/






























































































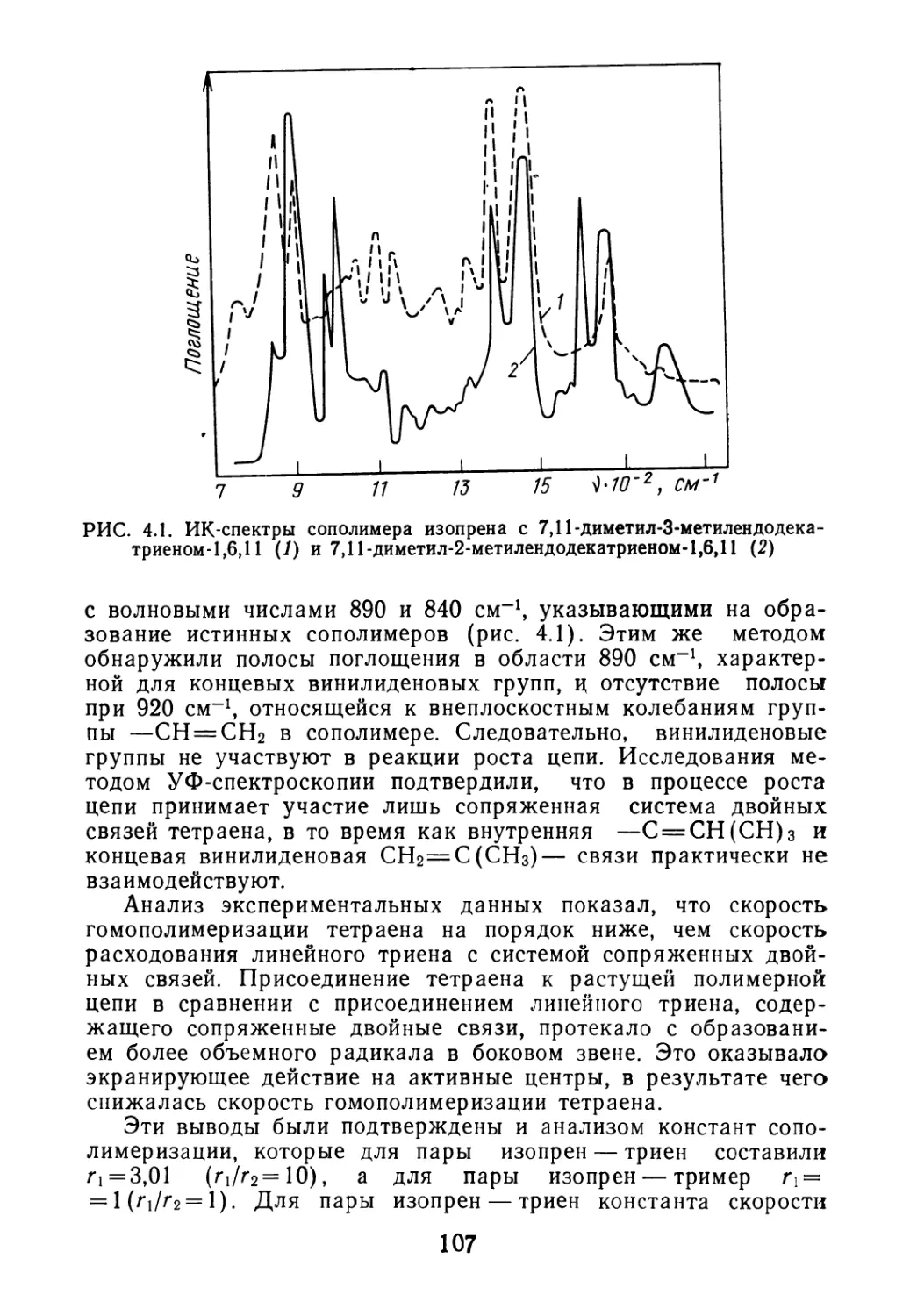
















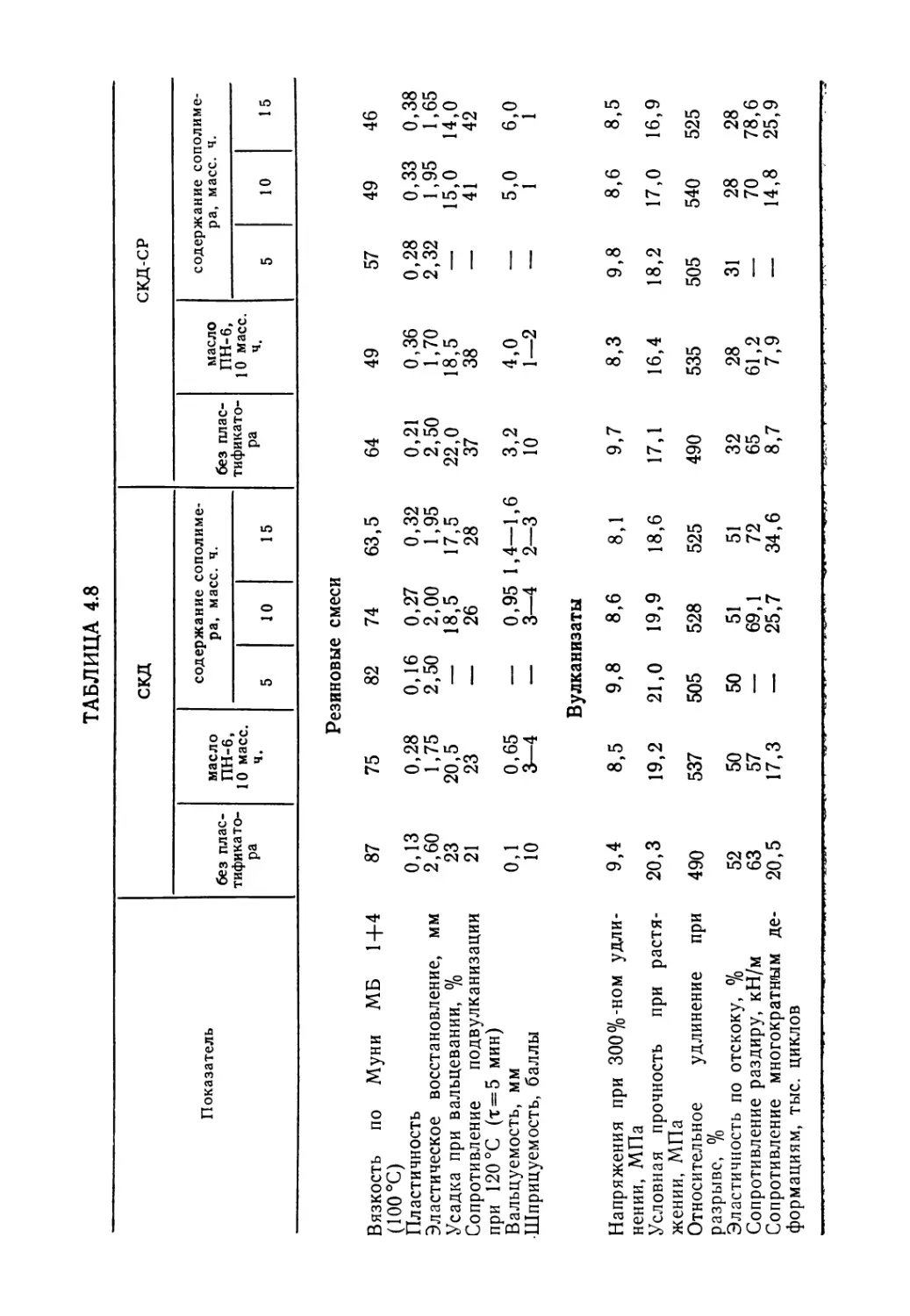








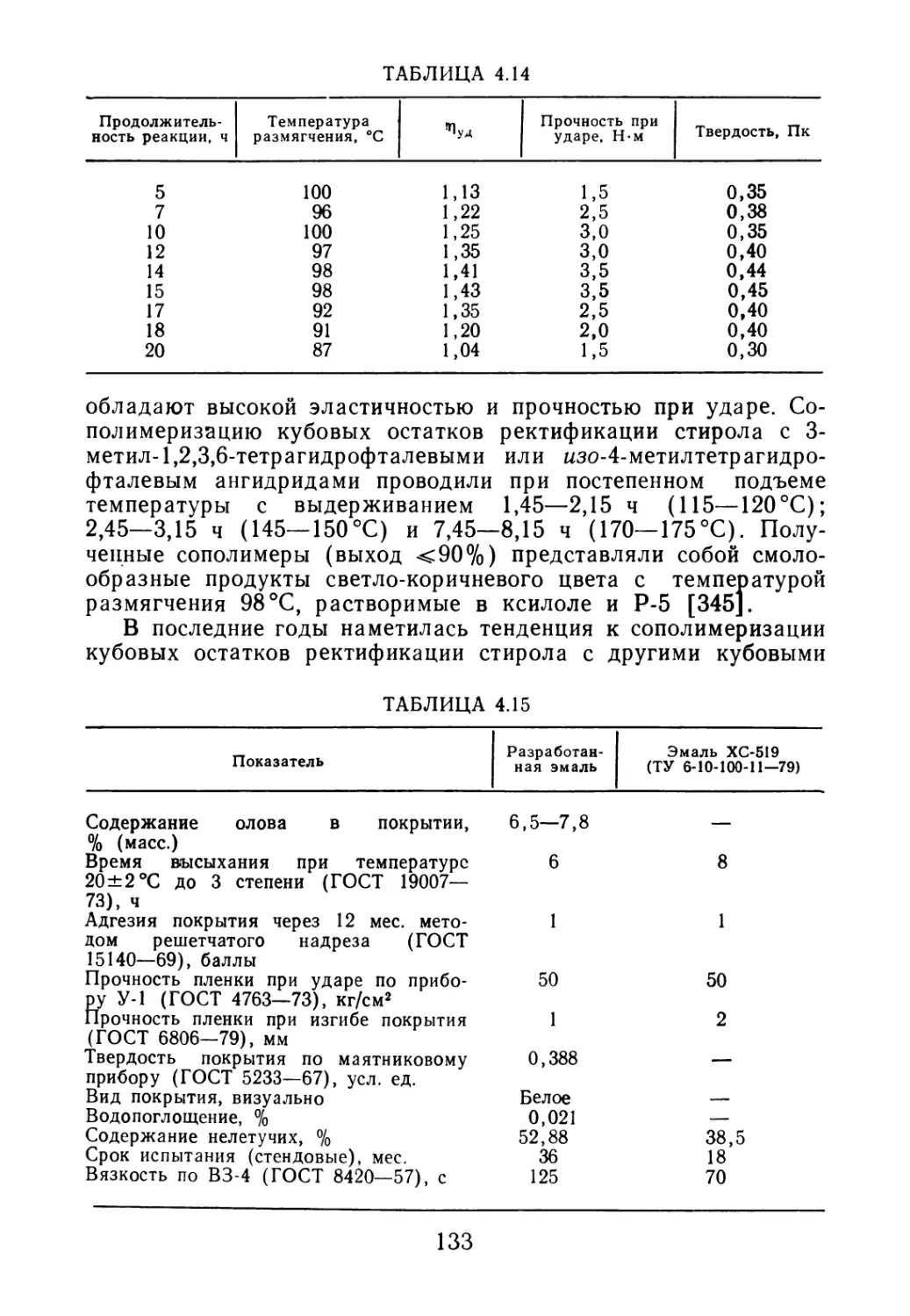




































































































Author: Черкашин М.И.
Tags: технология минеральных масел технология нефти и аналогичного сырья химическая промышленность нефтехимия нефтедобывающая промышленность
ISBN: 5—7245—0309—3
Year: 1989




Text
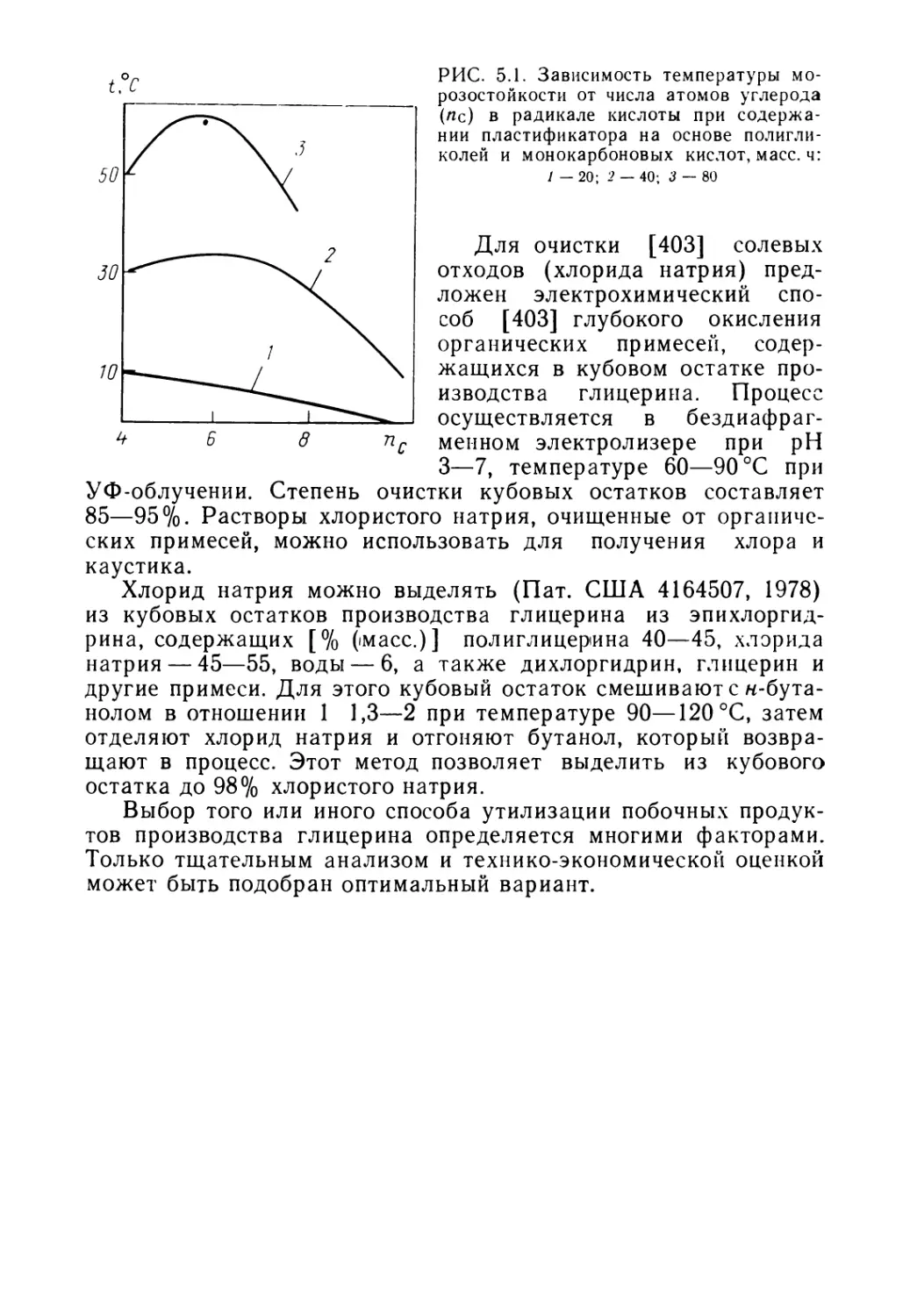
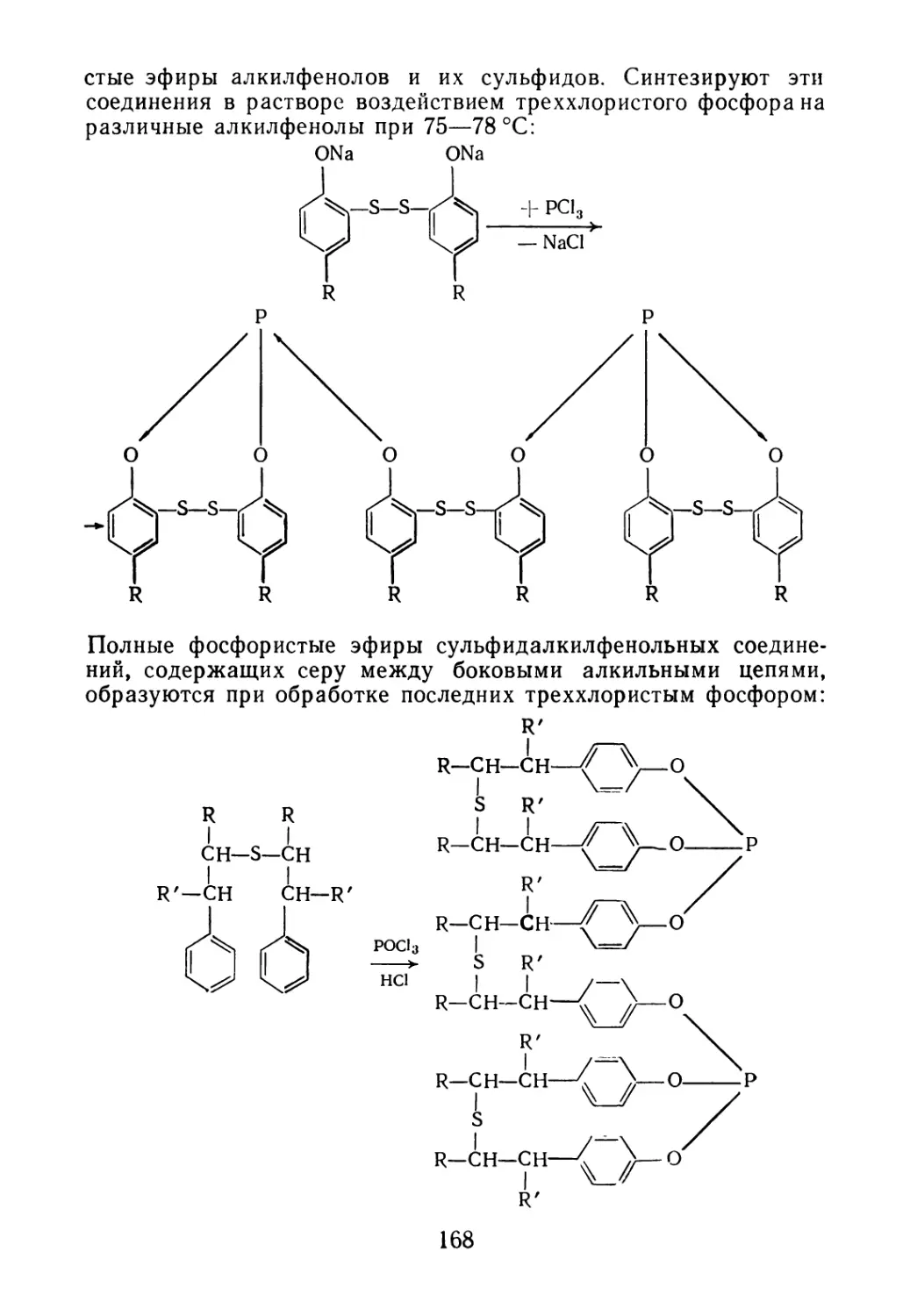
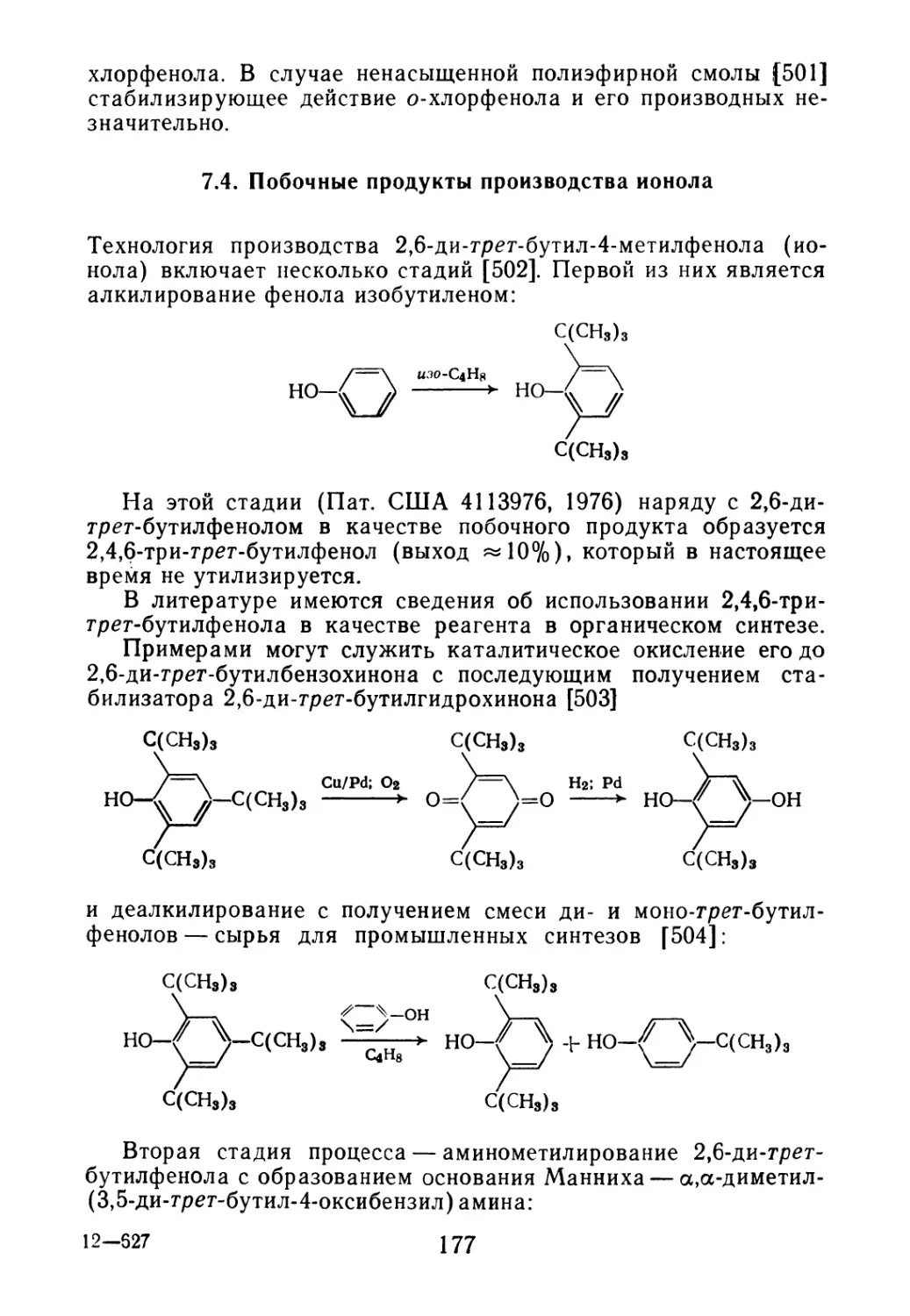
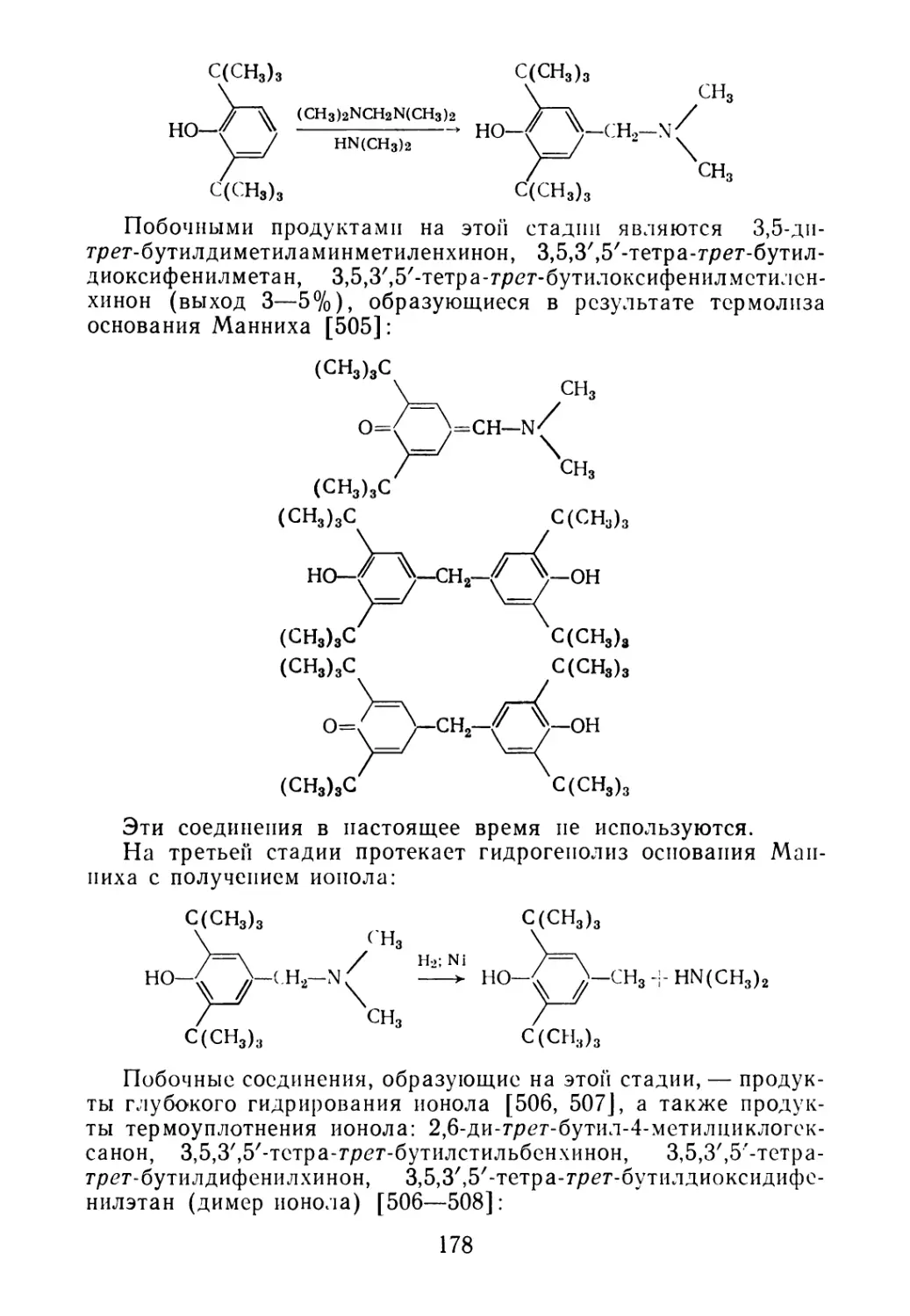
отходы
и побочные продукты
нефтехимических
производств -
СЫРЬЕ
ДЛЯ ОРГАНИЧЕСКОГО
СИНТЕЗА
отходы
и побочные продукты
нефтехимических
производств -
СЫРЬЕ
ДЛЯ ОРГАНИЧЕСКОГО
СИНТЕЗА
Под редакцией профессора М. И. Черкашина
МОСКВА «ХИМИЯ » 1989
ББК 6П7.50
0 878
УДК 665.652.7/.8.004.8
Авторы: С. С. Никулин, В. С. Шеин, С. С. Злотский, М. И. Черкашин,
Д. Л. Рахманкулов
Рецензент В. В. РАБОТНОВ (МНХП)
0 878 Отходы и побочные продукты нефтехимических произ-
водств— сырье для органического синтеза/С. С. Никулин,
В. С. Шеин, С. С. Злотский и др.; Под ред. М. И. Черкаши-
на.— М.: Химия, 1989.—240 с.: ил.
ISBN 5—7245—0309—3
Приведены состав, строение и свойства различных побочных продуктов,
являющихся ценным сырьем для органического синтеза и в производстве
полимерных продуктов. Комплексно рассмотрены проблемы снижения от-
ходов и создания процессов их переработки с учетом механизма реакций,
лежащих в основе этих процессов.
Для инженерно-технических и научных работников предприятий хими-
ческой, нефтехимической и нефтеперерабатывающей промышленности.
О
2804010000—042
--------------42—89
050(01)—89
ББК 6П7.50
ISBN 5—7245—0309—3
© Издательство «Химия», 1989
ОГЛАВЛЕНИЕ
Введение 5
Глава 1. ОТХОДЫ ПРОИЗВОДСТВА ДИЕНОВЫХ И ВИНИ Л АРО-
МАТИЧЕСКИХ МОНОМЕРОВ 7
1.1. Свойства и состав кубовых остатков ректификации бута-
диена-1,3 7
1.2. Состав побочных продуктов синтеза изопрена 10
1.3. Отходы производства винилароматических углеводородов 16
Глава 2. ДИЕНОВЫЕ ОЛИГОМЕРЫ — ПОБОЧНЫЕ ПРОДУКТЫ
СИНТЕЗА ПОЛИМЕРОВ 19
2.1. Образование олигомеров в процессе синтеза полимеров 20
2.2. Олигомеры изопрена 26
2.3. Олигомеры бутадиена 30
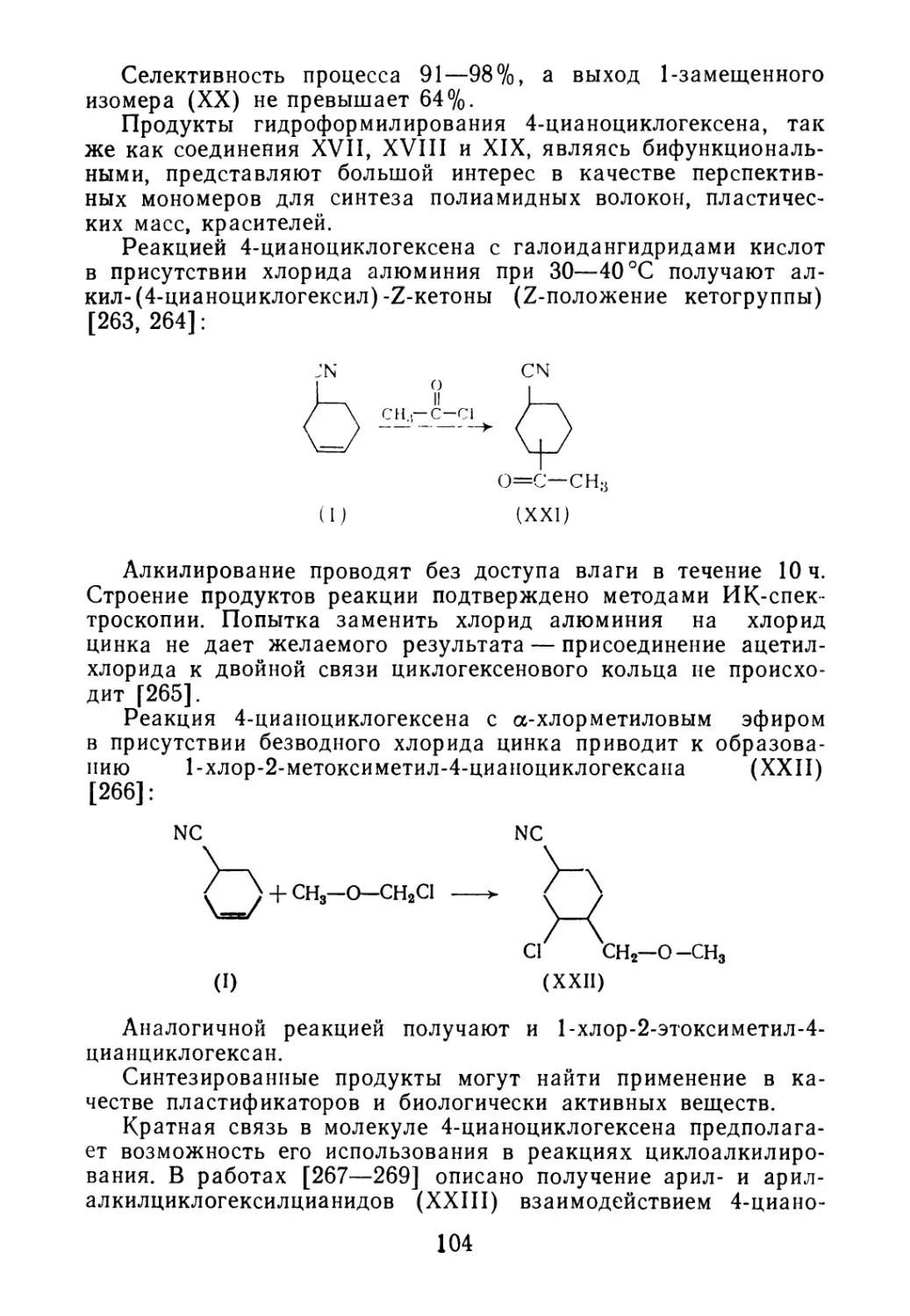
2.4. 4-Цианоциклогексен — побочный продукт производства бу-
тадиеннитрильных каучуков 34
2.5. Снижение концентраций олигомеров в каучуке 35
Глава 3. ИСПОЛЬЗОВАНИЕ ДИЕНОВЫХ ОЛИГОМЕРОВ В ОРГА-
НИЧЕСКОМ СИНТЕЗЕ 38
3.1. Синтез винилароматических углеводородов из димера бу-
тадиена 38
3.2. Эпоксидирование олигомеров бутадиена 39
3.3. Взаимодействие винилциклогексена с фенолами, нафтола-
ми и их производными 47
3.4. Модификация полимеров диеновыми олигомерами 57
3.5. Диеновый синтез на основе отходов нефтехимических
производств 60
3.6. Взаимодействие диеновых олигомеров с серной кислотой
и серой 67
3.7. Термическая переработка органических отходов 75
3.8. Галогенирование диеновых олигомеров 80
3.9. Металлорганический синтез на основе низкомолекулярных
производных диенов 83
3.10. Диспропорционирование и изомеризация диеновых оли-
гомеров 88
3.11. Окисление отходов производства синтетического каучука 89
3.12. Синтезы на основе 4-цианоциклогексена 95
Глава 4. СИНТЕЗ ПОЛИМЕРОВ И СОПОЛИМЕРОВ ИЗ ПОБОЧ-
НЫХ ПРОДУКТОВ И ОТХОДОВ НЕФТЕХИМИИ 105
4.1. Олигомеры изопрена 106
4.2. Олигомеры бутадиена 113
4.3. Кубовые остатки ректификации бутадиена 126
4.4. Кубовые остатки производства винилароматических угле-
водородов 128
3
Глава 5. ПРИМЕНЕНИЕ ПОБОЧНЫХ ПРОДУКТОВ 11РОИЗВОДСТ-
ВА ОДНО- И МНОГОАТОМНЫХ С! II IP К)В 137
5.1. Побочные продукты производства бутиловых спиртов 137
5.2. Побочные продукты производства гликолей 143
5.3. Побочные продукты производства глицерина 147
Глава 6. ПРИМЕНЕНИЕ ПОБОЧНЫХ ПРОДУКТОВ ПРОИЗВОДСТ-
ВА СИНТЕТИЧЕСКИХ ЖИРНЫХ КИСЛОТ И ФТАЛЕВОГО
АНГИДРИДА 151
6.1. Побочные продукты производства синтетических жирных
кислот 151
6.2. Побочные продукты производства фталевого ангидрида 157
Глава 7. ПРИМЕНЕНИЕ ПОБОЧНЫХ ПРОДУКТОВ ПРОИЗВОДСТ-
ВА ЗАМЕЩЕННЫХ ФЕНОЛОВ 160
7.1. Побочные продукты производства фенола и ацетона 160
7.2. Применение п-кумилфенола 165
7.3. Побочные продукты производства 2,4-дихлорфенола 173
7.4. Побочные продукты производства ионола 177
7.5. Побочные продукты производства дифенилолпропана 179
Глава 8. ПРИМЕНЕНИЕ ОТХОДОВ, ПРОМЕЖУТОЧНЫХ И ПОБОЧ-
НЫХ ПРОДУКТОВ ПРОИЗВОДСТВА ИЗОПРЕНА 181
8.1. Отходы производства изопрена 181
8.2. Применение 4,4-диметил-1,3-диоксана 183
Глава 9. СОСТАВ И ОБЛАСТИ ПРИМЕНЕНИЯ ПОБОЧНЫХ ПРО-
ДУКТОВ ПРОИЗВОДСТВА АРОМАТИЧЕСКИХ УГЛЕВО-
ДОРОДОВ 197
9.1. Применение побочных продуктов производства стирола 197
9.2. Применение побочных продуктов производства алкилбен-
золов 203
Глава 10. КОМПЛЕКСНОЕ ИСПОЛЬЗОВАНИЕ ПОБОЧНЫХ ПРО-
ДУКТОВ ПРОИЗВОДСТВА НЕКОТОРЫХ ПРИСАДОК 209
10.1. Применение побочных продуктов производства алкил-
салицилатных присадок 210
10.2. Применение побочных продуктов производства некото-
рых алкилфенольных присадок 212
10.3. Пути использования нефтешламов процесса производ-
ства присадок 213
217
Библиографический список
ВВЕДЕНИЕ
Нефтехимическое производство базируется на переработке нефти, природных
и попутных газов методами полимеризации, поликонденсации, пиролиза, де-
гидрирования, алкилирования, гидратации, окисления и др. Продуктами про-
мышленного нефтехимического синтеза являются углеводороды (олефины,
ароматические углеводороды, ацетилены и др.), высокомолекулярные соеди-
нения, а также органические вещества других классов (спирты, кислоты, эфи-
ры). В настоящее время широкое распространение получили высокомоле-
кулярные соединения. Среди полимерных материалов одно из важнейших
мест занимают синтетические каучуки, основные потребители которых —
шинная и резинотехническая промышленность. Организация производства син-
тетических каучуков неразрывно связана с выпуском мономеров, являющим-
ся самым материале- и энергоемким процессом. На его долю приходится до
75% общих затрат. Выход побочных продуктов достигает 1 т на 1 т целе-
вых мономеров.
Синтез полимеров методами ионной и радикальной (со)полимеризации
мономеров также сопровождается второстепенными процессами, приводящи-
ми к образованию побочных продуктов и отходов. В производстве изопре-
нового каучука, получаемого полимеризацией изопрена на катализаторах
Циглера — Натта, образуются низкомолекулярные продукты — димеры и три-
меры изопрена. При этом фракция димеров изопрена составляет 30—40%
(масс.) от всей массы кубового остатка (0,2—0,3% на 100% каучука). Ана-
логичная картина наблюдается и при производстве бутадиенового каучука.
Общее количество образующихся в процессе полимеризации бутадиена низко-
молекулярных производных (димеров, тримеров и т. п.) достигает 0,2% на
100% каучука. При этом основным побочным продуктом является димер
бутадиена — 4-винилциклогексен.
Аналогичные процессы протекают и в ряде других нефтехимических про-
изводств с участием диеновых углеводородов, например в производстве
этилиденнорборнена.
Таким образом, наряду с основными продуктами нефтехимического син-
теза образуется большое количество побочных и промежуточных продуктов,
которые до настоящего времени практически не находят применения и часто
рассматриваются как отходы. Эта проблема становится еще более актуальной
в связи с ростом в последние годы промышленных мощностей и тенденцией
к их дальнейшему увеличению.
В связи с этим необходимо одновременно искать пути снижения выхода
побочных продуктов и методы их переработки в ценные промышленные про-
дукты. Решение этих задач позволит снизить нормы расхода углеводородно-
го сырья на тонну готовой продукции и улучшить экологическую обстанов-
ку. Поэтому разработка новых технологий и технологических процессов пере-
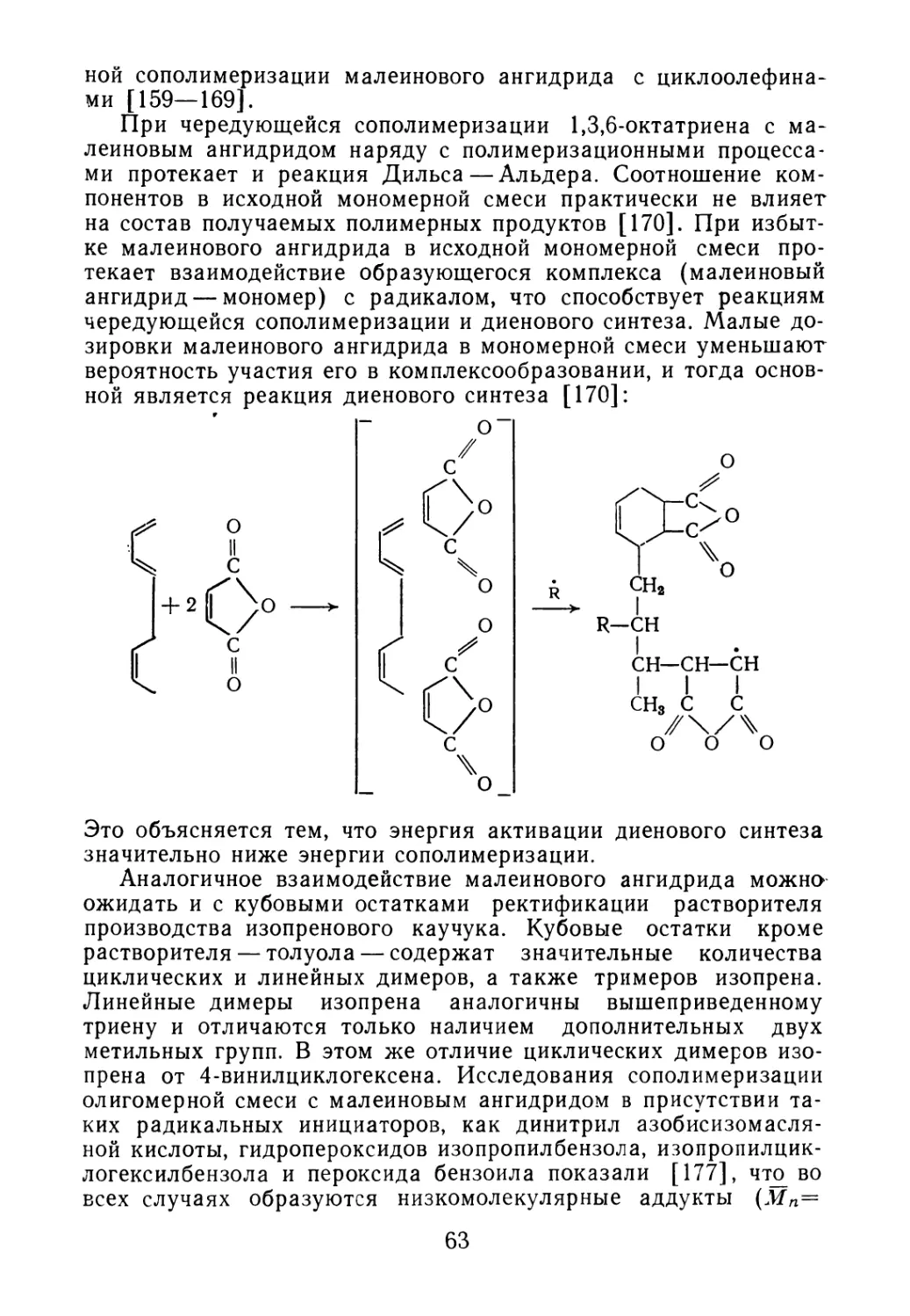
работки вторичного сырья является важнейшей проблемой промышленности
нефтехимического синтеза.
Сложности поисков путей использования промышленных отходов и по-
бочных продуктов органического и нефтехимического синтеза заключаются
в том, что все они, как правило, состоят из смеси углеводородов, различаю-
щихся по своему составу, строению и реакционной способности. Разделение
же такой смеси на индивидуальные соединения зачастую очень сложная про-
изводственно-техническая задача, требующая больших технико-экономических
затрат. Поэтому наиболее перспективным является использование отходов
производства без разделения их на отдельные компоненты.
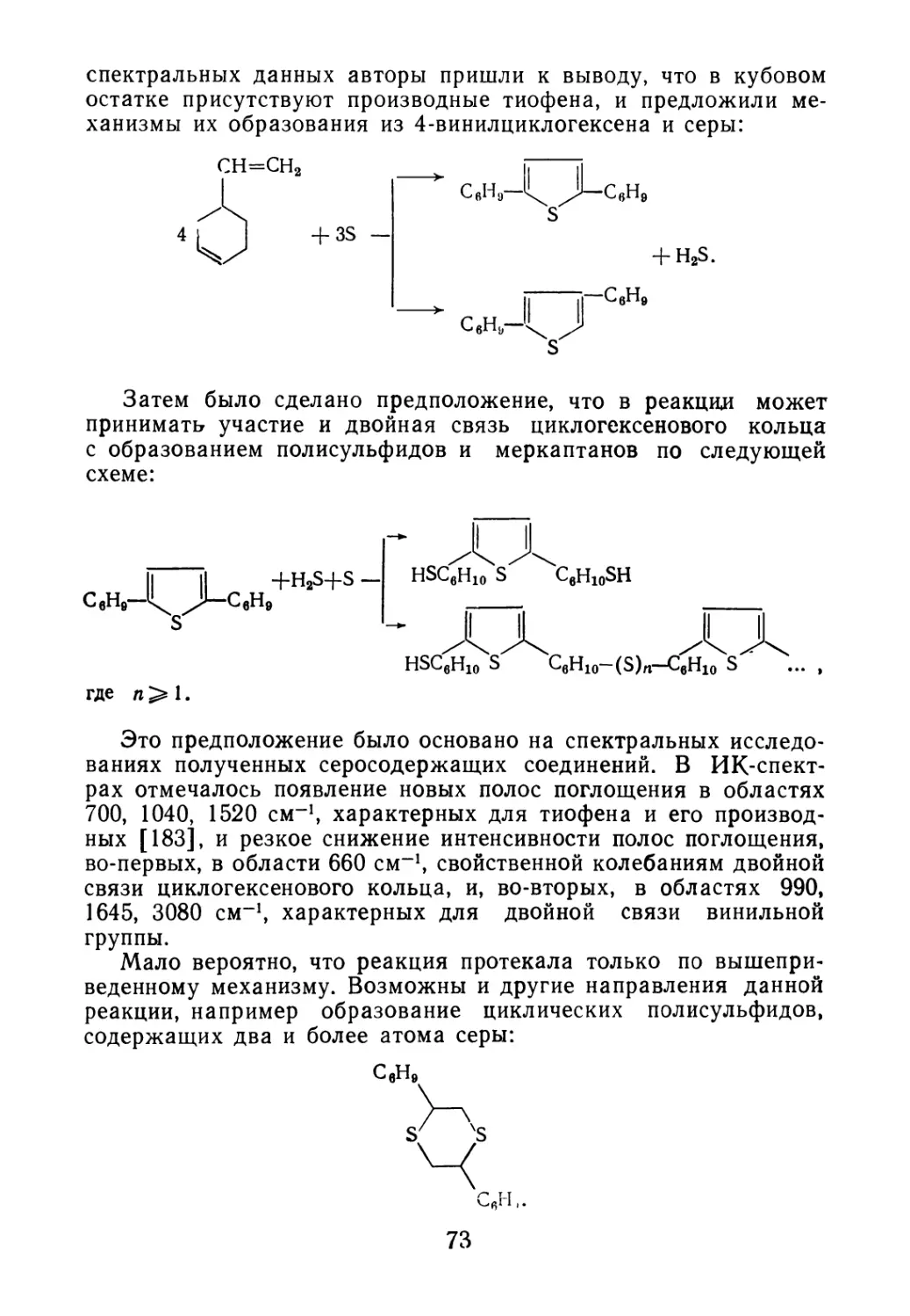
5
В настоящее время в промышленности уже осуществлены технологиче-
ские процессы, позволяющие использовать производственные отходы для по-
лучения пленкообразующих веществ, качество которых не уступает пленкам,
полученным на основе масла, а по некоторым показателям даже превосходит
их. Примером такого производства является получение низкомолекулярных
полимеров на основе отходов синтеза бутадиена и кубовых остатков ректи-
фикации стирола, которое действует в промышленных масштабах. Широкое
использование отходов нефтехимического производства позволяет резко со-
кратить потребление натуральных растительных масел в лакокрасочной про-
мышленности и расширить ассортимент выпускаемых синтетических пленко-
образующих.
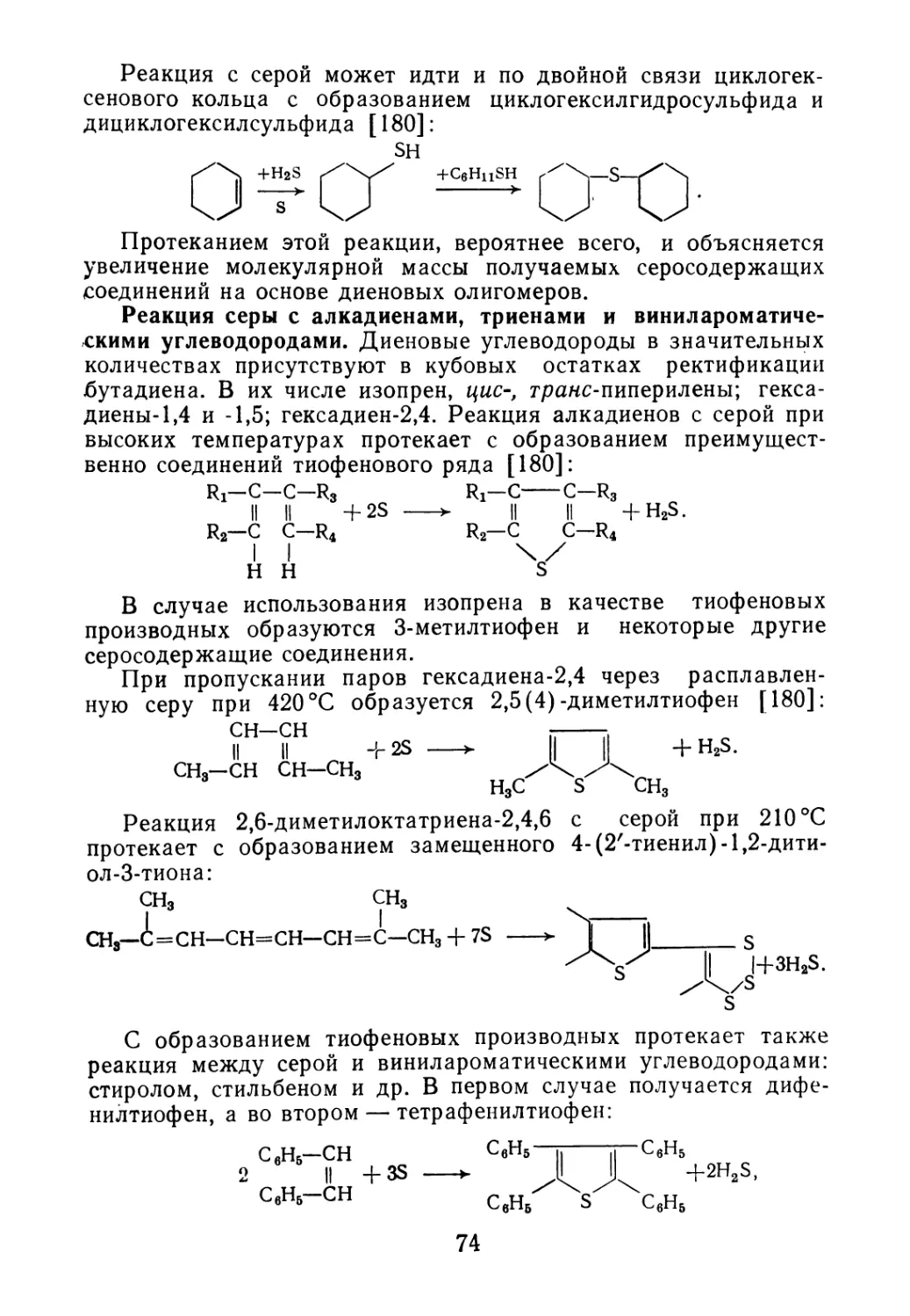
В последние годы появились сообщения [1—6] о переработке отходов про-
изводства и побочных продуктов органического и нефтехимического синтезов
для получения соединений, используемых в различных полимерных компози-
циях. Однако в настоящее время еще имеется ряд производственных отходов
и побочных продуктов органического и нефтехимического синтезов, которые
не нашли своего квалифицированного применения в народном хозяйстве.
В этой книге рассмотрены отходы и побочные продукты многотоннажных
производств основного органического и нефтехимического синтеза, а также
побочные продукты, образующиеся при получении высокомолекулярных со-
единений. К ним прежде всего относятся побочные продукты синтезов диено-
вых и винилароматических мономеров, а также синтетических полимеров. При-
ведены данные по применению побочных продуктов производства одно- и
многоатомных спиртов, жирных кислот, фталевого ангидрида и замещенных
фенолов. В книге обобщены имеющиеся литературные данные по строению,
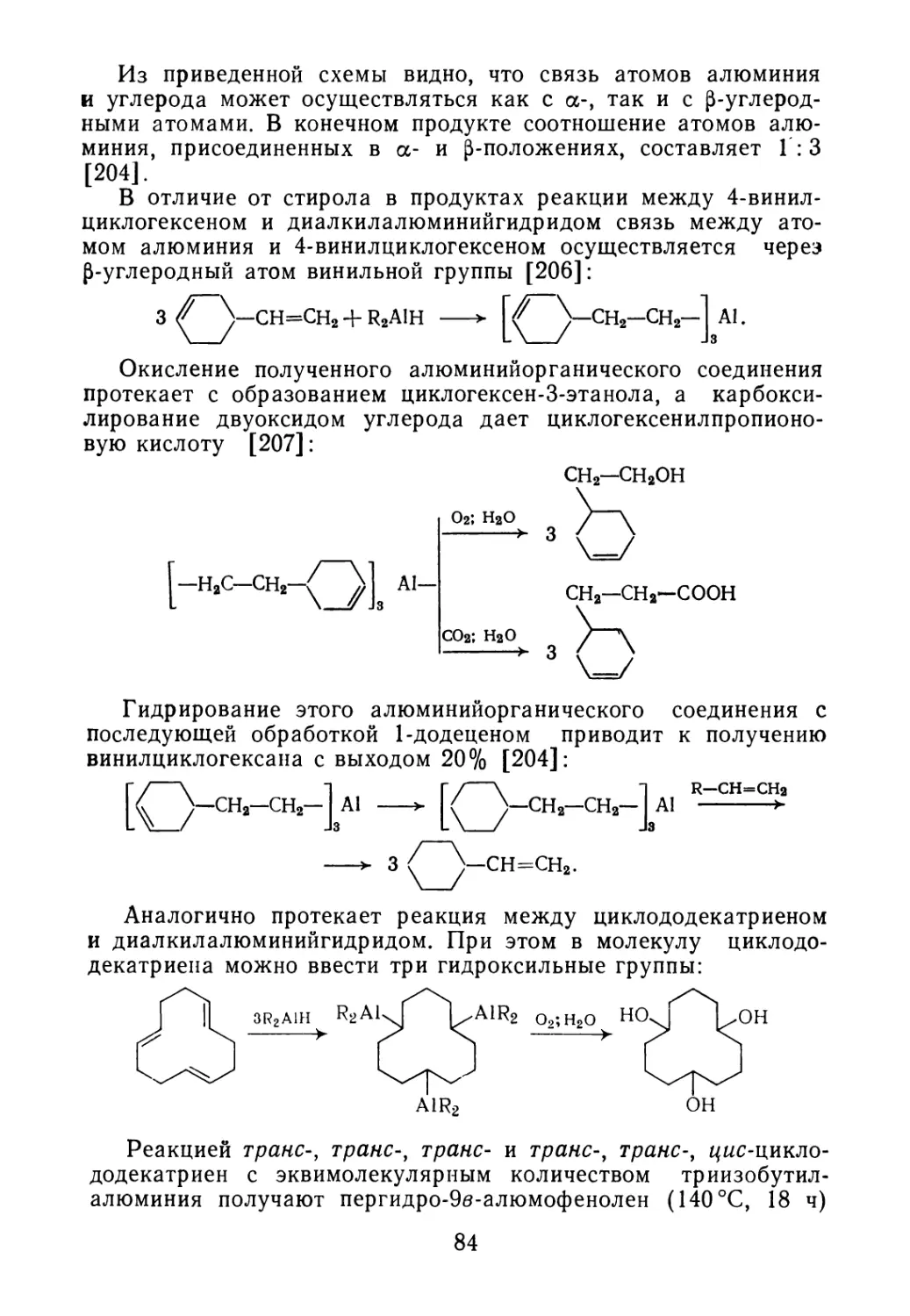
составу и свойствам отходов и побочных продуктов различных производств,
указаны возможные направления их использования в качестве исходных
продуктов для получения новых веществ и материалов. Кроме того, книга
может быть использована в качестве справочного пособия.
Авторы будут признательны за все рекомендации и предложения по ка-
честву приведенного материала, интерпретации данных и метода изложения.
ГЛАВА 1
Отходы производства диеновых
и винилароматических мономеров
Производство мономеров — важнейшая стадия схемы получения
синтетических полимеров. К числу мономеров, наиболее широ-
ко используемых в промышленности синтетического каучука, от-
носятся: бутадиен-1,3, изобутилен, изопрен, стирол, а-метилсти-
рол, акрилонитрил, хлоропрен. Одним из основных требований,
предъявляемых к мономерам, является их чистота. Высокая сте-
пень очистки достигается ректификацией. При этом основной
продукт выделяется из углеводородной смеси, а в кубовой части
ректификационных колонн концентрируются высококипящие
соединения.
Описание технологических схем производства бутадиена, изо-
прена, стирола и других мономеров изложено в монографиях
[7—10]. Рассмотрим стадию ректификации мономеров.
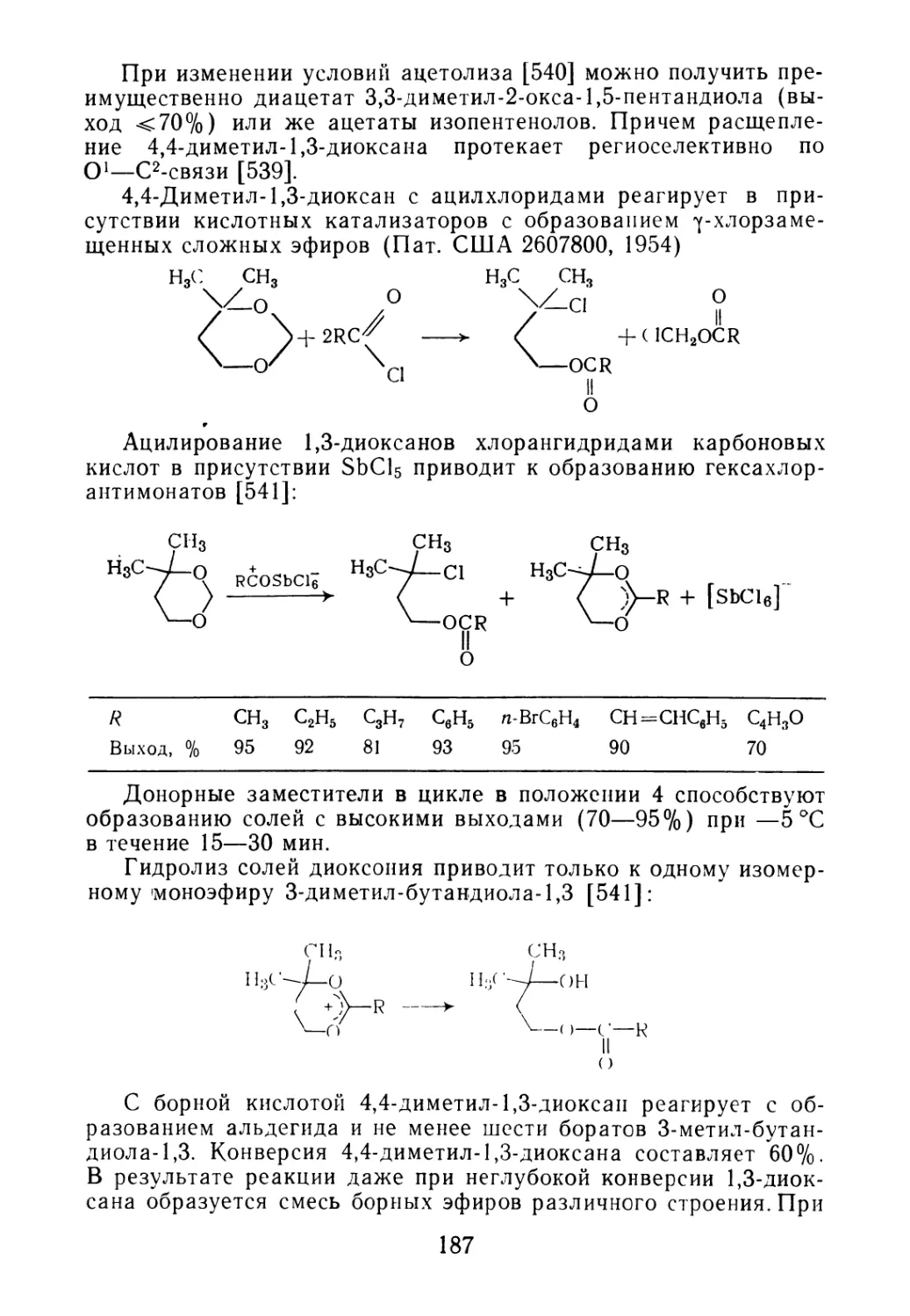
1.L Свойства и состав кубовых остатков ректификации
бутадиена-1,3
Бутадиеновая фракция после очистки в отмывной колонне по-
дается насосом на ректификацию (рис. 1.1). В ректификацион-
ной колонне 1 бутадиен отделяется от содержащихся в нем вы-
сококипящих углеводородов. Из верхней части ректификацион-
ной колонны 1 пары бутадиена поступают в дефлегматор 3
и далее в конденсатор 4. Конденсат из дефлегматора 3 и час-
тично из конденсатора 4 в виде флегмы возвращается на колон-
ну /, а бутадиен-ректификат поступает в сборник 5, откуда на-
правляется на полимеризацию. Кубовый остаток, содержащий
высококипящие продукты и еще значительные количества бута-
диена-1,3, подается на дополнительную его отгонку в колонну 6,
обогреваемую выносным кипятильником 7 Из верхней части
колонны 6 обогащенная бутадиеном фракция после конденсации
в дефлегматоре 8 и конденсаторе 9 возвращается в колонну /,
а кубовый остаток, углеводороды С5—Се, эфиры, амилены и дру’
гие продукты направляются на дальнейшую переработку.
Исследование кубового остатка показало, что в его состав
входит фракция С5—Се, состоящая из изопрена, транс- и цис-
пиперилена, гексадиенов. Кроме вышеперечисленных соедине-
ний в состав кубового остатка входят пентены, диэтиловый
7
РИС. 1.1. Схема очистки бутадиена:
1, 6 — ректификационные колонны; 2, 7 — кипятильники; 3, 8 — дефлегматоры; 4, 9 — кон-
денсаторы; 5 — приемник
эфир, метилциклопентан, толуол, этилбензол и другие углеводо-
роды, общее содержание которых колеблется от 60 до 80%.
Ниже приводятся состав непредельных соединений, входящих
в кубовый остаток ректификации бутадиена, полученного по
способу Лебедева, и некоторые их физико-химические свойства
(табл. 1.1).
Присутствие в кубовом остатке реакционноспособных моно-
меров— изопрена, пиперилена, гексадиенов создает предпосыл-
ки к использованию его для получения, ‘ например, низкомоле-
кулярных полимеров.
ТАБЛИЦА 1.1
Компонент Содержание, % (масс.) ’С ^кип' С Плотность
Изопрен 5-8 —145,95 34,067 20 0,6809 4
Ч^с-Пиперилен 2—4 —140,82 44,068 0,68592**
транс-Пиперилен 4—7 —87,47 43,032 0,67102**
Гексадиен-1,5* 6—9 — 140,68 59,46 20 0,6912“
Гексадиен-2,4 6—9 — 82,0 20 0,7108“
* Гексадиен-1,4 находится в смеси с гексадиеном-1,5, его содержание 6—9% (масс.).
** Плотности (г/см3) и показатели преломления измерялись при 25 °C, величины по-
казателей преломления для цис- и транс-пиперилена составили 1,43291 и 1,42669 соответ-
ственно.
8
ТАБЛИЦА 1.2
Вещество ,°с Содержание, % (масс.)
углерод водород | кислород
Бензол 80,6 92,2 7,8
Метилэтилкетон 79,6 66,7 11,1 22,2
Ацетон 56,4 62,5 10,6 26,9
2,3-Диметилбутадиен-1,3 69,0 87,8 12,2 —
Ч^с-З-Метил пентен-2 70,2 85,5 14,5 —
тра«с-3-Метилпентен-2 67,8 85,4 14,6 -—
Кроме синтеза бутадиена по способу Лебедева существуют
и другие промышленные способы получения бутадиена, напри-
мер дегидрированием бутана и бутиленов, пиролиз нефтяных
фракций и др. [7—14]. Во всех названных способах очистка це-
левого продукта осуществляется на ректификационных колоннах
и при этом в кубах концентрируется углеводородная фракция С5
и выше. Однако углеводородный состав кубового остатка в зна-
чительной степени определяется способами получения бутадиена.
По данным, приведенным в работе [14], в состав кубового
остатка после выделения бутадиена из бутилен-бутадиеновой
фракции входят следующие компоненты:
Углеводородные фракции с tK °C Содержание, % (масс.) Число компо- нентов (по хро- матограмме)
<80,1 17,5—31 ,5 20
<110,6 7,0—16,0 5
110,6 20,0—30,0 1
<138,4 7,0—9,0 3
138,4 12,0—33,0 1
>138,4 13,0—24,0 6
Разделением кубового остатка на лабораторной ректифика-
ционной установке извлекли фракцию с температурой кипения
30—78 °C. Последующее разделение углеводородов проводили
методом препаративной газожидкостной хроматографии с ис-
пользованием в качестве сорбента бензамида. Однако идентифи-
цировать этим способом все соединения, входящие в состав дан-
ной фракции, не удалось. Характеристика выделенных и иденти-
фицированных продуктов [14] приведена в табл. 1.2.
Кроме приведенных выше соединений в производстве бута-
диена образуются и ацетиленовые углеводороды, содержание
которых в товарном бутадиене не должно превышать 5-10_3%
(масс.) [ГОСТ 38-3—71], так как они являются сильными яда-
ми и замедлителями процессов полимеризации. Снижение со-
держания ацетиленовых углеводородов в бутадиене достигается
в промышленности в основном двумя способами: селективным
9
гидрированием ацетиленовых углеводородов, содержащихся во
фракции С4, и концентрата ацетиленовых углеводородов, вводи-
мых из колонны десорбции. В обоих случаях теряется значи-
тельное количество бутадиена. Однако малое количество обра-
зующихся ацетиленовых соединений не требует выделения их
в качестве конечных продуктов.
1.2. Состав побочных продуктов синтеза изопрена
Изопрену, как исходному мономеру в синтезе каучуков, отводит-
ся одно из ведущих мест.
Из литературных источников известно много способов полу-
чения изопрена [7—10, 15—17]. Наибольшее распространение
получили следующие: из изобутилена и формальдегида; катали-
тическое дегидрирование изопентана; каталитическое дегидриро-
вание изоамиленов; димеризация пропилена с последующей де-
метанизацией метилпентенов; из ацетона и ацетилена.
Синтез изопрена из изобутилена и формальдегида в настоя-
щее время широко применяется в промышленном масштабе.
Процесс при этом может осуществляться как в одну, так и в две
стадии.
При двухстадийном процессе на первой стадии осуществля-
ют конденсацию изобутилена с формальдегидом в присутствии
серной кислоты с образованием 4,4-диметилдиоксана-1,3:
СН3 СН3
I h2so4 I
СН3—С=СНг + 2СНгО --->- СН3 С СН2 СНг-
О—СН2—О
На второй стадии происходит отщепление воды и формальде-
гида от 4,4-диметилдиоксана-1,3 с образованием изопрена:
СН3 СН3
сн3—с—сн2—сн2 —> сн2=с-сн=сн2 + сн2о + Н2О.
I I
о—сн2—о
Одностадийный синтез изопрена осуществляют по следующей
схеме:
СН3 СН3
сн3—с=сн2+сн2о —> сн2=с—сн=сн2+н2о.
Изопрен, полученный такими способами, имеет высокую сте-
пень чистоты (99,5%) и не содержит пентадиен. Наряду с этим
образуется значительное количество высококипящих побочных
продуктов, которые распределяются между масляным и водным
слоями. Их физико-химические свойства приведены в табл. 1.3
10
ТАБЛИЦА 1.3
Фракция Молекулярная масса Пределы выки- пания при атмо- сферном давле- нии, °C Плотность (d429) С Средняя теп- лоемкость (25—90 °C) Дж/(г-°С) Температура, °C
вспышки воспламене- ния самовоспла- менения
Высококипящие побочные продукты масляного слоя
Эфиры метилбу- тандиола 138 130—185 0,975 1,435 2,11 74 81 —
Эфиры диоксано- вых спиртов 145 185—240 0,998 1 ,446 1,87—2,04 90 96 —
Диоксановые 150 240—260 1,063 1,456 1,76 115 127 325
Тяжелый кубовый 196 260—370 1,070 1,467 продукт Высококипящие побочные продукты 1,86 128 [ водного слоя 140 325
Преддиольная 120 133—199 1,032 1,456 — — — —
Дибльная 105 199-205 0,990 1,444 — — — —
Диоксановые спирты 147 245—260 1,098 1,463 — — — —
[16]. Выход этих высококипящих побочных продуктов достигает
220 кг/т 4,4-Диметилдиоксана-1,3. В таблицах указаны составы
исходного сырья и продуктов разделения высококипящих соеди-
нений масляного [16] (табл. 1.4) и водного слоев (табл. 1.5).
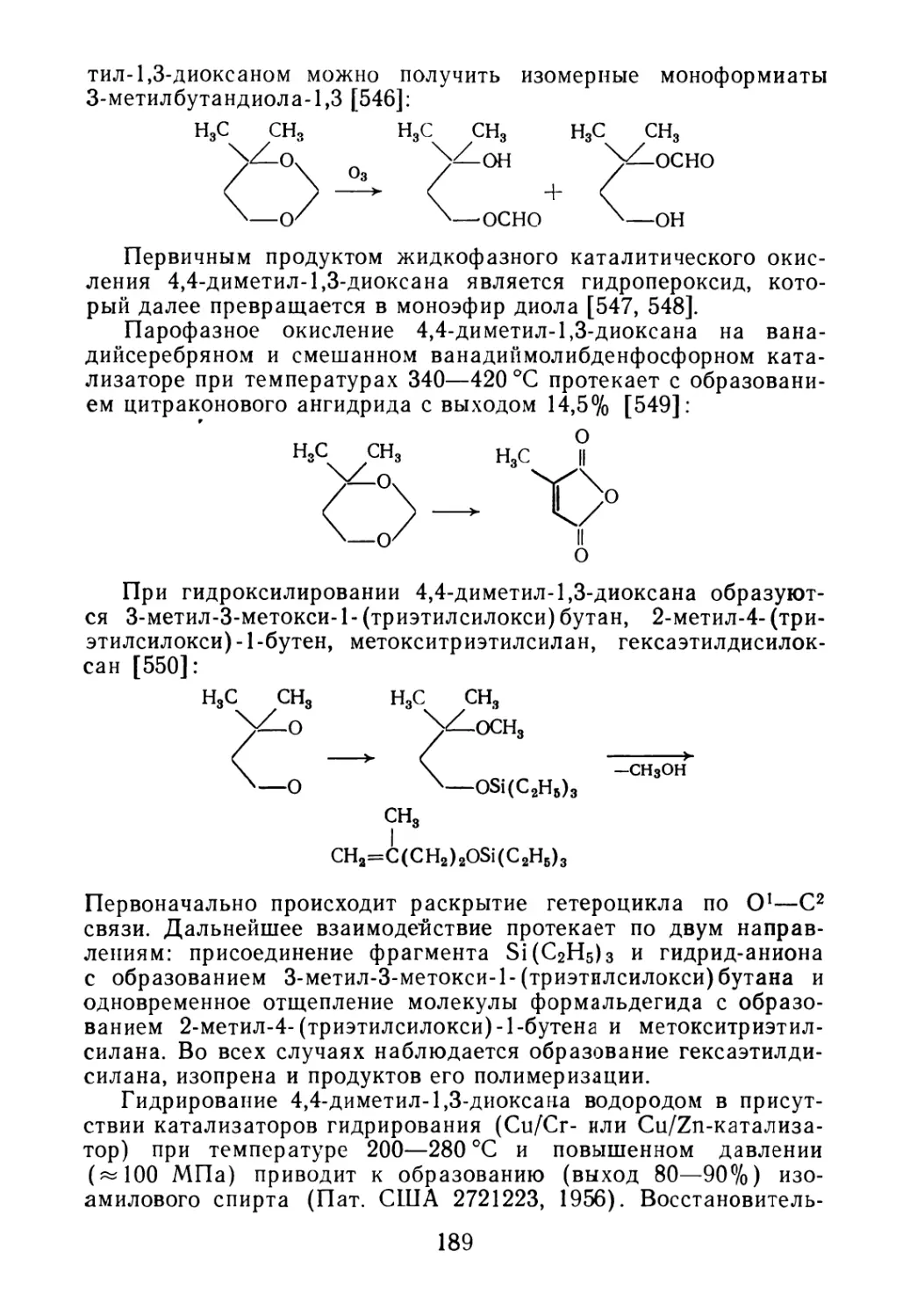
Процесс переработки высококипящих побочных продуктов
синтеза изопрена условно можно разбить на ряд стадий
(рис. 1.2) [16].
На первой стадии процесса предусматривается отделение
широкой фракции, содержащей диоксановые спирты, от тяже-
лого кубового остатка. Выделение широкой фракции из высоко-
кипящих побочных продуктов масляного (или водного) слоя
осуществляют на колонне 2, в которую побочные продукты по-
дают через подогреватель 1. Из верхней части колонны 2, обо-
греваемой выносным кипятильником 3, пары широкой фракции
поступают в конденсатор 4, откуда конденсат сливают в ем-
кость 5. Из емкости 5 часть широкой фракции в качестве флег-
мы возвращают в подогреватель /, а часть подают в колонну 6,
обогреваемую выносным кипятильником 7 В колонне 6 из ши-
рокой фракции выделяют эфиры диоксановых спиртов (или
диольную фракцию), отбирают их из верхней части колонны 6,
конденсируют в аппарате 8 и собирают в сборник 9. Из кубо-
вой части колонны 6 углеводородную фракцию подают в колон-
ну 10 для выделения фракции эфиров диоксановых спиртов
(или диольной фракции). Колонну 10 обогревают выносным
11
РИС. 1.2. Принципиальная технологическая схема установки ректификации:
/ — подогреватель; 2, 6, 10 — ректификационные колонны; 3, 7, // — кипятильники; 4,
8, 12, 15 — конденсаторы; 5, 9, 13, 16 — емкости; 14 — испаритель
кипятильником 11. Пары этой фракции конденсируют в аппара-
те 12 и собирают в сборник 13. Кубовую жидкость колонны 10
направляют в испаритель 14, где выделяют фракцию диоксино-
вых спиртов, пары которой конденсируют в теплообменнике 15,
и конденсат собирают в емкость 16. Тяжелый кубовый продукт
из испарителя 14 объединяют с кубовой частью подогревателя 1.
Строение и свойства побочных продуктов, образующихся
в процессе получения изопрена через 4,4-диметилдиоксан-1,3,
подробно изучены в работах [18—25]. В настоящее время ве-
дутся работы по подбору катализатора разложения 4,4-диметил-
диоксана-1,3. Исследования в этой области показали [18], что
при замене катализатора КДВ-15 на КФ-70 кроме известных
побочных продуктов выделяется новый — метилизопропилкетон.
Идентификацию веществ осуществляли различными методами
по относительному времени удерживания на 2—3 фазах, мето-
дом ИК-спектроскопии, по температуре кипения и по показате-
лю преломления.
Выделение индивидуальных веществ проводили на препара-
тивном хроматографе с колонкой 6 мХ18 мм, заполненной
20%-ным полиэтиленгликолем (ПЭГ), нанесенным на диатомит
при 65 °C. Дополнительную очистку проводили на полупрепара-
тивной колонке 2 мХ15 мм, заполненной 15% сквалана или
15% ПЭГ-1500 на хезосорбе, силинизированном гексаметилди-
силадоном. Результаты исследований приведены в табл. 1.6.
Строение большинства выделенных соединений было под-
тверждено результатами исследования их ПК-спектров, которые
хорошо согласуются с данными, приведенными в [26].
При производстве изопрена в качестве отхода получают пи-
12
ТАБЛИЦА 1.4
Компонент Состав, % (масс.)
исход- ное сырье фракция эфи- ров метилбу- тандиола фракция эфи- ров диокса- новых спир- тов фракция ди- оксановых спиртов
4,4-Диметил-1,3-диоксан 0,4 2,6 —
4-Метил-4-винил -1,3-диоксан 0,4 2,4 —— —
5- (1 -Пропен-2-ил) -1,3-диоксан 0,5 3,2 — —
Эфир трет-бутанола и 3-метил- 1,3-бутандиола 6,9 43,2 — —
Эфир метанола и 3-метил- 1,3-бутандиола 3,5 22,2 — —
Изомер эфира метанола и 3-метил-1,3-бутандиола 0,1 0,8 — —
4-Метил-4-гидрокситетрагид- ропиран 2,5 13,1 2,0 —
Формаль метанола и 3-ме- тил-1,3-бутандиола 3,2 12,5 5,6 —
Эфир метанола и 4-метил- 4-гидроксиэтил-1,3-диоксана (диоксанового спирта II) 1,0 4,5
Эфир метанола и 4,4-диме- тил-5-гидроксиметил-1,3-ди- оксана (диоксанового спир- та III) 0,3 1,4
З-Метил-1,3-бутандиол 2,1 — 9,8 —
Эфир трет-бутанола и диок- санового спирта III 1,9 — 8,8 —
Неидентифицированные про- дукты 2,1 — 8,9 —
Эфир ТБ и диоксанового спирта II 6,3 — 29,4 0,3
10-Метил-1,3,7-пергидронафта- лин 0,5 — 2,3 0,1
1,3,9-Триоксапиро- (5,5) -ун- декан 4,6 — 19,5 1,1
5- (2-Гидроксипропил) - 1,3-ди- оксан (диоксановый спирт I) 10,3 — 5,5 26,6
Диоксановый спирт II 18,9 — 2,3 51,9
Диоксановый спирт III 5,3 — — 13,6
Прочие высококипящие побоч- ные продукты 29,2 — — 6,4
периленовую фракцию, которая является одним из самых мно-
готоннажных отходов (0,1 т/т изопрена). Годовые ресурсы по
пипериленовой фракции составляют около НО тыс. т в год.
Однако только около 20 тыс. т в год используют в производстве
жидкого каучука (заменителя олифы), а остальное количество
экспортируется либо сжигается. Поэтому вопрос использования
пипериленовой фракции до настоящего времени остается откры-
тым [27].
13
ТАБЛИЦА 1.5
Компонент исходное сырье Состав, % (масс.) фракция диоксано - вых спиртов
фракция преддиоль- ная фракция диольная
Эфиры трет-бутанола и 3-метил- 1,3-бутандиола 1,0 11,8 — —
4-Метил-4-гидрокситетрагидропиран 3,0 33,5 0,7 —
Метилбутандиол 25,0 31,8 94,6 2,6
Эфиры трет-бутанола и диоксаново- го спирта II 1 ,о 0,3 4,0 0,2
Эфиры метанола и 3-метил-1,3-бу- тандиола 2,0 22,4 0,4 —
Диоксановый спирт I 4,0 — — 7,5
Диоксановый спирт II 34,0 — — 67,5
Диоксановый спирт III 8,0 — — 15,2
Прочие высококипящие побочные продукты и неидентифицированные соединения 22,0 0,2 0,3 7,0
Перспективным направлением утилизации пиперилена явля-
ется реакция Дильса — Альдера. Однако цис-пиперилен не всту-
пает в диеновые конденсации или вступает с большим трудом
[28], а траис-пиперилен легко конденсируется с различными
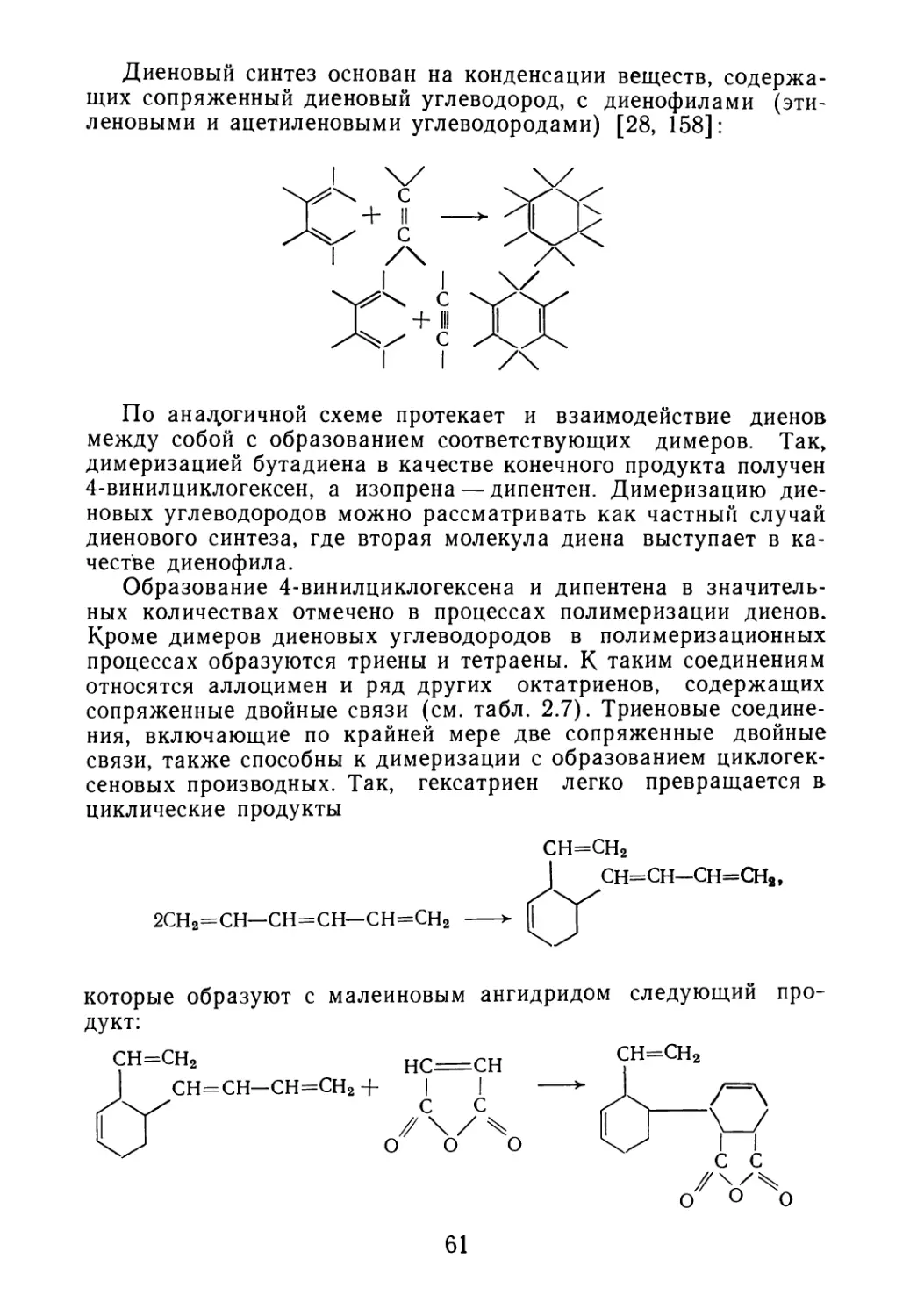
диенофилами. Диеновый синтез протекает по классической схеме
с высоким выходом аддуктов.* Примерами таких синтезов служат
реакции пиперилена с малеиновым ангидридом, акриловыми мо-
номерами и др.:
Полученные аддукты могут найти применение в клеевых
композициях, для улучшения адгезионных свойств и др.
14
ТАБЛИЦА 1.6
Идентифицированное вещество Относительное время удерживания °C п 20 nD
ПЭГ-400 ПЭГ-1500 по литера- турным данным экспе- римент по литера- турным данным экспе- римент
идентифи- цируемый компонент выделен- ный препа- ративно идентифи- цирован- ный компонент выделен- ный препа- ративно по литера- турным данным выделен- ный препа- ративно
2,4,4-Триметилпентен-1 — — — 7,63 — — 101,5 106,0 1,4086 —
З-Метилпентадиен-1,3 — — -— 4,45 — — 78,3 78,4 1,4528 1,4528
н-Масляный альдегид 1,43 1,40 2,26 2,30 — — 74,8 — 1,3791 —
Изовалериановый альдегид 1,80 1,80 3,80 3,80 1,85 1,83 92,5 — 1,3908 —
Диметилвинилкарбинол 4,30 4,25 2,70 2,68 — — 97,0 — 1,4170 1,4170
Метилизопропенилкетон — — — — 2,90 2,80 98,0 — 1,4220 1,4225
4-Винилдиоксан-1,3 12,50 12,40 20,00 20,00 — — 144,9 — 1,4440 —
Триметилкарбинол 1,80 1,80 1,30 1,28 1,56 1,56 82,9 — 1,3954 —
Толуол 2,80 2,80 11,30 11,00 3,57 3,59 110,6 — 1,4969 —
Метилентетрагидропиран 2,80 2,75 11,10 10,80 — — 108,8 — 1,4492 —
Метилдигидропиран 4,10 4,06 15,80 15,80 — — 118,5 — 1,4475 —-
4,4-Диметилдиоксан-1,3 6,70 6,70 18,00 18,00 — — 133,0 — 1,4231 —
Изобутенилкарбинол 14,75 14,80 — — — — 129,9 — 1,4340 —
Ацетон (стандарт) Неидентифицированные соединения 1,00 1,00 1,00 — — — —
Расщепление высококипящих продуктов кубовых остатков
может быть использовано для получения дополнительных коли-
честв изопрена, изобутилена и формальдегида [29, 30]. Опти-
мальный температурный режим расщепления при использовании
катализатора типа КАС-3 составляет 450—475 °C [29], а при
использовании твердого катализатора, состоящего из оксида
алюминия и фосфата кальция, — 315—360 °C.
В оптимальных условиях суммарный выход изопрена, изо-
бутилена и формальдегида достигает 57% [16,5, 29,5 и 11,0%
(масс.) соответственно] от массы органического вещества ис-
ходной смеси. За счет каталитического разложения побочных
продуктов синтеза изопрена выход изопрена может быть увели-
чен на 8—10% [29].
Перспективным направлением является использование высо-
кокипящих побочных продуктов синтеза изопрена (температура
кипения 250—380 °C, температура вспышки 128—155 °C, кислот-
ное число 0,3—0,6 мг КОН/г) в качестве пластификаторов ре-
зиновых смесей на основе бутадиеннитрильных каучуков. При
этом отмечено повышение теплостойкости и динамической вы-
носливости вулканизатов [31].
В работе [32] для удешевления резиновых смесей при со-
хранении физико-механических свойств резин рекомендовано
использовать продукт взаимодействия фульвеновой фракции
с муравьиной кислотой в количестве 1,5—7,0 масс. ч. на
100 масс. ч. ненасыщенного каучука.
Перспективным направлением является и использование ку-
бовых остатков процесса очистки изопрена, представляющих
собой смолу с температурой размягчения 30—60 °C и содержа-
щую 35—70% (масс.) фульвена и продуктов его полимеризации,
для модификации бутадиенстирольного пленкообразующего.
При этом снижается токсичность целевого продукта и улучша-
ются свойства получаемых покрытий [33].
Наряду с этим анализ имеющихся литературных данных по-
казывает, что полный состав побочных продуктов, образующих-
ся в производстве изопрена, до настоящего времени не известен.
Это создает определенные трудности в поиске путей наиболее
перспективного использования этих отходов.
1.3. Отходы производства винил ароматических углеводородов
Винилароматические мономеры широко используются для полу-
чения полимерных материалов. Состав отходов производства
винилароматических мономеров в значительной степени зави-
сит от выбора способа получения последних. При этом различен
и подход к переработке отходов. Это обусловлено тем, что тех-
нология переработки одних отходов зачастую неприемлема для
переработки других.
16
Кубовые остатки ректификации алкилбензолов. Отходы, об-
разующиеся в производстве винилароматических углеводородов,
относятся к многотоннажным. Среди них — отходы производства
этилбензола (или изопропилбензола), образующиеся при алки-
лировании бензола этиленом (изопропиленом) [34—37]. После
выделения из продуктов алкилирования этилбензола в кубовой
части ректификационных колонн концентрируются высококипя-
щие продукты [35], основными из которых являются
[% (масс.)]:
Дибутилбензолы 3,0
Дифенилметан 2,2
Тетраэтилбензол 28,2
Пента-, гексаэтилбензолы 58,0
и ароматические углево-
дороды
По данным, приведенным в работе [36], полиалкилбензоль-
ная смола со стадии алкилирования бензола этиленом (а) или
пропиленом (б) имеет следующий состав [% (масс.)]:
а б
1,3,5-Триэтилбензол 4,4
1,2,4-Триэтилбензол 2,9
Диизопропилбутилбензолы 3,4
1,1-Дифенилэтан 13,2
Тяжелый осадок 42,5
1,4-Диизопропилбензол 4,8
1,3-Диизопропилбензол 5,5
1,3,5-Триизопропилбензол 5,1
1,2-Дифенилпропан 6,6
1,1-Дифенилпропан 4,9
Алкилиндан (алкилтетра- 6,0
ли и)
Алкилдифепилпропаны 9,1
Тяжелый осадок 31,0
Кубовый остаток ректификации этилбензола (изопропилбен-
зола) представляет собой маслянистый или смолообразный про-
дукт темно-коричневого цвета и, как видно из приведенных дан-
ных, имеет сложный состав. В нем присутствует ряд неиденти-
фицированных соединений, и не совсем ясен состав тяжелого
остатка. Наиболее вероятно, что тяжелый остаток — смесь низ-
комолекулярных полимеров на основе этилена (пропилена), об-
разующихся при алкилировании бензола соответствующим оле-
фином в присутствии катализатора Фриделя — Крафтса. В со-
став полимерных продуктов могут входить и производные аро-
матических углеводородов.
Таким образом, различие в технологии получения кубовых
остатков производства алкилароматических углеводородов и,
следовательно, их составов создает значительные трудности
в поиске путей наиболее рационального использования этих
побочных продуктов.
2—627 17
Кубовые остатки ректификации винилароматических моно-
меров. При выделении и очистке стирола [38, 39] в процессе
ректификации накапливаются кубовые остатки, переработка ко-
торых чрезвычайно важна. В их состав входит большое количе-
ство различных органических соединений, в том числе и моно-
мерный стирол, полное извлечение которого на ректификацион-
ных колоннах не достигается. Кубовые остатки ректификации
стирола относятся к многотоннажным отходам. На 1 т товар-
ного стирола приходится около 20 кг отходов. Состав кубовых
остатков ректификации стирола в значительной степени опре-
деляется технологическим оформлением процесса и применяемы-
ми ингибиторами [40—45]. По данным, приведенным в рабо-
те [43], кубовые остатки содержат следующие соединения
[% (масс.)]:
Стирол
а-Метилстирол
Дибензил
1 -Фенил-З-трет-бутил-
циклогексан
22,40 Фенантрен
7,10 Полистирол
2,77 Остаток
2,68
3,10
45,20
2,53
В зависимости от условий фракционирования печного масла
содержание стирола в кубовых остатках ректификации может
изменяться от 10 до 50%, а полистирола—15—70% [45].
В настоящее время повышенный интерес проявляется к ме-
тоду получения стирола дегидратацией метилфенилкарбинола
[46, 47]. Состав кубового остатка, образующегося в этом про-
цессе, отличается от остатка при синтезе стирола из этилбен-
зола. Кубовый остаток процесса дегидратации включает стирол,
а-метилстирол, бензальдегид, ацетофенон, метилфенилкарбинол
и смолу, состоящую из полистирола, тяжелого остатка и инги-
биторов. Вопрос о переработке его пока не решен.
а-Метилстирол является менее распространенным мономе-
ром, чем стирол. Один из основных способов получения а-метил-
стирола — дегидрирование изопропилбензола. При этом в каче-
стве отхода образуется смолообразная масса, которая накап-
ливается в кубовой части ректификационных колонн. Она, так
же как и кубовые остатки ректификации стирола, содержит
а-метилстирол и другие органические соединения [48].
ГЛАВА 2
Диеновые олигомеры — побочные
продукты синтеза полимеров
Каталитические системы Циглера — Натта нашли широкое при-
менение в процессах получения стереорегулярных полимеров,
обладающих рядом ценных свойств. Однако наряду с основным
процессом стереоспецифической полимеризации протекают и по-
бочные, нежелательные процессы. Таким процессом при получе-
нии диеновых каучуков является олигомеризация, приводящая
к образованию соответствующих линейных и циклических диме-
ров и тримеров. Димеры и тримеры диеновых углеводородов
в небольших количествах образуются при полимеризации как
в растворе, так и в эмульсии, что приводит к повышенному рас-
ходу мономеров, увеличению содержания летучих веществ, по-
явлению характерного неприятного запаха товарной продукции.
Кроме того, образующиеся низкомолекулярные олигомерные
производные диеновых углеводородов, как правило, могут при-
нимать участие в формировании и росте цепи за счет двойных
связей, оказывая тем самым влияние на свойства получаемого
каучука. Поэтому при проведении процесса стереоспецифиче-
ской полимеризации большое внимание уделяют очистке воз-
вратного растворителя, содержащего вредные для полимериза-
ционного процесса продукты, в том числе димеры и тримеры
диеновых углеводородов. Для возврата растворителя проводят
его тщательную очистку от этих вредных примесей [49—53J,
которые концентрируются в кубовой части ректификационных
колонн. Кубовый остаток, содержащий растворитель и непре-
дельные соединения, еще практически не нашел применения.
Низкомолекулярные производные диеновых углеводородов
образуются и в ряде органических синтезов. Так, димер бута-
диена— 4-винилциклогексен — образуется в качестве побочного
продукта в производстве этилиденнорборнена [54], являющего-
ся третьим сомономером тройных этиленпропиленовых каучу-
ков [51].
Механизм олигомеризации диеновых углеводородов при их
полимеризации и сополимеризации рассматривался в ряде ра-
бот [51—57]. Однако и до настоящего времени нет полной яс-
ности в механизме олигомеризации диенов при их полимериза-
ции и сополимеризации.
В литературе описано 14 димеров и 10 тримеров бутадиена,
28 димеров и 14 тримеров изопрена [56]. При этом некоторые
из них имеют по несколько изомеров.
2"
19
2.1. Образование олигомеров в процессе синтеза полимеров
Образующиеся в процессе полимеризации диеновые олигомеры
одновременно с растворителем (особенно димеры) отгоняются
при выделении каучуков из раствора методом водной дегазации.
При этом димеры диеновых углеводородов отгоняются вместе
с растворителем на стадии дегазации более чем на 90%, а три-
меры— на 30—45% [56—58]. Следовательно, в каучуке, посту-
пающем на сушку, содержатся в основном тримеры диеновых
углеводородов. Это приводит к повышению пожароопасности и
концентрации вредных веществ в воздухе. Поэтому одно из ос-
новных требований к промышленности синтетического каучука —
исключение процесса олигомеризации диеновых углеводородов
при их полимеризации.
В литературе имеются сведения о каталитических системах,
позволяющих снизить процесс олигомеризации диеновых углево-
дородов [59, 60]. Однако в ряде случаев отмечается образова-
ние новых побочных продуктов. Например, при полимеризации
бутадиена в присутствии кобальтового катализатора в качестве
побочного продукта образуется бутентолил. При этом, естествен-
но, возникает необходимость переработки данного отхода.
В последние годы появились сведения о новых каталитиче-
ских системах, позволяющих исключить олигомеризационные
процессы при полимеризации и сополимеризации диенов [61 —
63]. К их числу относят трехкомпонентную систему, состоящую
из нафтената никеля, триизобутилалюминия и трехфтористого
бора. Однако замена действующих каталитических систем на
новые сопряжена с перестройкой некоторых узлов технологиче-
ской схемы либо разработкой новой технологической схемы,
а также с решениями ряда дополнительных проблем, включаю-
щих очистку сточных вод и др. Поэтому в реальных производст-
венных условиях идут по пути модификации действующих ката-
литических систем, благодаря чему удается если не полностью,
то хотя бы частично снизить образование побочных продуктов.
Процесс олигомеризации в значительной степени определяет-
ся режимом технологического процесса. Поддержание заданного
режима позволяет снизить выход олигомерных продуктов. По-
этому в процессе синтеза полидиенов необходимо: строго выдер-
живать заданный технологический режим процесса полимериза-
ции, соотношение компонентов в каталитическом комплексе и
его дозировку в> шихту; обеспечивать интенсивное перемешива-
ние шихты с раствором каталитического комплекса для пред-
отвращения местных концентрационных перепадов и использо-
вания разбавленных растворов катализатора; строго контроли-
ровать содержание микропримесей в растворителе и мономере.
Кроме того, на выход олигомеров большое влияние оказывает
и природа применяемого растворителя [56].
20
Технологический процесс синтеза полимеров предусматрива-
•€т многократное использование углеводородных растворителей.
Отгоняемый из полимеризата углеводородный растворитель со-
держит значительные количества вредных для процесса полиме-
ризации примесей. Так, электронодонорные примеси приводят
к увеличению количества диеновых олигомеров и общему сни-
жению конверсии диенов [64], а наличие в полимеризационной
системе кислородсодержащих соединений приводит к понижению
характеристической вязкости полимера, уменьшению его пла-
стичности и увеличению количества геля. В табл. 2.1 приведены
данные о влиянии примесей на свойства цис-1,4-полиизопрена
л его ненаполненных вулканизатов. В ненаполненных вулкани-
затах на основе каучуков отмечаются повышенные напряжения
при 500%-ном удлинении, снижение прочностных показателей.
Эти зависимости наиболее отчетливо проявляются при полиме-
ризации диена на двухкомпонентной каталитической системе.
Присутствие в каталитической системе амиленов приводит к по-
вышению пластичности, снижению прочности ненаполненных
вулканизатов при 100 °C, а пипериленов — к значительному
снижению характеристической вязкости полимера. Количество
цис-1,4 звеньев в сополимере не изменялось. Олигомеризацион-
вые процессы, протекающие при стереоспецифической полимери-
зации мономеров, приводят к значительным потерям массы при
сушке. Так, на показатель потери массы при сушке цис-1,4-по-
лиизопрена [66] в значительной мере влияет содержание олиго-
меров в каучуке, из которых 10—20% составляют димеры и
50—70%—тримеры изопрена. Содержание этих олигомеров в
каучуке в значительной степени определяется природой и кон-
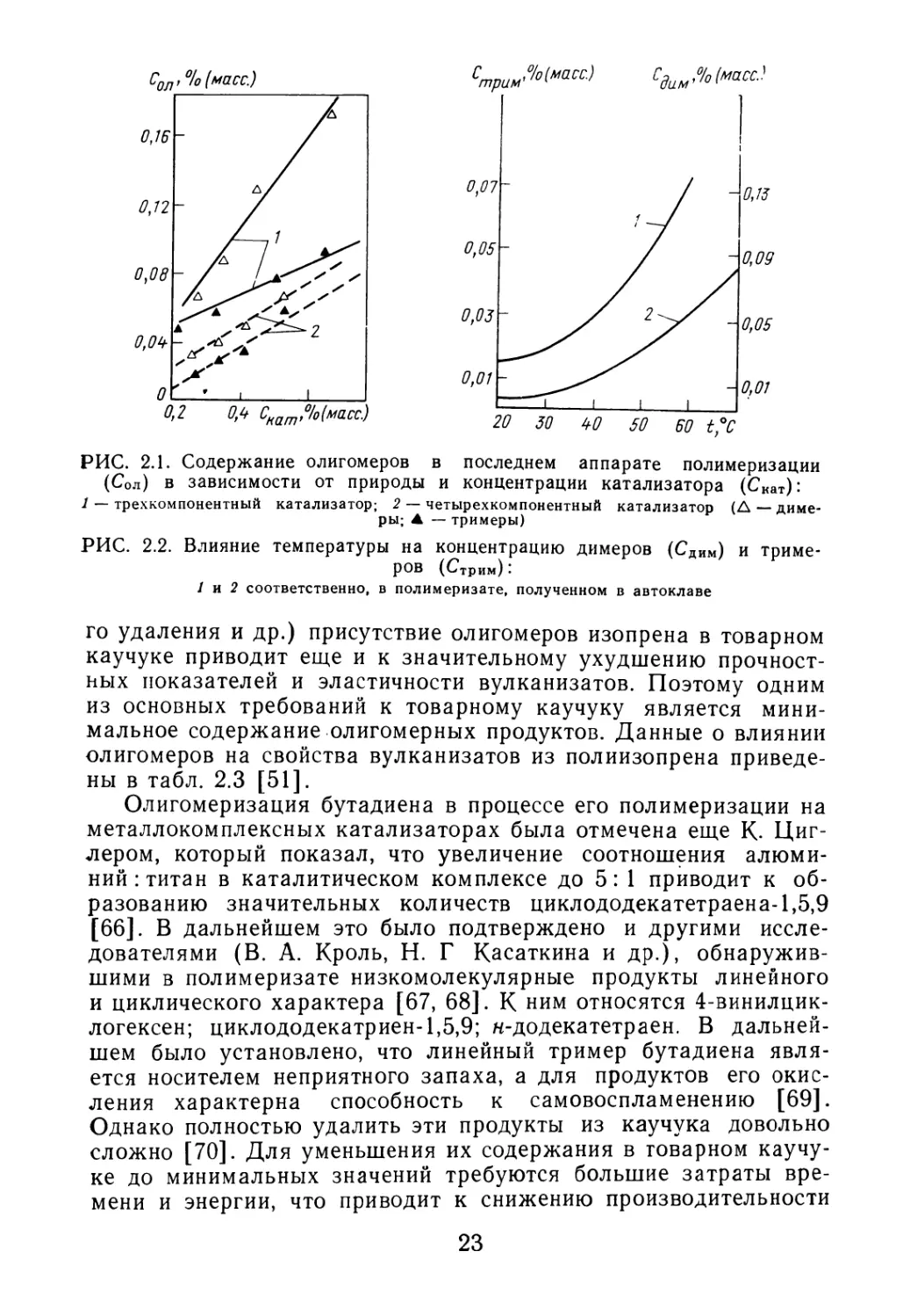
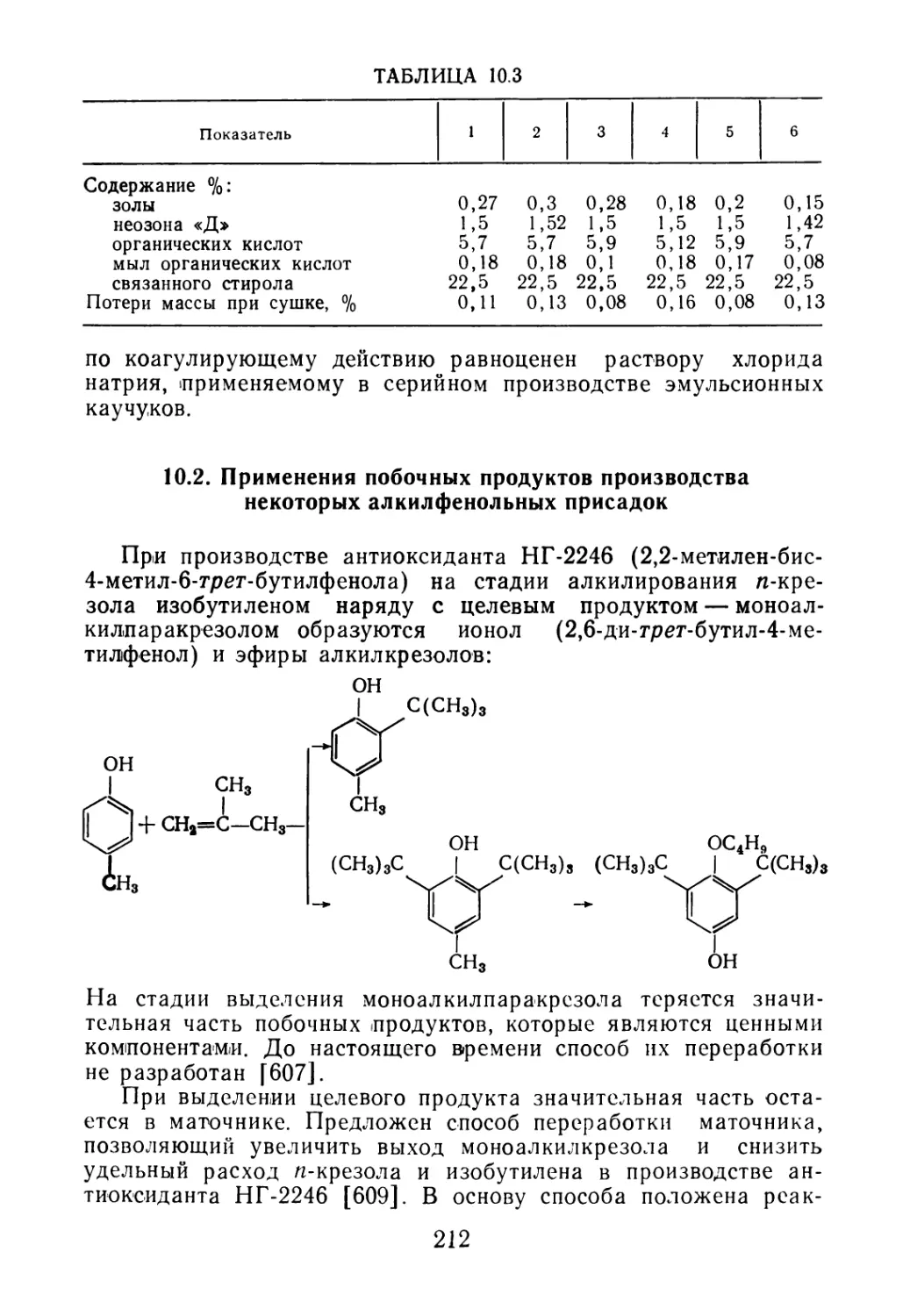
центрацией применяемого каталитического комплекса (рис. 2.1).
Использование четырехкомпонентной каталитической системы
в малых концентрациях в растворе приводит к уменьшению
олигомеризации изопрена.
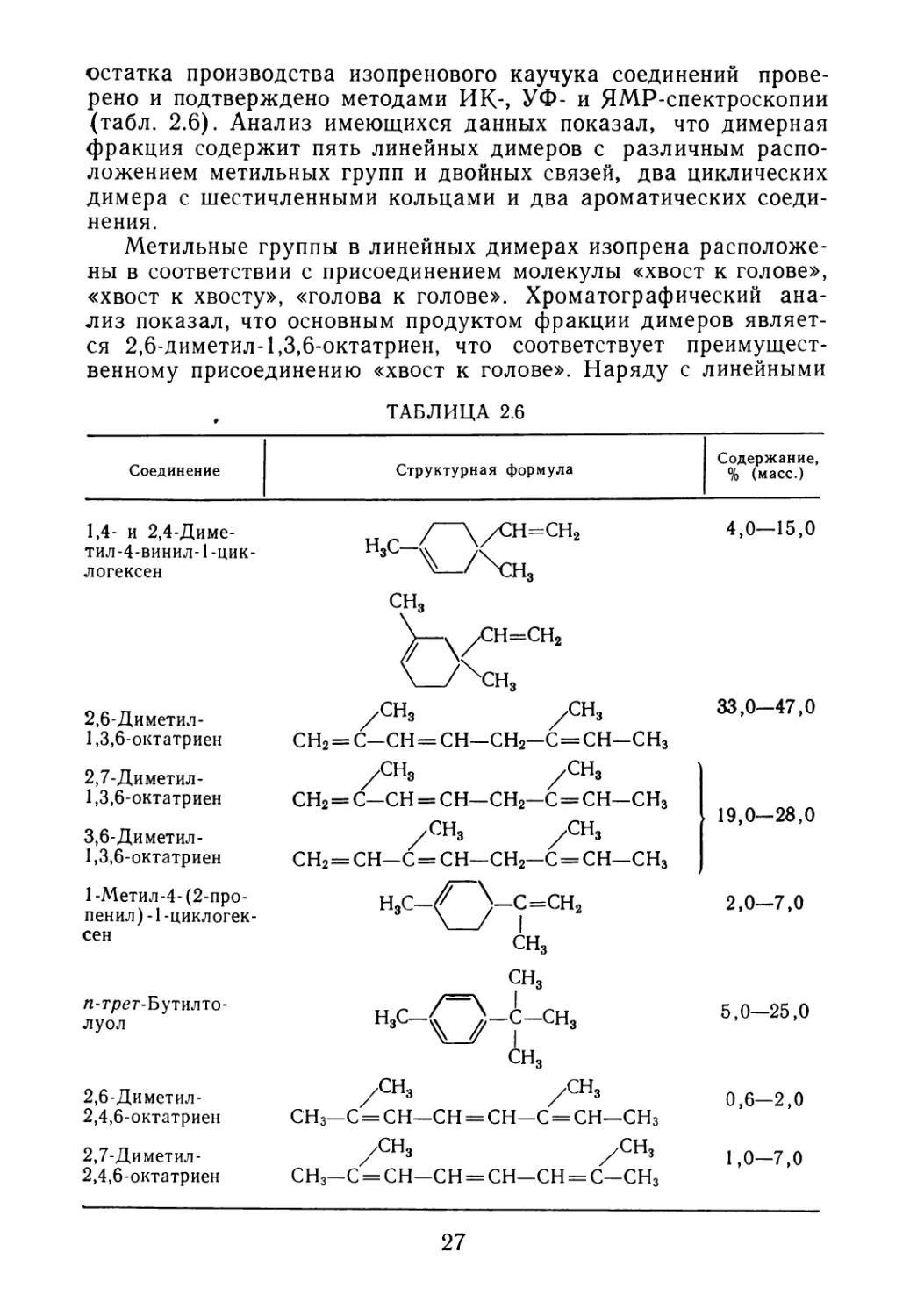
Температура процесса полимеризации также влияет на вы-
ход димеров и тримеров изопрена [65], количество которых
возрастает с повышением температуры (рис. 2.2). Увеличение
выхода олигомеров изопрена отмечается и в реальных произ-
водственных условиях. Ухудшение теплообмена из-за роста кон-
версии изопрена приводит к повышению температуры в каждом
последующем аппарате полимеризационной батареи. Сведения
о содержании олигомеров и температурных режимах в батарее
аппаратов полимеризации приведены в табл. 2.2.
Замена ароматических растворителей на алифатические при
полимеризации изопрена в растворе приводит к значительному
повышению выхода олигомеров изопрена (с 4—6 до 8 10%).
Кроме перечисленных выше недостатков (повышенный расход
мономера на тонну каучука, появление характерного запаха,
повышенный расход пара на дегазацию для их наиболее полно-
21
ТАБЛИЦА 2.1
Показатель Без при- месей Примеси
трет-бу- тил мети- ловый спирт ацетон метилакро- леин мети- лаль «-бутило- вый спирт триметил- карбинол диметил- ацетилен 2-метил- бутен-2 пиперилен цис- транс-
Двухкомпонентный катализатор Содержание примесей в — 0,02 0,015 — — 0,15 0,10 0,05 75 5,0 75 цзо-С5Н8, % (масс.) Свойства полимера: пластичность 0,20 0,14 0,20 — — 0,20 0,13 0,22 0,31 0,37 0,26 нерастворимая часть, 2,00 28,00 3,00 — — 2,0 16,0 Отсутствие 4,0 Отсутст- % (масс.) вие Условное напряжение 2,4 4,7 3,3 — — 3,2 5,8 2,4 1,7 3,1 2,4 при 500 %-ном удлине- нии, МПа Условная прочность при растяжении, МПа при 22 °C 32,2 30,9 31,2 — — 35,7 31,5 33,8 30,2 30,7 35,7 при 100°С 23,8 17,4 20,7 — — 23,1 17,0—23,0 28,8 21,0 10,9 25,2 Модифицированный четырехкомпонентный катализатор Содержание примеси в — 0,02 0,01 0,01 0,008 0,10 0,02 0,02 100 1,0 — цзо-С5Н8, % (масс.) Свойства полимера: пластичность 0,19 0,19 0,19 0,19 0,16 0,20 0,12 0,16 0,30 0,24* нерастворимая часть, 0,66 0,80 0,72 1,60 0,80 0,90 — 1,32 0,80 1,57* % (масс.) Условное напряжение 2,0 2,6 2,4 2,4 2,6 2,4 2,7 2,0 2,5 2,0* при 500%-ном удлине- нии, МПа Условная прочность при растяжении, МПа при 22°C 32,0 29,8 30,1 34,0 31,0 30,8 31,9 34,2 31,4 31,4* при 100 °C 24,9 23,1 27,0 24,2 24,3 12,9—25,4 13,1—23,0 26,7 20,7 22,2* * Сумма цис- и транс-пипериленов.
РИС. 2.1. Содержание олигомеров в последнем аппарате полимеризации
(Сол) в зависимости от природы и концентрации катализатора (Скат):
1 — трехкомпонентный катализатор; 2 — четырехкомпонентный катализатор (А — диме-
ры; ▲ — тримеры)
РИС. 2.2. Влияние температуры на концентрацию димеров (Сдим) и триме-
ров (СТрим):
1 и 2 соответственно, в полимеризате, полученном в автоклаве
го удаления и др.) присутствие олигомеров изопрена в товарном
каучуке приводит еще и к значительному ухудшению прочност-
ных показателей и эластичности вулканизатов. Поэтому одним
из основных требований к товарному каучуку является мини-
мальное содержание олигомерных продуктов. Данные о влиянии
олигомеров на свойства вулканизатов из полиизопрена приведе-
ны в табл. 2.3 [51].
Олигомеризация бутадиена в процессе его полимеризации на
металлокомплексных катализаторах была отмечена еще К. Циг-
лером, который показал, что увеличение соотношения алюми-
ний: титан в каталитическом комплексе до 5: 1 приводит к об-
разованию значительных количеств циклододекатетраена-1,5,9
[66]. В дальнейшем это было подтверждено и другими иссле-
дователями (В. А. Кроль, Н. Г Касаткина и др.), обнаружив-
шими в полимеризате низкомолекулярные продукты линейного
и циклического характера [67, 68]. К ним относятся 4-винилцик-
логексен; циклододекатриен-1,5,9; н-додекатетраен. В дальней-
шем было установлено, что линейный тример бутадиена явля-
ется носителем неприятного запаха, а для продуктов его окис-
ления характерна способность к самовоспламенению [69].
Однако полностью удалить эти продукты из каучука довольно
сложно [70]. Для уменьшения их содержания в товарном каучу-
ке до минимальных значений требуются большие затраты вре-
мени и энергии, что приводит к снижению производительности
23
ТАБЛИЦА 2.2
Показатель Аппарат полимеризационной батареи
1 " 1 111 1 IV 1 V I VI
Температура, °C 17 42 49 53 55 57
Конверсия изопре- на, % Содержание в по- лимеризате, % (масс.): 22,5 62,3 82,6 89,7 93,5 94,3
димеров 0,0045 0,0144 0,0412 0,0659 0,1210 0,1290
тримеров 0,0016 0,0055 0,0234 0,0410 0,0650 0,0606
Доля димеров от суммы образую- щихся во всех полимеризатах, % (масс.) 3,82 11,75 31,17 57,48 96,31 100,00
Доля тримеров от суммы образую- щихся во всех 2,85 10,19 31,40 61 ,67 97,08 100,00
полимеризатах,
% (масс.)
процесса в целом [70]. Влияние способов крошкообразования
на эффективность отгонки олигомеров из полибутадиена пока-
зано в табл. 2.4.
Наличие олигомеров бутадиена в полимеризате определяют
методом газожидкостной хроматографии [71], а их количество
[х, % (масс.)] рассчитывают по формуле:
х = (a- Snp)/5СТ,
где а — содержание олигомеров в стандартной смеси, % (масс.); Snp, SCT—
площади пиков олигомеров в анализируемом образце и стандартной сме-
си, мм2.
Проверка этого способа на искусственных смесях олигодие-
на с 4-винилциклогексеном и циклододекатриеном-1,5,9 в рас-
ТАБЛИЦА 2.3*
Содержание оли- гомера в поли- изопрене, % (масс.) Условная проч- ность при рас- тяжении, МПа Относительное удлинение при разрыве, % Остаточное удлинение, % Эластичность по отскоку, % при 20 °C
27,5/22,0 900/1130 6/14 71
— 23,8/18,3 930/1325 8/14 67
5 21,0/16,3 970/1210 6/12 65
10 19,5/15,5 1060/1330 6/14 62
5% масла ПН-6 26,0/21,8 1300/1150 8/12 66
* В числителе — показатель, । определенный при 20 “С, в знаменателе — при 80 °C.
2 4
творе толуола показала, что ошибка определения не превышает
11,1%, чувствительность метода по димеру составляет 0,01%,
а по тримеру — 0,002%.
На стадии извлечения бутадиенового каучука из раствора
методом водной дегазации одновременно с растворителем выде-
ляется около 98% димеров бутадиена и около 40—45% триме-
ров [69]. Общий выход олигомеров бутадиена достигает 0,3%
(масс.) на бутадиен.
Олигомеризация бутадиена в процессе его стереоспецифиче-
ской полимеризации в значительной степени определяется при-
родой галогенида титана, молярным соотношением компонентов
катализатора, концентрацией каталитического комплекса и сте-
пенью превращения бутадиена. Влияние первых двух факторов
из перечисленных выше на выход и состав тримеров бутадиена
показано в табл. 2.5 [56, 72]. Содержание линейного тримера
при отношении Al/Ti, равном 2, практически не зависит от при-
роды галогенида титана (4,5—6,5%). Увеличение концентрации
каталитического комплекса приводит к повышению относитель-
ной доли циклических тримеров в полимеризате. На выход ли-
ТАБЛИЦА 2.4
Показатель процесса дегазации Способ крошкообразования
«фильера» паровой инжектор система с использованием метода сброса давления
с обычным перегревом с увеличен- ным пере- гревом
Производительность по каучуку, % 100,0* 120,0 210,0 270,0
Удельный расход пара, т/т каучука Ввод острого пара на дегазацию, % от обще- го количества: 11,5 9,2 8,0 7,0
в систему крошкооб- разования 0,0 10,0—15,0 35,0—40,0 50,0—55,0
в секции аппарата второй ступени Степень дегазации кау- чука от олигомеров, % (масс.) : 100,0 85,0—90,0 60,0—65,0 45,0—50,0
после первой ступени 90,3/2,9** 93,0/10,7 95,6/40,2 96,8/60,0
после дегазации 97,7/17,2 99,3/24,7 100,0/53,1 100,0/72,8
* Производительность дегазационной системы по каучуку в случае использования
для крошкообразования «фильеры» принята за 100%.
** Степень дегазации каучука от олигомеров выражена дробным числом: числитель —
степень дегазации каучука от димеров бутадиена, знаменатель — степень дегазации кау-
чука от тримеров бутадиена.
25
ТАБЛИЦА 2.5
Катализатор Соотношение Al/Ti Выход на ис- ходный бу- тадиен, % Количество ли- нейного триме- ра на прореаги- ровавший бута- диен, 7о Состав тримеров, % (масс.)
Г Г ? « 7 ч о 1=( я i i i о о goods g. о сх транс-, транс-, цмс-цикло- додекатри- ен-1,5,9 н-додекатет- раен-2,4,6,10
полимера тримера
ТП44-ТИБА 2 76 5,7 6,5 0 7 93
4 78 3,0 3,1 6 14 80
6 68 0,3 0,2 0 50 50
TiBn+THBA 2 68 7,9 5,3 5 45 50
4 58 8,0 0,7 61 33 6
6 51 0,6 0,0 66 34 0
TiCl4+THBA* 2 57 34,8 4,5 21 67 12
4 12 1,7 0,0 77 23 0
* При соотношении Al/Ti = 6 отмечалось наличие следов полимера.
нейного тримера большое влияние оказывает температура про-
цесса. С повышением ее выход линейного тримера растет, а цик-
лического— изменяется мало. С увеличением конверсии бута-
диена отмечается преимущественное образование линейного
тримера. На образование циклических тримеров большое влия-
ние оказывает и природа галогенида. Так, при переходе от
Til4 к TiCl4 содержание циклических тримеров в полимеризате
возрастает.
2.2. Олигомеры изопрена
При полимеризации изопрена на каталитической системе TiCl4 —
Al (изо-С4Н9)з образуются низкомолекулярные олигомерные про-
дукты [73, 74].
Идентификация этих соединений показала, что основными
побочными продуктами являются линейные димеры изопрена:
диметил-1,3,6-октатриены с различным расположением метиль-
ных групп и циклические углеводороды (дипентен, производные
винилциклогексена и др.). Фракция димеров изопрена состав-
ляет 30—40% (масс.) от массы кубового остатка, а остальное —
растворитель, толуол и высококипящие соединения [75].
Для определения состава образующихся олигомеров изопре-
на и идентификации каждого из них исходный кубовый остаток
ректификации растворителя разделяли в две стадии. На первой
стадии на промышленной колонне выделяли фракцию, обога-
щенную этими компонентами, которые на второй стадии подвер-
гали более тонкому разделению на лабораторной колонке [75].
Каждый из выделенных олигомеров изопрена содержал не ме-
нее 95% основного вещества. Строение выделенных из кубового
26
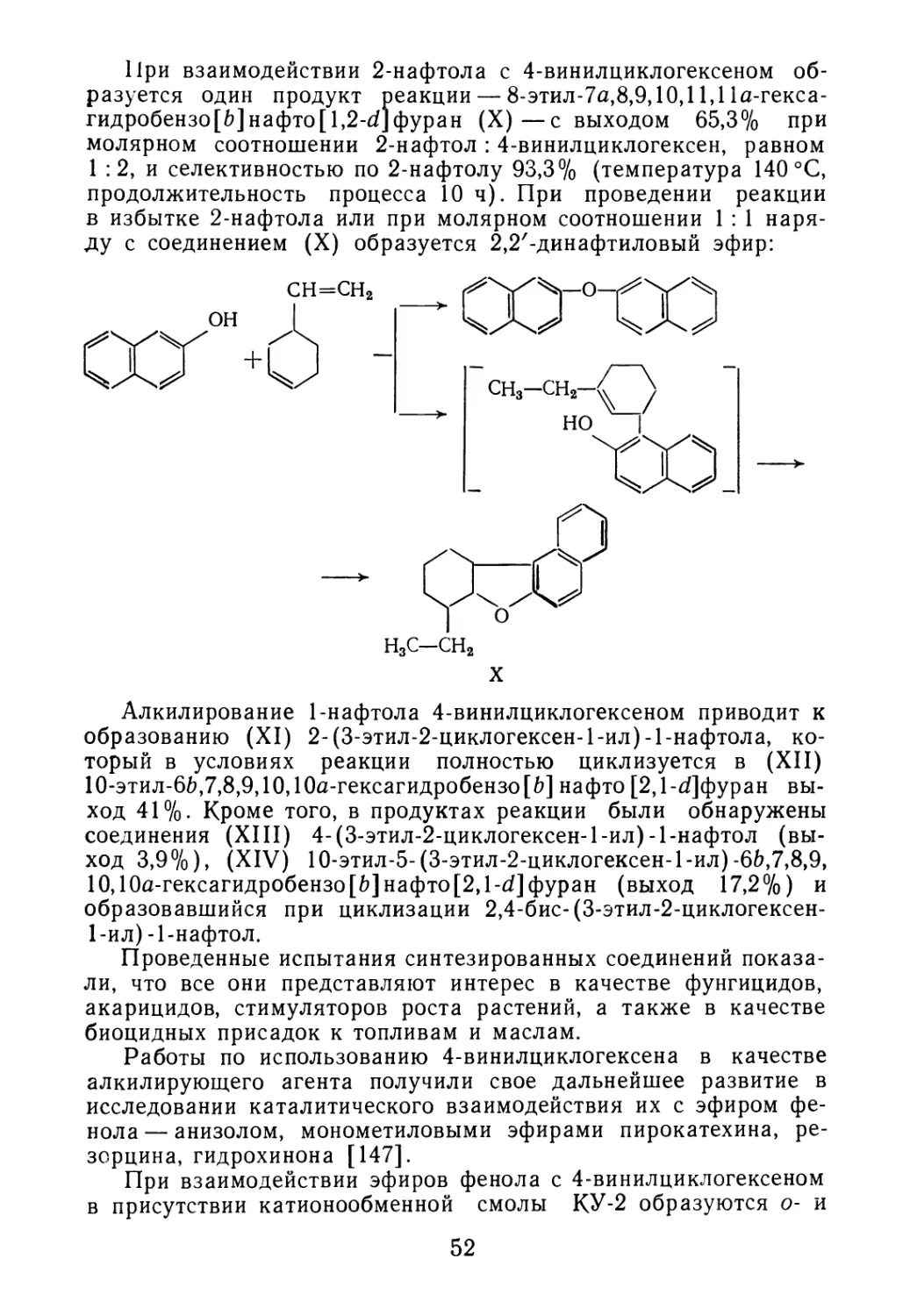
остатка производства изопренового каучука соединений прове-
рено и подтверждено методами ИК-, УФ- и ЯМР-спектроскопии
(табл. 2.6). Анализ имеющихся данных показал, что димерная
фракция содержит пять линейных димеров с различным распо-
ложением метильных групп и двойных связей, два циклических
димера с шестичленными кольцами и два ароматических соеди-
нения.
Метильные группы в линейных димерах изопрена расположе-
ны в соответствии с присоединением молекулы «хвост к голове»,
«хвост к хвосту», «голова к голове». Хроматографический ана-
лиз показал, что основным продуктом фракции димеров являет-
ся 2,6-диметил-1,3,6-октатриен, что соответствует преимущест-
венному присоединению «хвост к голове». Наряду с линейными
ТАБЛИЦА 2.6
Соединение Структурная формула Содержание, % (масс.)
1,4- и 2,4-Диме-
тил-4-винил-1 -цик-
логексен
СН3
2,6-Диметил-
1,3,6-октатриен
2,7-Диметил-
1,3,6-октатриен
3,6-Диметил-
1,3,6-октатриен
1-Метил-4-(2-про-
пенил) -1 -циклогек-
сен
и-трет-Бутилто-
луол
2,6-Диметил-
2,4,6-октатриен
2,7-Диметил-
2,4,6-октатриен
/СН3 /СН3
сн2 = С-СН = СН—СН2-С=СН-СНз
/СН3 /СН3
сн2=С-СН=СН—СН2-С=СН—СНз
/СН3 /СНз
СН2=СН-С = СН—СН2-С = СН—СНз
н,с-<^\-с=сн2
СНз
СН3
/=\ I
Н3С —с СН3
сн3
/СН3 /СНз
СНз—С = СН—СН = СН—С = СН—СНз
/СН3 /СНз
СНз—С = СН—СН = СН—СН = С—СНз
4,0—15,0
33,0—47,0
19,0—28,0
2,0—7,0
5,0—25,0
0,6—2,0
1,0—7,0
27
димерами с двумя сопряженными связями и одной изолирован-
ной в небольших количествах присутствуют линейные димеры
с тремя сопряженными связями. Такие соединения, по предполо-
жению авторов [75], являются продуктами вторичных превра-
щений диметилоктатриенов-1,3,6. Появление ароматических сое-
динении можно объяснить протеканием реакций алкилирования
толуола в процессе приготовления каталитического комплекса.
Структура одного из соединений авторами [75] не установ-
лена, хотя и было отмечено наличие ароматического ядра.
В работе [76] исследована методом ЯМР-спектроскопии
структура нескольких линейных димеров изопрена: 2,6-диметил-
1,3,6-октатриена; 2,7-диметил-1,3,6-октатриена; 3,6-диметил-1,3,6-
октатриена и 2,7-диметил-2,4,6-октатриена. Спектры !Н 3%-ных
растворов в тетрахлоруглероде и 13С 20—30% растворов в дей-
терохлороформе вышеперечисленных олигомеров снимали при
комнатной температуре на частотах 270 и 67,88 МГц соответст-
венно. Для всех линейных олигомеров изопрена определены хи-
мические сдвиги, константы спин-спинового взаимодействия и
мультиплетности сигналов ЯМР !Н и 13С.
На основании данных ПМР-спектроскопии установлено, что
димер изопрена — 2,7-диметил-2,4,6-октатриен — состоит из двух
изомеров с цис- и транс-конфигурацией центральной двойной
связи.
Каждый из исследованных димеров изопрена обладает комп-
лексом своих, ему присущих, химических сдвигов в протонных
и углеводородных спектрах.
Протонные сигналы димеров изопрена соотносили с данными
ЯМР-каталогов и исследований, опубликованных в работах
[77—79].
Олигомеризация изопрена, с одной стороны, является побоч-
ной реакцией в процессе синтеза каучука, с другой стороны —
источником ценных продуктов для народного хозяйства линей-
ных и циклических терпенов, а также более сложных производ-
ных— сесквитерпенов. В последние годы интерес к димерам и
тримерам изопрена значительно возрос. Это объясняется ^тем,
что они являются представителями одного из интереснейших
классов органических соединений — терпеноидов. В литератур-
ных источниках описан ряд перспективных синтезов димеров и
тримеров изопрена [80—86].
Классическим примером такого синтеза является реакция
димеризации изопрена в дипентен [87], который представляет
собой рацемат лимонена:
Н2С
2СН2=С—СН=СН2
сн3
н3с
^С-\ -у-СНз-
28
Лимонен один из важнейших и наиболее часто встречаю-
щихся в природе монотерпенов. Он является главной составной
частью терпеноидных фракций лимонного и апельсинового ма-
сел, масел укропа, тмина и др.
В качестве алициклического терпеноида в отходах производ-
ства СКИ присутствует аллооцимен:
Впервые термическую димеризацию изопрена осуществил
К. Бушарда в 1875 г. [75]. В дальнейшем термическая димери-
зация изопрена была подробно рассмотрена И. Н. Назаровым
с сотр. [76]. Они установили, что в этих условиях образуются
четыре шестичленных и два восьмичленных циклических диме-
ра изопрена:
По мере повышения температуры димеризации изопрена от
100 до 290 °C количество низкокипящей фракции, содержащей
смесь димеров (I) и (II), повышается с 19,2 до 42,6% (масс.),
а количество средней фракции — смесь димеров (III) и (IV) —
уменьшается с 80,8 до 57,4% (масс.).
Термическая димеризация изопрена в газовой фазе [82]
позволила получить два циклических димера изопрена — 2,4-ди-
метил-4-В1ИНилциклогексен-1 и дипентен. На ИК-спектрах синте-
зированных димеров были обнаружены полосы поглощения
двойной связи циклогексенового кольца в области 1629 см-1, ко-
лебания циклогексенового кольца в области 1135 и 1095 см-1,
деформационные колебания групп —СНз, —СН = СН2 и изопро-
пенильной в областях (см-1) 1460, 1300, 1380 соответственно.
Присутствие этих монотерпеноидов в отходах производства
СКИ подтверждено исследованиями [73—76]. В табл. 2.7 при-
ведены основные свойства выделенных и охарактеризованных
олигомеров изопрена из кубового остатка стадии очистки возв-
ратного растворителя производства СКИ.
29
ТАБЛИЦА 2.7
Димер Температура кипения, °C Давление, кПа Показатель преломления Плотность при 20 °C, г/см3
1,4-Димстил-4-винилцик- 44 1,2
логсксен-1 160—161 101,3 1,4658 0,8329—0,5331
2,4-Димстил-4-винилцик- логексен-1 48 1,3 1,4630 —
1 -Метил-4-изопропенил- 58 1,3 — —
циклогексен-1 174—175 101,3 1 ,4743 0,8454
2,6-Диметилоктатри- ен-1,3,6 57—58 1,6 1,4794 0,7966
2,7-Диметилоктатри- ен-1,3,6 54 1,3 1,4786 —
п-трет-Бутилтолуол 112-113 101,3 — 0,8614
94 3,3 1,4935 —
Экспериментальным путем подобран оптимальный режим
синтеза димеров изопрена: температура 520—530 °C и объемная
скорость 150—200 ч-1 (рис. 2.3 и 2.4). Выход димеров изопрена
в этих условиях на поданное сырье составляет 35—40%
(рис. 2.3), а на вступившее в реакцию — 75—80% (рис. 2.4).
2.3. Олигомеры бутадиена
При получении бутадиенового каучука образуются побочные
продукты, которые впервые наиболее подробно были описаны
Т. Вильке [88]. Димеризация бутадиена в присутствии катали-
РИС. 2.3. Выход димеров изопрена (В) на поданный изопрен в зависимости
от температуры и объемной скорости (v)
РИС. 2.4. Выход димеров изопре-
на (В) на изопрен, вступивший в
реакцию в зависимости от темпе-
ратуры и объемной скорости (v)
30
заторов Циглера — Натта протекает по следующим основным
направлениям: взаимодействие по типу 1,4-1,2- или 1,4-1,4-при-
соединения или по реакции диенилирования с миграцией водо-
рода [89]. В качестве побочных продуктов образуются винил-
циклогексен, циклооктадиен-1,5, октатриен-1,3,6, метилгепта-
триен-1,4,6, а также циклические и линейные тримеры—цикло-
додекатриен-1,5,9, н-додекатетраен [2, 55]:
^2^—СН=СНа
/\/\---------> ZX/4/4 •
Все вышеперечисленные соединения были выделены из ку-
бового остатка ректификации возвратного растворителя произ-
водства СКД [90, 91]. При этом ряд диеновых олигомеров имел
по несколько цис- и транс-изомеров, которые различались по
своим свойствам. Свойства основных побочных продуктов, выде-
ленных из кубового остатка ректификации растворителя, приве-
дены в табл. 2.8 [56].
Качественный и количественный анализы олигомеров бута-
диена показали, что фракция димеров бутадиена составляет
50—70% от общей массы олигомеров, а содержание н-додека-
тетраена достигает 50—70% от суммы тримеров [56]. Строение
всех вышеприведенных побочных продуктов синтеза СКД было-
доказано при помощи спектральных исследований.
ТАБЛИЦА 2.8
Продукт Температура плавления, °C Температура кипения, °C Давление, кПа Показатель преломления
4-Винилциклогексен* Циклооктадиен-1,5: — 129,5—130,5 101,3 —
цис-форма —70,1 150,8 100,6 —
транс-форма — 62 — — —
транс-, транс-, трансЛ1ркло- 34 96 1 ,3 1,5005
додекатриен-1,5,9 — 241—244 101,3 —
транс-, транс-, цис-Цикло- додекатриен-1,5,9 — 16,8 98 1,3 1,5078
транс-, цис-, цис-Циклодо- декатриен-1,5,9 —84—9 106 1,3 1,5129
н-Додекатетраен-2,4,6,10 43,5—44 * Плотность 4-винилциклогексена составляет 82 0,832 г/см3. 0,3 1,5897
31
Значительные количества винилциклогексена в качестве по-
бочного продукта образуются и в ряде других органических и
нефтехимических синтезов с участием бутадиена, например в
производстве этилиденнорборнена, являющегося третьим моно-
мером тройных этиленпропиленовых каучуков [52, 54]. Синтез
этилиденнорборнена осуществляют в две стадии. На первой ста-
дии получают винилнорборнен из бутадиена и циклопентадиена:
На этой стадии наряду с синтезом винилнорборнена протекает
и ряд побочных реакций, одной из которых является димериза-
ция бутадиена с образованием винилциклогексена:
сн=сн2
V_/+=C-C= ------->
Вторая стадия процесса — каталитическая изомеризация ви-
нилнорборнена в этилиденнорборнен [54].
Другим синтезом, где в значительных количествах образует-
ся винилциклогексен, является получение 4-метилциклогексена
[92]. Экспериментально установлено, что оптимальными усло-
виями проведения процесса являются атмосферное давление,
температура 550 °C, объемная скорость 45 ч-1 и объемное соот-
ношение С3 С4 = 4 1. В этих условиях выход 4-метилциклогек-
сена составляет 38,85%, а винилциклогексена—14,45%. Варьи-
руя соотношением Сз: С4, можно влиять на состав продуктов ре-
акции. При изменении соотношения Сз: С4 от 1:1 до 5: 1 кон-
центрация 4-метилциклогексена повышается с 15,8 до 45,95%,
а винилциклогексена уменьшается с 21 до 13,1%.
Винилциклогексен с высоким выходом получают из бутадие-
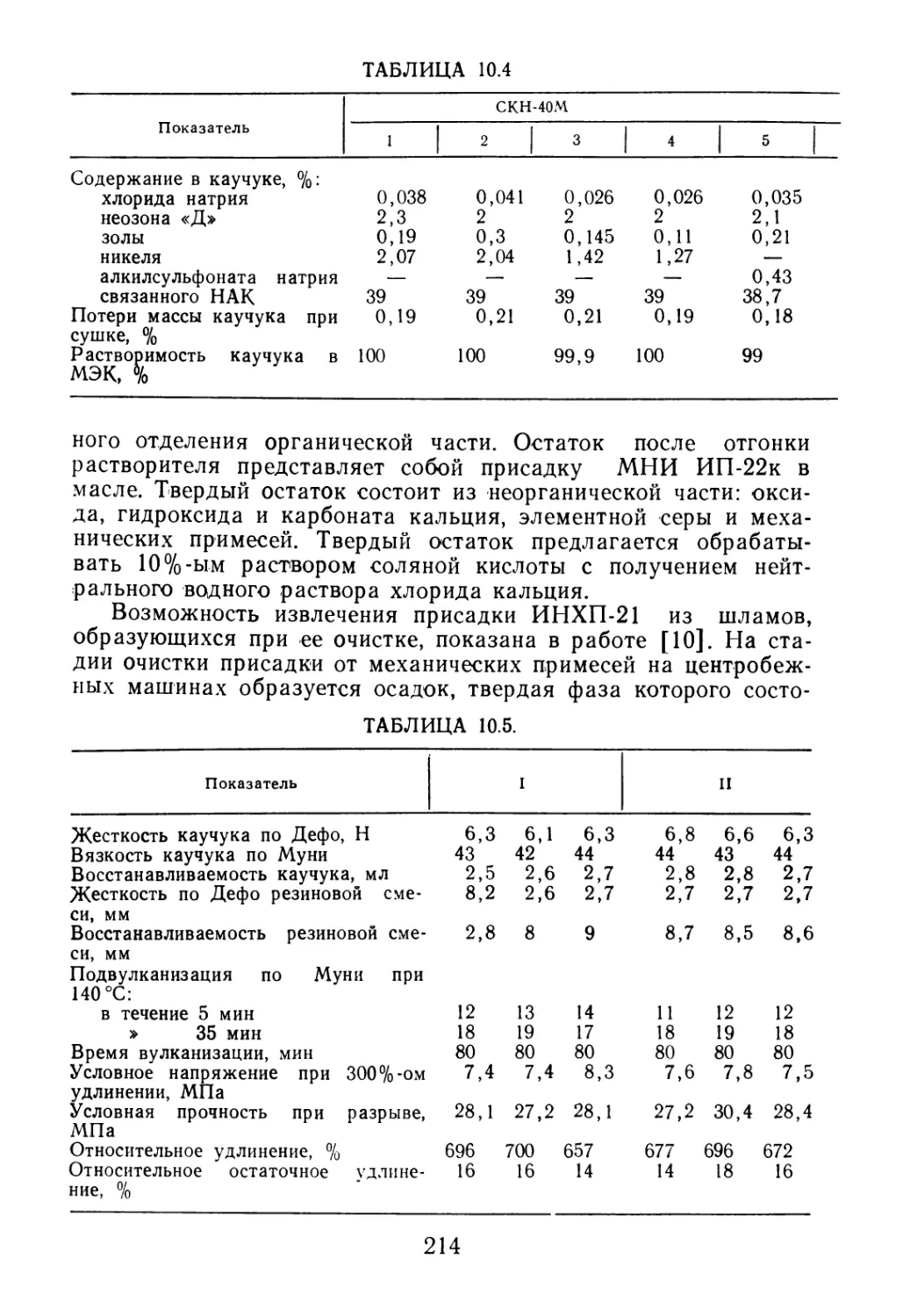
на в газовой фазе (рис. 2.5) [82]. Бутадиен концентрацией 97—
'98% под давлением азота из мерника через ротаметр 1 и испа-
ритель 2 подается в реактор, обогреваемый печью 3. Продукты
синтеза из реактора 3 подаются в куб колонны 4 для отделения
непрореагировавшего бутадиена, который из верхней части ко-
лонны 4 поступает в конденсатор 5. Продукт димеризации бута-
диена из кубовой части колонны 4 направляется на ректифика-
цию в колонну 6. С верхней части колонны 6 отбирают винил-
циклогексен-сырец концентрацией 80—85%. Выход 4-винилцик-
логексена на разложенное сырье, вступившее в реакцию (опти-
мальные условия синтеза), составляет 77% (рис. 2.6), а на про-
пущенное сырье — 47% (температура 440°C, объемная скорость
55 ч-1) (рис. 2.7).
32
Известен ряд способов получения винилциклогексена, где
термическая димеризация бутадиена осуществляется в присут-
ствии различных каталитических систем [93—96]. Примером
могут служить никельсодержащие каталитические системы [56,
95, 96], выход винилциклогексена при их использовании состав-
лял не более 90%.
Для получения тримеров бутадиена широко используются со-
единения титана [97—101], обеспечивающие выход циклододе-
катриена-1,5,9 более 80%. Находят применение и каталитические
системы, содержащие высшие алюминийорганические соедине-
ния, и комплексы никеля с формилкамфорой, оксихинолином,
биссалицилальэтилендиамином [ 102]. Циклотримеризация на
этих каталитических системах протекает селективно с образова-
нием в основном циклододекатриена-1,5,9. Выход полимеров и
низших олигомеров бутадиена невысок.
В Японии разработан способ получения циклододекатриена-
1,5,9 и циклооктадиена из бутадиена в неполярном растворителе
при температуре 0—100 °C в присутствии никельорганического
катализатора. Процесс проводили периодически под давлением
до 1 МПа. Изменение соотношения компонентов катализатора
влияло на состав продуктов реакции. По данным работы пилот-
ной установки на получение 1 т олигомеров бутадиена необхо-
димо: бутадиена— 1,15 т, водяного пара — 6,4 т, воды — 33 м3/т,
электроэнергии—180 кВт-ч [56].
В процессе полимеризации образование диеновых олигоме-
ров нежелательно, а целенаправленный синтез димеров и три-
меров бутадиена — очень перспективен. Это объясняется прак-
1 — ротаметр; 2— испаритель; 3 — реактор с печью; 4 — отгонная колонна; 5 конденса-
тор; 6 — ректификационная колонна
3-627
33
РИС. 2.6. Выход винилциклогексена (В) на бутадиен, вступивший в реакцию,
в зависимости от температуры и объемной скорости (у)
РИС. 2.7. Выход винилциклогексена (В) на поданный бутадиен в зависимо-
сти от температуры и объемной скорости (у)
тическим применением диеновых олигомеров в качестве исход-
ного сырья в производстве новых видов полиамидных волокон,
пластмасс, пластификаторов. Например, олигомер бутадиена
циклододекатриен-1,5,9 является исходным сырьем для получе-
ния ценных продуктов: 1,10-декандикарбоновой кислоты и
(о-додекалактама, гексабромциклододекатриена и др.
В настоящее время разрабатываются промышленные спосо-
бы получения из диеновых углеводородов соответствующих ди-
меров и тримеров.
2.4. 4-Цианоциклогексен — побочный продукт производства
бутадиеннитрильных каучуков
Бутадиеннитрильные каучуки практически из всех выпускаемых
спецкаучуков являются самыми распространенными. История их
производства насчитывает около 60 лет. Производственные
мощности бутадиеннитрильных каучуков возросли с 1950 по
1980 г. более чем в 5 раз [103]. Одним из основных способов
получения этих каучуков является эмульсионная сополимериза-
ция бутадиена с акрилонитрилом [51, 52, 103, 104]. В производ-
стве бутадиеннитрильных каучуков в качестве побочных про-
дуктов образуются соединения, представляющие собой аддукты
диеновой конденсации бутадиена-1,3 с акрилонитрилом (мета-
криловой кислоты) —4-цианоциклогексен (1-метилциклогексен-
3-карбоновая кислота). Количество этих продуктов значительно
и колеблется в зависимости от применяемой рецептуры от 2 до
10% (масс.) каучука. До настоящего времени они еще не нашли
своего применения и сжигаются в специальных установках или
применяются как добавки к моторному топливу. Это приводит
к потере ценного органического сырья и загрязнению окружаю-
щей среды.
34
В чистом виде отход производства бутадиеннитрильных кау-
чуков — 4-цианоциклогексен-З представляет собой прозрачную
бесцветную, легкоподвижную жидкость с характерным запахом,
напоминающим запах горького миндаля. Хорошо растворяется
в органических растворителях (кетонах, спиртах, ароматических
углеводородах и др.). В обычных условиях растворимость 4-циа-
ноциклогексена в воде невысока и составляет менее 0,5 г на
100 г воды. Основные физические свойства 4-цианоциклогексена:
Температура кипения, °C: при 101,3 кПа 192 [105]
при 0,7 кПа 59,5—60,5 [106]
при 2,9 кПа 92,5 [107]
Плотность при 20 °C, г/см3 0,9510 [108]
0,9540 [107]
Показатель преломления при 1,4744 [Ю8]
20 ”С 1,4738 [Ю7]
Дипольный момент, D 3,41 [Ю9]
В литературе имеются сведения о целенаправленных синте-
зах 4-цианоциклогексена, основным из которых является реак-
ция Дильса— Альдера [ПО]:
^СИ-СН CN
сн2 сн2 >
СН2=СН—CN
Эта реакция лежит в основе большинства промышленных спосо-
бов получения циклоолефинов с функциональными группами
[111—115].
Реакционная способность двойной связи в 4-цианоциклогек-
сене определяется в основном прочностью циклогексенового
кольца и не зависит от положения заместителя (аксиального
или экваториального). 4-Цианоциклогексен можно рассматри-
вать как бифункциональное соединение, т. е. с двумя изолиро-
ванными функциональными группами, поэтому целесообразно
сгруппировать реакции, типичные для этого соединения: по нит-
рильной группе, по двойной связи циклогексенового кольца.
2.5. Снижение концентраций олигомеров в каучуке
На основании данных, рассмотренных выше, можно сделать
вывод о том, что в процессе синтеза каучука одновременно про-
текают две реакции: стереоспецифическая полимеризация моно-
мера с получением высокомолекулярного полимера и олигоме-
ризация диеновых мономеров по катионному механизму с обра-
зованием низкомолекулярных производных диенов [116].
3* 35
Снижения содержания олигомеров в товарном каучуке мож-
но достичь разными способами. Наиболее интересным и перспек-
тивным из них является связывание олигомеров на завершаю-
щей стадии процесса полимеризации диенового углеводорода.
С этой целью [П7] в полимеризат на завершающей стадии до-
полнительно вводятся винилароматический мономер — стирол и
катализатор литийорганические соединения (дилитийполиди-
винил, н-бутиллитий, втор-бутиллитий). Ввод этих компонентов
в полимеризат СКД осуществляется в количествах (%): сти-
рол К бутиллитий— 0,06 при остаточном содержании бутадие-
на в полимеризате 10—15%. Процесс протекает при 55—60 °C
в течение 1,5 ч [117]. При таких условиях я-додекатетраен со-
полимеризуется с бутадиеном и стиролом на 100%, а циклооли-
гомеры— на 14—22%. Влияние условий обработки полимеризата
на содержание в нем я-додекатетраена показано в табл. 2.9.
Для получения каучука с конечной вязкостью 40—50 ед. по
Муни следует уменьшить на 6—10% количество добавляемого
титанового каталитического комплекса. Это приводит к получе-
нию каучука с повышенной молекулярной массой, но введение
на завершающей стадии стирола и бутиллития позволяет сба-
лансировать значение молекулярной массы и получить каучук,
имеющий такую же среднюю величину молекулярной массы, как
и у контрольного.
Известно [10, 104], что для промышленного получения кау-
чука СКД в качестве титанового компонента используется ди-
хлордииодтитан. Поэтому снижение подачи титанового катали-
затора до 10% позволяет уменьшать расход дорогого и дефицит-
ного иода, идущего на приготовление дихлордииодтитана.
Основные свойства резиновых смесей и вулканизатов на ос-
нове опытного каучука мало отличаются от серийного СКД, а
по некоторым показателям даже превосходят их (табл. 2.10).
Анализ составов побочных продуктов синтеза диеновых угле-
водородов, винилароматических, а также низкомолекулярных
продуктов, образующихся в процессе синтеза диеновых каучу-
ТАБЛИЦА 2.9
Номер опыта Введено в полиме- ризат, % (масс.) Содержание остаточного бутадиена в полиме- ризате, '% (масс.) Содержание к-додекатет- раена в полимеризате, % (масс.) Количество связанного н-додекатет- раена, % (масс.)
стирол бутиллитий до прогрева после про- грева
1 0 0 10—15 0,035 0,115 0
2 0 0,06 0 0,036 0,017 52,7
3 4 0 1,0 0,06 0,06 10—15 0 0,035 0,055 0,011 0,005 68,5 90,9
5 1,0 0,06 10—15 0,047 Следы 100,0
36
ТАБЛИЦА 2.10
Показатель Контрольный каучук Опытный каучук
Резиновые смеси
Пластичность 0,42 0,44
Эластическое восстановление, мм 1 ,о 0,9
Вязкость по Муни МБ 1+4 (100 °C) 50 52
Когезионная прочность, МПа 0,18 0,18
Вулканизаты
Условное напряжение при 300%-ном удлинении, МПа 4,2 4,4
Условная прочность при растяжении, МПа 14,5 16,0
Относительное удлинение при разрыве, % 736 720
Относительное остаточное удлинение после раз- 16 15
рыва, % Твердость по ТМ-2 43 44
Эластичность по отскоку, % 43 43
Коэффициент теплового старения (сут при 100 °C): по прочности при растяжении 0,77 0,85
по относительному удлинению при разрыве 0,75 0,84
Озоностойкость [концентрация озона 0,000056% (об.)]: время до появления трещин, мин 75 76
коэффициент озоностойкости по прочности 0,72 0,74
при растяжении Теплообразование по Гудричу, °C 81 82
Коэффициент морозостойкости при —45 °C 0,48 0,46
Усталостная выносливость резин при многократ- 260 360
ном растяжении, тыс. циклов
ков, показывает, что все они содержат в своем составе ряд цен-
ных органических соединений и могут найти применение как
для получения полимерных материалов, так и в органическом и
нефтехимическом синтезах. Основная сложность при использова-
нии этих отходов заключается в их большой неоднородности
широких колебаниях как по качественному, так и по количест-
венному составу. Кроме того, в состав побочных продуктов вхо-
дит ряд соединений, строение и содержание которых в кубовых
остатках еще до конца не выяснено, что также усложняет поиск
путей наиболее эффективной переработки данных отходов.
ГЛАВА 3
Использование диеновых олигомеров
в органическом синтезе
ЗЛ. Синтез винил ароматических углеводородов из димера
бутадиена
Одним из основных промышленных способов получения стирола
является дегидрирование этилбензола. В работе [118] этилбен-
зол получают парофазным каталитическим дегидрированием ви-
нилциклогексена (димера бутадиена). Реакцию проводят при
100—550 °C на катализаторе AI2O3, содержащем оксиды щелоч-
ных металлов (чаще всего Na или/и К) в количестве 0,1—30%.
Катализатор готовят пропиткой оксида алюминия раствора-
ми гидроксидов щелочных металлов (Na или К) с последующим
прокаливанием при 450—750 °C.
На основании данных газожидкостной хроматографии можно
сделать вывод, что на состав конечных продуктов синтеза боль-
шое влияние оказывает температура процесса (табл. 3.1). По-
вышение температуры процесса приводит к снижению выхода
этилбензола при одновременном увеличении выхода стирола и
некоторых низкокипящих продуктов. Таким образом, высокие
температуры способствуют протеканию процесса дегидрирования
этилбензола и 4-винилциклогексена в стирол. Однако кроме
основной реакции дегидрирования возможен и ряд побочных
реакций, о чем свидетельствует повышенное содержание низко-
кипящих веществ в продуктах синтеза, образование которых
можно объяснить протеканием деструкционных процессов при
высоких температурах:
сн2—сн3 сн=сн2
В работе [119] предложено получать стирол путем окисли-
тельного дегидрирования 4-винилциклогексена в газовой фазе в
стационарном или псевдоожиженном слое катализатора. В каче-
стве катализатора рекомендуется использовать силикагель, ок-
сид алюминия, неорганические силикаты и т. п., модифициро-
38
ТАБЛИЦА 3.1
Температура, °C Селективность, %
по этилбензолу по стиролу по низкокипящим веществам
100 100
130 100 — —
300 99,9 0 0
350 99,1 0 0,2
400 97,4 0 1,6
453 88,1 3,7 8,1
500 61,7 20,6 15,0
550 10,2 27,0 36,0
Примечание. Процесс дегидрирования проводили на А12Оз, содержащем 12% К,
скорость подачи 4-винилциклогексена — 3 ч-1, конверсия 4-винилциклогексена — 100%.
ванные добавками Sn, Sb, Fe, Zn, Си, Co, Те в различных ком-
бинациях при г-атомных соотношениях:
Sn Sb
Sn Sb: Те
Sn Sb Fe
Sn Sb (Zn-f-Cu+Co)
1:0,01—10,0
1:0,01—10,0:0,001—0,5
1:0,01—10,0:0,001—5,0
1:0,01—10,0:0,01—0,5
Процесс проводят при температуре 250—600 °C, атмосферном
давлении, объемной скорости подачи реагентов в реактор 100—
10 000 ч-1 и молярном соотношении кислород: 4-винилциклогек-
сен, равном 0,7: 10, в присутствии инертных разбавителей: азо-
та, водяного пара. При скорости газового потока 1240 ч-1 и тем-
пературе 400 °C конверсия 4-винилциклогексена составляет
67,1%, селективность по стиролу достигает 67,9%; по этилбен-
золу—4,1%; по бензолу—0,3%.
Таким образом, 4-винилциклогексен в качестве исходного
сырья может применяться для синтеза промышленно важного
мономера — стирола.
3.2. Эпоксидирование олигомеров бутадиена
Эпоксидные соединения, начиная со времени их открытия, при-
влекают к себе пристальное внимание химиков.
Самым простым эпоксидным соединением является оксид
этилена, который можно рассматривать как ангидрид этиленгли-
коля. Циклическая формула оксида этилена подтверждена изу-
чением ее электронограмм [120].
Исследованием ИК-спектров поглощения монозамещенных
а-оксидов жирного ряда установлено существование двух макси-
39
ТАБЛИЦА 3.2
Гидропероксид Конверсия гидроперокси- да, % Выход оксида на прореаги- ровавший гидропероксид, % (мол.)
Изопропилбензола 92,0 99,8
трет-Пентила 94,4 99,8
трет-Бутила 89,9 99,7
мумов поглощения, характерных для оксидного кольца при 917
и 840 см-1. Дальнейшие исследования в этом направлении по-
казали наличие у оксида этилена характеристической полосы
в области 1250 см-1 и двух полос в области 770—950 см-1, ко-
торые определяются строением оксидной молекулы в целом.
Из литературных источников известен ряд способов получе-
ния эпоксидных соединений: из этиленхлоргидрина, высших гли-
колей, дигалоидных производных, окислением непредельных
соединений. Наибольший практический интерес представляет
каталитическое окисление олефинов, так как в отличие от хлор-
гидринного метода для него не требуется затрат хлора и
щелочи.
Для снижения температуры окисления олефинов рекомендо-
вано применять различные катализаторы, представляющие со-
бой металлы (медь, серебро, платина, осьмий, оксид хрома
и др.) или их оксиды.
Эпоксидирование 4-винилциклогексена. В качестве эпоксиди-
рующих агентов широкое применение находят гидропероксиды.
Эпоксидирование 4-винилциклогексена органическими гидропе-
роксидами можно представить в виде уравнения:
С=С( +ROOH
В работах [121, 122] для эпоксидирования 4-винилциклогек-
сена использовали гидропероксиды изопропилбензола, трет-бу-
тила, трет-пентила. Эпоксидирование с высокой скоростью и се-
лективностью протекало в присутствии молибденового катализа-
тора при 90—95 °C, исходном соотношении 4-винилциклогексен
гидропероксид, равном 3:1, концентрации катализатора — мо-
либденовой кислоты—1% (масс.) в течение 2 ч (табл. 3.2).
Из эпоксидов, полученных окислением 4-винилциклогексена
гидропероксидом изопропилбензола, выделить образующийся
оксид винилциклогексена с содержанием основного вещества
более 92% оказалось практически невозможным. Это связано
с образованием в процессе синтеза а-метилстирола, который
трудно отделить от оксида винилциклогексена ректификацией.
40
Применение в качестве эпоксидирующих агентов гидроперокси-
дов трет-бутила и трет-пентила позволило получить оксид ви-
нилциклогексена с чистотой 95—98%.
Исследования механизма эпоксидирования 4-винилциклогек-
сена органическими гидропероксидами показали, что процесс
протекает в основном по двойной связи циклогексенового коль-
ца [121, 123]. Однако при этом было отмечено образование не-
которого количества оксида винилциклогексена и по двойной
связи винильной группы [122]:
1,2-Эпокси-4-винилциклогексан
Q-ch-ch2
1,2-Эпокси-4-этилциклогексен-1.
Добавками небольшого количества воды можно ингибиро-
вать процесс окисления винильной группы и получать только
1,2-эпокси-4-винилциклогексан. Содержание 4-(1,2-эпокси)-этил-
циклогексена-1 в продуктах реакции может изменяться в зави-
симости от применяемого катализатора от 0 до 12%. Наиболее
активными катализаторами являются резинат и гликолят мо-
либдена.
Использование для эпоксидирования 4-винилциклогексена
пероксобензимидокислоты приводит к получению реакционной
смеси, содержащей примерно равное количество обоих изоме-
ров [124]. Оба изомера выделены из смеси, и их строение ис-
следовано спектральными методами.
Изомерные эпоксиды 4-винилциклогексена, синтезированные
разными авторами, имели следующие физико-химические кон-
станты [121 —125]: 1,2-эпокси-4-винилциклогексан: /КИп = 38 °C/
/0,67 кПа; 171,9°С/101,31 кПа; nD20= 1,4701; d420 = 0,9606; эпок-
сидное число, %: получено 12,19; вычислено 12,90; MRd полу-
чено 36,028; вычислено 36,172 [122] и по данным [125] /Кип =
= 43,5—44,5 °С/0,93 кПа; nD2Q = 1,4682; d420 = 0,9 5 74.
1,2-Эпокси-4-этилциклогексен-1: /КИп = 39—40 °С/0,67 кПа;
181,3 °С/101,31 кПа; Ио20 = 1,47 83; d420 = 0,9772; эпоксидное чис-
ло, %: вычислено 12,90; получено 12,90; MRD получено 35,940;
вычислено 36,147 [122].
Дальнейшее исследование эпоксидирования 4-винилциклогек-
сена в присутствии молибденовой кислоты нашло свое отраже-
ние в работе [126], в которой был определен порядок реакции
по катализатору, равный 0,66, в интервале концентраций молиб-
деновой кислоты 0,0003—0,003 моль/л.
41
Порядок реакции по гидропероксиду трет-пентила и 4-винил-
циклогексену определяли на основании экспериментальных дан-
ных дифференциальным методом по уравнению
. / dA \ ( и \
М -~Г- =1й/ед+л I 1g Д-J-1g В ,
где кд константа скорости реакции; А и В — концентрации гидропероксида
и олефина; х и у — порядки реакции по гидропероксиду и олефину.
В данном случае реакция эпоксидирования имеет первый
порядок по гидропероксиду трет-пентила и нулевой —по 4-ви-
нилциклогексену, т. е. при постоянной концентрации катализа-
тора общий порядок реакции равен единице. Однако в некоторых
случаях при большой конверсии скорость реакции уменьшается
и реакция отклоняется от кинетического уравнения первого
порядка. Так протекает эпоксидирование 4-винилциклогек-
сена гидропероксидом трет-пентила. Логарифмическая зависи-
мость концентрации последнего от времени (молярное соотно-
шение гидропероксид трет-пентила : 4-винилциклогексен= 1 :2—
20) представляет собой ломаную линию с двумя прямолинейны-
ми участками. Константы скорости определены по наклону этих
прямых (fei, й2), они приведены в табл. 3.3.
При этом константа скорости первого участка более чем в
два раза превышает константу скорости второго участка. Такой
характер зависимости дает основание предположить, что в ходе
реакции протекает ступенчатая дезактивация катализатора.
Температура процесса оказывает заметное влияние на ско-
рость эпоксидирования 4-винилциклогексена. Повышение ее
с 60 до 90 °C приводит к значительному уменьшению индукцион-
ного периода, при 90 °C он практически полностью отсутствует.
На основании экспериментальных данных были рассчитаны кон-
станты скорости реакции эпоксидирования 4-винилциклогексена
гйдропероксидом трет-пентила при различных температурах
(табл. 3.4).
На основании вышеприведенных данных можно сделать вы-
вод, что эпоксидирование 4-винилциклогексена протекает с вы-
сокими скоростью и выходом целевых продуктов.
Монооксид винилциклогексена также может быть получен
эпоксидированием 4-винилциклогексена эквимолекулярным коли-
ТАБЛИЦА 3.3
Константа скорости, мин-1 Концентрация гидропероксида трет-пентила, моль/л
2,94 | 1,78 1 ».29 | 1,00 | 0,43
ki 0,0080 0,0080 0,0080 0,0085 0,0100
ki 0,0040 0,0025 0,0035 0,0030 0,0035
Примечание. Концентрация молибденовой кислоты ра — 80 °C. — 0,003 моль/л, температу-
42
ТАБЛИЦА 3.4
Константа скорости, мин-1 Температура, °C Энергия ак- тивации, кДж/моль
60 | 70 1 80 | 90
/г 2 0,0032 0,0012 0,0054 0,0020 0,0082 0,0037 0,0160 0,0090 62,7 71,2
Примечание. Концентрация молибденовой кислоты 0,003 моль/л.
чеством хлорноватистой кислоты, используемой в виде водного
раствора. Образующееся моноэпоксидное соединение
О>
у-сн -сн2
экстрагировали эфиром и выделяли фракцию с т. кип. 77—
85 °С/4,40—4,80 кПа [127].
Хлорноватистая кислота в количестве 2 моль при взаимодей-
ствии с 1 моль 4-винилциклогексена образует диоксид винил-
циклогексена
который представляет собой масло желтого цвета с т. кип. 76—
80 °С/0,20—0,27 кПа.
Эти эпоксидные соединения могут быть использованы в ка-
честве исходных продуктов для получения полимерных материа-
лов. Диэпоксидные соединения применяются для получения со-
полимеров с акриловыми мономерами, их эфирами. Для полиме-
ризации диэпоксидных соединений используют катализаторы
Фриделя — Крафтса (BF3, SnCl2 и др.). При этом образуются
твердые неплавкие продукты, которые используются в качестве
заливочных смол, в производстве слоистых материалов и др.
Реакция раскрытия цикла протекает по двум направлениям —
по вторичному и по третичному атомам углерода:
--> Na+OCHa-CH-OH Ch2-ch-r
СН2—СН—R + NaOH — r
> ОН—СН2—СН—R
I
O'Na+
--->• НО—СН—СН2—ОСН2—CHO’Na+ и т. д.
I I
R R
Эпоксидирование циклододекатриена. Эпоксидирование три-
мера бутадиена транс-, тракс-^ис-циклододекатриена-1,5,9, так
же как и винилциклогексена, изучалось в присутствии молиб-
43
ТАБЛИЦА 3.5
Концентрация катализатора, 10-* моль/моль гидропероксида Температу- ра, °C Конверсия гид- ропероксида, % Выход в расчете на прореагиро- вавший гидропероксид, % (мол.)
оксид циклодо- декатриена-1,5,9 циклододеканол
7.5 90 93.0 95,2 92,0 5.0 90 91,7 94,6 94,5 2,5 90 90,8 93,2 92,5 2.5 80 83,2 96,2 96,1 2.5 100 96,4 76,8 91,5 Примечание. Молярное соотношение циклододекатриен-1,5,9 гидропероксид = — 3 1, продолжительность реакции эпоксидирования 120 мин.
денсодержащего катализатора [128]. Выход целевых продуктов
в значительной степени зависит от молярного соотношения реа-
гентов (циклододекатриена и гидропероксида циклодецила).
Так, при молярном соотношении реагентов, равном 1 1, конвер-
сия гидропероксида составляет 56% (за 20 мин), а при 5:1 —
90%. Увеличение молярного соотношения исходных реагентов
от 1 1 до 5: 1 способствует повышению селективности образо-
вания оксида с 54 до 97%.
Однако процесс эпоксидирования винилциклогексена не ог-
раничивается образованием только соответствующих оксидов.
Кроме оксида в качестве побочных продуктов образуются цик-
лодекан, циклодеканол и др. При этом температура процесса
и концентрация молибденсодержащего катализатора оказывают
заметное влияние на выход оксида циклододекатриена и цикло-
додекана (табл. 3.5) [128].
Спектральные исследования исходного циклододекатриена и
полученных продуктов показали, что исходный циклододекатри-
ен имеет интенсивную полосу поглощения в области 970 см-1,
характерную для неплоскостных деформационных колебаний
транс-двойной связи и плоскостные деформационные колебания
цис-двойной связи в области 703 см-1. Кроме того, в спектре
оксида циклододекатриена отмечно появление сильной полосы
в области 872 см-1, характерной для эпоксид-группы, и сниже-
ние интенсивности полосы в области 970 см-1, свидетельствую-
щей о том, что эпоксидирование циклододекатриена протекает
избирательно по одной из трех имеющихся двойных связей.
При дальнейшем эпоксидировании полученного оксида цик-
лододекатриена гидропероксидом трет-бутила (исходное соотно-
шение 4 1) был получен диоксид циклододекатриена, образова-
ние которого доказано с помощью ИК-спектроскопии.
Диоксид циклододекатриена представляет собой смесь двух
изомеров: 1,2,5,6-транс, транс-диэпоксициклододецена-9 (I) и
и 1,2,9,10-транс, транс-диэпоксициклододецена-5 (II):
44
о
•О
\—z_0 \—/
(I) (II)
В работе [129] было предложено следующее определение
полос поглощения ИК-спектров синтезированного диоксида цик-
лододекатриена. Наряду с интенсивной полосой поглощения
в области 872 см-1, характерной для эпоксид-группы в транс-
положении, появляется сильная полоса поглощения в области
906 см-1, характеризующая наличие другой эпоксидной группы
в транс-положении молекулы циклододекатриен. Однако автора-
ми [129] было отмечено появление полосы в области 865—
850 см-1, характерной для эпоксид-группы в цис-положении.
Характеристика синтезированных продуктов:
оксид циклододекатриена: /Кип=1П—112°С/0,67 кПа; d420=0,9793; nD20=
= 1,5060 [128] и /кип = 100—101 °С/0,40 кПа; d420=0,9747; nD20= 1,5045 [129].
диоксид циклододекатриена: /Кип=137—138°С/0,33 кПа; d420= 1,0717;
1,5055 [128] и /кип=П8—120°С/0,13 кПа; d420= 1,0655; nD20= 1,5051 [129].
Таким образом, при эпоксидировании кубового остатка рек-
тификации возвратного растворителя производства бутадиеново-
го каучука, в котором содержится смесь винилциклогексена,
циклододекатриена и других олигомеров бутадиена, можно
ожидать получение смеси соответствующих моно- и диоксидов,
перспектива использования которых весьма реальна.
Восстановление циклододекатриена и его оксида. Как уже
отмечалось, циклододекатриен является исходным сырьем для
получения 1,10-декандикарбоновой кислоты и ш-додекалактама.
Однако синтез вышеперечисленных продуктов отличается спосо-
бами переработки циклододекатриена [130—134].
Восстановление оксида циклододекатриена [128, 129] до
циклододеканола проводили водородом в жидкой фазе на пал-
ладиевых катализаторах. Восстановление протекает стадийно
через образование промежуточных эпоксициклодеценов:
1,2-транс-
Эпокси-тронс-,
цис-циклодо-
1екадиен-5,9
Авторы [128,
оксидного цикла
1,2-транс-
Эпокси-трамс -,
цис-Ц4клодо-
децен-5
1,2-транс-
Эпокси-цнс-,
циклодо-
децен-9
1,2-тронс-
Эпоксицикло-
додекан
восстановление
129] отметили, что заметное
начинается лишь при температурах свыше
45
100 °C. Наряду с циклододеканолом в продуктах гидрирования
обнаружен циклододеканон, образующийся, вероятнее всего, за
счет побочных реакций изомеризации [135].
Максимальный выход циклододеканола (90%) был достиг-
нут с применением никель-хромового катализатора при темпе-
ратуре 130 °C, давлении водорода 5,0 МПа и продолжительно-
сти процесса 100 мин. Порядок реакции по катализатору, опре-
деленный дифференциальным методом, имеет дробное значение
и равен 1,5. В интервале исследованных давлений от 3,5 до
7,0 МПа скорость поглощения водорода не зависит от давления,
т. е. реакция имеет нулевой порядок по водороду.
Повышение температуры на заключительной стадии восста-
новления до 180—200 °C приводит к образованию циклододека-
нона с выходом 88%.
Характеристика продуктов реакции:
эпоксициклододекан: /КИп = 130 °С/0,80 кПа; ^420 = 0,9530; nD20= 1,4808.
циклододек анол: /кип = 130°С/0,53 кПа; ^ПЛ — 80,5—81,5 °C, а по данным
[136] /пл = 80°С.
Жидкофазное гидрирование циклододекатриена в циклододе-
кан подробно изучено на промышленном никель-хромовом ка-
тализаторе [137]. Исследования проводили на модельной уста-
новке проточного типа при давлении 5,0 МПа в интервале тем-
ператур 162—167 °C и объемной скорости по жидкому сырью
1,2—40 ч-1. При обработке экспериментальных данных были
получены кинетические уравнения, описывающие макрокинетику
реакции [137].
Эпоксидирование олигомеров изопрена. Эпоксидированию
олигомеров изопрена, образующихся в процессе полимеризации
изопрена на катализаторах Циглера — Натта, до настоящего
времени должного внимания не уделялось. В литературе имеют-
ся сведения лишь по эпоксидированию некоторых производных
изопрена, например дипентена, являющегося монотерпеном и
часто встречающегося в природе, и 2,6-диметил-2,4,6-октатриена,
известного как алициклический терпен — аллоцимен.
Присоединением кислорода к аллоцимену был получен диок-
сид — 2,6-диметил-2,3,6,7-диэпоксиоктен-4 [127].
Каталитическим гидрированием данного оксида может быть
получен насыщенный двухатомный спирт:
СН3 СН3
СН3—С—СН—СН=СН—С---------СН—СН3,
он сн3 он
сн3—с—сн2—сн2—сн2—сн—сн—сн3 •
<1н3
46
Окисление лимонена — дипентена было проведено Прилежае-
вым в 1908 г. Ему удалось присоединить кислород к ненасыщен-
ным связям лимонена в эфире (или хлороформе) при 0 °C в при-
сутствии надбензойной кислоты. Этим способом был получен
ряд моно- и диоксидов органических соединений, в том числе
и лимонена:
монооксид лимонена
(6ош= 130 - 131 °С/3,33 кПа)
диоксид лимонена
(^кип — 146 — 147 °С/6,67 кПа).
Под действием оксида алюминия при 200 °C оксид лимонена
изомеризуется в карвеол и дигидрокарвон [127]. В процессе
изомеризации карвеол частично полимеризуется, а дигидрокар-
вон превращается в карвенон.
Таким образом, анализируя вышеизложенное, можно сделать
вывод, что окисление и других непредельных соединений, содер-
жащихся в кубовом остатке ректификации возвратного раство-
рителя производства СКИ, будет протекать по аналогичному
механизму с образованием соответствующих моно- и диоксидов.
Это объясняется сходством в строении непредельных соедине-
ний, присутствующих в кубовом остатке, аллоцимена и дипенте-
на (см. табл. 2.7). Можно предположить, что данная смесь
эпоксидных соединений найдет применение в получении смол,
пригодных для технического использования.
3.3. Взаимодействие винилциклогексена с фенолами,
нафтолами и их производными
Алкильные производные фенола находят самое широкое приме-
нение. Одним из основных направлений их использования яв-
ляется производство стабилизаторов. Фенольные стабилизаторы
эффективно защищают от старения разнообразные полимерные
материалы. Многие фенольные стабилизаторы не токсичны и
могут применяться в производстве изделий, контактирующих с
пищевыми продуктами, и др. В качестве алкилирующих аген-
тов наибольшее распространение получили стирол и изобути-
лен [138].
Выпускаемые в настоящее время стабилизаторы на основе
фенола еще не полностью удовлетворяют все возрастающим
потребностям техники. Это приводит к необходимости создания
новых стабилизаторов. Одним из путей является поиск новых
алкилирующих агентов. Если учесть, что в стоимость стабилиза-
тора входит стоимость сырья и материалов, то становится оче-
47
видным, что в качестве алкилирующих агентов необходимо под-
бирать дешевое и доступное сырье. Именно к такому виду
сырья и относятся отходы нефтехимических производств.
В работе [139] в качестве алкилирующего агента рекомен-
довано использовать олигомеры изобутилена, являющиеся отхо-
дами производства мономера — исходного сырья в производстве
полиизобутилена и бутилкаучука. Алкилирование проводили на
модифицированных алюмосиликатных катализаторах. Синтези-
рованный в этих условиях /1-трет-октилфенол служит основой
для получения стабилизатора типа ВС-1.
Для алкилирования фенола и его производных могут быть
использованы и другие ненасыщенные соединения [138], в том
числе и те, которые образуются в качестве побочных продуктов
в ряде органических синтезов или при получении полимерных
материалов. Именно к таким продуктам и относится 4-винилцик-
логексен.
В работах [140—146] исследовано взаимодействие 4-винил-
циклогексена с двухатомными фенолами, галоидфенолами и
нафтолами.
Алкилирование резорцина 4-винилциклогексеном осуществ-
ляли в присутствии катионообменной смолы КУ-2, получая при
этом моно- и дизамещенные продукты. На первой стадии про-
цесса в качестве монопроизводных резорцина образуется смесь
продуктов замещения в положение 4, которая состоит из 80%
6-этил-5а,6,7,8,9,9а-гексагидро-3-дибензофуранола (I) и 20%
6-этил-5а,6,7,8,9,9а-гексагидро-1-дибензофуранола (II). На вто-
рой стадии моноалкилированный резорцин взаимодействует со
второй молекулой 4-винилциклогексена, в результате чего обра-
зуются 4,8-диэтил-1,2,3,4,4а,7а,8,9,10,11,11 а, 126-додекагидробен-
зо-[1,2-6:5,4-64-бисбензофуран (V) и 1,8-диэтил-1,2,3,4,4а,7а,
8,9, 10, 11, 11а, 12а-додекагидробензо-[1,2-6 ЗЛ-Ь^-бисбензофу-
ран (VI). Содержание соединений V и VI в полученной смеси
составляло 60 и 40% соответственно. Авторами установлено,
что при молярном соотношении резорцин 4-винилциклогексен,
равном 2 1, выход монозамещенных продуктов I и II составил
30,5% (селективность по 4-винилциклогексену—69,2%, темпера-
тура 128 °C, продолжительность процесса 5 ч).
Наибольший выход (19%) дизамещенных соединений V и VI
был достигнут при молярном соотношении резорцин: 4-винил-
циклогексен, равном 1:2 (селективность по резорцину 28,2%,
температура 128 °C, продолжительность процесса 7 ч). Влияние
температуры и молярного соотношения компонентов на выход
продуктов моно- и дизамещения показано в табл. 3.6, откуда
видно, что приведенные значения температур и молярного соот-
ношения реагентов являются оптимальными.
По аналогичной схеме протекает и алкилирование 5-метил-
резорцина, пирокатехина, гидрохинона 4-винилциклогексеном.
48
ТАБЛИЦА 3.6;
Температура, °C Молярное соот- ношение резор- цин : 4-винил- циклогексен Состав алкилата, % (масс.) Конверсия**, % Селективность**, %
непрореаги- ровавший резорцин непрореаги- ровавший 4-винилцик- логексен продукты монозаме- щения продукты дизамеще- ния смола продуктов монозаме- щения И Ф ° EJ >>2 tt сз О СО К Q.X X ЕЧХ
128 5:1 65,8 2,2 14,5 2,2 15,3 86,7 50,5 10,2
128 3:1 57,0 6,6 21,0 3,2 12,2 73,1 57,7 11,7
128 2:1 48,7 13,9 28,8 3,3 5,3 57,9 74,9 11,7
128 1:1 33,7 29,1 24,0 5,4 7,8 41 ,3 58,2 17,5
128 1:2 15,6 45,3 23,1 9,3 6,7 53,8 64,6 17,4
128 1:3 5,5 51 ,8 17,1 10,8 14,8 78,2 51,3 21,7
128 1:5 — 54,3 12,2 12,3 21,2 100,0 36,4 24,6
100 2:1 64,6 31,2 1,9 0,2 2,1 5,2 55,3 7,3
115 2:1 60,6 26,4 7,5 0,9 4,6 19,8 56,9 9,1
140 2:1 48,3 13,6 23,3 3,0 11,8 58,7 59,7 10,3
100 1:2 31,6 63,4 2,2 0,8 2,0 6,3 50,7 12,0
115 1:2 29,4 61,2 4,9 2,2 2,3 12,9 56,9 17,1
140 1:2 14,5 44,7 20,0 8,2 12,6 57,0 52,5 14,4
* Продолжительность реакции 3 ч, концентрация КУ-2 — 30% (масс.), растворитель —
о-ксилол 5 моль на 1 моль.
*♦ В опытах с избытком фенола расчет конверсии и селективности производили по
отношению к винилциклогексену, а в опытах с избытком винилциклогексена — по отно-
шению к фенолу.
В первом случае в качестве монозамещенных продуктов полу-
чены 1-метил-6-этил-5а,6,7,8,9,9а-гексагидро-3-дибензофуранол и
3-метил-6-этил-5а, 6, 7,8,9,9а-гексагидро-1-дибензофуранол. Со-
держание их в реакционной смеси составило 60 и 40% соот-
ветственно. Кроме этих соединений в реакционной массе был
обнаружен один продукт 2,4-дизамещения — 5-метил-1,8-ди-
этил-1,2,3,4,4а,7а,8,9,10,11,11 а, 12а-додекагидробензо- [ 1,2 3,4-Ь1]
бисбензофуран. Содержание в алкилате моно- и дизамещенных
соединений при исходном молярном соотношении 5-метилрезор-
цин 4-винилциклогексен, равном 2 1, составило соответственно
37,5 и 5,6% при селективности по 4-винилциклогексену 81,8 и
16,7% (температура 128°C, продолжительность процесса 5 ч).
Во втором случае при взаимодействии пирокатехина с 4-ви-
нилциклогексеном был получен и выделен единственный про-
дукт — 6-этил-5а,6,7,8,9,9а-гексагидро-4-дибензофуранол. При
молярном соотношении пирокатехин : 4-винилциклогексен, рав-
ном 2 1, выход целевого продукта составил 36,9% при селек-
тивности по 4-винилциклогексену 66,9% (температура 140°С,
продолжительность процесса 3 ч).
С высоким выходом целевых продуктов протекает алкилиро-
вание гидрохинона 4-винилциклогексеном в присутствии катио-
4—627 49
сн=сн2
V VI
нита КУ-2 с образованием двух алкильных производных: 6-этил-
5а,6,7,8,9,9а-гексагидро-2-дибензофуранола и 4,10-диэтил-1,2,3,
4,4а,6&,7,8,9,10,10а,12й-додекагидробензо-[1,2-& : 4,5-й1] бисбен-
зофурана. Максимальный выход этих продуктов (54,4 и 18,6%
соответственно) достигался при молярном соотношении гидро-
хинон : 4-винилциклогексен, равном 1 :2, продолжительности
процесса 7 ч и температуре 140 °C.
При алкилировании n-хлорфенола 4-винилциклогексеном в
качестве конечного продукта было выделено только одно соеди-
нение (IX) 3-хлор-4-этил-1,2,3,4,4а,9&-гексагидробензофуран.
Наибольший выход целевого продукта (36%) был достигнут
при мольном соотношении п-хлорфенол винилциклогексен, рав-
ном 1:2 (селективность по и-хлорфенолу 63,3%, температура
128 °C, продолжительность процесса 3 ч):
IX
Конечными продуктами взаимодействия о-хлорфенола с 4-ви-
нилциклогексеном являются 6-хлор-4-этил-1,2,3,4,4а,9&-гексагид-
родибензофуран и 2-хлор-4- (3-этил-2-цикло-гексен-1-ил) фенол
(выход 13,1 и 5,1% соответственно):
4'
51
При взаимодействии 2-нафтола с 4-винилциклогексеном об-
разуется один продукт реакции — 8-этил-7а,8,9,10,11,11а-гекса-
гидробензо[6]нафто[1,2-б/]фуран (X)—с выходом 65,3% при
молярном соотношении 2-нафтол : 4-винилциклогексен, равном
1 :2, и селективностью по 2-нафтолу 93,3% (температура 140 °C,
продолжительность процесса 10 ч). При проведении реакции
в избытке 2-нафтола или при молярном соотношении 1 : 1 наря-
ду с соединением (X) образуется 2,2'-динафтиловый эфир:
Алкилирование 1-нафтола 4-винилциклогексеном приводит к
образованию (XI) 2-(З-этил-2-циклогексен-1 -ил)-1 -нафтола, ко-
торый в условиях реакции полностью циклизуется в (XII)
10-этил-66,7,8,9,10,10а-гексагидробензо[6] нафто [2,1-й]фуран вы-
ход 41%. Кроме того, в продуктах реакции были обнаружены
соединения (XIII) 4-(3-этил-2-циклогексен-1-ил)-1-нафтол (вы-
ход 3,9%), (XIV) 10-этил-5- (З-этил-2-циклогексен-1 -ил) -66,7,8,9,
10,1 Оа-гексагидробензо[6] нафто[2,1 -d] фуран (выход 17,2%) и
образовавшийся при циклизации 2,4-бис-(З-этил-2-циклогексен-
1-ил) -1-нафтол.
Проведенные испытания синтезированных соединений показа-
ли, что все они представляют интерес в качестве фунгицидов,
акарицидов, стимуляторов роста растений, а также в качестве
биоцидных присадок к топливам и маслам.
Работы по использованию 4-винилциклогексена в качестве
алкилирующего агента получили свое дальнейшее развитие в
исследовании каталитического взаимодействия их с эфиром фе-
нола— анизолом, монометиловыми эфирами пирокатехина, ре-
зорцина, гидрохинона [147].
При взаимодействии эфиров фенола с 4-винилциклогексеном
в присутствии катионообменной смолы КУ-2 образуются о- и
52
л-замещенные анизола: 4-(3-этил-2-циклогексен-1-ил) -анизол
(XV) и 2-(3-этил-2-циклогексен-1-ил)-анизол (XVI) с выходом
10,4 и 17,2% соответственно. Кроме вышеперечисленных обра-
зуются также продукты гидрирования и дегидрирования л-ал-
кениланизола, продукты его бис-замещения XVII—XXI.
На основании проведенных исследований сделан вывод, что
наибольший выход целевых продуктов достигается при мольном
соотношении анизол : 4-винилциклогексен, равном 2:1, дозиров-
ке катализатора КУ-2 30% (масс.) на реагенты, температуре
128 °C, продолжительности реакции 8 ч. Уменьшение дозировки
катализатора до 20% снижает выход продуктов алкилирования.
Повышение дозировки до 50% (масс.) также вызывает сниже-
ние выхода продуктов моно- и ди-замещения при одновременном
увеличении количества смол, образующихся в результате реак-
ции полимеризации. Увеличение продолжительности процесса
с 4 до 8 ч приводит к повышению выхода продуктов моно- и
ди-присоединения (концентрация катализатора КУ-2 — 30 % >
температура 128°C). При дальнейшем увеличении продолжи-
тельности реакции с 8 до 10 ч отмечено снижение выхода про-
дуктов моно-замещения с 34 до 20% и увеличение выхода про-
дуктов ди-присоединения с 11 до 19%.
(XXI)
ТАБЛИЦА 3.7
Продолжитель- ность реакции, ч Молярное соот- ношение ани- зол : винилцик- логексен Состав алкилата, % (масс.) Конверсия 4-ви- нилциклогек- сена, % Селективность, %
непрореаги- ровавший анизол непрореаги- ровавший 4-винилцик- логексен продукты моно-заме- щения продукты ди- замещения смола продукты моно-заме- щения продукты ди- замещения
4 2:1 44,1 12,7 27,2 9,2 5,3 59,0 67,4 15,0
6 2:1 42,7 11,5 30,5 9,9 5,4 62,8 71,1 15,3
8 2:1 39,7 9,0 34,2 11,4 5,7 70,0 71,4 15,9
10 2:1 39,3 9,4 31,5 14,4 6,3 68,7 65,0 20,5
12 2:1 36,9 9,0 29,0 19,1 6,7 70,0 62,0 26,5
8 1:1 26,7 29,8 18,1 9,6 15,8 38,9 44,8 13,2
8 4:1 62,9 47,6 19,3 10,6 3,8 73,3 63,6 23,2
8 1:2г 13,4 39,4 25,0 9,4 12,8 60,2 61,5 15,4
Примечание. Температура марную загрузку реагентов. 128 °C, концентрация КУ-2 —30% (масс.) i ia сум-
Меняя мольное соотношение анизол: 4-винилциклогексен
0,5т-4 : 1, можно влиять на состав продуктов моно- и ди-заме-
щения (табл. 3.7). Увеличение содержания анизола в исходной
мономерной смеси приводит к возрастанию конверсии по винил-
циклогексену до 73,3% (соотношение анизол : 4-винилциклогек-
сен = 4: 1), снижению смолообразования до 3,8% и значительно-
му уменьшению выхода продуктов моно-замещения (с 34,2 до
ТАБЛИЦА 3.8
Продолжитель- ность реакции, ч Состав алкилата, % (масс.) Конверсия, % Селективность, %
непрореаги- ровавший гваякол непрореаги- ровавший 4-винилцик- логексен нейтральные продукты моно-заме- щения нейтральные продукты ди-замеще- ния кислые про- дукты а ч о S нейтральных продуктов моно-заме- щения нейтральных продуктов ди-замеще- ния кислых продуктов
Молярное соотношение 2 : 1
3 54,0 18,2 0,7 — 10,1 14,5 50,0 2,0 — 31,5
5 53,1 14,6 1,8 — 16,2 15,0 52,7 5,4 — 49,1
7 50,0 12,0 2,0 — 21,0 16,0 61,1 4,5 — 54,5
10 44,8 6,5 1,2 — 27,7 20,7 78,7 2,3 — 56,5
12 43,4 5,1 1,0 — 27,0 24,3 83,3 1,1 — 52,2
Молярное соотношение 1 :2
5 18,8 46,7 2,2 3,5 17,0 12,0 47,5 7,0 7,0 50,8
7 13,7 40,5 3,8 6,2 21,8 14,0 61,6 8,8 9,4 50,0
10 11,8 38,3 3,0 8,5 20,5 18,0 67,1 7,4 11,8 43,4
12 И,1 37,2 1,8 7,2 18,2 23,0 70,1 4,1 7,1 35,9
Примечание. Температура 128 ’С, концентрация КУ-2 — 30% (масс.] * на суммар-
ную загрузку реагентов.
55
19,3%). При эквимолярном соотношении исходных реагентов
отмечено резкое возрастание смолообразования (до 15,8%), что
объясняется полимеризацией 4-винилциклогексена.
По аналогичной схеме протекает и взаимодействие мономети-
ловых эфиров пирокатехина (гваякола), резорцина и гидрохи-
нона с 4-винилциклогексеном. Алкилирование гваякола 4-винил-
циклогексеном протекает с образованием как моно-, так и ди-за-
мещенных продуктов:
Н3СО С2Н5
На первой стадии происходит преимущественная ориентация
заместителя в n-положение по отношению к группам —ОН и
—ОСНз. По данным ПМР-спектроскопии установлено, что соот-
ношение образующихся продуктов в ходе синтеза 2-метокси-
4- (3-этил-2-циклогексен-1-ил) фенола и 2-метокси-5-(3-этил-2-
56
циклогексен-1-ил) фенола составляет 70 30, т. е. в продуктах
реакции преобладает n-изомер по отношению к ОН-группе. Вы-
делить из продуктов реакции 4-метокси-6-этил-5а,6,7,8,9,9а-гек-
сагидробензофуран авторам не удалось вследствие его цикли-
зации.
На второй стадии протекает взаимодействие моноалкинил-
гваяколов со второй молекулой 4-винилциклогексена с образо-
ванием (XXII) 4-метокси-2-(3-этил-2-циклогексен-1-ил)-6-этил-
5а,6,7,8,9,9а-гексагидродибензофурана. Проведенными исследо-
ваниями установлено, что выход n-алкенилгваяколов (табл. 3.8)
при увеличении продолжительности синтеза с 3 до 10 ч возра-
стает с 10,1 до 27,7%. Дальнейшее увеличение продолжитель-
ности реакции (12 ч) приводит к снижению выхода целевых
продуктов за счет протекания побочных реакций полимеризации
и смолообразования.
В случае взаимодействия монометилового эфира резорцина
с винилциклогексеном наблюдается преимущественная ориента-
ция заместителя в о-положение по отношению к ОН-группе.
Авторы [147] предположили, что на выход продуктов большое
влияние оказывает также продолжительность процесса. Увели-
чение продолжительности опыта с 3 до 10 ч способствует повы-
шению конверсии с 56,5 до 91,6% (соотношение эфир : 4-винил-
циклогексен = 2 1) и с 56,2 до 89,6% (соотношение эфир 4-ви-
нилциклогексен= 1 2). При этом выход нейтральных продуктов
реакции возрастает с 16,0 до 33,0%, а кислых проходит через
максимум 20,0% (5 ч), а затем начинает снижаться.
Монометиловый эфир гидрохинона в реакции с 4-винилцикло-
гексеном проявляет меньшую активность, чем гваякол или мо-
нометиловый эфир резорцина, что связано с несогласованной
ориентацией групп ОН— и —ОСНз в бензольном ядре. Основ-
ным продуктом, образующимся в процессе синтеза, является
4-метокси-6-этил-5а,6,7,8,9,9а-гексагидродибензофуран.
Синтезированные соединения могут найти применение в ка-
честве регуляторов роста растений, ингибиторов.
3.4. Модификация полимеров диеновыми олигомерами
Одним из интересных и перспективных направлений использо-
вания диеновых олигомеров и некоторых их производных явля-
ется модификация ими полимерных материалов, особенно обла-
дающих малой ненасыщенностью, например бутилкаучука. Кро-
ме того, диеновые олигомеры применяются как дополнительные
мономеры при получении насыщенных полимеров — этиленпро-
пиленовых каучуков.
Использование винилциклогексена и его монооксида в каче-
стве модификаторов создает хорошие предпосылки для получе-
57
Свсоп'О1^масс}
РИС. 3.1. Влияние концентрации и типа диена в реакционной смеси
(Св реакц.см) на его содержание в сополимере (Св СОп) при /=20°С, в катали-
тической системе A1RC12—VC14, где R — С2Н5; мзо-С4Н9):
1 — винилциклогексен; 2 — циклооктадиен; 3 — дициклопентадиен
РИС. 3.2. Температурная зависимость эластичности по отскоку (Эо, %) трой*
ных сополимеров этилена, пропилена и винилциклогексена
ния полифункционального бутилкаучука, содержащего двойные
связи или эпоксид-группы в циклогексеновом кольце. По мнению
авторов [148—155], данная модификация бутилкаучука усили-
вает вулканизующую способность каучуков, обеспечивает их
повышенную термостойкость, а также улучшает физико-механи-
ческие свойства вулканизатов.
Выше уже отмечалось, что для получения этиленпропилено-
вых сополимеров, вулканизующихся серой, в их полимерную
цепь необходимо ввести небольшое количество двойных связей
(добавка третьего мономера). В качестве третьих мономеров
широко используются дициклопентадиен, циклооктадиен и др.
[156]. Определенный интерес для использования в качестве
третьего мономера представляет и 4-винилциклогексен.
На основании проведенных исследований было установлено
[157], что влияние концентрации 4-винилциклогексена в полиме-
ризационной смеси на его содержание в сополимере выражено
менее резко, чем в случае применения циклопентадиена или цик*
лооктадиена (рис. 3.1). Тройные сополимеры этилена, пропиле-
на, 4-винилциклогексена, полученные в присутствии каталитиче-
ской системы А12(С2Н5)зС1з—VC14, представляли собой про-
дукты белого цвета, не содержащие кристаллической фазы
(рис. 3.2). Температура стеклования колебалась в пределах от
—55,5 до —60 °C и зависела от содержания звеньев этилена и
пропилена. На молекулярную массу тройных сополимеров
этилена, пропилена и 4-винилциклогексена, составляющую
58
ТАБЛИЦА 3.9
Бутилкаучук
Непредель-
ность, %
Вязкость
по Муни
Молекуляр-
ная масса
по Штаудин
геру
Исходный 0,4—0,8 71
Модифицированный 4-винилциклогек- 2,3—2,6 33
сеном
Модифицированный оксидом винил- — 48
циклогексена*
43000
46000
47000
♦ Эпоксидное число составляет 1,9—2,0.
1,2-104—3,8;104, влияло содержание 4-винилциклогексена в ис-
ходной смеси: чем выше содержание 4-винилциклогексена в ис-
ходной мономерной смеси, тем ниже значение молекулярной
массы.
Прививку 4-винилциклогексена и его оксида на бутилкаучук
осуществляли в углеводородном растворителе в присутствии не-
больших количеств инициаторов пероксидного типа (пероксид
бензоила, гидропероксид кумола и др.). Прививку проводили
при 80—90 °C в течение 3—6 ч. Модифицированный бутилкау-
чук выделяли из раствора и сушили в условиях вакуума при
температуре 60 °C. Свойства исходного, бутилкаучука модифи-
цированного 4-винилциклогексеном и оксидом винилциклогексе-
на [149, 150], приведены в табл. 3.9.
Проведенные испытания на вулканизатах на основе приви-
того и исходного бутилкаучука показали (табл. 3.10), что вул-
канизаты из исходного бутилкаучука имеют низкие модуль, ус-
ловную прочность при растяжении, относительную остаточную
деформацию после разрыва, что значительно ограничивает воз-
можности применения его в шинной промышленности. В то же
время модифицированный бутилкаучук отвечает всем необходи-
ТАБЛИЦА 3.10
Бутилкаучук Свойства вулканизатов, Т=131°С
время вулкани- зации, мин условное на- пряжение при 300%-ном удлинении, МПа условная прочность при растя- жении, МПа относитель- ное удлине- ние при раз- рыве, % относитель- ное остаточ- ное удлине- ние после разрыва, %
Модифициро- 20 М 19,4 800 52
ванный 40 4,3 20,8 738 43
Исходный 20 1,5 18,5 783 37
40 2,7 18,8 700 32
59
ТАБЛИЦА 3.11
Показатель Каучуки
1 1 2
Жесткость по Дефо, Н 11,5 8,5
Условное напряжение при 300 %-ном удлинении, МПа 14,5 14,8
Условная прочность при растяжении, МПа 28,1 22,4
Относительное удлинение, %:
при разрыве 470 440
остаточное после разрыва 19 20
Эластичность по отскоку, % 52 44
Твердость по ТМ-2 60 —
Сопротивление раздиру, кН/м 52 47
Коэффициент теплостойкости при 100 °C:
по сопротивлению разрыву 0,48 0,43
по относительному удлинению при разрыве 0,75 0,62
Примечание. Сажевые вулканизаты этиленпропиленовых каучуков с 4-винилцик-
логексеном (1) и с дициклопентадиеном (2).
мым требованиям. Единственный показатель модифицированных
бутилкаучуков, о котором невозможно судить по имеющимся
сведениям, газопроницаемость.
Сравнительные испытания сажевых вулканизатов этиленпро-
пиленовых каучуков с 4-винилциклогексеном (1) с сажевыми
вулканизатами этилена, пропилена и дициклопентадиена (2) по-
казали, что по всем основным физико-механическим показателям
они равноценны (табл. 3.11).
Таким образом, использование 4-винилциклогексена в каче-
стве третьего мономера в этиленпропиленовых каучуках явля-
ется весьма перспективным. Однако работы в этом направлении
широкого распространения не получили несмотря на то что
4-винилциклогексен образуется в качестве побочного продукта
в ряде органических синтезов, а также при получении бутадие-
нового каучука СКД.
3.5. Диеновый синтез на основе отходов нефтехимических
производств
Диеновый синтез является одним из важнейших как с практи-
ческой, так и с теоретической точки зрения процессов органи-
ческой химии. На его основе могут быть легко получены цен-
ные карбо- и гетероциклические соединения, находящие практи-
ческое применение в качестве полупродуктов в ряде органиче-
ских синтезов, в производстве лакокрасочных материалов,
эмульгаторов, детергентов и др. Реакция диенового синтеза бы-
ла открыта в 1928 г.
60
Диеновый синтез основан на конденсации веществ, содержа-
щих сопряженный диеновый углеводород, с диенофилами (эти-
леновыми и ацетиленовыми углеводородами) [28, 158]:
По аналогичной схеме протекает и взаимодействие диенов
между собой с образованием соответствующих димеров. Так,
димеризацией бутадиена в качестве конечного продукта получен
4-винилциклогексен, а изопрена — дипентен. Димеризацию дие-
новых углеводородов можно рассматривать как частный случай
диенового синтеза, где вторая молекула диена выступает в ка-
честве диенофила.
Образование 4-винилциклогексена и дипентена в значитель-
ных количествах отмечено в процессах полимеризации диенов.
Кроме димеров диеновых углеводородов в полимеризационных
процессах образуются триены и тетраены. К таким соединениям
относятся аллоцимен и ряд других октатриенов, содержащих
сопряженные двойные связи (см. табл. 2.7). Триеновые соедине-
ния, включающие по крайней мере две сопряженные двойные
связи, также способны к димеризации с образованием циклогек-
сеновых производных. Так, гексатриен легко превращается в
циклические продукты
2СН2=СН—СН=СН—СН=СН2 --->
сн=сн2
сн=сн—сн=сн2,
которые образуют с малеиновым ангидридом следующий про-
дукт:
61
Подобно гексатриену протекает и димеризация диметил-1,3,
6-октатриенов и диметил-2,4,6-октатриенов:
СН3 СН3
2 /С=СН—СН=СН—С=СН—СН3 -------->
CH3Z
Сц
СНз
4-Винилциклогексен, образующийся в процессе полимериза-
ции бутадиена, содержит изолированные двойные связи и в ана-
логичных реакциях участия не принимает.
В настоящее время проявляется повышенный интерес к сопо-
лимеризации малеинового ангидрида с различными непредель-
ными соединениями в присутствии радикальных инициаторов.
Особый интерес в этом плане представляют а-олефины и цик-
лоолефины, являющиеся малоактивными мономерами в ради-
кальных процессах. При сополимеризации непредельных соеди-
нений образуются чередующиеся сополимеры, что связано с об-
разованием комплекса и перераспределением заряда между
а-олефином и малеиновым ангидридом. Чередующиеся низко-
молекулярные соединения выделяются и в результате радикаль-
62
ной сополимеризации малеинового ангидрида с циклоолефина-
ми [159—169].
При чередующейся сополимеризации 1,3,6-октатриена с ма-
леиновым ангидридом наряду с полимеризационными процесса-
ми протекает и реакция Дильса — Альдера. Соотношение ком-
понентов в исходной мономерной смеси практически не влияет
на состав получаемых полимерных продуктов [170]. При избыт-
ке малеинового ангидрида в исходной мономерной смеси про-
текает взаимодействие образующегося комплекса (малеиновый
ангидрид — мономер) с радикалом, что способствует реакциям
чередующейся сополимеризации и диенового синтеза. Малые до-
зировки малеинового ангидрида в мономерной смеси уменьшают
вероятность участия его в комплексообразовании, и тогда основ-
ной является реакция диенового синтеза [170]:
Это объясняется тем, что энергия активации диенового синтеза
значительно ниже энергии сополимеризации.
Аналогичное взаимодействие малеинового ангидрида можно
ожидать и с кубовыми остатками ректификации растворителя
производства изопренового каучука. Кубовые остатки кроме
растворителя — толуола — содержат значительные количества
циклических и линейных димеров, а также тримеров изопрена.
Линейные димеры изопрена аналогичны вышеприведенному
триену и отличаются только наличием дополнительных двух
метильных групп. В этом же отличие циклических димеров изо-
прена от 4-винилциклогексена. Исследования сополимеризации
олигомерной смеси с малеиновым ангидридом в присутствии та-
ких радикальных инициаторов, как динитрил азобисизомасля-
ной кислоты, гидропероксидов изопропилбензола, изопропилцик-
логексилбензола и пероксида бензоила показали [177], что во
всех случаях образуются низкомолекулярные аддукты (Мп=
63
= 500—1200). При этом отмечалось уменьшение молекулярной
массы получаемых продуктов с увеличением дозировки малеи-
нового ангидрида в олигомеры. Однако, учитывая, что исходный
кубовый остаток имеет сложный состав и включает смесь линей-
ных и циклических димеров и тримеров изопрена, установить
точный состав и строение конечных продуктов не представляет-
ся возможным.
Циклогексеновое кольцо содержится в побочных продуктах
производства СКИ и СКД. Поэтому интересно рассмотреть со-
полимеризацию циклогексена в присутствии пероксидного ини-
циатора. Сополимеризация малеинового ангидрида с циклогексе-
ном с добавками пероксида бензоила протекает с образованием
чередующихся аморфных сополимеров [159]. Процесс сополиме-
ризации проходит через стадию образования комплексов с пере-
носом заряда. Обработка реакционной массы метанолом и соля-
ной кислотой приводит к получению стабильного аддукта:
hvi; СН3ОН; НС1
COOCH3
COOCH3
Чередование мономерных звеньев при сополимеризации ма-
леинового ангидрида с винильными мономерами одни авторы
объясняют стабилизацией переходного состояния при взаимодей-
ствии мономера с полимерным радикалом, а другие — донорно-
акцепторным взаимодействием, обеспечивающим попарное вхож-
дение мономера в полимерную цепь.
В работах [162, 163] были изучены некоторые закономерно-
сти в образовании комплексов с переносом заряда между ма-
леиновым ангидридом и винилциклоалкеном. Образование ком-
плексов между сомономерами подтверждается появлением но-
вых полос поглощения в электронных спектрах. На основании
найденных полос Хпзмакс была рассчитана энергия переноса
электрона от донора к акцептору.
Сополимеризация низкомолекулярных производных бутадие-
на— димеров и тримеров, содержащихся в кубовом остатке рек-
тификации возвратного растворителя, с малеиновым ангидри-
дом в присутствии радикальных инициаторов показала, что в
качестве конечного продукта образуются низкомолекулярные
61
аддукты (Л/п = 500—1200). Однако состав и строение полученных
аддуктов установлен не был, так как данный кубовый остаток
так же, как и кубовый остаток производства СКИ, имеет слож-
ный состав.
На основе циклического димера бутадиена (4-винилцикло-
гексена) был получен перспективный мономер, содержавший
эпоксидную группу в циклогексеновом кольце и винильную
группу, — 1,2-эпокси-4-винилциклогексан. Сополимеризацию
данного мономера с винилхлоридом в присутствии радикальных
инициаторов проводили в эмульсии или суспензии, получая со-
ответствующие сополимеры. При этом выход сополимеров и их
молекулярная масса в значительной степени определялись со-
держанием эпоксидного мономера в реакционной массе, что
объяснялось доминирующим влиянием реакции передачи цепи
на эпоксидный мономер. Присутствие звеньев эпоксидного моно-
мера в полимерной цепи поливинилхлорида приводило к значи-
тельному повышению термостабильности полимера. Константы
сополимеризации, рассчитанные для пары 1,2-эпокси-4-винилцик-
логексан — винилхлорид, составили Г1 = 0,20 и г2 = 3,14, а для
пары малеиновый ангидрид — 4-винилэпоксициклогексан соста-
вили Г1 = 0,004 и г2 = 0,14. Сополимеризацию малеинового ангид-
рида с эпоксидным мономером проводили в тетрагидрофуране,
диоксане, ацетоне при 60°C [171—173].
Чередующиеся низкомолекулярные полимерные продукты
были получены также и на основе малеинового ангидрида, сти-
рола (Ст) с 4-цианоциклогексеном (4-ЦДГ) [164], который
является побочным в синтезе бутадиеннитрильных каучуков и
других синтезах с участием бутадиена и акрилонитрила. Сопо-
лимеризация вышеприведенной трехкомпонентной системы обус-
ловлена способностью исходного мономера образовывать ком-
плексы с переносом заряда. При этом отмечалось появление
двух комплексов стирола и 4-цианоциклогексена с малеиновым
ангидридом [170]:
МА + Ст <- МА.. Ст, ki =! 0,326 моль/л;
k2
МА + 4-ЦЦГ МА. . 4-ЦЦГ, k2 0,051 моль/л.
Значения константы комплексообразования для соответст-
вующих комплексов определяли методами ЯМР и УФ-спектро-
скопии. При этом необходимо отметить, что гомополимеризация
малеинового ангидрида и 4-цианоциклогексена в условиях трой-
ной сополимеризации не наблюдается. Реакционная способность
комплексов малеинового ангидрида со стиролом и 4-цианоцик-
логексеном в радикальной сополимеризации определяется раз-
личием в значениях ki и &2. Классические уравнения бинарной
и тройной сополимеризации оказываются мало пригодными для
5—627
65
описания данных систем, так как не учитывают донорно-акцеп-
торного взаимодействия.
Кроме винилциклогексена, содержащего изолированные
двойные связи, который образуется в ряде синтезов с участием
бутадиена, известен винилциклогексен, содержащий сопряжен-
ную двойную связь и способный принимать участие в диеновом
синтезе. Конденсация данного винилциклогексена с диенофила-
ми, например с малеиновым ангидридом, протекает энергично
уже при смешении компонентов. При этом образуются син-цис-
и анти-цис-изомеры, дающие при омылении соответствующие
кислоты:
Конденсация винилциклогексена с несимметричными диено-
филами протекает с образованием в основном о-изомера:
Подробное описание конденсации этого изомера винилцикло-
гексена приведено в монографии [28] и в нашем случае рас-
сматриваться не будет, так как образование такого соединения
в процессах получения каучуков и нефтехимических синтезах
не отмечалось.
Таким образом, полимеры, полученные из побочных продук-
тов нефтехимических и полимерных производств, содержащие
звенья малеинового ангидрида, являются перспективными, обла-
дают рядом практически ценных свойств и могут найти примене-
ние в производствах лакокрасочных материалов, анионных по-
лиэлектролитов, эмульгаторов, загустителей и др.
66
3.6. Взаимодействие диеновых олигомеров
с серной кислотой и серой
Взаимодействие диеновых олигомеров с серной кислотой. Хоро-
шо известно, что олефиновые углеводороды энергично взаимо-
действуют с серной кислотой. При этом в качестве конечного
продукта образуются алкилсульфаты, нашедшие широкое при-
менение в качестве поверхностно-активных веществ синтетиче-
ских моющих средств. Сульфатирование олефинов протекает
при воздействии на них 96—98% серной кислоты, температуре
10—20 °C, в результате чего образуются вторичные алкилсуль-
фаты:
R—СН=СН2 + H2SO4 <=>: R—СН—СН3.
OSO3H
После сульфатирования продукты реакции подвергаются
нейтрализации раствором гидроксида натрия:
R—СН—СН3 + NaOH > R—СН—СН3 + Н2О.
I I
OSOgH OSOgNa
Кроме вышеприведенной основной реакции сульфатирования
протекают и побочные, приводящие к образованию вторичного
диалкил сульфата:
R—СН=СН2 + H2SO4 > R—СН— О—SO2—О—СН— R.
СН3 (Ь3
Вторичные алкилсульфаты также находят применение в ка-
честве поверхностно-активных веществ в процессах эмульсион-
ной сополимеризации и др. [174, 175]. Сульфатирование олефи-
нов является промышленно важным процессом и в производст-
ве спиртов — гидролиз моно- и диалкилсульфатов:
R—CHOSO3H + /R—СН \ SO4+ ЗН2О ---> 3R—СН—ОН + 2H2SO4.
Ан8 \ СН3/ з СН3
В данном случае кроме основной реакции сульфатирования
может протекать и (со) полимеризация олефиновых углеводоро-
дов, смолообразование. Это объясняется тем, что серная кисло-
та является катализатором катионного типа [176] и процесс
инициирует катионами водорода:
Н+ +
Н—С—С
Кубовые остатки ректификации возвратного растворителя
производств бутадиенового и изопренового каучуков содержат
ряд непредельных соединений, которые могут служить исходны-
5'
67
ТАБЛИЦА 3.12
Соотношение оли- гомеры : серная кислота Содержание олигомеров бутадиена в исходном очищенном толуоле, % (масс.)
4-винилциклогексен циклододекатри- ен-1,5,9 н-додекатетра- ен-2,4,6,10
1:0,0 9,830 1,540 2,710
1:0,2 3,420 0,260 0,009
1:0,4 1,200 0,140 0,004
1:0,6 0,260 0,008 0,002
1:0,8 0,090 0,006 0,001
1:1,0 0,030 0,003 0,001
1:1,2 0,000 0,001 0,000
Примечание. Время реакции 60 мин, концентрация серной кислоты 93%.
ми продуктами для вышеприведенных синтезов. Однако до на-
стоящего времени процесс взаимодействия диеновых олигомеров
с серной кислотой глубоко не изучался. Подбором соответствую-
щих условий взаимодействия диеновых олигомеров с серной кис-
лотой можно достичь высокоэффективного их связывания. Ис-
следования, проведенные в данном направлении, подтвердили
правильность выбранного направления [177]. При этом степень
очистки растворителя в значительной степени определяется до-
зировкой и концентрацией применяемой серной кислоты. Уве-
личение дозировки серной кислоты (или соотношения олигоме-
ры : серная кислота) и ее концентрации приводит к снижению
содержания диеновых олигомеров в очищенном растворителе
(табл. 3.12, 3.13). Применением 93%-ной серной кислоты и соот-
ношения олигомеры: кислота, равном 1 1,2, достигается прак-
тически полное связывание н-додекатетраена и 4-винилцикло-
гексена, а содержание циклододекатриена снижается до
0,001% (масс.).
Применение 60—75%-ной серной кислоты для очистки раст-
ворителя оказалось малоэффективным. На основании этого сде-
лан вывод, что для высокой степени очистки растворителя от
ТАБЛИЦА 3.13
Концентрация сер- ной кислоты, % (масс.) Содержание олигомеров в очищенном толуоле, % (масс.)
4-вин ил циклогексен циклододекатри- ен-1,5,9 н-додекатетра- ен-2,4,6,10
61 9,050 0,700 0,060
75 4,920 0,180 0,030
85 0,000 0,007 0,000
93 0,000 0,001 0,000
Примечание. Время реакции 60 мин, соотношение олигомеры бутадиена : сер-
ная кислота = 1 1,2.
68
ТАБЛИЦА 3.14
Показатель Толуол Толуол+4,Q% (масс.) очищен- ного толуола
Содержание звеньев в полибутадиене, %:
1,4-цис- 90,5 91,0
1,4-транс- 4,4 4,2
1,2- 5,1 4,8
Вязкость по Муни МБ 1-{-4 (100 °C) 48 45
Пластичность 0,52 0,54
Хладотекучесть, мм/ч Содержание, % (масс.): 10,0 15,0
гель 0,00 0,00
циклододекатриен-1,5,9 0,00 0,00
линейные олигомеры 0,05 0,04
Условное напряжение при 300%-ном удлине- нии, МПа 9 Условная прочность при растяжении, МПа 7,8 8,2
20,3 20,5
Относительное удлинение при разрыве, % 610 600
Относительное остаточное удлинение после 8,0 6,0
разрыва, % Эластичность по отскоку, % 52 52
диеновых олигомеров необходимо использовать кислоту с кон-
центрацией не менее 80%. Однако применение и высококонцент-
рированных кислот для очистки растворителя также нежела-
тельно. Так, в случае 98%-ной серной кислоты часть толуола
вступает в реакцию сульфирования с образованием л-толуол-
сульфокислоты.
Аналогичные закономерности наблюдаются и при очистке
толуола от олигомеров изопрена.
После обработки кубовых остатков ректификации возврат-
ного растворителя серной кислотой толуол из реакционной
массы отгоняли в виде мутного азеотропа. После чего промыва-
ли подщелоченной водой до нейтральной реакции, высушивали
над оксидом алюминия и вводили в количестве 4% (масс.) в ос-
новной растворитель перед подачей его на приготовление шихты.
Из проведенных испытаний следует, что добавка очищенного
толуола в основной растворитель не оказала влияния ни на ско-
рость полимеризации, ни на свойства получаемого каучука
(табл. 3.14).
Толуол, очищенный по предлагаемому способу, может быть
использован также и для приготовления раствора антиоксидан-
та, для очистки оборудования и др.
Для очистки толуола от диеновых олигомеров может быть
использована и 85%-ная серная кислота, являющаяся отходом
промышленности синтетического каучука. Однако в растворите-
ле при этом остается еще значительное количество циклододе-
катриена [до 0,02% (масс.)], хотя и в таком виде он может ути-
69
лизироваться. Для увеличения связывания диеновых олигомеров
в серную кислоту целесообразно добавлять концентрированную,
повышая тем самым ее содержание до 90—95%.
В качестве побочного продукта при сернокислотной очистке
кубового остатка образуется вязкий смолообразный продукт,
который по данным ИК-спектроскопии не содержит винильных
групп. Однако подробное описание его состава, строения и
свойств отсутствует. Можно только предположить, что он пред-
ставляет собой сложную смесь продуктов полимеризации и суль-
фатирования. Попытка использовать этот кубовый остаток в ка-
честве добавки в бутадиеновый каучук не дала положительного
результата. Введение его даже в небольших количествах приво-
дило к резкому снижению прочностных показателей вулканиза-
тов на основе бутадиенового каучука.
Сульфатирование ненасыщенных соединений, содержащихся
в отходах нефтехимических производств, поц действием других
агентов: серного ангидрида, его комплексов, хлорсульфоновой
кислоты и др. также весьма перспективно. Однако в литератур-
ных источниках это не отражено.
Общую схему сульфатирования ненасыщенных соединений
серным ангидридом и образующихся при этом продуктов [1781
можно представить в таком виде:
R' ,R"
R^ I 41
+ ' • ;
so3
9—SO2
OC4H8O so3
R< /R"
p—cf
HZ | pH
R SO2
О
/R"
/С—cf <
Rx| |Ni
о so2
o2s—о
K' | | П
"O3S—О SO3H
Н2О|
R< /R"
cC
RX | |XH
HO SO3H
RC ZR"
C—C<
1.1 H
P SOoH
Таким образом, работы по сульфатированию непредельных
соединений, содержащихся в отходах производства, необходимо
продолжать. На их основе могут быть получены как соответст-
вующие сульфопроизводные, так и спирты. Последние могут
найти применение в различных органических синтезах.
Взаимодействие диеновых олигомеров с серой. Использова-
ние серы широко распространено в органической химии и химии
70
высокомолекулярных соединений. Взаимодействием серы с ор-
ганическими соединениями получают и такие необходимые для
народного хозяйства продукты, как присадки к смазочным мас-
лам, лекарственные средства, красители, пестициды и др.
[179, 180].
Реакция серы со смесью диеновых олигомеров, содержащих-
ся в кубовом остатке, мало изучена. В работе [181] была пока-
зана возможность очистки растворителя (толуола) от олигоме-
ров изопрена серой. Максимальное связывание олигомеров
изопрена серой достигалось при их массовом соотношении
1:0,5—1,5, температуре 160—190°С и продолжительности про-
цесса 3—8 ч. Содержание толуола в отгоне составляло 99%.
В кубовом остатке концентрировалась маслообразная жид-
кость красно-коричневого цвета с молекулярной массой 500—
1500. Подробный состав серосодержащих продуктов на основе
олигомеров изопрена в работе не приводился.
Однако если учесть, что в состав кубового остатка ректифи-
кации возвратного растворителя входили моноциклические тер-
пены (лимонен), то реакция его с серой могла протекать с обра-
зованием тритионов, в молекулах «которых имелись тиофено-
вые циклы [179]:
Кроме того, реакция лимонена с серой могла сопровождать-
ся и образованием изопропилтолуола, n-цимола, оптически ак-
тивных циклических сульфидов [180]:
71
При реакции лимонена с серой выделен был сульфид общей
формулы CioHieS следующего строения:
Образование смолообразных продуктов отмечено при нагре-
вании винилциклогексена с серой и водным раствором аммиа-
ка при 200—220 °C.
Продукты взаимодействия терпенов с серой могут найти
применение в качестве ускорителей вулканизации, лаков, смол
и др. [180]. Строение полученных смолообразных продуктов не
приводится [180].
Очистка возвратного растворителя, толуола, производства
СКД от олигомеров бутадиена серой показала, что определяю-
щими факторами содержания растворителя в отгоне является
исходное соотношение олигомеры : сера, продолжительность про-
цесса и температура [182]. Максимальное связывание олигоме-
ров бутадиена серой достигалось при массовом соотношении
олигомеры : сера, равном 1 1 —1,5, и продолжительности очист-
ки 3—8 ч (табл. 3.15). Оптимальный температурный режим про-
цесса составлял 160—200 °C.
Так же как и при очистке толуола от олигомеров изопрена
серой в кубовой части оставалась маслянистая жидкость красно-
бурого цвета с молекулярной массой 500—1300. На основании
ТАБЛИЦА 3.15
Массовое от- ношение оли- гомеры бута- диена : сера Продолжительность реакции, ч
1.0 3,0 5,0 8,0 10,0
Содержание 4-винилциклогексена в толуоле, % (масс.)
1:0,10 14,20 12,80 11,20 10,60 9,90 1:0,25 12,10 9,50 8,40 7,90 7,80 1:0,50 11,20 4,70 3,20 2,90 1,90 1:1,00 10,20 1,00 0,35 0,14 0,11 1:1,50 9,6 0,25 0,08 0,00 0,00 Примечание. Температура процесса 160 °C.
72
спектральных данных авторы пришли к выводу, что в кубовом
остатке присутствуют производные тиофена, и предложили ме-
ханизмы их образования из 4-винилциклогексена и серы:
С,н9-J-CeH,
S
+ H2s.
с6н9
Затем было сделано предположение, что в реакция может
принимать участие и двойная связь циклогексенового кольца
с образованием полисульфидов и меркаптанов по следующей
схеме:
U
II || +H2S+S -
C.H,-\ JJ-CeHe
S
HSCeH10 S CeH10SH
' АД A
HSCeHio s C6Hio~(S)n—СвНю S
Это предположение было основано на спектральных исследо-
ваниях полученных серосодержащих соединений. В ИК-спект-
рах отмечалось появление новых полос поглощения в областях
700, 1040, 1520 см-1, характерных для тиофена и его производ-
ных [183], и резкое снижение интенсивности полос поглощения,
во-первых, в области 660 см-1, свойственной колебаниям двойной
связи циклогексенового кольца, и, во-вторых, в областях 990,
1645, 3080 см-1, характерных для двойной связи винильной
группы.
Мало вероятно, что реакция протекала только по вышепри-
веденному механизму. Возможны и другие направления данной
реакции, например образование циклических полисульфидов,
содержащих два и более атома серы:
СвН9
73
Реакция с серой может идти и по двойной связи циклогек-
сенового кольца с образованием циклогексилгидросульфида и
дициклогексил сульфида [180]:
SH
Протеканием этой реакции, вероятнее всего, и объясняется
увеличение молекулярной массы получаемых серосодержащих
соединений на основе диеновых олигомеров.
Реакция серы с алкадиенами, триенами и винилароматиче-
скими углеводородами. Диеновые углеводороды в значительных
количествах присутствуют в кубовых остатках ректификации
бутадиена. В их числе изопрен, цис-, тракс-пиперилены; гекса-
диены-1,4 и -1,5; гексадиен-2,4. Реакция алкадиенов с серой при
высоких температурах протекает с образованием преимущест-
венно соединений тиофенового ряда [180]:
Ri-C-C-R3 Ri—С---С—R3
II II + 2S ---> || || +H2S.
r2—с c-r4 r2—c c-r4
i A V
В случае использования изопрена в качестве тиофеновых
производных образуются 3-метилтиофен и некоторые другие
серосодержащие соединения.
При пропускании паров гексадиена-2,4 через расплавлен-
ную серу при 420 °C образуется 2,5(4)-диметилтиофен [180]:
СН—СН
II II -*г 2S
СН3—СН СН—сн3
+ H2s.
Н3С S
'СН3
Реакция 2,6-диметилоктатриена-2,4,6 с серой при 210 °C
протекает с образованием замещенного 4-(2'-тиенил)-1,2-дити-
ол-3-тиона:
СН3 СН3
СН3—i=CH-CH=CH—СН=С—СН3 + 7S
------S
II l+3H2S.
s
С образованием тиофеновых производных протекает также
реакция между серой и винилароматическими углеводородами:
стиролом, стильбеном и др. В первом случае получается дифе-
нилтиофен, а во втором — тетрафенилтиофен:
СвН6-СН
2 II +3S
С,Н6— СН
Свн8 |. .1 свн5
[1^ +2H2S,
С«Н6 S CeH5
74
Реакцию между серой и винилароматическими углеводоро-
дами проводят при высоких температурах (190—250°C). Инте-
рес к этому синтезу не случаен. Это объясняется тем, что сти-
рол, стильбен, метилстиролы в больших количествах присут-
ствуют в кубовом остатке ректификации стирола. Однако, так
же как и в других синтезах с участием серы, реакция не оста-
навливается на образовании только тиофеновых производных
и сопровождается восстановлением стирола выделяющимся се-
роводородом до этилбензола:
+h2s
СбН5-СН=СН2 ---> C6H5-C2H6 + S.
Стильбен в аналогичных условиях образует толуол:
СвН6—СН=СН—CeH5 + 2H2S --> 2CeH5—СН3 + 2S.
Так же протекают реакции серы и с другими виниларома-
тическими углеводородами [184].
Полученные серосодержащие продукты могут применяться
в различных областях народного хозяйства. Так, на основе
отходов производства бутадиенового каучука и серы получают
высококачественные лаки.
3.7. Термическая переработка органических отходов
Расширение нефтехимических производств неизбежно сопро-
вождается увеличением количества образующихся отходов.
В связи с этим появляется острая необходимость в разработке
технологических процессов, позволяющих утилизировать эти
отходы. Одним из них является термическая переработка, спо-
собствующая получению значительных количеств ценных жид-
ких и газообразных продуктов [185—192]. В работе [185]
предложена схема каталитической переработки отходов про-
изводства бутадиена и изопрена (рис. 3.3). Выход продуктов
пиролиза был рассчитан исходя из переработки 100 тыс. т от-
ходов в год (табл. 3.16).
По варианту I предложенной схемы все эти отходы реко-
мендуется использовать в качестве котельного топлива. Однако
такое направление их использования является наименее перс-
пективным.
75
По варианту II исходное сырье направляется на разделение.
Выделяющиеся при этом фракции с т. кип. <75 °C и т. кип.
>160 °C в количествах «26,9 и 30 тыс. т соответственно мо-
гут найти применение в качестве добавок к мототопливу и
сырья для производства технического углерода, используемого
в качестве конечного товарного продукта.
Фракцию с температурой 75—160 °C подают на комплекс-
ную переработку, включающую деалкилирование и экстрактив-
ную ректификацию. В качестве товарных продуктов получают
«10,2 тыс. т бензола и 30,1 тыс. т деароматизированной фрак-
ции с т. кип. <160°С, которую используют также в качестве
котельного топлива.
На первом этапе вариантов III и IV предусматривают так-
же пиролиз исходных отходов. Образующийся пирогаз направ-
ляют на выделение низших олефинов (С2Н4; С3Н6; С4Н8) и бу-
тадиена. Смесь оставшихся газообразных продуктов (Н2,
76
2СлН2п, СО и др.) в количестве —10,2 тыс. т используют в ка-
честве топлива. Фракцию с температурным интервалом 75—
160 °C в количестве — 29,5 тыс. т по варианту III подвергают
гидродеалкилированию и экстрактивной ректификации, полу-
чая при этом —23,8 тыс. т бензола и деароматизированную
фракцию с температурой кипения 160 °C в количестве
— 5,6 тыс. т. По варианту IV фракцию с температурой 75—
160°C направляют' на гидрирование и далее на выделение ин-
дивидуальных ароматических соединений (бензола —7,6 тыс. т;
толуола —9,7 тыс. т; ксилолов —7,1 тыс. т и этилбензола
— 2,8 тыс. т). Количество получаемой деароматизированной
фракции с т. кип. 160 °C составляет около 5,6 тыс. т.
В табл. 3.17 приведены относительные технико-экономичес-
кие показатели процессов переработки отходов. Условно за
100% прийята экономия за счет использования отходов в ка-
честве котельного топлива (I вариант). При сравнении
установлено, что вариант II не дает значительных преиму-
ществ. Наиболее высокая экономия достигается при пиролити-
ческом, разложении (варианты III, IV) смеси отходов — 403 и
424%. Эти направления и являются наиболее перспективными.
При пиролизе жидких отходов, образующихся при произ-
водстве бутадиена по способу Лебедева (из этилового спирта)
и содержащих углеводородную смесь из пентадиенов, гексадие-
нов и др., установлено, что максимальный выход бутадиена
ТАБЛИЦА 3.16
Компоненты Варианты переработки
I 1 п 1 111 IV
Пирогаз — — 28 000 28 000
Товарные продукты газоразделения:
с2н4 — — 17 800 17 800
С3Нб — — 3290 3290
с4н8 — — 1490 1490
с4нб — — 4990 4990
Топливный газ (Н2, СН4, С2Нб, — — 10 200 10 200
С3Н8, С2Н2, С4Н10, СО)
Газы гидродеалкилирования — 2786 3425 —
Жидкие продукты 100 000 97214 64 315 67 740
Фракция 75 °C — 26 930 1605 1605
Фракция 75—160 °C — 40 264 29 551 32 976
Фракция 160 °C — 30 020 33 159 33 159
Деароматизированная фракция — 30 130 5666 5666
160 °C
Бензол — 10 137 23885 7627
Толуол — — — 9757
Ксилолы в сумме — — — 7143
Этилбензол — — — 2783
Технический углерод — — 4260 4260
77
[6,5% (масс.)] и пропилена [5,5% (масс.)] наблюдается при
700 °C [189].
Термическое разложение диеновых олигомеров в толуоле
проводили в интервале температур 650—750 °C, концентрации
олигомеров в толуоле 15% (масс.), объемной скорости подачи
сырья 0,18—0,72 ч-1 в присутствии азота, подаваемого в реак-
тор в количестве не более 5 мл/мин.
Материальные балансы процесса разложения олигомеров в
растворе толуола показали (табл. 3.18), что увеличение объ-
емной скорости подачи сырья приводило к возрастанию выхода
пироконденсата. Повышение температуры в зоне реакции уве-
личивало выход газообразных продуктов, что особенно заметно
на малых объемных скоростях подачи исходного сырья [191].
Анализ выделенных из пироконденсата фракций показал, что
высокая степень очистки толуола от олигомеров бутадиена до-
стигалась при 700 и 750 °C. В оптимальных условиях (700 °C,
объемная скорость 0,5—0,7 ч-1) 75—85% поданного толуола
могло быть возвращено в технологический процесс.
ТАБЛИЦА 3.17
Показатель Варианты переработки*
II 1 111 IV
Затраты на переработку, % в том числе:
фракционирование 100 68 68
комплексная переработка (гидродеалкили- рование и экстрактивная ректификация) 172 132 —
пиролиз — 187 187
газоразделение — 126 126
гидрирование — — 66
многоступенчатая экстрактивная ректифи- кация — — 132
Итого: Экономический эффект от реализации про- дуктов переработки, % в том числе: 272 513 579
бензол 142 300 —
технический углерод — 30 30
топливный газ и газы гидродеалкилирова- ния 6 28 22
товарные продукты газоразделения — 383 383
фракция 75 °C 54 3 3
деароматизированная фракция 160 °C 30 6 6
фракция 160 °C 150 166 166
жидкие ароматические углеводороды — — 393
Итого: 382 916 1003
Экономия, % ПО 403 424
• При расчете приняли, что экономия при осуществлении 100%. варианта I составляет
78
ТАБЛИЦА 3.18
Содержание, % (масс ) Объемная скорость подачи сырья, ч-1
0,18 0,36 0,54 0,72
При 650 °C
Газ 3,50 2,76 2,53 2,20
Пироконденсат 92,70 94,04 94,67 95,18
Кокс+потери 3,80 3,20 2,80 2,62
При 700 °C
Газ 4,91 4,12 3,98 3,74
Пироконденсат 88,94 90,13 90,72 91,23
Кокс+потери 6,15 5,75 5,30 5,03
При 750 °C
Газ 6,90 5,80 5,38 5,23
Пироконденсат 85,73 86,52 87,42 87,72
Кокс+потери 7,73 7,68 7,20 7,05
Термическое разложение жидких отходов производства
СКД (кубовый остаток ректификации возвратного растворите-
ля, содержащий олигомеры бутадиена, его фракция с темпера-
турой 112—160 °C и смесь с гексен-гексадиеновой фракцией)
осуществляли на лабораторной установке [191] при температу-
ре 700°С с разбавлением сырья водяным паром [192]. Разло-
жение олигомерной смеси в данных условиях приводит к полу-
чению газа, содержащего [% (масс.)]: бутадиена—27,7; этиле-
на— 24,2; пропилена—16,4. При этом выходы газообразных
продуктов были невысокие и составляли 16%, и выходы целе-
вых продуктов также резко снижались (бутадиена до —4,5%)
[187].
Наиболее высокий выход бутадиена [ — 7,1% (масс.)] до-
стигался при пиролизе смеси олигомеров, выкипающих в интер-
вале 112—160 °C с гексен-гексадиеновой фракцией. Концентра-
ция бутадиена в газе повышалась до 16—18% (масс.), что
создавало хорошие предпосылки для выделения бутадиена из
газов пиролиза на имевшемся заводском оборудовании.
Проведенные технико-экономические расчеты показали, что
даже для небольшой действующей установки, перерабатываю-
щей до 10 тыс. т отходов в год, себестоимость полученного бу-
тадиена примерно в 2 раза ниже существующей [187].
Как уже было показано, диеновые олигомеры в качестве
побочных продуктов образуются в процессе стереоспецифиче-
ской полимеризации диеновых углеводородов. Основная масса
диеновых олигомеров вместе с растворителем отгоняется на
стадии выделения каучука из раствора методом водной дега-
зации, а небольшая их часть вместе с растворителем остается
79
в каучуке, который со стадии выделения поступает в сушиль-
ные агрегаты. Отходящие из сушильных агрегатов газы со-
держат в своем составе ароматические углеводороды в коли-
честве 0,5—10 мг/л; олигомеры бутадиена (винилциклогексен,
циклододекатриен и др.)—0,01—0,002 мг/л; свободный и свя-
занный хлор — 0,01—0,00015; водяные пары — 20—30 мг/л и
др. [193—195].
Таким образом, отходящий из сушилок газ содержит значи-
тельное количество ароматических углеводородов, олигомеров
бутадиена и других продуктов. Поэтому целесообразно выде-
лять эти углеводороды из газовой смеси и направлять их на
переработку. Однако в настоящее время с целью борьбы с за-
грязнением окружающей среды органическими соединениями
эти газы направляются на каталитическое дожигание, являю-
щееся весьма дорогостоящим процессом. Аналогичным спосо-
бом очищаются и газообразные отходы производства диеновых
мономеров [196].
3.8. Галогенирование диеновых олигомеров
Галогенирование является одним из. перспективных способов
переработки углеводородов. Введение галоида в углеводороды
осуществляют двумя способами: присоединением галогенирую-
щих агентов по двойным связям органического соединения и
замещением различных атомов или групп на галоген. Галоген-
содержащие углеводороды широко используются в народном
хозяйстве. Галогенпроизводные фенолов — три-, тетра-, пента-
хлорфенолы применяются в качестве фунгицидов, репеллентов
и др. [197]. Органические соединения с низким содержанием
галогена используются в качестве растворителей, а с высоким
содержанием — в различных полимерных композициях для
придания им повышенной огнестойкости (антипирены).
Для повышения огнестойкости полимерных материалов, в
том числе и резин, могут найти применение и галогенпроизвод-
ные диеновых олигомеров [198—201].
В работе [198] описан способ получения легко выделяемого
из реакционной смеси и имеющего высокую температуру плав-
ления (>180 °C) антипирена на основе циклододекатриена.
В основе процесса лежит бромирование транс-, транс-, цис- и
транс-, транс-, транс-циклододекатриена (или их смеси) с
растворителями — монометилацеталкиленгликолем и низшим
спиртом — при температуре <40 °C. Образующийся 1,2,5,6,9,10-
гексабромциклододекан выпадает в осадок, который промыва-
ют и сушат.
Бромирование циклододекатриена с высоким выходом ко-
нечного продукта (более 90%) протекает при использовании
смеси, содержащей эквимолекулярное количество брома и цик-
80
лододекатриена, при температуре, не превышающей 40 °C, с
последующим выдерживанием реакционной массы при 70 °C
[199]:
Процесс бромирования олигомеров бутадиена протекает с
хорошей скоростью при 20—25 °C в спиртовом растворителе
при интенсивном перемешивании и постепенном прибавлении
брома в реакционную массу [200].
Аналогично протекает и присоединение брома к молекуле
4-винилцикглогексена с образованием тетрагалогенпроизводных
[201]:
СН=СН2 СНВг—СН2Вг
Вг
Кроме основной реакции присоединения галогена по крат-
ной связи протекает и ряд побочных реакций. Для их предот-
вращения рекомендуется использовать смешанные растворите-
ли [200, 201].
На реакцию галогенирования большое влияние оказывает
температура процесса. В зависимости от температуры галоге-
нирование олефинов может протекать с образованием продук-
тов как присоединения, так и замещения. Хлорирование олефи-
нов при низких температурах ведет к преимущественному при-
соединению хлора по двойной связи (I), а высокие температу-
ры способствуют реакции замещения (II):
I СпН2п + СЬ ----> СЛН2ПС1.
СпН2пС1 • С12 --> СПН2ПС12 4~ С1 • и т. д.
II СПН2„ 4~ CI • -* CnH2n-i 4~ НС1
СпН2п-14" С12 --СпН2пС1 4-СЬ и т. д.
Реакция I является экзотермической ( — 108,9 кДж/моль),
и энергия активации составляет 4,2—8,4 кДж/моль. Энергия
активации реакции II несколько выше — — 25,1 кДж/моль.
Следовательно, для реализации реакции II необходимо приме-
нение более высоких температур (термическое инициирование)
или катализаторов (А1С13, FeCI3 и др.).
6—627 81
Таким образом, изменяя условия синтеза, можно влиять на
конечный состав продуктов галогенирования.
При жидкофазном или газофазном хлорировании, например
моноциклических терпенов, являющихся отходом производства
СКИ, в зависимости от условий реакции образуются продукты
за счет присоединения хлора по двойной связи и замещения,
а также смешанные производные, содержащие как присоеди-
ненный галоген по двойной связи, так и замещенный. В качест-
ве примера можно привести взаимодействие хлора с дипенте-
ном:
На этой схеме приведены лишь некоторые из возможных
продуктов галогенирования дипентена. Аналогичные продукты
образуются при хлорировании 1,4(2,4)-диметил-4-винил-1-цик-
логексена.
Галогенирование линейных димеров изопрена так же, как
и циклических, приводит к получению соединений за счет при-
соединения галогена по двойным связям и замещения, что зна-
чительно увеличивает общее его содержание в конечном про-
дукте.
Синтезированные галогенпроизводные диеновых углеводоро-
дов могут быть использованы в качестве добавок, повышаю-
щих огнестойкость полиэфирных, полиамидных волокон, резин
и других полимерных материалов.
Гидролизом галогенпроизводных диеновых углеводородов
можно получать соответствующие спирты, имеющие хорошие
предпосылки к использованию в народном хозяйстве. Для ус-
корения процесса рекомендуется использовать добавки некото-
82
рых солей и оксидов. К ним относятся оксиды магния, свинца,
карбонаты, которые препятствуют накоплению кислоты и тем
самым смещают равновесие реакции вправо:
R—С1 4- НОН --> R—ОН 4- НС1.
Г алогенпроизводные диеновых олигомеров, содержащие
циклогексеновые кольца, могут быть использованы для получе-
ния соединений, представляющих большой практический инте-
рес. Примером могут служить производные пирокатехина, при-
меняемые в качестве эффективных ингибиторов процессов окис-
ления и полимеризации непредельных углеводородов, стабили-
заторов полимерных материалов и др.
3.9. Металлорганический синтез на основе низкомолекулярных
' производных диенов
Химии элементорганических соединений в настоящее время
уделяется особое внимание. Это связано прежде всего с широ-
ким использованием многих элементорганических соединений в
промышленности. Так, бурное развитие химии органических
соединений титана и алюминия обусловлено открытием в
1954 г. Циглером стереоспецифической полимеризации непре-
дельных соединений под действием алюминийорганических со-
единений и галоида титана [51, 52, 202]. Кроме того, металлор-
ганические соединения применяются и в органических синтезах.
С их помощью можно вводить в молекулы органических соеди-
нений различные функциональные группы, осуществлять синтез
циклических углеводородов и др. [203, 204].
Самым распространенным способом получения алюминий-
органических соединений является прямой синтез алюминий-
триалкила из водорода, алюминия и а-олефинов. В Советском
Союзе в промышленных масштабах реализовано производство
триэтил- и триизобутилалюминия [205].
Прямой синтез высших алюминийорганических соединений
имеет ряд существенных недостатков, поэтому их получают
переалкилированием низших триалкил- или диалкилалюминий-
гидридов высшими олефинами.
Синтезы на основе алюминийорганических соединений.
В реакцию переалкилирования легко вступают циклические
соединения, имеющие винильную группу в бензольном ядре
[204], Примером такого синтеза может служить реакция меж-
ду стиролом и диалкилалюминийгидридом:
6'
83
Из приведенной схемы видно, что связь атомов алюминия
и углерода может осуществляться как с а-, так и с ^-углерод-
ными атомами. В конечном продукте соотношение атомов алю-
миния, присоединенных в а- и ^-положениях, составляет 1:3
[204].
В отличие от стирола в продуктах реакции между 4-винил-
циклогексеном и диалкилалюминийгидридом связь между ато-
мом алюминия и 4-винилциклогексеном осуществляется через
p-углеродный атом винильной группы [206]:
—ch=ch2+r2aih —>
СН2—СН2— А1.
Окисление полученного алюминийорганического соединения
протекает с образованием циклогексен-3-этанола, а карбокси-
лирование двуоксидом углерода дает циклогексенилпропионо-
вую кислоту [207]:
о2; н2о
со2; н2о
Гидрирование этого алюминийорганического соединения с
последующей обработкой 1-додеценом приводит к получению
винилциклогексана с выходом 20% [204]:
г /—\ "I r-ch=ch2
—СНа—СН2—j Al > СН2—СН2—j Al ------>-
---> 3 ( \-СН=СН2.
Аналогично протекает реакция между циклододекатриеном
и диалкилалюминийгидридом. При этом в молекулу циклодо-
декатриена можно ввести три гидроксильные группы:
Реакцией транс-, транс-, транс- и транс-, транс-, цис-и^кзю-
додекатриен с эквимолекулярным количеством триизобутил-
алюминия получают пергидро-9в-алюмофенолен (140 °C, 18 ч)
84
J204J. Разложение водой пергидро-9в-алюмофенолена протека-
ет с перегруппировкой и образованием смеси полициклических
углеводородов:
°— ’ О'сп-
При окислении кислородом воздуха и при взаимодействии
с диоксидом углерода пергидро-9в-алюмофенолена отмечено
образование соответствующего спирта и кислоты:
Интересно реагируют алюминийорганические соединения с
непредельными алифатическими углеводородами, содержащи-
ми две двойные связи. Так, реакция гексадиена-1,5 с алкила-
люмогидридом протекает с образованием циклического углево-
дорода — метиленциклопентана [203] :
—> f/\=CH2.
R2AIH
Z\/\Z + R2AIH ->
ch2air2
Однако если проводить реакцию переалкилирования в при-
сутствии катализатора тетрабутоксититана, то она протекает
с образованием бисалюминийалкильного производного [204]:
^\/\Z+R2AlH ----------------> R2Al/\/\/\AlR2.
Реакции германийорганических соединений с диенами. В от-
личие от других металлорганических соединений германийорга-
нические соединения изучены крайне мало. Однако интерес к
этим соединениям в последние годы резко возрос. Это связано
с установлением биологической активности некоторых герма-
нийорганических соединений.
В работах [208—211] исследовалось гидрогерманилирова-
ние пиперилена, 2,7-диметил-1,3,7-октатриена и некоторых дру-
гих непредельных углеводородов:
^\Z\+ HGe(C2H6)2Br
+ HGe(C2H6)2Br
/\=/\Ge(C2H6)2Br,
Соответствующие продукты были получены с выходами 45 и
85%.
О конфигурации двойной связи судили по наличию харак-
терных полос поглощения в области 700—720 см-1 в ИК-спект-
рах.
85
Аналогично протекает реакция и триэтилгермания с выше-
указанными диеном и триеном:
/\^\ + HGe(C2H6)3 ---> Z\=/\Ge(C2H5)3,l
+ HGe(C2H6)3
./Ge(C2H5)8.
На основе синтезированных алкенилдиэтилбромгерманов
могут быть получены дигерманоксаны с выходом ~30%:
Кон(5%)
RgGeBr -------->- RgGeOGeRg.
Взаимодействие диэтилдигидрогермана с олефинами, содер-
жащими кроме сопряженной и изолированную двойную связь,
приводит к образованию германоцикланов:
/\/\/\/ + H2Ge(C2H6)2
Выход продуктов с внутримолекулярной циклизацией до-
стигает 50% (120 °C, 5 ч).
Взаимодействие 2,7-диметил-1,3,7-октатриена с эфиратом
трихлоргермана сопровождается получением только линейных
продуктов 1,4-присоединения с выходом 50—60%:
+ HGeCl3-2(C2H5)2O ---------->
2(СзНб)2О
Е-Конфигурация одной из двойных связей доказана ЯМР-
спектроскопией !Н, в спектре которого сигналы протонов ме-
тильной группы, находящейся при втором атоме углерода, про-
являются в виде синглета при 1,72 м. д. Авторами [211] отме-
чено, что сигналы метиленовых протонов при атоме германия
сдвигаются в более слабое поле до 2,3 м. д. из-за наличия трех
атомов хлора при атоме германия.
На основе линейных продуктов 1,4-присоединения может
быть осуществлен синтез полиалкенилгермантранов:
Особый интерес представляет исследованная авторами [211]
реакция гидрогерманилирования димеров бутадиена 4-винил-
циклогексена и изопрена (лимонена) с использованием раз-
86
.личных гидрогерманов и соединений переходных металлов.
Так, в присутствии никелевой каталитической системы (три-
этил-, диэтилбромгерман и гидридпентаэтилгерман) при взаи-
модействии с 4-винилциклогексеном (150 °C, 5 ч) образуется
продукт ^-присоединения с выходами 25, 58 и 28% соответст-
венно. При контакте с диэтилгерманом дополнительно выделя-
ется еще и ди-аддукт.
где R= —-С2Нб; GeEt3; Вг; Cl; Н.
Как было установлено в [2П] лимонен в присутствии нике-
левой каталитической системы практически не взаимодейство-
вал с гидрогерманами. Реакция лимонена с триэтил- и диэтил-
бромгерманами протекала в присутствии H2PtCl6 (150 °C, 8—
10 ч). Однако выходы целевых продуктов были небольшие.
В первом случае выход составил — 10%:
СНз—С=СНа CH3-CH-CH2-Ge(C2H5)8
+ HGe(C2H5)3
а во втором случае отмечалось образование двух продуктов,
которые соответствовали присоединению по винильной группе
и по двойной связи циклогексенового кольца с общим выходом
25%:
сн3—с=сн2
2
+ 2 HGe(C2H5)2Br----►
СН3—СН—СН2—Ge(C2H5)3Br СН3— С=СН2
4Ge(C2H5)3Br
Полученные германийсодержащие циклические диолы явля-
ются биологически активными соединениями, стимулирующими
рост волос и обладающими ранозаживляющим действием.
87
3.10. Диспропорционирование и изомеризация
диеновых олигомеров
Реакцией диспропорционирования можно получать перспектив-
ные циклические углеводороды, полимерные соединения и др.
Процессы диспропорционирования и изомеризации непредель-
ных соединений хорошо изложены в ряде монографий [203,
204, 212].
Диспропорционирование 4-винилциклогексена на алюмомо-
либденовом катализаторе протекает с образованием этилена и
1,2-бис (циклогексен-З'-ил-l') -этилена:
СН=СН2 СН=СН2
СН2=СН2 +
Межмолекулярное диспропорционирование цис-, транс-,
цис- и транс-транс, тракс-циклододекатриена-1,5,9 на алюмини-
евом катализаторе сопровождается образованием сложной сме-
си циклических углеводородов, среди которых обнаружены цик-
логексадекатетраен-1,5,9,13; циклоэйкозапентаен-1,5,9,13,17; цик-
лотетраэйкозагексаен-1,5,9,13,17,21 и циклооктаэйкозагептаен-
1,5,9,13,17,21,25. Эти углеводороды оказались весьма перспек-
тивными для производства душистых веществ [213].
Авторами [204] сделано предположение, что олефины, не
способные вступать в реакцию переалкилирования с алюми-
нийорганическим соединением, претерпевают различные хими-
ческие превращения. Так, при нагревании 4-винилциклогексена
с трис-[2-(циклогексен-3)-этил]-алюминием, взятым в коли-
честве 10—20% (масс.), выделяют с выходом 60% бицикло-
[3,2,1]-октен-2 (150°С, 10 ч):
Изомеризация 4-винилциклогексена в присутствии натрия на
оксиде алюминия (20 °C) сопровождается образованием 4-
этилиденциклогексена-1:
СН2СН— ( \ ► сн3—сн=/ V
Под влиянием амида калия в жидком аммиаке 4-винилцик-
логексен уже при 40°С легко превращается в этилбензол, яв-
88
ляющийся исходным сырьем для получения такого широко
распространенного мономера, как стирол:
/--KNH2; (NH3) „ „ / \
СН2СН—/ » СНз—СН=^ " ►
---> СН3—СН2—> СН3—СН2—/~V
\__/ Нг \==/
Тример бутадиена — циклододекатриен может служить цен-
ным исходным сырьем для получения полицикланов, в частнос-
ти алкиладамантанов. На первом этапе этого .процесса прово-
дят переалкилирование триизобутилалюминия и изомеризация
циклододекатриена в трициклододецены (200—210°C), а затем
восстановление и изомеризацию полученных продуктов в при-
сутствии бромида алюминия (60 °C) [214]:
Алкиладамантаны могут быть получены и через пергидро-
9в-алюмофенолены, которые при разложении водой дают смесь
трициклических олефинов [215, 216].
Можно ожидать, что аналогичные процессы будут протекать
и при изомеризации димеров изопрена, представляющих собой
смесь терпеноидов и являющихся отходом производства изо-
пренового каучука. Возможности такой изомеризации показаны
в монографии [87].
3.11. Окисление отходов производства синтетического
каучука
В кубовых остатках ректификации возвратного растворителя
производства синтетического каучука содержатся циклические
органические соединения, содержащие непредельные связи.
89
На их основе возможно проведение синтеза с образованием
соединений с функциональными концевыми группами, которые
представляют значительную ценность в органическом синтезе
и получении полимерных материалов. Одним из таких отходов
производства бутадиенового каучука является тример бутадие-
на — циклододекатриен, который может использоваться в про-
изводстве полиамидного волокна [56, 203, 217]. Необходимый
для полимеризации лауролактам можно получить из циклодо-
декатриена по следующей схеме:
Как с теоретической, так и с практической точки зрения
интересно получение из тримера бутадиена дикарбоновых кис-
лот. При этом восстанавлив<ают циклододекатриен до циклодо-
декана, а затем последний окисляют азотной кислотой при
60—180 °C. Выход декандикарбоновой кислоты в этих услови-
ях составляет — 50% [203]. Выход дикарбоновых кислот мож-
но увеличить, если в качестве окислителя кроме азотной кис-
лоты использовать ди- и тетраоксид азота. В этом случае вы-
ход дикарбоновой кислоты повышается до 67% (50 °C, 60 ч).
Схема получения декандикарбоновой кислоты из циклодо-
декатриена приведена в работе [203].
Принципиальная технологическая схема непрерывного про-
цесса окисления циклододекана в дикандикарбоповую кислоту
приведена на рис. 3.4 [218].
На первом этапе осуществляют окисление циклододекана в
реакторе /, снабженном перемешивающим устройством, а вто-
90
РИС. 3.4. Технологическая схема процесса окисления циклододекана в де-
кандикарбоновую кислоту:
/, 2 — реакторы; 3 — сепаратор; 4 — кристаллизатор; 5 — фильтр
рую стадию процесса окисления проводят в реакторе вытесне-
ния 2. Выделяющиеся в процессе окисления газообразные про-
дукты (в основном из оксидов азота) через сепаратор 3 час-
тично возвращают ц реактор 7, а остальные направляют на
улавливание. Продукты окисления охлаждают до 50—70 °C,
отфильтровывают выпавшие в кристаллизаторе 4 кристаллы
декандикарбоновой кислоты. Последние промывают водой и су-
шат при 70—90 °C. Очистку дикарбоновой кислоты проводят
перегонкой в вакууме при 215—225 °C и остаточном давлении
0,7—2,7 гПа. Существуют и другие способы окисления цикло-
додекана [203].
Окисление винилциклогексена 62 %-ной азотной кислотой с
добавками 0,05% NH4VO3 при 65—90 °C приводит к образова-
нию бутантрикарбоновой-1,2,4 кислоты (выход — 70%) [219]:
СН=СН2
> ноос—сн2—сн2—сн—сн2—соон.
соон
Эта кислота может быть использована в производстве поли-
конденсационных полимеров. При наличии в молекуле двух
функциональных групп образуются полимеры линейного строе-
ния. Увеличение числа функциональных групп до трех, как это
имеет место в данном случае, и более приводит к появлению
полимеров пространственного строения.
Озонолиз циклоолефинов протекает с расщеплением цикла
и образованием бифункциональных соединений. В частном слу-
91
чае при озонолизе циклододекатриена в качестве конечного
продукта была получена декадиен-3,7-дикарбоновая-1,10 кисло-
та [203]:
+Оз(Н2О2)
—► ноос—сн2—сн2—сн=сн—сн2—сн2—сн=сн—сн2—сн2—соон.
Селективное озонирование одной из трех двойных связей
циклододекатриена с целью получения со-аминододекановой
кислоты проводят в смешанном растворителе, состоящем из
высококипящего парафинового масла, уксусной кислоты и ук-
сусного ангидрида. Полученная со-аминододекановая кислота
является исходным сырьем для синтеза додекалактама, ис-
пользуемого в качестве мономера для получения найлона-12
[220].
Ниже приводим общую схему получения со-аминодекановой
кислоты, а также полимера на ее основе [198], состоящую из
следующих стадий: озонирование, каталитическое разложение,
восстановительное аминирование, подкисление, поликонденса-
ция.
3H2;Ni;
130 °с; 4,9МПа
сно
соон
СН NH г т
2 2---------> h2n-(ch2)11~-conh-(ch2)11-“ -соон.
СООН (л-1)Н2О 2 К L
Аппаратурное оформление процесса озонирования циклодо-
декатриена приведено на рис. 3.5 [220].
Кислородсодержащий газ после осушителя 10 поступает в
озонатор 11. Далее озонокислородпая смесь направляется в
реактор 3 первой стадии озонирования, куда вводят также
циклододекатриен и растворитель. Газообразные продукты из
реактора 3 подают па очистку в аппараты 1 и 2, откуда они
частично возвращаются в цикл, а частично выводятся из про-
цесса.
92
РИС. 3.5. Технологическая схема получения аминододекановой кислоты:
1, 2 — очиститель; 3, 5, 7, 12, 14, /5 — реактор; 6, 8 — испаритель; 4, 15, 16 — сепаратора
9— ректификационная колонна; 10 — осушитель; 11— озонатор; 13 — нейтрализатор; П —
экстрактор; /9 — аппарат для промывки
Реакционную массу из реактора 3 насосами подают через-
подогреватель и сепаратор 4 в первый из трех последовательна
соединенных реакторов 5 с одновременной дозировкой в него
катализатора (смесь уксусного ангидрида и ацетата натрия).
Из последнего реактора 5 реакционную массу направляют в
испаритель 6, где под вакуумом из продуктов реакции удаляют
растворитель, возвращая его в реактор 3. Для удаления уксус-
ного ангидрида продукты реакции обрабатывают в реакторе 7
уксусной кислотой (в избытке). Выделившийся уксусный анги-
дрид одновременно с уксусной кислотой отделяют в испари-
тель <$, окончательную очистку продуктов реакции проводят на
ректификационной колонне 9 и возвращают в процесс.
Полученный ненасыщенный альдегид, представляющий со-
бой вязкую жидкость и содержащий побочные продукты синте-
за, направляется в реактор 12, а затем в нейтрализатор 13, где-
при О °C его обрабатывают последовательно водными раствора-
ми аммиака и щелочи. Продукты реакции направляют на гид-
рирование в батарею реакторов 14. Гидрирование проводят на
никелевом катализаторе при 130—180°C и давлении 4,9 МПа.
Избыточный аммиак и воду удаляют из продуктов реакции в
сепараторе 16 и возвращают в реактор 12.
Водный раствор натриевой соли со-аминододекановой кисло-
ты обрабатывают ксилолом в экстракторе 17 для выделения
образовавшегося в процессе синтеза диамина. После экстрак-
ции диамина водный раствор со-аминододекаповой кислоты
подкисляют в реакторе 18 серной кислотой, отделяют выпав-
шую (о-аминододекановую кислоту и промывают ее водой в
93
аппарате 19. При необходимости повторной перекристаллизаци-
ей можно получить со-аминододекановую кислоту с более вы-
сокой степенью очистки. В дальнейшем со-аминододекановую
кислоту направляют на производство мономера — лактама.
Анализ технологической схемы показывает, что циклы по
растворителю и уксусному ангидриду замкнуты, т. е. они мно-
гократно используются в процессе. Это позволяет значительно
снизить потери этих соединений и создает хорошие предпосыл-
ки к созданию безотходной технологии. Кроме того, в данном
процессе образуется небольшое количество сточных вод, не
содержащих биологически неразлагаемых веществ, и, следова-
тельно, легко поддается очистке.
Расходные показатели сырья и энергоресурсов при получе-
нии найлона-12 из циклододекатриена по способу итальянской
•фирмы «SNIA Viscosa» на 1 т конечного продукта составляют:
1075 кг циклододекатриена; 200 кг аммиака; 740 м3 кислорода;
490 м3 водорода; 360 кг щелочи; 380 кг серной кислоты;
390 кВт-ч электроэнергии; 20 т водяного пара.
Достоинством этого способа является его мобильность, т. е.
без каких-либо дополнительных технологических изменений он
может быть использован для получения дикарбоновых кислот,
диаминов, диолов и др. продуктов, являющихся ценным исход-
ным сырьем в органическом синтезе и в производстве полимер-
ных материалов.
Представляет интерес также способ получения диальдеги-
дов окислением или озонолизом циклических побочных продук-
тов производства синтетического каучука. Такие непредельные
альдегиды, содержащие двойные связи в молекулах, находят
применение в ряде органических синтезов, например для по-
лучения душистых веществ.
В этом случае диальдегиды получают путем подбора усло-
вий селективного окисления (озонирования) по двойной связи
циклоолефина [203]:
Оз; Н2 (Pd на CaCo34-PbO)
онс—сн2—сн2
С=С
сн=сн2
Н Н
сн2—сн2—сно
С=С
Н
СН2—СН2
Н
О3; H2(Pd на СаСОз + РЬО)
----------------> онс—сн2—сн—сн2-сн2—СНО.
I
сн=сн2
94
Конечным продуктом селективного расщепления двойной
связи циклододекатриена является транс-, транс-ды&кг^ъеп-
4,8-диаль-1,12, а винилциклогексена — З-винилгександиаль-1,6.
Из транс-, транс-, транс-циклододекатриена озонолизом
удалось получить транс-, транс-11 -формилундекадиен-4,8-овук>
кислоту:
НОС—СН2—СН2 Н Н СН2—СН2— С ООН
н сн2—сн2 н
Исходя из вышеизложенного, можно сделать вывод о боль-
шой перспективности синтеза продуктов окисления циклооле-
финов. Следует отметить, что окисление 4-винилциклогексена
и циклододекатриена протекает в одинаковых условиях. Эта
позволяет проводить процесс без предварительного разделения
кубовых остатков ректификации возвратного растворителя про-
изводства каучука СКД, в состав которых и входят в качестве
основных непредельных соединений 4-винилциклогексен и цик-
лододекатриен-1,5,9.
Можно предположить получение аналогичных результатов-
и при окислении побочных продуктов производства изопрено-
вого каучука, которые, так же как и в производстве каучука
СКД, концентрируются в кубовой части ректификационных ко-
лонн при очистке возвратного растворителя. Однако подтверж-
дения этому в имеющихся литературных данных получено не
было.
3.12. Синтезы на основе 4-цианоциклогексена
Синтезы на основе 4-цианоциклогексена с участием нитрильной
группы. Нитрильная группа обладает высокой полярностью.
Этим объясняется е$ склонность к участию в реакциях присо-
единения. Она способна поляризовать а,^-ненасыщенные крат-
ные связи и, подобно карбонильной группе, повышать кислот-
ность а-водородных атомов.
Одной из наиболее характерных реакций нитрилов являет-
ся гидролиз CN-группы, который может быть осуществлен в
присутствии катализатора — минеральных кислот, оснований,,
оксидов и гидроксидов металлов, — и без него [221, 222]. Со-
став продуктов реакции зависит от природы катализатора и
условий проведения процесса.
95
Кислотный гидролиз 4-цианоциклогексена (I) приводит к
образованию циклогексен-3-карбоксиамида (II) или циклогек-
сен-3-карбоновой кислоты (III):
Н20; Н+
Н2О; Н+
-------->
Гидролиз 4-цианоциклогексена в присутствии концентриро-
ванных растворов, [84,5—96% (масс.)] серной кислоты описан
в работах [108, 223]. При температуре 50—100 °C процесс за-
канчивается на стадии образования (II), и количество образо-
вавшейся (III) невелико. Увеличение температуры и продол-
жительности реакции ведет к повышению выхода (III). По
данным [Ю8], выход (III) при 140°C и времени реакции 8 ч
составляет 82% от теоретического. Снижение температуры ре-
акции до 30—40 °C, использование значительного (до пятикрат-
ного) избытка концентрированной серной кислоты и увеличение
'продолжительности процесса до 18 ч способствуют одновремен-
ному гидролизу нитрильной группы и гидратации двойной свя-
зи циклогексенового кольца, что ведет к образованию 4-окси-
циклогексанкарбоновой кислоты (IV) [224]:
88%-ная H2SO4, Н2О
NH4HSO4
(I) (IV)
Оксикислота (IV) и ее гидразид могут использоваться в
качестве мономеров для получения химических волокон, плас-
тических масс.
Гидролиз 4-цианоциклогексена в присутствии серной кисло-
ты и спиртов приводит к образованию сложных эфиров цикло-
гексен-3-карбоновой кислоты (V):
ROH, H2SO4
---------->
(I) (V)
Этим способом были получены метиловый [108], бутило-
вый, амиловый, гексиловый, гептиловый, октиловый, нонило-
вый [225] эфиры циклогексен-3-карбоновой кислоты. В работе
96
[226] показано, что промежуточным продуктом в этой реакции
является циклогексен-3-карбоксамид, это подтверждено дан-
ными ИК-спектроскопии.
Эфиры (V^ служат пластификаторами полимерных матери-
алов [227]. Химическая модификация эфиров (V) путем бро-
мирования или эпоксидирования двойной связи циклогексено-
вого кольца позволяет расширить области их применения. На-
пример, синтезированный [127] аллиловый эфир 3,4-эпокси-
циклогексанкарбоновой кислоты может найти применение в ка-
честве мономера и реакционноспособного пластификатора.
Эфиры карбоновой кислоты (III) и к-алканолов Се—С9, эпок-
сидированные перуксусной кислотой, могут использоваться в
качестве активных разбавителей эпоксидно-каучуковых компо-
зиций [228]. Наряду с эпоксидированным амидом (II) они мо-
гут применяться в качестве эффективных пластификаторов-ста-
билизаторов поливинилхлорида [229]. Производные кислоты
(III) широко используются в синтезе циклоалифатических
эпоксидных смол, о чем более подробно будет сказано ниже.
Взаимодействием нитрилов со слабощелочными растворами
пероксида водорода (реакция Радзишевского) получают ами-
ды. При использовании ненасыщенных нитрилов реакция со-
провождается одновременным эпоксидированием двойных свя-
зей и образованием эпоксамидов [221]. Иногда продуктом ре-
акции является смесь ненасыщенного амида и эпоксинитрила.
Эпоксидирование соединений, в которых нитрильная группа
под действием пероксида водорода не гидратируется, может
идти и в отсутствие щелочных добавок. Так, в работе [230]
было исследовано взаимодействие 4-цианоциклогексена с 50 % -
ным пероксидом водорода в присутствии оснований (pH 9,5—
10,0) й без них. В первом случае был выделен транс-\4-эпок-
сициклогексанкарбонитрил-1 (VI) с выходом до 60%:
NC NC
О
(I) (VI)
Во втором случае вместо ожидаемого эпоксиамида (внут-
римолекулярное окисление) образовалась смесь (II) и (VI):
NC H2NOC NC
(I) (И) (VI)
7-627
97
Выход (VI) при этом не превышал 45%.
Эпоксинитрил (VI) может применяться в качестве реакци-
онноспособного разбавителя, полупродукта в синтезе 4-циано-
циклогександиола-1,2 и ^-цианадипиновой кислоты [101, 231,
232].
4-Цианоциклогексен, подобно другим непредельным нитри-
лам, легко гидролизуется в щелочной среде. При кипячении
его в 50%-ном метанольном растворе гидроксида калия с вы-
соким выходом образуется циклогексен-3-карбоновая кислота
(III) [233].
При щелочном плавлении 4-цианоциклогексена наряду с
гидролизом функциональной группы происходит изомеризация
двойной связи и деструкции углеродного скелета молекулы
[234, 235]:
NC
66%-ный КОН; 320 °C
ноос соон
(VII)
Эта реакция, известная как синтез Варрентраппа, является
одним из удобных препаративных методов получения пимели-
новой кислоты (VII). Конденсация (VII) с гексаметиленди-
амином (или другими аминами) протекает с образованием по-
лимерных продуктов, сходных с полиамидами типа найлона и
капрона [236, 237].
Восстановление нитрилов является многоступенчатым про-
цессом и протекает по следующей схеме:
2Н+ Н+ 2Н+
r_C=N > R—CH=NH > R-CH2—NH2 -------------> RCH3.
nh3
Практически процесс восстановления нитрилов заканчива-
ется на стадии образования аминов.
Наряду с традиционными способами восстановления нитри-
лов различными реагентами (металлического натрия в присут-
ствии спиртов, хлорида олова, гидридов металлов) в послед-
нее время широко применяется каталитическое гидрирование
молекулярным водородом. Преимуществом этого метода явля-
ется высокая скорость, простота осуществления реакции и вы-
деления продуктов восстановления. Избирательное гидрирова-
ние нитрилов при наличии в молекуле других функциональных
групп, которые также могут быть восстановлены, зависит от
его строения, условий проведения реакции и природы катали-
затора. В зависимости от режима процесса восстанавливается
либо только нитрильная группа, либо нитрильная группа и
кольцо (в случае ароматических и гетероциклических нитри-
лов). В случае если двойная связь и CN-группа в молекуле ни-
трила удалены друг от друга на большее расстояние, чем у
98
а,^-ненасыщенных нитрилов, то нитрильная группа гидрирует-
ся легче, чем двойная связь [222].
При взаимодействии 4-цианоциклогексена с молекулярным
водородом в присутствии гетерогенных катализаторов возмож-
ны следующие направления реакции:
Pd/C
Cu(CrO4)2
Ni/Сг
Ni или (и)Со
N1 или (и) Со
ch2nh2
(j
(VIII)
j^]-CH2-N-CH2-[^^j|
(IX) Н
CN
a
(X)
(XI)
ch2nh2
a
(XII)
Селективное гидрирование нитрильной группы проводят при
температуре 130—150°С и давлении — 14 МПа. Подбором со-
ответствующих условий проведения процесса можно изменить
его направление в сторону образования либо 4-аминометилцик-
логексена-1 (VIII), либо бис-амина (IX) [237—239].
Побочные реакции образования вторичных и третичных
аминов сильно снижают выход первичных аминов. Для подав-
ления побочных реакций при гидрировании 4-цианоциклогексе-
на до гексагидробензамипа (XII) (150—200°C; <200 МПа; ка-
тализаторы па основе никеля или кобальта) предложено [240]
проводить процесс в присутствии аммиака [<30% (масс.)].
В работах [240—242] приведены результаты исследований
по гидрированию 4-цианоциклогексена и метилцианниклогексе-
на на различных гетерогенных катализаторах. Отмечено, что ис-
черпывающее гидрирование 4-цианоциклогексена протекает на
никель-хромовом катализаторе. При использовании такого ка-
тализатора гидрирование идет по двойной связи циклогексено-
7'
99
вого кольца о сохранением нитрильной группы. Образование
циклогексанкарбонитрила (X) подтверждено данными ИК-
спектроскопии.
Амины (VIII) и (IX) являются полупродуктами в синтезе
азотсодержащих соединений, например эпоксидных смол. По-
лученные на их основе эпоксидные смолы обладают комплек-
сом технически важных свойств: сочетанием низкой вязкости
и большой жизнеспособности; высокими прочностными свойст-
вами отвержденных материалов; стойкостью к действию ульт-
рафиолетовых лучей, температуре и давлению [242].
При взаимодействии нитрилов с реактивом Гриньяра, как
правило, образуются кетоны. В литературе имеются сведения
получении циклогексенилкетонов (XIV) реакцией 4-цианоцик-
логексена с алкил- и арилмагнийгалогенидами [243]:
NC R—C=N—MgX R—С=О
У--\ R-MgX^ Н3О+
(I) (XIII) (XIV)
где X — галоген; R — алкил, арил.
Однако, как указывается в [244], реакция с фенил-, бензил-
и циклогексилмагнийбромидами останавливается на стадии
образования иминоаниона (XIII).
Циклогексенилкетоны предполагается использовать для
синтеза биологически активных препаратов, а также в качест-
ве пластификаторов полимерных материалов.
В присутствии основного катализатора (амида натрия)
[245] 4-цианоциклогексен реагирует с н-алкилбромидами с об-
разованием нитрилов I-алкилциклогексен-3-карбоновой кисло-
ты, омылением которых могут быть получены амиды соответст-
вующих кислот, обладающие антиэпилептической активностью.
4-Циапоциклогексен вступает в реакцию с дициандиамидом
с образованием замещенных триазинов (XV) [246, 247]:
R
I
ыи /С\
R—CN + NC-NH—> N у
\ С С
NH, / X / \
HaN N NHa
(XV)
Синтез осуществляют нагреванием нитрила и дициандиами-
да в запаянной ампуле в среде растворителя, содержащего
активный водород (3 ч; КОН). Целевой продукт — 2-замещен-
ный 4,6-диамино-1,3,5-триазин — получают с выходом — 80%.
100
Одним из ценных свойств 4-цианоциклогексена является
способность к комплексообразованию с металлами переменной
валентности. Это свойство было использовано [248] для полу-
чения комплексов одновалентной меди, нашедших применение
в процессе крашения синтетических материалов (сополимеров
акрилонитрила) кислотными красителями. Окраска, получен-
ная с помощью медных комплексов, отличается большой насы-
щенностью.
Синтезы на основе 4-цианоциклогексена с участием двойной
связи циклогексенового кольца. 4-Цианоциклогексен, как и все
непредельные соединения, активно вступает в реакции присо-
единения с различными реагентами. Продукты присоединения
представляют большой практический интерес в качестве полу-
продуктов в синтезе полимеров, красителей, инсектицидов и
лекарственных препаратов.
Окисление в молекуле 4-цианоциклогексена довольно легко
идет по двойной связи. При этом в зависимости от условий
проведения процесса и применяемого окислителя реакция мо-
жет протекать с расщеплением или сохранением цикла. В пер-
вом случае получаются ди- и трифункциональные соединения
(в основном карбоновые кислоты), а во втором — эпоксиды,
спирты, карбонильные и другие кислородсодержащие соедине-
ния.
При окислении 4-цианоциклогексена концентрированным
раствором азотной кислоты образуется смесь двухосновных ки-
слот с малым числом атомов углерода (щавелевая, янтарная и
др.). Этот метод был использован для получения р-цианадипи-
новой кислоты [106]. Однако выход целевого продукта оказал-
ся невысок [—13% (масс.)], что объясняется одновременным
протеканием гидролиза по нитрильной группе:
NC
HNOa
СООН—СООН
С ООН—СН2—С Н—С N—(СН2) 2—С ООН
СООН—(СН2)4—соон.
При изучении процесса окисления азотной кислотой 4-Y-
циклогексенов, где Y-Me, —Н, —СООН, —CN, было установле-
но, что с увеличением электронодонорных свойств заместителя
возрастает индукционный период процесса, а содержание про-
изводных адипиновой кислоты в реакционной смеси уменьшает-
ся. Понижение температуры реакции (катализатор —NH4VO3 и
NaNO2 или без него) сопровождается увеличением выхода ща-
велевой кислоты до 87% и уменьшением выхода цианадипино-
вой до 7%. Другие дикарбоновые кислоты образуются в незна-
чительном количестве [106, 249].
101
Окислением 4-цианоциклогексена кислородом воздуха в при-
сутствии оксидов и гидроксидов бора [250] получают реакци-
онную смесь, содержащую нитрилы З-оксициклогексен-4-карбо-
новой и З-оксоциклогексен-4-карбоновой кислоты. Условия про-
ведения процесса не определены.
Жидкофазное окисление кислородом воздуха, несмотря на
простоту осуществления процесса, имеет ряд существенных не-
достатков. Разделение полученной в результате реакции слож-
ной смеси продуктов сопряжено со значительными технологи-
ческими трудностями. Кроме того, выход целевых продуктов
(эпоксидов) довольно низкий.
В настоящее время большое значение приобрел способ
эпоксидирования олефинов органическими пероксидами (гидро-
пероксидами этилбензола, изопропилбензола, трет-бутила и
Др)-
В отличие от жидкофазного окисления кислородом возду-
ха этот процесс характеризуется высокой селективностью.
В качестве катализатора чаще всего используются соединения
молибдена, вольфрама, ванадия, кобальта и других переход-
ных металлов. При эпоксидировании 4-цианоциклогексена
трет-бутилгидропероксидом применяются ацетилацетонат мо-
либдена [251], магний-силикатный катализатор [252]. Борид-
ные катализаторы [253—256] отличаются высокой селектив-
ностью (>90%), однако выход эпоксида не превышает 40% от
теоретического.
Эпоксидирование 4-цианоциклогексена концентрированными
(70—85%) растворами перуксусной кислоты протекает с вы-
соким выходом 3,4-эпоксициклогексанкарбонитрила-1 [257],
который, как показано в работе [109], имеет транс-строение:
CN CN
/ ( СН3СО3Н(Н+) / (
О
Этот же эпоксинитрил, являющийся полупродуктом в син-
тезе 4-цианоциклогександиола-1,2 и ^-цианадипиновой кислоты
[106], с почти количественным выходом получен эпоксидиро-
ванием 4-цианоциклогексена пербензойной кислотой. 3,4-Эпок-
сициклогексанкарбонитрил-1 может применяться в синтезе по-
лиамидов, красителей, лекарственных препаратов.
4-Цианоциклогексен вступает в реакцию гидроформилирова-
ния. Процесс проводят в присутствии катализаторов: скелет-
ный никель и кобальт, никель на кизельгуре [258—260], На
первом этапе образуется 4-цианоциклогексаналь-1 (XVI), ко-
102
торый затем может быть
(XVII):
превращен в карбоновую
кислоту
(I) (XVI) (XVII)
Процесс окисления полученного альдегида до кислоты про-
водят при температуре 40—50 °C и небольшом давлении кисло-
родом воздуха [258].
Гидрирование 4-цианоциклогексаналя в смеси с метанолом
и гидроксил амином приводит к образованию 1,4-диметиламино-
циклогексана (XVIII) [259]:
(XVIII)
В работе [260] 4-аминометилциклогексанметанол (XIX) по-
лучают оксосинтезом 4-цианоциклогексена над скелетным нике-
лем при исходном соотношении СО Н2=1 1, давлении
14,0 МПа и температуре 130°C:
Из литературных источников известно о получении 1-заме-
щенных циклогексенов [261, 262]. Реакцию проводят при тем-
пературе 180—450°C в присутствии оксидного катализатора
пературе 180—450 °C в присутствии
(А12О3—В20з) и воды:
NC
NC
AI2O3/B2O3; Н2О
(D (XX)
103
Селективность процесса 91—98%, а выход 1-замещенного
изомера (XX) не превышает 64%.
Продукты гидроформилирования 4-цианоциклогексена, так
же как соединения XVII, XVIII и XIX, являясь бифункциональ-
ными, представляют большой интерес в качестве перспектив-
ных мономеров для синтеза полиамидных волокон, пластичес-
ких масс, красителей.
Реакцией 4-цианоциклогексена с галоидангидридами кислот
в присутствии хлорида алюминия при 30—40 °C получают ал-
кил- (4-цианоциклогексил) -Z-кетоны (Z-положение кетогруппы)
[263, 264]:
JN CN
6 ф
О=с—СН3
(XXI)
Алкилирование проводят без доступа влаги в течение 10 ч.
Строение продуктов реакции подтверждено методами ИК-спек-
троскопии. Попытка заменить хлорид алюминия на хлорид
цинка не дает желаемого результата — присоединение ацетил-
хлорида к двойной связи циклогексенового кольца не происхо-
дит [265].
Реакция 4-цианоциклогексена с а-хлорметиловым эфиром
в присутствии безводного хлорида цинка приводит к образова-
нию 1-хлор-2-метоксиметил-4-цианоциклогексана (XXII)
[266]:
Аналогичной реакцией получают и 1-хлор-2-этоксиметил-4-
цианциклогексан.
Синтезированные продукты могут найти применение в ка-
честве пластификаторов и биологически активных веществ.
Кратная связь в молекуле 4-цианоциклогексена предполага-
ет возможность его использования в реакциях циклоалкилиро-
вания. В работах [267—269] описано получение арил- и арил-
алкилциклогексилцианидов (XXIII) взаимодействием 4-циано-
104
циклогексена с бензолом, толуолом, гг-ксилолом в присутствии
безводного хлорида алюминия:
В работе [270] предлагают вести этот процесс при 50—
150 °C, 20,0 МПа в присутствии фторида бора, который в смеси
с циклоолефином вводят в реактор, заполненный оксидами
алюминия, бора, титана.
Арил- и алкилциклогексилцианиды обладают люминесцент-
ными свойствами и могут применяться в композициях жидких
кристаллов [271].
ГЛАВА 4
Синтез полимеров и сополимеров
из побочных продуктов и отходов
нефтехимии
С точки зрения авторов получение полимерных материалов из
отходов и побочных продуктов нефтехимических производств
является очень перспективным. Однако реализация этого про-
цесса в промышленных условиях имеет свои особенности, свя-
занные с низкой реакционной способностью некоторых олефи-
новых углеводородов, входящих в состав кубовых остатков.
Для увеличения конверсии повышают дозировки катализато-
ров, дополнительно вводят в полимеризационную систему моно-
меры— акриловые, винилароматические, способные не только
повысить общий выход полимерных продуктов, но и положи-
тельно влиять на свойства получаемых сополимеров.
Для определения механизма полимеризации необходимо
иметь представление о полимеризационной способности непре-
дельных соединений, содержащихся в побочных продуктах.
В последние годы был опубликован ряд работ по полимериза-
ции и сополимеризации побочных продуктов и отходов нефте-
химических производств в присутствии ионных и радикальных
инициаторов. При этом выбор той или иной каталитической си-
стемы во многих случаях определялся спецификой основного
105
производства, имеющимися активаторами, их стоимостью,
свойствами получаемых сополимеров.
В настоящее^ время в промышленных масштабах осуществ-
лены синтезы низкомолекулярных сополимеров на основе отхо-
дов производства синтетического каучука. Продукты синтезов
могут применяться в качестве пластификаторов, мягчителей,
использоваться в полимерных композициях для улучшения тех-
нологических свойств каучуков, а также для изготовления
пленкообразующих материалов в лакокрасочной промышлен-
ности.
4.1. Олигомеры изопрена
Как отмечалось в гл. побочными продуктами синтеза поли-
изопрена являются линейные и циклические непредельные уг-
леводороды, в частности терпены. В нашей стране осуществле-
но промышленное производство политерпенов [272, 273] из
скипидара на алюмосиликатном катализаторе (выход 53—
54%). Выход политерпенов можно увеличить используя в ка-
честве катализатора сильнокислотный катионит КУ-23 [274].
Продолжительность реакции полимеризации в этом случае со-
ставляет 1,5 ч (выход 94%) [274].
Полимеры с небольшой молекулярной массой можно полу-
чать и из d-лимонена — одного из компонентов кубового остат-
ка ректификации растворителя производства СКИ. При этом
в качестве исходного мономера необязательно использовать ли-
монен [275—277]. Как было показано в работе [275], этот
же полимерный продукт образуется при полимеризации а-пи-
нена в присутствии катионных катализаторов. Объясняется это
изомеризацией а-пинена в d-лимонен и последующей полимери-
зацией последнего. Структура полученного полимера не иссле-
довалась. Однако высказывалось [270] предположение, что
процесс полимеризации протекает по винильной группе. Это
утверждение основывалось на определении (методом озоноли-
за) количества двойных связей, содержание которых составля-
ло 0,4—0,5 на мономерное звено. Исследования [276] подтвер-
дили это предположение.
Изучение сополимеризации изопрена с 7,11-диметил-З-ме-
тилендодекатриеном-1,6,11 —линейного тримера изопрена (об-
разование его в процессе получения синтетического изопреново-
го каучука не установлено) проводили [278] на каталитичес-
кой системе TiCl4—АЦизо-С^Н^з при температуре 20°C, пол-
ном отсутствии воздуха и влаги, концентрации мономеров
— 10% (масс.). Методом ИК-спектроскопии установили, что
при исходном мольном соотношении изопрен: 7,11-диметил-З-
метилендодекатриен-1,6,11, равном 7 1 и степени конверсии
<30% наблюдается постоянство отношения оптических полос
106
РИС. 4.1. ИК-спектры сополимера изопрена с 7,11-диметил-З-метилендодека-
триеном-1,6,11 (/) и 7,11-диметил-2-метилендодекатриеном-1,6,11 (2)
с волновыми числами 890 и 840 см-1, указывающими на обра-
зование истинных сополимеров (рис. 4.1). Этим же методом
обнаружили полосы поглощения в области 890 см-1, характер-
ной для концевых винилиденовых групп, ц отсутствие полосы
при 920 см-1, относящейся к внеплоскостным колебаниям груп-
пы —СН = СН2 в сополимере. Следовательно, винилиденовые
группы не участвуют в реакции роста цепи. Исследования ме-
тодом УФ-спектроскопии подтвердили, что в процессе роста
цепи принимает участие лишь сопряженная система двойных
связей тетраена, в то время как внутренняя —С = СН(СН)3 и
концевая винилиденовая СН2=С(СН3)— связи практически не
взаимодействуют.
Анализ экспериментальных данных показал, что скорость
гомополимеризации тетраена на порядок ниже, чем скорость
расходования линейного триена с системой сопряженных двой-
ных связей. Присоединение тетраена к растущей полимерной
цепи в сравнении с присоединением линейного триена, содер-
жащего сопряженные двойные связи, протекало с образовани-
ем более объемного радикала в боковом звене. Это оказывало
экранирующее действие на активные центры, в результате чего
снижалась скорость гомополимеризации тетраена.
Эти выводы были подтверждены и анализом констант сопо-
лимеризации, которые для пары изопрен — триен составили
Г1=3,01 (Г1/г2=10), а для пары изопрен — тример Г1 =
= 1 (ri/r2 = l). Для пары изопрен — триен константа скорости
107
присоединения изопрена к активному центру в три раза пре-
вышала константу скорости присоединения сомономера к этому
типу активных центров, тогда как для второго случая наблюда-
лось практическое равенство константы, что, однако, не исклю-
чало влияния предпоследних звеньев на рост цепи [278].
Сополимеризацию изопрена с его мечеными линейными ди-
мерами изучали в растворе изопентана при температуре 30 °C
в присутствии катализатора, состоящего из триизобутилалюми-
ния и тетрахлорида титана [279]. Меченые димеры были син-
тезированы из меченого изопрена, полученного реакцией три-
тийбромирования по следующей схеме:
2Р+5Вг24-6Т2О «=± 2ТРО3+ЮТВг
—20°С
СН2=С—СН=СН2 + ТВг -----> т—СН2—С=СН—СН2—Вг.
<^н3 ёнз
Последующим дегидробромированием получали меченый
изопрен:
120 °с
СН2Т—С=СН—СН2—Вг ------> СНТ=С—СН=СН2 + NaBr + Н2О.
| NaOH |
СН3 СН3
Димеризацию полученного изопрена проводили в присутст-
вии катализатора, состоящего из триизобутилалюминия и тет-
рахлорида титана, при мольном соотношении Al : Ti = 2 (60°С,
34). Полученная смесь димеров изопрена имела следующий
состав [% (масс.)]: 2,6-диметил-1,3,6-октатриен— 60; 2,7-ди-
метил-1,3,6-октатр иен— 10; 1 и 2,4-диметил-4-этенил-1-цикло-
гексен — 30, была использована для сополимеризации.
Молекулярную массу полимера М определяли вискозимет-
рическим методом в толуоле при 25 °C и рассчитывали по фор-
муле:
lgM = 5,063+ 1,28 (lg£),
где £— характеристическая вязкость, д-л/г.
Все полученные полимеры были радиоактивными [279].
Удельная радиоактивность полимеров зависела от исходного
состава мономерной смеси. Снижение содержания димеров изо-
прена в исходной мономерной смеси с 10 до 0,33% (масс.) при-
водило к уменьшению содержания радиоактивных продуктов в
сополимерах с 1,0 до 0,05% (масс.) соответственно. Увеличе-
ние глубины полимеризации способствовало обогащению со-
полимера радиоактивными продуктами. При этом отмечено, что
в процесс сополимеризации вступали только димеры изопрена
(табл. 4.1).
108
ТАБЛИЦА 4.1
Содержание ди- мера в исходной смеси с изопре- ном, % (масс.) Конверсия, изопрена, % Молекулярная масса полимер- ных продуктов Содержание ра- диоактивных звеньев в по- лимере, % (масс.) Среднее число звеньев изопре- на на звено ди- мера в макро- молекуле поли- изопрена
10,00 41,5 354 600 0,95 210
10,00 48,0 308 000 1,04 192
5,00 46,0 238 400 0,55 365
5,00 51,0 200 900 0,58 344
2,00 80,6 245 300 0,36 355
0,88 74,0 358 300 0,14 1424
0,88 72,00 354 600 0,14 1409
0,33 73,0 373 600 0,05 3924
0,33 75,0 358 300 0,05 4053
Введение в полимерную цепь димеров изопрена существен-
но влияло на скорость кристаллизации образцов. Уменьшение
количества звеньев димера изопрена в полимере до 1,5% сни-
жало скорость кристаллизации образцов почти в два раза, а
повышение их до 6,5% приводило к тому, что образец не крис-
таллизовался в течение двух суток.
Процесс сополимеризации изопрена с 1,3,6-октатриеном изу-
чали [280] при воздействии каталитической системы, состоя-
щей из тетрахлорида титана и триизобутилалюминия [мольное
соотношение Al/Ti = 1; температура процесса 25 °C; раствори-
тель— толуол; концентрация мономеров в растворе <10%
(масс.)].
Все полученные сополимеры представляли собой кау-
чукоподобные продукты. Температура стеклования (/с) зависе-
ла от состава полученных сополимеров. Для сополимера, содер-
жавшего [% (масс.)]изопрена — 60, триена — 40, tc =—19°С;
изопрена — 88, триена—12, tc = —40 °C. Для полиизопрена и
полиоктатриена температура стеклования составляет —58 и
—5 °C соответственно. Характеристическая вязкость, измерен-
ная в растворе гексана (25°C), изменялась в интервале 1—
3 д-л/г в зависимости от состава сополимера и понижалась с
увеличением содержания 1,3,6-октатриена в исходной мономер-
ной смеси.
В ИК-спектрах полученных сополимеров присутствуют поло-
сы поглощения в‘ областях 840 и 970 см-1, характерных для
цис- и транс-конформаций в изопреновых фрагментах и транс-
звеньев октатриенового фрагмента.
Константы сополимеризации изопрена (rj с 1,3,6-октатрие-
ном (г2) были рассчитаны методом Файнмана — Росса (а),
линейным графическим методом Келена — Тюдоша (б) и ме-
109
тодом Файнмана — Росса с последующей переиндексацией
(в):
Метод 5
расчета
Г\ 2,97 2,90 3,08 ср. 3,01±0,3
Г2 0,27 0,26 0,27 ср. 0,27±0.05
Были рассчитаны также [280] элементарные константы ско-
ростей роста [fe-Ю2, л/(моль-с)]: £Ии = 2,18; £ит = 0,73;
&тт = 0,21; А>ти = 0,76.
Установлено, что взаимодействие 1,3,6-октатриена с расту-
щей полимерной цепью протекало по типу 1,4-присоединения,
характерного для 1,3-диенов. Более высокая реакционная спо-
собность изопрена объяснялась стерическими затруднениями,
обусловленными особенностями строения 1,3,6-октатриена.
Полимеры с высокими адгезионными свойствами, высокой
теплостойкостью и хорошей совместимостью с другими поли-
мерами получали сополимеризацией олигомеров изопрена —
2,4-диметил-4-винил-1 -циклогексена, диметил-1,3,6-октатриена —
с алифатическими ненасыщенными углеводородами: бутадие-
ном, пипериленом, изопреном; ароматическими непредельными
углеводородами; стиролом, винилтолуолом; ненасыщенными
ациклическими углеводородами; циклопентадиеном, дицикло-
пентадиеном [281], а также с монотерпеновыми углеводорода-
ми [282]. Реакцию проводили в присутствии 0,05—15% (масс.)
катализатора Фриделя — Крафтса (А1С13, TiCl4) с возможными
добавками галоидалкилов в ароматическом углеводородном ра-
створителе в интервале температур от —50 до 100 °C.
Взаимодействием фенолов и крезолов с сополимером [281]
получали продукт, имевший невысокую молекулярную массу
(Мп = 960, выход 81%), который в смеси с сополимером этилена
и винилацетата (1 4) применяли в качестве клеев для метал-
лов с высокой стойкостью к отслаиванию.
Определенный интерес представляет исследованный в рабо-
те [283] процесс сополимеризации непредельных соединений,
содержащихся в кубовом остатке ректификации возвратного
растворителя производства СКИ-3.
Проведенные исследования показали, что на основе олиго-
меров изопрена в присутствии катализаторов Фриделя —
Крафтса могут быть получены низкомолекулярные полимерные
продукты, представляющие собой маслообразные жидкости.
При этом установлено, что на выход целевых продуктов основ-
ное влияние оказывали: дозировки стирола, катализатора и
продолжительность процесса.
Повышение дозировки хлорида алюминия в исходные моно-
меры и увеличение продолжительности процесса приводило к
110
резкому возрастанию выхода полимерных продуктов (рис. 4.2).
Была сделана попытка представить полученные эксперимен-
тальные данные в виде экспоненциальной зависимости;
В t= [ 1 — exp ( — а£)т°.4) ] • 100,
где В — выход полимерных продуктов, %; D — дозировка катализатора,
масс. ч. на олигомеры изопрена; т — продолжительность процесса, ч; а — эм-
пирический коэффициент, ч~!.
Сравнение экспериментальных данных с расчетными, полу-
ченными на основе вышеприведенного уравнения, позволило
сделать вывод, что они удовлетворительно согласуются между
собой при значении эмпирического коэффициента а = 25,5 ч-1.
При этом наилучшая сходимость экспериментальных и расчет-
ных данных наблюдалась при применении дозировок хлорида
алюминия более 1,0% (масс.) на мономеры.
Существенное влияние на выход полимерных продуктов и
стабилизацию процесса сополимеризации оказывала дозировка
стирола в исходные олигомеры изопрена. Увеличение стироль-
ной добавки в олигомеры изопрена приводило к повышению
выхода полимерных продуктов. Однако на увеличение молеку-
лярной массы получаемых сополимеров добавка стирола не
оказывала сильного влияния. Молекулярная масса получаемых
сополимеров при увеличении содержания стирола в исходной
мономерной смеси с 10 до 100% (масс.) изменялась с 700—
1300 до 1800—2500. Все полученные продукты представляли
собой маслянистые жидкости коричневого цвета.
Использование в процессе полимеризации олигомеров изо-
прена других катализаторов (тетрахлорида титана, олова) ока-
залось не эффективным. Выходы полимерных продуктов сни-
жались на 18—30%.
Испытания полученных полимеров в качестве добавки в
бутадиеновый каучук в количестве 5,0 масс. ч. на 100 масс. ч.
каучука показали, что введение указанного продукта повышает
пластичность резиновых смесей, снижает вязкость по Муни и
по своей эффективности приближается к общеизвестной группе
пластификаторов. Свойства резиновых смесей и вулканизатов
111
ТАБЛИЦА 4.2
Показатель Контрольный образец бу- тадиенового каучука Содержание стирола в ис- ходной смеси олигомеров изопрена, % (масс.)
0 25 50 100
Вязкость по Муни МБ 1+4 (100 °C) 43 44 41 43 41
Условная прочность при растяжении, МПа Относительное удлинение, %: при разрыве 17,5 16,4 17,7 18,1 18,3
510 520 530 515 500
остаточное после разрыва 10 9 10 8 8
Эластичность по отскоку, % 53 51 55 54 52
Массовая доля золы, % (масс.) Потери массы при 105 °C, % (масс.) 0,17 0,15 0,16 0,19 0,17
0,20 0,15 0,18 0,21 0,19
Массовая доля нафтам-2, % (масс.) 1,0 1,0 1,0 1,0 1,0
на основе бутадиенового каучука с добавками полимера из-
олигомеров изопрена приведены в табл. 4.2.
Применение более высоких дозировок полимера (до
15 масс. ч. на 100 масс. ч. каучука) на основе олигомеров
изопрена и стирола в резиновых смесях на основе бутадиено-
вого каучука позволило улучшить технологические свойства
смесей; по физико-механическим показателям опытные и конт-
рольные вулканизаты отличались незначительно [284—286].
Применение радикальных инициаторов для полимеризации
побочных продуктов синтеза СКИ оказалось мало эффектив-
ным. Выходы полимерных продуктов [286, 287] при использо-
вании в качестве радикальных инициаторов гидропероксидов
изопропилбензола, изопропилциклогексилбензола, пероксида
бензоила и динитрила-азо-бис-изо-масляной кислоты были не-
большие и не превышали 30—35% даже при высоких дозиров-
ках инициатора [(до 3,5% (масс.)].
Повышение общего выхода полимерных продуктов в случае
применения радикальных инициаторов возможно только при
введении в кубовые остатки активных мономеров, например
стирола. Ощутимый эффект наблюдался при высоких дозиров-
ках стирола [более 600% (масс.) на олигомеры]. Общий выход
сополимеров удалось повысить до 80% в присутствии 3,5%
(масс.) гидропероксида изопропилциклогексилбензола (130°C,
12 ч). Все синтезированные полимерные продукты имели не-
высокую молекулярную массу (Мп = 600—1500), возраставшую
с увеличением дозировки стирола до 5000 [286, 288]. В процес-
се полимеризации олигомеров изопрена образовались также
нерастворимые полимерные продукты, которые в процессе син-
теза выделялись из раствора в виде осадка.
112
Низкие выходы полимерных продуктов при наличии ради-
кальных инициаторов объясняются присутствием в кубовом
остатке ингибиторов процесса сополимеризации.
Структура получаемых сополимеров [283—288] не изуча-
лась из-за сложности анализа. Это связано прежде всего с тем,
что исходная мономерная смесь состоит из разнообразных оле-
финовых соединений (см. разд. 2.2), которые в процессе сопо-
лимеризации входят в состав полимерной цепи.
4.2. Олигомеры бутадиена
В литературе имеются сведения о получении полимеров и со-
полимеров из олигомеров бутадиена. Подробно рассмотрены
процессы полимеризации и сополимеризации олигомеров бута-
диена на катионных катализаторах и каталитических системах,
состоящих ,из галогенида титана и алюминийорганических со-
единений [289—292]. Из исследуемых олигомеров бутадиена
наиболее изучен димер бутадиена — 4-винилциклогексен. Ряд
работ [277, 293—299] посвящен изучению структуры и свойств
полимеров на основе 4-винилциклогексена, полученных с ис-
пользованием катионных катализаторов и катализаторов Циг-
лера — Натта.
Полимеризация 4-винилциклогексена в зависимости от при-
роды каталитической системы протекает по двойной связи ви-
нильной группы с образованием полимера со структурой
по механизму циклополимеризации, с получением полимера со
структурой
или по смешанному механизму.
Полимеризацию 4-винилциклогексена [293] проводили на
каталитических системах: BF3; BF3—(С2Н5)2О; TiCI4—А1(изо-
С4Н9)3. Полимерный продукт с приведенными зыше структура-
ми с выходом 20% был получен на каталитической системе —
тетрахлорид титана — триизобутилалюминий (20°C, 14 сут.).
Растворимая в бензоле фракция составляла 45% (масс.) от
8-627 ИЗ
общей массы получаемого полимера fa представляла собой
продукт 100%-ной циклизации. /
На каталитической системе а=71С13—А1(изо-С4Н9)3 полу-
чали полимер, структура которого/состояла из звеньев, соеди-
ненных через винильную группуУСтроение полученного поли-
мера подтверждается данными ИК-спектроскопии. Полученный
полимер [294] имел кристалл10ескую структуру и температуру
плавления >400°C. /
Были проведены исследования полимеризационной способ-
ности 4-винилциклогексену [296] на каталитических системах
TiCl4—А1(изо-С4Н9)3; aATiCl3—А1(изо-С4Н9)3; a = TiCl3. На
каталитической системе TiCl4—Al (изо-С4Н9)3 масимальный вы-
ход поливинилциклогексена (70%) был достигнут при проведе-
нии полимеризационного процесса при 100 °C в течение 36 ч.
В ИК-спектрах синтезированных полимеров присутствовали
полосы поглощения при 655, 1050, 1080, 1660 и 3020 см-"1, отно-
сившиеся к колебаниям эндоциклической двойной связи. На ос-
нове данных ЯМР-спектроскопии вычислили процент циклиза-
ции (Ц) полимера по уравнению:
100 (de- b)
Ц“ (de — д + а) 1
где а, в — количество $р3-связанных водородов в насыщенном и ненасыщен-
ном звеньях поливинилциклогексена, соответственно; с — количество $р2-свя-
занных водородов в ненасыщенном звене поливинилциклогексена; d — отно-
шение ($р2/$р3)-связанных водородов, определенное методом ЯМР-спектро-
скопии.
Рассчитанное по этой формуле [296] содержание фракций
полимера 100%-ной циклизации составило 45% (масс.). Таким
образом, повышение температуры процеса полимеризации ви-
нилциклогексена на каталитической системе TiCl4—Al (изо-
С4Н9)3 способствовало протеканию реакции по типу 1,5-диена
с образованием бициклической структуры.
Исследования показали, что термоокислительная деструк-
ция поливинилциклогексена начиналась при 220 °C, а при
480 °C полимер практически полностью деструктировал.
При полимеризации 4-винилциклогексена на каталитической
системе a = TiCl3 — А1(изо-С4Н9)3 образовывался полимер ли-
нейного строения, т. е. полимеризационпый процесс протекал
преимущественно по двойной связи випильной группы, что под-
тверждалось данными ИК-спектроскопии. Отсутствие в ИК-
спектре полимера полос поглощения, характерных для виниль-
ной группы (области 995, 1645 и 3080 см-1)» и присутствие ин-
тенсивных полос (области 655, 1050, 1080, 1660 и 3020 см-"1),
свойственных эндоциклической двойной связи, доказывали ли-
нейность строения полученного полимера.
Изучение термоокислительной деструкции полученного по-
лимера показало, что при нагревании образцов полимера на
114
воздухе наблюдается появление пика при 180 °C, объясняемого
окислением полимера по двойной связи циклогексенового коль-
ца, а начало интенсивной деструкции отмечается при 280—
320 °C.
Сохранение двойной связи в циклогексеновом кольце поли-
винилциклогексена важно при его последующей модификации.
Сохранение двойной связи в циклогексеновом кольце позволяет
увеличить адгезионную способность сополимеров 4-винилцикло-
гексена. Известно, что полимеры винилциклогексана обладают
хорошими диэлектрическими свойствами. Однако одним из су-
щественных их недостатков является малая адгезионная спо-
собность. Введение звеньев 4-винилциклогексена в молекулу
полимера винилциклогексана улучшает адгезионные свойства
полимера без ухудшения диэлектрических показателей.
Полимеризацией 4^винилциклогексена в присутствии а-
ТЮз получали полимерный продукт с малой ненасыщенно-
стью и низкой циклизацией, определенной по данным ЯМР-
спектроскопии. Полимеры представляли собой аморфные белые
вещества с температурой размягчения 200—205 °C. Деструкция
полимера наступала при 370 °C и завершалась при 480 °C»
[296].
Исследования по сополимеризации 4-винилциклогексена с
винилциклогексаном на каталитических системах ТЮз—
А1(изо-С4Нэ)з и TiCU—А1(изо-С4Н9)з приведены в монографии
[299]. На первой каталитической системе сополимеризация про-
текала только по винильной группе молекулы 4-винилциклогек-
сена. Уменьшение содержания винилциклогексана в исходной
мономерной смеси с 4-винилциклогексеном приводило к обога-
щению сополимера звеньями 4-винилциклогексена. На второй
каталитической системе полимеризация 4-винилциклогексена
протекала не только по винильной группе (fep), но и по меха-
низму циклополимеризации (£ц). При этом вклад циклополи-
меризации значительно меньший, чем в случае гомополимери-
зации 4-винилциклогексена на этих каталитических системах.
В данном случае имело место протекание двух конкурирующих
реакций [299]:
8‘
115
и степень циклизации определялась соотношением скоростей
этих реакций. 7
Сравнение значений констант сополимеризации Г1=0,2±
±0,026 и г2 = 3,8 ±0,266, определенных нелинейным методом
наименьших квадратов, показалоУчто 4-винилциклогексен обла-
дает повышенной реакционной способностью по сравнению с
винилциклогексаном. Более высокая реакционная способность
4-винилциклогексена объяснялась [299] наличием несопряжен-
ной двойной связи в молекуле 4-винилциклогексена, а близость
произведения Г[Г2 к единице (0,84) свидетельствовала об обра-
зовании статистических сбполимеров.
Сополимеризация 4-винициклогексена с акрилонитрилом в
присутствии C2H5AICI2 протекала с образованием чередующих-
ся сополимеров, состав которых в значительной степени опре-
делялся температурой процесса. При этом 4-винилциклогексен
входил в полимерную цепь преимущественно в виде бицикличе-
ских мономерных звеньев [300].
Сополимеры на основе 4-винилциклогексена и бутена-1 бы-
ло предложено получать [301] В присутствии катализаторов
Циглера — Натта при 20—100 °C. В этих условиях были полу-
чены сополимеры бутена-1 с 0,1—2,0% (мол.) 4-винилциклогек-
сена, обладающие следующими характеристиками: (Mwl
1Мп) >13; полупериод кристаллизации (77 К)—1,9—3,1 мин;
степень кристалличности свежеприготовленных образцов 30,5—
31,8 и 41,9—43,5 — состаренных; изотактичность 79—82%.
Перспективным оказалось использование 4-винилциклогек-
сена в качестве третьего мономера при получении этиленпро-
пиленовых сополимеров. Сополимеризацию проводили на ката-
литических системах, состоявших из сесквихлоридов алюминия
и VC14 (или VOCI3); УС14(изо-С4Н9)2С1; VC14—А1(С2Н5)з;
VC14—А1(изо-С4Н9)2С1. Полимеры, полученные в присутствии
каталитической системы сесквихлорид алюминия — VCh, имели
равномерное распределение двойных связей в сополимерах и
обладали высокими механическими показателями. Прочность
вулканизатов в зависимости от молекулярной массы составля-
ла 20,0—29,6 МПа. Применение двух других каталитических
систем обеспечивало получение сажевых вулканизатов с высо-
ким сопротивлением разрыву (17,5—20,2 МПа), однако при
этом они имели высокие остаточные удлинения (82—91%).
Наихудшими показателями обладали сополимеры, полученные
в присутствии каталитической системы А1(С2Н5)2С1—VC14,—
более 50% получаемого сополимера не содержало кратных свя-
зей и вулканизаты имели очень низкие сопротивления разрыву
(4 МПа). Все синтезированные сополимеры [302] были аморф-
ными, температура стеклования их колебалась в пределах от
—55,5 до —60°С, а молекулярная масса — от 12-104 до
38-104.
116
ТАБЛИЦА 4.3
Показатель Сажевые вулканизаты
сополимер эти- лена, пропилена, дициклопента- диена этиленпропиле- новые каучуки с 4-винилцикло- гексеном
Жесткость по Дефо, Н 8,50 11,50
.Условное напряжение при 300%-ном удли- нении, МПа 14,8 14,5
Условная прочность при растяжении, МПа 'Относительное удлинение, %: 22,4 28,1
при разрыве 440 470
остаточное после разрыва 20 19
-Эластичность по отскоку, % 44 52
Твердость по ТМ-2 — 60
Сопротивленйе раздиру, кН/м 47 52
Коэффициент теплостойкости при 100 °C:
по сопротивлению разрыву 0,43 0,48
по относительному удлинению 0,62 0,75
Сравнительные испытания сажевых вулканизатов этилен-
пропиленовых каучуков с 4-винилциклогексеном с сажевыми
вулканизатами сополимера этилена, пропилена и дициклопен-
тадиена показали, что по основным физико-механическим по-
казателям они равноценны (табл. 4.3).
Процесс сополимеризации бутадиена и изопрена с линейны-
ми и циклическими полиенами изучался на каталитических си-
стемах Циглера — Натта [303—307]. Благодаря этим исследо-
ваниям была найдена возможность использовать диеновые ди-
меры для модификации каталитического комплекса, а также
для получения полимеров с различным содержанием непре-
дельных связей в боковом звене, что создавало хорошие пред-
посылки к осуществлению полимераналогичных превращений.
Сополимеризация бутадиена с его линейными олигомерами:
октатриеном-1,3,6, триметилгептатриеном-1,4,6, додекатетрае-
ном-1,3,6,10 в присутствии каталитической системы на основе
TiI2Cl2—Al (шю-С4Н9)з протекала со статистическим распреде-
лением мономерных звеньев в цепи (Г1Г2->1). Сопоставление
значений констант сополимеризации ц и г2 указывало на более
высокую активность бутадиена в сравнении с линейными поли-
енами (табл. 4.4).
На основе экспериментальных данных был сделан вывод,
что в процессах сополимеризации относительная реакционная
способность полиена не зависит от размера ненасыщенного
радикала, находящегося рядом с сопряженной системой двой-
ных связей. Введение триметилгептатриена-1,4,6 в реакцион-
ную массу, содержавшую бутадиен, приводило к уменьшению
117
ТАБЛИЦА 4.4
Линейный олигомер п1гг
Система TH2CI2—Al (изо-С4Нэ)з, [Al/Ti-8] Триметилгептатриен-1,4,6 5,30 0,20 1,06 26,5 Октатриен-1,3,6 3,84 0,26 1,00 14,8 Додекатетраен-1,3,6,10 3,65 0,23 0,84 15,8 Система TiCl4—Al (изо-С4Нэ)з, [Al/Ti-3] Триметилгептатриен-1,4,6 2,88 0,34 0,97 8,4 Октатриен-1,3,6 1,95 0,37 0,72 5,3
скорости сополимеризации и снижению молекулярной массы
образующихся сополимеров.
Из анализа результатов спектральных исследований следу-
ет, что в реакции участвовала только система сопряженных
двойных связей диеновых олигомеров. Об этом свидетельство-
вало отсутствие полосы поглощения в УФ-спектрах при 227—
229 нм. По данным ИК-спектроскопии, с увеличением мольной
доли триена в сополимере изменялась интенсивность полос
поглощения в областях 970, 910, 735 см-1 ih одновременно воз-
растала интенсивность полосы поглощения в области 1380 см4,
что связано с ростом в полимерной цепи числа триеповых
фрагментов. Кроме того, увеличение содержания в сополимере
триметилгептатриена-1,4,6 с 0 до 67% (масс.) снижало долю
1,4-ф/с-структур с 92 до 39,5%, а содержание 1,4-тракс-струк-
тур и 1,2-звеньев возрастало с 3,7 до 33,8% и с 4,3 до 26,7%,
соответственно.
Добавление к бутадиену менее активного мономера (линей-
ного олигомера), наряду с уменьшением скорости сополимери-
зации, приводило и к увеличению в сополимере доли транс- и
уменьшению ^uc-звеньев (табл. 4.5). Такие изменения брутто-
микроструктуры могут быть связаны как с влиянием менее
активного мономера на содержание 1,4-^ис-звеньев в бутадие-
новом фрагменте сополимера, так и с вхождением в цепь сопо-
лимера триеновых фрагментов с отличающейся от бутадиено-
вой части конфигурацией звеньев.
Циклические олигомеры бутадиена (4-винилциклогексен,
циклододекатриен-1,5,9) в интервале молярных соотношений
4-винилциклогексен/дихлордииодтитан-15—200 и циклододека-
триен-1,5,9/дихлордииодтитан-15—200 не влияли на скорость
полимеризации бутадиена, молекулярную массу, микрострукту-
ру и температуру стеклования полибутадиена, т. е. их добавка
практически не влияла на природу и концентрацию активных
центров, а также на соотношение констант скоростей роста и
обрыва цепи.
118
Изопрен так же как и бутадиен вступал в реакцию сополи-
меризации с линейными олигомерами: октатриеном-1,3,6, три-
метилгептатр иеном- 1,4,6,2,6-диметилоктатриеном-1,3,6, 7,11-ди-
метил-З-метилендодекатриеном-1,6,11. При этом образовыва-
лись истинные сополимеры, что подтверждалось данными тур-
бодиметрического титрования. Константы сополимеризации
изопрена с линейными олигомерами, рассчитанные по диаграм-
ме состава сополимера от состава мономерной смеси, приведе-
ны в табл. 4.6.
В процессе сополимеризации изопрена с октатриеном-1,3,6
и триметилгептатриеном-1,4,6 на каталитической системе
TiCl4—Al (изо-С4Н9)з диен является более активным мономе-
ром. При сополимеризации с 2,6-диметилоктатриеном-1,3,6 и
7,11-диметил-З-метилендодекатриеном-1,6,11 константы сополи-
меризации выравнивались. Сходство констант сополимеризации
[307] объяснялось подобием структур триена и тетраена с изо-
преном.
Увеличение содержания диеновых олигомеров в реакцион-
ной смеси приводило к уменьшению скорости сополимериза-
ТАБЛИЦА 4.5
Линейный олигомер Содержание линейного олигомера в сополиме- ре, % (мол.) Содержание звеньев, % (масс.) (Д-л)/г
цис- транс- гвинильные
Система TiI2Cl2—А1(изо-С4Н9)з Октатриен-1,3,6 0 92 4 4 2,7 8 76 19 5 1,5 17 73 21 6 1,2 24 67 29 5 1,0 37 66 28 6 Триметилгептатриен-1,4,6 3 91 4 5 2,6 10 85 8 7 15 72 14 14 2,4 30 57 24 18 67 40 34 26 1,6 Додекатетраен-1,3,6,10 4 85 9 6 2,3 7 78 15 7 10 78 15 7 1,9 11 77 15 8 21 73 21 6 1,4 30 65 28 7 1,1
Система TiCl4—АЦнзо-СДЬЬ
Триметилгептатриен-1,4,6 0 64 32 4
27 55 35 10
41 35 38 27
Октатриен-1,3,6 7 59 38 3
15 52 43 5
20 42 54 4
37 41 56 3
119
ТАБЛИЦА 4.6
Линейный олигомер Г1Г2 । Г1/Г2
Система TiCl4—Al (изо-С4Н9); ь [Al/Ti-1]
Октатриен-1,3,6 3,01 0,27 0,81 И,1
Триметилгептатриен-1,4,6 2,00 0,37 0,74 5,4
2,6-Д иметилоктатриен-1,3,6 1,00 1,00 1,00 1,0
7,11-Диметил-З-метилендодека- 1,00 1,00 1,00 1,0
триен-1,6,11
Система VOCI3—А1 (изо-С4Н9) з, [A1/V-2]
Октатриен-1,3,6 1,00 1,00 1,00 1,0
Триметилгептатриен-1,4,6 0,75 2,03 1,52 0,37
ции, при этом наибольшее снижение отмечалось при введении
тетраена. Снижение скорости сополимеризации связано, веро-
ятнее всего, с тем, что каталитическая система TiCl4—Al (изо-
С4Нэ)з выполняла роль цис- или транс-регулятора присоедине-
ния полиена к полимерной цепи.
При сополимеризации изопрена с дипентеном на каталити-
ческой системе TiCl4—А1(изо-С4Н9)з отмечалось резкое умень-
шение выхода сополимеров с увеличением содержания димера
изопрена в исходной мономерной смеси. Добавка 50% (масс.)
дипентена уменьшала скорость полимеризации изопрена при-
мерно в 6 раз. Характеристическая вязкость получаемых про-
дуктов повышалась с 2,4 до 3,6 (g-л/г). Вероятнее всего это
связано с тем, что дипентен, имея малореакционноспособные
двойные связи, способен координироваться на активном цент-
ре и затруднять тем самым подход к нему изопрена, что и
приводит к снижению скорости полимеризации.
Сополимеризация изопрена с триенами на цис- и транс-ре-
гулирующих каталитических системах — галогенах титана и
ванадия в сочетании с алюминийорганическим соединением —
показала [307], что большей активностью обладает триметил-
гептатриен-1,4,6 (по сравнению с октатриеном-1,3,6). Отмеча-
лась инверсия активностей, в результате чего менялась после-
довательность реакционноспособности этих триенов. Наблюдае-
мый, эффект связывали с наличием группы СНз в молекуле
триметилгептатриена-1,4,6, которая увеличивала его активность
в сравнении с октатриеном-1,3,6 по отношению к изопрену, так-
же имевшему СН3-группу. Еще большее сближение реакцион-
ной способности отмечалось при сополимеризации изопрена с
2,6-диметилоктатриеном-1,3,6 (п-г2-1).
Сополимеризация бутадиена с октатриеном-1,3,6 и додека-
тетраеном-1,3,6,10 на каталитической системе TiI2Cl2—Al (изо-
С4Н9)3 протекала с выравниванием реакционной способности
120
триена и тетраена. При этом длина ненасыщенного радикала
не оказывала заметного влияния на скорость полимеризации.
Аналогичная картина наблюдалась при сополимеризации [307]
изопрена с 2,6-диметилоктатриеном-1,3,6 и 7,11-диметил-З-ме-
тилендодекатриеном-1,6,11 в присутствии TiCl4—Al (изо-С4Н9)з.
Таким образом, диеновые олигомеры, образовавшиеся в
процессе синтеза полимерных материалов, в зависимости от
их> строения оказывали далеко не однозначное влияние на ки-
нетические параметры и характер протекающих полимеризаци-
онных процессов. Циклические олигомеры бутадиена и изопре-
на (4-винилциклогексен, циклододекатриен-1,5,9, дипентен) не
вступали в реакцию сополимеризации [303, 307] с сопряжен-
ными диенами и не оказывали влияния на микроструктуру по-
лимерной цепи, ее молекулярную массу, температуру стеклова-
ния и скорость процесса полимеризации (в случае бутадиена).
Присутствие в полимеризационной системе дипентена приводи-
ло к снижению скорости полимеризации изопрена.
Линейные диеновые олигомеры в отличие от циклических
проявляли большую активность в процессах полимеризации.
Они вступали в реакцию сополимеризации с исходными диена-
ми по имеющимся сопряженным связям независимо от катали-
тической системы. Изолированные двойные связи оказывались
незатронутыми, что делало эти полимерные продукты перспек-
тивными для полимераналогичных превращений.
Сополимеризацию циклододекатриена-1,5,9 с метилметакри-
латом проводили [308] в присутствии 0,5% (мол.) пероксида
бензоила при 80 °C. Повышенный интерес к циклододекатрие-
ну-1,5,9 вызван вероятностью его применения в качестве плас-
тификатора, антиокислителя мономера при сополимеризации с
винильными производными [309—311].
При изучении полимеризационной способности циклододе-
катриена-1,5,9 установлено, что наибольшую активность прояв-
ляет одна из двух транс-двойных связей. Это было подтверж-
дено исследованиями мономеров и сополимеров методом ПК-
спектроскопии. В ИК-спектре присутствовали полосы поглоще-
ния в областях 968 и 1655—1665 см-1, характерные для цис- и
одной транс-двойных связей, а полоса в области 705 см-1, ха-
рактерная для другой транс-двойной связи, отсутствовала
[312, 313].
Константы сополимеризации, определенные методом Файн-
мана — Росса, для системы метилметакрилат — циклододекат-
риен-1,5,9 составили: Г1 = 4,84, г2 = 0,2. Сравнение этих величин
показало, что радикал метилметакрилата с большей скоростью
(в 4,8 раза) реагирует со своим мономером, чем с мономером
циклододекатриена-1,5,9, а радикал циклододекатриена-1,5,9 с
большей скоростью (в 5 раз) реагирует с мономером метилмет-
акрилата, чем со своим.
121
ТАБЛИЦА 4.7
Показатель Температура, °C
120 140 160 200
Степень превращения, % (масс.) димеров 29,4 59,0 80,6 100 тримеров 49,6 80,0 94,1 100 Динамическая вязкость при 20°C, 16,0 8,75 7,08 5,57 Пас Иодное число, г 12/100 г сополимера 109 140 137 119 Молекулярно-массовая характеристи- ка MurlO"3 8,3 4,5 2,8 3,5 Л/п-10-2 5,9 3,3 2,8 2,7 Mw/Mn 14,1 13,5 10,0 12,8
Как уже отмечалось, при очистке возвратного растворителя
производства бутадиенового каучука в кубах ректификацион-
ных колонн концентрируется смесь олигомеров бутадиена в
растворителе. Выделение индивидуальных соединений из кубо-
воц части сложно, трудоемко и экономически невыгодно. По-
этому было предложено [314] получать полимерные продукты
из смеси этих олигомеров на гетерогенном катализаторе при
температуре 160—200 °C, давлении 0,3—0,6 МПа и скорости
подачи сырья 0,12—0,50 ч-1. В полимеризационном процессе
использовалась кубовая жидкость ректификации толуола сле-
дующего состава [% (масс.)]: 4-винилциклогексен—13,40;
циклододекатриен-1,5,9 — 3,40; н-додекатетроен — 3,80; низко-
молекулярный полимер — 0,52; толуол — 78,88. Практически
100%-ная конверсия олигомеров бутадиена была достигнута за
10 часов. Повышение концентрации олигомеров бутадиена в
исходной мономерной смеси способствовало возрастанию выхо-
да сополимера. Существенное влияние на степень превращения
олигомеров бутадиена, а также на молекулярно-массовые ха-
рактеристики получаемых сополимеров оказывала температу-
ра процесса (табл. 4.7).
Спектральные исследования полученных сополимеров пока-
зали, что интенсивность полос поглощения в ИК-спектрах
групп —СН = СН2 в области 990 см-1 и = СН2-групп в области
910 см-1 значительно ниже, чем в исходном 4-винилциклогек-
сене, а наличие полос поглощения в области 740 см-1 и 967-1
указывало на присутствие цис- и тра^с-звеньев в полученном
сополимере [314].
Полученный сополимер был испытан в качестве пластифи-
катора бутадиенового каучука. Введение сополимеров иа осно-
ве олигомеров бутадиена в бутадиеновый каучук существенно
122
улучшало такие показатели резиновых смесей, как способность
к вальцеванию и шприцеванию [314]. Вулканизаты с добавка-
ми этого сополимера по напряжениям при 300%-ном удлине-
нии, прочности при растяжении и эластичности были равноцен-
ны резинам, содержавшим такое же количество в качестве мо-
дифицирующей добавки масла ПН-6, а по сопротивлению раз-
диру и многократным деформациям превосходили их. Таким
образом, сополимер на основе олигомеров бутадиена улучшал
не только технологические показатели смесей, но и динамичес-
кие характеристики вулканизатов (табл. 4.8).
Одним из основных недостатков этого процесса является
его периодичность. Наряду с полимеризацией на гетерогенном
катализаторе протекают процессы, дезактивирующие его актив-
ность. Поэтому для восстановления активности катализатора
необходимо^ проводить его регенерацию или заменять новым.
Кроме того, процесс полимеризации проводят при высоких
температурах (160—200°C), что требует больших энергети-
ческих затрат.
Представляет интерес исследование радикальной сополиме-
ризации олигомеров бутадиена. Изучена [315] радикальная со-
полимеризация смеси олигомеров бутадиена, содержащихся в
кубовом остатке ректификации толуола, в присутствии иници-
аторов: динитрила-азо-бис-изоизомасляной кислоты, гидропер-
оксидов изопропилбензола и циклогексилизопропилбензола и
пероксида бензоила. Для сополимеризации использовались ку-
бовые остатки ректификации толуола без какой-либо предва-
рительной подготовки и имевшие следующий состав [%
(масс.)]: 4-вииилциклогексен—19; тримеры бутадиена и дру-
гие высококипящие продукты—14; толуол — 67.
На основе проведенных исследований установлено, что на
выход полимерных продуктов оказывала влияние концентрация
олигомеров бутадиена в толуоле. Однако даже в случае высо-
коконцентрированных растворов достичь высокого выхода по-
лимерных продуктов не удалось (табл. 4.9). Максимальный вы-
ход сополимеров ( — 30%) был достигнут при использовании в
качестве инициатора гидропероксида изопропилбензола за
12 ч. Снижение дозировки инициатора с 3,5 до 1,7% (масс.)
на олигомеры бутадиена уменьшало выход сополимеров на
30—50%.
Все синтезированные сополимеры представляли собой мас-
лянистые жидкости темно-коричневого цвета, имеющие невысо-
кую молекулярную массу (Л7„ = 500—1200).
Состав кубового остатка изменяется в широких пределах,
поэтому воспроизводимость результатов была невысока.
Для стабилизации процесса, повышения выхода сополиме-
ров и их качества [316—319] исследовали сополимери-
зацию олигомеров бутадиена со стиролом на вышеприведенных
123
ТАБЛИЦА 4.8
Показатель скд скд-ср
без плас- тификато- ра масло ПН-6, 10 масс. ч. содержание сополиме- ра, масс. ч. без плас- тификато- ра масло ПН-6, 10 масс. ч. содержание сополиме- ра, масс. ч.
5 10 15 5 10 15
Резиновые смеси
Вязкость по Муни МБ 1+4 87 75 82 74 63,5 64 49 57 49 46
(100 °C) Пластичность 0,13 0,28 0,16 0,27 0,32 0,21 0,36 0,28 0,33 0,38
Эластическое восстановление, мм 2,60 1,75 2,50 2,00 1,95 2,50 1,70 2,32 1,95 1,65
Усадка при вальцевании, % 23 20,5 — 18,5 17,5 22,0 18,5 — 15,0 14,0
Сопротивление подвулканизации при 120 °C (т=5 мин) 21 23 — 26 28 37 38 — 41 5,0 42
Вальцуемость, мм 0,1 0,65 — 0,95 1,4—1,6 3,2 4,0 — 6,0
Шприцуемость, баллы 10 3—4 — 3—4 2—3 10 1—2 — 1 1
Вулканизаты
Напряжения при 300%-ном удли- 9,4 8,5 9,8 8,6 8,1 9,7 8,3 9,8 8,6 8,5
нении, МПа Условная прочность при растя- 20,3 19,2 21,0 19,9 18,6 17,1 16,4 18,2 17,0 16,9
жении, МПа Относительное удлинение при 490 537 505 528 525 490 535 505 540 525
разрыве, % Эластичность по отскоку, % 52 50 50 51 51 32 28 31 28 28
Сопротивление раздиру, кН/м 63 57 — 69,1 72 65 61,2 — 70 78,6
Сопротивление многократным де- 20,5 17,3 — 25,7 34,6 8,7 7,9 — 14,8 25,9
формациям, тыс. циклов
ТАБЛИЦА 4.9
Концентрация оли- гомеров в толуоле, % (масс.) Выход сополимеров, % (масс.) Концентрация оли- гомеров в толуоле, % (масс.) Выход сополимеров, % (масс.)
Инициатор — гидропероксид
изопропилбензола [3,5% (масс.) на
олигомеры]
Инициатор — динитрил-азо-бис-
изоизомасляная кислота
[3,0% (масс.) на олигомеры]
33*** 19,8
25 18,2
17 15,9
10 14,2
Инициатор — пероксид бензоила
[3,5% (масс.) на олигомеры]
33* 24,8
25 21,9
17 19,5
10 17,3
33** 29,4
Инициатор — гидропероксид
циклогексилизопропилбензола
[3,5% (масс.) на олигомеры]
33* 22,7
25 19,6
17 17,1
10 16,9
33** 26,8
33*** 21,6
25 19,5
17 17,0
10 16,6
*** Температура 145, 160, 80 °C соответственно.
инициаторах. Наиболее эффективным инициатором из рассмот-
ренных явился гидропероксид изопропилбензола. На выход
полимерных продуктов большое влияние оказывали дозировки
инициатора, стирола и продолжительность процесса (рис. 4.3).
От концентрации стирола зависела также молекулярная масса
получаемых сополимеров. Увеличение содержания стирола
приводило к возрастанию молекулярной массы. При соотноше-
нии олигомеры стирол^равном 1:1, образовывались масло-
образные продукты с Мп = 600—1300. При соотношении 1 3
полимерные продукты светло-коричневого цвета с Мп=1500—
3000, а при дальнейшем увеличении соотношения (до 1 :9)
получали твердые полимеры с молекулярной массой до 15000.
Спектральные исследования полученных сополимеров пока-
зали, что в ИК-спектрах присутствуют полосы в областях
1660—2000, 1604, 1495, 760 и 699 см-1, характерные для моно-
замещенного бензольного кольца, причем их интенсивность по
вышается с увеличением содержания стирольных звеньев в со-
полимере. Интенсивности полосы поглощения, соответствую-
щей веерным колебаниям связи С—Н в группе тр«кс-СН = СН2
при 967 см-1, а также полос при 1650—1670 см-1, возрастают
с увеличением содержания олигомеров бутадиена в сополиме
ре. Кроме того, с увеличением содержания стирола в сополи-
мере резко снижается интенсивность полосы поглощения в
области 655 см-1, характерной для колебаний двойной связи
циклогексенового кольца. Присутствие этой полосы свидетель-
125
РИС. 4.3. Влияние концентраций сти-
рола и гидропероксида изопропил-
бензола (Сг, %) на выход сополиме-
ров ((Вс, %):
1 — полимер на основе олигомеров бута-
диена; соотношение олигомеры бутадие-
на : стирол: 2 — 1:1; 3 — 1:3; 4—1:6;
5—1 9
ствует о протекании полиме-
ризации 4-винилциклогексена
по винильной группе. Эти
выводы подтверждаются и
исследованиями методом
ПМР-спектроскопии.
Введение полученных со-
полимеров в резиновые смеси
на основе бутадиенового каучука [316] в количествах до
2 масс. ч. не оказывало заметного влияния на изменения сво*
ств основного бутадиенового каучука (табл. 4.10).
Синтезированы сополимеры [320, 321] на основе олигоме-
ров бутадиена с акриловыми мономерами, пипериленом, изо-
бутиленом, бутадиеном. При этом так же, как и в случае со
стиролом, небольшие добавки в каучуки (до 2 масс, ч.) не ока-
зывали существенного влияния на свойства готовых изделий.
При дальнейшем увеличении их дозировки в резиновые смеси
(до 15 масс, ч) отмечалось улучшение технологических свойств
резиновых смесей и некоторых технических характеристик ре-
зины (сопротивление раздиру, морозостойкость, устойчивость
к многократному растяжению и др.).
Однако на основании имеющихся литературных данных
нельзя сделать окончательный вывод о влиянии полимерных
продуктов на свойства резиновых смесей и резин, полученных
на их основе. Поэтому работы в этом направлении перспектив-
ны и их целесообразно продолжать.
4.3. Кубовые остатки ректификации бутадиена
Основными непредельными соединениями, содержащимися в
кубовом остатке ректификации бутадиена, являются диеновые
углеводороды (изопрен, гексадиены, пиперилен и др.), пред-
ставляющие собой полимеризующуюся часть кубового остатка
и составляющие 23—37% от его общей массы. В промышлен-
ных условиях на алюмосиликатном катализаторе освоено про-
изводство на основе кубовых остатков ректификации бутадиена
низкомолекулярных сополимеров, которые находят широкое
применение в производстве композиционной олифы [322].
Одним из основных недостатков такой олифы является ее тем-
126
ТАБЛИЦА 4.10
Показатель Контрольный образец бу- тадиенового каучука Соотношение олигомеры : сти- рол
1 1 1 : з 1 9
Вязкость по Муни МБ l-f-4 (100 °C) 46 42 45 44
Пластичность 0,53 0,56 0,54 0,54
Условная прочность при растя- жении, МПа 19,8 19,7 20,1 20,4
Относительное удлинение при разрыве, % 585 620 600 605
Относительное остаточное удлине- ние после разрыва, % 6 6 6 6
Массовая доля золы, % (масс.) 0,11 0,12 0,10 0,13
Потери массы при 105 °C, % (масс.) 0,15 0,18 0,14 0,15
ный цвет. Установлено, что на цветность получаемых сополиме-
ров большое влияние оказывают кислородсодержащие соедине-
ния (эфиры, кетоны, альдегиды), присутствующие в кубовых
остатках ректификации бутадиена в значительных количест-
вах [до 10% (масс.)]. Эти соединения в значительной степени
интенсифицируют процессы смолообразования, что подтвержда-
ется на примере полимеризации модельной системы.
Сополимеры на основе кубовых остатков ректификации то-
луола [322], полученные на алюмосиликатном катализаторе,
обладают более светлым цветом, невысокой прочностью при
изгибе и более высокой продолжительностью высыхания (72—
96 ч). На основании этого при приготовлении композиционной
олифы было рекомендовано [322] использовать смесь полимер-
ных продуктов из кубовых остатков ректификации толуола и
бутадиена в соотношении 1 6—8.
Использование для технологического процесса алюмосили-
катного катализатора требует применения высоких температур,
его периодической регенерации или замены новым.
Перспективным направлением является радикальная сопо-
лимеризация непредельных соединений, содержащихся в кубо-
вом остатке ректификации бутадиена. В работе [323] изуча-
лась сополимеризация данных соединений со стиролом в при-
сутствии гидропероксида изопропилбензола. Для оценки влия-
ния стирола сделана попытка представить экспериментальные
данные в виде экспоненциальной кривой вида:
В = [1 — ехр ( — а/?т) ] • 100,
где В — выход полимерных продуктов, % (масс.); а — эмпирический коэффи-
циент, ч”1; D — массовое отношение стирол олигомеры; т — продолжитель-
ность процесса, ч.
127
ТАБЛИЦА 4.11
Количество стирола, % (масс.) Выход полимерных продуктов, %
расчет лабораторная установка пилотная установка расчет лабораторная установка пилотная установка
Время реакции 1 ч Время реакции 5 ч
200 14,8 15,0 15,0 55,1 54,5 54,2
300 21,3 21,2 20,5 69,9 69,3 68,2
400 27,4 27,1 26,5 79,8 77,9 76,2
Время реакции 3 ч Время реакции 8 ч
200 38,1 37,9 37,0 72,8 72,1 70,8
300 51,3 51,1 50,3 85,3 84,4 82,0
400 61,7 60,6 60,0 92,3 91,2 85,0
Установлено, что экспериментальные и расчетные данные
удовлетворительно согласуются между собой при значении эм-
пирического коэффициента а = 0,08 (табл. 4.11).
Реакция с удовлетворительной скоростью протекает в тече-
ние 6—8 ч. Затем отмечено резкое ее замедление из-за побоч-
ных реакций дезактивации катализатора. Для достижения мак-
симальной конверсии мономеров (0,05—0,1% по остаточному
стиролу) необходима дробная подача катализатора на строго
определенных стадиях сополимеризации. Это обеспечивает по-
лучение низкомолекулярных сополимеров с характеристической
вязкостью 0,05—0,2 (д-л/г) [323].
Добавка полученных сополимеров на основе кубовых остат-
ков ректификации бутадиена в резиновые смеси на основе дие-
новых каучуков (СКД, СКС и др.) в количестве 5—12 масс. ч.
на 100 масс. ч. каучука позволяет улучшить их технологичес-
кие свойства и повысить некоторые показатели вулканизатов
(сопротивление раздиру, динамические показатели) [324, 325].
Таким образом, низкомолекулярные сополимеры, получен-
ные на основе кубовых остатков ректификации бутадиена мо-
гут служить эффективными заменителями растительного сырья,
используемого в производстве олиф, а также в полимерных
композициях.
4.4. Кубовые остатки производства винилароматических
углеводородов
В производстве винилароматических мономеров образуется
ряд побочных продуктов. Наибольший интерес представляют
кубовые остатки ректификации этилбензола и стирола [326—
347].
128
Кубовые остатки ректификации этилбензола — отходы про-
изводства этилбензола — содержат полиалкилбепзолы, которые
было предложено использовать [331, 332] для получения этил-
бензола. Переалкилирование полиалкилбензольной смолы про-
водили при 85—100 °C в присутствии бензола. Соотношение
бензол полиалкилбензольная смола равно 2—7 1—3. В ка-
честве катализатора использовался комплекс, содержавший
10—12%хлорида алюминия, 25—30% полиалкилбензолов, 50—
60% бензола, хлорид водорода (газ) или вещества, способные
к его выделению [331].
Переработка кубовых остатков ректификации этилбензола
проводилась [332] при 50—55 °C, массовом соотношении бен-
зол хлорид алюминия кубовый остаток, равном 5—15 1 :1.
При этом 75% (масс.) кубового остатка переходило в углево-
дороды с меньшей молекулярной массой и образовывалось до
15% (масс.) этилбензола.
Полиалкилбензольная смола может быть использована
[333] для полной или частичной замены масла ПН-бш в про-
изводстве маслонаполненных диеновых каучуков и в качестве
мягчителя в различных резиновых смесях на основе диеновых
каучуков.
При этом, как показали проведенные испытания, по всем
основным показателям резиновые смеси и вулканизаты, содер-
жащие кубовые остатки ректификации этилбензола не уступали
серийным образцам.
Кубовые остатки ректификации стирола содержат непре-
дельные соединения (стирол, метилстиролы, стильбен и др.),
поэтому их можно использовать для получения полимерных
материалов, применяемых в лакокрасочной промышленности.
Первые попытки использования кубовых остатков ректифика-
ции стирола в этих целях были предприняты в 1958—1959 гг.
[326]. Из этих кубовых остатков выделяли низкомолекулярную
полистирольную смолу (выход 18—40%) и на ее основе гото-
вили лак, обладающий удовлетворительными физико-механиче-
скими свойствами.
В дальнейшем была разработана технология получения
пленкообразующих веществ сополимеризацией кубовых остат-
ков ректификации стирола с малеиновым ангидридом [327].
Сополимеризацию проводили без дополнительного использова-
ния каких-либо каталитических систем. Твердый смолообраз-
ный продукт, получаемый после вакуумной отгонки непроре-
агировавших мономеров (выход 95%), имел коричневый цвет
и хорошо растворялся в ароматических углеводородах, соль-
венте. Раствор этого сополимера в ксилоле или Р-5 с добавка-
ми пластификатора обладал свойствами лака (качество удов-
летворительное) для этого исходное сырье должно иметь сле-
дующий состав:
9—627
129
Стирол, % (масс.)
а-Метилстирол, %
(масс.)
Ингибитор, г/т
Полимер
>20
>7
<740
Нс регламентируется
Продукты отгона содержали 85—90% стирола, который
возвращали в процесс. Исследования по составу получаемых
продуктов показали, что в процессе сополимеризации образу-
ются сополимеры стирол — малеиновый ангидрид, а-метилсти-
рол, малеиновый ангидрид, транс-стильбен, малеиновый ангид-
рид и полистирол [334, 335].
Особенностью этого процесса является использование
качестве ингибиторов полимеризации стирола в кубовых остат-
ках ректификации ди-(n-гидроксифенил)-амина с гидрохиноном
с получением лака с удовлетворительными свойствами. Смесь
диоксима 1,4-бензохинона с гидрохиноном в качестве ингиби-
тора нарушает процесс термической сополимеризации кубовых
остатков ректификации стирола. В этом случае образуется про-
странственно-структурированный сополимер, который неприго-
ден для использования в лакокрасочной промышленности. От-
рицательное влияние на процесс сополимеризации оказывает
диоксим 1,4-бепзохинона. Для его дезактивации в реакционную
массу вводят хлориды металлов (FeCh, TiCh или ZnCb), в
присутствии которых диоксим 1,4-бензохинопа окисляется до
п-динитрозобензола.
Исследования радикальной сополимеризации кубовых остат-
ков ректификации стирола с малеиновым ангидридом в бензо-
ле или метилэтилкетоне в присутствии пероксида бензоила при
60—80°C показали, что наличие нафталина или олигостирола
ускоряет этот процесс (табл. 4.12). В случае тройной сополи-
меризации малеинового ангидрида, стильбена и стирола наблю-
дается экстремальный характер зависимости скорости реакции
от соотношения мономеров с максимумом значения начальной
ТАБЛИЦА 4.12
Содержание нафта- лина, % (масс.) Скорость сопо- лимеризации, 10-ь моль/л-с Содержание олп- гостирола, % (масс.) Скорость сопо- лимеризации, 10~б моль/л-с
0,99 0,99
0,1 1,33 5 1,30
0,5 1,56 15 1,65
1,5 2,64 25 2,10
2,5 7,50 35 2,30
— — 45 2,50
Примечание. Растворитель — бензол; инициатор — пероксид бензоила [0,5%
(масс.)]; соотношение м алеиновый ангидрид : стильбен = 1; [М]=0,2 моль/л.
130
РИС. 4.4. Зависимость скорости тройной
сополимеризации [ьг.’с, моль/(л-с)] ма-
леинового ангидрида, стирола, стильбе-
на от состава мономерной смеси
[См, % (мол.)] при суммарной концен-
трации мономеров, моль/л.
1 — 0,4; 2 — 0.5; 3 — 0.6; 4 — 0.7
скорости при соотношении ма-
леиновый ангидрид стильбен
стирол = 2 1 1 (рис. 4.4.)
На основании известных ура-
внений комплексно-радикальной
сополимеризации рассчитан
вклад комплекса с переносом за-
ряда в радикальную сополимеризацию малеинового ангидрида
со стильбеном [336]. Значение отношений констант скорости эле-
ментарных стадий в реакции роста цепи с учетом свободных и
комплексно-связанных мономеров составило: feic/£i,2 = 437,5;
^2c/^2,i = 300,0.
Константы сополимеризации для пары малеиновый ангид-
рид — стильбен, рассчитанные по методу Файнмана — Росса,
составляют Г\ =0,044 и г2 = 0,015.
Для улучшения физико-механических показателей покрытий
в реакционную массу одновременно с кубовыми остатками рек-
тификации стирола и малеинового ангидрида было рекомендо-
вано вводить каучук СКИ-3 и проводить процесс в присутствии
гидропероксида изопропилбензола [337].
Несмотря на широкие пределы качественного и количест-
венного составов кубовых остатков ректификации стирола,
обеспечить требуемый состав сырья не всегда возможно. Это
связано с продолжительностью работы катализатора и состоя-
нием систем выделения [338—341]. Кроме того, технология
процесса получения винилароматических углеводородов посто-
янно совершенствуется. Увеличение общей конверсии этилбен-
зола с внедрением новых катализаторов привело к снижению
селективности процесса. Природа и дозировка ингибиторов по-
влияла на содержание стирола [<20% (масс.)] в кубовых ос-
татках ректификации стирола и па молекулярную массу поли-
мера; молекулярная масса уменьшилась с 45000—70000 до
20000—36000. Все это отразилось на свойствах плепкообразую-
щего. Потребовались дополнительные корректировки процесса
сополимеризации, а в некоторых случаях и разработка новых
способов получения сополимеров. В табл. 4.13 приведены сос-
тавы кубовых остатков ректификации стирола, получаемые па
предприятиях [338].
По основному показателю пригодности кубовых остатков
ректификации стирола для синтеза лака — содержанию стиро-
9*
131
ТАБЛИЦА 4.13
Предприятие Содержание, % (масс )
стирол полимер тяжелый остаток
Воронежский завод СК 8,4 54,5 37,1
ПО «Салаватнефтеоргсинтез» 3,0 14,4 82,6
Шевченковский завод пластмасс 17,59 67,94 14,46
Узловский завод пластмасс 46,9 18,9 34,2
ПО «Нижнекамскнефтехим» 36,0 45,5 18,5
ла >20% (масс.) —только 53,2% всей массы остатков отвеча-
ют этому требованию. Следовательно, для решения проблемы
полной переработки КОРС и создания безотходного производ-
ства необходимо изыскивать новые пути, не зависящие от со-
держания стирола.
Было предложено заменить дефицитный малеиновый ангид-
рид на менее дефицитный фталевый ангидрид [342]. Дозиров-
ку его осуществляли в количестве 4—7% (масс.) в зависимос-
ти от содержания стирола и процесс проводили при постоянном
увеличении температуры от 130 до 175 °C в течение 20—25 ч.
Полученный сополимер использовали в виде 35—40%-ного ра-
створа в ксилоле с добавкой пластификатора в качестве лака,
свойства которого приведены в табл. 4.14. Кроме этого, сопо-
лимер можно применять и для приготовления антикоррозион-
ных и антиабразивных покрытий.
Для улучшения свойств олифы «Оргтехстрой-5» в качестве
связующего для олифы применяют кубовые остатки ректифика-
ции стирола [343], обработанные при 100 °C молотой известью.
Полученная олифа обладала следующими показателями: цвет
по йодометрической шкале — 650 ед. ИМШ; вязкость по Энг-
глеру — 8,0; высыхание от пыли — 4 ч; полное высыхание —
10 ч.
На Сумгаитском заводе СК разработана и внедрена в про-
изводство технологическая линия получения лаков на основе
кубовых остатков ректификации стирола, которые используют-
ся для создания защитных покрытий судов, морских эстакад
и др. Из сополимеров, па основе кубовых остатков ректифика-
ции стирола и малеинового ангидрида получены оловооргани-
ческие производные, обладающие повышенной биологической
активностью [344].
На основе указанного сополимера разработана рецептура
эмали, обладающей высокой адгезией и прочностью пленки
при ударе (табл. 4.15).
Полученные на основе кубовых остатков ректификации сти-
рола и ангидридов двухосновных кислот полимерные пленки
132
ТАБЛИЦА 4.14
Продолжитель- ность реакции, ч Температура размягчения, °C ^УД Прочность при ударе, Н-м Твердость, Пк
5 100 1,13 1,5 0,35
7 96 1,22 2,5 0,38
10 100 1,25 3,0 0,35
12 97 1,35 3,0 0,40
14 98 1,41 3,5 0,44
15 98 1,43 3,5 0,45
17 92 1,35 2,5 0,40
18 91 1,20 2,0 0,40
20 87 1,04 1,5 0,30
обладают высокой эластичностью и прочностью при ударе. Со-
полимеризацию кубовых остатков ректификации стирола с 3-
метил-1,2,3,6-тетрагидрофталевыми или изо-4-метилтетрагидро-
фталевым ангидридами проводили при постепенном подъеме
температуры с выдерживанием 1,45—2,15 ч (115—120°C);
2,45—3,15 ч (145—150°С) и 7,45—8,15 ч (170— 175°С). Полу-
ченные сополимеры (выход <90%) представляли собой смоло-
образные продукты светло-коричневого цвета с температурой
размягчения 98°C, растворимые в ксилоле и Р-5 [345].
В последние годы наметилась тенденция к сополимеризации
кубовых остатков ректификации стирола с другими кубовыми
Показатель Разработан- ная эмаль Эмаль ХС-519 (ТУ 6-10-100-11-79)
ТАБЛИЦА 4.15
Содержание олова в покрытии, % (масс.) 6,5—7,8 —
Время высыхания при температуре 20±2 °C до 3 степени (ГОСТ 19007— 73), ч 6 8
Адгезия покрытия через 12 мес. мето- дом решетчатого надреза (ГОСТ 15140—69), баллы 1 1
Прочность пленки при ударе по прибо- ру У-1 (ГОСТ 4763—73), кг/см2 Прочность пленки при изгибе покрытия (ГОСТ 6806—79), мм 50 50
1 2
Твердость покрытия по маятниковому прибору (ГОСТ 5233—67), усл. ед. 0,388 —
Вид покрытия, визуально Белое —
Водопоглощение, °/о 0,021 —
Содержание нелетучих, % 52,88 38,5
Срок испытания (стендовые), мес. 36 18
Вязкость по ВЗ-4 (ГОСТ 8420—57), с 125 70
133
остатками, например, с альдегидсодержащей фракцией кубово-
го остатка после ректификации продуктов гидроформилирова-
ния олефинов с температурой кипения 130—270 °C [346]:
Согласно данным ИК-спектроскопии в альдегидной фракции
преобладают карбонильные соединения (сильная полоса в об-
ласти 1720 см-1), способные вступать в реакцию сополимериза-
ции с непредельными соединениями, содержащимися в кубо-
вых остатках ректификации стирола с образованием эфирных
мостиков [346].
Было| предложено [326] использовать лак, полученный на
основе кубовых остатков ректификации стирола, для модифи-
кации эпоксидных смол. При использовании в качестве от-
вердителя смолы полиэтилепполиамида жизнеспособность смо-
лы (композиции) возрастает в 3—4 раза, а время высыхания
уменьшается до 2—3 ч. Покрытия, полученные( на основе этой
композиции, обладают высокой стойкостью к воде, к кислым,
щелочным и солевым растворам. Кроме того, снижается стои-
мость покрытия и сокращается расход дефицитных эпоксидных
смол.
Было предложено [347] для повышения адгезионных
свойств покрытий использовать ацилированные кубовые остат-
ки ректификации стирола с малеиновым ангидридом. Ацили-
рование проводили в присутствии катионных катализаторов с
добавкой а-метилстирола в растворе бензола. Модифицирован-
ные кубовые остатки ректификации стирола в толуольных ра-
створах образуют прозрачные и гладкие пленки на металличес-
ких и стеклянных подложках. Наличие карбоксильной группы
обеспечивает резкое повышение адгезионных свойств покрытий.
Таким образом, сополимеры на основе кубовых остатков
ректификации стирола могут применяться в различных отрас-
лях народного хозяйства: при изготовлении клеевых компози-
ций [348], лаков, пластмасс, строительных объектов. Сополи-
меры из этих кубовых остатков используются для повышения
механической прочности, светостойкости и снижения электри-
зуемости покрытий полов в композиции па основе синтетическо-
го каучука, наполнителя и пигмента [349]. Эти сополимеры
вводят в состав покрытий для увеличения адгезии к мокрой
поверхности бетона и металла [350]. Раствор смолы на осно-
ве кубовых остатков ректификации стирола используют для
приготовления безрулопноп кровли.
134
Низкомолекулярный полистирол, выделенный из кубовых
остатков ректификации стирола, может использоваться при
приготовлении цинковых красителей [351]. Такие краски обла-
дают хорошими протекторными свойствами в сравнении с крас-
ками на основе блочного полистирола.
Учитывая то обстоятельство, что далеко не весь кубовый
остаток ректификации стирола может быть использован для
получения полимерных материалов, предложены [352—357]
другие направления в его переработке. Одним из таких на-
правлений является термическая деструкция полимерных про-
дуктов, содержащихся в этих кубовых остатках. При деструк-
ции полистирольной смолы в качестве целевого продукта со-
бирается фракция с температурой кипения <150 °C, содержа-
щая до 58%', непредельных соединений, из которых выделяют
ароматические углеводороды, а остаток используют в качестве
сырья каталитического крекинга или компонента зеленого мас-
ла [327—330].
Непредельные углеводороды из стирольной смолы можно
выделить и путем отгонки с водяным паром [352], Однако
лаки, полученные на основе оставшейся полистирольной смолы,
обладают низкими показателями, что делает их применение
бесперспективным в народном хозяйстве.
Полистирольный лак на основе отходов стирольного произ-
водства предложено получать [353] выделением (вакуумной
перегонки) целевой фракции с температурным интервалом
120—200 °C. Последняя образуется при деполимеризации кубо-
вых остатков в трубчатой печи при температурах (°C): 350—
400, 400—450, 500—550. Полученный этим способом стирол по-
лимеризуют при ПО—130°C в присутствии пероксидного ини-
циатора (выход полимера «70%). Однако полимеры такого
типа в лакокрасочной промышленности практического примене-
ния не находят. Это связано с тем, что они дают хрупкие плен-
ки с низкой атмосферостойкостью.
Рассматриваемые до настоящего момента процессы деструк-
ции осуществляются в основном высокотемпературным спосо-
бом, т. е. без применения катализаторов. Использование ката-
лизаторов для деполимеризации полимерных продуктов кубо-
вых остатков ректификации стирола позволяет повысить про-
изводительность процесса. Так, в присутствии отработанного
окисного железо-хромо-калиевого катализатора деполимериза-
ция полимерных продуктов кубовых остатков до стирола проте-
кает с высокой скоростью при 500—550 °C [354].
Продукты пиролиза стирольной смолы, содержащие транс-
стильбен, могут быть использованы для синтеза антибиотиков
[355].
Тяжелый остаток или стиролсодержащую смолу, включаю-
щую в свой состав серу и трет-бутилкатехин, применяют как
135
ТАБЛИЦА 4.16
Соотношение вода : изо- пропилбензол Скорость по- дачи смолы, г/мин Выход а-ме- тилстирола на пропу- щенную смо- лу, % Состав газа, % (об.)
СО2 о2 непредель- ные СО н2
1,3:1 0,8 При 26,3 550 °C 18,1 5,5 0,3 0,9 42,6
1,5:1 0,9 27,8 26,5 0,3 0,7 0,4 54,1
1,8:1 0,7 18,3 14,1 5,3 0,3 0,4 45,3
1,8:1 0,9 26,0 25,5 1 0,8 0,8 56,9
2,0:1 0,7 14,5 15,5 5,1 0,2 0,6 46,0
2,8:1 0,7 26,9 25,0 1,0 0,8 1,4 59,0
3,0:1 1,0 18,9 18,9 1,1 0,2 0,6 26,8
1,4:1 0,7 18,7 16,4 7,4 0,3 0,9 38,6
1,5:1 0,7 15,4 17,4 1,0 0,1 2,2 47,4
2,0:1 0,7 При 15,2 600 °C 27,7 0,4 0,6 0,5 59,6
4,5:1 0,7 13,4 18,9 3,5 0,2 1,0 36,3
0,9:1 0,5 25,6 21,3 1,6 0,7 0,2 60,0
2,2:1 0,5 При 23,3 610°С 33,3 2,1 1,5 0,2 52,6
2,8:1 0,4 29,1 27,2 0,6 0,7 0,9 59,8
1,2:1 0,5 19,6 26,0 1,0 0,5 0,9 59,6
1,4:1 1,2 При 19,8 650 °C 27,0 0,3 0,6 0,6 64,0
2,1:1 0,5 23,3 23,4 0,4 0,4 4,3 66,0
3,0:1 0,4 23,6 18,3 0,4 0,2 8,0 60,1
0,3:1 0,5 5,44 16,9 1,7 0,3 21,0 39,9
2,2:1 0,5 При 10,75 700 °C 17,0 1,4 0,2 12,5 57,5
3,3:1 0,4 6,4 22,9 1,4 0,4 5,1 60,0
составную часть сырья замедленного коксования нефтяных ос-
татков [356].
Переработку кубовых остатков (смол), содержащих серу и
ее производные, осуществляют [357] путем их гидрирования
и выделения серы в виде сероводорода. При этом образуется
смесь органических соединений, состоящих из бензола, этил-
бензола, толуола и других более высококипящих соединений.
Утилизировать кубовый остаток ректификации а-метилсти-
рола можно двумя способами [358]: крекингом на катализато-
ре Р-1 в присутствии водяного пара при температуре 550—
700 °C и добавкой образовавшихся продуктов к изопропилбен-
зольной шихте и последующим дегидрированием.
Крекинг проводят на лабораторной установке проточного
типа. При этом наилучшие результаты (по выходу «-метилсти-
рола) получают при проведении процесса при 610 °C, скорость
136
подачи смолы 0,4 г/мин, разбавлении водяным паром 2,8 1
(табл. 4.16).
Использование кубового остатка в качестве добавки к изо-
пропилбензольной шихте в количествах до 2,0% не снижало
выхода целевого а-метилстирола в расчете как па поданный,
так и вступивший в реакцию на изопропилбензол.
Таким образом, кубовый остаток ректификации а-метилсти-
рола может служить в качестве дополнительного источника
получения а-метилстирола. Благодаря этому удается значи-
тельно повысить общий выход товарного а-метилстирола, сни-
зить расход углеводородного сырья в расчете на 1 т готовой
продукции, уменьшить загрязнение окружающей среды и др.
ГЛАВА 5
Применение побочных продуктов
производства одно- и многоатомных
спиртов
5.1. Побочные продукты производства бутиловых спиртов
Процесс получения бутиловых спиртов оксосинтезом включает
две стадии [359]. На первой происходит образование масляных
альдегидов из пропилена, оксида углерода и водорода в присут-
ствии катализатора:
1--> СН3СН2СН2СНО
СН3СН=СН2+СО + Н2 —
I--> СН3СН(СН3)СНО
На второй — альдегиды гидрируются до спиртов:
сн3сн2сн2сно —> сн3сн2сн2сн2он
СН3СН(СН3)СНО ----> СН3СН(СН3)СН2ОН
В производстве бутиловых спиртов образуются побочные про-
дукты, что обусловлено протеканием вторичных реакций [360].
Эти реакции описаны во многих работах, поэтому состав обра-
зующихся продуктов хорошо известен. Их можно разделить
на две группы: легкокипящая фракция и кубовые остатки (45 и
136 кг на 1 т бутиловых спиртов соответственно). Последние
представляют собой смесь высококипящих компонентов. Они ис-
пользуются в качестве флотореагентов на обогатительных фаб-
137
риках, пройдя предварительно стадию смешения с дизельным
топливом.
Количество побочных продуктов составляет до 10—20% в
расчете на превращенное сырье [361]. Использование такого
количества отходов имеет огромное народнохозяйственное зна-
чение.
Легкокипящие побочные продукты производства бутиловых
спиртов предлагается применять для удаления фенола из сточ-
ных вод процесса полукоксования при переработке углей [362].
Для этих целей были использованы легкокипящие фракции, ото-
гнанные от бутанолов нормального и изостроения (плотность
при 20 °C соответственно равна 0,800 и 0,810). Их составы при-
ведены ниже:
к-бутанол изобутанол
Содержание, % (масс.):
эфиры 54—60 50—60
н-бутанол 40—45 —
изобутанол — 40—50
вода 3—6 5—10
Степень выделения фенола этим методом достигает ’97%.
Применение легкокипящих побочных продуктов производства
бутиловых спиртов для экстракции фенолов из воды процесса
полукоксования экономически выгодно и не уступает по степени
удаления фенола широко распространенному для этих целей бу-
тилацетату.
Способы утилизации высококипящих побочных продуктов
процесса гидроформилирования пропилена различны. Высоко-
кипящую фракцию производства бутиловых спиртов после от-
гонки целевого продукта и смешения с дизельным топливом или
керосином применяют в качества флотореагента при флотации
угля (Пат. ЧССР 105336, 1972).
Известна [363] технология формилирования пропилена, в ко-
торой кубовый остаток производства бутиловых спиртов исполь-
зуют в качестве растворителя, что обусловлено требованиями,
предъявляемыми к процессу дистилляции целевого продукта.
Высококипящие продукты, получаемые при оксосинтезе спир-
тов из олефинов, рекомендуется добавлять в топливо двигателей
внутреннего сгорания в количестве 0,05—1,75% (масс.) для
предупреждения образования в карбюраторе отложений льда и
твердых гидратов углеводородов (Пат. США 2843463, 1958). Эти
добавки можно вводить в топливо непосредственно или в виде
растворов и смесей с растворителями. Их можно применять
также в качестве антидетонаторов, антиокислителей и др.
Кубовый остаток (начальная температура кипения
>210 °C) в композициях с высокомолекулярными кислотами
138
[5—Ю% (масс.)] может служить в качестве основы консистент-
ной смазки (Пат. США 2653132, 1952).
Предложено использовать побочные продукты гидроформи-
лирования пропилена для производства селективных гербици-
дов; добавлять в количестве 0,3—10% (масс.) к кислому гудро-
ну для стабилизации и уменьшения вязкости; использовать для
очистки металлических поверхностей при открытых разработках,
например, для предупреждения примерзания и прилипания по-
род к поверхностям ковшей экскаваторов [364].
Химическая переработка побочных продуктов производства
бутиловых спиртов позволяет значительно расширить области
их применения. Так, высококипящую фракцию подвергают по-
вторному гидрированию на опытной проточной установке на
катализаторе ГИПХ-105, что позволяет получать до 10% бути-
ловых и 1-8% высших жирных спиртов от проектной мощности
производства [365]. Высшие спирты, полученные из кубового
остатка, используются для синтеза пластификатора марки
ДАФ-789. Пластификатор ДАФ-789, полученный из спиртов ку-
бового остатка производства бутиловых спиртов, отвечает всем
требованиям ГОСТ 8728—66:
ДАФ-789 ГОСТ 8728— 66, I сорт Пластификатор из спиртов гидрогени- зата кубового остатка
Внешний вид Прозрачная однородная маслянистая жидкость
Цвет по йодометрической шкале 3 1
Плотность, б/420 Функциональные числа, мг КОН/г: 0,975 0,985
кислотное число 0,1 0,06
число омыления 280—300 315
Температура вспышки, °C 200 195
При замене никельхромового катализатора на сплавной алю-
моникельтитановый [366] для повторного гидрирования кубового
остатка производства бутиловых спиртов выход бутанолов [367]
увеличивается с 61—65% до 75—78%.
При повторном гидрировании высококипящих соединений
производства бутиловых спиртов возникает необходимость уда-
лять из реакционной смеси органические кислоты и крошку ка-
тализатора, которые корродируют оборудование, сокращают
проходимость продукта через реактор и снижают активность
катализатора. Для устранения этих недостатков была предло-
жена принципиальная технологическая схема переработки кубо-
вого остатка производства бутиловых спиртов [368]. Использо-
вание этой схемы в промышленных масштабах позволяет полу-
чать бутиловые спирты (выход 51—52%), отвечающие всемтре-
139
бованиям, предъявляемым к товарным бутиловым спиртам
[% (масс.)]: изобутанол — 20—25 и н-бутанол— 75—80.
Определены оптимальные условия гидрирования побочных
продуктов процесса гидроформилирования пропилена [369], по-
зволяющие получать дополнительное количество бутиловых
спиртов: 1-я стадия — катализатор алюмоцинкхромовый, темпе-
ратура 285—300 °C, скорость подачи сырья 1,5 ч-1; 2-я стадия —
катализатор никельхромовый, температура 150—155 °C, скорость
подачи сырья 1,7 ч-1; или катализатор — никель на кизель-
гуре, температура 175—180 °C, скорость подачи сырья 1,3 ч-1.
После первой стадии процесса получения бутиловых спиртов
предлагается (А. с. СССР 364587, 1973) разделять альдегиды и
кубовый остаток и в целях увеличения выхода бутилового
спирта подвергать раздельному гидрированию оба этих потока.
Гидрирование кубового остатка осуществляется на цинкхромо-
вом катализаторе при давлении 10,0—30,0 МПа, температуре
280—360 °C и скорости подачи сырья 0,2—1,0 ч-1.
Ацетали — высококипящие продукты производства бутило-
вых спиртов оксосинтезом — восстанавливают до соответствую-
щего спирта при 140—240 °C под давлением водорода 5,0—
50,0 МПа и оксида углерода 3,0—50,0 МПа в присутствии воды
и металлического кобальта. Это значительно увеличивает выход
целевого продукта (Пат. НРБ 177337, 1962).
Наряду с высококипящими продуктами в процессе получения
бутиловых спиртов выделяется значительное количество изомас-
ляного альдегида, не нашедшее дальнейшего применения. Из-
вестно, что при гидроформилировании пропилена на первой ста-
дии процесса получают [359] смесь масляного и изомасляного
альдегида (соотношение 4 1 соответственно). Часть изомасля-
ного альдегида применяют в производстве изобутилового спир-
та, ио основное его количество сжигается [361].
Существует [370—372] три способа превращения изомасля-
ного альдегида и кубового остатка в исходную смесь пропилена,
оксида углерода и водорода (синтез-газ): парциальное окисле-
ние, риформинг в паровой фазе и каталитическое расщепление.
Парциальное окисление осуществляют в интервале темпера-
тур 130—150 °C. Побочными продуктами сжигания крекинг-газа
являются метай и небольшое количество сажи.
Каталитический риформинг изомасляного альдегида в паро-
вой фазе приводит в основном к образованию газовых смесей,
содержащих >85% (масс.) водорода. Эти смеси перерабатыва-
ют в дальнейшем в водород, используемый для получения спир-
тов. В процессе образуется также сажа, загрязняющая и отрав-
ляющая катализатор. Если риформинг проводить в присутствии
избыточного количества диоксида углерода, то образуется экви-
молярная смесь (СО + Н2), которая после отделения избытка
СО2 используется в реакции гидроформилирования.
140
Каталитическое расщепление изомасляного альдегида осу-
ществляют путем его пропускания вместе с водой через слой
катализатора. Декарбоксилирование на никелевом катализаторе
протекает уже при 300 °C; более высокая температура и избы-
ток воды благоприятствуют снижению выхода сажи. Крекинг
при температурах 650—680 °C способствует образованию пропи-
лена, оксида углерода и водорода. Степень конверсии альдегида
составляет 35—40%. В качестве катализатора применяются так-
же медь, палладиевые сетки (200 °C), соединения радия, плати-
ны и рутения. При использовании этих катализаторов степень
конверсии достигает 80%. После прохождения реакционной зоны
газ охлаждают, непрореагировавший альдегид и воду направ-
ляют в рецикл, а продукты возвращают на оксосинтез.
При производстве бутиловых спиртов одним из побочных
продуктов'является бутилформиат, который образует азеотроп-
ные смеси с бутанолом и растворителями, применяемыми в про-
цессе оксосинтеза. В целях увеличения выхода бутилового спир-
та возможна переработка такого азеотропа. При этом превра-
щение бутилформиата в бутиловый спирт сочетают с выделени-
ем из азеотропа спирта и очисткой растворителя от указанных
соединений.
Расщепление бутилформиата (Пат. ФРГ 1817051, 1972) на
катализаторах (А12О3 или дегидрирующие металлы) протекает
по следующей схеме:
Катализатор
С4Н9ОСОН -------►- С4Н9ОН + со
Кубовый остаток продуктов гидроформилирования пропилена,
содержащий изобутилформиат, бутилформиат, изобутиловый
спирт, н-бутиловый спирт, высококипящие продукты конденса-
ции и 20% (масс.) воды, пропускают при 250—350 °C над А12О3.
В результате содержание н-бутилового спирта в смеси увеличи-
вается в два раза при одновременном значительном уменьшении
доли побочных продуктов.
Возможна очистка бутиловых спиртов от бутилформиатов в
колонне дистилляции в присутствии водяного пара (Пат. ФРГ
1197867, 1966). В описанных условиях ректификации степень
удаления бутилформиатов составляет 95%. При этом получают
спирты с содержанием 0,06% (масс.) бутилформиатов.
Узкокипящие фракции спиртов, получаемые оксосинтезом,
можно очищать (Пат. США 2867651, 1960) с помощью веществ
основного характера — боргидридами щелочных или щелочно-
земельных металлов. В этом случае происходит гидролиз бу-
тилформиатов до соответствующих спиртов. Процесс очистки
осуществляют добавляя боргидрид при разгонке спирта или про-
пуская спирт через колонну, заполненную А12О3 с нанесенным
на него боргидридом, либо перемешиванием спирта с суспензией
141
или раствором боргидрида в воде. Время контакта зависит о г
концентрации боргидрида, количества примесей, температуры и
колеблется от нескольких минут до 1—5 ч. Процесс проводят
при 150 °C и давлении 0,68 МПа. Для получения спиртов, осо-
бенно высокой степени чистоты, обработку боргидридом сочета-
ют с гидрированием или обработкой активным углем (или
А12О3) и соединениями (например, NaHSO4), образующими с
альдегидами комплексы.
В качестве веществ основного характера, под действием ко-
торых происходит гидролиз бутилформиатов, можно использо-
вать щелочи, карбонаты и алкоголяты щелочных металлов
(А. с. СССР 432119, 1974).
С целью упрощения технологии процесса и увеличения вы-
хода целевого продукта — бутилформиата — из кубового остат-
ка производства бутиловых спиртов предлагается нагревать вы-
сококипящие соединения в присутствии катализатора — оксида
алюминия, предварительно обработанного гидроксидами щелоч-
ных металлов или солями щелочных металлов органических и
неорганических кислот (А. с. СССР 425891, 1974).
Побочные продукты оксосинтеза, которые невозможно пре-
вратить в целевые, можно выделять с целью их дальнейшего
использования. Органические кислоты можно получать (Пат.
США 2945050, 1969) из побочных высококипящих продуктов
оксосинтеза, для чего предлагается обрабатывать кубовые ос-
татки водными растворами щелочей для омыления сложных
эфиров. При этом выход Се- и Сю-кислот в расчете на кубовый
остаток достигает 20% (кислотное число 0,54; эфирное — 61,7;
гидроксидное — 73,5).
Другой способ получения карбоновых кислот из спиртосодер-
жащего остатка дистилляции реакционной смеси состоит в
сплавлении кубового остатка с 10%-ным избытком NaOH и по-
следующим смешением полученного расплава с водой. Водный
слой отделяют, обрабатывают СО2 и получают смесь карбоно-
вых кислот, содержащих в углеродной цепи более 8 атомов
(Пат. Японии 3449, 1965). По указанному методу выделения
карбоновых кислот из высококипящих продуктов оксосинтеза
получают около 18% Се- и С16-кислот в расчете на кубовый
остаток.
Можно окислять высококипящие продукты оксосинтеза
спиртов концентрированной азотной кислотой при 25—150 °C и
71 кПа в присутствии катализатора (V2O5, олеаты и стеараты
Со, Си, Mg и др.) или без него при продолжительности окисле-
ния более 3 мин с целью получения соответствующих кислот.
После дистилляции выделяют до 30% цзо-С7Н]5СООН (Пат.
Англии 919455, 1962).
Исследования [373] показали, что содержащийся в кубовом
остатке от ректификации продукта гидроформилирования пропи-
142
ТАБЛИЦА 5.1
Кислота (экстрагент) Количество кисло- ты, моль/моль Со Время экстракции, мин Степень извлечения кобальта, % (масс.)
Муравьиная 3,0 480 92,0
2,9 240 89,5
Уксусная 2,8 10 88,4
3,6 180 91,6
Масляная 2,1 3 98,3
3,5 10 92,1
лена кобальт достаточно полно извлекается водными раствора-
ми органических кислот (табл. 5.1). Полученные при декобаль-
тизации водные растворы солей кобальта можно использовать
для получения кобальтовых солей высших органических кислот.
Высококипящий кубовый остаток (начало кипения >150 °C
при 1,33 кПа) может служить сырьем для производства пласти-
фикатора вискозной пряжи [374]. Побочный продукт приобре-
тает пластифицирующие свойства в результате его сульфирова-
ния при 20—25 °C хлорсульфоповой кислотой и дальнейшей
нейтрализации.
Применение побочных продуктов производства бутанолов без
химической переработки — наиболее простое и возможности их
использования в различных областях техники будут расширять-
ся по мере изучения свойств отходов. Однако более целесообраз-
ной представляется квалифицированная химическая переработ-
ка побочных продуктов с получением индивидуальных кислород-
содержащих веществ. Наибольшая потребность в настоящее
время испытывается в бутанолах, 2-этилгексаноле и высших
жирных спиртах. Поэтому в промышленность, вероятно, будут
внедряться способы химической переработки, направленные на
повышение выходов именно этих спиртов.
5.2. Побочные продукты производства гликолей
В производстве гликолей гидратацией оксида этилена после от-
гонки из реакционной массы этилен- и диэтиленгликолей в ка-
честве кубовой жидкости остаются побочные продукты — смесь
гликолей сложного состава (полигликоли) [361]. Они представ-
ляют собой высококипящую вязкую темную массу со следую-
щими физико-химическими характеристиками: J42o = 1,1310;
/zD20= 1,4498; ЛГ=194; v20 = 63,6 мм2/с; у5о=16,7 мм2/с; у1Оо =
= 5,1 мм2/с [29].
Состав полигликолей [% (масс.)]: этиленгликоль—15,6; ди-
этиленгликоль— 17,5; триэтиленгликоль — 65,1; высокомолеку-
лярные гликоли—1,8 [361]. Количество побочных продуктов
143
производства гликолей — полигликолей — составляет более
70 кг на тонну готового продукта [361]. Вторичное использова-
ние такого огромного количества отходов имеет важное значе-
ние. Полигликоли могут найти непосредственное применение
после отгонки целевого продукта для приготовления антифриза
и производства цемента.
Химическая обработка кубового остатка производства глико-
лей позволяет намного расширить области использования поли-
гликолей. Они являются сырьем для органического синтеза
сложных эфиров — качественных пластификаторов и синтетиче-
ских масел [375]. Вязкостно-температурные характеристики
смазочных масел и пластификаторов на основе полигликолей
выше аналогичных свойств соединений, широко используемых в
практике [376]. Наиболее перспективными пластификаторами и
смазочными маслами являются полигликолевые эфиры монокар-
боновых кислот. Эти эфиры характеризуются низкой температу-
рой текучести (<—70 °C), хорошими вязкостно-температурны-
ми свойствами, стойкостью в отношении образования осадков
при окислении и хорошей совместимостью со многими полимер-
ными материалами [377, 378].
Исследования в области химии сложных эфиров полиглико-
лей [379—381] позволили выбрать оптимальные условия реак-
ТАБЛИЦА 5.2
Показатель Марка
36/1* 36/1 К* ЛЗ-5**
Кислотное число, мг КОН/г Температура, °C: 0,4—0,6 2,2—5,5 0,3—0,5
вспышки 195—205 195—210 176—175
застывания Вязкость кинематическая, мм2/с: —60 —60 —40
20 °C — 9,5—11,5
100 °C 3,1—3,5 3,5—3,7 —
—40 °C Смазывающая способность на 4ШМ: 1800—2500 2550—3900 —
Р, МПа 14,0 —
d, мм 0,8—0,9 —
Испаряемость (200 °C, 1ч), % Характеристики масла при окисле- нии (200 °C, 10 ч): 1,0—1,9 — —
масса осадка, г 0,02—0,03 — —
кислотное число после окисления, мг КОН/г 0,8—1,7 — —
вязкость кинематическая при 40 °C, мм2/с 2200—3370 — —
Содержание диэтиленгликолевого эфира, % (масс.): * <34,5; ** 100.
144
ТАБЛИЦА 5.3
Масло Количество загущающей присадки, % (масс.) V, мм2/с при 50 °C —30 °C ^заст, °C ^всп °C
Диэтиленгликолевый эфир — 4,61 520 —52 170 жирных кислот С5—С9 и: вининол 25,0 51,63 4320 —50 162 полиметакрилаты (на нор- 10,5 49,99 1660 —52 168 мальном спирте) полиметакрилаты (на изо- 10,0 52,44 — —32 — спиртах) Примечание. Масла выдерживают испытания на воздушный удар при 20 МПа.
ции этерификации: 140 °C, 4 ч, катализатор — 5% (масс.), избы-
ток жирных кислот 5—10% (масс.). В этих условиях выход эфи-
ров превышает 93%.
Диэтиленгликолевые эфиры на основе монокарбоновых кис-
лот фракций С5—Се и С7—С9 получают в промышленных мас-
штабах [382]. Свойства масел, в состав которых входит диэти-
ленгликоль, приведены в табл. 5.2.
Масла [383] на основе диэтиленгликолевых эфиров монокар-
боновых кислот фракции С5—Сю по устойчивости и воздействию
удара равноценны высоковязким нефтяным маслам (МК-22,
МС-20) (табл. 5.3).
Одним из возможных путей утилизации полигликолей явля-
ется превращение их в сложные диэфиры нафтеновых кислот.
Определено, что оптимальными условиями образования сложных
эфиров нафтеновых кислот и полигликолей являются: темпера-
тура 220—240 °C, продолжительность реакции 6 ч, мольное со-
отношение нафтеновые кислоты : гликоли, равное 2,2:1. В этих
условиях выход эфиров составляет >96% [384]. Изучение фи-
зико-химических свойств [384] показало, что полигликолевые
диэфиры нафтеновых кислот (фракция 120—240 °C) могут найти
применение в качестве компонентов синтетических смазочных
материалов. Их свойства при соотношении 2,1 1 (числитель) и
2,2 1 (знаменатель) приведены ниже:
Функциональное число, мг КОН/г:
кислотное 2,5
омыления 205/212
эфирное 202,5/209,5
Температура, °C:
застывания —40/—36
вспышки 260
Иодное число, г 12/100 г 0/0,47
10—627
145
ТАБЛИЦА 5.4
Показатель Пальмовое масло Смазка СТП-1 Смешанный эфир поли- гликоля
Температура плавления, °C 34 42 38,5
Испаряемость (200 °C, 1 ч), % 23,4 28,0 17,0
Испытания на 4ШМ: б/изн, мм (числитель) и f (знаме- натель) при 20 °C 0,091/0,125 0,092/0,125 0,090/0,100
при 100 °C 0,091/0,115 0,100/0,089 0,096/0,090
при 150 °C 0,101/0,075 0,100/— 0,096/—
Испытания при прокатке: коэффициент вытяжки после про- пуска: I 1,45 1,47 1,48
II 1,78 1,80 1,80
удельная сила трения в 1-ом про- 3,4 3,0 3,0
пуске, кг/мм2 загрязненность полосы, мг/м2 350 309 300
Известно, что в отечественной промышленности для холодной
прокатки тонкой стальной полосы используются гидрированное
кориандровое масло и сложные эфиры синтетических жирных
кислот, в частности триэтиленгликолевый эфир фракции С17—C2i
и олеиновой кислоты [375]. За рубежом для этих целей широко
применяются пальмовое масло и смазки, полученные непосред-
ственно переработкой пищевых жиров [385, 386]. Показано
[374], что полигликоль — кубовый остаток производства глико-
лей— может служить сырьем при производстве смазок для про-
катки стального листа. Синтезирован смешанный эфир олеино-
вой кислоты фракции Сп—С2о и полигликолей и проведено срав-
нительное испытание полученного продукта, пальмового масла
и смазок СТП-1. Испытания показали, что смешанный эфир по
своим свойствам не уступает маслу и смазке СТП-1 (табл. 5.4).
Полигликоли можно использовать (Пат. США 4400552, 1975)
для обезвреживания токсичных галоидированных соединений:
полихлорированных бифенолов, гексахлорциклогексана, три-
хлорбензола, тетрахлорбензола, гексахлорбензола, 1,2-дихлор-
фенола и др. Действием едких щелочей на полигликоли полу-
чают соединения, которые реагируют (20—120°C) с галоиди-
рованными соединениями, удаляя из них почти весь галоид.
Таким образом, побочные продукты производства гликолей
могут применяться в качестве смазок и пластификаторов. Под-
робное изучение физико-химических свойств этих продуктов по-
зволит найти новые рациональные пути их использования.
146
5.3. Побочные продукты производства глицерина
В настоящее время распространен хлорный способ получения
глицерина [46, 387]. На первой стадии газофазным хлорирова-
нием пропилена получают хлористый аллилхлорид:
С12
СН2=СНСН3 -----> СН2=СНСН2С1 + НС1
Далее (при гипохлорировании аллилхлорида образуются ди-
хлоргидрины глицерина:
СН2=СНСН2С1
н2О; С121-> С1СН2СН(ОН)СН2С1
-НС1 I----НОСН2СНС1СН2С1
На третьей стадии получают эпихлоргидрин щелочным гид-
ролизом дихлоргидрина глицерина:
2С1СН2СН(ОН)СН2С1 + Са(ОН)2-+2Н2С-СНСН2С1 + СаС12 + 2Н2О
О
На последней стадии эпихлоргидрин гидролизуют в присут-
ствии карбоната натрия до образования глицерина:
Н2О 0,5Na2CO3; 0,5Н2О
Н2С---СНСН2С1 ---> НОСН2СН(ОН)СН2С1--------------->
----> НОСН2СН(ОН)СН2ОН + NaCl д- 0,5СО2
Наряду с основным процессом получения глицерина проте-
кают дополнительные реакции и образуются побочные продук-
ты. К ним относятся [387] хлорсодержащие соединения: дихло-
риды— со стадии хлорирования пропилена; трихлорпропан,
2,3-дихлорпропилен, 1,2,3-трихлорпропан и смесь тетрахлорпро-
пиловых эфиров — со второй и третьей стадии процесса получе-
ния глицерина. На четвертой стадии кроме хлористого натрия
образуются кубовые остатки дистилляции глицерина [360].
Основным их компонентом являются полиглицерины со сте-
пенью поликонденсации >5, причем содержание диглицерина в
смеси достигает 90% [388].
Количество побочных продуктов производства глицерина со-
ставляет 10—20% от объема готовой продукции [361], поэтому
очень важно организовать их вторичное использование.
Проблема утилизации отходов глицерина включает два ас-
пекта: использование хлорсодержащих соединений, образую-
щихся на первых трех стадиях, и утилизация кубовых остатков,
богатых полиглицеринами и хлоридом натрия.
В промышленном масштабе осуществлена переработка лишь
дихлоридов аллила, используемых в качестве пестицидов (пре-
парат «ДД»), и трихлорпропана, который отделяется от других
10’
147
побочных веществ и выпускается в качестве товарного продук-
та [387].
Проблеме утилизации токсичных хлорорганических остатков
уделялось большое внимание в последние годы [390—395]. Об-
щее количество побочных продуктов производств хлорсодержа-
щих веществ в 1981 г. в ФРГ составило около 60 тыс. т [390].
Способ переработки хлорсодержащих отходов зависит от
типа производства. Они используются в процессе получения це-
левых хлорсодержащих соединений либо сжигаются (на суше
или на судах в открытом море) с обязательным улавливанием
образующегося НС1.
Разработана [391] конструкция реактора и схема сжигания
побочных продуктов производства хлорорганических веществ,
характеризующаяся низким расходом энергии и высокой эффек-
тивностью. Процесс основан на мокром окислении шлама возду-
хом в вертикальном трубчатом реакторе. Схема предложена
американской фирмой «Вертех триэйтмент системз». В реакторе
удаляется более 99% присутствующих побочных токсичных ве-
ществ. Соляную кислоту можно использовать для получения
хлорида кальция из известняка. Хлорид кальция можно приме-
нять при бурении нефти [393].
Существует [394] метод обработки побочных продуктов, со-
держащих хлоргидрин. Кубовые остатки нейтрализуют газами
с установки синтеза хлорида водорода, затем охлаждают и сме-
шивают с отходами от других производств. Побочные продукты
обрабатывают аммиаком, фосфорной кислотой и активным шла-
мом, осуществляют аэрацию механическим способом в аэрато-
рах и добавляют флокулянт. После фильтрации шлам направ-
ляют в отвал, а фильтрат возвращают в процесс.
Предложен способ [396] обезвреживания галогенсодержащих
углеводородов, основанный на разрыве прочной связи С—CI,
под влиянием отрицательно заряженных атомов кислорода в от-
сутствие воды. Процесс проводят в аппарате небольшого объ-
ема, пропуская электрический ток через раствор побочных га-
лоидсодержащих продуктов в безводном растворителе. При
этом молекула растворенного кислорода приобретает избыточ-
ный электрон. Процесс может быть осуществлен в промышлен-
ном масштабе заменив дорогостоящее захоронение в грунт.
Хлорсодержащие побочные продукты целесообразно перера-
батывать [397] методом исчерпывающего хлорирования и окси-
хлорирования. Этот процесс позволит предотвратить загрязнение
окружающей среды и получить ценные продукты (СС14, С2С14).
Определены оптимальные условия получения СС14 и С2С14
методом исчерпывающего хлорирования отходов производства
глицерина [состав, % (масс.): аллилхлорид — 3,0; трихлорпро-
пан — 62,0; эпихлоргидрин — 9,6; дихлоргидрин — 8,0; тетрахлор-
пропиловый эфир—17,2]. Реакцию проводят при температуре
148
500 °C, молярном соотношении хлор фракция трихлорпропана,
равном 6:1, и времени контакта 4—5 с. При хлорировании в
этих условиях [398, 399] суммарное содержание СС14 и С2С14 в
катализате достигает 92%, а выход их на прореагировавший
продукт достигает 84%.
Предлагается получать [400, 401] СС14 и С2С14 также мето-
дом окислительного хлорирования побочных продуктов произ-
водства эпихлоргидрина при температуре 490 °C, молярном со-
отношении фракция трихлорпропана : хлор : воздух, равном 1
: 5,6 : 3,25, и времени контакта 7 с. Фракцию трихлорпропана
рекомендуется вводить в реактор ниже места подачи воздуха,
что приводит к подавлению процесса горения образовавшимися
тетрахлоруглеродами в зоне окислительного хлорирования.
В этом случае выход суммы СС14 и С2С14 достигает 80%.
Кубовый остаток производства глицерина можно использо-
вать для синтеза пластификаторов путем этерификации полигли-
церинов синтетическими монокарбоновыми кислотами [402].
Эффективность сложных эфиров на основе полиглицеринов
и монокарбоновых кислот фракции С4—Сю в качестве пластифи-
каторов поливинилхлорида характеризуется [388] критической
температурой растворения, температурой стеклования и некото-
рыми другими показателями. Установлено, что наименьшую
критическую температуру растворения поливинилхлорида (или
наибольшую совместимость с полимером) имеют пластификато-
ры с длиной цепи неполярной части молекулы С4_6. Дальнейшее
увеличение длины цепи снижает совместимость и критическая
температура растворения повышается до 183 °C. Гомогенный
раствор поливинилхлорида в пластификаторе с радикалами Сю
не удается получить даже при более высоких температурах.
Температура стеклования образцов с пластификаторами —
сложными эфирами на основе полиглицеринов и кислот С4_9 —
резко снижалась в области среднего содержания добавок (30—
40 масс, ч.), что свидетельствовало о молекулярном характере
взаимодействия этих пластификаторов с поливинилхлоридом.
Морозостойкость поливинилхлорида, пластифицированного син-
тезированными сложными эфирами, изменяется экстремально
при большом содержании пластификатора (рис. 5.1, кри-
вые 2, 3).
Установлено, что оптимальной длиной углеводородных ради-
калов в пластификаторах этого класса является С5_7, т. е. плас-
тификаторы, синтезированные на основе полиглицеринов с ва-
лерьяновой, капроновой и энантовой кислотами. Содержащие
их образцы по основным свойствам не уступают, а по морозо-
стойкости даже превосходят образцы поливинилхлорида, плас-
тифицированного широко применяемым для этих целей диок-
тилфталатом. При этом стоимость рекомендуемых пластифика-
торов на 30—35% ниже стоимости диоктилфталата.
149
РИС. 5.1. Зависимость температуры мо-
розостойкости от числа атомов углерода
(пс) в радикале кислоты при содержа-
нии пластификатора на основе полигли-
колей и монокарбоновых кислот, масс, ч:
1 — 20; 2 — 40; 3 — 80
Для очистки [403] солевых
отходов (хлорида натрия) пред-
ложен электрохимический спо-
соб [403] глубокого окисления
органических примесей, содер-
жащихся в кубовом остатке про-
изводства глицерина. Процесс
осуществляется в бездиафраг-
менном электролизере при pH
3—7, температуре 60—90 °C при
УФ-облучении. Степень очистки кубовых остатков составляет
85—95%. Растворы хлористого натрия, очищенные от органиче-
ских примесей, можно использовать для получения хлора и
каустика.
Хлорид натрия можно выделять (Пат. США 4164507, 1978)
из кубовых остатков производства глицерина из эпихлоргид-
рина, содержащих [% (<масс.)] полиглицерина 40—45, хлорида
натрия — 45—55, воды — 6, а также дихлоргидрин, глицерин и
другие примеси. Для этого кубовый остаток смешивают с «-бута-
нолом в отношении 1 1,3—2 при температуре 90—120 °C, затем
отделяют хлорид натрия и отгоняют бутанол, который возвра-
щают в процесс. Этот метод позволяет выделить из кубового
остатка до 98% хлористого натрия.
Выбор того или иного способа утилизации побочных продук-
тов производства глицерина определяется многими факторами.
Только тщательным анализом и технико-экономической оценкой
может быть подобран оптимальный вариант.
ГЛАВА 6
Применение побочных продуктов
производства синтетических жирных
кислот и фталевого ангидрида
6.1. Побочные продукты производства синтетических
жирных кислот
Процесс получения синтетических жирных кислот методом жид-
кофазного окисления парафиновых углеводородов в присутствии
кислорода ,и марганец-натриевого катализатора сопряжен с об-
разованием значительного количества сточных вод и кубовых
остатков, содержащих органические и неорганические кислоты
и их соли, спирты, альдегиды, кетоны, сульфат натрия, соли
кальция, марганца, железа и другие продукты органического
синтеза. В связи с этим возникает необходимость создания тех-
нологических процессов переработки сточных вод и кубовых
остатков производства синтетических жирных кислот с выделе-
нием загрязняющих компонентов и последующим их использо-
ванием.
Кислые сточные воды образуются на стадии окисления пара-
фина при охлаждении и промывке отработанного воздуха [404].
В кислых стоках содержится до 15% (масс.) низкомолеку-
лярных кислот Ci—С4. Их состав [404] в усредненном потоке
кислых вод [% (масс.)]: муравьиная — 5,48; уксусная — 5,04;
пропионовая — 2,40; масляная — 1,98.
Кроме кислот Ci—С4 в кислых сточных водах в небольших
количествах присутствуют карбоновые кислоты с большим чис-
лом углеродных атомов, соединения эфирного и карбонильного
характера, нейтральные органические продукты. Количество
кислых стоков (кислотное число 140—150 мг КОН/г) составляет
430—450 кг на 1 т исходного парафина.
Деструктивные методы очистки кислых сточных вод (биоло-
гическое и термическое окисление, электрохимическая очистка),
связаны с разложением присутствующих в кислых стоках орга-
нических соединений [405, 406]. Общим недостатком этих мето-
дов является потеря ценных низкомолекулярных кислот. Поэто-
му широкое распространение получили в последнее время в
СССР и за рубежом регенеративные методы очистки кислых
сточных вод — солевой (порошковый) [407—410], экстракцион-
ный [411—416], азеотропная ректификация [417—420] и др. Эти
методы связаны с извлечением низкомолекулярных кислот или
их производных.
151
Аммонийные соли кислот Q—С4 можно использовать в ка-
честве азотного удобрения [421]. Внесение этих солей в почву
в количествах, равных по азоту аммиачной селитре, обеспечива-
ет одинаковый с последней прирост урожая ячменя и картофеля.
Обработка посевов бобовых культур водными растворами, со-
держащими 0,01—0,02% (масс.) кислот Q—С4, более чем в
пять раз уменьшает количество и массу сорняков; на 13% повы-
шает урожайность культурных растений.
Положительные результаты получены при использовании
нейтрализованного аммиаком водного конденсата в качестве
деэмульгатора при обессоливании и обезвоживании нефтей
[422].
Явление адсорбционного понижения прочности твердых ма-
териалов может быть использовано в случае применения вод-
ного конденсата для улучшения свойств углей (А. с. СССР
294999, 1971). В результате обработки угля нейтрализованным
водным конденсатом (соотношение кальциевые соли кислот
Ci—С4:уголь=1 1000) его прочность уменьшается на 14—
36%, а коэффициент размолоспособности увеличивается на
8—27%.
В процессе добычи нефти и газа для повышения эффектив-
ности кислотной обработки призабойной зоны нефтяных, газо-
вых и нагнетательных скважин в рабочие растворы соляной
кислоты вводится до 5% (масс.) уксусной кислоты. Показана
(А. с. СССР 251984, 1970) возможность замены уксусной кисло-
ты в рабочих растворах соляной кислоты водным конденсатом.
Добавка последнего замедляет скорость взаимодействия раство-
ров соляной кислоты с карбонатными породами и увеличивает
суммарный объем его полезной работы. Аналогично уксусной
кислоте низкомолекулярные кислоты, содержащиеся в водном
конденсате, предупреждают выпадение гидроксидов железа и
алюминия в пространстве пласта, снижают набухаемость глини-
стых пород и коррозионное действие рабочей жидкости на кон-
струкционные элементы оборудования скважин (А. с. СССР
258985, 1970).
Водный конденсат можно использовать в процессе получения
красного глиняного кирпича [423], для промывок теплоэнерге-
тического оборудования (А. с. СССР 286617, 1970), а также в
качестве низкомолекулярной основы загустителя синтетического
солидола.
Перспективным потребителем смеси низкомолекулярных кис-
лот является кормопроизводство [405]. Включение аммонийных
солей низкомолекулярных кислот в дефицитный по протеину
рацион скота позволило снизить затраты питательных веществ
на 0,4 корм. ед. на 1 кг привеса и на 25% уменьшить содержа-
ние перевариваемого протеина в корме. Добавка в рацион нат-
риевых солей кислот Ci—С4 значительно повышает молочную
152
продуктивность коров (А. с. СССР 320968, 1971). Внесение вод-
ного конденсата [1,8—2,4% (масс.)] в комбинированный силос
обеспечивает более высокую сохранность питательных веществ
по сравнению с обычным силосованием.
Аммонийные соли кислот Q—С4 могут быть использованы
для получения препарата, предотвращающего прилипание и
примерзание сыпучих материалов к поверхностям транспортных
средств и сосудов [405].
Исследована возможность использования низкомолекуляр-
ных кислот и их солей в производстве смазок для холодной об-
работки металлов, моющих средств для очистки металлической
поверхности от пестицидов, а также в процессах выработки кож
и в качестве биогенных добавок [424].
Использование концентрата низкомолекулярных кислот [60—
70% (масс.*)] дало лучшие результаты по сравнению с водным
конденсатом. Изучение возможности применения концентрата
кислот проводилось в основном в тех же отраслях народного
хозяйства, где были получены положительные результаты от ис-
пользования водного конденсата. Установлено, что концентрат
низкомолекулярных кислот может использоваться для удаления
отложений с поверхностей теплообменников, консервирования
кормов, получения синтетического солидола и растворителей,
обработки нефтяных скважин и т. д. [405].
Основным препятствием широкого использования концентра-
та низкомолекулярных кислот является большое количество
примесей. Поэтому в соответствии с требованиями потребите-
лей целесообразно очищать кислоты от нежелательных компо-
нентов и выделять индивидуальные кислоты — ценные реагенты
в органическом синтезе [425, 426].
Сульфатные стоки производства представляют собой 8—
12%-ный раствор сульфата натрия с примесью органических ве-
ществ [406]. Установлено [427—429], что органические примеси
сульфатных вод в основном содержат кислоты Ci—С6 и дикар-
боновые кислоты С4—Си. Степень использования сульфатных
стоков составляет менее 20% [406]. Утилизация их в настоящее
время заключается в извлечении сульфата натрия, который при-
меняется на предприятиях целлюлозно-бумажной, стекольной,
химической, текстильной, фармацевтической и других отраслей
промышленности [430, 431].
Основная причина технологических затруднений в перера-
ботке и применении сульфата натрия заключается в примесях
органических соединений и металлов, содержащихся в сульфат-
ном стоке.
Метод выделения сульфата натрия из сточных вод состоит
в упаривании их до концентрации 24% с последующим обезво-
живанием в аппаратах КС или РКСТ [427, 428]. В процессе
упаривания часть органических соединений улетучивается в ат-
153
ТАБЛИЦА 6.1
Количество добавки
к массе цемента
Предел прочности
на сжатие, МПа
Состав смеси
% (масс.)
после про-
парки
после 28 сут
выдержки
при н. у.
Без добавки —
С добавкой (см. с. 155) 0,10
0,15
0,20
— 0,140—0,144 1,96—20,59
3,0 14,42 20,59
4,5 15,00 19,32
6,0 14,81 18,63
мосферу. Для устранения этого выпаренную часть подвергают
обезвреживанию термическим окислением в топках дорогостоя-
щих котлов-утилизаторов. В целях экономии предлагается сна-
чала очищать сточные воды от примесей, а потом подвергать их
упариванию [432]. Обезвоживание сульфатного стока, предвари-
тельно очищенного от органических примесей и соединений ме-
таллов, позволяет получать качественный сульфат натрия, не
обладающий запахом, с содержанием основного вещества
98% (масс.) [432].
Определены [433, 434] оптимальные параметры выделения
сульфата натрия из сульфатных стоков синтетических жирных
кислот в аппаратах КС: предварительное упаривание сульфат-
ного стока при pH 5,0—6,0; сушка нейтрализованного стока при
pH 6,0—7,0, температурах кипящего слоя 125 °C и агента суш-
ки 700 °C; прокаливание готового сульфата натрия при 400—
600 °C.
Термообработка гранулированного сульфата натрия приво-
дит к практически полному разрушению органических примесей
и улучшению химического состава продукта. Выделенный таким
образом сульфат натрия был испытан на заводах-потребителях
по производству тарного стекла, результаты — положительные.
На заводах по производству синтетических жирных кислот,
не имеющих сульфатных установок, обезвоживание сульфатного
стока с целью его утилизации целесообразно проводить спосо-
бом глубокого упаривания [435]. Этот метод позволяет получить
сульфат натрия следующего состава [% (масс.)]: сульфат нат-
рия— 92,34; растворимые органические примеси — 0,88; хлори-
ды— 1,39; металлы — 0,49; нерастворимый осадок — 0,065;
вода — 4,82.
В процессе производства синтетических жирных кислот обра-
зуется значительное количество лактонов, концентрирующихся
главным образом в сточных водах. Лактоны предлагается [436]
использовать в качестве исходного сырья для получения натрие-
вых солей оксикислот — пластификаторов для бетона [432].
Результаты испытаний бетонных смесей с добавкой побочных
154
продуктов производства синтетических жирных кислот приведе-
ны в табл. 6.1. Введение смесей натриевых солей оксикислот
[состав, % (масс.): фракция С5—С2о синтетических жирных кис-
лот— 25—38; лактоны и эфиры — 0,5—1,2; углеводороды —
12—17; вода] значительно повышает пластичность бетонной
смеси М-200.
В производстве синтетических жирных кислот одним из ос-
новных продуктов является кубовый остаток, образующийся
(выход 30%) при дистилляционной перегонке кислот и содержа-
щий в основном кислоты фракции С20 и выше.
Кубовый остаток находит широкое применение в производст-
ве литейных крепителей [437, 438]. Его свойства оказывают
влияние [439] на связующую способность литейного крепителя.
Для определения характера этого влияния кубовый остаток
разделяли на смолистые и несмолистые компоненты, из кото-
рых, в свою очередь, выделяли омыляемые и неомыляемые про-
дукты.
Уменьшение кислотного числа кубового остатка приводит к
возрастанию прочности крепителя на его основе, т. е. она обу-
словлена, в основном, и наличием смолистых соединений в ку-
бовом остатке [439].
У несмолистых соединений, наоборот, с уменьшением кис-
лотного числа кубового остатка прочность крепителя, получен-
ного на их основе, убывает. Прочность крепителя из омыляемых
веществ ниже прочности крепителя как из смолистых, так и не-
смолистых его составляющих.
Кубовые остатки производства синтетических жирных кислот
могут использоваться в качестве пластификаторов кумароновых
плиток — вида отделочного материала для настилки полов [440].
Плитки, изготовленные на основе кубового остатка, обладают
повышенной прочностью на изгиб (табл. 6.2).
Кубовые остатки с кислотным числом 70—100 мг КОН/г
можно применять в дорожном строительстве в качестве эмульга-
тора при изготовлении прямых битумных эмульсий.
Была предпринята попытка [441] выделения синтетических
жирных кислот фракции Ci8—С23 из кубового остатка дистилля-
ТАБЛ1ЩА 6.2
Пластификатор
Сопротивление
на изгиб, кг/см2
Сопротивление
на разрыв, кг/см2
Кубовый остаток"
Высокомолекулярные кислоты
Смолистые вещества
70 40
95 36
92 26
Кислотное число 57,8 мг КОН/
155
РИС. 6.1. Зависимость выхода кислот Cie—С2з
(Вк, %) от кислотного числа (к. ч.) кубового
остатка
цией при температуре <330°C, и ос-
таточном давлении от 0,27 до 1,07 кПа
(рис. 6.1).
Синтетические жирные кислоты
фракции Cie—С2з применяются для
получения смазочных материалов.
Соединения, входящие в состав ку-
бовых остатков производства синтетических жирных кислот, —
кислоты нормального строения, дикарбоновые кислоты и не-
которые другие вещества, плохо растворимые в топливах и
обладающие относительно низким защитным действием. С це-
лью улучшения качества защитных присадок к топливам на
основе кубовых остатков синтетических жирных кислот было
проведено [442] разделение/ их на компоненты с помощью об-
работки метиловым спиртом при различных температурах.
Разработана схема разделения и олигомеризации кубового
остатка синтетических жирных кислот для получения димеров
ненасыщенных кислот, хорошо растворяющихся в углеводород-
ных топливах. Испытания полученных продуктов в качестве
присадок к гидроочищенному топливу Т-7 показали значитель-
ное улучшение его защитных свойств.
Кубовые кислоты фракции С20—С25 могут быть использованы
для синтеза высших жирных спиртов, которые применяются в
производстве депрессаторов высокопарафинистых нефтей [443].
Требования к высшим жирным спиртам на основе кубовых кис-
лот приведены ниже:
Функциональное число, мг КОН/г:
кислотное 0,3
эфирное 2,0
гидроксильное 150—165
карбонильное 1,0
Иодное число, г 12/100 г 1,0—2,0
Содержание спиртов, % (масс.):
С18 Ю
С22 65
Ряд компонентов кубовых остатков производства синтетиче-
ских жирных кислот — ненасыщенные, окси- и кетокислоты, по-
лимерные соединения и кислоты изостроения представляют зна-
чительный интерес как соединения, обладающие высокими по-
верхностно-активными свойствами. Вместе с тем в кубовых ос-
татках содержатся соединения, имеющие невысокие поверхност-
но-активные свойства, например, неомыляемые, эфирсодержа-
156
щие соединения и др. Разделение кубового остатка рекоменду-
ется проводить [444] экстракцией метанолом или ацетоном при
30 °C, соотношении кубовый остаток растворитель, равном 1 :4.
Полученные при этом соединения можно использовать в качест-
ве защитных присадок среднедистиллятных углеводородных
топлив.
Изучена [445] возможность синтеза виниловых эфиров син-
тетических жирных кислот фракции С2о—С25, выделенных из
кубовых остатков.
На основе исследования физико-химических свойств сложных
эфиров кубовых кислот и полигликолей (кубового остатка про-
изводства гликолей) сделаны рекомендации по использованию
их в качестве компонентов основы технологических смазок для
пропитки и волочения металлов [446].
6.2. Побочные продукты производства фталевого ангидрида
Процессы получения фталевого ангидрида из нафталина и
о-ксилола протекают по следующим схемам [447]:
Протекание побочных реакций обусловливает образование
дополнительных продуктов, в основном, 1,4-нафтохинона [с5—
8% (масс.)] и малеинового ангидрида [<7—10% (масс.)] [447г
448]. При этом выход побочных продуктов при окислении о-кси-
лола выше, чем при окислении нафталина.
Побочные продукты концентрируются в кубовом остатке рек-
тификации целевого продукта и в отходных газах, представляю-
щих собой отработанный воздух, содержащий диоксид углеро-
да, фталевый и малеиновый ангидриды и 1,4-нафтохинон. При
окислении 1 т сырья образуется около 25 000 м3 отходящих га-
зов, в которых содержание органических веществ колеблется
в пределах 1,0—1,5 г/м3 [449]. В настоящее время отходящие
газы обезвреживают методом водной промывки. При этом зна-
чительные объемы сточных вод сбрасываются в водоемы, что
влечет за собой загрязнение окружающей среды и потери цен-
ных реагентов для органического синтеза. В связи с этим возни-
кает задача утилизации органических веществ, входящих в со-
став отходящих газов.
157
Водная промывка отходящих газов может рассматриваться
[447] как возможность преимущественного получения концен-
трированных растворов малеиновой кислоты (200—300 г/л) при
одновременной 100%-ной степени обезвреживания выбросов.
Характеристика получаемого в промышленных условиях про-
дукта утилизации малеиновой кислоты приведена ниже:
Внешний вид Жидкость бурого цвета без инород-
ных включений, практически без за-
паха
Плотность при 20 °C, г/см3 1,10—1,20
Содержание сухого вещества, 30—40
% (масс.)
Вязкость динамическая при 20°C, 0,3—1,0
Па-с
pH 6,0—7,5
Такой раствор может быть использован в качестве крепите-
ля при изготовлении стержней, применяемых при получении от-
ливок из чугуна и стали, вместо широко используемого крепи-
теля 4ГУ (А. с. СССР 309767, 1971), а также в качестве интен-
сификатора помола цемента [450].
Предложен (Пат. США 4391617, 1982) эффективный метод
извлечения малеинового ангидрида и непрореагировавшего наф-
талина из отходящих газов производства фталевого ангидрида.
Из-за сублимации кристаллических побочных продуктов в газо-
вом потоке на выходе содержится значительное количество
твердых веществ. Для выделения их предлагается установить
горизонтальные трубчатые теплообменники попеременно: один —
на конденсацию, другой — на плавление и подавать в них хо-
лодный или горячий воздух (например, отходящий газ). Этот
процесс пригоден для извлечения из газового потока любого
кристаллического вещества.
С целью выделения нафталина и фталевого ангидрида газо-
образные продукты каталитического окисления нафталина
быстро охлаждают до температуры ниже точки росы контакти-
рованием в теплообменнике с жидким нафталином. Охлажден-
ные продукты обрабатывают жидким нафталином и получают
раствор нафтохинона и фталевого ангидрида в нем (Пат. Япо-
нии 53—9270, 1977). Метод позволяет улавливать ценные про-
дукты, содержащиеся в отходящих газах производства фталево-
го ангидрида.
В литературе имеются сведения по совершенствованию про-
цесса очистки отходящих газов производства фталевого ангид-
рида. Однако способы утилизации органических побочных про-
дуктов не приводятся [451—453].
Кубовый остаток, образующийся в процессе вакуумной дис-
тилляции фталевого ангидрида [454], представляет собой тем-
но-коричневую массу следующего состава [% (масс.)]: летучие
158
компоненты — 55—65, смолистые вещества — 32—42, неоргани-
ческие примеси — 3. Предлагается [454] обрабатывать кубовый
остаток каменноугольными маслами и использовать получен-
ную смесь в качестве энергетического топлива или сырья для
получения технического углерода. Разработана и осуществлена
в промышленном масштабе технологическая схема утилизации
кубового остатка производства фталевого ангидрида этим ме-
тодом.
Предлагается (А. с. СССР 521273, 1976) выделять фталевый
ангидрид из кубовых остатков с использованием метода дистил-
ляции. С целью увеличения степени извлечения целевого про-
дукта процесс ведут в присутствии керосина.
Для разделения смеси (заявка Японии 53—34740, 1978), со-
держащей нафтохинон, фталевый ангидрид и фталевую кислоту
промывают ,водой и подвергают фильтрации или центрифугиро-
ванию для отделения нафтохинона и фталевой кислоты. Далее
влажный осадок обрабатывают инертным органическим раство-
рителем при температуре 60 °C. В этих условиях растворитель
не смешивается с водой и растворяет нафтохинон и не раство-
ряет фталевую кислоту. В результате нафтохинон экстрагируют
растворителем, а фталевую кислоту отделяют в виде кристал-
лов, содержащих небольшое количество растворителя. Фталевая
кислота содержит воду, что исключает необходимость ее допол-
нительной очистки путем перекристаллизации.
Предложена (Пат. Японии 53—9209, 1977) схема технологи-
ческого процесса выделения нафтохинона из продуктов катали-
тического газофазного окисления нафталина. Смесь, содержа-
щая нафтохинон, фталевый ангидрид, малеиновый ангидрид и
нафталин, взаимодействует с водным раствором основания для
удаления фталевого и малеинового ангидрида и нафтохинона.
Нафтохинон можно выделить из фталевого ангидрида (за-
явка ФРГ 2654405, 1977). С этой целью смесь фталевого ангид-
рида и нафтохинона обрабатывают (15—600 мин) 1,4,4,а,9д —
тетрагидроантрахиноном, взятым в количестве 0,3—3 моль на
1 моль нафтохинона, при температуре 200—300 °C и отсутствии
кислорода. Фталевый ангидрид удаляют перегонкой.
Для отделения побочных продуктов производства фталевого
ангидрида (фталевой кислоты, малеинового ангидрида и нафто-
хинона) от целевого продукта смесь обрабатывают конденси-
рующим агентом — резольной фенолформальдегидной смолой,
взятой в количестве 0,5—2,0% (масс.) при температуре 200—
230 °C. Полученную смесь выдерживают при температуре 250—
280 °C в течение 1—3 ч, после чего подвергают дистилляции
(А. с. СССР 539026, 1976).
159
ГЛАВА 7
Применение побочных продуктов
производства замещенных фенолов
7.1. Побочные продукты производства фенола и ацетона
Широко используется в мировой практике кумольный метод по-
лучения фенола [455]. Его преимущества состоят в небольшом
расходе дешевого сырья и совместном получении двух ценных
продуктов — фенола и ацетона. Производство фенола и ацетона
кумольным методом включает стадии получения изопропилбен-
зола, синтеза гидропероксида изопропилбензола и монокислот-
ного разложения его на фенол и ацетон:
А1С13 о2
СвНв+ СН3СН=СН2 ---> СвН5СН(СН3)2 -->
н+
---> СвН5С(СН3)2 —> свн6он + сн3созн3
ООН
Разнообразие реакций, протекающих при синтезе фенола по
рассматриваемой технологии, неизбежно приводит к образова-
нию побочных продуктов в количестве 0,1—0,2 т на 1 т фено-
ла [455].
Фенольная смола является основным побочным продуктом
производства фенола и ацетона кумольным методом. Известен
ее состав [456] [% (масс.)]: фенол — 6—12; диметилфенилкар-
бинол — 0—7; а-метилстирол и димеры — 7—23; ацетофенон —
7—9; и-кумилфенол — 32—43; остаток неидентифицированный —
2—15. Соотношение компонентов меняется в широких пределах
в зависимости от условий переработки. Примерная схема про-
цесса, выключающая побочные реакции образования основных
компонентов фенольной смолы [457], приведена на с. 161.
Фенольная смола, получаемая в процессе производства фе-
нола и ацетона, может найти различное применение. Представ-
ляется интересным непосредственное использование фенольной
смолы в промышленности строительных материалов взамен ши-
роко потребляемого товарного фенола. Однако изготовление
строительных материалов с применением фенольных смол в
промышленном масштабе реализовано лишь частично. Для
широкого внедрения в производство требуются дополнительные
исследования в этом направлении.
В некоторых случаях рационально использовать фенольную
смолу для компаундирования топочных мазутов и изготовления
консервантов древесины [458, 459].
160
Широкое распространение нашло применение фенолформаль-
дегидной смолы в качестве ингредиента резиновых смесей бла-
годаря комплексу усиливающих свойств, а также с целью улуч-
шения технических свойств сырых резиновых смесей. Введение
10—20 масс. ч. смолы на 100 масс. ч. каучука увеличивает стой-
кость вулканизатора к действию масел, бензина и других рас-
творителей в 1,5 раза, усиливает сопротивление тепловому ста-
рению по относительному удлинению, повышает его теплостой-
кость и твердость.
Увеличение содержания фенолформальдегидной смолы в ре-
зиновых смесях повышает сопротивление разрыву в 3—4 раза,
а относительное удлинение в 1,5—2 раза.
Используется фенольная смола в производстве деэмульгато-
ров нефтяных эмульсий, при изготовлении фенолформальдегид-
ной смолы, применяемой в качестве связующего в производстве
пресс-порошков, линолеума и рулонных кровельных материа-
лов [460—462].
Известно много способов утилизации фенольной смолы, свя-
занных, в основном, с химической переработкой и выделением
ценных компонентов. Так, а-метилстирол может использоваться
в производстве различных полимерных материалов, ацетофе-
нон— душистых веществ и красителей, кумилфенол — маслорас-
творимых смол, антиоксидантов и антисептиков [463].
Общий недостаток этих решений определяется необходи-
мостью выделения чистых продуктов и организации их после-
дующей переработки. Это усложняет технологический процесс
и делает производство фенола зависимым от потребления полу-
чающихся побочных продуктов. Поэтому более рационально
ориентироваться на переработку всей массы побочных продук-
тов с их использованием в основном технологическом процессе:
---> Водный слой
---► Отстаивание ---->
----►- Гидрирование фенольной смолы ---►-
---------------------------------------> Этилбензол
----> Ректификация-------> Фенол
Кумол
Технологические трудности и значительные капитальные за-
траты при переработке фенольной смолы делают в определен-
ных условиях экономически рациональным ее сжигание [464].
Предложены [12] различные способы переработки фенольной
смолы: периодическая термодеструкция (I), деструкция на дви-
жущемся теплоносителе (II) и гидрирование (III) (табл. 7.1).
Как видно, наиболее перспективный способ — гидрирование фе-
162
ТАБЛИЦА 7.1
Продукт Выход продуктов, кг/т
I и ш
Этилбензол 29 82 211
Изопропилбензол 180 160 316
а-Метилстирол 105 31 —
Фенол 223 138 241
Ацетофенон 95,5 95,5 —
вольной смолы. В случае его реализации удается извлечь до
85% ценных химических продуктов [% (масс.)]: этилбензола —
15; изопропилбензола — 49; фенола — 25 и др. Однако указан-
ный процесс до сих пор не реализован в промышленности из-за
сложности удаления солей из смолы и сравнительно небольшо-
го срока работы катализатора.
В фенольной смоле содержатся соли — сульфат и фенолят
натрия. Их количество достигает 2%. Соли образуются в про-
цессе нейтрализации реакционной смолы при разложении гид-
ропероксида изопропилбензола в присутствии серной кислоты и
10%-ного раствора щелочи. Они отравляют алюмокобальтмолиб-
деновый катализатор. Было предложено [465] разлагать гидро-
пероксид кумола в присутствии катионообменных смол КУ-2—8,
КУ-25 и др. Применение их увеличило процент разложения гид-
ропероксида и пробег катализатора возрос до 1 400 ч.
Радикальной схемой переработки фенольной смолы и ее сме-
сей с метилстирольной фракцией является деструктивная гидро-
генизация на аминокобальтмолибденовых катализаторах при
350 °C и 4,90 МПа [466—469]. В этом методе смола на 80%
вступает в реакцию с получением ценных продуктов: фенола,
кумола и этилбензола (образующегося при гидрировании аце-
тофенона). Такое решение не только позволяет освободиться от
побочных продуктов, но и на 8—10% уменьшает расход сырья.
Разработана промышленная установка по гидрогенизацион-
ной переработке фенольной смолы, на которой при расходе 1,8%
водорода (в расчете на массу исходной смолы) получают
59,4% углеводородов С8—С9 (в основном этилбензол и кумол)
и 21,7% фенола [469]. Разработана технология гидрогенизаци-
онной переработки продуктов сернокислотного разложения ку-
милгидропероксида после отгонки ацетона. Этот процесс позво-
ляет вообще не получать фенольную смолу, выход высококипя-
щих соединений не превышает 3% от массы продуктов гидроге-
низации [470].
Одним из простых решений является термическая деструкция
образующейся фенольной смолы. При нагревании до 350—500 °C
И* 163
РИС. 7.1. Принципиальная схема переработки фенольной смолы термоката-
литическим разложением:
1 колонка для отгонки фенольной смолы;2 — колонка отгонки смеси фенола и а-метил-
сгирольной фракции от ацетофеноновой фракции; 3 — реактор разложения фенолов
происходит деструкция димера и полимеров а-метилстирола с
образованием мономерного соединения и превращение кумилфе-
нола в фенол и а-метилстирол [471—473].
Изучена возможность переработки фенольной смолы термо-
каталитическим разложением (А.с. СССР 121796, 1959) в при-
сутствии фенолята алюминия (рис. 7.1). Оптимальными усло-
виями являются: температура разложения 230—250 °C, остаточ-
ное давление 200—400 мм рт. ст., расход катализатора по алю-
минию до 0,4% (масс.).
Известны также способы переработки фенольной смолы ка-
талитическим разложением в присутствии железных стружек
(А.с. СССР 191575, 1955), минеральных и органических кислот
(Пат. Англии 979134, 1969).
Изучено каталитическое разложение фенольной смолы на
стационарном катализаторе — активном оксиде алюминия
[462]. Предложен двухстадийный метод переработки фенольной
смолы в промышленном масштабе, включающий перегонку фе-
нольной смолы с перегретым водяным паром и каталитическое
разложение дистиллята на активном оксиде алюминия.
Выделение фенола, углеводородной (а-метилстиролизопро-
пилбензольной) фракции и сложного фенола перегонкой реко-
мендуется проводить по непрерывной схеме [474, 475]. При про-
ведении этого процесса значительно уменьшаются потери цен-
ных компонентов.
Осуществлены опытно-экспериментальные пробеги на про-
мышленной установке по изучению возможности производства
битума, кокса, фенольной фракции, ароматических углеводоро-
164
дов и котельного топлива методом окисления и коксования сме-
си фенольной и генераторной смолы [476].
Показана возможность получения полиоксибензиламинов
(олигомеров) на основе фенольной смолы [477, 478].
Предложен метод утилизации фенольной смолы при понижен-
ном давлении [479]. Метод основан на дистилляции летучих
компонентов смолы с выделением а-метилстирола и частично
ацетофенона. Этим способом можно переработать смолу, содер-
жащую относительно большой процент фенола. Дистилляцион-
ная фракция фенольной смолы по этому методу содержит 10—
20% ацетофенона, который применяется в качестве растворите-
ля эфиров целлюлозы и смол, в косметической промышленно-
сти. Модифицируя дистилляцию смолы можно получить фрак-
цию следующего состава [% (масс.)]—ацетофенона — 70, ку-
милфенола — 20, фенола—10. Остаток после переработки смо-
лы, как и сама фенольная смола, используется для модифика-
ции асфальтов [479]. Для предотвращения разжижения асфаль-
та при добавлении фенольной смолы предлагается смесь ас-
фальтов Д-50 и Д-70 с 5%-ной добавкой смолы подавать на
окисление, которое проводится при пропускании потока горя-
чего воздуха через нагретую до 140—160 °C смесь.
Одним из способов утилизации фенольной смолы является
конденсация фенольной смолы с СНгО в присутствии соляной
кислоты, приводящая к образованию фенолформальдегидных
смол новолачного типа. Такие смолы могут применяться в ка-
честве связующего материала в производстве пластмасс, резино-
технических изделий, в качестве усилителей резины, а также
связующих агентов в новолачных пресс-порошках [480].
Таким образом, при правильной организации переработки
фенольной смолы можно намного увеличить объем выработки
товарного фенола и других весьма ценных продуктов, необходи-
мых для народного хозяйства.
7.2. Применение п-кумилфенола
В последние годы большое внимание уделялось методам перера-
ботки фенольной смолы, основанным на выделении п-кумилфе-
нола и ацетофенона. В некоторых из них смола предварительно
обогащается п-кумилфенолом при помощи каталитического
взаимодействия димеров а-метилстирола с фенолом с повыше-
нием содержания n-кумилфенола до 60%.
Основными направлениями использования п-кумилфенола
являются: синтез смол; добавки к полимерам; биологически ак-
тивные вещества (в чистом виде и в виде производных).
Кумилфенол, как и фенол, имеет способность к образованию
смол типа новолачных. Смолы, полученные исключительно из
165
л-кумилфенола, находят применение главным образом при со-
здании клеев и покрытий.
Разработан способ получения (Пат. Румынии 55538, 1975)
фенолформальдегидной смолы непосредственно из кумилфеноль-
ной фракции, выделенной из смолы дистилляцией под вакуумом
в интервале температур 50—220 °C.
Известен способ получения новолачной смолы из смеси фе-
нола и л-кумилфенола (массовое соотношение 4,5:1) и форма-
лина в присутствии щавелевой кислоты (Пат. ФРГ 1801327,
1971). Образовавшаяся смола после смешения с наполнителя-
ми и добавления уротропина может использоваться как пресс-
материал. Продукт поликонденсации смеси фенола и л-кумил-
фенола (4:1) с формалином 55%-ной концентрации в присут-
ствии аммиака в растворе этанола был применен в качестве
грунтового слоя для стали под лаки, устойчивого на растрески-
вание и удар (Пат. США 3351605, 1978). Смесь кумилформаль-
дегидной резольной смолы с другими резолами — термореактив-
ная фенольная смола — затвердевает без добавления катализа-
тора (Пат. Японии 7317391, 1979). Композиция эпоксидной смо-
лы с кумилфенолформальдегидной новолачной смолой и добав-
кой уротропина (Пат. Японии 7444958, 1979) обладает высокой
механической прочностью.
Известен метод получения фенолодревесных смол (Пат. ПНР
99901, 1978). Метод основан на непосредственной конденсации
фенольной смолы с древесиной.
Процесс идет в две стадии. На первой стадии гидролизная
древесина обрабатывается раствором серной кислоты при 120—
180 °C и 0,3—1,0 МПа, а на второй стадии происходит взаимо-
действие древесины с фенольной смолой с добавкой аммиака
или уротропина с образованием смолы.
Кумилфенолформальдегидная смола может использоваться
в смеси с другими смолами (соотношения различные), напри-
мер, с эпоксидными (Пат. Японии 7308272, 1979). Эти смеси
затвердевают уже при температуре окружающей среды, приме-
няются как пресс- и изолирующие материалы.
В процессе конденсации можно использовать непосредствен-
но фенольную смолу с большим содержанием л-кумилфенола.
Смола, полученная поликонденсацией в присутствии основных
или кислых катализаторов, может быть использована как компо-
нент покрытия, заменитель алкидных смол, модифицированных
растительными маслами [481]. Единственное нежелательное
свойство этой смолы—термореактивность, которая, по всей ве-
роятности, обусловливается дифункциональностью л-кумилфе-
нола.
В результате конденсации фенольной смолы с формалином
образуются растворимые в маслах смолы, использующиеся в
производствах лаков и красок [482].
166
Поликонденсаты на основе смесей фенола с л-кумилфенолом
могут применяться как пресс-материалы с хорошими термореак-
тивными свойствами.
Кумилформальдегидные смолы, полученные в присутствии
основных катализаторов, могут использоваться как модифици-
рующие добавки к неопреновым клеям (Пат. Франции 1548955,
1984). Содержание смолы в клее может достигать 40% в расче-
те на неопрен.
Смолы с хорошими электроизоляционными свойствами (Пат.
Франции 1557726, 1982) образуются при поликонденсации СН2О
с фенолом и чистым л-кумилфенолом или выделяются из фрак-
ции, полученной в результате дистилляции фенольной смолы с
высоким содержанием л-кумилфенола. Такие смолы хорошо
растворяются в спиртовом растворе после отвердевания смолы
имеют очень хорошие изоляционные свойства и прочны на раз-
рыв. Материалы на основе этих смол используют для импрегни-
рования бумаги и других материалов.
Имеются сведения о возможности использования производ-
ных кумилфенола в качестве добавок к синтетическим смолам,
стабилизаторов клейкозы, регуляторов молекулярной массы в
процессе получения термодинамических поликарбонатов из ок-
сифенола и СОС12 (Пат. Японии 7534393, 1980). Такие карбо-
наты обладают лучшей термостабильностью, чем используемый
регулятор — л-третзамещенный бутилфенол.
п-Кумилфенол и его производные используются как ускори-
тели отверждения эпоксидных смол, селективные разбавители
эпоксидных, фенольных и фурановых смол, пластификаторы
полиуретанов (Пат. Японии 7620993, 1981). Для этих целей
можно также применять непосредственно кумилфенольную
фракцию (Пат. ФРГ 2440959, 1973).
Синтез 2,6-ди (третбутил) -4-кумилфенола алкилированием
л-кумилфенола третбутилом осуществлен в присутствии кислот-
ного катализатора. Это соединение используется в различных
синтезах, а также в качестве инсектицида. Он применяется как
эффективный антиокислитель и стабилизатор каучуков, поли-
меров и вулканизаторов (Пат. ЧССР 150822, 1971). Из л-кумил-
фенола получены соединения — основания Манниха, испытан-
ные в виде катализаторов образования синтетических смол из
изоцианатов (Пат. США 3680868, 1979).
Фосфорные производные л-кумилфенола применяются как
стабилизаторы полипропилена (Пат. Японии 7401935, 1979),
термостабилизаторы синтетических каучуков, полученных на ос-
нове бутадиена и стирола (Пат. Японии 7420928, 1979).
Из кумилфенола можно синтезировать различные фосфор-
и серосодержащие органические соединения, использующиеся в
качестве стабилизирующих и многофункциональных присадок к
маслам. В первую очередь к ним относятся полные фосфори-
167
стые эфиры алкилфенолов и их сульфидов. Синтезируют эти
соединения в растворе воздействием треххлористого фосфора на
различные алкилфенолы при 75—78 °C:
ONa ONa
R R
Полные фосфористые эфиры сульфидалкилфенольных соедине-
ний, содержащих серу между боковыми алкильными цепями,
образуются при обработке последних треххлористым фосфором:
РОС13
---->
НС1
R'
168
Фосфорсодержащие соединения можно получить также пу-
тем обработки алкилсульфидфенильных соединений сульфидом
фосфора:
Конденсация тиоэфира с хлорангидридом алкилфенилфос-
фористой кислоты протекает по реакции:
Конденсация сульфидалкилфенола с хлорангидридом проте-
кает следующим образом:
ОН ОН
R R
169
R R
На базе кумилфенола также возможен синтез фосфор- и се-
росодержащих органических соединений:
СН3
Синтез полного кумилфенольного эфира фосфористой кисло-
ты осуществляют взаимодействием сульфидкумилфенола с хлор-
ангидридом кумилфенилфосфористой кислоты. Необходимо от-
метить, что хлорангидрид кумилфенилфосфористой кислоты об-
разуется непосредственно перед реакцией конденсации и полу-
чение его является первой стадией при синтезе полного эфира
фосфористой кислоты:
СНз
170
н с—-с—сн3 н3с-с-сн3
Синтезированные на базе кумилфенола эфиры представляют
собой твердые смолоподобные продукты светло-коричневого цве-
та, хорошо растворимые в минеральных смазочных мас-
лах [483].
Эфиры испытывали на ЧШМ в смеси с минеральными и син-
тетическими маслами [484]. Результат испытаний показал, что
добавка синтезированных соединений к указанным маслам уве-
личивает прочность их масляной пленки. Добавка к синтетиче-
скому маслу этих эфиров в количестве 3% (масс.) увеличивает
предельную нагрузку почти вдвое.
Содержание хлора в составе органического соединения спо-
собствует увеличению смазывающих свойств масел при высоких
нагрузках, поэтому синтезировали органические соединения,
в состав которых, кроме серы и фосфора, входит и хлор. Более
прочное положение атома хлора непосредственно в ароматиче-
ском ядре. Синтезированные присадки, содержащие хлор в аро-
матическом ядре, удовлетворяют жестким эксплуатационным
требованиям к работе двигателей, для которых они предназна-
чаются.
Фосфор- и серосодержащие соединения резко снижают кор-
розию металлических пластинок при испытании на аппарате
Пинкевича. Они обладают сильными антиокислительными свой-
ствами. При добавлении незначительного количества некоторых
из указанных соединений резко увеличивается стабильность ма-
171
сел, уменьшается количество осадка и кислотное число после
окисления. Имеется возможность использования и-кумилфено-
ла и его производных в качестве биологически активных ве-
ществ. и-Кумилфенол и его натриевая соль оказывают сильное
биологическое действие на грибы, уничтожающие древесину и
подобные материалы [485]. Благодаря своей активности препа-
рат не только обеспечивает защиту древесины, но и повышает ее
стойкость к заболеваниям. Для этого были использованы лаура-
ты кумилфенола и 2-нитро- и 2-хлорпроизводные. Осуществлен
синтез и других производных, обладающих потенциальной био-
логической активностью:
где R = H, СН3; Ri = H, СН3; R2=OCH3, ОС2Н5, ОН, С6Н5, NHNH2; Х=Н, С1,
алкил.
Водная эмульсия или аэрозоли кумилфенола могут использо-
ваться как фунгицид для тканей, кожи и дерева (Пат. Англии
985279, 1969) биологически активных препаратов [57]. С этой
же целью могут быть применены и производные кумилфенола,
например натриевые соли 4(а,а-диметилбензол)-3,5-диметилфе-
нола и 2-хлор-4-(а,а-диметилбензил) фенола.
п-Кумилфенол в количестве 0,2% (масс.), добавленный к
трансформаторному маслу, предотвращает окисление масла и
приостанавливает образование твердых продуктов окисления и
кислых соединений [486].
n-Кумилфенол может использоваться в производстве лаков
и красок, а также как носитель в процессе окраски (можно с
добавлением эмульгатора) полиэфирных волокон дисперсион-
ными материалами (Пат. Бельгии 7302215, 1973). Добавление
0,2—0,3% (масс.) п-кумилфенола к типографской краске из
гидрофобного синтетического материала увеличивает адгезион-
ную способность краски (Пат. СССР 525730, 1980).
С помощью и-кумилфенола, растворенного в керосине с до-
бавлением 5—10% амилового спирта можно проводить экстрак-
цию цезия, рубидия, калия и лития из щелочных растворов.
Степень экстракции зависит от концентрации п-кумилфенола,
щелочи и экстрагируемого металла [487, 488]. Экстрагирующая
способность щелочных металлов из водных растворов их солей
росла в ряду Na<K<Li<Pb<Cs [487]. Для этого процесса
использовались растворы кумилфенола или кумола. Такой спо-
соб экстракции удобен для выделения щелочных металлов из
растворов, образующихся при переработке слюды.
172
На основе n-кумилфенола разработаны синтезы ряда органи-
ческих соединений, например, его четвертичной аммонийной
соли [489], этил- и фенилфосфатов кумилфенола [490] и др.
В настоящее время постоянно ощущается недостаток орга-
нических полупродуктов для химической промышленности. Было
бы неэкономично пренебречь таким ценным продуктом как фе-
нольная смола, и, в частности, находящимся в ней и-кумил-
фенолом.
7.3. Побочные продукты производства 2,4-дихлорфенола
2,4-Дихлорфенол является исходным продуктом для синтеза гер-
бицидных препаратов на основе 2,4-дихлорфеноксиуксусной кис-
лоты [491]. При хлорировании фенола [492, 493] образуется
сложная смесь [% (масс.)]: 2,4-дихлорфенол — 88—92; 2,6-ди-
хлор- и 2,4/5-трихлорфенолы — 5—8 и о- и и-хлорфенол— 0,02—
0,8. о-Карбоксиметилирование этой смеси в щелочной среде мо-
нохлорацетатом натрия приводит к образованию комплекса
хлорзамещенных феноксиуксусных кислот, часть которых обла-
дает свойствами гербицидов. Разделение побочных продуктов
производства 2,4-дихлорфенола позволит получить гербициды
высокого качества, а также ценное сырье для органического
синтеза.
Выделение индивидуальных хлорфенолов из их смеси явля-
ется трудоемким процессом. Существует несколько вариантов
его осуществления.
2,6-Дихлорфенол со степенью чистоты >98% можно выде-
лить (выход 75—85%) из смеси с изомерным 2,4-дихлорфено-
лом. Для этого смесь нагревают до 60 °C, образуется гомоген-
ная жидкость, которую смешивают с водой. При охлаждении
этой смеси до 40 °C происходит кристаллизация 2,6-дихлорфено-
ла и его легко выделяют из водной смеси (Пат. США 3336400,
1960).
Возможно выделение индивидуальных хлорфенолов на хро-
матографической колонке. Приведены времена удерживания
[494] для различных изомеров хлорфенолов, полученных таким
образом на колонке, заполненной сефадексом; в качестве рас-
творителя элюирования использовался хлороформ.
Анализ кинетики хлорирования фенола и свойств отдельных
хлорфенолов позволил разработать [491] технологию получения
индивидуальных хлорфенолов из их смеси. Эта методика осно-
вана на различии степеней индивидуальных веществ. В реакци-
онной массе присутствует в основном только одно из трудноот-
делимых веществ. Так, при степени хлорирования, соответст-
вующей плотности реакционной массы р440= 1,2 г/см3, содержа-
ние n-хлорфенола в продуктах реакции составляет 50% (масс.),
в то время как количество трудноотделимой от него примеси
2,4-дихлорфенола не превышает 1 —1,5% (масс.). Степень чис-
173
тоты получаемого по предложенной технологии и-хлорфенола
достигает 99%. По этому же методу удается выделить
99,5%-ный о-хлорфенол, 99%-ные 2,6- и 2,4-дихлорфенолы.
Предложен способ [492] селективного выделения побочных
продуктов производств 2,4- и 2,6-дихлорфенолов и 2,4,6-три-
хлорфенола в виде соответствующих хлорфенолятов натрия из
обезвоженных растворов хлорфенолов в ароматических углево-
дородах. Оптимальные условия процесса — массовое соотношение
толуол: смесь хлорфенолов, равное 3:1, продолжительность
осаждения хлорфенолятов натрия 10—20 мин, температура
осаждения 20—30 °C.
Побочный продукт хлорирования фенола — 2,6-дихлорфе-
нол— трудно отделить ректификацией от целевого продукта
из-за близости температур кипения. Отделение облегчается при
получении 2,4-дихлорфенола хлорированием фенола в раствори-
теле (П ат. США 275998, 1967).
При выделении о-, м- или и-фенола из смеси хлорированных
фенолов используют разницу констант диссоциации хлориро-
ванных фенолов и свойства анионита (Пат. Японии 9379,1958).
В качестве анионита применяют амберлит-400 или амбер-
лит-410, дауэкс-1 или дауэкс-2. Недостатком метода является
использование разбавленных растворов. Кроме того, применяе-
мый в качестве растворителя метанол токсичен, пожаро- и взры-
воопасен.
Метод кислотно-щелочной диссоциативной экстракции по-
зволяет достаточно легко получать индивидуальные 2,4- и 2,6-
дихлорфенолы практически любой степени чистоты (А. с. СССР
250159, 1969). В качестве экстрагента предлагается использо-
вать растворы гидроксида натрия, при этом исходную смесь
можно подавать на экстракцию в виде раствора.
Значения селективности [495] разделения смеси 2,4- и 2,6-
дихлорфенолов (р) определялись по формуле:
₽ = 2.6-ДХф/^2,4-ДХф)э/(^2,6-ДХф/^2,4-ДХф)р»
где С2,к- ДХФ и С2.в-ДХФ- концентрации соответственно 2,4- и 2,6-дихлорфе-
нолов (или изомерный состав растворенных фенолов); индексы «э» и «р»
указывают, что отношения берутся для фаз экстракта и рафината.
Использование в качестве экстрагентов различных органиче-
ских растворителей влияло на величину селективности, но во
всех случаях значение ее оставалось достаточно высоким:
Растворитель
Хинолин 3,6
Трис (хлорэтил) фосфат 7,0
Перхлорэтилен 8,8
Тетрахлористый углерод 8,9
Ацетофенон 9,4
Бензол 9,8
174
Побочные хлорфенолы можно извлекать экстракцией октило-
вым спиртом из водных растворов [496]. Показано, что на ко-
личественные характеристики экстракционного процесса влияет
число атомов хлора в молекуле органического соединения. На-
пример, коэффициент распределения трихлорфенола при экст-
ракции октиловым спиртом более чем в 150 раз превышает ана-
логичную величину для фенола, что делает возможным разде-
ление технической смеси 2,4-дихлорфенола на компоненты и
дальнейшее их использование в органическом синтезе.
Целесообразно (Пат. ФРГ 1109701, 1976) включить в техно-
логическую схему получения 2,4-дихлорфенола стадию восста-
новления водородом 2,6-дихлорфенола и 2,4,6-трихлорфенола до
фенолов и использовать регенерированный продукт в качестве
сырья:
2СвН3С12ОН + Н2 =4 2CeH4OH + 2НС1
Побочные продукты производства 2,4-дихлорфенола являют-
ся полупродуктами в синтезе биологических активных веществ.
2,4,6-Трихлорфенолы можно использовать для синтеза о-метил-
о- (2,4,6-трихлорфенил) -тиофосфорной кислоты, которая приме-
няется для уничтожения бактерий, грибов, насекомых, клещей,
тли, мух, и т. д. ( Пат. США 2887506, 1958).
2,6-Дихлор-3-метокси-4-нитрофенол и его соли щелочных
металлов в тепличных условиях (доза 22,4 кг/га) проявили вы-
сокую активность в качестве послевсходовых гербицидов (Пат.
США 3657358, 1970). Алкиловые эфиры 2,6-дихлор-4-нитрофено-
ла обладают гербицидными, фунгицидными и нематоцидными
свойствами (Пат. США 3554181, 1966). Новый гербицид (Пат.
США 3554181, 1966), содержащий 2,4,6-трихлорфенил-4-нитро-
фениловый эфир, наиболее эффективен против сорняков риса в
момент прорастания; малотоксичен для человека, теплокровных
рыб и ракушек; сохраняет токсичность для сорняков в почве
25—30 дней. 4,4-Дихлор-2-оксидифениловый эфир, внесенный в
почву, предотвращает рост сорняков и обладает избирательным
действием (Пат. Японии 16177, 1966). Было установлено так-
же, что высокой гербицидной активностью обладают хлорфен-
оксипиридины [497].
Гербициды (Пат. Японии 9000, 1968), содержащие в качест-
ве активных компонентов 2,4,6-трихлор-4-нитро, 2,4-дихлор-6-
алкил-4-нитро-, 2-алкил-4-хлор-4-нитро- или 2,4-динитро-4-хлор-
дифениловые эфиры с удачным подбором заместителей в аро-
матическом ядре проявляют синергизм, обладают невысокой
токсичностью к полезным растениям и характеризуются широ-
ким спектром гербицидной активности. 2,4,6-Трихлор-4-нитроди-
фениловый эфир и 2-метил-4,6-дихлор-4-нитродифениловый эфир
применяются для борьбы с сорняками на рисовых полях; не
175
токсичны для рыб (Пат. Англии 1016648, 1968). Соединения об-
щей формулы 2,4,6-С1зС6Н2ОСбН3МО2С1з активны против сорня-
ков (Пат. Японии 48—26208, 1972). Ряд гербицидных препара-
тов, действующими веществами в которых являются 2,2-динит-
ро-4-хлордифениловый или 4,4-динитро-2-хлордифениловый эфи-
ры, могут быть получены на основе о- и n-хлорфенолов. Такие
гербициды активны против двудольных и однодольных сорня-
ков, находящихся в стадиях выходов и прорастания семян (Пат.
Японии 19840, 1966). Разработан (Пат. Японии 6355, 1966) спо-
соб получения гербицидов на основе 2,4,6-трихлорфенола общей
формулы 2,4,6-Cl3C6H2OC6H4NO2-4, проявивший активность в
борьбе с сорняками на полях риса и проса. 2,6-Дихлор-п-нитро-
фенол используют в качестве фунгицида [498].
n-Хлорфенол выступает в виде реагента в органическом син-
тезе. Установлено [499], что алкилирование n-хлорфенола диизо-
бутиленом в присутствии H2SO4 приводит к получению в качест-
ве основного продукта 2-трет-бутил-4-хлорфенола; одновременно
в ходе реакции в зависимости от условий проведения процесса
образуются в различных соотношениях 2-трет-октил-4-хлорфе-
нол, а также тримеры и тетрамеры изобутилена. Подобраны оп-
тимальные условия ведения процесса, позволяющие практически
количественно превращать диизобутилен в 2-трет-бутил-4-хлор-
фенол [500].
В процессе окисления (Пат. Японии 55-30781, 1979) дигало-
генпроизводных одноатомных фенолов Н2О2 в среде кетонов в
присутствии 0,0001% НС1О4 или производных гетерополикислот
образуются многоатомные фенолы:
Многоатомные фенолы используются в синтезе красителей,
антиоксидантов и лекарственных препаратов.
Двухосновные фенолы из о- и п-хлорфенолов можно получить
щелочным гидролизом в присутствии катализатора при темпе-
ратуре 300 °C (А. с. ЧССР 182598, 1979). Выход двухосновных
фенолов по этому методу составляет 20—35%.
Установлено, что о-хлорфенол и его производные, содержа-
щие различные алкильные группы в п-положении к гидроксиль-
ной группе, являются эффективными ингибиторами термоокис-
лительной деструкции полипропилена [501]. Стабилизирующая
активность их возрастает с увеличением углеводородного ра-
дикала. Наибольший эффект получен в случае 4-фторгептил-2-
176
хлорфенола. В случае ненасыщенной полиэфирной смолы [501]
стабилизирующее действие о-хлорфенола и его производных не-
значительно.
7.4. Побочные продукты производства ионола
Технология производства 2,6-ди-трет-бутил-4-метилфенола (ио-
нола) включает несколько стадий [502]. Первой из них является
алкилирование фенола изобутиленом:
изо-С4 Hr
С(СН3)3
С(СНз)з
На этой стадии (Пат. США 4113976, 1976) наряду с 2,6-ди-
трет-бутилфенолом в качестве побочного продукта образуется
2,4,6-три-трет-бутилфенол (выход «10%), который в настоящее
время не утилизируется.
В литературе имеются сведения об использовании 2,4,6-три-
трет-бутилфенола в качестве реагента в органическом синтезе.
Примерами могут служить каталитическое окисление его до
2,6-ди-трет-бутилбензохинона с последующим получением ста-
билизатора 2,6-ди-трет-бутилгидрохинона [503]
Cu/Pd; о2
С(СНз)з
С(СНз)з
Н2; Pd
и деалкилирование с получением смеси ди- и моно-трет-бутил-
фенолов — сырья для промышленных синтезов [504]:
Вторая стадия процесса — аминометилирование 2,6-ди-трет-
бутилфенола с образованием основания Манниха — а,а-диметил-
(3,5-ди-трет-бутил-4-оксибензил) амина:
12—527
177
С(СН3)з
С(СН3)з
(CH3)2NCH2N(CH3)2
HN(CH3)2
С(СН3)з
С(СН3)3
Побочными продуктами на этой стадии являются 3,5-ди-
трет-бутилдиметиламинметиленхинон, ЗДЗ'Д-тетра-трет-бутил-
диоксифенил метан, ЗДЗ'Д-тетра-трет-бутилоксифенилмстилсн-
хинон (выход 3—5%), образующиеся в результате термолиза
основания Манниха [505]:
(СН3)3С
(СН3)3С
(СН3)3С С(СН3)3
(СНз)3С С(СН3)3
(СН3)3С С(СН3)з
(СН3)3С С(СН3)3
Эти соединения в настоящее время не используются.
На третьей стадии протекает гидрогенолиз основания Маи-
пиха с получением ионола:
С(СН3)з
Побочные соединения, образующие на этой стадии, — продук-
ты глубокого гидрирования ионола [506, 507], а также продук-
ты термоуплотнения ионола: 2,6-ди-трет-бутил-4-метилциклогек-
санон, ЗДЗ'Д-тстра-трет-бутилстильбенхинон, ЗДЗ'Д'-тетра-
тр^т-бутилдифенилхинон, ЗДЗ'Д-тетра-трет-бутилдиоксидифе-
нилэтан (димер ионола) [506—508]:
178
С(СН3)3
С(СН3)3
С/С14 i С/СЯ.к
С(СН3)3 С(СН3)3
С(СН3)3
С(СН3)3
В промышленном процессе в зависимости от условий выход
побочных продуктов гидрогенолиза колеблется от 2 до
10% (масс.) [508].
Четвертая стадия — выделение ионола ректификаций. При
этом образуются побочные продукты окислительной конденса-
ции— димер ионола, стильбенхинон. Примерный состав [508]
смеси [% (масс.)]: хинон — 5—15, бисфенолы — 65—90, ионол —
3—15. Эту смесь предлагается применять в качестве антиокис-
лительной присадки к топливам (А. с. СССР 578326, 1977).
7.5. Побочные продукты производства дифенилолпропана
Дифенилолпропан получают конденсацией фенола с ацетоном
[509]:
О СН3
2 НО—4- СН3ССН3 ------> НО—С—ОН
\=/ - н2о \=/ | \=/
СН3
Наряду с основным процессом протекают побочные реакции,
приводящие к образованию целого ряда продуктов: о-/г-изомер
12* 179
(22—40%) и о-о-изомер дифенилолпропана (2%), и-изопропе-
нилфенол (2%), п-изопропилфенол (1%). Выход побочных со-
единений достигает до 100 кг на 1 т дифенилолпропана [509].
Многие из побочных продуктов реагируют друг с другом или
с одним из реагентов, давая дополнительные количества самых
различных соединений. Эти реакции хорошо изучены [509].
В связи с тем, что в ряде производств к дифенилолпропану
как к сырью предъявляются весьма высокие требования, возни-
кает задача очистки дифенилолпропана и получения высокока-
чественного продукта. Существует [509] много способов очист-
ки: основанные на свойстве дифенилолпропана образовывать
кристаллические аддукты с различными веществами; путем ще-
лочно-кислотного переосаждения дифенилолпропана-сырца;
экстракцией примесей органическими растворителями; перекри-
сталлизацией из растворителей и очистка дистилляцией выде-
лившихся веществ.
Пути применения побочных продуктов при очистке дифени-
лолпропана делят [509] на три группы: 1) использование в раз-
личных синтезах без какой-либо предварительной обработки;
2) превращение их в дифенилолпропан; 3) разложение на более
легкокипящие компоненты (фенол, n-изопропилфенол и др.).
Последние, например фенол, можно вновь использовать в син-
тезе дифенилолпропана.
Без предварительной обработки побочные продукты можно
использовать для синтеза термореактивных фенолформальдегид-
ных смол, которые применяются для склеивания дерева и мо-
дификации фенольных клеев (Пат. ПНР 57393, 1968).
Превращение побочных продуктов в дифенилолпропан про-
водят в присутствии тех же кислотных катализаторов (НС1,
H2SO4, ионообменной смолы) в условиях, аналогичных произ-
водству целевого продукта. Имеются [509] способы, при кото-
рых специальной обработкой реакционной массы превращают в
дифенилолпропан только один из побочных продуктов — и-изо-
мер дифенилолпропана. Предприняты [104] попытки выделения
индивидуальных компонентов из смеси побочных продуктов с
целью их раздельного использования.
Предложенные [509] способы разложения продуктов (гид-
рогенизация, крекинг) основаны на том, что в молекулах побоч-
ных продуктов связь оксифенильных радикалов с четвертичным
углеродным атомом алифатической цепочки непрочная и разру-
шается при высокой температуре и под действием кислот, щело-
чей и других агентов.
ГЛАВА 8
Применение отходов, промежуточных
и побочных продуктов производства
изопрена
Синтез 4,4-диметил-1,3-диоксана — промежуточного продукта в
производстве изопрена — осуществляют конденсацией изобути-
лена с формальдегидом в присутствии кислотных катализаторов
[510—512]. Этот процесс осуществлен в промышленности [513].
Селективность образования 4,4-диметил-1,3-диоксана составляет
76—80% пб формальдегиду и 65—70%—по изобутилену. В по-
бочных продуктах присутствуют изомерные 1,3-диоксаны, яв-
ляющиеся продуктами конденсации высших олефинов, которые
содержатся в исходной изобутиленовой фракции. В небольших
количествах образуются ненасыщенные спирты. Основными по-
бочными продуктами в процессе получения 4,4-диметил-1,3-диок-
сана являются триметилкарбинол и высококипящие вещества.
Триметилкарбинол может служить исходным сырьем в синтезе
4,4-диметил-1,3-диоксана [514]. Возврат его в реакционный блок
повышает селективность процесса по изобутилену до 85% [513].
Состав высококипящих побочных продуктов производства
4,4-диметил-1,3-диоксана сложен и насчитывает более 50 компо-
нентов. В табл. 8.1 приведены состав и содержание основных
продуктов этой фракции [513]. Выход их составляет более
200 кг на 1 т 4,4-диметил-1,3-диоксана.
Термокаталитическое расщепление 4,4-диметил-1,3-диоксана
осуществляют при 300—390 на кислотных катализаторах
[515, 516].
Селективность промышленного процесса по изопрену состав-
ляет 80—85%, при этом до 10% 4,4-диметил-1,3-диоксана пре-
вращается в исходный изобутилен. Выход кокса, как правило,
не превышает 1—2% на превращенный 4,4-диметил-1,3-диоксан.
Состав побочных продуктов [517, 518] стадии расщепления 4,4-
диметил-1,3-диоксана при 370—390 °C представлен в табл. 8.2.
8.1.Отходы производства изопрена
Побочные продукты производства изопрена находят разнообраз-
ное применение. В частности, их можно вернуть в цикл, где они
будут служить дополнительным сырьем в производстве изопре-
на (Пат. Японии 7508015, 1972).
Разработана технология выделения вакуумным фракциони-
рованием 3-метилбутандиола-1,3 [<95% (масс.)] и диоксановых
181
спиртов [75—85% (масс.)] из побочных продуктов производства
изопрена. Каталитическим гидрогенолизом получены первичные
С5—С6-спирты и гликоли (Пат. Японии 9411805, 1974). Разрабо-
тана методика выделения 4-метил-4-гидроксиметил-1,3-диоксана
(Пат. Японии 7245343, 1973) и пиридиновых оснований [103] из
побочных продуктов производства 4,4-диметил-1,3-диоксана.
3-Метилбутандиол-1,3, ненасыщенные спирты и гликоли пред-
ставляют интерес как реагенты в органическом синтезе. Фрак-
ция диоксановых спиртов может быть использована в качестве
эффективного растворителя лаков нитроцеллюлозы смол [513].
Гидролизом и алкоголизом диоксановых производных высоко-
кипящих побочных продуктов получены полигидроксилирован-
ные продукты, которые можно применять в качестве раствори-
телей, пластификаторов, тормозных жидкостей и антифризов
(Пат. Японии 8545165, 1986). Добавление этих продуктов к нит-
рильному каучуку улучшает его механические свойства и моро-
зостойкость (Пат. Японии 7395434, 1974).
Широко используются высококипящие побочные продукты в
виде эффективного флотореагента обогащения руд цветных ме-
таллов [519] и более полного извлечения угля (А.с. СССР
914089, 1982), а также ингибитора коррозии (А.с. СССР 218909,
1968). Введение этих продуктов в промывочную жидкость повы-
шает ее смазочные, противоизносные и антикоррозионные свой-
ства (А.с. СССР 666194, 1979). Состав для предотвращения
гипсово-углеводородных отложений включает высококипящие
отходы производства 4,4-диметил-1,3-диоксана (А.с. СССР
ТАБЛИЦА 8.1
Продукт Содержание, % (масс.)
в масляном слое в водном слое
4-Мстил-4-випил-1,3-диоксан 1,1—1,5
4-Метил-4-оксиметил-1,3-диоксан 14,0 34,0
4,4-Диметил-5-оксиметил-1,3-диоксан 4,0—6,0 6,0—8,0
5- (2-Окси-2-пропил) -1,3-диоксан 5,7—9,0 6,0
1,3,9-Триоксаспиро-5,5'-ундекан 2,2—3,2 —
5- (1 -Пропен-2-ил) -1,3-диоксан <1,2 —
З-Метил-1,3-бутандиол 1,6 24,0—25,0
1-Метокси-З-метилбутанол-З 1 2-Метил-2-метоксибутанол-4 J <2,0 —
1 -трет-Бутокси-З-метилбутанол-З <9,0 0,1
4-Метил-4-(2-метоксиэтил) -1,3-диоксан <0,4 —
4,4-Диметил-5-метоксиметил-1,3-диоксан 0,5—2,0 .—
4-Метил-4- (2-трет-бутоксиэтил) -1,3-диоксан <9,3 <0,4
4,4-Диметил-5-трет-бутоксиметил-1,3-диоксан <2,0 <0,3
10-Метил-1,3,7-триоксапергидронафталин (цис) <0,7 —
4-Метпл-4-окситетрагидропиран <7,0 <3,0
4-Метил-4-окси-3-оксиметилтетрагидропиран 1,0 3,2—3,6
182
ТАБЛИЦА 8.2
Продукт Состав, % (мол.)
4,4-диметил- 1,3-диоксан 4,5-диметил- 1,3-диоксан 4-этил- 1,3-диоксан 4-винил- 1,3-диоксан
Углеводороды Q—С3 0,1 — — —
Изобутилен 10,0 — —— —
«-Бутилены — 3,0 1,5 —
Дивинил — — — 7,0
Амилены 0,2 4,0 6,0 —
Изопрен 82,5 45,0 — —
Пиперилен 0,5 — 71,0 20,0
Циклопентадиен — — — 3,0
4-Метилдигидропиран 1,6 — — —
Изоамиленовые спирты 1,6 8,0 11,0 —
Изовалериановый альде- — 7,0 1,5 —
гид
Олигомеры изопрена 0,5 — — —
(зеленое масло)
Кокс 1,0 1,0 2,0 28
Прочие — 27,0 7,0 42
514011, 1976). На основе последних разработан способ получе-
ния реагента для буровых растворов (А. с. СССР 1002334,1983).
На основе этих побочных продуктов получены полимерные мате-
риалы (Пат. Франции 149124, 1968), применяемые как добавки
к краскам, клеям, типографским краскам (Пат. Японии
7483792, 1975), присадки к каучукам (Пат. Японии 7483794,
1975), пластификаторы поливинилхлоридных смол (Пат. Фран-
ции 1413070, 1966) и отвердители смол. Из высококипящих по-
бочных продуктов синтезированы ненасыщенные сложные эфи-
ры, которые могут быть использованы как основа для лаков и
красок, образуются пленки, затвердевающие на воздухе (Пат.
Бельгии 611014, 1963).
8.2. Применение 4,4-диметил-1,3-диоксана
4,4-Диметил-1,3-диоксан является крупнотоннажным промежу-
точным продуктом производства изопрена. В последние годы
проведены обширные исследования, посвященные превращени-
ям 4,4-диметил-1,3-диоксана под действием различных реаген-
тов [520].
Гидролиз 4,4-диметил-1,3-диоксана [521—525] в присутствии
кислотных катализаторов приводит к образованию 3-метил-бу-
тандиола-1,3 (выход <50%). Изучена кинетика этого процесса
в среде вода — спирт [0—40% (масс.)]. С увеличением концен-
трации спирта в бинарном растворителе равновесие реакции
183
гидролиза смещается в сторону 4,4-диметил-1,3-диоксана. При
использовании различных спиртов обнаружилось, что константа
скорости гидролиза изменяется [526] в ряду:
изо-С4Н9ОН > «зо-С3Н7ОН > С2Н5ОН > СН3ОН
Предложен механизм гидролиза, согласно которому основ-
ными показателями, определяющими скорость процесса, явля-
ются: мономолекулярный распад протонированных молекул
1,3-диоксана; распад промежуточных частиц, состоящих из про-
тонированных молекул 1,3-диоксана и аниона кислоты [527]:
медленно
СН3 СН3
Си ГМ
АН" + НбО2+ + СН2(ОН)2 +
—+ОНАН
В продуктах взаимодействия 4,4-диметил-1,3-диоксана с ме-
тиловым спиртом обнаружены З-метилбутандиол-1; 3-метил-
З-метоксибутанол-1 и продукты дегидратации диола: З-метил-З-
бутен-1-ол; 3-метил-2-бутен-1-ол и изопрен [528].
Первичным продуктом взаимодействия 4,4-диметил-1,3-диок-
сана с метанолом является 3,3-диметил-4,6-диокса-1-гептанол
[528, 529]. Как показали опыты, использование в качестве ка-
тализатора H2SO4, катионита КУ-2 [528] или других катионооб-
менных смол существенно не влияет на результаты метанолиза
диметилдиоксана. Во всех случаях получены одни и те же про-
дукты с сопоставимой селективностью. В табл. 8.3 приведены
результаты метанолиза диметилдиоксана в присутствии H2SO4,
из которой следует, что существенное влияние на направление
процесса оказывают количество катализатора и продолжитель-
ность опыта (А. с. СССР 722893, 1980).
184
ТАБЛИЦА 8.3
Время реак- ции, ч Выход, % Конверсия 4,4-диметил- 1,3-диоксана, %
изопентолы спиртоэфир диол
175,0:238,4:2,1*
1,5 1,6 2,8 48,3 12,5
3,0 3,1 6,5 69,4 31,8
5,0 4,3 9,2 82,5 42,3
8,0 6,8 14,7 75,2 60,7
168,4:236,7 4,0*
1,0 7,5 10,3 35,2 32,6
2,0 9,0 12,1 47,2 53,7
4,0 11,2 14,9 60,8 68,8
10,0 17,1 28,2 41,2 90,4
175,8:240,0:8,3*
2,0 6,1 10,7 54,3 53,0
4,0 9,3 20,5 40,2 84,3
6,0 11,2 29,7 32,7 91,8
9,0 13,3 45,4 20,9 96,1
♦ Соотношения 4,4-диметил-1,3-диоксан : метиловый спирт: серная кислота.
Определены кинетические параметры метанолиза 4,4-диме-
тил-1,3-диоксана [529]. Особенно велико влияние на результат
реакции температуры проведения процесса. С более высококи-
пящими спиртами идет преимущественно дегидратация диола до
диолефина. Так, из 4,4-диметил-1,3-диоксана и бутилового спир-
та [530] при 108—ПО^С в течение 8,5 ч получены изопрен (вы-
ход 50%) и бутилаль (выход 88%):
СН3 Сн3
С4Н90Н I
I |^гн -------> СН2=С-СН=СН2+СН2(ОС4Н9)2
Ох/О
При взаимодействии 4,4-диметил-1,3-диоксана с этилмеркап-
таном выход 3-метилбутандиола-1,3 не превышает 35% Это
связано с его превращениями в бутенолы и в изопрен [531 —
534]. Последний взаимодействует с этилмеркаптаном с образо-
ванием 4-тио-2,3-диметилгексена- 1 [531]:
СН. СН.
I I
СН2=С—CH=CH2+C2H5SH ► сн2=с—сн—сн3
I
sc2H5
185
Кислотное расщепление 1,3-диоксанов в присутствии фенолов
в зависимости от условий может быть направлено как в сторо-
ну образования гликолей, так и продуктов их дегидратации.
При 80—100 °C в условиях равновесного фенолиза [535] об-
разуется гликоль (выход 13%):
н3с сн3 н3с сн3
Повышение температуры способствует образованию диено-
вых углеводородов [535] — З-метил-З-бутен-1-ол и 2,2-диметил-
хроман, наряду с фенолсодержащими смолами [536].
Высоких значений (<80%) выход 3-диметил-(бутандиола)-
-1,3 достигает в реакции с гликолями:
СН3
I
4- сн2=сн—с=сн2
Реакцию проводят при 50—80 °C в течение 4—8 ч в присутствии
кислотных катализаторов [H2SO4, НС1, Н3РО4, ВЕз-0 (С2Н5)2,
КУ-2] [537]. Выход изопрена в этих условиях не превышает
20%.
Ацетолиз 4,4-диметил-1,3-диоксана уксусным ангидридом при
нагревании до 70 °C приводит к образованию (выход 65—88%)
диацетата 3,3-диметил-2-окса-1,5-пентандиола [538, 539], кото-
рый в условиях реакции переэтерифицируется в диацетат 3-ме-
тил-1,3-бутандиола [538], а затем разлагается с образованием
ацетатов изопентенолов [540]. Катализаторами реакции явля-
ются КУ-2, FeCl3 либо H2SO4:
ОСН2ОСОСН<
/Ч^ОССНз н3с сн3 н3с сн3|| о сн3 > СН2=С—СН2СН2ОАс ~ сн3 1 ► СН,—С—СН2СН2ОАс 1 О Ас (СН3)зС(СН2)2(ОАс)2
186
При изменении условий ацетолиза [540] можно получить пре-
имущественно диацетат 3,3-диметил-2-окса-1,5-пентандиола (вы-
ход <70%) или же ацетаты изопентенолов. Причем расщепле-
ние 4,4-диметил-1,3-диоксана протекает региоселективно по
О1—С2-связи [539].
4,4-Диметил-1,3-диоксан с ацилхлоридами реагирует в при-
сутствии кислотных катализаторов с образованием у-хлорзаме-
щенных сложных эфиров (Пат. США 2607800, 1954)
Ацилирование 1,3-диоксанов хлорангидридами карбоновых
кислот в присутствии SbCl5 приводит к образованию гексахлор-
антимонатов [541]:
сн3 сн3
Н3С-/_ ci НзС-J-O
< + < й-R + [sbcie]
'—OCR '—О
II
о
R СН3 С2Н5 С3Н7 С6Н5 и-ВгС6Н4 СН=СНСвН5 С4Н3О
Выход, % 95 92 81 93 95 90 70
Донорные заместители в цикле в положении 4 способствуют
образованию солей с высокими выходами (70—95%) при —5 °C
в течение 15—30 мин.
Гидролиз солей диоксония приводит только к одному изомер-
ному моноэфиру 3-диметил-бутандиола-1,3 [541]:
СН3
н3с-4-о
СНо
4 ()—с—R
II
О
С борной кислотой 4,4-диметил-1,3-диоксап реагирует с об-
разованием альдегида и не менее шести боратов 3-метил-бутан-
диола-1,3. Конверсия 4,4-диметил-1,3-диоксана составляет 60%.
В результате реакции даже при неглубокой конверсии 1,3-диок-
сана образуется смесь борных эфиров различного строения. При
187
избытке борной кислоты в продувах реакции преобладает ус-
тойчивый к гидролизу эфир [542];
Реакцию проводят, нагревая ацеталь и твердую борную кислоту
(мольное соотношение 1,5 1; ПО—140 °C) и выводя из сферы
реакции альдегид и воду, могут быть добавлены азеотропообра-
зующие растворители — гексан, толуол.
Взаимодействие 4,4-диметил-1,3-диоксана с N-бромсукцин-
имидом [543] приводит к образованию 3-бромзамещенных фор-
миатов с выходом 29%:
В смеси преобладает изомер с первичной сложноэфирной
группой (соотношение изомеров 7 1). Бромированием 4,4-диме-
тил-1,3-диоксана диоксандибромидом при 0°С получен только
один изомер: З-бром-З-метилбутилформиат [543]:
В реакции 4,4-диметил-1,3-диоксана с диоксандииодидом при
0—80 °C получен 2-иод-4,4-диметил-1,3-диоксан (А. с. СССР
363697, 1973). При взаимодействии 4,4-диметил-1,3-диоксана с
окислительно-восстановительной системой [Fe (II) +Н2О2+
+ Fe(III)] в воде при 5—10 °C в инертной атмосфере (аргон)
параллельно образуются изомерные моноэфиры 3-метилбутан-
диола-1,3. В отсутствии пероксида водорода синтез моноэфиров
не происходит [544, 545]:
Fe2+; Н2О2; Fe3+
При соотношении изомеров, равном И 1 и преобладании
изомера с первичной формиатной группой суммарный выход
моноэфиров составляет 78%. Взаимодействием озона с 4,4-диме-
188
тил-1,3-диоксаном можно получить изомерные моноформиаты
3-метилбутандиола-1,3 [546]:
Первичным продуктом жидкофазного каталитического окис-
ления 4,4-диметил-1,3-диоксана является гидропероксид, кото-
рый далее превращается в моноэфир диола [547, 548].
Парофазное окисление 4,4-диметил-1,3-диоксана на вана-
дийсеребряном и смешанном ванадиймолибденфосфорном ката-
лизаторе при температурах 340—420 °C протекает с образовани-
ем цитраконового ангидрида с выходом 14,5% [549]:
При гидроксилировании 4,4-диметил-1,3-диоксана образуют-
ся З-метил-З-метокси-1- (триэтилсилокси) бутан, 2-метил-4- (три-
этилсилокси)-!-бутен, метокситриэтилсилан, гексаэтилдисилок-
сан [550]:
СН3
I
CH2=C(CH2)2OSi(C2H6)3
—CH3OH
Первоначально происходит раскрытие гетероцикла по О1—С2
связи. Дальнейшее взаимодействие протекает по двум направ-
лениям: присоединение фрагмента Si(C2H5)3 и гидрид-аниона
с образованием З-метил-З-метокси-1- (триэтилсилокси) бутана и
одновременное отщепление молекулы формальдегида с образо-
ванием 2-метил-4-(триэтилсилокси)-1-бутена и метокситриэтил-
силана. Во всех случаях наблюдается образование гексаэтилди-
силана, изопрена и продуктов его полимеризации.
Гидрирование 4,4-диметил-1,3-диоксана водородом в присут-
ствии катализаторов гидрирования (Cu/Сг- или Cu/Zn-катализа-
тор) при температуре 200—280 °C и повышенном давлении
(—100 МПа) приводит к образованию (выход 80—90%) изо-
амилового спирта (Пат. США 2721223, 1956). Восстановитель-
189
ное расщепление 4,4-диметил-1,3-диоксана наблюдается в при-
сутствии соединений меди. Выход изоамилового спирта состав-
ляет 55% [551]. /
При фотохимическом превращении 4,4-димстил-1,3-диоксана
образуются формиаты изоХерных амиловых спиртов, суммар-
ный выход которых составляет 15% [552]:
Н3С СН3
\/ О
/~ и\ |--► НСОО(СН2)2СН(СН3)2
\_о/ '—> НСООС(СН3)2(СН2)2
Радикальная изомеризация 4,4-диметил-1,3-диоксана под
действием перекиси трет-бутила при 130 °C протекает селектив-
но с образованием формиата изоамилового спирта [553, 554]:
Н3С СН3
/ \ ----> НСООСН2СН2СН(СН3)2
\—0/
Взаимодействие 4,4-диметил-1,3-диоксана с этилацетатом в
присутствии кислотных катализаторов приводит к сложной сме-
си продуктов [555]:
Н3С сн3
\zLoch2oc2h5
\—ОССН3
II
СН3
I
СН2=ССН.СН2ОСОСН3
сн3
/С^СНСН2ОСОСН3
CH3Z
Одновременно происходит превращение 4,4-диметил-1,3-диокса-
на в дигидропиран (выход 47%) и изопрен (выход 10%).
4,4-Диметил-1,3-диоксаи является исходным сырьем в синте-
зе З-метил-З-бутеп-1-ола и терпеновых его производных.
Обменное кислотно-катализируемое взаимодействие цикли-
ческого ацеталя с линейными протекает с образованием 2-R'-
4,4-диметил-1,3-диоксанов [556]:
R'CH(OR)2
CH2(OR)2
190
ТАБЛИЦА 8.4
Исходный реагент Время, ч Полученный 1,3-диоксан Выход, %
2,4-Диметил-1,3-диоксан* 4,0 2.4,4-Триметил-1,3-диоксан 90 2,4-Диметил-1,3-дноксолан** 4,0 То же 87 2-Метил-1,3-диоксан*** 3,5 » 95 2,4-Диметил-1,3-диоксан4* 3,5 » 97 2-Этил-4-метил-1,3-диоксан 4,0 2-Этил-4,4-димстил-1,3-дпоксан 93 2-Пропил-4-метил-1,3-диоксан 5,0 2-Пропил-4,4-Диметил-1,3-ди- 91 2-Изопропил-4-метил-1,3-дпок- 4,0 оксан сан 2-Изопропил-4,4-диметил- 97 2-Бутил-4-метил-1,3-диоксан 6,0 1,3-диоксап 2-Изобутил-4-метил-1,3-диоксан 4,0 2-Бутил-4,4-диметил-1,3-ДИоксан 90 2-Амил-4-метил-1,3-диоксан 7,0 2-Изобутил-4,4-диметил- 97 2-Гексил-4-метил-1,3-диоксан 7,0 1,3-диоксан 2-Октил-4-метил-1,3-диоксан 5,0 2-Ампл-4,4-диметил-1,3-диоксан 97 2-Гексил-4,4-диметил-1,3-ди- 88 оксан 2-Октил-4,4-дн мстил -1,3-ди- 87 оксан .♦ /=75°с. *♦ f = 85°C. *** / = ио °C. 4* В остальных опытах / = 115 °C.
В качестве гомогенных катализаторов применялись H2SO4, HCI,
BF3-O(C2H5)2, и-толуолсульфокислота, гетерогенных — КУ-2.
Аналогично циклическим ацеталям 4,4-диметил-1,3-диоксаи
реагирует с образованием новой пары ацеталей [557]:
В равновесных условиях выход продуктов не превышает 25—
40%. При непрерывном выводе низкокипящего формаля из зоны
реакции их выход может быть доведен до 75—95%. В качестве
катализаторов реакции могут использоваться H2SO4, НС1, и-то-
луолсульфокислота, КУ-2, BF3O (С2Н5)2. Лучшие результаты
были получены на КУ-2 и BF3-O(C2H5)2.
Результаты реакции 4,4-диметил-1,3-диоксана с циклически-
ми ацеталями приведены в табл. 8.4.
В присутствии кислотных катализаторов при температуре
60—120 °C происходит перециклизация 4,4-диметил-1,3-диоксапа
[559—563]:
191
Изучено влияние температуры^/продолжительности реакции,
природы и количества катализатора на селективность образо-
вания дигидропирана, результаты исследований при температу-
ре 100 °C приведены в табл. 8:5. Если воду не выводить из зоны
реакции, основными продуктами являются дигидропираны и
тетрагидропиранол [195] /
В качестве побочного процесса протекает гидролиз; из реакци-
онной массы выделяют гликоли, ненасыщенные спирты и диено-
вые углеводороды:
СН2=ССН3(СН2)2ОН —।
(СН3)2С=СНСН2ОН —I
—► сн2=ссн8сн=сн2
Конденсация СН2О с ненасыщенными соединениями приводит
к образованию высокомолекулярных соединений.
В присутствии альдегидов под действием кислотных катали-
заторов (H2SO4, КУ-2) происходит как переацетализация 4,4-
диметил-1,3-диоксана, так и образование соответствующих ди-
гидропиранов [564, 565]:
где
R7—СНз, С2Н6, С3Н7, C^Hg, wso-C^Hg, CgHg,
4,4-Диметил-1,3-диоксан реагирует с кетонами в присутствии
кислотных катализаторов с образованием 2,2-диалкилзамещен-
ных дигидропиранов [566]:
192
(33-42%)
где 1) R, = R2 = CH3; 2) R’ = R2 = C2H5; 3) R^CHs; R2 = C2H5, C3H7; «зо-С3Н7>
C4H9, U30-C4H9.
В присутствии концентрированной серной кислоты 4,4-диме-
тил-1,3-диоксан в реакции с нитрилами образует замещенные
5,6-дигидро-1,3-оксазины (выход 33—50%) [567]:
+ RCN
H2SO4
где R=tCH3, СН=»СН2, СвН5.
Аналогично протекает взаимодействие 4,4-диметил-1,3-диоксана
с тиоцианатами [568]:
^-Бутиролактон получен реакцией с оксидом углерода над
комплексным катализатором (Пат. ГДР 2657346, 1977), состоя-
щим из родия, кобальта и никеля.
Как правило, кислотно-катализируемые реакции 4,4-диметил-
1,3-диоксана сопровождаются процессами дегидратации произ-
водных 3-метил-1,3-бутандиола и образованием изопрена. При
определенных условиях изопрен становится основным продуктом
реакции.
ТАБЛИЦА 8.5
Катализатор Количество катализато- ра, г Время реак- ции, ч Конверсия 4,4-диметил- 1,3-диоксана Селективность, %
дигидропи- рана пиранэла
H2SO4 1,74 1,5 36,2 15,4 4,2
КУ-2 2,32 3 54,1 9,5 3,3
п-ТСК 2,61 3 27,8 6,3 2,4
13—627
193
Превращения 4,4-диметил- 1,3^(иоксана под действием
личных аминов в присутствии
но представить схемой: /
раз-
мож-
гидрохлоридов суммарно
n-C6H5NH2
----------->
3NH2OH; НС1
----------->
---> СН2—СН—С=СН2 + 2Н2О+
СН3
I
---> СН2=СН—С=СН2 + 2Н2О +
Выход изопрена зависит от структуры исходного амина и со-
ставляет 75—80% при концентрации катализатора 5—6% (масс.)
[569]. Наилучшие выходы изопрена достигаются при мольном
соотношении амин: диоксан в пределах 0,2—0,4: 1,0 (табл. 8.6).
Решающее влияние на селективность образования диена ока-
зывает природа амина.
При использовании 4,4-диметил-1,3-диоксана большое значе-
ние имеют его физико-химические свойства [570, 571]. В стап-
ТАБЛИЦА 8.6
Амин Конверсия 4,4-ди- метил-1,3-диоксана Селективность об- разования изопрена
Бутиламин 40,3 68,5
Дибутиламин 34,5 67,4
Трибутиламин 51,8 12,6
Октиламин 42,3 66,9
Дециламин 42,8 66,4
Ароматический амин 58,3 93,3
Метиланилин 53,4 78,6
Диметиланилин 51,0 62,8
'Пирролидон 77,0 65,2
а-Метилпирролидон 61,0 63,6
Морфолин 45,0 95,4
Этилендиамин 60,5 71,4
Гидроксиламин 88,6 87,3
Неорганический амин 98,6 97,9
Аммиак 58,9 50,2
194
дартных условиях 4,4-диметил-1,3-диоксан представляет собой
бесцветную прозрачную жидкость с характерным запахом:
/кип (°С/давление, кПа) 132,5/98,67
плотность, р420, г/см3 0,9631
показатель преломления, nD20 молекулярная рефракция: 1 ,4230
найдено 30,08
вычислено 30,89
поверхностное натяжение (при 20,0± ±0,1 °C), 10-3 Н/м 28,9
теплота испарения, кал/г-моль 9,400
Для процессов очистки, выделения и регенерации 4,4-диме-
тил-1,3-диоксана особую роль играют фазовые равновесия, азео-
тропообразрвание с различными растворителями [572—574].
4,4-Диметил-1,3-диоксан обладает хорошей растворяющей
способностью по отношению к высокомолекулярным соединени-
ям, углеводородам. Это позволяет использовать его в качестве
растворителя нитро- и ацетилцеллюлозы [575], лакокрасочных
материалов! и смол [576]. 4,4-Диметил-1,3-диоксан является пер-
спективным синтетическим растворителем для лакокрасочной
промышленности, поскольку способен растворять смолы различ-
ной структуры: алкидные, эпоксидные, поливинильные, целлюло-
зы, сложпоэфирные [577]. В работе он используется при изго-
товлении нитроцеллюлозных, ацетилцеллюлозных, глицерофта-
левых, фенольных, мочевиноформальдегидных, акриловых, эпо-
ксидных и полиуретановых лаков. Применение 4,4-диметил-1,3-
диоксана в лакокрасочных покрытиях на основе эпоксидных,
перхлорвиниловых и фенолформальдегидных смол позволяет
повысить прочность покрытия на истирание и обеспечивает по-
вышенную долговечность, озоно- и светостойкость [579]. На его
основе разработан растворитель для нейлона, а также состав
для выведения пятен (А.с. СССР 753958, 1980).
Изучены растворимость полярных и неполярных каучуков
в циклических ацеталях [580]. 1,3-Диоксаны растворяют глав-
ным образом полярные каучуки. В то же время, применяя 1,3-
дноксаны в смеси с известными растворителями, можно добить-
ся полного растворения и неполярных каучуков.
4,4-Диметил-1,3-диоксан обладает высокой растворяющей
способностью (А. с. СССР 1027373, 1983) по отношению к мас-
лам и парафинам. Поэтому его можно применять при депара-
финизации смазочных масел либо в чистом виде, либо в смеси
с ароматическими растворителями.
Водные растворы 4,4-диметил-1,3-диоксана [1—5% (масс.)]
могут быть использованы в качестве реагента для очистки при-
забойной зоны пластов в газовых скважинах [581]. Он входит
в состав для удаления асфальто-смолистых и парафинистых
13*
195
отложений (А. с. СССР 1209829, 1986). Возможно применение
4,4-диметил-1,3-диоксана в качестве растворителя в электроли-
тических ячейках (Пат. Франции 1490726, 1968).
Смесь 4,4-диметил-1,3-диоксана с водой (4 1) используется
в качестве экстрагента для определения содержания глицерина
в полиэтилене высокого давления (А. с. СССР 532041, 1977).
В результате повышается точность анализа и сокращается вре-
мя его проведения.
Разработаны методы получения смол различного состава и
свойств на основе 4,4-диметил-1,3-диоксана (Пат. Франции
1363211, 1964). Обработкой 4,4-диметил-1,3-диоксана борной
кислотой или оксидом бора получены термостабилизирующие
добавки к смолам (Пат. Франции 88251, 1960). Сополимеры
глицерина и фталевого ангидрида модифицированы добавлени-
ем 4,4-диметил-1,3-диоксана в ходе полимеризации. Он исполь-
зуется также и как компонент катализаторов при полимериза-
ции диенов [582, 583].
Применение оксидата 4,4-диметил-1,3-диоксана в качестве
инициатора полимеризации и одновременно пластификатора по-
лиметилметакрилата позволяет отказаться от необходимости ис-
пользования взрывоопасных инициаторов и дорогостоящих плас-
тификаторов и обеспечивает получение оргстекла с рядом улуч-
шенных физико-механических характеристик, например, повы-
шенной ударной вязкостью, светостойкостью, снижением термо-
окислительной деструкции полиметилметакрилата [584].
4,4-Диметил-1,3-диоксан проявляет свойства синергетической
добавки для стабилизации тетрагидрофталевого ангидрида
((А. с. СССР 649715, 1979) 1,1,1-трихлорэтана.
Введение 4,4-диметил-1,3-диоксана в количестве 0,5 —
2,0% (масс.) в смазочные масла для авиационных двигателей
снижает коксообразование (Пат. США 3248327, 1965). Добавле-
ние алкилзамещенных 1,3-диоксациклоалканов в дизельные топ-
лива приводит к более полному сгоранию топлива и уменьшению
выброса в атмосферу вредных веществ [585], при добавлении
3% (масс.) 4,4-диметил-1,3-диоксана в бензин теплота сгорания
увеличивается на 1—3%. Его добавки улучшают технико-эконо-
мические показатели термокаталитической переработки углево-
дородного сырья (А. с. СССР 1172436, 1985).
Производные 1,3-диоксана в количестве 5—40% (масс.) вво-
дят в состав присадок для понижения температуры застывания
высокопарафинистых нефтей и нефтепродуктов (А. с. СССР
453422, 1985). Применение предлагаемой композиции приводит
к синергетическому эффекту, выражающемуся в значительном
понижении температуры застывания нефтей.
4,4-Диметил-1,3-диоксан проявляет высокую эффективность
как реагент для флотации угля [586]. Его использование позво-
лило повысить извлечение горючей массы в концентрат до
196
96,8% [586] при одновременном снижении расхода реагента в
6 раз по сравнению с парафиновыми и ароматическими углево-
дородами.
4,4-Диметил-1,3-диоксан ингибирует комплексообразование
алканов с мочевиной [287]. Возможно его использование в ка-
честве реагента для подавления жизнедеятельности сульфатвос-
станавливающих бактерий (А.с. СССР 988776, 1983), протрави-
теля семян (А.с. СССР 245489, 1969) и добавки в процессе
микробиологического получения гиббереллиновой кислоты
(Пат. Японии 7414689, 1981).
ГЛАВА 9.
Состав и области применения побочных
продуктов производства ароматических
углеводородов
9.1. Применение побочных продуктов производства стирола
В производстве стирола дегидрированием этилбензола и при
комбинированном получении стирола и а-метилстирола совме-
стным дегидрированием смеси алкилбензолов на стадии ректи-
фикации образуются кубовые остатки, которые сжигают или
перерабатывают совместно с пиролизной смолой. Основными
компонентами, входящими в состав кубовых остатков!, являют-
ся полимеры, продукты уплотнения, образующиеся при высо-
котемпературном дегидрировании, а также остаточные моно-
меры— стирол и а-метилстирол. Содержание связанного (по-
лимер) и свободного мономера в кубовых остатках достигает
90—95%, их выход составляет 25—50 кг на 1 т получаемого
мономера, что соответствует образованию 3—5 тыс. т отходов
в год на современном агрегате по производству стирола мощ-
ностью 125 тыс. т/год [588—590].
Только 53,2% всей массы современных кубовых остатков
ректификации стирола могут быть использованы для произ-
водства лака [591]. Для решения проблемы утилизации кубо-
вых остатков и создания безотходного производства стирола
необходимо изыскание новых путей переработки кубовых ос-
татков, учитывающих низкое содержание в чих остаточного
стирола.
197
ТАБЛИЦА 9.1.
Компонент Температура толуола, °C Температура водя- ного пара, °C
450 | 500 | 550 | 600 550 | 600
Бензол 0,02 0,04 0,06 0,06 0,02 0,02
Этилбензол 0,05 0,08 0,10 0,10 0,10 0,10
Этилтолуол 0,27 0,28 0,30 0,28 0,32 0,30
Стирол 7,4 7,8 8,0 8,0 7,9 8,0
а-Метилстирол 87,6 87,0 86,5 86,3 86,7 86,5
п-Пропилбензол 0,13 0,14 0,14 0,14 0,16 0,16
Изопропилбензол 0,23 0,26 0,30 0,32 0,30 0,32
Смолы 4,3 4,4 4,6 4,8 4,5 4,6
В работе [588] выявлена температурная область процесса
разложения кубовых остатков на мономеры и изучен состав
продуктов разложения. Табл. 9.1 отражает состав (в %) про-
дуктов термодеструкции кубовых остатков ректификации при
совместном производстве стирола и а-метилстирола (объемная
скорость 30%-го раствора в толуоле 10 ч-1; теплоноситель —
толуол или водяной пар; массовое соотношение теплоноситель
раствор равно 1 1) [588]. Так (табл. 9.1), при 450 °C обеспе-
чивается практически полная регенерация мономеров из кубо-
вых остатков комбинированного производства стиролов. Даль-
нейшее повышение температуры приводит лишь к незначитель-
ному изменению состава: снижается количество а-мстилстиро-
ла в продуктах деполимеризации и увеличивается содержание
стирола, бензола, этил- и изопропилбензолов. Количество тяже-
лых смол в продуктах деструкции остается постоянным в ши-
роком интервале температур. Установлено также, что замена
толуола водяным паром существенно не влияет на состав про-
дуктов деструкции.
В табл. 9.2 приведен состав (в %) продуктов термодсст-
рукции кубовых остатков индивидуального производства сти-
рола (объемная скорость 50%-го раствора в толуоле 10 ч
теплоноситель — толуол или водяной пар; массовое соотноше-
ние теплоноситель раствор равно 1 1) [588].
Предложенный способ термической деполимеризации кубо-
вых остатков может быть использован при создании малоот-
ходной технологии производства стирола и а-метилстирола.
Для рекуперации стирола кубовый остаток предлагается
подвергать термическому разложению при 400—500°C в при-
сутствии катализатора — оксида меди (II), осажденного на
асбесте [А. с. СССР 956497; БИ, 1982, № 33].
Кубовый остаток ректификации стирола можно использо-
вать для получения сополимера с малеиновым ангидридом, ко-
торый находит применение в качестве противокоррозионных
покрытий [Пат. СРР 71349; РЖХ, 1983, 12С307П].
198
ТАБЛИЦА 9.2.
Компонент Температура толуола, °C Температура водяного пара, °C
500 | 550 | 600 600
Этилбензол 0,24 0,44 0,6 о,з
Этилтолуол 0,71 0,70 0,67 0,67
Стирол 59,9 59,7 58,6 59,0
а-Метилстирол 4,6 4,5 4,5 4,5
п-Пропилбензол 0,19 0,2 0,19 0,2
Изопропилбензол 0,09 0,10 0,09 0,10
м- и и-Метилстиролы 0,47 0,47 0,47 0,46
а-Метилстирол 0,60 0,59 0,58 0,57
Смолы 33,2 33,3 34,3 34,2
* Состав исходных кубовых остатков (в %): стирол —31,6; а-метилстирол — 3,1; этил
толуолы — 0,7; н-пропилбензол — 0,2; изопропилбензол — 6,1; м- и п-метилстирол— 0,5;
а-метилстирол — 0,6; смолы — 33,2; полимеры — 30.
Изучена [592] сополимеризация малеинового ангидрида с
мономерными компонентами, входящими в состав кубового ос-
татка ректификации стирола, инициированная перекисью бен-
зоила при 60—80 °C в среде бензола и метилэтилкетона. Най-
дено, что электронодонорные компоненты, такие как нафталин
и олигостирол, входящие в состав кубового остатка ректифи-
кации стирола, ускоряют радикальную сополимеризацию.
Проведено исследование [593] возможности синтеза плен-
кообразующего материала с удовлетворительными выходом и
физико-механическими показателями на основе сополимеров
кубовых остатков ректификации стирола и малеинового ангид-
рида. Для улучшения качества пленкообразующего в состав
кубового остатка вводили 8—75% полистирола, количество ма-
леинового ангидрида варьировали в пределах 8—25%. Под-
держивали ступенчатый температурный режим сополимериза-
ции: 2 ч при 120 °C; 3 ч при 150 °C; 15 ч при 175 °C; затем ва-
куумировали смесь 60 мин. Предлагается оптимальный состав
искусственной смеси, обеспечивающий максимальные показате-
ли качества лакокрасочного материала (в %): дивинилбензол —
отсутствие, кристаллическая фаза—5, стирол — 20, треххло-
ристое железо—1, полистирол — 74, малеиновый ангидрид—5.
Для произвольных составов кубовых остатков ректификации
стирола с завышенным содержанием кристаллических соедине-
ний и дивинилбензола рекомендовано разбавление сырья мо-
номерным стиролом.
В работе [594] разработана технология получения пленко-
образующего на основе кубового остатка путем совместной по-
лимеризации его с малеиновым ангидридом. Для синтеза ис-
пользовали кубовые остатки, содержащие ингибитор полимери-
зации стирола — гидрохинон с м-оксидифениламином. Полиме-
199
ризацию проводили блочным способом при 180 °C 14—18 ч без
катализатора. В настоящее время в производстве стирола при-
меняются в качестве ингибиторов диоксим-п-хинон с гидрохи-
ноном или с n-нитрофенолом. В этом случае полимеризацию
рекомендуется проводить в присутствии хлорида металла
(TiCl3, FeCl3, ZnCh). Полученная таким образом смола имеет
удовлетворительные физико-химические показатели и пригодна
для .приготовления лака с хорошими декоративно-защитными
свойствами. Она имеет следующие характеристики:
Продолжительность высыхания при 20 °C, мин:
от «пыли» 10
полное 60
Твердость по маятниковому прибору 0,5
Прочность при изгибе, мм, не более 1
Адгезия по методу «решетки», баллы 1
Водостойкость (244) Не меняет внешнего вида
Химическая стойкость в 20%-ых растворах Не менее 48
H2SO4, HNO3, НС1, ч Светостойкость (облучение дуговой лампой в течение 6 ч) Без изменений
В работе [595] с целью утилизации кубового остатка рек-
тификации стирола проведено исследование по ацилированию
его малеиновым ангидридом в присутствии катионных катали-
заторов и добавок а-метилстирола в растворе бензола. Найде-
но, что ацилированный кубовый остаток из растворов толуола
на металлических и стеклянных подложках образует прозрач-
ные и гладкие поликристаллы, а наличие в его составе карбок-
сильной группы способствует резкому повышению адгезионной
способности. Методами газожидкостной хроматографии опре-
делены расход стирола и состав кубового остатка в процессе
ацилирования в зависимости от условий реакции.
В работе [594] предлагается лак Дикорс на основе сополи-
меров кубовых остатков ректификации стирола и димерных
фракций продуктов гидроформилирования пропилена, обладаю-
щий повышенной прочностью при ударе, твердостью, улуч-
шенной пластичностью. Покрытия, образованные этим лаком,
водостойки, выдерживают воздействие нефтепродуктов и раст-
воров щелочей. Лак Дикорс рекомендуется для защиты метал-
лических и бетонных сооружений, оборудования, трубопрово-
дов, мелиоративных систем и других коммуникаций, для ок-
раски дачных домиков, изгородей, гаражей. Лак Дикорс позво-
ляет экономить растительное сырье, расширить ассортимент
технических лаков, снизить стоимость покрытий за счет замены
дорогостоящего малеинового ангидрида на продукт отходов
производства и исключения дибутилфталата.
200
Хлорированные кубовые остатки ректификации стирола
добавляют в количестве 5—13% в полимерные композиции
для крепления металлических штанг в! горнодобывающей про-
мышленности в целях повышения скорости отверждения и сни-
жения токсичности [А. с. СССР 834028; Б. И., 1981, № 35].
Предлагаемая композиция имеет следующие показатели: пре-
дел прочности при сжатии — 50 (40—80) МПа (в скобках по-
казатели известной композиции); при изгибе—12 (8—20)
МПа, при растяжении — 6 (5—12) МПа. Замена в ранее из-
вестной композиции хлорметилэтилового эфира на хлорирован-
ные кубовые остатки ректификации стирола резко улучшает
условия труда вследствие меньшей токсичности последнего.
Описан способ получения из кубовых остатков дисперги-
рующих добавок [Пат. США 4096089; РЖХ, 1982, 6И771П].
Термическая сополимеризация кубового остатка ректифика-
ции стирола и альдегидсодержащей фракции кубового остатка
ректификации продуктов гидроформилирования олефинов с
температурой кипения 130—270 °C позволяет получить смолу,
которая может быть использована для получения лаков и олиф
[А. с. СССР 1100279; Б. И., 1984, № 24].
Кубовый остаток в количестве 3—8% может входить в со-
став полимербетонной смеси, что придает бетону повышенную
подвижность и прочность. Предлагаемая полимербетонная
смесь имеет более высокую прочность (в сравнении с извест-
ными) при сжатии, изгибе и растяжении [А. с. СССР
2780029; Б. И., 1981, № 31].
Кубовые остатки в количестве 1—3% предлагается вводить
в состав электропроводного клея, что повышает его стойкость
к действию агрессивных сред [А. с. СССР 861379; Б. И., 1981,
№ 33].
В работе [А. с. СССР 257496; Б. И., 1969; № 36] изучены
ингибирующие свойства рекуперируемой серы и условия выде-
ления ее из кубового остатка ректификации стирола. Показано,
что при содержании в растворе кубового остатка 8—13% серы
и использовании толуола в качестве растворителя при темпе-
ратуре 10—20°C она легко осаждается и сохраняет свои инги-
бирующие свойства. По предложенной технологической схеме
рекуперации ингибирующей серы и стирола из кубового остат-
ка степень рекуперации серы составляет 80—85%. Удельный
выход стирола на единицу массы кубового остатка при темпе-
ратуре 500—550°C и объемной скорости подачи раствора ку-
бового остатка 1—5 ч-1 составляет 0,3. Регенерационные циклы
серы и стирола на заводе с годовой мощностью по стиролу
300 тыс. т/год обеспечивают экономический эффект от сокраще-
ния потребления серы и непроизводительных потерь стирола
более 0,5 млн. руб/год. Результаты экспериментальных иссле-
дований использованы при расчетах процессе рекуперации серы
201
и стирола из кубового остатка и основного технологического
оборудования — трубчатого мономеризатора, дискового крис-
таллизатора.
Иногда целесообразно отгонять легколетучие продукты из
кубовых остатков, образующихся в процессе ректификации
стирола. Обычно для этих целей применяют дистилляционные
кубы, недостатками которых являются малая производитель-
ность и то, что процесс отгонки в них сопровождается допол-
нительными потерями продукта в результате длительного воз-
действия температур [596].
В работе [597] изучена возможность использования для
этой цели роторного тонкопленочного испарителя. Приведены
схемы экспериментальной установки для отгонки стирола из
кубовых остатков и тонкопленочного роторного испарителя.
Дистилляции подвергали кубовый остаток, основными легко-
летучими компонентами которого являлись стирол и а-метил-
стирол. Максимальная производительность по дистилляту на
предлагаемой установке достигала 200—250 кг/ч с 1 м2 поверх-
ности теплообмена. Хроматографический анализ состава дис-
тиллята показал, что в нем содержатся оба основных легколе-
тучих компонента — стирол и а-метилстирол в соотношениях,
определяемых составом исходной смеси и режимом работы
аппарата. Содержание стирола в кубовом остатке может быть
доведено до величины, составляющей всего несколько процен-
тов от количества стирола в исходном продукте. Этот метод
позволяет снизить потери стирола с кубовыми остатками в слу-
чае сжигания последних. Если же кубовые остатки после
испарителя используют в качестве полупродуктов для дальней-
шей переработки, количество стирола в них может быть увели-
чено за счет повышения производительности аппарата. Отог-
нанные легколетучие компоненты должны быть сконденсиро-
ваны и направлены на повторную ректификацию. Возможен
вариант, когда пары из испарителя подаются на ректифика-
цию в отдельную колонну [598].
Предложен [А. с. СССР 1046237; Б. И., 1983, № 37] способ
выделения стирола из фракции Се пироконденсата или из про-
дуктов дегидрирования этилбензола азеотропной ректификаци-
ей в присутствии в качестве разделяющего агента — морфоли-
на. По этому способу в куб ректификационной колонны загру-
жают смесь стирола, этилбензола и морфолина (давление в
колонне 798 Па, флегмовое число 5). После выхода колонны
на режим отбирают дистиллят (азеотропную смесь этилбензо-
ла и морфолина). В кубе получают 99,9%-й стирол с выходом
97%. На ректификационной насадочной колонне проводят
опыты по разделению бинарной смеси этилбензол — стирол
простой и азеотропной ректификацией в присутствии морфоли-
на и уксусной кислоты. Флегмовое число во всех опытах 5.
202
Описан способ выделения стирола из продуктов дегидриро-
вания этилбензола адсорбционной очисткой этих продуктов
ионитом на основе сополимера стирола и дивинилбензола
[А. с. СССР 925926; Б. И., 1982, № 17].
Предложен [Пат. США 4288234; РЖХ, 1982, 13П257П]
способ очистки отходящего газа процесса производства стиро-
ла из этилбензола, содержащего водород, стирол, этилбензол,
толуол, от ароматических углеводородов. Для этого отходя-
щие газы предварительно промывают этилбензолом и охлаж-
дают с целью конденсации ароматических углеводородов, сни-
жая их концентрацию до 0,5—10%, затем отходящие газы при
температуре 2—52°C, контактируют с углеводородной фракци-
ей, адсорбирующей ароматические углеводороды. Получают
отходящий газ с содержанием ароматических углеводородов
менее 0,2%. В качестве углеводородной фракции используют
тяжелый побочный продукт процесса производства стирола ал-
килированием бензола, содержащий дифенилэтан, полиэтил-
бензол, 5% которого выкипает при 316—538 °C. Ароматические
углеводороды, адсорбированные углеводородной фракцией,
выделяют дистилляцией при 52—149 °C. Очищенный по этому
методу отходящий газ содержит более 90% водорода и менее
0,05% ароматических углеводородов.
Для очистки сточных вод и водных растворов от стирола в
них предлагается вводить воздух, в результате чего происхо-
дит выдувка этих примесей [591].
9.2. Применение побочных продуктов производства
алкилбензолов
Алкилирование бензола олефинами с целью получения раз-
личных алкилбензолов осуществляется в присутствии А1С1з,
который образует с молекулами бензола каталитический комп-
лекс. При этом происходит последовательное замещение атомов
водорода с образованием смеси продуктов разной степени ал-
килирования, что приводит к образованию побочных продуктов
и снижает выход целевого продукта. Кроме того, нежелательны
смолообразование, деструкция алкильных групп и полимериза-
ция олефинов. Смолообразование состоит в получении конден-
сированных бензолов с высокой температурой кипения: дибен-
зилалканов, трибензилинданов, дибензилолефинов и др. Дест-
рукция алкильных групп ведет к побочному образованию ал-
килбензолов с более короткой алкильной группой. Взаимодей-
ствие, вероятно, происходит на стадии расщепления карбоний-
иона, образовавшегося из алкилирующего агента:
RCH-CIbR RCH = CH2 С5Н5—СН—CH3 + C6H5R.
R
203
Образование полимеров происходит в результате последо-
вательного взаимодействия карбоний-иона с олефином:
+ С2Н4 + С2Н4 -L
СНз—СН2 СНз—СН2—СН2—СН —> СНз—СНг—СНг—СН2-СН2—СН2
И т. д.
В работе [599] с помощью ИК-спектроскопии, спектров!
ЯМР и хроматографического анализа установлено, что в сос-
тав этилбензольной смолы входит более тридцати высших по-
лиалкилбензолов (три-, тетра-, пента- и гексаалкилбензолов),
содержащих главным образом этильные группы, а также длин-
ноцепочечные полимерные и конденсированные ароматические
углеводороды (в %):
До диспро- После дис-
Углеводород порциониро- пропорцио-
вания нирования
Бензол+парафиновые углеводороды — 73,6
Этилбензол — 15,6
Изопропилбензол — 0,5
1,3,5-Триметилбензол — Следы
втор-Бутил — и изобутилбензол — Следы
о-, м~, и /г-Диэтилбензолы ] Следы
о-, м~, и п-Диизопропилбензолы > 1,3,4,5-Тетраметилбензолы j 1,2 3,0
3,4 3,0
Триэтилбензолы 1,0
Дибутилбензолы 4,4 | 0,7
Тетралин 0,4
Пентаметилбензолы+Xi—9,6+1,7 1,8 0,2
Нафталин+Х2 2,7
Триизопропилбензол 0,4 1,8
Тетраэтилбензол+дифенилметан 28,4 —
а- и [}-Метилнафтены 0,7 —
Г ексаметилбензол 2,2 —
Дифенил+Хз 2,7 —
Дифенилэтан 0,8 —
Пентаэтилбензол 18,9 —
Гексаэтилбензол 9,3 2
Антрацен+фенантрен+Х4 16,6 0,6
(Xi, Х2, Х3, Х4 — неидентифицированные соединения.)
Предложен [Заявка ФРГ 3437615 РЖХ, 1986, 2П22ОП]
способ переработки, содержащей пропилен реакционной смеси,
образующейся при алкилировании бензола пропиленом в при-
сутствии катализатора Н3РО4. Способ основан на отделении
пропилена и части бензола с помощью ступени дросселирова-
ния и одной ректификационной колонны, bi которой пропан и
вода отделяются от бензола. Оставшийся бензол и боковые по-
гоны с температурой кипения кумола выделяют в последова-
тельно подключенных двух ректификационных колоннах. От-
деление кумола от боковых погонов с температурой кипения
выше, чем у кумола, проводят в четвертой ректификационной
204
колонне, нижний продукт из которой с существенно понижен-
ным содержанием бензола перегоняют вместе с боковыми по-
гонами.
Разработан [А. с. СССР 1004330; РЖХ, 1986, 4Н135П1]
способ разделения продуктов алкилирования и А1С1з контак-
тированием алкилата и А1С1з с макропористым КУ-23 в Н-
форме. Запатентована [Пат. ГДР 227559; РЖХ, 1986, 12Н99П]
энергосберегающая схема разделения продуктов алкилирова-
ния бензола пропиленом в присутствии катализатора А1С13.
Из средней части колонны выделения непрореагировавшего
бензола пары отводят в колонну выделения побочного этилфе-
нола, которая работает без нагревательных элементов. По этой
технологии можно вырабатывать 99,8%-й кумол.
Один из основных отходов процесса жидкофазного алкили-
рования белзола— кубовый остаток, утилизация которого яв-
ляется важной проблемой нефтехимической промышленности.
Кубовый остаток ректификации моноалкилбензола, состоя-
щий главным образом из ди- и триалкилбензолов с алкилами
Си—С1з с примесью моно- и тетраалкилбензолов и имеющий
температуру кипения более 250 °C при 101,3 кПа, может ис-
пользоваться в качестве термостойкой жидкости, например
теплоносителя, который целесообразно стабилизировать извест-
ными антиоксидантами, применяемыми для стабилизации ма-
сел и полиалкенов [А. с. ЧССР 201878; РЖХ, 1984, 1П339П].
Известен ряд способов переработки кубового остатка с це-
лью получения этилбензола и других ценных продуктов [А. с.
СССР 887557, БИ, 1981, № 45]. В их основу положена реак-
ция переалкилирования полиалкил ароматических углеводоро-
дов, содержащихся в кубовом остатке по схеме
СбН6_пКпЛ' (п—1)СбНб —/гСбНбИ.
Для достижения высокой эффективности процесс рекомен-
дуется осуществлять либо в высокотемпературном режиме в
присутствии катализаторного комплекса, либо с использовани-
ем свежего хлорида алюминия и 5—10-кратного количества
бензола.
Особый интерес представляют процессы, которые позволя-
ют использовать алкилароматические углеводороды до момента
попадания их в кубовый остаток, т. е. на отдельных стадиях
алкилирования. Так, для снижения количества кубового остат-
ка в производстве изопропилбензола наиболее целесообразным
считается раздельное проведение реакций алкилирования и
переалкилирования [600], что достигается извлечением воз-
вратных полиалкилбензолов из потока, поступающего на ал-
килирование, и подачей их на стадию переалкилирования вмес-
те с реакционной массой из алкилатора.
205
ТАБЛИЦА 9.3.
Исходная —
После приготовле- тх-т;
ния 1и,и
Т7 5,9
После реакции ал- р-^-
килирования 5,9
После реакции пе- —у-
реалкилирования
88,6 0,6 0,8 0,1 0,6
79,7 0,5 18,1 0,1 0,2
72,2 0,3 14,6 ~ 0,5
71,9 0,2 24,8 0,4
66,5 0,2 11,3 “ 0,6
78,2 0J2 20,2 _ 0,1
75,6 б~2 16,6 0,2
0,3 5,6 4,0 — —
0,9 0,5 Следы — —
б“б 4?2 63 С2 ЗТТ
0,1 2,6 Следы — —
бГЗ 4/7 10,8 5^6 14~7
0,2 1,1 Следы ____ ___
6/1 2~Гб 4~7
Предложено [А. с. СССР 335927; БИ, 1975, № 30] исполь-
зовать реакцию переалкилирования для регенерации катализа-
торного комплекса. Добавление к последнему бензола и этил-
бензола в массовом соотношении 1 1 и 0,05% дифенилметана
позволило увеличить скорость алкилирования бензола этиленом
в два раза.
Описан способ переалкилирования ароматических углеводо-
родов в жидкой или газовой фазе на катализаторе, содержа-
щем кислотные цеолиты [А. с. ЧССР 212946; РЖХ, 1984,
22Н136П]. Толуол, содержащий 0,01% галогенированных угле-
водородов, диспропорционирует на предложенных катализато-
рах при 440°C и 2,5 МПа, объемной скорости Н2 1 ч-1 и мо-
лекулярном соотношении Н2 толуол, равном 10 1, и образует
продукт, содержащий (в %) низкокипящие продукты—1, не-
насыщенные вещества 2; бензол—13, толуол — 73,5, аромати-
ческие углеводороды С8 и С9 — 9,4.
С целью снижения количества отходов производства этил-
бензола изучено влияние компонентов, входящих в состав ку-
бового остатка, ва активность катализаторного комплекса.
Проведенные в лабораторных условиях исследования состава
углеводородной реакционной смеси для приготовления катали-
заторного комплекса на различных стадиях синтеза этилбензо-
ла (анализируемый слой — углеводородный — числитель, ката-
лизаторный — знаменатель) показывают (табл. 9.3), что уже
на первой стадии промышленного процесса, т. е. на стадии
приготовления катализаторного комплекса, при добавлении к
смеси бензола и полиалкилбензолов происходит перераспреде-
ление алкилбензолов между углеводородным и катализатор-
ным слоями.
206
ТАБЛИЦА 9.4.
Исходная х I II
С, % 1 X с, % 1
13,0 0,5 10,7 20 2,4
9,2 0,5 6,2 20 2,4
4,7 1,0 4,3 10 3,8
5,4 1,0 4,9 10 3,7
Как видно из табл. 9.3, количество смолистых продуктов
даже в свежеприготовленном комплексе составляет 3—4%, в
рециркулируемом промышленном комплексе достигает 25%.
Снижение активности катализаторного комплекса обычно
объясняют присутствием влаги в исходном сырье. Проведенные
исследования влияния добавок воды (I) и кубового остатка
(II) на удельную электропроводность катализаторного комп-
лекса (х-103, Ом-1 см-1) показывают, что смолистые продук-
ты, содержащиеся в кубовом остатке, играют определенную-
роль в этом нежелательном процессе (табл. 9.4), отрицательно
влияя на активность катализатора реакции алкилирования.
Основность веществ, входящих в состав этилбензольной
смолы, значительно выше, чем у бензола и этилбензола, по-
этому хлорид алюминия в силу высокой реакционной способ-
ности образует с ними прочные комплексы, следовательно, ка-
тализатор необратимо выводится из зоны реакции. Высшие
полиалкилбензолы и дифенилалканы, которые всегда сопутст-
вуют продуктам жидкофазного алкилирования и содержание
которых зависит от технологических параметров процесса, мо-
гут вступать в реакцию диспропорционирования с образовани-
ем целевого продукта. Принципиальная возможность такого
процесса показана в работе [2]. Установлено, что кубовый ос-
таток на 70—80% может превращаться в ароматические угле-
водороды с температурой кипения ниже 260 °C. Однако созда-
ние дополнительной установки, а также использование свежего
катализатора и большого избытка бензола делает процесс
трудноосуществимым в промышленных условиях.
Наибольший интерес представляет введение бензола в ка-
тализаторный комплекс после алкилатора. Опыты показывают,
что в этом случае не только уменьшается количество смолы,
которая на конечной стадии процесса попадает в кубовый ос-
таток, но и повышается активность катализатора, т. е. проис-
ходит регенерация катализаторного комплекса.
В работе [601] изложены возможные пути химической пе-
реработки ароматических толуолов и ксилолов — побочных
продуктов алкилирования бензола. Рассмотрены процессы гид-
родеалкилирования толуола в бензол, диспропорционирования
207
толуола в бензол и ксилолы, окисления толуола в бензойную
кислоту /и затем в фенол и терефталевую кислоту, хлорирова-
ния толуола.
Предложен [А. с. СССР 1087505; БИ, 1984, № 15] способ
получения этилбензола и изопропилбензола с низким расходом
бензола на побочные процессы, заключающийся В! алкилирова-
нии бензола этиленсодержащим газом при молекулярном соотно-
шении этилен : бензол, равном 0,35 0,5, при температуре 75—
105 °C и давлении в! присутствии каталитического комплекса
АЬОбСвНбСгНз-НО с последующей конденсацией отходящих
газов в конденсаторах-холодильниках при времени пребывания
в них 3—5 с абсорбцией остаточных газов, содержащих бен-
зол, циркулирующей смесью диэтилбензола и бензола при
10—15 °C, ректификацией продуктов алкилирования и выделе-
нием целевых продуктов и побочного диэтилбензола, который
направляют в стадию адсорбции. С целью снижения расхода
бензола на побочные процессы при конденсации и абсорбции
в стадии конденсации остаточных газов, содержащих бензол,
поддерживают скорость прохождения газов 0,67—0,4 м/с при
содержании бензола в рециркулирующей смеси 10—20% и
кратности циркуляции 5—10.
Описан [Заявка Японии 58—216128, РЖХ, 1984, 23Н106П]
селективный способ получения моноалкилбензолов с алкилиру-
ющими агентами типа олефинов, спиртов и (или) галогеналки-
лов в присутствии катализатора, содержащего ионообменный
цеолит типа модернита, соответствующего формуле М2О-А12Оз-
-(9—10)SiO2-6H2O (M-Na, К или/и эквивалент Mg или Са).
Известны способы регенерации катализаторов алкилирова-
ния бензола. Так, предложен [А. с. СССР 1076425, БИ, 1984,
№ 8] способ выделения А1С1з из реакционной массы алкили-
рования бензола олефинами обработкой реакционной массы
водным раствором А1С1з с получением водного слоя, содержа-
щего А1С1з и органического слоя — продукта алкилирования
бензола олефинами. Для повышения степени выделения А1С1з
в качестве водного раствора А1С1з используют раствор, содер-
жащий 200—300 г/л А1С13, а полученный органический слой
доочищают при пропускании через колонну, заполненную гид-
рофильной насадкой.
Предложен способ [А. с. СССР 1034760, БИ, 1983, № 30]
регенерации твердого фосфоркислотного катализатора для ал-
килирования бензола олефинами, заключающийся в обработке
дезактивированного катализатора растворителем при повышен-
ной температуре. Для отделения растворителя используют 40—
90%-й водный раствор ортофосфорной кислоты при массовом
соотношении катализатор растворитель, равном 1 1—3, 80—
100 °C и атмосферном давлении, а отделенный от растворителя
катализатор нагревают до 180—300 °C.
208
ГЛАВА 10
Комплексное использование побочных
продуктов производства некоторых
присадок
Присадки представляют собой сложные химические соеди-
нения, как правило, состоящие из неорганических и органичес-
ких веществ. В качестве исходных соединений в производстве
присадок применяют алкилфенолы, спирты, крезолы, олефи-
ны, органические и минеральные кислоты, масла, формальде-
гид, щелочи/хлорид натрия, пятисернистый фосфор, гидроксид
бария, аммиак, бензин и ряд других веществ [602].
Производство присадок—многостадийный процесс, вклю-
чающий промежуточные стадии: алкилирование, фосфорсерне-
ние, сульфирование, карбоксилирование, хлорирование, конден-
сацию, полимеризацию [603], а также вспомогательные:
отделение промежуточных продуктов реакции от непрореагиро-
вавших веществ, нейтрализацию, сушку, удаление растворите-
лей и катализаторов, промывку и пропарку оборудования и
т. д.
Большинство процессов производства присадок сопровожда-
ется образованием побочных продуктов или отходов, не нахо-
дящих квалифицированного применения. При этом на многих
стадиях производства образуется значительное количество вы-
сокоминерализованных токсичных сточных вод.
В литературе имеется мало сведений о химическом составе
и свойствах сточных вод производства присадок. Результаты
исследований химического состава сточных вод производства
некоторых присадок свидетельствуют о том, что такие стоки
отличаются многокомпонентным составом с высоким содержа-
нием органических и минеральных загрязнений, концентрация
которых зависит от степени чистоты исходного сырья и способа
проведения синтеза основного продукта [604—606]. Состав
сточных вод зависит от источника загрязнения и изменяется со
временем, причем суточное содержание различных ингредиен-
тов изменяется в три и более раз, что связано с периодич-
ностью процессов.
Количество образующихся сточных вод в производстве при-
садок соответствует в среднем 0,5—8 м3 на 1 т целевого про-
дукта.
14-627
209
ТАБЛИЦА 10.1
Стадия разложения алкил- Промывочные воды гото- вого продукта
Показатель салицилата натрия (кислые стоки) I промывка II промывка
Окраска (визуально) Запах (органолепти- чески) Прозрачность, см Сухой остаток, г/л ХПК, г О2/л ВПК, г О2/л Растворенный кислород, мг/л Хлориды, г/л Фенолы, мг/л Нефтепродукты (эфиро- извлекаемые), г/л Желтоватая Кислотный 10 1 72 10,6 5,1 0 226,5 29,8 Светло-розовая Специфический 20 20 0,25—0,9 1,5—2,1 113,2 98,5 1,6 1,8 0,25 0,2 0 0 63,8 58,5 25 14,2 0,3 0,2
10.1. Применение побочных продуктов производства
алкилсалицилатных присадок
Алкилсалицилатные присадки в общем объеме производства
составляют 10%. К ним относятся алкилсалицилат кальция
(АСК) |И многозольный алкилсалицилат кальция (МАСК),
а также бариевый аналог АСК — алкилсалицилат бария
(АСБ). Алкилсалицилатные присадки представляют собой
концентраты кальциевых солей алкилсалициловых кислот в
масле-разбавителе, эффективно улучшающие моющие свойства
моторных масел.
Процесс производства алкилсалицилатных присадок много-
стадийный. При этом используется большое количество компо-
нентов: фенол, олефины, полимердистиллят, минеральные и ор-
ганические кислоты, формалин, бензин и др. При проведении
технологического процесса на различных стадиях образуется
несколько видов сточных вод, которые различаются по степени
загрязненности как органическими, так и неорганическими со-
единениями (табл. 10.1) [607].
На 1 т присадки образуется 5—10 м3 стоков, утилизация кото-
рых является актуальной задачей.
Разработана [608] принципиальная схема очистки вод про-
изводства алкилсалицилатных присадок (АСК, МАСК, АСБ).
Способ обезвреживания сточных вод обеспечивает получение
15—20%-го раствора хлорида натрия. Возможность примене-
ния данного раствора в процессе водоподготовки для регенсра-
210
ТАБЛИЦА 10.2
Время отбора проб, ч Раствор NaCl or очистки с i очных вод производства алкилсалици- латных присадок Раствор технического NaCI
щелочность, мг-экц/л чССТКОСТЬ, мг-экв/л щелочность, мг-экв/л жесткость, мг-экв/л
1 3 250 3,0 300
2 3 30 3 50
3 3 30 2,9 50
4 2,9 35 2,8 50
5 2,9 35 2,8 40
6 2,9 35 2,8 40
7 2,8 35 2,8 40
8 2,8 40 2,7 40
9 2,8 45 2,7 65
10 2,8 45 2,7 65
11 2,7 30 2,7 65
12 2,7 30 2,6 65
13 2,6 30 2,6 65
14 2,7 30 2,6 65
15 2,6 30 2,6 70
16 2,6 50 2,5 70
17 2,6 100 2,5 100
18 2,6 220 2,4 270
19 2,5 400 2,3 340
Примечание. Время работы фильтра, регенерированного раствором, получен-
ным в результате очистки сточных вод алкилсалициловых присадок, составляет 194 ч,
что соответствует среднему пробегу фильтров, регенерированных раствором техническо- го хлорида натрия.
ции Na-катионных фильтров котельной нефтеперерабатываю-
щих заводов отражают результаты, приведенные в (табл. 10.2)
[608].
Изучена возможность использования полученного раствора
хлорида натрия для коагуляции латексов в процессе выделения
каучуков эмульсионной полимеризации [607]. Это обусловлено
сравнительно большим расходом хлорида натрия при выделе-
нии каучуков. Испытывались бутадиен-стирольный латекс ка-
учука СКС-30 АРКП и латекс бутадиен-нитрильных каучуков
СКН-40 CM, СКН-40М. По химическому составу опытные и
контрольные каучуки СКН-40М, СКС-30 АРКИ, СКН-40 СМ
близки и соответствуют нормам ГОСТ -и ТУ
В табл. 10.3 приведен химический состав опытных (/—3)
и контрольных (4—б) образцов каучука СКС-30 АРКП [607],
а в табл. 10.4 —каучуков СКН-40М и СКН-40 СМ (/, 2, 5 —
7 — опытные, 3, 4, 8—10 — контрольные образцы).
Результаты испытаний показали, что по пластическим свой-
ствам опытные (I) и контрольные (II) образцы СКС-30 АРПК
каучуков идентичны (табл. 10.5). Раствор хлорида натрия, яв-
ляющийся отходом производства алкилсалицичатных присадок,
211
ТАБЛИЦА 10.3
Показатель 1 2 3 4 5 6
Содержание %:
золы 0,27 0,3 0,28 0,18 0,2 0,15
неозона «Д» 1,5 1,52 1,5 1,5 1,5 1,42
органических кислот 5,7 5,7 5,9 5,12 5,9 5,7
мыл органических кислот 0,18 0,18 0,1 0,18 0,17 0,08
связанного стирола 22,5 22,5 22,5 22,5 22,5 22,5
Потери массы при сушке, % 0,11 0,13 0,08 0,16 0,08 0,13
по коагулирующему действию равноценен раствору хлорида
натрия, применяемому в серийном производстве эмульсионных
каучуков.
10.2. Применения побочных продуктов производства
некоторых алкилфенольных присадок
При производстве антиоксиданта НГ-2246 (2,2-метилен-бис-
4-метил-6-трет-бутилфенола) на стадии алкилирования и-кре-
зола изобутиленом наряду с целевым продуктом — моноал-
килпаракрезолом образуются ионол (2,6-ди-трет-бутил-4-ме-
тилфенол) и эфиры алкилкрезолов:
На стадии выделения моноалкилпаракрезола теряется значи-
тельная часть побочных продуктов, которые являются ценными
компонентами. До настоящего времени способ их переработки
не разработан [607].
При выделении целевого продукта значительная часть оста-
ется в маточнике. Предложен способ переработки маточника,
позволяющий увеличить выход моноалкилкрезола и снизить
удельный расход я-крезола и изобутилена в производстве ан-
тиоксиданта НГ-2246 [609]. В основу способа положена рсак-
212
ция переалкилирования полиалкилфенолов в присутствии фено-
ла или крезола.
Внедрение данного способа на опытно-промышленной уста-
новке ‘производства антиоксиданта НГ-2246 позволило сокра-
тить расход я-крезола и изобутилена на 25—30%, что сущест-
венно снизило себестоимость антиоксиданта, но значительные
затраты электроэнергии на нагревание маточника до 100 °C
(при более низкой температуре реакция практически не идет)
затрудняют широкое распространение предложенного метода.
В настоящее время во ВНИИПКнефтехим проводятся исследо-
вания по разработке эффективных методов переработки отхо-
дов производства антиоксиданта НГ-2246 с целью обеспечения
безотходного производства.
Предложен способ переработки сточных вод и твердых от-
ходов с получением продукта «Агидол-2ВП». Результаты иссле-
дований физико-механических свойств резин, полученных на
основе известных и исследуемых антиоксидантов!, проведенных
во ВНИИПКнефтехим, указывают на целесообразность его ис-
пользования в стандартной рецептуре резин на основе нату-
рального и стирольного каучуков (табл. 10.6) [607].
Испытания показали, что резины с продуктом «Аги-
дол-2ВП» по некоторым свойствам несколько уступают рези-
нам с НГ-2246, но превосходят резины с неозоном Д.
Внедрение способа переработки отходов производства с по-
лучением антиоксиданта «Агидол-2ВП» позволит рационально
использовать сырье и материалы, предотвратить загрязнение
окружающей среды и дополнительно получить более 100 т в
год продукта взамен дорогостоящих антиоксидантов!.
10.3. Пути использования нефтешламов процесса
производства присадок
В отечественной и зарубежной литературе широко освеще-
ны вопросы переработки таких отходов нефтехимической и
нефтеперерабатывающей промышленности, как отработанные
масла, кислые гудроны, нефтешламы, грунты, загрязненные
нефтепродуктами и т. д. При этом очень мало данных об ути-
лизации шламов по очистке присадок. На практике в боль-
шинстве случаев шламы после очистки присадок сбрасываются
в отвалы [607]. Имеются лишь единичные сообщения по пере-
работке шламов производства присадок.
Так, в процессе получения присадки МНИ ИП-22к на ста-
дии отделения механических примесей вместе с ними удаляет-
ся часть готовой присадки. С целью утилизации присадки и
обезвреживания твердых отходов производства сотрудники
МИНГ им. И. М. Губкина предложили способ переработки
шлама [610]. Отходы экстрагируют бензином «калоша» до пол-
213
ТАБЛИЦА 10.4
Показатель СКН-40М
1 1 2 1 3 1 4 1 5 1
Содержание в каучуке, %: 0,026 0,035
хлорида натрия 0,038 0,041 0,026
неозона «Д» 2,3 2 2 2 2,1
золы 0,19 0,3 0,145 0,11 0,21
никеля 2,07 2,04 1,42 1,27 —
алкилсульфоната натрия — — — — 0,43
связанного НАК 39 39 39 39 38,7
Потери массы каучука при сушке, % 0,19 0,21 0,21 0,19 0,18
Растворимость каучука в мэк, % 100 100 99,9 100 99
ного отделения органической части. Остаток после отгонки
растворителя представляет собой присадку МНИ ИП-22к в
масле. Твердый остаток состоит из неорганической части: окси-
да, гидроксида и карбоната кальция, элементной серы и меха-
нических примесей. Твердый остаток предлагается обрабаты-
вать 10%-ым раствором соляной кислоты с получением нейт-
рального водного раствора хлорида кальция.
Возможность извлечения присадки ИНХП-21 из шламов,
образующихся при ее очистке, показана в работе [10]. На ста-
дии очистки присадки от механических примесей на центробеж-
ных машинах образуется осадок, твердая фаза которого состо-
ТАБЛИЦА 10.5.
Показатель I П
Жесткость каучука по Дефо, Н 6,3 6,1 6,3 6,8 6,6 6,3
Вязкость каучука по Муни 43 42 44 44 43 44
Восстанавливаемость каучука, мл 2,5 2,6 2,7 2,8 2,8 2,7
Жесткость по Дефо резиновой сме- си, мм 8,2 2,6 2,7 2,7 2,7 2,7
Восстанавливаемость резиновой сме- си, мм Подвулканизация по Муни при 140 °C: 2,8 8 9 8,7 8,5 8,6
в течение 5 мин 12 13 14 11 12 12
» 35 мин 18 19 17 18 19 18
Время вулканизации, мин 80 80 80 80 80 80
Условное напряжение при 300%-ом удлинении, МПа 7,4 7,4 8,3 7,6 7,8 7,5
Условная прочность при разрыве, МПа Относительное удлинение, % 28,1 27,2 28,1 27,2 30,4 28,4
696 700 657 677 696 672
Относительное остаточное удлине- ние, % 16 16 14 14 18 16
214
CKH-40 CM
6 1 7 1 8 1 9 1 10
0,05 0,029 0,023 0,027 0,035
2,3 2 2,4 2,1 2
0,3 0,24 0,15 0,095 0,14
— — —
0,51 0,6 0,4 0,62 0,42
38,7 38,7 38,7 38,7 38,7
0,19 0,20 0,19 0,17 0,21
97,4 96,8 96,4 99,4 98,8
ит из сульфида и гидроксида бария, а жидкая — из присадки
растворителя. Испытаниями по экстрагированию осадка бензи-
ном БР-1 была показана возможность извлечения основной
массы присадки из осадка. Суспензию после экстрагирования
разделяют на сепараторе ИСБ, фугат используют для разбав-
ления присадки перед очисткой от механических примесей.
Осадок после экстрагирования содержал сульфид и гидрок-
сид бария. Установлена возможность разложения осадка сла-
бым раствором серной кислоты. После разложения осадка
проводилась нейтрализация избытка кислоты 10%-ым раство-
ром извести при температуре 20—30 °C и непрерывном переме-
шивании. В работе [611] (предложена схема утилизации при-
садки и обезвреживания шлама.
В работе [612] предлагается минеральный остаток, полу-
ченный в результате многоступенчатой экстракции бензином
присадки из шлама, использовать в производстве спеццементов.
При производства присадки ЦИАТИМ-338 образуются от-
ТАБЛИЦА 10.6
Антиоксидант Условная прочность, МПа Относитель- ное удлине- ние Коэффициент старения по
относитель- ному удли- нению условной прочности (484-100 СС)
Резина на основе натурального каучука
НГ-2246 182 630 0,88 0,52
Неозон «Д» 166 631 0,69 0,54
Агидол-2ВП 204 638 0,7 0,55
Резина на основе стирольного каучука
НГ-2246 36 355 0,66 0,7
Неозон «Д» 38 382 0,47 0,38
Агидол-2ВП 38 412 0,52 0,48
215
ходы — шламы, содержащие 40% органической части, в основ-
ном самой присадки, и 60% смеси сульфида и тиосульфида
бария. В работе [613] рассмотрена возможность практического
извлечения солей бария из отходов. Предложена технологичес-
кая схема переработки отходов, которая позволяет полностью
использовать ценные компоненты отходов производства с эко-
номическим эффектом около 350 тыс. руб. в год. При этом
значительно уменьшается количество отходов.
Опыт промышленной эксплуатации схем утилизации шла-
мов очистки сульфонатных присадок на установках ПО «Омск-
нефтеоргсинтез» и «Новополоцкнефтеоргсинтез» подтвердил
возможность переработки шламов и использования полученных
продуктов [618].
Так, при производстве высокощелочной сульфонатной при-
садки НМС для нормального проведения процесса карбоната-
ции необходим избыток гидроксида кальция (30—100% от тео-
ретического в зависимости от условий проведения реакции).
Избыток Са(ОН)2 взаимодействует с СО2, при этом образуется
кристаллический СаСОз, который в дальнейшем при центрифу-
гировании отделяется от присадки как механическая примесь
(шлам) и после регенерации из него бензина выводится на
свалку. В шлам уходит значительное количество присадки (по-
тери составляют 10—15% на исходный продукт при односту-
пенчатом центрифугировании). Для улучшения технико-эконо-
мических 'показателей на стадии центрифугирования было
предложено использовать шлам для нейтрализации кислого
масла, 'получаемого в процессе сульфидирования, взамен из-
вести-пушонки [607].
Для опытов были взяты кислое масло и шлам со следующи-
ми показателями:
Кислое масло
Кислотное число общее, мг КОН/г 55,5
Шлам
Содержание, %:
растворителя 40
кальция 25
воды 4,3
Сульфатная зольность, %, 79
Качество полученного нейтрального сульфоната:
сульфатная зольность, % 55
Содержание, %:
растворителя 28,5
воды 1,5
Вязкость кинематическая при 100 °C, м2/с 63,5
Щелочность (фугованная проба), мг КОН/г 0,8
Степень чистоты, мг О4“2/100 г:
нефугованная проба 8050
фугованная проба 210
216
Использование шлама после центрифуг при нейтрали mmiu
позволило значительно сократить расход гидроксида кальция
и масла сырья, поскольку со шламом на стадию нейтрализа-
ции возвращается непросульфированное масло, которое в даль-
нейшем служит как разбавитель присадки.
Разработан метод комплексной переработки шлама и кис-
лого гудрона производства сульфонатных присадок. Метод за-
ключается в смешении шлама с жидким органическим тепло-
носителем (экстрактом фенольной очистки масел, мазутом,
прямогонным гудроном) в отношении 1 1 и нагревании до
120 °C для отгона толуола. Отогнанный толуол (более 98%)
возвращается в производственный цикл, а шлам при интенсив-
ном перемешивании смешивается с кислым гудроном в отноше-
нии 1 : 1,3. Полученный продукт характеризуется следующими
показателями [607]:
Плотность, г/см2 3 0,99-1,01
Вязкость ВУ (100 °C) 20—58
Теплоемкость, кДж/кг 14 600—19 300
Температура размягчения, °C 57—70
Вспышка в открытом тигле, °C 165—230
Реакция водной вытяжки, pH 5,7—8,7
Содержание серы, % 4—7
Показана возможность использования нейтрального продук-
та в качестве компонента при производстве дорожных вяжу-
щих материалов или в составе спецтоплив.
Сдерживающими факторами при использовании отходов
является отсутствие достоверной информации о количественных
и качественных характеристиках образующихся отходов, чет-
кой системы организационных мер по поиску возможных по-
требителей, цен на отходы и недостаточно эффективный эконо-
мический контроль за использованием отходов. Необходимость
осуществления мероприятий по рациональному использованию
отходов диктуется экономическими и экологическими задачами.
БИБЛИОГРАФИЧЕСКИЙ СПИСОК
1. Забористое В. Н. Утилизация отходов промышленности синтетического
каучука. М.: ЦНИИТЭнефтехим, 1984. 72 с.
2. Кроль В. А., Ривин Э. М., Щербань Г Т. Свойства и применение диено-
вых олигомеров. М.: ЦНИИТЭнефтехим, 1984. 40 с.
3. Щербань Г Т., Жукова М. И., Никулин Н. А., Обрубов В. А. Ресурсо-
сбережение при очистке отходящих газов прэмышленности СК. М.:
ЦНИИТЭнефтехим, 1988. 64 с.
217
4. Василенко Г Д., Грунин Г Н. Переработка и использование отходов
производства присадок к смазочным материалам. М.: ЦНИИТЭнефтехим,
1987. 52 с.
5. Патрушева Е. Н., Пимкина В. А. Внедрение безотходной технологии
в нефтепереработке и нефтехимии. М.: ЦНИИТЭнефтехим, 1979. 47 с.
6. Ривин Э. М., Наровлянский А. К., Петров Л. Н. Пути использования пи-
перилена//Основной органический синтез и нефтехимия: Сб. науч. тр.
ЯПИ. Ярославль: ЯПИ, 1981. № 15. С. 8—17.
7. Крючков А. //.//Общая технология синтетических каучуков. М.: Химия,
1969. С. 143—177.
8. Литвин О. ^.//Основы технологии синтеза каучуков. М.: Химия, С. 43—
58.
9. Кирпичников П. А., Береснев В. В., Попова Л. М. Альбом технологиче-
ских схем основных производств промышленности синтетического каучука.
Л.: Химия, 1976. 112 с.
10. Кирпичников П. А., Лиакумович А. Г., Победимский Д. Г., Попова Л. №.//
Химия и технология мономеров для синтетических каучуков. Л.: Химия,
1981. 264 с.
11. Бакша Ю. М., Гурьянова Р. Н., Данилова Н. К-, Гельбштейн А. //.//Про-
мышленность СК. 1976. № 1. С. 6—8.
12. Корпачева А. Л., Горшков В. А., Кириллова Г. А., Павлов С. Ю.//Там же.
1976. № 11. С. 1—2.
13. Тушканов С. Н., Рожкова А. Я.//Там же. 1970. № 11. С. 1—5.
14. Храпкова Э. И., Калиничева Л. А.//Там же. 1970. № 1. С. 12—13.
15. Большаков Д. А., Бушин А. И., Кирнос Я. Я. и др.//Там же. 1977. № 1.
С. 3—5.
16. Блажин Ю. М., Ватина Л. К-> Елчанинова Б. В. и др.//Там же. 1980.
№ 6. С. 9—11.
17. Огородников С. К., Идлис Г. С. Производство изопрена. Л.: Химия, 1973.
296 с.
18. Дружинина Е. А., Гагина Э. А., Силкина А. Г. и ^.//Промышленность
СК. 1978. № 11. С. 18—20.
19. Фарберов М. И., Кутьин А. М., Кашинский Г. И.//ЖОХ. 1960. Т. 30.
С 883
20. Гершкович Ж-, Дувалма М.//Там же. 1962. Т. 32. Вып. 12. С. 3987—
3997.
21. Евдокимова С. П., Немцов М. С., Исакова И. Ф.//Нефтехимия. 1966.
Т. 6. № 1. С. 75.
22. Евдокимова С. П., Немцов М. С., Исакова И. Ф.//Там же. 1967. Т. 7.
№ 2. С. 235.
23. Кузнецова Е. В., Тургель Е. О., Тараненко С. А.//ЖАХ. 1973. К° 8.
Вып. 28. С. 1582—1587.
24. Артюхов И. М., Безмозгин Э. С., Петров В. И. и др.//Промышленность
СК. 1975. № 12. С. 3—5.
25. Смолин Ю. И., Вальдман Д. И., Дербишер В. Е. и др.//Там же. 1980.
№ 4. С. 2—4.
26. Клаузен Н. А., Семенова Л. П. Атлас инфракрасных спектров каучуков
и некоторых ингредиентов резиновых смесей. М.: Химия, 1965. 362 с.
27. Лоншакова Т И., Лиакумович А. Г. Перспективы использования пипери-
лена. М.: ЦНИИТЭнефтехим, 1982. 87 с.
28. Онищенко А. С. Диеновый синтез. М.: АН СССР, 1963. 650 с.
29. Артюхов И. М., Безмозгин Э. С., Петров В. Н. и ^.//Промышленность
СК. 1975. № 12. С. 3-5.
30. Заявка 2514762 ФРГ МКИ С 07 С 11/18. Способ обработки высококи-
пящих побочных продуктов, образующихся при получении изопрена.
31. А. с. 1028690 СССР, МКИ С 08 L 9/02. Резиновая смесь.
32. А. с. 990767 СССР, МКИ С 08 L 9/00. Вулканизуемая резиновая смесь
на основе ненасыщенного каучука.
218
33. А. с. 1087535 СССР, МКИ С 08 F 236/10, С 08 С 2/02. Способ получения
модифицированного бутадиенстирольного пленкообразующего.
34. Белов П. С. Основы технологии нефтехимического синтеза. М.. Химия,
1982. 280 с.
35. Липович В. Г., Полубенцева М. Ф. Алкилирование ароматических углево-
дородов. М.: Химия, 1985. 272 с.
36. А. с. 887557 СССР, МКИ С 07 С 5/27, С 07 С 2/66. Способ переработки
кубового продукта процесса каталитического алкилирования бензола эти-
леном.
37. Адельсон С. В., Вишнякова Т. П., Паушкин Я- В. Технология нефтехими-
ческого синтеза. М.: Химия, 1985. 607 с.
38. Черный И. Р. Производство мономеров и сырья для нефтехимического
синтеза. М.: Химия, 1973. 264 с.
39. Юкельсон И. И. Технология основного органического и нефтехимического
синтеза. М.: Химия, 1981. С. 442—445.
40. Брянцева Ю. В., Корчагина О. Н., Золотарева 3. В. и др.//Каучук и ре-
зина. 1959. № 4. С. 32—35.
41. Юкельсон И. И., Гугняева Л. И., Концова Л. В.//Лакокрасочные мате-
риалы л их применение. 1970. № 5. С. 18—20.
42. Алиев Д. А., Алиева С. Г., Дубровко Р. В.//Нефтеперераб. и нефтехимия.
1964. № 12. С. 32—36.
43. Юкельсон И. И., Гугняева Л. И., Концова Л. В., Леутина В. //.//Лако-
красочные материалы и их применение. 1967. № 2. С. 18—19.
44. Юкельсон И. И., Бутенко Т. Р., Бердутин А. Я.//Там же. 1979. № 4.
С. 9—10.
45. Сафонов В. С., Гладышев Н. Г., Дубровина В. А., Правдивцева 3. А.//
Хим. пром. 1983. № 11. С. 649—651.
46. Лебедев Н. Н. Химия и технология основного органического и нефтехи-
мического синтеза. М.: Химия, 1981. С. 442—445.
47. Слюсарева Л. Ю., Береговых В. В., Львов С. ^.//Промышленность СК.
1979. № 5. С. 3—5.
48. Румянцев А. В., Алексеева Н. А., Гимадеева Л. И., Туманова Г. А.//Там
же. 1968. № 4. С. 17—18.
49. Могилевич М. М., Туров Б. С., Морозов Ю. Л. Жидкие углеводородные
каучуки. М.: Химия, 1983. С. 6—19.
50. Долгоплоск Б. А., Маковецкий К- Л., Тинякова Е. И., Шараев О. К.
Полимеризация диенов под влиянием л-аллильных комплексов. М.: Нау-
ка, 1968. 159 с.
51. Кирпичников П. А., Аверко-Антонович Л. А., Аверко-Антонович Ю. О.
Химия и технология синтетического каучука. Л.: Химия, 1975. С. 246—
312.
52. Соболев В. М., Бородина И. В. Промышленные синтетические каучуки.
М.: Химия, 1977. С. 118—125. ' '
53. Сотников И. Ф., Ривин Э. М., Пожидаев В. .4.//Промышленность СК.
1969. № 5. С. 12—15.
54. Баснер М. Е., Бажанов Ю. В., Нефедова А. И., Титова Л. Ф.//Там же
1978. № 11. С. 2—4.
55. Юрьев В. П., Салимгареева И. М., Толстиков Г. А.//Химия и физикохи-
мия мономеров. Уфа: БФАН СССР, 1975. С. 5—21.
56. Кроль В. А., Ривин Э. М., Щербань Г Т. Свойства и применение диено-
вых олигомеров. М.: ЦНИИТЭнефтехим, 1984. 40 с.
57. Брикенштейн М. А., Белов Г П., Герасина М. П. Разработка научных
основ технологии процессов полимеризации, олигомеризации и димериза-
ции этилена на комплексных катализаторах. М.: ИХФ АН ССС?. Черно-
головка. 1983. 30 с.
58. Баркова Т. В., Дроздов В. А., Карпова Л. В и ^.//Промышленность
СК. 1978. № 12. С. 14—16.
59. Стереорегулярная полимеризация./'Под ред. У. Солтмана. М.: Мир, 1981
4.1. С. 17-89.
219
60. Ривин Э. М., Дымент JJ. О., Кузнецова Б. А. Синтетические каучуки об-
щего назначения. М.: ЦНИИТЭнефтехим, 1982. 61 с.
61. Синтетический каучук/Под ред. И. В. Гармонова. Л.: Химия, 1983.
С. 140—141.
62. Васышак М. О.//Труды МИТХТ. 1974. Т. 4. № 1. С. 72—77.
63. Васышак, М. О., Аносов В. И., Домогатская М. //.//Промышленность СК.
1978. № 4. С. 7—9.
64. Сире Е. М., Поспелова Л. М., Евдокимова 3. Х.//Там же. 1982. № 4.
С. 6—9.
65. Баркова Т. В., Дроздов В. А., Карпова Л. В. и др.//Там же. 1978. № 12.
С. 14—16.
66. Ziegler K.//Kngew. Chem. 1964. V. 76. N 13. P. 545—553.
67. Касаткина H. Г.//Вестник ЛГУ. 1969. № 16. С. 140.
68. Кроль В. А., Парфенова Г А., Бурова Г. В.//ДАН СССР. 1969. Т. 185.
№ 2. С. 398—400.
69. Кроль В. А. Исследование процессов стереоспецифической полимериза-
ции бутадиена, разработка и реализация промышленного способа получе-
ния СКД: Автореф. дис. д-ра хим. наук: 05.17.04. М.: ИНХС АН
СССР, 1972. 36 с.
70. Баженов В. Д., Коноваленко Н. А., Щербань Г. Т., Аносов В. Я.//Про-
мышленность СК. 1981. № 12. С. 5—7.
71. Климова Т. А., Караблина И. В., Щеглова Л. Н.//Там же. 1983. № 6.
С. 21—23.
72. Кроль В. А., Парфенова Г. А., Карелина Р. Я.//Высокомол. соединения.
1974. Т. Б16. № 16. С. 746—748.
73. Soltman W.f Gibbs W., Lal J.//У Am. Chem. Soc. 1958. V. 80. N 21.
P. 5612—5622.
74. Adams H.t Smith R.t Binder K.//Ind. Eng. Chem. 1958. V. 50. P. 1507—
1510.
75. Бурова Г. В., Гаромонов И. В., Дроздов В. А. и др.//Промышленность
СК. 1979. № 9. С. 9—12.
76. Хачатуров А. С., Абраменко Е. Л., Дроздова В. А//ЖВХО. 1982. Т. 27.
№ 4. С. 472—474.
77. Cunliffe A. V., Grinter R.t Harris В. К.//У Magn. Reson. 1970. V. 2.
Р. 200.
78. Albriksten Р., Harris R. K.//Acta Chem. Seand. 1973. V. 27. P. 1875.
79. Ohloff G., Seibl J„ Kocits E.//Annal. Chew. 1964. V. 675. P. 83.
80. Онищенко А. С. Диеновый синтез. M.: АН СССР, 1963. 650 с.
81. Назаров И. Н., Кузнецова А. И., Кузнецов Н. В.//ЖОХ. 1955. Т. 25.
С. 307.
82. Мономеры и продукты нефтехимического синтеза: Сб. тр. МИНХиГП.
М.: Химия, 1967. № 72. С. 23—28.
83. Заявка 2300750 Франция, МКИ С 07 С 13/27, 3/035. Способ получения
тримеров изопрена.
84. Заявка 1429036 Великобритания, МКИ С 07 С 11/21, С 07 С 3/10.
Триены-1,3,7 и способ их получения.
85. Заявка 50—24924 Япония, МКИ С 07 С 11/21. Способ получения алло-
цимена.
86. Заявка 50—24922 Япония, МКИ С 07 С 11/21. Способ получения тримеров
изопрена.
87. П. де Майо. Терпеноиды. М.: ИЛ, 1963. 494 с.
88. Вилке Т.//Успехи химии. 1964. Т. 33. № 6. С. 677—706.
89. Кроль В. А., Парфенова Г А., Бурова Г. В. и (^.//Промышленность СК.
1972. № 8. С. 9—12.
90. Кроль В. А., Парфенова ГА., Лапшина А. Н. и др.//ЖПХ. 1972. Т. 45,
№ 8. С. 1803—1808.
91. Сотников И. Ф., Кудрявцев Л. Д., Пожидаев В. А. и др.//Промышлен-
ность СК. 1970. № 12. С. 9.
220
92. Паушкин Я. М., Шоноров В. И., Пономаренко В. И. и др.//Там же. 1969.
№ 11—12. С. 3—5.
93. Заявка 3045229 ФРГ, С 07 С 13/20, 2/44. Способ термической димериза-
ции бутадиена.
94. Габиров Ф. И., Габиров И. И., Бабаев Н. М., Новрузов С. А. К вопросу
получения 4-винилциклогексена термической димеризацией бутадиена.
Баку: 1983. 7 с. Деп. в АзНИИНТИ 4.11.83, № 128 Аз.
95. Заявка 2548428 ФРГ, С 07 С 3/035. Способ димеризации бутадиена.
96. Hillyer J. С., Smith I. V.//Ind, Eng. Chem. 1953. V 45. N 5. P. 1133.
97. A. c. 730669 СССР, МКИ С 07 С 13/27. Способ получения 1,5,9-цикло-
додекатриена.
98. Пат. 2034183 ФРГ, МКИ С 07 С 13/277, 2/46, В 01 31/14. Способ полу-
чения циклододекатриена-1,5,9.
99. А. с. 887559 СССР, МКИ С 07 С 13/277. Способ получения циклододе-
катриена-1,5,9.
100. Заявка 3021795 ФРГ, МКИ С 07 С 13/277, С 07 С 2/46. Способ получе-
ния циклододекатриена-1,5,9.
101. Заявка 302Г840 ФРГ, МКИ С 07 С 13/277, С 07 С 2/46. Способ получения
циклододекатриена-1,5,9.
102. Юрьев В. П., Толстиков Г, А., Иванов Г Е., Антонов А. А.//Нефтехимия.
1974. Т. XIV. № 2. С. 232—235.
103. Ривин Э. М., Моисеев В. В., Кузнецова Б. А. и др. Бутадиеннитрильные
каучуки. М.: ЦНИИТЭнефтехим, 1982. 68 с.
104. Синтетический каучук/Под ред. И. В. Гаромонова. Л.: Химия, 1983.
С. 300—310.
105. Nakagawa Kiyoshi, Sawai Michimasa, Ishii Yasutaka, Ogawa Masaya//
Bull. Chem. Soc. Jap. 1977. V. 50. N 9. S. 2487—2488.
106. Atsasa Masataka, Sydzuki Joiti//}. Chem. Soc. Jap. In. Chem. Soc. 1971.
V. 74. N 3. P. 401—406.
107. Рзаев 3. M., Садых-заде С. И., Брыскина Л. В.//Высокомол. соединения.
1974. T. Б16. № 1. С. 8—11.
108. Хамлеуди X., Мехтиев С. И., Полчаев О. А.//Азерб. хим. журнал. 1981.
№ 2. С. 68—71.
109. Анастасъева А. П., Верещагин А. Н., Арбузов Б. А.//Изв. АН СССР.
Сер. хим., 1970. № 7. С. 1485—1491.
110. Вассерман А. Реакция Дильса — Альдера. М.: Мир, 1968. 134 с.
111. Янц Данкан//Природа. 1953. Т. 171. № 4360. С. 933.
112. А. с. 659567 СССР, МКИ С 07 С 121/48. Способ получения 4-цианцик-
логексена-1 и 1-метил-4 (3)-цианциклогексена-1.
113. Пат. 49—173 Япония, МКИ С 07 С 69/74. Получение продуктов цикли-
ческой конденсации.
114. Онищенко А. С. Диеновый синтез. М.: АН СССР, 1963. 650 с.
115. Muller И. К, Hamm G.//Wiss, z. Techn. Hochsch. О. Gueriche Magdeburg.
1970. V. Al4. N 8. S. 877—884.
116. Guzin D., Chavin J., Leterbre GJ/Emop. Polym. Joum. 1967. V. 3. P. 367.
117. Гостев M. M., Тертышник Г. В., Рысляев Г Ф., Кохановская Н. И.//
Каучук и резина. 1980. № 7. С. 10—12.
118. Пат. 4375571 США, НКИ 585—431. Process for the preparation of ethyl-
benzene from 4-vinylcyclohexene.
119. Пат. 4165441 США, НКИ 585—444. Process for the preparation of styrene.
120. Малиновский M. С. Окиси олефинов и их производные. М.: ГНТИ, 1961
554 с.
121. Тепеницына Е. Н., Дормидонтова С. Н., Фарберов М. И.//Уч. зап. Ярос-
лав. технол. ин-т/Ярославль, 1971. Т. 27. С. 35.
122. Дормидонтова С. Н., Тепеницына Е. Н., Сомова Г //.//Основной орга-
нический синтез и нефтехимия: Сб. науч. тр. ЯПИ. Ярославль- ЯПИ,
1974. Вып. 1. С. 52—56.
123. Sheng М. V.. Zajacek J. GJ/У Org. Chem. 1971. V 36. Р 1839
124. Carlson R. G„ Belin N. S.//Ibid. 1971. V 36. P. 3832.
221
125. Мухамедова Л. А., Малышко Т. М., Шагидуллин Р. В.//ДАН СССР.
1965. Т. 160. С. 1323.
126. Дормидонтова С. Н., Тепеницына Е. Н., Сомова Г. Н.//Основной орга-
нический синтез и нефтехимия: Сб. науч. тр. ЯПИ. Ярославль: ЯПИ,
1975. Вып. 2. С. 27—32.
127. Пакен А. М. Эпоксидные соединения и эпоксидные смолы. Л.: ГНТП.
1962. 964 с.
128. Кошель Г Н., Антонова Т. Н., Фарберов М. И. и ^.//Основной органи-
ческий синтез и нефтехимия: Сб. науч. тр. ЯПИ. Ярославль: ЯПИ, 1975.
Вып. 3. С. 25—30.
129. Захаркин Л. И., Корнева В. В.//ДАН СССР. 1960. Т. 132. С. 1078.
130. Цысковский В. /(.//Пластмассы. 1969. № 6. С. 18—20.
131. А. с. 144844 СССР, Кл. 12о, 25. Способ получения циклододеканона.
132. Бышкиров А. Н., Камзолкин В. В., Сокова К- М. и др.//Нефтехимия.
1961. Т. 1. С. 527—534.
133. Захаркин Л. И., Корнева В. В., Куницкая Г //.//Изв. АН СССР. Сер.
хим. 1961. № 9. С. 1908—1909.
134. Пат. 3607923 США, НКИ 260—531. Process for preparing dibasic acids.
135. Антонова T. H., Кошель Г Н., Коленкова А. //.//Основной органический
синтез и нефтехимия: Сб. науч. тр. ЯПИ. Ярославль: ЯПИ, 1979. Вып. 12.
С. 112—116.
136. Березин И. В., Быковченко В. Г Мелузова Г. ^.//Нефтехимия. 1961.
Т. 1. С. 535.
137. Проскурин И. В., Ермакова В. В., Парамонова 3. И. Изучение макроки-
нетики процесса гидрирования циклододекатриена-1,5,9 на никель-хро-
мовом катализаторе. М., 1983. 10 с. Деп. в ОНИИТЭХИМ. 27.05.83,
№ 574хп.
138. Горбунов Б. И., Гурвич Я. И., Маслова И. П. Химия и технология ста-
билизаторов полимерных материалов. М.: Химия, 1981. 368 с.
139. Данишевский В. Н. Автореф. дис. канд. хим. наук: 02.00.13. Баку:
ИНХП АН АзССР. 1981. 22 с.
140. Белов П. С., Меликян В. Р., Тонконогов Б. П., Львова Е. М//Нефтспе-
реработка и нефтехимия. 1977. № 3. С. 28—29.
141. Белов П. С., Меликян В. Р., Тонконогов Б. П., Львова Е. М.//Т&3. докл.
всесоюз. конф. «Превращения углеводородов на кислотно-основных гете-
рогенных катализаторах». Грозный, 1977. С. 147—149.
142. Белов П. С., Тонконогов Б. П., Меликян В. Р., Львова Е. М.//Т&3. докл.
польско-советского науч, симпозиума «Новейшие успехи химии гетеро-
циклических соединений кислорода». Торунь, 1977. С. 100.
143. Тонконогов В. //.//Нефть и газ: Сб. науч. тр. МИНХиГП. М.: МИНХиГП,
1977. С. 114—115.
144. Тонконогов Б. //.//Материалы Всесоюз. совещ. «Повышение качества
нефти и продуктов ее переработки». Москва, 1977. С. 40.
145. Белов П. С., Меликян В. Р., Тонконогов Б. П.//Тез. докл. III Всесоюз.
науч. конф, по химии и технологии фурановых соединений. Рига, 1978.
С. 82.
146. Тонконогов Б. П. Исследования в области взаимодействия фенолов с
4-винилциклогексеном: Автореф. дис. канд. хим. наук: 02.00.13. М.:
МИНХиГП, 1979. 24 с.
147. Марио Родригес де Сантьяго. Исследование реакции взаимодействия эфи-
ров фенолов с 4-винилциклогексеном: Автореф. дис. канд. хим. наук:
02.00.13. М.: МИНХиГП, 1982. 25 с.
148. Мехтиев С. Д., Грифель Б. Ю., Шигаев И. У и др.//Азерб. хим. журнал.
1972. № 3. С. 8.
149. Яблонский О. П., Мехтиев С. Д., Трифель Б. Ю. и др.//Тгм же. 1973.
№ 1. С. 66.
150. Садых-заде С. И., Трифель Б. Ю., Шигаев И. У. и др.//Там же. 1973.
№ 5. С. 58.
222
151 Садых-заде С. И., Грифель Б. Ю., Шигаев И. У. и др.//Высокомол. соеди-
нения. 1973. Т. А15. № 12. С. 2782.
152. Мамедов Ф. В., Шагаев И. У., Грифель Б. Ю. и ^.//Изучение и опти-
мизация процесса привитой сополимеризации ВЦГ с БК в присутствии
перекиси бензоила: Уч. зап./Азерб. ин-т нефти и химии. Баку: АЗИНЕФ-
ТЕХИМ, 1975. Сер. IX. № 6. С. 112.
153. Юсуфов О. Г Шихализаде П. О., Грифель Б. /О.//Электронно-микро-
скопические исследования бутилкаучука, модифицированного непредель-
ными циклическими соединениями: Ученые записки/Азерб. ин-т нефти
и химии. Баку: АЗИНЕФТЕХИМ, 1976. Сер. IX. № 8. С. 71.
154. А. с. 293815 СССР, МКИ С 08 d 5/02. Способ получения привитых сопо-
лимеров.
155. А. с. 327212 СССР, МКИ С 08 F 255/08. Способ получения привитого
сополимера.
156. Догадкин Б. А., Донцов А. А., Шершнев В. А. Химия эластомеров. М.:
Химия, 1981. 374 с.
157. Лившиц И. А., Коробова Л. М., Ковалева Г. А., Рейх В. //.//Каучук и
резина. 196§. № 7. С. 1—3.
158. Вассерман А. Реакция Дильса — Альдера. М.: Мир, 1968. 134 с.
159. Рзаев 3. М. Полимеры и сополимеры малеинового ангидрида. Баку: Элм,
1984. 158 с.
160. Антонова Л. Ф., Реймер Л. Г.//Химия и физикохимия высокомолекуляр-
ных соединений: Сб. науч. тр. БФ АН СССР. Уфа: БФ АН СССР, 1976.
185—196 с.
161. Tsuchida Е., Tsumudzi Г., Готово SJ/У Chem. Soc. Japan. Ind. Chem.
Soc. 1969. V 72. P. 765.
162. Стоцкая Л. Л., Орешкина Г А., Башкатова С. Г., Кренцель Б. А.//Тез.
докл. конф. «Полимер-71». Варна, 1971. С. 61.
163. Башкатова С. Т„ Ожерелев В. И., Клейнер В. И. и др.//Высокомол.
соединения. 1972. Т. А14. № 12. С. 2640—2646.
164. Рзаев 3. М., Садых-заде С. И., Брыкина Л. В.//Там же. 1974.
165. Новое в чередующейся сополимеризации: Сб. науч. тр. ИНХС АН СССР.
М. 1984, С. 102—120.
166. Заявка 3049432 ФРГ, МКИ С 08 F 2/06. Verfahren zur Herstellung von
radikalisch initiierten Copolymerisaten aus Maleinsaureanhydrid und Ole-
finen bzw. deren Gemische.
167. Заявка 3046648 ФРГ, МКИ С 08 F 8/44. Verfahren zur Herstellung von
wassrigen Salzlosungen der Copolymeren aus Maleinsaureanhydrid und
Olefinen bzw. deren Gemische.
168. Заявка 3046647 ФРГ, МКИ С 08 F 8/44. Verfahren zur Herstellung von
wassrigen Salzlosungen der Copolymeren aus Maleinsaureanhydrid und
Olefinen bzw. deren Gemische
169. Пат. 55—41257 Япония, МКИ С 08 L 13/00, С 08 К 3/20.
170. Леплянин Г. В., Подгородецкая В. А., Рафиков С. Р., Ибрагимова Ф. Ш.//
Изв. АН СССР. Сер. хим. 1978. № 5. С. 1073.
171. Duck W., Locke J. М., Thomas М. E.//Polym. 1968. V. 9. Р. 60.
172. Choshi Y., Tsuji A., Akazoma G„ Mirai K//L Chem Soc. Japan. Ind. Chem.
Soc. 1968. V. 71. N A58. P 908.
173. Николаев А. Ф., Андреева M. А.//Высокомол. соединения. 1968. T. АЗ.
С. 502—506.
174. Шейхет Ф. И. Материаловедение химикатов, красителей и моющих
средств. М.: Легкая индустрия, 1969. С. 150—154.
175. Науменко И. В. Синтетические жирозаменители, поверхностно-активные
вещества и моющие средства. М : ГОСИНТИ, 1960. С. 76—82.
176. Кеннеди Дж. Катионная полимеризация олефинов. М.: Мир, 1978. 430 с.
177 Гостев М. М.. Никулин С. С., Шеин В. С., Акиньшина А. //.//Промышлен-
ность СК, пшн и РТИ. 1984. № 6. С. 7—9.
178. Джильберт 3. Е. Сульфирование органических соединений. М • Химия,
1969.414 с
223
179. Оаэ Сигэру. Химия органических соединений серы. М.: Химия, 19-’
512 с.
180. Реакция серы с органическими соединениями/Под ред. М. Г. Воронкова.
Новосибирск: Наука, 1979. 368 с.
181. Никулин С. С., Шеин В. С.//Промышленность СК, шин и РТИ. 1986.
№ 10. С. 6—7.
182. Глазков С. С., Никулин С. С., Шеин В. С., Черкашин М. И.//Изв. ВУЗов
СССР. Химия и хим. технол. 1986. № 10. С. 133—136.
183. Казицина Л. А., Куплетская Н. ^.//Применение УФ-, ИК- и ЯМР-спект-
роскопии в органической химии. М.: Высшая школа, 1971. 251 с.
184. Пат. 45941 ПНР, kl. 22h 71/01. Sposob otrzymywania wysokowartoscio-
wych lakierdw asfaltowych.
185. Печуро H. С., Меркурьев A. H., Аксенова T. ^.//Промышленность CK.
1980. № 2. C. 5—7.
186. Печуро H. E., Меркурьев A. H., Былинкин A. H. Термическая переработка
жидких органических отходов производства дивинила двухстадийным
дегидрированием н-бутана//Основной органический синтез и нефтехимия:
Сб. науч. тр. ЯПИ. Ярославль: ЯПИ, 1977. Вып. 7. С. 39—42.
187. Печуро Н. С., Меркурьев А. Н., Торловский В. Н., Комаров В. //.//Про-
мышленность СК. 1973. № 6. С. 14—16.
188. Печуро Н. С., Меркурьев А. Н., Горловский В. Н. Пиролиз жидких от-
ходов производства изопрена//Основной органический синтез и нефтехи-
мия: Сб. науч. тр. ЯПИ. Ярославль: ЯПИ, 1976. Вып. 5. С. 99—103.
189. Печуро Н. С., Меркурьев А. Н., Горловский В. Н. и ^.//Промышлен-
ность СК. 1973. № 5. С. 9—11.
190. Печуро Н. С., Меркурьев А. Н., Комаров В. П. и др.//Там же. 1975. № 10.
С. 12—13.
191. Печуро Н. С., Комаров В. П., Меркурьев А. Н., Кузовлева Н. И.//Нефте-
химия. 1970. Т. X. № 3. С. 350—358.
192. Печуро Н. С., Меркурьев А. Н., Комаров В. //.//Нефть и газ. 1971. № 8.
С. 57.
193. Аланова Г. Г., Мягкова А. А., Орлова Л. //.//Промышленность СК. 1971.
№ 4. С. 8—10.
194. Аланова Г. Г., Мягкова А. А., Орлова Л. Н.//Гзм же. 1971. № 11.
С. 6—8.
195. Шаталов В. П., Хромыл Б. С., Григорьев В. Б., Еремеев В. И.//Т&М. же.
1971. № 8. С. 7—9.
196. Шпанцева Л. В., Орлянский В. В., Жолобова Л. И.//Гам же. 1978. № 10.
С. 15—16.
197. Харлампович Г. Д., Чуркин Ю. В. Фенолы. М.: Химия, 1974. 376 с.
198. Заявка 59—7124 Япония, МКИ С 07 С 23/02, С 07 С 17/06. Preparation
of Hexabromcyclododecane.
199. Пат. 49—24474 Япония, МКИ С 07 С 23/02, С 07 С 17/02. Способ полу-
чения кристаллического гексабромциклододекана.
200. Пат. 49—24475 Япония, МКИ С 07 С 23/02. Способ получения кристал-
лического гексабромциклододекана бромированием транс-, транс-, цис-щ\к-
лододекатриена.
201. Пат. 1449976 Англия, НКИ С 2 С (МКИ С 07 С 17/02). Bromination of
ansatarated hydrocarbons in mixed solyents.
202. Чирков H. M., Матковский П. E., Дьячковский Ф. С. Полимеризация на
комплексных металлоорганических катализаторах. М.: Химия, 1976. 416 с.
203. Фельдблюм В. Ш. Синтез и применение непредельных циклических угле-
водородов. М.: Химия, 1982. 207 с.
204. Юрьев В. П., Толстиков Г А., Кучин А. В. Реакция гндроалюминирова-
ния олефинов//Химия и физикохимия мономеров: Сб. науч. тр. БФ АН
СССР. Уфа: БФ АН СССР, 1975. С. 22—36.
205. Хананашвили Л. М., Андрианов К. А. Технология элементоорганических
мономеров и полимеров. М.: Химия, 1983. 414 с.
224
206. Юрьев В. П., Гайлюнас Г. А., Толстиков Г. А.//ЖОХ. 1972. Т. 42. С. 2111.
207. Толстиков Г. А., Юрьев В. П. Алюминийорганический синтез. М.: Наука,
1979. 290 с.
208. Салимеареева И. М., Богатова Н. Г Жебаров О. Ж- и др.//ДАН СССР.
1981. Т. 261. № 1. С. 118—120.
209. Рафиков С. Р., Салимеареева И. М., Богатова Н. Г. и др.//Изв. АН
СССР. Сер. хим. 1982. № 4. С. 920—925.
210. Салимеареева И. М., Богатова Н. Г., Панасенко А. А. и др.//Там же.
1983. № 7. С. 1605—1612.
211. Богатова Н. Г Реакции присоединения германийорганических соединений
к 1,3-диенам: Автореф. дис. на соискание уч. степени канд. хим. наук.
02.00.03. Уфа: БФ АН СССР, 1984. 18 с.
212. Фельдблюм В. Ш. Димеризация и диспропорционирование олефинов. М.:
Химия, 1978. 208 с.
213. Пат. 3865888 США, НКИ 260—666.
214. Толстиков Г. А., Юрьев В. П., Салимеареева И. М., Кучин А. В.//Изв.
АН СССР. Сер. хим. 1974. № 7. С. 1631—1632.
215. Юрьев В. fl., Толстиков Г. А., Салимгареева И. М. и др.//ЖОХ. 1973.
Т. 43. С. 2071.
216. Толстиков Г. А., Юрьев В. П., Кучин А. В. и др.//ДАН СССР. 1974.
Т. 215. № 6. С. 1397—1399.
217. Теддер Дж., Нехватал А., Джубб А. Промышленная органическая химия.
М.: Мир, 1977. 626 с.
218. Щетинская О. С., Седова С. М., Весельчакова Т. Л., Гольдман А. М.//Хни.
пром. 1976. № 7. С. 486—493.
219. Заявка 2218314 Франция, МКИ С 07 С 51/32. Preparation de lacide
/г-butane-tricarboxylique.
220. Росси П. П., Майони Ф.> Далл Аста Дж.//Нефть, газ и нефтехимия за
рубежом. 1979. № 8. С. 96—99.
221. Зильберман Е. П. Реакции нитрилов. М.: Химия, 1973. 447 с.
222. The Chemistry of the cianogroup/Ed. Z. Rappoport. London — New York —
Sydney — Toronto: Intersci. Publ., 1970. P. 258—270.
223. Лычкин И. П., Тарасевич Т. В., Квашнина С. П., Тиме Т. А. Сернокислот-
ная гидратация 4-цианоциклогексена. Воронеж, 1983. 8 с. Деп. в
ОНИИТЭхим, Черкассы. 29.06.83, № 674хп.
224. Пат. 3535367 США, НКИ 260—468. Process for synthesis of 4-hydro-
xycyclohexenecarboxylic acid and derivatives thereof.
225. Лычкин И. П., Тарасевич Т. В., Квашнина С. П., Тиме Т. А.//Изв. ВУЗов
СССР. Химия и хим. технол. 1985. Т. 28. Вып. 4. С. 23—25.
226. Лычкин И. П., Тарасевич Т. В., Квашнина С. П., Тиме Т. А. Получение
гексилового эфира циклогексен-3-карбоновой кислоты из ее нитрила. Во-
ронеж, 1982. 10 с. Деп. в ОНИИТЭхим, Черкассы. 11.03.82, № 305хп.
227. Гордон Г Я.//Стабилизаторы синтетических полимеров. М.: ГНТИ, 1963.
С. 250.
228. Тарасевич Т. В., Сухов В. С., Квашнина С. П., Мухина К. М.//Пластмас-
сы. 1985. № 6. С. 23—24.
229. Алферова И. К. Синтез эпоксидных соединений — стабилизаторов и мо-
номеров для пластмасс. М.: ЦНИИПИ, 1965. 171 с.
230. Ученые записки Азерб. ун-та. Сер. хим. Баку: Азерб. ун-т, 1971. № 3.
Q 52_______55
231. Пат. 169036 СССР, МКИ С 07 С, С 08 G. Способ получения эпоксидных
соединений.
232. Баточ А. Е., Белая Э. С. Циклоалифатические эпоксидные смолы. М.:
ЦНИИТЭхим, 1978. 42 с.
233. Bergmann Herman//J. Appl. Chem. 1953. V 3. N 1. P. 42—48.
234. Физер Л., Физер M. Органическая химия. Т. 2. М.: Химия, 1970. 799 с.
235. Установление структуры органических соединений физическими и хими-
ческими методами: Пер. с англ./Под ред. Я. М. Варшавского и И. Ф. Лу-
ценко. Кн. I. М.: Химия, 1967. 531 с.
15—627
225
236. Юкельсон И. И. Технология основного органического синтеза. М.: Химия,
1968. 846 с.
237. Коршак В. В., Виноградова С. В. Неравновесная поликонденсация. М.:
Наука, 1972. 696 с.
238. Гасса Фахид, Мехтиев С. Я.//Изв. ВУЗов СССР. Нефть и газ. 1980.
№ 4. С. 52—54.
239. Аль-Фахид Гасса Ибрагим. Исследование реакции селективного гидриро-
вания ненасыщенных алифатических и алициклических нитрилов: Авто-
реф. дис. канд. хим. наук 02.00.13. Баку: АЗИНЕФТЕХИМ, 1980.
24 с.
240. Пат. 1072241 ФРГ, Кл. 12 о 25. Способ получения гексагидробензил-
амина.
241. А. с. 182728 СССР, МКИ С 07 С. Способ получения вторичных нафте-
новых аминов.
242. Пат. 3143570 США, НКИ 260—563. Получение алициклических диаминов.
243. Ученые записки Казанского гос. ун-та//Сб. науч. тр./Казанский гос. ун-т.
Казань: 1953. Т. 113. № 8. 200 с.
244. Klein /.//Bull. Res. Council. Israel. 1962. V All. N 1. P 11.
245. Пат. 1048575 ФРГ, Кл. 12 о 25. Ferfahren zur Herstellung von Cyclohexen-
carbonsaureamiden.
246. Пат. 223 Япония, НКИ 16 E 72. Получение 1,2,5,6-тетрагидробензгуан-
амина.
247. Oda Pehay, Morita Iosimi//J. Chem. Soc. Japan. Ind. Chem. Soc. 1961.
V. 64. N 4. P. 659—661.
248. Пат. 3051736 США, НКИ 260—438. Комплексы одновалентной меди.
249. Adamaca Masataky, Sudzyky loty//S. Chem. Soc. Jap. Ind. Chem. Soc.
1971, V. 74, № 5, P. 1037—1039.
250. Пат. 4339 Япония, НКИ 16 С 865. Самоокисление нитрила циклогексен-3-
карбоновой кислоты.
251. Пат. 33055 Япония, НКИ 16 С 865. Способ получения цианоциклогекса-
нола.
252. Пат. 3642833 США, НКИ 260—348.5. Эпоксидирование олефинов.
253. Пат. 1517908 Англия, МКИ С 07 D 301/02. Боридный катализатор для
эпоксидирования олефиновых соединений.
254. Пат. 4038290 США, МКИ С 07 D 301/20. Боридный катализатор для
эпоксидирования эпоксидных соединений.
255. Пат. 4038292 США, МКИ С 07 D 301/20. Боридный катализатор эпок-
сидирования олефиновых соединений.
256. Пат. 4038291 США, МКИ С 07 D 301/20. Боридный катализатор эпокси-
дирования олефиновых соединений.
257. Султанов Р. А., Байрамов Г. К., Садых-заде//ЖОХ. 1968. Т. 4. № 5.
С. 789—791.
258. Пат. 55—39542 Япония, МКИ С 07 С 121/46. Получение 4-цианоцикло-
гексанкарбоновой кислоты.
259. Пат. 3143570 США, НКИ 260—563. Получение алициклических диаминов.
260. Пат. 3137727 США, НКИ 260—563. 4-Аминометилциклогексанметанол.
261. Пат. 23378 Япония, НКИ 16 С 86. Способ получения 1-замещенных цик-
логексенов-1.
262. Пат. 3088967 США. НКИ 260—464. Получение производных 1-циклогск-
сена.
263. А. с. 302335 СССР, МКИ С 07 С 119/00. Способ получения алкил-(4-циа-
ноциклогексенил) -Х-кетонов.
264. Пишнамаз-заде Б. Ф., Гусейнов И. A.//JXMA Азерб. ССР. 1969. Т. 25.
№ 5. С. 24—26.
265. Тр. АН Лит. ССР//С6. науч. тр./АН Лит. ССР Вильнюс: 1962, Б, 3 (30),
с. 115—120.
266. А. с. 247282 СССР, МКИ С 07 С. Способ получения 1-хлор-2-алкоксиме-
тил-4-цианоциклогексена.
226
267. Сахновский В. Б. Исследование в области синтеза и некоторых превра-
щений арилзамещенных цианоциклогексанов: Автореф. дис. на соискание
уч. степени канд. техн, наук: 02.00.03. Баку: ИНХП. 1970. 24 с.
268. Мусаев Р. М., Сахновская Е. Б., Мамедов Ф. А.//С6. тр./Ин-т нефтехим.
процессов АН Азерб. ССР. Баку: 1975. Вып. VI, С. 208—215.
269. Colonge Icon Dannis Henry//Ви\\. Soc. Chim. France. 1961. N 12.
P. 2238—2241.
270. Пат. 2995611 CHIA, НКИ 260—671. Алкилирование ароматических угле-
водородов.
271. Сб. тр. ин-та нефтехим. процессов АН Азерб. ССР/AH Азерб. ССР. Ба-
ку: 1975. Вып. VI, С. 200—207.
272. Бардышев И. И. Полимеры терпенов — новый продукт лесохимической
промышленности//ГЛХП. 1979. № 6. С. 23—24.
273. Страх Л. К. Получение полимеров терпенов из экстракционного скипи-
дара//Там же. 1981. № 6. С. 27—28.
274. Выродов В. А., Старцева Л. Г., Степанова Г. А. Химическая переработка
древесного угля сырья: Сб. науч. тр. ЛТА. Ленинград, 1984. С. 97—100.
275. Roberts W. J., Day A. R.//J. Am. Chem. Soc. 1950. V. 72. P. 1266.
276. Madena M., Bates R. B., Marvel C. S.//Ibid. 1965 V. АЗ. P. 949.
277. Кеннеди Дж. Катионная полимеризация олефинов. М.: Мир. 1978.
С. 185—186.
278. Монаков Ю. Б., Глухов Е. А., Кудашев Р. X. и ^.//Промышленность СК.
1979. № 6. С. 4—6.
279. Гармонов И. В., Дроздов В. А., Нильва С. Я. и др.//Там же. 1978. № 1.
С. 6—8.
280. Кудашев Р. X., Глухов Е. А., Монаков Ю. Б. и др.//ДАН СССР. 1977.
Т. 232. № 5. С. 1085—1087.
281. Пат. 55—127403 Япония, МКИ С 08 F 8/00. Получ. клейких полимеров.
282. Пат. 57—36108 Япония, МКИ С 08 F 240/00, С 08 F 4/06. Получение
липких смолообразных полимеров.
283. Никулин С. С., Шеин В. С.//Промышленность СК, шин и РТИ. 1986.
№ 10. С. 8—10.
284. Никулин С. С. Использование отходов производства СКИ в резиновой
промышленности.//Тез. докл. Всесоюз. конф. «Повышение качества про-
дукции и внедрение ресурсосберегающей технологии в резиновой про-
мышленности». Ярославль, 1986. С. 149.
285. Никулин С. С., Шеин В. С., Седых В. А., Черкашин М. И.//Тез. докл.
семинара «Перспективные эластомеры и эластомеролигомерные компози-
ции для резинотехнических изделий». М.: ЦНИИТЭнефтехим, 1986. С. 15.
286. Никулин С. С., Шеин В. С.//Тез. докл. I Всесоюз. конф. «Пути повыше-
ния эффективности использования вторичных полимерных ресурсов». Ки-
шинев, 1985. С. 190.
287. Никулин С. С., Шеин В. С. Исследование в области синтеза полимерных
продуктов из олигомеров изопрена в присутствии радикальных инициато-
ров. Воронеж: 1985. 9 с. Деп. в ЦНИИТЭнефтехим 4.04.85 № 43нх.
288. Никулин С. С. Влияние дозировки стирола на процесс радикальной со-
полимеризации олигомеров изопрена, являющихся отходами производства
каучука СКИ. Воронеж: 1984. 8 с. Деп. в ЦНИИТЭнефтехим 4.04.85
№ 44нх.
289. Пат. 596008 Англия, Polymerization of 4-vinylcyclohexene.
290. Пат. 2543092 США, НКИ 260—666. Dimerization of vinylcyclohexene.
291. Пат. 2576515 США, НКИ 260—666. Polymerization of vinylcyclohexene.
292. Пат. 5396 Япония, НКИ 2 В 11. Способ получения высокомолекулярного
полимера 4-винилциклогексена-1.
293. Butler G. В., Miles М. L.//S. Polym. Sci. 1965. V АЗ. Р 1609—1615.
294. Marconi W Gesca S., Fartuna G. D.//Ibid. 1964. V. B2. P 301.
15:
227
295. Валитов Р. Б. Исследование реакции димеризации и содимеризации ди-
енов из промышленных фракций Сз—С5. Автореф. дис. канд. хим.
наук 02.00.13. М.: МИНХиГП, 1967. 20 с.
296. Алиева А. Г., Стоцкая Л. Л., Кренцель Б. А.//Высокомол. соединения.
1973. Т. А15. № 5. С. 1005—1010.
297. А. с. 415276 СССР, МКИ С 08 f 19/00. Способ получения карбоцепных
сополимеров.
298. Алиева Л. Г. Исследование процессов гомо- и сополимеризации 4-винил-
циклогексена. Автореф. канд. хим. наук 02.00.06 М.: ИНХС АН СССР,
1975. 31 с.
299. Кренцель Б. А., Клейнер В. И., Стоцкая Л. Л. Высшие полиолефины.
М.: Химия, 1984. 184 с.
300. Стоцкая Л. Л., Алиева А. Г. Гомо- и сополимеризация а-олефинов на
комплексных катализаторах. М.: 1983. С. 64—71.
301. Пат. 4405772 США, МКИ С 08 F 210/08. Сополимер бутена-1 с 4-винил-
циклогексеном.
302. Лившиц И. А., Коробова Л. М., Ковалева Г А., Рейх В. //.//Каучук и ре-
зина. 1969. № 7. С. 1—3.
303. Кудашев Р. X., Глухов Е. А., Монаков Ю. Б. и др.//ДАТА СССР. 1977.
Т. 232. № 5. С. 1085—1087.
304. Фархиева И. Т., Кудашев Р. X., Монаков Ю. Б. и др,//Высокомолекул.
соединения. 1977. Т. Б19. № 9. С. 674—676.
305. Монаков Ю. Б., Глухов Е. А., Фархиева И. Т. и др.//Гем же. 1980.
Т. А22. № 2. С. 385—389.
306. Монаков Ю. Б., Глухов Е. А., Кудашев Р. X. и др.//Гем же. 1981.
Т. Б23. № 7. С. 524—527.
307. Глухов Е. А. Особенности сополимеризации бутадиена и изопрена с ли-
нейными и циклическими полиенами на каталитических системах Цигле-
ра— Натта: Автореф. дис. канд. хим. наук: 02.00.06. Уфа: БФ АН
СССР, 1982. 20 с.
308. Вдовин А., Шевчик //.//Высокомол. соединения. 1969. Т. Б11. № 7.
С. 509—512.
309. Пат. 3005805 США, НКИ 260—79.5. Rubber with cyclic triene therein.
310. Пат. 3007974 США, НКИ 260—648. Chlorination products of cyclodode-
catriene process.
311. Пат. 1201328 ФРГ, НКИ 12 о 19/01. Verfahren zur Herstellung von Mischo-
ligomeren aus 1,3-Dienen und vinylsubstituierten aromatischen Verbindung.
312. Breil H.t Wilke G.//Macromol. Chem. 1963. V. 69. P. 18.
313. Takahashi H.//Г Organ. Chem. 1963. V. 28. P. 1409.
314. Забористое В. H.t Аносов В. И., Антонова Н. Г., Домогатская М. И.//
Каучук и резина. 1983. № 6. С. 23—25.
315. Никулин С. С., Шеин В. С., Мисин В. М., Черкашин М. //.//Промышлен-
ность СК, шин и РТИ. 1985. № 6. С. 6—7.
316. Никулин С. С., Глазков С. С., Сергеев Ю. А. и др.//Гам же. 1985. № 10.
С. 1—3.
317. Никулин С. С., Сергеев Ю. А., Шеин В. С. Полимеризация и сополимери-
зация олигомеров бутадиена со стиролом в присутствии органических
гидроперекисей. Воронеж: 1984. 9 с. Деп. в ЦНИИТЭнефтехим 17.09.84
№ 76нх.
318. Никулин С. С., Шеин В. С., Сергеев Ю. А. Полимеризация и сополиме-
ризация олигомеров бутадиена со стиролом в присутствии динитрилазо-
бисизомасляной кислоты. Воронеж: 1984. 8 с. Деп. в ЦНИИТЭнефтехим
17.09.84, № 78нх.
319. Никулин С. С., Шеин В. С.//Тез. докл. I Всесоюзн. конф. «Пути повыше-
ния эффективности использования вторичных полимерных ресурсов». Ки-
шинев, 1985. С. 68.
320. Глазков С. С., Никулин С. С., Кузаев А. И. и др.//Гез. докл. научно-
техн. конф. «Разработка и внедрение безотходных технологий, использо-
228
вание вторичных ресурсов — пути повышения эффективности производ-
ства». Киров, 1986. С. 27.
321. Никулин С. С., Глазков С. С., Сергеев Ю. А. и др.//Тез. докл. семинара
«Перспективные эластомеры и эластомеролигомерные композиции для
резинотехнических изделий». М.: ЦНИИТЭнефтехим, 1986. С. 14—15.
322. Сергеев Ю. А., Никулин С. С., Шеин В. С.//Лакокрасочные материалы
и их применение. 1986. № 4. С. 15—16.
323. Сергеев Ю. А., Никулин С. С., Шеин В. С.//Тез. докл. «Динамика процес-
сов и аппаратов химической технологии». Воронеж, 1985. С. 20.
324. Никулин С. С., Сергеев Ю. А., Шаповалова Н. Н., Кузаев А. И.//Тез. докл.
научно-техн. конф. «Разработка и внедрение безотходных технологий,
использование вторичных ресурсов — пути повышения эффективности
производства». Киров, 1986. С. 17.
325. Никулин С. С., Сергеев Ю. А., Шаповалова Н. Н. и др.//Тез. докл. семи-
нара «Перспективные эластомеры и эластомеролигомерные композиции
для резинотехнических изделий». М.: ЦНИИТЭнефтехим, 1986. С. 62.
326. Юкельсон И. И., Бутенко Т. Р., Ржевская К. И. Утилизация отходов
производства стирола. Воронеж. 1979. 9 с. Деп. в ОНИИТЭХИМ. Чер-
кассы 11.03.79 № 2438.
327. Мамедов Ш. А.//Изв. АН АзССР. 1961. № 26. С. 37.
328. Пат. 3271349 США, НКИ 260—33.6. Обработка отходов производства
стирола.
329. Алиев С. Г., Шихализаде П. Д., Гаджиев Р. К.//Азерб. нефтяное хозяйст-
во.. 1973. № 12. С. 33—34.
330. Алиев Д. А., Алиева С. Г., Лоткова Л. М., Дубровко Р. В.//Нефтепере-
раб. и нефтехимия. 1963. № 7. С. 32—34.
331. А. с. 887557 СССР, МКИ3 С 07 С 5/27, С 07 С 2/66. Способ переработки
кубового продукта процесса каталитического алкилирования бензола эти-
леном.
332. А. с. 988801 СССР, МКИ3 С 07 С 15/073, С. 07 С 4/26. Способ получения
этилбензола.
333. Никулин С. С., Смирнов В. С.//Тез. докл. Всесоюз. конф. «Повышение
качества продукции и внедрение ресурсосберегающей технологии в рези-
новой промышленности». Ярославль, 1986. С. 150.
334. Гугняева Л. //.//Исследования в области химии и технологии органиче-
ского синтеза: Сб. науч. тр. ВТИ. Воронеж: ВТИ, 1972. С. 29—32.
335. Гугняева Л. //.//Исследования в области химии и технологии органиче-
ского синтеза: Сб. науч. тр. ВТИ. Воронеж: ВТИ, 1972. С. 33.
336. Зейналов И. П., Бабаев А. И., Рзаев 3. А1.//Промышленность СК. 1981.
№ 12. С. 10—11.
337. Легачева В. В., Бутенко Т. Р., Аканова Т. В.//Там же. 1983. № 1.
С. 15—16.
338. Фоменко А. И., Терехин Р. М.//Там же. 1983. № 2. С. 5—7.
339. Фоменко А. И., Терехин Р. М. К вопросу рекуперации кубового остатка
ректификации стирола. Воронеж: 1981. 6 с. Деп. в ОНИИТЭХИМ
19.04.82 № 443хп.
340. Легачева В. В., Ржевская К. //.//Промышленность СК, шин и РТИ.
1985. № 2. С. 6—9.
341. Лычкин И. П., Фоменко А. И., Усов Е. Н., Иншаков В. И. Влияние серы
как ингибитора на накопление полимера в кубовом остатке ректификации
стирола в производственных условиях. Воронеж: 1982, 8 с. Деп. в
ОНИИТЭХИМ 21.06.82 № 731хп.
342. Юкельсон И. И., Бутенко Т. Р., Ржевская К. //.//Лакокрасочные мате-
риалы и их применение. 1982. № 2. С. 51—52.
343. А. с. 181218 СССР, Кл. 22 h, 2. Способ получения синтетической олифы.
344. Зейналов И. П., Рзаев 3. М., Бабаев А. //.//Промышленность СК. 1983.
№ 3. С. 9—10.
345. А. с. 737407 СССР, МКИ3 С 08 F 212/08. Способ получения сополимеров.
229
346. А. с. 1100279 СССР, МКИ3 С 08 F 212/08, С 09 D 3/727. Способ получе-
ния пленкообразующих сополимеров.
347. Курбанова Р. А., Алиева Д. Н., Шукюрова М. Б., Бабаева Л. М. Иссле-
дование процесса и продуктов ацилирования КОРС.//Мономеры и поли-
меры. Баку, 1983. С. 136—139.
348. А. с. 437794 СССР, МКИ3 С 09 3/14. Клеевая композиция.
349. А. с. 456815 СССР, МКИ3 С 08 F 33/02, С 08 D 7/00. Композиция для
изготовления материала покрытия пола.
350. А. с. 553270 СССР, МКИ3 С 09 D 3/58, С 09 D 5/08. Состав для покры-
тия.
351. Ханларова А. Г., Негреев В. Ф., Гаджиева К. Г. и др.//Лакокрасочные
материалы и их применение. 1960. № 6. С. 16—21.
352. Алиев Д. А., Алиева С. Г., Дубрава Р. В.//Нефтепереработка и нефтехи-
мия. 1964. № 12. С. 32—36.
353. Щеголь Ш. Г., Златкин Б. С., Сватиков В. /7.//Бюл. АзИНТИ. 1962.
№ 3. С. 34.
354. А. с. 1035017 СССР, МКИ3 С 07 С 15/46. Способ получения стирола.
355. Мамедов Ш. А., Агаев А. С. Пути рационального использования стироль-
ной смолы Сумгаитского завода СК.//С6. науч.-техн. информ. Сер. неф-
тепереработка. 1961. № 2. С. 22.
356. Пат. 3617514 США, НКИ 208—131. Использование остаточных продуктов
получения стирола при замедленном коксовании.
357. Пат. 3501545 США, НКИ 260—674. Способ регенерации ароматических
соединений из серосодержащих кубовых остатков, получаемых при очи-
стке стирола.
358. Румянцев А. В., Алексеева И. А., Гимадеева Л. И., Тумасова Г. А.//
Промышленность СК. 1968. № 4. С. 17—18.
359. Рудковский Д. М., Трифель А. Г Алексеева К. А.//Оксосинтез. Л.: Гос-
топтехиздат, 1963. С. 51.
360. Алексеева К. А., Гордина Н. Я., Рудковский Д. М., Трифель А. Г.//
Карбонилирование ненасыщенных углеводородов. Л.: Химия, 1968. С. 80.
361. Амиров Я. С., Варфоломеев Д. Ф., Мухутдинов Р. X., Тищенко В. Е.
Рациональное использование вторичных ресурсов нефтехимии и охрана
окружающей среды. Уфа: Башкнигоиздат, 1976. 113 с.
362. Лаврентьев В. Г Лапан А. П., Титов И. П. и др.//Кокс и химия. 1975.
№ 1. С. 36.
363. Алексеева К. А., Трифель А. Г., Рудковский Д. М.//Карбонилирование
ненасыщенных углеводородов. Л.: Химия, 1968. С. 134.
364. Дельник В. В.//Хим. пром. 1976. № 6. С. 20.
365. Валитов Н. X., Кутлугужина И. X., Носаль Г //.//Нефтепереработка и
нефтехимия. 1971. № 1. С. 28.
366. Валитов Н. X., Сисин М. Ф., Захаров М. С. и др.//Там же, 1970. № 6.
С. 36.
367. Валитов Н. X., Носаль Г. И.//Там же. 1969. № 8. С. 31.
368. Валитов Н. X., Носаль Г. И., Кутлугужина И. Х.//Т&М же. 1973. № 6.
С. 23.
369. Алексеева К. А., Гордина Н. Я., Кагна С. Ш. и ^.//Гидроформилирова-
ние. Л.: Химия, 1972. С. 130.
370. Falbe I., Hahn D.//Chem. Ztg. 1972. Bd 96, N 3, S. 164.
371. Falbe L, Tummes H., Hahn D.//Angew. Chem. 1970. Bd 82. S. 181
372. Falbe /.//Ibid. 1972. Bd 84. S. 172.
373. Алексеева К. А., Высоцкий M. П., Высокинская А. Т. и ^.//Гидрофор-
милирование. Л.: Химия, 1972. С. 109.
374. Варфоломеев Д. Ф., Куковицкая Л. Б., Биккулов А. В. и ^.//Нефтепере-
работка и нефтехимия. 1973. № 1. С. 22.
375. Динцес А. И., Дружинина Л. В. Синтетические смазочные материалы.
М.: Гостоптехиздат, 1958. 300 с.
376. Синтетические смазочные материалы и жидкости: Пер. с англ./Под ред.
Р. С. Гундерсона и А. В. Харта. Л.: Химия, 1965. 385 с.
230
377 Lisman W. A.//Eng. Possibl. of Sintetic Lubricat. (SAE Trans). 1953. N 61.
P. 309.
378. Moreton D. //.//Lubricat. Eng. 1954. V. 10. N 2. P. 65.
379. Куковицкий M. M., Биккулов A. 3., Аглиуллина A. X. и др.//Докл. нефте-
хим. секции. Уфа: УНИ, 1971. Вып. 7. С. 31.
380. Куковицкая Л. Б., Биккулов А. 3., Куковицкий М. М., Хамаев В. X.//
Докл. нефтехим. секции. Казань: КХТИ, 1973. Вып. 9. С. 103.
381. Биккулов А. 3., Куковицкая Л. Б., Хамаев В. Х.//Докл. нефтехим. сек-
ции. Уфа: УНИ. 1971. Вып. 7. С. 41.
382. Куковицкий М. ^.//Нефтепереработка и нефтехимия. 1965. № 9. С. 14.
383. Крейн С. Е„ Макашева О. //.//Химия и технология топлив и масел. 1973.
№ 6. С. 58.
384. Биккулов А. 3., Хамаев В. X., Куковицкая Л. ^.//Нефтепереработка и
нефтехимия. 1974. Вып. 2. С. 71.
385. Billigman /., Fichte W.//Stahl und Eisen. 1958. N 6. S. 344.
386. Johnson W. /?.//Schwantbart Blast Furnace, and Steel Plant. 1955. V. 43.
N 3. P. 415.,
387. Мищенко Г. Л., Вацуро К. В. Синтетические методы органической химии.
М.: Химия, 1982. 440 с.
388. Корчагина В. И., Парток Н. И., Тюрин Б. К. и др.//Пластические массы.
1981. № 8. С. 55.
389. Schulse /., Weiser M//Chem. Ind. 1985. N 2. P. 105.
390. Schulse /., Weiser MJ/Ibid. 1985. N 3. P. 175.
391. Rappe GJ/Chem. Eng. 1985. N 8. P. 44.
392. Schulse I., Weiser MJ/Chem. Ind. 1984. N 11. P. 547.
393. Giltenan E. FJ/Chem. Business. 1984. V. 6. N 9. P. 42.
394. Hyland R. W., Parr I. LJ/Chem. Process. 1981. V. 44. N 9. P. 102.
395. Holmes ^.//Processing. 1981. V. 27. N 3. P. 37.
396. Holmes DJ/CzhL Geol. 1981. 34. N 6. P. 116.
397. Дубовой Л. И., Адитайс Э. P.f Берлин Э. P., Трегер Ю. AJ/Хмм. пром.
1982. № 11. С. 658.
398. Муганлинский Ф. Ф., Ахмедов Ф. Ш.//Изв. ВУЗов СССР. Сер. нефть и
газ. 1971. № 11. С. 53.
399. Гусейнов М. М., Муганлинский Ф. Ф., Мамедов Д. М., Ахмедов Ф. U1J/
Азерб. хим. журнал. 1971. № 1. С. 90.
400. Мамедов Д. В., Гусейнов М. М., Ахмедов Ф. Ш., Муганлинский Ф. ФJ/
Азерб. нефтяное хозяйство. 1971. № 6. С. 41.
401. Мамедов Д. BJ/Сб. тр. АЗИНЕФТЕХИМ. Баку, 1971. С. 8.
402. Тиниус К. Пластификаторы. М.: Химия, 1964. 301 с.
403. Тюрин Б. К., Бикбулатов И. X., Нурутдинов С. X. и дрJ/Тез. докл.
6 Всесоюз. конф, по электрохимии. Москва, 1982. Т. 2. М.: МГУ, 1982.
228 с.
404. Мацаренко В. А., Лебединская Н. Л., Дудкова Л. И., Гущина Л. И.//
Хим. пром. 1969. № 1. С. 12.
405. Филипов Н. А., Учватов Ф. Ф.//Очистка и утилизация кислых сточных
вод. М.: ЦНИИТЭнефтехим, 1985. 40 с.
406. Мацаренко В. А., Новикова В. Н., Лубянова А. А.//Максимальное по-
вторное использование сточных вод в производстве СЖК. М.: ЦНИИТЭ-
нефтехим, 1986. 39 с.
407 Матушкин К. М.//Нефтеперераб. и нефтехимия. 1975. № 10. С. 40.
408. Маньковская Н. /С//Маслобойно-жировая промышленность. 1954. № 1.
С. 23.
409. Кудряшов А. И., Макаров С. В., Бочкарев Ю. А. и др.//Нефтеперераб.
и нефтехимия. 1971. № 8. С. 39.
410. Сухотерин Н. С.//Труды науч.-техн, совещания по производству СЖК
и моющих средств. Белгород. Белгородкнигоиздат, 1959. С. 278.
411 Фомин В. В., Руденко Г И.//ДАН СССР. 1973. № 1. С. 162.
412. Чарыков А. К., Потанова Т М.//Вестник Ленинградского университета
1977. Вып. 1. № 4. С. 24.
231
413. Тезикова Л. Л.//Экстракционный метод очистки сточных вод. М.:
ЦНИИТЭнефтехим, 1976. С. 38.
414. Алферова Л. Л.//Маслобойно-жировая промышленность. 1959. № L
С. 28.
415. Якушин М. }7.//Хим. пром. 1969. № 7. С. 504.
416. Трофимов А. Н., Коган В. ^.//Гидролизная и лесохимическая промыш-
ленность. 1972. № 7. С. 11.
417. Деревянко Р. Ш.//Хнм. пром. 1983. № 2. С. 81.
418. Струнникова Г. А.//Гидролизная и лесохимическая промышленность.
1972. № 5. С. 25.
419. Алферова А. А., Бондарева Т. Н., Мацаренко В. Л.//Маслобойно-жиро-
вая промышленность. 1963. № 11. С. 35.
420. Шерстнева В. А., Г ужина Л. И., Мацаренко В. А.//Там же. 1964. № 7.
С. 78.
421. Цисковский В. К., Прокофьев Н. В.//Хим. пром. 1969. № 10. С. 737.
422. Болътин И. М., Донской Б. В., Тертыяная Т. Д.//Нефтеперераб. и нефте-
химия. 1972. № 1. С. 56.
423. Чередникова А. В.//Тез. докл. III Всесоюз. совещания по синтетическим
жирозаменителям, поверхностно-активным веществам и моющим средст-
вам. Шебекино, 1965. С. 127.
424. Перегудин Ю. Ф.//Очистка промышленных выбросов и техника безопас-
ности на химических предприятиях. 1975. № 3. С. 9.
425. Макаров С. В., Кудряшов А. И., Кононова Н. Л.//Нефтеперераб. и неф-
техимия. 1975. № 10. С. 31.
426. Кондратенко В. А., Суркова А. Ф., Морошкин Ю. Р.//Там же. 1982.
№ 12. С. 26.
427. Михайлов В. К., Петряков Н. Н., Панов Ю. П. и др.//Там же. 1974. №3.
С. 47.
428. Михайлов В. К., Грищенко А. С., Соколов В. П. и др.//Там же. 1975.
№ 8. С. 39.
429. Дороднова В. С., Игонин П. Г.//Там же. 1975. № 5. С. 38.
430. Жужкова Л. В.//Вторичные материальные ресурсы в нефтеперерабаты-
вающей и нефтехимической промышленности. М.: ЦНИИТЭнефтехим.
1981. С. 14.
431. Правилова Г. А.//Бум. пром. 1974. № 1. С. 15.
432. Грищенко А. С., Алферова Л. А., Рабинович Л. М., Михайлов В. К.//
//Нефтеперераб. и нефтехимия. 1981. № 6. С. 55.
433. Богданов Ю. С., Байдин И. И., Елагин В. Т Миненко А. И.//Там же.
1980. № 3. С. 38.
434. Миненко А. И., Богданов Ю. С.//Там же. 1979. № 1. С. 51.
435. Богданов Ю. С., Байдин И. И., Елагин В. Т., Миненко А. И.//Там же.
1981. № 1. С. 38.
436. Фролов А. Е., Толкачев С. П., Матушкин К. М.//Там же. 1978. № 3.
С. 43.
437. Оробченко Е. В., Прянишникова Н. Ю., Берлинская М. //.//Маслобойно-
жировая промышленность. 1961. № 2. С. 83.
438. Оробченко Е. В., Вепринская М. Н., Прянишникова Н. Ю.//Там же. 1962.
№ 8. С. 56.
439. Коваль Л. П., Соколов В. П., Паримский А. И., Беседина И. Д.//Нефте-
перераб. и нефтехимия. 1975. № 5. С. 36.
440. Маньковская Н. К., Удовенко С. А., Романова Л. Л.//Химия и техноло-
гия топлив и масел. 1962. № 2. С. 22.
441. Дроздов А. С., Волкова Л. Д., Уварова Л. В.//Нефтеперераб. и нефтехи-
мия. 1981. № 7. С. 60.
442. Лыков О. П., Вишнякова Т. П., Кульбацкая Л. А., Рудикова Ю. В.//Там
же. 1982. № ГС. 48.
443. Миньков В. А., Сокина В. Е., Макаров С. В., Поборцев Э. /7.//Там же.
1982. № 8. С. 42.
232
444. Лыков О. П., Вишнякова Т П., Цигал Л. В., Саркисян Р. Л.//Там же.
1983. № 2. С. 37.
445. Согшна В. Е., Закордонец О. П., Миньков В. А., Поборцев Э. //.//Там же.
1981. № 3. С. 35. n v
446 Куковицкая Л. Б., Биккулов А. 3., Куковицкий М. М., Хамаев В. X.//
//Там же. 1984. № 2. С. 16.
447 Русьянова Н. Д., Жилина И. Б„ Костромин А. С. и др.//Кокс и химия.
1977 № 2. С. 30.
448. Агаев А. С., Наджафов Ю. Б., Кобылкина Т //.//Химия и технология
топлив и масел. 1976. № 11. С. 44.
449. Евзельман И. Б., Харлампович Г. Д.//Хим. пром. 1973. № 6. С. 27.
450. Евзельман И. Б., Кругликов А. Л.//Цемент. 1968. № 1. С. 16.
451 Голик В. П., Соловьев Г И., Филипов М. //.//Хим. технология. 1985.
№ 6. С. 57.
452. Цыбенко Л. В.//Гам же. 1985. № 4. С. 63.
453. Геворкян А. А., Ермакова М. П., Амитин А. В., Горелик А. Г.//Хим. пром.
1980. № 7. С. 44.
454. Кульбачный В. Г Гореславский А. С., Пластун А. Л.//Кокс и химия.
1979. № 7. С. 31.
455. Харлампович Г. Д., Чуркин Ю. В. Фенолы. М.: Химия, 1974. 375 с.
456. Бочаров Ю. //.//ЖВХО. 1961. Т. 6. № 1. С. 74.
457. Pavlikowska Z).//Przemyst. chemistry. 1981. N 2. Р 76.
458. Rivoltah U., Battiston J., D’Alfons Н.//Г Chim. e. Ind. 1983. 65. N 9.
P. 545.
459. Пуджадо П. P., Сапазар Д. P., Барджер К. В.//Переработка углеводоро-
дов. 1976. № 3. С. 22.
460. Гречухина А. Л.//Нефтеперераб. и нефтехимия. 1972. Вып. 1. С. 37.
461. Рыбакова Ю. А. Пластические массы и синтетические смолы. 1973.
Вып. 12. С. 60.
462. Magidko К., Ciberowzki S.//Przemyst. chemistry. 1982. N 8. P. 254.
463. Воронцов H. H. Основы синтеза промежуточных продуктов и красителей.
М.: Госкомиздат. 1955. 839 с.
464. Granfield /.//Oil and Gas International. 1971. II. N 5. P 81.
465. Гапегов В. А., Рахимов В. P., Казымова А. М.//Хим. пром. 1972. № 8.
С. 630.
466. Воль-Эпштейн А. Б., Жарова М. Н., Суровцева В. В.//Там же. 1962.
№ 2. С. 12.
467. Воль-Эпштейн А. Б., Жарова М. Н., Беренц А. Д.//Нефтеперераб. и неф-
техимия. 1965. № 10. С. 37.
468. Воль-Эпштейн А. Б., Жарова М. //.//Нефтехимия. 1967. Т. 1. X? 2.
С. 13—17.
469. Бочаров Ю. //.//Нефтеперераб. и нефтехимия. 1967. № 3. С. 23.
470. Воль-Эпштейн А. Б., Гагарин С. Г Каталитические превращения алкил-
фенолов. М.: Химия, 1973. 273 с.
471. Kharasch М., Fono A., Nudenberg W.//5. Org. Chem. 1951. N 16. P. 113.
472. Безмозгин Э. С.//Хим. пром. 1963. № 8. С. 17.
473. Безмозгин Э. С. Термокаталитические методы переэаботки углеводород-
ного сырья. Л.: Химия, 1969. 283 с.
474. Амиров Я. С., Абызгильдин Ю. М., Русанович Д. А., Тищенко В. Е.
Вопросы рационального использования отходов нефтепереработки и неф-
техимии. Уфа: УНИ, 1976. 142 с.
475. Брук А. Ю.//Нефтеперераб. и нефтехимия. 1962. № 7. С. 29.
476. Жульман А. С.//Там же. 1970. № 7. С. 31.
477 Игонин Р. Л.//Нефтеперераб. и нефтехимия. 1973. № И. С. 39.
478. Игонин Р А.//Там же. 1975. № 1. С. 34.
479. Pawlikowska D.//Przemyst. chemistry. 1978 N 2 P 59
480. Банатенко В. А., Гусев В. И., Иванов Б. Е., Абдулхакова Н. И. Материа-
лы науч.-техн. конф. Казанского завода орг. синтеза. Казань: Изд-во га-
зет и журналов Татарского обкома КПСС. 1969. С. 92.
233
481 Lazazov L., Angelova G., Angelov S., Konstantinov K.//Chim. i. Ind. 1977.
N 3. P. 111.
482. Мещерякова 3., Остроумова .//.//Химическая наука и промышленность,
1959. № 4. С. 346.
483. Кулиев А. М. Присадки к смазочным маслам. Баку: АЗИНЕФТЕХИМ
1970. 318 с.
484. Листов В. А. Методы исследования нефтей и нефтепродуктов. М.: МГУ,
1975. 261 с.
485. Dziedzic G.//Referat Wygtoszony па Lesjd Nawkowy Institutu chemii i
Technologii Organonicznej. 1978. Bd. VI. S. 22.
486. Селезнев А. Г., Дороминская С. М.//Изв. ВУЗов СССР. Нефть и газ.
1971. № 1. С. 51.
487. Гоган В. С., Фаворская Л. В., Преснепугова В. А., Вайнберг Т. Н.//Тех-
нология минерального сырья. 1975. № 2. С. 31.
488. Букин В. Г Гранат А. В., Плющев В. Н. и др.//ЖНХ. 1976. № 21.
С. 1876.
489. Гаккель Н. Я., Гаджибаниев А. А.//Азерб. хим. журнал. 1975. № 2.
С. 106.
490. Сирунин В. О., Иванова Р. С. Фосфорорганические соединения и поли-
меры. Казань: КХТИ, 1976. С. 49.
491. Мельников Н. Н. Химия и технология пестицидов. М.: Химия, 1974,
765 с.
492. Шакиров Л. Г., Биккулов А. 3., Смолянец Е. Ф., Гимальдинова Г Г.//
Хим. пром. 1987. № 2. С. 75.
493. Соболев А. С., Зубарев С. В., Квятковский В. С., Игошев А. Д.//Химия
в сельском хозяйстве. 1968. № 8. С. 54.
494. Condrat В.//}. Chromatog. 1982. V. 237. N 1. Р. 148.
495. Лукашенок В. И., Игошев А. Д., Зубарев С. В., Пешекина 3. И.//Хим.
пром. 1972. № 11. С. 22.
496. Коренман Я. И.//ЖПХ. 1973. Т. 56. № И. С. 1095.
497. Fujikawa Kan-ichi, Kondo Katsuhide, Yokomichi Isao et al.//Kgv. and Biol,
chem. 1970. V 34. N 1. P. 68.
498. Романенко О. А., Степаненко H. И., Ряховская А. И.//Химическая про-
мышленность. Серохлорная промышленность. М.: ЦНИИТЭНЕФТЕХим,
1980. № 1. С. 17.
499. Касьянов В. В., Энрике А. Наварро, Муганлинский Ф. Ф., НижникМ. А.//
Изв. ВУЗов СССР Химия и химическая технология. 1977. Т. 20. № 1L
С. 1623.
500. Копылов В. В.//Успехи химии. 1970. Т. 39. Вып. 3. С. 471.
501. Крючкова В. Е., Шостак Т. С., Осьмак Л. П. и др.//Укр. хим. журнал.
1979. Т. 45. № 7. С. 678.
502. Гершанов Ф. Б., Гурвич Я. А., Долидзе В. И. и ^.//Промышленность
СК. 1973. № 12. С. 7.
503. Хидекель М. Л. Комплексообразование в катализе (проблемы кинетики
и катализа). М.: Наука, 1968. С. 126.
504. Горбунов Б. Н., Гурвич Я. А., Маслова А. П. Химия и технология стаби-
лизаторов полимерных материалов. М.: Химия, 1981. С. 214.
505. Пантух Б. И., Матвеева Ж. А., Шалимова 3. С.//Промышленность СК.
1983. № 10. С. 11.
506. Захарова И. В., Лиакумович А. Г., Мичуров Ю. И. и др. Каталитические
реакции в жидкой фазе.//Материалы IV Всесоюз. конф, по каталитиче-
ским реакциям в жидкой фазе, Алма-Ата: КГУ, 1974. С. 774.
507. Шалимова 3. С., Толстых Э. В., Пантух Б. И., Тимофеева В. Ф.//Тез.
докл. I-ой Всесоюз. конф. «Катализ и каталитические процессы произ-
водства химико-техчологических препаратов». М. 1985. Т. 2. С. 130.
508. Козина С. М., Тимофеева В. Ф., Шалимова 3. С. и др.//Тез. докл. респ.
конф. «Ускорение научно-технического прогресса в нефтехимической про-
мышленности». Уфа, 1981. С. 49.
509. Верховская 3. Н. Дифенилолпропан. М.: Химия, 1971. 196 с.
234
510. Галуткина К. А., Немченко А. Г., Аростолова И. В., Карадеева Л. Н.//
Хим. пром. 1980. № 2. С. 124—125.
511. Сафаров М. Г., Огородников С. К., Ибатуллин У. Г и др.//ЖОХ. 1981.
Т 17 5 С 922__925
512. Закошанский В. М., Йдлис Г С.//ЖПХ. 1985. Т. 58. № 8. С. 1918—
1920.
513. Огородников С. К., Идлис Г С. Производство изопрена. Л.: Химия, 1973.
С. 230.
514 Апьок И., Барток М., Караханов Р. А., Шуйкин Н. И.//]Азв. АН СССР.
Сер. .хим. 1968. № 10. С. 2354—2357.
515. Новикова Л. А., Иванова И. Г., Чудинова П. Н., Балагана Г М.,
Шарф В. 3.//Нефтехимия. 1984. Т. 24. № 2. С. 221—224.
516. Иванова И. Г., Китаев Л. Е., Новикова Л. А. и др.//Нефтехимия. 1984.
Т. 24. № 5. С. 650—656.
517. Соколовская И. В., Лестева Т. М., Буданцева Л. С.//Промышленность
СК, шин и РТИ. 1985. № 5. С. 3—6.
518. Милькина Т. И., Кузнецова Е. В., Моршакова Т. М. и др.//Нефтехимия.
1984. Т. 24. № 6. С. 822—828.
519. Безродная Р. М., Гурвич С. М//Цвет. металлургия. 1967. № 3. С. 21.
520. Рахманкулов Д. Л., Караханов Р. А., Злотский С. С. и др. Химия и тех-
нология 1,3-диоксациклоалканов.//Итоги науки и техники. Технология
органических веществ. Т. 5. М.: ВИНИТИ, 1979. 287 с.
521. Фарберов М. И., Шемякина Н. К.//ЖОХ. 1956. Т. 26. № 10. С. 2749—
2754.
522. Апьок И., Барток М., Караханов Р. А., Шуйкин Н. И.//Усп. химии. 1969.
Т. 38. № 1. С. 72.
523. Баснина Т. Д.//Нефтехимия. 1966. Т. 6. № 3. С. 463—467.
524. Закошанский В. М., Идлис Г С., Светлова Л. М., Огородников С. К.//
ЖОХ. 1971. Т. 7. № 8. С. 1563—1569.
525. Закошанский В. М., Идлис Г С., Огородников С. К.//Там же. 1974. Т. 10.
№ 5. С. 1076—1081.
526. Рывкина Л. А., Ранкин В. Ю., Синицын А. В., Полякова М. А.//Там же.
1983. Т. 19. № 9. С. 1957—1961.
527. Закошанский В. М., Рывкина Л. А., Ранкин В. Ю., Идлис Р. С.//Там же.
1984. Т. 20. № 9. С. 1893—1898.
528. Сафаров М. Р., Исагулянц В. И., Сафарова В. Р. Каталитический синтез
и превращения гетероциклических соединений. Гетероген. катализ. Уфа:
УНИ 1976 С 225________228
529. Сафаров М. Г., Сафарова В. Г., Рафиков С. Р.//ДАН СССР. 1976. Т. 226.
№ 6. С. 1347—1350.
530. Рахманкулов Д. Л., Максимова Н. Е., Меликян В. Р., Исагулянц В. И.//
//ЖПХ. 1974. № 1. С. 223—236.
531. Рахманкулов Д. Л., Максимова И. Е., Меликян В. Р., Исагулянц В. И.
Каталитический синтез и превращения гетероциклических соединений.
Рига: Зинатне, 1976. С. 217—224.
532. Рахманкулов Д. Л., Максимова И. Е., Меликян В. Р. Химия высокомо-
лекулярных соединений. Нефтехимия. Уфа: УНИ, 1973. С. 60—61.
533. Рахманкулов Д. Л., Максимова Н. Е., Кантор Е. А. Получение водорода,
углеводород/Под ред. Веселова В. В. Киев: Наукова думка, 1979.
С. 21—24.
534. Рахманкулов Д. Л. Каталитический синтез органических соединений се-
ры. Новосибирск: НГУ 1979. С. 167—173.
535. Максимова Н. Е., Кантор Е. А., Рахманкулов Д. Л. и др. //ЖПХ. 1977
Т 50. ЛЬ 7 С. 1612—1616.
536. Lesage J., Peinado M//Bull. Soc. Chim. Fr. 1969. N 8 P 2881—2889
537 Мусавиров P С., Кантор E. А., Рахманкулов Д. Л.//ЖПХ. 1978 T. 5
№ 10. C. 2295—2300.
538. Cerveny L., XX’urzlova A. Ruzicka V.//Sb. VSCHT Prage. 1983. V 23.
235
539. Bailey W. F., Rioera A. D.//J. Org. Chem. 1984. V. 49. N 25. P. 4958—
4964.
540. Ториков Д. M., Шахова Ф. А.//Науч.-тем. сб. Уфим. нефт. ин-та. Уфа.
1975. № 22. С. 134—138.
541. Косулина Т. П., Кромачевская Е. В., Фалина Л. А. и др.//Химия гете-
роцикл. соед. 1983. № 4. С. 464—466.
542. Девекки А. Б., Идлис Г. С., Огородников С. /С//ЖОХ. 1981. Т. 17. №10.
С. 2238—2239.
543. Рахманкулов Д. Л., Исагулянц В. И., Злотский С. С.//ЖПХ. 1972. Т. 45.
№ 7. С. 1642—1644.
544. Зорин В. В., Трифонова В. Н., Злотский С. С., Рахманкулов Д. Л.//ЖОХ.
1984. Т. 20. № 4. С. 869—871.
545. Зорин В. В., Злотский С. С., Шувалов В. Ф. и др.//ДАН СССР. 1977.
Т. 236. № 1. С. 106—109.
546. Брудник Б. М., Спирихин Л. В., Курамшин Э. М. и др.//ЖОХ. 1980.
Т. 16. С. 1281—1284.
547. Kuramshin Е. М„ D’yachenko V. A., Zlotskii S. S., Rachmankulov D. L.//
Oxid. Commun. 1983. V 4. N 1—4. P. 3—12.
548. Курамшин Э. M., Дьяченко В. А., Злотский С. С., Рахманкулов Д. Л.//
Нефтехимия. 1982. Т. 22. № 5. С. 620—622.
549. Милман И. А., Дзилюма Э. Е., Евграшин В. М.г Страутиня А. К.//Изв,
АН Латв. ССР. Сер. хим. 1977. № 3. С. 374—375.
550. Миронов И. В., Мельницкий И. А., Сираева И. Н. и др.//ЖОХ. 1981.
Т. 51. № 12. С. 2700—2704.
551. Девекки А. В., Кошелев Ю. Н., Малов Ю. И. и др.//ЖОХ. 1983. Т. 53.
№ 11. С. 2652—2653.
552. Rachmankulov D. L., Karakhanov R. A., Zlotskii S. S. et al.//Acta Phys.
Chem. 1973. V. 19. N 1—2. P. 171—176.
553. Эстрина Г Я., Агишева С. А., Курамшин Э. М. и ^.//Нефтехимия. 1980.
Т. 20. № 2. С. 264—267.
554. Батырбаев Н. А., Зорин В. В., Злотский С. С., Рахманкулов Д. Л.//
//ЖОХ. 1982. Т. 52. № 1. С. 134—139.
555. Рахманкулов И. Л., Кантор Е. А., Рахманкулов Д. Л.//ЖПХ. 1978. Т. 51.
№ 10. С. 2321—2325.
556. Мусавиров Р. С., Кантор Е. А., Имашев У. Б., Рахманкулов Д. Л.//ЖПХ.
1977. Т. 50. № 7. С. 1609—1612.
557. Мусавиров Р. С., Кантор Е. А., Рахманкулов Д. Л.//Тгм же. 1976. Т. 49.
№ И. С. 2513—2515.
558. Мусавиров Р. С., Недогрей Е. /7.//Изв. ВУЗов СССР. Химия и хим. тех-
нол. 1984. Т. 27. № 9. С. 1032—1035.
559. Рахманкулов Д. Л., Кантор Е. А., Мусавиров Р. С. и др.//ДАН СССР.
1977. Т. 233. № 2. С. 383—385.
560. Романов Н. А., Кантор Е. А., Мусавиров Р. С. и др.//ЖПХ. 1980. Т. 53.
№ 9. С. 2140—2143.
561. Романов Н. А., Кантор Е. А., Мусавиров Р. С., Рахманкулов Д. Л.//ЖОХ.
1977. Т. 15. № 5. С. 1059—1064.
562. Кантор Е. А., Романов Н. А., Мусавиров Р. С. и ^.//Каталитические
реакции в жидкой фазе. Алма-Ата: 1978. С. 177.
563. Романов Н. А., Кантор Е. А., Рахманкулов Д. Л.//Материалы Всесоюз.
научи, конф. «Химия и технология фурановых соединений». Рига: Зинатне.
1978. С. 115—116.
564. Романов Н. А., Кантор Е. А., Караханов Р. А., Рахманкулов Д. Л.//
//ЖПХ. 1983. № 2. С. 333—337.
565. Романов Н. А., Кантор Е. А., Мусавиров Р. С. и др.//ЖОХ. 1981. Т 17.
№ 8. С. 1762—1767.
566. Лапука Л. Ф., Кантор Е. А., Романов Н. А. и др.//Химия гетероцикл,
соед. 1980. № 6. С. 744—747.
567. Геворкян А. А., Токмаджян Г Г.//Арм. хим. журн. 1977. Т. 30. № 3.
С. 269—270.
236
568 Геворкян А. А., Токмаджян Г Г Саакян А. А.//Там же. 1977. Т. 30,
№ 9. С. 748—751.
569. Рахманкулов Д. Л., Максимова И. Е., Паушкин Я. М.//ДАН СССР.
1974. Т. 214. № 6. С. 1323—1325.
570. Качалова Р В., Немцов М. С.//ЖПХ. 1968. Т. 41. № 10. С. 2315.
571. Ольков П. Л., Сафаров М. Г.//Труды Уфим. нефт. ин-та. Уфа, 1971.
Вып. 9. С. 50—55.
572. Лестева Т. М., Черная В. И., Буданцева Л. С. Евстигнеев А. Ю.//Про-
мышленность СК. 1983. № 3. С. 2—5.
573. Лестева Т. М., Рогозильникова Л. А., Черная В. И., Буданцева Л. С.//
//Промышленность СК. 1984. № 9. С. 3—5.
574. Лестева Т. М., Буданцева Л. С., Черная В. М„ Евстигнеев А. Ю.//Т\.ро-
мышленность СК. 1983. № 11. С. 2—5.
575. Геллер Б. Э., Пшедецкая В. К-, Рахманкулов Д. Л. и др.//ЖПХ. 1976.
Т. 49. № 3. С. 614—617.
576. Мандель Р. Б., Поройкова Ю. А., Грицевич А. Г., Беканова Д. Н.ЦЛ.з-
кокрасочны£ материалы и их применение. 1975. № 4. С. 77—78.
577. Гамбарделла Е. //.//Лакокрасочные материалы и их применение. 1960.
№ 6. С. 27—31.
578. Ritter R.//Rev. Inst. Fr. Petrole Ann. Combust. Liquids. 1968. V. 23. N 10.
P. 1285—1303.
579. Рахманкулов Д. Л., Пименова В. T., Пшиялковский Б. //.//Труды Уфим.
нефт. ин-та. Уфа, 1974. Вып. 16. С. 259.
580. Рахманкулов Д. Л., Злотский С. С., Бакиева Ф, ^.//Проблемы нефтепе-
реработки и нефтехимии. Уфа: УНИ, 1973. С. 164.
581. Пешкин О. В., Мархасин В. Я.//Физико-хим. разраб, нефт. месторожд.,
Уфа: УНИ, 1982. С. 95—101.
582. А реет-Якубович А. Л., Аносов В. И., Басова Р. В. и др.//Высокомол.
соед. Сер. А. 1982. Т. 24. № 7. С. 1530—1534.
583. Кристальный А. В., Золотарев В. Л., Нахманович Б. И. и др.//Высоко-
мол. соед. Сер. Б. 1983. Т. 25. № 4. С. 290—292.
584. Шаульский Ю. М., Леплянин Г. В., Курамшин Э. М. и др.//Высокомол.
соед. Сер. А. 1984. Т. 26. № 5. С. 962—966.
585. Kuehn М., Tominga R.//Staub. 1965. Bd. 25. N 3. S. 86.
586. Петухов В. H., Обух Л. В. Сб. науч. тр. Магнитогорского горно-метал.
ин-та, 1975. С. 87—94.
587. Сыркин А. М., Рахманкулов Д. Л., Хакова Л. Д.//Нефтехим. синт. тех.
прогр. Уфа: УНИ, 1976. С. 122.
588. Сафронов В. С., Гладышев Н. Г Дубровина В. А., ПравдивцеваЗ. А.//
Хим. пром. 1983, № И. С. 9—11.
589. Лычкин И. П., Кокрева Л. И., Бутенко Т. Р. Ионный обмен и хромато-
графия. Л.: Наука. 1984. С. 150—155.
590. Юкельсон И. И., Бутенко Т. Р., Бердутин А. Я.//Лакокрас. материалы
и их прим. 1979. № 4. С. 9—13.
591. Воль-Эпштейн А. Б., Дьякова М. К., Суровцева В. В.ЦИзв. АН СССР,
отдел, хим. наук. 1960. № 12. С. 2230—2233.
592. Зейналов И. П., Бабаев А. И., Рзаев 3. ^.//Промышленность синтетиче-
ского каучука. 1981. № 12. С. 10—12.
593. Легачева В. В., Ржевская К. //.//Промышленность синтетического каучу-
ка, шин и резиновых технических изделий. 1985. № 2. С. 6—9.
594. Брянцева Ю. В. и др.//Каучук и резина. 1959. № 4. С. 32—35.
595. Курбанова Р. А., Алиева Д. Н., Шукюрова М. Б., Бабаева Л. М. Моно-
меры и полимеры: Тез. докл. научно-технич. конфер. Баку, 1983. С. 136—
139.
596. Ржевская К. И., Федотова Л. В., Школьник А. Е., Рассадин В. Г.//Нефте-
перераб. и нефтехимия. 1985. № 10. С. 26—28.
597 Марченко А. Н., Плавник В. П./ДЬ/м. пром. 1974. № 8. С. 54—56.
598. Фудзии Юдзи, Фудзиэ Коити, Кубота Х«росм//Мидзисёри гадзюцу 1978-
Т. 19, № 8. С. 719; РЖХ, 1982, 6И437.
237
599. Соркина В. И., Полубенцева М. Ф., Дуганова В. В., Липович В. Л//Хим.
пром. 1986. № 6. С. 17—21.
600. Ахмедова Р. 3., Лобкина В. В., Далин М. Л.//Нефтеперераб. и нефтехи-
мия. 1976. № 4. С. 34—37.
601. Gardos G., Horvath J.//Magy kem. Lapia. 1984. 39, N 5. P 229—233.
602. Грунин Г H., Гордаш Ю. Т., Василенко Г Д., Литовченко Н. Р.//Нефте-
перераб. и нефтехимия: Сб. тр./Киев: Наукова думка. 1981. № 20. С. 57—
64.
603. Кулиев А. М. Химия и технология присадок к массам и топливам. М.:
Химия, 1972. 384 с.
604. Грунин Г Н., Василенко Г Д., Тихонрук И. Ф., Панюхно В. Г. Химия,
технология и применение новых смазочных материалов: Сб. тр. ВНИИПК-
нефтехим. М.: ЦНИИТЭнефтехим. 1979. С. 21—23.
605. Василенко Г Д., Грунин Г Н.>, Строян Т. С., Марушевский Г Б. Синтез,
технология и применение присадок к смазочным материалам: Тез. докл.
II Всес. конфер. М.: ЦНИИТЭнефтехим. 1982. С. 179—185.
606. Василенко Г Д., Филинов Ю. П., Грунин Г Н.ь Мельник И. Ю. Синтез,
технология и применение присадок к смазочным материалам: Тез. докл.
III Всес. конф. М.: ЦНИИТЭнефтехим, 1982. С. 180—183.
607. Василенко Г. Д., Грунин Г. Н. Переработка и использование отходов про-
изводства присадок к смазочным материалам; Тем. обзор. М.: ЦНИИТЭ-
нефтехим, 1987. С. 51—55.
608 . Грунин Г. И., Гордаш Ю. Т., Василенко Г Д. Очистка особо загрязнен-
ных сточных вод производства присадок с утилизацией полученных про-
дуктов//Совершенствование процессов очистки сточных вод и подготовки
оборотной воды нефтеперерабатывающих и нефтехимических производств:
Тез. докл. Всес. научно-технич. совещ. Уфа. — М.: ЦНИИТЭнефтехим,
1980. С. 129—132.
'609 . Белов П. С., Гусев В. К-, Исангулянц В. И., Грачева В. И.//Хим. пром.
1969. № 9. 665 С.
•610 . Парфенова В. А., Ляпина В. А., Исангулянц В. Я.//Нефтеперераб. и неф-
техимия. 1974. № 3. С. 52—57.
611. Гордаш Ю. Т., Корбецкая В. И., Руденко Л. И., Пуствит В. £\//Нефтепе-
рераб. и нефтехимия: Сб. М.: ВО «Нефтехим», 1973. Вып. 5. С. 256—
258.
512. Копосов В. И., Гречко А. И., Шутова А. В. Утилизация шламов после
очистки присадок на основе дитиофосфатов бария//Вопросы химии и тех-
нологии смазочных материалов: Сб. трудов ВНИИПКнефтехим. М.:
ЦНИИТЭнефтехим. 1981. вып. 17. С. 113—116.
513. Карпов И. Ф.//Нефтеперераб. и нефтехимия. 1969. № 8. С. 21—25.
514. Антонов В. Н„ Генин Г. В.//Нефтеперераб. и нефтехимия. 1971. №9.
С. 15—18.
Производственное издание
С. С. НИКУЛИН, В. С. ШЕИН,
С. С. ЗЛОТСКИЙ И ДР.
Отходы и побочные продукты
нефтехимических производств —
сырье для органического синтеза
Редактор М. Б. ЛЕБЕДЕВА
Художник Е. В. БЕКЕТОВ
Художественный редактор К. К. ФЕДОРОВ
Технические редакторы С. Ю. ТИТОВА,
Н. Ю. БЕЛЯКОВА
Корректор Е. В. ФАРРАХОВА
ИБ № 2209
Сдано в наб. 31.10.88. Подп. в печать 06.03.89. Т-08396. Фор-
мат бумаги 60X 90’/ie. Бумага тип. № 2. Печать высокая.
Гарнитура Литературная. Усл. печ. л. 15,0. Усл. кр.-отт. 15,0.
Уч.-изд. л. 16,95. Тираж 2350 экз. Заказ № 627.
Цена Л-^к. *
Ордена «Знак Почета» издательство «Химия».
107076, Москва, Стромынка, 21.
Московская типография № И Союзполиграфпрома при Го-
сударственном комитете СССР по делам издательств, поли-
графии и книжной торговли.
113105, Москва. Нагатинская ул., д. 1.
ВНИМАНИЮ СПЕЦИАЛИСТОВ!
Издательство «Химия»
готовит к выпуску
в 1990 году книгу:
Темкин О. Н.г Шестаков Г. К., Трегер Ю. А. Аце-
тилен: Химия. Механизмы реакций. Технология. —
25 л. — 4 р. 80 к.
В книге описаны наиболее перспективные промыш-
ленные синтезы на основе ацетилена и его производ-
ных, применяемые при этом катализаторы. Подробно
рассмотрены кинетика и механизм реакций присоеди-
нения, оксихлорирования, карбонилирования, цикло-
и линейной олигомеризации, алкинольный синтез и др.
Предназначена для научных и инженерно-техниче-
ских работников институтов и предприятий нефтехими-
ской и химической промышленностей, а также для
студентов химико-технологических факультетов и ву-
зов.
В магазинах, распространяющих научно-техниче-
скую литературу, можно оформить предварительный
заказ на интересующие Вас издания.