимости от количества остатков моносахари-
дов, входящих в молекулы олигосахаридов, последние делят на дисахариды,
трисахариды и т. д. Наибольший интерес из олигосахаридов представляет
группа дисахаридов—соединений, которые широко рас рос ранены в приро-
де; многие из них имеют огромное практи еское значение.
По химическому строению дисахариды являются гликозидами моносаха-
ридов, агликонами которых служат также остатки моносахаридов. Дисахари-
ды, у которых остаток моносахарида, служащий агликоном, присоединяется
к гликозидному радикалу основного моносахарида по месту своего гликозид-
ного гидроксила, называют гликозидо-гликозидами. Если же связь между
агликоном и основным моносахаридом осуществляется за счет спиртового
гидроксила агликона, то дисах; риды называют гликозидо-глюкозами. Это
иллюстрируют формулы трегалозы и мальтозы:
Трегалоза
(a-D -глюкопиранозидо-a-
D -глюкопиранозид)-дисахарид
типа гликозидо-гликозида
Мальтоза
(а-О-глюкопиранозидо-4-а-О~
глюкопираноза)-дисахарид
типа гликозидо-глюкозы
314
В соответствии со своим химическим строением дисахариды типа трега-
лозы (гликозидо-гликозиды) и типа мальтозы (гликозидо-глюкозы) облада-
ют существенно различными химическими свойствами: первые не дают ника-
ких реакций, свойственных альдегидной или кетонной группе, т. е. не окисля-
ются, не восстанавливаются, не образуют озазонов, не вступают в реакцию
поликонденсации (не осмоляются), не мутаротируют и т. п. Для дисахаридов
типа мальтозы все упомянутые реакции, наоборот, весьма характерны. При-
чина этого различия вполне понятна из сказанного выше о двух типах
структуры дисахаридов и свойствах входящих в их состав остатков моноса-
харидов. Она заключается в том, что только в дисахаридах типа мальтозы
возможна кольчатоцепная таутомерия, в результате чего образуется свобод-
ная альдегидная или кетонная группа, проявляющая свои характерные
свойства:
У дисахаридов типа тре алозы, где оба гликозидных гидроксила использо-
ваны для образования гликозидной связи между остатками моносахаридов,
раскрытие цикла невозможно.
По спиртовым гидроксилам оба типа дисахаридов дают одинаковые реак-
ции: образуют простые и сложные эфиры, взаимодействуют с гидратами
оксидов металлов [например, растворяют Си (ОН)2 ] и т. д.
В природе существует большое число дисахаридов; существенное значение
среди них имеют упомянутые выше трегалоза и мальтоза, а также сахароза,
целлобиоза и лактоза.
Сахароза представляет собой дисахарид типа гликозидо-гликозида, соста-
вленный из остатков a-D-глюкопиранозы и P-D-фруктофуранозы. При гид-
ролизе сахароза распадается на два моносахарида—a-D-глюкозу и P-D-фрук-
тозу (см. с. 128). Сахароза—один из самых распространенных в природе
и практически наиболее важных дисахаридов. Она больше известна под назва-
нием тростникового или свекловичного сахара, так как из сока этих двух
растений практически получают весь сахар. Сахароза содержится также в соке
многих других растений, но в сравнительно небольшой концентрации, хотя
в соке клена, березы и пальмы содержание ее довольно высоко.
Сахароза хорошо кристаллизуется (гпл= 184° С), легко растворяется в воде
и незначительно в спирте. Растворы ее не мутаро ируют, как это и свойствен-
но гликозидо-гликозидам, но вращают плоскость п ляризации света вправо
на [a]‘D = 66,5°. Концентрацию сахара в растворе обычно измеряют при
помощи специального поляриметра, шкала которого градуир вана в вели-
чинах, показывающих процентное содержание сахара в растворе.
Так как при гидролизе сах; розы получающиеся из нее моносахариды
имеют противоположные углы удельного вращения (в равновесном состоянии
+ 52,5° у глюкозы и —92° у фруктозы), суммарный угол вращения после
гидролиза становится отрицательным. В связи с этим гидролизованную саха-
розу называют инвертным сахаром, а сам процесс гидролиза—инверсией (от
лат. inversio—переворачивание, перестановка).
1 Знак [a]D обозначает удельное вращение [а], измеренное в поляризованном свете, длина
волны которого соответствует длине волны спектральной линии натрия (£>=588 нм).
315
Мальтоза относится к дисаха идам типа гликозидо-глюкоз и составлена из двух
остатков a-D-глюкопиранозы, соединенных гликозидной связью в положениях 1,4
(см. с. 314). Связь такого типа называют а-1,4-гликозидной связью. Мальтозу
получают при гидролитическом расщеплении крахмала с помощью специфическо-
го фермента (см. с. 329), которым очень богаты прорастающие зерна ячменя. Так
как проросшие, а затем высушенные и измельченные семена ячменя называют
солодом, а английское название солода—malt, то образующийся из крахмала при
участии фермента солода дисахарид и получил наименование мальтозы.
Мальтоза кристаллизуется из водных растворов в виде моногидрата, не
имеющего резко выраженной точки плавления. Водные растворы мальтозы
(она очень хорошо растворима в воде) мутаротируют, так как за счет коль-
чатоцепной таутомерии постепенно идет превращение исходной формы снача-
ла в полуциклическую, а затем в другие таутомеры мальтозы, содержащие
вместо a-D-глюкопиранозы ее P-модификацию или а- и Р-глюкофуранозные
циклы. Равновесный угол удельного вращения мальтозы равен +136°.
Мальтоза образуется в растительных ц животных организмах как проме-
жуточный продукт при гидролизе крахмала и может быть обнаружена в сво-
бодном виде. При гидролизе мальтоза распадается на две молекулы глюкозы.
Целлобиоза, как и мальтоза, состоит из двух остатков D-глюкозы и от-
носится к дисахаридам типа гликозидо-глюкоз. Строение ее молекулы отлича-
ется от строения мальтозы только тем, что гликозидная связь между остатка-
ми моносахаридов начинается от гликозидного гидр< ксила, находящегося в Р-
положении. Следовательно, молекула целлобиозы характеризуется наличием
Р-1,4-гликозидной связи:
Целлобиоза
ф-С-глюкопиранозидо- 4-D-
глюкопираноза)
Целлобиоза отлично кристаллизуется, хорошо растворима в воде и мало
растворима в спирте, мутаротирует: [a]D= +34,6°, легко окисляется и являет-
ся хорошим восстановителем, образует характерный озазон, при гидролизе
дает исключительно D-глюкозу.
Целлобиоза широко распространена в растительном мире; она найдена
в прорастающих семенах, косточках абрикосов, пасоке деревьев. Целлобиоза
образуется при ферментативном гидролизе целлюлозы (клетчатки), составля-
ющей оболочки растительных клеток.
Лактоза является р-О-галактопиранозидо-4-О-глюкопиранозой:
Лактоза
316
Она содержится в значительном количестве в молоке (5—8%) и называется
поэтому молочным сахаром. Однако лактоза присуща не только млекопита-
ющим (хотя ее роль в их питании бесспорна), так как она найдена, апример,
в пыльцевых трубочках ряда растений. Лактоза плохо растворима в воде,
может быть получена путем упаривания молочной сыворотки. Кристаллизует-
ся в виде моногидрата с t„„ = 202° С. Ввиду того что остаток глюкозы облада-
ет свободным гликозидным гидроксилом, возможна мутаротация, и лактоза,
следовательно, может существовать в двух формах. Равновесная смесь ее а-
и P-формы имеет удельное вращение + 52,2°. В отличие от других дисахаридов
кристаллы лактозы не гигроскопичны.
Полисахариды. К полисах! ридам относятся вещества, построенные из
большого числа остатков моносахаридов или их производных. Если полисаха-
рид содержит остатки моносахарида одного вида, его называют гомополи-
сахаридом. В том случае, когда поли ахарид составлен из моносахаридов двух
видов или более, регулярно или нерегулярно чередующихся в молекуле, его
относят к гетерополисахаридам.
К числу наиболее важных природных гомополисахаридов принадлежат
крахмал, гликоген (животный крахмал), клетчатка, декстран и хитин. Первые
четыре полисахарида при гидролизе дают только D-глюкозу, последний—
высвобождает производное D-глюкозы—N-ацетил-глюкозамин. Кроме того,
известно много других гомополисахаридов. Так, полисахарид инулин при
гидролизе дает только фруктозу, маннан—маннозу, галактан—галактозу,
арабан—арабинозу и т. п. Названия этим полисахаридам даны по конечным
продуктам их расщепления.
К числу важнейших природных гетерополисахаридов относят гиалуроно-
вую и хондроитинсерную кислоты, гепарин, капсулярные полисахариды бакте
рий, агарозу, порфираны и каррагинаны красных водорослей, альгиновые кис-
лоты бурых водорослей, структурные полисахариды простейших.
Биологическое значение полисахаридов разнообразно. Многие из них (крах-
мал, гликоген, инулин и др.) являются в растительных и животных организмах
запасными питательными веществами. Некоторые полисахариды (например,
хондроитинсерная кислота, капсулярные полисахариды и клетчатка) несут
исключительно опорные и защитные функции. Ряд полисахаридов (маннаны,
галактаны и др.) используется и как строительный, и как питательный матери-
ал. Гиалуроновая кислота, составляющая межклеточное вещество тканей жи-
вотных, наряду со структурной функцией регулирует распределение жизненно
необходимых веществ в тканях. Гепарин предотвращает свертывание крови
в организме человека и животных. Во многих случаях полисахариды дают
очень прочные комплексы с белками, образуя гликопротеины, выполняющие
в организме ряд ответственных функций.
Химическое строение полисахаридов однообразно. Все они являются поли-
гликозидами, в молекулах которых за счет кислородных мостиков объединя-
ются сотни, тысячи, а иногда и десятки тысяч остатков моносахаридов и их
производных. При этом непременным партнером в образовании кислородных
мостиков служит гликозидный гидроксил моносахаридов, а другим партнером
оказывается один из спиртовых гидроксилов моносахарида, чаще всего в по-
ложении 4 (у линейных полисахаридов) или 4 и 6 (у разветвленных форм).
Полисахариды сравнительно легко гидролизуются при кипячении с разбав-
ленными растворами кислот или при инкубации с соответствующими фермен
тами. Щелочи не гидролизуют полисахаридов. Так, гликоген можно нагревать
в течение нескольких часов с 30%-ным раствором щелочи без какого-либо
разложения его. На этом основан метод количественного определ ния глико-
гена. Характерно, что в ряде случаев при ферментативном гидролизе таких
317
распространенных полисахаридов, как крахмал и клетчатка, образуются диса-
хариды (мальтоза и целлобиоза соответственно), тогда как при неспецифиче-
ском кислотном гидролизе образуется моносахарид (D-глюкоза).
Крахмал—один из наиболее распространенных запасных полисахаридов
растений. Он интенсивно накапливается в результате фотосинтеза и отклады-
вается в семенах, клубнях и других частях раст ний. Семена и клубни содержат
40—70% крахмала, другие части растений—от 4 до 25%. При кислотном
гидролиз крахмал распадается с образованием D-глюкозы, являющейся его
структурным элементом, и небольшого количества глюкозо-6-фосфата, так
как все виды крахмала содержат немного (0,02—0,16%) фосфора. Установле-
но, что глюкоза в составе крахмала находится в виде a-D-глюкопиранозы.
Природный крахмал состоит из двух различных фракций, отличающихся по
своему строению и свойствам. Примерно 20% крахмала составляет амилоза (от
греч. амилон—крахмал). Остальное приходится на вторую фракцию, получи-
вшую название амилопектина (от греч. пектос—студнеобразный). Такая терми-
нология отражает некоторые свойства этих двух видов крахмала. Амилопек ин
с трудом растворяется в горячей воде, причем раствор получается вязкий
кра мальный клейстер) и при охлаждении застывает в студневидную массу.
Амилоза же хорошо растворима в теплой воде и не образует клейстера.
Пользуясь этим обстоятельством, амилозу отделяют от амилопектина, много-
кратно извлекая ее теплой водой. С этой же целью используют способность
амилозы осаждаться под действием бутилового спирта при насыщении послед-
ним горячего раствора, содержащего смесь амилозы и амилопектина. Применя-
ют и хроматографические методы. Например, после пропускания диспергирован-
ного рахмала через колонку с фосфатом кальция и последующего промывания
фосфатным буфером амилоза элюируется а амилопектин остается на сорбенте.
Молекулярная масса амилозы и амилопектина различна: у не деградирова-
вших в процессе выделения препаратов амилозы она составляет от 100000 до
400000, а у амилопектина превышает, как правило, 20-10б. Эти данные
получены методом гельфильтрации через сефадекс G-200 и сефарозу 2В. При
этом картофельный крахмал, например, разделе на 9 фракций с молекуляр-
ными массами от 7 • 10б до 73 • 10б. Видимо, значения молекулярных масс
рассматрива мых здесь и ниже полисахаридов зависят от примененного спо-
соба выделения. В тех случаях, когда нативность препарата не утрачена,
молекулярные массы очень высоки. Соответственно коэффициент поликонден-
сации a-D-глюкопиранозы в молекулах амилозы оценивается в несколько
сотен, а у амилопектина—в несколько десятков и даже сотен тысяч.
Различна и хими еская структура амилозы и амилопектина. Молекулы
первой, как правило, строго линейны. В них остатки a-D-глюкопиранозы
связаны друг с другом—исключительно а-1,4-глюкозидными связями, т.е.
кислородные мостики возникают за счет гликозидного гидроксила 1-го угле-
родного атома одной молекулы a-D-глюкопиранозы и спиртового гидр ксила
при 4-м угл родном атоме другой:
Амилоза
318
В соответствии с таким строением амилозу можно характеризовать как
а-1,4-глюкан. Таким образом, амилоза представляет линейный полиса-
харид, молекулы которого имеют нитевидную структуру (рис. 103 и 104).
Остатки a-D-глюкопиранозы в составе амилозы имеют конформацию
типа лодки. В этом случае структурная формула амилозы принимает
следующий вид:
Лодкообразная конформация a-D-глюкопиранозных остатков в молекуле
амилозы способствует спирализации полигликозидной цепи. При этом один
виток спирали включает 6—7 остатков глюкозы. При длине каждого остатка
глюкозы, равной 0,5 нм, возникает спираль диаметром около 1 нм (рис. 103).
Допускают, что молекулы амилозы, как и других линейных полисахаридов,
Рис. 103. Спиральная конформация молекулы амилозы (Л), точки приложения действия
a-амилазы при гидролизе (£) и структура адсорбционного комплекса между спирализо-
. ванными участками крахмала и молекулами иода (В)
319
Рис. 103. Продолжение
могут на том или ином протяжении взаимодействовать друг с другом, образуя
вторичные структуры биспирального типа с взаимозакрученными полисаха-
ридными цепями.
Амилопектин имеет сферические молекулы с радиусом вращения от 82 до
255 нм. Их сферическая форма обеспечивается тем, что молекула составлена
из множества (несколько сотен) коротких полигликозидных цепочек, каждая
из которых в среднем содержит 20 остатков a-D-глюкопиранозы. В пределах
каждой короткой цепи глюкозные остатки соедин ны а-1,4-глюкозидными
связями. Друг с другом цепи соединяются посредством а-1,6-глюкозидных
связей. Строение разветвленного участка молекулы амилопектина таково:
320
A—амилоза; Б—амизопектин; В—гликоген; каждый кружок обозначает остаток глюкозы; Г—
современная гроздевидная модель молекулы амилопектина (а—компактная, псевдокристалличе-
ская область; б—менее компактная, аморфная область; точкой обозначена редуцирующая группа);
Д—модернизированная модель молекулы гликогена, демонстрирующая наличие спрятанных поли-
гликозид ных цепей; Е—строение частичкового гликогена; центральная линия—полипептидная
цепь, к которой через радикалы оксиаминокнслот присоединены 0-частицы, образующие а-частицу,
построенную по типу сложной костянки; Ж—объемная модель фрагмента молекулы амилопектина
(Р—редуцирующий конец; Н—нередуцируюшие концы)
Общая структура молекулы амилопектина в соответствии с ранними данными
показана на рис. 104, Б. На этом же рисунке приведена более современная модель
молекулы амилопектина (Г), выведенная на основании детального исследования
продуктов ферментативного гидролиза и рентгенографического анализа этого
полисахарида. Она получила название гроздевидной, так как по расположению
в ней полиглик зидных звеньев весьма напоминает гроздь винограда. Степень
полимеризации остатков a-D-глюкозы в звеньях, обозначенных утолщенными
линиями, достигает 45, тонкими -.-15. Протяженность каждой псевдокристалли-
ческой области составляет 6 нм, аморфной—втрое меньшую величину.
11—3502
321
Рис. 104. Продолжение
Считают, что в составе амилопектина a-D-глюкопираноза также находится
в лодкообразной конформации. Вследствие этого отдельные участки полигли-
козидных цепочек, составляющих молекулу амилопектина, видимо, спирализо-
ваны подобно амилозе (рис. 104, ж).
Обе фракции крахмала дают окрашивание с иодом в растворе К1, однако
амилоза окрашивается в чисто синий цвет, а амилопектин—в фиолетовый.
Реакция крахмала с иодом не связана с имическим взаимодействием между
ними, а состоит в образовании комплексов адсорбционного типа. Так как
ведущую роль в возникновении этих компл ксов играют спиральные участки
молекул крахмала того или другого типа, то различие в тональности окраски
вполне естественно. Предполагают, что молекулы иода втягиваются внутрь
полигликозидной спирали, где и замыкаются соответствующие связи, дающие
начало цветным комплексам (рис. 103, в).
При кратковременном нагревании порошкообразного крахмала его гигант-
ские молекулы распадаются, образуя смесь более простых полисахаридов мень-
шей молекулярной массы—декстрине . Декстринизация крахмала при нагрева-
нии сопровождается повышением его растворимости в воде. Обработанный
таким образом крахмал называют растворимым. Распад молекул крахмала до
декстрине! особенно интенсивно идет при нагревании крахмального клейстера
с 10%-ным раствором H2SO4. При дальнейшей обработке молекулярная масса
декстринов прогрессивно падает и конечным продуктом распада является D-
глюкоза. Крупномолекулярные декстрины окрашиваются иодом в красный цвет,
низкомолекулярные—окраски с иодом не дают. Таким образом, гидролиз
крахмала проходит ступ :нчато и может быть выражен сл здующей схемой:
+ НгО +Н2О +НгО
НО [С6Н10О 5]х Н—НО [С 6Н10О 5], Н-*ш НО [С6Н10О 5] z Н—>хС6Н 12О 6,
Крахмал ЭритрОдекстрины Ахродекстррны ГЛЮкоза
где x>y>z.
322
Декстринизация и осахаривание крахмала широко применяются в спирто-
вой промышленности, при производстве клея и т. д.
Так как крахмал составлен из оптически активных остатков a-D-глюкопи-
ранозы, его растворы вращают плоскость поляризации света вправо
([a]DS= 4-195°).
Пшкоген служит резервным питательным веществом в организме человека
и животных, вследствие чего за ним сохраняется название «животный крах-
мал». Однако он найден также в грибах, дрожжах и зернах кукурузы, что
ставит под сомнение его название «животный». Содержание гликогена в пече-
ни животных достигает 20%, а в мышцах—4%. Распадаясь до простых
продуктов довольно сложным путем, который называется гликогенолизом
(см. с. 351), гликоген обеспечивает потребность организма в энергии и мета-
болитах. Таким образом, его биологическая роль весьма велика.
1ликоген сравнительно хорошо растворяется в горячей воде, хотя некото-
рые виды натурального гликогена труднорастворимы. Подобно крахмалу,
гликоген дает цветную реакцию с иодом, причем тон окраски (красно-фиолето-
вый или красно-коричневый) свидетельствует о том, что гликоген ближе
к амилопектину, нежели к амилозе. Действительно, гликоген и амилопектин
весьма похожи. Так, молекулярная масса некоторых фракций нативного гли-
когена близка к молекулярной массе амилопектина, хотя в целом гликоген
в этом отношении отличается крайней гетерогенностью: препараты гликогена
из печени животных содержат фракции с М от 10 млн. до 3 млрд, с преоблада-
нием среди них молекул с М от 200 млн. до 600 млн. При неполном гидролизе
гликогена образуются декстрины, а при полном—D-глюкоза. Как и амилопе-
ктин, гликоген оптически активен, причем удельное вращение его растворов
(+196°) весьма близко к таковому крахмала.
Причины близости свойств гликогена и амилопектина заключаются
в сходстве строения их молекул: оно по существу одно и то же; различие
состоит лишь в том, что в молекуле гликогена средняя длина коротких цепей,
которые сочетаются здесь а-1,6-гликозидными связями, равна 12 остаткам
a-D-глюкопиранозы. Таким образом, молекула гликогена несколько плотнее,
компактнее, чем молекула амилопектина (см. рис. 104). Гликоген низших жи-
вотных ближе к амилопектину, так что во многих случаях трудно провести
резкую границу между первым и вторым, хотя накапливающиеся данные
о структуре гликогена (см. рис. 104, Д и £)- определенно свидетельствуют
о большей симметричности его молекулы, о наличии в ней значительного
числа спрятанных (не выходящих на поверхность молекулы) полигликозидных
звеньев (это часть звеньев В на рис. 104, Д), а также о существовании у глико-
гена макромолекулярной структуры. Последняя характерна для так называ-
емого частичкового гликогена, впервые выделенного А. Лазаревым (1942)
в мягких условиях методом дифференциального ультрацентрифугирования.
Он представлен огромными, глобулами, названными a-частицами (диаметр до
200 нм) и напоминающими ягоды шелковицы или малины. В свою очередь,
a-частицы составлены из Р-частиц, а те, в свою очередь,—из у-частиц с диаме-
тром 20—40 нм (см. рис. 104, Е).
Клетчатка—основной структурный полисахарид растений. Листья содер-
жат 15—30% клетчатки, древесина—50—70%, стебли волокнистых растений
(например, льна) еще больше, а волокна (волоски на семенах) хлопка пред-
ставляют собой почти чистую клетчатку. Само название этого полисахарида
подчеркивает его большую роль в построении клеточных стенок; в связи
с этим распространен также термин целлюлоза (от лат. cellula—клетка).
Подсчитано, что при сжигании всей клетчатки, содержащейся в составе расте-
ний, количество СО2 в атмосфере возросло бы наполовину.
323
Клетчатка отличается очень малой растворимостью в подавляющем боль-
шинстве агентов; лишь аммиачный раствор Си(ОН)2 и концентрированный
раствор Ca(SCN)2 при нагревании заметно растворяют ее. Устойчивость
клетчатки к действию растворителей объясняют тем, что ее длинные ни-
тевидные молекулы, взаимодействуя друг с другом, образуют прочные
мицеллы, которые, в свою очередь, собраны в фибриллы, располагающиеся
вдоль оси волокна. Так, тончайшие элементарные волокна хлопка диаметром
20 нм состоят из множества молекул целлюлозы (диаметр 0,6—0,7 нм),
упакованных очень плотно. Отрыв индивидуальных молекул клетчатки от
упомянутых устойчивых агрегатов весьма затруднен, и только немногие
вещества, способные нарушать межмолекулярные связи в мицеллах, рас-
творяют клетчатку.
При гидролизе клетчатки в присутствии специфического фермента, най-
денного у ряда бактерий, некоторых видов насекомых, плесневых грибков
и в прорастающих семенах, образуется целлобиоза. Однако при гидролизе
в присутствии кислот почти с количественным выходом возникает 0-D-
глюкопираноза; она и является основным структурным элементом молекулы
целлюлозы:
Целлюлоза
Тип связи остатков друг с другом аналогичен таковому в молекулах
амилозы, но в отличие от последней клетчатка является Р-полигликозидом,
так как остатки глюкозы соединены друг с другом Р-1,4-гликозидными свя-
зями. Как у амилозы, в молекулах клетчатки нет разветвлений, они построены
строго линейно, но намного длиннее молекул амилозы. Число остатков D-
глюкозы в молекуле клетчатки очень велико и достигает нескольких тысяч,
что соответствует молекулярной массе нативной целлюлозы в 10—20 млн.
Эти, на первый взгляд, небольшие отличия в строении амилозы и клетчатки
приводят к резкой разнице в их свойствах.
Более глубоко различие между амилозой и клетчаткой вскрывается при
сопоставлении строения упомянутых полисахаридов с учетом конформации
составляющих их остатков D-глюкозы. Установлено, что P-D-глюкопираноза
в составе клетчатки находится в креслообразной конформации:
Это исключает возможность спирализации полигликозидной цепи, и молекула
целлюлозы сохраняет строго линейное строение.
Раствор клетчатки в медно-аммиачном реактиве обнаруживает небольшую
оптическую активность ([а]/>=3,21°).
324
Как и у других полисахаридов, в молекулах клетчатки остается свободным
большое число спиртовых гидроксилов (при 2, 3 и 6-м углеродных атомах
каждого остатка P-D-глюкопиранозы). По этим ОН-группам возможны соот-
ветствующие химические реакции. Среди них особенно важны те, что ведут
к получению производных, широко применяемых в ионообменной хромато-
графии для разделения аминокислот, пептидов, белков, нуклеотидов и нукле-
иновых кислот. К их числу относятся карбоксиметилцеллюлоза (КМ-целлю-
лоза) и диэтиламиноэтилцеллюлоза (ДЕАЕ-целлюлоза).
КМ-целлюлозу получают, обрабатывая щелочную целлюлозу монохлор-
уксусной кислотой:
HO[C6H9O4(ONa)]xH+xClCH2COOH—>HO[C6HqC)4(OCH2COOH)]xH+xNaCl
Целлюлоза Монохлоруксусная Карбоксиметил-
кислота целлюлоза
В результате целлюлоза обогащается СООН-группами, придающими ей
свойства катионообменника.
ДЕАЕ-целлюлозу синтезируют, действуя на целлюлозу Р-хлорэтилдиэтил-
аммоний хлоридом в щелочной среде:
НО[С6Н9О4(ОН)]хН+х[С1СН2СН2—NH(CH2CH3)2]Cl+2xNaOH->
Целлюлоза Хлорид р-хлорэтилдиэтиламмония
-> НО [С 6Н 9О 4—О—СН 2—СН 2—NH (СН 2СН з) 2] х Н+2xNaCl+2хН 2О
Диэтиламиноэтилцеллюлоза
Диэтиламиноэтил-группировки придают ДЕАЕ-целлюлозе свойства ани-
онообменника.
. Декстран—полисахарид, продуцируемый некоторыми видами бакте-
рий. Молекулярная масса его огромна: различные препараты декстрана
обнаруживают значения М от 12 млн. до 1 млрд. Молекула декстрана
состоит из сравнительно коротких полигликозидных цепочек по 10—12
остатков a-D-глюкопиранозы в каждой. Остатки a-D-глюкозы в них (см.
с. 31) соединены а-1,6-гликозидными связями, а между собой цепи соединяют-
ся дополнительными 1,4-гликозидными связями. При обработке декстрана
эпихлоргидрином получают сефадексы—материалы, отличающиеся сетча-
той структурой, прекрасно набухающие и используемые в качестве молеку-
лярных сит.
Хитин—главная составная часть покровных тканей насекомых и рако-
образных. Этот полисахарид широко распространен в природе; подсчитано,
что только крабы ежегодно синтезируют 1 млн. т хитина. Будучи освобож-
ден от белков или СаСО 3, в комплексе с которыми он участвует в построе-
нии покровных тканей упомянутых животных, хитин представляет белое
вещество, напоминающее бумажную массу. Хитин отличается очень плохой
растворимостью, и только муравьиная кислота и насыщенные растворы
некоторых солей способны перевести его в частично растворенное состоя-
ние. Видимо, поэтому до сих пор не получено точных данных о молекуляр-
ной массе хитина.
Элементарной структурной единицей хитина является Ы-ацетил-Р-В-
глюкозамин, соединенный Р-1,4-гликозидными связями в линейную мо-
лекулу:
325
Структура хитина, как можно видеть из приведенной выше формулы,
весьма напоминает таковую целлюлозы. Рентгенограммы этих двух полисаха-
ридов очень похожи.
1иалуроновая кислота—важнейшая составная часть межклеточного веще-
ства тканей животных. Особенно высоко ее содержание в коже, стекловидном
теле глаза, сухожилиях и т. п. Гиалуроновую кислоту можно получить из
указанных тканей путем экстракции разбавленными щелочами, трихлоруксус-
ной кислотой или фенолом. После осаждения из экстракта спиртом гиалуро-
новую кислоту освобождают от прочно связанного с ней белка переваривани-
ем последнего ферментами.
Молекулярная масса гиалуроново-белкового комплекса достигает несколь-
ких миллионов. Однако препараты гиалуроновой кислоты отличаются сравни-
тельно низкими значениями молекулярных масс (270000—500000). Видимо,
в процессе выделения происходит деградация молекул гиалуроновой кислоты.
Являясь гетерополисахаридом, гиалуроновая кислота содержит в своем
составе две различные структурные единицы—Ы-ацетил-Р-П-глюкозамин и Р-
D-глюкуроновую кислоту в отношении 1:1. Они соединены друг с другом
попеременно Р-1,3- и Р-1,4-гликозидными связями:
Гиалуроновая кислота
Гйалуроновой кислоте в тканях животных присущи не только структурные
функции. Пронизывая ткани в качестве межклеточного вещества, гиалуроно-
вая кислота регулирует поступление в клетки тех соединений, которые или
нужны для жизнедеятельности клетки, или являются ее продуктом. Эта функ-
ция гиалуроновой кислоты в значительной степени осуществляется при уча-
стии фермента—гиалуронидазы, свойства которой интенсивно изучаются.
Хондроитинсульфат—непременная составная часть хряща, костной ткани,
сухожилий, сердечных клапанов и других подобных тканей животных. Содер-
жание его, например, в хряще носовой перегородки составляет 20—40%.
Хондроитинсульфат выделить в чистом виде трудно, так как он прочно связан
с белком—коллагеном. Вероятно, при выделении идет сильная деградация
молекул хондроитинсульфата, так как молекулярная масса его препаратов не
превышает 50 000, тогда как М его комплекса с белком составляет от 4 до 50 млн.
В виде очищенного препарата хондроитинсульфат пр< дставляет собой
белое вещество, распадающееся при гидролизе до глюкуроновой кислоты
326
и N-ацетилгалактозаминсульфата, которые соединены друг с другом 0-1,3-
и 0-1,4-гликозидными связями, аналогичными связям в гиалуроновой кислоте:
Хондроитинсульфат
Сульфогруппа, связанная с остатком N-ацетилгалактозамина в хондро-
итинсульфате, может занимать также 4-е положение. Эту разновидность хонд-
роитинсульфата обозначают как хондроитинсульфат А. Есть и другие виды
хондроитинсульфатов, отличающиеся от указанных выше форм некоторыми
деталями строения.
Тёпарин—специфический гетерополисахарид, препятствующий свертыва-
нию крови у животных и человека. Он обладает также антилипемическим,
антимитотическим и регуляторным по отношению к ряду ферментов действи-
ем. Гепарин содержится в печени (до 100 мг/кг ткани), легких, селезенке,
щитовидной железе, крови и, вероятно, в других тканях и органах; он получен
в кристаллическом состоянии. Молекулярная масса гепарина, выделенного из
разных объектов, колеблется от 4000 до 20000. Определения молекулярной
массы гепарина из разных объектов гравиметрическим, вискозометрическим
и гельфильтрационным методами дали более узкие ее пределы: 11 000—12900.
Молекула гепарина состоит из остатков глюкуроновой кислоты и а-глюкоза-
мина в виде двойного производного серной кислоты:
Под действием ряда ферментов (гепариназа, дисахаридсульфоэстераза,
сульфамидаза, сульфоэстераза и др.) гепарин распадается до составляющих
его структурных элементов и продуктов их деградации, т. е. в конечном счете
до глюкуроновой кислоты и глюкозамина. Период полураспада гепарина
в организме кроликов составляет 17,5 + 6,5 мин, собак—34+13,5 мин.
В настоящее время широко развернулись работы по выделению, очистке
и изучению состава, строения и функций ряда других гомо- и гетерополисаха-
ридов. К их числу относятся гемицеллюлозы, пектиновые вещества, глюко-
маннаны и галактоманнаны высших растений, полисахариды водорослей
(агар, каррагинаны, альгиновые кислоты, галактаны, маннаны, ламинарии
и др.), внеклеточные (ксантан, пуллулан и др.) и капсулярные полисахариды
бактерии и, наконец, полисахариды простейших (парамилон и др.). Как химия,
так и особенно биохимия многих из них представляет большой интерес.
327
ОБМЕН УГЛЕВОДОВ
Нередко функцию углеводов в обмене веществ сводят только к энергети-
ческому обеспечению химических реакций. Это далеко не так. Бесспорно, что
при распаде (окислении) углеводов в организме идет высвобождение энергии,
которая запасается далее в макроэргических связях АТФ, и что АТФ, син-
тезированная сопряженно с окислением углеводов, поставляет энергию для
осуществления химических процессов и для других нужд организма. Однако
углеводы вы! олняют еще одну важнейшую функцию в процессе обмена
веществ—они являются источником большого числа органических соедине-
ний, которые служат исходными продуктами для биосинтеза липидов, белков
и нуклеиновых кислот. В углеводах, образующихся в процессе первичного
биосинтеза органического вещества, связывается углерод и запасается энергия.
Распад углеводов. Пути распада полисахаридов и дисахари-
дов. Полисахариды и олигосахариды распадаются до более простых соедине-
ний посредством реакций двух типов: гидролиза и фосфоролиза. Классическим
примером распада первого типа является гидролиз крахмала, второго—фос-
форолиз гликогена.
Ступенчатый характер гидролиза крахмала рассмотрен ранее. Реакция гид-
ролиза крахмала ускоряется амилазами—специфическими ферментами, относя-
щимися к подклассу гидролаз гликозидов (класс гидролаз). В зависимости от
характера фермента разрыв гликозидных связез между остатками a-D-глюкопира-
нозы в молекуле крахмала и присоединение по месту разрыва элементов воды (Н
и ОН-группа) может происходить в различных позициях. Соответственно этому
конечными продуктами гидролиза крахмала оказываются либо глюкоза, либо
мальтоза, либо олигосахариды. Естественно, что в процессе постепенного укороче-
ния молекулы крахмала в результате гидролитического отщепления моносахарид-
ных, дисахаридных или олигосахаридных звеньев на какой-то ступени распада
в качестве промежуточных продуктов возникают декстрины. Наличие и динамику
их образования легко установить, прослеживая изменение окраски крахмала с ио-
дом от синей до красно-бурой в процессе ферментативного гидролиза крахмала.
В природе найдено несколько различных амилаз.
Епокоамилаза, или у-амилаза (а-1,4-глюкан глюкогидролаза), ускоряет реак-
цию гидролиза 1,4-связей в молекуле крахмала, олигосахаридов и даже дисахари-
дов (например, мальтозы), последовательно отщепляя остатки глюкозы от невос-
станавливающегося (не содержащего свободной альдегидной группы или гликозид-
ного гидроксила) конца молекулы. Механизм ее действия можно представить
следующей схемой:
Спиртовый гидроксил
(иевосстанавливающий
конец молекулы)
Глюкоамилаза
Гликозидный гидроксил
(восстанавл ивающий
конец молекулы)
328
Цифрами I, II, III, IV и т. д. здесь обозначены последовательные реакции
гидролиза 1,4-связей в молекуле 1,4-глюкана при каталитическом воздействии
глюкоамилазы. Если в молекуле полисахарида есть разветвление, то действие
у-амилазы прекращается. Глюкоамилаза ускоряет гидролиз не только крах-
мала, но и гликогена.
Глюкоамилаза распространена повсеместно; она открыта Е. Л. Розенфельд
(1959) в тканях животных, где ярко представлена, как, впрочем, и в плесне-
вых грибах. Из ряда источников глюкоамилаза выделена в гомогенном состоя-
нии. Ее молекулярная масса в большинстве случаев близка к 100000 (у-амилаза
из почек человека—97 000, из печени быка —107 000, из печени крысы —114 000,
из гриба аспергилла—всего 62 000). Для глюкоамилазы из печени быка доказа-
на мультимерная структура молекулы: 4 су ъединицы по 26000 каждая. Обнару-
жено 2 вида глюкоамилаз—кислая (оптимум pH 4,8—5,0, локализована в лизо-
сомах, Кы для гликогена—5,45 • 10-3 М) и нейтральная (оптимум pH 6,0—6,5,
локализована в микросомальной фракции клетки и в гиалоплазме, Кы для
гликогена—16,25 • 10-3 М). Отсутствие кислой глюкоамилазы у человека связа-
но с тяжелым наследственным заболеванием—гликогенозом; оно состоит
в накоплении гликогена в клетках печени, мышц и других органов. Глюкоамила-
за иммобилизована и в этом виде применяется в промышленном масштабе для
гидролиза крахмала до глюкозы; созданная у нас установка позволяет вести
процесс в течение 3 месяцев без заметной потери активности фермента.
Р-Амилаза (а-1,4-глюкан мальтогидролаза) ускоряет реакцию гидролиза
крахмала по 1—4 связям,'последовательно отщепляя остатки мальтозы, начи-
ная с нередуцирующего конца молекулы крахмала; ее действие, как и у-
амилазы, прекращается в точках разветвления в молекуле кр хмала:
___+НгО________________________+Н2О и т.д.
f-Амилаза
Мальтоза, освобождающаяся при гидролизе крахмала под действием р-
амилазы, получается в р-форме:
a-форма мальтозы f-Форма мальтозы
Предполагают, что образование P-формы мальтозы, т. е. изменение про-
странственной конфигурации молекулы по месту образующегося гликозид-
ного гидроксила при 1-м углеродном атоме остатка глюкозы, происходит
в момент гидролиза а-гликозидной связи. В соответствии с этим фермент
и получил свое название. P-Амилаза характер га только для высших растений,
329
• Восстанавливающий конец моле килы
крахмала 3
* кюцы nDJm°-
о-о связь W
ою связь 1-6
из которых она выделена и получена
в виде кристаллов. Молекулярная
масса 0-амилазы из батата равна
201000(4 x 50000).
а-Амилаза (а-1,4-глюкан 4-глю-
каногидролаза) ускоряет гидролиз
1,4-гликозидных связей в молекуле
крахмала без какого-либо опреде-
ленного порядка, в результате чего
сначала возникают олигосахариды,
которые тоже подвержены действию
а-амилазы, если они содержат три
или более остатков a-D-глюкозы.
Таким образом, a-амилаза является
эндоамилазой, так как она атакует
внутренние гликозидные связи в мо-
.Лк Точки приложения действия
ft V различных амилаз
Рис. 105. Схема ферментативного гидролиза
крахмала под действием амилаз разных типов
(пояснение в тексте)
лекуле крахмала и для ее действия
не имеет значения близость или уда-
ленность нередуцирующих концевых
остатков полигликозидных фрагме-
нтов молекулы крахмала. В качестве
главного конечного продукта гидро-
лиза крахмала при участии а-амила-
зы образуется мальтоза, так как
в дисахаридах 1,4-связи под действием a-амилазы не гидролизуются.
Если действие a-амилазы распространяется на спирализованный участок
молекулы крахмала или молекулы амилозы, то гидролиз 1,4-гликозидных
связей осуществляется латерально, вдоль одного бока спирализованного 1,4-
глюкана. Поскольку каждый виток спирали содержит 6—7 остатков a-D-
глюкопиранозы, в результате гидролиза возникают гекса- или гептасахариды.
Такой механизм действия a-амилазы показан на рис. 103.
Пщролиз крахмала с помощью a-амилазы во всех случаях приводит к об-
разованию a-формы мальтозы (см. с. 316), откуда ведет свое происхождение
название фермента. a-Амилаза найдена у всех растительных и животных
видов; кроме крахмала она гидролизует также гликоген. Молекулярная масса
a-амилазы из поджелудочной железы равна 45000, из солода—59000, из
мучного хрущака—68000, из двух видов аспергилла—51000 и 58000, из
бактерий—96000 (4 x 24000). Выяснена первичная и третичная структура а-
амилазы из аспергилла.
Амило-1,6-глюкозидаза ускоряет реакцию гидролиза 1,6-связей в молекуле
крахмала (амилопектина), расщепляя, таким образом, молекулу крахмала
в точках разветвления полиглюкозидной цепи и образуя олигосахариды с тем
или иным числом остатков a-D-глюкозы в них в зависимости от длины
отщепляемой цепи. Амило-1,6-глюкозидаза характерна для животных тканей.
Однако это не значит, что ее нет в растительном мире: у растений этот же
фермент называют Я-энзимом, а у микроорганизмов—изоамилазой.
Общая схема гидролиза крахмала при участии перечисленных выше фер-
ментов приведена на рис. 105.
Характерная особенность амилаз—отсутствие абсолютной специфичности
действия. При их участии гидролизуются различные соединения: амилоза,
амилопектин, гликоген, олигосахариды и родственные им вещества, по-
строенные из остатков a-D-глюкопиранозы и содержащие в молекулах 1,4-
и 1,6-связи. По-видимому, все амилазы являются металлопротеинами (содер-
330
Рис. 106. Третичная структура а-амилазы, продуцируемой Aspergillus orizae (така-амилаза
А)
Молекула представляет эллипсоид (8,0 4,5 -4,5 нм), содержит 452 аминокислотных остатка, чередование которых выясне-
но, а также олигосахарид, составленный из 2 остатков глюкозамина и 6 остатков маннозы- Имеет ясно выраженную
двухдоменную структуру; на границе доменов располагается активный центр (Л), включающий радикалы аспарагиновой
кислоты н гистидина, н Са2+-связывающий центр (С); остаток тирозина, нависающий над активным центром, принимает,
как и Са2+, участие в связывании субстрата. Третичная структура фиксируется четырьмя —S—S-мостиками (S1—S4).
8а-спиралей обозначены прямоугольниками (Н1—Н8), а 8 р-слоев—стрелками,. Разрешение—0,3 нм
жат Zn2+ и Са2+), некоторые из них мультимеры; предполагают, что ионы
металлов способствуют становлению молекулы мультимера из протомеров.
Активные центры амилаз образованы радикалами гистидина, аспарагино-
вой и глутаминовой кислот, а также тирозина (рис. 106). Последнему приписы-
вают функцию связывания субстрата, а первым трем—каталитическую функ-
цию. В соответствии с представлениями о структуре активного центра амилаз
предполагают на 1-м этапе следующий механизм распада гликозидных связей
при их посредстве:
331
На 2-м этапе в активный центр фермента входит молекула воды, присое-
динение ОН-группы которой обеспечивает распад возникшей сложноэфирной
связи и высвобождение радикала дикарбоновой аминокислоты; протон воды
присоединяется к депротонированному на 1-м этапе радикалу гистидина:
В результате восстанавливается структура активного центра фермента, и он
готов к осуществлению следующего каталитического акта в гидролизе глико-
зидной связи. Есть достаточно оснований считать, что не только амилазы, но
и все другие гликозидазы (см. ниже) действуют в соответствии с приведенным
выше механизмом.
Особую роль в осуществлении гидролиза гликозидных связей играют
имидазольные радикалы гистидина, присутствующие в каталитическом центре
амилаз. Именно при их посредстве идет перенос протонов, столь необходи-
мый для обеспечения каталитического акта. Такая функция радикалов гисти-
дина характерна и для каталитических центров ряда других гидролаз (см.
с. 229, 263 и 406).
Аналогично крахмалу и гликогену гидролизуются другие природные поли-
сахариды: целлюлоза—при участии целлюлазно о комплекса ферментов (со-
стоит из эндо-1,4-Р-глюканазы, экзоцеллобиогидролазы, целлобиазы и экзо-
1,4-Р-глюкогидролазы , инулин—с помощью инулиназы (2,1-0-В-фруктан
фруктан гидролазы), хитин—хитиназы, ксилан—ксиланазы, гиалуроновая
кислота—гиалуронидазы, пектиновые вещества—комплекса пектинолитиче-
ских ферментов (пектинэстеразы, полигалактуроназы и др.) и т. п. Во всех
случаях полисахариды распадаются до соответствующих моносахаридов или
их производных (иногда до дисахаридов), из которых составлены молекулы
указанных полисахаридов. Некоторые из гидролаз, перечисленных выше, ши-
роко распространены в природе, другие (целлюлаза, инулиназа)—присущи
лишь определенным видам растений, животных и микроорганизмов. Особое
внимание в последние годы привлекают целлюлазы, хитиназы, 1,3- и 1,6-0-
глюканазы и лихеназы (гидролизуют гетерополисахариды с перемежающими-
ся 1,3- и 1,4-гликозидными связями), так как они деструктируют растительные
и микробные остатки, являющиеся экологически опасными отходами произ-
водства, позволяя при этом получать глюкозу и N-ацетилглюкозамин. Неко-
торые из них, например, хитиназы из Bacillus cereus, множественны (68, 52
и 38 кДа), выделены и очищены до гомогенного состояния; они ускоряют
реакцию гидролиза хитина при pH=4 и t = 60° С до хитобиозы, но не до
К-ацетил-0-В-глюкозамина.
В свою очередь, дисахариды, возникающие в ряде случаев при гидролизе
полисахаридов (мальтоза при гидролизе крахмала, целлобиоза при гидролизе
целлюлозы) или существующие в организме в свободном виде (лактоза,
332
сахароза, трегалоза и т. п.), гидролизуются при каталитическом воздействии
а- и Р-гликозидаз до индивидуальных моносахаридов. Все гликозидазы, за
исключением трегалазы (а, а-трегалоза-глюкогидролазы), отличаются широ-
ким спектром специфичности, ускоряя гидролиз фактически любых гликози-
дов, являющихся производными того или иного а- или Р-моносахарида. Так,
а-глюкозидаза ускоряет реакцию гидролиза а-глюкозидов, в том числе маль-
тозы; р-глюкозидаза—р-глюкозидов, в том числе целлобиозы; Р-галактозида-
за— Р галактозидов и среди них лактозы и т. д. Примеры действия а и Р-
глюкозидаз были приведены ранее.
Гликозидазы, кроме гидролазной активности, как правило, обладают также
гликозилтрансферазным действием и ускоряют процессы переноса гликозиль-
ных остатков на те или иные субстраты. Так, например, целлобиаза из
аспергилла (М = 142 000) при действии на целлобиозу образует не только
P-D-глюкозу, но и трисахариды, т. е. переносит остаток p-D-глюкозы с одного
дисахарида на другой, что особенно ярко выражено в начале ферментативного
процесса. Сейчас все более укрепляется мнение, что трансгликозилирование
является одним из существенных элементов распада полисахаридов, олигосаха-
ридов и дисахаридов, сопутствующих их гидролизу.
Второй тип распада полисахаридов и олигосахаридов представлен реакци-
ями фосфоролиза. Если попытаться выразить реакцию фосфоролиза в виде
суммарного хими еского уравнения, то она сводится к присоединению элемен-
тов фосфорной кислоты (водорода и кислотного остатка) по месту разрыва
гликозидной связи между остатками моносахаридов в молекулах олиго- или
полисахаридов:
Глюкоза
Опираясь на это формальное представление, указанную реакцию расцени-
вали на первых порах как процесс, полностью аналогичный реакции гид-
ролиза, с тем лишь отличием, что роль воды в данной реакции играет Н3РО4.
Поэтому она и была названа реакцией фосфоролиза. Эта реакция ускоряется
специфическими ферментами—фосфорилазами, которые относятся к под-
классу гликозилтрансфераз и подподклассу гексозилтрансфераз. Они были
открыты Я. О. Парнасом, К. Кори и Г. Кори более полувека тому назад.
Фосфорилазы ускоряют процесс переноса гликозильного остатка с невос-
станавливающего конца молекулы полисахарида или олиг сахарида на неор-
ганический фосфат. Фосфоролитическому расщеплению подвергаются толь-
ко 1,4-связи (см. уравнение реакции на с. 128 и 334).
Представленный выше процесс многократно повторяется, и ступенчатый
распад а-1,4-глюкана сопровождается выделением большого числа молекул
глюкозо-1-фосфата. В случае строго линейного полигликозида фосфоролиз
идет до конца, в случае разветвленных полисахаридов—останавливается
в точках разветвлений полисахаридной цепи.
333
глюкана
фосфат
Реакция фосфоролиза полисахаридов широко предс авлена в природе.
Именно так идет распад гликогена, когда он вступает на путь гликогенолиза
(см. ниже). Аналогично этому посредством реакции фосфоролиза значитель-
ная часть крахмала превращается в глюкозо-1-фосфат при использовании
запасов крахмала для нужд растительного организма. Отмечены также случаи
фосфор лиза дисахаридов. Так, некоторые бактерии содержат мальтозофос-
форилазу, ускоряющую реакцию распада мальтозы на глюкозо-1-фосфат
и глюкозу при взаимодействии ее с Н3РО4.
Фосфорилазы—мультимеры высокой молекулярной массы. Наиболее изу-
чена фосфорилаза из мышц кролика. Ее молекула (М=400 000) состоит из
четырех протомеров (4x100000), каждый из которых представлен полипеп-
1 тидной цепью из 841 аминокислотного остатка. Последовательность их рас-
положения установлена; в частности, выяснено, что в положении 14 находится
серин, фосфорилированный по ОН-группе радикала, а в положении 679—ли-
зин, с e-аминогруппой которого альдиминной связью соедин н пиридоксаль-
фосфат, являющийся простетической группой фосфорилазы. Электронно-мик-
роскопически выяснена и третичная структура протомеров, их топография,
локализация субстратного и каталитического центров, пространственная ком-
поновка мономеров в тетрамере (рис. 107). Им нно в -таком состоянии, т. е.
в виде тетрамера, гликоге фосфорилаза из мышц кролика проявляет высокую
активность в переносе гликозильных остатков на неорганический фосфат. Ее
называют фосфорилазой а.
Будучи дефосфорилирована по 14-му остатку фосфосерина каждого из
протомеров (это происходит при участии протеинфосфатазы), тетрамерная
молекула фосфорилазы а распадается на две равные части (т. е. на два
димера), называемые фосфорилазой Ь, и частично теряет' ка алитическую
активность. После фосфорилирования протомеров в неполностью активных
димерах фосфорилазы b тетрамерная структура восстана ливаетс i и снова
идет реакция фосфоролиза. Фосфорилирование димеров осуществляется при
участии киназы фосфорилазы b (о киназах см. с. 124), переносящей фосфат
с АТФ на радикал остатка серина, находящийся в 14-м положении в полипеп-
тидной цепи мономера. Становление тетрамера зависит от присутствия Mg2+,
участвующего в образ вании ионных связей между протомерами, в том числе,
вероятно, за счет фосфатных групп:
334
Киназа фосфорилазы Ь
2 фосфорилаза Л+4 АТФЧ~ ~ '* Фосфорилаза а+4 АДФ
(не полностью активна) Протеинфосфатаза (полностью активна)
Фосфорилаза оказалась ферментом, исследование которого позволило
впервые детально изучить вопрос о регуляции активности ферментов за
счет реакций фосфорилирования—дефосфорилирования (рис. 108). Дело
в том, что действие киназы фосфорилазы Ь, в свою очередь, тоже регулиру-
ется при помощи такого же механизма. Этот фермент имеет огромную
молекулярную массу—1272000 Да. Он построен из 4 субчастиц
с М = 318 000, каждая из которых, в свою очередь, составлена из трех
протомеров; два из них (А и В) являются регуляторными, а один, С, несет
каталитическую функцию, т. е. ускоряет реакцию фосфорилирования фос-
форилазы Ь. Однако протомер С активен лишь в том случае, когда фосфо-
рилирован регуляторный протомер В и дефосфорилирован регуляторный
протомер А. Такой способ регуляции активности фермента получил назва-
ние регуляции за счет фосфорилирования второго регуляторного центра
(рис. 109).
Фосфорилирование протомера В, как, впрочем, и протомера А, когда
киназа фосфорилазы b инактивируется, в свою очередь, осуществляется при
участии киназы, ко орая является, таким образом, киназой киназы фосфори-
лазы Ь. Крайне существенно, что она активна только в присутствии ал-
лостерического регулятора—циклического аденозинмонофосфата (цАМФ),
возникающего из АТФ в присутствии аденилатциклазы, деятельность которой
контролируется гормонально (см. рис. 108). Естественно, что попеременное
дефосфорилирование протомеров А и В при посредстве протеинфосфатазы
тоже является условием регуляции активности киназы фосфорилазы Ь. Таким
образом, активность фосфорилаз, как и, следовательно, процесс фосфоролиза
в целом, тонко регулируется.
А
Рис. 107. Четвертичная структура гликогенфосфорилазы мышц кро-
лика (Л) и третичная структура протомера (£):
А—при вращении модели от О до 180° ясно прослеживается наличие четырех протомеров
и их взаимное расположение в мультимере; Б—детали пространственного расположения
полипептидной цепи опущены и показан лишь общий контур протомера; 1—N-концевая
аминокислота; 841 —С-концевая аминокислота; /4—остаток серинфосфата; 679—остаток
лизина с присоединенным к нему пиридоксальфосфатом; пунктирным кружком обозначена
локализация активного центра, косой штриховкой—субстратного центра, черным прямо-
угольником—АМФ-связывающиЙ центр (АМФ—активатор фермента), треугольником
и точкой в зоне активного центра—места связывания нуклеозидов и глюкозы (оба—
ингибиторы)
335
цАМФ
АТФ
РРн
Протеинкиназа * т
(неактивная)
Киназа АТФ
фосфорилазы Ъ
Фосфорилаза b
Протеинкиназа
(активная)
,АДФ Киназа
—v фосфорилазы Ъ
I (активная)
Рн
Гликоген
4АТФ’4АДФ
Фосфорилаза а
Глюкозо-
1-фосфат
Рис. 108. Регуляция активности гликогенфосфорилазы при посред-
стве протеинкиназных и протеинфосфатазных реакций (пояснение
в тексте)
В последние годы появились новые данные о структуре киназы фосфорила-
зы b и регуляции ее активности. Полагают, что ее субчастица (М = 316 000)
состоит из 4 протомеров (а—138000; 0—117000; у—44000 и 5—17000),
а полная молекула—из 4 суб частиц (М = 1 250000). Каталитическую функцию
несет протомер у, регуляторную—протомеры а, 0 и 5, причем последний
является Са2+-зависимым и отождествляется в Са2+-связывающим регуля-
торным белком кальмодулином.
Превращение моносахаридов. Как показано выше, при распаде
олиго- и полисахаридов возникают свободные монозы и их фосфорные эфиры.
Дальнейший обмен моносахаридов идет такими путями, что используются
только фосфорные эфиры моносахаридов, свободные же монозы фосфорили-
руются, т. е. превращаются в фосфорные эфиры.
Фосфорилирование свободных моносахаридов—обязательная реакция на
пути их использования для нужд организма. Оно приводит к возникновению
более реакционноспособных, чем свободные моносахариды, фосфорных эфиров
моносахаридов и поэтому часто рассматривается как реакция активирования.
Фосфорилирование моносахаридов существляется при взаимодействии их
с АТФ и ускоряется специфическими (и неспецифическими) фосфотрансфераза-
ми, которые называются также киназами. Фосфорилирование глюкозы, напри-
мер, идет согласно схеме:
336
Каталитический центр открыт,
киназа фосфорилазы b активна
М-145000
Дефосфорилирован
М-45000
М-128000
Фосфорили-
рован
1ЛЛ*'1
Фосфорилирован
Инактивация
Активация
^<^Ротеиифосф«2?'
Мф~ЗВВИСИ>А&*
**назы фосф0?1'*'
Каталитический центр
закрыт, киназа фосфорилазы •
неактивна
дефосфори-
пировал
Рис. 109. Регуляция активности киназы фосфорилазы b (пояснение в тексте)
Глюкокиназа (гексокиназа)
Глюкоза+АТФ-----------------------------► Епокозо-6-фосфат+АДФ
(АТФ: D-глюкоза
6-фосфотрансфераза)
Уравнение этой реакции приведено выше (см. с. 124).
В природе обнаружено более двух десятков индивидуальных фосфотранс-
фераз, переносящих остатки фосфорной кислоты с АТФ на те или иные
моносахариды. Молекулы многих из них, как выяснено в последнее время,
построены из 2 субъединиц. Так, гексокиназа из дрожжей при М = 102 000
состоит из 2 субъединиц с М = 51000 каждая и обнаружено 2 ее изозима;
галактокиназа из эритроцитов человека при М = 53000—из 2 субъединиц по
27000 каждая; рибулокиназа из кишечной палочки при М = 98 000—из 2 субъ-
единиц по 50000 каждая и т. п. Изучение третичной структуры субъединицы
Рис. 110. Конформационные изменения субъединицы молекулы гексокиназы в процессе
возникновения ферм нт-субстратного комплекса:
А—до присоединения глюкозы (открытая конформация); Б—после присоединения глюкозы (закрытая конфор-
мация). Достаточно легко различимы два главных домена субъединицы, перемещение которых обеспечивает измене-
ние ее конформации
337
дрожжевой гексокиназы выявило ее двухдоменную структуру и локализацию
связывания АТФ и глюкозы на дне щели между дом нами, где, собственно,
и протекает процесс переноса фосфата при непременном участии Mg2+. Выяс-
нены и более тонкие детали этого процесса, в частности переход субъединицы
из открытой конформации в момент сорбции субстратов в закрытую в процес-
се осуществления реакции (рис. 110).
Рассмотрим еще несколько примеров действия киназ. Так, фруктокиназа
ускоряет реакцию образования фруктозо-6-фосфата:
+ АТФ
0-D -Фруктоза
но он
Фруктокиназа
(АТФ'- D-фруктоза-
6-фосфотрансфераза)
Фруктозо-6-фосфат
Аналогично протекает взаимодействие между рибозой и АТФ:
^-D-Рибоза
/?-О-Рибозо-5-фосфат
Следовательно, практи ески любой моносахарид может быть переведен
в фосфорный эфир. Именно в виде фосфорных эфиров и осуществляется
дальнейший обмен моносахаридов.
Важн йшей особенностью фосфорных эфиров моносахаридов является их
способность к изомеризации. Последняя может осуществлятьс как за счет
стереоизомеризации (т. е. изменения пространственного расположения атомов
и атомных групп в молекуле), так и в результате внутримолекулярного перено-
са атомов Н. Одним из прим ров подобного рода превращений служи пере-
ход глюкозо-6-фосфата в фруктозо-6-фосфат, интенсивно осуществляемый
в мышечной ткани и приводящий к установл нию равновесия между этими
двумя эф рами в отношении 2:1:
Глю козо -6 -фосфат
(a-D-Глюкопиранозо-
6-фосфат)
Фруктозо-6-фосфат
(/J-D-фруктофуранозо-
6-фосфат)
Приведем еще некоторые характерные в этом смысле реакции. Так, рибозо-
5-фосфат может превращаться в рибулозо-5-фосфат, а последний, в свою оче-
редь,—в ксилулозо-5-фосфат:
338
л
С—н
H-i-он
сн,он
н-с-он
H-i-он
Рибозофосфат-
нзомераза
он
СН,—О—Р=О
и г ли Рибулоз ос т-
п ъ ип з-»г,ж№дм
Н-С-ОН он
СН,-О-Р=О
I
он
СН.ОН
С«=О
I
но-с-н
I
н-с-он он
I I
сн,-о-р=о
I
он
D-Рвбозо-б-фосфат
D-Рнбулозо-Б-фосфат
О-Ксалулмо-Б-фосфи
В первом случае происходит внутримолекулярный перенос водорода, во
втором—изменение расположения в пространстве атома Н и ОН-группы при
3-м углеродном атоме. И тот и другой процессы ускоряются ферментами,
принадлежащими к классу изомераз.
Особенно легко процесс перестройки одного моносахарида в другой
протекает, если фосфорный эфир моносахарида соединен с уридин-5'-фос-
фатом. Примером может служить превращение галактозы в глюкозу и об-
ратно:
УрИПИидифосф1тглюхоза(УДФ-глюкоза)
Уридинднфосфатгалактоза(УДФ-галактоза)
Если учесть, что в организме не только фосфорные эфиры моносахаридов,
но и свободные моносахариды могут превращаться друг в друга, то появля-
ется возможность перехода от любой гексозы или пентозы к любой другой,
изомерной ей. Это имеет огромное значение. В природе и синтез и распад
моносахаридов, как и весь процесс обмена углеводов, протекает через некото-
рые их формы, занимающие в данном процессе ключевые позиции. Это,
в первую очередь, фосфорные эфиры двух моносахаридов: глюкозо-6-фосфат
и рибулозо-5-фосфат.
Обмен глюкозо-6-фосфата. 1люкозо-6-фосфат образуется в орга-
низме разными путями. Во-первых, он может синтезироваться путем фосфо-
рилирования глюкозы за счет ее взаимодействия с АТФ. Во-вторых, он
образуется в результате реакции изомеризации фосфорных эфиров других
изомерных ему гексозофосфорных эфиров. В-третьих, он получается из глю-
козо-1-фосфата, который представляет собой продукт фосфоролиза олиго
и г олисахаридов. Две первые реакции рассмотрены в предыдущем разделе.
Что асается преобразования глюкозо-1-фосфата в глюкозо-6-фосфат, то эта
реакция протекает в два этапа при участии фермента фосфоглюкому азы
Молекулярная масса фермента из мышц кролика равна 62000; молекула
фосфоглюкомутазы состоит из двух субъединиц с М = 31 000 каждая. Актив-
ный центр ее включает в свой состав остаток фосфосерина с которого
339
остаток фосфорной кислоты передается на глюкозо-1-фосфат с образованием
глюкозо-1,6-дифосфата и дефосфорилированного фермента. Последний, вза-
имодействуя с глюкозо-1,6-дифосфатом, снова превращается в фосфопроте-
ин, однако получает остаток фосфорной кислоты, присоединённой к 1-му
углеродному атому глюкозы с высвобождением соответственно глюкозо-6-
фосфата:
В цепь—COv
^СН—CHjOH +
В цепь—NFr
Фосфоглюкомутаза
дефосфорилированная
В цепь—СО\
В цепь—NH^
ОН
СН—СН2—О—Р=О
ОН
Фосфоглюкомутаза
Равновесие рассмотренной реакции сильно сдвинуто в сторону образо-
вания глюкозо-6-фосфата. Поэтому в тканях содержание глюкозо-1-фосфа-
та не превышает 3—4% от общего количества гексозомонофосфатов в ор-
ганизме.
1люкозо-6-фосфат подвергается в организме разнообразным превращени-
ям. Некоторая доля erq распадается в конечном счете до СО2 и Н2О. При этом
многократно повторяются реакции окисления (дегидрогенизации) как самого
глюкозо-6-фосфата, так и продуктов его дальнейшего распада. Сопряженно
с передачей атомов водорода, снятых с глюкозо-6-фосфата и возникших из
него субстратов, на кислород (с образованием молекул воды) осуществляется
синтез АТФ из АДФ и неорганического фосфата, т. е. запасается, аккумулиру-
ется энергия в составе макроэргических связей молекул АТФ. Кроме того,
некоторое количество молекул АТФ синтезируется здесь же иным путем.
Следовательно, распад глюкозо-6-фосфата служит энергетическим целям: яв-
ляется источником энергии для организма.
Вместе с тем значительная часть промежуточных продуктов, возника-
ющих в процессе обмена глюкозо-6-фосфата, используется для синтеза ами-
нокислот (белков), нуклеотидов (нуклеиновых кислот), глицерина и высших
жирных кислот (триглицеридов, фосфатидов), стеролов (стеридов) и т. п.
В частности, как описано выше, для синтеза аланина используется пировино-
градная кислота (см. с. 276), являющаяся непременным промежуточным
продуктом при распаде глюкозо-6-фосфата по дихотомическому пути
(см. следующий раздел). Другие промежуточные продукты распада глюкозо-
340
6-фбсфата: 3-фосфоглицериновая кислота и фосфоенолпировиноградная кис-
лота (см. ниже) идут на синтез фенилаланина, тирозина, триптофана и сери-
на: Включаясь в цикл трикарбоновых и дикарбоновых кислот (см. рис. 117),
пировиноградная кислота, превращаясь в щавелевоуксусную и а-кетоглута-
ровую, дает начало аспарагиновой и глутаминовой кислотам, а из них—
ряду других аминокислот. Рибозо-5-фосфат, образующийся при апотомиче-
ском распаде глюкозо-6-фосфата (см. схему 6), служит для синтеза гистидина
и, в еще большей степени, для синтеза пиримидиновых и пуриновых нукле-
отидов (см. гл. VI).
Таким образом, глюкозо-6-фосфат обеспечивает организм и энергией
и строительным материалом для синтеза новых органических соединений,
используемых в процессе жизнедеятельности.
Распад глюкозо-6-фосфата осуществляется преимущес венно двумя путя-
ми. В одном случае на определенной стадии происходит распад шестиуглерод-
ной молекулы на две трехуглеродные, т. е. пополам. Этот путь получил
название дихотомического распада. Второй путь состоит в потере глюкозо-6-
фосфатом 1-го углеродного (головного) атома и именуется апотомическим
распадом. Есть еще третий путь, содержащий элементы первого и второго.
Рассмотрим каждый из них.
Дихотомический путь распада глюкозо-6-фосфата. Всту-
пая на дихотомический путь распада, глюкозо-6-фосфат прежде всего пре-
терпевает изоме изацию и превращается в фрук озо-6-фосфат, который
далее фосфорилируется по 1-му углеродному атому, образуя фруктозо-1,6-
дифосфат:
но—р—он
2
НО—р—он
о
но—р—он
о
Н
он
Н ОН
Глюкозофос- ОН,
Н фат-изоме-
раза
Н
ОН Н
он
н н
СН2ОН
Н Н
АТФ
сн.
Фосфофрукто-
киваза
он
н но
н н
СН2
о
о-23-Глюкопиранозо-
6-фосфат
P-U-Фруктофуранозо-
6-фосфат
но—р-он
Фруктозо-1,6-
дифосфат
Фосфофруктокиназа, представляющая собой белок с М = 140000 у бак-
терий и от 360000—400000 (во всех случаях—тетрамер) до 800000 (гек-
самер или октамер) у эукариот, является самым «медленным» из всех
ферментов, обслуживающих дихотомический распад углеводов. Эта прак-
тически необратимая реакция лимитирует весь процесс. В то же время
активность глюкозофосфатизомеразы, например, в дрожжах, в 500 раз
превышает активность фосфофруктокиназы и на 1—2 порядка выше, чем
у других ферментов дихотомического распада, т. е. это один из самых
«быстрых» ферментов обмена глюкозо-6-фосфата. Строение и механизм
действия фосфофруктокиназы из термофильной бактерии детально изучены
(рис. 111).
341
Рис. 111. Строение половины молекулы фосфофруктокиназы из термофильной бактерии
(3-я и 4-я субъединицы не показаны)
Молекулярная масса субъединицы, составленной из 316 аминокислотных остатков, равна 33900; третичная структура
характеризуется наличием 0-слоев (обозначены А—К) и а-спиралей (обозначены 1—13), а также существованием
двух доменов (1 и II); на границе доменов располагается активный центр, связывающий фруктозо-6-фосфат, АТФ
я Mg2+; вблизи С-концевой аминокислоты II домена находится аллостерический центр (АДФ—активатор, фосфо-
енолпировиноградная кислота—ингибитор), обеспечивающий регуляцию активности фосфофруктокиназы при по-
средстве ряда эффекторов, что является важнейшей особенностью этого фермента. В эпимолекуле между субъедини-
цами есть полость диаметром 0,7 нм, т. е. тетрамерный фермент имеет трубчатую структуру. Ф6Р—фруктозо-6-
фосфат
Концентрация фруктозо-1,6-дифосфата поддерживается на строго опре-
деленном уровне при посредстве сложного комплекса регуляторных процес-
сов: снижается под действием фруктозо-1,6-дифосфатазы (М = 140000,
4 х 35 000) и возрастает под влиянием фосфофруктокиназы. Однако актив-
ность первой тормозится, а второй—побуждается в присутствии фруктозо-
2,6-дифосфата, который, в свою очередь, синтезируется в печени при по-
средстве 6-фосфофрукто-2-фосфокиназы. Более того, последняя реакция
цАМФ-зависима, а сам этот фермент в печени бифункционален: один его
домен ускоряет синтез, а другой—распад фруктозо-2,6- дифосфата
(рис. 112). В дрожжах 6-фосфофрукто-2-фосфокиназа и фруктозо-2,6-дифос-
фатаза не образуют бифункционального фермента и являются самостоя-
тельными энзимами.
Последнее соединение (фруктозо-1,6-дифосфат) и подвергается далее
дихотомическому распаду на две фосфотриозы, превращающиеся друг в
друга:
342
он
Альдолаза (фруктозо-1,6-дифосфат-
триозофосфат-лиаза)
Триозофосфат-
изомераза
он
:н2—о—р=0
СНОН он
сн2он
Диоксиацетон-
фосфат (95%)
Г лицеральдегид-
3-фосфат (5%)
/
Оба фермента, ускоряющие приведенные выше реакции, получены в кри-
сталлическом состоянии. Альдолаза из мышц кролика характеризуется
М = 160000 (4x40000), а триозофосфат-изомераза—53000 (2x26500). Выяс-
нена первичная структура последней и параметры ее вторичной структуры
Рис. 112. Регуляция содержания ключевого метаболита дихотомического рас-
пада углеводов— фруктозо- ,6-дифосфата (Ф1,6Р2):
6РФ-1 -киназа— фруктозо-6-фосфат-1 -киназа; 6РФ-2-киназа—фруктозо-б-фосфат-2-киназа; ФбР—фрук-
тозо-6-фосфат; Ф2,6Р2—фруктозо-2,6-дифосфат; Ф1,6Р2-фосфатаза—фруктозо-1,6-дифосфатаза (Осталь-
ные пояснения в тексте)
343
(52% а-спиралей и 24% 0-слоев). Хотя при дихотомическом расщеплении
фруктозо-1,6-дифосфата (видимо, расщепляется его открытая форма) получа-
ется равное количество обеих фосфотриоз, в состоянии равновесия между
ними преобладает фосфодиоксиацетон.
В дальнейший обмен вступает только 3-фосфоглицериновый альдегид. По
мере расходования убыль этого соединения восполняется за счет фосфодиок-
сиацетона, который практически нацело в него превращается. Следовательно,
из каждой молекулы фруктозо-1,6-дифосфата фактически возникает две моле-
кулы 3-фосфоглицеринового альдегида, претерпевающего далее распад в соот-
ветствии со следующей схемой:
?н
:нучэ-р=о
ГТ
CHOH+HS-фермент (глицеральдегид-3- CHOH
фосфатдегидрогеназа)
Г лицеральдегид-
3-фосфат
CHOH Фосфогаацераткааа»
й С^он АТФ
о
♦ З-Фосфоглнцерн-
S-фермевт
Ан
1,3-Днфосфо-
Q.
3
новая кислота
t СНаОН н,О
♦U <?Н /0
\нс—о—р=о , "
Еаоааза
(2-фосфоглв-
иерат-гадрс-
анвва)
глицериновая
кислота
2-Ф осфое иол-
пир онниоград-
ная кислота
Енолпиро-
внноградная
кислота
ПирОВННО'
градиая
кислота
(пируват)
ХХ^Р=О ь- фермент
2-Фосфоглице-
риновая
кислота
Схема 5. Обмен 3-фосфоглицеринового альдегида
Все реакции, происходящие при обмене 3-фосфоглицеринового альдегида,
осуществляются ферментативным путем. Характерная их особенность состоит
в том, что на каждую молекулу распадающегося 3-фосфоглицеринового аль-
дегида синтезируются две молекулы АТФ из АДФ и остатков фосфорной
кислоты, поступающих сначала от 1,3-дифосфоглицериновой кислоты, а затем
от 2-фосфоенолпировиноградной кислоты (схема 5). Таким образом, уже здесь
запасается энергия, выделяющаяся в процессе постепенного окисления фосфо
344
251
/ / 'К >г<^зоо
>£vk Znff \U»r U)
Рис. 113. Структура субъединицы глицеральдегид-3-фосфатдегидрогеназ :
а-спиралн обозначены цилиндрами; фрагменты полипептидной цепи в Р-слоях—стрелками; в верхней части рисунка
нумерация их числовая (с^, а2 и т. д.; Pi. Рг и т. д), а в нижней, в области нуклеотидсвязывающего домена и активного
центра—буквенная (аА, ав и т. д.; рА, рв и т. д.); здесь же толстыми линиями показан кофермент—никотинамидаде-
ниндинуклеотцд (НАД), причем его восстанавливаемая никотинамидная часть сближена с цису^ к которому
присоединяется субстрат, что создает необходимые условия для окисления последнего
глицеринового альдегид . Конечным продуктом распада глюкозо-6-фосфата
является пировиноградная кислота (ПВК). В зависимости от объекта и усло-
вий, в которых идет обмен углеводов, дальнейшая ее судьба различна [см.
ниже).
Наиболее сложной из всех приведенных выше реакций на пути от 3-
фосфоглицеринового альдегида до ПВК является реакция окисления фосфо-
глицеринового альдегида в фосфоглицериновую кислоту. Остановимся на ней
345
Рис. 114. Механизм пируваткиназной реакции при сопряже-
нии окисления с фосфорилированием на уровне субстрата
Ион металла участвует в образовании тройного мостикового комплекса (фосфо-
енолпируват—металл—АДФ), обеспечивающего фосфорилирование АДФ; в ак-
тивном центре фермента координирующей функции иона металла способствует
радикал гис, а превращению енолпировиноградной кислоты в пировиноградную—
донор протопов ХН; ион металла закрепляется в активном центре также радика-
лом У и гидратируется. Препараты пируваткиназы, выделенные из разных источ-
ников, существенно не отличаются по молекулярной массе (она близка к 200 000);
молекула фермента в подавляющем большинстве случаев тетрамерна. Третичная
структура субъединицы мышечной пируваткиназы характеризуется наличием трех
доменов, на границе двух из них расположен активный центр
несколько подробнее. Реакция ускоряется глицеральдегцд-3-фосфатдегцдроге-
назой, полученной в кристаллическом состоянии из дрожжей, термофильных
бактерий и мышц кролика, омара и свиньи. Молекулярная масса фермента
в большинстве случаев равна 144000. Молекула фермента состоит из четырех
субъединиц с М = 37 000. Первичная структура их выяснена у глицеральдегид-
3-фосфатдегидрогеназы из пекарских дрожжей, термофильной бактерии
и мышц свиньи и омара, а третичная—у фермента из термофильной бактерии
и мышц омара (рис. 113).
Каждая субъединица несет одну молекулу НАД+ и 4 свободные HS-груп-
пы, принадлежащие остаткам цистеина. Сначала фосфоглицериновый аль-
дегид присоединяется к ферменту по радикалу остатка цистеина, занимающего
149-е положение в полипептидной цепи; в активный центр фермента входят
также остаток аргинина, находящийся в 231-м положении, и остаток гистиди-
на, занимающий 176-ю позицию. Затем в действие вступает НАД+, отни-
мающий от субстрата атом водорода в виде гидрид-иона (схема 5), и гмс17в
активного центра, снимающий другой атом водорода с ОН-группы тиополу-
ацеталя в виде протона; радикал гистидина удерживает снятый протон в тече-
ние непродолжительного времени и потом высвобождает его в среду (схема 5
и рис. 113).
В этот момент связь между остатком 3-фосфоглицериновой кислоты и фер-
ментом становится макроэргической обозначена значком ~, схема 5). Указан-
346
ная связь спонтанно распадается в присутствии Н3РО4 с образованием 1,3-
дифосфоглицериновой кислоты. Остальные реакции идут в основном при
участии соответствующих киназ. Важно отметить, что в момент отщепления
воды от 2 фосфоглицериновой кислоты (схема 5) также возникает макроэр-
гическая связь у остатка фосфата, что делает возможной дальнейшую киназ-
ную реакцию с образованием АТФ. Такой путь биосинтеза АТФ называется
субстратным фосфорилированием, которое возможно лишь потому, что в ак-
тивном центре фосфоглицераткиназы и пируваткиназы при участии Mg2+
сближаются концевой фосфат АДФ и переносимый на него фосфат, связанный
макроэргической связью в 1,3-дифосфоглицериновой или 2-фосфоенолпирови-
ноградной кислоте. В частности, эта важнейшая реакция фосфорилирования
АДФ на уровне окисляемого субстрата детально изучена у пируваткиназы
(рис. 114).
Апотомический путь распада глюкозо-6-фосфата. При апотомическом рас-
паде глюкозо-6-фосфата не происходит его превращения в фруктозо-1,6-дифос-
фат в результате введения в молекулу второй фосфатной группы. Распад
глюкозо-6-фосфата в этом случае начинается реакцией окисления его в 6-
фосфоглюконолактон. Окисление состоит в отнятии двух атомов водорода от
1-го углеродного атома глюкозо-6-фосфата. Акцептором Н служит НАДФ+,
являющийся коферментом глюкозо-6-фосфатдегидрогеназы, ускоряющей эту
реакцию:
Глюкозо-6- Никотянзмнд-
фосфат аденнндниуклео-
твдфосфат окис-
ленный (НАДФ+)
6—фосфо—
глюкоио-
8-лактон
Никотииаыид-
аденнндииук-
леотидфосфат
восстановленный
(НАДФН+Н*)
1люкозо-6-фосфатдегидрогеназа, открытая более полувека тому назад
О. Варбургом и сотр. и В. А. Энгельгардтом и А. П. Бархаш, выделена из
различных источников и характеризуется либо димерной, как, например,
в молочной железе крысы: М = 130000 (2x63000) или эритроцитах человека:
М = 204 000 (2x101400), либо тетрамерной, как, например, у нейроспоры:
М = 206 000 (4 x 57000), в надпочечниках быка: М = 284 000 (4 x 64 000) или
347
грене тутового шелкопряда: М=232 000 (4x54000), структурой. Она сущест-
вует в виде множествен ых форм, которыми особенно богаты эритроциты
человека, а ее активность задается соотношением НАДФ+/НАДФН. Выяснена
первичная структура глюкозо-6-фосфатдегидрогеназы из Bacillus megaterium
(М = 118 000; 4 х 29 500); в ее субъединице —262 аминокислотных остатка.
6-Фосфоглюконолактон при участии фермента глюконолактоназы гидроли-
зуется до 6-фосфоглюконовой кислоты, которая претерпевает окислительное
декарбоксилирование и превращается в рибулозо-5-фосфат:
С
6-фосфо-
н—с—-ОН I глюконо-
| I лактоназа
но—с—Н О + н2о^ :*
н—с—он]
с—он
н-с-он
но-с—н
Фосфоглю конат-
дегидрогеназа
(декарбоксилирующая)
СН2ОН
+ СО2
н-с
он
СН2—о—Р=о
он
H-с—он
н-с—он
сн2—о-
НАДФ* НАДФН+Н+Н—С—ОН
Г
н-с-он
он
он
он
2
6-Фосфоглюкоио-Л-
лактон
6-Фосфоглюкоиовая
кислота
Рнбулозо-5-
фосфат
Фосфоглюконатдегидрогеназа (декарбоксилирующая) представлена белком
с М = 100000, состоящим из двух равных субъединиц, независимо от источника
выделения (печень овцы и крысы, эритро хиты человека, дрожжи).
Даль ейший обмен рибулозо-5-фосфата—одного из центральных веществ
в углеводном обмене— ротекает весьма сложно (схема 6). Многократно
изомеризуясь, в частности, переходя в рибозо-5-фосфат и ксилулозо-5-фосфат,
а также вступая в транскетолазные и трансальдолазные реакции, заключающи-
еся в переносе двууглеродных и трехуглеродных фрагм нтой от одного фос-
форного эфира к другому, рибулозо-5-фосфат снова превращается в глюкозо-6-
фосфат. Подсчитано, что из 6 молекул рибулозо-5-фосфата получается 5 мо-
лекул глюкозо-6-фосфата. Таким образом, суммарный эффект всех реакций,
осуществляющихся при апотомическом распаде глюкозо-6-фосфата, сводится
к тому, что из каждых 6 его молекул одна полностью распадается. Это можно
выразить :ледующим уравнением:
6 Молекул глюкозо-6 фосфата4-12 НАДФ+ 4-7Н2О—»6СО2 +12 НАДФН+
4-12Н+ + Н3РО4 + 5 молекул глюкозо-6-фосфата
После сокращения 5 молекул глюкозо-6-фосфата в левой и правой частях
уравнения остается следующее:
1люкозо-6-фосфат+12 НАДФ+ +7Н2О—»6СО2 +12 НАДФН +
+ Н3РО44-12Н+
Как следует из схемы 6, важнейшее значение в апотомическом распаде
углеводов имеет превращение фосфопентоз. Поэтому этот путь обмена угле-
водов называют также пентозофосфатным циклом. Центральной реакцией
в нем является перенос двууглеродных фрагментов, осуществляемый при
каталитическом воздействии транскетолазы. Этот фермент (у нас в стране)
детально изучен Г. А. Кочетовым с сотр. Транскетолаза из пекарских дрож-
жей (М = 160 000) построена из двух субъединиц (2x75000), каждая из кото-
рых содержит в качестве кофермента тиами пирофосфат, присоединенный
348
со2
со2
со2
со.
6 молекул
глюкозо-
6-фосфата
Рибулозо-
5-фосфат
(С5)
Рибулозо-
5-фосфат
(С5)
Рибулозо-
5-фосфат
(С5)
Рнбулоэо-
5- фосфат
(С5)
Рибулозо-
5-фосфат
(С5)
Рибулозо-
5-фосфат
(С9)
Кейлулозо-
5-фосфат
(С5)
Рибозо-
5-фосфат
(С5) >
Седо-
гептулозе-/
7-фосфат
(С7) '
Ксилулозо-
5-«фосфат
(С5)
3-фосфо-
«глицер иво-
вый альде-
гид (С3)
Ксилулозо-
б-фосфат
(С5) '
3-фосфо-
глицернво-‘
вый альде-
гид (Сз)
Рнбозо-
б-фосфат
f (Cs)
Седо-
\гептулозо-
7'фосфат
/ (Ст)
Ксвлулозо-
б-фосфат
(С5)
Эритрозо-
«4-фосфат
(С4)
3-фосфо- *
глицерино-
вый альде-
гид (С3)
Фруктозо-
6-фосфат
Н20
Фруктозо-1,6-дифосфат
фруктозо-
6-фосфат
Фруктозо-
6-фосфат
Фруктозо-6-фосфат
Б молекул
гл ю к озо-
Б-фосфата
Н3Р04
Фруктозо-
6-фосфп
Эрвтрозо-
4-фосфаТ|/^
(С4) I
л 3-фосфо-
глицерино-
вый альде-
гид (Сз)
Схема 6. Превращения глюкозо-6-фосфата при апотомическом распаде (пояс-
нения в тексте) исходя из распределения 14С-промежу точных соединений
в экстрактах ацетоновых порошков печени крысы и листьев и корешков гороха
(В. Хоррекер и др., 1951). В 1978—1983 гг. усложнена Дж. Вильямсом и др.
введением в нее арабинозо-5-фосфата, диоксиацетонфосфата и октулозо-1,8-
дифосфата в качестве метаболитов, которые на схеме не показаны
к белковой части соответственно через радикал триптофана и пирофосфат-
ную группировку (рис. 115). Именно при посредстве тиаминпирофосфата
и осуществляется перенос двууглеродных фрагментов. Он идет с помощью
\й__ -группы тиазолового цикла (рис. 115). В каждой из субъединиц 20%
а-спиралей, 40% P-слоев и 40% полипептидной цепи в виде клубка. Для
становления димерной формы фермента и его функции важен Са2+. Транске-
толаза из эритроцитов человека резко отличается от дрожжевой. Будучи
очищена в 70000 раз, она имеет М = 140 000 и не нуждается ни в тиамин-
пирофосфате, ни в Mg2+ для проявления активности.
Постепенно укрепляется мнение, что транскетолазные и другие трансфераз-
ные реакции в обмене углеводов являются одними из наиболее древних. По
349
мере эволюции к ним присое-
динились дегидрогеназные про-
цессы (см. выше две первые
Рис. 115. Схема связывания тиаминпирофосфата с апо-
транскетолазой в активном центре фермента (поясне-
ния в тексте)
реакции окисления в апотоми-
ческом распаде углеводов)
и апотомический путь обмена
углеводов приобрел закончен-
ный вид. Но в дальнейшем ди-
хотомический путь распада уг-
леводов стал преобладающим,
и сейчас пен озофосфатный
цикл в целом, о обенно у выс-
ших животных, занимает
скромное место.
Путь Этнера—Дудорова.
Кроме дихотомического и апо-
томического путей обмена глюкозо-6-фосфата существует еще один путь,
характерный для микроорганизмов (у некоторых из них при его посредстве
распадается от 30 до 50% глюкозы), названный в честь его первооткрывателей
(1952). Первые стадии распада глюкозо-6-фосфата по этому пути, вплоть до
образования 6-фосфоглюконовой кислоты, полностью повторяют апотомиче-
ский путь. Но далее 6-фосфоглюконовая кислота окисляется без декарбок-
силирования в 2-кето-6-фосфоглюконовую кислоту, которая, восстанавливаясь
по 3-му углеродному атому, переходит в 2-кето-3-дезокси-6-фосфоглюконо-
вую кислоту, претер евающую альдолазное расщепление:
СООН
I
С=О
I 2-Кето-З-дезоксн-
6-фосфоглюконат-
'-•г альдолаза
Н-С-ОН
н-с-он он
СН„-О-Р=О
он
2-Кето-З-дезокси-
6-фосфоглюконовая
кислота
соон
I
с=о +
I
сн3
Пировиноград-
ная кислота
СН2—О—Р=О
I
он
З-Фосфоглииерииовый
альдегид
Оба конечных продукта распада поступают в общий метаболический фонд
и п двергаются обычным пре ращениям.
Обмен пировиноградной кислоты (ПВК). Проследим теперь судь-
бу ПВК, возникающей в качестве конечного продукта при дихотомическом
распаде глюкозо-6-фосфата и другими путями. В за исимости от места и усло-
вий протекания процесса в организме, наличия или отсутствия в последнем тех
или иных ферментных систем и т. п. судьба эта различна.
Если процесс идет в анаэробных условиях или при недостаточном снабже-
нии кислородом, то простейший вариант обмена ПВК заключается в ее
восстановлении до молочной кислоты. Донором атомов водорода при этом
служит НАДН, образующийся в процессе окисления 3-фосфоглицеринового
альдегида при дихотомическом распаде глюкозо-6-фосфата (см. схему 5) и во
многих других случаях. Эта реакция ускоряется лактатдегидрогеназой. На
этом ферменте впервые детально был разработан вопрос об изозимах
350
(см. С; 101). Изозим типа ММММ характерен для анаэробных тканей и обес-
печивает процесс превращения ПВК в молочную кислоту; изозим типа НННН
локализован в тканях с высоким аэробиозом и превращает в них молочную
кислоту в ПВК. 1ибридные формы (НМММ, ННММ и НННМ) обладают
промежуточной активностью. Так достигается очень тонкая регулировка на-
правления ферментативного процесса и соотношения в тканях молочной
и пировиноградной кислот.
Таким образом, в анаэробных условиях каждая молекула Тлю озо 6 фосфа-
та дает две молекулы молочной кислоты, ко' орая в этом случае представляет
конечный продукт реакции. Если исходным углеводом для образования глю-
козо 6-фосфата, а затем молочной кислоты служи глюкоза, то процесс назы-
вают гликолизом. Если же исходным углеводом, дающим начало глюкозо-6-
фосфату । через глюкозо-1-фосфат) и потом молочной кислоте, является глико-
ген, то процесс называют гликогенолизом. Учитывая, что и в том и в другом
случае на промежуточных стадиях дихотомического распада синтезируется
АТФ, гликолиз и гликогенолиз служит средством быстрого получения энергии
в анаэробных условиях.
При переключении в аэробные условия от 1/5 до 1/6 общего количества
молочной кислоты, возникшей при гликогенолизе, и, вероятно, вся молочная
кислота, образовавшая я при гликолизе, окисляются до СО2 и Н2О. От 4/5 до
5/6 общего количества молочной кислоты гликогенолитического происхожде-
ния идет на ресинтез гликогена путем обращения реакции гликогенолиза.
Энергия для этого черпается из реакций окисления, идущих в аэробных
условиях.
Таким образом, в анаэробных условиях ПВК, образующаяся при дихото-
мическом распаде углеводов, становится акцептором гидрид-ионов (Н-)
и протонов (Н+), снимаемых глицеральдегид-3-фосфатдегидрогеназой с 3-фос-
фоглицеринового альдегида. Регенерация окисленной формы НАД+ вслед-
ствие передачи гидрид-ионов на ПВК поддерживает течение гликолитического
процесса. Последний неизбежно остановился бы, если бы все коли ество
НАД+ оказалось насыщенным атомами водорода, ибо глицеральд гид-3-фос-
фатдегидрогеназа не смогла бы осуществлять свою функцию.
Полный набор ферментов гликогенолиза характерен для мышц и печени
животных. Однако если в первых превалирует распад гликогена, то для второй
более показателен его биосинтез.
У некоторых организмов, в частности в дрожжевых клетках,, содержится
мощная декарбоксилаза пировиноградной кислоты, способная в анаэробных
условиях превращать ПВК в у сусный альдегид и СО2:
,О
Пнруввтдекарбоксилам Л
сн3-со-соон----------------------- сн3-с5 + со»
хн .
Пировиноградная Уксусный
кислота альдегид
Начальная фаза реакции декарбоксилирования ПВК при участии дрож-
жевой пируватдекарбоксилазы (М=. 185 000, состоит из двух субъединиц,
каждая из которых несет молекулу тиаминпирофосфата в качестве коф рмента
и Mg2+ в качестве кофактора) рассмотрена ранее. Уксусный альдегид,
образующийся при распаде оксиэтилтиами пирофосфата (см. с. 161), восста-
навливается за счет НАДН при участии другого фермента—алкогольде-
гидрогеназы, от [ичаюшейся тоже очень высокой активностью в дрожжевых
клетках:
351
-О Алкоголь-
# ---------►
СН3-СЧ + НАДН + Н* ---------------- СН3—СН8—ОН + НАД*
Х„ дегидрогеназа •*
Н
Уксусный Этиловый спирт
альдегид
Механизм данной реакции, равно как и структура алкогольдегидрогеназы,
также детально рассмотрен выше (см. рис. 53).
Так как конечным продуктом обмена углеводов в этом случае оказывается
этиловый спирт, этот процесс называется спиртовым брожением. Как и при
глик лизе, акцептирование атомов Н при брожении ацетальд гидом поддер-
живает течение реакции окисления 3-фосфоглицеринового альдегида, т. е.
является условием осуществления процесса в целом.
Кроме спиртового брожения, у микроорганизмов существует еще ряд спе-
цифических путей утилизации трехуглеродных соединений, возникающих в ре-
зультате дихотомического распада углеводов. Сюда относятся молочнокислое
и пропионовокислое брожение, ацетоноэтиловое и ацетонобутиловое броже-
ние, маслянокислое брожение и др.
В аэробных условиях ПВК окисляется. Реакция ускоряется мультиэнзим-
ной системой, называемой пируватдегидрогеназным комплексом Она идет
в соответствии с уравнением
СН3-СО-СООН + НАД++HSKoA Пвд-вдтцрогытный '
комплекс
---► СО2+СНз - СО ~ SKoА + НАДН + Н+
Ацетил-жоэнзям А
Характерно, что в результате реакции окисления ПВК в образующейся
молекуле ацетил-КоА возникает макроэргическая связь. Она способствует его
энергичному обмену в дальнейшем.
Структура пируватдегидрогеназного комплекса (ПДГК) и первая фаза
ускоряемой при его посредстве реакции окислительного декарбоксилирования
ПВК рассмотрены ранее (см. рис. 46 и уравнение реакции на с. 161). Как видно
из этого уравнения, первая фаза процесса состоит в декарбоксилировании
ПВК. Эта реакция ускоряется пируватдекарбоксилазо", которая входит в со-
став мультиэнзимного комплекса в количестве 12 димерных молекул (см. Et
на рис. 46, Г); каждая из них несет две молекулы тиаминпирофосфата в качест-
ве кофермента. Естественно, что оксиэтильный радикал, возникающий после
декарбоксилирования ПВК, остается связанным с пируватдекарбоксилазой
в виде оксиэтилтиаминпирофосфата (рис. 116, стадия 1).
Далее оксиэтильный радикал окисляется в ацетильный радикал, который
переносится сначала на липоевую кислоту, а затем на коэнзим А. Оба эти
процесса (окисление и перенос ацетильного радикала) ускоряются вторым
компонентом пируватдегидрогеназного комплекса: липоа ацетилтрансфера-
зой (М = 70000). Она сосредоточена в центральной части комплекса в виде
24 молекул (см. Е2 на рис. 46, Г), упакованных, согласно современным дан-
ным, в виде куба, по 12 граням которого расп латаются 12 димерных молекул
пируватдекарбоксилазы, а по 6 плоскостям—6 димерных молекул
(М = 112 000) дигидролипоилдегидрогеназы 'см. Е3 на рис. 46,/). Общая моле-
кулярная масса ПДГК кишечной палочки 4,6 млн. Да, а у высших организмов
7—8 млн. Да.
352
о
Каждая молекула липоат-ацетилтрансферазы в качестве простетической
группы содержит молекулу липоевой кислоты, соединенную с апоферментом
через е-аминогруппу радикала лизина. Такое присоединение липоевой кислоты
обеспечивает ее подвижность в составе пируватдегидрогеназного комплекса
(длина «ножки»—1,5 нм) и беспрепятственный контакт как с пируватдекар-
боксилазой, так и с дигидролипоилдегидрогеназой при условии использования
не менее двух остатков липоевой кислоты (рис. 116, стадии 2, 3 и 4);
12—3502
353
Тиаминпиро-
фосфат
,СН2
hs/|
сн2
CH3CO~S\ |
ХСН-(СН2)4СООН
Ацетиллипоевая
кислота
HSKoA
CH3CO~SKoA
СН„
hsH
<?Н2.
HS4
хсн—(СН2)4СООН
Дигидролипоевая
кислота
Окислительное декарбоксилирование ПВК завершается следующими
(рис. 116, стадии 5 и 6) двумя реакциями. При посредстве третьего компонента
мультиэнзимного комплекса, т. е. с помощью дигидролипоилдегидрогеназы
(6 димерных молекул, содержащих по 2 молекулы ФАД в качестве кофермей-
та), дигидролипоевая кислота переходит в липоевую:
уСН
HSZ |
СН, + НАД*
HSv |
ХСН—(СНй)4-СООН
Днгадролнпоил-
дегидрогеказа
ZCH2
У|
I СН, + НАДН + н*
SxCH-(CHs)4-C00H
Поскольку коферментом дигидролипоилдегидрогеназы является ФАД, то
конечно, именно он снимает непосредственно два атома Н с дигидролипоевой
кислоты и передает их на НАД+. Поэтому в приведенном выше суммарном
уравнении окислительного декарбоксилирования ПВК в качестве акцептора
атомов Н выступает НАД+ (рис. 116). Пируватдегидрогеназный комплекс
активен в дефосфорилированном состоянии: цАМФ-независимая протеинки-
наза фосфорилирует и инактивирует его.
Ацетил-коэнзим А далее конденсируется со щавелевоуксусной кислотой
(ЩУК), которая всегда есть в клеточном содержимом. Образуется лимонная
кислота и освобождается коэнзим А. Каталитическую функцию в этой
реакции выполняет конденсирующий фермент. Предполагают, что реакция
идет в несколько стадий, которые могут быть выражены следующими
уравнениями:
СООН
I
с=о
сн2
соон
Кетоформа
щавелево-
уксусной
кислоты
соон
I
с-он
сн
соон
Енольная форма
щавелево-
уксусной
кислоты
354
НО—С-СООН + СНЭ—C~SKoA
V"H ______— Ацетил-
коэнзим А
СООН
Енольная
форма ЩУК
н
н2
—соон +
C~SKoA
СН2
соон
Цитрил-КоА
„ C~SKoA
Цитрат- синтаза |
(цитрат-конденсирую- СН2
..^ фермент) >но_1_соон .
I
^Н2
СООН
(jlOOH
сн2
н2о —*-но—с—соон
<*н2
соон
Лимонная кислота
Цитрил-КоА
HSKoA
Коэнзим А
Образованием лимонной кислоты открывается специфи ес ий цикл хими-
ческих реакций, приводящих к постепенному ее окислению до ЩУК, которая
снова конденсир ется с ацетил-коэнзимом А, так что образуется вновь лимон-
ная кислота. По существу, следовательно, идет окисление ацетильных оста-
тков до СО2 и Н2О. Этот цикл реакций получил название цикла трикарбоиовых
и дикарбоиовых кислот, так как именно эти кислоты являются главными его
компонентами (рис. 117). Его называют также циклом Кребса—по имени
первооткрывателя, удостоенного за это Нобелевской премии в 1959 г. Таким
образом, в конечном счете, ПВК окисляется до СО2 и Н2О.
Ферменты цикла трикарбоновых и дикарбоновых кислот (ЦТДК), ускоря-
ющие единый метаболический многоступенчатый процесс окисления ацетиль-
ных груйп, возможно собраны в специфически построенный комплекс (метабо-
лой), локализованный между расположенными друг против друга поверх-
ностями внутренней мембраны митохондрий (рис. 118). В метаболоне, как
полагают, осуществляется эстафетная передача промежуточных продуктов
цикла от одного фермента к другому без их высвобождения в матрикс мито-
хондрии. Поэтому процесс идет с большой скоростью. Рядом с метаболоном
ЦТДК располагаются пируватдегидр геназный комплекс и, вероятно, метабо-
лой Р-окисления высших жирных кислот, поставляющие ему CH3CO~SKoA.
Общая схема распада углеводов. Все сказанное выше о путях
распада углеводов и о механизме реакций, осуществляющихся в процессе их
деструкции, можно представить в виде следующей общей схемы (см. с. 357).
Из схемы видно, что глюкозо-6-фосфат занимает в этих процессах цент-
ральное место, а из промежуточных продуктов дальнейшего его распада
узловые позиции принадлежат ПВК и рибулозо-5-фосфату.
Какая же роль в общем обмене углеводов организма отводится рассмот-
ренным здесь путям распада углеводов: брожению гликолизу и дыханию,
апотомическому и дихотомическому, анаэробному и аэробному?
Зависимости между ними сложны и определяются как видовыми осо-
бенностями, так и условиями жизнедеятельности организмов. Например,
объем гликолиза в тканях находится в прямой зависимости от поступления
355
соон
СООН
Щиелевоуксусиая кислота
(енольная форма)
НАД’Н'Ж*
Малатдегидрогенам
(М-72000; 2X37000)
t’vooH (Sk
снон
соон «ь/Л- *’
Яблочная
кислота
н/
НАД
20%
ФП
‘COOH^S?^
CH*>
сн
соон
<!:н,
ён’
соон
tf-кетоглутарат- . а-Кетоглутаровая
•ЙмЖс(нй!г%йоооо) / (2'““™^овм)
HS-KoA-—
НАД±
ГДФ+Н,РО4 (f~SK0A
-X сн,
со,
>НАДН+Н +
СН
II
СН
соон
£оон
СукцИнил-КоА
Фумаровая
кислота
Янтарная
HS-KoA
ГТФ(ГТФ+А ДФ г=ГДФ+АТФ)
кислота
Рис. 117. Цикл трикарбоновых и дикарбоновых кислот (пояснения в тексте)
Рис. 118. Гипотетическая структура комплекса
ферментов (метаболона) цикла трикарбоновых
и дикарбоновых кислот (молекулярная масса
метаболона ЦТДК ~ 8 млн Да; высота—20 нм,
диаметр—50 нм):
а—вид прямо; б—вид сбоку; в—вид сверху; 1—цитратсин-
таза, 2—аконитаза; 3—изоцитратдегидрогеназа; 4а, 46, 4в—
а-кетоглутаратдегидрогеназный комплекс (а-кетоглутаратде-
гидрогеназа, транссукцинил аза и липоамиддегидрогеназа соот-
ветственно); 5—сукцинаттиокиназа; 7—фумараза; 8—малат-
дегидрогеназа; 9—аспартатаминотрансфераза; 10—нуклео-
зиддифосфаткиназа; П—заякорнвающие метаболой белки,
включающие сукцинатдегидрогеназу
356
Полисахариды
г_ Олигосахариды
Гидролиз j г
Моносахариды
ФосФооилиА.
рооание Г” 1 — " -ч
Гексозофосфаты Пентозоф ".фс
шпы
-f-
Т взаимопревра-
j щение
Глюкозо-6-фосфат ,
Глюкозо-
фосфат
^SS^^pacmS
Рибулозо-5- фосфат
Пировиноера. дная
кислота
брожение Гликолиз Дыхание
\_______v ->*______________
Анаэробно
Дыхание
->-------------*
Аэробно
Схема 7. Пути распада углеводов (пояснение в тексте)
кислорода: последний подавляет процесс образования молочной кислоты
пастеровский эффект). Даже в различных тканях и органах одного и того же
организма соотношения путей распада углеводов могут быть различными.
Тем не менее можно установить и некоторые общие закономерности. Так,
у годавляющего большинства организмов аэробный путь распада углеводов
в общем превалирует над анаэробным, а дыхание подавляет гликолиз и броже-
ние. Дихотомическому распаду углеводов принадлежит в целом более видное
место, чем апотомическому. В значительной мере эти соотношения путей
деструкции углеводов зависят от их энергетического эффекта. Последнее впол-
не естественно, так как одной из функций углеводов, в ряде случаев главной,
является обеспечение организма энергией, выделяющейся при их анаэробном
и аэробном распаде.
Синтез углеводов. Синтез простых углеводов. Простые углеводы
возникают главным образом при первичном биосинтезе органического веще-
ства на Земле. Этот процесс осуществляется автотрофными организмами—
растениями, а также фотосинтезирующими и хемосинтезирующими бактери-
ями. Первичный синтез органического вещества в природе идет путем восста-
новления СО2 атмосферы с одновременным формированием ор анических
молекул, содержащих цепи углеродных атомов. В связях между атомами
углерода и других элементов образующихся органических соединений заклю-
чена энергия, поэтому их новообразование сопровождается ее поглощением.
В общем виде процесс первичного новообразования ор анического вещества
принято изображать в виде следующей схемы (см. с. 358).
Тетеротрофные организмы, использующие для построения составных частей
своего тела уже готовые органические вещества, не обладают способностью
к первичному биосинтезу органических молекул, но могут образовывать их за
счет перестройки органических соединений пищи. Естественно, что в числе
новообразуемых гетеротрофами соединений находятся и простые углеводы;
однако как исходные материалы для их построения, так и первичные источ-
ники энергии здесь принципиально отличаются от таковых у автотрофов.
357
Восстановитель Энергия (*'-') Оксид углерода(ТУ)
(HhC ^ЧСО2)
СН2О
Элементы минераль-
ного питания (N,S, Р-
и т. п.)
Мономеры
(моносахариды, аминокислоты, нуклеотиды,
органические кислоты, фосфорные эфиры
органических соединений и т.п.)
Полимеры
(полисахариды, белки, липиды, нуклеино-
вые кислоты и др.)
Схема 8. Возможные этапы первичного биосин-
теза органического вещества
Рассмотрим механизм первичного биосинтеза простых углеводов у автотроф-
ных 1 организмов. В простейшем случае у хемосинтезирующих бактерий источни-
ком энергии, которая трансформир ется в стабильную энергию химических связей
между атомами углерода, служат реакции окисления неорганических соединений,
проходящие с выделением того или иного количества энергии (табл. 24).
Таблица 24
Энергетический эффект окислительных реакций у хемосинтезирующих бактерий
Уравнение реакции Количество энергии, выделившейся на 1 г/моль окисленного вещества, кДж Вахтерш
Na2S2O3+2*/2О2+Н2 - Na2SO4+H2SO4 882 Серобактерии
S+l I/2O2+H2O-H2SO4 493 »
NH3 +1 */2O2 -* hno2+H2O 276 Нитрифицирующие
бактерии
H2 + 1/2^2 —* H2O 234 Водородные бактерии
HzS+VzOz-HjO+S 171 Серобактерии
2FeCO3 + 3H2O+ llIOi -»2Fe(OH)3+2CO2 167 Железобактерии
HNOj + VzOz-HNOs 71 Нитр филирующие бак-
терии
Фотосинтезирующие бактерии и зеленые растения используют для первич-
ного синтеза органических веществ энергию световых лучен, которая, напри-
мер, для 6 х 10й квантов красного света (в основном поглощаемого зелеными
органами равна примерно 167 кДж.
Хемосинтез, т. е. ассимиляция СО2 микроорганизмами за счет энергии,
выделяемой при окислении неорганических соединений, впервые открыт в кон-
це прошлого столетия С. Н. Виноградским.
1 Автотрофный—сам себя питающий, т. е. развивающийся на среде, свободной от других
организмов и продуктов их жизнедеятельности.
358
Первичный акт, посредс вом которого энергия, освободившаяся при окис-
лении неорганических соединений хемосинтезирующими бактериями или вос-
принятая фотосинтезирующими организмами, превращается в доступную для
использования в химическом синтезе форму, состоит в трансформации этой
энергии в энергию макроэргической связи АТФ. Иначе говоря, энергетическое
обеспечение синтеза простых углеводов начинается с синтеза АТФ из АДФ
и неорганического фосфата. Можно предполагать, что процесс хемосинтетиче-
ского и фотосинтетического фосфорилирования идет, в общем, аналогично
окислительному фосфорилированию (см. гл. X), т. е. перенос электронов при
хемосинтезе и фотосинтезе вовлекает ряд энзиматических систем мембранного
аппарата бактериальных и растительных клеток, результатом чего является
возникновение мембранного пот нциала—истинного двигателя реакции фос-
форилирования аденозиндифосфорной кислоты: АДФ+НзРО4-»АТФ+Н2О.
Открытие в составе сопрягающих мембран (см. гл. X) разнообразных
организмов присутствия сопрягающих факторов схожей структуры, обеспечи-
вающих протекание этой реакции, является доказательством единства путей
акцептирования энергии в живых системах.
Одновременно с синтезом АТФ идет и вторая важнейшая для первичного
биосинтеза органических веществ реакция—высвобождение атомов водорода,
необходимых для восстановления СО2. Многие детали этого процесса неясны,
однако установлено, что донором атомов Н в реакциях хемосинтетического
и фотосинтетического восстановления СО2 является в подавляющем большин-
стве случаев вода, а промежуточным акцептором их—НАДФ+, т. е.
2НАДФ + + 2Н2О -»2НАДФН+2Н + + О2.
Восстановление СО2 непосредственно не идет. Оно осуществляется после
связывания СО2 в результате реакции карбоксилирования уже достаточно
сложного органического соединения—рибулозо-1,5-дифосфата, который об-
разуется путем фосфорилирования рибулозо-5-фосфата—продукта апотоми-
ческого распада глюкозы, всегда присутствующего в клеточном содержимом
или возникающего из рибозо-5-фосфата при посредстве рибозофосфатизоме-
разы (М = 54000). Именно на этом этапе расходуется АТФ, необходимая для
первичного биосинтеза углеводов:
он
сн2он сн2—о—р=о
। I I
с=о с=о он
I I
н—с—он н—с—он
Фосфорибуло-
Н—С—ОН ОН+АТФ — Н—С—ОН ОН+АДФ
I I киназа । ।
СН2—О—Р=О СН2—О—Р=О
I I
он он
Рибулозо-5-фосфат
Рибулозо-1,5-дифосфат
I
Фосфорибулокиназа, ускоряющая эту первую реакцию на пути ак ептиро-
вания СОг, открыта А. Вейсбахом с сотрудниками еще в 1954 г., но свойства ее
изучены лишь в оследнее десятилетие; фермент широко представле у фото-
и хемоавтотрофов, высоко специфичен, абсолютно зависим от присутствия
359
двухвалентных катионов, особенно Mg2+, обладает высокой молекулярной
массой (240 000; тетрамер) и образует прочный комплекс с другими фермента-
ми, участвующими в карбоксилировании рибулозо-1,5-дифосфата.
Механизм реакции карбоксилирования рибулозо-1,5-дифосфата достаточ-
но ясен. Сначала это соединение, преобразуется в енольную форму:
он
сн,—о—р=о
I I
с—он он
он
н—с—он ун
сн2—о—Р=О
он
Кетоформа рибулозо-
1.5-дифосфата
Енольная форма
рибулозо-1.5-дифосфата
Она присоединяет СОг, и возникший промежуточный продукт расщепляет-
ся на две молекулы фосфоглицериновой кислоты:
он
CHj-O—р=о
Z^^c-OH он~
. / II
с + C-OJH
II I »
О^Н-с-онюн
^НтО-Р=О
in
Промежуточное
соединение
З-Фосфоглицери-
новая кислота
(две молекулы)
Оксид угле- Енольная форма
рода(1У) рибулозо-1,5-
дифосфата
Приведенные выше уравнения представляют, конечно, лишь грубую схему
тех тонких процессов, которые происходят во время карбоксилирования рибу-
лозо-1,5-дифосфата в активном центре специфического фермента, ускоряюще-
го многостадийные преобразования при акцептировании СОг- Этот самый
распространенный на Земле фермент называют рибулозодифосфаткарбоксила-
зой (РДФК). Его систематическое название—З-фосфо-О-глицерат карбокси-
лиаза димеризующая. К настоящему времени он выделен из нескольких
десятков объектов (высшие растения, зеленые водоросли, сине-зеленые водо-
росли, фототрофные и хемоавтотрофные бактерии). При молекулярной массе
в 500 000—600 000 он обладает четвертичной структурой (8 больших субъеди-
ниц с М = 50000—60000 и 8 малых с М = 12000—15000), которая изучена
методами рентгеноструктурного анализа и электронной микроскопии
(рис. 119). Большие субъединицы являются каталитическими и несут центры
связывания рибулозо-1,5-дифосфата и СОг, а малы —регуляторными и име-
ют ряд аллостерических центров для соединения с эффекторами (Mg2+, фрук-
тозо-6-фосфат, HS—содержащие соединения и др.). Субъединицы РДФК син-
360
Рис. 119. Структура рибулозодифосфаткарбоксилазы:
четвертичная—высших растений (а) и водородных бактерий (б—вид сбоку, в—вид сверху), изученная
электронно-микроскопически; третичная—малой субъединицы высших растений, изученная методом рент-
геноструктурного анализа (г). Остальные пояснения—в тексте
хронно синтезируются: большие—на рибосомах 70S в хлоропластах, ма-
лые—на рибосомах 80S в цитоплазме, спонтанно собираясь в нативный
мультимер.
В значительной мере выяснены структура и работа активного центра
РДФК; предполагают, что рибулозо-1,5-дифосфат связывается в нем за счет
взаимодействия фосфатных групп с радикалами лизина, енолизация суб-
страта идет при участии HS-групп (их насчитывается 96), акцептирование
СОг по енодиольной группе сопровождается переносом протонов при помо-
щи гистидиновых остатков. РДФК активируется светом при помощи свето-
чувствительного белкового фактора (М=4000—8000) и ингибируется моле-
кулярным кислородом, инициирующим оксигеназную (см. гл. X) функцию
(т. е. окисление рибулозо- 1,5-дифосфата в 3-фосфоглицериновую кислоту
и 2-фосфогликолевую кислоту) этого уникального фермента, обеспечива-
ющего фиксацию СОг и первичный биосинтез углеводов в космических
масштабах.
Как отмечено выше, центральную роль в осуществлении фотосинтеза
играет трансформация энергии света в разность потенциалов мембраны
фотосинтетического центра и сопряженный с этим синтез АТФ. Недавно,
используя методы спектроскопии, рентгеноструктурного анализа и молеку-
лярной генетики, удалось получить детальную картину событий, проис-
ходящих при фотосинтезе и выявить пространственное расположение
и роль белков и пигментов, участвующих в этом процессе. За эту работу
немецкие ученые Р. Хубер, И. Дайзенхофер и X. Михель удостоены Нобе-
левской премии 1988 г.
Возникшая в результате рассмотренных выше процессов фосфоглицерино-
вая кислота восстанавливается в 3-фосфоглицериновый альдегид:
он он
СН,—0—^=0 СН,—О—Р=О
I Лн I о'н
2СНОН + 2НАДФН 4- 2Н* s» 2СНОН + 2НАДФ* 4- 2Н,0
if
Х)Н хн
З-Фосфоглицера- З-Фосфоглицернно*
новая кислота вый альдегид
Данная реакция тоже идет более сложным путем, нежели показано в при-
веденном выше уравнении. Она представляет собой обращение процесса пе-
рехода 3-фосфоглицеринового альдегида в 3-фосфоглицериновую кислоту,
361
осуществляющееся при дихотомическом распаде глюкозы. Напомним, что
указанный процесс ускоряется глицеральдегид-3-фосфатдегидрогеназой и идет
через 1,3-дифосфоглицериновую кислоту с тем лишь отличием, что содержа-
щаяся в хлоропластах форма этого фермента специфична в большей степени
к НАДФН, чем к НАДН, отличается очень высокой молекулярной массой
(600 000) и специально приспособлена для восстановления фосфоглицериновой
кислоты в фосфоглицериновый альдегид. Это значит, что при восстановлении
3-фосфоглицериновой кислоты в 3-фосфоглицериновый альдегид будет рас-
ходоваться АТФ (см. уравнение реакции на схеме 5, с. 344).
Следовательно, на этой стадии биосинтеза углеводов снова исполь-
зуется АТФ, образовавшаяся при фотосинтетическом или хемосинтетиче-
ском фосфорилировании и энергетически обеспечивающая новообразова-
ние углеводов. Здесь же, как следует из приведенного выше уравнения
реакции восстановления 3-фосфоглицериновой кислоты, осуществляется
в сущности восстановление акцептированного СОг при посредстве
НАДФН, возникшего одновременно с АТФ на первой фазе фотосинтетиче-
ского процесса.
Дальнейший путь синтеза углеводов из 3-фосфоглицеринового альдегида,
так же как и только что рассмотренная реакция восстановления 3-фосфо-
глицериновой кислоты, представляет обращение дихотомического пути распада
углеводов: фосфоглицериновый альдегид переходит в фосфодиоксиацетон; при
катали ическом воздействии альдолазы из упомянутых фосфотриоз синтези-
руется фруктозо-1,6-дифосфат, переходящий далее в глюкозо-6-фосфат. На
этом этапе биосинтеза углеводов действует особый фермент—фруктозо-1,6-
дифосфатаза (молекулярная масса фермента из растений и фото- и хемосин-
тезирующих организмов—130000—190000; две субъединицы; отличается аб-
солютной специфичностью), обеспечивающая переход от фруктозо-1,6-дифос-
фата к фрук озо-б фосфату, так как реакция:
фруктозо-6-фосфат+АТФ=фруктозо-1,6-дифосфат 4- АДФ
практически необратима. Фруктозо-6-фосфат, как известно, легко превращает-
ся в фосфорные эфиры других моносахаридов, что в итоге обеспечивает синтез
всего набора природных моноз, а из них—дисахаридов и полисахаридов. Ход
реакции первичного новообразования углеводов в процессе фотосинтеза дан
на схеме 9.
Что касается синтеза простых углеводов организмами-гетеротрофами, то
исходными веществами для этого могут служить продукты распада липидов,
белков и других органических соединений. Центральным звеном в переходе
этих соединений в простые (а далее и сложные) углеводы является образование
ПВК, возникающей или непосредственно (например, при распаде аминокис-
лот), или при посредстве глиоксилового цикла—при декарбоксилировании
щавелевоуксусной кислоты (см. с. 395). Переход от ПВК к углеводам осущест-
вляется путем обращения процесса дихотомического распада углеводов, сле-
довательно, из каждых двух молекул ПВК образуется одна молекула фрукто-
зо-1,6-дифосфата. Однако здесь есть одна особенность: переход от ПВК к фос-
фоенолпировиноградной кислоте идет обходным путем через
щавелевоуксусную кислоту вследствие необратимости реакций превращения
фосфоенолпирувата в ПВК. Так эта реакция протекает в печени, почках
и других тканях животных, в листьях и корнях растений и у микроорганизмов.
Только в мышцах действие пируваткиназы обратимо, и фосфоенолпировиног-
радная кислота возникает из ПВК и АТФ.
Кроме рибулозо-1,5-дифосфатного пути, существует еще два механизма
акцептирования СОг, занимающих подчиненное положение по отношению
к первому.
362
ЛДФ + Н3РО4
2НАДФ++2Н2О
Свет
Хлорофилл
С АТФ
2НАДФН+2Н++О2
IH
<JH2OH
со
н-с—он
^п2—и
АТФ АДФ СО
- *• Н-с—он
Фосфорибуло- |
ОН (j)H киназа Н—С—ОН
;н2—о—р=о
Ан СО2
он
Рибулозо-5-
фосфат
РДФК
ОН
I
CHj— О—Р=О
ОН
Рибулозо-1,5-
дифосфат
• 2 НСОН ОН
СООН
3 -Фосфоглицериво-
аая кислота
?н
сн—о—Р=о
2 HCOH Ан +
АоОН
3-фосфоглицери -
новая кислота
?Н
(JH— О—Р=О
2 НАДФН+2Н^*г2 H<j:OH ОН + 2 НАДФ+2 Н2О
Ппщеральде- С^^
гид-3-фосфат- з-фосфоглицерииовый
дегидрогеназа альдегид
?н
сн2—О—Р=О
неон
Триозо-
фосфат-
изомераза
ОН
хн
3-Фосфо глицериновый
альдегид
СН,—О—Р=0
1о Ан
Ан2он
Фосфодиокси-
ацетон
Ф рукто зо -1.6 - ди фосфат
i
Углеводы
Схема 9. Механизм биосинтеза углеводов в растениях
Один из них состоит в карбоксилировании фосфоенолпировиноградной
кислоты:
сн2 он
II I
С—0~ Р=О+СО2 + Н2О
I I
соон он
Фосфоенолпйруват-
жарбоксилаза
соон
I
сн2
I
СО+НзРО*
I
соон
Фосфоенол-
пяруват
Щавслевоуксусная
кислота
Фермент, ускоряющий эту реакцию, выделен из растений (обладающих
Сд-типом фотосинтеза), хемосинтезирующих и гетеротрофных бактерий (М®
400000; четыре субъединицы).
363
Другой механизм акцептирования СОг сводится к карбоксилированию аци-
льных производных коэнзима А (ацетил-КоА, пропионил КоА, сукцинил-КоА).
Например:
—ч Пируватсинтаза
СНз — СО ~ КоА 4- СОг+Ферредоксин (восст.) -
----- СН3—СО—СООН+HSKoА 4- Ферредоксин (окисл.)
Донорами атомов Н в указанных реакциях служат ферредоксины—желе-
зосодержащие белки негеминовой природы с Мж6000 у бактерий и около
17000 в хлоропластах. Они содержат несколько атомов Fe, собранных в кла-
стер и присоединенных, с одной стороны, к радикалам цистеина белковой
части, а с другой—связанных с атомами лабильной серы.
Продукты, возникающие по фосфоенолпируват- и ацилкоэнзим-А-кар-
боксилазному механизму (пировиноградная, а-кетоглутаровая и щавелевоук-
сусная кислоты), используются для биосинтеза структурных элементов бел-
ков, углеводов, нуклеиновых кислот и липидов или обмениваются далее
в цикле трикарбоновых и дикарбоновых кислот, усиливая, таким образом,
метаболические возможности организма. Это послужило основанием для
создания препарата-карбостимулина (NaHCO3—25 г, MgSO4-7H2O—3 г,
MnSO4-7H2O—0,05 г, ZnSO4-7H2O—0,05 г, цитрат натрия—7 г), находя-
щего благодаря работам школы акад.’ М. Ф. 1улого все более широкое
при ененйе для повышения продуктивности животных, в медицинской прак-
тике и т. п.
Синтез олигосахаридов. Долгое время предпринимались безуспеш-
ные попытки доказать, что синтез олигосахаридов, в частности дисахари-
дов, представляет собой реакцию обращения их гидролиза. Однако, несмот-
ря на многочисленные эксперименты такого рода, никто не сумел подоб-
рать условия, при которых удалось бы направить вспять реакцию гид-
ролиза, например сахарозы, протекающую при участии р-фруктофуранози-
дазы (сахараза).
Оказалось, что биосинтез олигосахаридов осуществляется путем реакций
трансгликозилирования. Перенос гликозильного остатка на один моносахарид
идет с фосфорного эфира другого моносахарида и ускоряется специфической
гликозилтрансферазой.
Исходными соединениями, с которых в процессе синтеза олигосахаридов
гликозильный остаток энергично переносится на моносахарид, служат нукле-
озиддифосфатсахара (НДФ-сахара).
Они были открыты Л. Лелуаром с сотр. (1950) и очень быстро привле-
кли к себе внимание как наиболее вероятные метаболиты в биосинтезе
углеводов. В настоящее время известно свыше 50 представителей НДФ-
сахаров.
Ясна и причина их особого значения в качестве доноров гликозильных
остатков: при гидролизе НДФ-сахаров изменение уровня свободной энергии
значительно выше, чем при гидролизе других доноров, а нуклеотидная часть
их молекул способна обеспечить избирательность гликозилтрансферазной ре-
акции.
В случае синтеза сахарозы, например, специфический фермент—сахарозо-
синтаза, ускоряет реакцию переноса остатка глюкозы с уридиндифосфат-
глюкозы на фруктозу (см. с. 365).
Долгое время считали, что именно так синтезируется сахароза в растениях
(животные не синтезируют сахарозу, а лишь используют ее). Однако оказа-
364
ОН н
f-D-Фруктоза
ОН
УДФ (уридиндифосфат) Сахароза
лось, что эта реакция ввиду ее обратимости служит для поддержания равнове-
сия между сахарозой и УДФ-глюкозой и даже используется для наработки
УДФ-глюкозы во время ее энергичного использования для биосинтеза крах-
мала и гликогена (см. следующий раздел этой главы). Этот процесс, в част-
ности, идет в больших масштабах в клубнях растений, запасающих крахмал.
Сахароза же в действительности синтезируется при помощи гликозилтранс-
феразной реакции из УДФ-глюкозы и фруктозо-6-фосфата:
УЛ Р- глюкоза+Фруктозо-6-Р _Сахарозо!роарат синтаза^
(УДФ-глюкоза-. Т)-фруктозо-
6-фоарат -2-а-глюкозил-
трансфграза)
------а^ДФ +Сахарозо-6-дюарат
^^^^Фоофшпаза
Сахароза h}po4
Как видно из уравнения реакции, сахарозофосфат-синтаза (молекулярная
масса фермента из проростков пшеницы—380000) д йствует в паре с фосфа-
тазой, которая обеспечивает молниеносное отщепление остатка фосфорной
кислоты от сахарозо-6 фосфата, чем полн стью сдвигает реакцию вправо.
Этот процесс идет в листьях растений, и образовавшаяся сахароза оттекает из
них в клубни и корни. Аналогично синтезируется главный дисахарид насеко-
мых, высших и низших грибов и микобактерий—трегалоза.
Будучи широко распространены в природе, НДФ-сахара синтезируются из
фосфорных эфиров моносахаридов и соответствующих нуклеозидтрифосфа-
тов путем нуклеотидилтрансферазных реакций, например:
365
Известно более двух десятков нуклеотидилтрансфераз, ускоряющих реак-
цию переноса тех или иных нуклеотидных остатков на соответствующие
фосфорные эфиры моносахаридов с освобождением пирофосфата. Наряду
с фосфорными эфирами моносахаридов и нуклеозиддифосфатсахарами роль
доноров гликозильных остатков в реакциях биосинтеза олигосахаридов могут
выполнять сами олигосахариды, а также декстрины.
Из сказанного выше ясно, что биосинтез олигосахаридов идет путем
переноса гликозильных остатков на моносахариды с разнообразных субстра-
тов при участии в каждом конкретном случае соответствующих гликозилтранс-
фераз. При этом новообразование химических связей сопряжено с распадом
их в тех соединениях, с которых идет перенос гликозильных остатков.
Синтез полисахаридов. Подобно синтезу оли осахаридов новообра-
зование полисахаридов также идет путем трансгликозилирования. Полная ана-
логия существует и в характере субстратов, с которых переносятся гликозиль-
ные остатки на конец растущей цепи олисахарида. Ими могут быть фосфор-
ные эфиры моноз, НДФ-сахара и ол госахариды. Реакции переноса остатков
моносахаридов в процессе биосинтеза полисахаридов ускоряются о ответству-
ющими гликозилтрансферазами. Приведем несколько примеров.
Синтез ами. озы, целлюлозы и подобных им 1,4-глюканов может проис-
ходить путем переноса гликозильных остатков с глюкозо-1-фосфата или ана-
логичных фосфорных эфиров моносахаридов. Эта реакция представляет собой
обращение реакции фосфоролиза указанных соединений (см. с. 333). Однако
более существенное значение имеет другой путь: биосинтез из НДФ-сахаров
при участии соответствующих трансгликозидаз. Это уравнение реакции нара-
щивания молекулы а-1,4-глюкана на один остаток глюкозы представлено на
с. 367.
Как следует из приведенного уравнения реакции, перенос гликозильного
остатка идет на невосстанавливающий конец молекулы синтезируемого поли-
сахарида. Эта реакция может повторяться многократно, что обеспечивает
ступенчатый синтез молекул полисахаридов, содержащих огромное число
остатков моносахаридов. Характерная особенность реакций такого типа со-
стоит в необходимости затравки, т. е. наличия в реакционной среде небольшого
количества молекул полисахарида. Он как бы предопределяет тот тип связи,
который возникает в процессе трансгликозилирования, и, следовательно, син-
тезируется полисахарид, одноименный с «затравочным». Роль затравки при
биосинтезе некоторых разветвленных полисахаридов, таких, например, как
частичковый гликоген, могут играть полипептцдные цепи (см. рис. 104), содер-
жащие олигосахаридные звенья.
Установлено, что при биосинтезе различных полисахаридов субстратами (а
точнее, коферментами соответствующих трансгликозидаз) служат разные
НДФ-сахара. Так, целлюлоза образуется с помощью гуанозиндифосфатглюко-
зы, полисахарид дрожжей (маннан)—с помощью гуанозиндифосфатманнозы
366
а-1,4-Глюкан, содержащий в молекуле на один
остаток глюкозы больше
и т. п. В связи с этим по-новому встает вопрос о специфи ности реакции
биосинтеза полисахаридов. Нуклеотидная часть НДФ-сахаров—это, по мне-
нию Н. К. Кочеткова, «рукоятка», при помощи которой фермент располагает
определенный сахар в нужном для осуществления реакции положении; она
нужна также для «узнавания» гликозилтрансферазой соответствующего НДФ-
сахара.
Конкретным представителем гликозилтрансфераз этого типа может слу-
жить гликогенсинтетаза. Она открыта Л. Лелуаром и сотр. (1957) в печени
крысы, а сейчас обнаружена у многих животных и микроорганизмов. ТЪмоген-
ный (без примеси прочно связанного с ним гликогена) фермент имеет
М = 330—340 тыс. Да (4x85000), содержит около 23% a-спиралей и легко
агрегирует. Соединяясь с гликогеном, образует комплексы с М = 5 — 6 млн.,
содержащие до 10 молекул фермента, локализованных по месту нередуциру-
ющих концов наращиваемых олигосахаридных цепей.
Активность гликогенсинтетазы регулируется за счет реакций фосфорилиро-
вания (снижение) и дефосфорилирования (возрастание). Предп лагают, что ее
367
фосфорилирование осуществляется той же цАМФ-зависимой протеинкиназой,
что и фосфорилирование гликогенфосфорилазы. Это подчеркивает весьма
тонкие взаимоотношения систем, регулирующих синтез и распад гликогена.
Донором гликозильных остатков при синтезе полисахаридов могут слу-
жить и олигосахариды. Изучены реакции переноса остатков глюкозы на
растущий конец цепи синтезируемого полисахарида с мальтозы, сахарозы
и других олигосахаридов. Реакция аналогична описанной выше. Однако раз-
нообразие типов связей, которые могут возникать при переносе гликозильных
остатков с олигосахаридов на новообразуемый полисахарид, гораздо больше.
Здесь перенос может идти не только на 4-й, но и на 6-й углеродный атом
остатка моносахарида, что обеспечивает синтез 1,6-глюканов, а возможно,
и полисахаридов разветвленного строения.
В последнее время внимание привлечено еще к одному источнику глико-
зильных и олигосахаридных остатков при биосинтезе полисахаридов и особен-
но полисахаридной части гликопротеинов и гликолипидов. Это—полиизо-
пренилмонофосфат и дифосфатсахара, называемые также долихолфосфат и до-
лихолдифосфатсахарами (от названия пЬлиизопреноидного спирта—долихола,
с числом изопреноидных остатков от 6 до 24). Особенно хорошо эти соеди-
нения представлены у микроорганизмов, хотя они, несомненно, принимают
участие в новообразовании гликопротеинов у животных и растений:
СН2ОН
он
„К” ^И-О—Р—О—(СН,—СН==С—СН7)„Н
А он о ^н3
Долихолмонофосфатглюкоза (л—18— 23)
Встраиваясь полиизопреноидной частью в липофильную мембрану, они
о еспечивают беспрепятственную переброску моносахаридных звеньев для
достройки олигосахаридных фрагментов пептидогликанов и гликолипидов,
происходящую при помощи соответствующих гликозилтрансфераз.
Особое значение реакции синтеза полисахаридов при участии долихолфос-
фатсахаров имеют для сборки пептидоглюканов клеточных стенок бактерий.
Именно при их участии идет перенос углеводных фрагментов на пептидные
группировки новообразуемого пептидоглюкана. Новым аспектом является
участие долихолдифосфатолигосахаридов в N-гликозилировании белков при их
посттрансляционной модификации..
Для реакций синтеза полисахаридов характерно также, что осуществляется
перенос не только остатков моносахаридов, но и полигликозидных фрагментов
с одного полисахарида на другой или же в пределах одной и той же молекулы.
Реакции этого типа изучены у нас А. Н. Петровой и ею же впервые получен из
скелетных мышц кролика гомогенный препарат соответствующего фермента
(1970).
Примером может служить перенос части полиглюкозидной цепи у 1,4-
глюкана из положения 4 в положение 6, катализируемый а-1,4-глюкан-вет-
вящим ферментом, систематическое название которого—а-1,4-глюкан: а-1,4-
глюкан 6-а-(а-1,4-глюкано)-трансфераза. По данным А. Н. Петровой, этот
фермент для осуществления реакции ветвления полисахарида нуждается в при-
сутствии РНК с М« 10000. Нуклеотидный состав и первичная структура этой
РНК (31 н. о., из них 1/3 минорных) изучены, хотя механизм активирования
ею фермента до конца не ясен. Мнение о том, что она является рибозимом,
оказалось ошибочным.
368
Приводимая ниже упрощенная схема иллюстрирует действие а-1,4-глюкан-
ветвящего фермента:
Так возникают в организме разветвленные молекулы полисахаридов (крах-
мала, гликогена и т. п.).
Использование промежуточных продуктов распада углеводов для синтеза
других органических соединении. Выше было о мечено, что одна из функций
углеводов в обмене веществ состоит в образовании продуктов распада, кото-
рые служат исходными веществами для синтеза многих других молекул. Из
числа продуктов распада углеводов в этом смысле важны фосфоглицериновая
кислота, фосфоенолпировиноградная кислота, пир< виноградная кислота, аце-
тил-коэнзим А, эритрозо 4-фосфат, рибулозо-5-фосфат, а также партнеры
цикла трикарбоновых и дикарбоновых кислот: щавелевоуксусная и а-кето-
глутаровая кислоты. Они служат исходными соединениям для синтеза амино-
кислот, высших жирных кислот, глицерина, нуклеотидов и ряда других моно-
меров, используемых для построения белков, липидов, нуклеиновых кислот
и других биополимеров. Конкретные примеры превращений перечисленных
выше соединений можно найти в предыдущих главах (см. разделы о синтезе
аминокислот, пуриновых и пиримидиновых оснований и др.).
ГЛАВА IX
ЛИПИДЫ И ИХ ОБМЕН
ОБЩАЯ ХАРАКТЕРИСТИКА И КЛАССИФИКАЦИЯ ЛИПИДОВ
К липидам относят природные органические соединения, не растворимые
в воде, но растворимые в жирорастворителях (бензин, петролейный эфир,
серный эфир, ацетон, хлороформ, сероуглерод, метиловый и этиловый спирты
и т. п.), являющиеся производными высших жирных кислот и способные
утилизироваться живыми организмами.
Название одной из групп липидов, а именно—жиров (от греч. липос—
жир) взято для обозначения класса в целом. Липиды—сборная группа ор-
ганических со динений и поэтому не имеют единой химической характеристи-
ки. Однако в известной мере их можно рассматривать как класс органических
соединений, болыпинств из которых принадлежит к сложным эфирам много-
атомных или специфически построенных спиртов с высшими жирными кисло-
тами. В зависимости от состава, строения и роли в организме сложилась
следующая классификация липидов.
1. Простые липиды представлены двухкомпонёнтными веществами—слож-
ными эфирами высших жирных кислот с глицерином, высшими или полицикли-
ческими спиртами. Сюда относятся: жиры (триглицериды)—сложные эфиры
высших жирных кислот и трехатомного спирта—глицерина; воски—слож-
ные эфиры высших жирных кислот и высших спиртов; стерцды—сложные
эфиры высших жирных кислот и полициклических спиртов—стеролов.
2. Сложные липцды имеют многокомпонентные молекулы, компоненты
которых соединены химическими связями различного типа. К ним принад-
лежат: фосфолипиды, состоящие из остатков высших жирных кислот, глицери-
на или других многоатомных спиртов, фосфорной кислоты и азотистых
оснований той или иной природы; гликолипиды, включающие в свой состав
наряду с многоатомным спиртом и высшей жирной кислотой такж углеводы.
В последнее время открыты две новые группы липидов, в составе которых
представлены и простые и сложные липиды. К ним относятся* диольные
липцды—простые и сложные эфиры двухатомных спиртов с высшими спирта-
ми и высшими жирными кислотами, содержащие в ряде случаев фосфорную
кислоту, азотистые основания и углеводы; орнитинолипиды, построенные из
остатков высших жирных кислот, аминокислоты орнитина (или лизина)
и включающие в некоторых случаях двухатомные спирты.
Простые и сложные липиды легко омыляются. Однако в суммарной фрак-
ции липидов, выделенной из природного материала экстракцией жирораст-
ворителями, всегда присутствуют вещества, обладающие такой же раство-
римостью, как и липиды, но не способные омыляться. Они называются
неомыляемой фракцией липидов. В ее состав входят свободные высшие жирные
кислоты, высшие спирты и полициклические спирты (стеролы), производные
стеролов—стероиды, жирорастворимые витамины, высшие гомологи пре-
370
дельных углеводородов и другие соединения. Это дало повод некоторым
авторам рассматривать неомыляемую фракцию липидов как одну из групп
липидов. Однако для такого расширения границ класса липидов нет достаточ-
ных оснований.
Из указанных веществ жиры, стериды, фосфолипиды и диольные липиды
распространены повсеместно, их участие в построении клеточных структур
и в биохимических процессах весьма велико. Воски представляют в этом
смысле менее важную группу соединений. Долгое время считали, что гликоли-
пиды присутствуют только в нервной ткани, однако впоследствии их нашли
в хлоропластах растений. Орнитинолипиды присущи микроорганизмам.
Липиды обладают способностью образовывать со многими другими органиче-
скими соединениями (особенно с высокомолекулярными—белками, углеводами)
комплексы, которым в настоящее время придают большое значение в осуществле-
нии ряда важнейших биохимических функций. В виде таких комплексов, особенно
с белками, липиды входят в состав цитоплазматических мембран, субклеточных
частиц и бактериальных мембран. Так, в ядрах клеток липиды составляют около
15% от сухого вещества, в митохондриях— ~ 20, в эндоплазматическом ретикулу-
ме— ~30 и в гиалоплазме— ~ 10%. Только в гиалоплазме в составе липидов
преобладают триглицериды (~70%), тогда как в остальных субклеточных
элементах более 90% приходится на фосфолипиды, стериды и гликолипиды.
Широко известно значение липидов, особенно жиров, как субстратов для
окисления и обеспечения организма энергией: при распаде 1 г жира до СО2
371
и Н2О ее выделяется 38,9 кДж, тогда как при распаде 1 г углеводов или
белков—всего 16,1 кДж. Естественно, что при окислении липидов возникают
метаболиты, широко вовлекаемые в биосинтез других соединений (см.
гл. XIII). Важнейшей функцией липидов является также структурная. Образуя
матрикс мембран в виде двойных липидных слоев, липиды являются основой
любой биологической мембраны. Из рис. 120 видно, что липидный бислой
образует ее самую су ественную часть, составляя от 15 до 50% ее сухого
вещества. Перечисленные функции липидов (энергетическая, з пасная, постав-
щика метаболитов и структурная) получили название канонических.
Благодаря участию в деятельности мембранного аппарата клетки реализу-
ются такие важнейшие биологические функции липидов, как регуляция дея-
тельности ряда гормонов и активности ферментов (сейчас известно несколько
сотен липидзависимых ферментов), влияние на процессы транспорта метабо-
литов и макромолекул, контроль реакций биологического окисления и энерге-
тического обмена, связь с репликацией ДНК и ее матричной активностью,
компартментализация обменных процессов в клетке вплоть до формирования
мембранных машин (хлоропластов, митохондрий), участие в межклеточных
взаимодействиях (особенно в эмбрио- и онтогенезе), обеспечение молекуляр-
ной памяти и пиктографического механизма записи информации. Перечислен-
ные функции липидов характеризуют как неканонические. За выяснение неко-
торых из них большой группе советских ученых (Е. М. Крепе, Л. Д. Бергель-
сон, Р. П. Евстигнеева и др.) в 1985 г. присуждена Государственная премия.
Свойства мембран как надсистем регуляции клеточного метаболизма, их
конформационные перестройки, изменение их вязкости зависят от соотноше-
ния различных видов липидов в мембране, степени окисленности последних,
состояния межмембранного и внутримембранного переноса липидов и т. п.
Все названные явления столь важны для понимания процессов жизнедеятель-
ности, что биохимия мембран постепенно перерастает в биологию мембран,
объясняющую ряд фундаментальных закономерностей в развитии организма.
ПРОСТЫЕ ЛИПИДЫ
Жиры. Жиры исключительно широко распространены в природе: они
входят в состав организма человека, животных, растений, микробов и даже
некоторых вирусов. Содержание их в некоторых биологических объектах,
тканях и органах достигает 90%.
Термин жиры употребляют в двух смы лах. Те вещества, которые называют
жирами в обыденной жизни (говяжий жир, сливочное масло и т. п.), не
представляют химически определенных соединений, так как сложены из многих
составляющих: смесей различных триглицеридов, свободных высших жирных
кислот, пигментов, ароматических соединений, а часто и клеточных структур.
В этом смысле, следовательно, жир представляет понятие более морфологиче-
ское или технологическое. В частности, растительные жиры принято называть
маслами, а морфологически обособленные жиры животных—салом. Из раз-
ных источников выделено свыше 600 различных видов жиров среди которых
насчитывается 420 видов жиров растительного происхождения, 80 видов жиров
сухопутных животных и более 100 видов жиров обитателей водоемов.
С точки зрения состава под жирами подразумевают строго определенные
соединения, а именно: сложные эфиры высших жирных кислот и трехатомного
спирта—глицерина.
В связи с этим химики предпочитают употреблять название триглицериды
(формулы триглицеридов см. ниже).
372
В составе природных триглицеридов найдено более пятисот органических
кислот, и список их расширяется с каждым годом. Среди них большая доля
п инадлежит высшим жирным монокарбоновым кислотам, т. е. кислотам с чис-
лом углеродных атомов в молекуле, равным 16 и более. Высшие органические
кислоты, обнаруженные в триглицеридах, часто содержат двойные связи и ок-
сигруппы в углеводородном радикале. В табл. 25 приведен список, формулы
и температуры плавления кислот, наиболее часто встречающихся в составе
жиров.
Таблица 25
Некоторые монокарбоновые кислоты, выделенные из природных жиров
Наименование и формула Температура плавления» °C Кем и когда открыта
Насыщенные кислоты Масляная (С4Н8О2) -5,3. Шеврале (1820)
СН3—(СН2)2—соон Изовалериановая (С5НюО2) -51,0 Шеврале (1817)
(СН3)2—СН—СН2—СООН Капроновая (С6Н12О2) -4,0 Шеврале (1820)
СНз—(СН2)4—СООН Каприловая (C8Hi6O2) + 16,0 Лерч (1844)
СНз—(СН2)6—СООН Каприновая (СюН20О2) +31,3 Шеврале (1820)
СНз—(СН2)8—СООН Лауриновая (С12Н24О2) +43,5 Марссон (1842)
СНз—(СН2)10—СООН Миристиновая (С14Н28О2) +54,4 Плайфар (1841)
СНз—(СН2)12—СООН Пальмитиновая (С16Н32О2) + 62,9 Шеврале (1816)
СНз—(СН2)14—СООН Стеариновая (С18Н36О2) +69,6 Шеврале (1816)
СНз—(СН2)16—СООН Арахиновая (С20Н4оО2) +75,4 Гёссман (1854)
СНз—(СН2)18—СООН Бегеновая (С22Н44О2) +80,0 Фолкер (1848)
СНз—(СН2)20—СООН Лигноцериновая (С24Н48О2) +84,2 Гелл и Герман (1880)
СНз—(СН2)22—СООН Церотиновая (С26Н52О2) +87,7 Броди (1848)
СНз—(СН2)24—СООН Ненасыщенные кислоты с одной двойной связью Пальмитоолеиновая (Ci6H30O2) цис-+0,5 Гофштадгер (1854)
СН3—(СН2)5—СН=СН—(СН2)7—СООН Олеиновая (С18Н34О2) транс-+31,0 цис-+16,0 Шеврале (1815)
СНз—(СН2)7—СН=СН—(СН2)7—СООН Эруковая (С22Н42О2) цис-+33,5 Дерби (1849)
СН3—(СН2)7—СН=СН—(СН2), 1—соон транс-+60,0
Нервоновая (С24Н46О2) цис-+40,5 Цумото (1927)
СН3—(СН2)7—СН=СН—(СН2)1з—СООН транс-+60,5
373
Продолжение табл. 25
Наименование н формула Температура плавления, °C Кем и когда открытд
с несколькими двойными связями Линолевая (С18Н32О2) цис- —43,0 Сакк (1844)
СНз—(СН2)3—(СН2—СН=СН)2—(СН2)7—СООН Линоленовая (С18Н30О2) транс- —13,0 -75,0 Хазура (1887)
СНз—(СН2—СН=СН)з—(СН2)7—СООН Арахидоновая (С2оН3202) -49,5 Хартли (1909)
СНз—(СН2)4—(СН=СН—СН2)4—(СН2)2—СООН с тройной связью Тарариновая (С18Н32О2) + 50,0 Арнауд (1892)
СНз—(СН2)10—С=С—(СН2)4—СООН Циклические кислоты Гиднокарповая (Ci6H28O2) +59,4 Пауэр (1904)
сн-сн I уСН—(CHJh,—соон СН.-СН, Чальмугровая (С18Н32О2) +68,3 Мосс (1879)
сн-сн \м-(СЩ1Г-СООН cHj-Zh, Оксикислоты Рицинолевая (С18Н34Оз) СН,—(СН2)5—СН (ОН)—СН2—СН= цис-от +5,0 Саалмюллер (1848)
СН—(СН2)7—СООН до +16,0
Как видно из данных таблицы, в составе природных жиров вначале были
найдены почти исключительно кислоты нормального строения с четным числом
углеродных атомов в молекуле. Лишь изовалериановая кислота, обнаружен-
ная в жире печени дельфина, имела нечетное число атомов углерода и развет-
вленную цепь.
В последнее время разработаны новые высокоэффективные методы раз-
деления (тонкослойная и газовая хроматография) и установления структуры
(инфракрасная спектрофотометрия) высших жирных кислот. В результате
в составе натуральных жиров обнаружен ряд новых представителей высших
жирных кислот—циклических, с нечетным числом атомов углерода и развет-
вленным углеродным скелетом. Последние, в частности, резко понижают тем-
пературу плавления жиров, обладают антибиотическими свойствами и видо-
вой специфичностью. Одним из представителей их является, например, мико-
левая кислота, выделенная из туберкулезных бактерий:
С25 Н51—СН2—СН—СН (ОН)—сн (ОН)—сн—соон
I I I
с6н13 с16н33 с24н49
474
Наиболее часто и в наибольшей пропорции в природных жирах встречается
олеиновая кислота (в большинстве жиров ее более 30%), а также пальмитино-
вая кислота (от 15 до 50% в большинстве случаев). Поэтому олеиновую
и пальмитиновую кислоты относят к категории главных жирных кислот,
содержащихся в жирах. Остальные жирные кислоты присутствуют в природ-
ных жирах, как правило, в небольшом количестве (несколько процентов)
и лишь в некоторых видах природных жиров их содержание измеряется
десятками процентов. Так, масляная и капроновая кислоты хорошо представ-
лены в некоторых жирах животного происхождения, а каприловая и кап-
риновая кислоты — в кокосовом масле. Лауриновой кислоты много в лав-
ровом масле, миристиновой—в масле мускатного ореха, арахиновой, бегено-
вой и лигноцериновой—в арахисовом и соевом маслах. Полиеновые 1ысшие
жирные кислоты—линолевая и линоленовая—составляют главную часть
льняного, конопляного, подсолнечного, хлопкового и некоторых других рас-
тительных масел. Стеариновая кислота содержится в значительном количестве
(25% и более) в некоторых твердых животных жирах (жир баранов и быков)
и маслах тропических растений (кокосовое масло).
Животные и растительные жиры отличаются некоторыми особенностями.
Животные жиры более разнообразны по набору высших жирных кислот,
входящих в их состав. В частности среди последних чаще встречаются высшие
жирные кислоты с числом углеродных атомов от 20 до 24. В составе рас-
тительных жиров очень высока доля ненасыщенных высших жирных кислот (до
90%), и из предельных лишь пальмитиновая кислота содержится в них в ко-
личестве 10—15%.
Среди триглицеридов различают простые и смешанные. Первые являются
сложными эфирами глицерина и одной из высших кислот, например:
СН2—О—СО—(СН2)14—СН3 СН2—О—СО—(СН2)7—СН=СН—(СН2)т—сн3
СН—О—СО—(СН2)14—СН3 СН—О—СО—(СН2)7—СН=СН—(СН2)7—сн3
СН2—О—СО—(СН2)14—СН3 СН2—О—СО—(СН2)7—СН=СН—(СН2)7—сн3
Трипальмитин Триолеин
Вторые построены из остатка глицерина и остатков разных высших жир-
ных кислот:
СН —О—-СО—(СН2)14—СН3 СН2—О—СО—(СН2)14—СН3
СН—О—СО—(СН2)16—СН3 СН—О—СО—(СН2)16—сн3
СН2—О—СО—(СН2)т—СН=СН—(СН2)т—СН3 СН2—О—СО—(СН2)16—сн3
Пальмитостеароолеин Пальмнтодистсарин
В природных жирах, представляющих собой смеси разнообразных три-
глицеридов, доля простых триглицеридов незначительна, тогда как процент-
ное содержание смешанных триглицеридов может быть очень высоким. Так,
из 8 азличных триглицеридов, найденных в составе свиного сала, лишь
1% приходится на долю трипальмитина и 3%—триолеина. Остальные шесть
триглицеридов свиного жира являются смешанными, из них преобладают
пальмитодиолеин (53%) и пальмитостеароолеин (27%). В кокосовом и паль-
мовом маслах найдены стеародипальмитин, олеодипальмитин, миристоди-
пальмитин, миристодилаурин, пальмитодимиристин и лауродимир стин. Та-
ким образом, природные жиры, выделенные из того или иного объекта,
375
Остаток
линолевой
кислоты
Остаток, .
стеариновой
кислоты
Остаток ,
арахивонооои
кислоты
Остаток
глицерина • атом углерода
атом кислорода
• простая связь
сэ двойная связь
Рис. 121. Структура и форма молекулы триглицерида
представляют всегда сложные
смеси растворенных друг
в друге разнообразных тригли-
церидов.
Физические свойства три-
глицеридов зависят от характе-
ра высших жирных кислот,
входящих в состав их молекул.
Особенно наглядной становит-
ся эта зависимость при рас-
смотрении температур плавле-
ния триглицеридов: если в со-
ставе триглицерида преобла-
дают насыщенные (твердые)
жирные кислоты, то и тригли-
церид твердый; если преобла-
дают ненасыщенные (жидкие)
кислоты, температура плавле-
ния триглицерида низкая и при обычных условиях он жидкий.
Такую же зависимость можно обнаружить у натуральных жиров: при
наличии преимущественно насыщенных триглицеридов в составе жира темпе-
ратура плавления последнего высокая, ненасыщенных—низкая. Бараний жир,
например, имеет температуру плавления примерно на 10° С выше, чем свиной,
потому что в нем содержится на несколько процентов меньше пальмитодиоле-
ина (46 и 53% соответственно) и больше олеодипальмитина (13 и 5% соот-
ветственно). Низкая температура плавления многих растительных масел нахо-
дится в полном соответствии с весьма значительным содержанием непредель-
ных кислот в составе их триглицеридов. Например, триглицериды жидкого
при обычных условиях подсолнечного масла (гпл=— 21° С) включают 39%
олеиновой и 46% линолевой кислоты, тогда как твердое растительное масло
бобов какао (/пл=30—34° С) имеет в своем составе 35% пальмитиновой и 40%
стеариновой кислот.
Триглицериды образуют оптические и геометрические изомеры, так как во
многих случаях обладают асимметрическим углеродным атомом в остатке
глицерина и одной или несколькими двойными связями в радикалах кисло-
тных остатков. Многочисленные комбинации, которые могут осуществляться
за счет перестановки ацильных группировок в смешанных триглицеридах,
дают начало семействам структурных изомеров. Характерно, что непредель-
ные высшие жирные кислоты в триглицеридах находятся, как правило, в цис-
конфигурации, что сказывается на форме молекулы (рис. 121).
Таким образом, в природе потенциально может существовать громадное
число индивидуальных триглицеридов, чем обеспечивается их видовая и иная
специфичность. Однако, как обстоит дело в действительности, неизвестно:
проблема пространственной изомерии и видовой специфичности жиров почти
не изучена.
Воски. Воски—группа простых липидов, являющихся сложными эфирами
высших спиртов и высших монокарбоиовых кислот. Натуральные воски кроме
упомянутых сложных эфиров содержат некоторое количество свободных
высших спиртов и высших кислот, а также немного углеводородов всегда
с нечетным числом углеродных атомов (от 27 до 33) и красящих и душистых
веществ. Общее количество этих примесей может достигать 50%. Воски
встречаются как в животном, так и в растительном царстве, где выполняют
главным образом защитные функции. Так, в растениях они покрывают тонким
376
слоем листья, стебли и плоды, предохраняя их от смачивания водой и про-
никновения микроорганизмов. От качества воскового покрытия зависят,
в частности, сроки хранения фруктов. Под покровом из пчелиного воска
храни ся мед и развиваются личинки пчелы. Другие виды животного воска
(ланолин) предохраняют волосы и кожу от действия воды. Все воски пред-
ставляют собой твердые вещества разнообразной окраски—чаще всего жел-
той или зеленоватой (в зависимости от происхождения); температура их
плавления—от 30 до 90° С. В составе восков найдены как в свободном
состоянии, так и в виде сложных эфиров несколько десятков высших жирных
кислот и спиртов. Вот некоторые из них:
Кислоты
Пальмитиновая
Лигноцериновая
Церотиновая
Монтановая
Мелиссиновая
Лацериновая
Спирты
Цетиловый
Цериловый
Монтановый
Мирициловый
СН3—(СН2)14—СООН
СН3—(СН2 )22—СООН
СН3—<СН2)24—СООН Л
СН3—(СН2)26—соон
сн3—<СН2 )28—соон
СН3—<СН2)30—соон J
Источник
Пчелиный воск, спермацет
Воск пальмы
Пчелиный воск, воск
листьев и фруктов
СН3-<СН2)14^-СН2ОН
СН3-<СН2)24—СН2ОН
СН3—-(СН2)26—СН2ОН 1
СН,—(СН,),а—СН2ОН J
Спермацет
Пчелиный воск
Пчелиный воск, воск
листьев и фруктов
В пчелином воске и воске, покрывающем поверхность листьев растений
и фруктов, найдены спирты и кислоты с 32 и 34 атомами углерода в молекуле.
Один из видов японского воска содержит в своем составе дикарбоновые
высшие кислоты. В составе рыбьего и китового жира обнаружены непредель-
ные высшие спирты—олеиловый и др. Непредельные кислоты в восках встре-
чаются сравнительно редко.
Спермацет—воск животного происхождения, добываемый из спермацето-
вого масла черепных полостей кашалота путем вымораживания и отжимания,
состоит в основном (на 90%) на пальмитиновоцетилового эфира:
СН3—(СН2)14—СО—О—СН2—(СН2)14—СН3. Спермацет—твердое веще-
ство, его ?пл=41—49° С.
Фракция, оставшаяся после выделения спермацета из спермацетового мас-
ла кашалота, представляет жидкий воск. Он состоит из смеси жидкого оле-
иново-олеилового эфира: СН3—(СН2)7—СН=СН—(СН2)7—СО—О—СН2—
—(СН2)7—СН=СН—(СН2)7—СН3 с другими жидкими эфирами, компо-
нентами которых являются либо олеиновая кислота, либо олеиловый
спирт.
В пчелином воске преобладает пальтимитиново-мирициловый эфир:
СН3—(СН2)14—СО—О—СН2—(СН2)28—СН3. Для него характерно также
высокое содержание свободных высших жирных кислот (до 13,5%)
и углеводородов (до 12,5%). Температура плавления пчелиного воска
равна 62—70° С.
Воск пальмы Copernicia cerifera, произрастающей в Бразилии, называемый
карнаубским воском, является в основном церотиново-мирициловым эфиром:
СН3—(СН2)24—СО—О—СН2—(СН2)28—СН3. Карнаубский воск—вещество
желтовато-серого цвета, покрывающее листья пальмы. Он защищает растение
от потери влаги.
377
Экстракцией органическими раство ителями бурого угля или торфа удается
выделить монтанный воск, в состав которого входят монтаиовая кислота и ее
эфиры (/„л=72 — 74° С).
Воски более устойчивы к действию света, окислителей, нагреванию и дру-
гим физическим воздействиям, а также хуже гидролизуются, чем жиры. Извес-
тны случаи, когда пчелиный воск сохранялся тысячелетиями. Именно поэтому
воски выполняют в организме защи ные функции.
Стериды. Большую группу простых липидов составляют стериды—слож-
ные эфиры специфически построенных циклических спиртов (стеролов) и выс-
ших жирных кислот. Стериды образуют омыляемую фракцию липидов.
В природе гораздо более широко, чем стериды, представлена фракция
неомыляемых, свободных стеролов и родственных им соединений. Так, в ор-
ганизме человека лишь 10% стеролов этерифицировано и находится в виде
стеридов, а 90% свободно и образует неомыляемую фракцию. Соотношение
стеролов и стеридов в разных тканях и жидкостях организма различно: печень
содержит их поровну, а в желчи содержатся только свободные стеролы.
Стеролы построены довольно сложно. В основе их молекулы лежит цикли-
ческая группировка атомов, состоящая из восстановленного фенантрена (пол-
ностью восстановленный фенатрен называют пергидрофенантреном) и цикло-
пентана. Эта циклическая группировка называется циклопентанопергидрофе-
нантреном или стераном. Стеран, несущий боковую цепь углеродных атомов
и две СН 3-группы (при 10-м и 13-м углеродных атомов цикла), называют
холестаном:
Цнклопентано-
пергндрофенантрен
Холестан
Углеродные атомы в этих углеводородах обозначают исходя из нумерации,
принятой для фенантрена (1—14-й атомы углерода); затем нумеруют четвер-
тый цикл и только после этого переходят к нумерации атомов углерода
в боковых цепях. Циклы принято обозначать прописными буквами латинского
алфавита.
Будучи окислен в положении 3 (кольцо А), холестан превращается в поли-
циклический спирт—холестаиол, дающий начало классу стеролов:
Однако не следует думать, что в природе стеролы возникают при восста-
новлении фенантрена. Выяснено, что их биосинтез идет путем циклизации
378
полиизопреноидо >, которые, по существу, и являются (см. ниже предшествен-
никами стеролов.
Характерное ядро холестанола повторяется во всех стеролах с незначи-
тельными вариациями. Они сводятся либо к возникновению между 5—6-м
и 7—8-м атомами углерода кольца В или 22—23-м атомами углерода
боковой цепи двойных связей, либо к появлению в положении 24 (в бо-
ковой цепи) радикала, который может иметь строение—СН3; = СН2;
— С2Н5; = СН—СН3 и т. п. Ниже приведены формулы наиболее важных
природных стеролов:
Холестерол (от греч. холе—желчь) является основным стеролом живот-
ных и человека, т. е. относится к разряду зоостеролов. Эргостерол характе-
рен для грибов. Ситостерол и стигмастерол типичны для растений (фитосте-
ролы): первый содержится, например, в соевом масле, а второй—в масле
зародышей семян пшеницы. Фукостерол обнаружен у бурых водорослей.
Наличие того или иного стерола часто специфично для определенного класса
или семейства животных или растений. Например, губки содержат ряд
уникальных стеролов с 28 и 29 атомами углерода в молекуле, а морские
звезды и голотурии—специфические стелласт ролы. Замечено, что чем при-
митивнее организм, тем более разнообразный набор стеролов для него
характерен. Человеку свойствен только один—холестерол. В настоящее
время известно более 60 зоостеролов и почти 140 фитостеролов (А. Кульман,
1989).
379
Среди стеролов широко распространены конформационные изомеры. На-
пример, известен изомер холестанола—копростанол, у которого иное взаимо-
расположение в пространстве составляющих его циклов:
При рассмотрении приведенных выше формул холестанола и копро-
станола легко заметить, что у первого циклы А и В расположены по
отношению друг к другу в транс-, а у второго—в цис- положении. Хо-
лестанол в небольших количествах присутствует в тканях животных (наряду
с большим количеством холестерола). Копростанол найден в экскремен ах
животных, так как в виде копростанола восстановленный холестерол вы-
водится из организма.
Характерной особенностью конформационных природных изомеров
стеролов является то, что у них ОН-группа при 3-м углеродном атоме
расположена по ту же сторону цикла, что и СН3-группа при 10-м
углеродном атоме. Такая конформация обозначается как £, а проти-
воположная ей—как а.
Стеролы—кристаллические вещества, хорошо растворимые в хлорофор-
ме, серном эфире и горячем спирте, практически не растворимые в воде;
устойчивы к действию гидролизующих агентов. В организме животных стеро-
лы окис яются и дают начало целой плеяде производных, носящих общее
название стероиды. Сюда относятся многие соединения, из которых упомянем
лишь несколько характерных представителей:
7-Дезоксихолевая кислота
Холевая кислота
Тестостерон
Холевые кислоты—важнейшие ингредиенты желчи, обеспечивающие нор-
мальный ход всасывания жирных кислот в кишечнике человека и животных.
Эстрадиол и тестостерон—соответственно женский и мужской половые гормо-
ны, оказывающие огромное влияние на процессы жизнедеятельности (см.
гл. XII).
Сложные эфиры зоо- и фитостеролов с высшими жирными кислотами
образуют группу омыляемых веществ—стеридов:
380
+ Н2О
сн3—(СН2)15-СООН +
CHj—(СН2)14—COO^-^ ' V
Пальмитохолестерид Пальмитиновая Холестерол
(холестеролпальмитат) кислота
Из высших жирных кислот в составе стеридов обнаружены в основном
пальмитиновая, стеариновая и олеиновая кислоты. Однако в стеридах, пред-
ставляющих, например, главную составную часть ланолина (фракция жира
овечьей шерсти), найдены миристиновая, арахидоновая и церотиновая кисло-
ты, а также специфические высшие жирные кислоты с разветвленной углерод-
ной цепью—ланопальмитиновая, ланостеариновая и др.
Все стериды, так же как и стеролы,—твердые, бесцветные вещества (от
греч. стереос—твердый). В природе, особенно в составе животных организ-
мов, они встречаются в виде комплексов с белками, функциональное значение
которых сводится к транспорту стеролов, стероидов и стеридов, а также
к участию в образовании биологических мембран. При увеличении содержа-
ния стеролов и стеридов в составе липидной части мембран уменьшается
проницаемость последних, возрастает их вязкость, ограничивается их подвиж-
ность, ингибируется активность ряда ферментов, встроенных в мембрану.
Стериды и стеролы регулируют й другие процессы в организме. Некоторые из
производных стеролов являются канцерогенными веществами, тогда как дру-
гие (например, естостеронпропионат) используют для лечения некоторых
видов рака. Стериды и стеролы в больших количествах входят в состав
нервной ткани человека и животных; значение и функции их здесь активно
исследуют.
СЛОЖНЫЕ ЛИПИДЫ
Фосфолипиды. Фосфолипиды—сложные эфиры многоатомных спиртов
с высшимй жирными кислотами, содержащие остатки фосфорной кислоты
и связанные с нею добавочные группировки (азотистые основания, аминокис-
лоты, глицерин, инозит и др.).
Из многоатомных спиртов в составе различных фосфолипидов найдено
три: глицерин, миоинозит и сфингозин:
НО НО/
н/1—Ж он
I/н /нМ
\QHZ Н/ сна—(сн2)12—сн=сн—сн—сн—СН2ОН
НОЗЬиЦ/н НО NH„
хн он
Миоинозит (оптически Сфингозин
неактивная мезоформа; (транс-1,3-диоксн-2-амино-октадецен-4)
пунктиром обозначена
ось симметрии)
В соответствии с этим фосфолипиды делят на три группы: глицерофосфоли-
пиды, инозитфосфолипиды и сфингофосфолипиды. 1лицерофосф< липидь час-
то называют фосфатидами, так как их можно рассматривать как производные
фосфатидной кислоты (см. ниже), а инози фосфолипиды—фосфоинозитидами.
381
В качестве высших жирных кислот в молекулах фосфолипидов содержатся
пальмитиновая, стеариновая, линолевая, линоленовая и арахидоновая кисло-
ты, а также лигноцериновая, нервоновая и др. (см. табл. 25).
В зависимости от типа фосфолипида в построении его молекулы принима-
ют участие один или два остатка высшей жирной кислоты. Фосфорная же
кислота входит, как правило, в состав фосфолипидов в количестве одной
молекулы.
Лишь некоторые виды инозитфосфолипидов содержат два и более остатка
фосфорной кислоты.
Азотсодержащие составляющие фосфолипидов разнообразны. Наиболее
часто встречаются этаноламин, холин и серин (см. ниже). Из химического
строения фосфолипидов ясно, что в их молекулах есть участки, способные
диаметрально противоположно взаимодействовать с молекулами раство-
рителя.
Углеводородный радикал остатка (или остатков) высших жирных кислот
формирует лиофобную часть, а остатки фосфорной кислоты и азотистого
основания, способные ионизироваться,—лиофильную. Благодаря этой осо-
бенности фосфолипиды, видимо, участвуют в обеспечении односторонней
проницаемости мембран субклеточных структур.
Фосфолипиды—твердые вещества жироподобного вида; они бесцветны, но
быстро темнеют на воздухе вследствие окисления по двойным связям входя-
щих в их состав непредельных кислот. Хорошо растворяются в бензоле,
петролейном эфире, хлороформе и т. п. Растворимость в спирте, ацетоне
и серном эфире у разных групп фосфолипидов различна. В воде они не
растворимы, но могут образовывать стойкие эмульсии, а в некоторых случа-
ях—коллоидные растворы.
Фосфолипиды найдены в животных и растительных организмах, но осо-
бенно много содержит их нервная ткань человека и позвоночных животных.
У беспозвоночных содержание фосфолипидов в нервной системе в 2—3 раза
ниже. Много фосфолипидов в семенах растений, сердце и печени животных,
яйцах птиц и т.п. Специфическими фосфолипидами обладают-микроорга-
низмы.
Фосфолипиды легко образуют комплексы с белками и в виде фосфоли-
попротеинов присутствуют во всех клетках живых существ, участвуя глав-
ным образом в формировании клеточной оболочки и внутриклеточных
мембран.
[лицерофосфолипиды, или фосфатиды,—сложные эфиры глицерина выс-
ших жирных кислот, фосфорной кислоты и азотистого основания. Их рассмат-
ривают как производные фосфатидной кислоты, откуда и происходит само
название этой группы фосфолипидов:
СН2—О—СО—R
Jh—О—СО—R
Jh,-o—Р=0
нсГ \)Н
Фосфатидная кислота
В зависимости от характера азотистого основания среди фосфатидов раз-
личают фосфатидилхолин (лецитины), фосфатидилколамин (кефалины), фос-
фатидилсерин и фосфатидилтреонин:
382
СН,—О—CO—(СН,)14—СН3
Лецитин (фосфатндилхолнн)
CHg—О—CO—(CHs)xe—сн3
<!н—О—CO—(CH»)I4—CH9 '
СН,—О—P—о—CHg—CHj—nh8
Кефалин (фосфатидилколв мн
СН3-О-СО—(СН2)1в—сн3
СН-О—СО—(СН,),—СН=СН—(СН»),—сн8
СН,—О—Р—О—СН»—СН—СООН
(Z N+H;j
Фосфатицилсернн
СН,—О—СО—(СН»)И—СН3
СН—О—СО—(СН2),—СН^СН—(СН,),—СН9
<(:н2—о—р—о—сн—сн—соон
/XI I
О о- CH3 N*HS
Фосфатидилтреоннн
Из них лецитины наиболее распространены в природе. Наличие в лецити-
нах холина в качестве азотистого основания впервые было установлено
К. С. Дьяконовым (1867).
Три первых вида азотсодержащих фосфатидов, видимо, могут переходить
друг в друга, так как они отличаются лишь строением азотистых оснований,
между которыми возможна, например, такая генетическая связь:
НО—СН,—CH—NH,-----------------------► НО—СН,—СН,—NH,-----------------
I Декарбоксилироваим Коламин Метилирование
СООН (етанолаыин)
Серин
СН.П
НО—СН,—СН,—N4—СН,
Хсн.
ОН-
Холин
Некоторые фосфатиды, открытые сравнительно недавно, не содержат азо-
тистого основания, место которого в молекуле в этом случае занимают
глицерин и его производные:
о он
СН,—О—СО—R СН,ОН
СН-О—СО—R' СНОН
СН,—О—СО—R СН,—О—Р—О—СН,
СН—О—СО—R* (!нон
I,—О—Р—О—СН,
/х
О он
Фосфггидилглнцернн
:н,
1-0—СО—R'
СН,—О-СО—R"
о он
Дифосфатшилглицерни (кардмолшмш)
Фосфатидилглицерин является обязательной составной частью хлоро-
пластов и в небольших количествах присутствует в бактериальных клетках
и тканях животных. Кардиолипин—одно из существенно необходимых
383
соединений в составе митохондриальных мембран, особенно в митохондриях
сердечной мышцы; он найден не только у животных, но и в растениях
и у бактерий. И тот и другой способны аминоацилироваться по остатку
глицерина с образованием аминоацилфосфатидил- и аминоацилдифосфатидил-
глицеринов (их другое название—липоаминокислоты), создающих в хлоро-
пластах, где они найдены (М. И. Молчанов, 1964), резерв аминокислот для
биосинтеза белков.
Обладая асимметрическим строением (2-й углеродный атом остатка гли-
церина всегда асимметричен), фосфатиды оптически активны и образуют
соответствующие стереоизомеры. Вместе с тем им свойственна изомерия за
счет перестановки остатков высших жирных кислот из а- в 0-положение или
наоборот.
Инознтфосфолипиды (фосфоинозитиды) представляют группу фосфоли и-
дов, строение которой выяснено в последние 10—15 лет благодаря интенсив-
ным работам по их химическому синтезу, осуществленным сотрудниками,
принадлежащими к школам Н. А. Преображенского и Л. Д. Бергельсона.
Простейший монофосфоинозитид построен так:
Как можно видеть, он похож на фосфатид, у которого азотистое основание
заменено остатком инозита.
При наличии двух или трех остатков фосфорной кислоты в молекуле
инозитфосфолипида все они связаны с остатком инозита. Инозитфосфолипи-
ды такого типа выделены из мозга и получены синтетически. Особенно высоко
их содержание в миелиновых оболочках нервных волокон спинного мозга.
Скорость обмена фосфатных групп в фосфоинозитиддифосфатах намного пре-
вышает таковую в других фосфолипидах. Благодаря наличию диссоциирован-
ных фосфатных групп фосфоинозитиддифосфаты обеспечивают перенос ионов
через биологические мембраны. Они же являются источником вторичных по-
средников, отщепляющихся в ответ на гормональный и иные сигналы (см.
с. 456 и 474). Многие осфоинозитиды содержат углеводы, амины, аминокис-
лоты и сфингозин.
Сфингофосфолипиды в отличие от рассмотренных выше фосфатидов и ино-
зитфосфолипидов содержат остаток высшей жирной кислоты, соединенный
с двухатомным аминоспиртом (сфингозином) пептидной связью:
сн3—(сн2)12 н
✓С==С\
HZ н—с*—он
СН3—(СН2)22— со—NH—с*-н о щ
СН2—О—Р—о—СН2— СН2— N—СН3
А- сн>
Сфинголипид
384
Остальные компоненты сфингофосфолипидов—фосфорная кислота и хо-
лин—присоединены так же, как у фосфатидов. Именно в сфинголипидах
в значительных количествах обнаружены лигноцериновая и нервоиовая кисло-
ты, менее характерные для остальных групп фосфолипидов.
Представители этой группы фосфолипидов, видимо, более характерны для
животного, чем для растительного мира. Однако из фосфолипидов раститель-
ного происхождения (из кукурузного зерна) выделен аминоспирт, весьма
похожий на сфингозин:
СН3-(СН2)13-СН-СН-СН-СН2ОН
I I I
он он nh2
Фитосфингозян
Этот же спирт найден в дрожжах и грибах, а недавно—в мозге и почках
человека, что указывает на возможность существования аналогичных фосфо-
липидов в растительных и животных объектах.
Сфинг фосфолипиды не растворимы в серном эфире, что используют при
их отделении от фосфатидов. Они характеризуются также трудной раство-
римостью в ацетоне и большей устойчивостью к действию окислителей, чем
фосфатиды.
Сфинголипидам свойственны весьма сложные пространственные конфигу-
рации, связанные с возможностью оптической изомерии (два асимметричных
углеродных атома в молекуле) и цис-транс-изомерии по месту двойной связи.
Этим объясняется их органная и видовая специфичность. Кроме того, установ-
лено, что органная специфичность сфинголипидов зависит от качественного
состава высших жирных кислот: так, для сфинголипидов мозга характерно
присутствие нервоновой кислоты.
Етиколипнды. Вторую группу сложных липидов образуют гликолипиды.
Они характеризуются тем, что полярная моно- или олигосахаридная состав-
ляющая (глюкоза, галактоза, глюкозамин, галакт замин, их N-ацетильные
производные и др.) через остаток многоатомного спирта (глицерин, сфинго-
зин) соединяется с неполярными радикалами высших жирных кислот (паль
митиновой, стеариновой, олеиновой, лигноцериновой, нервоновой, цереброно-
вой и др.) гликозидной и сложноэфирной связями.
Примерами являются моногалактозилглицерин и цереброн соответст-
венно:
МЬноплактоашдашдипяцера1
(глцероглашлшпш)
Цереброн (от лат. cerebrum-
мозг) (гликосфинголипид)
13—3502
385
Глицерогликолипиды и гликосфинголипиды широко распространены в.при-
роде. Особое внимание сейчас привлекают гликосфинголипиды, содержащие
олигосахаридную компоненту, например глобозиды:
СН3 — (ОУц н
н' \
н-с-он
R-CO-NH—СН
СН2ОН
СН2ОН ^2
Н1/Ь°\н
Н н он
н nh—со—га.
NH-СО—СН3
N-mmui-ra»-/ -1*3-Га*-м -1-*4-Гаи* j»-l-М-Глю-je -1-*1-цер»мид
Уже изучено ПО глобо-, лакто- и ганглиосфинголипидов. Они регулируют
рост клеток, являются маркерами трансформации нормальных клеток в рако-
вые, обеспечивают компактизацию бластомеров во время дробления яйца,
взаимодействуют с белковыми токсинами и выполняют ряд других важней-
ших функций.
В последнее время показано, что они участвуют в процессах рецеп-
ции гормонов, факторов роста, лейкинов, в регуляции роста и диффе-
ренцировки клеток, являются иммуномодуляторами и вторичными по-
средниками.
Диольные липиды. В природных липидах сложноэфирные связи с остатками
высших жирных кислот и фосфорной кислотой, равно как и простые эфирные
связи с остатками высших спиртов и углеводов, могут образовывать двух-
атомные спирты—этандиол, а также пропандиолы, бутандиолы и пентан-
диолы.
Такие липиды называют диольными липидами. Они широко распростра-
нены среди растений, животных и микроорганизмов в качестве минорных
компонентов липидной фракции, хотя у морских беспозвоночных и рыб неред-
ко составляют главную часть запасных липидов. Они же являются структур-
ными элементами мембран эндоплазматической сети, но не митохондрий.
Замечено, что они обмениваются энергичнее, чем липиды, где функцию спирта
выполняет глицерин. Известно 9 групп диольных липидов, выделенных из
различных объектов.
Орнитинолипиды. Первые указания о существовании этого нового вида
липидов относятся к 1963 г. В течение двух последующих десятилетий струк-
тура многих из них была расшифрована и высказаны предложения об их
функциональной роли.
386
Орнитинолипиды характерны для микроорганизмов. Их непременными
составными частями являются высшие жирные Р-оксикислоты и аминокис-
лота орнитин (или лнзин); в зависимости от типа строения могут присут-
ствовать также остатки этиленгликоля или 1,3-пропандиола. Наиболее
распространенные в природе орнитинолипиды построены следующим об-
разом:
R—СО NH2
I I
о (СН2)3
I I
R—СН—СН2—СО—NH—СН—СООН, где R—алкил (Сп—С15)
Остаток высшей жирной Остаток орнитина
Р-оксикислоты (со
свободной или ацилиро-
ванной ОН-группой)
Установлено, что орнитинолипиды в микробной клетке способны заменять
фосфатидилэтаноламин в качестве структурного элемента цитоплазматиче-
ских мембран. Это особенно ярко проявляется в условиях фосфатного го-
лодания.
ОБМЕН ЛИПИДОВ
Обмен жиров (триглицеридов). 1идролиз жиров. Первой фазой обмена жиров
(триглицеридов) является гидролиз, в результате которого освобождаются
глицерин и высшие жирные кислоты. Реакция гидролиза триглицеридов уско-
ряется гидролазой эфиров глицерина—л и п а з о й.
Гидролиз триглицеридов идет ступенчато: сначала распадаются две внеш-
ние сложноэфирные связи (a-связи). Уравнение этой реакции приведено выше
(см. с. 129). Так осуществляется, например, гидролиз триглицеридов в кишеч-
нике человека и животных при каталитическом воздействии липазы подже-
лудочной железы (М=48000, мономер). Р-Моноглицериды всасываются
стенкой кишечника и либо идут на ресинтез триглицеридов уже в кишечной
стенке, либо распадаются далее под действием неспецифических эстераз,
способных ускорять реакции гидролиза сложных эфиров вторичных спиртов.
Примером может служить гидролиз Р-моноглицерида в присутствии али-
эстеразы печени:
сн2—он 1 Алиэстераза СН—О—СО—С15Н 31 + н 2О I (карбок- ’ силэсте- СН 2 ОН раза) 0-Моноглицерид сн2—он - сн—он+с 15н31соон Пальмитиновая СН 2 ОН кислота Ешцерин
387
В растительном царстве липазы широко распространены в семенах
и вегетативных органах растений. Специфичность их к а- и Р-глицеридам
не установлена, а из дрожжевых грибков выделена липаза, атакующая
в равной мере и а- и p-связи (М = 55000, мономер, содержит 7% углево-
дов). Среди липаз из микроскопических грибов зафиксированы множе-
ственные формы. Различают простые липазы, которые каталитически уско-
ряют освобождение высших жирных кислот из свободных триглицеридов,
и липопротеинлипазы, способствующие гидролизу связанных с белками
липидов.
Третичная структура дрожжевой липазы выяснена. Ее полипептидная цепь
(430 аминокислотных остатков) сложена в глобулу (7x7x5 нм), в центре
которой находится активный центр, включающий остаток гистидина. Выска-
заны предположения и о структуре активного центра панкреатической липазы:
ведущую роль в нем играют радикалы гистидина, серина, дикарбоновых
аминокислот и изолейцина. Как и в случае других гидролаз, радикал гистиди-
на служит для переноса протонов, а радикал серина—для акцептирования
ацильной группы, высвобождающейся в момент распада сложноэфирной свя-
зи в молекуле триглицерида. Радикал изолейцина взаимодействует с углеводо-
родным радикалом остатка высшей жирной кислоты и способствует закрепле-
нию молекулы триглицерида в активном центре фермента (рис. 122). Выясне-
но, что активность липаз регулируется путем их фосфорилирования—
дефоо орилирования:
Триглицерид . -»» Глицерин + Высшие жирные кислоты
Активная липаза
(фосфорилирована)
Протеинкиназа
(неактивная) '
Протеинкиназа
(активная)
АДФ
АТФ
Неактивная липаза
(дефосфорилирована)
цАМФ
Аденилат-
циклаза
(активируется
гормонально)
РР
АТФ
Распад глицерина и высших жирных кислот. В обмене жиров
характерно широкое использование продуктов их распада для ресинтеза.
Поэтому значительная часть 0-моноглицеридов, глицерина и свободных выс-
ших жирных кислот, освобождающихся при гидролизе триглицеридов, ис-
пользуется для ресинтеза триглицеридов же, но несколько иного состава
и строения, характерного для того или иного организма (если для этого
используются пищевые жиры) или органа (если идет перестройка жиров
в пределах организма).
Так как новообразованные жиры неизбежно отличаются от распавшихся
триглицеридов по строению и соотношению остатков высших жирных кислот
(в соответствии с их видовой или тканевой специфичностью), то часть выс-
ших жирных кислот и некоторая доля глицерина подвергаются дальнейшей
деструкции.
388
CH2-O-CO—R
CH— O—CO—R
CH3
. XCH2 CH,
•. \ /
HN ZCH
4 Ch
r-co{-o-ch2
*
OH
co
SA- • • "OOC~CH2—CH асп(глу)
CO
иле x
CH2
NH
NH-CH-CO
CO
cep
HN —CH
гис
CH2 CH)
NH. CH
CH2—O— CO—R'
CH—O—CO—R
I
CH2OH
HOH---X
R—CO i
CH2
CH
4 CO—NH—CH— co
co
l=N4 "OOC— CH2— CH
---HN,
CH3 —CH2/CH3
CH
. . . NH—CH ОИ
иле 4 a, CH2
Рис. 122. Механизм гид- гп
релиза триглицеридов г,н
(пояснения в тексте) сю
NH
co
HN-CH^
асп(глу)
Етицерин независимо от того, поступил ли он на ресинтез жиров или будет
претерпевать дальнейший распад, прежде всего фосфорилируется. Донором
остатка фосфорной кислоты в этой реакции служит АТФ. Процесс ускоряется
соответствующей фосфотрансферазой:
сн2—он
Глицеролкиназа
СН—ОН+АТФ------------------
1 М=217000
I (4 x 55000)
СН2—ОН
СН2ОН
I
СН—ОН он+АДФ
СН2—О—Р=О
I
он
Глицерин
«-Глицерофосфат
Глицерофосфат в основном идет на синтез новых молекул триглицеридов,
но часть его окисляется с образованием диоксиацетон-фосфата:
о
389
сн2—он
:н—он
НАД+ НАДН+Н+
Г лицерол-3-фосфвт-
дегидрогеназа;
М-68000
(2X34000)
г
:н2—он
=о
н2—о—
“ОН
/ОН
Р=о
Х)Н
а-Глицерофосфат
Диоксиацетонфосфат
Диоксиацетонфосфат изомеризуется в 3-фосфоглицериновый альдегид, ко-
торый затем вступает в обменные реакции, рассм этренные ранее (см. гл. VIII).
Наибольший интерес в процессах обмена продуктов гидролиза триглице-
ридов представляет судьба высших жирных кислот. Первые гипотезы относи-
тельно механизма их распада были высказаны в начале нашего столетия
(Ф. Кнооп, 1904). В дальнейшем они были уточнены и развиты благодаря
работам лабораторий Ф. Линена, Д. 1рина, С. Очоа, Г. Ларди и А. Лениндж -
ра. Современные данные по этому вопросу сводятся к следующему. Считают,
что высшие жирные кислоты разрушаются преимущественно путем Р-окисле-
ния. Ненасыщенные высшие жирные кислоты (олеиновая, линолевая, линоле-
новая и др.) предварительно восстанавливаются до предельных кислот. Окис-
ление предельных высших жирных кислот осущ ствляется ступенчато, путем
отщепления от их молекул двууглеродных фрагментов. Все реакции многоста-
дийного окисления ускоряются специфическими ферментами, причем начиная
с третьей фазы (см. ниже) они собраны в виде метаболона с М=260 000 Да.
Первая фаза распада высших жирных кислот заключается в активировании
их путем образования соединения с коэнзимом А (КоА), содержащего макро-
эргическую связь. Последняя, видимо, сп собствует более гладкому протека-
нию реакций окисления образовавшегося соединения, которое называю- ацил-
коэнзимом А (ацил-КоА). Взаимодействие высших жирных кислот с КоА
ускоряется специфическими лигазами—ацил-КоА-синтетазами трех видов спе-
цифичных соответственно для кислот с коротким, средним и длинным углево-
дородными радикалами. Они локализованы в мембранах эндоплазматической
сети и в наружной мембране митохондрий. По-видимому, все ацил-КоА-
синтетазы являются мультимерами; так, фермент из микросом печени имеет
М = 168 000 и состоит из 6 идентичных субъединиц с М=28000.
Уравнение реакции активирования высших жирных кислот перед их окисле-
нием таково:
сн, о о
СпН,,—СООН+АТФ + HS—СН,—CHa—NH-CO—СН,-€Н,—NH—СО-СН-С—СН,—О-
Стеарииовая ОН
Стеариновая
кислота
>н
Ацил Коэна и-А-
сннтетаэа
Коэнзим A (HSKoA)
Mg ,К
ОН <j>H
НО—Р—О—Р—ОН
+ АМФ +
НО'
нс> н
о о
Пирофосфат
СН,
+ C„HaCO~S—CHf-CH,—NH—СО-СН,—сн2—N н-со—сн—с
он сн.
О
О
Стеарил-КоА
.О.
IV ' HlZ
HOXH NeefH
о—Р—о он
НОХ
390
Образующийся в этой реакции пирофосфат энергично расщепляется до
Н3РО4 при участии пирофосфатазы, что обеспечивает смещение равновесия
всего процесса вправо.
Вторая фаза распада высших жирных кислот состоит в окислении ацил-
КоА при посредстве ацил-КоА-дегидрогеназы, содержащей флавинадениндину-
клеотид (ФАД, см. с. 120) в качестве кофермента:
С15Н31—СН2—СН2—CO~SKoA
Стеарил-КоА
ФАД ФАД-Н2
Ацил-КоА-
дегилрогеназа
С15Н31\ /И
^с=с'
НХ XCO~SKoA
а, Д-Дегидростеарил -КоА
Существует, по меньшей мере, три ацил-КоА-дегидрогеназы, предпо-
читающие короткие, средние и длинные ацильные радикалы соответст-
венно.
Третья фаза окисления высших жирных кислот состоит в присоединении
молекулы воды по месту двойной связи дегидроацил-КоА. Эта реакция уско-
ряется соответствующими гидролназами. Так как присоединение воды (гид-
ратация) идет по двойной связи (двойная связь условно обозначается частицей
ен), то эти ферменты по современной номенклатуре называют еноил КоА-
гидратазами. Один из них специфичен к цисформам дегидроацил-КоА, дру-
гой—к транс-формам:
С15Н31—С—Н
II
Н—С—CO~SKoA+H2O
Транс-дегидростеарил-КоА
С15Н31—СН—СН2—CO~SKoA
ОН
L-p-Оксистеврил-КоА
Четвертая фаза распада высших жирных кислот заключается в еще одном
окислении путем отнятия двух атомов водорода от Р-углеродного атома
(отсюда и весь рассматриваемый здесь механизм носит название Р-окисления).
Как и во второй фазе про jecca, снятие атомов водорода осуществляется
оксид редуктазой, но с НАД+ в качестве кофермента. Фермент специфичен
лишь к L-форме Р-оксиацил-КоА:
С15Н31—СН—СН2—CO-SKoA
он
1--/?-Оксисте рил-КоА
Р-Оксивцил-КоА-
дегидрогенвза
НАД+ НАДН+Н+
CijHji—С СН2—CO~SKoA
Р -К етостеарил -КоА
Наконец, последняя, пятая фаза распада сводится к переносу предобразо-
ванной в молекуле Р-кетоацил-КоА новой ацильнои группировки на молекулу
КоА. Этот процесс ускоряется соответствующей ацилтрансферазой, которую
предпочитают называть тиолазой, так как сама реакция, по существу, представ
ляет расщепление С—С-связи с npi соединением по месту разрыва элементов
HS-группы тиолиз):
391
Рис. 123. «Спираль» окисления высших жирных кислот
3-Кетоацил-КоА тиолаза
HSKoA+С J 5Н з j—СО—СН 2—СО ~ SKoA
Прообразованный
остаток паль-
митиновой
кислоты
С15Н31—CO~SKoA+CH3—CO~SKoA
Пальмитил-КоА
Ацетил-КоА
В результате описанных выше реакций молекула высшей жирной кислоты
(стеариновой в нашем примере) укорачивается на два углеродных атома
и образуются пальмитиновая и уксусная кислоты в виде производных
КоА (пальмитил- и ацетил-КоА). Этот процесс многократно повторяется
(рис. 123). Окончательным продуктом Р-окисления высших жирных кислот
с четным числом углеродных атомов является ацетил-КоА, а с нечетным—
пропионил-КоА.
Если бы ацетил-КоА накапливался в организме, то запасы HSKoA скоро
исчерпались бы и окисление высших жирных кислот остановилось. Но этого не
происходит, так как КоА быстро освобождается из состава ацетил-КоА. К этому
приводит ряд процессов: ацетил-КоА включается в цикл трикарбоновых и дикар-
боновых кислот (см. рис. 117) или весьма близкий к нему глиоксилевый цикл (см.
ниже), или, наконец, ацетил-КоА используется для синтеза полициклических
спиртов (стеролов) и соединений, содержащих изопреноидные группировки и т. п.
P-Окисление высших жирных кислот протекает в митохондриях. Естествен-
но, что, поскольку в них же локализованы ферменты дыхательного цикла,
ведущие передачу атомов водорода и электронов на кислород сопряженно
с окислительным фосфорилированием, Р-окисление высших жирных кислот
может явиться источником энергии для синтеза АТФ.
В некоторых случаях высшие жирные кисло ы представляют единственные
вещества, окисление которых служит источником энергии для окислительного
{юсф рилирования (биосинтез белка в шелкоотделительной железе шелко-
пряда, полет насекомых).
В бесструктурной части клеточного содержимого тоже есть ферментные
системы, способные окислять высшие жирные кислоты. Окисление, идущее
392
здесь, осуществляется по а-углеродному атому и называется а-окислением.
В нем принимают участие Н2О2 и фермент—пероксидаза:
Пероксидам жирных кислот
СН3-(СН3)хэ-СНг-СООН 4- 2НА-------------------------►
(пальмитат: Н,О,-оксидо>
редуктаза)
Пальмитиновая кислота
------► со»+ СН3-(СНа)1я—+ зн,о
Пентадециловый
альдегид
Альдегид высшей жирной кислоты окисляется при посредстве дегидрогена-
зы в высшую жирную кислоту, и процесс повторяется снова:
о .о
Альдегиддегидрогеназа
СН3—(СН2)В—+ Н2О-------------- — ----СН3—(СН2)В—с.
хн
НАД(Ф)‘Г НАД(Ф)Н+Н
Пентадециловая
кислота
Так укорачиваются цепи высших жирных кислот, содержащих в своем
составе от 15 до 18 углеродных атомов. Указанный дополнительный путь
а-окисления высших жирных кислот характерен только для растений. Сущест-
вует также ферментная система, обеспечивающая со-окисление, т. е. окисление
по CHj-группе радикала высших жирных кислот. Она изучена в микросомной
фракции печени и у микроорганизмов. Сначала под действием монооксигена-
зы (см. гл. X) возникает ю-оксикислота, а затем—дикарбоновая высшая жир-
ная кислота. Последняя укорачивается с любого конца посредством реакций
Р-окисления.
Обмен ацетил-КоА. Выше отмечалось, что ацетил-КоА быстро
расходуется, высвобождая свободный HSKoA. Следовательно, в реак-
циях Р-окисления высших жирных кислот HSKoA и его ацильные про-
изводные, будучи коферментами, выполняют каталитическую функ-
цию.
Одним из процессов, в результате которого осуществляется регенера-
ция HSKoA, является образование ацетоуксусной кислоты. Этот путь
преобразования ацетил-КоА широко представлен в митохондриях пече-
ни. Сначала две молекулы ацетил-КоА конденсируются с образовани-
ем Р-кетобутирил-КоА и выделением одной молекулы свободного
HSKoA:
Ацетал-КоА:
CH3-CO~SKoA+CH3-CO~SKoA --------------♦
/ ацетнл-КоА-
ацетилтранс-
фераза
----> HSKoA+CHj-CO-CH2-CO~SKoA
Далее HSKoA высвобождается из состава Р-кетобутирил-КоА. Известно
несколько реакций, приводящих к такому результату. Среди них преобладает
реакция, в которую вовлекается еще одна молекула ацетил-КоА:
393
I сн
CH2—CO~SKoA + CH3—CO^SKoA + НОН £|(£.имети^глУтаР^1
' '“v—/ коэнзим А-синтаза
Д-Кетобутирил-КоА Ацетил-КоА
---► НООС—СН2—-С—СН2—-CO~SKoA + HSKoA
ОН
Д-Окси-Д-метилглутарил- КоА
Образовавшиеся в результате реакции конденсации р-окси-Р метилглута-
рил-КоА п едставляет весьма важное соединение, так как из него может
синтезироваться мевалоновая кислота—ключевое соединение в синтезе стеро-
лов и изопреноидов (см. с. 402). Однако в данном случае (т. е. в митохондриях
печени) Р-окси-Р-метилглутарил-КоА распадается на ацетоуксусную кислоту
и ацетил-КоА:
СН3
। _ Оксиметилглутарил-КоА-лиаза
HOOC-CH2-C-CH2-CO~SKoA -------------------—----------»
I
он
Р-окси‘р-метилглутарил-КоА
-------> CH3-CO-CH2-COOH+CH3CO~SKoA
Ацетоуксусная кислота Ацетйл-КоА
В результате перечисленных выше реакций из двух молекул ацетил-КоА
синтезируется одна молекула ацетоуксусной кислоты и высвобождаются две
молекулы HSKoA.
Другой распространенный путь обмена ацетил КоА—это взаимодействие
с енольной формой щавелевоуксусной кислоты с образованием цитрил-КоА,
т. е. вступление в цикл трикарбоновых и дикарбоновых кислот. При гидролизе
цитрил-КоА высвобождается HSKoA, а лимонная кислота обменивается далее
в соответствии с ранее рассмотренной схемой (см. рис. 117). Такой путь
обмена ацетил-КоА характерен для митохондрий подавляющего большинства
тканей—почек, мышц и т. Д., за исключением печени.
Известно еще много процессов, которые ведут к высвобождению HSKoA
из состава ацетил-КоА. Ацетил-КоА является универсальным донором аце-
тильных групп для реакций ацетилирования. Существует более десяти специ-
фических ацетилтра сфераз обеспечивающих ускорение реакций переноса аце-
тильных остатков (синтез ацетилхолина, N-ацетилглюкозамина и т. п.) Во
всех случаях выделяется свободный HSKoA.
Высвобождение HSKoA из состава ацетил-КоА может сопровождаться
накоплением щавелевоуксусной кислоты. Это происходит в том случае, когда
ацетил-КоА обменивается при посредстве так называемого глиоксилевого
цикла. В значительной мере химические процессы, протекающие при о уществ-
лении глиоксилевого цикла, совпадают с таковыми цикла дикарбоновых
и трикарбоновых кислот (см. рис. 117). Все идет одинаково, вплоть до об-
394
разования изолимонной кислоты. Однако в глиоксил вом цикле изолимонная
кислота расщепляется на янтарную и глиоксилевую кислоты:
соон кюн к—соон - 1 см^ Изопит0ат-лиа1а соон lz -* С + \н соон сн, СООН
соон
Изолимонная Глиоксилевая Янтарная
кислота кислота кислота
Янтарная кислота тем же путем, как и в цикле дикарбоновых и трикар-
боновых кислот, превращается в щавелевоуксусную кислоту. Тлиоксилевая же
кислота конденсируется с новой молекулой ацетил-КоА и образует в конечном
счете свободный HSKoA и яблочную кислоту:
СООН
Lo
СН,—СО—SKoA + С .
^Н фермент
Глиоксилевая
кислота
соон соон
снон снон
I Н,0 I + HSKoA
СН, ч сн,
CO-SKoA * СООН
Яблочная
кислота
Последняя, дегидрируясь, дает начало одной молекуле щавелевоуксусной
кислоты. Таким образом, при помощи глиоксилевого цикла ацетил-КоА пре-
вращается в щавелевоуксусную кислоту и свободный HSKoA. Этот процесс
имеет огромное значение для обеспечения в организме синтеза углеводов за
счет распадающихся высших жирных кислот.
Обмен пропионил-КоА. Пропионил-КоА, являющийся конечным
продуктом Р-окисления высших, жирных' кислот с Нечетным числом углерод-
ных атомов, превращается в сукцинил-КоА путем двух последовательных
реакций:
СН3—СН2—CO~SKoA + со2
АТФ АДФ+ЩРО,
Пропионил-КоА-
карбоксилаи
(биотинпротеин)
СН3— СН—CO~SKoA;
СООН
Метилмалонил-КоА
I f \ f----------“Ч Метилмалонил-КоА-
СН3—СН—CO~SKoA ——----------------------
3 | КоА-кароонилмутата
(Bp -протеин)
СООН
НООС—СН2—СН2—CO-SKoA
Сукцинил-КоА
Далее сукцинил-КоА утилизируется через цикл трикарбоновых и дикарбо-
новых кислот.
Синтез высших жирных кислот. Долгое время считали, что синтез
высших жирных кислот идет путем обращения реакции Р-окисления высших
жирных кислот. Однако после того как было обнаружено, что для его осущест-
вления необходим не только ацетил-КоА, но и СО2 (из которых при посред-
стве АТФ-зависимой реакции возникает малонил-КоА), а сам процесс ускоря-
ется синтетазой высших жирных кислот, локализованной в растворимой фрак-
ции клетки, эта точка зрения была оставлена. В 60-е годы огромную роль
395
в расшифровке механизма биосинтеза высших жирных кислот сыграли работы
Ф. Линена и сотр.
Современные представления о биосинтезе высших жирных кислот в ор-
ганизме представлены на схеме 10.
Начальный этап биосинтеза высшей жирной кислоты, приводящий к
синтезу малонил-КоА, ускоряется полифункциональным ферментом
(М=225 000 Да), содержащим домен биотинкарбоксилазы, биотин-карбоксил-
проводящий домен и домен транскарбоксилазы. Первый домен обеспечивает
ускорение реакции карбоксилирования биотина (рис. 124), который через ра-
дикал лизина присоединен ко второму, биотин-карбоксилпроводящему доме-
ну. Обладая высокой степенью подвижности, карбоксилированный биотин
переносит СО2 в активный центр третьего домена—транскарбоксилазы, кото-
рый снимает с него СО2 и непосредственно передает его на ацетил-КоА,
образуя малонил-КоА:
СО2 + СН3—CO~SKoA + АТФ + Н2О
Ацетйл-КоА
Ацетил-КоА-карбоксилаза
(биотинпротеии); Mg2+
СН2—СО~5КоА
АДФ+Н3РО4
СН3—CO—SKoA
Ацетил-КоА \
СО2
HS КоА
СООН
„ Малонил-КоА
Конденсирующий
фермент
НАДФН+Н+
СН3—СО—СН2—CO~SKoA
.30
CHj—СН—СН2—CO~SKoA
ОН
Р -Оксибутирил-КоА
Р -Оксиацил -КоА-
дегндратаэа
Н2О
СНз—СН=СН—CO-SKoA
Кротонил-КоА
Еноилредуктаза
CHj—СН2—СН2—CO—SKoA
Бутирил-КоА
Схема 10. Механизм биосинтеза высших жирных кислот
Многократное повторение наращивания радикала на два ато-
ма углерода приводит к синтезу кислот, содержащих 16 и бо-
лее углеродных атомов.
396
остатков
Рис. 124. Механизм биосинтеза малонил-КоА (пояснения в тексте)
В мономерном состоянии ацетил-КоА-карбоксилаза не активна и при-
обретает способность карбоксилировать CH3CO~SKoA только после
соединения мономеров в нитевидный олигомер с молекулярной массой
в несколько сотен миллионов и длиной около 500 нм. Процесс олиго-
меризации регулируется аллостернчески присоединением лимонной кис-
лоты.
Кроме того, активность ацетил-КоА-карбоксилазы регулируется ее фосфо-
рилированием (снижение) и дефосфорилированием (повышение). Таким обра-
зом, интенсивность работы цикла трикарбоновых и дикарбоновых кислот
и уровень протеинкиназных и протеинфосфатазных реакций предопределяет
объем биосинтеза высших жирных кислот, последующие стадии которого
осуществляются при посредстве второго полнфункционального энзима—син-
тетазы высших жирных кислот.
Этот комплекс у высокоорганизованных форм (млекопитающие, птицы,
насекомые) характеризуется М=400 000—560000, а у низкоорганизованных
(микобактерии, низщие грибы, жгутиковые)—1,4-0б—2,3-10б. В нем сосре-
доточены все каталитические активности, необходимые для обеспечения мно-
гоступенчатого биосинтеза высших жирных кислот, а также ацилпроводящий
домен, функция которого состоит в передвижении ацильной группы от
одного субдомена к другому в строгом соответствии с химизмом этого
процесса. Представление о работе синтетазы высших жирных кислот дают
рис. 125 и 126.
В первом случае (рис. 125, синтетаза из печени цыпленка) каждая его
полипептидная цепь длиной около 2300 аминокислотных остатков образует
3 домена и 8 субдоменов, с каждым из которых связана определенная
функция. Однако один из субдоменов, а именно—обладающий Р-кетоацил-
синтетазной активностью, работает только в паре с другой такой же полипе-
птидной цепью, расположенной по отношению к первой по правилу «голова
к хвосту». Он перебрасывает ацетильную (первый цикл синтеза) или аци-
льную (последующие циклы) группу со своего остатка цистеина (рис. 125) на
малонильный остаток, закрепленный на HS-группе пантетеиновой «руки»
Домен I
элоигащ цепи)
Домен II
(восстановление)
Домен Ш
(тиолиз)
Рис. 125. С роение и механизм действия синтетазы высших жирных кислот пене
ницыпленк :
цифрами обозначены молекулярные массы (в кДа) субдоменов. Остальные пояснения—в тексте
ацил-переносящего субдомена соседней субъединицы. Возникший Р-кего-
ацильный остаток при помощи той же пантотеиновой руки перемещается по
остальным трем субдоменам домена II (восстанавливающего Р-кетоацил
в ацил). Далее ацетил (ацил) трансфе азный домен элонгационного домена
I посылает эту ацильную группу на HS-группу остатка цистеина 3-кетоацил-
синтетазного субдомена и начинается новый цикл удлинения цепи, но уже на
соседней субъединице синтетазы высших жирных кислот. По достижении
ацильным радикалом длины в 16 атомов углерода он отщепляется тио-*
эстеразой в виде ацил КоА.
Во втором случае (синтетаза из дрожжей) принцип согласованной и вза-
имозависимой работы субъединиц синтетазы высших жирных кислот остается
в силе (рис. 126, пс яснения в подрисуночной подписи).
398
Рис. 126. Структура син*
тетазы высших жирных
кислот дрожжей
Фермент представляет а6 рб комп-
лекс с М=2469000 Да. Каждая из
субъединиц обладает своим набо-
ром каталитических активностей.
АСР—ацилпереносящий домен,
обладающий пантотеийовой «ру-
кой» [обозначена SH (P)J. SH—
(С)—цистеиновый остаток Р-кето-
ацилсинтетазы, с которого вдет
перенос ацетильной (первый цикл)
и ацильных (последующие циклы)
групп на малонильный остаток,
закрепленный на HS-группе панто-
теиновой «руки». Вверху—сверну-
тая; внизу—развернутая форма
фермента. Механизм действия
полностью аналогичен таковому
синтетазы высших жирных кислот
из печени цыпленка
Синтез триглицеридов. Из глицерина и высших жирных кислот при
каталитическом воздействии липазы in vitro удается получить триглицерид.
В связи с этим полагали, что и in vivo липаза может проявлять не только
гидролитическое, но и синтезирующее действие и что с ее помощью могут
возникать триглицериды путем обращения реакции гидролиза. В последние
годы доказана принципиаль о иная схема биосинтеза триглицеридов, в ко-
торой отправными веществами служат ацил-КоА и фосфоглицерин, а фер-
ментами—ацилтрансферазы. Учитывая общую тенденцию в эволюции на-
ших представлений в сторону совершенно четкого разграничения и опреде-
ленного различия путей распада и синтеза основных классов органических
соединений в биологических объектах (см. синтез и распад белков, нукле-
иновых кислот, полисахариде в), несомненно, что синтез жиров путем об-
ращения гидролиза вряд ли широко представлен в природе и что главный
путь новообразования триглицеридов заключается в осуществлении реакций
трансацилировайия.
Как упомянуто выше, исходными веществами при синтезе триглицеридов
по редством реакций трансацилирования являются а-фосфоглицерин и разно-
образные ацил-КоА. Первый возникает при фосфорилировании глицерина или
при восстановлении фосфодиоксиацетона. Прямое фосфорилирование глице-
рина характерно для почек животных, а также микроорганизмов. Восстановле-
ние ф эсфодиоксиацетона идет в мышцах, слизистой кишечника и т. п. Вторые
синтезируются либо путем активирования высших жирных кислот, либо путем
новообразования из ацетил-КоА (см. выше). Вначале посредством реакций
трансацилирования синтезируется фосфатидная кислота:
399
он
2CltH31-CO~SKoA 4- Н0-СН,-СН(ОН)-а^-0-1’=О
он
Пальштсл-КоА «-Глицерофосфат
Глшерофосфст-
•цялтрансфераза
СН,—О—СО—С,»НМ
-----► СН-О-СО-СцН,! + 2HSKOA
СН,—О—Р=О
1,2-Дипальмитил-З-фосфоглнперин (фосфатидная кислота)
При участии фосфатазы фосфатидная кислота гидролизуется с образовани-
ем диглицерида и фосфорной кислоты:
СН,—О—СО—СцН,, Фосфатядвт-
СН—О—СО—С15Н„ + Н,О фосфогцдрола»*
СН,—О—Р«=О
НО ОН
^г-Дапвльиитнл-З-фосфоглщерии
СН,-О-СО-С]6Н31
СН-О-СО-СцН,! + Н,РО*
£н,-он
Дипалыштаи
Диглицерид вступает снова во взаимодействие с ацил-КоА и образуется
триглицерид. И эта реакция ускоряется трансацилазой:
СН2— О — СО — С15Н31 Дяглиперид-
C15H3I - СО ~ SKoA + СН - О - СО - С15Н31 ацилтранс^раза
СН2ОН
Дяпальмитин
СН2-О-СО-С15Нз1
I
-------> CH-O-CO-CjjHn+HSKoA
I
СН2-О-СО-С15Нз1
Трнпальмшин
Ферменты, ускоряющие синтез триглицеридов в соответствии с приве-
денными выше уравнениями, найдены в печени, слизистой оболочке кишеч-
ника, жировой ткани и т. п. Интересной особенностью всех указанных
ферментов является их липопротеиновая (за исключением глицеролкиназы)
природа. Они ведут синтез триглицеридов на мембранах эндоплазматиче-
ской сети клетки. По мере своего возникновения триглицериды мигрируют
и поглощаются жировыми включениями клетки. Из тканей, активно син-
тезирующих триглицериды (например, печень), они переходят в ткани, где
нет активного синтеза (например, кровь). В организме животных существу-
ет обычно несколько жировых депо с медленно обменивающимися тригли-
церидами.
400
Механизм биосинтеза триглицеридов через ф сфатидные кислоты в качест-
ве мета олитов не явля :тся единственным. В слизистой оболочке кишечника
синтез триглицеридов идет из 0-моноглицеридов при посредстве весьма актив-
ной моноглицеридтрансацнлазы:
сн2-он
I Моноглицерид-
СН—О—CO-R'+R"—CO~SKoA----------------------
I ацилтрансфераза
СН2ОН
р-Моноглицерид
CH2-O-CO-R"
СН -О -СО - R'+HSKoA
СН2ОН
Диглицерид
Само собой разумеется, что диглицерид превращается далее в триглицерид
при каталити еском участии диглицеридтрансацилазы (см. предыдущее урав-
нение). Моноглицеридный тип биосинтеза энергетически вдвое выгоднее фос-
фатидного пути. Кроме того, недавно обнаружена диоксиацетонфосфатацил-
трансфераза, которая может открывать еще один путь биосинтеза ацилглице-
ринов.
Обмен стеридов. Вступая на путь распада, стериды сразу же гид-
ролизуются на жирную кисло у и стерол. Поскольку стериды представляют
собой в химическом отношении сложные эфиры высших жирных кислот
и полициклических спиртов (стеролов), то реакция гидролиза ускоряется
холестеролэстеразой, действующей также на сложные эфиры других стеролов
(см. с. 381).
Холестеролэстераза выделена из поджелудочной железы ряда животных
и человека; она представлена мономером с М = 65000—69000, склонным
к олигомеризации (М = 300 000—800000).
Что касается высших жирных кислот, высвобождающихся из состава
стеридов при гидролизе, то они далее могут либо использоваться для
ресинтеза липидов, в том числе и стеридов, либо распадаться до ацетил-
КоА и потом—до СО2 и Н2О. Поэтому рассмотрим, как протекает далее
обмен стеролов—второго компонента, образующегося при гидролизе сте-
ридов.
Распад стеролов. Та часть стеролов, которая не используется для ресин-
теза стеридов, подвергается видоизменению. Простейшее видоизменение
состоит в восстановлении стеролов по двойным связям. Так, холестерол
у человека и высших животных превращается в дигидрохолестерол (холеста-
нол), который в виде конформера (копростанола—см. с. 380) выводится из
организма:
Более сложный характер носит видоизменение стеролов путем окисления.
Сначала появляются ОН-группы в положениях 7 и 12 циклопентанопергид-
рофенантренового цикла, а затем окисляется боковая цепь, в которой возника
ет СООН-группа (положение 24). В результате образуются холевые кислоты.
Подсчитано, что до 80% холестерола превращается в печени в различные
401
холевые кислоты. При более полном окислении стеролов могут Возникну ь
стероцдные гормоны (см. с. 444). Таким образом, часть стеролов превращ ВТ-
ся в процессе окисления в различные с единения, выполняющие в организме
важные функции.
Синтез стеролов и стеридов. Механизм биосинтеза стеролов долгое
время оставался загадочным, хотя давно было известно, что стеролы бес-
препятственно синтезируются у большинства органических форм (исклю-
чение составляют, например, насекомые). Лишь применение метода меченых
атомов позволило расшифровать этот оказавшийся довольно сложным
процесс, основные этапы которого, видимо, совпадают у самых различных
организмов.
Синтез стеролов осуществляется из ацетил-КоА в качестве исходного
вещества. Первые стадии биосинтеза совпадают с реакциями, которые были
описаны выше при рассмотрении обмена ацетил-КоА. Напомним, что в ре-
зультате двух последовательно протекающих при этом реакций из трех моле-
кул ацетил КоА образуется одна молекула Р-окси-Р-метилглутарил-КоА. Это
соединение ферментативным путем восстанавливается в мевалоновую кислоту;
восстановление идет по макроэргической связи и сопровождается выделением
свободно о HSKoA:
Оксиметилглутарил-КоА- Т?3
« 10 7 Л редуктаза « V 1 ® .
НООС—CH2—С—CHj—CO~SKoA ---------—- НООС—CH2 —CH2— i2OH +HSKoA
0Н 2НАДФН+Н+ 2НАДФ+ ОН
fl-Окси-Р-метилглутарил-КоА Мевалоновая (0, в-диояси-Р-метилвялв-
риановая кислота
Оксиметилглутарил-КоА-редуктаза из микросом печени крысы имеет
М = 32000, но молекулярная масса втрое выше (97092 Да, 887 аминокислот-
ных остатков) у фермента из яичников китайского хомячка Обе активны
только в дефосфорилиро анном состоянии, тогда как протеинкиназ ая реак-
ция их полностью инактивирует. Этот фермент считают ключевым при биоси-
нтезе стеролов, так как он успешно конкурирует за субстрат с ферментами
других метаболических путей.
Мевалоновая кислота дважды фосфорилируется по 8-гидроксильн й груп-
пе. Донором остатков фосфорной кислоты в этих реакциях служит АТФ.
Процесс ускоряется специфическим фосфотрансферазами:
сн3 СНз 9Н
« 1?| V 9 Мсвалонаткииаза I _ |
НООС—СН2—С—CH2—СН2ОН ------** НООС—CH2—С—CH2—СН2—О—Р«=О
ОН АТФ АДФ он он
Пирофосфомевалоновая кислота декарбоксилируется. Одновременно
Осуществляется реакция дегидратирования и образуется изопентенилпиро -
фосфат:
402
Пирофосфомевалоиат-
декарбоксилаза
(дегидратирующая)
АТФ АДФ+Н3РО4
CHj
о
ОН ОН
iHJOOC—CHj—С—СН2—СН2—О—Р—О—Р—ОН
(fc)
Пирофосфомевалоновая кислота
СН3 ОН ОН
CH,==(L-CH,—СН2—О—I—О—Р—ОН + со2
II II
о о
Изопеитенилпирофосфат
Изопеитенилпирофосфат при участии фермента—изопентенилпирофосфат-
изомеразы превращается в диметилаллилпирофосфат (см. с. 135).
Из двух вышеупомянутых соединений: изопентенилпирофосфата и диметил-
аллилпирофосфата—и идет синтез стеролов. Сначала указанные соединения
образуют геранилпирофосфат:
Ди мети лалли л-
траисфераза
Диметилаллилпирофосфат
Изопеитенилпирофосфат
<рн ОН , ОН <j>H
НО—р—О—Р—ОН + СНз—С=СН-€Н,4-СН2—С=СН-СН3—О-Р—о-p—ОН
8 8 к । Ч. 8 8
Пирофосфат Ге ранил пирофосфат
Эта реакция ускоряется ферментом—диметилаллилтрансферазой, обеспечи-
вающей перенос диметилаллильного радикала на раскрывающуюся двойную
связь молекулы изопентенилпирофосфата. Одновременно выделяется пирофо-
сфат, получающий атом водорода от близлежащей метиленовой группы. Этот
же фермент катализирует реакцию переноса радикала геранила от геранил-
пирофосфата к следующей молекуле изопентенилпирофосфата:
Ди метал аллил-
трансфераза
Н4Р2О7
Пирофосфат
Фарнеэилпмрофосфат
403
Указанные реакции трансалкилирования поддерживаются сопутствующим
гидролизом пирофосфата при участии пирофосфатазы:
ОН ОН ОН
HO-i-O-j>-OH+H2O 2ГФОСФСТ' 2НО-t-ОН
II фосфогидролаза ц
О о 6
Две молекулы фарнезилпирофосфата соединяются по месту присоединения
пирофосфатных группировок с отщеплением последних. Источником атомов
водорода при образовании молекул пирофосфата является НАДФН. Уравне-
ние данной реакции можно представить в виде следующей схемы:
он он
Н(СН2—с=сн-сн2)3-о-р-о—р-он
сн3 <1 он
Ф арнезилл н рофосфат
он он
HO-g-O-I-O-(СН2-СН=С-сн2)3н
о о сн3
Фарнезилпирофосфат
CHj-C=CH-CH2|cH2-C=<;H-CH2|CH2-C=CH-CH2jCH2-CH=C-CH2icH2-CH=C-CH2+CH2-CH=C-CHj
СН3 СН2 СНз СНз сн3 сн3
Сквален
В результате реакции получается непредельный углеводород—сквален, соста-
вленный из 6 изопреноидных группировок. Процесс ускоряется сквален-синтазой,
изучение которой затруднено ее тесной ассоциацией с эндоплазматической сетью
и ее фосфолипидной составляющей. Однако выяснено, что при извлечении фер-
мента из микросомальной фракции без применения детергентов (с использовани-
ем ультразвуковой дезинтеграции и т. п.) его М=450 000, а с использованием
детергента—54500. Сквален-синтаза обладает двумя центрами связывания фар-
незилпирофосфата, расположенными, возможно, на разных субъединицах.
Молекула сквалена легко принимает пространственную конфигурацию,
близкую к пространственной конфигурации стеролов, и столь же легко окисля-
ется по крайней двойной связи с образованием сквален-2, 3-оксида с помощью
сквален-эпоксидазы, относящейся к подклассу моноксигеназ (см. с. 419). Про-
тонирование эпоксидной группы приводит к сдвигам электронной плотности
в системе двойных связей сквалена и замыканию (показано стрелками) шести-
членных и пятичленных циклов, характерных для стеролов. Схема указанного
перехода скваленоксида в стерол представлена ниже:
404
Ее особенность состоит в том, что при замыкании кольца С стерола
неизбежно происходит переброска СН3-группы из положения 8 в положе-
ние 13 и элиминирование протона от 9-го углеродного атома цикла.
Так протекает процесс в эндоплазматической сети клеток печени. Что
касается растений и других организмов, то циклизация сквален-2,3-оксида
идет у них с помощью других циклизующих ферментных систем и с иными
конечными продуктами, нежели ланостерол.
Путем преобразования ланостерола и других первичных продуктов цикли-
зации возникают разнообразные индивидуальные стеролы, характерные для
животного и растительного царства. Преобразование это многоступенчато;
например, только удаление двух метильных групп при 4-м углеродном атоме
кольца А (путем их окисления и последующего декарбоксилирования) осуще-
ствляется в 12 стадий.
Биосинтез стеридов протекает путем переноса остатка высшей жирной
кислоты от молекулы ацил-КоА на место водорода ОН-группы стерола при
каталитическом воздействии холестерол-ацилтрансферазы:
Источником ацильных групп при биосинтезе стеридов может также слу-
жить фосфатидилхолин. Так, например, синтезируются холестериды лимфы
и плазмы крови у человека при участии фосфатидил» лин-холестерол-ацил-
трансферазы.
В заключение подчеркнем, что диметилаллилпирофосфат и изопентенил-
пирофосфат служат универсальными исходными соединениями для биосин-
теза ряда других полиизопреноидов—каротиноидов, каучука и т. п.
Обмен фосфатидов. Пути распада фосфатидов. Современные пред-
ставления о путях распада фосфатидов в организме основаны главным об-
разом на тщательном изучении превращений, которые свойственны фосфати-
дам вне организма при воздействии на них теми или иными ферментами.
Поэтому когда говорят о путях распада фосфатидов, то имеют в виду скорее
возможные, чем действительные, пути их деструкции. Непосредственно в био-
логических объектах эти пути исследованы еще недостаточно. Однако извес-
тно, что время полужизни фосфатидилглицерина и дифосфатидилглицерина
у бактерий составляет 1 и 2 ч соответственно, а период полужизни фос-
фоинозитидов и сфингомиелинов в мозге крысы—12,5 и 40 суток соответст-
венно.
Фосфатиды распадаются на составляющие их структурные единицы—
высшие жирные кислоты, фосфорную кислоту, азотистые основания и глице-
рин—гидролитическим путем. Реакции гидролиза, приводящие к разрушению
сложноэфирных связей в молекуле фосфатида, ускоряются ферментами —
фосфолипазами, которые относятся к подклассу эстераз (класс гидролаз).
В зависимости от того, гидролиз какой из четырех имеющихся в молекуле
фосфатида сложноэфирных связей ускоряет фосфолипаза, ее характеризуют
как фосфолипазу А, В, С или D (схема 11).
Из схемы видно, что фосфолипазы А! и А2 ускоряют реакцию отщепления
а- и Р-ацильных радикалов в молекуле фосфатида; они характерны для
животных и локализованы первая в эндоплазматической сети, а вторая—в
405
CHs-O—CO—R'
СН2ОН
Диглицерид
(jHj-OH
СН—о—СО—R1
+/СН3
HO-CHjCH-N—СН,
2 4 I3
+/СН'
0=T>-0-CHjCH-N-CH
Ан
Фосфохолин
CHtO-CO-R’
I 2
CH-O-CO-R'
I +/Сн3
CHz-O-p-OCH^-CH'Nt-CHo
(Го- 2 2 Хсн3
Фосфатид (лецитни)
С
Холин
Фосфолипаза At
r'-coohX|
CHj-O-CO-R’
CH-O-CO-R"
СН5-0 -CHjCHj-N сн3
о О- V"
Лнзофосфатид
(лизолецитии)
|Фосфо-
^|липаза А2
’СООН
CH2OH+R'COOH+R"COOH сн2ОН кислота
СНОН Высшие жирные 1-----------
I ки слоты
HaO
Липаза
CH2OH
Глице-
рин
НО О~
3 Фосфатидная
кислота
H2O
V Фосфатидат-
* фосфогидролаза
!HjO-CO-R'
H-O-CO-Rj
снон у сн3 СН-О-С
CH5O-P-O-ClfcCJfeNH:H3 СН,ОН
О ^Н, Диглнцерид
а-Глицерофосфохолин
HjO
.СН _
HO-CHrCH—N—СН,'*"' Глицерофосфо-
Z Z \ и
^сн3
СН2ОН
снон (j)H
CHj-O-P=O
он
а -фосфоглица рии
Н2О'^1фосфомоио-
1 эстераза
холии-диэстераза
Холин
Н2О
Липаза
^h2oh+r'cooh+r"cooh
CHOH Высшие жирные
CHjOH кислот“
Глицерин
СН20Н-СН(ОН)-СН2ОН + Н3РО.
Глицерин
Схема 11. Пут распада фосфолипидов
митохондриях, образуя при гидролизе Р ацил-лизолецитин и tx-ацил-лизолеци-
тин соответственно. Фосфолипазы А могут секретироваться и присутствуют,
например, в ядах змей. У животных есть также фосфолипаза В, де ствующая
на обе связи. Путь распада фосфатидов, открывающийся действием фосфоли-
пазы С, присущ микроорганизмам, а фосфолипазы D—растениям.
Наиболее детально изучены, включая первичные и вторичные (рис. 127, Л)
структуры, фосфолипазы А2 из ядов змей и других источников. При молекуляр-
ных массах в 11 000—15 000 и м нимум 4 дисульфидных мостиках они имеют
активный центр, содержащий радикалы гистидина и аспарагиновой кислоты
и работающий по схеме, характерной для ускорения гидролитических реакций
(см. с. 332). Установлено также что фосфолипазы функционируют в виде
димеров, где одна субъединица осуществляет каталитический акт, а вторая—
удаление отщепленного остатка высшей жирной кислоты (рис. 127, Б).
406
A
А П
а -спираль Л-изгиб
д-слои
Б
Рис. 127. Гипотетическая схема .гидролиза фосфатидов фосфолипазой А2 из яда среднеази-
атской кобры:
А—вторичная структура фосфолипазы Д2; Б1—схема гидролиза—на l-й ступени реакции (кх) возникает фермент-
субстратный комплекс, в котором ацильная группа при а-углеродном атоме остатка глицерина располагается в субстрат-
ном центре, а остаток фосфорной кислоты с присоединенным к нему азотистым основанием—в каталитическом центре
фосфолипазы А2. На 2-й ступени (fc2) к фермент-субстратному комплексу присоединяется вторая молекула фермента,
связывающая в своем субстратном центре ацильный радикал при 0-углеродном атоме остатка глицерина молекулы
фосфатида. На 3-й ступени реакции (fcj) осуществляется гидролиз сложноэфирной связи при ^-углеродном атоме
и удаление из зоны реакции ацильного радикала высшей жирной кислоты. На 4-й ступени (fc4) димер фосфолипазы А2
распадается: с одним из протомеров остается связанным а-лизофосфатид, с другим—высшая жирная кислота. На 5-й
ступени (fc5) высвобождаются конечные продукты реакции
Особо следует подчеркнуть, что действие фосфолипаз на мембраны субкле-
точных частиц несомненно приводит к существенным сдвигам в функциональ-
ной активности последних. Этой роли фосфолипаз в обмене веществ и его
регуляции придают в последнее время все большее значение.
Дальнейший обмен продуктов распада фосфатидов—высших жирных кис-
лот и глицерина, был освещен ранее. Поэтому рассмотрим здесь лишь после-
дующие превращения холина.
Одной из важнейших реакций, в которую вступает холин, по крайней мере,
в нервной ткани животных, является реакция его ацетилирования. Источником
ацетильной группы при этом служит ацетил-КоА, сама реакция ускоряется
специфическим ферментом—холин-ацетилтраисферазой:
✓О
. .._ . + Холиивцетялтрлисферхм
СН3-С + НО—СН,—СНз—N (СНэ)з --------------------—>
4S—КоА Холин
Ацетил-коэнзим А
СНэ-Ск +
ХО-СН,-СН,-N(CH3)3 + HSKoA
Ацетилхолин Комины А
Ацетилхолин физиологически активен, так как он участвует в передаче
нервных импульсов. Именно поэтому, видимо, фосфатиды, в частности холин-
фосфатиды. являются непременной составной частью нервной ткани.
Другой реакцией, имеющей существенное значение в обмене веществ,
считают реакцию окисления холина в бетаин, который, в свою очередь, служит
отличным донором СН3-групп в реакциях трансметилирования (см. с. 170):
407
н3сч
Н3С—N—СН2— СН2—ОН
H3CZ
Холин
Холии-
легидрогеназа
ФАД ФАД Н2
HjC\
Н,с—N—СН2—cf
Н3С^ Н I
Бетаинальдегид
Н,С< уО Бетаинальдегид- Н3Ск
H3c\i<H2-c^ + н2о > н3с^Лсн2-соо- + н+ -
н3сх хн / ч НзС/
Бетаинальдегид НАД НАДН+Н Бетами
Бетаин, вступая в реакцию трансметилирования с гомоцистеином, образу-
ет метионин:
\H3C1 v Бетани-гомоцистеин-
H;c-^N-CH2-COO- + (h)S-CH2-CH2-CH-€OOH . ^нлгранефер». ..
H3CZ '' NH2
Бетаин Гомоцистеин
Нзс\
—* \-СН2—СООН + Н3С— S—СН2—СН2—СН—СООН
Нзс NH2
N,N-Диметилглицин Метионин
Как было указано выше, метионин в виде S-аденозилметионина является
универсальным источником метильных групп в реакциях трансметилирования
(см. с. 270).
Не исключено, что диметилглицин теряет оставшиеся две метильные груп-
пы при атоме азота и превращается в глицин.
Механизм биосинтеза фосфатидов. Как и во многих ранее отмеченных
случаях, биосинтез фосфатидов протекает совсем иным путем, чем обращение
реакций их гидролиза. Первые стадии биосинтеза фосфатидов совпадают
с таковыми синтеза триглицеридов. Все идет одинаково вплоть до образова-
ния фосфатидной кислоты, а из нее—диглицерида. Однако дальше в случае
биосинтеза фосфатидов на свободную ОН-группу диглицерида присоединяет-
ся остаток фосфохолина, который переносится из состава цитидиндифосфат-
холина (ЦДФ-холин) (см. верхнее уравнение реакции на с. 409).
Указанный путь биосинтеза фосфатидов был открыт Е. Кеннеди и С. Вей-
сом (1956). Реакция ускоряется специфическим ферментом—ЦДФ-холин-
1,2 диацилглицеролхолинфосфотрансферазо"[, локализованной в цитозоле
(М=200 000) и олигомеризующейся в присутствии диацилглицеринов с семи-
кратным возрастанием активности, которая таким образом саморегулируется.
Аналогично этому идет перенос остатка фосфоэтаноламина с ЦДФ-этанол-
амина на диглицерид при участии также специфического фермента. Следова-
тельно, такой путь биосинтеза вполне обоснован для лецитинов (холинфос-
фатидов) и кефалинов (коламинфосфатидов).
Возникает вопрос, каким же образом синтезируется в организме столь
сложное соединение, как ЦДФ-холин? Механизм его биосинтеза таков (см.
нижнее уравнение реакции на с. 409).
ЦМФ, взаимодействуя с АТФ, переходит снова в ЦТФ и, соединяясь
с фосфохолином, опять образует ЦДФ-холин. Следовательно, ЦДФ-холин
выполняет в этом процессе каталитическую функцию, перенося остатки фос-
фохолина на диглицерид. Если сравнить этот процесс с синтезом олиго-
408
+/СН3
НО—сн2—СН2—N—CHj
ЧСН3
Холин
АТФ АДФ он
< J I +/’
Холинкиназа^~ HO-P-O-CHj-CH-N-CHj
О СН3
Холиифосфат
Холиифосфотраис-
_____фераза
(ЦДФхолии: 1,2-диацил-
глицерол холиифосфо-
трансфераза)
Диглицерид
Цитидиндифосфатхолин
Цитидии монофосфат
и полисахаридов, то там подобную функцию выполняла УДФ-глюкоза в от-
ношении остатка глюкозы. Это дает основание утверждать, что в реакциях
биосинтеза соединения типа нуклеозиддифосфатрадикалов играют выдающу-
юся роль как доноры тех или иных остатков органических соединений. Воз-
можно, эта роль нуклеозидцифосфатпроизводных связана с их способностью
409
превращать стабильную энергию макроэргической связи между остатками
фосфорной кислоты в подвижную энергию возбуждения электронов взаимо-
действующих молекул, что и обеспечивает про екание реакции.
Ферменты и промежуточные продукты приведенного выше цикла реакций
обнаружены в большинстве тканей животных, печени птиц, моркови и т. д.
Особенно ярко они представлены в мозге. Впрочем, у дрожжей, например,
синтез фосфатидилхолина идет главным образом за счет метилирования
фосфатидилэтаноламина; это—второй путь биосинтеза лецитина, открытый
Дж. Бретеном и Г. Гринбергом (1960). Фосфатидилглицерин, дифосфатидил-
глицерин, фосфатидилинозит и фосфатидилсерин синтезируются через цитидин-
дифосфатидилглицерины, возникающие из ЦТФ и диглицеридов под воздей-
ствием ЦДФ-диглицеридсинтазы (М = 114 000, димер). Взаимодействуя с
глицеролфосфатом, инозитом и серином, ЦДФ-диглицериды образуют пере-
численные выше фосфатиды при участии соответствующих ферментов.
Что касается обмена некоторых других видов липидов (сфинголипиды,
гликолипиды и т. п.), то он осуществляется в соответствии с теми принципами,
которые были отмечены при рассмотрении реакций обмена триглицеридов
и фосфатидов. Распад сфинголипидов, гликолипидов и т. п. осуществляется
при участии гидролаз, а даль ейший обмен продуктов их гидролиза—путем
типовых реакций деструкции соединений соответствующих классов: углево-
дов, высших жирных кислот и др., рассмотренных ранее. Биосинтез сфинголи-
пидов и гликолипидов протекает при широком участии разнообразных ацил-
и гликозилтрансфераз.
Перенос липидов между мембранами. В последние годы намети-
лось новое направление в изучении обмена липидов. Оно касается достаточно
энергично протекающего процесса межмембранного переноса липидов, особен-
но фосфолипидов, из митохондрий в эндоплазматическую сеть и обратно, из
мембранной фракции клетки в липосомы, от липосом одного состава к липо-
сомам другого состава, от внутреннего липидного слоя мембраны к внешнему
и наоборот и т. п. Значение этого динамично протекающего обновления
и видоизменения липидного состава мембран огромно, так как при его
посредстве регулируется метаболическая активность мембранного аппарата
клетки и субклеточных структур.
Важно, что межмембранный перенос липидов осуществляется специфиче-
скими белками, имеющими повсеместное распространение. Так, например, из
цитозоля клеток печени быка выделен белок, переносящий фосфатидилхолин
от одних мембран к другим. Молекулярная масса этого белка, связывающего
и переносящего одну молекулу фосфатидилхолина, равна 22000, р7=5,8, в нем
190 аминокислотных остатков, 38% которых полярны.
ГЛАВА X
БИОЛОГИЧЕСКОЕ ОКИСЛЕНИЕ
Совокупность окислительных реакций, происходящих в биологических
объектах и обеспечивающих их энергией и метаболитами для осуществления
процессов жизнедеятельности, называется биологическим окислением.
Функции этого важнейшего биологического явления даже несколько шире,
чем это отмечено в приведенном выше определении, так как при посредстве
реакций биологического окисления разрушаются также вредные продукты
обмена веществ и проникшие в организм чуждые соединения. Более того,
состояние окислительно-восстановительных процессов в клетках сказывается
на регуляции обмена веществ в них:
Детоксикация ксено-
биотиков
Синтез важнейших
(ключевых)
метаболитов
Функции
биологического
окисления
Устранение шлаков (вредных
клетки продуктов обмена) 1
Регуляция обмена
Веществ
Энергетическое
обеспечение
поддержи- свечения химичес- осмоти- электри- механичес-
ния (биолю ми- кик син- ческих ческих кой работы
темпера— несценции) тезоО явлений процессов
туры тела
Разнообразные реакции биологического окисления ускоряются многочис-
ленными ферментами, относящимися к классу оксидоредуктаз. Эти ферменты,
как правило, встроены в биологические мембраны, причем очень часто в виде
ансамблей.
ИСТОРИЯ РАЗВИТИЯ ПРЕДСТАВЛЕНИЙ О МЕХАНИЗМАХ
РЕАКЦИЙ БИОЛОГИЧЕСКОГО ОКИСЛЕНИЯ
Разобраться в столь сложном явлении, как биологическое окисление, воз-
можно лишь после ознакомления с теми концепциями, которые постепенно
складывались по мере исследования этой проблемы.
Хотя более двухсот лет тому назад (1774—1777) А. Лавуазье считал
дыхание очень медленным горением продуктов питания в организме,
сходным со сгоранием угля, первая попытка выявить молекулярный механизм
411
биологического окисления была предпринята лишь в следующем столетии
Хр. Ф. Шёнбайном (1845—1868). Им была выдвинута идея о том, что необ-
ходимым условием протекания биологических окислительных процессов явля-
ется активирование кислорода. Естественно, что конкретные пути этого
активирования выглядели фантастически (предполагалось существование от-
рицательно-активной формы кислорода, тождественной озону и положитель-
но-активной, названной антиозоном), но сама идея была плодотворна.
Хр. Ф. Шёнбайн впервые высказал мысль о том, что биологическое окисление
есть каталитический процесс. Ему же удалось экспериментально доказать
образование Н2О2 при биологическом окислении.
На рубеже XIX и XX вв. наш соотечественник А. Н. Бах и независимо от
него в Германии К. Энглер и В. Вилд выдвинули гипотезу об образ вании
пероксидов органических соединений как первом этапе биологического окисле-
ния. При этом молекула кислорода переводилась в активированное состояние
за счет разрыва двойной связи в ней при посредстве «внутренней колебатель-
ной энергии» самого окисляемого соединения, обладающего кратной связью,
и при участии ферментов—оксидаз в соответствии с такой, например, схемой:
Перекись Баха
Возникшие пероксиды органических соединений, как и пероксид водорода,
могут окислять другие вещества при каталитическом воздействии пероксида-
зы—фермента, достаточно в то время уже изученного:
Пероксидаза
R'H2+H2O2-----------------------> R'+2H2O
Субстрат (или пероксид Субстрат (или Н2О+оксид
восстановленный органического окисленный органического
соединения) соединения)
Пероксид водорода может распадаться и иным путем, при участии фермен-
та каталазы, о котором в этот период тоже накопилось довольно много
сведений:
____ л Каталаза ___ _ л
2Н2О2 “ " > 2Н2О+О2
Вероятно поэтому, А. Н. Бах придавал большое значение участию в биоло-
гическом окислении пероксидазы и каталазы, полагая, что распределение
Н2О2 между пероксидазой и каталазой может служить для регуляции этого
процесса, например, в растениях.
В последующее время названным ферментам в окислительно-восстанови-
тельных процессах отводили более скромную роль.
Однако в свете данных об активных состояниях кислорода, о существова-
нии супероксид-ионов и ферментов, принимающих участие в их обмене,—
супероксиддисмутаз, значение, придаваемое Н2О2, каталазе и пероксидазе
в реакциях биологического окисления, стало неуклонно возрастать.
Принципиально иной подход к расшифровке механизмов реакции биологи-
ческого окисления был намечен в трудах В. И. Паллацина, а вслед за ним—
Г. Виланда. На основании опытов с дыхательными хромогенами (под ними он 5
подразумевал бесцветные вещества растительного происхождения, способные
412
в присутствии оксидаз присоединять кислород и переходить при этом в пиг-
менты, которые, в свою очередь, могли передавать присоединенный кислород
окисляемому субстрату, одновременно обесцвечиваясь), В. И. Паллада а впер-
вые (1912) высказал идею о том, что биологическое окисление есть перенос
водорода от окисляемого вещества навстречу кислороду с образованием воды
в качестве конечного продукта. К этой идее В. И. Палладии пришел после
того, как в одном из опытов обнаружил, что метиленовый синий, не содержа-
щий в своем составе кислорода, может играть роль дыхательного хромогена,
снимая атомы водорода с окисляемого субстрата:
Стало ясно, что дыхательные хромогены являются не переносчиками кислоро-
да, а акцепторами водорода.
Концепция В. И. Палладана довольно быстро получила подтверждение.
Благодаря трудам Т. Тунберга, Д. Самнера, Г. Сомерса, В. Мак-Шена и дру-
гих были выделены и охарактеризованы разнообразные дегидрогеназы, уско-
ряющие реакции окисления тех или иных субстратов при участии кофермен-
тов, являющихся акцепторами снимаемых с них атомов водорода.
Таким образом, в начале нашего столетия сложились две концепции биоло-
гического окисления: активирования кислорода и активирования водорода. Их
противоборство продолжалось недолго: в 1925 г. Д. Кейлин в тканях ряда
насекомых, а затем и в других аэробных биологических объектах открыл
цитохромы—те недостающие ферменты, которые позволили несколькими
годами позже связать активирование кислорода и водорода воедино. Этому
способствовало обнаружение О. Варбургом (1928) цитохромоксвдазы, получи-
вшей в то время наименование «дыхательного фермента Варбурга». Именно
цитохромоксидаза оказалась тем ферментом, который непосредственно ак-
тивирует кислород, а цитохромы- ферментами, снимающими электроны
с водорода и передающими их цитохромоксидазе. Так впервые возникло
пред тавление об ансамблях ферментов дыхательной цепи, обеспечивающих
реакции биологического окисления. В частности, оксидоредуктазная цепь,
главной составной частью которой являются цитохромы, получила название
цитохромной системы:
Ферменты, находящиеся в конце таких оксидоредуктазных цепей и непо-
средственно переносящие электроны на кислород, получили название терми-
нальных оксидаз.
413
Позже (1947—1966) было показано, что цитохромной системой не исч р-
пывается перечень ферментных систем, способньх активировать и водород,
и кислород с последующим образованием из них молекул воды. Таких систем
несколько. Про тейшая из них наряду с пероксидазными реакциями—глико-
латоксндазная:
Гликолат: 'кислород О
оксидоредуктаза %,
HO-СН»—СООН + О,------------------------- С-СООН + Н4О.
(оксидам L-2-окси- /
кислот) pj
Гликолевая кислоте Глиоксилевая кислота
Она представлена в растениях, у животных, грибов и бактерий. Тликолаток-
сидаза из листьев шпината—флавопротеин с М=270 000 (8x37000). Четвер-
тичная структура октамера гликолатоксидазы и третичная структура субъеди-
ниц недавно выяснена; имея около 10 нм в диаметре, октамер, составленный
из 4 димеров, обладает полостью диаметром 6 нм. Гликолатоксидаза содер-
жит флавинмовонуклеотид (см. с. 120) в качестве кофермента; при его посред-
стве дегидрируется гликолевая кислота (активирование водорода). Вместе
с тем гликолатоксидаза способна активировать кислород и передавать на него
атомы водорода с восстановленного флавинмононуклеотида с образованием
Н2О2, т. е. ей присуща флавопротеиноксидаэная функция. Образовавшийся
пероксид водорода распадается при участии каталазы.
В настоящее время изучено более двух десятков оксидаз флавопротеиновой
природы, содержащих ФМН и ФАД в качестве коферментов (оксалатоксидаза,
глюкозооксидаза, оксидазы L аминокислот, ксантиноксидаза и др.).
Другой тип ферментных систем, обеспечивающих н епосредстенное окисле-
ние субстратов с передачей атомов водорода на кислород, представлен медьсо-
держащими оксидазами. Так как концентрированные рас воры этих ферментов
имеют синий цвет, их называют «синими оксидазами». Характерным предста-
вителем этой группы оксидаз является аскорбато сидаза—белок
с М = 130000—140000, содержащий 8 атомов Си на молекулу и состоящий из
двух равных субъединиц. Она открыта А. Сцент Дьердьи (1928) и очищ на
X. Таубером (1938). Уравнение реакции кисления аскорбиновой кислоты при-
ведено на с. 171.
Развитие представлений об оксидоредуктазных системах, участвующих
в осуществл нии биологического окисления, сопровождалось уточнением их
функций классификации и номенклатуры входящих в их состав ферментов.
Напомним (см. с. 117), что те дегидрогеназы, которые обеспечивают непо-
средственное дегидр рование субстратов, называются первичными. В отличие
от них дегидрогеназы, получающие атомы Н от восстан вленных коферм н-
тов первичных дегидрогеназ (НАДН, НАДФН, ФМН -Н2, ФАД • Н2 и др.) или
от пр< межуточных акцепторов, на которые были переданы атомы водорода
с первичных дегидрогеназ, отнесены к категории вторичных дегидрогеназ.
Любые дегидрогеназы (и первичные, и вторичные), передающие атомы водо-
рода на определенные акцепторы, называют редуктазами. Как отмечено ранее,
все оксидоредуктазы, переносящие атомы водорода или электроны непосред-
ственно на кислород, называют оксидазами.
В связи с этим было обращено внимание на окислительно-восстановитель-
ные системы, в которых между дегидрогеназами и молекулярным кислородом
действует посредник. Атомы Н с восстановленной дегидрогеназы сначала
поступают на окисленную молекулу посредника а потом уже с нее—на О2.
Обе реакции ускоряют специфическими ферментами. Конечным продуктом
414
реакции является вода. Наиболее часто роль посре ников играют хиноны
и аскорбиновая кислота. При этом во взаимодействии с посредником участвует
только восстановленный кофермент, например НАДН или НАДФН.
В случае фенолхиноновой системы схема процесса такова:
деза
Первые данные о существовании совершенно нового пути биоло ического
окисления, не похожего на все изученные ранее, были сообщены Е. Андре
и К. Хоу (1932), открывшими в семенах сои особый фермент—липоксидазу.
Он ускорял реакцию прямого присоединения атмосферного кислорода по двой-
ным связям полиненасыщенных высших жирных кислот. Применение 18О2
и Н218О для экспериментального изучения аналогичных реакций позволило
в 1955 г. одновременно и независимо Г. Мэзону с сотр. и О. Хайаиши с сотр.
доказать наличие нового подкласса оксидоредуктаз—оксигеназ и изучить
механизмы включения молекулярного кислорода при их посредстве в различ-
ные органические молекулы.
Согласно современным данным, липоксигеназа имеет молекулярную массу
от 60000 до 100000 в зависимости от объекта вь деления, расп остранена не
только в растительн м, но и в животном мире, существует в виде ряда
молекулярных форм. В случае линолевой кислоты реакция идет в соо ветст-
вии с уравнением:
СН3—(СН2)4—с н=сн—сн2—сн=сн—(С н2)7—соон
Линолевая кислота
СН3—(СН2)4—сн—СН=СН — СН=СН—(СН2)2— соон
ООН
13 -Гидроперокси- цис-9-транс -11-октадек адиенооая
кислота
Липоксигеназы принимают участие в биосинтезе проста ландинов, лейко-
триенов и тромбоксанов из арахидоновой кислоты (см. с. 463).
Новую главу в учении о биоло ическом окислении сое авило открытие
В. А. Энгельгардтом (1931) сопряжения реакций окисления органических со-
единений с фосфорилированием АДФ. Определяя сод ржание аденазинпиро-
фосфорной кислоты, условно названной им пирофосфатом, в энергично дыша-
щих эритроцитах голубя, он установил, что в атмосфере азота оно падает,
а в атмосфере кислорода — возрастает до исходной величины. Повторение
периода анаэробиоза с возвращением к аэробиозу сопровождалось новым
415
циклом «распад—ресинтез» пирофосфата. Это означало, чтс существует ка-
кой-то механизм, при помощи которого энергия, выделяющаяся при окисле-
нии (в присутствии кислорода), не рассеивается, а используется для связыва-
ния, деминерализации неорганического фосфата, который обнаруживается
в макроэргических связях аденозинтрнфосфата: АДФ + Н3РО4->АТФ + Н2О.
Позже идеи В. А. Энгельгардта были более детально разработаны
В. А. Белицером с сотр., внесшими существенный вклад в развитие проблемы
сопряжения окисления с фосфорилированием в период ее становления. В част-
ности, В. А. Белицером было введено понятие: коэффициент окислительного
фосфорилирования, т. е. отношение количества молей синтезированной АТФ
к количеству молей кислорода, использование которого обеспечивало этот
синтез. Будучи вычислено после ряда измерений, значение коэффициента
оказалось в среднем близким к 3,0, что свидетельствовало о том, что перенос
двух атомов водорода (а в дальнейшем—двух электронов) по дыхательной
цепи ферментов сопровождается синтезом трех молекул АТФ, т. е. существует
три пункта сопряжения окисления с фосфорилированием АДФ.
Исследование тонких механизмов сопряжения окисления с фосфорилировани-
ем, являющееся фундаментом биоэнергетики, продолжается уже более полусто-
летия и еще далеко от завершения. Ниже будет освещено его нынешнее состояние,
отражающее всю сложность современных проблем биологического окисления.
КЛАССИФИКАЦИЯ ПРОЦЕССОВ БИОЛОГИЧЕСКОГО ОКИСЛЕНИЯ
И ИХ ЛОКАЛИЗАЦИЯ В КЛЕТКЕ
Приведенный выше краткий исторический очерк содержит достаточно
конкретного материала для того, чтобы стало понятным существование двух
типов биологического окисления.
1. Свободное окисление, не сопряженное с фосфорилированием АДФ, не
сопровождающееся трансформацией энергии, выделяющейся при окислении,
в энергию макроэргических связей. При свободном окислении высвобожда-
ющаяся при сопряженном с окислением распаде химических связей энергия
переходит в тепловую и рассеивается.
По типу свободного окисления идут все без исключения оксигеназные
реакции, все окислительные реакции, ускоряемые пероксидазами или сопровож-
дающиеся образованием Н2О2, многие реакции, катализируемые оксидазами.
Процессы свободного окисления сосредоточены в цитозоле, в мембранах
эндоплазматической сети клетки, в мембранах лизосом, пероксисом и ап-
парата Гольджи, на внешних мембранах митохондрий и хлоропластов. Они
идут также в ядерном аппарате клетки.
2. Окисление, сопряженное с фосфорилированием АДФ. Этот тип биологиче-
ского окисления осуществляется двумя способами.
Если макроэргическая связь возникает в момент непосредственного окис-
ления субстрата, а затем тем или иным путем передается на фосфатный
остаток, который, в свою очередь, используется для фосфорилир! вания АДФ,
т. е. синтеза АТФ, то такой вид биологического окисления называют окисле-
нием, сопряженным с фосфорилированием АДФ на уровне субстрата. Ранее
такой способ называли фосфорилирующим окислением или субстратным фос-
форилированием.
Если атомы водорода с коферментов дегидрогеназ, принимающих участие
в окислении субстратов, передаются в оксидоредуктазную цепь, где сопряжен-
но с переносом протонов и электронов на молекулярный кислород происходит
416
а тивирование неорганического фосфата и при его посредстве фосфорилирова-
ние АДФ с образованием АТФ, то такое сопряжение окисления с синтезом
АТФ называют сопряжением на уровне электронотранспортной цепи. Понятно,
что сам окисляемый субстрат в этом случае непосредственного участия в ак-
тивировании неорганического фосфата не принимает. Ранее этот вид биологи-
ческого окисления называли окислительным фосфорилированием и отождеств-
ляли с дыханием.
Сопряжение окисления с фосфорилированием, т. е. генерирование АТФ для
нужд клеток, идет главным образом на внутренних мембранах митохоцдрий.
Именно здесь осуществляется сопряжение окисления с фосфорилированием на
уровне электронотранспортной цепи. Что касается субстратного фосфорили-
рования, то оно сосредоточено в растворимой части клетки.
Фотосинтетическое и хем синтетическое фосфорилирование АДФ, сопро-
вождающееся биосинтезом АТФ, также происходит путем сопряжения перено-
са электронов в электронотранспортных цепях с активированием неорганиче-
ского фосфата. Механизм этого сопряжения близок к таковому при окис-
лительном фосфорилировании в митохондриях, что подчеркивает единую
природу процессов, приводящих к синтезу АТФ у гетеротрофных и аутотроф-
ных организмов.
СВОБОДНОЕ ОКИСЛЕНИЕ
Реакции свободного окисления органических соединений в живой природе,
равно как и ферментные системы, ускоряющие их, многообразны, и многие из
них рассмотрены в предыдущих главах и в начальном разделе этой главы.
Этим путем непосредственно окисляются не только многочисленные природ-
ные и неприродные субстраты, но и восстановленные коферменты (НАДН,
НАДФН, ФАД • Н2 и др.), образовавшиеся при действии первичных и вторич-
ных дегидрогеназ.
Хотя реакции свободного окисления идут и в цитозоле, и на мембранах
различных субклеточных структур, средоточием их являются мембраны эн-
доплазматической сети клетки. Так как последние при гомогенизации клеток
и фракционировании субклеточных частиц гомогената дают фракцию микро-
сом, которая может быть получена в виде препарата, то сейчас активно
изучаются организация и функции микросо дальней дыхательной цепи. Ее
первая особенность сводится к тому, что, несмотря на наличие ферментов цепи
переноса электронов, ни в одном пункте этой цепи не происходит сопряжения
с фосфорилированием АДФ. Вторая особенность заключается в своеобразии
структуры и функциональной активности цитохромов b 5 и Р-450), входящих
в ее состав. В частности, цитохром Р-450 (Л/% 50000, гемопротеин, первичная
структура более десятка его форм расшифрована) обладает множеством (сот-
ни, а может быть, и тысячи) форм, возникающих в ответ на введение (или
попадание) в организм того или иного класса ксенобиотиков, подобно тому,
как антитела синтезируются в ответ на присутствие антигенов; поэтому
цитохром Р-450 считают своего рода «мембранным иммуноглобулином».
Наконец, третья особенность состоит в высоком сродстве терминальной
оксидазы микросомальных цепей к кислороду, позволяющая ей конкурировать
за кислород с митохондриальной цитохромоксидазой. Поэтому, например,
в клетках печени доля микросомального окисления эндогенных субстратов
составляет 40%, а митохондриального—60%.
Наиболее своеобразными и почти не затронутыми в предыдущих главах
являются реакции свободного окисления, идущие при участии оксигеназ. Этот
14—3502
417
подкласс оксидоредуктаз содержит ферменты, ускоряющие включение в окис-
ляемый субстрат либо двух (диоксигеназы), либо одного (монооксигеназы)
атома молекулярного кислорода.
Свободное окисление при участии диоксигеназ. Одной из наиболее изученных
диоксигеназ является пирокатехаза (катехол: кислород-1,2-оксидоредуктаза
дециклизующая); ее М = 85000. Концентрированные растворы пирок техазы
красного цвета, так как она содержит в активном центре два прочно связанных
атома Fe, которые, согласно О. Хайаиши, соединяются с молекулярным кис-
лородом в комплекс, где кислород далее активируется: Fe2+ +O2-»Fe2+O2-»
->Fe3+O£.
Затем в активном центре пирока ехазы после присоединения субстрата
возникает тройной комплекс, преобразование которого приводит к включе-
нию молекулярного кислорода в пирокатехин:
Аналогично действует катехол-2,3-оксйгеназа (М = 140 000, 1 атом Fe),
3,4-диоксигеназа протокатеховой кислоты (М = 700 000, 8x90000), образую-
щая р карбоксимуконовую кислоту, триптофаноксигеназа (М = 123 000), при-
соединяющая молекулярный кислород по пирролы ому кольцу инд шильного
радикала, и другие диоксигеназы. Во всех случаях молекулярный кислород
активируется за счет присоединени электрона, теряемого двухвалентным
железом активного центра, а возникший анионный свободный ради ал кисло-
рода (О2_) атакует и оксигенирует субстрат. В результате расшифровки
механизма действия диоксигеназ стал ясен один из способов активирования
молекулярного кислорода терминальными оксидазами.
Одной из биологически важных диоксигеназных реакций является превра-
щение Р-каротина в витамин А:
Ретиналь (витамин А)
Прямое присоединение 18О2 по 15,15'-связи в p-каротине впервые доказано
Б. Б. Вартапетяном и сотр. (1966).
Свободное окисление при участии монооксигеназ. Ввиду высокой лабиль-
ности монооксигеназ, как, впрочем, и диоксигеназ, выделение их сопряжено
с большими трудностями. Тем не менее некоторые из них получены в кристал-
418
лическом состоянии. Монооксигеназы характеризуются молекулярными мас-
сами от 65000 до 200000, отсутствием, как правило, в их составе ионов
тяжелых металлов и наличием коферментов флавиновой природы. Кроме
того, многие из них требуют участия в реакции монооксигенир вания так
называемого парного донора, от которого поступают атомы водорода на один
из атомов молекулярного кислорода (второй внедряется в окисляемый суб-
страт).
Простейший пре ставитель монооксигеназ—фенолгидроксилаза (фенол-2-
монооксигеназа):
+ ог + надфн + н*
Февол- ,
гадроксилаэа
Монооксигеназы принимают большое участие в окислении аминокислот
(лизин-, аргинин- и триптофанмонооксигеназы, фенилаланин- и тирозингид-
роксилаза), оксикислот (салицилатгидроксилаза), полиизопреноидных соеди-
нений (сквален-эпоксидаза, см. с. 404).
Механизм действия монооксигеназ выяснен недостаточно. Предполагают,
что активной формой кислорода может быть связанный с ферментом перок-
сид водорода или его эквивалент. Между тем показано, что некоторые из
монооксигеназ (фенолаза из грибов, фёниланин-4-гидроксилаза) содержат Си+
в своем составе. В этих слу аях возможен такой механизм активирования
кислорода:
Си+ -------------Lz-Cu—------X
+ 0=^=0 —Монооксигеназа ~О-т-
Си+ -------------^Си—________'
Си-+-----х
“О-;- О~ + субстрат + НАДН + Н+ —
Cu^t-----'
Си+
+ субстрат + НАД+ + Н2О
Си+ гидроксилированный
О~
Здесь, как и в случае диоксигеназ, решающую роль в активировании
молекулярного кислорода играет передача на него электронов с металла,
входящего в состав фермента. Не исключено, что этот способ вовлечения
молекулярного кислорода в процессы оксидоредукции в клетке является об-
щим для оксидаз, участвующих как в свободном, так и в сопряженном
с фосфорилированием биологическом окислении.
ОКИСЛЕНИЕ, СОПРЯЖЕННОЕ С ФОСФОРИЛИРОВАНИЕМ АДФ
Субстратное фосфорилирование Примерами сопряжения окисления с фос-
форилированием на уровне субстрата могут служить реакции окисления 3-
фосфоглицеринового альдегида в 1,3 дифосфо лицериновую кислоту, 2-фос-
фоглицериновой кислоты—в 2-фосфоенолпировиноградную, а-кетоглутаро-
вой кислоты—в янтарную кислоту (здесь фосфорилируется ГДФ—см.
рис. 117). С возникающих при этом соединений фосфат, связанный макроэр-
гической связью, легко передается на АДФ (или ГДФ). Один из примеров
419
Ацетил-КоА
НАДН + Н* НАД*
Окислительное фосфорилирование
Рис. 128. Локализация синтеза АТФ при субстратном и окислительном
фосфорилировании (пояснения в тексте)
такого сопряжения и механизм переноса активированного фосфата на АДФ
детально рассмотрены выше (см. рис. 114). Однако п средством реакций
субстратного фосфорилирования образуется сравнительно небольшое количе-
ство АТФ (рис. 128).
Окислительное фосфорилирование. Главная масса АТФ у аэробных организ-
мов синтезируется путем окислительного фосфорилирования в митохондри-
ях—этих поистине энергетических фабриках клетки.
Атомы водорода, снятые с субстратов в цикле дикарбоновых и трикарбоно-
вых кислот, при р-окислении высших жирных кислот, при пируватдегидрогеназ-
ной, глутаматдегидрогеназной и некоторых других реакциях поступают в дыха-
тельную цепь ферментов внутренней мембраны митохондрий. Универсальным
донором атомов водорода для дыхательной цепи ферментов служит НАДН.
Если при окислении субстрата возникает НАДФН, то осуществляется реакция
420
Рис. 129. Компоненты дыхательной цепи митохондрий:
Eq—окислительно-восстановительные потенциалы компонентов дыхательной цепи; А£о—разность потенциалов между компонентами дыхательной цепи в точках сопряжения
с фосфорилированием АДФ (подчеркнуты толстой черной линией); I, II, III—точки сопряжения. Дыхательная цепь митохондрий более сложна, чем это показано на рисунке. При
определенных условиях из нее можно выделить 4 комплекса, каждый из которых характеризуется своей молекулярной массой, полипептидным составом и значениями
окислительно-восстановительных потенциалов (£о). Комплекс I: М = 700—900 кДа, 25 полипептидов; комплекс II: М==140 кДа, 4—5 полипептидов; комплекс III: М=250 кДа,
9—10 полипептидов; комплекс IV: М = 160—170 кДа, 8 полипептидов. Дыхательная цепь примыкает к АТФ-синтазному комплексу (комплекс V: М = 500 кДа, 12—14
полипептидов), показанному на рис. 130 и 131. Таким образом, с трансформацией энергии в митохондриях связано около 70 различных полипептидов н 6 типов фосфолипидов
сопрягающей мембраны (см. рис. «Блочная структура митохондриальной цепи» на форзаце в начале учебника)
НАДФН + НАД+ "№,,“1"""НАДФ* 4-НАДН
Следовательно, и в этом случае атомы Н перед поступлением в дыхательную
цепь передаются на НАД* (рис. 128).
Другим первичным источником атомов водорода и электронов в дыхатель-
ной цепи служит восстановленный флавопротеин, если он выполняет роль
первичной дегидрогеназы, как, например, в случае окисления янтарной кисло-
ты в цикле трикарбоновых и дикарбоновых кислот (см. рис. 117). Флавопроте-
ин, но несколько иной природы, служит передаточной инстанцией для атомов
водорода и электр нов от НАДН к убихинонпротеину дыхательной цепи.
На рис. 129 представлена дыхательная цепь ферментов митохондриальной
мембраны. Естественно, что она открывается НАДН, с которого атомы
Н передаются на первый белковый компонент дыхательной цепи—флаво-
протеин, несущий флавинмононуклеотид (ФМН) в качестве кофермента.
Остальные компоненты дыхательной цепи располагаются в порядке возраста-
ния их нормальных (измерены при 1 М концентрации и температуре 25° С, что
обозначают индексом Ео и pH 7,0 и маркируют значком ') окислительн -
восстановительных потенциалов (Е’о), обеспечивающих упорядоченную пе-
редачу атомов водорода и электронов по такой редоксцепи.
Самой примечательной особенностью дыхательной цепи ферментов явля-
ется наличие в ней участков, где соседние компоненты резко ’ отличаются
значениями окислительно-восстановительных потенциалов (Д£о):
Нормальны окислительно-восстановительные потенциалы соединений,
участвующих в биологическом окислении
Окисленная форма Число пере- даваемых электронов Восстановленная форма
Ацетат+СО2 2 Пируват -0,70
Сукцинат+СО2 2 а-Кетоглутарат -0,67
Ацетат 2 Ацетальдегид -0,60
о2 1 о2- -0,45
Ферредоксин (окисл.) 1 Ферредоксин (восст.) -0,43
2Н* 2 н2 -0,42
Ацетоацстат 2 Р-Гйдроксибутират -0,35
НАД+ 2 НАДН+Н* -0,32
НАДФ+ 2 НАДФН+Н* -0,32
ФМН-протеин 2 ФМН Н2-протеин -0,30
Липоат (окисл.) 2 Липоат (восст.) -0,29
1,3-Дифосфоглицерат 2 Г ицеральдегид-3-Р+Рн -0,29
Глутатион (окисл.) 2 Глутатион (восст.) -0,23
ФАД 2 ФАД Н2 -0,22
Ацетальдегид 2 Этанол -0,20
Пируват 2 Лактат -0,19
Оксалоацетат 2 Малат -0,17
а-Кетоглутарат+NH4 2 Глутамат -0,14
Метиленблау (окисл.) 2 Метиленблау (восст.) 0,01
Фумарат 2 Сукцинат 0,03
Коэнзим Q 2 Коб-Н2 0,04
Цитохром fc(Fe3+) 1 Цитохром b (Fe2 *) 0,07
Дегидроаскорбат 2 Аскорбат 0,08
422
Продолжение
Окисленная форма Число пере- даваемых электронов Восстановленная форма £0, В
Цитохром Ci(Fe3+) 1 Цитохром Cj(Fe2+) 0,23
Цитохром c(Fe3+) 1 Цитохром c(Fe2+) 0,25
Цитохром a (Fe3 +) 1 Цитохром a(Fe2+) 0,29
1/2О2 + Н2О 2 Н2О2 0,30
Феррицианид 2 Ферроцианид 0,36
Нитрат 1 Нитрит 0,42
Цитохром а3 (Fe3+) 1 Цитохром а3 (Fe2 +) 0,55
Fe3 + 1 Fe2+ 0,77
1/2О2+2Н + 2 Н2О 0,82
Именно здесь происходит сопряжение окисления с фосфорилированием АДФ
(рис. 129), так как разность энергетических уровней электрона, транспортиру-
емого с огромной скоростью (около 1 мс от одного переносчика к другому),
вполне достаточна для синтеза макроэргической связи и составляет 51 кДж
для I, 36—для Пи 80,7 кДж для ZZZ точки сопряжения. В целом интенсивность
окислительного фосфорилирования определяется энергетическим зарядом,
т. е, соотношением моно-, ди- и трифосфатов аденозина:
„ 0,5 [АДФ] + [АТФ]
[АМФ ] + [АДФ ] + [АТФ ]
Внутренняя двухслойная мембрана
Наружная двухслойная
мембрана
Кристы (впячивания внутренней
мембраны)
Матрикс (пространство, замкнутое
внутренней мембраной)
Межмембранное пространство
(между внешней и внутренней
мембраной митохондрии)
Рис. 130. Структура митохондрии (Л) и схема расположения ферментов дыха-
тельной цепи и АТФ-синтазного комплекса в ее внутренней мембране (£):
ФМН ^флавопротеин с флавинмононуклеотидом в качестве кофермента; FeS—железосерные белки;
Q—убихинонпротеин; Ь, с и с—цитохромы; а а —цитохромоксвдаза
423
•20Н"
20Н“
Рис. 131. Трансмембранный перенос электронов и протонов и сопряжение его с синтезом
АТФ (обозначения те же, что на рис. 129)
Мембраны, несущие ферменты переноса электронов и сопряженного с ним
фосфорилирования, называются сопрягающими. К ним относятся: внутренняя
мембрана митохондрий, мембрана тилакоидов хлоропластов зеленых расте-
ний, мембрана хроматофоров фотоси езирующих бактерий и клеточные
мембраны аэробных бактерий, обладающих дыхательным типом энергетики.
Они характеризуются толщиной в 7,0—9,0 нм, преобладанием белков над
липидами (2:1), низким содержанием холестерола и наличием кардиолипина;
примерно третья часть входящих в их состав белков принадлежит ферментам
дыхательной цеди, собранным в ансамбли (рис. 129),— в каждой митохондрии
клеток печени крысы, например, их содержится несколько тысяч.
Наиболее полно изучены структура и функция сопрягающей мембраны
митохондрий. Наряду с дыхательным ансамблем ферментов в ней находится
АТФ-синтазный комплекс, ответственный за образование АТФ. Каким же
образом расположены они в митохондрии? Ответ на этот вопрос дает рис. 130.
И тот и другой ферментные комплексы локализованы во внутренней мембране
митохондрий (рис. 130, Б), причем АТФ-синтазный комплекс представлен так
называемыми грибовидными выростами, которые усеивают внутреннюю ме-
мбрану и обращены в сторону матрикса митохондриальных частиц.
Проблема сопряжения окисления с фосфорилированием необыкновенно
сложна и далека еще от окончательного разрешения. Ранние гипотезы по
этому вопросу: гипотеза химических переносчиков (Е. Слейтер, 1953) и кон-
формационная гипотеза (П. Бойер, 1964) представляют в настоящее время
лишь исторический интерес, хотя отдельные элементы той и другой в некото-
рой мере присутствуют в общепризнанной сейчас хемиосмотической гипотезе
П. Митчела, поддержанной и развитой в нашей стране благодаря трудам
В. П. Скулачева и сотр.
Согласно хемиосмотической гипотезе, именно структурные и функцио-
нальные особенности сопрягающей мембраны (этот термин введен в био-
энергетику В. П. Скулачевым) обеспечивает биосинтез АТФ. В процессе
функционирования дыхательной цепи ферментов в сопрягающей мембране
митохондрий, не проницаемой ни для НАДН, ни для протонов, происходит
активный перенос шести Н+ из матрикса в межмембранное пространство
(рис. 131) на каждую пару электронов, проходящих по электронотранспортной
цепи. По поводу механизма этого переноса высказаны разные мнения. Сущ-
ность некоторых из них ясна из рассмотрения рис. 131. Предполагают также,
что перенос протонов идет при участии протонных транслоказ—специфи-
ческих белков, локализованных в сопрягающей мембране и обеспечивающих
424
При М=140 000 цитохром с-оксидаза состоит из 7 субъединиц, содержащих 2 атома Си и 2 атома
Fe (в составе гема, показан овальным кружком), соединенных координационными связями с атома-
ми азота радикалов гис. Так как атомы Си и Fe цитохрома а а3 находятся на расстоянии 3,5 нм, то
возможен только тоннельный перенос электронов между ними. Цитохромоксвдаза, изменяя кон-
формацию при переносе электронов, либо сама выполняет функцию протонной помпы, либо для
этого служит тесно прилегающий к ней белок. Как видно из рисунка, механизм активирования
молекулярного кислорода цитохромоксидазой вполне аналогичен таковому у диоксигеназ и некото-
рых монооксигеназ, т. с. осуществляется за счет передачи на кислород электронов с Fe2+ и Си+.
перенос протонов сопряженно с транспортом электронов при помощи бел-
кового комплекса, как, например, в случае цитохромоксидазы (рис. 132) или
НАДН: ^-оксидоредуктазы.
В результате создается трансмембранная разность потенциалов, так как
с внешней стороны внутренней мембраны митохондрий, в межмембранном
пространстве, накапливаются Н+, а на внутренней ее стороне, в матриксе,—
ОН- (см. рис. 131). Возникает градиент электрохимического потенциала Н+
(его обозначают как Лцн+)- Он складывав ся из градиента электрического
потенциала—Аф и градиента концентрации водородных ионов—ДрН и явля-
ется движущей силой синтеза АТФ.
Естественно, что ионы Н, накопившиеся в межмембранном пространстве
митохондрии, перенесенные туда за счет потерянной электронами энер ии
в процессе их транспорта по дыхательной цепи ферментов и перехода с более
высокого энергетического уровня в окисляемом субстрате на более низкий
энергетический уровень в активированной молекуле кислорода (см. рис. 129),
стремятся вернуться в митохондриальный матрикс. Энергезированная, электриче-
ски заряженная внутренняя мембрана митохондрий способна деэнергезироваться,
разрядиться. Этот процесс осуществляется при помощи протонной АТФазы.
Протонная АТФаза (Н+-АТФаза)—липопротеиновый комплекс, гидроли-
зующий АТФ сопряженно с трансмембранным переносом водородных ионов
против их электрохимического градиента (Дцн+)- Энергия для такого переноса
Н+ против градиента их концентрации черпается за счет энергии распада-
ющейся макроэргической связи в молекуле АТФ при ее гидролизе. В условиях
нарушения целостности комплекса FbFt (рис. 132) митохондрий, Н+-АТФаза
ускоряет именно этот процесс, обеспечивая обратный транспорт Н+ и со-
здание Лрн+- Но в составе энергезированной митохондриальной мембраны,
при нормальном состоянии комплекса Fo’Fi, функция протонной АТФазы
425
Рис. 133. Строение протонной АТФазы (пояснели в тексте)
сводится не к переносу ионов водорода из матрикса в межмембранное про-
странство, а, наоборот, к транспорту протонов внутрь митохондрии, к снятию
электрохимического градиента Н+ и, само собой разумеется, к синтезу (сопря-
женно с переносом Н+ с внешней стороны сопрягающей мембраны на ее
внутреннюю сторону) АТФ. Поэтому ее называют также АТФ-синтазой, что
подчеркивает ее истинную функцию в митохондриальной мембране.
АТФ-синтаза (прото 1ная (АТФаза) представлена двумя белковыми ком-
плексами, состоящими, в свою очередь, из субъединиц (рис. 133, А). Первый из
них, полностью «утоплен ый» в сопрягающую мембрану и пронизывающий ее
насквозь, состоит из трех видов гидрофобных полипептидных цепей (с раз-
личающимися в зависимости от объекта молекулярными массами порядка
19000—24000, 13500—18000 и 5400—8400 в соотношениях 1:2:5 или близких
к этому) и обозначается как Fo. Его функция состоит в доставке протонов из
межмембранного пространс ва, куда он открывается, ко второму белковому
комплексу, плотно к нему примыкающему.
Второй комплекс включает в свой состав пять различнь х белков и выступа-
ет, будучи частично погруж иным, из сопрягающей мембраны в виде грибо-
426
видного выступа. Это Fj-фактор или сопрягающий фактор, непосредственно
ответственный за биосинтез АТФ. Его молекулярная масса, слегка варьирую-
щая в зависимости от объекта выделения, составляет в среднем 368000,
а субъединицы представлены полипептидами с М ~ 57 000 (а), 53 000 (Р), 34 000
(у), 17000 (8) и 10000 (е). По данным ряда авторов, субъединичный состав
сопрягающего фактора подчиняется формуле а303убЕ. Полагают, что катали-
тический центр, ускоряющий реакцию синтеза АТФ из АДФ и Н3РО4, нахо-
дится на Р-субъединице, а а-субъединица прикрывает его от воздействия
ингредиентов митохондриального матрикса. Есть также мнение, что Е-субъ-
единица регулирует деятельность протонной АТФазы, ингибируя ее способ-
ность гидролизовать АТФ. Аналогично построены и действуют хлоропластная
и бактериальная Н+-АТФазы.
Каким же образом возникает АТФ при посредстве АТФ-синтазы? На этот
вопрос еще нет исчерпывающего ответа, но предложено несколько заслужива-
ющих внимания концепций.
Первая из них сводится к предположению, что протоны, поступающие по
протонпроводящему каналу Fo, активируют неорганический фосфат (Рв), связан-
ный активным центром Р-субъединицы, отнимая от него ОН-группу (реакция
элиминирования воды). Одновременно ОН-группа концевого фосфата АДФ,
также присоединенного к активному центру р-субъединицы, теряет протон за
счет взаимодействия с ОН “-группой матрикса (где ОН “-группы накапливают
ся в результате переноса Н+ в межмембранное пространство, см. рис. 131).
Активированные фосфат и АДФ со диняются и образуют АТФ (рис. 133, Б, 1).
Вторая концепция состоит в допущении, что ионы Н в активном центре
сопрягающего фактора активируют фосфат и карбоксильную группу одной из
Субъедин ц Fj-фактора, в результате чего во никает фосфоэнзим, где фосфат
присоединен мак{ оэргической связью. При последующем взаимодействии
АДФ и фосфоэнзима образуется АТФ (рис. 133, Б, 2). Здесь в видоизмененном
состоянии представлена гипотеза переносчиков.
Третья конц пция исходит из предположения, что роль транслоцируемых
в Ft-фактор протонов состоит в изменении конформации /^-фактора. Обладая
не менее чем двумя центрами связывания АДФ и неорганического фосфата,
-фактор способен синтезировать АТФ из АДФ и Рн, если связывающий
центр находится в закрытом состоянии. При этом АДФ и Рн в нем находятся
в окружении аминокислотных радикалов, способствующих отнятию молекулы
воды и синтезу АТФ (рис. 133, Б, 3, правая сторона Fj-фактора). При трансло-
кации протонов центр связывания открывается, и АТФ из него поступает
в матрикс (левая сторона Fi-фактора на рис. 133, Б,3), а ее место занимают
АДФ и Рн. Новый цикл конформационных перестроек переводит этот центр
связывания в закрытое состояние с одновременным высвобождением готовой
АТФ из другого связывающего центра, переходящего в открытое состояние.
Легко заметить, что в этом объяснении механизма биосинтеза АТФ использо-
ваны идеи конформационной гипотезы сопряжения окисления с форфорилиро-
ванием.
В последнее время появились новые точки зрения на механизм АТФ-
синтазной реакции. А. Д. Виноградов предложил кинетическую модель,
в соответствии с которой синтез АТФ в АТФазном комплексе и гидролиз
АТФ Fj-ЛТФазой идут разными путями и каталитически ускоряются раз-
ными формами фермента, причем синтазные центры локализованы на
а-субъединице, а гидролазные—на 0-субъединице. Л. Ф. Дмитриев обосно-
вал вариант химической гипотезы, где роль интермедианта играет энергизи-
рованный липидный радикал сопрягающей мембраны и учитывается роль
электрохимического потенциала и внутримембранных протонов в процессе
427
сопряжения окисления с фосфорили-
рованием.
Кроме биосин еза АТФ, электрохи-
мический потенциал А|1Н+, возникаю-
щий на сопрягающей мембране и пере-
водящий ее в энергетически заряжен-
ное, энергезированное состояние,, яв-
ляется источником энергии для меха-
нической работы (например, для дви-
жения жгутиков у мутантов бактерий,
утративших синтез АТФ за счет сопря-
жения окисления с фосфорилировани-
ем, вращения хлоропластов у харовых
водорослей, вбуравливания цианобак-
терий в ил и т. п.), для поддержания
осмотического давления и переноса ве-
ществ против градие гга концентрации,
для теплопродукции при утрате мито-
хондриями дыхательного контроля
(например, при переохлаждении жи-
Рис. 134. Тигантские разветвленные митохон- ВОТНЫХ, В период испарения цветами
дрии в клетках почечных канальцев эфирных масел для привлечения насе-
комых и др.), для обращения вспять
переноса электронов в дыхательной цепи ферментов, для синтеза пирофосфата
и полифосфатов (как макроэргических соединений).
Источником энергезированного состояния мембраны может быть также
возникновение электрохимического потенциала AjiNa+, что характерно для
некоторых морских бактерий. У них ApNa+ используется для приведения
в движение жгутиков, создания солевых градиентов и, что самое важное, для
синтеза АТФ при посредстве №+-зависимой АТФазы (Ка+-АТФ-синтазы).
В связи с этим возникает вопрос о статусе других металлозависимых АТФаз
(Na+, К+-АТФазы, Са2+-АТФазы) и не исключено, что эта область исследова-
ний станет в перспективе «горячей точкой» биоэнергетики.
Сказанное, по мнению В. П. Скулачева, наводит на мысль о том, что
возникновение Ацн+, а в ряде случаев ApNa+ на сопрягающей мембране есть
универсальный способ запасения энергии в клетке, предшествовавший ее кон-
сервированию в макроэргических связях АТФ.
Более того, современные наблюдения говорят о том, что в клетках, например
мышечных, существует митохондриальный ретикулум, при помощи которого
митохондрии связаны в единую цепь или представляют собой одну гигантскую
разветвленную митохондрию. По ее энергезированной мембране Ацн+ может
передаваться на значительные расстояния для того, чтобы в нужном месте
обеспечить синтез необходимого количества АТФ или выполнить иные функции,
присущие мембранному электрохимическому потенциалу (рис. 134).
ЭНЕРГЕТИЧЕСКИЙ БАЛАНС РАСПАДА УГЛЕВОДОВ
И ТРИГЛИЦЕРИДОВ
Если суммировать уравнения реакций, которые осуществляются при бро-
жении, гликолизе или полном окислении глюкозы по апотомическому или
дихотомическому пути, то легко прийти к следующим результатам.
428
Неполное окисление
Брожение
С6Н12Об + 2Н3РО44-2АДФ —>2С2Н5ОН 4- 2СО2+2АТФ+2Н2О
Гликолиз
СбН12О6 + 2Н3РО4+2АДФ —»2СН3 - СН (ОН) - СООН+2АТФ 4- 2Н2О
Полное окисление
Апотпомический путь
СбН! 2Об+АТФ + 7Н2О 4-12НАДФ+—> 6СО2+АДФ4-Н3РО4 4-
4-12НАДФН4-12Н+
Дихотомический путь
СбН12О64-4Н3РО44-4АДФ4-ЮНАД+4-2ФАД4-2Н2О—►
—> 6СО24- 10НАДН4- ЮН+ 4-2ФАДН24-4АТФ
Если принять, что все атомы водорода, снятые с субстратов при полном
окислении, будут направлены в дыхательную цепь ферментов и их дальнейшая
передача на кислород пройдет сопряженно с фосфорилированием, то в каждом
случае должно образоваться определенное, но разное число молекул АТФ.
Размеры синтеза АТФ, или, что то же самое, энергетический эффект распада
углеводов, выразятся для каждого пути распада следующим образом:
Пути распада углеводов Синтезируется
молекул АТФ
Брожение........................ 2
Гликолиз ....................... 2
Дыхание
апотомический путь..............• 35
дихотомический путь......... 38
Таким образом, для организма наиболее выгоден в энергетическом :мысле
обмен углеводов, идущий по пути дыхания,—он является источником мак-
симального числа молекул АТФ, запасаемых в организме. В этом случае
наилучшим образом консервируется энергия. Брожение и гликолиз дают
ничтожное количество АТФ и могут лишь кратковременно или в особых
условиях заменять окислительный распад углеводов.
Если сопоставить изменение уровней свободной энергии при брожении,
гликолизе и дыхании с количеством энергии, запасенной в макроэргических связях
АТФ в этих же случаях, то оказывается, что при спиртовом брожении запасается
27,8%, при гликолизе—32,8, при полном окислении по апотомическому пути—
39,6, а по дихотомическому пути—43,0% энергии, способной выделиться при
сгорании глюкозы. Таким образом, не только общий уровень запасаемой энергии,
но также и доля ее от общего количества высвобождаемой энергии оказывается
максимальной в случае полного распада глюкозы по дихотомическому пути.
Каков же энергетический эффект окисления другого класса органических
соединений—триглицеридов, окислительный эаспад которых сопряжен с фос-
форилированием? Проведем соответствующие расчеты для реакции окисления
тристеарина.
В результате гидролиза тристеарина получается одна молекула глицерина
и три молекулы стеариновой кислоты. При окислении глицерина до СО2
429
и Н2О на фосфорилирование глицерина расходуется одна мол( куда АТФ; по
три молекулы АТФ возникают сопряженно с окислением фосфоглицерина
в фосфодиоксиацетон и 3-фосфоглицеринового альдегида в 3-фосфоглицерат.
Две молекулы АТФ синтезируются при окислении 3 фосфоглицеринового
альдегида в пировиноградную кислоту (за счет 1,3-дифосфоглицериновой
и 2-фосфоенолпировиноградной кислот) и 15 молекул АТФ образуются при
окислении пировино радной кислоты в цикле трикарбоновых и дикарбоновых
кислот. Таким образом, за счет окисления глицерина (при условии, что все
атомы Н, снятые дегидрогеназами, идут в дыхательную цепь ферментов,
функционирующую сопряженно с окислительным фосфорилированием) об-
разуется 22 молекулы АТФ.
Каждый акт р-окисления дает две пары атомов водорода, которые тоже
могут быть направлены в дыхательную цепь, сопряженную с фосфорилирова-
нием, что приносит 5 молекул АТФ (две за счет передачи в дыхательную цепь
атомов водорода с ФАД-Н2 и три—с НАДН+Н*, см. рис. 129). На каждый
остаток стеариновой кислоты требуется 8 актов р-окисления; значит, распад
одной молекулы стеариновой кислоты до ацетил-КоА будет эквивалентен
синтезу 40 молекул АТФ, тогда как распад трех молекул (соответственно
одной молекуле тристеарина)—синтезу 120 молекул АТФ. Памятуя о том, что
при активировании каждой молекулы высшей жирной кислоты расходуется
одна молекула АТФ, эту цифру следует уменьшить на 3. Таким образом,
чистый синтез АТФ в процессе окисления трех молекул стеариновой кислоты
до ацетил-КоА будет составлять 117 молекул.
Если допустить, наконец, что все ацетильные остатки из молекул ацетил-
КоА (их получается 27 из каждой молекулы тристеарина) подвергнутся полно-
му окислению при помощи цикла трикарбоновых и дикарбоновых кислот, то
при условии сопряжения с фосфорилированием это даст 324 молекулы АТФ
(при окислении каждого ацетильного остатка в цикле снимается 4 пары
атомов водорода, чему соответствует 4x3 = 12 молекул АТФ; 12x27 = 324
молекулы АТФ). Таким образом, всего при распаде тристеарина может быть
запасено 22+117+324=463 молекулы АТФ. Это составит 42,2% от полного
количества энергии, выделяющейся при сгорании тристеарина, что сопостави-
мо со степенью запасания энергии при окислении углеводов. Поэтому жиры
наряду с углеводами являются в организме главным источником энер ии.
ГЛАВА XI
ВОДНЫЙ И МИНЕРАЛЬНЫЙ ОБМЕН
ВОДНЫЙ ОБМЕН
Содержание и распределение воды в организме и клетке. Ранее было
отмечено (см. гл. I), что вода составляет около 3/4 биомассы Земли. Ее
содержание в организмах примерно в 5 раз превышает количество воды во
всех реках земного шара. У разных организмов и особенно в различных
тканях животных и растений содержание воды колеблется в значительных
пределах. Так, в биологических жидкостях (пасока деревьев, кровь, лимфа,
слюна, желудочный сок животных и т. п.) содержится от 88 до 99% воды,
тогда как в древесине растений или костной ткани животных ее количество
измеряется 20—24%.
Чем моложе организм или орган, тем выше в нем содержание воды.
Отличной иллюстрацие этому является постеп иное обезвоживание организ-
ма человека и животных в процессе старения, сопровождающееся характер-
ным смо щиванием кожных покровов. Аналогично этому происходит посте-
пенное снижение содержания влаги в расте иях по мере их вегетации. Кроме
того, у древесных растений верхушечные (молодые) листья всегда значительно
богаче водой, чем приствольные (более старые) листья.
Большая часть воды в орг низме локализована в его клетках. Эту воду
называю* внутриклеточной. В противоположность этому вода, сосредоточен-
ная в межклеточном пространстве или входящая в состав биологических
жидкостей, называв' я внеклеточной. Так, в организме человека 2/3 составляет
внутриклеточная, а г/3—внеклеточная вода. По-видимому, содержание воды
в клетках в какой-то мере коррел рует с интенсивностью процессов жизнеде-
ятельности в них. Так, содержание воды в активно делящихся клетках достига-
ет 80% и в некоторых случаях даже 90%.
Долгое время считали, что, по крайней мере, в растительной клетке глав-
ная масса воды сосредоточена в вакуоли. Однако более тщательные определе-
ния показали, что лишь около 1/3 внутриклеточной воды приходится на
вакуоль а остальная ее часть локализована в протоплазме и клеточной стенке.
Из субклеточных частиц наименьшим содержанием воды отличаются ядры-
шко и липидные включения; ядерный сок, матрикс митохондрий и гиалоплаз-
ма достаточно богаты водой тогда как в липопротеиновых структурах эндо-
плазматического ретикулума ее содержание невысоко.
Состояние воды. В простейшей бактериалыой клетке на одну молекулу
нуклеиновой кислоты приходится около 100000, на одну белковую молеку-
лу—примерно 40000, а на каждую молекулу липидов—приблизи ельно
1500 молекул воды (см. гл. I). Таким образом, молекулы органических соеди-
нений в клетке постоянно окружены молекулами воды и, конечно, взаимод й-
ствуют с ними. Нельзя упускать из вида также взаимодействие молекул воды
друг с другом и неорганическими катионами и анионами.
431
Молекула воды полярна и обладает определенным дипольным моментом.
Вследствие этого молекулы воды соответствующим образом ориентируются
по отношению друг к другу, образуя структурированную систему. Попадая
в поле действия иона, молекулы воды образуют вокруг него гидратную
оболочку. Указанное взаимодействие сопровождается разрушением структуры
самой воды (эффект разупорядочения). Если вновь возникающая упорядочен-
ность в расположении молекул воды вокруг иона меньше, чем в самой
структурированной воде, то наблюдается так называемая отрицательная гид-
ратация, заключающаяся в том, что молекулы воды вблизи иона обладают
большей подвижностью, чем в чистой воде. В частности, отрицательная
гидратация свойственна ионам К, Rb, Cs, Cl и I, а положительная—ионам Na,
Li, Са и Ва. Аналогичные явления происходят также по катионным и анион-
ным центрам органических молекул, в том числе и макромолекул. Таким
образом, известная часть молекул воды в клетке находится в связанном
состоянии за счет участия в процессах гвдратации.
Не менее важную роль в структурировании самой воды и ее взаимодейст-
вии с макро- и микромолекулами играют водородные связи. Благодаря им
еще некоторая часть молекул воды в клеточном содержимом переводится
в связанную форму.
Предполагают, что вблизи гидрофобных участков макромолекул вода
ор авизуется в льдоподобную структуру, сп собствующую поддержанию тре-
тичной структуры ряда биополимеров, особенно белков. Естественно, что
и эту воду следует причислить к категории связанной воды.
Наконец, вода может захватываться внутрь как отдельных макро олекул
при формировании их третичной структуры, так особенно и надмолекулярных
биокомплексов, столь характерных для клеточного содержимого.
Сказанное выше описывает случаи перевода воды в связанное состояние на
молекулярном уровне. Однако при более глубоком исследовании состояния
воды в субклеточных структурах вырисовываются некоторые новые возмож-
ности. Так, хорошо известна высокая пористость рибосом, содержащих в на-
тивном состоянии воды в 2,7 раза больше, чем сухие рибосомы. Белково-
липидные мембраны митохондрий, лизосом, эндоплазматического ретикулу-
ма и других субклеточных структур изолируют от внутриклеточного про-
странства значительные емкости, в которых наряду с другими веществами
сосредоточена вода. Аналогичную роль выполняет ядерная оболочка. Таким
образом, в составе субклеточных структур также связано значительное количе-
ство воды. Ее иногда называют иммобильной водой.
Следовательно, есть все основания выделять две категории воды в организ-
ме: свободную и связанную. Последняя в зависимости от типа связанности
с большей или меньшей силой удерживается организмом. В соответствии
с этим ее, в свою очередь, подразделяют на слабосвязанную воду (вода диффу-
зионных слоев гидратационных оболочек, структурированная вода и т. п., т. е.
вода, которая служит растворителем и замерзает при температурах, близких
к 0° С) и прочносвязанную воду (вода 1-го гидратационного слоя, как «чулком»
охватывающая молекулу, почти не способная быть растворителем и замерза-
ющая при температурах—25° С и даже—269° С). Не исключено, что внутри-
клеточная вода непрерывно претерпевает регулируемые белками пульсацион-
ные переходы из упорядоченного в разупорядоченное состояние, взаимосвя-
занное с выталкиванием из клетки отслуживших метаболитов (шлаков)
и всасыванием необходимых веществ.
С современной точки зрения, вода организма не может рассматриваться
как инертная среда, заполняющая пространство между макромолекулами,
субклеточным истицами, клетками и т. д. Напротив, она должна расцени-
432
ваться в качестве структурного элемента как клеточного содержимого, так
и организма в целом. Только в этом случае может быть полностью оценено
и понято ее огромное значение в процессах жизнедеятельности, ее способ-
ность, по выражению А. Сцент-Дьердьи, быть «матрицей жизни».
Роль воды в процессах жизнедеятельности. В результате многочисленных
опытов было установлено, что вода в жизни организма играет важнейшую
роль. Так, полная утрата жиров и понижение наполовину содержания белков
в организме в результате голодания не столь опасны, как потеря 20% воды.
Продолжительность жизни голодающих собак, например, можно повысить
в 10 раз, если снабжать их водой.
Это объясняется тем, что вода в организме выполняет весьма существен-
ные и разнообразные функции. Вместе с другими соединениями она участвует
в качестве основного компонента в формировании единой внутриклеточной
структуры, благодаря которой достигается характерная для живого тонкая
упорядоченность процессов. Будучи составной частью субклеточных структур,
вода в значительной мере способна регулировать их функциональную актив-
ность: например, от степени набухания митохондрий зависит интенсивность
протекающего в них процесса окислительного фосфорилирования, от насыще-
ния водой рибоссгм—поддержание их структуры и способности осуществлять
белковый синтез в т. п. Закономерности связывания воды с биологически
активными веществами лежат в основе регуляции и саморегуляции ряда биохи-
мических и физиологических процессов. Поэтому, как справедливо указывает!
А. Сцент-Дьердьи, «нельзя говорить о белках, нуклеиновых кислотах и нуклео-
протеидах и о воде так, как если бы это были две различные системы. Они
образуют единую систему, которую нельзя разделить на компоненты без
разрушения ее сущности». Например, только при содержании воды в количе-
стве 0,6 г на 1 г сухой ДНК последняя сохраняет нативные свойства.
Являясь той основной средой, в которой внутри клетки распределены
молекулы разнообразных биополимеров, вода непосредственно участвует
в формировании золей и гелей протоплазмы. Переход золей в гели и обратно
сопровождается часто явлением тиксотропии (от греч. тиксис—прикоснуться
и троп—поворот, изменение), т. е. разжижением геля под влиянием механи-
ческих сил (например, встряхивания) и обратным его застыванием. Этому
сопутствует резкое изменение вязкости, тесно связанное с рядом физиологиче-
ских и биологических явлений: мышечным сокращением, дифференциацией
некоторых внутриклеточных структур, клеточным делением, движением про-
топлазмы и т. п.
Обладая низкой вязкостью, подвижностью и способностью растворять
большое число неорганических и органических соединений, вода выполняет
в организме транспортные функции. Она же служит для выведения из организ-
ма продуктов распада. Указанный перенос веществ осуществляется как в боль-
ших масштабах—по специальным транспортным системам (кровеносная
и лимфатическая система животных, проводящие пучки ксилемы и флоэмы
растений), так и в небольших дозах—через клеточные и внутриклеточные
мембраны.
Вода в живой природе является средой, в которой протекают многочислен-
ные и разнообразные химические процессы. Большая диэлектрическая посто-
янная воды обеспечивает электролитическую диссоциацию веществ, способ-
ных распадаться на ионы. Однако роль воды в биохимических процессах не
ограничивается функцией растворителя. Во многих случаях она сама является
непосредственным участником химических реакций. Таковы реакции гидроли-
за, гидратации и дегидратации, окисления и многие реакции синтеза, идущие
либо с поглощением, либо с выделением воды.
433
Высокая теплопроводность воды и значительное поглощение теплоты
в процессе ее испарения являются основой использования воды растениями
и животными для регуляции и выравнивания температуры тела. Этому же
служит достаточно высокая теплоемкость воды.
Обмен воды. Благодаря испарению через кожу с выдыхаемым воздухом,
с выделяемой мочой и калом человек теряет в среднем 2600 мл воды в сутки.
Опыты показали, что 6/у указанной убыли воды восполняется за счет ее
поступления с пищей. Долгое время дефицит воды, составляющий примерно
350 мл, оставался неизвестным. Однако позднее было выяснено, что это
количество влаги организм человека получает в результате образования воды
при обмене веществ. Эта вода, образующаяся в процессе жизнедеятельности
организма, получила название эндогенной воды в отличие от поступающей
в организм извне экзогенной воды. В эмбриогенезе вредной черепашки, напри-
мер *74 часть воды, а при прев ащении куколки тутового шелкопряда в бабоч-
ку почти вся вода имеет эндогенное происхождение.
Эндогенная вода синтезируется в результате окисления органических со-
единений. П дсчитано, что из 100 г жиров при полном окислении их получает-
ся 107,1 г воды, углеводов — 55,5 и белков—41,3 г. Тот или иной уровень
образования эндогенной воды характерен, по-видимому, для всех организмов.
Не менее, а, видимо, даже более энергично осуществляется обмен воды
в растениях. Так, в течение вегетационного периода одно растение кукурузы
или подсолнечника испаряет до 200 кг воды. В жаркий день количество
проходящей через лист воды в 2 раза пр вьппает массу самого листа. В засуш-
ливых условиях скорость восходящего потока воды в древесине достигает
25 м/мин.
В организме вода доставляется к каням и отводится от них в составе
биологических жидкостей. Проникновение воды в клетки и обратно осуществ
ляется через поры клеточных мембран, диаметр которых 0,3—0,4 нм. По
поводу механизма пр никновения воды через клеточную мембрану высказано
несколько предположений. Считают, что перенос воды идет активно за счет
взаимодействия дипольных молекул ее с полярными веществами, из которых
состоит стенка поры. Есть мнение, что обмен молекулами зоды между внут-
риклеточным и внеклеточным пространством осуществляется за счет свобод-
ной диффузии. Полагают также, что решающая роль в проникновении воды
в клетку и выходе ее обра но принадлежит осмотическим явлениям. Но так
или иначе указанный процесс протекает достаточно энергично, и время полу-
обмена воды в клетках ряда тканей составляет 30—90 с. Оно значительно
превышает время полуобмена органических молекул или ионов в аналогичных
опытах.
Регуляция обмена воды. Некоторые катионы специфически влияют на
адержку и отдачу воды кле ками и тканями. Так, Na+ вызывает накопление
воды в клетках и тканях, тогда как К+ и Са2+ оказывают прямо проти-
воположное действие.
В организме животных мощное влияние на баланс воды оказывают гормо-
ны: диуретический гормон, выделяемый передней долей гипофиза, способству-
ет усиленному выделению воды из организма с мочой (диурез), а антиди-
уре ический гормон (вазопрессин—см. с. 449), образуемый задней долей
гипофиза, повышает обратное всасывание воды в почечных канальцах и резко
сокращает диурез. Указанная регуляция осуществляется, таким образом, на
уровне органа—почек. Однако само соотношение в организме упомянутых
выше гормонов регулируется, по-видимому, центральной нервной истемой.
Существуют еще некоторые механизмы регуляции водного обмена при
посредстве тканей и органов. Так, в коже и печени животных и человека
434
располагаются депо воды, и при ее избыточном поступлении здесь накаплива-
ются водные резервы. В растениях запас ая вода концентрируется в меж-
клеточных пространствах, а уровень ее испарения регулируется у тьичным
аппаратом.
МИНЕРАЛЬНЫЙ ОБМЕН
Огромную роль в разви ии представлений об обмене минеральных соеди-
нений сыграли работы К. А. Тимирязева, а его исследование о значении цинка
в питании растений (1872) было одной из первых работ, положивших начало
учению о микроэлементах. Наверное, это не было случайностью, так как
согласно современным данным Zn является рекордсменом по числу Zn-
зависимых ферментов—их насчитывается уже 120. В блестящих работах
В. И. Вернадского (1922) по азана теснейшая связь состава организмов с со-
ставом земной коры, а в трудах его ученика—А. П. Виноградова (1939)
развита идея о биогеохимических провинциях суть которой сводится к установ-
лению тончайшей взаим связи элементов в системе почва—растения—жи-
вотные. Следствием избытка или недоста ка того или иного элемен а в биогео-
химической провинции являются эндемии—заболевания рас ений и живот-
ных. На современном этапе развития учения о минеральном обмене все более
пр слеживается связь м жду биологической активностью и строением атома
и, следовательно, местоположением его в периодической системе элементов
Менделеева.
Изучение роли минеральных элементов в обмене веществ недавно привело
к возникновению новой науки—бионеорганической химии (или неорганической
биохимии), предмет которой охарактеризован выше (см. Введение).
Участие минеральных веществ в формировании третичной и четвертичной
структуры биополимеров Наиболее фундаментальный механизм участия мине-
ральных соединений в пре цессах жизнедеятельности связан прежде всего с их
способностью со диняться с высокомолекулярными веществами—белками
и нуклеиновыми кисл >тами. В результате указанного взаимодействия ионы
металлов гаряду с другими фак орами обеспеч вают поддержание о ределен-
ной пространственной конфигурации биополимеров, которая далеко не безразли-
чна для проявления биологической активности макромолекул. Таким образом,
нормальное осуществление белками ферментативной, гормональной и других
функций, бес репятственна реализация информации, заключенной в нукле-
иновых кислотах, образование надмолекулярных комплексов, формирование
субклеточных частиц и т. п, немыслимы без участия катионов и анионов.
Приведем несколько примеров для иллюстрации высказанных выше общих
положений. В формировании активной формы гормона инсулина (см. с. 451)
выдающаяся роль пр надлежит Zn2+. Возможно, что отсутствие должного
уровня гормональной активности у син етического инсулина связано с ка-
кими-то тонкими особенностями структуры которые препятствуют возник-
новению биологически активного комплекса протомеров с Zn2+. Та или иная
конформация молекул высокополимерной РНК в огромной степени определя-
ется ионной силой раствора, а многие двухвалентные катионы (Cr, Ni, Fe, Zn,
Мп и др.) принимаю непосредственное участие в формировании спиральной
струк уры нуклеиновых кислот.
Очень яркой иллюстрацией участия катионов в образовании надмо-
лекулярных структур является зависимость процесса ассоциации—диссо-
циации рибосом от концентрации Mg2+ (см. с. 285); он же стабилиз рует
третичную структуру тРНК. Деятельность подавляющего большинства
435
ферментов, принимающих участие в свободном и сопряженном с фосфорили-
рованием окислении органических соединений, немысл ма без участия ионов
Fe и Си.
Число подобных примеров можно было бы умножить. Все они свидетельст-
вуют о том, что становление структуры биополимеров и биокомплексов
в значительной мере определяется наличием или отсутствием достаточного
количества тех или иных катионов и анионов в клеточном содержимом. Поэтому
естественно, что минеральные вещества оказывают огромное влияние на ход
ферментативных процессов, обмен различных соединений, формообразователь-
ные процессы и т. п. Роль их в жизнедеятельности организмов очень велика.
Участие минеральных веществ в ферментативном катализе. Действие более
четвертой части известных в настоящее время ферментов тем или иным
образом связано с металлами. В большинстве случаев ионы металлов вступа-
ют в непрочную связь с апоферментом, образуя с ним легко распадающийся
комплекс. В виде комплекса с металлом фермент проявляет максимальную
активность, приобретая со тветствующую пространственную конфигурацию.
Таким образом, здесь ионы металлов выступают как организаторы третичной
структуры фермента, в частности как организаторы активных центров фермен-
тов. Именно так обстоит дело при взаимодействии ионов одновалентных
металлов более чем с 60-ю и ионов цинка более чем с 30-ю ферментами
животного, растительного и бактериального происхождения. Велика роль
отдельных катионов в формировании ферментов—мультимеров, где связь
между отдельными протом рами осуществляется при участии ионов метал-
лов. К числу таких катионов относятся Mg2+, Mn2+, Zn2+, Са2+ и др.
Особенно подробно в указанном смысле изучена а-амилаза. В присутствии
Са2+ стабилизируются третичная и четвертичная структуры этого фермента,
устойчивые по отношению к пептидгидролазам желудочно-кишечного тракта.
ТЬраздо реже ионы металлов образуют с белком-ферментом прочное со-
единение. В этом случае металл не отделяется от фермента при диализе или
пропускании через ионообменник, т. е. фермент представляет собой истинный
металлопротеин. Так, например, осуществляется соединение ионов Си и Fe
в железосерных белках и синих оксидазах (см. с. 414). В какой-то мере упомя-
нутым металлопротеинам подобны селенопротеины, привлекающие внимание
в последнее время (в их числе такие ферменты, как формиат- и ксантиндегид-
рогеназа, тиолаза, глутатионпероксидаза и др.).
Достаточно широко распространено участие ионов металлов в действии
ферментов путем вступления ионов металлов в состав простетической группы
ферментов. Классическим примером ферментов подобного типа могут слу-
жить цитохромы.
Наконец, многие катионы (Mg2+, Mn2+, Zn2+ и др.) активно участвуют
в ферментативном катализе, связывая на короткое время очень непрочными
связями либо субстрат и фермент (при возникновении фермен* -субстратного
комплекса), либо кофермент с апоферментом (в двухкомпонентных ферментах).
Прим ры первого рода весьма многочисленны; они характеризуют одно из
самых старых представлений о возможной роли катионов в ферментативном
катализе. Образование тройного фермент-металл-субстратного комплекса от-
мечено при действии аргиназы, пептидгидролаз, карбоксилаз и многих других
ферментов. Примером второго рода может служить присоединение флавино-
вого кофермента к апоферменту при посредстве ионов Fe, Мо, Си и Zn.
В табл. 26 приведены некоторые данные об активировании катионами
металлов некоторых ферментативных процессов.
Благодаря тому что минеральные соединения тесно связаны с фермента-
тивными реакциями, они практически влияют на все стороны обмена веществ.
436
Рассмотрим конкретно, в чем выражается зависимость обмена нуклеиновых
кислот, белков, углеводов и липидов от содержания тех или иных катионов
и анионов.
Минеральные соединения и обмен нуклеиновых кислот. Ряд катионов прини-
мает непосредственное участие в поддержании вторичной и третичной структур
ДНК и РНК. В частности, эта функция приписывается ионам Fe, Си, Мп, Zn,
Со и Ni, обнаруженным в чистейших препаратах ДНК и РНК. В присутствии
некоторых из перечисленных катионов повышается температура плавления
нуклеиновых кислот, что свидетельствует о более высокой стабильности их
молекул. Предполагают, что указанная стабилизация достигается благодаря
возникновению межмолекулярных сшивок через ионы металлов по типу
«слоеного пирога». Аналогично этому ионы металлов принимают участие
в строении нуклеопротеинов, связывая молекулы белков и нуклеиновых кис-
лот. Вместе с тем обнаружено, что тяжелые металлы (медь, ртуть, кадмий
и цинк) изменяют основные этапы реализации генетической информации
(синтез ДНК, РНК и белка) у бактерий, водорослей и простейших.
Таблица 26
Ферменты, активируемые катионами металлов
Класс ферментов Наименование ферментов Наименование металлов, активирующих фермент
I. Оксидоредуктазы Аскорбатоксидаза Си
Полифенолоксидаза Си
Ксантиноксидаза Мо
II. Трансферазы Ацетилтрансфераза Mg, К
1ёксокиназа Mg, Мп
Аминоацилтрансфераза Мп
III. 1идролазы Аргиназа Mg, Со
Аденозинтрифосфатаза Мп
Карбоксипептидаза Zn
Фосфатаза Со
IV. Лиазы Альдолаза Zn, Со
Карбоксилаза Мп, Си, Zn, Со
V. Лигазы Аминоацил-тРНК-синтетаза Mg, Zn
Ацил-КоА-синтетаза Mg
VI. Изомеразы Фосфоглюкомутаза Мп, Со
Почти все ферменты, ускоряющие реакции распада и синтеза нуклеиновых
кислот, нуклеотидов, нуклеозидов, пуриновых и пиримидиновых оснований,
активируются ионами металлов. Особенно велика роль Mg2+: он активирует
ДНК-полимеразу и РНК-полимеразу, полинуклеотидфосфорилазу, нуклеоти-
дазу, РНКазу, ДНКазу и ряд других ферментов нуклеинового обмена. Ионы
Са и Ва увеличивают, a Zn уменьшают активность РНКазы. Недавно установ-
лено, что Zn2+ необходим для функционирования ДНК-полимераз и обратной
транскриптазы. Распад ДНК под действием ДНКазы повышается в присут-
ствии Mn2+, Са2+, Fe2+ и Со2+. Ионы Мп активируют фосфодиэстеразы,
ионы Мо—ксантиноксидазу, а ионы меди тормозят действие последней. Se4+
избирательно ингибирует транскрипцию рибосомальных генов; аналогично
этому соединения La, Рг, Nd и Sm тормозят биосинтез РНК в ядрах клеток
печени.
Интересные данные получены о роли бора в обмене нуклеиновых кислот.
В отличие от всех других элементов он не является кофактором или
437
активатором ферментов. Однако при отсутствии бора сильно тормозится
новообразование нуклеиновых кислот и усиливается их распад вследствие
повышения активности РНКазы. Предполагают, что влияние бора на обмен
нуклеиновых кислот связано с его участием в окислительном фосфорилирова-
нии и синтезе нуклеозидтрифосфатов—исходньх соединений для биосинтеза
нуклеиновых кислот. Не меньшее значение для сопряжения окисления с фос-
форилированием имеет Mg2+, так как комплекс Mg2+- АДФ непосредственно
участвует в АТФ-с нтазной реакции (см. гл. X).
В последнее время все большее значение придают роли ионов Си и Мо
в обмене нуклеиновых кислот.
Роль минеральных элементов в обмене белков. Распад и синтез белковых тел
в значительной степени зависят от ряда минеральных элементов. Ионы Мп,
Fe, Zn, Со и Ni повышают активность пептидгидролаз и аргиназы, г. е.
участвуют в деструкции белков. Биосинтез белков идет при непосредственном
участии ионов К, Mg и Мп. Первый и второй необходимы для поддержа i я
рибосом в функционально активном состоянии, причем от концентрации Mg2+
зависит уровень помех в кодон-антикодоновом взаимодействии. Третий обес-
печивает осуществление пептидилтрансферазной реакции при сборке полипеп-
тидной цепи. Ионы Ni влияют на высвобождение многих пептидных гормонов
по завершении их биосинтеза, а ионам Са принадлежит центральная роль
в функционировании сократительных белков. Си2+-содержащий белок мито-
хондрий—митохондрокупреин, представляет депо меди для син еза цито-
хромоксидазы.
Вопрос о роли минеральных элементов в обмене аминокислот разработан
еще недостаточно, однако следует отметить большое значение ионов Со
в биосинтезе метионина, Mg и Мп—в реакциях обмена аминокислот, связан-
ных с переносом одноуглеродных фрагментов (синтез серина из глицина,
цитруллина—из орнитина и карбамилфосфата и др.), Fe—в превращении
фенилаланина в тирозин, Zn—во включении глицина в глутатион печени,
Se—в окислении HS-групп радикалов цистеина в белках.
Участие минеральных соединений в обмене углеводов и липидов. Различные
катионы принимают активное участие в распаде и синтезе как непосредственно
углеводов и липидов, так и продуктов их деструкции, претерпевающих окон-
чательное разрушение в цикле трикарбоновых и дикарбоновых кислот.
Центральным элементом в гликолитическом процессе является Mg: он
активирует большинство ферментов гликолиза. В ряде случаев (гексокиназная
реакция, переход от 1,3-дифосфоглицериновой кислоты к 3-фосфоглицерино-
вой кислоте и от 2-фосфоглицериновой кислоты к фосфоенолпировиноградной
кислоте) он может быть заменен Мп. Значение Са2+ для поддержания струк-
туры а-амилазы отмечено ранее; подчеркнем лишь что лантаниды, например
Lu3+, столь же успешно активируют а-амилазу, как и Са2+. Новообразование
углеводов в процессе фотосинтеза невозможно без участия магния (составная
часть хлорофилла), марганца (участвует в фотосинтетическом фосфорилирова-
нии) и железа (необходимо для биосинтеза хлорофилла). Ряд других элемент в
(Си, Zn, Мо, Со и В) не безразличен для хода фотосинтеза.
Распад липцдов, как простых, так и сложных, активируется Са2+, так как
этот катион положительно влияет на деятельность липазы, фосфолипаз
А и С и липопротеинлипазь. р-Окясление ацил-КоА идет более энергично
в присутствии ионов Си и Fe. При синтезе ацетил-КоА, фо фохолина и холин-
фосфатидов необходим Mg2+.
Заключительный этап аэробного распада углеводов и липидов при посред-
стве цикла трикарбоновых и дикарбоновых кислот (дых ние) осуществляется
при активном участии Мп2+, который активирует почти все ферменты цикла
438
Кребса. Аналогичное действие присуще Mg2+ и в некоторых случаях Со2+
и Zn2+.
Обмен минеральных веществ. Минеральные вещества, посту ающие в рас-
тительный или животный организм и необходимые для ос ществления в нем
тех или иных функций, задерживаются в организме, образуя в подавляющем
большинстве случаев специфические соединения. Концентрирование элементов
в живой природе видоспецифично и наследственно. Так, свыше 150 растительных
видов (пасленовые, лютиковые и др.) накапливают Li, плауны—А1, морские
водоросли—I (в виде производных тирозина) и поливалентные металлы (в
количестве 108 т ежегодно), обыкновенный мухомор—Se (в виде Se-цистеина,
где Se заменяет S) и т. п.
Из макроэлементов Са- и Р у высших животных образуется фосфат каль-
ция—основа костной ткани. Сера в значительной мере в тупает в состав
органических соединений (HS-группы аминокислот, пептидов и белков, HO3S-
группы в гетерополисахаридах). Фосфор также весьма часто присутствует
в виде органических производных (фосфорные эфиры, фосфопротеины и пр.).
Для Mg, К и Na характерна ионная форма существования, уравновешенная
в основном анионами хлора, фосфата и карбоната. Mg и Fe широко представ-
лены в составе хлорофилла и гемоглобина.
Микроэлементы в большинстве своем вступают во взаимодействие с бел-
ками и нуклеиновыми кислотами либо йепосредственно, либо предварительно
включаясь в состав простетических групп органической природы.
Хотя, по-видимому, и не все да, но в большинстве случаев минеральные
вещества, бесполезные для организма, не усваиваются. Примером может
служить кремнекислота: при 5 %-ном уровне ее соде жания в скармливаемой
животным зеленой массе растений (на сухое вещество) кремнекислота не
усваивается животными, полностью выводится с экскрементами и использует-
ся как инертный носитель для расчета усвояемости корма. Вместе с тем
появились данные о возможном участии кремниевой кислоты в созревании
коллагена и образовании протеогликанов, а также о положительном влиянии
ряда кремнийорганических соединений на продуктивность животных.
Обмен ряда минеральных элементов протекает весьма энергично. Это
особенно ярко выявляется в тех случаях, когда элемент экскретируется из
организма в составе какого-либо нормального продукта жиз едеятельности.
Так, у млекопитающих большие количества Са и Р выводятся из организма
в процессе лактации. Но и в составе клеток отдельные элементы обменивают-
ся достаточно интенсивно, что выражается в изменении их концентрации
в субклеточных структурах и компартментах клетки, а также активном перено-
се через мембраны. С участием разнообразных бактерий Hg, As, Те, Tl, Au, Pb,
Sn и Cd метилируются с образ ванием крайне токсичных продуктов (Hg, Pb),
гибельных для животных в нанограммовых количествах, хотя человек может
переносить дозу метилртути в 0,03 мг ежедневно без каких-либо последствий.
Характерной особенностью обмена минеральных элементов является, с од-
ной стороны, взаимозаменимость ряда из них и, с другой—антагонизм дей-
ствия. Так, в ферментативных процессах, там, где К+, NH4 или Rb+ выступа-
ют как активаторы (например, при действии альдегиддегидрогеназы из дрож-
жей), Na+, Li+ или Cs+ являются ингибиторами. В таких же отношениях
находятся Mg2+ и Са2+, Мп2+ и Zn2+, Ni2+ и Cu2+ и т. п. Однако для ионов
Ni, с одной стороны, и Zn и Fe—с другой, характерен синергизм действия.
Изменение валентности элемента в процессе его обмена сопровождается
резкой сменой его физиологической активности.'Так, Сг3+ стимулирует белко-
вый, углеводный и липидный обмен у человека и животных, а Сг6+ блокирует
окислительное фосфорилирование в их митохондриях.
439
Значение минеральных веществ в сельском хозяйстве. Широкое участие
минеральных соединений в построении тканей животных и растений, в обес-
печении структурных особенностей биополимеров и в разнообразных биохи-
мических процессах в организме определяет их громадное значение для рас-
тениеводства, животноводства и медицины.
Выдающаяся роль в разработке теоретических аспектов минерального
питания и практического приложения их в сельском хозяйстве принадлежит
К. А. Тимирязеву и его ученику Н. Д. Прянишникову. Намеченная ими широ-
кая твор 1еская программа проникновения химии в сельское хозяйство, полу-
чившая название химизации, энергично проводилась в жизнь в нашей стране.
Занимая второе место в мире по производству минеральных удобрений,
наша химическая промышленность освоила выпуск ряда новых их видов
(нитрофоска, мочевина, обесфторенные фосфаты и др.). Принципиально важен
переход на концентрированные удобрения, без балластных веществ. Ряд про-
блем возник в связи с необходимостью упорядочения применения удобрений
в сельскохозяйственном производстве (проблема нитратов и нитритов, засоле-
ние почв, использование отстойных масс с полей орошения, цветение водо-
емов и др.), а также техногенном загрязнении среды минеральными элемен-
тами в концентрациях, губительных для окружающей природы. Их решение
может быть достигнуто лишь при посредстве мероприятий в рамках глобаль-
ных экологических программ.
База для производства калийных и азотных удобрений безгранична. Одна-
ко залежей фосфоритов хватит всего на несколько десятилетий, но есть
надежда, что удастся найти и использовать фосфаты вулканического проис-
хождения. Что касается микроэлементов, то современная химическая про-
мышленность полностью удовлетворяет в них запросы сельскохозяйственного
производства. Важнейшее значение имеет переход на использование плохо
растворимых, медленно гидролизуемых и долго сохраняющихся в почве удоб-
рений (такими свойствами, например, обладает соль MgNH4PO4). Планирует-
ся освоить выпуск комплексных удобрений (особенно на основе мочевины,
обладающей способностью к комплексообразованию) и сложных смесей удоб-
рений.
В животноводстве широко применяют подкормку сельскохозяйственных
животных соединениями Na, К, Са, Р, Си, I, Со, Мп, Zn и других элементов.
При этом животные лучше усваивают корм, прибавляю в массе. Продук-
тивность животноводства при использовании полностью сбалансированных
по минеральным элементам и другим составным частям рационов возрастает
в среднем на 10%. При откорме крупного рогатого скота включение в рацио-
ны макроэлементов дает 11—14%, а микроэлементов—12—15% прироста
живой массы по ср внению с контрольными группами животных.
ГЛАВА ХП
ГОРМОНЫ И ИХ РОЛЬ В ОБМЕНЕ ВЕЩЕСТВ
ОБЩЕЕ ПОНЯТИЕ О ГОРМОНАХ
Более ста лет тому назад, в 1849 г., физиолог А. Бертольд впервые осуще-
ствил опыт, результаты которого показали, что последствия кастрации можно
устранить путем пересадки семенников. Несколькими годами позже (1855)
английский врач Т. А. Аддисон установил, что разрушение надпочечников
является причиной возникновения бронзовой (аддисоновой) болезни. К концу
XIX столетия было выявлено еще несколько случаев мощного воздействия на
организм веществ, продуцируемых в тех или иных органах или тканях, глав-
ным образом в железах внутренней секреции. Это дало основание
Э. Н. Старлингу в 1905 г. предложить термин гормоны (от греч. гормао—
побуждаю, возбуждаю, поощряю) для обозначения химических соединений,
вырабатываемых жел зами внутренней секреции и оказывающих сильнейшее
влияние на процессы обмена веществ и функционирование органов и тканей.
Позже выяснилось, что вещества с аналогичной функцией образуются и вне
желез внутренней секреции.
Тормоны интегрируют обмен веществ, т. е. регулируют соподчиненность
и взаимосвязь разнообразных химических реакций в различных органах и тка-
нях и организме в целом. Само возникновение гормонов в процессе эволюции
живой материи, несомненно, связано с дифференциацией ее, с обособлением
тканей и органов, деятельность которых должна быть тонко координирована.
В свою очередь, деятельность желез внутренней секреции, продуцирующих
гормоны, находится под контролем центральной нервной системы. Классиче-
ским примером этого служит гипофиз—железа внутренней секреции, явля-
ющаяся непосредственно составной частью мозга.
НОМЕНКЛАТУРА И КЛАССИФИКАЦИЯ ГОРМОНОВ
В настоящее время известно несколько десятков гормонов. Хотя хи-
мическая природа подавляющего большинства их выяснена и, следовательно,
каждому из них может быть дано точное химическое наименование, пред-
почитают пользоваться тривиальными названиями гормонов. Это происходит
по двум причинам. Во-первых, химическая номенклатура многих гормонов
необычайно громоздка и сложна, а в некоторых случаях (гормоны пептидной
и белковой природы)—практически неприемлема. Во-вторых, тривиальное
название, как правило, отражает либо функцию, либо происхождение
гормона.
Такое же двойственное положение сложилось и в отношении классифика-
ции гормонов. С одной стороны, давно применяется классификация, основан-
ная на происхождении гормонов. Согласно этой классификации, гормоны
объединяют в группы, каждая из которых получает название по имени железы
441
£ Таблица 27 Классификация сигнальных веществ (химических посредников) (по П. Карльсоиу)
Химически природа Происхождение
Нервные клетки1 Нейросекреторные клетки Клетки эндокринных желез Диффузные клетки Иные клепай2
Амины, аминокислоты Ацетилхолин Норадреналин Дофамин Глутамат 4-Аминобутират Тироксин Мелатонин (ТЧ-ацетил-5-мет- окситриптамин) Гистамин Серотонин
Пептиды Вещество Р Эндорфины Энкефалины Окситоцин Вазопрессин Соматостатин Гипоталамические рилизинг-факторы Инсулин Глюкагон Паратирин (паратгормон) Кортикотропин. Кальцитонин Гастрин Секретин Панкреозимин Желудочные ин- гибирующие пептиды Кинины Ангиотензин
Белки * Пролактин Соматотропин Тиротропин Гонадотропины
Стероиды и родственные соеди- нения Альдостерон Кортизол Половые гормоны Экдистероиды Ювенильные гормоны Простагландины
1 Вещества, перечисленные здесь, являются главным образом нейротрансмиттерами и нейромодуляторами.
2 Вещества, поименованные в этом столбце, часто называют медиаторами.
внутренней секреции где данная группа гормонов синтезируется. Между тем
существует множество гормонов, особенно открытых в последнее время,
биосинтез которых не приурочен к железам внутренней секреции. Химическое
же строение и тех и других прекрасно изучено. Поэтому наиболее целесообраз-
на химическая классификация гормонов, выделяющая среди них, по крайней
мере, три группы:
1. Стероидные гормоны. К этой группе принадлежат гормоны, являющиеся
производными стеролов. Они синтезируются в надпочечниках, семенниках,
яичниках и некоторых других органах и тканях (печень, плацента и т. п.).
2. Пептидные гормоны. Сюда входят все гормоны, химическая структура
которых представлена полипептидами той или иной молекулярной массы.
Местом их биосинтеза служат нейросекреторные клетки мозга, гипоталамус,
гипофиз, щитовидная и паращитовидная железы, поджелудочная железа, пе-
чень и слизистая органов пищеварения.
3. Прочие гормоны. К ним относят все остальные гормоны, не являющиеся
стероидами или полипептидами. Примерами гормонов этого типа у человека
и животных могут служить тироксин, адреналин, простагландины и др.,
синтезирующиеся в щитовидной железе, надпочечниках, репродуктивных ор
ганах и некоторых тканях. Сюда же относятся и фитогормоны (ауксины,
гетероауксин, гиббереллины, цитокинины и др.), возникающие в клетках выс-
ших и низших растений, а также ювенильные гормоны членистоногих.
Вместе с тем в последнее время предпр 1нята мног обещающая попытка
классифицировать гормоны, исходя не только из их хими1 еской природы, но
и учитывая их происхождение и физиологическую, в основном сигнальную
функцию (табл. 27). Эта классификация более адекватно отражает предназ-
начение гормонов в организме.
Рассмотрим строение, функци , механизм действия и пути биосинтеза
отдельных групп гормонов в соответствии с их химической классификацией.
СТЕРОИДНЫЕ ГОРМОНЫ
Строение и функциональная активность стероидных гормонов. Как указано
ранее, стероиды (см.гл. IX) представляют производные полициклических спир-
тов—стеролов, у которых укорочена (окислена) боковая цепь (20—27-й атомы).
Из биологических объектов выделено несколько десятков стероидов, но толь-
ко примерно двадцати из них свойственна гормональная функция. Так, в кор-
ковом слое надпочечных желез продуцируется более сорока различных стеро-
идов, которым присвоено общее название—кортикостероиды. Восемь из них
являются стероидными гормонами коры надпочечников. Наибольшее распро-
странение и значение в организме имеют кортикостерон, 17-оксикортикостерон
( идрокортизон) и альдостерон (в сумме они составляют 4/5 всех кортикосте-
роидов коры надпочечников):
443
В мужских и женских половых железах, а также в надпочечных железах
синтезируется 10 стероидов, обладающих свойствами половых гормонов.
Важнейшее значение из них имеют тестостерон (мужской половой гормон)
и эстрадиол (женский половой гормон):
Тестостерон Эстрадиол
Кортикостерон—кристаллическое соединение с /пл=182° С. Оптически ак-
тивен ([a]D=+223°). В норме в надпочечниках человека в течение суток
образуется 0,84—4,0 мг кортикостерона. При недостаточном поступлении
кортикостерона в кровь, а следовательно, в ткани организма наступают
нарушения в обмене углеводов, белков, липидов и минеральных элементов:
сокращаются запасы глико ена в мышцах и печени, падает содержание глюко-
зы в крови, усиливается распад белков до аминокислот, растет содержание
остаточного азота в крови, усиливаются липолитические процессы, нарушают-
ся нормальная экскреция и обратное всасывание Na+ и К* в канальцах почек,
падает кровяное давление. Все это приводит к сердечной недостаточности,
развитию отеков, мышечной слабости, развитию пигментации покровов
(бронзовая болезнь) и другим патологическим явлениям. При избытке кор-
тикостерона резко усиливаются анаболические процессы, что тоже приводит
к ряду отклонений от нормы.
17-Оксикортикостерон (гидрокортизон или кортизол)— кристаллы
с /дл=220° С. Раствор его отклоняет плоскость поляризации света на значение
[<x]D= + 167°. В течение суток в надпочечниках синтезируется от 4,9 до 7,9 мг
кортизола. В норме содержание кортизола в периферической крови человека
составляет 11 мкг %. При его дефиците развиваются те же нарушения обмена
веществ, что и при недостаточности кортикостерона, за исключением обмена
минеральных элементов, на который кортизол оказывает незначительное
влияние. Наиболее резко выражено при этом нарушение обмена углеводов,
вследствие чего кортизол относят к разряду глюкокортикостероидов. Особенно
характерны изменения в обмене веществ при избытке кортизола: резко усили-
вается превращение аминокислот в углеводы, возрастает, синтез гликогена
и жиров, повышается содержание глюкозы в крови (стероидный диабет).
В результате развивается ожирение туловища, появляется «буйволиный» (жи-
ровые складки) затылок, лицо становится круглым и красным, тогда как
мышцы атрофируются, скелет становится хрупким, а конечности—худыми.
Альдостерон—кристаллическое вещество с <пл=219° С и оптической актив-
ностью растворов, характеризующейся [a]D= 4-88°. В надпочечниках за сутки
образуется 0,15—0,4 мг альдостерона. На его долю приходится всего 1—2%
от суммы кортикостероидов, тогда как кортикостерон и 17-оксикортикостерон
содержится в ней в количестве 75%. В венозной крови человека альдостерон
присутствует в концентрации 0,08 мкг %.
При недостаточности альдостерона развивается бронзовая болезнь. Одна-
ко особенно характерно при этом резкое нарушение минерального обмена—
пад ние обратного всасывания Na+ в почечных канальцах и, как следствие
этого, усиление задержки в организме К+. Поэтому альдостерон считают
минералокортикостероидом. Его способность повышать обратное всасывание
Na+ в канальцах почек в 300 раз выше, чем у кортизола, являющегося.
444
глюкокортикостероидом. В то же время альдостерон не оказывает почти
никакого влияния на обмен углеводов.
Тестостерон—кристаллы с гпл = 155° С. Оптически активен ([a)D= + 117°).
Синтезируется в семенниках, где его содержание у человека составляет
21,6 мкг % в сыром веществе. Он обусловливает нормальный рост мужских
половых органов и раз итие вторичных половых признаков у мужчин. При
дефиците тестостерона у взрослых особей снижается биосинтез белков, раз-
вивается ожирение, утрачивается волосяной покров. Тестостерон, как и другие
мужские половые гормоны, является типичным анаболическим стероидом,
увеличивающим задержку азота в организме и усиливающим интенсивность
новообразования белков.
Эстрадиол кристаллизуется в двух модификациях: в виде перистых листо-
чков с гпл=175—177° Сив форме призматических игл с ?пл= 178,5—179,5° С.
Вырабатывается в яичниках в количестве 1 мг в сутки. Вызывает течку и раз-
растание слизистой матки у животных. При недостаточности эстрадиола
нарушаются циклы менструации, происходят самопроизвольные выкидыши,
развивается ожирение. Эстрадиол оказывает влияние на обмен углеводов,
белков и нуклеиновых кислот.
Механизм действия стероидных гормонов. Как видно из приведенных выше
данных о функциональной активности рассмотренных стероидных гормонов,
их влияние распространяется на многочисленные и разнообразные химические
процессы в тканях организма. Следовательно, в отличие от ферментов или
витаминов гормоны изменяют скорость протекания не какой-то конкретной
химической реакции или группы сходных реакций, а затрагивают в обмене
веществ некие фундаментальные процессы. Последние, в свою очередь, сказы-
ваются на самых различных сторонах обмена веществ. Указанный подход
лежит в основе современных представлений о механизме действия гормонов.
Какие же фундаментальные процессы контролируются стероидными гор-
монами? По этому поводу в последние годы получены интересные сведения.
Оказалось, что стероидные гормоны взаимодействуют с клетками-мишенями,
мощно и избирательно регулируя в них синтез, в первую очередь, информаци-
онных РНК. Последние немедленно обеспечивают наработку специфических
белков, оказывающих влияние на обмен веществ и как следствие этого на
физиологические и иные процессы. Список этих белков, в том числе каталити-
чески активных, сейчас достиг уже нескольких десятков.
Не следует думать, что стероидные гормоны непосредственно взаимодей-
ствуют с генетическим аппаратом клетки и прямо дерепрессируют ДНК.
Регуляция при их посредстве биосинтеза мРНК идет более сложным путем:
с хроматином вступает в контакт гормон-рецепторный комплекс. Он представ-
ляет собой комплекс стероидного гормона и белка-рецептора, образующийся
в цитоплазме клетки-мишени, куда проникает соответствующий гормон и где
всегда имеется специфичный к нему рецепторный белок (рис. 135). Более того,
после присоединения стероидного гормона к белку-рецептору гормон часто
преобразуется (например, восстанавливается по двойной связи в кольце А),
а белок-рецептор изменяет свою конформацию. В последнее время появились
данные о том, что рецепторный белок в норме постоянно соединен с веществом-
модулятором его третичной структуры: присоединение стероидного гормона
ведет к потере модулятора и изменению конформации гормон-рецепторного
комплекса. Именно в новой конформации гормон-рецепторный комплекс
транслоцируется в ядро, где связывается с акцепторным участком хроматина,
переводя ДНК в этой зоне хроматина в транскрипционно-активное состояние.
Важно подчеркнуть, что лишь строго определенный стероидный гормон
«узнает» свою клетку-мишень, а в ней—свой специфический белок-рецептор.
445
Рис. 135. Механизм действия стероидных гормонов (пояснения в тексте)
В свою очередь, возникший гормон-рецепторный комплекс сугубо избиратель-
но атакует в ядре клетки ту область хроматина, которая содержит фрагмент
ДНК, ответственный за синтез индивидуальной мРНК и, следовательно, белка
с только ему присущей функциональной активн >стью. Так обеспечивается
специфичность действия различных стероидных гормонов, в основе которой
отчетливо вырисовывается ключевая роль цитоплазматических рецепторных
белков, передающих гормональный сигнал генетическому аппарату клетки.
Белки-рецепторы интенсивно изучаются. Так, рецепторный белок для гид-
рокортизона имеет М = 67000, а для эстрадиола—М = 200 000. В последнем
случае он состоит из двух субъединиц одна из которых ответственна за
связывание гормона, а вторая—акцепторного участка хроматина. В яйцево-
дах курицы, где сосредоточены клетки мишени эстрадиола, ядра которых
содержат до 500 мест акцептирования гормон рецепторного комплекса каж-
дое, в опытах по индукции б осинтеза яичного альбумина показано, что число
молекул мРНК возрастает в несколько тысяч раз. В результате воздействия
эстрадиола в яйцеводах резко повышается синтез яичного альбумина. Пред-
полагают, что гормон-рецепторный комплекс фиксируется на ядерной ДНК
при модулирующем действии лабильно связанных негистоновых белков хро-
матина, некоторые из которых, возможно, сами являются рецепторами стеро-
идных гормонов.
Для других стероид рецепторных комплексов зафиксировано от 8000 до
23 000 мест связывания на 1 пг ДНК и протяженность в 700—800 н. п., зани-
маемая гормон-рецепторным комплексом.
Было бы не совсем правильно сводить механизм действия стероидных
гормонов к стимулированию биосинтеза только мРНК в тканях-мишенях,
хотя это, конечно, центральное звено в регуляции обмена веществ при их
посредстве. Накапливаются данные о том, что стероидные гормоны влияют на
биосинтез ДНК, вызывая ампли икацию генов и как следствие этого, усиле-
ние биосинтеза других видов РНК (рРНК, гистоновых мРНК и т. п.). Появи-
лись также сведения о возможном трансмембранном механизме действия стеро-
идных гормонов; так, например, экдизон (см. в конце этой главы инициирует
образование вторичных посредников из фосфоинозитидов (см. рис. 139 на с. 475).
Время полужизни молекул гормонов редко превышает 1 ч, поэтому для
поддержания регуляции обмена веществ в организме на должном уровне их
содержание должно поддерживаться путем непрерывного синтеза. »
Биосинтез стероидных гормонов. Существует два пути биосинтеза стероид-
ных гормонов. Сведения о том и другом получены методом меченых атомов.
Первый путь состоит в превращении холестерола (схема 12) в стероидные
гормоны путем окисления его в прогестерон и, далее, в другие производные
последнего. Этот путь реализ ется в надпочечных железах под влиянием
446
I
Кортикостерон
Альдостерон
Тестостерон
Эстрадиол
Схема 12. Возможные пути биосинтеза стероидных гормонов
адренокортикотропного гормона (см. ниже). Второй путь заключается в био-
синтезе стероидных гормонов из ацетил-КоА, минуя холестерол. По всей
вероятности, он осуществляется в соответствии с уравнением, приведенным
в гл. IX при рассмотрении механизма новообразования холестерола. Однако
в этом случае окисление боковой цепи (20—27-й атомы) наступает ранее
образования холестерола.
Как видно из схемы 12, при биосинтезе стероидных гормонов из того или
иного предшественника центральное положение занимает прогестерон—уже
в значительной степени окисленное производное, где при 3-м углеродном
атоме кольца А сформирована кетогруппа, а между 4-м и 5-м углеродными
атомами этого же цикла—двойная связь, тогда как боковая цепь при кольце
D укорочена уже до двууглеродного фрагмента, также содержащего кетогруп-
пу в положении 20.
t Дальнейшее окисление СН3-группы боковой цепи при кольце D (20-й и 21-й
углеродные атомы) приводит к возникновению 11-дезоксикортикостерона,
447
который, в свою очередь, дает начало кортикостерону (дальнейшее окисление
в кольце С по 11-му атому углерода) и альдостерону (окисление СН3-группы
в 18-м положении).
Напротив, окисление прогестерона по кольцу D и образование ОН-группы
в положении 17 приводит к 17-оксипрогестерону. Последний дает начало ряду
стероидных гормонов: при полном окислении боковой цепи в кольце D воз-
никает андростендион и, далее, эстрадиол и тестостерон, а при окислении
углеродного атома, занимающего положение 11 (кольцо С), синтезируется 17-
оксикортикостерон (см. схему 12).
ПЕПТИДНЫЕ ГОРМОНЫ
Структура и функции пептидных гормонов. Сейчас известно несколько де-
сятков природных пептидных гормонов, и список их постепенно пополняется.
Благодаря широкому использованию методов бурно развивающейся бел-
ковой химии в последние годы ряд пептидных гормонов получен в го-
могенном состоянии, изучен их аминокислотный состав, выяснена первичная
(а в случае белковых гормонов—вторичная, третичная и четвертичная) струк-
тура и некоторые из них приготовлены синтетическим путем. Более того,
большие успехи, достигнутые в области химического синтеза пептидов, по-
зволили искусственно получить множество пептидов, являющихся изомерами
или аналогами ^натуральных пептидов. Изучение гормональной активности
последних принесло исключительно важную информацию о взаимосвязи
структуры пептидных гормонов с их функцией. Примеры этого будут при-
ведены ниже.
Важнейшими пептидными гормонами являются окситоцин, вазопрессин,
гастрин, глюкагон, инсулин, адренокортикотропин, меланоцитостимулирую-
щий гормон, паратгормон, тиреотропин и гормон роста. В последние годы
открыто и изучено большое семейство нейрогормонов, опиоидных пептидов
и рилизинг-факторов. Рассмотрим строение и функции перечисленных гор-
монов.
Окситоцин—9-членный пептид, выделенный В. дю-Виньо с сотр. из задней
доли гипофиза. Этот же ученый в 1953 г. (одновременно с Г. Туппи) пред-
ложил полную структурную формулу окситоцина:
CtH4- ОН СНз
NHj СН2 СН—СН2—СНз
СН2—СН—СО—NH—СН—СО—NH—СН
I 1 2 Л1
Н3С—СН—СНз
448
Как видно из приведенной выше формулы, окситоцин содержит цикл,
замыкающийся в результате возникновения дисульфидной связи между 1-м
и 6-м остатками цистеина в его молекуле.
Окситоцин уже через 20—30 с после внутривенного введения в ко-
личестве всего лишь 1 мкг вызывает у рожениц сокращение мышечных
волокон, расположенных вокруг альвеол молочных желез. Это приводит
к выделению молока. Кроме того, по мере приближения родов усиливается
чувствительность к окситоцину мышц матки, сокращающихся под его
воздействием. Поэтому данный гормон способствует нормальному про-
теканию родов.
Окситоцин синтезирован; создано также много его изомеров и аналогов.
Это дало возможность детально выяснить значение каждого аминокислотного
остатка и их сочетаний для функциональной активности гормона. Оказалось,
что размыкание дисульфидного мостика в молекуле окситоцина сопровожда-
ется полной его инактивацией. Важнейшее значение для биологического дей-
ствия окситоцина имеет остаток амидированной аспарагиновой кислоты (по-
ложение 5). Ацилирование амидных групп (в 4-м, 5-м и 9-м положениях) ведет
к понижению активности окситоцина. Наличие свободной ОН-группы у оста-
тка тирозина способствует проявлению полной физиологической активности.
Устранение боковой цепи тирозина из состава окситоцина приводит к появле-
нию у него способности ингибировать действие других гипофизарных гормо-
нов. Замена изолейцина (положение 3) на лейцин, норлейцин, валин или
аллоизолейцин убеждает в том, что боковая цепь изолейцина служит для
специфической связи гормона с рецептором. Присоединение по свободной
ЫН2-группе остатка цистеина (положение 1) других аминокислот или корот-
ких пептидов приводит к удлинению срока действия гормона в организме по
сравнению с контролем.
Вазопрессин—9-членный пептид, выделенный тоже из задней доли гипо-
физа. Заслуга в выяснении его структуры и разработке метода его синтеза
принадлежит В. дю-Виньо (1953—1956). Строение вазопрессина весьма напо-
минает структуру окситоцина (см. рис. 22 на с. 50).
Как можно легко заметить, за исключением позиций 3 и 8, структура
вазопрессина полностью повторяет структуру окситоцина. Так построен вазо-
прессин, выделенный из гипофиза человека, обезьяны, крупного рогатого
скота, лошади, верблюда, овцы и собаки. В молекуле вазопрессина, получен-
ного из гипофиза свиньи, вместо остатка аргинина (8-е положение) располага-
ется остаток лизина. Это свидетельствует о наличии видовой специфичности
в строении вазопрессина.
Будучи сходен с окситоцином по структуре, вазопрессин, в известной мере,
похож на него и по функциональной активности: он стимулирует сокращение
гладких мышц сосудов. Однако указанная функция не является для вазопрес-
сина главной. Основное действие его направлено на регуляцию водного об-
мена. Вазопрессин обеспечивает должный уровень ресорбции воды в дисталь-
ных канальцах почек у высших позвоночных, регулируя водный баланс ор-
ганизма и осмотическое давление плазмы крови. Естественно, что он способен
повышать кровяное давление, и это отражено в его названии.
Гйстрин— 17-членный пептид, выделяемый слизистой входной части желуд-
ка. Его первичная структура расшифрована, и он получен синтетически:
1 2 3 4 5 6 7 8
гли —гли —про —три —мет —глу —глу —глу —
9 10 11 12 13 14 15 16 17
глу —глу —ала —тир —гли —три —мет —асп —фен—NH2
15—3502
449
Гастрин стимулирует секрецию желудочного сока. Его активность в 500 раз
превышает в этом отношении активность ранее известного стимулятора соко-
отделения—гистамина. Гастрин действует возбуждающим образом почти ис-
ключител но на образование соляной кислоты и лишь в небольшой мере
повышает выработку пепсина. Он способствует выделению секре а поджелу-
дочной железы и усиливает тонус и сокращение мышц желудка и тонкого
кишечника. У1 азанная специфическая гормональная активность гастрина поч-
ти исключительно связана с наличием в его молекуле С-концевого тетрапеп-
тида (14—17-й остатки).
Поскольку аналогичная ситуация воспроизводится у многих пептидных
гормонов, а также белков с иной биологической активностью, огромный
интерес представляет выяснение назначения остальной части полипептида, не
участвующей непосредственно в осуществлении гормональной, биокаталити-
ческой или иной функции.
Ппокагон—29-членный пептид, синтезирующийся в а-клетках островковой
части поджелудочной железы. Первое упоминание об этом гормоне восходит
к 1923 г., когда И. Мурлин с сотр. обнаружил его присутствие в препаратах
инсулина. В 1953 г. Ф. Штрауб полупил глюкагон в виде гомогенного кристал-
лического препарата, а несколько позже была выяснена его первичная струк-
тура. Она такова:
1234567 89 10
1йс— сер— глн— гли— тре— фен— тре— сер— асп— тир—,
20 19 18 17 16 15 14 13 12 11
глн— ала— арг— арг— сер— асп— лей— тре— лиз— сер—'
I 21 22 23 24 25 26 27 28 29
I— асп— фен— вал— глн— три— лей— вал— асн— тре—
Избыточное выделение глюкагона поджелудочной железой или и кусствен-
ное введение его в организм животны и человека приводит к кратковременному
повышению содержания глюкозы в крови—гипергликемии. Это действие глюка-
гона объясняется тем, что он способствует превращению менее активной формы
фосфорилазы печени в более активную (см. с. 334). В результате под действи-
ем фосфорилазы а усиливается распад ликогена в печени и возрастает содержа-
ние глюкозы (в виде глюкозо-1-фосфата) в крови. Естественно, что запасы
гликогена в печени при этом сокращаются, а процесс гликогенолиза в организме
усиливается. Таким образом, глюкагон способствует деструкции углеводов.
Инсулин—белок, вырабатываемый в Р-клетках поджелудочной железы.
Его строение детально изучено. Инсулин был первым белком, у которого
Ф. Сангером (см. с. 61) была выяснена первичная структура. Он же явился
первым белком, полученным путем хими еского синтеза.
Впервые наличие в железе гормона, влияющего на углеводный обмен, было
отмечено Мерингом и О. Миньковским (1889). Позднее Л. В. Соболев (1901)
установил, что источником инсулина в поджелудочной железе служи остро-
вковая часть ее, в связи с чем в 1909 г. этот гормон, не будучи еще индивиду-
ализирован, получил наименование—инсулин (от лат. insula—остров).
В 1922 г. Ф. Бантинг и Г. Бест впервые приготовили активный препарат
инсулина, а к 1926 г. были разработаны способы его выделения в высокоочи-
щенном состоянии, в том числе в виде кристаллических препаратов, содержа-
щих 0,36% Zn.
Молекулярная масса кристаллического инсулина равна 36000. Его молеку-
ла представляет собой мультимер, составленный из шести протомеров и двух
450
Глюкоза
Рис. 136. Структура инсулина и его рецептора:
I—третичная структура протомера; А1—А21—цепь А; В1—ВЗО—цепь В; пунктиром показана эона
связывания с рецептором; II—четвертичная структура гексамерной молекулы; каждый блок соответствует
протомеру; один блок отсутствует, чтобы показать взаимодействие ионов цинка (пунктирные линии) с тремя
радикалами гистидина и иона кальция—с тремя радикалами глутаминовой кислоты (оба радикала в цепи
В, в положениях 10 и 13 соответственно); и тот и другой ион связаны также с тремя молекулами воды
каждый (на рисунке не показано); III—транспортные системы, чувствительные к регуляторному действию
инсулина на клетки-мишени: 1—стимуляция переноса глюкозы; 2—стимуляция Na+, К+-насоса; 3—повы-
шение интенсивности Na+/Н+-обмена; 4—ингибирование Са2+-насоса; 5—стимуляция транспорта амино-
кислот; б—повышение активности системы №+/Са2+-обмена; Р—рецептор; Г—гормон; IV—гипотетиче-
ская структура рецептора инсулина (а2р2-цепн, связанные дисульфидными мостиками; а—95000, 0—135000
Да). Это гликопротеин (М=460 кДа), встроенный в плазматическую мембрану клетки; наружу экспонирова-
ны а-субъединицы, а Р-субъединицы пронизывают мембрану; фрагмент полипептидной цепи Р-субъединицы,
локализованный в цитозоле, обладает тирозинкиназной активностью, в частности по отношению к фосфа-
тазе; последняя, будучи активирована, дефосфорилирует ряд ферментов углеводного обмена и изменяет их
активность
атомов Zn. Протомеры образуют димеры, которые взаимодействуют с ими-
дазольными ядрами радикалов гпс10 цепи В и способствуют их агрегации
в гексамер (рис. 136). Распадаясь, мультимер дает три субчастицы с молеку-
лярной массой 12000 каждая. В свою очередь, каждая субчастица расщепляет-
ся на две равные части с М = 6000. Все перечисленные модификации ин-
сулина—протомер, димер и гексамер—обладают полной гормональной ак-
тивностью. Поэтому часто молекулу инсулина отождествляют с протомером,
обладающим полной биологической активностью (М = 6000), тем более, что
в физиологических условиях инсулин существует в мономерной форме. Даль-
нейшее фрагментирование молекулы инсулина (с М = 6000) на цепь А (из
451
21 аминокислотного остатка) и цепь В (из 30 аминокислотных остатков) ведет
к утрате гормональных свойств.
Инсулины, выделенные из поджелудочной железы различных животных,
почти идентичны по первичной структуре (см. табл. 8). При недостаточном
уровне биосинтеза инсулина в поджелудочной железе человека (в норме еже-
суточно синтезируется 2 мг инсулина) развивается характерное заболевание—
диабет, или сахарное мочеизнурение. При этом повышается содержание глю-
козы в крови (гипергликемия) и растет выведение глюкозы с мочой (глюкозу-
рия). Одновременно развиваются различные вторичные явления—падает со-
держание гликогена в мышцах, замедляется биосинтез пептидов, белков и жи-
ров, нарушается минеральный обмен и т. п.
Введение инсулина путем инъекции или per os в виде препарата, ин-
капсулированного в липосомы, вызывает противоположный эффект: по-
нижение содержания глюкозы в крови, повышение запасов гликогена
в мышцах, усиление анаболических процессов, нормализацию минераль-
ного обмена и т. д. Все перечисленные выше явления представляют резуль-
тат изменения под воздействием инсулина проницаемости для глюкозы
клеточных мембран, на поверхности которых выявлены высоко- и низкоаф-
финные Са2+-зависимые инсулиновые рецепторы. Повышая уровень про-
никновения глюкозы внутрь клетки и субклеточных частиц, инсулин усили-
вает возможности ее использования в тех или иных тканях, будь то биосин-
тез из нее гликогена или дихотомический или апотомический ее распад
(рис. 136).
При взаимодействии инсулина с рецептором клеточной мембраны воз-
буждается активность протеинкиназного домена инсулинового рецептора
(рис. 136, IV), что сказывается на внутриклеточном метаболизе углеводов,
липидов и белков. Сущность этого ясна из рассмотрения рис. 136, III. Для
инсулина не типичен аденилатциклазный механизм действия.
Адренокортикотропный гормон (АКТГ)—39-членный пептид, продуци-
руемый передней долей гипофиза. Он открыт в 1928 г., но лишь немногим
менее чем через три десятилетия удалось расшифровать первичную струк-
туру сначала АКТГ овцы (Ч. Ли и сотр., 1955), а затем свиньи (П. Белл
и сотр., 1956). В настоящее время известна также первичная структура
АКТГ быка, человека и акулы. Приведем в качестве примера строение
АКТГ человека:
1 2 3 4 5 6 7 8 9 10
Сер- го тир— 19 сер — 18 мет — 17 глу— 16 гис— 15 фен— 14 арг— 13 три— 12 гли— 11
—вал— 21 про - 22 -арг— 23 арг— 24 лиз— 25 лиз— 26 гли— 27 вал— 28 про — 29 лиз—* 30
*— лиз— 39 вал— 38 тир— 37 про- зе асп— 35 ала— 34 гли— 33 глу— 32 асп— 31 глн—
фен — глу — лей — про — фен — ала — глу — ала — сер
. Как показал К. Гофман, видовая специфичность АКТГ обусловлена чере-
дованием аминокислотных остатков в позициях 25—33. Участок молекулы
АКТГ между 1-ми 13-м аминокислотными остатками абсолютно необходим
для обеспечения активности гормона. Между 14—20-м аминокислотными
остатками располагается якорная площадка гормона (15—18-й остатки, т. е.—
лиз—лиз—арг—арг—). Пептидная группир вка из 19 аминокислотных
остатков (позиция 21—39) может быть удалена без каких-либо последствий
452
для активности гормона, но она определяет его иммунологическую специфич-
ность.
АКТГ оказывает разностороннее действие: повышает активность фосфори-
лазы, липазы и глюкозо-6-фосфатдегидрогеназы, усиливает синтез белков
и рибонуклеиновых кислот и др. Однако главная его функция в организме
сводится к регуляции интенсивности и объема биосинтеза кортикостероидов
надпочечными железами. В свою очередь, падение в крови концентрации
кортикостероидов ниже определенного уровня стимулирует выработку АКТГ
в передней доле гипофиза. Известно также, что АКТГ стимулирует главным
образом биосинтез глюкокортикостероидов, изменяя, таким образом, соот-
ношение между различными кортикостероидами, продуцируемыми надпочеч-
ными железами.
Есть основания полагать, что АКТГ, с одной стороны, повышает биосин-
тез кортикостероидов, обеспечивая более высокий уровень в организме
НАДФН, необходимого для их новообразования из холестерола. С другой
стороны, АКТГ активирует мембранно-связанную аденилатциклазу, т. е. био-
синтез цАМФ (см. ниже), сказывающийся на интенсивности протеинкиназных
реакций при стероидогенезе.
Меланоцитостимулирующий гормон (МСГ)—18-членный пептид, образу-
емый средним (а у ряда видов, например у свиньи, задним) отделом гипофиза
животных. МСГ человека представлен 22-членным пептидом, очень похожим
на МСГ животных.
Хотя сведения о существовании в среднем отделе гипофиза МСГ относятся
к 1932 г., впервые он был получен в 1955 г. из задней доли гипофиза свиньи
Дж. Поратом, разработавшим детальный метод выделения гипофизарных
пептидов. Он был назван а-МСГ. В том же году был выделен также из задней
доли гипофиза сходный с ним МСГ, обозначенный Р-МСГ.
Строение молекулы а-МСГ свиньи, выясненное Дж. Гаррисом и П. Россом
(1956), таково:
1 2 3 4 5 6 7 8 9
Асп— глу— гли— про— тир— лиз— мет— глу— гис—
18 17 16 15 14 13 12 11 10
асп— лиз— про— про— сер— гли— три— арг— фен—
Сейчас известна также первичная структура а- и Р-МСГ человека, обезья-
ны, быка, лошади, верблюда и акулы, а- и Р-МСГ свиньи синтезированы.
Кроме того, получено около двух десятков аналогов МСГ.
Механизм действия Р-МСГ на меланоциты у низших позвоночных в насто-
ящее время рассматривают с точки зрения влияния Р-МСГ на переход гранул,
содержащих меланин, из состояния геля в состояние золя. Это сопровождается
распределением пигмента в клетке и ее потемнением. Смысл изменения степе-
ни потемнения покровов у низших позвоночных сводится к адаптации их
к окружающим условиям. Роль МСГ у высших позвоночных неясна.
Паратгормон—белок, синтезируемый паращитовидными железами. Впер-
вые на гормональные свойства кислого экстракта из паращитовидных желез
указал Дж. Коллип (1925). Однако в индивидуаль ом состоянии паратгормон
получен значительно позже благодаря усилиям Г. Аурбаха и X. Расмуссена
(1959—1964). Он о :азался белком с М = 9500, состоящим из 84 аминокислот-
ных остатков. Первичная структура паратгормона быка была окончательно
выяснена в 1970 г. Дж. Поттсом с сотр.:
453
Ала— вал— сер— глу— иле— глн— фен— мет— гис— 10 асн— лей—
1—глу— вал— {—три— лей— г—арг— три— '—асп— гли— г—лиз— глн— '—сер— лей— Аналогично, с 20 арг— глу— мет— сер— сер— арг— лиз— лиз— лей— глн— 40 ала— иле— сер— ала— гли— 50 сер— сер— глн— арг— про— 60 гис— сер— глу— вал— лей- 70 гли— глу— ала— асп— лиз— 84 глн— про— лиз— ала— небольшими отличиями (всего лей— гис— 30 асп— вол- лей— ала— арг— лиз— вал— асн— ала— асп— 80 лиз— иле— несколько J лиз— гли—> гис— асн-t вал— фен-1 лиз— лиз—\ асп— глу— вал— асп—I лей— вал-1 аминокислотных
замен) построены паратгормоны человека и ;виньи.
Паратгормон регулирует содержание катионов кальция и анионов фосфор-
ной и лимонной кислот в крови. При длительном дефиците солей кальция
в пище или при нарушении всасывания солей кальция в кишечнике содержание
их в крови понижается. Это ведет к повышению синтеза и выделения паращи-
товидными железами гормона, который мобилизует соли кальция (в виде
цитратов и фосфатов) из костной ткани. Поддержание нормального уровня
Са2+ в крови достигается усилением под влиянием паратгормона экскр ции
фосфатов почками, в результате чего замедляется отложение фосфата кальция
в костях.
Биологическая активность паратироидного гормона обеспечивается N-
концевым фрагме том длиной не менее 28 аминокислотных остатков, причем
особо важны первые две N-концевые аминокислоты.
ТЬчкой приложения действия паратгормона являются рецепторные белки
плазматической мембраны клеток-мишеней и зависимая от них а тивация
аденилатциклазы, в результате чего в клетках повышается содержание цАМФ
(см. ниже). Это сопровождается усилением протеинкиназных реакций, активи-
рованием мембранно-связанной Са2+-зависимой АТФазы, что приводит к пе-
рераспределению кальция в компартментах клетки и, в конечном счете, между
, тканями и органами.
Тиреотропин—белок, выделяемый передней долей гипофиза. Он представля-
ет собой гликопротеин с М = 28 300, составленный из двух неравных субъединиц
(М = 13600 и 14700), исключительно богатых дисульфидными мостиками (5
и 6 со тветственно). Первичная структура тиреотропина быка и свиньи выясне-
на. При недостатке тиреотропина (гипофункция гипофиза) ослабляется дея-
тельность щитовидной железы, она умен шается в размерах, а содержание
в крови выделяемого ею гормона—тироксина (см. ниже)—сокращает я вдвое.
Таким образом тиреотропин стимулирует деятельность щитовидной желе-
зы. В свою очередь, выделение тиреотропина регулируется по принципу
обратной связи гормонами щитовидной железы. Следовательно, деятельность
двух упомянутых желез внутренней секреции тонко координирована.
Введение тиреотропина вызывает множественные сдвиги в обмене веществ:
через 15—20 мин повышается секреция гормонов щитовидной железы и уси-
ливается поглощение ею иода, необходимого для синтеза этих гормонов
(см. ниже); повышается поглощение кислорода щитовидной железой, возрас-
тает окисление глюкозы, активируются обмен фосфолипидов и новообразова-
ние РНК. Сейчас выяснено, что механизм действия тиреотропина, как и мно-
гих других пеп идных гормонов, сводится к активир ванию аденилатциклазы,
расположенной в непосредственной близости от рецепторного белка, с кото-
454
рым связывается тиреотропин. Как следствие этого, в щитовидной железе
ускоряется ряд процессов, в том числе и биосинтез тире идных гормонов.
ГЬрмон роста (соматотропный гормон, СТГ)—белок, секретируемый перед-
ней долей гипофиза позвоночных животных. Наличие его в экстрактах из
гипозифа было отмечено еще в 1921 г. Г. Эвансом и Дж. Лонгом, однако лишь
через два десятилетия (1944) он был получен в виде очищенного препарата,
а через несколько лет после этого (1948)—в кристаллическом состоянии.
В ависимости от вида животного (бык, овца, свинья, крыса и др.) молеку-
лярная масса кр сталлического препарата гормона роста колеблется от 20 000
до 22000.
В гипофизе человека содержится от 3,7 до 6,0 мг СТГ. Обладая М = 21000, он
представлен одной полипептидной цепочкой из 191 аминокислотного остатка.
Первичная структура ее выяснена Ч. Ли и сотр. (1969) и уточнена Г. Найлом с сотр.
(1973). Известна также первичная структура СТГ быка и овцы (по 191 аминокислот-
ному остатку). Изучена последовательность аминокислотных остатков в гормоне
роста многих рыб (байкальского омуля, кеты, форели, угря, трески, лосося, щуки
и др.) и выявлена множест енность генов, кодирующих слегка различающиеся
варианты полипептидных цепей СТГ даже у одного и того же вида рыб. Эти данные
открывают новое направление в химии гормонов пептидной природы, раскрываю-
щее причину множес венности не только у СТГ, но и во многих других случаях.
Третичная струк ура СТГ человека характеризуется сближенностью С-
и N-концевых аминокислот, наличием четырех мощных спиралей, охватыва-
ющих половину его полипептидной цепи и присутствием неорганизованной
последовательности, составляющей вторую половину молекулы. В узнавании
рецептора СТГ участвуют его 1-ая и 2-ая а-спирали и часть неупорядоченной
петли, соединяющей 1-ую и 2-ую спирали; вслед за этим по 1-ой и 3-ей
спиралям присоединяется вторая молекула рецептора, что обеспечивает его
димеризацию и передачу гормонального сигнала. Содержание СТГ в крови
человека в норме колеблется в широких пределах—от 0—3 мкг/мл после
ночного голодания до 100 мкг/мл после приема сахара.
Гормон роста обладает ярко выраженным анаболическим действием и влия-
ет на все клетки организма, повышая в них уровень биосинтетических процес-
сов. Он усиливает биосинтез белков, ДНК, РНК и гликогена, но способствует
мобилизации жиров из жировых депо и ускоряет распад высших жирных кислот
и глюкозы. СТГ улучшает функции почечных канальцев и нормализует мине-
ральный и водный обмен организма. Все это способствует росту организма, но
в конечном счете действие СТГ гораздо шире, нежели только регуляция роста.
Исследования на молекулярном уровне показали, что СТГ стимулирует
деятельность РНК-полимераз и полирибосомного аппарата клетки. Как сви-
детельствуют опыты с меченым фосфором, самым ранним эффектом действия
гормона роста является синтез в ядрах клеток предшественников мРНК
и рРНК. Вместе с тем велико его влияние и на проницаемость клеточных
стенок, так как фонд внутриклеточных аминокислот в присутствии СТГ
значительно возрастает, что способствует новообразованию белков. Возмож-
но, первопричиной всех этих явлений служит все же активирование мембран-
но-связанной аденилатциклазы. Вместе с тем показано, что СТГ повышает
содержание в крови особых стимулирующих рост фак оров—соматомеди-
нов—белков с М»7000. Механизм их действия активно изучают.
ГЬрмои ожирения—лептин (от греч. leptos—тонкий) открыт группой ис-
следователей во главе с Дж. Фридманом в 1992 г., хотя первые сведения
о мутациях гена «оЬ» (obese—ожирение), вызывающих ожирение у мышей,
были получены в начале 50-ых гг. Этот белковый гормон, экспрессирующийся
в адипоцитах в виде 167-членного предшественника, экскретируется в кровь
455
в качестве активного пептидного гормона, составленного из 145 аминокислот-
ных остатков, последовательность которых выяснена. При парэнтеральном
и внутривенном введении его в количестве нескольких микрограммов на
животное в день происходит повышение энергетического обмена, снижение
потребления пищи, увеличение двигательной активности и снижение веса
мышей вследствие уменьшения запасов жира в организме. После прекращения
введения лептина все исследованные параметры достаточно быстро возвраща-
ются к исходному уровню. Предполагают, что гормон ожирения секретирует-
ся адипоцитами в кровь в изменяющихся количествах в зависимости от
потребностей организма. Какие клетки или ткани играют роль «липостата»
и каков механизм взаимодействия гормона с рецептором и метаболических
изменений, происходящих в ответ на рецепцию, остается неизвестным. Заме-
тим, что вопрос об использовании лептина для лечения ожирения у человека
потребует специального исследования.
Механизм действия пептидных гормонов. Пептидные гормоны не проникают
внутрь клеток-мишеней и взаимодействуют с белковыми рецепторами, располо-
женными на их поверхности, в плазматической мембране. Поэтому их механизм
действия принципиально отличается от такового стероидных гормонов.
Подавляющее большинство гормонов пептидной природы в результате
связывания с рецепторным комплексом клеточной мембраны возбуждает ак-
тивность аденилатциклазы, встроенной в эту же мембрану:
Возникающее при этом соединение—циклический аденозинмонофосфат, от-
крытое в 1957 г. одновременно двумя группами исследователей—Е. Сатерлэн-
дом с сотр. и Д. Маркхэмом с сотр., оказалось тем веществом, которое передает
гормональный сигнал метаболическим системам клетки, т. е. является, по сущест-
ву, вторичным посредником в передаче этого сигнала (первичный посредник—
рецепторный белок, воспринимающий гормональный сигнал). Дело в том, что
цАМФ является аллостерическим регулятором протеинкиназ, при участии кото-
рых фосфорилируются гистоны и негистоновые белки хроматина (это сказыва-
ется на метаболической активности генома клетки и, в частности, на уровне
биосинтеза мРНК), рибосомальные белки и белковые факторы трансляции (это
отражается на интенсивности новообразования белков в рибосомальном аппа-
рате клетки), многие ферменты (что предопределяет степень их активности)
и т. п. Поскольку это затрагивает фундаментальные стороны обмена веществ,
то вполне объяснимы биохимические и физиологические явления, наблюдаемые
при недостатке или избытке пептидных гормонов. Ряд конкретных примеров
такого механизма действия пептидных гормонов был рассмотрен ранее при
изучении реакции фосфоролиза гликогена, липолитических процессов и др.
Передача гормонального сигнала аденилатциклазе осуществляется в ре-
зультате работы гормон-рецепторного комплекса, структура которого пред-
ставлена на рис. 137. Ключевыми в системах сопряжения гормонального
456
Рис. 137. Механизм действия гормо-
нов пептидной природы:
Р—рецептор гормона; G—регулярный белок, спо-
собный связывать гуаниловые нуклеотиды (отсюда
название—G-белок), сопрягающий мембранный ре-
цептор с системой вторичных посредников; АЦ—
аденилатциклаза, Г—гормон. При взаимодействии
рецептора с гормоном сигнал передается регулятор-
ному G-белку, в котором связанная с ним ГДФ
заменяется на ГТФ. Это приводит к активированию
аденилатциклазы н синтезу цАМФ
и иных сигналов с образова-
нием вторичных посредников
являются G-белки. Они могут
стимулировать (Gs=белки)
и ингибировать (Сгбелки) пе-
редачу сигналов. Многие из
них выделены в гомогенном
состоянии, расшифрован их
субъединичный состав: содер-
Мембраны клеточной оболочки
ГТФ
гдф
???
глюкагон, паратгормон, тиреотропин,
лютропин, фоллитропин, АКТГ, каль-
цитонин, -меланотропин, вазопрес-
син, адреналин
? Наружный слой
мембраны
Внутренний слой мембраны
АТ9 цМ19+РР
Протеинкиназы
Рибосомы
юоосооск
Хроматин ферменты и другие
белки
жать всегда а (М = 39—52 тыс. Да), Р (35 тыс. Да) и у (8—10 тыс.
Да). Выяснена первичная структура ряда субъединиц и их функциональная
активность (рис. 138). Что касается рецепторов, обеспечивающих сопряжение
действия сигнала с гетеротримерными G-белками, то сейчас их уже известно
более сотни. Они не только обеспечивают передачу сигнала к G-белкам,
но и способны умножать этот сигнал до тех пор, пока на них действует
лиганд, так как каждый рецептор может активировать множество G-белков,
пока с ним продолжает оставаться связанной сигнальная молекула. Таким
образом G-белок-сопряженные. взаимодействия образуют многовекторную
и многоуровневую сеть передачи и обработки сигнальной информации.
Рецепторы, несмотря на функциональные различия, обладают высокой сте-
пенью структурной гомологии, так как каждый из них включает 7 транс-
мембранных доменов, представленных ос-спиральными конформациями по-
липептидной цепи.
Концентрация цАМФ, а также другого вторичного посредника (цГМФ)
контролируется фосфодиэстеразами циклических нуклеотидов, которые уско-
ряют гидролиз цАМФ и цГМФ и прекращают их регуляторное действие.
Активность же фосфодиэстеразы цАМФ ингибируется 2', 5'-олигоаденилатом.
Так создается многозвенная система регуляции протеинкиназных реакций
в клетке:
илиго-z -аденилат
457
Рис. 138. Структура и механизм действия G-белка:
I—под воздействием рецептора гормона ГДФ отделяется от G-белка и заменяется на ГТФ, а сам
G-белок диссоциирует на а-субъединицу и комплекс Р- и у-субъедиииц; II—а-субъединица сближа-
ется с аденилатциклазой и активирует ее, что обеспечивает образование цАМФ; III—медленно
работающий ГТФазный центр гидролизует ГТФ, что приводит к утрате а-субъедиинцей ее ак-
тивирующего воздействия на аденилатциклазу и инактивации последней; синтез цАМФ прекраща-
ется; IV—а-субъединица высвобождает аденилатциклазу и ассоциируется с комплексом 0- н у-
субъединиц; система приходит в исходное состояние и готова снова принять и реализовать гормо-
нальный сигнал
Однако некоторые из гормонов пептидной природы действуют не по
аденилатциклазному механизму. Например, инсулин, связываясь с белковым
рецептором (М=460 000, гликопротеин, состоящий из 4 субъединиц) плазмати-
ческой мембраны клетки-мишени, изменяет ее проницаемость (см. рис. 136).
В результате этого усиливается проникновение в клетку субстратов (глюкоза,
аминокислоты и др.) и в ней на полную мощность включаются в работу
соответствующие ферменты. Аналогичным образом действует окситоцин: обра-
зование гормон-рецепторного комплекса сопровождается усилением переноса
Са2+, что инициирует сокращение мышечных волокон альвеол молочных желез.
Биосинтез пептидных гормонов. Биосинтез всех пептидных гормонов, за
исключением рилизинг-факторов (см. ниже в этом разделе), протекает в соот-
ветствии со схемами, известными для биосинтеза белковых тел. Однако есть,
по крайней мере, три особенности в биосинтезе пептидных гормонов, которые
следует отметить.
Первая из них состоит в том, что, как правило, пептидные гормоны
возникают путем протеолитической фрагментации высокомолекулярных пред-
шественников—препрогормонов. При этом от них сначала отщепляются
458
сигнальные пептиды (см. с. 300), способствовавшие выведению предшествен-
ников гормонов из железистых клеток, а потом—пептидные фрагменты про-,
гормонов.
Вторая особенность касается биосинтеза пептидных гормонов, молекулы
которых состоят из нескольких пептидных цепочек, как, например, у инсулина.
Выяснено, что соединение пептидных цепочек друг с другом идет при участии
инсулин: глутатионтрансгидрогеназы:
2SS-1 гтатиои
(Н8)2-цепь А + (Н8)2-цепь В--' '** ЦеПЬ ^“^h“uenb В
4Н8-глутатиои
Впрочем, в последнее время выявлено, что вообще замыкание SS-мостиков
в биологически активных белках осуществляется ферментативным путем.
Поэтому, возможно, эта реакция не столь специфична для пептидных гормонов.
Третья особенность заключается в том, что биосинтез пептидных гормонов
регулируется в ряде случаев нейрогормонами, продуцируемыми особым участ-
ком головного мозга—гипоталамусом. Они представлены олигопептидами,
и их насчитывается около десяти. Структура 4 из них выяснена. Так как они
либо усиливают синтез пептидных гормонов гипофиза, попадая в него по
капиллярной системе прямо из гипоталамической области, либо ослабляют
его, их называют либеринами и статинами соответственно (сохраняется и пер-
воначальное название, а именно—рилизинг-факторы, т. е. облегчающие вы-
свобождение тропных пептидных гормонов). Так, благодаря титанической
работе, проведенной двумя группами ученых (Р. Тиллемин с сотр. и Э. Шелли
с сотр.; половина Нобелевской премии 1977 г. по физиологии и медицине),
в конце 70-х годов была установлена структура тиролиберина—трипептида,
резко повышающего выделение гипофизом тиреотропина:
Пироглу-гис-про (NH2)
Не следует забывать также, что пептидные гормоны с тропными функци-
ями подчиняются регуляции по принципу обратной связи теми конкретными
гормонами, биосинтез которых они ускоряют.
Открытие рилизинг-факторов послужило толчком для мощного всплеска
работ по изучению регуляторных функций пептидов. Наиболее выдающимся
их результатом оказалось обнаружение семейства опиоидных, эндогенных
пептидов (эндорфины и энкефалины), являющихся индукторами ощущений
удовольствия, приятного настроения, состояния эйфории в результате их
прямого морфиноподобного воздействия на опиатные рецепторы центральной
нервной системы. Они же оказывают болеутоляющее действие, влияют на
кровяное давление, двигательную и дыхательную функцию, температуру тела
и т. п. Будучи пептидами протяженностью от 5 до 31 аминокислотного
остатка, опиоидные пептиды возникают из проопиокортина (М = 29 кДа, 265
аминокислотных остатков, синтезируется в мозге) путем его селективного
гидролиза. Так как характерная для них последовательность с N-конца моле-
кулы—тпир-гли-гли-фен- довольно распространена в пищевых белках, они
могут возникать при деструкции последних.
459
Открытие опиоидных пептидов и ряда других нейропептидов поставило
проблему возможных социальных последствий развития этой области эндо-
кринологии; его расценивают сейчас как «нейропептидную революцию».
ПРОЧИЕ ГОРМОНЫ
S,
Из гормонов, не являющихся по своей химической природе стероидами
или пептидами, у человека и животных хорошо известны адреналин, тироксин
и простагландины, а также аналогичные им соединения; в растительном
царстве широко представлены ауксины, гиббереллины и цитокинины.
Адреналин—гормон, синтезируемый в мозговом веществе надпочечных
желез. О его существовании известно более столетия. В 1901 г. адреналин был
выделен из экстракта надпочечников в кристаллическом состоянии Такамине,
Альдрихом и И. Фюртом. Двумя годами позже Ф. Штольц дал окончательное
доказательство его струк уры путем синтеза. Адреналин оказался 1-(3,4-
диоксифенил)-2-метиламиноэтанолом:
ОН
*снон
I
h3c-nh-ch2
Это бесцветный кристаллический порошок с /пл = 215—126° С. Обладая асим-
метрическим атомом углерода (отмечен звездочкой), адреналин существует
в виде двух оптических изомеров. Из них левовращающий ([ot]D2O= —53,5°) по
гормональному действию в 15 раз активнее правовращающего. Именно он
синтезируется в надпочечниках.
В мозговом слое надпочечников человека, весящих 10 г, содержится около
5 мг адреналина. Кроме того, в них же найдены гомологи адреналина: норад-
реналин (0,5 мг) и изопропиладреналин (следы). Формулы их приведены ниже
при рассмотрении путей биосинтеза этих гормонов.
Адреналин и норадреналин есть также в крови человека. Содержание их
в венозной крови составляет 0,04 и 0,2 мкг% соответственно. Предполагают,
что адреналин и норадреналин в виде соли с АТФ в небольших количествах
откладываются в окончаниях нервных волокон, высвобождаясь в ответ на их
раздражение. В результате этого устанавливается химический контакт между
окончанием нервного волокна и клеткой или между двумя нейронами.
Все три вещества—адреналин норадреналин и изопропиладреналин —
оказывают мощное влияние на сосудистую систему организма. Кроме того, они
повышают уровень обмена углеводов в организме, усиливая распад гликогена
в мышцах. Это объясняется тем, что фосфорилаза мышц под опосредствован-
ным аденилатциклазой действием адреналина переходит из неактивной формы
(фосфорилаза в) в активную форму (фосфорилаза а) (см. с. 334).
Таким образом, адреналин в мышцах выполняет ту же функцию, что глюкагон
в печени, обеспечивая запуск аденилатциклазной реакции после взаимодействия
с поверхностным гормональным рецептором клетки-мишени (см. рис. 137).
Биосинтез адреналина и его гомологов осуществляется из тирозина в соот-
ветствии со следующей схемой:
460
Все указанные р акции осуществляются при участии соответствующих
ферментов.
Тироксин—гормон, возникающий в щитовидной железе. Первые сведения
о тиреоидном гормоне относятся к концу прошлого столетия (Ф. Блюм, 1896),
но лишь сравнительно недавно Е. Кендаль, Дж. Рош и Р. Питт-Риверс устано-
вили, что непосредственным действующим началом, выделяемым железой
в кровь, являются иодтиронины:
L-Тироксин
(3,5, з’.б’-тетраиод-
тироиин)
Схема 13. Пути биосинтеза адреналина и его гомологов
Связываясь с переносчиком—а2-глобулином крови, они попадают в клет-
ки тканей организма, где и проявляют свое действие. В течение суток в кровь
поступает около 1 мг тироксина, что в три раза превышает суточную потреб-
ность в нем. В тканях осуществляется дальнейшее видоизменение иодтирони-
нов, в результате чего из них возникают более активные в биологическом
отношении вещества, являющиеся, возможно, истинными носителями гормо-
нальной активности.
Перечисленные выше соединения в разной степени проявляют нижеследу-
ющую биологическую активность. При их недостатке (гипофункция щитовид-
ной железы) замедляется обмен веществ, развивается ожирение, задерживается
рост у детей, приостанавливается психическое развитие. Все эти явления часто
объединяют под общим названием—кретинизм. При избытке тироксина и его
461
производных (гиперфункция щитовидной железы) резко усиливается обмен
веществ, учащается пульс, повышается раздражимость, падает масса, развива-
ется пучеглазие. Весь комплекс перечисленных нарушений принято называть
базедовой болезнью (по имени врача Базедова, детально описавшего гиперфун-
кцию щитовидной железы).
Механизм действия тиреоидных гормонов окончательно не установлен.
Однако ясно, что он сводится к индукции биосинтеза ферментов, так как
уровень продукции более 100 из них изменяет я под влиянием L-тироксина.
Есть достаточно оснований считать, что белки-рецепторы тиреоидных гормо-
нов сосредоточены непосредственно в хроматине ядра; связываясь с ними,
иодтиронины изменяют метаболическую активность последнего. Вместе с тем
установлено, что тиреоидные гормоны повышают активность аденилатцик-
лазы и содержание цАМФ в клетках-мишенях, т. е. действуют через рецепторы
аденилатциклазной системы (см. рис. 136).
В биосинтезе тиреоидных гормонов есть черты большого своеобразия. Клю-
чевой реакцией является конденсация двух молекул дииодтирозина в молекулу
тетраиодтиронина. Процесс осуществляется в щитовидной железе при участии
особого белка—тиреоглобулина (М=660 000; 2 x 330000). Полипептидная цепь
каждой из субъединиц его включает «2600 аминокислотных остатков и является
претендентом на самую длинную из известных природных полипептидных цепей.
Установлено, что иодированный тирозин не вступает в полипептидную
цепь тиреоглобулина в процессе его биосинтеза. В специфической области
щитовидной железы, которую называют коллоид, при посредстве тиреоид-
пероксидазы в присутствии эндогенной Н2О2 идет включение атомов иода
в N-концевой остаток тирозина полипептидной цепи субъединицы тирео-
глобулина. Затем осуществляется реакция конденсации свободного дииодтиро-
зина с радикалом связанного дииодтирозина:
К-концевой тироксин
462
Вместе с тем возможна конд нсация N-концевого моноиодтирозина со
свободным дииодтирозином или моноиодтирозином. Это приводит к синтезу
Л-концевых трииодтиронина и дииодтиронина.
В результате реакции гидролиза N-концевые тироксин, трииодтиронин или
дииодтиронин отщепляются и поступают в железу, а затем—в кровь. Как
показано выше, синтез тироиодных гормонов стимулируется тиреотропином.
Простагландины—соединения с широким спектром гормонального дей-
ствия. Первые данные об их существовании относятся к 1930 г., хотя свое
название они получили только в 1957 г., когда С. Бергстрем и сотр. выделили их
в кристаллическом состоянии. Простагландины оказались производными поли-
еновых 20-углеродных жирных кислот, из которых они синтезируются во многих
тканях человека и животных, преимущественно в репродуктивных органах.
В зависимости от строения циклической части молекулы различают природные
простагландины А, В, С, D, Е, F,G и Н, а число двойных связей в боковых цепях
перечисленных типов простагландинов обозначают цифровыми индексами:
Простагландин С2
В настоящее время известно около 30 природных простагландинов и син-
тезировано около 500 их аналогов. За работы по изучению простагландинов
С. Бергстрему (выделение), Б. Самуэлсону (выяснение структуры) и Дж. Вейну
(синтез аналогов) в 1982 г. присуждена Нобелевская премия.
Действие простагландинов отличается крайне разнообразными физиологи-
ческими и фармакологическими эффектами. В связи с этим они находят все
463
более широкое применение при создании принципиально новых лекарствен-
ных средств. Видимо, это связано с тем, что механизм их действия сводится
к усилению или ослаблению влияния многих других гормонов на фундамен-
тальные стороны обмена веществ на генетическом, аденилатциклазном и дру-
гих уровнях. Постепенно укрепляется мнение, что простагландины являются
модуляторами гормон-рецепторных комплексов, т. е. способны изменять их
активность, опосредуя действие гормонов.
Ауксины—гормоны, стимулирующие рост целых растений и отдельных их
частей. Открыты в период 1924—1928 гг. Н. Г. Холодным во время его рабо-
ты в Германии. Они содержатся в корнях, стеблях и листьях. Известно два
ауксина—а и fe:
Н(ОН)—СН2—СН(ОН)—СН(ОН)—соон
Ауксин а
н3с—сн2—сн
сн—сн2—сн3
н
:н3
Н3С—СН2—СН
I
СН
н—сн2—сн:
Нз
СН(ОН)—сн2—со
—сн2—соон
Ауксин b
Это жирорастворимые вещества, в ничтожных количествах ускоряющее рас-
тяжение клеток и рост растений.
Гетероауксин—гормон, содержащийся в растениях и образуемый дрожжа-
ми, плесневыми грибами и бактериями. В 1934 г. Ф. Кегль установил, что он
представляет собой Р-индолилуксусную кислоту:
соон
Гстсрояукош
Особенно хорошо ускоряет рост корней, в связи с чем его широко ис-
пользуют практически при размножении растений черенкованием.
Гиббереллины—гормоны, образуемые грибком из рода фузариум, были
выделены в 20-е годы нашего столетия в Токийском университете под руковод-
ством Т. Ябута. Сейчас известно несколько десятков гиббереллинов. Струк-
турная формула одного из них представлена ниже:
Гибберелловая кислота
Гиббереллины инициирут прорастание семян, ускоряют рост растений,
стимулируют начало цветения и т. п.
464
Этилен (СН2=СН2). Его гормональная активность подмечена в 1901 г.
Д. Н. Нелюбовым (С.-Петербургский университет) и переоткрыта в 20-е годы
нашего столетия. Он ускоряет созревание плодов, вызывает укорочение
и утолщение стебля растений, препятствуя полеганию хлебов, способствует
опадению листьев и плодов. В Уэльском университете (Великобритания)
М. Холлом и в Висконсинском университете (США) Т. Бликкером секвениро-
ваны гены рецепторов этилена и интенсивно ведутся эксперименты по выясне-
нию механизма действия этого простейшего гормона растений. Недавно вы-
яснено, что в растениях этилен возникает при окислении 1-аминоциклопропан-
1-карбоновой кислоты при участии этиленобразующего фермента (М = 35 кДа,
оптимум pH = 7,2, оптимум t=26° С).
Кинетин—гормон, относящийся к группе цитокининов, случайно обнару-
жил ассистент Висконсинского университета Ф. Скуч в продуктах распада
ДНК (1940—1950). Он резко ускоряет клеточное деление, способствует биоси-
нтезу нуклеиновых кислот и белков. Его химическая природа такова:
Кинетии (N&-фурфу рил-
аминопурии)
В последнее время предпринимают очень активные попытки выя нить
механизм действия гормонов растений. Особенно интересные данные получе-
ны при изучении механизма действия гетероауксина, гиббереллина и кинетина.
Оказалось, что перечисленные вещества воздействуют на один из фундамен-
тальных процессов в живой природе—метилирование ДНК и тем самым
могут контролировать транскрипцию, т. е. экспрессию генов. Кроме того,
в цитоплазме растительных клеток найде ы белки, похожие на рецепторы
стероидных гормонов животных и способные под влиянием фитогормонов
вовлекаться в модуляцию транскрипции генов. Отсюда проистекают все те
разнообразные влияния, которые названные соединения оказывают на раз-
витие растений, индуцируя или активируя синтез белков, необходимых для
прохождения определенных физиологических процессов в растительном ор-
ганизме.
ПРИМЕНЕНИЕ ГОРМОНОВ
Гормоны широко используют в практической медицине для лечения бо-
лезней, вызванных гормональной недостаточностью. В частности, их приме-
няют для лечения аддисоновой болезни, болезни щитовидной железы и диа-
бета. Стероидные гормоны и их многочисленные синтетические аналоги
применяют с успехом для лечения ревматических осложнений, ожогов, глаз-
ных болезней. Половые гормоны и их производные, в частности тестостерон-
пропионат, используют для лечения грудных желез. Адренокортикотропный
гормон и кортизон благоприятно действуют при острой лейкемии (рак
крови).
Под влиянием эстрогенов и анаболических стероидов привес откарм-
ливаемых животных увеличивается на 16—20% при экономии расхода
корма на 1 кг привеса на 8—12%. Применение соматотропного и тире-
оидных гормонов в молочном животноводстве сопровождается приростом
465
удоев на 10—25% и возрастанием жирности молока. Эти же препараты
стимулируют рост шерсти у овец. Стероидные гормоны важны для регуля-
ции репродуктивных циклов у животных. Естественно, что применение пере-
численных препаратов в мясном и молочном животноводстве должно со-
провождаться жестким контролем остаточных гормональных препаратов
в готовых продуктах.
Одно из самых п рспективны направлений в практическом использовании
фитогормонов—применение для регулирования роста и развития растений
ауксинов, гетероауксина, гиббереллинов, кинетина и многих десятков более
активных, чем перечисленные, природных и синтетических препаратов. На
базе тех представлений, развитию которых дали толчок исследования по
регулированию роста растений, возникли практические способы борьбы с сор-
няками при посредстве гербицидов, заде жки прорастания семян при их
хранении, предотвращения преждевременного сбрасывания плодов фруктовы-
ми деревьями, предуборочного удаления листьев у хлопчатника при помощи
дефолиантов и т. п.
Особенно большой интерес представляет практическое использование гор-
монов и их аналогов для создания Ш и IV поколений инсектицидов. Дело в том,
что насекомые, уничтожающие ежегодно до 15% урожая и конкурирующие
с человечеством за источники питания, обладают гормонами, которых нет
у теплокровных животных и человека. К их числу относятся ювенильные
гормоны, поддерживающие фазовое состояние насекомых:
сн, сн,
н :-с—сн—сн,—сн,—с=>сн—сн,—сн,—сн=сн—соосн,
Юмвялыш* горио, III
Их называют гормонами молодости, так как обработанные ими насекомые
не в состоянии перейти к следующей фазе развития (ш пример, личинка не
превращается в куколку, а куколка—в бабочку) и погибают. Такой же и даже
более мощный эффект оказывают аналоги ювенильных гормонов—ювеноиды.
Их синтезировано сейчас более двух тысяч, и некоторые из них отобраны для
уничтожения насекомых. Это и есть инсектициды Ш поколения, действие
которых основано на нарушении гормональной регуляции обмена веществ
у насекомых. Ювеноиды совершенно безвредны для человека и сельскохо-
зяйственных животных.
Оказалось также, что действие ювенильных гормонов у насекомых уравно-
вешивается другими их гормонами—экдизонами, относящимися к стероид-
ным гормонам:
Д-экдизон
466
И на ювенильные гормоны насекомых, и на экдизоны оказывают сильное
влияние блокирующие их действие вещества—антигормоны. Например, дей-
ствие экдизона блокируется дифторбензуроном (техническое название—ди-
милин):
Дифторбензурон
^[(4-хлорфснил)амино] рбонил]-2>дифтор0еяз*мид
Они дали начало четвертому поколению инсектицидов. Есть все основания
полагать, что исследование нейрогормонов насекомых приведет к создан ю
пятого поколения средств борьбы с насекомыми.
Чем глубже исследуют ученые строение и механизм действия гормонов,
тем ближе подходят они к разработке глубоко научных и наиболее эффектив-
ных способов управления ростом и развитием животных и растений, а также
здоровьем человека.
ГЛАВА ХШ
ВЗАИМОСВЯЗЬ И РЕГУЛЯЦИЯ ОБМЕНА
ВЕЩЕСТВ
Было бы большой ошибкой думать, что обмен различных классов ор-
ганических соединений осуществляется независимо друг от друга. Между тем:,
в такую ошибку легко впасть, так как по необходимости изучение превраще-
ний веществ, принадлежащих к различным классам, ведется раздельно. Поэ-
тому, рассм трев вопросы обмена белков, нуклеиновых кислот, углеводов
и липидов, необходимо разобраться во взаимосвязях между этими процессами
и закономерностями их регуляции.
ВЗАИМОСВЯЗЬ ПРОЦЕССОВ ОБМЕНА ВЕЩЕСТВ
Если обратиться к первичному биосинтезу органического вещества, то
легко убедиться в том, что первым стабильным соединением, которое образу-
ется в результате фиксации СО2 на рибулозо-1,5-дифосфате, является 3-фосфо-
глицериновая кислота. Уже от этого простейшего соединения начинаются цепи
реакций, ускоряемых ферментами, в результате которых синтезируются угле-
воды, аминокислоты, глицерин, высшие жирные кислоты, полиизопреноиды,
стеролы и другие соединения. Из аминокислот, СО2 и NH3 возникают пурино-
вые и пиримидиновые основания. Следовательно, прямым продолжением
первичной фиксации СО2 сразу являются многообразные процессы создания
мономеров, из которых далее строятся биополимеры (полисахариды, белки,
нуклеиновые кислоты и т. п.), разнообразные липиды и многие другие ор-
гани еские соединения, входящие в состав растений, животных и микробов.
Однако уже у автотрофов наряду с прямым, первичным биосинтезом
органических веществ осуществляется новообразование органических соедине-
ний одних классо t за счет таковых других классов. Такого рода превращения
достигают своего расцвета у гетеротрофов, где не только в процессе питания,
но и в ходе жизнедеятельности идет перестройка белков, нуклеиновых кислот,
углеводов, липидов и многих других соединений через ключевые метаболиты
промежуточного обмена, в первую очередь через пировиноградную кислоту
(ПВК), а-кетоглутаровую и щавелевоуксусную кислоту (ЩУК) и ацетил-КоА
(схема 14).
Следовательно, взаимопереходы между отдельными классами органиче-
ских соединений—естественное, неизбежное и крупномасштабное явление
в живой природе.
Рассмотрим несколько подробнее, как они осуществляются в ряде конкрет-
ных случаев.
Взаимосвязь обмена нуклеиновых кислот и белков выражается прежде всего
в том, что новообразование как нуклеозидтрифосфатов, так и самих нукле-
иновых кислот зависит от наличия в клеточном содержимом соответствующе-
го набора белков-ферментов (ДНК- и РНК-полимераз, лигаз, топоизомераз,
468
Белки
Углеводы
Липиды
Схема 14. Взаимосвязь обмена главных классов органических соединений
Нуклеотиды
Нуклеиновые
кислоты
С02 С,- NHj
фраг-
менты
а также ферментов биосинтеза пуриновых и пиримидиновых циклов). Кроме
того, именно аминокислоты (аспарагиновая—в случае пиримидиновых нукле-
отидов и 1 лицин, аспарагиновая кислота и глутамин—в случае пуриновых
нуклеотидов) служат основными исходными соединениями для построения
пиримидинового и пуринового колец. Вместе с тем новообразование белков по
матричной схеме невозможно без участия всех видов РНК и, естественно,
ДНК, на которой в качестве матрицы возникают рибонуклеиновые кислоты.
Поэтому многие исследователи полагают, что в истории развития жизни на
Земле биосинтез белков представлял процесс первичный, а биосинтез нукле-
иновых кислот—явление вторичное, призванное в основе своей обслуживать
биосинтез белка. В свою очередь, в процессе уриколиза образуется глиок-
силовая кислота. Путем переаминирования она может превращаться в глицин,
и, следовательно, известная часть этой аминокислоты может возникать за счет
распадающихся пуриновых оснований.
Взаимосвязи в обмене нуклеиновых кислот и углеводов многообразны.
Во-первых, в процессе апотомического распада углеводов образуется рибозо-
5-фосфат, из которого возникает 5-фосфорибозил-1 -пирофосфат, служащий
совершенно незаменимым соединением для биосинтеза пуриновых и пирими-
диновых нуклеотидов. Именно 5 фосфорибозил-1-пирофосфат принимает на
себя недостроенную молекулу пиримидина и именно на 5-фосфорибозил-1-
пирофосфате начинает строиться имидазольный цикл будущего пуринового
кольца. Таким образом, р,D-рибоза и Р,D-дезоксирибоза, являющиеся непре-
менными составными частями пуриновых и пиримидиновых нуклеотидов,
поступает в нуклеиновые кислоты за счет распадающихся углеводов.
Во-вторых, в известной мере, и распад нуклеиновых кислот может служить
источником соединений, служащих для биосинтеза углеводов, так как вы-
свобождаемая при гидролизе пуриновых и пиримидиновых нуклеотидов ри-
боза, включаясь в общий круговорот углеводов в ор анизме, может пе-
реходить в рибозо-5-фосфат, из которого легко строится глюкозо-6-фосфат.
Взаимопереходы последнего в фосфорные эфиры других моносахаридов ши-
роко известны.
В-третьих, распадающиеся углеводы поддерживают на определенном уров-
не субстратное и окислительное фосфорилирование АДФ, т. е. обеспечивают
биосинтез АТФ. Последняя абсолютно необходима для превращения нуклео-
зидмонофосфатов в нуклеозидтрифосфаты—субстраты для полимераз. Сле-
довательно, от наличия в организме сахаров и интенсивности их распада
зависит объем биосинтеза нуклеиновых кислот. Лишь у фотосинтезирующих
и хемосинтезирующих организмов эта зависимость может быть ослаблена за
счет использования АТФ, озникшей в результате фотосинтетического и хемо-
синтетического фосфорилирования.
469
Наконец, в четвертых, биосинтез углеводов в значительной мере зависит
от нуклеинового обмена. Эта зависимость выражается в том, что известная
часть уридинтрифосфорной кислоты используется для биосинтеза УДФ-глю-
козы—важнейшего продукта, гликозидные остатки с которого переносятся
на нередуцирующий конец молекулы синтезируемого глюкана. Аналогична
роль гуанозиндйфосфатглюкозы в биосинтезе целлюлозы и ряда других
нуклеозиддифосфатсахаров в новообразовании тех или иных гексозанов
и пентозанов. Все это совершенно по-новому ставит вопрос о зависимости
специфического биосинтеза сложных углеводов от обмена соединений нукле-
отидной природы.
Формы связи обмена нуклеиновых кислот и липидов разработаны мало. Ни
те ни другие не [вляются непосредственными исто’ никами соединений кото-
рые могли бы использоваться для построения нуклеиновых кислот за счет
липидов или наоборот. Следовательно, «субстратная» форма связи не харак-
терна для обмена нуклеиновых кислот и липидов, хотя, конечно, через посред-
ство углеводов и белков в конце концов может осуществлять я частичный
переход от первых ко вторым и обратно.
Что касается иного типа взаимосвязей обмена нуклеин эвых кислот
и липидов, то они выявляются более отчетливо. При распаде пиримидино-
вых оснований возникает Р-аланиа—аминокислота, используемая для био-
синтеза коэнзима А, столь необходимого как для новообразования, так
и для деструкции высших жирных кислот. Несомненно, что Р-окисление
высших жирных кислот—составных частей большинства липидов—служит
источником для поддержания на достаточном уровне синтеза нуклеозид-
трнфосфатов, если указанное окисление сопряжено с фосфорилированием
и новообразованием АТФ. Так же, как и в биосинтезе углеводов, большую
роль в биосинтезе некоторых липидов играют нуклеозиддифосфатсоединения,
для образования которых расходуются соответствующие нуклеозидтрифос-
фаты. Так, для биосинтеза ЦДФ-холина или ЦДФ-коламина—важнейших
метаболитов в синтезе фосфатидов—необходим ЦТФ—метаболит нукле-
инового обмена.
Связующим звеном в обмене белков и углеводов при переходе первых во
вторые и особенно вторых в первые служит ПВК. Являясь главным конечным
продуктом дихотомического распада углеводов, ПВК служит исходным веще-
ством для биосинтеза аланина, валина и лейцина. При ее карбоксилировании
образуется щавелевоуксусная кислота, из которой строится новая группа
аминокислот—аспарагиновая кислота, треонин, метионин, изолейцин и ли-
зин. Вступая в цикл трикарбоновых и дикарбоновых кислот, ПВК использует-
ся для 6i осинтеза а-кетоглутаровой кислоты, из которой образуются глута-
мина вая кислота, пролин и аргинин. Предшественник ПВК—3-фосфоглицери-
новая кислота—я ляется исходным соединением для синтеза серина, глицина,
цистина и цистеина.
Наконец, промежуточные продукты апотомического и дихотомического
распада углеводов незаменимы в синтезе остальных постоянно встречаю-
щихся в белках аминокислот: на рибозо-5-фосфате строится имидазольное
кольцо гистиди а, а из эритрозо-4-фосфата и фосфоенолпировиноградной
кислоты синтезируется шикимовая кислота, из которой образуются фени-
лаланин, тирозин и триптофан. Таким образом, у аутотрофов из углеводов
при наличии источника аммиака в организме могут синтезироваться все
аминокислоты, постоянно встречающиеся в белках. Естественно, что из них
образуются белки, и, следовательно, переход углеводов в белковые тела
представляет основной вид взаимосвязи обмена указанных двух классов
соединений.
470
Возможен и обратный процесс. Многие аминокислоты (аланин, фенилала-
нин, тирозин, гистидин, триптофан, серин, цистеин) содержат в своем составе
трехуглеродный фрагмент, из которого в процессе распада указанных амино-
кислот возникают ПВК и ее дериваты. Дезаминирование глутаминовой и ас-
парагиновой кислот ведет к образованию а-кетоглутаровой и щавелевоуксус-
ной кислот соответственно, которые при посредстве цикла трикарбоновых
и дикарбоновых кислот переходят в ПВК. Такова же судьба пролина, который
легко превращается в глутаминовую кислоту, а из нее—в пировиноградную.
Следовательно, подавляющее большинство аминокислот может явиться в ор-
ганизме источником для образования ПВК. От последней несложен переход
к углеводам посредством в основном обращения реакций дихотомического
распада фруктозо-1,6-дифосфата.
Из других форм взаимосвязи обмена белков и углеводов привлекают
внимание две. Многочисленные белки-ферменты обслуж вают процессы рас-
пада и синтеза углеводов в организме. В свою очередь, распад углеводов,
сопряженный с синтезом АТФ из АДФ и неорганического фосфата, энергетиче-
ски обеспечивает белковый синтез в клетке.
Взаимосвязь обмена белков и липидов выражается в том, чо распад липи-
дов, как и распад углеводов, обеспечивает, с одной стороны, исходные соеди-
нения для биосинтеза аминокислот (а из них белков) и, с другой стороны, не
менее, а может быть, более, чем углеводы, поддерживает образование белков
энергетически.
Одним из основных продуктов распада липидов, в частности высших жир-
ных кислот, возникающих при гидролизе триглицеридов, фосфатидов или
стеридов, является ацетил-КоА. Включаясь в цикл трикарбоновых и дикарбо-
новых кислот, он обеспечивает синтез а-кетоглутаровой кислоты, превращение
которой в аминокислоты рассмотрено выше. Поступая в глиоксилевый цикл,
ацетил-КоА служит для расширенного воспроизводства в организме щавеле-
воуксусной кислоты, а из нее—ПВК. Из обеих названных кислот также
синте ируются аминокислоты.
Обмен глицерина, высвобождаемого при гидролизе триглицеридов, через
углеводы ведет к таким аминокислотам, как гистидин, фенилаланин, ирозин
и триптофан. Следовательно, все постоянно встречающиеся в белках амино-
кислоты могут синтезироваться за счет распадающихся липидов.
В известной мере, возможен синтез липидов за счет распадающихся белков.
В предыдущем разделе было показано, что при распаде ряда аминокислот
образуется ПВК. При ее окислительном декарбоксилировании возникает
ацетил-КоА—исходное соединение для синтеза высших жирных кислот,
стеролов и других составных частей липидов. ПВК может также превратить-
ся в фосфоглицерин (путем обращения реакций дихотомического распада
углеводов)—другой важный компонент липидов. Однако такого рода пере-
ход вряд ли широко осуществляется в нормальных условиях жизнедеятель-
ности.
Энергетическая роль липидоц, особенно триглицеридов, общеизвестна. По-
тенциальные возможности для синтеза АТФ сопряженно с окислением высших
жирных кислот огромны. Известны случаи, когда распад липидов является
единственным источником энергии для биосинтеза белка (например, при
синтезе фиброина и серицина шелка в шелкоотделительной железе коконо-
прядущих насекомых).
Говоря о взаимосвязи обмена белков и липидов, нельзя обойти вопрос
о влиянии последних на процесс биосинтеза белков. Твердо установлено, что
рибосомальный синтез белка протекает во много раз энергичнее, если рибосо-
мы связаны с липопротеиновыми мембранами.
471
Углеводы и липиды очень легко взаимопревращаются в организме; связу-
ющими соединениями при этих переходах служат ПВК в ацетил-КоА.
Пировиноградная кислота—основной продукт дихотомического распада
углеводов, при окислительном декарбоксилировании дает ацетил-КоА, кото-
рый служит для синтеза высших жирных кислот, стеролов, каротиноидов
и других полиизопреноидов. Столь же легко осуществляется переход от
углеводов к фосфоглицерину, необходимому для синтеза простых и сложных
липидов.
Ацетил-КоА и глицерин—главные продукты распада липидов—служат
исходными соединениями для синтеза углеводов. Ацетил-КоА при посредстве
глиоксилевого цикла переходит в ПВК, а из нее—в углеводы путем обраще-
ния реакций дихотомического распада последних.
Превращение глицерина в углеводы идет через 3-фосфоглицериновый аль-
дегид, а затем описанным выше способом.
Сказанное не исчерпывает всего многообразия взаимосвязей обмена бел-
ков, нуклеиновых кислот, углеводов, липидов и других соединений. Между
ними существуют более сложные, нежели простое использование в качестве
субстратов, формы взаимозависимости. Так, вещества, образующиеся в про-
цессе обмена соединений одного класса, оказывают глубочайшее влияние на
обмен веществ, относящихся к другому классу. Превосходным примером
подобного типа взаимосвязи обмена белков и нуклеиновых кислот может
служить образование информационной РНК, служащей матрицей для биосин-
теза специфических белков, с одной стороны, и блокирование синтеза иРНК
определенного вида белками—с другой.
Общеизвестно, что никакие реакции обмена невозможны без специфиче-
ских белков-ферментов, и в этом смысле белковый обмен определяет ход
превращений соединений, относящихся к другим классам. Решающее значение
имеет ход окислительного фосфорилирования и создание резервов АТФ
в клетке. От уровня последней в клеточном содержимом зависит, в свою
очередь, весь ход обмена веществ, ибо АТФ обеспечивает энергетические
потребности биосинтеза соединений всех классов. Число подобных примеров
глобальной взаимозависимости и взаимообусловленности обмена белков, нук-
леиновых кислот, углеводов, липидов и других соединений огромно. В сово-
купности они и составляют учение о регуляции обмена веществ. Но каждый из
них в отдельности подчеркивает ту или иную форму взаимосвязи обмена
веществ в организме.
РЕГУЛЯЦИЯ ОБМЕНА ВЕЩЕСТВ
Приведенные выше данные о взаимосвязи и взаимозависимости обмена
белков, нуклеиновых кислот, углеводов и липидов убеждают в том^ что обмен
веществ представляет собой стройный ансамбль многочисленных и тесно
увязанных друг с другом химических процессов. Ведущая роль в этом бесчис-
ленном множестве взаимодействий принадлежит белковым телам. Благодаря
их каталитической функции осуществляется все это великое множество хими-
ческих процессов распада и синтеза. С помощью нуклеиновых кислот поддер-
живается строгая специфичность при биосинтезе макромолекул, т. е. в конеч-
ном счете видовая специфичность в строении важнейших биополимеров. Бла-
годаря обмену углеводов и липидов в организме постоянно возобновляются
запасы АТФ—универсального донора энергии для химических преобразова-
ний. Эти же вещества поставляют простейшие органические молекулы, из
которых строятся биополимеры и другие соединения. В результате совершает-
472
ся непрерывный процесс самообновления живой материи, обслуживаемый
теми биохимическими механизмами, изучение которых составляет предмет
общей биохимии.
Слаженность биохимических превращений, их теснейшая связь и взаимо-
обусловленность, возможность быстрой мобилизации одних соединений для
синтеза других, возможность взаимопереходов от одного класса органических
соединений к другому, всеобщая соподчиненность биохимических механизмов
как никогда ярко выступают, когда мы оцениваем обмен веществ в целом.
Общий ход биохимических процессов в организме, регулируемый внутрен-
ними и внешними факторами, представляет единое неразрывное целое, и сам
организм в этом смысле выглядит как самонастраивающаяся, саморегулиру-
ющаяся система, поддерживающая свое собственное существование путем
обмена веществ.
Тем не менее, исходя из методических соображений, регуляцию жизненных
процессов принято рассматривать на метаболитном, оперонном, клеточном,
организменном и популяционном уровнях. Каждый из них характеризуется
своими закономерностями регуляции обмена вещества, действующими в сня-
том виде на каждом последующем уровне организации живой материи. Харак-
терно, что чем выше уровень регуляции обмена веществ, тем яснее выступает
иерархическая, блочная система управления, суть которой сводится к слежению
за поступлением сигнала и ответом на него.
Метаболитный уровень регуляции. Слаженность обмена веществ в организ-
ме, в значительной мере, определяется концентрацией разнообразных метаболи-
тов—низкомолекулярных соединений, представляющих собой продукты тех
или иных химических превращений в биологических объектах или поступа-
ющих в них в процессе питания.
Формы регуляции обмена веществ при участии метаболитов крайне
многообразны. Простейшая из них сводится к ускорению или замедлению
биохимических процессов за счет недостатка или избытка тех соединений,
которые являются участниками соответствующих реакций. Так, объем бел-
кового синтеза у гетеротрофов лимитируется поступлением незаменимых
аминокислот и интенсивностью синтеза полузаменимых аминокислот. На
этом, в частности, базируется микробиологический метод количественного
определения содержания аминокислот в белковых гидролизатах и иных
средах.
Более сложный характер носит регуляция обмена веществ за счет кон-
курентных взаимоотношений тех обменных процессов, которые замыкаются на
общие метаболиты, относящиеся, как правило, к категории ключевых: пиро-
виноградную, щевелевоуксусную и а-кетоглутаровую кислоты, ацетил-КоА,
глюкозо-6-фосфат. Многочисленные примеры такого рода приведены в раз-
деле о взаимосвязи обмена веществ в начале этой главы.
Несомненно, велика роль в регуляции обменных процессов ряда низкомо-
лекулярных соединений, относящихся к разряду биологически активных,—
витаминов, антивитаминов, коферментов, гормонов, антигормонов, вторичных
по редников и др.
Метаболиты, взаимодействуя с ферментами, способны активировать или
ингибировать их активность; примером первого рода является неоднократно
упоминавшееся активирование протеинкиназ при действии на них цАМФ; не
меньшую роль в регуляции обмена веществ играет другой вторичный посред-
ник—цГМФ, активирующий фосфолипазу А2 и С, а также участвующий
в биосинтезе простагландинов из арахидоновой кислоты. Источником новой
группы вторичных посредников, как показано недавно, являются фосфоинози-
тиды (см. с. 384):
473
Гормональный
сигнал
СН2-О—со-R
СН—О—СО —R
О
о
НО-Р-ОН он
Серотонин
Ацетилхолин
Гкстамия
Брадикинин
Вазопрессин
f Экдизон
Фосфолипаза С
н2о
Днацил-
глицерин
(ДАТ)
Инозит -1,4,5-
-* трифосфат
(пу
Фосфатидил-*
инозит -4,5-
-дифосфат
(РП\)
Гормональный сигнал воспринимается рецептором, передающим его G-
белку, который активирует фосфолипазу С. И диацилглицерин, и инозит-
1,4,5-трифосфат являются вторичными посредниками, передающими гормо-
нальный сигнал далее: первый—протеинкиназе С (М = 67—83 кДа, активи-
руется Са2+, переносит фосфат с АТФ на радикалы серина и треонина,
насчитывает не менее 7 форм — a, pi, 011, у, 5, е, Q, фосфорилирующей около
20 ферментов и ряд белков (белки цитоскелета, белки-рецепторы и др.),
а второй—в эндоплазматический ретикулум, из которого высвобождается
Са2+, возбуждающий активность Са2+/кальмодулин-зависимой протеинки-
назы, тоже обеспечивающей фосфорилирование функционально значимых
белков (рис. 139). К числу вторичных посредников относится также оксид
азота (NO)—мощный фактор гомеостаза. Он образуется в различных клет-
ках и тканях, в том числе и в тромбоцитах человека^ из L-аргинина за счет
окисления азота аминогруппы гуанидиновой группировки последнего под
действием Ь-аргинин-ИО-синтазы. Она существует в трех формах, две из
которых являются конституивными, а одна—индуцибельной, причем в ин-
дукцию экспрессии гена последней вовлекаются липополисахариды и ядер-
ный белковый фактор. Возникший так оксид азота активирует гуанилатцик-
лазу, что приводит к повышению концентрации цГМФ, функции которого
в регуляции обмена веществ отмечены выше (см. предыдущую стр.). Кроме
того, непосредственно сам оксид азота является нейротрансмиттером и ци-
тотоксическим агентом.
Примером второго рода является ретроингибирование (ингибирование по
пр нципу обратной связи) активности фермента, стоящего в начале многосту-
пенчатого превращения субстрата конечным продуктом реакции, что де ально
разработано при изучении регуляции биосинтеза пиримидиновых нуклеотидов
(см. с. 239):-
Уридин-
монофосфат
(УМФ)
Цитйдин-
Трифосфат
(ЦТФ)
Аспарагиновая________ Карбамил- Дигидро- Оротовая Оротидин-
кислота —аспарагиновая оротоваякислота —— моно-
Карбамил - кислота кислота фосфат
трансфераза
Ретроингибирование ,
474
I
Гормон
эндоплазматический ^2+ ретикулум
Рис. 139. Фосфоинозитидный путь регуляции обмена веществ (пояснения в тексте)
Явления такого же порядка происходят во многих других случаях ал-
лостерического изменения активности ферментов. Вместе с тем метаболиты
могут быть и изостерическими (конкурентными) ингиб торами ферментов
(см. с. 112). Высказано предположение, что метаболоны (см. с. 355) регули-
руются сигналами, передаваемыми через вторичные посредники; так, напри-
мер, считают, что гликолитический метаболой регулируется потоком ионов
Са, поступающих в микрокомпартменты клетки, где он локализован. Нако-
нец, метаболиты являются индукторами и корепрессорами в системах, дей-
ствующих на оперонном уровне регуляции обмена веществ.
Оперонный уровень регуляции. Опероном называется упорядоченная ком-
пактная совокупность цистронов (вместе со знаками начала и конца), считыва-
емая как единое целое в процессе синтеза мРНК на ДНК. В случае моноцист-
ронного оперона на нем синтезируется мРНК, предназначенная для биосинтеза
в рибосомальном аппарате клетки одного единственного белка, в случае
полицистронного (до полутора десятков цистронов)—ряд мРНК, на которых
рибосомаль ым путем создается семейство различных белков (чаще всего
ферментов), необходимых для осуществления многостадийного биохимиче-
ского процесса в клетке. Первичное представление о моноцистронном оперо-
не, равно как и о знаках начала и конца его считывания, дает рис. 87 на с. 259.
Сейчас ясно, что на уровне оперона регулируется главным образом объем
биосинтеза ферментов за счет изменения количества молекул мРНК, возника-
ющих в процессе транскрипции. Это оказывае решающее влияние на ход
обменных процессов, мощными двигателями которых являются ферменты.
Вместе с тем нельзя упускать из виду и то обстоятельство, что на уровне
оперона регулируется синтез мРНК для новообразования гистонов, негисто-
новых и рибосомальных белков, а также ряда других протеинов, не облада-
ющих каталитической активностью, но являющихся регуляторами метаболи-
475
Индукция
Оперой
Ферменты
Репрессор,
инактивированный
индуктором
Индуктор
Репрессия
Рис. 140. Модель оперона
ческой активности генома, деятельности трансляционного аппарата клетки
и других фундаментальных процессов обмена веществ.
Если рассмотреть несколько детальнее регуляцию на оперонном уровне
объема биосинтеза ферментов в клетке, то вырисовываются два пути: индук-
ция и репрессия. Сущность и того и другого ясна из рассмотрения рис. 140.
Биосинтез фермента может быть индуцирован низкомолекулярным метаболи-
том—индуктором, который, соединяясь с репрессорным белком (он запрещает
транскрипцию), освобождает зону оператора (знак начала оперона), что со-
провождается присоединением РНК-полимеразы и началом синтеза про-
мРНК, а затем, после ее посттранскрипционной модификации,—фермента.
Ферменты, биосинтез которых регулируется этим путем, называются ин-
дуцибельными. К ним относятся 0-галактозидаза, рибулокиназа, тирозиназа,
аспарагиназа и многие другие. Добавление индуктора (как правило, субстрата
индуцибельного фермента) резко повышает объем синтеза фермента. Напри-
мер, при добавлении 0-галактозида (лактоза) в культуральную среду кишеч-
ной палочки у последней синтез 0-галак озидазы возрастает в 10000 раз.
476
В отличие от этого ферменты, биосинтез которых стопорится под влиянием
низкомолекулярного метаболита—корепрессора (рис. 140) (переводит репрес-
сорный белок, не способный в норме оккупировать зону оператора, в активное
состояние), называются репрессибельными. К их числу принадлежат орнитин-
карбамилтрансфераза (ее корепрессор—аргинин), глутаминсинтетаза, уреаза
(их корепрессор—NH4) и ряд других.
В последнее время показано, что индукция и репрессия могут быть генера-
лизованными, т. е. контролироваться не каким-либо одним конкретным индук-
тором или корепрессором, а целой группой сходных с тем и другим соедине-
ний. Накапливаются также сведения о том, что один-единственный эффектор
(например, ppGpp—3'-пирофосфо-гуанозин-5'-дифосфат) может индуцировать
биосинтез целого семейства ферментов (например, ферментов биосинтеза
гистидина и ряда других аминокислот). Поэтому проблема индукции и репрес-
сии биосинтеза ферментов достаточно сложна, особенно в отношении индук-
ции синтеза ферментов, не свойственных данному организму (например, фер-
менты детоксикации инсектицидов у насекомых).
Конечно, только индукцией и репрессией синтеза ферментов не исчерпыва-
ется регуляция обмена веществ на уровне генети еского аппарата клетки. Как
репликация самой ДНК, так и синтез на ней в качестве матрицы разнообраз-
ных РНК, в том числе и мРНК, что, в значительной мере, предопределяет ход
обмена веществ в клетке, зависит от множества других событий. Среди
них—метилирование ДНК; фосфорилирование и ацетилирование гистонов
и негистоновых белков, входящих в состав хроматина; взаимодействие с хро-
матином гормон-рецепторных комплексов; аденилирование белков, участву-
ющих в деятельности репликационного аппарата и др. Все они связаны
с изменением метаболической активности генома, регуляцией его функций
в целом.
Клеточный уровень регуляции. К регуляторным процессам на уровне клетки
относятся: ядерно-цитоплазменные отношения; посттранскрипционная и пост-
трансляционная модификация макромолекул; транспорт веществ через мемб-
раны субклеточных частиц и мембраны эндоплазматической сети; макромоле-
кулярные (белок-белковые, белково-нуклеиновые, углеводно-белковые и липид-
белковые) взаимодействия и др. Все они носят фундаментальный характер
в регуляции обмена веществ.
Ядерно-цитоплазменные отношения сводятся к взаимозависимому контро-
лю синтеза важнейших функционально активных биополимеров. Так, малые
белковые субъединицы рибулозо-1,5-дифосфат-карбоксилазы, при посредстве
которой осуществляется важнейш й процесс акцептирования СО2 в раститель-
ной клетке (см. с. 360), синтезируются в цитоплазме, а большие субъеди-
ницы—в хлоропластах. Биосинтез первых кон ролируется, следовательно,
ядерным аппаратом клетки, вторых—хлоропластным геномом, локализован-
ным в цитоплазме. В целом, из 800—1000 белков, необходимых для функцио-
нирования хлоропластов, лишь около 15% кодируется геномом этих клеточ-
ных органелл. Кроме рибулозо-1,5-дифосфат-карбоксилазы, при участии двух
генетических систем растительной клетки (ядерной и хлоропластной) форми-
руются тилакоидные мембраны, АТФазный и РНК-полимеразный комплексы
хлоропластов. Аналогичный ядерно-цитоплазматический контроль характе-
рен также для синтеза белковых субъединиц таких важнейших каталитически
активных систем, как протонная АТФаза и цитохромоксидаза, белков внутрен-
ней и внешней мембран митохондрий, белков хлоропластных и митохондри-
альных рибосом и т. п. Таким образом, только при согласованной деятель-
ности генома ядра и геномов митохондрий, хлоропластов и других субклеточ-
ных структур, при согласованной работе белоксинтезирующих систем
477
перечисленных субклеточных частиц возникают сложные белково-ферментные
комплексы клетки, обеспечивающие фундамен альнь направления обмена
веществ.
Посттранскрипционная и посттраисляциониая модификация макромолекул—
второй важ ейший регуляторный процесс на клеточном уровне. Возникающие
при транскрипции предшественники рибонуклеиновых кислот после ряда пре-
образований (метилирование, отщепление и присоединение олигонуклеотид-
ных фрагментов и т. п.) превращаются в функционально активные РНК. Эти
процессы детально изучены при созревании мРНК, рРНК, тРНК (см. гл. VI).
В целом они предопределяют интенсивность белкового синтеза в клетке.
Однако и белки, образующиеся при рибосомальном синтезе, тоже подвергают •
ся посттрансляционной модификации (метилирование, отщепление пептидны
фрагментов, присоединение углеводной составляющей при биосинтезе глико-
протеинов и т. п.). В результате из полипептидов-предшественников получают-
ся активные ферменты, гормоны, биологически активные пептиды и др. Есте-
ственно, что от уровня осттрансляционно“[ модификации прямо или опос-
редованно зависят многие обменные процессы в клетке.
В поел дние годы особое значение придают ковалентной модификации
ферментов, так как по этому принципу регулируется активность не менее 100 из
них. Кроме фосфорилирования по радикалам серина, треонина и тирозина
соответствующими протеинкиназами (см. гл. IX, XII) большая роль принадлежит
аденилированию и уридилированию, а также АДФ-рибозилированию ферментов.
Аденилирование и уридилирование например, детально изучено Е. Стедма-
ном у глутаминсинтетазы:
%
с— сн2-сн2- сн— СООН+ NH,
нох I
nh2
АТФ
АДФ + НзРО4
О
с— сн2— сн2— сн- соон
H2N NH2
Глутаминовая кислот*
Глутамия-
синтетаза
(М-600 кДа,
12X50 000)
Глутамин
Активна в биосинтезе глу амина деаденилированная глутаминсинтетаза;
по мере присоединения к каждой из ее 12 субъединиц остатков адениловой
кислоты (при этом возможно существование 382 форм фермента) активность
ее падает. Аденилирование идет по радикалу тирозина:
478
В свою очередь, аденилилтрансфераза активна лишь в том случае, когда
она связана с регуляторным белком, не уридилированным по остатку
тирозина; если же регуляторный белок уридилирован (реакция идет ана-
логично аденилированию, но с УТФ в качестве донора уридилатного
остатка), то аденилилтрансфераза ускоряет реакцию деаденилирования
глутаминсинтетазы и переводит ее снова в более ак ивное состояние.
В случае АДФ-рибоз лирования на гуанидиновый фрагмент радикала ар-
гинина фермента (или H2N-rpynny лизина или аспарагина) переносится оста-
ток АДФ-рибозы из состава НАД+; при этом высвобождается молекула
никотинамида. Эту реакцию ускоряет НАД+-аза, обладающая АДФ-рибозил-
трансферазной активностью:
К посттрансляционной модификации белков тесно примыкает протео пани-
ческая деградация белков, особенно ферментов. Благодаря ей в клетке непре-
рывно идет противобор во двух процессов—распада ферментов и их син-
теза, определяющее в конечном итоге количество того или иного фермента
и ход ускоряемой им реакции.
Посттрансляционная модификация является источником возникновения
множествен ых форм ферментов в клетке, как, например, в случае 1 лутамин-
синтетазы. Наряду с генетически детерминированными изоферментами они
обеспечивают тончайшие юансы в регуляции обменных процессов, так как
каждый из них подобен отдельному инструменту в оркестре, вучание которо-
го составляет симфонию жизни. В последнее время также активно разрабаты-
вают проблему контроля транскрипции ядерного и митохондриального гено-
ма продуктами цитоплазмы у животных i растений.
Перенос веществ через мембраны ядра, митохондрий, лизосом, эндоплаз-
матической сети и других субклеточных элементов, равно как и через клеточ-
ную оболочку,— один из существенных механи мов регуляции обмена веществ
и многих физиологических функций организма на клеточном уровне. В пере-
носе ионов ведущая роль принадлежит циклическим пептидам и депсипеп-
тидам, низкомолекулярных веществ—сп циальным ферментам (транслока-
зам) (см. рис. 43), высокомолекулярных соединений, в частности белков,—сиг-
альным пептидам и их взаимодействию с белково-липидной частью
мембран. Недавно обнаружена и изучена особая категория белков, названных
поринами: они образуют в мембранах поры и активно переносят по ним
различные соединения (см. рис. 101 и 102).
479
Рис. 141. Белково-нуклеиновые вза-
имодействия при созревании и перено-
се мРНК
Изучению процесса транспорта веществ
уделяется все большее внимание в м мбра-
нологии. В конечном итоге поступление ме-
таболитов внутрь субклеточных частиц
и компартментов клетки, равно как и вынос
их оттуда самым непосре ственным обра-
зом, сказывается на скорости биохимиче-
ских превращений. Конечно, проблема про-
странственного разобщения обменных реак-
ций в клетке не сводится только к созданию
градиентов концентрации метаболитов.
Сейчас привлечено внимание к существова-
нию связанных с субклеточными частицами
ферментов, причем в этом случае их актив-
ность значительно возрастает («адсорбцион-
ный механизм» регуляции ферментов, со-
провождаемый эстафетной передачей про-
межуточных продуктов каталитического
процесса).
Роль макромолекулярных взаимодейст-
вии в регуляции обмена веществ на клеточ-
ном уровне особенно отчетливо выявилась
при изучении белково-нук еиновых взаимодействий. Оказалось, что про-мРНК,
синтезируемые в ядре, сначала соединяются с ядерными белками (инфор-
матиками), причем уже здесь идет отбор про-мРНК: лишь часть из них
превращается в рибонуклеопротеины (308-частицы и их ансамбли), а осталь-
ные, несущественные в данный момент для метаболических потребностей
клетки, разрушаются. В составе 308-частиц про-мРНК созревают и транспо-
нируются через ядерную оболочку, теряя при этом белковую часть. Однако
в момент выхода в цитоплазму мРНК снова соединяются с обладающими
высоким сродством к ним цитоплазматическими белками, образуя инфор-
мосомы. В составе последних мРНК сохраняются в латентном состоянии
вплоть до поступления в рибосомы. Вполне понятно, что рассмотренный
каскадный механизм переноса информации из ядра и цитоплазму самым
существенным образом сказывается на ходе обмена веществ (рис. 141). Ис-
следования в этой области, проведенные у нас под руководством академиков
Г. П. Георгиева и А. С. Спирина, удостоены Государственной премии СССР.
Разнообразные и важные для регуляции обменных процессов белково-нукле-
иновые взаимодействия идут на уровне генетического и рибосомного аппарата
клетки.
В течение последнего десятилетия получены принципиально новые данные
о контроле обмена веществ на уровне белково-нуклеиновых взаимодействий.
Обнаружено, что основная роль универсальной и наиболее изученной протеин-
киназы эукариотических клеток, а именно—казеинкиназы типа II, состоит
в регуляции степени маскирования—демаскирования-мРНК взаимодейству-
ющими с ней белками. При этом молекулам как самой мРНК, так и особому
классу иных РНК отводится роль регулятора активности [ротеинкиназ : чем
сильнее фосфорилированы РНК-связывающие белки, тем ниже их сродство
к мРНК и тем выше ее способность включаться в трансляционные процессы.
Не менее впечатляющие результаты получены при изучении липид-белковых
взаимодействий. Для регуляции обмена веществ особенно важны те из них,
что развертываются в составе белково-липидных мембран клетки. Именно
они предопределяют уровень активности мембранно-связанных ферментов,
480
степень проницаемости мембран для метаболитов, возможность трансмемб-
ранного переноса макромолекул и осуществления ряда других процессов,
зависящих от физико-химических параметров мембранного аппарата.
Изучение углеводно-белковых взаимодействий открыло новую страницу
в развитии представлений о специфических контактах на уровне макромоле-
кул, субклеточных частиц и клеток. Они основаны на существовании белков
(лектинов), избирательно узнающих углеводные компоненты, что инициирует
процессы, имеющие важнейшее значение для регуляции метаболиза.
Что касается белок-белковых взаимодействий, то их значение для регуляции
обмена веществ на клеточном уровне наиболее существенно. Они сводятся
к становлению ферментов-мультимеров, образованию мультиэнзимных ком-
плексов, формированию ансамблей ферментов и метаболонов, возникновению
гормон-рецепторных комплексов в случае гормонов пептидной и белковой
природы. Соответствующие конкретные примеры рассмотрены ранее.
До недавнего времени практически не было данных по биологически
значимому углевод—углеводному взаимодействию. Сейчас ситуация диамет-
рально противоположна. Открыты закономерности межклеточной адгезии,
опосредуемой или инициируемой углевод-углеводным узнаванием, причем
последнее является быстрым первичным процессом, предваряющим белок-
белковую адгезию. Установлены особенности контактов мембранного аппара-
та, где высока плотность встроенных гликолипидов, получены поразительные
данные о функциональной роли и регуляционных потенциях последних.
Организменный уровень регуляции. Главный механизм регуляции обмена
веществ на уровне организма—гормональный (см. гл. XII). Осуществляясь
гуморальным путем у животных и через системы проводящих путей у расте-
ний, он, в свою очередь, направляется сигналами нервной системы у первых
и внешней среды—у вторых. Таким эбразом, здесь осуществляется естествен-
ный и логический переход из области биохимии в сферу физиологии.
Популяционный уровень регуляции. Так же, как и предыдущий, этот уровень
регуляции лежит на грани биохимии и физиологии, постепенно перерастая
в новую науку—химическую экологию. Поэтому сейчас логичнее говорить об
уровне регуляции метаболизма в экосистемах, имея в виду глобальные аспек-
ты химических взаимодействий в живой природе. Суть его сводится к мощ-
ному влиянию химических соединений, вырабатываемых и выделяемых одни-
ми особями, на обмен веществ и поведенческие реакции других особей. Оно
реализуется через рецепторные системы или ткани-мишени организма реципи-
ента. Выше (см. гл. IV) приведены соответствующие примеры, касающиеся
антибиотиков и телергонов. Однако перечень веществ, участвующих в хими-
ческих внутри- и межвидовых взаимодействиях особей, гораздо более широк
и непрерывно возрастает. Среди них: фитонциды—антибактериальные веще-
ства, вырабатываемые здоровыми растениями (важную роль в их исследова-
нии сыграли работы Б. П. Токина и его учеников); фитоалексины—защитные
соединения, образующиеся в ра тениях в ответ на бактериальное или грибко-
вое заражение; новые виды антибиотиков, фитогормонов, нейрогормонов
и т. п. Их всестороннее изучение, глубокое раскрытие сути и механизмов
существующих в природе биохимическ х связей крайне существенно для раз-
работки экологической стратегии, столь необходимой человечеству в наше
время.
16—3502
ГЛАВА XIV
КВАНТОВО-МЕХАНИЧЕСКИЕ АСПЕКТЫ БИОХИМИИ
При рассмотрении строения и свойств ряда соединений, а также [екоторых
биохимических процессов в предыдущих главах сделана попытка привлечь
квантово-механические представления к объяснению ряда явлений и закономе-
рное ей. В частности, такой подход использован при описании пептидной
связи (зависимость ее свойств от делокализации и сопряжения электронов)
и вторичной структуры белка (вклад л-электронов в поддержание «-спираль-
ной конформации) (см. гл. II), механизма действия пиридоксалевых фермен-
тов (смещение электронной плотности в фермент-субстратном комплексе—
см. гл. III), природы цис-транс-изомерных превращений ретиналя (зависи-
мость этого явления от значений порядка связей в сопряженной системе),
структуры и свойств тиаминпирофосфата (причина повышенной электронной
плотности у 2-го углеродного атома тиазольного цикла) и повышенной реак-
ционной способности изоаллоксазина в 1-м и 10-м положениях (у них мак-
симальны индексы свободных валентностей) (см. гл. IV), при обсуждении
вопроса о сущности жизни, при изучении природы макроэргических связей
(неустойчивость системы сопряжения электронов) (см. гл. V), структуры
и свойств пиримидиновых и пуриновых оснований (зависимость между поряд-
ком связи и реакциями присоединения), стэкингвзаимодействий в молекулах
ДНК (их изменение при контактах молекул воды с протон-донорными и про-
тон-акцепторными центрами азотистых оснований) (см. гл. VI), механизма
активирования молекулярного кислорода в процессе биологического окисле-
ния (см. гл. X) и некоторых других случаях.
Как можно видеть из этого перечисления, в данном руководстве использова-
ны лишь некоторые простейшие понятия квантовой биохимии, оформившейся
как самостоятельное научное направление в начале 60-х годов нашего столетия.
Оно возникло на стыке квантовой химии и молекулярной биологии, а в более
широком плане—на границе физики и биологии, перерастая постепенно в увле-
кательную и перспективную новую науку—физико-химическую биологию.
Логика и смысл возникновения и развития квантовой биохимии состоят
в том, что от простейших представлений об элементарном составе, порядке
расположения и пространственной локализации атомов в молекулах органиче-
ских соеди ений (имеющих, естественно, биологическое значение) она обеспе-
чивает переход посредством квантово-механических расчетов к данным о рас-
пределении в них электронной плотности и подвижности электронного облака,
а также к энергетическим характеристикам, говорящим об устойчивости моле-
кул и их способности отдавать и принимать электроны, т. е. в конечном счете
об их реакционной способности, связи структуры и биологической функции.
Квантово-механические расчеты дают достаточно отчетливые представле-
ния об энергетических индексах молекул (энергия делокализации электронов,
энергия высшей заполненной молекулярной орбитали, энергия низшей свобод-
ной молекулярной орбитали, энергия возбуждения), равно как и о структур-
482
ных индексах молекул (электронные заряды атомов, порядок связи, индекс
свободной валентности).
Знание уровней энергии делокализации электронов (разность между на-
блюдаемой энергией молекулы и энергией, рассчитанной на основании кано-
нической структурной формулы) необходимо для более глубокого понимания
механизма действия коферментов и других биологически активных соедине-
ний, обладающих системой сопряженных двойных связей; значения энергии
делокализации электронов важны, кроме того, для оценки устойчивости водо-
родных связей между комплемент рными пуриновыми и пиримидиновыми
основаниями, а также существенны при рассмотрении вопроса об установле-
нии равновесия в разнообразных таутомерных превращениях моносахаридов,
кетокислот, азотистых оснований и т. п. Данные об энергии высшей заполнен-
ной и низшей незаполненной орбиталей прямо взаимосвязаны с электрон-
донорными и электрон-акцепторными свойствами молекул соответственно
и используются для характеристики оксидоредуктазных систем клетки. Сведе-
ния об энергии возбуждения, т. е. разности энергии электрона до и после
перехода на новую орбиту, неоднократно обсуждались в связи с вопросом
о полупроводниковых свойствах белков и нуклеиновых кислот.
Что касается структурных индексов молекул, то они наиболее часто учиты-
ваются при рассмотрении связи структуры и функции органических соедине-
ний. Электронные заряды атомов, характеризующие вероятность пребывания
электрона вблизи данного атома на всех присущих ему и заполненных ор-
би алях, специфически распределяются, например, у легко гидролизуемых
субстратов, в том числе макроэргических (см. данные об электрондефицитных
связях в молекулах АТФ—гл. V). Порядок связи, характеризующий относи-
тельную близость рассматриваемой связи к стандартной простой или двой-
ной, помогает понять реакционную способность определенных участков моле-
кулы не только, например, в пиримидинах и пуринах, но и в более сложных
соединениях (классическим примером здесь является выявление высокореакци-
онной так называемой К-области в молекулах канцерогенных ароматических
углеводов). Индексы свободной валентности атомов (как показатель остаточ-
ной, нереализованной валентности у данного атома) указывают на наиболее
реакционно-способные центры в молекуле, по которым может происходить
присоединение свободных радикалов, атомов водорода (см. структуру изоал-
локсазинового кольца в молекуле витамина В2).
Рассмотрение современного состояния квантово-механических подходов
к анализу биохимических процессов показывают, что наиболее плодотворные
результаты получены при толковании механизмов ряда ферментативных реакций
(аминотрансферазная, оксидоредуктазная, лиазная и др.), природы макроэргиче-
ских связей, канцерогенности ароматических углеводов, особенностей структуры
и функции нуклеиновых кислот, связи структуры и фармакологического эффекта
молекул. Так, например, при квантово-химических расчетах энергий электронных
переходов для различных ионных форм пиридоксаль-5-фосфата, пиридокса-
мин-5-фосфата и оксимпиридоксаль-5-фосфата, образующихся в процессе амино-
трансферазной реакции, получены хорошие совпадения с таковыми, выведенны-
ми на основании спектроскопических данных что способствовало выяснению
природы промежуточных кофермент-субстратных комплексов в этой реакции.
Исходя из гипотезы, что треугольник из атомов N—О—О в составе обладаю-
щих противоопухолевым действием производных антрахинона ответствен за
названную биологическую активность, синтезирован эффективный новый канце-
ростатический препарат: 1,4-дигидрокси-5,8-бис-2-(гидроксиэтил) аминоэтила-
ми но-9,10-антрацендион. Это еще раз подчеркнуло плодотворность примене-
ния методов квантовой биохимии для понимания механизма биологической
483
активности канцерогенных и антиканцерогенных веществ, начало чему было
положено в классической работе А. Пюльмана и Б. Пюльмана, касающейся
выявления способности канцерогенных углеводородов взаимодействовать
с клеткой через К-область:
В какой-то мере, данные квантовой биохимии оказались полезными для
объяснения свойств белков и пептидов, а также порфириновых соединений.
Вместе с тем необходимо реально представлять себе, что квантовая биохи-
мия находится в самом начале своего пути. Существуют немалые трудности
при квантово-химических расчетах электронной структуры не очень сложных
молекул, не говоря уже о полных молекулах ДНК, РНК и белков—важней-
ших природных биополимеров. Ее расчеты справедливы, по-видимому, лишь
для фиксированных молекулярных структур, но в них не учитываются еще
множественные конформационные состояния макромолекул, неизбежно от-
ражающихся на распределении электронных плотностей. Нерешенным остает-
ся вопрос о распределении зарядов в исследуемом соеди ении в зависимости
от характера растворителя и взаимодействия его молекул с атомными группи-
ровками изучаемого вещества, иначе говоря—вопрос о корректности элек-
тронных характеристик для вещества в той биологической жидкости, где,
собственно, и происходят биохимические реакции.
Еще более трудную, но, конечно, центральную задачу квантовой биохимии
представляет изучение межмолекулярных взаимодействий, оценка и сопоставле-
ние значений энергии в возникающих комплексах, где метод молекулярных
орбиталей явно недостаточен из-за невозможности учитывать электронную
корреляцию. Поэтому сейчас возлагают надежды на вантово-статистические
подходы, где, по существу, уже рассматривается статистика молекул, а не
электронов. Вместе с тем методом ЛКАО (линейной комбинации атомных
орбиталей) получены вполне удовлетворительные результаты в «квантовой
фармакологии» (например, предсказаны новые нейтролептики). И наконец, все
построения современной квантовой биохимии осуществляются без учета того,
что взаимодействия молекул в живых системах идут на более высоком,
качественно новом уровне их организации. Таковы, например, пиктографический
способ распознавания молекул в процессе их рецепции (узнавание образа, а не
конкретных атомных или электронных структур), а также принципы, закладывае-
мые в новое научное направление—топобиологию, т. е. учение о биологических
взаимодействиях, определяемых пространственным расположением биологиче-
ских структур, дающим ключ к пониманию сложнейших проблем морфогенеза.
Сюда же следует отнести наметившиеся тенденции рассматривать простран-
ственную организацию некоторых биологических структур, например, белкового
цитоскелета, как компьютерную систему, осуществляющую непрерывную пере-
работку информации и выдачу необходимых команд для обеспечения нормаль-
ной жизнедеятельности клетки. В связи с этим вряд ли оправдан основной
методологический принцип квантовой биохимии—свести явления жизни к «са-
мореализации потенциальных возможностей электронных состояний атомов».
484
РЕКОМЕНДУЕМАЯ ЛИТЕРАТУРА
ОБЩИЕ РУКОВОДСТВА'
Альберте Б., Брей Д., Льюис Дж., Рефф М., Робертс К., Уотсон Дж. Молекулярная биология
клетки—М.: «Мир», 1994. Т. 1—3.
Биополимеры / Под ред. Ю. Иманиси.—М.: Мир, 1988. 544 с.
Бохински Р. Современные воззрения в биохимии.—М.: Мир, 1987. 543 с.
Гильберт С. Биология развития.—М.: Мир, 1991. Т. 1—3.
Досон Р., Эллиот Д., Эллиот У., Джонс К. Справочник биохимика.—М.: Мир, 1991. 543 с.
Ленинджер А. Биохимия.—М.: Мир, 1985. Т. 1—3.
Марри Р., Греннер Д., Мейс П., Родуэлл В. Биохимия человека.—М.: Мир, 1993. Т. 1 и 2.
Молекулярная биология клетки/Пер. с англ. Под ред. Г. П. 1еоргиева.—М.: Мир, 1986.
Т. 1—2.
Овчинников Ю. А. Биоорганическая химия.—М.: Просвещение, 1987. 816 с.
Перспективы биохимических исследований/Под ред. Дж. Туза и С. Пренсиса.—М.: Мир,
1987. 188 с.
Рис Э., Стенберг М. От клеток к атомам.—М.: Мир, 1988. 142 с.
Страйер Л. Биохимия.—М.: Мир, 1984. Т. 1—3.
Campbell Р. N., Smith A. D. Biochemistry illustrated. Second edition.—Churchill Livingstone Inc.
Edinburg, London, Melbourn, New 'Ybrk, 1988. 288 p.
Zubay G. Biochemistry. Addison-Wesley publiching company, 1983. 1268 p.
Methews Ch. K., Van Holde К. E. Biochemistry. The Benjamin / Cummings publishing company, Inc,
1990. 1129 р.
ВВЕДЕНИЕ
Биотехнология /Под ред. И. Хиггинса, Д. Беста, Дж. Джонса.—М.: Мир, 1988. 479 с.
Бланко М. А. Меченые атомы в биохимии.—М.: Наука, 1988. 117 с.
Браунштейн А. Е. На стыке химии и биологии/Под ред. А. А. Баева.—М.: Наука,
1987. 239 с.
Воспоминания о В. А. Энгельгардте / Под ред. А. А. Баева.—М.: Наука, 1989. 336 с.
Гинтер В. К. Медицинская генетика: достижения и перспективы//Биология в школе, 1993.
№ 5. С. 8—12.
Блинов Е. П. Химическая микробиология.— М.: Высшая школа, 1989. 448 с.
Конарев В. Г. Биохимия, молекулярная биология и проблемы растениеводства//Сельскохо-
зяйственная биология, 1987. № 11. С. 18—24.
Молекулярные и клеточные аспекты биотехнологии / Под ред. С. Г. Инге-Вечтомова.—Л.:
Наука, Ленинградское отд., 1986. 256 с.
Перспективы биоорганической химии и молекулярной биологии / Под ред. Ю. А. Овчиннико-
ва.—М.: Наука, 1986. 365 с.
Сассон А. Биотехнология: свершения надежды.—М.: Мир, 1987. 411 с.
Шамин А. Н. История биологической химии. Формирование биохимии.—М.: Наука, 1991.
Глава I. ХИМИЧЕСКИЙ СОСТАВ ОРГАНИЗМОВ
Биогеохимический круговорот веществ в биосфере/Под ред. В. А. Ковда.—М.: Наука,
1987. 142 с.
Вадковская ГГ. К., Лукашов К. И. Химические элементы и жизнь в биосфере.—Минск:
Вышейш. школа, 1981. 175 с.
Вернадский В. И. Биосфера и ионосфера.—М.: Наука, 1989. 262 с.
485
Дьяков В. М. Кремний в жизни и науке.—М.: Знани 1989. 32 с.
Минеев В. Г. Агрохимия и биосфера.—М.: Колос, 1984. 248 с.
Моисеев Н. Как приблизиться к ноосфере//Химия и жизнь, 1989. № 6. С. 5—9; № 7.
С. 28—33; № 8. С. 10—16.
Ноздрюхина Л. Р. Биологическая роль микроэлементов в организме животных и человека.—
М.: Наука, 1977. 184 с.
Осидзе Д. Ф. Значение микоплазм в вирусологии и их роль в патологии животных.—М.:
ВНИИТЭИСХ, 1970. 75 с.
Пири Н. Химическое многообразие и проблема происхождения жизни//Сб.: Возникновение
жизни на Земле.—М.: Изд-во АН СССР, 1956. С. 79—87.
Уолд Д. Почему живое вещество баэиру тся на элементах II и III периодов периодической
системы//Сб.: 1оризонты биохимии. М.: Мир, 1964. С. 102—113.
Чернов В. М., Чернова О. А. Последовательности, гомологичные сайтам узнавания эукари-
отической топоизомеразы I в геномах микоплазм//Молекулярная биология, 1996. Т. 30. В. 2.
С. 286—292.
Глава II. БЕЛКИ
Абелев Г. Н. Основы иммунитета // Соросовский образовательный журнал, 1996. № 5. С. 4—
10.
Барковский Е. В., Бандарин В. А. Предсказание структурных участков глобулярных белков по
их первичной структуре//Б эорганиче ая химия, 1979. Т. 5 № 1. С. 24—34.
Борисов В. В. Каждый белок—свой сюже //Химия и жизш 1990. № 2. С. 41—51.
Васильев Ю. М. Клетка ка: архитектурное чудо. Часть I. Живые нити. Часть 2. Цитоскелет,
способный чувствовать и помнить//Соросовский образовательный журнал, 1996. № 2. С. 36—43;
№4. С. 4—10.
Долгих Д. А., Кирпичников М. П., Птицин О. Б., Чемерис В. В. Белковая инженерия искусствен-
ных белков//Молекулярная биолог я, 1996. Т. 30. В. 2. С. 261—271.
Иванюшина В. А., Морнин А. Б., Киселев О. ГЕ Молекулярные шапероны: новые белки—новые
функции//Молекулярная биология, 1991. Т. 25. В. 4. С. 869—880.
Курочкина Л. П., Месянжинов В. В. Фолдинг белка в клетке//Успехи биологической химии,
1996. Т. XXXVI. С. 49—86.
Лопатин С. А., Варламов В. П. Новые тенденции в металлохелат аффинной хроматографии
белков//Биоор хкаг <имия, 1995. Т. 31. № 3. С. 259—266.
Меклер Л. Б., Идлис Р. Г. Общий стереохимический генетический код—путь к биотехнологии
и универсальной медицине XXI века уже сегодня // Природа, 1993. № 5. С. 29—69.
Наградова Н. К. Внутриклеточная регуляция формирования нативной пространственной
структуры белков//Соросовский образовательный журнал, 1996. № 7. С. 11—18.
Поглазов Б. Ф., Бурнашова С. А. емышечные двигательные системы//Биологическая химия
(Итоги науки и техники ВИНИТИ АН СССР), 1989. Т. 29. 176 с.
Практическая химия белка /Под ред. А. Дарбре.—М.: Мир, 1989. 621 с.
Скоупс Р. Методы очистки белков.—М.: Мир, 1985. 358 с.
Степанов В. М. Молекулярная биология. Структура и функция белков.—М.: Высшая школа,
1996. 335 с.
Чипенс Г. И. Мутации в структурах гомологичных белков подтверждают существование кода
взаимодействия аминокислот//Биоорганическая химия, 1991. Т. 17. № 9. С. 1284—1287.
Шульц Г., Ширмер Р. Принципы структурной организации белков.—М.: Мир, 1982. 354 с.
Якубке X Ешкаите X. Аминокислоты пептиды, белки.— М.: Мир, 1985. 455 с.
Koenig М„ Monaco A., Kunkel L. The complete sequence of dystrophin predicts a rod-shaped
cytoskeletal protein // Cell, 1988. V. 53. P. 219—228.
Sanger F. Sequences, sequences and Sequences//Annual review of biochemistry, 1988. V. 57.
P. 1—28.
Глава III. ФЕРМЕНТЫ
Андреева H. С. ЗачЬм и почему пепсин стабилен и активен при pH 2? // Молекулярная биоло-
гия, 1994. Т. 28. В. 4. С. 1400—1404.
Бекман Э. М„ Григорьев М. Ю. Рибозимы: РНК—РНК-взаимодействия и эндонуклеотическое
расщепление//Успехи биологической химии, 1988. Т. XXIX. С. 113—121.
Березин И. В. Исследования в области ферментативного катализа и инженерной энзимоло-
гии.—М.: Наука, 1990. 382 с.
Березов Т. Т. Применение ферментов в медицине//Соросовский образовательный журнал,
1996. № 3. С. 23—27.
Болдырев А. А. Регуляция активности мембранных ферментов//Соросовский образователь-
ный журнал, 1997. № 6. С. 21—27.
486
Валуева Т. А., Мосолов В. В- Белки—ингибиторы ротеолитических фер ентов у растений//
Пр кладнав биохимия и микробиология, 1995. Т. 31. В. 6. С. 579—589.
Диксон М.. Уэбб Э. Ферменты.—М.: Мир, 1982. Т. 1—3.
Иммобилизованные клетки и ферменты. Методы/Под ред. Дж. Вудворда.—М.: Мир,
1988. 215 с.
Квеситадзе Г. И. Ферменты микроорганизмов, живущих в экстремальных условиях.—М.:
Наука, 1990. 52 с.
Наградова Н. К. Белок-белковые взаимодействия в функционировании НАД-зависимых дегид-
рогеназ. 44-е Баховское чтение.—М.: Наука, 1990. 55 с.
Прист Ф. Внеклеточные фермента микроорганизмов.—М.: Мир, 1987. 117 с.
Ренненберг Р. Элексиры жизни.—М.: Мир, 1987. 151 с.
Сорочинский В. В.. Курганов Б. И. Ферментные электроды//Биотехнология (Итоги науки
и техники ВИНИТИ АН СССР), 1988. Т. 13. 207 с.
Филиппович Ю. Б., Коничев А. С. Множественные формы ферментов насекомых и проблемы
сельскохозяйственной энтомологии,—М.: Наука, 1987. 166 с.
Фридрих П. Ферменты: четвертичная структура и надмолекулярные комплексы.—М.: Мир,
1986. 374 с.
Чек Т. Кто бы мог подумать, что РНК способна работать ферментом//Химия и жизнь 1988.
№ 12. С. 31—33.
Чернов И. Н. Ферменты в клетке и пробир е//Соросовский образовательный журнал, 1996.
№ 5. С. 28—34.
Enzyme Handbook / Eds. D. Schomburg, M. Salzmann, D. Stephan. Springer-Verlag: Berlin, Heidel-
berg, New York, London, Paris, Tokyo, Hong Kong, Barcelona. 1990— 995. Vol. 1—10.
Srere P. A. Complexes of sequential metabolic enzynes//Annual review of biochemistry, 1987.
V. 56. P. 89—124.
Глава IV. КОФЕРМЕНТЫ. ВИТАМИНЫ И НЕКОТОРЫЕ ДРУГИЕ
БИОАКТИВНЫЕ СОЕДИНЕНИЯ
Алекперов У. К. Антимутагены и охрана генофонда.—М.: Знание, 1989. 26 с.
Букин Ю. В., Плихтях И. Л., Драудин-Крыленко В. А. и др. Аскорбиген и его произг шые
депо-формы аскорбиновой кислоты// Б ооргаиическая химия. 1987. Т. 13. № 4. С. 539—545.
Витамины / Под ред. М. И. Смирнова. -М.: Медицина, 1974. 496 с.
Витамин U (S-метилметионин). Природа, свойства, применение//Сб. под ред. В. Н. Букина
и В Е. Анисимова.— М.: Наука, 1973. 152 с.
Дмитровский А. А. Метаболизм Р-каротина и витамина А и биохимические спекты и приме-
нения // Автореф. дисс. д. б. н., М., 1993. 68 с.
Душейко А. А. Витамин А. Обмен и функции: Киев: Наукова думка, 1989 288 с.
Карнаухов В. Н. Биологические функции каротиноидов.—М.: Наука, 1988. 239 с.
Киршенблат Я. Б. Телергоны—химические средства взаимодействия животных.—М.: Наука,
1978. 142 с.
Натане Д. 1ён1 цветного зрения//В мире науки, 1989. № 4. С. 16—25.
Познанская А. А., Корсова Т. Л. Аденозилкобаламинзависимые ферменты//Успехи биологи-
еской химии, 1988. Т. XXVIII. С. 66—101.
Поплинская В. А. Цитоструктура и орфогенез наружных сегментов палочек//Онто енез,
1295. Т. 26. № 1. С. 1—21.
Стайер Л. Молекулы зрительного возбуждения // В мире науки, 1987. № 9. С. 16—25.
Суслова В. А. и др. Организация гена р-субъединицы фоторецепторной ФДЭ цГМФ челове-
ка//Биоорган ческая химия, 1996. Т. 22. № 4. С 256—263.
Труды международной конференции по ретиналь-содержащим белкам / Под ред. Ю. А. Овчин-
никова.—М.: Наука, 1989. 325 с
Филиппов В. В. Функции и синтез биотина в живом организме.—М.: Наука, 1985. 216 с.
Юркевич А. М., Рудакова И. П. Структура, свойства и механизм действия кобаламиновых
коферментов//Биоорганическая химия (Итоги науки и техник ВИНИТИ АН СССР), 1985.
Т. 5. 196 с.
Глава V. ОБЩЕЕ ПОНЯТИЕ ОБ ОБМЕНЕ ВЕЩЕСТВ
И ЭНЕРГИИ В ОРГАНИЗМЕ
Варшавски! И. А. М ханизм онто- и герои генеза//С огенез, 1995. Т. 26. № 6. С. 481—488.
Заварзин Г. А, Глобальные аспекты микробиологии//И вестия АН СССР. Сер. биол., 1981.
№ 6. С. 824—834.
Крисаченко В. С. Памяти В, И. Вернадского//Успехи современной биологии,-1984. Т. 98. В. 2.
С. 312—319.
487
Мирзоян Э. Н. Теория живой материи В. И. Вернадского//Журнал общей биологии, 1994.
Т. 55. № 1. С. 13—28.
Михайловский Г. Е. Контрапункт биологической термодинамики // Химия и жизнь. 1979. № 2.
С. 19—25.
Нусинов М. Д., Марон В. И. Эволюция вещества во Вселенной и происхождение жизни на
Земле. Природа.—1991. № 4. С. 26—31.
Шайтан К. В. Конфор анионная динамика и новые подходы к физическим механизмам
элементарных актов переноса массы, трансформации энергии и передачи информации в биомик-
ромолекулярных структурах//Молекулярная биология, 1994. Т. 28. В. 3. С. 670—676.
Шварц С. С. Эволюция биосферы и экологическое прогнозирование // Вестник АН СССР,
1976. № 2. С. 61—73.
Энгельгардт В. А. К новым рубежам в познании основ явлений жизни//Вестник АН СССР,
1976. № 2. С. 73—86.
Глава VI. НУКЛЕИНОВЫЕ КИСЛОТЫ И ИХ ОБМЕН
Агол В. И., Богданов А. А. Структура и биосинтез нуклеиновых кислот.—М.: Высшая
школа, 1989.
Балыкин С. Г. Динамика структуры хроматина//Молекулярная биология (Итоги науки и тех-
ники ВИНИТИ АН СССР). М„ 1988. Т. 26. С. 3—121.
Баев А. А. Геном человека. Общий взгляд.—М.: ВИНИТИ, 1989. 45 с.
Богданов А. А., Лаврик И. Н., Докудовская И. С., Донцова О. А. Структура и функция
5SpPHK в рибосоме//Молекулярная биология, 1995. Т. 29. В. 6. С. 1218—1227.
Гвоздев В. А. Механизмы регуляции активности генов в процессе транскрипции // Соросовский
образовательный журнал, 1996. № 1. С. 23—31.
Георгиев Г. П. Гены высших организмов и их экспрессия.—М.: Наука, 1989. 254 с.
Глазков М. В. Петельно-доменная организация генов в эукариотических хромосомах // Моле-
кулярная биология, 1995. Т. 29. В. 5. С. 965—992.
Женодарова С. М. Нуклеозимы и минизимы//Молекулярная биология, 1994. Т. 28. В. 3.
С. 506—510.
Збарский И. Б. Организация клеточного ядра.— М.: Медицина, 1988. 368 с.
Зенгер В. Принципы структурной организации нуклеиновых кислот.—М.: Мир, 1987. 584 с.
Иванов В. А., Ильин Ю. В. Обратные транскриптазы и их биологическая роль // Молекулярная
биология, 1995. Т. 29. В. 2. С. 258—280.
Иванов В. И., Минченкова Л. Е. A-форма ДНК: в поисках биологической роли//Молекуляр-
ная биология, 1994. Т. 28. В. 6. С. 1258—1271.
Итоговая конференция «Теном человека-94» (Черноголовка, 9—11 марта 1994 г.)//Генетика,
1995. Т. 31. № 5. С. 733—736.
Кнорре Д. Г. Биохимия нуклеиновых кислот//Соросовский образовательный журнал, 1996.
№ 3. С. 11—16.
Копылов А. М. Еще один шаг к миру РНК//Биохимия, 1995. Т. 60. В. 1. С. 159—161.
Копылов А. М. Кто мы? Откуда мы? Куда идем? (Хроника конференции в г. Дублине, Ирлан-
дия)//Биохимия, 1995. Т. 60. В. 6. С. 987—989.
Краевский А. Л., Куханова М. К. Репликация ДНК у эукариот // Молекулярная биология (Ито-
ги науки и техники ВИНИТИ АН СССР), 1986. Т. 22. С. 5—132.
Лещинская И. Б. Генетическая инженерия//Соросовский образовательный журнал, 1996. № 1.
С. 32—39.
Льюин Б. Гены.—М.: Мир, 1987. 544 с.
Мазин А. Л. Энзиматическое метилирование ДНК как механизм старения//Молекулярная
биология, 1994. Т. 28. В. 1. С. 21—51.
Молекулярная биология. Структура и биосинтез нуклеиновых кислот / Под ред. А. С. Спири-
на.—М.: Высшая школа, 1990. 352 с.
Никифоров В. Г. РНК-полимераза бактерий: сравнительные исследования//Успехи микро-
биологии, 1987. Т. 21. С. 105—150.
Пташне М. Переключение генов: регуляция генной активности и фаг X.—М.: Мир, 1988. 158 с.
Радмар М., Вагнер Р. Высокая точность репликации ДНК//В мире науки, 1988. № 10.
С. 16—23.
Рысков А. П., Гордон И. О. Геномная дактилоскопия//Биология в школе, 1993. № 5. С. 13—19.
Свердлов Е. Д. Новости в исследованиях генов и геномов//Биоорганическая химия, 1996.
Т. 22. № 4. С. 316—320.
Стейн Д. А. Большая роль малых РНП // В мире науки, 1988. № 8. С. 20—27.
Турпаев К. Т., Васецкий Е. С. Ядерные белковые факторы, взаимодействующие со специфиче-
скими последовательностями ДНК//Генетика, 1990. Т. 26. № 5. С. 804—816.
Фаворова О. О. Сохранение ДНК в ряду поколений: репликация ДНК // Соросовский об-
разовательный журнал, 1996. № 4. С. 11—17.
488
Флорентьев В. А. Конформационные возможности ДНК//Биоорганическая химия, 1982. Т. 8.
№ 7. С. 885—899.
Франк-Каменецкий М. Д. Самая главная молекула: (о молекуле ДНК).—М.: Наука,
1988. 176 с.
Щелкунов С. Н. Вирус натуральной оспы (ВНО)—источник новых медицинских препара-
тов//Соросовский образовательный журнал, 1995. № 1. С. 28—31.
Хейме Б., Хиггинс С. Транскрипция и трансляция.—М.: Мир, 1987. 259 с.
Adams R., Knowler J., Leader D. The biochemistry of nucleic acids. Tenth edition.—L.,N.—Y.:
Chapman and Hall, 1986. 526 p.
Fraser С. M. et al. (38 persons). Genomic Sequence of a Lyme disease spirochaete, Borrelia
•burgdorferi B31 //Nature, 1997. Vol. 390. N 6660. P. 580—586.
Kunst F. et al. (150 persons). The complete genome sequence of the Gram-positive bacterium Bacillus
Subtilis//Nature, 1997. Vol. 390. N 6657. P. 249—256.
West S. C. DNA helicases get physical//Nature, 1996. Vol. 384. № 6607. P. 316—317.
Глава VII. ОБМЕН БЕЛКОВ
Бродский В. Я., Нечаева Н. В. Ритм синтеза белка.— М.: Наука, 1988. 239 с.
Высоцкая В. С., Гарбер М. Б. Регуляция экспрессии генов рибосомных белков Escherichia crei //
Успехи биологической химии, 1995. Т. XXXV. С. 67—95.
Зайцева Г. Н„ Энтелис Н. С. Особенности структуры транспортных РНК и генетического
кода митохондрий//Успехи биологической химии, 1989. Т. XXX. С. 106—129.
Инге-Вечтомов С. Г. Цитогены и прионы: цитоплазматическая наследственность без ДНК? //
Соросовский образовательный журнал, 1996. № 5. С. 11—18.
Киселев Л. Л., Вольфсон А. Д. Аминоацил-тРНК-синтетазы высших эукариот//Успехи биоло-
гической химии, 1995. Т. XXXV, С. 3—65.
Киселев Л. Л., Фролова Л. Ю. Терминация трансляции у эукариот//Молекулярная биология,
1995. Т. 29. В. 6. С. 1286—1293.
Кордюм В. А. Самовоспроизводящиеся белки, некоторые следствия и прогнозы. Биополиме-
ры и клетки, 1990. Т. 6. № 4. С. 5—20.
Лаврик О. Н. Механизмы специфического отбора аминокислот в биосинтезе белка // Соросов-
ский образовательный журнал, 1996. № 4. С. 18—23.
Лим В. И., Спирин А. С. Стереохимический анализ транспептидации, транслокации и сво-
рачивания растущего пептида на рибосоме//Успехи биологической химии, 1988. Т. XXVIII.
С. 3—19.
Минченко А. Г. Загадки митохондриального белкового синтеза. Биохимия, 1989 Т. 54. В. 12.
С 1071—2075.
Мосолов В. В. Механизмы контроля протеолиза//Успехи биологической химии, 1988.
Т. XXVIII. С. 125—144.
Мосолов В. В., Валуева Т. А. Растительные белковые ингибиторы протеолитических фермен-
тов.—М.: Институт биохимии им. А. Н. Баха РАН, 1993. 207 с.
Рязанов А. Г., Спирин А. С. Компартментализация биохимических процессов на полирибо-
сомах и других субклеточных структурах//Успехи биологической химии, 1988. Т. XXIX.
№ 3—43.
Спирин А. С. Молекулярная биология. Структура рибосомы и биосинтез белка.—М.: Высшая
школа, 1986. 303 с.
Спирин А. С. Регуляция трансляции мРНК-связывающими факторами у высших эукариот//
Успехи биологической химии, 1996. Т. XXXVI. С. 3—48.
Спирин А. С. Рибосомальная транслокация: факты и модели // Успехи биологической химии,
1986. Т. XXVII. С. 3—29.
Спирин А. С. Энергетика и динамика белоксинтезирующего аппарата//Успехи биологической
химии, 1989. Т. XXX. С. 3—24.
Спирин А. С., Четверил А. Б., Воронин Л. А. и др. Биосинтез белка и перспективы бесклеточ-
ной биотехнологии//Природа, 1991. № 5. С. 10—19.
DNA makes RNA makes protein / Ed. by T. Hunt, S. Prentis, J. Tooze.— Elsevier biomedical press.:
Amsterdam—New Y>rk—Oxford, 1983. 284 p.
Глава VIII. УГЛЕВОДЫ И ИХ ОБМЕН
Бовин Н. В. Углевод-углеводное взаимодействие//Биохимия, 1996. Т. 61. В. 6. С. 968—983.
Горбач 3. В. Пентозофосфатный путь превращения углеводов: структура, функции, регуляция /
/Успехи современной биологии, 1988. Т. 105. В. 1. С. 35—46.
Дайзенхофер И., Михель X. Фотосинтетический реакционный центр пурпурной бактерии.—
М.: Знание, 1990. № 2. С. 3—24.
489
Ермолаев J1. П. Регуляция глюконеогенеза онтогенезе.—М.: Наука, 1987. 168 с.
Игнатов В. В. Углеводоузнающие белки-лектины //Соросовский образовательный журнал,
1997. № 2. С. 14—20.
Клесов А. А. Биохимия и энзимология гидролиза целлюлозы // Биохимия. 1990. Т. 55. Выл. 10.
С. 1731—1765.
Кулаева О. И. Хлоропласт и его полуавтономность в клетке//Соросовский образо> ательный
журнал, 1997. № 7. С. 2—9.
К рганов Б. И., Любарев А. Е. Гипотетическая структура комплекса ферментов гликолиза //
Молекулярная биология, 1988. Т. 22. № 6. С. 1605—1613.
Лахтин В. М. Молекулярная организация ле тинов//Молекулярная биология, 1994. Т. 28.
В. 2. С. 245—273.
Ливанова Н. Б. Особенности четве тичной структур! а регуляторных свойств изозим кина-
зы фосфорилазы//Успехи биологической химии, 1988. Т. XXVIII. С. 44—65.
Линхард Г. Э., Слот Я. У., Джеймс Д. Э., Мьюклер А. Как клетки поглощают глюкозу // В
мире науки, 1992. № 3. С. 22—28.
Любарев А. Е., Курганов Б. И. Гипотетическая структура комплекса ферментов цикла трикар-
боновых кислот//Молекулярная биология, 1987. Т. 21. № 5. С. 1286—1296.
Себякин Ю. Л., Евстигнеева В. П. Полиизопренолфосфосахара в биосинтезе углеводсодер-
жащих биополимеров//Известия АН СССР. Сер. биол., 1985. № 2. С. 485—498.
Скулачев В. П. Биологический преобразователь энергии: тертеж с точностью до атома //
Химия и жизнь, 1989. № 3. С. 35—39.
Степаненко Б. Н. Химия и биохимия углеводов (полисахариды).—М.: Высшая школа,
1978. 256 с.
Степаненко Б. Н. Современны! тро лемы биохимии углеводов.—М.: Наука, 1979. 54 с.
Тиунова Н. А. Хитинолитические ферменты микроорганизмов //Успехи биологической химии,
1989. Т. XXX. С. 199—219.
Трачук Л. А. Хитиназы Bacillus cereus: выделение и характеристика//Биохимия, 1996. Т. 61.
В. 2. С. 357—368.
Хемосинтез /Под ред. М. В. Иванова.—М.: Наука, 1989. 256 с.
Шарон Н., Лис X. Углеводы в клеточном узнавании//В мире науки, 1993. №2/3.
С. 104—ИЗ.
Глава IX. ЛИПИДЫ И ИХ ОБМЕН
лес нко А. В. Функциональная роль Липидов в экспресса клеточных онкогенов//Успехи
биологической химии, 1993. Т. XXXIV. С. 85—102.
Бергельсон Л. Д. Биологические мембраны.—М.: Наука, 1975. 183 с.
Брокерхоф X., Дженсен Р. Липолитически< ферменты.—М.: Мир, 1978. 395 с.
Бурлакова Е. Б. Роль липидов в процессе передачи информации в клетке / Биохимия липидов
и их роль в обмене веществ. М., 1981. С. 23—34.
Дембицкий В. М. Алкоксилипиды—реликты органического мира: образование, биотрансфор-
мация, эволюция//Биологические науки, 1991. № 2. С. 5—25.
Дятловицкая Э. В. Сфинголипиды и злокачественный рост//Биохимия, 1995. Т. 60. В. 6.
С. 843—848.
Климова Т. А. З-гидрокси-З-метилглутарил-СоА-редуктаза в тканях животного организма//
Успехи биологическо! химии, 1989. Т. XXX. С. 181—198.
Корнелли Т. В. Липиды микробактерий и родственных микроорганизмов.—М.: Изд-во Моск,
ун-та, 1984. 158 с.
Костецкий П. В., Архипова С. Ф., Владимирова Р. Р. Консервативные и вариабельные участки
аминок слотных последовательностей фосфолипаз А, змей//Молекулярная биология, 1991.
Т. 25. В. 5. С. 1345—1355.
Курик М. В. Холестерин: семь точек зрения //Химия и жизнь. 1987. № 1. С. 66—70.
Никитин В. М., Бабенко М. А. Липиды и липидны Обмен в онтогенезе //Успех! современной
биологии, 1987. Т. 104. В. 3. С. 331—345.
Номенклатура липидов //Молекулярная биология, 1992. Т. 26. В. 3. С. 648—662.
Сим Э. Биохимия мембран.— М.: Мир, 1985. ПО с.
Хакомори С. И. Гликосфинголипиды//В мире науки, 1986. № 7. С. 12—23.
Цырлов И. Б., Ляхов В. В. Роль микросомных фосфолипидов в организации и регуляции
активности монооксигеназной системы//Успехи современной биологии, 1977. Т. 84. № 1.
С. 3—21.
Швец В. И., Степанов А. Е„ Крылова В. Н., Гулак П. В. Миоинозит и фосфоинозитиды.—М.:
Наука, 1987. 247 с.
Шулындин А. А Эволюционное разнообразие фосфолипидных структур // УЬпехи со ременной
биологии, 1983. Т. 95. В. 1. С. 16—32.
490
Пава X. БИОЛОГИЧЕСКОЕ ОКИСЛЕНИЕ
Арчаков А. И. Оксйгеназы биологических мембран.—М.: Наука, 1983. 54 с.
Вартапетян Б. Б. Кислород и структурно-функциональная организация р стителъной «Лет-
ки.—М.: Наука, 1985. 88 с.
Виноград) А. Д. Каталитические свойства ATP-синтетазы митохондрий//Биохимия» 1984.
Т. 49. Выл. 8. С. 1220—1238.
Дмитриев Л. Ф. О механизме сопряжения окисления и фосфорилироаан «//Молекулярная
биология, 1986. Т. 20. В. 4. С. 1111—1125.
Звягильская Р. А.. Котельникова А. В. Дыхательная цепь [рожж вых митохондрий//Успехи
биологической химии.—М.: Наука, 1989. Т. XXX. С. 154—180.
Коте ьникс i А. В. Пути превращени энергии мембранными генераторами В процессе эво-
люции//Успехи биологической химии.—М.: Наука, 1985. Т. XXVI. С. 153—168.
Кулаев И. С. Неор анические полифосфаты и их роль на разных этапах эволюции//Соросов-
ский образовательный журнал, 1996. № 2. С. 28—35.
Николс Д. Биоэнергетика. Введение в хемиосмотическую ‘ ор ю.—М.: Мир, 1985. 190 с.
Рэкер Э. Биоэнергетические механизмы: новые взгляды.—М.: Мир, 1979. 216 с.
Скулачев В. П. Законы биоэн ргетики//Соросовский образовательный журнал, 1997. № 1.
С. 9—14.
Скулачев В. П. Кислород в живой клетке: добро и зло//Соросовс ий обр зовательный жур-
нал, 1996. № 3. С. 4—10.
Скулачев В. П. Рассказы о биоэнергетике.—М.: Молодая гвардия, 1982. 191 с.
Скулачев В. П. Энергетика биологических мембран.—М.: Наука, 1989. 564 с.
С улачев В. П. Эволюция биологических механизмов запасай я энергии//Соросовский об-
разовательный журнал, 1997. № 5. С. 11—19.
Сырцова Л. А. О механизме сопряжения в биологичес их системах трансформации энергии//
Успехи биологической химии, 1989. Т. XXX. С. 130—153.
Таджибаев Ш. С., Наценка Е. Г. БйохиМИчес ие основы профилактики цитото сичвского дей-
ствия ксенобиотиков//Украинский биохимический журнал, 1992. Т. 64. № 3. С. 3—11.
Тихонов А. Н. Молекулярные преобразователи энергии в живой клетке//Соросовский об-
разовательный журнал, 1997. № 7. С. 10—17.
Тихонов Aj Н. Трансформация энергии в хлоропластах—энергообраз ощих органеллах рас-
тительной клетки//Соросовский образовательный журнал, 1996. № 4. С. 24—32.
Энгельгардт В. А. Корреляция «ежду дыханием и превраще ями пирофосфата в эритроци-
тах птиц//Молекулярная биология, 1994. Т. 28. В. 6. С. 1218—1228.
Глава XI. ВОДНЫЙ И МИНЕРАЛЬНЫЙ ОБМЕН
Аксенов С. И. Вода и ее роль в регуляции биологических процессов.—М.: Наука, 1990. 116 с.
Александров С. Ртуть в рыбе//Химия и жизнь, 1991. № 1. С. 82—84.
Андроникашвили Э. Л. ДНК вблизи абсолютного нуля//Химия и жизнь, 1986. № 3. С. 27—35.
Верхова О. А., Сорока В. Р. Биологическая роль лантанидов//Успехи совре енной биологии,
1980. Т. 90. № 3. С. 365—381.
Вишняков С. И., Левантовский С. А., Рыжкова Г. Ф. Биолог ск (ействие хрома в зависи-
мости от его валентности//Биологические науки, 1992. № 9. С. 105—108.
Дорожко О. В., Ротшильд Е. В. Микроэлементы в изнеДе тьно 'и патогенных и некото-
рых других микроорганизмов//Успехи современной биологии, 1985. Т. 99. В. 2. С. 313—318.
Кальницкий Б. Д. «Новые» незаменимые микроэлементы в питании животных (обзор)//Сель-
скохозяйственная биология, 1986. № 6. С. 64—68.
Левицкий Д. О. Кальций и биологические мембраны.—М.: Высшая школа, 1990. 124 с.
Мирошниченко Н. С., Шуба М. Ф. Роль ионов кальция в процесса сокращения и расслабле-
ния скелетной мышцы //Доклады АН СССР, 1987. Т. 296. № 4. С. 1001—1004.
Расмуссен Г. Цирк гяция кальция и внутриклеточная передача внешних сигналов//В мире
науки, 1989. Ns 12. С. 36—43.
Сигуля Е. Е. Ванадий, ферменты и сердце//Химия и жизнь, 1989. № 6. С. 84—85.
Строчкова Л. С. О некоторых механизмах проникновенш микроэл ентов в клетку и ИХ
локализации//Успехи современной биологии, 1990. Т. 101. В. 1. С. 101—117.
Строчкова Л. С., Юрова А. В., Жаворонков А. А. Влияние никеля на организм животных
и Че, овека//Успехи современной биологии, 1987. № 1. С. 142—155.
Торшин С. П., Удельнова Т. М., Ягодин Б. А. Микроэлементы, экология и здоровье человека//
Успехи современной биологий, 1990. Т. 109. Вып. 2. С. 279—292.
Усенко Е. В. Влияние тяжелых металлов на интенс вность синте а нуклеиновых кислот и бел-
ка в клетках про- и эукариот//Автореф. дисс. к. б. н., Харьков, 1991.
Шавловский Г. М., Логвиненко Е. М. Роль железа в регуляции биосинтеза белков у микроор-
ганизмов//Успехи микробиологии, 1988. Т. 22. С. 108—132.
491
Глава XII. ГОРМОНЫ И ИХ РОЛЬ В ОБМЕНЕ ВЕЩЕСТВ
Ашмарин И. П., Обухова М. Ф. Регуляторные пептиды, функционально-непрерывная совокуп-
ность//Биохимия, 1986. Т. 51. В. 4. С. 531—545.
Бакл Дж. Гормоны животных.—М.: Мир, 1986. 86 с.
Варфоломеев С. Д. Простагландины—новый тип биологических регуляторов//Соросовский
образовательный журнал, 1996. № 1. С. 40—47.
Рребенщиков Ю. Б., Машковский Ю. Ш. Физико-химические свойства, структура и функцио-
нальная активность инсулина // Биоорганическая химия (Итоги науки и техники
ВИНИТИ АН СССР), 1986. Т. 7. 296 с.
Дёрфлинг К. Гормоны растений.—М.: Мир, 1985. 303 с.
Кефели В. И. Биохимия гормонов растений//Успехи биологической химии, 1991. Т. XXXII.
С. 222—238.
Кулаева О. Н. Как регулируется жизнь растений//Соросовский образовательный журнал,
1995. № 1. С. 20—27.
Кулинский В. И. Передача и трансдукция гормонального сигнала в разные части клетки //
Соросовский образовательный журнал, 1997. № 8. С. 14—19.
Кусе ь С. И., Стойка Р. С. Молекулярные механизмы в действии полипептидных факторов
роста.—М.: Наука, 1984. 240 с.
Мертвецов Н. П. Регуляция экспрессии генов стероидными гормонами.—Новосибирск: На-
ука, 1990. 264 с.
Нейрохимия/Под ред. И. П. Ашмарина и П. В. Стукалова.—М.: Изд-во института биомеди-
цинской химии РАМН, 1996. 469 с.
Панков Ю. А. Белок «оЬ»—продукт экспрессии гена ожирения и некоторые аспекты развития
современной эндокринологии//Биохимия, 1996. Т. 61. В. 6. С. 984—992.
Розен В. Б. Основы эндокринологии.—М.: Высшая школа, 1984. 336 с.
Сергеев П. В. Стероидные гормоны.—М.: Наука, 1984. 240 с.
Серебряный А. М., Крашенинникова Г. А., Бахмина Л. В. Этиленобразующий фермент расте-
ний//Биохимия, 1995. Т. 60. В. 7. С. 1005—1016.
Судаков С. К. Нейропептиды в центральных механизмах пищевого поведения//Успехи со-
временной биологии, 1988. Т. 105. В. 1. С. 100—116.
Суханов А. А. Механизм гормональной регуляции роста опухолевых клеток//Успехи биоло-
гической химии, 1995. Т. XXXV. С. 97—134.
Ткачук В. А. Введение в молекулярную эндокринологию.—М.: Изд-во МГУ, 1983. 256 с.
Филиппович Ю. Б., Кутузова Н. М. Гормональная регуляция обмена веществ у насекомых //
Биологическая химия (Итоги науки и техники ВИНИТИ АН СССР), 1985. Т. 21. 226.С.
Яглов В. В., Ломоносова Г. А. Диффузная эндокринная система. Итоги и перспективы ис-
следования//Успехи современной биологии, 1985. Т. 99. В. 2. С. 264—276.
Karlson Р. Introduction: the concept of hormonal systems in retrospect and prospect//Nova acta
Leopoldina, 1984. NF 56. N 255. P. 9—20.
Глава XIII. ВЗАИМОСВЯЗЬ И РЕГУЛЯЦИЯ ОБМЕНА ВЕЩЕСТВ
Буланова Е. Г., Будагян В. М. Протеинфосфатазы: структура и функции//Молекулярная био-
логия, 1994. Т. 28. В. 5. С. 991—999.
Васнецова А. Л., Гладышев Г. П. Экологическая биофизическая химия.—М.: Наука, 1989.
134 с.
Замятнин А. А. Компьютерная биохимия эндогенных регуляторных олигопептидов//Успехи
биологической химии, 1996. Т. XXXVI. С. 87—112.
Иткес А. В., Туницкая В. Л., Северин Е. С. Регуляция биологической активности клетки сис-
темой вторичных мессенджеров: сАМР, 2', 5'-олигоаденилат i и кальция//Успехи б ологической
химии.—М.: Наука, 1985. Т. 26. С. 125—152.
Капков Д. В., Турапов О. А., Степанов А. С. Казеиновые киназы типа 2: структура и локали-
зация в клетках эукариот//Успехи биологической химии, 1995. Т. XXXV. С. 135—159.
Курганов Б. И. Принципы интеграции клеточного метаболизма // Молекулярная биология,
1986. Т. 20. В. 2. С. 369—377.
Магакян Ю. А. Гиперрепликация ДНК//Общие проблемы физико-химической биологии (ито-
ги науки и техники ВИНИТИ АН СССР), 1990. Т. 15. 220 с.
Малыгин А. Г. Регулярность в структуре сети реакций метаболизма//Биохимия, 1989. Т. 54.
В. 6. С. 883—894.
Остроумов С. А. Введение в биохимическую экологию.—М.: Изд-во МГУ, 1986. 176 с.
Реутов В. П. Цикл окиси азота в организме млекопитающих//Успехи биологической химии,
1995. Т. XXXV. С. 189—228.
Северин Е. С., Кочеткова М. Н. Роль фосфорилирования в регуляции клеточной актив-
ности.—М.: Наука, 1985. 288 с.
492
Степанов А. С. Белковые факторы маскирования и демаскирования мРНК в клетках эукари-
от//Автореферат дисс. докт. биол. наук, М., 1996. С. 1—44.
Троицкий Г. В. Постсинтетическая модификация белка /Украинский биохимический журнал,
1985. Т. 57. № 3. С. 81—97.
Харборн Дж. Введение в экологическую биохимию.—М.: Мир, 1985. 311 С.
Хохлов А. С. Низкомолекулярные микробные ауторегуляторы.—М.: Наука, 1988. 270 с.
Худяков Ю. Е. Последовательность Шайна-Дальгарно и эффективность инициации трансля-
ции//Молекулярная биология, 1985. Т. 19. № 3. С. 702—716.
Глава XIV. КВАНТОВО-МЕХАНИЧЕСКИЕ АСПЕКТЫ БИОХИМИИ
Баренбойм Г. М., Маленков А. Г. Биологически активные вещества. Новые принципы поис-
ка.—М.: Наука, 1986. 363 с.
Бушелев С. И., Степанов Н. Ф. Электронная структура и биологическая активность молекул//
Новое в жизни, науке и технике, 1989. № 5. 30 с.
Войтюк А. А., Близнюк А. А. Квантово-химическое исследование влияния молекул воды на
нуклеиново-белковые взаимодействия//Молекулярная биология, 1989. Т. 23. В. 5. С. 1289—1294.
Димогло А. С., Чумаков Ю. Н., Берсукер И. Б. Квантово-химическое изучение взаимодейст-
вия соединений платины (II) с основаниями ДНК//Теоретическая и экспериментальная химия,
1981. Т. 17. № 1. С. 88—92.
Кларк Т. Компьютерная химия.—М.: Мир, 1990. 384 с.
Камерницкий А. В. Молекулы-пиктограммы//Химия и жизнь, 1987. № 4. С. 42—45.
Кузьмин В. Е., Коновороцкий Ю. П. Взаимосвязь структурных и топологических характерис-
тик молекул//Журнал структурной химии, 1985. Т. 26. № 4. С. 14—21.
Либерман Е. А. Как работает живая клетка//Новое в жизни, науке и технике. Сер. биол., 1990.
№ 4. 64 с.; № 11. 55 с.
Морозов Ю. В., Савин Ф. А. Электронная спектроскопия и электронное строение молекул //
Молекулярная биология, 1985. Т. 19. № 1. С. 132—144.
Нефёдов В. И., Вовна В. И. Электронная структура органических и элементоорганических
соединений.—М.: Наука, 1989. 199 с.
Рувре Д. Г. Химию прогнозирует топология//В мире науки, 1986. № 11. С. 14—22.
Соловьев Л. Г. Живое энергетически выгоднее неживого//Химия и жизнь, 1990. № 2. С. 40.
Пюльман Б., Пюльман А. Квантовая биохимия.—М.: Мир, 1965. 654 с.
Хубер Р. Перенос энергии в биологических структурах//Новое в жизни, науке и технике. Сер.
хим., 1990. № 1. С. 3—32.
Эдельман Дж. М. Топобиология//В мире науки, 1989. № 7. С. 24—33.
'Эрнст Е. Субатомная биология: электронная биология, биополупроводимость//Биофизика,
1975. Т. XX. В. 3. С. 540—546.
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Авидин 175, 221
Автотрофы 358*
Авитаминозы 52—170
Агароза 31, 141, 190, 317
Агликон 309, 314
Агматин 268*
Аценилаткиназа 243
Аденилатциклаза 141, 335*, 388*, 456*, 460*
Адениловая кислота 175, 198, 251 * (
Аденилосукцинатлиаза 244*
Аденилосукцинатсинтетаза 244*
Аденин 192*, 200*, 232*
Аденин-амииогидролаза 232*, 233*
Аденин ; [юсфорибозилтрансфераза 245*
S-аденозилгомоцистеин 271*
S-адёнозилметионин 270*, 271, 408
S -аденози плетионинсин аза 271
Аденозин 232, 233
Аденозиндифосфат (АДФ) 124*, 137*, 138*,
243, 415*, 419*, 423*
Аденозинкобаламин 166
денозин он фосфат (АМФ) 196*, 251*, 274*
Аденозинтрифосфат (АТФ) 21,124*, 136*, 137*,
183*—188*, 238—245*, 253*, 270*, 334*,
388*—402, 420*—429*, 456
Аденозин-З'-фосфат 197
Аденозин-5'-фосфат 197*, 243*, 244*, 281, 422*
Адреналин 271, 460*
Адренокортикотропный гормон (АКТГ) 447,
452*
АДФ-рибозилтрансферазы 282
3'-азидо-2', З’-дидезокситимидин 240*
Азидотимидин 240*
Аконитаза 355*, 356*
Аконитовая кислота 355*
Акриламид 28*, 141
Акрилгидразин 141
Актин 91, 92*
Актиномицин 175
Акцептирующий конец тРНК 214*, 215*, 281,
283
В-аланил-В-аланин-синтетаза 176
Аланин 40*, 42*, 90, 276*, 340
D-аланин 175
а-Аланин 115, 116*, 144, 175
р-Аланин 234, 268
Аланинаминотрансфераза 140
Аланиндегидрогеназа 276
а-Аланин: 2-оксбглутарат-аминотрансфераза 115
Аланин-рацемаза 175
Алиэстераза 387*
Алкогольдегидрогеназа 119*, 145*, 351*
Аллантоин 234*, 235, 236
Аллантоиназа 234
Аллантоиновая кислота 233, 234*
Аллостерическая регуляция 74, 99, 294, 302*,
335*, 397, 487
Альбумин сывороточный 88*
Альбумин яичный 52*, 446
Альгиновые кислоты 317
льдегиддегидрс наза 393*
Альдегид-лиазы 133
Альдолаза 133, 343
Альдостерон 443*, 444, 447*, 448
Амидазы 131
Амидирование 272, 273
Амидовый черный 28*
Амидосинтетазы 272
Амидофосфорибозилтрансфераза 240, 244
Амилазы 114, 130, 140, 141, 328, 330*—332*,
438
Амило-1,6-глюкозидаза 330*
Амилоза 318*, 319*, 322*
Амилопектин 318, 319, 320*, 321*, 322*
Аминирование 275, 276
Аминоациладенилаты 137, 268, 281*, 294
Аминоацилдифосфатидилглицерин 384
Аминоацилпроводящий белок (АПБ) 294, 295*
Аминоацил-тРНК 137, 282, 284*, 288, 292, 293,
214*, 281*
Аминоацил-тРНК-сИНтетазы 44, 213, 220, 281,
282, 283*, 284*
Аминоацилфосфатидилглицерин 384
Аминогидролазы 232
Р-Аминоизомасляная кислота 235
Аминокислоты:
— активный транспорт 47*, 263, 264*
— активирование 280—285
— биосинтез 125*, 275, 276*
— заменимые 276, 277
— продукты распада 272—275
— незаменимые 276—277
— объемные модели 41, 42*
— оптическая активность 39, 41
— превращения 265—277
у-Аминомасляная кислота 268*
Звездочкой отмечены страницы, на которых помещены рисунки, таблицы или формулы.
494
6-аминопенициллановая кислота 143
Аминопептидазы 131
Аминотрансферазы 124
Амплификация генов 255
ц-АМФ-независимая протеинкиназа 354
Амфолины 29
Анаболизм 180
Анамеры 309
Андростендион 446, 447
Антаманид 50*
нтибиотики 175
Антивитамины 174
Антиген 85
нтигормонг 167
Анти! одон 284, 285, 291, 292
Антикодоновая петля тРНК, 214*, 285
Антимицин 175
Антитела 85
аг Антитрипсин 114*, 145
Алотранскетолаза 350*
Апофермент 98, 117, 147
Арабан 317
Арахидоновая кислота 159*, 415, 473
Аргиназа 132, 269, 436
Аргинин 41, 42, 269
а-Аргинин 144, 268, 269, 271, 274*
Аргининмоноксидаза 419
Аргининсукцинат-лиаза 274*
Аргининсукцинат-синтетаза 274*
Аргининфосфат 271
Аргинин-янтарная кислота 274*
Аскорбатоксидаза 171, 414*
Аскорбатредуктаза 414*
Аскорбиген 171
Аскорбиновая кислота 170, 171*, 415*
Аспарагин 132*, 140, 272*, 273, 296*
Аспарагиназа 131, 132, 140
Аспарагиновая кислота 40*, 125*, 132*, 134*,
144*, 268*, 274*, 275, 276*
Аспарагинсинтетаза 171*, 273
Аспартатаминотрансфераза 127, 128*, 140,
355*, 356*
Аспартат-аммиак-лиаза 134, 143, 144, 275
Аспартатдекарбоксилаза 268
Аспартат-карбамилтрансфераза 100*, 236, 239
Ассимиляция 180
АТФ-геиерирующие вещества 188
АТФ-синтаза (протонная АТФаза) 425, 426, 427
АТФ-синтазный комплекс 423
Ауксины 464
Аутосплайсинг 259
Аффинная хроматография 97, 190, 221
Ацетанилид 279
N-Ацетил алак озаминсульфат 326*
N-ацетил-р, D-глюкозамин ПО*, 317, 326*, 332
Ацетил-коэнзим А 128*, 138, 145, 161, 173,
352*, 354*, 364*, 392*, 396*, 407*, 438, 447*,
468*, 471
— обмен 183*, 393*, 472
Ацетил-КоА: ацетил-КоА-ацетилтрансфераза 394*
Ацетил-КоА-карбоксилаза 395*, 396*
N-ацетилмураминовая кислота 110
N-ацетилмурамовая кислота 313
Ацетилсерин 112*
Ацетилхолин 92, 93, 103, 104, 112, 128*,
169,407*
Ацетилхолинэстераза 92, 102, 104, 112
Ацетоуксусная кислота 394*
Ацил-коэнзим А (ацил-КоА) 390*, 405*
Ацил-коэнзим А-дегидрогеназа '391 *
Ацил-коэнзим А-синтетаза 138, 390*
а-Ацил-лизолецитин 406*
р-Ацил-лизолецитин 406*
Ацилтрансферазы 128, 399, 410
Ацилфосфаты 186
Бацитрацин 294, 296
Белки 23
— агрегированное состояние 76
— аминокислотный состав 38, 45*
— аналогичные пептидные группировки 64
— биосинтез 278, 280*, 285, 290*—296*
— вторичная структура 65, 67*
— гидролиз 38, 56*, 57*, 262*
— денатурация 24, 80
— доменная структура 73, 74, 81
— изоэлектрическая точка 46, 52
— классификация 81, 82*
— методы выделения 24*—34*
— молекулярная масса 34*—37
— надвторичная структура 74, 76, 257*
— номенклатура 80, 81
— олигомерная структура 76
— первичная структура 54*, 55*
— посттрансляционная модификация 300*,
301*
— регуляция биосинтеза 301, 302*
— ренатурация 80
— а-спиральная конформация 66, 67*, 71*, 72*
— Р-структура 69*, 71*, 72*, 81, 82*
— твердофазный синтез 47*, 48
— транслокация 300*, 301*
— третичная структура 68—74*
— четвертичная структура 76—78, 435
Белок-активатор 259*
Белок-репрессор 259*, 302*, 476*, 477
Бензилпенициллин 143*
Бензохинон 120*
Бетаин 407*, 408*
Бетаинальдегид 407*
Бетаинальдегиддегидрогеназа 407*
Бетаин-гомоцистеин-мегил-трансфераза 408*
Биогенные амины 177, 267
Биологические мембраны 371 *, 372*, 381
Биотин 173*, 396*, 397*
Биотинпротеин 395*, 396*, 397*
Биофлавоноиды 173
Бомбикол 176*
Бромелаин 131
Бутирил-КоА 396*
Вазопрессин 50*, 434, 449*
Валин 40*, 42*, 90, 265, 276, 295*, 296*
Вирус табачной мозаики 76*, 77*, 94
Викасол 158*
Витамерия 150
В тамерь; 150
Витамин А, Ап А2 151
— В,; В2, В3 159—163
— В6, В12, В15, В , В 164—170
— С 170
— D, D2, D3 154—156
495
— Е 156, 157
— F 159
— Н 173
— К, Кп К2, К3 157, 158
— Р 172
— РР 163, 271
— U 174
Витамины 148, 149*, 150*
Витамин К-зависимая карбоксилаза 158
Вода 209, 431
— иммобильная 431
— обмен 434
— роль в процессах жизнедеятельности 433
— регуляция 434
— содержание в организме 16
— состояние 431, 432
— экзогенная 434
— эндогенная 434
Водородные связи 43*, 66*, 77, 104, 200*,
205*, 209*
Воски 376, 377*
Вторичный посредник 384, 446, 456, 473*,
474*, 475*
D-Галактоза 313, 339*
a-D-Галактоза 339*
P-D-Талактоза 316, 339*
Галактан 318
Р-Галактозидаза 210
Галактокиназа 140
Гастрин 449*, 450
Гексокиназа 124, 338*, 337*
Гель-фильтрация 31
Гем 77*, 114, 123
Тёмин 10
Гёминфермент 114
Тёмованадины 89
Гемоглобин 37, 52*. 62*. 64, 76, 77*, 78*, 89, 221
Гемоцианины 89
Гемэритрины 89
Тён-оператор 302*
Тен-регулятор 302*
Тён-репрессор 302*
Тёны 190, 204*, 302*
ранилви эф 4>ат 403*
Тёпарин 317, 327*
Гепариназа 327
Гётероауксин 464*, 465
Гётерополисахариды ЗГ7, 326*
Гётеротрофы 357
Гиалуронидаза 140, 326, 332
Гиалуроновая кислота 140, 317, 326*, 332
Гиббереллины 464*, 465
Гибберелловая кислота 464*
Гидрогеназа 145, 146*
Гидрокортизол 443, 444, 446
Гидролазы 117, 129, 138, 142, 147, 230
Гидролиазы 391
Гидрофобные взаимодействия 209
Гипервитаминозы 149
Пшерхромный эффект 211
Гиповитаминозы 149
Гипоксантин 232*, 233*
Пшпуровая кислота 279
Гистамин 267*
Тиствдин 40*. 42*, 77, 228, 229, 265, 267*, 283
Гистидиндекарбоксилаза 267*
Гистоны 64*, 81, 83, 84*, 210, 221, 229, 456
Гликоген 317, 322, 323*, 328, 369*, 321*
Гликогенолиз 334, 351
Гликогенсинтетаза 367, 368
Гликогенфосфорилаза 128, 368, 335*, 336*
Гликозидазы 129, 333
а-Гликозидазы 130, 333
Р-Гликозидазы 130, 140, 147, 333
Гликозидо-гликозиды 314, 315
Гликозидо-глюкозы 316
Гликозилы 312, 314
N-Гликозилирование 368
Гликозилтрансферазы 128*, 364, 367, 410
Гйиколат: кислород-оксидоредуктаза 414*
Глйколатоксидаза 414
Гликолатоксидазная система 414*
Гликолевая кислота 414*
Гликолакгиловые кислоты 313
Гликолиз 351, 355, 438
Гликолипиды 385*, 386*
Гликопротеины 175, 317
Глиоксиловая кислота 233, 234*, 395*, 414*
Тлицеральдегид-З-фосфатдегидрогеназа 344,
345*, 346, 350, 361, 362*
Глицерин 97, 340, 381*, 389*, 472
— распад 388*
Тлицеролкиназа 389*
Глицерол-З-фосфатдегидрогеназа 390*
Глицерофосфат 182*, 389*, 400*, 410
Глицерофосфат-ацил-трансфераза 400*
Глицерофосфолипиды 382*, 405*, 408*
а-Тлицерофосфохолин 405*
Тлицерофосфохолин-диэстераза 405 *
Глицин 40*, 43*, 90, 235, 241, 243, 271*
Тлицин-метилтрансфераза 271 *
Глобозиды 386*
ГЬобулины 80
Глутаматдегидрогеназа 100, 266*
у-Гпутамилсинтетаза 264*
у-Глутамилтрансфераза 141, 264*
у-Глутамилциклотрансфераза
Глутамин 240, 243, 272, 273, 279, 296*
Глутаминаза 132, 133
Глутаминдегидрогеназа 276
Глутаминовая кислота 100, 115*, 126*, 144, 235,
239, 264, 266*, 268*, 269*, 272, 273, 276*, 283
Глутаминсинтетаза 2.72 273
Глутатион 48*, 49*, 97, 145, 147, 171, 264*, 279
Глутатионсинтетаза 264*
Глюкагон 450*, 460*
а-1,4-Глюкан 333*, 366*, 367*, 369*
1, 3- и 1,6-Тлюканазы 332
и-1,4-глюкан-ветвящий фермент 368, 369*
Глюкоамилаза 140, 328*, 329, 330*
Глюкоза 124*, 129*, 144, 145*, 172*, 193, 307,
308*—313*, 328, 339*
a-D-глюкозамин 225, 327*
Глюкозо-1,6-дифосфат 312*, 340*
Глюкозоизомераза 143
Глюкозооксидаза 140, 145, 162
Глюкозо-1-фосфат 129*, 182*, 312*, 333*; 334,
339, 340*, 364*
Глюкозо-6-фосфат 124*, 182*, 145,.312*, 338*,
351, 355*
— обмен 339, 340*
496
— распад 341*, 343*, 347*, 350*
Глюкозо-1-фосфатаза 129
Глюкозо-6-фосфатдегидрогеназа 347*, 348
Ппокозофосфатизомераза 341 *
Глюкокиназа 337*
Глюкокортикостероиды 444
Глюконо лактоназа 348*
a-D-Глюкопираноза 135*, 309*, 313, 315, 316,
318, 323*, 325
P-D-Глюкопираноза 135*, 309*, 313, 324*
a-D-Глюкопиранозо-!-фосфат 128*
О-Глюконовая кислота 145*, 313*
P-D-Глюкуроновая кислота 326*, 327*
Гомоцистеин 408*
Гбмополисахариды 317
Гормоны 83, 131, 262, 441
— классификация 443*
— номенклатура 441
— применение 465, 466*
— пептидные 112, 305, 443*, 448
-----биосинтез 458, 459
-----механизм действия 456, 457*
— стероидные 145, 402, 443*
-----механизм действия 445*
— ювенильные 466*
Грамицидин 50*, 175, 294, 295*
Гуаниловая кислота 198, 230
Гуанилрибонуклеазы 229, 230
Гуанин 192*, 200*, 221, 232*
Гуанин-аминогидролаза 232*
Гуанозин 231*
Гуанозиндифосфат (ГДФ) 243*, 291, 293, 419*
Гуанозиндифосфатглюкоза 366*
Гуанозиндифосфатманноза 365*, 366
Гуанозинмонофосфат 196*
Гуанозинмонофосфат (ГТФ) 153, 188, 196*, 239,
243*, 266, 290, 293, 419*
Гуанозин-З'-фосфат 230, 231*
Гуанозин-5'-фосфат 243*, 244*
Гулоза 170*
Гулоновая кислота 170*
Дегидроаскорбиновая кислота 170*, 414*
Дегидроацилкоэнзим А 391 *
Дегидрогеназа a-глутаминовой кислоты 266
Дегидрогеназы 98, 117, 118*, 164, 266, 413*
2,3-Дегидро-а-гулоновая кислота 170*
а, Р-Дегидростеарил-КоА 391 *
7-Дегидрохолестерол 154
Дезаминирование 232—234, 265*, 266—267
Дезоксиаденозинмонофосфат (дАМФ) 197*
Дезоксиаденозинтрифосфат (дАТФ) 239, 244
Дезоксигуанозинмонофосфат (дГМФ) 197*
Дезоксигуанозинтрифосфат (дГТФ) 239, 244*
11-Дезоксикортикостерои 446*, 447
Дезоксирибоза 192, 193*, 206, 231, 238, 244, 249
Дезоксирибонуклеазы 140, 196, 224, 225
Дезоксирибонуклеиновая кислота 19, 194
— белкоаые факторы биосинтеза 251
— биосинтез 246*, 253*—257, 446
— вторичная структура 205*—209*, 247
— конформационные состояния 206*, 207*,
208
— локализация в клетке 195
— метилирование 465
— молекулярная масса 195*
— нуклеотидный состав 199*
— палиндромные структуры 210
— первичная структура 201*, 203*, 204*
— репарация 231
— свойства 211
— третичная структура 210*, 211*, 437
— уровни суперспирализации 210, 212*
— ферменты биосинтеза 249, 250
Дезоксирибонуклеозиддифосфат 239
Дезоксирибонуклеозид-З'-фосфат 225
Дезоксинуклеозидтрифосфаты 248*
Дезоксирибонуклеотиды 96*
Дезокситимидинмонофосфа (дТМФ) 197*,
239, 244*
Дезокситимидингрифосфат (дТТФ) 239*, 240,
243
7-Дезоксихолевая кислота 380*
Дезоксицитидинмонофосфат (дЦМФ) 197*,
238*
Дезоксицитидинтрифосфат (дЦТФ) 238*, 239
Декарбоксилазы 133*, 160, 267
Декарбоксилирование 267, 268
Декстран 317, 325
Декстрины 322, 328
Депрессия генома 85
Десмолазы 116
Детергенты 26*, 285
Диаминоксидазы 272
Диаминопимелиновая кислота 144*
Диацилглицерин 474*
Дигестаза 140
Дигидролипоевая кислота 354*
Дигидролипоилдегидрогеназа 352*, 353*
Дигидрооротаза 338, 339
Дигидрооротовая кислота 338, 339
Дигидропиримидиназа 234*
Дигидроуридиловая кислота 214
Дигидроуридиловая петля тРНК 214, 215*
Дигидроурацил 234*
Дигидроурацилдегидрогеназа 234*
Диг церидацилтран ра 00*, 401
Дигидрохолестерол 378*, 380*, 401*
Диглицерид 401*, 405*
Диглицерид-ацетилтрансфераза 401 *
Диизопропилфосфосерин 112*
Диизопропилфторфосфат 112*
Дииодтирозин 462*
Дииодтиронин 461 *, 462
Диметилаллилпирофосфат 135*, 403*
Диметилаллилтрансфераза 403*
М6-Диметиладенин 192*
Ы2-Диметилгуанин 192*
N, N-Диметилглицин 407, 408*
6, 7-Диметилизоалло сазин 162*
Диоксиацетонфосфат 390*
Диоксиацетонфосфатацилтрансфераза 401 *
3, 4-Диоксигеназа протокатеховой кислоты 418
Диоксигеназы 418
12, 13-Диоксиолеиновая кислота 415*
Диоксифенилаланин (ДОФА) 460*
Диоксифенилсерин 460*
1, 2-Дипальмитил-З-фосфоглицерин 400*
Дипальмитин 400*
Дипептидазы 131
ипепт гидролазы 131
Дисахаридсульфоэстераза 327
497
Дисахариды 314
Диссимиляция 180
Дитиотреитол 97*
Дифосфатидилглицерин 383, 405
1,3-Дифосфоглицериновая кислота 183*, 187,
342, 344*, 419, 438
Дифторбензурон 467
Диэгиламиноэтилцеллюлоза (ДЕАЕ-целлюло-
за) 324, 325*
ДНК-глюкозидазы 231
ДНК-гираза 248*
ДНК-зависимая РНК-полимераза 44, 63
ДНК-закручивающий белок 251
ДНК-лигаза 251, 252*, 255
ДНК-полимераза 44, 246, 249, 250*, 254*,
255*, 437
ДНК-раскручивающий белок 251, 254*
ДНК-связывающий белок 251, 254*
Долихолдифосфатсахара 368
Домен 73, 81, 102, 341*, 342*
Дыхательный фермент Варбурга 414
Еноил-КоА-гидратазы 391*
Енолаза 344*
Енолпировиноградная кислота 344*
Железопорфирины 147
Железосерные белки 421*, 423*
Жирные кислоты высшие 159, 340, 372*, 374,
375, 377*
------биосинтез 395*
------распад 388*—394*
Жиры 372
Зимаза 147
Зимоген 131
Изоаллоксазин 161, 162
Изовалериановая кислота 374
Изозимы 100*, 101*, 124, 140, 341
Изолейцин 40*. 42*, 90, 142, 265, 276
Изолимонная ислота 355*, 395*
Изомеразы 117, 134, 147, 339*
Изомеризация 338*, 339*
Изопентенилпирофосфат 135*, 403*, 446*
Изопентенилпирофосфат-изомераза 135*, 403*
Изопропиладреналин 460*
Изостерия 112
Изоферменты 83
Изоцитратдегидрот та а 140, 355*, 356*
Изоцитрат-лиаза 395*
Изоэлектрофокусирование 97
Иминоглутаровая кислота 266*
Иммуноглобины 85, 86, 87*
Инверсия 315
Инвертаза 315
Инве] ный сахар 315
Ингибиторы ферментов 111*, 112*, ИЗ*
Р-Индолилуксусная кислота 464*
Индукция 476*, 477
Инозин-5'-фосфат 242*, 243, 244*
Инозит-1, 4, 5-трифосфат 474*
Инозитфосфолипиды 384*
Инсулин 51*, 59*, 61*. 64, 65*. 77*. 145, 435,
450*, 451*, 452,458*
Инсулин: глутатионтрансгидрогеназа 459*
Интерлейкины 88
Интерфероны 88, 145, 205
Инулин 317, 332
Инулиназа 332
Информосомы 302
Иодтиронины 461 *
г
Кадаверин 133*, 268*
Са2+-Кальмодулин-зависимая протеинкиназа
474,475*
Кальпаины 263
Кальпостатин 263
Кальцитонин 154*
Кальциферол 154
Карбамид-амидогидролаза 115
N-Карбамил-р-аланинамидогидролаза 234*
Карбаматкиназа 235, 273
Карбамиласпарагиновая кислота 236
Карбамилфосфат 184*, 236, 273, 274*
Карбаминовая кислота 234
карбоксилазы 136, 436
Карбоксиметилцеллюлоза (КМ-целлюлоза) 324,
325*
Карбоксипептидазы 132*, 141
Кардиолипин 383*. 384, 423
Карнитин 169
Карнозин 49*
Р-Каротин 418*
Р-Каротин-15, 15'-оксигеназа 418*
Каррагинаны 317
Катаболизм 180
Ка алаза 100*, 140, 412*
Катепсины 128
Катехол-2,3-оксигеназа 418
Р-Кетоацил-коэнзим А 391*
3-Кетоацил-КоА-тиолаза 391*
Р-Кетобутирил-КоА 394*
2-Кетоглюкоиовая кислота 145
2-Кето-3-дезокси-6>фосфоглюконатальдолаза
350*
2-Кето-6-дезоксифосфоглюконовая кислота
350*
а-Кетоглутаратдегидрогеназа 355*, 356*
а-Кетоглутаровая кислота 115, 126*, 266*,
355*, 419*, 468*, 471
2-Кето-6-фосфоглюконовая кислота 350*
Кефалины 383*, 408
Киназа киназы фосфорилазы Ь 335*, 336
Киназа фосфорилазы Ъ 334*, 335*
Киназы 336*
Кинетин 465*
Клетчатка 318, 323, 324*
Код белкового синтеза 175,296*, 297*, 298,299
Кодон 282, 290, 291
Кодон-антикодоново взаимодействие 292,293 *
Козимаза 147
Коллаген 90, 171, 326,439
Коллагеназа 131,140
Комплементарные основания 201 *, 206*, 247*
Копростанол 380*, 401
Корепрессор 476*
Кортикостероиды 443
Кортикостерон 443*, 444, 446*, 447
Кофермент 98, 117, 147
Коэнзим А 128, 138, 145, 163, 352*. 354* 390*,
394*, 402*
498
Крахмал 140, 141,318,319,321 *, 322*, 328,369*
Крахмалфосфорилаза 128
Креатин 167, 187
Креатинсинтаза 140
Креатинфосфат 167, 183*, 187
Кристаллины 90
Кротонил-КоА 395*
Ксантин 232* 233*, 234
Ксантиноксидаза 162, 233, 437
Ксантозин-5'-фосфат 243
Ксантозин-5'-фосфат: L-глутамин-амидолигаза
244*
Ксилулозо-5-фосфат 338*, 348*
Кэп 222*, 258
Лактаза 147, 316*
Лактатдегидрогеназа 101, 116, 140, 350
Лактоза 210
Лактозный оперон 209*
Ланостерол 404*
Левулоза 313
Леггемоглобин 60*, 63
Лейцин 40*, 42*, 90, 265, 276, 294, 295*
Лектины 307
Лецитины 383*, 406*, 409*
Лиазы 117, 132, 147, 139
Либерины 459
Лигазы (синтетазы) 117, 136, 147,272, 390*
Лигноцериновая кислота 385
Лизин 41*, 42*, 133*, 144*, 171, 251*, 353*, 387
Лизиндекарбоксилаза 133*
Лизинмонооксигеназа 419
Лизоамидаза 140
Лизоцим 24, 62*, 72*. 70*, 83*, 88, 97, 99*,
ПО* 140, 299*, 313
Лизосомы 97, 138, 262
Лимонная кислота 354*, 355*, 394
Линолевая кислота 159, 375,415*
Линоленовая кислота 159, 375
Липаза 114, 129, 130, 131, 241* 387, 388*,
405*, 438
Липиды 370
— диольные 370, 386*
— обмен 387*, 395*, 399, 405
— перенос 384, 410
— простые 370, 372
— сложные 370, 381
Липоат-ацетилтрансфераза 352*, 353*
Липоамиддегидрогеназа 355*, 356
Липоевая кислота 147, 250*, 251 *, 252*
Липоксигеназа 140,415*
Липоксидаза 415*
Липопротеинлипазы 388*, 438
Р-липотропин 24, 89
Лихеназы 332
Люмихром 162
Малатдегидрогеназа 90, 355*, 356*
Мальтаза 130*, 314*, 315, 328, 329*, 330, 334
Мальтозофосфорилаза 334
Малонил-КоА 396*, 397*
Малонил-КоА-карбоксилаза 396*, 397*
Макроэргические связи 182, 184—186, 346*,
352* 359*, 416
Макроэргические соединения 182,186
D-Манноза 307*, 313
Маннаны 313, 317, 366
Мевалонаткнназа 402*
Мевалоновая кислота 394, 402*
Меланоцитостимулирующий гормон (МСГ)
453*
Меркаптуровые кислоты 271
Метаболизм 180
Метаболиты 17, 302, 372, 473
Метаболой 101, 139, 355*, 390, 475
Металлопротеин 436*
1-Метиладенин 192*
2-Метиладенин 192
3-Метиладенин-ДНК-глюкозидаза 231
Метилбензохинон 158*
а-Метилглюкозид 109*, 111, 312
Р-Метилглюкозид 109*, 111, 312
1-Метилгуанин 192*
7-Метилгуанин 192*
Метилкобаламин 165
Метилмалонил-КоА 394*
Метилмалонил-КоА-КоА-карбонилмутаза
394*
S-Метилметионин 174
№-Метилтетрагидрофолиевая кислота 239
Метилтрансфераза 270, 282
5-Метилцитозин 191*
Метионил-тРНК 290*, 291
Метионил-тРНК-формилтрансфераза 169*
L-Метионин 135*, 144, 167, 168, 169, 267, 270*,
276,408*, 438
Мет-энкефалин 50*
Микобациллин 294, 296
Миколевая кислота 374*
Миксомиозин 91
Минеральные вещества 16, 435, 436*, 437
----значение в сельском хозяйстве 440
----обмен 439
Минералокортикостероиды 444
Минорные основания 191*, 192, 213, 214, 258
Миогемэритрин 82*
Миоглобин 39, 52*, 59*, 70*, 90, 62*, 71*,
72*, 82*
Миозин 91, 221, 92*
Миоинозит 381 *
Митомицин 175
Митохондрии 97, 139, 417, 422*, 423*, 428*
Множественные формы ферментов 101, 140,
348
Молочная кислота 100*, 313, 350, 351
Молочный сахар 317
Р-Моноглицерид 387*, 401*
Моноглицеридтрансацилаза 401 *
Монодегидроаскорбиновая кислота 171
Монооксигеназа 393, 404, 418, 419*
Моносахариды 307, 336
Мочевая кислота 233*, 234*, 235
Мочевина 10, 115, 131, 132*, 145, 233, 235, 234*,
269*, 273, 274*
Муконовая кислота 418*
Мультимер 100*
Мультиэнзимные комплексы 138
Мультиэнзимы 101
Мурамовая кислота 313*
Мутагены 177
Мутаротаза 135
Мутаротация 310*, 313, 316
499
Негистоновые белки 85, 301, 446, 456
Нейрогормоны 459, 467
Неогуаниловая кислота 213*
Нервоновая кислота 385
Никотинамид 118*, 163
Никотинамидадениндинуклеотид (НАД+) 116,
118*, 119, 147, 164, 171, 234, 236, 252*,
266, 346, 354*, 390*, 391*, 392*. 407*, 414*,
419*, 420*
Никотинамндадениндинуклеотидфосфат
(НАДФ+) 119, 147, 164, 236, 266, 347*, 359*,
362*, 393*, 414*, 419*, 420*
НАДН: Q-оксидоредуктаза 424*
Никотинамидрибозофосфат 252*
Никотиновая кислота 163*
Нистатин 175
Новобиоцин 175
Норадреналин 460*
З'-Нуклеазы 224*
5'-Нуклеазы 224*
Нуклеиновые кислоты 189
----биосинтез 246*
----водородные связи 202*
----выделение 189, 190
----коэффициент конденсации 199
----коэффициент специфичности 202
----механизм воспроизведения первичной-
структуры 247*—249*
-------------------------------пути распада 224*
-------------------------------строение и свойства 189
-------------------------------химизм реакций образования 246*, 248*,
249*
-------------------------------химический состав 190—193
Нуклеозид-аминогидролазы 230*, 238
Нуклеозидфосфаты 131*
— биосинтез 235—245
— обмен 231*—234
Нуклеозиддифосфаткиназа 355*
Нуклеозиддифосфатсахара 129, 147, 364*, 365*,
366
[уклеозидтрифосфаты 246
Нуклеозиды 197, 231
[уклеотид аминогидролаз 233
Нуклеотидилтрансферазы 129, 139, 247*, 248
З'-Нуклеотидаза 231 *
Нуклеотиды 196*, 197*, 340
Нуклеосома 85, 211 *, 212*, 254*
Обмен веществ 180
----взаимосвязь 468*—471
----уровни регуляции:
----клеточный 477—481
----— метаболитный 473
----оперонный 475—476*
----организменный 481
----популяционный 481
----общее понятие 180
Окисление биологическое 411, 413
----функции 411 *
----классификация 416
----свободное 417—419*
----сопряженное с фосфорилированием АДФ
417, 419—429
Оксалоацетатдекарбоксилаза 133*
Р-Оксиацил-КоА 391*
Р-Оксиацил-КоА-дегидрогеназа 391*
Р-Оксибутирил-КоА 396*
Оксигеназы 415
Оксидазы 33, 97, 412*, 414
Оксидазы медьсодержащие 414
Оксидазы терминальные 413, 417
Оксидоредуктазы 117, 141, 147—162, 175, 233,
391*, 411
17-Оксикортикостерон 443*, 444, 447*, 448
Оксилизин 171
Олеиновая кислота 375*
Олигомицин 175
Олигосахариды 314, 328, 368
Р-Окси-Р-метилглутарил-КоА 394*, 402*
Оксиметилглутарил-КоА-синтаза 394*
Оксиметил-КоА-редуктаза 402
5-Оксиметилцитозин 191
17-Оксипрогестерон 447*, 448
Оксипролин 171
Окситирамин 460*
Окситоцин 448*, 449, 458
Оксиэтилтиаминпирофосфат 160*, 161*, 351,
352*
2-Оксоглутаровая кислота 115*
Октил-Р, D-глюкопиранозид 115*
Оперон 209*, 221, 302*, 475, 476*
Опсин 92, 152
а-Орнитин 132*, 144, 268*, 269*, 274*, 294*,
295*, 386
Орнитин-карбамилтрансфераза 273
Орнитинолипиды 370, 386, 387
Оротатредуктаза 236
Оротат-фосфорибозилтрансфераза 237
Оротидин 237
Оротидин-5'-фосфат 237*, 238*
Оротидин-5'-фосфатдекарбоксилаза 237
Оротовая кислота 236*
Офтальмовая кислота 236*
Палиндромы 209*, 210, 255
Р-Пальмитилглицерин 129*
Пальмитиновая кислота 129, 375
Пальмитодистеарин 375*
Пальмитостеарооле 375*
Пальмитохолестерид 405*
Пангамовая кнслота 167*
Пантотеин 294*, 295*
Пантотенатсинтетаза 136
Пантотеновая кислота 136, 162*, 268
Папаин 114, 131
Паратгормон 155*, 453*, 454
Пенициллановая кислота 143*
Пенициллин 143, 144, 175
Пенициллинамидаза 143
Пепсин 52*, 54*, 63, 114, 131, 140
Пептидазы 130, 262
Пептид-гидролазы 130, 436
Пептидогликаны 314
Пептидил-пептидогидролазы 130
Пёптидилтрансферазная реакция 296*, 438
Пептидил-тРНК 288, 293*
Пептиды 46, 262
— биологически активные 47*, 48,49*, 113,263
— психотропные 263
— опиоидные 177, 459
— сигнальные 113, 299*, 300*, 459, 479
— твердофазный синтез 46, 47
500
Пептидилэстеразная активность 294
Переаминирование 124*, 266, 276
Пероксидаза 393*, 412*
Пиридинпротеин 118, 119
Пиридоксаль 114, 133, 164*
Пиридоксальфосфат 124*, 126, 133, 147, 164*,
266, 267, 334*
Пиридоксамин 164*
Пиридоксаминофермент 126*
Пиридоксальфермент 114, 124
Пиридоксол 164*
Пиримидин-аминогидролаза 233
Пиримидиновы основания 170, 190*—191, 228
----распад 232*, 233*—235
----обмен 235*—240*
Пировиноградная кислота 100*, 133*, 137*,
161, 174, 187, 276, 340, 344*, 350*, 355*, 468,
470, 471
Пироглутаминовая кислота 264*
Пирокатехаза 418*
Пирокатехин 418*
Пирофосфат 247*, 251*, 272, 281, 365*, 390*,
404*, 137*
Пирофосфатаза 391, 404*
Пирофосфомевалонат- декарбоксилаза 403*
Пнрофосфомевалоновая кислота 403 *
Пируватдегцдрогеназный комплекс 100*, 352*,
354*
Пируватдекарбоксилаза 133*, 351*—353*
Пируваткарбоксилаза 136
Пируваткиназа 347*
Пируватсинтаза 364*
Плазми 140
Пластеины 278, 279
Пластохинон 121*
Полиамины 211
Полиаденилатный фрагмент 222, 258
Полиизопренилдифосфатсахара 368*
Полиизопренилмонофосфатсахара 368*
Полинуклеотидфосфорилаза 230
Полисахариды 314, 317
Полиферментный комплекс 100*, 353*
Полифункциональные ферменты 396*, 397*,
398*
Порины 89
Полиуридиловая кислота 221, 297
Правила Чаргаффа 200—202
Праймосома 254*
Праймер 249, 254*, 256, 257
Праймаза 254*
Препрогормон 458
Прогестерон 446*, 447
Про ормон 459
Проламины 80
Пролин 41 ♦, 42*, 269*, 294*, 295*
Проколлаген 11
Промотор 257*
Проопиокортин 459
Пропионил-КоА 392*, 395*
Пропионил-КоА-карбоксилаза 395
Простагландины 159, 415, 463*
Простетическая группа 98
Протамины 81, 221
Протеазы 114
Протеинкиназы 113, 124, 282, 292, 388*, 456,
474, 475*
ротеиназы ИЗ, 130, 139, 262
Протеиноиды 80
Протомер 100
Протромбин 158
Процессинг 258
Псевдоглобулин 81
Псевдоферомоны 176
Пвсевдоуридиловая петля тРНК 214, 215*
Псевдоуридин 213*
Птеридин 167*
Птероевая кислота 168*
Птероилглутаминовая кислота 167*
Пурин-аминогидролаза 233
Пуриновые основания 169, 189, 191*
----распад 232*—235
----обмен 240—245
Пуромицин 175
Путресцин 268*
Рацематы 307
Рацемизация 142*
Ревертаза 255, 256
Регуляторный G-белок 457, 458*
Редуктазы 117, 414
Репарация 231, 254, 255
Репликативная вилка 254*, 255
Репликация 175, 206, 249, 254*, 255
Ретикулин 90
Ретиналь 151*, 152, 418
Ретинол 151*
Ретин олгидрогеназа 152*
Ретинол-изомераза 135
Ретинолсвязывающий белок 152
РестриКтазы 63, 225*, 226, 227
Рибит 162, 313
Рибоза 192, 236, 238, 244, 253*, 307*, 338* .
P-D-Рибоза 193*
Рибозилтрансферазы 231
Рибозимы 97, 258*, 259*
Рибозо-1-фосфат 231 *, 232
Рибозо-5-фосфат 237*, 240, 243, 348*, 359*
Р, П-Рибозо-5-фосфат 338*, 339*
Рибозофосфатизомераза 359
Рибокиназа 338*
Рибонуклеаза 24, 70*. 82*. 97,99*, 141, 213,224,
226, 228* 229*
Рибонуклеиновые кислоты (РНК) 194
----биосинтез 256, 257*, 258*, 446
----вирусные 196
----информационные (иРНК) 196, 220, 221*,
222, 258, 259
----молекулярная масса 194*, 195
----нуклеотидный состав 211*, 215, 221, 222
----регуляция биосинтеза 258, 259*
----рибосомальные (рРНК) 195, 218, 220*,
259*
----содержание и локализация в клетке 195
----транспортные (тРНК) 195, 215*, 216*—
220*, 255, 258, 437
Рибонуклеотиды 196
Рибосома 213, 220, 221, 268, 279, 285
— состав 286*
— строение и функции 220*, 285, 287*, 288*,
289*
— транслирующая 288*, 292
Рибофлавин 161*
501
Рибофлавинфосфат 119*, 147, 237, 422*, 423*
a-D-Рибофураноза 309*
P-D-Рибофураноза 309*
D-Рибулоза 308*
Рибулозо-1, 5-дифосфат 359*, 360*
Рибулозодифосфаткарбоксилаза (РДФК) 360,
361 ♦
Рилизинг-факторы 113, 131, 177, 263, 459
Рибулозо-5-фосфат 312*, 339*, 355*, 359*
РНК-зависимая ДНК-полимераза 255, 256
РНК-метилазы 258
РНК-полимераза 100*, 246*, 257*, 437
РНК-предшественники 258
Рифамицин 175
Родопсин 63, 92, 152, 153
Рутин 172*, 173*
Салицилаттидроксилаза 419
Сахараза 130
Сахароза 105, 128*. 130*, 142, 145, 182*
315, 364
Сахарозо-6-глюкозилтрансфераза 128*
Сахарозосинтаза 364*, 365*
Сахарозо-6-фосфат 365*
Сахарозофосфорилаза 128
Секвенатор твердоф ньй 56*, 58
Серин 40*, 42*, 112*, 169, 267, 334*, 341, 381,
383*, 410
L-Серин-гидролиаза 134
Серотонин 268
Сефадекс 141, 325
Сефароза 141, 190, 221
Сигналопептидаз 300
Синигрин 171*
Синтазы 134
Сш гтав высших жирных кислот 296, 40б,
397*, 398
Синтетазы 136
Ситостерол 379*
Сквален 404*, 446*
Сквален-2, 3-оксид 404*
Сквален-оксид-циклаза 404*
Сквален-синтаза 404*
Сквален-эпоксидаза 404*, 419
Соленоид 111,212*
Соматомедины 455
Соматосз гин 50*
Соматотропный гормОи (СТГ) 455
D-Сорбит 312*, 313
Сплайсинг 259*
Сплайсосома 259
Спермацет 377*
tie мид! 211, 268
Спермин 211, 268
Спирть высшие 177*
Статины 459
Стеарил-КоА 390*
Стеариновая кислота 375
Стекинг-взаимодействия 209
Стелластеролы 379
Стеран 378*
Стериды 340, 378*, 380*, 401*, 405*
Стероиды 380*, 443
Стеролы 154, 378 * 380, 401 *, 402*, 443
Стигмастерол 379*
Стрептокиназа 140
Стрептомицин 175
Субтилизин 131
Сукцинат дегидрогеназа 355*, 356*
Сукцинил-КоА 355*, 395*
Сукцинаттиокнназа 355*, 356
Сульфамидазы 327
Сульфоэстераза 372
Сфингозин 381*, 384, 385*
Сфингофосфолипиды 384*, 385
Таурин 270
Таутомерные превращения 201, 308*, 309*, 310
Телергоны 176
Тестостерон 380*, 444
Тиамин 133*, 443*, 444, 447*
Тйаминпирофосфат 133, 160*, 348*, 349*,
350, 351
Тиаминпирофосфаткиназа 160
Тиаминтрифосфат 160*, 161 -
Тимидилат-синтаза 239
Тимидинтрифосфат (ТТФ) 239
Тймин 159, 169, 191*, 200*
Тйогюкозидаза 172*
Тиогликозиды 172
Тнолаза 391 ♦
Тиолиз 391
оредоксин 239
Тиоредоксинредуктаза 239
Тиреоглоб ш 76, 462
Тиреоидпероксидаза 462
Тиреотропин 454, 459, 462
L-Тирозин 143, 269*, 271, 286* 341, 460*
Тирозингидроксилаза 419
Тирозин-фенол-лиаза 143
Тйроксин 76, 461 *, 462*, 463
Тиролиберин 50*, 459
Тйроцидин 294, 295*
а-Токоферилхинон 156*
Токоферол 156*
Топоизомеразы 211
Токсины 88, 306
Трансаминазы 266
Трансацилаза 00
Трансгидрогеназа 119
Трансгликозилирование 364, 366
Трансгликозидазы 130, 237, 366
Транскетолаза 348, 349*
Транскриптон 256
Транскрипция 157, 175, 206, 256, 280, 465
Транслоказы 89*, 424, 479
Трансляция 222, 290, 292, 293*, 300, 457
Трансметилирование 407*, 408
Транспептидированне 279, 293*
Транс-ретиналь 154*
Транс-ретинол 135, 154*
Трансдуцин 154
Транссукцинилаза 355*, 356
Трансферазы 117, 123, 142, 147
Трансформация энергии 188*
Трегалоза 115*, 314*, 365
Трегалозо-1-глюкогидролаза 115
Треонинальдолаза 271
Треонин 9, 40*. 42*, 144, 267, 271*
Триглицериды 372, 375*, 376*
— биосинтез 399*, 400*
— распад 388*
502
Трииодтиройин 461 *, 462
язомера а 82*, 134, 343
Триолеин 375*
Трипальмитин 375*
Трипсин 131, 140
Триптофан 9, 41*, 42*, 143, 144, 164, 271,
276. 341
Триптофаноксигеназа 418, 419
Триптофансинтаза 143
Тубулин 91
Убиквитин 262
Убихиноны 120*, 123, 157, 158
Убихинонпротеин 120, 421*, 422, 423*
Углеводы 306
— биосинтез 357*, 358, 363*, 364*, 366*
— изомерия оптическая 307, 309
— конформация 310*, 311
— обмен 328
— простые 307*—313
— пути распада 328, 336, 341, 342*, 347, 350*,
355*, 357*
— сложные 312, 314—327
— таутомерия 308*, 309*, 310*, 315
Углерод-азот-лиазы 134
Углерод-кислОрод-лиазы 133
Углерод-углерод-лиазы 133
Уггьтрацентрифугирование 36, 37*, 190
Уратоксидаза 140*, 234*
Урацил 191*, 192*, 200*, 230, 231*, 234, 236
Урацил-ДНК-гликозидазы 230
Уреаза 33, 98, 111, 114, 115, 131, 145
Уридиловая кислота 198, 259
Уридин 231*
Уридиндифосфат 239*, 367*
Уридиндифосфатглюкоза 182*, 339*, 364*,
366*, 367*
Уридинмонофосфат 196*, 238*
Уридин-5'-фосфат 237*, 238*
Уридинфосфорилаза 231*
Уридинтрифосфат 188*, 239*
Уриколиз 234
Урокиназа 140
Фаллоидин 50*
Фарнезилпирофосф т 403*, 404*
Феназин етосу >фат 146*
Фенилаланин 41, 42*, 140, 144, 269*, 276, 294*,
296*, 297, 341
L-Фенилаланин-аммиак-лиаза 140
Фенилаланингидроксилаза 419
Фенолаза 419
Фенолгидроксилаза 419
Фенолоксидаза 414*
Фенолхиноновая система 414*
Ферменты 83, 95
— единица активности 107
— ингибирование активности 93, 110
— индуцибельные 476
— кинетика действия 105
— классификация 116, 117
— локализация в клетке 138
— методы выделения и очистки 97
— механизм действия 102—104
— молекулярная активность 107
— номенклатура 114, 115
— применение 139
— регуляция активности 74
----аденилирование 478*
----АДФ-рибозилирование 478, 479*
----аллостерическая 74, 99, 294, 302*, 335*,
397, 456
----протеолитичес ая деградация 263, 479
----уридилирование 478*
----фосфорилирование- дефосфорилирова-
ние 335*, 336*, 388*, 397
— репрессибельные 477
— свойства 107-
— специфичность 109, 111
— иммобилизованные 141*, 142*—145*
— термолабильность 107, 108
— удельна а тивность 107
Феромоны 176
Ф рредоксин 24, 97, 364*
Фибриноген 158
Фиброин шелка 65*, 68, 221
Филлохинон 157*
Фитосфингозин 385*
Фицин 131
Флавинадениндинуклеотид (ФАД) 120*, 147,
171, 354*, 391 *, 407*, 414, 415*, 422*
Флавинмононуклеотид (ФМН) 119*, 147, 236,
422*, 423*
Флаводоксин 82*
Флавопротеины 119, 120, 233,-266, 414, 421*,
422*, 423*
Фолиевая кислота 167*
Формилметионин 290*
N-Формилтетрагидрофолиев я кислота 169*,
241
Формилми онил-тРНК 169*, 290, 291*—293
Фосфатаза 141
Фосфатаза щелочная 141
— кислая 141
Фосфатазы 129,400*
Фосфатидатфосфогидролаза 400*, 405*
Фосфатидилглицерин 383*, 405
Фосфатидилинозит-4, 5-дифосфат 474*
Фосфатидилтреонин 382*, 383
Фосфатидилхолин 382, 383*, 405, 410
Фосфатидилэталонамин 382*, 410
Фосфатидная кислота 382*, 399*, 405*
Фосфатиды 381, 382, 383*
— гидролиз фосфолипазой А2 407*
— механиз биосинтеза 408, 409*
— обмен 405
— пути распада 406*
Фосфоглицераткиназа 347*
Фосфоглицерат-фосфомутаза 135, 344*
а-Фосфоглицерин 405*
2-Фосфоглицериновая кислота 135*, 341*,
344*, 347*, 419, 438, 468
З-Фосфоглицериновая кислота 135*, 188*,
341*, 347*, 359*, 361*, 438
З-Фосфоглицериновый альдегид 134*, 343*,
344*, 361*, 390, 419
Фосфоглюкомутаза 339, 340*
Фосфоглюконатдегидрогеназа 348*
6-Фосфоглюконовая кислота 348*, 350*
6-Фосфоглюконолактон 347*, 348*
6-Фосфоглюконол ктоназа 348*
Фосфодиоксиацетон 134*, 343*, 344
503
Фосфодиэстеразы циклических нуклеотидов 457*
2-Фосфоенолпировиноградная кислота 187,
341*, 344, 363*, 419, 438
Фосфоенолпируваткарбоксилаза 363*
Фосфоинозитиды 382*, 384*, 405*, 446, 473*,
475*
Фосфолипаза А 406*, 407*, 473
Фосфолипаза С 406*, 473, 474*
Фосфолипиды 170, 381, 382*
Фосфомевалонаткиназа 402*
Фосфомоноэстераза 405*
Фосфопротеинфосфатазы 281
5-Фосфорибозиламин 240*, 241 *
5-Фосфорибозилглицинамид 241 *
Фосфорибозилглицинамид-синтаза 241 *
Фосфорибозилг лицинам ид-формилтрансфера-
за 241 *
5-Фосфорибозил-1-пирофосфат 237, 240*, 245*,
469
Фосфорибозилтрансферазы 245
Фосфорилаза а 334, 335*, 336*, 460
Фосфорилаза b 334, 335*, 336*, 460
Фосфорилирование 312, 335*—337*, 388*, 403
— окислительное 417, 420*, 421*—425*, 438,
468
— субстратное 347*, 355*, 416, 420*, 469, 346*
Фосфоролиз 128, 230, 231, 328, 333*
Фосфорибулокиназа 359*
Фосфосерин 334*, 339*
Фосфотрансферазы 123, 273, 389*, 402
Фосфофруктокиназа 341 ♦, 342*
Фосфохолин 405*, 408*, 438
Фрагменты Оказаки 250, 254*, 255*
Фруктоза 145, 310*, 313*, 364*, 308*
Фруктозо-1, 6-дифосфат 312*, 338*, 341*
Фруктозо-2, 6-дифосфат 342*, 362, 133*
Фруктозо-1, 6-дифосфат-альдолаза 140
Фруктозо-1,6-дифосфатаза 342*, 362
Фруктозо-2, 6-дифосфатаза 342*
Фруктозо-6-фосфат 341*, 362
Фруктозаны 313
₽-Фруктофуранозидаза 142, 364
Фукостерол 379*
Фумараза 355*, 356*
Фумарат-гидратаза 133*, 356*
Фумаровая кислота 134*, 144, 274*, 275, 355*
Хеликаза 254*
Хемиосмотическая гипотеза Митчела 424*
Хемосинтез 358*
Химотрипсин 131, 262*, 263*
Химотрипсиноген 64*, 99*, 131, 140, 72*
Химические посредники 442*
Хинонредуктаза 415*
Хиноны 120, 415
Хитин 317, 325*, 332
Хитиназа 332
Хлорокруорины 89
Хлорамфеникол 175
Холевая кислота 270, 279, 401
Холекальциферол 154*
Холестан 378*
Холестанол 378*, 380*, 400*
Холестерол 154, 379*, 400*, 405*, 423, 446*
Холестерол-ацилтрансфераза 405
Холестеролэстераза 400*
Холин 102*, 128*, 167,169, 382, 383*, 405*, 407*
Холин-ацетилтрансфераза 128*, 407*
Холиндегидрогеназа 407
Холинкиназа 409*
Холинорецепторные белки 91, 92, 93
Холинфосфотрансфераза 409*
Холинфосфат 409*
Холофермент 147
Хондроитинсульфат 317, 326*
Хроматин 84, 85, 211, 212*, 445*
Хромосомы 210, 211*, 212*
Хромогены дыхательные 412, 413*
ЦДФ-диглицеридсинтаза 410
Целлобиоза 316*, 324*
Целлюлазный комплекс ферментов 332
Целлюлоза 141, 323, 324*, 332, 366
Центр аллостерический 99*
— ка 1лит ческг 98*
— субстратный 99
Цереброзиды 385*
Цереброн 385*
Церулоплазмин 89
Цианокобаламин 165*
Цикл глиоксилевый 362, 392, 394*
— у-глутамилтрансферазный 264*
— орнитиновый 273*
— пентозофосфатный 348, 349*
— трикарбоновых и дикарбоновых кислот
(Кребса) 341, 355*
D-Циклосерин 176*
Циклоспорин 294, 296
Цинковый палец 256*
Цис-ретиналь 152, 153*
Цис-ретинол 135, 153*
Цистеамин 97*
Цистеин 40*, 42*, 97, 134*, 267, 269, 270*,
293, 346*
Цистеиновая кислота 270*
Цистеинредуктаза 270*
Цистин 270
Цистрон 259*
Цитидицдифосфатидилглицерин 410*
Цитидиндифосфатхолин 147, 408*, 409*
Цитидинмонофосфат 196*, 238*, 408, 409*
Цитидинтрифосфат 188, 238*, 408*, 409*
Цитозин 191* 200*, 221, 230, 232, 234
Цитозин-аминогидролаза 233*
Цитохромная система 121, 122, 139, 175, 413*
Цитохромоксидаза 90, 123, 413*, 417, 423*,
424*, 438
Цитохромы 64,90,114, 140,171,413*, 421 *, 423*
Цитохром Ь5 71*, 417
— с 24, 62*, 76, 97, 121*, 122*, 421*, 423*
— Р-450 417
Цитраль 176*
Цитрат-синтаза 354*, 355*
Цитрил-КоА 355*, 394*
Цитруллин 274*
Щавелевоуксусная кислота 126, 137*, 174, 275*,
355*, 362, 395, 468*, 470
Экдизон 9, 446, 466*
Экзонуклеазы 224
Экзопептидазы 130
504
Экзорибонуклеазы 230
Экспрессия генов 465
Эластаза 114*, 140
Электрофорез 97, 190, 195, 203
Эндонуклеазы 222, 224, 226
Эндопептидазы 130
Эндорибон леазы 230
Эндорфины 8, 459
Энергетический баланс углеводов и тригли-
церидов 428—430
Энергия, превращения в живой природе 181
— свободная 181
Энкефалины 8, 50*, 459
Энхансер 259*
Эпимолекулы 76
Эргостерол 154*, 379*
Эритромицин 175
Эстрадиол 444*, 445*, 446*
Эстеразы 129, 387*, 405
Этаноламин 382, 383 *
Ювеноиды 466
^блочная кислота 144, 355*, 395*
Янтарная кислота 114*, 356*, 395*, 419*, 420*
ОГЛАВЛЕНИЕ
Предисловие..................................................................... 3
Введение........................................................................ 5
Глава I. Химический состав организмов........................................... 15
Глава II. Белки................................................................. 23
Элементарный состав белков...................................................... 23
Выделение белков................................................................ 24
Очистка белков.................................................................. 32
Оценка гомогенности белка....................................................... 34
Молекулярная масса белков....................................................... 34
Форма белковых молекул.......................................................... 37
Аминокислотный состав белков.................................................... 38
Пептиды......................................................................... 46
Структура белковой молекулы..................................................... 49
Свойства белков................................................................. 78
Номенклатура и классификация белков............................................. 80
Характеристика отдельных групп белков........................................... 83
Глава III. Ферменты............................................................. 95
Общее понятие о ферментах....................................................... 95
Методы выделения и очистки ферментов....................................'...... 97
Строение ферментов........................................................... 98
Механизм действия ферментов................................................... 102
Кинетика ферментативных реакций................................. 105
Свойства ферментов ........................................................... 107
Номенклатура ферментов....................................................... 114
Классификация ферментов и характеристика некоторых групп...................... 116
Локализация ферментов в клетке................................................ 138
Применение ферментов.......................................................... 139
Глава IV. Коферменты, витамины и некоторые другие биоактивные соединения...... 147
Коферменты.................................................................... 147
Витамины..................................................................... 148
Другие биоактивные соединения................................................. 174
Гл а ва V. Общее понятие об обмене веществ и энергии в организме.............. 178
Глава VI. Нуклеиновые кислоты и их обмен...................................... 189
Строение и свойства нуклеиновых кислот........................................ 189
Обмен нуклеиновых кислот...................................................... 223
Биосинтез ДНК и РНК....................................................... 246
Глава VII. Обмен белков....................................................... 261
Значение белкового обмена..................................................... 261
Распад белков и аминокислот................................................... 262
506
Биосинтез белков................................................................ 278
Перенос новообразованных белков через мембраны.................................. 299
Посттрансляционная модификация белков........................................... 300
Регуляция белкового синтеза..................................................... 301
Биосинтез белков и незаменимых а инокислот для практических целей............... 303
Глава VIII. Углеводы и их обмеи................................................ 306
Строение углеводов............................................................. 306
Обмен углеводов................................................................ 328
Глава IX. Липвды и их обмеи................................................. 370
Общая характеристика и классификация липидов................................. 370
Простые липиды............................................................... 372
Сложные липиды............................................................... 381
Обмен липидов................................................................ 387
Глава X. Биологическое окисление................................................ 411
История развития представлений о механизмах реакций биологического окисл ния.... 411
Классификация процессов биологического окисления и их локализация в клетке....... 416
Свободное окисление.............................................................. 417
Окисление, сопряженное с фосфорилированием АДФ................................... 419
Энергетический баланс распада углеводов и триглицеридов.......................... 428
Глава XI. Водный и минеральный обмен.......................................... 431
Водный обмен.................................................................. 431
Минеральный обмен............................................................. 435
Глава XII. 1Ърмоны н их роль в обмене веществ................................. 441
Общее понятие о гормонах...................................................... 441
Номенклатура и классификация гормонов......................................... 441
Стероидные гормоны............................................................ 443
Пептидные гормоны............................................................. 448
Прочие гормоны................................................................ 460
Применение гормонов........................................................... 465
Глава XIII. Взаимосвязь н регуляция обмена веществ............................ 468
Взаимосвязь процессов обмена веществ.......................................... 468
Регуляция обмена веществ...................................................... 472
Глава XIV. Квантово-мехаиическне аспекты биохимии............................. 482
Рекомендуемая литература.................................................... 485
Предметный указатель.......................................................... 494
Учебное издание
Юрий Борисович ФИЛИППОВИЧ
ОСНОВЫ БИОХИМИИ
Лицензия ЛР № 064411 от 22.01.96
Сдано в набор 20.03.97 г. Подписано в печать 17.08.99 г. Формат 70х ЮО1/^. Бумага
газетная. Гарнитура ’’Таймс”. Печать высокая. Уел. печ. л. 41,67. Уч.-изд. л. 36,85.
Тираж 5000 экз. Заказ № 3502.
Издательская фирма ’’Агар”
103045, Москва, Костянский пер., д. 6
Тел./Факс (095) 956-76-15
232-29-90
Набрано и отпечатано в Государственном ордена Октябрьской Революции, ордена
Трудового Красного Знамени Московском предприятии ’’Первая Образцовая
типография” Государственного комитета Российской Федерации по печати.
113054, Москва, Валовая, 28
с. 51, нижняя строка
с. 58, рис. 27
с. 160, формула
тиаминпи рофосфата
с. 220, конец 2-го абзаца
с. 255, подпись к рис. 85,
3 строка снизу
с. 270, 2-ое уравнение
реакции
с. 322, конец 2-го абзаца
с. 414,2-ой абз., 6 стр.
с. 415, 5 строка снизу
с. 426, 11 строка снизу
с. 489, Часть VII, обмен
белков, Высоцкая и др.
Суперобложка
(об авторе, 15 строка
снизу)
ОПЕЧАТКИ
Неправильно
(см. с. 53)
Отсутствует формула де-
каноильной группировки
ОН
—р—ОН
и
он
EF — G — связывающими
фосфори диэфи рн ых
АТФ г
VL-метионин-
(рис. 103, в)
(см. с. 120)
аденазинпирофосфорной
(прогонная (АТФаза)
Escherichia crei
и Х(ФРГ) энтомологи-
ческого конгресса
Правильно
(см. с. 54)
деканоильная
[СН3-(СН2)8-СгО|
группировка
ОН
---Р---ОН
О
EF - G — связывающими
фосфодиэфирных
АТФ г
L-метионин-
(рис. 103, В)
(см. с. 119—120)
аденози нпирофосфорной
(протонная АТФаза)
Escherichia coli
и Х(ФРГ) Международ-
ных биохимических кон-
грессов; XIV (Австралия)
Международного энто-
мологического конгрес-
са
РАЗМЕР ГАПЛОИДНОГО ГЕНОМА
Размер пшлоядноге гевома (число нуклеотидных пар)
"... отваляло костввыяи, в касяшящее время фвяяикмшческую задачу —
определять явмую аервачяую ааруюяуру геяама чемвека " (гл.6).
БЛОЧНАЯ СТРУКТУРА МИТОХОНДРИАЛЬНОЙ ЦЕПИ
ВНУТРИКЛЕТОЧНАЯ ЛОКАЛИЗАЦИЯ ГЛАВНЫХ МЕТАБОЛИЧЕСКИХ ПУТЕЙ
Ядро
Репликация ДНК;
Синтез тРНК, мРНК
Микротельца
Окисление аминокислот;
деградация стеролов;
реакция при участии
каталазы и пероксидазы
и некоторых
ядерных белков
Хлоропласты
(растения)
Фотосштез
Ядрышко
Синтез рРНК
Рибосомы
Синтез белка
Плазматическая
мембрана
Энергозависимые
транспортные
системы
ретикулум
Эндоплазматический
Комплекс Гольджи
Созревание
гликопротеинов и
других компонентов
мембран и
секреторных
гранул
Свободное окисление,
синтез липидов,
транспорт веществ
Гранулы гликогена
Синтез и распад-
гзшкогена
Цитозоль
Дихотомический
и анатомический
распад углеводов;
глюконеогенез;
ВЖК; активирование
аминокислот и
синтез нуклеотидов
Митохондрии
ПТДК;
окислительное
фосфорили-
рование;
окисление
ВЖКвПВК
катаболизм
аминокислот
Липосомы
Гидролиз
при участии
рибонуклеаз,
фосфатаз и др.
" С современной точки зрения клетка представляется
высокоорганизованной системой, в отдельных частях
которой осуществляется строго определенные
биохимические процессы ’’ (гл. 3).
о о Возраст (млн лет)
Губки
CS-- Кишечнополостные
Круглее черви
Плоские черви
Брюхоногие моллюски
Двустворчатые моллюски
Головоногие моллюски
Кольчатые черви
Паукообразные
Ракообразные
Насекомые и многоножки
Иглокожие
Щетннкочелюстные
Оболочники
Бесчерепные
Бесчелостные
Хрящевые рыбы
Костные рыбы
Амфибии
Рептилии
Птицы
Млекопигаюише
ЭНДОКРИННЫЕ ОРГАНЫ ЦАРСТВА ЖИВОТНЫХ
Мезозой Цеяозой
ОБ АВТОРЕ
Ю. Б. Филиппович — заведующий кафедрой
органической и биологической химии Москов-
ского педагогического государственного уни-
верситета, доктор биологических наук, по-
четный профессор, заслуженный деятель на-
уки Российской Федерации — широко известен
в нашей стране как выдающийся ученый и пе-
дагог. Им лично и в ряде случаев совместно с
сотрудниками кафедры создан комплект учеб-
ников и учебных пособий, в полной мере обеспе-
чивающих подготовку по биологической химии
студентов, обучающихся по направлению хи-
мия и(или) биология. Комплект состоит из
взаимосвязанных системой ссылок пяти руко-
водств общим объемом 92,6 печл. (учебник,
практикум, задачник, спецкурс, сводные во-
просы для самоконтроля) и огранично вписыва-
ется в современную триаду (бакалавриат —
магистратура — аспирантура), повышая мо-
тивацию к обучению и гарантируя самостоя-
тельность студентов в выборе литературы
для дальнейшего углубленного изучения молеку-
лярных основ жизни. Постоянно совершенст-
вуясь в процессе переизданий, комплект результативно и эффективно используется в практике
высшей педагогической школы в течение трех десятилетий (начиная с выхода в свет его учебника
’’Основы биохимии ” в 1969 г.). На посту председателя Научно-методического совета по химии
при Управлении учебных заведений Министерства высшего и среднего образования (1969—1991) им
осуществлена экспериментально обоснованная работа по совершенствованию учебных планов фа-
культетов и отделений, готовящих учителей химии и биологии.
Основываясь на высочайшем научном потенциале (опубликовано 6учебников, 5монографий, 467
статей в научных журналах, получено 12 авторских свидетельств, подготовлено 3 доктора и 46
кандидатов наук), им разработана и реализована концепция о взаимопроникновении и
взаимообусловленности химии и биологии как в области фундаментальных наук, так и
химических и биологических дисциплин в средней и высшей школе, внедрены рейтинговая система
изучения курса биохимии и прогрессивный вариант студенческих экзаменов в форме научного
доклада. Ю. Б. Филиппович — участник V (СССР), VII (Япония), VIII (Швейцария), IX (Швеция)
и X (ФРГ) энтомологического конгресса; VI, VII, VIII, IX, X, XI и XV конференций Федерации
Европейских Биохимических Обществ; спецсимпозиумов по ферментам (Югославия) и
дифференцировке и функциям клеток (Греция) и др. На базе накопленных научных данных по его
инициативе в 1968 г. открыта первая в России научно-иследовательская лаборатория биохимии
насекомых, пользующаяся международным признанием.
За плодотворную деятельность в сфере народного образования Ю. Б. Филиппович награжден
значком ’’Отличник народного просвещения РСФСР” (1966 г.), значком "Отличник просвещения
СССР) (1979г.) и орденом ’’Знак почета”(1978г.). Активный участник войны 1941—1945гг.,
кавалер двух орденов ’’Отечественной войны I степени ” и ряда медалей. Трижды лауреат премий
за лучшую научную работу университета (1986, 1987 и 1992 гг.). Удостоен премии
Правительства Российской Федерации (1999 г.) как автор уникального учебника "Основы
биохимии” (3-е издание, М., "Высшая школа”, 1993 г., 40 п.л., 141 рис., 14 схем, 27 таблиц,
двухцветная печать, более 200 наименований рекомендуемой литературы) и за огромный вклад в
разработку образовательных программ, учебников и учебно-методических пособий,
педагогическое мастерство и высокие результаты педагогической деятельности.