/
































































Author: Ластовский Р.П.
Tags: производство органических веществ органические соединения химия химическая промышленность химические реакции
Year: 1965




Text
ГОСУДАРСТВЕННЫЙ КОМИТЕТ
ХИМИЧЕСКОЙ ПРОМЫШЛЕННОСТИ ПРИ ГОСПЛАНЕ СССР
МЕТОДЫ ПОЛУЧЕНИЯ
ХИМИЧЕСКИХ РЕАКТИВОВ
И ПРЕПАРАТОВ
Выпуск 12
ВСЕСОЮЗНЫЙ НАУЧНО-ИССЛЕДОВАТЕЛЬСКИЙ ИНСТИТУТ
ХИМИЧЕСКИХ РЕАКТИВОВ И ОСОБО ЧИСТЫХ ХИМИЧЕСКИХ ВЕЩЕСТВ
МОСКВА — 1965
СОДЕРЖАНИЕ
Алкилдихлорфосфины. И. П. Комков, К. В. Караванов, С. 3. Ивин 5
Алкилдихлорфосфипоксиды. С. 3. Ивин, К. В. Караванов,
В. В. Лысенко, Г. И. Дрозд................................. 8
Алкилдихлорфосфинселениды, С. 3. Ивин, И. Д. Шелакова ... 17
Алкилдихлорфосфинсульфиды. И. П. Комков, С. 3. Ивин,
К. В. Караванов........................................... 20
Алкил(арил)тетрафторфосфины. И. 77. Комков, С. 3.. Ивин,
К. В. Караванов, Л. Е. Смирнов ......... .... 23
5-Ацетилпсевдокумол /И. Т. Разумовская, А. И. Белякова,
Г. И. Корельская.......................................... 27
N-Бензилэтилендиамин. Р. 7/. Ластовский, И. Д. Колпакова,
Л. В. Криницкая, Т. И. Иванова............................ 31
2,7-Бис-[М,Г4-ди-(карбоксиметил)-аминометил]-4,5-дийодфлуоресце-
ина динатриевая соль. В. В. Сидоренко, Т. П. Коноплева,
Н. В. Лапшина . ..................... 34
4,4’ Бис-[2,4-бис-(2-кгрбокси.метил амино)- 1,3,5-триазин и л-6-амино]-
2,2’-стильбеидисульфокислоты гексаиатриевая соль Р. П. Лас-
товский, В. Я. Темкина, Г. Ф. Ярошенко, Л. /И. Самылова 37
р-Бромэтиламин бромистоводородный. Р. П. Ластовский,
И. Д. Колпакова, Л. В. Криницкая, Л. Д. Завьялова . . . 41
р-Бромэ!илиминодиацетонитрил. Р. И. Ластовский, И. Д. Колпа-
кова, Л. В. Криницкая..................................... 44
Винилметилхлорфосфинат. Ю. Г. Гололобов, Т. Ф. Дмитриева,
Л. 3. Соборовский.......................................... 46
Винилхлорфосфаты. 10. Г. Гололобов, Т. Ф. Дмитриева,
Л. 3. Соборовский.......................................... 48
2 Гидразинохинолин. В. М. Дзиомко, И А. Красавин, И. И. Ми-
рошкина.................................................... 50
Диалкилхлорфосфины. И. П. Конков, К В. Караванов, С. 3. Ивин 54
Диалкилхлорфосфинсултфиды. С. 3. Ивин, К. В. Караванов . . 57
Диалкилтрифторфосфины. 77. И. Комков, С. 3. Ивин, К. В. Кара-
ванов ..................................................... 59
Диангидрид пйромеллиювой кислоты. М. Т. Разумовская,
Л. А. Егорова.............................................. 61
Дихлорангидрид 2,3-дихлориропилфосфиновой кислоты. Л. 3. Со-
боровский, Ю. М. Зиновьев, Л. И. Мулер................. 64
Дихлорангидрид циклогексилфосфиновой кислоты. Л. 3. Соборов-
ский, Ю. М. Зиновьев....................................... 66
Диэтилвииилфос(} ат. Ю. Г. Гололобов, Т. Ф. Дмитриева,
Л. 3. Соборовский.......................................... 68
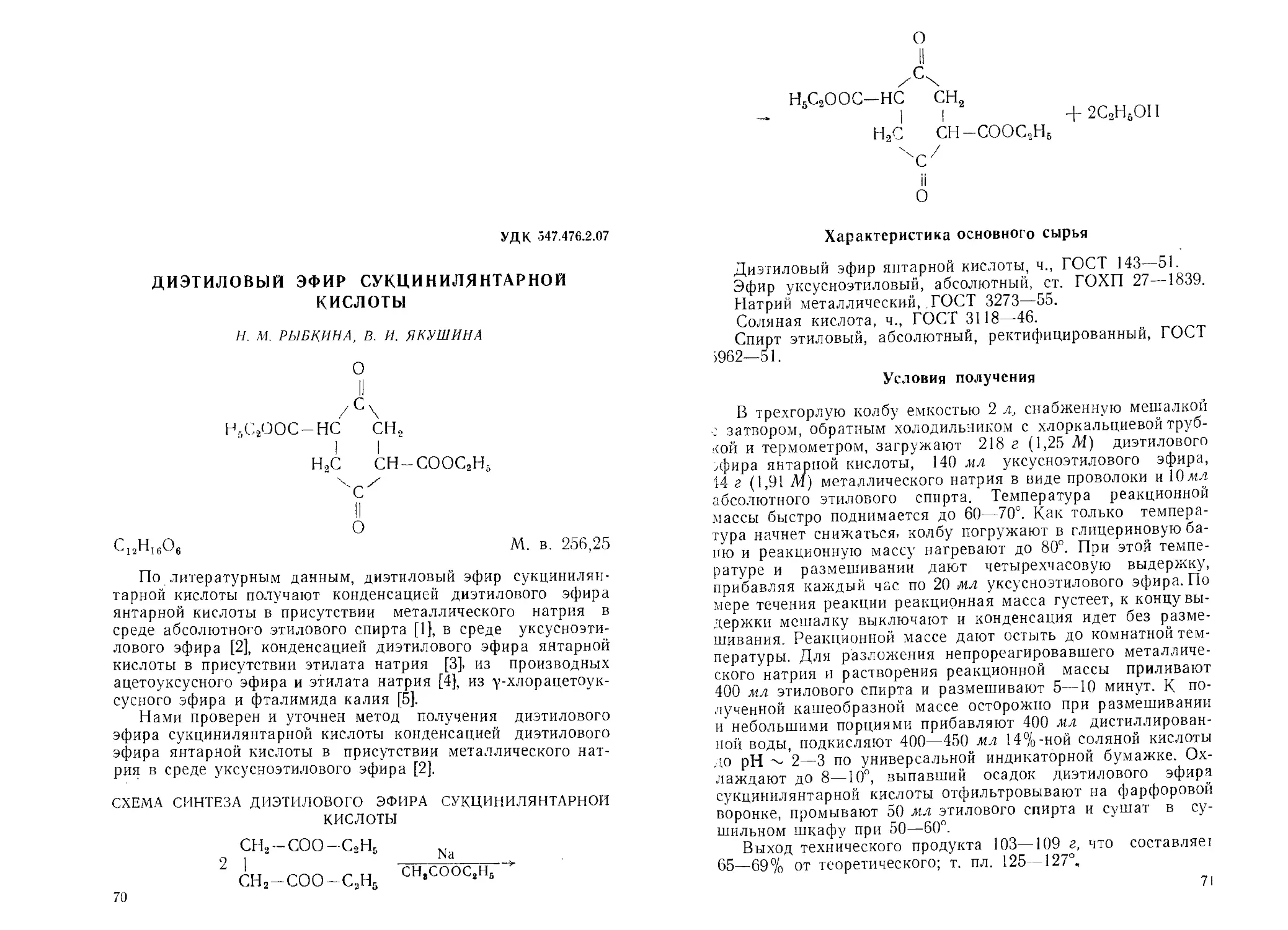
Диэтиловый эфир сукцинилянтарной кислоты. И. М. Рыбкина,
В. И. Якушина ....................... 70
3
Комплексные соединения алкилтетрахлорфосфинов с хлористым
алюминием. И. П. Комнов, К. В. Караванов, С. 3. Ивин . 73
Комплексные соединения диалкилтрихлорфосфинов с хлористым
алюминием. С. 3. Ивин, К. В. Караванов ......... 76
2 Метокси-4-амииотолуол-М, N-диуксусная кислота. Р. П. Ластов-
ский, И. Д. Колпакова, Л. В. Криницкая, Г. И. Иванова,
Л. Д. Завьялова............................................ 7!)
Натриевые соли О-алкиловых эфиров алкилфосфивовой кислоты.
И. Д. Шелакова, С. 3. Ивин.................................. 81
₽-Нафталинсульфохлорид. Б. М. Болотин, Д А. Драпкина,
В. Г. Брудзь, Т. Е. Оськина . . . ................... 84
4’-Нитро-2 амино-4-метокси-5-метилазобензол. Р. П. Ластовский,
И. Д. Колпакова, Л. В. Криницкая, Л. Д. Завьялова, Т. И.
Иванова .................................................... 86
4’-Нитро- 2 -амино- 4 -метокси-5-метилазобензол - N, N-диуксусная
кислота и ее диметиловый эфир. Р. П. Ластовский,
И. Д. Колпакова, Л. В. Криницкая, Т. И. Иванова............ 89
.и-Оксифенилиминодиуксусная кислота. Р. П. Ластовский,
В. Я. Темкина, Г. Ф. Ярошенко, И. П. Фадеева................ 93
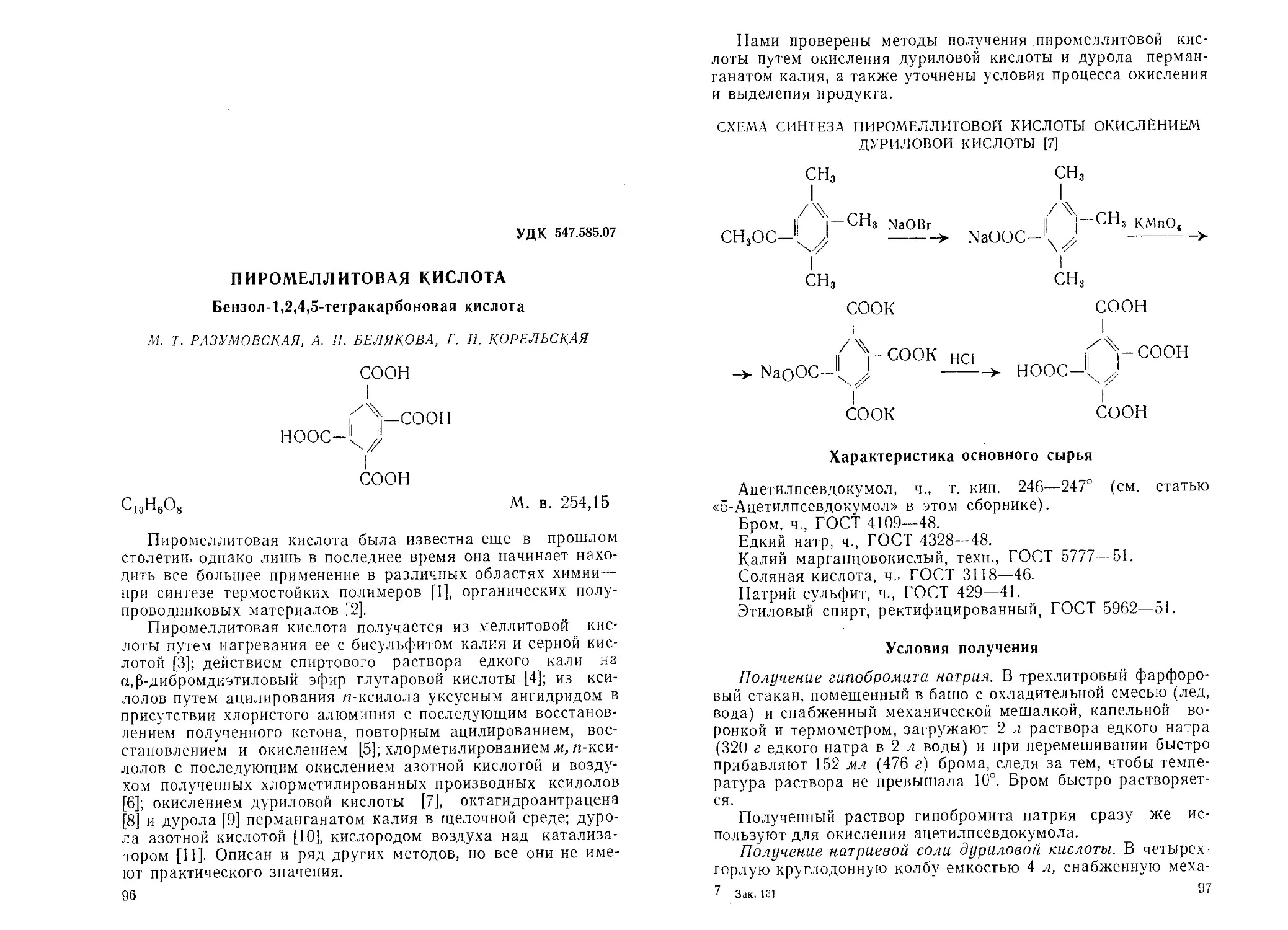
Пиромеллитовая кислота. М. Т. Разумовская, А. И. Белякова,
Г. И. Корельская............................................ 96
Получение алкилдихлорфосфинов восстановлением алкилдихлор-
фосфиноксидов металлами. И. П. Комков, К. В. Караванов,
В. Г. Груздев, С. 3. Ивин.................................. 101
Стильбексон. Р. П. Ластовский, В. В. Сидоренко, Н. В. Лапшина,
Г. П. Коноплева............................................. ЮЗ
3,3',4,4’-Тетраметнлдифенилоксид. М. Г. Разумовская, А. И. Беля-
кова, Л. А. Егорова........................................ 105
Тетрамид пиромеллитовой кислоты. М. Т. Разумовская, А. И. Бе-
лякова, Л. А. Егорова..................................... 108
Тетрацианбензол. М. Т. Разумовская, А. И. Белякова, Г. И. Ко-
рельская................................................. 111
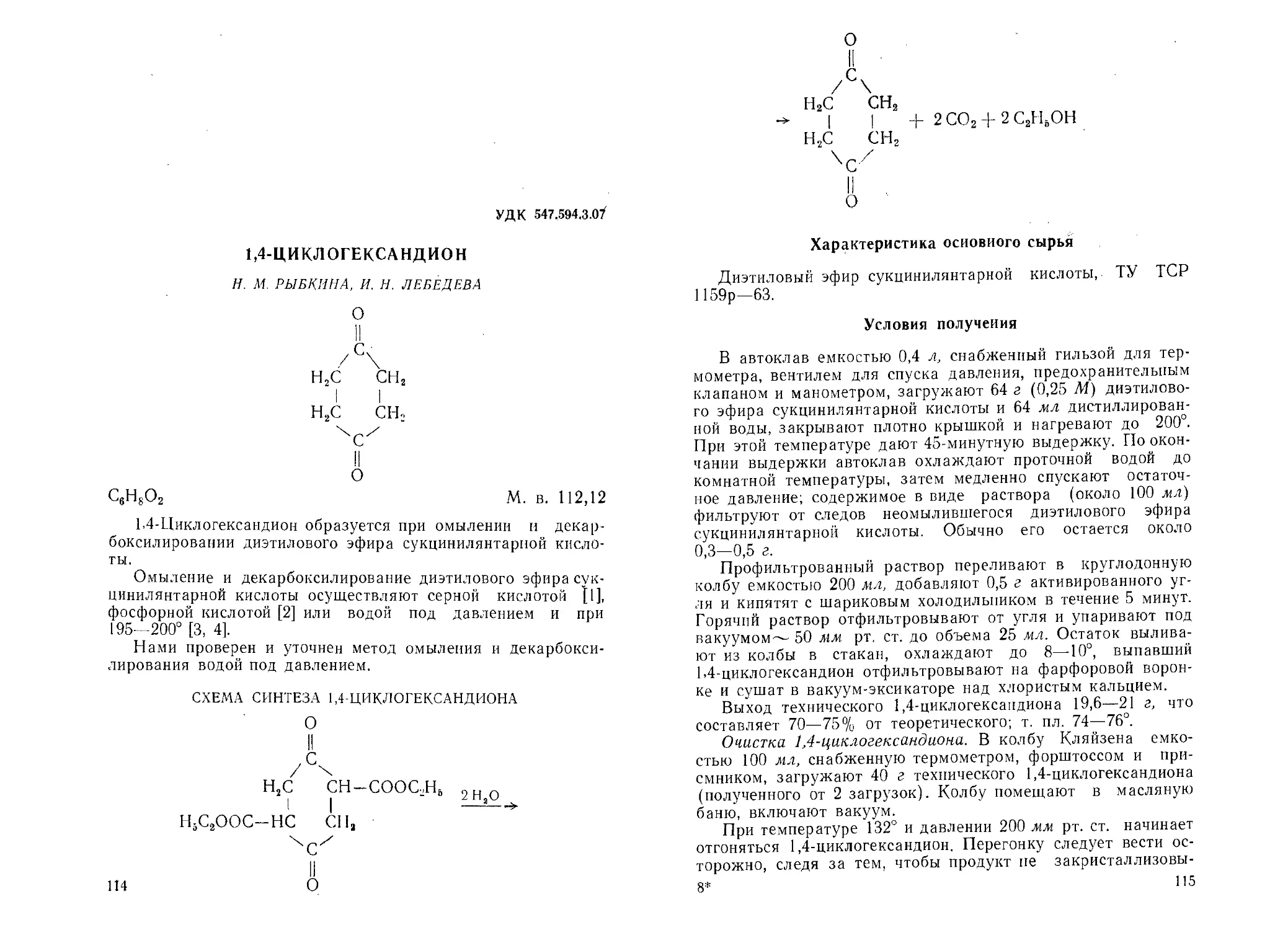
1,4-Циклогександион. Н. М. Рыбкина, И. Н. Лебедева ...... 114
О-2-Хлоралкилалкилхлорфосфивиты. С. 3. Ивин, К. В. Караванов 117
5-2-Хлоралкил-этил(алкил)хлорфосфиниты. С. 3. Ивин, И.Д. Ше-
лакова ................................................... П9
1-Хлор-З-бромпропан. Ю. Г. Гололобов, Э. Б. Гуськова,
Л. 3. Соборовский ..................... 121
а-Хлор-ф-этилмеркаптоэтилацетат. Л. 3. Соборовский, Ю. Г. Го-
лолобов ................................................... 123
Хрнзоидин-N, N'-диуксусная и хризоидин-N, N, N', N'-тетрауксус-
иая кислоты. Р. ГТ. Ластовский, И. Д. Колпакова, Л. В. Кри-
ницкая, Л. Д. Завьялова, Т. И. Иванова..................... 125
jl-Хлорэтиламии хлористоводородный. Р. ,П. Ластовский,
И. Д. Колпакова, Л. В. Криницкая, Т. И. Иванова............ 128
Этилмеркаптоацетальдегид. Л. 3. Соборовский, Ю. Г. Гололобов 130
Алфавитный перечень соединений, описанных в настоящем сборни-
ке ........................................................ 133
УДК 661.718.07
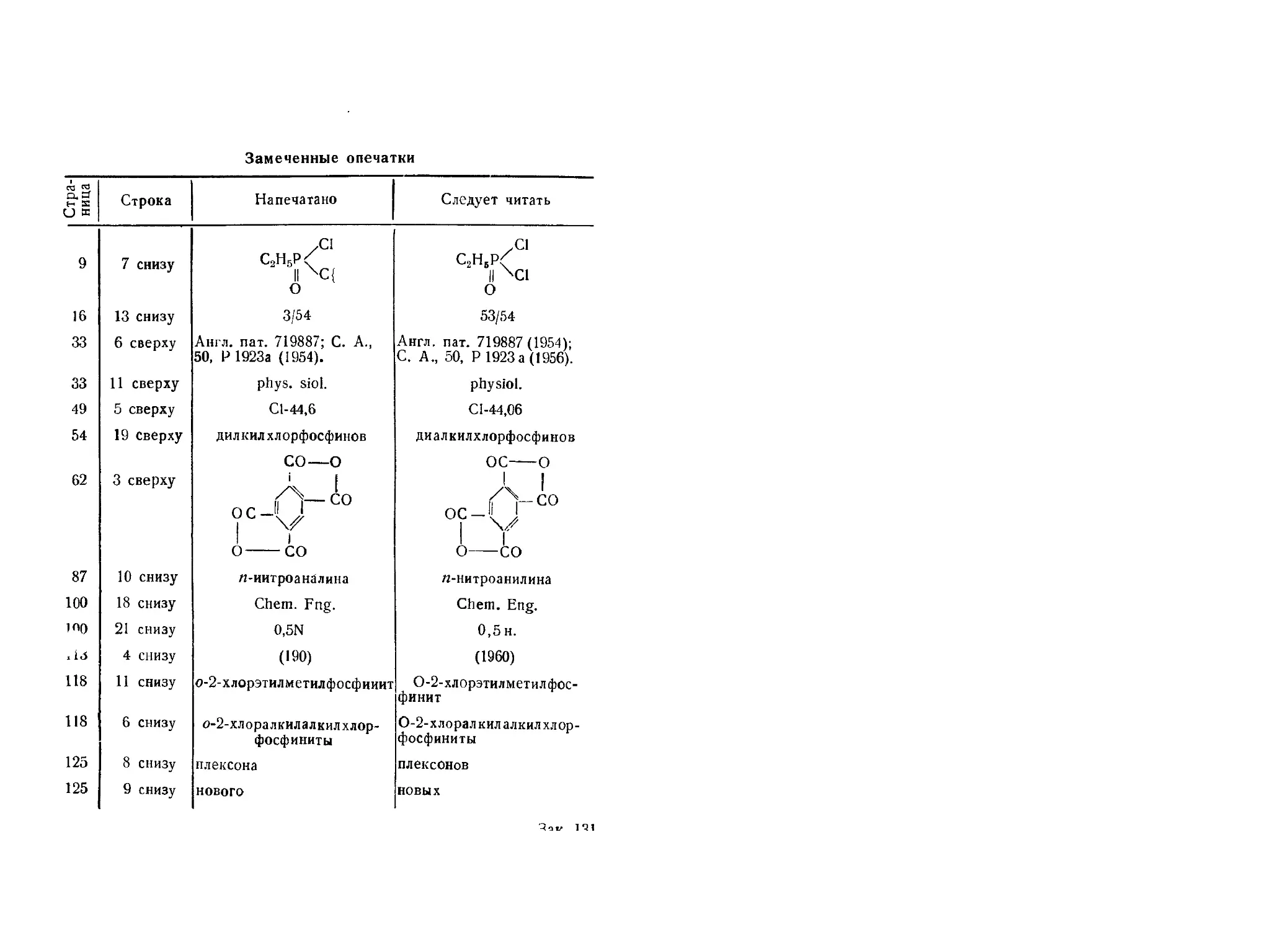
АЛКИЛДИХЛОРФОСФИНЫ
И. П. КОМКОВ, к. В. КАРАВАНОВ, С. 3. ИВИН
RPC13
Алкилдихлорфосфины являются исходными полупродук-
тами в синтезе различных фосфорорганических соединений.
В литературе описано несколько способов их получения.
Одним из первых описан метод, основанный па реакции диал-
килртути с треххлористым фосфором [1]. Алкилдихлорфосфи-
ны также могут быть получены алкилированием треххлори-
сгого фосфора с помощью тетраэтилсвинца [2], кадмийоргани-
ческих соединений [3], прямым алкилированием красного фос-
фора [4], взаимодействием треххлористого фосфора с углево-
дородами [5], восстановлением комплексных соединений ал-
килтетрахлорфосфинов с хлористым алюминием: а) порош-
кообразной сурьмой или цинком в диэтилфталате (диэтил-
фталат использовался для связывания треххлористого фос-
фора и как растворитель) [6]; б) фенилдихлорфосфином в
присутствии хлорокиси фосфора [7].
Перечисленные методы являются, как правило, малоудоб-
ными, но главным .недостатком для большинства из них яв-
ляются низкие выходы алкилдихлорфосфинов, а также срав-
нительно дорогие или малодоступные исходные реагенты.
Нами [8] разработан другой способ получения алкилди-
хлорфосфинов, заключающийся в восстановлении комплекс-
ных соединений алкилтетрахлорфосфинов и хлористого алю-
миния металлами и неметаллами в присутствии свежепрока-
ленного хлористого калия вместо диэтилфталата. По этому
методу выход алкилдихлорфосфинов достигает 70%.
5
ЭТИЛДИХЛОРФОСФИН
С2Н5РС13 М. в. 130,94
СХЕМА СИНТЕЗА ЭТИЛДИХЛОРФОСФИНА
КС!
С,НбРС14-А1С13 + AI —-> С2Н5РС12 + А1С13-КС1
Характеристика основного сырья
Комплексное соединение этилтетрахлорфосфина с хлори-
стым алюминием, получают по методике, описанной в насто-
ящем сборнике (см. статью «Комплексные соединения алкил-
тетрахлорфосфинов с хлористым алюминием» в этом сборни-
ке) .
Алюминий в виде алюминиевой пудры (сухой).
Калий хлористый, х. ч., безводный, свежепрокаленный,
ГОСТ 4234- 48.
Условия получения
В колбу Кляйзена емкостью 0,75 л с прямым холодильни-
ком, термометром и присоединенной к горловине колбочкой
помещают 100 г (0,3 М) комплексного соединения этилтетра-
хлорфосфина с хлористым алюминием (см. примечание 1) и к
слегка расплавленному комплексному соединению из присое-
диненной колбочки постепенно небольшими порциями (см.
примечание 2) присыпают смесь, состоящую из 5,4 г (0,2 М)
алюминиевой пыли и 45 г (0,6 М) прокаленного хлористого
калия с одновременным осторожным нагреванием (см. при-
мечание 3) реакционной массы и постоянным встряхиванием
содержимого колбы. Реакция протекает со значительным вы-
делением тепла. После внесения всего количества смеси про-
водят отгонку образовавшегося продукта.
Выход этилдихлорфосфина 26 г, что составляет 67% от те-
оретического; т. кип. 113—114° при 752 мм; d^°—1,2592;
rfy—1,4930 (см. примечание 4).
По аналогичной методике может быть получен метилди-
хлорфосфин.
Примечания;
I. Комплексное соединение этилтетра хлорфосфина с хлористым алю-
минием гигроскопично, поэтому следует избегать излишнего соприкосно-
вения его с воздухом.
2. Смесь хлористого калия и алюминиевой пудры присыпают неболь-
шими порциями. В случае присыпания больших количеств смеси реакция
протекает бурно, с выделением тепла, что может привести к выбросу ре-
акционной массы. Очередную порцию смеси присыпают после того, как
прореагирует предыдущая.
6
3. Нагревание проводят таким образом, чтобы через склянку Тищен-
ко с раствором перманганата, поставленную на конце прибора, как мож-
I'o меньше проскакивало пузырьков. Комплексное соединение, нагретое
выше температуры его плавления, разлагается на составные части.
4. Этилдихлорфосфин легко окисляется на воздухе, в связи с чем его
пе р чение и перегонку желательно вести в токе азота пли какого-либо
другого инертного газа.
По описанной методике можно получать и другие алкил-
дихлорфосфины.
Вместо алюминиевой пудры в качестве восстановителей
можно использовать металлический натрий, красный фосфор,
магний, кальций, железо, цинк, медь и др. (в виде пыли).
Литература
1. Fг. Gulchard. Вег., 32, 1572 (1899).
2. М. S. Kharasch и др. J. Organ. Chem., 14, 429 (1949).
3. R. В. Fox. J. Amer. Chem. Soc., 72, 4147 (1956).
4. Л. 3. С о б о p о в с к и й, Б. М. Гладштейн. Авт. свид. 130513;
Бюлл. изобр., № 15 (1960).
5. I. Р 1 a n f е 11 i, Qriim. J. Amer Chem. Soc, 84, 5. 851 (1962).
6( 1. L. Ferron. В. I. Perry. Nature (Eng), 188, 4746, 227 (1960).
7, Паршалл. J. Inorg. and Nucl. Chem., 12, 3— 4, 372 (i960).
8. И. П. Комков, К. В. К a p а в а н о в, С. 3. Ивин. Ж. общ. хи-
мии, 28, 11, 2963 (1958).
Поступила в июне 1£64 г.
УДК 661.718.1.07
алкилдихлорфосфиноксиды
Дихлорангидриды алкилфосфиновых кислот
С. 3. ИВИН К. в. КАРАВАНОВ, В. В. ЛЫСЕНКО, Г. И. ДРОЗД *
/С1
RP(
Г Cl
о
Алкилдихлорфосфиноксиды широко используются как
промежуточные реагенты для синтеза разнообразных фос-
форорганических соединений. Однако до последнего времени
способы их синтеза оставались малодоступными или трудно-
воспроизводимыми. Так, описан способ, основанный на реак-
ции разложения комплексных соединений хлористого алкила,
треххлористого фосфора и хлористого алюминия водой [1]
или спиртами [2].
Существенным недостатком этого способа является очень
бурная, трудноуправляемая реакция комплексов с водой или
спиртами, сопровождающаяся зачастую гидролизом образую-
щихся алкилдихлорфосфиноксидов или хлорэфиров алкил-
фосфинатов. Лишь при осуществлении серии опытов удается
воспроизвести этот способ и выделить из комплексов алкил-
дихлорфосфиноксиды.
Другие описанные в литературе способы синтеза алкил-
дихлорфосфиноксидов связаны с использованием сравнитель-
но труднодоступных исходных реагентов, например алкилтет-
рахлорфосфинов [3—5], алкилфосфиновых кислот или их эфи-
ров [6, 7].
Поскольку комплексные соединения алкилтетрахлорфос-
финов и хлористого алюминия являются доступными реаген-
* Г. И. Дрозд принимал участие в разработке способа одновременно-
го получения алкилдихлорфосфиноксидов н хлористых ацилов.
8
тами, нами на их основе разработаны удобные способы одно-
временного получения алкилдихлорфосфиноксидов и хлори-
стых ацилов, 1,2-дихлоралканов или соединений, содержащих
нитрильную группу.
Так, при взаимодействии комплексного соединения алкил-
тетрахлорфосфина и хлористого алюминия с монокарбоновы-
ми кислотами в присутствии хлористого калия, связывающего
хлористый алюминий, образуются алкилдихлорфосфиноксиды
с выходом до 60% и хлористые ацилы с выходом до 75% [8].
При нагревании смеси комплексного соединения алкилтет-
рахлорфосфина и хлористого алюминия с амидами карбоно-
вых кислот в присутствии соединений, связывающих хлори-
стый алюминий, например пиридина или свежепрокаленного
хлористого калия, образуются алкилдихлорфосфиноксиды с
выходом до 70% и соединения, содержащие цианогруппу с
выходом до 95% [9].
При взаимодействии комплексного соединения алкилтет-
рахлорфосфина и хлористого алюминия с алкиленоксидами в
присутствии хлористого калия, вводимого в реакцию для свя-
зывания хлористого алюминия, образуются алкилдихлорфос-
финоксиды с выходом до 75% и 1,2-дихлоралканы с выходом
до 98%, рЮ].
СОВМЕСТНОЕ ПОЛУЧЕНИЕ ЭТИЛДИХЛОРФОСФИНОКСИДА
И ХЛОРАНГИДРИДА ПРОПИОНОВОЙ кислоты
С2Н5РОС12
С,НБР<
pci
о
М. в. 146,94
7$
СН3СН2С%
С1
С3Н5ОС1 ДА. в. 92,52
СХЕМА СИНТЕЗА
z.0 КС1 7С1
С2Н6РС14-А1С1з + CH3CH2Cf-----------> С2Н6Р(
'ОН |РС{
о
/ZO
+ СН3СН,С< + НС1 + А1С13-КС1
4 Cl
Характеристика основного сырья
Комплексное соединение, этилтетрахлорфосфина и хлори-
стого алюминия, готовят из хлористого алюминия, треххло-
ристого фосфора и хлористого этила по методике, описанной
в настоящем сборнике (см. статью «Комплексные соединения
9
алкилтетрахлорфосфинов с хлористым алюминием» в этом
сборнике).
Кислота пропионовая, обычная, продажная.
Калий хлористый, х. ч., безводный, свежепрокаленный,
ГОСТ 4234—48.
Условия получения
В трехгорлую колбу емкостью 250 мл, снабженную мешал-
кой с глухим затвором (см. примечание 1), позволяющим соз-
давать в системе разрежение, капельной воронкой и прямым
холодильником с приемниками и ловушкой (на выходе), ох-
лаждаемой смесью сухого льда с ацетоном, помещают тща-
тельно перемешанную смесь 100 г (0,298 Л1) комплексного
соединения этилтетрахлорфосфина и хлористого алюминия
(см. примечание 2) и 21,23 г (0,298 Л4) хлористого калия.
К содержимому колбы (см. примечание 3) при периодиче-
ском встряхивании в течение 1,5 часа прикапывают 22,07 г
(0,298 Л4) пропионовой кислоты. По мере ее добавления на-
блюдается разогревание реакционной массы до 40—45° и пре-
вращение ее в темную вязкую массу, из которой выделяется
хлористый водород.
По окончании прикапывания пропионовой кислоты вклю-
чают механическую мешалку и перемешиваемую реакцион'
ную массу нагревают на глицериновой бане до 100—120° в те-
чение часа. По мере нагревания происходит резкое увеличение
выделения НС1 хлористого водорода с одновременной отгонкой
хлорангидрида пропионовой кислоты в приемник. По оконча-
нии выделения перемешивание реакционной массы прекра-
щают. Приемник с хлорангидридом пропионовой кислоты за-
меняют другим, к системе подключают вакуум и оставшуюся
часть продуктов реакции отгоняют в вакууме 10—15 мм при
температуре бани 100—120° (см. примечание 4).
После вторичной перегонки получают 23 г этилдихлор-
фосфиноксида, что составляет 52,5% от теоретического выхо-
да; т. кип. 48—50’ при 1 мм или 175° при 760 мм; d^20—1,3766;
Нд - 1,4641.
Выход хлорангидрида пропионовой кислоты 18 г, что сос-
тавляет 65,4% от теоретического; т. кип. 78—81° при 760 мм;
di20 — 1,0645; Яд - 1,4057.
Примечания:
1. Герметичность затвора, позволяющую подводить к системе разре-
жения 10—15 мм, достигают уплотнением затвора отрезком вакуумного
каучука.
2. Комплексное соединение этилтетрахлорфосфина и хлористого алю-
миния должно быть сыпучим и не прилипать к стенкам эксикатора, в ко-
тором хранится.
10
3. Механическая мешалка включается по окончании прикапывания
пропионовой кислоты, когда в реакторе образуется достаточно подвижная
сиропообразная масса ^ерно-серого цвета.
4. Перед подключением к системе вакуума мешалку удалять нс сле-
дует во избежание попадания в реакционную массу влаги воздуха. Склян-
ку Тищенко отсоединяют от системы и в случае использования масляного
насоса после ловушки устанавливают колонку с гранулированной едкой
щелочью для поглощения хлористого водорода с целью защиты насоса.
СОВМЕСТНОЕ ПОЛУЧЕНИЕ МЕТИЛДИХЛОРФОСФИНОКСИДА И
АЦЕТИЛХЛОРИДА
СН3ОРС12
С1
CHs-p(
;ЛС1
О
М. в. 132,89
М. d. 78,49
СХЕМА СИНТЕЗА
ZzO КС1
СН3РС14.А1С1з + сн3с< --------->
он
/С1 ,0
-> СН3Р. + СН3С< + А1С13-КС1
il 'Cl 4 Cl
о
Характеристика основного сырья
Комплексное соединение метилтетрахлорфосфина и хлори-
стого алюминия, готовят из хлористого метила, треххлористо-
го фосфора и хлористого алюминия по методике, описанной в
настоящем сборнике (см. статью «Комплексные соединения ал-
килтстрахлорфосфииов с хлористым алюминием»).
Кислота уксусная, ледяная, х. ч., ГОСТ 61—51.
Калий хлористый, х. ч., безводный, свежепрокаленный,
ГОСТ 4234—48.
Условия получения
В трехгорлую колбу емкостью 250 мл, снабженную мешал-
кой, капельной воронкой и обратным холодильником, поме-
щают смесь 100 г (0,311 М) комплексного соединения метил-
тетрахлорфосфииа с хлористым алюминием и 23,2 г (0,311 М)
хлористого калия. При периодическом встряхивании к смеси
прикапывают 18,66 г (0,311 М) уксусной кислоты, после чего
включают механическую мешалку и нагревают смесь на гли-
цериновой бане до 70—80°. Затем реакционную массу в тече-
11
ние 3 часов выдерживают при температуре 100—120°. По
окончании нагревания обратный холодильник заменяют пря-
мым и продукты реакции отгоняют в вакууме 20 мм (см. при-
мечание) при температуре бани 110—120°.
После вторичной перегонки получают 25 г метилдихлор-
фосфиноксида, что составляет 60,44% от теоретического вы-
хода; т. кип. 160—163° при 760 мм; т. пл. 32°.
Выход ацетилхлорида равен 21,3 г, что составляет 84,05°/о
от теоретического; т. кип. 50—52е при 760 мм; d420— 1,1047;
по - 1,3901.
Примечание.
Ацетилхлорид собирают в ловушке, охлажденной смесью сухого льда
с ацетоном.
Остальные требования те же, что и при получении этилдихлорфос-
финоксида.
СОВМЕСТНОЕ ПОЛУЧЕНИЕ МЕТИЛДИХЛОРФОСФИНОКСИДА И
АЦЕТОНИТРИЛА [9]
/С1
СН3Р( СН8С = N
II ХС1
о
СН3РОС12 М. в. 132,91 C2H3N М. в. 41,05
СХЕМА СОВМЕСТНОГО СИНТЕЗА МЕТИЛДИХЛОР-
ФОСФИНОКСИДА И АЦЕТОНИТРИЛА
СН3РС1+• А1С1з + CH3CONH2 4- 2C6H5N
,С1
СН3Р( + СН3С\ ф 2НС1 + A1C13-2C6H5N
II ХС1
О
Характеристика основного сырья
Комплексное соединение метилтетрахлорфосфина и хло-
ристого алюминия (см. выше).
Ацетамид, ч., ГОСТ 684—51.
Пиридин, ч., перегнанный, абсолютный, ГОСТ 2747—44.
Условия получения
В трехгорлую колбу емкостью 250 мл, снабженную ме-
шалкой с глухим затвором (см. примечание 1), капельной во-
ронкой и обратным холодильником, соединенным с ловушкой
(охлажденной до минус 78° смесью сухого льда и ацетона),
помещают 50 г (0,155 А!) комплексного соединения метилтет-
рахлорфосфина с хлористым алюминием и 9,2 г (0,155 Л4)
обезвоженного ацетамида. При охлаждении колбы до минус
20° (при более высокой температуре реакция протекает очень
12
(см.
вы-
81’
бурно) медленно прикапывают 2.4,6 г (0,31 Л!) абсолютного
пириДина. Происходит сравнительно бурная реакция, содер-
жимое колбы приобретает коричневый цвет. По окончании
прикапывания включается мешалка, колбу постепенно нагре-
вают до 100 — 110° и выдерживают при этой температуре до
прекращения выделения хлористого водорода (5—6 часов),
определяемого с помощью водного аммиака. При этом боль-
шая часть ацетонитрила отгоняется и собирается в охлажден-
ной ловушке. Затем перемешивание прекращают, обратный
холодильник заменяют прямым, соединенным с приемниками.
Из реакционной колбы отгоняют фракцию, кипящую в интер-
вале 40—75° при 20 мм. Эту фракцию переносят в колбу Кляй-
зена и перегоняют вторично. При этом выделяется 14 г ме-
тилдихлорфосфиноксида, что соответствует 67,4 % от теорети-
ческого выхода; т. кип. 162—163° при 760 мм; т. пл. 32°.
Далее продукты, сконденсировавшиеся в ловушке
примечание 2) разгоняют из колбы Кдяйзена. При этом
деляется ацетонитрил в количестве 4,3 г (67%); т. кип.
при 760 мм- г%20— 0,7792; п™-1,3468.
Этот метод может быть распространен на получение
личных алкилдихлорфосфиноксидов, исходя из соответствую-
щих комплексных соединений алкилтетрахлорфосфинов с
хлористым алюминием и амидов карбоновых кислот.
Примечания:
1. Мешалка с глухим затвором устроена следующим образом: на муф-
ту затвора одет вакуумный каучук, в который вставлена мешалка, сма-
занная глицерином нли другой смазкой. Такой затвор позволяет созда-
вать в системе вакуум с остаточным давлением 3—5 мм рт. ст.
2. Продукты, сконденсировавшиеся в ловушке перед разгонкой, осто-
рожно нагревают до 25—36* для удаления газообразного хлористого во-
дорода.
раз-
СОВМЕСТНОЕ ПОЛУЧЕНИЕ МЕТИЛДИХЛОРФОСФИНОКСИДА
И 1,2-ДИХЛОРЭТАНА [10]
С1СН2-СН2С1
2
С1
сн,р;
||ХС1
о
СН3РОС12 М. в. 132,91 С2Н4С12 М. в. 98,95
СХЕМА СОВМЕСТНОГО СИНТЕЗА МЕТИЛДИХЛОРФОСФИНОКСИДА
И 1,2-ДИХЛОРЭТАНА
СН3РС14-А1С13 + СН2-СН, + КС1 ->
С1
CH3Pf + С1СН2-СН2С1 + AlClg-КС1
II Cl
о
13
Характеристика основного сырья
Комплексное соединение метилтетрахлорфосфина и хлори-
стого алюминия (см. выше).
Окись этилена, перегнанная.
Калий хлористый, х. ч., свежепрокаленный, ГОСТ
4234—48.
Условия получения
В четырехгорлую колбу емкостью 250 мл, снабженную
мешалкой с глухим затвором, термометром, барботером, опу-
щенным до дна и обратным холодильником, соединенным с
ловушкой (охлажденной до минус 78° смесью сухого льда и
ацетона), помещают 50 г (0,155 Л1) комплексного соединения
метилтетрахлорфосфина с хлористым алюминием и 11,5 а
(0,155 М) хлористого калия. В реакционную смесь, охлаж-
денную до минус 15—20°, пропускают медленным током через
счетчик пузырьков, наполненный инертной жидкостью, 6,8 г
(0,155 М) окиси этилена. По окончании пропускания окиси
этилена и без прекращения перемешивания температура в
колбе медленно повышается до комнатной. Перемешивание
продолжают еще 1 час. Затем обратный холодильник заме-
няют прямым, соединенным с приемником, и перемешивание
прекращают. Через отвод охлажденной ловушки к системе
подключают вакуум 20 мм рт. ст. Из реакционной колбы от-
гоняют фракцию, кипящую в интервале 40—75° при 20 мм.
При этом низкокипящие продукты собираются в охлажден-
ной ловушке. Из полученной фракции повторной разгонкой
и.з колбы Кляйзена выделяют 14,8 г метилдихлорфосфинокси-
да, что составляет 72% от теоретического выхода; т. кип.
162—163° при 760 мм; т. пл. 32°. Продукты, сконденсировав-
шиеся в ловушке, разгоняются из колбы Кляйзена.
Выход 1,2-дихлорэтана 14,7 г, что составляет 96,1% от
теоретического; т. кип. 83—84°; %20— 1,2533; «д — 1,4438.
-лот метод может быть распространен на различные комп-
лексные соединения алкилтетрахлорфосфинов с хлористым
алюминием и другие алкиленоксиды.
СОВМЕСТНОЕ ПОЛУЧЕНИЕ МЕТИЛДИХЛОРФОСФИНСУЛЬФИДА
И 1,2-ДИХЛОРЭТАНА
О
сн3р; С1Снг-сн2С1
II ХС1
S
CH3SPC12 М. в. 148,97 С2Н4С12 М. в. 98,95
14
СХЕМА СОВМЕСТНОГО СИНТЕЗА МЕТИЛДИХЛОРФОСФИНСУЛЬ-
ФИДА И 1,2-ДИХЛОРЭТАНА
СН3РС14 • AlClg + СН2-СН2 КС1
xsz >
,С1
-> сн3р; + cich2-ch2ci+aici3-kci
II ХС1
S
Характеристика основного сырья
Комплексное соединение метилтетрахлорфосфина и хло-
ристою алюминия (см. выше).
Этиленсульфид, т. кип. 54,9° при 760 мм, получают из ро-
данистого калия и окиси этилена [11].
Калий хлористый, х. ч., безводный, свежепрокаленный.
I ОСТ 4244—38.
Условия получения
В четырехгорлую колбу емкостью 200 мл, снабженную ме-
ханической мешалкой, термометром, капельной воронкой и
обратным холодильником, соединенным с ловушкой, охлаж-
денной смесью сухого льда с ацетоном, помещают тщательно
перемешанную смесь 100 г (0,321 М) комплексного соедине-
ния метилтетрахлорфосфина с хлористым алюминием и 23,94 а
(0,321 Л4) свежепрокаленного хлористого калия.
К смеси, охлажденной до минус 5°, в течение часа прика-
пывают 19,26 г (0,321 М) этиленсульфида, после чего баню с
охлаждающей смесью убирают. Температура реакционной
массы через 20—30 минут самопроизвольно поднимается до
комнатной.
Затем содержимое колбы при перемешивании нагревают
на водяной бане при температуре 50—55° в течение 1,5 часа и
при 80—85° еще 30 минут.
По окончании нагревания мешалку выключают и реакци-
онную массу в реакторе охлаждают до комнатной температу-
ры. После этого мешалку вынимают из реактора (см. приме-
чание 1), обратный холодильник заменяют прямым и продук-
ты реакции выделяют перегонкой в вакууме 3—5 мм (см.
примечание 2).
После вторичной перегонки получают 29,65 г (61,9%) ме-
тилдихлорфосфинсульфида; т. кип. 150° при 760 мм;
d420 — 1,4221; л*’-1,5530.
15
Выход 1,2-дихлорэтана 6 г, что составляет 18,8% от тео-
ретического; т. кип. 84° при 760 мм; d420— 1,2533; Лд —1,44 38.
Примечания:
1, После прекращения нагревания и перемешивания мешалка подни-
мается над поверхностью реакционной массы, так как по мере охлажде-
ния последняя затвердевает.
2. Часть продуктов реакции, и в основном 1,2-дихлорэтан, собирает-
ся в ловушке охлажденной смесью сухого льда с ацетоном.
Литература
1. А. М. К 1 п п е а г, Е. А. Р е г г е п. J. Chem. Soc., 3437 (1952).
2. F. W. Н о f f m а п, T. С. S i ш a n s, Z. I. G 1 a n z. J. Amer.
Chem. Soc., 72, 3570 (1957).
3. A. M i c h a e 1 i s, F. Kammerer. Ber., 8, 1306 (1875).
4. A. Michaelis. Ber., 6, 816 (1873).
5 1. Linder. В r u g g e r, J e n k e r. Mh. Chem., '3/54, 263
(1929).
6. G. M. Kosolapoff. Organophosphorus Compounds, N.-Y., 1950.
7. M. И. К а б а ч н и к, П. А. Российская. Изв. АН СССР, Отдел
хим. наук, ,515 (1946).
8. К- В. Караванов, С. 3. Ивин, В. В. Лысенко,
Г. И. Дрозд. Ж, общ. химии (в печати).
9. С. 3. Ивин, К- В. Караванов, В. В. Лысенко. Авт. свид.
163615; Бюлл. нзобр., № 13 (1964).
10. С. 3. И в и н, К- В. Караванов, В. В. Лысенко. Авт. свид.
163617, там же.
И. И. В. Б р а з. Ж- общ. химии, 21, 688 (1951).
Поступила в июне 1964 г.
УДК 549.31.07
АЛКИЛДИХЛОРФОСФИНСЕЛЕНИДЫ
С. 3. ИВИН, И. Д. ШЕЛАКОВА
R-PSe С12
В литературе описан способ получения галоидангидридов
селенопроизводных кислот фосфора. Штрекер и Гроссман [1]
описали хлор- и бромангидриды диароксиселенофосфорной
кислоты, полученные ими присоединением металлического се-
лена к соответствующим галоидангидридам диароксифосфо-
ристой кислоты,
Алкилдихлорфосфинселениды в литературе не описаны.
Указанные соединения нами получены по аналогии синтеза
галоидангидридов диароксиселенофосфорной кислоты, т. е.
присоединением металлического селена к алкилдихлорфосфи-
нам. Присоединение селена к алкилдихлорфосфинам проте-
кает сравнительно легко, однако образующиеся алкилдихлор-
фосфинселениды оказались в присутствии кислорода неустой-
чивыми и в процессе реакции легко разлагались с выделени-
ем селена в виде красного осадка и алкилдихлорфосфинокси-
дов, Выделить образующийся алкилдихлорфосфинселенид
оказалось возможным лишь в том случае, если и получение,
и перегонку в вакууме проводить в токе азота или какого-
либо другого инертного газа.
Кроме того, нами разработан новый способ получения ал-
килдихлорфосфинселенидов путем взаимодействия металли-
ческого селена с комплексным соединением алкилтетрахлор-
фосфина и хлористого алюминия. Реакция протекает с обра-
зованием алкилдихлорфосфинселенида в токе азота. Наличие
в реакционной колбе воздуха или кислорода приводит к об-
разованию сложной смеси продуктов, выделить из которой ал-
килдихлорфосфинселенид практически не удается.
В чистом виде алкилдихлорфосфинселениды могут дли-
тельное время храниться в атмосфере азота в запаянных ам-
пулах,
2 Зак. 131 17
МЕТИЛДИХЛОРФОСФИНСЕЛЕНИД
Z/Se
CH3-P^C1
CHjPSeClg
M. в. 195, 87
СХЕМА СИНТЕЗА МЕТИЛДИХЛОРФОСФИНСЕЛЕНИДА [2]
х5е
CH3PCl2 + Se-> СН3Р%-С1
'С1
Характеристика основного сырья
Метилдихлорфосфин, получают по методике, описанной в
литературе [3].
Селен металлический, безводный, ч., ГОСТ 5455—50.
Азот, СТУ 36—13—748—61, осушенный над серной кисло-
той.
Условия получения
В колбу Кляйзена с елочным дефлегматором, термомет-
ром, прямым холодильником и трубкой для ввода сухого азо-
та помещают 83 г (0,71 Л4) метилдихлорфосфина и 56 а
(0,71 М) порошкообразного селена. Реакционную смесь про-
дувают азотом и нагревают при 80° в течение 2 часов. Обра-
зовавшийся метилдихлорфосфинселенид перегоняют в вакуу-
ме (присоединение селена и перегонку вещества проводят в
токе азота).
Выход продукта 80 г, что составляет 67% °т теоретическо-
го; т. кип. 74—75° при 25 „млцй^18 — 1,7540; п'^ — 1,5970.
Этот метод может быть использован для получения дру-
гих алкилдихлорфосфинселенидов.
C2II5PSeCl2
ЭТИЛДИХЛОРФОСФИНСЕЛЕНИД
xSe
СоН5 —Р—Cl
4 Cl
М. в. 209, 90
СХЕМА СИНТЕЗА ЭТИЛДИХЛОРФОСФИНСЕЛЕНИДА ИЗ
КОМПЛЕКСА И СЕЛЕНА [2]
^5е
С,Н5РСЦ-А1С13+ Se + KCl -> С2Н5 - P-^-Cl + КС1- А1С13
ХС1
18
Характеристика основного сырья
Комплексное соединение этилтетрахлорфосфина и хлори-
стого алюминия, получают по методике, описанной в Литера-
туре [4].
Селен металлический, безводный, ч., ГОСТ 5455—50.
Калий хлористый, безводный, свежепрокаленный, ГОСТ
4234—48.
Азот, СТУ 36—13—748—61, осушенный над серной кисло-
той.
Условия получения
В колбу Кляйзена с прямым холодильником, термометром
и трубкой для ввода сухого азота помещают смесь, состоя-
щую из 50 г (0,15 Л4) комплексного соединения этилтетра-
хлорфосфина и хлористого алюминия, 11,7 г (0,15 Л4) селена
и 11,1 г (0,15 М) хлористого калия. Смесь хорошо перемеши-
вают встряхиванием и выдерживают в токе азота 1 час, затем
нагревают до 200°, при этом комплекс расплавляется. Обра-
зовавшийся продукт отгоняют в вакууме 40—50 мм при
100—130° в токе азота.
Выход этилдихлорфосфинселенида после повторной пере-
гонки 15,6 г, что составляет 50% от теоретического; т. кип.
95° при 25 мм; d^— 1,6750; п* - 1,5850.
По описанной методике, исходя из соответствующих комп-
лексных соединений и селена, можно получить и другие ал-
килдихлорфосфинселениды.
Литература
1. Strecker, Grossman. Вег., 4 >. 63 (1916).
. 2. С. 3. И в и н, И. Д. Ш е л а к о в а. Ж. общ. химии, 31, 12. 4053
(1961).
3. И. П. Комков, К. В. Караванов, С. 3. Ивин. Ж. общ. хи-
мии, 28, 11, 2963 (1958).
4. 1. Р. Clay. J. Organ. Cheru., 16, 6, 892 (1951) .
Поступила в июне 1961 г.
УДК 547.569.2.07
АЛКИЛДИХЛОРФОСФИНСУЛЬФИДЫ
И. П. КОМКОВ, С. 3. ИВИН, к. В. КАРАВАНОВ
RP—CI
ХС1
Алкилдихлорфосфинсульфиды обычно получают длитель-
ным нагреванием в запаянных трубках (до 120—125°) смеси
алкилдихлорфосфинов с серой [1], действием пятисерпистого
фосфора на алкилдихлорфосфиноксиды [2] или при взаимо-
действии сероводорода с комплексными соединениями алкил-
тетрахлорфосфинов с хлористым алюминием [3].
Недостаток первых двух способов в том, что исходные
реагенты для их осуществления являются сравнительно мало-
доступными. По третьему способу алкилдихлорфосфинсуль-
фиды получают с низкими выходами.
Нами разработан простой способ, позволяющий получить
алкилдихлорфосфинсульфиды с хорошим выходом из вполне
доступных реагентов. Способ основан на взаимодействии ком-
плексных соединений алкилтетрахлорфосфинов и хлористого
алюминия с серой, роданистым калием и этиленсульфидом в
присутствии свежепрокаленного хлористого калия [4, 5].
ЭТИЛДИХЛОРФОСФИНСУЛЬФИД
CsHsPSCl3 М. в. 163, 0
СХЕМА СИНТЕЗА ЭТИЛДИХЛОРФОСФИНСУЛЬФИДА
КС1 .S
C,H6PC14-A1C1s 4-S---^С.,Н5Р—С1 + А1С13.КС1
"С1
20
Характеристика основного сырья
Комплексное соединение этилтетрахлорфосфина с хлори-
стым алюминием, получают по методике, описанной в насто-
ящем сборнике (см. статью «Комплексные соединения алкил-
тетрахлорфосфинов с хлористым алюминием» в этом сборни-
ке).
Сера, продажная, черенковая (сухая).
Калий хлористый, х. ч., безводный, свежепрокаленный.
ГОСТ 4234—48.
Калий роданистый, ч., обезвоженный, ГОСТ 4139—48.
Условия получения
Первый способ. В колбу Кляйзсна емкостью 0,5 л загру-
жают хорошо перемешанную смесь, состоящую из 100 г
(0,32 М) комплексного соединения этилтетрахлорфосфина с
хлористым алюминием, 9,6 г (0,3 УИ) серы и 22,2 г (0,32 М)
свежепрокаленного хлористого калия. Колбу помещают в
глицериновую баню, которую медленно, в течение 3 часов, на-
гревают до 170° (см. примечание). Образовавшийся продукт
реакции отгоняют в вакууме 15—20 мм.
Выход этилдихлорфосфинсульфида 26,3 г, что составляет
61% от теоретического; т. кип. 68—69° при 20 мм;
d420— 1,35 32; «20-1,5435.
Аналогичным путем можно получить и другие алкилди-
хлорфосфинсульфиды при нагревании соответствующих ком-
плексных соединений с серой, а также с сульфидами алюми-
ния, калия, сурьмы, фосфора в присутствии хлористого калия.
Хлористый калий применяется для связывания хлористого
алюминия.
Примечание.
Нагревание реакционной массы ведут так, чтобы как можно меньше
проскакивало пузырьков газа через склянку Тищенко, поставленную на
конце прибора.
Второй способ. В колбу Кляйзена емкостью 0,3 л с пря-
мым холодильником и приемниками помещают хорошо пере-
мешанную смесь, состоящую из 50 г (0,152 М) комплексного
соединения этилтетрахлорфосфина с хлористым алюминием,
19,5 г (0,2 Af) роданистого калия. Смесь нагревают под ваку-
умом 20 минут таким образом, чтобы продукты реакции при
этом отгонялись. При повторной перегонке собранной жидко-
сти получают 18,5 г этилдихлорфосфинсульфида, что состав-
ляет 74% от теоретического; т. кип. 68° при 20 мм (178° при
760 мм); — 1,3539; п™ - 1,5431.
По литературным данным, т. кип. 69° при 20 мм;
<Л20 — 1,3532 [1].
21
Хлористый калий для связывания хлористого алюминия в
этой реакции не применяют, он образуется в процессе синте-
за.
Литература
1. В. М. Плец. Органические соединения фосфора, М., Оборонгпз,
1940, стр. 176—177.
2. М. И. К а б а ч н и к, Н. Н. Годовиков. Докл. АН СССР, НО,
217 (1956).
3. А. М. К innear, Е. А. Реггеп. J. Chem. Soc., 3437 (1952).
4. И. П. Комков, К. В. Караванов, С. 3. Ивин. Ж. общ. хи-
мии. 28, 2960 (1958).
5. К. В. К а р а в а н о в, С. 3. Ивин. Ж. общ. химии, 35, 78 (1965)
Поступила в июне 1964 г.
УДК 661.718.1.07
АЛКИЛ(АРИЛ)ТЕТРАФТОРФОСФИНЫ
И. П. КОМКОВ, С. 3. ИВИН, к. в. КАРАВАНОВ, Л. Е. СМИРНОВ
R(Ar)PF4
Алкил (арил) тетрафторфосфины являются реакционноспо-
собными соединениями. Они легко вступают в реакцию со
многими веществами, образуя различные производные алкил
(арил) ортофосфиновой кислоты и могут быть использованы в
качестве полупродуктов для получения различных фосфор-
органических соединений.
В литературе описан способ получения алкил(арил)тет-
рафторфосфинов, заключающийся во взаимодействии ал-
кил (арил) дихлорфосфинов с трехфтористой сурьмой или
трехфтористым мышьяком. Выход алкил(арил)тетрафторфос-
финов достигает 60—70%. Так, в 1959 г. Ягупольский и Ива-
нова [1] описали получение фенилтетрафторфосфина фтори-
рованием фенилдихлорфосфина трехфтористой сурьмой.
Алкилтетрафторфосфины впервые были получены нами [2]
действием фторидов тяжелых металлов или безводного фто-
ристого водорода на алкил (арил) дихлорфосфины. При про-
ведении указанных реакций происходит окисление трехва-
лентного фосфора до пятивалентного с одновременным вос-
становлением до свободного состояния элемента—носителя
фтора. Реакция протекает по схеме
3 RPC12 + 4 SbF3 -> 3 RPF4 + 2SbCl3 + 2 Sb.
В случае применения фтористого водорода получают не-
стойкие алкилдифторфосфины, которые затем диспропорцио-
нируются в алкилтетрафторфосфины и циклические соедине-
ния фосфора.
Позже в литературе [3] было описано получение алкил-
тетрафторфосфинов действием трехфтористой сурьмы на ал-
килдихлорфосфины.
23
Нами разработан кроме вышеуказанного [2] и другой
удобный способ получения алкилтетрафторфосфинов из дос-
тупных исходных веществ. Метод основан на фторировании
трехфтористой сурьмой, трехфтористым мышьяком, фтори-
стым калием и фтористым водородом комплексных соедине-
ний алкилтетрахлорфосфинов с хлористым алюминием. Наи-
более удобным фторирующим агентом оказался безводный
фтористый водород. Он реагирует с моноалкильными комп-
лексными соединениями при комнатной и даже более низкой
температурах с образованием алкилтетрафторфосфинов:
HF
RPC14-A1C13---> RPF4 + НС1 + A1F3-
Однако для достижения высоких выходов необходим из-
быток фтористого водорода.
ПОЛУЧЕНИЕ ЭТИЛТЕТРАФТОРФОСФИНА ИЗ КОМПЛЕКСНЫХ
СОЕДИНЕНИЙ
C,H5PF4 М. в. 136,02
СХЕМА СИНТЕЗА ЭТИЛТЕТРАФТОРФОСФИНА
HF
С2Н6РС14-А1С13---> C2I15PF4H-HC1+A1F3
Характеристика основного сырья
Комплексное соединение этилтетрахлорфосфина с хлори-
стым алюминием, получают по методике, описанной в насто-
ящем сборнике (см. статью «Комплексные соединения алкил-
тетрахлорфосфинов с хлористым алюминием» в этом сборни-
ке).
Условия получения
Первый способ. В У-образную алюминиевую трубку диа-
метром 15 мм и длиной 500 мм помещают 100 г (0,3 А4) ком-
плексного соединения этилтетрахлорфосфииа с хлористым
алюминием (см. примечание 1). Затем через трубку пропус-
кают 420 г (2,2 М) фтористого водорода (см. примечание 2).
Реакция протекает со значительным выделением тепла. Этил-
тетрафторфосфин при этом выносится из трубки током обра-
зующегося хлористого водорода и улавливается в трех после-
довательно соединенных ловушках, охлажденных до минус
78°. После пропускания фтористого водорода систему в тече-
ние часа продувают током сухого воздуха.
Сконденсированный этилтетрафторфосфин в ловушках пе-
регоняют в токе азота (см. примечание 3). Получают 17,5 г
24
этилтетрафторфосфина, что составляет 58% от теоретического
выхода; т. кип. 34—34,5° при 760 мм; d^-°— 1,3158.
По литературным данным, т. кип. продукта 34—35° при
760 мм [3].
Второй способ. В колбу Кляйзена емкостью 150 мл поме-
щают смесь, состоящую из 50,75 г (0,15 Л1) комплексного сое-
динения этилтетрахлорфосфина с хлористым алюминием,
62,6 г (0,35 Л4) трехфтористой сурьмы и 11,2 г (0,15 Л4) хло-
ристого калия. Смесь хорошо перемешивают и нагревают
пламенем горелки до начала реакции, протекающей затем с
большим выделением тепла. Образующийся этилтетрафтор-
фосфин при этом отгоняют в ловушку, охлажденную до ми-
нус 40—50°. При перегонке получают 10 г (50%) этилтетра-
фторфосфина с т. кип. 34°.
Описанными методами, исходя из соответствующих комп-
лексных соединений, можно получить различные алкилтетра-
хлорфосфины.
Примечания:
1. Комплексное соединение этилтетрахлорфосфина с хлористым алю-
минием легко гидролизуется, в связи с чем следует загружать У-образную
трубку как можно быстрее.
2. Фтористый водород вызывает ожоги на коже, поэтому необходимо
соблюдать осторожность.
3. Этилтетрафторфосфин гидролизуется на воздухе.
ПОЛУЧЕНИЕ ЭТИЛТЕТРАФТОРФОСФИНА ИЗ
АЛКИЛДИХЛОРФОСФИНОВ
СХЕМА СИНТЕЗА
3 С,Н6РС12 + 4 SbFs -> 3 C2H5PF4 -% 2 SbCl3 + 2 Sb.
Характеристика основного сырья
Этилдихлорфосфин, перегнанный, получают по одному из
описанных в настоящем сборнике методов (см. статью «По-
лучение алкилдихлорфосфинов восстановлением алкилди-
хлорфосфиноксидов» в этом сборнике).
Трехфтористая сурьма, ч., МРТУ 6-09-524—63, безводная
свежепрокаленная.
Условия получения
В колбу Кляйзена с елочным дефлегматором помещают
54,6 г (0,3 Л4) трехфтористой сурьмы и из капельной воронки
медленно прибавляют 30 г (0,22 Л4) этилдихлорфосфина. Ре-
акция протекает с разогреванием. После прибавления всего
количества этилдихлорфосфина содержимое колбы оставляют
25
на 30 минут при комнатной температуре, а затем образовав-
шийся этилтетрафторфосфин перегоняют.
Выход этилтетрафторфосфина 21,9 г, что составляет 70%
от теоретического; т. кип. 34—34,5° при 760 мм; d420—1,3074.
По описанной методике, исходя из различных алкилди-
хлорфосфинов, можно получить различные алкилтетрафтор-
фосфины.
Литература
1. М. М. Ягупольский, Ж- М. Иванова. Ж. общ. химии, 29,
3766 (1959).
2. И. П. Комков, С. 3. Ивин, К- В. Караванов, Л. Е. Смир-
нов. Ж. общ. химии, 32, 301 (1962).
3. Schmutzler. R. chem. and Ind., 43, 1868 (1952).
Поступила в июле 1964 г.
УДК 547.535.3'032.07
5-АЦЕТИЛ ПСЕВДОКУМОЛ
2,4,5-Триметилацетофенон
М. Т. РАЗУМОВСКАЯ, А. И. БЕЛЯКОВА, Г. И. КОРЕЛЬСКАЯ
СН3
О )
II г Vch3
СН3-С-1 II
СН3
СдНнО
М. в. 162,23
Ацетилпсевдокумол применяется для синтеза дуриловой,
пиромеллитовой кислот и других органических продуктов.
По литературным данным, ацетилпсевдокумол получают
по реакции Фриделя—Крафтса конденсацией псевдокумола с
хлористым ацетилом в присутствии хлористого алюминия в
сероуглероде [1], лигроине [2] и в других индифферентных
растворителях; окислением а-хлор-|3-(2,4,5-триметилфенил)-
а-пропилена водным раствором перманганата калия в ацето-
не [В].
Нами проверены и уточнены условия конденсации псевдо-
кумола с хлористым ацетилом, а также отработаны условия
реакции конденсации псевдокумола с ангидридом уксусной
кислоты.
В данном сообщении описываются оба метода получения
ацетилпсевдокумола.
27
СХЕМА СИНТЕЗА АЦЕТИЛПСЕВДОКУМОЛА ПУТЕМ
КОНДЕНСАЦИИ ПСЕВДОКУМОЛА С ХЛОРИСТЫМ
АЦЕТИЛОМ
СН3СОС1+А1С13 СН3СОС1-А1С13
сн3 сн3
1 о '
ЛгСНз + СН3СОСЬ А1С1а -> II fVCHa+HCl
Чх сн3-с-^ х
СН3 А1С1’СН3
сн3
0 А
II А-сн8
СН3-~С-^;! +«н2о->
I сн3
А1С13
сн3
0 А
-> CH _Ll АСНз + А1(ОН)з + ЗНС1
3 ч/
I
СНз
Характеристика основною сырья
Псевдокумол, ч., ВТУ РУ 902—58.
Ацетил хлористый, ч., ГОСТ 5829—51.
Алюминий хлористый, безводный, ВТУ МХП 3500—52.
Углерод четыреххлористый, ч., ГОСТ 5827—57.
Кислота соляная, ч., ГОСТ 3118—46.
Кальций хлористый, плавленый, ч., ГОСТ 4460—48.
Уксусный ангидрид, ч., ГОСТ 5815—52,
Условия получения
Получение комплекса. В четырехгорлую колбу емкостью
3 л, снабженную мешалкой с глицериновым затвором, ка-
пельной воронкой и обратным холодильником, защищенным
хлоркальциевой трубкой, помещают 1125 мл четыреххлори-
стого углерода и 175,5 г (1,32 М) хлористого алюминия.
Смесь интенсивно перемешивают, охлаждают до 8—12° и
28
быстро прибавляют НО мл (1,4 Л1) хлористого ацетила, а за-
тем в течение 1 —1,2 часа — раствор 170 мл (1,25 М) псевдо-
кумола в 225 мл четыреххлористого углерода, следя за тем,
чтобы температура реакционной смеси не поднималась выше
25° (см. примечание 1). Реакционную смесь продолжают пе-
ремешивать при комнатной температуре до прекращения
обильного выделения хлористого водорода, что указывает на
конец реакции. Реакция длится около 3,5 часа (см. примеча-
ние 2).
Цвет реакционной массы в процессе реакции постепенно
меняется от желтого до оранжево-красного вследствие обра-
зования комплексного соединения хлористого алюминия с
ацетилпсевдокумолом.
Разложение комплекса. Реакционный раствор охлаждают
до 8—10° и при перемешивании прибавляют 10%-ный рас-
твор соляной кислоты с такой скоростью, чтобы температура
смеси не превышала 25° (см. примечание 1).
При добавлении кислоты реакционная масса приобретает
светло-желтую окраску и из раствора выпадает хлопьевид-
ный осадок гидрата окиси алюминия. Кислоту (650—700мл)
прибавляют до полного растворения осадка (см. примеча-
ние 3).
Затем мешалку останавливают и реакционную смесь вы-
держивают при комнатной температуре в течение 30 минут.
Нижний органический слой отделяют, промывают водой до
нейтральной реакции на лакмус и сушат над хлористым каль-
цием в течение 3—4 часов. После отгонки четыреххлористого
углерода (см. примечание 4) остаток перегоняют в вакууме,
собирая фракцию продукта при 132—133715—16 мм.
Выход 5-ацетилпсевдокумола 150—170 г, что составляет
74—84% от теоретического.
По литературным данным, т. кип. продукта 137—138° при
20 мм; 247- 248° при 760 мм [1, 2].
СХЕМА СИНТЕЗА АЦЕТИЛПСЕВДОКУМОЛА ПУТЕМ
КОНДЕНСАЦИИ ПСЕВДОКУМОЛА С УКСУСНЫМ АНГИДРИДОМ
(СН3СО)2О -% /zAlCls -> СН3СОС1/гА1С13 + СН3СООА1С1а
СНз
// \
| || ° 3 -с СН3СОС1-А!С1а
I
СН3
сн3
I
° \ '|ГСНз % на
СНз-Г Y
Aids СНз
29
сн3
0 Jx
CH J! I ГСНз +«H2o ->
CH3-C—
А1С13СНз
CH3
0
-* II f |ГСНз + А1(ОН)з + 3HC1
CH3-C-’4J
CH3
Конденсация псевдокумола с уксусным ангидридом прово-
дится в том же приборе, что и конденсация псевдокумола с
хлористым ацетилом.
К 1500 мл четыреххлористогб углерода при перемешива-
нии добавляют 333 г (2,5 А4) хлористого алюминия. Смесь
интенсивно перемешивают, охлаждают до 8—12° и быстро
приливают 94 мл (1 Л4) уксусного ангидрида, а затем в тече-
ние 25—30 минут — раствор 135 мл (1 М) псевдокумола в
200 мл четыреххлористого углерода, следя за тем, чтобу тем-
пература реакционной массы не поднималась выше 25° (см.
примечание 1). После прибавления раствора реакционную
смесь, не прекращая перемешивания, выдерживают около
4 часов при 25°.
Вследствие образования комплекса СНзСОС6Н2.(СН3)зА1С1з
реакционная масса приобретает красно-коричневую окраску.
Дальнейшее разложение комплекса и выделение ацетил-
псевдокумола ведут, как описано выше.
Выход 5-ацетилпсевдокумола 108—118 г, что составляет
67—73% от теоретического.
П р и м е ч а н п я:
1. При более высокой температуре происходит осмоленпе значитель-
ной части продукта.
2. Увеличение длительности процесса приводит к осмолению значи-
тельной части продукта и снижению его выхода.
3. Кислота применяется с небольшим избытком против необходимого
количества для удаления алюминия в виде растворимых солей.
4. Четыреххлористый углерод можно регенерировать и использовать
вновь.
Л итература
1. A d. С 1 a u s. J. Р г a k t. Chem., (2), 41, 509 (1890).
2. А. К 1 a g е s, Р. А 11 е n d о г f. Вег., 31, 1005 (1898).
3. К. A n v е г s, A. Kockritz. Liebigs Ann. Chem., 352, 312 (1904).
Поступила в июле 1961 г.
ИРЕА
УДК 547.415.1.07
N-БЕНЗИЛЭТИЛЕНДИАМИН
Р. П. ЛАСТОВСКИИ, И. Д. КОЛПАКОВА, Л. В. КРИНИЦКАЯ,
Т. И. ИВАНОВА
ch,nhch2ch2nh3
I
C9HuN2 М. в. 150, 22 .
N-Бензилэтилендиамин является промежуточным продук-
том для синтеза бензилэтилендиамин-М,М,М7-триуксусной
кислоты, а также ряда других синтезов.
Бензилэтилендиамин может быть получен восстановлени-
ем основания Шиффа, в свою очередь полученного взаимо-
действием бензальдегида или толуола с избытком этиленди-
амипа [1—5].
Бензилэтилендиамин получается также в качестве побоч-
ного продукта при синтезе дибензилэтилендиамипа конден-
сацией дихлорэтана с избытком бензиламина (наряду с
М,Ь1,М'-трибензилэтилендиамином) [6], а также действием со-
ляной кислоты на М,№'-дибензолсульфонил-М,М'-дибензил-
этилендиамин [7]. Существует еще ряд способов получения
бензилэтилендиамина с использованием в качестве сырья
Р-гуанидинэтанола [8], 1-бензил-5-гомопиперазипа [9] и дру-
гие.
Наиболее доступным методом синтеза этого вещества яв-
ляется конденсация этилендиамина с хлористым бензилом с
предварительной защитой аминогрупп (ацетилирование этил-
ацетатом и конденсация с бензолсульфохлоридом) и последу-
ющим разложением ацетилсульфониламида соляной кислотой
[10, 11, 12, 13]. Синтез N-бензилэтилендиамина возможен пу-
тем непосредственной конденсации хлористого бензила с из-
31
бытком этилендиамина с последующим выделением продукта
экстракцией бензолом [14, 15, 16].
Нами проверен и уточнен последний метод синтеза.
СХЕМА СИНТЕЗА N-БЕНЗИЛЭТИЛЕНДИАМИПА
СН2С1
NaOH
+ nh2ch2ch2nh2--------->-
X/
ch2nhch2ch2nh3
-> + NaCl %-H2O
X/
Характеристика основного сырья
Хлористый бензил, ч., ТУ МХП 50—49.
Этилендиамин, 100%-ный, перегнанный.
Условия получения
В четырехгорлую колбу, снабженную механической ме-
шалкой, термометром, обратным холодильником и капельной
воронкой, помещают 600 г (10 УИ) 100%-ного этилендиамина,
нагревают до 80° и через капельную воронку при перемеши-
вании постепенно в течение 3 часов добавляют 253 г (2 М)
хлористого бензила с такой скоростью, чтобы температура в
колбе не поднималась выше чем на 2—3% После прибавления
хлористого бензила, продолжая перемешивание, реакцион-
ную массу выдерживают в течение 8 часов при температуре
80°, затем переносят ее в стакан, добавляют 140 мл (2 М)
40%-ного едкого натра, отфильтровывают выпавший осадок
хлористого натрия и из фильтрата экстрагируют продукт тре-
мя порциями бензола по 750 мл. Полученный бензольный
экстракт сушат над едким кали, отгоняют бензол. Высушен-
ный над металлическим натрием остаток перегоняют в ваку-
уме.
Получают 207 210 г (69—70%) N-бензилэтилендиамина
в виде жидкости с т. кип. 130—135° при 6 мм; т. пл. хлоргид-
рата (из спирта) 253°.
По литературным данным, т. кип. продукта 136—140°при
5 мм [15]; т. пл. хлоргидрата (из спирта) 253°.
N-Бензилэтилендиамин растворим в воде, спирте, эфире,
бензоле. На воздухе слегка желтеет.
Найдено, %: N—18,64; 18,72.
C9HuN2. Вычислено, %: N—18,70.
32
Л итература
1. J. Van Alphen. Recueil trav. chim , 54, 595 (1935).
2. Z. Eckstein, Л. Lukasiewicz. Bulk Acad. Poton. sci., ser scf,
Chem., Qeol. et Geograph, 7, 11, 789, 797 (1959).
3. Z. Eckstein, A. Lukasiewicz. Przem. Chem., 39, 367 (I960).
4. Англ. пат. 719887; С. A., 50, P 1923a (1954).
5. С. В e n к о, M. F i s 1 e r. Croat. Chem. Acta, 30, 4, 243 (1958).
6. A. Frost, S. Chaberek, A. E. Martell. J. Amer. Chem. Soc.,
71. 3842 (1949).
7. L. В lei er. Ber„ 32, 1825 (1899).
8. H. S c h о 11 e. Z. phys. siol. Chem., 174, 119 (1928).
9. S. C. D 1 с к e r m a n n, A. J. В c s о z z i. J. Organ. Chem., 19, 1855
(1954).
10. S. R. Aspin all. J. Anier. Chem. Soc., 63, 852 (1941).
11. A. J. H i 11, S. R. A s p i n a 11. J. Amer. Chem. Soc., 61, 82'2 (1939).
12. L. H. Amundsen, R. J. Longley. J. Amer. Chem. Soc., 62, 2811
(1940).
13. R. S. S11 r e i b e r, R. L. S h r i n e r. J. Amer. Chem. Soc., 56, 1618,
(1934).
14. F. L i n s к e r, R. L. E v a n s. J. Amer. Chem. Soc.. 67, 1581 (1945).
15. A. E. Fr о s t, A. A. С a r 1 s о n. J Organ. Chem., 24, 1581 (1959).
16. A. J. В r u n o, S. C h a b e г e к, A. E. .Martel 1. J. Amer Chem.
Soc,, 78, 2723 (1956).
Поступила в сентябре 1964 г.
ИРЕА
3
сак. 131
УДК 547.633.6.07
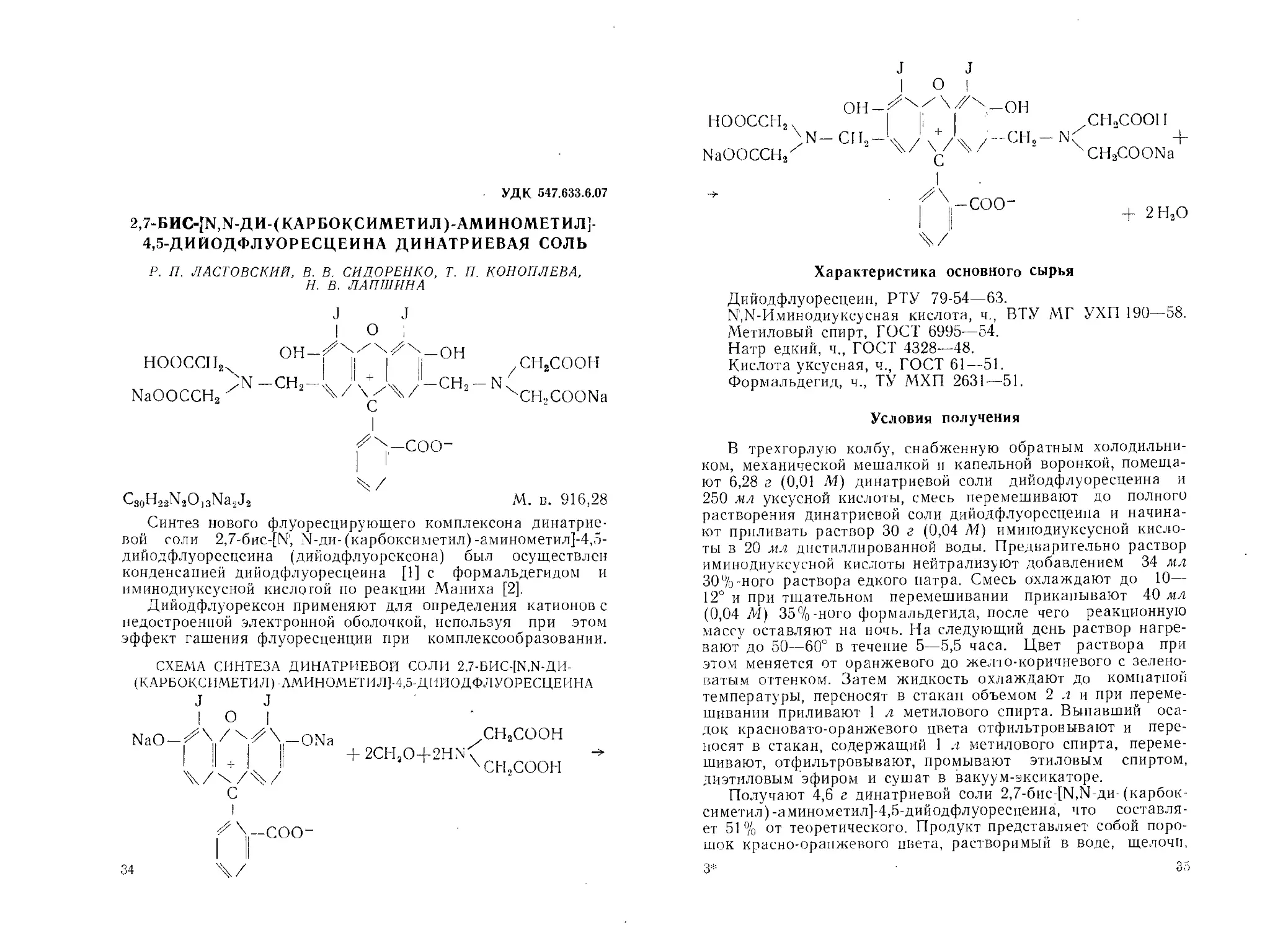
2,7-BHC-[N,N-ДИД КАРБОКСИМЕТИЛ )-АМИНОМЕТИЛ]
4,5-ДИЙОДФЛУОРЕСЦЕИНА ДИНАТРИЕВАЯ СОЛЬ
Р. П. ЛАСТОВСКИЙ, В. В. СИДОРЕНКО, Т. П. КОНОПЛЕВА,
И. В. ЛАПШИНА
О
нооссп2х
NaOOCCH3 х
ОН-Х\^\Х\_ОН
N -СН2~\ / \Ja / ~СН2 - N (
С
СН2СООН
CH.COONa
C3oH23N30]3Na2J2
М. в. 916,28
Синтез нового флуоресцирующего комплексона динатрие-
вой соли 2,7-6hc-[N', N-ди-(карбоксиметил)-аминометил]-4,5-
дийодфлуоресцеина (дийодфлуорсксона) был осуществлен
конденсацией дийодфлуоресцеина [1] с формальдегидом и
иминодиуксусной кислотой по реакции Маниха [2].
Дийодфлуорексон применяют для определения катионов с
недостроенной электронной оболочкой, используя при этом
эффект гашения флуоресценции при комплексообразовании.
СХЕМА СИНТЕЗА ДИНАТРИЕВОЙ СОЛИ 2.7-БИС-[М.М-ДИ-
(КАРБОКСИМЕТИЛ) АМИНОМЕТИЛ]-4,5 ДИЙОДФЛУОРЕСЦЕИНА
J J
I О |
NaO-X\ / xZ\-ONa
+ 2CHaO--|-2HN
СН2СООН
ХСН2СООН
f \-coo-
34
X/
J J J ° 1
нооссн2х СНоСООП
NaOOCCH/ >n-cii3-L/\+Л /-Chh-N^ “ 4- 7 Y xCH3COONa 1 |/Х-соо- + 2НзО 4/
Характеристика основного сырья
Дийодфлуоресцеин, РТУ 79-54—63.
М'.Х-Иминодиуксусная кислота, ч., ВТУ МГ
Метиловый спирт, ГОСТ 6995—54.
Натр едкий, ч„ ГОСТ 4328—48.
Кислота уксусная, ч., ГОСТ 61—51.
Формальдегид, ч., ТУ МХП 2631—51.
УХП 190—58.
Условия получения
В трехгорлую колбу, снабженную обратным холодильни-
ком, механической мешалкой и капельной воронкой, помеща-
ют 6,28 г (0,01 Л4) динатриевой соли дийодфлуоресцеина и
250 мл уксусной кислоты, смесь перемешивают до полного
растворения динатриевой соли дийодфлуоресцеина и начина-
ют приливать раствор 30 г (0,04 М) иминодиуксусной кисло-
ты в 20 мл дистиллированной воды. Предварительно раствор
иминодиуксусной кислоты нейтрализуют добавлением 34 мл
30%-ного раствора едкого натра. Смесь охлаждают до 10—
12° и при тщательном перемешивании прикапывают 40 мл
(0,04 Л1) 35%-ного формальдегида, после чего реакционную
массу оставляют на ночь. На следующий день раствор нагре-
вают до 50—60° в течение 5—5,5 часа. Цвет раствора при
этом меняется от оранжевого до желто-коричневого с зелено-
ватым оттенком. Затем жидкость охлаждают до комнатной
температуры, переносят в стакан объемом 2 л и при переме-
шивании приливают 1 л метилового спирта. Выпавший оса-
док красновато-оранжевого цвета отфильтровывают и пере-
носят в стакан, содержащий 1 л метилового спирта, переме-
шивают, отфильтровывают, промывают этиловым спиртом,
диэтиловым эфиром и сушат в вакуум-эксикаторе.
Получают 4,6 г динатриевой соли 2,7-бис-Щ,1Ч-ди-(карбок-
симетил)-аминометил]-4,5-дийодфлуоресцеина, что составля-
ет 51% от теоретического. Продукт представляет собой поро-
шок красно-оранжевого цвета, растворимый в воде, щелочи,
За
плохо растворимый в ацетоне, нерастворимый в спирте, бен-
золе, серном эфире и хлороформе.
CsgH22N2O13Na2Ja. Найдено, %: С—39, 51; Н—2,5.
Вычислено, %: С—39,3; Н-2,5.
Литература
1. Химические реактивы и препараты (справочник), М., Госхимнздат,
1953, стр. 289.
2 G. Schwarzenbach, G. Anderegg, R. S а 11 m a n. Helv.
(him. acta, 35, 1785 (1952).
Поступила в сентябре 1964 г. ИРЕА
УДК 547.541.2.07
4,4' БИС-[2,4-БИС-(2-КАРБОКСИМЕТИЛАМИНО)-1Д5-
ТРИАЗИНИЛ 6 АМИНО]-2,2 -СТИЛЬБЕНДИСУЛЬФО-
КИСЛОТЫ ГЕКСАНАТРИЕВАЯ СОЛЬ
Р. П. ЛАСТОВСКИЙ, В. ,ff. ТЕМКИНА, Г. Ф. ЯРОШЕНКО,
Л. М. САМЫЛОВА
NHCH2COONa
нс-f V_nh-c<n с>
х Ч-с/
SO3Na
NHCH2COONa
SO3Na
NH СН„С OONa
НС-/ 4—NH—С
N=C
Cag^aNjaO^NagSa
NHCH2COONa
M. в. 952, 62
Производные бис- (триазиниламино) -стильбена в послед-
ние годы нашли широкое применение в качестве флуорес-
центных красителей [1—5].
4,4/-Бис-[2,4-бис-( 2-карбоксиметиламино )-1,3,5-триазинил-
6-амино]-2,2'-стильбендисульфокислоты гексанатриевая соль
является не описанным в литературе соединением, которое
образует комплексы с рядом катионов. Соединение получено
по аналогии с триазинтриуксусной кислотой [6]. Синтез ком-
плексона состоит из нескольких стадий. Первая стадия за-
ключается в конденсации натриевой соли*диаминостильбен-
37
дисульфокислоты с 2 молями хлористого цианура при 0°, вто-
рая стадия - в замещении двух атомов хлора на аминоаце-
татную группу при 45°, третья — в замещении оставшихся
атомов хлора при 95°. Продукт выделяется при подкислении
раствора.
СХЕМА СИНТЕЗА СОЕДИНЕНИЯ
НС-/’ V- NH.,
SO3Na
,i SO3Na
НС—/ ~/-NH2
Cl
I
,N = C,
2C1-C< >N
Cl
I
---о /N = cx
HC—/ NH- C'
\==./ x'N-cZ
SOsNa I
1 SO3Na ,
V—N-Gx
HC —/ /-NH-C^ ;N
XN=CZ
Cl
HSNCH,COOH
---------->
45°; 95°
Na2CO3
NHCHsCOONa
HC—/ NH -Cx/N-C\n
/=X ^'N-C^
Мл I
3 NHCH2COONa
SO3Na
HC-/- NH - cf
NHCH3COONa
I
n-ca
4N
N=CX
NHCH2COONa
38
Характеристика основного сырья
4,4/-Диаминостильбен-2,2<-дисульфокислота, ч., МРТУ
6-09-78—62.
Цианур хлористый, ч.
Глйкокол (амииоуксусная кислота), ч., ГОСТ 5860—51.
Условия получения
В колбу емкостью 500 мл с мешалкой, термометром, ка-
пельными воронками для растворов соды и диаминостильбен-
дисульфокислоты помещают 60 мл воды, охлаждают до тем-
пературы 0° и при размешивании добавляют суспензию
3,72 г (0,019 М) хлористого цианура в 3,8 мл 1%-ного рас-
твора смачивателя ОС-20, предварительно охлажденного до
0°. К реакционной массе добавляют еше 40 мл воды, охлаж-
денной до 0—1°. Затем при 0—5° и быстром размешивании в
течение 10—15 минут прикапывают из капельной воронки
раствор натриевой соли диаминостильбендисульфокислоты
(см. примечание), поддерживая pH 6—7 по универсальной!
индикаторной бумажке добавлением из капельной воронки
10%-ного раствора соды. Реакционную массу выдерживают
при температуре 0—5° в течение 30 минут.
После выдержки добавляют 3,53 г (0,042 Л1) аминоуксус-
ной кислоты, предварительно нейтрализованной 10%-ным рас-
твором соды до pH 7.
Затем заменяют капельную воронку для диаминостиль-
бендисульфокислоты обратным холодильником и медленно
нагревают на глицериновой бане в течение 1,5 часа до 95—
97°, поддерживая pH 7, а в конце реакции—pH 8 добавлени-
ем 10%-ного раствора соды.
Реакционную массу выдерживают при 95—97° в течение
5 часов, затем раствор фильтруют, подкисляют концентриро-
ванной соляной кислотой до pH 2 и охлаждают до 0—2°. Вы-
павший осадок желтого цвета фильтруют, промывают 85 %-
ным метиловым спиртом до отсутствия ионов хлора и сушат
в сушильном шкафу при 60°.
Полученный продукт представляет собой гексанатриевую
соль 4,4/-бис-[2,4-бис-(1,2-карбоксиметиламнно)-1,3,5-триази-
нил-6-амино]-2,2'-стильбендисульфокислоты.
Выход 6,8 г, что составляет 66% от теоретического.
Найдено, %: С-35,5; 35,31; Н-3,26; 3,09;
N—17,29; 17,15; Na- 14,79; 14,86.
C28H.,2NI2OHNa6S2. Вычислено, %: С —35,30; Н — 2,53; N — 17,64;
Na-14,5.
Примечание.
Раствор 3,82 г (0,01 М) диаминостильбендисульфокислоты в 50 мл
воды нейтрализуют в течение 30—40 минут 10%-ным раствором соды до
pH 7—8, затем фильтруют и охлаждают до 0—Г.
39
Литература
1. Dennis, A. W. Adams, R. И. Wilson, Англ. пат. 624051
(1949).
2. H. Ishikawa, Т. N i in и г а. Япон. пат. 7172 (1959).
3. В. Н. Garioll, J. Е. Jones. Пат. США 2875058 (1959).
4. Н. На ursermann, R. Keller. Textil — Rundschau, 16, 176
(1961).
5. Noguchi’Tamehiko, Vuki Gesei Kagaku Куока i.
Schi, 19, 920 (1961).
6. P. П. Ластовский, H. M. Дятлова, И. Д. Колпакова.
Ж. аналит. химии. 15, 4, 419 (1960).
Поступила в сентябре 1961 г. ИРЕА
УДК 66.062.412.07
р-БРОМЭТИЛАМИН БРОМИСТОВОДОРОДНЫЙ
Р. И. ЛАСТОВСКИЙ, И. Д. КОЛПАКОВА, Л. В. КРИНИЦКАЯ,
Л. Д. ЗАВЬЯЛОВА
BrCH2CH2NH2-HBr
CaHcNBr-HBr М. в. 204,90
Р-Бромэтиламин бромистоводородный широко использует-
ся как промежуточный продукт в органическом синтезе.
В литературе описано получение бромэтиламина бромгид-
рата по реакции Габриэля взаимодействием фталимида калия
с бромистым этиленом с последующим гидролизом N'-(|3-
бромэтил)-фталимида [1, 2], взаимодействием бромистого во-
дорода с этиленимином [3, 4], нагреванием моноэтаноламина
с дымящей бромистоводородной кислотой в запаянной труб-
ке при 170° [5] и, наконец, бромированием моноэтаноламина
бромистоводородной кислотой уд. веса 1,48 [6, 7].
Нами проверен и уточнен последний из указанных спосо-
бов получения 0-бромэтиламина бромгидрата с использова-
нием бромистоводородной кислоты уд. веса 1,38, что позволи-
ло избежать стадию укрепления бромистоводородной кисло-
ты.
СХЕМА СИНТЕЗА ₽ -БРОМЭТИЛАМИНА БРОМИСТОВОДОРОДНОГО
HOCH2CH2NH3 + 2НВг BrCH2CH2NH2-HBr4-H2O
Характеристика основного сырья
Моноэтаноламин, ч., перегнанный, СТУ 12-10-112—62.
Бромистоводородная кислота, ч., уд. в. 1,38; ГОСТ 2062—
43.
41
Условия получения
В трехгорлую колбу емкостью 2 л, снабженную механи-
ческой мешалкой, капельной воронкой и обратным холодиль-
ником, помещают 1776 г (8,8 Л1) бромистоводородной кисло-
ты уд. веса 1,38 и постепенно при охлаждении и перемешива-
нии из капельной воронки добавляют 166 а (2,7 Л1) свежепе-
регнанного охлажденного моноэтаноламина. После оконча-
ния прибавления моноэтаноламина меняют холодильник на
нисходящий и на масляной бане при ПО—113° отгоняют пер-
вую порцию дистиллята в количестве 430 мл. Затем холо-
дильник меняют на обратный и нагревают со стеканием флег-
мы 1 час. После этого отбирают еще 130 мл дистиллята, а
раствор вновь нагревают с обратным холодильником 1 час.
Эту операцию повторяют, отбирая порции дистиллята в 50,
40, 25, 15 и 10 мл. Температура отгонки при этом постепенно
увеличивается. Последние 10 мл дистиллята отгоняются при
169°. Затем реакционную массу снова нагревают с обратным
холодильником в течение 3 часов и отгоняют еще 370 мл ди-
стиллята. Таким образом, общий объем отогнанного дистил-
лята 1070 мл. Оставшуюся в колбе горячую сиропообразную
массу переносят в литровый стакан, охлаждают до 60° и до-
бавляют 550 мл ацетона. После выдержки на холоду в тече-
ние ночи выделившийся осадок растирают в фарфоровой
ступке с несколькими порциями ацетона (всего 1 л), затем от-
фильтровывают, промывают еще 1 л ацетона, отжимают и
полученные белые кристаллы сушат на воздухе.
Фильтрат собирают, концентрируют до объема 100 мл и
охлаждают, при этом из раствора выпадает вторая порция
продукта, которую также отжимают, промывают небольшим
количеством свежего ацетона и сушат на воздухе.
Общий выход p-бромэтиламина бромистоводородного
400 г, что составляет 72% от теоретического. Продукт со-
держит основного вещества 98,9—99,1% (определяется арген-
тометрическим титрованием бромистого водорода).
Для очистки бромгидрата (3-бромэтиламина его перекрис-
таллизовывают из смеси этилового спирта и этилацетата (4:1)
в соотношении 1:1,5; т. пл. перекристаллизованного бромгид-
рата 0-бромэтиламина 172°.
По литературным данным, т. пл. вещества 174—175° [6];
172,5—173° [5].
Р-Бромэтиламин бромистоводородный гигроскопичен, слег-
ка расплывается на воздухе, растворим в воде, плохо раство-
рим в ацетоне.
Литература
1. S. Gabriel. Вег., 21, 566 (1888).
2. О. Seitz. Вег., 24, 2624 (1891).
42
3. S. Gabriel. Ber., 21, 1049 (1888).
4. S. G a b r i e 1, R. S t e 1 z n c r. Ber., 28, 2929 (1895).
5. S. Gabriel. Ber., 50. 826 (1917).
6. F. Cortese. J. Amer. Chem. Soc.., 58, 191 (1936).
7. Синтез органических препаратов, Сб. 2, М., ИЛ, 1949, стр. 126.
Поступила в сентябре 1964 г.
ИРЕА
УДК 547.239.2.07
р -БРОМЭТИЛИМИНОДИАЦЕТОНИТРИЛ
Р. П. ЛАСТОВСКИЙ, И. Д. КОЛПАКОВА, Л. В. КРИНИЦКАЯ
CH2CN
BrCHaCH2N(f
'CHoCN
CeH8N3Br
M. в. 202,05
Р-Бромэтилиминодиацетонитрил может быть использован
как промежуточный продукт для синтеза бромэтилиминоди-
уксусной кислоты и ряда других органических соединений.
В литературе описано получение р-хлорэтилиминодиацето-
нитрила в качестве промежуточного продукта при синтезе
p-хлорэтилиминодиуксусной кислоты [1] конденсацией р-хлор-
^тилимина с формальдегидом и цианистым натрием.
По этому методу нами проведена конденсация бромгидра-
та p-бромэтиламипа с формальдегидом и цианистым натри-
ем.
СХЕМА СИНТЕЗА ₽ -БРОМЭТИЛИМИНОДИАЦЕТОНИТРИЛА
BrCH2CH2NH2 HBr + 2СНаО + 2NaCN
,CH,CN
BrCH2CH2N ( + 2Н,0 + NaCl + NaBr
^CH2CN
Характеристика основного сырья
Р-Бромэтиламин бромгидрат (см. статью «р-Бромэтил-
амин бромистоводородный» в этом сборнике).
Формалин, 35%-иый раствор.
Натрий цианистый, ч., ВТУ 3962—53.
Соляная кислота, ч., ГОСТ 3118—46, уд. в. 1,18.
44
Условия получения
Синтез должен проводиться в хорошо действующем вы-
тяжном шкафу.
Реакцию проводят в четырехгорлой колбе емкостью 1 л,
снабженной механической мешалкой с ртутным затвором,
обратным холодильником, термометром и капельной ворон-
кой. Обратный холодильник через промежуточную емкость
соединен со склянкой, содержащей 7%-ный раствор желез-
ного купороса для поглощения избыточного количества вы-
деляющегося в результате реакции цианистого водорода.
В колбу загружают 143,5 г (0,7 М) бромгидрата р-бром-
этиламина и при охлаждении прибавляют 120 г (1,4 М)
35%-ного раствора формальдегида и 70 мл (0,76 М) концен-
трированной соляной кислоты уд. в. 1,18. В капельную во-
ронку помещают 32%-ный раствор [70,6 г (1,44 Л4)] циани-
стого натрия и постепенно в течение 3,5 часов при переме-
шивании и охлаждении приливают в колбу так, чтобы тем-
пература в колбе не превышала 15°.
После окончания прибавления цианистого натрия рас-
твор перемешивают еще 30 минут при 15°, а затем выдер-
живают двое суток при комнатной температуре.
Выпавший белый блестящий кристаллический осадок
фильтруют, промывают двумя порциями воды по 150 мл, су-,
шат в эксикаторе над хлористым кальцием и кристаллизуют
из этилового спирта в соотношении 1:2.
Выход p-бромэтилиминодиацетонитрила равен 81 г, что
составляет 57% от теоретического; т. пл. продукта 59—60°.
Р-Бромэтилиминодиацетонитрил нерастворим в воде, рас-
творим в этиловом спирте.
Л итература
1. G. Schwarzenbach, G. Anderegg, W. Schneider,
H. Senn. Helv. chim. acta, 38, 1147 (1955).
Поступила в сентябре 1964 г.
ИРЕА
УДК 546.183.07
ВИНИЛМЕТИЛХЛОРФОСФИНАТ
Ю. Г. ГОЛОЛОБОВ, Т. Ф. ДМИТРИЕВА, Л. 3. СОБОРОВСКИЙ
/ОСН^СН,
СН3Р(
'[ ХС1
о
С3Н6О2РС1
М. в. 140,50
Винилметилхлорфосфинат, как и другие хлорангидриды
а-алкениловых эфиров алкилфосфиновых кислот, описан в
самое последнее время [1, 2]. Метод получения винилметил-
хлорфосфината основан на взаимодействии эквимолярных
количеств дихлорангидрида метилфосфиновой кислоты, ацет-
альдегида и триэтиламина.
СХЕМА СИНТЕЗА ВИНИЛМЕТИЛХЛОРФОСФИНАТА
СНЯР
II
II
о
С1
С1
+ СНзСНО 4-(C2H6)3N ->
,осн=сна
-> СН3Р< 4- (C3H6)3N.HC1
II ХС1
о
Характеристика основного сырья
Дихлорангидрид метилфосфиновой кислоты (см. соответ-
ствующую статью настоящего сборника).
Ацетальдегид, ч., ГОСТ 2633—51.
Триэтиламин, ч., ВТУ РУ 796—53.
46
Условия получения
В автоклав на 750 мл загружают последовательно 50,5 г
(0,5 Af) охлажденного (0°) триэтиламина (см. примечание!),
66,5 г (0,5 М) дихлорангидрида метилфосфиновой кис-
лоты и 22 г (0,5 АГ) охлажденного (0°) ацетальдегида. Ав-
токлав закрывают и смесь встряхивают (см. примечание 2),
при этом наблюдается разогревание реакционной массы до
90—100° (см. примечание 3). После падения температуры
до комнатной смесь либо обрабатывают эфиром, фильтруют
и перегоняют, либо нагревают в вакууме (1 мм) до полной
отгонки жидкой части. Перегонкой в вакууме получают око-
ло 30 г винилметилхлорфосфината; т. кип. 63—64° при8лыц
de°— 1,2476; п® — 1,4492.
Найдено, %: С1—25,54; 25,17.
С8НвО2РС1а. Вычислено, %: 0- 25,26.
Примечания:
1. Триэтиламип высушивают щелочью и перегоняют над металличе-
ским натрием.
2. Применение автоклава с мешалкой повышает выход целевого ве-
щества.
3. Введение в данную реакцию высших альдегидов не приводит к
столь значительному повышению температуры реакционной массы. В этих
случаях для завершения реакции необходимо смесь нагревать при 100—
110° (2—4 часа), причем через некоторое время обычно наблюдается рез-
кий подъем температуры (до 120—140°). В этот момент реакционную мас-
су следует охладить, поскольку в противном случае происходит значитель-
ное осмоление продукта.
Литература
1. Ю. Г. Г о л о л о б о в, Т. Ф. Дмитриева, Л. 3. Соборов-
с к п й. Авт. свид. 154544; Бюлл. изобр., № 10 (1963).
2. Ю. Г. Г о л о л о б о в, Т. Ф. Д м и т р и е в а, Л. 3. Соборов-
с к н й. Ж. общ. химии, 866 (1964).
Поступила в июне 1964 г.
УДК 547.37.07
ВИНИЛХЛОРФОСФАТЫ
Ю. Г. ГОЛОЛОБОВ, Т. Ф. ДМИТРИЕВА, Л. 3. СОБОРОВСКИИ
Винилхлорфосфаты получают взаимодействием соответ-
ствующих количеств хлорокиси фосфора, ацетальдегида и
триэтиламина [1]. Описан дивинилхлорфосфат, синтезирован-
ный реакцией дивинилэтилфосфита с пятихлористым фосфо-
ром [2].
ВИНИЛДИХЛОРФОСФАТ
(СН,=СНО) Р С1.->
С2Н3О,РС12
М.в. 160,92
СХЕМА СИНТЕЗА ВИНИЛДИХЛОРФОСФАТА
POC13+CH3CHO+(C.H6)3N^(CH.,=CHO) PC1..+(C,H5)3N.HC1
II
О
Характеристика основного сырья
Хлорокись фосфора, техническая.
Ацетальдегид, ч., ГОСТ 2633—51.
Триэтиламин, ч., ВТУ РУ 796—53.
Условия получения
В автоклав емкостью 0,5 л помещают 76,7 г (0,5 Л1) хлор-
окиси фосфора, 50,5 г (0,5 Л1) триэтиламина и 22 г (0,5 Л1)
ацетальдегида. Автоклав встряхивают. Протекает экзотерми-
ческая реакция, температура реакционной массы поднимает-
ся до 90—100°, и давление достигает 2—3 атмосфер. После
падения температуры до комнатной автоклав вскрывают и от
48
реакционной массы под вакуумом (2—3 мм) отгоняют жид-
кую часть. Перегонкой получают около 30 г винилдихлорфос-
фата с т. кип. 36—40° при 30 мм; d420 — 1,41 1 1 ; Лд — 1,4450.
Найдено, %: 0—44,2; 43,7.
С2Н8О2РС12. Вычислено, %: С1— 44,(6.
Винилдихлорфосфат представляет собой бесцветную под-
вижную жидкость, темнеющую при хранении. Водой быстро
гидролизуется.
ДИВИНИЛХЛОРФОСФАТ
(СН2=СНО).,Р"
С4Н0О3рС1 М. в. 168,51
СХЕМА СИНТЕЗА Д14ВИН14ЛХЛОРФОСФАТА
Р0С13 + 2СН3СНО + 2(C2H6)3N -
(СН2=СНО)2Р-С1 + 2(C2H5)3N.HC1
О
Условия получения
В автоклав емкостью 0,5 л помещают 46 г (0,3 М) хлор-
окиси фосфора, 70,5 г (0,7 М) триэтиламина и 26,4 г (0,6 М)
ацетальдегида. .Закрытый автоклав встряхивают. Начинается
реакция, вследствие чего температура поднимается до 90—
100°, при этом давление в автоклаве достигает 2—4 атмос-
фер. После обработки реакционной массы по приведенной
методике получают около 30 г дивинилхлорфосфата с т. кип.
58-59" при 11 мм; 1,2408; ri% — 1,4319.
По литературным данным, т. кип. продукта 60—61° при
12 мм; d^0 —1,2442; л2’— 1,4350 [2].
Найдено, %: С1 -21,58; 21,57.
О
II
(СН2=СНО)2Р- С1. Вычислено, %: С1- 21,06.
Примечай и е.
См. примечания 1 и 2 в статье «Винилметилхлорфосфинат» этого
сборника.
Литература
1. Ю. Г. Гололобов, Т. Ф. Дмитриева, Л. 3. С о бор о в-
с к п й. Авт. заявка 809290/23—4.
2. И. Ф. Луценко, 3. С. К Р а й ц, М. В. П р о с к у н и н а. Докл.
АН СССР, 148, 4, 846 (1963).
Поступила в июне 19М г.
4 Зак. 131
49
УДК 547.831.05
2-ГИДРАЗИНОХИНОЛ ИН
2-Хинолилгидразин
В. М. ДЗИОМКО, И. А. КРАСАВИН, Н. И. МИРОШКИНА
Z\/ X
I I! \
^/xn/;/\Nh-nh2
C9H9N3 М. в. 159,20
Лучшим способом получения 2-гидразинохинолина являет-
ся нагревание 2-хлорхинолина с избытком гидразин-гидрата
[1, 2]. Оптимальные условия синтеза 2-гидразинохинолина
(выход 94%), а также и исходных веществ — 1-метилкарбо-
стирила (выход 89%) и 2-хлорхинолина (выход 85—90%)
разработаны Перкином и Робинсоном [2]. Другие методы,
например нагревание 2-хинолинсульфокислоты с гидразин-
гидратом в автоклаве в присутствии хлористого цинка (выход
64,5%) [3] и взаимодействие 2-хлорхинолин-1 -оксида с гид-
разин-гидратом [4], не имеют практического значения из-за
недоступности сырья. Опубликован метод количествен-
ного анализа 2-гидразинохинолина [5].
Нами проверена методика получения 1-метилкарбостири-
ла, 2-хлорхинолина и 2-гидразипохинолина [2] с использова-
нием хинолина коксохимического происхождения и получены
удовлетворительные результаты, хотя недостаточная чистота
такого хинолина отразилась на выходах и качестве продук-
тов. Поскольку применение больших объемов эфира (при
экстракции 1-метилкарбостирила) сопряжено с определенной
опасностью, он был заменен бензолом.
50
ПОЛУЧЕНИЕ МЕТОСУЛЬФАТА 1-МЕТИЛХИНОЛИНИЯ
Внимание! Диметилсульфат очень ядовит, поэтому все ра-
боты с ним необходимо проводить в хорошо действующем
вытяжном шкафу (см. примечание 1).
В трехгорлую колбу емкостью 500 мл, снабженную ме-
шалкой, обратным холодильником и капельной воронкой, по-
мещают 32,3 г (0,25 М) свежеперегнанного хинолина (см
примечание 2) и прибавляют к нему при размешивании за
7—10 минут 32,8 г (0,26 М) диметилсульфата (см. примеча-
ние 3). Для регулирования бурно протекающей реакции кол-
бу периодически охлаждают с помощью водяной бани. Под
конец содержимое колбы затвердевает. Выход количествен-
ный. Для использования в следующей стадии полученный
метосульфат 1-метилхинолиния растворяют в 300 мл воды.
ПОЛУЧЕНИЕ 1-МЕТИЛ КАРБОСТИРИЛА
| |! | _+2K3Fe(CN)0+3KOH
4/\n^CH3SO4
I
СН3
4- 2K4Fe(CN)e + CH3KSO4 + 2H3O
В плоскодонной колбе емкостью 3 л готовят раствор 198 г
(0,6 М) феррицианида калия (красной кровяной соли; см.
примечание 4) в 1000 мл воды, смешивают его с раствором
метосульфата 1-метилхинолиния в 300 мл воды и приливают
320 мл бензола. Затем прибавляют за 20—30 минут раствор
50,5 г (0,9 М) едкого кали в 450 мл воды небольшими порци-
ями, периодически перемешивая содержимое колбы вручную
круговыми движениями, но не взбалтывая (см. примечание
5). Реакция жидкости должна быть сильно щелочной. Через
30 минут бензольный слой отделяют, а водный экстрагируют
бензолом (500 мл) в 2—3 приема. Все экстракты соединяют,
4- 51
сушат над прокаленным карбонатом калия, растворитель от-
гоняют, а остаток перегоняют в вакууме при 152—156°/3 мм.
Получают 25—26,5 г (63—67%) 1-метилкарбостирила,
который застывает в приемнике в светлую массу (см. приме-
чания 6 и 7).
ПОЛУЧЕНИЕ 2-ХЛОРХИНОЛИНА
Х\/Х /\/Х
| || | +PCL— | (I | +РОС13 + СН3С1
X/xn/Xq
СНз
В круглодонной колбе емкостью 500 мл со шлифом сме-
шивают 23,9 г (0,15 М) 1-метилкарбостирила и 48 г (0,23 М)
растертого в порошок пятихлористого фосфора. Прибавляют
24 г (0,157 Л1) свежеперегнанной хлорокиси фосфора, присое-
диняют обратный 8-шариковый холодильник, защищенный
хлоркальциевой трубкой и нагревают колбу на масляной ба-
не при 170—180э в течение 9—12 часов. Реакционная масса,
вначале густая, к концу нагревания разжижается. Тогда за-
меняют холодильник на нисходящий и отгоняют хлорокись
фосфора при пониженном давлении (водоструйный насос) при
температуре бани не выше 170°. Остаток охлаждают, вылива-
ют на 250 г толченого льда, помещенного в круглодонную
колбу емкостью 2 л, осторожно подщелачивают добавлением
10%-ного раствора едкого натра и отгоняют 2-хлорхинолин с
водяным паром. Отгонку продолжают до тех пор, пока дис-
тиллят станет прозрачным (около 2,5—3 л). Дистиллят экст-
рагируют бензолом (3—4 раза по 100 мл). Экстракт сушат
безводным сульфатом натрия и растворитель отгоняют. Оста-
ток (около 22 г) перегоняют в вакууме при 124—130°/5 мм
(см. примечание 6). Выход 2-хлорхинолина 17—18 г (69—
775%). Он застывает в белую массу, плавящуюся при 37—
38°.
ПОЛУЧЕНИЕ 2-ГИДРАЗИНОХИНОЛИНА
/х/Х /Х/Х
| I! I +2(N2H4.H2O)—I 1| | +
X/\nXxc1 X/XnXxNHNH3
+ N2H4-HC1 + 2H2O
В колбе с обратным холодильником на шлифе кипятят
смесь 16,4 г (0,1 М) 2-хлорхинолина и 75 г (1,5 М) гидразин-
гидрата на масляной бане в течение 1,5—2 часов, затем отго-
няют избыток гидразина при пониженном давлении, нагре-
52
вая колбу на водяной бане. К остатку добавляют 100 мл го-
рячей воды и нагревают в течение 15 минут на водяной бане.
Охлаждают колбу ледяной водой, через несколько часов от-
сасывают кристаллы, промывают небольшим количеством хо-
лодной воды и высушивают в вакууме. Получают 13,8—13,9 г
(86,5—87,1%) 2-гидразинохинолина в виде светло-красных
кристаллов с т. пл. 138—139°. После перекристаллизации из
бензола (150—160 мл) образуется 8,5—9,5 г вещества с т. пл.
141—141,5° (см. примечание 8).
Примечания:
1. Попадание диметилсульфата на кожу или вдыхание его паров мо-
жет привести к тяжелым последствиям. Специфическим нейтрализатором
является концентрированный раствор аммиака, который необходимо иметь
под рукой для обезвреживания диметилсульфата, остающегося в реакци-
онных сосудах, при попадании его на резиновые перчатки или при слу-
чайном разливе.
2. Применяли хинолин коксохимического происхождения. Если ис-
пользовать синтетический хинолин, полученный по реакции Скраупа, то
выходы на всех стадиях повышаются до 90%, а качество веществ улуч-
шается.
3. Применяли перегнанный в вакууме нейтральный диметилсульфат.
После внесения капли его в пробирку с 1—2 мл воды последняя должна
иметь pH 6—7.
4. Применяли продажный феррицианид калия квалификации «чистый».
Авторы оригинальной работы [2] использовали свежеперекристаллизован-
ный феррицианид, но такая очистка нецелесообразна из-за больших по-
терь этой соли.
5. Если применяется синтетический хинолин, то следует воспользо-
ваться механической мешалкой, что улучшит условия экстракции и повы-
сит выход. При употреблении коксохимического хинолина сильное разме-
шивание или взбалтывание часто способствует образованию стойкой эмуль-
сии и пленок, избавиться от которых очень трудно. В этом случае невоз-
можность четкого разделения слоев приводит к значительному снижению
выхода.
6. Перегонку проводили из специальной колбы [6].
7. Чтобы получить чистый 1 -метилкарбостирил, его можно перекрис-
таллизовать из петролейного эфира, из которого он выделяется в бесцвет-
ных иглах с. т. пл. 73—74°.
8. После повторной перекристаллизации из бензола можно получить
вещество с т. пл. 142,5—143°. По литературным данным, т. пл. продукта
134—135° [1]; 142—143“ [2].
Литература
1. W, Marckwald, Е. М е у е г. Вег., 33, 1885 (1900).
2. W. Н. Perkin, i u n„ R. Robinson. J. Chem. Soc., 103, 1973
(1913).
3. Y. Suzuki. Yakugaku Zasshl, 81, 1146 (1961); Chem. Abstrs, 56,
3450 c (1962).
4. S. К a tn 1 у a. Yakugaku Zasshl, 81, 1743 (1961); Chem. Abstrs, 57,
16556 b (1962).
5. H. Me Kennis, 1 r. A. S. Yard. US Dept. Com. Office Tech.
Serv., PB Rept, 143, 914 (1957); Chem. Abstrs, 55, 17375g (IK 1).
6. В. M. Дзиомко, И. А. Красавин, О. В. Иванов. Методы
получения химических реактивов и препаратов, вып. 9, М„ ИРЕА, 1964,
стр. 9.
Поступила в декабре 1964 г ИРЕА
53
УДК 661.718.07
ДИАЛкилХЛОРФОСФИНЫ
И. П. КОМКОВ, к. в. КАРАВАНОВ, С. 3. ИВИН
RoPCI
Диалкилхлорфосфины являются исходными полупродук-
тами в синтезе различных фосфорорганических соединений.
В литературе описан способ их получения, основанный на
разложении триалкилдихлорфосфинов [1, 2]. Диалкилхлор-
фосфины получают прямым алкилированием красного фос-
фора хлоралканами [3] и восстановлением комплексных сое-
динений диалкилтрихлорфосфинов с хлористым алюминием
порошкообразной сурьмой и цинком в диэтилфталате. Ди-
этилфталат применяют как растворитель и для связывания
хлористого алюминия [4].
Перечисленные методы обладают существенными недос-
татками: метод, основанный на термическом разложении три-
алкилдихлорфосфинов часто не воспроизводится; прямое ал-
килирование красного фосфора требует определенного обору-
дования, а выходы дилкилхлорфосфинов сравнительно низки.
Нами разработан удобный способ получения алкилдихлор-
фосфинов с хорошим выходом из доступных веществ. Метод
основан на восстановлении комплексных соединений диалкил-
трихлорфосфинов и хлористого алюминия с помощью алюми-
ниевой пыли, красного фосфора, натрия и других металлов в
присутствии свежепрокаленного хлористого калия [5].
Следует отметить, что наиболее удобным восстановителем
оказался алюминий в виде алюминиевой пудры. Хлористый
калий применяется для связывания хлористого алюминия, что
значительно повышает выход диалкилхлорфосфинов.
С4Н10РС1
54
ДИЭТИЛХЛОРФОСФИН
с2нв
>РС1
С2н/
М. в. 124,55
СХЕМА СИНТЕЗА ДИЭТИЛХЛОРФОСФИНА
КС1
(СаНб)аРС13-А1С1в 4-А1 (С2Н5)2РС1 +А1С13-КС1
Характеристика основного сырья
Комплексное соединение диалкилтрихлорфосфина с хлори-
стым алюминием, получают по методике, описанной в насто-
ящем сборнике (см. статью «Комплексные соединения диал-
килтрихлОрфосфинов с хлористым алюминием» в этом сбор-
нике).
Алюминий в виде алюминиевой пудры (сухой).
Калий хлористый, х. ч., безводный, свежепрокаленный,
ГОСТ 4234—48.
Условия получения
В колбу Кляйзена емкостью 0,75 л с прямым холодильни-
ком помещают 98,7 г (0,3 М) комплексного соединения ди-
этилтрихлорфосфина с хлористым алюминием (см. примеча-
ние 1). Затем к слаборасплавленному комплексному соедине-
нию из присоединенной к горловине колбы Кляйзена колбоч-
ки постепенно небольшими порциями (см. примечание 2) при-
сыпают смесь, состоящую из 5,4 г (0,2 М) алюминиевой пыли
и 37,2 г (0,5 М) свежепрокаленного хлористого калия. Реак-
ционную массу при этом осторожно нагревают (см. примеча-
ние 3) и постоянно встряхивают. Реакция протекает со значи-
тельным выделением тепла.
После внесения всего количества смеси алюминиевой пы-
ли и хлористого калия (см. примечание 4) проводят отгонку
диэтилхлорфосфина нагреванием колбы пламенем горелки.
Выход диэтилхлорфосфина 25,6 г, что составляет 66% от
теоретического; т. кип. 61—62° при 72 мм; сЦ20—1,0233;
«о—1,4720.
Примечания:
1. Комплексное соединение этплтетрахлорфосфина с хлористым алю-
минием гигроскопично; следует избегать длительного соприкосновения с
воздухом.
2. Смесь хлористого калия с алюминиевой пудрой присыпается не-
большими порциями во избежание сильного саморазогревания в результа-
те бурно протекающей реакции. Последнее может привести к выбросу ре-
акционной массы. Очередную порцию смеси следует присыпать после того,
как прореагирует предыдущая порция.
3. Нагревание проводят так, Чтобы через склянку Тищенко, постав-
ленную на конце прибора, как можно меньше проскакивало пузырьков
газа. Комплексное /соединение, нагретое выше температуры плавления
(160°), разлагается на составные части.
4. Отгонку диэтилхлорфосфина необходимо проводить немедленно по-
сле присыпания последней порции смеси хлористого калия с алюминиевой
пудрой, в противном случае образовавшийся диэтилхлорфосфин может
разложиться.
55
5. Диэтплхлорфосфпн легко окисляется на воздухе, поэтому его полу-
чение п перегонку необходимо вести в токе азота пли другого инертного
газа.
По описанной методике, исходя из комплексных соедине-
ний диалкилтрихлорфосфинов и хлористого алюминия, можно
получить различные диалкилхлорфосфины с одноименными
или разноименными алкильными радикалами.
Литература
I. Т. N. С о 11 i е, F. R е g п о ! a s. J. Chem. Soc., 107, 368 (1(| 15).
2. В. М. Плец. Органические соединения фосфора, М., Оборонгпз,
1910, стр. 155.
3. Л. 3. Соборовский, Б. М. Гл ад штейн. Авт. свид. 130513;
Бюлл. пзобр., № 15 (1960).
4. I. L. Ferron. Canad. J. Chem., 39, 4 , 842 (1961).
5. И. П. Комков, К. В. Караванов, С. 3. Ивин. Ж. общ. хи-
мии, 28, 2963 (1958).
Поступила в июне 1164 г.
УДК 547.569.2
ДИАЛКИЛХЛОРФОСФИНСУЛЬФИДЫ
С. 3. ИВИН, К. В. КАРАВАНОВ
r2psci
Диалкилхлорфосфинсульфиды являются исходными про-
дуктами в синтезах ряда фосфорорганических соединений.
Обычно они получаются нагреванием в запаянных труб-
ках смеси диалкилхлорфосфинов с серой [1] или действием
пятисернистого фосфора на диалкилхлорфосфиноксиды [2].
Однако эти способы имеют тот недостаток, что исходные ди-
алкилхлорфосфины и диалкилхлорфосфиноксиды, используе-
мые при их получении, являются малодоступными.
Нами разработан [3] способ получения диалкилхлорфос-
финсульфидов с хорошим выходом из доступных реагентов—
комплексных соединений диалкилтрихлорфосфинов с хлорис-
тым алюминием и серы или роданистого калия. Метод осно-
ван на нагревании указанных реагентов в присутствии свеже-
прокаленного хлористого калия для связывания хлористого
алюминия. (При нагревании комплексного соединения с ро-
данистым калием хлористый калий для связывания хлорис-
того алюминия не используется, так как он образуется в про-
цессе реакции).
ДИЭТИЛХЛОРФОСФИНСУЛЬФИД
С3Н6 к z.S
/Р<
с2н/ С1
C4H1OPSC1 М. в. 156, 61
СХЕМА СИНТЕЗА ДИЭТИЛХЛОРФОСФИНСУЛЬФИДА
С,Н-. КС1 С2НЕХ xS
^РС13-А1С13 4- S----> ;р< 4-А1С13-КС1
С2Н/ Сан/ ХС1
Характеристика основного сырья
Комплексные соединения получаются по методике, опи-
санной в настоящем сборнике (см. статью «Комплексные сое-
динения диалкилтрихлорфосфинов с хлористым алюминием»
в этом сборнике).
Сера, обычная, черенковая (сухая).
Хлористый калий, х. ч., безводный, свежепрокаленный,
ГОСТ 4234—48.
Условия получения
В колбу Кляйзена емкостью 0,5 л загружают хорошо пе-
ремешанную смесь, состоящую из 98,7 г (0,3 Л4) комплексно-
го соединения диэтилтрихлорфосфина с хлористым алюмини-
ем, 9,6 г (0,3 Л4) серы и 22,3 г (0,3 М) прокаленного хлори-
стого калия. Колбу помещают в глицериновую баню, которую
медленно, в течение 3 часов, нагревают до 200° (см. приме-
чание 1). Затем образовавшиеся продукты реакции отгоняют в
вакууме 10 мм. При повторной перегонке получают 22,9 г ди-
этилхлорфосфинсульфида, что составляет 48,9% от теорети-
ческого выхода; т. кип. 99—100° при 11 мм; d420—1,1542,
п™ —1,5325.
Аналогичным путем можно получить и другие диалкил-
хлорфосфинсульфиды при нагревании соответствующих ком-
плексных соединений с серой, а также с сульфидами алюми-
ния, мышьяка, сурьмы, фосфора в присутствии свежепрока-
ленпого хлористого калия.
Хлористый калий в данных случаях применяется для свя-
зывания хлористого алюминия, что значительно повышает
выход диалкилхлорфосфинсульфидов.
Примечание.
Нагревание реакционной массы ведут так, чтобы как можно меньше
проскакивало пузырьков газа через склянку Тищенко, поставленную на
конце прибора.
Литература
1. В. М. Плец. Органические соединения фосфора, М., Оборонгиз,
1940, стр. 176.
2. М. И. К а б а ч н и к, Н. Н. Годовиков. Докл. АН СССР, 110,
217 (1956).
3. С. 3. И в и и, К. В. Караванов. Ж. общ. химии, 28, 2958 (1958).
Поступила в июне 1964 г.
УДК 661.718.07
ДИАЛКИЛТРИФТОРФОСФИНЫ
И. П. КОМКОВ, С. 3. ИВИН, К. В. КАРАВАНОВ
r3pf3
Диалкилтрифторфосфины являются бесцветными реак-
ционноспособными соединениями. Они легко вступают в реак-
цию со многими веществами, образуя различные производ-
ные диалкилфосфиновых- и диалкилортофосфиновых кислот.
Впервые диалкилтрифторфосфины были получены нами
[I] при фторировании диалкилхлорфосфинов трехфтористой
сурьмой и трехфтористым висмутом.
ДИЭТИЛТРИФТОРФОСФИН
/РРз
с3н/
c4hI0pf3
М. в. 146, 09
СХЕМА СИНТЕЗА ДИЭТИЛТРИФТОРФОСФИНА
3(С2Н5)2РС1 -F3SbF3-> 3(С2Н5)2 PF3-F SbCl3 + 2Sb
Характеристика основного сырья
Диэтилхлорфосфин, перегнанный, получают по методике,
описанной в настоящем сборнике (см. статью «Диалкилхлор-
фосфнны»).
Трехфтористая сурьма, ч., безводная или свежепрокален-
ная, МРТУ 6-09-524—63.
59
Условия получения
В колбу Кляйзена с елочным дефлегматором, прямым хо-
лодильником, термометром и капельной воронкой помещают
45,4 г (0,254 М) трехфтористой сурьмы, к которой из капель-
ной воронки медленно прибавляют 29,2 г (0,2 М) диэтилхлор-
фосфина. Реакция протекает с разогреванием. После прибав-
ления всего количества диэтилхлорфосфина содержимое кол-
бы выдерживают 20—30 минут при комнатной температуре,
а затем проводят отгонку диэтилтрифторфосфина. Получают
25,3 г-диэтилтрифторфосфина, что составляет 62,8 °/о от теоре-
тического выхода; т. кип. 104—105° при 760 мм; dt20—1,1486;
п2° — 1,3650.
По описанной методике, исходя из различных диалкил-
хлорфосфинов, можно получать различные диалкилтрифтор-
фосфины.
Вместо трехфтористой сурьмы можно использовать трех-
фтористый висмут.
Литература
1. И. П. Комков, С. 3. Ивин, К. В. Караванов, Л. Е. Смир-
нов. Ж- общ. химии, 32, 301 (1962).
Поступила в июве 1961 г.
УДК 547.585.07
ДИАНГИДРИД ПИРОМЕЛЛИТОВОИ кислоты
Диангидрид бензол-1,2,4.5-тетракарбоновой кислоты
М. Т. РАЗУМОВСКАЯ, Л. А. ЕГОРОВА
со-о
СъН2Ов
М. в. 218, 12
Диангидрид пиромеллитовой кислоты является сырьем для
получения тетрацианбензола и других термостойких полиме-
ров [1]; в смеси с фталевым и малеиновым ангидридом он ис-
пользуется для получения эпоксидных смол [2], а также для
выделения /г-ксилола из смеси ксилолов [3].
Впервые диангидрид пиромеллитовой кислоты был полу-
чен Эрдманом в 1851 г. при сухой перегонке меллитовой кис-
лоты [4].
Диангидрид пиромеллитовой кислоты получают из пиро-
меллитовой кислоты дегидратацией с водоотпимающими реа-
гентами [5] или нагреванием в вакууме выше ее температу-
ры плавления [6], а также окислением дурола кислородом
воздуха над катализатором [7, 8).
Ниже описывается препаративный лабораторный метод
получения чистого диангидрида пиромеллитовой кислоты де-
гидратацией ее уксусным ангидридом.
61
СХЕМА СИНТЕЗА ДИАНГИДРИДА ПИРОМЕЛЛИТОВОИ КИСЛОТЫ
соон
Г^-СООН
HOOC-t
соон
СН3СО \
+ 2 >О->
СНзСО
со - о
со
(>r р I +4СН3СООН
I
о—со
Характеристика основного сырья
ПироМеллитовая кислота, безводная, ч. (см. статью «Пи-
ромеллитовая кислота» в этом сборнике).
Уксусный ангидрид, ч., ГОСТ 5815—52.
Условия получения
В круглодонную двухгорлую колбу емкостью 1 л, снаб-
женную обратным холодильником и термометром, загружают
101,6 г (~0,4 М) безводной пиромеллитовой кислоты и 500мл
(5,3 А4) уксусного ангидрида. Реакционную смесь нагревают
до 120—125° и выдерживают при этой температуре в течение
1,5 часа, затем обратный холодильник заменяют нисходящим,
температуру реакционной массы повышают до 130—135°. При
этой температуре отгоняют смесь уксусной кислоты с избыт-
ком уксусного ангидрида. Отгонку прекращают, когда отто-
пится 350 мл этой смеси. Остаток охлаждают ледяной водой
до 10—12°. Выпавшие бледно-желтые кристаллы отсасывают,
промывают холодной водой до нейтральной реакции и сушат
при 130—140° до постоянного веса.
Выход диангидрида пиромеллитовой кислоты 70—74 г, что
составляет 81—85% от теоретического; т. пл. 285—286°.
По литературным данным, т. пл. продукта 286° [6]; 285—
287° [1].
Найдено, %: С — 55,/9; .55,60; Н —0,81; 0,92.
С10Н2Ов. Вычислено, %: С—55,05; Н - 0,92.
62
Литература
1. A. Epstein, В. S. Wild у. J. Chem. Phys., 32, 324 (1960);
Chem. Week., 88, 46 (1961).
2. R. В. F e i 1 d, C. F. Robinson. Ind. Eng. Chem., 49, 3, 369
(1957).
3. Brennstoff- Chemie, 43, 9, 263 (1962).
4. O. Erdman. Liebigs Ann. Chem., 80, 281 (1851).
5. Philippi, Seka. Monatsh. Chem., 43, 617 (1920); Beilst., доп. 11,
17/19, 705(1934).
6. G. Schroeter. Ber., 57, 2023 (1924); Пат. США 2937189, 1960;
РЖхим, 14 л, 142 (1961). •
7. Пат. США 2509855; С. А., Р3873 d (1951).
8. Пат. США 2576625; С. А., Р5087 i (1952).
Поступила в июне 1В64'. ИРЕА
УДК 661.718.07
ДИХЛОРАНГИДРИД
2,3-ДИХЛОРПРОПИЛФОСФИНОВОЙ кислоты
2,3-Дихлорпропилдихлорфосфиноксид
Л. 3. СОБОРОВСКИЙ, Ю. А-f. ЗИНОВЬЕВ, Л. И. МУЛЕР
С1СН2СНС1СН2Р
II С1
С3Н6ОРС1
М. в. 229, 89
Дихлорангидрид 2,3-дихлорпропилфосфиновой кислоты
может быть использован для получения соответствующих
эфиров, которые после дегалоидирования превращаются в
производные пропенилфосфиновой кислоты. Таким образом,
названный дихлорангидрид оказывается удобным реагентом
для синтеза различных фосфорсодержащих материалов, зна-
чение которых в последнее время неуклонно растет.
Дихлорангидрид 2,3-дихлорпропилфосфиновой кислоты
получают из доступных реагентов при окислительном хлор-
фосфинировании хлористого аллила треххлористым фосфором
и кислородом [1, 2].
СХЕМА СИНТЕЗА ДИХЛОРАНГИДРИДА
2,3-ДИХЛОРПРОПИЛФОСФИНОВОЙ кислоты
СН2 — СНСН2С1 4- 2РС13 + О2 - С1СН.2СНС1СН2РОС12 + РОС1
Характеристика основного сырья
Хлористый аллил, ч., ТУ МХП 1917—49.
Треххлористый фосфор, ч., ГОСТ 91- 41.
Кислород, ГОСТ 5583—58.
61
Условия получения
В цилиндрический реактор, снабженный шоттовским бар-
ботером, помещают 76,5 г (1 М) хлористого аллила и 412 г
(3 Л4) треххлористого фосфора (см. примечание 1). Через
смесь пропускают сухой кислород до тех пор, пока реакция
не прекратится. Внешним охлаждением температуру в реак-
ционной смеси поддерживают в пределах 0—5°.
Отходящие из реактора газы — непрореагировавший кис-
лород, содержащий примесь паров хлористого аллила, трех-
хлористого фосфора и хлорокиси фосфора, пропускают через
конденсатор для их улавливания и затем промывают после-
довательно водой и 5—10%-ным раствором щелочи.
После прекращения выделения тепла (окончание реакции)
из реакционной массы отгоняют в вакууме 50 мм сначала
непрореагировавший хлористый аллил, затем образовавшую-
ся хлорокись фосфора (см. примечание 1). Полученный про-
дукт перегоняют в вакууме.
Выход дихлорангидрида 2,3-дихлорпропилфосфиновой кис-
лоты 170—180 г, что составляет 75—80% от теоретического;
т. кип. 103—107° при 3 мм; с/420—1,5 891; и™ —1,5073 (см.
примечание 2).
Прпмечани я:
1. Наряду с окислительным хлорфосфинированием происходит пря-
мое окисление треххлористого фосфора в хлорокись фосфора (2РС1з + О2 ->
— 2 РОС1з), поэтому в продуктах реакции количество образовавшейся
хлорокиси фосфора значительно превышает теоретическое.
2. В полученном дихлорангидриде возможно присутствие небольшого
количества изомерного дихлорангидрида 1,3-дпхлорпропилфосфиновой
кислоты.
Литература
1. Л. 3. Соборовский, Ю. М. Зиновьев, Л. И. М у л е р.
Докл. АН СССР, 109, 98 (1956).
2. Л. 3. Соборовский, Ю. М. Зиновьев, Л. И. Мулер.
Ж- общ. химии, 29, 3947 (1959).
Поступила в июне 19S4
О Зак. 131
УДК 661.718.07
ДИХЛОРАНГИДРИД
циклогексилфосфиновой кислоты
Циклогексилдихлорфосфиноксид
Л. 3. СОБОРОВСЕИИ, Ю. М. ЗИНОВЬЕВ
С /--р(
—z II 'Cl
О
C6HjiOPCl2 М. в. 201,05
Дихлор ангидрид циклогексилфосфиновой кислоты может
служить доступным реагентом для синтеза разнообразных
фосфорорганических соединений. Простейшим путем для его
получения является окислительное хлорфосфинирование
циклогексана треххлористым фосфором и кислородом [1, 2. 3].
СХЕМА СИНТЕЗА ДИХЛОРАНГИДРИДА
ЦИКЛОГЕКСИЛФОСФИНОВОЙ КИСЛОТЫ
/~\ /С1
С6Н.12 + 2РС13 + О2 \_/+ РОС1Н-НС1
h U1
о
Характеристика основного сырья
Циклогексан, ч., ВТУ У 153—51.
Треххлористый фосфор, ч., ГОСТ 91—41.
Кислород, ГОСТ 5583—58.
Условия получения
В цилиндрический реактор, снабженный шоттовским бар-
ботером, помещают 168,32 г (2М) циклогексана и 137,4 г
66
(1 М) треххлористого фосфора. Через смесь пропускают су-
хой кислород. Сразу же начинается реакция, сопровождаю-
щаяся значительным выделением тепла. Температуру в реак-
торе наружным охлаждением поддерживают в пределах
5—10°. По мере прохождения реакции интенсивность выделе-
ния тепла постепенно снижается и полностью прекращается
после ее завершения.
Отходящие из реактора газы (непрореагировавший кисло-
род и образовавшийся хлористый водород), содержащие пары
циклогексана, треххлористого фосфора и хлорокиси фосфора,
пропускают через конденсатор для их улавливания и затем
промывают последовательно водой и 5—10%-ным раствором
щелочи.
После прекращения выделения тепла из реакционной мас-
сы отгоняют сначала непрореагировавший циклогексан, за-
тем образовавшуюся хлорокись фосфора (в вакууме 50мм,
см. примечание 1). Полученный продукт перегоняют в ваку-
уме при 92—9572 мм. Остаток незначительный (см. приме-
чание 2). При повторной перегонке выделяют чистое вещест-
во. Выход 95—100 г, что составляет 45—50% от теоретическо-
го; т. кип. 93—9472 мм; т. пл. 39—40°.
Примечания:
1. Наряду с приведенной выше реакцией при окислительном хлорфос-
финировании всегда происходит прямое окисление треххлористого фосфо-
ра (2 РС1з + О2-»2 РОС1з). Поэтому в продуктах реакции количество об-
разовавшейся хлорокиси фосфора значительно превышает теоретическое,
определяемое уравнением.
2. Проведение реакции возможно и при иных соотношениях реаген-
тов. Применяя меньшее отношение СеНцрРСЬ, в продуктах реакции полу-
чают заметное количество тетрахлорангидрида циклогексилдифосфиновой
кислоты—C6Hio(POCl2)2—вещества с т. кип. 192—19570,5 мм, застываю
щего в белые кристаллы. При большем значении указанного отношения
образования тетрахлорангидрида циклогексилдифосфиновой кислоты прак-
тически ие происходит.
Литература
1. Л. 3. С о б о р о в с к и й, Ю. М. Зиновьев, М. А. Энглин.
Докл. АН СССР, 67, 293 (1949); Авт. свид. 117901; Бюлл. изобр., Я» 3
(1959).
2. J. С I а у t о н, W. Jensen. J. Amer. Chem. Soc., 70, 3880 (1948).
3. R. Graf. Ber., 85, 9 (1952).
Поступила в июне 1964 г.
3*
УДК 546.183.07
ДИЭТИЛВИНИЛФОСФАТ
Ю. Г. ГОЛОЛОБОВ, Т. Ф. ДМИТРИЕВА, Л. 3. СОБОРОВСКИЙ
СДДзОдР
(С2НбО)2Р-ОСН=СН2
о
М. в. 180,14
Диэтилвинилфосфат получают из триэтилфосфита и хлор-
ацстальдсгида [1]. Нами показано [2], что диэтилвинилфосфат
с высоким выходом может быть получен из диэтилфосфорной
кислоты и дивинилртути.
СХЕМА СИНТЕЗА ДИЭТИЛВИНИЛФОСФАТА
(С2Н6О)2Р—ОН + Hg(CH=CH2)3
II
О
-> (С2Н5О)2Р-ОСН=СН2 + Hg 4- сн2 - сн2
Характеристика основного сырья
Диэтилфосфорная кислота, получают гидролизом диэтил-
хлорфосфата [3].
Дивинилртуть, получают по прописи [4] (см. примечание!),
Условия получения
Нагревают смесь 15,4 г (0,1 Л1) диэтилфосфорной кислоты
и 25,5 г (0,1 А4) дивинилртути до 80—90°. При достижении
указанной температуры начинается реакция — выделяется
этилен и металлическая ртуть. После прекращения выделения
68
газа смесь перегоняют в вакууме. Получают диэтилвинил-
фосфат с выходом 90% от теоретического; т. кип. 59°при 1 мм;
d^0—1,0926; л» —1,4109.
По литературным данным, т. кип. продукта 79° при 6 лш
[1] (см. примечание 2).
Примечания:
1. Дивинилртуть обладает отвратительным запахом, ощущаемым в
ничтожных концентрациях. Разрушение дивинилртути достигается обра-
боткой крепкой соляной кислотой.
2. Описанный метод пригоден для получения виниловых эфиров раз-
личных кислот фосфора, но при введении в реакцию тиокислот фосфора
выходы заметно снижаются.
Л итература
1. J. F. А 11 е п, О. И. Johnson. J. Ашег. Chem. Soc., 77. 2871
(1955).
2, Ю. Г. Г о л о л о б о в, Т. Ф. Дмитриева, Л. 3. С о б о р о в-
с к и й. Авт. свид. 163618; Бюлл. изобр., Л’д 13 (1964).
3. Toy. J. Amer. Chem. Soc., 70. 3882 (1948).
4. G. F. Reynolds, R. E. D e d s s у. H. H. Jaffe. J. Organ. Chem.,
23, 1217 (1958).
Поступила в июнэ 1964 г.
УДК 547.476.2.07
ДИЭТИЛОВЫЙ ЭФИР СУКЦИНИЛЯНТАРНОЙ
кислоты
Н. М. РЫБКИНА, В. И. ЯКУШИНА
С12Н16О6
о
II
/с\
Н5С2ООС-НС сн„
I I
Н2С СН-СООС2Н5
о
М. в. 256,25
По литературным данным, диэтиловый эфир сукцинилян-
тарной кислоты получают конденсацией диэтилового эфира
янтарной кислоты в присутствии металлического натрия в
среде абсолютного этилового спирта [1J, в среде уксусноэти-
лового эфира [2], конденсацией диэтилового эфира янтарной
кислоты в присутствии этилата натрия [3]. из производных
ацетоуксусного эфира и этилата натрия [4], из у-хлорацетоук-
сусного эфира и фталимида калия [5].
Нами проверен и уточнен метод получения диэтилового
эфира сукцинилянтарной кислоты конденсацией диэтилового
эфира янтарной кислоты в присутствии металлического нат-
рия в среде уксусноэтилового эфира [2].
СХЕМА СИНТЕЗА ДИЭТИЛОВОГО ЭФИРА СУКЦИНИЛЯНТАРНОИ
КИСЛОТЫ
СН2--СОО-С2Н5
2 I
СН2-СОО-С3Н5
Na
СН,СООС2Н6
70
о
Н5С2ООС-НС сн2
| ( 4- 2СоН501 I
Н2С СН-СООСоНб
Характеристика основною сырья
Диэтиловый эфир янтарной кислоты, ч., ГОСТ 143—51.
Эфир уксусноэтиловый, абсолютный, ст. ГОХП 27—1839.
Натрий металлический, ГОСТ 3273—55.
Соляная кислота, ч., ГОСТ 3118—46.
Спирт этиловый, абсолютный, ректифицированный, ГОСТ
>962—51.
Условия получения
В трехгорлую колбу емкостью 2 л, снабженную мешалкой
•с затвором, обратным холодильником с хлоркальциевой труб-
кой и термометром, загружают 218 г (1,25 М) диэтилового
зфира янтарной кислоты, 140 мл уксусноэтилового эфира,
14 г (1,91 М) металлического натрия в виде проволоки и 10мл
абсолютного этилового спирта. Температура реакционной
массы быстро поднимается до 60—70°. Как только темпера-
тура начнет снижаться, колбу погружают в глицериновую ба-
ню и реакционную массу нагревают до 80°. При этой темпе-
ратуре и размешивании дают четырехчасовую выдержку,
прибавляя каждый час по 20 мл уксусноэтилового эфира. По
мере течения реакции реакционная масса густеет, к концу вы-
держки мешалку выключают и конденсация идет без разме-
шивания. Реакционной массе дают остыть до комнатной тем-
пературы. Для разложения непрореагировавшего металличе-
ского натрия и растворения реакционной массы приливают
400 мл этилового спирта и размешивают 5—10 минут. К по-
лученной кашеобразной массе осторожно при размешивании
и небольшими порциями прибавляют 400 мл дистиллирован-
ной воды, подкисляют 400—450 мл 14%-ной соляной кислоты
до pH 2—3 по универсальной индикаторной бумажке. Ох-
лаждают до 8—10°, выпавший осадок диэтилового эфира
сукцинилянтарной кислоты отфильтровывают на фарфоровой
воронке, промывают 50 мл этилового спирта и сушат в су-
шильном шкафу при 50—60°.
Выход технического продукта ЮЗ—109 г, что составляв!
65—69% от теоретического; т. пл. 125—127°,
71
Для дальнейшей очистки продукт перекристаллизовыва-
ют из 1680 мл кипящего этилового спирта в присутствии ак-
тивированного угля. Получают 79—84 г чистого диэтилового
эфира сукцинилянтарной кислоты (49—54% от теоретическо-
го выхода); т. пл. 126—128°.
По внешнему виду препарат представляет собой вещество
белого цвета с зеленоватым оттенком. По литературным дан-
ным, т. пл. продукта 128° [6]; 126—127° [3].
Литература
1. А. У с п е н с к и й, И. Тюрин. Ж. русского физ-хим. об-ва, 51,
269 (1919).
2. Н. Lieberman. Liebigs Ann. Chem., 404, 287 (1919).
3. P. Хаузер, Б. E. Хадсон. Органические реакции, т. 1, М., ИЛ,
1948, стр. 368.
4. Somme lei, С о и го их и др. Bull. Soc. chim. France, 29, 405
(1921).
5 В. M. Родионов, М. А. Губарева. Ж. общ. химии, 23, 1833
(1953).
6. Bellst, 10, 894, Е 1 434.
Поступила в мае 1964 г. ИГЕА
УДК 661.718:541.49:546.621'131
КОМПЛЕКСНЫЕ СОЕДИНЕНИЯ
АЛКИЛТЕТРАХЛОРФОСФИНОВ С ХЛОРИСТЫМ
АЛЮМИНИЕМ
и. п. комков, к. В. КАРАВАНОВ, С. 3. ИВИН
RPCI4 • А1С13
Комплексные соединения алкилтетрахлорфосфинов и хло-
ристого алюминия являются реакционноспособными соеди-
нениями, они находят все более широкое использование для
синтеза разнообразных фосфорорганических препаратов.
Комплексные соединения впервые были получены
В. А. Плотниковым в 1916 г. [1] при взаимодействии броми-
стого алкила, бромистого алюминия и пятибромистого или
трехбромистого фосфора.
В 1951 г. независимо друг от друга Клей [2], Киннер и
Перрен [3] описали получение комплексных соединений, осно-
ванное на взаимодействии хлористых алкилов, треххлористо-
го фосфора и хлористого алюминия. Авторы отмечают необ-
ходимость соблюдения при их получении следующих условий:
а) строгий контроль температурного режима реакции; б) су-
хое, безводное состояние аппаратуры (предпочтительно авто-
клавов) и реагентов; в) определенная последовательность
смешения реагентов.
Эти требования приводят к сравнительно сложному аппа-
ратурному оформлению и к препаративным трудностям.
Нами предложен более простой и удобный в аппаратурном
оформлении способ получения комплексных соединений. Он
заключается в том, что хлористый алюминий, треххлористый
фосфор и хлористый алкил смешиваются и выдерживаются в
запаянной трубке или в любой закрытой металлической ем-
кости. Особенно удобными для получения комплексов оказа-
лись баллончики из-под сжатого воздуха или кислорода емко-
стью 0,7 л.
73
КОМПЛЕКСНОЕ СОЕДИНЕНИЕ ЭТИЛТЕТРАХЛОРФОСФИНА
С ХЛОРИСТЫМ АЛЮМИНИЕМ
С2Н6РС14-А1С13 М. в. 335, 18
СХЕМА СИНТЕЗА КОМПЛЕКСА
С2Н6С1 + РС(3 4- А1С1з - С2Н5РС14 • А1С13
Характеристика основного сырья
Хлористый этил из баллонов или ампул без предвари-
тельной перегонки.
Фосфор треххлористый, ч., перегнанный, ГОСТ 91—41.
Алюминий хлористый, ос. ч., безводный, ВТУ МХП
3500—52 или свежевозогнанный.
Условия получения
В охлажденный до минус 40° автоклав (баллон) емкостью
0,7 л (см. примечание 1) загружают 133,3 г (1 А1) хлористо-
го алюминия, 137,3 г (1 М) треххлористого фосфора и через
15 минут приливают 96,7 г (1,5 М) хлористого этила (см.
примечание 2). Быстро закрывают автоклав (баллон) и по-
мещают его на трясучку. Через некоторое время реакционная
смесь самопроизвольно разогревается до 50—80°. Встряхива-
ние продолжают до тех пор, пока реактор не охладится до
комнатной температуры (примерно один час) и оставляют на
ночь. Содержимое в автоклаве (баллоне) закристаллизовы-
вается в однородную массу. Затем автоклав (баллон) поме-
щают в водяную баню, нагретую до 40—50°, выпускают из-
быток хлористого этила в охлажденную ловушку через вен-
тиль, вскрывают автоклав (баллон) длинным шпателем, раз-
мельчают осадок, быстро переносят его (см. примечание 3) в
вакуум-эксикатор и выдерживают под вакуумом 10 мм 1 час.
Выход комплексного соединения этилтетрахлорфосфина с
хлористым алюминием 331,1 г, что составляет 99% от теоре-
тического; т. пл. 230—240°.
Примечания:
1. Автоклав (баллон) должен быть сухим и при охлаждении защи-
щен от попадания влаги (хлоркальциевая трубка).
2. Хлористый этил необходимо приливать охлажденный до минус 70°
и быстро закрыть автоклав.
3. Комплексное соединение легко гидролизуется, в связи с чем пере-
носить его из реактора необходимо как можно быстрее.
В хорошо закрытой таре (без доступа воздуха) комплексное соеди-
нение может храниться годами.
По описанной методике можно получить комплексные сое-
динения с различными алкильными радикалами при фосфо-
ре.
74
Л итература
1. В. А. Плотников, ж. русского физ.-хим. об-ва, 48, 189 (1916).
2. Р. Clay. J. Organ. Chem., 16, 6, 899 (1951).
3. А. М. К inn ear, Е. A. Per ген. J. Chem. Soc., 3437 (1952).
Поступила в июле 1964 г.
УДК 661.718:541.49:546.621'121
КОМПЛЕКСНЫЕ СОЕДИНЕНИЯ
ДИАЛКИЛТРИХЛОРФОСФИНОВ С ХЛОРИСТЫМ
АЛЮМИНИЕМ
С. 3. ИВИН, к. в. КАРАВАНОВ
R2PC13
Комплексные соединения диалкилтрихлорфосфинов с хло-
ристым алюминием впервые пытались получить Киннер и
Перрен [1]. Так, при взаимодействии этилдихлорфосфина с
хлористым этилом и хлористым алюминием ими были выде-
лены твердые соединения. Однако строение этих соединений
не изучалось и авторы не были уверены в том, что получили
диалкильные комплексы.
В 1957 г. Бидл с сотрудниками [2) описали взаимодействие
фенилдихлорфосфина с хлористыми алкилами в присутствии
хлористого алюминия, приводящее к образованию твердых
продуктов, из которых при действии большого количества во-
ды были получены фенилалкилфосфиновые кислоты. Более
подробной характеристики продуктам реакции фенилдихлор-
фосфина, хлористого алкила и хлористого алюминия сделано
не было.
Комплексные соединения диалкилтрихлорфосфинов и хло-
ристого алюминия нами получены взаимодействием алкилди-
хлорфосфинов, хлористых алкилов и хлористого алюминия.
Выход комплексных соединений достигает 98—99%. Комплек-
сные соединения представляют собой белые кристаллические
вещества, легко вступающие в реакции со многими реагента-
ми [3].
КОМПЛЕКСНОЕ СОЕДИНЕНИЕ ДИЭТИЛТРИХЛОРФОСФИНА С
ХЛОРИСТЫМ АЛЮМИНИЕМ
(С2Н6)2РС13-А’С13 М. в. 328, 79
СХЕМА СИНТЕЗА КОМПЛЕКСА
7б С2Н5РС13 + е2НБС1 + А1С13 (С2НБ)2РС13 • А1С13
Характеристика основного сырья
Этилдихлорфосфин, перегнанный, получен по методике,
описанной в настоящем сборнике (см. статью «Алкилдихлор-
фосфины») .
Хлористый этил из баллонов или медицинский, без пред-
варительной перегонки.
Алюминий хлористый, безводный, ВТУ МХП 3500—52 или
свежевозогнанный.
Условия получения
В охлажденный до минус 40° автоклав (баллон) емкостью
1 л (см. примечание 1) загружают 133,3 г (1 М) хлористого
алюминия, 130,9 г (1 М) этилдихлорфосфина (см. примеча-
ние 2) и через 15 минут приливают 96,7 г (1,5 М) хлористого
этила (см. примечание 3). Быстро закрывают автоклав (бал-
лон) и помещают его на трясучку. Через некоторый проме-
жуток времени реакционная смесь самопроизвольно разогре-
вается до 50—80°. Перемешивание продолжается примерно
один час до охлаждения автоклава (баллона) до 15—20°, по-
сле чего реактор оставляется на ночь. По мере охлаждения
содержимое в автоклаве (баллоне) закристаллизовывается в
однородную белоснежную массу. Затем автоклав (баллон)
помещают в водяную баню, нагретую до 40—50°, и выпускают
избыток хлористого этила в охлажденную ловушку. Автоклав
(баллон) вскрывают и длинным металлическим шпателем
осадок размельчают и быстро переносят (см. примечание 4)
в вакуум-эксикатор, где выдерживают под вакуумом 10—
15 мм 1 час.
Получают 325,1 г комплекса диэтилтрихлорфосфина с хло-
ристым алюминием, что составляет ~ 99% от теоретического
выхода; т. пл. 155—160°.
Примечания:
1. Автоклав (баллон) должен быть сухим и при охлаждении защи-
щен от попадания влаги (хлоркальциевой трубкой). Вместо автоклава с
успехом используются баллончики из-под воздуха пли кислорода емко-
стью 0,75—1 л.
2. Этилдихлорфосфин легко окисляется на воздухе и при его загруз-
ке автоклав необходимо заполнить азотом.
3. Хлористый этил, охлажденный до минус 40°, необходимо приливать
быстро, автоклав (баллон) немедленно закрыть.
4. Комплексное соединение легко гидролизуется, в связи с чем кон-
такт его с воздухом необходимо ограничить. В хорошо закрытой таре ком-
плекс может храниться продолжительное время.
Другие комплексные соединения диалкилтрихлорфосфинов
и хлористого алюминия с одноименными или разноименными
алкильными радикалами получаются по описанной методике
с выходом 98—99%.
При использовании хлоралкилов с большим числом ато-
мов углерода получение диалкильных комплексов можно осу-
77
ществлять нагреванием реагентов в колбе с обратным холо-
дильником.
Литератур-®
1. Л. М. Kinnear, Е. A. Perren. J. Chem. Soc., 3437 (1952).
2. Р. В i е 11 е, I. Кеппе di, J. W 111 a n s. Chemistry and industry,
45, 1471 (1957).
3. С. 3. И в и и, К. В. Караванов. Ж. общ. химии, 28, 2958 (1958)
Поступила в июне 1964 г.
УДК 547.292.07
2-МЕТОКСИ-4-АМИНОТОЛУОЛ-!\’,И-ДИУКСУСНАЯ
КИСЛОТА
Р. 77. ЛАСТОВСКИЙ, И. Д. КОЛПАКОВА, Л. В. КРИНИЦКАЯ,
Т. И. ИВАНОВА, Л. Д. ЗАВЬЯЛОВА
,СН2СООН
I 'СНаСООН
^/И-ОСНз
СН3
C12H16NO5
М. в. 253,28
2-Метокситолуол-4-амино-М,М-диуксусная кислота синте-
зирована и предложена ИРЕА в качестве нового комплексо-
на и как промежуточный продукт для получения 4'-нитро-2-
амино-4-метокси-5-метилазобензол-Г\т,М-диуксусной кислоты.
Синтез вещества нами осуществлен взаимодействием 2-меток-
си-4-аминотолуола с монобромуксусной кислотой.
СХЕМА СИНТЕЗА 2-МЕТОКСИ-4-АМИНОТОЛУОЛ-М,М-ДИУКСУСНОИ
КИСЛОТЫ
nh2
J^J-OCH:
+ 2BrCH2COOH + 2NaOH
СНз
79
СН2СООН
N\
I XCH3COOH
nr., 4-2NaBr 4- 2H2O
CH3
Характеристика основного сырья
4-Амино-2-метокситолуол, т. пл. 56—57° (синтезирован в
НИОПиК).
Монобромуксусная кислота, ч., ТУ МХП 3667—55.
Условия получения
В трехгорлую колбу, снабженную механической мешалкой,
термометром и обратным холодильником, загружают 5 г
(0,0365Л4) 2-метокси-4-аминотолуола, смачивают его 10 мл
дистиллированной воды и в течение 15 минут при перемеши-
вании приливают раствор 15,3 г (0,11 М) монобромуксусной
кислоты, предварительно нейтрализованной (до pH 7) 20 % -
ным раствором едкого натра. Конденсацию проводят при пе-
ремешивании при 80°, поддерживая pH 7—8 по универсаль-
ной индикаторной бумажке прибавлением 20%-ного раствора
едкого натра. Реакцию считают законченной, если после при-
бавления последней порции щелочи pH раствора не изменя-
ется в течение 30 минут. После этого раствор перемешивают
при 80° в течение часа, горячим фильтруют и после охлажде-
ния подкисляют 10%-пым раствором соляной кислоты до pH
1,5—2. Выпавший осадок на следующий день фильтруют и
сушат при температуре 80°.
Полученный комплексон очищают перекристаллизацией из
воды в соответствии 1:5 (1:8) или из 40 %-кого этилового спир-
та в соотношении 1:2.
При перекристаллизации из воды получают 4,7 г продукта,
что составляет 50% от теоретически возможного выхода; по
внешнему виду это светло-желтый порошок с т. пл. 145—147°;
при перекристаллизации из спирта выход 4,3 г, что составля-
ет 45% от теоретического; по внешнему виду это светло-ко-
ричневый порошок с т. пл. 154—156°. 2-Метокси-4-аминотолу-
ол-N,N-диуксусная кислота нерастворима в холодной воде,
растворима в горячей, а также в 40%-ном этиловом спирте
при нагревании.
Найдено из воды. %: N-5.53; 5,64; С—56,94; 57,24; Н—6,57; 6,44.
Найдено из спирта, %: N—5,61; 5,52; С—57,01; 56,94: Н—5,80; 6,03.
C12H15NO5. Вычислено, %; N -5,53; С—56,90; Н—5,92.
Поступила в сентябре 1V54 г. ИРЕА
УДК 661.718.07
НАТРИЕВЫЕ СОЛИ О-АЛКИЛОВЫХ ЭФИРОВ
АЛКИЛФОСФИНОВОЙ КИСЛОТЫ
Я. Д. ШЕЛАКОВА, С. 3. ИВИН
-P-^OR'
XONa
Натриевые соли О-алкиловых эфиров алкилфосфиновых
кислот находят широкое применение в лабораторной практи-
ке при синтезе различных производных алкилфосфиновых
кислот [1, 2, 3]. Нами получены натриевые соли О-алкиловых
эфиров путем взаимодействия О-алкилового эфира алкилфос-
финовой кислоты с алкоголятами или металлическим натри-
ем в эфире.
НАТРИЕВАЯ СОЛЬ О-ЭТИЛОВОГО ЭФИРА МЕТИЛФОСФИНОВОЙ
КИСЛОТЫ
о
CH3P^ONa
Хос2н6
C3H8O3PNa
М. в. 146,05
СХЕМА СИНТЕЗА НАТРИЕВОЙ СОЛИ О-ЭТИЛОВОГО ЭФИРА
МЕТИЛФОСФИНОВОЙ КИСЛОТЫ
О О
I. СНзР^ОН + C2H6ONa CH3P^ONa + С2Н6ОН
ХОС,Н5 ОС2Н6
6 Зак.131
81
II. 2CH3P^OH 4-2Na-> 2CH3P^ONa + H2
ос,н5 хос,н.
Характеристика основного сырья
О-Этиловый эфир метилфосфиновой кислоты, т. кип. 115°
при 1 мм; di20—1,0800; получен путем прибавления эквимо-
лекулярного количества воды к хлорангидриду О-этилового
эфира метилфосфиновой кислоты.
Спирт этиловый, абсолютный, ГОСТ 5962—51.
Натрий металлический, ТУ МХП 1664—50.
Эфир этиловый, абсолютный, ГОСТ -6265—52.
Условия получения
Способ 1. В трехгорлую колб5' с механической мешалкой,
обратным холодильником и капельной воронкой помещают
2,3 г (0,1 М) металлического натрия и 60 мл абсолютного эти-
лового спирта. К полученному раствору этилата натрия мед-
ленно при перемешивании прибавляют 12,4 г (0,1 М) О-эти-
лового эфира метилфосфиновой кислоты. По окончании при-
бавления спирт отсасывают из реакционной колбы в вакууме
водоструйного насоса. Полученную натриевую соль О-этило-
вого эфира метилфосфиновой кислоты промывают эфиром и
высушивают в вакууме.
Выход 13,2 г, что составляет 90% от теоретического.
Способ 2. В трехгорлую колбу с механической мешалкой,
обратным холодильником и капельной воронкой помещают
2,3 г (0,1 М) мелкодисперсного металлического натрия и 60 мл
абсолютного эфира. К полученной взвеси металлического нат-
рия в эфире медленно при перемешивании прибавляют 12,4 г
(0,1 М) О-этилового эфира метилфосфиновой кислоты. Обра-
зовавшуюся натриевую соль отфильтровывают и высушивают
в вакууме.
Натриевая соль О-этилового эфира метилфосфиновой кис-
лоты представляет собой белое мелкокристаллическое веще-
ство с т. пл. 232°. Оба способа позволяют получить достаточ-
но чистые натриевые соли О-алкиловых эфиров алкилфосфи-
новых кислот с хорошими выходами.
Ниже приводится таблица исходных веществ и полученных
натриевых солей О-алкиловых эфиров метилфосфиновой кис-
лоты и их температуры плавления.
82
Исходные вещества Т. кип. Полученная Na-соль T. пл., °C
/° СН,Р^-ОН ХОС2Н6 115°/1 мм 1,080 Z CHaP~-ONa OCjH5 232°
СН8Р^ОН OC8H7i 118°/1 мм 1,054 //* СН3Р(—ONa XOC8H,Z 234п
СН3Р^ОН ОС4Н9 1 120°/1 мм 1,079 о CH,P^ONa 4"OC4H„ I 238°
Литература
1. Pelchowlcz, Leader. J. Chem. Soc, № 16, 3320 (GJ5) (1963).
2. Pianka, Polton. Англ, пат. 867780 (1961).
3. Michalski, Wiczorkowski, W a s r a k. Pliszka Roczn. Chem.
33, I, 247 (1959).
Поступила в июне 1964 г.
УДК 547.653.1.07
[3 НАФТАЛИНСУЛЬФОХЛОРИД
Б. М. БОЛОТИН, Д. А. ДРАПКИИА, В. Г. БРУДЗЬ, Т. Е. ОСЬКИНА
C10H7ClOaS
М. в. 226,68
0-Нафталинсульфохлорид представляет интерес в качест-
ве исходного продукта в ряде синтезов. В промышленности
его получают из натриевой соли р-нафталинсульфокислоты и
пятихлористого фосфора с последующей кристаллизацией из
хлороформа. Синтез довольно длительный, а выход около 45%
от теоретически возможного.
Нами разработаны оптимальные условия получения
p-нафталинсульфохлорида по описанному [1] общему способу
получения хлорангидридов кислот. Реакцию ведут в средеди-
метилформамида, а в качестве хлорирующего агента исполь-
зуется хлористый тионил. В этих условиях синтез (3-нафталип-
сульфохлорида протекает быстро и сразу получается качест-
венный продукт с хорошим выходом.
СХЕМА СИНТЕЗА р-НАФТАЛИНСУЛЬФОХЛОРИДА
ПРг30* +soci, -
/\_cnri
- | II I 2 +SO2+NaCl
Ч/W
84
Характеристика основного сырья
fl-Нафталинсульфокислоты натриевая соль, ч,, ВТУ РУ
1189—55.
Диметилформамид ч., ВТУ РУ 1193—56.
Хлористый тионил, ч., ВТУ МХП 3591—52.
Условия получения
В двухлитровый фарфоровый стакан, снабженный пропел-
лерной мешалкой и термометром, помещают 375 мл диметил-
формамида и при размешивании присыпают 230 г (1 Л1) нат-
риевой соли р-нафталинсульфокислоты. Затем, продолжая
интенсивное размешивание, в течение 5 минут приливают 76 мл
(1,1 34) тионилхлорида; температура смеси при этом не дол-
жна превышать 40°. По окончании прибавления тионилхлори-
да реакционную массу перемешивают еще 20 минут, затем ох-
лаждают до минус 3° и приливают 1400 мл ледяной воды с
такой скоростью, чтобы температура реакции не поднималась
выше 10°. Выпавший продукт отфильтровывают на воронке
Вюхнера и сушат в сушильном шкафу при 60° до постоянного
веса.
Выход p-нафталинсульфохлорида равен 182 г, что состав-
ляет 80% от теоретического; т. пл. 76,2—77,Г.
Литература
1. Н. Н. В о s s h а г d, R, Могу, М. Schmid, Н. Z olinger. Helv.
chim. acta, 42, 1653 (1959).
Поступила в июле 1964 г.
ИРЕА
УДК 547.556.31.07
4' НИТРО-2-АМИНО-4-МЕТОКСИ-5-МЕТИЛАЗОБЕНЗОЛ
Р. П. ЛАСТОВСКИЙ, И. Д. КОЛПАКОВА, Л. В. КРИНИЦКАЯ,
Л. Д. ЗАВЬЯЛОВА, Т. И. ИВАНОВА
Ч / Ч /
I Н3С |
no2 осн,
C14HuN4O3
М. в. 286,29
Азокраситель 4z-HHTpo-2-aMHHO-4-MeTOKCH-5-MeTHfla3o6eH-
зол был получен в ИРЕА в качестве промежуточного продук-
та для синтеза комплексона по методике, аналогичной полу-
чению красителя 2'-хлор-4'-нитро-2-амино-4-метокси-5-метил-
азобензола (прочно-коричневая фау-база), разработанной в
НИОПиК в 1958 году, сочетанием л-нитроанилина с 4-амино-
2-метокситолуолом в среде метилового спирта в присутствии
серной кислоты (моногидрата).
Нами уточнены условия синтеза и разработан метод выде-
ления и очистки красителя.
СХЕМА СИНТЕЗА 47-НИТРО-2-АМИНО-4-МЕТОКСИ-5-
МЕТИЛ АЗОБЕНЗОЛА
I. O2N-/ 4_NH2-]-NaNO24-2H2SO4
-> J" O.2N-^-^-N = n]+HSO4- + 2H2O ф-NaHSO,
86
zch3
_ /
и. Jo2n—n—nJ i’hso4-+^z ^-ОСн3 ch,qh,h2so^
i=
nh2
X.CH3
-> O2N-^ 4-N^N-^ V-OCH3 + H2SO4
nh2
Характеристика основного сырья
Натрий азотистокислый, ч., ГОСТ 4197—48.
Серная кислота, уд. в. 1,83 (купоросное масло).
Серная кислота, уд. в. 1,84 (моногидрат).
п-Нитроанилин, ч., ТУ 1461—46.
4-Амино-2-метокситолуол, т. пл. 56—57° (синтезирован в
НИОПиК).
Условия получения
Получение п-нитродиазобензола. В трехгорлую колбу ем-
костью 100 мл, снабженную механической мешалкой, обрат-
ным холодильником и термометром, загружают 32 мл кон-
центрированной серной кислоты (уд. в. 1,83) и при энергич-
ном размешивании прибавляют в течение 25—30 минут 4 г
98%-ного или 3,92 г 100%-ного (0,057 М) сухого хорошо рас-
тертого нитрита натрия. Смесь нагревают на водяной бане в
течение 20—25 минут до 60—65°, размешивают при этой же
температуре еще 30 минут и затем охлаждают до 25°. К при-
готовленной нитрозилсерной кислоте медленно в течение часа
добавляют при размешивании 7,4 г (0,055 М) сухого м-нитро-
анилина и размешивают 30 минут при 20—25°. Пробой с йод-
крахмальной бумажкой убеждаются в наличии избытка азо-
тистой кислоты, который затем осторожно удаляют добавле-
нием п-нитроаналина.
Получение азокрасителя. В стакан емкостью 250 мл поме-
щают 130 мл метилового спирта и при размешивании вносят
8,6 г (0,063 М) 4-амино-2-метокситолуола и 11 мл серной кис-
лоты (моногидрата). Охлаждают смесь до 0° и постепенно в
течение 25—30 минут добавляют раствор п-нитродиазобензо-.
ла. Реакционную массу перемешивают 10—12 часов при
5—10°.
Конец реакции азосочетания определяют пробой со щелоч-
ным раствором Р-соли: в момент соприкосновения капли сме-
87
си с раствором соли на фильтровальной бумаге не должно
быть (или очень слабое) окрашивания.
Выпавший осадок фильтруют, отжимают на воронке, про-
мывают 16 мл метилового спирта, а затем водой до слабо-
кислой реакции по конго. Полученный коричневый порошок
сушат при 98—100° и тщательно растирают.
Получают 9,65 г технического красителя, что составляет
46,5% от теоретически возможного выхода.
Продукт очищают водным спиртом в нейтральной среде.
Для этого в трехгорлую колбу с механической мешалкой, об-
ратным холодильником и термометром загружают 9,65 г кра-
сителя и 400 мл 75 %-ного этилового спирта, нейтрализуют до
pH 6 -7 добавлением 20%-ного водного раствора едкого нат-
ра, нагревают до температуры кипения и кипятят при непре-
рывном размешивании 3 часа. После охлаждения фильтру-
ют, промывают 200 мл дистиллированной воды, отжимают и
сушат при 98—100° до постоянного веса.
Получают 6,45 г красителя, что составляет 67% от техни-
ческого продукта; т. пл. 178°.
4'-Нитро-2-амино-4-метокси-5-метил азобензол нерастворим
в воде, растворим в 40 %-ном этиловом спирте, в метиловом
спирте и при нагревании в 96%-ном этиловом спирте.
Найдено, %: N—20,11: 20,00; С-58,83; 58,59; Н-4,88; 4,91.
Вычислено, %: N -19,59; С—58,65; Н—4,89.
Поступила в сентябре 1964 г,
ИРЕА
УДК 547.292.07
4Z НИТРО-2-АМИНО-4-МЕТОКСИ-5-
МЕТИЛАЗОБЕНЗОЛ N.N-ДИУКСУСНАЯ КИСЛОТА И
ЕЕ ДИМЕТИЛОВЫЙ ЭФИР
Р. П. ЛАСТОВСКИЙ, И. Д. КОЛПАКОВА, Л. В. КРИНИЦКАЯ,
Т. И. ИВАНОВА
—N = ЧН2СООН
Ч\ 4\_Nf
L J ,1 чсн,соон
Ч/ /Ч/
I Н3с I
NO, ОСН3
C18H18N4O, М. в. 402,36
—N = N—г- /СНзСООСНз
М! ХСН2СООСН3
I НзС 1
NO2 ОСНз
C30H22N4O, М. в. 430,41
4'-Нитро-2-амино-4-метокси-5-метилазобензол-Ы, N'-диуксус-
ная кислота синтезирована и предложена ИРЕА в качест-
ве нового комплексона. В виде промежуточного продукта син-
тезирован и выделен диметиловый эфир этой кислоты.
Синтез диметилового эфира 4'-нитро-2-амино-4-метокси-5-
метилазобензол-N, N-диуксусной кислоты осуществлен сочета-
нием n-нитроанилина с 2-метокси-4-аминотолуол-Ы, N-диук-
сусной кислотой с одновременной этерификацией карбоксиль-
ных групп метиловым спиртом в присутствии серной кислоты
(моногидрата).
Сочетание проводили по методике, аналогичной получению
азокрасителя 2'-хлор-4/-нитро-2-амино-4-метокси-5-метилазо-
89
бензола (прочно-коричневая фау-база), разработанной в
НИОПиК в 1958 году.
Нами уточнены условия синтеза, разработан метод выде-
ления и очистки диметилового эфира комплексона, омылени?
эфира с образованием 4'-нитро-2-амино-4-метокси-5-метилазо
бензол-Ы.Н-диуксусной кислоты, ее выделения и очистки.
СХЕМА СИНТЕЗА 4'-НИТРО-2-АМИНО-4-МЕТОКСИ-5-
МЕТИЛАЗОБЕНЗОЛ-Ы.ЬКДИУКСУСНОИ КИСЛОТЫ И ЕЕ
ДИМЕТИЛОВОГО ЭФИРА
I. O2N-^ ^-NH2 + NaNO2 + 2H2SO4-
-> [O2N-^ ^-N = N] + HSO4-+ NaHSO4 + H2o
II. [O2N-^ = n] + hso4- +
ch3
। ОС pi 2CH8OH, HgSO4 ;
^П/сНаСООН
N<
CH2COOH
CH3
/
- O2N-^ ^-OCH3 + H2SO4 + 2H2O
= Г/СНЛЗООСНз
N(
'CH3COOCH3
CH3
III. O2N-^ ^-OCH3 +2H2O^-»
= ПСН.СООСНз
N<
CH2COOCH3
—N = N-7 zCII2COOH
r \ Nz
-* XCH2COOH +2CH3OH
I H3c I
NOa OCH3
90
Характеристика основного сырья
Натрий азотистокислый, ч., ГОСТ 4197—48.
Серная кислота, уд. в. 1,83 (купоросное масло).
Серная кислота, уд. в. 1,84 (моногидрат).
zi-Нитроанилин, ч., ТУ МХП 1461—46.
2-Метокси-4-аминотолуол-М, N-диуксусная кислота, синте-
зирована в ИРЕА.
Спирт метиловый, ч., ГОСТ 6995—54.
Условия получения
Получение п-нитродиазобензола дано в статье «4'-Нитро-
2-амино-4-метокси-5-метил азобензол».
Получение диметилового эфира 4'-нитро-2-амино-4-меток-
си-5-метилазобензол-П,М-диуксусной кислоты. В стакан ем-
костью 150 мл помещают 65 мл метилового спирта и при раз-
мешивании вносят 7,5 г (0,0296 М) 2-метокси-4-аминотолуол-
Н,Ы-диуксусной кислоты и 5,5 мл серной кислоты (моногид-
рата). Смесь охлаждают до 0° и постепенно в течение 25—
30 минут добавляют к пей раствор диазосоединения. Реакци-
онную массу перемешивают 10—12 часов при 5—10°.
Конец реакции азосочетания определяют пробой со ще-
лочным раствором Р-соли: в момент соприкосновения капли
смеси с раствором соли на фильтровальной бумаге не должно
быть окрашивания (или очень слабое).
Выпавший осадок кирпично-красного цвета фильтруют,
отжимают, промывают 8 мл метилового спирта и водой до
слабокислой реакции по конго и затем сушат при 50—60°.
Выход пригодного для синтеза комплексона диметилового
эфира 4/-нитро-2-амино-4-метокси-5-метилазобензол-М,Ы-ди-
уксусной кислоты 5,55 г, что составляет 51—52% от теорети-
ческого.
Для очистки диметиловый эфир комплексона дважды пе-
реосаждают водой из диметилформамида, промывают водой
и сушат при 98—100°; т. пл. продукта 147°.
Диметиловый эфир 4/-нитро-2-амино-4-метокси-5-метилазо-
бензол-М,,Ы-диуксусной кислоты нерастворим в воде, креп-
ких и разбавленных щелочах, плохо растворим в разбавлен-
ных минеральных и органических кислотах, в холодном эти-
ловом спирте. Растворим в концентрированных кислотах, в
горячем этиловом спирте, в метиловом спирте, ацетоне, хлоро-
форме, четыреххлористом углероде, бензоле и в других орга-
нических растворителях.
Найдено, %: N— 12,24; 12,48,С 56,57; 56,43; Н-5,05; 4,75.
C20H22N4O7. Вычислено, %: N —13,0; С - 55,9; Н-5,12.
Получение 4'-нитро-2-амино-4-метокси-5-метилазобензол-
N, N-диуксусной кислоты. В трехгорлую колбу, снабженную
91
механической мешалкой, обратным холодильником и термо-
метром, загружают 1 г (0,023 Л1) диметилового эфира комп-
лексона, добавляют 18,5 мл (0,023 2И) 10%-ного раствора ед-
кого натра и перемешивают при 90° в течение часа. Охлаж-
денный раствор фильтруют и подкисляют 10%-ным раствором
соляной кислоты до pH 2. Выпавший темно-коричневый оса-
док фильтруют, промывают большим количеством дистилли-
рованной воды до отсутствия ионов хлора и сушат при 60°.
Получают 0,75 г комплексона, что составляет 80%, считая на
загруженный эфир.
4'-Нитро-2-амино-4-метокси-5-метилазобензол-М, N-диуксус-
ная кислота нерастворима в воде, растворима в щелочах.
Найдено, %: N—13,87; 14/4; С- 53,94; 54,30; Н—5,^3; 4,99.
C1SH1SN4O,. Вычислено, %: N—13,93; С—53,90; Н—4.51.
Поступила в сентябре 19G4 г. ИРЕА
УДК 547.466.22.07
м -ОКСИФЕНИЛ ИМИ НОДИУКСУСНАЯ КИСЛОТА
Р. П. ЛАСТОВСКИЙ, В. я. ТЕМКИНА, Г. Ф. ЯРОШЕНКО,
И. П. ФАДЕЕВА
Ci0HnNO5
Ггон
| ,СН2СООН
N\
СН2СООН
М. в. 225, 20
л-Оксифенилиминодиуксусная кислота является неописан-
ным в литературе изомером оксифенилиминодиуксусных кис-
лот [1, 2}. Этот комплексон образует хелатные соединения с
рядом катионов, а также является мономером для получения
новой формальдегидной ионообменной смолы.
Продукт представляет собой серый порошок, хорошо рас-
творимый в воде, спирте, нерастворимый в ацетоне, бензоле,
хлороформе.
Синтез л1-оксифенилиминодиуксусной кислоты осущест-
влен нами взаимодействием .w-аминофепола с монохлоруксус-
ной кислотой при pH 7—8. Комплексон выделен в чистом виде
переводом его в свинцовый комплекс и последующим осажде-
нием свинца сероводородом.
СХЕМА СИНТЕЗА М-ОКСИФЕНИЛИМИНОДИУКСУС1ЮИ КИСЛОТЫ
// \ __ ОН
| . + 2С1СН2СООН 4-2NaOH ->
Ч/
I
nh2
93
он
-+| II + 2NaCl + 2Н,0
Ч/
| /СНаСООН
N\
ХСН2СООН
Характеристика основного сырья
лг-Аминофенол, ч., ВТУ 3929—53.
Монохлоруксусная кислота, ч., ГОСТ 5836—51.
Натр едкий, х. ч., ГОСТ 4328—48.
Уголь активированный, ГОСТ 8703-58-АР—3.
Кислота соляная, х. ч., ГОСТ 3118—46.
Свинец уксуснокислый, ч., ГОСТ 1027—51.
Железо сернистое, ТУ МХП 87—52.
Условия получения
В трехгорлую колбу, снабженную обратным холодильни-
ком и мешалкой, помещают 50 г (0,46 М) л-аминофенола и
100 мл дистиллированной воды. К реакционной массе прибав-
ляют при размешивании 130 г (1,375 Л4) монохлоруксусной
кислоты, предварительно нейтрализованной 20%-ним раство-
ром едкого натра до pH 7.
Реакцию проводят при размешивании и температуре 80°,
поддерживая pH 7—8 добавлением 20%-ного раствора едкого
натра в течение 4—5 часов (до прекращения изменения pH
раствора). Затем к горячему раствору добавляют 2—3 г ак-
тивированного угля, перемешивают в течение 10—15 минут и
фильтруют,
Охлажденный до комнатной температуры фильтрат под-
кисляют концентрированной соляной кислотой до pH 2. Кри-
сталлизацию вызывают трением стеклянной палочки о стенку
стакана после длительного (15 часов) охлаждения раствора
при 0°.
Полученный объемистый осадок Na-соли л-оксифепилими-
нодиуксусной кислоты фильтруют и промывают 50 мл ацето-
на; сушат без доступа света сначала на воздухе, затем в су-
шильном шкафу при 40—50°.
Получают 50 г технического продукта, содержащего 12—
14% хлористого натрия.
Для очистки комплексона от хлористого натрия 50 г тех-
нического продукта растворяют в 20 мл дистиллированной
£Оды и добавляют 10%-ный раствор ацетата свинца до пол-
ноты осаждения свинцового комплекса.
Осадок фильтруют и промывают дистиллированной водой
до отсутствия ионов хлора.
94
К полученному осадку свинцового комплекса прибавляют
100 мл дистиллированной воды и пропускают через получен-
ную суспензию сероводород для осаждения сульфида свинца.
Выпавший осадок отфильтровывают, а фильтрат, содержа-
щий Na-соль .и-оксифенилиминодиуксусной кислоты, упари-
вают досуха на водяной бане и затем сушат в сушильном
шкафу при 50°.
Выход продукта 26 г, что составляет 25% от теоретичес-
кого.
Найдено, %: С-48,13; 48,19; Н —3,61; 3,59; N -5,44;
5,53; Na - 10,1; 10,23.
Ci0H10O6NNa. Вычислено, %; С — 48,5; Н- 4,05; N — 5,66; Na — 9,3.
Комплексон выделяют в виде кислоты ионообменным ме-
тодом.
Найдено, %: С-53,0; 53,13; Н-4,61; 4,58; N— 6,44; 6,51-
С10НцОвЫ. Вычислено, %: С — 53,3; Н — 4,9; N—6,2.
Примечание.
При обработке реакционной массы ацетатом свинца с последующим
осаждением сульфида и выделением комплексона путем упаривания
фильтрата выход может быть повышен до 40%.
Литература
1. Р. П. Л а с т о в с к п й, В. Я. Темкина, Л. М. С а м ы л о в а. Ме-
тоды получения химических реактивов и препаратов, вып. 6, М., ПРЕД,
1962, стр.' 67.
2. Р. П. Л а с т о в с к п й, В. Я. Темкина, Л. М. С а м ы л о в а.
Там же, 1962, стр. 68.
Поступила в сентябре 1961 г.
ИРЕА
УДК 547.585.07
ПИРОМЕЛЛИТОВАЯ КИСЛОТА
Бензол-1,2,4,5-тетракарбоновая кислота
М. Т. РАЗУМОВСКАЯ, А. И. БЕЛЯКОВА, Г. И. КОРЕЛЬСКАЯ
соон
/'S-соон
ноос-1^
I
соон
C10HeO8 М. в. 254,15
Пиромеллитовая кислота была известна еще в прошлом
столетии, однако лишь в последнее время она начинает нахо-
дить все большее применение в различных областях химии—
при синтезе термостойких полимеров [1], органических полу-
проводниковых материалов [2].
Пиромеллитовая кислота получается из меллитовой кис-
лоты путем нагревания ее с бисульфитом калия и серной кис-
лотой [3]; действием спиртового раствора едкого кали на
а.р-дибромдиэтиловый эфир глутаровой кислоты [4]; из кси-
лолов путем ацилирования л-ксилола уксусным ангидридом в
присутствии хлористого алюминия с последующим восстанов-
лением полученного кетона, повторным ацилированием, вос-
становлением и окислением [5]; хлорметилированием м, «-кси-
лолов с последующим окислением азотной кислотой и возду-
хом полученных хлорметилированных производных ксилолов
[6]; окислением дуриловой кислоты [7], октагидроантрацена
[8] и дурола [9] перманганатом калия в щелочной среде; дуро-
ла азотной кислотой [10], кислородом воздуха над катализа-
тором [11]. Описан и ряд других методов, но все они не име-
ют практического значения.
96
Нами проверены методы получения.пиромеллитовой кис-
лоты путем окисления дуриловой кислоты и дурола перман-
ганатом калия, а также уточнены условия процесса окисления
и выделения продукта.
СХЕМА СИНТЕЗА ПИРОМЕЛЛИТОВОЙ КИСЛОТЫ ОКИСЛЕНИЕМ
ДУРИЛОВОЙ КИСЛОТЫ [7]
СНз
СН3ОС
сн3
СН3
NaOOC-
~СН, кмпо4
СН3
COOK соон
i I
/Vcook НС1 ifVcOOH
-> NaoOC--------------> HOOC-1^ J
I I
COOK COOH
Характеристика основного сырья
Ацетилпсевдокумол, ч., т. кип. 246—247° (см. статью
«5-Ацетилпсевдокумол» в этом сборнике).
Бром, ч., ГОСТ 4109—48.
Едкий натр, ч., ГОСТ 4328—48.
Калий марганцовокислый, техн., ГОСТ 5777—51.
Соляная кислота, ч., ГОСТ 3118—46.
Натрий сульфит, ч., ГОСТ 429—41.
Этиловый спирт, ректифицированный, ГОСТ 5962—51.
Условия получения
Получение гипобромита натрия. В трехлитровый фарфоро-
вый стакан, помещенный в баню с охладительной смесью (лед,
вода) и снабженный механической мешалкой, капельной во-
ронкой и термометром, загружают 2 л раствора едкого натра
(320 г едкого натра в 2 л воды) и при перемешивании быстро
прибавляют 152 мл (476 г) брома, следя за тем, чтобы темпе-
ратура раствора не превышала 10°. Бром быстро растворяет-
ся.
Полученный раствор гипобромита натрия сразу же ис-
пользуют для окисления ацетилпсевдокумола.
Получение натриевой соли дуриловой кислоты. В четырех-
гордую круглодонную колбу емкостью 4 л, снабженную меха-
7 Зак. 131 97
нической мешалкой, капельной воронкой, обратным холодиль-
ником и термометром, загружают 162 г (1,0 Л4) ацетилпсевдо-
кумола, нагревают до 96—98° и прибавляют в течение 1 —1.5
часа полученный раствор гипобромита натрия. По окончании
прибавления всего количества гипобромита натрия реакцион-
ный раствор выдерживают 3 часа при 100°,
Горячий водно-щелочной раствор, содержащий натриевую
соль дуриловой кислоты и натриевые соли бромистоводород-
ной и бромноватистой кислот, отделяют декантацией от бро-
моформа и четырехбромистого углерода.
Получение пиромеллитовой кислоты. В открытый сосуд ем-
костью 6—7 л, снабженный механической мешалкой и термо-
метром, загружают полученный раствор натриевой соли ду-
риловой кислоты ~ 2,2—2,3 л (~2,8 кг), 2 л воды, нагревают
до 40—45° и небольшими порциями (по 20 г) прибавляют
800—900 г (5,1—5,7 Л4) перманганата калия (см. примеча-
ние 1). Температура реакционной смеси повышается до 75—
85° и поддерживается в этих пределах 7—8 часов до оконча-
ния окисления (см. примечание 2). После прибавления всего
количества перманганата калия реакционную смесь перемеши-
вают 1,5 часа при 100°, охлаждают до 60е. Избыток перманга-
ната калия удаляют добавлением 15—20 .мл. этилового спирта.
Двуокись марганца отфильтровывают, промывают на фильтре
(4 раза по 300 мл) горячей водой. Основной фильтрат соеди-
няют с промывными водами, упаривают до 300 мл, охлажда-
ют до 10° и подкисляют 150—170 мл соляной кислоты уд. в.
1,175 до pH 1. Одновременно с кислотой для удаления избыт-
ка брома добавляют 60 мл насыщенного раствора сульфита
натрия. Нейтрализация и высаживание пиромеллитовой кис-
лоты сопровождаются бурным вспениванием и разогревани-
ем реакционной смеси (см. примечание 3). Пиромеллитовая
кислота выпадает в виде белых кристаллов монокалисвойсо-
ли. Кислый раствор оставляют стоять на ночь при комнатной
температуре. Выпавший осадок белого цвета отсасывают и
сушат при ПО—120° до постоянного веса.
Выход монокалиевой соли пиромеллитовой кислоты
180—195 г. Полученную соль растворяют при нагревании в
10%-ном растворе соляной кислоты (из расчета 10 г монока-
лиевой соли на 100 мл соляной кислоты), фильтруют через
кислотоупорный фильтр. Фильтрат охлаждают водой и льдом;
выпавший осадок отсасывают, промывают на фильтре дистил-
лированной водой до отсутствия иона хлора в промывных во-
дах, сушат при 110—120° до постоянного веса. Маточник от
перекристаллизации упаривают и после повторной перекри-
сталлизации получают дополнительно 10—12 г' чистой пиро-
меллитовой кислоты.
Выход пиромеллитовой кислоты с содержанием основного
98
вещества не ниже 98% (см. примечание 4) 126—129 г, что со-
ставляет 49,6—51%, считая на ацетилпсевдокумол.
Температура плавления безводной пиромеллитовой кисло-
ты 273—275°, что соответствует литературным данным [2, 7, 8}
СХЕМА СИНТЕЗА ПИРОМЕЛЛИТОВОИ КИСЛОТЫ ОКИСЛЕНИЕМ
ДУРОЛА
СНз
/%.сн
il I
H3C-V
сн3
COOK
КМпО4
Водно-
пиридино-
вая среда
I
С^-СООК IIC1
kooc--?xJ —>
।
COOK
соон
/^-соон
HOOC-^J
соон
Характеристика основного сырья
Дурол, ч„ ВТУ РУ 462 -51.
Пиридин, ч., ГОСТ 2747—44.
Калий марганцовокислый, техн., ГОСТ 5777—51.
Соляная кислота, ч., ГОСТ 3118—46.
Этиловый спирт, ректифицированный, ГОСТ 5962—51.
Условия получения
В четырехгорлую круглодонную колбу емкостью 3 л, снаб-
женную механической мешалкой с затвором (последний за-
полняют на '/з вазелиновым маслом), обратным холодильни-
ком, термометром и воронкой для сыпучих веществ, загружа-
ют 200 мл пиридина, 1000 мл воды и 33,5 г (0,25 М) дурола.
Смесь нагревают па водяной бане до 85—90° и неболь-
шими порциями по 10 г при интенсивном перемешива-
нии вводят перманганат калия в количестве 650 г.
(-—4,1 Л4) в течение 11 часов (примечание 1). После прибав-
ления первых 325 г перманганата калия в реактор добавляют
500 мл горячей воды и заканчивают окисление введением ос-
тального количества перманганата калия при 95°. Окисление
считают законченным, если введенная новая порция перман-
ганата калия не раскислится при 95° в течение часа. Избыток
7< 99
перманганата калия удаляют прибавлением 15—20 мл этило-
вого спирта. Горячую реакционную смесь декантируют и от-
фильтровывают от двуокиси марганца на воронке Бюхнера.
Для полноты выделения продукта двуокись марганца промы-
вают на фильтре горячей водой, 6 раз по 150 мл. Основной
фильтрат (~1500 мл) и промывные воды (900 мл) соединяют
вместе, переносят в перегонную колбу и упаривают на кипя-
щей водяной бане в вакууме от водоструйного насоса до объ-
ема 800 мл. Полученный фильтрат переливают в двухлитро-
вый стакан, охлаждают до 10° ледяной водой и при переме-
шивании постепенно прибавляют к нему около 100 мл кон-
центрированной соляной кислоты до кислой реакции по уни-
версальной индикаторной бумаге (см. примечание 3). Кислый
раствор оставляют стоять в течение ночи при комнатной тем-
пературе. Выпавший белый мелкокристаллический осадок от-
сасывают на фарфоровой воронке и тщательно отжимают.
Сырой продукт в количестве 300—350 г перекристаллизовы-
вают из 600—650 мл 10%-ного раствора соляной кислоты, как
описано выше.
Выход чистой пиромеллитовой кислоты равен 45—47 г, что
составляет 71—74% от теоретического с содержанием основ-
ного вещества 99%; т. пл. 282—283е.
Примечания:
1. Каждую последующую порцию перманганата калия прибавляют
после раскисления предыдущей (темно-малиновая окраска раствора ог
КМпО4 должна перейти в бурую от образовавшейся МпО2. Проба на вы-
тек) .
2. Окисление заканчивается, когда внесенная новая порция перманга-
ната калия не раскисляется при нагревании в течение часа.
3. Во избежание вспенивания с выбросом реакционной массы кислоту
прибавляют небольшими порциями.
4. Содержание основного вещества определяют титрованием 0,5 N
раствором щелочи с 1%-ным спиртовым раствором фенолфталеина.
Литература
1. Chem. Week., 88, 46 (1961); Р. .1. Brennan. Chem. Fng., 70, 2, 68
(1 63); Chem Age, 88, 2267, 955 (1962).
2. A. Epstein, B. S. Wildy. J. Chem. Phys., 32, 324 (I960).
3. O. S 11 b e rr a d. .1. Chem. Soc., 89, 1795 (1906).
4. F. Feist. Ber., 44, 137 (1911); Farmer. 1 n с о 1 d. J. Chem. Soc.,
119, 2011 (1916).
5. Philippi. Ann., 428, 300 (1922).
6. Англ. пат. 771086, 1957; C. A., 16519g (1957); J. Braun, 1. Nelles.
Ber., 67, 1094 (1934).
7. E. Clar. Ber., 75, 1335 (1942); W. H. Mills. J. Chem. Soc., 101,
2191 (1912).
8. G. Sell roeter. Ber., 57, 2023 (1924); J. Braun, G. Zemke.
Ber., 57, 681 (1942).
9. B. Fish wick. J. Chem. Soc, № 3, 1198 (1957).
10. Англ. пат. 771086, 1957; С. A., 16549 g 11957).
11. Пат. США 2509855, 1950; С. А., 3873 g (1951). Пат. США 2576625,
1951; С. А., Р5087 i (1952).
Поступала в июне 1964 г.
ИРЕА
УДК 661.718:542.941:546.3
ПОЛУЧЕНИЕ АЛКИЛДИХЛОРФОСФИНОВ
ВОССТАНОВЛЕНИЕМ АЛКИЛДИХЛОРФОСФИНОКСИ-
ДОВ МЕТАЛЛАМИ
И. П. КОМКОВ, к. в. КАРАВАНОВ, В. Г. ГРУЗДЕВ, С. 3. ИВИН
,С1
RP^ -f" металлы (Al, Mg, Zn, Fe, Cd, Ca) ->
II XC1
О
RPC12 + RP (O)Clj-MeO
В литературе описан способ получения алкилдихлорфос-
финов восстановлением дихлорангидридов алкилтиофосфино-
вых кислот (алкилдихлорфосфипсульфидов). Эта реакция
проводится при непрерывном встряхивании в запаянной ам-
пуле 66 часов при температуре 140° с выходом 20% [!]•
Нами разработан более простой способ получения алкил-
дихлорфосфинов из доступных исходных продуктов с выходом
до 50-55% [2].
МЕТИЛДИХЛОРФОСФИН
СН3РС12 м. в. 116, 91
СХЕМА СИНТЕЗА МЕТИЛДИХЛОРФОСФИНА
,С1
сн3р; +Zn CH3pci2 + CH3p(O)ci,-ZnO
II ci
о
101
Характеристика основного сырья
Метилдихлорфосфиноксид, перегнанный, т. кип. 163° при
760 мм, получают по описанной в настоящем сборнике мето-
дике (см. статью «Алкилдихлорфосфиноксиды»).
Цинковая пыль, 85%-ная.
Условия получения
В двугорлую круглодонную колбу емкостью 0.75 л, снаб-
женную насадкой Кляйзена (длина елочного дефлегматора
26 см), прямым холодильником с приемниками, помещают
32,6 г (0,5 М) цинковой пыли (см. примечание 1) и к ней по-
сле продувки прибора азотом из капельной воронки неболь-
шими порциями приливают 66,5 г (0,5 М) расплавленного ме-
тилдихлорфосфиноксида (т. пл. 32°) (см. примечание2). Одно-
временно с добавлением метилдихлорфосфиноксида колба на-
гревается пламенем горелки до температуры отгонки метил-
дихлорфосфина. Реакцию и отгонку продукта ведут в слабом
токе азота (см. примечание 3).
Выход метилдихлорфосфина 29,3 г, что составляет 50% от
теоретического; т. кип. 80—82° при 760 мм; Л.2С— 1,3041;
п2" — 1,4940.
Примечай и я:
1. Вместо цинковой пыли можно использовать магниевые стружки,
однако в этом случае, иногда реакция идет со вспышками и выход ниже.
Можно применять железные опилки, реакция протекает спокойно и с хо-
рошим выходом метилдихлорфосфина. Реакция проходит спокойнее и
лучше, когда металлы сухие и меньше содержат окисей.
2. Если метилдихлорфосфиноксид приливать большими порциями да-
же при комнатной температуре, реакция может протекать с выделением
большого количества тепла и выбросом реакционной смеси.
3. Метп.тдихлорфосфин легко окисляется при соприкосновении с воз-
духом.
По описанной методике при наличии алкилдихлорфосфин-
оксидов можно получить различные алкилдихлорфосфины.
Литература
I. Пат. США 3024278 (1962).
2 Авт. свпд. 160184; Бюлл. нзобр., № 3 (1964).
Поступила в i'io:ie 1961 г.
УДК 547.425.5.07
СТИЛЬБЕКСОН
Динатриевая соль 4,4/-диаминостильбен-(Ы, N, N', N'-
тетракарбоксиметил)-2,2'-дисульфокислоты
Р. П. ЛАСТОВСКИЙ, В. В. СИДОРЕНКО, Н. В. ЛАПШИНА,
Т. И. КОНОПЛЕВА
НООСН2СЧ сн,соон
)N- f ^-сн=сн-<
HOOCELC7 \="' >СН»СООН
3 I
SO3Na NaO3S
М. в. 646,51
Стильбексон впервые получен и предложен ИРЕА для ко-
личественного люминесцентного определения железа [I, 2].
Чувствительность метода — 0,005 мкг железа в 5 мл раствора.
СХЕМА СИНТЕЗА СТИЛЬБЕКСОНА
H2N-^ ^-СН = СН-^ ^-NH2+4C1CH2COOH +
SO3Na NaO3S
НООСН2С v x-
4-4NaOH /-CH = CH-f
H00CHacz x=| y
SO3Na NaO3S
.CH2COOH
-N( 4-4NaCH-4H2O
CH2COOH
103
Характеристика основного сырья
Диаминостильбендисульфокислота, ч., МРТУ 6-09-78—62.
Монохлоруксусная кислота, ГОСТ 5836—51.
Натр едкий, ч.д.а., ГОСТ 4338—48.
Этиловый спирт, ректифицированный, ГОСТ 5963—51.
Соляная кислота, ч., ГОСТ 3118—46.
Условия получения
В трехгорлую колбу емкостью 1 л, снабженную механиче-
ской мешалкой, обратным холодильником и капельной ворон-
кой, помещают 37 г (0,1 М) диаминостильбендисульфокисло-
ты, 24 г (0,6 М) едкого натра, растворенного в 100 мл воды.
Реакционную массу тщательно перемешивают, нагревают на
водяной бане до температуры в бане 50°. Затем прикапывают
раствор 56 г (0,6 Л4) монохлоруксусной кислоты в 100 мл ди-
стиллированной воды. После этого температуру в бане повы-
шают до 60—70° (см. примечание) и реакцию ведут в течение
8 часов, поддерживая pH 10—11 (малиновое окрашивание по
фенолфталеину). По окончании нагрева реакционную массу
охлаждают до комнатной температуры (20°) и при перемеши-
вании подкисляют концентрированной соляной кислотой до
pH 1. Выпавший осадок отфильтровывают и промывают
200 мл 70%-ного этилового спирта и сушат в сушильном шка-
фу при 70°.
Выход стильбексона 40 г, что составляет 61% от теорети-
ческого.
Найдено, %; N--3,75; 3,79.
C2lH2liN2O]4Na.,Si. Вычислено, %: N 3,76.
Примечание.
Реакционная масса представляет собой суспензию коричневого цвета;
по мере прибавления щелочи окраска раствора переходит в темно-корич-
невую.
Литература
1. Р. П. Ластовский, В. В. Сидоренко, Е. А. Б о ж е в о л ь-
н о в, С. У. Крейн гольд. Ж. аналит. химии, 18, 11, 1356 (1963).
2. Р. П. Ластовский, В. В. Сидоренко, Е. А. Б о ж е в о л ь-
н о в, С. У. Крейн гольд. Ж. аналит. химии, 19, 2, 268 (1964).
Поступила в сентябре 1864 г.
ИРГА
УДК 547.562.07
3,3 ,4,4' ТЕТРАМЕТИЛДИФЕНИЛОКСИД
3,3',4,4'Тетраметилдифениловый эфир
М. Т. РАЗУМОВСКАЯ, А. И. БЕЛЯКОВА, Л. А. ЕГОРОВА
сн3-^“^-о-^~^-сн3
1=
сн3 сн8
С1вН18О М. в. 226,31
З. З'ДД'-Тетраметилдифенилоксид является промежуточ-
ным продуктом при синтезе органических полимерных соеди-
нений, обладающих электрофизическими свойствами.
В литературе имеется одно сообщение о получении 3,3',
4,4/-тетраметилдифенилоксида конденсацией 3,4-диметилфе-
нола с 4-бром-о-ксилолом по Ульману [1]. В свою очередь
4-бром-о-ксилол получается прямым бромированием о-ксило-
ла в присутствии катализаторов [2] или диазотированием
о-ксилидина по Зандмейеру [3].
Нами проверен указанный метод получения З.З'ДД'-тет-
раметилдифенилоксида, уточнены условия ведения конденса-
ции, выделение и очистка продукта.
При получении 4-бром-о-ксилола был использован метод
прямого бромирования [2]. В качестве катализатора применя-
ли железные опилки. Отработаны условия выделения и очи-
стки продукта.
СХЕМА СИНТЕЗА 3,3',4,4'-ТЕТРАМЕТИЛДИФЕНИЛОКСИДА
Вг
I
/X Fe /Ч
|| | + Вг2 --> || , + НВг
^^СНз Х|/АСН3
СН3 СН3 105
Br
ОН
/Ч /V кон
сн3 сн3
СН3-^ Ч-О-^ \-сн3
Г =Г
СНз СН3
+ КВг%-Н2О
Характеристика основного сырья
о-Ксилол, ч., ВТУ МХП 2940—51.
Бром, ч., ГОСТ 4109—48.
Железные опилки, ч.
Натр едкий, ч., ГОСТ 4328—48.
Магний сернокислый плавленый, ч., ГОСТ 4460—48.
3, 4-Диметилфенол, ч., СТУ 77-20—61.
Кали едкое, ч„ ГОСТ 4203—48.
Медь металлическая в порошке, ч., ЦМТУ 4451—54.
Спирт метиловый, ч., ГОСТ 6995- 54.
Четыреххлористый углерод, ч., ГОСТ 5827—51.
Условия получения
Получение 4-бром-о-ксилола. В четырехгорлую колбу ем-
костью 1,5 л, снабженную механической мешалкой, обратным
холодильником с газоотводной трубкой, термометром, капель-
ной воронкой, загружают 500 г о-ксилола (4,72 М) и 12 г чи-
стых железных опилок. Смесь охлаждают до минус 5° и в те-
чение 3,5 часа прибавляют 660 г (4,13 Af) брома, следя за
тем, чтобы температура реакционной массы не поднималась
выше 0°. По окончании прибавления брома реакционную
смесь оставляют стоять на ночь при комнатной температуре,
затем выливают в 1000 мл воды. Нижний органический слой
отделяют, промывают последовательно водой (500 ли), 3%-
ным раствором едкого натра (2 раза по 500 ли) и снова во-
дой до нейтральной реакции по универсальной индикаторной
бумаге. Сушат серпокислым магнием (20 г) в течение 6 ча-
сов. Выход 700—735 г, что составляет 91,4—96%, считая на
бром. Высушенный продукт разгоняют в вакууме, применяя
для разгонки колбу с 'небольшим елочным дефлегматором
(15 см). 4-Бром-о-ксилол отбирают в интервале 92—94° при
14—15 мм. Получают 500—520 г вещества (65—68% от тео-
ретического выхода).
106
Получение 3,3г,4,4'-тетраметилдифенилоксида. В четырех-
горлую круглодонную колбу емкостью 2,5 л, снабженную ме-
ханической мешалкой, нисходящим холодильником Либиха и
термометром, загружают 500 г (4,0 А4) 3,4-диметилфенола,
который расплавляют при температуре 60—65°, и в расплав-
ленный продукт при перемешивании небольшими порциями
вносят 200 г (3,5 М) мелкоизмельченного едкого кали. При
добавлении едкого кали температура реакционной массы са-
мопроизвольно поднимается до 120° и затем доводится посте-
пенно до 180—185°. При этой температуре через нисходяший
холодильник отгоняют 60 мл воды. После отгона воды содер-
жимое колбы охлаждают до 150°, добавляют сначала 2,5 г
порошкообразной меди, затем в течение 60 минут 462,5 г
(2,5 М) 4-бром-о-ксилола. Реакционную массу перемешива-
ют при 148—150° в течение часа, охлаждают до 55—60° и из
полученной массы экстрагируют четыреххлористым углеро-
дом (4 раза по 250 мл) ЗЛ'ЗЛ'-тетраметилдифенилоксид.
Вытяжки соединяют вместе, промывают последовательно
3%-ным раствором едкого натра (4 раза по 250 мл), затем
водой до нейтральной реакции по универсальной индикатор-
ной бумаге, сушат прокаленным сернокислым магнием (20 г)
в течение 6 часов. Четыреххлористый углерод отгоняют при
атмосферном давлении, остаток перегоняют в вакууме, соби-
рая фракцию при 158—160°/4—6 мм рт. ст. или 170—
172°/7—8 мм. рт. ст. Полученное масло охлаждают водой
(14—15°); выпавшие кристаллы белого цвета отсасывают и
перекристаллизовывают из метилового спирта. Выход чистого
3,3',4,4'-тетраметилдифенилоксида равен 300—350 г, что со-
ставляет 52—60% от теоретического; т. пл. 56—58°.
Найдено, %: С- 84,65; 84,61; Н-8,02; 7,72.
С1вН,аО. Вычислено, %: С-84,9; Н—7,97.
По литературным данным, температура плавления трижды
перекристаллизованного из метанола 3,3',4,4'-тетраметилди-
фенилоксида 58--59,2 [1].
Литература
1. С. S. М а г v е I, J. U. R a s s w е 11 с г. J. Amer. Chem. Soc., 80
1196 (1958).
2. В. Виза н с к и й, С. Ансбахе р. Синтез органических препа-
ратов, сб. 4, 1949, стр. 93.
3. Т. Sandmeijer. Вег., 17, 1633 (1884); В г a n d, I. u d w i g, Ber-
lin. J. prakt. Chem., 2, 110,34 (1928),
Поступила в сентябре 1964 г.
ИРЕА
УДК 547.585.05
ТЕТРАМИД ПИРОМЕЛЛИТОВОЙ КИСЛОТЫ
М. Т. РАЗУМОВСКАЯ, А. И. БЕЛЯКОВА, Л. А. ЕГОРОВА
conh2
/^-conh2
HsNOC-II I
2 w
conh2
CjoHjo04N4
M. B. 250,21
Тетрамид пиромеллитовой кислоты применяется для син-
теза тетрацианбензола и новых термостойких полимерных ма-
териалов [1].
Тетрамид пиромеллитовой кислоты получают взаимодей-
ствием диангидрида виромеллитовой кислоты с мочевиной в
среде трихлорбензола [2] или обработкой диангидрида пиро-
меллитовой кислоты концентрированным (25—28%-ным) рас-
твором аммиака в течение 5—6 суток [3].
Нами проверен и уточнен метод получения тетрамида пи-
ромеллитовой кислоты взаимодействием диангидрида пиро-
меллитовой кислоты с мочевиной в среде хлорбензола, кото-
рый более доступен, чем трихлорбензол, и отработан новый
метод получения тетрамида пиромеллитовой кислоты сплав-
лением безводной пиромеллитовой кислоты с мочевиной.
Последний метод отличается простотой и быстротой вы-
полнения и позволяет сократить продолжительность процесса
до 6 часов.
В данной статье описываются оба метода.
108
СХЕМА СИНТЕЗА ТЕТРАМИДЛ ПИРОМЕДДИТОВОИ КИСЛОТЫ
СООН
f\-COOH
ноос-!^ J
соон
нн3
+ 2СО^
'NH,
conh2
^\-conh2
H2NOC-^J
I
CONH2
2 COj H2O
Характеристика основного сырья
Диангидрид пиромеллитовой кислоты, ч. (см. соответст-
вующую статью в этом сборнике).
Мочевина, ч., ГОСТ 2081—43.
Хлорбензол, ч., ТУ МХП 2877—54.
Аммиак водный, 25%-ный, ГОСТ 9—40.
Условия получения
Получение тетрамида пиромеллитовой кислоты, взаимо-
действием диангидрида пиромеллитовой кислоты с мочевиной
в среде хлорбензола. В круглодонную колбу емкостью 1 л<
снабженную обратным холодильником и термометром, загру-
жают 109 г (0,5 А4) диангидрида пиромеллитовой кислоты,
100 г (1,66 (И) мочевины и 650 мл хлорбензола. Смесь нагре-
вают до 130—132° и выдерживают при этой температуре в те-
чение 7 часов, затем охлаждают до комнатной температуры,
хлорбензол сливают с осадка, а осадок заливают 300 мл 25%-
ного раствора аммиака и перемешивают при комнатной тем-
пературе в течение 4 часов до образования белой однородной
массы. Осадок отфильтровывают, промывают 3 раза по 100 ль?
12,5%-ным раствором аммиака, водой до нейтральной реак-
ции по универсальной индикаторной бумаге и сушат при
80—85° до постоянного веса.
Выход продукта 80 г, что составляет 80% от теоретическо-
го, считая на диангидрид пиромеллитовой кислоты или 64%,
считая на пиромеллитовую кислоту.
Найдено, %: N—22,06; 22,80.
CJ0HI0O4N4. Вычислено, %; N—22,4.
109
Получение тетрамида пиромеллитовой кислоты сплавле-
нием пиромеллитовой кислоты с мочевиной. В двухгорлую
круглодонную колбу емкостью 0,5 л, снабженную обратным
холодильником и термометром, загружают 108 г (0,425 М)
безводной пиромеллитовой кислоты и 130 г (2.16 М) мочеви-
ны. Смесь нагревают до расплавления (135—140°) и выдер-
живают при этой температуре в течение 6 часов. Образовав-
шуюся твердую массу охлаждают до комнатной температуры,
залипают 350 мл 25%-ного раствора аммиака и перемешива-
ют в течение 1,5 часа до образования белой однородной мас-
сы. Осадок отфильтровывают, промывают 2 раза по 50 мл
концентрированным раствором аммиака, затем водой до ней-
тральной реакции по универсальной индикаторной бумаге,
сушат при 80—85° до постоянного веса.
Выход тетрамида пиромеллитовой кислоты 72—81 г, что
составляет 68—76% от теоретического.
Найдено, %: X' — 22,14; 22,25.
C10Hll(O4N4. Вычислено, %: N-22,4.
Литература
1. Е. A, Low ton, D. D. Me P i t c 11 i e. J.‘Organ. Chem., 24,26(1959)-
J. Freeman, E. Traunor, J. Mi g 1 a r e s e, R. Zunn. SPE Trans;
actions, 216—221, July (1962).
2. A. Epstein, B'. S. W i 1 d y. J. Chem. Phys., 32, 2, 324 (1960).
3. H. Meyer, H. Steiner. Monatsh. Chem., 35, 391 (1914).
Поступила в июле 1961 г.
ИРЕА
УДК 547.52.07
ТЕТРАЦИАНБЕНЗОЛ
Тетранитрил пиромеллитовой кислоты
М. Т. РАЗУМОВСКАЯ, А. И. БЕЛЯКОВА, Г. И. КОРЕЛЬСКАЯ
C10H2N4 М. в. 178,15
Тетрацианбензол применяется для синтеза органических
полимеров с ценными электрофизическими свойствами.
В литературе имеется лишь два сообщения о получении
тетрацианбензола: дегидратацией тетрамида пиромеллитовой
кислоты фосгеном с выходом 66% от прореагировавшего тет-
рамида пиромеллитовой кислоты [1] и хлористым тиопилом с
выходом 28% [2]. В последнем случае подробности описания
синтеза отсутствуют.
Нами отработана методика получения тетрацианбензола
дегидратацией тетрамида пиромеллитовой кислоты хлорис-
тым тионилом с выходом 50—55% от теоретического.
СХЕМА СИНТЕЗА ТЕТРАЦИАНБЕНЗОЛА
CONH2
J^/conh2
|| | + 4SOC12
H2NOC I
conh2
111
CN
A/CN
_> II I + 8HC1 + 4SO2
/\A
NC |
CN
Характеристика основного сырья
Тетрамид пиромеллитовой кислоты, ч. (см. статью «Те-
трамид пиромеллитовой кислоты» в этом сборнике).
N, N-Диметилформамид, ч., ВТУ РУ 1193—55.
Тионил хлористый, ч., ВТУ МХП 3591—52.
Уксусная кислота, ледяная, х. ч., ГОСТ 61—51.
Спирт этиловый, ректифицированный- ГОСТ 5962—51.
Условия получения
В трехгорлую колбу емкостью 1 л, снабженную механиче-
ской мешалкой, обратным холодильником, капельной ворон-
кой и термометром, загружают 50 г (0,2 М) тетрамида пиро-
меллитовой кислоты и 400 мл диметнлформамида. Смесь пе-
ремешивают 10 минут при комнатной температуре, нагрева-
ют до 40—45° и при постоянном перемешивании прибавляют в
2 приема 120 мл (1,6 М) хлористого тиопнла с такой скоро-
стью, чтобы температура реакционной массы была не выше
50°. После прибавления первой порции (60 мл) хлористого
тионила реакционную массу выдерживают 2 часа при 48—50°,
затем при этой же температуре прибавляют остальное коли-
чество хлористого тионила.
После прибавления всего количества хлористого тионила
реакционную массу перемешивают 9 часов при 50°. К этому
времени в реакционном растворе остается незначительное ко-
личество (~ 1 г) тетрамида пиромеллитовой кислоты и реак-
ционный раствор приобретает коричневый цвет.
В горячем виде реакционную массу отфильтровывают от
непрореагировавшего тетрамида пиромеллитовой кислоты и
небольшими порциями выливают на мелкоизмельченный лед.
Выпавший осадок отсасывают, промывают холодной водой до
нейтральной реакции и перекристаллизовывают из ледяной
уксусной кислоты следующим образом: 40 г сырого техниче-
ского продукта растворяют при нагревании в 400 мл ледяной
уксусной кислоты. Раствор доводят до кипения, кипятят в те-
чение 7 минут и фильтруют в горячем виде. Фильтрат охлаж-
дают до 15° и выдерживают 1 час. Выпавшие кристаллы от-
сасывают, промывают водой, 2%-ным раствором едкого нат-
ра и снова водой до нейтральной реакции на лакмус, затем
112
20 мл спирта. Сушат при 80—90° под лампой инфракрасного
света до постоянного веса.
Из маточного раствора при разбавлении его холодной во-
дой в отношении 1 : 1 получают дополнительное количество
(~2г) тетрацианбензола.
Выход чистого тетрацианбензола 18—20 г, что составляет
50—55% от теоретического; т. пл. 263—265°.
По литературным данным, т. пл. продукта 258—260° [2];
264—267° [1].
Найдено, %: С-67,08; 67,28; Н—1,11; 1,07; N-30.98; 31,19.
CjoHjNp Вычислено, %: С—67,42; Н—1,12; N—31,46.
Литература
1. A. Epstein, В. S. Wildy. J. Chem. Phys., 32, 2, 324 (1970).
2. Е. A. Lowton, D. D. Me Pitchie. J. Organ. Chem., 24, 26
(1959).
Поступила в нюне 196-1 г. ИРЕА
8 Зак. 131
УДК 547,594.3.0/
1,4-ЦИКЛОГЕКСАНДИОН
Н. М РЫБКИНА, И. Н. ЛЕБЕДЕВА
Н2С СН2
I I
Н2С сн2
CeHsO2
М. в. 112,12
1.4-Циклогександион образуется при омылении и декар-
боксилировании диэтилового эфира сукцинилянтарной кисло-
ты.
Омыление и декарбоксилирование диэтилового эфира сук-
цинилянтарной кислоты осуществляют серной кислотой [1],
фосфорной кислотой [2] или водой под давлением и при
195 -200° [3, 4].
Нами проверен и уточнен метод омыления и декарбокси-
лирования водой под давлением.
СХЕМА СИНТЕЗА 1,4-ЦИКЛОГЕКСАНДИОНА
О
II
Сх
Н2С СН-СООС,Н6 2На0
Н5С2ООС-НС СП,
II
114 О
о
II
/с\
Н2С сн2
| | + 2 СО2 + 2 С2НдОН
Н2С СН2
XCZ
О
Характеристика основного сырья
Диэтиловый эфир сукцинилянтарной кислоты, ТУ TCP
1159р—63.
Условия получения
В автоклав емкостью 0,4 л, снабженный гильзой для тер-
мометра, вентилем для спуска давления, предохранительным
клапаном и манометром, загружают 64 г (0,25 М) диэтилово-
го эфира сукцинилянтарной кислоты и 64 мл дистиллирован-
ной воды, закрывают плотно крышкой и нагревают до 200°.
При этой температуре дают 45-минутную выдержку. По окон-
чании выдержки автоклав охлаждают проточной водой до
комнатной температуры, затем медленно спускают остаточ-
ное давление; содержимое в виде раствора (около 100 мл)
фильтруют от следов неомылившегося диэтилового эфира
сукцинилянтарной кислоты. Обычно его остается около
0,3—0,5 г.
Профильтрованный раствор переливают в круглодонную
колбу емкостью 200 мл, добавляют 0,5 г активированного уг-
ля и кипятят с шариковым холодильником в течение 5 минут.
Горячий раствор отфильтровывают от угля и упаривают под
вакуумом— 50 мм рт. ст. до объема 25 мл. Остаток вылива-
ют из колбы в стакан, охлаждают до 8—10°, выпавший
1>4-циклогександион отфильтровывают на фарфоровой ворон-
ке и сушат в вакуум-эксикаторе над хлористым кальцием.
Выход технического 1,4-циклогексапдиона 19,6—21 г, что
составляет 70—75% от теоретического; т. пл. 74—76°.
Очистка 1,4-циклогександиона. В колбу Кляйзена емко-
стью 100 мл, снабженную термометром, форштоссом и при-
емником, загружают 40 г технического 1,4-циклогександиона
(полученного от 2 загрузок). Колбу помещают в масляную
баню, включают вакуум.
При температуре 132° и давлении 200 мм рт. ст. начинает
отгоняться 1,4-циклогександион. Перегонку следует вести ос-
торожно, следя за тем, чтобы продукт не закристаллизовы-
8* 115
вался в форштоссе. По окончании перегонки приемник" нагре-
вают на водяной бане, расплавленный продукт переливают в
фарфоровую ступку, дают охладиться и растирают.
Выход продукта 35 г или 61—65%, считая на диэтиловый
эфир сукцинилянтарной кислоты; т. пл. 76—78°.
По литературным данным, т. пл. препарата 77—78° [2].
Литература
1. А. Успенский, И. Тюрин. Ж. русского физ.-хим. об-ва, 51, 209
(1919).
2. W. Doe ring, A. S a v i g h. J. Organ. Chem., 26. 1365 (1961).
3. H. Meerwein. Liebigs Ann. Chem., 398, 248 (1913).
4. I. R. Vincent. J. Organ. Chem., 9, 003 (1939).
Поступила в мае 1964 г.
ИРЕА
УДК 861.718.07
0-2-ХЛОРАЛ КИЛАЛКИЛХЛОРФОСФИНИТЫ
С. 3. ИВИН, к. в. КАРАВАНОВ
,С1
RP<
OCH2CHCIR'
О-2-Хлоралкилалкилхлорфосфиниты являются исходными
полупродуктами в синтезе различных производных трех-, а
также пятивалентного фосфора.
До последнего времени О-алкилалкилхлорфосфиниты в
виду отсутствия метода их получения не были описаны в лите-
ратуре. Нами предложен простой и доступный метод получе-
ния этих соединений [1], заключающийся во взаимодействии
адкилдихлорфосфинов с алкиленоксидами.
Аналогичная работа была опубликована другими автора-
ми [2].
О-2-ХЛ ОРЭТИЛ МЕТИЛХЛ ОРФОСФИНИТ
.о
сн3р;
х ОСНоСНоС!
С3Н7РС1,
М. в. 144, 96
СХЕМА СИНТЕЗА О-2-ХЛОРЭТИЛМЕТИЛХЛОРФОСФИНИТА
.С!
СН3РС1.,+СН9 - СН, -> СН3Р<
\п/ ХОСН2СН2С1
Характеристика основного сырья
Метилдихлорфосфип, свежеперегнанный, получают по ме-
тодике, описанной в настоящем сборнике (см. статью «Полу-
117
чснис алкилдихлорфосфинов восстановлением алкилхлорфос-
финоксидов металлами»).
Окись этилена, безводная, перегнанная.
Этиловый эфир, абсолютный, ГОСТ 6265- 52.
Условия получения
В колбу Кляйзена с елочным дефлегматором, прямым хо-
лодильником и газовводной трубкой помещают 11,7 г (0,1 Л!)
метилдихлорфосфина и 35—40 мл абсолютного эфира (см.
примечание). Затем при температуре 0—5° в колбу вводят
5,3 г (0,12 М) окиси этилена. По истечении 20—25 минут по-
сле пропускания всего количества окиси этилена эфир отго-
няют. Образовавшийся О-2-хлорэтилметилхлорфосфинит пе-
регоняют в вакууме при 12 мм (см. примечание). Получают
продукт с т. кип. 59—60° при 12 мм; d418—1,2699; —1,495.
Выход 10,2 г, что составляет 64% от теоретического.
Примечание.
Метилдихлорфосфин и образующийся о-2-хлорэтилметилфосфинит лег-
ко окисляются на воздухе, поэтому все операции по получению последнего
проводят в токе азота или какого-либо другого инертного газа.
По описанной методике, исходя из различных алкилди-
хлорфосфинов и алкеленоксидов. можно получить различные
О‘2-хлоралкилалкилхлорфосфиниты.
Литература
1. С. 3. Ивин, К- В- К а Р а в а н о в. Ж- общ. химии, 29, 3456 (1959).
2. Г. К а м а й, В. С. Ц и в у н и н. Химия и применение фосфороргани-
ческих соединений, М., АН СССР, 1962, стр. 317.
Поступила в июне 1964 г.
УДК 661.718.07
S-2-ХЛОРАЛ КИЛ-ЭТИЛ (АЛ КИЛ )ХЛОРФОСФИНИТЫ
С. 3. ИВИН, И. Д. ШЕЛ А КО В А
SCH2 CH C1R
R-P^
ХС1
Получение S-2-хлоралкил-этилхлорфосфинитов нами осу-
ществлялось путем взаимодействия алкилдихлорфосфинов с
алкиленсульфидами [1]. Реакция указанных реагентов начи-
нается немедленно, трехчленное тиоКисное кольцо алкилен-
сульфида раскрывается с образованием соответствующего
S-2-хлоралкил-этил (алкил) хлорфосфинита по схеме •
С1
R — HC - CHR + R'PCla-^R'-P
х / XS-CH-CHC1
XSZ | I
R R
Были проведены реакции метил- и этилдихлорфосфинов с
этилен- и пропиленсульфидами. Реакция протекала энергично
и практически заканчивалась образованием соответствующих
соединений по окончании прибавления тиокисей к алкилди-
хлорфосфину.
Полученные вещества представляют собой бесцветные под-
вижные жидкости, легко перегоняющиеся в вакууме. При по-
лучении S-2-хлоралкил-этил (алкил) хлорфосфинитов необхо-
димо соблюдать осторожность, так как это вещества кожно-
го действия.
S -2 ХЛОРЭТИЛ-МЕТИЛХЛОРФОСФИНИТ
,С1
СН3Р(
xSCH2CHaCl
CROUPS
М. в. 177,03
119
СХЕМА СИНТЕЗА S-2-ХЛОРЭТИЛ-МЕТИЛХЛОРФОСФИНИГА
,ci
сн3рс12 + сн2-сн? -> сн3р; ,
\с/ 'SCH2CH2qi
Характеристика основного сырья
Метилдихлорфосфин, получают по методике, описанной в
литературе [2].
Тиокись этилена, получают по методике, описанной в лите-
ратуре [3].
Условия получения
В трехгорлую колбу с обратным холодильником, капель-
ной воронкой и механической мешалкой помещают 18 г
(0,15 Л4) метилдихлорфосфина, к которому постепенно, при
перемешивании прибавляют 9 г (0,15 М) этиленсульфида. За-
тем реакционную смесь нагревают при 60° в течение 20 ми-
нут. Образовавшийся продукт перегоняют в вакууме при
102—103715 мм.
Выход S-2-хлорэтил-метилхлорфосфинита 19 г, что состав-
ляет 70% от теоретического; d420— 1,2830; га™— 1,5760.
Все остальные опыты по расщеплению алкиленсульфидов
алкилдихлорфосфинами осуществляли аналогично.
S-2-Хлорпропил-метилхлорфосфинит. Из 18 г (0,15 М) ме-
тилдихлорфосфина и 11 г (0,15 М) пропиленсульфида полу-
чили 18 г (62%); т. кип. 110—111° при 16 мм; d4w—1,2105;
я® — 1,5225.
S-2-Хлорэтил-этилхлорфосфинит. Для реакции взяли 31 г
(0,25 М) этилдихлорфосфина и 15 г (0,25 М) этиленсульфи-
да.
Получено 28 г (61%) вещества; т. кип. 125° при 26 мм',
</420— 1,2240; га™ — 1,5508.
S-2-Хлорпропил-этилхлорфосфинит. Из 20 г (0,15 М) этил-
дихлорфосфина и 11 г (0,15 М) пропиленсульфида получено
18,5 г (60%) вещества; т. кип. 94—95° при 3 мм; rf42n—1,1805;
га™ — 1,5160.
Литература
1. С. 3. Ивин, И. Д. Шел а ко в а. Ж- общ. химии (в печати).
2. И. П. Комков, К. В. Караванов, С. 3. Ивин. Ж. общ. хи-
мии, 28, 11, 2963 (1958).
3. Г. И. Б р а з. Ж. общ. химии, 21, 2, 688 (1951).
Поступила в июне 1964 г.
УДК 547.213.07
1-ХЛОР-З-БРОМПРОПАН
Ю. Г. ГОЛОЛОБОВ, Э. Б. ГУСЬКОВА, Л. 3. СОБОРОВСКИЙ
С1СН3СНаСН2Вг
С3Н0С1Вг М. в. 154,36
1-Хлор-З-бромпропаи является исходным продуктом в ря-
де синтезов, в частности при получении ряда химеотерапев-
тических препаратов, например плазмоцида, аминазина и
других [1].
Обычно 1-хлор-З-бромпропан получают при взаимодейст-
вии хлористого аллила с бромистым водородом в присутствии
катализатора перекиси бензоила [2]. Нами показано, что эта
реакция протекает с выходом, близким к количественному, в
присутствии газообразного кислорода,
СХЕМА СИНТЕЗА 1-ХЛОР-З-БРОМПРОПАНА
С1СН,СН=СН2 + НВг -> С1СН2СН3СН3Вг
и2
Характеристика основного сырья
Хлористый аллил, технический.
Бромистый водород, получают действием брома на крас-
ный фосфор в присутствии 46 %-пой бромистоводородной кис-
лоты [3].
Кислород (из баллона).
Условия получения
Через помещенный в колбу хлористый аллил при комнат-
ной температуре пропускают кислород в течение 10—15 ми-
нут. Затем при охлаждении (температура реакционной массы
121
минус 10—20°) и перемешивании вводят бромистый водород
до прекращения поглощения последнего. Перегонкой полу-
чают с выходом, близким к количественному, 1-хлор-З-бром-
пропан; т. кип. 139—141°.
Приведенная методика позволяет получать также 1,3-ди-
бромпропаи из бромистого аллила и бромистого водорода с
выходами до 95% [4].
Литература
1. Н. Н. Дыханов, И. И. Коротки на. Медицинская промышлен-
ность СССР, 4, 26 (1957).
2. М. S. К h а г a s с 11, F. R. М а у о. J. Amer. Chem. Soc., 5>, 2468
(1933).
3. М. Ф. Ш о с т а к о в с к и й. Ж. прикл. химии, 2, 682 (1936).
4. I. U г u s h 1 b а г а, М. Т a k е Ь а у a s li i. Bull. Chem. Soc. J арап, 12
173 (1937).
Поступила в июне 1964 г.
УДК 661.731.7.07
tz-ХЛОР- р-ЭТИЛМЕРКАПТОЭТИЛАЦЕТАТ
Л. 3. СОБОРОВСКИЙ, К). Г. ГОЛОЛОБОВ
СН,С OCHC1-CH.,SCJI6
'll
CeHnO,SCl
М. в. 182,GO
а-Хлор-р-этилмеркаптоэтилацетат служит исходным про-
дуктом для синтеза различных соединений и описан сравни-
тельно недавно [1]. Метод его получения основан на взаимо-
действии винилацетата с этилсульфенилхлоридом. Необходи-
мый для реакции этилсульфенилхлорид получают из диэтил-
дисульфида и хлора и вводят в реакцию без очистки.
СХЕМА СИНТЕЗА -ХЛОР- ₽-ЭТИЛМЕРКАПТОЭТИЛАЦЕТАТА
CH3C-OCH=CHa + C2H5SCl -> CHsC-OCHCl-CH2SC?Hs
Характеристика основного сырья
Винилацетат (см. примечание 1).
Диэтилдисульфид (см. примечание 2).
Хлор (из баллона).
Условия получения
В колбу, снабженную мешалкой, термометром, капельной
воронкой и барботером, помещают 44 г (0,36 А4) диэтилди-
сульфида, после чего в последний при минус 70° пропускают
25,6 г (0,36 А4) хлора. К полученному таким образом этил-
сульфенилхлориду прибавляют по каплям при перемешива-
123
нии и охлаждении 62 г винилацетата (0,72 М). Температуру
реакционной массы поддерживают около минус 30°. После
прибавления винилацетата смесь перегоняют.
Выход а-хлор-р-этилмеркаптоэтилацетата 83,4% от теоре-
тического; т. кип. 109° при 19 мм; rf420— 1,1603; —1,4770.
Найдено. %: С-39,55; Н-5.85; S -18,28; CI-19,60;
40,02; 5,55; 18,12; 19,45.
C.HjASC). Вычислено, %: С-39,57; Н-6,0.1; S—17,52; С1-19,45.
Примечания:
1. Винилацетат чистый (перед введением в реакцию перегоняют).
Использование неперегнанного винилацетата (если он сухой) не оказыва-
ет существенного влияния на выход продукта.
2. Диэтилднсульфид получен окислением этнлмеркаптана.
Литература
1.Л. 3. Соборовскпй, Ю. Г. ГоДолобов. Авт. свнд. 149425;
Бюлл. изобр., № 16 (1962).
Поступила в июне 1904 г.
УДК 547.292.07
ХРИЗОИДИН-Х.Х-ДИУКСУСНАЯ И ХРИЗОИДИН-N, N,
N', N-ТЕТРАУКСУСНАЯ КИСЛОТЫ
Р. П. ЛАСТОВСКИИ. И. Д. КОЛПАКОВА, Л. В. КРИНИЦКАЯ,
Л. Д. ЗАВЬЯЛОВА, Т. И. ИВАНОВА
^-nhch3cooh
= [—
nhch2cooh
Хризоидип-М.М'-диуксусиая кислота
CifiHieN4O4
М. в. 328, 32
,СН2СООН
)-N=N-(
—' — ХСН2СООН
,сн.?соон
NC
сн2соон
Xpn30iunii'N',N,N',N'-TCTpayKcy<'Hafi кислота
CMH20N4O8 М. в. 444, 40
Хризоидиндиуксусная и хризоидинтетрауксусная кислоты
синтезированы в ИРЕА и предложены в качестве нового ком-
плексона. Могут быть применены в полярографическом ана-
лизе. Наличие в этих комплексонах полярографически ак-
тивной азогруппы, принимающей участие в комплексообра-
зовании, позволяет проводить определение элементов, вос-
станавливающихся в сильноотрицательной области поляро-
графического спектра.
Оба комплексона синтезированы конденсацией хризоиди-
на с монобромуксусной кислотой в слабощелочной среде.
125
СХЕМА СИНТЕЗА ХРИЗОИДИН-МК-ДИУКСУСНОЙ КИСЛОТЫ
- N=N~
г/
• Н Cl + 2BrCI 12СООН + 3NaOI I ->
nh2
—N=N— ч
i Г \-NHCH2COOH , охт D , , „„
|| | || 4-2NaBr + NaCl |-ЗН2О
NHCHjCOOH
Характеристика основного сырья
Хризоидин (2,4-диаминоазобензол-хлоргидрат), ч., ВТУ
ГКХ 168—60.
Монобромуксусная кислота, ч., ТУ МХП 3667—-55.
Условия получения
В трехгорлую колбу, снабженную механической мешал-
кой, обратным холодильником и термометром, загружают
раствор 58 г (0.42 Л1) монобромуксусной кислоты, предва-
рительно нейтрализованной 20%-ним раствором едкого натра
до pH 7—8, и постепенно прибавляют суспензию 17,4 г
(0,07 Л4) хризоидина в 40 мл воды (см. примечание). Кон-
денсацию проводят при 80°, поддерживая при этом нейтраль-
ную среду (pH 7—8) по универсальной индикаторной бумаж-
ке постепенным добавлением 20%-ного раствора едкого
натра. Реакция считается законченной, если pH среды не из-
меняется в течение 30 минут после добавления последней
порции щелочи. После этого раствор выдерживают при этой
же температуре еще 1 час. Отфильтрованный и охлажденный
раствор подкисляют 10%-пым раствором соляной кислоты до
pH 2, устанавливаемого по потенциометру ЛП-58.
Выпавший осадок фильтруют и тщательно промывают
большим количеством дистиллированной воды (1:25). Нерас-
творившийся темно-коричневый осадок хризоидиндиуксусиой
кислоты очищают неоднократным переосаждением водой из
днметилформамида. Выход продукта 2,5 г, что составляет
11 % от теоретического.
Хризоидин-N, N'-диуксусная кислота нерастворима в воде,
диоксапе, бензоле, растворима в щелочах, диметилформами-
де.
Найдено, %: N— 17,17; 17,19.
C|6H16N4O4. Вычислено, %; N — 17,08,
126
СХЕМА СИНТЕЗА ХРИЗОИДИН -N, N, N/ N'- ТЕТРАУКСУСНОИ
КИСЛОТЫ
N=N —
NJH
| II I II 2 -НС14ВгСН2СООН %- 5NaOH->
Ч/ х/
NHS
-N—N-? ХН.СООН
-> | || I || ^СН СООН 4NaBr + NaCl %- 5Н2О
4/ Ч/ ‘
| >СН2СООН
N\
4 СН2СООН
Условия получения
В трехгорлую колбу, снабженную механической мешал-
кой, обратным холодильником и термометром, загружают
раствор 58 г (0,42 Л1) монобромуксусной кислоты, предвари-
тельно нейтрализованной 20%-ним раствором едкого натра
(pH 7—8), и постепенно добавляют суспензию 17,4г (0,07 М)
в 40 мл воды хризоидина (см. примечание). Конденсацию ве-
дут в условиях, аналогичных условиям получения хризоидин-
диуксусной кислоты.
После окончания первой стадии добавляют еще 39 г
(0,28 М) нейтрализованной монобромуксусной кислоты и
продолжают конденсацию в тех же условиях. Реакцию счи-
тают законченной, если pH среды не изменяется в течение
30 минут после добавления последней порции щелочи. После
этого реакционную массу выдерживают 1 час при 80°. От-
фильтрованный и охлажденный раствор подкисляют 10%-ным
раствором соляной кислоты до pH 2, устанавливаемого по по-
тенциометру ЛП-58.
Выпавший темно-коричневый осадок хризоидинтетраук-
сусной кислоты фильтруют, сушат при 80° и перекристаллизо-
вывают из воды. Выход продукта 2 г, что составляет 6,5% oi
теоретического.
Хризоидинтетрауксусная кислота растворима в воде, в
спирте, в диметилформамиде, нерастворима в бензоле, в хло-
роформе, в четыреххлористом углероде, в петролейном эфи-
ре.
Найдено, %: N-12,86; 12,97; С-54,13; 54,18; Н—4,8; 4,8.
C2oHsoN4Os. Вычислено, %: N—12,61; С-54,10; Н—4,54.
Примечание.
Хризоидин предварительно следует перекристаллизовать из воды в
соотношении 1:5.
Поступила в сентябре 1£Г64 г, ИРЕА
127
УДК 547.333.1.07.
(З-ХЛОРЭТИЛАМИН ХЛОРИСТОВОДОРОДНЫЙ
Р. П. ЛАСТОВСКИЙ, И. Д. КОЛПАКОВА, Л. В. КРИНИЦКАЯ,
Т. И. ИВАНОВА
C1CH2CH,NH2HC1
CaHgNCl.HCl ’ М. в. 115,99
p-Хлорэтиламин хлоргидрат широко используется как
промежуточный продукт в органическом синтезе.
В литературе описано получение хлорэтиламина хлоргид-
рата по реакции Габриэля взаимодействием фталимида ка-
лия с хлористым этиленом с последующим гидролизом N-(p-
хлорэтил)-фталимида, [Г 2], а также конденсацией хлоргид-
рата моноэтаноламина с хлористым тионилом [3]. Последний
способ нашел применение в производстве. В качестве рас-
творителя для хлорирования этаноламинов обычно рекомен-
дуют хлороформ [4]. Нами проверен и уточнен способ полу-
чения p-хлорэтиламина хлористоводородного хлорированием
хлоргидрата моноэтаноламина хлористым тионилом с ис-
пользованием в качестве растворителя сухого бензола и хло-
роформа.
СХЕМА СИНТЕЗА fl -ХЛОРЭТИЛАМИНА ХЛОРИСТОВОДОРОДНОГО
HOCH2CH2NH24-HC1 -> 1IOCH2CH2NH2-HC1
HOCH2CH2NH2-HC1 + SOC!2 ->
C1CH2CH2NH2.HC1 + so2] +HC1 I
Характеристика основного сырья
Моноэтаноламин, ч., СТУ 12-10-112—62.
Соляная кислота, ч., уд. в. 1,19, ГОСТ 3118—46.
Хлористый тионил, ч., ВТУ МХП 3591—52.
128
Условия получения
Получение хлоргидрата моноэтаноламина. К 200 г (3,3 М)
моноэтаноламина при охлаждении льдом постепенно прибав-
ляют 112 мл (3,3 М) концентрированной соляной кислоты.
Затем добавляют еще 35 мл (1 М) той же кислоты и упари-
вают раствор в чашке на паровой бане.
Оставшийся после упаривания осадок охлаждают, промы-
вают 50 мл этилового спирта, отжимают и высушивают при
60°.
Получают 105 г хлоргидрата моноэтаноламина, что сос-
тавляет 33% от теоретического; т. пл. 70—75° (вещество пла-
вится в пределах 1°).
Получение хлоргидрата ^-хлорэтиламина. В сухую четы-
рехгорлую колбу, снабженную механической мешалкой, тер-
мометром, обратным холодильником с хлоркальциевой труб-
кой и капельной воронкой, помещают 97>5 г (1 А4) хлоргид-
рата моноэтаноламина и постепенно через капельную ворон-
ку при охлаждении приливают раствор 96 мл (1 М) хлори-
стого тионила в 100 мл сухого бензола или хлороформа; при
этом температура в колбе не должна превышать 5°. После
окончания прибавления тионилхлорида реакционную массу
перемешивают 3 часа при 30°. Выделившиеся газы, непроре-
агировавший хлористый тионил и растворитель отсасывают
насосом.
Оставшуюся хлористоводородную соль 0-хлорэтиламина
кристаллизуют из смеси, состоящей из 100 мл абсолютного
этилового спирта и 50 мл этилового эфира. Перекристаллизо-
ванный продукт в виде белых игольчатых кристаллов промы-
вают холодным абсолютным этиловым спиртом и сушат в
вакуум-эксикаторе над хлористым кальцием.
Получают 25,7 г |]-хлорэтиламина хлористоводородного с
т. пл. 145° (22% от теоретического выхода).
По литературным данным, т. пл. продукта 144° [4].
p-Хлорэтиламин хлористоводородный — гигроскопичное
вещество, темнеющее и расплывающееся на воздухе. Рас-
творим в воде, нерастворим в холодном абсолютном спирте и
эфире.
Литература
1. S. Gabriel. Вег., 21, 566 (1888).
2. О. Seitz. Вег., 24, 2624 (1891).
3. К. Ward. J. Amer. Chem. Soc., 57. 5, 914 (1935).
4. Л. 3. С о б о p о в с к и й, Г. Ю. Эпштейн. Химия и технология
боевыг химических веществ, М.. Оборонгиз, 1938, стр. 384.
Поступила в сентябре 196! г. ИРЕА
9 3:>к. 131
УДК 547.281.2.07
ЭТИЛМЕРКАПТОАЦЕТАЛЬДЕГИД
Л. 3. СОБОРОВСК.ИИ, Ю. Г. ГОЛОЛОБОВ
C2H5SCH2CHO
C4H8OS М. в. 104,14
Описанный ранее метод получения этилмеркаптоацеталь-
дегида основан на взаимодействии ацеталя бромацетальде-
гида с натриевым меркаптидом [1] и последующим омылени-
ем ацеталя этилмеркаптоацетальдегида до свободного альде-
гида.
Нами показано [2], что необходимый в ряде синтезов в ка-
честве исходного соединения этилмеркаптоацетальдегид мо-
жет быть получен гидролизом доступного а-хлор-р-этилмер-
каптоэтилацетата.
СХЕМА СИНТЕЗА ЭТИЛМЕРКАПТОАЦЕТАЛЬДЕГИДА
СН3С-ОСНС1 -CH2SC3H6-> C2H5SCH,CHO 4- HCI 4СН3СООН
Характеристика основного сырья
а-Хлор-|3-этилмеркаптоэтилацетат (см. соответствующую
статью в этом сборнике).
Условия получения
В круглодонной колбе смешивают 73 г (0,4 Л4) а-хлор-р-
этилмеркаптоэтилацетата с 500 мл воды и смесь при переме-
шивании кипятят 2—4 часа. После охлаждения реакционную
массу экстрагируют эфиром, эфирную вытяжку сушат суль-
130
фатом натрия и перегоняют. Получают около 15 г этилмер-
каптоацетальдегида, т. кип. 47° при 18 мм; d420— 1,0400;
— 1,4766.
Найдено, %; С-46.26; 46,53; Н-7,39; 7,52; S -29,60; 29,32.
C4HaOS. Вычислено, %: С—46,2; Н-7/4; S—30,75.
Литература
1. Е. L. Wick и др. J. Agric. and Food Chem., 9, 289 (1961),
2. Л. 3. С о б о p о в с к п й, Ю. Г. Гололобов. Авт. свид. 150502;
Бюлл. изобр., № 19 (1962).
Поступила в июне 1961 г.
8*
АЛФАВИТНЫЙ ПЕРЕЧЕНЬ СОЕДИНЕНИЙ, СПИСАННЫХ
В НАСТОЯЩЕМ СБОРНИКЕ
Алкилдихлорфосфипы....................................... 5
Алкилдихлорфосфиноксиды..................................... 8
Алкилдихлорфосфинселепиды................................... 17
Алкилдихлорфосфинсульфиды ................................... 20
Алкил(арил)тетрафторфосфнны.................................. 23
5-Ацетилпсевдокумол ......................................... 27
N-Беизилэтилендиамип . . . . ..................................... 31
2,7-Bhc-[N,\t -ди-( карбоксиметил )-аминометил]-4,5-дийодфлуорес-
цеина динатриевая соль........................................ 34
4,4'-Бис-[2.4-бис-(2-карбоксиметиламино)-1,3,5-триазинил-6-амино]-
2,2'-стильбендисульфокислоты гексаиатриевая соль.............. 37
З-Бромэтиламин бромистоводороднын................................. 41
р-Бромэтилимиподиацстонитрил...................................... 44
Винилдихлорфосфат........................................... 48
Винилметилхлорфосфинат...................................... 46
Винилхлорфосфаты.......................................... 48
2-Гидразинохинолин .......................................... 50
Гипобромит................................................... 97
Диалкилтрифторфосфины........................................ 59
Диалкилхлорфосфины........................................... 51
Диалкилхлорфосфинсульфиды.................................. 57
Диангидрид пиромеллитовой кислоты............................ 61
Дивинилхлорфосфат............................................ 49
Диметиловый эфир 4’-нитро-2-амиио-4-м етокси-5-метил азобензол -
N.N-диуксусной кислоты .................................. 89
Дихл орангидрид 2,3-дихлорпронилфосфиновой кислоты........... 64
Дихлорангидрид-циклогексилфосфиповой кислоты................ 66
Диэтилвинилфосфат.......................................... 68
Диэтиловый эфир сукциниляитарпой кислоты..................... 70
Диэгилтрифторфосфин.......................................... 59
Диэтил.хлорфосфин............................................ 54
Диэтил.хлорфосфипсульфид.................................... 57
Калиевая соль пиромеллитовой кислоты ....................... 98
Комплексные соединения алкилтетрахлорфосфинов с хлористым
алюминием................................................. 73
Комплексные соединения диалкилтрихлорфосфинов с хлористым
алюминием................................................. 76
133
Комплексное соединение диэ сил трихлор |юсфииа с хлористым алю-
минием .................................................... 76
Комплексное соединение этилтетрахлорфосфипа с хлористым алю-
минием .................................................... 74
Мегилдихлорфосфин............................................. 101
Метилдихлорфосфинселенид........................................ 18
1-Метилкарбостирил ............................................. 51
2-Метокси-4-аминотолуол-М, N-диуксусная кислота................. 79
Метосульфат 1-метилхинолиния.................................... 51
Натриевые соли О-алкиловых эфиров алкилфосфиновой кислоты . 81
Натриевая соль дуриловой кислоты ............................... 97
Натриевая соль изобутилового эфира метилфюрфосфииовой кис-
лоты ...................................................... 83
Натриевая соль изопропилового эфира метилфосфиновой кислоты 83
Натриевая соль О-этилового эфира метилфосфиновой кислоты . . 81
р-Нафталинс.ульфохлорид...................................... 84
4'-Нитро-2-амино-4-метокси-5-метнлазобензол............... 86
4'-Нитро-2-амипо-4-метокси-5-метилазобензол-Н,Н-днуксуспая кис-
лота ...................................................... 89
я-Нитродиазобензол..................•.......................... 87
лт-Оксифенилиминодиуксусиая кислота...................... 93
Пиромеллитовая кислота................................... 96
Получение алкилдихлорфосфинов восстановлением алкилдихлор-
фосфиноксидов металлами.................................. 101
Совместное получение метилдихлорфосфиноксида и ацетилхлорида 11
Совместное получение метилдихлорфосфиноксида и ацетонитрила 12
Совместное получение метилдихлорфосфиноксида н 1,2-дихлор-
этана ..................................................... 13
Совместное получение метилдихлорфосфинсульфида и 1,2-дихлор-
этана...................................................... 14
Совместное получение этилднхлорфосфиноксида и хлорангидрида
пропионовой кислоты ........................................ 9
Стильбексон.................................................. ЮЗ
3,3',4,4'-Тетраметилдифенилоксид ............................. 105
Тетрамид пиромеллитовой кислоты .............................. 108
Тетрацианбензол ................•............................. 111
О-2-Хлоралкилалкилхлорфосфиниты .............................. 117
8-2-Хлоралкил-этил(алкил)хлорфосфиш1ты......................... 1Ю
1-Хлор-З-бромпропан .......................................... 121
Хлоргидрат моноэтаноламина.................................... 123
S-2-Хлорпропил-метилхлорфосфипит......................... . . 120
S-2-Хлорпропил-этилхлорфосфинит............................... 120
2-Хлорхинолин .................................... •........... 52
fi-Хлорэтиламии хлористоводородный............................ 123
а-Хлор-₽-этилмеркаптоэтилацетат .............................. 123
О-2-Хлорэтилметилхлорфосфинит ................................ 117
S-2-Хлорэтил-метилхлорфосфинит................................ 119
S-2-Хлорэтил-этилхлорфосфинит............................. 120
Хризоидин-Н.И'-диуксусная кислота............................. 125
Хризоидии-N, N, N', N'-тетрауксусная кислота ................. 125
1,4-Циклогександион......................................... 114
Этилдихлорфосфин................................................ 6
134
Этилдихлорфосфинселенид...................................... 18
Этилдихлорфосфинсульфид ..................................... %
Этилмеркаптоацетальдегид.....................•.............. 18°
94
Этилтетрафторфосфин.....................................
Замеченные опечатки
Стра- ница Строка Напечатано Следует читать
9 7 снизу ,С1 С2НБР< II ХС{ О .С1 С2НВР( II ХС1 о
16 13 снизу 3/54 53/54
33 6 сверху Англ. пат. 719887; С. А., 50, Р 1923а (1954). Англ. пат. 719887 (1954); С. А., 50, Р 1923 а (1956).
33 11 сверху phys. siol. physiol.
49 5 сверху Cl-44,6 Cl-44,06
54 19 сверху дилкилхлорфосфинов диалкилхлорфосфинов
СО—О ОС О
62 3 сверху ОС-А-™ 1 Y /V- СО ос Il 1 1 Y
о со О—со
87 10 снизу л-иитроаналина л-нитроанилина
100 18 снизу Chem. Fng. Chem. Eng.
W0 21 снизу 0,5N 0,5 н.
id 4 снизу (190) (1960)
118 11 снизу о-2-хлорэтилметилфосфииит О-2-хлорэтилметилфос- финит
118 6 снизу о-2-хлоралкилалкилхлор- фосфиниты О-2-хлорал кил алкил хлор- фосфиниты
125 8 снизу плексона плексонов
125 9 снизу нового НОВЫХ
3™ 1Q1