






















































































































































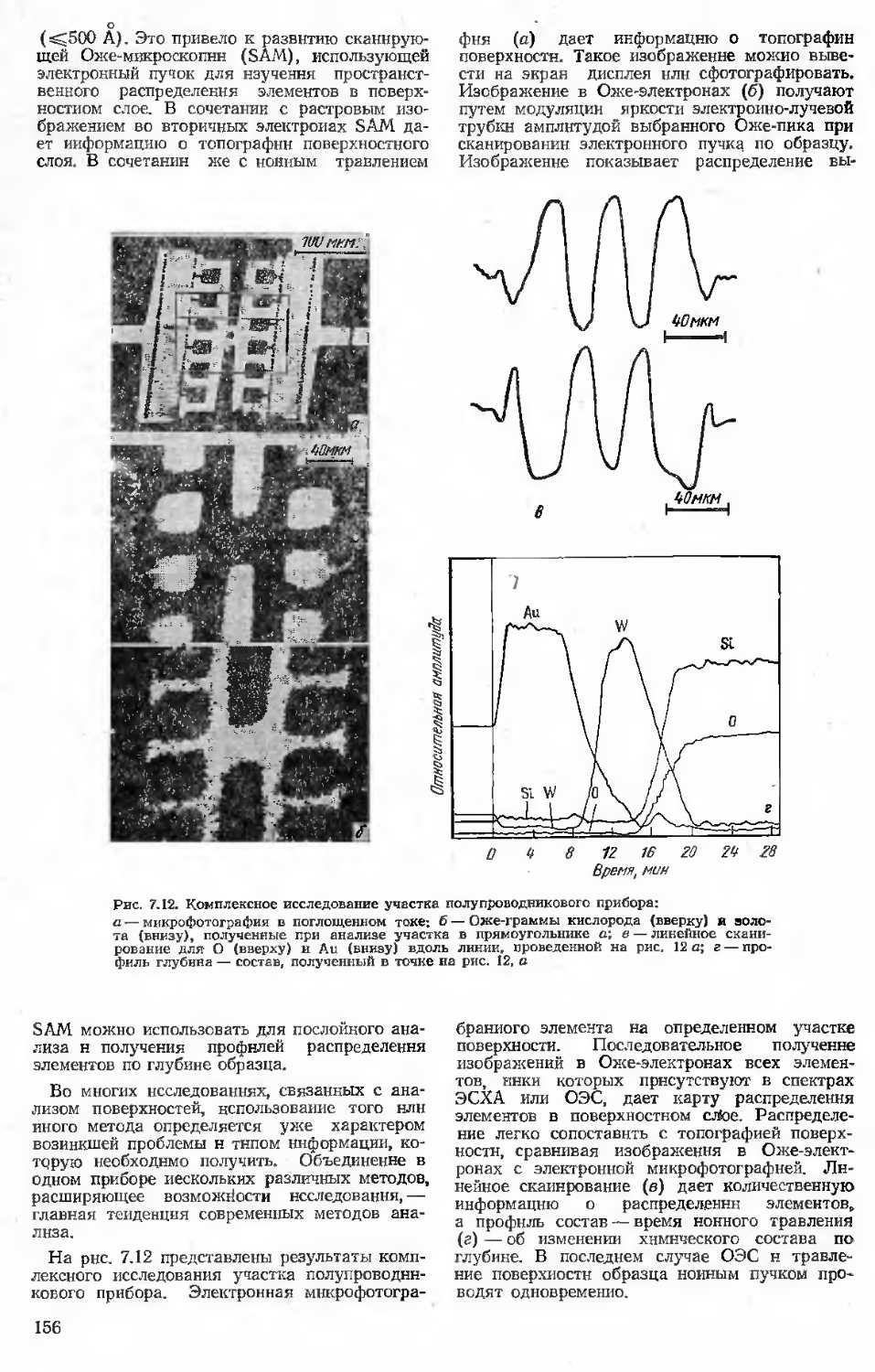




































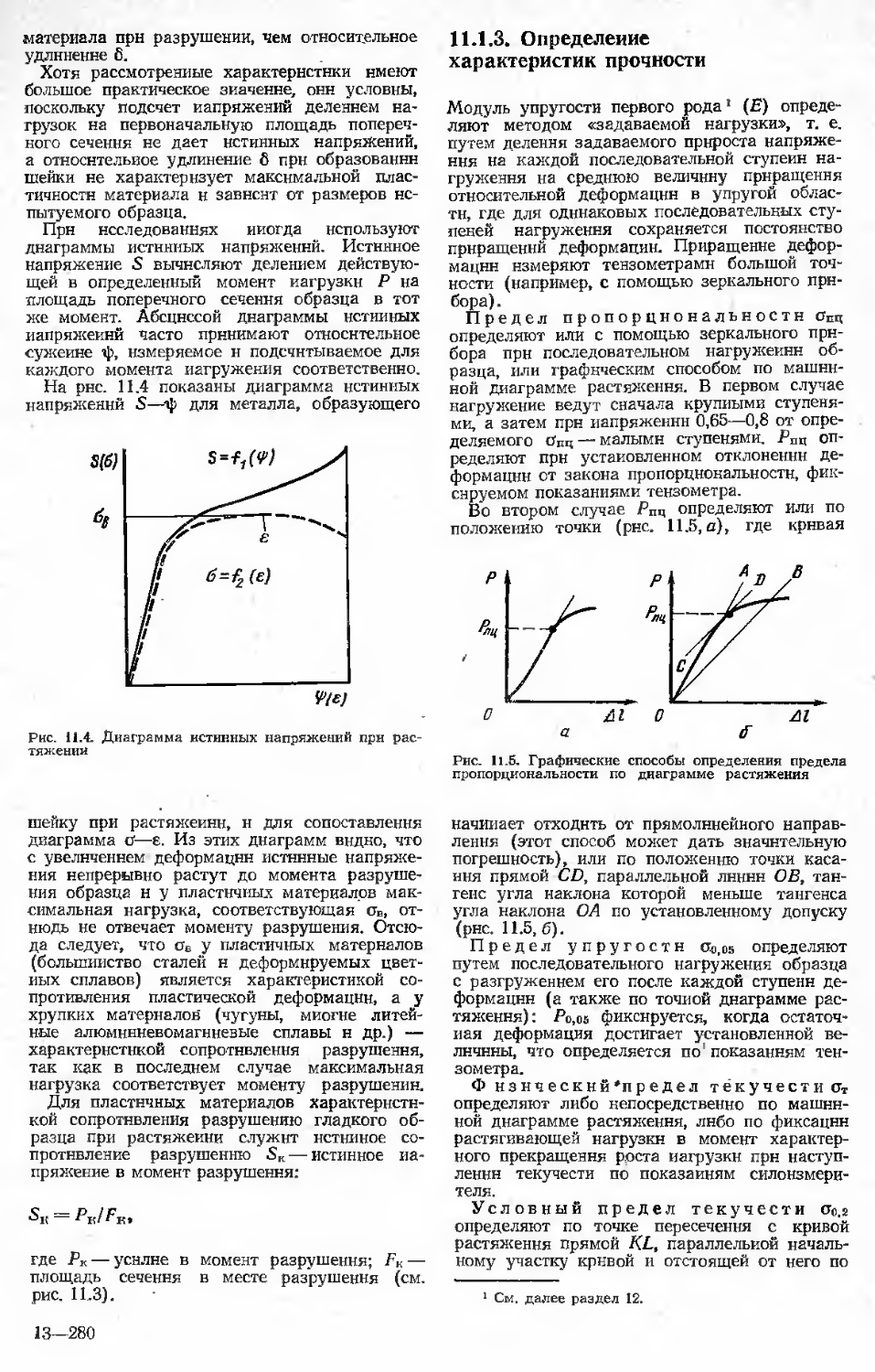





























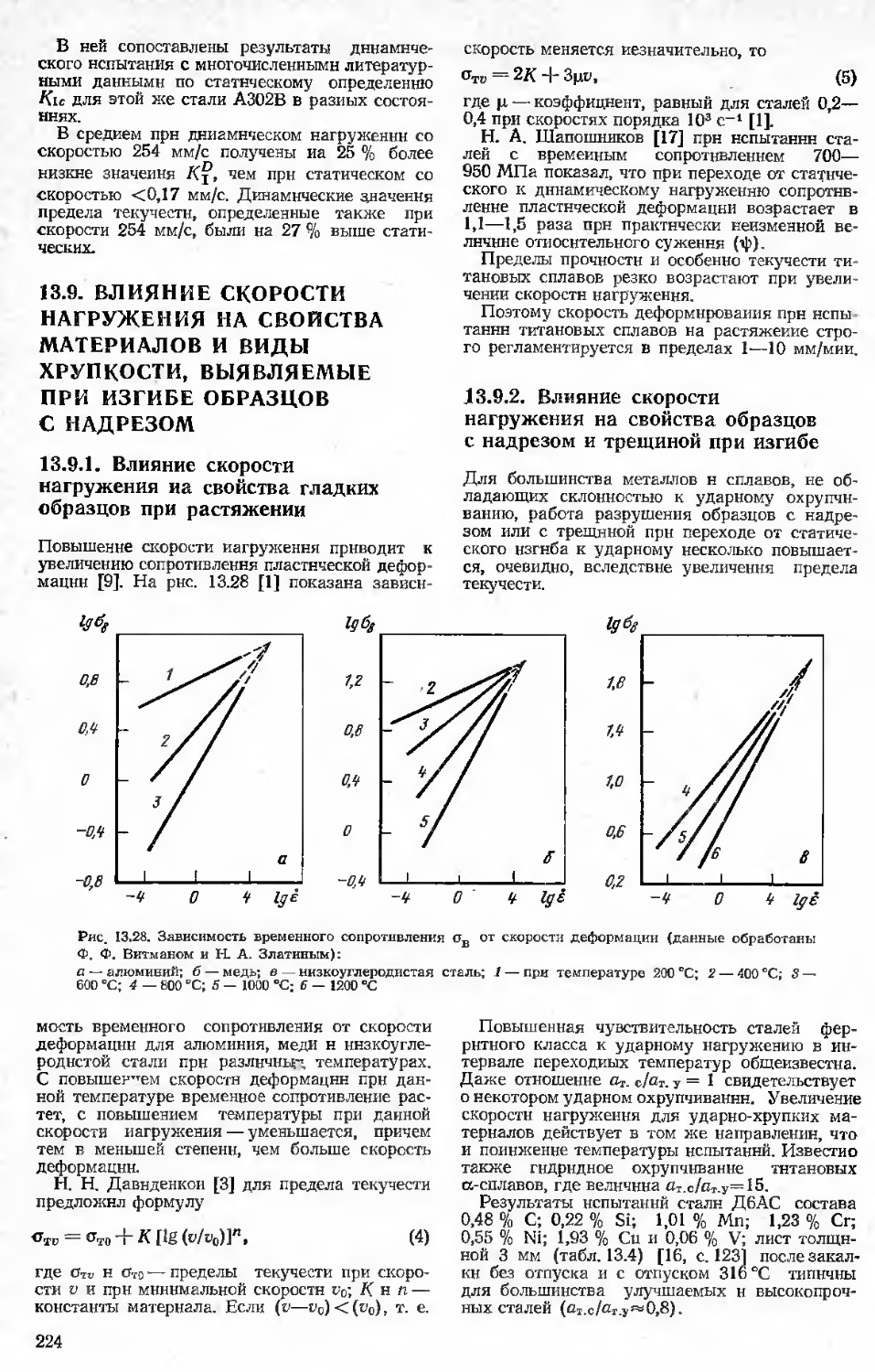



























































































































Author: Бернштейн М.Л.
Tags: металлургия черных металлов железо, чугун и сталь испытания материалов товароведение силовые станции общая энергетика свойства и структура молекулярных систем технология обработки без снятия стружки в целом: процессы, инструмент, оборудование и приспособления обработка стали
Year: 1983




МЕТАЛЛОВЕДЕНИЕ И ТЕРМИЧЕСКАЯ ОБРАБОТКА СТАЛИ
иими
В ТРЕХ ТОМАХ
Издание третье, переработанное и дополненное
Под редакцией
М.Л.Бернштейна и А.Г.Рахштадта
ТОМ I
Методы испытаний и исследования
МОСКВА ’’МЕТАЛЛУРГИЯ” 1983
УДК 669.14:620.18+539.2+621.78(083)
Авторы первого тома
Докт. физ.-мат. наук Б. С. БОКШТЕИН, докт. техн, наук Ю. Г. ВЕКСЛЕР, докт. техн, наук М. И. ВИНОГРАД, канд. техн, наук Б. А. ДРОЗДОВСКИЙ, инж. Г. М. КАЗИЧ-КИНА, канд. техн, наук Б. А. КЛЫПИН, инж. В. Г. КОСТОГОНОВ, докт. техн, наук Б. Г. ЛИВШИЦ, канд. физ.-мат. наук А. С. ЛИЛЕЕВ, канд. физ.-мат. наук Ю. Я. ЛИ-ЛЕЕВА, канд. техн, наук Л. Г. КОРШУНОВ, ннж. Е. Е. ПОПОВА, докт. техн, наук А. Г. РАХШТАДТ, докт. техн, наук О. Н. РОМАНИВ, докт. техн, наук 10. А. СКА-КОВ, канд. техн, наук М. П. УСИКОВ, докт. техн, наук \л, М, УТЕВСКИИ\, докт. техн, наук [Я. Б. ФРИДМАН\, канд. техн, наук К. С. ЧЕРНЯВСКИЙ, докт. техн, наук Л. М. ШКОЛЬНИК, докт. техн, наук Е. А. ШУР.
УДК 669.14 : 620.18+539.2+621.78 (083)
Металловедение н термическая обработка стали: Справ, нзд. — 3-е изд., перераб. н доп, В 3-х т. Т. I. Методы испытаний н нсследовання/Под ред. Бернштейна М. Л., Рах-штадта А. Г. М.: Металлургия, 1983. 352 с.
Третье издание (второе — в 1962 г.) переработано и дополнено материалами, отражающими современные достижения науки. В первом томе изложены методики современного металловедческого анализа: микроскопического, электронно-микроскопического, рентгеноструктурного, микрорентгеноспектрального. Оже-спектрографин, мессбауэровского и других способов исследования строения металлов н сплавов. Приведены -основные положения статистической обработки экспериментальных результатов.
Для инженерно-технических н научных работников предприятий, лабораторий и научно-исследовательских организаций различных отраслей промышленности. Ил. 375, Табл. 113. Бнблиогр. список: 430 назв.
М
2605000000—092
040(01)—83
32—83
© Издательство «Металлургия», 1983 Щ
СОДЕРЖАНИЕ
Стр.
Стр.
Предисловие к третьему изданию . . 7
Предисловие к первому тому .... 8
Термическая обработка в металлургии 9
1. Методы исследования макро- и микроструктуры (А. Г. Рахштадт, Б. А. Клы-пин)................................... 15
1.1. Исследование макроструктуры (макроанализ) .............................15
1.2. Исследование микроструктуры (микроанализ) .............................17
1.2.1. Приготовление микрошлифов 17
1.2.2. Методы световой микроскопии 24
1.2.3. Методы микроскопического исследования металлов ... 26
1.2.4. Основные типы и конструктивные особенности металлографических микроскопов . . 28
1.2.5. Количественные анализаторы структуры..........................31
1.2.6. Исследование микроструктуры
при повышенных температурах 33
1.2.7. Основные методы микроанализа ................................83
Библиографический список ... 47
2. Просвечивающая электронная микроскопия (М. П. Уснков, Л. М. Утевский) 47
2.1. Конструкция микроскопа н принципы его работы....................47
2.2. Методы электронно-микроскопического исследования металлов и сплавов................................50
2.2.1. Косвенный метод ... 50
2.2.2. Полупрямой метод . . 50
2.2.3. Прямой метод ..... 51
2.3. Сведения, получаемые прн исследовании металлов и сплавов методом тонких фольг...........................54
2.3.1. Микродифракцнонный фазо-
вый анализ.....................54
2.3.2. Определение ориентировки кристаллов, разориентировки зерен * и субзерен н ориентационных соотношений ... 54
2.3.3. Структурные особенности фазовых превращений ... 56
2.3.4. Диффузное рассеяние электронов ........ 58
2.3.5. Изучение дислокационной структуры..........................59
2.3.6. Прямое научение процессов, происходящих в тонкой фольге 60
2.4. Тенденции и перспективы развития метода.................................61
Библиографический список ... 61
3. Растровая электронная микроскопия (Е. А. Шур)............................62
3.1. Растровый электронный микроскоп 62
3.1.1. Принцип работы .... 62
3.1.2. Классификация РЭМ ... 63
3.1.3. Место РЭМ в микроскопии 63
3.1.4. Конструкция РЭМ .... 64
3.1.5. Формирование контраста изображения ..........................66
3.2. Методика исследования металлов с помощью РЭМ........................... 67
3.8. Применение РЭМ в металловедческих исследованиях .
3.3.1. Металлография ....
3.3.2. Фрактография..............
3.3.3. Локальный анализ .
3.3.4. Изучение кристаллографической и дислокационной структуры металлов ...................
3.3.5. Другие области использования .............................
3.3.6. Перспективы развития РЭМ . Библиографический список .
4. Стереологии (количественная металлография) (К- С. Чернявский) .
4.1. Основные понятия и принципы
4.2. Объекты и техника стереологнче-ского анализа .......................
4.2.1. Объекты стереологического анализа .........................
4.2.2. Анализ с участием оператора 4.2.3. Автоматический анализ
4.3. Характеристики размеров частиц
4.3.1. Распределение размеров частиц . ......
4.3.2. Средний размер D, число частиц в единице объема Nv
4.3.3. Погрешности стереологиче-ской реконструкции характеристик размеров частиц
4.4. Описание формы частиц
4.4.1. Непосредственная реконструкция .............................
4.4.2. Статистическая реконструкция
4.4.3. Факторы формы Кф . . .
4.5. Характеристики поверхностей раздела . .....
4.5.1. Протяженность (удельная поверхность) Sv....................
4.5.2. Кривизна..................
4.5.2.1. Средняя кривизна Н
4.5.2.2. Среднян гауссова кривизна К
4.5.3. Двугранные углы 0
4.6. Характеристики линейных элементов
4.6.1. Плотность Lv..............
4.6.2. Характеристики искривленности .............................
4.7. Объемная доля...................
4.8. Характеристики размещения элементов структуры в пространстве .
4.8.1. Ориентировка..............
4.8.2. Расстояния между элементами структуры........................
4.8.3. Связанность...............
4.9. Фрактографические характеристики 4.9.1. Поверхность разрушения .
4.9.2. След разрушения на случайном сечении .....................
Библиографический список .
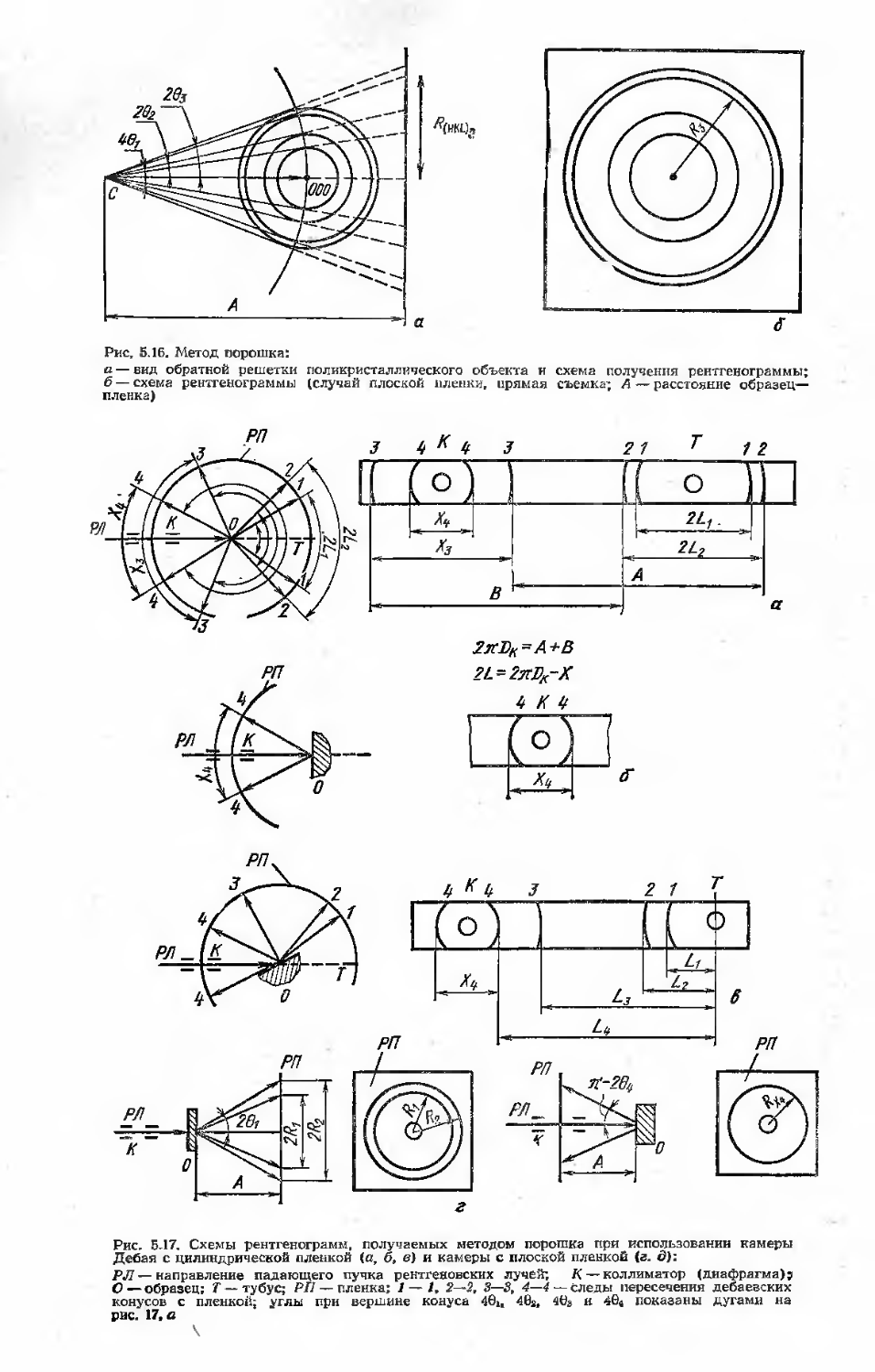
5. Рентгеноструктурный анализ (Ю. А. Скаков) .............................
5.1. Задачи рентгеноструктурного анализа в исследованиях и контроле качества металлических материалов
5.2. Принципы дифракционных методов анализа и аппаратура . . . .
5.2.1. Основные определения н формулы структурной кристаллографии. Структура кристаллов
68
68
69
70
70
72
72
72
75
75
77
78
81
81
83
86
87
87
88
88
88
89
89
89
90
90
91
91
92
92
93
93
93
94
94
94
95
95
3
Стр.
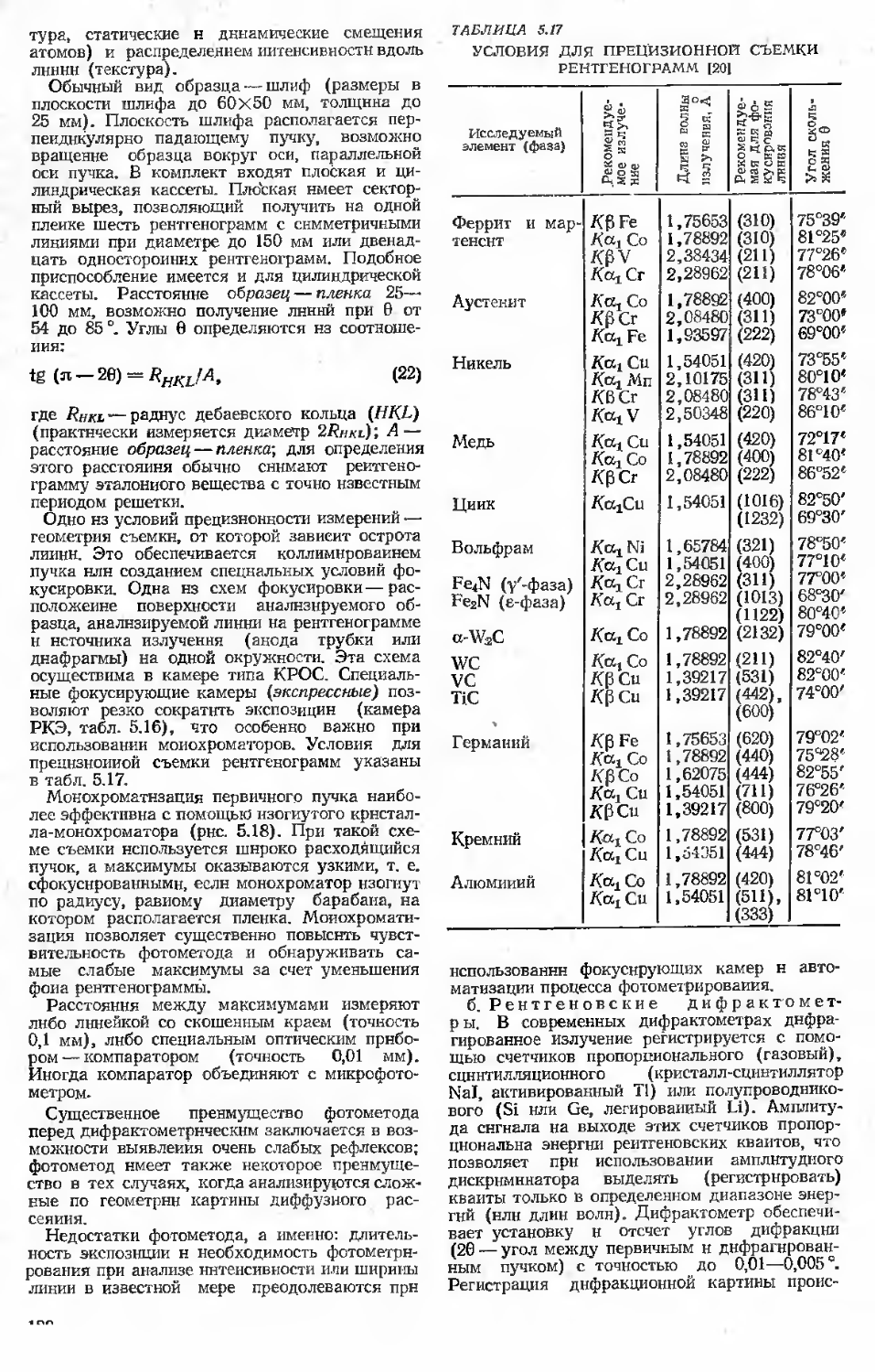
5.2.2. Геометрия дифракционных картин и обратная решетка кристаллов........................110-
5.2.3. Интенсивность рассеяния рентгеновских лучей кристаллом . 115
5.2.4. Техника получения и регистрация дифракционных картин. Аппаратура.........................116
5.2.5. Дозиметрия и техника безопасности в лабораториях рентгеноструктурного анализа . . 123
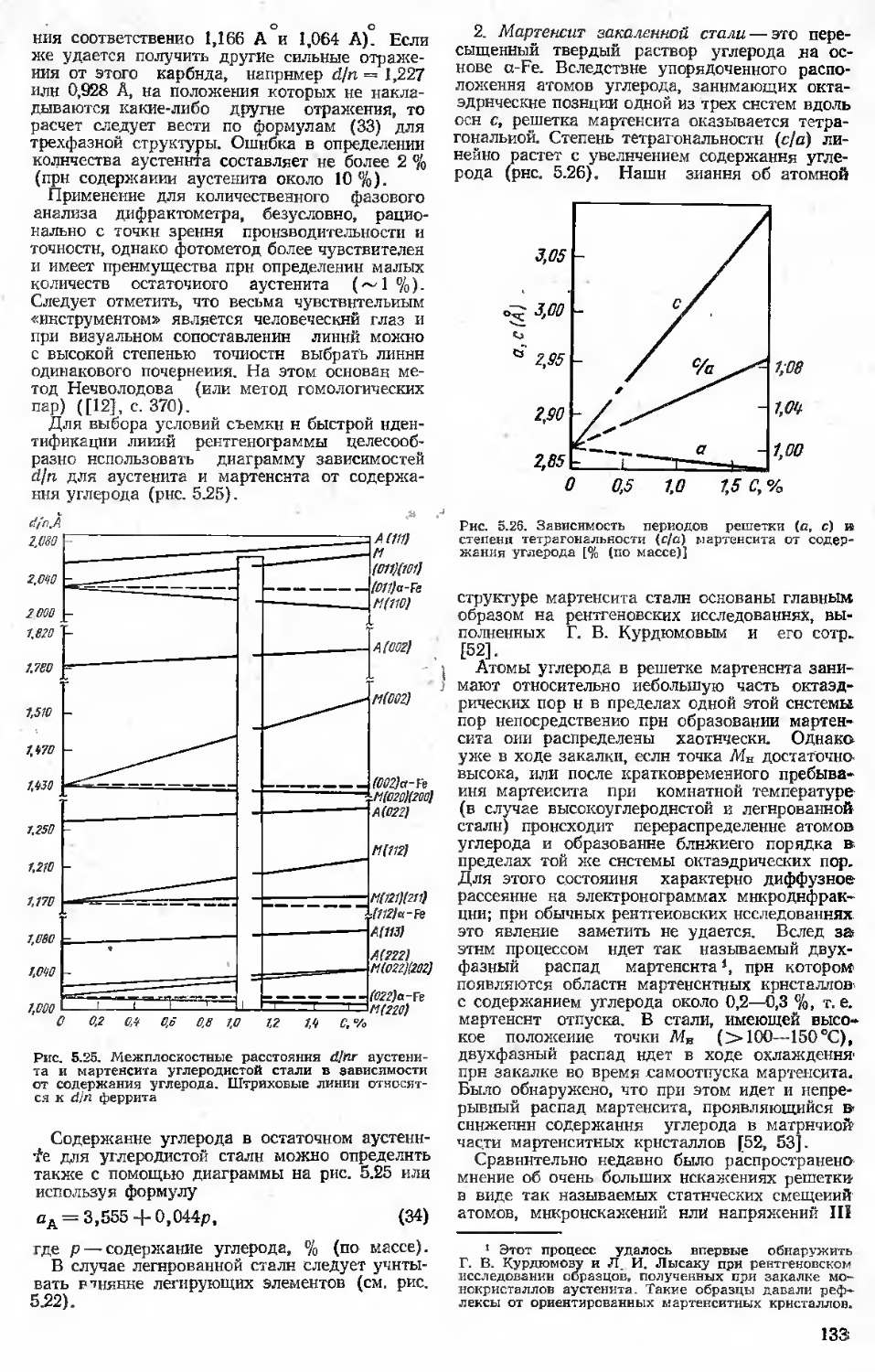
5.3. Анализ фазового состава . . . 124
5.3.1. Фазовый (качественный н количественный) анализ . . 124
5.3.2. Анализ твердых растворов . 127
5.3.3. Построение диаграмм состояния ...............................130
5.3.4. Анализ фазового состава стали после термической обработки ................................131
5.4. Анализ структурного состояния металлических материалов . . . . 136
5.4.1. Анализ текстур .... 136
5.4.2. Анализ напряжений . . . 138
5.4.3. Анализ субструктуры в отожженных н слабо деформированных материалах . . . 139
5.4.4. Анализ дефектов кристаллического строения по эффекту уширения линий рентгенограмм .............................141
Библиографический список ... 142
6. Рентгеноспекгральный микроанализ (В. Г. Костогонов).....................144
6.1. Устройство рентгеноспектрального мнкроаналнзатора......................144
6.2. Основные параметры метода . . 145
6.2.1. Определяемые элементы . . 145
6.2.2. Локальность РСМА . .146
6.2.3. Чувствительность метода (пре-
дел обнаружения) .... 146
6.2,4. Точность количественного РСМА...............................146
6.3. Возможности метода .... 146
6.4. О поправках при количественном РСМА..................................149
6.5. Подготовка образцов и эталонов . 150
Библиографический список . . . 150
7. Методы исследования поверхностей в металлах (Б. С. Бокштейн) . . . . 151
7.1. Введение..........................151
7.2. Принципы методов н их особенности 151
7.3. Аналитические характеристики и сравнение методов ..... 155
7.4. Некоторые методические н конструктивные требования.....................157
7.5. Примеры применения .... 158
7.5.1. Сегрегация примесей . . . 158
7.5.2. Коррозия н окисление . . 159
Библиографический список . . . 160
8. Ядерный гамма-резонанс (эффект Мессбауэра) (Б. С. Бокштейн) . . . 161
8.1. Введение.....................161
8.2. Сущность эффекта.............161
8.3. Методика съемки Я ГР-спектров поглощения .............................162
8.4. Параметры спектров н методика их
определения................ . 163
- Стр.
8.4.1. Сверхтонкая структура спектра 163
8.4.2. Изомерный сдвиг (положение) линии........................164
8.4.3. Ширина линии .... 165
8.4.4. Высота линии............165
8.5. Некоторые применения ЯГР-спектро-скопни..........................166
8.5.1. Электронная структура . . 166
8.5.2. Аналитические применения
(фазовый и химический анализы) ...........................167
8.5.3. Фазовые превращения и упорядочение ........................167
8.5.4. Динамика решетки . . . 168
8.5.5. Дефекты н диффузия . . 170
Библиографический список ... 171
9. Радиоспектроскопия (Ю. Я- Лнлеева) 171
9.1. Ядериый магнитный резонанс . 171
9.1.1. Условие возннкиовення ядер-ного магнитного резонанса (ЯМР)............................ 171
9.1.2. Ширина линии спектра ЯМР и времена релаксации . . . 173
9.1.3. Сдвиг резонансной частоты . 175
9.2. Квадрупольные эффекты и квадру-польный резонанс......................176
9.2.1. Квадрупольные взаимодействия, форма линии спектра ЯМР...............................176
9.2.2. Квадрупольный резонанс . . 177
9.2.3. Приложения ЯМР-спектроскопии к решению некоторых ме-таллофнзических задач . . 178
9.3. Электронный парамагнитный резонанс (ЭПР).........................179
9.3.1. Структура линий ЭПР . . . 179
9.3.2. Основные параметры линии ЭПР............................... 179
9.3.3. Применение метода ЭПР . . 131
9.4. Ферромагнитный н антиферромагнитный резонансы................182
9.4.1. Ферромагнитный резонанс (ФМР)........................182
9.4.2. Антиферромагнитный резонанс 182
Библиографический список . . . 183
9.5. Приложение....................184
10. Сведения о механических свойствах металлов (Я. Б. Фридман) .... 185
10.1. Деформация н разрушение . . 185
10.2. Основные стадии процесса деформации ................................185
10.3. Основные закономерности упругой и пластической деформации и разрушения ................................185
10.4. Хрупкое и пластичное состояние материалов..........................187
10.5. Характеристики механических свойств металлов .................... 187
10.6. Связь между различными механическими свойствами . , . 189
Библиографический список . . 190
11. Статические испытания металлов (Я. Б. Фрндмаи)....................190
11.1. Испытание на одноосное растяжение ..................................190
11.1.1. Машины и образцы для испытания на растяжение . 190
11.1.2. Диаграммы деформации при растяжении...............................191
4
11.1.3. Определение характеристик прочности
11.1.4. Определение характеристик пластичности ...................
11.2. Испытание на сжатие . . .. .
11.3. Испытание на изгиб . . . .
11.4. Испытание на кручение
11.5. Мнкромеханические испытания
11.6. Определение твердости . . . .
11.7. Механические свойства при длительных статических нагрузках
11.8. Испытание на двухосное растяжение ...............................
Библиографический список
12. Методы определения модулей упругости металлов (А. Г. Рахштадт, Е. Е. Попова) „.....................
12.1. Статические методы определения модулей упругости . . . .
12.2. Динамические методы определения модулей упругости...................
Библиографический список
13. Динамические испытания металлов (Б. А. Дроздовский).................
13.1. Понятие «динамические испытания» 13.2. Назначение динамических испытаний ................................
13.3. Высокоскоростные машины для динамических испытаний .
13.4. Копры для динамических (ударных) испытаний .....................
13.5. Образцы для обычных ударных испытаний ............................
13.6. Образцы для испытания падающим грузом (ИПГ) (DWTT) н взрывом
13.7. Испытание стандартных образцов с надрезом н трещиной на изгиб
13.7.1. Испытания на маятниковых копрах по ГОСТ 9454—78
13.7.2. Испытание образцов с трещиной иа статический изгиб
13.8. Методические вопросы динамических испытаний......................
13.8.1. Способы оценки хладноломкости прн динамических испытаниях ......................
13.8.2. Сопоставление испытаний образцов сечением 10X10 мм с V-образным надрезом н ИПГ............................
13.8.3. Точность определения нагрузки при ударных испытаниях с осцнллографнрова-ннем н определение динамической вязкости разрушения
13.9. Влияние скорости нагружения на свойства материалов н виды хрупкости, выявляемые при изгибе образцов с надрезом .... 13.9.1. Влияние скорости нагружения на свойства гладких образцов прн растяжении
13.9.2. Влияние скорости нагружения на свойства образцов с надрезом и трещиной при изгибе ........................
13.9.3. Виды хрупкости, наиболее отчетливо выявляемые при
Стр.
193
194
194
195
196
197
198
202
203
204
205
206
207
208
209
209
209
210
211
215
218
218
218
219
219
219
220
222
224
224
224-
испытании на изгиб образцов с надрезом
Библиографический список
14. Циклические испытания механических свойств (Л. М. Школьник)
Библиографический список
15. Определение сопротивления разрушению (О. Н. Романнв)...............
15.1. Общие сведения о сопротивлении разрушению и методах его оценки
15.2. Методы интегральной оценки сопротивления разрушению .
15.3. Испытания на вязкость разрушения ................................
15.4. Влияние различных факторов на вязкость разрушения сталей .
15.5. Оценка трещнностойкостн прн циклическом нагружении
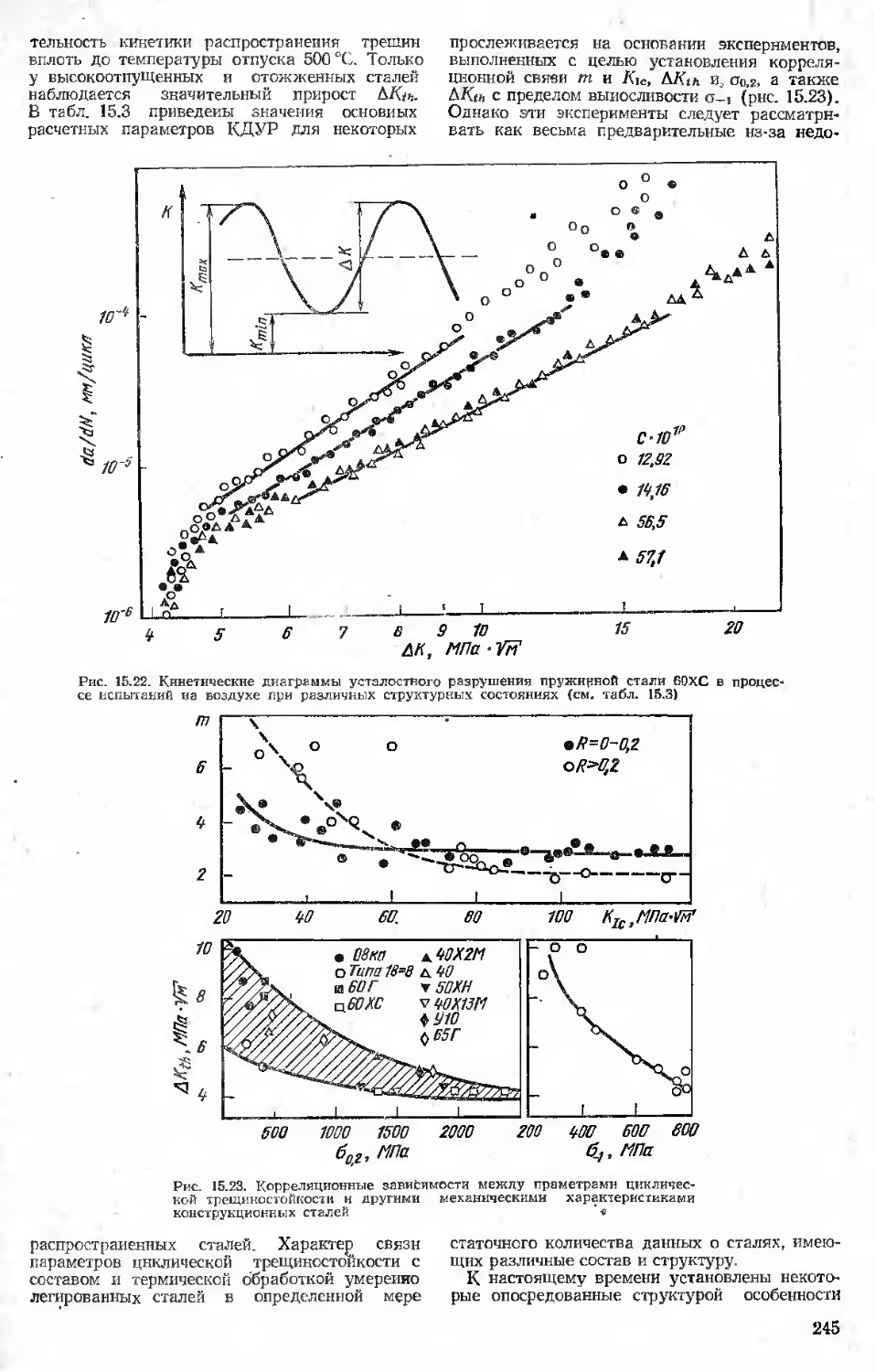
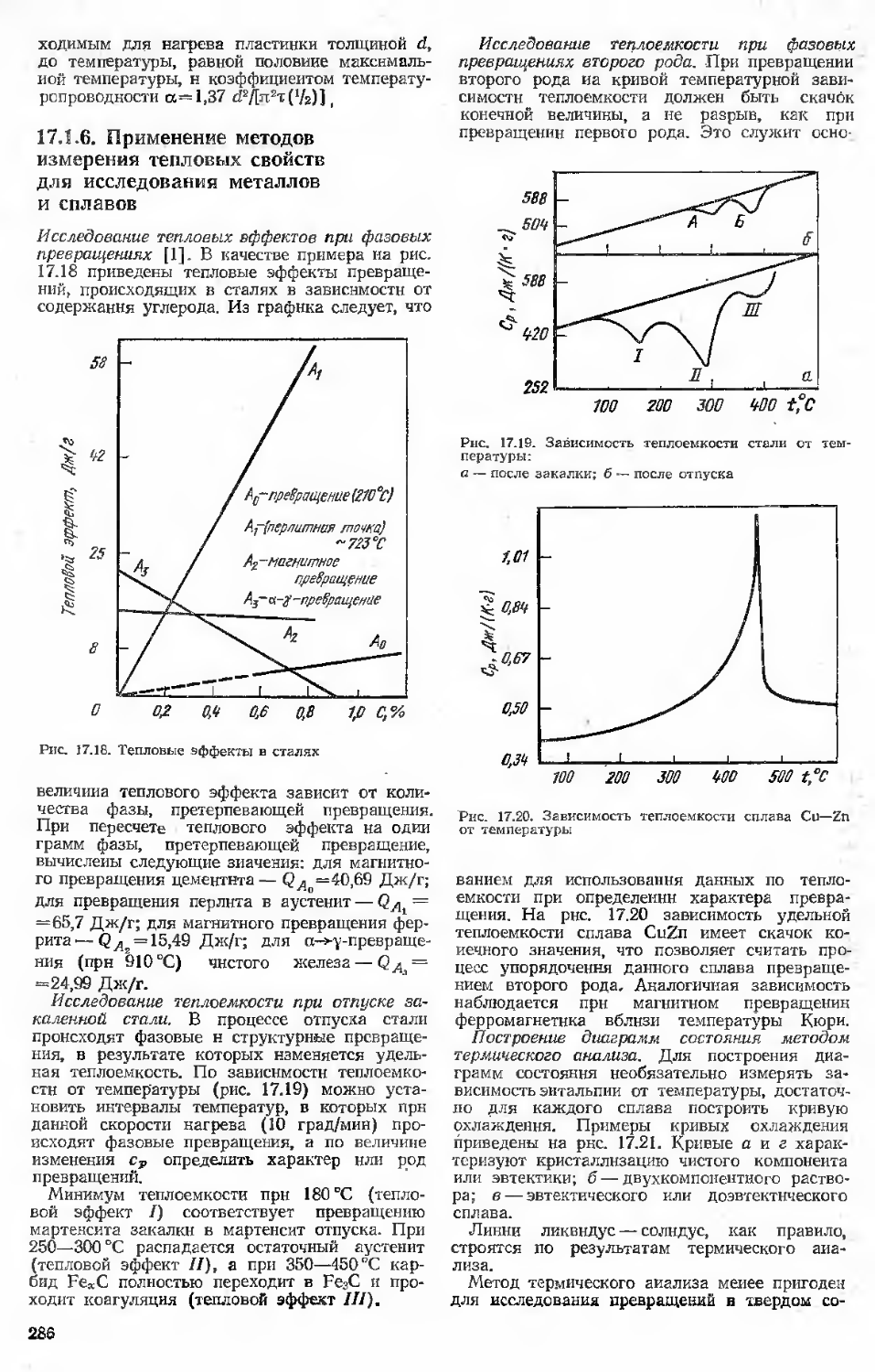
15.6. Статическая трещиностойкость. Учет воздействия рабочих сред Библиографический список
16. Специальные испытания (Ю. Г. Векслер, Л. Г. Коршунов)................
16.1. Класификацня и терминология
16.1.1. Коррозия...............
16.1.2. Изнашивание ....
16.2. Характеристика коррозионных процессов .............................
16.2.1. Химическая коррозия .
16.2.2. Электрохимическая коррозия ...........................
16,2.3. Влияние внешних факторов на коррозию ....
16.3. Характеристика трепня и процессов изнашивания.....................
16.4. Методы испытаний..............
16.4.1. Испытания на коррозионную стойкость ....
16.4.2. Испытания на кавитационную стойкость ....
16.4.3. Испытания в газовых потоках ...........................
16.4.4. Испытания иа изнашивание Библиографический список
17. Физические методы исследования (тепловые, объемные, электрические, магнитные) (Б. Г. Лившиц, А. С. Лиле-ев).................................
17.1. Тепловые свойства.............
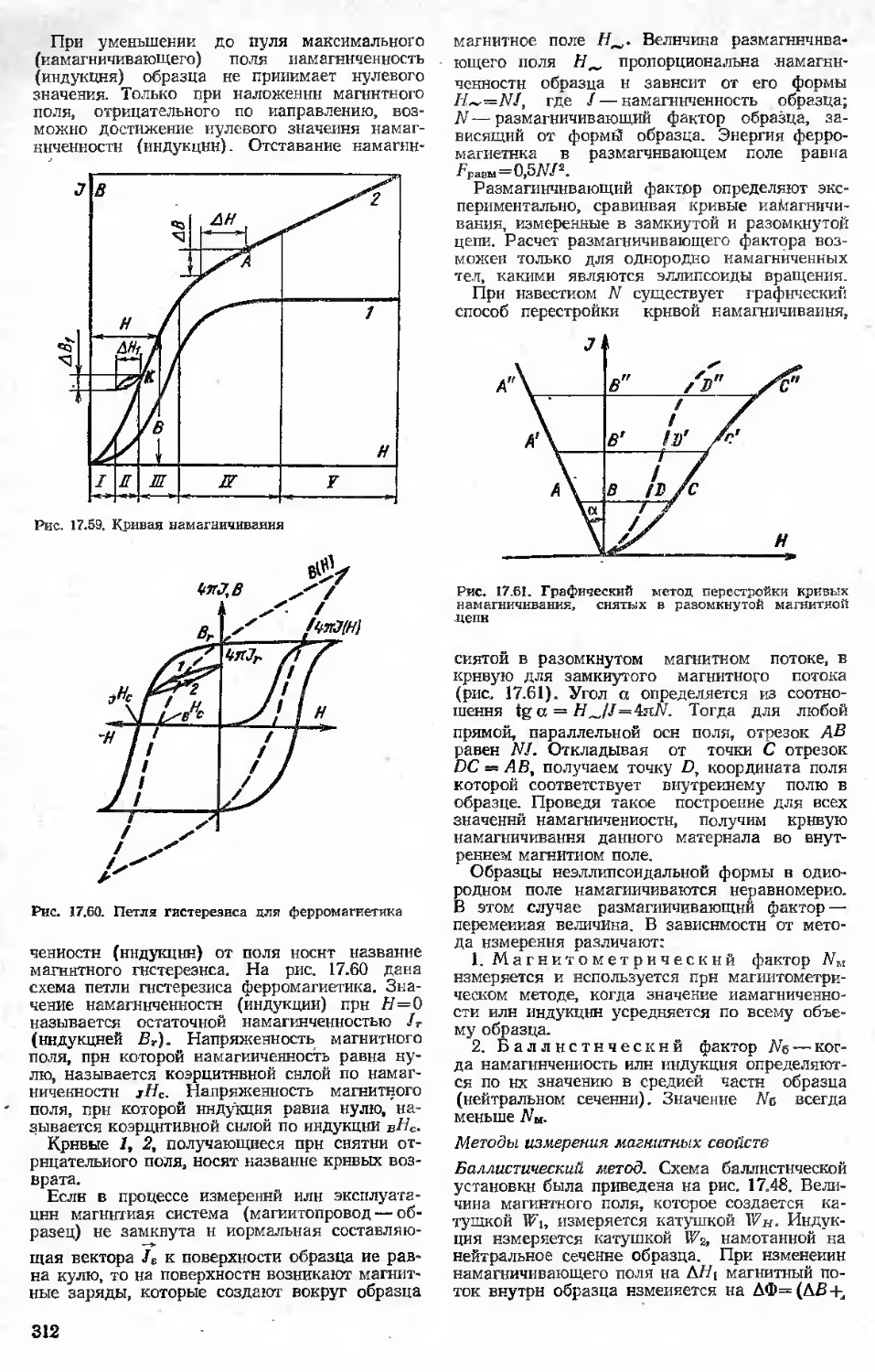
17.1.1. Эитальпня и теплоемкость
17.1.2. Методы измерения энтальпии и теплоемкости
17.1.3. Термический анализ
17.1.4. Теплопроводность .
17.1.5. Методы измерения теплопроводности .....
17.1.6. Применение методов измерения тепловых свойств для исследования металлов и сплавов ....................
17.2. Плотность и термическое расширение ................................
17.2.1. Плотность и методы ее измерения .......................
17.2.2. Термическое расширение и температурный коэффициент линейного расширения
17.2.3. Методы измерения коэффициента линейного расширения ...........................
Стр.
225
225
226
234
235
235
235
237
240
243
246
248
249
249
249
250
250
250
252
253
255
261
261
265
266
269
274
275
276
276
277
280
281
283
286
287
287
289
290
5
Стр.
Стр.
17.2.4. Применение методов изме рения плотности и термического расширения при исследовании металлов и сплавов 292
17.3. Электрические свойства . . . 292
17.3.1. Электрическая проводи-
мость и электрическое сопротивление металлов и сплавов......................292
17.3.2. Методы измерения электрических свойств .... 296
17.3.3. Термоэлектрические, гальваномагнитные и термомаг-иитные свойства . . . 298
17,3.4. Методы измерения термоэлектрических, гальваномагнитных н термомагннтных свойств 300
17.3.5. Сверхпроводимость . . 301
17.3.6. Применение методов измерения электрических свойств при исследовании металлов п сплавов.....................302
17.4. Магнитные свойства .... 305
17.4.1. Классификация магнетиков 305
17.4.2. Методы измерения напряженности магнитного поля 307
17.4.3. Измерение пара- и диамагнитной восприимчивости . 310
17.4.4. Ферромагнитные свойства и методы их измерения . 311
17.4.5. Магнитная анизотропия и методы ее измерения . . 314
17.4.6. Измерение магнитных свойств в переменных полях 316
17.4.7. Магнетокалорнческий эффект ...........................317
17.4.8. Применение методов измерения магнитных свойств при исследовании металлов и сплавов.......................318
Библиографический список . . 319
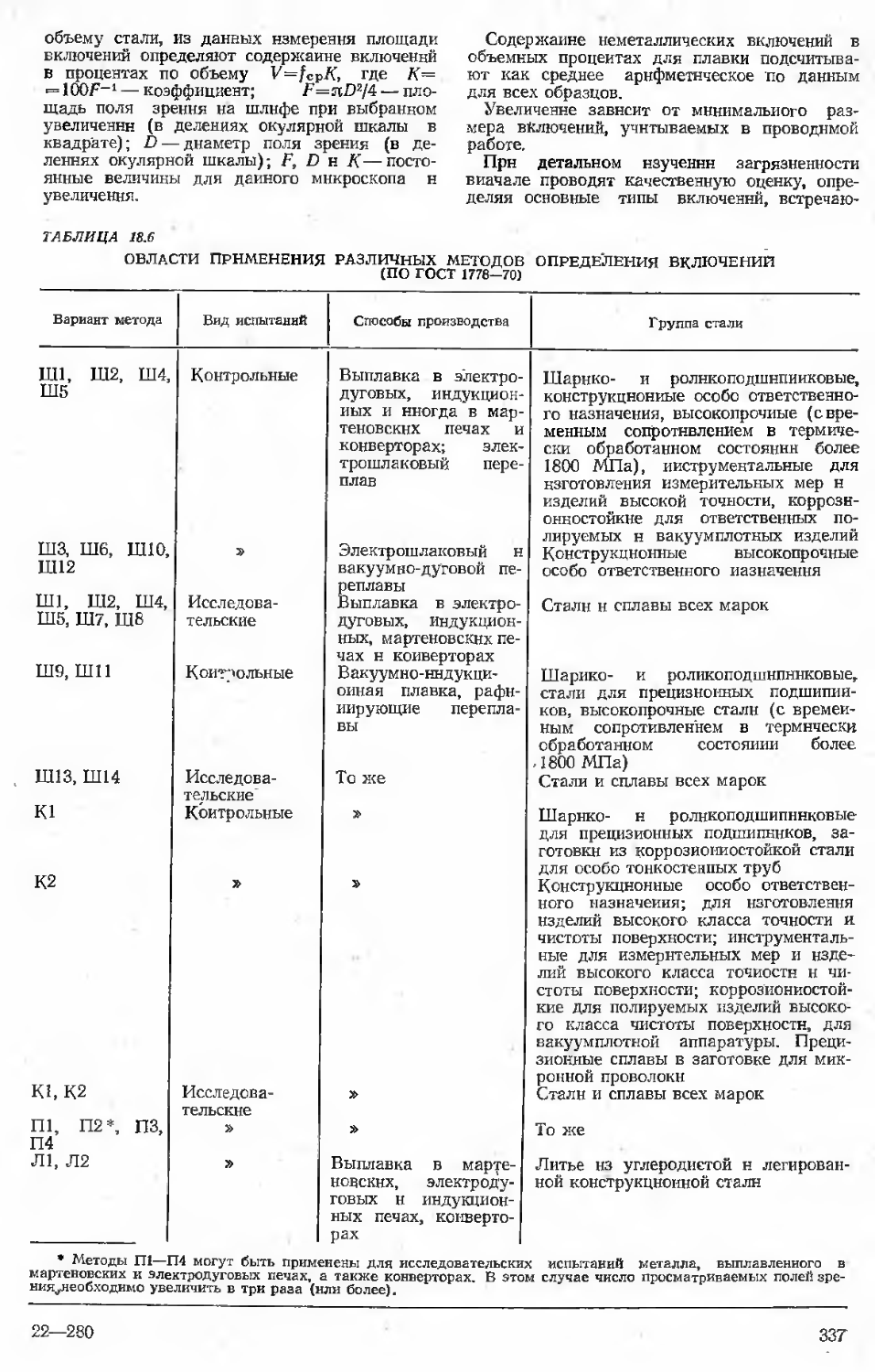
18. Металлургический контроль качества
(М. И. Виноград, Г. М. Казичкина) . 320
18.1. Общие положения контроля . . 320
18.2. Химический состав.........322
18.3. Внешний вид металлопродукции . 323
18.4. Испытание на осадку .... 325
18.5. Макроструктура стали .... 325
18.6. Волосовины .....*, 332
18.7. Неметаллические включения . . 333
18.8. Величина зерна............338
18.9. Глубина обезуглероженного слоя 338
18.10. Микроструктура стали . . . 341
18.11. Альфа-фаза в нержавеющей стали 345
18.12. Чувствительность к закалке . . 346
18.13. Испытания на высокотемпературную пластичность......................346
Библиографический список . . 347
Предметный указатель . . . 348
ПРЕДИСЛОВИЕ К ТРЕТЬЕМУ ИЗДАНИЮ
XXVI съезд КПСС в качестве главного направления развития черной металлургии выдвинул задачу дальнейшего повышения качества и увеличения выпуска эффективных видов металлопродукции. Успешное решение этой задачи во многом определяется достижениями в науке о металлах— в области теории их строения и фазовых превращений, теории сплавов, результатом чего явилось создание новых процессов термической обработки и новых сплавов. Все это нашло и находит свое отражение в металловедческой литературе — в отдельных научных и производственно-технических книгах и статьях.
Вместе с тем непрерывно возрастает потребность в справочных изданиях по металловедению, в том числе фундаментальных, которые способствуют практическому внедрению новейших достижений науки в производство. Именно это и послужило основанием для подготовки нового, третьего издания справочника «Металловедение и термическая обработка стали». В нем нашли отражение изменения, происшедшие в области методов исследования, а также в теоретическом и техническом металловедении за годы, прошедшие со времени выпуска двух первых изданий справочника, хотя третье издание и создано на базе предыдущих при принципиально той же композиционной структуре.
В предлагаемом читателю издании, в отличие от предыдущих, представлены такие новые и перспективные методы исследования, как количественный анализ структуры, диффузное рассеяние электронов, растровая электронная микроскопия, рентгеноспектральный анализ, Оже-элек-тронная спектроскопия, ядерный гамма-резонанс, радиоспектроскопия и др.; справочник дополнен разделом о способах оценки параметров, вязкости разрушения, живучести; отдельно освещены такие важные специальные испытания, как оценка износостойкости, кавитационной стойкости.
По-новому изложены и некоторые теоретические вопросы: процессы превращения при закалке и отпуске стали, теория диффузии и процессы рекристаллизации. В раздел «Основы термической обрабвтки» включены теория границ зерен, теория дисперсионного твердения и др. Учитывая развитие новых технологических процессов упрочнения и формоизменения, в третье издание справочника включены разделы о структурной наследственности, строении деформированных металлов и сверхпластичности.
Раздел, освещающий влияние легирования и термической обработки ' на структуру и свойства сталей различных классов, сокращен в связи с выходом в последние годы специальной справочной и монографической литературы по отдельным классам стали.
В третьем томе справочника рассматривается термическая обработка металлопродукции различных видов. В нем широко освещены современные методы термической обработки поковок, проката, листов, рельсов, труб, термомеханическая обработка металлопродукции и др.
Авторский коллектив надеется, что справочник «Металловедение и термическая обработка стали» сыграет свою положительную роль в решении задач, поставленных партией перед отечественной промышленностью.
ПРЕДИСЛОВИЕ К ПЕРВОМУ ТОМУ
Современное состояние развития металловедения характеризуется активным проникновением физических методов исследования в экспериментальную технику. Развивающиеся в последние годы расчетные методики определяют получение количественных результатов, необходимых для объективного описания структуры сталей и сплавов. Самым перспективным в будущем представляется направление, приводящее к установлению надежной количественной связи между параметрами структуры металлического сплава и значениями его свойств. Это и является основной задачей физического металловедения, решение которой лежит на пути совершенствования экспериментальных методов исследования, а также развития комплексного подхода, предусматривающего рациональное сочетание различных методик для получения адекватной картины связи структуры и свойств.
Предлагаемый читателю первый том справочника «Металловедение и термическая обработка стали» посвящен изложению методик изучения тонкого строения и структуры сталей и определению их разнообразных свойств (механических, физических, эксплуатационных). Такое построение многотомного справочника представляется правильным, если иметь в виду преимущественно экспериментальный характер науки о металлах. В этом томе, наряду с традиционными методами изучения структуры и свойств (макро- и микроанализ, рентгеновская дифрактометрия, электронная микроскопия, определение механических свойств при растяжении, ударе, циклическом нагружении и т.п.), рассмотрены развитые в последние годы тонкие методы структурых исследований (спектроскопические, резонансные, микроспектральные и др.) и методы определения сопротивления разрушению в различных условиях нагружения (параметры вязкости разрушения, кавитационное разрушение, износостойкость, сопротивление газбвой коррозии) в сочетании с подробным изложением методик фрактографического анализа. Все эти новые разделы отличают настоящее издание от предыдущих.
Следует подчеркнуть, что разделы, касающиеся традиционных методов анализа структуры и определения свойств, претерпели по сравнению с предыдущими изданиями существенную переработку. Приведены описания и основные конструктивные схемы новых приборов, выпускаемых отечественной и зарубежной промышленностью, новые методики пооперационной регистрации процессов и интерпретации конечных экспериментальных результатов. Читатель найдет также данные о целесообразности применения того или иного конкретного метода исследования в комплексе с другими для получения наиболее достоверных результатов, определяющих работоспособность стали и сплавов в реальных конструкциях.
Авторский коллектив стремился весь этот большой и весьма важный для практического использования в лабораториях заводов и научно-исследовательских институтов материал изложить кратко, но прн сохранении необходимого научного уровня; кроме того, учитывалось, что предлагаемый читателю материал должен быть подспорьем в практической деятельности инженеров и научных работников, занятых производством и использованием сталей и сплавов.
М. Л. Бернштейн
А. Г. Рахштадт
ТЕРМИЧЕСКАЯ ОБРАБОТКА В МЕТАЛЛУРГИИ
Выпуск металлопродукции повышенного качества металлургической промышленностью страны — важнейшая задача народного хозяйства. Решение ее должно определить едва ли не основную линию технического развития металлургии. Такую постановку задачи обусловливают следующие постоянно действующие факторы.
1. Экологические: необходимость рационального использования природных ресурсов при ограниченности запасов руды, коксующихся углей, минералов и топлива весьма остро ставит проблему получения н использования металлопродукции с более высокими механическими свойствами; это позволит уменьшить сечение выпускаемых металлических полуфабрикатов, более полно обеспечить машиностроение и строительство трубами, листами, фасонным и сортовым прокатом из того же количества сырья, что даст его экономию.
2. Технический прогресс: создание изделий новой техники, уникальных сооружений и магистральных трубопроводов, крупногабаритных сосудов, агрегатов ядерной энергетики, криогенной аппаратуры большого тоннажа — все это требует использования упрочняющей обработки для повышения комплекса механических свойств металлопродукции массового производства.
3. Энергетические: проведение упрочняющей обработки непосредственно в цикле металлургического производства с использованием тепла деформационного нагрева является важным резервом экономии топлива, так как исключается термическая обработка на машиностроительных заводах; одновременно это способствует улучшению состояния окружающей природной среды, что особенно важно, учитывая размещение машиностроительных заводов в жилых массивах; следовательно, этот фактор связан также с экологическим.
4. Технико-экономические: облегчение конструкций, повышение их долговечности и надежности, выпуск металлопродукции, полностью отвечающей требованиям стандартов, рациональное использование сырья, топлива и полуфабрикатов — не временное, а постоянное требование, которое всегда будет определять пути развития любой отрасли техники, в том числе и металлургии.
Таким образом, перед отечественной металлургической промышленностью стоит вполне определенная и очень важная задача — резко увеличить объем выпускаемой металлопродукции с упрочняющей термической и термомеханической обработкой. На это указывалось в ряде директивных решений, определявших пути развития металлургии в прошлых пятилетках; об этом говорится и в директивах партии на XI пятилетку и на период до 1990 года. Следует отметить, что эта задача решается еще недостаточными темпами, хотя пути ее решения благодаря опережающим практическое внедрение научным разработкам во многом ясны.
Применительно к классу строительных сталей, из которых изготовляют значительный объем проката (листов, полос, труб, фасонных профилей и др.), перспективным методом упрочнения является освоенная нашей промышленностью контролируемая прокатка, которая в большинстве случаев обеспечивает более высокий уровень механических свойств по сравнению с термической обработкой с отдельного нагрева. Это объясняется тем, что контролируемая прокатка, будучи вариантом термомеханического упрочнения с воздушным охлаждением, приводит к одновременному повышению прочности, пластичности, вязкости и хладостойкости. Такое уникальное сочетание свойств, получаемое только в результате термомеханической обработки, обусловлено тремя основными факторами: созданием развитой субструктуры в условиях регламентированной деформации в межфазной аустенито-ферритной области;
9
-формированием весьма дисперсных карбонитридов ниобия, упрочняющих сталь и стабилизирующих субструктуру; измельчением зерна. Немаловажную роль в повышении сопротивления разрушению ряда изделий, в частности листов, идущих на изготовление сварных труб магистральных трубопроводов, играет и создание текстуры при контролируемой- прокатке с окончанием деформации в межфазной области, когда сосуществующие фазы с различными решетками тормозят миграцию большеугловых границ и соответственно рекристаллизацию. Развивающаяся в поперечном направлении зародышевая трещина должна многократно менять свое направление из-за наличия текстуры, что и повышает энергоемкость процесса разрушения.
Дальнейшее развитие контролируемой прокатки представляется следующим:
1. Расширение опробования, а затем и использования этой технологии при производстве не только листов для газопроводных труб из малоуглеродистой стали, но и других видов металлопродукции, главным образом тех, которые представляют собой почти готовые изделия. Развитие работ по контролируемой прокатке рельсов, горячекатаных бесшовных труб, периодических профилей различного назначения, арматурной стали и т. п. полуфабрикатов, что является насущной задачей ближайшей перспективы.
2. Совершенствование состава сталей для обеспечения-возможности практического осуществления контролируемой прокатки металлопродукции указанных выше видов. Работы необходимо вести в направлении создания экономнолегированных сталей, с акцентом на микролегирование, имея в виду реализацию достижений отечественного металловедения. Речь идет о развитой советскими учеными идее наиболее полного использования субструктурного упрочнения, создаваемого регламентированной горячей (теплой) деформацией тогда, когда субструктура стабилизирована дисперсными выделениями упрочняющих фаз.
Другой интенсивно развивающийся в последние годы метод термической обработки строительных сталей — закалка холоднокатаных листов, используемых главным образом в автомобилестроении, на двухфазное состояние. Обработка проводится с отдельного нагрева в межкритическую феррито-аустенитную область, затем следует резкое охлаждение для получения так называемой дуальной структуры, представляющей собой ферритную матрицу с островками малоуглеродистого мартенсита (обычно в тройных стыках зерен), Стали с такой структурой имеют низкое отношение предела текучести к пределу прочности, что определяет хорошую штампуемость (важно для автомобилестроения), а после штамповки — высокую прочность благодаря деформационному упрочнению феррита и наличию мартенситных участков. Создание дуальной структуры после этой обработки при сохранении высокого уровня пластичности и вязкости позволяет уменьшить толщину листов, что уже дало значительную экономию металла в автомобильной промышленности некоторых стран, например США.
Развитие работ по созданию сталей с феррито-мартенситной дуальной структурой целесообразно в следующих направлениях:
а) освоение этой обработки применительно не только к листам, но и к металлургической продукции другого сортамента;
б) создание дуальной структуры не только в строительных, но и в машиностроительных сталях, идущих для изготовления силовых деталей машин и механизмов;
в) совмещение нагрева в межкритический феррито-аустенитный интервал с горячей деформацией; при последующей закалке будет создан естественно композиционный материал с вытянутыми «нитями» фаз, одни из которых обладают высокой прочностью, а другие — высокой вязкостью.
Термомеханическая обработка для создания деталей с дуальной структурой открывает весьма широкие перспективы целенаправленного изменения всего комплекса механических свойств на сталях сравнительно простого химического состава, без использования многокомпонентного легирования, а главное, при исключении дефицитных элементов. Следует, особо подчеркнуть, что разработка методов термической обработки на дуальную структуру основывается на разумной реализации известного в металловедении основного положения об определяющей роли структуры в достижении заданного уровня свойств. Структура в данном случае прямо регулируется температурой нагрева в межкритической области и выдержкой в ней, что и определяет требуемое соотношение фаз в каждом данном микрообъеме. Важным дополнительным регулирующим фактором является регламентированная деформация. Легирование в этом случае играет второстепенную, технологическую роль (выбор благоприятной скорости охлаждения, прокаливаемость) и может быть осуществлено, исходя из разумного сочетания недефицитных добавок и, главным образом, в направлении микролегировання.
Термическая обработка машиностроительных сталей в процессе их металлургического производства должна развиваться в двух направлениях. Первое направление относится к продукции, практически прямо используемой в машиностроении, без какой-либо существенной дополнительной обработки: рессорные полосы, трубы, гнутые и фасонные профили, буровые и нефтенасосные штанги и т.п. Эти изделия должны проходить термомеханическое упрочнение в потоке стана, а такие изделия, как валки холодной прокатки,—-при их изготовлении на металлургическом заводе. Приведем несколько примеров.
На опытных и опытно-промышленных установках, созданных по инициативе Московского института стали и сплавов, на металлургических заводах «Красный Октябрь» и «Днепроспецсталь», а также на Никопольском южно-трубном заводе прокатано несколько сот тонн термомеханически упрочненных прутков для штанг, рессорных полос и труб для их испытания у потребителя. Установлено, что:
1) термомеханически упрочненные нефтенасосные штанги, из неле-гнрованной стали марки 40 диаметром 19 мм могут заменить серийные штанги из стали марки 20НЗМ диаметром 22 мм. Таким образом, уменьшается масса комплекта для буровых скважин на 25 % и на каждой тонне стали экономится около 20 кг никеля и молибдена;
2) в два-три раза возрастает эксплуатационная стойкость буровых штанг для горно-рудной промышленности, причем это повышение стойкости используется в рамках самого Министерства черной металлургии;
3) аналогично — примерно в два раза — увеличивается и стойкость валков для станов холодной прокатки, что подтверждается работой валков многовалковых станов на установках для термомеханического упрочнения, эксплуатируемых на Ленинградском сталепрокатном н Магнитогорском метизно-металлургическом заводах и созданных Ижевским механическим институтом;
4) упрочненные термомеханической обработкой обсадные трубы из простой углеродистой стали Д попадают в категории прочности К и Е, т. е. могут заменить трубы тех же размеров из легированных сталей 36Г2С и 38ХНМ. Термомехаиическая обработка высокопрочных обсадных труб из стали 36Г2С позволяет уменьшить толщину стенки на 20 % при одновременном увеличении коэффициента запаса прочности на 15%.
Второе направление относится к используемой в машиностроении металлургической продукции, подвергаемой холодной или горячей обработке и упрочняемой окончательной термомеханической обработкой. Эта продукция либо поставлятся без термической обработки, либо проходит на металлургических заводах смягчающую термическую обработ-
ку, а также обработку для подготовки требуемой исходной структуры (которая затем так или иначе видоизменяется при окончательной термической обработке на машиностроительном заводе). Для этой целина металлургических заводах сооружены и сооружаются цеха и отделения, в которых осуществляют (правда, еще в недостаточном объеме) термическую обработку с отдельного нагрева. Это дорогостоящая операция, требующая больших капитальных вложений (при сооружении новых цехов) и больших энергетических затрат. Поэтому за рубежом все шире стремятся осуществлять термическую обработку полуфабрикатов этих видов с использованием тепла деформационного нагрева, упрощая при этом технологический процесс обработки, монтируя установки регулируемого охлаждения в потоке стана.
Особо следует подчеркнуть, что к широкому сортаменту металлургической продукции применима разработанная советскими учеными схема обработки на наследование термомеханического упрочнения.
В понимании природы упрочнения, достигаемого в результате термомеханической обработки, определяющим является факт наследственной передачи развитой дислокационной структуры горячедеформирован-иого аустенита образующемуся'при дальнейшем охлаждении мартенситу или бейниту. При этом необходимо учитывать особый характер возникающей в горячедеформированном аустените развитой сетки дислокационных субграниц динамической полигонизации, которые представляют собой особый вид полупроницаемых барьеров. Исследования, выполненные в нашей стране, прямо показали, что такие полупроницаемые барьеры сдерживают движущиеся дислокации, т. е. повышают прочность до определенного разумного ее значения; при возникновении у этих барьеров опасных с точки зрения создания «пиковых» напряжений скоплений дислокаций происходят прорыв полупроницаемых субграниц и релаксация напряжений путем передачи деформации в смежные объемы, что уменьшает вероятность образования трещин разрушения. Таким образом, становится очевидной научная основа термомеханического упрочнения: при регулировании температуры, скорости и степени горячей деформации в результате динамической полигонизации создаются условия для образования развитой сетки полупроницаемых суб границ. Это и определяет уникальное сочетание свойств, наблюдаемое только после термомеханической обработки, когда наряду с повышением прочности наблюдается и повышение сопротивления разрушению.
Смысл «наследования» упрочнения (созданного термомеханической обработкой) после соответствующей термической обработки вытекает из следующего. Высокие механические свойства после ТМО обусловлены повышенной плотностью несовершенств (дислокаций), являющейся результатом сочетания пластической деформации и фазовых превращений, и созданием их определенных конфигураций (фрагментированной субструктуры). Если при термической обработке после ТМО плотность несовершенств не будет заметно уменьшаться, а фрагментированная структура не исчезнет, то сохранятся и высокие механические свойства. Например, краткий смягчающий отпуск, при котором исключена рекристаллизация, приводит к распаду мартенсита (и делает возможной механическую обработку, например, резанием), но не вызывает существенного снижения плотности несовершенств и разрушения дислокационной структуры, так как отсутствует миграция поверхностей раздела (высокоугловых границ), характерная для развития рекристаллизации. Последующий скоростной нагрев под закалку с кратковременными выдержками обусловливает переход сх-фазы с повышенной плотностью несовершенств в у-фазу, которая также будет иметь высокую нх плотность (по тому же механизму наследования дислокаций, какой наблюдается при переходе из г. ц. к. в о.ц. к. решетку при так называемой прямой ТМО). Здесь применимы основные положения теории структурной на
22
следственности, разработанные академиком В. Д. Садовским. После заключительной закалки образуется мартенсит, сохраняющий (в той или иной мере) дополнительную «насыщенность» несовершенствами, а главное— в той или иной мере сохраняющий фрагментированность, что определяет «восстановление» высоких механических свойств, которые были получены в результате прямой ТМО.
На практике схема наследования термомеханического упрочнения была реализована при прокатке прутков для автомобильных пружин и рессорных полос. Было установлено, что в результате термомеханической обработки пружинной стали 60С2 стойкость пружин возрастает в 2—3 раза и обеспечивается экономия металла благодаря уменьшению потребности в запасных частях для грузовых автомобилей МАЗ и «Колхида» на 25 % (пружины опрокидывания кабины). Применение термомеханической обработки по схеме наследования для упрочнения пружин подвески автомобилей открывает перспективу уменьшения диаметра прутков-заготовок для пружин, что даст экономию металла и снижение массы автомобиля. Рессоры грузовых и легковых автомобилей, изготовленные из термомеханически упрочненных полос имеют на 25 % более высокую эксплуатационную стойкость.
Весьма перспективно использование регламентированной деформации для создания полигонизованной структуры применительно к коррозионностойким нержавеющим аустенитным сталям (типа Х18Н10Т). На Никопольском южно-трубном заводе при прокатке труб из стали 12Х18Н12Т по температурно-деформационным режимам, специально разработанным совместно с Московским институтом стали и сплавов, было достигнуто (в связи с созданием развитой полигонизованной субструктуры и мелкого зерна) повышение предела текучести на 70 % • Поскольку однородность структуры достигается непосредственно при регламентированной прокатке по разработанным режимам, отпадает необходимость в последующей термической обработке, к тому же нежелательной, так-как она приводит к падению прочности.
Испытания на коррозионную стойкость показали, что термомеханн-чески упрочненные трубы из аустенитных сталей обладают стойкостью, отвечающей существующим требованиям.
Применение термомеханической обработки к сталям этого класса позволит перейти на изделия (в данном случае трубы) с меньшей толщиной стенки, что даст существенную экономию высоколегированной стали, содержащей много никеля и хрома.
Приводимые выше соображения и примеры не означают, что на металлургических заводах следует заниматься только термомеханической обработкой и не развивать методы термической обработки с отдельного нагрева. Эти методы себя надежно зарекомендовали, и их следует развивать в тех случаях, когда они целесообразны. В большинстве своем, как было сказано, цель такой термической обработки — подготовить структуру либо для дальнейшего металлургического передела, либо, что чаще, для машиностроительных заводов, на которых будет затем производиться окончательная упрочняющая термическая обработка.
Внимание, которое уделено в этой статье термомеханической обработке определяется следующими соображениями, вытекающими из специфики самого металлургического производства:
а) схема высокотемпературной термомеханической обработки легко вписывается в цикл прокатного производства и требует в большинстве случаев небольшой корректировки температурно-деформационных параметров;
б) используется тепло деформационного нагрева и упрощается весь технологический процесс обработки;
в) создается уникальный комплекс свойств, когда одновременно с повышением сопротивления деформации благодаря созданию особой
13
дислокационной структуры с полупроницаемыми барьерами повышается и сопротивление разрушению.
До недавнего времени операции пластической деформации рассматривались в основном как связанные с формоизменением. Хотя и ранее было известно, что энергия, затрачиваемая на деформирование, больше энергии, выделяемой в процессе деформирования, после пластической деформации эту накопленную энергию из металла «изгоняли». Затем, приступая' к термической обработке вновь осуществляли процессы, приводящие к метастабильному состоянию, обеспечивающему высокую прочность. Несмотря на очевидную целесообразность совмещения обоих мощных факторов воздействия на структуру — пластической деформации и фазовых превращений, — такие комбинированные технологические процессы долгое время почти не имели распространения. Только понимание роли, которую играют несовершенства строения в процессах структурообразования и формирования многих важнейших структурночувствительных свойств (главным образом механических), металлов и сплавов, позволило создать фундамент для развития термомеханической обработки.
Теория и практика создания высокопрочных металлических сплавов свидетельствует о перспективности использования комбинированных способов воздействия на процессы структурообразования, а отсюда и на свойства этих материалов. В прогнозах на 1990—2000 гг. специалисты разных стран указывают, что в основе производства больших масс высокопрочных металлических материалов будет лежать совмещение процессов пластической деформации с фазовыми превращениями.
С. П. Ефименко, докт. техн, наук
1
= МЕТОДЫ ИССЛЕДОВАНИЯ МАКРО-
И МИКРОСТРУКТУРЫ
1.1. ИССЛЕДОВАНИЕ МАКРОСТРУКТУРЫ (МАКРОАНАЛИЗ) [1, 2]
Макроструктуру металла изучают путем просмотра поверхности специально подготовленных образцов (макрошлифов) или изломов невооруженным глазом илн при небольших увеличениях — до 30 раз.
Макроанализ дает представление об общем отроении металла и позволяет оценить его качество после различных видов обработки: литья, обработки давлением, сварки, термической н химико-термической обработки.
Этот анализ не определяет подробностей строения и часто является предварительным видом исследования; однако ои позволяет выбрать те участки, которые требуют дальнейшего микроскопического исследования. С помощью макроанализа можно определить:
1) нарушения сплошности металла: подусадочную рыхлость, центральную пористость, свищи, подкорковые пузыри, межкристаллитные трещины; трещины, возникшие при обработке давлением н термической обработке; флокены; дефекты сварки (в виде непровара, газовых пузырей);
2) дендритное строение, зону транскрнстал-лизации, размеры н ориентацию зерен в литом металле;
3) химическую неоднородность литого металла (ликвацию) и присутствие в нем грубых инородных включений;
4) волокнистую структуру деформированного' металла;
5) структурную нли химическую неоднородность металла, созданную термической, термомеханической или химико-термической обработкой;
6) вид излома: вязкий, хрупкий, нафтали-нистый, камневидиый и т. д.;
7) прокаливаемость (для инструментальных сталей, для которых требуется сохранение вязкой сердцевины).
Приготовление образцов для макроанализа. Макроанализ проводят на продольных н поперечных макрошлифах (темплетах) и изломах. В случае крупных цилиндрических деталей темплеты вырезают в радиальном, тангенциальном и Z-направлении. Большое значение для успешного выполнения макроанализа имеет правильный выбор наиболее характерного для изучаемого изделия сечення нли излома.
При использовании макроанализа для контроля качества металла число образцов, их размеры, место иырезки н Другие условия отбора проб указывают в стандартах и технических условиях на конкретные виды металлопродукции. В частности, макроструктуру прутков обычно контролируют на поперечных макрошлифах.
Поверхность макрошлнфов перед травлением необходимо подвергать торцеванию,
строганию нлн шлифованию. После механической обработки поверхность должна быть ровной н гладкой без значительного поверхностного наклепа н прижога металла. На поверхности макрошлифа не должно быть загрязнений, следов масла н т.п., поэтому ее перед травлением промывают (протирают) специальными составами.
Методы макротравлення подразделяют на три основные группы: глубокого травления; поверхностного' травления; отпечатков. Структура, выявляемая глубоким травлением, сравнительно слабо зависит от подготовки поверхности образца; поверхностное травление илн метод отпечатков требует более тщательной подготовки поверхности.
Для изучения изломов образцы, вырезанные в поперечном или в продольном направлении (по отношению к течению металла при формоизменении), надрезают, а затем разрушают по месту надреза на прессе нлн копре. Разрушение образца следует производить с максимальной скоростью и большой сосредоточенной нагрузкой, т. е. в условиях, исключающих смятие поверхности излома и образование ложных расслоений (в поперечных изломах).
Способы макроанализа различны в зависимости от состава сплава и задач, стоящих перед исследователем.
1. Выявление дефектов, нарушающих сплошность литой и деформированной стали. Для этой цели макрошлифы (темплеты) подвергают глубокому н реже поверхностному травлению. Операцию выполняют в вытяжном шкафу в ванне, изготовленной нз материала, не вступающего в реакцию с применяемыми травильными растворамн. В некоторых случаях травление осуществляют проткркой тампоном, смоченным в реактиве.
Наиболее широко применяемые реактивы н режимы глубокого травления приведены ниже.
— Коррозиоииостойкие, жаропрочные н другие стали аустенитного класса:
1. 100 мл НС1, 10 мл HNOg, 100 мл воды; /=60=70 °C; т=5=10 мин.
2. 100 мл НС1, 100 мл HNO3, 100 мл воды; /=60=70°C; т=5=10 мин.
3. 100 мл НС1, 100 мл HNO3, 100 мл воды; 11,0—11,5 г двухромовокислого калия; t— =20 °C; т=5=10 мии.
— Коррозиониостойкне, жаропрочные н другие стали феррнтиого или аустенитного класса: травление рекомендуется производить протиркой тампоном, смоченным в реактиве 100 мл HCI, 7 мл H2SO4, 20 г C11SO4 (безводной); /=20 °C; т= 15-7-25 мин; шлиф после травления промыть водой и 5—10 %-иым раствором хромпика.
— Все остальные стали: 50 %-ный водный раствор НС1; /=60=80 °C; т=5=45 мнн.
Образцы перед травлением рекомендуется подогревать до температуры раствора. Время травления должно быть более продолжитель-
15
иым (в рекомендованных пределах) для легированных н коррозионностойкнх сталей, для металла с повышенной твердостью, прн выполнении операции без подогрева или в менее нагретом растворе.
После травления образцы промывают в проточной воде н просушивают. Образцы, предназначенные для хранения, рекомендуется дополнительно обработать 10 %-иым спиртовым раствором аммиака или промыть спиртом, а затем покрыть бесцветным лаком.
После травления макрошлиф приобретает рельефную поверхность с отчетливо видимыми осями дендритов (литая сталь), лнкваци-онной неоднородностью, пористостью, трещинами и другими дефектами *, а также волокнистой структурой (деформированная сталь).
Для поверхностного травления широко используют реактив Гейна, содержащий (на 1000 мл воды) 53 г хлористого аммония (NH4C1) и 85 г хлорной медн (СиС12). Прн погружении макрошлифа в реактив (на 30—60 с) происходит обменная реакция: железо вытесняет медь из водного раствора, и она оседает на поверхности шлифа; на участках, недостаточно защищенных медью (поры, трещины, неметаллические включения), происходит травление. Затем макрошлиф вынимают, слой осевшей меди снимают тампоном под струей воды и протирают макрошлнф досуха, чтобы предохранить его от быстрого окисления на воздухе. Этот реактив хорошо выявляет характер* ликвации (особенно фосфора н углерода), волокнистую структуру деформированной низко -и среднеуглеродистой стали, а также сравнительно крупную пористость, например в сварных соеднненнях. Участки, обогащенные фосфором н углеродом, окрашиваются на макрошлнфах в более темный цвет. Однако реактивы поверхностного травления не могут заменить реактива глубокого травления при выявлении флокенов, а также трещин и пор, не выходящих непосредственно на поверхность металла.
Для выявления или уточнения природы дефектов макроструктуры используют также изучение изломов1 2. Этот способ позволяет обнаружить грубые раскатанные поры н газовые пузыри, грубую пятнистую ликвацию, подусадочную рыхлость, флокены, межкристаллитные прослойки, нафталинистый н камневндный изломы и другие дефекты.
Для исследования .макрошлнфов и изломов прн небольших увеличениях и для нх фотографирования часто применяют стереоскопический микроскоп МБС-2 с микрофотонасадкой МФН-5.
2. Определение химической неоднородности стали. Для этих целей используют методы поверхностного травления и отпечатков.
Для выявления ликвации фосфора наряду с описанным выше реактивом Гейна часто применяют реактивы Обергоффера и Стэда.
Реактив Обергоффера (3 мл НС1; 0,2 г СиС12-2Н2О; Зг FeCl3; 0,1г SnCl2; 10 мл спирта; 100 мл воды) обеспечивает медленное и равномерное формирование картины травления. Темплет подвергают травлению после
1 Описание типичных дефектов макроструктуры см. в разделе 18.
2 См. раздел 3.
тщательного шлифования, полирования и сушки. Обогащенные фосфором места остаются гладкими, в то время как обедненные им становятся шероховатыми из-за образования на них тонкой медной пленки. Контрастность выявления макроструктуры можно повысить, используя многократное промежуточное полирование и повторное травление.
При травлении реактивом Стэда (5 мл НС1; 25 г СиС12-2Н2О; 20 г MgCl2; 500 мл спирта; 100 мл воды) несколько капель реактива наносят на поверхность шлифа и примерно' через 1 мнн сливают. Этот процесс повторяют до достижения желаемой степени вытравливания структуры. Образование медного слоя на поверхности темплета начинается на свободных от фосфора участках и постепенно распространяется на участки феррита с незначительным содержанием фосфора. В результате длительного травления только участки с высоким содержанием фосфора остаются без медного покрытия.
Распределение серы определяют методом сериого отпечатка (метод Баумана) по ГОСТ 10243—75. Поверхность макрошлифа обрабатывают на шлифовальных шкурках № 12 и 8 (по ГОСТ 6456—75) и протирают тампоном, смоченным и спирте, для удаления загрязнений. При снятии отпечатков с высокосернистых (автоматных) сталей макрошлнфы предварительно протирают тампоном, смоченным в 2—5 %-ном растворе H2SO4, удаляя прн этом продукты первичной реакции.
Отпечатки снимают на бромсеребряную фотобумагу, соответствующую размерам темплета (уиибром по ГОСТ 10752—79). Листы фотобумаги выдерживают 5—8 мин на свету в 2—5 %-иом водном растворе серной кислоты, слегка просушивают между листами фильтровальной бумаги и накладывают эмульсионной стороной иа поверхность макрошлифа. С обратной стороны фотобумагу непрерывно проглаживают резиновым валиком или тампоном; не допуская ее смещения, до полного удаления пузырьков газа, образующимися при реакции. Отпечатки снимают при температуре около 20 °C в течение 3—15 мин в зависимости от легирования стали и содержания в ней серы.
Сернистые включения (FeS, MnS), имеющиеся в поверхностных участках металла, реагируют с серной кислотой, оставшейся иа фотобумаге: MnS (FeS)+H2SO4->H2S-1--Г MnSO4 (FeSO4).
Образующийся сероводород непосредственно против очагов выделения воздействует на кристаллики бромистого серебра фотоэмульсии: H2S 4-2AgBrr>Ag2S н-2НВг.
Темные участки сернистого серебра, образующиеся на фотобумаге, показывают форму и характер распределения сульфидов.
Отпечаток считается готовым при потемнении фотобумаги от светло-коричневого (иа легированной стали) до темно-корнчиевого цвета (на углеродистой стали с повышенным содержанием серы, а также фосфора). Снятую с микрошлифа фотобумагу промывают в проточной воде и обрабатывают фиксажем в течение 20—30 мин, после чего снова промывают примерно 10 мин в воде и просушивают. «р
Если в стали и чугуне содержится повышенное количество фосфора, то он в отдельных
участках из-за значительной ликвации может также участвовать в реакции с бромистым серебром, образуя фосфиды сереора темного цвета.
Присутствие свинца в стали и его скопления выявляют методом Брэгга по ГОСТ 10243—75. Подготовленный макрошлнф опускают в 10 %-ный раствор надсернокислого аммония,- выдерживают до получения серой окраски, промывают в проточной воде до удаления серого налета и высушивают. Бромсеребряную фотобумагу обрабатывают для удаления солей серебра фиксажем в течение 7—10 мнн, промывают в проточной воде и высушивают. Затем фотобумагу замачивают в течение 5—7 мнн в 5 %-ном водном растворе едкого натра, слегка просушивают фильтровальной бумагой и накладывают эмульсионной стороной иа образец. Протиркой ватным тампоном в течение 5 мин обеспечивают плотный контакт фотобумаги с поверхностью образца.
Готовый отпечаток погружают иа 10—15 с в 5 %-ный раствор сульфида натрия, промывают н высушивают. При наличии в стали свинца отпечаток приобретает светло-коричневый цвет с темными пятнами в местах ликвации. При отсутствии свинца цвет бумаги не изменяется.
Полученные отпечатки оценивают сравнением с внутризаводскими эталонами; проводят описание наблюдаемого распределения серы или свинца.
3. Определение неоднородности в структуре, созданной термической и химнко-термичес-кой обработкой. Толщину закаленного слоя устанавливают по виду нзлома: он более мелкозернистый, а прн закалке без перегрева фарфоровидиый. Чтобы более точно установить толщину этого слоя, образец шлифуют по излому (перпендикулярно оси) н травят в течение 3 мни в 50 %-ном растворе соляной кислоты при 80 °C. Закаленный слой приобретает более темную окраску.
Наружный цементованный н закалившийся слой также имеет в изломе более мелкое зерно, а прн цементации и закалке без перегрева матовый фарфоровидиый (шелковистый). По толщине этого слоя судят о глубине цементации.
Более точно толщину цементованного слоя можно определить шлифованием места нзлома (перпендикулярно оси) и травлением в реактиве состава: 2 г СиС12-2Й2О н 1 мл НС1 иа 100 мл спирта в течение 1—2 мнн. Мягкая нецементованная сердцевина покрывается красноватым налетом меди вследствие вытеснения ее железом из реактива, а цементованный слой остается нетронутым.
Обезуглероженный слой в изломе прутков поперек волокна отличается светлой крупнозернистой структурой.
4. Последовательность макроанализа. Прн необходимости полного макроскопического исследования, а также определения нарушений сплошности металла и дефектов строения целесообразно придерживаться следующей последовательности: сначала травить образец реактивом поверхностного' травления, затем снова шлифовать н определять распределение, серы по отпечатку на фотобумаге, .после чего' производить глубокое травление для определения нарушений сплошности.
2—280
1.2. ИССЛЕДОВАНИЕ МИКРОСТРУКТУРЫ (МИКРОАНАЛИЗ) [1—5]
В настоящем разделе рассматриваются методы исследования металлов с помощью оптических микроскопов (методы электронной микроскопии— см. раздел 2), позволяющих наблюдать структуру специально подготовленных образцои (микрошлифов) прн увеличениях от 30—50 до 1500—1800.
Микроанализ проводят с целью определения микроструктуры н фазового состава сталей и сплавов, оценки количества, размеров, формы и распределения различных фаз. Этот анализ позволяет установить связь химического состава, условий производства и обработки сплава с его микроструктурой и свойствами.
1.2.1. Приготовление микрошлифов
Процесс изготовления металлографических шлифов обычно включает следующие основные операции: 1) вырезку образца и подготовку поверхности; 2) шлифование; 3) полирование; 4) травление.
В некоторых случаях, например при изучении неметаллических включений в сталях, под микроскопом исследуют нетравленую поверхность шлнфа.. Наряду с травлением в растворах различного состава для выявления структуры используют другие методы.
Хорошо приготовленный микрошлиф должен отвечать ряду требований. Прежде всего он должен быть представительным для структуры н свойств изучаемого объекта (детали).
Вырезка, шлифование и полирование образца должны осуществляться таким образом, чтобы на его поверхности оставался минимальный слой деформированного металла. На поверхности шлифа не должно быть царапин, рисок, ямок н загрязнений. В процессе приготовления шлифа ие должно происходить выкрашивания неметаллических включений карбидных н других фаз. Кроме того, поверхность шлнфа должна быть достаточно плоской, чтобы его можно было рассматривать при больших унеличеннях. Последнее требование особенно важно' при изучении микрошлифов на автоматических количественных микроскопах, где анализ микроструктуры на сравнительно больших участках осуществляется без корректирования фокусировки. Требования к качеству шлифов, изучаемых на автоматических микроскопах для количественного анализа повышенные.
Вырезка образцов. Выбор числа образцов, места их вырезки н сечения материала, по которому проходит плоскость мпкрошлнфа, определяется целью металлографического исследования, размерами, формой и особенностями структуры изучаемого объекта.
При производственном контроле образцы следует отбирать из таких участков, которые дают наибольшую информацию о структурной неоднородности металла; места отбора шлифов обычно оговариваются соответствующими техническими условиями на конкретные виды металлопродукции.
Микроструктуру литых металлов и сплавов
17
(в фасоииых отливках) проверяют в различных сечениях — от самых больших до минимальных, так как различные участки обычно охлаждаются с различной скоростью, а структура многих литейных сплавов сильно зависит не только от состава, но и от скорости охлаждения. В этих случаях важно определить также сечение, по которому следует изготовлять микрошлнф. Часто выбирают плоскость, перпендикулярную поверхности отвода тепла, чтобы можно было определить структуру в периферийных и срединных слоях металла.
Для изучения микроструктуры слитка вырезают несколько образцов таким образом, чтобы можно было определить изменение структуры в поперечном сеченин н по высоте слитка.
При исследовании горяче- и холоднодефор-мированного «металла шлифы обычно изготовляют в плоскости, параллельной направлению течения металла при формоизменении (продольные шлифы), реже — в перпендикулярном направлении (поперечные шлифы). На продольных микрошлифах определяют деформацию, которую претерпели зерна металла и неметаллические включения. Если изделие подвергалось ковке или штамповке, то важно изучить участки наиболее сложной гибки нли большой вытяжки, а также объемы металла, на которые не распространялась деформация.
Структуру сплавов, прошедших термическую обработку, проверяют в поверхностных и более глубоких слоях с целью оценки ее однородности, наличия обезуглероженного или на-углерожениого слоя, глубины закаленного слоя и т. д.
Если образец имеет тонкий поверхностный слой, отличающийся от основного металла по структуре и фазовому составу (например, при нанесении покрытий или химико-термической обработке), то используют косые шлифы, плоскость которых расположена под острым углом к поверхности образца. Такне шлифы позволяют более детально исследовать структуру тонкого поверхностного слоя, облегчают измерение его микротвердостн или толщины.
Если требуется установить причину разрушения нли природу дефекта в металле, то образцы обычно вырезают таким образом, чтобы плоскость шлифа пересекала дефект или была вблизи места разрушения. Для сравнения исследуют также образцы из бездефектных участков металла.
Площадь поверхности образцов, используемых для приготовления шлифов, обычно восставляет 1—4 см1 2. Высота образца определяется удобством манипулирования прн шлифовании и полировании н обычно составляет 10—15 мм.
Вырезку образцов следует проводить, соблюдая определенные меры предосторожности, чтобы не вызвать изменения структуры из-за наклепа илн нагрева. Наиболее часто для вырезкн образцов в металлографических лабораториях используют отрезные станки с абразивными кругами. Для удовлетворительной резки, обеспечивающей отсутствие прижо-гов и значительного деформационного повреждения поверхности, важно выбрать соответствующий круг н режим резания. Для резки
сталей предпочтительнее использовать круги с абразивными частицами нз А12О3, а для резки цветных металлов — круги с частицами SiC. Грубозернистые круги обычно более быстро и с меньшим нагревом режут крупные сечения, а мелкозернистые позволяют получить лучшую чистоту поверхности н исключить прижог при резке деталей малого сечения (например, тонкостенных труб). Для резки мягких материалов обычно применяют твердые круги (с твердым связующим материалом), а для резки твердых материалов — мягкие круги.
Во всех случаях резку абразивными кругами следует проводить с использованием охлаждающей жидкости.
Подготовка поверхности. Наряду с резкой абразивными кругами получить плоскую поверхность (необходимую для шлифа) можно путем токарной обработки, фрезерования, шлифования нли опиливания.
Образцы малых размеров (лента, проволока) или сложной конфигурации после вырезки для изготовления шлифов помещают в пластмассы или легкоплавкие сплавы \ используя заливку или запрессовку в цилиндрические обоймы. Закрепление образцов в пластмассах обычно используют также прн изготовлении шлифов на станках для автоматического шлифования илн полирования, поскольку эти станки часто сконструированы для образцов определенного размера, устанавливаемых в специальных держателях, или для одновременной обработки нескольких шлифов, закрепляемых посредством пластмассы в общей обойме.
Материалы для заливки илн запрессовки образцов должны быть химически стойкими по отношению к применяемым реактивам для травления и обладать достаточно плотным сцеплением с поверхностью образца, чтобы исключить образование у кромки шлифа зазора, в который может проникать травитель, абразивные частицы или загрязнения. Кроме того, этн материалы должны быть достаточно' твердыми и износостойкими, чтобы обеспечить сохранение плоской поверхности шлифа в процессе его дальнейшей обработки и предохранить кромки шлнфа от закругления (завала), что особенно важно при исследовании структуры поверхностных слоев металла.
Для запрессовки образцов используют порошки из термореактнвиых н термопластичных смол. Процесс запрессовки в. этн материалы осуществляют в специальных прессах при совместном действии давления и иагрева. Термореактивные смолы могут быть удалены из пресс-формы при максимальной температуре формовки; термопластичные смолы для затвердевания необходимо охлаждать в пресс' форме почти до комнатной температуры. Наиболее часто для этих целей применяют термореактивные фенольные и эпоксидные смолы илн термопластичные акриловые смолы. Иногда в смолы вводят различные наполнители, например твердые частицы нли короткие стеклянные волокна для придания износостойкости и сохранения кромок шлифа без завалов, илн медный порошок для придания электро
1 Для заливки используют наиболее легкоплавкие сплавы, так как структура излучаемых образцов
может изменяться при нагреве.
18
проводности, если образцы в дальнейшем подвергают электролитическому полированию.
Если действие нагрева и давления прн запрессовке может привести к нежелательным изменениям структуры нлн вызвать деформацию образцов тонкого сечения, то можно использовать холодную заделку образцов с помощью эпоксидных, полиэфирных и акриловых смол. Этот метод не требует специального оборудования, позволяет одновременно монтировать большое число образцов, поэтому его широко применяют в металлографических лабораториях. Образцы устанавливают в металлические, пластмассовые или стеклянные кольца н заливают смесью смолы с отвердителем. Наиболее часто для холодной заделки шлнфов используют эпоксидные смолы. Они обладают достаточной твердостью, наименьшей объемной усадкой прн отвержденнн н хорошо соединяются с большинством металлических образцов.
Прн изготовлении шлнфов из тонкого листового материала применяют также зажимы (струбцины) в виде двух стальных пластин толщиной 4—6 мм, которые стягиваются болтами. В зажиме можно монтировать сразу пакет из нескольких образцов, которые иногда разделяют прокладками из медн илн другого мягкого материала, чтобы устранить зазоры по кромкам образцов.
Шлифование. После получения плоской поверхности образец шлифуют бумажной шлифовальной шкуркой вручную или иа шлифовальных станках. При ручном шлифовании шкурку помещают па плоское твердое основание (обычно толстое стекло). Образец прижимают шлифуемой поверхностью к шкурке и ритмично перемещают вперед н назад по прямой линии. Прн механическом шлифовании шкурку закрепляют на вращающемся круге с помощью зажимных колец или клеевого покрытия иа обратной стороне шкурки, а образец прижимают к шкурке вручную или устанавливают в зажимное приспособление станка.
Для сухого шлифования используют шлифовальную бумажную шкурку, соответствующую ГОСТ 6456—75. Маркировка шкурки включает ее тип (для металлов применяют шкурки типа I), способ нанесения абразивного материала, размеры листов (рулонов), марку бумаги-основы, марку абразивного материала, зернистость, тнп связки н класс износостойкости. Например, маркировка «1Э 620X50 П2 15А 25-Н М А» по ГОСТ 6456—-75 соответствует бумажной шлифовальной шкурке типа I с абразивным материалом, нанесенным электростатическим способом, шириной 620 мм, длиной 50 м; бумага-основа марки 0-200; абразивный материал— нормальный электрокорунд марки 15А зернистостью 25-Н на мездровом клее; класс износостойкости А.
Для мокрого шлифования используют водостойкую бумажную шлифовальную шкурку (ГОСТ 10054—75). Маркировка этой шкурки включает размеры листа (рулона), марку абразивного материала, зернистость н класс износостойкости. Например, маркировка «Водостойкая 310 X 230 64С20П А ГОСТ
10054—75» соответствует водостойкой шлифовальной шкурке размерами 310x230 мм из зеленого карбида кремния марки 64С зернистостью 20П, класс износостойкости А.
Марки абразивных материалов по ГОСТ 6456—75 следующие:
Электрокорунд нормальный — 16А; 15А; 14А; 13А
Электрокорунд белый — 25А; 24А; 23А
Электрокорунд легированный — 35А; 34А; 33А; 32А
Монокорунд — 45А; 44А; 43А
Карбид кремния зеленый — 64С; 63С
» » черный — 55С; 54С; 53С
Кремеиь — 81Кр
Стекло — 71 F-
Зернистость шлифовальных шкурок приведена в табл. 1.1.
Шлифование проводят, используя шкурку нескольких номеров с последовательно уменьшающейся зернистостью. Во время шлифования на каждой шкурке следует сохранять одно н ТО' же положение образца, чтобы все риски на его поверхности были параллельны. При переходе к шкурке следующего номера направление шлифования изменяют на 90° н проводят его до полного' удаления всех рисок, образовавшихся во время предыдущей операции.
ТАБЛИЦА 1J
РАЗМЕРЫ АБРАЗИВНЫХ ЧАСТИЦ
И ОБОЗНАЧЕНИЯ ЗЕРНИСТОСТИ БУМАЖНОЙ ШЛИФОВАЛЬНОЙ ШКУРКИ, ИСПОЛЬЗУЕМОЙ ДЛЯ ПРИГОТОВЛЕНИЯ МЕТАЛЛОГРАФИЧЕСКИХ ШЛИФОВ
Размеры абразивных частиц*1, мкм Обозначение зернистости Обозначение зернистости сходной водостойкой шкурки по стандартам FEPA*® (6]
по ГОСТ 6456—75 по ГОСТ 10054—7о*2 (водостойкая) прежнее*1 новое
320—250 25 60 Р60
200—160 16 16-П; 16-Н 80 Р80
160—120 12 12-П; 12-Н 100 Р100
120—100 10 Ю-П; 10-Н 150 Р120
100—80 8 8-П; 8-Н 180 Р150
80—63 6 6-П; 6-Н 240 Р180
63—50 5 5-П; 5-Н 280 Р240
50—40 4 4-П; 4-Н 320 Р320
40-28 М40 М40-П; М40-Н — Р400
28—20 — М28-П; М28-Н 400 Р600
20—14 '—- М20-П; М20-Н 600 Р1000
Указана крупность основной фракции абразивных частиц по ГОСТ 3647—80.
*2 Буквенные индексы П и Н обозначают содержание основной фракции абразивных частиц в пределах 55—60 й 40—45 % соответственно.
*3 Федерация западноевропейских производителей абразивных материалов.
*4 Аналогичные обозначения используются американскими фирмами.
2*
19
Важная задача шлифования — достижение минимальной толщины слоя деформированного металла, чтобы последние его следы можно было удалить последующим полированием. Глубина царапин и толщина слоя деформированного металла под царапинами уменьшаются с уменьшением размера абразивных частиц; при этом толщина слоя деформированного металла примерно обратно пропорциональна твердости образца н в 10—50 раз превышает размеры абразивных частиц [3]. Необходимо, чтобы на каждой ступени шлифования происходило полное удаление слоя деформированного металла, образовавшегося на предыдущей ступени.
Для оценки качества шлифования можно исследовать поверхность образца прн переходе от одной ступени к другой под микроскопом. После каждой ступени шлифования поверхность должна быть покрыта равномерными по величине и отчетливо видимыми царапинами; никаких следов царапин от предыдущего шлифования наблюдаться не должно.
Большое значение прн шлифовании имеет величина давления, приложенная к образцу. Давление должно быть достаточно высоким, чтобы обеспечить резание абразивом н нужную скорость шлифования. Недостаточное давление создает условия трения, не обеспечивающие эффективного' удаления металла. В то же время чрезмерное давление приводит к нежелательному выделению тепла, неравномерной величине царапин, вдавливанию абразивных частиц в металл, а также преждевременному износу и фрагментации абразива. Обычно оптимальное давление зависит от материала образца и определяется опытным путем.
После каждой ступени шлифования поверхность образца следует тщательно очищать во избежание переноса сравнительно крупных частиц абразива иа более мелкозернистый абразивный материал, используемый прн последующих ступенях шлифования. Для этой цели образцы обычно промывают в воде.
В последнее время в металлографических лабораториях наиболее часто используют мокрое шлифование на водостойких шлифовальных шкурках, обеспечивающее эффективное охлаждение и постоянную очистку шкурки н образца.
При шлифовании очень мягких металлов в ряде случаев шкурку предварительно смачн-иают в керосине или натирают парафином (например, при изготовлении мнкрошлифов из алюминия), чтобы свести к минимуму вдавливание абразивных частиц в поверхность шлифов.
В качестве промежуточной операции между тонким шлифованием н полированием используют притирку. В этом случае абразив наносят на шлифовальный (притирочный) круг, изготовленный из чугуна нли таких материалов, как дерево, свинец, нейлон, парафин, бумага нли специальная ткань. Абразив может быть запрессован в материал притирочного круга с помощью стальной плитки нли подаваться на круг в виде смеси абразивных частиц с водой различной консистенции (от жидкой дб пастообразной), а также специальных паст.
Хорошее качество обеспечивает, в частности, использование операции прнтнрки на бумажном круге (два слоя ватмана) после обработ
ки на шлифовальных шкурках зернистостью 3 илн 4 с нанесением пасты следующего состава: 5.00 г шлифовального порошка М28; 90 г стеарина; 40 г вазелина; 5—7 г керосина.
Полирование. Полирование служит для удаления мелких рисок, оставшихся после шлифования, и получения гладкой зеркальной поверхности шлифа. Применяют механический, электрохимический и химико-механический методы полирования.
Механическое полирование производят на вращающемся круге с натянутым полировальным материалом (фетр, сукно, драп илн специальная ткань), на который непрерывно илн периодически наносят очень мелкий абразив в виде суспензии в воде (желательно дистиллированной). В качестве абразивов применяют оксид хрома, оксид алюминия и оксид железа (крокус). Все более широкое использование находят полировальные алмазные пасты, которые наносят на специальную ткань нли бумагу (ватман).
Пасты состоят из алмазных микро порошков марок АСМ илн AM, связующих и поверхио-стно-актнвных веществ. В зависимости от зернистости пасты условно подразделяют на четыре группы, которые окрашивают в различные цвета.
Пасты выпускают нормальной (Н) н повышенной (П) концентрации с соответствующим содержанием алмазного микропорошка для каждой зернистости. Кроме того, имеются пасты, смываемые водой (В) н органическими растворителями (О) (спирт, беизнн н др.), а также смываемые как водой, так н органическими растворителями (ВО). По консистенции пасты подразделяют на мазеобразные (М) и твердые (Т).
Основные характеристики алмазных паст приведены в табл. 1.2.
Пример маркировки пасты из порошка марки АСМ зернистостью 5/3, нормальной концентрации, смываемой органическими растворителями, мазеобразной консистенции: «Паста алмазная АСМ 5/3 НОМ СТ СЭВ 206—75».
ТАБЛИЦА 1.2
ОСНОВНЫЕ ХАРАКТЕРИСТИКИ
ПОЛИРОВАЛЬНЫХ АЛМАЗНЫХ ПАСТ
Обозначение зернистости по ГОСТ 9206-70 Размеры абразивных частиц*, мкм Концентрация алмазного порошка. % (по массе) Цвет пасты и этикетки
Н п
60/40 60—40 10 20 Красный
40/28 40—28 7 14 »
28/20 28—20 7 14 Голубой
20/14 20—14 5 10
14/10 14—10 5 10 »
10/7 10—7 3 6 Зеленый
7/5 7—5 3 6 »
5/3 5—3 2 4 »
3/2 3—2 2 4 Желтый
2/1 2—1 1 2 »
1/0 До 1 1 2 »
* Для основной фракции, составляющей не менее 65 %; для пасты 1/0 — не менее 95 %.
20
Процесс полирования обычно состоит из двух или трех операций. Для грубого полирования используют абразивы с размером частиц 1 — 10 мкм н сравнительно твердые ткани без ворса нли с коротким ворсом. Для тонкого полирования применяют абразивы с размером частиц менее 1 мкм и мягкие ворсистые ткани. Скорость вращения полировальных кругов диаметром 200 — 250 мм прн грубом полировании обычно составляет 400 — 600 об/мив, а при топком — менее 300 об/мии.
Полировальный круг должен быть достаточно влажным. Для проверки влажности круга оценивают время, необходимое для сушки образца после его снятия с круга; обычно это время должно быть в пределах 5 — 8 с.
Важное условие получения качественных шлифов — тщательное соблюдение чистоты при полмровавии. Желательно, чтобы помещение для полирования было отделено от помещения, где производят резку н шлифование образцов. После каждой операции приготовления шлифа образец необходимо тщательно промывать под струей воды, чтобы исключить загрязнение полировального круга абразивными частицами н продуктами резаиия, внесенными с предыдущих операций. Если в образце имеются мелкие поры н трещины, а также если для полирования используются алмазные пасты, желательно применять ультразвуковую очистку.
Полирование осуществляют вручную нли на автоматических станках. При ручном полировании образец непрерывно перемещают от центра к периферии, что обеспечивает равномерное распределение абразива и однородный износ полировального материала. Кроме того, образец периодически вращают нли перемещают «восьмеркой», чтобы исключить образование «хвостов» около неметаллических включений и частиц выделившихся фаз.
Давление на образец определяют опытным путем. Как правило, оио незначительно и уменьшается по мере перехода от грубого полирования к более тонкому.
Прн автоматическом полировании одни или несколько образцов устанавливают в специальное приспособление станка, которое обеспечивает приложение требуемого давления к образцам и их вращение в направлении, обратном вращению круга.
Полирование считают законченным, когда иа поверхности шлифа под микроскопом не наблюдаются риски или царапины.
* * *
Для приготовления шлифов часто используют шлифовально-полировальные станки «Не-рнс» типа ЗЕ881 (рис. 1.1). Широкое применение в металлографических лабораториях находит оборудоиаине, выпускаемое фирмой «Kathenow» (ГДР): шлифовально-разрезные станки «Метасекар», обеспечивающие получение достаточно чистой и плоской поверхности реза прн диаметре сечения до 60 мм, станки для мокрого шлифования «Метасинекс» и полировальные станки «Монтазупал 201».
Ниже приведены некоторые типичные схемы приготовления шлифов.
Схема I (оптимальная):
— резка абразивным кругом с охлаждающей жидкостью;
— мокрое шлифование на шкурках зернистостью 5(4), М40, М28 и М20;
— полирование иа алмазных пастах зернистостью 7/5 и 3/2 (илн 1/0);
— окончательное полирование на тонком сукне (драп-велюр, биллиардное, сукно и т. п.)
Рис. 1.1. Шлифовально-полировальный станок «Neris» типа ЗЕ8Б1
с суспензией оксида хрома нли оксида алюминия (обычно для шлифов с низкой твердостью).
Схема II (прн отсутствии оборудования для мокрого шлифования и алмазных паст):
— обработка на наждачном камне зернистостью 40 нлн 46;
— шлифование иа шкурках 16, 12, 5, 4, 3;
— полирование на плотном сукне (например, шинельном) с суспензией оксида хрома;
— см. схему I, п. 4.
Схема III (для ускоренного приготовления шлифов):
— обработка на наждачном камне зернистостью 40;
— шлифование на шкурке 4 нли 3;
— притирка на бумажном круге (ватман) с пастой, содержащей шлифовальный порошок М28 (см. с. 20);
— полирование на алмазных пастах зернистостью 28/20, 14/10 и 7/5;
— см. схему I, п. 4.
Электролитическое полирование [4, 7, 8] основано иа использовании процесса анодного растворения металла, который при определенных условиях протекает с образованием гладкой блестящей полированной поверхности. Образец после механического шлифования погружают в качестве анода в электролизную ванну н выдерживают при заданном режиме (напряжении, плотности тока и температуре электролита) определенное время. Катодом обычно служит пластинка из нержавеющей стали.
Согласно одной из наиболее распространенных теорий при электрополированни у поверхности анода образуется вязкая пленка, состоящая из продуктов взаимодействия металла с электролитом. На выступах поверхности шлифа толщина пленки н соответственно ее электросопротивление меньше, чем во впади
нах, что обусловливает преимущественное удаление металла с выступов.
Основным преимуществом элечтрополиро-вания является отсутствие иа поверхности шлнфа деформированного слоя, образующегося при шлифовании или механическом полировании и часто не удаляющегося полностью при последующем травлении. Этот метод особенно подходит для полирования шлифов из мягких металлов н легко наклепывающихся сплавов. Кроме того, поскольку электрополирование устраняет наклеп, его применяют прн изготовлении образцов для измерения микротвердости, рентгеноструктурного анализа и электронно-микроскопического исследования. Возможность получения высококачественной зеркально отполированной поверхиостн непосредственно после сравнительно грубой механической обработки значительно ускоряет процесс приготовления шлнфов н позволяет экономить время и абразивные материалы. Однако электролитическое полирование имеет ряд недостатков, ограничивающих его применение: чувствительность к неоднородности химического состава, преимущественное растворение металла вокруг пустот и неметаллических включений, краевые эффекты (затрудняющих использование метода для образцов малых размеров) и т. п.
Метод электрополировання применим для всех чистых металлов, однофазных сплавов, а также тех гетерогенных сплавов, у которых анодное растворение отдельных структурных составляющих происходит с примерно одинаковой скоростью, в частности для большинства сталей.
Важнейшие факторы, влияющие иа процесс полирования: перемешивание электролита; исходное состояние поверхности образцов; размеры электродов и расстояние между ними. В ряде случаев без перемешивания ванны качественное полирование невозможно.
Составы электролитов и режимы электролитического полирования для некоторых материалов приведены ниже:
1. Углеродистая или низколегированная сталь. Состав ванны: 185 мл хлорной кислоты, 765 мл уксусного ангидрида, 50 мл воды; 7=30 °C; 6=4-5-7 А/дм2; £7=50 В; материал катода — железо или алюминий; т=4=10 мин.
2. Доэвтектоидная сталь. Состав ванны: 70 мл хлорной кислоты, 800 мл этилового спирта, 130 мл воды; £=35 °C; 6=250=400 А/дм2; £7=36=70 В; материал катода — нержавеющая сталь; т= 15=30 с.
3. Заэвтектоидная сталь. Состав ванны: 20 мл хлорной кислоты, 800 мл этилового спнрта; 7=35 °C; 6=400= 500 А/дм2; £7=110 В; материал катода — нержавеющая сталь; т= = 10=15 с.
4. Сталь мартенситного класса. Состав ван-пы: 135 мл ледяной уксусной кислоты, 25 г хромового ангидрида, 7 мл воды; £=18°C; 6= =90=250 А/дм3; £7=20 В; материал катода — нержавеющая сталь; т=4=6 мин.
5. Быстрорежущая сталь, углеродистая и легированная сталь, нержавеющая сталь. Состав ванны: 200 мл хлорной кислоты, 700 мл этилового спирта; 7=16=32 °C; 6=50=
=220 А/дм2; £7=54-15 В; материал катода — нержавеющая сталь; т=0,5=1,5 с, 10=15 с н 20=30 с соответственно для указанных сталей.
6. Нержавеющая сталь типа 18-8. Состав ванн:
а) 85 мл хлорной кислоты, 800 мл этилового спнрта, 115 мл воды; 7=25=30 °C; 6=50-4-=60 А/дм2; материал катода — нержавеющая сталь;
б) 420 мл ортофосфорной кислоты, 470 мл глицерина, 150 мл воды; £=100 °C; 6=15= =20 А/дм2; материал катода — нержавеющая сталь; т=84-15 мнн;
в) 133 мл ледяной уксусной кислоты, 25 г хромового ангидрида, 7 мл воды; £=18 °C; 6= 90-4-250 А/дм2; £7=20 В; т=4=6 мин.
7. Жаропрочная сталь типа 14-14-2. Состав ванны: 43 % серной кислоты (плотность 1800 кг/м3), 40 % ортофосфорной кислоты, 3 % хромового ангидрида. 14 % воды; £=70= 4-80 °C; 6=40-4-50 А/дм2; £7=84-12 В; т= = 10 мнн.
8. Высоко хромистая сталь. Состав ванны: 30 мл хлорной кислоты, 1000 мл ледяной уксусной кислоты, 10 мл воды; 7=20 =30 °C; 6= ' =24=26 А/дм2; £7=244-26 В.
9. Белый чугун. Состав ваниы: 85 мл хлорной кислоты, 800 мл этилового спирта, 115 мл воды; 7=20 = 30 °C; 6=50=600 А/дмэ; £7=110 В; т= 154-20 с.
10. Чугун с шаровидным графитом. Состав ванны: 70 мл хлорной кислоты, 800 мл этилового спнрта, 130 мл воды; 7=20=30 °C; 6= = 60=100 А/дм2; £7=10=15 В; т= 154-20 с.
Рис. 1.2. Прибор «Элнповист» для электролитического полирования
11. Серый чугун. Состав ванны: 200 мл хлорной кислоты, 700 мл этилового спирта, 100 мл глицерина, 35 % (по массе) щавелевой кислоты; 7=20=30°С; 6=30=40 А/дм2; £7=1,5 В.
12. Кремнистое железо (выявление дислока
22
ций). Состав ванны: 133 мл уксусной кислоты, 25 г хромового ангидрида, 7 мл воды; t— = 17-ь 19 °C; режим устанавливается опытным путем.
13. Никель. Состав вани:
а) 390 мл серной кислоты (концентрированной), 290 мл воды; t=35 °C; материал катода— никель; т—4-ьб мин;
б) 1 ч. хлорной кислоты, 2 ч. уксусной кислоты; £=18 °C; 6=30 А/дм2; £/=30 В; материал катода — нержавеющая сталь; т=1 мни.
14. Молибден (спеченный, литой илн сплавы молибдена). Состав ваниы: 100 мл серной кислоты (концентрированной), 700 мл метилового спирта; 1=21 °C; 6=204-50 А/дм2; £/=124-
4-18 В; материал катода — нержавеющая сталь нлн платина; х~30=90 с.
Следует отметить, что электролиты, содержащие хлорную кислоту, взрывоопасны; нельзя повышать температуру ваииы, энергично ее перемешивать, а также монтировать шлнфы в пластмассу.
В табл. 1.3 даны типичные дефекты электролитического полирования н указаны способы нх устранения.
Наряду с разнообразными лабораторными установками сравнительно простой конструкции для электролитического полирования используют специальные серийно выпускаемые приборы, например прибор «Элнповист» (рнс.
ТАБЛИЦА 1.3
ДЕФЕКТЫ ЭЛЕКТРОЛИТИЧЕСКОЙ ПОЛИРОВКИ И СПОСОБЫ ИХ УСТРАНЕНИЯ*
Дефект Возможная причина Способы устранения
Сильное разъедание центра образца Питтинг и разъедание краев образца Осадки на поверхности Грубая или матовая поверхность Волнистость илн риски на полированной поверхности Пятна на полированной поверхности Неотполированные места Образование рельефа от различных фаз Питтинг В центре образца не образуется полирующий слой Полирующий слой очень вязкий или очень тонкий Осаждение на аноде нерастворимых продуктов Полирующий слой не обеспечивает нормальных условий полировки 1. Недостаточная продолжи- тельность полировки 2. Интенсивность перемешивания слишком высока нли слишком низка 3. Плохая подготовка поверхности 4. Слишком большая продолжительность полирования Травление после отключения тока Газовые пузырьки Полирующий слой ие обеспечивает нормальных условий полировки 1. Очень большая продолжительность полировки 1. Повысить напряжение 2. Уменьшить интенсивность перемешивания 3. Применить более вязкий электролит 1. Уменьшить напряжение 2. Увеличить интенсивность перемешивания 3. Применить менее вязкий электролит 1. Выбрать другой электролит 2. Повысить температуру 3. Повысить напряжение 1. Повысить напряжение 2. Применить более вязкий электролит 1. Увеличить или уменьшить интенсивность перемешивания 2. Улучшить подготовку поверхности 3. Повысить напряжение, уменьшив продолжительность полировки 1. Извлекать образец немедленно после отключения тока 2. Выбрать менее активный электролит Увеличить интенсивность перемешивания 1. Повысить напряжение 2. Улучшить подготовку поверхности 3. Уменьшить продолжительность полировки 1. Улучшить подготовку поверхности 2. Понизить напряжение 3, Уменьшить продолжительность полировки 4. Испробовать различные электролиты
* По данным Американского общества испытания материалов (ASTM).
23
1.2) фирмы «Karl Zeiss, Jena» (ГДР), который позволяет быстро осуществлять процесс полирования при плавном регулировании напряжения и скорости циркуляции электролита; прн этом одновременно можно наблюдать поверхность образца под микроскопом.
Для сплавов, которые содержат фазы, не растворяющиеся или незначительно растворяющиеся прн электрополированнн, применяют комбинированный метод электролитически-ме-ханического полирования. В этом случае полирование производят на вращающемся диске нз коррозионностойкой стали, обтянутом полировочным сукном, в среде электролита с добавками абразива (рис. 1.3). При этом диск служит катодом, а образец — анодом.
Рис. 1.3. Схема электролитически-механической полировки:
/ — полировальный круг (катод): 2— образец (анод);
3 — электролит
Химико-механическое полирование производят полировальным кругом, иа который вместе с абразивом наносят химические вещества, способствующие более быстрой обработке. Для полирования черных металлов применяют тонкие шлифовальные пасты, а для полирования цветных и некоторых редких металлов — травящие химически активные реактивы (например, раствор желтой кровяной соли), ускоряющие процесс полирования, а в некоторых случаях также выявляющие микроструктуру без специального последующего травления.
После полирования, независимо от способа его выполнения, мнкрошлнф промывают водой, затем, если сплавы окисляются, спиртом и просушивают фильтровальной бумагой.
1.2.2. Методы световой микроскопии [1, 3—5, 10—13]
Принцип действия и устройство металлографического микроскопа. Для изучения микроструктуры металлов используют металлографические микроскопы (рнс. 1.4). Подготовленный соответствующим образом шлиф 1 помещают перпендикулярно оптической оси микроскопа в плоскости, совпадающей с передней главной фокальной плоскостью объектива 2. Шлиф освещается проходящим через объектив почти параллельным оптической осн пучком света, который формируется посредством осветительной системы, состоящей из источника (лампы) 3, коллекторной линзы 4, апертурной 5 и полевой 7 диафрагм, вспомогательных линз 6, 8 и полупрозрачной пластинки 9. Световые лучи, отражающиеся от участков поверхности шлифа, приблизительно нормальных оптической осн микроскопа, попадают в объектив, а те лучи, которые отражаются от неровностей поверхности, не попадают в его поле. На конечном изображении поверхности
шлнфа, создаваемом окуляром 10, все отражающие свет участки, перпендикулярные оптической оси микроскопа, оказываются светлыми, а участки, наклоненные к оси. — темными. Благодаря этому выявляются различные элементы структуры металла, например гранн-
Рис. 1.4. Принципиальная схема металлографического микроскопа
цы зерен, которые прн подготовке шлнфа обычно вытравливаются в канавки, нли частицы выделений, включения н поры.
Объектив создает обратное действительное увеличенное изображение образца в передней фокальной плоскости окуляра Si. Окуляр дополнительно увеличивает это изображение н дает окончательное мнимое увеличенное изображение образца Ss на расстоянии ~250 мм от глаз наблюдателя 11.
Прн фотографировании изображения илн его наблюдении на экране вместо «глазных» окуляров используют специальные фотоокуляры (нли гомалн), которые принимают световые лучи, идущие непосредственно нз объектива, н создают действительное первичное изображение на фотопластинке или экране.
Увеличение микроскопа равно произведению соответствующих увеличений объектива и окуляра. Основное увеличение обеспечивается объективом, оно может достигать 100. Увеличение окуляра обычно не превышает 20. Если необходимо точно определить увеличение проецируемого изображения, то в качестве объекта следует использовать пластинку с микрометрической шкалой (объект-микрометр), на которой нанесены через каждые 0,01 мм деления иа общей длине 1 мм.
Разрешающая способность микроскопа характеризуется минимальным расстоянием d между двумя соседними деталями структуры объекта, которые еще могут быть раздельно различимы. Ограничения разрешающей способности оптических приборов связаны с дифракционными явлениями и аберрациями элементов оптических систем. Максимальная разрешающая способность микроскопа соответствует условию:
d — Х/2л sin а = X/271, (1)
где X — длина волны света; п — показатель преломления среды между объектом н объективом (для воздуха п=1); о — угловая апер
тура объектива, равная половине угла, под которым виден зрачок объектива из точки предмета, лежащей на оптической осн. Величина А —п sin о, называется числовой апертурой и является важнейшей характеристикой объектива. Числовую апертуру объектива можно увеличить, заполняя пространство между объективом и объектом (шлифом) иммерсионным маслом (обычно кедровым) с п=1,52. Для этой цели используют специальные иммерсионные объективы.
Поскольку величина а практически не бывает больше ~72° н максимальное значение sin а я» 0,95, максимальное значение числовой апертуры для «сухого» объектива составляет 71 = 1-0,95=0,95, а для иммерсионного объек-тива А = 1,52 • 0,95= 1,44.
Для освещения объекта наиболее часто применяют белый свет, для которого можно принять Х=0,55 мкм. Согласно условию (1) максимальная разрешающая способность микроскопа равна: cf~0,55 :2 :1,44^0,2 мкм.
Чтобы использовать разрешающую способность объектива, т. е. увидеть те детали структуры объекта, которые разрешаются объективом, необходимо установить соответствующее увеличение микроскопа. Увеличение микроскопа /V называют полезным, если разрешаемые детали структуры можно наблюдать под углом зрения 2'—4'. Полезное увеличение находится в пределах Л'—50071 = 100071 (2).
Увеличение меньше 500А не позволяет различить все детали структуры, изображение которых формируется объективом прн апертуре А, а применение увеличений, превышающих 1000А, нецелесообразно, поскольку оно не дает каких-либо новых деталей в изображении структуры, а лишь приводит к ухудшению качества изображения.
При исследовании структуры металла объектив выбирают, исходя из необходимого полезного увеличения микроскопа, определяемого из выражения 7V=2OO/dz, где d' — минимальный размер интересующих деталей структуры (например, частиц какой-либо фазы), мкм; 200—разрешаемое расстояние для глаза наблюдателя, мкм.
Зная величину N, можно определить соответствующую числовую апертуру по формуле (2) н выбрать объектив, а затем окуляр.
Следует учитывать, что в практике металлографических исследований иногда приходится в ущерб разрешающей способности заботиться о повышении контрастности изображения и об увеличении глубины резкости, характеризуемой величиной вертикального смещения деталей микроструктуры, которое не приводит к потере фокусировки.
Эта величина обратно пропорциональна числовой апертуре и общему увеличению микроскопа, т. е. при более рельефной поверхности образца целесообразно использовать объективы с малой апертурой. Контрастность изображения растет до тех пор, пока общее увеличение микроскопа ие превзойдет полезного увеличения. Поэтому увеличение окуляра ие должно быть излишне высоким, так как это вызывает размытие изображения деталей структуры.
Объектив микроскопа состоит нз нескольких линз, установленных коаксиально. Система линз обеспечивает более илн менее полное устранение дефектов изображения (аберраций), к которым относятся хроматическая н
сферическая аберрации, астигматизм, кома, кривизна изображения и дисторсия.
Хроматическая аберрация обусловлена тем», что прн использовании немонохроматнческого света лучи с меньшей длиной волны преломляются лнизой сильнее, чем лучи с большей длиной волны; в результате возникают изображения разной величины, располагающиеся в различных плоскостях.
Сферическая аберрация связана с различным преломлением монохроматических лучей, проходящих через различные участки линзы. В случае световых лучков с довольно большим диаметром к сферической аберрации добавляются дефекты асимметрии (кома), в результате которых изображение отдельных деталей образца, располагающихся на некотором расстоянии от оси линзы, получается размытым. Вследствие астигматизма прн прохождении через лнизу пучка лучей от светящегося точечного источника, расположенного вие оптической оси, образуются две фокусные лнннн, находящиеся в разных плоскостях, а изображение точки в промежуточных плоскостях имеет форму круглого нлн эллиптического пятиа рассеяния.
Аберрация, называемая дисторсией, связана с различным увеличением деталей объекта, находящихся на разном расстоянии от оптической оси, так что изображения прямых линий оказываются искривленными и нарушается подобие в геометрической форме между предметом и его изображением. И наконец, возможно искривление изображения, прн котором точечные изображения, возникающие от плоского объекта, перпендикулярного оптической оси, лежат не иа плоскости, а на искривленной поверхности.
В зависимости от степени исправления аберраций и области спектра, в которой онн работают, объективы металломикроскопов делятся иа ахроматы, апохроматы, планахроматы и планапохроматы.
У ахроматических объективов исправлена сферическая аберрация, кома н хроматическая абберация для двух цветов, наиболее важных для визуального наблюдения; кривизна изображения не исправлена. Апохроматические объективы отличаются более высокой степенью исправления сферической аберрации и комы, а также обспечивают более правильную цветопередачу. В сочетании с компенсационными окулярами эти объективы дают высокое качество изображения и особенно подходят для больших увеличений н микрофотографирования. Планахроматы и планапохроматы скорректированы соответственно так же, как ахроматические н апохроматические объективы, н, кроме того, у них неправлена кривизна изображения.
Окуляры микроскопов, как и объективы, характеризуются собственным увеличением, а также степенью коррекции изображения. Современные металлом нкроскопы снабжаются окулярами с увеличениями от Б до 20. По роду н степени коррекции различают следующие основные типы окуляров: 1) простые, нли окуляры Гюйгенса, используемые обычно при визуальной работе с объективами-ахроматами с низкой или средней апертурой; 2) компенсационные окуляры, специально рассчитанные на исправление остаточных хроматических аберраций объективов-апохроматов и применяемые
25
с этими объективами; 3) фотоокуляры и го-малн, которые предназначены для микрофотографирования или проектирования изображения на экран.
Для четкого наблюдения микроструктуры важно создать определенные условия освещения шлифа. Контрастность изображения возрастает с увеличением интенсивности освещения. Поэтому с учетом сложного пути луча в микроскопе и значительных потерь света применяемые источники света должны обладать достаточной мощностью прн сравнительно малых габаритах. Для этих целей в современных металломикроскопах обычно используют Кварцевые лампы с йодным циклом (галогенные лампы), а для получения наибольшей интенсивности — ксеноновые лампы высокого давления. Для уменьшения потерь интенсивности падающего света в некоторых микроскопах вместо полупрозрачной пластинки в ход лучей вводят призму.
Увеличение светопропускання и повышение контрастности изображения достигаются также в результате применения просветленной оптики, обеспечивающей устранение рефлексов при отражении.
1.2.3. Методы микроскопического исследования металлов
Большинство металлографических исследований проводят с применением светлопольного (вертикального) освещения (см. рис. 1.4). Дли дополнительного повышения контрастности применяют другие виды освещения.
Метод косого освещения. При этом методе в создании изображения участвуют преимущественно косые лучи, не параллельные оптической оси системы. Повышение контраста прн косом освещении связано, во-первых, с увеличением роли дифрагированных на разных элементах структуры объекта- лучей в формировании изображения и, во-вторых, с образованием теней от рельефа поверхности объекта. Поэтому косое освещение целесообразно применять при достаточно резком рельефе поверхности шлнфа, так как только прн этом условии выступающие участки будут отбрасывать тень на остальную поверхность, которая дает меньшее отражение лучей. Косое освещение достигается обычно включением между объективом и полупрозрачной пластинкой призмы косого освещения или смещением, по отношению к оптической оси системы апертурной диафрагмы, вращением которой изменяется плоскость падения света на объект.
Метод темнЪпольного освещения. При темнопольном осиещении в отличие от светлопольного свет ие проходит через объектив. Пройдя через кольцевую диафрагму 1 (рис. 1.5), свет отражается от кольцевого зеркала 2, установленного иа месте полупрозрачной пластинки, и попадает на зеркальную отражающую параболическую поверхность специального конденсора темного поля 3, который устанавливается иа объектив или монтируется в одной оправе с ним (эпнобъектнв). Такая система создает косое освещение объекта, прн котором освещающий пучок имеет большую апертуру, чем в случае светлопольного освещения. Темнопольное изображение явлиется обратным по отношению к светлопольному
(углубления и выступы становятся светлыми на однородном темном фоне), поскольку в объектив попадают лучи, отраженные неровностями поверхности. Этот тип освещения дает высококоитрастные изображения, четко выявляет зернистую структуру металла, границы между отдельными фазами, натуральный цвет
Рис. 1.5. Схема темнопольного освещения
неметаллических включений и дефекты на отполированной поверхности мнкрошлифа (царапины, поры, трещины).
Исследование в поляризованном свете. -Поскольку большинство металлов, а также металлических и неметаллических фаз являются оптически анизотропными, в металлографиче ских исследованиях часто целесообразно использовать поляризованный свет. С этой целью перед коллекторной линзой помещают поляризатор (призму Николя или поляроид). Создающийся в поляризаторе плоскополяризованный свет после отражения от объекта проходит через анализатор, расположенный между объективом и окуляром или над окуляром. Если объект оптически изотропен, то при соответствующем взаимном положении поляризатора и анализатора («положение скрещения») можно добиться полного поглощения света. Однако если кристаллиты одной или разных фаз оптически анизотропны, то при скрещенных полярофильтрах полного поглощения ие происходит и отдельные кристаллы оказываются светлыми, т. е. получается видимое контрастное изображение. Эта преимущественная освещенность отдельных кристаллов объясняется эффектами эллиптической поляризации и вращением плоскости поляризации.
Структурные составляющие, которые имеют кубическую решетку, изотропны, поэтому их легко отличить от других составляющих. С помощью поляризованного света на нетрав-леиых образцах анизотропных материалов можно изучать их микроструктуру и определять размер зерна. Этот метод позволяет также наблюдать интерметаллические фазы в легированных сталях. Но наиболее часто метал
26
лографическое исследование в поляризованном свете применяют для идентификации неметаллических включений в сталях, так как этн включения имеют характерные цвета или изменяют цвет при вращении цредметного столика микроскопа. Для облегчения идентификации имеются специальные таблицы [II].
Конструкция металломикроскопа, приспособленного для исследования в поляризованном свете, предусматривает включение н выключение полярофильтров и вращение анализатора в пределах 0—90°.
Для изучения структуры металлов и природы неметаллических включений в поляризованном свете требуется высокое качество поверхности шлифа, отсутствие заметного рельефа и следов механической обработки.
Метод фазового контраста. Контрастность изображения рельефных структур может быть дополнительно повышена прн использования системы фазового контраста, имеющейся в некоторых металломикроскопах, илн отдельной фазовоконтрастной приставки к микроскопу. Неровности поверхности шлифа создают разность фаз отраженных световых лучей, которая усиливается системой, состоящей из кольцевой диафрагмы 1 и фазовой пластинки 2 (рис. 1.6). Кольцевую диафрагму устанавлн-
Рис. 1.6. Схема метода фазового контраста: L — источник света; S — объект
вают так, что ее изображение располагается в задней фокальной плоскости объектива и совпадает с кольцом фазовой пластинки, толщина которого иная, чем у остальной части пластинки. В результате этого световые лучи, проходящие через кольцо, сдвигаются по фазе (обычно на 90°) относительно лучей, дифрагированных поверхностью образца и проходящих через пластинку за пределами кольца. Помимо этого, кольцо фазовой пластинки поглощает значительную часть проходящего через него света, что обеспечивает оптимальный контраст н резкость изображения. Для введения системы фазового контраста в оптическую систему микроскопа включается лннза Бертрана, с помощью которой добиваются совмещения изображения апертурной диафрагмы с кольцом фазовой пластинки.
С помощью фазовокоитрастного метода удается обнаружить разность в уровнях рельефа
О
поверхности до -~50А. Этот метод особенно
полезно использовать для изучения границ зерен, двойников, линий скольжения и дисперсных выделений.
Метод интерференционного контраста. Небольшие изменения микрорельефа поверхности можно обнаружить с помощью интерференционного микроскопа или микроинтерферометра. Последний прибор позволяет, кроме того, количественно оценивать изучаемый рельеф, что особенно важно для исследования структурного механизма пластической деформации. Используют методы двухлучевой и многолучевой интерферометрии. В первом случае (интерферометр Лниника) свет от источника L расщепляется полупрозрачной пластинкой Т на два пучка (рис. (.7). Один пучок, отраженный от
Рис. 1.7. Принципиальная схема метода двухлучевой интер фером етрии: Ок — окуляр
пластинки Т, падает иа исследуемую поверхность Si, а Другой пучок, прошедший через пластинку Т, освещает эталонную оптически плоскую поверхность зеркала S2. Лучи, отравленные от поверхностей Si и S2, проходят через объективы 01 н О2 и образуют в плоскости S' накладывающиеся одно на другое изображения поверхностей Si и S2.
Прн наличии разности хода двух пучков должно возникать чередование максимумов и минимумов освещенности. Наблюдаемая интерференционная картина позволяет оценить глубину рельефа с точностью до V20 длины волны.
Прн использовании многолучевой интерферометрии образец помещают на эталонную поверхность тщательно отполированной и посеребренной стеклянной пластинки. Если осуществить плотный контакт образца и пластинки и осветить их монохроматическим световым пучком, то образуются очень тонкие интерференционные полосы. Чувствительность и точность метода увеличиваются в десятки раз и достигают ’/sso длины волны.
Необходимо учитывать, что применение методов фазового н интерференционного контрастов требует особо тщательной подготовки мнкрошлнфов. Шлнфы должны иметь высококачественную гладкую полированную (реже слабо протравленную) поверхность, на которой отсутствуют заметный рельеф и поверхностный иаклеп. Для удаления деформированного поверхностного слоя, особенно в случае легко наклепывающихся сплавов, после механического полирования целесообразно приме-
пять окончательное слабое электролитическое полирование.
В последних моделях металлографических микроскопов вместо описанных методов фазового и интерференционного контраста используется система дифференциального интерференционного контраста (система Номарского), позволяющая получать цветные объемные изображения структурных составляющих, которые трудно выявить обычными методами, а также исследовать без травления микрошлифы различных материалов.
1.2.4. Основные типы
и конструктивные особенности металлографических микроскопов
В зависимости от назначения металлографические микроскопы имеют различные пределы увеличения и позволяют использовать те или иные виды освещения, а также некоторые специальные методы металлографического исследования. Микроскопы, предназначенные для металлографического контроля металлопро
Рис. 1.8. Оптическая схема (с) и общий вид (б) микроскопа ММР-4:
1— источник света; 2— коллектор; 3— призма; 4— апертурная диафрагма; 5 — линза; 6 — зеркало; 7 — линза; 8 — полупрозрачная пластинка; 9 — полевая диафрагма; 10 — телеобъектив; 11 — панкра-тический окуляр; 12 — линзы оборачивающей системы; 13, 14—зеркала; 1S — призма; 16 — призмы бинокулярной насадки; 17 — кольцевая диафрагма; 18 — фазовая пластинка; 19 — линза Бертрана; 20 — кольцевая диафрагма темного поля; 21 — кольцевое зеркало; 22 — поляризатор; 23 — анализатор; 24 — набор светофильтров; 25 — зеркало; 26— экран; 27— линза; 28 — гомаль; 29, 30— зеркало; 31 — линза; 32 — гомаль; 33 — фотопластинка; 34 — линза; 35— фотопленка; 36 — револьверная головка; 37 — бинокулярная насадка; 38 — рукоятка панкратического окуляра; 39— экран; 40 — рукоятка включения фазовой пластинки; 41 — рукоятка переключения зеркал 13 и 25', 42 — рукоятка микрометрической подачи; 43 — рукоятка перемещения столика; 44— рукоятка включения поляризатора и анализатора; 45 — рукоятка включения темнопольного освещения
28
дукции в заводских условиях, оценки качества приготовления мнкрошлифов и других рядовых работ (рабочие микроскопы), обычно позволяют наблюдать и фотографировать структуры в светлом и темном полях и в поляризованном свете при увеличении до 1000—1500. Современные исследовательские микроскопы рассчитаны на предельное (достигаемое в видимом свете) увеличение и, как правило, являются универсальными, т. е. предусматривают возможность использования всех перечисленных выше методов исследования. Кроме того, металлографические микроскопы могут быть снабжены приспособлениями для измерения микротвердости, приставками для нагрева образца в вакууме и счетными устройствами для использования методов количественной металлографии.
Микроскоп ММР-4 (рис. 1.8). Рабочий металлографический микроскоп ММР-4 предназначен для наблюдения и фотографирования микроструктуры металлов в светлом поле при прямом и косом освещении, темном поле, поляризованном свете н методом фазового контраста.
В комплекте оптики микроскопа ММР-4 объ-ективы-планахроматы смонтированы на револьверной головке 36, обеспечивающей их быструю замену. Наряду с компенсационными окулярами с увеличением 10, установленными в бинокулярной иасадке 37, микроскоп снабжен паикратнческой системой линз, позволяющей изменять увеличение микроскопа в 2—3 раза вращением рукоятки 38 без дополнительной фокусировки. Общее увеличение микроскопа от 50 до 1500.
Оптическая схема микроскопа показана иа рис. 1.8, а. Свет от источника 1 (лампы накаливания с йодиым циклом типа КИМ9-75) проходит через коллектор 2 и призмой 3 проецируется в плоскость апертурной диафрагмы 4; далее линзой 5, зеркалом 6, линзой 7 н полупрозрачной пластинкой 8 изображение источника 1 и апертурной диафрагмы проецируется в плоскость опорного торца под объектив. Полевая диафрагма 9 помещается в фокальной плоскости второй осветительной лннзы 7 и проецируется ею в бесконечность, а после объектива — в плоскость предмета. Лучи, пройдя объектив и отразившись от шлифа, вновь проходят через объектов, пластинку 8 и телеобъективом 10 собираются в промежуточной плоскости, являющейся плоскостью предмета для паикратнческой системы 11. Затем лучн отражаются _от зеркал 13 и 14, проходят через линзы оборачивающей системы 12 и призму 15 н поступают в бинокулярную насадку 16.
Прн наблюдения в темном поле вместо линзы 7 и пластинки 8 в ход лучей включаются линза 20 (кольцевая диафрагма) и кольцевое зеркало 21. При работе в поляризованном свете в ход лучей одновременно вводятся поляризатор 22 н анализатор 23.
Прн использовании метода фазового контраста в осветительную систему включается кольцевая диафрагма 17, & в систему наблюдения — фазовая пластника 18.
Прн настройке системы фазового контраста между линзами оборачивающей системы включается линза Бертрана 19.
При фотографировании в ход лучей вместо зеркала 13 вводится зеркало 25; при наблюде
нии изображения на экране 26 зеркала 13 к 25 выключаются. Изображение проецируется линзой 27 и гомалью 28 с помощью зеркал 29 н 30 на экран 26 илн линзой 31 и гомалью 32 на фотопластинку 33 размерами 9X12 см илн с помощью линзы 34 на пленку 35 фотоаппарата «Зор кий -4К».
Горизонтальный микроскоп МИМ-8м. Исследовательский микроскоп МИМ-8м дает увеличение до 1350 при визуальном наблюдении и до ~ 1700 при фотографировании, обеспечивая высокую четкость изображения. В микроскопе применяют ахроматические н апохроматические объективы. Общий вид центральной части микроскопа МИМ-8м н его оптическая схема показаны на рис. 1.9 н 1.10.
Рис. 1.9. Микроскоп МЙМ-8М (центральная часть): 1 — поляризатор: 2 — апертурная диафрагма; 3 — осветительный тубус; 4 — передвижная рамка с полевой и кольцевой диафрагмой: 5 — столик микроскопа; € — винты перемещения столика; 7—объектив; 8— рукоятка призмы косого освещения; 9 — рукоятка анализатора; 10 — окуляр; 11 — фототубус; /2 — рукоятка перевода призмы визуального наблюдения для фотографирования; 13 — рукоятка механизма грубой подачи; 14 — барабанчик механизма микро-метренной подачи; 15 — рукоятка зажимного винта; 16 — винт перемещения апертурной диафрагмы
В настоящее время микроскоп МИМ-8м заменен более совершенной моделью МИМ-9. Этот микроскоп с увеличением при визуальном наблюдении от 20 до ~1700 позволяет использовать все виды освещения, включая фазовый н интерференционный контраст. В нем автоматизированы раздвижка меха фотокамеры и отработка экспозиции при фотографировании, а также грубая подача предметного столика.
Микроскоп ММУ-3 (рис. 1.11) !. Упрощенная модель металломикроскопа с нижним расположением предметного столика. С его помощью можно осуществлять визуальное наблюдение в светлом и темном полях и поляризованном свете при увеличениях 100, 300 и 500. В микроскопе имеется переходная втулка для стандартных мнкрофотонасадок МФН-12 (с фотокамерой «Зоркий-4»), МФН-8 (с пластиночной фотокамерой 9X12 см) или МФН-7 (с пластиночной фотокамерой 6,5X9 см).
’ Будет замерен усовершенствованной моделью METAM-PI.
29
Рис. 1.10. Оптическая схема микроскопа МИМ-8М:
7— источник света (лампа К-30, 170 Вт); 2— коллектор; 3 — теплопоглотитель (для предохранения поляризатора); 4— откидная линза (для работы в темном поле); 5—кольцевая диафрагма; 6— светофильтры; 7 — поляризатор; 8—гомаль или окуляр; 9—апертурная диафрагма; 10, /7—линза осветительного тубуса; 12—полевая диафрагма; 13— линза осветительного тубуса; 14— призма косого освещения; 75—полупрозрачная пластинка; 16 — кольцевое зеркало; /7 — объектив; 18 — анализатор; 19 — ахроматическая линза; 20 — призма визуального тубуса; 21 — призма фототубуса; 22—ахроматическая линза; 23— неподвижная призма визуального тубуса; 24 — конденсор темного поля
Рис. 1.11. Микроскоп ММУ-3
Наряду с отечественными микроскопами в исследовательских и заводских лабораториях широко применяются микроскопы фирмы «Karl Zeiss, Jena» (ГДР); особенно горизонтальный исследовательский микроскоп «Неофот-21» (рис. 1.12). Этот микроскоп снабжен высококачественными объективами — плаиа-хроматами и планапохроматамн, дает увеличение от 10 до 2000 и предусматривает различные виды освещения, включая фазовый контраст, а также имеет приспособление для измерения микротвердости. Микроскоп снабжен встроенным устройством автоматического экспонирования для крупноформатной камеры (съемка иа пластинки 13X18 см и 9Х Х12 см). Кроме того, возможна съемка с помощью малоформатной камеры на пленку 24X36 мм с использованием отдельного экс
позиционного автоматического устройства. Дополнительные удобства работы на микроскопе: быстрый переход с одного вида освещения иа другой,' быстрая смена объективов с помощью механизма быстрого подъема предметного столика и возможность изменения кратности увеличения без смены окуляров посредством специального переключателя. С помощью двух сменных опак-иллюминаторов можно осуществлять наблюдение н фотографирование в светлом и темном полях прн увеличениях от 10 до 50. Для облегчения металлографического контроля можно использовать дополнительное устройство, позволяющее одновременно наблюдать исследуемый шлнф и эталонные снимки (например, соответствующие различному номеру зерна) прн одинаковом увеличении и формате изображений.
Измерение микротвердости [11, 12]. Дополнительные данные о природе и свойствах различных структурных составляющих сталей и сплавов получают путем измерения мнкро-твердости. Для этой цели используют специальные приборы (обычно ПМТ-З и ПМТ-5) 1 илн приспособления к световым микроскопам. Наиболее распространенный метод измерения мнкротвердостн основан на измерении линейной величины диагонали отпечатка d от вдавливания алмазной пирамиды с углом между гранями 136° под нагрузкой 'от 0,02—2Н. В зависимости от твердости исследуемой фазы и величины нагрузки диагональ отпечатка может изменяться от нескольких до нескольких сот микрометров, что позволяет изучать структурные составляющие размером до ~ 10 мкм.
Величину нагрузки при измерении микротвердости выбирают, исходя нз размеров изучаемых структурных составляющих. Для правильного испытания необходимо, чтобы рас
1 Методы измерения микротвердости и приборы, используемые для этих целей, описаны в разделе 11.
стояние; между отпечатками и ст края отпечатка до границы зерна или частицы было ие менее 2d, что ограничивает размер отпечатка (величину нагрузки).- Однако в том случае, если испытуемая фаза находится в матрице с близкими механическими свойствами, допу-
чении диаграмм состояния измерение микро-твердостн часто позволяет установить пределы растворимости и более точно идентифицировать фазы, образующиеся прн изменении состава сплавов. Кроме того, намерения мнк-ротвердости часто проводят в исследованиях
Р>1с. 1.12. Микроскоп «Neophot 21»
скается большая величина отпечатка, так как следует учитывать, что с уменьшением нагрузки микротвердость обычно повышается, особенно в интервале нагрузок инже 0,2Н (это может существенно снизить точность измерении) .
На микротвердость металлов и сплавов могут в значительной мере влиять такие факторы, как подготовка поверхности образца, анизотропия свойств материала и микронеоднородность структуры, связанная, например, с ликвацией илн неравномерной степенью деформации различных зерен. Для исключения влияния наклепа поверхностного слоя шлифа, особенно в случае сравнительно мягких материалов, следует применять электролитическое полирование образцов.
Анизотропия механических свойств кристаллов может приводить к неодинаковой величине отпечатков иа различно ориентированных по отношению к плоскости шлифа кристаллитах, к различию диагоналей одного и того же отпечатка н к неодинаковой степени изогнутости различных сторон отпечатка. При количественном изучении отклонений отпечатков от правильной квадратной формы можно получить важную информацию об анизотропии пластической деформации кристаллов.
Применение метода измерения микротвердости в металловедческих исследованиях связано в основном с проблемами оценки свойств в идентификации отдельных фаз и структурных составляющих, имеющих малый объем. Этот метод широко используют прн исследовании поверхностных покрытий и слоев, а также влияния различной механической, термической или химико-термической обработки иа поверхностные свойства материалов. Прн изу-
диффузии, взаимодействия металлов с различными средами, ликвации и других процессов.
1.2.5. Количественные анализаторы структуры [3, 5j
В последние годы все больше внимания при исследоваини н контроле качества сталей н сплавов уделяется установлению связи между их свойствами и количественными характеристиками микроструктуры, например размером зерна, содержанием различных фаз и неметаллических включений, их распределением по размерам, форме и т. д. Более широкому применению весьма трудоемких методов количественной металлографии1 в значительной мере способствовали разработка специальных автоматических приборов для количественного анализа изображений.
Одними нз первых и наиболее известными из таких анализаторов являются приборы типа «Кваитнмет» фирмы «Cambridge Instruments» (Англия). В этих приборах использован принцип линейного анализа. Получаемое в обычном вертикальном микроскопе с автоматическим перемещением предметного столика изображение структуры с фокальной плоскости окуляра вводится в телевизионную камеру, сигналы с которой подаются одновременно на детектор и экран контрольного телевизора (рис. 1.13). Детектор выделяет и оде-ннвает импульсы, соответствующие оптической отражательной способности исследуемых
См. раздел 4.
структурных составляющих, н с помощью ЭВМ преобразует этн импульсы в выбранные измеряемые параметры микроструктуры, которые регистрируются показывающим прибором н печатающим устройством. Контрольный
Рис. 1.13. Блок-схема прибора «Квантнмет»:
1—образец; 2— микроскоп; 3 — эпидиаскоп; 4 — телевизионная камера; 5— контрольный телевизор; 6 — детектор; 7 — ЭВМ; 8 — показывающий прибор; В — печатающее устройство
телевизор служит для наблюдения микроструктуры, выбора полей н контроля результатов измерений. Вместо шлифов можно анализировать фотографии или негативы с помощью эпидиаскопа.
Модель «Квантимент 360», предназначенная в основном для контроля качества металлопродукции в производственных условиях, отличается высокой производительностью н простотой в обслуживании. Она позволяет с высокой точностью распознавать оксидные и сульфидные включения в сталях, определять количество включений н объемную долю (прн содержаниях до 0,01 % и размерах до 1 мкм), классифицировать по размерам и получать необходимые статистические характеристики. На этом приборе можно также определять средний размер и распределение по размерам зерен светлой фазы (феррита, аустенита ит. п). За 2 мин прибор производит измерения на 2000 полей с выдачей результатов на печатной ленте.
Более совершенная модель «Кваптимет 720» сконструирована по модульному принципу (т. е. может состоять из различных узлов-модулей, предназначенных для выполнения определенных задач) и обеспечивает гораздо более широкие возможности количественного анализа структуры. Этот прибор автоматически выбирает заданные структурные составляющие, оценивают их количество, классифицируют по размерам, форме,, ориентации, оптической плотности и т. д. С помощью специального «светового пера» можио корректировать анализируемое изображение, исключая из него те нли иные элементы (например, дефекты приготовления шлифа, отдельные включения и др.) или, наоборот, «дорисовывая» структуру (например, иеныявленные при травлении границы зерен).
Многие металлографические лаборатории оснащены структурными анализаторами «Эпи-квант» фирмы «Karl Zeiss, Jena» (рис. 1.14), которые также работают на основе метода линейного анализа. Числовые данные измерения получают путем автоматического механического сканирования образца, осуществляемого посредством перемещения предметного столика. При этом на выбранном формате
анализируется 25 линий, расположенных на одинаковых расстояниях друг от друга. Образец освещают с помощью стабилизированного источника света; отраженный от него свет попадает в фотоэлектронный умножитель. Вдоль линии сканирования от отдельных структур-
Рис. 1.14. Количественный анализатор структуры «Эпиквант»
ных составляющих получают сигналы различной амплитуды и продолжительности. Регистрируется каждый переход через границу зерна или межфазную границу, причем измеренные длнмы отрезков можио разделить по размерам на 13 классов. Логическая система распознавания образов позволяет прн автоматическом подсчете отличать границы зерен от дефектов изготовления шлифов (например, рисок).
Прибор «Эпикваит» дает возможность определять содержание различных фаз, неметаллических включений, пор и т. д., оценивая одновременно до трех структурных составляющих, а также устанавливать величину зерна. Достаточная точность измерений на приборе обеспечивается при содержании фазы более 1 %-
В связи с механической системой сканирования прибор значительно уступает по производительности анализаторам типа «Кваитн-меит». Однако благодаря высокой чувствительности фотоэлектронного умножителя ои позволяет выделять и оценивать отдельные структурные составляющие (например, нитриды и карбонитриды титана и. ниобия в сталях), которые нельзя отличить от других структурных составляющих иа приборах «Квантимет».
При использовании автоматических количественных анализаторов структуры необходимо иметь в виду, что они могут учитывать нежелательные детали изображения. Поэтому к анализируемым шлифам предъявляются высокие требования в отношении чистоты поверхности, отсутствия дефектов изготовления, четкости выявления структурных составляющих наряду со специфическим требованием плоско-лараллельностн, обеспечивающей постоянство фокусировки микроскопа.
32
1.2.6, Исследование микроструктуры при повышенных температурах [3, 4, 13]
Для непосредственного изучения структурных изменений, происходящих прн нагреве н охлаждении материалов, применяют высокотемпературные микроскопы. С нх помощью можно осуществлять прямое наблюдение процессов рекристаллизации, роста зерен, фазовых превращений, а также некоторых поверхностных явлений. С целью расширения исследовательских возможностей высокотемпературные микроскопы часто используют в комплекте с устройствами, позволяющими одновременно подвергать образцы различным видам нагружения (растяжению, сжатию, изгибу, ползучести, усталости), измерять мнкротвердость и регулировать в широких пределах скорости деформации, иагрева и охлаждения. Такне установки позволяют получать ценную информацию о механизмах пластической деформации и разрушения, взаимосвязи между структурой и свойствами исследуемых материалов.
Для исследования иа высокотемпертурном микроскопе используют образцы с тщательно отполированной поверхностью. Образцы нагревают в вакуумной камере прн остаточном давлении 1,3—1,3-10~2 Па нли в среде разреженного инертного газа. Выявление структуры прн таких исследованиях основано на эффекте «вакуумного травления», обусловленном избирательным испарением фаз в вакууме, различной скоростью испарения для участков с различной кристаллографической ориентировкой, а также повышенной скоростью испарения в участках, расположенных у границ зерен и других дефектов кристаллического строения. В результате вакуумного травления на полированной поверхности появляется характерный микрорельеф, отображающий структуру материала при высокой температуре н ее изменение в процессе нагрева. Например, используя последовательные кратковременные выдержки при иагреве стали в вакууме до определенных температур в интервале 900—1300 °C, можно наблюдать миграцию границ зерен и изучать кинетику роста зерна аустенита. к
Нагрев в вакууме позволяет также исключить образование на поверхности образца окисной плеикн, препятствующей наблюдению структуры. Степень вакуума, требуемого для полного предотвращения окисления зависит от исследуемого материала. Например, прн -остаточном давлении 1,3 Па на поверхности низкоуглеродистых сталей, нагретых до 800 °C, не наблюдается окисления, а на поверхности сталей, содержащих хром, прн тех же условиях образуется тонкий окисный слой.
Для получения заданной температуры образца обычно используют радиационный нагрев от внешнего источника тепла нлн контактный электронагрен. В некоторых установках применяют индукционный или электроннолучевой нагрев. Выбор метода нагрева определяется требуемыми максимальными температурами и скоростями нагрева образцов, которые могут достигать 2500 н 1000 °С/с соответственно. Для увеличения скорости охлаждения образца используют охлаждающие устройства, посредством которых
3—280
осуществляется, например, обдув образца холодным инертным газом.
Наблюдают за структурой образца через смотровое стекло вакуумной камеры. В связи с этим в высокотемпературных микроскопах применяют специальные длиннофокусные зеркально-линзовые объективы. Максимальное увеличение, получаемое прн использовании таких объективов, обычно не превышает 500, что ограничивает возможности оптических высокотемпературных микроскопов* 1.
Для защиты смотрового стекла от осаждения конденсата испаряющихся с поверхности образца веществ обычно используют набор тонких кварцевых стекол, которые прн помощи специальных приспособлений перемещаются внутри вакуумной камеры против смотрового стекла. Испарение о поверхности образцов можно1 устранить путем иагрева в атмосфере инертных газов. Однако недостатком этого метода является необходимость тщательной очистки газов от примесей кислорода, паров воды и других веществ во из-бежание окисления и загрязнения поверхности.
Ряд установок для высокотемпературной микроскопии разработан Институтом машиноведения АН СССР. Последней серийной установкой, изготавливаемой Фрунзенским заводом КИП, является установка «АЛА-ТОО» (типа ИМАШ-20-75). Установка предназначена для прямого наблюдения, фотографирования и киносъемки микроструктуры различных материалов при. нагреве (охлаждении) и деформации растяжением в вакууме 6,5-10-2. В установке предусмотрены две системы нагрева образца: нагрев пропусканием тока (до 1500 *С) и радиационный нагрев (до 1200°C). Микроструктуру образца исследуют с помощью высокотемпературного' микроскопа МВТ-71 с максимальным увеличением 410. Образец можно исследовать прн воздействии на него постоянной нагрузки или прн растяжении с постоянной скоростью перемещения активного захвата.
Из зарубежных установок для высокотемпературной металлографии наибольшее распространение в отечественных лабораториях получили микроскоп MeF с вакуумной камерой «Вакутерм» фирмы Reichert (Австрия) и установка НМ-4 фирмы «Union Optical» (Япония), снабженная устройствами для растяжения, сжатия, изгиба и измерения микро-твердости.
1.2.7. Основные методы микроанализа2
Определение и Сценка микроструктуры сталей. В задачу микроанализа обычно входит определение основных структурных составляющих стали (феррита, перлита, мартенсита, карбидов и др.), нх количества, морфологии и распределения. Применяемые увеличения
1 Значительно большие увеличения (до 10 000) можно получить на фотоэмиссионных микроскопах при нагреве образца до 2000 °C.
1 Дополнительные сведения о применении методов оценки микроструктуры, неметаллических включений, величины зерна и обезуглероживания приведены в разделе 18.
микроскопа зависят от степени дисперсности структурных составляющих.
Для более быстрой оценки микроструктуры прн металлографических исследованиях и контроле различных сталей используют эталонные шкалы микроструктур, приведенные в соответствующих стандартах нлн технических условиях с указанием реактивов для травления н увеличений. Эти шкалы позволяют оценить микроструктуру образца в баллах в зависимости от содержания определенных структурных составляющих их размеров, степени дисперсности, неравномерности распределения и других признаков. По эталонным шкалам определяют, например, количество феррита, перлита и других фаз стали, дисперсность мартенсита н перлитных структур, собственно структуру перлита, степень развития карбидной неоднородности, количество нитридов и т. д. Перечень стандартных шкал используемых для оценки микроструктуры различных сталей, приведен ниже:
ГОСТ 8233 — 56. Сталь. Эталоны микроструктуры
1. Пластинчатый перлит (степень дисперсности); Х1000; 10 баллов
2. Зернистый перлит (степень дисперсности); Х1000; 10 баллов.
3. Мартенсит (размеры игл); Х1000; 10баллов.
4. Включения нитридов (количество); ХбОО; 10 баллов.
5. Карбидная сетка (степень развития); Х500; 6 баллов.
6. Карбидная неоднородность; ХЮО; 6 баллов.
7. Соотношение перлита н феррита; хЮО; 10 баллов (0— 100 %).
8. Соотношение мартенсита и троостнта; Х500; 10 баллов (0—100 %).
9. Соотношение зернистого и пластинчатого перлита; ХБОО; 10 баллов (0—100%).
ГОСТ 5640 — 68. Сталь. Металлографический метод оценки микроструктуры листов и ленты
1. Структурно-свободный цементит (количество, форма, распределение), Х360 — 400;
6 баллов.
2. Перлит в малоуглеродистой деформированной стали (форма, количество н распределение); Х360— 400; 6 баллов.
3. Полосчатость феррито-перлитной структуры (количество н сплошность феррнтных полос); Х100, 6 баллов.
4. Видманштеттова структура (количество и размеры игольчатого феррита); ХЮО; 6 баллов.
ГОСТ 1435 — 74. Сталь инструментальная углеродистая
1. Микроструктура в отожженном состоянии (дисперсность н соотношение зернистого и пластинчатого перлита); ХбОО; 10 баллов.
2. Оценка остатков цементитной сетки; Х500; 5 баллов.
ГОСТ 5950 — 73. Сталь инструментальная легированная
1. Микроструктура в отожженном состоянии (дисперсность н соотношение зернистого н пластинчатого перлита); ХБОО; 10 баллов. ГОСТ 19265-—73. Сталь инструментальная быстрорежущая
1 и 2. Карбидная неоднородность; Х100; 8 баллов.
ГОСТ 801 — 78. Сталь подшипниковая
1. Карбидная сетка; Х500; 5 баллов.
5. Структурная полосчатость; Х100; 5 баллов.
6. Карбидная ликвация; Х100; 5 баллов.
8. Микроструктура в отожженном состоянии (дисперсность и соотношение зернистого н пластинчатого перлита); Х500; 10 баллов. ГОСТ 11878 — 66. Сталь аустенитная, методы определения содержания а-фазы
Шкала для определения (количества) сс-фазы; ХЗОО; 5 баллов.
ГОСТ 3443 — 77. Отливки из чугуна с различной формой графита. Методы определения структуры
Набор шкал для оценки формы, размеров и распределения включений графита, а также структуры металлической основы.
Анализ неметаллических, включений [10, 14, 15]. В зависимости от химического состава, технологии выплавки и разливкн сталь может содержать включения различных видов (окис-лы, сульфиды, нитриды), различающиеся по размерам, форме н распределению. Полный анализ неметаллических включений состоит из определения нх химического состава, структуры и количественной оценки загрязненности металла различными включениями. Сначала устанавливают основные типы включений, встречающихся в данном образце. Для точного определения состава н структуры включений обычно используют (полностью или частично) комплекс методов, в который входят металлографический анализ с определением микротвердости, рентгеноструктуриый, микрорентгеноспектральиый, электронно-
микроскопический н петрографический методы. Однако металлографический метод наиболее прост и удобен и во многих случаях позволяет достаточно надежно идентифицировать включения без использования других методов.
Неметаллические включения размером 5>1 мкм в литых н деформированных сталях изучают на тщательно приготовленных не-травлениых микрошлифах. Для улучшения условий подготовки шлифов из малоуглеродистых сталей применяют предварительную-термическую обработку образцов, повышающую твердость и устраняющую выкрашивание включений.
При изучении включений на металлографическом микроскопе используют светло- н темнопольное освещение, а также поляризованный свет. Определяют такие признаки включений, как форма, цвет, прозрачность, степень анизотропии, деформируемость, микротвердость, взаимодействие с определенными химическими реактивами и др. Сопоставляя данное включение с эталонами и используя-класснфнкационные таблицы, его идентифицируют.
Исследование включений под микроскопом на шлнфе дополняется изучением выделенных электролизом нз металла включений в проходящем или отраженном свете и определением их оптических констант, т. е. определением их минералогического состава петрографическими методами [15]. Наиболее важная-характеристика включений — показатель преломления N. Схема определения природы включений н характерные признаки основных неметаллических включений в сталях приведены в табл. 1.4 н на рис. 1.15.
34
Схема определения включений под мегйпломикроскопом
Наблюдение нетравленого шлифа в темном поле
,, л л ,, . SiO,., Ai2O3,Zr02, AIN, MnS, 2FeO • SiO2,
MnO, Cr, Оз ,1С-О MnO, TiO,, FeO-TiO3, FeO- Cis 03, FeO Al, 03 2MnO> S102, MnO• SiO,, 3Al, 03 2810,,
< mcao .ngjo, ( MgO- Al2 03, MnO-Ai3 03,
большинство силикатных стекол
в ТОНКИХ слоях
FeO, FeO-VjOj, FeO-Fe2O3, FeS, FeS-MnS, TiN,Ti(CN), ZrN, Zr(CN), VN, TiO, й-—Ti203, TijOj
I Окрашенные
Розовые, бурые / Красные
Зеленые _________/ ___________
ЕеО,ТЮ2,ТЮ
МпО, Сг2 О3
FeO—MnO, FeO-Cr, О3
Al,O3,ZrO5, FeO-AI2O3, mCaO nSiOj, AIN, MnS, некоторые силикатные стекла
Слабо окрашенные или бесцветные
Бледно-желтые
„„„ Бесцветные
или серо-зеленые
Аняаотротшые Изотропные
Наблюдение в поляризованном свете
Наблюдение в светлом поле (травление)
Наблюдение в поляризованном свете
Изотропные Анизотропные
МпО
Ст, Оз
Анизотропные
Наблюдение в светлом поле
Стандартные реактивы не действуют
Проверка свойств по таблице
FeO-Cr203
Рис. 1.16.
SiO, , 3 Al2 O3,2S1O2 , MnO SlOj , Mg0-Al,O3, MnO- AI2O3, некоторые силикатные стекла
Наблюдение в поляризованном свете
Анизотропные /
/ Изотропные Анизотропные
Alj О3, ZrO, , AIN, mCaO>nSiO,
FeO-AIj О3, MnS, силикатные : стекла
Наблюдение в светлом поле
Вытравливаются 3 %-ной Н, SO4
FeO—MnO
Проверка свойств по таблице
Розовые до коричневого
Зеленовато-желтые до коричневого
SiO2—кварц, ЗА13О3 * 2810ч
MaO SiO/ ’
Изотропные
Глобули, _ темный крест "
Кристаллики
2FeO-SiO,
2MnO-SiO} |
Химический, петрографический анализы
Силикатные стекла
MgO-Al,O3 MnO-AIjOj
FeO V, 03, FeO • Fe3 О3, TiO FeS-MnS, TIN, Ti(CN), j ZrN, Zr(CN), VN
Наблюдение в светлом поле
Проверка , свойств по таблице
Правильные кристаллы различной окрики
TIN, TiO, ZrN, VN, FeO-V} O3,
Светло-желтые до розового
^TiNTiO
Лимонножелтые
Белые с розовым оттенком
Светло-серые с розовым оттенком
Наблюдение в светлом поле
"П— Бледно-фиолетовые
Розово-фиометовьге до фиолетово^ сиреневого
Серо-голубые
~T Темно-коричневый
Ti(CN)|
Серые с коричневым оттенком
FeS-MnS
FeO
ТАБЛИЦА 1.4
ХАРАКТЕРНЫЕ ПРИЗНАКИ И СВОЙСТВА НЕМЕТАЛЛИЧЕСКИХ ВКЛЮЧЕНИЙ В СТАЛИ
Название включения Химическая формула Кристаллическая система и форма, наблюдаемая на шлифе Расположение Полируемость Пластичность Твердость HV
1 2 3 4 5 6 7
Простые
Закись железа (вюстит) FeO Кубическая; обычно округлые зерна (гло-були), которые после деформации вытягиваются Отдельные включения, иногда по границам зерен Хорошая Слабо деформируются 430
Закись марганца (манганозит) МлО Кубическая; кристаллы неправильной формы, иногда дендритного строения, после деформации слегка вытягиваются вдоль ее направления Располагаются^ скоплениями » » 280
Твердый раствор закиси железа и закиси марганца FeO—МпО Кубическая; при большом содержании МпО октаэдры или кристаллы непра- вильной формы, иногда приближающейся к дендритной; при большом содержании FeO могут встречаться в виде округлых зерен (глобулей) Б большинстве случаев скоплениями Хорошая, не выкрашиваются » 440
Оксид титана TiO Кубическая; мелкие идиоморфные кристаллы (квадраты) — — — —
Полутора-ОКСИД титана a-TI«Os Гексагональная; округлые зерна Располагаются чаще скоплениями — — —
Твердый раствор Т i TI аОл (аносовят) Диоксид титана (рутил) «л TIOa Ромбическая Тригональная — — — —
Ильменит (кричтонит) Оксид алюминия (корунд) FeO—TiO, a-AltOs Тригональная; мелкие кристаллы неправильной формы, иногда с закругленными контурами Гексагональная; в большинстве случаев мелкие зерна неправильной формы, иногда правильной гексагональной формы; реже крупные зерна Скоплениями или в виде изолированных аерен. после деформации строчками В большинстве случаев скоплениями, после Деформации строчками Хорошая Плохая, выкрашивают -си. оставляя «хвосты» Не деформируются 630 Очень высокая
36
Отража-1 тельная способ-| ность Оптические свойства Химические свойства Другие свойства
при обыкновенном свете в поляризованном свете в проходящем свете (после выделения из стали)
в светлом поле в темном поле
8 9 10 И 12 13 14
скислы.
Сред- Серые с коричне- Совершенно не- Изотроп- Непрозрачны; в Вытравливаются: Образуют
няя вым оттенком по краям прозрачны (обычно темнее, чем основной фон); окружены тонкой светящейся линией ны виде темных гло-булей 3 %-ной H2SO4; насыщенным спиртовым раствором SnCb; 10 %-ной HCI; кипящим раствором КМпСи в 10 %.ной H2SO4; 5 %-ным водным раствором C11SO4 непрерывный ряд твердых растворов с МпО
Низ- Темно-серые, в В тонких слоях Изотроп- Неправильной Вытравливаются: Образует
кая тонких слоях за- прозрачны; собст- ны; в тон- формы; при ко- насыщенным рас- непрерывный
метны внутренние венный цвет изу- ких слоях сом освещении твором SnCIa в ряд твердых
отсветы мрудно-зеленый зеленые зеленые. IV—2,19 спирте; 20 %-ным НС1; 20 %-ным NaOH; раствором KMnCU в HiSOi растворов с FeO
Сред -няя Цвет изменяется с повышением содержания Мп О от серого до серофиолетового с красным отсветом в центре Прозрачны; прозрачность увеличивается с увеличением содержания МпО. Собственный цвет кроваво-красный с различными оттенками Изотропны; прозрачны; цвет красный с различными оттенками Вытр а вливаются: 3 %-ной H2SO4; насыщенным спиртовым раствором SnCh; 10 %-ной НС1; 20 %-ным спиртовым раствором HF; раствором КМпО« в 10 %-ной H2SO4. Щелочной пикрат натрия и 20 %-Ный NaOH окрашивают включения
Высокая Золотисто-желтые, подобны нитридам титана Изотропны Черного цвета непрозрачны По форме и цвету сходны с TIN; можно различить только при рентгеноструктурном анализе
Слабая Цвет от бежевого до коричневого — Анизотропны Черного цвета; непрозрачны, сильно анизотропны —
Серые — Цвет черный; непрозрачны; анизотропны
Светло-серые в Мелкие темные призмы; в тонких слоях прозрачны. Высокий показатель преломления: /Vo=2,567; Ne =2,824. Чрезвычайно высокое двупреломление Очень тугоплавкие
Средняя Темно-серые с фиолетовым оттенком В тонких слоях прозрачны; цвет от розового до бурого Анизотропны; светятся яркорозовым цветом Вытравливаются 5 %-ным водным раствором C11SO*
Низ- Цвет темно-серый Прозрачны; блед- Прозрач- Мелкие, прозрач- Стандартные ре- —
кая до черного (с фиолетовым оттенком) но-желтого цвета ны; анизотропны. Эффект анизотропии проявляется слабо, особенно у мелких включений ные зерна неправильной формы; 7VD=1,768; Ne= = 1,759 активы не действуют
37
Название включения Химическая формула Кристаллическая система и форма, наблюдаемая на шлифе Расположение Полируемость Пластичность Твердость HV
I 2 3 4 5 6 7
Оксид хрома СггОа Гексагональная; кристаллы шестигранной или неправильной формы (чаще округлой) с неровными краями и шероховатой поверхностью Отдельные зерна Плохая, легко выкрашиваются и оставляют «хвосты» Не деформируются Очень высокая (—1500)
Диоксид кремния (кремнезем): а) кварц SiOa Существует в модификациях а и р. Кристаллическая система Р-кварца—тригональная, а-кварца— гексагональная. Крупные зерна неправильной угловатой формы (обломки) Отдельные включения Плохая, легко выкрашиваются » Высокая
б) тридимит SiO£ Существует в а-, |3-и ^-модификациях; кристаллическая система модификаций: а—ромбическая; ₽—тригональная; V—гексагональная Очень низкая
в) кристобалит SiOs Существует в а- и ₽-модификациях. Кристаллическая система модификаций: а — кубическая; р — тетрагональная То же
Диоксид циркония (бадделеит) ZrOg Известны две полиморфные модификации a-ZrOs квадратной системы и g-ZrO2 моноклинной. Мелкие зерна неправильной или округлой формы Чаще скоплениями; после деформации строчками Хорошая Не деформируются Слона Средняя же окислы
Магнезиальная шпинель MgAljOi (MgO-Al.0,) Кубическая; правильная кристаллическая форма (ромбы, треугольники, трапеции И др.) — -- — —
Марганцевая шпинель (галаксит) №А12О.| (MnO A120i) Кубическая — — — —
Железная шпинель (герценит) FeA!2O4 (FeO-AlA) Кубическая; кристаллы правильной кубической формы — — — —
Хромовая шпинель (хромит) FeCr2O4 (FeO-CrsO3) Кубическая; почти всегда кристаллы правильной формы (треугольники, ромбы. квадраты и т. д.) Преимущественно скоплениями; в деформированном металле в виде строчек Хорошая Не деформируются 570
Феррована-диевая шпинель FeVgO4 (FeOVjOJ Кубическая; в большинстве случаев правильной кристаллической формы (прямоугольники, треугольники и т. д.) Преимущественно скоплениями; после деформации строчками Плохая, легко выкрашиваются * 930
Магнетит Fe-Fe2O, (FeO-Fe8O3) Кубическая; неправильной формы — — —
38
Продолжение табл. 1.4
й К , М та ю g.S3£ £•« о о Гу С t? р и н о £ Оптические свойства Химические свойства Другие свойства
при обыкновенном свете в поляризованном свете в проходящем свете {после выделения из стали)
в светлом поле в темном поле
8 9 10 11 12 18 14
Низкая » Темно-серые с фиолетовым оттенком Цвет темно-серый до черного Прозрачны; .в очень тонких слоях имеют зеленую окраску Прозрачны; ярко светятся беложелтым цветом Анизотропны; в очень тонких слоях имеют зеленую окраску Слабо анизотропны г ' Прозрачны; Wn=2,5 Бесцветны; пока-ватель преломления (3-кварца: lVg=l,55; N р= = 1.54; а-кварца: ^=1,54; lVp=l,53 То же Стандартные реактивы не действуют При 870 °C а-кварц переходит в •р-тридимит
— __ — — Бесцветны; /V =1,473; lVp=l,469 — Температура перехода а-тридимита в р-кристобалит 1470 °C
Цвет темносерый Серого цвета Темно-серые Прозрачны; цвет бледно-желтый Анизотропны Бесцветны; показатель преломления а-кристо-балита: Af=l,486; й-кристобали-та: ЛГ0= 1,487; = 1,484 Зерна неправильной нли округлой формы; бесцветны, анизотропны; 2Vg>2,19„- Wp=2,14±0,02 > Стандартные реактивы не действуют Температура плавления 1710 СС
(группе шпинелей)
— — Мелкие кристаллы октаэдрической формы; бесцветны, прозрачны, изотропны; W = 1,718-ь 1,720 — Под микроскопом подобны корунду
— — Мелкие кристаллы октаэдрической формы; прозрачны, бесцветны, изотропны: М=1,8 —
Низкая Темно-серый —• — В тонких слоях прозрачны, цвет зеленоватый; А?= = 1,8 —
Средняя Серые со слабым фиолетовым оттенком В тонких слоях прозрачны; цвет красный, краснокоричневый до темного в толстых слоях Изотропны В ТОНКИХ слоях прозрачны, цвет от красного до бурого; !V=2,i2 Стандартные реактивы не действуют
» Светло-серые с розоватым оттенком. особенно хорошо заметным при диафрагмировании Непрозрачны; по границам окаймлены тонкой светящейся линией » •— Вытравливаются: 2 %-ным водным раствором (ЫШаСОз; 5 %-ным водным раствором CuSO<; 10 %-ным СггОз —
Темно-коричневые частицы с ноздреватой по? верхностью М=2,42 Вытравливаются НС1 при кипячении
39
Название включения Химическая формула Кристаллическая система и форма, наблюдаемая на шлифе Расположение Полируемость Пластичность Твердость
I 2 3 4 5 6 7
Силикаты и сила
Силикаты железа — фаялит 2 FeO-SiOj Ромбическая; встречаются в виде глобу-лей, часто вместе с выделениями кремнезема и закиси железа Отдельные зерна Хорошая Деформируются слабо 350-700
Силикаты марганца — тефроит 2 MnO-SiO2 Ромбическая; в большинстве случаев в виде глобулей Я* Деформируются хорошо —
Метасиликат марганца (родонит) MnO-SiO2 Триклинная; в большинстве случаев в виде глобулей > То же —
Алюмосиликат (муллит) 3 A12Os-2S1O2 Ромбическая; часто в виде призм и игл » Не деформируются —
Силикаты кальция CaO S!08 (CaSiO3): 2CaO-SiO£ (CaeSiO4); 3 CaO-SiOs Глобули разных размеров > Плохая. легко выкрашиваются То же 400—600
Кварцевое стекло SiO2 Аморфное; включения глобулярной формы различных размеров То же » 700
Стекла железо -марганцево-силикатные Различные соотношения FeO—MnO и sio2 Аморфны; глобулярной формы с выделениями FeO—МпО и SiO2 Хорошая От высокой до низкой в зависимости от содержания SiO2 400—700
Стекла алюмосиликатные Стекла циркониево-сяликатные Различные соотношения Al20.,, SiO2 и FeO Различные соотношения Zr02, SIO2 и FeO Аморфный; глобулярной формы Аморфны; включения глобулярной формы с выделениями SiO2 и ZrOs Плохая, часто выкрашиваются Средняя При деформации хрупко разрушаются То же 1000—1300 400—600
Стекла кромово-силикатные Различные соотношения Cr20s, SiO2 и FeO Аморфны; включения глобулярной формы с выделениями хромитов. —
Сульфид -железа FeS Гексагональная; обычно в виде скоплений округлых зерен Преимущественно скоплениями и иногда по границам зерен в виде сетки Хорошая Легко деформируются, вытягиваются в направлении деформации Суль 240
Сульфид марганца MnS Кубическая; как правило, кристаллы квадратной формы Отдельные зерна или скоплениями » Слабо деформируются —
40
Продолжение табл. 1.4
Отражательная способность Оптические свойства Химические свойства Другие свойства
при обыкновенном свете в поляризованном свете в проходящем свете (после выделения из стали)
в светлом поле в темном поле
8 9 10 11 12 13 14
катные стекла
Сред- Темно-серые i Прозрачны; цвет Анизо- Прозрачны; цвет Вытравливаются Температура
няя от зеленовато-желтого до темно- и ярко-красного с кольцевыми отсветами тропны; прозрачны от зеленовато- желтого до коричневого; Nm= =1,877; ДГр=1,835 HF плавления 1205 °C. Образуют непрерывный ряд твердых растворов с силикатами марганца
Низкая Прозрачны; цвет от розового до коричневого Анизотропны Прозрачны: цвет от розового до коричневого; Af = 1,820; Wm= =1.807; IV =1,785 Температура плавления 1300 °C. Образуют непрерывный ряд твердых растворов с силикатами железа
Прозрачны; бесцветны или розового цвета » W =1,730; N = S =1,733 * —
» Прозрачны; бесцветны; N„ = -М54; 4= = 1,644; Np =1,642 -— —
Темно-серые, имеют шероховатую поверхность Прозрачны, светятся Изотропны — Вытравливаются: 20 %-ным NaOH; 5 %-ным раствором CuSCU и кипящим раствором КМпО4 в 10 %-ной H2SO4 —
Сред- Черные с блестящей точкой в центре и кольцевыми отсветами Ярко светятся, очень прозрачны Изотропны; с характерным черным «крестом» Прозрачные гло-були и глобули с темным ядром; АГ= 1,458 Вытравливаются 20 %-ным спиртовым раствором HF; 5 %-ным раствором CuSO4
Серые, с увеличе- В большинстве Изотроп- В большинстве Вытравливают- —
няя нием содержания SiOj цвет меняется до темного случаев прозрачны, Цвет от ярко-красного до темного ны случаев прозрачны, изотропны; N изменяется в зависимости от состава ся HF
Низкая Темно-серые Прозрачны, цвет светло-желтый х> Прозрачны, бесцветны » —
Средняя Серые Прозрачны; белые, блестящие Изотропны; бесцветны То же > » —
X фиды » Прозрачны, серожелтого цвета, включения FeO. Сг2О3 ярко-красного цвета Изотропны; прозрачны Прозрачны; бурожелтого цвета » > —
Высокая Светло-желтые Непрозрачны Непрозрачны; бледно-желтого цвета, ярко анизотропны Непрозрачны; анизотропны .Вытравливаются: 3 %-ноЙ H2SO4; 10 %-ной НС1; раствором КМпСЦ в 10 %-ной H2SO4 Температура плавления 1170-1185 °C, Встречаются только в стали с очень малым содержанием марганца, с MnS образуют ряд твердых растворов. Могут содержать в раство- ре до 13 % Ni
срад- Серо-голубые Слабо прозрачны. Изотроп- Изотропны; про- Вытравливают- Температура
няя Наблюдаются зеленые внутренние рефлексы, особенно с иммерсией ны зрачны, цвет темно-зеленый ся слабыми кислотами (5—10 %) плавления 1620 °C. Образуют непрерывный ряд твердых растворов с FeS
41
Название включения Химическая формула Кристаллическая система и форма, наблюдаемая на шлифе Ра сположение Полируемость Пластичность Твердость HV
1 2 3 4 5 6 7
Сульфад железа и марганца (твердый раствор) Fes—MnS Зерна округлой формы Отдельные зерна или скоплениями; иногда по границам верен металла Хорошая Легко деформируются, вытягиваются в направлении деформации
Нитриды и
Нитрид титана TIN Кубическая; кристаллы правильной формы (квадраты, прямоугольники) Отдельные зерна или скоплениями; после деформации строчками Плохая. легко выкрашиваются Не Деформируются Высокая
Карбонитриды титана Ti (CN); (TiCTiN); [Tk (CNS)] То же То же ТО же То же
Нитрид циркония ZrN > » Хорошая 300
Карбонитрид циркония Zr (CN) Кубическая; включения наблюдаются чаще в виде зерен неправильной формы » Плохая Высокая
Нитрид ванадия VN Кубическая; кристаллы правильной формы Хорошая *
Нитрид алюминия AIN Гексагональная; наблюдаются в виде шестигранников, треугольников, прямоугольников Скоплениями Плохая 850
После определения основных видов включений, встречающихся в данной стали, проводят количественную оценку загрязненности металла по этим видам включений, используя методы ГОСТ 1778 — 70 (см раздел 18).
После просмотра шлнфа в нетравлеиом виде для определения распределения, формы н размеров неметаллических включений с использованием наблюдения в темном поле и в поляризованном свете, а также после специального травления этих включений приступают к изучению основной структуры стали нли сплавов.
Методы травления микрошлифов [2, 4, 7, 9]. Для микроанализа мнкрошлифы подвергают травлению реактивами, различающимися по своему воздействию на поверхность металла. В табл. 1.5 приведены наиболее широко распространенные реактивы — растворы кислот, солей и щелочей, которые вызывают избирательное растворение металлических, или других фаз, а также нх пограничных участков вследствие различия физнко-хнмических свойств. В результате на поверхности микрошлифа образуется рельеф н прн наблюдении под микроскопом более сильно растворившиеся участки из-за теин нли более низкого коэффициента отражения (из-за растравленной,
шероховатой поверхности) представляются более темными, а иерастворившнеся более светлыми.
Для травления по этому способу шлнф погружают полированной поверхностью в соответствующие реактивы, состав которых зависит от состава стали, ее обработки, цели изучения (табл. 1.5); продолжительность погружения устанавливается экспериментально. В ряде случаев применяют электролитическое травление, которое в определенных условиях позволяет более четко выявить структурное состояние стали. Составы реактивов для электролитического травления и его режимы диаы в табл. 1.6.
Структуру, наблюдаемую под микроскопом или фиксируемую фотографическим методом, либо сопоставляют со специальными шкалами н эталонами, либо подвергают специальному изучению для решения конкретных практических илн научных задач часто (особенно в последние годы) с использованием статистических методов оценки.
Помимо описанных методов травления, на практике также используют метод теплового травления, заключающийся в нагреве мнкро-шлифа в окислительной атмосфере. Вследствие образования разной по толщине и соста-
42
Продолжение табл. 1.4
Отражательная ' способность Оптические свойства Химические свойства Другие свойства
при обыкновенном свете в поляризованном свете в проходящем свете (после выделения из стали)
в светлом поле в темном поле
8 9 10 11 12 13 14
Средняя От серо-голубого до светло-желтого в зависимости от содержания MnS Непрозрачны Изотропны Изотропны; непрозрачны Вытравливаются: 3 %-ной H2SO4; 10 %-НОЙ НС1; 20 %-ноЙ HF; 10 %-ной хромовой кислотой Встречаются в углеродистой стали почти всех марок и в низколегированной стали
карбонитриды
Высокая От светло-желтого до розового Непрозрачны; окаймлены по границам светящейся линией Изотропны; непрозрачны Изотропны; прозрачны не- Обычные реактивы не действуют Температура плавления 3000 °C
От розового с фиолетовым оттенком до фиолетово-сиреневого в зависимости от содержания углерода То же То же То же То же —
Лимонно-желтый 2> Изотропны Вытравливаются: 20 %-ным NaOH; щелочным раствором пикрата натрия; 20 %-яым раствором HF Температура плавления 2985+50 СС
X» Бледно-фиолетовый » Стандаратные реактивы не действуют
Очень высо -кая Белый с бледно-розовым оттенком » Вытравливаются кипящим 10 % -ным водным раствором Сг2О3 Температура плавления 2050 °C
Низ- Темно-серый Прозрачны, цвет Сильно Прозрачны, бес- Стандартные ре- Температура
кая бледно-желтый анизотропны цветам, сильно анизотропны; АГ =2,17 ± 0.2; =2,13 н активы не действуют плавления 2160—2200 °C
ву окисной пленки на фазах разного состава отдельные участки шлифа (после охлаждения) приобретают различную окраску вследствие известного интерференционного эффекта. Тепловое травление позволяет дифференцированно определять фазовый состав сплавов в тех случаях, когда метод химического или электролитического травления мало' эффективен.
Наконец, используется и метод вакуумного травления, основанный на избирательном испарении фаз разного состава или разной скорости испарения в объеме зерен или по их границам даже в однофазных сплавах при нагреве шлифов в вакууме (см. раздел 1.2,6). В этом случае не требуется дополнительного травления микрошлифов и микроструктуру можно наблюдать или непосредственно прн температуре иагрева или после охлаждения. Для изучения структуры сплавов, отличающихся очень низкой коррозионной стойкостью при химическом травлении и в то же время низкой упругостью паров при нагреве в вакууме, микрошлифы подвергают катодному травлению.
В общем случае микроаиалнз используют для определения величины зерна, фазового состава и структуры сплавов в условиях равновесия, например после отжига, для характе
ристики неравновесных структур, особенно после закалки и отпуска, для определения характера обработки металлов, определения состояния поверхностных слоев в результате Обезуглероживания илн после химико-термн-ческой обработки, для изучения особенностей распределения избыточных фаз и др.
Определение величины верна (по ГОСТ 5639—65). Величину действительного зерна, т. е. зериа, которое образовалось при принятой обработке, определяют на мпкрошлнфах, путем травления!.
Прн исследовании н контроле сталей часто определяют величину не действительного, а «наследственного» зерна, т. е. величину зерна аустенита после нагрева прн определенных условиях (температуре, времени выдержки и скорости охлаждения), указанных в соответствующих стандартах. «Наследственное» зерно характеризует чувствительность стали к росту зерна при нагреве для термической обработки и оказывает значительное влияние на многие свойства стали, в частности на ее сопротивление разрушению.
1 Для выявления зерна в углеродистых и легированных сталях применяют реактивы № 1 и 2. а в аустенитных сталях —№ 9 (см. табл. 1.5), Ка 3, 6’ (см. табл. 1.6)
43
ТАБЛИЦА 1.5
РЕАКТИВЫ ДЛЯ МИКРОЙССЛЕДОВАНИЯ СТРУКТУРЫ СТАЛИ И ЧУГУНА
к» Наименование реактива Состав реактива Назначенье и особенности применения
Для травления углеродистых низколегированных сталей и чугунов
1 Спиртовой раствор азотной кислоты (реактив Ржешо-тарского) I—5 мл азотной кислоты (плотность 1420 кг/№), 100 мл этилового или метилового спирта Реактивы окрашивают перлит в темный цвет, выявляют границы зерен феррита, структуру мартенси-
2 Спиртовой раствор пикриновой кислоты (реактив Ижевского) 3—5 г пикриновой кислоты (кристаллической), 100 мл этилового или метилового спирта та и продуктов отпуска. Применяются также для выявления структуры азотированной и цементованной стали. С увеличением количества азотной кислоты возрастает скорость травления. Продолжительность травления — от нескольких секунд до минуты
3 Раствор пикриновой кислоты с добавкой поверхностноактивных веществ (ПАВ) Насыщенный водный раствор пикриновой кислоты; 1—5 % ПАВ типа Синтонол или моющих веществ «Прогресс», «Астра» и др. Для выявления границ зерен аустенита в закаленной стали. Время травления при 20 °C — от 5 до 30 мин, при 50—70 °C — 0.5—6 мни
4 Раствор пикриновой и азотной кислот 2 г пикриновой кислоты, 2—4 мл азотной кислоты (плотность 1480 кг/мв), 100 мл этилового спирта Для выявления границ зерен в закаленной углеродистой стали
Для травления высоколегированных сталей
5 Раствор азотной и соляной 10 мл азотной кислоты (плотность Для выявления структуры высоко-
6 кислот в глицерине Царская водка 1420 кг/м8), 20—30 мл соляной кислоты (плотность 1160 кг/м8), 30 мл глицерина 3 ч. соляной кислоты (плотность хромистой, быстрорежущей и аустенитной марганцевой стали в закаленном состоянии рекомендуется попеременное травление и полирование Для выявления структуры нержаве-
7 Солянокислый раствор хлор- 1190 кг/м8); 1 ч. азотной кислоты (плотность 1480 кг/м3) 5 г хлорного железа, SO мл соля- ющих сталей и сплавов. Перед употреблением реактивы надо выдержать 20—30 ч Для исследования структуры высо-
8 ного железа Раствор хромовой и соляной ной кислоты (плотность 1190 кг/м8), 100 мл воды 50 мл соляной кислоты (плотность коникелевой нержавеющей аустенитной стали Для выявления структуры терми-
9 кислот Солянокислый раствор мед- 1190 кг/м3), 50 мл хромовой кислоты (10 %-ной) 10 г сернокислой меди, 50 мл соля- чески обработанной аустенитной стали Для выявления структуры сложно-
10 ного купороса (реактив Мербле) Раствор хромпика в соляной ной кислоты (плотность 1190 кг/м3), 50 мл воды 45 мл соляной кислоты (плотность легированной аустенитной стали Для выявления структуры жаро-
11 и азотной кислотах Для Щелочной раствор пикрата 1190 кг/м3), 5 мл азотной кислоты (плотность 1560 кг/м8), 12 г двухромовокислого калия выявления карбидов, фосфидов, вольфр 2 г пикриновой кислоты, 25 г едко- прочных никелевых сплавов амидов Для выявления цементита, который
12 натрия Щелочной раствор красной го натра, 100 мл воды 10 г красной кровяной соли, 10 г окрашивается в темный цвет; карбиды хрома и вольфрама не окрашиваются. Реа актив применяется в кипящем состоянии Применяется в горячем состоянии
13 кровяной солн (реактив Мураками) Солянокислый раствор хлор- едкого кали. 100 мл воды Для выявления линий, напряжений 40 мл соляной кислоты, 5 г хлор- для выявления хромистых карбидов, вольфрамидов в быстрорежущей и других сталях. Этот же реактив выявляет фосфиды в фосфид-ной эвтектике. Фосфид окрашивается в темный цвет Для выявления напряжений и дне-
ной меди ной меди, 30 мл воды. 25 мл этилового спирта персионного твердения стали. Реактив применяется иа холоду. Время травления до 10 с
Для выявления границ зерен аустенита используют следующие методы.
Метод цементации. Этот метод применяют для цементуемых сталей, а таже для неце-ментуемых сталей, содержащих до 0,3 % С. Образцы подвергают нагреву в плотно закрытом железном ящике прн 930±10°С, 8 ч (после полного прогрева) в цементующей среде, чаще в смеси из 40 % ВаСОз н 60 %
древесного угля нли из 30 % ВаСОз н 70 % древесного угля. После охлаждения вместе с ящиком до 600 °C (скорость охлаждения для углеродистой стали 100 °С/ч, для легированной 50°С/ч), а затем на воздухе изготовляют мнкрошлнф; поверхностный слой сошлифовы-вают на глубину не более 2 мм и проводят травление в реактивах № 1,2 н ,11 (см. табл. 1.5). Величину зерна определяют в за-
44
ТАБЛИЦА 1.6
РЕАКТИВЫ ДЛЯ ЭЛЕКТРОЛИТИЧЕСКОГО ТРАВЛЕНИЯ ШЛИФОВ
№ Наименование реактива Состав реактива Назначение и особенности применения
1 Щелочной раствор пикриновой кислоты 2 г пикриновой кислоты, 20 г едкого натра, 100 мл воды Для углеродистой и низколегированной стали; окрашивает карбидную фазу. Условия травления: температура 100 °C, напряжение 6 В, продолжительность 20 с. Катод из нержавеющей стали
2 Солянокислый раствор хлорного железа 0.Б г хлорного железа, I г соляной кислоты (плотность 1190 кг/м3), 98,5 % (по массе) метилового спирта Для углеродистой стали. Условия травления: плотность тока 5 А/дм2, температура 20—30 °C. Катод из нержавеющей стали
3 Раствор щавелевой кислоты 2—10 г щавелевой кислоты, 100 мл воды Для выявления карбидов и основной структуры нержавеющей стали и никелевых сплавов. Режим травления: температура 20—30 °C, напряжение 3—6 В. Катод — нержавеющая сталь
4 Раствор хромовой кислоты 10 %-ная хромовая кислота Для выявления карбидов и о-фазы в аустенитной или ферритной нержавеющей стали. Условия травления: температура 20— 30 °C, напряжение 3 В. Катод из нержавеющей стали
5 Раствор азотной кислоты 1 ч. азотной кислоты, 1 ч. воды Для выявления границ зерен в аустенитной нержавеющей стали. Условия травления: температура 20—30 °C, напряжение 1,5 В, время травления до 2 мин. Катод из нержавеющей стали
6 Раствор едкого натра 400 г едкого натра, ЮОО мл воды Для нержавеющей стали. Окрашивает о-фазу и карбиды, но не вытравливает феррит. Условия травления: температура 20—30 °C, напряжение 1,5—2 В. Катод из нержавеющей стали
7 Раствор азотной и уксусной кислот 2 ч. азотной кислоты (плотность 1560 кг/м3), 1 ч. ледяной уксусной кислоты, 17 ч. воды Для никелевых сплавов; особенно пригоден для выявления зерна. Условия травления: температура 20—30 °C, напряжение 1,2 В, продолжительность травления 20— 60 с.
эвтектоидной зоне цементованного' слоя по цементнтной сетке, окаймляющей границы бывших зерен аустенита.
Метод окисления. Полированные шлнфы нагревают в вакууме или в сухой инертной атмосфере в муфельной печи на 20—30 °C выше обычных температур закалкн с выдержкой 3 ч. Затем в печь подают воздух на 30—60 с, после чего образцы охлаждают в воде. Перед закалкой рекомендуется провести нагрев в расплавленной буре прн 930—950 °C в течение 30—40 с, а затем охладить в воде. После указанных обработок шлнфы полируют н подвергают травлению в 15 %-ном растворе соляной кислоты в этиловом спирте. Г раннцы зерен выявляются по сетке окислов.
Метод травления границ зерен аустенита. Его обычно применяют для сталей, закаленных на мартенсит (или бейнит). Образцы нагревают в тех же условиях, как и по предыдущему методу, охлаждают в масле илн в воде, а затем подвергают отпуску в течение 15—30 мин прн 225—250 СС для углеродистой н низколегированной стали н 500—550 °C для высоколегированной стали. После изготовления микрошлифа проводят травление в реактиве 3 (табл. 1.5).
Метод сетки феррита (для сталей с содержанием углерода до 0,6 %) или цементита (для зазвтектоидных сталей). Образцы нагревают в тех же условиях, что и в последних двух случаях, а затем охлаждают: углеродистые стали с 0,3—0,5 % С на воздухе, стали с 0,5—0,6 % С со скоростью 50—100 °С/ч, а легированные н заэвтектоидные стали со скоростью 20—30°С/ч. Затем образцы охлаждают в воде н изготовляют микрошлифы. Прн выявлении зерна по сетке феррита травление
проводят в реактиве' 1 (табл. 1.5), а по сетке цементита—в реактивах 1,2 илн 11 (табл. 1.5).
Метод сетки троостита для сталей, близких по составу к эвтектоидным. Образцы нагревают, как и в случае метода окисления, а затем охлаждают путем погружения в воду одной половины образца; вторая половина охлаждается на воздухе. В переходной зоне образца образуется структура, состоящая из мартенсита, окаймленного сеткой троостита; последняя выявляется травлением в спиртовом растворе азотной илн пикриновой кислоты.
Для определения размера зерна сравнивают1 наблюдаемую микроструктуру прн увеличении 100 со стандартными шкалами2 или подсчитывают число зерен, приходящихся на единицу поверхности шлифа, илн вычисляют средний условный диаметр зерен нли их число в 1 мм3 металла. Расчеты этих параметров оценки величины зерна для стандартных номеров (баллов) приведены в табл. 1.8.
Число зерен3 подсчитывают на матовом стекле микроскопа илн по микрофотографии в пределах площади, органиченной окружностью. диаметром 79,8 мм. Прн увеличении 100 это соответствует площади на шлифе 0,5 мм2. Общее число зерен подсчитывают по
1 Указанные ниже методы определения величины зерен не следует применять для наклепанных или частично рекристаллизованных металлов,
2 Шкалы составлены таким образом, что при увеличении 100 номер зерна N соответствует формуле п=8Х2^« где п—количество зерен на 1 мм2 площади шлифа. Если размер зерна выходит за пределы номеров 1—10, то пользуются другими увеличениями, пересчитывая их по табл. 1.9.
3 Число верен должно быть не менее "50. Если их меньше, то используют меньшее увеличение.
45
ТАБЛИЦА 1.8
ХАРАКТЕРИСТИКИ (ПАРАМЕТРЫ) СТРУКТУРЫ СТАЛИ С РАЗНОЙ ВЕЛИЧИНОЙ ЗЕРНА
Номера зерен Средняя площадь сечения зерна, мм Среднее число зерен на площади 1 мм4 шлифа Среднее число зерен в 1 mms Средний диаметр зерна, мм
по расчету условный
—3 1,024 1 1 1,00 0,875
—2 0,512 2 2,7 0,694 0,650
—1 0,256 4 8 0,500 0,444
0 0,128 8 21 0,352 0,313
1 0,064 16 64 0,250 0,222
2 0,032 32 179 0,177 0,167
3 0,016 64 512 0,125 0,111
4 0,008 128 1 446 0,088 0,0788
5 0,004 256 4 096 0,060 0,0553
6 0,002 512 11 417 0,041 0,0391
7 0,001 1 024 32768 0,031 0,0267
8 0,0005 2048 92 160 0,022 0,0196
9 0,00025 4 096 262 122 0,015 0,0138
10 0,000125 8 192 737 280 0,012 0,0099
11 0,000062 16 384 2 097 152 0,0079 0,0069
12 0,000031 32 768 5930 808 0,0056 0,0049
13 0,000016 65 536 16 777 216 0,0039 0,0032
14 0,000008 131 072 47 448 064 0,0027 0,0023
ТАБЛИЦА 1.9
ПЕРЕСЧЕТ НОМЕРА ЗЕРНА НА СТАНДАРТНОЕ УВЕЛИЧЕНИЕ (ХЮО) ПРИ ИСПОЛЬЗОВАНИИ УВЕЛИЧЕНИЙ ОТ 25 ДО 800
Увеличение Номера зерен
-3 —2 —1 0 1 2 3 4 5 6 7 8 9 10 п 12 13 14
25 1 2 3 4 5 6 7 8 9 10
50 1 2 3 4 5 6 7 8 9 10
200 1 2 3 4 5 6 7 8 9 10
400 1 2 3 4 5 6 7 8 9 10
800 1 2 3 4 5 6 7 8
формуле m1O0=m+O,5mb где т — число зерен внутри круга; т\ — число зерен, пересекающихся окружностью. Число зерен, приходящееся на I мм2 поверхности шлифа,- равно Л4= =2-mlW). Прн увеличении, отличном от 100, Л'1=2(§-/1С0)2/пд, где g—применяемое увеличение и те— число зерен, подсчитанное прн этом увеличении. Указывают среднее число зерен (7Иср), выводя его из числа зерен для трех характерных полей зрения.
Среднюю площадь зерен (Sep) н диаметр зерен (tiCp) вычисляют по формулам:
SCp = 1 / МСр; dcp = 1 / \ МСр.
Величины равноосных зерен характеризуют средним условным диаметром, который определяют ПО1 изображению на матовом стекле микроскопа или по микрофотографии. С этой целью- проводят произвольно несколько прямых линий так, чтобы каждая линия пересекала ие менее 10 зерен. После этого подсчитывают число пересечений на длину всех линий (включают и те зерна, которые полностью не пересечены у концов линий). Затем, разделив суммарную длину всех линий на число пересечений н учитывая выбранное увеличение, получают средний условный диаметр зерен.
Для количественной характеристики структуры, в частности размеров зерен, применяют статистические методы н строят кривые рас
пределения нли гистограммы *. С помощью окуляр-микрометра определяют число зерен, размер которых находится в пределах определенного числа делений окуляр-микрометра (например, от 0,5 до 1,5; от 1,5 до 2,5; от 2,5 до 3,5 и т. д.); по этим данным и строят гистограммы. Если кривая имеет ряд минимумов и максимумов, то проводят ее сглаживание, подсчитывая число зерен в более крупных интервалах размеров, т. е. увеличивают интервалы группировки.
На основании кривой распределения подсчитывают средний днаметр зерна: dcp= =/eXmx/2/n, где т — частота группы (т. е. число зерен, приходящихся на определенный интервал шкалы окуляр-микрометра); х — диаметр в делениях шкалы (т. е. определенный интервал шкалы); k — средняя цена деления шкалы, мкм.
Средняя площадь зерен (Scp) может быть вычислена, исходя из формулы определения dep, если принять, что зерно имеет форму шара (X=jtD2/4):
Scp = k2 2 (лЕ>2/4) m/Ym.
Отсюда число зерен (Дг), приходящихся на 1 мм2, определяют по формуле N=l/Scp.
1 Гистограмму можно получить быстро с помощью количественных микроскопов, например тип» «Квантимет».
46
БИБЛИОГРАФИЧЕСКИЙ СПИСОК
1. Лаборатория металлографии. 2-е над./ Панченко Е. В., Скаков К). А., Кример Б. И. и др. М.: Металлургия, 1965. 439 с.
2. Беккерт М., Клемм X. Справочник по металлографическому травлению. М.: Металлургия, 1979. 335 с.
3. Приборы н методы физического металловедения. Вып. 1. М..- Мир, 1973. 427 с.
4. Металлография железа. Т.1—3: Пер. с англ. Мл Металлургия, 1972. 127 с; 104 с.; 75 с.
5. Испытание материалов. Справочник. Пер. с нем. М.: Металлургия, 1979. 447 с.
6, Mathesius Н. А. — Praktische Metallo-. graphie, 1977, Bd 14, № 1, S. 46—48.
7. Попилов JI. К-, Зайцева JI. П. Электро-полироваиие и электротравление металлографических шлнфов. М.: Металлур-издат, 1955. 311 с.
8. Жаке П. Электролитическое и химическое полирование. М.: Металлургиздат, 1959. 139 с.
9. Коваленко В. С. Металлографические реактивы 3-е изд.; Справочник. М.: Металлургия, 1970. 133 с.
10. Червяков А. Н., Киселева С. А., Рыль-никова А. Г. Металлографическое определение включений в стали. 2-е изд. М.; Металлургиздат, 1962. 248 с.
11. Мотт Б. В. Испытание на твердость мнк-ровдавлнваннем. М: Металлургия, 1960. 338 с.
12. Григорович В. К. Твердость и микро-твердость металлов. М.: Наука, 1976. 230 с.
13. Лозинский М. Г. Тепловая микроскопия материалов. М.: Металлургия, 1976.
303 с.
14. Виноград М. И., Громова Г. П. Включения в легированных сталях. М.: Металлургия, 1972. 215 с.
15. Литвинова Т. В., Пирожкова В. П., Петрова А. К. Петрография неметаллических включений. М.: Металлургия, 1972. 184 с.
2
==== ПРОСВЕЧИВАЮЩАЯ ЭЛЕКТРОННАЯ МИКРОСКОПИЯ
Просвечивающая электронная микроскопия — одни из наиболее информативных методов исследования структуры металлов и сплавов, в котором сочетаются возможности получения в одном эксперименте изображений с высоким О
разрешением (вплоть до ~ 1 А) и дифракционных картин одного и того же участка образца (этот участок, как правило, монокристаллический). Применение дифракционной электронной микроскопии (метода тонких фольг) оказало решающее влияние на формирование современных представлений о механизмах фазовых превращений (мартенситных превращений, распада пересыщенных твердых растворов и др.), строения границ, о процессах пластической деформации, разрушения и рекристаллизации, о структурных проявлениях радиационных повреждений, о тонкой химической неоднородности сплавов н т. д.
Применяются электронные микроскопы различных типов: растровые (сканирующие), эмиссионные, отражательные. Все они успешно используются при решении металловедческих задач; наиболее широкое распространение в физическом металловедении получили электронные микроскопы просвечивающего типа с электромагнитными линзами.
2.1. КОНСТРУКЦИЯ
МИКРОСКОПА
И ПРИНЦИПЫ ЕГО РАБОТЫ
Просвечивающий электронный микроскоп состоит из электронной пушки и системы электромагнитных лниз, заключенных в колонну, откачиваемую до остаточного давления ие вы
ше 10~2—10~3 Па. Оптическая система микроскопа такая же, как у светового микроскопа, но содержит дополнительные ступени увеличения (дополнительные лиизы). Осветительная система микроскопа состоит из электронной пушки и, как правило, двухлинзового конденсора. Электронная пушка (трехэлектродная электростатическая линза) состоит из катода, которым служит раскаленная вольфрамовая нить —- источник электронов, фокусирующего электрода, имеющего отрицательный потенциал относительно катода и анода с высоким положительным потенциалом по отношению к катоду. Силовое поле между катодом и фокусирующим электродом работает как собирающая линза, а силовое поле между этим электродом и анодом — как рассеивающая лииза. В результате и целом пушка работает как слабо рассеивающая лииза. Первая электромагнитная лииза двухлиизового конденсора создает уменьшенное изображение источника электронов, вторая перебрасывает это изображение в плоскость, близкую к плоскости объекта, что позволяет освещать на объекте лишь минимально необходимый . участок. Малый апертурный угол падающего электронного пучка обеспечивается конденсорными диафрагмами.
Объективная линза, расположенная ниже плоскости объекта, фокусирует рассеянные объектом электроны в плоскости изображения этой линзы, формируя первичное изображение. Следующая, промежуточная, лииза перебрасывает это изображение в предметную плоскость проекционной лиизы, а та в свою очередь формирует конечное изображение объекта на флюоресцирующем экране или фотопластинке.
47
Подвижная апертурная диафрагма с отверстием диаметром 10—50 мкм расположена в задней фокальной плоскости объективной линзы; оиа позволяет выбрать из всех рассеянных электронов более илн менее узкий пучок и лишь его использовать для формирования изображения, что обеспечивает контраст изображения (как абсорбционный, так н дифракционный). Кроме того, апертурная диафрагма способствует получению большей резкости изображений, уменьшая влияние сферической аберрации. Малая угловая апертура объективной линзы обеспечивает и большую глубину резкости, необходимую для получения резких снимков иа фотопластинках, расположенных значительно ниже экрана, на котором фокусируется изображение. Наличие подвижной апертурной диафрагмы позволяет получать темнопольные изображения путем смещения падающего электронного пучка или диафрагмы таким образом, чтобы через нее проходили только рассеянные электроны. Тогда те участки объекта, которые сильнее рассеивают электроны, будут иа изображении более светлыми. При исследованиях необходимо выбирать оптимальные размеры апертурной диафрагмы, поскольку с их уменьшением возрастают контрастность и резкость изображения, но падает его яркость.
Фокусировка изображения осуществляется путем изменения фокусного расстояния объективной лннзы, изменение увеличения — путем изменения фокусных расстояний промежуточной н проекционной линз, а также сменой полюсных наконечников последней. Изменение фокусных расстояний линз достигается изменением силы тока в обмотках линз.
Общее увеличение микроскопа определяется произведением увеличений объектной, промежуточной н проекционной линз н может варьироваться от нескольких тысяч до примерно 500000 и более в микроскопах лучших конструкций.
Для юстировки электронной пушки, кон-денсорных и изображающих линз, т. е. выведения их на единую оптическую ось микроскопа, в современных электронных микроскопах предусмотрены электромагнитные отклоняющие системы; эти системы позволяют быстро юстировать пучок и получать резкие светло-н темнопольные изображения. Особенно это важно при исследовании ферромагнитных материалов, поскольку собственное магнитное поле образца — фольги — влияет на магнитное поле объективной лннзы, и ее юстировку приходится проводить прн каждом изменении положения данного образца в процессе наблюдения в микроскопе.
В современных электронных микроскопах имеется приставка для уменьшения загрязнения объекта (металлической фольги) продуктами разложения электронами паров углеводородов, неизбежно присутствующих в колонне микроскопа (вакуумные масла, смазки). Загрязнения возникают на облучаемом электронами участке объекта так быстро, что уже через 2—3 мин дальнейшее наблюдение н получение качественных снимков становится невозможным. Приставка представляет собой наполняемый жидким азотом металлический сосуд Дьюара, расположенный вблизи камеры объекта. Внутренняя стейка сосуда Дьюара соединена с помощью медного стержня — хла-
допровода — со специальными экранами той илн иной формы, находящимися внутри колонны микроскопа в непосредственной близости от объекта. Использование такой приставки позволяет в несколько десятков раз-уменьшнть скорость роста углеродного (почти аморфного) слоя на объекте, поскольку конденсация паров углеводородов и а холодных поверхностях экранов резко снижает пар-циоиальное давление этих паров в камере объекта.
Загрязнение объекта значительно уменьшается н прн применении пористых адсорбентов-цеолитов, способных поглощать крупные молекулы углеводородов. Цеолит в виде гранул-помещают в плоские коробочки, покрытые мелкой сеткой из нержавеющей стали; коробочки устанавливают в камере объекта.
Для металловедческих исследований обычно применяют микроскопы с ускоряющим напряжением 100—200 кВ. Такие микроскопы позволяют просматривать объекты толщиной до —0,2—0,6 мкм, причем предельная толщина, зависит от типа материала (его атомной массы). Существуют также высоковольтные и сверхвысоковольтиые электронные микроскопы с ускоряющим напряжением 500, 1000,1500 и даже 3500 кВ. Такие микроскопы обеспечивают изучение объектов толщиной до нескольких микрометров н, кроме того, обладают рядом других преимуществ.
Благодаря тому что в конструкции электронного микроскопа имеются промежуточная линза н специальная селекторная диафрагма, расположенная в плоскости изображения объективной линзы, можно- получать мнкродиф-ракцнонные картины от участка, выделяемого этой диафрагмой. После установки селекторной диафрагмы уменьшают силу тока в промежуточной лннзе до такой величины, при которой предметная плоскость этой лнизы совпадает с задней фокальной плоскостью объективной линзы (т. е. с плоскостью, в которой формируется дифракционная картина от кристаллического объекта), и удаляют апертурную диафрагму. В результате иа конечном экране с помощью проекционной линзы фиксируется дифракционная картина от выбранного селекторной диафрагмой участка объекта. Величина участка, выделяемого для микро дифракций, определяется размером селекторной диафрагмы, деленным на увеличение объективной лннзы. Уменьшение величины участка ограничено снижением яркости изображения дифракционной картины и величиной несоответствия между участком объекта, от которого-эта картина получена, н участком, вырезаемым селекторной диафрагмой. Нанлучшее соответствие между изображением и микродифрак-цнониой картиной достигается при условии, что селекторная диафрагма устанавливается прн таком режиме работы промежуточной линзы (увеличении микроскопа), прн котором края диафрагмы резко сфокусированы (т. е. достигается точное совпадение плоскости промежуточного изображения, формируемого объективной линзой, с плоскостью расположения селекторной диафрагмы); лишь после этого фокусируют изображение объекта. Тем не менее даже прн выполнении этих условий из-за сферической аберрации объективной лннзы указанное несоответствие все же может достигать десятых долей микрометра. Практически
48
в100-кВ электронном микроскопе (для которого характерна определенная величина сферической аберрации объектива) хорошее соответствие между участком, выделенным диафрагмой и видимым иа изображении, н участком, от которого наблюдается мнкродифрак-ционная картина, сохраняется при диаметре селекторной диафрагмы ие менее 1 мкм. Прн меньших размерах диафрагмы в образоваинн дифракционной картины принимают участие дифракционные пучки, сформированные участками объекта, которые находятся вне участка, выделенного для микродифракции. Поэтому при получении мнкродифракцин от участков площадью менее ~1 мкм2 (напрнмер, от мелких кристалликов разной природы в фольге или реплике) для установления надежного соответствия между изображением н микро-дифракционной картиной необходимо использовать темнопольиые изображения в свете рефлексов, присутствующих на микрозлек-ронограмме.
Существенная конструктивная особенность современных электронных микроскопов — наличие в камере объекта гониометрического столика. Известно много конструкций гониометра, но все оин обеспечивают наклон объекта на угол ~20° (иногда и до 60°) относительно горизонтальной осн, расположенной вблизи плоскости помещенного в микроскоп объекта. С помощью гониометра можно получать различные дифракционные картины от одного и того же участка, изменять контраст на изображении, проводить пространственные измерения и др. Гониометр предварительно калибруют, т. е. определяют точное положение проекции осн наклона иа конечном экране и цену деления угломерного устройства [6, 7).
Теоретическая разрешающая способность любого оптического прибора имеет величину порядка длины волны излучения, используемого для освещения объекта. Длина волны электронов А (в ангстремах): А= 12,236: 1 V, где V — ускоряющее напряжение, В.
Для 100-кВ электронного микроскопа А= D
=0,037 А. Прн ускоряющих напряжениях свыше 500 кВ прн определении А необходимо вносить релятивистскую поправку, снижающую темп уменьшения А с повышением V; величина релятивистской поправки достигает ~ 30 % прн напряжении 1000 кВ.
Малая длина волны электронов означает, что теоретический предел разрешения электронного микроскопа очень высок. Однако практически он ие реализуется из-за дефектов электронной оптики — хроматической и сферической аберрации и астигматизма линз.
Хроматическая аберрация возникает из-за различной скорости (т. е. длины волны) электронов, формирующих изображение, непостоянства (во времени) фокусных расстояний линз, а также из-за потерь энергии электронами прн прохождении объекта. Первые два источника хроматической аберрации можно уменьшить путем высокой стабилизации ускоряющего напряжения и силы тока в линзах (в современных приборах относительные колебания высокого напряжения не превышают величины порядка 10“6, а токов в линзах — порядка 10~5), третий источник — путем использования более тонких объектов. Сферическая аберрация возникает в результате того, что
изображение формируется электронами, проходящими иа разных угловых расстояниях от оптической оси линз. Величина сферической аберрации линзы пропорциональна а3, где а — половина апертурного угла лучей, пропускаемых линзой. Поэтому эта величина больше в более сильных линзах, таких как объективная и проекционная. Минимальное значение сферической аберрации достигается наложением строгих ограничений на геометрию полюсных наконечников лииз; кроме того, к уменьшению сферической аберрации приводит н увеличение ускоряющего напряжения, поскольку изображение при этом формируется электронами, в меньшей степени отклоненными от оптической осн лииз.
Возникновение астигматизма связано с нарушением магнитной илй геометрической симметрии лиизы. Уменьшение астигматизма достигается точной механической обработкой ее магнитных деталей (прежде всего полюсного наконечника), а также внешней корректировкой с помощью специального стигматора, который обеспечивает восстановление симметрии магнитного поля лннзы. В современных микроскопах стигматорамн снабжены объективная и вторая конденсорная лннзы.
Указанные несовершенства электронной оптики вынуждают резко органичивать апертурные углы (т. е. использовать только приосевые пучки электронов) и приводят к тому, что реальная разрешающая способность на 2—3 порядка хуже теоретической.
Различают предельное разрешение по линиям и по точкам. Разрешение по линиям определяют путем разрешения на изображении кристаллических плоскостей вещества с известной структурой. Такие изображения получают путем пропускания через апертурную диафрагму первичного' и одного' из дифрагированных пучков (метод прямого разрешения см. 2.2.3). Разрешение по точкам определяют по максимальному полезному увеличению микроскопа, при котором удается различить на изображении (фотопластинке) две характерные точки, отстоящие одна от другой на 0,1—0,2 мм. Условия формирования изображения рассмотренных двух типов таковы, что разрешение по линиям всегда немного выше, чем по точкам.
В электронных микроскопах последних конструкций (японская фирма JEOL) методом прямого разрешения удалось получить рекорд-
С
ное разрешение ~1 А, которое позволило прямо «видеть» атомы таких металлов, как медь и золото. Паспортное разрешение лучших отечественных и ‘зарубежных микроскопов О
2—10 А, однако' «рабочее» разрешение несколько хуже (20—30 А) и зависит от ряда факторов: типа объекта, механической устойчивости прибора, его юстировки, наличия загрязнений и т. д.
Разрешение дифракционной картины (дифракционная ошибка) зависит от апертуры падающего электронного пучка (угла сходимости пучка на объекте). Малая длина волны электронов и, следовательно, малая величина-брэгговских углов приводят к тому, что дифракционная ошибка весьма существенна даже при типичных для электронного микроскопа малых углах сходимости электронного пучка-
4—280
4&>
порядка нескольких угловых минут. Поэтому точность определения межплоскости ых расстояний из электронно-дифракционных данных обычно примерно на порядок меньше, чем из рентгенографических. Дифракционную ошибку можно несколько снизить путем использования подвижных конденсорных диафрагм меньших размеров (50—100 мкм вместо обычных 300—500) н уменьшения угла сходимости пучка за счет уменьшения силы тока второго конденсора («разведения» пучка). При этом, естественно, уменьшается яркость изображения.
2.2. МЕТОДЫ ЭЛЕКТРОННОМИКРОСКОПИЧЕСКОГО ИССЛЕДОВАНИЯ МЕТАЛЛОВ И СПЛАВОВ
2.2.1. Косвенный метод
Методически наиболее прост косвенный метод исследования с помощью тонких реплик (слепков), получаемых с поверхности образца [1, 2]. Поскольку микрорельеф протравленного шлифа отражает микроструктуру образца и его химическую неоднородность, изучение такого рельефа прн больших электронно-оптических увеличениях дает определенную информацию о тонких деталях микроструктуры. Прн изучении пластической деформации, мартенситных превращений н микрорельефа разрушенных образцов самостоятельное значение имеет наблюдение микрорельефа на нет рае леном шлифе.
Одноступенчатые (негативные) реплики приготовляют путем конденсации из паров углерода, кварца, титана и других веществ непосредственно иа поверхность исследуемого образца, а двухступенчатые (позитивные) — яа предварительно изготовленный оттиск (обычно из полистирола) исходного рельефа образца. Конденсацию (напыление) проводят в вакууме. В качестве репликн для ряда исследуемых материалов (медн, алюминия и др.) можно использовать окисную пленку, которая образуется прн электролитическом или химическом оксидировании поверхности образца. Неплохие результаты дает применение лаковых реплик, получаемых путем нанесения на поверхность шлнфа тонкого слоя лака (4 %-ного раствора коллодия в амилацетате) .
Негативные реплики отделяют либо механически (с использованием желатины), лябо травлением образца под репликой, предварительно насеченной на квадратики. Первичные оттиски двухступенчатых отпечатков отделяют от образца механически, а позитивные репликн от оттиска — растворением его.
В практике электронно-микроскопических исследований косвенным методом наиболее широко применяются негативные репликн, полученные конденсацией из паров, поскольку они лучше всего воспроизводят рельеф шлифа. При этом чаще всего используют углеродные реплики, обладающие высокими прочностью, устойчивостью под электронным пучком, отсутствием собственной структуры и хорошей контрастностью.
Важный методический прием, повышающий контрастность изображения репликн, — оттенение рельефа достаточно тяжелым метал
лом — хромом, титаном, золотом, платиной н др. Ойо прежде всего необходимо для таких слабоконтрастных реплик, как лаковые. Оттенение проводят путем напыления в вакууме небольшого количества металла с таким расчетом, чтобы средняя толщина его слоя не превышала нескольких сотен ангстремов. Объектом напыления может быть либо готовая реплика, либо поверхность шлифа еще до получения с него реплики.
Косвенный метод исследования применяется ограниченно из-за трудности однозначно интерпретировать эффекты контраста иа изображении н идентифицировать различные структурные составляющие, нз-за частого возникновения артефактов, связанных с деформацией реплики прн ее отделении от объекта н прн различных манипуляциях с ией. Кроме того, разрешение электроино-мнкроско-пнческих изображений лимитируется разрешением самой реплнки, которое в лучшем случае достигает нескольких десятков ангстремов. В то же время развитие растровой (сканирующей) электронной микроскопии позволяет примерно с тем же разрешением прямо изучать поверхностный рельеф металлического образца, а также по рентгеновскому характеристическому излучению определять химический состав различных структурных составляющих и даже наблюдать картину распределения того или иного химического элемента по поверхности объекта. Поэтому практическая значимость косвенного метода невелика и в настоящее время ограничена электронной фрактографией.
2.2.2. Полупрямой метод
При исследовании гетерофазных сплавов используют полупрямой метод [1,3], т. е. косвенное (с помощью реплнк) электронномикроскопическое исследование основной фазы (матрицы) и прямое электронно-микроскопическое и микродифракционное исследование второй фазы, частицы которой извлекаются из матричной фазы в реплику.
Для получения репликн с извлеченными частицами шлиф исследуемого сплава протравливают таким образом, чтобы растворялась только матричная фаза; после этого на шлиф наносят пленку (например, углеродную), как при изготовлении негативного отпечатка. Далее образец с напыленной пленкой, насеченной лезвием на квадратики, протравливают по режиму, обеспечивающему растворение основы и сохранение второй фазы, до полного отделения пленки с частицами этой фазы (частицы фиксируются в пленке в тех положениях, которые оии занимали в образце). Изготовленная таким способом реплнка позволяет изучать поверхностный рельеф шлифа, как и в случае косвенного метода, и, кроме того, получать четкие электронно-микроскопические изображения частиц второй фазы, определять их размеры, форму, распределение н (с помощью мнкродифрак-ции) атомно-кристаллическую структуру1.
1 Нужно учитывать, что при извлечении частицы, особенно в случае ее упругого сопряжения с решеткой матрицы, возможно изменение ее упруго-напря- । жеиного состояния, которое может повлечь за собой даже изменение атомно-кристаллической структуры.
50
Прн электронно-микроскопическом изучении реплик с извлеченными частицами весьма полезно выполнять стереосъемку такого препарата, которая дает наглядное пространственное представление о взаимном расположении частиц в матрице. Особенно большой (н полезный для анализа) стереоэффект возникает при стереосъемке реплики, полученной от излома.
Рис. 2.1. Сталь 20ХГ2. Закалка и отпуск при 630 °C, 1 ч + 500 СС, 24 ч. Углеродная пленка с фиксированными карбидными частицами: крупные округлые частицы по границам зерен — тригональный карбид хрома СггС5, мелкие вытянутые частицы внутри зерен — цементит FesC. ХЮ ООО
Преимущества полупрямого метода по сравнению с косвенным привели к его широкому применению для исследования гетерофазных сплавов. С помощью полупрямого метода получены важные результаты об особенностях карбндообразования прн отпуске легированных сталей, о структуре ряда стареющих сплавов, о процессах разрушения металлов н сплавов н др.
Типичный пример электронной микрофотографии, которая получена с репликн, содержащей частицы второй фазы, приведен на рис. 2.1.
Полупрямой метод незаменим при исследованиях, связанных с идентификацией различных дисперсных фаз в металлической матрице прн малой объемной доле этих фаз (например, неметаллических включений в стали).
Отсутствие собственной структуры у репликн (в дифракционном смысле) позволяет получать дифракционные картины от отдельных частиц, находящихся в такой «бесструктурной» реплике; подобные дифракционные картины весьма сложно выявить и зафиксировать, если частица находится в металлической матрице (прн прямом исследовании), поскольку интенсивное рассеяние электронов матрицей «забивает» слабые рефлексы от малой частицы.
2.2.3. Прямой метод
Этот метод электронно-микроскопического исследования дает наибольшую информацию о структуре объекта, которым служит тонкая металлическая пленка (фольга), прозрачная или полупрозрачная для электронов. Получают ее либо путем осаждения из паров в вакууме (илн электролитически из раствора), либо путем утонения массивных образцов.
Метод получения тонких металлических пленок конденсацией в вакууме аналогичен методу получения реплик напылением (например, углеродных). Металл, из которого необходимо получить тонкую пленку, помещают в испаритель (обычно свернутая в спираль вольфрамовая или молибденовая проволока, через которую пропускают электрический ток). Пар конденсируется на специальной подложке, в качестве которой обычно используют легко растворимые в воде кристаллы NaCl нлн КС1. После отделения от подложки тонкие пленки готовы для исследования. Онн однородны по толщине и прак-ч тическн свободны от загрязнений, неизбежно присутствующих в массивных металлических образцах. Именно на таких пленках толщиной D
порядка 100 А было получено рекордное раз-О
решение менее 1 А.
Использование пленок, конденсированных в вакууме, ограничено чистыми металлами и однофазными сплавами; в последнем случае для обеспечения химической однородности по сечению пленок необходим длительный гомогенизирующий отжиг полученных пленок.
Структура и свойства тонких пленок, полученных путем конденсирования в вакууме, иные, чем массивных образцов. Поэтому результаты наблюдений, проводимых на таких пленках, имеют значение прежде всего для пленочных материалов (используемых в блоках памяти ЭВМ, полупроводниковых устройствах и т. д.). На тонких пленках получены фундаментальные данные о явлении эпитаксии н характере сопряжения кристаллов различных материалов при последовательной их конденсации один на другом. Подробно результаты электронно-микроскопических исследований тонких конденсированных пленок приведены в работе (4]. На рнс. 2.2 приведен типичный пример электронной микрофотографии тонкой пленки, полученной испарением в вакууме.
Другим, причем более распространенным, объектом прямого электронно-микроскопического исследования является тонкая фольга, получаемая утонением массивных образцов. Методы утонения — механическое многократное расщепление по плоскости спайности (для слоистых материалов); ионное травление (ионная бомбардировка); расплющивание капли расплава на холодной полированной поверхности1; химическая и электролитическая полировка. Последний метод наиболее универсален н его используют чаще всего.
Исходной заготовкой для химического нлн
1 При этом надо учитывать, что структура фольги отвечает состоянию материала, охлажденного с очень большими скоростями; этот метод вообще применяется для получения аморфных материалов. Прим. рвд.
Рис. 2.2. Текстурованная пленка алюминия, полученная конденсацией из паров на кристалле КС1. Зерна ориентированы плоскостью (111) параллельно подложке. Х50 ООО:
1— высокоугловая граница; 2— малоугловая граница вращения — гексагональная дислокационная сетка;
3 — малоугловая граница наклона — дислокационная стенка (В. М. Косевич и др.)
электролитического утонения могут быть прокатанная лента толщиной до <~0,5 мм или вырезаемая из массивного образца пластинка толщиной 0,2 — 0,5 мм (пластникн вырезают абразивным камнем или иа станках — элект-роэрозионном или анодномеханическом). Желательно, чтобы заготовка имела форму диска и диаметр, подходящий для последующей установки объекта в объекте держателе. В противном случае необходима будет опасная с точки зрения возможных механических повреждений операция по вырезке из утоненной пластинки образца нужных размеров. Наиболее удобны для изготовления дисковых заготовок цилиндрические образцы диаметром 3 — 4 мм.
Заготовки-диски сошлнфовывают на тонкой абразивной бумаге до толщины 0,1—0,15 мм, а затем подвергают химической или электролитической полировке. Химическую полировку чаще используют для предварительного утонения, однако существует много примеров, когда утонение этим методом используют на заключительной стадии приготовления тонких фольг. Преимущество химической полировки перед электролитической — возможность получать более протяженные и однородные по толщине участки фольги. Кроме того, при химической полировке нежелательный во многих случаях нагрев образца происходит в меньшей степени.
Электролитическая полировка осноиана иа аиодном растворении металлов; образец в форме диска — анод—устанавливают в специальном пинцете с отверстием, диаметр которого несколько меньше диаметра диска, и опускают в электролит между двумя металлическими пластинами — катодами. Пинцет предохраняет края образца от растворения. Электролит, как правило, находится и водоохлаждаемом сосуде с двойными стенками. Процесс утонения ведут прн таком режиме, чтобы поверхность образца сохранялась блестящей, без видимых на ней продуктов раство
52
рения. При электрополировке образец нагревается, температура локальных его участков может достигать 200—250 °C. Поэтому при приготовлении фольг из материалов, для которых нагрев нежелателен (например, из закаленной стали), следует использовать электролиты с низкой электропроводностью, в которых электрополировка идет при небольших плотностях тока. Хорошие результаты с точки зрения быстроты ведения процесса и качества получаемых фольг, а также возможности автоматизации процесса дает струевая электрополнровка (струя электролита подается самотеком или с помощью насоса, илн под небольшим избыточным давлением воздуха иад электролитом).
Электрополнровку ведут до тех пор, пока в центральной части утоняемого образца не образуется отверстие (или несколько отверстий) диаметром 0,2—0,8 мм. По краям таких отверстий находятся более или менее протяженные участки фольгн, пригодные для просмотра в электронном микроскопе. При меньших размерах отверстий в готовой фольге площадь пригодных для исследования участков мала, а при больших — тонкие участки, как правило, «съедаются» электролитом, н края таких отверстий непрозрачны для электронов.
Подробные сведения о конструкциях установок для приготовления тонких фольг из различных материалов с помощью химической и электролитической полировки, а также методических приемах и режимах содержатся в монографиях [5—7].
Контраст иа электроино-мнкроскопическом изображении тонкой фольгн определяется упругим н неупругнм рассеянием электронов. Неупругое рассеяние ограничивает толщину образца, который можно изучать иа просвет; оно ответственно за абсорбционный контраст, возникающий прн прохождении электронами участков фольгн, имеющих различную массовую толщину (иапрнмер, темные крупные включения в тонкой фольге). Упругое рассеяние вызывает дифракцию и вносит основной вклад в контраст на изображении реальных кристаллов, содержащих различного типа дефекты, выделения второй фазы н т. д.
Дифракционный контраст возникает вследствие различия в интенсивностях иеотклоиен-иого и дифрагированных пучков, обусловленного локальным изменением условий дифракции, в том числе из-за присутствия в фольге дефектов (дислокации, дефекты упаковки и т. п.). Поскольку изображение формируется либо прямо прошедшим пучком (светлопольное), либо дифрагированным (темнопольиое), приближение к брэгговскому положению каких-либо кристаллических плоскостей данного участка фольги приводит к потемнению иа светлопольном изображении этого участка и посветлению на темиопольном.
Малая величина углов Вульфа—Брэггов для быстрых электронов н соответственно меньшая, чем в случае рентгеновских лучей, чувствительность интенсивностей прямого н дифрагированных лучей к небольшим искажениям кристаллической решетки приводит к тому, что контраст на электронно-микроскопических изображениях дислокаций получается шириной всего лишь в несколько десятков илн сотен ангстремов. Поэтому удается различать от-
дельные дислокационные и другие дефекты при значительной их плотности (например, дислокации в сильно деформированном материале при их плотности вплоть до 1011 см~2). Различные типы дифракционного контраста — деформационного, ориентационного, контраста типа полос смещения (как на дефекте упаковки), контраста вследствие различия структурных факторов — детально описаны в работах [6, Ю].
Особый случай дифракционного контраста возникает при прямом изображении периодичности кристаллической решетки исследуемого материала. Такое изображение получается в результате интерференции прямо прошедшего н дифрагированных пучков, пропущенных через апертурную диафрагму. Картина периодического распределения элементов структуры (в пределе отдельных атомов), на которых происходит рассеяние электронов, тем информативнее, чем больше пучков пропускается через апертурную диафрагму. Для получения картин прямого разрешения кристаллической решетки необходимо, во-первых, чтобы микроскоп имел достаточно высокую разрешаемую способность и, во-вторых, чтобы объект был достаточно тонким, плоскопараллельным и имел прямые сквозные структурные каналы, параллельные прямому пучку.
Прн исследованиях методом тонких фолы чрезвычайно богатую информацию дает мик-роднфракция. Мнкродифракционная картина позволяет не только выбирать и правильно интерпретировать дифракционный контраст на соответствующем электронно-микроскопическом изображении, ио и проводить фазовый анализ, определять кристаллографические н кристаллогеометрические особенности наблюдаемых структур н т. п. Особенности микродифракцн-онных картин (МДК), получаемых в электронном микроскопе при просмотре тонких фолы:
1. МДК обычно точечная («монокристаллическая»), поскольку участок фольги, от которого она получена, содержит одни или несколько кристаллов одной н той же фазы или разных фаз.
2. МДК представляет собой правильную сетку рефлексов, соответствующую практически плоскому сеченню обратной решетки исследуемого кристалла (малой длине волны электронов X отвечает большой по сравнению с периодами обратной решетки радиус сферы отражения: jR=X-1); появлению множества рефлексов одновременно способствуют малая толщина фольги, наличие в ней несовершенств и локальных изгибов, сходимость падающего электронного пучка, многократное рассеяние электронов.
3. Интенсивность рефлексов иа МДК сравнима с интенсивностью прямо прошедшего пучка, поэтому в результате взаимодействия последнего с дифрагированными пучками по мере прохождения нх сквозь кристалл (многократного рассеяния) происходит усреднение интенсивностей рефлексов. Вследствие этого затрудняется определение структурного фактора по относительным интенсивностям отражений с использованием обычных формул кинематической теории.
4. На МДК благодаря более слабой, чем в
случае рассеяния рентгеновских лучей, зависимости рассеяния от атомного номера Z можно получать заметное рассеяние электронов на легких атомах в присутствии много более тяжелых (например, сверхструктурные рефлексы в упорядоченных растворах внедрения).
5. На МДК от достаточно толстых и совершенных кристаллов наблюдаются так называемые кикучи-лнинн, которые возникают в результате когерентного рассеяния электронов, первоначально иеупруго рассеянных образцом; каждое семейство кристаллографических плоскостей, примерно параллельных падающему электронному пучку, дает на МДК пару кнку-чн-лнний: светлую (на позитиве), расположенную вблизи брэгговского рефлекса от этого семейства плоскостей, и темную — вблизи следа первичного пучка. Поскольку кикучн-линии параллельны следам на МДК соответствующих плоскостей, с их помощью можно очень точно определять ориентировку кристалла, малые угловые разорнентировки субзерен и т. д.
6. Многократное рассеяние приводит к возникновению на МДК экстра-рефлексов, если луч, дифрагированный иа одном семействе плоскостей, может вторично дифрагировать иа другом семействе плоскостей (с иным меж-плоскостиым расстоянием) илн иа плоскостях того же семейства, но несколько иначе ориентированных; эксгра-рефлексы часто возникают на МДК от двойниковых и двухфазных структур, что усложняет анализ таких МДК, несколько создается ложное впечатление о периодичности, в действительности не существующей.
Последняя из перечисленных особенностей МДК позволяет получать на соответствующем электронно-микроскопическом изображении муаровый узор. Такой узор возникает в результате интерференции прямо прошедшего пучка и пучка, дважды дифрагированного: а) на семействах параллельных плоскостей (Л1М1) и и (Л2&2Ы с близкими межплоскостиыми расстояниями di и d2 в двух наложенных кристаллах (параллельный муаровый узор); б) иа семействах плоскостей (hkl) двух наложенных одинаковых кристаллов, взаимно слегка развернутых (муаровый узор вращения). В обоих случаях иа электроино-мнкроскопическом изображении возникает система регулярных полос с периодом D=did^\di—da] для параллельного муарового узора нли D^djz для муарового узора вращения, где в — угол взаимного поворота решеток в радианах.
Полосы муара параллельны отражающим плоскостям в случае параллельного муара и перпендикулярны им в случае муара вращения. Поскольку периоды муара обычно составляют величины порядка нескольких десятков н даже сотен ангстремов, для наблюдения муарового узора не требуется высокого разрешения.
Разнообразные картины муара возникают на электронно-микроскопических изображениях двухслойных пленок, полученных последовательной конденсацией [4], на изображениях тонких фольг двухфазных материалов с полностью нли частично когерентными выделениями нлн фольг, содержащих наклонно расположенные малоугловые дислокационные суб-граннцы и т. п. [6, 7].
2.3. СВЕДЕНИЯ, ПОЛУЧАЕМЫЕ ПРИ ИССЛЕДОВАНИИ МЕТАЛЛОВ
И СПЛАВОВ МЕТОДОМ
ТОНКИХ ФОЛЬГ
2.3.1. Микродифракционный фазовый анализ
Поскольку обычно, зиая химический состав и режим обработки исследуемого материала, можно заранее предположить возможность присутствия фаз с известными кристаллическими структурами, типичная задача фазового мнкродифракционного анализа в металловедении— экспериментальная проверка таких пред-положеиий, т. е. соответствия МДК какому-то сечению обратной решетки одной или нескольких из ожидаемых фаз.
Как уже отмечалось, МДК, как правило, получаются точечными, как от монокристаллов. Однако иногда, иапрнмер при хаотическом распределении большого числа мелких кристалликов в матричном кристалле, на МДК фиксируются сплошные или состоящие нз отдельных рефлексов кольца от этих кристалликов; расчет таких МДК аналогичен расчету рентгеновских дебаеграмм. Подобный расчет применяют также в том случае, когда несколько кристалликов одной и той же фазы дают на МДК лишь одно-два отражения, не образующих какого-либо сечения обратной решетки данной фазы.
Если кольца на МДК (сплошные илн состоящие из отдельных пятен) принадлежат кристалликам одной и той же фазы, то для идентификации последней необходимо:
1) точно измерить диаметры колец 2rt;
2) по формуле di^’kLjri вычислить соответствующие межплоскостиые расстояния (XL — постоянная прибора);
3) выявить среди вероятных в данном случае фаз такую, для которой межплоскостиые расстояния н их последовательность совпадают с экспериментально определенными.
О
Постоянная прибора XL (А-мм), определяющая масштаб дифракционной картины на МДК, колеблется от снимка к снимку, при .этом колебания составляют примерно несколько процентов (даже для МДК, последовательно получаемых от одного и того же участка при наклоне объекта). Поэтому необходимо проводить эталонирование каждой МДК, если требуется различить фазы с одинаковыми решетками и близкими периодами. Удобным «внутренним» эталоном является точечная дифракционная картина от матрицы с известной структурой.
Таким образом, путем достаточно точного расчета последовательного ряда экспериментальных значений di н сравнения нх с табличными значениями для каждой из предполагаемых фаз (см. раздел 6) можно идентифицировать фазу, от которой получена МДК-
Обычно для идентификации фазы по кольцевым электронограммам достаточно приблизительно оценить периоды ее решетки, а затем, вычислив отношения периодов, надежно определить кристаллическую симметрию ис-
следуемой фазы. Действительно, соотношение ri: r2: rs: - ' =gi: g2 :
: й'з • • Sr. (gi — модуль вектора обратной решетки, однозначно характеризует тип
структуры, поскольку величины записанных отношений gi (или йД1) зависят от значений периодов решетки и типа ее структуры.
Микродифракционный фазовый анализ существенно облегчается, если кристаллик нлн система одинаково ориентированных кристалликов анализируемой фазы (с известной структурой) дают на МДК правильную сетку рефлексов, отвечающую в масштабе XL какому-либо плоскому сечению обратной решетки. Тогда следует отобрать по темнопольным изображениям рефлексы, принадлежащие одному кристаллику или группе одинаково ориентированных кристалликов исследуемой фазы, измерить радиусы-векторы этих рефлексов н углы между ними и отыскать из числа предполагаемых в данном случае фаз ту обратную решетку, плоское сечение которой представляет собой МДК- Последняя стадия описанной процедуры практически состоит в сравнении экспериментальной электронограммы с различными сечениями ряда обратных решеток. Поэтому необходимо иметь такие сечения для всех возможных фаз или уметь быстро их строить аналитически пли с помощью стандартных стереографических проекций решеток этих фаз. Методы построения обратных решеток и их сечеинй подробно изложены в работах [6-8].
2.3.2. Определение ориентировки кристаллов, разориентировки зерен и субзерен
и ориентационных соотношений
Определение ориентировки кристалла сводится к конкретному инднцнрованню на МДК двух рефлексов, не принадлежащих одному и тому же систематическому (т. е. проходящему через центр МДК) ряду. Это нидицирование однозначно связывает осевые векторы решетки с реперными направлениями — падающим электронным пучком и кантом кадра МДК на фотопластинке.
Процедура определения ориентировки кристалла:
1) измеряют на МДК радиусы-векторы двух-трех рефлексов, ближайших к следу первичного пучка;
2) находят тип индексов этих рефлексов на основании заранее заготовленных сечений обратной решетки илн табличных значений межплоскостных расстояний;
3) по взаимному расположению рефлексов с учетом углов между ними принимают какой-либо одни нз возможных вариантов конкретного нндицироваиия рефлексов;
4) находят индексы осн зоны [ниш] (общего направления для всех отражающих плоскостей, дающих данную дифракционную картину), антипараллельной пучку, как векторное произведение двух любых конкретно проиндн-цированных векторов отражения (илн векторов обратной решетки) [gi • g2], причем вектор g2 должен быть повернут относительно
54
вектора gi против часовой стрелки, если смотреть на фотопластинку со стороны эмульсии.
Установленные таким путем индексы оси зоны неоднозначно определяют ориентировку кристалла в пространстве, поскольку из-за центросимметричностн МДК относительно следа первичного пучка (узла ООО) возможен другой вариант правильного индицирования рефлексов, при котором индексы оси зоны получатся [zway] вместо [zww], что означает поворот решетки кристалла (и осевых направлений) на 180° вокруг направления оси зоны. Поэтому для однозначного (и правильного) определения ориентировки кристалла необходимо использовать дополнительные данные: узор кикучи-линнй, отдельные рефлексы на МДК, не принадлежащие данной сетке рефлексов, или получение еще одной МДК при наклоне образца на известный угол относительно известной осн (осуществляется с помощью гоинометра [7]). Такие данные ие требуются только в том случае, если ось зоны является осью симметрии четного порядка решетки кристалла.
Описанная процедура нахождения ориентировки кристалла универсальна; она пригодна и для работы с кристаллами гексагональной сингонии, если для обозначения направлений н плоскостей использовать системы с тремя индексами.
Особенности формирования МДК в электронном микроскопе прн работе на просвет (вытянутость узлов обратной решетки в направлении, перпендикулярном плоскости фольги, эффективное увеличение угла сходимости неот-клонениого пучка нз-за многократного рассеяния электронов по мере прохождения их сквозь фольгу) приводят к тому, что одна и та же (по геометрии, но ие по интенсивностям) точечная МДК сохраняется в интервале углов наклона образца ±5° и более (для очень тонких фольг). Поэтому ориентировку кристалла рассмотренным иыше способом можно определить именно с такой точностью. Эта точность во многих практических случаях недостаточна, и для ее повышения применяют ряд приемов, с помощью которых устанавливают величину углового отклонения направления пучка электронов от оси зоны, соответствующей фиксируемой иа МДК сетке рефлексов [7].
При точном определении ориентировки соседних зерен илн субзереи можно найти их угловую разориеитнровку. В случае зерен угловая разориеитировка велика и соседним зернам отвечают различные дифракционные картины. После однозначного и возможно более точного (с помощью кикучн-линий или по наличию сильных рефлексов [7]) установления ориентировок полученные иаправлення помещают в центр одной н той же стереографической проекции. Затем находят полюсы осевых направлений решеток обоих зерен и вектор разорнеитировки, определяющий ось наклона и величину угла, на который необходимо повернуть решетку одного нз зерен до совмещения ее с решеткой второго зерна. Возможно и аналитическое решение этой задачи. Существуют программы для ЭВМ, позволяющие быстро и с заданной точностью иайти вектор разориен-
тнровки, исходя из тех же исходных дифракционных данных. По величине этого вектора можно сделать вывод о типе р азориентировки зерен (специальная, произвольная) и о типе нх границ. Это имеет важное значение прн изучении выделения второй фазы по границам зерен, межзеренного разрушения и т. д.
Субзерна разориентироваиы относительно друг друга иа небольшие углы (<10°) и поэтому на МДК, полученной от двух соседних субзерен, обычно присутствует одна сетка рефлексов, причем они могут быть «размазаны» в азимутальном направлении, т. е. по концентрическим дугам относительно следа первичного пучка. Поэтому азимутальную составляющую разориентировки двух субзерен Да можно легко определить путем измерения иа МДК угла между однотипными рефлексами (если субзериа дают раздельные рефлексы) с помощью транспортира или по формуле Аа= =Да/[г/1лг|, где Да — расстояние между рефлексами; гм( — раднус-вектор рефлекса, мм.
Горизонтальную составляющую разориенти-ровкн (относительно оси, лежащей в плоскости МДК) можно определить путем аиалнза различия в узоре кикучи-линий от каждого нз субзерен или применяя специальную темно-польиую методику (подробнее см. в работе [7]).
Точное определение кристаллографической ориентировки фольги при ее исследовании в электронном микроскопе позволяет устанавливать ориентационные соотношения кристаллов разных фаз. Если ориентационное соотношенне между фазами известно, то задача сводится к нахождению конкретного варианта этого соотношения (такие задачи приходится решать, например, при исследовании крнсталлострук-турных особенностей мартенситных превращений). Для решения указанной задачи достаточно установить точную ориентировку одной пары соседствующих кристаллов обеих фаз (в простейшем случае получить одну МДК с двумя сетками рефлексов от обеих фаз. После этого отыскивают пары взаимно параллельных плоскостей и лежащих в них направлений, входящих в ориентационное соотношение и описывающих найденную взаимную ориентировку кристаллов. Для этого можно использовать стандартные стереографические проекции, отвечающие данному ориентационному соотношению, илн матрицы размерного и структурного соответствия, вычисленные для различных конкретных вариантов этого соотношения [7].
Если же орнеитациоиное соотношение между фазами неизвестно, то устанавливают точную взаимную ориентировку трех-четырех пар кристаллов исследуемых фаз. Этн ориентировки наносят на стереографическую проекцию и отыскивают однотипные взаимно параллельные плоскости н направления решеток обеих фаз по возможности с малыми индексами. Если такие плоскости и направления иайтн не удается, то, следовательно, ориентационной связи между решетками фаз иет. На рнс. 2.3 приведена структура отпущенной стали, в которой выделение цементита из мартенсита произошло в соответствии с ориентационным соотношением Исайчева (Багаряц-кого).
Рис. 2.3. Сталь 50Н29. Закалка с 1150 ”С в щелочи + охлаждение в жидком азоте + отпуск при 250 СС, 2 ч:
а — темнопольная электронная микрофотография участка полностью двойникового мартенситного кристалла в пучке матрицы 101^. Х60 000; б—темнопольное изображение в пучке Fe3C 2Н^. Х60 000; в — электронограмма отв, ориентировка [131]ки ИЧ]Ц
2.3.3. Структурные особенности фазовых превращений
1. Мартенситные превращения. При исследовании мартенситных превращений можно получить детальные крнсталлоструктуриые данные об отдельных мартенситных кристаллах и их ансамблях, а именно: о морфологии кристаллов, их габитусе, ориентационных соотношениях с исходной фазой, внутренней структуре кристаллов (двойники, дефекты упаковки, дислокации), строении межфазной поверхности, направлении и величине деформации формы прн превращении и др. Для получения многих нз перечисленных данных необходима точная кристаллографическая привязка изучаемых плоских или линейных дефектов структуры, т. е. нахождение Миллеровских индексов этих дефектов путем сопоставления микроизобра-жений с соответствующей МДК. Существует ряд способов точной кристаллографической привязки различных элементов структуры; все они связаны с пространственными измерениями исследуемого участка объекта на электронных микрофотографиях, полученных прн различных положениях его (наклонах с помощью гониометра) относительно первичного электронного пучка. Подобные стереомнкроскопн-ческие измерения, проводимые на мнкроизо-бражениях анализируемой структуры, подробно изложены в работе [7].
Некоторые из отмеченных выше данных можно получить н прн исследовании нормальных полиморфных превращений (происходящих, например, в сплавах железа). Типичный пример структуры мартенсита приведен иа рнс. 2.4.
2. Распад пересыщенных твердых растворов. При исследовании стареющих сплавов можно установить следующие структурные особенно
сти: морфологию, размеры и распределение частиц второй фазы, ее кристаллическую структуру н ориентационные соотношения с матрицей на разных стадиях распада, структуру межфазной поверхности и др. Особое значение имеет анализ дифракционного контрас-
Рис. 2.4. Структура мартенсита закаленной быстрорежущей стали типа Р6М5. X10 000 (В. А. Казаковце-ва)
та, который возникает на изображениях структуры двухфазного сплава и обусловлен либо наличием искажений в матрице вблизи выделения, либо изменением дифракционных условий при переходе от решетки матрицы к решетке второй фазы. Изучение контраста на выделениях позволяет получить данные о ха-
56
Рис. 2.5, Типичные структуры стареющих сплавов с полностью или частично когерентными выделениями второй фазы:
а — сплав XH77T2IO (типа «нимоник»), старение при 800 °C, 10 ч; видны равноосные когерентные частицы 'Р'-фазы Nb(Ti, Al), вызывающие появление деформационного контраста в матрице вокруг них за счет центросимметричных полей искажений, X100 000; б — сплав Fe—0,07 % N, старение при 100 °C, 1 ч; деформационный контраст на изображении пластинчатых
Рис. 2.6. Сплав Fe — 25 % (ат.) А1 в упорядоченном состоянии (сверхструктура DO3). Темнопольное изображение в сверхструктурном рефлексе 100 0 ц видим антифазные домены. Х75 000 (А. М. Глезер)
рактере сопряжения решеток выделяющейся фазы и матрицы (когерентное, полу когерентное, некогереитиое). Классификация контраста от гетерофазных структур приведена в работах [6, 7].
Весьма плодотворным оказалось применение темиопольной методики в сочетании с анализом диффузного рассеяния, возникающего иа МДК (см. далее, раздел 2.3.4), а также ме то да прямого разрешения решетки для нссле доваиия начальных («зонных») стадий распада. Рассмотренными методами были получены прямые данные о размерах и распределении в матричной решетке кластеров растворенных атомов вплоть до прямого «видения» последних. Примеры электроиио-микроскопи-ческнх изображений структур стареющих сплавов приведены иа рнс. 2.5.
3. Упорядочение. Прн исследовании упорядоченных сплавов можно получить информацию о структурном типе упорядочения (в том числе в разбавленных твердых растворах внедрения), о взаимном расположении упорядоченных и неупорядоченных областей, их форме, особенностях дефектов решетки, доменной структуре упорядочения и др. Специфический дифракционный контраст на изображениях упорядоченных структур связан с возникновением сверхструктурных отражений и с наличием в структуре антифазных доменов, разделенных антифазными границами (АФГ). Поскольку интенсивность сверхструктуриых рефлексов пропорциональна степени дальнего порядка, по контрасту на темнопольных изображениях в сверхструктурных отражениях пря определенных условиях можно судить о степени упорядочения. Наличие контраста на
когерентных выделений нитрида Fe<N, расположенных параллельно падающему электронному пучку й вызывающих смещения в матрице, перпендикулярные плоскости пластинки. Х60 000; в — сплав ЖС6КП. старение 950 °C, 500 ч + испытания на ползучесть при 950° в течение 500 ч (о=100 МПа); видны ряды краевых эпитаксиальных дислокаций на межфазных поверхностях {100} крупных стержневых частиц -/-фазы Nis(Ti, Al). X 50 000
57
АФГ и его исчезновение в зависимости от условий дифракции позволяют определить вектор смещения на АФГ н тем самым установить их природу — термическую или сдвиговую, Детальный обзор результатов электронно-микроскопических исследований упорядоченных сплавов приведен в работе [8]. На рис, 2.6 представлена типичная электронная микрофотография структуры упорядоченного сплава с антифазными доменами.
2.3.4. Диффузное рассеяние электронов
На МДК от монокристаллов часто наблюдаются эффекты диффузного (ие брэгговского, но когерентного) рассеяния электронов.
I. Тепловое диффузное рассеяние. Это рассеяние связано с тепловыми колебаниями атомов (ионов), составляющих кристаллическую решетку; анализ эффектов теплового диффузионного рассеяния в пространстве обратной решетки позволяет получить данные об атомных конфигурациях, возникающих в кристаллах при тепловых колебаниях. Такими конфигурациями являются плотноупакованиые цепочки атомов (<110> в г. ц, к. решетке, < 111 > в о. ц. к. решетке).
2. Диффузное рассеяние, связанное с размерами кристалла. Малые размеры кристалла, облучаемого электронами, вызывают размытие всех рефлексов, включая узел ООО, в направлении этого малого размера. Если кристалл представляет собой тонкую пластинку, то рефлексы вытягиваются в направлении, перпендикулярном плоскости пластинки, образуя «обратные стержни». Для пластинок, расположенных параллельно первичному электронному пучку, характерно возникновение на МДК тяжей, длина которых в одну сторону от рефлекса равна /-1, где t—толщина пластинки. Для пластинок, расположенных наклонно к первичному электронному пучку, характерно некоторое смещение рефлексов из своих «нормальных» положений, причем направление и величина смещения зависят от точности выполнения дифракционных условий для этих рефлексов, толщины пластинки и угла наклона ее по отношению к первичному пучку. Большая протяженность обратных стержней (малая толщина пластинок) может привести к появлению на МДК экстра-рефлексов, которые соответствуют точкам «прокола» сферы отражения стержнями от узлов обратной решетки, лежащих вне выявляемого на данной МДК ее плоского сечення. В случае кристалла игольчатой формы в обратном пространстве возникает пластина интенсивности, перпендикулярная оси иглы; на МДК от такого кристалла обычно наблюдаются тяжн, являющиеся следами плоскости обратной пластины на плоскости МДК.
3. Эффекты ближнего порядка. Твердые растворы часто проявляют тенденцию к упорядочению или расслоению. При тенденции к упорядочению возникает диффузное рассеяние, максимумы интенсивности которого близки к положениям сверхструктуриых рефлексов для полностью упорядоченного сплава При тенденции к расслоению диффузное рассеянне возникает вблизи основных рефлексов (включая узел ООО), причем оно симметрично распределено в обратном пространстве, т. е. подобно
дифракционным эффектам, связанным с размерами и формой кристалла. Анализ диффузного рассеяния, обусловленного наличием ближнего порядка (расслоения), состоит в построении нзодиффузных поверхностей (поверхностей равной интенсивности диффузного рассеяния) в обратном пространстве кристалла и в последующем сопоставлении экспериментального рассеяния с теоретически рассчитанным для различных конфигураций атомов разного сорта. Это позволяет выбрать правильную атомную модель структуры твердого раствора с ближним порядком. Разновидностью ближнего порядка в твердых растворах является так называемый ближний порядок смещений, возникающий в результате образования в исходной решетке атомных конфигураций, симметрия которых отличается от симметрии решетки матрицы Наличие ближнего порядка смещения приводит к появлению диффузного рассеяния вблизи рефлексов и между ними. Анализ подобного диффузного рассеяния, возникающего вблизи точек фазовых переходов, с помощью метода волн статических смещений дал возможность установить типы атомных конфигураций и пути их получения в некоторых твердых растворах (в о. ц. к. растворах иа основе Ti и Zr вблизи о. ц. к,—и о. ц. к.~-> ->-г. п. у. превращений [9], в углеродистом мартенсите на стадии образования когерентного е-карбида и др.).
4. Эффекты, связанные с выделениями второй фазы. Диффузное рассеяние, возникающее па начальных стадиях распада пересыщенных
Рис. 2.7. Стели У14. Закалка с 1000 °C в воде Элект-ронограмма от участка све?к со бра во в а иного кристалла мартенсита; ориентировка [100]
твердых растворов, аналогично диффузному рассеянию для ближнего расслоения (которое и является самой ранней стадией распада). Для анализа его применимы методы, указанные в п. 3. Прн когерентном выделении второй фазы из твердого раствора около матричных рефлексов возникает диффузное рассеяние, топология которого зависит от индексов рефлексов и упругих свойств решетки матрицы. Отличительная особенность этого рассеяния— отсутствие размытия узла 000. Установив форму размытых узлов в пространстве
58
обратной решетки, можно опредстять таи искажения исходной атомной решетки (сдвиговой, дилатационный) при выделении второй фазы и найти компоненты тензора упругой деформации, необходимой для перестройки решетки матрицы в решетку фазы выделения. На МДК от модулированных структур с той илн иной степенью корреляции в расположении частиц второй фазы вблизи основных рефлексов возникают (в соответствующих направлениях) пятна-сателлнты. По величине
О
расстояния (d, А;) от основного рефлекса до пятна-сателлита можно определять длину о
волны (к, А) модулированной структуры в направлении от основного рефлекса на сателлит: A=d-1.
На рис. 2.7 в качестве примера приведена МДК от свежеобразованного кристалла мартенсита высокоуглеродистой стали с характерным диффузным рассеянием, возникающим в результате ближнего расслоения по углероду.
2.3.5. Изучение дислокационной структуры
Можно выделить два основных направления электронно-микроскопических исследований дне локационной структуры металлов и сплавов.
1. Изучение свойств и поведения отдельных дислокаций. Анализ контраста на изображении дислокации в разных дифракционных условиях^ позволяет определить ее вектор Бюргерса (fe). Для этого используют правило, согласно кютороыу_дисяокацпя становится невидимой, если ее b лежит в отражающих плоскостях, формирующих единственный _снльный рефлекс (§) на МДК (критерий g-b=Q). Зная b и кристаллографическую привязку дислокационной линии, видимой на изображении, можно установить тип дислокации (краевая, винтовая или смешанная). Существуют приемы, с помощью которых можно определять тип призматических петель (вакаксиоякые петли или петли нз внедренных атомов). Применение теынопольной методики в слабом пучке (формируемом плоскостями, находящимися далеко от отражающего положения), а также метода прямого разрешения дает возможность видеть расщепление полной дислокации на частичные, даже когда оно составляет лишь несколько десятков ангстремов и менее, н вычислять энергию дефекта упаковки. Наиболее полная информация об особенностях формирования диз токационной структуры и поведения отдельных дислокаций при пластической деформации может быть получена при исследовании монокристаллов, когда имеется возможность рассчитать величину напряжения для любой системы скольжения н сопоставить ее с возникающей структурой. Удобным методическим приемом в этом случае является получение ориентированно вырезанных фольг (параллельно плоскости скольжения, перпендикулярно вектору Бюргерса дислокаций первичной системы скольжения и т. д.), на которых можно изучать те илн иные детали образующейся при деформации дислокационной структуры. Метод топких фолы дает также возможность исследовать взаимодействие между дислокациями и возникающие при этом дне-
Рис. 2.3. Примеры дислокационной структуры сплавов. возникающей при пластической деформации:
а — сплав Fe — 0.3 % Ti. деформация растяжением. й—В%; ячеистая дислокационная структура. X-10 000 (Л. Г. Орлов); б —сплав Х20Н80 (нихром), деформация растяжением, 6=2 %; плоские скопления дислокации вблизи границы зерен. ХЗО ООО; в — сплав ХН78ТЮ. деформация 2 % зя 1 ч при 700 °C (с= -40 МПа); неправильные геликоидальные дислокации и пространственно искривленные дислокации, свиде-тельс.вующне о протекании при ползучести переползания дислокаций. Х35000
локационные конфигурации, взаимодействие с точечными дефектами, выделениями второй фазы, границами зерен и субзерен и т. д. Важное значение имеет изучение роли дислокаций прн различных фазовых превращениях,
2. Изучение дислокационных структур. Исследуют структуры, возникающие в металлах и сплавах прн холодной и горячей пластической деформации, в том числе прн термомеханической обработке, ползучести, полигонизации и рекристаллизации, прн облучении быстрыми частицами и др. Можио определять плотность дислокаций (в интервале от 107 до 10“ см-2), изучать особенности формирования дислокационных структур в сплавах с различной энергией дефектов упаковки в зависимости от температуры и скорости деформации и уровня приложенного напряжения (характер распределении дислокаций в материале, образование дислокационных конфигураций и пр,). Существуют специальные приемы исследования сложных дислокационных структур (с плотностью дислокаций >10и см-2), возникающих при сильной пластической деформации нли в результате мартенситного превращения [7].
Подробное описание методики исследования дислокационных структур анализа контраста иа дислокациях дано в монографиях [6,7,10]. Типичные примеры изображения дислокационной структуры приведены иа рис. 2.8
2.3.6. Прямое изучение процессов, происходящих в тонкой фольге
Важное достоинство метода — возможность прямого наблюдения протекания ряда процессов, происходящих прн нагреве, охлаждении илн деформации тонкой фольги в колонне электронного микроскопа.
1. Опыты с нагревом образца. Многие электронные микроскопы снабжены приставкой для иагрева образца в колонне микроскопа. Такая приставка представляет собой встроенный в столик объекта электронагреватель н позволяет просматривать объекты в процессе нагрева нлн при постоянной температуре вплоть до —1200 °C (прн этом необходимо принимать меры предотвращения окисления объекта, особенно прн высоких температурах нагрева). Приставку для нагрева можно использовать прн исследовании динамики процессов выделения и растворения второй фазы, упорядочения и разупорядочения, полигонизации и др, Одиако следует иметь и виду, что кинетика (но не механизм) этих процессов, протекающих в тонкой фольге, может отличаться от таковой в массивных образцах, так что подобные исследования целесообразно проводить, используя высоковольтные микроскопы, позволяющие просматривать более толстые образцы. Быстрый локальный нагрев образца легко осуществить электронным пучком; для этого достаточно убрать подвижную конден-сорную диафрагму илн изменить режим работы первой коиденсориой линзы (увеличить диаметр пучка на объекте). Таким путем можно разогреть небольшой участок, примерно равный по площади первичному пучку на объекте,
до разных температур — вплоть до 500— 600 °C, прн этом достигаемая температура в течение некоторого отрезка времени пропорциональна продолжительности облучения широким электронным пучком. С помощью этого простого метода нагрева были проведены исследования процессов упорядочения в твердых растворах внедрения (Та—О), бейннтного распада остаточного аустенита закаленной высокоуглеродистой стали, обратного мартенситного превращения в сплавах Fe—N и Fe—Al—С.
2. Опыты с охлаждением образца. Некоторые электронные микроскопы снабжены приставкой, позволяющей охлаждать исследуемый образец в колонне микроскопа до температуры жидкого азота. Основная часть такой приставки— сосуд Дьюара, заливаемый жидким азотом, и связанный с ним хладопровод (обычно медный стержень), находящийся в контакте со столиком объекта, теплоизолированным от металлических частей колонны микроскопа. Температуру объекта измеряют с помощью термопары; небольшая нагревательная печь, встроенная в столик, служит для получения температур, промех<уточных между температурой жидкого азота и комнатной. Последние модели приставок для охлаждения позволяют производить и наклон образца.
Так можно наблюдать различные процессы, протекающие прн низких температурах, в частности мартенситные превращения, С помощью приставки для охлаждения были проведены исследования предмартенситного состояния исходной фазы вблизи мартенситной точки (в сплавах Ni—Ti, никелевых сталях н др.), кинетики упорядочения и ближнего расслоения в мартенсите азотистой и углеродистой сталей соответственно.
3. Опыты с деформацией образца. Приставки для деформации в колонне микроскопа служат для прямого изучения процессов пластической деформации и разрушения, происходящих в тонкой фольге. Поскольку эти процессы в тонкой фольге протекают иначе, чем в массивных образцах, для подобных исследований также целесообразно использовать высоковольтные микроскопы. В деформируемом металле (деформация достигалась с помощью приставок, а иногда благодаря возникновению напряжений при облучении тонкой фольги электронным пучком) удалось наблюдать зарождение дислокаций на различных структурных несовершенствах (границах зерен, двойников, межфазных поверхностях, полигональных стенках и др.), движение дислокаций, расщепление полных дислокаций иа частичные с образованием дефектов упаковки, коалесценцию и разрушение полигональных стеиок и сеток, зарождение и распространение трещин н многое другое. Существует приставка к высоковольтному микроскопу, позволяющая деформировать образец в микроскопе в широком интервале температур с записью диаграммы растяжения. Деформация монокристаллических образцов с помощью такой приставки дает возможность установить температурную зависимость скорости движения дислокаций (винтовых и краевых) прн разных уровнях приложенного напряжения.
2.4. ТЕНДЕНЦИИ
И ПЕРСПЕКТИВЫ РАЗВИТИЯ МЕТОДА
Отметим основные тенденции и перспективы развития метода дифракционной микроскопии.
Высоковольтная электронная микроскопия. Создание высоковольтных микроскопов (1000 кВ и более) дало следующие преимущества, связанные с повышением ускоряющего напряжения:
а) увеличение проникающей способности электронов, что позволило просвечивать более толстые фольги;
б) уменьшение размеров участка на образце, от которого можно получить мнкроэлектроио-грамму;
в) получение более богатых рефлексами дифракционных картин и повышение яркости изображения;
г) возможность получения электронограмм от более грубых включений;
д) возможность получения высокоразрешающих многолучевых изображений прн использовании апертурных диафрагм большого диаметра (методом прямого разрушения);
е) приближение дифракционной картины к плоскому сечению обратной решетки благодаря уменьшению кривизны сферы отражения (за счет уменьшения длины волны электронов).
Увеличение толщины просвечиваемой фольги позволяет уменьшить влияние внешних ее поверхностей на внутреннюю структуру, поэтому последняя точнее отвечает структуре массивного образца; кроме того, более надежно можно относить результаты исследования динамики различных процессов прн прямом наблюдении в микроскопе (деформации, фазовых превращений) к массивным образцам. С увеличением толщины фольги также значительно повышается точность пространственных измерений, необходимых для кристаллографического анализа. Возможность получения дифракционной картины от отдельной, даже очень крупной, частицы н уменьшение размеров участка, от которого можио получить электронограмму, существенно облегчает проведение фазового анализа сплавов.
Дополнительное преимущество высоковольтной электронной микроскопии состоит в возможности изучения радиационных повреждений, возникающих при бомбардировке исследуемого объекта электронами, обладающими энергией, превышающей пороговую.
Высоковольтные микроскопы благодаря своим достоинствам дол йены найти широкое применение в практике металловедения, несмотря иа громоздкость, сложность обслуживания н высокую стоимость.
Метод прямого разрешения. Улучшение разрешающей способности электронных микроскопов в сочетании с увеличением нх ускоряющего напряжения обусловливает возможность широкого применения метода прямого разрешения для исследования кристаллических, в том числе металлических объектов. Электронно-микроскопические изображения, полученные этим методом, дают наиболее наглядное представление (в пределе — иа атомном уровне) о структуре реального объекта. Несомненно, что использование метода прямого разрешения позволит выяснить детали атомного строения
границ зерен, межфазных поверхностей, различного рода структурных несовершенств кристаллической решетки и др. Принципиальное значение имеет применение этого метода для установления атомного механизма фазовых превращений (полиморфных, мартенситных, упорядочения, старения и т. д.) в металлических системах, особенно, если проводить прямые наблюдения в микроскопе, используя соответствующие приставки.
Развитие аналитических методов в электронной микроскопии. Современный электронный микроскоп все более становится аналитическим прибором благодаря разработке и применению различных приставок н прежде всего приставок для локального химического анализа. Наиболее распространена приставка для анализа характеристического спектра рентгеновских лучей, возникающих прн взаимодействии быстрых электронов с исследуемым образцом. Трудности количественного определения содержания того или ниого элемента связаны с необходимостью эталонирования экспериментальных спектров (для эталонирования необходимо точно знать толщину фольги, объемную долю исследуемой фазы и т. д.). В приборах новейших конструкций локальность определения химического состава, ограниченная размерами падающего иа образец электронного пучка, достигает десятков ангстремов. Поэтому весьма перспективны растровые (сканирующие) электронные микроскопы просвечивающего типа, снабженные такой приставкой; наличие интенсивного электронного зоида малого диаметра (до —-10 А) позволяет с большой локальностью определять химический состав исследуемого участка объекта, в том числе, и содержание легких элементов.
Другой весьма перспективный метод локального химического анализа — анализ спектра энергетических потерь электронов, прошедших через исследуемый объект. Спектр потерь также характеристический для каждого элемента перноД1гческой системы. Анализатором служит электростатическая лниза, помещаемая между проекционной линзой и конечным экраном; степень отклонения электронов этой линзой зависит от нх скорости (энергии), что и позволяет с помощью специальных электронных схем получать спектры энергетических потерь. Описываемый метод очень чувствителен и имеет локальность, соответствующую максимально полезному увеличению микроскопа. Кроме того, этим методом легче, чем рентгеиоспектраль-ным, проводить анализ иа легкие элементы.
Таким образом, современным н перспективным можио считать высоковольтный электронный микроскоп с ускоряющим напряжением 1000 кВ и более и с разрешением порядка 1—2 А, снабженный электронно-оптической системой для сканирования электронного пучка н приставками для исследования динамики различных процессов в образце и одновременного высоколокального и высокочувствительного химического анализа.
БИБЛИОГРАФИЧЕСКИЙ СПИСОК
I. 2/тевский Л. М. — Вкн.: Металловедение и термическая обработка стали. Т. 1: Справочник. М: Металлургиздат, 1961, с. 165—175.
61
2. Лаборатория металлографии. 2-е изд./ {Панченко Е. В., Скоков Ю. А. Кример Б. И. и др. М.: Металлургия, 1965. 439 с.
3. Утевский Л. М.—Заводская лаборатория, 1952, № 6, с. 697.
4. Структура межкристаллитных н межфазных граииц/досевпч В. М., Иевлев В. М., Палатник Л. С., Федоренко А. И. М.: Металлургия, 1980. 256 с.
5. Томас Г. Электронная микроскопия металлов. М.: ИЛ, 1963. 351 с.
6. Электронная микроскопия тонких кристаллов: Пер. с англ./ Хирш П., Хови А., Никльсон Р. и др. М.: Мир, 1968. 574 с.
7. Утевский Л. М. Дифракционная электронная микроскопия в металловедении. М.: Металлургия ,1973. 583 с.
8. Скоков Ю. А., Глезер А. М.—Итоги науки и техники. Металловедение и терми-мическая обработка. Выл. 9. М.:
ВИНИТИ, 1975, с. 5—72.
9. Тяпкин. Ю. Д.—Итоги науки и техники. Металловедение и термическая обработка. Вып. 11. М.: ВИНИТИ, 1977, с. 152— 213.
10. Амелинкс С. Методы прямого наблюдения дислокаций. М.: Мир, 1968. 440 с.
3
.^=-= РАСТРОВАЯ ЭЛЕКТРОННАЯ МИКРОСКОПИЯ
3.1. РАСТРОВЫЙ
ЭЛЕКТРОННЫЙ
МИКРОСКОП
Растровый электронный микроскоп (РЭМ)1 формирует изображение объекта при сканировании его поверхности электронным зондом. Это -один из наиболее универсальных и перспективных приборов для исследования микроструктурных характеристик металлов. По темпам развития и количеству моделей РЭМ опережает просвечивающие электронные микроскопы (ПЭМ). Число микрофотографий, выполненных -на РЭМ, приблизилось к числу микрофотографий, полученных с помощью светового микроскопа (СМ), и значительно превосходит ПЭМ. По растровой электронной микроскопии имеется несколько монографий [1—5] н обзоров {6—9], ежегодно издаются труды конференций н семинаров [10—12].
3.1.1. Принцип работы
В РЭМ поверхность исследуемого массивного образца облучается стабильным во времени тонко сфокусированным (диаметр до 5—10 нм) электронным зондом, совершающим возвратно-поступательное движение по линии или развертывающимся в растр (совокупность близко расположенных параллельных линий, вдоль некоторых зонд обегает выбранный участок иа поверхности образца). При взаимодействии .зонда с веществом образца в каждой точке поверхности происходит ряд эффектов, которые регистрируются соответствующими датчиками (рис. 3.1).
Эти эффекты служат основой для получения разнообразной информации о внутреннем строении исследуемых объектов. Сигналы от датчиков после усиления модулируют локальную .яркость электронно-лучевой трубки (ЭЛТ), развертка которой синхронна со смещением электронного пучка. При этом каждой точке на поверхности образца соответствует определенная точка на экране ЭЛТ, а ее яркость определяется интенсивностью сигнала из соответствую
1 В соответствии с ГОСТ 21006—75 применение термина «сканирующий электронный микроскоп», являющегося синонимом РЭМ, не допускается.
щей точки образца. Контраст на экране ЭЛТ зависит от изменения интенсивности излучения по площади сканирования на поверхности образца. Увеличение РЭМ определяется соотношением амплитуд развертки луча по экрану ЭЛТ н зоида по поверхности образца.
Рис 3.1. Эффекты, возникающие при взаимодействии электронного пучка с веществом и используемые для формирования изображения в РЭМ:
1 — электронный пучок; 2 — образец; 3 — отраженные электроны; 4—вторичные электроны; 5 — ток поглощенных электронов; €—катодолюминесценция; 7 — рентгеновское излучение; 8 — Оже-электроны; 9 — наведенный ток,- 10 — прошедшие электроны
Вторичные, отраженные н Оже-электроны, а также рентгеновское излучение генерируются в определенных объемах внутри образца и несут разнообразную информацию о рельефе, химическом составе и кристаллографической ориентации объемов, прилегающих к поверхности (рнс. 3.2). Зона выхода вторичных электронов, с помощью которых обычно формируется изображение и РЭМ, ограничена малой областью вокруг места падения зоида. Это позволяет достигать довольно высокой разрешающей способности (указывается в технических характеристиках приборов, табл. 3.1). Области, в которых генерируются отраженные электроны,
62
ТАБЛИЦА 3.i
КЛАССИФИКАЦИЯ РАСТРОВЫХ ЭЛЕКТРОННЫХ МИКРОСКОПОВ
Тип РЭМ Разрешающая способность, нм Ускоряющее напряжение, кВ дополнительные приставки и устройства Рентгеновские спектрометры, Оже-спектрометры Типичные представители
Упрощенные н ми-нн-РЭМ 20—50 I—15 (5-50) Отсутствуют Отсутствуют РЭМ-200, РЭМН-2 (СССР); JSM-15, MSM-5 (Япония)
Универсальные 7—12 1—60 Имеются Имеются РЭМ-100-75, РЭМ-1, Зонд-6 (СССР); JSM-50A, JSM-35 (Япония); Stereoscan 180, 150 (Англия)
РЭМ с автоэмнс-сионной пушкой 4—9 0,2—20 Вакуум 10~6— —10-8 Па Cwikscan (Австрия)
Специализированные Зависит от специализации 1—50 Не обязательны » Camebax (Франция); JCXA-50A (Япония); Biosem (США)
Просвечивающие 0,1—0,3 (1,5-7) 100—200 То же » ST-100 (ФРГ); НВ-5 (Англия); JSEM-200, JEM-100C/AS1D (Япония)
рентгеновское н катодолюмннесцентное излучение, намного больше объема, облучаемого электронным зондом. Поэтому и разрешающая способность при работе РЭМ в соответствующих режимах меньше, чем во вторичных электронах.
разрешающая спс-сооность в отваженных электронах I
Пространственная разрешающая способность В рентгеновском излучении
Рис. 3.2. Области генерации и пространственное разрешение в отраженных, вторичных и Оже-электронах, а также в рентгеновском излучении, образующихся в РЭМ:
/-Оже-электроны; 2 — вторичные электроны; 3— отраженные электроны; 4—характеристическое рентгеновское излучение; 5 — непрерывное рентгеновское излучение; 6 — вторичная флуоресценция за счет рентгеновского излучения
3.1.2. Классификация РЭМ
Классификация РЭМ возможна по нескольким признакам. В зависимости от разрешающей о способности различают РЭМ 1 класса (<150А), II класса (200—500А), III класса (>500А). По ускоряющему напряжению РЭМ подразделяют на приборы с напряжением <5 кВ, 5— 50 кВ (самые распространенные) и >50 кВ (используются для исследования образцов на просвет в просвечивающих растровых электронных микроскопах—ПРЭМ). В зависимости от количества дополнительных приспособлений микроскопы делятся иа универсальные, к которым выпускаются многочисленные приспособления, и специализированные. К последним относятся также РЭМ, предназначенные для контроля в производственных условиях. Возможна классификация РЭМ по типу катода электронной пушки: с термокатодом из вольфрамовой проволоки, с катодом косвенного нагрева нз гексаборнда лантана и с автоэмисси-онным катодом. Основной является классификация по разрешающей способности.
3.1.3. Место РЭМ в микроскопии
РЭМ занимает промежуточное положение между световыми микроскопами (СМ) и просвечивающими электронными микроскопами (ПЭМ). В табл, 3.2 и 3.3 приведена сравнительная характеристика этих микроскопов по разрешающей способности, глубине фокуса и другим показателям. Не следует рассматривать СМ, <РЭМ и ПЭМ как конкурирующие приборы. Скорее они дополняют друг друга н наиболее перспективно комплексное их использование в металловедческих исследованиях.
Можно указать следующие преимущества РЭМ, определяющие бурное развитие растровой электронной микроскопии:
— высокая разрешающая способность;
— большая глубина фокуса в сочетании с
63
ТАБЛИЦА 3.2 РАЗРЕШАЮЩАЯ СПОСОБНОСТЬ И ГЛУБИНА ФОКУСА МИКРОСКОПОВ РАЗНЫХ ТИПОВ
Тип микроскопа | Увеличение Разрешающая способ- ность Поле зрения Глубина фокуса
СМ РЭМ
СМ 1 0,2 мм 100 мм -
СМ, РЭМ* 10 0,02 » 10 » 0,1 мм 10 мм
СМ Л РЭМ* 102 2 мкм 1 » 1 мкм 1 »
СМ, РЭМ, 108 0,2 » 0,1 » — 10 мкм
ПЭМ
РЭМ*, ПЭМ 104 200 А 10 мкм — 1 »
РЭМ*, ПЭМ 105 20 » 1 » — —
ПЭМ 10й 2 » 0,1 » — —
* Сравнительные данные приведены для РЭМ с
экраном 10X10 см (наблюдение на расстоянии 25 см).
наглядностью изображения (во вторичных электронах) дает возможность исследовать объекты с ярко выраженным рельефом поверхности;
— простота подготовки объектов исследования, которая обеспечивает высокую производительность РЭМ и исключает артефакты;
— простота изменения увеличений от малых -до больших обеспечивает высокую прицельность исследования на РЭМ;
ТАБЛИЦА 3.3
— возможность проведения реитгеноспек-трального и катоде люминесцентного анализов, электронной спектрометрии, изучения магнитных н электрических микрополей, дифракционных эффектов н т. д.
— возможность проведения исследований в статическом и динамическом режимах позволяет успешно изучать непосредственно в РЭМ процессы, протекающие при механическом нагружении металлов, нагреве, охлаждении, воздействии среды и т. д.;
— электронно-зондовая система н поточечный принцип формирования изображения более гибкие, чем традиционная оптическая система ПЭМ, позволяет использовать микроЭВМ для автоматизации количественного анализа изображения и обработки результатов измерений.
К недостаткам РЭМ можио отнести высокую стоимость, ограниченную разрешающую способность, невозможность выявления структуры внутри образца, отсутствие цветного сигнала, необходимость помещения образца в вакуум, радиационные повреждения некоторых материалов в процессе исследования, затруднения при изучении диэлектриков (электризация, приводящая к неуправляемой эмиссии электронов).
3.1.4. Конструкция РЭМ
На принципиальной схеме РЭМ (рис. 3.3) можно выделить следующие основные системы: электронно-оптическую 1—10, формирующую электронный зоид и обеспечиващую его сканн-
СРАВНИТЕЛЬНАЯ ХАРАКТЕРИСТИКА МИКРОСКОПОВ РАЗНЫХ ТИПОВ [3]
Характеристики СМ РЭМ ПЭМ
Разрешение: рабочее доступное при высокой квалификации оператора требующее особых приемов , . Глубина фокуса . ...... Режим работы: иа просвет отражение дифракция прочие Образец: приготовление диапазон и тип Максимальная толщина образца для исследования Среда Полезное пространство . .... Поле зрения Сигнал . . Стоимость 5 мкм 0,2 » 0,1 » Малая + + + Некоторые Как правило, простое Разнообразный, оригинал или его реплика Массивный Разнообразная Маленькое Достаточно большое Только изображение Низкая 0,2 мкм 10 нм 0,5 » Высокая + + 4- Много Простое Разнообразный, оригинал или его реплика Средний Обычно вакуум Большое Достаточно большое Поддается обработке Высокая 10 нм 1 » 0,2 » Умеренная + + Нет Сложное, воз- можны артефакты Только тонкий или реплика Очень тонкий Вакуум Маленькое Ограниченное Только изображение Высокая
Примечание. Основные недостатки выделены полужирным шрифтом, преимущества—курсивом.
64
ТАБЛИЦА 3.4
ТЕХНИЧЕСКИЕ ХАРАКТЕРИСТИКИ ЭЛЕКТРОННЫХ ПУШЕК РАЗНЫХ ТИПОВ
Тип электронной пушки Вакуум, Па Температура катода, К Яркость ЙСТОЧ-Бкика (Д/сма) ср SMl ИВДМ Эффективный размер источника, НМЙМ'- Долговечность, ч
Термокатод из вольфрамовой проволоки lO-3—io-i 2650—2900 104—105 100000 30—100
Катод из гексаборида лаитана . . 10—4—10г-3 1750—2000 106—10“ 2000 100—500
Автоэмисснонная пушка 10-8—10-в 300—1000 10?—ю» 10 300—1000
Рис. 3.3. Принципиальная схема РЭМ:
/ — катод; 2 — фокусирующий электрод; 3 — анод: 4—«граничивающая диафрагма; 5 — первая конден-сорная линза; 6 — вторая конденсорная линза; 7 — отклоняющие катушки; 3 — сткгматор; 9 — конечная (евъективная) линза; 10 •—диафрагма, ограничиваю* щая размер пучка; 11 — детектор рентгеновского излучения (кристаллднфракционный нли энергетический); 12— усилитель фотоумножителя; 13 — генераторы развертки; 14 — образец; 15— детектор вторичных электронов; 16 — к отклоняющим катушкам; 17 — управление увеличением; 18 — ЭЛТ
рование по поверхности образца 14; систему, формирующую изображение, 11—18; вакуумную автоматизированную систему; устройства точной механики (шлюзы, держатели образцов, устройства разнообразного воздействия на образцы и т. д.).
Электронно-оптическая система состоит из электронной пушки, электромагнитных лииз, диафрагмы и катушек отклоняющей системы. Электронная пушка—источник электронов— состоит нз катода 1, фокусирующего электрода 2 н анода 3. Анод заземлен, а катод н фокусирующий электрод соединены с источником высокого напряжения (обычно 10—30 кВ, иногда -1,5 кВ).
При использовании электронных пушек нз тонкой вольфрамовой проволоки пучок электронов создается в основном за счет термо-эмнссин. Электронные пущкц с катодами из острозаточенных стержней гексаборнда лантана, окруженных нагревательной спиралью, н автоэмнссионные пушкн с холодным катодом имеют большую яркость н меныпнй эффективный размер катода, одиако стабильность получаемого пучка обеспечивается только прн высоком н сверхвысоком выкууме (табл. 3.4).
Эмитированные катодом электроны ускоряются н формируются в пучок, проходящий через диафрагму 4, конденсорные линзы 5 н 6 и объективную линзу 9, которые существенно уменьшают изображение источника электронов, фокусируя его на поверхности образца. Характеристикой электронно-оптической системы РЭМ является ее уменьшение (которое может доходить до 5000—10000 раз) н величина аберраций (сферической, хроматической, дифракционной н астигматизма).
Внутри объективной линзы находятся две пары отклоняющих катушек 7, которые соединены с генератором 13, обеспечивающим синхронную развертку электронного зонда и луча ЭЛТ 18 в квадратный растр. Развертка осуществляется в двух взаимно перпендикулярных направлениях, число строк в кадре составляет 500—1000. Примениют быструю развертку (как в телевизионных системах) и медленную. В последнем случае ЭЛТ для визуального наблюдения должны обладать длительным послесвечением в отлнчне от ЭЛТ для фотографирования. Время сканирования изменяется от нескольких секунд (прн визуальном наблюдении) до минут (при фотографировании). Стиг-матор 8 используют для коррекции астигматизма, вызванного ассимметрней магнитного поля линзы.
В каждой точке на поверхности образца электронный пучок находится в течение ограниченного времени, определяемого скоростью развертки. В результате взаимодействия электронов пучка с образцом возникают: отраженные электроны больших энергий (>50 эВ); низкоэнергетические вторичные электроны; рентгеновское излучение н излучение в ультрафиолетовой, видимой н инфракрасной областях (см. рис. I). Формирование изображения в РЭМ происходит в результате улавливания специальными детекторами электронов 15 и излучений И, испускаемых образцом, усиления этих сигналов н использования их для управления яркостью на экране ЭЛТ. Яркость каждой точки иа экране ЭЛТ определяется сигналом
5—280
66
детектора, получаемым в результате взаимодействия электронов зонда с соответствующей точкой на поверхности образца. Управление увеличением 17 осуществляется за счет изменения отношения амплитуд развертки луча по экрану ЭЛТ и электронного зонда по образцу. Увеличение можно плавно менять от 20 до 100000. Обычно используют увеличения <20000. Предельно высокие увеличения используют как вспомогательные для улучшения фокусировки и устранения астигматизма.
В качестве детектора вторичных и отраженных электронов, создающих очень небольшой ток вторичной эмиссии (10~п—10~,sA), в РЭМ используют детектор Эверхарта — Торилн (сцинтиллятор-фотоумножитель) (рис. 3.4). Ос-
Рис. 3.4. Схема детектора эмиттированных электронов Эверхарта — Торн ли:
1 — электронный зонд; 2 — эмиттнрованиые поверхностью образца электроны; 3 — образец; 4— сетка;
5 — сцинтиллятор; 6 — световод; 7 — фотоумножитель
новная часть детектора — сцинтиллятор 5, в котором прн попадании электронов с высокой энергией (10—15 кэВ) генерируется световое излучение. Сцинтиллятор размещен внутри коллектора, закрытого в передней части сеткой 4, потенциал которой относительно образца 3 можно менять от —50 до +250 В и тем самым регулировать соотношение собранных отраженных н вторичных электронов. Положительный потенциал служит для сбора вторичных электронов. Поскольку энергия вторичных электронов недостаточна для активации сцинтиллятора, оии ускоряются под действием высокого напряжения, равного 12 кВ. Световое излучение, создаваемое в материале сцинтиллятора, проходит по световоду 6 и попадает на фотоумножитель 7, где преобразуется в электрический сигнал.
Переход от изображения во вторичных электронах к изображению в отраженных электронах осуществляется при изменении условий сбора электронов с поверхности образца за счет использования других детекторов, расположенных непосредственно над образцом. Сбор вторичных электронов обеспечивается прн положительном потенциале сетки н размещении коллектора вне прямой видимости поверхности образца. Прн этом под действием электростатического поля траектории низкоэнергетических вторичных электронов отклоняются, обеспечивая большой телесный угол сбора вторичных электронов, в том числе нз затененных участков (глубоких впадин на поверхности и т. д.). Это позволяет выявлять больше деталей на поверхности н определяет получение полутонов на изображениях во вторичных электронах. Сбор отраженных высокоэнергетических электронов обеспечивается при отрицательном по
тенциале сетки н размещении коллектора в направлении прямой видимости поверхности образца. Прн этом затененные участки остаются невидимыми. Для изображений в отраженных электронах характерны резкие тенн. Изображения в отраженных электронах используют для улучшения контраста прн изучении относительно гладких образцов при небольших увеличениях.
В качестве детектора отраженных электронов также используют твердотельный или по-* лупроводниковый детектор, эффективность которого повышается прн больших токах пучка.
3.1.5. Формирование контраста изображения
Из информации, получаемой с помощью РЭМ (табл. 3.5), основными являются сведения о локальных изменениях топографии н химического состава поверхности. Соответственно выделяют топографический н композиционный контрасты. Топографический контраст обусловлен изменением интенсивности эмиссии вторичных электронов и коэффициента отражения для отраженных электронов при изменении угла наклона элемента поверхности к первичному пучку. Прн этом коэффициент вторичной эмиссии меняется сильнее, чем коэффициент отражения, что определяет преимущества использования вторичных электронов для изучения топографии поверхности.
ТАБЛИЦА 3.5
ИНФОРМАЦИЯ, ПОЛУЧАЕМАЯ С ПОМОЩЬЮ РЭМ
Свойства Способ формирования контраста
Топография поверх- Вторичные электроны (ос-
пости новной), отраженные электроны, катодолюминесценция
Химический состав Рентгеновское излучение (основной), отраженные электроны, Оже-электроны, катодолюминесценция
Толщина образца Прошедшие электроны
Электрические и магнитные локальные поля в образце Контраст напряжения
Электрические свой- Внутренние токи и напряже-
ства образца (особенно полупроводников) НИЯ
Характерная черта топографического контраста в РЭМ — повышенная яркость изображения острых вершин н выступов рельефа (краевой эффект), вызванная увеличением выхода электронов с этих участков. Снижение разрешающей способности н потеря отдельных деталей изображения усугубляются при этом за счет более эффективного улавливания коллектором электронов, вылетающих из выступов рельефа.
Существует очевидное сходство изображений, получаемых в световом и растровом микроскопах (прн использовании отраженных электронов впаднны кажутся темными, выступы— светлыми и отбрасывающими тени), несмотря на существенные различия в механизме формирования контраста.
Прн наблюдении в световом микроскопе объект обычно освещен под разными углами, а
66
изображение формируется в глазу наблюдателя. В РЭМ объект освещен очень узким электронным зондом, вторичные электроны поступают на коллектор под самыми различными углами, поэтому изображение выглядит так, как будто источник света расположен на месте коллектора электронов, а наблюдение проводится со стороны электронной пушки (рассеянное освещение при использовании вторичных электронов, косое — прн использовании отраженных электронов).
Композиционный контраст возникает при сканировании электронным зондом объектов с
Рис. 3.5. Использование парного детектора для разделения топографического н композиционного контрастов:
/ — электронный зонд; 2 — электронная пушка; 3 — отклоняющая катушка; 4 — магнитная линза; 5 — образец; 6 — отраженные электроны
локальными изменениями химического состава прн изменении коэффициентов вторичной эмиссии и отражения электронов. Эффективность отражения зависит от атомного номера мишени сильнее, чем эмиссия вторичных электронов, поэтому использование отраженных электронов в этом случае предпочтительнее. С увеличением атомного номера элемента коэффициент отражения электронов растет, поэтому места, обогащенные более тяжелыми элементами, отражают больше электронов н выглядят на изображении более светлыми.
Разрешающая способность изображений в С
отраженнных электронах (1000 А) существенно выше, чем в характеристическом рентгенов-
О
ском излучении (10000 А): в первом случае ограничиваются определением качественного распределения элементов. Однако прн этом можно выявить локальные неоднородности химического состава не только на поверхности образца, ио н на некоторой глубине до 1000 А. Композиционный контраст дает возможность изучать электрически непроводящие объекты с нанесенными электропроводными покрытиями, подповерхностные включения и т. д.
Для того чтобы разделит^ эффекты, вызываемые изменением химического состава и топографии, используют парный детектор А-В (рнс. 3.5). Интенснвиость сигналов, возникающих в детекторах, расположенных симметрично над образцом 5, зависит от композиционного н топографического контраста. Сигналы, вызванные композиционным контрастом, в обоих детекторах эквивалентны, а вызванные топографическим — обратны, так как оба детектора вндят поверхность с различных сторон. Суммирование сигналов от детекторов (А+В) увеличивает эффект композиционного контраста н уничтожает влияние топографии, а вычитание сигналов от этих детекторов (А—В) подавляет композиционный контраст н лучше выявляет топографию поверхности (см. рнс. 3.5).
Таким образом, в первом случае получают изображение, зависящее только от состава объекта, а во втором — только от его топографии,
3.2. МЕТОДИКА
ИССЛЕДОВАНИЯ
МЕТАЛЛОВ С ПОМОЩЬЮ РЭМ
Основное требование к образцу — соответствие его размеров размерам камеры образцов, чтобы его можно было передвигать по всем трем осям, вращать и наклонять по отношению к электронному зонду. Такое перемещение обеспечивает обследование всех поверхностей образца, кроме той, которой он приклеен Специальным токопроводящим клеем к предметному столику. После нанесения клея на образец его необходимо тщательно высушить. В непросохшем клее в вакууме образуются пузырьки, что загрязняет образец н колонну микроскопа. При исследовании изломов или шлифов крупных деталей последние приходится разрезать на части. Прн этом необходимо предохранить поверхности изломов от загрязнения, окисления и механических повреждений.
Поверхность образцов, исследуемых в РЭМ, должна быть чистой, так как неэлектропроводные окислы н разнообразные загрязнения под действием электронного зонда заряжаются. Перед исследованием в РЭМ образцы должны быть очищены с помощью различных растворителей в ультразвуковой камере в сочетании с осторожной механической очисткой. Иногда обычную механическую полировку при изготовлении шлифов заменяют электролитической нлн применяют в сочетании с ней, чтобы устранить искаженный поверхностный слой металла.
Прн изучении в РЭМ диэлектриков (например, продуктов коррозии нлн пластиковых реплик) эффектов накопления заряда можно избежать напыляя иа образец тонкое электропроводное покрытие, снижая ускоряющее на
51
67
пряжение нли используя метод однокадровой экспозиции.
Прн исследовании шлифов н изломов в РЭМ иногда полезно менять наклон образца по отношению к электронному зонду и детектору для усиления яркости н контраста изображения. Однако следует учитывать, что прн этом искажаются истинные размеры наблюдаемого образца, особенно, если он обладает развитым рельефом. Поэтому полезно получать микрофотографии в РЭМ, ориентируя поверхность образца перпендикулярно электронному зонду н наклоняя ее в сторону детектора. Изменение контраста изображения и «теней» прн вращении образца вокруг нормали к поверхности также облегчает понимание особенностей рельефа.
Большая глубина фокуса, высокая разрешающая способность н обилие полутонов на изображении, полученном в РЭМ, создают впечатление объемности и часто позволяют правильно представить себе пространственную конфигурацию деталей исследуемого объекта. При сложном рельефе, характерном для изломов, не всегда удается получить трехмерную реконструкцию по одной плоской проекции. В таких* случаях Для усиления эффекта объемности изображения проводят съемку стереопар исследуемого участка, изменяя его наклон по отношению к зонду на 5—10° в зависимости от увеличения. Изменение угла наклона образца обычно производят механическим способом с помощью гониометра, однако эту операцию также можно проводить, наклоняя зонд н ие изменяя прн этом положения образца. Стереопары рассматривают с помощью простейших стереоскопов, в которых впечатление объемности создается за счет эффекта параллакса. Количественную оценку деталей рельефа на микрофотографиях (измерение глубины, высоты и углов наклона) осуществляют с помощью стереокомпараторов по методикам, используемым в картографии. Имеются сообщения о получении стереоизображений непосредственно в РЭМ (на двух экранах в реальном масштабе времени).
Электронная обработка сигналов облегчает расшифровку мелких деталей изображения в РЭМ и позволяет изменять яркость и контраст, получать негативное изображение, подавлять светлый или темный фон, а также проводить некоторые специальные операции. Основные методы обработки сигнала:
— подавление постоянной составляющей позволяет растянуть переменную составляющую (основной носитель информации) иа весь динамический диапазон фотографии, включающий все возможные градации серых оттенков;
— нелинейное, или гамма-усиление узкого диапазона сигнала, близкого к минимальному, для выявления интересующей информации иа затененных участках;
— дифференцирование сигнала по времени приводит к усилению высокочастотной составляющей за счет низкочастотных н способствует оконтуриванию элементов изображения;
— У-модуляция, прн работе в режиме которой сигнал, обычно изменяющий яркость луча, модулируется по Y-коордннате изображения, т. е. электронный луч постоянной яркости на экране ЭЛТ отклоняется в вертикальном направлении от нулевого положения иа величн--ну, пропорциональную интенсивности сигнала.
В результате каждая горизонтальная строка растра превращается в волнистую линию, совокупность которых образует изображение. На экране ЭЛТ получается осциллографическая запись яркостного сигнала по ширине исследуемой области образца. Прн работе в этом режиме могут быть обнаружены тонкие различия в яркости участков, недостаточно выявляемые прн работе в других режимах, а контраст изображений объектов со слаборазвитым рельефом повышается.
Прн измерении размеров микроструктурных элементов на микрофотографиях, полученных с помощью РЭМ, необходимо учитывать, что возможны значительные искажения размеров. Исследование [13] метрических свойств растровых электронно-микроскопических снимков, проведенное с помощью контрольной сетки, содержащей 200 линий на 1 мм в обоих направлениях, показало основные источники геометрических искажений: несовершенство сканирования; неточность юстировки отклоняющих катушек; оптическая днсторцня фотопрнставки; деформация фотоматериала; сферичность экрана ЭЛТ н др. Для микрофотографий, полученных с помощью РЭМ, характерно различие размеров вдоль осей X и У.
3.3. ПРИМЕНЕНИЕ РЭМ
В МЕТАЛЛОВЕДЧЕСКИХ ИССЛЕДОВАНИЯХ
3.3.1. Металлография
Благодаря высокой разрешающей способности РЭМ целесообразно нзспользовать для металлографического нсследовання сплавов, обладающих дисперсными структурами. Прн этом четко выявляется строение эвтектических и эвтектоидных смесей; размеры, форма и распределение дисперсных частиц второй фазы н т. д.
На рис. 3.6 в качестве примера показано изображение (во вторичных электронах) морфологических особенностей порошка железо—медь, полученного распылением нз жидкого состояния.
Прн приготовлении образцов для растровой электронной микроскопии можно использовать металлографические методы, применяемые при подготовке микрошлнфов для наблюдения с помощью СМ. Наибольшие различия при этом заключаются в способах травления микрошлнфов. В световой металлографии структура выявляется за счет разности скоростей коррозии отдельных структурных составляющих и за счет различия продуктов химического взаимодействия травителя с образцом, осаждающихся иа определенных элементах структуры. Реактивы, образующие на отдельных структурных составляющих тонкие пленки, изменяющие отражательную способность образца, непригодны для РЭМ. Для образцов РЭМ используют только реактивы, образующие рельеф на поверхности микрошлнфов.
Для получения более глубокого рельефа н полного использования глубины фокуса РЭМ время травления микрошлнфов следует увеличить. Информация, полученная, например, при изучении внутренних поверхностей ямок трав-
68
Рис. 3.6. Морфологические особенности порошка Fe—Си. полученного распылением из жидкого состояния, при различных увеличениях:
С—ХЗОО, б—ХЗООО (Е. А. Войтехова)
ления, значительно больше, чем прн наблюдении тех же ямок в СМ.
Большая глубина резкости изображения в РЭМ может быть использована для выявления формы и тонкого строения включений в металлической матрице, которую равномерно стравливают до глубины 10—20 мкм. Применяя данную методику для изучения форм роста графита в чугуве, удалось показать [7], что включения шаровидного графита в заэвтекти-ческом чугуне образованы перекрывающими друг друга чешуйками, а пластинчатые включения также состоят нз аналогичных чешуек, смыкающихся в ветви, исходящие из центров, сходных по строению с включениями шаровидного графита. Весьма перспективно применение для исследования металлов в РЭМ селективно действующих травителей. В реактивах, применяемых прн травлении микрошлифов для исследования в РЭМ, содержание кислот, как правило, увеличено.
В растровой электронной микроскопии успешно применяется ионное травление, обеспечивающее высокую чистоту получаемой поверхности и выявление тонких деталей структуры.
Ионное травление в установках типа ВУП-2К или непосредственно в РЭМ осуществляется с использованием ионов инертных газов (агрощ иеон, гелнй), бомбардирующих поверхность образца (ускоряющее напряжение 10—15 кВ).
При анализе неметаллических включений используется и катодолюминесценция: образование квантов света с различной длиной волны в результате рекомбинации избыточных электронно-дырочных пар. Иногда на этот процесс может расходоваться до трети всей энергии первичного пучка электронов. Процесс катодолюминесценции происходит в некоторых полупроводниках н диэлектриках н усиливается прн понижении температуры.
3.3.2. Фрактография
Это основная область применения РЭМ и металловедении. Фрактографический метод исследования предусматривает получение качественной или количественной информации о строении изломов прн визуальном их рассмотренииг а также с использоианием СМ, РЭМ, ПЭМ и других приборов. Именно в области фрактографии преимущества РЭМ перед другими микроскопами проявляются наиболее заметно. Основные области применения РЭМ во фрактографии: контроль качества металлов; изучение механизма разрушения при различных видах, нагружеиня; установление причин эксплуатационных разрушений деталей машин и элементов конструкций.
Прн контроле качества металлов очень важно, кроме прочностных, пластических и энергетических параметров, определить характер разрушения и сопоставить его с эталонными шкалами изломов. Фрактографический метод — основной прн определении критических температур вязко-хрупкого перехода. Выявление структурных составляющих и изломе особенно целесообразно при малом их содержании благодаря избирательности процесса распространения-трещины. Важной инженерной задачей является установление продолжительности развития усталостной трещины и условий нагружения по фрактографическим показателям (расстоянию между бороздками), а также определение места н причины зарождения разрушения (характера технологических н эксплуатационных дефектов).
Особенность современного состояния растровой фрактографии заключается в переходе от качественного описания изломов [14, 15] к количественному. Наиболее часто производят измерение линейных размеров (шаг бороздок; ширина зоны вытягивания; размер ямок, фасеток, граней в вязком, хрупком, межзерениом и внутрнзеренном изломах) и измерение доли, той илн иной составляющей от общей поверхности излома.
Перспективные направления растровой фрактографии:
— расширение использования методов фотограмметрии, определеине размеров в третьем направлении, а также построение профилей и карт методом стереопар;
— повышение прицельности метода, исследование соответствующих мест на смежных частях изломов (метод сопряжения двух профилей);
— автоматизация измерений с применением' анализаторов изображений.
"Рис. 3.7. Типы разрушения металлов:
.я —хрупкое внутризеренное. х500; б — хрупкое меж-1зеренное. XIOO; в — вязкое ямочное. X -5000; а—усталостное. > 6000
Большое значение имеет также стандартизация и унификация терминологии, методов и трактовки получаемых результатов фрактографических исследований [16].
На рнс. 3.7 в качестве примера показаны наиболее типичные виды изломов металлов: вну-тризереиный скол с ручьистым узором (а), межзеренное хрупкое разрушение (б), ямочный рельеф прн вязком разрушении (в) и бороздки в усталостном изломе (г).
3.3.3. Локальный анализ
Наибольшие аналитические возможности РЭМ связаны с определением локальной химической неоднородности исследуемых материалов с помощью отраженных электронов, Оже-электронов и локального рентгеноспектральиого анализа.
Рентгеновские микроанализаторы [17] появились независимо от РЭМ, одиако сейчас уже трудно представить себе РЭМ без приставок для рентгеновского микроанализа (РМА). Оба прибора очень хорошо дополняют друг друга.
Современные РЭМ позволяют проводить осциллографическую запись распределения анализируемого элемента по любой строке растра и получать изображение в характеристическом рентгеновском излучении.
Интересные данные можно получить, используя Оже-электроны. Этот метод особенно перспективен для анализа легких элементов с Z<15, когда рентгеновский анализ затруднен. Локальность по глубине при этом составляет О
2—10 А (т. е. порядка нескольких атомных о
слоев), а по поверхности 200—300 А.
С помощью Оже-электронов можно получить информацию о строении тонких поверхностных слоев объекта (например, сегрегации элементов у границ зерен прн межзерениом разрушении). Особая чувствительность метода Оже-электрон-иой спектроскопии вызывает необходимость получения в таких приборах сверхвысокого вакуума (10~7—10-9 Па).
3.3.4. Изучение кристаллографической и дислокационной структуры металлов
Такого рода исследования с помощью РЭМ возможны благодаря эффекту каналирования электронов.
Эффект каналирования основан на аномальной адсорбции электронов прн определенных углах наклона электронного зонда к кристаллографическим плоскостям. Прн этом электроны проникают глубоко в кристалл, проходя между рядами атомов (вдоль «каналов»); выход вторичных электронов снижается и образуются темные линии. Используя различные углы наклона электронного зонда к поверхности образца, можно получить картины каналирования (псевдо-Кнкучи линии) — сетку темных линий, пересекающих светлый фон в различных направлениях. Локальность метода составляет О
500 А по глубине и 1 мкм по поверхности. Набор картин каналирования электронов, полученных прн наклоне кристалла во всех возможных направлениях относительно зонда, может быть объединен в карту каналирования. Сравнивая экспериментально полученные картины каналирования с атласом карт, рассчитанных
ла ЭВМ, определяют кристаллографическую ориентировку зерна н параметры кристаллической решетки.
При определенных условиях сканирования можно обнаружить дефекты кристаллической решетки и их плотность, отличить более искаженные фасетки внутризеренного скола от менее искаженных фасеток межзерениого разрушения, выявить границы пластически деформированной зоны вблизи вершины трещины, вдавливания индентора и т. д.
На рис. 3.8, а в качестве примера приведено изображение в режиме каналнроваиня электронов скола поликристаллического молибдена.
Рис. 3.8. Скол поликристаллического молибдена: а~в режиме каналирования электронов. Х35; б — картина каналирования от одного из зерен (Е. А. Войтехова)
В поле зрения три зерна с различной интенсивностью освещенности, обусловленной тем, что в каждом зерне существует собственная картина каналирования, присущая кристаллографической ориентировке зерна. На рис. 3.8, б приведена картина каналирования от одного из зерен.
На рнс. 3.9 показана микроструктура длительно отожженной прн температуре 1200 °C стали 0Х18Н10Т с зерном н крупными выделе
ниями карбидов в отраженных электронах и в режиме каналирования электронов (б)_ В последнем случае выявляется зеренный контраст за счет различной окраски, так как в-каждом зерне существует определенная картина каналирования.
Рис. 3.9. Карбиды в длительно отожженной стали 0Х1ВНЮТ1
а — в отраженных электронах. X100: о — в режиме каналирования электронов. Х100 (Е. А. Войтехова)
3.3.5. Другие области использования
РЭМ успешно применяют для изучения послойной структуры окислов вплоть до границы раздела металл — окисел, изменений поверхности в результате износа, кавитации, эрозии, контактной усталости, схватывания н других внешних воздействий. Установлению механизма разрушения в каждом отдельном случае способствует исследование продуктов износа, также возможное с помощью РЭМ. Широкие возможности имеет РЭМ для исследования порошков и композиционных материалов на разных стадиях их изготовления. Кроме обычных статических наблюдений, РЭМ может быть успешно применен для проведения динамических экспериментов in situ, когда непосредственно bi
7£
микроскопе протекают различные процессы (пластическая деформация, усталость, износ, (коррозия, воздействие магнитного и электрического поля) и изменяется строение поверхности •образца. При этом используют ЭЛТ с быстрой разверткой н видеомагнитофоны. Для иссле-дования динамических периодических и быстрых процессов (например, распространения пьезоволн, ультразвуковых волн, движения доменов) используется электронная стробоскопия.
3.3.6. Перспективы развития РЭМ
Основные тенденции развития РЭМ:
— повышение разрешающей способиостн О
(достигла 30—100 А в режиме вторичных электронов н 1000 А в режимах отраженных и поглощенных электронов);
— увеличение числа специальных приставок (в лучших микроскопах до 60, а их стоимость превышает стоимость основной системы РЭМ); расширение использования стереоизображения;
— слияние' электронной микроскопии (просвечивающей и растровой) и рентгеноспектрального микроанализа. Использование для наблюдения в РЭМ топографического, композиционного и ориентационного контраста;
— автоматизация анализа изображений для повышения скорости обработки данных и увеличения точности получаемых результатов. Использование специализированных мини ЭВМ позволяет улучшить качество изображения, упростить его интерпретацию и автоматизировать управление РЭМ [18]. При этом можно обнаруживать и оценивать доли структурных составляющих на заданном поле любой конфигурации, сравнивать и определять степени расхождения топографии объекта исследования с данными о топографии, заложенными в память ЭВМ, измерять размеры структурных составляющих (ширину, длину, площадь, периметр, расстояние между составляющими) по заданной программе с выбором объекта измерения.
БИБЛИОГРАФИЧЕСКИЙ СПИСОК
1. Кальнер В. Д., Зильберман А. Г. Практика микрозоидовых методов исследования металлов и сплавов. М.: Металлургия, 1981. 216 с.
4
- — - СТЕРЕОЛОГИЯ (КОЛИЧЕСТВЕННАЯ
4.1. ОСНОВНЫЕ ПОНЯТИЯ И ПРИНЦИПЫ [1—4]
Современная стереологня — область прикладной математики, формулирующая характеристики геометрической структуры объектов различного происхождения и разрабатывающая способы их определения — реконструкции на основании простых измерений, производимых иа плоскости наблюдения. Геометрическая структура — совокупность точечных, линейных, поверхностных и объемных элементов, определенным об-
2. Практическая растровая элекронная микро-скопня:/Пер. с англ. Под ред. Гоулдстей-на Дж., Яковица X. М.: Мир, 1978. 656 с.
3. Heart в L W. S., Sparrow J. Т., Cross Р. М. The Use of the Scanning Electron Microscope. Oxford.- Pefgamon Press, 1972. 278 p.
4. Thornton P. R. Scanning Electron Microscopy. L.: Chapman and Hall, 1968. 368 p.
5. Wells О. C. Scanning Electron Microscopy. N.—Y.: McGraw-Hill, 1974, 421 p.
6. Васичее Б. H., Кабанов A. H.—Изв. AH СССР Сер. физ., 1977, т. 41, № 7, с. 1513— 1524.
7. Епанчинцев О. Г.—Заводская лаборатория, 1975, № 2, с. 206—216.
8. Спивак Г. В., Сапарин Г. В., Быков М. В.— УФН, 1969, т. 99, с. 635—662.
9. Шабер О.—В кн.: Приборы и методы физического металловедения. Вып. 2. М.: Мир, 1974, с. 65—130.
10. Материалы IX Всесоюзной конференции по электронной микроскопии и II Всесоюзного симпозиума по растровой электронной микроскопии.—Изв. АН СССР. Сер. физ., 1974, т. 38, № 7, с. 1362—1553.
11. Применение в металловедении просвечивающей и растровой электронной микроскопии. Материалы семниара-конференцин. М.: МДНТП, 1976. 169 с.
12. Материалы конференции «РЭМ-78»./Науч-ный совет АН СССР по электронной микроскопии. Звенигород: 1978. 14 с.
13. Мельник В. Н., Любимов И. Л,—Заводская лаборатория, 1977, № 3, с. 288—291.
14. Гордеева Т. А., Жегина И. П. Анализ изломов прн оценке надежности материалов. М.: Машиностроение, 1978. 200 с.
15. Metals Handbook. Fractography and Atlas of Fractographs. V. 9. Ohio: ASM, Metals Park, 1974. 499 p.
16. Классификация видов поверхностей разрушения (изломов) металлов. Расчеты и испытания на прочность в машиностроении. Рекомендации. М.: Госстандарт СССР, 1974. 45 с.
17. Локальные методы анализа материалов/ Боровский И. Б., Водоватов Ф. Ф-, Жуков А. А. н др. М.: Металлургия, 1973. 296 с.
18. Васичее Б. Н. Мурашко Г. ТА.—Изв. АН СССР. Сер. физ., 1974, т. 38, № 7, с. 1398—1402.
МЕТАЛЛОГРАФИЯ)
разом размещенных в пространстве. Применительно к металлам и сплавам элементам геометрической структуры соответствуют: точечным — общие вершины четырех зерен («четверные точки»), узлы дислокационной сетки1; линейным — общие ребра трех зерен («тройные стыки»), сегменты дислокаций и (условно) тонкие стержневые выделения и включения; поверхностным — границы внутри- и межфазного раздела зерен, границы двойников, выделений и т. п., объемным — собственно
’ Без учета возможного расщепления.
72
зерна (частицы), выделения, двойники, поры и т. д.
Различают однофазную структуру, состоящую из частиц илн зерен, одного компонента (феррит, аустенит), н матричную, образованную зернами матрицы н частицами избыточных фаз или структурных составляющих (аустенит н карбиды, перлит и избыточный цемеитнт н т. д_), находящихся в различной степени взаимного контакта — от полного разобщения (отдельные случайные включения) до полной связанности (сплошные прослойки остаточного аустенита). С точки зрения способа размещения элементов структуры в пространстве оиа может быть изотропной н ориентированной. Реальная структура металлов и сплавов — полиднсперс-ная иемоиоморфная (т. е. состоящая из частиц разного размера н различной формы) система.
Основные этапы стереологического анализа: получение первой представительной выборки структуры — образца, пробы; препарирование — получение объекта стереологн-ческого исследования (вторая представительная выборка-); получение изображения объектов структуры на плоскости наблюдения оптического прибора; измерения на плоскости наблюдения — первичные измерения; преобразование первичных измерений в характеристики пространственной структуры — сте-реологическая реконструкция.
В металловедении первая представительная выборка — образец, вырезанный из массивного объема (полуфабриката, готового изделия) компактного (литого, деформнроваииого, спеченного, термически обработанного) металла илн сплава; проба, отобранная от партии од-нокомпонентиого порошкового материала илн порошковой смесн; поверхности разрушения (излом) массивного объекта. Условие представительности такой выборки — воспроизведение характерных особенностей структуры (неоднородности, преимущественной ориентировки, наследственной агломерации).
Основные объекты металловедческого исследования— шлиф нли реплика с него, экстракционная реплика, монослой частиц порошка, участок поверхности разрушения. Условия представительности определяются особенностями структуры и методом последующей реконструкции. Общее требование — достаточная протяженность (достаточный объем выборки) и воспроизведение особенностей структуры об
ТАБЛИЦА 4.1
разца (пробы) без искажений (погрешности препарирования).
Изображение объекта получают с помощью различных видов микроскопии — световой и электронной: просвечивающей, эмиссионной и растровой. Основное требование к данному этапу анализа — перенести без искажения (инструментальные погрешности) структурное содержание объекта на плоскость наблюдения.
Первичные измерения — по существу третья представительная выборка системой точек нли линий (измерительной системой) на плоскости наблюдения. Общее условие представительности — достаточное число измерений; дополнительные требования зависят от типа структуры и способа последующей реконструкции.
Принципиально возможны два способа сте-реологнческой реконструкции — непосредственная и статистическая. Непосредственная реконструкция методом последовательных сечений — построение пространственной модели структуры на основании изображений ее на последовательных по глубине сечениях — шлифах в металлографическом световом микроскопе (СМ), эмиссионном (ЭМ) илн растровом (РЭМ) электронном микроскопе нли на репликах в просвечивающем электронном микроскопе (ПЭМ). Последовательные сечения с минимальным шагом 1 получают строго параллельным последовательным механическим нли электролитическим полированием образца. Некоторые характеристики пространственной структуры определяют непосредственно и а модели, другие—на представляющем ее графе. Непосредственную реконструкцию методом стереопар проводят в основном для поверхностей разрушения в РЭМ или ПЭМ н частил, порошковой пробы в РЭМ. На изображениях одного и того же участка структуры, полученных с одинаковым увеличением прн двух различных углах наклона объекта относительно пучка электронов, измеряют горизонтальный параллакс (разность координат идентичных точек на двух изображениях) и на его основе рассчитывают соответствующие высоты.
Статистическая реконструкция — преобразование первичных измерений х(Л) в характеристики геометрической структуры X(V) с по-
1 Полное восстановление структуры требует исчезающе малого шага между сечениями. Практически добиться этого можно сочетанием одновременной электролитической полировки и кииематографирова-ния [3, 5]
ГЕОМЕТРИЧЕСКИЕ ВЫБОРКИ В ПРОСТРАНСТВЕ СТРУКТУРЫ И ПЕРВИЧНЫЕ ИЗМЕРЕНИЯ
Выборка в пространстве структуры Измерения на плоскости наблюдения
базовая система измеряемые величины
Сечение плоскостью Сканирование плоскостью Сеченне линиями Сканирование прямой Бросание точки 'Плоскость наблюдения Плоскости последовательных сечений Система линий Сканирующая прямая, система параллельных прямых Система точек Числа, линейные размеры и площади элементов структуры Числа касаний параллельных плоскостей с поверхностями раздела в структуре Числа пересечений с элементами структуры, длины отрезков, отсекаемые границами-' изображений Числа точек касания с элементами структуры Число точек, попавших на изображения частиц данной фазы
73
ТАБЛИЦА 4.2
ОБОЗНАЧЕНИЕ ПЕРВИЧНЫХ ИЗМЕРЕНИЙ И ХАРАКТЕРИСТИК СТРУКТУРЫ ДЛЯ РАЗЛИЧНЫХ БАЗОВЫХ СИСТЕМ
Базовая система Измеряемые и расчетные величины
V S А L Р. Т. I N
Объем стуктуры V у** v V sv — - TV Nv
Система плоскостей площадью А * — — аа ^А РА* ТА' 1А ^А
Система линий длиной L —* —Г- — Ll Pl
Система точек числом Р — — —• — п** —
* Величины, отнесенные к единице площади, называются ареальными; ареальная доля Ад; ареальная протяженность Тд; ареальные числа Рд, Т, /д, Nд.
* * Величины Vy, Ад. Lp и Рр называются соответственно объемной, ареальной, линейной и точечной долями; Sg — поверхностная доля.
ОСНОВНЫЕ СТЕРЕОЛОГИЧЕСКИЕ ХАРАКТЕРИСТИКИ
Характеристика Размерность Обозначение Объект*1 Реконструкции Необходимость задания формы
непосредственная статистическая
Распределение разме- ММ“3 Ш, Ф, ЭР — + +
ров частиц Число всех частиц в еди- ММ~s п Ш, Ф, ЭР + + +
иице объема Средний поперечник ча- МКМ; ММ Ш,Ф,ЭР,П — + +
стиц в системе D^l*3 ш — + —
Факторы формы — Кф III, п — +
Удельная поверхность мм2/мм3 sv Ш, Ф — + —
Кривизна поверхностей: средняя ММ—1 н III, Ф ——« 4- —
гауссова к ш + •—~
Двугранный угол град, рад е ш — +
Плотность линейных мм/мм3; Pv Ф, ЭР Ш, Ф 4“ 1 + —
элементов Кривизна линейных эле- i I I I k ф — 4- —
ментов Кручение линейных эле- ММ—1 t ф — 4- —
ментов Объемная доля м№/мм5 Ш, Ф — + —
Степень орнеитироикн — (X III — 4- —
То же — cos (Z, п) ш — 4- —
^Среднее межцентровое мкм; мм Рц Ш, ЭР — 4- +
расстояние Оредиее свободное рас- мкм; мм L ш — 4- —
•стояние Топологические инва- мм—3 Gv, Nrvon ЭР ш 4~ + +
рианты Координационное число —• Nciaja ш — 4- +
Ш — шлиф или реплика; Ф — фольга; ЭР — экстракционная реплика; Г — порошковая проба.
*г d — средний поперечник изображений частиц.
*s I — средняя хорда, отсекаемая границами сечений частиц.
74
мощью оператора геометрической вероятности р(У, А) [3]. Первичные измерения иа плоскости наблюдения, содержащей сечения нлн проекции элементов структуры, имитируют мысленную выборку в пространстве геометрической структуры с помощью базовых систем поверхностей, линий и-точек [3, 4]. Основное условие представительности этой выборки — статистическая однородность, чаще всего выражающаяся как случайность (в смысле ориентировки и трансляции) взаимного положения элементов геометрической структуры и базовой системы. В табл. 4.1 приведены основные виды выборок н соответствующие нм первичные измерения.
Первичные измерения и реконструированные характеристики часто представляют в виде величии, удельных по отношению к базовой системе. Для обозначения таких величин используют систему Международного общества стереологни (МОС): заглавная латинская буква соответствует измеряемой величине, индекс (также в виде заглавной латинской буквы) — базовой системе. Принято обозначать: V—объемы, S — поверхности, А — плоскости, L — линии, Р—числа точек пересечения и точечных элементов структуры, Т — числа точек касания, I — числа точек перегиба, JV— числа объемных, плоских и линейных элементов структуры. Основные виды обозначений удельных велнчнн представлены в табл. 4.2.
Стереологнческие характеристики бывают по своему смыслу зависящими (метрические) и не зависящими (топологические инварианты) от размеров и формы частиц; однако часто зависимость от формы возникает из-за введения допущений прн измерении илн расчете (табл. 4.3). Адекватное описание структуры дают те характеристики, определение которых инвариантно к форме частиц.
4.2. ОБЪЕКТЫ И ТЕХНИКА
СТЕРЕОЛОГИЧЕСКОГО АНАЛИЗА
4.2.1. Объекты
стереологического анализа
На рис. 4.1 и в табл. 4.4 и 4.5 дана характеристика изображений структуры основных
объектов, изучаемых с помощью различных оптических приборов. Некоторые погрешности первичных измерений иа плоскости наблюдения являются следствием специфических особенностей изображений, обусловленных препа-
Рис. 4.1. Объекты стереологического исследования [31:
а — исследуемый представительный объем; б — шлиф (Ш) и одноступенчатая реплика (Р); в — «толстая» (Ф1) и «тонкая» (Ф2) фольги; г — экстракционная реплика (стрелкой отмечена частица, которая может удалиться до экстракции или сместиться из исходного положения при экстракции); д— порошковая проба при наблюдении в просвечивающих световом и электронном микроскопах; 1 — плоскость случайного сечения; 2, 3 — граничные поверхности фольги; 4 — поверхности шлифа и реплики; 5 — плоскость наблюдения; С — сечения; П — проекции; Э. П — экранированные проекции; Н. И ~ проекции из положения наибольшей устойчивости; Э. Р — экстракционная реплика
ТАБЛИЦА 4.4
ХАРАКТЕРИСТИКА ИЗОБРАЖЕНИЯ СТРУКТУРЫ НА ПЛОСКОСТИ НАБЛЮДЕНИЯ
______________________РАЗЛИЧНЫХ ОБЪЕКТОВ [3}___________________
Метод исследования Объект Изображение элементов структуры
Оптическая микроскопия Просвечивающая электронная микроскопия Эмиссионная электронная микроскопия Растровая электронная микроскопия Шлиф Порошковая проба Реплика Фольга Экстракционная реплика Порошковая , проба Шлиф Порошковая проба Шлиф Случайные сечения Проекции всех частиц в монослое в поло-женин наибольшей устойчивости Случайные сечения Проекции из объема фольги, случайные сечения поверхностями фольги Проекции частиц, экстрагированных случайной плоскостью Проекции всех частиц в мопослое в положении наибольшей устойчивости Случайные сечения Объемные изображения частиц Случайные сечения
75
ТАБЛИЦА 4.5
УСЛОВИЯ ФОРМИРОВАНИЯ КОНТРАСТА И РАЗРЕШАЮЩИЕ СПОСОБНОСТИ МЕТОДОВ ИССЛЕДОВАНИЯ СТРУКТУРЫ
Метод исследования Исследуемый объект Характер контраста Минимальное разрешаемое расстояние
Оптическая микроскопия Шлиф Порошок Обусловлен рельефом от травления * Абсорбционный 0,2—0,5 мкм 0,2—0,5 мкм
Просвечивающая электронная микроскопия Реплика Экстракционная реплнка, порошок Фольга Обусловлен рельефом от травления Абсорбционный Дифракционный, абсорбционный 100 А 20 А ЮА
ЭЭМ в режиме вторичной эмиссии и растровая Шлиф Обусловлен рельефом от травления 100 А О
электронная микроскопия Порошок Обусловлен естественным рельефом 100 А
ЭЭМ в режиме термо- н фотоэмиссии Шлиф Обусловлен кристаллографической анизотропией работы выхода электронов I00A
* Возможны также фазовый и ориентационный контрасты, например в результате теплового травления.
рированием и условиями наблюдения [2, 3].
Шлиф (реплика). Препарирование связано с двумя источниками погрешностей — образованием рельефа (полировка, травление) и вырыванием и или замазыванием частиц второй фазы (полировка). Наличие рельефа снижает точность определения ареальных величин Na и Аа (поверхность шлифа отличается от плоскости) и размеров частиц (рис. 4.2). Эта погрешность существенна для определения D,
Рис. 4.2. Влияние рельефа шлифа на кажущееся число и размеры частиц [2]
Nv и Vv и мало влияет на измерение Sv. Замазывание и вырывание частиц (рис. 4.3) приводит также к кажущемуся уменьшению Na и Аа.
Рис. 4.3. Схемы вырывания неметаллических включений при шлифовке и «замазывание» их при полировке на продольном (а) и поперечном (б) шлифах J2]
Фольга. Операции препарирования (резкал шлифовка) могут заметно исказить структуру, особенно дислокационную, вследствие нагрева и деформации. Другая причина перестройки дислокационных конфигураций — существенно большая велнчииа отношения поверхности к объему по сравнению с массивным образцом [3, 6]. Дифракционный контраст обязывает применять гониометрическое устройство для выведения всех элементов структуры в отражающее положение [6, 7]. Геометрические источники погрешностей — взаимная экранировка частиц при проецировании на плоскость наблюдения, пересечение некоторых частиц граничными поверхностями. Последним эффектом можно пренебречь прн (t — толщина фольги; D — средний поперечник частиц). Эффект экранировки сказывается иа определении Nv и D прн больших Vv [3, 8, 9].
Экстракционная реплика. Возможные источники погрешностей: значительный рельеф, вызванный глубоким травлением; неполная экстракция; экстракция посторонних частиц («осколков», зерен абразива); экстракция в смещенном положении. Причиной неполной экстракции является также вырыв частиц при приготовлении шлнфа.
Способы предотвращения погрешностей — предварительная очистка шлнфа лаковой репликой; напыление углерода на образец перед снятием экстракционной репликн [10, II].
Монослой частиц порошка — дисперсия частиц на предметном стекле (СМ), тонкой (например, коллодиевой) подложке (ПЭМ) или непосредственно на объектном столе (РЭМ). Источники погрешностей: агломерация частиц; несоблюдение условий проецирования из положения наибольшей устойчивости для отдельных частиц. Разделение агломератов (дезагрегацию) лнбо вызывают диспергированием (гидромеханическим, ультразвуковым, электро
76
статическим) [12, 13], либо производят непосредственно на изображении — мысленно прн ручнбм измерении или с помощью специального устройства, оптимизирующего изображение, при автоматическом анализе [3, 14].
4.2.2. Анализ
с участием оператора
Первичные измерения производит оператор, наблюдая изображение структуры либо в СМ, ПЭМ, ЭЭМ нли РЭМ, либо на. фотографии, полученной в одном из этих приборов. Измерительные базовые системы накладывают на плоскость наблюдения, т. е. помещают в окуляр СМ (окулярные вставки), наносят на флюоресцентный экран ПЭМ илн ЭЭМ, на экран дисплея РЭМ или на фотографию. Основные типы измерительных систем представлены иа рис. 4.4, а выполняемые с их помощью операции первичных измерений — в табл. 4.6.
Для определения площадей и диаметров методом сравнения чаще всего используют измерительные шаблоны (рис. 4.5); для повышения точности таких измерений рекомендуется квадратная сетка [3, 15].
Подсчет точек, попавших на изображение, эквивалентен измерению площади (одна точка соответствует площади №, где h — шаг сетки).
Для реализации приближения формы поперечник изображения (сечения илн проекции) частицы определяют нз условия равенства площадей изображения и фигуры выбранной формы. Этого можно достигнуть сравнением с шаблонами (см. рнс. 4.5) нли с большей точностью точечным методом [14, 15]. В послед
ТАБЛИЦА 4.6
нем случае возможна предварительная градуировка эквивалентный диаметр de— число точек Р. В частности, для сферического приблн-
Рис. 4.4. Окулярные линейки (а) и сетки (б) различных типов [2]
ження (фигуры изображения — круги) с учетом увеличения М н шага сетки h:de= =ZhPlJ2l .
Помимо de, в качестве линейной меры индивидуального изображения используют: случайные, максимальные и минимальные хорды; диаметр Фере dF — проекцию изображения на
ВОЗМОЖНОСТИ ВЫПОЛНЕНИЯ ПЕРВИЧНЫХ ИЗМЕРЕНИЙ С ПОМОЩЬЮ РАЗЛИЧНЫХ ИЗМЕРИТЕЛЬНЫХ СИСТЕМ*
Первичные измерения Измерительная система Манипуляции с объектом**
сетка линейка пере- крестие
Подсчет случайных точек Р (для + — — —
определения Рр) — + Перемещение вдоль ортогональных прямых
Подсчет структурных точек: Расса, Раар , I; чисел изображении элементов структуры (для определения Ра, Ia, Na) —
Подсчет точек пересечения Р (для 4- — — —.
определения Pl) — + + Перемещение в перпендикулярном к лннейке направлении (по окончании измерений на лииейке) Перемещение в горизонтальном направлении прн измерении н в вертикальном между сериями измерений
Подсчет точек касания Т (для + — — .—•
определения Та ) + Перемещение в направлении, перпендикулярном линейке, в процессе измерений
Измерение хорд 1, диаметров d + — — —
(для определения £ь, L, 1, d) + Перемещение перпендикулярно лннейке по окончании серин измерений
* Рассмотрена техника измерений на одном поле.
** Т. е. с приводом предметного столика в СМ или держателя образца в ПЭМ. ЭЭМ и РЭМ.
77
прямую заданного направления; диаметр Мар, тина йм — хорду, параллельную заданному направлению и делящую площадь частицы на две равновеликие части. Этн величины приемлемы для нахождения оценки среднего разме-
рите. 4.5. Окулярные вставки (а, б) и накладной шаблон (в) для оценки площадей сечений методом сравнения (а — для округлых неметаллических включений, б —для полиэдрических зерен) [2]
ра частиц в системе и неприемлемы для классификации частиц по размерам [12, 14].
Один из источников погрешностей анализа с участием оператора — конечные размеры элементов измерительных систем (рис. 4.6). Другой источник — неправильный выбор увеличения — сохраняется и прн автоматическом анализе. Например, недостаточное увеличение в обоих случаях приводит к кажущемуся снижению ареальных величия Na и А а, долей Рр и н к кажущемуся возрастанию d и I.
Для механизации первичных измерений предложены ручные счетчики, сумматоры, путп-интеграторы н другие устройства [1, 3, 4]. По
следние модели Видеоплан («Leitz», ФРГ), МОП («КопДоп», Австрия) весьма эффективно ускоряют измерения, классификацию и даже обработку результатов.
Рис. 4.6. Влияние ширины секущих (а) и линий сетки (б) на измерение [2]
4.2.3. Автоматический анализ [3, 16 18]
В основу разработки автоматических анализаторов изображения ААИ положен принцип эквивалентности сканирования точкой и измерения с помощью статической системы (см. табл. 4.6). В настоящее время большинство ААИ — устройства со сканированием электронным лучом в плоскости изображения («Квантнмет-720», Великобритания; «Мнкровндеомат», ФРГ; «Омникон» и «Омннмет», США); значительно реже применяется сканирование объектного стола («Эпиквант», «Морфоквант», ГДР). ААИ со сканированием электронным пучком обладают большим быстродействием, что позволяет использовать их совместно с ЭВМ специального назначения.
Современный ААИ состоит из трех основных систем: ввода сигналов изображения — видеосигналов; обработки этих сигналов (процессора) и выдачи информации. Кроме того, в ААИ последних конструкций предусмотрено программное устройство, управляющее работой всех систем, а также система оперативного взаимодействия с ААИ (рис. 4.7).
Картина проекции нли сечения структуры на плоскости наблюдения — чередование различных градаций яркости — от абсолютного белого до абсолютного черного тона. Детектирующее и сканирующее устройства преобразуют этн колебания яркости в видеосигналы. Детектирующее устройство — мишень передающей
Рис. 4.7. Принципиальная схема ААИ [3]t
i — вистема ввода изображения; 2 — процессор; 3 —система выдачи информации; 4 — система оперативного взаимодействия; 5 — программное устройство
78
электронно-лучевой трубки (ЭЛТ) типа видикон нли плюмбикон. Сканирующее устройство «осуществляет развертку («считывание») детектированного изображения и преобразование -его в видеосигналы. В новейших ААИ для повышения разрешающей способности н увели-
Рис. 4.8. Работа дискриминатора [3]:
а — первичный видеосигнал; б — сигнал после дискриминации; в — квантование сигнала
чения отношения снгнал/шум применяют развертку из 720 строк в кадре со скоростью 10,5 кадр/с без чересстрочности.
Дискриминатор получает на входе видеосигнал— изменение электрического импульса Ф от значения Фтш, соответствующего темному фону (например, матрице в двухфазной структуре), ДО Фтах, соответствующего светлому объекту (частице второй фазы), и снова до Фш1л, что свидетельствует о прохождении сканирующим пятном изображения частицы; уровень дискриминации Фд = (Фшах—Фш1п)/2 (рис. 4.8). Для введения импульса с дискриминатора в управляющее устройство — цифровой компьютер, работающий на двоичном коде 0—1, предусмотрено квантование импульса (см. рис. 4.8). Квантование превращает изображение в матрицу «точек» изображения (ТИ), расстояние между которыми по горизонтали— интервал квантования б, а по вертикали— расстояние между строками в кадре. ТИ имеют определяемые масштабом общего увеличения размеры, в которых можио выражать длины хорд и площади изображений частиц. Работа логических схем ААИ похожа на работу оператора, накладывающего на изображение структуры квадратную измерительную сетку.
Логические схемы выполняют все основные операции сбора первичной информации — счета изображений частиц и точек и определения размеров (длин хорд, площадей, периметров и диаметров), а в некоторых моделях ААИ также производят классификацию изображений по их форме (рнс. 4.9).
Система выдачи информации обычно включает цифровое табло, цифропечатающее устройство, графопостроитель. Одна нз основных частей системы оперативного взаимодействия с ААИ — дисплей — видеоконтрольное устройство, на экране которого воспроизводятся исследуемое изображение и все операции, про
водимые как самим оператором (настройка дискриминатора), так и логическими схемами ААИ. Часто используется «световое перо», позволяющее оператору выбирать для счета и измерений детали изображения. В ряде конструкций ААИ предусмотрено устройство для оптимизации изображения — амендер, позио-
Рис. 4.9. Классификация изображений по различным параметрам [3]:
а — исходное изображение; б —сепарация; в — классификация
ляющее оператору несколько изменять характер изображения структуры — заполнять пустоты», «собирать» разрозненные детали изображения нли, наоборот, «разделять» сгустки деталей.
В новейшем поколении ААИ (например, анализатор «ТАС», фирма «Leitz», ФРГ), работа которых построена иа принципах теории случайных точечных множеств [19, 20], первичные измерения производятся с помощью операций, похожих на операции оптимизации В этих приборах точка изображения — «струк-турирующнй элемент» СЭ (правильный многоугольник, круг) — используется для измерения ие путем заполнения строк между границами изображений или наложения сетки ТИ, а в результате непосредственного взаимодействия с контурами изображений. Так выполняются операции «эрозия» — обкатывание СЭ по контуру изображения его периферией и «обратная» ей «дилатация» — прохождение центра СЭ по контуру изображения (рнс. 4.10). Другие операции — «вскрытие», «закрывание», «скелетоннзация» и т. д. — являются комбинациями двух основных в различной последовательности [20].
Точность автоматического анализа зависит от величин погрешностей, вносимых прибором, и от качества объекта. Специфическими, для ААИ являются погрешности устройства ввода видеосигналов и системы обработки нзображе-
79
ния. Помимо геометрического разрешения, определяемого числом ТИ в кадре, разрешающая способность ААИ характеризуется чувствительностью к градапиям яркости—способностью к переносу контраста. Ограниченность
изображения прн неправильной установке уровня дискриминации (рис. 4.11). * Другими источниками ошибок являются: высокочастотные «шумы», неравномерное распределение освещенности (затенение), геометрические ис-
Рис. 4,10. Операция со структурирующим элементом — эрозия Ц9]
с ,
Рис. 4.11. Погрешности в определении размеров, вызванные ограниченным разрешением ААИ и неправильной установкой порога дискриминации I3J:
I — сканирующее пятно; II — изображение; а — идеальный сигнал (С — действительная длина хорды); б — реальный сигнал (Д — ошибка, связанная с ограниченным разрешением); в — неправильный выбор порога дискриминации; г — «пороговая» ошибка С
указанной чувствительности вместе с конечными размерами сканирующего пятиа обусловливают искажение прямоугольного видеоимпульса в трапециевидный, что может привести к ошибке в определения размера элемента
Рис. 4.12. Коррекция краевой ошибки с помощью двойной рамки кадра [3].-
1 — изображение не учитывается; 2 — изображение учитывается; 3 — предохранительная зона; 4—внешняя рамка; Б — внутренняя рамка; ТАС — адресн । (точка «антисовпадения»)
кажения в электронно-оптической системе ЭЛТ.
Краевой эффект — ошибка, связанная с пересечением изображений границами поля зрения. В современных ААИ эта погрешность компенсируется с помощью «раздвижной рамки». Кадр имеет две рамки — внешнюю н внутреннюю, размеры которой могут быть изменены оператором иа величину, кратную размеру одной ТИ. Логические схемы ААИ производят измерения только на тех изображениях, адресная точка которых — точка аитисовпадения ТАС— находится в пределах внутренней рамки; очертания частиц могут выходить за эту рамку — в «предохранительную» зоку (рис. 4.12). Способы учета краевого эффекта с помощью специальных расчетов изложены в работах [3, 21].
Автоматический анализ предъявляет повышенные требования к качеству объекта. На изображении совершенно недопустимы посторонние детали (например, задиры, риски, выкрашивания на шлифе). Препарирование должно обеспечивать однородный контраст однотипных деталей (изображений частиц одной фазы, границ зерен) и значительное различие в контрасте между частицами различных фаз (структурных составляющих). Для объекта-шлифа весьма существенно отсутствие «завалов», макрорельефа и других источников неравномерной передачи яркости и фокусировки.
4.3. ХАРАКТЕРИСТИКИ РАЗМЕРОВ ЧАСТИЦ
4.3. L Распределение размеров частиц
В общем виде стереологнческая реконструкция непрерывного и «дискретного» (разбиение на классы) распределений может быть соответственно представлена:
NA(x}dx = J p(JD,x)Nv (D) dD; (1)
D=x
k
<2>
где x нли Xi — первичные измерения на плоскости наблюдения (площади Аг, случайные хорды Lt или диаметры di изображений); D или Dj—'Средние поперечники индивидуальных частиц (классы размеров); p(D, х) илн pji — множитель геометрической вероятности, характеризующий вклад частиц размеров D^>x (класса в изображения размера х (класса г); k — число классов.
Средний поперечник D — усредненное по всем возможным ориентировкам частицы расстояние между параллельными плоскостями, касательными к крайним ее точкам; в случае сферической формы D совпадает с диаметром частицы; связь между D и характерными размерами ряда простых форм представлена в работах [1,3, 22].
Для стереологнческой реконструкции распределения размеров приходится предполагать, что все частицы имеют одинаковую форму, и задавать эту форму. В формулах (1) и (2) приближение формы косвенно выражают члены D или D} и р(Ь, х) илн pji.
Случайные сечения (шлифы, реплик и). Для реконструкции распределения размеров разработаны методы непосредственного дифференцирования, основанные на формуле (I), а также методы последовательных подстановок и матричные, которые базируются на формуле (2). В методах последовательных подстановок используется прямая матрица рц
7=1
ТАБЛИЦА 4.7
СТЕРЕОЛОГНЧЕСКАЯ РЕКОНСТРУКЦИЯ РАСПРЕДЕЛЕНИЯ РАЗМЕРОВ СФЕРИЧЕСКИХ ЧАСТИЦ НА ОСНОВАНИИ РАЗЛИЧНЫХ ПЕРВИЧНЫХ ИЗМЕРЕНИИ НА ПЛОСКОСТИ
НАБЛЮДЕНИЯ СЛУЧАЙНОГО СЕЧЕНИЯ (ШЛИФА, РЕПЛИКИ)
Первичные измерения Деление на класоя* Расчетная формула
Хорды число хорд длиной U на единице длины секущей, > Диаметры di, ареальные числа сечений частиц диаметра di, NA(di) Диаметры или площади сечений частиц; ареальное число сечений диаметра di нли площади аг, Na (di) нли NA(ai) Равномерное с интервалом А Логарифмическое в соответствии с табл. 4.8 Равномерное с интервалом А= =Dt&^!k, k — число классов Логар нфмическое в соответствии с табл. 4.8 „ 4 Г#ц<4) АГ. (/£+1)1 Nv Г" X —L— — — - v этД2 |_ 2 г—1 2 £-1-1 J (Спектор, 1950 г. [23]); Nv(Dtf = ~ [3,4501 NL(lt)-2,1769 (Салтыков, 1968 г. [1, 2]) Nr(D.) = Kf NA &) ~ NA - - Коэффициенты приведены в табл. 4.9 ^(Пг)**=П7'1[1 ,6461 NA (d£) -0,4561 NA{d^-—0,1162 X Na (d^) - 0,0415 Na (d£-s) - - 0,0173 Na (d^) - 0,0079 Na (d^) - - 0,0038 Na (d^) — 0,0018 Na (d^7) — — 0,0010 NA (dj-g) — 0,0003 tfx(df-9) — - 0,0002 Na (d^) - 0,0002 NA (d£-u)J
* На плоскости наблюдения находят наибольшую хорду /гаа1. наибольший диаметр d^^ илн сечение наибольшей площади и полагают, что Zmax или ^юах =^гшах’ или amax="^max ’ исхо«я иа
этого производят деление на размерные классы.
** Формула реконструкции, основанной на измерении площадей сечений а., аналогична, т. е.
Ny (О.) = DT1 [1.646ШЛ (а.) - 0,4561 НА (с.^) - ... - 0,0002 NA (о.-^)].
6—281
81
« по формуле (2) последовательно вычисляется Nv(Dj) сначала для j=k, i=k из соотношения результат подстав-
ляют в аналогичное соотношение прн i—k—1; f=k, k—1 и т. д. Матричные методы требуют нахождения обратной матрицы р (}-=р~^ Матрицы рц для сферы и некоторых других тел, часто используемых для аппроксимации формы частиц, приведены в книге (3].
Метод последовательных подстановок весьма трудоемок и связан с катастрофическим накоплением ошибок прн малых i. Метод непосредственного дифференцирования в чистом виде (Каи и Фулмэн, 1956) практически неприменим из-за естественной дискретности первичных измерений. Вместо этого применяют графическое дифференцирование [4, с. 65], связанное с заметной погрешностью, или используют конечные разности (Спектор, 1950 [23];' Бокштигель, 1966 [24]). Основные методы реконструкции распределения размеров сферических частиц, применяемые в настоящее время, представлены в табл. 4.7.
Для нахождения распределения Nv(Di) поперечников Di кубических частиц С. А. Салтыковым (1968) предложена формула (табл. 4.8):
WV(D() =Dp [2,433ЛГл(<1г) — 0,97IWz X
>: (*4-1) - 0,270^ (О,-_2) - 0.097W, (ам) -— 0.04W, (а;_4) — О,О2977л (а,_5) — - 0.019^ (aM) - 0,012/7л (aw) --0,007ЛГл - 0,007/7, (0,_9)].
Первичные измерения — площади сечений Qi. Прн отыскании максимального сечения следует помнить, что оно должно иметь не только наибольшие размеры, но н форму -Прямоугольника с отношением сторон а!а\^2= 1/1,414; по отношению к нему и производят деление на
классы в соответствии с табл. 4.8. Для этого же сечения находят Стах=3/2-а.
Более сложные метода стереологнческой реконструкции размеров частиц другой формы рассмотрены в работах [3, 4, 25, 26].
Фольги. Для приближенного расчета распределения поперечников сферических частиц Nv(Di) иа основании данных о распределении проекций NA(di) используют формулу NA{di) ^tNv(Di=di), где f—толщина фольги, определяемая независимо.
Отсутствие учета экранировки и пересечения (см. п. 2.1) в последней формуле связано с погрешностью, величина которой тем больше, чем выше объемная доля частиц.
Экстракционные реплики. Задача стереоре-конструкции решена для частиц сферической формы. На плоскости наблюдения измеряют диаметры di и ареальные числа NA(di) изоб-рахсеннй частиц, рассчитывают средний диаметр d и дисперсию ag стандартными методами. Находят величины Nv и Г по формулам:
Nv=(NATd) [1 + (аа/3)!]; (3)
O = d/[l + (a2/d)2], (4)
где
i
Реконструкция распределения размеров частиц заключается в пересчете частостей FA(di) =NaJNa в частости Fv(Di)D = =NV /Nv по формуле Fv(Di) = (1Va/Nv)X
v I
x(dj)FA(di)&d, где Ad=&—dj-i— ширина класса размеров.
Монослой частиц (порошок). Для этого объекта специальная операция статистической реконструкции ие производится. Она ие требуется для сферических частиц, поскольку для них dt=Di, и при достаточном объеме выборки F(A}(di) ^-F(v)(Di).
Для частиц несферической формы часто ограничиваются измеренным на плоскости на-
ТАБЛИЦА 4.8
ЛОГАРИФМИЧЕСКОЕ ДЕЛЕНИЕ НА КЛАССЫ РАЗМЕРОВ И СООТВЕТСТВУЮЩЕЕ
РАСПРЕДЕЛЕНИЕ ХОРД, ДИАМЕТРОВ И ПЛОЩАДЕЙ СЕЧЕНИЙ ШАРА (ПО САЛТЫКОВУ [1])
Номер класса Размерные интервалы Относительные частости *. %
“V^niax- ^max oi^°max f f (d.) или f (а.)
1 1,0000—0,7943 1,0000—0,6310 36,904 60,749 (41,1)
2 0,7943—0,6310 0,6310—0,398! 23,285 16,833 (16,4)
3 0,6310—0,5012 0,3981—0,2512 14,692 8,952 (11,1)
4 0,5012—0,3981 0,2512—0,1585 9,270 5,200 (7,9)
5 0,3981—О,, 3162 0,1585—0,1000 5,849 3,134 (5,8)
6 0,3152—0,2512 0,1000—0,0631 3,690 1,926(4,4)
7 0,2512—0,1995 0,0631—0,0398 2,329 1,195(3,5)
8 0,1995—0,1585 0,0398—0,0251 1,469 0,747 (2,7)
9 0,1585—0,1259 0,0251—0,0158 0.927 0,469 (2,1)
10 0,1259—0,1000 0,0158—0,0100 0,585 0,2294 (1,7)
11 0,1000—0,0794 0,0100—0,0063 0,369 0,185
12 0,0794—0,0631 0,0063—0,0040 0,233 0,117
13 0,0631—0,0501 — 0,147 —
14 0,0501—0,0398 — 0,092 —
15 0,0398—0,0316 — 0,059 —
В скобках приведены относительные частости f{a}) для площадей сечений куба
ТАБЛИЦА 4.9
КОФФИЦИЕНТЫ а. В ТАБЛИЦЕ 4.7
Количество сечений зерен для групп 1—7
Количество зерен для групп 1—15 Na (1) ^A & (3) ^(4) na <5> HA (6)
Kv (1) +1.0000 —0,1547 —0,0360 —0,0130 —0,0061 —0,0033 —0,0020
Nv (2) +0,5774 —0,1529 —0,0420 —0,0171 —0,0087 —0,0051
(3) +0,4472 —0,1382 —0,0408 —0,0178 —0,0093
Nv (4) +0,3779 —0,1260 —0,0386 —0,0174
(5) +0,3333 —0,1161 —0,0366
(6) +0,3015 —0,1081
Nv (7) +0,2773
Xv (8) *
Xv (9)
^v(I0)
^(11)
^(12)
M13)
2Vv(14)
М/ (15)
Nv +1,0000 +0,4227 +0,2583 +0,1847 +0,1433 +0,1170 +0,0988
Количество сечений зерен для групп 8—15
Количество зерен для групп 1—15 NA(8) KA (9) NA (10) KA (И) HA (12) NA (13) Na (14) NA (15)
(1) —0,0013 —0,0009 —0,0006 —0,0005 —0,0004 —0,0003 —0,0002 —0,0001
Nv (2) —0,0031 —0,0021 —0,0015 —0,0010 —0,0009 —0,0006 —0,0006 —0,0004
(3) —0,0057 —0,0037 —0,0026 —0,0018 —0,0013 —0,0010 —0,0007 —0,0007
Nv (4) —0,0095 —0,0058 —0,0038 —0,0027 —0,0020 —0,0016 —0,0012 —0,0009
Nу (5) —0,0168 —0,0094 —0,0059 —0,0040 —0,0028 —0,0021 —0,0016 —0,0013
Nv (6) —0,0346 —0,0163 —0,0091 —0,0058 —0,0041 —0,0028 —0,0022 —0,0016
tfy (7) —0,1016 —0,0329 —0,0155 —0,0090 —0,0057 —0,0040 —0,0029 —0,0022
Nv (8) +0,2582 —0,0961 —0,0319 —0,0151 —0,0088 —0,0056 —0,0039 —0,0028
Nv (9) +0,2425 —0,0913 —0,0301 —0,0146 —0,0085 —0,0055 —0,0039
^v(10) +0,2294 —0,0872 —0,0290 —0,0140 —0,0083 —0,0054
tfp(H) +0,2182 —0,0836 —0,0280 —0,0136 —0,0080
(12) +0,2085 —0,0804 —0,0270 —0,0132
(13) +0,2000 —0,0776 —0,0261
^(14) +0,1925 —0,0750
7+(15) +0,1857
Nv +0,0856 +0,0753 +0,0672 +0,0610 +0,0553 +0,0511 +0,0472 +0,0441
блюдення распределением диаметров или площадей проекций в положении наибольшей устойчивости F{Ay(di) или Г(А](сц).
4.3.2. Средний размер Р,
число частиц в единице объема Nv
Случайные сечения. Средний размер (поперечник) частиц в системе D и полное число в
единице объема Nv связаны формулой Na~ =DNV.
Ареальное число сечений частиц Мл определяют на плоскости наблюдения. Величину D часто приближенно оценивают на основании замеров диаметров сечеинй di или случайных хорд li, отсекаемых границами сечеиий:
6®
83
"л
о=а=(л?х1)2<».лгл(й.);
х>=7=^2 /Лх<'Д
<—1
Обе оценки являются смещенными; для различных распределений сферических частиц D= 1,51-5-1,75/, причем оценка по I не требует приближения формы [3, 27].___
Для оценки R [где /?=(D/2)] по методу Фуллмэна [28] на плоскости наблюдения измеряют di и вычисляют d~l=N^1 ^Na (dj) dT1.
i
Ниже приведены расчетные формулы для мономорфных систем частиц различной формы:
для сферических частиц /Уу=(2/з1)ДО.д4-1; ^=n/4dr’;
для круглых дисков пренебрежимо малой толщины Z7v= (8/л2) Mid-1; R=3iftd~l.
Оценку средних размеров вытянутых зерен в текстурованном материале производят по методу ДеГоффа на нескольких сечениях [4, с, 142]. В случае линейной ориентации_(вы-
тяжка, волочение): jR=ji/4dy1;
Х(/% !NAs)-, где индексы «||» н «_1_» относятся соответственно к продольным и нормальным сечениям;
н L—средине значения радиуса н длины осн зериа — волокна.
В случае плоскостио-лннейной ориентации частицы считают трехосными эллипсоидами с постоянным отношением осей: Dj — в направлении прокатки; Du — в поперечном направлении в плоскости прокатки; Ьз— в высотном направлении перпендикулярно плоскости прокатки; трн сечения имеют соответственно индексы 12, 23 и 13. Измеряют ареальные числа изображений частиц на каждом из них: АГд, NA& н и d13J —среднюю обратную длину оси тех эллиптических сеченнй, которые вытянуты в направлении 3 в плоскости сечения 13. Расчетные формулы: Nv= (^^[N^J N А^) ^17 > £’i=(^a/^a)31^2^131’ D2=
D3=3\J2, d13 .
Способы нахождения средних поперечников и характерных размеров в системах частиц другой формы рассмотрены в работах [3, 4, 25, 29].
По методу «обратных диаметров» Салтыкова [1, 2] в системе сферических частиц могут быть определены Nv, D и дисперсия Nv— = (2/k)1F1-Na « 0,6366 d3 Wa; Г =3i/2 d3*» «1,5708/d-1;
o- = ]/’(4/3i)dD —(D)2.
По методу «укрупненных показателей» Салтыкова fl, 2] для системы сферических частиц, распределение размеров которых подчиняется логарифмически нормальному закону, находят Nv—Qti2(Na/2Pl)5Pp; D=NaINv-,
= |<2Р£/п^г —(D)2 ,
где первичные измерения: Na — ареальное число сечений частиц; PL — число точек пересечения контуров изображений частиц единицей длины случайной секцией; РР— точечная доля сечений частиц.
Методы расчета моментов /распределения Nv(D) — среднего размера, дисперсии, асимметрии без стереологической реконструкции—• с помощью графического дифференцирования кривой Nb[li) рассмотрены в работах [3; 25, с. 193—194; 30].
Фольга. Если пренебречь эффектами экранировки н пересечения (см. п. 2.1), то величины D н Nv можно определять непосредственными -Измерениями на плоскости наблюдения: D^d; Nv^NA]t.
Если же необходимо учитывать экранировку, то расчет следует вести по формуле
^=»W(< + 5); к'а=^а + ^а- (5)
где 2 —толщина фольгн (определяется независимо); МА — число экранированных изображений.
Один нз методов приближенного определения Ма — расчет по формуле, справедливой прн Vv<0,3; MA=3Vv(tID)NA/2, где Vv — объемная доля (методы определения см. в п. 7).
Эффект экранировки может быть учтен также определением Nv по эмпирической формуле Хиллиарда:
NA = Nv ехР (“ mNVi )-1 (6)
Для этого определяют Na к d прн нескольких толщинах фольги t и строят график 1п(Ма+М)—t. Отрезок, отсекаемый на ординате при 2 — 0, равен In Nv-
Экстракционная реплика. Для сферических частиц расчет Nv и D проводят по формулам (3) и (4).
Монослой частиц (порошок). Величина Nv для этого объекта не имеет смысла. Средний размер D находят усреднением dj_ по числу частиц D=dn, по их поверхности D—dg н по объему D=dy [12, 31]. Соответствующие расчетные формулы имеют внд: dn — ^dini,l^ ni‘,
i i L i
4.3.3. Погрешности стереологической реконструкции характеристик размеров частиц
Основные источники погрешностей: ошибки измерения (см. п. 2.2, 2.3); произвольное допущение о форме частиц; несовершенство метода стереологической реконструкции; недостаточное число первичных измерений; малое число размерных интервалов.
В настоящее время иет способов оценить погрешность, вносимую произвольным заданием единой формы частиц, для реальной ие-мономорфиой полндисперсной системы. Качественные оценки влияния сферического приближения в случае гипотетических матричных структур, содержащих частицы одинаковой, но не сферической формы, показали недопустн-
84
мость сферического приближения не только для явно иесферических частиц (кубов, треугольных призм), но н для таких частиц, форма которых отличается от сферической незначительно (сферы с фасетками, кубы со скругленными углами нт. п.).
Совершенство методов реконструкции оценивалось по отношению к модельным полиднс-персным системам сферических частиц для объектов — случайных сечений. Прн одинаковом типе первичных измерений наибольшую точность обеспечивают матричные методы, наименьшую — методы последовательных подстановок. Из первичных замеров наибольшую точность позволяет получить измерение площадей, наименьшую — измерение хорд (табл. 4.10).
ТАБЛИЦА 4.10
ЧИСЛО ПЕРВИЧНЫХ ИЗМЕРЕНИЙ,
НЕОБХОДИМОЕ ДЛЯ ПОЛУЧЕНИЯ ЗАДАННОЙ ТОЧНОСТИ РЕКОНСТРУКЦИИ
РАСПРЕДЕЛЕНИЯ РАЗМЕРОВ СФЕРИЧЕСКИХ ЧАСТИЦ
Ошибка, % Число измерений
хорд диаметров
1 42630 12490
2 10658 3235
5 1704 516
10 426 129
По данным Салтыкова [1], оптимальное число размерных интервалов 10—15; недопустимо использовать меньше пяти размерных интервалов.
4.4. ОПИСАНИЕ ФОРМЫ ЧАСТИЦ
4.4.1. Непосредственная реконструкция
Наиболее точный способ реконструкции формы частицы — построение ее пространственной модели; для этой цели существуют три принципиальные возможности:
— последовательные сечения для построения пространственных моделей частиц в однофазной и двухфазной структурах;
— последовательные сечения с целью получения обобщенной характеристики формы од-
Рис. 4.13. Различные случаи (а—в) прохождения четверной точки на последовательных сечениях однофазной структуры [3]
нофазной структуры в виде удельных чисел вершин (четверных точек), ребер (тройных стыков), граней н собственно зерен:?/®,
Wy, Ny (рис. 4.13 и 4.14).
4.4.2. Статистическая реконструкция
Если справедливо допущение о моиоыорфно-стн системы частиц, то по распределению форм нх сечений можно реконструировать истинную форму самих частиц. Распределение форм сечений некоторых правильных многогранников показано в табл. 4.11.
Реконструировать форму частиц в ввде эллипсоидов (трехосных и вращения) с постоянным отношением осей можно с помощью графиков на рис. 4.15 и 4Д6. Для этого на плоскости наблюдения необходимо измерить н классифицировать величины отношений осей нх эллиптических сечений.
Рис. <14. Выявление вершин (четверных точек), граней и ребер на последующих сечениях однофазной структуры [3]:
а — исследуемый участок структуры (зерно 5 в окружении зерен 1^1}; б — последовательные се-
— выделение частиц путем вытравливания матрицы [2, 3], «острого» травления [2] или «горячей» фильтрации [2] и их наблюдение в РЭМ с применением методики стереопар для измерений на индивидуальных частицах;
Эллипсоиды вращения, используемые для обобщенного описания формы частиц без граней, бывают двух типов — вытянутые и сплюснутые. Для определения типа эллипсоида необходимо сравнить длины главных осей иан-
85
ТАБЛИЦА 4.11
ЧАСТОТА ВСТРЕЧАЕМОСТИ (%) РАЗЛИЧНЫХ СЛУЧАЙНЫХ СЕЧЕНИЙ ПРАВИЛЬНЫХ
МНОГОГРАННИКОВ
1 Число сторон се-чеиия. Куб Тетраэдр Октаэдр Пентагон-додека-эдр** Ромбододекаэдр** Кубоктаэдр**
3 27,98 71,15 7 4,0 7,3
4 48,68* 28,85 44,8 14 13,4 13,4
5 18,70 —. — 16 16,2 11,8
6 4,64 — 55,2 24 29,9 31,2
7 — — 29 19,1 18,3
8 — — — 15 16,3 13,1
9 — — — 2 1.1 3,8
10 —’ — 1 — 1.1
* Имеются две группы четырехугольных сечений куба: трапеции и параллелограммы, которым соответствуют относительные частости 68,2 н 31,8 % в пределах общей относительной частости 48,68 % для четырехугольных сечений.
** Эти многогранники часто используют для описания формы частиц в равновесной однофазной структуре.
Рис. 4.16. Распределение случайных сечений трехосного эллипсоида с отношением осей 2 1,5 : 1 (У) и сходных трех- н двухосных эллипсоидов (2 — 2:1;
3 — 2 : 1,25 : 1; 4 — 1 : 1.75 : 2; 5 — 1 : 2)
Отношение осей эллиптических сечений
Рис 4Л5, Распределение случайных сечений трехосного эллипсоида с отношением осей 3:2:1 (/) и близких к нему эллипсоидов вращения (2—3: 1 + +2:1; 3 — 6: 1+3:1; 4 — 3: Г, 5 — 3: 1+1:3; 6 — 1 : 3)
тайные для некоторых простых тел, представлены в табл. 4.12.
ТАБЛИЦА 4.12
ФАКТОРЫ ФОРМЫ НЕКОТОРЫХ ТЕЛ (2]
Тело ЛФ1 ^фз
Шар .... Пентагондо- 1,000 0,0531 1,000
декаэдр . . . 0,955 0,0462 0,867
Кубоктаэдр . . 0,954 0,0473 0,842
Цилиндр . . . 0,935 0,0455 0,778
Октаэдр . . . 0,920 0,0462 0,695
Куб 0,896 0,0417 0,667
Тетраэдр . . . Пластины с со- 0,820 0,0359 0,448
отношением
сторон: 1:5:5. . . . 0,769 0,0281 0,390
1 : 10 : 10 . . . 0,659 0,0182 0,238
1 : 20 : 20 . . . 0,546 0,0106 0,133
1 : 50 : 50 . . . 0,414 0,0047 0,057
больших сечений: неравиоосного — эллипса н равноосного — круга. Если диаметр равен длине меньшей осн, то частицы — вытянутые эллипсоиды, а если он равен большей осн, то частицы — сплюснутые эллипсоиды.
4.4.3. Факторы формы Кф
Факторы формы — безразмерные величины, представляющие собой комбинацию размерных характеристик частицы, взятых в соответствующих степенях. К нх числу, в частности, относятся: Кф^г.гУ1'3 */^1/2; Кф^УЯ/£2-
•=PpNaI$P2l\ K<s>3=6V[SD, где V — объем тела; S— его поверхность; D — поперечник; коэффициенты в формулах связаны с нормировкой; для шара К<ы=Кфз=1-
Комбинированные факторы формы, рассчи-
Из рассмотренных факторов только Кф2 (ДеГофф [4, с. 135]) может быть определен путем статистической реконструкции измерений удельных величин на случайном сечении; Кф! н Кф2, как и многие другие факторы формы, можио определить только измерением на индивидуальных частицах, для чего требуется непосредственная реконструкция 5.
Другие факторы формы — отношения характерных размеров, например для вытянутых частиц-цилиндров [4, с. 144]. Кф.ч=/?/Г=
» где знакн _!_ и -|| относятся соответственно к ареальным числам изображений
1 Безразмерная комбинация линейных измерений
на изображениях индивидуальных частиц на плоско-
сти наблюдения может оказаться эквивалентной фак-
тору формы только для объектов, изображения час-
тиц которых представляют собой проекции, т. е. для фольги, экстракционной реплики и монослоя частиц
86
частиц на поперечном н продольном сечениях.
Адекватное опнсанне формы частиц с цн-.ливдрической симметрией _дает фактор (/?/£); в общем случае (/?/£) (/?/£); однако (R/L) не может быть определено на основании измерений удельных величии иа случайном сечении. Способ реконструкции отношения осей Ыа частиц в виде эллипсоидов вращения по отношениям осей b'ja' их изображений на плоскости наблюдения случайного сечения весьма трудоемок и требует применения ЭВМ [3, 33].
Для предельно обобщенного описания формы частиц полностью округлых (сферы, эллипсоиды) н ограненных, а также для выявления различных форм перехода между ними (кубы со скругленными углами, сфероиды с фасетками) предложен фактор формы [32]: КФ5=Р2РЯ7Р?Щ» гДе н PL- соответственно число пересечений единицей длины случайной секущёи прямолинейных границ изображений частиц и суммарное число пересечений со всеми границами этих изображений.
Точность определения' факторов формы Кф реальных систем частиц практически не может быть регламентирована, поскольку для количественного описания с помощью Лф чаще всего используется обобщенная произвольно заданная форма частицы. В рамках этих ограничений нормы точности нахождения определяются эмпирически как числа первичных измерений, необходимых для получения воспроизводимых результатов, а также для нахождения статистически значимых различий между структурными состояниями.
4.5. ХАРАКТЕРИСТИКИ
ПОВЕРХНОСТЕЙ РАЗДЕЛА
4.5.1. Протяженность (удельная поверхность) Sy
Сечения (шлифы, реплики). Для полностью изотропной структуры измерения проводят на
случайном сечении, а расчет Sv выполняют по формуле (Салтыков, 1945): SV=2PL (7), где Pl— числа точек пересечений единицы длины случайной секущей с границами изображений частиц на плоскости наблюдения случайного сечения.
Прн наличии выраженной текстуры измерения проводят для ориентированных сечений (табл. 4.13).
Расчет по формулам (8)—(10) —см. табл. 4.13 — является приближенным, поскольку предполагает, что реальная ориентированная структура представляет собой суперпозицию полностью ориентированной и полностью изотропией частей.
Полную удельную поверхность ориентированной системы, имеющей ось симметрии, можно определить по формуле (7), если на изображение на плоскости наблюдения наложить базовую систему в виде специального контура (Спектор, 1960), периметр которого равен учетверенной длине малой его осн. Координаты этого контура:
±х........
±У........
±х........
±У........
0,00 0,10 0,20
1,57 1,54 1,50
0,60 0,80 0,90
1,17 0,87 0,62
0,40 1,38
1,00 0,00
Малая ось контура должна быть параллельна осн симметрии.
Точность метода определения Sv с помощью случайных секущих зависит от соблюдения условий равной вероятности любой взаимной ориентировки секущих и системы поверхностей. В случае изотропной структуры это условие сводится к выбору достаточной протяженности одного случайного сечения, т. е. достаточно большого числа точек пересечения Р (табл. 4.14). Прн наличии ориентировки в структуре может возникнуть дополнительная погрешность из-за недостаточного числа сечений и систем секущих различной ориентировки.
ТАБЛИЦА 4.13
ОПРЕДЕЛЕНИЕ УДЕЛЬНОЙ ПРОТЯЖЕННОСТИ СИСТЕМ ОРИЕНТИРОВАННЫХ ПОВЕРХНОСТЕЙ (ПО САЛТЫКОВУ [1, 2])
Тил преимущественной ориентировки (текстуры) Ориентация сечения по отношению к элементам текстуры Ориентация системы секущих по отношению к элементам текстуры Первичные измерения Расчетная формула
оси плоскости осн плоскости
Линейная (волочение) ’'Параллельная То же — Параллельная Перпендикулярная — Аз Г" 1- i= = 0,429РД|| + +1,571 Р£± (8)
Плоскостная (прокатка) Плоскостно-линейная (прокатка) Параллельная То же Перпендикулярная То же Перпендикулярная То же Перпендикулярная То же Перпендикулярная То же Параллельная Перпендикулярная То же » Параллельная Перпендикулярная Параллельная Перпендикулярная Параллельная Перпендикулярная = Н = Н — -1 Щ >-4 ч щ Щ Щ а, а, а, а, а, а. ^ = *<+ + % (9) = 0,429Р£|| + +0,571 P£j + +PL± (10)
87
ТАБЛИЦА 4.14
МИНИМАЛЬНОЕ ЧИСЛО ТОЧЕК ПЕРЕСЕЧЕНИЯ Р ДЛЯ ПОЛУЧЕНИЯ ОТНОСИТЕЛЬНОЙ ОШИБКИ.
НЕ ПРЕВЫШАЮЩЕЙ Ео [2]
и н о сс Число точек пересечения Р при" доверительной вероятности
0,5 0,6 0,7 | 0,8 0.9 0,95
1 4543 7090 10754 16410 27060 38416
2 1136 1773 2689 4103 6765 9604
3 505 778 1195 1823 1007 4268
4 284 443 672 1026 1691 2401
5 182 284 430 656 1082 1537
6 126 197 299 456 752 1067
7 93 145 219 335 552 784
8 71 111 168 256 423 600
9 56 88 133 203 334 474
10 45 71 108 164 271 384
И 38 59 89 136 224 317
12 32 49 75 114 188 267
13 27 42 64 97 160 227
14 23 36 55 84 138 196
15 20 32 48 73 120 171
Фольга. Без учета эффектов экранировки н пересечения удельная поверхность может быть определена по формуле Sv=2PpJt, где — точечная доля проекций всех поверхностей раздела, находящихся в объеме фольги, на плоскость наблюдения.
Более строгое определение Sv следует проводить по формуле Sv~—41п[1—Рр— —2Vv]ft, где Vv — объемная доля частиц, удельная поверхность которых Sv измеряется; Vv измеряется независимо, как правило, на шлифе (реплике) того же материала.
Общая количественная оценка точности определения для фольги невозможна, так как требует знания истинных условий экранировки и пересечения. Нормы точности определяются эмпирически из условия получения воспроизводимых результатов. Приближенный учет экранировки для фольги рассмотрен в работе [8].
4.5.2. Кривизна
Согласно дифференциальной геометрии каждый элемент dS аналитической (без «особых» точек) поверхности характеризуется двумя главными значениями кривизны kt и k2, которые соответствуют двум кривым, образованным в результате сечения элемента aS ортогональными плоскостями, параллельными нормали к dS. Если векторы ki н kz направлены к центру кривизны, то элемент dS выпуклый; если векторы ki и k2 направлены от центра, то элемент вогнутый; если направление ki и k2 различно, то элемент седлообразный.
Существуют два параметра кривизны поверхностей— средняя (1/2) (kt+ks) н гауссова (Л1&2) кривизна. Стереологическимн характеристиками кривизны системы поверхностей являются величины Н и К, получаемые соответственно усреднением средней и гауссовой кривизны всех поверхностей системы.
4.5.2.1. Средняя кривизна Н
Случайное сечение (шлиф, реплика). Для определения величины /7 на плоскости наблюдения случайного сечения измеряют [2, 3, 34]: Та — ареальное число касаний сканирующей прямой с контурами изображений частиц; Pl~ число точек пересечения этих контуров с единицей длины случайной секущей. Расчетная формула
H = 2Ta№l. (11)
Прн подсчете ТА и Pl могут быть отдельно учтены участки контуров, выпуклых (4-) и вогнутых (—) по отношению к частицам дайной фазы (рис. 4.17), н соответственно по фор-
Рнс. 4.17. Схема измерения средней кривизны граничных поверхностей методом подсчета касаний при сканировании прямой А [2]
муле (II) рассчитаны значения кривизны выпуклых н вогнутых граничных поверхностей: H+~2T.a+/siPl+; Н-—2Та_/пРl_, г также суммарные значения кривизны с учетом и без учета знака Я=2(7'л+—Тд_)/(Р£+4-Рд_);
==27z++7a_)/(Pl++Pe_) .
Способ определения Н по формуле (11) является общим для структур любого типа; другие способы, пригодные для некоторых типов структур, рассмотрены в работах [3, 35].
Фольга. Для определения средней кривизны на плоскости наблюдения фольги измеряют [35]: Рь—'Число пересечений единицы длины случайной секущей с контурами изображений частиц; Рр — точечную долю частиц, кривизна поверхностей которых измеряется. На шлифе, реплике той же структуры измеряют объемную долю этих частиц Vv; независимо определяют толщину фольги t. Расчет ведут по формуле 7/=[jrPi./(Pjp—Vv)]—п/2С
В случае малых объемных долей Vv и «больших» толщин f(f^slOr) допустимо вычислять среднюю кривизну по приближенной формуле Д=пРь/Рр-
4.5.2.2. Средняя гауссова кривизна К
Определение К требует сочетания методов последовательных и случайных сечений [34]. На
88
последовательных сечениях определяют Tv— удельные числа касаний поверхностей частиц со сканирующей плоскостью. Первичные измерения иа плоскости наблюдния случайного сечения Ръ — числа пересечений контуров изображений частиц единицей длины случайной секущей. Расчет проводят по формуле К~ =пГ vjPc.
4.5.3. Двугранные углы 0
В стереологии двугранным называется угол 0 между граничными поврехностямн двух контактирующих зерен одной фазы (рис. 4.18).
В однофазной равновесной структуре 0=120°.
Для оценки дисперсии ое двугранных углов в однофазной_структуре относительно среднего значения 0=120° используют формулу [36] <т6 = 1,11 (<уф-485).
Для оценки норм точности определения 0 можно использовать соотношение между ошибкой е и объемом выборки N (число углов <р, измеренных на плоскости наблюдения с доверительной вероятностью 95 %, полученное в работе [37]):
W.......... 25 50 75 100 250 500 1000
е, % ... 40 30 23 20 12 9 6,2
Фольги и экстракционные реплики. Двугранные углы 0 можно1 определить непосредственна измерением на плоскости наблюдения, если выполнены следующие условия: 1) сфероидальная (линзообразная) частица находится на границе двух зерен матрицы; 2) частица имеет некогерентные границы с матрицей, т. е. контраст преимущественно абсорбционный; 3) исключены эффекты экранировки и пересечения; 4) плоскость границы зерен матричной фазы параллельна пучку электронов.
При соблюдении указанных условий истинное значение 0 можно определить, измерив диаметр D и толщину t линзообразной частицы [38]: tg(0/4) =f/D.
Четвертое условие легко выполняется для фольгн, поскольку возможен наклон ее по отношению к электронному пучку; в случае экстракционной реплики указанное требование выполняется не всегда и для точных расчетов используют методику, изложенную в работах [3, 38].
Рис. 4.19. Распределение углов <р на сечении для различных двугранных углов 6 [3]
Рис. 4.18. Сечения <р двугранных углов 6 [3] с—контакт двух зерен фазы «. погруженных в матрицу |5; 6— вторая фаза (матрица) на стыке трех зерен матрицы (фазы); в — вторая фаза (матрица) иа границе двух зерен матрицы (фазы)
Случайные сечения (шлифы, реплики). Для определения двугранных углов 0 иа основании измерений плоских углов <р между границами случайных сечений частиц используют методику Харкера и Паркера (1948) — сравнение расчетных гистограмм (рис. 4.19) с экспериментальными.
4.6. ХАРАКТЕРИСТИКИ ЛИНЕЙНЫХ ЭЛЕМЕНТОВ
4.6.1. Плотность Lv
Плотность (удельную протяженность) изотропно расположенных линейных элементов определяют на случайных сечениях и фольгах.
89
В первом случае используют формулу Lv— “ 2РА, где РА — ареальное число точек пересечения линейных элементов случайной плоскостью («тройных стыков», если линейные элементы ребра частиц; ямок травления, если дислокации).
В случае фольги определяют Pl—число точек пересечения единицы длины случайной секущей о проекциями элементов на плоскости наблюдения; расчеты ведут по формуле Lv—2PL/t, где t — толщина фольги (измеряется независимо).
Для оценки погрешности бэ, связанной с экранировкой при высоких плотностях Lv, используют формулу [39]: &Btt2atLv, где а— ширина изображения дислокации нлн другого линейного элемента.
Плотность Lv в системах линейных элементов с преимущественной ориентировкой определяют на ориентированных сечениях [1, 2].
В случае частичной линейной ориентации (нитевидные неметаллические включения, ребра полиэдров в однофазной структуре после волочения) jLv=Pa,||где РАц и Ра — соответственна ареальные числа точек пересечения линейных элементов плоскостями поперечного н продольного шлифов.
Для частично плоскостной ориентации (ребра полиэдров однофазной структуры после прокатки) Lv = 1 ,57РА ±4-0,43Ра ц .
Ареальную плотность линейных элементов, полностью расположенных в некоторой плоскости, параллельной плоскости сечения, находят по формуле Аа=(п/2)Рг, (12), где Pl— число точек пересечения единицы длины случайной секущей с линейными элементами.
4.6.2, Характеристики искривленности
Искривленность системы лнннй в пространстве (дислокаций, ребер ограненых частиц) характеризуется средней кривизной k н кручением t. Кривизна элемента кривой описывает искривленность в плоскости, содержащей касательный t и нормальный п векторы к элементу кривой; кручение 1^ элемента d7. характеризует поворот этой плоскости прн движении вдоль dL. Характеристики k н t—
результат усреднения величин н tk по всей длине линейных элементов структуры. Обе характеристики рассчитываются на основании первичных измерений иа плоскости наблюдения фольги [40]: k^siT aLZPl', t—nlA^Pi,, где Та — ареальное число' точек касания сканирующей прямой с проекциями линейных элементов на плоскости наблюдения; I а — ареальное число точек перегиба на проекциях линейных элементов; Pl — число точек пересечения единицы длины случайных секущих с проекциями линейных элементов.
4.7. ОБЪЕМНАЯ ДОЛЯ
Случайные сечения {шлиф, реплика). Принцип Кавальери-Акера-Глаголева, выражающийся соотношением Vv=Aa=:Ll—Pp (13), определяет соответственно планиметрический, линейный и точечный методы нахождения объемной доли Vv частиц даииой фазы иа случайном сеченни.
На точности определения объемной доли существенно сказываются систематические погрешности, связанные с препарированием, в частности образование рельефа высотой t, которое может быть учтено введением соответствующей поправки в формулу (13) [9]:
—Svt/4;, где Vv и Vv —соответственно «истинное» н найденное по формуле (13) значение объемной доли.
Систематическую ошибку, связанную с недостаточным разрешением («размытием») контуров изображения частиц, объемная доля которых определяется, находят из выражения ДУу/ Vv^&La/Aa, где 6 — минимальное разрешаемое расстояние; La — ареальная плотность границ изображений, вычисляемая по формуле (12).
Случайную абсолютную ошибку в определения Vv планиметрическим методом можно оценить по формуле [1]*
а точность линейного, н точечного методов — с помощью табл. 4.15.
1 В формуле: t — нормированное отклонение; N— число измеренных сечений; А — средняя площадь сечений; Од — дисперсия площадей сечений.
ТАБЛИЦА 4.15
МИНИМАЛЬНОЕ ЧИСЛО ТОЧЕК ИЛИ ОТРЕЗКОВ, НЕОБХОДИМОЕ ДЛЯ ПОЛУЧЕНИЯ ВЕРОЯТНОЙ АБСОЛЮТНОЙ ПОГРЕШНОСТИ, НЕ ПРЕВЫШАЮЩЕЙ 8,
ПРИ ТОЧЕЧНОМ И ЛИНЕЙНОМ АНАЛИЗАХ [21
Содержание фазы, %
Абсолк погреш е, % 1 99 2 98 3 97 4 96 5 95 10 90 15 85 20 80 25 75 30 70 35 65 40 60 45 55 50
о,1 4400 8700 12065 17055 21109 39996 56661 71104 83325 93324 101101 106656 109989 111111
0,2 -—. 2178 3233 4266 6277 10000 14165 17776 20831 23331 25275 26664 27497 27775
0,3 — 968 1438 1897 2347 4446 6299 7904 9263 10374 11239 И 856 12222 12350
0,4 — • 809 1068 1321 2502 3545 4448 5213 5838 6325 6672 6881 6950'
0,5 -—» — 518 684 846 1612 2270 2848 3338 3738 4050 4282 4406 4460
1 -—- 1 — —, — 400 567 711 833 933 1011 1067 1100 1111
2 — ь — —. — 142 178 208 233 253 267 275 278
3 — — — 79 93 104 112 119 122 124
4 — 58 63 67 60 70
9С
Фольга. Определение объемной доли Vv частиц данной фазы с помощью измерений на плоскости наблюдения возможно только в том случае, если в результате независимого определения известна удельная поверхность Sv этих частиц: VF=—1п(1—Аа)—Svt/4, причем измерение А а может быть заменено измерением Рр или
Экстракционная реплика. Для определения объемной доли Vv сферических частиц необходимо подсчитать на плоскости наблюдения ареальное число проекций частиц Na, определить средний нх размер d и рассчитать дисперсию 03-(см. п. 3.1). Расчет проводят по формуле 140] Vt-= (d24-o|)-
6 «
Для логарифмического деления на классы л — используют формулу Vv=ln —Л^а+21п<1₽+ _____________ о
Ч-21п2с~> , где dg—среднее геометрическое
значение диаметра проекций.
4.8. ХАРАКТЕРИСТИКИ
РАЗМЕЩЕНИЯ ЭЛЕМЕНТОВ
СТРУКТУРЫ В ПРОСТРАНСТВЕ
4.8.1. Ориентировка
Степень ориентировки различных элементов структуры можно оценить приближенно, исходя из представления о структуре как о
суперпозиции полностью ориентированной и полностью изотропной частей (табл. 4.16 н 4.17).
Степень ориентации граничных поверхностей, имеющих осевую симметрию, можно оценить с помощью среднего косинуса нормалей д
cos(/, п) (Спектор, 1955), т. е. средневзвешенного по всей площади граничных поверхностей косинуса угла между нормалями п к поверхностям н осью симметрии I текстуры. Эту величину определяют по формуле (1,2]
cos(/, n)=Pr/Sv, где Pl — число точек пересечения границ сечений с единицей длины системы прямых, параллельных оси симметрии; Sv — полная удельная протяженность граничных поверхностей (способы определения см. в п. 5).
Распределение ориентировок может быть представлено графически и аналитически. Графическое представление — построение «розы чисел пересечения» [1,2], т. е. пространственной фигуры в сферических координатах р, в, <р, где модуль раднуса-вектора |р| равен числу пересечений Pl следов границ единицей длины секущей в данном направлении; в — угол между системами секущих иа плоскости сечения, ориентированной под углом ср. Методика построения «розы чисел пересечения» подробно рассмотрена Салтыковым [1]. Аналитические методы требуют нахождения отио-
ТАБЛИЦА 4.16
ОПРЕДЕЛЕНИЕ СТЕПЕНИ ОРИЕНТИРОВКИ ГРАНИЧНЫХ ПОВЕРХНОСТЕЙ С ПОМОЩЬЮ
ОРИЕНТИРОВАННЫХ СЕЧЕНИИ (ПО САЛТЫКОВУ (1, 2])
Тип преимущественной ориентировки (текстуры) 1 Ориентация сечения по отношению к элементам текстуры Ориентация системы секущих по отношению к элементам текстуры <и ™ л S gs Расчетная формула
осн плоскости} оси плоскости £ ф й р. С s
Линейная (волочение) Плоскостная (прокатка) Парал- — Парал- — = н = о о о а, а, а. рч~рч
лельиая То же Перпендикуляр- лельная Перпендикулярная ] Параллельная 0,273PLjl+PLi а PL^
Плоскостнолинейная (прокатка) Параллельная ная То же » Параллельная Перпендикулярная Параллельная Pl\\ __ 1,571(7^ -%)
То же » Перпендикулярная Перпендикулярная рч ^RH 0,429Р£[| 4-0,571/^ -К£±
Г Перпендикулярная То же » » » Параллельная Перпендикулярная рч рг р^-рч
m 0,429₽£|i+0,571P£1+PL1
91
ТАБЛИЦА 4.17
ОПРЕДЕЛЕНИЕ СТЕПЕНИ ОРИЕНТИРОВКИ ЛИНЕЙНЫХ ЭЛЕМЕНТОВ С ПОМОЩЬЮ ОРИЕНТИРОВАННЫХ СЕЧЕНИИ (ПО САЛТЫКОВУ [4, 2])
Тип преимущественной ориентировки (текстуры) Ориентация сечения по отношению к элементам текстуры Первичные измерения Расчетная формула
оси плоскости
Линейная Параллельная '— % 1 1 -1 ‘хз
Перпендикулярная —
Плоскостная — Параллельная %
— Перпендикулярная % “ Pa±~^4PAii
снтельной плотности нормалей (Спектор, 1955) или применения Фурье-трансформации (Хиллиард, 1969); оин рассмотрены в книгах 11-3].
4.8.2. Расстояния между элементами структуры
Расстояния между частицами «второй» фазы могут быть определены двояко — между их центрами (межцентровые расстояния £ц) и между их граничными поверхностями, т. е. по матрице («свободные» расстояния £).
Общие формулы определения средних межцентровых и средних свободных L расстоянии: £ц=аА/'^‘1^3 (14); £=4(1—Vv)/Sv.
Определение £ нивариантио к форме частиц; для нахождения £ц требуется задание формы
и схемы размещения частиц в структуре (сх~ -1) [3].
В табл. 4.18 приведены первичные измерения и расчетные формулы определения межчастичных расстояний для различных объектов.
4.8.3. Связанность
Строгое количественное описание связанности частиц «второй» фазы (выделений, включений) в матричной структуре а+₽, не зависимое от формы и размеров этих частиц, получают с помощью топологических инвариант [3,41]: jV£°n— числа связанных наборов частиц в единице объема структуры; Gv—связности, т. е. удельного числа контактов (границ) частиц, разрыв которых еще не приводит к увеличению Связь между топологическими инвариантами дает теорема
ТАБЛИЦА 4.18
ОПРЕДЕЛЕНИЕ МЕЖЧАСТИЧНЫХ РАССТОЯНИЙ ДЛЯ РАЗЛИЧНЫХ ОБЪЕКТОВ
Объект Первичные измерения Расчетные формулы Оценка погрешности (коэффициент вариации)
Случайное сечение (шлнф, реплика) Экстракционная реплика Фольга Рр> Рц NA,~d. 0d Рр £=2(1— PpVPL рлГ , /05 VI)-1/3 М >+ — 1 d L d ) JJ _ 4 £« - t — толщина фольги (±1/3) IV; N — число замеров для определения Na± ± V Р; Р — число замеров для определения Р& (±,ФгЫМ] Nt и Лг2 — числа замеров для. определения d(o-) и Na Г 1 У, -!_1 ±[^1 (а 1+ n, ]; Ni и — те же числа Формула приближенная, справедлива прн весьма малых значениях Vv частиц (см. п. 7)
* Величину определяют нз общей формулы (14), найдя предварительно Ny по формуле (5) нлн (6).
92
Эйлера: Gv=MvKa—^„+^(15), где Nva и Nva<x — удельные чнсла частиц фазы а и контактов между ними сш.
В полностью разобщенной матричной структуре Мка=М™п, Gv—0; в полностью связанной структуре Л'р511 = 1, примером является однофазная полиэдрическая структура.
Нахождение величин NVa н NvKa с помощью статистической реконструкции требует введения приближения формы частиц, кроме того, реконструкция статистическими методами невозможна нн для Gv, нн для М™п-Поэтому первичные данные для решения уравнения (15) получают непосредственной реконструкцией с помощью последовательных сечений. В общем виде процедура сводится к построению для каждого сечення плоского графа, в котором вершины соответствуют частицам фазы а, а стороны — нх границам аа, н установлению связи между отдельными плоскими графами иа основании известных их свойств [3,42]. Число отдельных пространственных графов равно N™n, а величина Gv определяется уравнением (15).
Методика определения Gv н с помощью последовательных сечений с применением теории графов изложена в работах [3,4, 25].
Связанность может быть приближенно оценена статистическими методами как среднее число Мка^а контактов каждой нз частиц с соседними — координационное число. Расчетная формула для случайных сечений:
м =-------
t л2
/NA PL о + 2Р,
Na Р,
\ лсх / Чха
ТАБЛИЦА 4.19
КРИТЕРИИ КАЧЕСТВЕННОЙ ОЦЕНКИ СВЯЗАННОСТИ ЧАСТИЦ ФАЗЫ а В МАТРИЧНОЙ СТРУКТУРЕ а+0
Координационное число Характер структуры фазы
Полностью разобщенные частицы
Отдельные наборы (сростки) контактирующих частиц малой протяженности
^аа/а~^ »й 1,5<СМаауе. <2 Протяженные (от границы до границы образца) наборы частиц (цепочки)
Протяженные цепочки и разветвленные наборы частиц
Однократно связанная разветвленная структура (все частицы находятся в попарном контакте) Многократно связанная структура
где N Аа и —ареальные числа изображений частиц н их контактов (границ) иа плоскости наблюдения; -Рпаа н — чнсла точек пересечения единицы длины случайной секущей с изображениями межчастичных и межфазных границ. Формула выведена в допущении о сферической форме частиц и круговой форме нх контактов.
Вероятностные критерии оценки связанности по величине Мка;а приведены в табл. 4.19.
Степень развитости контакта в связанной структуре частиц оценивают величиной смежности С, которая определяется иа случайном сеченнн независимо от формы частиц: Ска—
+2Рц аа)«
где Рида и — числа точек пересечения единицей длины случайных секущих границ изображений частиц иа плоскости наблюдения случайного сечения.
4.9. ФРАКТОГРАФИЧЕСКИЕ
ХАРАКТЕРИСТИКИ
4.9.1. Поверхность разрушения
Изображение поверхности разрушения получают иа плоскости наблюдения растрового (собственно образец) нлн просвечивающего (реплика) электронного микроскопа. Для оценки ареальной доли разрушения данного вида (интер- или транскристаллнтного) 5яинтер’ Стране нлн ареальной доли фазы Ssa иа плоскости наблюдения измеряют соответствующие величины ареальных долей Л^интсг- Л^транс« поскольку согласно
работам Эль-Суданн [43] для излома SBi — ~аАг
Прн измерении Аа используют соотношение
4.9.2. След разрушения на случайном сечении
На следе трещины на шлифе или реплике измеряют величины ходов Li трещины по, данной структурной составляющей, например для двухфазной структуры сс+₽: La ; Laa\ Соответствующие характеристики — средние ходы L н линейные доли LL1 например,
^La (2 Лх.)/Чл.тр» i
где ^сл.тр ~ S ^ct. “Ь ^^аа. +
I i 1
В общем случае нз-за неслучайности сечения поверхностью излома Аа^
^Vva, т. е. ии след, ни поверхность разрушения нельзя использовать для определения объемной доли фазы в материале в целом.
93
БИБЛИОГРАФИЧЕСКИЙ СПИСОК
1. Салтыков С. А. Стереометрическая металлография. 3-е изд. М.: Металлургия. 1970. 376 с.
2. Салтыков С. А. Стереометрическая металлография. Стереологня металлических материалов. М.: Металлургия, 1976. 272 с.
3. Чернявский К. С. Стереологня в металловедении. М.: Металлургия, 1977. 280 с.
4. Quantitative Microscopy. N.-Y.: McCraw Hill, 1968. 422 p.
’5. Чернявский К. С., Головашкина Н. В. — Заводская лаборатория, 1972, Ne 4, с. 434— 439.
Б. Электронная микроскопия тонких кристаллов: Пер. с англ./Хирш П., Хови А., Николсон Р. и др. М: Мир, 1968. 574 с.
7. Утевский Л. М. Дифракционная электронная микроскопия в металловедении. М.: Металлургия, 1973. 584 с.
8. Hilliard J. Е. — Trans. А1МЕ, 1962, v. 224, р. 906—917.
9. Underwood Е. Е.—-J. Microscopy, 1972, v. 95, pt. 1, р. 25—44.
10. Mukherjee Т., Stumpf W. E., Sellars С. M. — J. Mater. Sci., 1968, v. 3, p. 127—135.
11. Stumpf W. E., Sellars С. M.— Metallography, 1988, v. I, p. 26—34.
12. Herdan G. Small Particle Statistics. L.: Butterworth, 1960. 418 p.
13. Коузов П. А. Основы анализа дисперсного состава промышленных пылей и измельченных материалов. М.: Химия, 1974. 280 с.
14. Чернявский К- С. — Заводская лаборатория, 1978, № 9, с. 1096—1104.
15. Кауе В. Н., Murphy К. — Powder Technology, 1970/71, v. 4, р. 203—213.
16. Beadle С. — Pract Metallography, 1970, v. 7. р. 249—256.
17. Cole М. — Microscope, 1971, v. 19, p. 87-ЮЗ.
18. Fisher C. — Pract. Metallography, 1969 v. 6, p. 659—672.
19. Serra J.—J. Microscopy, 1972, v. 95, pt. 1, p. 93—103.
20. Serra J. — В ки.: Quantitative Analysis of Microstructures in Materials Science, Biology and Medicine (Proceedings of the Symposium), Stuttgart: Riederer, 1978, p. 9—24.
21. Bockstiegel G.— Pract. Metallogr., 1972, v. 9, p. 329—341.
22. Myers E. J. — Proc. 1st Intern. Congress Stereology. Vienna: Springer, 1963, y. 15/1— 15/10; Stereology. N. Y. — Berlin: Springer, 1968, p. 187—188.
23. Спектор А. Г. — Заводская лаборатория, 1950, № 2, с. 173—181.
24. Bockstiegel G. — Z. Metallic, 1966. Bd 57, S. 647—656.
25. Stereology. N. Y.—L. — Berlin: Springer, 1968. 340 p.
26. DeHoff R. T. — Trans. AIME, 1962, v. 224, p. 474—481.
27. Thompson A. W. — Metallography, 1972, v. 5, p. 366—369.
28. Fullman R. L. — J. Metals, 1953, № 3, p. 447—462.
29. Quantitative Methoden der Morphometrie. Heidelberg: Springer, 1988. 340 p.
30. Hilliard J. E. — Trans. AIME, 1968, v. 242, p. 1373—1379.
31. Чернявский К С. — Заводская лаборатория, 1978, № 12, с. 1488—1491.
32. Warren R. — J. Inst. Metals, 1972, v. 100, p. 176—181.
33. Ondracek G., Schulz B. — Pract. Metallography, 1973, v. 10, p. 16—25; 67—74.
34. DeHoff R. 7. —Trans. AIME, 1967, v. 239, p. 617—621.
35. Cahn I. W. — Trans. AIME, 1967, v. 239, p. 610—616.
36. Дувалян A. B. — Заводская лаборатория, 1971, № 8, с. 939—941; 952—953.
37. Stickels C. A., Hucke E. E.—Trans. AIME, 1967, v. 230, p. 796—801.
38. Martin A. C., Sellars С. M. — Metallography, 1970, v. 3, p. 259—273.
39. Brandon G., Komem Y. — Metallography, 1970, v. 3, p. 111—126.
40. Ashby M. F., Ebeling R. — Trans. AIME, 1966, v. 236, p. 1396—1404.
41. Чернявский К. С. — Заводская лаборатория, 1971, № 8, с. 935—939.
42. О ре О. Теория графов; Пер. с англ. М.: Мир, 1968. 223 с.
43. El-Soudani S. М. — Proc. IVth Intern. Congress Stereology MBS Spec. Publ. № 431, 1976, p. 273—276; Metallography, 1974, v. 7, № 4, p. 271—311.
5
7 РЕНТГЕНОСТРУКТУРНЫЙ АНАЛИЗ
5.1. ЗАДАЧИ
РЕНТГЕНОСТРУКТУРНОГО
АНАЛИЗА В ИССЛЕДОВАНИЯХ
И КОНТРОЛЕ КАЧЕСТВА
МЕТАЛЛИЧЕСКИХ МАТЕРИАЛОВ
‘Основной задачей рентгеноструктурного анализа является определение кристаллической структуры вещества. К этой физической (нлн крнсталлохимической) задаче примыкает обширная область прикладного рентгеноструктурного анализа, связанная с материаловедением: идентификация вещества по его крис
таллоструктурным характеристикам, что очень важно для современных технических материалов, в частности для сложнокомпоиентных и многофазных металлических сплавов.
Прикладной рентгеноструктурный анализ включает определение разного рода нарушений кристаллической структуры в реальных веществах (дисперсности и блочного строения кристаллитов, дислокации, дефектов упаковки и пр.), а также анализ атомной структуры частично упорядоченных и некристаллических объектов (например, металлические стекла).
Имеется ряд особенностей кристаллической структуры (или ее нарушений), которые мо
94
гут быть использованы для разработки и контроля качества новых материалов с заданными механическими или физическими свойствами н для контроля условий работы материалов в конструкциях. К этим особенностям относятся фазовый состав сплавов, текстура, зональные напряжения, плотность дислокаций и др.
Методы рентгеиоструктуриого анализа используются для решения многих задач физики твердого тела. Анализ динамики решетки и энергетических характеристик взаимодействия компонентов сложных веществ и некоторые другие применения рентгеиоструктуриого анализа имеют значеине в физике твердого тела и для физического металловедения — в решении вопросов стабильности микроструктуры, возможности образования тех илн иных фаз и фазового равновесия.
Цели настоящей главы — дать представление о возможностях рентгеиоструктуриого анализа для металловедов, необходимые начальные знания основных идей, определения и термины, достаточные для чтения современной литературы по физическому металловедению, для выбора н дальнейшего изучения конкретных методик рентгеиоструктуриого анализа с использованием специальных руководств по реитгеноструктурному анализу (РСА)1.
Ниже приводится основная н специальная литература по РСА. Все разделы кристаллографии систематически и подробно изложены в учебнике [1]. Книга [2] отличается более формальным подходом, ее достоинство — использование специального математического аппарата. Фундаментальный характер имеет коллективный труд «Современная кристаллография» [3].
Наиболее полное изложение теории дифракционных методов анализа дано в книгах: Гинье[4], Г. С. Жданова [5], Джеймса [6], А. И. Китайгородского [7]. Отдельным вопросом теории посвящены книги Д. М. Васильева [8] (общее описание методов, геометрия дифракции), В. И. Ивероновой и Г. П. Ревке-вич [9] (теория рассеяния, интенсивность дифракции), Я. С. Уманского [10] (теория рассеяния, диффузное рассеяние).
Вопросы теории рентгеноструктурного аиа-
1 Для решения некоторых иа указанных выше задач используются также методы электронографии и нейтронографии, однако самым универсальным и массовым является метод рентгенографии.
лиза и применения рентгеиоструктуриого анализа рассматриваются в учебниках Б. Я. Пи-неса [11], Я- С. Уманского [12] и А. А. Русакова [13]- Практическим аспектам реитгеноструктурного анализа н лабораторной технике посвящены книги [14,15]. Обзор методов реитгеноструктурного анализа в применении к физическому металловедению содержится в коллективной монографии [16]. Справочный материал приведен в Приложении к книге [14], а также в специальных справочниках [17 — 21].
5.2. ПРИНЦИПЫ ДИФРАКЦИОННЫХ МЕТОДОВ АНАЛИЗА И АППАРАТУРА
5.2.1. Основные определения и формулы структурной кристаллографии.
Структура кристаллов
Пространственные решетки (ПР), или решетки Броеэ, — наиболее общий (абстрактный) образ внутреннего строения кристалла (рис. 5.1). ПР получаем, если исключим все особенности химической природы составляющих его частиц — форму, размер н состав молекул, атомов нлн ионов и вместо частиц будем рассматривать точки (узлы решетки) — центры тяжести частиц. По взаимному расположению узлов ПР все многообразие кристаллов сводится к 14 типам. ПР, или решетка Бравэ, характеризуется прежде всего группой трансляций (три) нли параллелепипедом повторяемости— элементарной ячейкой (ЭЯ) (см. рис.
5.1). Параллельным переносом (трансляцией) элементарной ячейки в трехмерном пространстве н строят ПР. Трансляция — одна нз операций симметрии, поэтому решетки Бравэ можно называть также трансляционными группами2. Симметрия относительного располо-
2 Здесь и далее понятие группы совпадает с математическим термином «группа»: множество объектов или совокупность элементов, удовлетворяющих определенным положениям математической теории групп [1—3]; в данном случае этими элементами являются элементь! симметрии. Математически строго выводятся в кристаллографии для трехмерного пространства 14 трансляционных групп, 32 точечные группы и 230 пространственных групп.
Рнс. 5.1. Пространственная решетка кристалла
ТАБЛИЦА 5.1
ОСНОВНЫЕ ЭЛЕМЕНТЫ СИММЕТРИИ И ИХ ОБОЗНАЧЕНИЯ
С Е 2 Наимено вания Обозначения
1 Плоскость симметрии (зеркальное отражение) кг или Р
2 Поворотные осн симметрии разного порядка 1, 2, 3, 4, 6 или Ln
3 Центр ниверсни (нлн центр симметрии) 1 или С
4 Инверсвонные оси разного порядка 2, 3, 4, 6 или £nf(T=C, W)
5 Трансляция t
6 Плоскости скользящего отражения а, Ъ, с, п, d
7 Винтовые осн симметрии разного порядка н смещений на разную долю /1 1 трансляции — t, \ X о 2 4 5 \ 3 6’6/ У & f CD W б, I-. ю w Г 1 ' в) 4» к> н.
Примечания: 1—4 — элементы симметрии, присущие как точечным, так и пространственным группам; 5—7—элементы симметрии, присущие пространственным группам.
ження узлов ПР проявляется в существовании поворотных осей симметрии и единичных (т. е, не повторяющихся в данной ПР) направлений. Симметрия расположения узлов ПР полностью сохраняется в симметрии той части ПР, которая называется элементарной
ТАБЛИЦА 5.2
ячейкой (см, рнс. 5.1). ЭЯ должна иметь вид параллелепипеда с максимальным числом прямых углов между ребрами н максимальное число равных ребер, при этом объем параллелепипеда должен быть минимальным. Ребра ЭЯ соответствуют трансляциям ПР или осевым единицам (а, Ь, с); значение трансляций по модулю называют периодами ПР (а, Ь, с)* Форма ЭЯ определяет кристаллографическую систему координат, для которой в качестве направления осей берут направления ребер ЭЯ с соответствующими осевыми единицами.
По форме ЭЯ н соответственно по совокупности элементов симметрии ПР делятся иа семь сингоний, или систем (рнс, 5.2, табл, 5,1 и 5.2), Эти сингонии в свою очередь подразделяются на три категории, различающиеся по числу единичных направлений: высшая (кубическая), средняя (гексагональная, тетрагональная, ромбоэдрическая), низшая (ромбическая, моноклинная, трнклиииая) сингонии. Из 14 решеток Бравэ семь простых (нли примитивных), т. е. таких, которые строятся осевыми трансляциями к узлам в вершинах параллелепипедов повторяемости, а семь сложных, т. е. таких, которые строятся трансляциями к точкам, находящнмси либо в центрах граней ЭЯ (базо- и гранецентрированные ячейки), либо в центре объема ЭЯ (объемноцентрированные ячейки, см. рнс. 5,2). Сложные ячейки характеризуются так называемым базисом. Базис представляет координаты минимального числа узлов, трансляцией которых строится пространственная решетка (табл, 5.3), В применении к кристаллическим структурам сложных веществ определение базиса включает координаты частнц с указанием нх химической природы. Целесообразно оставить понятия пространственная, решетка илн кристаллическая решетка за решетками Бравэ (абстрактный, математический образ кристалла), а для действительных струк-
® Термин «постоянная решетки» применять яе следует.
КРИСТАЛЛИЧЕСКИЕ СИСТЕМЫ* (ИЛИ СИНГОНИИ)
Сингония Соотношения между периодами Углы между координатными осями Определяющий элемент симметрии* Число единичных направлений
Кубическая а=р=у-90с 4X3 Нет
Гексагональная а=Ь^=с а=р—90°, у=120° 6 илн в 1
Тетрагональная а=Ь^-с а=₽=Т=90° 4 или 4 1
Ромбоэдрическая (илн трнго- а=Ь—с а=:|3=т^90о 3 илн 3 1
иальная)
Ромбическая (нлн орторомбн- а^Ъфс 3X2 нли 3
ческая) 3X2
Моноклинная а=т=90°; ₽^90° 2 илн 2 Множество
Триклинная а=рЬ^с <х=/=рт^‘у¥’90° 1 илн 1 Все
* Цифра, выделенная полужирным шрифтом, означает порядок поворотной или инверсионной оси симметрии, цифра перед знаком умножения — число осей симметрии; 1 — отсутствие элементов симметрии (ось первого порядка); 1 — центр инверсии.
96
Решетка Бравэ
оЗъенноценгнрироВинноя гранецентрированная (П
(F)
Сингония
примитивная
(Р)
базоцентрировинная ______ (О _____
Р2/ГП
С2/т
Fm3m
ТтЗт
РтЗт
Рис. 5.2. Трансляционные группы (четырнадцать) — решетки Бравэ
с Ь
Рттт
Сттт
Jmmm
Fromm
РЗт
Р4/ттт
1Щттт
Рб/ттт
7—280
97
ТАБЛИЦА U.S
ХАРАКТЕРИСТИКИ НЕКОТОРЫХ КРИСТАЛЛИЧЕСКИХ СТРУКТУР МЕТАЛЛОВ И ПОЛУПРОВОДНИКОВ
Тип решетки Число атомов на элементарную ячейку Базис решетки Координационное число Доэффи циент заполнения. %
Примитивная кубическая , . 1 [[000]J 6 52
Объемноцентрированная ку- ГГ 1 1 1 *п 68
бическая (о. ц, к.) Гранецентрированная кубиче- 2 [о°О; — —- — 11 LL 2 2 2 JJ ГГоАА 1 1 О 1 А 1 8
ская (г. ц. к.) 4 LL 2 2 2 2 о A All 2 2 JJ ГГ 1 2 1 11 12 74
Гексагональная компактная , 2 к - - —J] ГГЛ 1 I I,1 12 74
Кубическая тнпа алмаза . . . 8 [[«»; т Т 0; Т 0 Т: o-L А. А А А. 2 2 4 4 4 1 3 11 1 3 4 4 4’4 4 4 ’ 3 3 _3_Т1 4 4 4 JJ 4 38
тур (атомных, ионных, молекулярных) применять термин кристаллическая структура (КС).
Кристаллические структуры чистых металлов (а также многих металлических сплавов— твердых растворов) имеют атомный характер и узлы решетки Бравэ представляют центры атомов (точнее, положительных ионов) — частиц, имеющих сферическую симметрию. Исходя из принципа плотной шаровой упаковки, действующего в случае ионной н металлической химической связи, определяется атомный (металлический) радиус как половина расстояния между центрами соприкасающихся атомов (ионов) (табл. 5,4). Простой расчет позволяет оценить коэффициент заполнения, т, е. долю (в процентах) объема решетки кристалла, занятого атомами или ионами (см, табл. 5.3).
У металлов наиболее распространены решетки, отличающиеся высокой компактностью: /- н F-решетки кубической сингонии (о. ц. к. и г. ц. к,), а также гексагональная компактная решетки (гекс, к) — рис. 5.3. В табл, 5,3 наряду с кристаллическими структурами атомного характера, в которых каждому узлу решетки Бравэ соответствует один атом (нон), включены две кристаллические структуры — кубическая тнпа алмаза и гексагональная компактная, которые приводятся к решеткам Бравэ, если узлы взять в центрах тяжести пары соседних атомов.
Совокупность элементов симметрии континуума (пп. 1—4,' табл. 5.1), действующих на точку, определяет точечную группу нли дауэв-ский класс симметрии кристалла. Поскольку одна точка объекта при таких преобразованиях
симметрии остается неподвижной, точечные группы описывают симметрию тела конечного размера, например симметрию многогранников. Важным является применение точечных групп
Рис. 5 3. Плотная и плотнейшие упаковки атомов: а — двухслойная ABABA... и гекс. комп, ячейка; б — трехслойная АВСАВСА... и г. ц. к. — ячейка; в—> о. ц. к, — ячейка
98
66
OS S-SX S tS я о л> о о о и ы ц -О S сл л я tr м о я а с». » » 5 <о 2 Я Я Магний Литий Кремний Лантан Кобальт xxxssg> W fi> Ci-rt X P Z Я 2 ja s О Я »g Я 5»3 S “*“* Железо "I’-inCJ® 0> Ф Si о я 8--Q ы и n = 2 b ? g x » s д=я я-a ч Я t> => я Ванадий Бор Алюминий Барий Бериллий Элемент
wZ 2?О □ о* "О cf з £ era И IS 9 5Й С*) ьч 0» pL-i *•* с 71 п> 03 < — < И g’S’b M Символ 1
25 29 42 25 41 50 to w at •—* to ND »—-4 .₽»-U о со 00 -J СО СО О1 -j CJ GO -"J 03 ю w CI 13 56 4 co Атомный номер
54,93 63,.54 95,95 58,59 92,91 118,70 24,32 I 6,94 28.06 138,92 58,94 197,2 114,76 192,2 112,41 39,096 40,08 55,85 209,00 183,92 59,72 72,60 178,6 СП О Oi 10,82 26.97 137.36 9,013 A. Атомный вес
Я -й-ззЯ г 1 1 ш Т» * О>-й"5> 1 a-* 5 9 X 4 4 111114 -d 7Э o> я _4 1 1 1 1 1 1 Я I 1 I фазы П penpal
51 111 als 1 £ 1 I § 4>-cj н- 1811 1 И ё W § I1 I I 1 1 1 1 1 О О Кения
1260 1083 2620 1455 1950 231,89 at s 1412 826 8 1163 155 2350 320,9 62,3 810 1535 271,3 3370 29,75 958,5 2130 1710 659,7 850 1350 Температура плавления, °C
t»a i -csp i i os30 я । тоятаа [ ^я-е xs> » И 1 1 I » И 1 H 1 'З^о^г.а 1 1 I Go Сим. вол
J*>>» >>>>>> CO bJ >>>»>>>>> И W r“ to »> Wt>3w О структурный тип О
ihiW4’ r> *t> ‘ТЭ-м’ТЗ --.“a -3? S’ °* ? °* a S' gs§3a§ssa r> Ci S Ta cn S’ •ч ь. “nJ* "чЭ Э i? !г> 3? -г, й* > щ з 3“>ч 3 5 9 Э й 3 iS" СО 2 2, a to 1 ЕтЗт Jni'im PG/mmc O пространственная группа руктура
QI o> 03 ND CO CO W W w o> 00 От ^"Ьостэ co w<o 3,94 4,39 2,514 3,554 5,4307 3,754 5,296 3,086 3,6093 3,2084 4,745 3,1652 3,526 5,6576 3,1952 3,50 2,8664 3,656 3,571 2,93 4,0786 4,585 3,8312 2,97311 5,344 5,560 5,057 3,0399 8,75 10,12 10.12 4,0494 5,009 2,283 - а
11 II 1 1 1 1 II 1 1 1 1 Illi 1 1 1 1 1 1 1 1 1 1 1 1 1 4,520 1 I 1 1 1 1 I s <а- Териоды
£i i-Si 11 j 111 nd 5,2103 6,823 6,063 at o 3» 1i|i€i1111| bi£l 1 § § 1 1 л ^cn co 'Eg §1 । w Г» >о
111111111 11 1 1 1 Illi 11 1 111111111111111S . 88. 1 ot5 1 1 *1 w I 1 Осевой угол
крин, » » 430 18 22 25 Комн. 18 0 25 -196 18 18 f- s ao v S oo 1-» о *-* юО tO‘-*totc}^toaitoCoto»-»k-*N0K> 02 at оз to сп tp о о <=» >z? <=> оэ си aio И СП О cnaoSSS coqo ai СП 3 tr -э л> CD &* Й S г~1 Я ТЗ я tJg »с» s *о * СО
2 58 20 4 2 4 2 4 2 2 8 4 Л, Ю М Л 4* 1О 1 to to to co оо ta to ьо 3 о Число узлов в ячейке
ай Шз11 *Ь 8 1,55 1,34 1,87 1—• LO Qi 1,44 1,66 1,35 1.56 2,36 1,97 1,26 Wass 1,34 0,91 1,43 2,21 1,13 Атомный радиус для К. Ч.=42*5
ТАБЛИЦА 5.4 ___ ______ СТРУКТУРА МЕТАЛЛИЧЕСКИХ И НЕКОТОРЫХ НЕМЕТАЛЛИЧЕСКИХ ЭЛЕМЕНТОВ
О , Продолжение табл. 54
° 1 1 2 3 1 4 1 5 1 « 1 8 1 9 10 1 11 12 13 14 15 16 17
Осмий Палладий Os Pd 76 46 190,2 106,7 — — 2700 1553 АЗ Al Pv/mmc FrnZm 2,734 3,8902 — 4,320 — 18 18 2 4 1,35 1,37 1,38 1,82
Платина Pt 78 195,23 a* 1773,5 Al 3,9231 22
Празеодим Pr 59 140,23 a; 0 792 940 a АЗ Pfymmc 3,672 — 11,835 18 4
Рений Родий Re Rh 75 45 186,31 102,91 3000 1985 £ Al АЗ Al Fm3m Рб/mmc Ftn3m 5,151 2,7609 3.8034 111 4,4583 —♦< Комн 20 18 4 2 4 1,37 1,34
Ртуть • Рубидий Hg Rb 80 37 200,61 86,46 -9 — —38,87 38,70 A10 A2 R3tn Im3m 2.9925 5,710 — — 70°44' -46 22 1 9 1,50 2,48 1,34 1,75 1.6
Рутений Свинец Ru Pb 44 82 101,7 207,21 —• — 2450 327,4 A3 Al Ро/тгпл Fm'Atn 2.706 4,9503 — 4,282 — Комн, 18 2 4
Селен Se 34 78,96 Vi a*1 217 V A8 P321 4,3640 wvw 4,9594 22 3
32,056 a Afe P\/n 9,05 9,07 11,61 90°46' Комн 26
Сера S 16 a, 0*‘ —* 119 a. A17 Fdda 10,44 12,84 24.37 Комн 128
Серебро Ag 47 107,88 960,5 ’ £ Al Fm'irn 10,92 4,0662 11,04 9,27 93e4' 103 18 48 4 1,44 2,15
Стронций Sr 38 87,63 a-+0 215 770 a Al Fm3tn 6,085 b - Комн 4
0*7 605 1 P A3 PG/min 4,32 7,06 248 2
Сурьма Таллпй 3b T1 51 81 121,76 204,39 a-ff? 231 630,5 303,5 V a A2 A7 A3 lm3tn R3m Pfi/mmo 4,85 4,505 3,4564 111 5,531 57°7' —I 614 „ 22 18 2 2 2 1,61 1.71
Тантал Теллур Титан Ta Те Ti 73 52 22 180,95 127,61 47,90 822,5 2996 450 1668 £ a A2 A2 A8 A3 IrnUm !m3m P321 Pfymmt 3,882 3,3058 4,5559 2.950 Illi 5,9268 4,686 Illi 262 18 18 18 2 2 3 2 1,46 1,70 1.46
Торий Th 90 232,05 ct->-0 1425 1845 P Ct A2 Al lm3m Fm3m 3,605 5,0843 — — — 900 22 2 4 1.80
Углерод C 6 12,01 o «V» 5000 P A2, A9*3 Ini'Atn P321 4.12 2,4614 — 6,7014 1400 18 2 4 0,77
Д4*з Fd3m 3,568 — 18 8
a*4 tn R3m 2,461 — 10,064 — 18 G
Уран U 92 238,07 a->P 660 1133 a A20 Cmcm 2,656 5,877 4,955 18 4 1,53
₽*? 775 •— P Ab P4nm 10.758 w 5,656 700 30
Фосфор P 15 30,98 a, U*’ 44,1 V a A2 A16 /m3n Cm~a 3,49 3,32 4^39 10,52 Комн. 2 8 1,3
Хром Cr 24 52,01 a, p, v*1 1845 3 a 42 /1432 /тЗт 11,31 2,884 — — Комн. 18 66 2 1,27
P 43 Рб/ттс 2,717 4,418 18 2
Цезий Церий Cs Ce 55 53 132,91 140,13 700 28,5 804 V a 412 A2 Al /43m Zm3m Fm3m 8,717 6,067 5,143 1 1 1 — Комн. 18 22 58 2 4 2,68 1,83
Цинк Цирконий Zn Zr '30 40 66,38 91.22 a-*0 862 419,49 1845 0 Ct 43 43 43 Po/mmc P6/mmc Ptymnu 3,65 2,656 3,232 — 5,96 4,947 5,147 — Комн, 22 ' 18 2 2 2 1,39 1,60
6 A2 fm3rn 3.62 867 2
примечание. Периоды решеток даны с точностью до единицы в последнем знаке.
Метастабильная фаза.
*5 а-графит,
*3 Алмаз.
. *4 Д-графит.
ния меж™Мпен^ для координационного числа (K.4J-12 приведены по данным 13, т. 2, с. 70]. При понижении К.Ч. атомные радиусы, как расстоя-
ния между центрами соседних атомов в плотной упаковке, уменьшаются: для К,Ч-18 на 2%, для К. ч.«6 на 4 %, для К. Ч.-4 на 12 %.
для описания огранки кристаллов, а также симметрии физических свойств кристаллов. Всего имеются 32 кристаллографические точечные группы, которые также, как и трансляционные группы, могут быть разбиты иа семь сингоний *.
Следующим приближением к описанию внутренней структуры кристаллов являются пространственные (или федоровские) группы — совокупности элементов симметрии, действующих на систему трансляций или на ячейку Бравэ (элементы симметрии дисконтинуума— пп. 5—7 в табл. 5.1). Всего получается 230 пространственных групп (Пр. гр.). Символ Пр. гр. включает символ ячейки Бравэ н далее символы осей симметрии или нормалей к плоскостям симметрии вдоль так называемых главных трансляционных направлений, которые для разных сингоний выбираются по-разному, а именно: для кубической: [001], [111], [ПО]; для гексагональной, тетрагональной, ромбоэдрической: [OOlf, [010], [ПО]; для ромбической [001], [010], [100]; для моноклинной [010]; в триклиииой сингонии нет осей симметрии нлн плоскостей симметрии.
Действие совокупности элементов симметрии на вектор трансляции зависит от положения точки — конца этого вектора по отношению к этим элементам — н определяет кратность точек разного положения. С получаемыми таким образом правильными системами точек связаны координаты базиса для разных сортов атомов так, чтобы кратность точек соответствовала стехиометрическим соотношениям для вещества. Рентгеноструктурный анализ в общем случае позволяет установить так называемую рентгеновскую илн дифракционную группу— симметрию обратной решетки кристалла, которая отличается от симметрии кристалла (Пр. гр.) тем, что всегда содержит центр инверсии.
Кристаллические структуры металлических элементов были указаны в табл. 5.4, структуры некоторых фаз, встречающихся в сталях, указаны в табл. 5.5 и 5.6 *. Подробное описание
1 В двумерном пространстве (для плоскости) получается 10 точечных групп. Эти точечные группы описывают возможную симметрию дифракционных картин — рентгенограмм и электронограмм.
♦ В табл. 5.4—5.6. а также ниже в табл. 5.18 даны обозначения структурных типов, принятые в справочном издании Strukturberichte (Ewald Р. Р., Herman С. Strukturbericht der Zeltschrift fCr Kristafiograp-hy, Bd 1—7, Leipzig, 1031—1943).
В основе обозначения — химический состав: для обозначения структур простых веществ (элементов) используют букву A (Al — г, ц. к., А2 —о. ц. к., АЗ — г. п.« А4—кубическая типа алмаза и т. д.), для обозначения бинарных соединений равноатомного состава ЛУ — букву В, для соединений ХУ, — букву С, для 'хп^пг ~ букву Ддя Фаз металлических сплавов в свое время было введено обозначение L (от немецкого Legierungen); цифры не имеют специального содержания и отражают хронологическую последовательность учета данного структурного типа; дополнительная буква в индексе первоначально носила характер временного обозначения.
Периоды решетки, межплоскостные расстояния, атомные и ионные радиусы, а также длины волн рентгеновского излучения в структурном анализе обычно измеряют в ангстремах (внесистемная специальная единица, временно разрешенная к исполь-
О _____о
зованию; I А=10 см) или в нанометрах (I нм— =s=l0-9 см). Ранее иногда применяли еще одну специальную единицу: Х-единнцу, нли килоикс (1 кХ — о
=1,00202 А); эта единица была введена после того, как была замечена ошибка в исходных данных, использованных при первых определениях длин волн.
ТАБЛИЦА 5.5
КРИСТАЛЛИЧЕСКИЕ СТРУКТУРЫ НЕКОТОРЫХ КАРБИДНЫХ И НИТРИДНЫХ ФАЗ
Структурный тип В1 (г. ц.к.) и L\
Фаза 0 at A Фаза 0 0, A
TiC 4,328 TiN 4,244
ZrC 4,698 ZrN 4,577
VC 4,165 VN 4,139
NbC 4,471 6-NbN 4,392
TaC 4,455 TaN 4,149
CrN 4,149 MosN 4,169
4,27 WSN Fe4N 4.126 3,801
Структурный тип Bh (примитивная гексагональная ячейка)
Фаза а, А О с, А
y-MoC 2,90 2,81
wc 2,906 2,839
MoN 2,86 2,80
Структурный тип Bi (гексагональная ячейка)
Фаза О а, А ю с, А
у'-МоС 2,93 10,97
e-NbN 2,96 11,27
Структурный тип U3 (плотноупакованная гексагональная)
Фаза 0 a. A i‘ 0 c, A
Мо2С 3,003 4,729
Та2С 3,106 1,945
Nb2C 3,127 4,972
w2c 2,990 4,690
Cr2N 2,760 4,479
e-FesN (Fe2N) 2,764 4,420
e-FexC 2,720 4,320
Структура с ромбическими искажениями, обусловленными упорядочением
Фаза 0 a, A b, A 0 G A
Cr2N 2,840 4,63 4,33
C-FeEN 5,511 4,83 4,425
Структурный тип D84 (кубическая сложная)
Фаза СГдзСб
a, A 10,680 10,586
Продолжение табл. 5.5
Структурный тип DOп (ромбическая, Пр. гр. Pbnm)
Фаза О а, к О А с, А
FesC—цементит 4,524 5,088 6,794
MnsC 4,530 5,080 6,772
Структурный тип С7Сз, Пр. гр. C6/mmc (сложная гексагональная)
Фаза О a, A о с, К
Сг9Сз 14,010 4,532
Мп3Са 13,838 4,539
Структурный тип Е93, Пр. гр. Fd3m
(тип «TizNi» или «карбид быстрорежущей
стали» или ^-карбид), кубическая сложная
Mo3FesC к a p б н д ы WgCofiC до Nb3CrsC
MofiNisC W8Co4C WeCoeC Ta2 (V, Fe)3 C
WsFesC до W8Fe4C Mo4Fe2C Мо4СогС Mo4NisC W^CoaC Пг-к a p б н д ы СП, (Ti, (Ti, (V, (Cr, Co)g C Ta3 (Cr, Cu)3C Nb)4 NiaC Ta) Co2C Ta)4 №2C W)4 Ni?C
этих структур н условий образования соответствующих фаз имеется в работах [20—29].
Кристаллографические индексы направлений (и плоскостей) определяют положение в пространстве кристалла семейства параллельных прямых (н плоскостей), проходящих через узлы ПР. Основной характеристикой кристаллографического направления является период идентичности (/w,c>w) — расстояние между соседними узлами; основная характеристика кристаллографической плоскости (точнее, семейства плоскостей) — межплоскостное расстояние (dhhi).
Символ направления [wvw] включает три взаимно простых целых чнсла, пропорциональных координатам любого узла, который лежит на узловой прямой, проходящей через начало координат (рис. 5.4).
Символ плоскости (hkl) включает три взаимно простых целых чнсла, обратных отрезкам, отсекаемым на координатных осях плоскостью, ближайшей к началу координат, н измеренным в долях осевых единиц (илн обратно пропорциональных отрезкам, отсекаемым любой плоскостью данного семейства) (рис. 5.5, а). В гексагональной сннгонн для обозначения узловых плоскостей часто пользуются координатной системой нз четырех осей (рис. 5.5,6), при этом кристаллографически идентичные
плоскости имеют численно одинаковые индексы (hkil) с разными знаками н в разной последовательности в зависимости в зависимости от ориентировки плоскости. Независимые три индекса; h, k, /; i==—(ft+fc)-
Рис. 6.4. Некоторые важные направления и нх кристаллографические индексы (в двойных квадратных скобках записаны координаты точек, не совпадающих с узлами решетки)
Рис. 5.5. Некоторые важные плоскости и их кристаллографические индексы:
а—индексы Мнллеоа; б — четырехосная система координат и индицированные в гексагональной решетке (индексы Миллера — Бравэ)
Семейства плоскостей (ft|Mi), (hik^ нт.д., имеющие общее направление [nvw], образуют
102
ТАБЛИЦА s.e
КРИСТАЛЛИЧЕСКИЕ СТРУКТУРЫ НЕКОТОРЫХ ФАЗ СИСТЕМ МЕТАЛЛ—МЕТАЛЛ
А. Фазы с координацией атомов по типу плотнейшей упаковки на основе структурного типа Си (А1)
Тип АиСиз. Пр. гр. РтЗт, структурный тип Ll%
Фаза О а, А Фаза О af А
AlNi3 3,567 AlZr3 4,372
FeNi3 3,552 FePts 3,88
NisSi 3,60 Fe3Pt 3,75
Тип АиСи. Пр-гр. P4[mmm, структурный тип Lio
Фаза С a, A О с, A
A1TI 3,99 4,07
FePd 3,85 3,72
MnNi 3,74 3,52
Тип TiAl3. Пр. гр. 14/mmm, структурный тип DOzz
Фаза -© a, A О b. A c/a
TiAU 3,85 8,60 2,234
VAla 3,780 8,322 2,202
TaAI3 3,842 8,553 2,226
VNi3 3,542 7,173 2,036
Б. Фазы с координацией атомов плотнейшей упаковки на основе структурного типа Mg(A3)
Тип MgCds. Пр. гр. Рб^ттс структурный тип DO^, гексагональная - структура
с последовательностью слоев АВ...плотнейшей упаковки
фаза D a. A О G A 2 c/a
MoCos 5,12 4,12 1,61
WCo3 5,12 4,12 1,61
AiTig 5,77 4,62 1,602
Тип TiNis. Пр. гр. Рб^ттс, структурный тип DOzs., последовательность слоев ABAC...
Фаза о a. A О c, A c/a
TiNig 5,109 8,319 1,628
Продолжение табл. 5.6
Тип РиА1&. Пр.гр. Р6т2, гексагональная структура с последовательностью слоев АВСАСВ...
Фаза О д, A © A О £•< A
VCo3 5,032 12,27 2,438
(или в гексагональной сингонии 5,032, с/с= 1,675
Тип TiCua. Пр. гр. Pmmn, структурный тип DOa
Фаза О а» A Ь, A О с, А
NbNis 5,116 4,260 4,565
MoNi, 5,064 4,224 4,448
TaNi3 5,114 4,250 4,542
Ta12, Б^i 76 5,070 4,228 4,460
В. Фазы с координацией атомов по типу о. ц. к. структурного типа W (А2)
Тип ^-латуни (или CsCl). Пр.гр. РтЗт, структурный тип В2
Фаза О a, A | фаза О a, A
FeA! 2,909 CuBe 2,707
FeCo 2,857 NiAl 2,887
FeTi 2,976 NiBe 2,621
FeV 2,910 1 NiTi 3,015
CoAl 2,870 RuHf 3,225
CoTi 2,991 RuTi 3,070
Тип ^-латуни в тройных системах металлов переходных групп; пример: Ti&o (Ре, Х)^
Состав, % (ат.) О О Э лектронная
Ti | Re | X a, A c, A кон центраци я (s-f-d). эл/ат
х~Os
50 50 — 3,118 — - - 5,5
50 41 9 3,108 — 6,0—6,1
50 25 25 3,092 —. 6,0—6,1
X=Ir
50 40 10 3,105 6,0—6,1
60 20 30 3,096 6,0-6,1
50 15 35 3,048 3,190 6,1
60 5 45 2,983 3,317
50 — 50 2,925 3,446 6,5
XsdPt
60 40 10 3,106
50 30 20 3,106 — ~6,2
* Dwight A. E., Beck P. A. — Trans. Metallurg.
Sos. AIME. 1S69, 245, р. 389—390.
103
Продолжение табл. 5.6
Tun FesAl (или BiF2). Пр. ep.FmSm, структурный тип D0%
Продолжение табл. 6.6
Фазы типа Ti2Ni. Пр.гр. Fd3m, структурный тип Е93
Фаза.......... FesAl FeaSi Cu3AI
а, А.......... 5,78 5,64 5,84
Г. Фазы на основе плотнейших упаковок с многократным замещением атомов (при существенном различии атомных радиусов)
Фазы Лавеса типа MgZn2. Пр.гр. P62fmmm, структурный тип С14
Фаза О a. A О c. A c/a
FeBe2 4,212 6,853 1,626
TiCrB 5,108 8,232 1,611
NbCr2 4,93 8,12 1,65
4,925 8,062 1,637
NbMn2 4,879 7,902 1,620
TaMn2 4,884 7,947 1,634
TiFe2 4,779 7,761 1,624
NbFe2 4,830 7,882 1,632
TaFea 4,827 7,838 1,624
Mol;e2 4,73 7,72 1,632
WFe2 4,744 7,720 1,627
NbCog 4,834 7,853 1,624
(или NbCo18) TaCo2 4,794 7,827 1,632
Nb (Cr, Si)s 4,898— 7,972— 1,626—
4,925 8,068 1,638
Mo (Co, Si)E 4,70 7,67 1,63
Mo (Ni, Si)E 4,70 7,66 1,63
KNa„ 7,495 12,295 -—
VBe2 4,394 7,144 —
Фазы Лавеса типа MgCu2. Пр.гр. Fd3m, структурный тип С15
Фаза О c, A Фаза О a, A
HfV, 7,386 1 TaCr2 6,961
TiBe, 6,448 TaFeNi 6,717
TiCr2 6,943 Muj 0Nii egSi0 66 6,686
ZrCr2 7,207 ZrS0MoBNie6 6,906
bibCr2 6,990
Фазы Лавеса типа MgNi2. Пр.гр. Рб^ттс, структурный тип С36
Фаза a, A © c, A c/a
HfCr2 5,057 16,34 - -
NbCOs 4,738 15,46 3,262
Ta^Cog 2 4,72 15,39 —
HfFeP 4,968 16,167 —
фаза © аУ А Фаза О а> А
Ti2Co 11,30 Ti4Fe2O** 11,28
Ti2Ni 11,33 Ti4Cu2O** 11,47
^Г0,70^О0,15^0,1б 12,34 TisCu 11,24
* Сплав гетерогенный. ** Фазы, стабилизированные кислородом; см. так-
же табл. 5.5 (карбиды — тип ч или 41)-
Д. Сигма-фазы и другие родственные фазы, образованные переходными элементами (или с участием переходных элементов) при близких атомных радиусах
и-фаза. Тип (CrFe). Пр. гр. P4zlmmm, структурный тип В8ъ
Фаза О а, А О С, А
CrFe 8,799 4,544
Р-фаза
Фаза О а, А О Ь, А О с, А
Cri8Mo4aNi40 9,070 16,983 4,752
R-фазы
фаза О a, A © c, A
Сг 18 яМо30 дСоб1 я 10,90 19,34
TifiMn7;Si19 10,87 19,23
Мо8МП78$119 10,65 19,18
V37Fe41SiS2 10,79 19,23
V45Co4f)Si1B 10,82 19,10
Mo45NiBgSi2 11,04 19,32
MoB3Ni 24Si18 11,06 19,87
р-фазы. Пр. гр. R3m, структурный тип D8$
Фаза О A* a p a, A** О £. A
MocFe7 8,99 30° 38,6 4,761 25,91
WeFe7 9,04 30° 30,5 4,746 25,78
MoeCo7 8,97 30° 47
WeCo7 8,94— 30° 42 4,751 25,67
9,01 30° 40 4,723 25,48
NbNi — 4,893 26,64
TaNi — 4,921 26,905
Ta44MoeNi60 — — 4,888 26,68
104
Продолжение табл. 5.6
cos <р = (ht hz + ki kz -J- li l2)/
Фаза 0 a, A* a © a, A** 0 c9 A
NbCo ... . 4,928 26,30
NbFe -— — 4,926 26,80
(Nb19Fe£0)
Nb6 (Си, Al), •—- 5,029 27,36
TaFe — 4,911 26,98
TaCo — ‘— 4,90 26,42
1 MoeFe, 1 —- •— 4,741 25,63
AlOfjCoy -—• — 4,767 23,65
Mo45CoS5 — — 4,725 25,42
M°6oNi46Si6; — 4,731 25,70
Aio4gNis,Sii5 (fila= 5,43)
* В ромбоэдрической системе.
•* В гексагональной системе.
зону [wwj; для всех плоскостей зоны справедливо условие зональности:
hu-\- kv~\- lw — 0. (1)
Индексы оси зоны определяются через индексы принадлежащих ей двух плоскостей: u=kil2—k2li; v=lths—tzhi; w~hik2—h2kb
Углы ср между разными плоскостями в кубической сингонии приведены в табл. 5.7 и могут быть рассчитаны по формуле
/]/ ^2 + ^2 + 1% •
(2)
Формулы для других сингоний приведены в табл. 5.8.
Период идентичности, нлн расстояние между соседними кристаллографически идентичными точками кристалла, — узлами пространственной решетки вдоль направления [дош]. В кубической сингонии:
LUvw ~ аУ и* п2 4г w2.
(3)
Связь периодов решетки с межплоскостнымн расстояниями d выражается в квадратичных формах существенно различных для разных снигоний. В кубической сингонии: d~z~(hz+ +kz+l2)/a\ (4) (2)
Квадратичные формы для всех сингоний даны в табл. 5.9*.
Непосредственно получаемый в дифракционном (рентгеновском) эксперименте ряд межплоскостных расстояний dhht (или d/n, где п—-порядок отражения) вместе с интенсивностями отражений характеризует данное кристаллическое вещество н лежит в основе рентгеновского фазового анализа. Кристаллическая
* В табл. 5.9 вместо индексов плоскостей (hkl) использованы индексы интерференции (HKL)i H^nh, K—nk, L=nl, где n— порядок отражения; =
dfikllri
ТАБЛИЦА 5.7
УГЛЫ МЕЖДУ НЕКОТОРЫМИ ВАЖНЫМИ ПЛОСКОСТЯМИ КРИСТАЛЛА КУБИЧЕСКОЙ СИНГОНИИ
(hskilti) Угол между плоскостями (fcifejG) и (ft2fc2Zs) в кубическом кристалле
100 100 0° г 90° -- .
110 45° 90° — .— —
111 54° 44' -— —
210 26° 34' 63°26' 90° —-
211 35° 16' 65° 64' 1
221 48° 11' 70° 32' * — - —„
311 25° 14' 72° 27' — — - -
110 110 0° 60° 90° —-
111 35° 16' 90° —
210 18° 26' 50° 46' 71° 34' .
211 30° Г 54° 44' 73°13' 90°
221 19° 28' 45° 76° 22' 90° .
311 31° 29' 64° 47' 90° — —
111 111 0° 70° 32' — -- .
210 39° 14' 75° 2' — .
211 19° 28' 61 ° 62' 90°
221 15° 48' 54° 44' 78° 54' . -
311 29° 30' 58° 30* 79° 58' — ~ —.
210 210 0° 36°62' 53° 8' 66° 25' 78° 28' 90°
211 24° 6' 43° 5' 56° 47' 79° 29' 90° —
221 26° 34' 41 ° 49' 63° 24' 63° 26' 72°39' 90°
311 19° 17' 47° 36' 65° 8' 62° 15' —
211 211 СР 33° 33' 48°11' 60° 70° 32' 80° 24’
221 17° 43' 35° 16' 47° 7' 65° 54' 74° 12' 82° 12'
311 10° 8' 42° 24' 60° 30' 75° 45' । 90° —
221 221 СР 27°16' 38° 57' 63° 37' 83° 37' 90°
311 311 0° 35° 6« 50° 29' 62° 58' 84° 47' —
105
ТАБЛИЦА 5.8
ФОРМУЛЫ ДЛЯ ВЫЧИСЛЕНИЯ УГЛОВ (ф) МЕЖДУ ПЛОСКОСТЯМИ В РАЗНЫХ сингониях
Сингония
Угол ф между плоскостями и h9kslx
Кубическая
Тетрагональная
Ромбическая
Гексагональная
______________+ fejfeg 4- _____________ +^i + if К^2 4~ ^2 +
1 3 С2
^1^2 4“ 4 7“ (^1^2 4“ ^e^i) 4~ ~7 7” ^1/2
2 4 c2
СО5ф —------ —------------------------------ —
+ h A + ~ i\ J/^1! + ^2 + ^2^2 Ь Z2
Рис. Б.6. Сферические и стереографические проекции: а — направлений (ОК и ОМ); б—плоскости (Р); о — центр комплекса, сферы сферической проекции и круга стереографической проекции; N. S — точки зре-
структура данного вещества определяется по результатам индицнровання отражений н прежде всего по способу, которым удается это сделать, т. е. установить связь ряда значений d с индексами hkl (нли HKL, разную для разных снигоний (уравнение (4), табл, 5.9],
Кристаллографические проекции (КП) используют для наглядного представления н анализа элементов симметрий и для решения задач, связанных с анализом ориентировки кристалла. В основу построения КП положен кристаллографический (нли точечный) комплекс (КК), который получается параллельным переносом направлений (узловых прямых) и плоскостей до пересечения в одной точке (в любом узле ПР). Сферическая проекпия получается прн пересечении элементов КК с поверхностью сферы, центр которой совмещен с центром комплекса. Для построения стереографической проекции (СтП) выбирают одну из плоскостей, проходящих через центр сферической проекции (О на рнс. 5.6). Сферическая проекция служит лишь промежуточным этапом в построении стереографической проекции, которая изображается на плоской поверхности н вмещает проекции всех элементов КК в ограниченной площади — внутри круга проекции (Q на рис. 5.6). В СтП направления изображаются точками (А", М" на рис, 6, а), плое-
ния при построении стереографической проекции; Q — плоскость стереографической проекции; М' — сферическая проекция направления ОМ; М" — стереографическая проекция того же направления; К' и
— то же, для направления OK; SPh SP? и т. д. — лучи зрения, используемые при построении стереографической проекции плоскости Р
106
Рнс. 5.7. Сетка Вульфа [на схеме показано определение угла между направлениями А и В в стереографической проекции и построение полюса (Р) плоскости, в которой лежат направления А и В]
ТАБЛИЦА 5.9
ФОРМУЛЫ для РАСЧЕТА МЕЖПЛОСКОСТНЫХ РАССТОЯНИЙ И ОБЪЕМА
ЭЛЕМЕНТАРНОЙ ЯЧЕЙКИ U
Сингония Межплоскостное расстояние Объем ячейки V
Кубическая 1 Я2 + К2 + £2 d2 “ о2 а3
Тетрагональная 1 Н2 + Z<2 I2 d8 = а2 + са а2с
Ромбическая Ромбоздрнче- J Н2 К2 £2 d2 ~ а2 + Ь2 + с2 1 d2, ' abc а3 У 1 — 3 cos2 а + 2 cos8 а
ская (fl2 +/<2 + £2) sin2 а + а2 (1—3 cos2 а + + 2 (AZ( 4- К£ 4 HL) (cossg — cosa) + 2 cos8 а)
107
Продолжение табл. 5.9
Сингония
Межплоскостное расстояние
Объем ячейки V
Гегсагоиальная
Моноклинная
Триклинная
1 = 4 (/72 + + К2) _L^
d2 ~ 3 с2 ~с2
d2 a2sinap В2 £a 2HL cos р
с2 sin2 р ас sin2 Р
^7 ~ Isn^2 + «22^2 + $зз£2 + 4- 2s12HK + 2s23K£ + 2s13ff£]
а2с= 0,866 a2c
abc sin p
abc X
X TZ1 — cos2 a — cos2 p — cos2 у + + 2 cos a cos P cos у
Примечание: 5М*=&VsIn« a: s(2°a&c-(cos a cos P—cos ?); s22=a’c2 sin® ₽; s23“a2&c(cos {3 cos y—cos a); b2 sins y; sja=a6®c(cos cos a—cos £),
700
320
720
711
5Й
531
•311
321
211
*533
351
770
151
171
131
132 153°
IK^3
310
210-
551 •
9 *331
231
1Q8
711
* 511
*301
•201 9^73 9312
701
— 272 •
111 _«•
® 535 ^3315
335 •
355 * J72^_
172Z
*123 9 775
^3 *735
012*
135
732 ^355
Т71
031
151
351 231
553
321
*371
511
021
2Z19
9331
*551
121
9 *353
010
Т?1
130
120
230
320
210
Стандартные проекции для кубического
Рис. 5.8.
а— (001); б—(011)
зз/^3!3
9533
9271 513
310
210
320
110
*311
?30
1ZO
*531
321
•351
010
031
011
J21
*132
*753 021
ej31
@ 151
• 171
*551 ®331 lS531
*211
^513 z^-x
9312 *333
313 л535 711
* 212
315^ ^13 335 355
9102
9103
9112 9122
•715 *335
^717 ^013012
013 плЛ ® • 001^2
*117
•115
*113 9103
J12 -*10Z_
^335 ^15 315*213
213 701 313 9333^
212 _ *533
513 *312
• *211
117 135 /ту
*.115 •
J12 9355
9335
9171
*151 ^131
•121
353
231
553 ®
*331
® 551
*321
*153
9132
130
©
351
•201
•311
*531
9301
511
9511
*711
711
310
100
a
кристалла
(гномостереографические п роекции
кости — меридианами внутри круга проекции (Р иа рис. 6,6), в частности, прямыми линиями, проходящими через центр проекции (диаметры),— в случае плоскостей КК, перпендикулярных плоскости СтП, и кругом, совпадающим с основным кругом СтП — в случае плоскости, параллельной плоскости СтП (плоскость Q). Особенно часто предметом анализа являются ориентировки плоскостей; построение их проекций упрощается, если за основу брать так называемый полярный (илн обратный) кристаллографический комплекс (ПКК), образованный нормалями к плоскостям, которые исходят из одной точки — центра комплекса, В этом случае проекция называется гномостереографической (ГСтП); проекция плоскостей в ГСтП имеют вид точек (как стереографические проекции направлений),
Измерение углов между направлениями и между плоскостями — одна из самых важных задач, решаемых с помощью СтП (я ГСтП), С этой целью применяют сетку Вульфа (рнс. 5.7), которая представляет собой стереографическую проекцию сеткн меридианов и широтных линий (параллелей) на поверхности сферы, проведенных с интервалами 2° (тонкие
013
103
315
213л
117
*115
113 ©
101
302
*573*
/02
315 ,203 ®
313 515
535 в ® ’
* 212
312
533 * '
• 211
* 3/1
553 < ।
о227
513
201
*301
511
•* *711
331
310
551
213
511*
317
221
553
515
211 а
533
551
357
531 о
371
371
*531
210
320 •
^711* *511 ~ „
27/ ® 311 ^11 111
Г , 3S '
201 *_
513
513 *- ~ «
302 515*
*317
313*
212 ~
535 9
н? 115 113
155*
155^^^ • 032 *153
*133 * „qj
- 153 *
031
3S- v>353. 1,2’ №
*533 *221 231 151* *171
371 *331Л *351 W 171
а20 V \5S1 ^.,30 „у
• 511 * _ «75
711* 310гЦ'3г° *^*5 *137
77 • 531 *331*23?т
£*/00 *321 *221 <
11$ 115
б
линия) и 10 ° (жирные линии). Плокостью проекции при этом является одна из меридиа-нальных плоскостей сферы. Для измерения углов между направлениями сфереографн-ческие проекции этих направлений должны лежать на одном меридиане Чтобы получить такое совмещение, приходится вращать сетку Вульфа относительно стереографической проекции анализируемого кристалла (нли наоборот), причем центр сетки Вульфа должен совпадать с центром анализируемой проекции. Широтные линии используют для отсчета углов (см, рис. 5,7).
При работе со СтП (илн ГСтП) используют стандартные сетки (СС) —гиомостереографи-ческие проекции разных плоскостей илн стереографические проекции разных направлений при определенных (стандартных) ориентировках кристалла (рис. 5.8 и 5.9). В случае кубических кристаллов этн два вида СС совпадают (см. рнс. 5.8). Для кристаллов других сингоний этн проекции различны. Из эксперимен-
1 Этот меридиан — стереографическая проекция плоскости, в которой лежат оба рассматриваемых направления (А и В — на рис. 7).
%2 335
231
131
351
710
230
151
• J51
353
121
9113
а115
*117
351
553
221
331
551
355 122*
*
©755
132
£53
- *132 122 15$
» •
755
/23
*112 *135 *
*012
013
_ *353 •. 7
*55 ф -_ 355
212*535 335
313 в ..
Э 213 с
*315
*203
*315 *103
*121.
-.Г, I20 130 •
~ 1Z3
135 * 012 г * 023
• 135
1?Ь
*112
/13 *335 */22
355 • ‘
S1/* ^935^111
^353 * '
001
551 \
_ 331
231
*031
*021 753
*032
75$
° 733 355
011
023
• - 135
/23 _ _
' 335
712
кристаллографических плоскостей На плоскости проекции);
7010
2130
1120
?230
3210
0110
1100
1320
1210
2310
2021
1121
1011
3211
1231
1122
2310
1321
1211
1212
1210
1212
1320
1125
1101
1123
0221
1100
3211
1011
3210
"2021
2110
1010
"2023
11012
"1013
41014
"1012
2023
1123
1125
0115
1213
1215
1104
05_ л!Г1103
1104
7213
I2T5
115
0114
Ы013
Рис. 5.9. Гяомостереографическая проекция плоскостей (гексвгонального кристалла при с/а=>1,63)
"1015 fyO7o
та (из гониометрических измерений на ограненном кристалле, из анализа лауэграммы или из измерений с помощью текстур-днфракто-метра для текстурованного поликристалла) получают полюсную фигуру—гномостереографические проекции разных граней или плоскостей на выбранную плоскость проекции в случае монокристалла или гномостереографические проекции одного определенного типа плоскостей {hkl} текстуроваииого поликристалла; в последнем случае плоскость проекции выбирают в соответствии с направлениями действия внешних сил (или формой образца)
Часто необходимо определять индексы точек на полюсной фигуре. Такая задача решается отождествлением этих точек с точками СС. Поскольку анализируемая проекция может относиться к некоторой произвольной ориентировке кристалла, операции совмещения анализируемых точек с точками СС должен предшествовать поворот кристалла нлн изменение плоскости проекции, что производится с помощью сетки Вульфа. Если ось поворота совместить с вертикальным (меридианальным) диаметром сетки Вульфа, то точки анализируемой проекции будут перемещаться по параллелям
’ См. ниже «Анализ текстур*.
(широтным линиям) сетки Вульфа. Обычно используют СС и сеткн Вульфа с диаметром основного круга 20 см*. Прн этом точность угловых измерений оказывается равной примерно 1—2°. Для достижения более высокой точности используют сеткн диаметром 30 см. Анализируемую полюсную фигуру обычно строят на кальке.
5.2.2. Геометрия дифракционных картин и обратная решетка кристаллов
Широко распространенное истолкование картины дифракции рентгеновских лучей от трехмерного кристалла как «отражения» от семейств параллельных атомных плоскостей позволяет легко найти зависимость направления максимумов интенсивности дифрагированных лучей от соотношения длины волны излучения (X) н межплоскостных расстояний (dhu) в кристалле (уравнение Вульфа—Брэггов):
sin 9 = n№dhM. (5)
* Сетка Вульфа н ряд СС диаметром 20 см имеются в Приложении (Атласе) к книге [14].
ПО
Это истолкование кажется весьма наглядным н .уравнение Вульфа—Брэггов, безусловно, справледливо. Однако само это истолкование не описывает физического явления н не дает непосредственной связи геометрнн дифракционной картины с параметрами трехмерной структуры кристалла. Дифракция рентгеновских лучей (а также электронов и нейтронов) на кристалле описывается в рамках общей теории дифракции как случаи фраувго-феровой дифракции иа трехмерной решетке, когда период дифракционной решетки очень мал и можно считать, что по сравнению с этим периодом расстояния источник лучей — кристалл (решетка) и кристалл—место регистрации дифракционной картины бесконечно велики Поскольку рентгеновские лучн рассеиваются электронами вещества, зная распределение электронной плотности р(г), с помощью преобразования Фурье для трехмерного пространства можно найти распределение максимумов интенсивности или, точнее, значения структурного фактора интенсивности:
р (К) = J р (г) exp [2ju (г /<)1 drt (6)
^кр
где г — радиусы-векторы в пространстве кристалла; К — радиусы-векторы в пространстве, которое можно называть пространством изображения нлн Фурье-пространством кристалла. Это реально существующее пространство диф-
представлеиия уравнения Вульфа—Брэггов в векторной форме. На схемах рис. 5.10 показан переход от обычного представления этого уравнения как «зеркального отражения» при определенной величине межплоскости ого расстояния dhhi (рис. 5.10, а) к векторному представлению (уравнеине Лауэ) (рнс. 5.10, б и в). Схема рис. 5.10, в отличается от рис 5.10, б только тем, что вместо единичных _векторов для падающего (Ко) и отраженного (К') лучей взяты векторы, равные по модулю 1/Л, тогда как модуль результирующего вектора равен jr*| кь.Прн этом dnKL—dhki/п, HKL— индексы интерференции: H=nh- K=nk, L~nlf где (hkl) —индексы отражающей плоскости; п— порядок отражения. Единичный вектор К (см. рнс. 5.10,б) илн вектор г* (см. рнс.5.1(\в) совпадают по направлению с нормалью (N) к плоскостям (hkl).
Итак, вектор обратной решетки г* по модулю равен обратной величине межплоскостного расстояния |r*| =\/dhki (при л=1) н направлен по нормали к соответствующей плоскости ПР. Схема рис. 5.11, а отличается от схемы рис. 5.10, в только тем, что_ изображает не один, а несколько векторов г*, исходящих нз точки О; следы соответствующих плоскостей ПР (Р на рис. 5.10, б, в) здесь уже ие показаны. Концы векторов г* упираются в точки— узлы ОР,
Рис. 5.10. Графическое представление уравнения Вульфа—Брэггов:
с—обычная б —векторная форма, /С—в —то же, что б, но для (К—Ко)/Л=»
—Klh—r* при |r| —d//Kjb
ракцнонной картины кристалла, в котором векторы К, соответствующие отличным от нуля значениям F(K), характеризуют направления дифрагированных лучей. Трехмерной периодичности пространственной решетки кристалла отвечает также трехмерная периодичность распределения интенсивности [или F(K)], которую принято называть обратной решеткой кристалла (ОР). Обратную решетку следует представлять как систему узлов — математических точек, если кристалл имеет идеальную структуру и неограниченные размеры.
Понятие об ОР, а также о связи ОР и ПР кристалла по существу следует из графического * 10
• Периоды решетки кристаллов имеют порядок о
10 * см (А); упомянутые здесь расстояния обычно порядка 1—10 см (см. п. 5.2.4 и табл, 5.16>.
Следующая схема (рнс. 5.11, б) представляет уже плоское сечение ОР, включающее узлы высших порядков отражения (п>1). Легко видеть, что тройки значений индексов HKL являются координатами узлов ОР, еслн за направления координатных осей принять направления векторов Гд01, г010 и 400. Этн векторы характеризуют элементарную ячейку ОР (случай примитивной ПР):
а “Поо» =г010» с “rooi* (7)
Вектор ОР определяется через координаты соответствующего узла ОР:
?hk! = Ha+Kb*+ (8)
Периоды ПР с периодами ОР кристалла (рис. 5,12) связаны уравнениями;
111
c* — [fe cV(a [fe cl); fe* — la c]/(fe la cl);
c* = la fel/(c [a fel). (9)
Из (9) следует, что
(a a*) = (fe b*) = (c c*) == 1 (10)
и (a fe*) = (a c*) = (fe c*) =,.. = О; (10а)
| a* | = fee sin a/V; | fe* | = ac sin p/V;
c* = afesiny/V. (11)
Здесь V — объем элементарной ячейки ПР кристалла; а, f>, у—углы между координатными осями кристалла.
б 022 012 002 072 022
• • • в •
021 011 001 011 021
С 020 ~ОТО ООО 010 020
• • • •
021 011 OOf 011 02f
• • • • •
022 072 002 012 022
Рис. Б.11. Обратная решетка кристалла (О— начало координат):
—>*
а — векторы и узлы, ближайшие к началу координат; б — плоское сечение обратной решетки (зона [100], примитивная. ОР, соответствующая примитивной ПР)
Рис. 5.12. К построению элементарной ячейки обратной решетки кристалла (общий случай)
Для кубических, тетрагональных н ромбических ПР
|о‘| = 1°Г; 1Н=1Я-’; М = 1<Г'-
(Па)
Симметрия ОР соответствует симметрия ПР кристалла; различие симметрии связано преж
де всего с тем, что ПР может не иметь центра инверсии. ОР всегда имеет центр инверсии — начало координат ОР (см. рис. 5.11). Трансляционная симметрия при переходе от сложных ПР к ОР изменяется: F-ячейке Бравэ в ПР соответствует /-ячейка в ОР; наоборот, /-ячейке в ПР соответствует F-ячейка в ОР1.
Присутствие центра инверсии в ОР объясняет отмеченное раньше (п. 5.2.1) ограничение в определении Пр. гр.: непосредственно по дифракционной картине устанавливается дифракционная группа. Центр инверсии ие всегда явным образом присутствует в расположении дифракционных максимумов (рефлексов) на рентгенограммах только потому, что рентгенограмма представляет собой сложное сечение и некоторую проекцию (в соответствии с геометрией укладки фотопленки) определенной области ОР.
Связь рентгенограмм с ОР кристалла следует из схем рис._5.10, в н рнс. 5.13: вектор падающего луча Ко упирается в начало коордн-
Гис. 5.13. Сфера отражения (радиус К т) и сфера ограничения (радиус 2J7.) в обратной решетке
нат ОР—точку О; в этом направлении откладывается длина А-1; далее с центром в точке С (начало вектора Kolf) проводится сфера радиусом А*"1*. Те точки ОР, которые окажутся на поверхности этой сферы, удовлетворяют условию Вульфа—Брэггов подобно точке Q на рис. 5.10, в. Для них Ki/A—Ко/А= гЯК£-СФе' ра радиусом А-1, построенная иа векторе Л’о/А н проходящая через начало координат ОР, называется сферой отражения (Сф. О). Очевидно, прн изменении ориентировки кристалла относительно падающего пучка в отражающее положение попадают другие плоскости кристалла, нли, иначе говоря, на поверхность Сф. О попадают другие узлы ОР. Сфера радиуса 2/А (см. рис. 5-13) определяет ту область углов ОР, которые прн данном значении А дифрагирующего излучения могут оказаться на рентгенограмме. Эта сфера называется сферой ограничения.
1 Хотя здесь ОР рассмотрены как картина дифракции. не следует думать, что этим ограничивается значение этого представления. Ряд других свойств кристалла также описывается в Фурье-пространстве кристалла. Используемая в физике твердого тела ОР, например при описании электронной структуры, отличается от рассмотренной здесь ОР только по масштабу (Х2л).
* На рис. 5.13 —окружность радиусом (сечение сферы).
112
Как следует из рис. 5.13, прн случайной ориентировке монокристалла при постоянной и большой длине волны (т. е. прн малом радиусе кривизны Сф. О) для большого н совершенного кристалла (когда узлы ОР—точки) мала вероятность образования значительного числа рефлексов на рентгенограмме (или попадания узлов ОР иа поверхность Сф. О). Чтобы получить достаточно информативную рентгенограмму, используются следующие методы:
1. Съемка в характеристическом излучении (т. е. прн постоянной длине волны 7.) кристалла, вращающегося (или колеблющегося) вокруг определенной оси, обычно совпадающей с важным направлением в кристалле, — метод вращения (колебания или качания). Регистрация производится на плоскую или свернутую по цилиндру фотопленку; в последнем случае ось цилиндра совпадает с осью вращения кристалла. Из схемы дифракции в представлений ОР (рис. 5.14) легко понять, что рефлексы на
Рис. 5.14. Метод вращающегося кристалла в представлении обратной решетки (А — ось вращения кристалла и связанной с ним обратной решетки. Z=-0, 1. S — слоевые линии, соответствуют плоскостям обратной решетки кристалла, перпендикулярным оси вращения)
рентгенограмме должны располагаться по слоевым линиям, которые связаны с узлами ОР, принадлежащими определенным плоскостям ОР (трн из них показаны на рнс. 5.14). Метол в основном применяется для определения кристаллической структуры1.
2. Съемка неподвижного кристалла в полихроматическом излучении (сплошной спектр или спектр торможения рентгеновских лучей) — метод Лауз. Регистрация обычно производится на плоскую пленку, которую располагают после образца и на которой регистрируются рефлексы, соответствующие небольшим вульф-брэгговским углам (0<45°). Вариантом данного метода является обратная съемка (метод эпиграмм), когда пленку располагают между рентгеновской трубкой н образцом н на ией регистрируются рефлексы, соответствующие вульф-брэгговскнм углам 0>45°. Метод не
1 Вариант метода — метод КФОР (камера фотографирования обратной решетки), который позволяет получать на рентгенограмме неискаженные сечения обратной решетки. Методы вращения и КФОР удобны для анализа диффузного рассеяния, связанного с нарушениями кристаллической структуры, например при исследовании процессов выделения из пересыщенного твердого раствора [12, с. 166 и 385J.
подвижного кристалла при металловедческих исследованиях используется в основном для определения кристаллографической ориентировки — расположения кристаллографических направлений (или плоскостей) образца по отношению к внешней координатной системе (форма кристалла, направление внешних воздействий и т. д.)_ Как видно из рис. 5.15, при непод-
Рис. 5.15. Метод неподвижного кристалла (метод Лауэ) в представлении обратной решетки
вкладом образце симметрия рентгенограммы должна передать (с известным искажением) симметрию ОР относительно направления вектора падающего пучка Ко- Выбор вида съемки определяется размером н характером образца, прямая съемка применима для кристалла достаточно малого размера или для кристалла в виде фольги. Оптимальная толщина кристалла зависит от линейного коэффициента ослабления (р,) и принимается примерно равной толщине слоя половинного ослабления в сантиметрах: 1/р.. В случае толстых образцов (монокристалл илн крупнокристаллический поликристалл) применяется метод эпиграмм. Съемка неподвижного кристалла может оказаться полезной для выявления несовершенств кристаллического строения (анализ астеризма для деформированных и отожженных кристаллов, диффузное рассеяние в случае стареющих с’плавов,'тепловое рассеяние и пр)1.
3, Съемку поликристаллических образцов производят в монохроматическом — характеристическом илн Специально монохроматизиро-ванном — излучения; образец имеет обычно вид тонкого столбика нлн плоского шлифа, может быть неподвижным или вращаться (вокруг осн цилиндра нли вокруг осн, перпендикулярной плоскости шлнфа); применяются передняя и обратная съемки на плоскую пленку с очевидными ограничениями для фиксируемых углов 0 (рис. 5.16), а также съемка на цилиндрическую пленку (классический метод порошка илн метод Дебая), позволяющая фиксировать любые углы 0 (рнс. 5.17). Разнообразие возможностей применения метода для поли-кристаллических образцов обусловлено, с одной
* При определении кристаллической структуры съемку лауэграмм используют для анализа симметрии кристалла и его целесообразного ориентирования для последующего получения рентгенограмм вращения.
€—280
Рис, Б.16. Метод порошка:
а — вид обратной решетки поликристаллнческого объекта и схема получения рентгенограммы; б — схема рентгенограммы (случай плоской пленки, прямая съемка; Л — расстояние образец— пленка)
Рис. 5.17, Схемы рентгенограмм, получаемых методом порошка при использовании камеры Дебая с цилиндрической пленкой (а, б, в) и камеры с плоской пленкой (г. д):
РЛ — направление падающего пучка рентгеновских лучей; К — коллиматор (диафрагма)? О —образец; Г —тубус; РЛ —пленка; 7 —2—2, 3—3, 4—4 —следы пересечения дебаевских конусов с пленкой; углы при вершине конуса 4б>, 40s, и показаны дугами на рис. 17» а
\
стороны, высокой прецизионностью измерений периода решетки и связанных с ннм характеристик, а с другой стороны, тем. что кристаллические материалы в технике в основном поликрнсталлнческие*.
5.2.3. Интенсивность рассеяния рентгеновских лучей кристаллом2
При рентгеноструктуриом анализе обычно ис-
О пользуют лучи с длинами волн от 0,5 до 2,5 А. Под воздействием переменного электрического поля этой электромагнитной волны электрически заряженные частицы вещества (ядра н электроны) совершают вынужденные колебания с частотой падающего излучения. Колеблющийся заряд — осциллятор — порождает электромагнитные волны с частотой падающих колебаний, но с отставанием от ннх по фазе на л/2. Эти волны распространяются во все стороны, т. е. происходит рассеяние падающей волны иа рассеивающих центрах (РЦ). Таким образом, падающая плоская волна порождает на каждом РЦ сферическую волну той же частоты, но отстающую по фазе на л/2. Интенсивность рассеяния зарядом е и массой т под углом 26 к направлению падающей волны:
/29 ~ A2Q —
ел т2 с4 Z?2
1 1 4~ coss 26 _
2 4
(12)
(/о—интенсивность падающей волны).
Так как масса ядра значительно больше массы электрона, рассеяние на ядре менее интенсивно, чем на электроне. Таким образом, основным РЦ для рентгеновских лучей является электрон.
Лучи, рассеянные всеми электронами атома, интерферируют между собой, Даная результирующую амплитуду:
Аат = %AS3l exp (— iAcp j), (13)
где Acpj—разность фаз лучей, рассеянных j-тым электроном и электроном в начале координат;
A J
где Ki н Ко —единичные векторы в направлении рассеянной н падающей волн; q — расстояние /-того электрона от начала координат. Тогда, обозначив (Кг—Koi/k-k, получим для амплитуды рассеяния атомом AaT=ABnf(k), где /(7г) = —= S ехр[— 2л£ К г J называют
Аэп /=2
атомной функцией рассеяния.
Аналогичный подход к рассеянию атомами одной элементарной ячейки кристалла дает амплитуду лучей, рассеянных элементарной ячейкой (структурная амплитуда):
F(k) = Аяч/ Адл — 2/jexp [— 2xt(k rj)]t
* 1 * * * V (14)
1 В методе поликристалла задача определения кристаллической структуры не имеет общего способа
решения для веществ, относящихся к низшим сингониям.
2 Составлено А. Н. Ивановым.
* Так как заряд электрона «размазан» в объеме атома о с плотностью р(г), более строго выражение t (А) = J с(г) ехр[—2ni(ф
V
где суммирование ведется по всем атомам базиса.
Наконец, интерференция волн, рассеянных кристаллом, который содержит К2, N3 элементарных^ ячеек в направлениях (соответственно) а, Ь, с, приводит к результирующей амплитуде:
укр = 2 F3 ехР 2то" $ г/)1> (1Б)
/
где Г/ — расстояние от начала координат до центра /-той ячейки со структурной амплитудой Fj. Так как k—'Некоторый вектор в ОР, то его можно_разложить на орты в ОР; И=1ца*+ +h2b*+h3c*, аналогично для вектора в ПР;
г = пг a -j- п2 Ъ 4- ё;
Wi—1 Ns—1
укр = /? 2 exp (—Sra/i! nJ 2 X 0 о
Ars—1
X exp (— 2stih2 n2) 2 exP (— 2лЙ3 n2); о
X
ЬР\ 1 2 3/ Kp Sin2 Л/lj
sin2 лКа h2 sin2 nNa h2
sin2 лЬ> sin2
max t?2 s*nS F sin2 К sin2 ttNs L ‘kp ' F
(16)
(17)
sin2 nH sin2 лК sin2 л£
Интенсивность должна быть максимальной (интерференционный максимум), если /ц—//, Л2=К, Л3—£; /У, К, L — целые числа, т. е. условие максимума интенсивности:
k = 2(1 ;= = [fa* + р _
А
радиус-вектор ОР). При соблюдении приведенных выше условий разность фаз лучей, рассеянных всеми ячейками кристалла, кратна 2л н 1™х ~F2№ (здесь N=NM).
Расходимость н немонохроматичность первичного пучка и некоторые другие инструментальные н физические факторы не позволяют экспериментально измерить /™рах, теоретически определяемую из выражения (17).
Экспериментальному определению поддается так называемая интегральная интенсивность:
292
Лф = $I(hlhMdv* = L f /(26)d(26), v* 2'0Х
(18)
где L — «масштабный» фактор, или фактор перехода от пространства ОР кристалла к пространству его рентгенограммы (фактор Лоренца).
Для сравнения экспериментально измеренной и рассчитанной интегральных интенсивностей следует учесть уменьшение интенсивности вследствие поглощения рентгеновских лучей в веществе н вследствие тепловых колебаний атомов. Тогда выражение для интегральной
8s
115
интенсивности монохроматических лучей, рассеянных малым кристаллом объема v:
е9
нр = Л) а 4
1 т2с4
х-Ц-л(е)о, vL
1 4- cos2 20 2sin226
F2X
(IS)1
а для поликристалла (иа единицу длины дебаевского кольца на расстоянии R от образца):
ЛхОЛШф — /0 Лг2 С4 №
1 -|- cos2 26 sin2 6 cos В
(20)
где Vm — объем элементарной ячейки; А (В) — абсорбционный фактор; Phkl— фактор повторяемости (число семейств плоскостей в совокупности {HKL}); структурная амплитуда
FHKl = ехР (— Mi) ехР [— 2jrZ (//я^ +
i
+ Kpj 4- Lqj)]
(m, P> (i— координаты /-того узла базиса, а ехр (—Mj) — член, учитывающий ослабление интенсивности иследствие тепловых колебаний атомов в /-том узле решетки).
Значения всех множителей в формулах (19) и (20) приводятся в справочной литературе по реитгеноструктуриому анализу.
1 Формулы (19) и (20) получены в «кинематическом» приближении, которое не учитывает взаимодействия падающей и рассеянной волн. Для подавляющего большинства металловедческих задач это справедливо. Учет этого взаимодействия приводит к формулам динамической теории рассеяния (см. [91).
5.2.4. Техника получения и регистрация дифракционных картин. Аппаратура1 *
Источником рентгеновских лучей для структурного анализа служат электронные отпаянные трубки (табл. 5.10)*. На анод этих трубок нанесен слой определенного металла (Cr, Fe, Со, Ni, Си, Mo, Ag). Используется характеристическое излучение К-серии. В ряде случаев применяют фильтры, чтобы исключить излучение Кр (табл. 5.11) нлн монохроматоры (табл. 5.12). Дифрагированное излучение (дифракционная картина) регистрируется либо иа рентгеновскую фотопленку (табл. 5.13), либо с помощью детекторов, в которых используется ионизационное или сцинтилляционное действие рентгеновских лучей (табл. 5.14).
Рентгеновские аппараты состоят из устройства для электрического питания трубки (одной или Двух), устройств для согласованного расположения н движения анализируемого образца, источника излучения (трубки), системы диафрагм и детектора излучения или фотопленок. В зависимости от способа регистрации различают аппараты для фотографического метода н дифрактометры (табл. 5.15) [19, 29 — 31].
а. Рентгеновские камеры. Этн камеры составляют принадлежность аппарата для рентгеноструктурного анализа с фото'ре-гистрацией (например, современного аппарата УРС-2,0, см. табл. 5.15 и 5.16), однако прак-
1 Составлено совместно с А. Н. Ивановым.
* Используются также электронные трубки с вра-
щающимся анодом, которые дают особенно большую мощность излучения, необходимую для исследования быстропротекающих процессов [19. кн. 2, с 41]. В самые последние годы находит применение рентгеновское излучение от мощных ускорителей — синхротронов [32].
ТАБЛИЦА 5.10
ХАРАКТЁРИСТИКИ НЕКОТОРЫХ РЕНТГЕНОВСКИХ ТРУБОК ДЛЯ СТРУКТУРНОГО АНАЛИЗА
Тип трубки Число окон Фокус, мм Предельная мощность на аноде, кВт
истинный проекция (угол выхода а=6°)
БСВ-4 4 0 3,-0 (круг) 3X0,3 (эллипс) 0,16—0,07
БСВ-5; БСМ-1* 2 0 0,02—0,04 (круг) 0,02—0,004
БСВ-6 2 2,5X5 2,5X0,25 (а=2°) 0,45—0,20
БСВ-7 (острый фокус) 2 0,1X2 0,1 ХОД (а—3°) 0,110—0,025
БСВ-8 3 1X12 2 пучка 1X1,2; 1 пучок 0,1X12 1,0—0,5
БСВ-9 3 2X12 2 пучка 2x1,2; 1 пучок 0,2X12 1,5—0,7
БСВ-10 (БСВ-11) 3 0,45X8 2 пучка 0,45X0,8; 1 пучок 0,05X8 0,8—0,3
БСВ-19** (БСВ—22)** з' 0,4X8 2 пучка 0,4X0,8; 1 пучок 0,04X8 1 1,2—0,6
БСВ—23** 3 1X10 2 пучка 1X1 1,5—0,7
БСВ—24** 3 1,6X10 2 пучка 1,6X1,0; 1пучок ОДбхЮ 2,0—1,0
Примечание. Трубки изготавливаются с анодами из W, Ag, Мо, Си, Ni, Со, Fe и Сг. Минимальная мощность соответствует трубкам с анодом из Fe, а максимальная — из Си и Мо.
* Трубка с масляным охлаждением и теми же параметрами, что и БСВ-5, ** Трубки с коаксиальным вводом высоковольтного кабеля.
Ы6
ТАБЛИЦА 5.11
ДЛИНА ВОЛН ХАРАКТЕРИСТИЧЕСКОГО ИЗЛУЧЕНИЯ К-СЕРИИ (А) И МАТЕРИАЛЫ ДЛЯ ФИЛЬТРАЦИИ Кр -ЛУЧЕЙ ВАЖНЕЙШИХ АНОДОВ
Элемент Атомный номер Линии К-край поглощения и материалы фильтра
а. 6,
Сг 24 2,29351 2,28962 2,08480 2,269 V
Fe 26 1,93991 1,93597 1,75653 1,896 Мп
Со 27 1,79278 1,78892 1,62075 1,743 Fe
Си 29 1,544390 1,540513 1,39217 i,488Ni
Мо 42 0,713543 0,709261 0,632252 0,689 Zr
Ag 47 0,563774 0,559363 0,49701 >0,509 Pd
Примечание. При расчете дифракционных максимумов, являющихся слившимися Кк >к-дублетами, используют среднее значение длины волны вычисляемое из соотношения (^К’о,)Ср=(2^а1+^а2}/3.
ТАБЛИЦА 5.12
ХАРАКТЕРИСТИКИ НЕКОТОРЫХ КРИСТАЛЛ-МОНОХРОМАТОРОВ
Кристалл Отражающая плоскость Интенсивность Механические свойства Примечания
Si02 — кварц (1011); d=3,35 А Средняя Упругий, можно гнуть Общее применение
СаСОз—кальцит (200); б?=3,04 А » Пластически деформируется Хорошее разрешение
NaCl (200); d=2,82 А Сильная В воде пластически деформируется Гигроскопичен
LiF — фтористый литий (200); d-2,01 А Очень сильная В теплой воде пластически деформируется Общее применение
Пентаэритрит (002); d=4,40 А То же Очень легко пластически деформируется Длинноволновое излучение
Графит (002); <2=3,352 А — Общее применение
ТАБЛИЦА 5.13
ОТНОСИТЕЛЬНАЯ ЧУВСТВИТЕЛЬНОСТЬ И РАЗРЕШАЮЩАЯ СПОСОБНОСТЬ __________________РЕНТГЕНОВСКИХ ПЛЕНОК [20] _______________
Тип пленки Относительная чувствительность при разных о длинах волн А)* Разрешение, линий/мм Общая характеристика
0.45 (Ag) 0,71 (Мо) 1,54 (Си) 1,93 (Fe) 2,23 (Сг)
РТ-1 100 100 100 100 100 70 Высокочувствитель-
РТ-2 33 38 41 55 59 75 ные, зерно эмульсин
РТ-3 32 32 35 48 53 — — I мкм
РТ-4 18 19 22 — — 100—130 Малочувствительные,
РТ-5 8 9 9 — зерно эмульсин — 0,4 мкм
* В скобках указан материал анода рентгеновских трубок.
ТАБЛИЦА 5.14
ОСНОВНЫЕ ХАРАКТЕРИСТИКИ ДЕТЕКТОРОВ РЕНТГЕНОВСКОГО ИЗЛУЧЕНИЯ
Тип детектора* Эффективность, % Мертвое время, мкс Собственный фон, имп/мии Амплитудное разрешение, % (Для Си-/<а)
Си-К^ Мо-Кк
Пропорциональный БДП-3) (БДП-2, 60 30 1 5—15 15—20
Сцинтилляционный БДС-13, БДС-8) . (БДС-6, 90 95 1 5—20 40—50
Полупроводниковый (СРП-1) 80 80 1—5 — 3-5
* В скобках указаны марки выпускаемых промышленностью детекторов для структурного анализа.
117
ТАБЛИЦА S.15
ОСНОВНЫЕ ТИПЫ ОТЕЧЕСТВЕННЫХ РЕНТГЕНОВСКИХ АППАРАТОВ ДЛЯ СТРУКТУРНОГО
АНАЛИЗА
Тип Назначение Потребляемая мощность, кВ-А Масса, кг установочная площадь, м3 Особенности
УРС-0,02 Острофокусный источник для работы фото метод ом 0,14 45 1 Естественное охлаждение трубки
УРС-0,1 То же 0,5 170 1,5 —
УРС-2,0 Получение рентгенограмм фотометодом 5 800 5 Поставляется с комплектом камер, может питать две трубки одновременно
АРТВА-2,0 АРТВА-5,0 Мощный источник излучения для фотометода и дифрактометрии 5 10 1450 5 7 Применяется разборная трубка с вращающимся анодом
ДРОН-2,0* Универсальный дифрактометр общего назначения 5,5 1200 12
ДРОН-3* То же 6 1500 10 Может работать в согласовании с ЭВМ типа М-6000
ДРОН-УМ1 » 6 1200 <15 Управление ЭВМ«Искра»
ДАРН-2,0 Дифрактометр для определения макронапряже-ннй в крупногабаритных металлических объектах 5,5 1000 10 Реализован метод «б1п2Ф» н фокусировка по Зееману—Болину
ДАРТ-2,0 Дифрактометр для автоматического построения полюсных фигур н исследования текстур — —— 13 Кроме полюсных фигур, прибор выдает измеренную интенсивность . на цнфропечать илн перфоратор
ДРФ-2,0 Для проведения качественного и количественного фазового анализа поликристаллов 5 800 2 Кожух трубки жестко закреплен на гониометре
ДАРМ-2,0 Получение полного набора интегральных интенсивностей монокристаллов 5 1600 20 Работает по командам программ, рассчитанных на ЭВМ и нанесенных на перфоленту
ДАРК-2,0 Получение полного набора структурных множителей с использованием 512-канального координатного детектора 8 700 (без ЭВМ) 50 (с ЭВМ) Управляется ЭВМ типа М-6000, которая ведет и первичную обработку данных
ДРД-4 Анализ радиоактивных образцов 5,5 900 — Используется монохрома-тизация дифрагированного излучения
ДРПНК-2,0 Многоканальный дифрактометр для экспрессного определения количества фаз в различных отраслях промышленности 1500 . 10 Имеет пять измерительных каналов
ДРАМ-2,0 Измерение малоуглового рассеяния (растворы, пленки, волокна, фольги) 5 1000 10
* На дифрактометр могут монтироваться высокотемпературные (ГПБТ-1500. УРВТ-2000) и низкотемпературная (УНРТ-180) приставки.
118
ТАБЛИЦА 5.16
КАМЕРЫ ДЛЯ РЕНТГЕНОСТРУКТУРНОГО АНАЛИЗА [30J
Тип Назначение Важнейшие характеристики
РКД Получение дебаеграмм Камера светонепроницаемая. 35-мм пленка прижимается к внутренней цилиндрической поверхности камеры диаметром 57,3 мм. Регистрация углов 0=44-84° Центровка образца производится с помощью магнитного держателя — диска
РКЭ Экспрессная съемка поликрнс-таллических образцов фокусирующим методом Фокусировка пучка, исходящего непосредственно из фокуса трубки. Пятикадровая плоская кассета. Два интервала углов регистрации: малые 0=104-4-30° илн большие 0=604-88°. Расстояние от оси наклонов образца до пленки 50—150 мм
КРОС Измерение параметров ячейки и величины напряжений в полн-крнсталлнческих образцах Камера с оптической скамьей и двумя секторными кассетами: дисковой и цилиндрической. Расстояние от образца до кассеты 25—100 мм. Возможно вращение образца и кассеты как совместное, так и независимое
РКУ-114 Прецизионные измерения на монокристаллах и на поликристаллах Камера светонепроницаема и позволяет получать снимки с неподвижного, вращающегося илн колеблющегося образца. Положение н величина интервала колебаний могут устанавливаться и заменяться в процессе съемки. Диаметр камеры 114,6 мм. Величина углов отражения 4—86°
РКСО-2 Съемка монокристаллов. Получение лауэграмм и эпиграмм. Возможно исследование текстур Имеются две плоские кассеты: одна для охвата’ углов 0 =24-30°, другая 60—87°. Расстояние образец— пленка 40 мм. Имеются две гониометрические головки: ГГ-2 с возможностью наклона по дугам головки на ±60° и ГГ-3 с возможностью плавного наклона по дугам на ±15°
тически комплект камер в лаборатории складывается в соответствии с кругом задач исследований- в данной лаборатории. Наиболее часто в металловедческих исследованиях применяются камеры для анализа поликристаллов и среди иих камеры Дебая (например, тип РКД) и камеры обратной съемки (например, тип КРОС). Для всех камер общими конструктивными элементами являются: коллиматор “система диафрагм, задающая угол расходимости и форму падающего на образец пучка; держатель образца; кассета, где помещается пленка.
Камера РКД предназначена прежде всего для фазового анализа поликрнсталлнческих объектов в виде порошка, проволоки, фольги нли шлифа.
Наиболее выгодный тип образца — цнлнндр (столбик) диаметром 0,4—0,8 мм, длиной несколько миллиметров. При таком образце получают систему линии, соответствующих углам 6 от 4 до 84°, которые симметрично располагаются относительно выходного (тубус) и входного (коллиматор) отверстий (см. рис. 5.17).
В случае съемки столбика углы рассчитывают по формуле
ег = 2£г(57,3/2Дк), (2!)
где 2Lt — расстояние между линиями, симметричными относительно выходного отверстия; Вк— диаметр камеры, т. е. цилиндра, по которому свернута пленка.
Наиболее целесообразно асимметричное расположение пленки (когда ее концы сходятся
между отверстиями тубуса и коллиматора — рис. 5.17, а). Прн этом оказывается возможным определить эффективный диаметр камеры путем измерения расстояний между линиямя двух пар, симметричных около отверстия тубуса и отверстия коллиматора (Д и В). Для определения углов 0 (при 0>45°) измеряется расстояние X.
При так называемом стандартном диаметре Ок=57,3 мм: В(град)=2£^ (мм)/2.
Используются также цилиндрические камеры диаметром 86 и 114 мм. Образец-столбик во время съемки может вращаться, это позволяет получать сплошные лнннн даже в случае крупнозернистой структуры. Образец в виде шлифа — пластинки размерами до 10х12х Х5 мм — размещают так, чтобы ось камеры лежала в плоскости шлифа. Прн измерении периода решетки плоскость шлифа должна находиться под углом 90° к падающему пучку, при фазовом анализе (см. рис. 5.17, б, в)— под углом—35°, прн этом получают практически весь набор возможных отражений.
Камера КРОС предназначена для получения от поликристаллического образца ограниченного числа линий (одна-две) при большом угле 0, обеспечивающих (при известной структуре объекта) наиболее точное измерение периода решетки и связанных с ним характеристик материала — состава твердого раствора, зональных напряжений, а также характеристик структуры материала, связанных с шириной линии (дисперсность кристаллитов, плотность дислокаций), интенсивностью (мозаичная струк-
119
тура, статические н динамические смещения атомов) и распределением интенсивности вдоль линии (текстура).
Обычный вид образца — шлиф (размеры в плоскости шлифа до 60x50 мм, толщина до 25 мм). Плоскость шлифа располагается перпендикулярно падающему пучку, возможно вращение образца вокруг оси, параллельной оси пучка. В комплект входят плоская и цилиндрическая кассеты. Пло'ская имеет секторный вырез, позволяющий получить на одной пленке шесть рентгенограмм с симметричными линиями при диаметре до 150 мм или двенадцать односторонних рентгенограмм. Подобное приспособление имеется и для цилиндрической кассеты. Расстояние образец — пленка 25— 100 мм, возможно получение линий при В от 54 до 85 °. Углы 0 определяются нз соотношения;
tg (я-20) = Кн^М, (22)
где Кнкь — радиус дебаевского кольца (HKL) (практически измеряется диаметр 2Rhkl); А — расстояние образец — пленка; для определения этого расстояния обычно снимают рентгенограмму эталонного вещества с точно известным периодом решетки.
Одно нз условий прецизионности измерений — геометрия съемки, от которой зависит острота линии. Это обеспечивается коллимированием пучка нлн созданием специальных условий фокусировки. Одна нз схем фокусировки — расположение поверхности анализируемого образца, анализируемой линии на рентгенограмме и источника излучения (анода трубки или диафрагмы) на одной окружности. Эта схема осуществима в камере типа КРОС. Специальные фокусирующие камеры (экспрессные) позволяют резко сократить экспозиции (камера РКЭ, табл. 5.16), что особенно важно при использовании монохроматоров. Условия для прецизионной съемки рентгенограмм указаны в табл. 5.17.
Моиохроматнзация первичного пучка наиболее эффективна с помощью изогнутого кристалла-монохроматора (рнс. 5.18). При такой схеме съемки используется широко расходйщийся пучок, а максимумы оказываются узкими, т. е. сфокусированными, если монохроматор изогнут по радиусу, равному диаметру барабана, на котором располагается пленка. Моиохромати-зация позволяет существенно повысить чувствительность фотометода и обнаруживать самые слабые максимумы за счет уменьшения фона рентгенограммы.
Расстояния между максимумами измеряют либо линейкой со скошенным краем (точность 0,1 мм), либо специальным оптическим прибором-компаратором (точность 0,01 мм). Иногда компаратор объединяют с микрофотометром.
Существенное преимущество фотометода перед дифрактометрическим заключается в возможности выявления очень слабых рефлексов; фотометод имеет также некоторое преимущество в тех случаях, когда анализируются сложные по геометрии картины диффузного рассеяния.
Недостатки фотометода, а именно: длительность экспозиции и необходимость фотометрн-рования при анализе интенсивности или ширины линии в известной мере преодолеваются прн
ТАБЛИЦА 5.17
УСЛОВИЯ ДЛЯ ПРЕЦИЗИОННОЙ СЪЕМКИ РЕНТГЕНОГРАММ [201
Исследуемый элемент (фаза) .Рекомендуемое излучение Длина полны О излучения, А Рекомендуемая для фокусирования 1 линия Угол скольжения 0
Феррит и мар- K₽Fe 1,75653 (310) 75°39*
тенент Со I,78892 (ЗЮ) 81°25«
KPV 2,38434 (211) 77°26е
Кс^ Cr 2,28962 (2П) 78°06*
Аустенит Kat Со 1,78892 (400) 82°00*
КрСг 2,08480 (ЗП) 73'W*
Каг Fe 1,93597 (222) 69°00?
Никель Kat Си 1,54051 (420) 73°55*
Kcq Мп 2,10175 (ЗП) 80°10*
КВСг 2,08480 (ЗП) 78W
KatV 2,50348 (220) 8640'
Медь Кс^ Си 1,54051 (420) 72°17*
Каг Со 1,78892 (400) 81°40г
КрСг 2,08480 (222) 86о52е
Циик KtXjCu 1,54051 (1016) (1232) 82°50' 69°30'
Вольфрам KtXj Ni 1,65784 (321) 78°50'
Кс^Си 1,54051 (400) 77°10г
Fe4N (у'-фаза) Ка3 Cr 2,28962 (ЗН) 77'00*
Fe2N (е-фаза) Cr 2,28962 (1013) (1122) 68с30' 80°40*
a-WsC Kcq Co 1,78892 (2132) 79с00*
WC Кщ Co 1,78892 (211) 82°40'
VC KpCu 1,39217 (531) 82°00*
TiC KP Си 1,39217 (442), (600) 74°00'
Германий Kp Fe 1,75653 (620) 79';02'
KccjCo 1,78892 (440) 75W
KpCo 1,62075 (444) 82°55'
К«! Си 1,54051 (7Н) 76°26'
KpCu 1,39217 (800) 79°20'
Кремний Kctj Co 1,78892 (531) 77°03'
Ка, Cu 1,54351 (444) 78°46'
Алюминий Kcq Co 1,78892 (420) 81°02'
Kaj Cu 1,54051 (5U), (333) 81°10'
использовании фокусирующих камер и автоматизации процесса фотометрироваиия.
б. Рентгеновские дифрактометры. В современных дифрактометрах дифрагированное излучение регистрируется с помощью счетчиков пропорционального (газовый), сцинтилляционного (кристалл-сцинтиллятор Nal, активированный Т1) или полупроводникового (Si нли Ge, легированный Li). Амплитуда сигнала на выходе этих счетчиков пропорциональна энергии рентгеновских квантов, что позволяет прн использовании амплитудного дискриминатора выделять (регистрировать) кванты только в определенном диапазоне энергий (илн длин волн). Дифрактометр обеспечивает установку н отсчет углов дифракции (20 — угол между первичным н дифрагированным пучком) с точностью до 0,01—0,005 е. Регистрация дифракционной картины проис-
о
Рис. 5.18. Схемы съемки в фокусирующей камере с применением монохроматора (а — на отражение, б — на просвет):
Ф — фокус рентгеновской трубки; М — кристалл-монохроматор; О — образец; П пленка
•ходит последовательно при изменении угла между отражающими плоскостями и первичным пучком, а ие одновременно для всех углов дифракции, как в рентгеновской камере. Нанбо-
Рис. 5.19. Оптическая схема (фокусировка по Брэггу— Брентано), обычно используемая в дифрактометрии:
1 — генераторное устройство; 2 — трубка; 3 — диафрагмы первичного пучка; 4 — образец; 5 — счетчик; 6 — диафрагма счетчика
лее часто применяют дифрактометры с фокусировкой по Брэггу—Брентано (плоский образец) — рис. 5.19.
Современные отечественные дифрактометры общего иазиачения типа ДРОН позволяют регистрировать днфрактограммы прн:
а) непрерывном автоматическом изменении угла дифракции 0 (счетчик вращается с вдвое большей угловой скоростью, чем образец) с записью интенсивности иа лейте электронного потенциометра;
б) шаговом (с различным заданным интервалом) перемещении образца и детектора, прн этом интенсивность регистрируется в течение заданного времени илн до заданного числа квантов и результаты- выводятся на цнфропе-чать нли иа перфоленту вместе со значением угла дифракции;
в) непрерывном движении счетчика и образца в заданном интервале углов дифракции; суммарное число набранных квантов выводится иа цифропечать или перфоленту. Некоторые приборы комплектуются с ЭВМ, управляющей работой дифрактометра по заданной программе и обрабатывающей полученные результаты, т. е. работают автоматически.
Сменные приставки позволяют анализировать крупнозернистые образцы (вращение образца в собственной плоскости), получать прямые н обратные полюсные фигуры, вести съемки прн высоких (до 2200 °C) н низких (до —180 °C) температурах, исследовать и ориентировать монокристаллы.
В дифрактометре предусмотрена монохрома-тизация излучения отражением от кристалла на первичном и дифрагированном пучке. Последнее более эффективно, поскольку позволяет избавиться и от флуоресцентного излучения образца.
Дифрактометрию целесообразно применять тогда, когда необходимы точные количественные, измерения интегральной интенсивности нлн распределения интенсивности в зависимости от угла дифракции1, а также анализ интенсивности диффузного фона. Наиболее часто дифрактометрия используется при количественном фазовом анализе, прецизионном измерении периодов решетки н определении величины напряжений, при анализе формы н ширины интерференционного максимума, анализе текстур.
При дифрактометрических исследованиях возможны ошибки в определении углового по
1 Поэтому в дифрактометрах стабилизируют питающее напряжение и ток через трубку, чтобы интенсивность излучения была достоянной.
121
ложения максимумов н в их профиле (распределение интенсивности в функция угла дифракции). Основные источники ошибок: эксцентриситет образца (ось поворота для изменения углов дифракции не лежит в плоскости образца), конечная глубниа проникновения лучей в образец, вертикальная расходимость пучка (для отечественных дифрактометров, в которых ось поворота для изменения углов дифракции вертикальна), конечная ширина приемной щели детектора, погрешность в определении нулевого положения счетчика. Этн ошибки могут быть устранены нли их влияние резко уменьшено прн правильной юстировке прибора и выборе оптимальной геометрии прямого и дифрагированного излучений.
Кроме того, следует учитывать ошибки в определении интенсивности из-за статистического распределения во времени чнсла квантов, испускаемых рентгеновской трубкой (статистические ошибки счета).
Амплитудное разрешение полупроводникового детектора, т, е. его способность различать кванты с малой разницей в энергии (AS), позволяет получать дифракционную картину от полн- нлн монокристалла прн постоянном угле дифракции и использовании полихроматического излучения.
Уравнение Вульфа — Брэггов 2dhhi sin может быть переписано с учетом hcl^=E:
dhki — hc/2E sin 9 = 6,199/E sin 6; (23)
это выражение дает возможность, иайдя (с помощью многоканальных анализаторов) энергию квантов (£), образовавших прн интерференции дифракционный максимум, определить dtiM. Точность зависит от минимального значения ЛЕ для полупроводникового детектора. У современных счетчиков точность составляет &d/d=0fi5 + 0,005.
Метод — скоростной: рентгенограмма получается за 10—15 с, что позволяет изучать кинетику фазовых превращений, изменения при деформации и т. п.
Кроме того, использование полупроводникового детектора н одноканального анализатора в стандартных дифрактометрах позволяет обойтись без применения кристаллических монохроматоров при том же примерно отношении интенсивности сигнала к фону, но со значительно большей светосилой.
Дифрактометр ДРОН-2,0 (дифрактометр общего назначения). Состоит нз следующих узлов:
— генераторного устройства [высоковольтный источник питания мощностью 2 кВт при максимальном напряжении на трубке 50 кВ и максимальном токе трубки 60 мА (ВИП-2-50-60)];
— стойки с защитным кожухом трубки и механизмом юстировки трубки (например, тип БСВ-12 с фокусом 1X12 мм, БСВ-14 с фокусом 0,04X8 мм);
— гониометра ГУР-5 (радиус 180 мм) с приставками .для анализа текстур, для анализа монокристаллов и для съемки неподвижных образцов;
— измерительного и регистрирующего устройства ЭВУ-1-4;
— устройства для вывода информации (УВИ-ЗМ-1) на самопишущий потенциометр, цифропечать нли перфоратор;
— блока автоматического управления (БАУ);
— блока детектирования (сцинтилляционный н пропорциональный счетчики).
Пределы измерения углов 6 от —90 до +165°, ошибка измерения ±0,005 °.
Стабилизация высокого напряжения и анодного тока не хуже 0,1 %.
Основные преимущества дифрактометрического метода — возможность полной автоматизации работы, включая обработку результатов измерений, что все в большей степени реализуется в современных приборах.
Отечественная промышленность приступила к серийному выпуску дифрактометров, управляемых вычислительной машиной — ДРОН-УМ!. В них применен управляющий и вычислительный комплекс «Искра», новый гониометр ГУР-8 (радиус 192 мм) с комплектом приставок, аналогичных приставкам для ГУР-5, источник питания ВИП-2-50-60М, обеспечивающий поддержание установленного анодного тока и напряжения на трубке со стабильностью не хуже 0,03 %. Программное обеспечение позволяет полностью автоматизировать сбор н обработку измерений, а также контролировать работоспособность дифрактометра.
Подготовка образцов для рентгеноструктурного анализа в общем случае состоит в том, чтобы придать нм оптимальные для данного вида съемки размеры и форму, не нарушая структурного состояния. При подготовке шлифов необходимо удалить поверхностный слой (обычно 0,1—0,2 мм), искаженный механической обработкой. Для этого шлиф протравливают в разбавленных растворах сильных кислот нлн электролитически по режимам травления или полировки, используя те же реактивы, которые применяются в металлографической практике1.
Толщина слоя, участвующего в формировании рентгеновской дифракционной картины, для стали составляет десятки микрометров. В ряде случаев, например прн фазовом анализе в камере РКД, наиболее целесообразно использовать образцы в виде порошка, который наклеивают обычно цапон-лаком иа тонкую стеклянную нить нлн набивают в капилляр, например из коллодия. Прн этом рассеяние рентгеновских лучей веществом связки должно быть относительно мало, образец-цилиндр должен иметь возможно меньший диаметр (0,4—0,8 мм). В схемах съемки от шлифа (камера КРОС, дифрактометр) порошком заполняется путем прессования со связкой нли без связки специальная кювета. Порошок, приготовленный нз монолитного образца, просеивают через сита (80—320 меш). Наклеп от иапнливання, сверления нли дробления и истирания в ступке снимают отжигом. При приготовлении образца не должны образовываться загрязнения (частицы материала напильника и т. п.), иначе возникнут их линии на рентгенограмме.
Глубина анализируемого слоя и анализ поверхностных слоев. Ограниченная проникающая способность рентгеновских лучей является достоинством метода рентгеноструктуриого анализа прн изучении структуры поверхностного слоя нлн изменения структуры по глубине. Толщину слоя вещества, участвующего в фор-
• Может оказаться удобным снятие наклепа путем отжига, но при этом не следует допускать фазового превращения, образования окисла йли обезуглероженного слоя.
122
мнровании рентгеновских линий при съемке на отражение, можно оценить экспериментально. Например, если объект исследования железо, то на поверхности образца можно наносить слои никеля заданной толщины (конденсацией из паров и др.) н определять их толщину, прн которой в дифракционной картине исчезнут линии основы Для оценки толщины работающего слоя рассчитывают долю интенсивности лучей, рассеянных слоем х, исходя из коэффициента ослабления р, (зависит от порядкового номера z, плотности вещества образца р и длины волны Л [12, 14, 20]) и направления лучей, падающих на образец и дифрагированных, по отношению к поверхности шлифа (соответственно а и £):
G(x) — 1 —exp (—px(l/sintx4- 1 /sin £)]. (24)
На рнс. 5.20 Дана зависимость G(x) для случая обратной съемки (камера КРОС, «%=
Рис. S.20. Доля интенсивности рассеянного излучения в зависимости от толщины «отражающего» слоя (условия указаны в тексте}
=90 °) образца стали в Со-излученин; р= =26—90“^47и. Видно, что 95 % интенсивности связано с отражающим слоем 0,025 мм н из инх—50 %—со слоем всего лишь 0,005 мм. Слой толщиной 0,025 мм примерно равен поперечнику одного зерна прн размере его, соответствующем баллу 8. Эффективную толщину слоя (х) можно оценить в случае обратной съемки (камера типа КРОС, а=90°, Р=20—90°) по формуле
х — U sin p/р, (1 4- sin р), (25а)
а в случае дифрактометра х= U sin ct/2j.i. (256).
Величины U для выбранного значения G(x) приведены ниже [20]:
6(х).......... 0,50 0,75 0,95
U............. 0,69 1,39 3,0
Если глубина слоя с изменяющейся структурой материала (наклеп при поверхностной обработке, химико-термическая обработка и т. д.) существенно превосходит толщину отражающего слоя, то проводят послойный анализ, при этом удаление слоев не должно заметно нарушать структуру. Изменяя жесткость излучения (величины р.), а также угол падения лучей на образец (угол а), можно изменять
эффективную толщину в значительных пределах. При переходе от излучения Сг к излучению КаМо коэффициент ослабления ц н, следовательно, толщина слоя изменяются почти в 10 раз. Для случая съемки Fe иа излучении КаСо при переходе от се» 6 к tx»l° толщина слоя изменяется от 10—15 до 0,5 мкм. При использовании малых а (скользящий пучок) формулы для оценки отражающего слоя усложняются [20].
5.2.5. Дозиметрия и техника безопасности в лабораториях рентгеноструктурного анализа
Доза излучения — это поток излучения на единицу площади. Такое определение имеет ясный физический смысл, однако действие рентгеновских лучей на человеческий организм1 прн равной энергии существенно зависит от качества (жесткости) излучения. Биологическое действие вызывает именно та часть энергии, которая поглощается. Поэтому введено понятие поглощенная доза (ПД), или доза, измеряемая энергией (поглощенной) на единицу массы (Дж/кг). Специальной единицей ПД является рад (1 рад=100 эрг/г=10~2 Дж/кг). В расчетах поглощенной дозы учитывают средний состав мягкой биологической ткани: 76,2 % О; 11,1 % С; 10,1 % Н; 2,6 % (по массе) N. В нормах радиационной безопасности используют понятие эквивалентная доза (Экв. Д), которое с помощью коэффициента качества учитывает зависимость неблагоприятных биологических последствий облучения от качества (жесткости) излучения. Специальной единицей Экв. Д является бэр, равный 1 рад/Q, где Q — коэффициент качества; для рентгеновских лучей Q=l. Нормами радиационной безопасности (НРБ—76) устанавливается предельно допустимая доза (ПДД) — наибольшее значение индивидуальной Экв. Д за год, которое при равномерном воздействии в течении 50 лет не вызовет в состоянии здоровья персонала неблагоприятных изменений, обнаруживаемых современными методами. Для лиц, непосредственно работающих с источниками ионизирующих излучений в условиях облучения всех частей тела, установлена ПДД, равная 5 бэр в год [33].
Для измерения дозы рентгеновского и гамма-излучения было предложено использовать ионизирующее действие этого излучения при поглощении лучей в воздухе, т. е. полный заряд нонов одного знака, возникающих в воздухе при полном торможении всех вторичных электронов, которые были образованы фотонами. Таким образом получают экспозиционную дозу (ЭД), характеризующуюся величиной заряда иа единицу массы (Кл/кг). Специальной единицей ЭД является рентген; 1Р= =0,285 Кл/кг. Доза 1Р отвечает заряду ионов в одну электростатическую единицу в 1 см3 атмосферного воздуха при 0 °C н 760 мм рт. ст. (масса 0,001293 г). В случае мягкой биологической ткани н рентгеновских лучей 1Р= =0,93 рад» 1 рад; 1 Р»1 бэр.
1 Биологическое действие ионизирующих излуче-
ний и связанные с этим общие законодательные положения описаны в учебниках (например,
с. 481—482}.
123
Приведенная выше ПДД (5 бэр за год) соответствует 0,1 Р за неделю или мощности дозы около 3 мР/ч1.
Безопасные условия работы обеспечиваются защитными устройствами современной аппаратуры и строгим выполнением работающими необходимых правил. Организационно-технические мероприятия включают: правильную установку аппарата, а также дополнительных устройств для защиты работающих от облучения в соответствии с особенностями эксплуатации аппаратуры в данной лаборатории, определение с помощью дозиметров безопасных зон и зон повышенной опасности возле аппарата, периодический дозиметрический контроль, составление рабочих инструкций по работе, включающих правила техники безопасности и мероприятия при аварийной обстановке, специальное обучение и периодическое проведение профосмотра персонала. Работа на аппаратах с отключенными илн снятыми защитными устройствами запрещена.
Прн работе с аппаратами для рентгеноструктурного анализа имеется опасность поражения работающих электрическим током и ионизирующим излучением. Защита от поражения электрическим током обеспечивается защитными ограждениями н системой блокировок. Однако следует иметь в виду, что блокировочные устройства (дверцы и пр.) разрывают цепь высоковольтного питания и разряжают емкости в высоковольтной схеме, но прн этом обычно не происходит обесточивания цепей низкого напряжения (220—380 В).
Современные рентгеновские трубки для структурного анализа, дают весьма значительную интенсивность излучения: у окошка трубки она может достигать порядка 105 рентген в минуту. Если у окошка трубки камера не установлена, то окошко должно быть закрыто специально установленной на кожухе трубки задвижкой. Камера (нли система гониометра) имеет коллимационное устройство, вырезающее узкий пучок, направляемый на объект исследования. Установка камеры (гониометра) возле трубки и специальные защитные устройства должны преграждать пути для вредного рассеянного излучения. При установке камеры (и образца) под пучком флюоресцирующий экран иа выходе лучей нз камеры должен быть закрыт свинцовым стеклом, во всяком случае глаз человека ие должен попадать под прямой рентгеновский пучок.
5.3. АНАЛИЗ
ФАЗОВОГО СОСТАВА
5.3.1. Фазовый (качественный и количественный) анализ
. Рентгеноструктурный метод определения фазе- . вого (вещественного) состава является прямым, поскольку в его основе лежат характеристики атомногкрнсталлической структуры вещества. Данные этого анализа вместе с обычно имеющимися сведениями о его химическом составе однозначно характеризуют фазовый состав в пределах чувствительности метода.
• По данным НРБ—76. естественный фон излучения на территории СССР соответствует мощности экспозиционной дозы 4—20 мкР/ч 133].
Для'доказательства присутствия той нлн иной фазы в сплаве необходима регистрация нескольких характерных отражений. На использовании этих же отражений основано определение природы фаз (качественный фазовый анализ). Для получения по возможности более точных значений межплоскостных расстояний din и интенсивностей соответствующих отражений, а также в связи с тем, что объекты анализа чаще всего мелкокристаллические й поликрнсталлические вещества — целесообразно использовать метод порошка (камера Дебая, камера Ги.нье илн дифрактометр). В необходимых случаях прибегают к измельчению объекта и отбору оптимальной для анализа фракции порошка. Прн этом необходимо предупреждать ялн учитывать возможные прн препарировании объекта изменения его фазового состояния, например в результате пластической деформации н нагрева при механическом размельчении и отжиге, а также изменение фазового состава осадков при химическом или электрохимическом высаживании илн прн последующих операциях препарирования.
Качественный фазовый анализ. Рентгеноструктурные дапные для идентификации фазы можно иайтн в справочниках, построенных по алфавиту или по химическому (минералогическому) признаку.
Первый этап идентификации фазы проводят по трем интенсивным линиям, располагающимся в таблице поиска в порядке убывания интенсивности. При этом для каждой фазы указывают иомер соответствующей карточки (или таблицы), которая содержит полную характеристику фазы. В основе поиска могут быть сами рентгеноструктурные данные. Для этого используют таблицы, в которых указанные выше данные о d/п для наиболее интенсивных линий расположены по группам с убывающим значением d]n для первой (самой интенсивной) линии и далее (внутри групп) по подгруппам с убывающим значением djn второй линии. Этот принцип составления поисковых таблиц был предложен в 1938 г. В. И. Михеевым [35] я независимо от него Ханевальтом [36]. В настоящее время наиболее широко для идентификации фаз используют Рентгенометрический определитель минералов В. И. Михеева [35], картотеку Американского общества испытания материалов (ASTM) [37] илн продолжающееся издание Объединенного Комитета по порошковым дифракционным стандартам (ICPDS) [38] (издаются материалы в виде картотек, кинг, включающих карточки и указатели (ключи), а также в виде микрофишей и магнитной записн).
Определитель В. И. Михеева [35] включает данные почти для 1000 веществ .(главным образом минералов). Издания ICPDS к 1977 г. охватывали около 32000 веществ, из которых 22350 неорганических. Отдельно - изданы два тома карточек и том указателя для металлов и сплавов [39].
Сводная таблица рентгеноструктурных характеристик в [35] содержит din н интенсивности пяти наиболее сильных линий. Практически большинство веществ находят по самой сильной н второй по интенсивности ЛИНИН, но в некоторых случаях требуются и третья, н четвертая линии.
Для поиска с помощью картотеки ASTM [37] необходимо знать межплоскостные рас-
стояния для трех самых сильных линий с точ-о
иостью не хуже 0,01 А н их интенсивности, оцененные по 100-балльной шкале. Имея в виду возможные колебания интенсивности и ошибки в их оценке, указатель составлен так, что каждое вещество упоминается трижды: поочередно каждая линия записывается первой в соответствующей группе. В таблицах [38] к указанным трем добавляются еще пять следующих по интенсивности линий.
Прн анализе двух- и многофазных смесей без данных о химическом составе нельзя рассчитывать на успешную идентификацию.
Поиск существенно затрудняется нз-за низкой точности при измерениях din и ошибок в оценке интенсивности, особенно при близких интенсивностях характерных линий. Предпочтительнее использовать дифрактометрические данные, чтобы получить надежные данные об интенсивности. При фотометоде целесообразнее съемка в фокусирующих камерах. При диаметре камеры 114 мм ошибка Дй для О
d= I -:~3 А прн съемке по схеме Дебая составля-О
ет 0,002—0,010 А, а прн съемке по схеме О
Гинье 0,001—0.003 А. При составлении таблиц поиска возможные экспериментальные ошибки учитываются перекрытиями значений din (в О
области около 2 А это перекрытие составляет о о о
0,01 А, а в области 5 А равно 0,05 А), т. е. одно и то же вещество приводится дважды — в соседних главных группах.
В заключение проводят ннднцированне всех имеющихся линий с помощью основной таблицы, что необходимо не только для контроля идентификации фазы, но н для того, чтобы убедиться в отсутствии другой фазы, а при наличии второй фазы—выделить лнннн этой фазы для ее последующей идентификации.
Выявление второй фазы ограничивается чувствительностью метода рентгеноструктурного анализа, которая определяется возможностью регистрации нескольких характерных (т. е. самых интенсивных) линий. Эта чувствительность зависит от рассеивающей способности атомов элементов, составляющих вторую фазу, от ее кристаллической структуры, а также от степени искажения решетки. Чем меньше порядковый номер элемента (z), ниже симметрия решетки (меньше фактор повторяемости в выражении интенсивности), тем ниже чувствительность. Для остаточного аустенита в стали эта чувствительность около 1 %, а для цементита — несколько процентов. Чувствительность качественного фазового анализа существенно улучшается при использовании монохроматизации. снижающей уровень интенсивности фона.
Для идентификации фаз, присутствующих в сплавах в очень малых количествах, широко используют методы химического нлн электрохимического растворения и соответствующей обработки осадка.
Была предложена [40] методика рентгеноструктурного анализа отдельных частиц неметаллических включений размером 5=40 мкм, заранее отмеченных на шлифе; методика предусматривает извлечение частицы из металла и ее размещение иа специальном держателе образца в рентгеновской камере.
Прн исследовании многофазных материалов
по методу Дебая полезна съемка без вращения образца. При этом характер лниий (размы-тяе, точечиость, текстурные максимумы) может дать основание для группировки линий по принадлежности к разным фазам.
Качественный фазовый анализ упрощается, если известен химический состав и типы возможных фаз. В табл. 5.18 приведены рентгеноструктурные данные для идентификации некоторых фаз, встречающихся в сталях и в сплавах на железной основе.
Разрабатываются информационно-поисковые системы для рентгеновского фазового анализа поликристаллических сплавов или смесей. Система, описанная в [41], составлена на базе ЭВМ БЭСМ-4М и состоит из ряда программ, решающих отдельные задачи: отбор возможных эталонных фаз, расчет критериев достоверности существования именно дайной фазы в образце н воспроизведение фазового состава по отобранным эталонам. Анализ может быть проведен для образца известного химического состава. Если последний известен, то это облегчает отбор возможных эталонов.
Программа не учитывает изменения величин межплоскостиых расстояний н интенсивностей отражения для фаз, связанных с легированием и образованием твердых растворов. Очевидно, в этом случае достоверность определения фазового состава будет хуже. Задача может быть решена в случае известного химического состава сплава. Это позволяет предположить, а затем н подтвердить с помощью теоретического нндицнровання и расчета интенсивностей присутствие фазы определенного структурного типа.
Количественный фазовый анализ. Такой анализ проводят обычно с помощью дифрактометров. Для случая двух фаз, имеющих близкие коэффициенты ослабления рентгеновских лучей, можно определить коэффициент пропорциональности (К) в уравнении, связывающем отношение интенсивности линий, выбранных для анализа, с отношением количеств этих фаз:
/£/7, = КХ27(1-Хй). (26)
Величину К определяют экспериментально для смеси с известным соотношением фаз нли теоретическим расчетом где Ki н
К2— 'факторы интенсивности рентгеновских отражений данных линий анализируемых фаз). В других случаях требуется построение градуировочных графиков и применение эталонных веществ.
При выборе условий съемки и эталонных веществ следует предупреждать возможные наложения линий анализируемых фаз. Точность определений зависит от многих факторов— конкретных объектов, условий съемки н требующейся экспрессностн. В наиболее благоприятных условиях погрешность в определении количества фаз оказывается близкой к погрешности в измерениях интенсивности, т. е. до 1—2 %.
К проведению количественного фазового анализа может быть приспособлена конструкция дифрактометра: несколько упрощено гониометрическое устройство н добавлены счетно-решающие устройства (дифрактометр ДРФ-2).
Для проведения серийных анализов сложных многофазных сплавов (или смесей) целе-
125
ТАБЛИЦА 5.18
РЕНТГЕНОСТРУКТУРНЫЕ ДАННЫЕ ДЛЯ ИДЕНТИФИКАЦИИ ФАЗ В СТАЛЯХ (НИТРИДЫ, КАРБИДЫ. БОРИДЫ, ОКИСЛИ, СУЛЬФИДЫ)*1
Фаза Кристаллическая система, структурный тип*5 ——•— •“* J -о Меж плоскостные расстояния, А Относительная интенсивность
A1N Г, В4 3,70 2,37 2,49 100 70 60
Fe2N о 2,11 1,17 0,93 100 43 36
FeoN г 2,09 2,19 1,61 100 25 25
Fe4N к 2,19 1,14 1,90 100 83 77
CrN К, В1 2,40 2,07 1 100 100 —
Cr2N г 2,10 2,38 2,22 100 25 25
NbN076 т 2,49 1,53 1,32 100 80 80
E-NbN г, Bi 1,30 1,02 1,87 100 100 90
TiN К, В1 2,12 2,44 1,50 100 77 56
e-TiN т 2,28 2,46 2,21 100 80 80
г 2,15 1,41 1,29 100 80 80
VN к, bi 2,47 2,14 1,62 100 100 70
ZrN К, В1 2,64 2,28 1,61 100 100 80
Co2W4C К, Т)2 ) 2,16 2,57 2,29 100 57 48
CosW8C к, tfc Е93 2,13 2,26 2,77 100 61 37
Fe4(Mo, W)2 C К Г 2,12 2,25 1,30 О.С С. С.
Сг3С2 О, В54о f 2,30 2,24 1,95 100 100 100
{ 2,30 2,53 2^24 100 60 60
СГ7С3 г (Тр) 2,03 2,28 2,11 100 50 60
К, £>84 2,38 2,17 2,05 100 100 100
0-Fe3C О, D0„ 2,01 2,06 2,38 100 70 65
e-Fe^C Г 2,08 2,16 1,60 100 60 60
Mo2C г, £'э 2,28 1,50 1,35 100 35 35
NbC(Nb4C3) К, £1 2,56 2,22 1,57 100 100 100
TiC К, £1 2,18 2,51 1,54 100 80 50
VC (V4CS) К, В1 2,40 2,07 1,47 100 100 50
WC Г, Bh 1,87 2,51 2,83 100 80 70
wsc г, jL's 2,27 1,49 1,07 100 60 60
1,35 1,00 2,35 100 100 70
ZrC К, в\ 2,70 2,34 1,65 100 75 50
AlBa Г, С32 2,04 2,61 1,51 100 40 30
CoB О, В27 2,19 1,85 1,20 100 100 100
Co2B Т, С16 2,50 1,97 1,19 100 100 100
FeB О, В27 2,19 2,01 1,90 100 100 100
Fe2B Т, С16 2,01 2,12 1,63 100 25 18
6-M0B*3 Т, Во 2,12 2,72 2,28 100 90 90
0-MoB*4 о, Bf 2,13 2,10 2,49 100 67 65
M0B24 Г, С32 2,00 2,64 3,07 100 90 42
Mo2B Т. С16 2,20 2,37 1,33 100 90 90
а-А12О8 (корунд) Г, D5i 2,09 2,55 1,60 100 92 81
3Al2Os-2SiO2 (муллит) О 3,38 2,20 3,41 100 75 /1
2CaO- AlsO3-SiO2 (re- т 2,85 1,75 2,43 100 100 70
леннт) 2,41 70 70
CaO-MgO-SiO2 (moh- О 1,81 1,59 2,66 100 80
тичеллнт) 2,65 1,81 1,59 100 90 90
3(Ca, Fe, Mn,Mg)O- к 2,61 1,56 1,27 100 80 80
-(Al, Fe, Cr)2O3- 1,62 60
-3SiO2 (гранаты) Сг2О3 (двуокись хрома) Г, D51 2,67 2,48 1,67 100 96 90
FeO (вюстит) к, в\ 2,15 2,49 1,52 100 80 60
cc-Fe2O3 (гематит) ГЦр),^! 2,69 2,51 1,69 100 80 80
T-Fe2O3 к 2,62 1,48 2,59 100 53 34
C-Fe2Os г 2,19 1,69 2,63 100 100 80
FesO.j (магнетит) К, 2,53 1,48 2,97 100 70 60
Fe3O4 (магнетит) О 1,28 1,09 1,05 100 100 100
MgAl.2O4 к, B\i 2,44 2,02 1,43 100 58 58
MgO AlgOg / 2,42 1,42 4,63 100 100 80
( 2,02 80
MnO A12O3 (галаксит) к 1,40 2,50 2,39 100 50 50
2,93 17 58
MnO (манганозит) К, В! 2,22 2,57 1,57 100 62
126
Продолжение табл, 5.18
Фаза Кристаллическая система, структурный тип*2 О Межплоскостные расстояния, А Относительная интенсивность
MnO-SiO2 (родонит) 2,94 2,60 2,97 2,76 100 80 90 80
SiO2 (а-кварц) г 3,34 4,26 1,82 100 35 17
SiO2 (а-кристобалит) Т 4,04 2,49 2,85 100 18 14
SiO2 (а-кристобалит) К 4,14 2,53 1,64 100 90 70
ZrO2-SiO2 (циркон) т 3,30 1,71 4,43 2,52 100 40 45 45
FeS (пирротит) г, 88i 2,09 2,98 2,67 100 90 90
MnS(cz), (Мп, Fe)S К, 51 2,61 1,85 1,51 100 48 19
CrS (0) м 2,06 2,02 1,71 100 100 100
CrS(₽') г, 581 2,02 2,60 1,31 100 90 90
TiS Г 2,20 1,71 1,12 100 100 90
MnSe ф) К, 53 3,36 2,08 1,76 100 100 100
CaS К, 51 2,85 2,01 1,64 100 68 2!
Основная часть данных табл. 5.18 —из справочника [20].
•2К — кубическая, Г—гексагональная, Т—тетрагональная, Тр—тригональная (или ромбоэдрическая), О — орторомбическая [или ромбическая), М — моноклинная; структурные типы указаны по системе Strukturbe-richte.
*а Низкотемпературный.
** Высокотемпературный.
сообразно применение многоканальных дифрактометров, в которых число одновременно работающих устройств для регистрации соответствует числу анализируемых линий илн фаз.
Дифрактометр ДРПМК-2,0 имеет четыре канала с малогабаритными сцинтилляционными счетчнкамн СРС-7 для измерения интенсивности анализируемых линий и пятый канал для измерения интенсивности фона. В этом дифрактометре поверхность образца (шлиф нлн кювета с порошком) располагается горизонтально, а счетчики устанавливаются в заданные по углам Вульфа—Брэггов положения так, что их приемные Щели лежат на окружности, которая касается поверхности объекта и проходит через источники излучения (фокусировка Зеемана—Болина). Методы количественного фазового анализа при помощи фотографической регистрации подробно описаны в литературе [12—15, 20].
5.3-2. Анализ твердых растворов
Методы рентгеноструктурного анализа позволяют определять следующие характеристики атомной структуры твердых растворов: тип твердого раствора (замещения, внедрения, вычитания); тип сверхструктуры и степень дальнего порядка; тип и параметры ближнего порядка; состав и концентрационные неоднородности твердого раствора (ликвация, ранние стадии распада, К-состояние).
Тип твердого раствора можно определить, зная число атомов на элементарную ячейку растворителя по (чистый металл или химическое соединение) и число атомов на ячейку данного твердого раствора п.
Для раствора замещения п—п0, для раствора внедрения л>п0, Для раствора вычитания п<па. В одной н той же системе могут быть разные типы твердого раствора. Для промежуточных фаз, например для р-фазы.
раствор Ni в NiAl — раствор замещения, а раствор А1 в NiAI — вычитания. Один и тот же сложнокомпонентный сплав может быть н твердым раствором замещения, и раствором внедрения (например, аустенит или мартенсит в легированных углеродсодержащих сталях). Наконец, при закалке нз жидкого состояния нли иониой имплантации в случае двухкомпонентных систем возникают твердые растворы замещения и внедрения одновременно.
Величина па является характеристикой данной кристаллической структуры: у о. ц. к. решетки п0 = 2, у г. ц. к, По=4. Для определения п необходимы рентгеновские прецизионные измерения периодов решетки, измерения плотности и химический (элементный) анализ. Число п определяют, сравнивая массу атомов (Асртп — средняя масса) н измеренный объем элементарной ячейки (Vm) и измеренную плотность (р, г/см3):
п — Уяч р/^ср тв (27)
(для кубической сннгоннн Vm~as илн а2с — для тетрагональной; пгя=1,66• 10~24 г—масса атомов водорода или, точнее, ‘Де массы атома кислорода; если химический состав выражен в атомных процентах (pi,..., р%):
Азр — (Pi А НН Ра Ай )/100.
Анализ сверхструктур) и определение степени дальнего порядка требуют индицирования дифракционной картины и измерений интенсивности определенных линий. В большинстве случаев упорядочение сопровождается появлением так называемых сверхструктурных линий. Структурный фактор интенсивности запрещает определенные отражения (HKL) для простых веществ или твердых растворов со статистическим распределением атомов в узлах решетки [9, 11—13].
Прн упорядочении атомов в растворах замещения иа основе металлических решеток о. ц. к. по типу CsCl (или р-латуни) сверх-
127
структурные линии имеют индексы 100, 111, 210 и т. д. (сумма индексов.— нечетное число). Прн упорядочении по типу FesAl (а также CuaMnAl, NaTI) на рентгенограммах, кроме указанных, появляются линии, которым в системе координат исходной неупорядоченной решетки пришлось бы приписывать дробные индексы ^например,• Это свидетельствует об удвоении периода решетки в упорядоченном состоянии (%п«2а0), т. е. возникает «сверхячейка».
При упорядочении иа основе г. ц. к. решетки металлов, кроме появления сверхструктурных линий 4, может наблюдаться расщепление (или аномальное расширение) некоторых линий в связи с изменением формы ячейки. Тет-рагональность обязательно возникает .прн упорядочении по типу CuAu и иногда CusAu; ячейка становится ромбоэдрической прн упорядочении по типу CuPt.
Для вычисления степени порядка необходи- • мо знать атомные факторы рассеяния компонентов сплава и измерить соотношение интенсивности структурной н сверхструктурной линий.
В случаях, когда упорядочение связано с появлеинём тетрагоиальностн, мерой степени порядка может служить отношение da.
Сверхструктурные линии оказываются слишком слабыми или не выявляются, если атомные факторы рассеяния (илн порядковые номера z) для компонентов мало различаются. В этих случаях можно попытаться увеличить интенсивность сверхструктурных линий, используя специальный подбор излучений [42] нли с помощью нейтронографии (см. [12, с. 421]). Анализ упорядочения твердых растворов внедрения с помощью рентгеновских лучей возможен, если образуется сверхячейка или изменяется форма элементарной ячейки, например мартенсит в углеродсодержащей стали, интрнд FcicNe.
Данные о структуре некоторых упорядоченных твердых растворов приведены выше в табл. 5.5 и 5.6. Обзор исследований сверхструк-тур дан в работе [43].
Ближний порядок в истинном смысле (илн жидкоподобный, статистический [10, 29, 44]) проявляется в модуляции интенсивности фона диффузного рассеяния (рис. 5.21), который меняется в зависимости от типа ближнего порядка. Этот порядок может быть типа расслоения (сегрегации) при положительном знаке энергии смещения н преимущественных соседствах одноименных атомов н типа упорядочения прн отрицательном знаке энергии смещения и преимущественных соседствах разноименных атомов. Характер н степень отклонения относительного расположения атомов компонентов твердого раствора от равномерно-статистического определяется знаком и величиной параметра ближнего порядка сс/=1— —Паб‘/Св21 (28).
Здесь Пав i — число атомов Б на i-той координационной сфере; Св — концентрация компонента В в сплаве; 2,— координационное число. Анализ диффузного рассеяния прн наличии ближнего порядка осложняется наложе-
' Сверхструктурные линии в случае г. ц. к. решетки имеют так называемые смешанные индексы, т. е. четные и нечетные (100, ПО, 210 и т. д.)_
нием так называемого размерного эффекта н эффекта теплового рассеяния (статические и динамические смещения атомов). Чтобы наделено определить знак н величину параметра ближнего порядка (ос,) для нескольких пер-
Рнс, Б.21. Ход изменения интенсивности диффузного рассеяния (фона рентгенограммы):
1 — твердый раствор с хаотическим распределением атомов по узлам решетки; 2 — твердый раствор при наличии ближнего порядка типа упорядочений: 3— то же, тина расслоения
вых координационных сфер, необходимо разделить эти эффекты, поэтому предпочтительно исследование на образцах-монокрнсталлах.
Анализ распределения интенсивности по всей ячейке ОР позволяет определить бинарные параметры ближнего порядка для ряда координационных сфер. Если эти параметры соответствуют равновесным состояниям, то непосредственно можно получить термодинамические характеристики растворов — энергии упорядочения или распада, активности компонентов, особенности критических флуктуаций вблизи точки фазового перехода второго рода, а также исследовать характеристики электронной структуры металлических сплавов, радиусы поверхности Ферми [45, 46]. Преимуществом рентгеновского метода является то, что он применим и для концентрированных растворов, когда нз-за малости длины свободного пробега электронов другие методы неэффективны. Рентгеновское определение термодинамических характеристик твердых растворов — эффективный метод анализа диаграмм состояния бинарных систем.
Неоднородности химического состава твердых растворов, обусловленные действием кинетических факторов при кристаллизации расплавов (дендритная ликвация) илн прн превращениях в твердом состоянии (незавершенные процессы выравнивания концентрации после растворения нли начальные стадии распада твердых растворов) в ряде случаев могут быть выявлены методами рентгенострук-туриого анализа. Важнейшая особенность получаемой при этом информации — ее статистический характер, обусловленный самим методом.
Рентгеновски определяемый период решетки твердого раствора фиксируется как результат интерференции рентгеновских лучей, рассеянных атомами, которые относятся к области, рассеивающей как единое целое, так называемой области когерентного рассеяния (ОКР). Размеры ОКР, даже в сильно искаженном кристалле, имеют порядок десятков нанометров. Именно поэтому получаются резкие реф
128
лексы (лнннн) рентгенограмм от неупорядоченных твердых растворов с хаотическим расположением атомов, сильно отличающихся по размерам. Этот принцип также объясняет отсутствие существенных изменений положения рефлексов прн образовании зон Гннье—Престона (зоны Г,-—П.) в распадающихся твердых растворах (зоны Г.—П. по размеру на несколько порядков меньше ОКР).
Только в том случае, когда дифракционная картина является результатом суммирования отражений для разных ОКР — разных участков, не однородных по составу-кристаллита (дендритная ликвация, диффузионный слой), прн достаточной амплитуде изменений состава твердого раствора и достаточно резкой зависимости периода его решетки от состава возможен дифракционный эффект — размытие (±2 АО) линий, возрастающее с увеличением угла в по закону тангенса: kCICf^&ala = — etg 6Дв.
Известно, что прн определенном (достаточно большом) переохлаждении процесс кристаллизации твердого раствора нз расплава может идти безднффузионным путем. Доказательством этого может служить острота дифракционной линии (например, £ разрешение Ка —
-дублета [12]), если кристаллизация при малых переохлаждениях дает размытые ливни. Неоднородности, связанные с незавершенным процессом распада твердого раствора, т. е. зоны Гинье—Престона, модулированная структура, К-состояння, двухфазный распад, могут давать разные дифракционные эффекты [12, 47].
На данных измерений периода решетки практически не сказывается зонная стадия распада, и образование зон Г.—П. можно заметить лишь по эффектам диффузного рассеяния в ближайших окрестностях узлов обратной решетки матричного твердого раствора. Если неоднородности структуры, обусловленные образованием зон, носят регулярный характер (модуляции рассеивающей способности нлн модуляции межплоскостных расстояний), то диффузное рассеяние концентрируется, образуя сателлиты возле основных рефлексов, и легко выявляется даже прн съемке рентгеновской картины поликристаллов [47, 48]. В остальных случаях выявление зон Г.—П. возможно либо прн рентгеновском анализе монокристаллов илн крупнокристаллических поликристаллов (нз-за малости размера ОКР в поликристаллах и наложений эффектов диффузного рассеяния), либо методом электронной дифракции в просвечивающем электронном микроскопе, где область дифракции всегда ограничена малой частью монокристалла (метод микродифрак-цин, см. раздел 2). В некоторых сплавах зоны Г.—П. имеют координацию атомов, отличную от координации атомов в матричном твердом растворе (например, зоны Гинье—Престона— Багаряцкого в сплавах Al - Mg—Si), илн упорядоченную структуру (например, зоны Г.—П.П или фаза 6" в сплавах AI—Си). Прн этом эффекты рассеяния должны наблюдаться в точках ОР, соответствующих этой структуре. По характеру распределения диффузного рассеяния можно судить о форме зон и в простейших случаях (при действии только форм-фактора) оценивать их размеры. К-состоянне связывается с процессами упорядочения и выде
ления в упорядочивающихся системах при нестехиометрических составах сплавов [29, 43] и характеризуется высокоднсперсной структурой типа зон Г.—П. нли областей локального дальнего порядка1, также проявляющихся в эффектах диффузного рассеяния.
Рентгеновский анализ роста когерентных частиц новой фазы прн распаде пересыщенных твердых растворов основан на анализе так называемых кулоновских полей смещения в матрице, когда смещения атомов в матричном твердом растворе, обусловленные различиями атомных объемов матрицы и выделяющейся фазы, убывают с расстоянием (г) от частицы по закону 1/г2 [45, 49].
По теории М. А. Кривоглаза [45] прн наличии таких полей появляется диффузное рассеяние Д, а интенсивность правильных отражений 1С ослабляется /о«е”ь; показатель экспоненты
Рис. 5.22. Концентрационные зависимости периода решетки a-твердых растворов в бинарных сплавах железа (по [20]):
а — Fe—Au, Fe—Nb. Fe—Mo, Fe—Cu, Fe—Мп; 6 — Fe—Sb, Fe—Sn, Fe—W. Fe—Zn, Fe—Ni, Fe—Be, Fe—Si
1 См. раздел 26.
129
Здесь р — атомная доля выделяющейся фазы; D — диаметр частицы; VB — атомный объем нсдеформированного (изолированного от матричной фазы) выделения; —сумма квадратов индексов интерференции; п — в предельных случаях малых и больших локальных искажений соответственно равно 2 и 8/s- При больших искажениях интенсивность диффузного рассеяния Л сгущается в сравнительно узкие распределения, максимумы которых смещены по отношению к правильным отражениям. При правильные отражения /0 исчезают и остаются только диффузные максимумы Ц. На значение периода решетки твердого раствора, измеренного по положению максимумов /о, влияют упругие межфазовые деформации, поэтому эти значения могут изменяться в направлении, противоположном направлению изменения концентрации твердого раствора. Анализ изменений интенсивности /0 в результате старения прн известных характеристиках выделяющейся фазы (D н AVb/Vb) позволяет оценить общий объем выделений, а прн известных характеристиках микроструктуры (D, Р) оценить поля искажений и контролировать стадию когерентных выделений [49, 50].
Особый тип распада пересыщенных твердых растворов — ячеистый, илн двухфазный распад. Его признак — сосуществование твердых растворов двух концентраций — исходного и конечного (стабильного илн метастабильного) составов — и соответственно наличие двух систем линий изоморфных фаз в рентгеновской картине.
Точные измерения периодов решетки твердых растворов позволяют судить о его составе прежде всего в случае двухкомплектных систем, а также некоторых более сложных систем. На рис. 5.22 приведены концентрационные зависимости периода для двухкомпонентных систем, содержащих железо.
5.3.3. Построение диаграмм состояния
Применение рентгеноструктурного метода для анализа стабильного или метастабильного фазового равновесия основано иа том, что получаемые данные при наличии самой общей характеристики химического состава системы в целом однозначно характеризуют фазовые состояния в случае кристаллических фаз, а также химический состав фаз, если известна зависимость периодов кристаллической решетки от состава. Информацию о составе фаз, образующих дисперсные области в гетерогенной системе, невозможно получить обычным методом химического анализа, а современные инструментальные методы микроанализа1 имеют ограничения в отношении локальности и точности.
Принимая во внимание внешние и внутренние факторы фазового равновесия, при анализе фазового состояния рентгенографическими методами необходимо выполнять съемки при изменении температуры и давления в необходн-^ мых для данной системы пределах, если не удается надежно фиксировать закалкой высо-
1 Рентгеновские микроанализаторы типа МАР и др. (см. раздел 6).
котемпературиые фазы нли фазы высокого давления.
Промышленность выпускает установку для рентгенодифрактометрического анализа прн температурах от 80 до 300 К (УРНТ-160). Камеру-криостат устанавливают иа гониометре дифрактометра типа ДРОН-1 или ДРОН-2; пониженные температуры создаются с помощью жидкого азота.
Требования к образцам и процедурам дифрактометрических измерений остаются общими. Прн оценке точности измерений dfri (или периодов решетки) следует учитывать погрешность установки заданной температуры (±3°С) и точность ее поддержания в криостате (ие ниже ±0,3°C).
С помощью высокотемпературной установки УВД-2000 можно проводить рентгенодифрак-тометрнческий анализ прн температурах от комнатной до 2000 °C в вакууме, до 1800 °C в инертном газе и до 1200 °C на воздухе. Камера-печь устанавливается на гониометр дифрактометра типа ДРОН, нагрев осуществляется печью сопротивления с молибденовым нли нихромовым нагревателем. Точность поддержания температуры не хуже ±4 °C.
Для рентгенографического анализа при высоких гидростатических давлениях (до 20 кбар) в Институте физики высоких давлений АН СССР разработаны камеры типа поршень-цилиндр с бериллиевыми сосудами. При более высоких давлениях до 160—200 кбар рентгеновский анализ проводят в камерах с наковальнями из твердого сплава. Исследуемый образец помещают и узкий канал (диаметр 0,1—0,2 мм) таблетки из аморфного оора, через которую создаются условия квазигндроста-тического давления. Аморфный бор мало поглощает рентгеновские лучи и дает очень слабые собственные диффузные линии на рентгенограммах.
Исследования малых образцов (0,25 X Х0.15 мм) при комнатной температуре и давлениях до 120 кбар возможны с помощью приставки к дифрактометру ДРОН-1 [51].
В Институте физики высоких давлений АН СССР разработаны варианты камер для получения дсбаеграмм в интервале давлений от 0 до 100 кбар при регулируемой температуре 25—500°C и в интервале давлений 0—130 кбар при температурах от 25 до —160 °C [51]. Применение низкотемпературных камер в ряде случаев облегчает фиксирование путем закалки фаз, полученных в условиях высоких давлений.
Определение фазового состава — первая и очевидная, но не единственная задача исследования фазового равновесия методами рентгеноструктурного анализа.
Для построения диаграммы состояния важно установить границы фазовых областей. Наиболее точнцй метод определения границы растворимости основан на анализе зависимости периода решетки твердого раствора от состава сплава. В соответствии с правилом фаз в двухфазной области двухкомпонентной системы при постоянной температуре составы равновесных фаз должны оставаться постоянными. Практически работа по построению границы растворимости сводится к прецизионному определению периодов решетки для:
а) серии образцов с однородной структурой, полученной, в частности, путем закалки,
130
•которая позволяет построить зависимость периода решетки от состава;
б) одного-двух образцов заведомо двухфазных сплавов после обработки, фиксирующей равновесное состояние для заданных температур; это состояние обеспечивается быстрым охлаждением (закалкой) после достаточно Длительного отжига; для ускорения диффузионных процессов иногда целесообразна предварительная пластическая деформация, например при приготовлении порошковых образцов.
Ошибки в определении предела растворимости [АС, % (ат.)] связаны с ошибками в измерении периода решетки (Аа), а также температуры (АГ) в ходе отжига двухфаз-го образца. Влияние этих ошибок тем больше, чем круче зависимость периода от состава твердого раствора н чем больше изменения растворимости с температурой:
АС = (дС/да) Ла + (ЭС/й7)АТ. (30)
В случае иекубических решеток вместо периода может быть взято какое-либо межплос-
Fe 5 10 15 20 25 30 35 50
St, °/о (ат.)
Рис. 5.23. Линии равных значений периода для твердых растворов в тройных системах [20(:
а — Fe—Сг—Si (о ц. к. фаза); б — Fe—Сг—V (о. ц. к. фаза, сечение при 700 °C)
костное расстояние d/п или отношение периодов {с/а при тетрагональной илн гексагональной решетке).
Если система трехкомпонентная и более сложная, то метод анализа зависимости периода решетки твердого раствора от состава сплава также применим, поскольку иа границе фазовых областей ход этой зависимости должен изменяться (рнс. 5.23). Особое значение имеет этот метод в определении направления конод в двухфазных областях трехкомпонентных систем.
Очевидно, данный метод непрнмеиим при слишком слабой зависимости периода решетки от состава твердого раствора (например, для системы А!—Ag, где изменение периода ре-
О
шетки составляет 10-4А иа 1 %). В некоторых случаях может оказаться эффективным так называемый метод исчезающей фазы, основанный на экстраполяции к нулю зависимости количества второй фазы от состава сплава [13].
5.3.4. Анализ фазового состава стали после термической обработки 1
Рентгенографически определяются следующие характеристики фазового состава термически обработанной стали: количество остаточного аустенита и содержание в нем углерода; содержание углерода в мартенсите и количество распавшегося мартенсита; природа карбидных фаз, а также других промежуточных фаз в сложнолегированных сталях и сплавах.
1. Рентгенографический анализ остаточного аустенита в стали в - отличие от магнитного и металлографического дает возможность измерять его количество в слое определенной толщины детали произвольной формы. При магнитном методе определяют среднее содержание аустенита по всему объему специально изготовленного образца, а при металлографическом исследуют только поверхность. При рентгеновском исследовании, кроме того, решается и другая задача: определение содержания углерода в аустените. Это имеет значение прн выяснении условий полноты закалки, обезуглероживания поверхностного слоя и прн анализе результатов химико-термической обработки. В последнем случае возможен послойный анализ.
Аустенит представляет собой твердый раствор внедрения углерода в y-Fe, металлические компоненты легированной стали растворяются в аустените по типу замещения. Атомы углерода статистически распределены по октаэдрическим порам г. ц. к. решетки.
При определении количества остаточного аустенита следует учитывать возможность наложения иа сравниваемые линии аустенита н мартенсита линий от других фаз (карбиды). Зная примерное соотношение фаз в стали и dfri для этнх фаз, выбирают условия съемки (излучение и анализируемые линии), обеспечивающие оптимальное сочетание точности и экспрессности анализа.
Использование линии аустенита (111)ка и линии мартенсита (ПО)—(011), (101) не может обеспечить высокой точности анализа
1 Составлено с участием Н. В. Еднерал.
131
из-за близости положения этих линий, особенно в случае расщепления мартенситной линии— дублета (110)—(Oil), (101)—н наложения иа этн линии интенсивных линий от карбидных фаз FesC и Сг23Сб. При мягком излучении (Ка Сг) число подходящих для анализа линий ограничено. Прн жестком (/Са Мо) возрастает число взаимно накладывающихся линий. Наиболее подходящими для анализа являются линии аустенита (ЗИ)кю или (222) ка и мартенсита (112—-211, 121)ка илн (220—202, 022) к а на железном илн кобальтовом излучении. В этом случае анализ проводят на дифрактометре типа ДРОН в режиме сканирования по точкам и сравнением интегральных интенсивностей. Анализ сталей, содержащих большое количество карбидной фазы, проводят так же, как и трехфазной смеси. Особенность определения количества аустенита состоит в том, что коэффициенты ослабления рентгеновских лучей (р) для основных фазовых составляющих стали (аустенита и мартенсита) практически одинаковы.
Можно выделить трн случая:
а. При отсутствии значительного количества карбидной фазы для определения количества остаточного аустенита сравнивают линии (ЗП)ка аустенита и (112—211, 121) мартенсита на Со- нлп Fe-нзлученни; при этом используют фильтр ^-излучения в связи с наложением отражения (ЗИ)кр аустенита на отражения (2П)«а мартенсита. Однако если количество аустенита мало и провести съемку линий (311)ка аустенита с фильтром затруднительно, то можно снимать эту линию без фильтра. С этой целью, используя какой-либо образец с достаточно большим содержанием аустенита, предварительно снимают линию (311 )ка с фильтром и без фильтра. При дальнейшей работе снимают эту линию без фильтра н учитывают полученную таким образом поправку на ослабление. Эта рекомендация относится и к рассмотренному ниже анализу трехфазной смеси структуры — аустенит, мартенсит и цементит.
Интегральную интенсивность линии (HKL) фазы, занимающей х % облучаемого объема, определяют по формуле
1(ИLP <е) Р HKL Хф’
(31)
где Уяч. $ — объем элементарной ячейки фазы; коэффициент К одинаков для всех линий рентгенограммы (Ка - илн /Ср -линий); LP(6) = (1+ cos2 20) /sin2 6 cos 0 — произведение факторов Лоренца и углового [см. также уравнение (20)].
Количество остаточного аустенита (хд) в бинарной системе можно определить по отношению интенсивности линий, принадлежащих к двум фазам. Для увеличения точности определения целесообразно выбирать линнн, расположенные по углам 0 близко друг к другу хА=(1-ЬйВ)-1, (32)
где В =/М//А; k — коээфнцнент, который заранее рассчитывается для данной комбинации фаз, исходя нз факторов интенсивности выбранных линий.
б. В присутствии в структуре стали значительного количества цементита для анализа следует взять линнн (311)ка аустенита и (220—022, 202) ка мартенсита. Линия (121, 211—И2)кд мартенсита в данном случае ие пригодна для анализа, так как вблизи макси-
Рис. 5.24. Теоретически рассчитанная штрих-диаграмма закаленной стали У12 для ИеКд-излучения в области углов 20 для линий 211-112 мартенсита; показаны положения сильных линий карбидов FesC и Сг2аСе.
мума этой линнн наблюдается линия высокой нитеиснвностн (233) ка от цементита (рис. 5.24). Если в расчете можно пренебречь количеством цементита, то используют формулу (32). Если количество цементита велико и удается зафиксировать его интенсивные отражения, например (312) к а или (113) + (122) то расчет следует проводить по формулам для трехфазной структуры (33):
хк~ “Ьki
~ \ W( ^А ^2 ;ц);
ХЦ = *8 W(^A + + ^2 Al)»
Г'
(33)
где /А , /м» — интегральные интенсивности
соответствующих линий:
k Ум^/1РД(ГР>А
(^)М
к Уц«д
5Ц /ц (^)ц
в. Если в стали присутствует карбид хрома типа Сг23Се, то количество остаточного аустенита определяют по линиям (220—202, 022) к для мартенсита и (222) « а для аустенита по формуле (32) для двухфазной структуры. Применять формулы расчета для трехфазной -структуры не имеет смысла, так как прн съемке массивного образца от карбидных включений Сг23С6 обычно фиксируется только одно наиболее сильное отражение (5П)ка, соответствующее межплоскостному расстоянию о
2,04 А, которое накладывается на отражение (110).кд мартенсита.
Линии (112—121, 211)кд мартенсита и (ЗИ)кд аустенита не следует использовать для количественного анализа, так как на этн линии накладываются сильные отражения линий карбида Сг23С6 (межплоскостные расстоя
132
ния соответственно 1,166 А и 1,064 А). Если же удается получить другие сильные отражения от этого карбида, например d/n = 1,227 илн 0,028 А, на положения которых не накладываются какие-либо другие отражения, то расчет следует вести по формулам (33) для трехфазной структуры. Ошибка в определении количества аустенита составляет не более 2 % (прн содержании аустенита около 10%).
Применение для количественного фазового анализа дифрактометра, безусловно, рационально с точки зрения производительности и точности, однако фотометод более чувствителен и имеет преимущества прн определении малых количеств остаточного аустенита (~ 1 %). Следует отметить, что весьма чувствительным «инструментом» является человеческий глаз и при визуальном сопоставлении линий можно с высокой степенью точности выбрать линии одинакового почернения. На этом основан метод Нечволодова (или метод гомологических пар) ([12], с. 370).
Для выбора условий съемки и быстрой идентификации линий рентгенограммы целесообразно использовать диаграмму зависимостей dfn для аустенита и мартенсита от содержания углерода (рнс. 5.25).
й'/'л.Л 2.080
2,040
2000
1,820
1.780
1,510
1,470
1,430
1.250
1,210
1,170
1,080
1,040
1,000
О 0.2 0,4 0.6 0.8 1,0 1,2 1,4 С. °Л>
Рис. 5.25. Межплоскостные расстояния d]nr аустенита и мартенсита углеродистой стали в аависимости от содержания углерода. Штриховые линии относятся к din феррита
Содержание углерода в остаточном аустените для углеродистой стали можно определить также с помощью диаграммы на рис. 5.25 ила используя формулу
сА— 3,555 + 0,044р, (34)
где р— содержание углерода, % (по массе).
В случае легированной стали следует учитывать ртнянне легирующих элементов (см. рис. 5.22).
2. Мартенсит закаленной стали — это пересыщенный твердый раствор углерода на основе o-Fe. Вследствие упорядоченного расположения атомов углерода, занимающих октаэдрические позиции одной из трех систем вдоль осн с, решетка мартенсита оказывается тетрагональной. Степень тетрагональностн (с/а) линейно растет с увеличением содержания углерода (рнс. 5.26). Наши знания об атомной
Рис. 5.26. Зависимость периодов решетки (а, с) в степени тетрагональностн (с/а) мартенсита от содержания углерода [% (по массе)]
структуре мартенсита стали основаны главным образом на рентгеновских исследованиях, выполненных Г. В. Курдюмовым и его сотр. [52].
Атомы углерода в решетке мартенсита занимают относительно небольшую часть октаэдрических пор н в пределах одной этой системы пор непосредственно прн образовании мартенсита оии распределены хаотически. Однако уже в ходе закалки, если точка Ms достаточновысока, или после кратковременного пребывания мартенсита при комнатной температуре-(в случае высокоуглероднстой и легированной стали) происходит перераспределение атомов углерода и образование ближнего порядка в пределах той же системы октаэдрических пор. Для этого состояния характерно диффузное рассеяние на электронограммах мнкроднфрак-цни; при обычных рентгеновских исследованиях, это явление заметить не удается. Вслед за этим процессом идет так называемый двухфазный распад мартенсита*, прн котором появляются области мартенситных кристаллов-, с содержанием углерода около 0,2—0,3 %, т. е. мартенсит отпуска. В стали, имеющей высокое положение точки Мп (>100—150°C), двухфазный распад идет в ходе охлаждения1 прн закалке во время самоотпуска мартенсита. Было обнаружено, что при этом идет и непрерывный распад мартенсита, проявляющийся в-снижении содержания углерода в матричной части мартенситных кристаллов [52, 53].
Сравнительно недавно было распространено мнение об очень больших искажениях решетки? в виде так называемых статических смещений атомов, мнкронскажений нлй напряжений III
1 Этот процесс удалось впервые обнаружить Г. В. Курдюмову и Л. И. Лысаку при рентгеновском исследовании образцов, полученных при закалке монокристаллов аустенита. Такие образцы давали рефлексы от ориентированных мартенситных кристаллов.
133
рода, которые как будто проявлялись в закономерном ослаблении линий рентгенограммы. Отношения интенсивности компонент тетрагонального дублета (ПО—011, 101; 200—020, 002 и т д.) теоретически должны равняться отношению факторов повторяемости, т. е. 2. Специально поставленные эксперименты, как и теоретические расчеты, показали [53], что аномальные отношения интенсивности нельзя объяснить только статическими смещениями и основной причиной этой аномалии являются частичный распад мартенсита уже в процессе закалки и образование мартенсита отпуска, если точка Мк выше 100—150 °C.
Определение содержания углерода в мартенсите основано на приведенной выше (см. рис. 5.26) зависимости периодов его решетки от содержания углерода [р, % (по массе)]. Аналитически эта зависимость выражается уравнениями:
« = 2,866 —0,015р; с = 2,866 + 0,118р;
с/а = 1 + 0,0467р. (35)
Практически содержание углерода удобно •определять по междублетпому расстоянию, которое в общем случае обусловлено содержанием углерода и не зависит от содержания легирующих элементов, растворяющихся в мартенсите по типу замещения. У мартенсита в некоторых легированных сталях были отмечены аномальные значения тетрагоиальиостн, а прн отпуске — образование фазы с ромбической •структурой [54]. В сталях, легированных никелем и титаном или алюминием, аномально высокая тстрагональность мартенсита (и тетра-гоиальность при отсутствии углерода) объясняется образованием в аустените когерентных частиц упорядоченной фазы (Ni3Ti). В этом случае рекомендуется [55] определять содержание углерода по зависимости объема элементарной ячейки от его концентрации, по,
Zift мин
Ав, мин
02 0,6 1,0 1,4 0,2 0,6 10 1,4
С, % С. %
Рис. 5 27. Зависимости междублетного расстояния от содержания углерода в мартенсите:
а — Д6 (101)— (НО) для излучений хрома (Г) и железа (2); б — &G (2Ю— (112) для излучения кобальта
естественно, при постоянном содержании легирующих элементов, образующих раствор замещения.
Использование дублета (110)—(011, 101 )к а нецелесообразно из-за наложения линии остаточного аустенита (111)ка. Преимущество использования дублетов (200, 020—002) ка и (211, 121—И2)ка состоит в том, что измеряемая величина (Д-2 0) существенно больше (рис. 5.27), чем для дублета (110—011, 101)ка. Осложнения в расчетах связаны с тем, что прн содержании углерода <0,6 % тетрагональный дублет не разрешается, а в тех случаях,
когда он разрешается, на положение одной из линий дублета влияет присутствие мартенсита отпуска. Тстрагональность решетки мартенсита отпуска невелика, междублетное расстояние по расчету для линий (121, 211— 112) в FeKa -излучении составляет всего 202 Таким образом, можно говорить о расположении трех линий: две линии исходного мартенсита (мартенсита закалки) и одна линия мартенсита отпуска (рис. 5.28). В работе [55] ре-
4$°51‘
в=48°ОЗ'
49°34’
49°55‘
(112)огп)^ОГР
(211)зак
Рис. 5.28. Наложение линий мартенсита мартенсита отпуска для стали 120Н10,
закалки и излучение
комендуется определять содержание углерода в мартенсите закалки по положению линии (002) ка (соответственно по зависимости периода с от содержания углерода), считая, что линия мартенсита отпуска накладывается на отражение (200—020)ка исходного мартенсита. Для расчета доли мартенсита отпуска (хМотП) может служить рнс. 5.29. Параметр у определяется как отношение площади сложного максимума (020—200) к площади максимума (002).
При большом содержании углерода (2>1 %), когда эффект перекрытия «хвостов» линий мал, можно разделить линии («вычесть» из сложной линии ливню мартенсита отпуска [53]). При меньших содержаниях углерода можно разделить линии с помощью ЭВМ. В работе [56] описана программа (Спектр-24) и приведены требования к съемке линий анализируемого дублета. Расчет дает положения центров тяжести н интенсивности двух составляющих дублета исходного мартенсита н линия мартенсита отпуска.
Для анализа сложных профилей используют также метод моментов [57]. В применении к рентгенограмме мартенсита предложен метод моментов 3-го порядка. При однородном мартенсите свежезакалепной стали междублетное расстояние можно определить через третий момент сложной линнн (взКСП) и эталонной лннни (нГ) » та же сталь после отжига) и заранее известных весов синглетов сложной линии А1 н Аг’.
в = 3/( гГ" -iff)/а, А2 (А, - А2) . (36)
Подходящими для анализа этим методом являются линии 200, 020—002 (в этом случае наложение линии от мартенсита отпуска мало сказывается на величине рз). После определения 6 (и количества углерода) можно выделить линию мартенсита отпуска из сложного профиля линии (200, 020) графическим путем. Проверка показала хорошую надежность методики при содержании мартенсита отпуска до 70%.
.134
Для случая, когда на рентгенограмме тетрагональный дублет не разрешается, предлагались разные аналитические методики разделения компонент дублета и методы оценки содержания углерода по ширине сложной линии. Прн этом обычно предполагалось, что линии
Рис. 5.29. График для определения доли мартенсита отпуска (х) в зависимости от отношения интегральной интенсивности суммарного максимума (200— 020) -р{ 200 ) к максимуму (002) 3 [55]
дублета экспериментально не выявляются в связи с малым междублетиым расстоянием С. В действительности, это связано не только с малостью б, но и с тем, что именно в стали с невысоким содержанием углерода мартенсит
Рис. 5.30. Положение линии (222) К^Си в зависимости от содержания углерода в мартенсите (Р, %) и степени тетрагональности J58]
распадается в ходе закалки и оказывается неоднородным (за счет как двухфазного, так и непрерывного распада). В этом случае целесообразно оценивать среднее содержание углерода в твердом растворе и использовать для этого положение центра тяжести линии, которая не испытывает расщепления вследствие тетрагональности, например лннин (222). В работе [58] предложено использовать линию (222) в СиКа -излучении *, при этом для a-Fe 2 0 « 137°05'. О точности метода позволяет судить график рнс. 5.30, ошибка в измерении 6 па 5' дает ошибку а определении величины отношения с!а 0,0017, что соответствует менее чем 0,05 % (по массе) С. Вариант этого метода в работе [59] включает использование Др -излучения Со. Преимущества этого варианта связаны с тем, что линия Kg в отличие от (^aiLa2 ) должна быть симметричной, поэтому асимметрия профиля (222) кр сразу
1 Линия оказывается в прецизионной области углов 6, что касается фона вторичного излучения, то современные методы регистрации па дифрактометра позволяют с ним не считаться.
указывает на неоднородность состава мартенсита. Действительно, несимметричной лилии, полученной от массивного образца закаленной стали У12, соответствует линия с двумя пиками — мартенсита закалки и мартенсита отпуска, полученная от электролитически выделен* него порошка мартенситных фаз нз того же образца (рис. 5.31). При использовании данного метода надо быть уверенным в отсутствие заметного влияння на положение линии (222) зональных напряжений. Для проверки этого надо использовать наклонную съемку [60].
Рис. 5.31. Профиль линии (222) Kg Со образца же-
леза (2) и профили линии мартенсита стали У12, полученные от массивного образца (2) и от электролитического осадка (3). Положения ликов определя-
ются содержанием углерода [59]
3. Карбидные фазы наряду с ферритом, мартенситом и аустенитом входят в число основных фаз стали н определяют особенности их термической обработки н эксплуатационные свойства стали.
Карбид железа Fe3C (цементит, или 0-фаза) имеет ромбическую кристаллическую решетку (см. табл. 5.5). Координация атомов железа в структуре цементита близка к гексагональной. С этим, в частности, были связаны трудности выявления карбида иизкоотпущен-ной стали (е-карбида), который действительно-имеет гексагональную компактную упаковку атомов железа с неупорядоченным расположением атомов углерода (тип e-FesN). До енх пор дискуссионными являются вопросы о содержании углерода в Е-карбиде и об образовании при распаде мартенсита углеродистой стали других карбидных фаз. Е-карбид образуется при низкотемпературном распаде мартенсита не только в углеродистых (при содержании углерода более 0,3—0,4 %), ио и в легированных сталях, в которых стабильными могут быть специальные карбиды (хрома, молибдена и др.).
Растворимость легирующих элементов стали-в цементите обычно невелика и только в отдельных случаях превышает их среднее содержание в стали. При значительном содержании карбидообразующих элементов образуются специальные карбиды. В сталях, содержащих хром, это Сг2зС6, Сг7С3, СГ3С2, которые имеют сложные структуры, в сталях с титаном, ванадием, ниобием или цирконием — карбиды состава МеХ с г. ц. к. решеткой (фазы внедре-
135-
ння, тип NaCl), в сильно легированных инструментальных сталях, содержащих молибден, вольфрам, кобальт — «карбиды быстрорежущей стали» (фазы т)), в составе которых может быть примерно от 20 до 7—8 % (ат.) С (Fe2W2C до Fe6W6C) (см. табл. 5.5). Только в отдельных случаях прн очень большом содержании карбидов илн прн специальных условиях съемки (монохроматическое излучение, монокристаллы) удается провести исследование карбидных фаз в массивных образцах. Общий способ исследования этих (а также ннтерме-таллидных) фаз в стали состоит в выделении осадка, в котором присутствуют только этн фазы путем электролитического растворения металлической матрицы. Осадок исследуют рентгенографически и подвергают химическому анализу. При аккуратной работе удается провести не только качественный анализ (т. е. установить природу карбидных фаз), но и количественный (по отношению массы осадка к массе растворенной части образца).
5.4. АНАЛИЗ СТРУКТУРНОГО СОСТОЯНИЯ МЕТАЛЛИЧЕСКИХ МАТЕРИАЛОВ
5.4.1. Анализ текстур1
Текстурой называют преимущественную ориентировку кристаллитов в пространстве, возникающую прн направленном внешнем воздействии илн ориентирующем действии среды
•’ Составлено совместно с Э. И. Спектор.
В случае аксиальной текстуры, образующейся прн одноосном воздействии (волочение, растяжение, сжатие), определенные кристаллографические направления [ицад] в кристаллитах ориентируются вдоль направления действия внешней силы («ось текстуры»). Прн текстуре прокатки определенные кристаллографические плоскости (hkl) устанавливаются вдоль плоскости прокатного листа, а направления [но ад] вдоль направления прокатки (НП); текстура прокатки записывается как (hkl) [ноад]. В реальных условиях часто бывает не одна, а несколько преимущественных ориентировок кристаллитов в пространстве, т. е. возникает так называемая многокомпонентная текстура.
Текстуру обычно анализируют с помощью прямых и обратных полюсных фигур. Прямой полюсной фигурой (ППФ) называется стереографическая проекция нормалей к определенным плоскостям (hkl) для всех кристаллитов данного материала. ППФ строят в координатах самого образца (рнс. 5.32). Для текстуры прокатки плоскость проекции обычно устанавливается параллельно плоскости прокатного листа, а центр ППФ совпадает с направлением нормали к плоскости листа (НН).
Обратная полюсная фигура (ОПФ) представляет собой стандартный стереографический треугольник, на котором около проекций различных кристаллографических направлений (нлн полюсов) стандартной проекции монокристалла указана так называемая полюсная плотность (Phki)- Полюсная плотность определяется как вероятность совпадения данного кристаллографического направления с заданным физическим направлением самого образца (например, с направлением осн волочения
{/7/J
Рис. 5.32. Полюсная фигура для плоскостей { 111) прокатанного никеля (плоскость проекции параллельна плоскости прокатки НП — направление прокатки, ПН — поперечное направление, НН — направление нормали к плоскости листа)
336
для проволоки или с нормалью к плоскости листа) (рис. 5.33).
Прн рентгенографическом изучении текстур фотометодом, используемым в основном для исследования аксиальных текстур, анализируют расположение и протяженность текстурных максимумов вдоль дебаевских колец для отражений с малыми индексами (точнее, с малым фактором повторяемости). Днфрактомет-
Рис. Б.ЗЗ. Обратная полюсная фигура для области сварного шва алюминиевого сплава (сварка магнито-импульсным методом при давлении 9 ГПа)
рические методы построения ППФ основаны иа анализе характера изменения интенсивности для определенных интерференций (HKL) прн изменении положения образца и соответственно положения отражающих плоскостей (HKL) в объеме образца, создающих интерференционную картину. Во всех случаях счетчик устанавливается под углом 20 к падающим лучам и в процессе записи кривой остается неподвижным. Съемку проводят на отражение (метод наклона нлн метод поворота) и на просвет тонкого образца (толщина несколько сотых долей миллиметра).
Положение плоского образца, установленного в центре гониометра, может быть изменено (рнс. 5.34) путем поворота этого образца на
Рис. Б.34, Схема движения образца при дифрактометрическом анализе текстуры
угол а вокруг горизонтальной осн у в случае метода наклона (наклон к плоскости гониометра) илн вокруг вертикальной оси гониометра в случае метода поворота н прн съемке на просвет. Угол а обычно изменяют от 0 до 50— 70°. Кроме того, образец поворачивают в своей плоскости на угол (3 (0—360°) вокруг оси х, перпендикулярной плоскости образца.
ППФ строят с иомощью радиальной сетки Болдырева. Значения интенсивности в каждой
точке (для углов а, р) дифракционной кривой переносят иа плоскость проекции. Точки одинаковой заданной интенсивности соединяют линиями уровня. При съемке на отражение удается построить только центральную часть ППФ, а на просвет — периферийную ее часть. Для получения полной ППФ приходится проводить сложный пересчет интенсивностей, полученных разной съемкой, что достаточно* трудоемко.
Для построения прямой полюсной фигуры (ППФ) методом наклона, который обладает рядом преимуществ по сравнению с методом поворота, и для съемки на просвет используют специальный держатель — приставку ГП-2 илн ГП-15 к дифрактометру типа ДРОН. Имеются специальные текстурдифрактометры, позволяющие автоматизировать процедуры изменения полох<ення образца, угла а, расчета (введение поправок на интенсивность) н самого построения ППФ на бумаге (текстурднф-рактометр ДАРТ-2,0).
. Для определения текстуры прокатки по ППФ ее поочередно накладывают иа разные стандартные проекции монокристалла до совмещения областей сгущения нормалей с выходами анализируемых нормалей на стандартной проекции. Для гексагональных материалов требуются дополнительные расчеты [61, 62].
Обратную полюсную фигуру (ОПФ) строят на основании измерений интегральной интенсивности отражений для разных плоскостей (hkl) для текстурованного и эталонного образцов. С помощью дифрактометра в условиях фокусировки по Брэггу—Брентано полюсные плотности для разных плоскостей (Рьм), рассчитанные нз соотношения отражений интенсивностей (HKL) образца и эталона, наносят около соответствующего полюса (hkl) в стандартном треугольнике. Точки с одинаковым значением Рьм соединяют уровнями.. Индексы полюсов ОПФ, около которых Phki максимальна, указывают на наиболее вероятную кристаллографическую ориентировку нормали к плоскости образца.
Для анализа аксиальной текстуры достаточно иметь одну ОПФ, построенную для плоского образца, вырезанного перпендикулярно оси-текстуры; для анализа текстуры прокатки получают ОПФ для двух образцов, вырезанных перпендикулярно НН (параллельно плоскости, листа) н перпендикулярно НП. Анализ таких ОПФ по сравнению с анализом ППФ позволяет проводить приближенный, полуколнчест-венный анализ компонент текстуры. Наиболее существенный недостаток анализа ОПФ связан с тем, что не удается проанализировать. Phki Для достаточно большого чнсла полюсов (особенно для кристаллов с кубической решеткой).
Способ построения обратной полюсной фигуры (ОПФ) с помощью ЭВМ, предлохсенный Бунге [62—64], состоит в математической обработке экспериментальных дифрактометрических* кривых, позволяющей получить значения Phki примерно в 50 равномерно расположенных точках ОПФ. Исходными данными являются дискретные (через 5° по углу а) значения интенсивности, полученные по трем-четырем интерференционным кривым /(а), которые, снимают методом наклона, но при быстром вращении по углу (3, точнее при усреднении по углу р. Угол а меняют до 50—60°, ско
137
рость вращения по углу 0 равна 60 об/мин. Обычно анализируют отражения (111), (200) и (220) для г. ц. к. и (НО), (200) и (211) для о. ц. к. решеток. Принципиальная возможность перехода от значения интенсивности в определенном участке (кольце) прямой полюсной фигуры к определенной точке ОПФ основывается на том, что в усредненных по углу 0 кривых /(а) содержится информация о возможных кристаллографических углах между анализируемым полюсом ОПФ и искомым внешним направлением в образце, для которого строится ОПФ.
Общий недостаток прямых и обратных полюсных фигур — их двухмерность. Действительные положения кристаллитов в пространстве трехмерной характеристики можно изучать с помощью трехмерных функций распределения ориентировок [63]. Эти функции рассчитывают на основании полных трех прямых полюсных фигур и строят в эйлеровых координатах (ср, Ф,, Ф2); связанных определенным способом с осями образца (например, с осями НН, НП и ПН прокатанного листа). Целесообразно использовать нейтронографические данные, поскольку этот метод дает информацию о текстуре в больших объемах образца.
Применение синхротронного рентгеновского полихроматического излучения и усовершенствованных методов дискриминации в измерениях интенсивности даст возможность одновременно получать несколько ППФ илн достаточное количество исходных данных для непосредственного построения ОПФ.
5.4.2. Анализ напряжений
Остаточные напряжения (после обработки давлением, резких тепловых воздействий, фазовых превращений, сопровождающихся изменениями объема) уравновешиваются в объеме всей детали (или значительной ее части), вследствие этого детали по объему имеют зоны напряжений разного знака. Это объясняет термин зональные напряжения. Термин макронапряжения имеет смысл только в связи с представлением о микро напряжениях как напряжениях, уравновешивающихся в микрообъемах — внутри зерен как основного элемента микроструктуры. Рентгеновский эффект от этих напряжений — размытые линии, возрастающие с увеличением угла 6 по закону tg6. Однако в последнее время стало ясно, что деформации решетки, создающие эффект рентгеновских микронапряжений, связаны с упругими полями дислокаций. В связи с этим целесообразно отказаться от термина микронапряжения (а следовательно, и от термина макронапряжения) и пользоваться термином микродеформации решетки, имея в виду поле упругих деформаций от дислокаций, нлн выражать эту характеристику через плотность дислокаций и их распределение.
Рентгеновский метод измерения зональных напряжений выгодно отличается от механических методов прежде всего тем, что относится к числу неразрушающих. Метод основан на прецизионном измерении межплоскостных расстояний. Эти измерения могут быть проведены на самых малых участках поверхности детали. Прн исследовании деталей сложной формы, а также громоздких конструкций пре
имущество имеет фотометод; дифрактометрический метод обеспечивает экспрессиость анализа и высокую точность.
Самый простой способ оценки напряжений — съемка рентгенограммы испытуемого образца и рентгенограммы образца того же материала, свободного от напряжений (эталон), в камере типа КРОС. Далее измеряют разницу в ПОЛО-
Рис 5.35. Эллипсоид деформации е (с) и главные напряжения Ci и Os для плосконапряженного состояния (б). Напряжения вдоль направления <р а = = О| cos2 tp 4- sin2 q>
женин линий Аб=0ялс(оор)—Ол/кцат). В условиях обратной съемки при нормальном падении рентгеновских лучей на поверхности образца величина /_\0 определяется изменением d/n для плоскостей (HKL), расположенных почти параллельно поверхности образца. Величину напряжений, действующих в этом направлении (о ||), рассчитывают по формуле o'и = (E/vJclg ОДО (37), где v — коэффициент Пуассона.
Недостаток метода в том, что измерения проводят для случайного направления. В общем случае однородная деформация тела описывается эллипсоидом деформации (рнс. 5.35). Рентгенографически анализируется напряженное состояние в тонком поверхностном слое материала. В этом случае надо принять напряжение, нормальное к поверхности Os, равным нулю. Деформация в некотором направлении (рис. 5.35, с) определяется через сумму главных напряжений Oi+o2 и напряжение о :
eW (1 + v) Е-1 sin1 ф — vE-1 (ох 4- о2).
(38)
Измеряя деформации для направлений с разными ф прн постоянном ср, молено найти о(;, и 01+02. Практически задача сводится к съемкам рентгенограмм при разных наклонах образца, вычислению и построению и анализу зависимости от З1п2ф (рис. 5.36).
138
Эта последняя операция дала название методу — метод sin2 ф. В этом методе можно обойтись без съемки эталона, если принять, что прн перпендикулярной съемке d ~do, где de — мсжплоскостное расстояние в отсутствие напряжений и е^, = (dHKL^—dИЕ1 .
Обычно ограничиваются определением напряжения в заданном направлении оф, однако существуют способы определения величин и па-
Рис. 5.36. Зависимость деформации е от угла ф (чш2ф) при <P=const
правлений действия главных напряжений и о2 [65]. Метод sin2-ф в варианте фотометода осуществляется с помощью специально сконструированной камеры [66]. Прн использовании дифрактометра (типа ДРОН) держатель образца должен давать возможность вращения образца вокруг оси гониометра независимо от движения счетчика. Специализированный дифрактометр ДАРН-2,0 предназначен для определения напряжений в крупногабаритных конструкциях. Точность определения зависит от точности измерения смещений дифракционных линий. В случае стали для линии (310) в излучении Л'аСо при ошибке А(20) = = ±2,5' ошибка в определении о составляет ±20 МПа [67].
5.4.3. Анализ субструктуры в отожженных и слабо деформированных материалах
Представление о мозаичной структуре нлн субструктуре реальных кристаллов возникло, когда обнаружились расхождения между экспериментальными значениями интенсивности дифрагированных лучей и теоретическими расчетами по динамической теории. Согласно этим представлениям реальный кристалл состоит из областей правильного строения — блоков, повернутых по отношению друг к друзу на небольшие углы. Развитие теории дислокаций и прямые наблюдения подтвердили реальность модели мозаичного кристалла. Было показано, что разсриситировка блоков определяется плотностью дислокаций, образующих малоугловые границы. Подобная полигонизо-ваилая субструктура наблюдалась с помощью дифракционной электронной микроскопии во многих металлических материалах после отжига, которому предшествовала холодная деформация, после деформации прн повышенных температурах и др.
Тенденция к расположению дислокаций в виде вертикальных стенок зависит от склонности дислокаций к расщеплению, т. е. от энергии дефектов упаковки, и поэтому в разных металлах
и сплавах выражена в разной степени. В общем виде задача анализа субструктуры формулируется как оценка плотности дислокащш и характера нх распределения. Методы рентгеновского анализа субструктуры хорошо отожженных металлических материалов (плотность дислокаций < 106 см-2) можно разделить на две группы: методы локального характера (рентгеновская топография, метод микропучков) и методы статистического характера (измерения интенсивности пли распределения интенсивности рассеянного излучения). При плотности дислокации > 109 см-2 применим только анализ распределения, интенсивности для дифракционной линии (или так называемый анализ уширения липни). Достоинство методов первой группы—-простота или однозначность интерпретации наблюдаемой картины. Для методов второй группы интерпретация наблюдаемых эффектов в терминах модели мозаичной структуры или дислокационных представлений не всегда достаточно определенна. Однако поскольку решение основной задачи физического металловедения — установление- связи между характеристиками структуры и свойствами, прежде всего механическими, требует статистических характеристик структуры, следует отдать предпочтение именно методам второй группы1.
Рис. 5.37. Схемы к объяснению эффектов экстинкцин-« —первичной; б—вторичной (До—падающий луч;
—прошедший луч в прямом направлении; XI — дифрагированный луч: Л —дважды дифрагированный Л}4)
а. Размеры блоков; использование эффекта экстинкции. Явление первичной экстинкции состоит в том, что дифрагированные лучи, проходя через кристалл, испытывают повторное «отражение» от тех же кристаллографических плоскостей: дважды отраженные лучи распространяются в направлении падающего (первичного) пучка с разностью фаз, равной л (рис. 5.37). Процесс двойного (и многократного) отражения приводит к перекачке энер-
> Систематическое описание методов анализа субструктуры можно, найти в учебниках [10, 12. 13] » обзорных статьях [68, 69].
139
гнн рентгеновского излучения от направления прямого пучка к направлению дифрагированного пучка н обратно. Этот эффект зависит от рассеивающей способности кристалла, отнесенной к одной плоскости q. В связи с этим можно оценить эффект экстинкции как поправку к вычисленной из кинематической теории интенсивности отражения с малыми индексами —Qu
Л __ Qt Z th (пд)г
’ — I * Ja (OVJ
/2 Q2 \ Л<71 /
Здесь Л и /2—измеренные значения интегральной интенсивности линии с малыми индексами (/7K£)i и линии с большими индексами (HKL)?, .th(nqi)/nqi — поправочный множитель для интенсивиостн линии с малыми индексами (HK£)i; п — число плоскостей в пределах блока нлн, точнее, в пределах области когерентного рассеяния (ОКГ).
Опнсаиная методика уже в рамках представлений о мозаичной структуре кристаллитов имеет недостаток: она не учитывает эффект вторичной экстинкции. Сущность этого эффекта ясна нз схемы рис. 5.37, б. Видно, что дифрагированный луч от одного блока в пределах того же кристаллита встречает одинаково ориентированный другой блок и испытывает повторное отражение. Вероятность этого эффекта тем больше, чем меньше утлы раз-©рнентнровки блоков. Влияние обоих эффектов экстинкции на экспериментально определяемую интегральную интенсивность 7Э учитывается формулой Дарвина:
^»ксп = Q/2 (н + gQ);
Q = Q0[Zft (nq)!nq). (40)
Здесь Qo — интегральная отражательная способность на единицу объема в кинематическом приближении; Q — то же, но с поправкой на эффект первичной экстинкции; ц — линейный коэффициент ослабления; g—коэффициент вторичной экстинкции.
Дальнейшее уточнение приводит к более сложной зависимости:
Y = 73ксп//кин = (1+ cos» 20)—1 [(1 + TQo)-1^ + cos» 26 (1 + yQo cos2 * 26)—1. (41)
где Y=0,6(D2A)ctg 0-bg/p, и D—ndHK^
Выражение (41) дает возможность по измеренной величине 7ЭКсв найти у для каждой лннни (НК£) н, построив зависимость у от ctg0, определить размер ОДР (D) н средний угол нх разорнентнровки
«-(гУгТя)'1 (42)
В рамках мозаичной модели кристалла величины D н S позволяют определить плотность дислокаций p—3bjDb (43), где b— модуль вектора Бюргерса дислокаций.
Трудности в применении описанной методики связаны с влиянием текстуры в случае массивных образцов: текстура в идеально мозаичном деформированном образце, как правило, не совпадает с текстурой в отожженных образцах. Применение данной методики неоправдан©, если субструктура не соответствует модели мозаичного кристалла.
Для случая хаотического распределения дислокаций была предложена [70] методика оценки их плотности, основанная на представлении
о двух структурных составляющих: окрестности дислокаций (цилиндрические области радиуса гд вокруг каждой дислокации), которые дают вклад в интенсивность линий, определяемый по кинематической теории, и остальной части кристалла, т. е. областей правильного строения, интенсивность отражения от которых вычисляется, исходя из динамической теории
Твксп = 7 кин “i- дин 0 ’ Уд)» (44)
Уд=рлГд—объем «кинематической» области материала, т. е. объем материала, связанного с дислокациями.
Радиус области кинематического рассеяния можно определить через экстннкцноииые расстояния гд « /экст/3:
л Уячтс* .
#0кет— sin 0,
где Уяч — объем элементарной ячейки; F — амплитуда рассеяния.
Например, для отражения (110) a-железа в излучении -Со величина ^экст—4,8- 10"4 см и Гд« 1,6-10~4 см.
Исследование отожженных после холодной прокатки образцов низкоуглеродистой стали показало хорошее соответствие результатов анализа, выполненного по описанной методике, и данных, полученных металлографическим методом с выявлением ямок травления (р^ ж 107 см-2).
б. Разориентировка блоков; использование двойного вульф-брэгговского отражения. Рассеяние рентгеновских лучей под малыми углами обычно связывают с неоднородностями в распределении электронной плотности. Однако в случае кристаллического вещества дополнительный эффект рассеяния рентгеновских лучей может быть обусловлен тем, что часть дифрагированных лучей, встречая на своем пути соответствующим образом ориентированные области кристалла, испытывает повторное отражение и идет в направлении прямого пучка, несколько отклоняясь от него в зависимости от разорнентировкн областей в пределах угла в. Величина 7(e) зависит от функции г (kb), которая в свою очередь определяется разориентнровками субзерен в кристаллитах:
7 (е) = Ar(kb)/e. (45)
Здесь А — коэффициент, зависящий от материала объекта, излучения н инструментальных факторов; k— параметр, который характеризует угловой интервал распределения ориентировок субзерен; предполагается определяющая роль в двойном вульф-брэгговском отражении наиболее сильного отражения [например, отражения (Ш) в случае г. ц. к. структуры].
Функцию г (kb) задают (например, как гауссово распределение) нлн выбирают, сопоставляя экспериментальные и теоретические кривые. Методом двойного вульф-брегговского отражения выявлялись разориеитировкн порядка минут н десятков минут прн деформации растяжением никеля [71] и ползучести алюминия [72].
1 Основание для такой модели дают исследования
контраста возле дислокаций, наблюдаемого с помо-
щью рентгеновской топографии и дифракционной
электронной микроскопии.
в. Разорнентировка блоков; применение дифрактометра. Дифрактометр позволил существенно повысить точность н экспрессность анализа ориентировок при использовании методов, которые первоначально применялись с фотографической регистрацией [15, 68]. В процессе поворота (илн качания) образца прн неподвижном детекторе в отражающее положение вводятся новые области кристалла, так что счетчнк регистрирует распределение интенсивности как фувкцию угла поворота, что связано с распределением субзерен по нх ориентировкам (кривые качания). Прн этом сле-
Рнс. Б.38. Дифрактометрическая кривая качания образца поликристаллической меди после деформации 6%
дует ограничить горизонтальную расходимость падающего пучка н устранить влияние немо-нохроматнчностн применяемого излучения. Схема двукрнстального спектрометра (монохроматор— первый кристалл с предельно совершенной структурой) позволяет определить разорнентнровку соседних блоков с угловым разрешением до 10 с. При исследовании по-лнкристаллических металлов н сплавов такая прецизионность обычно не требуется. На рис. 5.38 показана типичная кривая качания, полученная [73] на дифрактометре УРС-50ИМ с гониометром ГУР-4 н трубкой БСВ-6 без применения монохроматора. Горизонтальная расходимость пучка ограничивалась диафрагмой у трубки 0,03 мм, расстояние трубка — гониометр 100 мм. Таким образом, горизонтальная расходимость 0,4'. Диафрагма у счетчика была выбрана такой, чтобы регистрировать только Кщ -отражения. Для отожженного образца получены разориентнровкн до 0,5-10~2 рад; после деформации 6—18-10~2 рад.
5.4.4. Анализ дефектов кристаллического строения по эффекту уширения линий рентгенограмм 1
Рентгенограмма металла с малой плотностью кристаллических дефектов, регистрируемая на дифрактометре, т. е. дифрактограмма, характеризуется наличием дифракционных максиму
1 Составлено А. Н. Ивановым.
мов конечной угловой ширины, а не бесконечно узкими отражениями, как это следует нз теории. Дело в том, что экспериментальные условия съемки (расходимость падающего пучка, его немонохроматнчиость, дублеты поглощение лучей в образце ие в поверхностном слое, а в слое толщиной несколько десятков микрометров) приводят к уширению области существования максимумов. Это так называемое инструментальное ушн-ренне.
Прн введении в образец дефектов, протяженных хотя бы в одном направлении, наблюдается дополнительное уширение угловой области отражения. Если распределение интенсивности в функции угла дифракции 0, т. е. профиль линии, обозначить как F(20), то физической шириной линии (уширением) называется величина
₽ = Jf (26)d26/Fmax. (46)
При одновременном влиянии инструментальных и физических факторов суммарный профиль линии //(26) является сверткой функций физического F(20) н инструментального G(26) уширения, т. е.
Н (26) = J F (20) G (26 — у) dy. (47)
Коэффициенты разложения в комплексной форме этих профилей в ряд Фурье просто связаны друг с другом: С„ =С? (48). По-
этому, зиая профиль линии исследуемого образца [Щ26)] и эталона-образца, в котором ушнреиие вызвано только инструментальными факторами [6(20)], можно определить набор значений С%, а по ним и величину р. Ту же задачу можно решить, используя и метод аппроксимации [14, 15].
Физическое уширение линий (3 может быть вызвано несколькими причинами:
1. Дисперсностью кристалликов; их размер (D) от 50 до 1500 А вызывает уширение ₽= =&%/£) cosO (49).
Эффект дисперсности может быть связан с образованием в материале блоков малого размера, границей которых является стенка дислокаций, н с дефектами упаковки.
2. Смещением атомов (нонов) нз узлов идеальной решетки под действием дальнодей-ствующнх упругих нолей дефектов (дислокации, дислокационные скопления типа «pile пр» и границы ячеек н т. п.)_ Эти смещения, трактуемые как мнкродеформация е, вызывают уширение p=4etg0 (50).
Если в изучаемом материале действуют оба фактора уширении, то суммарный физический профиль лнинй является также сверткой функции уширения от дисперсности А4(26) и микродеформаций N(20), т. е. F(26)= | /И(20)#Х XW-y)dy (51).
Для разделения этих эффектов применяют уже упоминавшиеся методы аппроксимации нлн гармонического анализа [10, 14, 74], которые используют для анализа профиля обязательно двух линий, в общем случае отличающихся лишь порядком отражения (например, HKL и 2H2K2L).
Анализ позволяет определить размер кристаллитов нлн блоков D, среднюю величину микродеформацин, концентрацию дефектов
141
упаковки в материале (в случае о. ц. к. структуры). Аналогичные результаты можно получить, используя метод вторых нли четвертых центральных моментов [57].
Следует отметить, что в основе описанных методов анализа уширения линий лежит модель кристалла, разбитого на упруго деформированные области когерентного рассеяния, поэтому они применимы только тогда, когда в изучаемом металле имеются физически ограниченные области малого размера. В массивных материалах такие области, как правило, не обнаруживаются прямыми электронно-мик-роскопическими методами. В этом случае анализ уширения можно провести на основе теории рассеяния рентгеновских лучей дефектными кристаллами, разработанной М. А. Кривоглазом [9, 45]. Он показал, что в кристаллах, содержащих прямолинейные хаотически распределенные дислокации, дислокационные скопления типа «pile пр» и границы ячеек, физическое уширение меняется пропорционально tg0 и корню квадратному из плотности этих дефектов. В частности, для прямолинейных дислокаций плотностью р уширение равно
(In cj I pig6 (52), где —константа, значение которой зависит от соотношения краевых и винтовых компонент, величины вектора Бюргерса, упругих постоянных кристалла и индексов отражающей плоскости; величина qhki может быть вычислена достаточно точно; cv=F(.0[//^p—численный коэффициент, который характеризует эффективный радиус Дг, поля смещений дислокаций при данном характере их распределения. Для практических расчетов можно считать cv=5-е8 (меньшие значения для кристаллов с низкой энергией дефектов упаковки) [75].
При приближенных расчетах (с точностью до полупорядка) можно воспользоваться формулой
4 ctg20 Р 5 ’
где h — вектор Бюргерса; уширение р выражено в радианах; формула получена в предположении, что коэффициент Пуассона v=0,3, а кристалл является упруго изотропным.
Основным критерием возможности проведения расчетов по теории М. А. Кривоглаза является соотношение уширений двух линий разных порядков отражения, т. е. ]WPi~ ^tgOg/tgGi (в пределах точности эксперимента).
Многочисленные измерения, проведенные на деформированных массивных материалах (не порошках), показали', что угловая зависимость уширения пропорциональна tg 0.
(53)
БИБЛИОГРАФИЧЕСКИЙ СПИСОК
I. Шаскольскач М. П. Кристаллография. М.: Высшая школа, 1976. 389 с.
2. Васильев Д. И. Физическая кристаллография. М.: Металлургия, 1981. 246 с.
3. Современная кристаллография. Т. 1. М.: Наука, 1979. 383 с.
4. Гинье А. Рентгеновская кристаллография. М.: Физматгиз. 1961. 602 с.
5. Жданов Г. С. Основы рентгеиоструктуриого анализа. М.: Гостехиздат, 1940. 446 с.
6. Джеймс Р. Оптические принципы дифракции рентгеновских лучей. М.: ИЛ, 1950. 572 с.
7. Китайгородский А. И. Рентгеноструктурный анализ. М.: Гостехиздат, 1950. 650 с.; Рентгеноструктурный анализ мелкокристаллических и аморфных тел. М.: Гостехиздат, 1952. 588 с.
8. Васильев Д. М. Дифракционные методы исследования структуры. М.: Металлургия, 1977. 247 с.
9. И веронова В. И., Ревкевич Г. П. Теория рассеяния рентгеновских лучей. М.: Изд-во МГУ, 1978, 274 с.
10. Уманский Я- С. Рентгенография металлов. М.: Металлургия. 1967. 235 с.
11. Пинес Б. Я. Лекцин по структурному анализу. Харьков. Изд. ХГУ. 1957. 455 с.
12. Уманский Я. С. Рентгенография' металлов и полупроводников. И.: Металлургия, 1969. 496 с.
13. Русаков А. А. Рентгенография металлов. М.: Атомиздат, 1977. 479 с.
14. Горелик С. С., Расторгуев Л. И., Сканов Ю. А. Рентгенографический и электронно-оптический анализ. М.: Металлургия, 1971. 368 с.
15. Рентгенография в физическом металлове-депии/Под ред. Багаряцкого Ю. А. Мл Металлургиздат, 1961. 368 с.
16. Митчелл С. ЛЕ—В кил Приборы и методы физического металловедения. Вып. 1.: Пер. с англ./Под ред. Вейнберга Ф. М.: Мир, 1973, с. 332—423.
17. Миркин Л. И. Справочник по рентгеноструктурному анализу поликристаллов. Мл Физматгиз, 1961. 863 с.
18. Миркин Л. И. Рентгепоструктурный анализ. Получение и измерение рентгенограмм: Справочное руководство. Мл Наука, 1976. 326 с.
19. Рентгенотехника: Справочник в 2-х кн./Под ред. Клюева В. В. Мл Машиностроение, 1980. 431 н 383 с.
20. Миркин Л. И. Рентгеноструктурный контроль машиностроительных материалов: Справочник. Мл Машиностроение, 1979. 134 с.
21. Смитлз К. Дж. Металлы. Справочник. Мл Металлургия, 1980. 446 с.
22. Шуберт К. Кристаллические структуры двухкомпоиентных фаз: Пер. с нем. Мл Металлургия, 1971. 531 с.
23. Интерметаллические соединения. Пер. с англ./Под род. Корнилова И. И. Мл Металлургия, 1970. 439 с.
24. Гольдшмидт X. Дж. Сплавы внедрения.: Пер. с англ./Под ред. Чеботарева Н. Т. Мл Мир, 1971. Вып. 1. 423 с.; вып. 2. 463 с.
25. Андриевский Р.. Уманский Я. Фазы внедрения. Мл Наука, 1977. 239 с.
26. Нарита К. Кристаллическая структура неметаллических включений в стали: Пер. с япенск./Под ред. Арсентьева П. П. М.: Металлургия, 1969. 191 с.
27. Теслюк 41. Ю. Металлические соединения со структурами фаз Лавеса. Мл Наука. 1968. 136 с.
28. Матюшенко Н. И. Кристаллические структуры двойных соединений (соединения элементов с Be, В, С, N, Mg, Al. Si, Р): Справочник. М.: Металлургия, 1969. 302 с.
29. Уманский Я. С., Скаков К). А Физика ме-
142
таллов. Атомное строение металлов и сплавов. М.: Атомиздат, 1978. 350 с.
30. Уманский М. М. Аппаратура для рентгеноструктурных исследований. М.; Физматгиз, 1960. 358 с.
31. Хейкер Д. ЛЕ, Зевин Л. С. Рентгеновская дифрактометрия. М.: Физматгиз, 1963. 380 с.
32. Хейкер Д. ЛЕ Рентгеновская дифрактометрия монокристаллов. Л.: Машиностроение, 1973. 256 с.
33. Кулинасов Г. Н., Скринский А. Н. — УФЫ, 1977, т. 122, № 3, с. 369.
34. Нормы радиационной безопасности НРБ— 76. М.: Атомиздат, 1978. 56 с.
35. Михеев В. И. Рентгенометрический определитель минералов. М.: Госгеолтехиздат, 1957. 370 с.
36. Hanawalt J. D. — Conference on Applied Crystallography Proc., v. 1, Kozubnik. Po-’ land, Ed. Siles. Univers. Katowice. 1979, p. 54—70.
37. ASTM Card File (Diffraction Data Cards and Alphabetical and Grouped Numerical Index of X-ray Diffraction Date). Philadelphia: Ed. ASTM, 1969.
38. Powder Diffraction Fili. Inorganic Sets 1—5, 6—10, 11—15, 16—18. Swarthmore. Pennsylvania: Ed. JCPDS, 1977.
39. Selected Powder Diffraction Data for Metals and Alloys. Swarthmore, Pennsylvania: Ed. JCPDS, 1977.
40. Шевченко Ж- А., Спектор А. Г. — Заводская лаборатория, 1965, № 7, c. 814—816.
41. Бурова E. M., Жидков H. П., Зильберман А. Г. и др. — Кристаллография, 1977, т. 22, вып. 6, с. 1182—1190.
42. Кацнельсон А. А., Салонов В. М.— Аппаратура и методы рентгеновского анализа. Вып.. IX/СКБ РА. Л.: 1971, с. 78—81.
43. Скаков Ю. А., Глезер А. М.— Итоги науки н техники. Серия металловедения и термическая обработка. Т. 9л М.: ВИНИТИ, 1975, с. 5—72.
44. Иверонова В. И., Кацнельсон А. А. Ближний порядок в твердых' растворах. М.: Наука, 1977. 255 с.
45. Кривоглаз М. А. — В кн.: Металлы, электроны, решетка. Киев: Наукова думка, 1975, с. 355—388.
46. Наумова М. М., Семеновская С. В. — ФТТ, 1971, т. 18, №2, с. 181.
47. Захарова М. И. Атомно-кристаллическая структура и свойства металлов и сплавов. М.: Изд-во МГУ, 1972. 215 с.
48. Чуистов К- В. Модулированные структуры в стареюших сплавах. Киев; Наукова думка, 1975.
49. Гитгарц М. И.-—Металлофизика. Вып. 51. Киев: Наукова думка, 1974, с. 45—59.
50. Климанек П. И., Мирский А. Л., Скоков Ю. А. — Изв. вузов. Черная металлургия, 1978, № 11, с. 127—131.
51. Верещагин Л. Ф., Кабалкина С. С. Рент-геноструктурпые исследования при высоком давлении. М.: Наука, 1979 . 351 с.
52. Курдюмов Г. В., Утевский Л. М., Эн
тин Р. И. Превращения в железе и стали. М.: Наука, 1977. 286 с.
53. Еднерал Н. В., Зюлина Н. Р., Скаков Ю. А. — ФММ, 1975, т. 39, вып. 5, с. 1049—1053.
54. Лысак. Л. И., Николин Б. И. Физические основы термической обработки стали. Киев: Техшка, 1975. 350 с.
55. Лысак Л- И., Устинов Р. И. — Заводская лаборатория, 1975, № 12, с. 1473.
56. Штремель М. А., Капуткина Л. 41. — ФММ, 1972, т. 32, с. 991—998.
57. Каган А. С. — Заводская лаборатория, 1980, № 5, с. 406—414.
58. Брусиловский Б. А.. Заброцкий В. К-, Иванов Ф. И. — Заводская лаборатория, 1976, т. 42, № 1, с. 27—29.
59. Тананко И. А., Белозеров В. В., Махати-лова А. И. — МиТОМ, 1979, № 10, с. 12— 14.
60. Фукс М. Я., Гладких Л. И. — Заводская лаборатория, 1965, № 8, с. 978—980.
61. Вассерман Г., Гревен И. Текстуры металлических материалов. М.: Металлургия, 1969. 664 с.
62. Бородкина М. М., Спектор Э. Н. Рентгенографический анализ текстуры металлов и сплавов. М.: Металлургия, 1981. 271 с.
63. Bunge Н. I. Matematische Metoden der Тех-turanalyse. Berlin: Akademie-Verlag, 1969. 330 s.'
64. Бабареко A. A. — Итоги науки и техники. Серия Металловедение и термическая обработка. Вып. 13. М.: ВИНИТИ, 1980, с. 79—148.
65. Комяк Н. И., Мясников Ю.' Г. Рентгеновские методы и аппаратура для определения напряжений. Л.: Машиностроение, 1972. 87 с.
66. Васильев Д. М., Комяк Н. И., Кострова В. Н. и др. — Заводская лаборатория, 1965, № 8, с. 972—978.
67. Васильев Д. ЛЕ, Комяк Н. И., Кострова В. И. — Аппаратура и методы рентге-* невского анализа. Вып. 11/СКБ РА. Л.: 1972, с. 3—12.
68. Ровинский Б. М., Лютцау В. Т., Ханон-кин А. А. — Аппаратура и методы рентгеновского анализа. Вып. 9/СКБ РА. Л.: 1971, с. 3.
69. Skakov Yu. А. (Скаков Ю. А.) — Kristall und Technik, 1977, Bd 12, H. 3, S. 283—293.
70. Иванов A. H., Климанек П. И., Скаков Ю. А., Фомичева Е. И. — В кн.: Всесоюзное совещание «50 лет отечественного рентгеновского приборостроения» ЛНПО «Буревестник». Л.: 1978, с. 185—186.
71. Пигин В. М., Крутовский Ю. Н., Бекре-нев А. Н. — Ученые записки Петрозаводского государственного университета, 1971, т. VI, с. 226.
72. Бетехтин В. И., Мышляев М. М. — ФММ, 1967, т. 24, с. 1069.
73. Кайбышев О. А., Эпштейн Г. Н., Казачков И В. — Заводская лаборатория, 1967, т. 23, с. 1528—1531.
74. Штремель М. А. — Кристаллография, 1969, т. 14, с. 34—43.
6
-=- РЕНТГЕНОСПЕКТРАЛЬНЫЙ МИКРОАНАЛИЗ
Рентгеноспектральный микроанализ (РСМА) используется для исследования распределения компонентов и примесей в сталях' н сплавах [1—10], прн этом обеспечивается разрешение порядка микрометров.
Методом РСМА определяют химический состав микрообластей на металлографическом шлифе. Информация такого рода необходима прн изучении дендритной ликвации, микросег-регацни, диффузии, идентификации включений и фазовых составляющих в сплавах и др.
РСМА основан иа регистрации рентгеновскими спектрометрами эмиссионного рентгеновского излучения, возбужденного пучком электронов с энергиями 1—50 кэВ, сфокусированных иа образце в пятно диаметром ~ 1 мкм. Измеряя длину волны и интенсивность характеристического рентгеновского излучения, отнесенную к интенсивности эталона, определяют, какие элементы присутствуют в выбранном мнкрообъеме и каковы нх концентрации. Относительная интенсивность t-того элемента определяется измерением интенсивностей (за вычетом интенсивностей фона) А для образца н /^т для эталона, содержащего 100 % i-того элемента (прн использовании в качестве эталона химического соединения с известным содержанием i-того элемента ki= > гле —расчетная интенсивность анализируемого элемента в эталоне).
Метод был разработан в конце 40-х — начале 50-х годов иезависнмо друг от друга Кастеном и И. Б. Боровским.
6.1. УСТРОЙСТВО
РЕНТГЕНОСПЕКТРАЛЬНОГО МИКРОАНАЛИЗАТОРА
Принципиальная схема рентгеноспектрального микроаналнзатора (электронного микрозонда), предложенная Кастеном, показана на рнс. 6.1. Все применяемые в настоящее время микрозонды состоят нз следующих основных частей:
1) электронно-оптической системы для получения сфокусированного пучка электронов (электронная пушка н электронные линзы);
2) рентгеновских спектрометров и детекторов рентгеновского излучения для измерения длин волн и интенсивностей возбуждаемых характеристических рентгеновских линий;
3) светового микроскопа для выбора исследуемого участка на образце.
Сигналы с детекторов излучения через усилители подаются на амплитудные анализаторы импульсов и регистрируются интенсиметра-ми, пересчетными схемами пилн печатающим устройством типа телетайп (анализ по точкам). Выходной сигнал с интенснметра может быть подан на самопишущий потенциометр, и прн непрерывном перемещении образца под электронным зондом на диаграммной ленте самописца получают кривые распределения определяемых элементов вдоль выбранной прямой. В приборах также имеется возможность сканировать электронный луч в двух взаимно
перпендикулярных направлениях нлн электронный луч в одном направлении, а образец в перпендикулярном и подавать сигнал с каждой точки исследуемой площади шлифа на электронно-лучевую трубку. Фотографирование с
Рис. 6.1. Схема рентгеноспектрального микроанализатора:
1 — нить накала; 2— цилиндр Венельта; 3—анод; 4—конденсорная линза; 5 — источник света; 6— окуляр; 7 — объективная линза; 8 — отражательный объектив; 9 — детектор излучения; 10 — кристалл-анализатор
экрана электронно-лучевой трубки в процессе сканирования позволяет получать распределение ннтенснвности характеристического излучения, а следовательно, кабину распределения того нлн иного элемента по выбранной плоша-дн шлифа.
Сканирование с регистрацией на электроннолучевой трубке тока поглощенных или отраженных электронов дает качественную оценку среднего атомного номера составляющих на. разных участках шлнфа, а сканирование с регистрацией тока вторичных электронов позволяет судить о рельефе исследуемой поверхности. Последнее обстоятельство дает возможность использовать микрозонды и как растровые электронные микроскопы.
Современные микрозонды имеют разные-приставки, позволяющие получать дополнительную информацию об образце. В табл. 6.1 приведены основные технические данные некоторых микрозондов разных марок. Как правило, микрозонды являются одновременно и
144
ТАБЛИЦА 6.1 ТЕХНИЧЕСКИЕ ДАННЫЕ НЕКОТОРЫХ МИКРОАНАЛИЗАТОРОВ
Прибор Ускоряющее напряжение, кВ Диаметр зонда, мкм Определяемые элементы Угол выхода рентгеновского излучения Число рентгеновских спектрометров Разрешение в растровом режиме, им Другие приспособления
марка страна, фирма
Camebax micro Франция, «Сатеса» 1—50 0,06— 1,2 в—и 40 4 7 ЭДС, миннкомпь-ютер PDP-11, ка-тодолюмннесцент-ная приставка, охлаждение образцов, другие приставки
Superprobe-733 Япония, «JEOL» 0—50 0.005— 1,1 в—и 40 5 7 ЭДС, миникомпьютер и другие приставки
MS/46 Франция, «Сатеса» 1—40 1 в—и 18 4 Анализатор для количественной металлографии и другие приставки
МР-4 СССР, «Буревестник» 1—50 1 в—и 40 4 — ЭДС
MAP-2 СССР, Красногорский механический завод 10—50 1 в—и (кроме OhF) 17—32*1 2 — —'
ЭММА-4 СССР, Сумской завод электронных микроскопов 10— 100 0,3 В—и 45 1*2 1—2*3 Детектор ультра-мягкого излучения
ЭММА-2 То же 29—70 0,3 В—и 50 2 4*з —
•* Переменный.
•2 Восемь кристаллов-аналнваторов.
В просвечивающем режиме.
растровыми электронными микроскопами, что позволяет изучать поверхности изломов с одновременным качественным илн полуколичест-венным РСМА структурных составляющих. Кроме спектрометров волновой дисперснн (брэгговских), микрозонды имеют энергетические дисперсионные спектрометры (ЭДС), которые позволяют наблюдать энергетический спектр всех элементов одновременно, начиная с Z=ll (Na). Это значительно сокращает продолжительность качественного анализа.
Оснащение приборов компьютерами облегчает труд оператора благодаря автоматизации ряда операций и математической обработке результатов эксперимента, а также ускоряет анализ.
6.2. ОСНОВНЫЕ ПАРАМЕТРЫ МЕТОДА
6.2. L Определяемые элементы
Все элементы от бора (Z=5) до урана {Z— — 92); на некоторых приборах можно определять также бериллий (Z—4). Определение ряда элементов в присутствии других в ряде случаев затруднено.
Рентгеновский спектр любого вещества в отличие от оптического спектра сравнительно
прост, состоит нз малого числа линий, слабо зависящих от типа химических связей, что делает РСМА достаточно быстрым и надежным,
.ТАБЛИЦА 6.2
ХАРАКТЕРИСТИЧЕСКИЕ ЛИНИИ НЕКОТОРЫХ ЭЛЕМЕНТОВ С БЛИЗКИМИ ЗНАЧЕНИЯМИ
ДЛИН ВОЛН (А)
Элемент Линия X, А Элемент Линия A, A
С Ка 44,5 Pt 6,05»
Ni* ^01 43,6 Мо 4 5,406
N 31,6 S 5,372
Ti 31,4 Zr 5,385
Al* ка 25,02 Ti 2,752
О «а 23,57 Ba 2,776
Мп Lv 22,3 As 1,177
Р 6,157 Pb 1,175
Zr 6,079
* 3-й порядок отражения.
10—280
145
Но имеются случаи, когда на линии /(-серии одного элемента накладываются линии L- или Af-серии другого элемента (табл. 6.2).
При наложении линий определение одних элементов в присутствии других требует использования специальных методических приемов. Например, на линию Ка серы практически накладывается линия La молибдена. В этом случае нельзя получить значение относительной интенсивности ks, так как прибор зарегистрирует суммарную интенсивность £s+ +АМо. Можно приближенно оценить величину k&, если измерить АМо по линии МоЛ'а 4 о
(Л=0,709 А) и затем вычесть из измеренной суммарной относительной интенсивности (feg -f-+йМо) значение k^Q. Ошибка в полученног таким образом значении k5 связана с тем, что нс учитывается разница коэффициентов поглощения в образце излучений МоАа н Мо£а . Аналогично поступают и при анализе карбонитридов Ti(C, N).
6.2.2. Локальность РСМА
Локальность РСМА, т. е. эффективный объем вещества, в котором возбуждается характеристическое рентгеновское излучение, определяется в первую очередь диаметром зонда на образце. При анализе монолитных образцов линейная локальность (диаметр пятна на образце) не может быть лучше 1—2 мкм. Это объясняется тем, что электроны успевают пройти в образце расстояние 1—3 мкм прежде, чем их энергия станет недостаточной для генерации характеристического рентгеновского излучения. Согласно Кастену эффективный размер пятна нз-за рассеяния электронов определяется выражением S=O,O33(FJ’7—£^7)A/pZ, где Ео и Ек, выраженные в кэВ, соответственно энергия падающих на образец электронов, определяемая заданным ускоряющим напряжением, и энергия возбуждения характеристического рентгеновского излучения элемента с атомным номером Z и атомной массой А; р—-плотность образца. Размер пятна существенно зависит от энергии электронов. Так, для чистого алюминия (£^=1,5 кэВ) размер пятна равен 6 мкм при £о=ЗО кэВ и 1,5 мкм при Ео=1О кэВ. Обычно работают при напряжениях в интервале 10—20 кВ. Нецелесообразно уменьшать диаметр зонда до величнн, меньших 0,3—0,5 мкм, так как при заданном ускоряющем напряжении пучки меньшего диаметра нз-за рассеяния электронов будут возбуждать рентгеновские лучи с той же эффективной площади образца. Количественный РСМА можно проводить при размерах фаз ~5 мкм. Минимальный объем частиц в экстракционных репликах, которые удается анализировать па микрозонде, составляет 0,2— 0,3 мкм3. На электронном микроскопе-мнкро-аиализаторе (ЭММА) в экстракционных репликах или в фольгах определялся состав равномерно распределенных частиц с минималь-
О
ными размерами 30—60 А, а для отдельных выделений размерами 100X100X600 А [14].
6.2.3. Чувствительность метода (предел обнаружения)
Минимальная концентрация элемента, определяемая на микрозонде, зависит от отношения интенсивности сигнала, получаемого при регистрации излучения чистого элемента, к интенсивности фона и составляет сотые доли процента для элементов с Zz> 11; для легких элементов (бор, углерод, азот, кислород) чувствительность анализа 1—5 %. В отдельных случаях, увеличивая время счета и повышая стабильность работы прибора, удается добиваться более высокой чувствительности. В работе [15] показано, что, выбрав оптимальные условия проведения РСМА, можно изучать распределение примесных элементов в сталях с чувствительностью 0,002; 0,003; 0,002; 0,002; 0,0007; 0,0005 и 0,0008 соответственно для Al, Si, Р, S, Са, Tf и Pb.
Локальная чувствительность — абсолютная массовая чувствительность, отнесенная к единичному определению в микрообъеме и выраженная в граммах, — может быть оценена для каждого отдельного случая в зависимости от локальности, чувствительности и анализируемого вещества. Например, для вещества с плотностью 10 г/см3 при чувствительности 0,05 % и диаметре электронного зонда 1 мкм объем зоны возбуждения равен ~8 мкм3 и локальная чувствительность 4Х ХЮ"14 г.
6.2.4. Точность количественного РСМА
Точность количественного РСМА определяется инструментальными ошибками, квалификацией оператора и точностью внесения поправок. Инструментальные ошибки зависят от точности установки угла 0, соответствующего максимуму интенсивности рентгеновского излучения, стабильности прибора, времени счета, качества поверхности исследуемого шлифа, а при анализе малых содержаний, близких к пределу чувствительности, от правильности записи интенсивности фона.
Точность расчета поправок для большинства элементов, за исключением легких (Zell), обычно составляет 2—5'%.
6.3. ВОЗМОЖНОСТИ МЕТОДА
В зависимости от задач, которые решают методом РСМА, и характера информации, которую желательно получить прн этом, можно вести анализ по точкам, сканированием вдоль выбранной прямой нли сканированием по площади участка мнкрошлифа (табл. 6.3).
Прн анализе по точкам имеется возможность для каждого определяемого элемента в зависимости от его концентрации в исследуемом микрообъеме, подбирая необходимое время счета импульсов, обеспечить проведение количественных определений с заданной точностью. При анализе неметаллических включений и фазовых составляющих, даже при методически правильном его выполнении, часто могут появляться трудно определяемые источники ошибок. Обычное требование к размерам анализируемого участка (^5 мкм) является необходимым, но не достаточным. Аналнзиру-
146
ТАБЛИЦА 6.3
ОСНОВНЫЕ МЕТОДИКИ РЕНТГЕНОСПЕКТРАЛЬНОГО МИКРОАНАЛИЗА И ОБЛАСТИ ИХ ПРИМЕНЕНИЯ
Методика Информация Области применения
Анализ в точке Анализ вдоль линии, дискретный То же, непрерывный Анализ по площади шлифа с получением картины распределения элементов Качественные и количественные сведения о содержании элементов Количественные характеристики концентрационной кривой Полуколичественные и количественные данные о концентрационной кривой Качественные и пол у количественные данные о распределении элементов Идентификация фаз. Обнаружение отдельных элементов Контроль степени гомогенизации (изучение дендритной ликвации) Изучение диффузионных процессов Изучение характера распределения элементов в многокомпонентных системах, переходных слоях п пр.
То же, с выдачей числовых характеристик Фазовый состав Количество фаз Усредненный анализ заданных об- ластей Количественный объемный анализ Стереологический анализ Количественный анализ гетерогенных областей
слое включение может иметь размер вдоль направления электронною зонда меньше области возбуждения, тогда на результатах анализа сказывается влияние матрицы (рис. 6.2, а). Возможно также влияние включений на анализ матрицы (рис. 6.2,6). Поэтому по апалп-
Рис. 6.2. Область возбуждения рентгеновских лучей.-а — эффект матрицы; б — эффект включения: 1 — электронный зонд; 2— включение; 3—область возбуждения
зу единичных включений, если в них обнаружен элемент основы, нельзя делать выводы об нх количественном составе. Следует провести анализ ряда идентичных включений. Эффект матрицы (или включения) молено уменьшить, также снизив ускоряющее напряжение (прн этом несколько уменьшится область возбуждения рентгеновских лучей).
При непрерывном анализе вдоль выбранной прямой получают пол у количественные данные об изменениях концентрации элементов в диффузионных зонах (поверхностные покрытия, сварные соединения и пр.). В диффузионной зоне наряду с изменением концентрации по глубине наблюдается микронеодно-родпость распределения элементов. Из-за этого при работе с точечным зондом отмечаются значительные колебания интенсивности рент
JO*
геновского излучения, возбуждаемого в этих микрообластях. Это затрудняет обработку концентрационных кривых. В таких случаях проводят усреднение интенсивностей рентгеновскою излучения вдоль линии, перпендикулярной градиенту концентрации (параллельной фронту диффузии). Для этого с помощью электростатической или электромагнитной развертки зонд развертывают в линию шириной 0,5—1 мкм и длиной 40—60 мкм. Под таким зондом образец относительно медленно перемещают в направлении градиента концентрации, при этом интенсивность аналитических линий непрерывно регистрируется на диаграммной ленте самопишущего потенциометра. При соответствующем выборе скорости перемещения образца, постоянной времени прибора и скорости движения диаграммной лепты можно получить результаты, полностью совпадающие с результатами, полученными при анализе по точкам.
Анализ по площади шлнфа с получением изображения в характеристическом рентгеновском излучении дает наглядные фотографии распределения элементов иа выбранном участке шлнфа размерами от 30X30 до 400Х Х400 мкм (увеличение па экране электроннолучевой трубки соответственно —2000 и —150). Качественные результаты, получаемые при этом, позволяют приближенно судить о составе разных участков сложных включений, выделений по границам зерен и т. д (рис. 6.3). При сканировании хорошо обнаруживаются отдельные включения или участки с большой разницей в концентрации Малые количества элементов этим методом обнаружить нельзя, так как при сканировании продолжительность регистрации в каждой точке невелика, что приводит к ошибке счета.
Ток, проходящий через образец, также используется для модулирования яркости свечения электронно-лучевой трубки. Изображения, полученные с помощью тока поглощенных электронов (или в «поглощенных электронах»), обладают более высокими чувствительностью
147
Рис. 6.3. Распределение элементов по площади включения сложного состава (в характеристическом рентгеновском ) излучении). Х800:
а — А1; б — S1; в — Ti; г—Сг: д — Мп; е — Fe
н контрастом и могут давать картину распределения элементов. Часто первоначально проводят анализ по площади шлифа, а затем ведут количественный анализ в отдельных выбранных точках.
Прн анализе по площади можно проводить
количественный металлографический анализ, дифференцируя фазы по химическому составу. Это особенно важно в тех случаях, когда выделения мало отличаются по отражательной способности и трудно различимы на приборах типа «Квантимет» нлн «Эпнквант».
148
На микрозонде MS/46 фирмы «Сатеса» количественный металлографический анализ проводят следующим образом. Электронный зонд совершает возвратно-поступательные перемещения по оси х; одновременно образец линейно перемещается по осн у, в результате чего зонд иа шлнфе проходит пилообразную траекторию, пересекая встречающиеся по пути частицы. Концентрация фазы на площади сканирования определяется из отношения времени, в течение которого зонд находится на исследуемой фазе, к времени общего сканирования. Для этого используется генератор импульсов (60 Гц). Прн сканировании определяется общее число импульсов за время сканирования исследуемой площади на образце и число импульсов генератора за время, когда зонд находится на анализируемой фазе. Из сигнала от счетчика спектрометра дискриминатором выделяется лишь та часть, которая отвечает заданной интенсивности характеристического излучения, зависящей от концентрации элемента. Дискриминаторы трех каналов настраивают на разные уровни характеристического излучения одного элемента илн на излучение различных элементов. Анализ • ведется в режиме сканирования по площади с фоторегнстрацией (изучение характера распределения) нлн с регистрацией на соответствующих пересчетных устройствах (получение количественных характеристик),
Микрозондовый количественный металлографический анализ позволяет различать объекты (фазы, выделения, области) по трем независимым параметрам — длине волны характеристического рентгеновского излучения, его интенсивности и по одновременному присутствию до трех компонентов в фазе, Прн этом, как и другими методами количественной металлографии, можио определить объемный процент фаз, установить нх распределение по площади шлифа, определить площадь, занимаемую этими фазами, и средний размер выделений. Существенный недостаток метода — его низкая производительность: прн максимально возможной площади сканирования (для прибора MS/46 2,4 мм2) продолжительность сканирования составляет 1,5—2 ч [иа .приборе «Эпи-квант» прн больших площадях (8 мм2) — 5 мин].
Место РСМА средн других методов определяется его высокой локальностью. Электрохимические методы фазового анализа и анализа неметаллических включений с анодным растворением образца, как видно нз табл. 6,4, являются интегральными. Для того чтобы
ТАБЛИЦА 6.4
анализ давал правильное представление о фазовом составе или о виде и количестве неметаллических включений в металле, необходимо, чтобы «иавеска» была не менее определенной. Прн использовании интегральных методов- можно «потерять» единичные, порой довольно крупные включения, иногда оказывающие существенное влияние на эксплуатационные свойства металла. ,
РСМА — мнкрометод, или дифференциальный метод. С его помощью получают сведения о составе отдельных частиц, границ зерен илн мнкрообластей. Этот метод не может заменить интегральных методов фазового анализа или анализа неметаллических включений, по результатам которых судят об усредненном фазовом составе и о количестве и составе неметаллических включений.
Метод РСМА предполагает предварительное металлографическое исследование. Объект исследования — металлографический шлнф, на котором металлографическими методами выявлены какие-либо неоднородности структуры: включения, фазовые составляющие, границы зерен, переходные зоны и т. д. Исследование на микрозонде случайных, единичных включений без предварительного глубокого металлографического анализа может ввести в заблуждение технологов прн оценке и выборе технологических вариантов выплавки сталей и сплавов. Приступая к исследованию на микрозонде неметаллических включений, следует знать, насколько типичны и представительны выбранные для микроанализа включения. РСМА должен использоваться в комплексе современных методов исследования.
Применение РСМА наиболее эффективно в тех случаях, когда он используется для решения тех научных н практических задач, которые не могут быть решены никакими другими аналитическими методами. В подавляющем большинстве случаев ввиду большой методической сложности всех этапов анализа и обработки получаемых результатов проведение конкретных анализов эквивалентно выполнению исследовательской работы.
6.4. О ПОПРАВКАХ
ПРИ КОЛИЧЕСТВЕННОМ РСМА
Интенсивность рентгеновского излучения зависит от ряда факторов. Измеренные относительные интенсивности характеристического излучения i-того элемента (ki) связаны с концентрацией соответствующего элемента (с<) соот
КОЛИЧЕСТВА ВЕЩЕСТВА. УЧАСТВУЮЩИЕ В АНАЛИЗЕ СТАЛИ ДЛЯ РАЗНЫХ МЕТОДОВ АНАЛИЗА
Метод Размеры исследуемой области «Невеска» для анализа, г
Электрохимический с анодным растворением образца: анализ неметаллических включений фазовый анализ Спектральный эмиссионный анализ Рентгеновский флюоресцентный анализ Лазерный спектральный микроанализ РСМА Образец 012 мм, 1= = 120 мм 0=10 мм, /=60 мм 0—4 мм 0 — 4 мм 0—30 мкм Диаметр зоида —1 мкм 100 0,1—1,0 2-10-3 5-10-2 2-10—8 ~10~14
149
ношением Лг=с#Д(сь с2,сп), где сь ...» сп— концентрации содержащихся в образце элементов, a ft (сь с%, ...» сп) — поправочная функция, состоящая из трех вводимых при анализе поправок на атомный номер fj(-Z), ла поглощение fi{x) и на дополнительное флюоресцентное возбуждение fi(v).
Имеется много методов расчета поправок для перевода относительных интенсивностей в концентрации во массе анализируемого элемента. Разные методы расчета отличаются степенью точности, границами применимости для разных приборов и продолжительностью операций введения поправок. Наиболее полные обзоры известных методов даиы в работах [6, 12, 13]. Для приборов, снабженных ЭВМ, фирмы, как правило, прилагают несколько вариантов программ внесения поправок. В литературе приводятся также методики ускоренного расчета поправок прн проведении количественного анализа [10, 11].
Прн пол у количествен ном анализе пользуются калибровочными кривыми.
6.5. ПОДГОТОВКА ОБРАЗЦОВ
И ЭТАЛОНОВ
Подвергаемая анализу поверхность образцов должна быть совершенно плоской и не иметь рисок и рельефа. Изменение плоскостности только на 1° для образцов с коэффициентом поглощения 1000 при угле выхода рентгеновских лучей 20° на приборе приводит к изменению интенсивности на 3% [10]. Канавка в магнии глубиной 0,5 мкм при использовании такого же прибора приводит к ошибке в определении концентрации иа 10 % [5].
Для обеспечения необходимой плоскости шлифа целесообразна полировка алмазной пастой. При подготовке шлифов нельзя использовать абразивные материалы, содержащие элементы, входящие в состав исследуемых сплавов или включений (например, нельзя пользоваться окисью хрома, если проводится анализ на хром, окисью алюминия, если определяют содержание алюминия, н т. д.).
Следует избегать травления, так как оно может привести к образованию рельефа и к изменению состава поверхностного слоя. Если фазы, которые надо анализировать, выявляются травлением, иа травленом шлифе возле исследуемого участка наносят отпечатки индентором микротвердомера, фотографируют эго место, а затем полируют образец так, чтобы отметки сохранились.
Если в состав сталей или сплавов входят мягкие компоненты (например, свинец в медных сплавах, сплавы А1—Si и пр.), то может происходить их «намазывание» па поверхность шлнфа, делая се непредставительной. Во избежание этого рекомендуется проводить мокрую полировку. «Намазывание» может быть и при подготовке шлифов, залитых в третник, сплав Вуда и пр. При подготовке шлифов, залитых в пластмассу, следует удалить пластмассу с поверхности образца, так как под действием электронною пучка она испаряется и загрязняет детали, расположенные внутри колонны.
При анализе неэлектропроводящих объектов (шлаки, огнеупоры, крупные неметаллические включения, неметаллические покрытия и пр.) на поверхность шлифа напыляют плен-
ки электропроводящих материалов: углерода о с
(50—80 А), алюминия (~50 А) или меди С
(~30 А). Хорошие результаты (для многократных исследований) дает напыление серебром или золотом. На алюминиевых пленках быстро образуется окисная пленка, углеродные пленки часто отслаиваются в процессе работы. Тем не менее чаще всего напыляют углеродные пленки. При количественном анализе проводят одновременное напыление на образец и эталоны.
Эталоны должны быть чистыми без окисных пленок. При использовании в качестве эталонов сплавов или химических соединений следует убедиться в их гомогенности. Для получения гомогенных эталонов используют проплавление их участков пучком лазера [16]. Хорошие результаты дает изготовление эталонов переплавом с закалкой из жидкого состояния.
БИБЛИОГРАФИЧЕСКИЙ СПИСОК
1. Бирке Л. Рентгеновский микроанализ с помощью электронного зонда. М.: Металлургия, 1966. 216 с.
2. Birks L. Electron probe microanalysis. 2 ed. Chem. Anal. N. ¥.: Wiley, 1671, v. 17. 190 p.
3. Боровский И. Б. — В кн.: Локальные методы анализа материалов. М.: Металлургия, 1973, с. 109—139.
4. Боровский И. Б. — В кн.: Оптика рентгеновских лучей и микроанализ. Л.: Машиностроение, 1976, с. 57—61.
5. Браун Л., Треш X. — В кн.: Приборы и методы физического металловедения, Вып. 2. М.: Мир, 1974, с. 221—270.
6. Электронно-зондовый микроанализ/Под ред. Боровского И. Б. М.: Мир, 1974. 260 с.
7. Рид С. Электронно-зондовый микроанализ. М.: Мир, 1979. 423 с.
8. Практическая растровая электронная мнк-роскопия/Под ред. Гоулдстейна Дж. и Я косица X. М.: Мир, 1978. 656 с.
9. Гиммельфарб Ф. А., Шварцман С. Л. Современные методы контроля композиционных материалов. М.: Металлургия, 1979. 246 с.
10. Кальнер В. Д.. Зильберман А. Г. Практика микрозопдовых методов исследования металлов и сплавов. М.: Металлургия, 1981. 214 с
11. Физические основы рснтгспоспектралыюго локального анализа. М.: Наука, 1973. 31! с.
12. Сидоров Л. Ф.. Донников С. Г.. Лукьянченко Е. М. — Аппаратура и метойы рентгеновского анализа. Вып. 7. Л.: Машиностроение, 1970. с. 217—251.
13. Сидоров А. Ф. — Аппаратура и методы рентгеновского анализа. Вып. 23. Л.: Машиностроение, 1980, с. 152—188.
14. Васичее Б. И. Электронно-зондовый микроанализ тонких пленок. М.: Металлургия, 1977 239 с.
15. Смирнова В А., Батырев В А.—Заводская лаборатория, 1973, № 6, с. 699—701.
16. Боровский И. Б.. Городским Д. Д., Шара-феев И. М., Морящее С. Ф. — Заводская лаборатория, 1982, № 2, с. 30—31.
150
7
=^== МЕТОДЫ ИССЛЕДОВАНИЯ ПОВЕРХНОСТЕЙ В МЕТАЛЛАХ
7.1. ВВЕДЕНИЕ
За последнее десятилетне заметно возрос интерес к исследованию поверхности твердых тел и происходящих иа ней процессов. Речь идет о поверхностях раздела твердое тело — газ (адсорбция, катализ, атмосферная коррозия, поверхностная диффузия и растекание, износ), твердое тело — жидкость (коррозия, жидкометаллчческая хрупкость), о внутренних поверхностях раздела в металлах (межкристаллитная внутренняя адсорбция, диффузия по границам зерен и фаз, микролегирование, хрупкость, межкристаллитная коррозия, стабильность композиционных материалов) и о процессах в тонких пленках и на границе раздела пленка — матрица (защитные покрытия, микроэлектроника). Можно без преувеличения утверждать, что физика поверхностных явлений— это сейчас одна нз самых (если не самая) быстро развивающихся областей физики твердого тела.
Помимо важности указанных проблем, рост исследований в очень большой мере обусловлен использованием новых методов исследования поверхности. Основной отличительной чертой этих методов является высокое разрешение по глубине (локальность), позволяющее получать информацию от слоя глубиной от одного до нескольких десятков межатомных расстояний. Широкое внедрение этих методов в лабораторную практику связано прежде всего с массовым производством высоковакуумного оборудования и совершенствованием измерительной аппаратуры.
Все методы основаны на взаимодействии первичного излучения (теплового, рентгеновского, электрического и магнитного поля, потока фотонов, электронов, ионов, нейтральных атомов н молекул и т. д.) с веществом и анализе рассеянного или (чаще) вторичного излучения [1]. Таких методов известно несколько десятков, однако наибольшее распространение получили четыре: Оже-электр онна я спектроскопия (ОЭС или AES: Auger Electron Spectroscopy) часто в сочетании с дифракцией медленных электронов (ДМЭ нли LEED— Low Energy Electron Difraclion); фотоэлектронная спектроскопия, чаще называемая электронной спектроскопией для химического анализа (ЭСХА или ESCA — Electron Spectroscopy for Chemical Analysis); ионный микроанализ илн масс-спектрометрия вторичных иолов (МСВИ или SIMS: Secondary Ion Mass-Spectrometry). Все чаще в одном приборе объединяют различные методы. Такое объединение расширяет возможность исследователя и увеличивает объем получаемой информации.
Помимо этих методов, о которых подробнее будет сказано ниже, следует упомянуть еще о двух.
Первый метод — просвечивающая электронная микроскопия (см. раздел 2), дающая кар-
О тину поверхности с разрешением до 10 А, и растровая электронная микроскопия (50— 100 А) — см. раздел 3.
Недавно появились приставки к просвечивающим электронным микроскопам, которые позволяют судить о химическом составе анализируемых областей (см. раздел 6) по результатам анализа энергии вторичного рентгеновского излучения либо потерь энергии проходящих электронов вследствие иеупругого рассеяния, зависящего от присутствия примесей.
Второй метод—автоионная микроскопия — предложен Мюллером [1, с. 401'—463] в 1951 г. Это—уникальный метод, позволяющий наблюдать изображение поверхности твердого
С
тела с атомным разрешением (2—3 А). Образец— тонкое, заточенное травлением острие из исследуемого металла. Поверхность его кончи-О
ка — почти полусфера радиусом 200—2000 А — радиально проектируется на люминесцентный экран. Для создания изображения используется явление десорбции полем (10® В/см), испарения полем (при высоких температурах) и чаще всего ионизации полем постороннего газа (гелия, неона нлн водорода). Кристаллические грани поверхности острия изображаются на экране с увеличением порядка 10е и разреше-
О
нием 2—3 А, так что можно различить отдельные атомы, в том числе междоузельные и адсорбированные, а также вакансии, радиационные повреждения, дислокационные полосы скольжения и границы зерен. Была показана возможность обнажения глубинных слоев образца путем контролируемого послойного испарения полем [2, 3]. Сочетание полевого ионного микроскопа с масс-спектрометром сделало его прибором ие только структурного, но и химического анализа.
7.2. ПРИНЦИПЫ МЕТОДОВ
И ИХ ОСОБЕННОСТИ
При облучении поверхности электронами в зависимости от вида анализируемых частиц (рис. 7.1) различают следующие методы:
ДЦЗ,Э0С,ДБЭ,Д0БЭ, ДНМЭ (электроны)
ЗСИД,НСЭПЗ(иоНы)
ЗСД, ДПР](нейтральные атомы или молекулы)
СХИ, СПП (ротоны)
Рис. 7.1. Методы, основанные на электронном облучении
ОЭС, ДМЭ; ДБЭ — дифракция быстрых электронов; ДОБЭ — дифракция отраженных быстрых электронов; ДНМЭ — дифракция неупруго отраженных медленных электронов; ЭСИД — электронно-стнмулироваиная нонная десорбция; МСЭПЗ — масс-спектрометрия с
151
электронным поверхностным зондом; ЭСД — электронио-стнмулнрованиая десорбция; Д ПМ — десорбция поверхностных молекул; СХИ — спектроскопия характеристического излучения; СПП — спектроскопия пороговых потенциалов [1, с. 60—101].
Если энергия первичных электронов такова, что длина их волны соизмерима с периодом «поверхностной решетки», то упруго отраженные электроны дадут дифракционную картину. Следовательно, надо зарегистрировать нх пространственное распределение. Это — метод ДМЭ, поскольку речь идет о медленных электронах с энергией 20—200 эВ. Аппаратура для получения дифракционных пятеи несложна, чего нельзя сказать об анализе дифракционной картины и возможностях однозначной трактовки структуры поверхности. Возможности эти значительно улучшаются, если одновременно с пространственным распределением измеряется число отраженных частиц в каждом пятие как функция энергии первичных электронов, что однако усложняет эксперимент.
Метод ОЭС основан на энергетическом анализе вторичных Оже-электроиов. Эффект Оже (обнаружен в 1925 г.) назван по нменн открывателя французского физика П. Оже (Auger). Падающий (первичный) электрон возбуждает атом, переводя электрон с внутренней (К, L) оболочки на более высокий (внешний) уровень. Возбужденный атом может вернуться в основное состояние одним из двух способов:
1. Выбитый электрон возвращается с более высокого (внешнего) уровня на вакантное место во внутренней оболочке атома. Прн этом испускается характеристический рентгеновский фотон (рис. 7.2,а). Вероятность такого про-
Рентген-(ротон Внешний уровень
Внутренние уровни
ё падающий
( первичный)
• электрон
е вторичный Оже-электрон
Внешний уровень
Внутренние уровни
ё падающий
( первичный)
/ электрон
Рис. 7.2. Схема энергетических уровней, иллюстрирующая возникновение рентгеновских фотонов (а) и Оже-электронов (б):
L £i, К — электронные оболочки
цесса пропорциональна квадрату атомного номера (Z2), поэтому для легких элементов она мала.
2. Другой электрон с более высокого (внешнего) уровня вылетает из атома (рис. 7.2,6). Этот вторичный электрон и есть Оже-электрон. Оже-эффект не сопровождается испусканием фотона н является безызлучательным перехо
дом. Относительная вероятность Оже-эффекта больше для легких элементов.
Кинетическая энергия Оже-электроиа определяется разницей двух энергий: энергии внутренней оболочки, с которой был выбнт электрон, н энергии уровня, с которого вылетел Оже-электрон. Таким образом, Оже-электроны являются характеристическими: их энергии характерны для данного элемента. Анализ энергий Оже-электронов, как и анализ характеристического рентгеновского излучения, позволяет определить элементный состав исследуемых образцов.
В методе ОЭС используется пучок электронов с энергиями, достаточными для возбуждения внутренних уровней изучаемых атомов, но не слишком большими. С ростом энергии первичного пучка, во-первых, растет вероятность испускания рентгеновского фотона (для энергий =С2 кэВ доля Оже-электронов >90%); во-вторых, ухудшается разрешение по глубине (увеличивается зона возбуждения). Поэтому обычно энергия падающих электронов находится в интервале 0,1—3 кэВ.
Электроны с энергией 1—3 кэВ все же проникают в материал на значительную глубину. Однако длина свободного пробега Оже-электрона в твердом теле невелика, что н определяет разрешение по глубине. Оно зависит от изучаемого материала и, как правило, находится в интервале от 2 до 10 атомных слоев (рнс. 7.3), причем основной вклад в сигнал
Энергии электронов, эВ
Рис. 7.3. Зависимость глубины выхода Оже-электронов и фотоэлектронов от их кинетической энергии
дают первые два-три слоя. Получение таких пучков — задача несложная. Сравнительно просты и хорошо известны устройства для энергетического анализа электронов (стандартная аппаратура ДМЭ). Сложность в том, что приходится измерять малое число Оже-электронов на большом фоне неупруго рассеянных первичных электронов. На кривой зависимости общего числа эмитируемых электронов от их энергии N(E) Оже-пйки очень слабы и мало заметны.
В 1968 г. Харрис предложил с помощью сравнительно простых электронных устройств дифференцировать кривые N(E) по энергии (рнс. 7.4). Это резко увеличило чувствительность метода и позволило выделить в спектре сигнал адсорбированных частиц, количество которых составляет около 1 % моноатомного слоя. На рнс. 7.5 показан Оже-спектр очищенной поверхности олова Кроме линий олова, обнаружены остаточные загрязнения углеродом, хлором и кислородом.
152
Разрешение по поверхности в методе ОЭС, определяемое диаметром первичного пучка электронов (в лучших спектрометрах от 5 мкм О
до 500 А), иа несколько порядков хуже разрешения по глубине.
В последнее время метод ОЭС стал одним йз самых распространенных методов анализа
о 200 400 000 800 1000
Энергия электронов, эВ
кислорода н их форма сравнительно слабо различаются для атома кислорода, молекулы н атомов в составе различных молекул (СОг, SiO2 и др.). В этом отношении более полезным является метод ЭСХА [1, с. 60—101].
При облучении поверхности фотонами в инфракрасном, видимом, ультрафиолетовом и рентгеновском диапазонах длин волн в зависимости от вида анализируемых частиц различают следующие методы (рнс. 7.6): ЭСХА;
ЖП,КРС,ЭС, РФС((ротоны)
/ ФД (нейтральные атомы
Фотоны / или молекулы)
РФС,ЗСХ/\ (электроны)
Рис. 7.6. Методы, основанные на облучении фотонами
Рис. 7.4. Энергетические спектры серебра (энергия первичных электронов 1 кэВ)
состава поверхностей твердых тел. Основные преимущества метода — высокая чувствительность прн проведении элементного анализа
О
приповерхностной области (5—30 А), быстрота получения спектра и возможность обнаружения всех элементов с Z>2. Оже-спектр дает количественную информацию о составе приповерхностного слоя, в некоторых случаях—'Сведения о химических связях атомов в ием, в сочетании с ионным травлением позволяет получить профили распределения элементов по глубине, а в сочетании с ДМЭ— сведения о «составе и топографии [1, с. 200—275; 7].
Для получения количественных данных, т. е. возможности связать амплитуду Оже-пика илн его интегральную площадь с количеством данного элемента, необходима калибровка по эталонному образцу, а также выверенная и контролируемая работа анализатора. Если хотят определить, получен ли сигнал от изолированного атома нли от атома, входящего в «состав соединения, т. е. получить информацию о химических связях, то требуется высокое энергетическое разрешение, достигающее 0,3 % •от измеряемой энергии. Такое требование связано с тем, что, например, энергии Оже-пиков
ИКП—нифракрасиое поглощение; КРС — комбинационное рассеяние света; ЭМ. — эллипсометрия (так же, как и КРС, видимого света) ; РФС — рентгеновская фотоэлектронная спектроскопия; ФД — фотодесорбция. Отметим, что фотоны минимально возмущают поверхность и не заряжают ее. Основные трудности связаны с получением интенсивных пучков в нужном спектральном интервале; здесь оказались полезны лазеры — монохроматические источники большой интенсивности. Кроме того, как правило, малы сечения реакций взаимодействия фотонов с поверхностью, однако совершенствование измерительной аппаратуры позволяет добиваться достаточной чувствительности.
Под действием рентгеновского излучения возникает эмиссия электронов внутренних оболочек (фотоэффект). Кинетическая энергия этих электронов равна разнице между энергией падающего фотона и энергией связи. Они, следовательно, характеризуют атомы и их валентное состояние. С помощью спектрометра определяется зависимость числа электронов от их кинетической энергии. Такой метод получил название рентгеновской фотоэлектронной спектроскопии (РФС) или’ ЭСХА, поскольку в основном он применяется для химической идентификации поверхностных компонентов и позволяет определять все элементы с Z>2. В этом отношении он весьма близок к ОЭС высокого разрешения, отличаясь лишь тем, что вместо электронов поверхность облучают рентгеновскими фотонами. Рентгеновское излучение обладает более высокой проникающей способностью, однако в диапазоне энергий, которым пользуется метод ЭСХА (несколько килоэлек-1 тронвольт), разрешение по глубине, определяемое длиной свободного пробега, электрона примерно такое же, как в ОЭС, н составляет 3—30 А, хотя нижний предел редко бывает О
меньше 10 А.
В режиме ЭСХА анализ малых областей поверхности затрудиеи, поскольку рентгеновские лучн трудно сфокусировать и облучению подвергается большой участок поверхности с линейным размером от 2 до 10 мм. Поэтому методом ЭСХА трудно также определить про
153
странственное распределение компонентов. В отношении же исследования химических связей метод ЭСХА точнее ОЭС, поскольку энергия фотоэлектронов однозначно связана с валентным состоянием атома.
На рис. 7.7 показан ЭСХА-спектр поверхности чистого (вверху) и окисленного (внизу)
Рис. 7.7. ЭСХА-спектр поверхности чистого (вверху) и окисленного (внизу) олова
Рис. 7.8. ЭСХА-спектр поверхности свежеприготовленного (слева) и пролежавшего пять дней на воздухе (справа) алюминия
олова. Линии в спектре окисленного олова расщеплены. Линии олова в спектре окисла смещены на 1,4 эВ в сторону более низких энергий (химический сдвиг позволяет сделать вывод о валентности элемента в соединении). Отношение высот (или площадей) линий окисленного и чистого олова является мерой толщины окисленного слоя. На рнс. 7.8 показан ЭСХА-спектр поверхности алюминия: свежеприготовленного (слева) и пролежавшего пять дней на воздухе при комнатной температуре (справа). Линия Al (III) в окисле смещена по отношению к линии А1 (0) почти С эВ. Видно, что отношение высот пике А1 (II!)
к А1 (0) выросло; окисел был и на поверхности свежеприготовленного алюминия, но его толщина стала больше. В современных ЭСХА-спектрометрах разрешение по энергии >0,3 % от измеряемой кинетической энергии.
При облучения поверхности .ионами в зависимости от вида анализируемых частиц (рис. 7.9) различают следующие методы.- МСВИ;
СИР, МСВИ (ионы)
Рис. 7.9. Методы, основанные на ионном облучении
СИР — спектрометрия ионного рассеяния; ИНС — ионно-нейтрализационная спектроскопия; ИМАР— ионный микрозонд с анализом рентгеновских лучей; ПИР — рентгеновское излучение, создаваемое протонами. Ионные пучки вызывают наибольшие изменения в поверхностном слое [Г, с. 60—101]; это — разрушающий метод контроля.
Как и прн анализе электронов, в этом случае можно измерять число, энергетическое и пространственное распределение эмитируемых ионов. Однако в случае эмиссии ионов появляется и новая аналитическая возможность — анализ вторичных ненов по массам. Этот метод и называют МСВИ. Масс-спектр вторичных ионов характеризует химический состав поверхностного слоя. Большое число вторичных ионов значительно усложняет спектр, однако делает его и более информативным. Кроме того, метод МСВИ обладает очень высокой чувствительностью (пг;? ’ меняющейся в широких пределах) ч г ->ляет определять все элементы, включая водород, гелий, а также различные изотопы элементов и молекулярные осколки [7]. На рис. 7.10 показан МСВИ-
спектр, иллюстрирующий наличие примесей в серебре.
Разрешение метода МСВИ по глубине определяется глубиной проникновения первичных ионов в образец и глубиной выхода вторичных
О ионов и составляет в среднем 10—100 А. Разрешение зависит ст энергии и природы первичных ионов (чаще всего Аг+, О?' и другие с
154
энергией от нескольких сот до нескольких тысяч эВ при силе тока от 10^12 до 10~5 А), скорости ионного травления (плотности тока первичных ионов), материала образца и т. д. и может меняться в довольно широких пределах.
Изменяя плотность первичного ионного тока, можно использовать ионный пучок для масс-спектрометрии тонкого слоя (при малой
Рис. 7.11. Распределение бора в SiO2
плотности) и для ионного травления (при большой плотности). Чередуя эти операции, можно получать концентрационные профили. На рис. 7.11 показано распределение бора по глубине в SiCK Предел обнаружения 10,с ато-мов/см3. Глубина проникновения растет с ростом энергии падающего пучка.
Разрешение метода МСВИ по поверхности меняется в широких пределах в зависимости от конструкции иоииой пушкн и составляет от 10 мм2 до 10 мкм2
7.3. АНАЛИТИЧЕСКИЕ ХАРАКТЕРИСТИКИ И СРАВНЕНИЕ МЕТОДОВ
Основные аналитические характеристики методов ОЭС, ЭСХА и МСВИ приведены в табл. 7.1.
Метод ЭСХА особенно удобен, если требуется анализ поверхности без разрушения образца и сверх того нужна информация о химическом состоянии. Чувствительность метода в общем невелика (1—10~2 %) при измерении в течение 2—3 мин и регистрации с помощью измерителя скорости счета и самописца. Применение многоканального анализатора и ЭВМ позволяет проводить измерения в течение многих часов — соответственно растет нижний предел обнаружения и чувствительность повышается на порядок.
Метод ОЭС обладает высокой чувствительностью (в среднем на 1—2 порядка выше ЭСХА при том же времени измерения), особенно для элементов с низким атомным номером и низкой энергией Оже-электронов, что повышает скорость анализа. Важна возможность объединения Оже-спектрометра и растрового электронного микроскопа для получения информации о химическом составе и микротопографии поверхности.
Наибольшие трудности при количественном анализе методом ОЭС вызывает нарушение линейной связи между интенсивностью Оже-пиков и количеством анализируемых атомов в приповерхностном слое. Необходимо вводить коэффициенты обратного рассеяния для учета Оже-эффекта под действием рассеянных электронов, вторичных электронов и т. д. Метод ОЭС пригоден для количественного анализа монослоев и в меньшей степени — для анализа слоев большей толщины.
Чувствительность метода МСВИ очень сильно колеблется в зависимости от природы анализируемого элемента и условий опыта (на шесть порядков). В первом приближении за меру чувствительности можно принять потенциал ионизации: чем ои ниже, тем легче обнаруживается примесь. В этом случае чувствительность достигает 10~е и даже 10~е. Щелочные металлы обнаруживаются очень легко и встречаются в каждом спектре.
На интенсивность спектра вторичных ионов сильно влияет матрица; даже незначительные загрязнения поверхности металла кислородом, хлором, фтором уменьшают интенсивность сигнала вторичных ионов на несколько порядков. Частично влияние матрицы ослабляют, предварительно насыщая поверхность кислородом или применяя ноны кислорода в качестве первичных, — это стабилизирует эмиссию вторичных ионов.
Среди рассматриваемых метод ОЭС обладает наилучшим поверхностным разрешением
ТАБЛИЦА 7.1.
АНАЛИТИЧЕСКИЕ ХАРАКТЕРИСТИКИ МЕТОДОВ ИССЛЕДОВАНИЯ ПОВЕРХНОСТИ
Характеристика ОЭС ЭСХА МСВИ
Чувствительность анализа, % Точность анализа, % Возможность обнаружения элементов Возможность изотопного анализа Возможность анализа без разрушения образца , о Разрешение по глубине, А Разрешение по поверхности (линейный размер) . Возможность получения информации о химической связи . Возможность изучения топографии поверхности . 5—10 Z>2 + 3—30 о 500 А—5 мкм + 1—10“2 5—10 Z>2 ++ 3—30 2—10 мм 10~2—10“8 (10—1вг) 5—10 Все + 10—100 3 мкм—3 мм ++
* Совместно с ДМЭ.
155
(^500 А). Это привело к развитию сканирующей Оже-микроскопии (SAM), использующей электронный пучок для изучения пространственного распределения элементов в поверхностном слое. В сочетании с растровым изображением во вторичных электронах SAM дает информацию о топографии поверхностного слоя. В сочетании же с ионным травлением
фня (а) дает информацию о топографии поверхности. Такое изображение можно вывести на экран дисплея нлн сфотографировать. Изображение в Оже-электронах (б) получают путем модуляции яркости электронно-лучевой трубки амплитудой выбранного Оже-пика при сканировании электронного пучка по образцу. Изображение показывает распределение вы-
Рис. 7.12, Комплексное исследование участка полупроводникового прибора:
а — микрофотография в поглощенном токе; б — Оже-граммы кислорода (вверку) и золота (внизу), полученные при анализе участка в прямоугольнике а-, в — линейное сканирование для О (вверху) и Ап (внизу) вдоль линии, проведенной на рис, 12 а; г — профиль глубина — состав, полученный в точке на рис. 12, а
SAM можно использовать для послойного анализа н получения профилей распределения элементов по глубине образца.
Во многих исследованиях, связанных с анализом поверхностей, использование того нлн иного метода определяется уже характером возникшей проблемы и типом информации, ко-торую необходимо получить. Объединение в одном приборе нескольких различных методов, расширяющее возможности исследования, — главная тенденция современных методов анализа.
На рнс. 7.12 представлены результаты комплексного исследования участка полупроводникового прибора. Электронная мнкрофотогра-
браниого элемента на определенном участке поверхности. Последовательное получение изображений в Оже-электронах всех элементов, инки которых присутствуют в спектрах ЭСХА или ОЭС, дает карту распределения элементов в поверхностном слйе. Распределение легко сопоставить с топографией поверхности, сравнивая изображения в Оже-электронах с электронной микрофотографией. Линейное сканирование (в) дает количественную информацию о распределении элементов» а профиль состав — время ионного травления (а) — об изменении химического состава по глубине. В последнем случае ОЭС и травление поверхности образца ноиным пучком проводят одновременно.
156
7.4. НЕКОТОРЫЕ
МЕТОДИЧЕСКИЕ
И конструктивные
ТРЕБОВАНИЯ
*
Преимущества комплексного подхода реализуются при выполнении двух наиболее важных условий:
1. Возможность перемещения образца внутри прибора (от одного детектора к другому) позволяет избежать загрязнений поверхности, искажающих конечные результаты. Можно анализировать одни н тот же участок поверхности.
2. Проведение анализа в условиях высокого вакуума 10“6—10~8 Па. При такой малой глубине анализа, как в методе ОЭС, например, чрезвычайно возрастает роль атомов и пленок газа, адсорбкрованного на исследуемой поверхности. Они усложняют Оже-спектр, снижают его интенсивность и могут совершенно исказить —• не только количественно, но даже качественно — результаты анализа. Поэтому анализ необходимо проводить на максимально
Комбинированный прибор содержит следующие основные элементы: источник облучения, анализатор, устройство для приготовления и манипуляции с образцами, вакуумную систему.
На рис. 7.13 показана в качестве примера схема спектрометра ESCA/AES "модели PHJ-548 фирмы «Physical Electronics Industries». Источник возбуждения для ЭСХА — рентгеновская трубка с алюминиевым (1486,6 эВ), магниевым (1253,6 эВ) илн двойным катодом. Фирма выпускает также систему ESCA/SAM с приставками для LEED (электронная оптика н блок управления) и SIMS, состоящими из квадрупольного масс-спектрометра с энергетическим фильтром и электронных блоков управления. Система травления образцов используется в качестве источника облучения для метода SIMS. Смена образцов обеспечивается манипулятором с карусельным держателем, в котором можно закрепить до 12 образцов далее больших размеров н неправильной формы. Манипулятор позволяет менять положение образца, а также использовать устройства для нейтрализации заряда (во избежание накопления заряда на поверхности прн работе с не-
Рис. 7.13. Схема спектрометра ESCA/AES модели РН-548
чистой поверхности, которую получают непосредственно в вакуумной камере, прн разрушении образца, высокотемпературном нагреве, ионной бомбардировке с последующим отжигом. Сверхвысокий вакуум необходим для получения н анализа свежеприготовленной поверхности, особенно при проведении комплексного исследования. Важно подчеркнуть, что сверхвысокий вакуум создают безмаслянымн средствами откачки во избежание загрязнения исследуемой поверхности углеродом. Обычно применяют турбомолекулярные насосы нлн комбинации турбомолекулярных, магнитных, электроразрядных и тнтаиовых испарительных насосов.
проводящими материалами), для нагрева и охлаждения образцов н др. Сопоставление характеристик различных спектрометров можно иайти в [7].
В качестве дополнительного оборудования со спектрометром могут быть использованы ЭВМ; камера для шлюзования н обработки образцов; приспособление для разрушении образцов н т. д. ЭВМ, помимо контроля за работой всех приборов, обработки данных и запоминания информации, позволяет выводить результаты на экран дисплея нли графопостроитель в виде зависимости амплитуды пика или концентрации от глубины.
Шлюзовая камера позволяет в течение 2—
157
3 мин вводить образцы в рабочую камеру со сверхвысоким вакуумом и обеспечивает возможность обработки (нагрева, охлаждения) образцов в широком интервале температур (от —150 до +700 °C) в атмосфере реактивных газов перед введением образцов в рабочую камеру.
Специальное приспособление дает возможность разрушать образец в высоком вакууме для анализа поверхности свежего излома. Это приспособление необходимо для исследования состава и структуры внутренних поверхностей раздела (границ зерен, межфазных границ) в твердых телах.
7.5. ПРИМЕРЫ ПРИМЕНЕНИЯ
Из имеющихся в настоящее время примеров применения локальных методов исследования поверхностей к решению прикладных задач рассмотрим касающиеся только следующих областей: сегрегации примесей на поверхности, границах зерен, межфазных границах; коррозии (включая межкристаллитную) и окисления. Имеются работы по контролю поверхностей раздела в композиционных материалах [7], идентификации атомных структур и выделяющихся на поверхности фаз [5], поверхностной диффузии и поверхностных реакций, адгезии и износа. Много работ посвящено исследованию поверхности катализаторов в связи с их активностью [6] и материалам полупроводниковой техники [8]. Все результаты, приведенные ниже, получены методом ОЭС, иногда в сочетании с другими методами.
7.5.1. Сегрегация примесей
Равновесную поверхностную сегрегацию в разбавленных твердых растворах исследовали для многих систем. Для нее справедлива изотерма адсорбции Лангмюра [11]:
с __ Г сехр(<?/Р7)
2 1+сехр(<?/ЯТ) '
где Г—количество прнмесн на поверхности (моль/см2); 2— максимальное количество атомов, соответствующее полному заполнению одного монослоя (порядка 10~8 моль/см2); 0 — доля мест, занятых атомами примеси на поверхности; с — атомная доля примеси в объемном растворе; q — теплота адсорбции, т. е. теплота нли энергия, необходимая для перемещения одного моля примесн из объема на свободную поверхность.
Равновесную поверхностную сегрегацию О в сплавах Nb—О и Та—О наблюдали в температурном интервале 800—1700 °C. Теплота адсорбции кислорода уменьшается от 71 до 45 кДж/моль (Nb—О) и от 79 до 28 кДж/моль (Та—О) прн повышения концентрации кислорода от 0,1 до 2,3 % (ат.). Измерения, проведенные методом ОЭС в сочетании с ионным травлением [10], показали, что сегрегация ограничена приповерхностной областью глубиной 1—5 монослоев.
Исследования сегрегации углерода на поверхности никеля показали, что сегрегация имеет различный характер при разных температурах. По кривым зависимости 0/(1—0) от Г-1 авторы [11] определили трн теплоты сегрегации, соответствующие различным степеням
покрытия поверхности, что говорит об изменении химической связи на поверхности.
ОЭС чрезвычайно успешно применяли и применяют для идентификации примесей, сегрегирующих на границах зерен. Уже первые опыты [13] с образцами низколегированной стали (3,5 % N1; 1,6 % Сг; 0,4 % С и 0,03 % Sb)
£ J
X S на AI3I 5140 о S на Ре -0,0Sb ц Р ни AISI 5140 0 Sb на AZSZ 3340 a Sb на Fe-£ZSb • Р на Fe-Ni-Cr-C-P v зъ на Ve-0,6 Sb Зп на Fe-Ni-Cr-C-Sn
5 10
Гл у Зина
Рис. 7.14. Зависимость концентрации элементов иа поверхности излома от средней гл5'бины слоя, удаленного ионным распылением
о*
показали, что при обработке, приводящей к развитию отпускной хрупкости, наблюдается сегрегация сурьмы на границах зерен.
Приведенная выше формула оказалась справедливой для различных примесей, способных к сегрегации также и по границам зерен [12]. Исследования проводили на свежеприготовленных зернограничных изломах. Методом ОЭС и ионного травления определяли глубину сегрегации. Было показано, что все примеси, в том числе Sb, Р, Sn, концентрируются в пределах узкой приграничной зоны (1—4 атомных слоя) ([14], рис. 7.14). Степень обогащения границы примесью обратно пропорциональна объемной концентрации и изменяется в широких пределах (табл. 7.2). В большинстве случаев степень развития отпускной хрупкости можно было прямо связать с сегрегацией на границах зерен (в первую очередь фосфора и его химических аналогов — сурьмы, мышьяка и др.).
Интересно, что ширина зоны у границ ’зерен, в которой наблюдается сегрегация никеля, при развитии отпускной хрупкости гораздо больше (порядка нескольких сот ангстрем) и ие уменьшается по мере травления. Это означает, что сегрегация никеля неравновесная. Методом ОЭС была обнаружена корреляция между сегрегацией никеля н сурьмы, следовательно, их взаимодействие играет какую-то роль в процессе сегрегации.
Известно, что вольфрам, полученный методами порошковой металлургии, часто охрупчивается при отжиге в районе 825 °C. Было высказано предположение, что охрупчивание связано с сегрегацией кислорода иа границах зерен. Однако ОЭС не обнаружила сегрегации кислорода [15] на поверхности излома, зато обнаружила сегрегацию фосфора, локализованного в приграничной зоне толщиной менее О
20 А. Следует отметить, что вольфрам такого типа ранее даже не аттестовали по содержанию фосфора. На рпс. 7.15 приведены Оже-
158
ТАБЛИЦА 7.2
СЕГРЕГАЦИЯ ПРИМЕСЕЙ НА ГРАНИЦАХ ЗЕРЕН ЖЕЛЕЗА И ЕГО СПЛАВОВ, ПО ДАННЫМ ОЭС [HJ
Сплав Примесь, % (по массе) Сегрегирующий элемент, % Коэффициент обогащения, Р=Г/2с
Fe 1,5—5,0Sb Sb 150—95
Fe 0,003—0,005S s 15 700—8110
Fe 0,05—0,09—0,2P P 211—202—134
Fe 0.05P p 201
0,003S S 7270
Fe 0,02P; 0,003S s 4550
Fe 0,02—0,06Sb s 7170—9300
0,003S Sb 312—<850
Fe 0,25—1,0—4, OSn Sn 323—190—106
Fe—0,3C—4Ni—2,2Cr 0.02P p 2180
Ni 11
Fe—0,04C 0,02Te Те 20 000
Fe—0,4C—3,5Ni—1,7Cr 0,06Sb Sb 2039
Cr 20
Ni 13
Fe—0,3C—3,7Ni 0,048Sn Sn 820
Ni 2
Fe—0,4C—1,7Cr 0,06P P 536
Cr 13
Fe—0,4C—3,5Ni—1,7Cr 0,06P P 590
Cr 19
Ni 20
Fe—3,5Ni—1,6Cr 0.05P P 788
Ni 14
спектры излома (вверху) и участка, удаленного от него на несколько десятков микрон (внизу).
Как правило, съемка Оже-спектров является заключительной частью исследования нли
Рис. 7.15. Оже-спектр вольфрама:
а — излом по границе зерна; б— участок, удаленный на 30 мкм
же за ней следует еще ионное травление. Вначале исследователь получает обычные н электронные (в поглощенном токе нли во вторичных электронах) микрофотографии изучаемой
поверхности и выбирает точки съемки Оже-спектров.
Так была обнаружена сегрегация сурьмы иа межфазной поверхности раздела Fe — графит в чугуне (0,07 % Sb в сплаве и <40 % на границе). Образуя барьерный слой толщиной О
20—40 A, Sb препятствует диффузии растворенного углерода к графиту, что приводит к появлению цементита в феррите.
Было также обнаружено, что на поверхности трещин в литой стали возникает равновесная сегрегация S (до 40 % (ат-) при содержанки в стали 0,06 % S) толщиной в несколько атомных слоев. Сегрегация не связана с наличием по границам дендритов (а именно там возникают трещины) и толстой окисной
О
пленки (до 3000 А), которая, по-видимому, возникает уже при охлаждении (т. е. имеет вторичный характер).
7.5.2. Коррозия и окисление
Добавка небольших количеств циркония значительно улучшает коррозионные свойства некоторых металлов. Так, добавка 0,01 % Zr к жидкому Bi предупреждает разъедание контейнеров из низколегированной стали для хранения и перевозки Bi.
Добавка 1 % Zr резко уменьшает коррозию по границам зерен Nb под действием жидкого лития. В работе [16] методом ОЭС был исследован промышленный сплав Nb—1 % Zr после нагрева на различную температуру и охлаждения. Было показано, что при температуре ~ 700 °C возникает и сохраняется прн охлаждении приблизительно постоянная концентрация N, О и Zr, притом довольно значительная [для Zr пря 1200—1500 °C ~30 % (ат.)]. Ион
159
ное травление показало, что сегрегация ограничена несколькими атомными слоями. На поверхности образуются устойчивые соединения Zr с О и N, чем н объясняются антикоррозионные свойства.
О 200 ЧОО 600 800 100012001400 Энергия, эВ
Рис. 7.16. Оже-спектры поверхности прокорродировав-шегося участка стального образца, покрытого цинком, и того же участка после удаления с него слоя о
в 200 А
Рис. 7.17. Оже-граммы Са (а) и К (б) соответствуют участку рис. 16 (верх)
После нанесения на сталь гальванического цинкового покрытия некоторые образцы сильно корродировали. С прокорроднровавшего
участка и после удаления слоя в 200 А были сняты Оже-спектры (рис. 7.16, верхний и нижний соответственно). Видно, что Оже-спектры «плохого» участка содержат Са, К, Mg и Si. На рис. 7.17 хорошо видно, что Са н К сосредоточены в участке, сильно подверженном коррозии.
Имеется много ОЭС данных, что межкристаллитная коррозия (например, аустенитных сталей) связана с сегрегацией примесей (S, Р, Sb); по-вндимому, это утверждение относится и к межкристаллитной коррозии под напряжением; совсем недавно с помощью ОЭС установлено, что равновесная сегрегация Р, N и некоторых других примесей сильно повышает восприимчивость сталей к водородной хрупкости.
БИБЛИОГРАФИЧЕСКИЙ СПИСОК
1. Методы анализа поверхностей: Пер. с англ./Под ред. Зандерны А. М.: Мир, 1979. 582 с.
2. Мюллер Э. В. — УФН, 1962, т. 77, с. 481— 552.
3. Мюллер Э., Цонь Т. Автонониая микроскопия (принципы н применения). М.: Металлургия, 1972. 360 с.
4. Полашегг X. Анализ поверхностей тонких пленок — основы и результаты. Leybold-Heraeus, 1979. 19 с.
5. The Structure and Chemictry of Solid Surfa-ces/Ed. G. A. Somorjai. N. Y.: Wiley, 1969.
6. Somorjai G. A. — Journ. of Catalysis, 1972, v. 27, № 3, p. 453—456.
7. Гимёльфарб Ф. А. Шварцман С. Л. Современные методы контроля композиционных материалов. М.: Металлургия, 1979. 248 с.
8. Протопопов О. Д. Оже-спектроскопия в применении к исследованиям поверхности сложных эмиттеров. М_: Институт электроники, 1970. 79 с.
9. Бакштейн Б. С., Швиндлерман Л, С. Эффект внутренней адсорбции в твердых телах. Черноголовка: ИФТТ, 1978. 25 с.
10. Joshi A., Strongin М. — Scripta Met., 1974, v. 8, № 4, р. 413—424.
11. Gsett L. С., Blakely М. — J. Vacuum Sci. and Technology, 1975, v. 12, № 1, p. 237— 241.
12. Гликман E. 3.—Доклады III Всесоюзного совещания по взаимодействию между дислокациями и атомами примесей. Тула: ТПИ, 1976, с. 83—91.
13. Marcus A. L., Palmberg Р. W.—Trans. AIME, 1969, v. 245, р. 1164.
14. Hondros S. D., Seah M. P. — Intern. Met Review, 1977. Dec., p. 262—301.
15. Joshi A., Stein D. F. — Metal. Transactions, 1970, v. 1, № 9, p. 2543—2546.
16. Joshi A., Varma M. N., Strongin M. — Metal. Transactions, 1974, v. 5, № 4, p 861— 863.
8
ЯДЕРНЫИ ГАММА-РЕЗОНАНС (ЭФФЕКТ МЕССБАУЭРА)
8.1, ВВЕДЕНИЕ
Многие современные фнзнческне методы исследования металлов основаны на изучении взаимодействия объекта с каким-либо вндог^ электромагнитных волн. Помимо классических (оптических, рентгеновских н электронно-микроскопических) методов, используются ядерный магнитный и электронный парамагнитный резонанс [1]; методы исследования поверхности (Оже-электронная спектроскопия и дифракция медленных электронов); электронная спектроскопия для химического анализа; ионный микрозонд [2] и др. Во всех случаях изучается поглощение, рассеяние падающих нлн испускание вторичных электромагнитных волн (или пучка электронов, ионов) частицами исследуемой системы. Прн некоторых эиергнях падающего излучения, совпадающих с энергиями соответствующих переходов в системе, интенсивность эффекта возрастает — такие методы являются резонансными. В частности, резонанс у-кван-тов на атомных ядрах заключается и резком возрастании вероятности поглощения (илн рассеяния) у-квантов с энергией, соответстную-щей возбуждению ядерных переходов.
Эффект Мессбауэра, открытый в 1958 г.,— резонансное поглощение у-квантов решеткой (системой связанных ядер) без потери энергии иа отдачу.
Гамма-резонансный (ГР) спектр представляет собой зависимость ннтенсивностн у-квантов, излученных источником и прошедших через поглотитель нлн рассеянных нм, от относительной скорости источника нли поглотителя. Основное достоинство получающегося спектра — чрезвычайно узкая линия поглощения (рассеяния). Отношение ширины лннни к энергии излучаемого у-кванта, т. е. разрешающая способность, обычно составляет 10~"— 10-13, что в абсолютных величинах соответствует точности определения энергии 10“®— 10~в эВ. Возможность измерения столь малых энергетических сдвигов оказалась весьма полезной для изучения различных сверхтонких взаимодействий в твердых телах. Благодаря этому применение эффекта Мессбауэра положило начало развитию метода исследования твердых тел — ядерной гамма-резонансной (иногда просто гамма-резонансной) спектроскопии, метода ЯГРС илн ГРС [3, 4].
К достоинствам метода (помимо высокой разрешающей способности) относятся чувствительность к быстрым динамическим процессам [5] с характерным временем 10~7—10-юс. К недостаткам — прежде всего низкая химическая чувствительность, требующая использования относительно большого количества вещества (0,01—I г/см2), также необходимо применять особые мессбауэровские ядра (что очень сужает число возможных объектов исследования) и проводить исследования (во многих случаях) при низких температурах,
11—-280
8.2 СУЩНОСТЬ ЭФФЕКТА
Как уже было подчеркнуто, наиболее важная особенность ЯГР — уникальная разрешающая способность. Ннжннй возможный предел измерения энергии задается соотношением неопределенностей, согласно которому (A£“)minTS>fe, где т — среднее время жизни ядра в возбужденном состоянии; Л=6,59-10_,е эВ-с — постоянная Планка.
Для низколежащих состояний ядер т=10-15“ -т-10-11 с, а энергия излучаемого у-кванта (£0) колеблется в пределах 104—10® эВ. Поэтому разрешающая способность (АЕ/Ео) оказывается очень высокой н доходит до 10“12 (I19Sn), 10—13 и даже io~le (67Zn). Эффект
Мессбауэра — это весы, на которых можно заметить изменение массы железного груза в 1 кг на 10“10 г. Недаром в одной нз самых впечатляющих раниих работ — работе Паунда и Ребкн — авторы с помощью эффекта Мессбауэра «взвесили» у-квант.
В определении эффекта (см. выше) существенны слова «без потери энергии на отдач у».
Если излучают н поглощают свободные ядра, то вследствие отдачи центр линии испускания (зависимости числа излученных у-квантов от их энергии) соответствует энергии Еа—R, а линнн поглощения (рис. 8.1).
Рис. 8.1. Смещение линий испускания (Г) и поглощения (2) из-за потери энергии на отдачу
Таким образом, энергии излученного н поглощенного у-квантов отличаются на 2R — две энергии отдачи: R—Eq /(2тс2), где т — масса ядра; с — скорость света.
Как видно из рис. 8.1, резонансное поглощение осуществляется только в тех случаях, если линнн испускания н поглощения перекрываются, т. е. /?<Г, где 2 Г — естественная ширина линнн (ширина линнн на половине высоты), определяющая минимальную измеряемую энергию (Afmin) :Г—h/x.
Для мессбауэровского изотопа железа 5’Fe:£c = 14400 эВ; /?=1,9-10-3 эВ; Г= =4,6-10~9 эВ. Поскольку Г -сД, то линии не
161
ТАБЛИЦА 8.1
ХАРАКТЕРИСТИКИ НЕКОТОРЫХ
МЕССБАУЭРОВСКИХ ИЗОТОПОВ (м. и.)*
fcrFe tiNi e7Zn il9Sn 121Sb 125Te 181Ta 182 W Ifibpf >07Au
2,2 1,2
4,1
8,6
57,25 6,99 99,99 26,4 33,8
100
14,4 67,4 93,0 23,8 37,1 35,6 6,25
100,1 98,9 77,3
98
5,3 9400
18
3,5
I,4 6800
1,4 0,17
1,8
0,46 8,6 0,005
2,5
13
33 0,007 33 270
25
1,95
40,0
69,3 2,56
6,12
5,44
11,6
29,6
26,9
16,3
* Oo — сечение резонансного поглощения.
23,6 7,21
1,18 13,3 11,8 2,86 17,0 2,4
61,0 0,44
соответственно в возбужденном и основном состояниях. Прошедшие через поглотитель у-кван-ты регистрируются детектором (пропорциональный' счетчик; сцинтилляционный кристалл с фотоэлектронным умножителем; полупроводниковые детекторы излучений), сигнал которого должен быть пропорционален энергии детектируемых частиц. Выход детектора соединен с одноканальным амплитудным анализатором, который выделяет нз всех излучаемых у-кван-тов те, энергия которых близка к Ео (участок спектра, соответствующий резонансному переходу).
Исследование сверхтонких (слабых) взаимодействий в твердых телах с помощью ЯГР-
перекрываются н резонансного поглощения у-квантов не происходит. Таким образом, отдача приводит к тому, что количество у-кван-тов, прошедших через поглотитель, не зависит от их энергии.
Однако если излучающее и поглощающее ядра связаны в кристаллической решетке (нлн во всяком случае конденсированной фазе), то при определенных условиях энергию отдачи воспринимает не отдельное ядро, а кристалл в целом, масса которого на много порядков превосходит массу ядра. Появляется конечная вероятность испускания (поглощения) у-кван-тов без отдачн. В спектре этому процессу соответствует несмещенная линия естественной ширины. Иначе говоря, максимум испускания и поглощения соответствует энергии Ео, не смещенной иа R (штриховая лнння на рис. 8.1), а ширина линии на половине высоты равна 2й./т. Вероятность такого «безотдачного» процесса и есть вероятность эффекта Мессбауэра.
Условия, прн которых велика вероятность резонанса без отдачн, следующие: достаточно жесткая связь атомов в решетке (высокая дебаевская температура), сравнительно большая масса ядра и не слишком жесткое излучение. Этими требованиями определяется перечень возможных излучателей. Для наблюдения эффекта необходимо иметь так называемый мессбауэровский изотоп, обладающий у-перехо-дом с низкой энергией (ниже 150—200 кэВ) н достаточно большим временем жизни в возбужденном состоянии. В настоящее время известно большое количество мессбауэровских изотопов. Однако практически для металловедческих целей используются очень немногие: 57Fe (наиболее часто); 119Sn н ряд других. Некоторые параметры ианболее распространенных мессбауэроискнх изотопов приведены в табл. 8.1.
спектросколнн означает измерение изменения энергии ядерного перехода (разницы энергий основного и возбужденного состояний ядра) в поглотителе по сравнению с источником, если в одном нз иих (обычно поглотителе) такое взаимодействие существует, а в другом нет. Если энергия перехода (Со) изменилась в поглотителе по сравнению с источником на ДЕ, то судить об этом можно с помощью эффекта Допплера, приведя источник в движение по отношению к поглотителю. Максимум (центр) линии резонансного поглощения "будет находиться прн и^0 (рнс. 8.2), причем &Е[Ей=ъ1с> где с — скорость света.
Рис. 8.2. Применение эффекта Допплера для измерения линии поглощения:
1 —• источник; 2 — поглотитель; 3 — детектор
8.3. МЕТОДИКА СЪЕМКИ
ЯГР-СПЕКТРОВ ПОГЛОЩЕНИЯ
Съемка Я ГР-спектра поглощения осуществляется с помбщью источника излучения и поглотителя, содержащих мессбауэровские ядра
Таким образом, источник должен иметь возможность двигаться относительно поглотителя либо с постоянной скоростью, которую можно дискретно изменять, либо с переменной, меняющейся по известному закону (например, линейному).
В спектрометрах первого типа источник в течение определенного времени движется с постоянной скоростью, а анализатор (обычно одноканальный) регистрирует сигнал детектора (количество прошедших через поглотитель у-кваитов). Затем устанавливается другое значение скорости и измерение повторяется. Спектр поглощения представляет собой результат таких последовательных измерений.
В спектрах второго типа сигнал детектора, соответствующий различным значениям скорости, регистрируется в различных каналах многоканального анализатора.
162
Я ГР-спектрометры с переменной скоростью дают возможность автоматически регистрировать весь спектр поглощения в выбранном интервале скоростей (обычно около ±10 мм/с). Спектрометры с постоянной скоростью дешевле, проще в эксплуатации н позволяют с высокой точностью исследовать небольшие участки спектра.
Промышленность выпускает различные мессбауэровские источники (S7Co в хроме, палладии, нержавеющей стали) активностью от 4-107 до 4-10® Бк. Наиболее распространенным (и единственным для ЯГР-спектроскопии железа, его сплавов и соединений) является изотоп 57Fe, возникающий в результате распада материнского изотопа 57Со с периодом полураспада для мессбауэровского перехода 14,4 кэВ, равным 270 дн. Чаще всего используют источники 57Со в платине, палладии, хроме. нержавеющей стали 1Х18Н9Т.
Образцы (поглотители) обычно имеют вид тонких фольг нли порошков. Оптимальная толщина образца определяется разными факторами.' вероятностью эффекта (зависит от чнсла мессбауэровских ядер; см. ниже), необходимостью избежать сильного поглощения и т. д. При работе с чистым железом оптимальная толщина фольги 20—30 мкм, что соответствует 16—25 мг/см2 железа илн 0,3—0,5 мг/см2 изотопа 57Fe (2,2 %). Промышленность выпускает железо (металлическое или окись), обогащенное стабильным изотопом 57Fe (<g:95%). Хорошо приготовленные источник и поглотитель (в виде фольги) обеспечивают получение узкой линии поглощения шириной 0,20—0,25 мм/с (2Г—0,19 мм/с).
При больших значениях скорости (п«) резонансное поглощение исчезает. Величина с™ зависит от выбранного изотопа и условий опыта. Удобно строить спектр в координатах: отклонение от нерезонансного поглощения (е) — скорость (vt), причем
е (fi) = ----~,
где N(Vi) — интенсивность илн число у-квантов, прошедших через поглотитель и зафиксированных в i-том канале анализатора, соответствующем скорости vt;
-^7V(Ooc).
Площадь экспериментального спектра определяется суммированием по всем точкам п
(скоростям, каналам): SSKCn = 2 е (&г) -
Г=1
Прн работе с многоканальным аналнзато- ’ ром получают зависимость N (или е) не от скорости, а от номера канала анализатора. Поэтому необходимо знать, какой скорости (в мм/с) соответствует один канал, т. е. провести градуировку. В качестве стандартного поглотителя (во всяком случае для ЯГР-спектроскопии сплавов и соединений железа) обычно используют нитропруссид натрия — Na2[Fe(CN)ENO]’Н2О. Он стабилен, имеет большую вероятность эффекта при комнатной температуре (5—10 %) и узкую линию (дублетную). Расстояние между линиями дублета 1,705 мм/с используется для градуировки каналов анализатора по скорости.
В СССР распространен мессбауэровский
спектрометр (ЯГРС-4М) с одноканальным анализатором. Рабочий диапазон скоростей ±20 см/с. Спектрометр работает в режиме постоянных ускорений (сигнал скорости имеет форму пилы или трапеции) и постоянных скоростей (с отсчетом нулевой скорости). Ширина линии на поглотителе нз нитропруссида толщиной 0,1 мг/см2 с источником 67Со в хроме <0,3 мм/с. Величина эффекта по 57Fe с нитропруссидом при комнатной температуре ^8 %. Спектр выводится на цифропечатающую машину БЗ-15.
Спектрометр может работать н с многоканальным анализатором, как и большинство зарубежных устройств, например венгерский МТА-512.
Для работы при повышенных н пониженных температурах н в вакууме спектрометр МТА-512 снабжен дополнительными блоками: вакуумной системой, вакуумной печкой, криостатом.
8.4. ПАРАМЕТРЫ СПЕКТРОВ
И МЕТОДИКА ИХ ОПРЕДЕЛЕНИЯ
2’
Простейший спектр поглощения — одиночная линия лоренцовой формы: e(^ = 8(r0)/
где е(щ)—величина эффекта; п0 — скорость, соответствующая центру линий; Г — ширина линнн в единицах скорости.
В общем случае ЯГР-спектр поглощения может быть сложным. Ниже рассмотрены основные параметры спектра: число линий (сверхтонкая структура) спектра, положение центра каждой линии, ширина линии и ее высота, характеризующая интегральную интенсивность резонанса.
8.4.1. Сверхтонкая структура спектра
Сверхтонкая структура (СТС), т. е. появление в спектре нескольких лнннй, максимумы которых находятся при различных скоростях, является следствием расщепления уровней энергии ядер источника и поглотителя илн одного из ннх. Среди различных причин, вызывающих расщепление, следует выделить магнитное дипольное (ядерный эффект Зеемана) и электрическое квадрупольное расщепление.
В магнитном поле Н энергетический уровень ядра со спином I и магнитным моментом р расщепляется на (27+1) равноудаленных подуровней с энергией Ет=—т(\х] /Г)Н, где т — квантовое число, определяющее проекцию спина ядра на направление магнитного поля (дп=—1, 0, +/). Если расстояние между подуровнями Д£т>2Г, то спектр разделяется иа серию линий; в противном случае линия остается нерасщеп-ленной, но уширяется.
Если расщепление происходит только в поглотителе, то правила отбора для магнитных переходов (Дт=0,1) приводят к возникновению в спектре поглощения шести линий (секстета). На рнс. 8.3 показаны спектр no
il*
163
глощения и схема расщепления уровней в металлическом железе (в источнике — ядра &IFe в парамагнитной нержавеющей стали, расщепление отсутствует). Для поликрнстал-лнческих образцов в отсутствие анизотропии вероятности эффекта Мессбауэра относитель-
имодействие квадрупольного момента ядра (Q) с градиентом напряженности электрического' поля (q), создаваемого электронной оболочкой. Для аксиально симметричного поля eq=Vzz, где V — потенциал и е — заряд электрона. Vzz и q не равны нулю, если ядро
-5.6-4,2-2.8-14 0 1,4 2,8 4,2 5,6
Скорость, мм/с
Рис. 8.3. Спектр поглощения (с) и магнитное расщепление (б) перехода 14,4 кэВ в 67Fe
ные интенсивности линий секстета подчиняются соотношению 3:2:1:1:2:3. Появление анизотропии меняет соотношение интенсивностей (эффект Гольданского—Карягина).
Величину внутреннего поля (А/м) иа ядрах поглотителя можно иайти по формуле //=2,46-10й (^6—01), где -Ге и — скорости (мм/с), соответствующие максимумам линий в спектре. Так, в металлическом железе щ—01—10,66 мм/с, а Н~ 2,63-10’ А/м и антипараллельна магнитному моменту атома.
Изменение расстояния между линиями спектра одного и того же изотопа в различных соединениях характеризует изменение эффективного поля. Так, величина поля на ядрах 67Fe в FesO3 примерно в 1,5 раза больше, чем в металлическом железе (около 4,38-10’ А/м).
Рис. 8.4. Квадрупольное расщепление и дублет, соответствующий переходу в немагнитном материале
Заметим, что система линий сверхтонкого магнитного расщепления в чистом железе используется для градуировки скорости: расстояние между линиями 2—4 или 3—5 в спектре составляет 3,92,, а между крайними 10,66 мм/с.
Если форма ядра не является сферически симметричной, то возникает сверхтонкое вза-
находнтся в положении с неку.бнческой симметрией. Следовательно, квадрупольное расщепление позволяет судить о симметрии собственного электронного распределения в кристалле либо о ее нарушении под влиянием примесных атомов и других дефектов. В отсутствие магнитного расщепления для ядер со спином /=3/2 возникают два подуровня, сдвинутые на величину квадрупольного расщепления ДЕе = ± (e2<?Q)/4. Таким образом, в Я ГР-спектре Для ядерных переходов 8/г~> ->’/г в немагнитных материалах возникает дублет (две линии) чисто квадрупольного рас-
Рис. 8.5. Изменение положения линий прн совместном действии магнитного и осесимметричного электрического полей:
a — взаимодействие отсутствует; б — магнитное дипольное взаимодействие; в — то же и электрическое квадрупольное взаимодействие
щеплення (рнс. 8.4). Величину ДЕв находят по расстоянию между линиями дублета. Наложение электрического квадрупольного и магнитного дипольного взаимодействий меняет относительное расположение шестерки линий СТС (рнс. 8.5).
8.4.2. Изомерный сдвиг (положение) линии
Изомерный сдвиг линии (б) находят из ЯГР-спектра как положение центра линии логло-
164
щення (в единицах скорости) относительно линии стандартного поглотителя (например, нитропруссида натрия или металлического железа). Появление изомерного сдвига связано с различием в локальном химическом окружении излучающего и поглощающего ядра; по-
ХЮ-23 м2); fa — вероятность поглощения V-квантов без отдачн.
Во-вторых, уширение может быть следствием неразрешенного электрического илн магнитного взаимодействия, если А£9 или А^ги^Г.
Рис. 8 6 Схема смещения энергетических уровней ядра при возникновении изомерного сдвига б (У —источник (s); // — поглотитель (а}; / — основное состояние; 2 — возбужденное состояние)
этому сдвиг иногда называют химическим. Прн этом плотность s-электронов на обонх ядрах (т. е. в точках, где расположены оба ядра) оказывается разной, н различное кулоновское взаимодействие ядра и электронов приводит к сдвигу резонансной энергии (рнс. 8.6). Итак, изомерный сдвиг:
«=Л!Не(0)|^„-|%(0)|вст)-
Здесь |ф8(0)|2— плотность «-электронов на ядре в атомных единицах; коэффициент А связан с разностью радиусов ядра в основном и возбужденном состоянии. Для ядер &7f?e А——1",08-10~27 см/с ат. ед. Для метал-личе ского a-Fe | ф8 (0) |2 = 11882,5 ат. ед.
Наложение изомерного сдвига на магнитное расщепление сдвигает все линии СТС на 6 влево нли вправо по осн скоростей в зависимости от знака сдвига. При изменении температуры источника или поглотителя возникает также температурный (допплеровский) сдвиг линии поглощения (бт). Обычно
8.4.3. Ширина линии
Уширение мессбауэровской линии (АГ), если ее форма близка к лореицевой, находят как разницу между экспериментально измеренной шириной линии на половине высоты н суммарной шириной линий источника н поглотителя. К уширению* приводят трн вида причин.
Во-первых, причины методического характера: конечная толщина поглотителя-, аппаратурное уширение (непостоянство скорости, недостаточное разрешение н др.). Влияние конечной толщины можно качественно оценить по формулам (6]:
(0,25са прн 0 < са < 5;
9
0,29сд — 0,005Сц при 4 < са < 10,
где Ca=naGofa — эффективная толщина поглотителя; па=па— число резонансных ядер поглотителя на единицу площади; а — содержание мессбауэровского изотопа; для природного (необогащенного) железа, например а—0,022; о0 — сечение мессбауэровского поглощения у-квантов (для ’ S7Fe Ос—15Х
В-третьнх, к уширению приводят релаксационные процессы (электронные флуктуации, релаксация электронного спнна, диффузионное движение атомов), если время релаксации меньше или сравнимо с временем х<из-ни возбужденного состояния ядра. Соответственно уширение третьего типа может дать информацию о процессах, которыми оно вызвано.
8.4.4. Высота линии
Высота линнн характеризует интегральную интенсивность эффекта и позволяет найти вероятность перехода без отдачн (/ илн fa — в источнике и поглотителе соответственно) —• величину, непосредственно связанную с колебаниями атомов, динамикой их движения в кристалле.
Как показано в работе [7], для источника без самопоглощения относительная высота одиночной лнннн в спектре
в (0) = af [1 — exp (— са/2) /0 (са/2) J, где а — доля резонансных квантов, проходящих через поглотитель; f — вероятность испускания у-квантов без отдачи; 10(х) —
функция Бесселя кулевого порядка от мнимого аргумента; ca=n0cr0fe (обозначения см. предыдущую формулу).
Экспериментально определяемая площадь под наблюдаемой в спектре одиночной линией S= =ла/К(со), где К(са) =саехр(—са{2) [10(са/2) 4-+/|(со/2)]; /1(3с)—фувкцня Бесселя первого порядка от мнимого аргумента.
Этн результаты применимы и к хорошо разрешенным линиям миоголинейчатого спектра.
В случае «тонких» поглотителей (са<С 1):
е (0) = (ос<т0 па н S = лао0 па ffa.
Формулы позволяют найти одну из вероятностей (/ или fa), если известна другая, и число резонансных ядер на квадратный сантиметр поглотителя (лс). Значение а определяют из амплитудного спектра источника либо вместе с f как произведение af из опытов с «черным» поглотителем. Для такого поглотителя Са-^-оо, поэтому значения е и 5 оп
166
ределяются af. В качестве «черного» поглотителя используют какое-либо вещество, дающее широкую линию в спектре н поглощающее практически все мессбауэровские у-кванты вблизи резонанса.
Хотя величина f (или fa) дает интегральную характеристику колебательного спектра, однако оиа относится к резонансным мессбауэровским ядрам. Таким образом, если ввести последние в локальные области кристалла, то можно получить информацию об атомах в этих областях. Указанное соображение справедливо н для сведений, которые получаются при измерении других параметров Я ГР-спектра. Наряду с очень высокой чувствительностью эта особенность метода является принципиальным достоинством ЯГР-спектроскопнн.
Учитывая сложность Я ГР-спектров, для получения наиболее точных результатов при определении параметров все чаще используют ЭВМ.
Для обработки спектра на ЭВМ каждой Л-той линии спектра приписывают лоренцеву форму нли для чнсла отсчетов Ащ в t-том канале (для скорости :
Здесь индекс «О» относится к каналу (скорости), соответствующему центру A-той линии, а индекс «со» — к каналу (скорости), далекому от резонанса.
Для спектра, состоящего из п линий, Л',— = i fe=l
Затем минимизируют сумму квадратов отклонений точек экспериментального спектра (/Vi) от теоретического (в рамках какой-либо модели) по всем каналам (чаще всего методом наименьших квадратов). Процедуру минимизации проводят несколько раз, пока погрешность ие станет меньше N.
Для ускорения счета иногда используют программу сравнения экспериментального спектра с набором стандартных спектров известных веществ.
8.5. НЕКОТОРЫЕ ПРИМЕНЕНИЯ
ЯГР-СП ЕКТРОСКО ПИИ
Первоначальный период бурного энтузиазма в отношении ЯГР-спектроскопни сменился в середине 80-х годов периодом более спокойного н систематического освоения нового'метода для исследования свойств твердых тел. Если говорить о металловедческих аспектах проблемы, то выяснилось, что наиболее полезную информацию ЯГРС дает прн изучении магнитной структуры металлов и сплавов [8], электронного распределения (зарядового состояния, типа связи) [9], фазовых превращений, включая процессы упорядочения, и динамики решетки [5].
Ниже мы рассмотрим некоторые применения ЯГРС.
8.5.1. Электронная структура
Измерение параметров ЯГР-спектров позволяет находить эффективные магнитные поля, градиенты электрических полей, действующих на ядра, электронную плотность на ядре. Этн величины полезны для обсуждения вопросов об электронной структуре атомов в чистых веществах, твердых растворах и соединениях, о зарядовом состоянии н характере связей резонансного атома. Поскольку мессбауэровские ядра можно вводить в большое число различных матриц с различным химическим окружением, удалось накопить значительный экспериментальный материал о величине и направленности связей, соотношении в них ионной и ковалентной составляющих, образовании гибридных связей и т. д.
Систематическое исследование изомерных сдвигов (ИС) в различных соединениях железа показало, что величина сдвига существенно различается для двух- и трехвалентного железа. Если принять за ноль ИС в металлическом железе, то для ионов Fe3+ 6=0,6— ч-0,7, а для ионов Fe2+ 1,4ч-1,5 мм/с.
Столь сильное различие ИС ионов Fe2+ и Fes+ делает ИС незаменимым для определения зарядового состояния нонов в металлических сплавах и металлургических шлаках. Так, в работе [10] было изучено распределение нонов Fe2+ и Fe3+ между окта- и тетраполо-жениями в силикатных шлаках. Анализ ЯГР-спектров показал, что в шлаках типа СаО — SiO2 — Fe2Os от 45 до 70 % ионов Fe3+ иаходится в октаположениях, хотя в соответствии с принятыми представлениями теории шлаков трехвалентиые амфотерные катионы должны иметь. преимущественно тетраэдрическую конфигурацию. В шлаках типа Na2O— SiO2— Fe2O3 все ноны Fes+ действительно находятся в тетраположениях.
Увеличение положительного значения 6 для ноиа Fe2+ по сравнению с Fes+ означает уменьшение плотности s-электроиои на ядре поглотителя. Предполагая, что электронная конфигурация у этих ионов в соединениях такая же, как у свободных ноиов, т. е. 3s2 3d5 4s° и 3s2 3d6 4s° соответственно, приходим к выводу, что Sd-электроиы уменьшают плотность s-электронов иа ядре тем сильнее, чем нх больше. Собственная плотность Зй-электронов на ядре равна нулю.
В металлах нлн в соединениях, где связь не является чисто ионной, внешние электроны образуют конфигурацию 3<F 4sx, содержащую долю х от s-электронов в состоянии 4s. Было показано наличие корреляции между х и уменьшением электроотрицательности. Так, в соединениях FeO, FeS и FeSe величина х равна соответственно 0,1; 0,2 и 0,3 (1=6). ИС для металлического железа соответствует конфигурации валентных электронов 3d74s1 (у изолированного атома железа — 3d6 4s2).
По-видимому, величина ИС позволяет судить о доле ковалентных связей в соединениях данного тнпа. Такая корреляция была, например, установлена в работе [И]. Была получена линейная зависимость между уменьшением ИС на ядрах Fe в фазах Лавеса типа Z?F2 (R — =Ti, Nb, Та, W, Zr) и увеличением числа единичных ковалентных связей атомов железа (по Полингу [2]). Фазы Лавеса с участием f-ne-
166
реходных металлов не укладывались в эту зависимость.
Дополнительные сведения об электронном распределении можно получить из данных о величине градиента q. Большие квадруполь-ные расщепления наблюдали только у соединений двухвалентного железа (до 2—3,5 мм/с); в соединениях трехвалентного железа они малы (A£s=0,2 4-0,6 мм/с)—пять внешних электронов симметрично распределены в наполовину заполненной d-оболочке.
Следует подчеркнуть, что отсутствие достаточно строгой теории металлической связи делает рассмотрение электронных структур и обсуждение характера связи иа основе ЯГР-спектров в основном качественным.
8.5.2. Аналитические применения (фазовый и химический анализы)
Различные соединения мессбауэровских изотопов (в частности, железа) характеризуются разными значениями ИС, квадрупольного н магнитного расщепления. Поэтому возможна их идентификация в сложном Я ГР-спектре. В табл. 8.2 приведены значения ИС (б), эффективного магнитного поля (Дэф) и квадрупольного расщепления (ДЕ5) для некоторых соединений железа.
Пользуясь этими данными, анализируют железосодержащие минералы. Практически полезным оказался фазовый анализ руд с помощью «эталонных» источников, дающих спектры испускания, характерные для какого-либо соединения. Естественно, что максимальную интенсивность в спектре поглощения будут иметь линнн именно этого соединения, если оно присутствует в поглотителе. Имея набор таких источников, достаточно измерить
ТАБЛИЦА 8.2
ПАРАМЕТРЫ ЯГР-СПЕКТРОВ ^Fe В НЕКОТОРЫХ МЕТАЛЛАХ И СОЕДИНЕНИЯХ
Металл или соединение T, к б*, мм/с ^эф’ 10™’ А/м
cc-Fc 300 0,26 2,63
4 0,26 2,69
a-Fe 300 0,18
?-FegO3 300 0,60 4,02
a-FegO3 300 0,62 4,10
Fe3O4 300 0,63 3,98
FeO** 300 1,33 '
FesC 300 0,55 1,66
FeS** 300 0,87 2,45
FeS2* 300 0,57 —
Fe2Zr 85 0,09 1,54
Fe2U 85 0,10 0,29
Fe2W 300 0,01 —
Fe2Mo 300 0,00 —
Co 300 0,30 2,47
Ni 300 0,30 2,11
Cr 4 0,07 0,28
Cu 300 0,48 —
* ИС дан по отношению к нитропруссиду.
** Д£^, мм/с. Для FeO, FeS, FeSj составляет 0,3; 0,32; 1,38 соответственно.
эффект с каждым из источников ej и решить систему линейных уравнений:
п
ei =2
л=1
где е,-й — величина эффекта на Л-том поглотителе с i-тым источником; — содержание в руде Л-того компонента; л — общее число компонентов. При этом для каждого источника ие кужно снимать весь спектр, а достаточно измерить эффект при двух скоростях—• нулевой и бесконечной, т. е. выводящей систему из резонанса, что сильно упрощает дело. Таким же образом можно анализировать фазовый состав сплавов на основе железа — определять количество остаточного аустенита, ннтерметаллидов н т. д.
Для тонких поглотителей относительная высота лиинн е(0) и экспериментально определяемая площадь под наблюдаемой в спектре линией а пропорциональны па-—числу резонансных атомов иа 1 см2 поглотителя: е(0) — —k\tta и S—k2na.
При условии предварительной калибровки (определения коэффициентов kt и йа) последнее уравнение можно использовать для определения химического содержания элемеюа (железа, олова).
8.5.3. Фазовые превращения и упорядочение
Возможность идентификации различных фаз в Я ГР-спектре позволяет следить за фазовыми превращениями. Важно подчеркнуть, что параметры ЯГР-спектра чувствительны к локальному окружению излучающего нли поглощающего атома, отражая влияние иа него ближайших соседей (в пределах нескольких координационных сфер). В этом смысле метод Я ГР — локальный и позволяет изучать про-
-0,4 0 0,4 -0,4 0 0,4
V, см/с
Рис. 8.7. Мессбауэровский спектр закаленного (сг) и отпущенного (б) образца углеродистой стали
цессы перераспределения атомов прнмеси в твердых растворах (возникновения ближнего порядка, образования сегрегаций), ранние стадии выделения зародышей избыточных фаз и т. д.
На рис. 8.7 показан ЯГР-спектр образца углеродистой стали, закаленного (а) н отпущенного (б). В обоих случаях можно видеть шесть линий ферромагнитного мартенсита и центральная линия (Л) парамагнитного остаточного аустенита. Прн отпуске интенсивность центральной линии уменьшается. При анализе спектров более сложных сплавов удается не только разделить линии мартенсита или фер-
167
рнта и аустенита, но н цементита, е-карбида н некоторых ннтерметаллндов.
В работе [13] исследовали сегрегации при старении железохромнстых сплавов (от 20,1 до 46,5 % Сг). После закалки с 1100 °C в воде наблюдали обычные 6 линий магнитной СТС. Расстояние между ними соответствовало эффективному полю, меньшему, чем 2,63Х XIО' А/м в чистом железе: 2,31-10' А/м (20,1 % Сг) и 1,59-Ю7 А/м (46,5 % Сг). После старения (500 °C, 150 ч) поле во всех сплавах увеличилось примерно до 2,39-107 А/м, что соответствует появлению областей, обогащенных железом (—12 % Сг). Одновременно появилась слабая несмещенная лнння, соответствовавшая парамагнитной фазе, обогащенной хромом. Интенсивность этой линии росла с увеличением содержания хрома.
В работе [14] были изучены процессы выделения в пересыщенных твердых растворах Си—Fe [<£3,5 % (ат.)] после различных термической и механической обработок. ЯГР-спек-тры сплава, содержащего 0,6 % (ат.) Fe после закалки с 875 °C и старения (600 °C, 5 мин) представляли собой наложение двух линий с различными ИС: первая соответствует железу в твердом растворе, вторая — у-выделениям. Расстояние между линиями составляло 0,33 мм/с и совпадало с ИС между медноже-лезным источником и нержавеющей сталью в качестве поглотителя. Прн увеличении времени старения доля у-выделений росла и достигла 60 %.
После 50 % деформации в спектре появлялись 6 пиков tx-мартенсита. Аналогичный результат получали и прн холодной деформации некоторых нержавеющих сталей.
При отжиге в вакууме (850 °C, 2 ч, от 10-4 до 10-2 мм рт. ст.) закаленного и состаренного сплава с 2,2 % Fe выделения y-Fe в Си окислялись до FeO. Об этом можно было судить по появлению в спектре дублета квадру-польною расщепления (рис. 8.8, а—г). При
Рис. 8.8. Расщепление, связанное с внутренним окислением (а—г) и восстановлением до «-состояния (£) у-выделений железа из твердого раствора в сплаве Си-i-2,2 % Fe (ат.):
а — отжиг, 850 °C, 2 ч; 3-10”4мм рт. ст.; б — то же. 3-10'г*8 мм рт. ст.; в — то же. 3-10 2 мм рт. ст.; е—чистое FeO; Q — отжиг S0O СС, 1,5 ч в водороде
отжиге в водороде окисел восстанавливался до чистого железа (рнс. 8.8, д).
Возможность исследования ближнего порядка связана с 'тем, что если в кристалле вокруг атома железа имеется несколько различных атомных конфигураций, то результирующий спектр будет суперпозицией «частных» спектров, соответствующих определенным локальным окружениям мессбауэровских ядер.
В бинарных сплавах локальное окружение определяется количеством атомов ч примеси («г) в i-той координационной сфере, н каждый «частный» спектр соответствует определенному набору чисел {п«}. При анализе обычно принимают гипотезу аддитивности, т. е независимого влияния каждого атома примеси на параметры спектра. Это положение, по-вн-димому, справедливо для малых концентраций примеси.
Наиболее чувствительны к локальному окружению внутреннее поле (Н) (в магнитных материалах) и ИС (6). Для данного набора {п,}: Я=Дс+Хн1-ДД1-; 6=60+X/ifA6t, где Но i i
и 60 — поле и ИС, соответствующие отсутствию примесей; АНг- и Абг- — изменения, вносимые одним атомом примеси в /-той координационной сфере.
В результирующий спектр каждый «частный» спектр входит со статистическим весом, пропорциональным вероятности данного локального окружения. Эту вероятность (Р) можно найти из биномиального распределения: P{tu, п2,...} — ДС/ р”* (1 — Р»)^^* Здесь
i i i
Zi — координационное число i-той сферы;
— биномиальные коэффициенты:
C% = Zl/(n!(Z — п)|);
Pi — вероятность замещения данного узла /-той сферы атомом прямеси.
В полностью неупорядоченном сплаве р<— =с — атомной доле примеси. В упорядоченном сплаве pi можно выразить через параметры порядка Cd: Pi=c(14-<Xi).
Таким образом, выделяя в экспериментальном спектре «частные» составляющие н идентифицируя нх, можно в принципе найти как параметры порядка для разных координационных сфер (tti), так и определить влияние атомов примеси на энергетические параметры (&Hi, А6{). Задача существенно упрощается для разбавленных растворов, в которых можно, во-первых, пренебречь вероятностью обнаружить два и более атомов прнмеси в одной координационной сфере и, во-вторых, учитывать взаимодействие только с ближайшими соседями. В этом случае результирующий спектр разлагают на две составляющие, интенсивности которых относятся как 1 — pZ к pZ. Первая соответствует отсутствию примесей (Но, бо); вторая — одному атому примеси в первой координационной сфере (Hi~ =Н0+&Н и б|=60-|-Аб).
Таким образом были изучены процессы возникновения ближнего порядка в большом числе систем на основе железа: с С, Al, Si [15], V [16], Мп, Ni и др.
8.5.4. Динамика решетки
Колебания атомов в твердых телах существенно влияют на вероятность эффекта Месс
168
бауэра. Температурная зависимость f нли fa определяется фактором Дебая — Валлера:
/ — ехр (— 21F) — | <С ехр ( i k и) > |2 ~
ехр [—< (й
Последнее приближенное равенство получено в гармоническом приближении теории кристаллов, когда тепловые смещения излучающего или поглощающего атома (и) подчиняются гауссову распределению.
Таким образом, по температурной зависимости f или fa можно непосредственно оценить средний квадрат тепловых смещений <х2> в направлении у-кванта. Поскольку £=2л/А, где X —длина волны у-кванта, то < (ku) —k2<Zx^^>= 4п2 <. х2 > !У?. Чем
меньше О'2>, т. е. чем сильнее связан атом в решетке, тем больше f (нлн fa).
В дебаевском приближении
2F= 6R/kT (1/4 -Н776)Ф (6/7)],
где k=l,4-10“23 Дж/К— постоянная Больцмана; &=(hvm)/k—дебаевская температура; Vm — максимальная частота в дебаевском спектре; 7?=0,19-10“2 эВ — энергия отдачи для 57Fe и
.С Mi
ехрЮ-1 ' о
При высоких температурах (Г»0, х^>1) 21^~6Д7’/й02, а при низких (Т<^6, х<С1):
2W = ЗЯ/2А0 [1 + 2л2/3 (7/6J2 + .. J.
Дебаевская модель применима только для наиболее простых кристаллов. Если в элементарной ячейке содержится более одного атома, даже если атомы кристалла одинаковы по массе, необходимо учитывать аигармощфм колебаний. Так, в работе [17] это показано прн исследовании динамики движения атомов Р—Sn (рис. 8.9); сплошная линия иа рисунке
Рис. S.9. Температурная зависимость In fa для белого олова
соответствует гармоническому приближению.
Если в элементарной ячейке находятся разные по массе атомы, расчет /(Г) еще усложняется. В случае небольшого различия снло-иых постоянных взаимодействия примесь — растворитель (у) и растворитель — раствори
тель (у'.)» можно приближенно охарактеризовать колебания примесного атома эффективной дебаевской температурой: 0Эф= 6 КМу7 /(ТИ'у), где 0 — дебаевская температура растворителя; М и М' — массы атомов растворителя и примеси. Однако в работе [18] было, например, показано, что введение в медную матрицу как тяжелого (золото), так и сравнительно легкого (железо) атома приводит к существенному (примерно в 1,5 раза) изменению силовых постоянных. Такне же эффекты описаны для Ан в Pt, Fe в W и Pt и др. Следует также иметь в виду существование значительной анизотропии колебаний атомов в кристалле. На рис. 8.10 приведены ЯГР-спект-
Рис. 8.10. ЯГР-спектры 67Со в монокристалле Zn (Г»» =300 К)
ры S7Co в монокристалле Zn [19] с осью с, параллельной и перпендикулярной направлению у-квантов. Площадь в первом случае меньше; это соответствует удвоению среднеквадратичного смещения вдоль осн с при 300 К.
Уменьшение вероятности эффекта характерно также для атомов, находящихся на поверхности твердого тела. Они слабее связаны с решеткой, частоты их колебаний меньше. Соответственно увеличивается среднеквадратичное смещение (для атомов иа поверхности серебра — вдвое; кобальта на поверхности вольфрама— в 1,9—2,5 раза в зависимости от направления и т. д.) и падает вероятность эффекта. Аналогичный эффект наблюдали для
О
ультрамалых частиц (С 100 А) [5].
К сильному изменению f могут приводить фазовые переходы. Например, прн плавлении f-И). Однако резкое падение f наблюдают только для чистых и кристаллических веществ. В аморфных веществах часто наблюдают область предплавлевия, где f уменьшается постепенно.
Предпереходные аномалии были обнаружены также для структурных фазовых переходов первого рода в твердом состоянии, в системах Fe — Ni, Fe — Мп и др. Это переходы тнпа мартенситного превращения, характеризующиеся малой теплотой и кристаллографической возможностью кооперативных смещений атомов. В работе [20] были получены температурные зависимости fa в сплавах Fe — Мп и обнаружено уменьшение fa прн охлаждении сплава примерно на 50° выше тем
169
пературы мартенситного превращения (рис, 8.11). В этом же (только еще более широком, около 200 °C) интервале температур было обнаружено аномальное увеличение коэффициента диффузии и пластичности. Это позволило авторам указать, что для объяснения эффекта недостаточно обычного предположения о не»
Рис. 8.11. Температурная зависимость fa для сплавов Fe—Мп:
слева — 10 % Мп, М$ «=170 °C; справа — 8 % Мп, =290 °C
стабильности («смягчении») некоторых колебательных мод вблизи точки перехода. В работе предложена модель предпереходиого состояния, согласно которой вблизи точки перехода атомы наряду с малыми колебаниями около положений равновесия исходной фазы совершают кооперативные (число атомов порядка 102) смещения в положения, отвечающие метастабильной фазе. Среднее время жизни атомов в этих положениях ~10~п с, что на два порядка превышает средний период тепловых колебаний.
8.5.5. Дефекты и диффузия
Необходимым условием изучения дефектов структуры с помощью эффекта Мессбауэра является связь излучающего илн поглощающего ядра с дефектом. Так, появление квадрупольного дублета в кубических кристаллах закиси железа нестехиометрического состава показывает, что даже в решетках с кубической симметрией наличие катионных вакансий, связанных с ионом железа, приводит к нарушению этой симметрии и изменению спектра. Аналогичная ситуация будет, если излучающее нли поглощающее ядро связано с примесным атомом.
Таким образом, возникает принципиальная возможность изучения вакансий и внедренных атомов я различных их ассоциаций. Эта возможность до сих пор была реализована только на ионных кристаллах, где удалось определить энергию активации перемещения катионных вакансий и отношение доли изолированных атомов железа к связанным с вакансиями.
Таких работ для металлов проведено ие было, если не считать упоминавшегося выше изучения f н среднеквадратичных смещений поверхностных атомов, а также сегрегаций [21] и выделения избыточных фаз на границах зерен.
Движение атомов в твердых телах уширяет спектральную линию, однако эффект можно, по-виднмому, заметить только вблизи точки плавления нз-за малости коэффициента диффузии [22]. В работе [23] исследовали диф
фузию S7Fe в Au и Си. На рис. 8.12 прведены спектры 57Со в Ап при 952 и 769 °C с нитропруссидом в качестве поглотителя. На рис. 8.13 показано уширение линии АГ, логарифм кото* рого линейно (в соответствии с теорией) зависит от обратной температуры ~ехр(~£//?П.
Рис. 8.12. ЯГР-спектры Б7Со в Au при разных температурах
Одиако уширение линии оказалось вдвое меньше теоретического, что, по-видимому, является следствием не учтенной теорией корреляции в движении атомов примеси. Как было показано в работе [22], анизотропия уширения различна для разных механизмов диффузии. Одиако таких измерений проведено не было.
Заметные эффекты уширения были обнаружены также прн диффузии атомов вдоль поверхности в дисперсных системах с высокоразвитой поверхностью (силикагели, цеолиты, ионообменные смолы н др.) [5].
Для таких систем с ультрамалыми частицами размером порядка и менее 100 А наблюдали эффект исчезновения магнитной СТС при низкой температуре (ниже точки Кюри) за счет явления суперпарамагнетизма [24]. Эффект возникает, если время флуктуаций магнитного момента, экспоненциально зависящее от объема частицы т=тоехр(КК/й7'), становится меньше времени ларморовской прецес-
170
син ядра. В последней формуле т0«1О~10— 1С'11 с; V—объем частицы; К—константа магнитной анизотропии однодоменной частицы.
БИБЛИОГРАФИЧЕСКИЙ СПИСОК
1. Методы испытания, контроля н исследования машиностроительных материалов. Т. I./Под ред. Туманова А. Т. М.; Машиностроение, 1971. с. 512—551.
2. Методы анализа поверхности: Пер. с англ./ /Под ред. Зандерны А. М/. Мнр, 1979. 582 с.
3. Вертхейм Г. Эффект Мессбауэра: Пер. с англ. М.: Мнр, 1966. 172 с.
4. Шпинель В. С. Резонанс гамма-лучей в кристаллах. М.: Наука, 1969. 382 с.
5. Суздалев И. П. Динамические эффекты в гамма-резонансной спектроскопии. М,: Атомиздат, 1979. 192 с.
6. Margulies S. — Z. Phys., 1963, Bd 176, № 1, S. 63—66.
7. Быков Г. М., Фам Зуи Хиен— ЖЭТФ, 1962, т. 43, № 3, с. 909—920.
8, Кривоглаз М. А. — Серия Итоги науки. Физика твердого тела. М.: ВИНИТИ, 1965, с. 5—125.
9. Химические применения мессбауэровской спектроскопии; Пер. с англ./Под ред. Гольданскога В. И. М.: Мир, 1970. 501 с.
10. Pargamin L., Lapis С. Н. Р., Flinn Р. А.— Met. Trans., 1972, v. 3, р. 2093—2105.
11. Бокштейн Б. С., Дьяконова Н. П., Ни-
Кольский Г. С., Ягодкин Ю. Д. — Ж ФХ, 1977, т. 60, № 9, с. 2166—2170.
12. Pauling L. Theory oi Alloy Phases. Ohio: Press, 1956. 408 p.
13. Yamamoto H. — Jap. Journ. Appl. Phys., 1964, v. 3, № 12, p. 745—748.
14. Gonser U., Grant R. W., Muir A. H., Wie-dersich H. — Acta Met., 1966, v. 14, № 3, p. 259—264.
15. Stearns M. B. — Phys. Review, 1963, v. 129, № 3, p. 1136—1144.
16. Бокштейн Б. С., Гугля E. Б. — Изв. вузов. Черная металлургия, 1979, № 5, с. 98— 100.
17. Boyle A. J. F., Bunbury D. S. P., Edwards C., Ball H. E.— Proc. Phys. Soc., 1961, v. 77, № 493, p. 129—135.
18. Cohen S. S., Nussbaum R. H., Howard D. G. — Phys. Rev., 1975, v. 12, № 10, p. 4095—4101.
19. Housley R. M., Nussbaum R. H.— Phys. Rev., 1965, v. 138A, № 3, p. 753—755.
20. Бокштейн Б. С., Клингер JI. M., Разумовский И. M. — ФТТ, 1977, т. 19, вып. 2, с. 476—479.
21. Бокштейн Б. С., Войтковский Ю. Б.. Жуховицкий А. А., Пауткина Г. Н. — ФТТ, 1968, т. 10, вып. 12, с. 3699—3701.
22. Кривоглаз М. А., Репецкий С. Н. — ФТТ, 1966, т. 8. вып 11, с. 2908—2915.
23. Knauer R. С., Mullen J. С. — Phys. Rev., 1968, v. 174, № 3, р. 711—713.
24. Крупянский Ю. Ф., Суздалев И. П. — ЖЭТФ, 1973, т. 65, № 6, с. 1715—1726.
9
:-----РАДИОСПЕКТРОСКОПИЯ
Приведены основные сведения о методах радиоспектроскопии, использующих резонансные эффекты при взаимодействиях между электромагнитным полем и веществом в виде атомов илн ионов. Принцип резонанса состоит в избирательном поглощении энергии электромагнитного излучения электронной илн ядер-ной подсистемой вещества, помещенного в постоянное магнитное поле. В связи с пространственным квантованием результирующих магнитных моментов (спиновых илн орбитальных) под действием постоянного магнитного поля возникают зеемановские уровни, переходы между которыми используются в методах, изложенных ниже.
9.1. ЯДЕРНЫЙ МАГНИТНЫЙ РЕЗОНАНС
9.1.1. Условие возникновения ядерного магнитного резонанса (ЯМР)
Методом ЯМР могут быть исследованы лишь те элементы периодической таблицы Менделеева, ядра которых обладают отличным от нуля магнитным моментом р. Эти ядра в дальнейшем будут называться ядернымн диполя
ми; величины магнитных моментов некоторых нз них приведены в таблице.
Согласно классической теорни_ электромагнетизма на магнитный момент р, находящийся в магнитном поле напряженностью Но, действует вращающий момент с, величина н направление которого определяются равенством Взаимосвязь магнитного и меняющегося во времени механического момента позволяет записать уравнение движения отдельного ядра: йр/с!/=у[|л//о], где у — отношение магнитного момента р, к собственному моменту количества движения ядра Ih.
В неподвижной системе координат производная по времени может быть выражена через величины, относящиеся к вращающейся с угловой _скоростью со системе координат: d\xldt=dpldt+ [top].
Из двух ^предыдущих формул получаемурав-неиия д|л/д/=у[р,Я0] + [шр] илн dpfdt— =у[р,(Я0+<в/у)].
Уравнения одинаковы по виду, если ввести понятие эффективного поля #»фф=== (//о+ы/у), где Но — напряженность внешнего поля; ей — частота вращения системы координат вокруг общей для обеих рассматриваемых систем осн z. При равенстве кулю напряженности эффективного поля магнитный момент во вращающейся системе координат неподвижен dpldt—
171
ФИЗИЧЕСКИЕ ХАРАКТЕРИСТИКИ ИЗОТОПОВ НЕКОТОРЫХ ЭЛЕМЕНТОВ [13]
Изотоп Содержание в естественной смеси. % Спин Резонансная частота для В=1Т. 10е Гц ьн/н, % Цвадрупольный момент. ЦТ-28 ы- Магнитный момент
*н 99,9 1/2 42,57 +2,7930
’Li i 92,6 3/2 16,547 0,0261 —0,012 +3,2559
»Ве 100,0 3/2 5,983 — 4-0,020 —1,1774
?3Na 100,0 3/2 11,262 0,112 +0,110 +2,2171
27А1 100,0 5/2 11,094 0,162 +0,156 +3,6408
зяК 93,1 3/2 1,987 0,285 +0,140 +0,3910
61у 100,0 7/2 11,193 0,580 — +5,1478
взСц 69,1 3/2 11,285 0,232 —0,160 +2,2262
6БСн 30,9 3/2 12,090 0,237 —0,150 +2,3845
eeGa 60,0 3/2 10,218 0,449 +0,180 +2,0170
71Ga 40,0 3/2 12,984 0,449 +0,110 +2,5610
86Rb 72,2 5/2 4,111 0,650 +0,300 +1,3532
87Rb 27,8 3/2 13,932 0,653 +0,140 +2,7501
B3Nb 100,0 9/2 10,407 0,850 .— +6,1660
107 Ag 51,4 1/2 1,722 0,522 — —0,1110
10BAg 48,6 1/2 1,981 0,522 — —0,1290
116In 95,8 9/2 9,329 0,786* +0,760 +5,5000
138Cs 100,0 7/2 5,585 1,490 —0,003 +2,5771
13BLa 99,9 7/2 6,014 0,630 +0,300 +2,7760
i8iia 100,0 7/2 5,095 1,100 +6,000 +2,1000
1S7Re 62,9 5/2 9,684 -— +2,600 +3,1755
lB3Jr 61,5 3/2 • 0,860 2,000 + 1,500 +0,1700
187 Au 100,0 3/2 0,691 1,400 +0,600 +0,2000
gcejj 70,4 1/2 24,570 1,560 — +1,6272
• Сдвиг Найта для жидкого индия при 443 К; в твердом состоянии часто наблюдается каадруполыгый резонанс.
=0. По отношению к покоящейся системе отсчета магнитный диполь ц прецессирует с угловой частотой (&а~уНо, называемой ларморовской. Отсюда, кстати, видно, что ядра с магнитным моментом, равным кулю (ц=0), не подвергаются прецессии (а>0—0).
Рис^9.1 Ларморовская прецессия магнитного момента ц во внешнем постоянном магнитном поле Но
Если перпендикулярно плоскости, в которой лежат векторы ц н Но, приложено вращающееся магнитное поле напряженностью Hlt причем |/?i| ТО—на Днполь Р будет
действовать пара снл [p+i], Рассмотрим два случая.
1. Поле Ht вращается с угловой частотой,
отличной от частоты ларморовской прецессии. Тогда пара сил [pJA] в соответствии с относительными фазами двух движений вокруг направлений векторов Но и Н] вызывает лишь иебольшие_возмущения прецессионного движения около Но.
2. Поле Ht_вращается синхронно.с прецессией диполя ц (рнс. 9.1), наступает резонанс, при этом угловая частота вращающегося магнитного поля становится равной угловой частоте ларморовской прецессии. Другими словами, резонанс наблюдается прн наложении дополнительного вращающегося магнитного' поля Hi в плоскости, перпендикулярной к направлению постоянного магнитного поля Но. Следует отметить, что обычно для наблюдения резонанса применяется не вращающееся, а изменяющееся по частоте линейно поляризованное поле 2Н\ cos (at с заданным направлением. Простейший способ наблюдения ДМР приводится ниже.
Образец, атомные ядра которого обладают магнитным моментом, помещается между полюсами постоянного магнита с напряженностью Но. Стремление магнитных моментов ядер ориентироваться по направлению поля приводит к появлению в образце результирующего макроскопического магнитного момента, прецессирующего под влиянием магнитного поля с угловой частотой у Нс. Образец, помещенный в небольшую катушку, ось которой перпендикулярна Но, находится в слабом переменном магнитном поле Hi, создаваемом высокочастотным генератором задающих колебаний. Когда частота, заданная генератором,
172
близка к частоте прецессии, поле Hi отклоняет магнитный момент от направления вектора Но. Это приводит к резонансному поглощению энергии высокочастотного поля образцом (системой ядерных спинов), в результате падает высокочастотное напряжение на контуре, включающем в свою схему катушку с образцом. Регистрация резонансной частоты и величины падения высокочастотного напряжения на колебательном контуре — задача, обычная для радиотехники, и многочисленные варианты ее решения можно найтн в монографиях по ЯМР [1—6].
Основополагающие работы в этой области выполнены двумя группами ученых, которые одновременно провели решающие эксперименты и опубликовали их в 1946 г. [7, 8]. Теория ядериого магнитного резонанса впервые наиболее полно была изложена в 1948 г. Блом-бергеном, Перселом и Паундом в ставшей классической работе [9].
Прогресс ЯМР был ускорен развитием радиотехники. В настоящее время в серийно выпускаемых спектрометрах ЯМР используют магнитные поля 5-Ю5—20-105 А/м; ЭВМ для обработки данных; широкий температурный интервал исследований (4—500 К) (см. Приложение) ,
9.1.2. Ширина линии спектра ЯМР
и времена релаксации
Рассмотрим поведение изолированного ядра в постоянном магнитном поле Но [10]- Предположим ядерное спиновое число />0, следовательно, ядро обладает магнитным моментом. Магнитное квантовое число т этого ядра может принимать любые (27+1) значения нз последовательности 7, I—1, I—2,...,—(/—1), —/. На рнс. 9.2 приводится случай, когда
Рис. 9.2. Схема энергетических уровней ядра со спином % в магнитном иоле Н
/=®/2 н существуют четыре возможные проекции на направление приложенного поля Нй. Энергетические уровни, связанные с ними, равно удалены друг от друга. Из квантовой механики следует, что разрешены лишь те переходы, при которых магнитное квантовое число т изменяется иа ±1. Разность между соседними уровнями (y.Ho)/Z=/iVo, где Vo — частота электромагнитного поля. Очевидно, в поле Но переходы будут происходить только прн характерной для каждого ядра частоте Vo—ixHo/Ih, если не учитывать влияния окружения.
Образец, помещенный в электромагнитное поле Ни направленное перпендикулярно внеш
нему магнитному полю Но, прн определенных условиях (см. 9.1.1) способен поглощать электромагнитную энергию, что соответствует переходам между соседними уровнями ядерных спннов. Поскольку вероятности перехода «снизу вверх» и «сверху вниз» одинаковы, а заполнение нижнего уровня больше, то наложенное поле Hi стремится выравнять населенность верхнего и нижнего энергетических уровней ядерной спиновой системы. Наглядно такое выравнивание можно представить как «нагрев» спиновой системы под влиянием радиочастотного поля [Hit Момент выравнивания основного и возбужденного состояний называется «насыщением». Однако существуют два механизма перехода ядер в состояние с более низкой энергией без испускания излучения. Этн механизмы принято обозначать как спин-спнновую н спин-решеточную релаксации. Прн спнн-спнновой релаксации ядро одного атома, находящееся в возбужденном состоянии, передает часть своей энергии другому ядру. Спнн-решеточная релаксация включает перенос энергии к «решетке» (все, что составляет окружение резонирующих ядер: растворитель, электроны системы, атомы илн ноны, отличающиеся от исследуемых). Энергия, отданная «решетке», переводит ядро в пижиее энергетическое состояние. Таким образом, в то время как радиочастотное поле уменьшает избыток яд'ер, находящихся на нижнем - энергетическом уровне, взаимодействие с «решеткой» восстанавливает этот избыток до его первоначальной величины. Возвращение к первоначальному равновесному распределению ядер по энергетическим уровням происходит не мгновенно, скорость этого процеса характеризуется временем спни-решеточной релаксации Л.
Если система спинов в целом находится в термодинамическом равновеснн с «решеткой» прн спиновой температуре Т, то согласно уравнению теплового баланса количества переходов снизу вверх н обратно должны быть равны; W+N+ = W-N-, где Л/+, N- — число ядер в единице объема с низкой и высокой энергией соответственно; W+, W- — вероятности перехода между уровнями т=+’/2 н т=
—V2. При равновесии отношение Лч/ЛГ-равно фактору Больцмана: —ехр((2ц,Но)/кТ)^1+2цНо/кТ. Поэтому можно записать каждую из вероятностей через среднее значение вероятностей перехода и Й7_: П7+ = №(1—iiHofkT); «7_=Г(1 +
+p,H0/kT). Под влиянием радиочастотного поля спиновая температура системы непрерывно возрастает и скорость изменения избытка ядер (п) в нижнем энергетическом состоянии на единицу объема составляет dnldt—2N~W- — 2N+w±. Множитель 2 появляется потому, что n~N+ — N~ при каждом переходе меняется на два. Совместное решение двук последних уравнений дает оконча-тельный результат dn/dt=2W(no —п), где По=(НцНо)/ЬТ — число ядер в единице объема прн условии термодинамического равновесия между системой спинов и решеткой; N— —N++N-.— общее число ядер в единице объема.
В последнем уравнении W имеет размерность [с-1], что позволяет принять 2F=7’pI>
173
тогда интегрирование дает: nD — n—(nD— —па)е 71 . Это решение при 7=0 удовлетворяет условию п=па — начальное значение. Разность (ис—п) характеризует отклонение величины п от равновесного значения п0 в момент t. Таким образом, через время t—T\ (время спин-решеточной релаксации) отклонение п от равновесного значения п0 уменьшается в е раз. Практически Л изменяется от Ю-4 до 104 с.
Второй механизм перехода ядер в низкоэнергетическое состояние обусловлен взаимодействием магнитных моментов ядер с локальными магнитными полями напряженностью Нл, создаваемыми соседними ядрами. Направление вектора каждого резонирующего ядра зависит от расположения соседних ядер и их магнитного квантового числа т. Напряженность магнитного поля, задаваемого магнитным моментом р., убывает обратно пропорционально кубу расстояния. Для оценки этой величины подставнм р—р,0—5-0,508Х ХЮ-27 А-м2 (ядерный магнетон), расстояние до ближайших соседей примем равным 10-10м, тогда величина Ня=5-103 А/м. Таким образом, поля, в которых будут находиться два произвольно взятых ядра, могут отличаться на 5-I03 А/м. В металлах, например, где взаимодействием спинов пренебречь нельзя, четкого выполнения условия резонанса ие будет, частоты ларморовской прецессии распределятся в интервале (cd+Ло). Для описания спектра ЯМР в этом случае удобно ввести понятие времени спни-спинового взаимодействия.
Предположим, что в единице объема находится ТУ ядер. За единицу времени dN ядер поглотят излучение с частотой, лежащей между о и co+dio. Для записи взаимосвязи dN с шириной частотного интервала do вводится g(co)—вероятность поглощения кванта электромагнитного излучения спиновой системой: оо оо
dN—Ngfti^do, причем f dN=N, J g((3))do— О о
= 1. Линия ЯМР симметрична относительно частоты ®=Wp, когда величина g(w) максимальна. Частотный интервал между точками спектра ЯМР, в которых величина поглощения становится равной половине максимального значения |д(со)]тах/2, называется шириной линии ЯМР (6ш0). Функция §(<£>) имеет размерность времени, поэтому можно записать 7’2= [g(<d)]niav/2, где Т'2 — время спин-спнновой релаксации, определяется диполь-дипольным взаимодействием н связано непосредственно с шириной линии ЯМР, как быо^
1 (рис. 9.3).
Подводя итоги сказанному о временах релаксации и Т2, необходимо указать иа их роль в реальных материалах. В монографии [12] можно найти описание оператора, являющегося суммой членов, каждый из которых отражает характерный для данного соединения тип взаимодействия резонирующих ядер между собой, так н со своим окружением. Например, рассмотрим жесткую решетку, т. е. случай, когда ядра неподвижно закреплены в пространстве. Как видно из рнс. 9.1, прецессию магнитного ядра ц можно разложить на
вращающуюся вдоль вектора поля Hi н статическую (вдоль поля Но) компоненты. При этом прецессия ядра (точнее, вращающаяся компонента) создает в месте расположения соседнего ядра переменное поле, вызывающее его резонансный переход. Из соображений сохранения энергии следует, что возбужденное ядро совершает обратный переход, ие изменяя
Рис. 9.3. Линия поглощения ЯМР
общую энергию спиновой системы. В результате подобных переходов уменьшается время жизни отдельных спиновых состояний, ‘ что соответствует расширению линии спектра ЯМР за счет спнн-спинового взаимодействия (ширина линии определяется временем релаксации Г2). По мере ускорения движения атомов в решетке этот тип взаимодействия играет все меньшую роль. Сохранение общей энергия обусловливается переходом энергии спиновой системы в энергию движения атомов н характеризуется спин-решеточиой релаксацией, а ширина линии определяется в основном временем релаксации Tt. Отсюда следует, что Т’гсТ’ь
Большинство структурных задач, исследуемых методом ЯМР, нуждается в построении модели решетки соединения [13]. Поэтому остановимся на влиянии спин-спниового взаимодействия как следствии данного расположения ионов решетки. Статическая компонента локального магнитного поля диполя /, обладающего моментом ц, на расстоянии (рис. 9.1) записывается в внде:_ц(3cos2 G,j— —1)г})3, где 0(г- — угол между г^-и/70. Длина вектора магнитного момента ядра уй[7(7-!-+ 1)]1/2. тогда величина локального поля представляется выражением: уй [7 (7 +1) ]1 X X(3cos26,-/—1)г^3. Условия жесткости решеткг н идентичность ядер в ней дают возможность оценить среднеквадратичную величину локального поля по всем соседним ядрам j: у>гИ51 (7+ + 1)S(3 cos2eij— 1)2Г^6.
Ван-Флек [14] предложил формулы расчета второго и четвертого моментов линии спектра ЯМР. В монокристалле, содержащем только один сорт магнитных ядер, выражение для второго момента S, равного среднему квадрату ширины линии спектра ЯМР, обычно записывают в виде;
174
s = ~ г (I +1) fMo «-’• (3 cos2ew-
где pc — единица магнитного момента, определяемая соотношением еИ/2тр (т$ — масса протона); N— число магнитных моментов ядер в системе, по которому проиодится суммирование; g—ix/ncI — фактор расщепления илн ^-фактор. Для поликрнсталлического образца линия поглощения — сумма сигналов поглощения от всех изотропно ориентированных кристаллических зерен, а второй момент линии спектра ЯМР равен среднему значению вторых моментов каждой частицы. Учитывая, что среднее значение (3cos20,j— 1)2=2/5, предыдущее уравнение приобретает внд:
s = V / U + 1) S2 Й W-1Гц6.
Для полноты изложения необходимо привести формулу расчета второго момента для случая, когда кристаллическая решетка содержит ядра нескольких типов. Для дополнительного члена уравнения
V1.2 Л”1 sti I, ( + 1) gj (3 cos2 eif - 1) rj/ о
вводится среднее значение по углам между направлением магнитного поля Но и вектором гэ/, содержащим ядра /- н f-типа. Окончательное выражение второго момента линии спектра в ЯМР для поликрнсталлического образца принимает вид:
3
s = -7- / (/ + 1) g2 W-1 2 ц Гц6 +
4
9.1.3. Сдвиг резонансной частоты
Один из важных параметров линнн спектра ЯМР — сдвиг резонансной частоты спектра. Впервые в 1949 г. в работе Найта [15] было экспериментально обнаружено, что линия какого-либо элемента ЯМР в металлическом образце наблюдается прн большей напряженности поля по сравнению с такими же ядрами в неметаллическом образце.
Кратко суть явления можно сформулировать следующим образом: электроны проводимости за счет парамагнетизма Паулн создают в месте расположения ядра среднее постоянное магнитное поле, что вызывает смещение резонансной частоты, называемое сдвигом Найта. Условие резонанса, о котором говорилось в разделе 1.1, записывается следующим образом:
v0 = — уН0/2л,
где Но — напряженность магнитного поля, в котором находится исследуемое ядро. Это соотношение не зависит от материала, окружающего ядра, поэтому можно сравнивать между собой резонансы данных ядер в различных соединениях. Из этого же уравнения следует, что измерения частоты при постоянном
внешнем магнитном поле дают ту же информацию, что и измерения поля при неизменной частоте. Частота может быть измерена достаточно точно, поэтому на практике для обозначения сдвига резонансной линии обычно пользуются либо величиной Av=Vq— Vv, где Vo и vr — резонансные частоты исследуемого металла (см. таблицу) и эталонного соединения, либо величиной относительного сдвига Av/vo.
Следует различать сдвиги резонансной частоты в неметаллических соединениях и металлах [16]. В неметаллах орбитальное движение электронов создает небольшое _поле, направленное против внешнего поля Но, кроме того поляризованные парамагнитные ноны илн комплексы соединения наводят внутренние поля, совпадающие по направлению с Но. Оба этн эффекта можно рассматривать как магнитную экранировку резонирующих ядер, определяющую сдвиги, которые принято называть химическими. Величины химических сдвигов относительно резонансной частоты малы н составляют приблизительно 10~4 %.
Характерная особенность относительных сдвигов резонансной частоты в металлах — постоянство величины ЕН/Н0 для различных изотопов для одного н того же элемента в образце, несмотря на то, что ядра изотопов какого-либо элемента обладают различными магнитными моментами, что свидетельствует об отсутствии влияния свойств самих ядер.
Созданная Найтом теория объясняет появление сдвига резонансной частоты наличием неспаренных s-электронов металла. Учитывая плотность неспаренных электронных спииои, можно записать выражение для величины найтовского сдвига:
(Vo — Vr)/vr = Av/vr = = (8яХ^о|¥Т0)|2)/з где — доля магнитной восприимчивости, обусловленная электронами проводимости; Vo— атомный объем; |ф^(0) |г — средняя плот-
ность вероятности у ядра для всех электронных состояний на поверхности Ферми, выраженная через волновую функцию электронов.
Анизотропия резонансного сдвига возникает вследствие нарушения простой симметрии решетки металлов (фазы внедрения, замещения, сплавы или чистые металлы после деформации). Наглядно анизотропию линии спектра ЯМР можно продемонстрировать на порошковых металлических образцах. Если каждому нз кристаллитов соответствует симметричная линия, положение которой зависит от ориентации кристаллической осн по отношению к вектору Но, то во всем объеме образца содержатся кристаллиты практически всех ориентаций. Рассмотрим два крайних случая ориентаций кристаллитов: 6—0 н 6=л/2 (6 — угол между осью симметрии и направлением поля Но). Для 6=0 (гексагональная илн тетрагональная ось) полное добавочное поле, вызванное электронами проводимости в месте расположения ядра, можно представить в следующем виде:
Д/7И -^Hvs^BVQN(EF)qFH^
где рв — магнитен Бора; N(EF)—плотность одноэлектроиных состояний на поверхности
Фермн, отнесенная к единице объема; qr — мера анизотропии в распределении заряда, связанная с волновой функцией электронов проводимости.
Для 0=л/2 аналогичное выражение выглядит следующим образом:
Д«х “ ЛН.,г - Рв v» N (ef
Для произвольного угла добавочное поле равно; АН—ЛЯ у cos8 6-1-Л//, sin26.
Если в направлении оси z плотность электронного заряда больше, чем под прямым углом к ней (решетка кадмия), т. е. qF>0, тогда А/7 || больше, чем АЙХ. Поэтому резонанс для кристаллитов, ориентированных под углом 0=0, наблюдается прн более высокой частоте vr. Аналогично А77х соответствует vt (рнс. 9,4). Для qF<0 (решетка таллия, напрн-
Рис. 9 4. Теоретическая форма линии спектра ЯМР металлического порошка (штриховая линия — форма, обусловленная анизотропным сдвигом Найта; сплошная линия — то же, но с учетом других источников уширения)
мер) получаются обратные соотношения, однако максимум линии всегда приходится на АН х.
В поликристаллическом образце А// усредняется до АНИЭ; относительный изотропный сдвиг Найта измеряют в точке, отстоящей от А/7 к на 2/з интервала между А/7 ц и А// х. В литературных источниках для упрощения описания сдвигов резонансных кривых пользуются относительной величиной A=Av/v0 или АД/Но, кроме того, если нет специальных указаний, под сдвигом Найта подразумевается изотропный сдвиг резонансной линии. Сдвиг резонансной частоты в переходных металлах н нх сплавах представляет возможность для изучения электронного строения [17]. Перечислим основные компоненты, из которых складывается иайтовскнй сданг: K=Ks+Ko5p+Kd. Первое слагаемое суммы отражает относительный сдвиг Найта, вызванный контактным взаимодействием резонирующего ядра с электронами в S-зоне; Лобр возникает при взаимодействии ядерного момента с токами, индуцированными прецессией электронов внутренних оболочек атома вокруг поля Но; сдвиг резонансной частоты относительно v0 (Ка) появляется из-за поляризации d-электронамн внутренних S-оболочек атома, а также электронов проводимо
К®
сти. Последнее слагаемое пропорционально числу неспаренных d-электронов, зависит от температуры н может иметь отрицательное значение.
Ниже приведены в качестве примера доли различных вкладов К Для 05Мо в сдвиг Найта, %:
Контактный сдвиг ........ 0,09
Сдвиг за счет поляризации остова . —0,11
Орбитальный сдвиг.................. 0,59
Полный сдвиг линии ЯМР .... 0,57
Видно, что наиболее значительна орбитальная часть найтовского сдвига.
9.2. КВАДРУПОЛЬНЫЕ ЭФФЕКТЫ И КВАДРУПОЛЬНЫЕ! РЕЗОНАНС
9.2.1. Квадрупольные взаимодействия, форма линии спектра ЯМР
Остановимся кратко на квадрупольных возмущениях лнинн ЯМР, наиболее часто встречаю
щихся в металлах. Ядра со спином об
ладают электрическим квадрупольным моментом н взаимодействуют со своим электронным окружением, если его симметрия меньше, чем кубическая, в системе энергетических уровней возникает расщепление. Энергия электрического квадрупольного взаимодействия достаточно мала по сравнению с магнитным взаимодействием в поле Но, так что ее можно рассматривать как слабое возмущение магнитных уровней. Для порошкообразных образцов форма линии напоминает рассмотренную прн описании анизотропного сдвига Найта (см. 9.1.3). Отличие состоит в том, что для мо-
Рис. 9.5. Квадрупольное расщепление первого порядка линии спектра ЯМР металлического порошка (/=72)
нокрнсталла резонансная линия содержит 27-компоненты, нх расположение в спектре симметрично частоте, соответствующей отсутствию квадрупольного взаимодействия. Таким образом, центр тяжести спектра не смещается.
Из рис. 9.5 н уравнений, приводимых далее,
следует, что при переходе т—1/2-»- —1/2 частота vm=v0; резонанс, наблюдаемый при vg, называется центральной компонентой лнпии поглощения. При переходах w^al/2 возникают (27—1) Сателлитные линии, расположенные симметрично центральной компоненте. Появление сателлитов не зависит от величины 'внешнего поля, а их интенсивность относится к интенсивности центральной компоненты как 4: 3 (I=sh — одна пара сателлитов); 9:8:5 (7=s/2— две пары сателлитов); 16 :15 :12: 7 (Z=7/z— трн пары сателлитов).
Иную картину можно наблюдать при 7>*/г, когда ивадрупольное взаимодействие достаточно сильно н возмущение первого порядка не описывает явление с достаточной точностью, а во втором н высших порядках прослеживается зависимость расщепления от угла 0 (для порошков центральная составляющая линии поглощения /н— l/2-^т——1/2 сильно размыта н ее регистрация затруднена). Для описания расщепления спектра включающего в себя 21 составляющих, вводится понятие константы квадрупольного взаимодействия &Qq!h н определяется ориентация главных осей н степень осевой симметрии тензора градиента электрического поля в местах расположения ядер. Частота перехода иа соседний магнитный уровень в первом приближении теории возмущений, развитой Паундом [18], равна:
= Vo 4- [(2т — 1)(3 cos2 0 — 1 )Х
X 3es ?Q]/87 (27 — I) ft, где 0 — угол между осью симметрии электрического поля н Но; Vo — невозмущенная резонансная частота; Q—квадрупольный момент ядра, находящегося в неоднородном электрическом поле осевой симметрии.
Следует отметить, что приведенное выше равенство справедливо для решеток с осью симметрии третьего и более высоких порядков (тетрагональных, ромбоэдрических н гексагональных структур). В металлах с кубической или тетраэдрической симметрией решетки градиент электрического поля на узлах равен нулю, поэтому квадрупольное расщепление отсутствует. Электрический градиент eq — величина второй производной электростатического потенциала вдоль осн симметрии в месте нахождения ядра, который создается всеми зарядами, расположенными вне ядра: eq~ =£/в/(3 cos2<p/—1)гу3, где <р/ — угол между осью симметрии и радиусом-вектором г{, который соединяет центр тяжести ядерного заряда с зарядом в/; / — обозначение всех внешних зарядов по отношению к данному ядру.
9.2.2. Квадрупольный резонанс
Известен ряд твердых тел, в которых электрическое поле, создаваемое кристаллической решеткой, вызывает достаточно большое квад-рупольиое расщепление ядерных уровней без наложения внешнего магнитного поля [19]. Высокочастотное поле индуцирует резонансные переходы между этими уровнями, что и лежит в основе метода ядерного квадрупольного резонанса (ЯКР) или чисто квадрупольного резояанса. Это аналог ядерного магнитного резонанса, но расщепление энергетических уровней имеет электрическое происхождение.
С классических позиций вектор магнитного момента ядра, обладающего электрическим квадрупольным моментом и находящегося в электрическом окружении с осевой симметрией, наклонен к осн z под угло^ В (рис. 9.6). Энергия квадрупольного взаимодействия записывается следующим образом; £=
Z
Рис. 9.6. Прецессия ядерного квадруполя вокруг оси симметрии электронного окружения
•=e2Q*<72Z(3cos20—1)/8, где cos2 6=ст2//(7+1)— квантовомехаиическая аналогия косинуса угла 0 получена на основании принципа пространственного квантования. Для угла прецессии 0, близкого к нулю, cos 0=1, следовательно, ст2= /(/+1), поэтому можно ввести среднюю величину квадрупольного взаимодействия:
(3 cos2 0-—1) — 2е2 Q;
fi2Q* = 2e2Q (7 + 1)/(27 — I).
Тогда выражение для энергии принимает вид:
3m2 ~ 7 (7 + 1) 7(27-1)
С помощью этого уравнения можно вычислить уровни энергии ЯКР в твердых телах, выражая их через постоянную квадрупольного взаимодействия e2Q^z. Переходы * ядерных спинов между этими уровнями и обусловливают явление ядерного квадрупольного резонанса. Важно отметить, что поляризующий_фактор в ЯМР — внешнее магнитное поле До, а в ЯКР — неоднородное электрическое поле, для которого величина энергии квадрупольного расщепления Е~т2, где т^—проекция спииа I на ось квантования.
На рис. 9.7 показаны энергетические уровни для целых н полуцелых значений 7. Приведем некоторые значения энергий этих уровней и частот перехода между ними:
7=1. Уровень энергии (e2Q<7zz)/4 двукратно вырожден, ст=±1; уровень энергии —(e2QQZ3)/2, ст—0; частота перехода v= =3(е2С9гг)/4.
/=3/г- Уровень энергии (e2Q<7zt)/4 двукратно вырожден, т—±3/2, уровень энергии —(e2Q?zz)/4 двукратно вырожден, ст=±‘/2; частота перехода v= (e2Qtfzz) /2.
7==s/s- Переходы возникают прн двукратно вырожденных уровнях энергии (см. рис. 9 7) иа двух частотах v 1 c=3(e2Qfo)/20 н
~2^~2
i2—28Q
177
v з в == 3 (е3(&гг)/ГО; частоты соотносятся как 2:1.
Z=7/s- Частоты резонансных переходов относятся как 3:2-И.
Чаще всего градиент электрического поля ие имеет осевой симметрии, что нарушает ука-
Пол у целое!
Целое I
Рис, 9.7. Энергетические уровни, возникшие при взаимодействии ядерного электрического квадруполя с аксиально симметричным электрическим полем
занные соотношения частот переходов. Классическим примером служит работа [201, где при изучении резонанса 1271 (7—5/г) обнаружилось нарушение отношения частот 2:1 на несколько процентов, что дало возможность авторам обсуждать отклонение электростатического потенциала от осевой симметрии. Для характеристики степени асимметрии тензора градиента электрического поля вводят параметр асимметрии: = | (qxx—qvv)lqZz\, где х,у, г направлены так, что цхх<цт<Цгг; следовательно, границы 1) определены в интервале 0<т|<1. Обычно параметр асимметрии дается в процентах, В [21] приведены таблицы значений квадрупольных моментов, констант квадрупольного взаимодействия, параметров асимметрии и измеренных частот поглощения.
Следует отметить, что применение метода ЯКР целесообразно, если константа квадрупольного взаимодействия eQqzz>l МГц. При меньших значениях обычно применяются другие резонансные методы (ЯМР, эффект Мессбауэра— см. раздел 8). На рис. 9.8 представлены соотношения между резонансными методами для случая I—3/2. Как уже было показано (см. раздел 9.2.1) центральная компонента, наблюдаемая на частоте v0(m=
, 1 и
=-f——*т =—~2~1> окружена сателлитами
/ , 3 И
т=± — -—>т— ±—— .
к 2 2 J
Ширина наблюдаемых квадрупольных линий составляет 10~2—10-3 резонансной частоты. Как н в ЯМР, существует два источника уширения линии ЯКР: 1) магнитное дипольное взаимодействие между соседними ядрами: 2) малость времени релаксации 7\.
Кроме этого, ширина линии ЯКР растет по мере увеличения степени несовершенства кристаллической решетки, в результате которого градиенты поля соседних узлов решетки различны. Изучение кристаллов показало, что наличие крутильных колебаний вызывает, во-первых, появление дополнительных градиентов
Рнс. 9.8. Расщепление уровней анергии в результате квадрупольных и магнитных взаимодействий
электрического поля (следовательно, возникает еще одна возможность уширения линнн), во-вторых, усреднение градиента электричес-ского поля по кристаллу (следовательно, имеет место сдвиг резонансной частоты). На этом основании как стационарную, так и импульсную методики ЯКР успешно применяют при изучении молекулярной подвижности н фазовых переходов.
9.2.3. Приложения ЯМР-спектроскопии к решению некоторых металлофизических задач
Структурные исследования. Для нахождения положений внедренных атомов изучаемого изотопа в металлах и их сплавах задаются структурной моделью двух встроенных друг в друга подрешеток, одна из которых состоит из атомов металла, Другая — из атома примеси. Таких моделей может быть несколько н для каждой из них методом Ван-Флека рассчитывают теоретически второй момент резонансной линнн (Av“eop). Экспериментально второй момент линии (Av|KCn) определяют по спектру ЯМР, предполагая, что в выбранном температурном интервале подрешетку из внедренных атомов можно считать жесткой. Отличие между Av|.eop и Av|KCri , не превышающее 3 % от абсолютной величины, позволяет правильно выбрать модель структуры изучаемого соединения.
Количественный анализ состава. Определение малых добавок в фазах внедрения, замещения н т. д. методом ЯМР относится к методам экспресс-аиализа в производственных условиях. Такой анализ может быть проведен оперативно и с достаточной точностью путем сравнения интенсивности сигнала ЯМР от образца и эталона (например, при анализе водорода). Сравнение необходимо вести по интенсивности спектра, отнесенной к единице объема изучаемого образца.
178
Диффузия. Изучение подвижности атомов методом ЯМР основано на том, что движение отдельных ядер усредняет локальные внутренние поля, сужая спектр ЯМР н изменяя его форму. При достаточно низких температурах позиции, занимаемые ре* зонирующим ядром в структуре металла илн сплава, отвечают минимуму потенциальной энергии. Повышение температуры, приложенной к образцу, вызывает рост собственной энергии ядра до величины, сравнимой с потенциальным барьером, переход через который обеспечивает диффузионное движение. Средняя частота диффузионных перескоков между двумя соседними позициями vc пропорциональна числу ядер, способных преодолеть потенциальный барьер t/0, определяющий энергию активации процесса диффузии: vc“ =тоехр(—U/kT). Если частота перескоков настолько велика, что Vc^Av, где Ат=уА/7/2л— ширина линии для жесткой решетки, то по температурному сужению линии можно найти величину потенциального барьера [22].
Фазовые переходы в твердых телах. Если исследуемый образец содержит ядра со спином />’/2, то наблюдение квадрупольных эффектов иа монокристаллах позволяет определить положение главных осей тензора градиента электрического поля внутри кристалла и параметр его асимметрии. Эти данные позволяют обнаружить выделение повой фазы н особенности ее структуры, оценить степень статических искажений кристаллической решетки и распределение электронной плотности вблизи включений. Метод ЯМР эффективно применяется прн изучении начальных стадий фазовых переходов, т. е. его чувствительность выше, чем у традиционных методов исследования.
9.3. ЭЛЕКТРОННЫЙ ПАРАМАГНИТНЫЙ РЕЗОНАНС (ЭПР)
9.3.1. Структура линий ЭПР
Честь открытия ЭПР принадлежит советскому ученому Е. К. Завойскому, который в 1944 г. опубликовал первые резонансные кривые. Суть явления можно сформулировать следующим образом: парамагнитный резонанс — резонансное поглощение радиочастотного поля веществом, содержащим парамагнитные частицы (молекулы, атомы, ионы, слабо связанные с атомом электроны, обладающие постоянным магнитным моментом), при наложении статического поля #о [22, 23]. Из-за различия ориентаций магнитных моментов отдельных частиц по отношению к направлению поля основной энергетический уровень парамагнитных частиц расщепляется иа ряд зеемановских подуровней. Воздействие осциллирующего магнитного поля вызывает переходы электронов между подуровнями, что приводит к появлению одной или нескольких линий резонансного поглощения.
У электронов (спин */2) квантовое число спинового углового момента может принимать значения ms— ±’/2, поэтому в отсутствие магнитного поля наблюдается двукратное вырож-.12*
дение спиновых энергетических состояний, которое снимается при наложении магнитного поля. В состоянии с низшей энергией (тБ— =—’/2) магнитный момент ориентирован по полю, тогда как в состоянии с более высокой энергией (тв= + }/2) магнитный момент направлен противоположно полю. Переходы между этими расстояниями могут возбуждаться переменным полем с частотой v согласно резонансному условию hv—gpB^. Мно« житель g определяет вклады орбитального момента и спина в магнитный момент, и его значение изменяется от атома к атому. Для свободного электрона, яе имеющего орбитального момента g—2,002322, а для свободного, парамагнитного атома его величина определяется фактором Лайде. Таким образом, энергии двух групп электронов в магнитном поле Н равны соответственно —и +gVcnH/2, а создаваемое поле расщепления g[isH.
В общем случае g зависит от ориентации иона (или молекулы), содержащего иеспарен-ный электрон, относительно магнитного поля. Одиако для идеальной кубической структуры (октаэдрическая илн тетраэдрическая решетки) g не зависит от ориентации кристалла в целом, другими словами, является изотропным. В кристаллическом окружении с более низкой симметрией значение g анизотропно. Прн условии, что ось высшего порядка кристалла совпадает с иаправлеиием магнитного поля, вводится обозначение g ц, аналогично этому значения g вдоль осей х и у кристалла, перпендикулярные полю, будут равны между собой и потому обозначаются одинаково gx. Если направление магнитного поля составляет угол б с осью 2, то в общем виде значение g может быть определено таким образом: g~— =g^l cos26 +gx$ sin20.
На определении соотношений значений g по различным направлениям основаны структурные задачи, которые нельзя решить рент-геиоструктуриым методом нз-за недостаточной чувствительности к малым искажениям симметрии [25].
9.3.2. Основные параметры линии ЭПР
Рассмотрим условие резонанса для метода ЭПР. Атом, в свободном состоянии имеющий отличный от нуля результирующий магнитный момент, помещен в постоянное магнитное поле Н. Каждый энергетический уровень атома характеризуется квантовым числом полного момента. Прн этом полный магнитный момент количества движения J равен векторной сумме орбитального L и спинового 5 моментов: У=£+5. Соответствующее квантовое число полного момента J при принимает (25+1) значений нз набора J=£+5, L+S—1, L—S. Прн £<5 сущест-
вует (2£ + 1) значений равенства J—S+L, S+L—1, ..., 5—L. Если приложенное внешнее магнитное поле слабее внутренних полей атома, то оно не_иарущает спин-орбитальной связи, а вектор / прецессирует относительно Но. Проекции вектора / принимают целочис-
179
ленные значения в единицах fz~rn]ti, где результирующие квантовые числа mj имеют 2/4-1 возможных значений 1—1, ....
О, ..., —J (зеемановское расщепление). Для атома, помещенного в переменное магнитное поле с частотой v, существует возможность переходов между соседними подуровнями, что обеспечивает резонансное поглощение энергии атомом (нли ионом) тэпр —у/7о=§>
В полях порядка 102—108 А/м резонансные частоты лежат в интервале 107—1011 Гц, т. е. в радиочастотной (107—Ю9 Гц) и микроволновой (IO10— 10п Гц) областях. Условие резонанса можно продемонстрировать _на примере «рецессии магнитного момента J в статическом поле Нс, направленном вдоль оси 2. Если вдоль оси х наложить переменное поле Н, то вращающий момент [.W] будет искажать ларморовскую прецессию вокруг осн г. Прн (й^ьщь средний по времени вращательный момент [УД] t стремится к нулю, в противном случае (при | © |=| ©ь |) векторы /к Н вращаются синхронно, т. е. величина [//7] t отлична от нуля и постоянна.
Другими словами, вектор / то приближается к оси 2 (отдача энергии), то удаляется от иее (поглощение энергии). Следует_ помнить, что, кроме статического поля Но, на магнитные моменты парамагнитных ионов в кристалле действуют внутренние поля, которые также влияют на резонансную частоту. Например, в иоиах группы железа резонансная частота определяется в основном кристаллическим полем, энергия взаимодействия с которым на 4 порядка больше энергии взаимодействия с внешнем полем Но.
Прн термодинамическом равновесии между системой атомов н окружением уровни с более низкой энергией заселены больше, что создает возможность поглощения энергии под влиянием переменного магнитного поля резонансной частоты. Полученная извне энергия электромагнитного поля настолько быстро передается, что прн используемых в ЭПР частотах отношение заселенностей нижнего (гц) н
характеризуются величиной Т2 — временем спин-спнновой релаксации и Л — временем спин-решеточиой релаксации.
Уравнение ДУдпр^Т^ + ^Г1 лишь прибли? знтельно устанавливает связь между шириной линии спектра и временами релаксации, одиако хорошо иллюстрирует соотношение между различными подсистемами, где ТГ>Т^. Время Тг сильно зависит от температуры (Л~7’-1) и равно ~ 10-ev— ю-2 с прИ изменении температуры от 20 °C до 4 К. Время Т% практически не зависит от температуры н имеет порядок 10~10 с. Отсюда можно сделать н практические выводы: уширение лнннн спектра ЭПР за счет спни-решеточной релаксации можно предельно уменьшить, проводя эксперимент прн достаточно низких температурах (=^20 К). Для изучения спин-решеточиой релаксации необходимо использовать большие внешние поля прн переменном поле малой частоты, когда можно пренебречь спин-спиновой релаксацией (т^СТТГ1). И, наоборот, для исследования спнн-спнновой релаксации подходят высокие частоты.
Следует выделить случай, когда зеемановское расщепление энергетических уровней намного больше энергии спин-спннового взаимодействия. Возникает перекрестная релаксация (кроссрелаксация), заключающаяся в обмене зеемановской энергией между двумя нли более парамагнитными частицами. Время кросс-релаксацин Т21 связано с 7*1 н 7g следующим образом: 72<7’21<7’i.
Процессы спии-спиновой релаксации включают два основных типа: днполь-дипольное магнитное и обменное электростатическое взаимодействия. Диполь-дипольное магнитное взаимодействие возникает нз-за того, что каждый парамагнитный нон находится в магнитном поле, представляющем собой сумму внешнего стационарного поля и полей, наведенных соседними ноиамн. Вследствие хаотической ориентации ионов это суммарное поле отличается по величине от внешнего и резонанс наблюдается в некотором интервале полей (частот) около среднего значения. Шн-
Ркс. fi.9. Схема релаксационных процессов в парамагнетике при ЭПР; Н© — квант внешнего электромагнитного поля
верхнего (п2) уровней практически не отклоняется от равновесного значения: ngjrii — =ехр(—где ДЕ*—энергетическая разность двух магнитных подуровней.
Восстановление теплового равновесия в парамагнетиках в общем случае можно представить как двуступенчатый процесс (рнс. 9.9) .-1) равновесие устанавливается внутри спиновой системы (система магнитных моментов всех парамагнитных частиц): 2) спиновая система отдает энергию кристаллической решетке (в жидкостях энергия, полученная от спиновой системы, активизирует броуновское движение).
Скорости этих восстановительных процессов
ISO
рнна липни определяется «добавкой» к величине внешнего магнитного поля, которую вносит магнитное окружение иона. Дальнодействие влияния соседей пропорционально г-8, поэтому при среднем расстоянии между парамагнитными нонамн порядка 5-Ю-8 см ширина лнннн спектра ЭПР составляет 4-104 А/м. Прн увеличении расстояния между магнитными ионами (что имеет место в образцах с малой концентрацией магнитных примесей) наблюдается сверхтонкая структура — сложный спектр, состоящий из лнннй поглощения в индивидуальном для каждого резонирующего нона поле.
Эффект, обратный диполь-дипольному ушн-
рению, заключается в обменном взаимодействии двух соседних парамагнитных ионов с учетом принципа Паулн. Такой обмен ведет к сужению резонансных линий. Развитый ВанФлеком метод моментов резонансных кривых позволяет судить о преимуществе того нли иного механизма взаимодействия в исследуемых образцах.
Процессы спнн-решеточной релаксации возникают как следствие передачи энергии от магнитных степеней свободы парамагнитных нонов в колебательную энергию кристаллической решетки. Чем быстрее происходит этот обмен, тем больше неопределенность (размытость) энергетических уровней и тем шире спектральная линия. Следует упомянуть о двух основных механизмах спнн-решеточной релаксации.
1. Механизм Валлера объясняет изменение ориентации магнитного момента парамагнитного иона во внешнем поле под влиянием колебаний решетки н нарушением магнитного взаимодействия между соседними колеблющимися ионами,
2, Механизм Кронинга—Ван-Флекса связывает тепловые колебания решетки с появляющимся электрическим внутрикрнсталлнческим полем, которое посредством спнн-орбитально-го взаимодействия вызывает переходы в спиновой системе.
Обсуждаемое до сих пор равновесное (или близкое к нему) распределение спннов возможно прн достаточно активном релаксационном выравнивании заселенности энергетических уровней. Если же мощность радиочастотного поля велнка и релаксационные процессы не в состоянии одновременно уменьшить заселенность верхних уровней (п2) синхронно с поглощаемой извне энергией, то наступает так называемое насыщение, которое обнаруживается по уменьшению эффекта ЭПР.
Характерные особенности ЭПР — тонкая и сверхтонкая структура спектра. Тонкая структура спектра ЭПР проявляется в возникновении группы линий, положение и интенсивность которых сильно зависит от ориентации монокристалла во внешнем поле. Анализ спектра в этом случае сводится к вычислению элементов матрицы [git]. Линии ЭПР часто имеют сверхтонкую структуру (СТС), вызванную взаимодействием между электронным и ядерным магнитными моментами. На величину СТС — расщепления влияют: 1) величина ядерного магнитного момента; 2) спиновая плотность электронов в непосредственной близости от ядра; 3) анизотропный эффект (анизотропия g н величины константы сверхтонкого взаимодействия).
Подводя итоги, перечислим факторы, влияющие на форму линии ЭПР: I) релаксационные процессы (характеризуются временами Л и Тг); 2) анизотропия д; 3) наличие тонкой н сверхтонкой структуры; 4) явление насыщения; 5) скнн-эффект; 6) неоднородность внешнего поля Но.
9.3.3. Применение метода ЭПР
Ионные кристаллы. Использование метода ЭПР для исследования ионных кристаллов, содержащих переходные металлы, сравнением величин внутреннего электростатического поля окружения парамагнитного нона с внутри
атомным взаимодействием позволило получить информацию о расположении парамагнитных центров и их взаимодействии. В качестве иллюстрации ниже приводится несколько примеров.
1. Характер расщепления кристаллическим полем энергетических уровней иоиов редкоземельных металлов, парамагнетизм которых обусловлен глубоколежащими 4/:-электронами, определяется симметрией этого поля. Воздействие кристаллического поля на парамагнитные ионы незначительно по сравнению со спин-орбитальиой связью, н величины интервалов между подуровнями составляют —> 102 см-1. За исключением ионов Cd+3, Ей*3 (наличие S-состояний) во всех редкоземельных металлах наблюдается сильная спин-решеточная связь, что позволяет наблюдать спектры ЭПР лишь прн низких температурах (см. раздел 9.3.2).
2. Для большинства нонов группы железа кристаллическое поле сильнее спии-орбитальной связи н поэтому полностью снимает вырождение орбитального уровня. Интервалы между подуровнями ~ 10й см-1. Подробный обзор результатов работ, выполненных для нонов с электронной конфигурацией dn, можно найти в книге [26], где авторы обобщают интерпретацию полученной ЭПР информации для п = 3, 4, 5, 8, 9 н п — 1, 2, 6, 7.
3. Изучение ионов Pd (^-конфигурация), Pt (5</п-конфигурация) и И (5/-конфигурация) методом ЭПР доказало необходимость учета ковалентной связи с соседними атомами. Основной вклад в кристаллическое поле вносит аксиально-симметричное поле электронов, связывающих комплекс типа уранила (ПОг)4-2.
Металлы. Источник парамагнетизма во всех металлах>—электроны проводимости, а в переходных металлах еще и незаполненные d- и ^-оболочки. Малое количество работ, выполненных ЭПР на металлах, можно объяснить наличием иногда непреодолимых трудностей, таких как:
1) уширение и искажение линии за счет скин-эффекта;
2) неизбежно присутствующее небольшое количество ферромагнитных включений, которое усложняет расчеты величин констант взаимодействия с кристаллическим полем;
3) значительные спин-рещеточные взаимодействия вынуждают проводить эксперименты при возможно более низких температурах.
Результаты исследований сплавов Си—Мп, Ag—Мп, Mg—Мп показали, что воздействие кристаллического поля практически полностью экранируется электронами проводимости.
Полупроводники, дефекты кристаллической решетки [25, 27]. Широкое применение полупроводников в современной технике продиктовало использование метода ЭПР для решения практических задач. Несколько примеров, демонстрирующих подобные исследования, приведено ниже:
1. При изучении спнн-решеточной релаксации в кремнии, легированном фосфором, были рассмотрены различные типы взаимодействия спннов. Установлено, что можно определять концентрации примесей в кремнии, не используя явления массопереноса. Кроме того, сильная зависимость вероятности релаксации от температуры подсказала применение системы
181
в качестве точного термометра в гелиевом диапазоне температур.
2. ЭПР применен для изучения электрически активных химических примесей и исследования дефектов, образующихся прн облучении кремниевых полупроводников с проводимостью n-тнпа электронами с энергией 1,5 МэВ. Показано, что при получении кремния методом вертикальной зонной плавки основные уровни, образующиеся при облучении, расположены гораздо глубже в запрещенной зоне н собственная проводимость проявляется прн значительно меньших дозах, чем в кремнии, выращенном в кварцевых тиглях.
3. Явление захвата свободных носителей в фотопроводнике позволяет объяснить многие люминесцентные свойства, в частности фотопроводимость соединений типа ZnS. Так как обнаружение процесса захвата проведено непосредственно при комнатной температуре, то предложено аппаратуру ЭПР использовать для измерения фотопроводимости.
9.4. ФЕРРОМАГНИТНЫЙ
И АНТИФЕРРОМАГНИТНЫЙ РЕЗОНАНСЫ
9. 4.1. Ферромагнитный резонанс (ФМР) [28]
Ферромагнитный резонанс (электронный магнитный резонанс в ферромагнетиках) представляет собой процесс избирательного поглощения энергии электромагнитного поля на частотах, совпадающих с собственными частотами прецессии магнитных моментов электронной системы во внутреннем эффективном магнитном поле.
Пользуясь уже описанным ранее классическим приближением (см. раздел 1.1) при записи условия ферромагнитного резонанса (ырез — = уН0), следует иметь в виду большую (порядка 0,1 Т в ферромагнетиках) [29] спонтанную намагниченность, которая приводит к большому резонансному поглощению (в 103 больше, чем в парамагнетиках). Кроме того, магнитные взаимодействия между электронами, участвующими в спонтанном моменте, создают сильные внутренние поля магнитной анизотропии. Эго означает, что эффективное поле, а следовательно, н частота резонанса будут зависеть от симметрии кристалла, формы образца, характера расположения во внешнем поле кристаллографических осей кристалла. Существование отдельных областей (доменов) с различными направлениями самопроизвольной намагниченности в объеме образца заставляет работать в условиях резонансного насыщения, когда внешнее поле разрушает доменную структуру и в первом приближении можио весь образец представить как «одиодомен-ную» структуру с однородной намагниченностью. Строго говоря, только поверхности второго порядка (сфера, эллипсоид, бесконечный круговой цилиндр и т. п.) не вносят неоднородности в общую намагниченность образца. Внутреннее магнитное поле в ферромагнетике (кроме указанной кристаллографической магнитной анизотропии) зависит как от величины, так и от ориентации внешних и внутренних упругих напряжений. Пере
чень- факторов, влияющих на условие резонанса в ферромагнетике, необходимо закончить неоднородностью высокочастотного поля, вызванной скин-эффектом. Глубина проникновения полей сантиметрового диапазона (СВЧ) в металл составляет 10~5—10“4 см.
К причинам уширения линии ФМР (как и в описанных ЯМР и ЭПР) относят спин-спнно-вый и спин-решеточиый механизмы релаксации. Наиболее узкая линия ФМР в совершенных монокристаллах (ДЯ=42,2 А/м) зарегистрирована в соединении YgFesOis (иттриевый феррит со структурой граната). Кроме влняняя дефектов, в этом кристалле ширина линнн ФМР определяется дипольным (магнитостатическим) взаимодействием и магнитострикцией. При введении редкоземельных примесей наблюдается максимум на кривой температурной зависимости ширины линии н анизотропия спектра ФМР: изменение ширины линнн в зависимостн от ориентации осн легкого намагничивания кристалла.
Особенности магнитного резонанса в металлических ферромагнетиках обусловлены наличием в них электронов проводимости. Благодаря скин-эффекту индуцированная высокочастотным магнитным полем намагиичивае-мость неоднородна по объему образца, что ведет к ушнренню линии ФМР-поглощения. По порядку величины ширина линии равна Ato = =[хве (а/б) 2]h, где 0е = (2g- рВА) /а27И0 х — константа порядка температуры Кюри; А — параметр обменного взаимодействия; Мо — намагниченность насыщения; а — постоянная решетки; 6 — глубина проникновения электромагнитного поля в металл. Количественные оценки показывают, что обменное взаимодействие электронов расширяет линию ФМР примерно до 103 А/м прн комнатной температуре, а прн охлаждении возрастает как Vct (ст — удельная электропроводность образца).
Существование электронов проводимости как подсистемы со степенями свободы, которые нельзя описать с помощью плотности магнитного момента М и электропроводности ст, создает новый тип релаксации — спин-элек-тронной. Частота испускания и поглощения спиновых волн электронами проводимости оценивается в 10s™10s Гц, что соответствует ширине линии ~103—104 А/м.
В настоящее время ие существует строгого и последовательного учета всех особенностей ФМР. Однако отдельные аспекты резонансных явлений в ферромагнитных материалах широко премеяяются в технике. Например, развитие представлений о линейных н нелинейных эффектах прн ФМР дало основу для создания новых СВЧ устройств: вентилей, ферритных генераторов и усилителей, параметрических преобразователей н ограничителей мощности.
9. 4.2. Антиферромагнитный резонанс
Электронный антиферромагнитный резонанс (ЭАФР) •—электронный спиновой резонанс в антиферромагнетикам— явление избирательного резонансного поглощения энергии электромагнитных волн, наблюдаемое при частотах, близких к собственным частотам прецессии магнитных моментов магнитных подрешеток антиферромагнетнка [29], Понятие «магннт-
182
на я подрешетка» вводится для описания магнитной структуры кристалла, обладающего атомным магнитным порядком. Магнитная подрешетка состоит из атомов (илн нонов) с параллельным магнитным моментом и принадлежит к одной и той же группе решеток Браве. Антиферромагнетикн, для которых магнитные моменты атомов направлены вдоль
Рис. 9J0. Прецессия векторов намагниченности двух подрешеток феррита
или против оси антиферромагнетизма, можно представить как две магнитные подрешетки, причем каждая нз ннх объединяет одинаково направленные магнитные моменты.
При Но~О прецессия магнитных моментов двух подрешеток /ь /2 происходит во внутренних эффективных полях магнитной анизотропии ±НА, направленных вдоль естественной осн антиферромагнетизма (рис. 9.10). Частоты резонанса для подрешеток зависят как от величины эффективного поля обменных сил (молекулярного поля Вейса) Нт, так и от На, удерживающего вектора J2 вдоль оси г: <0^2“ л- Для обычных в ан-
тиферромагиетиках значений Нт?х 108-ь 10s А/м и НА~ 105-5-10е А/м наблюдение ЭАФР возможно в миллиметровом диапазоне (частоты порядка 1O1Z—1013 Гц), что соответствует полям 106—107 А/м [27].
Наложение внешнего поля {H^G} снимает вырождение резонансных частот и их расщепление приводит к новой зависимости для условий резонанса
“1.2-г[2//„Лл+(“«о/2П,/2±
±уД,(1 —а/2),
где а = % у /х± — отношение статической магнитной восприимчивости в иапраилении оси z к аналогичной величине в перпендикулярном направлении. Следует отметить, что уравнение в простом виде справедливо лишь для кристаллов с высокой степенью симметрии и значительно усложняется прн изучении ннако-снмметричных решеток (ромбические и др.).
Из уравнения следует, что метод ЭАФР пригоден для исследования обменных взаимодействий и сил магнитной анизотропии в антиферромагнетиках. Методика эксперимента в основном повторяет технику ЭПР.
БИБЛИОГРАФИЧЕСКИЙ СПИСОК
1. Эндрю Э. Ядерный магнитный резонанс: Пер. с англ. М.: ИЛ, 1957, 296 с.
2. Поггл Дж.. Шнейдер Б., Бернстейн Г. Спектры ядерного магнитного резонанса высокого разрешения: Пер. с англ. М.: ИЛ, 1963. 592 с.
3. Леше А. Ядерная индукция: Пер. с нем. М.: ИЛ, 1963. 684 с.
4. Шумиловский Н. Н., Скрипко А. Л., Король В. С., Ковалев Г. В. Методы ядерио-го магнитного резонанса. М.—Л.: Энергия, 1966. 138 с.
5. Уо До/с. Новые методы ЯМР в твердых телах: Пер. с англ. М_: Мир, 1978. 178 с.
6. Хеберлен У., Меринг М. ЯМР высокого разрешения в твердых телах: Пер. с англ. М.: Мир, 1979. 512 с.
7. Purcell Е. М., Torrey Н. С., Pound К. V. — Phys. Rev., 1946, v. 69, р. 37.
8. Bloch Г., Hansen В/. W., Packard M. — Phys. Rev., 1946, v. 69, p. 127; v, 70, p. 474.
9. Bloembergen N., Purcell E. M., Pound R. V- — Phys. Rev., 1948, v. 73, p. 679.
10. Сликтер Ч. Основы теории магинтиого резонанса. 2-е изд.: Пер. с англ. М.: Мир, 1981. 448 с.
11. Гольдман М. Спиновая температура и ядерный магнитный резонанс в твердых телах: Пер. с англ. М.: Наука, 1972. 245 с.
12. Абрагам А. Ядерный магнетизм: Пер. с англ. М_: ИЛ, 1963. 551 с.
13. Киттель Ч. Введение в физику твердого тела: Пер. с англ. М.: Наука, 1978. 792 с.
14. VW-V/ec/г /. Н. —Phys. Rev., 1948, v. 74, р. 1168.
15. Knight W. D. — Phys. Rev., 1949, v. 76, p. 1259.
16. Роуланд T. Дж. Ядериый магнитный резонанс в металлах: Пер. с англ. М_: Металлургия, 1964. 115 с.
17. Винтер Ж- Магнитный резонанс в металлах: Пер. с англ. М.: Мир, 1976. 288 с.
18. Pound R. V. — Phys. Rev., 1950, v. 79, p. 685.
19. Гречишкин В. С. Ядерные квадрупольные взаимодействия в твердых телах. *М.: Наука, 1973. 263 с.
20. Dehmelt Н. G. — Am. J. Phys., 1951, v. 130, р. 356; 1954, v. 22, р. 110.
21. Абрагам А., Блини Б. Электронный парамагнитный резонанс переходных групп.
Т. 1: Пер. с англ. М.: Мнр, 1972. 361 с.
22. Бузник В. М. Ядерный магнитный резонанс в ионных кристаллах. Новосибирск: Наука, Сиб. отд., 1981. 250 с.
23. Альтшулер С. А., Козырев Б. М. Электронный парамагнитный резонанс соединений элементов промежуточных групп. М.: Наука, 1972. 450 с.
24. Сверхтонкие взаимодействия в твердых телах: Сб. М.: Мнр, 1970. 368 с.
25. Мейльман М. Л., Самойлович М. И. Введение в спектроскопию ЭПР активированных монокристаллов. М.: Атомиздат, 1977, 268 с.
26. Керрингтон А., Мак-Лечлан Э. Магнитный резонанс и его применение в хнмнн: Пер. с англ. М.: Мир, 1970. 447 с.
27. Электронный спиновой резонанс в полупроводниках: Сб. М.: ИЛ, 1962. 380 с.
28. Ферромагнитный резонанс: Сб. М.: Физматгиз, 1961. 343 с.
29. Вонсовский С. В. Магнетизм. М.: Наука, 1971, 1031 с.
183
9.5. ПРИЛОЖЕНИЕ
ПЕРЕЧЕНЬ НАИБОЛЕЕ ИЗВЕСТНЫХ РАДИОСПЕКТРОМЕТРОВ И ИХ ТЕХНИЧЕСКИХ ХАРАКТЕРИСТИК
Прибор Область применения, получаемая информация Диапазон рабочих частот. МГц Индукция поля, т Температурный интервал. К Время прохождения спектра, с ЭВМ-оснащение
JEOL W-40 (Япония) ЯМР-спектры многих ядер в диа- и парамагнитных соединениях 10,5— 40,0 1—2 80—500 60—1800 I. Программы для обработки спектров 2. Накопитель слабых сигналов
VARIAN Associates V-4300B ЯМР-спектроскопня в твердых и жидких веществах для решения физических и химических задач 40—60 0,9-1,4 80—500 30—1800 1. Автоматизированное управление записью спектра 2. Программированная обработка спектров
BBUKER SXP 4-100/15** Импульсный ЯМР-спектрометр, пригоден для изучения структурных параметров, кинетических процессов н внутренних полей в твердых телах: феррн-, ферро-, антиферромаг-нетикн 4—100 100—500 0,01— 1200 1. Система преобразования Фурье 2. Накопитель слабых сигналов 3. Набор про- грамм для обработки данных
ЯМР-213 (СССР) Изучение молеку- лярной подвижности н фазовых переходов в твердых телах; исследования кристаллической и электронной структуры кристаллов 30—100 5,5 4,2—300 150— 1200
РЯ-2308 (СССР) Малогабаритный радиоспектрометр высокого разрешения ДЛЯ учебных н исследовательских лабораторий 60 1,4 150—400 180—600 Накопитель слабых сигналов
РЯ-2301 (СССР) ЯМР-спектры низкого разрешения для изучения металлов и сплавов полупроводников и диэлектриков: фазовые переходы, времена релаксации, диффузионные процессы 2,4—40 0,3—1,0 100—500 0,1— 1000
РЭ-1301 (СССР) Количественный и качественный анализы парамагнитных веществ в твердом или жидком состоянии 0,001— 0,4 • 30—100
Ферромагнитный резонанс в тонких пленках Исследование нелинейных эффектов в широком диапазоне частот 20—4000 3,5-10-2 80—600 60—900
164
10
- - — СВЕДЕНИЯ о МЕХАНИЧЕСКИХ СВОЙСТВАХ МЕТАЛЛОВ
10.1. ДЕФОРМАЦИЯ
И РАЗРУШЕНИЕ
Элементы машин и конструкций, изготовленные нз металлов, их сплавов н других материалов, под действием приложенных к ним внешних усилий претерпевают деформацию. Деформацией (в механике) называется процесс изменения взаимного расположения каких-либо точек твердого тела в результате механического воздействия (рис. 10.1). При соот
Рис. 10.1. Изменение расстояния между точками А и В при различных видах деформации;
а — растяжение; б — сжатие; в >— сдвиг
ветствующих условиях нагружения деформация может закончиться разрушением, т. е. полным или частичным нарушением сплошности тела. Деформация может быть обратимой, т. е. исчезать после снятия нагрузки, вызвавшей ее, и необратимой — оставаться после удаления сил, под действием которых она возникла. Обратимая деформация называется упругой, а необратимая — пластической (остаточной) деформацией.
10.2. ОСНОВНЫЕ СТАДИИ ПРОЦЕССА ДЕФОРМАЦИИ
Наиболее наглядное представление о различных стадиях процесса деформации можно получить, рассматривая диаграмму деформации тела под воздействием возрастающей нагрузки. Такая диаграмма обычно строится по результатам опыта в координатах деформация— сила (рис. 10.2). Для металлов и их сплавов диаграмма деформации имеет два характерных участка: в начальной стадии нагружения до определенной нагрузки макроскопическая деформация возрастает по линейному закону (закон Гука), а затем зависимость между силой и деформацией становится, криволинейной. Кривая деформации практически обрывается в тот момент, когда происходит лавинное разрушение тела и вследствие этого нагрузка очень быстро спадает.
Если на первой стадии нагружения приостановить рост силы, а затем снять ее, то практически деформация в макромасштабе полностью исчезает. При снятии же нагрузки иа второй стадии исчезает только упругая часть деформации.
Б соответствии с этим весь процесс деформации целесообразно разделить на три последовательно проходящие одна за другой стадии:
1) стадия упругих деформаций; зависимость между силой и деформацией определяется за-
Начало пластических деформаций S отдельных зернах
Начало разрушения Разрушение '(конец}
Упруго-пластические деформации
(/. Начало массоВь/х пластических
Упругие деформаций 'деформации
Деформация
Рис. 10.2. Схема процесса деформации
коиом Гука н зависит от упругих свойств материала;
2) стадия упруго-пластических деформаций; зависимость между силой и деформацией определяется кривой, характер которой зависит от свойств материала, условий нагружения и выбора координат диаграммы деформации;
3) стадии разрушения.
Такое разделение процесса деформации условно, поскольку указанные стадии невозможно четко разграничить. Так, в области практически линейной зависимости между силой и деформацией, т. е. в макроскопически упругой области, металлографическими и рентгеновскими методами обнаруживается пластическая деформация отдельных зерен полнкрнсталли-ческого металла. Эта неоднородность деформации сохраняется и в пластической области. Поэтому задолго до полного разрушения даже довольно грубыми методами (например, наблюдая поверхность тела через бинокулярную лупу), можно обнаружить на отдельных его участках трещины разрушения (см. рис. 10.2).
Однако приведенное выше разделение процесса деформации необходимо и целесообразно, поскольку оно дает возможность разграничить основные закономерности поведения материалов прн механическом нагружении.
10.3. ОСНОВНЫЕ
ЗАКОНОМЕРНОСТИ УПРУГОЙ, ПЛАСТИЧЕСКОЙ
ДЕФОРМАЦИИ
И РАЗРУШЕНИЯ
Упругая деформация, как уже отмечалось, подчиняется закону Гука, который для одноосного растяжения записывается так:
185
е || = а/Е,
где е у — относительная продольная деформация (с || =А///); сг—нормальное напряжение в поперечном сечении; Е—модуль упругости первого рода; I — длина образца.
Поперечная деформация е Д6/6, где b — ширина образца; отношение ц иоснт
название коэффициента Пуассона (рнс. 10.3, а).
тельствуют о том, что пластическая деформация при различных способах нагружения осуществляется путем сдвигов, т. е. скольжением дислокаций, причем максимальные макроскопические сдвиги происходят по плоскостям действия максимальных касательных напряжений (рнс. 10.4);
2) процесс развития пластической деформации для данного материала макроскопически описывается полученной из опыта кривой деформации. Во многих случаях при построе-
Рис. 10.3. Закон Гука для различных напряженных состояний; а — одноосное; б — объемное; в — чистый сдвиг
Для трехосного растяжения закон Гука записывается в следующей форме:
1
ei — г, to — И to + Os)J!
с
1
еа = -у to — Р- to + os)l;
еа = [сг8 — F to +^в)];
Е
где О|, аг, Оз—главные напряжения; еь ег, еа — относительные линейные деформации по направлениям действия соответствующих главных напряжений (рнс. 10.3,6).
Закон Гука для сдвига: у=т/С, где т—касательное напряжение; у — относительный сдвиг (угол сдвига); G— модуль упругости второгорода (рис. 10.3,в).
В приведенных выражениях фигурируют три характеристики упругих свойств материала Е, G и ц, между которыми существует следующая зависимость:
G-£/[2(14-|x)).
Эти величины целиком характеризуют поведение изотропного материала в упругой области и принадлежат к числу достаточно стабильных механических свойств; оии мало зависят от небольших изменений температуры, изменения содержания легирующих элементов при сохранении основы сплава н характера термической обработки. Развитие пластической деформации характеризуется следующими основными закономерностями:
1) непосредственные наблюдения (появление линий Людерса, выявление пачек скольжения металлографическими методами) и сопоставление результатов испытания металлов при различных напряженных состояниях свиде-
нии этой диаграммы в координатах, отражающих истинные характеристики внутренних сил и деформаций с учетом изменения размеров (например, в координатах истинные макси-
Рис. 10.4. Плоскости максимальных сдвигов яри пластической деформации:
а — растяжение; б — сжатие; в — кручение
мольные касательные напряжения tmax — истинные максимальные сдвиги gmax), удается получить кривую, которая приближенно действительна для любого напряженного состояния (рис. 10.5);
3) если приостановить нагружение на какой-то стадии пластического деформирования и затем сиять нагрузку, то линия нагрузки пойдет по прямой, параллельной первоначальному прямолинейному участку диаграммы, поскольку при снятии нагрузки исчезает только упругая деформация (закон Гука). При повторном нагружении кривая идет также параллельно начальному прямолинейному участку до тех пор, пока нагрузка не достигнет значения, при котором началась разгруз
186
ка (закон Герстнера). Кривая деформации при дальнейшем нагружении пойдет так же, как если бы она шла без промежуточной разгрузки (рнс. 10.6);
4) в упруго-пластической Стадии нагружения объем материала практически не изменяется.
Рис. 10.5. Диаграмма деформации силумина при сжатии (/). кручении (2} и растяжении (3) (Н. М Бескоровайный и Я. Б. Фридман)
Рис. 10.6. Ход деформация в упруго-пластической стадии прн промежуточной разгрузке и дальнейшем нагружении
Рис. 10.7. Виды разрушения образцов при различных напряженных состояниях
Наиболее распространенная точка зрения на макроскопическое разрушение исходит из признаков двойственного характера сопротивления разрушению: каждый материал в зависимости от условий деформации может разрушаться от действия растягивающих (нормальных) напряжений (путем отрыва) или касательных путем поперечного нли продоль
ного среза или сдвига. Тот или иной вид разрушения определяется соотношением указанных напряжений и соотношением сопротивлений материала разрушению путем отрыва и среза при данных условиях нагружения (рнс. 10.7). Однако природа разрушения, определяющаяся его микроскопической картиной, значительно сложнее и недостаточно изучена.
10.4. ХРУПКОЕ И ПЛАСТИЧНОЕ СОСТОЯНИЕ МАТЕРИАЛОВ
В завнсимостн от характера нагружения один н тот же материал может находиться и в пластичном, н в хрупком состояниях. При пластичном состоянии разрушению материала предшествует большая или меньшая пластическая деформация. При хрупком состоянии материал разрушается без макроскопически заметной пластической деформации. Очевидно, что абсолютно хрупкого разрушения, особенно для металлов, нет, поскольку разрушению предшествует та или иная пластическая деформация. Поэтому хрупким (точнее малопластичным) разрушением называют разрушение с относительно малой пластической деформацией (рис. 10.8). Назовем факторы,
Рис. 10.8. Диаграммы деформации при хрупком (чугун). малопластичном и пластичном (сталь 20) разрушении при растяжении
способствующие переходу материала в хрупкое состояние:
1) изменение напряженного состояния —-уменьшение доли касательных напряжений по отношению к нормальным (например, переход от гладкнх к надрезанным образцам);
2) понижение температуры для хладноломких материалов;
3) изменение скорости нагружения для материалов, чувствительных к скорости нагружения.
10.5. ХАРАКТЕРИСТИКИ
МЕХАНИЧЕСКИХ СВОЙСТВ
МЕТАЛЛОВ
Величины, характеризующие сопротивление деформации (выраженные в напряжениях) нли разрушению (выраженные в напряжениях с учетом длины трещин), деформацию (выраженную, например, относительным удлинением) и вязкость (выраженную работой деформации или разрушения), носят название механических свойств.
На поведение материала существенно влияет характер приложения внешней нагрузки (статический, длительный статический, циклн-
187
ческий, динамический и т. и.). Имеют значение также напряженное состояние н способ нагружения (растяжение, сжатие, изгиб, кручение и др.).
Механические испытания могут быть разделены на следующие группы:
1. Однократные кратковременные статические испытания. Прн этих испытаниях строится кривая деформации (рис. 10.9, а) в ко-
рне. 10.9 Способы выражения результатов испытания в вависимости от характера приложения нагрузки:
а — статическое растяжение; б — испытания на ползучесть (о —предел ползучести при допуске вПр за время тПр). е—усталостные испытания;
в — ударные испытания (Д—работа разрушения)
ординатах, соответствующих характеру Напряженного состояния, и определяются упругие постоянные (£, G, р) характеристики сопротивления пластической деформации (например, при растяжении апц, Ст или g0>2, аЕ, SK) и характеристики пластичности (например, при растяжении 6 н ф).
2. Испытания на ползучесть при постоянной нагрузке. При этих испытаниях строится кривая (рис. 10.9,6), показывающая нарастание пластической деформации во времени, и определяется предел ползучести по заданному допуску на деформацию.
3. Испытания на релаксацию при постоянной деформации. Прн этих испытаниях строится кривая, показывающая снижение нагрузки во времени.
4. Испытания на усталость ври нагружении, изменяющемся по величине н направлению.
5. Динамические испытания ударным нагружением нлн нагрузкой, возрастающей с большой скоростью. Прн ударных испытаниях основной характеристикой является работа, затрачиваемая на разрушение образца. Эта работа графически может быть изображена площадью диаграммы деформации при ударе (рис. 10.9, а). С увеличением скорости деформации затрудняется процесс пластической деформации н кривая деформации идет круче (рис. 10.10).
6. Испытания на разрушение (см. раздел 15). Прн этих испытаниях, как однократных кратковременных или длительных, так и циклических, используют образцы с предварительно созданной усталостной трещиной. Испытания на разрушение могут быть только сравнительными (качественными), например испытание на ударный изгиб образцов с усталостной трещиной, и количественными, дающими воз
можность приближенной оценки размера допускаемого дефекта в конструкции, например определение критического коэффициента ии-
fietpopнация
Рис, 10.10. Ход кривой деформации при различных скоростях нагружения:
1— ударное нагружение; 2— статическое нагружение
Рис. 10.11. Ход кривой деформации при различных температурах [диаграмма истинных напряжений отожженной яри 6Б0 °C меди, испытанной при разных температурах (Е. М. Шевандин)]
Уисло циклов N 00 разрушения, 10 6
Рис. 10.12. Кривые усталости закаленной и кизкоот-пугценной стали ШХ15 (И. В. Кудрявцев):
1—на воздухе при +20 °C (О_______*=840 МПа); 2—в
ацетоне при —75°C (С_^=845 МПа); 3— в масле при + 100 °C (o^i->623 МПа); 4 — в воде прн +100 °C -=(50 МПа)
168
тенсивности напряжений Ктс нлн скорости роста усталостной трещины.
На механические свойства металлов оказывают влияние внутренние факторы состояния металла, как-то: состав, исходная структура, ее изменение в результате технологических режимов обработки н др., а также внешние
Рис. 10-13. Диаграмма растяжения при 20 °C монокристаллов свинца со скоростью 240 %/мин в неактивной (/) и активной (2) средах (В. И. Лихтман)
(температура испытания, воздействие коррозионной среды, воздействие адсорбционноактивных веществ). Влияние последних состоит в следующем:
С повышением температуры испытания пластическая деформация, как правило, облегчается (за исключением особых случаев хрупкости при нагреве, например, синеломкости и т. п_). Об этом свидетельствуют: изменение хода кривых деформации прн изменении температуры испытания (рис. 10.11), рост ползучести при повышении температуры, уменьшение пластически деформированной зоны образцов, испытанных прн пониженных температурах.
Воздействие коррозионной среды может привести и чаще всего приводит к снижению прочности (рнс. 10.12) и пластичности металлов.
Адсорбционно-активные вещества облегчают пластическое деформирование и разрушение металлов, что иллюстрируется диаграммой растяжения (рис. 10,13).
10.6. СВЯЗЬ МЕЖДУ РАЗЛИЧНЫМИ МЕХАНИЧЕСКИМИ СВОЙСТВАМИ
В основу попыток построения теории механических свойств были положены взгляды о двойственной природе разрушения материалов. Каждый материал, обладая двумя видами сопротивления разрушению — сопротивлением срезу н сопротивлением отрыву, в зависимости от характера напряженного состояния может разрушаться путем среза (вязко) и путем отрыва (хрупко). ,
Зависимость поведения материала от вида напряженного состояния свидетельствует о необходимости обобщенной оценки механических свойств металла. Простейшим примером
такой оценки может служить установление связи между твердостью по Бринеллю н временным сопротивлением.
Одной из попыток дать обобщение механических свойств является построение диаграммы механического состояния материала. При построении этой диаграммы принимаются следующие исходные положения:
1) для каждого материала ход процесса деформации прн разных способах нагружении характеризуется так называемой обобщенной кривой в координатах /тВх (максимальные истинные касательные напряжения) — gm&x. (максимальные истинные сдвиги), следовательно, переход в пластическую область определяется постоянным для данного материала значением предела текучести t?;
2) разрушение путем среза определяется при разных способах нагружения постоянным для данного материала значением сопротивления срезу /cpj
3) разрушение путем отрыва определяется значением сопротивления отрыву Si>TP, характеризующим в приведенных напряжениях предельно растягивающее удлинение. Диаграмма механического состояния состоит из двух частей (рнс. 10.14). Слева строится собствен-
Рис. 10.14. Диаграмма механического состояния
но диаграмма механического состояния в координатах /max —Стах (<$тах )» ГДе S“ax — наибольшее приведенное растягивающее напряжение, характеризующее максимальные упругие удлинения S^ax — Si—p,(S2-bS3), где Si, S2 и S3 — главные истинные напряжения; р, — коэффициент Пуассона, равный 0,25 — 0,30. На диаграмме указаны линии, соответствующие текучести, срезу и отрыву, и прямолинейные лучи (пунктирные), тангенс угла наклона которых выражает в соответствующем масштабе «коэффициент жесткости» а— при рЭЗЛНЧНЫХ способах НЗГруЖС-НИЯ.
В правой части диаграммы строится «обобщенная кривая» деформации. Диаграмма позволяет сделать следующие выводы:
1) характер разрушения (отрыв нлн срез) определяется способом нагружения. Для данного материала при растяжении луч напряженного состояния раньше достигает линии отрыва, а при кручении — линии среза, что н определяет различие в видах разрушения (отрыв прн растяжении и срез при сжатян и кручении);
2) при разрушении путем отрыва не используется максимально возможная (при других
189
видах нагружения) пластичность данного материала: разрушение происходит хрупко (кривая деформации обрывается в точке Л), Для одного и того же напряженного состояния изменение соотношения tcV/Sorp может привести к изменению характера разрушения. Так, если линия отрыва займет положение, показанное пунктиром, то при кручении будет отрыв. Исходные положения, принятые при построении диаграммы механического состояния, соблюдаются в ряде случаев лишь с большим приближением, поэтому эта диаграмма не может служить основанием для количественных расчетов. Например, всестороннее давление повышает прочность и пластичность. «Обобщенные» кривые расходятся у сложных, особенно у структурно неустойчивых и резко анизотропных материалов. Следует подчеркнуть, что разрушение путем отрыва и путем среза не является независимым одно от другого. Например, предшествующая разрушению пластическая деформация может изменять сопротивление отрыву н срезу, нормальные напряжения могут влиять на сопротивление срезу и т. п.
Необходимо также отметить, что в настоящее время стало ясным, что сопротивление отрыву как характеристика сопротивления хрупкому разрушению, выраженная в виде напряжения, ие является постоянной характеристикой данного материала, все попытки определить эту «константу» не привели к положительным результатам. Сопротивление отрыву предусматривает одновременный отрыв по всему сечению образца. Хрупкое разрушение и в эксплуатации и даже в лабораторных испытаниях цдет от точки к точке нз какого-то начального очага. Поэтому характе
ристикой хрупкого разрушения не может быть одно напряжение, оно должно включать и напряжение, и длину трещины. Применяемый в настоящее время критерий хрупкого разрушения Eic—ус I (раздел 15) является более постоянной величиной для данного материала, чем сопротивление отрыву.
Несмотря на все указанные недостатки, диаграмма механического состояния используется во многих случаях для качественной оценки поведения материалов при различных условиях нагружения,
БИБЛИОГРАФИЧЕСКИЙ СПИСОК
1. Фридман Я. Б- Механические свойства металлов. 4.1. 3-е изд., перер аб. и доп. М<: Машиностроение, 1965. 472 с.
2. Бадан А. Пластичность и разрушение твердых тел: Пер. с англ. М.: ИЛ, 1954. 64 с.
3. Бернштейн М. Л., Займовский В. А. Структура и механические свойства металлов. М.: Металлургия, 1970. 472 с.
4. Мак Лин Д. Механические свойства металлов: Пер. с англ. М.: Металлургия, 1965. 431 с.
5. Некоторые проблемы прочности твердого тела: Сб. М.: Изд-во АН СССР, 1959. 386 с.
6. Вопросы прочности крупных деталей машин. Кн. П2./Под ред. Кудрявцева И. В. М.: Машиностроение, 1976.
7. Биргер И. А., Шорр Б. Ф,, Шнейдеро-вич Р. М. Расчет иа прочность деталей машин. 2-е нзд., испр. и доп. М: Машиностроение, 1966. 626 с.
11
--СТАТИЧЕСКИЕ ИСПЫТАНИЯ МЕТАЛЛОВ
При статических испытаниях определяют свойства, характеризующие: 1) упругость; 2) сопротивление начальным пластическим деформациям; 3) сопротивление значительным пластическим деформациям; 4) пластичность.
Для полного выявления механических свойств испытания проводят при различных способах нагружения (растяжение, кручение, сжатие, изгиб и т.п.).
Силами инерции движущихся частей испытательной машины прн статических испытаниях обычно пренебрегают н определяют величину усилий методом статической тарировки (равновесия). Деформацию определяют путем измерения размеров образцов до и после деформации микрометром илн штангенциркулем, а более точно по показаниям механических нлн электрических тензометров, укрепленных на испытуемом образце.
11.1. ИСПЫТАНИЕ
НА ОДНООСНОЕ РАСТЯЖЕНИЕ
Испытание иа растяжение (одноосное напряженное состояние) характеризуется коэффициентом «жесткости» а=/п1вж/5дах =0,5.
Одноосность напряженного состояния сохраняется только до образования шейки (до достижения максимальной нагрузки). Для многих чугувов и литейных легких сплавов растяжение является «жестким» способом испытания, приводящим к отрыву. Для большинства отожженных и улучшенных сталей, а также для деформируемых цветных сплавов растяжение — «мягкий» способ (разрушение путем среза),
11.1.1. Машины и образцы для испытания на растяжение
Машины для испытания иа растяжение состоят из механизмов:
1) для нагружения (деформирования) образца;
2) для передачи растягивающей силы и центровки образца;
3) для измерения растягивающего усилия.
По методу нагружения машины подразделяются на два основных типа:
1) с механическим приводом, в которых деформация образца осуществляется переме--щением траверсы вдоль винта-шпинделя пу
190
тем вращения гайки, соединенной с траверсой;
2) с гидравлическим приводом, в которых деформация осуществляется перемещением штока поршня гидравлического цилиндра. Для небольших машин предпочитают механический привод, для мощных — гидравлический. На образец усилия передаются с помощью захватов. Чтобы уменьшить эксцентриситет приложения нагрузки, захваты снабжают само-центрирующимися шаровыми опорами (хотя значительное трение в них не обеспечивает вполне точной центровки) или лучше шарнирами Гука или применяют специальные аксиаторы.
Усилия измеряют следующими способами:
1) уравновешиванием приложенной силы подвижным грузом («взвешиванием» силы методом десятичных весов);
2) уравновешиванием силы маятниковым рычагом, который с изменением силы изменяет угол наклона;
3) с помощью гидравлического измерителя (гидравлической месдозы по показаниям специального манометра);
4) с помощью электронного силоизмерителя, состоящего из упругого растягиваемого стержня с наклеенными на нем тензорезисторамн
L
Рис. 11.1. Образцы для испытания на растяжение
и усилителя, передающего сигнал на ленту машины или двухкоординатный самописец.
На большинстве разрывных машин можно производить также испытание на сжатие и на изгиб, для чего имеются специальные приспособления (реверсоры). Образцы для испытания на растяжение изготовляют согласно ГОСТ 1497—73. Форма образцов цилиндрическая (чаще) илн призматическая (рис. 11.1). Обычно образцы на концах снабжены головками, форма н размер которых соответствуют захватам машины. Образцы без головок, устанавливаемые в клиновые зажимы с острыми насечками, применяют только для испытания очень пластичных материалов. В образцах из хрупких материалов (инструментальные стали, чугун, силикаты, цементы) переходы от головок к цилиндрической части выполняют в виде галтелей большого радиуса; часто применяют образцы с постоянным радиусом кривизны по всей длине (без цилиндрической части). Места вырезки образцов указываются в соответствующих стандартах нли технических условиях.
ТАБЛИЦА 11.1
ПРОПОРЦИОНАЛЬНЫЕ ЦИЛИНДРИЧЕСКИЕ
ОБРАЗЦЫ
Номер образца Рабочая часть образца, мм
короткая длинная
/о 1 1о 1
1 25 125 >160 250 >275
2 20 100 >120 200 >220
3 15 75 > 90 150 >165
4 10 50 > 60 100 >110
5 8 40 > 48 80 > 88
6 6 30 > 36 60 > 66
7 5 25 > 30 60 > 55
8 4 20 > 24 40 > 44
9 3 15 > 18 30 > 33
Для испытания применяют цилиндрические образцы согласно табл. 11.1. Допускаемые отклонения от номинальных размеров образцов даиы в табл. 11.2. Для плоских образцов ГОСТ 1497 — 73 предусмотрены 26 типов длинных и 26 типов коротких образцов.
ТАБЛИЦА 1-1.2
ПРЕДЕЛЬНЫЕ ОТКЛОНЕНИЯ ОТ НОМИНАЛЬНЫХ РАЗМЕРОВ ЦИЛИНДРИЧЕСКИХ ОБРАЗЦОВ
Номинальный диаметр, мм Допускаемые отклонения по диаметру, мм Допускаемая разность наибольшего и наименьшего диаметра на длине рабочей части образца, мм
До 10 ±0,1 0,03
Св. 10 до 20 ±0,2 0,04
Св. 20 ±0,25 0,05
11.1.2. Диаграммы деформации при растяжении
Зависимость между силами и деформациями записывается с помощью механического илн электронного диаграммного аппарата машины или двухкоординатного самописца в виде кривой растягивающая сила Р — абсолютное удлинение образца Д/. На рис, 11.2 показаны
Al Al Al
Рнс. 11.2. Диаграммы растяжения:
о —диаграмма большинства металлов в пластичном состоянии с постепенным переходом в пластическую область (легированные стали, медь, бронза); б—диаграмма некоторых металлов в пластичном состоянии со скачкообразным переходом в пластическую область в виде «зуба» или площадки текучести (нпз-коугяеродпстые стали, (3-латуни, некоторые отожженные марганцовистые и алюминиевые бронзы); в — диаграмма металлов, находящихся в хрупком состоянии при растяжении (закаленные и низкоогпущен-ные стали, чугун, силумин)
типичные диаграммы растяжения. Для получения удельных механических характеристик данного материала, не зависящих от размеров образцов, диаграмма деформации прн растяжении строится в координатах — растягивающее напряжение о — относительное удлинение е;
g=P/Fq; г = Ы/16,
где Р—растягивающая сила; Fo — исходная площадь поперечного сечення образца; Л/ — абсолютное удлинение; /0 — расчетная длина образца до испытания.
Диаграмма о—в отличается от диаграммы Р—AZ только масштабом и поэтом у при приемо-сдаточных испытаниях часто механические свойства определяют по первичной диаграмме Р—Д/ (рнс. 11.3).
Рис. 11.3. Характерные участки и точки диаграммы растяжения. На рисунке совмещены диаграммы с постепенным и резким переходом в пластическую область
В упругой области нагружения, где имеется прямая пропорциональность между удлинением образца н соответствующей нагрузкой (участок О—РПЦ> см. рис. 11.3), основной характеристикой является модуль продольной упругости (модуль первого рода, или модуль Юнга) »:
Дх=сг/е = р/0/гйАг.
В настоящее время более точным считается определение модуля упругости динамическим способом (см. раздел 12).
Сопротивление начальным (малым)5 пластическим деформациям, возникающим прн переходе из упругой области в упруго-пластическую и отличаемым по отклонению от линейной зависимости, характеризуют следующие величины.
Предел пропорциональности Опц — условное напряжение, соответствующее отклонениям от линейного хода кривой деформации (от закона Гука), задаваемым определенным допуском, например увеличением тангенса угла наклона кривой деформации к
1 Размерность модуля упругости, так же как и сопротивления малым и большим пластическим деформациям. кгс/мм2 — прн нагрузке в килограммси-лах и МПа — при нагрузке в ньютонах.
8 Речь идет о макроскопических деформациях. Микроскопические пластические деформации в отдельных зернах могут появляться значительно раньше.
осн напряжений на 25 или 50 % прн переходе от прямолинейного участка к криволинейному:
апц ~ Рпц/РО I
где Рац — нагрузка при пределе пропорциональности.
Предел упругости3 сто.ов — условное напряжение, соответствующее появлению остаточных деформаций заданной величины (0,05 %):
°Ь.О5 = Р O.t>5^P0»
где Ро.оз — нагрузка на пределе упругости.
Предел текучести (физический) о» — условное напряженке, соответствующее наименьшей нагрузке «площадки текучести», когда деформация образца происходит без увеличения нагрузки:
Оу = P^/Ffj,
где Рт — нагрузка при пределе текучести,
Предел текучести (условный)
Оо,2—условное напряжение, при котором остаточная деформация достигает величин 0,2 %:
Оо,а ” РqjJPо»
где Ро,2 — нагрузка прн условном пределе текучести.
Сопротивление значительным пластическим деформациям пластичных металлов характеризуется пределом прочности (временным сопротивлением) ов. ств — условное напряжение, соответствующее наибольшей нагрузке, выдерживаемой образцом:
Ов ~ Р^1 P()t
где Рв — максимальная нагрузка, достижение которой практически совпадает с началом образования шейкн в образце нз пластичного материала (переход от равномерной деформации всей рабочей части образца к сосредоточенной деформации в одном сеченни).
Пластичность металлов характеризуют следующие величины:
а) относительное удлинение (прн разрыве) б — отношение прироста длины образца после разрыва к первоначальной расчетной длине:
6 = [(/K”W/W 1°°%,
где /к — длина образца на расчетном участке после разрыва.
Величина б зависит от базы 10, по которой определяется б. Чем больше /0, тем мецьшеб. Индекс у б (62,5; 65; бю) указывает иа кратность испытуемого образца (1оМо);
б) относительное сужение $п р и разрыве) ф — отношение наибольшего (вместе разрыва) уменьшения поперечного сечения образца к первоначальной площади поперечного сечення:
t = (Го —
где FK — площадь поперечного сечення образца в месте разрыва.
Относительное сужение ф как более локальная характеристика лучше оценивает вязкость
8 Предел упругости при исследования микропла-стических деформаций определяют с допуском на остаточную деформацию до 10 6—10"-
192
материала прн разрушении, чем относительное удлинение 6.
Хотя рассмотренные характеристики имеют большое практическое значение, они условны, поскольку подсчет напряжений делением нагрузок на первоначальную площадь поперечного сечення не дает истинных напряжений, а относительное удлинение 6 прн образовании шейки не характеризует максимальной пластичности материала н зависит от размеров испытуемого образца.
Прн исследованиях иногда используют диаграммы истинных напряжений. Истинное напряжение X вычисляют делением действующей в определенный момент нагрузки Р на площадь поперечного сечення образца в тот же момент. Абсциссой диаграммы истинных напряжений часто принимают относительное сужение -ф, измеряемое н подсчитываемое для каждого момента нагружения соответственно.
На рнс. 11.4 показаны диаграмма истинных напряжений X—ф для металла, образующего
Рис. 11.4. Диаграмма истинных напряжений прн растяжении
11 Л.3. Определение характеристик прочности
Модуль упругости первого рода1 (Е) определяют методом «задаваемой нагрузки», т. е. путем деления задаваемого прироста напряжения на каждой последовательной ступени нагружения на среднюю величину приращения относительной деформации в упругой области, где для одинаковых последовательных ступеней нагружения сохраняется постоянство прнращеннй деформации. Приращение деформации измеряют тензометрами большой точности (например, с помощью зеркального прибора).
Предел пропорциональности оПц определяют или с помощью зеркального прибора прн последовательном нагружении образца, или графическим способом по машинной диаграмме растяжения. В первом случае нагружение ведут сначала крупными ступенями, а затем прн напряжении 0,65—0,8 от определяемого Опт; — малыми ступенями. Рпц определяют прн установленном отклонении деформации от закона пропорциональности, фиксируемом показаниями тензометра.
Во втором случае РПц определяют или по положению точки (рнс. 11.5, а), где кривая
Рис. 11.5. Графические способы определения предела пропорциональности по диаграмме растяжения
шейку при растяжении, и для сопоставления диаграмма о—е. Из этих диаграмм видно, что с увеличением деформации истинные напряжения непрерывно растут до момента разрушения образца н у пластичных материалов максимальная нагрузка, соответствующая сгв, отнюдь не отвечает моменту разрушения. Отсюда следует, что ов у пластичных материалов (большинство сталей и деформируемых цветных сплавов) является характеристикой сопротивления пластической деформации, а у хрупких материалов (чугуны, многие литейные алюмннневомагнневые сплавы и др.) — характеристикой сопротивления разрушения, так как в последнем случае максимальная нагрузка соответствует моменту разрушении.
Для пластичных материалов характеристикой сопротивления разрушению гладкого образца при растяжении служит истинное сопротивление разрушению Хк— истинное напряжение в момент разрушения:
^к — РЕ/Рк»
где Рк — усилие в момент разрушения; FK — площадь сечення в месте разрушения (см. рис. 1L3).
начинает отходить от прямолинейного направления (этот способ может дать значительную погрешность), или по положению точки касания прямой CD, параллельной лнннн ОБ, тангенс угла наклона которой меньше тангенса угла наклона ОА по установленному допуску (рнс. 11.5,6).
Предел упругости а0>05 определяют путем последовательного нагружения образца с разгруженнем его после каждой ступени деформации (а также по точной диаграмме растяжения): До,os фиксируется, когда остаточная деформация достигает установленной величины, что определяется по' показаниям тензометра.
Физический'предел текучести стт определяют либо непосредственно по машинной диаграмме растяжения, либо по фиксации растягивающей нагрузки в момент характерного прекращения роста нагрузки прн наступлении текучести по показаниям силонзмери-теля.
Условный предел текучести ст0,2 определяют по точке пересечения с кривой растяжения прямой KL, параллельной начальному участку кривой и отстоящей от него по
См. далее раздел 12.
13—280
горизонтали иа расстоянии ОК=0,2 (Zo/ЮО) в соответствии с принятым допуском (рис. 11.6).
Временное сопротивление ав (предел прочности) определяют по максимальной ординате кривой растяжения нли по максимальной нагрузке, отмечаемой снлоизмернте-лем.
Рнс. 11.6. Определение предела текучести 2 по диаграмме растяжения
части. Измеренную длину расчетной части подсчитывают из соотношений:
I = Ьс + се; be — ab + ас,
где ab — длина участка от места разрыва а до крайней метки Ь, в сторону короткой части образца до метки с; се — длниа участка, охватывающего от метки с в сторону разрыва до метки е столько делений, сколько нх содержится от метки с до крайней метки f (до головки Б). .. s
Рис. 11.8. Определение относительного удлинения в относительного сужения
Рис. 11.7. Виды разрушения образцов при растяжении:
а — разрушение с образованием шейки (низкоуглеродистая сталь); б — разрушение путем отрыва (чугун); в — разрушение путем среза (алюминиевые сплавы, магний деформированный)
Истинный предел прочности SK (прн разрушении), илн истинное временное сопротивление, определяют по конечной ординате кривой растяжения.
Характер разрушения определяют по виду излома образца (рис. 11.7).
11.1.4. Определение характеристик пластичности
Относительное удлинение образца, разрушившегося в средней части расчетной длины, определяют по расчетной длине образца, отмечаемой до испытания рисками, симметрично относительно средней части и по абсолютному удлинению, которое измеряется после разрыва по нанесенным ранее рискам как разность I—/0 (рис. 11.8). Для получения сопоставимых результатов прн разрушении образца иа крайних участках расчетной длины предварительно наносят равные деления через 5 илн 10 мм и определяют Ы из предположения, что разрыв произошел в средней
Относительное сужение цилиндрического образца определяется по среднему значению диаметра в месте разрыва, подсчитанному по двум замерам в двух взаимно перпендикулярных направлениях. В случае испытания плоских образцов (ГОСТ 1497—73), как правило, ие рекомендуется определение величины относительного сужения. В случае необходимости определения ф площадь поперечного сечения FK в месте разрыва определяют путем умножения наибольшей ширины образца в месте разрыва т на наименьшую толщину п.
Относительное сужение после разрыва определяют по формуле
= 100%.
11.2. ИСПЫТАНИЕ НА СЖАТИЕ
Сжатие — один нз видов «мягких» нагружений [с малым участком растягивающих поперечных деформаций и вовсе без участия растягивающих напряжений; при р=0,25; а= = (JmaxAS^ax) =2], поэтому применение испытаний иа сжатие целесообразно для материалов хрупких при растяжении (чугун, цемент и т. п.). Испытания можно проводить на специальных машинах и иа большинстве обычных разрывных машин, которые снабжены приспособлением для испытания на сжатие (реверсоры). Во избежание перекоса одну из опор (обычно верхнюю) делают шаровой, аиа сжимающих плоскостях для центровки тонкими лнннямн намечают центры, совпадающие с общей центральной осью сжатия (рис. 11.9).
Прн испытании образцов малого размера применяют специальные направляющие приспособления (рис. 11.10).
Образцы для испытания на сжатие металлов чаще всего имеют цилиндрическую форму с отношением высоты н диаметру 1—2. По ГОСТ 2055—81 для чугуна рекомендуются образцы диаметром 10—25 мм и высотой, равной диаметру. Применяются также образцы диаметром £)=6 мм и высотой Б=6 мм; Б=10 мм и Н=15 мм. Иногда используются
194
образцы с отношением HID—& с утолщенными концами (рис. 11.11). Торцовые поверхности образцов должны быть строго параллельны н отшлифованы. Влияние треиня иа торцах существенно сказывается на результатах испытаний, поэтому они зависят от состояния поверхности торцов н диаметра образцов.
Предлагаемые способы уменьшения трення (смазка торцов, применение конических насадок с углом, равным углу трення) не дают
Рис. 11.11. Образцы для испытания на сжатие: а — нормальный; б — восьмикратный
Рис. 11.9. Установка образца при испытании на сжатие.
1 — опорная подушка; 2— опора с шаровой поверхностью; 3 — головка машины; 4— образец
Рис. 11.10. Направляющее приспособление для сжатия мелких образцов:
1 — направляющая втулка; 2 — сжимающий шток; 3 — образец; 4 — опора
желаемых результатов. Многие хрупкие материалы (чугун, литые алюминиевые сплавы), разрушающиеся прн растяжении путем отрыва, прн сжатии разрушаются срезом (скалыванием) (рис. 11.12).
Высокопластнчные материалы (иизкоуглеро-дистая сталь, свинец н др.) не удается разрушить прн сжатии, так как онн сплющиваются без разрушения. Продольное разрушение путем отрыва прн сжатии очень хрупких материалов (органическое стекло, мрамор н др.) наблюдается лишь при очень тщательной смазке торцов. Прн наличии трення разрушение происходит по комическим поверхностях (см. рис. 11.12). Прн сжатии (как и прн растяжении) мохено определять пределы упругости, пропорциональности и текучести, но из-за методических трудностей эти величины определяют редко.
Испытание на сжатие преимущественно используют для образцов нз чугунных отливок, причем определяют предел прочности и реже •относительное укорочение.
а
я в
Рис. 11.12. Разрушение образцов нз различных материалов при сжатии:
а — чугун: б — закаленная сталь; в — то же, когда трение на торцах уменьшено
11.3. ИСПЫТАНИЕ НА ИЗГИБ
Прн изгибе узкого образца возникает неоднородное напряженное состояние, изменяющееся от одноосного растяжения [а— — (*max/Sraax) =0^] одноосного сжатия [о= — Gmax/S^jaj.) =2].
Целесообразность проведения испытаний на изгиб обусловлена прежде всего широким распространением изгиба в практике нагружения конструкций. В большинстве случаев испытания проводят сосредоточенной нагрузкой на образец, лежащий на двух опорах (рнс. 11.13, а), и реже двумя ровными, енмметрич-
Рис. 11.13. Схема испытания на изгиб:
а — сосредоточенный изгиб; б — чистый изгиб (максимальное напряжение где —
момент сопротивления сечения образца)
но приложенными сосредоточенными нагрузками, создающими на участке между инмн чистый изгиб с постоянным моментом (рис. 11.13,6).
13*
195
Испытание на изгиб можио проводить почти иа всех машинах, пригодных для испытания на сжатие. Большинство универсальных машин снабжено раздвигающимися опорами для испытания на изгиб. Нагрузки в опорах и в местах приложения усилий передаются иа образец через роликоподшипники для уменьшения трения при деформации изгиба. Образцы имеют большей частью призматическую форму с прямоугольным сечением. Чтобы избежать смятия в опорах, желательно по возможности увеличить поверхность контакта и уменьшить изгибающую силу. Последнее может быть достигнуто прн достаточно большой величине пролета.
Для испытания чугуна используют образцы двух различных типов; диаметром 30 и данной 650 мм, а также диаметром 30 и длиной 340 мм. Расстояние между опорами принимают соответственно 600 н 300 мм. Отношение пролета к размерам поперечного сечення образца и форма сечения существенно влияют на результаты испытаний. Помимо напряжений, соответствующих различным нагрузкам, при испытании определяют стрелу прогиба образца f (см. рис. 11.13), полученную на диаграммном аппарате машины илн с помощью прогибомера.
На рнс. 11.14 показаны типичные диаграммы изгиба. Прн изгибе хрупких материалов
Рис. 11.14. Типичные диаграммы изгиба:
а — пластичный материал; б — малопластичный материал; в — хрупкий материал
максимум нагрузки часто совпадает с появлением первой трещины. Иногда образование трещины сопровождается резкими срывами на ниспадающей ветви диаграммы.
Характерной особенностью испытания иа изгиб является то, что гладкие образцы из пластичных материалов (медь, алюминий, железо и их сплавы в отожженном, а часто и в улучшенном состояниях) не могут быть доведены до разрушения, так как образцы изгибаются до соприкосновения концов, не разрушаясь. Поэтому испытания иа изгиб гладких образцов с определением предела прочности н максимальной стрелы прогиба применяют прежде всего для малолластичных при растяжении материалов (чугунов, инструментальных сталей). В этом случае предел прочности
подсчитывают по обычной формуле
®шах = ЭДспах/^»
где Мшах — максимальный изгибающий момент; w—bhzl§ — для прямоугольного сечения; аэ=л£/3/32—для круглого сечення.
При изгибе можно определять пределы упругости, пропорциональности и текучести с точным замером деформаций. Различают два вида условных пределов текучести прн изгибе, подсчитанных по - определенному допуску на остаточное удлинение крайних волокон (например, 0,2 %):
1) реальный с вычислением истинных напряжений по формуле
2
smax = --[2M + 0(rfWde)]t
где 6, h — ширина и высота прямоугольного сечения; 26 — угол взаимного поворота двух сечений образца, расположенных иа участке чистого изгиба; dMjdQ — определяется графически как тангенс угла наклона касательной к кривой;
2) номинальный с вычислением условных напряжений по формуле
^тах = -^тах/т
При изгибе устраняется важный недостаток испытания на растяжение — влияние перекосов. Поэтому величины сопротивления разрушению у хрупких материалов более точно определяются при изгибе.
11.4. ИСПЫТАНИЕ НА КРУЧЕНИЕ
При кручении цилиндрического образца возникает напряженное состояние чистого сдвига, которое характеризуется равенством абсолютных значений Ттах, Отах, Cmin (рИС. 11.15) И
Рис. 11.16. Распределение напряжений при кручении цилиндрического образца: а — в площадках прямоугольных элементов на поверхности; б — в поперечном сечении
коэффициентом «жесткости» а=0,8 (в упругой области).
Пластическая деформация образца прн кручении происходит равномерно по его длине до образования первых видимых трещин, при этом диаметры поперечных сечений остаются прямыми н сохраняется цилиндрическая форма образца, что дает возможность надежно подсчитать напряжение н деформации. Образцы обычно имеют цилиндрическую форму; нормальным образцом для испытания иа кручение по ГОСТ 3565—58* принимается образец
196
с диаметром рабочей части б!о=Ю мм н расчетной длиной 4=100 и 50 мм.
Прн кручении образца в испытательной машине с помощью диаграммного аппарата илн непосредственно по показаниям приборов регистрируется угол повороту одной головки образца относительно другой при возрастающих значениях крутящего момента.
В ГОСТ 3565—58 приводится методика определения следующих механических свойств при кручеинн:
1) модуля сдвига (модуля упругости второго рода) G;
2) относительного максимального сдвига Утах»
3) предела пропорциональности тПц;
4) условного предела прочности тв;
5) истинного предела прочности fK;
6) условного предела текучести те.з;
7) характера разрушения при кручении.
Модуль упругости второго рода G определяется прн нагружении в упругой области по формуле
6 = 32M/0/jtdJ<p,
где М — крутящий момент; Iq — расчетная длина, мм; <р — угол закручивания, рад, на участке длиной /о, регистрируемой с помощью зеркального прибора.
Относительный максимальный сдвиг:
Ушах — ‘Ртах d/2l.
Предел пропорциональности тПц — условный предел текучести То.з и условный предел прочности тв — подсчитывают по формуле т= = 16M/nd3 по соответствующим значениям
Рис. 11.16. Диаграмма кручения
крутящих моментов Мт, 7И0,з и Мв, определяемым по диаграмме кручения (рис. 11.16).
Мад — соответствует ступени нагружения, при которой приращение угла закручивания равно полуторной величине среднего приращения указанного угла в прямолинейной части диаграммы кручения. М>,з — соответствует остаточному сдвигу 0,3 %. Мв — разрушающий крутящий момент.
Истинный предел прочности tK вычисляют по формуле
4
/к = —ЛЗЛ4в + <р(4ВД0)],
* В настоящее время введен в действие ГОСТ 3565—80, который дополнен испытаниями трубчатых образцов.
где <р — угол закручивания (рад); dM{d& — тангенс угла между касательной к кривой в точке Мв и осью 6 диаграммы, построенной в координатах М—0 (см. рнс. 15).
Характер разрушения при кручении определяют по поверхности излома. Прн срезе поверхность разрушения перпендикулярна оси образца, а при отрыве проходит по винтовой поверхности (рнс. 11.17).
Рис. 11.17. Разрушение образцов при кручении: с—путем среза (низкоуглеродистая сталь); б—путем отрыва (чугун)
Испытания иа кручение применяют в следующих случаях:
1) прн оценке пластичности хрупких при растяжении инструментальных сталей;
2) прн оценке пластичности н вязкости высокопластичных металлов н сплавов, образующих прн растяжении шейку;
3) прн необходимости четко разграннчнть-внд разрушения (отрыв илн срез);
4) при технологических испытаниях (например, при контроле проволоки, оценки обрабатываемости давлением и др.).
Чувствительность метода кручения к качеству обработки поверхности и наличию микро-трещни в закаленной стали определяют возможность применения этого метода для технологической оценки материалов.
11.5. МИКРОМЕХАНИЧЕСЦИЕ
ИСПЫТАНИЯ
Прн изучении сварных соединений, местной’ закалки, местного наклепа, степени неоднородности и аиизотропнн свойств материалов, а также в ряде других случаев необходима оценка свойств металлов в малых объемах.. Для этой цели служат микромеханнческне испытания. Размеры образцов приведены на рис. 11.18.
При испытании крупнозернистых металлов не следует брать образцы диаметром менее. 1,5 мм. Кроме цилиндрических, можно испытывать также плоские образцы нз листового материала. Цилиндрические образцы изготовляют так, чтобы следы обработки на образцах из хрупких материалов не были заметны при 25-кратиом увеличении, а на образцах остальных материалов при 5-кратном увеличении. Во избежание влияния иаклепа на результаты испытаний при механической обработке он должен сводиться к минимуму. Испытания микрообразцов можно проводить на специаль
197
ной машине ВИАМ (серийно не выпускается) илн на выпускаемых серийных машинах Р-05, Инстрон с максимальной нагрузкой 2 т и других машинах, имеющих шкалу с максимальной нагрузкой 100 кгс.
£4
Рис. 1!.18. Цилиндрические микрообразцы для испытания материалов на растяжение:
а — ов> 800 МПа; б — С? в =40*80 МПа; в — ов
С 40 МПа
11.6. ОПРЕДЕЛЕНИЕ ТВЕРДОСТИ
При определении твердости внешние нагрузки передаются на образец вдавливанием в его поверхность твердого наконечника в виде шарика, конуса или пирамиды, мало деформирующихся при испытаниях. Напряженное состояние, создаваемое прн определении твердости, характеризуется большим значением коэффициента «жесткости» (rf>2), что делает возможным применение метода твердости для испытания материалов, хрупких прн других способах нагружения. Испытанием на твердость оценивается в основном сопротивление значительным пластическим деформациям.
Определение твердости по Бринеллю вдавливанием шарика. За меру твердости по Бринеллю НВ (ГОСТ 9012—59) принято среднее сжимающее напряжение, вычисляемое условно на единицу площади поверхности отпечатка диаметром d и глубиной t, который получается прн вдавливании силой Р в килограмм-силах шарика диаметром D в миллиметрах (рис. 11.19):
НВ = P/F,
где F—площадь шарового сегмента (отпечатка), мм2, вычисляемая по формулам:
_ jtD2 лВ -------------
F — - — —- V D2—d2 , нли F-~-TtDt\
А А
Р — сила вдавливания, кгс.
Определение НВ по данным измерения d надежнее и проще. Диаметр отпечатка измеряют специальным измерительным микроскопом нлн измерительной лупой. Для ускорения и упрощения испытания составлены таблицы зависимости НВ от d. Твердость НВ вычисляют как среднее из ее определений как минимум по двум отпечаткам, а диаметр каждого отпечатка как среднее из двук его измерений
по двум взаимно перпендикулярным диаметрам.
Наибольшее распространение получили шарики диаметром 10 мм. Реже применяют шарики диаметром 2,5 и 5 мм, так как погрешность измерений при их использовании боль-
Рис. 11.19. Определение твердости при вдавливании шарика
ше. Шарики изготовляют из твердой закаленной стали типа ШХ15 с твердостью /7Г>850. Способом Бринелля испытывают металлы с твердостью не более НВ 450. Для шарика диаметром 10 мм прн стандартном испытании применяют нагрузку 3000 кгс. В случае шариков меньшего диаметра необходимо сохранять постоянным отношение P/D2=3000 : 100=30. Диаметр отпечатков должен находиться в пределах 0,2£><й<:0,6£>. Для сравнимости результатов испытаний стандартом регламентированы длительность нагружения и время выдержки под нагрузкой, так как некоторые металлы «текут» под постоянной нагрузкой во времени. Обычно продолжительность нагрузки составляет 30 с (табл. 11.3).
Для характеристики условий испытания приводят нагрузку, диаметр шарнка и длительность нагружения. Например, НВ 5/250/30— 200 означает, что число твердости 200 получено при испытании шариком диаметром 5 мм под нагрузкой 250 кгс и длительности нагружения 30 с. Для получения правильного отпечатка необходимы следующие условия: образцы должны иметь толщину не меньше, чем 10-кратная глубина отпечатка, а центр отпечатка должен быть удален от края образца не менее чем на 4d.
Несмотря на простоту испытания твердость НВ представляет собой довольно сложную механическую характеристику. Напряженное состояние прн вдавливании шарика неоднородно, поэтому величина НВ определяет некоторое усредненное сопротивление пластической деформации различно деформированных элементов объема. Для многих металлов твердость НВ хорошо коррелирует с временным сопротивлением. Для кованой н катаной стали Ов^О.Зб НВ-, для серого чугуна св = (НВ— —40) /6; для стального литья сгв= (0,3-5--*0,4)ЛВ.
Благодаря простоте и удобству измерения НВ, а также благодаря наличию устойчивой связи НВ и Ов (для металлов, дающих шейку) эта характеристика сыграла большую роль, хотя в последнее время широко внедряются более обоснованные методы определения твердости — прн вдавлнваннн кокуса илн пирамиды. В ряде случаев измерение твердости необходимо провести на крупных деталях, ко-
ТАБЛИЦА 11.3
УСЛОВИЯ СТАНДАРТНОГО ОПРЕДЕЛЕНИЯ ТВЕРДОСТИ ПО БРИНЕЛЛЮ
Материал Интервал ИВ Толщина испытуемого образца, мм P/D*. кгс/мм® Диаметр D, мм Нагрузка Р, кгс Выдержка под нагрузкой, с
Черные ме- таллы 140—450 От 6 ДО 3 » 4 » 2 Менее 2 30 10,0 5,0 2,5 3000 750 187,5 10
То же 140 Более 6 От 6 до 3 Менее 3 10 10,0 5,0 2,5 1000 250 162,5 10
Цветные металлы 130 От 6 до 3 » 4 » 2 Менее 2 30 10,0 5,0 2,5 3000 750 187,5 30
То же 35—130 От 9 до 3 » 6 » 3 Менее 3 10 10,0 5,0 2,5 1000 250 62,5 30
» 8—35 Более 6 От 6 до 3 Менее 3 2,5 10,0 5,0 2,5 200 62,6 15,6 60
торые невозможно установить на стационарный пресс Бринелля. В этом случае применяется метод измерения твердости по Бринеллю переносным твердомером статического действия, что регламентировано ГОСТ 22761—77.
Определение твердости при вдавливании конуса или пирамиды. Испытание на твердость измерением нагрузки прн заданной глубине вдавливания конуса в начале XX в. предложено П. В. Кубасовым, который провел соответствующие опыты на рельсовой стали. В дальнейшем твердость стали при вдавливании конуса Як начали определять по формуле HK—4P/std2, где d — диаметр отпечатка от конусного наконечника с углом прн вершине 90° (рнс. 11.20). форма отпечатков в этом
Рис. 11.20. Определение твердости при вдавливании конуса
иой пирамиды с углом 136° между противоположными гранями (ГОСТ 2999—75). Твердость по отпечатку пирамиды определяют делением нагрузки Р (кгс) на площадь поверхности отпечатка (мм2) (рис. 11.21):
Р________Р __ 1,8544Р
“ F -da/2cos22°_ d2 »
где d — диагональ отпечатка.
Величина HV ие изменяется при изменении нагрузки вследствие геометрического подобия
Рис. 11.21. Отпечаток при вдавливании пирамиды
случае геометрически подобна при всех нагрузках, поэтому твердость Нк ие зависит от величины нагрузки, что подтверждается н опытом. Для одних н тех же сталей Нк в среднем примерно на 8 % больше НВ. Конусы обычно изготавливают из победита или других твердых сплавов.
Несмотря на указанное преимущество, эта методика ие нашла широкого применения. Дальнейшим развитием методов испытания на твердость является метод вдавливания пирамиды (метод Виккерса, ГОСТ 2999—75). Испытание твердости в этом случае производится путем вдавливания алмазной четырехгран-
отпечатков. Благодаря большому углу в вершине пирамиды диагональ отпечатка достаточно велика даже при малой глубине вдав-лнваиня. Это увеличивает чувствительность метода и делает его особо пригодным для изучения также поверхностных слоев металла (при обезуглерожнваннн, поверхностном наклепе, хнмнко-термической обработке поверхности нли при малой толщине листов —до 0,3 мм). До НВ 350—400 величины HV и НВ равны. Прн большей твердости НВ ниже, чем HV. При вдавливании пирамиды степень деформации в переводе на усредненное удлинение составляет ~10. В табл. 11.4 приведены
199
ТАБЛИЦА 11.4
ТАБЛИЦА 11.5
УСЛОВИЯ ОПРЕДЕЛЕНИЯ ТВЕРДОСТИ .ПО ВИККЕРСУ*
Рекомендуемые нагрузки, кгс. при твердости
2—50 50—100 100—300 300—900
5—10
— — 5—10 10—20
5—10 26—10 10—20 ~-
10—20 —30 20—50 20—50
>20 >30 >50
* Минимальная толщина образца: для стали в 1,2 раза больше диагонали отпечатка, для цветных металлов — в 1,5 раза.
условия определения твердости по Виккерсу.
Определение твердости при вдавливании шарика или конуса с предварительным нагружением (по Роквеллу). Прн этом методе глубина отпечатка измеряется в самом процессе вдавливания, что значительно упрощает испытание. В зависимости от твердости материала применяют наконечники двух типов: стальные шарики диаметром 1,6 мм для испытания металлов малой и средней твердости при суммарной (основной и предварительной) нагрузке 100 кгс (шкала В) и алмазный конус с углом прн вершине 120° н радиусом закругления в вершине конуса 0,2 мм для испытания твердых металлов прн суммарной нагрузке 150 кгс (шкала С) и при суммарной нагрузке 60 кгс (шкала А). Нагрузка прилагается последовательно в две стадии (ГОСТ 9013—59): сначала предварительная, обычно равная 10 кгс, а •затем окончательная, равная большей частью 90 или 140 кгс. После приложения предварительной нагрузки индикатор, измеряющий глубину отпечатка, устанавливается на нуль. Когда отпечаток получен приложением окон-
ПРЕДЕЛЫ ИЗМЕРЕНИЯ ПО РОКВЕЛЛУ И ВИККЕРСУ
Шкала Число твердости Пределы измерения в единицах твердости по Роквеллу MR Соответствующие приближенные значения чисел твердости, измеренных алмазной пирамидой по Викерсу HV
В HRB 25—100 80—240
С HRC 20—67 240—200
А HRA 70—65 360—900
окружиостн шкалы, отвечает 2 мкм глубины вдавливания <
Деления циферблата пронумерованы в направлении, обратном движению стрелки. Поэтому число, показываемое стрелкой на циферблате, обратно пропорционально глубине вдавливания, т. е. прямо пропорционально твердости материала.
Циферблат имеет две шкалы — черкую и красную. Красная шкала смещена по отношению к черной на 30 делений. Прн испытании алмазным конусом отсчеты ведутся по черной шкале, а стальным шариком — по красной.
Пределы измерения твердости по указанным шкалам устанавливаются в соответствии с табл. 11.5.
Способ с применением предварительной нагрузки имеет ряд методических преимуществ (быстрота испытаний и др.) н поэтому очень удобен при массовом контроле. Для исследовательских работ предпочитают испытание пирамидой, дающее большую точность результатов. В табл. 11.6 сопоставлены значения твердости HV, НВ, HR (стандартные шкалы А, В и С).
Микротвердость (ГОСТ 9450—76). Для оценки свойств небольших объектов илн даже отдельных зерен металла применяется метод определения мнкротвердостн вдавливанием
Рис. 11.22. Положение наконечника при определении твердости с предварительным нагружением (Z—IV— порядок нагружения)
нательной нагрузки, основную нагрузку снимают н измеряют остаточную глубнку проникновения наконечника t под действием основной нагрузки (рис. 11.22). Эта глубина, выражаемая в условных единицах, характеризует твердость по Роквеллу HR. Число твердости отсчитывают по круговой шкале индикатора. Угловое перемещение стрелки индикатора на одно деление, соответствующее сотой части
алмазной пирамиды прн разных нагрузках. Метод дает возможность оценивать твердость отдельных структурных составляющих, более тонких поверхностных слоев, чем это достижимо с помощью прибора Виккерса. Прн этом
1 На приборе Суперроквелл одно деление шкалы соответствует 1 мкм. Нагрузки, используемые при испытаниях на этом приборе, равны 15, 30 и 45 кгс (150, 300 и 450 Н).
200
ТАБЛИЦА 11.6
ТАБЛИЦА ОРИЕНТИРОВОЧНОГО ПЕРЕВОДА ЗНАЧЕНИИ ТВЕРДОСТИ. ОПРЕДЕЛЯЕМЫХ РАЗЛИЧНЫМИ МЕТОДАМИ
Твердость HV Твердость НВ Твердость НВ по шкале । Твердость HV Твердость НВ Твердость НВ по шкале
диаметр отпечат- ка, мм НВ при испытании стандартным стальным шариком НВ при испытании шариком из карбида вольфрама С А В диаметр отпечат- ка, ММ НВ при испытании стандартным стальным шариком НВ при испытании шариком из карбида вольфрама С А в
1234 2,20 780 872 72 84 228 4,00 229 20 61 100
1116 2,25 745 840 70 83 — 222 4,05 223 — 19 60 99
1022 2,30 712 812 68 82 — 217 4,10 217 .— 17 60 98
941 2,35 682 794 66 81 — 213 4,15 212 — 15 59 97
868 2,40 673 760 64 80 — 208 4,20 207 — 14 59 95
804 2,45 627 724 62 79 — 201 4,25 201 — 13 58 94
746 2,50 601 682 60 78 — 197 4,30 197 — 12 58 93
694 2,55 578 646 58 78 192 4,35 192 <— 11 57 92
650 2,60 555 614 56 77 — 186 4,40 187 -— 9 57 92
606 2,65 534 578 54 76 — 183 4,45 183 —. 8 56 90
587 2,70 514 555 52 75 — 178 4,50 179 -—. 7 56 90
551 2,75 495 525 50 74 — 174 4,55 174 — 6 55 89
534 2,80 477 514 49 74 —, 171 4,60 170 — 4 55 88
502 2,85 461 477 48 73 — 166 4,65 167 — 3 54 87
474 2,90 444 460 46 73 — 162 4,70 163 — 2 53 88
460 2,95 429 432 45 72 — 159 4,75 159 •— 1 53 85
435 3,00 415 418 43 72 — 155 4,80 156 — — — 84
423 3,05 401 401 42 71 — 152 4,85 152 — — — 83
401 3,10 388 388 41 71 —- 149 4,90 149 — — — 82
390 3,15 375 375 40 70 — 148 4,95 146 — — — 81
386 3,20 363 364 39 70 — 143 5,00 143 — — — 80
361 3,25 352 352 38 69 — 140 5,05 140 — — — 79
344 3,30 341 341 36 68 — 138 5,10 137 — -— -—- 78
334 3,35 331 330 35 67 — 134 5,15 134 — — —- 77
320 3,40 321 321 33 67 — 131 5,20 131 W — — 76
311 3,45 311 311 32 66 — 129 5,25 128 — —. — 75
303 3,50 302 302 31 66 — 127 5,30 126 — —» — 74
292 3,55 293 — 30 65 — 123 5,35 123 — — — 73
285 3,60 285 * 29 65 -— 121 5,40 121 — — — 72
278 3,65 277 — 28 64 — 118 5,45 118 • — — 71
270 3,70 269 — 27 64 — 116 5,50 116 — — — 70’
261 3,75 262 — 26 63 — 115 5,55 114 — — •— 68
255 3,80 255 -Д— 25 63 ——- 113 5,60 111 — — — 67
249 3,85 248 —. 24 62 — НО 5,65 110 — — — 66
240 3,90 241 .— 23 62 102 109 5,70 109 — — — 65
235 3,95 235 — 21 61 101 108 5,75 107 — — 64
следует учитывать, что прн малых размерах отпечатка погрешность определения может до4 стигать 20%. Поэтому, чем меньше применяемая нагрузка, тем больше отпечатков должно быть сделано для получения уверенной средней величины мнкротвердостн.
Для определения микротвердости согласно ГОСТ 9450—76 прнменяютси четыре типа алмазных наконечников, приведенных в табл. 11.7. При методе восстановленного отпечатка число мнкротвердости определяют делением приложенной нагрузки в ньютонах (кило-грамм-снлах) иа условную площадь боковой поверхности полученного отпечатка в квадратных миллиметрах.
Испытания по методу невосстановленного отпечатка следует проводить, когда требуются дополнительные характеристики материала
(упругое восстановление, релаксация, ползучесть при нормальной температуре). При этом методе применяются те же наконечники, что-н при испытании по методу восстановленного, отпечатка. Число микротвердостн определяют делением приложенной нагрузки на условную площадь боковой поверхности отпечатка, соответствующую его измеренной глубине.
Формулы (5)—(8) получены из соотношений между размерами d нлн I н высотой h рабочей части алмазных наконечников (см. табл. 11.7). Для измерения микротвердости: применяют приборы по ГОСТ 10717—75. Приложение нагрузки должно быть плавным. Действие ее должно быть постоянным в течение установленного времени. Допускаемые погрешности нагружения не должны превышать 2 % от номинального значения для нагрузок 0,1 Н
201
ТАБЛИЦА 11.7
АЛМАЗНЫЕ НАКОНЕЧНИКИ, ПРИМЕНЯЕМЫЕ ПРИ ИЗМЕРЕНИИ МИКРОТВЕРДОСТИ И ФОРМУЛЫ ДЛЯ ПОДСЧЕТА МИКРОТВЕРДОСТИ»
Формулы Для подсчета микротвердости по методу
Наименование алмазных наконечников
восстановленного отпечатка
невосстановленного отпечатка
Четырехгранная пирамида с квадратным основанием (индекс «кв»)
,, Р 2sin«/2 1.854Р
77кв=----—---------~-------- ♦ (1)
кв S d2 d2 '
где d. — диагональ отпечатка; а, — пространственный угол в вершине
Трехгранная пирамида с основанием в виде равностороннего треугольника (индекс «тр»)
«ТР- s
3Psina 4,57
Четырехгранная пирамида с ромбическим основанием (индекс «рб»)
где /Тр — высота треугольника; ос — пространственный угол между гранью и высотой пирамиды
Р 2Ptg (а/2) cos(P/2) ИрС = — - ——г ~
‘рб
12.896Р
’ <3»
‘рб
#квЛ — с ~ о
_ 0.03784Р
^кв
где Акв'—глубина отпечатка
WTp- s ~
0.03797Р
— <6)
ЛТр
р
Нрб~ с ~ о
0,01385Р
Бицнлиндрический наконечник (индекс «ц»)
/рб — большая диагональ; ос и р — углы между гранями
Р SRsiaaP 4,168
(4)
‘ц *ц
где Рц — радиус цилиндра; /ц — длина линий сопряжения цилиндров; а — пространственный угол
О,07292Р -------- (8) h /е ц
• Р — нормальная нагрузка, /pg ««30.61/г pgj /ч«»3,8Бй мм.
приложенная к алмазному наконечнику. Н (кгс); й» 7,OOfcKB;/тр «=6.43Ятр;
н менее и 1 % от номинального значения для нагрузок более 0,1 Н.
Прн подготовке поверхности испытуемого изделия необходимо принять меры, исключающие возможное изменение твердости испытуемой поверхности. Шероховатость поверхности не должна быть грубее /?о=0,32 мкм. Рабочая поверхность и поверхность наконечника должны быть сухими (без смазки).
Прн испытании на микротвердость применяют нагрузки 0,049; 0,098; 0,196; 0,490; 0,981; 1,962; 4,905 Н, Минимальная толщина образца нлн слоя должна превышать глубину отпечатка не менее чем в десять раз.
11.7. МЕХАНИЧЕСКИЕ
СВОЙСТВА
ПРИ ДЛИТЕЛЬНЫХ
СТАТИЧЕСКИХ НАГРУЗКАХ
Механические свойства, определяемые прн длительных нагрузках, тесно связаны как с внутренними превращениями, которые ускоряются одновременным влиянием повышенной
температуры и нагружения, так и с воздействием внешней среды (окисление прн высокотемпературных испытаниях, воздействие коррозионной илн адсорбционно-активной среды) нлн диффузионных процессов (например, процесса диффузии водорода к месту разрушения).
Длительное нагружение металла в условиях высоких температур сопровождается двумя наиболее важными и характерными явлениями: ползучестью и развитием тепловой хрупкости. Ползучестью называется постепенная пластическая деформация металла прн постоянной нагрузке во времени. Тепловая хрупкость — уменьшение пластичности металла, определяемое по величине деформации в условиях постоянной нагрузки при высоких температурах.
Следует различать две основные группы механических свойств:
а) сопротивление пластической деформации прн длительных статических нагрузках [определение пределов ползучести (ГОСТ 3248— 81)];
б) сопротивление разрушению (длительная
прочность) н пластичность прн длительных статических нагрузках (ГОСТ 10145—81).
Обычно прн повышенных температурах образцы испытывают иа длительное статическое растяжение на специальных машинах. Груз прилагается к образцу через рычаги, обеспечивающие увеличение усилия в 10—30 раз. Для нагрева образца применяют электрические печн с терморегуляторами, поддерживающими постоянную температуру. Результаты испытаний изображают в виде кривой зависимости удлинения от времени. Типичные кривые приведены иа рис. 11.23. При малых нагрузках
Рис. 11.23. Типичные кривые ползучести
удлинение увеличивается практически только до определенной величины (/); при больших рост сначала замедляется, а затем приблизительно равномерен во времени (Я), наконец, при еще больших нагрузках удлинение увеличивается с возрастающей скоростью во времени вплоть до разрушения (///).
Для измерения удлинения применяют механические или более точные оптические приспособления. Основной величиной, характеризующей сопротивление пластическому деформированию прн длительном нагружении, является предел ползучести. Пределом ползучести считается то постоянное напряжение, которое вызывает за определенное время прн определенной постоянной температуре деформацию заданной величины (например, 1 % за 100 ч илн 1 % за 100000 ч) или определенную скорость деформации в течение заданного промежутка времени (например, 10~4 %/ч).
Пересчитывая результаты опытов при растяжении на сложные напряженные состояния, обычно сравнивают между собой пределы ползучести, выраженные в касательных напряжениях, т. е. принимают, что решающую роль в процессе ползучести играют касательные напряжения.
Испытания на длительную прочность проводят следующими способами:
1) определяют число часов до разрушения образца при данном напряжении;
2) прн данной температуре проводят серию испытаний прн различных постоянных напряжениях и затем строят зависимость продолжи
тельности жнзнн образца от напряжения (рнс. 11.24);
3) для каждой температуры испытания н для каждого постоянного напряжения определяют зависимость деформации от времени.
Рис. 11.24. Результаты испытаний на длительную прочность
Последний способ наиболее полный, однако ои значительно сложнее, так как требует приборов замера деформации, не чувствительных к сотрясениям.
11.8. ИСПЫТАНИЕ НА ДВУХОСНОЕ РАСТЯЖЕНИЕ [5, 6, 7]
В ряде конструкций, особенно работающих под внутренним давлением, листовой материал испытывает двухосное растяжение напряжениями Oi и аг- Оно может быть симметричным (cf[=a2) и несимметричным (ai>a2).
В упругой области диаграммы деформации имеют различный угол наклона при двухосном н одноосном растяжении вследствие различного значения модуля упругости:
Рис. 11.25. Диаграммы разрушения сплава Д16Т1 (лист толщиной 1,5 мм) при повторно-статическом двухосном растяжении внутренним давлением сферических сегментов разного радиуса кривизны со щелевым надрезом 0,3X10 мм, расположенным вдоль направления прокатки [7J. Частота нагружения п= =0,17 Гц. Номинальное напряжение Отду “100 Minai <W/amax J°-2- Кривизна:
7 — 0,12 см""1; 2 — 0.05 см”1; 3 — 0.0
при одноосном растяжении E,= (cr1/ei); (<y2/Oi=0);
прн симметричном двухосном растяжений £=(01/80(1—ц); (g2/Qi = 1)-
Прочность гладких образцов нз пластичных материалов, как правило, при двухосном растяжении выше, чем прн одноосном. Прочность гладких образцов из хрупких материалов обычно ниже при двухосном растяжении, чем при одноосном. Если образцы для испытания на двухосное растяжение имеют сквозную трещину, то чем больше кривизна образца (например, сферические сегменты), тем мень-
Рис. 11.26. Диаграмма циклического разрушения при симметричном двухосном растяжении сферических сегментов и плоских образцов из сплава Д16Т1 разной кривизны [7]:
1 — кривизна 0,12 см”1; 2— кривизна 0,05 см 1; 3 — кривизна 0,0
ше прочность прн однократном растяжении и тем больше скорость роста трещины прн циклическом растяжении (рнс. 11.25) одинаковым номинальным напряжением цикла. Это происходит за счет увеличения напряжений изгиба -берегов трещин с увеличением кривизны. При одинаковом значения коэффициента интенсивности напряжений цикла /Стах илн его размаха (Лтах--Кпнп—ДК), вычисленного по определенным формулам, с учетом влияния напряжений изгиба [7] скорость роста трещин усталости одинакова для различной кривизны, в том числе и для плоского образца, растягиваемого перпендикулярно направленными силами (рнс. 11.26).
* * *
При статических однократных кратковременных испытаниях обычно предполагается, что каждой величине напряжения соответствует -определенная величина деформации, причем скорость процесса деформации не учитывается.
Однако нз работ С. Н. Журкова и других вытекает, что знание одних критических величин напряжений без учета времени их действия часто недостаточно.
Последующими за работами С. Н. Журкова
исследованиями показано, что следует принимать во внимание не только суммарное время нагружения, ио н кинетику пластической деформации и разрушения, т. е. скорости н ускорения этих процессов. Как для пластической деформации, так и для разрушения в общем случае наблюдаются четыре кинетических периода:
I — инкубационный, скорость, и ускорение положительны; часто например, прн ползучести этот период не выявляется;
И — торможение, скорость положительна, а ускорение отрицательно вследствие упрочняющих процессов;
Ш — стационарный, скорость положительна, ускорение равно нулю;
IV — заключительный — ускоренный (иногда лавинный), скорость н ускорение положительны.
Переход от третьего к четвертому периоду соответствует переходу нз докритнческого состояния в закритическое.
Для пластической деформации и разрушения можно различать:
а) нарушение прочности в докритическом состоянии без уменьшения несущей способности;
б) нарушение прочности, начиная с появления закритического состояния, вызывающего снижение несущей способности, а при заданной нагрузке — положительное ускорение деформации и. разрушения.
Часто пластическая деформация и разрушение проходят одновременно, накладываясь друг на друга. Некоторые механические характеристики оценивают переход в закрнтнче-ское состояние (например, в пластической области оЕ при наличии шейки, в области разрушения без шейки).
Для ряда случаев работы материала следует, ие ограничиваясь докритнческнми характеристиками, оценивать также поведение металла, близкое к закритнческому, например:
1) испытывать образцы и детали с исходными трещинами;
2) проводить испытания прн различной степени релаксации внешней нагрузки, например с разным исходным запасом упругой энергии, с заданной нагрузкой н с заданными перемещениями и т. п.;
3) записывать изменение длины трещины во времени, в частности в заключительный период путем осциллографнровання;
4) проводить анализ строения изломов, выявляя на изломе границы между до- и закри-тическнм состояниями.
БИБЛИОГРАФИЧЕСКИЙ СПИСОК
1. Шапошников Н. А. Механические испытания металлов. 2-е изд. М.—Л.: Машгиз, 1954. 436 с.
2. Фридман Я. Б. Механические свойства металлов. Ч. 2. 3-е изд., перераб. и доп. М.: Машиностроение, 1965. 368 с.
3. Авдеев Б. А. Техника определения механических свойств металлов. 4-е изд. М.: Машиностроение, 1965. 488 с.
4. Тимошу к Л. Т. Механические свойста металлов. М.; Металлургия, 1971. 234 с.
5. Демина Н. И., Зилова Т. К., Фридман Я- Б.— Заводская лаборатория, 1969, № 1, с. 8—88.
6. Фридман Я- Б., Зилова Т. К., Ноеосильце-
204
ва Н. И.— ДАН СССР, 1967, т. 174, № 3, с. 572—575.
7. Соин В. Ю., Новосильцева II. И., Моро
зов Е. М — Физика и механика деформации и разрушения. Вып. 6. М.: Атомнздат, 1979, с. 98—103.
12
; МЕТОДЫ ОПРЕДЕЛЕНИЯ МОДУЛЕЙ УПРУГОСТИ
МЕТАЛЛОВ
При достаточно малых упругих деформациях соотношение между напряжением и деформацией подчиняется закону Гука. В общем виде эта зависимость, определяющаяся шестью составляющими напряжения, может быть представлена как
fa] = [c][E], (I)
где матрица [с] содержит 21 независимый элемент, представляющий коэффициенты (константы) упругости. Для металлов, как и для других кристаллических тел, упругие свойства могут быть рассмотрены не только в макроскопическом приближении, ио и в рамках микроскопической теории. Предположение о том, что силы межатомного взаимодействия, мерой которого и являются константы упругости, являются центральными, а атомы представляют собой центры симметрии, снижает число независимых элементов’ до 15, а учет симметрии пространственного расположения атомов для кристаллических решеток типа кубической снижает число независимых элементов до трех* а для изотропной среды — до двух.
Независимыми константами упругости кристаллов кубической симметрии являются Сц, С и и Сц Коэффициент Сц— мера сопротивления деформации, вызываемой скалывающими напряжениями в плоскости (100) в направлении <010>. Коэффициенты Сц и С12 не имеют такого прямого физического объяснения, однако их линейные комбинации
K=(Cu + 2C12)/3; (2)
G = (C11-C12)/2 (3)
могут трактоваться как объемная упругость (К), нлн сопротивление всестороннему сжатию, и как сопротивление деформации (G), вызываемой скалывающими напряжениями в плоскости (ПО) в направлении <П0>.
Когда металлы имеют поликрнсталлическое строение и деформация изучается в областях, значительно больших, чем размеры отдельных кристаллитов, эти металлы можно рассматривать как изотропную среду и описывать упругие свойства с помощью двух независимых модулей упругости — модуля всестороннего сжатия К и модуля сдвига G.
На практике наиболее распространенными видами нагружения являются линейное растяжение (сжатие) и простой сдвиг.
Модуль упругости прн линейном растяжении, нлн модуль нормальной упругости, Е связан с независимыми константами упругости через модули К и G.
E = 9KG/(3K4-G). (4)
Поперечная деформация, сопровождающая продольную при линейном растяжении, связа
на с последней через коэффициент Пуассона:
_ ед _ 1 3K-2G Е
11 в 2 ЗКЧ- G 2G ’ О
где ед — поперечная деформация; е—относительное удлинение.
Поскольку G и К всегда положительны, коэффициент Пуассона может меняться от —1 (при К—0) До 0,5 (прн G=0). Фактически р меняется от 0 до 0,5.
Таким образом, связь между модулями упругости изотропных материалов выражается следующими формулами:
Е = 2G (1+ р) = 9KG/(3K + G); (6)
G = E/[2(l+p)]; (7)
(8)
3(36 —Е) 3 (1 — 2р) 3(1—2р)‘ ' '
Модули упругости имеют различные величины в зависимости от того, протекает деформация и изотермических нлн адиабатических условиях.
Формулы (6)—(8) связывают между собой изотермические модули упругости. Однако часто деформация сопровождается изменением температуры тела в результате процесса деформирования и по другим причинам, а прн изменении температуры тела оио, даже в отсутствие внешних сил, деформируется вследствие теплового расширения.
Адиабатическими являются деформации, при которых не происходит теплового обмена между различными участками деформируемого тела, а также между этим телом и внешней средой. В этом случае адиабатические и изотермические модуля связаны следующими соотношениями
Ga;a; = G; (9)
1 1 762
= <10>
Лад Л Ср()
Е ~ F с о ’ ' 4
сад п cplJ
где а и р — линейный и объемный коэффициенты теплового расширения; ср — удельная теплоемкость при постоянном давления и р — плотность.
Количественно различие между адиабатическими и изотермическими модулями упругости не превышает 0,5 % и рассматривается в основном прн теоретическом анализе. В практике испытаний невозможно осуществить нн бесконечно медленного нагружения для определения изотермического модуля, нн мгновенного нагружения для определения адиабатического. Кроме того, эти модули определяются различными методами, прн которых различны
205
экспериментальные погрешности. В случае изотермических модулей эти погрешности соизмеримы с разницей в величине адиабатического и изотермического модулей.
12.1. СТАТИЧЕСКИЕ МЕТОДЫ ОПРЕДЕЛЕНИЯ
МОДУЛЕЙ УПРУГОСТИ
Медленное статическое деформирование меняет служить аналогом изотермического нагру-жения. Определяемый при статическом деформировании модуль упругости в литературе часто называют релаксирующим. Измеряют его прн различных, в том числе и значительных напряжениях, способных вызвать в металле необратимые изменения. Кроме того, прн статическом деформировании практически всегда успевают пройти релаксационные процессы, связанные с дополнительным удлинением растянутого образца прн его нагреве до температуры окружающей среды (в процессе быстрого растяжения образец охлаждается), а также с другими явлениями, обусловленными поведением несовершенств кристаллической решетки при деформировании. Разница между адиабатическим и изотермическим модулями связана лишь с первой причиной, тогда как разница между релаксирующим и нерелаксирующим модулями обусловлена еще и влиянием несовершенств кристаллической решетки — границ зерен, дислокаций, примесных атомов и др., обусловливающих внутреннее трение.
Чаще всего используют статические методы определения модулей упругости, точность которых достаточна для технических расчетов, особенно применительно к условиям работы деталей, близким к статическим. Обычные виды нагружения для определения модулей Е и G — растяжение н крученне. Модули упругости прн этом рассчитывают согласно закону Гука:
о — Ее; т = Gy. (1)
Для измерения упругих деформаций и напряжений применяют приборы, называемые тензометрами. Их подразделяют на механические, пневматические, оптические, электрические, комбинированные.
В механических тензометрах обычно используется рычажная нли зубчатая передача. Наиболее распространенные нз них — тензометр Гугенбергера и тензометр с индикатором часового типа [1]. Эти тензометры применяют для определения деформаций на большой базе, так как их передаточное число обычно равно 1 : 1000. Основное достоинство тензометров этого типа — простота устройства и обращения с ними.
Принцип работы пневматических тензометров заключается в том, что деформация базы тензометра передается на диафрагму, изменяющую площадь сечения для прохода воздуха. С изменением ее изменяется давление воздуха в пространстве между калиброванным отверстием, к которому подводится воздух под постоянным давлением, и диафрагмой. По изменению давления, определяемого уровнем жидкости в капилляре, судят о деформации, причем передаточное отношение пневматиче
ского тензометра составляет ~104. Использование этого тензометра, однако, требует тарировки, а линейный участок тарировочной кривой весьма ограничен.
Оптические тензометры снабжены микроскопами илн зеркальными устройствами. Самостоятельно микроскоп используется редко, однако в сочетании с приборами механического типа дает передаточное число от 2000 до 10000.
Из конструкций с зеркальными устройствами наиболее широко применяют тензометр Мартенса, подробно описанный в работах [1, 3]. В этом приборе, как н в других приборах такого типа, зеркало закреплено на подвижном контакте н поворачивается прн смещении контакта на угол, пропорциональный деформации. При этом смещается световой «зайчик», отражаемый зеркалом на достаточно удаленную шкалу. Обычно используемые оптические рычаги обеспечивают передаточное отношение порядка 500—1000; в комбинированных зеркально-механических тензометрах оно может быть увеличено.
В настоящее время широко применяются тензометры электрического типа (2]. Однако потенциометрические приборы для измерения больших деформаций, а также емкостные датчики, отличающиеся малым выходным сигналом, применяются в меньшей степени, поскольку они должны использоваться с усилителем и тщательной экранировкой всех частей прибора.
Приборы индукционного типа разных конструкций используют в углах, отверстиях и других труднодоступных местах деталей. Величина подводимого или индуцируемого в катушке соленоида тока может быть обусловлена ее положением относительно перемещаемого внутри сердечника, величиной воздушного зазора между сердечником и якорем, изменением магнитного потока, деформирующегося вокруг датчика в зависимости от положения диафрагмы из немагнитного металла, в котором возникают вихревые токи. Изменение индукции определяют мостовым методом нлн двухмостовым в случае дифференциального датчика. Такие датчики применяют для получения линейной зависимости выходящего тока от положения подвижного элемента датчика.
В фотоэлектрических приборах сочетаются механический и фотоэлектрический принципы. Сравнительно незначительная деформация на базе измерений механически увеличивается и передается для отклонения пластинки, закрывающей световой поток, направленный на фотоэлемент. При использовании высокочувствительных гальванометров, регистрирующих фототок, получают увеличение до 500000 раз. Специальные электронные лампы для непосредственного измерения деформации (сила анодного тока изменяется в зависимости от расстояния между электродами) имеют почти линейную характеристику при сдвоенном аноде и не требуют усилителя, что значительно упрощает нх эксплуатацию. Наиболее широкое распространение в настоящее время получили электрические тензометры сопротивления И, которые обладают достаточно линейной зависимостью электросопротивления от степени деформации, высокой тензочувстви-тельностью, малой длиной контакта с деталью или образцом н малой массой. Кроме того,
206
этн датчики можио использовать прн довольно высоких температурах. Датчики изготовляют из металлов и сплавов, а также полупроводников, поскольку у последних выше коэффициент тензочувствнтельностн К, определяемый из соотношения:
ДЯ/Яе = К. (13)
Для проволочного датчика можно записать: dot о
/С=14-2ИЧ-^-=1-|-2и + о. (14)
Даже в области упругих деформаций коэффициент теизочувствнтельностн меняется, так как удельное электросопротивление зависит от величины деформации. Для тензометров, применяемых для измерения деформаций вплоть до пластической области, важно, чтобы коэффициент v, связывающий приращение удельного электросопротивления с деформацией, был близок к единице.
В этом случае коэффициент тензочувствн-тельности примерно одинаков для упругой и пластической областей, что справедливо, например, для датчика из константана. Датчики электросопротивления выполняются в виде петель из тонкой проволоки, в виде решеток (получаемых травлением образца из тонкой фольги, наклеиваемой иа пластмассу), в виде прижимных датчиков многократного использования. Первые нз перечисленных тензодатчиков приводятся в контакт с испытуемой деталью путем приклеивания, что ведет к значительным затруднениям в их использовании — к необходимости тщательной подготовки поверхности и довольно длительному высыханию. Прижимные датчики укрепляют изолированно над базой, ограниченной с одного конца неподвижным двойным ножом, а с другого— простым подвижным. Для получения высокой точности ппедпочтительиее наклеиваемые тензодатчики. Помимо малой массы, большим достоинством электрических тензодатчиков является возможность использования их для исследования знакопеременных деформаций в динамических режимах.
12.2. ДИНАМИЧЕСКИЕ МЕТОДЫ ОПРЕДЕЛЕНИЯ
МОДУЛЕЙ УПРУГОСТИ
Преимущества динамических методов определения модулей упругости — нх более высокая точность по сравнению со статическими, а также гибкость методики, позволяющей проследить на одном и том же образце зависимость модулей упругости от различных факторов, в частности от температуры, без значительного силового воздействия.
Чтобы определять характеристики упругости динамическими методами, необходимо тем или иным способом установить скорость распространения упругих волн в материале. По методам определения этой скорости различают импульсные, резонансные и другие динамические методы [3]. При импульсных методах непосредственно измеряют время, в течение которого упругая волна проходит дю илн иное заданное расстояние. Скорость распространения упругой волны рассчитывают исходя из соотношения:
с —S/t, (15)
где S — путь, проходимый упругим возмущением за время т.
Резонансный метод основан на связи скорости распространения упругой волны с с ее длиной X и частотой собственных колебаний f образца:
с = А/. (16)
Значительно меньшее распространение по сравнению с рассмотренным получил метод, основанный на явлении преломления упругой волны прн переходе нз одной среды в другую: Cx/sin фх = c2/sin <р2, (17)
где Ci и с2 — скорости распространения волн в первой и во второй среде; ф{ — угол падения (в первой среде); ф2 — угол преломления (во второй среде).
В случае определения скорости распространения упругих воли этим методом необходимо точно знать скорость распространения упругой волны в одной нз сред.
Прн определении модулей упругости импульсными методами, независимо от того, используется ли прн этом «сквозное» прозвучи-вание илн локационный принцип, чаще всего возбуждают высокочастотные импульсы продольных илн поперечных колебаний с помощью пьезоэлектрических преобразователей. Импульсные методы широко применяются прн определении констант упругости монокристаллов и в дефектоскопии. Время прохождения импульсом заданного расстояния измеряют по развертке иа осциллографе, куда посылают сигналы датчик возбуждений и приемный датчик. Датчики имеют акустический контакт с образцом, что легко осуществимо прн температурах, близких к комнатной, но требует применения специальных переходников в случае экспериментов, проводимых прн повышенных температурах.
Резонансный метод определения модулей упругости широко распространен прн исследованиях температурных зависимостей модулей упругости полнкрнсталлических металлов. Собственную частоту колебаний измеряют обычно иа стержневых образцах постоянного сечення. Модуль упругости определяют как прн продольных, так и прн нзгнбных колебаниях. В случае продольных колебаний поперечные сечення стержня остаются плоскими, перпендикулярными его оси и смещаются вдоль осн стержня. Скорость распространения продольной упругой волны в стержне, поперечные размеры которого малы по сравнению с длиной волны А, связана с модулем упругости формулой
с = У~Ё/^, (18)
где р — плотность металла.
Наиболее низкие частоты собственных колебаний (первой формы) для консольно закрепленного стержневого образца А=4Z (на длине образца укладывается четверть длины волны), а для стержневого образца со свободными концами и закреплением в центральной точке А=2/ (полуволновые стержни).
Учитывая соотношение (16) можио записать для полуволнового стержня со свободными концами расчетную формулу модуля упругости:
Е = с2р = 4г2^р, (19)
где fi — собственней частота продольных колебаний стержня Длиной I.
Для более точного расчета значений модуля упругости необходимо введение поправки Рэлея, учитывающей дисперсию волны за счет поперечного смещения элементов стержня, сопровождающего волну растяжения — сжатия, идущую по образцу [4]. Поправку Рэлея можно вводить расчетным » н экспериментальным путем. С учетом поправки модуль упругости:
4 9 9 / р2 ц2 л2 d2\
(20>
где р — номер формы колебаний; ц —коэффициент Пуассона; d — диаметр стержня.
Экспериментальное введение поправки Рэлея целесообразно лишь для металлов и притом в диапазоне частот,' характеризующихся небольшим внутренним трением, н требует определения частот ие только первой формы колебаний, но и более высоких порядков. Определение собственных частот колебаний разных форм с одного установа образца позволяет изменять соотношение длины волны и диаметра образца. Далее экстраполяцией зависимости fip/p от р2 к нулевому значению можно определять собственную частоту колебаний с учетом поправки Рэлея. Для большей точности эксперимента необходимо измерять возможно большее число форм колебаний, проверяя при этом зависимость (fip/pf от (d/X)2, где flo— частота свободных колебаний стержня, полученная экстраполяцией зависимости fiF/p от р2 к р=0. Возможность экспериментального введения поправки Рэлея ограничена линейным участком этой зависимости.
При определении модуля упругости методом изгнбиых колебаний, помимо смещения поперечного сечения в плоскости изгиба, наблюдается его вращение вокруг осей, перпендикулярных этой плоскости.
При расчетах модуля упругости, не требующих большой точности, предполагается, что в процессе изгибных колебаний стержня поперечные сечения совершают лишь перемещения, перпендикулярные его оси, не участвуя во вращательном движении. В этом случае уравнение, связывающее частоту собственных колебаний с модулем упругости, имеет вид:
“ 2л/2
(21)
£=4р
где гя — радиус инерции поперечного сечения стержня относительно осн, лежащей в нейтральной плоскости и перпендикулярной осн стержня rI.=\/^J/S, где I—момент инерции сечения; S — площадь сечения; ai=xd— корни частотного уравнения изгибных колебаний стержня со свободными концами.
Первые четыре корня этого уравнения в случае гармонических колебаний соответственно равны: x0Z=O; xjZ=4,7300; x2Z=7,8532; %2l== = 10,9956.
Модуль упругости при этом рассчитывают по формуле
nl2fi \2
'«а1/
Более точные расчетные соотношения полу
(22)
чены с учетом инерции вращения и сдвига поперечных сечений стержня и приведены в работах [4].
Погрешность определения модуля методом изгибных колебаний по сравнению с методом продольных колебаний легко определить по формуле
2,757 ~~
100%,
(23)
где 2,757 — отношение квадратов корней х2Г Н X]/.
По мере увеличения l/d отношение стремится к 2,757, а погрешность определения модуля — к нулю. Прн практически удобных величинах Z/d=10-i-15 погрешность в определении модуля весьма заметна н находится в пределах 2—5 %.
Модуль сдвига (G) определяют методом крутильных колебаний, прн которых происходит движение поперечных сечений стержня вокруг его неподвижной осн. Полученные сечения прн этом остаются перпендикулярными осн стержня. Скорость распространения крутильных колебаний определяется формулой
= КG/p , (24)
причем в отличие от продольных колебаний эта скорость одинакова для безграничной среды и ограниченных тел определенной формы, т. е. дисперсии крутильных волн ие наблюдается.
Для цилиндрического стержня со свободными концами н закреплением в, центральной точке
G-4p/2/2.
(25)
Для вычисления модулей упругости и сдвига в мегапаскалях расчетные формулы принимают вид:
G = 0,4p*2/2, (26)
(MpZ2/,; (27)
Е = 0,1262 р (28)
В случае прямоугольного сечения
£ = 0,09455 p(z4f?//i2), (29)
где р — плотность, г/см3; Z — длина колеблющейся части образца, см; /—частота резонансных колебаний, кГц; d, h — поперечные размеры образца, см.
БИБЛИОГРАФИЧЕСКИЙ СПИСОК
1. Шапошников Н. Д. Основы механических испытаний металлов. М.: Машгиз, 1954. 443 с.
2. Рузга 3. Электрические тензометры сопротивления. М.: Мир, 1964. 355 с.
3. Испытания • материалов: Справочиик/Под ред. Блюменауэра X. М.: Металлургия,. 1979. 448 с.
4. Кузьменко В. А. Звуковые и ультразвуковые колебания прн динамических испытаниях материалов. Киев: Изд-во АН УССР, 1963. 152 с.
5. Тимошенко С. П. Колебания в инженерном деле. М_: Наука, 1967. 444 с.
13
ДИНАМИЧЕСКИЕ ИСПЫТАНИЯ МЕТАЛЛОВ
13.1. ПОНЯТИЕ «ДИНАМИЧЕСКИЕ ИСПЫТАНИЯ»
Динамический процесс деформации или разрушения может возникнуть как вследствие резкого возрастания внешней нагрузки, так и вследствие резкого понижения сопротивления тела, например при хрупком разрушении [1]. Поэтому, строго говоря, динамические испытания металла могли бы проводиться и при медленном, статическом приложении нагрузки. Однако подобный подход исключает возможность разделения испытаний на статические и динамические, так как это разделение оказывается зависящим от свойств материала, а ие от характера приложения нагрузки. Поэтому оговаривая, что это разделение условно, вод динамическими будем понимать испытания, прн которых скорость деформирования значительно больше, чем прн обычных статических механических испытаниях.
Современные разрывные машины позволяют перемещать захваты со скоростью до нескольких метров в секунду, поэтому граница между статическими и динамическими испытаниями становится еще более условной.
В проекте методических указаний Госстандарта СССР [2] динамическим испытанием принято считать нагружение при перемещении захватов "со скоростью более 10 мм/мин. При такой скорости нагружения на высокоскоростных машинах могут быть определены динамические свойства прн растяжении и сжатии и параметры динамической вязкости разрушения К®, аналогично тому, как это осуществляется прн статическом нагружении с малыми скоростями деформирования (ЛГ1с, Gjc) (см. раздел 15). Подобные испытания на высокоскоростных машинах имеют пока чисто исследовательское назначение из-за высокой стоимости оборудования, относительной сложности испытаний и недостаточной отработки методики.
Основным видом динамических испытаний, получившим широкое распространение, является ударное испытание надрезанных образцов на маятниковых копрах, главным образом на изгиб. Такие испытания проводят при начальной скорости удара 3—6 м/с. Перемещение ударяющего молота в процессе разрушения замедляется, причем тем в большей степени, чем ближе запас потенциальной энергии молота к величине работы, поглощенной и процессе разрушения образца. Между ударными испытаниями на копрах и динамическими испытаниями на высокоскоростных машинах имеется различие. Ударное испытание происходит при расходовании заданного запаса потенциальной энергии без подвода дополнительной энергии извне, из-за чего скорость деформирования в процессе деформации илн разрушения может существенно снизиться. При испытаниях на высокоскоростных машинах в процессе деформации н разрушения-об-
14—280
разца энергия подводится непрерывно, вследствие чего скорость перемещения захватов в течение всего испытания сохраняется постоянной.
13.2. НАЗНАЧЕНИЕ ДИНАМИЧЕСКИХ ИСПЫТАНИЙ
Ударный изгиб образцов с надрезом — самое простое и наименее трудоемкое нз всех механических испытаний. В то же время при испытании этого вида достаточно хорошо выявляются многие хрупкие структурные состояния металла, на которые приходится наибольшее число аварийных случаев разрушения в эксплуатации конструкции. Это было показано еще в 1901 г. Шарпи, впервые предложившим ударные испытания образцов с надрезом. Он разработал также конструкцию маятникового копра для этих испытаний, в основном сохранившуюся до настоящего времени. Благодаря простоте и малой трудоемкости ударные испытания образцов с надрезом получили широкое распространение в области металловедческих исследований и прн массовом контроле качества материалов. Ударные испытания применяются для определения критических температур хрупкости [3—6]. Минимально допустимые значения ударной вязкости включают в паспорта и технические условия на материалы. При этом обычно не учитывают, с какой скоростью данный материал нагружается в эксплуатации. Получаемая прн ударных испытаниях характеристика считается наиболее -консервативной и пригодной для оценки материала и прн статическом нагружении его в эксплуатации.
Во многих случаях такой подход оправдан, например в случае низколегированных, низкоуглеродистых сталей прн температурах в интервале хрупкости, однако, как будет показано ниже, это справедливо далеко не всегда.
Таким образом, первое и до настоящего времени основное назначение динамических (ударных) испытаний — простейшее н наименее трудоемкое определение опасной хрупкости металлов и сплавов для сравнительной оценки составов сплавов н технологических режимов.
Другое назначение динамических испытаний — определение механических свойств металлов и сплавов при повышенной скорости деформирования. Это часто необходимо для материала конструкций, испытывающих в эксплуатации нагружение с большой скоростью. Вероятно, наиболее целесообразно для этих делец, испытание на высокоскоростных машинах с постоянной в процессе испытания скоростью относительного перемещення захватов, причем не только образцов с надрезом на изгиб, но н образцов другой формы прн других способах иагруження.
Однако высокоскоростные машины еще ма-
20»
лодоступны для широкого использования. Поэтому и для второго назначения в ряде случаев также применяют испытание на маятниковых копрах.
Для сопоставления ударных испытаний со статическими либо используют работу разрушения образца для обоих видов нагружения, либо копер оборудуют снлоизмерительной аппаратурой; в этом последнем случае сопоставление более полное.
13.3. ВЫСОКОСКОРОСТНЫЕ МАШИНЫ
для ДИНАМИЧЕСКИХ ИСПЫТАНИЙ
Подобные машины выпускаются фирмами Шенк в ФРГ и МТС в США. Высокоскоростные машины фирмы Шенк типа гидронульс изготовляются с предельной нагрузкой от 40 До 200 кН и постоянной скоростью перемещения поршня, регулируемой в пределах от 0,005 до 10 м/с (последняя цифра, хотя и записана в проспекте, но не всегда достижима). Максимальная длина движения поршня с постоянной скоростью равна 100—250 мм. Схема работы машины показана на рнс. 13.1. Машина снабжена пьезокварцевым датчиком 1 для измерения нагрузки и индуктивным датчиком 2 для измерений перемещения поршня (см. рис. 13.1), работает по принципу обратной связи. Сигнал от датчиков нагрузки 1 и перемещения поршня 2 подается через усилители 3 и 4 на запоминающий осциллограф 5 и усилитель 6 для регулировки работы трехступенчатого серво-вентиля 7, управляющего через вспомогательные регуляторы 15 н 16 подачей масла нз помпы 8 в цилиндр пульсатора 13. Помпа приводится в действие электродвигателем 9 через пульты управления 10 и 11 н де
литель 12. Отсос масла из цилиндра производится дополнительной помпой 14.
Программа нагружения задается задатчиком 17. Для поддержания в процессе испытания постоянного давления к серво-вентилю подключены дополнительные аккумуляторы 18, находящиеся под давлением перед испытанием.
В процессе испытания запасенное в них давление используется для Дополнительной стабилизации скорости движения поршня.
Чтобы испытание проходило с постоянной скоростью, имеется устройство задержки во времени момента приложения нагрузки (рнс.
Рис. 13.2. Схема устройства задержки момента приложения нагрузки в машине Гидропульс фирмы Шенк:
1 — месдоза; 2 — дополнительный груз; 3 — поршень
210
13.2). До момента соприкосновения с тягой, соединенной с захватом, поршень проходит некоторый участок с и за это время приобретает заданную скорость, преодолевая инерцию. Для испытания материалов повышенной пластичности с целью большей стабильности скорости перемещения поршня рекомендуется к верхней части поршня прикреплять дополнительный груз (см. рис. 13.1), своей инерцией восполняющий потерн на пластическую деформацию.
13.4. КОПРЫ
ДЛЯ ДИНАМИЧЕСКИХ (УДАРНЫХ) ИСПЫТАНИЙ
Схема работы маятникового копра приведена на рис. 13.3. Маятник свободно качается иа шариковых подшипниках в точке О. Исходный угол подъема о. (точка А) может быть для данного копра постоянным или переменным.
Рис. 13.3. Схема работы маятникового копра
В последнем случае иа копре имеется храповик с несколькими зубцами и защелкой. Постоянный угол подъема имеет то преимущество, что все образцы испытываются с одной скоростью удара. Недостатком постоянного угла подъема является испытание в ряде случаев со значительной величиной остаточной энергии, что, по мнению Н. Н. Даввденкова [6], уменьшает точность испытания. В настоящее время большинство копров выпускают с постоянным начальным углом подъема маятника. Изменение запаса энергии достигается использованием молотов, разных по массе.
Полный запас потенциальной энергии равен высоте свободного взлета маятника от нижнего вертикального положения до точки Е иа угол Со, умноженной на его приведенный вес Р:
Дп — Р',
Ео — R + /? соз (180° — с$) —R-[-R (— cos а0)=
— Р — R cos а0 = Р (1 — cos а0).
Угол «о должен быть практически равен углу а, т. е. потери иа тренне в точке О должны быть невелики. Радиусом качания маятника Р называют расстояние от центра тяжести всего маятника, включая молот и штангу, соединяющую молот с осью качания, до осн качания в точке О.
Во избежание нагрузки на подшипники в точке О при ударе центр тяжести маятника должен совпадать с центром удара, т. е. с точкой соприкосновения молота с образцом. Правильность расположения центра тяжести, т. е. величина радиуса р контролируется определением полного периода качания f. Длина Р должна быть равна
Р = ^2, 0)
где g— ускорение свободного падения в дайной точке земного шара, где производятся испытания, которое может колебаться в пределах от 978 до 983 см/с2 [7] н прн расчетах принимается равным 981 см/с2; f — полный период качания, с; для определения f по секундомеру отсчитывают время Т полных 100 качаний маятника в пределах 10—15°; f = = 77100. Допускается лишь очень незначительное смещение центра удара (ие более Ю % Р) в сторону осн качания от р, полученного по формуле (1).
Приведенный к центру тяжести вес маятника Р определяют после проверки правильности расположения центра удара. Маятник устанавливают в горизонтальном положении В и через стержень, приложенный к центру удара (к ножу маятника), определяют весовое давление маятника на чашку весов. Вторая чашка уравновешивается гирями. Вес гирь и составит приведенный к центру удара вес маятника Р. Работа, израсходованная на деформацию и разрушение образца Дн, определяется разностью полной запасенной энергии Дп и остаточной энергии Ассч: Ап (илн К по ГОСТ 9454—67) == Ап—ДОст, где ДОст = ЕгР (см. рнс. 13.3).
ErP — (R — R cosебд) Р — PR (1 — cosat).
Отсюда
Дн = PR (1 — cos а0) — PR (1 — cos cq) —
= PR (cos Oj—cos a0) — PR (cos ax-J-cos §), (2) где = 180°—ag .
Конструктивно маятниковые копры изготовляют двух модификаций. В копре типа Шарпи (рис. 13.4) маятник передвигает стрелку циферблата, на котором нанесены угловые градусы, начиная с наибольшего угла подъема, нли вычисленные по формуле (2) значения работы разрушения образца. Стрелка посажена на продолжение осн качания маятника с некоторым трением, вследствие чего она останавливается на угле максимального взлета маятника прн данном испытании.
211
В копре Другой модификации ролик, соединенный с маятником (рис. 13.5), передвигает указатель вдоль вертикального стержня и показывает непосредственно высоту подъема маятника после разрушения образца. Шкала
Тогда части хрупко разрушившегося образца будут пролетать между опорами и ножом, ие задевая ни за опоры, ни за нож. В случае вязкого разрушения заклинивание может произойти из-за большой длины образца нлн
Рис. 13.4. Внешний вид копра типа Шарли
Рис. 13.5. Внешний вид копра типа Амслер
градуируется в кгс-м илн кгс-см от нуля сверху вниз.
По методу Изода (рис. 13.6) образец зажимают в тиски на уровне надреза и удар производят по свободному верхнему концу образца. Затраченную иа разрушение образца работу измеряют так же, как й на копре Шарпи.
При наиболее распространенном трехточечном изгибе для установки образца и проверки расстояния между опорами и центрального положения маятника служат шаблоны (рнс. 13.7). Обычно принятое расстояние между опорами 40 мм соответствует стандартной ширине (высоте) образца 10 мм. Во избежание заклинивания образца применяют специальные конструкции опор (рис. 13.8), которые препятствуют попаданию обломков образца между маятником и опорами. При хрупком разрушении половники образца могут оказаться между маятником и опорами (рис. 13.9) даже при использовании приспособлений, приведенных на рис. 13.8. Чтобы этого ие происходило, расстояние между опорами Llt толщина ножа маятника в месте удара е и ширина (высота) образца Н должны быть связаны неравенством: £1>2//+еЧ-2 мм.
большого угла заострения ножа молота. Зависимость между максимально допустимым углом заострения ножа <р и максимально допустимой длиной б лежащих на опорах концов образца (рис. 13.9, а) согласно [6J выражается формулой
К— (К sin <р 4- 2 cos ср)
° — 7Г~-----------' “ > (3)
2 sinip
где К = £(///; Lt — расстояние между опорами; Н — ширина (высота) образца. Прн Li = = 40 мм, А' = 10 мм, К = 4, при угле 40° величина б = 10 мм, т. е. образец не должен быть длиннее 60 мм. В основном следует выдерживать стандартную длину образца, равную 55 м прн угле <р не более 45°. Обычно его принимают равным 30—40°.
Копры указанных выше двух типов для испытания стандартных образцов на трехточечный изгиб изготовляют с различным предельным запасом энергии от 0,1 кгс-м (1 Дж) до 30 кгс-м (300 Дж).
В США получили распространение так называемые испытания падающим грузом (ИПГ)
212
(DWTT)1, которым подвергают образцы значительно большего сечеиия, чем стандартные, принятые у иас. Испытание этих образцов требует запаса энергии, значительно превышающего 300 Дж. Для этой цели ранее использовались вертикальные копры с запасом энергии до 1100 кДж, высотой до 12 м. В дальнейшем в США были сконструированы маятниковые копры с запасом энергии 8,7 кДж.
Рис. 13.6. Эскиз образца по Изоду и схема его испытания на копре Изода
В настоящее время японская фирма «Testing Machines MFGLTD» выпускает маятниковые копры с запасом рабочей энергии 490 кгс-м.
Маятниковые копры с большим запасом энергии имеют существенное преимущество перед вертикальными, хотя и дороже последних. Преимущества заключаются в большей точности измерений, значительно меньшем шуме прн испытаниях н большей надежности. Измерение затраченной работы в вертикальных копрах должно производиться или с помощью юсцнллографирования, что в достаточной мере сложно, илн методом осадки свинцовых крешеров, что существенно снижает точность испытания. t
Массовый контроль качества материалов с помощью ударных испытаний производят исключительно по величине работы, затраченной иа деформацию н разрушение образца. Однако уже давно предпринимались попытки получить прн ударных испытаниях величину максимальной разрушающей нагрузки и выявить характер ее изменения в процессе разрушения образца. Первые Опыты в этом направлении были проведены путем оптического измерения
1 Drop weight tear testing.
скорости маятника после разрушения образца. Этот принцип недавно был опробован [8] с использованием современных достижений электроники и ЭВМ.
Однако в большинстве работ используют прямой метод измерения давления ножа маятника на образец. Нагрузка записывается иа экране катодного (обычно двухлучевого) ос-
d’
Рис. 13.7. Шаблоны для испытания образцов на сосредоточенный изгиб на маятниковом копре:
а — шаблон для центрирования опор в 'отношении маятника {/—нож маятника; 2 — шаблон; 3— опора); б —шаблон для установки образца на опоры копра (/ — шаблон; 2 — образец; 3 — опоры; 4 — центрирующая призма на шаблоне)
Рис. 13.8. Устранение заклинивания образца изменением конструкции опор:
а. б — неправильно,- в — правильно
Рис. 13.9. Возможные случаи заклинивания образца при правильной конструкции опор:
а — при хрупком разрушении; б — правильный выбор угла копра маятника и длины образца; заклинивание в случае вязкого разрушения отсутствует; в — неправильный выбор угла ножа и длины образца; при вязком разрушении образец может заклиниться
циллографа в функции прогиба илн времени. Для получения осциллограммы на вход осциллографа подаются сигналы от датчика нагрузки и от датчика прогиба нлн времени. Иногда иа один луч подаются сигналы от датчика прогиба и нагрузки, а иа второй — сигнал от час
тотного генератора, который развертывается на экране синхронно с основной диаграммой и служит таким образом меткой времени.
Датчики нагрузки представляют собой пьезокварцевые пластинки или упругие элементы с наклеенными на них тензорезнстора-мн [9].
Датчиком прогиба обычно служит луч, падающий на фотоэлемент н постепенно перекрываемый шторкой, движущейся вместе с маятником. Датчиком иремени может служить частотный генератор. Чем ближе к месту удара маятника по образцу находится датчик нагрузки, тем точнее данные о нагрузке. Исходя из этого, располагать датчик на ноже маятника целесообразнее, чем на одной из опор, хотя это конструктивно менее удобно, так как в первом случае необходимо электрически связывать движущийся маятник с осциллографом, что значительно труднее, чем при расположении датчика иа неподвижной опоре.
В ряде случаев маятниковые копры оборудуют приспособлением для испытания на ударное растяжение (рис. 13.10). Упоры 3, при-
Рис. 13.10. Приспособление для испытания на растяжение на маятниковом копре
крепленные к опорам копра 2, воспринимают удар перекладины 4, в которую ввинчен одни конец образца 5. Другой конец образца ввинчен в тело маятника 1. Падающий маятник проходит между упорами 3, а поперечина ими останавливается. Образец, увлекаемый маятником, подвергается растяжению его инерционным усилием. Работа, затраченная на растяжение образца, определяется по углу взлета маятника (остаточной энергии) после разрушения образца так же, как прн изгибе.
Для того чтобы полностью устранить нагрузку на станину в некоторых случаях применяют двухмаятниковые копры. В этих копрах на одном маятнике помещают опоры, а на другом иож и прн точном размещении центра удара станина копра ие несет практически никакой нагрузки.
Для исследования поведения материала при скоростях, превышающих 5 м/с, применяют ротационные копры. Такой допер выпускала фирма «Veb Werkstoflpriifmaschinen» (ГДР). С его помощью можно испытывать образцы при скорости удара до 50 м/с. Максимальный запас энергии копра 300 кДж. Копер оборудован осциллографическим устройством для получения диаграмм нагрузка — прогиб.
13.5. ОБРАЗЦЫ
ДЛЯ ОБЫЧНЫХ УДАРНЫХ ИСПЫТАНИЙ
Как уже указывалось, в результате испытаний образцов с надрезом на маятниковых копрах определяют нлн всю работу (К по ГОСТ 9454-—78 илн Лн по ГОСТ 9454—60), затраченную на деформацию и разрушение образца данного типа или чаще удельную работу КС=К/50(ан=Дв/50), где <S0— площадь поперечного сечення образца в месте надреза до испытания. При осцнллографированнн диаграммы нагрузка—прогиб величина К (или Лн) выражается площадью под диаграммой (рис. 13.11). Различные материалы (/—4) могут иметь одинаковую полную (и удельную) рабо-
Позтому в большинстве стран за рубежом надрез Менаже с радиусом гн = 1 мм заменен V-образным надрезом с радиусом га = 0,25 мм н углом раскрытия 45°. Этот надрез был введен н в ГОСТ СССР 9454—60 (тип IV), но практического применения не получил.
Причиной этому послужило то, что образец типа IV не был признан основным, а его изготовление с жесткими допусками по радиусу надреза естественно значительно сложнее, чем образца типа I.
Еще большее приближение Ав к Лр может быть получено применением усталостной трещины вместо надреза. Подобное предложение было сделано в СССР в 1955—1959 гг. [12], а с 1961 г. [13] образцы с усталостной трещиной начали находить применение н за рубежом.
Определение работы разрушения образца 10X10 мм с усталостной трещиной позволяет
Ац~А3+Ар Ар
Прогиб
Рис. 13.11. Четыре разновидности диаграмм нагрузка — прогиб для четырех различных материалов (1, 2, 3 и 4) при одинаковой величине поглощенной энергии (схема)
ту, но различную форму диаграммы нагрузка— прогиб. Эта форма зависит от величины максимальной нагрузки и от соотношения между работой, затрачиваемой иа зарождение трещины Ая (в основном на упругое и пластическое деформирование образца в основании надреза данной остроты), и работой, затрачиваемой на разрушение Лр — развитие магистральной трещины н сопутствующее ему пластическое деформирование объемов металла, прилегающих к берегам трещины1.
Материалы 1 и 2 нлн 3 и 4 могут обладать одинаковой полной работой К (ЛЕ) прн разных значениях нагрузки и прогиба. В то же время материалы /, 3 и 2, 4 могут характеризоваться различными соотношениями между Ав и Ар при одинаковой величине Ав (К) и одинаковой максимальной нагрузке.
Величина АР, отражающая способность материала тормозить начавшееся разрушение, в меньшей степени зависит от остроты надреза, чем величина А3, и в большей степени характеризует надежность материала в случае возникновения в нем острой коррозионной, сварочной и особенно усталостной трещины.
Деление Ав на А3 и АР связано с рядом методических трудностей, что лишает ударное испытание основного его преимущества — простоты. Чем острее исходный надрез, тем меньше величина А3 и тем меньше величина Ав отличается от Ар.
* Такое разделение при статическом изгибе было введено в работе [10], а при ударном—в работе [11].
непосредственно оценить степень возможности хрупкого разрушения материала в деталях толщиной до 10 мм прн возннкновенин в ннх усталостной трещины.
Рис. 13.12. Эскиз образца с концентратором тина U
Кроме того, это испытание часто может изменять порядок расположения материалов по сравнению с испытанием образцов типа 1—10 (ГОСТ 9454—78) н в их качественном ряду, и сравнительная оценка ближе к оценке надежности материала по его способности тормозить начавшееся разрушение прн эксплуатации.
Действующий в настоящее время ГОСТ 9454—78 (взамен ГОСТ 9454—60, 9455—60 н 9456—60) предусматривает образцы с кон-
215
ТАБЛИЦА 13.1
СТАНДАРТНЫЕ ОБРАЗЦЫ НА УДАРНЫЙ ИЗГИБ ПО ГОСТ 9454—78 (размеры в мм)
Тип образца Длина, L Ширина, В Высота, Н Глубина надреза, h* Глубина концентратора, h Высота рабочего сечения.
Концентратор U радиусом 1±0,07
I 55 10+0,10 10 8+0,10
2 55 7,5+0,10 10 — — 8+0,10
3 55 5+0,05 10 — — 8+0,10
4 55 2+0,05 8 —. 6+0,10
5 55 10+0,10 10 - — * — 7+0,10
6 55 7,5+0,10 10 — -— 7+0,10
7 55 5+0,05 10 «- • — 7+0,10
8 55 10+0,10 10 — — 5+0,10
9 55 7,5+0,10 10 — — 5+0,10
10 55 5+0,05 10 — — 5+0,10
Концентратор \ т радиусом 0, 25 ±0,025
11 55 10+0,10 10 ,— . 8+0,05
12 55 7,5+0,10 10 —- — 8+0,05
13 55 5+0,05 10 — — 8+0,05
14 55 2+0,05 8 — — 6+0,05
Концентра тор Т (трещи на)
15 55 10+0,10 И 1,5 3,0 —
16 55 7,5+0,10 11 1,5 3,0 —
17 55 5+0,05 И 1,5 3,0
18 55 2+0,05 9 1,5 3,0 —
19 55 10+0,10 10 3,5 5,0 .—
20 140 25+0,10 25 10 12,5
* Предельные отклонения по длине ±0,6 мм, по высоте ±0,1, по глубине надреза ±0,1, по глубине концентратора ±0,6.
центраторами трех видов: U-образным с гн — = I мм, V-образным с ги = 0,25 и углом 45°, Т — усталостной трещиной.
Соотношения ширины, высоты образца н глубины надреза на образцах различных видов даны в табл. 13.1 и вис. 13.12—13.14.
Кроме приведенных в табл. 13.2 образцов стандартных типов, допускается использование образцов без надреза н с одной нлн двумя необработанными поверхностями, размеры
которых отличаются от указанных в таблице. Риски на поверхности надрезов видов U и V, видимые глазом, не допускаются.
Усталостные трещины, (концентратор вида Г) получают иа специальных вибраторах нли других усталостных машинах, допускающих повторный изгиб без вращения. Усталостный цикл может быть как знакопостоянным, так н
Рис. 13.14. Эскиз образца с концентратором типа Т (у сталостн ая трещин а):
а — общий вид; б — форма концентратора для образцов типов 15—19; в — форма концентратора дли образцов типа 20
216
Рис. 13.15. Внешний вид вибратора для получения трещин при консольном изгибе (асимметричный знакопостоянный и знакопеременный цикл)
Рис. 13.16. Внешний вид установки УТО-1 для получения трещин при трехточечном изгибе (асимметричный знакопостоянный цикл) (Ивановский ЗИП)
знакопеременным. Во втором случае можно получить трещины прн том же максимальном напряжении, но за меньШее число циклов. В случае знакопостоянного цикла целесообразно применять коэффициент асимметрии цикла не более 0,2. Изменение напряжения цикла и чнсла циклов до-получения усталостной трещины в широких пределах в большинстве случаен ие сказывается на величине работы разрушения. Тем ие менее во избежание чрезмерной пластической деформации в вершине трещины н возможности разрушения об
разца число циклов при получении трещины глубиной 1,5 мм должно быть ие менее 3000 н остаточный прогиб ие должен превышать 0,25 мм для образцов длиной 55 мм н 0,5 мм для образцов длиной 140 мм.
В настоящее время для получения усталостных трещин при ’ консольном изгибе и знакопеременном асимметричном цикле имеются чертежи вибратора, разработанного в ВИАМе (рнс. 13.15). Этот вибратор работает без измерения нагрузки цикла. Для получения трещин прн трехточечном изгибе н знакопостоян
217
ном цикле Ивановским заводом испытательных приборов выпускается установка для нанесения усталостных трещин на образцах модель УТО-1 — рнс. 13.16 [14]. В последней значительно проще установка образца (образцы не закрепляются болтом, а вкладываются между роликами), но время для получения трещин обычно несколько больше, чем на вибраторе ВИАМ, поскольку для достижения того же числа циклов, как прн знакопеременном цикле, следует увеличивать статическую подгрузку, а это иногда может привести к разрушению образца.
13.6. ОБРАЗЦЫ ДЛЯ ИСПЫТАНИЯ ПАДАЮЩИМ ГРУЗОМ (ИНГ) (DWTT) И ВЗРЫВОМ
На копрах большой мощности — иертикаль-ных до 1080 кДж или маятниковых до 10 кДж — в США производятся так называемые испытания падающим грузом (ИПГ) (илн DWTT). Эти испытания известны также под названием Dinamic Tear (DT), т. е. испытания на динамический изгиб (ИДИ).
Рис. 13.17. Эскиз образца для ИПГ (с) и схема (б) определения доли волокнистости в изломе (% В). В центре толщины штриховкой показан кристаллический участок излома
Метод применяют главным образом для оценки качества листов толщиной 3,2—19,1 мм из сталей ферритного класса, идущих на изготовление труб.
Испытание стандартизовано ASTM, стандарт Е436—74 [15]. Используется образец длиной 305 мм и шириной (высотой) 76 мм. Толщина образца может быть различной н обычно равна толщине листа, подлежащего испытанию.
Испытания проводят в интервале температур от —73 до 100 °C. Результатом испытания является определение степени волокнистости в изломе (%).
При анализе излома учитывают только его центральную (по ширине) часть. Участки излома на длине, равной толщине образца от осиоваиня надреза и от грани, противоположной надрезу, не учитывают (рис. 13.17). В начале введения этих испытаний для инициирования трещины применяли хрупкую наплавку. Затем был предложен прессованный надрез с
углом раскрытия 45° и радиусом в вершине надреза га = 0,25 1до°з26 мм- В исследованиях (например, [16, с. 199]) была установлена идентичность результатов прн этих двух способах инициирования трещины. Однако ASTM в 1974 г. был регламентирован прессованный надрез как более простой в изготовлении.
В исследовательских работах и при построении так называемых диаграмм анализа отношения Л1с/оо,2 н Ан/оо,2 при ИПГ определяют также и работу разрушения.
Однако в стандарте ASTM принято только определение доли волокна (в процентах) в изломе.
Кроме ИПГ, для оценки вязкости трубиой стали применяют также динамическое испытание взрывом. Испытание проводят вдавливанием путем взрыва квадратных заготовок, вырезанных из листа, в кольцевую матрицу. Для листов толщиной 16 мм применяют, например, квадраты размером 500X500 мм с насечкой нлн хрупкой наплавкой в их центре [16, с. 199].
13.7. ИСПЫТАНИЕ
СТАНДАРТНЫХ
ОБРАЗЦОВ С НАДРЕЗОМ
И ТРЕЩИНОЙ НА ИЗГИБ
13.7.1. Испытание на маятниковых копрах по ГОСТ 9454—78
Испытание проводят прн нормальной (20 ± ±10°C), повышенной (до 1000°C) или пониженной (до—100 °C) температуре.
Для охлаждения или нагрева образцов служат криостаты или печи. Охлаждение в кри остате производится смесью жидкого азота илн твердой углекислоты с не замерзающей прн температуре испытания нетоксичной жидкостью (этиловый спирт). Доля кислорода в жидком азоте ие должна превышать 10 % (по массе). Специальные испытания прн температуре ниже —100 ®С выполняют по ГОСТ 22848—77.
Для контроля температуры прн охлаждении используют термометры с погрешностью не более ±0,5%; для измерения температуры нагрева до 600 °C — термопары с погрешностью ±5 °C, а выше 600 °C — с погрешностью ±8 °C.
Для обеспечения требуемой температуры испытания образец должен быть заранее переохлажден нли перегрет. Интервал от момента извлечения образца из трностата или печн до момента удара допускается ие более 3—5 с
Прн выполнении этого условия величина перегрева (°C) может быть установлена по табл. 13.2. Перед началом испытания проверяют положение стрелки (копер типа Шарли) илн вертикального указателя (копер типа Амслер). При свободном паденнн маятника указатель должен показывать нуль с отклонением не более чем на 0,5 % от максимальной энергии копра. Образец должен свободно лежать на опорах и вплотную прилегать к задней вертикальной стенке гранью с надрезом. Образец устанавливают с помощью шаблона.
В случае испытаний при температуре, отличной от комнатной, допускается использование торцового ограничителя, который не должен
218
ТАБЛИЦА 13.2
ТЕМПЕРАТУРА ПЕРЕОХЛАЖДЕНИЯ И ПЕРЕГРЕВА
Температура испытания, °C Температура, °С
(—100) — (—60) 4—6*
(-60)-(40) 3-4*
(—40) — (+10) 2—3*
(+30)-(+200) 3—5
(+200) — (+400) 5—10
(+400) — (+500) 10—15
(+500) — (+600) 15—20
(+600) — (+700) 20—25
(+700) — (+800) 25—30
(+800) — (+900) 30—40
(+900) — (+1000) 40-50
* Температура переохлаждения.
мешать образцам свободно деформироваться. Полную работу разрушения образца (илн энергии, затраченной иа его разрушение) прн ударе К (ЛЕ) определяют непосредственно по шкале копра или по таблице (если шкала градуирована и градусах). Удельная работа КС (сн), («т.у) определяется делением полной работы на площадь сечення (нетто):
КС (аи) = К! Sq (Ак1 So),
где Sq—HiB (cm. табл. 13.1).
Измерение Hi в образцах с концентраторами U и V производят до испытания измерительным инструментом с погрешностью не более 0,05 мм.
Измерение Hi в образцах с трещиной (концентратор Т) производят по рис. 13.18 по из-
Рис. 13.18. Схема определения высоты сечения нетто образца с трещиной (Hi). Площадь, заштрихованная поперечной штриховкой, должна быть равновелика двум площадям с односторонней штриховкой:
1— надрез; 2 — усталостная трещина; 3—начальное положение визирной линии инструментального микроскопа; 4 — конечное положение визирной линии при измерении
лому после разрушения образца. Величину В для всех концентраторов измеряли до испытания.
13.7.2. Испытание образцов с трещиной на статический изгиб
Повышенная склонность к хрупкому разрушению образцов с трещиной в некоторых случаях проявляется не при ударном, а при стати
ческом нагружении. Поэтому для получения полной характеристики материала следует проводить параллельные испытания образцов с трещиной и на ударный, и на статический изгиб. Лучший способ для этого — испытание на высокоскоростных машинах с использованием и динамической (2—5 м/с), и статической (1—10 мм/мин) скорости перемещения захватов. Однако статические испытания можно проводить и иа обычных машинах при относительной скорости перемещения копра и опор в интервале 1—10 мм/мин. Рекомендуется использовать машины с податливостью не более 0,06 мм/кН. Машина должна давать возможность получать диаграмму нагрузка— прогиб с масштабом по оси деформации не менее 50:1 (желательно 100:1). Полная работа разрушения Ат. с равна площади под диаграммой нагрузка—прогиб в соответствующем масштабе. Удельная работа ат. с = = /1т. c/Sc. Величину So определяют так же, как и иа образцах, испытанных на удар.
13.8. МЕТОДИЧЕСКИЕ ВОПРОСЫ ДИНАМИЧЕСКИХ ИСПЫТАНИЙ
13.8.1. Способы оценки хладноломкости при динамических испытаниях
Низколегированные, низкоуглероднстые стали, содержащие в структуре избыточный феррит, обладают значительной хладноломкостью, т. е. их вязкость резко уменьшается прн понижении температуры. Вместе с тем эти стали широко применяются в трубопроводах, дорожных машинах, реакторах и других конструкциях, температура эксплуатации которых может снижаться до минус 50 — минус 70 °C. Поэтому определение критической температуры хладноломкости этих сталей очень важная задача.
Наиболее простой метод определения этой температуры — сериальные испытания на ударный изгиб надрезанных образцов прн различных температурах.
Прн массовых ударных испытаниях без ос-цнллографировання можно получить три характеристики:
1) работу, затраченную на разрушение образца (Ан, К);
2) долю волокнистости в изломе В (в процентах) ;
3) относительное утолщение (или сужение) поперечного сечения образца в месте разрушения; (Во—St) мм нли ДВ=(В2—Bi)/2, нли [(В2—Bl)/В2] 100 % (рнс. 13.19).
Последняя характеристика используется относительно редко.
Между работой, затраченной иа разрушение, и деформационной характеристикой часто наблюдается хорошая корреляция [17].
Типичная кривая зависимости волокнистости излома В и KCU(an) от температуры испытания для низкоуглеродистой стали с 0,59 % Ni приведена на рис. 13.20 [18]. Верхний порог хладноломкости более четко выявляется по величине В (%).
Следует отметить, что четкое определение В возможно только в случае низколегированных сталей. Характеристики К(ЛН) и В можно нс-
219
пользовать для оценки критической температуры. Критической считают температуру получения некоторой доли от максимального значения К£7(АН) илн В для данного материала; обычно эта доля составляет 50 %. При оцен-
й
Рис. 13.19. Схема определения деформационной характеристики при испытании образца с надрезом или трещиной
~80 -W 0 fyO 60 Температура испытания^С
Рис. 13.20. Сериальные диаграммы ударной вязкости образцов с надрезом гн = 1 мм (а) и доля волокна (%) н изломе (б). Низкоуглеродистая сталь с
ке только по величине KCU(as) иногда принимают некоторую минимальную величину этой характеристики, обычно 250 илн 300 кДж/м2. Кроме этих характеристик, отмечают верхний и инжннй пороги хладноломкости. Первый при оценке по доле волокнистости (в процентах) обычно соответствует температуре появления 10 % кристалличности в изломе, а прн оценке по величине КД(Лк) или KCU(пн) — температуре испытания, отвечающей максимальной величине этих характеристик. Ннжннй порог соответствует температуре достижения 90 % кристалличности в изломе и некоторой условно принятой минимальной величине KCU (ан).
Прн испытании взрывом определяют три характерные температуры:
1) температуру полной пластичности (ТПП), т. е. наименьшую температуру, прн которой трещина прн большой пластической деформации, только раскрывается, но не развивается;
2) температуру хрупкого разрушения (Тхр), т. е. начало хрупкого разрушения при пластической деформации ~5 %; трещина распространяется, но останавливается в области упругого сжатия;
3) температуру нулевой пластичности (ТИП), т. е. максимальную температуру, прн которой трещина пересекает весь образец при пренебрежимо малой пластической деформации.
Последнюю температуру определяют также прн ИПГ: она соответствует максимальной температуре, при которой разрушение происходит практически без изменения формы сечения образца.
13.8.2. Сопоставление испытаний образцов сечением 10X10 мм с V-образным надрезом и ИПГ
Образцы сечением ЮХЮ мм, даже с трещиной, из низколегированных ннзкоуглеродистых сталей при 20 °C обычно недостаточно чувствительны для выявления опасной хрупкости материала. В то же время прн эксплуатации деталей значительно большего сечения эта хрупкость может привести к серьезным авариям.
С целью приближения ударных испытаний к работе конструктивных элементов при эксплуатации в США были предложены описанные выше образцы для испытаний падающим грузом (ИПГ). Как видно нз табл. 13.3 [16, с. 199], критические температуры, определенные по виду излома (КТВИ) (50 % волокна в изломе) и по энергетическому критерию (КТЭ) (50 % работы разрушения от максимальной величины этой работы разрушения), прн ИПГ практически одинаковы.
В случае образцов сечением ЮХЮ мм КТВИ существенно ниже (иа 14—19), чем определенные прн ИПГ.
Ниже [16, с. 242] показано различие в энергии, затрачиваемой прн ИПГ образцов толщиной 25 мм и прн испытании стандартных образцов сечением ЮХЮ мм открытой и вакуумной плавок мартеиснтно-стареющей стали с пределом текучести 1300 МПа, Дж;
Открытая Вакуумная плавка плавка
Испытания стандартных ' образцов . . . ИПГ................
40 61
883 6082
ТАБЛИЦА 13.3
СОПОСТАВЛЕНИЕ КРИТИЧЕСКИХ ТЕМПЕРАТУР ХРУПКОСТИ ПРИ ИПГ И ПРИ ИСПЫТАНИИ ОБРАЗЦОВ 10X10 мм. НИЗКОУГЛЕРОДИСТАЯ
СТАЛЬ Х52 ТОЛЩИНОЙ 16 мм [16, с. 3171
Образцы Оценка температуры Полуфабрикат Критическая температура, °C
Для ИПГ толщи- КТВИ Лист 18
иой 16 мм Труба 27
ктэ Лист 21
Труба 27
ЮХЮ мм с V-об- ктви Лист 4
разным надрезом (тип 11 по ГОСТ 9454—78) Труба 8
220
На рис. 13.21 [16, с. 242] приведены результаты сериальных испытаний образцов, вырезанных из сосуда, хрупко разрушившегося прн 9 °C. В случае стандартных образцов 10X10 мм температура —18 °C соответствует полностью волокнистому излому, а в случае
2438
Сосуд-образец
ц 100 -
Л ^200 -
i
S №С -
^газсуыемм । сосуда 19ЛС)~
2000=
тип
- 2000
-140 -100 -60 -20 0 20
МДИ 1601L Н.=16мн)
i
12000 Т
10000'~~ §
8000 Й
- 6000 ?
- 4000
Теииеоа/лур/Азрллп гкитаетро<р\ v 16000
Температура г °C
Рис. 13.21. Сопоставление сериальных кривых по испытаниям образцов Шарли 10X10 мм на динамический изгиб (ИДИ, ИПГ) с температурой разрушения сосуда и температурой остановки трещины по Робертсону при напряжении 0.5Oq 2(точка А на диаграмме). Сталь А541 (о0 2=600 МПа)
ИДИ ( С=25мм)
Испытания по Шарпа
хрупкость стали, чем испытания стандартных образцов сечением 10ХЮ мм с V-образным надрезом.
С помощью ИПГ можно перейти к весьма приближенным, но все же количественным оценкам материала прн различных температурах; с большей степенью точности это, однако, достигается определением параметра вязкости разрушения Kic (см. раздел 15).
Для приближенной оценки влияния размера дефектов при различных температурах -и напряжениях служит так называемая диаграмма анализа разрушения (ДАР) или, как ее чаще называют, диаграмма Пеллнин (рис. 13.22) [16, с. 229]. Ннжняя кривая характеризует зависимость напряжений от температуры, при которой движущаяся трещина останавливается, т. е. материал в этих условиях способен противостоять развитию дефектов любого размера.
Последующие кривые соответствуют различным размерам дефектов прн напряжениях, равных данной доле от предела текучести. Горизонтальные участки кривых отвечают полностью хрупкому разрушению. Крайние правые точки этих участков соответствуют ТНП при ИПГ, а температура получения 50 % волокнистости примерно соответствует TXJ>.
Таким образом, имея эти две температуры, определенные экспериментально по данным ИПГ, можно сделать заключение, что при температуре, равной или меиьшей ТНП, если номинальные напряжения достигнут оо,2, даже малая трещина способна вызвать разрушение. Прн
игви, волокнистость 50°/>
Температура
Рис. 13.22. Диаграмма анализа разрушения (диаграмма Пеллини)
ИПГ при толщине образцов 25 мм — температуре нулевой пластичности. Температура этого хрупкого разрушения (9 °C) совпадает с точкой А на кривой прн ИПГ, находящейся по температуре несколько ниже, чем половина максимальной энергии по ИПГ.
Из приведенных примеров видно, что ИПГ в ряде случаев лучше выявляют опасную
Ухи даже трещины большой длины не-вызовут разрушения, если напряжение ниже предела текучести.
Необходимо, конечно, отметить, что диаграмма анализа разрушения (ДАР) основана только иа величине предела текучести и в разных материалах, ио с одинаковым пределом текучести согласно ДАР имеющаяся трещина долж
221
на одинаково тормозиться. Это далеко не бесспорно для широкого диапазона материалов. Однако для ограниченного круга материалов, для которых установлена связь параметров, показанная иа ДАР, испытания падающим грузом дают возможность получить ориентировочную зависимость между допустимым размером дефектов, номинальным напряжением н температурой эксплуатации. При этом необходимо проводить ИПГ на образцах, близких
Рис. 13.23. Влияние толщины образцов из стали А514 на величину затраченной энергии (штриховые линии) и вид излома (сплошные линии) при ИПГ
по толщине конструктивным элементам, для которых описывается допустимый размер дефектов.
Как видно из рнс. 13.23 [16, с. 229], изменение толщины образца из низколегированной низкоуглероднстой стали А514 от 6,4 до 25,4 мм приводит к повышению КТВИ прн ИПГ с —90 до —40 °C. По энергетическому критерию [50 % К МЕ)тах] повышение значительно меньше.
13.8.3. Точность определения нагрузки при ударных испытаниях с осциллографированием и определение динамической вязкости разрушения
Определение работы, поглощенной прн ударном испытании, планиметрированием осциллограмм нагрузка—прогиб и непосредственное ее измерение по отклонению маятника дают близкие результаты (рис. 13.24) [19]. Однако это не доказывает, что нагрузка при осцнллографиро-ванин измерена достаточно точно. Прн хрупком разрушении, т. е. прн малых значениях прогиба, даже прн существенном различии в максимальной нагрузке могут быть получены близкие значения работы, поглощенной при испытании .образцов. В то же время основным назначением измерения нагрузки при ударных испытаниях является определение параметра вязкости разрушения при динамическом нагружении Кр. Для определения этой характеристики необходимо существенно ограничить пластическую деформацию у вершины трещины, т. е. в
наибольшей степени приблизить разрушение к хрупкому. При хрупком разрушении, продолжающемся единицы и десятки микросекунд, возможны большие неточности в измерении нагрузки. Эти неточности тем больше, чем ближе период свободных колебаний упругого датчика
Рис. 13.24. Сравнение энергии, измеренной по стрелке маятника, с энергией, подсчитанной по площади осциллограммы при испытании образцов сечением
10X10 мм с V-образным надрезом
Рис. 13.25. Кривые ударного импульса при различных собственных частотах датчика:
сплошные линии — истинные кривые нагрузки; штриховые— реакция датчика
силоизмернтеля и всей нагружающей системы к продолжительности протекания процесса разрушения (рнс. 13.25) [20].
В работе [16, с. 175] показано, что прн малой жесткости датчиков нагрузки и большой
222
жесткости образцов могут быть существенные ошибки в измерении нагрузки прн хрупком разрушении. Этн ошибки связаны с влиянием инерционной нагрузки, которая возрастает с уменьшением времени до разрушения.
В работе [16, с. 208] для трехточечного изгиба стальных образцов сечением брутто 10Х Х10 мм с трещиной предложен поправочный множитель У, являющийся функцией времени, необходимого для достижения максимальной нагрузки при различных отношениях длины трещины к ширине (высоте) образца (рис. 13.26). По мнению автора [16, с. 100], умноже-
Рис. 13.26. Инерционная поправка при трехточечном изгибе 4). По оси абсцисс — время для достижения максимальной нагрузки для стального образца сечением 10x10 мм с V-образным надрезом
ннем полученной на осциллограмме нагрузки от датчика иа ноже маятника на этот поправочный множитель достигается уменьшение усилия вследствие завышения инерционной нагрузки. Однако этот поправочный множитель не учитывает соотношения упругих коистаит датчика и образца.
В работе [16, с. 100] дано более полное решение для инерционной поправки, приводящее к весьма сложной формуле н требующее ряда дополнительных экспериментальных данных. Вопрос о введении поправки осложняется еще тем, что при малом времени до разрушения на осциллограмме наблюдается ряд пиков, поскольку период собственных свободных колебаний системы становится соизмеримым с временем до разрушения. Уменьшение скорости нагружения в результате уменьшения высоты падения маятника обеспечивает получение нормальной осциллограммы (рис. 13.27).
Помимо недостаточной точности и относительной сложности измерения нагрузки, при определении вязкости разрушения с помощью ударного нагружения возникает вопрос об обоснованности использования малых образцов, применяемых на обычных копрах. Ойо может быть использовано иа высокопрочных материалах прн 20 °C, для которых толщина 10 мм уже достаточна, чтобы получить истинное значение /Clc.
В случае более пластичных материалов в работах [19] предлагается испытывать образцы сечением 10X10 мм с усталостной трещиной и боковыми надрезами.
Однако автор [16, с. 100] высказывает серьезные сомнения в том, что боковые надрезы адекватно компенсируют малый размер образца.
Определение вязкости разрушения прн ударном нагружении иа стандартных образцах се-
рпе. 13.27. Сопоставление осциллограмм при различной скорости нагружения (А. М. Воробьев, Б. А. Дроздовский):
а — 336 м/мин; б — 57 м/мин; в — 26 м/мни
чением 10x10 мм даже при точном измерении нагрузки имеет ограниченное применение — для высокопрочных материалов (стали с ов> >1800 МПа), титановых сплавов (ов> >1200 МПа), алюминиевых сплавов (ав> >600 МПа). Применение боковых надрезов вводит большую неопределенность, поскольку увеличение трехосности напряженного состояния за счет боковых надрезов зависит от глубины, остроты и угла раскрытия последних, а степень этой зависимости изменяется с изменением локальной пластичности материала.
Наиболее целесообразным является определение К® при виецентренном растяжении иа высокоскоростных машинах. Подобные испытания стандартных компактных образцов нз низколегированной низкоуглеродистой стали А302В проводились, например, в работе [16, с. 257].
223.
В ней сопоставлены результаты динамического испытания с многочисленными литературными данными по статическому определению Kic для этой же стали А302В в разных состояниях,
В среднем прн динамическом нагружении со скоростью 254 мм/с получены иа 25 % более низкие значения К&, чем прн статическом со скоростью <0,17 мм/с. Динамические значения предела текучести, определенные также при скорости 254 мм/с, были на 27 % выше стати-ческих.
13.9. ВЛИЯНИЕ СКОРОСТИ НАГРУЖЕНИЯ НА СВОЙСТВА МАТЕРИАЛОВ И ВИДЫ ХРУПКОСТИ, ВЫЯВЛЯЕМЫЕ ПРИ ИЗГИБЕ ОБРАЗЦОВ
С НАДРЕЗОМ
13.9.1. Влияние скорости нагружения иа свойства гладких образцов при растяжении
Повышение скорости нагружения приводит к увеличению сопротивления пластической деформации [9]. На рнс. 13.28 [1] показана зависн-
скорость меняется незначительно, то
°ти = 2/С + Зри, (5)
где р — коэффициент, равный для сталей 0,2— 0,4 при скоростях порядка 103 с-1 [1].
Н. А. Шапошников [17] прн испытании сталей с временным сопротивлением 700— 950 МПа показал, что при переходе от статического к динамическому нагружению сопротивление пластической деформации возрастает в 1,1—1,5 раза прн практически неизменной величине относительного сужения (ф).
Пределы прочности и особенно текучести титановых сплавов резко возрастают при увеличении скорости нагружения.
Поэтому скорость деформирования при нспы танин титановых сплавов на растяжение строго регламентируется в пределах !—10 мм/мии.
13.9.2. Влияние скорости нагружения на свойства образцов с надрезом и трещиной при изгибе
Для большинства металлов и сплавов, не обладающих склонностью к ударному охрупчиванию, работа разрушения образцов с надрезом или с трещиной прн переходе от статического изгиба к ударному несколько повышается, очевидно, вследствие увеличения предела текучести.
Рис. 13.28. Зависимость временного сопротивления ов от скорости деформации (данные обработаны Ф. Ф. Витманом иН А. Златиным):
а — алюминий; б — медь; в — низкоуглеродистая сталь; I — при температуре 200 °C; 2 — 400 °C; 3 — 600 °C; 4 — 800 °C; 5 — 1000 °C; 6 — 1200 °C
мость временного сопротивления от скорости деформации для алюминия, меди и ннзкоугле-роднстой стали прн различны, температурах. С повышением скорости деформации прн данной температуре временное сопротивление растет, с повышением температуры при данной скорости нагружения — уменьшается, причем тем в меньшей степени, чем больше скорость деформации.
Н. Н. Давнденкои [3] для предела текучести предложил формулу
= стт0 Д- к [1g (v/v0)]n, (4)
где Civ н ст — пределы текучести при скорости v и прн минимальной скорости и0; К и п — константы материала. Если (и—t'c) <(п0), т. е.
Повышенная чувствительность сталей ферритного класса к ударному нагружению в интервале переходных температур общеизвестна. Даже отношение а?. с/ат. у = 1 свидетельствует о некотором ударном охрупчивании. Увеличение скорости нагружения для ударно-хрупких материалов действует в том же направлении, что и поинженне температуры испытаний. Известно также гндрндное охрупчивание титановых сх-сплавов, где величина Дт.с/пт.у= 15-
Результаты испытаний стали Д6АС состава 0,48 % С; 0,22 % Si; 1,01 % Мп; 1,23 % Сг; 0,55 % Ni; 1,93 % Си и 0,06 % V; лист толщиной 3 мм (табл. 13.4) [16, с. 123] после закалки без отпуска и с отпуском 316 °C типичны для большинства улучшаемых н высокопрочных сталей (ат.с/Ят.у^0,8).
224
В случае отпуска при 208 °C происходит существенное охрупчивание при статическом изгибе (ат.с/ат.у=0,43). Это уменьшение сопровождается охрупчиванием излома (30 % волокна при статическом изгибе).
ТАБЛИЦА 13.4
СОПОСТАВЛЕНИЕ РАБОТЫ РАЗРУШЕНИЯ И КОЛИЧЕСТВА ВОЛОКНА В ИЗЛОМЕ ОБРАЗЦОВ С ТРЕЩИНАМИ ПРИ СТАТИЧЕСКОМ И УДАРНОМ ИЗГИБЕ*
Режим отпуска** 1 е" Ву,% О Й с о дт-с “т.у
Без отпуска 7,9 80 6,6 98 0,83
209 °C, 4 ч 7,8 60 3,3 30 0,43
316 °C, 1 ч 7,8 80 6,3 70 0,81
* а т у. а г с — работа разрушения соответственно при ударном и статическом изгибе; By, Ве — количество волокна в изломе при тех же условиях.
** Закалка при 927 °C на воздухе.
Еще в большей степени охрупчивание при статическом изгибе наблюдается прн (см-0)-титаиовых сплавов после закалки и старения, где величина От.сМт.у—0,18 [21].
13.9.3. Виды хрупкости, наиболее отчетливо выявляемые при испытании на изгиб образцов с надрезом
При испытании на изгиб образцов с надрезом выявляются следующие виды хрупкости: хладноломкость; синеломкость, илн тепловая хрупкость; обратимая и необратимая хрупкость, обусловленная структурным состоянием.
Необратимая отпускная хрупкость так же, как и обратимая, отчетливо выявляется в основном при ударном испытании [22]. Увеличение остроты надреза в результате использования образцов с трещиной повышает чувствительность выявления этой хрупкости [23].
В иеохрупченном состоянии работа разрушения при ударном изгибе большинства сред'не-и высоколегированных улучшаемых и высокопрочных сталей несколько выше, чем при статическом.
Существенное понижение работы разрушения при статическом изгибе по сравнению с ударным было также показано выше на примерах стали Д6АС и (а+Р)-титановых сплавов. В этих случаях статической хрупкости работа разрушения прн статическом изгибе может быть в 10—15 раз ниже, чем при ударном. По.-этому универсальную характеристику чувствительности данного материала к трещине можно получить при параллельном испытании и на ударный, и иа статический изгиб.
БИБЛИОГРАФИЧЕСКИЙ СПИСОК
1. Фридман Я. Б. Механические свойства металлов: Ч. 2. М_: Машиностроение, 1974. 368 с.
2. Расчеты и испытания на прочность в машиностроении. Методы испытания металлов. Определение характеристик разрушения при динамическом нагруженнн/Госстандарт СССР, ВНИИМаш, Институт машиноведения АН СССР. М.: 178. 32 с.
3. Давиденков Н. Н. Проблема удара в металловедении. М.—Л.: Изд-во АН СССР, 1938.116 с.
4. Шевандин Е. М., Разве И. А. Хладноломкость и предельная пластичность металлов в судостроении. Л.: Судостроение, 1965. 336* с.
5. Шевандин Е. М. Склонность к хрупкости низколегированных сталей. М.: Металлург-издат, 1958, 182 с.
6. Давиденков Н. И. Динамические испытания металлов. Л.—М.: ОНТИ НКТП СССР, Главная редакция по черной металлургии, 1936. 395 с.
7. Шокин П. Ф. Гравиметрия. М.: Изд-во геодезической литературы, I960. 312 с.
8 Маркочев В. М., Житенев В. В., Воробьев А. М.—Заводская лаборатория, 1979, № 10, с. 944—949.
9. Волошенко-Климовицкий Ю. Я- Динамический предел текучести. М.: Наука, 1965. 179 с.
10. Дроздовский Б. А. — Заводская лаборатория, 1946, № 4—5, с. 489—499.
11. Otani Z. — Tetsudo Gidzuku Капку Syre, 1957, 14, № 11, р. 503.
12. Дроздовский Б. А., Фридман Я. Б. — Заводская лаборатория, 1955, № 5, с. 579— 590; 1959, № 3, с. 320—328.
13. Or пег G. М., Hartbower С. Е. — Weld J., v. 40, № 9, 1961, р. 405S—421S.
14. Беликов В; С., Дроздовский Б. А., Смуш-кович Б. А., Можаев А. В. — Заводская лаборатория, 1979, № 3, с. 272—274.
15 Annual Book of ASTM Standards, 1974, E-436—74, Part 10, p. 425—457.
16. Ударные испытания металлов. M.: Мир, 1974. 317 с. '
17. Шапошников Н. А. Механические испытания металлов. М.—Л.: Машгнз, 1951. 383 с.
18. Георгиев М. Н. Вязкость малоуглеродистых сталей. М.: Металлургия, 1973, 234 с.
19. Serwer W. L., Tetelman A. S. — Eng. Fract. Meeh., 1972, № 4, p. 361—375.
20. Инженерные методы исследования ударных процессов. М.: Машиностроение, 1977 239 с.
21. Проходцева Л. В., Дроздовский Б. А. — Заводская . абораторня, 1975» № 9, с. 1122— 1127. сч
22. Садовский В. Д., Бородина Н. А.— Проблемы конструкционной стали. Кн. 12. М—Л.: Машгнз, 1949, с. 102—119.
23. Дроздовский Б. А., Фридман Я. Б. Влияние трещин на механические свойства конструкционной стали. М.: Металлургиздат, I960. 260 с.
225
14
"" .. ЦИКЛИЧЕСКИЕ ИСПЫТАНИЯ
МЕХАНИЧЕСКИХ СВОЙСТВ
Циклические испытания механических свойств материалов имеют важное значение, поскольку большинство деталей машин, транспортных и других конструкций в процессе службы претерпевают (воздействие переменных нагрузок. Под действием циклически изменяющихся переменных напряжений (деформаций) происходит процесс постепенного накопления повреждений, приводящий к критической степени искажения решетки в отдельных объемах (зернах) вследствие протекания циклической микропластической деформации, к созданию локальных пиковых напряжений, могущих вызвать разрыв межатомных связей, к образованию зародышевых усталостных трещин, их развитию и, .наконец, к разрушению.
Сопротивление усталости — свойство металла противостоять усталости. Совокупность последовательных значений переменных напряжений за одни период их ‘изменения составляет цикл напряжений. Число смей циклов напряжений в единицу времени дает представление о частоте нагружения.
Усталостные изломы имеют характерные признаки, отличающие их ст изломов другого рода (рнс. 14.1). Фокус излома — малая ло-
Рис. ИЛ. Схема усталостного излома вала."
1 — зона доломи: 2 — участок ускоренного развития трещины; 3—5— зона собственно усталостного развития (3 — зона избирательного развития; 4 — очаг разрушения; 5— фокус излома); 6 — ступеньки и рубцы; 7 — пасынковые трещины и вторичные ступеньки и рубцы, 8 — усталостные линии нли полосы; 9 — рубцы; JO —скос
кальная зона, близкая к точке, в которой возникает зародышевая (начальная) макроскопическая трещина усталости н откуда начинается ее развитие. Очаг разрушения — малая зона, прилегающая к фокусу излома и соответствующая объему, в котором располагалась начальная макроскопическая трещина усталости. Очаг разрушения характеризуется наибольшим блеском и наиболее гладкой поверхностью. Линии усталости на поверхности очага разрушения обычно отсутствуют. В зоне избирательного развитая трещины от очага разрушения, как из центра, расходятся линии усталости — следы фронта продвижения трещины. При больших напряжениях (или при
наличии протяженных по периметру концентраций напряжений) возникает несколько очагов разрушения. Прн слиянии нескольких трещин, развивающихся в параллельных плоскостях, на изломе образуются ступеньки и рубцы. Зона долома образуется на последней стадии разрушения н обладает признаками макрохрупкого разрушения.
Анализ усталостного излома занимает важное место прн установлении причин поломок деталей в эксплуатации. По усталостным полосам можно выявить очаг разрушения и проанализировать роль конструкционных, технологических н эксплуатационных факторов, определивших разрушение. Наряду с макрофрактографией широко применяется электронная фрактография (для определения механизма распространения усталостных трещин на различных стадиях их развития)1.
Опасность всякой трещины заключается в том, что она при определенных условиях иа-груження и длине, превышающей критическую, обусловливает возможность внезапного хрупкого разрушения.
Прн циклических испытаниях на усталость определяют следующие характеристики: предел выносливости, усталостную долговечность, чувствительность к концентрации напряжений, влиянию среды, температуры, частоты, асимметрии цикла и величины среднего напряжения цикла, к перегрузкам, масштабному фактору. Кроме того, оценивают степень повреж-денности металла при воздействии циклических нагрузок, скорость роста трещин, длительность инкубационного периода до появления трещины и длительность периода живучести.
По результатам испытаний на усталость производится:
— построение кривой усталости и определение предела выносливости;
— построение диаграмм предельных напряжений (Сешх—Ощ) и предельных амплитуд (сго—о,п) с учетом различных коэффициентов асимметрии цикла (/? = Omin/oinax);
— определение предела выносливости для заданного уровня вероятности разрушения.
Кривую распределения предела выносливости строят, исходя из кривых распределения долговечности иа вероятностной сетке, по осн абсцисс которой откладывают значения пределов выносливости, по оси ординат:
— вероятности в масштабе, соответствующем нормальному илн другому закону распределения;
— построение кривой усталости равной вероятности разрушения, характеризующей зависимость между максимальными или амплитуд^ нымн значениями напряжений цикла и долговечностью образцов, соответствующей заданной вероятности;
— определение среднего значения и среднего квадратического отклонения логарифма дол-
1 См. раздел 3.
226
РАЗМЕРЫ, мм, ГЛАДКИХ ОБРАЗЦОВ
Тип образца Размеры
I rf 5,0 7,5 10 12 15 20 25
R=5d 25 37,5 50 60 65 >90
II d 5,0 7,5 10 12 15 20 25
l~od 25 37,5 50 60 75 100 125
R 5,0 7,5 10 12 15 20 25
Ш,а ft <3,0 >3—5 5 7,5 10
(изгиб в плоско- b 10ft 5ft 10 15 20
сти размера ft) R 20h 10ft 20 30 40
III, б ft=3,04-20,0; ft—0,5ft-? 2ft; 7?>6ft
(изгиб в плоскости
размера Ь)
IV ft <0,3 >3,0—5,0 5,0 7,5 10
b 10ft 5ft 10 15 20
Z=5,65Ktft 17,9 12,6 40 60 80
/? 10ft 5ft 10 15 20
говечности на заданном уровне напряжений илн деформаций;
— определение среднего значения н среднего квадратического отклонения предела выносливости;
— построение кривой усталости в малоцик-ловой упруго-пластнческой области;
— построение диаграмм упруго-пластического деформирования и определение их параметров.
Основными критериями прн определении -предела выносливости и построении кривых усталости являются полное разрушение или появление трещин, протяженность которых по поверхности составляет 0,5—1,0 мм. Дополнительными критериями могут бытьг. резкое па
дение нагрузки илн частоты циклов, значительный рост деформации, резкий подъем температуры, а также характеристики, обусловленные возникновением и развитием усталостных трещин, обнаруживаемые электрическими, магнитными, токовихревыми, акустическими (ультразвук, акустическая эмиссия) и другими методами. В пределах одной серин испытаний критерии разрушения должны быть одинаковыми.
Испытания на усталость выполняют прн следующих схемах нагружения-, чистый изгиб при вращении; то же, в одной плоскости; поперечный изгиб прн вращении; то же, в одной плоскости; растяжение — сжатие; переменное кручение; внутреннее давление. Кроме того, проводят испытания прн комбинированных видах
кольцевой v-образной выточкой
ТАБЛИЦА 14.2
РАЗМЕРЫ КРУГЛЫХ ОБРАЗЦОВ ТИПА V С
D d Изгиб Растяжение—сжатие Кручение
MM p> MM to. град a о L/G p« MM 1 ®. ; град a a L/C P, MM (0, град a T L/G
10 5,0 2,5 2,5 2,00 80 1,33 11,17 2,00 80 1,48 15,67 2,00 80 1.17 17,50
12 7,5 3,75 2,25 1,09 70 1,68 11,17 — — — 15,67 — —— 17,50
15 7,5 3,75 3,75 1,09 70 1,75 11,7 1,33 70 1,95 15,67 0,92 70 1,45 17,50
17 7,5 3,75 4,75 1,09 70 1,75 11,17 —- — — 15,67 -— -— — 17,50
20 10 5,0 4,0 0,78 65 2,20 11,17 1,00 65 2,45 15,67 0,62 60 1.71 17,50
24 12 6,0 6,0 0,61 60 2,63 11,17 0,83 60 2,89 15,67 0,50 65 1,94 17,50
10 5,0 2,5 2,5 1,00 70 1,58 6,53 1,00 70 1,87 7,87 0,50 60 1,52 6,57
12 7,5 3,75 2,25 0,60 65 2,04 6,53 — — .— 7,87 - 1 —— — 6,57
15 7,5 3,75 3,75 0,60 65 2,18 6,53 0,87 65 2,60 7,87 0,30 60 1,96 16,57
17 7,5 3,75 4,75 0,60 65 2,18 6,53 —-—- — — 7,87 * — —»>-» 6,57
20 10 5,0 5,0 0,43 60 2,80 6,53 0,50 60 3,35 7,87 0,22 50 2,40 6,57
24 12 6,0 6,0 0,36 55 3,30 6,53 0,42 55 3,99 7,87 0,18 45 2,77 6,57
10 5,0 2,5 2,5 0,50 65 1,99 3,56 0,50 65 2,45 3,92 — — — —
12 7,5 3,75 2,25 0,32 60 2,58 3,56 — — — 3,92 • ° — — —•
15 7,5 3,75 3,75 0,32 60 2,83 3,56 0,33 60 3,58 3,92 — — —M
17 7,5 3,75 4,75 0,32 60 2,83 3,56 — — —- 3,92 — — — —,
20 10 5,0 5,0 0,23 50 3,73 3,56 0,25 50 4,65 3,92 — —- — —
24 12 6,0 6,0 0,19 45 4,42 3,56 0,21 45 5,55 3,92 — — — -—
15*
227
TunU
ТипШ^
Рис. 14.2. Рабочие части образцов для испытания циклической нагрузкой на усталость
нагружений (изгиб с растяжением нли кручением, растяжение с кручением и т. д.) н при контактных напряжениях. Наряду с испытаниями иа обычную многоцикловую усталость в ряде случаев выполняют испытания иа малоцикловую, высокочастотную, ударную, термическую, термо-механическую и коррозионномеханическую усталость.
Для испытаний на механическую усталость используют машины, установки и стенды с различным возбуждением переменных нагрузок или деформаций; гидравлическим, электро-гидравлическим, электродинамическим, пру
жинным, центробежными вибраторами, кривошипным, пневматическим, акустическим, компрессорным, а также путем подвески грузов. По механизму создания переменных нагрузок установки подразделяют на работающие в вынужденном и в автоколебательном режимах.
По ГОСТ 23026—78 испытания на усталость проводят на образцах круглого и прямоугольного сеченнй, гладких и с надрезом. Рабочие части образцов показаны иа рис. 14.2, а их размеры приведены в табл. 14.1—14.7. Образцы типов I—IV — гладкие, типов V—VIII — с надрезом. Круглые образцы изготавливают с
228
ТАБЛИЦА 14.6
ТАБЛИЦА 14.3
РАЗМЕРЫ ДОПОЛНИТЕЛЬНЫХ КРУГЛЫХ ОБРАЗЦОВ ТИПА V С КОЛЬЦЕВОЙ
V-ОБРАЗНОИ ВЫТОЧКОЙ
Й d а t Р а о при S а о
мм со, град , растяжении-сжатии изгибе при кр)
10 5,0 2,5 2,5 0,5 0,25 65 50 2,45 3,35 1,99 2,63 1,52 1,83
12 7,5 3,75 2,25 0,5 0,25 65 50 2,28 2,83 —
15 7,5 3,75 3,75 0,5 0,26 60 45 2,93 4,04 2,33 3,14 1,68 2,08
17 7,5 3,75 4,75 0,5 0,25 60 45 — 2,33 3,14 -—
20 10 5,0 5,0 0,5 0,27 50 40 3,35 4,65 2,63 3,56 1,83 2,30
30 15 7,5 7,5 0,5 45 4,05 3,14 2,08
V-образными н полукруглыми выточками (типы V н VIII). Плоские образцы изготовляют с симметричными V-образными боковыми надрезами (тип VI), с центральным поперечным отверстием (тип VII) н с двумя боковыми от* ТАБЛИЦА 14.4
РАЗМЕРЫ ОБРАЗЦОВ ТИПА VI С СИММЕТРИЧНЫМИ БОКОВЫМИ V-ОБРАЗНЫМИ НАДРЕЗАМИ
И | h | Ь [ а | t | р
мм
10 5,0 10 2,50 2,50 0,50 0,25 65 60 2,18 2,90 2,94 4,07
15 7,5 15 3,75 3,75 0,50 0,25 60 45 2,57 3,48 3,55 4,98
20 10 20 5,00 5,00 0,50 0,25 50 40 2,90 3,95 4,07 5,73
40 20 <10 10,0 10,0 0,5 40 — 5,73
верстиями типа «замочной скважины» (тип IX).
Образцы с надрезами используют главным образом для оценки чувствительности металла ТАБЛИЦА 14.5
РАЗМЕРЫ ОБРАЗЦОВ ТИПА VII С ЦЕНТРАЛЬНЫМ ПОПЕРЕЧНЫМ ОТВЕРСТИЕМ
h ъ d CXo при
ММ изгибе растяжении— сжатии
<3,0 3,0—5,0 W/i 5h b/10 b/10 2,08 2,28 2,73 2,73
РАЗМЕРЫ ОБРАЗЦОВ ТИПА VIII С КОЛЬЦЕВОЙ ВЫТОЧКОЙ
D а г i при ft.
мм изгибе растяжении-сжатии а при т чеини
6,00 5,00 0,50 0,50 1,89 2,18 1,46
9,00 7,50 0,75 0,75 1,89 2,18 1,46
12,00 10,0 1,00 1,00 1,89 2,18 1,46
к концентрации напряжений, а также для оценки влияния абсолютных размеров. Размеры образцов выбирают таким образом, чтобы параметр подобия усталостного разрушения L/G варьировался в возможно более широких пределах при заданном диапазоне изменения диаметров. Величина L — периметр рабочего сечения образца илн его часть, прилегающая к зоне повышенной напряженности. Для образцов типов I, П, V и VIII при изгибе с вращением, кручеинн и растяжении — сжатии £= =nd; для образцов типов III, IV и VI прн изгибе в одной плоскости, а также для образцов типа VI прн растяжении — сжатии L=2b; для образцов типов III, IV, VII и IX прн растяжении — сжатии L~2h.
Величина G— относительный градиент, равный G==G/Omax, где G=do/dx«Mg0; Отах — максимальное напряжение в зоне концентрации.
Рабочая часть образцов должна быть изготовлена не ниже III класса точности. Параметр шероховатости поверхности рабочей части образцов должен быть 0,32—0,16 мкм по ГОСТ 2789—73. '
При испытании образцов допускается мягкое (заданной иеличиной является размах нагрузки) и жесткое (заданной величиной является кинематически ограниченное перемещение) нагружения. Относительная влажность н температура окружающей среды при обычных испытаниях должны соответствовать ГОСТ 15150—69 (ддя исполнения Б и категории 4.2). В пределах от 10 до 300 Гц частота циклов не регламентируется.
Для построения кривой усталости и определения предела выносливости испытывают не менее 10—15 одинаковых образцов. База «испытания для определения предела выносливости принимается: 10-106 циклов для металлов и сплавов, имеющих практически горизонтальный участок на кривой усталости, н 100- 10е для металлов и сплавов, не имеющих такого участка. Для сравнительных .испытаний база соответственно принимается 5-Ю6 и 20-Ю6 циклов.
ТАБЛИЦА 14.7
РАЗМЕРЫ ОБРАЗЦОВ ТИПА IX С ДВУМЯ ОТВЕРСТИЯМИ ТИПА «ЗАМОЧНОЙ СКВАЖИНЫ»
В 1 1 <• 1 р
мм ап
40 10 . <10 3,0 1,5 2,44 3,15
229
Кривые усталости строят в полулогарифмических координатах (Отах—Ig к ИЛИ Оа—lg N) или в двойных логарифмических координатах (1g Стах—Ig N ИЛН Ig Oo— lg N).
Для построения семейства кривых усталости по параметру вероятиостн разрушения, а также кривой распределения предела выносливости и для оценки среднего значения н среднего квадратичного отклонения предела выносливости на 4—6 уровнях напряжений испытывают серии не менее чем из 10 одинаковых образцов. Для построения кривой распределения долговечности н оценки среднего значения и среднего квадратичного отклонения логарифма долговечности на заданном уровне напря-.жений испытывают серию не менее чем из 10 образцов до полного разрушения или до образования макротрещнн. Результаты испытаний подвергают статистической обработке.
Диаграммы предельных напряжений н предельных амплитуд строят с помощью семейства кривых усталости по результатам испытания не менее 3—4 серий одинаковых образцов при разных для каждой серии средних напряжениях или коэффициентах асимметрии цикла напряжений (рис. 14.3).
Рис 1-1.3. Диаграммы предельных напряжений (а) и пре цельных амплитуд, (б)
Из методов ускоренной оценки предела выносливости, основанных на использовании малого числа образцов, корреляционных зависимостей, характеристик упругости, а также косвенных и безобразцовых методов, следует выделить метод ступенчатого нагружения.
Метод ступенчатого нагружения по Локатн (ГОСТ 19533—74) предназначается для ориентировочной оценки пределов выносливости образцов и изделий машиностроения нз металлов и сплавов, кривые усталости которых имеют горизонтальный участок, т. е. разность пределов выносливости на базах 107 и 10е не превышает точности нх оценки. Метод не может быть применен для ускоренной оценки предела выносливости образцов и изделий при испытании на ударную, контактную и термическую усталость.- Предел выносливости определяют при ступенчатом увеличении нагрузки, используя не менее трех образцов (для усреднения полученных оценок). По результатам испытаний по ГОСТ 19533—74 подсчитывают сумму относительных долговечностей 2(щ/М), где значения долговечностей принимают из семейства предположительных кривых усталости, выбранных нз имеющихся экспериментальных данных. Образец или деталь нагружают начальным напряжением о0 и испытывают в течение циклов. Далее без пауз напряжение увеличивают на Ао до Gi н продолжают испытания прн этом уровне напряжений в тече
ние щ~по циклов, затем снова увеличивают напряжение и т. д. до разрушения образца. Число циклов при последнем уровне напряжения определяется разрушением образца.
Малоцикловая усталость—усталость металла, при которой образование макротрещии или полное разрушение происходит при повторнопластическом деформировании с разрушающим числом циклов до 5-104. При этом имеется в виду малоцикловая низкочастотная усталость. Область же высокочастотной малоцикловой усталости — это область циклических перенапряжений. При малоцикловой усталости в отличие от многоцикловой поглощаемая энергия уже с первых циклов нагружения расходуется на разрушение.
Малоцикловое нагружение сопровождается развитием общей нли местной (в вершине надреза или трещины) пластической деформации, величина и закономерности накопления которой определяют условия перехода к предельному состоянию и контролируют характер разрушения (квазистатйческий, усталостный). Переход к усталостному многоцикловому разрушению (число циклов прн высокой частоте нагружения) сопровождается резким падением интенсивности предельной пластической деформации.
Для испытания на малоцикловую усталость применяют образцы типов II и IV; допускается также использование образцов типов I и III, а также трубчатых образцов и образцов корсетного типа. При испытании на растяжение— сжатие образцов типов II и IV деформацию следует измерять в продольном направлении. При испытании образцов типов I и III допускается измерять деформацию в поперечном направлении.
Для пересчета поперечной деформации в продольную используют формулу Znp= =4(/у)поп+2(1г)поп, где (Л) поп— упругая составляющая поперечной деформации; (/п)поп — пластическая составляющая поперечной деформации.
Оценка циклической трещиностойкости. Анализ результатов циклических испытаний должен проводиться с учетом двустадийности процесса усталостного разрушения. Процессы возникновения трещин н нх развития подчиняются различным закономерностям. Распространение усталостной трещины илн период живучести может охватывать от 10 до 90 % общей долговечности образца или детали. Усталостные трещины, возникающие прн циклических нагрузках, постепенно разрастаясь, подготавливают условия для хрупкого разрушения. Скорость роста усталостных трещин— важная характеристика материала.
На иее оказывают влияние условия нагружения: уровень приложенных напряжений, асимметрия н частота цикла, характер напря-женного состояния образца, величина средних напряжений цикла, перегрузки и недогрузки; большое значение имеют конструктивные факторы и внешняя среда, в которой происходит испытание.
Процесс роста усталостной трещины делится на три периода. В первом периоде трещина развивается вдоль систем скольжения, находящихся в зоне действия максимальных касательных напряжений (рис. 14.4). При этом длина и скорость роста трещины невелики, в изломе отсутствуют бороздки. Во втором
230
периоде трещина растет нормально к приложенным напряжениям. Это период установившегося роста трещины, ее скорость пропорциональна размаху коэффициента интенсивности напряжении. Третий заключительным период— катастрофическое развитие трещины, которое заканчивается разрушением.
Рис. 14.4. Схеме развития трещин усталости:
1 — начальная стадия; 2 — магистральная трещина
ТАБЛИЦА 14.8
РАЗМЕРЫ, мм. ОБРАЗЦОВ ДЛЯ ОЦЕНКИ ЦИКЛИНЕСКОЙ ТРЕЩИНОСТОИКОСТИ
Тип образца ь н Ъл 11 D
X 100 400 300 33
150 600 400 40
200 800 —, 600 40
XI 80 320 .— 240 24
100 460 — 300 33
150 600 — 450 45
XII 50 60 62,5 27,5 12,5
100 120 125 55 25
150 180 167,5 82,5 37,5
200 240 250 ПО 50
XIII 60 ' " — — —-
100 .— — — —
150 — — —
200 — — — —
Для практики наибольший интерес представляет оценка скорости роста трещин на второй стадии ее развития.
Методы анализа усталостного разрушения и фиксации длины трещины в зависимости от принципов, положенных в их основу, подразделяются иа три группы:
1) методы непосредственного наблюдения — визуальный, волоконная оптика, оптический, фотоупругие покрытия, цветовые и др.;
2) физические методы — магнитный, ультразвуковой, акустический, вихретоковый, тензометрический й др.;
3) методы, основанные иа фиксации изменения свойств материала — электрического сопротивления, деформационные, внутреннего трення, магнитные и др.
Для испытания на циклическую трещнно-стойкость используют плоские образцы (рис. 14.5), размеры которых приведены в табл. 14.8.
Тип образца выбирают в зависимости от толщины материала;
Тип образца . х X
Толщина /, мм <5
Ширина образ-
ца Ь, мм . . . 100—200
XI- XII, хш XIV 5—20 >10 >10
80—150 —
Рекомендуется, чтобы толщина образца была равна толщине изделия, поскольку этот параметр оказывает существенное влияние иа скорость роста трещины. Наиболее широко применяют образец типа XI шириной 100 мм, вырезанный таким образом, чтобы трещина развивалась параллельно направлению прокатки (поперечные образцы). Для предупреждения разрушения образцов типов X и XI в зоне отверстий используют усиливающие накладки (иа штифтах нли приварке), а также делают корсетность в сечении надреза или углубляют сам надрез. Образец типа XIII испытывают иа опорах диаметром, равным ширине образца, при расстоянии между опорами, равном четырехкратной ширине образца. Прирост трещины обычно начинают измерять пос-
Продолжение
Тип образца Е а 2а L
X — — 10 — —
—— -— 15 —— •
— — 20 — —
XI — — 24 —
—— — 33 — —
— 45 .— —-
хи 2,5 20 — — —
5 40 — — —-
7,5 би —- —.
Ю 80 «— —
XIII — 70 — 2U0 225
•—- 20 400 450
— 30 — 600 685
— 40 — 800 900
ле образования у конца надреза усталостной трещины длиной 0,5 t (t — толщина образца в миллиметрах).
Скорость роста трещин представляют в зависимости от парметров цикла нагружения, выраженных через коэффициент интенсивности напряжений Д, отмечая, при каком коэффициенте асимметрии проводятся испытания.
Коэффициенты интенсивности напряжений Д устанавливают для разных типов образцов по формулам:
Тип X: K = YlPlQ'5/tb, где Гх = 1,77 + 0,227 (2//6) — — 0,51 (2//&2) + 2,7 (Z/6)3, Тип XI: Л = У2Р(0’5/», где 1,99 —0,41 (//&) + 18,7 (Z/&)2 — — 38,48 (1/Ь)3 + 53,85 (Z/&)a.
Тип XII; Д=У3Р/°’5/Й,
231
Тип ЕЕ
Рис. 14.Б. Образцы для оценки циклической тр ещииостойкости
где Уз = 29,6 — 185 (//6) + 655,7 (Z/*)2 — — 1017 (Z/6P +638,9 (//&)*.
Тип XIII: К = У4 Pp'5/lb,
где Уа = 1,93 —3,07 (//6) + 14,63 (7/й)2-
— 25,11 (//6)3+25,8(//й)4.
Графики зависимости Y-f(llb) для указанных типов образцов приведены на рис. 14.6. Образен типа XIV в виде круглой пластинки-диска с центральной сквозной трещиной (см. рнс. 14.5) отличается тем, что величина К не зависит от длины трещины в некотором диапазоне ее измерения — до 2(12—А) и определяется только величиной нагрузки. Размеры дискового образца: D>150 мм; /=1-е20 мм; 2а= (0,16-^0,20)D; а>2/; d<0,9c;
Диаграмму циклической треш иностой кости строят в двойных логарифмических координатах. Средний участок этой диаграммы описывается уравнением Пэриса1 у—С(ДА)П, где С и п— эмпирические постоянные для исследуемого материала (см. рис. 14.7).
Введение промежуточных пороговых значений АА или К позволяет провести четкую периодизацию кинетики роста трещин, выполнять расчеты для определения сопротивления началу ускоренного развития трещины, найти
1 По другим данным, для скорости роста трещин используют обозначение dljdN.
связь между периодами развития трещины и преимущественными механизмами разрушения, объяснить особенности роста трещии при изменении силового режима нагружения.
Параметр ААС или Кь — характеристика тре-щипостопкости, соответствующая возрастанию (или интенсивности возрастания) ускорения роста трещин и изменению интенсивности нарастания пластической деформации на фронте трещины.
На участке кривой, .соответствующем высо-коам плиту дней области, имеется перегиб, который позволяет установить пороговое значение ДАг>, характеризующее трещиностойкость материала на стадии докритического возрастания трещины. Область от ААЛ до ККЬ соответствует стадии ускоренного роста трещины, а область после A/\z, — ее быстрому росту. Область от ДА* до Kci названа «циклическим до-ломом». Значение Kef соответствует циклической вязкости разрушения.
Предложена (С. Е. Гуревич) формула скорости роста трещин при циклическом нагружении, в которую входят: размах приложенного коэффициента интенсивности напряжений АА"; среднее значение Ат; максимальное значение в цикле нагружения Атах*, размах порогового значения Д/Gzi и предельно возможное значение А.-,<
V — С [(ДА — ДА/л) Кгп/(^сж Атах)1п»
232
где С и п — постоянные уравнения Пэриса.
Для оценки циклической трещиностойкости используют следующие характеристики:
а) скорость роста усталостных трещин в зависимости от размаха -коэффициента интенсивности напряжений ДК (графически выра-
Рис. 14.6. Значение У в зависимости от Ifb для образцов типов Х-Х1П (Л-тарировка)
Рис. 14.7. Диаграмма циклической трещиностойкости
жается диаграммой циклической трещиностойкости — см. рис. 14.7);
б) параметры Сияв уравнении у=С(Д7<)'5. Эти параметры могут быть определены с использованием диаграммы циклической тре-щнностойкости (рис. 14.7), а также путем подстановки в указанное уравнение фактических значений скорости роста трещин и разма
ха коэффициента интенсивности напряжений, соответствующих двум каким-либо точкам диаграммы, и решения полученной системы уравнений, например:
v = 10—5 мм/цикл = С ^Д/<1(Г_Ёр
и v — 10—4 мм/цикл = С ^ДК10„^п;
в) размахом коэффициента интенсивности напряжений при различных скоростях роста трещин (например, при &=10-s; 10~4; 10~3 и 10-2 мм/цнкл);
г) стартовым илн пороговым значением размаха коэффициента интенсивности напряжений страхования магистральной трещины Д7(ст или ДКгл, соответствующим г>Ст—стартовой скорости роста трещин в образцах (принимается равной 10~5 мм/цикл или другой величине, которой вследствие ее малости можно пренебречь по условиям безопасной работы конструкции, для которой выбирается материал);
д) живучестью N™t характеризуемой числом циклов от момента появления усталостной трещины (например, длиной 0,5 кt, где t — толщина образца) до полного разрушения образца.
Все характеристики определяются прн заданном условиями эксперимента коэффициенте асимметрии цикла.
Высокочастотная усталость. ГОСТ 23026—78 предусматривает проведение испытаний образцов при циклических нагрузках при частотах, не превышающих 300 Гц. В необходимых случаях испытания проводят с частотой, доходящей до звукового днапазоиа. Частота более 2000 Гц достигается на магнитострикционных установках; от 300 до 2000 Гц —на электродинамических виброустановках. Применяют образцы специальной формы.
Контактная усталость — процесс накопления повреждений и развития разрушения поверхностных слоев материала под действием переменных контактных нагрузок,, вызывающих образование ямок выкрашивания (питтингов) или трещин и в конечном счете снижение долговечностей.
Для напряженного состояния при контактном нагружении характерна объемность (трехосное напряженное состояние) с большим градиентом I изменения величины ' напряжения, а также относительная мягкость (большое значение отношение величины касательных напряжений к величине нормальных растягивающих напряжений), вследствие чего деформируются даже те материалы, которые при обычных более жестких условиях нагружения не могут пластически деформироваться.
Испытания на контактную усталость выполняют по ГОСТ 25. 501—78 при нагружении по следующим схемам: качение без йроскальзы-вання; качение с постоянным проскальзыванием; качение с тяговым моментом; качение с тормозным моментом; прн пульсирующем контакте.
Испытания проводят при нормальной, повышенной или пониженной температуре, со смазкой или без нее, в различных агрессивных средах.
Для построения кривой контактной усталости и определения предела контактной выносливости испытывают не менее 12 образцов, из
233
них ие менее трех должны быть испытаны на уровне предела контактной выносливости.
Основным критерием разрушения при определении пределов контактной выносливости н построения кривых контактной усталости является возникновение на контактной поверхности нескольких ямок выкрашивания диаметром, равным половине малой полуоси контактной плошади, вычисленным для условно принятого значения максимального контактного напряжения. Прн обработке результатов испытаний рекомендуется учитывать накопленную остаточную деформацию. Для испытания каждого образца используют новую дорожку на обкатывающем контртеле. Частота циклов не регламентируется в пределах 1000—60000 циклов ъ минуту. База испытаний при определении предела контактной выносливости должна быть не ниже 107 циклов для металлов и сплавов с твердостью НВsg200; 5-Ю7 — для металлов и сплавов с /77?С<40, имеющих горизонтальный участок на кривой контактной усталости; 108 — для металлов и сплавов с H'RC>4£>, имеющих горизонтальный участок иа кривой контактной усталости; 2,0 • 108—5 - !08 — для металлов и сплавов, ие имеющих горизонтального участка на кривой контактной усталости.
Коррозионная усталость — процесс постепенного накопления повреждений, которые обусловлены одновременным воздействием циклических нагрузок н коррозионноактивной среды.
Характерные отличия коррозионной усталости от механической: отсутствие горизонтального участка на кривой усталости; наличие многоочагового разрушения, развивающегося из участков коррозионного повреждения; существенно более низкая прочность при растяжении, чем при сжатии; более сильная зависимость от частоты циклов; более частое возникновение межкристаллитного разрушения.
Основное требования к испытаниям на коррозионную усталость — проведение их в условиях, максимально приближающихся к условиям службы металла в конструкции. Не рекомендуется для ускорения испытаний применять среды с большей (чем в реальных условиях службы) коррозионной активностью.
Испытания проводят в жидких и газообразных средах, вакууме, тумане, при периодическом смачивании. К коррозионноусталостиым испытаниям непосредственно примыкают испытания на фреттинг-коррозионную усталость при совместном действия напряжений и фрет-тннг-коррозноиного процесса.
Ударная усталость — процесс накопления повреждений и развития трещин, вызванный ударными нагрузками. Оценивают предел выносливости прн ударном циклическом нагружении путем приложения к образцу многократных ударов нли наложением периодических ударов на плавную гармоническую нагрузку. Ударно-усталостную прочность рассматривают в качестве самостоятельной характеристики механических свойств материала. Используют механические, пневматические, гидравлические, электромагнитные установки.
Термическая усталость — малоцикловая низкочастотная усталость, которая характеризуется тем, что возбуждение переменных температурных остаточных напряжений в материале обусловливается циклическим изменением температуры.
В связи с большим числом факторов, оказывающих влияние на механические свойства металлов при циклических испытаниях, большое значение имеют испытания натурных деталей или их крупномасштабных моделей. Натурные детали и элементы конструкций существенно отличаются от образцов, изготовленных из того же материала или вырезаемых из деталей, по градиенту изменения структуры металла и механических свойств по сечению, эпюре остаточных напряжений, текстуре, состоянию поверхностного слоя и концентрации напряжений в переходных сечениях.
Испытания деталей при циклическом приложении иагрузкн должны выполняться при схеме н условиях нагружения, максимально приближающихся к эксплуатационным по уровню нагруженности, среде, температуре, частоте и параметрам цикла нагружения.
Критерием правильности выбора схемы нагружения и силовых условий испытания является воспроизведение эксплуатационного характера разрушения детали в том сечении, в котором возникают повреждения при эксплуатации. Если эксплуатационный характер разрушения не известен, то схему приложения сил и условия нагружения выбирают с таким расчетом, чтобы обеспечить возникновение разрушения в наиболее слабом по расчету сечеиии нли в любом другом сеченин, обусловленном задачей исследования.
БИБЛИОГРАФИЧЕСКИЙ СПИСОК
Иванова В. С,, Терентьев В. Ф. Природа усталости металлов. М.: Металлургия, 1975. 455 с.
Ногаев В. П. Расчеты на прочность при напряжениях, переменных во времени. М.: Машиностроение, 1977, 231 с.
Литвак В. И. Автоматизация усталостных испытаний натурных конструкций. М.; Машиностроение, 1972, 383 с.
Методы исследования сопротивления металлов деформированию и разрушению при циклическом 'иагружении/Грошенко В. Т., Грязнов Б. А., Отражало В. А. н др. Киев: Наукова думка, 1974. 254 с.
Степнов М. Н. Статистическая обработка результатов механических испытаний. М.: Машиностроение, 1972. 232 с.
Трощенко В. Т. Устал ось и неупру гость металлов. Киев: Наукова думка, 1971. 268 с.
Школьник Л. М. Методика усталостных испытаний. М.: Металлургия, 1978. 302 с.
Ярема С. Я-, Панасюк В. В., Попович В. В Метод испытания металлов на трещино-стойкость/АН УССР, Фнз.-мех. инет. Препринт № 9. Львов: 1978. 55 с.
15
ОПРЕДЕЛЕНИЕ СОПРОТИВЛЕНИЯ РАЗРУШЕНИЮ
15.1. ОБЩИЕ СВЕДЕНИЯ О СОПРОТИВЛЕНИИ РАЗРУШЕНИЮ
И МЕТОДАХ ЕГО ОЦЕНКИ
Все известные виды кратковременных и длительных механических разрушающих испытаний, в том числе широко распространенные испытания на статическое растяжение, ударную вязкость, ползучесть, усталость, прямо или косвенно дают меру сопротивления металлов разрушению в различных условиях эксплуатации. Однако только в течение двух последних десятилетий благодаря прогрессу в изучении механических и металловедческих аспектов проблемы разрушения были надлежащим образом осмыслены и приобрели самостоятельное значение специальные методы оценки сопротивления разрушению. Эти методы служат средством аттестации н ранжировки сплавов, а также диагностики разрушения. В последние годы получают также развитие основанные на различных характеристиках сопротивления разрушению расчеты несущей способности сплавов в изделиях.
Вследствие многообразия видов механических разрушений не может быть какого-то единого универсального метода оценки сопротивления разрушению. Процессы разрушения и соответствующие им оценочные виды испытаний следует в первую очередь разделять в зависимости от способа силового воздействия. В этой связи важная группа методов оценки сопротивления разрушению относится к кратковременным разрушающим испытаниям при статическом и динамическом нагружениях. Другая группа методов охватывает длительные испытания прн циклическом и статическом нагружениях, которым соответствует усталостное, нлн замедленное, разрушение.
Характер разрушения сплавов в изделиях может быть существенно различен в зависимости от условий нагружения (вид прикладываемой нагрузки, температура, окружающая агрессивная среда), а также состава и структуры сплава. Ввиду наибольшей опасности с точки зрения реальных условий технической эксплуатации металлических материалов главное внимание уделяется методам оценки сопротивления хрупкому разрушению, которое характеризуется рядом специфических механических и фрактографических признаков.
В любом процессе разрушения можно выделить три стадии. Первая (подготовительная) стадия связана с зарождением трещины. Вторая и третья стадии связаны с субкритиче-скнм, а затем критическим распространением трещины. В соответствии с таким характером процесса разрушения существуют различные интегральные методы оценки сопротивления разрушению сплавов, а также методы оценки сопротивления распространению трещин на
субкритической и критической стадиях. Последние методы достаточно хорошо разработаны и обоснованы иа базе механики- разрушения. Что касается первой (подготовительной) стадии, то вследствие физической сложности явлений, ее определяющих, и многогранности самой подготовительной стадии пока еще не существует обоснованных инженерных методов оценки сопротивления зарождению трещины.
Нашлн распространение два подхода к оценке сопротивления разрушению сплавов: 1) энергетический подход, базирующийся на оценке работы разрушения; 2) силовой подход, связанный с оценкой напряженного состояния и его экстремальных компонент в условиях разрушения. Более старым н распространенным является энергетический подход, который часто не требует уточнения конкретной ситуации (напряжений н деформаций) в очаге разрушения. Энергетический подход используется для оценки как общей работы разрушения, так и ее составляющих, связанных с зарождением и распространением трещины. В использовании силового подхода для оценки процессов образования (зарождения) трещины имеются большие трудности.
15.2. МЕТОДЫ ИНТЕГРАЛЬНОЙ ОЦЕНКИ СОПРОТИВЛЕНИЯ РАЗРУШЕНИЮ
Различные исследователи использовали в качестве показателей сопротивления разрушению разнообразные механические характеристики, оцениваемые при стандартных испытаниях на растяжение, сжатие, изгиб и т. п. Так, в частности, оо.2 рассматривается как мера сопротивления разрушению на стадии развития малых пластических деформаций, SK — как мера сопротивления разрушению на стадии развития шейки. Из традиционных характеристик сопротивления разрушению наиболее универсальной признана ударная вязкость, оцениваемая обычно по ГОСТ 5454—76 на призматических образцах с U-образным надрезом (образцы Мена-же), разрушаемых на маятниковых копрах.
Практика технического металловедения показала, что величина ударной вязкости при комнатной температуре испытаний не может служить мерой сопротивления разрушению материалов в различных ужесточенных условиях испытаний (например, при понижении температуры) и во многих случаях не может выявить различных структурных и металлургических факторов, ответственных за ухудшение эксплуатационных характеристик. Это обусловлено тем, что при вязком разрушении, которое обычно реализуется при комнатной температуре испытаний, чувствительность к структурным факторам, которые определяют охрупчивание, резко снижается. В то же время изменение условий нагружения, способствующее
235
хрупкому разрушению, позволяет четко выявить отрицательное влияние тех или иных структурных факторов. Такое изменение условий может быть прежде всего достигнуто путем снижения температуры испытаний, обеспечивающей в ряде о. ц. к. металлов выявле-
ние. 15.1. Зависимость температуры хладноломкости от «жесткости» напряженного состояния (Хейндльхо-фер)
нии их прочности в связи со снижением температуры отпуска (рис. 15.2). Это делает необходимым использование как макрофрактографи-ческого (А. П. Гуляев), так и микрофрактогра-фнческого метода - определения порога хладноломкости (для некоторых, в частности строительных сталей). Вместе с тем для высокопрочных сталей со структурой мартенсита такое определение теряет всякий смысл в связи с крайне вялым, практически неощутимым изменением как самой ударной вязкости, так и фрактографической картины разрушения.
В исследовательской практике за рубежом широко используют и другие методы интегральной оценки работы разрушения, в том числе оценку нулевой пластичности иа копрах с падающим грузом, испытания по Робертсону, испытания пластинчатых образцов при нагружении взрывом н др. Названные методы предусматривают испытания различных образцов, в том числе сварных образцов с надрезом, с наплавленным валиком, инициирующим трещину, и т. п. К сожалению, различные виды испытаний не дают, как правило, воспроизводимых результатов, поэтому выбор вида разрушающих испытаний должен опреде-
Рис. 15.2. Кривые хладноломкости:
а — нормализованные углеродистые стали: б — сталь ПГХ15 после отжига и закалки с последующим отпуском (температуры отпуску указаны на рисунке)
нис вязко-хрупкого перехода. Определяемая таким образом температура хладноломкости достаточно адекватно отражает склонность сталей к опасному хрупкому разрушению в различных экстремальных условиях эксплуатации. Температура хладноломкости, вопреки встречающимся ошибочным воззрениям, не может рассматриваться как константа материала и зависит от конфигурации и размеров образцов, остроты надреза и вида испытаний (рис. 15.1). Следует также отметить трудности в оценке склонности к хрупкому разрушению по температуре хладноломкости для сталей повышенной прочности в связи с пологим характером их кривых хладноломкости. Положение порога хладноломкости, четко детерминированное для иизкоуглеродистых сталей, становится трудноопределяемым при повышении содержания углерода и сталях и при повьпле-
ляться их максимальным приближением к эксплуатационным условиям.
Для некоторых методов разрушающих испытаний характерна тенденция к разделению полной работы разрушения А на составляющие зарождения и распространения трещины (Аз и Ар соответственно). Такое разделение достигается благодаря определению А и Ар, после чего считают, что АЭ=А—Ар.
По методу А. П. Гуляева АР определяют путем измерения работы ударного разрушения серии образцов с надрезом, радиус которого постепенно уменьшается. При этом Ар оценивают обычно путём линейной экстраполяции уменьшающихся значений А на нулевой радиус. Недостаток такого метода — неправомерность линейного экстраполирования.
В отечественной практике получил широкое распространение метод прямого измерения ра
236
боты Лр по методу Б. А. Дроздовского (часто обозначаемой также Атр). Испытываются образцы с нарощенной (наведенной) усталостной трещиной регламентированной длины в устье концентратора нормального ударного образца. Поскольку размеры стандартных ударных образцов в случае испытаний низкопрочных материалов весьма ограничены, величина Лтр включает значительную работу пластической деформации в условиях вязкого и полувязкого разрушения и не отражает, таким образом, поведения материала в экстремальных условиях хрупкого разрушения, которые реализуются в случае ужесточения условий испытаний (эксплуатации).
15.3. ИСПЫТАНИЯ
НА ВЯЗКОСТЬ РАЗРУШЕНИЯ
Испытания на вязкость разрушения (кратковременную трещиностойкость) принадлежат к наиболее апробированным и теоретически обоснованным, получающим широкое распространение в практике технического металловедения. Эти испытания базируются главным образом на линейной механике разрушения, которая берет свое начало от работ Гриффитса. Впервые на основании энергетического подхода он показал, что причиной резкого несоответствия реального и теоретического сопротивления разрушению твердых тел может быть присутствие в них малых дефектов (трещии), способствующих возникновению концентрации напряжений, достигающих в локальных объемах теоретической прочности. В развитие нден Грнффнтса Ирвин показал, что этн локальные напряжения в самом общем случае отрывного нагружения тела с трещиной пропорциональны так называемому коэффициенту интенсивности напряжений К, который может быть записан в виде:
А — о
ала
О)
где с — номинальное напряжение, оцениваемое в теле без учета трещины; а — длина трещины; а —параметр, зависящий от формы тела (образца) н геометрии трещнны.
Различают трн основных вида нагружения тел с трещинами (отрыв, продольный сдвиг н поперечный сдвиг, рис. 15.3), именуемых соот-
Рис. 15.3. Три основных способа деформирования плиты с трешиновидаым концентратором
ветственно I, И и III способами нагружения. Следуя такой индексации, коэффициенты интенсивности напряжений в окрестности вершины трещины записываются для указанных иа рнс. 15.3 случаев соответственно Kt, Kir н Кгп. Первый (отрывной) вид смещения берегов
трещины реализуется в случае наиболее опасного хрупкого разрушения, поэтому все инженерные методы оценки вязкости разрушения касаются этого способа.
Прн создании критериев трещиностойкости материалов Ирвин исходил из того, что прн достижении нестабильного спонтанного роста трещины коэффициент интенсивности напряжений достигает своего критического значения Кс. Вначале предполагали, что Кс является константой материала. Однако оказалось, что уровень этой характеристики зависит от толщины испытываемых изделий (например, пластины) н с увеличением толщины уменьшается в связи с изменением (трансформацией) в вершине трещины плосконапряженного состояния на наиболее опасное для реализации хрупкого разрушения плоскодеформнроваиное состояние, достигая при этом определенного минимального значения Kic (рис. 15.4). Имеются все основа-
Рис. 15.4. Изменение Кс и доли хрупкого излома в зависимости от толщины образца с трещиной
иия рассматривать вязкость разрушения при плоской деформации Kic в заданных температурно-скоростных условиях нагружения в качестве важного параметра, который может быть использован прн ранжировке материалов по нх сопротивлению хрупкому разрушению.
Кроме вышеприведенной силовой, Ирвин ввел также эквивалентную энергетическую интерпретацию критериев разрушения, вводя понятие интенсивности потери энергии в вершине трещин прн критических значениях этой интенсивности Gc и б?1с. Последние связаны с Кс и Kic следующими соотношениями:
Gc = К%Е; GIc = К\с (1 - (2)
где |Л — коэффициент Пуассона; Е— модуль Юнга.
Названные характеристики именуются еще сопротивлением распространению трещины. Их размерность (МПа • м] в отличие от Кс н Kic, имеющих непривычную размерность [МПа - р^м].
При приложении внешней нагрузки напряжения в вершине трещины в конечном счете превышают предел текучести материала и в окрестности трещины образуется пластическая зона радиусом гу, для которой упругий анализ напряжений теряет силу. Размер зоны определяется из выражений
'»=---------------- и ,— (3)
2ло^2 2jig0,2
237
соответственно в условиях плоской деформации н плосконапряжениого состояния. Важное условие правильности оценки вязкости разрушения, исходя нз формул теории упругости,— малость пластической зоны, длину которой по оси трещины обычно добавляют к исходной длине трещины, благодаря чему достигается корректировка оценок вязкости разрушения.
Определение вязкости разрушения. Согласно получившему уже значительную апробацию проекту ГОСТа, в основу которого положена рекомендация по стандартизации PC 3642—72 [4], вязкость разрушения определяют следующим образом.
Испытания проводят при внецентренном растяжении нлн изгибе на образцах, показанных соответственно на рис. 15.5, а и б. Для образ
Рис. 15.5. Типы образцов для определения К1С и соотношения их размеров:
а — для испытаний на изгиб; б — Для испытаний на внецентрениое растяжение; в — цилиндрический образец для испытаний на растяжение
цов характерен острый надрез, заканчивающийся наводимой усталостной трещиной. Ее наводят на специальных вибраторах нлн усталостных машинах типа пульсаторов при асимметричном цнкле. Длна надреза с трещиной должна быть а= (0,45н-0,55) W.
Во избежание недопустимого наклепа материала в вершине трещины максимальный уровень К в процессе циклического нагружения прн наведенин трещины ие должен превышать 0,6 искомого значения Кхс-
Специфика определеня вязкости разрушения заключается в том, что достоверность оценки Kic регламентирована размерами испытываемого образца, которые зависят от искомой ве-
0,007
17
0,006 26
0,009 4,5
0,008
10
0,010
1,5
трещиной иа
ЛИЧИНЫ Kic, подчиняясь условиям
а н В >2,5 (/С1с/о0 2)2. (4)
Поэтому выбирать размеры образцов приходится, исходя из ориентировочных значений Kic, которые задаются иа основании имеющегося опыта. При этом целесообразно назначать оценочную величину Kic по известным значениям п0,2 и Е исследуемого материала.
Ниже приведены ориентировочные соотношения параметров вязкости разрушения и предела текучести для конструкционных сталей (по литературным данным) : с^о/Е........... 0,005
(^Чд)2- ММ. . 36
0(1,2./ В.......
(#1С/о0,2)2. ММ .
Готовые образцы с наведенной подвергают разрушающим испытаниям стандартных (но жестких) испытательных машинах, при этом скорость нагружения должна находиться в пределах 0,6—2,5 МПа-'Км/с.
В процессе испытаний на двухкоординатвом самописце автоматически регистрируется диаграмма разрушения в координатах: прилагаемая нагрузка Р — смещение берегов трещины V. Запись смещения обеспечивается установкой на образце датчика раскрытия трещины (рис. 15,6),
На рнс. 15,7 показаны три характерных типа диаграммы разрушения. Для диаграмм типов И и III в качестве расчетной нагрузки Pq для определения вязкости разрушения принимают максимальную нагрузку в секторе, отсекаемом 5 %-ной секущей. 'Для диаграммы типа 1 в качестве Pq принимают В5 при условии, что Vi/Vs^0,25 (см, рнс. 15,7). По значениям Pq ti усредненной длине трещины определяют величину коэффициента интенсивности напряжений Kq.
При испытаниях на нзгнб:
Pq Г (а к 2,9 —
С ВГ3/2 L \
а \5/2 / а \7/2
— 37,6 {—
a V/2 .
—4,6
«7
+ 21,8
. „/а \9/2
+ 38,7 [ -— I \ R7 /
(5)
Для внецентренного растяжения:
I а \5/2 / а V/2
+ 655,71 — I -1017 — +
\W } \W }
I a W2T
+ 638,9^—J ]. (6)
Полученные таким образом значения Kq проверяют для условий (4) и в случае их выполнения принимают в качестве действительных значений Kic- Если размеры образцов окажутся недостаточными, то необходимо провести повторные испытания на образцах большей толщины.
Кроме рассмотренных выше типов образ
238
цов, б практике получили распространение и другие типы образцов, приведенные на рис. 15.5. Формулы для расчета К<у этих типов образцов, а также ряд других важных методических подробностей можно найти в руководствах [3, 5, 6].
также способ, основанный иа анализе кривых сопротивления распространению трещин, так называемый метод /?-кривых.
^-кривая представляет собой зависимость определенной характеристики сопротивления распространению трещины R от ее Длины.
Рис 15.6. Устройство двухконсольного датчика для измерения смещения и способ его крепления на образце 15]:
1 — фольговые тензодатчики; 2 — балансировочное сопротивление; 3 — клеммы двухкоординатного самописца
В качестве R может быть использована удельная энергия разрушения на единицу площади, сила движения трещины G н т. п. Возможные формы /?-кривых показаны на рнс. 15.8 соот-
Рис. 15.8. Характерные типы R-кривых
Рис. 15.7. Характерные типы диаграмм нагрузка — смещение и методика их обработки
В последние годы признан весьма перспективным цилиндрический образец с кольцевидной трещиной (см. рис. 15.5, в), который также рекомендован для проекта отечественного стандарта для оценки вязкости разрушения прн испытаниях на растяжение и изгиб. Как показано в работе [6], в силу специфики контура трещины прн использовании цилиндрических образцов могут быть существенно ослаблены жесткие требования относительно размеров образцов. Для цилиндрического образца с кольцевидной трещиной геометрические условия правомерности оценки Клс имеют вид: d > 1,6 (Kq/o0 2)2J В 2,3 (Kq (7)
Целесообразность апробации цилиндрического образца определяется также тем обстоятельством, что он создает единственную в своем роде возможность оценки трещиностойко-стн круглого проката с учетом его анизотропии, возникающей при обработке давлением.
Для оценки критических значений коэффициентов интенсивности напряжений используют
ветственно для идеально хрупкого (Л) и реальных материалов (Б, В), разрушающихся при существенной пластической деформации. В качестве характеристики нестабильного распространения трещины принимают ординату точки касания R-кривой с прямой, проведенной через начало координат (см. рис. 15.8). В настоящее время происходит накопление данных о ^-кривых для различных материалов и непрерывно совершенствуются основы этого перспективного метода [7].
Существенной трудностью при оценке трещн-ностойкости материалов по параметру К1С являются весьма жесткие геометрические условии (4), регламентирующие реализацию при разрушении плоскодеформированного состояния. Таким образом, надлежащая оценка Ктс возможна прежде всего для высокопрочных сталей, в то время как переход к наиболее распространенным сталям средней и особенно низкой прочности требует значительного увеличения размеров (толщины) испытываемых образцов (рис. 15.9) и мощности испытательных устройств. В итоге оценка трещиностойко-
239
сти многих конструкционных сталей по Кгс становится либо невозможной в силу отсутст-вия материалов требуемой толщины, либо затруднительной в связи с испытаниями образцов больших размеров. В этих случаях нужно ориентироваться прежде всего на возможность сравнительной оценки по параметру Кс различных материалов равной толщины. Одиако такой подход также ограничен сталями повышенной прочности (о.в>1200 МПа).
Рас 15.9. Номограмма для аыбора необходимой толщины образца при определении /<Jc
Рис. 15.10, Распределение напряжений у края трещины в соответствии с расчетной моделью Леонова — Панасюка п величина критического раскрытия 6К
Указанные трудности в оценке трещнностой-кости сталей низкой прочности могут быть преодолены при использовании в качестве меры трещиностойкостн материалов так называемого критического раскрытия трещины (КРТ) 6к. Понятие введено в работах М, Я. Леонова и В. В. Панасюка и получило наибольшее распространение в британской практике благодаря работам Уэллса—Дагдей-ла. Измерение 6К, соответствующего раскрытию трещины в момент ее спонтанного роста (рис. 15.10), производится с помощью специального устройства типа поворотной лопатки, а иногда также оценивается по смещению берегов трещины по показаниям двухконсольио-го датчика (см. рис. 15.6). Метод КРТ позволяет сопоставлять инзкопрочные стали в образцах идентичных размеров н испытанных в одинаковых условиях. В этом смысле КРТ можно рассматривать как характеристику, зависящую от размеров и конфигурации образца и способа нагружения, что делает КРТ менее информативным параметром, чем ДТе-
В работах В. С. Ивановой предложена мето
дика определения вязкости разрушения конструкционных сплавов по данным испытаний на усталость гладких образцов. Вследствие более жестких условий нагружения в микрообъемах при усталостных испытаниях метод позволяет определять вязкость разрушения на малогабаритных образцах разнообразных материалов, включая пластичные [8]. Испытывают образец (плоский или цилиндрический) на усталость до разрушения и затем по поверхности излома определяют глубину усталостной трещины If. Расчет получаемой в итоге вязкости разрушения при циклических нагрузках Д'р. ведут по формулам линейной механики разрушения в зависимости от геомегрин образца. Так, в частности, для наиболее распространенного случая испытаний цилиндрического образца при изгибе с вращением, когда имеет, место одностороннее распространение трещины, величина KfC = о . Важным условием правильности подсчета Kic является надлежащий выбор амплитуды напряжений, которая должна быть равной нли меньшей порогового напряжения. ок, характеризующего переход от мало- к многоцнкловой усталости, который наступает прн определенном числе циклов Для стали JVH=2-105.
В последние годы предпринимаются успешные попытки создания нового универсального метода оценки вязкости разрушения стали как низкой, так и высокой прочности по величине так называемого /-интеграла, представляющего собой изменение потенциальной энергия в упруго-пластическом континууме в процессе распространения трещины. При этом предварительно строят диаграмму нагрузка — податливость, которую затем перестраивают для определения /-интеграла. Имеется ряд лабораторных методик оценки /-интеграла, которые отличаются значительной трудоемкостью, существенно превосходящей трудоемкость определения KfC. Ввиду того, что пока отсутствует единый методический подход к измерению /-интеграла, приемлемый для инженерных расчетов, подробности различных вариантов имеющихся методик оценки /-интеграла здесь не приводятся.
15.4. ВЛИЯНИЕ РАЗЛИЧНЫХ ФАКТОРОВ НА ВЯЗКОСТЬ РАЗРУШЕНИЯ СТАЛЕЙ
Для характеристики работы сталей прн низких температурах представляют обычно большой интерес температурные зависимости Kic как параметра заведомо хрупкого, отрывного разрушения. Многочисленными испытаниями установлено, что для иизкопрочных (прежде всего ннзкоуглеродистых) сталей характерна пороговая температурная зависимость (рис. 15.11). По мере повышения прочности стали (за счет увеличения содержания углерода нлн прн использовании низкого отпуска после закалки) понижение температуры испытаний в сторону криогенных сопровождается лишь незначительным уменьшением ,и так очень низких исходных значений Д1С. Температурная чувствительность К1С находится в очевидной тесной зависимости с температурной
240
чувствительностью предела текучести. Это в свою очередь и определяет чувствительность Kic к скорости нагружения при испытаниях. Для низкопрочных сталей температурная зависимость вязкости разрушения при динамическом нагружении (Kia} сдвигается, существенно вправо по сравнению со статическими нс-
Рис. 15.11. Пространственная диаграмма температурных зависимостей вязкости разрушения термически упрочненных сталей с различным содержанием углерода
Рнс. 15.12. Температурные зависимости X (/) и
(2) мартенситио-стареющей стали Н18К8М6Т (Д) и стали типа 20 (Б)
пытаниями (рис. 15.12). В то же рремя для высокопрочных сталей эти зависимости обычно совпадают (например, сталь типа Н18К8М6Т, см. рис. 15.12). Эквивалентность влияния температуры и скорости испытаний на К\с и предел текучести подтверждается единой зависимостью Kic и /(id от о0,2 (ряс. 15.13). Согласно Кортену и Шумейкеру К\с cnejnyev рассматривать как функцию температуры Т и скорости деформациие вида Т 1п(А/в), где А — постоянная (см. рнс. 15.13).
Необходимо отметить, что в результате охрупчивания водородом нлн деформационного старения может наступить инверсия влияния упомянутых факторов, т. е. повышение температуры испытаний и снижение скорости нагружения может сказаться отрицательно иа уровне Kic.
Влияние различных металлургических и структурных факторов и состава сталей на Ктс изучено наиболее досконально на высокопрочных сталях1. Характер влияния содержания
ООО 500 600 700 800 000 2000 0000 6000 8000
бд^МПа Т\1п(А/ё),*0,555К
Рис. 15.13. Вязкость разрушения стали типа 20 как функция предела текучести (с) и параметра Т 1в (Л/е) (б):
1 — статическое нагружение; 2 — динамическое нагружение (в этом случае /<г<р
углерода н температуры отпуска достаточно четко прослеживается по пространственной диаграмме на рис. 15.11.
В ряде случаев прн испытаниях сталей со структурой тросстита и сорбита зафиксировано увеличение Ki< по мере уменьшения величины действительного аустенитного зерна. Такая тенденция обнаруживается наиболее четко, когда разрушение образцов с трещиной протекает по межзеренному механизму. Вместе с тем величина аустенитного зерна обычно не контролирует уровня Kic в структурах, образующихся после высокого отпуска, для которых распространение трещин идет по механизму коалесценций пор с образованием в изломе ямочно-вязкого рельефа. В ряде американских
Рис. 15.14 Влияние величины исходного зерна аустенита на вязкость разрушения закаленной стали 40Х (отпуск 200 °C)
и отечественных исследований неожиданно было обнаружено, что рост зерна прн перегреве стали ведет к значительному увеличению Клс высокопрочных конструкционных сталей типа 40Х и 40ХНМ при низком отпуске (рис. 15.14). Этот специфический случай нарушения структурной корреляции между Kic н ударной вязкостью свидетельствнет о том, что характер влияния некоторых факторов на хрупкость может в значительной мере зависеть от Типа
1 Вследствие методических трудностей (большие размеры образцов), возникающих при оценке трещи-ностойкости инзкопрочкых сталей, а также из-за склонности высокопрочных сталей к хрупкому разрушению.
16—280
241
концентратора, ответственного за разрушение1
Данные рис. 15.15 убедительно свидетельствуют о сильном влиянии серы и фосфора на вязкость разрушения сталей. Данные о влиянии других примесей не столь убедительны. Рафинирование сталей также способствует повышению Kic, причем это влияние уснлива-
жают вязкость разрушения сталей. Легирование, ведущее к образованию в сталях дисперсных фаз, затрудняя пластическое течение, ведет к уменьшению Kic. Это нашло подтверждение на сталях, обнаруживающих склонность к вторичному твердению (типа 20 МН, 40 МН и 45 ХМВ — см. рис. 15.17).
Рис. 15.15. Влияние суммарного содержания серы и фосфора на вязкость разрушения сталей типа 40ХЙ2М при температурах отпуска 200 и 370 °C
Рис. 15.17. Влияние вторичного твердения на вязкость разрушения стали типа 45ХМВ
Рис. 15.16. Влияние способа выплавки на вязкость разрушения сталей типа 40ХН2М:
/ — открытая плавка; 2 — вакуумно-дуговая плавка
ется в области высоких температур отпуска (рнс. 15.16). Согласно модели Спицнга неметаллические включения становятся центрами порообразования при разрушении сталей по вязкому мнкромеханнзму, а усредненная частота таких включений, соответствующая впоследствии частоте ямок в изломе, контролирует уровень Kic.
Характер влияния умеренного легирования конструкционных сталей на Kic остается в значительной мере подобным влиянию на порог хладноломкости. Присутствие в малых количествах в сталях хрома, ванадия, ниобия, титана н тантала обеспечивает измельчение зерна вследствие карбндообразующей способности названных элементов, что в свою очередь способствует увеличению Кгс. Никель и марганец в количествах до 1 % также измельчают зерно. Раскисление сталей алюминием сказывается благоприятно на Kic также вследствие измельчения зерна. Оказалось, что легирующие элементы, упрочняющие твердые растворы, снн-
! Ряд экспериментальных данных свидетельствует о том. что повышение К1с в результате перегрева стали при нагреве под закалку связано с «очищением» границ зерен от вредных примесей (от серы) в связи с растворением их в теле зерна при высокотемпературном нагреве. Прим. ред.
Принципиально новые возможности с точки зрения воздействия на Kic легирующих элементов открываются благодаря разработке мар-тенситно-стареющнх сталей (МСС) н метаста-бильных аустенитных сталей (МАС), именуемых трии-сталямн. Как видно нз табл. 15.1, в высокопрочных безуглерсдистых МСС с 18 % Ni после закалки и отпуска (старения) реализуются уровни Kic, не достижимые в традиционных сталях. Уникальное сочетание прочности и вязкости разрушения МСС обеспечивается за счет наличия высокопластичной матрицы, в которой при последующем старении эффективно реализуется иптерметаллндное упрочнение.
МАС обеспечивают высокий уровень Kic (до 160 МПа'Ум) при о0.2 до 2000 МПа за счет образования мартенсита в областях, подверженных пластической деформации в окрестности вершины трещины. Оптимальное сочетание свойств обеспечивается в МАС прн их комплексном легировании перечисленными ниже элементами в пределах 0,2—0,5 % С; 8— 14 % Сг; 8-32 % Ni; 0,5—2,5 %Мп; 2— 6 % Мо; с2 % Si.
ТАБЛИЦА Л5-!
ВЯЗКОСТЬ РАЗРУШЕНИЯ ВЫСОКОПРОЧНЫХ
КОНСТРУКЦИОННЫХ сталей
Сталь °о,2- мпа К1с, МПа-Ум
Н18К8МЗ 1380 110—175
Н18К7М5 1725 98—163
HI8K9M5 2070 90—140
Д6АС (тяпа 45Х2НМ) 1380 90—98
Hl 1 (типа 40Х5МФ) 1795 65—70
AISI 4340 (типа 40ХН2М) 1795 60—65
Н50 (типа 40Х5МФ) 1570 33
242
ТАБЛИЦА 15.2
МЕХАНИЧЕСКИЕ СВОЙСТВА И ВЯЗКОСТЬ РАЗРУШЕНИЯ СТАЛЕЙ 40С2Х и 60С2Х ПОСЛЕ ВТМО И КОНТРОЛЬНОЙ ЗАКАЛКИ (КЗ)*
*ОТП' С СВ’ МПа °0,2’ МПА 6. % Ф. % МПа-Ум
Сталь 40С2Х
200 2060 1765 12 53 78
1960 1620 9,5 30 67
1910- 1690 •12 55 90
ЗОП 1860 1570 10 37 78
Сталь 60С2Х
300 2400 2205 8 38 45
2160 1940 5 27 30
425 2160 2060 10 35 70
2010 1860 7 30 52
ГАП 1710 1570 11,5 40 73
oUv 1620 1450 И 36 55
* В числителе — ВТМО. в знаменателе — КЗ.
В связи с высокой стоимостью МСС и МАС в настоящее время предпринимаются попытки достижения лучших сочетаний прочности и вязкости разрушения в традиционных умеренно легированных сталях путем новых видов упрочняющих обработок.
Средн известных видов термомеханической обработки с точки зрения повышения сопротивления разрушению особо следует выделить
Рис. 15.18. Зависимость между и OR сталей типа
X .ГСНМ в интервале значений 0,25—0,5 % С после отпуска при 200 ®С:
1 — ВТМО; 2 — контрольная закалка
ВТМО, которая способствует существенному повышению хрупкой прочности сталей. В табл. 15.2 приведены данные, свидетельствующие о повышении прочности и вязкости разрушения после ВТМО сталей 40С2Х и 60С2Х. Из результатов, полученных К. Мазанцем для сталей типа ХГСНМ, следует, что имеется возможность повышения Mic путем ВТМО в области высокопрочных СОСТОЯНИЙ при Со,2= = 17004-2200 МПа (рис. 15.18).
К другим видам обработки, позволяющим повысить трещиностойкость сталей без уменьшения упрочнения, относятся отпуск под на
пряжением, скоростная термическая обработка иа сверхмелкое зерно (СМ3 — Грейндж, Р. И. Энтин).
Результаты анализа перспектив достижения высокого сопротивления разрушению у различных сталей могут быть проилллюстрирова-
500 1000 1500 2000 2500 3000
бог,НПа
Рис. 15.19. Сводная диаграмма конструктивной прочности различных сталей
иы сводной диаграммой конструктивной прочности Kic — Go,2, построенной для испытаний при комнатной температуре (рнс. 15.19). Диаграмма показывает преимущества термически улучшенных среднеуглеродистых сталей по сравнению с ннзкоуглеродистымн, потенциальные возможности МСС и МАС, а также термомеханической обработки и обработки на сверхмелкое зерно.
15.5. ОЦЕНКА
ТРЕЩИ НОСТОЙКОСТИ
ПРИ ЦИКЛИЧЕСКОМ
НАГРУЖЕНИИ
В соответствии с широко используемой инженерно-исследовательской практикой оценка сопротивления распространению трещин при циклическом нагружении сводится обычно к построению так называемой кинетической диаграммы усталостного разрушения (КДУР), устанавливающей зависимость между скоростью роста трещины v и коэффициентом интенсивности напряжений в вершине трещины (его амплитудным значением АК нли максимальным значением Кшах с учетом асимметрии цикла). Типичная КДУР строится в логарифмических координатах и имеет вид, показанный иа рис. 15.20. На диаграмме обычно различают трн участка {I—1II}. Важными параметрами, используемыми в расчетах на циклическую трещиностойкость, являются: а) пороговый коэффициент интенсивности напряжений Ktk, соответствующий верхнему значению Ктах, при котором трещина не развивается; б) циклическая вязкость разрушения К/с, соответствующая спонтанному разрушению (долому); и) коэффициенты Сит, характеризующие поведение
243
материала на втором (линейном участке) диаграммы в соответствии с уравнением Пэриса:
(в)
КДУР строят обычно иа основании слеженид (как правило, оптического) за длиной продви-
метрик цикла, температуры испытаний н воздействия внешней рабочей среды. Асимметрию цикла принято выражать посредством коэффициента асимметрии 2?=Лппп/Ктаах (обозначения в соответствии со схемой к рис. 15.22). В связи с воздействием фактора среды при испытаниях может проявляться также роль фактора частоты нагружения.
Для циклического нагружения образцов используют различные машины для испытаний иа усталость обшего и специального назначения. Наиболее распространенные виды нагружения — пульсирующие растяжение н плоский изгиб. Надо при этом учитывать, что испытаниям, обеспечивающим получение КДУР, предшествует этап наведения трещины в устье искусственного концентратора при нагрузках, предотвращающих развитие существенных зон пластической деформации, которые могли бы повлиять на кинетику распространения трещин в условиях монотонного роста ДЛ'. Принимая во внимание ряд регламентирующих условий, обеспечивающих воспроизводимость диаграмм от образца к образцу, прн испытаниях следует руководствоваться методическими указаниями, подготавливаемыми Госстандартом СССР [9].
Рнс. 15.20. Кинетическая диаграмма усталостного разрушения (КДУР)
Рис. 16.21. Типы (а—г) образцов, обеспечивающие постоянство коэффициента интенсивности напряжений в процессе субкритического роста трещины
гающейся причины по мере увеличения числа циклов АГ. Для таких испытаний пригодны разновидности образцов, используемых при оценке кратковременной трещнностойкостн (см. рис. 15.5). При испытаниях на циклический из-ги0 или растяжение таких образцов обычно наблюдается возрастание ДК по мере увеличения длины трещины. Вместе с тем заслуживают внимания образцы, обеспечивающие при постоянной нагрузке постоянство в определенном (достаточно значительном) интервале длины трещины. К таким образцам следует прежде всего отнести дисковый образец с центральной трещиной, предназначенный для испытаний на циклическое растяжение (рис. 15.21, в). Характер КДУР зависит от асим-
Рассмотрим основные данные о влиянии структурных факторов на кинетику распространения трещин усталости в конструкционных сталях. Хотя в самых общих чертах можно говорить, что характер влияния структурных факторов на кратковременную и циклическую трещи нестойкость аналогичен, в последнем случае обнаруживается ряд особенностей. По мере повышения температуры отпуска скорость роста трешины прн циклических нагрузках уменьшается (см. рис. 15.22). Это в наибольшей мере характерно для периодов испытаний, отвечающих второму и третьему участкам КДУР. Что касается припороговой области и значений &Ktn, то здесь отмечается практически полная структурная нечувстви
244
тельность кинетики распространения трешин вплоть до температуры отпуска 500 °C. Только у высокоотпущенных и отожженных сталей наблюдается значительный прирост &Kin. В табл. 15.3 приведены значения основных расчетных параметров КДУР для некоторых
прослеживается на основании экспериментов, выполненных с целью установления корреляционной свяеи т и Л']С, Atfih и, сго.г, а также ДК<й с пределом выносливости (рнс. 15.23). Однако эти эксперименты следует рассматривать как весьма предварительные нз-за недо-
Рис. 15.22. Кинетические диаграммы усталостного разрушения пружинной стали 60ХС в процессе испытаний на воздухе при различных структурных состояниях (см. табл. 15.3)
Рис. 15.23. Корреляционные зависимости между праметрами цикличес-
кой трещмностойкостн и другими конструкционных сталей
распространенных сталей. Характер связи параметров циклической трещиностоикости с составом и термической обработкой умеренно легированных сталей в определенной мере
механическими характеристиками '«
статочного количества данных о сталях, имею-
щих различные состав и структуру.
К настоящему времени установлены некоторые опосредованные структурой особенности
245
ТАБЛИЦА 15.3
РАСЧЕТНЫЕ ПАРАМЕТРЫ КИНЕТИЧЕСКИХ
ДИАГРАММ УСТАЛОСТНОГО РАЗРУШЕНИЯ
Сталь ШИ | Коэффициент асимметрии m с
60ХС 2200 —0,33 3,41 12,9-10“10
2100 —0,33 2,9 14,2-10-ю
1950 —0,33 2,4 56,5-10-ю
1350 —0,33 2,4 57,1-10-10
50ХН 1200 —0,33 2,2 68,5-10-ю
ШХ15 2100 —0,33 3,18 7,87-10—10
30ХГСН2А 1500 —0,33 2,73 1,82- IO-8
S15C 190 0,1 5,05 1,1-IO-13
(0,36Мп—0,15С) 180 0,1 4,45 3,6-10-13
SS41 (1,02Мп—0,39С) 250 0,1 3,3 5,8-10-11
S38C (0,67Мп—0.39С) 380 0 2,45 2,8-10—9
STY—R (0,62Мп—0,7С) 410 0,1 4,0 5,2-10-12
1.1N1—1Сг— —0,4Мо—0.2С 760 0,1 3,13 1,15-10-н
НТ80 (0,5Ni—lCr— —0,5Мо—0,1 С) 800 0 2,13 1,38-10—р
ЗСг—0,5Мо— —0,2С 840 0,1 3,0 2,15-10-ю
2,2Сг—1Мо— —0,19С 960 0,1 2,7 6,3-10-1°
кинетики усталостного разрушения сталей. У пнзкоотпущенных сталей скорость роста усталостных трещин резко интенсифицируется по мере роста коэффициента асимметрии цикла 7?; высокоотпущенные стали такой зависимости от R ие обнаруживают.
Усталостная трещиностой кость сталей со структурой мартенсита в результате предварительного перегрева при аустенитизации увеличивается, как и Kic. Однако в отличие от Kic присутствие в стали наряду с мартенситом структурно свободного феррита не уменьшает циклической трещиностойкости.
Благоприятное влияние на циклическую тре-щиностойкость оказывает уменьшение загрязненности сталей вредными примесями и снижение количества содержащихся в них неметаллических включений. Это подтверждено для структур, образующихся после низкого и после высокого отпуска. Исследования перлитных сталей показали преимущества обработки иа перлит с зернистыми карбидами по сравнению с пластинчатым перлитом.
15.6. СТАТИЧЕСКАЯ
ТРЕЩИНОСТОЙКОСТЪ. УЧЕТ ВОЗДЕЙСТВИЯ РАБОЧИХ СРЕД
Анализ статической трещиностойкости сплавов— неотъемлемая часть проблемы оценки склонности их к замедленному хрупкому разрушению. Наиболее универсальной разновидностью такого разрушения сплавов является нх растрескивание при воздействии коррозионных
сред. У высокопрочных закаленных сталей также наблюдается так называемое задержанное разрушение в результате развития внутренних дефектов под воздействием водорода н адсорбционно-активных примесей или вследствие миграции закалочных дефектов.
Оценку статической трещиностойкости сплавов проводят (аналогично циклической трещиностойкости) на образцах с предварительно наведенными усталостными трещинами, используя различные сравнительно простые устройства, позволяющие осуществлять мягкое (постоянная нагрузка) пли жесткое нагружение (постоянная деформация). Для испытаний пригодно большинство образцов, применяемых для оценки Aic н циклической трещнностойко-сти (см. рнс. 15.5, 15.21).
Для оценки кинетики субкритнческого роста трещин используют кинетические диаграммы растрескивания (КДР), дающие зависимости скорости роста трещины у от текущих значений К. КДР наиболее полно изучены для случаев воздействия различных рабочих сред. Типичная КДР представлена на рнс. 15.24. Ее
Рис. 15.24. Типичная кинетическая диаграмма растрескивания (I—III — стадии)
существенное отличие от типичной КДУР — наличие горизонтального второго участка (плато), свидетельствующего о постоянстве о при определенных значениях /С. Существует мнение, что четкое разделение КДР на различные участки обусловлено проявлением в пределах каждого из них доминирующего механизма субкритического роста трещины (водородное охрупчивание, анодное растворение, адсорбционное облегчение роста трещины). В связи с этим иа диаграммах конкретных пар металл— среда отдельные участки КДР могут отсутствовать. Универсальной особенностью коррозионных трещин является нх микроветвление, связанное с зернограничным ростом трещины и проявляющееся обычно в наибольшей мере в пределах плато (участок II).
Важным этапом построения КДР является определение пороговых значений К, получивших обозначение Kiscc или Kscc (для плоско-деформированного и плосконапряженного состояний соответственно). Считают, что Kxscc можно рассматривать как инвариантную характеристику исследуемой пары металл—среда. Для многих сталей, алюминиевых и титановых сплавов, испытываемых в традиционных коррозионных средах, Kiscc может снижаться до 0,25 Kic (табл. 15.4).
246
ТАБЛИЦА 15.4
ПОРОГОВЫЕ КОЭФФИЦИЕНТЫ ИНТЕНСИВНОСТИ НАПРЯЖЕНИИ ДЛЯ НЕКОТОРЫХ ПАР СТАЛЬ - СРЕДА
Сталь а0 2, МПа ^исп* °с Среда rz ISCC’_ МПа-Ум
45ХН2МФА 1630 25 Дистиллированная вода 12,4
60ХС 2190 25 » » 7,8
ЗООМ (типа 30ХН2МС2) 1740 23 * » 18,6
То же 1500 23 » » 18,0
» 1200 23 Аэрированная вода 7,0
Аустенитная сталь 1200 90 » » 7,1
0.5С—18Мп—5Сг
4340 (типа 40ХН2М) 1470 20 Дистиллированная вода 15,5
То же 1300 20 » » 27,9
4320 (типа 20ХН2) 1260 20 » » 27,9
20ГС2 1010 25 3,5 %-ный раствор NaCl 21,9
20ГС2 1030 25 То же 73,6
4340 (типа 40ХН2М) 1450 23 » 14,8
То же 1640 23 » 16,4
» » 1590 23 » 26,4
» » 1490 23 » 19,8
Hl 1 (типа 40Х5МФ) 1300 23 3,5 %-ный раствор NaCl 33
ЗООМ (типа 40ХН2МС2) 1500 23 То же 18
304L (типа Х18Н10) 210 130 42 %-иый раствор NaCl 8,1
То же 210 289 0,0 ( М растиор NaCl 8,0
Рис 15.25. Кинетические диаграммы усталостного разрушения пружинной стали 60ХС (Я =0.3; f=18 Гц):
Испытательная среда
1
2
3
4
Лабораторный воздух » »
Дистиллированная во да
» »
200
300
200
300
3,41 12.92
2.91 14.15
3.82 76,30
3,55 40,80
КДР следует снимать в условиях жесткой стабилизации состава среды н температуры испытаний (до 1—2°C). В ряде случаев представляет интерес использование температурных зависимостей кинетики СРТ для оценки с помощью уравнения Аррениуса энергии активации СРТ, что позволяет судить о доминирующем механизме и физико-химической природе процесса.
При изучении коррозионной усталости сплавов возникает также необходимость в построении КДУР на образцах с трещинами в условиях циклического нагружения и одновременного воздействия рабочих сред. Важнейшим параметром таких диаграмм являются пороговые коэффициенты интенсивности напряжений при воздействии среды ДКшс. Исследование
циклической трещнностойкостн закаленных сталей прн воздействие воды показало значительное увеличение и в припороговой области. Под воздействием воды величина &Ktnc становится структур но-чувствительной характеристикой (рис. 15.25) и существенно уменьшается по сравнению с ЛК/ь в инактнвной атмосфере (табл. 15.5), Коррозионная среда способствует увеличению скорости v в сталях со структурой мартенсита; при этом такая чувствительность особенно проявляется по мере роста К (см. рис. 15.25). При воздействии активных сред кинетика распространения трещин обнаруживает высокую чувствительность к частоте нагружения: с уменьшением частоты испытаний M(thc снижается.
247
ТАБЛИЦА 15.5
амплитудные значения пороговых коэффициентов интенсивности напряжении
Сталь д. МПа Испытательная среда Коэффициент асимметрии *Kth или ДКг/мА МПа-Км
60ХС 2200 Лабораторный воздух Дистиллировапаная вода —0,33 4,2 3,2
2100 Лабораторный воздух Дистиллированная вода —0,33 4,2 3,8
• 1950 Лабораторный воздух Дистиллированная вода —0.33 4,2 4,2
1350 Лабораторный воздух Дистиллированная вода —0,33 4,2 4,5
4340 (типа 45ХН2МА) 880 Лабораторный воздух 3,5%-ный раствор NaCl 0 15,2 15,3
1220 Лабораторный воздух 3,5 %-ный раствор NaCl 0 15,2 8,9
12N1—5Сг—ЗМо — Лабораторный воздух 3,5 %-ный раствор NaCl 0,2 22,3 10,9
12Ni(MCC) — Лабораторный воздух 3,5 % -ный раствор NaCl 0 27,1 16,3
9 Ni—4Со—0,25С — Лабораторный воздух 3,5 %-иый раствор NaCl 0 21,7 19,5
4340 (типа 45ХН2МА) — Аргон Дистиллированная вода 0,8 4,6 3,5
RQ 360А (0.6 Мп—0,6 Mo- О.18 С) — Лабораторный воздух Дистиллированная вода 0,8 7,8 6,0
* В числителе указаны значения в знаменателе — ДК^С.
БИБЛИОГРАФИЧЕСКИЙ СПИСОК
I. Фридман Я. Б. Механические свойства металлов. Т. 1. М.: Машиностроение, 1974-368 с.
2. Бернштейн М. Л., Зайжовский В. А. Механические свойства металлов. М.: Металлургия, 1979. 496 с.
3 Прикладные вопросы вязкости разрушения: Пер. с англ, М.: Мир, 1968. 562 с.
4. Рекомендация по стандартизации PC 3642—72. — Физико-химическая механика материалов, 1975, № 5, с. 5.
5. Браун У., Сроули Дж. Испытания высокопрочных материалов при плоской деформации: Пер. с англ. М_; Мнр, 1972. 248 с.
6. Панасюк В. В., Андрейкив А. Е„ Ковчик С. Е. Методы оценки трещииостойкостн конструкционных материалов. Киев: Науко-ва думка, 1972. 380 с.
7. Fracture Toughness Evaluation by R-curse Methods/ASTM STP 627. Philadelphia: 1973. 112 p.
8. Иванова В. С. Разрушение металлов. М.: Металлургия, 1979. 180 с.
9. Методические указания. Расчеты н испытания на прочность в машиностроении/Госу-дарственный Комитет стандартов СССР. Львов: .АН УССР. Физико-механический институт, 1979. 114 с.
10. Романив О. Н. Вязкость разрушения конструкционных сталей. М.: Металлургия, 1979. 176 с.
11. Нотт Дж. Ф. Основы механики разрушения: Пер. с англ. М.г хМеталлургня, 1978. 256 с.
12. Романне О. Н., Никифорчин Г. Н.—Заводская лаборатория, 1979, № 1, с. 84.
16
— СПЕЦИАЛЬНЫЕ ИСПЫТАНИЯ
16.1. КЛАССИФИКАЦИЯ
И ТЕРМИНОЛОГИЯ
Процессы поверхностного разрушения деталей машин под влиянием внешнего воздействия можно разделить на коррозию и изнашивание (ГОСТ 5272—68). Существуют различные классификации видов поверхностного разрушения деталей машин. Ниже приводится схема классификации видов поверхностного разрушения по Л. А. Урванцеву (рис. 16.1) [1].
мической коррозии процесс взаимодействия металлов со средой сопровождается возникновением электрического тока.
Электрохимическую коррозию в зависимости от условий протекания и свойств среды подразделяют на кислотную, щелочную, солевую (соответственно в растворах кислот, щелочей, солей, в расплавленных солях, щелочах, на воздухе или в газе); почвенную под воздействием блуждающих токов (например, у подземных сооружений); контактную (при контакте разнородных металлов); биокоррозию (под воздей-
Рис 16.1. Схема классификации видов поверхностного разрушения (по Л. А. Урванцеву)
16.1.1. Коррозия
Коррозионные процессы в зависимости от механизма реакций, протекающих на поверхности металла, подразделяются на химическую и электрохимическую коррозию [2, 3].
Химическая коррозия вызывается процессами, протекающими при химическом взаимодействии металла и агрессивной среды и не сопровождается возникновением электрического тока (химическая гетерогенная реакция жидкой нли газовой среды с поверхностью металла).
По химизму самих процессов на металлическую поверхность действуют: сухие газы и пары при невозможности конденсации жидкости на поверхности (газовая коррозия); жидкяе неэлектролиты (спирты, минеральные масла, органические соединения), расплавы металлов.
Электрохимическая коррозия является гетерогенной электрохимической реакцией. К ней относятси процессы, протекающие в водных растворах электролитов, влажных газах, расплавленных солях и щелочах. При электрохи-
ствием продуктов, выделенных микроорганизмами) н т. п.
Характер разрушения, кинетика н механизм процессов коррозии может резко изменяться под воздействием внешних факторов.
Коррозионное разрушение классифицируют по характеру повреждений и по условиям его возникновения (рис.. 16.2).
Общая (сплошная) коррозия. Распространяется на всю поверхность металла. Может быть равномерной (рис. 16.2, с) или (гораздо чаще) неравномерной (рис. 162,6).
Местная коррозия. Сосредоточивается на отдельных участках поверхности [имеет характер пятен (рис. 16.2,в), язв (рис. 16.2, а), точек (рис. 16.2, д) ит. п.] нли под поверхностью (рис. 16.2, е).
Избирательная коррозия. Подразделяется на компонентно-избирательную и структурно-избирательную (рис, 16.2, ж). Межкристаллитную (рис. 16.2, в) н ножевую (рис. 16.2, и) коррозию (разрушение околошовной зоны) следует отнести к избирательной.
249
Рис. 16.2. Виды коррозионного разрушения
Прн одновременном воздействии агрессивной среды и механических напряжений возникает особый внд разрушения — коррозионное растрескивание, а при воздействии переменных напряжений — коррозионная усталость.
16.1.2. Изнашивание
Изнашивание — это процесс разрушения и отделения материала с поверхности твердого тела и (или) накопление его остаточной деформации при треиии, проявляющейся в постепенном изменении размеров н (нли) формы тела (ГОСТ 23.002—78).
Износ — результат изнашивания-, определяемый в установленных единицах.
Характеристики износа: 1) линейный износ, U, мкм — изменение размера поверхности при износе, измеренное перпендикулярно ей; 2) скорость изнашивания у—dUfdt, мкм/ч—отношение величины износа ко времени, в течение которого он возник; 3) интенсивность изнашивания In =dU/dS — отношение величины линейного износа к пути трения dS, иа котором происходило изнашивание; ltl — безразмерная величина, если линейный износ н путь трення выражены в одних единицах.
В некоторых случаях целесообразно использовать интенсивность изнашивания по массе Ig. по объему lv или энергетическую интенсивность изнашивания lw\ 1g—&G/S, Iv—^VjS, где AG и ДУ—соответственно масса и объем изношенного на пути S материала. У/УУ—ЛV/FS, где Ур— работа силы трения; F — сила трения.
Многообразие условий работы деталей машин, а также материалов, используемых в технике, обусловливает различные виды взаимодействия поверхностей н, следовательно, различные виды изнашивания. Изнашивание классифицируют по кинематическим признакам, в соответствии с особенностями процесса разрушения н явлениями, вызвавшими износ, по условиям работы и др.
В табл. 16.1 приведена классификация видов изнашнвання материалов в зависимости от природы процессов, протекающих в зоне контакта, по ГОСТ 23.002—78.
Известны также классификации видов изнашивания, предложенные А. К. Зайцевым, В. А. Кисликом, Б. И. Костецким, М. М. Хрущевым и другими исследователями.
ТАБЛИЦА 16.1
ВИДЫ ИЗНАШИВАНИЯ
Признак классификации
Виды механического изнашивания
Виды коррозионно-механического изнашивания
Виды изнашивания прн действии электрического тока
Вид изнашивания
Абразивное, адгезионное, гидроабразивное (газоабразивное), эрозионное, гидроэрозион-ное (газоэрозионное), кавитационное, усталостное, изнашивание прн фреттинге Окислительное, изнашивание при фрет-тинг-коррозин Электроэрозионное
16.2. ХАРАКТЕРИСТИКА
КОРРОЗИОННЫХ ПРОЦЕССОВ
16.2.1. Химическая коррозия
Характерный признак химической коррозии — образование продуктов коррозии непосредственно в месте взаимодействия металла с агрессивной средой.
Газовая. коррозия в окислительных средах. Наиболее часто на практике наблюдается химическая коррозия при высоких температурах— газовая коррозия. Скорость газовой коррозия зависит от состава сплава, свойств образующихся продуктов коррозии, состава и свойств газовой среды, температуры н Др.
При газовой коррозии наиболее важен процесс окисления, в результате которого на поверхности образуются пленки окислов. Защитные свойства окисных пленок зависят о г их сплошности, прочности, плотности, коэффициента теплового расширения и адгезионных свойств. Условие образования сплошных окисных пленок — соотношение объемов окисла (Уд1СО) и исходного металла (Уме): если Уд[еО /Уме>1, то, как правило, образуются пленки, тормозящие процесс окисления (табл.
250
ТАБЛИЦА 16.2
СООТНОШЕНИЕ ОБЪЕМОВ ОКИСЛА И ИСХОДНОГО МЕТАЛЛА
Металл Окисел Металл Окисел
Металлы с пористы- Металлы co СПЛОШ-
ми пленками окислов ними пленками окис-
К Na к2о Na.2O 0,45 0,55 Th лов ThO2 1,35
Са CaO 0,64 Ti Ti2O3 1,48
Ва BaO 0,67 Zr ZrO2 1,56
Mg MgO 0,81 Zn ZnO 1,55
Метал ЛЫ co СПЛОШ- Ni Be NiO BeO 1,65 1,68
ними пленками OKUC- Cu Cu2O 1,64
Cd лов CdO 1,21 Si U Cr SiO2 uo2 Cr2O3 1,88 1,97 2,07
Al ai8o3 1,28 Fe Fe2O3 2,14
Sn SnO2 1,32 W WO3 3,35
16.2) , Однако нз-за возникновения напряжений в пленке, летучести окнслов и по другим причинам защитные свойства окисных пленок могут резко снижаться.
Скорость роста пленок определяется скоростью диффузии атомов среды и атомов металла. В случае образования пленок, не обладающих защитными свойствами (например, щелочные, щелочноземельные металлы), справедлив линейный закон, а для металлов и сплавов с защитными пленками — параболический * или логарифмический.
Важное значение для техники имеет механизм окисления железа и сплавов иа его основе в газах, содержащих кислород. Эти процессы подробно описаны в работах [2—6].
Окалина, образующаяся на железе, имеет сложное строение и состоит нз окислов FeO, FesO4 и Fe2O3. Соотношение толщин окисиых слоев зависит от температуры и условий окисления. FeO (вюстит) устойчив выше 570 °C, РезО4 (магнетит) — во всем интервале температур, a F2O3 (гематит) устойчив до температуры 1100 °C, выше которой начинается его диссоциация.
Присутствие водяного пара, углекислого газа и других агрессивных газов резко ускоряет окисление нелегированных сталей. Одновременно с окислением при высоких температурах происходит обезуглероживание, изменяется структура поверхностных слоев н снижаются прочностные свойства сталей.
Газовой коррозии подвержены также цветные металлы и сплавы.
На поверхности алюминия и его сплавов образуются пленки, обладающие высокими защитными свойствами (окислы тнпа AI2O3), и возможность применения этих сплавов определяется в основном не их жаростойкостью, а нх жаропрочностью при рабочих температурах.
Сплавы и а основе медн не обладают высокой жаростойкостью.
Никель н легированные сплавы на его основе имеют очень высокую стойкость против газовой коррозии, что свя
зано с защитными свойствами окисла никеля NiO и возможностью образования сложных окислов-шпннелей типа NiAl2O4, NiCr2O4.
Титан и его сплавы не обладают достаточной стойкостью в кислородсодержащих средах прн высоких температурах и могут взаимодействовать с различными компонентами газовых сред: окисью и двуокисью углерода, водяным паром, аммиаком и др. [6].
Молибден, ниобий и вольфрам обладают невысокой стойкостью в окислительных средах. Это связано с летучестью их основных окислов типа MoOg, Nb2Os, WO3 [6].
Основные методы защиты от газовой коррозии в окислительных средах: применение сталей н сплавов с высокой стойкостью прн заданных параметрах эксплуатации; защитные покрытия, наносимые термодиффузионным путем (алитирование, хромирование, силицирование, комплексное насыщение жаростойкими элементами), плазменным напылением, электронно-лучевым методом и др.; введение в рабочую среду ингибиторов, затрудняющих процессы газовой коррозии; конструктивные методы (снижение рабочей температуры поверхности детали, уменьшение скорости движения среды н др.); технологические методы (повышение чистоты поверхности деталей, применение термической обработки для создания тонких пленок, препятствующих коррозионному процессу, и др.).
Газовая коррозия в атмосфере водяного пара протекает интенсивнее, чем иа воздухе при той же температуре. Низколегированные стали окисляются в перегретом паре примерно в два раза сильнее, чем иа воздухе. Поэтому при выборе сталей для паросиловых установок жаростойкость желательно определять в перегретом паре, а не на воздухе [7, 8].
Водородная коррозия. Протекает при высоких температурах н давлениях, причем в этом случае водород взаимодействует с цементитом и обезуглероживает сталь.
Считается, что водород может диффундировать в сталь, образуя гидриды и раствор водорода в железе. Все эти процессы снижают прочностные и пластические свойства; особенно опасным является резкое падение ударной вязкости и параметров вязкости разрушения.
Поскольку скорость водородной коррозии сильно зависит от давления и температуры, при оценке возможности применения сталей в водородсодержащих средах определяют глубину обезуглероживания в зависимости от этих факторов, что позволяет прогнозировать их работоспособность. Растягивающие напряжения увеличивают скорость водородной коррозии.
Легирование сталей сильными карбидообразующими элементами (Cr, V, Ti, Мо, Nb и др.) препятствует обезуглероживанию н повышает стойкость против водородной коррозии. Например, в водородсодержащих средах стали типа 20, ЗОХМА применяют только прн температурах до 300 °C, а высокохромнстые стали — до 600 °C.
Карбонильная коррозия. Возникает под действием окиси углерода, например, в процессах получения спиртов при высоких температурах н давлениях. Заключается в образовании легко возгоняющихся веществ — карбонилов: Ме-Ь +п(СО)->Ме(СО)п.
Карбонилы — жидкости с низкой температурой кипения, их пары разлагаются на металл н
251
окись углерода. Так, пентокарбонил железа Fe(CO)5 имеет температуру кипения 102 °C, пары его при давлении 0,1 МПа и 140 °C полностью диссоциируют. Распад пентокарбоиила железа идет с увеличением объема в пять раз. Максимальное количество пентокарбоиила железа образуется при 200 °C и увеличивается с повышением давления до 30 МПа. Результатом коррозии является разрыхление поверхностного слоя, которое может проникать на глубину несколько миллиметров [7].
Чтобы избежать карбонильной коррозии, используют стойкие к ней высокохромистые стали (до 30 % Сг) и хромоникелевые аустенитные стали типа 25—20, 23—20, работающие при давлении до 35 МПа и температурах <:700°С. При более низких значениях температуры и давления можно использовать хромистые (13'— 17 % Сг) и аустенитные (типа 18-8) стали.
Сернистая коррозия. Протекает в средах, содержащих сероводород, сернистый газ, элементарную серу и другие серосодержащие вещества. Такой вид коррозии наблюдается на изготовленных из различных сталей деталях оборудования газо- и нефтеперерабатывающих заводов. Сернистые соединения могут взаимодействовать также с медью, образуя сульфидные и окисиые соединения. При взаимодействии с никелем образуется легкоплавкая эвтектика Ni—Ni3S2 с температурой плавления 625 °C, что может вызвать межкристаллитное разрушение. Кроме того, сернистый газ может окислять никель (3Ni4-SO2=NiS+2NiO).
Присутствие SO< в газе способствует превращению равномерной коррозии в язвенную.
Чтобы избежать сернистой коррозии ответственные детали н аппаратуру изготовляют из высокохромистых сталей. Дополнительное повышение стойкости против сернистой коррозии может быть достигнуто при легировании сталей кремнием и алюминием (3—5 %) [7, 8].
Коррозия в хлоре и хлористом водороде. Связала с возможностью протекания экзотермической реакции, приводящей к возгоранию металла. Такие реакции отмечены у алюминия (160°C), железа (300°C), меди (300°C). Серебро устойчиво до 425 °C, цирконий при 206 °C, а тантал прн 250 °C дают летучие соединения. Наиболее устойчивы никель и его сплавы, а также хромоникелевые аустенитные стали [7].
Особенности коррозионно-эрозионного разрушения металлов и сплавов в скоростных газовых потоках. При небольших скоростях газовых потоков влияние динамических эффектов на механизм и кинетику газовой коррозии незначительно. Однако при скоростях потоков, сравнимых со скоростью звука, кинетическая энергия газовых молекул растет пропорционально квадрату М-чнсла (М — число Маха, представляющее собой отношение скорости течения газа к местной скорости звука в газо-* образной среде) и становится сравнимой с тепловой энергией. Известно, что вблизи поверхности, обтекаемой скоростным газовым потоком, образуется пограничный слой изменения скорости, давления и температуры, в котором и определяют энергетическое воздействие среды на металл. Разрушение металлической поверхности в скоростных газовых потоках происходит вследствие механического, теплового и химического воздействия, интенсивность которых определяется составом газовой среды,
температурой, скоростью потока, условиями обтекания, режимом работы, а также составом, структурой н свойствами конструкционного материала.
Специфика динамического воздействия среды проявляется тем сильнее, чем выше температура и скорость газа.. Микррударное воздействие потока вызывает растрескивание и отслаивание окисных пленок, а также пластическую деформацию поверхностных слоев. Химическое воздействие характеризуется усилением неоднородности и локальности коррозионных процессов. Местное повышение температуры поверхности сопровождается рекристаллизацией и разупрочнением, изменяется состав и структура поверхностных слоев. Этн процессы значительно изменяют конструктивную прочность, долговечность и надежность всего изделия [9].
Коррозия в жидкометаллических средах. Протекает обычно в расплавах щелочных металлов (Li, Na, К) и тяжелых металлов (РЬ, Bi, Hg), применяемых в качестве теплоносителей в ядериой энергетике, а также в качестве нагревающей среды при термической обработке (РЬ), представляет собой сложное явление, в котором участвуют следующие процессы:
1) растворение твердого металла в жидком;
2) термический перенос массы;
3) межкристаллитное растворение;
4) взаимодействие с примесями в жидком металле;
5) образование твердых растворов н химических соединений.
Многие расплавы вызывают также охрупчивание конструкционных металлов и сплавов вследствие проявления адсорбционных эффектов (эффект Ребиндера), причем по мере убывания интенсивности влияния расплавы металлов располагаются следующим образом: свинец, висмут, кадмий, олово. С повышением температуры химическое воздействие расплавов усиливается, а эффекты адсорбционного понижения прочности и пластичности уменьшаются. Данные о характере разрушения, а также стойкости конструкционных металлов и сплавов в натрии, литнн, свинце, висмуте и их сплавах приведены в работах [2, 10, II].
16.2.2. Электрохимическая коррозия
Общие положения. При электрохимической коррозии одновременно протекают два процесса— окислительный (анодный), вызывающий растворение металла на одном участке, и восстановительный (катодный), связанный с выделением катиона из раствора, восстановлением кислорода и других окислителей на другом. В результате возникают микрогальванические элементы и появляется электрический ток, обусловленный электронной проводимостью металла и иоиной проводимостью раствора электролита.
Анодные н катодные процессы локализуются иа тех участках, где их протекание облегчено. Причины, вызывающие электрохимическую неоднородность поверхности, весьма многочисленны,- макро- и микронеоднородности металла; фазовая Н структурная неоднородность сплавов; анизотропия кристаллитов; неодиород-
262
ность н несплошность поверхностных пленок; неоднородность деформаций и напряжений. Кроме того, неоднородны и жндкне фазы, контактирующие с поверхностью. Классификация этих факторов дана в работах Н. Д. "Ромашова [3, 12]. 1
Энергетическая характеристика перехода ионов в раствор илн обратно — электродный потенциал. Стандартные (нормальные) потенциалы определены для большинства технических металлов (по отношению к раствору с активностью ионов, равной единице) н приведены в соответствующей литературе [2, 3, 13]. Однако в реальных процессах коррозии равновесные-условия обычно не достигаются, так как реакции на поверхности металла идут различными путями. Поэтому важно знать неравновесные электродные потенциалы металлов и сплавов в различных средах. Величина этих потенциалов зависит от температуры, концентрации раствора, состояния поверхности металла и других факторов; неравновесные электродные потенциалы определяются опытным путем [2, 7].
Согласно теории электрохимической коррозии разрушение металлов обусловлено работой множества короткозамкнутых гальванических элементов, образующихся вследствие неоднородности среды и металла. В данной среде металлы и сплавы с большей гомогенностью структуры и состава должны обладать более высокой коррозионной стойкостью по сравнению с гетерогенными металлами н сплавами, что часто подтверждается экспериментом.
Коррозионный процесс — весьма сложное явление, скорость которого зависит от многих факторов. Прн работе коррозионного элемента уменьшаются разность начальных потенциалов, что сопровождается уменьшением коррозионного тока. Этот процесс называется поляризацией. Различают анодную и катодную поляризацию.
Анодную поляризацию вызывают: перенапряжение ионизации металла (ноны металла медленее переходят в раствор, чем электроны отводятся в катодную область); концентрационная поляризация вследствие недостаточной скорости отвода перешедших в раствор ионов металла; возникновение анодной пассивности в связи с образованием пассивных пленок на поверхности металла. Поведение металлов и сплавов в условиях анодной поляризации характеризуется анодными поляризационными кривыми.
Процессы и вещества, способствующие уменьшению поляризации, называют деполяризаторами. Ими могут быть процессы механического характера, химические вещества, связывающие выходящие ионы в растворе в комплексные соединения.
Катодная поляризация сопровождается смещением потенциала электрода в отрицательную сторону и вызывается в основном малой скоростью электрохимической реакции соединения деполяризаторов с электронами (перенапряжение реакции катодной деполяризации) или недостаточной скоростью подвода к катодной поверхности деполяризаторов н отвода продуктов восстановления (концентрационная поляризация). Подробно виды катодной поляризации рассмотрены в работах Н. Д. То-машова [3, 12].
Коррозия металлов с водородной деполяризацией. Наблюдается при взаимодействии
большинства металлов н сплавов с кислыми средами или металлов с сильными отрицательными потенциалами в нейтральных растворах. Термодинамически водородная деполяризация возможна, когда равновесный потенциал металла отрицательнее потенциала водородного электрода (для нейтральной среды pH=7,0; £н=0,414 В).
Процесс водородной деполяризации проходит в несколько стадий: диффузия гидратированных ионов водорода к катоду; дегидратация, адсорбция и разряд ионов; рекомбинация ионов в молекулу и удаление молекул от поверхности катода. Считается, что контролирующей стадией является стадия разряда ненов водорода [2,3].
Коррозия металлов с кислородной деполяризацией. Происходит при наличии в электролите кислорода, когда процесс водородной деполяризации затруднен. Такой механизм наблюдается при коррозионном воздействии на металл воды, нейтральных растворов солей, атмосферы, а также в слабокислых средах при наличии кислорода. Кислородная деполяризация термодинамически более вероятна, чем водородная, так как равновесный потенциал деполяризации кислорода высок (+0,815 В).
Пассивность металлов. Состояние относительно высокой коррозионной стойкости, связанной с торможением анодного процесса в определенном интервале значений потенциала вследствие образования на металлической поверхности фазовых нлн адсорбционных слоев. Возможность перехода металлов в пассивное состояние (пассивация) зависит от природы металлов. Так, например, Al, Cr, Ni, Ti, AV, Mo легко пассивируются, при легировании этими элементами можно увеличить склонность сплавов к пассивации.
Переходу в пассивное состояние обычно способствуют окислительные среды, а некоторые металлы пассивируются кислородом воздуха, растворенным в среде. Изменение внешних факторов (концентрации среды, температуры и др.) может способствовать или препятствовать возникновению пассивного состояния. Депассивацию облегчают восстановительные процессы, механическое нарушение защитного слоя, действие некоторых активных ненов, повышение температуры и т. д.
С увеличением окислительной способности среды пассивация облегчается, однако в’некоторых случаях наступает явление перепасси-вацин — переход пассивированного металла в активное состояние (например, скорость коррозии нержавеющих сталей в растворах азотной кислоты прн концентрациях >80—90% резко возрастает).
В работе [12] было также установлено, что коррозионная стойкость может быть повышена путем легирования сплавов (в частности, нержавеющих сталей) катодными металлами Pt, Cu, Pd и др. Это явление связывается с возможностью облегчения пассивации вследствие дополнительной анодной поляризации сплава.
16.2.3. Влияние внешних факторов на коррозию
Скорость коррозионных процессов зависит от многих факторов, связанных как со свойствами, составом и строением металлического ма-
253
тернала, так и со свойствами среды н внешними воздействиями.
Влияние pH среды. Это влияние чаще всего проявляется в увеличении скорости коррозии с уменьшением pH, так как при этом облегчаются процессы водородной и кислородной деполяризации. Однако в кислотах, способствующих пассивации (напрнмер, HNO3), увеличение активности иоиов Н+ может замедлять коррозионный процесс. Уменьшает коррозию и увеличение активности кислот H2SO4 и Н3РО4, образующих при взаимодействии с металлами труднорастворимые соединения [FeSO4, Fe3 (РО4)2 и др.].
Увеличение pH щелочной среды (рН>7) обычно заметно сказывается только в области высоких концентраций ОН_(рН>12), что связывают с образованием растворимых соединений железа (типа NaFeO2, Na2FeO2).
Механические напряжения. Сильно влияют на кинетику и механизм разрушения металлов в агрессивных средах. В зависимости от вида напряжений и характера разрушения различают коррозию под напряжением, когда в результате действия внешних и внутренних, вернее, остаточных (после сварки, пластической деформации, термической обработки) напряжений изменяется скорость коррозионных процессов. В этом случае разрушение приобретает локализованный характер. В результате действия растягивающих напряжений и агрессивной среды может возникнуть весьма опасный вид разрушения — коррозионное растрескивание. Оно происходит при почти полном отсутствии заметной макропластической деформации и приводит к серьезным авариям. Наблюдается в агрессивных средах (аммиак, цианистый водород, растворы щелочей, нитратов, хлоридов, кислот и др.) [7,14].
В зависимости от свойств среды и сплава коррозионное растрескивание может протекать по-разному. Для объяснения механизма коррозионного растрескивания сплавов и сталей предложены различные гипотезы. Наиболее убедителен адсорбционно-электрохимический механизм.
Установлено, что с повышением температуры и увеличением концентрации среды коррозионное растрескивание усиливается. Прн интенсивной общей коррозии коррозионного растрескивания не наблюдается.
Считается, что наиболее склонны к коррозионному растрескиванию стали с мартенситной структурой; углеродистые феррите-перлитные и перлитные стали обнаруживают склонность к коррозионному растрескиванию только при высоких напряжениях (>Оо,г) и в сильно агрессивных средах (например, нитраты щелочных и щелочноземельных металлов). Хромистые аустенито-ферритные и ферритные стали менее склонны к коррозионному растрескиванию при отсутствии в них мартенсита.
Аустенитные хромоникелевые стали подвергаются коррозионному растрескиванию преимущественно в растворах хлоридов, в сероводороде, в воде с высоким содержанием кислорода. Склонность к этому виду разрушения зависит от уровня деформационного воздействия, содержания легирующих элементов и углерода и определяется в основном наличием и состоянием карбидов на границах зерен. Склонность к коррозионному растрескиванию аустенитых
сталей повышается с увеличением содержания серы.
Коррозионное растрескивание наблюдается также у легких цветных металлов и их сплавов (сплавы А!—Mg) и медноцинковых сплавах. Легирование и термическая обработка существенно снижают склонность этих сплавов к коррозионному растрескиванию.
Важное зачение в современной технике имеет явление коррозионного растрескивания под воздействием расплавов металлов [10, 11].
В случае воздействия металл коррозионной среды и переменных напряжений может развиваться процесс коррозионной усталости.
Коррозионная усталость. Явление, приводящее к преждевременному разрушению многих ответственных деталей (корабельные гребиые валы, штоки дизелей, рессоры, детали насосов н Др.). В отличие от усталостного разрушения на воздухе при коррозионной усталости разрушение имеет специфический вид хрупкого излома из многих очагов. В коррозионных средах усталостные трещины возникают в начале циклического нагружения н период нх развития составляет до 90% общего чнсла циклов до разрушения; на воздухе этот период составляет 10—30 %. Кривая коррозионной усталости непрерывно снижается с увеличением числа циклов нагружений, таким образом, можно говорить только об ограниченной выносливости, которую характеризуют условным пределом выносливости, отвечающим тому циклическому напряжению, при котором образец не сломался в коррозионной среде при заданном цикле нагружений [14].
Коррозионная усталость развивается в воде, растворах электролитов и других коррозионно-активных средах. Это явление связано с электрохимическими процессами, которым предшествует адсорбция ионов илн молекул, что может вызвать адсорбционную усталость. Адсорбция водорода на катодных участках стали при коррозии с водородной деполяризацией вызывает явление водородной усталости, которая проявляется при условии, что концентрация водорода в металле при циклическом нагружении ие падает ниже определенного минимального уровня, т- е. если десорбция водорода происходит медленнее, чем развитие усталостного процесса. Таким образом, в понятие коррозионной усталости входят также понятия адсорбционной и водородной усталости [14].
Анионитный состав, концентрация ионов. При одинаковых значениях pH могут очень существенно влиять на коррозию.
Рис. 16.3. Зависимость скорости коррозии железа от природы галоидного аниона |7]
Рис. 16.3 иллюстрирует коррозию железа в солях галоидных кислот, препятствующих пассивации большинства металлов (исключая Мо в хлористых и Mg во фтористых солях).
254
С увеличением концентрации соли скорость коррозии большинства металлов вначале растет, а затем уменьшается, что связано, с одной стороны, с увеличением электропроводности раствора, а с другой, с уменьшением растворимости кислорода.
Кислород. Растворенный кислород может и увеличить, и уменьшить скорость коррозии в зависимости от того, является ли он деполяризатором или способствует пассивации (рис. 16.4). В большинстве растворов солей, где кор-
Рис. 16.4. Влияние растворенного в воде кислорода на скорость коррозии железа [7]
розненный процесс идет с кислородной деполяризацией, с увеличением концентрации соли растворимость кислорода падает и процесс коррозии замедляется.
Температура. В большинстве случаев с увеличением температуры скорость электрохимической коррозии повышается. Однако при коррозии с кислородной деполяризацией зависимость носи г сложный характер и при уменьшении концентрации кислорода с увеличением температуры коррозия может замедляться.
Давление. Влияет на растворимость газов в электролитах; обычно с увеличением давления усиливаются процессы, идущие с кислородной деполяризацией.
Скорость движения среды. Изменяет условия подвода кислорода, ионов и условия образования и сохранения защитных пленок. В большинстве случаев увеличение скорости движения агрессивных сред усиливает процессы коррозии, а при достаточных скоростях возникают явления коррозионной эрозин и кавитации.
Основные представления о механизме кавитационного разрушения
Процесс кавитация можно представить как возникновение пузырьков пара или газа в тех областях потока, где давление паров жидкости ниже соответствующего дайной температуре, и их уничтожение (аннигиляция) в областях повышенного давления. Кавитационное разрушение — следствие механического нагружения микроударного типа, химического, теплового и электрического воздействия кавитационной зоны на металлическую поверхность.
Многие исследователи отводят основную роль в разрушении микроударному нагружению, которое обусловливает возникновение локальных пиков нагрузки порядка 1500— 2500 МПа при длительности импульса порядка 10~5с. Тепловое воздействие кавитации прояв
ляется в повышении температуры на локальных участках до 260—600 °C [15—17]. Участие процессов коррозии в кавитационном разрушении очевидно, однако доля коррозионных и эрозионных процессов меняется в широких пределах в зависимости от свойств среды я материала. Коррозионные среды всегда обладают адсорбционной активностью, так как в них содержатся поверхностно активные компоненты. Поверхностно активные вещества (ПАВ) двояко влияют на кинетику кавитационного разрушения; уменьшают микроударное воздействие, создавая на поверхности гидрофобный слой — буфер; облегчают деформацию и разрушение за счет проявления эффекта адсорбционного понижения прочности и пластичности (эффекта Ребиндера). Установлено, что влияние ПАВ зависит от интенсивности кавитационного воздействия: при высокой интенсивности добавки ПАВ ускоряют разрушение, а прн невысокой— несколько уменьшают скорость разрушения.
Установлено, что максимальная интенсивность кавитационного разрушения наблюдается в кислых средах и увеличивается с понижением pH среды. Увеличение агрессивности среды приводит к возрастанию роли коррозионного фактора, причем в тем большей степени, чем меньше коррозионная стойкость материала. В нейтральных, как и в щелочных средах, коррозионные процессы происходят с кислородной деполяризацией и на поверхности образуются плотные окисиые пленки, затрудняющие смачивание и изменяющие характер адсорбционных процессов. Одновременно изменяются и условия возникновения и смыкания кавитационных пузырей. Все это, как правило, увеличивает кавитационную стойкость сталей и сплавов.
Кавитация наблюдается не только в воде и растворах электролитов, но и в расплавах легкоплавких металлов, причем механизм н кинетика разрушения в этом случае существенно меняются, что связано с физико-химическими свойствами среды и высокой температурой. При смыкании кавитационных пузырьков в расплавах выделяется в несколько раз больше энергии, чем в воде, соответственно возрастает разрушающее действие кавитационной зоны. Так, кавитационное разрушение в ртути происходит в 10—20 раз быстрее, чем в воде. В то же время высокие температуры расплавов вызывают разупрочнение конструкционных материалов и резко усиливают процесс!: коррозии (растворение конструкционных материалов в расплавах, взаимодействие с примесями и др.).
Таким образом, кавитационное разрушение в расплавах протекает обычно с большой скоростью и сопровождается значительными изменениями конструктивной прочности и надежности материалов.
16.3. ХАРАКТЕРИСТИКА ТРЕНИЯ И ПРОЦЕССОВ ИЗНАШИВАНИЯ
Основные законы внешнего трения твердых тел
Внешнее тренне —- явление сопротивления относительному перемещению, возникающему между двумя телами в зонах соприкосновения поверхностей по касательным к ним, сопровождаемое диссоциацией энергии (ГОСТ 23.002— 78).
В зависимости от наличия относительного
движения различают: трение покоя и тренне движения. В зависимости от характера движения-— тренне скольжения и трение качения. В зависимости от наличия смазочного материала—трение без смазочного материала, трение со смазочным материалом.
С энергетической точки зрения тренне — диссипативный процесс, характеризующийся превращением внешней работы, затраченной на преодоление сил трения в тепловую (75— 100%), электрическую, химическую н другие виды эиергин. Небольшая часть работы трення затрачивается на увеличение внутренней энергии поверхностных слоев контактирующих тел (ие более нескольких процентов) [19, 20].
Основные положения, вытекающие из классических представлений о природе трения [21]: 1) сила трения пропорциональна нагрузке; 2) коэффициент трения определяется природой контактирующих тел и не зависит от геометрической площади нх касания, а также от условий трения (скорость, нагрузка и т. д.); 3) коэффициент трения покоя#выше коэффициента трения движения.
Исследования в области теории трення и опыт, накопленный практикой в течение последнего столетия, показали, что данные положения носят весьма приближенный характер и в ряде конкретных случаяев не выполняются. Было установлено, что коэффициент трения может изменяться в широких пределах в зависимости от условий трения (нагрузка, скорость, характер среды, состояние поверхности и др.).
Сложность процессов, протекающих в зоне контакта твердых тел, способствовала возникновению различных гипотез и теорий внешнего трения. Известны молекулярная, механическая, молекулярно-механическая, электрическая и другие теории трения. Наиболее глубокое развитие получила молекулярно-механическая теория внешнего трения, предложенная советским ученым И. В. Крагельским и независимо от него английским физиком Ф. Боуденом. Эта теория базируется на представлении о двойственной природе трения н дискретном характере контакта между реальными поверхностями твердых тел. Неровности на поверхности любого твердого тела обусловливают контакт на отдельных элементарных площадках (пятнах) касания. Общая площадь фактического контакта складывается нз суммы площадей ^бт-дельных пятен касания £ф = 2 н зависит i=l
от внешней нагрузки N, механических свойств контактирующих тел. Для неподвижного (статического) контакта S яз NIH, где Н — твердость наименее твердого компонента из двух контактирующих тел [22]. Для подвижного контакта прн треиии (аУ) /И, где а — коэффициент, зависящий от способности металла к упрочнению в процессе трения [31]. Чем выше способность металла к упрочнению, тем меньше а. При наличии упрочнения металла в зоне контакта 2-5-7. В случае разупрочнения поверхности, например в результате интенсивного фрикционного нагрева, а, очевидно, может принимать более высокие значения. Взаимодействие поверхностей при трении проявляется в формировании фактической площади контакта, а также в возникновении между ними связей, создающих сопротивление их взаимному перемещению. Согласно молекулярио-
механической теории поверхностные связи при тренин формируются главным образом вследствие упруго-пластнческой деформации и адгезионного взаимодействия их поверхностей. По Ф. Боудену, сила трения — это сумма сопротивлений срезу металлических соединений Fc н Fa — сопротивлений пластическому оттеснению (пропахиванию) менее прочного металла прн движении внедрившихся в него мнкроиеровно-стей более твердого: F=Fo4-Fh=GS$-|-tS, где 6 — сопротивление металла иа срез; — площадь фактического контакта; т — предел текучести менее твердого металла; S — площадь профилей царапин [22].
В соответствии с теорией И. В. Крагельского сила трения состоит из молекулярной и механической составляющих. Важная составная часть теории трения И. В. Крагельского —положение о фрикционных связях — пятнах касания, возникающих, существующих н разрушающихся пр» совместном действии нормальных н тангенциальных сил. Трение —• процесс преодоления фрикционных связей [23, 24].
Коэффициент трения для единичной движущейся неровности может быть определен из соотношения: /= (тс/А)-( p l Л'-< V hjr, где т0— удельная прочность молекулярных связей на сдвиг; Рт — фактическое давлейие в контакте; £ — коэффициент упрочнения молекулярных связей в условиях действия сжимающих напряжений; Ка;=0,55— для пластического контакта, при упругом контакте Kx~0,19ar (ссг— коэффициент гистерезисных потерь, равный отношению работы, превращенной в тепло, к энергии упругой деформации); h-—глубина внедрения неровности в поверхность коитрте-ла; г — радиус единичной неровности.
Формула обобщенного закона трения, полученная И. В. Крагельским, имеет внд: [= = (а5ф/)У)+р, где d и р — параметры, зависящие от молекулярных н механических свойств контактирующих поверхностей [23].
Изнашивание металлов при трении
Важнейшую роль в явлении изнашивания играют процессы упругой и пластической деформации, протекающие в активных слоях контактирующих тел. Пластическая деформация локализуется в микрообъемах поверхностного слоя металла, примыкающих к пятнам касания. В ходе .трення пятиа касания непрерывно перемещаются по поверхности трения, вовлекая последовательно всю поверхность в деформационный процесс. В результате этого на поверхности трення достигается высокая равномерность и высокая степень пластической деформации, которые обычно недостижимы в условиях объемного деформирования. Установлено, что некоторые микрообъемы материала в зоне контакта подвергаются действию всесторонних сжимающих напряжений, в результате чего может происходить пластическая деформация даже таких хрупких высокопрочных фаз, как карбиды, мартенсит.
Пластическая деформация (основной первичный процесс, обусловливающий возникновение износа в результате разрушения, являющегося конечным процессом) приводит к образованию в поверхностных слоях трущихся тел большого количества дефектов кристаллического строения (точечных дефектов, дислокаций, дефектов упаковки, двойников и др.), сильной фрагмен-
256
тацни зерен и текстурированию металла. Это обусловливает высокий уровень деформационного упрочнения поверхностных слоев металлов, который обычно .резко снижается с увеличением расстояния от поверхности трения.
За несколько последних десятилетий в СССР и за рубежом были проведены обширные исследования взаимодействия микронеровностей в зоне контакта трущихся тел. Было показано, что каждая единичная микронеровиость, внедренная в поверхность контртела, прн движении создает в нем две зоны напряжений, различ-
Рис. 16.5. Схема взаимодействия движущейся жесткой сферической неровности и идеально гладкой поверхности металла [21, 23):
/ — зона сжимающих напряжений: 2 — зона растягивающих напряжений
/. Упругое оттеснение. Данный вид напряженного состояния в зоне контакта возникает при трепни высокопрочных материалов, а также в период установившегося изнашивания. Разрушение поверхности носит усталостный характер (фрикционная усталость). Число циклов до разрушения велико. (п->оо). Для сталей Л//? <0,01; для цветных металлов h/R с <0,0001.
II. Пластическое оттеснение металла. Характеризуется остаточной деформацией поверхности после прохода микровыступа: сухое трение Л//? <0,1; граничное трение Л/А’<0,3.
При данном виде напряженного состояния возникает малоцикловая фрикционная усталость в поверхностных слоях металла. Наиболее часто это имеет место в период приработки, а также’ при треннн пластичных материалов (i<n<icp).
III. Микрорезание. Характеризуется образованием микростружкн; сухое трение Л/Р>0,1; граничное трение h/R >0,3.
Разрушение материала происходит за 1 цикл (п— 1).
Это аварийный вид разрушения поверхности, наиболее часто наблюдающийся при воздействии закрепленного абразива иа поверхность металла.
Рис. 16.6. Виды 'нарушения фрикционных связей [24]
ных по знаку (рнс. 16.5). Перед фронтом микронеровиости — в зоне сжимающих напряжений— возникает валик из деформированного металла. Позади микронеровиости в результате действия силы трения в материале контртела возникают растягивающие напряжения. Адгезионное взаимодействие происходит на границе зоны сжимающих напряжений с неровностью. По мере смещения микровыступа в пределах, ограниченных прочностью адгезионных связей на контакте, валик растет за счет материала, вытесненного прн смещении. Позади выступа соответственно растет деформация растяжения. Прн достижении критической величины перемещения неровности происходит разрушение молекулярных связей на контакте; прн этом неровность подминает валик или перескакивает его. Таким образом, материал контртела испытывает за один проход микровыступа последовательное воздействие сжимающих и растягивающих напряжений.
Виды нарушения фрикционных связей
И. В. Крагельский с сотр. показали, что в зависимости от прочности молекулярных связей на сдвиг по отношению к пределу текучести материала ti/g? н величины относительного внедрения неровностей сжатых поверхностей h/R возможны пять видов нарушения фрикционных связей и соответственно пять видов напряженного состояния в зоне контакта неровности с контртелом (рис. 16.6) [24].
IV. Схватывание пленок на поверхности материалов и их разрушение. Происходит прн трении с твердыми смазками, а также в случае образования на поверхности металлов прочных окисиых пленок. Это благоприятный вид разрушения фрикционных связей, так как процесс разрушения локализуется внутри поверхностной пленки; разрушение же основного материала происходит за очень большое число циклов («-*-оо/ Условие возникновения данного вида нарушения фрикционных связей — положительный градиент прочностных свойств в поверхностном слое материала dxldh>I\ где т — сопротивление сдвигу; h—расстояние от поверхности.
V- Схватывание металлических поверхностей. Наблюдается при отрицательном градиенте прочностных свойств в поверхностном слое материала dx/dh<fi. Аварийный вид нарушения фрикционных связей: разрушение материала происходит за один проход мнкровыступа (п=1).
Схватывание металлов при трении
Под схватыванием металлов обычно понимают явление образования металлических связей между участками поверхности контактирующих тел при сближении их на расстояния порядка межатомных [25]. Схватывание металлических поверхностей происходит прн различных условиях трения: между одинаковыми и различными материалами, на воздухе, в газовых и жидких средах прн высоких и отрица
17—280
257
тельных температурах. Наиболее интенсивно схватывание наблюдается в вакууме, в восстановительных н нейтральных средах. Повышение температуры в зоне контакта в общем случае способствует активизации схватывания [26, 27]. Установлено, что пластическая дефор-мация металла в зоне контакта — необходимое условие возникновения н развития узлов (мостиков) схватывания. Ряд исследователей считает, что пластическая деформация прн этом главным образом разрушает поверхностные пленки, загрязняющие металлические поверхности, а также способствует формированию и развитию контакта между вновь образовавшимися чистыми («ювенильными») поверхностями [22, 23].
Существуют, однако, данные, свидетельствующие о том, что пластическая деформация приводит поверхностные слон металла в возбужденное («активизированное») состояние, характеризующееся высокой плотностью дефектов кристаллического строения, текстурой, понижением работы выхода электрона и др. [20, 28]. Такая «активизация» поверхности контактирующих тел способствует развитию процесса схватывания.
Большое влияние на развитие схватывания оказывают тип кристаллической решетки, наличие илн отсутствие взаимной растворимости, а также деформационные характеристики контактирующих металлов. В работах Б. И. Ко-стецкого, Н. Л. Голеги показано, что в условиях трения могут наблюдаться две разновидности схватывания металлических поверхностей: атермическое (I рода) и тепловое (II рода) [20, 27]. Схватывание I рода развивается при малых скоростях скольжения (<0,5 м/с) и больших удельных давлениях в условиях невысокого фрикционного нагрева поверхностей (средняя объемная температура в поверхностных слоях ие превышает 100—150°C).
Схватывание I рода развивается в результате интенсивной пластической деформации поверхностных слоев металла, приводящей к разрушению окисных пленок и формированию контакта между’ чистыми поверхностями. В результате схватывания I рода происходит глубинное вырывание и перенос менее прочного металла на более прочный.
Схватывание II рода возникает прн больших скоростях скольжения (2?! м/с) и давлениях, когда интенсивно нагреваются поверхностные слон (до 400—1100 °C), что ведет к термическому разупрочнению поверхностных слоев металлов. В этих слоях, по-видимому, протекают диффузионные процессы, приводящие к изменению структуры и химического состава. Структурные изменения металлов при трении Деформация металла в зоне контакта сопровождается выделением тепла трення, вследствие чего поверхностные слои нагреваются. Температура нагрева определяется интенсивностью выделения тепла н скоростью отвода его нз зоны трения. Интенсивность тепловыделения в отдельном пятне касания определяется из соотношения [29]: g=(fNv)/AI, где f — коэффициент трения; А’ — нормальная нагрузка; v — скорость скольжения; А — площадь пятна; I — механический эквивалент тепла.
При треннн различают: 1) температуру на единичном пятне контакта (температуру вспышки); 2) среднюю температуру поверхности;
3) среднюю объемную температуру; 4) предельную суммарную температуру на поверхности трення [23].
На пятнах фактического контакта, размеры которых 1—10 мкм, возникают температурные вспышки (импульсы) длительностью 10-4— 1(W с. Температура вспышки может достигать температуры плавления контактирующих металлов. Температура вспышки оказывает большое влияние иа возникновение и развитие трибохимических реакций между поверхностью металла и окружающей средой. Расчет н экспериментальное измерение температуры вспышки весьма сложны.
Средняя температура поверхности характеризует температурные условия в объемах и на участках поверхности, соизмеримых по величине с размером зерен (порядка 10—100 мкм). В случае контакта одинаковых материалов прн круговой форме площадки касания (по Герцу) средняя температура поверхности может быть определена [30] по формуле: /Ср= =9 fNv/64I7.a, где X — коэффициент теплопроводности материала; а — радиус площадки касания.
Объемная температура характеризует температурные условия в объемах поверхностного слоя материала размером—0,1—1,0 мм и более. Объемную температуру легко определить экспериментально с помощью мнкротермопар [19].
Предельная температура на поверхности трення t% определяется как сумма температуры вспышки /вс н средней температуры /Ср поверхности: /2 — tw+tcp.
Большое количество дефектов кристаллического строения в поверхностных слоях трущихся тел, а также повышенные температуры обусловливают интенсивное развитие диффузионных процессов, приводящих к изменению структуры, химического и фазового состава материалов. Физико-химическое взаимодействие поверхности металла с окружающей средой приводит к образованию пленок так назьь ваемых вторичных структур. Как показано в работах Б. И. Костецкого, в зависимости от природы материалов и условий трення (нагрузка, скорость, характер среды и др.) на поверхности трення могут возникать два типа вторичных структур [20].
1. Пересыщенный твердый раствор кислорода или другого активного элемента среды в металле. Образующаяся на поверхности пленка данной структуры имеет ультраднсперсиое ориентированное строение. Предполагается, что она обладает «сверхпластичиостью», благодаря которой легко перемещается по поверхности трення.
2. Пленка окислов (нли других химических соединений) нестехиометрического состава с дефицитом кислорода.
Вторичные структуры, обладая высокой прочностью, теплостойкостью, хорошими антифрикционными свойствами, пониженной теплопроводностью хорошо защищают поверхность основного материала от разрушения (рис. 16.7) и способствуют развитию нормального изнашивания [20].
Если пленки вторичных структур обладают повышенной хрупкостью, то они интенсифицируют изнашивание. Установлено, например, что насыщение поверхности трення титановых
258
сплавов, сталей и .других металлических материалов водородом при трении в водорсдсодер-жащих средах (вода, авиационное топливо, некоторые типы смазок, глицерин и др.), а также в паре с пластмассами, деревом вызывает резкое охрупчивание поверхностного слоя.
Рис. 16.7. Влияние скорости скольжения (v) на толщину и состав возникающей окисной пленки, а также на интенсивность износа сталей [30]::
1—предельная толщина окисной пленки; v<v\—износ пленки Fe2O3; V|<v<w2— механический износ пленки Fe2Os; v2<-'v<vt— износ пленки Fe3O< и механический износ; v<<v<ve—износ пленки Fe3O4; v>vG—механический износ и износ пленки Fe^Gi
Интенсивное выкрашивание хрупкого наво-дороженного слоя металла способствует развитию особого вида ускоренного изнашивания поверхности трения, получившего название водородного (водородный износ) [32].
Диффузионные процессы в микрообъемах металла, примыкающих непосредственно к поверхности трения нли к пленкам вторичных структур, могут приводить к значительным структурным изменениям в этих микрообъемах. Фрикционный нагрев способствует протеканию в поверхностном слое процессов отпуска, возврата и рекристаллизации, что приводит к разупрочнению поверхности, снижению ее несущей способности, усилению схватывания.
В тяжелых условиях трения (высокие давления и скорости, отсутствие смазки), когда имеет место интенсивный фрикционный нагрев, в поверхностном слое сталей может происходить обратное а^у-превращение. Возникает так называемый аустенит трения.
Деформация, снижающая температуру Аси облегчает образование аустенита трения. И. М. Любарский с сотр. обнаружил на поверхности трения стали 20Х2Н4А аустенитный слой толщиной несколько микрометров. После прекращения трення в процессе охлаждения этот аустенит полностью нли частично распадался [33]. Аустенит трения в ряде случаев обладает повышенной устойчивостью н может сохраняться в структуре стали после охлаждения до комнатной и более низких температур. Это объясняется высоким уровнем его легиро-ванности, а также стабилизирующим влиянием деформационного н фазового наклепа. Поверхностный слой обогащается легирующими элементами в результате их диффузии нз глубинных слоев металла (термоднффузня, восходящая диффузия), а также из окружающей среды. Так, при термическом разложении смазки в зоне контакта поверхность металла может насыщаться углеродом н другими элементами, содержащимися в смазке. Аустенит
Ю*
трення, обладая повышенной прочностью, теплостойкостью, может увеличивать сопротивление стали изнашиванию [33, 34]. Образование аустенита прн трении и его ускоренное охлаждение (вторичная закалка) приводит к формированию белых (нетравящнхся) слоев на поверхности стальных деталей. Белые слои характеризуются высокой твердостью (Нц = — 10-J-20 ГПа) н хрупкостью. Структуры белых слоев н условий их возникновения при треннн изучали Б. Д. Грозии, К. В. Савицкий, И. М. Любарский и другие исследователи. Установлено, что белые слон характеризуются высокой дисперсностью структуры, химической неоднородностью н сложным фазовым составом. В них присутствуют аустеннт (20—80 %)р мелконгольчатый мартенсит н карбиды. В условиях динамического нагружения белые слом из-за высокой хрупкости интенсивно выкрашиваются, что ведет к ускоренному разрушению поверхности.
Кинетика процесса изнашивания
На основании многих исследований установлено, что изнашивание во времени протекает неравномерно н носнт многостадийный характер. При работе деталей н узлов машин обычно наблюдаются три периода изнашивания: приработка (/); установившееся (нормальнее) изнашивание (2); катастрофическое (аварийное) изнашивание (3) — рис. 16.8. Период приработки характеризуется повышенной скоростью изнашивания, постепенно снижающейся
Рис. 16.8. Зависимость износа (Q) и высоты микронеровностей (Ra) на поверхности трения от продолжительности работы (с)
во времени. Условия трення пары постепенно меняются, так как на поверхности трущихся тел формируется новый, характерный для конкретных условий нагружения рельеф н происходят структурные нзменення материалов. Площадь фактического контакта при этом возрастает, а среднее давление и'температура в зоне контакта снижаются [35]. Как только структура н рельеф на поверхности материалов становятся оптимальными для данных условий треиня, скорость нх изнашивания снижается до минимума.
Период установившегося изнашивания характеризуется относительным постоянством условий трения и скорости изнашивания. Коэффи-
259
циент трения практически не изменяется. В этот период в поверхностных слоях контактирующих тел устанавливается динамическое равновесие между процессами упрочнения н разупрочнения, образования новых структур и их разрушения [20, 33]. Структурные изменения выражены относительно слабо н в поверхностных слоях материалов сохраняется образовавшаяся и период приработки оптимальная структура (н соответствующий ей рельеф). Износостойкость деталей машин в период установившегося изнашивания, а также время наступления катастрофического изнашивания в сильной степени зависят от характера рельефа и структуры, образовавшихся на поверхности материалов в период приработки. Поэтому важно умение управлять процессами формирования рельефа н структуры на поверхности деталей машни в начальный период изнашивания (в период приработки). ‘
Период аварийного изнашивания наступает вследствие изменения зазоров в трущихся сопряжениях, нарушения установившейся геометрии контакта иди в результате изменений режима работы узла (машины). Прн этом условия трения изменяются: растут давление, температура, ухудшаются условия смазки в зоне контакта, которые вызывают в поверхностных слоях материалов нежелательные структурные нзменення. Динамическое равновесие между процессами упрочнения и разупрочнения, существовавшее в течение всего периода установившегося изнашивания, нарушается. Происходит снижение уровня прочности и несущей способности поверхности материалов, которое обусловливает развитие катастрофических видов нарушения фрикционных связей (схватывание металлических поверхностей, микрорезаине).
Требования, предъявляемые к износостойким материалам
Выбор и разработка износостойких материалов— весьма сложная задача, так как поведение материалов при трении обусловлено не только нх свойствами, но н конкретными условиями нагружения. В зависимости от условий трения н назначения узла меняется н комплекс требований к материалам. Опыт показывает, что при создании износостойких пар трения следует основываться на физических и механических свойствах контактирующих тел ц разделяющих нх пленок, покрытий [25]. Износостойкие материалы должны обладать высокой прочностью; высоким сопротивлением’ усталостному разрушению; теплостойкостью; способностью к образованию прн трении прочных пленок вторичных структур; способностью к хорошему удержанию смазки на поверхности.
Известны соотношения, связывающие параметры изнашивания материалов с их прочностными характеристиками. Для случая изнашивания металлов закрепленным абразивом Q^kNIH, где 7V — нагрузка; Н— твердость материала; Q— износ материала; k — постоянный коэффициент [6].
Для случая адгезионного изнашивания действует формула Арчарда [36]: №=(Л/3) (N/H), где W — объем изношенного металла; N — нагрузка; И — твердость; k — вероятность отделения частицы износа с пятна контакта (Л=Ю-2-г-Ю-7).
Для усталостного механизма изнашивания справедливо выражение [24]: п— (ов/£т)*, где п — число циклов, приводящее к отделению частиц износа с поверхности трения; ов — временное сопротивление разрыву; /г — постоянная Величина; t — показатель степени (изменяется в пределах 2—12); х— удельная сила трения.
Приведенные зависимости параметров изнашивания от механических свойств материалов носят весьма приближенный характер и справедливы лишь для определенных условий трения. Опыт показывает, что ни одна из механических характеристик, взятая в отдельности, не может служить надежным критерием при выборе износостойких материалов. Известно, например, что материалы, имеющие близкие значения твердости или предела текучести, но различную микроструктуру, обладают в ряде случаев, разным уровнем износостойкости [прочностные свойства (НВ, ов, о0,2 и Др.) характеризуют свойства макрообъемов материала, а сопротивление материалов изнашиванию сильно зависит от свойств микрообъемов и отдельных структурных составляющих].
Свойства, определяемые прн стандартных механических испытаниях, не могут характеризовать прочность материала в условиях напряженного состояния, возникающего в зоне трения. В связи с этим в последние годы разрабатываются методы оценки фрикционной прочности материалов — испытания на фрикционную усталость и на фрикционную теплостойкость [24, 27].
В процессе трення происходят изменения структуры н свойств материалов. Их сопротивление изнашиванию определяется не столько исходным уровнем прочностных свойств, сколько свойствами новых структур, возникающих на поверхности трения. Структура, формирующаяся на поверхности трення материалов, возникает вследствие упрочнения слоев металла по мере их износа за счет наклепа и диффузионных процессов. Развивая представления о направленности превращений, обеспечивающих упрочнение поверхности трения, Б. И. Костец-кий выдвинул положение о структурной приспосабливаемое™ материалов при тренин [20], т. е. перестройке исходной структуры материалов в новую в направлении максимального упрочнения. При этом происходит ориентация структуры относительно направления действия силы трения. Структурная приспосаблпваемость7 носит универсальный характер, так как она наблюдается при трепни любых материалов в определенном диапазоне внешних условий (скоростей, давлений и др.). В результате развития структурной приспосабливаемости все виды взаимодействия трущихся тел и окружающей среды локализуются в слоях вторичных структур.
При этом снижается интенсивность изнашивания, она приобретает нормальный характер. Благодаря структурной приспосабливаемости, можно максимально использовать способность материалов к упрочнению н положительное влияние внешней среды на трение н изнашивание, а также создавать новые материалы путем направленного их легирования и новые составы рабочих сред, обеспечивающих достижение высокой поверхностной прочности (и износостойкости) деталей машин.
260
Механизм упрочнения сталей н сплавов зависит от' природы легирования. Известно, например, что значительной износостойкостью прн трении с высокими давлениями и ударном нагружении обладает высокоуглеродистая марганцевая аустенитная сталь 110Г13Л. Повышенная износостойкость этой стали обусловлена ее способностью к интенсивному деформационному упрочнению. При треиин упрочнение связано с образованием в поверхностном слое большого количества дефектов кристаллического строения (дислокаций, дефектов упаковки, двойников деформации), а также с взаимодействием этих дефектов с атомами углерода, растворенного в аустените [38]. Перспективные износостойкие материалы — мета-стабнльные марганцевые и хромомаргаицевые аустенитные стали, содержащие 0,4—0,8 % (по массе) С. Образование на поверхности данных сталей мартенсита деформации, его ориентированное расположение по отношению к действию силы трения обусловливают интенсивное упрочнение поверхности. Вследствие этого нестабильные марганцевые н хромомар-ганцевые аустенитные стали обладают повышенной износостойкостью в условиях развития адгезионного н усталостного разрушения поверхности [21].
16.4. МЕТОДЫ ИСПЫТАНИЙ
16.4.1. Испытания на коррозионную стойкость
Задачи коррозионных испытаний: получение сравнительных данных о коррозионной стойкости материалов и покрытий в различных средах; изучение кинетики и механизма процессов коррозии. Коррозионное разрушение в значительной степени определяется условиями работы материалов (температурой, составом среды, режимом работы, напряжениями, скоростью движения среды, давлением н др.), поэтому ясно, что одни только лабораторные испытания не могут дать точную оценку поведения материалов в условиях эксплуатации [44, 46]. В связи с этим широко используются испытания в условиях, моделирующих эксплуатационные н эксплуатационных.
При испытаниях на коррозионную стойкость необходимо знать химический состав, свойства, структуру, технологию производства образцов. Онн должны иметь простую форму, чтобы обеспечивать необходимую точность замеров площади н удаление продуктов коррозии. Считают, что толщина образцов не должна быть очень большой, поэтому испытания обычно ведут на листовых образцах после прокатки (толщина 5—10 мм, размер 26X40 мм; диски диаметром 30 мм), илн на цилиндрических образцах (диаметр 10—20 мм, высота 40 мм). Одновременно испытывают не менее трех образцов, причем коррозионную стойкость контролируют на строго определенной поверхности, а остальную поверхность изолируют.
Качественными показателями коррозии является изменение внешнего вида образцов и жидкой среды (окраска, продукты коррозии, осадок). Микроскопические исследования позволяют получать данные о характере развития коррозионных процессов, роли структурных н фазовых составляющих; определять анодные н
катодные участки металлической поверхности; обнаруживать межкристаллитную коррозию; определять особенности коррозионного растрескивания. Для этих целей используют микрофотографирование и мнкрокиносъемку, в частности для исследования кинетики коррозионных процессов, развития коррозионных трещии н др.
К качественным методам оценки коррозии относят также испытания с использованием индикаторов, позволяющих определить анодные и катодные участки поверхности [5].
Для количественных измерений применяют массовый, объемный, электрохимический, магнитометрический, манометрический и другие методы [2, 44—46].
Массовый метод оспован иа определении массы после выдержки в течение заданного времени в рабочей среде, прн этом определяют либо прибыль, либо убыль массы. В последнем случае следует все продукты коррозии удалить либо механическим путем, либо травлением реактивами, которые растворяют только продукты коррознн. Например, продукты коррознн с алюминия удаляют обычно 5 %-иым раствором HNOg, а со стали, например, 10 %-ным раствором лимоннокислого аммония, нейтрализованного аммиаком.
Скорость коррозии [г/(м2’ч)] находят по формуле KM=(go—gi)/Sx, где gc— масса исходного образца, г; gi— масса образца после испытаний, г; S — площадь поверхности, мм2; т — время испытаний, ч.
Прн количественной оценке рекомендуется учитывать фактор неравномерности коррозии:
(Sr/So) 100 %, где SK—площадь образца, подвергшаяся коррозии, мм2; 50 — общая площадь образца, мм2.
Пересчет массового показателя коррознн на глубинный (мм/год) производится по формуле: Лл=8,76 Км/у, где Км — массовый показатель коррознн; у — плотность металла.
Согласно ГОСТ 13819—68 для оценки коррозионной стойкости металлов рекомендуется десятибалльная шкала (табл. 16.3).
Для определения глубинных показателей предложены различные приборы, основанные на применении индикаторов часового тнпа [2, 7]. Потеря массы н глубинный показатель коррознн наиболее распространенных сплавов приведены в табл. 16.4.
ТАБЛИЦА 16.3
ШКАЛА ОЦЕНКИ КОРРОЗИОННОЙ СТОЙКОСТИ
Группа стойкости Скорость коррозии металла, мм/год Валл
Совершенно стойкие <0,001 1
Весьма стойкие . >0,001—0,005 2
То же >0,005—0,01 3
Стойкие .... >0,01—0,05 4
» .... >0,05-0,1 5
Пониженностойкне >0,1—0,5 6
» >0,5—1,0 7
Малостойкие . . >1,0—5,0 8
» ... >5,0—10,0 9
Нестойкие . . . >10,0 10
261
о
X о й £ ф ед CJ ф ж ф м ед о о к
к эд о s £ 2 ж S « а ед CJ о № М •%* О ж S ед о и о ч ф о Б
о о о ь f—< о о сз ж о
ё г~ и № СО о Q К ж ж к
>> - -
Сц к 1—1 >— 1—1 > > >
Объемные методы. В случае протекания процесса коррозии с водородной деполяризацией количество растворенного металла пропорционально количеству выделившегося водорода, что позволяет определить скорость коррозии по количеству выделившегося водорода [2, 44, 46].
Скорость коррозии Ko6 = V/tS, где V — объем водорода; S — площадь поверхности образца, мм2; т —время испытания. Если процесс коррозии протекает частично с кислородной деполяризацией, то результаты испытания соответственно снижаются.
Известен также метод измерения количества поглощенного кислорода для процессов, идущих с кислородной деполяризацией [46].
Электрохимические методы. Позволяют изучать механизм и кинетику электрохимической коррозии и заключаются в измерении электродных потенциалов н снятии поляризационных кривых (рис. 16.S), которые исследуются либо гальваностатнческим, либо потенциостатическим методами.
Рис. 16.9. Потенциостатическая поляризационная кривая металла (схема):
АВ активное растворение металла; ВС — активнопассивное состояние; CD — устойчивое пассивное состояние; DE — перепассивация; — потенциал начала пассивации: & — потенциал конца пассивации; Ез — потенциал перепассивации; it — критический ток пассивации; 12 — ток полной пассивации
Гальваиостатический метод заключается в измерении стандартных потенциалов металла при пропускании токов заданной плотности, а потенциостатический— в измерении плотности тока прн постоянном во времени потенциале. Промышленность выпускает потенциостаты (П5827, П5827М, П5848), позволяющие проводить комплексные исследования этим методом.
Измерение механических свойств после коррозионных испытаний используется для оценки влияния рабочей среды и проводится обычно на стандартных (или специальных) образцах. Испытания позволяют определить влияние рабочей среды на характеристики прочности и пластичности.
Испытания на межкристаллитную коррозию (МКК) проводят в соответствии с ГОСТ 6032—75. Этот ГОСТ распространяется иа стали ферритного, аустенито-мартенситного, аусте-нито-ферритного и аустенитного классов; сплавы на железоникелевой основе, а также на двухслойные стали н биметаллические трубы с плакирующим илн основным слоем нз этих марок сталей.
262
Испытания по метод у AM в водном растворе меди сернокислой (160 г CuSO4 по ГОСТ 4165—68) или медного купороса по ГОСТ 19347—74, серной кислоты (100 мл плотностью 1,835 г/см3 по ГОСТ 4204—66) и 1000 мл воды с добавлением медной стружки проводят при температуре кипения в колбе с обратным холодильником илн в бачке с крышкой, снабженной холодильником, путем выдержки образцов в течение 24 ч для сталей марок 08Х17Т, 15Х25Т, 08Х17Н5МЗ, 08Х22Н6Т, 08Х21Н6М2Т, ОЗХ16Н15МЗБ, 09Х16Н15МЗБ, 08Х17Н13М2Т, 10Х17Н13М2Т, 10X17H13M3T, 08Х17Н15МЗТ, 04Х18Н10, 08Х18Н10, 08Х18Н10Т, 06Х18Н11, 03X18HI2, 08Х18Н12Т, 08Х18Н12Б; 15 ч для сталей марок 20Х13Н4Г9, 09Х15Н8Ю, 07Х16Н6, 09Х17Н7Ю, 08Х18Г8Н2Т, 10Х14Г14НЗ; 10Х14Г14Н4Т, 12Х17Г9АН4, 12Х18Н9,
12Х18Н9Т, 12Х18Н10Т, 12Х18Н10Е, 12Х18Н12Т, 07X21Г7АН5.
Образцы сталей перед испытанием обычно подвергают провоцирующему иагрену при температуре 650 °C в течение 1 ч с охлаждением иа воздухе. Размеры и методы отбора образцов определены ГОСТ 6032—75.
Для обнаружения МКК по окончании испытаний образцы загибают иа угол 90 °.
Изгиб следует прозводнть по ГОСТ 14019— 80 и ГОСТ 6996—66. Наличие трещин иа поверхности изогнутого образца (исключая продольные) свидетельствует о склонности стали к МКК- Аналогично оценивают склонность сталей к МКК и после испытаний по методам АМУ, В, ВУ.
Испытания по методу АМУ — в водном растворе меди сернокислой (50 г), серной кислоты (250 мл плотностью 1,835 г/см3) и воды (1000 мл) в присутствии медной стружки—проводят так же, как и по методу AM, но в течение 8 ч, т, е. этот метод является ускоренным.
Испытания по метод у В — в водном растворе медн сернокислой (НО г), серной кислоты (55 мл), воды (1000 мл) и цинковой пылн (5 г) — проводят в течение 144 ч при температуре кипения. Этот метод предназначен для сталей марки 03Х21Н21М4ГБ и сплавов на железоникелевой основе ‘ марок 06ХН28МДТ и 03ХН28МДТ.
Испытания по методу ВУ — в водном растворе железа сернокислого окисиого по ГОСТ 9485—74 (40 г на 1000 мм серной кислоты по ГОСТ 4204—66 (концентрация 50 %, плотность 1,395 г/см3) — проводят в течение 48 ч при температуре кипения.
Метод предназначен для сталей, указанных в методе В, н является ускоренным.
Испытания по методу ДУ — в водном растворе азотной кислоты (65 %-пая, плотностью 1,391 г/см3)—проводят циклами по 48 ч, причем после каждого цикла образцы промывают, высушивают и взвешивают и затем загружают вновь. Всего проводят 5 циклов.
Метод предназначен для контроля сталей марок 03X16H15M3, 03Х17Н14М2, 03Х18Н11, 03Х18Н12. Для обнаружения МКК определяют скорость коррозии за каждый 48-ч цикл испытаний. Если скорость коррозии превышает 0,5 мм/год или если на сварных образцах наблюдается ножевая коррозия, то нх следует считать не выдержавшими испытания.
Испытания по методу Б заключают
ся в анодном травлении (при плотности тока 0,65 А/см2) сталей в водном растворе серной кислоты (концентрацией 60 % по ГОСТ 4204—66), 5 мнн.
Метод предназначен для сталей 12X18Н9, 12Х18Н9Т, 04Х18Н10, 08Х18Н10, 08X18II10T, 12Х18Н10Т, 03Х18Н11, 06Х18Н11, 08XI8H12T, 12Х18Н12Т.
После анодного травления прн увеличении не менее 20—30 изучают микроструктуру поверхности: сетка протравленных границ свидетельствует о склонности стали к МКК.
Предложен метод оценки склонности МКК по изменению электросопротивления после испытания в кипящем растворе H2SO4+C11SO4. Определяется коэффициент МКК А'я=[(Л1— —Ro)/Ro] 100 %, где Ro — исходное электросопротивление; Ri — электросопротивление после кипячения в растворе.
При этом следует учитывать увеличение электросопротивления нз-за уменьшения сечення образца вследствие общей коррозии; Ri — —Ro=Polo (Sf1—^(Г1) ♦ где Ро — удельное электрическое сопротивление; /0—длина образца; S, — площадь сечения после коррозионного испытания; So — площадь сечення в исходном состоянии.
В качестве дополнительных методов контроля склонности легированных сталей к МКК используют методы измерения внутреннего трения, токовихревой, ультразвуковой и др.
Методы испытания на коррозионное растрескивание
Критерием склонности металлов к коррозионному растрескиванию обычно считают время до разрушения образцов прн определенных пороговых напряжениях, т. е. напряжениях, ниже которых не происходит растрескивания. Обычно строят диаграммы а—1g т (напряжение— время испытания) и с определенными допущениями интерполируют полученную кривую иа более ннзкне напряжения. Напряжение, ниже которого не происходит коррозионного растрескивания при выбранной базе испытаний, называется условным пределом длительной коррозионной прочности.
Для создания напряжений к образцу либо прикладывают постоянную нагрузку, либо создают постоянную деформацию. Конструкцию ванн выбирают в зависимости от среды, ее свойств, температуры. Испытания прн повышенных температурах и давлениях часто проводят на трубчатых образцах, когда рабочая среда находится во внутренней полости нлн воздействует на внешнюю поверхность образцов, помещаемых в автоклав [14].
Виды образцов и способы нх нагружения при испытаниях по схеме с постоянной деформацией приведены на рнс. 16.10 и 16.11. Для нагружения образцов (постоянная нагрузка) применяются нагружающие устройства в основном рычажного типа. Кинематическая схема установки УИП-6 приведена иа рнс. 16.12. Иногда при ускоренных испытаниях непрерывно повышают нагрузнн илн используют программированное нагружение.
Для изучения скорости развития зародившейся коррозионной трещины и определения условий, когда трещниа не растет, предлагается проводить испытания образцов с заранее наведенными трещинами в рабочей среде и
263
строить диаграммы Ki— {da]dr], где — коэффициент интенсивности напряжений в вершине трещины при раскрытии последней путем отрыва: da]dx — скорость распространения трещины. Это дает возможность установить значение коэффициента Kiscc (stress corrosion
Рис. 16.10. Образцы, используемые для испытаний на коррозионное растрескивание:
а — цилиндрический гладкий; б — цилиндрический с нулевой длиной рабочей части; в — цилиндрический с концентратором; г — трубчатый гладкий; д — трубчатый с концентратором; е —плоский; ж — плоский для одноосного растяжения; э — плоский с концентратором; « — призматический с концентратором
Рис. 16.11. Схемы трехточечного (а) и четырехточечного (б) нагружения образцов
cracking — коррозионное растрескивание), ниже которого роста трещины не происходит. Этот коэффициент можно определить нз диаграммы Xi—т (Ki — коэффициент интенсивности напряжений в вершине трещины в начале испытаний; т—время испытаний). В данном случае очень важно правильно выбрать базу испытаний н установить момент прекращения роста трещины при данных условиях, что обычно делают путем металлографических исследований у устья (вершины) трещины [48].
Методы изучения контактной, щелевой и газовой коррозии
Контактная коррозия возникает в результате образования макропар нз различных металлов или значительного различия электрохимических характеристик участков одного и того же металла.
Этот вид коррозии исследуется путем моделирования макропар, измерения нх коррозионных токов, построения коррозионной поляри
зационной диаграммы, определения электродных потенциалов пары.
Особенности щелевой коррозии также изучают путем моделирования щелей, зазоров, застойных зон я подобных конструктивных условий. Коррозионную стойкость оцсни-
Рис. 16.12. Схема крепления образца (а) и кинематическая схема (б) установки УИП-6:
1, 3 — самоцевтрирующиеся захваты; 2 — образец; 4 — редуктор с нагружающим винтом; 5 — сменные зубчатые колеса; 6 — электродвигатель; 7 — редукторы; В — штуцер; 5 —медные прокладки; 10 — печь
вают либо массовым методом, либо определяя коррозионный ток в щелн и сопоставляя его с коррозионным током металла в объеме электролита.
Газовую коррозию оценивают по уменьшению илн увеличению массы образцов. Углеродистые н низколегированные стали испытывают обычно в течение 200 ч, взвешивая через каждые 50 ч; для высоколегированных сталей в зависимости от назначения продолжительность испытаний может составлять 500—1000 ч.
Структуру окнсных пленок изучают оптическими и рентгеиоструктурными методами, методами авторадиографии [2, 3, 44—46, 49]. В последнее время для анализа поверхностных слоев используют Оже-спектроскопию (см. раздел 7). Особенно ценные сведения этот метод дает прн изучении адсорбции на твердой поверхности, а также образования фазовых поверхностных плеиок под действием сред и других поверхностных эффектов.
Испытания на газовую коррозию проводят в соответствующих газовых средах при заданных параметрах (температура, время). После
264
испытания стойкость определяют по снижению илн увеличению массы; исследуют структуру и состав окислов или других продуктов коррозии.
16.4.2. Испытания на кавитационную стойкость
Кавитационная стойкость как характеристика конструктивной прочности материалов. Поскольку кавитационное разрушение имеет специфический характер и природа этого явления очень сложна н многообразна, оценочная характеристика сопротивления металлов и сплавов кавитационному воздействию возможна лишь с помощью сравнительных испытаний.
В зависимости от условий возникновения кавитационной эрозии при эксплуатации желательно проводить испытания с возможно более полной имитацией реальных параметров работы деталей: свойств среды, температуры и ресурса испытаний, так как иначе характеристики кавитационной стойкости тердют смысл.
Многочисленные попытки установления связи между кавитационной стойкостью и какими-либо 'механическими, физическими и химическими свойствами успеха ие имели, хотя в рамках определенных групп материалов и удавалось получить определенные зависимости между кавитационной стойкостью в воде и такими характеристиками, как упругие свойства, твердость, временное сопротивление, предел текучести, пластичность нлн их производные [15, 16, 50].
Кавитационную стойкость оценивают по потерям массы (в граммах) Am образцов после определенного времени испытания, отнесенным к единице площади поверхности S (в квадратных миллиметрах), т. е. Am/S, или прн сравнительных испытаниях однотипных образцов— просто по весовым показателям Ат.
Важные данные о стойкости сплавов в условиях кавитации дают измерения рельефа поверхности с помощью профилографа — профилометра, изучение упрочнения поверхности путем измерения микротвердостн нли твердости.
При определении сопротивления материалов кавитационному воздействию большое значение имеет внешний вид разрушения н микроструктурное изучение испытуемой поверхности (желательно в местах разрушения). Для оценки применяют н такие показатели, как глубина разрушения и глубина упрочнения, которые определяют прн изучении поперечного сечения образцов нлн методами поэтапного стравливания.
Для определения кавитационной стойкости деталей, работающих под действием внешней нагрузки или при ползучести, используют такие показатели, как скорость ползучести в условиях кавитационного воздействия, время до разрушения, пластичность. Для оценки влияния кавитационных повреждений иа уровень механических свойств последние определяют либо после определенного времени кавитационного воздействия на рабочую зону образца, либо непосредственно в процессе кавитационного нагружения.
Лабораторные методы и установки для изучения кавитационной стойкости. Полное мо
делирование кавитационного воздействия возможно прн стендовых испытаниях изделий (турбин, насосов, двигателей внутреннего сгорания и др.), однако такие испытания очень длительны, дороги и позволяют определять стойкость материалов только для конкретной серии изделий.
Для изучения механизма кавитационного воздействия и влияния внешних факторов (скорости потока, количества растворенных газов, наличия твердых частиц и др.) используются гидродинамические трубы [16]. Однако интенсивность получаемой в них эрозии невелика, а испытания длительны и трудоемки.
В металловедении для изучения кавитационной стойкости сталей и сплавов наибольшее распространение получили струеударные и магнитострикционные установки.
В струеудариых установках образцы, укрепленные на колесе, пересекают струю (сплошную или распыленную), диаметр которой можно регулировать. Эрозия достигается прн соударении металлической поверхности с каплями воды (рнс. 16.13). Аналогичная схема используется и при изучении гидроабразивного
Рис. 16.13. Стенд для ударно-эрозионных испытаний схем:
/—электродвигатель: 2—по л у жестка я муфта; 3 — стальной диск; 4 — образец; 5 — сопло; 6 — водонапорный бак
износа. В этом случае образец соударяется с пульпой (водой со взвешенными в ней абразивными частицами). Результаты испытаний существенно зависят от диаметра струи жидкости и скорости вращения колеса с образцами: скорость эрозии резко возрастает с увеличением диаметра струи и скорости соударения. Весьма широко применяются для изучения кавитационной эрозии установки, где в качестве источника кавитационной зоны используются магнитострикционные вибраторы [15, 16].
Преимущество установок этого типа — возможность изучения кавитационной стойкости материалов в широком интервале температур (от криогенных до 1100°C), в различных рабочих средах (нейтральных, кислых, щелочных, поверхностно активных, в расплавах металлов, солей и др.), при различных внешних параметрах (давление, состав атмосферы, механические напряжения н деформация н т. п.)_ Все это привело к созданию серин установок, одинаковых по принципу магнитострикционного возбуждения кавнтацни, но с испытательными камерами и сопутствующей аппаратурой различной конструкции. На рис. 16.14 приведена схема установки для испытания кавита-
Слив раствора
Рис. 16.14. Установка для кавитационно-коррозионных испытаний:
1 — магнитостриктор; 5 — трансформатор колебаний; 3 — термопара; 4— рабочая камера; 5 — змеевик охлаждения; 6 — кислотостойкая резина; 7 — нагревательный элемент; 8 — образец; 9 — концентратор; 40 — уровнемер
цнонной стойкости материалов в активных средах, а иа рис. 16.15 — при кавитационномеханическом нагружении. При испытаниях с использованием магнитострикционного возбуждения кавитации образец либо навинчивают на никелевый (или пермендюровый) стержень и прн вибрации последнего он разрушается жидкой средой, либо помещают с зазором 5—10 мм под вибратор (диск или плоский образец). В последнем случае разрушаются и образец, и торец вибратора, который следует изготовлять из кавитационностойких материалов н заменять после определенного времени работы, чтобы сохранить постоянными условия испытаний.
16.4.3. Испытания
в газовых потоках
В связи с многообразием видов эрозионного разрушения используют различные методики и установки для оценки стойкости материалов. При конструировании испытательных установок стремятся сохранить близкими к реальным условиям основные параметры (скорость газового потока, температуру, давление н др.) и в то же время усилить эффект разрушения, чтобы сократить длительность испытаний.
Для оценки стойкости стволов стрелкового оружия и артиллерийских стволов испытания проводятся на базе соответствующего вооружения с некоторыми добавочными приспособлениями. Так, прибор Робертса — насадка на обрез винтовки, в которую (насадку) помещают образцы в виде стержня- (рнс. 16.16). Широко применяются также приборы типа экспериментальных стреляющих аппаратов
Рис. 16.15. Схема установки для кавитационно-механических испытаний:
1 — нагреватель; 2 — неподвижная тяга; 3 — упругий элемент; 4 — газовый редуктор; 5 — образец; б — термопара; 7 — магнитостриктор; 8 — индикатор зазора; 9 — уровнемер: 10 — камера; 11 — механизм нагружения; 12— индикатор деформации; 13подвижная тяга; 14 — редуктор; 15 — электродвигатель
266
(ЭСА), в которых испытывают трубчатые образцы под действием пороховых газов (рис. 16.17) (1].
Для изучения эрозии под действием газовых смесей различного состава в широком диапазоне давлений и температур используют установки, в которых образцы испытывают в манометрических бомбах. В этих приборах дав-
Рис. 16.16. Схема прибора Робертса для эрозионных испытаний [1]:
1 — образец; 2 — упор; 3 — нарезная втулка; 4 — конусообразный затвор; 5— обрез винтовки; 6 ~~ разрывной диск
ление может превышать 300—400 МПа [9]. Предложены установки для изучения газоаб-разнвной эрозии лопаток газовых турбин и других деталей, оборудования электростанций, работающих на твердом топливе. Методика испытаний состоит в определении изменений массы образцов после воздействия потока запыленного газа с определенными значениями температуры, концентрации абразивных час* тиц, скорости и содержания агрессивных при-
ских трубах (рнс. 16.19), где удается имитировать различные условия работы материалов в скоростных газовых потоках (обшивка сверхзвуковых летательных аппаратов, лопатки турбин турбореактивных двигателей, ракетные системы в плотных слоях атмосферы и т. п.) [9].
Принцип аэродинамической трубы положен также в основу установки, предложенной в работах [51, 52]. Установки типа газодинамического стенда позволяют исследовать комплекс свойств металлов, сплавов и защитных покрытий в скоростных воздушных н газовых потоках:
а) определять эрозионную стойкость в широком интервале температур и скоростей потока, в том числе эрозионную стойкость под напряжением;
б) изучать механические свойства при рас-
Рис. 16.18. Схема конструкции экспериментального сопла фирмы «Кертисс — Райт» [1]:
/ — изоляция из рефразила; 2— пирографит, тепло-проводящий в радиальном направлении; 3 — графит АТУ; 4— пирографит, теплоизолирующий в радиальном направлении (толщина 1.3 мм); ё—стельная оболочка (толщина 1,5 мм); б —стальной кожух сопла; 7 — стальная камера сгорания
Рис. 16.17 Схема экспериментального стреляющего аппарата ЭСА-4 fl]:
1 — упор; 2 — свинцовый буфер: 3 — направляющие; 4 — подвижный груз; 5 — стержень; 6 — крепящая гайка; 7 — образец; 8 — обтюрирующее устройство; 9 — отверстие для крешериого прибора; 10 — пороховой заряд; 11 — нить накала с воспламенителем: 12— корпус; 13 — поршневой затвор
месей. Эти испытания дают информацию о сравнительной эрозионной стойкости сталей, сплавов н защитных покрытий в условиях, близких к эксплуатационным. Недостаток* этих установок — трудоемкость н длительность испытаний, сложность аппаратуры.
Прн испытаниях материалов для теплозащитных экранов сопловых устройств н обшивки летательных аппаратов широко применяют установки на базе экспериментальных реактивных двигателей с различными сопловыми устройствами (рис. 16.18) [1].
Экспериментальные исследования эрозионной стойкости проводятся и в аэродинамиче-
тяженни непосредственно в газовых потоках;
в) определять характеристики ползучести н длительной прочности;
г) изучать характеристики термической усталости в широком диапазоне параметров испытания;
д) исследовать роль аэродинамических колебаний и состояние поверхности на свойства материалов в скоростных газовых потоках.
В газодинамическом стенде использована аэродинамическая труба непрерывного действия, в рабочей камере которой имеются системы для нагружения образцов, контроля температуры и параметров потока.
267
Рис. 16.19. Схема экспериментальной аэродинамической установки:
1 — компрессор ВКУ-100/230; 2 — накопительные баллоны; 3 — компрессор ВК-8/20; •? — регулирующая арматура; 5— форкамера; 6—сопло; 7 — образец; 8— рабочая камера; 9 —подвижная тяга; 10 — нагружающее устройство; 11 — диффузор; 12 — пульт управления; 13 — регистрирующий потенциометр; 14 — пирометр; 15 — измеритель деформации ИСД; 16 — упругий элемент; 17 — тяга; 18 — трансформатор
Испытания могут проводиться в потоке воздуха илн газа прн скоростях 0<М<3 н в диапазоне от комнатной до температуры плавления образцов.
При испытании образцов в скоростном воздушном потоке питание установки производится от компрессора через ресивер, а нагрев прн пропускании через образец тока.
Прн испытаниях в продуктах сгорания жидкого топлива газовый поток создается в камере сгорания. В этом случае образцы в рабочей камере нагреваются горячими газами. Температура газового потока регулируется по-, дачей топлива и воздуха.
Стойкость материалов оценивают либо по результатам измерений массы, либо по изменениям механических характеристик, либо по времени до появления трещин или времени до полного разрушения.
Поскольку большинство деталей, работающих в скоростных воздушных н газовых потоках, находится в сложном напряженном состоянии, то возникновение даже небольших повреждений поверхностных слоев, изменение их состава н структуры существенно отражаются на свойствах и эксплуатационной стойкости.
В работах [9, 51—53] приведены данные испытаний наиболее широко применяемых - в технике металлов н сплавов в скоростных воздушных н газовых потоках (СВП); некоторые результаты представлены в табл. 16.5.
Установить определенную связь между составом, фнзико-хнмнческимн, механическими свойствами н эрозионной стойкостью в газовых потоках не удается [8, 9, 51—53]. В результате испытаний в манометрической бомбе показано, что стойкость железа снижается при легировании никелем и хромом. Сплавы иа основе инкеля имеют низкую стойкость, более стойки сплавы кобальта и молибдена.
Л. А. Урванцевым [1] проанализировано
влияние тепловых и физических свойств на эрозионную стойкость. Автор считает, что при высоких тепловых нагрузках различия в сопротивлении эрозионному воздействию газов,
ТАБЛИЦА 16.5
СВОЙСТВА МЕТАЛЛОВ И СПЛАВОВ ПРИ ИСПЫТАНИЯХ НА ВОЗДУХЕ
Материал Температура испытаний, °C Неподвижный воздух, М=0 СВП. М=1.4 Снижение прочности под слиянием СВП, %
сгв, МПа с й и to % *9
Никель 600 195 46,5 140 39,0 28,0
750 116 46,0 90 16,0 29,2
Железо 600 115 32,5 60 17,0 48,0
Ниобий 1000 55 22,0 30 10,0 52,0
Титан ВТ-1-0 20 480 30,0 43 21,0 10,3
200 215 37,0 173 29,0 20,0
300 125 32,0 75 25,5 35,0
400 НО 22,0 69 19,0 40,2
Титановый 20 1120 10,0 1095 8,3 2,3
сплав ВТ-20 300 850 13,5 828 15,2 2.6
450 710 18,0 530 16,7 25,0
500 580 25,5 320 25,0 45,0
10XI8H9T 600 420 41,0 350 25,0 16,7
700 290 25,0 160 14,0 45,0
800 220 20,0 95 10,0 52,5
ХН77ТЮР 700 670 12,0 620 6,0 7,4
800 535 20,0 435 9,0 18,4
ХН60ВМТЮ 800 445 32,0 321 16,0 26,0
900 235 27,0 175 14,0 31,5
вн-з (ниобиевый' сплав) 1100 320 10,5 220 9,5 31,0
268
связанные с легированием различных сплавов, в значительной степени нивелируются: химический состав незначительно влияет на эрозионную стойкость. В некоторых условиях, например, жаропрочные сплавы хуже сопротивляются эрозии, чем стали. Однако прн менее напряженных условиях испытания эти сплавы становятся, наоборот, более эрозионностойки-ми. Высокая эрозионная стойкость обнаружена у твердых металлокерамических сплавов и сплавов на основе тугоплавких металлов. Л. А. Урванцев приходит к заключению, что эрозионная стойкость увеличивается при повышении температуры плавления и коэффициента теплопроводности материалов, а также при уменьшении коэффициента термического расширения.
Многочисленные исследования влияния структуры сплавов на эрозионную стойкость позволяют сделать вывод, что в общем случае гомогенные или мелкодисперсные структуры обладают более высокой стойкостью, чем грубые, гетерогенные. Особенно отрицательно сказываются крупные выделения малопрочных нли хрупких фаз по границам зерен.
Прн выборе материалов для деталей, работающих в скоростных газовых потоках, приходится учитывать многие, часто противоположные требования. Так, в большинстве случаев материалы -должны иметь высокую жаропрочность н минимальную плотность, высокие механические н хорошие технологические свойства, высокое сопротивление термической усталости и газовой коррозии и эрозии.
В каждом случае значение отдельных факторов может быть различным, поэтому материалы выбирают исходя из конкретных условий работы деталей.
Для лопаток высокотемпературных газотурбинных двигателей в настоящее время применяют жаропрочные сплавы на основе никеля (литые, деформируемые), на основе кобальта, а для менее высоких температур — жаропрочные аустенитные стали.
Для деталей проточной части реактивных установок все шире применяют сплавы на основе тугоплавких металлов [8], обычно с защитными покрытиями.
Широко используются эрозионностойкне материалы и сплавы на основе окислов, карбидов, боридов, нитридов и интерметаллических соединений тугоплавких металлов.
16.4.4. Испытания на изнашивание
Износостойкость и фрикционные свойства пар трения определяются природой контактирующих материалов н условиями трения. Большое число факторов, влияющих иа процесс изнашивания деталей машии, а также необходимость изучения особенностей этого влияния обусловливают значительный объем экспериментальных исследований. В настоящее время широко применяют испытания на треиие и изнашивание, проводимые в четыре этапа [23]" 1) обычные лабораторные испытания физических и механических свойств материалов; 2) испытания материалов иа трение н изнашивание на лабораторных установках; 3) стендовые испытания узлов треиия; 4) натурные (эксплуатационные) испытания машин и механизмов.
Задача 1-го этапа — определение физических н механических свойств материалов и прогнозирование по инм износостойкости. 2-й этап позволяет оценивать влияние этих свойств в сочетании с выбранными режимами трення на износостойкость материалов. Стендовые испытания (3-й этап) проводятся для оценки влияния конструктивных факторов на работоспособность пар трення. Натурные испытания (4-й этап) необходимы для оценки надежности и долго вечности механизмов в целом. Таким образом достигается последовательное приближение условий испытаний к реальным условиям работы деталей машин. Прн этом из большого числа перспективных (износостойких) материалов отбирают лишь действительно пригодные для данной пары трения.
1-ии 2-й этапы испытаний необходимы также для оценки износостойкости новых материалов и способов их обработки, а также для контроля качества материалов. Исходя из получаемых данных, конструкторы имеют возможность обоснованно подходить к выбору материалов для конкретных деталей н узлов.
Поэтапный характер испытаний способствует значительному сокращению объема дорогостоящих и длительных эксплуатационных испытаний. Ниже рассматриваются 2-й и 4-й этапы испытаний на изнашивание.
Испытание материалов на изнашивание на лабораторных установках. В процессе трения в поверхностных слоях контактирующих тел формируются новые, отличные от исходных структуры, определяющие сопротивление материалов изнашиванию в данных условиях нагружения.
Изменение условий трення (нагрузка, скорость, режимы смазки н др.) может привести к резкому изменению интенсивности изнашивания (рис. 16.20). Это связано с возникновением в поверхностном слое материалов критических температур, критических степеней деформации, приводящих к необратимым структурным изменениям в зоне контакта. Образование новой структуры приводит к изменению интенсивности н механизма нзнашива-
Рис. 16.20. Пространственная диаграмма зависимости величины износа нормализованных образцов из стали 45 от скорости скольжения V и удельного давления Р. Трение без смазки в паре с нормализованной сталью 10 [37]
269
ния. Сильное влияние различных факторов на интенсивность изнашивания обусловливает необходимость проведения испытаний в возможно более широком интервале нагрузок, скоростей, температур и др. (см. рис. 16.20).
При испытаниях с практическими целями необходимо, чтобы интервал изменения основных условий трения (нагрузки, скорости и др.) был шире, чем при эксплуатации деталей машин.
Прн испытаниях материалов на лабораторных установках должны воспроизводиться основные условия трення на поверхности, которые имеются прн эксплуатации деталей, прн том, что обеспечивается одни и тот же вид изнашивания [40]. Для правильного выбора методики и условий испытаний иа лабораторной установке необходимо подробно ознакомиться с условиями работы исследуемого узла трения (характер смазки, скорость скольжения, давление в зоне контакта, температура в поверхностном слое деталей и др.), а также установить основной механизм (вид) изнашивания пары. Выявить основной механизм изнашивания можно лишь прн тщательном изучении характера повреждений рабочей поверхности деталей, а также структурных изменений в их активных слоях. В тех случаях, когда иа лабораторных установках воспроизведение условий треиня при эксплуатации затруднено, используют следующие критерии правильности выбора условий испытаний [40]: 1) обеспечение одинаковой формы разрушения материала прн испытании на лабораторной установке н прн эксплуатации детали; 2) обеспечение одинакового характера повреждений поверхности, структурных изменений н микротвердо-стн поверхностного слоя материала, испытанного на лабораторной установке и в условиях эксплуатации.
В зависимости от характера относительного перемещения образцов все машины трения по кинематическому признаку можно разделить на два класса: I — однонаправленного движения; II — знакопеременного движения [24]. При однонаправленном и знакопеременном движении интенсивность и характер разрушения поверхности трения материалов могут заметно различаться. Внутри каждого класса машин различают две группы: 1) машины торцового трення; 2) машины трення по образующей. Испытания на машинах этих двух групп различаются по условиям формирования граничных смазочных слоев. Каждая группа машин включает две подгруппы установок: а) с коэффициентом взаимного перекрытия, стремящимся к единице (Kbs-^-I), б) машины с коэффициентом взаимного перекрытия, стремящимся к нулю (КЙЗ->0). Коэффициент взаимного перекрытия — отношение площадей трення контактирующих тел. Прн торцовом трении колец одинаковых размеров Квз — 1. При Тренин пальчиковых образцов по плоской поверхности диска Квз-И). Коэффициент взаимного перекрытия сильно влияет на температурные условия в поверхностных слоях пары 'Греция (эта характеристика введена А. В. Чичинадзе).
Таким образом, существует восемь типов лабораторных установок, позволяющих воспроизводить условия трения и изнашивания, возникающие при эксплуатации машин и механизмов.
Некоторые нз лабораторных установок, выпускаемых нашей промышленностью, рассматриваются ниже.
1. Установка УМТ-1. Предназначена для исследования трення и изнашивания материалов в широком интервале скоростей скольжения и нагрузок. Установка универсальная, так как
Рис. 16.21. Схема установки УМТ-1
позволяет проводить испытания прн однонаправленном и знакопеременном относительном движении образцов, а также по различным схемам контакта. Прн однонаправленном движении испытания осуществляются по схемам: палец — диск, кольцо по кольцу (торцовое тренне), вал — втулка. При знакопеременном движении (качании) испытания проводят по схеме вал — втулка.
Испытательная машина состоит (рнс. 16.21) из электрического асинхронного двигателя /, электромеханического привода 2 с бесступенчатой регулировкой скоростей вращения вала. На валу закреплено коитртело — образец (например, диск) 3, к плоской поверхности которого под действием силы Р прижимаются образцы 4, закрепленные в держателе 5. Держатель расположен в узле иагруження 6, который может перемещаться вдоль осн вращения вала с помощью привода 7. В процессе испытания измеряются следующие характеристики трепия: нагрузка на образец, скорость вращения вала, момент трення, температура в поверхностных слоях неподвижного образца. Момент трення и температура регистрируются на лейте записывающего прибора. Износ образцов определяется по уменьшению нх массы нли длины.
Машина имеет следующие основные технические характеристики:
Мощность привода, кВт .... 25
Скорость вращения вала, об/мнн 15—3000
Рабочая нагрузка, кН...........0,2—4,0
Момент треиня, Н-м............. 2—40
Диаметр диска, мм.............. 350
Диаметр пальчиковых образцов, мм............................. 5; 10
Длина пальчикового образца, мм 20
Наружный диаметр кольца, мм . 28
Внутренний диаметр кольца, мм . 16
Высота кольца, мм.................. 15
Диаметр втулки внутренний, мм 35—40
То же, наружный диаметр втулки, мм................................ 50
Длина втулки, мм ...... 10; 15; 20
2. Установка СМЦ-2. Предназначена для исследования изнашивания материалов прн тре
270
нин качеиня, тренни качення с проскальзыванием и трении скольжения (по классификации I, 2,6). При тренни качення и качения с проскальзыванием контакт осуществляется по наружной поверхности дисков (образцов). При трении скольжения схема контакта: вал— вкладыш. На верхний и нижний валы с установленными на них образцами 6 (рис; 16.22) вращение передается от асинхронного электрического двигателя 1 через клнноремениую
3. Установка Х4-Б. Предназначена для испытания материалов’ на изнашивание закрепленным абразивом. Схема контакта палец — диск (по классификации I, 1,6). Конструкция установки разработана М. М. Хрущевым [40].
На плоской поверхности вращающегося диска 1 (рнс. 16.23) закреплена абразивная шкурка. Цилиндрический образец 2, находящийся в держателе, прижимается грузом 3 к поверхности диска (шкурки). При вращении диска
Рис. 16.22. Схема установки СМЦ-2
передачу, контрпривод 2, муфту н валы 3—5. Нагрузка задается нагружающим пружинным устройством 8 через подвижную верхнюю бабку 7.
Момент трення измеряется с помощью индуктивного датчика по скручиванию упругого элемента и непрерывно регистрируется на ленте электронного потенциометра.
При испытании по схеме вал — вкладыш (трение скольжения) вращение на верхний образец не передается (он закрепляется неподвижно вместе с валом). Установка имеет набор сменных герметичных камер, позволяющих проводить испытания в жидких средах.
Основные технические характеристики установки:
Диаметр образцов (дисков), мм . 50
Скорость вращения вала, об/мин . 300, 500,
1000
Нагрузка иа образцы, кН . . . 5
Проскальзывание образцов, ,% « Ю—20
образец перемещается в радиальном направлении. Путь трення — Спираль, т. е. тренне (и изнашивание) образца, происходит только по свежей поверхности шкурки. Поверхность шкурки разделена на зоны с равной длиной пути трення образца. В этих зовах (через одну) испытываются образец и эталон. Износ определяется по уменьшению длины нли массы образца. Результат испытания выражается через относительную износостойкость е, равную отношению износа эталона к износу образца E = Qa/Qo.
На результат испытания большое влияние оказывает отношение твердости абразива к твердости испытуемого материала Прн износ образца очень мал илн вообще отсутствует (е->со), при 1,4 ч-1,6 из-
носостойкость сохраняет постоянные значения, т. е. уже ие завнснт от твердости абразива. В этом случае основой механизма изнашивания материала закрепленным абразивом является микрорезание.
Рис. 16.23. Схема установки для испытания материалов на изнашивание закрепленным абразивом Х4-Б
Технические характеристики установки:
Скорость вращения диска,
об/мин........................ 60
Диаметр диска, мм .... 250
Путь трения, м........... 15
Диаметр образца, мм ... . 2,0
Длина образца, мм..... 10—15
Нагрузка иа образец, Н . . . 3
Вид шкурки для материалов твердостью HV:
<1350 ........................ Электро-
корунд
<2000 ...................... Карборунд
Зернистость ........ 180
4. Установка 77МТ-1. Предназначена для испытания материалов на изнашивание прн возвратно-поступательном движении. Схема контакта палец — плоскость (по классификации II, 1,6). Верхний образец 2 (рис. 16.24) прижимается с помощью рычага 4 и груза к нижнему плоскому образцу I, закрепленному
на самоустаиавливающемся ползуне. Ползун может приводиться от кривошипного механизма 3. Испытания можно проводить в различных жидких средах. В этом случае нижний образец помещают в ванну 5. Износ верхнего образца определяется по уменьшению его массы или длины. Износ нижнего образца может быть определен с помощью профилографиро-вання поверхности.
Основные технические характеристики установки 77МТ-1:
Длина рабочего хода образца, мм........................... 30—70
Число двойных ходов в минуту 100, 200,
300, 400
Нагрузка на образец, Н . . . 100—600
Испытание деталей машин на изнашивание в условиях эксплуатации. На основании эксплуатационных испытаний определяют ресурс работы узла и делают окончательные выводы
Рис. 16.24. Схема установки для испытания материалов на накашивание при возвратно-поступательном движении 77МТ-1 ----
272
о правильности выбора материалов для исследуемой пары трення. Испытания этого вида характеризуются, как правило, большой длительностью и трудоемкостью, что связано с необходимостью выработан ресурса узла, с его периодической разборкой со сложностью измерения величины износа деталей. Поэтому для эксплуатационных испытаний отбирают предельно ограниченное число вариантов сочетаний материалов, которые в процессе предварительных лабораторных и стендовых испытаний показали наибольшую износостойкость по сравнению с другими материалами. Изнашивание в условиях эксплуатации оценивают по уменьшению размеров деталей нли по изменению выходных параметров сопряжения (утечка масла илн рабочей жидкости, температура, коэффициент трения н Др-)-
Оценка суммарного износа реальных деталей по изменению нх объема и .массы затруднена нли невозможна из-за больших габаритов, сложной формы, трудности полной разборки узла.
Изменение выходных параметров узла (сопряжения) дает лишь косвенные данные о величине износа. Наиболее полную информацию о величине износа и его распределении по поверхности трення дают дифференциальные методы измерения износа: микрометрирование, метод искусственных баз, метод поверхностной активации [41].
Метод микрометрнровання основан на измерении деталей до и после испытаний с помощью инструментальных микроскопов, микрометров, индикаторов и других приборов. Для определения малых величин износа и характера распределения его по рабочей поверхности деталей применяют метод профи-лографировання поверхности с помощью профилографов различных типов (ММК-1, Ка-либр-ВЭИ, модель 201, ИЗ П-5 н др.). Вертикальное увеличение профилографов составляет 400—200000, что позволяет с большой точно- -стью определять износ. О величине износа судят на основаинн сопоставления профилограмм, снятых с рабочей поверхности детали до и после испытаний.
Недостатки метода микрометрировання:
а) невозможность измерения износа в процессе испытания б^з остановки н разборки узла;
б) необходимость наличия базы (поверхности), относительно которой производится измерение размеров детали до и после испытаний.
От указанных недостатков в значительной степени свободен метод пневматического мнк-рометрировання, а также метод определения износа по расходу рабочей среды через зазоры в сопряжениях [42].
Мет,од искусственных баз состонт в том, что па рабочую поверхность деталей наносят углубления строго определенной геометрической формы (коиус, пирамида, сфера и др.). При изнашивании поверхностного слоя детали происходит уменьшение глубины н других размеров углубления, по которым судят о величине линейного износа данного участка поверхности.
Метод искусственных баз предназначен для оценки местного или локального линейного износа деталей. Система углублений позволяет оценивать характер распределения нз-
1В—280
иоса на рабочей поверхности детали. Углубления наносят на поверхность детали методом отпечатков и методом вырезных лунок. Отпечатки обычно наносят четырехгранной алмазной пирамидой с квадратным основанием, углом при вершине между противолежащими гранями 136° (такие пирамиды используют в твердомерах Виккерса ПМТ-3). Глубину отпечатка h вычисляют по формуле h=dl2V2tg(a/2), где а — угол прн вершине пирамиды между противолежащими гранями; d — длина диагонали отпечатка. Прн а=136° величина h =0,143 d.
Линейный износ определяют как разность глубины отпечатков до и после испытания:
—ft2=0,143(di—d2) при а=136°, где hi — глубина отпечатка до испытания; hz — то же, после испытания.
Использование отпечатков, наносимых с помощью прибора ПМТ-3, позволяет оценивать иитенснвиость изнашивания отдельных структурных составляющих материала (например, карбидной фазы), если размеры частиц позволяют наносить на них отпечатки мнкротвер-достн. Точность измерения линейного износа по отпечаткам микротвердостн составляет доли микрометров ( — 0,3 мкм). Размеры диагоналей наносимых отпечатков обычно составляют 1—10 мкм.
В производственных условиях прн измерении линейного износа в труднодоступных местах деталей больших размеров отпечатки наносят коническими кернами из твердого сплава илн закаленной стали или высверливают отверстия конической формы. В этом случае линейный износ определяют по формуле Дй = =0,5Adtg(90—а/2), где d— диаметр отпечатка; а — угол при вершине конуса.
При а=120° величина Aft=0,288Ad. Диаметры отпечатков измеряют, как н в случае определения твердости по Бринеллю, с помощью микроскопа МПБ-2.
Следует отметить, что на результат измерения размеров отпечатков влияет вспучивание металла по краям отпечатка. Поэтому перед измерением вспучивание удаляют шлифовкой нли проводят первое измерение после приработки деталей. Применение метода отпечатков затруднено, когда износ сопровождается пластической деформацией поверхностного слоя, приводящей к искажению формы и заплыванию отпечатков. Прн использовании метода мнкротвердости отпечатки довольно трудно обнаружить после испытания деталей. Форма отпечатков после снятия нагрузки на индентор заметно изменяется у материалов с высоким пределом текучести в результате упругого восстановления материала.
М. М. Хрущев и Е. С. Беркович разработали способ нанесения углублений — метод вырезных лунок. На поверхности детали вырезают с помощью вращающегося алмазного резца (в виде трехгранной призмы) углубление в форме остроугольной лункн (рис. 16.25). Глубину лунки определяют по формуле ft— —0,125(Z/r), где I — длина луякн; г — радиус вращения резца. Линейный износ определяют по уменьшению глубины лунки: Aft=0,125(/,—• —Преимущества методов вырезных лунок перед методом отпечатков — отсутствие выдавливания металла по краям лунки, из-
273
менення формы лунки, а также большая точность метода. Соотношение между глубиной и длиной лунки составляет 1:50 — 1 :80 против 1 :7 при методе отпечатков. Авторы метода вырезных луиок рекомендуют наносить луики следующих размеров: /=1-4-2 мм, h—
Рис. 16.25. Схема вырезания лунки
=20,8-~ 83,0 мкм. В ряде случаев более удобно наносить лунки абразивным (карборундовым или алмазным) диском [54]. Недостатки метода вырезных лунок — невозможность применения его для измереиня износа грубо обработанных поверхностей, а также для определения очень малого износа (порядка 1 мкм)
Метод поверхностной активации. Метод разработан В. И. Постниковым [43]. Основан на создании радиоактивного поверхностного слоя глубиной 0,05—0,5 мм в заданном участке поверхности детали посредством облучения его заряженными частицами (протонами, нейтронами, а-частнцами), ускоренными до энергии 10—20 МэВ. Облучение деталей осуществляется на специальном ускорителе (циклотроне). Одновременно с деталями активируются образцы, которые затем используются для построения тарировочного графика зависимости изменения радиоактивности поверхности от глубины изношенного слоя: где No— начальная ско-
рость счета импульсов; N — скорость счета импульсов после изнашивания поверхностно! о слои толщиной ДК Тарнровочный график строят на основании лабораторных испытаний активированных образцов, а затем используют для определения величины износа детали в процессе эксплуатации машины нли узла по уменьшению радиоактивности поверхности.
Радиоактивность (у-излученне) измеряют с помощью аппаратуры, в комплект которой входят: счетчик импульсов (сцинтилляционный илн газоразрядный); высоковольтный стабилизированный выпрямитель; дискриминатор; пересчетами прибор и регистрирующее устройство.
Чувствительность метода 1—2 мкм. Активность поверхности детали после облучения обычно не Превышает 10 Ки, что позволяет использовать данный Метод в производственных условиях и при эксплуатации машин без какой-либо специальной радиационной защиты.
Прн измерении износа крупногабаритных деталей применяют специальные активированные вставки, которые изнашиваются совместно с деталью. Материал вставки может отли
чаться от материала детали; подбирают его в соответствии с длительностью испытаний так, чтобы период полураспада изотопов в активированной вставке превышал длительность испытаний на изнашивание.
Основные преимущества метода поверхностной активации перед другими методами измерения величины износа деталей машин — высокая чувствительность, быстрота измерений, возможность измерений без остановки и разборки машины (узла), независимость свойств радиоактивных изотопов от температуры, давления, состояния поверхности н других параметров.
Недостатки, ограничивающие более широкое распространение данного метода: необходимость использования сложного оборудования для регистрации изменения радиоактивности детали; сложность подготовки поверхности деталей перед испытанием (необходимость облучения на ускорителе).
БИБЛИОГРАФИЧЕСКИЙ СПИСОК
I. Урванцев Л. А. Эрозия и защита металлов. М.; Машиностроение, 1966. 234 с.
2. Жук Н. П. Курс теории коррозии н защиты металлов. М.: Металлургия, 1976. 472 с.
3. Томатов Н. Д. Теория коррозии и защиты металлов. М.: Изд-во АН СССР, 1959. 591 с.
4. Эванс Ю. Р. Коррозия н окисление металлов: Пер. с англ. М.: Машгнз, 1962. 856 с.
5. Кубашевский О., Гопкинс Б. Окисление металлов и сплавов: Пер. с англ. М.: Металлургия, 1965. 428 с.
6. Кофстад П. Высокотемпературное окисление металлов: Пер. с англ. М.: Мир, 1969. 392 с.
7. Клинов И. Д. Коррозия химической аппаратуры н коррозиоииостойкне материалы. М.: Машиностроение, 1967. 468 с.
8. Химушин Ф. Ф. Жаропрочные стали и сплавы. М_: Металлургия, 1969. 749 с.
9. Контактная прочность металлических сплавов: Сб. Свердловск: УПИ нм. С. М. Кирова, 1972. 252 с.
10. Никитин В. И. Физико-химические явления прн воздействии жидких металлов иа твердые. М.: Атомиздат, 1967. 441 с.
11. Вествуд А., Прис К. Камдар М. — В ки.: Разрушение. Т. 3. М.: Мир, 1976, с. 635— 691.
12. Томатов Н. Д„ Чернова Г. П. Коррозия и коррозионностойкие сплавы. М.: Металлургия, 1975. 232 с. ‘
13. Химушин Ф. Ф. Нержавеющие стали. М.: Металлургия, 1967. 600 с.
14. Василенко И. И., Мелехов Р. К. Коррозионное растрескивание сталей. Киев: Нау-кова думка, 1977. 262 с.
15. Богачев И. Н. Кавитационное разрушение и кавитационностойкие сплавы. М.: Металлургия, 1972. 189 с.
16. Кнэпп Р., Дейли Дж., Хэммит Ф. Кавитация: Пер. с англ. М.: Мир, 1974. 687 с.
17. Garsia G. Г... Hatnmii F. G., Nistrom R. В. — ASTM, Special Technical Publ. № 408. ASTM, 1967, p. 239—283.
18. Словарь-справочник по трению, износу н смазке деталей машин. Киев: Наукова думка, 1979. 187 с.
274
19. Кузнецов В. Д. Физика резания и трения металлов и кристаллов. Избранные труды. М.: Наука, 1977. 310 с.
20. Поверхностная прочность материалов при трекни/Костецкий Б. И., Носовский У. Г., Караулов А. К и др. Киев: Техника, 1976. 292 с.
21. Мур Д. Основы и применение трнбоннки: Пер. с аигл. М_: Мнр, 1978. 487 с.
22. Боудэн Ф. П., Тейбор Д. Тренне и смазка твердых тел: Пер. с аигл. М_: Машиностроение, 1968. 543 с.
23. Крагельский И. В. Треиие и нанос. М_: Машиностроение, 1968. 480 с.
24. Крагельский И. В., Добычин М. Н., Ком-балов В. С. Основы расчетов на треиие н износ. М.: Машиностроение, 1977. 526 с.
25. Лихтман. В. И. — В ки.: О природе схватывания твердых тел. М: Наука, 1968, с. 30—32.
26. Носовский И. Г. Влияние газовой среды на износ металлов. Киев: Техника, 1968. 180 с.
27. Голего Н. Л. Схватывание в машинах и методы его устранения. Киев: Техшка, 1965. 231 с.
28. Рыкалин Н. Н., Шоршоров М. К-, Красу-лин Ю. Л. — Изв. АН СССР. Неорганические материалы, 1965, т. I, № 1, с. 29—36.
29. Block Н. — Proc. Gen. Disc, on Lubricating and Lubricants, Inst. Meeh. Eng., 1967, v. 11, Croup HI, p. 14—20.
30. Коровчинский M. B. — В кн.: Новое в теории трения. М.: Наука, 1966, с. 98—143.
31. Yoshimoto G„ Tsukizoe Т. — Wear, 1958, v. 1, № 6, р. 472—490.
32. Гаркунов Д. Н., Поляков А. А. — В кн.: Исследование водородного износа. М.: Наука, 1977, с. 3—12.
33. Любарский И. М. Повышение износоустойчивости тяжел она груженных шестерен. М.: Машиностроение, 1965. 132 с.
34. Любарский И. М., Палатник Л. С. Металлофизика трения. М.: Металлургия, 1976. 176 с.
35. Костецкий Б. И., Колесниченко Н. В. Качество поверхности и тренне в машинах. Киев: Техн1ка, 1969. 216 с.
36. Archard J. Е., Hirst W. — Proc. Roy. Soc. London, Ser. A., v. 236, 1956, p. 397—410.
37. Гриб В. В., Лазарев Г. Е. Лабораторные
испытания материалов на трение и износ. М.: Наука, 1968. 139 с.
38. Богачев И. Н„ Коршунов Л. Г., Хады-ев М. С. и др. — ФММ, 1977, т. 43, вып. 2, с. 360—387.
39. Коршунов Л. Г., Богачев И. Н., Аверин Ю. И. и др. — ФММ, 1980, т. 49, вып. 1, с. 113—120.
40. Хрущов М. М.. Бабичев М. А. Абразивное изнашивание. М,; Наука, 1970. 252 с.
41. Проникав А. С. Надежность машин. М: Машиностроение, 1978. 592 с.
42. Ясь Д. С., Подмокав В. Б., Дяденко Н. С. Испытания на тренне и нзнос. Киев: Тех-шка, 1971. 140 с.
43. Постников В. И. Радиоактивные изотопы в нсследованин и автоматизации контроля нзноса. М.: Машиностроение, 1967. 140 с.
44. Романов В. В. Методы исследования коррозии металлов. М: Металлургия, 1965. 280 с.
45. Новые методы нсследовання коррознн металлов. М.: Наука, 1973. 220 с.
46. Розенфельд И. Л., Жигелова К. Д. Ускоренные методы коррозионных испытаний металлов. Мл Металлургия, 1966. 347 с.
47. Лабораторные работы по коррозии и защите металлов//ома шов Н. Д., Жук Н. П., Титов В. А„ Веденеева М. Е. Мл Металлургия, 1971. 280 с.
48. l^et R. Р., Novak S. К., Williame D. Р. — Mater. Res. and Standarts, 1972, v. 23, p. 25—30.
49. Приборы н методы физического металловедения: Пер. с аигл. Вып. 2. Мл Мир, 1974. 363 с.
50. Preiser Н. S., Thiruvengadam A.t Conchman С. Proceedings of ASME, Simposium on Cavitation Research, Facilities and Techniques, Philadelphia, 1964, p. 146—166.
51. Векслер Ю. Г., Сорокин В. Г., Рожка А. Л. н др. — Проблемы прочности, 1978, К® 4, с. 32—46.
52. Векслер Ю. Г.. Богачев И. И., Сорокин В. Г. — Изв. АН СССР. Металлы, 1973, № 4, с. 139—143.
53. Богачев И. Н., Векслер Ю. Г., Сорокин В. Г. — В кн.: Влияние физико-химической среды иа жаропрочность металлических материалов. Мл Наука, 1974, с. 14—21.
17
—---— ФИЗИЧЕСКИЕ МЕТОДЫ ИССЛЕДОВАНИЯ
(ТЕПЛОВЫЕ, ОБЪЕМНЫЕ, ЭЛЕКТРИЧЕСКИЕ, МАГНИТНЫЕ)
Физические свойства сплавов определяются количеством и свойствами отдельных фаз, их структурой и субструктурой.
Прн изменении состава сплава, а также в результате различных на него воздействий (термических, механических, радиационных и т. п.) происходит изменение физических свойств. В основу физических методов исследования положено существование взаимосвязи между изменениями физических свойств И
процессами, пронсходнвшнмн в сплавах при их обработке илн в результате тех илн иных воздействий.
Условно физические свойства делят иа структурно-чувствительные и структурно-нечувствительные. Последние более существенно изменяются при изменении количества и состава фаз н меньше при изменении их структуры (намагниченность, температура Кюри) [1].
18*
Основными принципами исследования физических свойств сплавов в равновесных состояниях является установление зависимостей состав —‘ свойство или температура —• свойство. При исследовании кинетических характеристик процессов устанавливается зависимость свойство — время.
Выбор того или иного физического свойства для исследования н соответствующего метода измерений зависит от свойств и состояния исследуемого объекта. Приведенные ниже простые примеры решения металловедческих задач наглядно- иллюстрируют возможности того пли иного метода. Успехи физического металловедения за последние годы показывают, что все большее применение находят комплексные исследования, т. е. анализ изменения ряда свойств при сложном, иногда одновременном, воздействии разных факторов на исследуемый объект. В связи с этим большое внимание должно уделяться выяснению физической природы исследуемого свойства н его зависимости от состава, структуры, воздействия внешних факторов.
17.1. ТЕПЛОВЫЕ СВОЙСТВА
17.1.1. Энтальпия и теплоемкость
Эти свойства наиболее часто используются при исследовании структурных (главным образом фазовых) превращений в сплавах.
На них основан калориметрический анализ— определение скрытых теп лот превращений, теплоемкости и термический анализ — определение критических точек для построения диаграмм состояния.
Энтальпия (27, Дж) — функция состояния системы. При изобарическом процессе равиа количеству теплоты, необходимой для повышения температуры тела массой tn от абсолютного нуля до температуры Т-. Н=стТ.
Коэффициент пропорциональности с называется средней удельной теплоемкостью во взятом интервале температур и равен количеству теплоты, необходимому для повышения температуры тела массой в 1 г иа 1 К.
Истинная (удельная) теплоемкость, Дж/(г-К), для данной температуры прн постоянном давлении определяется выражением c~dHldTm. Теплоемкость грамм-атома вещества С=сЛ (Л—атомная масса). Разница величин теплоемкости при постоянном давлении Ср и теплоемкости при постоянном объеме Су [Дж/(К-моль)] определяется соотношением Ср—Су—p2Vr/x, где р — температурный коэффициент объемного расширения; V— атомный объем; Т — температура; % •— коэффициент всестороииего сжатия.
В теории теплоемкости Дебая предполагается [2], что энергия колебаний атомов дискретна и имеет определенный спектр частот. Средняя энергия линейного осциллятора при температуре Т равна hv!(eh^kT— 1), а не kT, как в классической теории (й — постоянная Планка; v— частота колебаний атома; k — постоянная Больцмана).
Полная энергия кристалла при температуре Т Wr
и — 9RT (T/eD)3 [ х3/(е*—I)dx,
о
где x=hxjkT\ Gd—характеристическая температура Дебая, определяемая через граничную (максимальную) частоту vd колебательного спектра атомов Gjo—Zivo/fc, характеризует силы межатомной связи.
Согласно правилу Дюлоига—Пти теплоемкость 1 моля элемента (металла) в кристаллическом состоянии (обусловленная колебаниями атомов) Су=дН!дТс^2Ъ Дж/(К-моль) яй яйЗТ? нс зависит от температуры [/?=* —8,3144 Дж/(К-моль) — универсальная газовая постоянная]. Уменьшение теплоемкости при понижении температуры представляет собой квантовый эффект.
Отсюда следует, что теплоемкость при низких температурах (T/0i>^O,l) равиа Су = «233,87? (7/0»)3.
При высоких температурах (7/0» 0,6)
Cu-3/? — 3/?(0D/7)2/20.
В табл. 17.1 приведены характеристические температуры Дебая и значения теплоемкости [3]. Видно, что прн температуре 293 К для всех металлов, кроме переходных, для которых значения теплоемкости значительно выше ожидаемых, выполняется правило Дюлонга— Птн.
При температурах, близких к абсолютному нулю, следует учитывать вклад электронной теплоемкости в общую теплоемкость. Средняя энергия электрона Е при температуре Т: £(7)=3/5£41-Н5л2/12)(£Г/£г)2], где EF — энергия Ферми. Вклад электронной доли в молекулярную теплоемкость C^n=Tt2fe/?7/2£F.
Несмотря, на малую величину' вклада теплоемкости электронного газа в общую теплоемкость металла, его можно обнаружить при измерениях вблизи абсолютного нуля, так как основной вклад в теплоемкость за счет колебаний атомов решетки при этих температурах стремится к нулю по закону Г3, т. е. быстрее, чем Cv. Таким образом, при очень низких температурах полная теплоемкость Су равна Су=а73+у7, где первое слагаемое — теплоемкость за счет изменения энергии колебания атомов, второе — теплоемкость электронного газа.
На рнс. 17.1 приведена зависимость теплоемкости серебра от температуры в координатах CvRT—Г2, в этом случае зависимость Cv (7) — прямая линия, а отрезок, отсекаемый
Рис. 17.1. Зависимость теплоемкости серебра от температуры
276
ТАБЛИЦА '17.1
ТЕМПЕРАТУРА ДЕБАЯ [4J И ТЕПЛОЕМКОСТЬ ПРИ ДЕБАЕВСКОЙ ТЕМПЕРАТУРЕ (CpD) 293
И ПРИ 293 к (Ср ) ДЛЯ МЕТАЛЛОВ
К Е Ф П) e£) . К 4 0 X о a. a О S OO ” ~ СЧ Cl s <u Й eD, к ё о о С.Л § 2 8 _ и S ф «=; e‘D. К Л § i 0 я co 02 л
Li 370 24,91 24,74 Nb 237 24,70 24,61 Dy 194 28,13
Be 1160 25,33 16,45 Mo 470 24,57 24,07 Но 184 27,17
Na 160 26,96 28,26 Tc — 24,74 24,28 Ег 195 -—. 28,13
Mg 406 25,12 24,91 Ru 600 24,49 24,07 Тт 168 — 27,05
Al 427 24,70 24,36 Rh 362,5 25,16 24,99 Yb 101 —. 26,75
Si 670 25,74 20,01 Pd 275 25,83 26,00 Lu 157 I—1 26,80
К 90 18,08 29,60 Ag 226 25,08 25,41 Hf 219 1— 25,74
Ca 220 27,46 26,38 Cd 219,5 24,16 25,96 Та 251 25,20 25,37
Sc 231 25,53 25,54 In 109 25,96 26,75 W 337 24,36 24,28
Ti 430 25,99 25,03 Sn 200 25,96 - 27,00 Re 415 25,54 25,79
V 432 26,79 24,91 Sb 204 24,28 25,25 Os 500 24,57 24.79
Cr 357 25,12 23,36 Те 153 21,89 25,75 Ir 420 25,12 25,12
Mn 450 25,12 26,29 Cs 39,2 25,75 27,59 Pt 229 25,24 25,87
Fe 445 25,12 24,99 Ba 96 — 27,63 Au 162,9 24,66 25,37
Co 445 25,12 24,83 La 142,6 26,29 27,84 Hg 71,8 — 26,59
Ni 441 26,37 26,08 Ce 119 25,16 26,96 TI 89 21,48 26,39
Cu 345 24,70 24,45 Pr 272 26,92 27,46 Pb [06,0 24,40 26,46
Zn 305 24,49 25,41 Nd 238,0 26,75 27,42 Bi 117 22,36 25,54
Ga 333 23,90 25,87 Pm — - ' 27,21 Po . — 25,46 26,38
Ge 370 23,65 23,36 Sm — 25,20 29,59 Th 163 25,87 27,33
Rb 55 25,12 30,89 Eu 90,4 — 27,67 Pa — —. 28,49
Sr 129 24,95 26,37 Gd 215 — — U 200 25,29 27,67
Y 235 22,36 26,54 Tb 217 — 27,67 Pu 60 24,91 32,11
Zr 275 25,16 25,37
иа оси ординат, соответствует коэффициенту у, который равен: у— n2kRj2EF.
В случае фазового превращения 1-го рода в металлах энтальпия II изменяется скачком на величину скрытой теплоты превращения Qu, а зависимость теплоемкости от температуры имеет разрыв, т. е. ее величина стремится к бесконечности. Прн превращениях 2-го рода наблюдается резкое увеличение энтальпии, а на температурной зависимости теплоемкости— пик конечной величины.
Деформация повышает величину энтальпии металла и его теплоемкость иа 1—2 %. Энергия, поглощенная металлом в результате воздействия холодной пластической деформации, выделяется при рекристаллизации.
Теплоемкость сплава С можно подсчитать по известным теплоемкостям компонентов Ci и С2 (правило Коппа—Неймана): C=piCi + +Р2С2, где pi и р2—количества компонентов в процентах по массе прн определении удельной теплоемкости и в атомных процентах при расчете атомной теплоемкости.
Правило Коппа—Неймана удовлетворительно (погрешность не более 4 %) выполняется для твердых растворов, химических соединений, гетерогенных структур.
Важной характеристикой прочности межатомных связей является теплота образования вещества. Наибольшей теплотой образования обладают стойкие химические соединения, меньшей — промежуточные фазы и твердые растворы (табл. 17.2),
17.1.2. Методы измерения энтальпии и теплоемкости
Метод Сайкса — Грузина [1]. Метод основан на измерении количества теплоты, необходимой для нагрева образца до определенной температуры (принцип обратной калориметрии). Образец 1 (см. рис. 17.2) массой m нагревается электрической спиралью 2 от температур Ti до Г2. Необходимое количество теплоты без учета тепловых потерь определяется как Q=/2/?t, где I— сила электрического тока проходящего через спираль сопротивлением R за время т. Средняя теплоемкость образца Cp=IzRx[m. Для определения истинной теплоемкости необходимо уменьшить МТ— =Т2—Т\. Для этого образец помещают в блок 3, находящийся в печи 4. Теплоемкость определяется уравнением СР— WI{mdTBldx), где W— мощность спирали; m — масса образца; dTJdx— скорость измерения температуры образца.
Уравнение справедливо в условиях теплового равновесия, когда температура блока (Ть) равна температуре образца (ТБ) (прн включении спирали 2). Поскольку Ть изменяется в соответствии с режимом печн 4, удобнее использовать выражение dTbldxA-d[ts—Tb)ldx— =dTs!dx,
Определение теплоемкости при температуре Т сводится к измерению мощности необходимой для выравнивания Ть и Т&, измере-
277
ТАБЛИЦА 17.2
ТЕПЛОТЫ ОБРАЗОВАНИЯ ХИМИЧЕСКИХ СОЕДИНЕНИЙ ПРОМЕЖУТОЧНЫХ ФАЗ И ТВЕРДЫХ РАСТВОРОВ (ПО Б. Г. ЛИВШИЦУ)
Система Состав, % (ат.) Теплота образования. кДж/МОЛЬ Формула соединения или фазовое состояние Система Состав. % (ат-) Теплота образования, кДж/моль Формула соединения или фазовое состояние
Химические соединения
AI—Са 25 4-53,59 А1яСа Hg—Na 33 г25,12 HgNa2
А1—Се 33 “-54,42 А12Се Li—Pb 30 -35,17 LiePb2
Al—Sn 50 --17,16 AlSn Li—Sb 33 -41,86 LiSb2
Bi—Са 60 --96,2 Bi2Ca Li—TI 50 -26,79 LiTl
Bi—Li 75 —58,6 BiLi3 Mg—Pb 33 — -17,58 MgPbp
Bi—Mg 60 —30,56 BiaMg8 Mg—Sb 40 -56,94 Mg3Sba
Bi—Na 75 --47,72 BiNa3 Mn—Sn 33 г23,44 MngSn
Ca—Si 50 4-234,4 CaSi Mn—Zn 57 -17,58 MgZna
Ce—Mg 50 4-27,21 CeMg Na—Sb 25 -50,24 NasSb
Co—Sb 50 4-20,93 CoSb Na—Sh 50 +5,12 NaSn
Co—Si 50 -1-50,24 CoSi Na—Те 50 -18,84 NaTl
Fe—Si 50 4-37,68 FeSi Ni—Si 33 46,47 NiaSi
Hg-K 50 4-27,21 HgK
Промежуток 1ные фазы
Ag-Cd 60 4-6,07 T Al—Co 50 4-56,52 6(CoAl)
Ag—Sb 60 4-7,53 a-Sb+e' Al—Mg 57 4-29,30 0
Al—Cu 67 4-21,77 T Al—Ni 50 +71,17 у (NiAl)
Au—Cd 50 4-16,32 ₽ Au—Zn 50 +23,02 p (AuZn)
Cd—Hg 50 4-4,8 0 Ca—Pb 33 +62,60 Y (Ca2Pb)
Cd—Sb 50 4-7,53 Y Ca—TI 50 +71,17 T (CaTl)
Cu—Sn 25 4-7,53 Y (Cu3Sn) Cd—Mg 50 +19,25 a(CdMg)
Cu—Zn 60 4-12,56 Y Ni—Sb 50 +33,49 T (NiSb)
Твердые j метеоры
Ag—Au 50 4-3,97 Pb—TI 80 +2,72 —I
Ag—Cu 50 4-5,23 —
пню скорости нагрева блока dTtJdx с помощью термопары 5 и скорости выравнивания температур блока и образца (ЦТь—Ts}fd% с помощью дифференциальной термопары 6.
Метод Смита (5]. Метод позволяет производить термический анализ, определять теплоемкость и скрытую теплоту превращения. Сущность метода заключается в том, что в процессе эксперимента градиент температуры
б/ на стенке контейнера 1 (рис. 17.3), в который вставлен образец 2, поддерживается постоянным. Контейнер изготовлен нз огнеупорного материала с малой теплопроводностью. В этом случае устанавливается некоторое стационарное состояние, при котором пустой контейнер нагревается на Ыс за время Атс. Количество теплоты, израсходованное на повышение температуры на А?с, составляет
Рис. 17.2. Схема метода Сайкса—Грузина
278
Рис. 17.3. Схема метода Смита
НЬъс—М<£стс, где Cc и mc — теплоемкость н масса контейнера.
Отсюда: 7/=(А/с/Атс)Сстс.
При нагреве контейнера с образцом Н= — (А^о/Ато) (Сотс+Сстс), где Со и т0 — теплоемкость и масса образца соответственно; А/о/Ато — скорость нагрева.
Для определения экспериментальных погрешностей контейнер нагревают с эталоном, теплоемкость (Сэ) и масса (me) которого известна, тогда Н~ (А/а/Атэ) (Сстс+Сятё). Решив совместно уравнения, получим:
Csms __ (Атэ/А/а — Лтс/А/с)
(Атд/А/р Атд/ А^р)
где Ат/Д£ — величина, обратная скорости иагрева. Таким образом, измерив скорости нагрева пустого контейнера, контейнера с образцом и эталоном (заданный градиент 6f контролируется дифференциальной термопарой 5), можно рассчитать теплоемкость образца при любой температуре Т.
Зиая -величину И прн температуре превращения, можно найтн скрытую теплоту превращения из Qntnj) = Нкт, где Qn — скрытая теплота превращения, Ат — время превращения.
Измерение Ат/Д/ и t (термопарой 4) позволяет построить дифференциальную кривую, т. е. провести термический анализ превращений в адиабатических условиях.
Импульсный метод [6]. Применяется для невысоких температур. В основе его лежит кратковременный нагрев образца (104—5Х X104 °С/с) импульсом электрического тока, тепловые потери при этом пренебрежимо малы. На рнс. 17.4 представлена схема устаиов-
Рис. 17.4. Схема импульсного метода измерения теплоемкости
ки для измерения теплоемкости импульсным методом. На образец 1 подается импульс тока от батареи 2. Сила тока и падение напряжения регистрируются делителями 3 и 5 и шлейфовыми осциллографами 4. Вращающийся цилиндр 8 с зеркалом 6 и шкалой 7 позволяют осуществлять временную развертку процесса.
По кривым изменения напряжения и силы тока можно определить теплоемкость образца при любой температуре.
Модуляционный метод измерения теплоемкости [6]. Если к образцу подводить мощность [Р), изменяющуюся по закону Р = Рс+ + Р sin сот, где сот — угол сдвига фаз; Ро— величина мощности (по модулю), то его температура при этом будет колебаться с частотой о> около среднего значения Т = Го+6.
Условие баланса мощностей приводит к уравнению й0/йт+й0/тс = Psin ыт[тс, где т — масса; с — удельная теплоемкость образца; k — коэффициент теплоотдачи образца.
Решение уравнения имеет вид: 0 = — 0о sin (гот—-ф); 0О = Р •sm<±[mc и; tgq = = mctojk (ф— угол сдвига).
Для адиабатических условий (ф=90°) легко рассчитать теплоемкость. Одни из методов определения температуры образца и амплитуды ее колебаний использует зависимость электрического сопротивления образца от температуры. По образцу пропускается ток /о+/Х Xsine>T(7<C/0). С учетом колебаний температуры образца и их отставаний по фазе от колебаний тока импеданс образца Z = Р+ (dR/dT) 0QX Xsin (сот—<р); переменная составляющая напряжения U~IR sin oz+I(dR/dT) 0osin(coT— —<p). Второе слагаемое связано с изменением сопротивления образца при колебаниях температуры (падение напряжения при протекании постоянного тока по изменяющемуся сопротивлению). Импеданс для переменного тока можно представить в комплексной форме:
z = Я + °о COS ф -н Zt^- е0 Sin <р, idi lai
что справедливо для активного сопротивления, шунтированного емкостью. Благодаря этому теплоемкость образца можно определить прн помощи мостовой схемы переменного тока, где одно плечо — образец, остальные — активные сопротивления, одно нз которых шунтируется емкостью. Из условия равновесия мостовой схемы получаем:
тс = [2ll/<^R2C2) (dR/dT),
где dR/dT—производная сопротивления образца по температуре; 2?2 — сопротивление; Сг — емкость.
Мостовая схема уравновешивается независимо от амплитуды колебаний температуры образца.
Схема установки для измерения теплоемкости модуляционным методом представлена на рис. 17.5. Погрешность метода в основном определяется надежностью данных о температурной зависимости электросопротивления образца, так как все остальные параметры (сила тока, частота) определяются достаточно точно.
Измерение энтальпии образцов модуляционным методом сводится к измерению температуры н электрического сопротивления, так как
72 - Рг п
С 2 f Л)
АЯ= medT = — —dR,
J w J RC
Ti Pi
Из-за влияния холодных контактов моста на образец при высоких температурах целесообразнее пользоваться потенциометрической схе-
279
Рнс. 17.5. Схема установки для измерения теплоемкости модуляционным методом
1 — образец; 2 — усилитель; 3 — осциллограф; 4 — генератор низких частот
Рис. 17.6. Потенциометрическая схема модуляционного метода
Рис. 17.7. Схема установки с применением ПБС
мой измерения (рнс. 17.6). Проволочный образец 1 нагревается током от выпрямителя 2 и генератора 3. Потенциальные провода 4, 5 соединены с центральной частью образца. Момент компенсации схемы устанавливается по индикатору 6.
Метод определения теплоемкости и энтальпии металлов в расплавленном состоянии. В этом случае измерения рекомендуется проводить в процессе плавки металла во взвешенном состоянии (ПВС) [7]. Схема калориметрической установки с использованием ПВС представлена на рис. 17.7. Установка состоит нз двух основных частей: камеры ПВС 1 и изотермического калориметра 2. Под индуктором 3 расположен водоохлаждаемый поворотный диск 4 с изложницей 5 для непредусмотренного слива металла. После выдержки образец 6 сбрасывают в приемное устройство калориметра 2. Прн этом диск с изложницей отводят в сторону, в результате отключается генератор н открывается вход в калориметр. Время падения образца 0,19 с. Заслонка калориметра автоматически закрывается через 0,30 с. Температура измеряется оптическим пирометром. Разброс'экспериментальных точек —1 %.
17.1.3. Термический анализ
Для исследования химических реакций и превращений, происходящих под влиянием нагрева или охлаждения сплавов, применяется метод дифференциального термического анализа, позволяющий измерять незначительные тепловые эффекты, н метод термогравиметрин, с большой точностью определяющий изменение массы исследуемого образца в процессе превращения [8].
Аппарат для термического анализа состоит из печи н трех термопар. Одна термопара служит для измерения температуры исследуемого образца. Две термопары включены по дифференциальной схеме, прн этом одна термопара (или спай) помещена в эталонное вещество, не претерпевающее изменений под влиянием тепла, но создающее с точки зрения теплопередачи условия, тождественные исследуемому веществу. Второй спай дифференциальной термо-пары помещен в исследуемый образец. При использовании такой схемы измерения температуры при нагреве печи равномерно повышается температура как образца, так и эталона до тех пор, пока в исследуемом металле не начнутся превращения. С этого момента изменение температуры исследуемого образца либо ускорится, либо замедлится в зависимости от того, сопровождается ли превращение выделением нлн поглощением тепла. Показания дифференциальной термопары определяются величиной теплового эффекта процесса. Показания термопары, измеряющей температуру образца, и дифференциальной термопары с помощью световой системы гальванометров проектируются на фоточувствительиую бумагу записывающего устройства.
Термогравиметрня позволяет с большой точностью проследить за изменением массы образца при нагреве и связать эти изменения с реакциями, происходящими в исследуемом веществе. Масса вещества измеряется автоматически иа аналитических весах. По кривой термо-гравиметрнческого (ТГ) анализа можно про-
280
следить за превращениями исследуемого металла и произвести расчеты с определенным количеством продуктов реакции. Трудности оценки кривой ТГ привели к созданию дифференциальной термографии. Кривая скорости изменения массы образца во времени (ДТГ) позволяет с большей надежностью судить о превращениях в исследуемом металле. Одновременное измерение изменения массы и энтальпии позволяет полнее анализировать проходящие прекращения. Совместные термический и термогравнметрнческий анализы осуществляются с помощью дериватографов.
Принципиальная схема дериватографа изображена на рис. 17.8. Исследуемый образец и эталонный металл помещаются в тигли 2 н 3
Рис. 17.8. Схема дериватографа
печи 1. Массу исследуемого металла определяют иа весах 7, на коромысло которых опирается тигель. Оптическая щель 6 на стрелке весов размещена так, что прн смещении стрелки световой луч от осветителя 8 запишет на светочувствительной бумаге 12 барабана 13 изменение массы исследуемого образца. Скорость изменения массы измеряется с помощью катушки 10, подвешенной к коромыслу весов н находящейся в поле постоянного магнита 9. Магнитное поле индуцирует в движущейся катушке ток, сила которого пропорциональна скорости движения коромысла, а следовательно, и скорости изменения массы исследуемого образца. Световой гальванометр 11а записывает на светочувствительной бумаге кривую ДТГ. Тигли 2 и 3 выполнены таким образом, что спаи 4 термопар 5 размещены внутри образцов. Кривая дифференциально-термического анализа описывается показаниями дифференциальной термопары, измеряющей разность температур эталонного и исследуемых образцов; через гальванометр Не луч осветителя 8 записывает показания термопары на светочувствительную бумагу 12. Температура печи записывается с помощью гальванометра 116.
ПЛ А. Теплопроводность
Измерения теплопроводности редко проводятся при исследованиях в силу сложности н невысокой точности. Однако она является очень важной технологической и эксплуатационной характеристикой материала.
В изотропном твердом теле распространение тепла подчиняется закону dQ=~xgradrdSdr,
где dQ— вектор, величина которого равна потоку тепла, перенесенного через сеченне площадью^, перпендикулярное dQ, за время dr; Т — температура; я — коэффициент теплопро* водности, Вт/(м-К); знак минус связан с тем, что тепло переносится в направлении, противоположном градиенту температуры. В кристалле, который ие обладает кубической симметрией, вектор dQ может быть не параллелен gradT н уравнение примет вид;
dQi = — кц (OT/dxj) dSdt,
где Яц — тензор второго ранга.
Высокая теплопроводность металлов объясняется тем, что перенос тепла в них осуществляется в основном передачей энергии электронами в отличие от неметаллических веществ, где эиергин переносится в основном тепловыми колебаниями атомов. Однако соотношение вкладов зависит от конкретных условий н материала, например в сверхпроводящих материалах относительные вклады этах механизмов различны в нормальном и сверхпроводящем состоянии. В общем случае теплопроводность является суммой решеточной н электронной теплопроводности: х=ахреш+6хе.
Теплопроводность, обусловленная переносом тепла электронами: Хе=пеСео2т/3, где Се—теплоемкость на один электрон; и —средняя тепловая скорость электронов; пеСе—Су — электронная доля молекулярной теплоемкости; пе —число электронов в единице объема.
Допуская, что поведение электронов подобно поведению молекул идеального газа;
Cev = Зй/2, а те о2/2 = ЗйТ/2.
Подставляяя значения для Су и о2 в выражение для получаем следующее соотношение между тепло- и электропроводностью: ха/о=ЗГ (£/е)2, которое является законом Видемана—Фраица— Лоренца: прн комнатной температуре для металлов отношение удельной теплопроводности, которая в основном определяется электронами, к электропроводности есть величина постоянная. Позднее Лоренц показал, что в широкой области температур отношение постоянно.
Прн выводе закона Видемана — Фраица —> Лоренса было допущено, что электроны ведут себя аналогично молекулам идеального газа. Однако с точностью до постоянной это выражение можно получить, допустив, что электроны подчиняются статистике Фермн—Днрака и их взаимодействие с нонами решетки иоснт дискретный характер. В этом случае
где £о — идеальное число Лоренца, равное 2,443-10"8 В2/К2.
Отклонение от закона Вндемана —Франца-— Лоренца в основном связано с непостоянством соотношения между значениями электронного и решеточного вклада в величину теплопроводности (подробнее см. [9]).
При очень низких температурах, близких к абсолютному нулю, теплопроводность х определяется чистотой металла и формой образца. Она растет с увеличением температуры аиа-
281
логично теплоемкости. Рост теплопроводности с температурой прекращается, когда становятся заметными процессы рассеяния электронов на колебаниях решетки и уменьшается длина свободного пробега электрона. При дальнейшем повышении температуры теплопроводность падает. Таким образом, зависимость к(Т) в отличие от температурной зависимости электрического сопротивления имеет максимум [10, II]. Положение максимума на зависимости х(Г) определяется дефектностью материала и соотношением электронного н решеточного вкладов.
Величину решеточной составляющей теплопроводности Хрещ МОЖНО ВЫЧИСЛИТЬ ПО формуле: хреШ=2,41 пСг^^где Cv—молярная теплоемкость; а — межатомное расстояние; d — плотность; Е — модуль упругости.
В чистых металлах прн очень высоких температурах значения чнсла Лоренца близки к идеальному. При низких температурах отношение х/о7 оказывается значительно ниже Lo и может составлять только малую его часть. Но прн Т->0К величина х/оГ опять возрастает до Lo. Температура, при которой отношение х/оГ приближается к £,&, неодинакова для раз-
Рис. 17.9. Зависимость и 276^ для образца с
различной концентрацией дефектов
ных металлов и сильно зависит от концентрации примесей и количества дефектов.
На рис. 17.9 представлены зависимости отношения nfaT от приведенной температуры для образцов с различной концентрацией дефектов (сплошные линии).
Примеси,- содержащиеся в сплавах, обычно подавляют электронную компоненту теплопроводности, уменьшая длину свободного пробега электрона, ио слабо влияют на решеточную компоненту. Последняя определяется главным образом фонон-фононным н электрон-фононным взаимодействием. В силу этого решеточную компоненту в полной теплопроводности для сплавов можно определить точнее, чем в чистых металлах, во-первых, потому, что в сплавах она относительно больше, во-вторых, электронную составляющую теплопроводности в сплавах можно оценить с большей точностью, используя результаты измерения электропроводности. Самый простой способ определения решеточной теплопроводности чистого металла состоит в экстраполяции результатов измерений для сплавов различного состава к нулевой концентрации примесей [9].
Для интерпретации полученных результатов измерения теплопроводности следует использовать данные, характеризующие индивидуальные свойства данного материала. Одиако следует отметить, что даже приближенные значения соотношения Видемана — Франца — Лоренца позволяют перенести закономерности, найденные для электропроводности, на явления теплопроводности, что очень удобно, так как измерение теплопроводности гораздо более сложно н менее надежно нз-за затруднений прн измерении величины теплового потока [9].
Как н электропроводность, теплопроводность металлов с кубической решеткой не зависит от кристаллографического направления. Для металлов с иекубнческой решеткой наблюдается анизотропия теплопроводности. Например, для монокристалла гексагонального кадмия параллельно главной оси симметрии к=83,1 Вт/ /(м-К), в перпендикулярном направлении — 4,1 Вт/(м-К).
Ниже приведены значения х чистых металлов прн 0 °C:
Металл..........
х, Вт/(м-К) . • .
Li
70
Na К Be
140 100 160
Mg Al
172 226
Ta Mo W
55 137 169
Fe Co Ni
94 70 62
Ir
60
Металл..........
x, Вт/(м-К) . . ,
Pd
70
Pt
70
Cu Ag Au
393 415 312
Zn Cd Hg Те Sn
H3 098 104 51 66
Pb Sb
35 19
Bi
10
Как указывалось выше, при рассмотрении температурной зависимости теплопроводности металлов необходимо учитывать изменение соотношения между вкладами электронной и решеточной теплопроводности в общее ее значение. При этом для различных металлов это отношение меняется неодинаково в зависимости от концентрации н подвижности электронов проводимости. Например, для меди вклад решеточной составляющей незначителен (см. табл. 17.3).
При высоких температурах (7’>0г) теплопроводность и электропроводность уменьшаются плавно с ростом температуры, а при температуре плавления резко падают. Исключение составляют висмут н сурьма, теплопроводность
ТАБЛИЦА 17.3
ОЦЕНКИ РЕШЕТОЧНОЙ ТЕПЛОПРОВОДНОСТИ ЧИСТОЙ МЕДИ И ЕЕ ВКЛАДА
В ПОЛНУЮ ТЕПЛОПРОВОДНОСТЬ
Температура К хполи‘ Вт/(м-К) хрепь Вт /(м-К) хреш/хполн
300 400 5 0,01
200 410 8 0,02
100 500 15 0,03
40 1600 30 0,02
10 18000 12—35 0,001
282
которых возрастает при температуре плавления.
Теплопроводность зависит от величины зерна металла и резко увеличивается с его ростом. Значения теплопроводности в зависимости от величины зерна для электролитического железа приведены ниже:
Среднее число
зерен на 1 см2 10 170 634
х, Вт/(м-К) 92,91 89,58 83,75
В ряду непрерывных твердых растворов теплопроводность понижается с увеличением процента легирующего компонента. Минимум теплопроводности сплавов, как правило, отвечает 60 % (ат.), а ее величина может быть в несколько раз ниже, чем теплопроводность компонентов (рнс. 17.10).
Прн образовании гетерогенных структур зависимость теплопроводности от объемной кон-
Рис. 17.10. Теплопроводность твердых растворов системы Ag—Ап
цеитрации компонентов почти линейная. Примером могут служить сплавы системы Sn—Bi (рис. 17.11).
Для техники существенное зиачеине имеет величина коэффициента температуропроводности ос, равная а—x/Сг, где х— Теплопроводность; Cv—теплоемкость на единицу объема.
Коэффициент температуропроводности в тепловых процессах характеризует скорость изменения температуры. Чем больше а, тем меньше разность температур отдельных участков
Рис. 17,11. Теплопроводность сплавов системы Sn—Bi
тела при одинаковых условиях нагрева н охлаждения.
17J.5. Методы измерения теплопроводности
Стационарные методы
Методы, в которых во время эксперимента поддерживается постоянный поток тепла или постоянная разность температур, называются стационарными. Эти методы можно использовать, когда известно, что тепловой поток от нагревателя распространяется вдоль образца.
Метод продольного потока тепла [9]. Схема эксперимента представлена на рис. 17.12. Пос-
ЛТ
L
Рис. 17.12. Схема измерения х методом продольного потока тепла
тоянный тепловой поток Q пропускается вдоль однородного стержня с площадью поперечного сечения S. Термопары прикрепляют к образцу иа расстоянии L одна от другой н измеряют разность их показаний (Д7’)_ Коэффициент теплопроводности определяется нз формулы Q^z.SA7/7.
Если разность Д7 не велика, то определяемое значение и будет соответствовать средней температуре между термопарами, даже если и сильно зависит от температуры.
На рнс. 17.13 приведена принципиальная схема установки для определения теплопроводности стационарным методом при продольном потоке тепла [1]. Испытуемый образец 1 ввинчивается в медный блок 2, в котором расположена спираль электронагревателя. Верхняя часть образца помещена в медный блок 5, который охлаждается водой. Зная расход воды и разность температуры воды прн ее входе 4 н выходе 5, подсчитывается количество теплоты, проходящее в единицу времени через сечение образца (при условии, что вся теплота без потерь уносится водой). Распределение температур по образцу измеряется термопарами 6, 7, 8. Для уменьшения радиационных потерь имеется трубчатый защитный экран 9. Температура по экрану распределяется так же, как н по образцу, так как внизу экран контактирует с нагревательным блоком, а вверху охлаждается водой до той же температуры, что н блок 3. Вариант этого метода — прибор (рнс. 17.14), в котором количество тепла, прошедшего по образцу,- определяется количеством электрической энергии, необходимой для нагрева образца, при этом нагреватель 1 расположен внутри образца 2. Холодный конец образца помещаетсн в ванну 3, в которой поддерживается постоянная температура, термопары размещаются в прорезях а—а и b—b.
283
Рнс. 17.13. Схема установки для измерения и методом продольного потока
Верхний предел по температуре этого метода определяется радиационными потерями тепла, его можно повысить с помощью радиационных щитов, вдоль которых создается такое же распределение температур, как и вдоль образца. Лучший способ увеличить область использования метода стационарного потока — проведение внутреннего нагрева образца, чтобы избежать тепловых потерь на излучение.
Метод Кольрауша [5]. Метод продольного потока используется и в приборе конструкции Кольрауша (рис. 17.15). Основное отличие метода состоит в том, что образец нагревается прн прнхождеини через него тока. Температура концов образца поддерживается постоянной. В этом случае температура равномерно возрастает от концов образца (АТ) и достигает максимума на середине. Теплопроводность можно вычислить по формуле х=£/2/8р(АГ)2, где U — разность потенциалов иа концах испытуемого образца; р-—удельное электрическое сопротивление образца.
Метод радиального потока тепла [9]. Метод продольного теплового потока осложняется тем, что, кроме тепловых потерь, необходимо учитывать тепловое сопротивление контакта, которое может быть весьма значительным, а в месте контакта возникает скачок температур. Во избежание этого применяется радиальный
Рис. 17.14. Схема прибора для измерения и с внутренним нагревом образца
поток тепла. Прн нагреве образца изнутри излучение и другие потерн не влияют на температуру поверхности. Если нагреватель расположен на осн полого цилиндра н выделяет одн-
Рис. 17.15. Схема прибора Кольрауша:
1 — образец; 2 — печь; 3 — рубашка водяного охлаждения; 4 — штатив; 5 — цанги
284
наковое количество теплоты вдоль всей длины, то тепловой поток на единицу длины в направлении радиуса цилиндра связан с температурами Тг и Тг на радиусах п и г2 формулой Q=2 гос [ГГ1—f r J /1п (гх/г2).
Формула справедлива, если рассматриваемая область находится достаточно далеко от концов цилиндра, так что можно пренебречь продольным потоком. Если образец состоит из набора колец, радиальный поток не изменится, нс нз-за теплового сопротивления продольный поток значительно уменьшается.
Метод Стакса—Чесмара [12]. На практике часто используются сравнительные методы, когда поток тепла может быть н продольным, и радиальным. По методу продольного потока сравнивают либо температурные градиенты образца из исследуемого материала и эталона с известной теплопроводностью, либо расстояния,
Рис. 17.16. Схема прибора Стакса—Чесмара
на которых температурные градиенты одинаковы. Во всех случаях через образцы должны проходить одинаковые потоки тепла. На рнс. 17.16 приведена схема прибора Стакса -Чес-мара. Поток тепла проходит от источника через набор чередующихся образцов из исследуемого н эталонного материала. В местах контакта исследуемого образца и эталона помещены термопары 1—6. По методу радиального потока стандартный и исследуемые образцы располагаются концентрически.
Нестационарные методы
Произвольное распределение температуры в образце меняется со временем, причем скорость изменения зависит от отношения коэффициента теплопроводности к теплоемкости на единицу объема (коэффициент температуропроводности а=х/Сг).
Существует несколько методов, основанных на измерении скорости изменения температуры в образце. Этн методы иосят название нестационарных.
Метод Ангстрема [12]. В методе Ангстрема (метод температурной волны) температура на
одном конце длинного цилиндрического образца меняется по синусоидальному закону. Измеряется затухание температурной волны и фазовый сдвиг температуры (рнс. 17.17). Если в точке х—0 отклонение температуры образца от средней температуры меняется во времени по закону Т'(0) = Г0 sin со/, то без учета тепловых потерь изменение температуры на расстоянии х определяется формулой
Т1 (0) = То ехр (— V(й/2ах) sin (art —
— V"(й/2ссх) .
В этих условиях коэффициент температуропроводности можно найти или по декременту затухания волны
I/Z (х) = ехр (— со/2ах ),
или по фазовому сдвигу ф (х) = }/ (й/2ах ,
т. е. а—<ох2/2(1пХ)2=сох2/2ф.
Этот метод можно использовать прн низких
Рис. 17.17. Схема прибора Ангстрема
температурах, когда легко устранить тепловые потери. Однако даже при наличии тепловых потерь выполняется соотношение а=ых2/ /(2ф In А)..
Метод кратковременного нагрева [9]. Метод кратковременного нагрева используется при продольном н радиальном потоке тепла. Потери на излучение можно сделать минимальными за счет ограничения времени эксперимента. Это достигается кратковременным нагревом одной поверхности тонкой пластинки с помощью лампы-вспышки нлн лазера. После локального нагрева образца измеряется температура поверхности, удаленной от источника тепла. Еслн пренебречь потерями тепла за время, необходимое для прогрева поверхности до максимальной температуры, то можно получить простое соотношение между временем необ-
285
ходимым для нагрева пластинки толщиной d, до температуры, равной половине максимальной температуры, н коэффициентом температуропроводности а—1,37 d2An2T(V2)J (
17Л.6. Применение методов измерения тепловых свойств для исследования металлов и сплавов
Исследование тепловых еффектов при фазовых превращениях [1]. В качестве примера иа рис. 17.18 приведены тепловые эффекты превращений, происходящих в сталях в зависимости от содержания углерода. Из графика следует, что
величина теплового эффекта зависит от количества фазы, претерпевающей превращения. При пересчете теплового эффекта на один грамм фазы, претерпевающей превращение, вычислены следующие значения: для магнитного превращения цементита — £2^=40,69 Дж/г; для превращения перлита в аустенит — Qyli — =65,7 Дж/г; для магнитного превращения феррита — Сд2 = 15,49 Дж/г; для а->у-превращения (прн 910 °C) чистого железа — QA — =24,99 Дж/г.
Исследование теплоемкости при отпуске закаленной стали. В процессе отпуска стали происходят фазовые н структурные превращения, в результате которых изменяется удельная теплоемкость. По зависнмостн теплоемкости от температуры (рис. 17.19) можно установить интервалы температур, в которых прн данной скорости нагрева (10 град/мин) происходят фазовые превращения, а по величине изменения ср определить характер нли род превращений.
Минимум теплоемкости прн 180 °C (тепловой эффект /) соответствует превращению мартенсита закалкн в мартенсит отпуска. При 250—300 °C распадается остаточный аустенит (тепловой эффект II), а при 350—450 °C карбид FeaC полностью переходит в FesC и проходит коагуляция (тепловой эффект III).
Исследование теплоемкости при фазовых превращениях второго рода. При превращении второго’ рода иа кривой температурной зависимости теплоемкости должен быть скачок конечной величины, а не разрыв, как при превращении первого рода. Это служит осно-
Рис. 17.19. Зависимость теплоемкости стали от температуры:
а — после закалки; б -- после отпуска
Рис. 17.20. Зависимость теплоемкости сплава Cu—Zn от температуры
ванием для использования данных по теплоемкости при определении характера превращения. На рнс. 17.20 зависимость удельной теплоемкости сплава CuZn имеет скачок конечного значения, что позволяет считать процесс упорядочения данного сплава превращением второго рода. Аналогичная зависимость наблюдается прн магнитном превращении ферромагнетика вблизи температуры Кюри.
Построение диаграмм состояния методом термического анализа. Для построения диаграмм состояния необязательно измерять зависимость энтальпии от температуры, достаточно для каждого сплава построить кривую охлаждения. Примеры кривых охлаждения приведены на рнс. 17.21. Кривые а к г характеризуют кристаллизацию чистого компонента или эвтектики; б — двухкомпонентного раствора; в — эвтектического или доэвтектического сплава.
Линии ликвидус — солндус, как правило, строятся по результатам термического анализа.
Метод термического анализа меиее пригоден для исследования превращений в твердом со
286
стоянии, так как тепловой эффект в этом случае меньше, и целесообразнее использовать дифференциальный термический анализ.
При применении обоих методов наиболее существен правильный выбор скорости охлаждения. При значительных переохлаждениях
Рис. 17.22. Зависимость теплоемкости олова от температуры:
1 — сверхпроводящее состояние; 2 — нормальное состояние
жидкости резко выделяется скрытая теплота кристаллизации и на кривой охлаждения может возникнуть площадка там, где ее не должно быть. Превращения в твердом состоянии происходят при больших переохлаждениях, поэтому необходимо следить за достижением термического равновесия.
Измерение теплоемкости при переходе металла в сверхпроводящее состояние. Когда по каким-либо причинам невозможно определить критическую температуру перехода образца из нормального в сверхпроводящее состояние прямым методом, проводят измерения теплоемкости. На рис. 17.22 приведена зависимость теплоемкости олова от температуры. Скачок конечной величины на кривой С(Т) соответствует превращению 2-го рода, каким и является переход в сверхпроводящее состояние (критическая температура для олова 3,7 К ПЗ]).
17.2. ПЛОТНОСТЬ
И ТЕРМИЧЕСКОЕ РАСШИРЕНИЕ
17.2.1. Плотность и методы
ее измерения
Плотность р—масса единицы объема данного материала p—m/V, где m — масса; V — объем тела.
Величина, обратная плотности, называется удельным объемом v н соответствует объему единицы массы: о—l/p=V/w.
Определение плотности сводится к определению массы н объема тела. Измерение массы тела с достаточной точностью производится обычно на аналитических весах, а объем определяют двумя методами: пикнометрическим и гидростатическим. [1].
Пикнометрический метод основан на определении объема жидкости, вытесненной при погружении в нее испытуемого образца. Невысокая точность (~1%) не позволяет применять его в исследовательских целях, поскольку изменение плотности металлов н сплавов в результате наклепа, термической обработки обычно =^1 %. Однако для технических целей пикнометрический метод успешно используется. Жидкость, в которую погружают исследуемый образец, должна обладать хорошей смачивающей способностью.
Возможны два варианта измерения объема вытесненной жидкости: измеряется увеличение объема жидкости в сосуде определенной формы, проградунрованном по объему; определяется масса жидкости, вытекшей из пикнометра после погружения образца. Взвешиваются; 1) образец на воздухе; 2) пикнометр, заполненный жидкостью; 3) пикнометр, заполненный жидкостью, с находящимся в нем образцом. Плотность образца определяется по формуле
Р
P ______ р [ ру [Р®---РйОЗд) + РВОЗД,
где Q — масса пикнометра с жидкостью; р — масса пикнометра с жидкостью и образцом; Р— масса образца; рж — плотность жидкости; Рвозд — плотность воздуха.
Более точный метод, пригодный для исследовательских целей, — гидростатический. По закону Архимеда сила тяжести погруженного в жидкость тела уменьшается на величину, равную весу вытесненной жидкости. Испытуемый образец взвешивается иа воздухе и в жидкости. Зная плотность жидкости рН1, плотность образца можио вычислить по формуле
Р = Р (Рж Рвозд)/(-f ~"Q) + Рвозд,
где Р—'Вес образца; Q —вес образца в жидкости.
Плотность металла тесно связана с его структурой и атомным строением. Объем, занимаемый 1 г-атомом вещества (Уд), определяется как Уд~А/р, где А— масса 1 грамм-атома.
Величина, полученная при делении Уд на число Авогадро (N=6,0228 -102S), соответствует объему, приходящемуся на один атом, vn. Следует иметь в виду, что в атомный объем входит доля пор решетки, зависящая от коэффициента компактности структуры. Поскольку va усредненный параметр, он, как и плотность, зависит от количества дефектов в кристалле. В табл. 17.4 приведены атомные объемы металлов.
При рассмотрении вопроса структурообразования и сил межатомного взаимодействия важно проследить изменение размеров атомов при образовании той или иной структуры. В этом случае правильнее пользоваться не
287
ТАБЛИЦА 17.4 ПЛОТНОСТЬ, АТОМНЫЕ ОБЪЕМЫ И КОЭФФИЦИЕНТЫ ВСЕСТОРОННЕГО СЖАТИЯ МЕТАЛЛОВ [4, 14]
Элемент Тип решетки р-10”3, кг/м® » -10® Элемент Тип решетки р-10“3, кг/ма D -10»’ а » м8 м*/н
Li Na О. ц. к. О. ц. к. 0,534 0,97 21,622 39,49 86 142 Nb О. ц. к. 8,58 18,077 5,7
К О. ц. к. 0,851 75,3 232 Та О. ц. к. 16,6 18,0 . 4,8
Rb О. ц. к. 1,53 92,6 328 Ст О. ц. к. 7,14 12,001 6,0
Cs О- ц. к. 1,873 116,0 365 Мо О. ц. к. 9,01 15,580 3,5
Be Г. п. 1,85 8,11 7,8 W О. ц. к. 19,3 15,655 2,9
Mg Г. п. 1,74 23,24 29,5 Мп Г. ц. к 7,3 12,8 7,9
Ca Г. ц. к. 1,55 43,632 57 Тс Г.п. 11,49 14,213 —
Sr Г. ц. к. 2,60 56,335 82,0 Re Г.п. 20,53 15,705 2,7
Ba О. ц- к. 3,5 62,99 102 Fe О. ц. к. 7,86 11,776 5,9
Al Г. ц. к. 2,70 16,603 13,4 Ru Г.п. 12,1 13,574 3,4
Sc Г.п. 2,90 25,002 — Os Г. п. 22,5 13,993 2,6
I Г.п. 4,50 33,012 — Co Г.п. 8,71 11,128 5,4
La Дв. г. п. 6,18 37,415 40,37 Co Г. ц. к. — 11,077 —
Ce Дв. г. п. 6,69 34,472 40,97 Rh Г. ц. к. 12,44 13,766 3,6
Pr Дв. г. п. 6,71 34,56 32,08 Ir Г. ц. к. 22,42 14,144 2,7
Nd Г. п. 6,96 34,181 30,02 Ni Г. ц. к. 8,96 10,939 5,3
Sm Ромбоэдрическая 7,60 33,12 33,36 Pd Г. ц. к. 12,2 14,724 5,3
Eu О. ц- к. 5,30 48,099 66,63 Pt Г. ц. к. 21,4 15,104 3,6
Gd Г. п. 7,80 33,103 25,59 Cu Г. ц. к. 8,93 11,807 7,2
Tb Г. п. 8,19 31,97 24,6 Ag Г. ц. к. 10,492 17,057 9,9
Dy Г. п. 8,35 31,522 25,52 Au Г. ц. к. 19,3 16,961 5,8
Ho Г.п. 8,65 31.12 24,72 Zn Г. п. 7,13 15,212 16,6
Er Г.п. 9,01 30,642 23,88 Cd Г. л. 8,65 21,581 20,0
Tm Г. п. 9,20 30,099 24,71 Hg Ромбоэдрическая 13,546 23,4 34,0
Yb Г. ц. к. 7,02 41,281 73,93 Gd Ромбическая 5,93 19,595 20,0
Lti Г п. 9,79 29,496 23,85 In Тетрагональная 7,28 26 ,.144 25,0
Ti Г.п. 4,6 17,665 8,0 TI Г. п. 11,86 28,583 34,8
Zr Г. п. 6,44 23,272 11,0 Si Алмгза 2,33 15,5 3,2
Hf Г.п. 13,3 22,321 9,0 Ge » 5,32 18,0 14,1
V О. ц. к. 5,87 13.814 6,1 Sn Pb » Г. ц. к. 7,29 11,34 27,047 30,326 18,8 23 ,7
атомным объемом, а радиусом или диаметром атома.
В зависимости от тнпа межатомной связи (металлическая, ковалентная, ионная) эффективный радиус атома-иоиа может изменяться почти в два раза. Формула для расчета радиуса атома, находящегося в окружении п атомов, предложена Полингом [14].- —
—/?(71)=0,3 Ign, где 7?(1)—радиус атома в связях единичной кратности, — радиус этого же атома в связях кратности п.
Атомные диаметры, рассчитанные, исходя из кратности межатомных расстояний в структурах элементов, — параметры структурно зависимые, тогда как атомные объемы гораздо меньше зависят от типа решетки. Из табл. 17.5 видно, что при полиморфном превращении г. п. решетки в г. ц. к. или о. ц. к. в г. ц. к. изменение атомного объема ^3 %.
Нагрев приводит к непрерывному увеличению объема металла и соответственно к уменьшению плотности. Согласно правилу Грюнай-зена общее увеличение объема в интервале температур от 7=0 К до температуры плавления составляет у многих металлов с г. ц. к. решеткой ~7 %. Объемный коэффициент рас
ширения (3 связан с линейным коэффициентом расширения а для кубических кристаллов соотношением р«3а.
При плавлении плотность большинства металлов понижается. Можно отметить, что у металлов с наиболее компактной решеткой прн плавленнн удельный объем увеличивается максимально. Элементы, имеющие в твердом состоянии некомпактную решетку (Sb, Bi, Ga, Si) с небольшим координационным числом (1—3), прн плавлении становятся более компактными, их плотность увеличивается, а координационное число составляет уже 8—10.
При горячей деформации плотность, как правило, увеличивается в основном благодаря заполнению пор н трещин.
Прн малых степенях холодной деформации происходит увеличение плотности, связанное с устранением внутренних макропустот, а с увеличением степени деформации плотность понижается нз-за накопления дефектов.
Прн всестороннем упругом сжатии плотность возрастает, а объем металла уменьшается вследствие сближения атомов.
Коэффициент всестороннего сжатия х—
ТАБЛИЦА /7Л
ИЗМЕНЕНИЕ АТОМНОГО ОБЪЕМА МЕТАЛЛОВ %) ПРИ ПОЛИМОРФНЫХ
ПРЕВРАЩЕНИЯХ ПРИ РАВНОВЕСНОЙ
ТЕМПЕРАТУРЕ [14]
Металл Г, п.-»Г. ц. к. О. ц. К.-+Г. И. к.
Li —0,4
Na —0,4 ——
Be —3,58 —
Са — 0,20
Sr * — —0,30
Тс —0,55 —-
Zr —0,66 —
Hf —1,05 —
Мп — 0,8
Fe-a - -* 1,06 (y^cx)
Fe-у — 0,48 (vX6)
Те —0,15 —.
La — 1,3
Се — 0,1 (?£6)
Pr 0,5 —
Nd 0,1 —
Yb —1,25
Th -o,i
Pu — —2,0
=—dVjVdP, где Р— давление всестороннего сжатия; V — объем.
Значение % уменьшается с ростом давления и увеличивается с повышением температуры. Коэффициент всестороннего сжатия (см. табл. 17.4) — одна из характеристик сил межатомной связи.
Прн образовании твердого раствора нз металлов, имеющих близкие кристаллические структуры, период решетки твердого раствора согласно соотношению Вегарда должен линейно изменяться в зависимости от концентрации компонентов, выраженной в атомных долях: a=xIat+x2a2, где at и а2-—параметры решетки чистых компонентов; x( и х2—атомные доли компонентов.
Однако это соотношение выполняется довольно редко. Считается, что более удовлетворительно выполняется соотношение между) атомными объемами компонентов (d_ , v п и твердого раствора на нх основе (do): va —
17.2.2. Термическое расширение и температурный коэффициент линейного расширения
Из зависимости потенциальной энергии взаимодействия атомов в решетке U, межатомного расстояния г (рис. 17.23) [2] видно, что изменение межатомного расстояния с ростом температуры (термическое расширение) будет наблюдаться только при несимметричной зависимости U (г) (см. рис. 17.23, сплошная кривая). С ростом температуры энергия взаимодействия U повышается и центр колебаний атома (го) смещается в сторону больших г.
Для получения аналитической зависимости
а от температуры необходимо воспользоваться таким соотношением для потенциальной энергии атома, которое отражало бы ее асимметричный характер прн смещении атома из положения равновесия: t/(x)=ax2—bx3, где а — коэффициент, характеризующий гармоническую связь; b — коэффициент, характеризующий ан-
Рис. 17.23. Зависимость энергии взаимодействия атомов от расстояния между ними
гармоническую связь; х — смещение атома из положения равновесия; г—/о+х.
После преобразований, которые подробно изложены в работе [2] для среднего отклонения (х), получено соотношение x—ZbkTfea?. Отсюда для теплового расширения dxjdT— =3bk/4a2.
Отсутствие зависимости от температуры в полученном выражении связано с использованием классической статистики и фактора £/(*)
/iT
Больцмана е . Если заменить kT на энергию квантового осциллятора E—hvl(E^kT— —1), то получим для термического коэффициента расширения температурную зависимость, аналогичную температурной зависимости теплоемкости [2].
Прн экспериментальном определении коэффициент термического расширения получают из соотношения 12—1х[1-Ьа(Т2—Л)], где 12, li — длины образца прн температурах Т2 н Гр
Отсюда средний коэффициент линейного расширения (а) равен: а=12—1\ЦТъ— Соответственно истияный коэффициент линейного расширения (схт) при температуре Т: UT—dlldTlr.
Лля кристаллов кубической системы Грю-найзеиом было найдено выражение для ат: ат = 12CV Q0/[6Q0 - (m - п + 3) U]^
т
где Cv — атомная теплоемкость; U — | CydT;
Ь
Qo=V/yx: V— объем 1 г-атома; %— коэффициент всестороннего сжатия; у=(п+2)/6; т, п — показатели степени в выражении потенциальной энергии атома в кристаллической решетке.
Из приведенной выше формулы следует, что при низких температурах, когда потенциальная энергия U мала, ат пропорционален теп-
19—280
289
ТАБЛИЦА 17.6
ПАРАМЕТРЫ ГРЮНАЙЗЕНА ДЛЯ КУБИЧЕСКИХ КРИСТАЛЛОВ
Металл Интервал температур. СС V6
Си 0—600 1,7—1,95
Ag 0—300 2,7—2,8
Аи 0—300 2,3—2,35
А1 0—400 2,3—2,7
Na 0—200 0,9—1,1
К 0—150 1,05—1,2
нне колебаний решетки реального кристалла от гармонических.
Средний коэффициент линейного расширения (а) металлов для интервала температур О—100 °C приведен в табл. 17.7.
Для металлов с кубической решеткой величина ат практически не зависит от кристаллографического направления. Для иекубическнх кристаллов эта зависимость значительна.
При образовании твердых растворов металлов коэффициент термического расширения линейно зависит от количества второго компонента. В случае твердого раствора на базе переходных металлов значение ссг ниже, чем определяемое по правилу аддитивности.
лоемкостн CVt следовательно, ход кривых ат и Су при изменении температуры должен быть одинаков, что и подтверждается экспериментально.
Одним из фундаментальных соотношений, отражающим влияние изменения объема кристалла на энергию межатомного взаимодействия, является параметр Грюиайзена [14]: ус=д In QD/d In V, где 6г> — температура Дебая; V — объем.
В табл. 17.6 приведены значения параметра Грюнайзена для некоторых металлов. Обычно Yg~2 н его величина характеризует отклоне-ТАБЛИЦА 17.7
КОЭФФИЦИЕНТЫ ЛИНЕЙНОГО РАСШИРЕНИЯ МЕТАЛЛОВ [4]
Металл cc-10e, К"1 Металл a-10е, K-1 Металл a -10’, к—1
Li 58 Ge 6,0 Ba 17,0
Be 10,97 As 4,7 Ta 6,57
В 8,0 Pb 90,0 W 4,4
Nr 71,0 Zr 5,83 Re 12,45
Mg 27,3 Nb 7,2 Os 5,7
iAl 23,8 Mo 4,9 Ir 6,58
Si 6,95 Ru 7,0 Pt 8,9
К 84,0 Rh 8,5 Au 14,0
Са 22,0 Pd H,7 Hg 181,79
Ti 7,14 Ag 18,7 TI 33,65
Сг 6,7 Cd 31,0 Pb 28,3
v-Мп 14,75 In 77 Bi 12,1
a-Fe 11,5 tx-Sn 46,6 Tb ПД
Co 12,5 B-Sn 22,2 u 40,0
Ni 13,3 Sb 10,8
Си 17,0 Те 17,0
Zn 38,7 Cr 97,0
Ca 18,3
17.2.3. Методы измерения коэффициента линейного расширения
Дифференциальный дилатометр Шевенара. Высокая чувствительность прибора достигается прн использовании механического и оптического усиления. Принципиальная схема представлена на рис. 17.24.
В дифференциальном дилатометре удлинение образца 1 н эталона 2, расположенных в кварцевых трубках 3, 4, передается через систему стержней 5—8 к точкам 9—10 треугольной пластинки 11, закрепленной неподвижно в точке 12, служащей центром вращения при перемещении точек 9 и 10. Зеркало 13 отражает луч света от осветителя иа экран.
Для нагрева образца и эталона на кварцевые трубки 3, 4 надвигается электрическая трубчатая печь. В качестве эталона используется пирос — Сг—Со—Ni-сплав с известным коэффициентом линейного расширения, постоянным в исследуемом интервале температур.
На рис. 17.25 представлена схема записи дилатометрической кривой. Если предположить, что прн нагреве будет расширяться только эталон, то пластинка 11 с зеркалом 13 (см. рис. 17.24) будет вращаться вокруг оси 10—12 и световая точка иа экране с координатами х, у будет смещаться нз точки О по штриховой кривой АО (см. рис. 17.25). Если расширяется только образец, то пластина И с зеркалом 13 будет вращаться вокруг оси 9—12, а световая точка на экране смещается вертикально вверх по штриховой прямой ОБ (см. рис. 17.25). Прн одновременном расширении образца и эталона световая точка смещается по равнодействующей указанных траекторий (сплошная линия ОС, см. рис. 17.25). Эта линия наклонена направо вниз, так как коэффициент термического расширения пнро-
Рис. 17.24. Принципиальная схема дифференциального дилатометра Шевенара
290
са больше, чем у сталей (исключение составляют аустенитные стали). Если при какой-то температуре произошло сжатие образца, то светящаяся точка на экране пойдет вниз (отрезок ЕЁ, см. рис. 17.25).
Смещение световой точки в горизонтальном направлении Ох пропорционально температуре пнроса, поэтому, зиая его температурный ко-
Рис. 17.25. Схема записи дилатометрической кривой
эффициент расширения, можно проградуировать горизонтальную ось по температуре. Для получения точных результатов необходимо учитывать линейное расширение кварцевой трубки.
Коэффициент линейного расширения образца рассчитывают по формуле
^обр ~ ^ЭТ 4- (Kl/К2) (ССэт {dxf,
где cto6p — коэффициент линейного расширения образца; аэт — коэффициент линейного расширения пироса; акв — коэффициент линейного расширения кварца; Ki н Л2—коэффициенты оптического увеличения прибора по оси абсцисс и оси ординат соответственно; dxjdy — первая производная исследуемой кривой.
Емкостной дилатометр [15]. Емкостной дилатометр основан иа изменении емкости конденсатора, одна нз пластин которого связана с исследуемым образцом и смещается при его удлинении. Это приводит к изменению емко-
Ё айраацу
Рис. 17.25. Схема емкостного дилатометра
сти конденсатора, последнюю измеряют путем подстройки всего контура. Удлинение образца определяется по формуле M=d&C/lC, где d — расстояние между пластинами; I — длина образца; ДС/С — изменение емкости. Метод позволяет измерять деформацию 2^10~7 см.
Емкостной метод был применен для измерения удлинения А/ при низких температурах [15]. Схема прибора приведена на рис. 17.26.
Средняя из трех пластин конденсатора Сиа скреплена с исследуемым образцом. Эта пластина может перемещаться относительно двух неподвижных. Конденсатор образует два плеча емкостного моста, на который со звукового генератора 1 подается ток напряжением 50 В, частотой 10 кГц. Мост балансируется переменными емкостями Ci н С2 и сопротивлением R. При возникновении смещения появляется напряжение разбаланса, которое после усилителя 2 н фазовращателя 6 подается на ламповый милливольтметр 3 и синхронный детектор 4, с детектора сигнал идет иа самописец 5. Для контроля чувствительности моста в одно его плечо включают эталонную емкость Сзт-
Относнтельная ошибка составляет 5 %, чувствительность установки 10~8. Установка позволяет измерять ат н магнитострикцию в интервале .2—too к.
Интерференционный дилатометр [15]. В основу прибора положен интерферометр Линника (рнс. 17.27). В интерферометр входит мик-
Рис. 17.27. Схема интерференционного дилатометра
роскоп 10, между объективом которого 6 н окуляром О помещена призма 5. На уровне призмы расположен объектив 7 с зеркалом 9, напротив объектива 7 помещается источник монохроматического света. Образец 7, находящийся в кварцевой трубке, помещен в печь 2, которая прикреплена к основанию корпуса 4 прибора. Свободный конец образца имеет насадку 8, иа которой укреплено зеркало 3. Интерференция возникает вследствие оптической разности хода двух лучей, появляющихся при разложении света призмой 5. Часть лучей проходит через призму, отражается зеркалом 9 и, попадая опять в призму, отражается в направлении окуляра. Другую часть лучей призма отражает на зеркало 3 и от него в окуляр. Перемещение образца вызывает сме
19*
291
щение зеркала 3, и интерференционная картина изменяется. Чувствительность прибора 3-10~6 см. Изменение длины образца определяется по формуле AZ—пк/2, где п—число интерференционных полос; Л — длина волны света.
Дилатометр Стрелкова [1]. Дилатометр Стрелкова основан на преобразовании поступательного движения образца, вызванного термическим расширением, во вращательное движение, которое измеряется с помощью авто-коллимационных труб с окуляр-мнкрометра-ми. Дилатометр предназначен для измерения удлинения (сжатия) прн температурах от комнатной до 1000 °C. По чувствительности он близок к интерференционному, мало чувствителен к вибрациям. Схема дилатометра приведена на рнс. 17.28. Расширение образ-
Рис. 17.28. Схема дилатометра Стрелкова
ца 1 через серьгу 2 и толкатели 3 передается к измерителю малых перемещений. На толкатели одето ярмо 4, которое притягивается к магниту W и зажимает ролик 5 с зеркалом 6, благодаря чему при расширении илн сжатии ролик образца катится без скольжения. Зеркало, закрепленное на ролике, позволяет измерять расширение образца.
Для измерения коэффициента термического расширения прн высоких температурах (1000— 2500 °C) рекомендуется использовать оптические компараторы, фиксирующие перемещение верхнего конца испытуемого образца.
17.2.4. Применение методов
измерения плотности
и термического расширения при исследовании металлов и сплавов
Часто невозможно определить места расположения атомов компонентов в кристаллической решетке твердого раствора по изменению межплоскостных расстояний.
В этом случае для установления типа образующегося твердого раствора измеряют зави
симость плотности от концентрации второго компонента. Например, период решетки в системе Ni—Al (/, рнс. 17.29) вблизи эквнатом-ного состава [50 % (ат.) Ni] имеет максимальное значение. Он уменьшается при увеличении содержания Ni, так как атомный диаметр Ni
И1,%(О77.)
Рис. 17.29. Зависимость периода решетки / и плотности 2 сплавов Ni—-Al От состава
меньше, чем у А1. Уменьшение периода решетки прн уменьшении содержания Ni связано с образованием твердого раствора вычитания, что подтверждается резким падением плотности сплавов, содержащих <50 % Ni (2, см. рнс. 17.29).
17.3. ЭЛЕКТРИЧЕСКИЕ
СВОЙСТВА
17.3.1. Электрическая проводимость и электрическое сопротивление металлов и сплавов
При отсутствии градиентов температуры и процессов диффузии носителей тока связь между плотностью электрического тока / в проводнике н напряженностью электрического поля Е определяется законом Ома: / = оЕ, где коэффициент пропорциональности сг называется удельной электрической проводимостью. В изотропном металле ст — скалярная величина, в анизотропном — симметричный тензор второго ранга. Физический смысл о: количество электричества, переносимое за единицу времени через единицу площади сечения проводника в электрическом поле Е единичной напряженности.
Величина, обратная удельной электрической проводимости, называется удельным электрическим сопротивлением: р = о-1 = rS//, где г — электрическое сопротивление проводника; S и I — площадь сечения и длина проводника соответственно.
Размерность р —Ом-м, размерность о — См/м (сименс на метр). В табл. 17.8 приведены величины удельного сопротивления металлов.
292
ТАБЛИЦА 17.8
УДЕЛЬНАЯ ЭЛЕКТРИЧЕСКАЯ ПРОВОДИМОСТЬ <т, УДЕЛЬНОЕ СОПРОТИВЛЕНИЕ р, ТЕМПЕРАТУРНЫЙ КОЭФФИЦИЕНТ СОПРОТИВЛЕНИЯ а р [4]
Элемент p. 108 Om-m <7, 6 Cm/w ap , 10я к—1 Элемент p, 10s Om-m <r, 10“® См/м <xp , 10s K*-11
Li 9,39 10,7 4,50 Sb 39 2,56 5,1
Бе 6,6 15,2 6,67 Ca 29,8 4,81 6,00
Na 4,74 21,1 4,34 Ba 60 1,67 3,6
Mg 4,0 25,0 3,90 La 56,5 1,77 2,2
Al 2,62 38,2 4,26 Ce 75,3 1,33 0,9
Si 10s—106 10-1—10—4 —1,8—1,7 Pr 70,5 1,42 1,71
К 8,0 15,0 5,81 Nd 64 1,56 1,68
Ca 3,8 25,0 3,33 Pm — — —
Sc 64 1,57 4,25 Sm 105 0,995 1,7
Ti 58 1,73 3,3 Eu 81,3 1,23 4,8
V 22 4,55 3,58 Gd 143 0,70 1,76
Cr 15 6,67 2,5 Tb 116 0,86 —
Mn 258 0,388 1,7 Dy 90 1,11 1,19
Fe 10 10 6,6 Ho 90 1,11 1,71
Co 6,5 15,4 6,6 Er 85 1,18 2,01
Ni 7,5 13,3 6,7 Tm 90 1,11 1,95
Cu 1,7 58,8 4,33 Yb 27 3,7 1,30
Zn 6,1 16,4 4,17 Lu 68 1,47 2,4
Ga 26 3,84 3,9 Hf 41 2,44 4,4
Ge 6,5-107 1,54-10—6 1,4 Ta 15 6,67 3,47
Rb 12,5 8,65 4,7 W 5,6 4,82
Sr 17 5,9 5,2 Re 19,0 5,28 3,11
Y 65 1,54 3,6 Os 8,2 12,2 4,2
Zr 46 2,18 4,4 Ir 5,2 19,25 4,1
Nb 15 6,67 3,95 Pt 11 9,10 3,92
Mo 5,0 20,0 4,33 Au 2,3 43,5 ‘ 3,96
Tc 16,9 5,9 — Hg 95,7 1,04 0,92
Ru 7,5 13,3 4,58 TI 15,0 6,65 5,17
Rh 5,1 19,6 4,35 Pb 19,0 5,27 4,2
Pd 13,0 7,7 3,68 Bi 109 0,92 4,2
Ag 1,62 62,0 4,1 Fr — — —
Cd 7,57 13,3 4,25 Th 19 5,26 4,0
In 8,45 14,9 4,9 U 32 3,1 2,1
Sn 11,3 8,85 4,4 Pu 146 0,685 0,21
Перенос электрического заряда в металлах осуществляется в основном валентными электронами. Основываясь иа модели свободных электронов, где валентные электроны ие взаимодействуют ии между собой, ин с ноиами решетки, а представляют идеальный газ, подчиняющийся классической статистике, теория Друде—Лоренца дает аналитическое выражение закона Ома в виде / = (е2Ат£)/т.
Сравнивая это выражение с приводимым выше для /, получим о — e^Nxltn, где е и т — заряд н масса электрона; А; — число свободных электронов в единице объема; т—время релаксации (в данной теории время между столкновениями электрона с ионами решетки). Однако классические представления о природе электрической проводимости ие позволяют объяснить все электрические свойства металлов.
Современная зонная теория электрической проводимости учитывает характер поведения электронов в ионном остове металлической решетки с позиций квантовой механики [16— 22]. Основные положения квантовой теории электрической проводимости:
1) дискретный (квантовый) характер энергетического спектра электрона;
2) применение принципа Паули, согласно которому два электрона не могут иметь одинаковые наборы квантовых чисел;
3) использование для определения энергетического состояния ансамбля частиц, состоящего из валентных электронов, статистику Ферми—Дирака, в соответствии с которой электроны не могут иметь произвольное значение энергии, а в одном энергетическом состоянии могут находиться лишь два электрона с противоположными спинами. Прн этом функция распределения электронов по энергиям f{E) = = [ехр(£—£1.)Д7'-|1]^1, где Ер — максимальная энергия электронов при Г=0 К (энергия Ферми), k — постоянная Больцмана. График f (£) представлен на рнс. 17.30. Согласно функции распределения прн достаточно высокой плотности электронов даже при То электроны, находящиеся вблизи уровня Фермн, имеют высокую энергию (£р~10 эВ), а следовательно, большую скорость (10й м/с);
4) образование энергетической зоны, а не отдельных энергетических уровней электронов каждого атома в 'результате взаимодействия электронов с прочими атомами прн объединении их в кристалл;
5) учет волновых свойств электрона. Электрическая проводимость — не движение частнп
293
и их столкновения с нонами решетки, а распространение и рассеяние электронных воли;
6) дискретность значений энергии электронов прн движении электронной волны по периодической решетке кристалла. Причина дискретности — образование стоячих элек-
нов в металле, вводится понятие эффективной массы электрона т*, которая определяется соотношением (т *)-* — (4п/й2) (d2£/d&2), где k — волновой вектор; Е — энергия электрона; h — постоянная Планка.
В общем случае т* — тензор второго ранга,
Рис. 17.30. Распределение Ферми—-Дирака электронов по энергиям
тронных волн в результате отражения электронных волн от плоскостей кристаллической решетки в соответствии с законом Вульфа— Брэггов: пК — 2d sin 6, где d — межплоскостное расстояние; 6 — угол падения электронной волны; А—длина волны электрона.
Таким образом, в энергетическом спектре электронов возникают зоны допустимых н запрещенных значений энергии, связанные с периодичностью кристаллической решетки.
Зонная теория позволяет объяснить большие различия (на десятки порядков) значений электрической проводимости различных веществ (парафин 1017 Ом-м, железо 10-7 Ом-м). В металлах верхняя зона разрешенных энергий не занята полностью н имеются свободные энергетические уровни, на которые могут перейти электроны проводимости. У изоляторов валентные электроны полностью заполняют верхнюю разрешенную зону, а следующая незанятая разрешенная зона отделена широкой запрещенной зоной (7—10 эВ). В полупроводниках с собственной проводимостью запрещенная зона узкая (0,1—1,0 эВ) и под действием теплового возбуждения некоторые электроны могут приобрести энергию, достаточную для перехода в свободную зону.
Учитывая вышесказанное, следует пересмотреть физический смысл величин, входящих в выражение для а.
Ускоряющему действию приложенного поля подвергаются все электроны. Однако в случае распределения Ферми—Днрака основную роль в процессе рассеяния играют электроны с энергией, близкой к энергии Ферми. Это означает, что т определяется временем релаксации электронов, находящихся вблизи поверхности Ферми т(Б».
Из-за волнового поведения электронов и их взаимодействия с решеткой в общем случае ускорение может быть направлено не параллельно приложенному полю. У свободных электронов полученный от поля импульс полностью переходит в изменение собственного импульса, а электроны в потенциальном поле решетки обмениваются импульсами с ионами кристаллической решетки. Чтобы сохранить справедливость закона Ньютона для электро-
Эффективная масса электрона может быть больше и меньше массы свободного электрона и даже отрицательной. В случае узкой разрешенной зоны электрон не может значительно ускориться и формально его эффективная масса — массы свободного электрона (на-
пример, электроны в d-зоне переходного металла группы Fe) [10].
Когда электрон находится близко к верхней границе разрешенной зоны, т. е. когда начинает выполняться условие отражения, и электрон в конечном счете теряет больший импульс, чем приобретает, то он как бы обладает отрицательной массой.
Таким образом, выражение для о следует записать в виде а = (е2№/т*)ъ(Ер).
Зависимость удельного сопротивления металлов от температуры в широком интервале описывается формулой Блоха—Грюиайзена:
X
р (Т) = В/хб J x*dx/(ex — 1) (1 — e~x)t
где В — постоянная, различная для разных металлов; х — Gd/T; Qd — характеристическая температура Дебая.
Прн низких температурах (7<^6С) сопротивление увеличивается пропорционально Г3, прн высоких (Г>1,5 0в)—пропорционально Т.
Зависимость Блоха—Грюиайзена хорошо соблюдается для простых металлов. Для переходных металлов (особенно ферромагнитных) прн низких температурах показатель степени прн Т может уменьшиться до 2. Это связано с тем, что, кроме рассеяния электронов на кристаллической решетке, существенный вклад вносят другие механизмы рассеяния (электрон-электронное рассеяние, переход s-электронов на d-уровни, влияние обменного взаимодействия). При Т> 1,500л в силу линейности температурной зависимости электросопротивления температурный коэффициент сопротивления (ар) имеет постоянный порядок величины и равен ~4-1(Н3 град-1, причем у переходных металлов он несколько больше.
Прн определении зависимости р от темпера-
294
ТАБЛИЦА 17.9
ЗНАЧЕНИЯ ИЮ~8 Ом-м/% (ат)1 СЕРИИ СПЛАВОВ ПРИ 20 °C [1. 13]*
Рпеткоренный компонент Значения для растворителей
Си Ag Аи А1 Fe
Mg 0,6 _— 0,5 (2,8) 1,30 0,2) 0,6 —
Al 1,25 (4,5) 1,95 (3,0) 1,87 (0,6) — 5,5
Si 3,95 (4,9) — — — 0,7 6,2
Р 6,7 — — — — — —
Ti 8,6 — — — 12,9 — 3,9 3,0
V 5,8 -— -— — 12,6 " 5,3 1,2
Cr 5,5 •— — « — 4,30 (0) 6,3 2,1
Мп 2,90 (0) 1,60 (0) 2,41 (0) 7,3 1,4
Fe 9,66 (20) 4,3 8,5 (3,1) 6,7 —
Co 6,3 — — 5,8 (2,8) 0,4 0,8
Ni 1,22 (0,3) — .—- 0,80 (0,1) 0,25 1,9
Cu — ' 0,07 — 0,45 (1,6) 0,8 —
Zn 0,30 (2,7) 0,63 (3.0) 0,94 0 0,35 —
Ga 1,40 (4,2) 2,36 (2,5) 2,2 — Ы6 -—
Ge 3,76 (2,8) 5,5 (2,2) 5,1 (1,8) - 6,6
As 6,8 -— 8,5 -— — 0,4
Zr 11,0 (0) •— — —
Rh 4,3 (1,1) — — 4,2 —’— —
Pd 0,86 (0,6) 0,44 (0,7) 0,41 (0,5) — 1,7
Ag 0,12 0,32 (1,3) 1,2 —
Cd 0,20 — 0,38 (3,4) 0,63 (2,9) 0,6
In 1,05 (2,4) 1,78 (3,4) 1,35 (4,0) ' —
Sn 2,88 (3,2) 4,36 (2,6) 3,3 (1,1) 0,9 8,0
Sb 5,4 7,2 6,8 0,9 —*
Ir 5,7 — — — — — —• -—
Pt 2,15 (0,7) 1,7 (4,4) 1,0 (0,3) *
Au 0,54 (1,0) 0,36 (0,7) •— -— — —
Hg — — 0,79 — 0,4 —
TI —- 2,26 (0,7) 1,7 — — —
Pb — —w~ 5,1 (2,4) —
Bi — — 7,4 6,5 — — ——
c — — — * — — 4,9
N — — — — — — — 6,3
* В скобках дана величина Л (атомные доли 1).
туры необходимо учитывать изменение сопротивления, вызванного искажением размеров проводника из-за температурного расширения
+«г • (1 + а р Д Г), где ат — температурный коэффициент линейного расширения в интервале температур ДГ [13]. Для ферромагнетиков вблизи температуры Кюри наблюдается аномалия зависимости сопротивления от температуры, причем прирост удельного сопротивления Др пропорционален квадрату спонтанной намагниченности [23] Др/р~72.
По мере понижения температуры удельное сопротивление металлов н сплавов стремится к некоторому постоянному значению — остаточному удельному сопротивлению р0. Оно сильно зависит от концентрации дефектов решетки (повышается с увеличением концентрации). Остаточное удельное сопротивление практически не зависит от температуры, так что р = р(Г)+р0, где р(7)—зависящая от температуры составляющая удельного сопротивления бездефектного (чистого) металла (правило Маттиссена).
Сопротивление большинства металлов прн всестороннем (гидростатическом) сжатии уменьшается. Поскольку при гидростатическом
обжатии уменьшается объем тела, коэффициент давления для удельного сопротивления вычисляют по формуле +Х/3, где
X — коэффициент всестороииего сжатия; схгр ==drjdP)l2 — коэффициент давления полного сопротивления. Ниже приведены величины ар^ прн 20 °C и Р — 10й Н/м2:
Металл р Металл
Be —0,163 Ti
Mg —0,467 Fe
Al —0,406 Ni
Металл
—0,112 Zr —0,033
—0,226 Nb —0,i37
—0,182 Mo —0,129
При упругой деформации растяжением и измерении электросопротивления вдоль растягивающей силы коэффициент давления равен: % =г~1(дг/дс) — (1+2ц,)/Е, где о — рас-с
тягивающее напряжение; Е—модуль Юнга; р — коэффициент Пуассона.
Ниже приведены значения коэффициента растяжения ао для удельного сопротивле-
ння прн 20 °C;
295
ТАБЛИЦА 17.10
ЗНАЧЕНИЯ агу (10~4 град“г) ПРИ ТЕМПЕРАТУРАХ О-ЮО3С ДЛЯ ТВЕРДЫХ РАСТВОРОВ f о
НА ОСНОВЕ МЕДИ, СЕРЕБРА И ЗОЛОТА
Растворенный металл Металл -основа Растворенный металл Металл-основа Раство. ренинй металл Металл-©снова
Cu Ag Au Cu Ag Au Cu Ag Au
Be 3,6 Al 1,6 1,7 1.5 Sb 0,95 0,72 .им—
Mg —2,3 1.4 —0,5 Ga 1,6 1,5 2,4 Ti 1,8 — —-
Zn — « 2,4 1,9 In 2,3 1,5 2,6 Cr —32 — —2,5
Cd —— 1.6 1.7 Sn 1,55 1,1 1,5 Mn —265 —1,9 —0,1
Fe —1.7 — — Co 0,3 —— —2,6 Ni 1,2 4,1
Pd —0,3 —0,5 — Pt 0,8 0,7 9,6
Металл . . Cu Ag Au Fe Pd
... . —0,175 0,286 0,387 0,142 0,137
гаемых, характеризующих нзменение сопротивления растворителя и остаточного сопротивления:
dp}dP = ар р (Т) ар р0,
JT иг
Пластическая деформация увеличивает удельное сопротивление на 2—6 % из-за искажения пространственной решетки кристаллов при наклепе. Увеличение концентрации вакансий и дислоцированных атомов также приводит к увеличению удельного сопротивления. При комнатной температуре прирост удельного сопротивления Др за счет вакансий и дислоцированных атомов согласно [22] составляет на 1% (ат,) соответственно (1,0-:-1,5) • 10-8 и (0,5-е 1,0) -10~8 Ом-м.
При образовании твердых растворов удельное сопротивление растет. Это общее правило, не зависящее от удельного сопротивления растворителя и растворенного металла. Максимальное значение удельного сопротивления достигается обычно прн концентрациях 50 % (ат.). Для твердых растворов переходного металла в простом максимум сопротивления может соответствовать концентрации, отличной от 50 % (ат.). Для разбавленных твердых растворов простых металлов удельное остаточное сопротивление р0, связанное с легированием (по правилу Нордгейма) р0 = с(1—с)£, где с — атомная доля растворенного вещества; С — коэффициент, характеризующий рассеивающее действие примеси.
Прн больших концентрациях растворенного вещества приведенное соотношение необходимо дополнить эмпирической поправкой А: ро = — е(1—А с) £ (значения А и £ для твердых растворов на основе Cu, Ag, Au, А1 н Fe приведены в табл, 17,9 [13]).
Коэффициент £ пропорционален квадрату разностей валентности растворителя и примеси AZ (правило «Пйнде): £ = а+6 (AZ)2, где а н b — константы.
Температурный коэффициент сопротивления твердого раствора приблизительно определяется соотношением dp/dT == ctp р(7) +аррс, где р(Т’) и ар —удельное сопротивление и температурный коэффициент сопротивления растворителя; р0 и <zp. —остаточное удельное сопротивление и его температурный коэффициент (табл, 17.10),
Аналогично коэффициент давления для удельного сопротивления твердых растворов можно представить в виде суммы двух сла-
где р(Т), ар
— удельное
сопротивление и
коэффициент давления удельного сопротивле-
ния
растворителя; р0 и аро
Р
— остаточное
удельное сопротивление и его коэффициент давления.
Удельное сопротивление двухфазных структур зависит от количественного соотношения фаз и от морфологии структуры.
Если пренебречь рассеянием электронов на дефектах структуры н предположить, что размеры зерен значительно превышают длину волны электрона, то электрическую проводимость можно рассматривать как аддитивное свойство и для двухфазного сплава а — OiVi-l -Ьст2(1—Vj), где Ст1 н 02 — электрические проводимости фаз; И — объемная доля первой фазы
17.3.2. Методы измерения электрических свойств
Метод амперметра и вольтметра. Метод позволяет измерять величину сопротивления. Метод прост н позволяет проводить нзмереиня сопротивления в рабочем режиме при постоянном и переменном токе (рис. 17.31), Когда Rx> >Ra используется схема а, когда R*<g.Rv— схема б. В первом случае Rx = Vvllx = — Vv!(Ia—UvJRv) ~Uv!Ia, во втором Rx— = Vx!Ia = UvIIa—Ra = VvIIa, где I a, Uv, Ra, Rv — сила тока, напряжение н сопротивления, указанные иа схемах.
Мостовые методы [22], Принципиальные схемы измерений представлены на рис. 17.32, Подбором сопротивлений Ri н /?2 (см, рис. 17.32, а) одинарного моста устанавливается равновесие токов в диагонали моста, тогда Rx ~ RnRi/Rs- Однако точно определить малые значения Rx (менее 1 Ом) нельзя, так как фактически определяется не Rx, а суммарное сопротивление между точками А н D (см, рнс. 17.32,а). Оно складывается из сопротивления образца Rx, сопротивления соединительных проводов, переходных сопротивлений (во-первых, в точках крепления соединительных проводов—А и D; во-вторых, в точках крепления
293
образца К и L). Когда сопротивление мало, может оказаться, что величина дополнительных сопротивлений сравнима или даже больше Rx- Принципиальное отличие двойного моста (см. рис. 17.32, б) от одинарного со-
Рис. 17.31. Схема метода амперметра и вольтметра
Рис. 17.32. Одинарная (а) и двойная (б) мостовые схемы
।
стоит в том, что в результате введения дополнительной параллельной ветви с сопротивлениями и R2 гальванометр Г подсоединяется ие между измеряемым сопротивлением и эталонным (образцовым) сопротивлением R&, а между сопротивлениями /?[ и R2 (точка D). Двойной мост позволяет измерять сопротивления 10~6 Ом.
Электрическую схему двойного моста можно разбить на две цепи — рабочую (токовую) и измерительную. Рабочая цепь: измеряемое сопротивление Rx- образцовое сопротивление jRn; источник постоянного тока (аккумуляторная батарея нлн выпрямитель); регулировоч
ный реостат; амперметр. Сопротивления Rx и Ri< соединены медной шиной (Z?o).
Измерительная часть: сопротивления Ri, Rz, RJt ₽2, Rk и Я*’» гальванометр Г (сопротивления R\ и Rz и сопротивления Rr и R'2 в свою очередь образуют две параллельные ветви измерительной цепи). Измеряемое и эталонное сопротивления входят одновременно в измерительную и токовую цепи. Сопротивление по двойной мостовой схеме измеряют нулевым методом, уравновешивая схему подбором сопротивлений Rt, Rt, R2 и R2. Когда мост уравновешен, гальванометр фиксирует отсутствие тока. В этом случае, воспользовавшись законами Кирхгофа и исключив нз полученных уравнений отношения токов, находим Rxz
/?, *0 ^2
— Rn n — т ~ х
х Ъ ^ + ^ + ^2
Влияние второго члена уравнения мояфо свести к минимуму, если выполняется равенство #1/#2 НЛН R^—Rf, R2~^2'
С этой целью сопротивления и связывают между собой так, что они автоматически изменяются одинаково. Одинаковые по величине сопротивления Я2 н Я2 устанавливаются заранее н в процессе уравновешивания моста остаются неизменными.
Уменьшить влияние второго члена уравнения на точность измерения Rx можно, применив соединительную шину с минимальным сопротивлением. При соблюдении этих условий расчетная формула приобретает вид: Rx = =RnRi/Rz-
Условия, прн которых достигается высокая точность измерения:
1) R] и Rz^>Rx н RN; 2) Rx^Rni 3) ~/?2; 4) для компенсации т.э.д. с. измерения проводят прн различных направлениях тока в образце.
Потенциометрический (компенсационный) метод [24]. Метод основан на компенсации неизвестной э. д. с. Ех известным падением напряжения иа эталонном сопротивлении Rn. На рис. 17.33 представлена схема измерения электросопротивления Rx с использованием потенциометра. Процесс измерения включает следующие операции:
I) установку рабочего тока /р потенциометра компенсацией э. д. с. нормального элемента (и.э.) разницей потенциалов от внутреннего источника потенциометра (сопротивлением R?et). В случае нулевого показания гальванометра Ip—Eka!Rn и в дальнейшем не изменяется;
2) установку тока, проходящего через образец, и эталонное сопротивление R9 (реостатом JR2);
3) измерение, которое осуществляется регулировкой сопротивления потенциометра R'. Rx=R"R9IR't где R" и R' значения сопротивления потенциометра (R) прн измерении Ux и 1/обр.
Измерение электрических, свойств в перемен
297
ных электрических полях [25]. Для измерения активных, реактивных н комплексных сопротивлений применяют мостовые схемы переменного тока, В последнее время переменные электрические поля широко используются для бесконтактных измерений электросопротивления (например, жидких металлов).
Рис. 17.34. Схемы мостов переменного тока
В случае, когда все плечи моста содержат активные и реактивные сопротивления, т. е. содержат комплексные сопротивления Zi, Zz, Z-л, Zt, условия равновесия схемы: ZiZ4=Z2Z3; <pi+<p4—фа+<Рз1 отсюда видно, что равновесие достигается при регулировке не менее двух параметров. В большинстве мостовых схем два плеча обычно имеют активное сопротивление, а два других — комплексное (одно из которых образцовое). На рис. 17.34 представлены мостовые схемы для измерения емкости (а) и индуктивности (б).
Для схемы рнс. 17.34, а <p2=<p4=0 и регулировкой Ro можно добиться фа.=ф0. Неизвестные параметры:
Rx — ВД/R&t Сх — CfiRzl
Для схемы б нз условий равновесия получаем:
Rx = RiRz/Ro н ~ C0RiRz.
Равновесие достигается регулировкой сопротивлений Ro и Ri, Ro и Т?2.
Для высоких частот (десятки мегагерц) применяют специальные мостовые схемы (Г-об-разные мосты). Преимущество таких схем — возможность заземления входной и выходной цепей, что облегчает экранировку элементов схемы. На рнс. 17.34, в представлен Т-образный мост для измерения индуктивного сопротивления. В условиях равновесия, когда сила тока, проходящего через нуль-прибор (НП), равна нулю:
Rx = (/?оыС%)-1; Lx = 2J^C0.
17.3.3. Термоэлектрические, гальваномагнитные
и термомагнитные свойства
Эмиссия электронов [2, 17]. Для того чтобы электрон покинул металл, необходимо затратить работу. Потенциал электрона вне металла принято считать равным нулю. Внутри металла потенциал электрона постоянен и глубина потенциальной ямы составляет IFo« 10 эВ. Прн Г=0К потенциал электрона в металле можно принять равным энергии Ферми (£₽). Тогда работа, необходимая для освобождения электрона нз металла (эффективная работа выхода IF): IF=IFC'—Ер.
В табл. 17.11 приведены значения W для ряда металлов.
Энергию, необходимую для отрыва электронов, можно сообщить нм различными способами: воздействием света (фотоэффект), нагревом (термоэмиссия) или электрическим полем (холодная или автоэлектронная эмиссия).
Термоэлектронная эмиссия. Плотность тока на поверхности металла в этом случае определяется уравнением Ричардсона: /= =AT2e~wfkT, где А — постоянная, не зависящая от температуры и природы вещества; W — работа выхода; Т — температура.
Холодная эмиссия. Если к проводнику приложить сильное однородное электрическое поле (108 В/м), направленное вдоль проводника, то существует значительная вероятность того, что электрон пройдет сквозь потенциальный барьер (туннельный эффект). Прн этом плотность тока выражается соотношением: /=* —BE2e~^k7\ где В и ₽ — константы, связанные с эффективной работой выхода электрона; Е — напряженность приложенного электрического поля.
Контактные явления. Если привести в соприкосновение два металла, то между ними возникнет ток эмиссии, который будет существовать до >тех пор, пока ие наступит равновесие электрохимических потенциалов. Оно характеризуется тем, что границы Ферми обоих металлов будут лежать на одном энергетическом уровне. Равновесие устанавливается путем перехода электронов нз металла с более высоким уровнем Ферми в другой металл, который заряжается при этом отрицательно. Возникшая разность потенциалов — контактное напряжение Вольта V&— равно:. Ув=>
298
ТАБЛИЦА 17.11
РАБОТА ВЫХОДА ЭЛЕКТРОНА W, КОЭФФИЦИЕНТ АБСОЛЮТНОЙ Т.Э.Д.С. 8 А ПРИ 293 К.
КОНСТАНТА ХОЛЛА 7?Н [41
Элемент wz, эВ ea‘ мкВ/грйД A -ic,c il м«/Кл Эле- мент w. эВ £a' мкВ/град мя/Кл Элемент w, эВ E/i’ мкВ/град ti м’/цл
Li 2,38 + 14,37 —2,0 I 3,3 +2,2 —1,25 Tb 3,15 —4,3
Be 3,92 — +7,7 Zr 3,9 — +0,212 Dy 3,25 — —2,7
Na 2,35 —4,4 —2,3 Nb 3,99 — +0,72 Ho 3,22 —— —
Mg 3,64 -0,4 —0,9 Mo 4,3 +5,9 +1,91 Er 3,25 —0,34
Al 4,25 —1,6 —0,38 Tc 4,4 — — Tm 3,10 ‘— —1,8
Si 4,8 — —10+^ Ru 4,6 — +22 Yb 2,6 +3,77
К 2,22 — 15,6 —4,2 Rb 4,75 +1 ,o +0,48 Lu 3,14 — —0,535
Ca 2,60 —8,2 —1,78 Pd 4,8 —9,54 —6,8 Hf 3,53 — +0,43
Sc 3,3 —3,6 —0,67 Ag 4,3 +1,4 —0,9 Ta 4,12 —5,0 +0,96
Ti 3,95 — —2,4 Cd 4,1 +2,8 +0,67 W 4,54 +1,5 +0,856
V 4,12 -— +0,76 In 3,8 4-2,4 —0,24 Re 5,0 — +3,15
Cr 4,58 +3,63 Sn 4,38 +0,1 —0,048 Os 4,7 II Отриц.
Mn 3,83 — +0,844 Sb 4,08 +210 Ir 4,7 +1,2 +0,318
Fe 4,31 —51,3 18,0 Cs 1,81 +0,2 +0,24 Pt 5,32 —4,4 —0,194
Co 4,41 —18,5 +3,6 Ba 2,49 ~— ..— Au 4,30 +1,1 —0,69
Ni 4,50 —18,0 —0,6 La 3,3 —0,8 Hg 4,62 —3,4 —0,73
Cu 4,40 +1,7 —0,52 Ce 2,7 +4,39 +1,92 TI 3,7 +0,4 +0,24
Zn 4,24 +2,9 +0,9 Pr 2,7 -— +0,709 Pb 4,0 —4,4 +0,09
Ga 3,96 — —0,63 Nd 3,2 *— +0,97 Bi 4,4 —70 —6104
Ge 4,76 +302,5 + 10» Pm 3,07 — 1 < Fr 1,8
Rb 2,16 —8,26 —5,9 Sm 2,7 — —0,2 Tb 3,3 1— —0,88
Sr 2,35 • — Eu 2,5 — — и 33 +5 +0,34
Cd 3,1 — —0,95 Pu — — +0,69
=6"I(lV'i--R7s), где Wi и 1Г2—эффективные работы выхода соответствующих металлов.
Напряжение Вольта можно измерить в отличие от напряжения Гальвани, Уг, пропорционального разности энергии Ферми металлов: Vr=e~i(EF~Ep2).
Термоэлектрические явления [1, 10]. Электрические н тепловые процессы тесно связаны между собой, изменение энергетического спектра электронов ведет за собой изменение фотонного спектра и наоборот.
Явление Зеебека (термоэлектричество). Если между контактами проводников А и В, составляющими контур, имеется разность температур, то в цепи возникает термоэлектродвнжу* щая сила (т. э. д. с.), которая определяется соотношением 6ав(Го) — [с?ГЛв(Г|Го)/с)7’] То, где Vab — разность потенциалов, возникающая в контуре.
Т. э. д. с. считается положительной, если с ростом температуры потенциал конца, связанного с контактом при температуре Tt тоже растет; еав— дифференциальная т.э.д, с. двух металлов.
Чистые металлы можно расположить в ряд, в котором любой последующий металл более отрицателен, чем предыдущий: Si, Sb, Fe, Alo, Cd, W, Au, Ag, Cu, Zn, Rb, Ir, TI, Cs, Ta, Pb, Mg, Al, Hg, Pt, Na, Pd, K, Ni, Co, Bi.
Зависимость Vab можно записать: Vab= —atA-bt^ct3, при этом Vab выражается в микровольтах; i — температура в градусах Цельсия. Величина Vab может быть подсчитана простым суммированием (правило аддитивности), если контур состоит из нескольких металлов. Абсолютное значение т.э.д.с. одного металла можно измерить, если в качестве второй ветвн термоэлемента использовать сверхпроводник. Величина абсолютной т.э.д.с,
еабс связана с параметрами электронной структуры следующим соотношением [1 ]:
бабе = (— We) (<? in p/^E)£==Sf ,
где е — заряд электрона; ЕР— энергия Ферми; k — постоянная Больцмана; р — удельное сопротивление.
Явление Пельтье. Обратимое выделение тепла на контакте двух проводников при прохождения тока. При изменении направления тока тепло будет поглощаться. Явление связано с тем, что в металле изотермический электрический ток сопровождается тепловым потоком Qn =Плв<7=ПА£ГТ, где Пав — коэффициент Пельтье; Qn—количество выделившейся теплоты; <? — количество прошедшего через контакт электричества; i — электрический ток; т — время. Явление Пельтье подчиняется правилу аддитивности.
Явление Томсона относится к отдельному проводнику, между двумя точками которого поддерживается постоянная разность температур АГ. При пропускании электрического тока между этими точками выделяется нли поглощается теплота: Q?~Lq&T=LixlS.Tt где L — коэффициент Томсона; От—выделяющаяся теплота; q — количество электричества; i— электрический ток; т — время*.
Термоэлектрические явления связаны соотношениями Кельвниа:
Tde/
LA = =
т
о
которые позволяют определить по одному ко-
299
эффициенту два остальных. Для расчета абсолютной т.э. д. с. в твердых растворах простых металлов молено применить правило Гортера — Нордгейма [1]: всш^вприм+роХ Х(®о'—®приы)/рспл, где бприы~ т. э. д. с. растворенного вещества; е0 и ро — т. э. д. с. и удельное сопротивление растворителя; есил н ре пл — т.э.д.с. и удельное сопротивление сплава.
Гальваномагнитные и термомагнитные эффекты [10. 26]. Под гальваномагнитными эффектами понимается совокупность эффектов, возникающих в проводниках под действием магнитного поля Н, когда в них протекает электрический ток плотностью /. Под термо-магнитными эффектами понимается совокупность эффектов, возникающих в электронных проводниках под действием магнитного поля //, когда_в них имеется тепловой поток плотностью Q. *
Гальвано- и термомагннтные эффекты могут быть трех типов в зависимости от взаимной ориентации векторов: Я, /, Q.
Наиболее широко используется из гальваномагнитных эффектов эффект Холла и магнетосопротивление.
Эффект Холла заключается в возникновении поперечной э. д. с. в проводнике с силой тока I при помещении его в магнитное поле Н. Возникающая э. д. с. Холла Ен равна Ея=#нШ1<1, где Rh — постоянная Холла; d— толщина образца.
Для образца шириной b можно записать: En~RuHjb, где / — плотность тока.
Поскольку в стационарном состоянии магнитная и электрическая силы, отклоняющие электрон, равны, т. е. Hev—E^elb, где е — заряд электрона; v — средняя скорость электрона. Принимая j~Nev, а Ен=НЬо, получаем Ен=N~ 1е~1, где АГ —число электронов в единице объема. Отсюда видно, что гальваномагнитные свойства позволяют определять плотность носителей заряда в металле.
Магнетосопротивление — изменение удельного сопротивления металла р в продольном илн поперечном магнитном поле. Согласно правилу Колера Ap/p=f(27/p), где f— функция, зависящая от физических характеристик и формы образца. Для неферромагннтных металлов магиетосопротивленне положительно в продольном и поперечном полях, причем в слабых полях Др/р~Ё2, в сильных Др/р~Е. Для ферромагнитных металлов поведение Др/р более сложное [23].
Эффект Иернста — Эттингсхаузена является поперечным термомагиитным эффектом. Заключается в возникновении э. д. с. в проводнике (теле), имеющем перепад температуры в направлении, перпендикулярном направлению магнитного поля.
Константа Нернста—Эттингсхаузеиа -связана с параметрами электронной структуры следующим выражением:
_ jt£sT2 / dr \ 3m*c \ dE )ef’
где k — постоянная Больцмана; т* — эффективная масса электрона; с—скорость света; Ер — уровень Ферми; т— время релаксации электрона; Е — энергия электрона.
При исследовании металлов и сплавов со смешанной проводимостью (электронно-дырочной) для определения концентрации эффективных носителей зарядов необходимо исследование одновременно не менее трех гальвано- и термомагннтных эффектов.
17.3.4. Методы измерения термоэлектрических, гальваномагнитных и термомагнитных свойств
Методы измерения т.э.д.с. [1]. Схема установки для измерения т. э. д. с. представлена на рнс. 17.35, Исследуемый образец 1 нагревается
Рис. 17.35. Схема установки для измерения т. э. д. с.
в печи О и охлаждается массивным медным кольцом, соединенным с термостатом S. К электрическим изолированным медным кольцам Ki и Ks подведены термопары. Между концами термопар измеряется т. з. д. с., соответствующая разнице температур ДТ. Точность измерений около 0,2 % при сравнительно больших т. э. д. с.
Для измерения малых э. д. с. можно использовать установку, схема которой представлена на рис. 17.36. Отличие этой схемы состоит
Рис. 17.36. Схема установки для измерения малых т. э. д. с,
в том, что элементы цепи Gb G9 сделаны из того же материала, что и образец р. Образец и элементы цепи Gi, G2, изолированные слюдяной фольгой, запрессованы в медные блоки 7\ н Т2. Все холодные концы помещены в термостат Го. ЕТ—Ti—Т2 измеряется дифференциальной термопарой А Г; разность потенциалов — прибором ДЕ. Точность измерения
при чувствительности прибора 10-7 В равна ±1 %-
’ Измерение э.д.с. Холла [15]. Методы измерения гальваномагнитных н термомагнитных свойств в целом сводятся к измерению или разности потенциалов нлн разности температур. Разность потенциалов измеряют потенциометрическим методом (см. раздел 17.3.2), а
Рис. 17.37. Схема установки для измерения эффекта Холла (Г — гальванометр, НЗ — нормальный элемент, Б — источник питания, К—ключ, А — амперметр)
температуру — термопарами. На рис. 17.37 приведена схема установки для исследования эффекта Холла. Образец / в форме пластины помещен между полюсами N п S магнита. Электрический ток к образцу проводится через контакты 2—2. Разность потенциалов между точками 3 и 5 (э.д.с. Холла) измеряется потенциометром ППТИ-1. Два контакта (4. 5) с одной стороны образца позволяют перед включением магнитного поля с помощью реостата 6 сравнять разность потенциалов между контактами 3 н 5. Для измерения малых э.д.с. Холла вместо потенциометра использу-
Рис. 17.38. Схема установки для измерения эффекта Нернста—Эттингсгаузена
ется фотоэлектронный усилитель. Эффект Холла для материалов с высоким сопротивлением измеряется, в переменных полях.
Измерение эффекта Нернста — Эттингсхау-зена [15]. Э. д. с. Нериста — Эттннгсхаузена измеряется компенсационным методом (рис. 17.38). Э. д. с. в точках 1 и 2 измеряется прн двух направлениях магнитного поля, создаваемого соленоидом 4 прн переключении ~ключа 3. Для измерения эффекта иа ферромагнитном материале одновременно измеряется намагниченность, для чего в центральной части образца х помещается измерительная обмотка 7. Измерение намагниченности проводят баллистическим методом. Температурный гра
диент создается с помощью двух электрических нагревателей 5, 6, расположенных вдоль длинных сторон образца. Установка позволяет исследовать термомагнитные эффекты в интервале 80—1000 СС.
17.3.5= Сверхпроводимость [16, 27]
Сверхпроводимость—явление отнюдь ве редкое: более 20 элементов переходят в сверхпроводящее состояние, а список сплавов включает тысячи наименований.
Исходным пунктом теории, объясняющей явление сверхпроводимости, служит наличие эффективного притяжения между электронами вблизи поверхности Ферми, следствием которого является возникновение пар электронов. Образование пар может происходить прн температурах ниже некоторой критической прн этом сопротивление металла становится нулевым.
Переход нз нормального состояния в сверхпроводящее является обратимым и происходит как фазовое превращение 2-го рода, сопровождающееся скачком теплоемкости. Ниже критической температуры сверхпроводящее состояние может быть разрушено наложением магнитного поля не меньше некоторого критического значения величина которого зависит от температуры.
В табл. 17.12 приведены значения критической температуры и критического поля 27к (при 7=0) для элементов, в которых наблюдалась сверхпроводимость [26].
Различают сверхпроводники I н П рода. На рнс. 17.39 показана зависимость намагниченности таких сверхпроводников ниже критической температуры Тк от величины приложенного внешнего поля. Прн T<zTu внешнее маг-
Рис. 17.39. Зависимость намагниченности сверхпроводников 7 и II рода от напряженности внешнего поля
нитное поле ие проникает в толщу массивного сверхпроводника (за исключением тонкого поверхностного слоя), т. е. вещество в сверхпроводящем состоянии является идеальным диамагнетиком Именно поэтому диамагнетизм вещества ниже Тк свидетельствует о переходе в сверхпроводящее состояние. Как видно нз рис. 17.39, в сверхпроводнике I рода сверхпроводящее состояние существует в полях, меньших Нк, н полностью исчезает прн достижении полем критической величины Нк. В сверхпроводниках II рода сверхпроводящее состояние существует в полях ниже HKi и полностью исчезает в полях, превышающих . При напряженности внешнего поля от 27Kf до /7Кя в сверхпроводнике II рода существует так называемое смешанное состояние, когда в
301
ТАБЛИЦА 17.12
ЗНАЧЕНИЯ Я„ и ДЛЯ СВЕРХПРОВОДЯЩИХ ЭЛЕМЕНТОВ
Элемент тк, К Як, кА/м* Элемент ГК’ K | HK, кА/м* Элемент Тк, к Нк. кА/м*
Be 0,026 Mo 0,916 7,165 W 0,0154 0,091
Al 1,175 8,352 Тс 7,78 112,284 Re 1,697 14,968
Ti 0,39 7,962 Ru 0,493 5,254 Os 0,655 5,175
V 5,31 85,582 In 3,405 22,413 Ir 0,14 1,512
Zn 0,87 4,379 Cd 0,56 2,356 hg(a) 4,15 32,723
Са 1,08 4,721 Sn 3,72 24,284 Hg(P) 3,949 26,991
Cs(p) 6,2 Sb 2,6 —- TI 2,39 14,013
Zr 0,53 3,742 La 4,88 64,332 Rb 7,23 63,934
Nb 9,25 156,854 Ta 1 4,47 66,164 Po Th 1,4 1,374 10,430
* Значения Як приведены для 7'=0К.
материале имеется смесь сверхпроводящих и нормальных областей.
Критическую температуру определяют, измеряя изменение удельного сопротивления или удельной теплоемкости в зависимости от температуры. В этом случае критическую температуру определяют из соотношения:
(£с Сц) Tr—
’ \2
-------- I к.
, дТ ) '
где Сс н Сн — теплоемкость в сверхпроводящем н нормальном состоянии; Тк — критическая температура; Нк — критическое поле; V — объем образца [28].
17.3.6< Применение методов измерения электрических свойств при исследовании металлов и сплавов
Исследование диаграмм фазового состояния [13]. В первом приближении зависимость электросопротивления от состава в области твердых растворов описывается параболическим законом, а в двухфазной области — линейным. Исследуя изменение сопротивления в зависимости от состава сплавов прн различных температурах, можно установить положение границ однофазных и двухфазных областей на диаграмме состояния. Для иллюстрации сказанного иа рис. 17.40 приведена наряду с диаграммой состояния Au—Cd зависимость электрической проводимости и температурного коэффициента сопротивления от состава сплавов при различных температурах. В области существования граничных растворов а и б электропроводность изменяется по кривым, близким к параболе. Промежуточные фазы «I, аг, р и у максимально упорядочены При стехиометрических составах Au3Cd, AuCd и Au3Cd соответственно, поэтому имеют высокую электрическую проводимость и высокий температурный коэффициент сопротивления. По мере повышения температуры проводимость этих сплавов приближается к значениям проводимости сплавов иестехиометрического состава из-за уменьшения степени дальнего порядка. В двухфазных областях а+р, р+у и
у+б зависимость проводимости от состава почти линейна, и ее значения лежат в интервале проводимостей фаз, ограничивающих гетерогенную область на диаграмме.
Исследование процессов упорядочения твердых растворов [1]. Классический пример — система Си—Аи (рис. 17.41).
А. А. Смирнов теоретически рассчитал остаточное сопротивление упорядочивающихся сплавов в зависимости от состава и степени дальнего порядка. Автор исходил нз предположения, что упорядоченный сплав прн абсолютном нуле, как и чистый металл, не имеет электрического сопротивления и что оно появляется только при нарушении порядка в расположении атомов. Учитывая связь между средним временем свободного пробега и вероятностью рассеяния электронов прн разупо-рядоченнн, автор пришел к следующему выражению Для остаточного удельного сопротивления: р'=п[с(1—с)—v(I—v)T]2J, где с —относительная атомная концентрация компонента А в сплаве; v — относительная концентрация узлов решетки, предназначенных для атомов этого компонента; а — коэффициент, зависящий от природы компонентов; ?] — степень дальнего порядка: 1}~(р—с)/(1—т), где р— число мест, занятых своими атомами.
Однако не для всех твердых растворов падение р столь четко выражено. На рис. 17.42 представлены зависимости удельного сопротивления р (б) н магнетосопротнвлення Ар/р (а) для сплавов системы Fe—Сг [29]. Образование сверхструктур в системе Fe-~Cr соответствует слабовыраженным точкам перегиба на кривой зависимости р от концентрации Сг. Гальваномагнитный эффект (Ар/р) оказывается более чувствительным свойством и изменяется прн этих концентрациях скачком, являясь, таким образом, свойством, дающим большую разрешающую способность при исследовании процессов упорядочения в данной системе.
Исследование концентрационных неоднородностей в твердых растворах В зависимости от знака и величины энергии смешения атомов в твердом растворе возможно образование различных концентрационных неоднородностей: областей ближнего порядка, дальнего порядка, расслоения. Поскольку при об-
302
Рис. 17.40. Диаграмма Au—Cd и зависимости температурного коэффициента а и электропроводности О от состава сплавов
разованин подобных сегрегаций изменяется структура сплава, основным методом исследования процессов, происходящих в твердых растворах, является анализ зависимостей электрических и гальваномагнитных свойств от температуры и концентрации.
Согласно эмпирическому правилу Кестера, если направление изменения константы Холла при легировании и термической обработке (отпуск после деформации или отжиг) имеет -один и тот же знак, то происходит ближиее упорядочение. Если знаки различны — ближнее расслоение. Сопротивление и константа
Холла уменьшаются, как правило, прн установлении в твердом растворе дальнего порядка. Однако в некоторых твердых растворах встречается аномальное поведение сопротивления прн термической обработке после деформации: удельное сопротивление имело минимальное значение после деформации и сильно возрастало после термической обработки. Это явление названо К-состояннем.
Позднее было показано, что К-состоянне определяется существоваинем микроскопических областей дальнего порядка, а повышение электросопротивления связано с рассеянием
303
электронных волн на границах этих областей. На рнс. 17.43 приведены зависимости константы Холла от температуры отпуска для сплава CugZnNi; в котором прн 150—200 °C возникает ближннй порядок, при 250—350 °C — дальний, проходя через К-состоянне [30].
Рис. 17.41. Зависимость удельного электросопротивления от состава в системе Си—Аи после закалки а а после медленного охлаждения (б)
Л/?# 'W *
о ______I____I______I___I____
0 20 W 60 во 100
Fe % (am.)
Рис. 17.42. Зависимость Др/р и р от состава в системе Fe—Cr
Кривые /, 2, 5 соответствуют различным исходным состояниям перед отпуском (деформированное, закаленное, отожженное).
Исследование процессов старения. Если сплав подвергается деформационному старению, связанному с образованием атмосфер примесных атомов вокруг дислокаций, то удельное сопротивление увеличивается с ростом концентрационной неоднородности в сплаве.
В случае, когда старение связано с распадом пересыщенного твердого раствора, следует иметь в виду) возможность двух механизмов старения: первый, когда распад твердого раствора происходит в результате образования зародышей метастабильиой нлн стабиль
ной избыточной фазы, и второй, когда распад протекает через стадию образования зои Г ияье—Престона.
Исследование старения можно проводить как путем измерения сопротивления при ие-
Рис. 17.43. Зависимость RK от температуры для сплава Сиг2пЬГ1 после:
1 — деформации; 2 — закалки; 3 — отжига
прерывном нагреве образца, закаленного из однофазной области, так и путем измерения сопротивления непосредственно при температуре старения. Наконец, часто измеряют электросопротивление прн 20 СС образцов, подвергнутых старению при разных температурах и затем быстро охлажденных [22].
На рнс. 17.44 представлена схема, описывающая часть диаграммы состояния, включаю-
Рис. 17.44. Изменение электросопротивления в процессе старения
щей исследуемый сплав (? (а), н ход кривой зависимости электросопротивления образцов, закаленных с различных температур после выдержки т прн каждой температуре (б). Уменьшение сопротивления соответствует выделению стабильной избыточной фазы р. Кривые (рнс. 17.44,6), обозначенные т,, т2, Тз, т4, соответствуют изменению электросопротивления прн различных временах выдержки перед закалкой, причем т:1>Т2>тз>Т4.
Если распад пересыщенного твердого раствора происходит через стадию образования зон Гннье—Престона, то зависимость электросопротивления от температуры будет более сложной [13]. Обычно при образовании зон Г.—П. электросопротивление снижается (стадия 7, рис. 17.45), а далее при повышении
304
температуры вновь возрастает вследствие растворения зон (стадия 2, рнс. 17,45), а затем наблюдается новое снижение за счет образования избыточной фазы (стадия 3, рнс. 17,45)
Рис. 17.43. Изменение электросопротивления при нагреве закаленного сплава Al—38Ag
Рис. 17.46. Зависимость электросопротивления от содержания углерода
Рис. 17.47. Зависимость р от содержания углерода в стали при тонкопластинчатом (Г), грубопластинчатом (?) и зернистом (3) цементите
и, наконец, снова возрастание уже в результате растворения этой фазы (стадия 4, рис. 17.45),
Исследование процессов, происходящих при
закалке и отпуске сталей. На рнс. 17.46 представлены зависимости р от содержания углерода в мартенсите (7), в закаленной (2) н отожженной стали (3) [22]. В процессе отпуска происходит распад мартенсита с образованием частиц карбидной фазы, прн этом наблюдается падение р. Другая причина падения электрического сопротивления — распад остаточного аустенита.
Прн анализе изменения электрического сопротивления в процессе отпуска необходимо учитывать, что р — структурно-чувствительное свойство и зависят не только от фазового состава, но и от морфологии частиц карбидных фаз, о чем свидетельствуют, в частности, данные о зависимости р от морфологии перлита (рис. 17.47).
17.4. МАГНИТНЫЕ СВОЙСТВА
Характеристикой интенсивности взаимодействия тела с данным магнитным полем является магнитный момент М [Д-м2], данного тела, являющийся_ суммой элементарных магнитных моментов:
Магнитный момент единицы объема 7, А/м, называется намагниченностью где
V — объем тела.
Связь между намагниченностью и магнитным полем устанавливается соотношением 7= —КыН, где Кы — магнитная восприимчивость (в системе СИ Км=4этх).
Магнитный поток Ф, Вб, является суммой потоков, создаваемых внешним полем Н и намагниченности 7: Ф—Хр,0(7+Н), где р0—магнитная постоянная, равная 4п-I0-7 Г/м.
Магнитный поток, проходящий через единицу площади, называется индукцией В, Т.
В=Ф/5 = ц0(/4-Я).
Связь между индукцией н полем выражается как В=р.аСсД, где |хабс— абсолютная проницаемость рабе—ЦоР, Г/м; р — магнитная проницаемость среды.
Магнитная проницаемость связана с магнитной восприимчивостью соотношением: Рабс = Ро(1 +Ам).
17.4.1. Классификация магнетиков
Имеется три типа магнитных эффектов: диамагнетизм, парамагнетизм и кооперативный магнетизм, нлн магннтоупорядоченное состояние. Последний включает ферромагнетизм, антиферромагнетизм и ферримагнетизм [23, 31].
Диамагнетизм. Диамагнетизм металлов н сплавов определяется в основном магнитной восприимчивостью решетки.
Количественное рассмотрение приводит к следующему выражению для диамагнитной восприимчивости на единицу объема:
-диа = _ МЧ*-
* 6m
где п — число атомов в единице объема; Z — атомный номер элемента; е и т — соответственно заряд и масса электрона; г — усредненное значение квадрата радиуса электронного облака; р.о — магнитная постоянная.
Диамагнитная восприимчивость определяет
20-286
305
ся пространственным распределением зарядов и не зависит от температуры. Вычисленные значения совпадают с опытными и имеют величину порядка 10~8. Диамагнитный эффект универсален и должен проявляться у каждого атома-, однако его может перекрывать парамагнитный эффект.
Электронный газ в металле обладает диамагнитной восприимчивостью, которая имеет квантово-механическую природу и составляет одну треть от величины парамагнитной восприимчивости электронного газа: ХдИа ~
Парамагнетизм. Связан с наличием у атома собственного магнитного момента, не зависящего от того, приложено внешнее магнитное поле или нет.
В отсутствие магнитного поля магнитный момент тела равен нулю вследствие беспорядочного распределения атомных магнитных моментов. Действие магнитного поля сводится к ориентированию магнитных моментов в одном направлении и преодолению дезориентирующего действия теплового движения. Отсюда следует, что парамагнитная восприимчивость зависит от температуры.
При малых полях и (нли) высоких температурах, т. е. когда glpsCkT, в квантовой н классической механике получаем: /— =n^p?Bl2l?>kTt где п —число атомов на единицу объема; g— фактор Ланде (характеризует отношение магнитного момента к механическому) ; / — квантовое число атома; k — константа Больцмана; цв — магнетон Бора (единица атомных магнитных моментов): рв= = p,0(eft/4nm), где р0 — магнитная постоянная; h — постоянная Планка; е и m — заряд и масса электрона.
Тогда восприимчивость х=н§‘2{-^72/зй7’= =с7'~1. Последнее выражение — температурная зависимость парамагнитной восприимчивости (закон Кюри).
Учет магнитного взаимодействия между атомами приводит к закону Кюри—Вейса: х= =с[(Т—G), где 6 — постоянная для данного вещества величина.
Свободный электрон в магнитном поле может быть ориентирован параллельно н антипараллельно полю. Это дополнительно расширяет энергетические уровни электронов проводимости в металле. В результате такого расщепления электроны проводимости обладают парамагнетизмом (парамагнетизм Паули).
Парамагнитная восприимчивость коллективизированных электронов определяется соотношением:
/”ра = (3npB)/2(l(,£f. [1 +п2 (М7Ев)2/12] ,
где ЕГ — энергия Ферми; п— число электронов проводимости в единице объема.
Поскольку для металлов kTjEF очень малая величина, следует ожидать, что парамагнетизм Паули почти не зависит от температуры.
Магнитоупорядоченное состояние (ферромагнетизм, антиферромагнетизм, ферримагнетизм) [23, 26]. Важная особенность ферромагнитных тел — существование в них областей, намагниченных до насыщения, т. е. доменов, даже в отсутствие магнитного поля. В размагниченном состоянии, когда суммар
ный момент тела отсутствует, векторная сумма магнитных моментов всех доменов равна нулю, хотя каждый домен намагничен до насыщения.
Ферромагнитное состояние — явление кван-товомехаиическое, не имеющее классического аналога.
Наличие спонтанной намагниченности связано с магнитным моментом спнна электрона на незаполненной внутренней оболочке атома. Согласно квантовомеханической теории ферромагнетизма параллельное расположение спинов обеспечивается квантовомеханнческими обменными силами, которые, несмотря на их электростатическое происхождение, не могут быть описаны с классических позиций.
Возникновение обменных эффектов связано с принципом неопределенности и принципом Паули.
Обменной силе соответствует энергия взаимодействия между двумя атомами I и j со спинами Si н Sj, т. е. обменная энергия Eff- — =—2ASiSj, где А — обменный интеграл, зависящий от взаимного перекрытия электронных оболочек, характеризующий различие в величине кулоновского взаимодействия прн параллельном (ферромагнитное упорядочение) и аитнпараллельном (антиферромагнитное) расположение спинов.
Поскольку обменные силы действуют на малом расстоянии, то, учитывая только первую координатную сферу, можно записать: zA~ —2й6с, где z — координационное число; k — константа Больцмана; 6с — температура Кюри.
Таким образом, по температуре Кюрн можно оцеинть величину обменного интеграла.
Критерии ферромагнитного упорядочения:
1. Наличие недостроенных d- или f-оболочек атома с большим азимутальным числом.
2, Радиус этих оболочек должен быть мал по сравнению с расстояниями между ядрами в решетке.
Следствие этих условий — положительная величина обменного интеграла, прн которой для ферромагнетиков энергетически более выгодна параллельная ориентация магнитных моментов спинов в недостроенных оболочках. Если величина обменного интеграла отрицательна и энергетически более выгодно аитнпа-раллельное выстраивание магнитных моментов спииов, то в металле реализуется антиферромагнитное упорядочение.
Антипараллельные моменты ие всегда взаимно компенсируются. В этом случае остается нескомпенсироваиный магнитный момент, обусловливающий слабый ферромагнетизм, отличающийся, однако, от истинного ферромагнетизма и называемый ферримагнетизмом.
Формальная теория ферромагнетизма дает для температурной зависимости намагниченности выражение: /s//c=^((A//o)/(7’/6c)), где — намагниченность насыщения при температуре Т\ Jo — намагниченность насыщения при Т—0; 6с—температура Кюри.
Однако для низких температур лучше согласуется с экспериментальными данными температурная зависимость намагниченности, полученная из теории спиновых воли Блоха: Js— —/0(1—аТ3^2), где а —числовая константа.
Прн высоких температурах вблизи темпера
туры Кюри экспериментально подтверждается зависимость: /в=/0 (1—77ес)1/2. Для температур выше температуры Кюрн можно вывести закон Кюри — Вейса для парамагнетиков у,=с[(Т—6с), где с — числовая константа.
17.4.2. Методы измерения напряженности магнитного поля
В зависимости от целей исследования, интервала магнитных полей и температур в качестве источников магнитного поля служат соленоиды постоянного тока для создания полей напряженностью до 105 А/м, прн наличии системы охлаждения до 106 А/м, электромагниты для полей до 4-10е А/м, постоянные магниты для полей до 2- 10е А/м. Все большее применение получают установки, в которых магнитное поле напряженностью до 107 А/м создается сверхпроводящим соленоидом.
Импульсные магнитные поля напряженностью до 8-107 А/м получают в результате кратковременного мощного токового разряда.
Баллистический метод определения напряженности магнитного поля, В измеряемое маг-
= (—юс?Ф) /5т, где w — число витков катушки; 5Ф/5т— изменение магнитного поля во времени через площадь витков катушки.
Так как дФ---дВ5 (S — площадь сечения катушки; дБ — изменение магнитной индукции в зазоре); E~iR {R — общее сопротивление баллистической цепи, i — сила тока в цепи), то из выражения для Е следует: i(h=dQ= =—(dBwS)/R, где Q — количество электричества, которое протекает через гальванометр.
Угол отклонения подвижной рамкн гальванометра а пропорционален Q, т. е. Q=Cq«, где Cq — постоянная гальванометра. Интегрируя выражение для dQ, получаем: С=С$7? = —bBSwIv.. Величина С измеряется в веберах на одно деление.
Поскольку сопротивление R в процессе измерения не меняется, то C=CqR обозначают как постоянную установки. Чтобы определить С, используется эталонная катушка взаимной индуктивности М, первичная обмотка которой включена последовательно в цепь гальванометра БГ: C=2p.0Mi/a.
Напряженность магннтнбго поля И (ампер на 1 м) будет определяться выражением: И— =Ca/(p,0Sw),
Рис. 17.48. Схема установки для определения вапряженвости магнитного поля н магнитной индукции баллистическим методом:
П) и П2 — переключатели в намагничивающей цепи, П3 и П4 — переключатели в измерительной цепи, — нагрузочное сопротивление цепи гальванометра
нитное поле помещается измерительная катушка таким образом, чтобы вектор напряженности магнитного поля был направлен перпендикулярно к плоскости витков катушки. Размеры катушки обусловлены объемом пространства, в котором определяют величину напряженности магнитного поля. Катушка соединена с баллистическим гальванометром.
Если катушку быстро удалить нз поля, то в ее витках возникает э. д. с. (Е) Е~
На рнс. 17.48 представлена принципиальная схема баллистической установки.
Измерение напряженности магнитного поля с использованием эффекта Холла [32]. В качестве материалов для датчиков Холла используются полупроводники, обладающие большой подвижностью носителей тока (Те, Bi, Ge, HgTe, InAs, InSb).
Принцип измерения напряженности магнитного поля состоит в измерении э. д. с. Холла,
307
пропорциональной напряженности пронизывающего датчик (£>) магнитного поля при постоянной силе тока, проходящего через датчик от батареи Б\. На рис. 17.49 приведена принципиальная схема установки измерения напряженности поля.
Рис. 17.49. Принципиальная схема установки для измерения напряженности поля с помощью эффекта Холла
Ряс. 17.50. Блок-схема измерителя напряженности магнитного поля Е-11-3:
1 — генератор; 2 — датчик Холла; 3 — переключатель диапазонов; 4 — делитель напряжения; 5 — усилитель;
6 — индикатор
С помощью нормального элемента НЭ, батареи Б н гальванометра Г реостатом Ri устанавливается рабочий ток потенциометра. Реостатом /?2 производится установка нуля в отсутствие магнитного поля. Для повышения точности измерений в приборе установлена схема термостатирования датчика Холла. Температура поддерживается с точностью ±0,25 °C. Приведенную схему можно применять н для измерения переменного поля промышленной нлн звуковой частоты. Для этого на датчик D подается ток такой же частоты, как и исследуемое поле. Во избежание сдвига фаз между током н полем в цепь включают реактивное сопротивление.
На рнс. 17.50 представлена схема измерителя напряженности магнитного поля Е-11-3. Прибор имеет два датчика, один из которых используется для измерения поля (=С1,ЗХ ХЮ6 А/м) в зазоре электромагнитов, второй—для измерения поля (<: 2,4-105 А/м) в соленоидах.
Измерение напряженности магнитного поля методом ядерного магнитного резонанса ('ЯМР). Если парамагнитное вещество поместить в постоянное магнитное поле, обеспечивающее парамагнитную поляризацию ядер, а в направлении, перпендикулярном направле
308
нию поля, включить высокочастотное переменное магнитное поле, частота которого близка к частоте ларморовской прецессии магнитного момента ядра, то магнитные моменты ядер будут прецессировать вокруг направления постоянного поля. При совпадении частот переменного поля со н ларморовской прецессии наступит резонансное поглощение энергии высокочастотного поля. Частота переменного магнитного поля в условиях резонанса связана с величиной постоянного магнитного поля И соотношением: (о—уН, где у — гиромагнитное отношение (у протона 336,166 А-1 •с~1-м).
Точность в определении И обусловлена точностью измерения резонансной частоты и достигает 10_® %.
На рнс. 17.51 приведена принципиальная схема промышленной установки Е-11-2 для нзме-
Рис. 17.51. Блок-схема измерителя напряженности магнитного поля Е-11-2
рения напряженности магнитного поля методом ЯМР [15].
Образец О (вода или глицерин) помещают в катушку £|, которая образует колебательный контур генератора 1. При резонансе в этом контуре будет дополнительное затухание. Для определения поглощения постоянное поле электромагнита (ЭМ) Н моделируется переменным полем частотой 25—50 Гц и амплитудой несколько ампер на метр, которое создается катушками £2 и генератором 5. Прн совпадении среднего значения поля И с резонансным значением модулирующее поле дважды за период проходит резонансное поглощение, осуществляя амплитудную модуляцию колебаний высокочастотного контура. После детектирования 2 и усиления 3 сигнал подается на осциллограф 4, где регистрируется как функция постоянного поля. ‘
Метод дает возможность изменять поле от 103 до 2-106 А/м. При неоднородности ^0,02 % иа сантиметр погрешность ^0,01 %.
Электродинамический метод [32]. В основу метода положен эффект взаимодействия магнитного поля и рамки с током. Если катушку с w витками и площадью S подвесить в однородном магнитном поле, то при пропускании тока I катушка повернется на некоторый угол 6. В момент равновесия К(0—sin <р, где К •— константа упругости нити; — угол между нормалью к плоскости катушки и направлением поля; (6—<р) — угол закручивания нити.
На этом принципе основан промышленный прибор ИМИ-1 для измерения напряженности
магнитных полей от 8- 10е до 1,3-106 А/м (погрешностью 4 %). Принципиальная схема прибора показана на рис. 17.52. Рамка 1 с катушкой Rt помещается в измеряемое магнитное поле. Измеритель 2 состоит из системы регулировочных резисторов Rs, R/„ Rs, R6, источ-
Рис. 17.52. Блок-схема измерителя напряженности магнитного поля ИМИ-1
. Жесткие потенциалометры, выполненные обычно в виде полукольца, используются для определения напряженности магнитного поля иа участках магнитной цепи, где напряженность считается постоянной.
На рнс. 17.53 представлена схема жесткого потенциалометра. Постоянная жесткого потен-цналометра равна К1=2ю£/л, величина пото-
Рис. 17.53. Схема жесткого потенциалометра
ника питания Б, ключа К н измерительного прибора mA с сопротивлением Rz. Рамка укреплена на осях и с помощью гибкого провода связана с измерителем. При измерении через рамку пропускается ток L Вращательный момент рамкн М=СВ1, где С — постоянная прибора. Противодействующий момент равен /ИПР==1Га, где а — угол отклонения рамки; №—удельный момент закручивания. При равновесии B—Wa/Ci. При измерениях стрелка, связанная с рамкой, устанавливается (регулировкой силы тока) на определенное ‘деление шкалы. Прибор может быть проградуирован в единицах поля.
Магнитные потенциалометры [32]. Потен-цналометр — плоская пластина, на которой намотана катушка с равномерно распределенными витками. Обмотку делают таким образом, чтобы начало и конец ее выводились в середине пластины и подключались к гальванометру. В зависимости от назначения потен-циалометры изготовляют гибкими и жесткими. Для измерения магнитных потенциалов (магнитного напряжения) между двумя точками пространства с различной напряженностью магнитного поля используют гибкие потенциа-лометры. Если концы гибкого потенцналомет-ра расположены в точках с разными магнитными потенциалами, то магнитный поток, пронизывающий витки 'потенциалометра: Ф — ~powSUn, где S — площадь потенциалометра; w — число витков на 1 см; Un — разность магнитных потенциалов между двумя точками.
Если удалить потенциалометр из магнитного поля, то возникнет поворот подвижной системы баллистического гальванометра, пропорциональный изменению потока. Отсюда Un= = (Съа)1К, где а—показания гальванометра; /С=ю5ц0— постоянная потенциалометра; Сь— баллистическая постоянная гальванометра. Для определения К используют катушку с известным числом витков, в которую вставлен потенциалометр. При этом концы потенцнало-метра соединяются между собой. Включение нли выключение тока в градуировочной катушке вызывает изменение потокосцепления в потенциалометре.
Рнс. 17.54, Схема феррозонда
косцеплення прн изменении Н: ЛФ=Лр0Д/А
Феррозонды [32]. Для измерения небольших по величине постоянных и переменных полей применяют феррозонды — стержни нз магнитно-мягкого материала, имеющие по две обмотки, одна пара нз которых Wj и создает переменный магнитный поток (поля возбуждения) Ф~ другая w2 н w2 является измерительной (рис. 17.54). Если по обмотке пропускать переменный синусоидальный ток, то магнитное поле сердечника будет изменяться по динамической симметричной петле и в обмотке появится э. д. с., содержащая, кроме основной частоты, высшне (нечетные) гармоники. Прн перемещении зонда в постоянное магнитное поле форма динамической петли изменится и она перейдет в несимметричный цикл. При неизменных величине и форме переменного тока в намагничивающей обмотке зонда изменяются величина н форма э. д. с. н наряду с нечетными гармониками возникнут четные. Амплитуда второй гармоники пропорциональна напряженности магнитного поля. Для облегчения выделения второй гармоники феррозонды делают в виде двух одинаковых сердечников, вторичные обмотки которых соедн-
309
няют так, чтобы при нулевом постоянном поле напряжение на концах вторичной обмотан было равно нулю.
17.4.3. Измерение пара-и диамагнитной восприимчивости [28]
Существуют две группы методов измерения восприимчивости слабомагиитиых веществ. Методы, основанные на определений изменения коэффициента взаимо- нлн самоиндукции катушки, когда в нее вводится исследуемый
разца 30—40 мг, позволяет проводить измерения до температур 1200 °C, для чего в прибор вмонтированы термопара Т и печь П.
Маятниковые весы [15]. Принципиальная схема маятниковых весов приведена на рис. 17.56. Образец 2, закрепленный на пластинке 4 в сосуде Дьюара 5, при включении поля втягивается нли выталкивается из полюсов 1 магнита в направлении, перпендикулярном плоскости чертежа. На пластину, которая при этом изгибается, наклеены тензометрические датчики А и В. Изгиб пластины вызывает изменение сопротивления тензодатчиков, включенных в мостовую схему. Возникает ее рас-
Рис. 17.55. Схема весов Фарадея—С екемнта (а) и профиль магнитных полюсов Д' и 5 и распределение градиента поля dHjdX (б);
ЭМ — магнитное поле
образец, и методы, при которых измеряется сила f, действующая на образец, помещенный в неоднородное магнитное поле: f— где m — масса образца; у — восприимчивость; dHjdz — градиент магнитного поля Н.
Метод Фарадея — Сексмита. Силу, действующую на образец, измеряют при помощи упругого кольца К, изготовленного нз фосфористой или бериллиевой бронзы. Схема установки приведена иа рис. 17.55, а. Смещение образца О при включении неоднородного поля приводит к упругой деформации кольца, в результате чего зеркала Mt и М? поворачиваются относительно первоначального положения. Луч света, проходящий оба зеркала, изменяет направление и на экране световой зайчик смещается. Величина смещения зайчика пропорциональна силе, деформировавшей кольцо. Пружины St и S2 демпфируют колебания держателя образца.
Неоднородное поле создается с помощью профильных конических наконечников электромагнита N и S. Профиль полюсов и распределение градиента поля приведены на рнс. 17.55,6.
Установка градуируется по веществу с известной восприимчивостью. Определение упругой постоянной кольца может проводиться грузами известной массы, которые располагаются на специальном столнке кольца Р. Описанный метод обладает чувствительностью по удельной восприимчивости 10~7 при массе об-
Рис. 17.56. Схема маятниковых весов
компенсация. Регулируя силу* тока в компенсационной катушке 3, возвращают образец в неходкое положение (отсутствие тока в диаго-
310
иали моста). Метод позволяет исследовать магнитную восприимчивость прн низких температурах.
В маятниковых весах для регистрации отклонения используют фотоусилители, оптические регистраторы. При нулевом методе возвращение образца в исходное положение может быть осуществлено механической или электрической системой. Чувствительность весов этого тнпа 10-7 при массе образца 30— 50 мг.
Крутильные весы [15]. Принципиальная схема крутильных весов приведена на рнс. 17.57.
Рис. 17.57. Схема крутильных весов
Пондемоторная сила, действующая на образец со стороны неоднородного магнитного поля ЭМ, создает момент кручения нити подвеса S, к которому прикреплено горизонтальное плечо D с исследуемым образцом О. Другое плечо С имеет демпфирующую систему F. Отклонение контролируется положением зеркала 3. На вертикальной штаиге В укреплена катушка К, которая находится в зазоре магнита М. Регулировкой силы тока в катушке К восстанавливается исходное положение образца. Пондемоторная сила определяется по величине силы тока в катушке К.
Частотные методы измерения восприимчивости [15]. Находят в последнее время все большее применение. Основаны на изменении добротности колебательного контура прн внесении в контур исследуемого образца.
Величина удельной магнитной восприимчивости определяется по формуле х== —Npe%e/(/V»p), где Хэ — магнитная восприимчивость эталонного образца; Ng — разность показаний частотометра при наличии в катушке эталонного образца и без него; ДО— то же, для исследуемого образца; рэ и р — плотность эталона н исследуемого образца.
На рис. 17.58 приведена блок-схема установки для измерения восприимчивости частотным методом.
В контур образцового генератора 1 (частота 3,184 МГц) вводят исследуемый образец. Опорный генератор 2 генерирует частоту 2,264 МГц, на выходе умножителей 3, 4 частоты 101 и 88 МГц соответственно. Смеситель -5 — три индуктивности с детектором нз кристаллического кварца. Усилитель низкой частоты 6 имеет полосу пропускания 5—5000 Гц. Измерение осуществляется частотометром 7 н осциллографом 8.
С помощью установки можно измерить удельную восприимчивость порядка 1СН6 с точностью до 1 %. Существенные недостатки этих методов — необходимость поддержания высо-
Рис. 17.58. Блок-схема измерения частотным методом
восприимчивости
кой стабильности частоты (до 10~8); в связи с этим возникают сложности в проведении температурных измерений.
17.4.4. Ферромагнитные свойства и методы их измерения
Кривая технического намагничивания
Исходное состояние ферромагнетика перед измерениями — размагниченное, достигается прн наложении знакопеременного магнитного поля с убывающей амплитудой нлн при нагреве образца выше температуры Кюри и охлаждении в отсутствие магнитного поля. В размагниченном состоянии ферромагнетик состоят нз отдельных доменов, в которых атомные магнитные моменты выстроены параллельно н лежат в одном нз направлений легкого намагничивания, а величина намагниченности в них равна или близка к намагниченности насыщения. Однако суммарный магнитный момент образца равен нулю, так как векторы намагниченности доменов в общем случае распределены в пространстве равновероятно и их геометрическая сумма равна нулю.
Величина, форма доменов н поведение ферромагнетика в магнитном поле будут определяться соотношениями различных видов энергии (обменной, кристаллической анизотропии и т. д.) при данной температуре н данном магнитном поле. На рис. 17.59 (кривая 1) приведена схема кривой намагничивания, на которой можно в общем случае выделить пять областей: I—область обратимого смещения доменных стенок; II— область Релея (частично необратимого смещения доменных стенок); III — область необратимого смещения; IV — область вращения векторов намагниченности; V — область парапроцесса.
Кривая 2 (см. рис. 17.59) построена в координатах магнитная индукция — поле (В—II). Отношение рполн=В/(р0/7) называется полной проницаемостью. Угол наклона касательной к кривой, проведенной из начала координат, равен максимальной проницаемости.
Начальная проницаемость ин= Нт [В/бхоХ ДЯ->0
X#)]-
Дифференциальная проницаемость в точке А Urn [AB/(|iDA/7)]. Прн уменьшении маг-Д/7->0
нитного поля в произвольной точке К и последующем увеличении индукция В опишет петлю. Величина = lirn [ДВ1/(р,0АЯ1)]
называется обратимей проницаемостью.
311
При уменьшении до нуля максимального (намагничивающего) ноля намагниченность (индукция) образца не принимает нулевого значения. Только при наложении магнитного поля, отрицательного по направлению, возможно достижение нулевого значения намагниченности (индукции). Отставание намагнн-
Рис. 17.59. Кривая намагничивания
Рис. 17.60. Петля гистерезиса для ферромагнетика
чениостн (нндукцнн) от поля носнт название магнитного гистерезиса. На рис. 17.60 дана схема петли гистерезиса ферромагнетика. Значение намагниченности (индукции) прн /7=0 называется остаточной намагниченностью Л (нндукцней Вг). Напряженность магнитного поля, прн которой намагниченность равна нулю, называется коэрцитивной силой по намагниченности зНс. Напряженность магнитного поля, прн которой индукция равна нулю, называется коэрцитивной силой по индукции ВВС.
Кривые /, 2, получающиеся прн снятии отрицательного поля, носят название кривых возврата.
Если в процессе измерений илн эксплуатации магнитная система (магиитопровод — образец) не замкнута н нормальная составляющая вектора 1е к поверхности образца ие равна кулю, то на поверхности возникают магнитные заряды, которые создают вокруг образца
магнитное поле /7^. Величина размагничивающего поля /7~ пропорциональна намагниченности образца н зависит от его формы где / — намагниченность образца;
М—размагничивающий фактор образца, зависящий от формй образца. Энергия ферромагнетика в размагчнвающем поле равна 7ра8Ы=0,5Ж
Размагничивающий фактор определяют экспериментально, сравнивая кривые намагничивания, измеренные в замкнутой и разомкнутой цепи. Расчет размагничивающего фактора возможен только для однородно намагниченных тел, какими являются эллипсоиды вращения.
При известном N существует графический способ перестройки кривой намагничивания,
Рис. 17.61. Графический метод перестройки кривых намагничивания, снятых в разомкнутой магнитной лепи
снятой в разомкнутом магнитном потоке, в кривую для замкнутого магнитного потока (рис, 17.61). Угол а определяется из соотношения tg а =/7~//==4л77. Тогда для любой прямой, параллельной осн поля, отрезок АВ равен Ш. Откладывая от точки С отрезок DC — АВ, получаем точку D, координата поля которой соответствует внутреннему полю в образце. Проведя такое построение для всех значений намагничениостн, получим кривую намагничивания данного материала во внутреннем магнитном поле.
Образцы неэллипсоидальной формы в однородном поле намагничиваются неравномерно. В этом случае размагничивающий фактор — переменная величина. В зависимости от метода измерения различают:
1. Магнитометрический фактор NM измеряется и используется прн магнитометрическом методе, когда значение намагниченности илн индукции усредняется по всему объему образца.
2. Баллистический фактор Ns — когда намагниченность илн индукция определяются по нх значению в средней части образца (нейтральном сечении). Значение N& всегда меньше #м-
Методы измерения магнитных свойств
Баллистический метод. Схема баллистической установки была приведена на рис. 17.46. Величина магнитного поля, которое создается катушкой W\, измеряется катушкой Wu. Индукция измеряется катушкой W%, намотанной на нейтральное сечение образца. При нзменеиин намагничивающего поля на A/7t магнитный поток внутри образца изменяется на ДФ=(Д£+Л
312
r-^/S.H)S. Это изменение регистрируется баллистическим гальванометром БГ. Метод определения баллистической постоянной описан выше, значение индукции находят по формуле B=C6a/(p0Sw), где Сб — баллистическая постоянная; tx — отброс гальванометра; 5 — площадь нейтрального сечення образца; w — число внтков катушки индукции, намотанной на образец.
Прн построении кривой намагничивания этим методом, как правило, индукцию измеряют при коммутации поля нз положительного I в отрицательное направление И (переключателем /7(), в результате чего изменение индукции равно удвоенному значению максимальной индукции данного частотного цикла. Увеличивая амплитуду намагничивающего поля реостатами Ri и Rz по точкам, соответствующим вершинам частных циклов, строят кривую намагничивания.
Магнитометрический метод [15]. Метод основан иа взаимодействии намагниченного исследуемого образца и подвижной магнитной стрелки. Намагниченность образца определяют
Рис. 17.62. Схема астатического магнетометра
по углу отклонения магнитной стрелки. Формула для расчета намагниченности: l=HR3a./ /(41V) —Са, где Н — горизонтальная составляющая напряженности земного поля; R — расстояние между центрами образца и стрелки; I — расстояние от шкалы до зеркала; V — объем образца; а,— угол отклонения светового зайчика; С — константа.
Основная часть магнетометра — магнитная стрелка — подвижный магнит, имеющий форму цилиндра. Магнитную стрелку подвешивают иа тонкой нити, к которой крепится зеркало; с его помощью отсчитывают угол поворота. Намагничивающую катушку располагают горизонтально так, чтобы ее ось проходила через центр стрелки. Для компенсации магнитного поля, действующего от намагничивающей катушки на стрелку с противоположной стороны, устанавливают точно такую же компенсирующую катушку с противоположным иапраиленнем ПОЛЯ.
Более широко применяется астатический магнетометр, подвижная часть которого состоит из двух стрелок с равными магнитными моментами (рис. 17.62).
Подвижная часть имеет демпфирующее устройство D. Катушка является намагничивающей и в ией расположен образец О. Катушка Кг служит для компенсации действия
катушки К| иа подвижную систему. Катушка Кз позволяет вести измерения нулевым методом и компенсирует воздействие образца на подвижную систему. Чувствительность установки зависит от длины нити подвеса н магнитного момента стрелок. Установка градуируется по току в катушке К3- Можно использовать эталонный образец с размерами, равными размерам исследуемого образца (Д — амперметр, R — реостат).
Вибрационный магнитометр [15j
На рис. 17.63 показана схема вибрационного магнитометра, разработанного на физическом
Рис. 17.63. Схема вибрационного магнетометра
факультете МГУ. Образец 1 и постоянный магнит 2 укреплены иа стержне 3, который колеблется с определенной частотой, например 79 Гц, перпендикулярно магнитному полю электромагнита 4. В измерительных катушках 5 и 6 возбуждаются э. д. с., пропорциональные магнитным моментам образца и постоянного магнита. Этн сигналы поступают в усилители 7, 8, фазовращающее устройство 9 н сумматор 10, где эти сигналы сравниваются; разность, пропорциональная изменению намагниченности образца, через катодный повторитель И поступает в индикатор 12. Вибрационная система состоит нз динамика 15, который питается от генератора звуковой частоты 13 и усилителя 14.
Система позволяет проводить измерения в широком интервале магнитных полей и температур. Точность измерения до I %. Имеются модели магнетометров, в которых колеблется не образец, а измерительные катушки.
Метод измерительного генератора [33]. Принцип измерения основан на возникновении э. д. с. прн вращении рамки нз проводника в магнитном поле. В зависимости от плоскости вращения рамки генератор может либо давать сигнал, пропорциональный напряженности однородного магнитного поля, т. е. используется как измеритель поля, нли при вращении рамки в однородном поле ие давать сигнала; в последнем случае сигнал возникнет только прн внесении в однородное поле образца, обладающего собственным магнитным моментом. В этом случае генератор работает как измеритель намагниченности.
На рис. 17.64 представлена схема генератора, измеряющего магнитный момент. Если катушка К вращается в однородном магнитном поле Н, то э. д. с. в ней индуцироваться не будет, так как плоскость внтков катушки параллельна силовым линиям однородного поля соленоида S. При внесении в поле образца р, обладающего собственным магнитным
313
моментом, силовые линии от образца пересекают витки катушки, а генератор начинает индуцировать сигнал, который с контактов т я f поступит иа прибор G. Величина сигнала пропорциональна магнитному моменту образца. Чувствительность установки регулируется скоростью вращения генератора Т. Сила тока
Рис. 17.64. Схема измерительного генератора
руется термопарой 8 и милливольтметром 9.
Те же физические процессы лежат в .основе метода «выдергивания» образца из магнитного поля. Для расширения интервала намагничивающих полей и уменьшения влияния неоднородности магнитного поля вместо одной катушки К используют две катушки, намотанные одна иа другую н подключенные навстречу друг другу. Число витков в катушке подбирают таким образом, чтобы произведение площади витков катушки на число витков nS было одинаковым. Помещение такой катушки в поле не вызывает отклонения гальванометра, а выдергивание нз нее образца (или вталкивание) приводит к появлению сигнала гальванометра, поскольку число силовых линий, пересекающих при этом верхнюю н ииж-июю катушки, будет разным. В этом случае сигнал гальванометра пропорционален магнитному моменту образца.
Рис. 17.65. Схема измерения намагниченности
методом вталкивания
в соленоиде / от источника Б регулируется реостатом /? н контролируется амперметром А.
Метод вталкивания образца в магнитное поле [1]. Этот метод широко применяется прн исследовании порошковых образцов. В основу метода положена установка Штеблейна, схема которой приведена на рнс. 17.65. В канал электромагнита 1 с обмоткой 2 вталкивается образец 7 таким образом, чтобы середина образца совпадала со средней точкой межполюсного расстояния, что определяется длиной толкателя 5 и регулируется упором 6 и втулкой 10. Перед вталкиванием образца в электромагните устанавливается необходимое поле н цепь гальванометра Гв замыкается. В момент вталкивания образец намагничивается, а силовые магнитные линии образца пересекают витки измерительной катушки 3 и в ней индуцируется э. д. с., пропорциональная магнитному моменту образца, которая отклоняет стрелку гальванометра Гв. Для измерения температурных зависимостей образец помещают в печь 4. Температура образца коитролн-
17.4.5. Магнитная анизотропия
и методы ее измерения
В результате спин-орбнтальиого изаимодействия в ферромагнетике возникает анизотропия в расположении атомных магнитных моментов относительно осей кристалла. В соответствии с этим в одних направлениях намагничивание проходит прн минимальных затратах энергии (направление легкого намагничивания), в других намагничивание затруднено (направления трудного намагничивания). Этот вид магнитной анизотропии называется магнитной кристаллографической анизотропией. Энергия кристаллографической анизотропии для кубических кристаллов описывается выражением:
где Ко, Ki, К2 — константы магнитной анизо-
314
тропик «1, а2, — косинусы углов между на-
правлением вектора намагниченности н кристаллографическими направлениями.
В случае мелкнх удлиненных частиц, когда возникновение доменной структуры в них невозможно из-за их малого размера, энергетически выгодное положение вектора намагниченности в частице определяется анизотропией ее формы. Величина константы анизотропии формы описывается выражением: /СфОрмы= — —где и — размагничивающие факторы вдоль и поперек длинной оси частицы; Js — намагниченность насыщения.
Если направление легкого намагничивания в кристалле обусловлено магинтоупругой энергией, связанной с явлением магнитострикции, то в этом случае константа анизотропии определяется величиной внутренних и внешних напряжений и константой магнитострикции X: Ам.у=роХ, где р — коэффициент, близкий к единице; о — напряжение; X — магнитострикция.
Магнитострикция — это изменение объема и формы ферромагнетика прн переходе его в ферромагнитное состояние и прн его намагничивании.
Изменение объема и формы областей спонтанной намагниченности приводит к возникновению внутренних упругих напряжений в многодоменном кристалле. При наложении внешнего магнитного поля происходит изменение ориентации векторов намагниченности, следовательно, в процессе намагничивания также изменяется упругая энергия.
Энергию магнитострикционной деформации для намагниченного до насыщения кубического кристалла можно записать: FK.y=(E/2)X X [Хюо4-3(Хц|—-Хюо) где
Е — модуль упругости; Хюо, — магнитострикции в направлениях [100] и [111]; tzb сс2, а.3 — косинусы углов между векторами намагниченности и осями кристалла
В композиционном материале, в котором возможно обменное взаимодействие между ферромагнетиком и антиферромагнетиком, при охлаждении с температуры выше температуры Нееля до низких температур возникает однонаправленная обменная анизотропия. В этом случае взаимодействие антиферромагнетика с ферромагнетиком приводит к асимметрии петли гистерезиса, т. е. к облегчению перемагничивания в одном направлении и существенному затруднению в противоположном.
В твердых растворах на базе ферромагнитных элементов после термической обработки в магнитном поле может происходить перераспределение атомов, связанное с нх магнитным взаимодействием. Это явление получило название направленного упорядочения [34].
Образование пар или комплексов атомов, выстроенных в одном преимущественном направлении, создает в материале направление легкого намагничивания и, следовательно, наведенную магнитную анизотропию.
Наконец, в тонких ферромагнитных пленках анизотропию магнитных свойств создает поверхность, роль которой для малых объемов становится существенной.
Реализация в материале того или иного вида анизотропии определяет поведение ферромагнетика в магнитном поле. Чем больше ве
личина магнитной анизотропии, тем большую энергию необходимо затратить на процесс намагничивания и перемагничивания материала, тем большее значение имеет коэрцитивная сила. В соответствии с этим магнитные материалы деляется на высококоэрцитивные (магнитно-жесткие), обладающие большим значением Нс и служащие источниками магнитного поля (постоянные магниты), магнитно-мягкие, обладающие способностью перемагничиваться в малых магнитных полях, н полужесткие материалы, обладающие средним значением коэрцитивной силы, используемые чаще всего как материалы для магнитной записи.
Процесс перемагничивания, обусловленный как магинтнымн константами, так и структурой материала, может определяться тремя основными механизмами: 1) зарождением и дальнейшим смещением доменной стенки; 2) вращением векторов намагниченности во всем объеме материала или отдельном домене; 3) отрывом уже существующей доменной стенки в материале от места ее закрепления.
В реальных материалах возможно совмещение нлн смена механизмов перемагничивания.
Метод определения констант анизотропии по кривым намагничивания. Величина свободной энергии вдоль основных кристаллографических направлений осей монокристалла <Ю0>, <110>, <111>, намагниченного до насыщения будет равна: Е[юо]=Ко; F(iio]=Ko+ (Ki/4); A]nij =/<o4 (Ki/3). По площадям, ограниченным осью ординат / и кривыми намагничивания монокристалла в указанных направлениях, находят величины rnooj; ^(поК Лш] и рассчитывают Ко и К по вышеприведенным формулам. Однако этот способ Довольно неточный.
Определение констант анизотропии при подходе к насыщению. Закон приближения намагниченности к насыщению для поликристалли-ческого образца записывается как dJjdH— =Х₽ + GW) + (Ж/3) 4- (С/НЧ, где В -=0,0763 (К2/Jj), С=0,0384 {Ку .ty — коэффициенты, обусловленные процессом вращения вектора спонтанной намагниченности под действием поля; Хр — восприимчивость парапроцесса; А — коэффициент, зависящий от величины остаточных внутренних напряжений.
По измеренной зависимости дифференциальной восприимчивости (dJtdH) строится зависимость: [(dlldH)—%р]Н3=АН+В.
Коэффициент А определяется графически. Коэффициент В определяется как тангенс угла наклона, а С как отрезок, отсекаемый на оси ординат прямой:
[(dJ/dH — %р) Н* — А] И2 = ВН± С.
Этот метод наиболее эффективен при изучений сплавов с высокой коэрцитивной силой.
Метод вращающих моментов. Прн помещении ферромагнитного монокристалла, имеющего форму шара или эллипсоида вращения, во внешнее магнитное поле иа него будет действовать механический момент, если направление поля не совпадает с одной нз осей легкого намагничивания. Механический момент определяется соотношением: М— VMi=HJs зш(ф— —<р) V, где Mi — механический момент иа единицу объема; ф — угол между осью кристалла и направлением поля; ф— угол между вектором намагниченности и осью кристалла. Мо
315
мент стремится повернуть кристалл так, чтобы <р был равен нулю. Для удержания кристалла в прежнем положении необходимо приложить равный по величине механический момент, создаваемый, например, упругой нитью. Устойчивое равновесие определится из условия минимума энергии анизотропии и энергии кристалла в магнитном поле: Г=/7аниз+^н:=Лс+ 4- 2Ki (txf а|+<Хз а? +«з «1) — HJS cos (ф — ср). Равновесие определяется равенством [dF(dH) = =0. Тогда из условия равновесия получаем: M\=dFnldty.
Если кристалл ориентирован так, что поле находится в плоскости (100), тогда Mj— —/<1 sin 4ф, а в плоскости (110): М\= ~Fi(2 sin 4<р+3 sin 4<р)/4.
Устройство анизометра показано на рнс. 17.66. Образец 1 в держателе 2 закреплен иа упругой ннтн 3. От осветителя 4 луч падает на
Рис. 17.66. Схема анизометра вращения
зеркало 5, укрепленное на штанге 6 держателя. При включении поля образец с держателем и зеркалом поворачивается н луч отклоняется по шкале. По положению луча отсчитывается угол поворота а, а затем определяется механический момент M-Cc/Jl, где С — упругая постоянная ннтн; I — длина инти; а— угол поворота.
17.4.6. Измерение магнитных свойств в переменных полях
Прн наложении на ферромагнитный образец переменного магнитного поля происходит отставание намагниченности от напряженности магнитного поля. Отставание увеличивается с частотой переменного поля.
В периодическом магнитном поле магнитное состояние ферромагнетика изменяется по дн-
316
иамической петле гистерезиса, которая значительно отличается от петли в статическом поле, Рост потерь на перемагничивание обусловлен возникновением потерь на вихревые токи. Прн исследовании магнитных свойств в переменных полях необходимо учитывать скии-эффект, т. е, эффект неполного проникновения магнитного поля в глубину образца. Глубина проникновения h поля в ферромагнетик определяется выражением: (pof) 112, где р—
проницаемость; a—удельная электрическая проводимость образца; f— частота магнитного поля.
В. К. Аркадьев [35] ввел понятие о комплексной проницаемости в переменных полях и разработал методы их определения по измеренным значениям эффективных магнитных проннцаемостей.
Отношение максимальной нидукцин Вт к максимальному значению напряженности магнитного поля Нт является амплитудной проницаемостью ферромагнетика р=Ато/р0Я„,.
Для аналитического выражения динамических петель В, К. Аркадьев предложил заменить нх эквивалентным эллипсом с одинаковой площадью. Тогда Н = Нт sin of; В = = Вт sin (©/—6) = Вт sin (tit—Вт COS (tit, где си —круговая (циклическая) частота; 6 — угол отставания индукции от поля (угол сдвига нлн угол потерь); tg6 = BmjBmi = р2/рг, Вт^ = <= Вт cos б; Вт^ = Bmsin б.
Комплексная магнитная проницаемость р — = pi—ip2, где pi = упругая про-
ницаемость, связанная с обратимыми процессами прн перемагничивании; рг = BmJ^Hmi— вязкая проницаемость, обусловленная процессами поглощения. Модуль комплексной проницаемости представляет собой амплитудную проницаемость
й = К bi + р! .
Упругая проницаемость связана с Вт соотношением pi — Вт cos бДцо/Ап), вязкая — с остаточной индукцией Р2=Аг/(р1:,Н?)!).
Магнитные свойства в переменных полях измеряются в основном индукционным методом. Для расчета р используют формулу р = ~ Дт/(Цо^7действ )2), где Ддейств==®1Д?; W— число витков намагничивающей обмотки; I — сила тока.
Амплитудная проницаемость может быть рассчитана из индуктивности L образца: р = = LI/hquPS, где I — длина образца; S — площадь сечения образца.
Мостовые методы [36]. Принципы мостовых измерений изложены в разделе 17.3. Для мостовой схемы с переменкой индуктивностью нлн емкостью, с помощью которой измеряется зависимость магнитной индукции от поля н амплитудная проницаемость, соотношения между измеряемыми величинами Lx, гх н магнитными параметрами р, Вт, Нт _следующне: Р = (BxTCdcp)/pgW2S; Вт = (i^x[A2)/Sw; —
= (1(0^2) fade р, где <5ср — средний диаметр тороида; S — площадь сечения торонда; w — число витков.
Погрешность метода 2—5 %.
Потенциометрический метод [33]. На рис. 17.67 приведена схема установки для измере-
ння магнитных свойств в переменном поле с помощью потенциометра. На образце О две обмотки — намагничивающая iVi и измерительная W2. Трансформатор Т служит для питания .потенциометра П и намагничивающей обмотки образца через автотрансформаторы АТ. В ка-
рие. 17.67. Схема потенциометрического метода
честве индикатора И может быть использован вибрационный гальванометр или телефон.
Измерение сводится к определению падения напряжения на эталонном сопротивлении и э. д. с. £2 в обмотке W2. Значение величин Ut и дает возможность рассчитать силу тока Z в обмотке По значениям £2 и I рассчитывают Вт, Вт, р, и потери на перемагничивание Рг. в по формулам:
Впг=£2/(1,11лш2/5); 27„ = (iBw1 KF)//;
Вгл — (г’м “4 V 2 )//,
где f — частота магнитного поля; Хд и — действительные и мнимые части токов; Яд и
—соответственно магнитные поля; w — число витков в обмотке. В этом случае = « BmHnfao ; р2 - ВтВт/ц0 +
» И = Вгп/ЦоНт-
Потери иа перемагничивание рассчитывают по формуле Рг. в — (iDE2wi)/w2.
Ошибка метода составляет около 3 %.
Ваттметрический метод определения полных потерь на гистерезис и вихревые токи [36]. Ваттметрический метод основан на измерении потерь мощности в трансформаторе с разомкнутой вторичной цепью (т. е. не потребляющий мощности), причем в качестве сердечника трансформатора используется испытуемый материала (аппарат Эпштейна). Принципиальная схема установки представлена на рнс. 17.68. В четыре секции трансформатора «ь п2 набирается образец из пластин, которые образуют магнитную цепь. В цепь первичной намагничивающей катушки П] включен амперметр А и токовая обмотка ваттметра Wi, в цепь вторичной обмотки трансформатора включены вольтметр V и обмотка напряжения ваттметра —W2. Полные потерн на гистерезис и вихревые токи Рг. в равны: Рг. в = (Р— £22/P2)wi/W2, где Р — показания ваттметра;
£2 — э.д.с. во вторичной цепи; £2—сопротивление вольтметра и обмотки напряжения ваттметра; wj н e>2 — число витков в обмотках
и Г2.
Максимальная индукция: Bm — E2!{4kw2JS)f где k — коэффициент формы кривой э. д. с., равный 1,11.
В практике электротехнических сталей стандартизовано значение удельных потерь Рг.в при Вт = 1 н 1,5 Тл.
Калориметрический метод определения удельных потерь. Абсолютный метод, в кото-
Рис. 17.58. Схема ваттметрического метода
ром мерой потерь энергии при перемагничивании является температура образца. Известно три калориметрических метода: смешения, ввода н протока тепла. Наибольшее распространен не получил метод ввода тепла. Прн исследовании больших потерь используются однокамерные калориметры, для малых потерь — дифференциальные.
Образец вместе с обмоткой погружают в среду калориметра, затем на обмотку подается намагничивающий ток и некоторое время образец подвергается перемагничиванию переменным полем. Величина Вт контролируется по показанию вольтметра. После прекращения пропускания тока и выравнивания температур среды и образца рассчитывают количество теплоты, полученное калориметром. В случае дифференциального калориметра во втором калориметре, идентичном первому, находится образец, аналогичный исследуемому, но нз ие-ферромагннтного материала. При включении переменного поля в обоих калориметрах между ними создается разность температур, которая компенсируется подводом к эталонному калориметру тепла от источника постоянного тока.
Потерн рассчитывают по формуле Рс. в = = х2г, где х—постоянный ток, подводимый к эталонному образцу; г — сопротивление цепи нагрева калориметра с эталоном. Ошибка метода 1—2 %.
17.4.7, Магнетокалорический эффект (26]
При адиабатическом намагничивании ферромагнетика происходит увеличение чнсла параллельных спннов сверх ферромагнитного технического насыщения, прн этом уменьшается энергия обменного взаимодействия и энергия по отношению к внешнему магнитному полю. Получающийся выигрыш в энергии не может «уйти» из системы в силу условий адиабатичности, поэтому увеличивается тепловая энергия элементарных магнетиков, т. е. тело нагревается. Наоборот, прн выключении поля в
317
адиабатически замкнутом ферромагнетике про-исходит разрушение упорядоченной параллельной ориентации спниов, которая осуществляется за счет внутренней тепловой энергии тела и приводит к его охлаждению. Это явление носит название магнетокалорнческого эффекта.
Величина эффекта незначительна, поскольку прн температурах ниже температуры Кк>-рн намагниченность / близка к намагниченности насыщения. Как следует нз термодинамики ферромагнетизма, величина магнетокалори-ческого эффекта АГ линейно увеличивается с ростом намагничивающего поля. При температуре Кюри величина АГ имеет максимальное значение и для чистого железа АГ «2°. На рнс. 17.69 приведены температурные зависимости магнетокалорнческого эффекта для Fe (кривые 1, 2, 3 измерены в магнитных полях 160, 398 и 637 кА/м соответственно). Интересно, что вблизи температуры Кюрн АГ изменяется пропорционально квадрату истинной намагниченности. Исторически это явилось экспериментальным доказательством существования областей спонтанной намагниченности. В настоящее время магнетокалорический эффект используют для получения сверхнизких температур; достигнутое значение температуры удалось понизить от 0,3 (откачка паров Не3) до 10“3 К.
17.4.8. Применение методов измерения магнитных свойств при исследовании металлов и сплавов
Термомагнитный анализ [31]. Для определения состава и фазового анализа используют структурно-нечувствительное свойство — намагниченность; обычно намагниченность насыщения единицы массы os. Установка для проведения термомагннгного анализа должна обеспечивать быстрый нагрев образца, чтобы в процессе иагрева и измерений не изменился фазовый состав сплава. Часто для термомагиитиого анализа используются весы Сексмита. В случае существования в сплаве магнитной фазы величина ос сплава будет пропорциональна количеству фазы, а исчезновение ферромагнитных свойств соответствует температуре Кюрн фазы. Прн наличии двух ферромагнитных фаз с различными намагниченностями oSi и oS2 н температурами Кюрн и 6^ на кривой о (Г) будет перегиб, соответствующий температуре 6С1- Поскольку намагниченность насыщения обладает свойством аддитивности, то намагниченность сплава о£ = cs vi4-os v2, где Vj н vg —доли (по массе) первой и второй фаз. Это позволяет, экстраполируя участок кривой о« (Г), расположенный прн температурах выше 6С , иа ось ординат, определить н oS£v2 н> зная oSi и cSz, определить соотношение фаз илн по н -V2 определить cs и oSo. На рис. 17.70 представлены результаты анализа кривой для сплава Fe—Si из четырех фаз, три нз которых ферромагнитны (т), и" н а). Количество четвертой фазы определяется по разности между массой сплава и vi+vs+vs.
Исследование изотермического превращения в сталях [1]. Для исследования кинетики распада переохлажденного аустенита широко используется анизометр Акулова, который пригоден для исследования любого превращения, которое сопровождается изменением намагниченности образца. Измерение /в сводится к определению магнитного момента образца, помещенного в однородное магнитное поле под некоторым постоянным углом к направле
318
нию И. Удлиненный образец закреплен на плоской пружине, по углу закручивания которой определяют 1а. Схема расположения образца в анизометре представлена на рнс. 17.71.
В случае Д<р«С<р JB =—CA<p/(VHsnnp), где V — объем образца; И — напряженность маг-
Рис. 17.71. Схема расположения образца в анизометре Акулова
Рис. 17.72. Зависимость 4ji/s сплавов Fe—W от концентрации W
нитного поля; С — упругая постоянная нити.
Количество распавшегося аустенита, поскольку он прекращается в ферромагнитную смесь феррита и цементита илн мартенсит, пропорционально намагниченности образца. Для быстрого достижения образцом температуры изотермического превращения анизометр снабжен сменными ваннами, нагретыми до различных температур.
Изучение диаграмм фазового равновесия [1]. Так как в однофазной области намагниченность насыщения монотонно изменяется с изменением концентрации компонентов, а в двухфазной области намагниченность сплава определяется правилом аддитивности, иа границе фазовых областей наблюдается перегиб кривой зависимости от концентрации. Указанное служит основанием для использования измерения намагниченности при построении фазовых диаграмм. На рис. 17.72 приведена в качестве иллюстрации зависимость 4зхЛ сплавов Fe—W от концентрации вольфрама. Коэрцитивная сила, будучи структурно-чувствительным свойством, резко изменяется при проявлении в сплаве второй фазы, следовательно, анализ изменения коэрцитивной Силы от концентрации также позволяет установить грани
цу фазовых областей. На рис. 17.73 приведена зависимость Нс от концентрации в системе Fe—Мо.
Граница двухфазной области 7 % Мо отмечена резким возрастанием коэрцитивной силы и изменением характера ее зависимости от концентрации.
Нс-10*А/м
Рис. 17.73. Зависимость Н с в системе Fe—Мо от концентрации Мо
Определение размера однодоменных частиц дисперсной ферромагнитной фракции [34]. Уменьшение размера ферромагнетика до размера, когда образование доменной структуры энергетически невыгодно, приводит к тому, что частица в любом поле остается миогодо-менной и перемагничивание осуществляется вращением ее вектора намагниченности полем. При дальнейшем уменьшении размера частиц энергия анизотропии ух<е не удерживает вектор намагниченности в направлении легкого намагничивания. Энергия анизотропии такой частицы KV (V — объем частицы; К — константа анизотропии) приближается к kT. Начиная с этого момента, вектор намагниченности частиц начинает флуктуировать и температурная зависимость намагниченности подчиняется закону Ланжевена для парамагнетиков. Поскольку магнитный момент ферромагнитной частицы велик по сравнению с атомным моментом парамагнетика, описанное явление названо «суперпарамагнетизмом». Вероятность самопроизвольного перемагничивания очень быстро увеличивается с уменьшением размера частиц: для сферической частицы Fe
О
диаметром 230 А время релаксации 0,1 с, О
для частицы диаметром 300 А Ю0 с. Переход к суиерпарамагнетнзму сопровождается резким падением Нс.
Наличие суперпарамагнетизма позволяет определить размер и количество мелкодисперсных выделений второй фазы. Размер включений определяется непосредственно по измеренной кривой намагничивания. Начальный наклон функции Ланжевена равен: J/HJ e=M/3kT, где 1е — намагниченность насыщения; / — магнитный момент ферромагнитной частицы. На сплаве меди с 2 % Со удалось этим методом изучать частицы выделяющейся фазы размером несколько ангстремов.
БИБЛИОГРАФИЧЕСКИЙ СПИСОК
1. Лившиц Б. Г. Физические свойства металлов и сплавов. Мп Металлургия, 1980. 320 с.
31Э
2. Шульце Г. Металлофизика: Пер. с ием. М.: Мир, 1971. 503 с.
3. Регель А. Р., Глазов В. М. Периодический закон и физические свойства электронных расплавов. М.: Наука, 1978. 305 с.
4. Свойства элементов: Справочник. Ч. I. Физические свойства. М: Металлургия, 1976. 599 с.
5. Методы испытания, коитроля н исследования машиностроительных материалов: Сир. пособие/Под ред. Туманова А. Т. Т. 1. М.: Машиностроение, 1969, 551 с.
6. Крафтмахер Я. А. Работы по физике твердого тела. Вып. 1. Новосибирск: Наука, 1967.211 с.
7. Глебовский В. Г., Бурцев В. Т. Плавка металлов во взвешенном состоянии. М: Металлургия, 1974. 175 с.
8. Серов Г. С., Крашенинников М. Г., Падерин С. Н. Физико-химические методы исследования металлургических процессов. М.: МИСиС, 1978. 92 с.
9. Берман Р. Теплопроводность твердых тел: Пер. с англ. М.: Мир, 1979. 285 с.,
10. Блатт Ф. Физика электронной проводимости в твердых телах: Пер. с англ. М.: Мнр, 1971. 470 с.
11. Рейсленд Дж. Физика фоионов: Пер. с англ. М.: Мир, 1975. 365 с.
12. Драбл Дж., Гоядсмид Г. Теплопроводность полупроводников: Пер. с англ. М.: ИЛ, 1963. 266 с.
13. Новиков В. Ю. Физические и механические свойства металлов. М.: МИСиС, 1976. 96 с.
14. Пирсон У. Кристаллохимия и физика металлов и сплавов: Пер. с англ. М.: Мнр, 1977. 419 с.
15. Чечерников В. И. Магнитные измерения. М.: Изд-во МГУ, 1969. 387 с.
16. Ашкрофт Н., Мермин В. Физика твердого тела. Т. I: Пер. с англ. М.: Мнр, 1979. 399 с.
17. Ашкрофт Н., Мермин Н. Физика твердого тела. Т. II: Пер. с англ. М: Мнр, 1979. 422 с.
18. Займан Дж. Физика металлов. Ч. I. Электроны: Пер. с англ. М.: Мир, 1972. 463 с.
19. Смит М. К. Основы физики металлов: Пер.
с англ./Под ред. Любова Б. Я. М.: Метал-, лургиздат, 1959. 456 с.
20. Киттель Ч. Квантовая теория твердых тел: Пер. с англ. М.: Наука, 1967. 491 с.
21. Брандт Н. Б., Чудинов С. М. Электронная структура металлов. М.: Изд-во МГУ, 1973. 331 с.
22. Кекаяо И. Б. Физические свойства металлов и сплавов. М.: МИСиС, 1979. 106 с.
23. Вонсовский С. В., Шур Я. С. Ферромагнетизм. М.: Наука, 1963. 264 с.
24. Машиностроение: Энциклопедический справочник. М.: Машгнз, 1948. 712 с.
25. Савенко В. Г. Измерительная техника. М: Высшая школа, 1974. 334 с.
26. Вонсовский С. В. Магнетизм. М.: Наука, 1971. 1031 с.
27. Сэнде М., Фейман Р., Лейтон Р. Фейма-новские лекции по физике. Т. 9. М.: Мир, 1967. 258 с.
28. Испытания материалов: Пер. с нем./Под ред. Блюменауэра X. М.: Металлургия, 1979. 448 с.
29. Белов К П. Упругие тепловые и электрические явления в ферромагнетиках. М.: ГИЗ Технико-теоретической литературы, 1957. 279 с.
30. Лившиц Б. Г., Чупятова Л. П., Л и леев А. С. — Изв. вузов. Черная металлургия, 1967, № 1, с. 123—127.
31. Физическое металловедение Т. 1/Под ред. Кана Р. М.: Мнр, 1967. 333 с.
32. Кифер И. И. Испытания ферромагнитных материалов. М.: Энергия, 1969. 359 с.
33. Чернышев Е. Т., Чернышева Н. Г., Чечу-рина Е. Н. Магнитные измерения на постоянном и переменном токе. М.: Стан-дартгнз, 1962. 183 с.
34. Магнитные свойства металлов и сплавов: Пер. с англ./Под ред. Вонсоеского С. В. М.: ИЛ, 1961. 445 с.
35. Аркадьев В. К- Электромагнитные процессы в металлах. Ч. I. Постоянное электрическое, магнитное поле. М.—Л.: Энергоиз-дат, 1934. 230 с.
36. Чернышев Е. Т., Чучурина Е. Н„ Чернышева Н. Г., Студенцов И. И. Магнитные измерения. М.: Комитет стандартов, 1969. 247 с.
18
—МЕТАЛЛУРГИЧЕСКИЙ КОНТРОЛЬ КАЧЕСТВА
18.1. ОБЩИЕ ПОЛОЖЕНИЯ КОНТРОЛЯ
Основной объект контроля качества — плавка стали. Качественная и высококачественная стали поступают к потребителю поплавочно после всех испытаний, предусмотренных соответствующими стандартами или техническими условиями.
Контроль качества начинается в процессе выплавки стали и заканчивается сдаточными испытаниями [1. 2]. Иногда металл подвергают предварительному плавочному контролю на одном илн нескольких слитках. По результатам плавочного контроля плавку назначают
для конкретного вида изделий (сортовой прокат, поковки и др.) н конкретного способа обработки (прокатка, ковка, штамповка).
Испытаниям некоторых видов (осмотр поверхности, ультразвуковой контроль и др.) подвергают все прутки, поковки и т. д. Большинство испытаний проводится выборочно — на нескольких образцах от плавки, прошедшей определенную технологическую операцию (прокатка иа данный размер, отжиг в одной садке н т. д.). Достоверность выборочного коитроля зависит от однородности металла партии — плавки н объема выборки. Однако большие выборки приводят к потерям металла, повышают трудоемкость испытаний и экономически нецелесообразны.
320
21—280
ТАБЛИЦА 18J
ВИДЫ ВЫБОРОЧНОГО КОНТРОЛЯ ДЛЯ НАИБОЛЕЕ РАСПРОСТРАНЕННЫХ МАРОК СТАЛЕЙ
8 Макроструктура твердость Механические свойства (по Прокаливаемость и чувствительность к закалке ф S а CJ S Я й сх ф । и a<u ф « Я g чая ть , сетка V я А ш 2 s г Й к g S
ГОСТ Химически! став макро-шлифы | ИЗЛОМЫ в S п 8 о ч о « в СОСТОЯНИИ по- ставки | после закалки и отпуска разрыв ударная ВЯЗКОСТЬ Длительна! прочность Осадка Изгиб Вытяжка Эриксену) S « Ч к Ч к н ¥ о о £ 2 Щ о Обезуглер ванне Величина з Структура лита в ото ной стали Карбидная однородно о, У >» н Е- s £3 58 Карбидная Структура бодный це а-фаза Межкристг пая корро
1050—74 «Сталь углеродистая качественная конструкционная» .... 4- -р«> + — + — -ре — 4-*‘ 4-*‘ 4-*1 +*' — — — — —
1577—81 «Сталь горячекатаная листовая качественная углеродистая и легированная конструкционная» + -pl — >— 4- 4- +* 4"*й — — *— -pi — — — — — —
4543—71 «Сталь легированная конструкционная» . ...... 4- + 4-*‘ + — 4- 4- - _)_*3 —|—*1 -pl — — — — — —
801—78 «Сталь шарико-и роликоподшипниковая» + -рю + — 4- 4- — — — 4* — — -pl 4- 4- — 4- 4- 4- 4- — — —
1435—74 «Сталь инструментальная углеродистая» + — + — + 4- — — — + — — 4- — 4* — _р. — — .р? — — —
5950—73 «Сталь инструментальная легированная» . 4- 4* — 4- — — — — — — — + — 4- 4-*' •— — — —
19265—73 «Сталь инструментальная быстрорежущая» . + + 4- *—• + + — — — — — — 4- — — 4- — — — — —
5949—75 «Сталь сортовая и калиброванная, коррозионностойкая, жаростойкая и жаропрочная» 4- 4- 4-*‘ 4- -р5 4-*5 4-*3 — — — — — — — — — — 4-*’
9045—80 «Сталь тонколистовая холоднокатаная малоуглеродистая, качественная для холодной высадки» 4-. — — — 4- — 4- — —1 — — 4- — — — 4- — —• 4- — — .
*' По требованию заказчика. *2 Для размеров от 4 до 60 мм. *5 Может гарантироваться без испытания. *4 Для стали, предназначенной для горячей штамповки, высадки. ш *5 Для некоторых марок. *6 Для марок типа 12Х18Н10Т. *7 Для размеров более 60 мм по соглашению. *8 Для стали категорий 2—5, Для стали категории 2—4. Разме-ю ры менее 30 мм — только излом. _____
Рис, 18.1. Схема разрезки квадратных темплетов со стороной 90 мы для различных видов испытаний:
1 — образец для поперечного макрошлифа (шлиф изготовляется со стороны, обращенной внутрь темпле-та); 2— образец для продольного излома; 3— образец для неметаллических включений; 4 — темплет для механических испытаний (вырезка образцов по ГОСТ 7564—73); 5— образец для продольного макрошлифа; 6 — ступенчатый образец для определения волосовин: 7 — образцы для испытания на межкристаллитную коррозию; 8 — образец для определения а-фазы
Каждая плавка (или партия) стали подвергается испытаниям, установленным соответствующими ГОСТами и техническими условиями. Дополнительные испытания проводятся по внутризаводским инструкциям.
В табл. 18.1 приведены виды выборочных испытаний, предусмотренных ГОСТами для широко применяемых марок стали. В стандартах на другие виды металлопродукции, например на сплавы сопротивления, электротехническую сталь, сталь или сплавы для постоянных магнитов, предусматриваются специальные испытания (электросопротивление, магнитные свойства, жаростойкость н т. д.), которые рассматриваются в других разделах справочника. Образцы для испытаний проката илн поковок отрезают на прессах и пилах горячей резки нлн в холодном состоянии — на пилах нлн абразивных отрезных станках, иногда автогеном. Испытания проводятся на образцах, отрезанных от прутков или заготовок, соответствующих верхней или средней части слитка, В случае ответственных назначений металла испытывают образцы из верхней, средней н нижней частей слитка. Для объективной оценки качества стали образцы для испытаний рекомендуется отбирать от худшего места в слитке. Однако по отношению к разным видам испытаний «худшими» могут быть разные участки: наибольшая загрязненность сульфидами и наибольшая ликвация наблюдаются обычно в верхней части слитка, а загрязненность оксидными включениями — в нижних участках. Расположение «худших» участков в слнтке зависит от способа разливки н условий затвердевания стали.
Поэтому место отбора образцов но .высоте слитка устанавливают на каждом заводе для данного вида продукции на основании исследований и данных массовых испытаний. Если надежных данных нет, то следует отбирать образцы от разных мест по высоте слитка (верх, середина, ннз).
Пример схемы разрезки проб для различных видов испытаний приведен на рнс. 18.1.
Методика проведения различных видов испытаний описана ниже.
18.2. ХИМИЧЕСКИЙ СОСТАВ
Отбор проб для проведения химического анализа производится по ГОСТ 7565—81. Для химического анализа плавки стали обычно во время разливки отливают в чугунную изложницу (стаканчик) пробу массой 0,2—0,5 кг. Методика химического анализа приведена в соответствующих ГОСТах. Состав плавки может быть также определен при анализе стружки от полного сечення прутка готового проката илн поковки. На металлургических заводах ряд элементов определяют спектральным методом по ходу плавки и в готовом прокате, при этом гарантируется соответствие результатов спектрального анализа данным химического анализа. Химический состав плавки стали указывают в сертификате на готовую продукцию.
18.3. ВНЕШНИЙ ВИД
МЕТАЛЛОПРОДУКЦИИ
Весь металл, поставляемый металлургическими заводами, подвергают внешнему осмотру. Размеры и правильность геометрической формы проверяют универсальными приборами илн шаблонами. Прн этом отклонения от номинальных размеров не должны превышать допустимых норм для данного вида продукции. Поставщик металла проверяет по размерам нлн 100 % прутков, поковок н т. д., или выборочно 5—10 %, но прн этом также гарантируется соответствие всей партии требованиям сортамента.
Слиткн, заготовку, сортовой прокат илн поковки подвергают внешнему осмотру для обнаружения дефектов поверхности. Поверхность слитков осматривают после выгрузки нх из изложниц перед загрузкой холодных слитков в методические печи. На заводах, работающих с передачей иа нагрев горячих слнтков, осматривают ие слитки, а поверхность заготовки после первого передела. В зависимости от состояния поверхности, характера н чнсла дефектов слиткн подвергают обднрке на токарных стайках нлн местной зачистке абразивами. Слитки, имеющие глубокие дефекты, обычно бракуют. Слитки высоколегированных сталей н сплавов, содержащих титан, подвергают сплошной обдирке.
При большом количестве дефектов поверхность заготовки или сляба подвергают сплошной зачистке на абразивных станках илн огневой зачистке [3]. Небольшие дефекты вырубают пневматическими зубилами илн зачищают абразивами.
Поверхность готовой продукции перед осмотром для выявления дефектов, уточнения нх характера н глубины подвергают местному «светленню» абразивными кругами или напильниками (для калиброванного металла). «Свет-ление» производят змейкой или кольцами. Расстояние между кольцами нлн шаг змейки 200—400 мм. Выявленные после светления дефекты отмечают и удаляют. Для более полного обнаружения дефектов, главным образом на заготовках, применяют травление в растворах кислот. После травления металл осматривают, отмечают дефекты н удаляют их.
Высоколегированную сталь, имеющую после горячей деформации закаленную структуру (мартенситную), перед травлением необходимо подвергать отжигу или высокому отпуску, чтобы избежать травильных трещин. Особенно чувствительны к образованию таких трещин быстрорежущие стали, а также стали 40X13 н 40X10С2М.
Пруткн с гладкой шлифованной поверхностью можно контролировать методами магнитной дефектоскопии. Этими методами обнаруживаются трещины, волосовины, закаты й другие дефекты. Магнитный контроль может приводить к неправильным заключениям, в тех случаях, когда ферромагнитный порошок осаждается вдоль структурных составляющих, отличающихся от основной массы металла (карбидные строчки, строчки a-фазы в аустенитной структуре и т. д.), особенно в случае использования для контроля магнитных полей высокой напряженности.
Места осаждения следует очерчивать н про
сматривать после удаления магнитного порошка. Трещины, закаты, волосовины имеют вид темных полосок, структурные составляющие ие обнаруживаются.
Применение магнитной дефектоскопии ограничивается ферромагнитными материалами. Для выявления дефектов немагнитных сталей н сплавов применяется метод флуоресцентной или люминесцентной дефектоскопии. Описание некоторых видов дефектов н нх причины имеются в литературе [1, 2, 4, 5, 9, 20, 22, 24, 26, 32].
Требования к чистоте поверхности металлопродукции изложены в ГОСТах и технических условиях на металлопродукцию. Термины и определения дефектов поверхности регламентированы ГОСТ 20847—75 «Прутки, полосы и профили горячекатаные и кованые из сталей и сплавов. Дефекты поверхности» и ГОСТ 21014—75 «Листы и ленты стальные. Дефекты поверхности и формы». Некоторые определения дефектов приводятся ниже.
Раскатанное загрязнение (рнс. 18.2)—вытянутое вдоль направления деформации за-
Рис. 18.2. Раскатанное загрязнение
Рис. 18.8. Раскатанный пузырь
грязненне слитка нлн литой заготовки шлаком или огнеупорами. На мнкрошлифах по месту дефекта видны грубые неметаллические включения, обезуглероживания нет.
Раскатанный пузырь (рис. 18.3) — прямолинейное нарушение сплошности поверхности вдоль направления деформации. Образуется нз наружного нли подповерхностного пузыря слитка илн литой заготовки. Часто дефект имеет групповое расположение. Полость дефекта заполнена окалиной. Стенки полости обезуглерожены, зона обезуглероживания резко ограничена н насыщена мелкими окислами. Полость дефекта расположена примерно под прямым углом к поверхности. Образуется вследствие попадания влаги при выпуске и
Рис, 18.4. Раскатанная трещина а и б
разливке илн при высокой газонасыщенности металла.
Раскатанная трещина (рис. 18.4) — разрыв («горячая трещина»), образовавшийся в слитке или литой заготовке при высоких температурах в период затвердевания из-за недостаточной прочности корки затвердевшего металла. Может вытягиваться вдоль направления деформации. Дефект заполнен окалиной. Стенки дефекта обезуглерожены, разрыв может быть разветвлен. Аналогичный дефект об
21*
323
разуется из трещин напряжения («холодная трещина») в слитке илн литой заготовке.
Раскатанная корочка (рис. 18,5)—местное отслоение металла, образовавшееся в результате раскатки завернувшихся корочек, заливки н брызг на поверхности слитка илн литой за-
верхиости. Образуется вследствие напряжений, вызванных структурными превращениями в твердом или хрупком металле. На микро-шлнфе трещина напряжения проходит по границам зерен, имеет тонкий конец и неокисле-на. Окисление н обезуглероживание дефекта
Рис. 18.5, Раскатанная корочка
Рис, 18.6. Рванины
Рис, 18.9. Трещины напряжений (а. б)
готовки. На мнкрошлифах в зоне дефекта наблюдается обезуглероживание н скопление неметаллических включений сложного состава, шлака и огнеупоров. Дефект может иметь небольшой наклон к поверхности нли идти почти параллельно ей.
Рванины (рнс. 18.6) — раскрытые разрывы, расположенные поперек нлн под углом к направлению наибольшей вытяжкн прн деформации. Образуются вследствие пониженной пластичности металла при деформации. Пониженная пластичность может быть связана с условиями выплавки металла или с’режимом нагрева под деформацию. На микрошлнфах в зоне дефекта наблюдаются разветвленные разрывы металла, В разрывах может быть окалина, а зоны у1 стенок дефекта могут быть насыщены окислами н нитридами, образовавшимися прн нагреве (нлн охлаждении) раскрытых разрывов.
Прокатная плена (рнс, 18,7) — отслоение металла языкообразной формы, соединенное с
Рис, 18.7. Прокатная плена
Рис. 18,8, Чешуйчатость
основным металлом одной стороной, образовавшееся вследствие раскатки рваннн нлн следов глубокой зачистки местных дефектов поверхности. Нижняя поверхность отслоения и металл под ним покрыты окалиной. На мнкро-щлнфе в зоне, прилегающей к дефекту, может наблюдаться обезуглероживание.
Чешуйчатость (рис. 18,8) — отслоения н разрывы в виде сетки, образовавшейся прн деформации вследствие пониженной пластичности или перегрева металла периферийной зоны. Снижение пластичности может быть вызвано образованием по границам зерен легкоплавких соединений или насыщением поверхности металла серой прн нагреве в среде сернистых газов. На микрошлнфах разрывы проходят по границам зерен н сопровождаются окислами н эвтектическими фазами.
Трещина напряжения (рнс. 18,9) — прямолинейный или зигзагообразный разрыв, идущий в глубь металла под прямым углом к по-
Рис. 18.10. Ус:
а — для крупных профилей; б — для мелких профилей
Рис. 18.11. Закат:
а — гладкий; б — зазубренный
происходят только прн последующем нагреве.
Ус (рнс. 18.10) — продольный выступ с одной нлн двух диаметрально противоположных сторон, образуется вследствие неправильной подачи металла в калибр, переполнения калибра или неправильной настройки валков п прн-валковой арматуры.
Закат (рнс. 18.11)—прикатанный продольный выступ металла с одной или двух диаметрально противоположных сторон, образовавшийся прн вдавливании уса или подреза. Дефект может иметь зазубренный край. На поперечном мнкрошлнфе дефект располагается под острым углом к поверхности. Конец дефекта ие разветвлен, заполнен ' окалниой и огибается волокном. Металл по дефекту обезуглерожен.
Шлифовочные трещины (рис. 18.12)—сетка паутинообразных нли отдельных произвольно
Рис. 18.12. Шлифовочные трещины
Рис. 18.13. Травильные трещины
направленных поверхностных разрывов, образовавшихся при шлифовании металла, обладающего высокой хрупкостью, твердостью и малой теплопроводностью. На мнкрошлнфе трещины распространяются по границам зерен.
324
Травильные трещины (рнс. 18.13)—разрывы, иногда в виде сеткн, образовавшиеся при травлении металла, имевшего напряжения от структурных превращений илн деформаций. Типичны для металла с мартенситной структурой. Трещины распространяются по границам зерен.
Рис. 18.14. Волосовины
Рис. 18.1 Б. Пузырь-вздутие
Волосовины (рнс. 18.14)—нитевидные не-сплошностн, расположенные вдоль направления деформации; образуются нз скоплений неметаллических включений нли из отдельных крупных полупластичных н пластичных включений, вытягивающихся вдоль направления деформации и образующих неглубокие дефекты различной протяженности. Длина волосовин зависит от степени обжатия и возрастает с увеличением ее.
Пузырь-вздутие (рнс. 18.15)—дефект поверхности листов в виде вспучивания металла, образующегося из-за загрязнения металла неметаллическими включениями нлн газовыми пузырями. Образование пузырей-вздутнй часто наблюдается прн насыщении водородом тонких листов в процессе травления. В местах прослоек (обычно силикатов) илн иесварнв-шихся газовых пузырей накопившийся водород
Рис. 18.16. Расслоение
образует вздутие. Пузырн-вздутия образуются также при быстром нагреве листов металла с прослойками включений.
Расслоение (рис. 18.16)—дефект на торцах и кромках листов представляет собой иесплош-ность, образующуюся нз-за несвариваемости металла вследствие загрязнения неметаллическими включениями (шлаком, огнеупором Н др.).
18.4. ИСПЫТАНИЕ НА ОСАДКУ
Для обнаружения наружных н подкорковых дефектов сталь, предназначенную для горячей нли холодной высадки илн штамповки, испытывают на осадку. Испытание иа осадку в холодном состоянии проводят по ГОСТ 8817—73, в горячем состоянии — на образцах высотой, равной двум диаметрам (нлн двойной толщине) прутков. Образцы нагренают до температуры ковкн и осаживают на ’/з первоначальной высоты.
Испытание иа осадку позволяет выявить н классифицировать такие дефекты, как раскатанные пузыри, закаты, трещины, риски н т. д. Дефекты, имеющие значительную глубину,
раскрываются прн осадке. Годным считается металл, иа образцах которого после осадки не обнаруживается дефектов, выходящих за пределы требований технических условий. Испытание на холодную нли горячую осадку дает представление о пригодности стали к холодной илн горячей деформации.
Испытание на осадку в холодном состоянии особенно важно для стали высокой твердости.
18.5. МАКРОСТРУКТУРА СТАЛИ
Контроль макроструктуры — основной внд испытаний для определения качества стали и обнаружения разнообразных металлургических дефектов. Макроструктуру контролируют на поперечных нлн продольных шлифах и изломах. Наиболее часто макроструктура контролируется на поперечных травленых' макрошлифах. Этот способ позволяет оценить все сечение прутков стали и благодаря травлению выявить крупные и мелкие дефекты (включая ликвацнонные) и особенности структуры. Оценка макроструктуры производится по ГОСТ 10243—75. Макроструктуру прутков и заготовок размером до 140 мм проверяют в полном сеченнн, а более 140 мм—иа перекованных пробах.
Темплеты для поперечных шлифов и продольных изломов вырезают перпендикулярно направлению прокатки илн ковки, для продольных шлифов — параллельно направлению прокатки, ковки, при этом плоскость шлнфа должна совпадать нлн быть близкой к осн контролируемого прутка. Длина продольных темплетов 100—150 мм. Рекомендуемая высота поперечных темплетов 15—40 мм. Пробы для контроля на флокены отбирают иа расстоянии не менее одного диаметра (стороны квадрата) от конца заготовки по окончании охлаждения нлн термической обработки каждой партии данной плавки. Если образец отрезан в горячем состоянии, сразу после прокатки нли ковкн, то его длина должна быть не менее четырех диаметров (сторон квадрата). Охлаждение или термическую обработку такого образца производят имеете с металлом контролируемой партии плавки.
Если партия стали не подвергается термической обработке, то после охлаждения вместе с партией металла перед отжигом пробы легированной н конструкционной стали выдерживаются прн 20 °C не менее 24 ч, а пробы высоколегированной стали (18Х2Н4ВА н др. )— не менее 48 ч. Темплет для коитроля макроструктуры вырезают нз середины образца.
Допускается производить контроль на флокены на продольных темплетах и продольных изломах, а также методом ультразвуковой дефектоскопии.
Поверхность темплетов перед травлением подвергается торцеванию, строганию, шлифованию (подробнее см. раздел 1).
Продольный излом контролируют иа темплетах (шайбах) толщиной 15—40 мм. Для этой цели используют и поперечные шлифы. Их надрезают на глубину, позволяющую получить излом высотой з>10 мм для прутков 2>80 мм н ;>5 мм — для прутков диаметром <80 мм. Надрезанные образцы закаливают в воде по режиму, принятому для данной марки стали, а затем ломают под прессом нли на копре.
325
Для поперечного излома прутки в состоянии поставки надрезают с одной илн двух сторон и надрезанный кусок отламывают.
Методика анализа макрошлифов приведена в разделе 1,
Поперечные шлифы оценивают путем сравнения с фотоэталонамн шкал или отдельных дефектов. ГОСТ 10243—75 предусматривает оценку (на шлифах) по пятибалльным шкалам следующих видов дефектов и особенностей структуры: центральной пористости, точечной неоднородности, общей пятнистой ликвации, краевой пятнистой ликвации, лнквацнониого квадрата, подусадочной ликвацнн, подкорковых пузырей, межкристаллитных трещин, послойной кристаллизации и светлой полоски (контур). В стандарте также приведены фотоэталоны макроструктуры с такими дефектами, как пузыри, корки, флокены, черновины, ковочные трещины, скворечники, корона и др.
Прн необходимости контроля на продольных шлифах и изломах оценку производят визуально путем сравнения с эталонами, согласованными между поставщиком н потребителем металла. Осмотр продольного излома позволяет уточнить и выявить усадочную раковину, центральную пористость, пузыри, сколы, флокены, строчки неметаллических включений, перегрев осевой зоны (черновины), камневидный, нафталиннстый, шиферный изломы и другие особенности структуры.
Внд излома зависит от термической обработки образцов (шайб) перед испытанием. В образцах (шайбах), закаленных иа высокую твердость, излом более гладкий и направлен по надрезу. Отпущенные нлн мягко закаленные шайбы дают неровный вязкий излом, отклоняющийся от плоскости надреза. На вязком изломе дефекты обнаружить легче, чем иа хрупком. Для получения высокой вязкости шайбу закаливают иа мартенсит и подвергают высокому отпуску.
При осмотре поперечного излома обнаруживаются усадочная раковина, внутренние разрывы, флокены, черный излом, черновины (осевой перегрев), ковочные трещины, обезуглероживание и др. Поперечный излом целесообразен для контроля высокоуглеродистых сталей, обладающих в состоянии поставки достаточной хрупкостью. Вязкие стали (12ХНЗА илн аустенитная нержавеющая) нельзя испытывать таким способом: в нзДоме часто образуются вырывы (ложные расслоения), связанные с условиями испытаний [4].
Расслоение (вырывы) в поперечных изломах возникают вследствие двухфазной (полосчатой) структуры- металла, а не в связи с нарушениями его сплошности.
Дефекты макроструктуры выявляются также и неразрушающими методами контроля. Наиболее эффективный из них — ультразвуковой контроль (УЗК). Общие требования к методам УЗК приведены в ГОСТ 12503—75. Способ УЗК находит все более широкое применение.
Примеры типичных внутренних дефектов стали, их описание, причины образования и меры предупреждения приведены ниже.
Усадочная раковина — полость, не заполненная металлом и расположенная обычно в центре верхней части слнтка. По контуру полость обогащена лнкватами (серой, фосфором) и неметаллическими включениями.
Образуется вследствие уменьшения объема при затвердевании стали. Усадочная раковина может быть уменьшена прн применении утепленных прибыльных надставок, термитных смесей, электрообогрева для сохранения в верхней части слитка жидкого металла, питающего усадочную полость; снижении скорости разлнвкн н температуры жидкого металла [4, 20, 22, 24].
Пузыри — округлые полости нлн каналы в литом металле (рнс. 18.17,а). Прн горячей деформации ие завариваются, если стенки покрыты окислами кремния н др. В деформированном металле иа поперечных шлифах имеют
Рис. 18.17. Пузыри:
а — в литом металле: б — подкорковые в деформированном металле.
вид черточек. Могут располагаться по сечению нли вблизи поверхности; в последнем случае называются подкорковыми пузырями (рис. 18.17, б).
Образуются прн высокой концентрации газов (кислорода, азота, водорода) вследствие выделения газовой фазы (СО, N2, Н2) в период кристаллизации, так как растворимость этих газов резко уменьшается прн переходе из жидкого в твердое состояние.
Для предупреждения образования пузырей в спокойной стали снижают до минимума содержание растворенных газов. Снижение содержания кислорода достигается раскислением, водорода — применением свежепрокаленной извести, сушкой материалов н разливочных ог-
иеупоров, азота — предохранением от контакта с воздухом [4, 19, 20, 22, 24].
Центральная пористость — группа мелких пор в центральной части прутков (рнс. 18.18). Встречается в средней или ннжней (для больших слитков) части слитков. Сопровождается ликватамн и неметаллическими включениями.
Рис. 18.18. Центральная пористость
Образуется нз осевой усадочной несплошно-стн слитка. Из-за присутствия включений нли малой степени обжатия прн деформация полностью не заваривается. Можно избежать, уменьшая отношение высоты слитка к диаметру, замедляя кристаллизацию (те же меры, что н для уменьшения усадочной раковины).
Рнс. 18.19. Ликвацнонный квадрат
Ликвационный квадрат—контур зональной ликвацнн, по форме соответствующий сечению слнтка (рнс. 18.19). Полоска ликвацнн располагается в средней части слитка (после зоны столбчатых кристаллов). Участки металла, соответствующие зоне столбчатых кристаллов,
травятся менее интенсивно, чем зона равноосных кристаллов в центре слитка. Наиболее сильно травится металл лнквацнонного контура (на границе между столбчатыми и равноосными кристаллами).
Образуется прн направленной кристаллизации металла от стенок изложницы в глубь слитка. В конце зоны столбчатых кристаллов повышается концентрация элементов, растворимость которых в твердой фазе значительно ниже, чем в жидкой (серы, кислорода). Прн затвердевании здесь выделяются сульфиды и концентрируются окислы. Образование лнква-циониого контура может быть связано с различной скоростью кристаллизации, размерами дендритов н различной степенью дендритной ликвации [6, 21, 22]. Центральная зона с более высокой степенью дендритной лнквацни может иметь повышенную травнмость. Уменьшение лнквацнонного контура достигается: снижением содержания ликвирующих примесей; ускорением затвердевания; созданием мелкокристаллических структур.
Точечная неоднородность — мелкие сильно травящиеся точки, расположенные по сечению образца (рнс. 18.20), за исключением крае-
Рис. 18.20. Точечная неоднородность
вой зоны. Точки часто располагаются по контуру ликвацнонной зоны в средней части мак-рошлнфа.
Образуется в участках, обогащенных примесями вследствие развития дендритной ликвации. Иногда наблюдается пересечение плоскостью макрошлнфа ликвационных усов. Меры предупреждения такие нее, как н для лнквацнонного квадрата.
Флокены — тонкие разветвленные трещины различных размеров (до 100 мм) в средней части прутков нли поковок (рнс. 18.21, с). Располагаются чаще произвольно по отношению к направлению деформации, но иногда только поперек нли только вдоль него. В продольных изломах имеют вид блестящих пятен (рнс. 18.21,а). Обычно встречаются в деформированном металле, иногда в литой стали.
Флокены образуются в стали в процессе охлаждения при температурах с200 °C. Раз
327
виваются нз микротрещин, возникновение которых обусловлено хрупкостью металла, высокой концентрацией водорода и напряжениями от структурных превращений в межосных участках дендритов [4, 7, 8]. Уменьшение содержания водорода достигается применением
Рис. 18.21. Флокены;
а — в поперечный макрошлиф; б — в излом
Рис. 18.22. Межкристаллитные трещины:
а — литой металл; б — излом деформированного металла
прн выплавке стали свежепрокалениой навести н просушенных материалов; после горячей деформации с помощью режимов замедленного охлаждения н специальной термической об
работки можно предупредить образование флокенов.
Межкристаллитные трещины и прослойки. Располагаются обычно в ’ Центральной зоне слитков. В изломе литых дисков (рнс. 18.22, а) трещины проходят по границам кристаллитов (блестящие). На макрошлифах деформированного металла выглядят как «паучок», в изломах— «сколы» (рис. 18.22,6). Дефект встречается в низко- и среднеуглеродистых легированных сталях (18Х2Н4ВА, 12Х2Н4А н др.).
Образуются прн кристаллизации металла вследствие выделения на границах кристаллов пленок окнслов илн сульфидов. Для уменьшения межкристаллитных трещин необходимо хорошее раскисление; прн выплавке долю отходов в шихте следует ограничивать. Улучшению качества металла могут способствовать добавки титана (до 0,1 %), гомогенизация слитков нлн заготовки прн 1150—1200 °C [4, 22].
Ковочные трещины. Располагаются в осевой зоне и могут иметь инд креста, трещины по днагоиалн, двух нлн более трещин, направленных от центра в стороны (рис. 18.23). В из-
Рис. 18.23. Ковочные трещины
ломе выглядят как грубые, окисленные расслоения.
Образуются вследствие высоких напряжений прн ковке металла, имеющего прн температурах деформации высокую прочность н малую пластичность [9]. Часто возникают прн обкатке на круг илн прн переходе с квадрата на круг. Для предупреждения образования обкатку на круг следует производить в фасонных бойках илн обжимах малыми подачами [27].
Внутренние разрывы — местные периодические поперечные разрывы в центральной зоне прутков, иногда значительных размеров (рнс. 18.24, а, б).
Образуются прн прокатке слитков малопластичного металла (быстрорежущей стали н др.), при калибровке круг — овал, прн высоких напряжениях в центральной зоне, прн перегреве слитков. Для предупреждения понн-
328
жают температуру нагрева н применяют другие калибровки. При последующих обжатиях с большими степенями, если отсутствует окисление, дефект заваривается.
Рванины и разваливание слитков — разрывы металла прн горячей деформации (ковка, про
1
« I
Рнс. 18.24. Внутренние разрывы:
а — продольный макрошлиф; б — поперечный излом
катка, штамповка, прошивка труб и т. д.). Разрывы могут приводить к полному разрушению слитков (рис. 18.25, а) или к образованию рванин по всей поверхности (рис. 18,25,6). Часто наблюдаются разрывы по углам заготовок.
Образование дефекта вызвано низкой пластичностью некоторых марок стали (нлн отдельных плавок данной марки). Рванины образуются и црн деформации металла, нагретого выше оптимальной температуры. Образования рванин можио полностью илн частично избежать: а) выдерживая оптимальный температурный интервал горячей механической обработки; б) уменьшая скорости деформации и степени обжатня; в) улучшая качество стали (лучшее раскисление и минимальное содержание вредных примесей) [9, 10, 17, 18].
Корка — тонкий слой затвердевшего металла, содержащий скопления неметаллических включений (рис. 18.26). Иногда металл расплавляется н иа месте корки остается только скопление включений.
Коркн образуются в момент заливки изложницы, затем (прн сифонной разливке) приподнимаются под напором струи металла (светлые коркн). Наиболее часто корки образуются
на верхней поверхности металла в изложнице (темные корки) н вследствие большей плотности твердого металла тонут в жидком металле, увлекая вместе с собой скопления включений; корки могут располагаться в любом месте
Рис. 18.25. Рванины и разваливание слитков:
а — разрушение при ковке ковшовой пробы; б — рванины на поверхности сляба
Рис. 18.26. Корки
слитка. Для предупреждения образования темных корок металл в прибыльной части слитка засыпают экзотермическими смесями [4, 22].
Черновины — группа разрывов во внутренней зоне прутков. На макрошлифах имеют вид темиотравящихся участков (рис. 18.27, а). Зона мелких надрывов в продольном изломе представлена иа рис. 18.27, б. На микрошлн-
фах наблюдаются типичные «микропоры» (рнс. 18.27,в). Дефект также называется «осевой пережог» или «осевой перегрев».
Образуются прн деформации преимущественно литого металла нз-за подплавлення участков металла, обогащенных ликватамн или со-
Рис. 18.27. Черновины (осевой перегрев):
а —- поперечный макрошлиф: б — излом; в — микропоры на шлифе. хЮО
Рис. 18.28. Титановая неоднородность
держащих ледебурит. Прочность металла резко снижается, образуются многочисленные разрывы. Снизив температуру металла, можно
прокатывать слитки данной плавки без дефектов [4, 22].
Титановая неоднородность — групповые скопления неметаллических включений (нитридов н окислов титана). На макрошлнфах — темные точки (рнс. 18.28), в продольных изломах— темные полоски. Встречаются в краевой зоне, ио могут быть расположены и по всему сечеиию. На поверхности и в продольном мак-рошлнфе образуют волосовины.
Образуются в виде корочек на поверхности вследствие контакта с воздухом стали, содержащей титан. Приварившиеся к поверхноств елнтка или погруженные в металл корочки создают зоны повышенной загрязненности окис-лами и ннтрндамн титана [26]. Для предупреждения следует проводить разливку в атмосфере аргона или в защитных устройствах (под колпаками н т. д.)_ Встречается неоднородность цериевая н др.
Скворечник — разрыв внутри деформированного металла, иногда выходящий на поверхность (рнс. 18.29).
Рис. 18.29. Скворечник
Рис. 18.30. Черный излом
Образуется из внутренней, обычно поперечной, трещины, возникшей прн посадке холодных елнтков или заготовок из твердых сталей в печь со слишком высокой температурой. Прн последующей деформации не заваривается, а раскрывается. Для предупреждения образования дефекта: а) не следует производить посадку иа нагрев елнтков с температурой ниже О °C; б) обеспечивать относительно медленный нагрев в интервале 500—800 °C. В аустенитной
области опасность появления трещин уменьшается.
Черный излом — зоны нлн полосы черного цвета на поверхности излома встречаются обычно в высокоуглеродистой стали, легированной кремнием (рнс. 18.30). Неоднородность в данном случае связана с условиями получения излома (форма надреза и способ разрушения), но может вызываться н неоднородным распределением графита.
холодной деформации; ж) закалка с отпуском прн 700 °C.
Волосовины — тонкие строчки (протяженность мм) раскатанных неметаллических включений нли их скоплений (рис. 18.31,а,б).
Окисные включения состоят из частиц огнеупоров, шлаков и др. или образуются в результате реакций кислорода с раскислителями и компонентами стали, а также при взаимодействии раскислителей н компонентов стали со
Рис. 18.31. Волосовины:
а—ступенчатый образец; б—продольный микрошлиф. XI00
Рис. 18.32. Послойная кристаллизация
Образование черного излома объясняется выделением графита. Этому процессу способствуют: а) высокое содержание углерода н кремния в стали; б) отсутствие хрома; в) раскисление алюминием; г) низкая температура конца горячей деформации; д) длительный отжиг прн температурах немного ниже Ас^ (700°C); е) высокий отпуск или отжиг после
шлаками и огнеупорами. Сульфиды образуются прн взаимодействии серы с компонентами стали, раскислителями н десульфураторамн, иитрнды — при реакции компонентов стали с азотом, растворенным в стали и азотом воздуха. Уменьшение количества н размеров неметаллических включений достигается применением высококачественных огнеупоров, снижением температуры металла, сливом металла без шлака, подбором оптимальных условий раскисления и десульфурации, продувкой газами, устранением контакта с воздухом Г10, 19, 28, 29, 31]. *
Послойная кристаллизация — чередующиеся концентрические слон металла в виде узких светлых и темных полос. Расположены ближе к поверхности (рнс. 18.32). Такая структура характерна для металла электрошлакового и вакуумно-дугового переплавов.
Образуется в процессе переплава электродов, прн колебаниях подводимой электрической мощности илн неуравновешенном движении металла [23, 25, 30].
Частицы короны — дефект макроструктуры металла, вакуумно-дугового переплава, представляющий скопления неметаллических включений (ГОСТ 22838—77). Встречается в высоколегированных сплавах, содержащих титан, часто имеет внд спирали (рнс. 18.33). В зоне дефекта наблюдается изменение состава металл а-осиовы н большая плотность окислов и
331
нитридов титана, интридов алюминия, а также окисли кальция н магния.
Образуются нз падающих в жидкий металл кусочков «короиы», расположенной над верхней частью слитка. Корона образуется в процессе переплава из брызг металла н металлн-
Рис. 18.33. Частицы короны
ческих паров, конденсирующихся на стенке кристаллизатора. На поверхности короны осаж-даются также окислы, а контакт тонкого слоя короны с атмосферой печн приводит к насыщению ее окисламн н нитридами. По мере наплавления слитка куски короны закручиваются и, подплавляясь, падают в металл. Для предупреждения дефекта следует вести плавку с небольшой длиной междугового промежутка, избегать нонизацнн н применять для электродов металл достаточной чистоты.
18.6. ВОЛОСОВИНЫ
В деформированном металле скопления неметаллических включений и отдельные пластичные включения вытягиваются вдоль направления деформации и образуют строчки, иногда имеющие значительную протяженность. В тех случаях, когда протяженность строчек I мм и более, дефект может быть отнесен к волосовинам н обнаруживается визуально илн с применением лупы с 10-кратиым увеличением. Если х<е дефект имеет небольшую протяженность (не более 1 мм), то его оценку производят под микроскопом методами, применяемыми для оценки неметаллических включений.
К волосовинам можно отнести и мелкие раскатанные пувыри, не заварившиеся в процессе прокатки (ковки) из-за прослойки включений.
Оценка степени загрязненности отдельных плавок стали волосовинами — сложная задача, так как распределены они в объеме металла неравномерно, встречаются довольно редко и имеют различную длику, которая зависит как от размера отдельного включения нли строчки, так и от степени вытяжки в процессе деформации. Поэтому загрязненность плавок волосовинами можно сравнивать только прн одинаковой степени вытяжки образцов. Для получения надежных результатов необходимо осматривать достаточно большие поверхности, что значительно повышает трудоемкость контроля. Степень загрязненности стали волосовинами, определенная на малом числе образцов лишь весьма приблизительно, характеризует плавку.
Контроль иа малом числе образцов эффективен только при условии, что плавки сильно различаются по загрязненности. Тем не менее контроль иа волосовины характеризует качество металла и используется для внутризаводского контроля и при проведении исследовательских работ. Контроль проводят иа 3—12 образцах от каждой плавки. Для определения .загрязненности волосовинами применяют цилиндрические (рис. 18.34), или плоские
Рис. 18.34. Круглые образцы для оценки волосовины:
А, Б — одноступенчатые; В — многоступенчатый
Рис. 18.35. Плоские образцы для оценки волосовин: А — одноступенчатый; Б — односторонний трехступенчатый; В — двусторонний трехступенчатый
(рнс. 18.35) образцы. Если металл предназначается для изделий цилиндрической формы, целесообразно контролировать его на цилиндрических образцах. Прн этом контролируемая поверхность должна быть расположена на той же глубине, что и рабочая поверхность изделия.
Контролируемую поверхность образцов шли фуют и осматривают визуально нлн с помощью 5—10-кратной лупы. Слабое травление образцов для снятия шлифованной поверхности облегчает осмотр. Сильное травление прн-
332
ТАБЛИЦА 18.2
ДИАМЕТРЫ СТУПЕНЧАТОЙ ОБТОЧКИ
В ЗАВИСИМОСТИ ОТ РАЗМЕРА ПРУТКОВ», мм
d 4i d3 di
16—20 13 9 5
21—25 18 15 10
26—30 23 18 10
31—40 28 22 15
41—50 38 28 15
51—60 48 32 15
61—80 58 40 20
81—100 76 55 30
101—120 96 70 40
121—150 116 85 50
* Обозначении см, на рис. 34, в. Длина каждой ступени 50 мм.
Степень загрязненности волосовинами оговаривается потребителем в технических условиях. Для сталей особо ответственного назначения существуют нормы технических условий (ГОСТ 4543—71). Согласно этим техническим условиям учитывают волосовины длиной 5>0,5 мм. Прн. этом на определенной площади деталей нормируется загрязненность металла волосовинами по следующим критериям:
1) допускаемое число волосовин;
2) максимальная длина волосовин, мм;
3) суммарная протяженность волосовин, мм.
Применение технических условий показало, что наиболее часто металл бракуется по критериям максимальной длины и суммарной протяженности волосовин.
Допустимые нормы (табл. 18.3) условны и определяют ответственность поставщика металла за степень загрязненности изделий волосовинами. Для конкретных деталей нормы могут изменяться н определяться специальными техническими условиями.
водит к растравливанию полосок структурных составляющих стали, в результате трудно отличить вытравленные структурные неоднородности (карбидные строчки или строчки феррита в аустенитной структуре) от волосовин.
Критерии оценки загрязненности плавки: количество волосовин, суммарная протяженность волосовин, нх максимальная длина и др.
Для контроля сортового проката применяют н трехступенчатые круглые образцы длиной 200—250 мм. Рекомендуемые диаметры обточки ступеней приведены в табл. 18.2. Последний проход прн обточке ступеней делается острым резцом. Глубина резания 0,5 мм, подача 0,25 мм/об. Просматривают каждую ступень и замеряют длину волосовин на ней.
Для облегчения обнаружения волосовин используется метод магнитной дефектоскопии. Намагниченные образцы погружают в жидкость со взмученным магнитным порошком. Порошок оседает вдоль волосовин, расположенных на поверхности и на небольшой глубине. Поскольку объем контролируемого металла увеличивается, точность метода повышается [10].
ТАБЛИЦА 18.3
18.7. НЕМЕТАЛЛИЧЕСКИЕ ВКЛЮЧЕНИЯ
Контроль стали на загрязненность неметаллическими включениями проводится по методикам ГОСТ 1778—70 «Сталь». Металлографические методы определения неметаллических Включений. Стандартом предусмотрены четыре метода:
метод III — сравнение с эталонными шкалами (для испытания деформированного металла) ;
метод К —подсчет количества -включений (для испытания деформированного и литого металла);
метод П — подсчет количества и содержания включений в объемных процентах (для литого и деформированного металла);
метод Л — линейный подсчет включений (для отливок).
Выбор метода контроля и нормы допустимой загрязненности предусматривается в стандартах и технических условиях на соответствующую металлопродукцию.
Наиболее широко распространен метод Ш,
НОРМЫ ЗАГРЯЗНЕННОСТИ ДЕТАЛЕЙ ПО ВОЛОСОВИНАМ»
Общая площадь контролируемой поверхности детали, CMS Количество допустимых волосовин в стали Максимальная длина волосовин, мм. в стали Суммарная протяженность волосовин, мм, в стали
качественной высококачественной особо высококачественной качественной и высококачественной особо высококачественной качественной высококачественной особо высококачественной
До 50 5 2 1 6 3 10 5 3
Свыше 50—100 6 3 2 7 3 10 8 5
» 100—200 8 4 2 8 4 20 10 6
» 200—300 10 6 3 9 4 30 15 8
» 300—400 11 8 4 10 5 40 20 10
» 400—600 ~ 12 9 5 12 6 60 30 18
» 600—800 13 10 5 14 6 80 40 24
» 800—1000 15 11 6 15 7 100 50 20
♦ Методика и нормы устанавлнзаются по соглашению сторон.
333
имеющий 14 вариантов (от Ш1 до Ш14), различающихся условиями испытаний (увеличение, диаметр поля зрения, способ оценки шлифов и критерии оценки плавок).
В основе всех вариантов лежит сравнение включений на шлифе с эталонными шкалами.
Для контроля на неметаллические включения от плавки стали отбирают не менее 6 образ-
Рис. 18.36. Отбор образцов для контроля на неметаллические включения методом Ш:
а — из круглого и квадратного профиля диаметром или толщиной до 40 мм; б — то же. свыше 40 до 80 мм; в — то же, свыше 80 до 120 мм включительно; J — плоскость реза; 2 — плоскость шлифа
цов — от разных прутков. Прн диаметре нлн толщине прутков не более 120 мм вырезают образцы в поставляемом размере, а при диаметре илн толщине более 120 мм образцы отрезают после перековки темплетов на круг илн квадрат размером 80—120 мм.
Загрязненность включениями по методу Ш контролируют на шлифах с продольным направлением волокна.
Образцы нз прутков вырезают, как показано на рнс. 18.36.
Длину образца выбирают таким образом, чтобы площадь шлнфа была 400±50 мм*. Для небольших прутков (диаметр нлн толщина <10 мм) можно уменьшить площадь шлнфа, ио не менее 200 мм2. Образцы для по-вида, имеющих балл 2 и более.
обработке по режимам, предусмотренным в технических условиях. После термической обработки сошлнфовывают припуск >0,5 мм и приготавливают шлиф. Допускается изготовление шлнфа на двух взаимно перпендикулярных плоскостях образца, вырезанного в виде четверти круга (квадрата). Каждая плоскость считается отдельным шлифом.
Метод Ш. Всю площадь шлнфа просматривают под микроскопом прн увеличении 100 и сравнивают с эталонными шкалами. В зависимости от площади, занятой включениями, эталонные шкалы имеют пять баллов. Площадь включений от балла к баллу увеличивается примерно в два раза.
Пятибалльная шкала классифицирует:
— оксиды строчечные (ОС)—включения мелких частиц, обычно корунда и шпинели, расположенные группами в виде строчек; в шкале приведены две разновидности строчек: нз более мелких кристаллов и более крупных, но в меньшем количестве;
— оксиды точечные (ОТ) — кристаллы простых илн сложных окислов (корунд, шпинель и др.), рассредоточенные по всей плоскости шлифа;
— силикаты хрупкие расположенные
вдоль направления деформации сплошные нлн прерывистые строчки кристаллических включений (силикаты, алюминаты, шпинели н скислы), частично сцементированные пластичными стекловидными силикатами, вытянутыми вдоль направления деформации. Имеются три разновидности эталонов СХ с различной степенью вытяжки н размерами кристаллов;
— силикаты пластичные (СП)—вытянутые вдоль направления деформации пластично деформированные силикаты. В шкале имеются две разновидности СП, различающиеся степенью вытяжки;
— силикаты недеформирующиеся (СП) — недеформирующиеся единичные нли групповые округлые (глобулярные) или неправильной формы включения силикатов, силикатных стекол, алюминатов, отдельные крупные кристаллы корунда, шпинели н др.;
— сульфиды (С)—пластичные, вытянутые вдоль направления волокна, единичные включения нлн группы включений; обычно — это двойной сульфид железа н марганца;
• — нитриды строчечные (НС) — две разновидности строчек: единичная строчка с высокой плотностью включений; несколько коротких строчек, желто-розовых кристаллов и карбонит-рндов титана. По этим же шкалам могут оцениваться бледно-розовые включения нитридов и карбоинтридов инобня;
— нитриды точечные (НТ) — произвольно распределенные по шлифу включения нитридов илн карбонитрндов титана, а также нитридов н карбонитрндов инобня;
— нитриды алюминия (НА) — мелкие кристаллы нитридов алюминия; шкала имеет две разновидности: строчки и произвольно расположенные по шлнфу кристаллы интрнда алюминия.
Загрязненность шлифов оценивается двумя способами: определением для каждого вида включений балла по наиболее загрязненному месту шлнфа (максимальный балл) и подсчетом на шлифе количества включений каждого вышення твердости подвергают термической
334
ТАБЛИЦА 18.4
КРИТЕРИИ ОЦЕНКИ ЗАГРЯЗНЕННОСТИ НЕМЕТАЛЛИЧЕСКИМИ ВКЛЮЧЕНИЯМИ
Варианты метода 111 Диаметр поля зрения, мм Шлиф Плавка
Ш1 0,75—0,85 По наиболее загрязненному месту шлифа (максимальный балл) Средний балл, подсчитанный как среднее арифметическое максимальных оценок каждого образца для каждого вида включений
Ш2 0,75—0,85 То же Средний и максимальный баллы и число образцов с баллом выше максимального, в процентах от общего числа образцов
П13 0,75—0,85 1,1—1,3 > Средний и максимальный баллы и число образцов с максимальным баллом
Ш4 Средний балл, подсчитанный как среднее арифметическое максимальных оценок каж-дого образца для каждого вида включений
Ш5 1,1—1,3 1,1—1,3 Средний и максимальный баллы и число образцов с баллом выше максимального, в процентах от общего числа образцов
Ш6 » Средний н максимальный баллы и число образцов с максимальным баллом
Ш7 0,75—0,85 Число полей зрения с баллом 2 н более по каждому виду включений Число полей зрения с баллом 2 и более раздельно по кислородным, сульфидным и нитридным включениям, отнесенное к площади 10 см2
Ш8 1,1—1.3 То же То же
IT.I9 0,38—0,48 По наиболее загрязненному месту шлифа (максимальный балл) Средний балл, подсчитанный как среднее арифметическое максимальных оценок каждого образца для каждого вида включений
Ш10 0,38—0,48 То же Средний и максимальный баллы и число образцов с максимальным баллом
ши 0,6—0,8 » Средний балл, подсчитанный как среднее арифметическое максимальных оценок каждого образца для каждого вида включений
Ш12 0,6—0,8 » Средний и максимальный баллы н число образцов с максимальным баллом
Ш13 0,38—0,48 Число полей зрения с баллом 2 и более Число полей зрения с баллом 2 и более раздельно по кислородным, сульфидным и нитридным включениям, отнесенное к площади 10 см2
Ш14 0,6—0,8 То же То же
Примечание. Оценка по вариантам 1111—Ш8 проводится при увеличениях SO—ПО, по вариантам Ш9-Ш14— при увеличениях 170—210.
Критерии степени загрязненности отдельных плавок при данном методе оценки шлифа могут быть различными и предусматриваются различными вариантами метода П1 (табл. 18.4).
Варианты Ш1—Ш6 н 1П9—Ш12 основаны иа оценке шлифов максимальным баллом. Этот способ наиболее распространен и используется уже в течение нескольких десятилетий для контроля и исследований. Оценка по наиболее загрязненному месту (максимальным баллом) позволяет довольно быстро просматривать большие площади шлифов н выявлять наиболее крупные включения, встречающиеся в стали довольно редко, но сильно снижающие эксплуатационные свойства изделий.
Способ оценки шлнфа количеством включений в полях зрения с баллом 2 и выше (варианты Ш7, Ш8, Ш13 и Ш14) используется преимущественно и исследовательских целях.
Как видно из табл. 18.4, критерии оценки плавок на основе максима’льиых оценок шлифов учитывают среднюю загрязненность плавки и число образцов с наиболее крупными включениями. Эти критерии объективно отра
жают содержание крупных включений, отрицательно влияющих на служебные свойства.
Метод Ш целесообразно использовать при оценке загрязненности металла, выплавленного в электродуговых н мартеновских печах, а также в конверторах. Он широко применяется для контроля конструкционных, теплоустойчивых, инструментальных, нержавеющих и других сталей.
Оценка загрязненности стали после элек-трошлакового н вакуумно-дугового переплавов также возможна методом Ш, но в этом случае целесообразно просматривать шлнфы прн увеличении 170—210.
Метод К. Число включений, имеющих размеры больше установленного, подсчитывают под микроскопом на иетравленых шлифах с продольным направлением волокна. Шлиф просматривают прн 170—180-кратном увеличении. Цена деления окулярной шкалы* 0,007± ±0,0005 мм. Число кислородных, сульфидных и нитридных включений определяют раздельно по группам размеров:
1-я группа — включения свыше 1 до 2 де-
335
ТАБЛИЦА 18.5
ГРУППИРОВКА ВКЛЮЧЕНИЙ ПО РАЗМЕРАМ В ДЕЛЕНИЯХ ОКУЛЯРНОЙ ШКАЛЫ
Группы включений Средняя значимость для групп по площади включений Размер включения в делениях шкалы Площадь включения, деления окулярной шкалы в квадрате
по диаметру по стороке квадрата
1 0,6—0,7 0,4—0,6 0,18—0,35
2 г/г >0,7—0,9 >0,6—0,8 >0,35—0,7
3 1 >0,9—1,3 >0,8—1,2 >0,7—1,4
4 2 >1,3—1,9 >1,2—1,7 >1,4—2,8
5 4 >1,9—2,7 >1,7—2,4 >2,8—5,6
6 8 >2,7—3,8 >2,4—3,4 >5,6—11,3
7 16 >3,8—5,4 >3,4—4,8 >11,3—22,6
8 32 >5,4-7,6 >4,8—6,7 >22,6—45,1
9 64 >7,6—10,7 >6,7—9,5 >45,1—90,2
10 128 >10,7—15,2 >9,5—13,4 >90,2—180,5
11 256 >15,2—21,4 >13,4—19,0 >180,5—361,0
12 512 >21,4—30,3 >19,0—26,9 >361,0—722,0
13 1024 >30,3—42,9 >26,9—38,0 >722,0—1444,0
Примечание. Группы построены по возрастанию площади включений в геометрической прогрессии, знаменатель 2. ,
лений окулярной шкалы включительно (7— 14 мкм);
2-я группа — включения свыше 2 до 3 делений окулярной шкалы включительно (15— 21 мкм);
3-я группа — включения свыше 3 до 4 делений окулярной шкалы включительно (22— 28 мкм);
4-я группа — включения свыше 4 до 5 делений окулярной шкалы включительно (29— 35 мкм);
5-я группа — включения свыше 5 до 6 делений окулярной шкалы включительно (36— 42 мкм).
Включения размером менее 1 деления (7 мкм) не учитывают. Число групп может быть увеличено в завнснмости от максимальных размеров включений в металле. С помощью окулярной линейки замеряют диаметр или толщину включений, близких в сеченнн по форме к кругу илн квадрату, или максимальный н минимальный размеры включений другой формы. Если отношение максимального размера включений к минимальному не превышает 2, то размер включения определяют как их среднее арифметическое.
Критериями оценки плавки служат: для варианта К1 число включений каждой группы на площади 24 см2 (6 шлифов площадью по 400 мм2 каждый); для варианта К2 — суммарное число включений (начиная со 2-й группы) на площади 24 см2
Учитывая особенности методов Ш н К, можно рекомендовать их для контроля стали разных способов производства [10].
Метод К наиболее удобен для оценки металла после рафинирующих переплавов (электро-шлакового, вакуумио-дугового и электроннолучевого) . Этот метод полезен также при оценке — загрязненности металла глобулярными включениями.
В настоящее время метод К применяют для контроля трубных заготовок из некоторых жаропрочных сталей после электрошлакового переплава и для прецизионных сплавов.
Метод П. Подсчет количества включений с учетом их размеров используется пре
имущественно прн проведении исследовательских работ. Для оценки деформированного металла применяют образцы с поперечным направлением волокна. Размер включений определяют с помощью окулярной шкалы. Каждое включение относят к определенной группе по размеру. В табл. 18.5 приведена группировка включений по размерам в делениях окулярной шкалы.
В зависимости от увеличения, применяемого при оценке, метод П имеет варианты: П1 — Х30О(Х280—300); П2 — Х400(Х400—420); ПЗ—Х500 (Х500—520); П4—ХбОО (ХбОО— 630).
В каждом поле зрения определяют размеры всех или некоторых видов включений в зависимости от цели исследования. Результаты подсчета фиксируются для каждого вида включений отдельно.
На каждом шлифе оценивается не менее 125 полей зрения прн увеличениях 300 и 400 и не менее 375 полей зрения прн увеличениях 500 и 600. Для контроля одной плавки просматривают иа шести шлифах соответственно 750 полей зрения (варианты П1 и П2) и 2250 полей зрения (варианты ПЗ и П4). Размер включения неправильной формы подсчитывают как среднее арифметическое нз его максимального и минимального размеров.
Критерии оценки загрязненности плавки по методу П:
1) содержание включений в процентах по объему;
2) число включений определенных размерных групп иа площади 100 мм2.
Для подсчета площади, занятой включениями иа шлифе, число включений каждой группы умножают на среднее значение площади включения данной группы. Полученные для всех групп произведения суммируют.
Среднюю площадь включения fcp в одном поле зрения вычисляют по формуле fcp=fM» где f — общая площадь включений; п — число полей зрения.
Основываясь на том, что отношение площади, занятой включениями, к общей площади шлнфа равно отношению объема включений к
336
объему стали, из данных измерения площади включений определяют содержание включений в процентах по объему V=fCpK, где К— =100Г-1 — коэффициент; F=nDzl4— площадь поля зрения на шлнфе при выбранном увеличении (в делениях окулярной шкалы в квадрате); D — диаметр поля зрения (в делениях окулярной шкалы); F, D и К—постоянные величины для данного микроскопа и увеличения.
Содержание неметаллических включений в объемных процентах для плавки подсчитывают как среднее арифметическое по данным для всех образцов.
Увеличение зависит от минимального размера включений, учитываемых в проводимой работе.
Прн детальном изучении загрязненности вначале проводят качественную оценку, определяя основные типы включений, встречаю
ТАБЛИЦА 18.6
ОБЛАСТИ ПРИМЕНЕНИЯ РАЗЛИЧНЫХ МЕТОДОВ ОПРЕДЕЛЕНИЯ ВКЛЮЧЕНИЙ (ПО ГОСТ 1778—70)
Вариант метода Вид испытаний Способы производства Группа стали
UI1, Ш2, Ш4, Контрольные Выплавка в электро- Шарнко- и роликоподшипниковые,
Ш5 дуговых, индукционных и иногда в мартеновских печах и конверторах; элек-трошлаковый переплав конструкционные особо ответственного назначения, высокопрочные (с временным сопротивлением в термически обработанном состоянии более 1800 МПа), инструментальные для изготовления измерительных мер и изделий высокой точности, коррозн-онностойкне для ответственных полируемых и вакуумплотных изделий
III3, Ш6, шю, » Электрошлаковый и Конструкционные высокопрочные
Ш12 вакуумно-дуговой переплавы особо ответственного назначения
П11, Ш2, Ш4, Исследова- Выплавка в электро- Стали н сплавы всех марок
Ш5, Ш7, Ш8 тельские дуговых, индукционных, мартеновских печах и конверторах
Ш9, Ш11 Контрольные Вакуумно-индукционная плавка, рафинирующие переплавы Шарике- и роликоподшипниковые, стали для прецизионных подшипников, высокопрочные стали (с временным сопротивлением в термически обработанном состоянии более ,1800 МПа)
Ш13,Ш14 Исследовательские’ То же Стали и сплавы всех марок
К1 Контрольные » Шарнко- и роликоподшипниковые для прецизионных подшипников, заготовки из коррозиониостойкой стали для особо тонкостенных труб
К2 Конструкционные особо ответственного назначения; для изготовления изделий высокого класса точности и чистоты поверхности; инструментальные для измерительных мер и изделий высокого класса точности н чистоты поверхности; коррозиониостой-кие для полируемых изделий высокого класса чистоты поверхности, для вакуумплотной аппаратуры. Прецизионные сплавы в заготовке для микронной проволоки
KI, К2 Исследовательские » Стали и сплавы всех марок
П1, П2 % ПЗ, П4 » » То же
Л1, Л2 » Выплавка в мартеновских, электроду-говых н индукционных печах, конверторах Литье нз углеродистой и легированной конструкционной стали
* Методы П1—П4 могут быть применены для исследовательских испытаний металла, выплавленного в мартеновских и электродуговых печах, а также конверторах. В этом случае число просматриваемых полей зре-ния^леобходимо увеличить в три раза (нли более).
22—280
337
щихся в данном металле. Последующий подсчет по размерным группам производится раздельно по типам включений. Например, если в металле присутствуют коруид, сульфиды и ннтрнды, то по этим типам включений и должен производиться подсчет. Если интересует общее количество кислородных включений, то корунд и силикаты (стекла) можно оценивать вместе.
Метод П рекомендуется для оценки загрязненности металла электрошлакового, вакуумного н электронно-лучевого переплавов. Отсутствие в металле после рафинирующих переплавов крупных включений и скоплений включений делает нецелесообразным применение методики оценки шлифов максимальным баллом. Она, хотя и позволяет выявить отсутствие крупных включений, но не дает четкого представления об общей загрязненности металла.
Метод П применяется и для оценки загрязненности металла мартеновской, электродуго-вой или конверторной стали, ио в этом случае, учитывая неравномерное распределение включений, следует просматривать в 2—3 раза больше полей зрения.
В табл. 18.6 приведены рекомендуемые стандартом области применения различных методов оценки плавок по загрязненности неметаллическими включениями.
В последние годы в металлографических лабораториях заводов и институтов осваиваются автоматические количественные телевизионные микроскопы (КТМ-360, КТМ-720, Эпнквант)1, позволяющие производить подсчет включений по группам размеров и определять долю площади (в процентах), занятую включениями. Эти приборы используют пока только для исследовательских работ и накопления данных о загрязненности стали различных марок н способов производства [11—14]. Преимущество этих приборов — повышение объективности и снижение трудоемкости контроля. Однако прн небольшой площади, просматриваемой на приборах, не всегда учитываются редко встречающиеся крупные включения. Если важно обнаружить такие включения, то целесообразнее использовать метод оценки шлифов максимальным баллом, который дает больше информации, чем автоматические микроскопы.
18.8. ВЕЛИЧИНА
ЗЕРНА
На металлургических заводах контролируют склонность стали к росту зерна аустенита и фактическую (действительную) величину зерна металла в состоянии поставки. В некоторых случаях величина зерна оценивается по излому (мелкозернистый илн крупнозернистый). Такой способ оценки весьма приблизителен и субъективен. Чаще, если величина зерна играет существенную роль, эту характеристику определяют металлографическими методами (ГОСТ 5639—65).
Образцы для определения * величины зерна отбирают от металла в состоянии поставки. Для определения склонности к росту зерна часто используют половинки разбитых ударных образцов, ковшовые пробы, образцы, ото
1 См. раздел 1 Справочника.
бранные на промежуточных стадиях горячей прокатки. Ковшовые пробы (—400 г) проковывают на квадрат 14—15 мм. Металлургический завод гарантирует соответствие результатов испытаний на склонность к росту зерна, выполненных на ковшовой пробе, требованиям, предъявляемым к готовому прокату.
Обычно склонность к росту зерна, определенная иа ковшовых пробах, оказывается несколько меньше, чем в готовом прокате (примерно иа 1 номер).
Для определения величины зерна рекомендуются образцы площадью до 150 мм2.
Склонность к росту зерна аустенита устанавливают при нагреве до определенной температуры. Применяют два стандартных режима нагрева: I — нагрев до 930±10°С, 8 ч; II — нагрев до 930± 10 °C, 3 ч.
Режим I применяют преимущественно для конструкционных цементируемых сталей (определение склонности к росту зерна в процессе цементации), режим II — в остальных случаях. Применяются н другие режимы нагрева, оговоренные в технических условиях.
Металлографические методы определения величины зерна приведены в разделе 1.
18.9. ГЛУБИНА
ОБЕЗУГЛЕРОЖЕННОГО
СЛОЯ
Глубину обезуглероженного слоя на металлургических заводах проверяют в готовой металлопродукции и на промежуточных переделах. Данные, получаемые на промежуточных переделах (горячая деформация, черный отжиг и т. д.), позволяют определять степень обезуглероживания на каждом этапе и подбирать условия последующей обработки, обеспечивающие выполнение требований потребителя к предельно допустимой глубине обезуглероживания. Обычно произвольно отбирают по 3—5 образцов от прутков партии металла нлн нз определенных участков садки (например, сверху и снизу) н др.
ГОСТ 1763—68 предусматривает следующие методы контроля: металлографические (М, Ml, М2); замера термоэлектродвнжущей силы (т. э. д. с.); замера твердости (Т); химический (X). Метод контроля готовой металлопродукции указывают в стандартах и технических условиях. Методика контроля на промежуточных переделах устанавливается заводом — производителем металла.
Сущность металлографических методов — определение глубины обезуглероженного слоя по структуре под микроскопом прн увеличении 63—150. Измерения проводят с помощью окуляр-микрометра с точностью ±0,02 мм.
Различают две зоны: полного ц частичного обезуглероживания.
Зона полного обезуглероживания имеет структуру феррита, зона частичного обезуглероживания — структуру, отличающуюся от основного металла. Общая глубина обезуглероживания складывается нз глубины зоны полного обезуглероживания н глубины зоны частичного обезуглероживания н измеряется по месту, имеющему наибольшую глубину в поперечном сечении от края шлифа до основной структуры металла (рис. 18.37). Различают
338
равномерное обезуглероживание (по всему периметру образца) н местное обезуглероживание (на отдельных участках по периметру образца) .
Метод М — определение глубины обезуглероженного слоя по структуре под микроскопом на поперечных травленых шлифах в состоянии поставки. Образцы вырезают так, чтобы плоскость шлнфа была перпендикулярна исследуемой поверхности. Схемы вырезки образцов представлены на рнс. 18.38.
Рис. 18.37. Схема замера глубины обезуглероженного слоя
Рис. 18.38. Схемы вырезки образцов для определения глубины обезуглероживания (метод М) от проката различных профилей:
а — диаметр илн толщина до 80 мм — все сечение; б — диаметр или толщина свыше 30 до 60 мм — половина сечения; в — диаметр или толщина свыше 60 до 100 мм — четвертая часть сечения; г — диаметр или толщина свыше 100 до 150 мм — шестая часть периметра круга или угол квадрата
Рекомендуемая площадь шлнфа — до 10 см2.
Травление проводят в реактивах, позволяющих отчетливо выявить структурные составляющие (2—4%-ный раствор азотной или пикриновой кислоты в этиловом спирте и др.).
Зона частичного обезуглероживания характеризуется: для доэвтектоидной стали феррито-перлитной структурой (содержание феррита больше, чем в основной структуре); для заэвтектондной стали — феррито-перлитной структурой или структурой пластинчатого перлита при основной структуре зернистого перлита.
У стали со структурой зернистого перлита зона частичного обезуглероживания обеднена карбидами по сравнению с основной структурой.
Метод М имеет ряд преимуществ: простота подготовки образцов, высокая точность оценки, контроль металла непосредственно и состоянии поставки.
Метод М применяется для сдаточного и промежуточного контроля углеродистых н легированных инструментальных сталей, шарнко- н роликоподшипниковых сталей, конструкционных углеродистых и легированных сталей (с содержанием ^0,3 % С), рессорно-пружинных и др.
Метод Ml не приводится, так как используется только в исследовательских работах для заэвтектондных сталей.
Метод М2 (метод Садовского) применяется для оценки глубины обезуглероженного слоя в быстрорежущих сталях по структуре под микроскопом (на поперечных травленых шлифах образцов, прошедших специальную термическую обработку).
Схема отбора образцов представлена иа рнс. 18.39. Образцы подвергают специальной
Рис. 18.39. Отбор образцов для определения глубины обезуглероживания методом М2:
с — прутки диаметром до 25 мм полное сечение (лыска 2—3 мм снимается до полного удаления обезуглероженного слоя); б — прутки диаметром более 25 до 40 мм — из половины сечения прутка; в — прутки диаметром более 40 мм
термической обработке, в результате в слое, обедненном углеродом, и в объеме металла формируются различные структуры. Рекомендуемые режимы термической обработки приведены в табл. 18.7. После нагрева до 820— 840 °C образцы переносят в раскисленную хлорбарцевую ванну и выдерживают 1—3 мин, затем охлаждают 10 мин в масляной или соляной ванне (первая ванна охлаждения), затем иа 10 мнн переносят в соляную нли евнн-цовую ванну (вторая ванна отпуска) и, наконец, охлаждают на воздухе. Объем ванны должен быть достаточным для поддержания температуры в заданных пределах. Травленый
22*
ТАБЛИЦА 18.7
РЕЖИМЫ ТЕРМИЧЕСКОЙ ОБРАБОТКИ ОБРАЗЦОВ НЕКОТОРЫХ МАРОК
БЫСТРОРЕЖУЩЕЙ СТАЛИ*
Марка стали Температура закалки (хлорбариевая ванна), °C Температура первой ванны охлаждения, °C
Р9 1220—1240 160—180
Р12 1240—1260 175—195
Р18 1270—1290 175—195
Р6МЗ 1210—1230 176—195
Р9Ф5 1230—1250 190—210
Р14Ф4 1240—1260 200—220
Р18Ф2 1270—1260 180—200
Р9К5 1220—1240 160—180
Р9КЮ 1220—1240 160—180
Р10К5Ф5 1230—1250 160—200
Р18К5Ф2 ' 1270—1280 180—200
* Температура второй ванны (отпуска) Б80—600 °C.
шлнф просматривают под микроскопом и измеряют глубину обезуглероженного слоя. Если термическая обработка не приводит к дополнительному обезуглероживанию по заточенной площадке' (лыске), то структура поверхностного н глубинных слоев одинакова. Зона полного обезуглероживания у поверхности характеризуется структурой феррита, а зона частичного обезуглероживания — темнотравящей-ся троостнтной нлн игольчатой троосто-мартеи-ситной структурой.
Метод М2 очень сложен, а результаты оценки сильно зависят от колебаний температуры в первой и второй ваннах, точность метода не велика, поэтому его часто заменяют методом т. э. д. с.
Метод т. э. д. с. Замер термоэлектродвижущей силы на обезуглероженной и необез-углероженной поверхностях образца.
Метод применяется для всех сталей. Особенно часто используется при контроле шлифованной стали, у которой обезуглероженный слой не допускается, нлн для прутков диаметром до 30 мм, когда допускается незначительная глубина обезуглероживания. В случае сталей, в которых обезуглероживание не допускается, с одной стороны образца (абразивным кругом илн другим способом) готовят площадку (лыску) длниой 10—15 мм, позволяющую удалять обезуглероженный слой.
В случае прутков, на которых допускается обезуглероживание, обезуглероженный слой снимают, а .затем готовят образец.
Схема установки для определения т. э. д. с. приведена иа рнс. 18.40. В установку входят: амперметр, автотрансфррматор и микроамперметр 4. Температура стержня 1 поддерживается в интервале 150—160° С. Установку градуируют по эталонам — образцам, в которых заранее металлографическим методом определено наличие илн отсутствие обезуглероживании. Для стали данной марки устанавливают разницу в показаниях прибора для обезуглероженного н необезуглероженного образцов.
Образец помещают на медное основание
прибора 2 так, чтобы лыска находилась точно под рабочей частью нагретого стержня /. По результатам 3—5 замеров иа лыске подсчитывают среднюю величину показаний. Такие нее замеры (5—10) производят в различных точках поверхности образца (вне лыски) и подсчитывают среднее значение. Образец считается необезуглероженным, если разница в показаниях прибора на лыске н на поверхности образца не превышает величины, установленной по эталонам.
Метод замера т. э. д. с. применяется для всех марок стали (обычно в качестве внутризаводского). В случае контроля готовой продукции
Рис. 18.40. Схема установки для определения глубины обезуглероживания методом т. э. д. с:
/ — нагреваемый медный стержень; 2 — медное основание; 3 — исследуемый образец; 4 — измерительный прибор
этим методом поставщик гарантирует соответствие результатов контроля нормам, установленным для методов М илн М2.
Наибольшее распространение метод получил для контроля шлифованной быстрорежущей стали взамен более сложного метода М2 (Садовского).
Метод замера твердости (Т) — измерение твердости образцов после термической обработки. Применяется для прутков нз шлифованной и калиброванной стали диаметром не более 30 мм. Образцы длиной 20— 50 мм подвергают термической обработке (в условиях, исключающих возможность дополнительного обезуглероживания) по режимам, установленным стандартами илн техническими условиями. Твердость (по Роквеллу) замеряют иа поверхности образцов для продукции, в которой не допускается обезуглероживание н после снятия допуска иа обезуглероживание для продукции, на которой обезуглероживание допускается. Образец считают необезуглероженным, если его твердость соответствует нормам для данной марки стали.
Метод Т можно использовать для всех сталей, но обычно его применяют только для калиброванной шарнко- и роликоподшипниковой стали.
Химический метод (X) — определение содержания углерода в стружке, снятой послойно с поверхности образца. Образцы отбирают от готового металла. Длина образцов должна обеспечивать достаточное для анализа количества стружкн, которую снимают слоями толщиной не более 0,1 мм. Содержание углерода в каждом слое определяют до получения химического состава, соответствующего марочному. Образец считается необезуглероженным,
340
если содержание углерода в первом слое соответствует марочному. Глубина обезуглероживания определяется по глубине снятой стружки до получения марочного состава.
Метод применяется только выборочно прн проверке других методов нлн проведении исследований. '
В заводских условиях глубину обезуглероженного слоя иногда контролируют визуально по излому (см. раздел 1). Этот способ используется для отожженных прутков (до 30 мм) нз углеродистой и легированной инструментальной стали. На прутке делают зарубку, по которой отламывают его конец длиной 30— 50 мм. Прн наличии обезуглероженного слоя в изломе видна кайма блестящих, обычно более крупных зерен. Появление каймы прн отжиге заэвтектоидных сталей связанно с тем, что обезуглероженный слой перегревается (из-за отсутствия избыточных карбидов) н в нем при охлаждении образуется крупнопластннча-тый перлит. Хрупкий излом проходит по границам зерен аустенита нлн перлитных колоний, вязкая мелкокристаллическая сердцевина имеет структуру зернистого перлита.
18.10. МИКРОСТРУКТУРА СТАЛИ
Ряд сталей в состоянии поставки контролируется на микроструктуру. Контроль осуществляется иа образцах готовой стали после дополнительной термической обработки (закалки с отпуском). Ниже приводятся контролируемые характеристики микроструктуры.
Структура перлита в отожженной стали. После отжига сталь приобретает структуру зернистого перлнта. Зерна карбидов нлн цементита могут различаться по размерам: от дисперсных (точечных) до относительно крупных, (4—5 мкм). Иногда в зернистой структуре перлнта встречаются участки пластинчатого нлн отдельные пластникн. Возникновение пластинчатого перлита связано с отклонениями от оптимального режима отжига. Для некоторых сталей (например, углеродистой эвтектоидной) получение однородной структуры зернистого перлита (без пластинок) требует применения сложных режимов термической обработки.
Тонкие, плотно прилегающие друг к другу пластинки перлнта н мелкие карбиды свидетельствуют о недогреве стали при отжиге, более грубые разобщенные пластинки — о перегреве. Структуру перлита контролируют обычно иа поперечных шлифах.
Шлнфы приготовляют для прутков размером до 25 мм по всему поперечному сечению; 26—40 мм — иа половине сечення; 41—60 мм — на четверти сечення. Шлнфы травят в 3—4%-иом растворе азотной кислоты, в этиловом спирте.
Контроль проводят прн увеличении 500. В ГОСТ 1435—74 для углеродистой инструментальной стали приводится шкала оценки перлита нз 10 баллов (рис. 18. 41). Баллы 1 и 2—структура недогретой при отжиге стали. Основа структуры балла 1— пластинчатый перлит. Балл 2 — смешанная структура зернистого и пластинчатого перлита. Баллы 3—7 — структура нормально отожженной стали с постепен
но увеличивающимися размерами цементита. В структуре баллов 8—10 — пластинчатый перлит, что характерно для перегретой стали. Балл 8 — небольшое количество пластинчатого перлита, балл. 10 — преобладание пластинчатого перлита.
В ГОСТ 5950—73 для инструментальной легированной стали приводятся шкала оценки перлита, немного отличающаяся от рассмотренной выше.
По стандарту для шарнко- и роликоподшипниковой стали структура перлнта оценивается по 10-балльной шкале, причем эталоны баллов 1—6 — это разновидности зернистой структуры с увеличивающимся размером карбидов, а эталоны 7—10 — смешанная структура зернистого и пластинчатого перлита. С изменением балла от 7 до 10 меняются количество и степень огрубления пластинчатого перлита.
Карбидная неоднородность. Представляет собой скопления эвтектических карбидов, образующихся нз расплава при кристаллизации последних порций стали, обогащенных углеродом и легирующими элементами.
В литом металле карбиды выглядят как эвтектические участки или сетка. В процессе нагрева и горячей деформации прн переделе слитков на заготовку и сортовой прокат карбидные скопления н сетка дробятся н превращаются в строчки, вытянутые вдоль направления деформации. Чем выше степень деформации, тем больше дробятся карбидные скопления.
Наиболее сильно карбидная неоднородность проявляется в сталях ледебуритного класса и значительно меньше — в заэвтектоидных сталях (ГОСТ 19265—73 и ГОСТ 801—78).
В быстрорежущей стали (ГОСТ 19265—73) карбидная неоднородность — элемент структуры, степень ее развития определяется по эталонным шкалам. Для оценки карбидной неоднородности от готового сортового проката отрезают шайбы толщиной 10—12 мм. Из них готовят продольные шлифы, проходящие через центр. Образцы закаливают (по режиму, предусмотренному стандартом для стали данной марки) и отпускают прн 680—700° С, 1 ч. В быстрорежущей стали карбидная неоднородность оценивается по трем шкалам при увеличении 100: для прутков из сталей с высоким содержанием вольфрама (PI2, Р18, Р18Ф2, Р14Ф4, Р18К5Ф2, Р10К5Ф5, Р9КЮ, Р9М4К8) по шкале № 1; для прутков ннзковольфрамо-вой и вольфрамомолнбденовой сталей (Р6МЗ, Р6М5, Р9К5, Р6М5К5) по шкале № 2. Характеристика шкал приведена в табл. 18.8. Карбидная неоднородность шайб оценивается по шкале № 3. Шкалы построены по принципу увеличения балла: Д— с уменьшением степени вытяжки и раздробленности карбидных скоплений; Б — с увеличением площади, занятой строчками карбидов. Считается, что качество быстрорежущей стали улучшается с уменьшением балла карбидной неоднородности.
Карбидная неоднородность в инструментальной легированной стали определяется прн увеличении 100 по двум шкалам (табл. 18.9). Шкала № 2 используется для оценки сталей ледебуритного класса (XI2, XI2M, XI2BM, Х12Ф1), шкала № 3 — для оценки сталей с меньшим содержанием углерода (ХВ4. 9Х5ВФ, 8Х6НФТ, Х6ВФ, 6Х6ВЗМФС, 8Х4ВЗМЗФ2). Карбидная неоднородность (карбидная ликвация) сталей 11ХФ, 13Х, 9Х,
341
Рис. 18.41. Шкала оценки структуры перлита б углеродистой инструментальной стали. Х500
ТАБЛИЦА 18.8
КАРБИДНАЯ НЕОДНОРОДНОСТЬ СОРТОВОЙ БЫСТРОРЕЖУЩЕЙ СТАЛИ
* СО сз Описание карбидной фазы Допустимая ширина полос или скоплений карбидов, мм. по шкале
№ 1 № 2
1 Тонкая полосчатая-структура с короткими разорванными полосами 2 1
2 Полосчатая структура, единичные нли множественные полосы. Карбиды внутри полос раздроблены 4 2
3 А. Полосчатая структура, единичные или множественные полосы. Карбиды внутри полос раздроблены 6 4
Б. Остатки карбидной сетки. Сетка с раздробленными карбидами 6 4
4 А. Полосчатая структура. Единичные илн множественные полосы. Карбиды внутри полос раздробленные 8 5
Б. Слабо выраженная разорванная сетка со скоплениями 8 5
5 А. Грубополосчатая структура, единичные илн множественные полосы 13 6
Б. Отчетливо выраженная разорванная сетка со скоплениями 13 6
6 А. Грубополосчатая структура, единичные или множественные полосы 15 9
Б. Явно выраженная разорванная сетка с малоразо-рванньши ячейками, скоплении 15 9
7 Сетка, разорванная в отдельных местах, н скопления 20 14
8 Малодеформиров энная разорванная сетка н скопления 25 18
Примечание. За полосу принимается скопление карбидов, длина которого больше или равна пятикратной ширине. Две лежащие рядом полосы принимаются за одну, если расстояние между ними меньше ширины более узкой полосы. По шкале № 1 оцениваются высоковольфрамовые сплавы, по шкале № 2 — низковольфрамовые и вольфрамомолибде-новые.
Рис. 18.42. Карбидная ликвация в стали ШХ15 (балл 3). X100
X, 12X1, 9ХС, В2Ф, ХГС, 9ХВГ, ХВГ, ХВСГ определяется по шкале 6А ГОСТ 8233- -56 (по требованию потребителя).
Карбидная неоднородность (карбидная ликвация) сталей ШХ15 и ШХ15СГ (рис. 18.42) оценивается при увеличении 100 по шкале ГОСТ 801—78. Для оценки обычно используют те же образцы, что н для определения неметаллических включений, но обязательно закаленные. Шкала 5-балльная н нмеет две раз-
Рис. 18.43. Структурная полосчатость стали ШХ15. Х100:
а — небольшая; б — значительная
343
ТАБЛИЦА 18.9
ОПИСАНИЕ КАРБИДНОЙ НЕОДНОРОДНОСТИ (ГОСТ 5950-73)
Баллы Шкала № 2 Шкала Xs 3
1 Равномерное распределение карбидов Равномерное распределение карбидов
2 Слабо выраженная полосчатость, тонкие строчки карбидов Слабо выраженная полосчатость
3 Строчечное расположение карбидов Полосчатость
4 Резко выраженная полосчатость, грубые строчки карбидов Резко выраженная полосчатость
5 Значительно деформированная, местами разорванная сетка карбидов Резко выраженная полосчатость со скоплениями
6 Деформированная сетка эвтектических карбидов Резко выраженная полосчатость со скоплениями, сильно деформированная разорванная сетка эвтектических карбидов
7 Сплошная деформированная сетка карбидов с участками эвтектики Деформированная сетка эвтектических карбидов, разорваияая в отдельных местах
8 Слабо деформированная сетка карбидов с участками эвтектики Сплошная деформированная сетка эвтектических карбидов
9 Слабо деформированная сетка с грубой карбидной эвтектикой Сплошная деформированная сетка со скоплениями карбидов
10 Структура, соответствующая литой стали Структура, соответствующая литой стали
Примечание. Шкала имеет два ряда эталонов микроструктур. Верхний ряд микроструктур предназначен для оценки карбидной неоднородности стали на образцах после закалки и отпуска, нижний — для оценки карбидной неоднородности отожженных образцов стали.
новндностн структуры, для каждого балла. С увеличением балла размер и число строчек эвтектических карбидов возрастают.
Структурная полосчатость. У сталей ШХ15 и ШХ15СГ представляет чередующиеся полосы несколько отличающейся структуры. Наблюдается в деформированном металле (рнс. 18. 43). Полосы образуются из осей и междуосных участков дендритов, имеющих различный состав. Междуосные участки и лнквацнонные полосы имеют более высокое содержание углерода, хрома и марганца. После закалки и травления иа продольных шлифах такие участки выглядят как полосы различной травимостн (в связи с различием структур). В зависимости от условий закалки (температура, скорость охлаждения) формируются различные структуры: а — в полосах, обогащенных углеродом и хромом, аустенитизация с достаточным растворением карбидов не проходит, поэтому в связи с недостатком в растворе легирующего элемента образуется троосто-мартеиснтная структура, а и полосах с меньшим содержанием углерода и хрома — структура мартенсита; б — в полосах, обогащенных углеродом и хромом, образуется мелкозернистый (бесструктурный) мартенсит с избыточными карбидами, в полосах, обедненных этими элементами, — крупноигольчатый мартенсит; в — в полосах, обогащенных углеродом н хромом, наблюдается структура мартенсита с карбидами, в обедненных — мартенсит с трооститом.
Неоднородность структуры сопровождается неравномерным распределением твердости, что нежелательно для подшипников. Структурная полосчатость оценивается по шкале, в которой с увеличением балла повышаются размеры и контрастность полос.
Карбидная сетка. В заэвтектондиых сталях избыточные карбиды выделяются в виде сетки
по границам зерен аустенита. Процесс проходит прн охлаждении стали с температуры конца горячей деформации до температуры Агг (образование перлнта). Карбидная сетка повышает хрупкость металла. При отжиге сетка частично рассасывается н сфероидизируется [15]. Остатки карбидной сетки в структуре регламентируются. Контролю подвергают углеродистые и легированные инструментальные, а также ролнко- н шарикоподшипниковые стали (ШХ15, П1Х15СГ). Контроль производят на поперечных шлифах для инструментальной стали и иа продольных для подшипниковых сталей. Образцы закаливают с температуры, принятой для данной марки стали.
Шлифы травят в 2—4 %-ном растворе азотной кислоты в этиловом спирте до потемнения, чтобы светлая сетка избыточных карбидов отчетливо выделялась иа темном фоне. Остатки скоагулнрованной сетки оценивают по 5-нлн 6-балльиой шкале (ГОСТ 5950—73, ГОСТ 1435—74)) и по шкале стандарта иа шарнко-н роликоподшипниковую сталь (ГОСТ 801—78).
Структурно-свободный цементит. Сталь тонколистовая холоднокатаная малоуглеродистая качественная для холодной штамповки (ГОСТ 9045—80) контролируется по микроструктуре иа структурно-свободный цементит (ГОСТ 5640—68). Из листа вырезают образцы по 2 от листа: один с края, другой из середины для приготовления продольных шлифов. Шлифы травят в 4 %-ном растворе азотной кислоты в спирте. Структуру оценивают прн увеличениях 360—400. Шкала эталонов построена в зависимости от количества, формы н расположения цементнтных частиц и включает трн ряда и шесть баллов. Ряд А построен по принципу образования цементит-ной сетки по границам зерен, ряд Б — по воз-
344
ТАБЛИЦА 18.10 ”
ОПИСАНИЕ МИКРОСТРУКТУРЫ СТРУКТУРНО-СВОБОДНОГО ЦЕМЕНТИТА ПО ШКАЛЕ № !
| Йомер балла 1 А в В
0 Равномерно распределенная точечная илн мелкоглобулярная сыпь из це-ментнтных частиц размером 1—2 мм Точечная нлн мелкоглобулярная сыпь нз цемеи-тнтных частиц, имеющих тенденцию к образованию однослойных цепочек Точечная или мелкоглобулярная сыпь, равномерно распределенная по полю шлнфа и имеющая некоторую ориентировку в направлении деформации
1 Неравноосные включения цементита размером 5 мм, расположенные равномерно в объеме зерен и их стыках Частицы цементита размером 1—2 мм, образующие однослойные цепочки Мелкоглобулярная сыпь с частицами размером 1—2 мм, ориентированная в направлении деформации
2 Относительно равномерно распределенные частицы цементита размерами >5 мм, имеющие склонность к залеганию в виде сетки по границам зерен и охватывающие не более */б периметра зерна феррита Однослойные н двухслойные цепочки из частиц цементита размерами до 3 мм Небольшие скопления частиц цементита размерами 1—2 мм, ориентированные в направлении деформации
3 Включения цементита, залегающие в виде сеткн по границам зерен и охватывающие до ‘/з периметра зерна феррита Однослойные и двухслойные цепочки из частиц размером до 5 мм Глобулярные частицы размером 2—3 мм в виде скоплений и разорванных полос, вытянутых в направлении деформации
4 Включения цементита, залегающие в виде сетки по границам зерен и охватывающие :С2/з периметра зерна феррита Двухслойные и трехслойные цепочки, проходящие через все поле зрения и состоящие из частиц цементита размерами более 5 мм Структура соответствует баллу 4 ряда Б
5 Включения цементита, образующие сплошную или почти сплошную сетку по границам зерен феррита Широкие многослойные цепочки, проходящие через все поле зрения и состоящие нз крупных (более 5 мм) частиц Структура соответствует баллу 5.ряда Б
растанию размеров частиц структурно-свободного цементита, образующих однослойные, двухслойные н многослойные цепочки различной протяженности, ряд В — по принципу перехода равномерно распределенной точечной сыпи в неравномерную полосчатую структуру. В табл. 18.10 приведено описание структур по баллам шкалы. Большое количество структурно-свободного цементита, особенно в виде сетки илн крупных частиц, понижает пластичность при штамповке.
18.11. АЛЬФА-ФАЗА
В НЕРЖАВЕЮЩЕЙ СТАЛИ
В структуре аустеннто-ферритной нержавеющей стали тнпа 12Х18Н10Т (ГОСТ 5949—75) присутствует некоторое количество а-фазы. Ее тем больше, чем выше концентрация фер-рнтообразующнх элементов (Cr, Ti, Si) и ниже концентрация аустеннтообразующих элементов (С, Ni).
Присутствие значительных количеств а-фа-зы снижает пластичность стали прн горячей деформации (особенно прн прошивке труб). В связи с этим (обычно для трубиых загото
вок) устанавливают нормы, ограничивающие содержание а-фазы.
Определение содержания a-фазы по ГОСТ 11878—66 предусматривает два метода: металлографический н магнитный. Стандарт распространяется на прутки размерами от 80 до 270 мм.
При применении металлографического метода a-фазу определяют на продольных шлифах в плоскости осн прутка от центра до середины раднуса (рис. 18.44). Контролируется не менее двух образцов от каждой плавки. Длина образцов в направлении осн прутка 2>10— 12 мм (припуск на шлифовку 0,5 мм). Шлиф подвергают электролитическому нлн химическому травлению. Электролитическое травление осуществляют в 10 %-ном водном растворе щавелевой кислоты прн комнатной температуре, плотность тока 0,03—0,08 А/см2, продолжительность травления 20—40 с. Реактив для химического травления: 20 мл воды, 20 мл соляной кислоты н 4 г медного купороса; температура комнатная; продолжительность 8— 10 с.
Оценка содержания a-фазы производится под микроскопом при увеличениях 280—320 (диаметр поля зрения 0,38—0,43 мм). Прн йро-смотре шлнфа определяют место с наибольшим
345
содержанием a-фазы и оценивают его, сравнивая с эталонами пятибалльной шкалы. Площадь, занимаемая a-фазой на фотоэталонах, возрастает от балла к баллу в геометрической прогрессии с коэффициентом 2.
Нормы содержания a-фазы устанавливаются стандартами и техническими условиями.
Рис. 18.44. Отбор образцов для определения а-фазы в нержавеющей стали
Определение содержания a-фазы магнитным методом производят иа поперечных темплетах высотой ^10 мм с помощью фазометров или ферритометров, проградуированных по эталонам, оценённым металлографическим способом. По диаметру прутка проводят ие менее 40 замеров. В зоне с максимальным содержанием a-фазы проводят ие менее 20 замеров. Содержание a-фазы на шлифе определяют как среднее арифметическое нз трех максимальных показаний прибора с последующим выражением его в баллах шкалы илн процентах.
Плавку оценивают: а) по максимальному значению оценок двух образцов; б) по среднему значению оценок двух нлн более образцов. Для каждого прибора строят градуировочную кривую в координатах показание прибора — балл а-фазы по данным не менее 10 измерений иа разных образцах для каждой из 5—6 точек. Сравнение металлографического и магнитного методов определения содержания a-фазы проведено в работе [16].
18.12. ЧУВСТВИТЕЛЬНОСТЬ
К ЗАКАЛКЕ
Углеродистую инструментальную сталь (ГОСТ 1435—74) проверяют на прокалнваемость. С этой целью прн разливке отливают контрольный слиток (~10 кг), который проковывают на квадрат 20+1,5 мм. Из средней части прутка вырубают три образца по 100 мм, иа которые наносят клеймо — иомер плавки и порядковый номер. Посередине каждого образца делают надрез глубиной 3—5 мм. Образцы отжигают прн 730—750 °C с выдержкой 2 ч, затем охлаждают с печью до 650 °C, 2—4 ч. Квадратные образцы вырезают нз сортового проката после отжига.
Испытание на прокалнваемость проводят и на круглых образцах (диаметр 21—23 мм, глубина надреза 5—7 мм).
Отожженные образцы зачищают от окалины (абразивами нлн травлением) и закаливают в воде с трех температур: 760 °C; 800 (вы
держка 20 мин) и 840 °C (выдержка 15 мин). После закалки образцы ломают на копре или под прессом. Прн осмотре изломов определяют глубину закаленного слоя, наличие перегрева нли трещин, а также иезакаленные изломы. По шкале (рис. 18.45) устанавливается груп-
Овозначения:
Незакаленныи.
О Вязкая сердцевина
Сквозная прокачиваемость
Трещины
Рис. 18.45. Шкала изломов закаленных образцов инструментальной углеродистой стали
па (балл) по глубине прокалнваемостн в миллиметрах (обозначена над изломами образцов, закаленных с 800 °C).
18.13. ИСПЫТАНИЯ
НА высокотемпературную ПЛАСТИЧНОСТЬ [9, 17, 18, 33]
Проба на ковкость — наиболее простой метод определения пластичности стали. Пробы массой 0,5—1 кг отбирают в конце плавки илн прн разливке стали и непосредственно после крнсталлизацнн подвергают ковке до получения квадратного (10—15 мм) стержня, который загибают на 180° до соприкосновения сторон. Если прн ковке и гибке на углах и гранях образца не появляется надрывов, то ковкость считается хорошей, прн незначительных надрывах — удовлетворительной, прн больших рванинах илн разрушении — неудовлетворительной. Отбор и контроль пробы в конце плавки или по ходу ее позволяют регулировать процесс раскнслеиия для получения наиболее высокой пластичности. Прн хорошей ковкости можно назначать плавку на прокатку, а прн удовлетворительной — на ковку нли прессование. Проба на ковкость позволяет сделать предварительные выводы о пригодности плавки к горячей деформации, но не дает возможности определить пригодность стали для прошивки труб, т. е. для деформации в условиях, требующих высокой пластичности.
Пробная деформация (прокатка, ковка) одного или двух слитков плавки выявляет воз
346
можность применения данного вида горячей деформации к конкретной марке нлн плавке стали (при оптимальном режиме иагрева слитков), но не позволяет оценить пригодность металла для прошивки труб. Оценка степени пластичности, необходимой для прошивки труб, проводится в процессе испытаний на кручение при высоких температурах (см. ниже).
Испытание на ударную вязкость при высоких температурах проводят для оценки пластичности прн этих температурах. Образцы для испытаний изготовляют из прокованных на квадрат 12 мм ковшовых проб, вырезают нз литого илн деформированного металла. Стандартные образцы типа I (ГОСТ 9454—78) перед испытанием на копре нагревают до 900, 1000, 1100, 1200, 1250 °C. По значению ударной вязкости примерно определяют вид последующей обработки слнтков илн заготовок. Прокатка слнтков может быть удовлетворительной, если при 900—1250 °C KCU^s 5*1000 кДж/м2, ковка, если KCU 5= 600 кДж/м2. Прессование возможно н при меньшнх значениях ударной вязкости, но не менее 400 кДж/м2.
Прокатка образцов на клин. Для этого испытания от двух или четырех слнтков квадратного сечения (35X35 мм или 45X45 мм, высота 850—900 мм) илн нз предварительно обжатого металла отрезают образцы длиной 200—250 мм, которые нагревают до 900, 1000, 1100, 1200 °C. Нагретые образцы прокатывают на клин в валках переменного сечення. Обжатне (отношение разности высот до н после прокатки к первоначальной высоте образца) меняется по длине клина от 0 до 0,8. При осмотре поверхности клнна определяют обжатне, при котором появляются рваннны иа металле. Такое обжатне называется пределом пластичности стали прн данной температуре. Металл высокой ковкости (прокатываемостн) имеет предел пластичности 5*5,6 (1-й класс), предел пластичности гС0,2 (5-й класс) характеризует нековкий металл, а промежуточные значения (2—4-й классы) — пониженную ковкость.
Кручение при высоких температурах. Метод отличается от других испытаний высокой скоростью деформации н тем, что пластичность определяется в направлении, перпендикулярном направлению волокна. Образцы для испытаний изготовляют из литого н деформированного металла. Методика испытаний приведена в ГОСТ 3565—58. Пластичность металла определяется числом оборотов до разрушения образца. Метод применяется для испытаний металла, идущего на изготовление бесшовных труб, преимущественно нз нержавеющих н жаропрочных сталей. Испытания на крученне широко используют при разработке новых сталей нли технологических процессов.
Растяжение при высоких температурах. Пластичность прн высоких температурах можно определить по относительному удлинению н относительному сужению поперечного сечення образцов при испытаниях на растяжение прн относительно небольших скоростях деформации прн 900—1300 °C.
Испытание на осадку. Проводится обычно на сернн цилиндрических образцов (отношение высоты к диаметру 2 :1 нлн 3:1). Образцы нагревают до 900—1300 °C н осаживают (под молотом нли прессом) на Уз первона
чальной высоты. О пластичности судят по появлению нлн отсутствию разрывов на поверхности образцов. В некоторых случаях предельную величину осадки, прн которой отсутствуют разрывы, определяют, изменяя силу удара молота (копра) или высоту перемещения пресса.
Описанные выше методы определения пластичности прн высоких температурах применяются прн внутризаводских испытаниях, при разработке новых технологических процессов илн сталей, режимов иагрева перед горячей деформацией, а также прн выборе вида деформации. В отдельных случаях такие испытания предусматриваются техническими условиями на заготовки, используемые для горячей механической обработки.
БИБЛИОГРАФИЧЕСКИЙ СПИСОК
1. Сперанский В. Г., Забалуев И. П. Контроль качества электростали. М.: Металлургия, 1964. 200 с.
2. Гурин В. Г. Технический контроль на металлургическом заводе. М.; Металлургиз-дат, 1956. 390 с.
3. Фастовский Б. Г. Огневая зачистка стали. М.: Металлургия, 1975. 224 с.
4. Калинина 3. М. Дефекты легированных сталей. Свердловск: Металлургиздат, 1960. 247 с.
5. Отделка сортового проката/Шефтель Н. И., Мурзин М. И., Аршавский В. 3. н др. М.: Металлургия, 1974. 407 с.
6. Голиков И. И. Дендритная ликвация в стали. М.: Металлургиздат, 1958. 206 с.
7. Поволоцкий Д. Л, Морозов А. Н. Водород н флокены в стали. М.: Металлургиздат, 1959. 183 с.
8. Виноград М. И. — Сталь, 1953, № 9, с. 827—828.
9. Дзугутов М. Д. Пластическая деформация высоколегированных сталей н сплавов. М.: Металлургия, 1977. 480 с.
10. Виноград М. И., Громова Г. П. Включения в легированных сталях н сплавах. М.: Металлургия, 1972. 216 с.
11. Павперова И. А., Виноград М. И., Киселева С. А. — Новые методы испытаний металлов: Сб. № 2. М_: Металлургия, 1973 (МЧМ СССР), с. 66—70.
12. Павперова И. А., Пикина О. Т. — Новые методы испытаний металлов: С б. № 4. М_: Металлургия, 1977 (МЧМ СССР), с. 131— 137.
13. Павперова И. А., Проклова А. Н. — Новые методы испытаний металлов: Сб. № 5. М.: Металлургия, 1977 (МЧМ СССР), с. 7—10.
14. Марченко В. Н., Литвиненко Д. А., Морга-лев В. Н., Павперова И. А. — Сталь, 1977, № 7, с. 655—658.
15. Комиссаров А. И., Хорош В. А., Худеньких А. А. —- Сталь, 1966, № 4, с. 359—360.
16. Киссина Л. Б., Потапов В. Д. — Сталь, 1969, № 2, с. 170—173.
17. Пластичность стали прн высоких темпера-турзх/Зуев М. И., Култыгин В. С., Виноград М. И. н др. М.: Металлургиздат, 1954. 101 с.
18. Чашников Д. И. Деформируемость судостроительных сталей при обработке давлением. Л.: Судостроение, 1974. 136 с.
19. Включения н газы в сталях н сплавах-
!Явойский В. И., Близнюков С. А., В ши каров А. Ф. н др. М.: Металлургия, 1979. 272 с.
20. Атлас дефектов стали. М.: Металлургия, 1979. 188 с.
21. Голиков И. Н., Масленков С. Б. Дендритная ликвация в сталях и сплавах. М.: Металлургия, 1977. 224 с.
22. Колосов М. И., Строганов А. И., Смирнов В. Д., Охримович Б. П. Качество слитка спокойной стали. М.: Металлургия, 1973. 408 с.
23. Сергеев А. Б., Швед Ф. И., Тулин И. А. Вакуумно-дуговой переплав конструкционной стали. М.: Металлургия, 1974. 192 с.
24. Ефимов В. А. Разливка и кристаллизация стали. М.: Металлургия, 1976. 552 с.
25. Волков С. Е., Волков А. Е., Забалу-ев Ю. И., Бураковский Г. А. Неметаллические включения и дефекты в электрошла-ковом слитке. М.: Металлургия, 1979. 136 с.
26. Бородулин Г. М., Мошкевич Е. И. Нержавеющая сталь. М.; Металлургия, 1980. 176 с.
27. Дзугутов М. Я. Напряжения и разрывы прн обработке металла давлением. М._- Металлургия, 1974. 280 с.
28. Явойский В. И., Бубенчик Ю. И., Окен-ко А. П. Неметаллические включения и свойства стали. М.: Металлургия, 1980 176 с.
29. Шульте Ю. А. Неметаллические включения в электростали. М.: Металлургия, 1964. 208 с.
30. Мошкевич Е. И., Стеценко Н. А., Спектор Я. И., Павперова И. А. и др. — Проблемы стального слитка. Труды III конференции. М.: Металлургия, 1969, с. 163—169.
31. Поволоцкий Д. Я. Алюминий в конструкционной стали. М: Металлургия, 1970. 129 с. .
32. Смоляренко Д. А. Качество углеродистой стали. 3-е изд. М.: Металлургия, 1977.
33. Булат . С. И., Тихонов А. С., Дубровин А. К. Деформируемость структурно-неоднородных сталей и сплавов. М.: Металлургия, 1975. 352 с.
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Аберрация 25, 49
Автоматический анализатор изображения 78
Альфа-фаза 345
Анализаторы структуры количественные 31
Антиферромагнитный резонанс 182
Атомная функция рассеяния 115
Атомный объем 288. 289
Аустенит остаточный 131
Ближний порядок, рентгеноструктурный анализ 128
Блоха — Грюиайзена зависимость 294
Бравэ решетки" 95
Баллера механизм 181
Величина зерна 338
Вероятная абсолютная погрешность 91
Видемана — Франца — Лоренца закон 281
Внутренние разрывы 328
Волосовины 325. 331—333
Восприимчивость:
определение 305
диамагнитная 305
методы измерения 310, 311
парамагнитная 306
Временное сопротивление 192. 194
Вторичная экстинкция 140
Вульфа — Брэггов уравнение 122
Вульфа сетка 107, 109
Вязкость разрушения*
определение 223, 237. 238
влияние величины зерна 241
— вторичного твердения 242
— легирования 242
— серы и фосфора 242
— способа выплавки 242
— температуры .240, 241
Гейна реактив 16
Геометрическая структура 72
Герстнера закон 186 »
Гномостереографические проекции 109, 110
Гортера— Нордгейма правило 296. 300
Границы зерен, выявление:
метод окисления 45
— сетки троостита 45
----феррита 45
— травления 45
— цементации 44
Граничные поверхности, определение степени ориентировки 91
Грюиайзена параметр 290
Гука закон 185, 186, 205, 206
Дебая температура 276, 277
— теория 276
ДеГоффа метод 84
Деполяризаторы 253
Дериватограф 281
Детекторы рентгеновского излучения 117
Дефекты кристаллического строения, анализ 141
— слитков 323
Деформация 185
— пластическая 185
• — стадии процесса 185
— упругая 185
Диаграммы:
изгиба 196
истинных напряжений 193
кинетическая растрескивания 246, 247
— усталостного разрушения 243, 246
конструктивной прочности 243
кручения 197
механического состояния 189
нагрузка — податливость 240
нагрузка — прогиб 215
разрушения 238, 239
растяжения 189, 191
состояния, построение 130
Диамагнетизм 305
Дилатометр:
емкостной 291
интерференционный 291
Стрелкова 292
Шевенара дифференциальный 290
Диполь-дипольное магнитное взаимодействие 180
Дифракция медленных электронов 152
Доза излучения 123
--- предельно допустимая 123
Допплера эффект 162
Дюлонга — Пти правило 270
Зеебека явление 299
Зеемановское расщепление 180
Зерно металла:
действительное 43
методы определения величины 43
«наследственное» 43
Зонная теория 293-, 294
Излом:
волокнистость 219
вязкий 15
камневидный 15
нафта линистый 15
поперечный, контроль 326
348
продольный, контроль 325, 326
усталостный 226
хрупкий 15
Измерительные шаблоны 78
Изнашивание:
аварийное 259, 260
виды 250
приработка 259, 260
при трении 256, 257
установившееся 259. 260
Изнашивание, испытания:
в условиях эксплуатации 272, 273
методом вырезных лунок 273, 274
— искусственных баз 273
— микром етрирования 273
— поверхностной активации 274
Изотопы, физические характеристики 172
Интегральная интенсивность 115
Инфракрасное поглощение 153
Ион но-нейтрализационная спектроскопия 154
Ионный анализ 151
— микрозонд с анализом рентгеновских лучей 154 Испытания:
динамические 488, 209, 21,0
микромеханические 197
на изгиб 195. 218
— кручение 196
— — при высоких температурах 347
— осадку 347
— ползучесть 188
— разрушение 188, 236
— растяжение двухосное 203
---одноосное 190
--при высоких темпреатурах 347
— сжатие 094
— твердость — См. Твердость
— ударную вязкость 347
— усталость 188
статические 188
Кавельери ~ Акера — Глаголева принцип 90
Кавитационная стойкость, методы испытания 265, 266
Кавитационное разрушение 255
Карбидная неоднородность 341, 343, 344
— сетка 344
Карбидные фазы, реНтгеноструктурный анализ 135
Квадрупольная энергия 177
Квадрупольные эффекты 177
Квадрупольный резонанс 177
Кватовомеханическая теория ферромагнетизма 306
Кельвина соотношения 299
Когерентные частицы, рентгеновский анализ 129
Контраст:
интерференционный 27
композиционный 67
топографический 66
фазовый 27
Коппа — Неймана правило 277
Корка, дефект слитков 329
Коррозионная стойкость, методы испытания 261—
264
— усталость 260, 254
Коррозионное растрескивание:
определение 250, 254
методы испытаний 263, 264
образцы 264
схема нагружения 264
Коррозия:
избирательная межкристаллитная 249
— ножевая 249
местная 249
.общая 249
показатели 261
скорость 254, 255, 261, 262
химическая — См. Химическая коррозия
щелевая 264
электрохимическая — См. Электрохимическая коррозия
Коэрцитивная сила 312
Коэффициент асимметрии цикла 244
— всестороннего сжатия 258, 259
— интенсивности напряжений 231—233
----виды 237—239
----критические значения 239
----пороговые значения 246, 247
— температуропроводности 283
—--тензочувствительности 207
— термического расширения 288—290
Краевой эффект 81
Кривизна поверхностей 88
----средняя гауссова 88
Кривые качания 141
— ползучести 203
— усталости 230
Кристаллическая структура 98
---- карбидов 101
--- нитридов 101
---фаз металл — металл 103
Кристаллические системы 96
Кристаллы-монохроматоры, характеристики 117
Кристаллографические индексы 102
• — проекции 106
Критическая температура 302
Кронинга — Ван-Флекса механизм 181
К-состояние 303. 304
Кюри — Вейса закон 306, 307
Ларморовская прецессия 172, 180
Лауэ метод ЛЗ
Лауэвский класс симметрии 98
Ликвационный квадрат 327
Линейные элементы 9Q, 92
Людерса линии 186 t
Магнетокалорический эффект 317, 318
Магнетосопротивление 300
Магнитная анизотропия 314, 315
— проницаемость 305
— — амплитудная 316
— -— комплексная 316
---обратимая 311
Магнитно-жесткие материалы 315
Магнитно-мягкие материалы 315
Магнитно-полужесткие материалы 315
Магнитострикция 316
Магнитоупорядоченное состояние антиферромагнитное 306 ---ферримагнитное 306 ---ферромагнитное 306
Магнитные свойства, методы измерения:
баллистический 312, 313
вибрационным магнетометром 313
вталкиванием в магнитное поле 314
измерительным генератором 313, 314
магнитометрический 313
Магнитный гистерезис 312
— момент 305
— поток 305
Макроанализ 15, 17
Макронапряжения 138
Макроструктура, контроль 325
Макрошлиф 15
Мартенситные превращения 56
Масс-спектроскопия вторичных ионов 151, 154, 155
Маттиссена правило 295
Маятниковые копры:
Амслера 212
вертикальные 213
заклинйвание образца 214
запас энергии 211
Изода 212, 213
испытания на ударное растяжение 214
— падающим грузом 213, 218
радиус качания 211
ротационные 214
схема работы 211
Щарпи 212
Межплоскостные расстояния, расчет 107
Межчастичные расстояния, определение 92'
Мессбауэра эффект 161, 178
Метод вращения 113
Метод порошка 114
— последовательных сечений 73
— прямого разрешения 61
— R-кривых 239
— ступенчатого нагружения по Локати 230
Методы исследования поверхностей 151
------применение 1*58
------ характеристики 155
Механические свойства 187
---при длительных статических нагрузках 202
Микроанализ 17
Микроанализаторы 144, 145
— локальность 145
Микродеформации 138
Микронапряжения 138
Микроскопия автоионная 151
Микроскопия световая:
контрастность изображения 26
поляризованный свет 26
светлое поле 26
темное поле 26
Микроскопия электронная просвечивающая:
диффузионное рассеяние электронов 58
изучение дислокационной структуры 59
косвенный метод 60
кристаллографический анализ 54
микродифр акционный фазовый анализ 54
полупрямой метод 50
приготовление образцов 51
прямой метод 51
Микроскопия электронная растровая:
349
изучение структуры 70
локальный анализ 70
перспективы развития 72
Микроскоп световой: высокотемпературный 33 полезное увеличение 24 разрешающая способность 24 типы 29—31
Микроскоп электронный просвечивающий:
ВЫСОКОВОЛЬТНЫЙ 61
приставки аналитические 61
— для деформации 60
--- нагрева 60
---охлаждения 60
разрешение 49
— прямое 61
увеличение 48
Микроскоп электронный растровый:
классификация 63
глубина фокуса 64
разрешение 64
электронные пушки, характеристики 65
Микроструктура, контроль 341, 342
Микротвердость 30, 200
— алмазные наконечники 202
— формулы для расчета 202
Микроструктура 33
— эталонные шкалы 34
Микрошлифы 17
— полирование 20
---механическое 20. 21
--- пасты 20
— — станки 21
---химико-механическое 20. 24
---электролитическое 20, 22. 52
— шлифование 18—21
Михеева определитель 124
Модель решетки соединения 174
Модуль сдвига 205, 205
— упругости, адиабатический 205
---изотермический 205
---релаксирующий 206
Модуль, метод определения:
изгибных колебаний 208
импульсный 207
резонансный 207, 208
Нагружение, виды 237
— частота 226
Найта сдвиг 175, 176
Намагниченность 305
Намагничивания кривая 311, 312
Напряжения:
зональные 138
истинные максимальные касательные 186
остаточные 138
рентгеноструктурный анализ 138
Напряженность магнитного поля, методы измерения: баллистический 307 потенциалометром 309
с использованием эффекта Холла 307. 308 феррозондом 309
электродинамический 308, 309
ядерного магнитного резонанса (ЯМР) 308
Нем еталлич еские включения:
карбонитриды 42, 43
методы контроля 333—338
нитриды 42, 43
— алюминия строчечные 434
---точечные 334
оксиды 36—39
— строчечные 334
— точечные 334
признаки 36—43
силикаты 40, 41
— недеформирующиеся 334
— пластичные 334
— хрупкие 334
схема определения 35
сульфиды 40, 41, 334
Неоднородность титановая 330
— точечная 327
Нернста — Эттинсгаузена эффект 300, 301
Обезуглероженный слой, зоны 338, 339
Обезуглероженный слой, методы определения глубины:
металлографический 338, 339
по твердости 340
терыоэлектродвижущей силы 340
химический 340, 341
Обергоффера реактив 16
Обменное электростатическое взаимодействие 180
Обратная решетка кристалла 111
Объекты металловедческого исследования 73
Оже-электроцная спектроскопия 151, 152, 155
Окалина 251
Ориентированные сечения 90
Относительное сужение (прн разрыве) 192, 194
— удлинение (при разрыве) 192, 194
Парамагнетизм 306
Пассивность металлов 253
Паули парамагнетизм 175
Пеллини диаграмма 221
Пельтье явление 299
Перлит, структура 341
Петля гистерезиса в переменном магнитном поле 316
----для ферромагнетика 312
Пластичность 192, 220
Плотность 287. 288
Ползучесть 202
Полинга формула 288
Полюсная фигура 136
Поляризация 253
Порог хладноломкости 235
Послойная кристаллизация 331
Потенциалометр 309
Предел выносливости 226
— ползучести 203
— пропорциональности 1192, 193
— текучести условный 192, 193
----физический 192, 193
— упругости 192, 193
Проба на ковкость 346
Пробная деформация 346, 347
Прокалнваемость, испытание 346
Прокатная плена 324
Пространственные (федоровские) группы 101
— решетки 95
Пуанта теория 177
Пуассона коэффициент 205
Пузыри 325, 326
Работа зарождения трещины 215
— полная 215
— разрушения 215, 219
— удельная 215, 219
Радиоспектрометры, технические характеристики 184
Радиоспектроскопия 171
Размагничивающий фактор:
баллистический 312
магнитометрический 312
Разрушение, виды 187, 220
— очаг 226
— поверхность 93
— след 93
Раскатанная корочка 324
Раскатанное загрязнение 323
Раскатанный пузырь 323
Рассеяние света комбинационное 153
Расслоение 325
Рванины 324
— разваливание слитков 329
Релаксация спин-спиновая 173
— спин-решеточная 173, 181
Рентгеновская фотоэлектронная спектроскопия 153
Рентгеновское излучение, создаваемое протонами 154
Рентгеноспектральный микроанализ 145
----основные методики 147
----подготовка образцов 150
---- поправки 149
----точность 446
----чувствительность метода 146
Рентгеновские дифрактометры 120
— камеры И6, 119
— пленки 117
— трубки 116
Рентгеноструктурный анализ 94
— — аппараты 418
----идентификация фаз 124—127
— — качественный 124
----количественный 125
----определение углерода в мартенсите 134
----доли мартенсита отпуска 135
----поверхностных слоев >122
---- сверхструктуры 127
----твердых растворов 127, 128
----техника безопасности 123
---- фазового состава 13!
Ричардсона уравнение 298
Рэлея поправка 208
Салтыкова метод обратных диаметров 84
—— «укрупненных показателей» 84
Сверхпроводимость 301, 302
Связанность второй фазы и матричной структуры 92, 93
Сдвиг резонансной частоты спектра 175
Сингония, типы 96
— кубическая, углы между плоскостями 105
Синеломкость 225
350
Системы ориентированных поверхностей, удельная протяженность 87
Скворечник 330
Случайные сечения 81, 83, 89, 90
Сопротивление усталости 226
Спектр ЯМР металлического порошка, теоретическая форма '176
Спектрометрия ионного рассеяния 154
Стандартные сетки 109
Статистическая реконструкция 73
Стереографические проекции 106
Стереологическая реконструкция:
распределение размеров частиц 81
формы частиц 85
характеристик размеров частиц, погрешности 84
Стерео логические характеристики 74, 75
Стереологический анализ:
объекты 75
основные этапы 73
первичные измерения 77
типы измерительных систем 77
Стереология, определение 72
Стереопара 68
Структура металлических и неметаллических элементов 99
Структурная неоднородность, определение 17
Структурная полосчатость 344
Структурно-свободный цементит 344, 345
Супермагнетизм 319
Сфера отражения 112
Сферические проекции 106
Съемка на отражение 137
---просвет '137
Твердость 198
— по Бринеллю 198
---Виккерсу 199
---Роквеллу 200
— таблица перевода 201
Твердые растворы, распад 56
Текстура 136
Тензометры 206
Тепловая хрупкость 202
Тепловые эффекты 286
Теплоемкость:
грамм-атома 276
удельная 276
— истинная 276
— средняя 276
Теплоемкость, методы измерения:
общие сведения 286, 287
в расплавах 280
импульсный 279
модуляционный 279
Сайкса — Грузина 277, 278
Смита 278, 279
Теплопроводность 231
— полная 282
— решеточная 282
— электронная 282
Теплопроводность, методы измерения:
Ангстрема 235
кратковременным нагревом 285
продольным потоком тепла 283. 284
радиальным потоком тепла 284. 285
Стакса — Чесмара 285
Термический анализ:
дифференциальный 280, 286, 287
термогравиметрия 280
— дифференциальная 281
Термомагнитный анализ 318
Термоэлектродвижущая сила абсолютная 299
---измерение 300
Термоэлектрические явления 299
Томпсона явление 299
Точечная группа 98
Травление:
вакуумное 43
глубокое (макрошлифа) 15
катодное 43
метод отпечатков 15.16
поверхностное (макрошлифа) 16
тепловое 42
химическое, реактивы 44
электролитическое 42
Трансляционные группы 97
Трансляция 95
Трение:
виды 256
коэффициент 256
сила 256
структурные изменения 258, 259
схватывание 257, 258
Трещиностойкосты
критерии 237
образцы 231, 232
оценка 230, 233, 239, 240
статическая 246
циклическая 244—246
Трещины:
ковочные 328
критическое раскрытие 240
межкристаллитные 328
напряжения 324
раскатанные 323
скорость роста 232
сопротивление распространению 237
травильные 325
шлифовочные 324
Туннельный эффект 298
Ударная вязкость 235
Упорядочение 67
Упругости константы 205
Ус 324
Усадочная раковина 326
Ускорение силы тяжести, значения 211
Усталость высокочастотная 233
— контактная 233. 234
— коррозионная 234, 247
— малоцикловая 230
— термическая 234
— ударная 234
Факторы формы 86
Ферми — Дирака распределение электронов 293, 294
Феррозонд 309
Ферромагнитный резонанс 182
Флокены 327, 328
Фрикционные связи 257
Фотодесорбция 153
Фрактография 69
Фуллмэна метод 84
Характеристические линии элементов 145
Характеристическое излучение К-серии 117
Хиллиарда формула 84
Химическая коррозия:
в расплавах 252
— хлоре и соляной кислоте 252
газовая 249—25!
карбонильная 251, 252
сернистая 252
эрозионное разрушение 252
Химическая неоднородность 16
Хладноломкость 219, 220, 225. 235
Холла эффект, измерение 300, 301
Хрупкость отпускная 225
Центральная пористость 327
Цикл напряжений 226
Циклический долом 232
Частицы короны (дефект) 331, 332
Черновины (осевой перегрев) 329, 330
Черный излом 331
Чешуйчатость 324
Электрическая проводимость 292, 293
— — удельная 292, 293
Электрические свойства, методы измерения:
амперметра и вольтметра 296
в переменных электрических полях 298
мостовые двойные 297
— одинарные 296
потенциометрический 297
Электрическое сопротивление:
влияние закалки и отпуска 305
— концентрационных неоднородностей 302, 30г
— состава сплава 302
— старения 304
— степени упорядочения 302
металлов 295
остаточное 295
твердых растворов 296
удельное 292, 293
Электродный потенциал 253
Электронная спектроскопия для химического анализа
(ЭСХА) 151, 153, 155
Электронный парамагнитный резонанс (ЭПР):
параметры линий 179
понятие 179
применение 181
структура линий 179
Электроны:
вторичные 62
каналирование 70, 71
Оже 62, 70
отраженные 62
эффективная масса 294
Электрохимическая коррозия:
био 249
351
влияние давления 255
— концентрации ионов 254, 255
— растворенного кислорода 256
— скорости движения среды 255
— температуры 255
кислотная 249
контактная 249. 264
почвенная 249
с деполяризацией водородной 253
----кислородной 253
солевая 249
щелочная 249
Элементарная ячейка 95, 107
Элементы симметрии 96
— структуры, ориентировка 91
----размещение в пространстве 91
Эллипсометрия '153
Эмиссия электронов термоэлектронная 298
----холодная 298
Энтальпия 276
Эрозионная стойкость, методы •испытания 266—269
Ядерный гамма-резонанс (ЯГР) 161
----источники 163
----применение 166
----разрешающая способность 161
ЯГР-спектр поглощения 162, 163
— — высота линий 165
—— изомерный сдвиг 164
— — параметры в металлах и соединениях 167
----сверхтонкая структура 163
----ширина линий 165
Ядерный магнитный резонанс, определение 171
------применение 178
Ячеистый (двухфазный) распад твердых растворов 130
Борис Самуилович Бокштейн, Юрий Генрихович Векслер, Мария Ипполитовна Виноград, Борне Александрович Дроздовский, Галина Михайловна Казичкнна, Борис Александрович Клыпии, Владимир Георгиевич Костогонов, Борис Григорьевич Лившиц, Алексей Сергеевич Лилеев, Юлия Яковлевна Лилеева, Лев Георгиевич Коршунов, Елена Евгеньевна Попова, Александр Григорьевич Рахштадт, Олег Николаевич Ро-манив, Юрий Александрович Скаков, Михаил Петрович Усиков, Лев Маркович Утев-ский, Яков Борисович Фридман, Кирилл Сергеевич Чернявский, Лев Михайлович Школьник, Евгений Авелевич Шур.
МЕТАЛЛОВЕДЕНИЕ И ТЕРМИЧЕСКАЯ ОБРАБОТКА СТАЛИ
СПРАВОЧНИК
Редакторы издательства Л. М. Гордон, М. Н. Новиков Художественный редактор А. И. Гофштейн Технический редактор Г. Б. Жарова Корректоры Ю. И. Королева, Н. П. Собко
ИБ № 1782
Сдано в набор 11.11.82. Подписано в печать 04.04.33. Т-06855. Формат бумаги 70X108l/i6. Бумага типографская № 2. Гарнитура литературная. Печать высокая. Усл. печ. л. 30,80. Усл. кр.-отт. 30,80. Уч.-изд. л. 44,21. Тираж 21 000 экз. Заказ № 280. Цена 2 р. 50 к. Изд. № 0372
Ордена Трудового Красного Знамени издательство «Металлургия». 119034, Москва, Г-34, 2-Й Обыденский пер., д. 14
Владимирская типография «Союзполиграфпрома» при Государственном комитете СССР по делам издательств, полиграфии и книжной торговли
600000, г. Владимир, Октябрьский проспект, д. 7